/















































Текст
fl
идДАНИрЭДбЁННО-^ЕХНИЧЕСКОИ: АКАДЕМИИ Р. К.’
;лПроф/гАГд. СОЛОНИНА,
$й
-гй
ЛАБОРАТОРНОЕ ПРИГОТОВЛЕНИЕ ВЗРЫВЧАТЫХ ВЕЩЕСТВ.
лет
:&8
'*>‘-9ОКИ-
JOTB
аиче-
., збяе
Явкой
Пособие для практически» -звппли# в лаборатории.
ано и
и
ЛЕНИНГРАД
1О'>5
ПРЕДИСЛОВИЕ.
Предлагаемое пособие впервые появилось 20 лет тому назад, как результат заведывания практическими занятиями по приготовлению взрывчатых вещеотв в химической лаборатории Военной Электротехнической Школы о 1889 Г од г Посте этого это пособие переиздавалось в ХВхц нужд Артиллерийской
Академии. В настоящее время пособие переработано и дополнено получением новых взрывчатых веществ и новых способов.
А. Солонина.
I октября 1026 года.
ОГЛАВЛЕНИЕ.
Введение............................ •
Сложные эфиры азотной кислоты...........
Пироксилин. У...........................
; V. Нитроглицерин. /.....................
О.-Д7и и а м и т ы. у.....................
. ; □ >'' Динитро глицерин.-^-».............
IМоИохлордин1?троглицерин.............,. . . .
Динитроацетин..........................
i > ' Динитроф©рмин.л/-......................
t Тетранитродиглицерин ... ..................
Динитро гликоль.........................
. Тетранитропентаэритрит................
Нитроманнит. . .............. ..... .
Нитрокрахмалы..................... . . .
Нлтрооахара...................
Нитросоединения..................... .
Тринитротолуол (тротил)./...............
..ТринитрОЙСИЛОЛ (ксилил). . . . . .-.V. . . ’ Тринитрофенилметилнитрамин (-тетрил).'/. . .
Тетранитрофенилметилнит рамин (пентил)./. . Тетранитроанилин (Те четыре) ....... Пикриновая кислота ............ Пикраты..... . . . .....................
Тринитрокрезол .............. ..........
Тринитрокрезоляты......................
' .Тринитронафталин / . . . .............
Динитронафталин. . ............... . » . .
Нитробензол.^...........................
Динитробензол.....................
Тринитробензол . . . ....................
^ононитрофекол . ... . .............
Страница
I
2
2 '
8
10
14 .. .
18 ' "Т-
21 . <<(
22 .-А,
22- :
25 ’
25 : 'Т А
26 • . .
29 -
33 .4
,35. -А'
37
44 ,
45 Ч'
47
48- 1
49-
57- •
60-
62
63
65 с
66 а
68 ’ (П
69 U
70- . ’ . |в
Ркксанитоодифениламин ............... . . ,. 72-.
Гремучие соли........................... 75
Гремучая ртуть. . . . .... . . . . . . . . . 76
Гремучее серебро..........................78
Свинцовая соль азотистоводородной кислоты ... 79
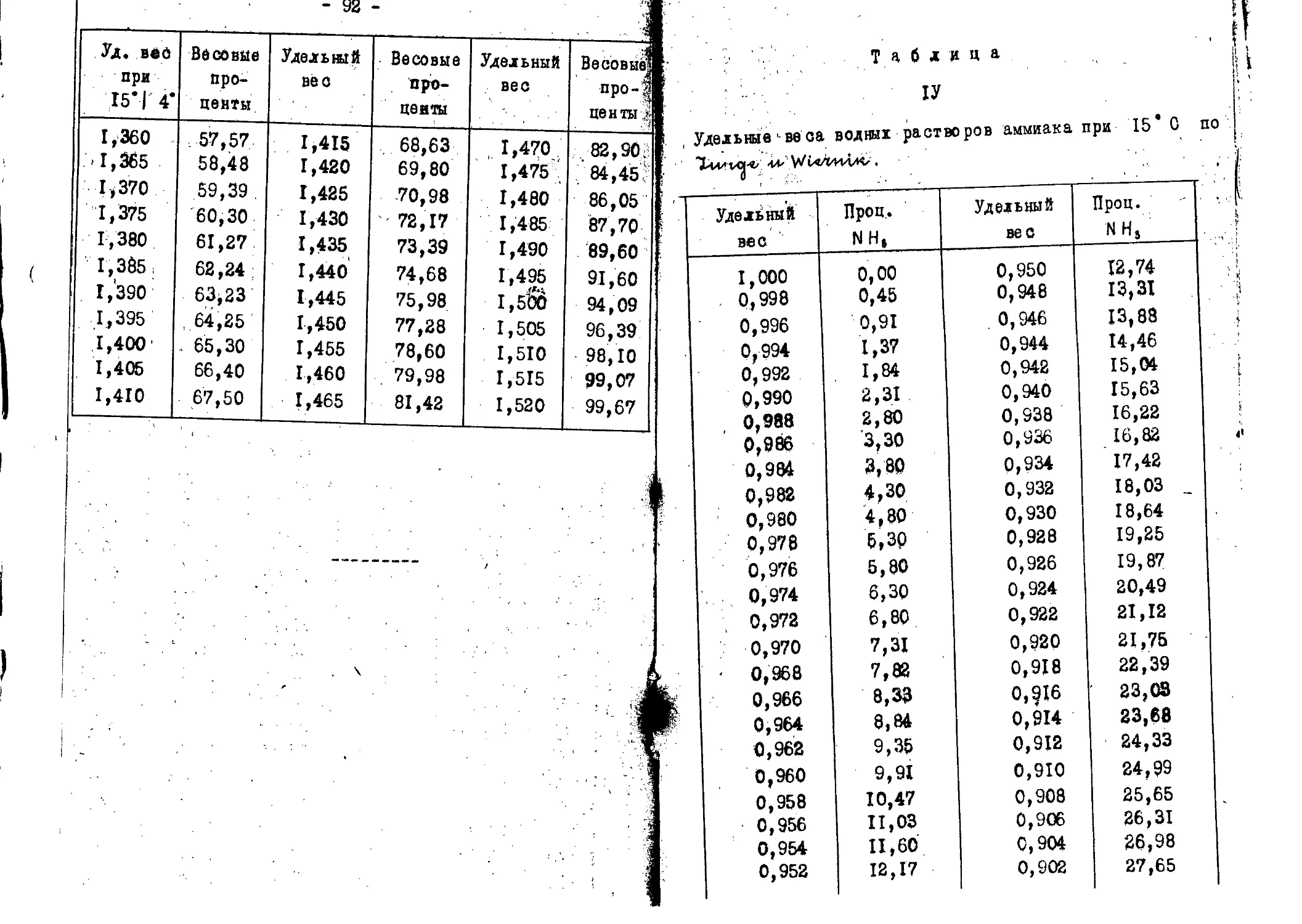
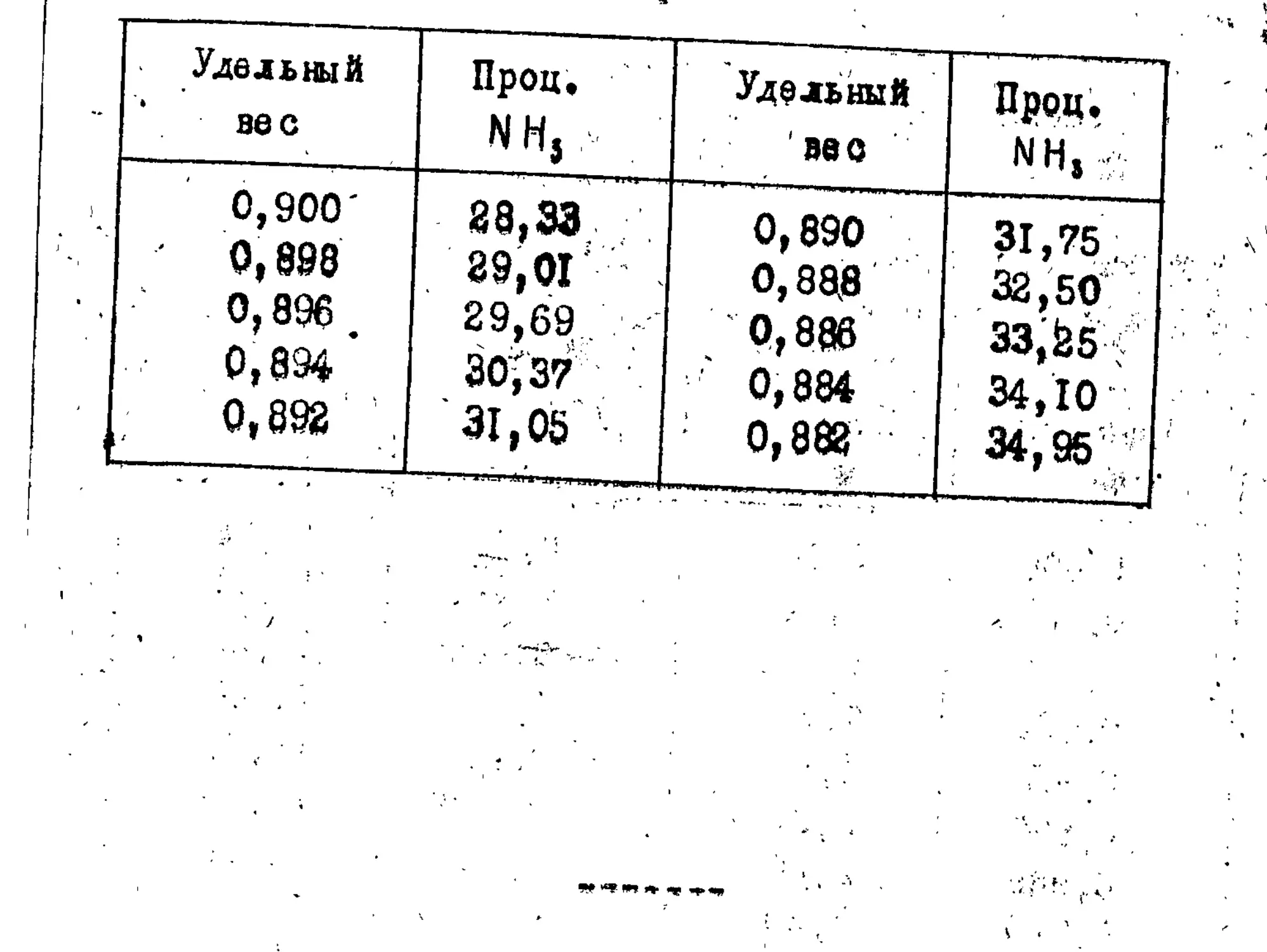
Таблицы...................................84
Ji
<1 >i
ВВЕДЕНИЕ. fl
Все употребляемые ныне в военной технике взрывчаты Я вещества можно разделить на 3 группы.
I-ая группа - механическая смесь тел, самих по себя не взрывчатых, взрывающая вследствие химического взаи.Я действия своих составных частей. Сюда относятся черня и бурые селитросероугольные пороха и т.п. '
П-ая группа - взрывчатые вещества, представляющие Я определенные химические соединения. Сюда относятся: пЯ роксидин, нитроглицерин, гремучая ртуть и т.д. Я
III—ън группа - смеси определенных химических соедЯ нений, т.е. П-й группы с другими взрывчатыми (I и НчЯ группы) или п не взрывчатыми телами. Сюда относятся 'Я взрывчатые желатины, динамиты, пикриновые пороха и т.Я
Здесь приводятся, главных образом, взрывчатые телаЯ П-й группы, которые в своей молекуле содержат одно в j® менно углерод, кислород и азот, так как эти вещества Ж имеют наиболее практическое значение. Из этого рода единений можно выделить 3 рода органических веществ:.»
I)сложные эфиры азотной кислоты, К
2)нитросоединения и Ж
3)гремучие соли. №
Так как в последнее время уже начинает йрименятьс!® свинцовая соль азотистовородоной кислоты, как замена,шН гремучей ртути, то поэтому в конце мною указано ее щИ лучение, а также и исходного продукта - азида натрия®
Последнее вещество, как не содержащее углерода нед подходит под вышеуказанные группы. |И
Проф.Л.А,Солонина - Лаборат.приготовл.вар.вещ. Л.|Я|
f WI
- a -
слохные э^йры азотной кислоты.
Эти эфиры получаются действием азотной кислоты на органические соединения, содержащие в молекуле гидрожси-лы (водные остатки ОН,характеризующие спирты), причем водород в гидроксила замещается группой нитро NO^ т.е. из спирта 1L(ОН) мы получаем сложный зфирК(0Л10г),От сюда видно, что максимальное количество групп 1М0г в сложном эфире зависит от максимального количества групп ОН в данном спирте. Чем больше в -сложном эфире групп Ногтем большими взрывчатыми свойствами обладает тело, следовательно, наиболее развиты взрывчатые свойства у полных эфиров многоатомных спиртов.
ПИРОКСИЛИН. (Расчет на 5 гр.ваты).
Пироксилин получается действием азотной кислоты HN05 на клетчатку(СьН|О05)х,. Для лабораторных опытов можно брать вату; для усиления действия азотной кислоты вси завляют серную кислоту Нг50, дейст вие серной кислоты про не всего обгоняется, тем, что последняя удаляет воду из сруга взаимодействия азотной кислоты на клетчатку; в з&-»исимости от крепости кислот, их отношения к количеству 1летчаткж. и т.п., получаются пироксилины, содержащие различное количество азота.
’Приняв по Вьелю т.е., действие азот
юй "-кислоты можно’выразить следующими уравнениями:
’t- ^Н,вОм + ^НМбГ;.СмН„О,(ОМОДгПН4О ; , j
-A'/;’' - ' -• - Л-, " •’ -'A " Й'Г ’-‘'д
Лабораторное приготовление рахлого пироксилина мы будем;* ю ^возможности, вести1 в тех условиях, как это производится на русских заводах; опишем производство 2-х сортов пи-юксилина: нерастворимого и растворимого, смесь которых
в настоящее время употребляется для фабрикации бездымн го пороха на наших заводах. За образец мн взяли фабрич ное приготовление пироксилина на Охтенских пороховых з водах И'изменили детали, сообразно лабораторным услови ям. Все лабораторное производство пироксилина можно ра| бить на следующие пять операций:
Пподготовка хлопка (в данном случае ваты), 2)приготовление кислотной смеси (азотной и серной), 3)Нитрование хлопка, 4)промывка пироксилина и
5)сушка пироксилина.
Подготовка хлопка.
Вата, которая употребляется для приготовления рыхло^ пироксилина, тщательно перебирается руками для отделен случайных примесей. Хорошо брать для этой цели белую г грбскопичвекую вату. ,
Вата сушится непрерывно в продолжении 2-3-х часов в сушильном шкафу при температуре около 100 С; при сушке' вата кладется тонким слоем в фарфоровую чашку или друг' открытый сосуд. Охлаждают высушенную вату в эксикаторе над серной кислотой в течении часа. .
Приготовление кислотной оде си.
Во время сушки модно приготовлять кислотную смесь. . счет будем вести на 5 гр. ваты. Чтобы получить нераств!
' римый пироксилин, на Охтенском пироксилиновом заводе.б< рут смесь, содержащую .моногидрата HNO}- 21,5% по весу, .- моногидрата - Нг50^- 70,5% по весу и воды 8% по: весу;
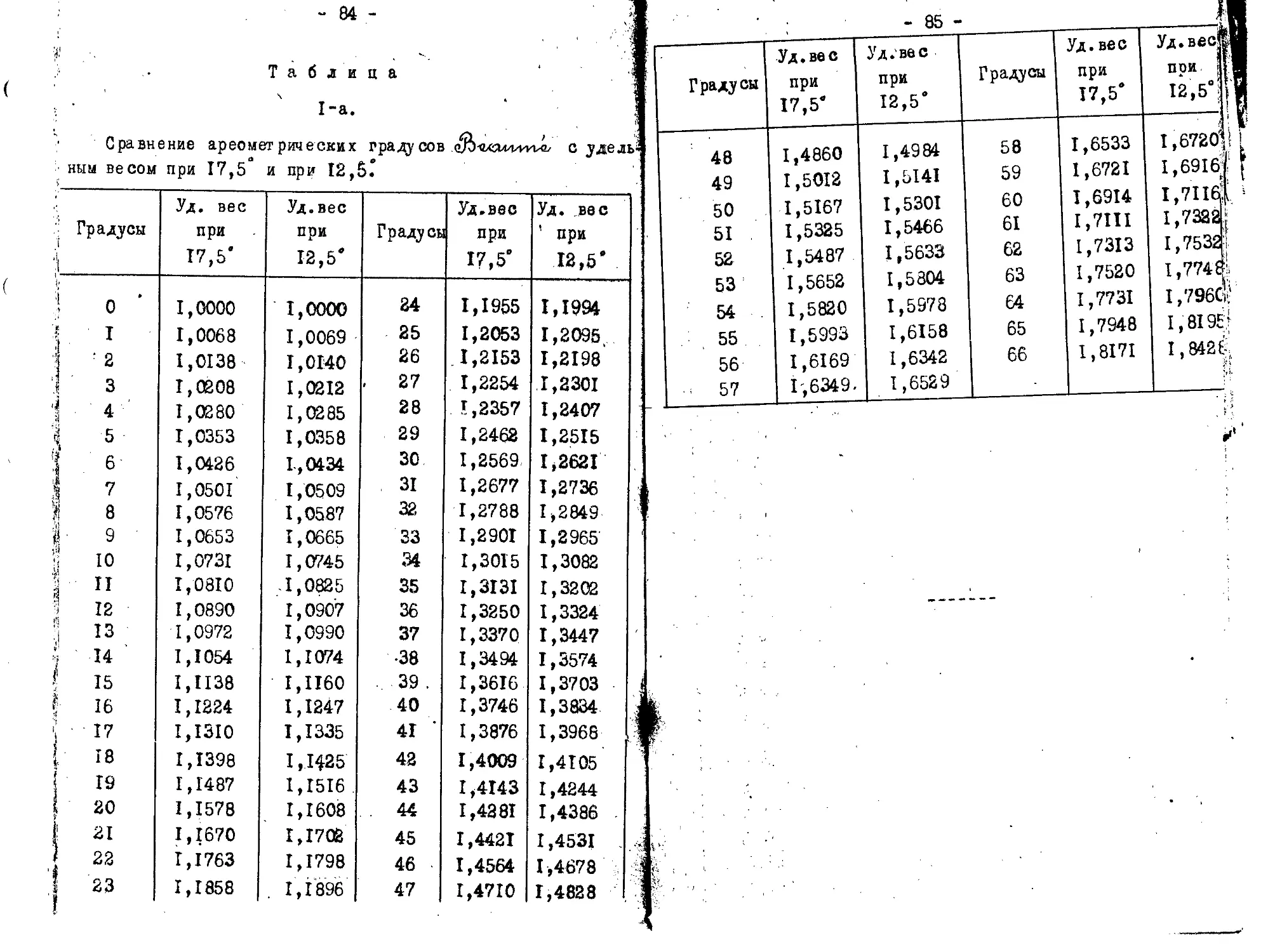
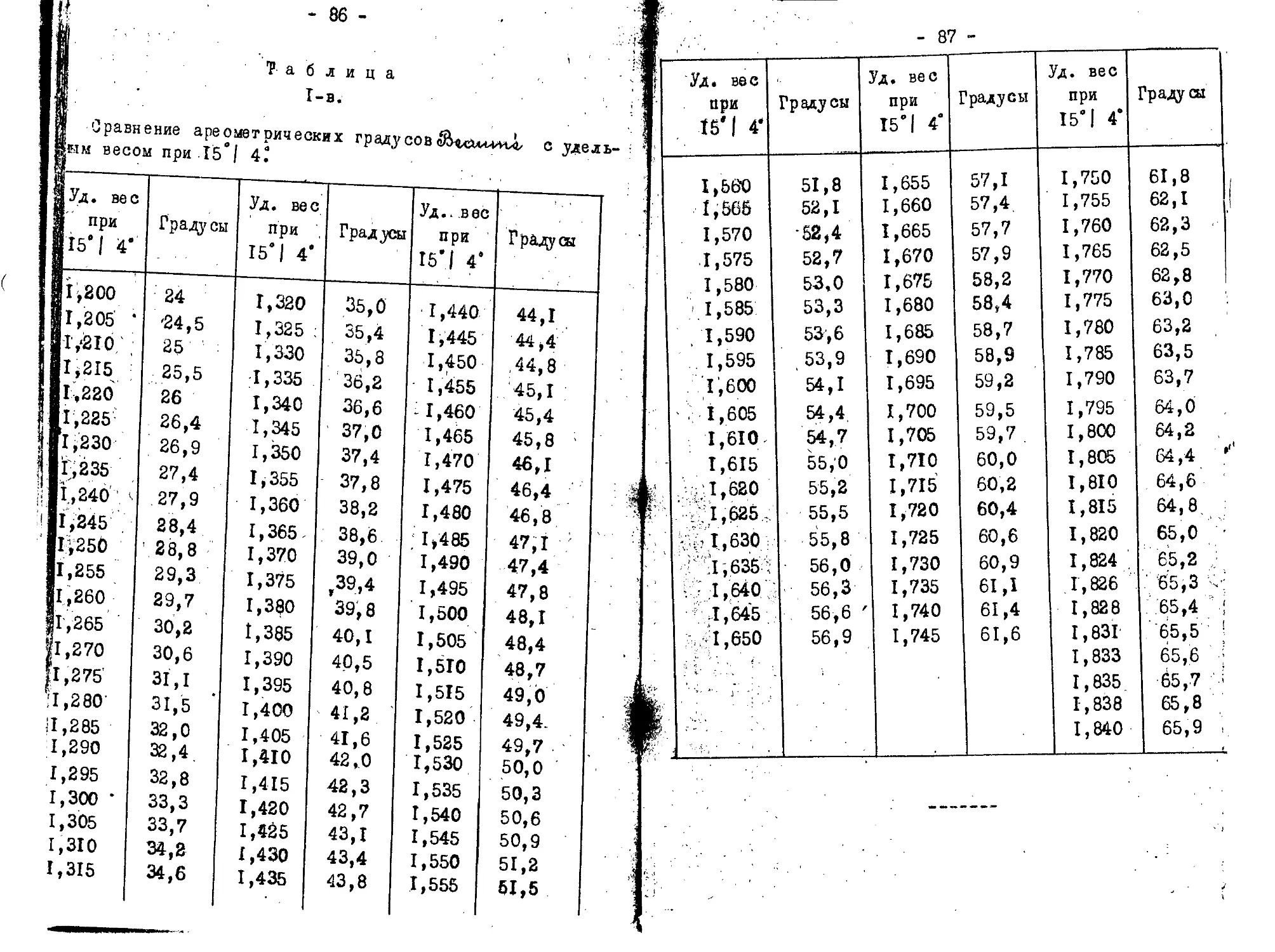
для растворимого пироксилина 18.С моногидрата НЫОЭ, 6$ моногидрата 11,50., и 14% воды. Смеси же берется на. за: де I килограмм (при работ© с небольшим количеством хло: г/ ка можно брать меньше). Расчет кислот производится nod. /определения удельного веса их при помощи таблиц (смотр:
в конце). Удельный вес кислот обыкновенно быстро оппеж
- 4 -
летая njhi помощи 1)весов или менее
чно 2) при помощи ареометра.
Веси e^Lo'tvtz-Wii.s^otvcvEz'ju,
Коромысло этих весов находится в равновесии, когда двешенный к нему, с помощью очень тонкой платиновой про лови, поплавок, состоящий из маленького термометра,сводке висит в воздухе.
Когда весы уравновешаны, острие противовеса точно сов .дает с острием штатива, при этом необходимо, чтобы осно ^ние весов было вполне горизонтально, что легко достига-ся с.помощью особого винта. Для определения удельного ’Са жидкости спускают в последнюю поплавок, которых конт >лируется одновременно температура и восст анавляют равно->сие помощью коньков.К прибору полпгается 4 конька. Каж-1Й из 2-х больших коньков равняется весу вытесненной по-гавком диет илиро ван ной воды в 15 С; средний по величине )нек весит в десять раз меньше большого, а самый малый сит в сто раз меньше большого. Подвешенные на одном из глений коромысла коньки показывают соответственное этим злениям число десятых, сотых или тысячных долей; часто потребляется еще конек для десятитысячных долей.
Нарушенное, при погружении поплавка в дистиллированию воду, рановесие весов восстанавливается, если подвесить большой конек, В этом случае весы показывают 1,000, .е., удельный вес воды. При жидкостях, которые плотнее оды (т.е. в данном случае) пользуются, двумя большими оньками, один большой конек подвешивается к крючку коро-ысла и устанавливается затем равновесие другими конька* и; если, напр., большой конек находится на 8-м, средний а 4-м и малый на 6-м делении,, то удельный вес жидкости-1,846; если большой и средний - на 6-м И малый на 9-м елении, то удельный вес х 1,669 и т.д. При жидюстях ме-ее плотных, чем вода употребляется лишь один большой коми и ставят перед запятой нуль; если, напр., большой ко-
"5 - "I
нек на 9-м, средний на 1-ом и малый на 6-м делениях, гоя удельный вес -- 0,747 и та. Для получения точных резулья татов при употреблении весов <уЦойл, МмЦэКслЖ'л, необходим мо, чтобы вместе с поплавком погружалось в жидкость н<в только закрученная часть платиновой проволоки, но и рав-Я ная ей 'по длине прямая часть. Этому условии легко уд о в лея ворить, отвертывая особый винт и перемещай верхнюю частгЯ весов выше или ниже. Для окончательной установки подклаЛ дывают под цилинд несколько листов бумаги. По окончании:» опыта обмывают водой поплавок и платиновую проволоку, с которой следует обращаться весьма осторожно и обсушиваю:» их с помощью пропускной бумаги, Я
Описанный поплавок не имеет определенного веса и об”«Я ма и смотря по большей или меньшей величине, вытесняет Я при погружении в жидкость большее или меньшее количеств^ иоследней. Вследствие этого каждый поплавок требует осо,В бенного разновеса, пригодного только для этого поплавкаЖ и, в случае поломки последнего, является необходимость И новом разновесе и в новой выверке корошсла относительна» противовеса. Когда определен удельный вес, то по таблиц® (в конце записок) легко определить % содержание моногид® ратов серной и азотной кислот. Ш
Рекомендуется осторожность при наливании и выливании® кислот из стеклянного цилиндра, так как брызги кислот,п®| падая на коромысло или на коньки, раз”едая их, изменяют® вес и таким образом производят трудно поправимую порчу iAi со в. ' К
Ареометры. ж
Ареометры бывают 2-х родов: с постоянным весом 1 с Я стоявным объемом. Аремометры с постоянным весом, которьИ чаще всего применяются, следующего устройства. ш
Они представляют из себя полые стеклянные цилиндры, И которым сверху прикреплены трубки со шкалой делений; п]К чем шкала делений ареометра для жидкостей, б<И
- 6
iee тяжелых, чем вода, приготовляется так: нуль излучается погружением в чистую воду, точка десять - погружением в 10% раствор поваренной соли при 17,5 С*;промежуток между нулем и десятью делится на десять ровных ча-•.тей, при чем деления эти продолжают дальше вниз за девятью. В ареометре нуль такой, как и•
о цифра 30 ставит-ся при погрудении в жидкость уд. в. |,850; одна тридцатая есть величина градуса и для жидко-.той тяжелее воды. Эти градусы наносятся вниз. В ареомет-е употребляемом в Аигдии, одному градусу
соответствует увеличение плотности на О,COS* В нормаль-ых ареометрах непосредственно читается уд. вес/ так как нигприготовляются погружением в ряд жидкостей, удельный ес которых был предварительно определен. Так как в од-? ой • и той ie жидкости ареометр может в зависимости от е'температуры, дЯть различные показания, то поэтому на реометре всегда обозначается температура, при которой го показания справедливы и, следовательно, надо всегда д ост свариться, при помощи термометра, в том, что темпе-дтура жидкости надлежащая; если же нет, то надо повы-дть или понизить погружением цилиндра, где налита испы-,уемам жнднэсть, в сосуде с теплой или холодной водой. : Пример расчета приготовления кислотной смеси.
Если Q. количество данной серной кислоты, содержа-эй сь % моногидрата Иг50„,
- количество азотной кислоты, содержащей в % мо-ргидратаН>Ч04
, Л - % содержание моногидрата Н950А требуемое для при-зтовления кислотной смеси,
- % содержание моногидрата НNO, для приготовления юл от ной смеси, )гда можно написать следующие уравнения
. CvGt ;=Jl(Q+Cp . . . . • 0) .
..............(%)
- 7 -
Так для нерастворимого пироксилина: ^ = 70,5% ,
то если взяли 100 гр. серной жслоты, содержащей 95,6% моногидрата HXSO^ или уд.в. 1,84, то по уравнениям (I) и (2) вычисляется, что надо прибавить 35,6 гр. азотной кислоты, с содержанием моногидрата 81,8%, что соответ-> ствует уд.в. 1,463; для приготовления же I кгр. смеси 737 гр. серной кислоты, уд.в. 1,84, и 263гр. азотной кислоты, ул.в. 1,46.
Нитрование хлопка. "
Разрезанная на возможно мелкие кусочки навеска ваты (5 грамм) опускается постепенно в стакан, куда налита охлажденная до комнатной температуры кислотная смесь, и затем'перемешивается стеклянной палочкой. Вата, напитан-, ная выше указанным количеством кислот, покрывается затеУ стеклянной пластинкой остается стоять в стаканчике: 12 часов - при приготовлении нерастворимого и 4 часа при;' ' приготовлении растворимого пироксилина. - ‘ ' i
Промывка пироксилина.
i
После нитрации пироксилин, насколько возможно, от ди- 1 мается от кислот и уже после этого подвергается прошв-1 ?е. Сначала промывают холодной водой в течении '4 часа. Лля этого пироксилин перемещают в большую стеклянную чая ку или стакан и пускают в сосус с пироксилином из под крана, при помощи каучуковой трубки, небольшую струю колодной воды; веда все время меняется, так как к посуде приспосабливается сифон. Во время промывки рекомендуется . помешивать пироксилин стеклянной палочкой.
После холодней прсмывки пироксилин отжимается и про- швается горячей.водой. Лля этого в колбе или лучше в ; жестяном цилиндре нагревают воду до кипения и пар при помощи трубки проводится в стакан или коническую колбу ’ с пироксилином, в которой раньше прилито такое воличест
- 8 -
> води, чтобы весь пироксилин был покрыт водой.
Горячая промывка употребляется 8 раз; I-я, 7-я и 8-я .з всяких примесей, а при 2-ой, 3-й, 4-й, 5-й и 6-й помывках прибавляется в коническую колбу, где произволен промывка, по 0,2 грамма (расчет на 5гр. пирокси-хна) соды. Пропускание пара продолжается за раз не ;нее Г/г часов; затем вода выливается из колбы, насыпа-ся, если нужно 0,2 гр. соды и опять пропускается пар
;Т.Д. v
Сушка пироксилина.
! Вода отжимается,насколько возможно, ручным прессом пироксилин кладется между листами пропускной бумаги| и мшится в сушильне при температуре не выше 50*0. Надо шить не менее 6-8 часов до постоянного веса. Охлаада-сяв эксикаторе над серной кислотой и взвешивается. :ход пироксилина при этом обыкновенно получается та-й, что из 5 граммов ваты получается около 8 граммов :рок силина.
НИТРОГЛИЦЕРИН.
( Расчет на 10 гр. глицерина ).•
сА(о н),+3 н N оъ=CSH « (о N 3 l\0
Глицерин С3Н$(ОН)5 уп стреляющийся для приго->вления нитроглицерина, должен быть свободен, по воз-ожности, от всяких примесей. Отсутствие жирных кислот ^называется отсутствием черного осадка от прилития )%-ного раствора ляписа, (в количестве равном об"ему шцерина; берется глицерина не более 2 к.с.); при чем ,1створ ставится в темноту на 10 мин. Допускается кОли-jjcTBO минеральных Примесей не более ОДХ»
Кальциевы соли (проба щавеле во аммиачная соль(МН^хСгОч), серная кислота и ее соли (проба хлористый барий ДОС) и хлористые соли (проба ляпис Jbc|NOi') совершен-
- 9 -
но не допускаются в глицерине. а|
Удельный вес глицерина должен быть близок к 1,26, при Я 15 С, т.е. содержание воды не более 2-3$. Если же он ме- Д нее, вследствие большого содержания воды, то повышеяие Я удельного веса достигается нагреванием до требуемого дД удельного веса при температуре 130. .Я
После поверки удельного веса и чистоты глицерина, при.» готовляется смесь азотной и серной кислот. Обыкновенно ,а берется на 3 весовых части азотной кислоты HN03 (удельное ‘ го веса I,5Q) - 5'весовых частей серной (уд.в.1,84), прите • чем на I часть глицерина требуется 8 частей кислотной ® . смеси, следоватахьно, при обработке Югр.глицерина надо Л« 30 гр. азотной кислоты и 50гр. серной.
Серная кислота вливается тонкой струей в стакан, где ® находится азотная кислота, охлажденная снегом или льдом® до О‘С; .при чем во время приливания серной кислоты произЯ водится помешивание смеси стеклянной палочкой. Когда ж смесь остыла до 0*С, то начинается приливание отвешен но-Д? го количества глицерина из капельной воронки в стакан, ко торый охлаждается снегом или льдом (лучше еще прибавить».' немного соли); приливать надо глицерин по каплям и.все, И время наблюдать за термометром, чтобы температура была выше 5’0. Если же температура поднимается выше, то перс®, стают капать и отходят от вытяжвого шкафа, где лроизво-Ж ' дится приготовление нитроглицерина, и предупреждают при^ж-сутствующих об опасности взрыва. Во все время капания Ж; смесь надо помешивать стеклянной палочкой.
Когда кончилось капание, надо до тех пор помешивать ж палочкой, пока смесь не примет температуру около 0*С. З.'Ж тем всю смесь из стакана выливают в сосуд, с большим кол/9 чеством холодной воды и продувают,для лучшей промывки,в S дух через нитроглицерин, опустившийся на дно сосуда. За ® тем, когда нитроглицерин отстоится, повторяют эту опера® цию, смывши предварительно большое количество воды сифож ном. Промывка эта производится около 10 раз (пока синяя ж
- 10 -
акмусовая бумажка не перестанет краснеть). Затем окон-ательно Промывают нитроглицерин IX раствором соды. Потом пять проиявают чистой водой для удаления соды, красная акмусовая бумажка не должна синеть ц на разделительной >ронкв отделяют нитроглицерин, который затем фильтруют; яя высушивания на фильтре кладут порошок, предварительно рока'ленной, измельченной и остуженной позареной соли.За--.а, переливают нитроглицерин в маленький стаканчик, ста-IT его в эксикатор, из которого затем выкачивается воз-,х. Нельзя пользоваться эксикатором с серной кислотой,а эязательно - эксикатором с хлористым кальцием. Все сосу-I, где могут остаться капли нитроглицерина, надо мыть ^очищенным древесным ( СН?ОН, метиловым) спиртом. Вы-эд нитроглицерина должен быть около двойного веса взято-э глицерина.
АННАМИТЫ.
Динамитом назвается, смесь какого либо пористого тела, эторое называется поглотителем (основанием), с нитрогли-зрином. Смотря по природе поглотителя, динамиты делятся 1 2 рода: на динамиты с недеятельным основанием ж с дея-иьным основанием.
1-й род динамитов - этой такой, у которых поглотитель > изменяется при взрыве; сюда относятся, напр., смесь эемнезема с нитроглицерином. Ко 2-му роду относятся та-че динамиты, у которых при взрыве поглотители учавствуют разложении, напр., смесь пироксилина с нитроглицерином.
Аннамиты с недеятельным основанием.
♦
; Пр и г о т о в л е н и • кизельгур диамата Н о б • л я (три сорта). Этот динамит полу ;втся поглощением нитроглицерина кивельгуром. Кизельгур * (оловянное сито в Змм. это очень пористое тело, предоставляющее обломки кремни- ' : ых оболочек инфузорий в продаже кизельгур
-п- . и,
встречается в виде комков и содержит 25-40% воды. Надо прежде всего кизельгур прокалить в фарфоровом тигле до ! красного каления; прокаливание производится до постоянно!! го веса. 1
Остывший в эксикаторе над серной кислотой кизельгур I измельчается в фарфоровой ступке и просеивается через би-| то с Отверстиями в 1,5-2мм. Затем берут чистую соду, про-|
IP ..
| ' каливают в тигле до постоянного веса, измельчают в фарфо-
ровоЙ ступке и тоже просеивают через сито с отверстиями
1,5-2мм. В ступке насыпают 24,5- гр. кизельгура И 0,5г₽*р соды, хороиенько перетирают пестиком и постепенно прилИ-. вают 75гр, нитроглицерина. Перемешивают стеклянной палбч-кой и протирают руками в каучуковых перчатках через оловянное сито с отверстиями в Змм.
Так получается I-й сорт,
Надо взять для 2-го сорта: нитро глицерина......... 50гр.
кизельгура. ....... .49,5гр.
о д ы.................0,5гр.
ад
для 3-го аорта: j
...............35 гр.
..............64,5гр.
...............0,5rp,j
с
j
Динамиты с деятельным основанием.
i
г“ч
Н о б ей
П
я
I
риготовление динамита
№2. Калийная селитра KNO3 измельченная в порошок,I сушивается до постоянного веса при IOOJ точно так же из- Ь мельченный древесный уголь сушится при ЮО’С. Затем, се- 11 литра и уголь, отдельно просеиваются через сито с Г4-2мм.‘| | отверстиями; отвешивают калийной седитры 71 гр. и угля О 10 гр.; отвешаиные вещества насыпаются в фарфоровую ступ-0 ку и тщательно перемешиваются с I гр. порошкообразного п«Ш.’ раффина. Затем, в ступку к смеси постепенно приливается 18гр. нитроглицерина, и все тщательно перемешивается и затем ружамж, в каучуковых перчатках, протирается через
> отверстиями.
- 12 -
Пржготовленйе гремучего студия.
Отвешивается 9,6гр. каллодмоиного хлопка, возможно лучше измельченного ж высушенного сначала при 40”я потом над хлористым кальцием«или серной кислотой до постоянно-го веса (в эксикаторе). v
В ступку кладут 4 гр. камфоры и тщательного пестиком расширяют хлопок с камфорой и затем смесь кладут в фарфо ровую чашку, которую нагревают осторожно на водяной бане до 70 С.,К смеси приливают постепенно 86,4гр. нитроглицв' рина' и, при нагревании смеси не выше 70J размешивают стеклянной палочкой, пока смесь не примет однообразной вид, для его нужно несколько часов.
! Безопасные для рудников нитроглицериновые взрывчатые вещества.
, В угольных рудниках возможны несчастные случаи, так ак могут после взрыва происходить пожары от взрыва медиа, угольной пыли и даже воспламенения пластов камеино-о угля.
По опытам произведенным во Франции надо для безопас-ости производить ’взрывы с такими веществами, чтобы зрывчатые вещества не выделями горючих продуктов при зрыве и чтобы не развивали температуру выше I900J если зрыв производится в породе при образовании галлерей и е выше I500J при взрыве в слоях камааного угля, К таким зрывчатым веществам относятся: 1)гризутины, 2)гризутиты, )карбониты и 4)желатиновые динамиты.
I) .!1 р и з у т и н ы. Гризутинами называются смеси с 'ммиачной селитрой. Например, во Франции применяется месь 11,75% нитроглицерина, 0,25% коллодионного хлопка, 8% аммиачной селитры; теитература при взрыве 1432.
2) . Гризутиты. Гризутитами называются смеси !' солями, содержащими кристаллизационную воду. Например,
- 13 -
в Бельглии применяется смесь из 44% сернокислого магния сЛ4^5ОА.7Нг0 ( 44% нитроглицерина и 12% целлулозы. В Германии - „ W смесь 52% нитроглицерина,13%
кизельгура и 35% кристаллической соды Nc^COj 1онгО.
3) . Карбониты, Серые смеси, подобные хлебному тесту, с мукой и различными солями. Например, по ейфскчХ'и карбонит^ I,- смесь 25% нитроглицерина, 30,5% нитриевой релитры, 39,5 муки и 5% двухромокислого кали.
Карбонит № 2. Смесь 30% нитроглицерина, 24,5%.натриевой селитры,40,5% муки и 5% двухромокислого кали.
УголМЫй карбонит, , Смесь 25% нитро-,;
глицерина, 34% калийной селитры, 1% баритовой селитры,. 1 38,5% древесной муки и 0,5% соды.
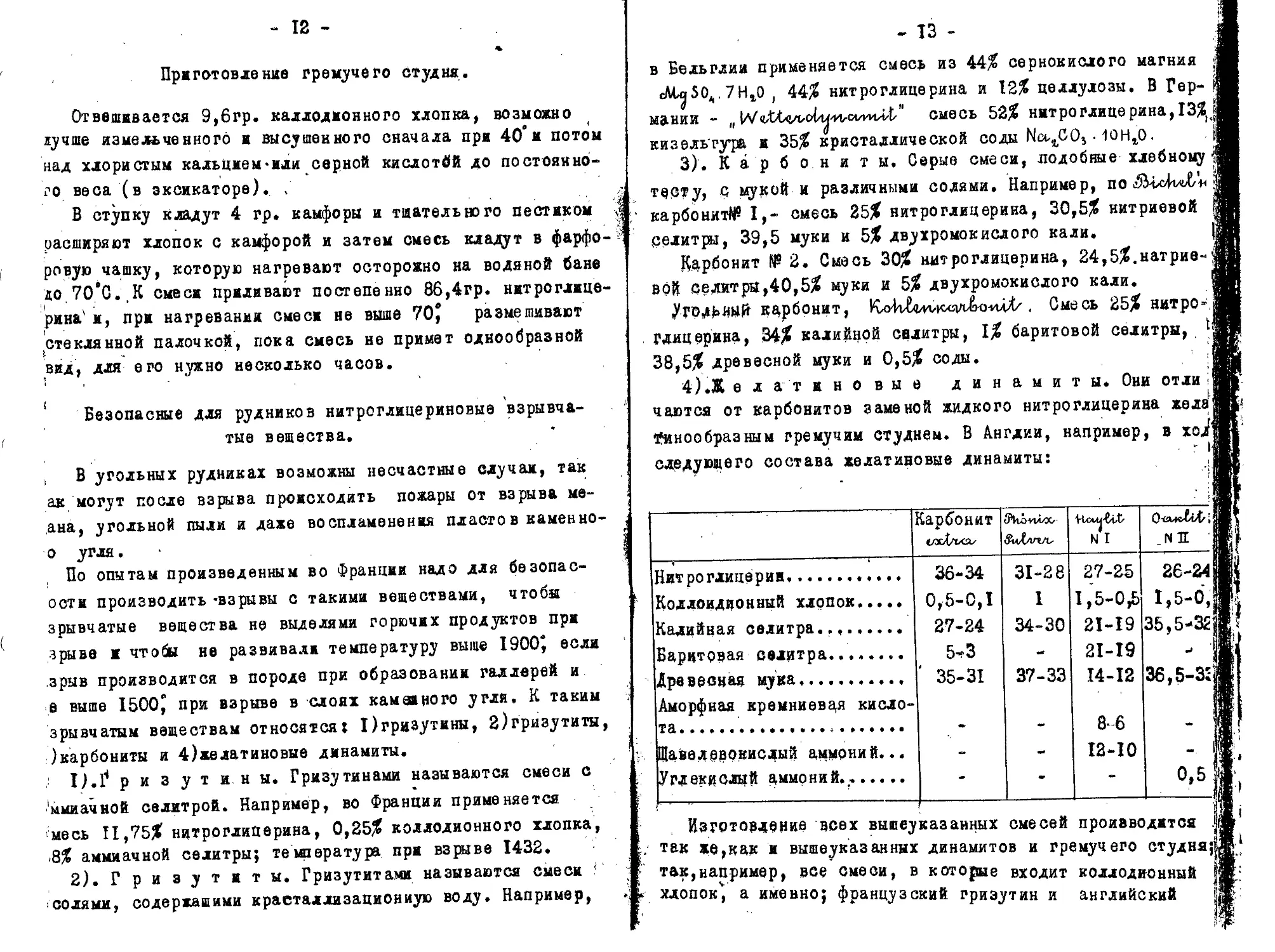
4) .Ж е л ат ивовые динамиты. Они отли ;! чаются от карбонитов заменой жидкого нитроглицерина желА1 Аинообразным гремучим студнем. В Англии, например, в хо/ следующего состава желатиновые динамиты:
{арбонит wcX/ixo/ SuX/in/is HcMj-Cvt N I _ N н !
Нитроглицерин, 36-34 31-28 27-25 26-241
Колдоидионный хлопок 0,5-0,! I 1,5-0,5 1,5-0,
Калийная селитра.., 27-24 34-30 21-19 35,5-32
Баритовая селитра. 5-3 21-19 «*
Древесная мука, 35-31 37-33 14-12 36,5-3;
Аморфная кремниевця кислота . * 8-6 — 1
Щавелевокислый аммоний... - - 12-10
Углекислый аммоний.. - • - 0,5
, Изготовление всех вышеуказанных смесей производится I так же,как и вышеуказанных динамитов и гремучего студня;|; так,например, все смеси, в которые входит коллодионный || хлопок, а именно; французский гриэутин и английский |
- 15
- 14 -
moBMoi так: сначала высушенный и измельченный коллодионный хлопок растворяют в нитроглицерине при нагревании на водяной бане не выше 70J а после полного растворения прибавляют постепенно высуженные и тон-
iKO измельченные смеси'солей и других веществ, просеянные через сито, при чем смесь все время нагревается не выше 70 и хорошо размешивается,
। • ДИНЙТРОГЛИЦЕР ИН,
C.H.fOHXONO,),
’ с1н!(он)ггн«о,=с,н!(онХо«о,),пн11)
(от 15 Августа 1903г.), динитроглиперив получается следуя щим образом: 10 весовых частей глицерина уд.виса 1,2$2 при охлаждении, смешиваются с 33 вес. частями азотной кислоты, уд.веса 1,50? лучше всего медленно приливать азотную кислоту в глицерин, при непрерывном помешивании? сначала образуется, главным образом, мононитроглицерин:
С3Н5(0Н), + НМ0}=С}Н5[0Н)г(0Н0^ +нго,
.который, при стоянии смеси в течение нескольких часов (до отаточно два часа), при температуре около + 10* 0, передо дит в динитроглицерин:
C^^O^^ONO^-HNOj-CjH^OHXOMOj^H^
После окончания нитрации разбавляют смесь 10-ю высовы-
ми частями холодной воды, нейтрализуют порошкообразным J углекислым кальцием (лучше всего известковым шпатом) до тех пор, пока жидкость не Примет удельного веем 1,58;,тог-Ча. динитроглицерин всплывает, его отделяют при помощи раз-Целительной воронки, промывают слабым раствором соды до ? Целочной реакции, затем чистой водой до нейтральной реак- ' ции:и сушат в эксикаторе.над хлористым кальцием. j ' _
Другой способ получения - несколько позже был вправо- ‘ >
в Неу б абель с бе р ге (фра нц. пате нт
3524486, e/Lict'. ^-«/S . vo-vviz. iVoLtb?' & •) |
поэтому способу глицерин при 18°- 20°С нитруется 4-7 ве4 сов, частями кислотной смеси, которая в 100 весовых ча•• j
стях содержит 8-12 вес. частей воды, 60-70 вес. частей моногидрата серной кислоты ^,,50^ и 15-32 вес. части моногидрата азотной кислоты НМО3 Я
Привожу примеры получения динитроглицерина по втому .Я]
СПОСОбу (Цр-ve. VtHv, ‘Diz. Sicovte^ t oUaXtez ]^И
жр>,э. w) S
J-й „пример: 100 грамм глицерина постепенво при Ж® помешивании, вливают в 500 гр. смеси: * Ж.
10% \0 *2215‘»Н<М03 + 67,5”»
наблюдая, чтобы температура была 18-20’С; маслянистой •ми’ жидкости или совсем не выделяется, или очень малое коли;|К чество? в последнем случае это исключительно нитроглице131 рин (тринитроглицерин); по отделении масла, кислотная смесь, в которой находится динитроглицерин, разбавляет-Ж ся водой; воды берется в 10 раз более по об”ему, чем ки’Ж* плотной смеси; при таком разведении водой, кислоты неЦ • , действубт на экстрактирующее веществе. Л
. После разведения водой, извлекают динитроглицерин !|р эфиром и этот экстракт нейтрализуют углекислым кальцием;':»’ после отгонки эфира динитроглицерин представляет масло-ML образную жидкость светложелтсго цвета удельного веса •|!J
около.1,5, лишь со следами тринитроглицерина. Можно такЦ же поступать иначе, а именно, раствор динитроглицерина Ж” нейтрализуют углекислым кальцием; выделившийся сульфат р. Ссб50йотфильтровывают на фарфоровой воронке Бухнера; ней® тральный раствор извиеяают эфиром к перегоняют под уменч ; шейным давлением. Зыход динитроглицерина около 60% Tool : ретичемкого, т.е. из расчета по уравнению: й
- - ^hXohX^hno^^hXohXonoJ.^k^ i
- 16 -
Можно брать и другие количества кислотной смеси, например, следующего состава (расче* на 100 грамм глице-эа):'
2) 400 гр. кислотной смеси, состава:
87. НгО+50,77. HNOj+61,37. Н45(\ .
3)700 гр. кислотной смеси состава
П7.Н4О*17,б7.НМ0}*70Л7.Н150Д(
Извлечение двунитроГлицерина эфиром можно заменить хло эоформом, бензолом или бензином.
Еще позже профессор Wi/U дал новый способ получения 'I Мая 1904г. патент I7575I) динитроглицерина: сна чала получается нитроглицерин (тринитроглицери) - ТОО гр. нитроглицерина размешивают с 1000 гр. серной кислоты, со держащей 70# моногидрата Мг504 пока не получится прозрач яый, или почти прозрачный, раствор. Процесс растворения идет без заметного выделения продуктов разложения (без выделения бурых паро в)Нераствори вшиися тринитроглицерин отделяется и прозрачный раЪтврр после достаточного охлаждения вливается в 4-5 литров холодной воды, затем извлекают эфиром, нейтрализуют й отгоняют эфир.
В последнее время и Novels (10 Ноября 1906г.
немецкий патент) дали очень удобный способ приготовления динитроглицерина: 92 гр. глицерина, удельного веса 1,26, смешиваются с 200гр. серной кислоты уд,‘веса 1,84, при чем смесь охлаждается до 15-20*0;. затем, приливается,при охлаждении до.30°0, азотная кислота, удельного веса 1,48 в количестве 150гр.; по окончании нитрации охлаждают смесь до 20* и оставляют смесь стоять, пока она, медленно нагреваясь, не примет температуру 30-35; тогда влива-(ют в воду, взятую, в двойном или тройном o6”eMe, ив раст • вора извлекают динитроглицерин эфиром или другим растворителем (хлороформом, бензолом или бензином).
- 17 -
_ й
Свойства динитроглиперина. J
e<Jc|e€o-544/ito^fc ,Я. ^'OocZc^ -7908 ?✓. f|
Динитроглицерин представляет прозрачную и светло-жел-| тую жидкость, без всякого запаха, удельного веса 1,47, J
при 15*0» При 15мм. давления перегоняется при 146*0.
При - 30* замерзает в стеклообразную массу. В воде п^ 15* растворяется в отношении 1:20; в разведенных кисло-^ так около 1:5. Динитроглицерин имеет жпчий вкус и таг| же ядовит, как и эринит ро глицерин. К высоким темпер ату- 1 рам он менее чувствителен, чем нитроглицерин; при зажиг^ иии огнем горит спокойно и без. детонации; к механически^ воздействиям(к толчкам или ударам)тоже менее чувствител^ чем тринитроглицерин; по Wt-tfA-o детонирует сухой три-s нитроглицерин от груза 2 килограммов с высоты 4 сайт., сухой динитрат - при ударе того же груза, но с высоты 7 сайт. Динитроглиц^рин хорошо растворяет и желатинизир;' ет различные, тела, напр., нитроклетчатку и нитрокрахмал j коллодионный хлопок, даже на холоду,вполне желатинизируй ется. Динитроглицерин смешивается во всех отношениях о;]' нитроглицерином и понижает значительно температуре заме1: зания; смесь из 50$ динитро глине рина и 50$ тринитро глшц рина не замерзает при -20 ; смесь 30$ динитроглицерина 70$ тринитроглицерина замерзает при -12*0.
( 5)-w« 1905г.№8) приводит для^
сравнения следующие данные для ди- и нитроглицеринов:
(см. стр.18)|
Проф.А.А.СоЛонина - Лаборат.приготовл.вар.вещ.
- 18 -
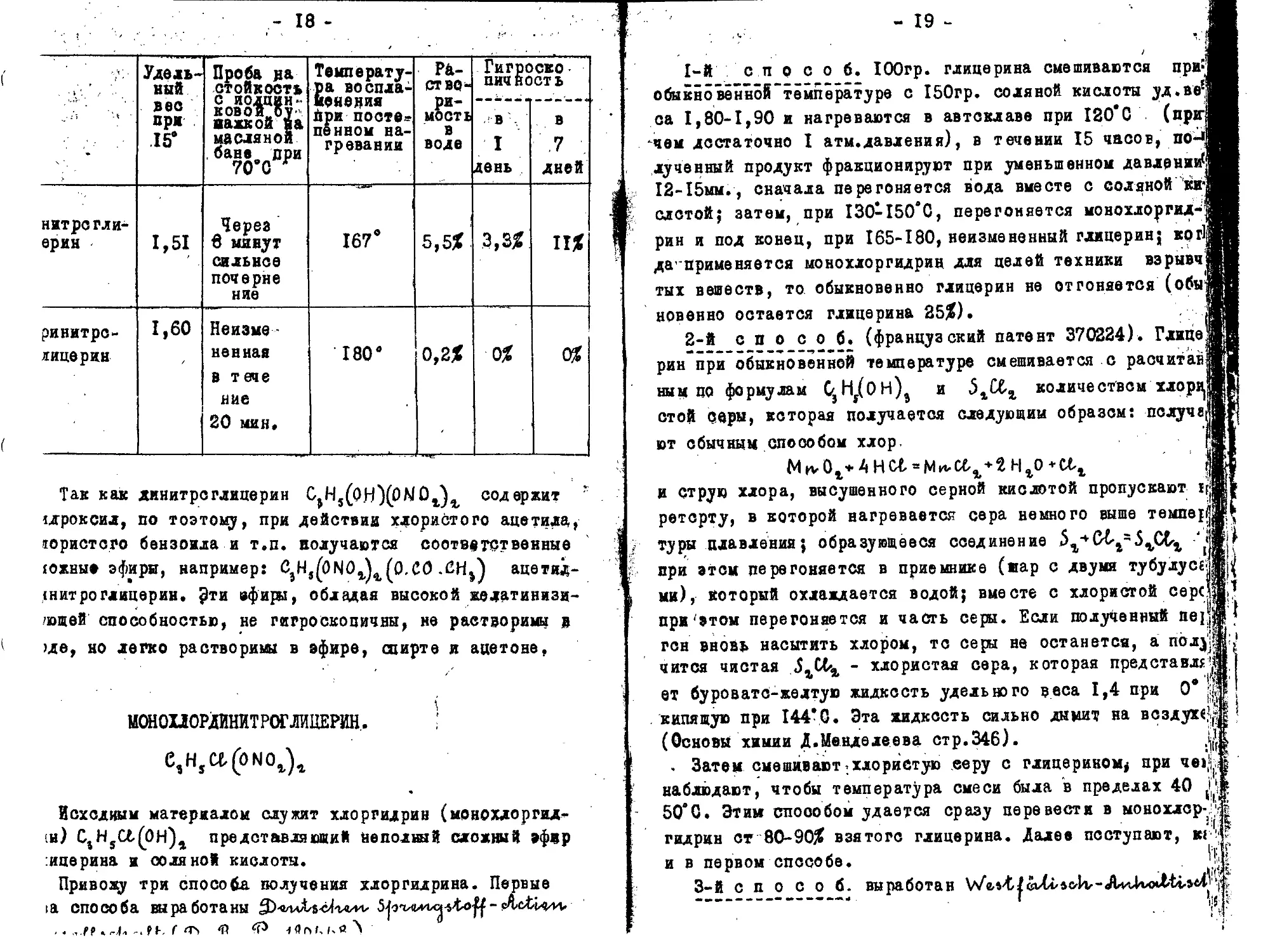
• 4 ‘ . . л - Удельный вес при .15’ Проба на стойкость С ИОДЦИН--ковоТгоу' иажкой яа масляной ,бане„ при 70’0 Г Температура воспламенения При посте» пенном нагревании Раст во-ри-мость в воде Гигроско• пич ноет ь
в I день в .7 дней
нитрс гли-ерин 1,51 Через в минут сильнее почерне ние 167е 5,5% 3,3% п%
ринитрс-яйцерин 1,60 Неизмененная в тече ние 20 мин. 180’ 0,2% 0% 0%
Так как динитрсглицерин С>Н5(0.Н)(0^О1)г содержит 1дроксил, по тоэтоыу, при действии хлористого ацетила, пористого бензоила и т.п. получаются соответственные южные эфиры, например: С}Н,£01\|Ог)Д0.СОацетид-<нитроглицерин. ?ти эфиры, обладая высокой желатинизи-/юней способностью, не гигроскопичны, не растворимы в ив, но легко растворимы в эфире, спирте и ацетоне,
МОН0110РДИНИТРОГЛИЦЕРИН, е, Hs Ct (о NO,,\
Исходным материалом служит хлоргидрин (монохдоргид-н) С4НsCt(0Н)4 представляющий Неполный сложный эфир :ицерина и соляной кислоты.
Привожу три способа получения хлоргидрина. Первые ia способа выработаны -ЛоЬ'илл.
.. „др. tl. f -(АоМхйА
-19-
1-й с и особ. ЮОгр. глицерина смешиваются при:^И обыкновённоЙ’тёмпературе с 150гр. соляной кислоты ^д.ве!^И са 1,80-1,90 и нагреваются в автоклаве при 120*0 (прю^В чем достаточно I атм.давления), в течении 15 часов, по->ИИ лученный продукт фракционируют при уменьшенном давленииЯИ 12-15мм., сначала перегоняется вода вместе с соляной ки'ЯВ слотой; затем, при 130-150*0, перегоняется монохлоргид-^^И рин и под конец, при 165-180, неизмененный глицерин; когшК даприменяется монохлоргидрин для целей техники взрывчЦд тых веществ, то обыкновенно глицерин не отгоняется (обыдд новенно остается глицерина 25%). . Жв
2-й спо с о б. (французский патент 370224). ГлицеЖИ рин при обыкновенной температуре смешивается с расчитан® ним ПО формулам С}Ну(ОН)э и количеством хлор^ИН
стой серы, которая получается следующим образам: получ8Ж j ют обычным способом хлор. 1Ж
и струю хлора, высушенного серной кислотой пропускают г Ж ретсрту, в которой нагревается сера немного выше темпв!|Ж ? туры плавления; образующееся соединение ^гСС-г
при этом перегоняется в приемнике (мар с двумя тубулусеЖ^ ми), который охлаждается водой; вместе с хлористой серсШ.1 при'этом перегоняется и часть серы. Если полученный пе;|ж | гон вновь насытить хлором, то серы не останется, а полуЖ,' чится чистая - хлористая сера, которая представлю w I ет буровато-желтую жидкость удельного веса 1,4 при О* кипящую при 144*0. Эта жидкость сильно дымит на воздух*^ Н (Основы химии Д.Менделеева стр.346). L
> Затем смешивают’ Хлористую серу с глицерином* при че1<;
наблюдают, чтобы температура смеси была в пределах 40
- 50*0. Этим способом удается сразу перевести в монохлср-1
гидрин от 80-90% взятого глицерина. Далее и в первом способе.
3-й способ, выработан We,»>t|cvU5c 4,
поступают, кг---
V - ‘С
- 20 -
сЛл£ . Пропускают хлористый водород
грез глицерин, вагретый от 70’до 100'; по окончании .тоузнается взвешиванием), перегоняют под уменьшенным селением; как и в первом способе.
При .этом способе, главным образом, получается *мо-«хлоргидрин СНг<г-СН(0Н)-СН40Н и небольшое лишь ко-ячество Jb хлоргидрина
ОН ОН-CH Ot-CH ОН . ' X * 4,<4dU3y
По J1. aB-O-«AV4/b-'(2<1Лл.с4г- о1см> W«/sowI ,
>.22) монохлогидрин нитрируют смесью азотной и рной кислоты так же, как при получении тринитроглицери-из глицерина, после нитрации образующийся монохлорди-троглицери после стояния быстро и хорошо отделяется от слогной смеси, постоянство продукта легко достигается омывкой. слабо щелочной водой.
Монохлординитроглиперин представляет светло-желтую, гко подвижную жидкость, слабо ароматического запаха, торая растворима в органических растворителях, как, на-имер,- вфире, спирте, ацетоне, хлороформе и т.п.; на* против воде и Кислотах не растворима; удельный вес, и 15’0 1,5408.
При нагревании, начиная с 180’ выделяются желтые пары около Т90* жидкЬсть закипает и перегоняется без датона-и и без вспышки; перегонка жидкости, при обыкновенном влении, происходит при 190-193’ лишь с небольшим равнением (бурые пары); при уменьшенном давлении пере го-етея совершенно без разложения; при давлении 15мм. пе-гоняется почти безцветная жидкость прм*121 123’ С» Осо-нно замечательна большая нечувствительность монохлор-нитроглицерина, к. толчкам,: ударам и трению. Удар груза . 2 килограмма с высоты 2 метров не производит детонацию, тда как тот же, груз динитро глицерин по, W-iJC н> взры-ет с высоты 7снм., а три нитро глицерин - с высоты 4снм.; убавление тринитроглицерина значительно увеличивает ветвит ель ноет ь к механическому воздействию.
- 21 -
От капсюля гремучей ртути происходит правильная и с<; вершенная детонация жонохлординитроглицерина. Отморозм 25-30 С не замерзает. Продукт не гигроскопичен и обла-J дает хорошей желатинизирующей способностью по отношен ши| к нитроцеллулозе. Монохлординитроглицерин может употре(.1 ля.ться, как примесь к нитроглицерину для понижения тем-j ; пературы замерзания, например, примесь 20% уже делает нитроглицерин не способным замерзать при 13“0, несмот]^ на введение кристалла нитре глицерина. . ,
Ip
I
ЛИНИТР О-АЦЕТИН. Tg
Исходным продуктом служит моноацетин С5ну(он)4(о&ос/н « который получается, по ; нагреванием Одина1,!!
ковйх об”емов ледяной уксусной кислоты и глицерина в т!‘« чейие 114 часов, при 100*0. Для отделения от образуюии']! ся диацетина и Триацетина обрабатывается смесь равным ’Я объемом воды и неоколыю раз взбалтывается с иебольши # ми количествами бензола; после того, как взбалтывание мД с бензолом удален триацетин, обрабатывается эфиром; /4 эфирный экстракт выпаривается на водяной бане и остато^ перегоняется иод уменьшенным давлением; моноацетмн пе'$ ре гоняется по при 2-Змм. давления, при 1301
132 О, в виде lycTOft жидкости, удельного веса при 15°С‘,У» г 1,2212 ( Q-«ziX<Uz( П-owMi/ovl- /Дъ /eVcn«X-i/je<k«z СФивлпЬв/ (2) ,57,l'f 417 и 118).
40 частей моноацетина приливается постепенно к смес^( 100 частей азотной кислоты, удельного веса 1,513 и 25 Р частей серной кислоты, с содержанием 25%50, при это«вди Г кислотная смесь охлаждается до 25°; по окончании нитраЯ; ? ции вливают смесь в ледяную воду,и промывают сначала .1!и‘• холодной водой и затем слабым нагретым до 70* растворов»: седы. При этом получается выход около 95% теоретическо^Ж
Я
- 22 -
с%н»(он)»(°^0&нэ) + 2Нм0зя Ч нЛог40*\(0й0С/Н») +
Тинжтроацетид представляет светло-желтую жидкость, уд. i 1,45 при 15*0, нерастворим в воде, бензоле и сероуг-*де и хорошо растворяется в азотной кислоте, этиловом тилов ом спиртах, ацетоне, эфире и нитроглицерине. От >а не взрывает. Хорошо^растворяет, даже на холоду, пж-илин с 13,4$ йдота; не замерзает при -20J даже при шивании и введении кристалла нитроглицерина.
Л И НИ Т Р О Ф О Р м и н.
CA(0NO.)(0tH0)
случается по рс.
192^ способом аналогичным получению динитроацетина, леей равных количеств глицерина и безводной щавелевой эты сначала нагревается до 100? затем, 20 часов при £50* и затем нитруется смесью азотной и серной кисло» ?ак же приготовленной, как и для нитрования динитро-1на); полученный продукт промывают водой и затем,сла->аствором соды, при чем лишь получается 30$ динитро-;на и 70$ трмнитроглицерина.
.кая смесь пригодна' для технического употребления,как ю замерзающая, почти не уступает в силе тринитрогли-у, мало чувствительна к ударам и толчкам
С/ООН I =оо, + мс-оон ОООН г
С3НГ(ОН)Э*НСООН’
- 23 -
О
ТЕТРАНИТРОДИГЛИЦЕРИН.
CjH^oNO^;
Исходным продуктом служит диглицерин (пироглицерин) ^Я s"Mx. 'Я
C3Hf(0H)t/U ;Я
который был получен из глицерина Хо-и/кишо ^c/UumXvs fM сЛйтлиь е£ ale, ^о-1^ь-Ьсрл<г (з) f>7 Зоо} . ’4^
Образование его можно проще всего представить следуиИ!
щим уравнением: 2 03 Ну (он\ = но+[с?н г(он)г]гО . . Яй
ж
Приведем два способа Csc-cv^'tv ,Йж]
1-й способ ^мхиХсд и J*fovоно'ся- (немецкий 1|ж
тени 12 Мая 1906г.) I часть глицерина смешивается с И'Ж =• 1|5 весов ч.воды (или лучше, соляной кислоты, не больж] 1|5 вес. части) и затем, при нагревании до 100} проиуаЯ ется в течение нескольких часов струя хлористого водор^3|' пока не будет более поглощения газа; во прекращении пр *| пускания хлористого водорода прибавляется к смени равн.;Л: по весу часть глицерина и все нагревается в колбе с обг, Г ным холодильником в течение 20 24 часов на водяной бан^Ц т.е. при 100- . д
2-й способ Глицерин, без разбавлен jJ-r
водой при 100* насыщают хлористым водородом (пока шивамие не укажет больше привеса) и затем нагревают n'Zl 150*. При такой конденсации всегда кроме диглицерина неизменного глицерина получается монохлоргидрин, дихло*,.; L ' гидрин, хлоргидрин ди глицерина, триглицерин и хлоргидр^Я-триглиперина. jj№
Обыкновенно, полученную смесь прямо нитруют, как ют чистый Глицерин, так как азотные эфиры этих побочнн^Я) продуктов, в особенности, различные нитрохлоргидрины^мЯ Ж
34' -
! адают низкой температурой замерзания.
I, Если желают получить .чистый диглицерин, то лучше все-^О, по VVlC'C'Vo и ^to4w«ztz'i^ (be^tsc/K- оЬ. s$. -ppwot
-^^ад^^Ьо^ие^л^т^.ХЗ^глицерин нагревать 7-8 часов, при -.90-295’, пои чем нагревание производится так, чтобы во-могла отгоняться, а высококипяшие продукты стекали бы обратно. При этом получается 6,0# диглицорина и, в зависимости от нагревания, 4-6# триглице рина и др. продуктов >нденраиии, остальное - неизменный глицерин.
С При перегонке под уменьшенным давлением 8-10мм., гли->рин перегоняется при I60-220j а продукты конденсации 1‘)и 220-270*; после нескольких перегонок под уменьшенном Явлением можно получить чистый диглицерин, который, при ;мм. давления, перегоняется без разложения при 240 -'0*0. Диглицерин очень вязкая прозрачная жидкость слад-^ватого вкуса удельного веса 1,33; вязкость диглицерина 'II раз более глицерина. Диглицерин легко нитруется в
X самых условиях, как и глицери:
“ <Z|H.N0> =&,Н *4 Ht0
fi Продукт нитрации О во многом напомина-
J тринитро глинерин; он растворим хорошо в органических .^творителях, как например, эфире, спирте и т.п.; дето-;.Ьует легко от удара или от гремучей ртути; в виде динара (напр., с кизельгуром), при детонации, производит ;toe же действие, как тринитроглицерии,- но в общем ме-] чувствителен, чем тринитроглицерин и значительно труд 'I замерзает, так как при довольно низких температурах 'кристаллизируется; смесь тринитроглицерияа с 20-25# ранит роди глицерина не замерзает при обычных зимних мо ах (в Германии).
V'l
- 25
ДИНИТРОГЛИКОЛЬ. -j
ОН,ONO,— ОН ONO, :«
X X w a# ; Jj»
CK.0H-CHt0H + aHN0%=OH%0N04-CH40N0il i • % . );gg|
10 грамм гликоля при размешивании по каплям приливаю!Ц’ при охлаждении льдом к смеси 24 грамм дымяшейся азотной, Ж кислоты и 48 грамм серной кислоты 66"<5Н.
Полученная маслянистая жидкость промывается сначала холодным 5% раствором соды и затем чистой водой до ней— тральной реакции. После отделения от воды в разделитель3^ ной воронка высушивается в эксикаторе над хлористым калР® .эк, днем при разряжении.
Динитрогликоль жидкость удельного веса по у • ж
1,496 при 15° (по l,AS) . э||
Демьянов дал новый способ получения динитро гликоля |Й действием азотного авгидрида на этилен Ж
2NA*cA=WONO1V2NOs • ¥ Пропускается чистый этилен через раствор азотного ангидрида в безводном эфире при сильном охлаждении льдо j с солью. По окончании реакции, что видно по тому, что этилен перестает поглошатъся, эфир отгоняется на вода-ной бане, а остаток промывается холодным 5# раствором соды и чистой водой до нейтральной реакции. i.i|:
ТЕТРАНИТРОПЕНТАЭРИТРИТ. , Z
4M,(ONOJ,
<^ьнгон),М Н«о5=<^см,омо,), .4 Н.,0
, л t '
10 грамм тонко измельченного и высушенного пентаэрИ|'
11. трита..всыпается при тщательном размешивании в стакан с,..
30 куб.сан. азотной кислоты уд. в. 1,5 охлажденной до>( 01 При нитрации надо следить, чтобы температура не ордц;' нимелась выше 30°. ;<'
- 26 -
Когда весь пентаэритрит прибавлен то продолжают ешь азмешивание до полного растворения твердого тела и за-^м^фдливают при размешивании такое количество крепкой зрйо¥ кислоты, пока дальнейшее прибавление .уже не вызнает осадка.
’< Отработанную кислоту отделяют декантацией и получение твердое тело промывает до нейтральной реакции, высу-(вают между листами фильтровальной бумаги и затем в экваторе над хлористым кальцием при разряжении.
л •
п
/ НИТРОМАННИТ.
V < <\Hs(o/vo^6HtO
; Способ получения профессора Соколова, расчет на Югр. ннита.
Маннит С()Н(^он)ь должен быть, по возможности, чистым, (?о поверяется определением температуры плавления. , ./ Перед определением точки плавления, измельченный в ;нкий порошок маннит сушится при температуре около 100° ^течении 5 часов и остуживается в эксикаторе'над серией 'рлотой. ' ч
VХимически чистый маннит плавится по Фавру и Дандольту /Д66? Продажный маннит,с температурой плавления 160-/5,-допускается для приготовления нитроманнита.
/ Производится определение температуры плавления следую-/«образом: маннит, при помоши маленькой ступки, расти-этся в тонкий порошок и насыпается в стеклянный капил- ' /э, при чем высота слоя насыпанного ваиества в трубочке / должна превышать 2 мм. Капилляр прикрепляется (при по-1и отрезка платиновой проволочки или каучуковой труб-* к термометру так, чтобы середина слоя вешества а ка-гляре находилась против середины шарика термометра; в
-27 Ж
наружную крлбу особого прибора наливается крепкая и ям-и стая серная кислота; термометр с прикреплениям к нему Ж капилляром, при помощи трубки вставляется во внутреннюю® воздушную баню. Нагревается прибор возможно маленьким ж пламенем Вунзеновой горелки так, чтобы пламя не касалосЛр стекла прибора. (i||
Если знают приблизительно температуру плавления ведЖ ства, напр., для маннита не ниже 160^ то не доходя . до предполагаемой температуры градусов на 10, быстро нагреМЬ вают, а уж затем - постепенно, и наблюдают состояние ве!,| щества при каждом градусе: для этого, ставя в сторону r-j||j релку, дают ртути подняться до остановки и т.д. пока вер щество не расплавится. Если вещество не чисто, то началок плавления будет на несколько градусов ниже окончательЯо'^ плавления всего вещества. Перед нагреванием не забывать^ поворачивать притертую йробку прибора так, чтобы расжжрД щийся воздух мог свободно выходить из колбы. По бхлажд&ж нии прибора надо завернуть так притертую пробку, чтобы *$. влажность не могла попадать в колбочку, содержащую сер-^. ь ную кислоту.
| Кроме вышеописанного прибора часто пользуются более г простыми,в которых нет внутренней пробирки, а прямо терМ мометр опускается в серную кислоту; в этих приборах мол® но не прикреплять капилляр каучуком или проволокой, а смочивши серной кислотой, приложить капилляр: он будет < держаться от смачивания серной кислотой.
Отвешенное количество маннита (Югр.) растирается в .сухой фарфоровой ступке и затем помемногу приливают 10О| i. грамм'охлажденной до 0’ чистой азотной кислоты НМО^удезнЖ ного веса 1,52. Во все время приливания растирают пестдЖ ком маннит в кислоте; полученная смемь -выливается в Ь® стакан, охлажденный льдом или снегом. Затем ступка опо-;^ лаекивается 100 граммами охлажденной до 0*0 серной кисл’у ты Нг$ Отдельного веса 1,82 и постепенно, при помвшивя^ нии стеклянной палочкой, выливается встакан со смесью jsj
28 -
!аннита и азотной кислоты. Обращать большое внимание на шательное охлаждение, так как при недостаточном охлаж-(внии могут выделяться краснобурые пары окислов азота и, )место нитроманнита, получаются продукты его окисления.
Нитроманнит начинает уже выделяться. тотчас по прилитии ерной кислоты в виде творожистой белой массы.'После при-ития серной кислоты, помешивают смесь стеклянной пало’ч-ой до охлаждения смеси около 0° и затем выливают содер> ание стакана в сосуд с большим количеством охлажденной о 0 воды; дают отстояться нитроманниту и сверху, при омощй сифона, сливают воду с растворенной в ней кисло-ой смесью, затем, еще раза три продежвают эту операцию, рибавляя каждый раз свежей воды. Затем фильтруют при потреблении водяного насоса и фарфоровой воронки.
По величине плоского круглого дна фарфоровой воронки грезывается такой же величины круглый бумажный фильрр, гобы лишь он прикрывал все те мелкие отверстия, которые деются на плоском дне воронки; фильтр предварительно :ачивают водой и прижимают; затем наливают жидкость и булус конической колбы соединяется каучуковым шлангом водяным насосом. Чтобы фильтр не провало, надо следить : тем, чтобы не было такого момента, когда нет жидко-и над фильтром. На воронке нитроманнит промывается раз лодной водой, затем однопроцентным раствором .соды ^COj и опять водой, пока красная лакмусовая бумажка перестанет синеть.
Выход около 21!4 грамма. Сырой еще нитроманнит раство-ют в возможно малом количестве нагретого почти до ки-.ния винного спирта С4НУОН , и оставляют прозрачный створ охлаждаться. Выделявшийся нитроманнит отфиль-овывают,' пользуясь, как раньше, фарфоровый воронкой насосом. Затем, кладут в фарфоровую чажечку и сушат я 70 * до постоянного веса и охлаждают в эксикаторе.
Сырой т.е. неочищенный нитроманнит плавится около
ICO* С. Очищенный перекристаллизацией из спирта - пла- . | вится при 112'113‘С без малейших признаков разложения, Удельный вас кристаллического нитроманнита при 0*0 . pa- (( вен 1,604; уд.вес расплавленного и отвердевшего - около М 1,5 ,446; 1,503; к 1,537 J при 0*. Прессованием можно
увеличить уд.вес до 1,806.
•с. 1 i ft
ч’| НИ ТР ОКРА ПА Л Ы. .
S 2-!>CzCxXt4 "/305v. с*и|о *270 |'Л
Тетранитрокрахмал.
<\.h,a(ono4.
о с об. 5гр. тонко измельченногф высушеннс крахмала растворяется, при помешивании и охлаждении (смесью снега с солью) в 60 гр.
с п
20*0
1-й го при при сильном крепкой азотной кислоты удельного веса 1,5 и затем при* ,J|
; бавляется 40гр. крепкой серной кислоты уд.веса 1,83. Во г время приливания серной кислоты надо хорошо размешивать^! I После охлаждения все выливается в избыток ледяной воды; отстаивается нитрокрахмал, который промывается чистой водой до нейтральной реакции.
Получается 6,61-6,73 гр. нитрокрахмала; полученный . W продукт обрабатывался избытком 96$ спирта, при чем частг'^ ие растворяется и таким образом удается получить два из< -j Ни мера. ; 1
' . 2-й способ. (Опыты
, 'У
20гр. высушенного при 100* до постоянного веса крах-мала вносятся, при помешивании при 20-25’ в 200 гр. азотной кислоты, уд. веса 1,501 и оставляется стоять
- 30 -
50 часов. Полученной желтый прозрачный раствор, в количестве ЮОгр., выливается, при помешивании в течении двух минут, в смесь, со стоя жую: из Ю8гр. азотной кисло-ты, уд.веса 1,59, 737 гр.серной кислоты, уд. веса 1,829 л 155 гр. волы. После 15 минут все выливается в ледяную ;'зоду и в течение !4 часа отстаивается, затем отфильтровывается, промывается чистой водой и высушивается, 'i 3-й спо с о б. 40 грамм высушенного при ЮО’крах-лала, при обыкновенной температуре растворяется в 400 Драммах азотной кислоты, удельного веса 1,501, несколько ’ней раствор стоит, за тел выливается в холодную воду,про-..ывается слабым раствором соды и высуживается.
При всех этих трех способах получения, нитрокрахмал .ущится после промывания чистой водой в эксикаторе над ерной кислотой до постоянного веса; для скорости вы су ш и-,ания лучше выкачивать воздух из эксикатора.
'Результаты этих трех опытов приведены ниже.
Яитрокрахмал Темпера-тура воспламенения Растворимость ,
в 96Х спирте в эфире '
<(-й способ: . ^растворимый изомер ‘ютворимый изомер й способ. й способ 1 178 4 172 ’ 175’ 170*‘ Не растворим. Растворим. Растворим Растворим. Легко растворим. Трудно ' растко рим. Не растворим. Не растворим.
31 -
f'i
Пентанитрокрахмал. 1
’’I
WH), *1
I часть рисового или картофельного крахмала смешивав! сн с 30 раз большим по весу количеством кислотной смеси состоящей щи I части крепкой азотной кислоты, удельного’.) веса 1,Б И 3-3,6 част, крепкой серной кислоты,удельногА$ веса 1,83. Тетранитрокрахмал отделяется из j нитрокрахмала растворением, после прошвки водою, ( то-зфирнОЙ смеси, при выпаривании эфира выделяется цент^Й нитрокрахмал, тогда как тетра нитро крахмал находится в*1” растворе.
Определение азрта дало 12,50-12,98&
полученногб|) дою, в спиУ|
и
£
% .
Гексанитрокрахмал.
слн„о.(омо,),
40 грамм высушенного при 100*С крахмала при обыкнрчеж
{ ной температуре вносится в 400 гр. азотной кислоты, ys»w веса 1,501 и оставляется стоять 24 часа. Затем в темени®' получаса размешивается с I2CO куб. сантиметрами серн0!Ж кислоты, 66°и выливается после стояния в боль-1|| шое количество вод$. Получается всегда с примесью пентаж . нитрокрахмала. 'Я
лает следующую таблицу нитрокрахма-
лов. (См. следующую стран.) ж
ПФ
Я
- 32 -
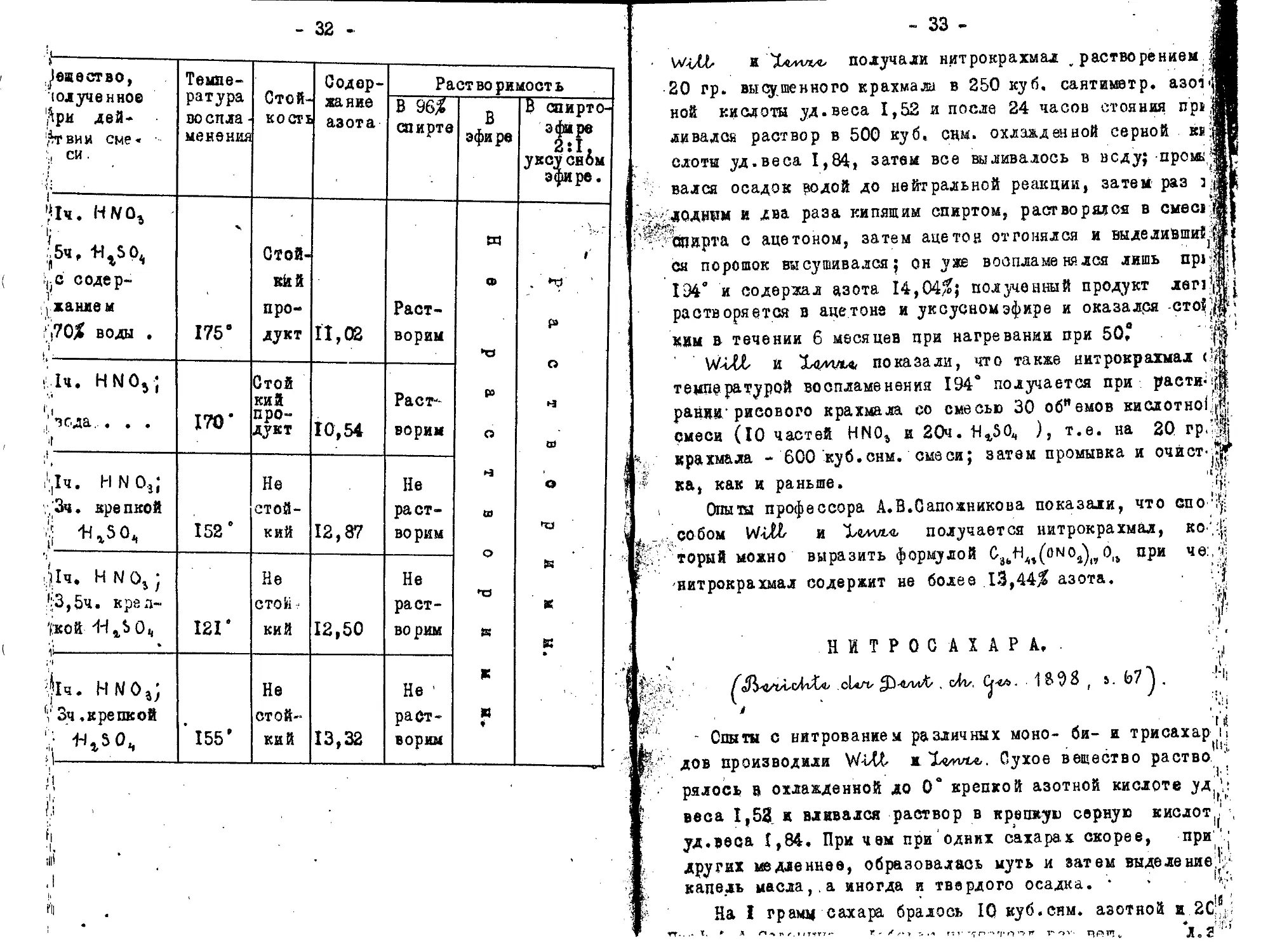
^еиество, юлученное ^рм дей- • Ат вин сме« J СИ ' > ‘ Температура во спла-ме нения Стой-i кость. Содер-хание азота Раст во римост ь
В 96% спирте В эфире В спирто-эфаре уксуснбм эфире.
•<1ч. Н1УОг 'i ;',5чг {-d содер- 1 хание м j70% воды , 1 175 е Стой-кйй продукт И, 02 Раст- ворим я а> 41 ₽3 О ч ш о 43 К К Я ♦ -t • *7 р> G Н В) о 43 ts К К •
1ч. hno5; •’i j. вода... . . ">r 170* Стой кий про-дукт 10,54 Растворим
с,1ч. HNO,; ,3ч. apeпкой | ^гЗО. 152 ° Не стойкий 12,87 Не раст-во ркм
Д1ч« Н N Оь ) :(!-3,5ч. крел-'(кой /H1S 0« 12Г Не сто Ji кий 12,50 Не растворим
\jl4. нмо%; V Зч. крепкой ' и^о- И ; ’ 155’ Не стойкий 13,32 Не 1 растворим
f|
I, ill’
,1
fll
33 -
w-iXL и получали нитрокрахмал v растворением
20 гр. высушенного крахмала в 250 куб. сантиметр. азо1>|И ной кислоты уд.веса 1,52 и после 24 часов стояния пр>Я1 ливадсв раствор в 500 куб. сим. охлажденной серной в>1и слоты уд.веса 1,84, затем все выливалось в веду; промгШг ? вался осадок водой до нейтральной реакции, затем раз ]11 лодным и два раза кипящим спиртом, растворялся в смею Ж Г- спирта с ацетоном, затем ацетон отгонялся и выделивши?.w ся порошок высушивался; он уже воспламенялся лишь пр*ж 194° и содержал азота 14,04%; полученный продукт лег1ж| растворяется в ацетоне и уксусномэфире и оказался -сто|^> хим в течении 6 месяцев при нагревании при 50“
WaZt и X<j>vwL<i, показали, что также нитрокрахмал <ч| температурой воспламенения 194е получается при расти-Ц ранни-рисового крахмала со смесью 30 об**емов кислотно!.!^ смеси (10 частей Н1ЧОг и 20ч. Иг50,ц ), т.е. на 20 гр.’|у ч крахмала - 600 куб.снм. смеси; затем промывка и очйст-^ : ка, как и раньше.
1 Опыты профессора А.В.Сапожникова показали, что спо'';у:
_ . собом W-M и Уллукь получается нитрокрахмал, ко‘,.|;
С торый можно выразить формулой СзьИ4,(омоДу 0(> при че:,’^
' нитрокрахмал содержит не более .13,44$ азота. ,1,
НИТРОСАХАРА. • '
л i!
oltzv ££)-аллЛ- , ой/. (j-гл. 1 8 9 8 ( 5. Ь7 J . '•
i '
Опыты с нитрованием различных моно- би- и трисахар и дов производили W-iZt aXewt-o. Сухое вещество раство рялось а охлажденной до 0° крепкой азотной кислоте уд( ; веса 1,53 в вливался раствор в крепкую серную кислот ' уд.веса 1,84. Причем при одних сахарах скорее, при1' других медленнее, образовалась муть и затем выделение^',-? капель масла,, а иногда и твердого осадка. • *
На I грамм сахара бралось IQ куб.снм. азотной и.2С^;
VY-. .• t. * .X П -s n , ттгт- r,r T- ov ЛоЗ ^1
куб.снм. сорной кислоты. После прибавления серной кисло
*1 ты смесь отстаивалась, пока больше не было заметно виде ?( ления осадка и затем все прошвалой ледяной водой. Полу д ченные сложные эфиры в большинстве случаев, растворимы i 1 ацетоне, уксусной кислоте, спирте (при кипении) и не-растворимы в воде.
< • Нитро сахара восставовляют легко Фелингову жидкость
j при нагревании. . ,
• ' Таким образом получены: < .
j Из рамнозы -• тетранитрорамноза
' : . СЛ(° N0.\0'
; с.н„о,Анмо,-е,н,(омо,),о^*н1о
,.р.омбические кристаллы с температурой плавления 135°0.
’А ...Из .арабинозы - тетранитроарабиноза:
1/; *' ^A(onos\o, ' .
' Д' ? C5Hlo0s*AHN0rC,5H4(0.N04\0* АНгО
। монокл. безцветн. кристаллы, температура плавления 85* ' ? •
, ’’ Из ксилозы смесь двух - одно жидкое, а другое тве] дое, с температурой плавления 14Ц " эерД ятно, тетранитроксилоза; '?'
с.ндомо^а
Из глюкозы пентанитроглюкоза - густое масло:
\ C.H,(»W0j,0, ,<
Из галактозы - пент&питро г-a лактоза: f
с АО* ° л° м
в виде двух изомеров; оба тела кристаллические, ОС - труд . но растворимое, точка плавления 115-П6* J5 - мокл. иг . лы, точка плавления 72-73'.'
Из маннозы « пентанитроманноза
’ ^Н,С^Оа)5О _
137-139 48-52’.
- 35 -ромбические кристаллы, с точкой плавления 81-82’С Из левулозы два изомера тринит ролевулоды:
O.H,0,(ONO%)S Оба тела твердые; сь ст. плавл. "
Из сорбинозы тринитросорбиноза . c.H.o.l»''0.),
т,плавя. 40-45.
Из тросникового (или свекловичного) сахар
сахара. -
окт 01 1
С„Н«0.♦ 8 н N0, = С„Н„(ONO,),0,• S НХО плавится при 28-29’; непостоянен.
НИТРОСОЕДИНЕНИЯ..
зольного) ряда; . ной- кислоты на
где под Н_ мы
i®-’*
Все, употребляемые в военном деле,врзывчатые нитрате' динения, относятся к нитросоединениям ароматич еского Ж они легко образуются прямым действие:?'^;
производные бензола:
Янкино,-Ян^о,у <|Н.О разумеем тот водород, который находит бензольном ядре, так как эти лишь водороды обыкновен ' меняются нитро группой NO^. Как при получении азотных так и при получении нитро соединений, употреляется азотно! еще и серная кислота; роль ее здесь та же са !‘!&
Отличие строения нитросоединений от азотных эфиро ж что азоты нитрогрупп в нитросоединанияз напо средстве м единены с углеродными атомами, а в азотных эфирах -средстве кислорода. 1,1'Jf
Приведем нитроглицери, как пример азотных эфиров 1,1 криновую кислоту, как пример нитросоединений: ’Г (,•
..т
is i
- 36 -
H-C-O-NO. j
I C
H - C-O-NO* I
H - & — 0 —NO* * АЛ
Нитроглицерин
Пикриновая кислота.
Этим строением нитросоединений к объясняется то, что 1итрогруппа здесь связана весьма прочно и обыкновенно ;зот при различных химических 'реакциях остается в частж-
ie органического вещества.
Например, при действии водорода в момент выделения группа нитро переходит в группу амидо; так из нитробен
зола получается анилин.
НС
НС
Прочность
золе, можно доказать ткм, что при нагревании с водой пр>|| '00^ в течении долгого времени, мы не обнаруживаем ника-^Л <ого взаимодействия воды на нитробензол'.
Азотные же эфиры легко бмыляются при действии щелочей^ ислот и даже воды и при этом отщепляется радикал азот ней, /J нслоты, с образованием соли। содержащей азот. > ' ।
Из. нитросоединений здесь приведено получение мо-, ди- j тринитробенаолов, трлнитротолуола, тринитрокфжлола, мо- 1 ди- и тринитронафталинов, гексанитродифениламина, 1
- 37--
тринитрофенилметилнитрамина (тетрила), тетранитрофвнил-f метилнитрамина (пентила), гексанитродифениламина (гексила).
a1*-
< - ТРИНИТРОТОЛУОЛ.
, (Тротжл ИЛИ тол).
МММ Я
Тротил может быть получен непосредственным действием Ш смеси серной и азотной кислот на толуол; так его ж полу^ ЧИЛ ВП6 рВЫе Ли*глД<лФ1- oLe/v 14Я, 17£^
Но этот способ неудобен и по имеющимся сведениям, го- $ товят теперь его заграничные заводы двумя ч способами:
1-й спо с о б. Сначала из толуола готовят моно тротолуол:
л !'
СЬН5СНЭ-И№3 = 3^(N°Oc,VHi0 затем последний перерабатывают в динитросоединение:
’^H,(MOt)tCH^H4O
ж,.наконец, из динитротолуола получают тротил: ебН5(мо4\снз+нмо5’еьн^о^сн^н,о
(Сообщение директора завода IW. ЛЛи-ихАо-хХ-
. 5c4»0*vez6^OK,cv. cb. &14ка/). b ’ll
Так готовят, например, в о, и теперь у
HaQ* ' W
2-й спо с о б. Сразу готовят из толуола динитро-||:|| соединение.
$ г -IE;
- 38 -
/|1тем,из последнего - тротил:
'МтИМ способом ГОТОВЯТ В еЯигйл4хЬоЬо-^'ъ .
*Гак как полученные сведения из заграницы отличались ,:1вей сжатостью, то поэтому, были проделаны специальные .fra для установки фабрикации тротила у нас в России на .нноком заводе взрывчатых веществ. Эти опыты, произве- ,, гые заведывающим химической лабораторией Охтенских 1 ж
>дов взрывчатых веществ Федоровым касались как полу* ,i'ia сразу из толуола тротила, так и двух указанных 'з способов.
У’пособ непосредственного получения дал неблагоприят-результаты, так как получился тротил со значитель-
примесью моно и динитротолуолов. Нитрация производи-
•'> аналогично нитрации фенола,но со значительно боль-
I количеством кислот и без сульфокислоты; нитрация зует много времени - до 48-ми часов и высокой, срав-'ильно,температуры - до -120*0.
,'рля всех опытов употреблялся толуол, т.кипения НО— 'jiC; кислоты для приготовления нитротолуола - азотная , < во всех же других случаях -= дымяшаяся с 91% мо-
;:драта HN0$ а серная - всегда с 95% моногидрата -. Тротил готовился, как на обыкновенных, так и на веских чистых кислотах, причем существенной разни-j^ieг наблюдалось.
^агрузка толуола - 50 грамм. ' 7^
,;-ый способ. В плоскодонную колбу, емкостью в г’»
-литр, навешивалось Г50 грамм урепкой серной кисло-и. туда же вливалось 100 грамм азотной кислоты, . Так как при смешении происходит разогравание^ то смеси остыть и затем вводится толуол в количестве ^рамм, небольшими порциями; при этом температура серимого колбы повышается; следует вести процесс так, температура поднималась не выше 50*С; поме кая-
~ 39
дого приливания толуола смесь хорошо взбалтывается.
При вышеупомянутых условиях эта операция требует ’/г ча-ЯВ са времени. W
Когда весь толуол прилет, то дают смеси этой стоять в ‘ 1в| течение 2-х часов при температуре 80«90’ С, для чего кол--|П ба нагревается на водяной бане. Ж
Мононитротолуол собирается в виде масла на поверхности)^ кислотной смеси. Содержимое колбы выливается в воду, ко- t торой по об”ему берется в три раза больше; тогда нитрото луол собирается на дне в виде густого желтоватого магсла, | которое отделяется на разделительной воронке, несолько |’Г раз промывается чистой водой и может служить для дальней шей переработки в динитросоединение. (<
Иа 5Огр. толуола получалось около 75 грамм мононитро-толуола. -у.
Для переработки нитротолуола в динитропроизводное, по лученные 75 грамм нитросоединения нагревают в колбе до 65*0, и к нему приливается постепенно смесь 60 грамм кре^М' кой азотной кислоты с 9IX моногидрата Н»МО4 и 120 граш У' крепкой серной кислоты с 95% моногидрата 44^50/, причем '' колба все время взбалтывается; когда вся кислотная смесь ,г будет введена, колба нагревается на водяной бане в тече-ние одного часа при 90’С и изредка взбалтывается.
По остывании смеси,динитросоединение выкристаллиэовы- 1 ; ваетсл на поверхности кислот; твердое тело отжимается от : । ^кислот на воронке Бюхнера и идет на дальнейшую перерабр-i!(, икУ> Г?' ?- 4 ij.i
” Из 75 грамм нитротолуола получается около 100 грамм й ^Динитрбтолуола с температурой плавления около 67*0. Полуп-;. ценное динитропроизводное в количестве 100 грамм раство-(,; ряется при 45-50*С в 400 граммах крепкой серной кисхотк^,: и затем туда же вливается 200 грамм крепкой азотной kmcj/.
ты с 91#’моногидрата HNO^ смесь тщательно перемешивав? ?! ся при нагревании до 95*0, пока не прекратится выделение^ бурых пород окислов азота, для чего требуется около-4 g
- 40 -
в времени, после чего смесь при остывании выделяет на верхности кристаллический тротил, который отжимается кислот на воронке Бюхнера, промывается сначала горя-ft водой (50-60*0); затем промывают еще холодной водой агаушивают нрй 60* и до постоянного веса - в вксикато-. Температура плавления полученного таким способом тро- j ta 73-77*0. Выход тротила около 120 грамм. j
". •
2-й с.по с о б, Зтот способ, по опытам Федорова, ;т очень хорошие результаты и поэтому может, давили* *¥> считаться более выгодным и кроме того,более удоб-i как для лабораторного получения^ так и для фабрика-> в больших количествах.
Приготовление динитротолуо~ . Триста грамм серной кислоты с 95% моногидрата,Нг50^| шивается с 200 Тр. крепкой азотной кислоты с 91% мо-идрата HNOj при смешивании температура поднимается / Ж □ этому дают остыть; над сосудом устанавливается стек*’^ ная воронка с краном, где помещается 100 гр. толуола,' М □рый постепенно вливается в смесь кисхот в течение х часов; при смешивании толуола с кислотами происхо^О» энергичная реакция с повышением’температуры, причем ' ’ □ стараться, чтобы температура не была выше 90°С.
эгда толуол вводится в смесь кислот, то для перемети-й туда уже «продувается воздух, высушенный пропускани-аэрез серную кислоту. ’ ' jg|§|
>сле окончания вливания толуола в смесь кислот, в
<е одного часа продувают сухой воздух. После охлажде-''-^^ динитротолуол кристаллизуется на пове рхности кислоту йЦи отжимают и пускают в дальнейшую переработку. Я
1ыход динитротолуола - около 200 гр,,с теммературой ления около 67*0. . ‘1
переработка дияитр ©толуола т р о т и л. В тот же сосуд помешают 100 грамм дини-олуола и 400 гр. крепкой серной кислоты с 95% моно- ’
-41 гидрата Нв504 и нагревается до 40-50*0; когда весь дини-^1 тротолуол растворится, то приливается 2ООгр. азотной ,ки-: слоты о 91# моногидрата HNO5 затем вся смесь нагревает-* ся до 95*0 и изредка продувается сухим воздухом для пере мешивания.
Когда окончится выделение бурых паров окислов азота,то । перестают нагревать, и, по охлаждении, ныкрасталлизовыва-1 ется тротил, который отжимается от кислот, промывается сначала гори чей водой (60-70*0), г - опять водой, но холодной и высушивается до постоянного веса при .0*0.
Получается при этом, обыкновенно, тротил с температу-рой плавления 77*0; но Федорову удавалось, при вышеупомя-ну mi условиях,- иногда получать тротил с температурой плавления;и ,801 Вообще же можно поднять температуру с J 67* до, 8Г4* Для тротила, выкристаллизовывая его из горя- г че го 5спирт а.
В ви ду того, что тротил находит себе в последнее время ’Обширное применение в технике взрывчаьых веществ, считаю необходимым сказать несколько слов о его свойствах.
Температура плавления полученного вышеуказанным спосо ?бом тротила и выкристаллизованного из спирта, а именно около 80*, показывает, что получается изомер следующего строения
Т8 т
<ь-
н-с
NO1 С
с-н.
C-NOt
ot-2,4,6 тринитротолуол
!<
r'i^ J, 1!®
Ь* Not-e
CH $
V 1 I $ у
Так как для этого изомера дает температу- { ||
ру плавления 82* С, а 81*52 и 78*84, съ Ji и j -
изомеры име&т температуру плавления 112 е и 104* В техни-J ке различают'два различных сорта тринитротолуола: с тем-
’ I? i.i Г.
42 •
натурой плавления 72’- 74’0 и с температурой плавления 79 Более же чистый, с температурой плавления 81-
У готовят кристаллизацией технического продукта о темпе* ?’|Урой плавления 77’- 791 По сообщению директора завода Jb.wVV. JLKv/v-
<j?UVtz Cj4L6-©tVyvt^b 5^ive<A6 AAs. S[oizjzW<^-C>to|-.|lv/.
j'7 № I стр.4. Перевод - Арт.журнал 1907 № 4 стр. 497) „
/нитротолуол, с температурой плавления 72* 74* С, придется исключительно, как примесь к различным взрывча-{веществам Вместо ранее употреблявшихся динитробензола, «Тротолуола и нитро Производных нафталина, так как три-ротолуол имеет два преимущества; - он обладает большей рй и безопасен в обращении.
Зажное значение имеет тринитротолуол, как подходящий зриал для приготовления, так называемых, безопасных печатях веществ ( ). В настоя -
'.время тринитротолуол с температурой плавления 72-74*, }ет главную роль более чем в двадцати взрывчатых веще- > 4х, тогда как 20 лет тому назад он был совершенно нечетен в промышленности взрывчатых веществ.
'{истый тринитротолуол, с температурой плавления 81 - ’•>
!>’ С, получаешй кристаллизацией из спирта, из продукта-зящегося при 78 - 79*С, уже 17-Г8 лет тому назад начал меняться как самостоятельное взрывачтое вещество и в /оящее время является серьезным конкурентом пикрино-(iкислоты, которая почти везде, как наиболее сильное бри-'•ное вещество, употреблялось для снаряжения артиллерий-' с снарядов (мелинит, шимозе и т.д.). .
1олгое время думали, что одно из важных преимуществ три-;отолуола, это отсутствие вредного влияния на здоровье них, как при фабрикации его, так и при всех дальней-работах с ним; тогда как пикриновая кислота своими па-.1, как при фабрикации, так и при плавке ее для снарядов, |.но действует на здоровье (ослабляет деятельность серд I расстраивает пищеварение), но в настоящее время это
I
1
- 43 -
ji
Сказалось неверным - пары тротила еще вреднее. .'J
.Хотя тротил не имеет гидроксила и поэтому не дает так U [егко, как пикриновая кислота,пикрато в, но от дейст- э>! 1ия щелочей вое же может давать тротилаты, так как Л
рт дейопгвия едкой щелочи в присутствии воды получается ‘ изомер, содержащий гидроксил, где водород и замещается . J
te та л л ом. : сга||!
;
CJ4jNOACH +Ь1сьНО+Н,О« Л !
I»
ЙЯ’: '
Особенно легко действует едкое кали в присутствии спир-Я
с.н JN оЛе н, 1-С Н ,0 к. = с н ,-e.H^N 0t)tN- о к.
•У
н • ’J* 3
• Эти металлические тротилаты очень непостоянны 1взрывают при значительно меньшей температуре, чем тротил и более чувствительны к удару.
1. Силой тротил немного уступает пикриновой кислоты - в ^отношении 34 : 37. Удельный вес в кристаллическом ви
16 и в порошке равен единице, но в сплавленном виде Достигает 1,5. Посредством сильного пресованния, что вполне применимо вследствие малой чувствитальности, можно достигнуть удельного веса 1,60 - 1,65, т.е. плотности сплавленной пикриновой кислоты.
!лом считает
i
I 11
i-
гр1
г
I >
работавший много лет с тринитротолуо-что его удельный вес, а может быть и си-у ’
и
I
б ’•
- 44 -
г^,«Можно повысить прибавкой к нему содержащих кислород j'oiecTB. >
^.Тринитротолуол беднее кислородом, чем пикриновая кисло-у :«|V4 поэтому при разложений его выделяется много угля.
- на построенном им заводе тринитротолуола^ dr. €W<xz приготовил (взят||||и г и ,Ге кт) желатинообразную смесь из динитротолуола, колло^^н ^йиого хлопка, тринитротдлу ола, бертолетовой соли и азеиЯ| ^•кйслдго свинца, удельного веса 2, причем сила,получение— ^‘вещества бодее Пироксилина и пикриновой кислоты.
Ц, \ -у
ТРИНИТР0КОИЛ01.
(Ксилил).
4
|f ,Z * ' ИУ-^Кядй
!'. . - ,;-S
М В России тринитроксилол начал впервые изготовляться в
Льших количествах на трех заводах во время войны 1914г. 111 способу, выработанному А.Соломина, причем сразу из к си-'|. '• ' - Л
(а.в одну нитрацию получался тринитроксилол. %
В стаканчик наливается 100 грамм азотной кислоты уд£ ,5 ’и 300 грамм серной кислоты уд.в. 1,84; после' размепГив^ i'i и охлаждения до комнатной температуры начинается" пр^ репейное приливание 25 грамм ксилола ( от.кил.137*138*')^ i • , -
к тщательном размешивании, причем стаканчик все ‘Время лаждается проточкой водой, Чтобы температура внутри ста- < Ла не поднималась выше 35’. Когда прилвание ксилола ззлдк-'> лось, то не прекращая размешивание поднимают температуру J
' - 4S - ilH
h OR
[ до 40е и затем дают остынуть. Дп
Содержимое стакана выливается в избыток холодной вод{0 й и выделявшийся ксилил отжимается на воронке Бюхнера, np^lJ мывается водой до нейтральной реакции, высушивается межЦ4 к листами’фильтровальной бумаги и затем в сушильном шкафу Й. * ?'прж 1001 .. з! i
'^‘:; . .Для очистки ксилил раст воряется в наименьшем ко лич ecJH ^ве-бензола при7:.оЬявяовенноЙ температуре и из раствора нЖ Оделяется цепким-спиртом, отжимается на воронке Бюхнерам !
высушивается; в сушильном шкафу при* IOOZ Очиденный,таким® ; образ ом ,коилил затвердевал, при 181*,3 0, тогда как тольвW промытый спиртом при 176*; обычно заводский ксилил проад! тый лишь?водой затрерл.евает при 164’ Д
, Ц|
.. ; '' к Т^даТРОфЁНИЛМЕТИЛНИТРАМИН. Ж
; • (Тетрил.) ' Ж*
VI- Гун
; xN0t '
скн,м(он,\*|рнмо,-С.Н,(иодной ^нв,ч«,ии,о а ' & ' •' ?->, 1 • . . . 1<й.
5CtH5N^H^bHN0J=5ebH^0^N<^5C0^8H,p4^. Л
Тетрил был впервые получен в 1883 году oZf^
окл 1/ИХ/и~ОИЛОС. рЬ«/ «Pouj-b -Л-ovb 2, 108) Д^ц
Икййиеу-сне си азотной и серной кислот, как на метиланили'^ иа, дииетиланилин.. Впоследствии появился еще целы If других- способов получения тетрила. 20 гр. диметилаи. ; ’ лина приливают постепенно, по каплям к 200гр. серной кв>‘
слоты уде лью го веса 1,$4, причем колба охлаждается вод;
После остываЯИя, полученный сульфодиметиланилин по кац^ • приливают к 400гр. дымящейся азотной кислоты, удельно:;,: веса 1,48 при помешивании палочкой и сильном охлаждении
- 46 - I
смесью льда с солью. I
Получается темнокоричневая жидкость, ее оставляют ст®
ять до следующего дня, затем нагревают до 60’С на водяне бане до окончания выделения окислов азота в течении пят; шести часов.
Когда смесь остынет, то" ее выливают в избыток холодж воды со льдом; полученное твердое тело проживают водой i отсасывают на воронке Бюхнера. Для очищения выкристаллизовывают из ацетона. Температура плавления 127%,
На заводе в ЗЫлг<Л& тетрил готовится следующие образе
Постепенно в течении одного часа приливается 46 кило: технического диметиланилина к десятикратному количеству серной кислоты уд.веса 1,84 причем температура поддерживается 40’0, Охлажденная сульфокислота в количестве 125 килогр. приливается при тщательном размешивании к 50клгр азотной кислоты, содержащей 92£ моногидрат з НмО3 в течении 3-4 часов, причем температура должна быть в пределах ел плт л - - ' ' В Н И
60 м
п
Д 1
Н 1
F «• лк
Температура застывания расплавленного тетрила, пол УЙМ ценного от Рей иск о-Вестфаль око го завода (Тройсдорф) onjffl до 126* й этот тетрил имел слабо-кислый запах, тогда продукт, полученвдй научно-технической лабораторией МоЦБ ского Ведомства имеет температуру плавления 127* и сов^Ц шенно не имеет запаха, . ж Г
js I
Тетрил чувствительнее к удару, чем тротил и пикринса';|| кислота, поэтому едва-ли его возможно применять непосре‘11 ственно для снаряжения артиллерийских снарядов; но такш ' как его чувствительность ничтожна по. сравнению с грецу^И ' ми составами, то благодаря большей силе, превосходящей ;№ пикриновую кислоту и тринитротолуол и благодаря тому, Ш рн является энергичных возбудителем детонации, его пр/'Й няют в капсюлях, где на 0,8гр, тетрила приходится сверМ всего 0,5 гремучей ртути, с 15$ бертолетовой соли.
Фугасное действие тетрила превосходит тринитротолуол^ пикриновую кислоту, а именно, Югр. вещества, ----------
< двумя граммами гремучей ртути дали раздутие свинцовых линдров.
. Для пикриновой КИСЛОТЫ п п
При опытах в бомбе Сарро и Вьеля давление газов дльЛ тетрила на 21% превышает давление для пикриновой кислс;Р' а время горения в 10 раз менее; при этом надо заметить ‘ . что.тетрил легче воспламеняется, чем пикриновая кислот^ • -ч
, взорвацн^-свинцовых $'
,!
-ф
t1:
70* С. и > в : о
с о б т о б л .а т щх§.
е возможен взрыв).
О ч а
енно обращать
ы температ
70’ и ч т О бы
тельное р а
а Р и и
Корда влито 1|2 - 2|3 сульфокислоты , 1 20 килогр. азотной кислоты.
Тетрил,в чистом виде, представляет порошок из ромбиче| ских кристаллов бледно-желтого цвета, не имеющий запаха] и трудно растворимый в холодном спирте и воде.
•ют
н е л *е
е,
ы ш с ь
(инач(
У Р а про
3 М 0 Щ И 'В<а«|
и 3 ? о
то оке прибавляТ
360 к.с.
тринитротолуола,,.., 370 к.с.
тетрила,............ 480 к,с.
и трудно растворимый
в холодном спирте и воде, NO».
С
НС
CNO*
сн
Л m-*N 0 * c"N''eH,
<гу
1
ТЕТРАНИТР0ФЕ1ИНМЕТИЛНИТРАМИН.
(Пентил).
% sjt
о
10 грамм диметиланилина растворяются в Й00 к.с. сер кислоты уд,в, 1,84, По охлаждении раствора охлаждающе!';^
-48-
^смесью до -2 е к нему при тщательном.размешивании приливает смесь 12 грамм азотной кислоты уд.в. 1,49 и 60гр. сараной кислоты уд. в. 1,84, причем все время сильно охлажда-^ют (до-2’), После приливами кислотной смеси оставляют • еще стоять полчаса. При отом'Получеиная смесь состоит из ^мета-.и пара нитродиметиланилинов. Эта смесь затем посте--1 пенно выливается при размешивании в 100 куб.сан,, охлаж-j денной азотной кислоты уд .веса 1,49 и оставляют смесь сто-/ять при комнатной .температуре в течении недели. Пентид вывери ста ллиз овивается постепенно из раствора^ а тетрил оста» ется в растворе. Пентил отсасывается на воронке с стеклянной ватой и промывается четыре раза: сначала 80%, затем ’40£, 20% серной кислотой и, наконец, водой до нейтраньной реакции.
,• Пентил менее стоек, чем тетрил, р Поте выкристаллизовывания из бензола, высушенного, фосфорным ангидридом, плавится при 146-147* При-кипячении ^водой разлагается с образованием тринитрометилнитрбамАис^м-- . и я " авт пои 240* фенола с температурой плавления I83J 220 . и взрывав при 44V.
ТЕТРАНИТРОАНИЛИН, .1 (Те четыре),
t c.H(N0.),NHl
Это взрывчатое вещество впервые получено ' <
’нитрованием метанитроаналина . -
1
iA,Солонина и В.Крюков выработали способ получения тетра- ? нитроанилина и показали, что можно нитровать как смесью .селитрл с серной кислотой, так щ одной азотной кислотой, причем как так и А,Солонина и В.Крюков
пользовались готовым метанитроанилином (подробности в
- 49 - |
,5 части курса Технологии взрывчатых веществ А.Солонина/|н Здесь приводится способ veuvSXuinb приготовления те,и транитроанилина из анилина: 26 гр. анилина растворяются Ж в 700 к.с. крепкой серной кислоты и при охлаждении до -5* осторожно приливается при перемешивании смесь 16куГ« онм. азотной кислоты уд.в. 1,49 и 20 куб.сим. крепкой сш серной кислоты. Затем смесь при охлаждении стоит два ч^Я са и после этого постепенно при помешивании присыпаетсяЯ П7 грамм высушенной и измельченной калийной селитры,njw
‘ этом необходимо ©ладить, чтобы температура не поднимЛ
Лась выше 50* Дают смеси стоять 24 часа и после этого ж j нагревают 1'4 часа при 50’ и после того дают день стоятф|< Тетранитроанилин выделяется в. виде желтого порошка, 0Г'-,Ш отфильтровывают на воронке со стеклянной ватой, затем '4® промывают 50% серной кислотой и, наконец, водой до ней1:Ц1 ральной реакции. Выход 26 грамм с температурой плавлбН1|| 207* Выкристаллизовывают из ледяной уксусной кислоты.
при 2.Ж г®
• • >
j
Выкристаллизованный тетранитроанилин плавится
ПИКРИНОВАЯ КИСЛОТА.
Син$(он)+знг<о5» сьнг(мог)3он + знго
I. Получение пикриновой кислоты из фенола
Г*:/';
по
Фенол, САН фон') должен быть, по возможности, чистым и. неЬдолжен быть жидйим, так как в последнем случав он со-'^ держит много воды.
Химически чистый фенол плавится при 42,5 - 43 кипит при I ный прозрачней кристаллический фенол годится ния-пикриновой кислоты.
К ТО гр. фенола приливают осторожно 45гр. конце нтри-. Проф.а.А.Солонина - Лаборат.приготовл.взр.вещ. Л.^
180-185° по , Продаж- t:
ический фенол годите# для получе' ,
50 -
юванной серной кислота; смешение производят в сосуде с ритертой пробкой и после прилития серной кислоты взбал-ывают несколько раз смесь и затем дают ей стоять до тех ор, пока проба смеси, разбавленная водой, не дает осадка фенола, так как фенол растворяется труднее сульфофе-оловой кислоты; в колбу нали дается 75гр. азотной кисло-л (уд.в. 1,4) и начинают из капельной воронки по немно-у капать сульфеноловую смесь и по временам взбалтывают олбу; при этом нитровании выделяются краснобурые пары сислов азота, которые отводятся при помощи трубки в от-эр?тие тяги.
Затем смесь нагревают в продолжении 1-2часов на водной бане, пока еще заметно-действие .азотной кислоты. Жид->сть теряет своей первоначальный темный цвет и окрашивайся в желтый.
Если, как это иногда бывает (обыкновенно, если ршибоч-> взято мало азотной кислоты), отделяется темно желтое • !.сло, состояще, главным образом, из динитрофенола, то ся окончательного превращения динитррфенола в пикриновую слоту, надо выпарить на песочной бане смесь др 2|3 о0и-(а и вновь заменить дымящейся азотной кислотой убыль дности. •
Жидкость опять нагревают на водяной бане до тех пор,' ка проба смейи, будучи разбавлена холодной вбдрй^ W делит желтных кристаллов, растворяющихся в чистой аиря-й воде без мути и следов масла. Наконец, через еще го-у-чую смесь, продувают воздух; для этой цели опускают сте-/нную трубку в смесь и, соединивши эту трубку 0 каучу->' зым шаром или мехом, продувают воздух до тех пор, flpjta ->естанут выделяться краснобурые пары окислов азота»
сь из колбы выливается в избыток холодной воды,' кото**
। через некоторое время, когда пикриновая кислота ося-, при помощи сифона сливают и заменяют свежей.
При помощи фарфоровой воронки и водяного насоса филь-гот пикриновую кислоту и, растворивши при нагревании в
- 51 - .
не боль гом количестве воды (20ч.воды на 1ч. кислоты), ост ДМ в ля ют пикриновую кислоту вы крист ал лиз о еы ваться. 1|М
Выкристаллизовавшуюся пикриновую кислоту фильтруют ( опять при помощи фарфоровой воронки и водяного насоса )||Я сушат на часовом стекле или в фарфоровой чашке при темг’^Я ратуре 35“С до постоянного веса. Охлаждают в эксикатор •» Ц и затем взвешивают. пМ й
Температура плавления пикриновой кислоты должна быт1'| близка к Ш. '-j.l ei
Хорошая пи!финовая кислота при нагревании в течении .га I трех часов при 100° не должна изменяться в цвете и не1| должна становиться тестообразной !
I i
2. Получение пикриновой кислоты из бензола.
За недостатком фенола во время великой войш 1914
/ как у нас, так и загранице^, готовили пикриновую кисло^Ж
. из бензола, получая сначала хлорбензол, динитрохлорбен:
и динитрофенол*
< . :1Ж
Ю CH/^NcvNO *2.Н-5О Л
’•**/• 5 . J * . В
| Получение хлорбензола. ,?й'
о О I» Ч w ,,,Ли
Р Удобно пользоваться хлором выпуская его из баллона ( п| г жидким хлором. За неимением баллона с жидким хлором no.-ffl чают хлор обыкновенно действием соляной кислоты на пврч ||
- Ой -
дсь марганца (пиролюзит) по уравнению; '1
М Ми/Ог-ьАНС^=М^(Хгг+^НгО+СЪг 1
l:f, В колбу вместимостью 1-2 литра (осторожно, чтобы но | разбить стекло) насыпается слой в 3-4 сантиметра кусоч- :^ов перекиси марганца величиной с орех. Колба закрывает- j ц.я каучуковой пробкой с тремя отверстиями. Через 'одно У;
^верстив вставляется капельная воронка, вместимостью -Уа
1цр0-200 куб. сантиметро в, через другое отверстие вставля- ; ^.гсв газоотводная трубка, а через третье стеклянная два j :^1за согнутая трубка, конец которой погружен, в. рту ть,на-^рдящую в цилиндре под крепким раствором едкого натра.
Для получения хлора выливают из капельной вороги Цепкую соляную кислоту и нагревают колбу. Хлор промывайся водой и высушивается серной кислотой или хлористым ’}льцием. В колбу,соединенную с обратным холодильником ^сыпались до верху железные стружки и. наливался до поло- : ,!,|‘1ны колбы бензол. Хлор пропускался через бензол, причем ^опускание хлора так регулировалось, чтобы температура Ч ^ржалась в пределах 30-40* В случае поднятия темпера- | t,|pj выше 40е колбу надо охлаждать, прекративнй ,струю ч$°Ра* Ч.Й
У.! Прекращают пропускание хлора, когда хлор заметно не, ] Дагирует, что видно по окрашиванию газа выделяющегося $ иолбы. Берут пробу жидкости и, если она будет иметь Цельный вес около 1,1, то больше хлора не пропускают, в ^отиэном случае продолжают пропускание хлора. (Удельный. г ;(;С бензола при 15* - 0,88). - ж
(I:, Затем жидкость из колбы промывается сначала раство~у|| Jm соды, затем чистой водой и отгоняют с дефлегматором^^ $бирают для дальнейшей работы фракцию 128-133. ,J||
$ Химически чистый хлорбензол кипит при 132е (бензол ки-Л| |’|.т при 80е) и имеет удельный вес 1,106 при 21. 'В
а . : *4. • : . ' '
f ' . ' у I
- ' ' : I
- 53 -
Нитрация хлорбензола. й
• -л' 4
CbH5&t+^NcbN0J + 2H45Oli = CbHf(NO#)jtCl, +2<NavH3O/) + ' ' ^1
В колбу вливают 77гр. серной кислоты 95$ и всыпают
Игр. натровой селитры, тщательно перемешивают и постер пенно при перемешивании и подогревании приливают 10 грЗ1: хлорбензола наблюдая, чтобы температура в колбе . нахо.Ч лась бы в пределах 85-95?
. Когда прилит весь хлорбензол, то постепенно при разг| шивании прибавляют еще Игр. натровой селитры и нагревг ют колбу тех пор, деет.
После
водой.
до 120-125’0 и выдерживают эту температуру пока взятая проба масла при остывании не затве
остывания, содержимое колбы промывается кипяще
Омыление.
С Н (NO.YCt+irl^cvHO = C H.flVO^ OWcv-»- NcvC^ + H.O / о "/*' • Эк *• I
В Стакан помещается 50гр. динитрохлорбензола и растяу 25гр.едкоРо натра в 33,5 к.с. воды. При размешивании с5*1 , держимое стакана нагревается до 100“ в течении часа. За । । тем в течении получаса через стакан пропускают водяной ^наблюдая, чтобы пар перешешивал хорошо содержимое стака,;, ।
'• ' ,'|Е
Нейтрализация (выделение динитрофенола). ;|:1'
В'стакан наливалась слабая серная кислота, полученн; '' разведением I весовой части 90$ серной кислоты в 5ч. во, ; в расчете на 10 куб., сантиметров раствора фенолята 5 к.< I ? разведенной'серной кислоты^
К/’ Фенолят постепенно приливается к серной кислоте при у
°*. I
I :
||>шем размешивании.
По охлаждении, поджученные кристаллы динитрофенола от-г ^.сываются на воронке Бюхнера и промываются золодной во-I пока промывные воды не будут давать с раствором ля-* ;Ыса осадка.
; Затем динитрофенол раскладывается на фильтровальной J гу|маге и сушится при 70-80“
Обыкновенно полученный динитрофенол плавится при 106-
11“ (Славянск). '
Химически чистый 2,4 динитрофенол плавится при ИЗ -ь* . а 2,6 динитрофенол при 63-64*0.
Получение тринитрофенола.
/NO^dH^NO^H^O^C.H^NO^OH+^HSO^^H^ 1
В стакан помещается 50 гр. динитрофенола и 140 гр. 95% •ной кислоты и затем стакан нагревается до 60“ и тща-:ьно размешивается; при продолжающемся размешивании •/ У||торожно,. небольшими порциями, прибавляется 50гр. чилйй-Йон селитры, наблюдая, чтобы температура находилась бы ,< . ^пределах 100-II5*. Когда вся селитра прибавлена подыма-| температуру до 125-130“ и держат ее в течении I-TJ4 ча-.^,в. Конец реакции узнается взятием пробы: I грамм
ьдцества должен растворяться без остатка в 20к.с. кипящей $ды. . л
По окончании нитрации все содержимое выливается посте--л)нно при размешивании в большой стакан, где налита хо-!^дная вода в количестве 500 куб.сим. После отстаивания '|кантацией сливается вода, которая заменяется свежей.Вй-(илившаяся пикриновая кислота отсасывается на воронке (нхнера и промывается холодной, водой, пока проба хлори- Йым барием не будет давать осадка. Отсосанная пикриновая • Лслота раскладывается на фильтровальной бумаге и сушится j •й’и 7о: ' 1
3. Получение пикриновой кислоты нитрацией бензола в присутствии ртутных солей.
(патент csu 194883 4 Авг.1906г.)
В колбу с обратно поставленных холодильником наливает: ся 40 грамм бензола, 66 грамм азотной кислоты уд.в. 1,48* и 5 грамм азотнокислой ртути. Колба нагревается на вода-1 Мой бане, причем время от времени производят взбалтываний Когда перестанут выделяться окисли азота еще продолжают ' нагревание при частом взбалтывании в течении часу и зате 1„' из колбы отгоняют нитробензол, остаток, содержащий пикрин1" ; вую кислоту выкристаллизовывается из горячей воды. j
Растворимость в воде пикриновой кислоты в 100 частых воды 5
при 0* 5* 10* 15’ 20° 25" 30' 4C'j
1,05 1,0*7 1,10 1,16 1,22 1,37 1,55 !,£•?!
при > 50е. 607 70° 80“ 90“ Ю0“ ')
2,53 3,17 3,89 4,66 5,49 6,33
'/ Г'ч
Обыкновенно получается 18 грамм пикриновой кислота дл растворения которой нужно 285 куб.снм. кипяцей воды, kot^.J рая при охлаждении до 15* должна выделить 14,7 гр. пикр^1 новой кислоты.
Больший выход пикриновой кислоты получается, если зна3) читешьно увеличить количество азотной кислоты, а именно,^ если на 40гр. бензола взять 135гр. азотной кислоты уд.в.!,|( 1,39 и 5 гр. азотнокислой ртути. Получается 38 грамм пик);; ринов ой ли слоты, 16 грамм нитробензола и 0,2гр. ортонит^ • фенола. ' , ’г
'Si
s J *
4. Получение пикриновой кислоты из анилина переходя через сульфаниловою кислоту.
(Патент W-«zn^ivo|-|-eAz'<su 125096. от 6 Сент. 1905г.)
Получается сначала из анилина сульфаниловая киаиота, с,н^нднзо9)*нго .
колбу наливается 100 гр. серной кислоты уд.в. 1,84 ж
йри взбалтывании постепенно приливаете» ЗОгр. свеже пере- ; угнанного анилина (темп.кип. анилина 182 - 184*), затем •1|»олба нагревается на масляной бане при 180 - 190“ до тех (|ор пока проба от прибавки едкого натра больше не выдел я-< У|т анилина; для этого требуется около 4-5 часов нагрева-ждя. ',
я После остывания содержимое колбы выливается в холодную ||оду и выделившиеся кристаллы сульфаниловой кислоты от- j Йильтровывакгс® на воронке Бюхнера, промываются водой и vi выкристаллизовываются из воды с двумя молекулами воды. ||ыход около 30-35 грамм. (При 10* растворяется 1ч. в 166 |.воды).
у 30 Грамм сульфаниловой кислоты взбалтывается с 250к.а ^оды и с 12 граммами азотистонатриевой соли.
'I После тщательного размешивания, осторожно при сильном jj-хлаждении ледяной водой, приливается около 100 куб.снм. п1хлаждэнного раствора 10 грамм серной кислоты удельного уеса 1,84 в 100 к.с. воды. ,2s
Nct,NO +H,30 «HNO. + Nc^HSO, , X ** *• л *1' •
st*
ebH.(NHaXH50J)+HNVCbH^1t5O,-2HiO,
•4
-ОН ^Н^ЗО^н^О »CfcH< *Nt ,
После приливания серной кислоты при охлакдении смесь;д время от времени размешивается в течении одного-двух иа» ’7 сов и оставляется стоять в течении двух часов при о бык-! \] вовенной температуре. Полученная пористая масса отфиль- тровывается на в о ранке Бюхнера, перемещается в стакан и1 ( затем при-размешивании и подогревании на водяной бане приливается 96 грамм азотной кислоты 40\й-ё/ (уд. в. I ,38) J 1
По окончании подливания азотной кислоты, производят нагревание до окончания выделения газов и затем оставля'н ют стоять в течении суток. ч;
Выделившаяся пикриновая кислота отфильтровывается на воронке Бюхнера, промывается холодной водой. !.'•
Из 100 весовых частей анилина получается обыкновенно ; около 220 весовых частей пикриновой кислоты.
t: I.
ПИКРАТЫ.
•г ‘I
Водород гидроксила ОН пикриновой кислоты: СЬН4(|УОД ‘-I может замещаться металлом, и тогда получается пикрат^та’/* как пикриновая кислота не имеет группы карбоксила-&^.®н . то она, собственно, неправильно называется кислотой, тсi1 но так же, как фенол называется карболой кислотой. 11
Правильное название пикриновой кислоты - это тринитрН фенол, а поэтому и пикраты, это, собственно, не соли, а’1; феноляты, т.е. соединения, аналогичные алкоголятам.
Известно много разных способов получения пикратов; ь из них рассмотрим два главных: I-й способ - получение ’>• пикратов из пикриновой кислоты и солей металлой, 2-й способ - получение пикратов из пикратов, полученных по 1 первому способу и солей металлов.
* 58 ~
I,*; I-й способ. Получение пикратов калия, натрия,ба-$.я, стронция и магния производится, в сущности, Одйнакс-
в стаканчике доводится вода до кипения и в ней раство-^ется пикриновая кислота (на 20ч. воды -» 1ч* пикриновой ’^слоты по весу). К горячему раствору прибавляются уГлеки-fy'jue соли соответствующих металлов.
f’!l Количество углекислых солей расчитывается по теории.
:';1 каждую частицу поташа 1£гС03,соды Мсл.гСО3 и углекислых: ба-& ’Йчя/С0. стронция и кальция СсиСО/ надо ПО 2 ЧАСТИЦЫ
I -1 ** *
^криновой кислоты; это видно из указания Состава Пикра-
1^В .
fl C.H^NO^OK.;
Й e^NO^OHo. ;
Я Пикрат бария содержит 5 частиц воды после высушивания Л постоянного веса при 50’ и теряет большую часть воды || высушивании при 100-12 (X
я
[С4Н»(«0МмЭ- •
$3иая атомные - веса элементов (С =» 12; N«I4; 0 = 16;
•^23; JHc|=24; Ccva40; 5^= 88; 137), легко
Числить весовые отношения; напр., на Югр. пикриновой ^лоты 2-СьНг(МОг)3ОН (вес частицы 2.(12.6+ 2+ 46 . 3+17)= (1’58) надо взять безводной соды Мсх^СО, , ’1-вес частицы .
2 + 12 + 16 . 3 = 106 всвго,^^.»ак.ЛЛ-1|о.(М)-/-чПосле прибавления указанного количества углекислых со-л1
.1'. доводят жидкость до кипения и помешивают стеклянной ,1'очкой; затем еще прибавляют одну-две крупинки углеки-1й соли и смотрят, - выделяется углекислота или нет. В 7леднем случае фильтруют горячий раствор ^ри помощи во-,.ки и водяного насоса; профильтрованный раствор ©ставится кристаллизоваться на сутки, или более, - в завися-
59 -
мости от выпавшаго осадка. Затеи пикраты отфильтровывают''' опять при помощи фарфоровой воронки и водяного насоса и расклады в аюися после отсасывания насосом маточного раствор ра, на часовом стекле или чашечке и сушатся до постоянно- / го веса при температуре не выше 50* 1
Пикрат аммония можно также бы было готовить, исходя из .:i углекислого аммония, но проще - к раствору в горячей воде :', пикриновой кислоты прибавить нашатырного спирта; прибавля ется столько нашатырного спирта, чтобы после 10 мин. кипеЛ ния раствора он еще выделял бы пары аммиака; проба - кра-J сной лакмусовой бумажкой. Затем раствор фильтруется и т.д/-. как и раньше.
Расчет ведется для пикрата аммония по формуле: V-
C6Ha(N04),0H+NH^CfcHt(N0t)30NH, 1
но, конечно, надо предварительно определять крепость на-шатырного спирта аре омет ром'и по таблице №4, находящейся
'1л1'
в конце записок, определяют процетное количество аммиака
2-й способ. Получение пикратов железа ртут!?.,
-Н^,цинка - 2+v и меди -Cur,
° К'раствору пикрата бария (1ч. безводного в 85 ч. воды,4''
нагретого почти до кипения, прибавляется насыщенный при’;1^ обыкновенной температуре раствор сернокислых солей железу или цинка, или ртути, или. меди, тоже нагретых пости до к>, пения. Еше горячий раствор профильтровывают для удаления " осеввего сернокислого бария при помощи фарфор’овой воро:1( ни и водяного насоса; при получении пикрата ртути необходимо особенно быстро фильтровать, так как в холодной вод!^
пикрат ртути мало растворим (1ч. пикрата ртути растворим/ лишь всего в 1200 ч.воды на холоду). ’ ’
С профильтрованным раствором поступают, как указано i)!
раньше при пикратах калия и т.д.
Для расчета количества сернокислых солей привожу сост | пикратов»
60 -
I мм. Ме.М^,<М«.о, M-K,aaS^&lHl(N0,)s0]i5H,0 , сернокислых солей;
Температуры взрывов стронция. . . бария .... железа. . . . свинца. . . . меди.............
Ликрата
- * -(i!
И
4 - * -
т
Z^S0^.7Ht0
И ZQ CwSOj,. 5Н*О &,50..7h'q
* *> Z
351’ 340 е 300’ 260*
310*
пикрата - " _
_ И _ И
_ И
цинка. . . . 315 аллюминия. . 320 калия. . . . 325 никкеля. . . 340 олова. ... 360'
и н И т ?
С.Н('СН!ХКО.)ЭОН. 3
»5HtO )
приготовления тринитрокрезола употребляется продаж-' --- ------------------- - г—’ изомеров орто, мета и
р
ОКРЕ
Й ••• & ' Для ж $й крезол, состоящий из смеси трех ^•ра: гн
с,
ЗОЛ,
а! нс
h! нс'
сон
сн
сн
не
сн5 С
сон
сн
сн
не
не
сн%-С
сн
сн
е он
'Ж - 61 - ' I
у® Этот крезол представляет жидкость, обыкновенно окра- }1’ 'решённую в буроватый цвет и перегоняется в пределах 190 - 'Ш •жЗОЗ’С. Получение трйнитрокрезола в общем похоже на по» ;иь уЖлучение пикриновой кислоты, но существенная разница в
жтом, что для получения тринитрокрезола надо затратить -'и •и гораздо более времени, так как от замедления процесса ' » ? нитрации значительно зависит успех работы. I®
Сначала надо приготовить сульфо крезоловую смесь:' ук.
' . СН3СьН^ОН) + Нг5О^СН}СкН3(+1503)0Нм-Нг0 '1
К ЮОгр. крезола в склянку с притертой пробкой прили- н вают тонкой струей 260 гр. серной кислоты 62-63’ па Боме.и Смесь сильно взбалтывают несколько раз и оставляют сто-ять на сутки (до тех пор пока проба в воде на-цело растворяется).
Азотная кислота, в количестве 800 гр. З7’по Боме, на- ' . ливается в реторту, а сульфокрезоловая смесь по каплям 1 ' выливается из капельной воронки.
з Сульфо крезоловую смеаь выливают по каплям в 5 приемов;' йд 1-ые 80 гр. выливают по каплям в продолжении 30 шн., во две© ярема капания взбалтывают реторту. Потом около минуты1^ ^продувают воздух; для этого, вынув пробку из тубулуса ре-1’ :торты, вставляют стеклянную трубку и, надев на нее. каучу-новую трубочку, при помощи каучуковых шаров или меха осто рожно продувают воздух через азотную кислоту. Затем, да-1 ют стоять; на стояние вместе с продувкой требуется около Ук часов.
Затем, в каждые выливают по каплям всего по 70гр. т.е. на 280 гр. требуется 6 час. Затем, вторично продува-^: >т воздух до прекращения выделения краснобурых паров оки- i слов азота. Вся смесь из ретортах выливается в сосуд с ^большим количеством холодной воды, которая после ртстаива 1 нил тринитрокрезола заменяется новой, и т.д. до тех пор, ^пока лакмусовая бумажка перестанет указывать присутствие кислот.
Затем, при помощи фарфоровой воронки и водяного насоса-
4^ 1
q -62 - Ж
^отсасывают твердый тринитрокрезол. Тринитрокрезол перемеЖ.
мщают из воронки в стаканчик, куда наливают воды, прибли-|Г Шзительно в 20 раз бодее, чем получено тринитрокрезола и,^ Шпомешивая палочкой, плавят тринитрокрезол, затем еще ели--мвают тщательно воду, а тринитрокрезол высушивают при 60* ,^до постоянного веса и охлаждают в эксикаторе над серной Шкислотой. Чтобы получить брлее чистый продукт, надо раст-'. вдворить тринитрокрёзол в горячем винном спирте С4Н5(рн) Йи выпавшие, по охлаждении спирта кристаллы, отфильтровы-кивать при 60*0, а потом ~ в эксикаторе над серной кисло-Ытой.
|| Заводский тринитрокрезол плавится при 82 - 92*, а выкри-горячего спирта при 102-104°.
Йсталлизованный из ы
-Ч
У
н
ТРИ
КРЕЗОЛЯТЫ
И Т Р о
1 СЦин водород ^гидроксиле, так
в
частице же, как и
тринитрокрезола, а именно в в пикриновой кислоте, может за» I мешаться металлами и получаются, так называемое, тринитрд> крезодяты.
Например, тринитрокрезолят натрия
C»CHaH(NOj:ON<>v
До 1900 рода были известны лишь тринитрокрезоляты аммония, калия', свинца и серебра. Тринитрокрезоляты натрия и меди получены впервые А.Солонина.
В 1901г. по/учецы и исследованы в лаборатории Военной Электротехнической Школы, тринитрокрезоляты бгрия и стронция Поручиком Лазаревым по указанию А.А.Солонийа.
г'- Можно тринитрокрезоляты получать теми же двумя спосо-^бами, как и пикраты, только тринитрокредол, вследствие ?р’плохой растворимости в воде (1ч. тринитрокрезола раство-^ряется в 449 ч. холодной и 123ч. горячей), не растворяет
I ’ - 63 - и
pg, а расплавляется на водяной бане в стаканчике под ело ; к ем воды. Затем поступают так же, как при получении пикра '
TOB.
*' Температура взрыва тринитрокрезолята бария около 318*?
Ь'
ТРИНИТРОНАФТАЛИН.
|о 5 V
V*
.' ?£• ?
= V-
Для приготовления тринитронафтадина сначала пр^^трв-i1 ляют моновитронафталин, а уж из него потом готовите^ триГ|? нитронафталии»
Нафталин, употребляющийся для получения мононитронаф-fy* талина, должен быть, по'возможности, чистым, что поверя-Ч-ется температурой плавления, которая для химически чистой го нафталина, по еЛфЫлл'и равна 80,06 0.
• По &иЛЛк',и поверяют чистоту нафталина тем, что ? в фарфоровом тигле расплавляют ГЛ грамма трёххлорцстой । сурьмы (темп, плавления 72*0) и затем прибавляют малень-i : Кими порциями небольшое количество нафталина; при нечистом нафталине получает к росное окрашивание.
В стакан помещают ЮОгр, тонко измельченного нафтали-^ на и приливают туда 600 гр. азотной кислоты, удельного веса 1,33 и затем в течении целого дня производят перемешивание пеФтМО! стеклянной палочкой или лучше лопаточкой, сначала до тех пор, пока он не растворяется, а S^SMfвозможно чаще; на ночь оставляют стоять и на другой день тоже чаще помешивают. Еще дня на два j
оставляют нитроваться и производят в эти дни по временам помешивание, Когда не мешают, то закрывают тщательно ста-
J1!
Н - 64 - . .
I кап стеклянной пластинкой. Таким образом на нитрацию J I Необходимо не менее 4~х дней, |
, | Затем, промывают получившийся нитропродукт струей хо-'] | лодной воды из под крана в* течение I часа, причем в на-; ! чале промывки производят помешивание стеклянной лопаточ~. I. I "кой, После этого продукт отсасывают на фарфоровой ворон-1 ; I ке, пользуясь водяным насосом и сушат при 40? После суш-, | ки охлаждают и ставят на сутри в эксикаторе над серной ;
I кислотой. Проба, мононитронафталина должна иметь темпе-| ратуру плавления 56-60*0, что близко к температуре ;|цдавления,рс. - мононитронафталина, который будучи хими-йчески чист, плавится при 58-61.
Н И0г л Л
Н Н
$ Высушенный мононитронафталин растирается в ступке С $22 граммами высушенной и тонко измельченной натровой сердит рой NcvNOj ; затем в стаканчик наливается 80 грамм ’ $ серной кислоты крепостью около 66* по Бома, J В зависимости от крепости серной кислоты, она долж-^ Й на быть остужена или нагрета; так, если серная кислота:^
;'г крепостью 65,7 по Боме, ТО остужается до 0е 0
I?, 65,6 W W W и ” 4е
(I " 65,5 н W W W • 8*
« 65,3 п W и и ” 16е
j и 65,2 и W W и ” 20’
крепостью 65,1 по Боме, то нагревается до 24 М
" 65,0 " * " " " ЙЯ“
I В серную кислоту сразу опускается смесь мононитронаф-талина о селитрой. Смеоь нагревается на водяной бане и производится размешивание стеклянной палочкой до появле- •! ния краснобурйх паров. После этого нагревание на водя ной бане продолжается еще в течение 2-х часов и в то же вре- 'Н' мя, по возможности, чаще продувают воздух. ВД
После окончания нагревания продувают воздух до тех пор, пока температура не понизится до 75е0. Затем прилй- Ж вают холодную воду (возможно более), помешивают палочкой несколько минут и оставляют стоять до .другого дня.
На другой день промывают холодной водой до тех пор, пока вода не будет давать кислой реакции на лакмусовую бу^-' мажку. Отсасывают на фарфоровой воронке и сушат часов 5, Г:' сначала при 60* а затем в эксикаторе над серной кислотой^?1 до постоянного веса. Точка плавления полученного продукта|!; 90-170* у.
Исследование, произведенное профессором Артиллерийской^ .Академии Академиком Ипатьевым, показывает, что при завод-<• ском получении тринитро нафталина в описанных выше услови- -I ях получается тринитрон афт ал ин, состоящий, главным обра-зом, из # « изомеров.
I'
и
ДИНИТРОНАФТАЛИН. 1J
Динитронафталин можно получать двумя способами: i
1)нитрацией нафталина
C„Ha.lHN05=e,.Hb(NOi)t>%H10 ;
2)нитрацией мононитронафталина
%H,(N01)a.HNO,.eiXroj)CH»0
Проф.А*А Солонина - Даборат.приготовл.взр.вещ. л«э ;
- 66 -
i || Получение по первому способу (/Jtcptivvb'):
; •$ 15 грамм нафталина небольшими порциями при размешива-; $ии прибавлялись в стакан с 50гр. крепкой дымящейся азот-; Мой кислотой, после прибавления нафталина смесь кипяти-• .йась около 4 часов, затем выливалась в избыток холодной ’ Воды, твердый осадок промывался водой, отфильтровывался ! *»а воронке Бюхнера и затем после высушивания между листа» ‘йи фильтровальной бумаги, высушивался еще в сушильном шка-уму в. течении 4 часов при 100*
Я Высушенный продукт обрабатывался спиртом до тех пор по-ла остаток не плавился ниже 2П. Остаток этот представля-
• динитронафталин с небольшой примесью Ji соединение Получение по второму способу:
грамм мононитронафгалина растворяется в 160 граммах
Верной кислоты 66“Ж>«, (уд.в. 1,84) и затем очень медленно Приливается 32 грамма смеси равных количеств крепких сер-|;юй и азотной кислот. В начале образуется темнокрасный раствор, который скоро светлеет с выделением окислов азота образуется желтая каша, из которой отжимают отработанную кисло туи затем про1А1вают теплой водой, затем слабо -Цалочной и под конец чистой водой.
I.'
Й . 40
НИТРОБЕНЗОЛ.
ж Хотя нитробензол и монотрофенолы не могут употреблять^ j Цзя, как самостоятельные взрывчатые вещества, но приводят-^ j жзя потому, что употребляются, как составная часть некого^ ।Мых динамитов и, взрывчатых' студней, например, взрывчатую,Л ’ жке ла тину (стр.225) мбжно
a.-
г ' ”67 “ .1
приготовить из 8Й весовых частей нитроглицерина,10 весо - вых частей нитробензола (или динитробензола) и 8част ,| растворимого пироксилина. Нитробензол:
C.H.NO,,
CjvHN0)-eAN'VHJ?
К 150 гр. крепкой серной кислоты, которая находится
колбе, вместимостью около ‘4 литра, приливают постепенно, *il при взбалтывании, 100 гр. крепкой азотной кислоты (удели веса 1,4). После того, как теплая смесь охладится до ко? j ватной температуры от погружения в холодную воду, приба^;; ляют постепенно, при частом взбалтывании, к смеси 50 гр ; бенасла» Если температура при этом будет стоять выше 60*~ то погружают при дальнейшем вливании бензола сосу/ Л воду на короткое время. К колбе приспособляют полни» ? щуюся вверх трубку и нагревают в течение часа, при част • взбалтывании, на Водяной бане, при температуре около 6<Г/*’ (термометр в воде).
После охлаждения отделяют нижний слой, который состс.. ит из серной и азотной кислот, на разделительной воронь., от верхнего слоя, который содержит нитробензол. ,
Последний несколько раз взбалтывают в разделительной •{ воронке с водой, причем теперь наблюдается то, что нитрс^ бензол образует нижний слой.
После прошвания переливают нитробензол в сухую колб}, и нагревают с хлористым кальцием на водяной бане, пока, и убившая сначала молочной, жидкость не сделается ясной. Н) i ^конец, его очищают пере гонкой из фракционной колбы с приспособленной к ней удлиненной трубкой. И
Точка кипения 206-2Q7? Выход 60-70 гр. »
ДИНИТРОБЕНЗОЛ,
ш< смеси 85 гр. крепкой'серной кислоты и 15 гр, дымящей-Жзотной кислоты прибавляют постепенно (под тягой) Югр. |вобензола и. нагревают, при частом взбалтывании, в, откры-Цколбе '/г часа на водяной бане. Немного остывшую смесь, В взбалтывании, выливают в холодную воду, затем застывший нитробензол отфильтровывают, промывают водой, растирают Влиняной тарелке и выкристаллизовывают из спирта, температура плавления 90® выход 10-12гр.
О'емпература плавления показывает, что при таким способе 4ации получается метадинитробензол, т.е.| следующего i|e ния
НС
CNO,
At
е-н
й
•5
НС
CNO,
А»
СН
|как ортодинитробензол плавится при П8’ а парадинитро (Нол - при 171.
- 69 ~
ТРИНИТРОБЕНЗОЛ.
CA(N0l)5 .
<\i<*3hno4’C hJno,V3h,o . >,•
Oto 9 Ь Э 4 X/ 9 X
w ' * a
1 Из многочисленных от особое получения тринитробензол 8^ (ив тротила и др.) указывается лишь способ непосредстве^ ной нитрации, выработанной еще в I9II году А.Солонина. yij
В колбу с обратным холодильником наливается 30 грамму азотной кислоты уд.в. 1,5 и 75 грамм олеума, содержащего, на 100 весовых частей моногидрата Нг50^ около 50 вест» частей сеяного ангидрида и затем всыпается 10 грамм мет;,-динитробензола, смесь сально взбалтывается и нагреваете сначала при 80е в течении 8 часов и затем 16 часов при 120** на масляной бане. После этого берется проба и есл'' окажется заметное количество динитробензола, то смесь?', продолжает еще нагреваться при I40-I50* до исяезновени(* дивитробензола. Затем содержимое разбавляется толчением^' льбом, затем осторожно холодной водой; тринитробензол он сасывается на воронке Бюхнера и выкристаллизовывается и-< спирта.
Температура плавления шкристаллиэованного продукта равна 121<-122в и поэтому тринитробензол представляет ct/ метрич еский изомер
CNO.
А/
- 70 -
JJ МОНОНИТРОФЕНОЛЫ.
I. ' СЛЙН^О,) .
; ! 80рр. натровой селитры растворяют, при нагревании, в
I 200 рр. воды и к охлажденному раствору прибавляют, при | помешивании, ЮОгр. концентрированной серной кислоты. ЖСмесь, находящуюся в стакане, охлаждают затем до 25е и ^прибавляют к ней по каплям из воронки с краном, припо-исто ян ном помешивании термометром, расплавленную при по -Моши нагревания смесь из 50 гр. кристаллине скоро фенола Ни 5 рр. спирта.
В Температуру поддерживают между 25-30“ погружая ста-Цкан с смесью в холодную воду на более или менее продол» I житель ное время. Если фенол застывает в воронке, то его снова расплавляют, подогревая слегка небольшим пламенем.
IT Реакционную смесь затем оставляют стоять около двух часов, часто перемешивают и разбавляют двойным объемом во-; |ды, причем реакционный продукт собирается на дне сосуда * |в виде темного масла. Большую часть водной жидкости ели--Ь|вают, масло промывают небольшим количеством воды и пере-' !роняют его водяными парами в 54 литра воды до тех пор, по-
|ка не будет более переходить О -нитрофенол.
J Так как о - нитрофенол засоряет трубку холодильника, ; I вто поэтому, когда получится много твердого тела в труб-Яке, выпускают воду из холодильника. Д
। Я? ; По .охлаждении, дистиллят фмьтруют; о -нитрофенол про>я ‘ Вмывают водой, отжимают на глиняной тарелке и высушивают i а в эксикаторе. Он не требует дальнейшего очищения, т.к. 1 i «выделяется в совершенно Листом виде. Что бы получить остав^ Цшийся в колбе нелетучий Ъ -нитрофенол, содердимое охлажЦ >8 • ,. ,<?
(Задают, погружая ее в холодную воду, отфильтровывают вод-/Ц
Жный раствор от нерастворимого остатка и кипятят фильтрат®
71
F !4 часа с животным углем, прибавляя постоянно воду, по м Г ре ее испарения. После этого животный уголь отфильтровы вают, а фильтрат оставляют стоять ночь в прохладном нес*
причем р -нитрофенол гыделяется в виде длинных,, почти'1 безцветных игл. Масло, оставшееся в перегонной колбе, к 1 пятят со смесью: из I об"ема концентрированной соляной1/ слоты и 2-х обпемов воды с некоторым количеством живота!* го угля; раствору дают несколько охладиться, затем фьльэ труют его и оставляют стоять на ночь. aii
Таким образом'получают вторую кристаллизацию.: Если в? делившиеся кристаллы не чисты, то их кристаллизуют еще 1 раз, с прибавкой животного угля, из разведенной соляной кислоты.
Температура плавления орто-нитрофенола 45.
Температура плавления пара-нитрофенола 114*
в*
А ;
I;
Р 5
И 1°
1
1 I
I
t
[I
, I )
- 72 -
Jr ft
Одноатомные фенолы бензольного ряда, в противоположу ность соответствующим углеводородам, очень легко нитруются. .
При нитровании бензола, например, для лучшего отщепления воды, должно прибавлять концентрированной серной кислоты, действие же одной азотной кислоты на,фенол уже настолько энергично, что кислоту для реакции должно разбавлять водой.
При нитровании феноле образуются одновременно О ж нитрофенолы, из которых первый - летуч с водынми парами;
। При нитровании £омологов фенола нитрогруппа входит [ также в 0- и jo- положения к гидроксильной группе, но никогда в vw -положение. Чтобы приготовитьvvu-нитрофенол должно взять ktu-.нитроанилин, продиазотировать его и. джа-зораствор прокипятить.
Нитрофенолы показывают все свойства фенолов. Вступле-
J нием отрицательной нитро-группы кислотный характер фе-$'нола- настолько усиливается, что нитрофенолы растворяют-ся не только в едких щелочах, но также и в углекислых.' Вследствие этого из щелочных растворов нитрофенолы нельг выделить углекислотой. Нитрофенолы обладают, кроме тоге Г еще свойствами нитро-соединений? при энергичном восста-;j, нов л ен и и они переходят, например, в амидофенолы и т.д.
ГЕКСАНИТРОДИФЕНИЛАМИН
Впервые получен 5<и4А-«лгоЬ сЯ/ил^м^'о-м^ нитров Ч пикрилнитроанилина, который получался по из
троанилина и тринитрсхлорбензола по следующим уравне 5*'
- 73 -
- ' > . S-' ' -ft
! .№
обыквовев^
2С6Н^М0г)МНг= NH(C(iHx,NO4)[CbHt(NO1)^ -j
♦CjH^NO^Ge- = CbH,N04NHtH'C^
Для этой цели эквивалетные количества нитроанилина jy тринитрохлорбензола растворяются отдельно в кипящем спи{ те. Спирта берется такое лишь количество, чтобы при кипе ' иии вещества перешли й раствор; некоторый избыток спирта / не вредит. и|
При смешивании выделяется пикрилнитроанилин, который : промывается горячим спиртом и горячей водой; высушиваете^ при I00t
Для нитрования
NH(CtH,NO,)[e,H,(HOj,].«H NO, = =NH[c4Ht(N0,),]t‘2Hs0.
размешивают постепенно пикрилнитроанмлин, при ____________
ной температуре, в избыток смеси крепкой (дымящейся) азе -ной и серной кислот (1:3). После стояния в течении сутот ’ смесь выливается в холодную воду и отфильтровывается; пр г мывается горячим спиртом для , удаления смолообразшх ве щёств и растворяется в возможно меньшем количестве при rfl пячении в уксусной кислоте и кипятится с ift
вотным углем.
После фильтрования при остывании выделяются из уксусн кислого раствора желтые кристаллы, которые промываются Н| i холоду водой и высушиваются. Температура плавления 378’ । [ > Опыты показали, что можно гексанитродифе!’!
«Ниламин готовить прямо из метилдифениламина и даже'из ди/ фениламина действием азотной кислоты.
Так, например, при нагревании метилдифениламина с азо< I ной кислотой наступает быстро энергичная реакция; когда I энергичная реакция окончится, то нагревают до окончания 1 | выделения краснобурых паров окислов азота. Затем смесь в : ливают в холодную воду, промывают горячей водой и выкри-
- 74 - ' И
^сталлизовывают из .уксусной кислоты. • -Я
j| Химическая фабрику взяла патент на полу-Я
$чение гексанитродифеламина. Этот способ хорош тем, что ’я |не получается смолообразных« продуктов и продукт совершен-11 рно не содержит серной кислоты. - :
Сначала получается 2,4 динитродифениламина из динитро-^бензола и анилина. При действии на спиртовый раствор ани-м ;$лина, насыщенного сероводородом, действуют ос> -динитро- | ^лорбензол, фильтруют и после выпаривания спирта выделя- • |ется динитродифениламин. Затем 2,4 динитродифениламин «обрабатывают с 32*5^ азотной кислотой и затем уже нагре-ивают с азотной кислотой 46вВч/.Полученный продукт промыва- . датся спиртом и водой.
1 Один из практических способов получения гексанитроди-жениламина описан в ’
-Wtd-lAtr /910 N 1 C’/wjO. 16 , * '
Д I весов.часть дифениламина постепенно при размешивании вводится в 10 весовых частей Серной кислоты 98# и ^затем размешивается до растворения. }
| Раствор медленно (в течении 3-х часов) вводится в 10 весовых частей азотной кислоты удельного веса 1,50. При ’рливании раствора азотная кислота должна хорошо охлаждаться и при вливании все время размешивают. После вливания в течении I часа сильно охлаждают. Всплывающие jtoMKa надо немедленно погружать вниз, иначе может произойти воспламенение.
£ Затем сосуд, где находится вещество, нагревается на ' , водяной бане, сначала слабо и когда кончится энергичное выделение бурых паров, то до кипения, пока не будет бо-лее выделяться азотистых газов и охлаждается.
j По, охлаждении смесь вливается в 60 вес.ч. воды (т.е., /-)сли взято Югр. дифениламина, то в 600 куб. сайт.), пе-^вшивается и после полного охлаждения фильтруется и за-/ем. еще промывается сначала холодной, а потом и горячей
- 75 -
I водой до нейтральной реакции, опять фильтруется на фар-
I форовой воронке и высушивается при 50 - 60*С. 100 й. ди-;'; ;
[ фениламйна дают 170 ч. гексанитропродукта. fi;
•fs
ГРЕМУЧИЕ СОЛИ.
Гремучие соли представляют из себя металлические про-If изводные, так называемой, гремучей кислоты, эмпирически,^ состав которой -HCNO.
Строение гремучей кислоты до настоящего времени еще J' достаточно хорошо выяснено. Существует несколько формул строения гремучей кислоты. Долгое время держались предп..^< ложения, что гремучие соли - не что иное, как металличе ские производные нитроацетонитрила
CHNO- ‘
। 1 г (•
CN
но в настоящее время доказано, что это предположение не^ верно, так как получен нитроацетонитрил и он не сходе»!
-гремучей кислотой'. Наиболее вероятны формулы:
Григоровича
Н-Св№0
'и Солонина
N=CH -и/ C = NH
й
О
О
s
- 76 -
ГРЕМУЧАЯ РТУТЬ
-*) ен.-сн,он+о = сн.сно+н,о, 5 % О % /
. ^NOH ,
x)ch.cho+hno »не< + н.о 7 4 1 хсно 4 '
. .✓МОН л-НОН з)нС^ +о =нег' 7 ,сно соон
4)hc^N0H +hno.=c^^^-cooh + н.о
7 ^СООН 3 4N0,
Xz.z,N0H zzN0H
—соон = С —Н + 00.
У XNOt х'М0г Х
б)с —НН = HeNO*HNO,
7 XNO, 1
Должно обратить внимание на чистому ртути и кислот. Ртуть очищается пропусканием несколько раз . маленькими капельками через слабую азотную кислоту и, затем фильтрованием через полотно или . замшу.
Азотная кислота,употребляемая для нитрации, должна быть прозрачной, не должна ордержатв^ серной поверяется пробой с Хлористым барием быть осадка от прибавки этого реактива.
По _ -------
1886, 1370), ЮОгр. 90^ спирта наливается в колбу. Затем в стаканчике растворяется при обыкновенной температуретуре Югр. ртути в 120 гр, азотной кислоты HNOj уд.веса 1,34, Раствор ртути в кислоте нагрева-
КИСЛОТЫ,ЧТО не должно 1 4? civ ota/v‘p-vwZ'Sft'Uz, otvwiz. S
ется до 70чи уже после этого маленькими порциями посте- “ пенно приливается к спирту, при непрерывном взбалтыва-: НИИ. S
* Прибор оставляют стоять и если через часа не начнет--г ся выделения белых паров, то можно нагреть колбу на водя-) ной бане до начала кипения, т.е. до появления пузырьков s
' , (обыкновенно обходятся без нагревания). J
Когда реакция кончилась, то дают гремучей ртути отсто-i яться в течении часа и затем выливают содержимое колбы в 8 стакан с холодной водой, где гремучая ртуть промывается 1 несколько раз холодной водой до тех пор, пока в промыв- Z ной воде лакмусовая синяя бумажка не перестанет краснеть. t
в
Затем отсасывают на фарфоровой воронке, при помощи водяного насоса, влажную гремучую ртуть. Сушится при 50' течении 6-ти часов и затем в эксикаторе над хлористым кальцием.
Удается, как показали мои опыты, хорошее получение грумечей ртутив, если брать на 50гр, ртути 460 гр. азотной кислоты уд.веса 1,38 и 500 куб.сайт, спирта около 96уС; причем ртутный раствор не нагревался. Этими способами гремучая ртуть получается всегда Серова то-желтоватой, ' >
Чтобы получить совершенно белую гремучую руть, надо прибавить меди и соляной кислоты ( 1905, 1348
Wok^vtz и ) или, как показали
мои опыты, монохлордстую медь CwCt-. Аля получения белой ^гремучей ртути монохлористую медь прибавляют к спирту в ^количестве I до 2 граммов на 50гр. металлической ртути. > Для очистки фабричной гремучей ртути ее растворяют в ! растворе цианистого калия (на 1ч. K-C-JT 3!4 части воды; причем берут на 1ч. серой гремучей ртути - 0,83ч. циа-i НИ6Т0Р0 налип), S профильтрованному раствору прибавляет--ж'оя постепенно азотная кислота (на I гр, гремуч ей 'ртути -.-«2,3 к.С, азотной кислоты уд.веса 1,38, растворенной в
\с
t
I
I
I
ft i J.
!
- 78 -
8,9 к.с. воды). .ХдИ
Виде лившийся белый, очень мелкий, порошок промывают,^
водой (раз десять) до тех пор, пока лакмусовая бумажкач не перестанет краснеть после сохранения гремучей ртути под водой в течении не скольских часов. ,
Другой способ очищения, впервые испытанный мною, за-. , ключается в растворении гремучей ртутит в пиридине (на. один грамм серой гремучей ртути берется 6,9, к,сайт. пи-ридина). i
После фильтрования прибавляется постепенно вода (на .
( 1гр. серой гремучей ртути 100 к.сайт,воды) и, когда ося- < дет выделившаяся гремучая ртуть, ее отфильтровывают,про- ; мывают холодной водой (на I гр, гремучей ртутд - 2 0 Ок. q, , промыв, роды) и затем, высушивают в эксикаторе над СсМХг . из которого выкачивают воздух до тезе pop, пока не из* чезнет запах пиридина и, затем, еде до постоянного ве-са»
ГРЕМУЧЕЕ СЕРЕБРО.
Реакция ведется по Гей Люссаку и Либиху так же, как . z и для подучейия гремучей ртутц.
В колбу наливают 27 гр, винного спирта 90Х и к нему 5 приливают сразу охлажденный до комнатной температуры pa-1 створ; | гр, сервера (химически чистого) в 20 гр. азот- ** ной кислота уд,веса 1,30-1^8, и нагревают до начала кд-., пения; затем приливают еже 27 гр. спирта 90$ д оставля-:.' дт рри комнатндй температуру. ., .
^адрше поступают так же, как и с гремучей ртутью, но не рекомендуется сушить в бане при повышенной температуре, а прямо в эксикаторе над хлористым кальцием (лучше,
|Ч;' * 79 " -Л
f ! если 'эксикатор из темного стекла), который надо. поста^ш .|
к вить в темное место. Ж'
Не сушить более I гр. гремучего серебра, так как, онснр < чрезвычайно чувствительно к слабым механическим Ьлияни-ф ям и от удара, даже, может взорваться под водою. 3;
I'
t'
Г
н t
Исходным продуктом служит обыкновенно натрийзид | который на заводах в настоящее время готовится двумя . ''I способами. У!
1«.й СПрррб, §трт прсоб представляет собствен-^ НО усовершенствованный и Способ q !
W oUsU С^углги, 2У.5-О8А '
Способ ЭТОТ можно разбить на две фазы: ^получение натрийамцда действием аммиака на натри1;.; 'и Ч
2)получение цатрийазида действием закиси азота на ,-f натрийамид,
Получение натрийамида i ?
ХООгр* натрия постепенно расплавляются в никкелевой чг'( , ке с перпендикулярными стенками (диаметр чашки 13 сану.; И вышина 8 вант,). Предварительно натрий после разрезе,!, ния прошваетря дважды В безводной этиловом эфире (не- ; регнанным над-натрием). Чашка, еще др вкладывания НИ”, 1 три», прме'дается в железный сосуд и воздух из сосуда ; , тесняется аммиаком, тщательно высушенным пропусканием через колонны с едким кали и обрезками натрия. Когда
- 80 -
iecb натрий расплавится, то пропускают через натрий ам-иак под небольшим давлением, для чего трубку из кото-ой выходит сухой аммиак опускают в расплавленный натрий, ри опускании аммиака температура должна быть '260-270* ля контроля прохождения аммиака ставят проданную склян-у с небольшим количеством высококипящаго нефтяного мала. Аммиак для этой цели удобнее всего получать нагре-анием- в медном сосуде хлористого аммония с двойным по осу количеством гашеной извести} смесь перед накалива-ием слегка смачивается. Обратить особенное внимание ,что-ы аммиак перед впусканием в расплавленный натрий хорошо >i сушив алея. Через 4Иг - 7 часов реакция окончивается. Ко-ец реакции легко'узнать испытанием выделяющегося газа," именное выделяющийся газ по окончании реакции не дол-эн гореть после промывания водой.
Полученный натрийамид охлажадется до комнатной темпе-1туры в струе аммиака и три четверти его вынимается из аки и хранится в эксикаторе над металлическим натрием.
Получение натрийазида. • ,
Nev N H^+NjO = NcsvNj + Н гО •Nciz NН,+ Н,0 = Nci/Ho *NH, ^Ncv-NH +N,0 = NevN.*NcvHO + NH< A J, э 3
На одну четверть полученного в никкелевой чашке нат-_й амида из ЮОгр, натрия, действуют закисью дзота при. гревании 190* и при размешивании. Отклонение от 190 е три градуса в ту или другую сторону не имеет значения, .кись азота, удобнее всего получать нагреванием смеси ' вес. частей натровой селитры NcvNO, 20 вес, частей~ка- , йной селитры KzNOs и 19:вес. частей сернокислого 'дыни я(ЫН<|\50/(. Закись азота перед высушиванией 'Пропускает-т через раствор железного купороса ^50^. Высушивание ад-.
- 01 -
киси азота должно быть'очень'тщательное, что достигается пропусканием через ряд склянок и колонн,с серной кисло» той и хлористым кальцием. Обыкновенно реакция кончается =1 через 5 часов. Конец реакции легко узнать, так как по И окончании действия закиси азота уже более не выделяется до аммиак и, следовательно, красная лакмусовая бумажка' не d будет ’синеть. Охлаждается в струе закиси азота. Если реакция доведена до конца, то проба полученного азида у‘ натрия с водой не должна выделять газов. Ш
Полученная смесь .натрийазида и едкого натра растворяЛ ется в врде и по охлаждении по степенно при помешивании нейтрализуется слабой азотной кислотой до слабо щелочной^ реакции, Такой раствор уже может применяться для получе-^ ния азида свинца. ;;
11-й способ. Этот способ основан на окислении гидразина азотистой кислотой. Для того, чтобы получить хороший ВЫХОД ol.dz. Оклугих- 4l, 2,314^
брал (до 80%) сернокислый гидразин прибавлял к нему эти-, лат натрия (количества взяты по теории) и в присутствии \ спирта кипятят всю эту смесь с амилнитритом с обратным ( холодильником, После окончания реакции сначала отгоняется спирт, а затем водяным паром производится отгонка об:.
разевавшегося амилового спирта и оставшегося амилнитрита Реакция объясняется тем, что азотистая кислота дей-
ствует на гидразин по уравнению:
t Ы,Н +HNO, = N,H+2-Н.0 I.
№ Если же прямо действовать на гидразин азотистой киалс . 1^ той, то хотя и получается А/$Н но выход очень мал воле; ( V ствие действия ' на N^H азртистой кислоты.
Можно вместо амилнитрина взять этилнитрит
cJUi okX. OkMMz. 41, 2811)«. 5 грамм гидрат a ’
£ 7 гидразина вмешивается с 37,5 куб.сайт. 4 нормального ра* | створанЗтрий метила та и зат ем прибавля ется 12,6куб.самт. | этилнитрита и 50 куб.сайт, эфира. Через. 24 часа стояния ;
' ' ' ’ Г;
Я , ГТПИГОТОВЛ. взр.ввщ. Л.6;
получается NouNj который промывается эфиром»
Способ d't-o-ttv менее уд обе н^ так как полученким pin створом нельзя прямо пользоваться для. получения азида1 свинца, ado раствор содержит сернокислый натрий, а надо л еде этот раствор подкислить слабой серной кислотой Я 07г гонять М5Н; отгон поглощается раствором едкого натра.
Под руководством А.Сояонина вдувателем Артиллерийский Академии Костедким был получен гидразинJ? из него азид натрия следующим образом:
;270. грамм едкого натра растворялось в одном литре, во-ЛЫи раствор при. охлаждении насыщался хлором до увеличения веса на 200 грамм. Полученный раствор медленна !?£&•? лив алея к 3*4 '.литрам тройного аммиака, в который пред» ’варительно . вливался раствор 10 гряж* столярного клея, в 200 граммах воды. Эту операцию надо вестипри возможно ' ^лучшем охлаждении и перемешиваний*
V Затем производится отгонка аммиака др ирнца еговыде-'ления, что требует 8-10 часов времени,
После удаления аммиака к раствору приливается дород! Гкйслота до нейтральной реакции и раствор на псловину вы-паривается, причем в осадке выпадают хлористый натрий, сернокислый аммоний и сернокислый натрий.
После отфильтровывания осадка фильтрат выпаривается до 1|10 первоначального облема и да де лившийся осадок отфвдьтровывается. Осадок обрабатывается рддбым раствором аммиака и опять фильтруется, Фильтрат добавляю? к= фильтрату, полученному при выпаривании до I/Ю» Собрани й ный таким путем фильтрат сильно подкисляется крепкой ; серной кислотой и охладив, отфильтровывают выпавший . , гидразинсульфат, который затем • высушивается,
, 150 грамм гидразинсульфата’ вмешиваются с 200 граммами порошкообразного едкого натра и после приливания 300 куб.сайт, кинного спирта нагреваются в колбе с обратным холодильником в течение 3-4 часов. После охлаждения- оед» док отсасывается на воронке Бюхнера и промывается с пир-
i - 83 -
; трк^ цока 9 Общем количество жидкости не достигнет 800 -f едб.,ОЛНТ.
К 800 куб.сайт. полученного раствора гидразина медле! НО приливают 120 грамм этилнитрита и затем быстро бООку-сант. безводного этилового эфира. Полученную смесь.силь) взбалтывают и затем оставляют на холоду стоять.12 час о г? затем 12 часов при обыкновенной температуре. -
Впавший а течении 24 часов азид натрия отфильтровыв^ ОТСя и промывается несколько раз спиртом и безводным эс£ ром и затем. высушивается при 80-90*
Получается аздд свинца eR&Na следующим образом:
,5гр, ватрийаэида растворяется в 50 куб. сант. воду (воли натрий азид продажный, например от 'cv
/необходимо. раствор профильтровать). Полученный раствор медленно по каплям при размешивании приливается красть ру 2§ PJU .азотнокислого свинпа в 250 к.с< вс-ды. После t .?0, как осадок отстоится, его промывают несколько раз Л дой, отстаивают на воронке Бюхнера и еще промывют 2 раг спиртом и один раз эфиром; высушивается при 100° в течс ЙИФ Ш? часа и ставится в эксикатор над хлористым кальци<
Обращать внимание, чтобы при осавдении азида свинца как раствор натрийализа, так и азотнокислого свинца не были бы теплыми. Ни в каком случае не следует заменять азотнокислый свинец уксуснокислым свинцом. Хранить ази, свинца;надо в темном месте или в темном эксикаторе над хлористым кальцием. Уничтожать остатки азида свинца npj ; .помощи крепкого раствора едкого натра. При работе о аз] ; дбм свинца надевать на глаза проволочную сетку и под с; ;??кой слюдяные очки.
84 -
Таблица
I-а.
Сравнение ареомет рических граду сов с удель^
ним весом при 17,5° и при 12,S’
• Градусы i Уд. вес при 17,5' Уд.вес при 12,5' Градусы Уд.вес при 17,5’ Уд. вес ’ при 12,5'
4 0 1,0000 1,0000 24 1,1955 1,1994
?• I 1,0068 1,0069 25 1,2053 1,2095.
; 2 1,0138 1,0140 26 .1,2153 1,2198
] 3 1,0208 1,0212 . 27 1,2254 1,2301
% 4 1,0280 1,0285 28 1,2357 1,2407
< 5 1,0353 1,0358 29 1,2462 1,2515
) •1 6 1,0426 1,0434 30 1,2569 1,2621
S 7 1,0501 I,0509 31 1,2677 1,2736
8 1,0576 1,0587 32 1,2788 1,2849
9 1,0653 1,0665 33 1,2901 1,2965
*d 10 1,0731 1,0745 34 1,3015 1,3082
1’ II 1,0810 .1,0825 35 1,3131 1,3202
'•jl 12 1,0890 1,0907 36 1,3250 1,3324
13 1,0972 1,0990 37 1,3370 1,3447
14 1,1054 1,1074 •38 1,3494 1,3574
15 1,1138 1,1160 , 39 . 1,3616 1,3703
1-•t 16 1,1224 1,1247 40 1,3746 1,3834
j 'l 17 1,1310 1,1335 41 ’ 1,3876 1,3968 ,
I 18 1,1398 I,1425 42 1,4009 1,4105
1 19 1,1487 1,1516 . 43 1,4143 1,4244
? 20 1,1578 1,1608 . 44 1,4281 1,4386 -
21 1,1670 1,1702 45 1,4421 I,4531 .
22 1,1763 1,1798 46 1,4564 1,4678
1 23 1,1858 1,1896 47 1,4710 1,4828
- 85 - $
Градусы Уд, ве с при 17,5* Уд .вес при 12,5е Градусы Уд. вес при 17,5е Уд. вес||И при |г| 12,5°J
48 1,4860 1,4984 58 1,6533 Г 1 1,672О[И
49 1,5012 1,5141 59 1,6721 I,69I6j J
50 1,5167 1,5301 60 1,6914 1,71161
51 1,5325 1,5466 61 1,7111 1,732Ш
52 1,5487 1,5633 62 1,7313 1,7532}
53 1,5652 1,5804 63 1,7520 1,774ф
54 1,5820 1,5978 64 1,7731 1,796$
,55 1,5993 1,6158 65 1,7948 1,8195;
56 1,6169 1,6342 66 1,8171 1,8426,
- 57 1,6349. 1,6529 У) Si-
- 36 - >.'Д
Таблица 1
jj . 1-1 3. ’ 1
| Сравнение аре омет рических градусов с удель- • ’
SHM весом при 15 | 4.
|Уд. вес Уд. вес Уд., .вес
1 ПРИ Гралу сы при , Градусы при Градусы
|l5e| 4* 15’1 4* 15’1 4’
|l,200 24 : 1,320 35,0 •1,440 44,1
11,205 • '24,5 1,325 : 35,4 1,445 44,4 '
Т,‘2Т0 ; 25 1,330 35,8 1,450 44,8
1)215 25,5 1,335 36,2 1,455 45,1
1,220 26 1,340 36,6 1,460 45,4
1,225 26,4 1,345 37,0 1,465 45,8 ;
1)230 . 26,9 1,350 37,4 1,470 46,1
I,2 35 27,4 1,355 37,8 1,475 46,4
1,240 27,9 1,360 38,2 1,480 46,8
1,245 28,4 1,365 38,6 1,485 47,1 ; ' -
1,250 28,8 1,370 39,0 1,490 47,4
1,255 29,3 1,375 ,39,4 1,495 47,8
«,260 29,7 1,380 39,8 1,500 48,1
|Г,265 30,2 1,385 40,1 1,505 48,4
|1,270 30,6 1,390 40,5 1,510 48,7
11,275' 31,1 1,395 40,8 1,515 49,0
'1,280' 31,5 1,400 41,2 1,520 49,4.
11,285 32,0 1,405 41,6 1,525 49,7
1,290 32,4 1,410 42,0 1,530 50,0
1,295 32,8 1,415 42,3 1,535 50,3
1,300 • 33,3 1,420 42,7 1,540 50,6
1,305 33,7 1,425 43,1 1,545 50,9
1,310 34,2 1,430 43,4 1,550 51,2
1,315 34,6 1,435 43,8 1,555 51,5
87 -
1 Уд. вес При 15'1 4’ Градусы Уд. вес при 15’1 4* Градусы Уд. вес при 15’1 4’ Граду си
1,560 51,8 1,655 57,1 1,750 61,8
1,565 52,1 1,660 57,4. 1,755 62,1
1,570 -52,4 1,665 57,7 1,760 62,3
• 1,575 52,7 1,670 57,9 1,765 62,5
1,580 53,0 1,675 58,2 1,770 62,8
- 1,585 53,3 1,680 58,4 1,775 63,0
, 1,590 53,6 1,685 58,7 1,780 63,2
1,595 , 53,9 1,690 58,9 1,785 63,5
1,600 54,1 Г, 695 59,2 1,790 63,7
' 1,605 54,4. 1,700 59,5 1,795 64,0
1,610- 54,7 1,705 59,7 . 1,800 64,2 ,
1,615 55,0 1,710 60,0 Г,8О5 64,4 *'
.1,620 55,2 1,715 60,2 1,810 64,6
1,525 55,5 1,720 60,4 1,815 64,8.
< - 1,630 55,8 1,725 60,6 1,820 65,0 '
1,635 56,0 1,730 60,9 1,824 65,2 ;<
V-.' 1,640 56,3 1,735 61,1 1,826 65,3
1,645 56,6 ' 1,740 61,4 1,828 65,4 '
1,650 56,9 1,745 61,6 1,831 65,5 '
г f - 1,833 65,6 '
' j • -' ' 1,835 65,7 -
Ч* \ - 1,838 65,8
1,840 65,9 , _ . . 1
аа -
*1
Таблица > '
л ‘ II. . • . 3
Удельный вес и содержание серной кислоты в водном ра-створе по Г1
Уд. вес при 15*1 4* Весовые проценты Уд. вес при 15*| 4° Be со вые проценты Уд. вес при 15’1 4* Весовые проценты
1,000 * 0,09 1,120 17,01 1,240 32,28
1,005 0,83 1,125 17,66 1,245 32,86 .
1,010 1,57 1,130 18,31 1,250 33,43
1,015 2,30 1,135 18,96 1,255 34,00
1,020 3,03 1,140 19,61 1,260 34,55
1,025 3,76 1,145 20,26 1,265 35,14
1,030 4,49 1,150 20,91 1,270 35,71
1,035 5,23 1,155 21,55 1,275 36,29
1,040 5,96 1,160 22,19 1,280 36,87
1,045 6,67 1,165 ' 22,83 1,285 37,45
1,050 7,37 1,170 23,47 1,290 38,03
1,055 8,07 1,175 24,12 1,295 38,61
1,060 8,77 1,180 24,76 1,300 39,19
1,065 9,47 1,185 25,40 1,305 39,77
1,070 10,19 1,190 26,04 1,310 40,35
. 1,075 10,90 1,195 26,68 1,315 40,93
1,080 11,60 1,200 27,32 1,320 41,50 ’
1,085 12,30 1,205 27,95 1,325 42,08 j
1,090 12,99 1,210 28,58 1,330 42,66
1,095 13,67 1,215 29,21 1,335 43,20
1,100 14,35 1,220 29,84 1,340 43,74
1,105 15,03 1,225 30,48 1,345 44,28
Г,ПО 15,71 1,230 31,11 1,350 44,82
1,115 16,36 1,235 31,70 1,355 45,35
t;..г;- :'?• -: ' - 89-
Уд. вес Весовые Уд. вес Весовые Уд. вес Весовые ц
, при про- при про- при про-; • Ks
15“| 4* . центы 15’1 4’ центы 15’1 4' цента; , F
1,360 45,88 1,510 60,65 1,660 73,64 ь
1,365 46,41 1,515 61,12 Г, 665 74,07 j
1,370 ; 46,94 Г, 520 61,59 1,670 74,51 j
1,375 47,47 1,525 '62,06 1,675 74,97 1
1,380 ! 48,00 1,530 62,53 1,680 75,42
1,385 48,53 1,535 63,00 1,685 76,86 р 1
>1,390 49,06 1,540 ’ 63,43 '1,690 76,30 i!
1,395 49,59 1,545 63,85 1,695 76,73 1
1,400 50,11 1,550 64,26 1,700 77,17 Я i
1,405 50,63 1,555 64,67 1,705 77,60
1,410 51,15 1,560 65,08 1,710 78,04 j
1,415 51,66 1,565 65,49 1,715 78,48 j
I,420 ’ 52,15 1,570 65,90 1,720 78,92
I*,425 52,63 1,575 66,30 1,725 79,36 j
1,430 53,11 1,580 66,71 1,730 79,80Ч
1,435 53,59 1,585 67,13 1,735 80,24 •
1,440 ' 54,07 1,590 67,59 1,740 80,68
1,445 54,55 1,595 68,05 1,745 si д2 ;
1,450 55,03 1,600 68,51 1,750 81,56
Г,455 55,50 1,605 68,97 1,755 82,00
1,460 55 „97 1,610 69,43 1,760 82,44
1,465 56,43 1,615 69,89 1,765 82,88 :
1,470 56,90 1,620 70,32 1,770 83,32
1,475 57,37 1,625 70,74 1,775 83,90 ;
1,480 57,83 1,630 71,16 1,780 84,50
1,485 58,28 ,1,635 71,57 1,785 85,10-
1,490 58,74 1,640 71,991 1,790 85,70
1,495 59,22 1,645 72,40 1,795 86,30
1,500 59,70 1,650 72,82 1,800 86,
1,505 60,18 1,655 73,23 1,805 87,60
90
Уд. вес ' при 15*1 4* Весовые процент Уд.: вёб при 15*, 4* Йеббййе fipo-центы W мв при 154 4* Вёбвйне .< Про-» У цен® ...
1,810 88,30 1,835 93,43 1,8420 98,20 ‘
1,815 89,05 1,840 95,60 1,8425 98,70
1,820 90,05 .1,8405 95,95 1,8400 99,20
1,825 91,00 1,8410 97,00 1,8395 99,45
1,830 92,10 1,8415 97,70 1,8390 99,70
1,8385 99,95
1
Jy : ' : .. r . ' , t . J /T -'j<' .Удел ь в растворе, ый вес и •- 91 ’ - •, г водном 1 В >
F& б 1 и tit. содержа ние Ц а. азотной кислоты в
Уд. вес Весовые Удельный Весовые Удельный Весовые V |в
" : ПРИ ' • про- вас про- вес про-
I5’| 4* центы центы Центы
1,000 0,10 1,120 20,23 1,240 38,29 к
1,005 1,00 1,125 21,00 1,245 39,05:
. i,oio ; 1,90 1,130 21,77 1,250 39,82i
1,015 2,80 1,135 22,54 1,255 40,58 •
i,oeo 3,70 1,140 23,31 1,260 41,34 * 1 < 1
1,025 4,60 1,145 24,08 1,265 42,I0',i ’ *
1,030 5,50 1,150 24,84 1,270 42,87 1
1,035 6,38 1,155 25,60 1,275 43,64
1,040 7,26 1,160 26,36 1,280 44,41
1*045 8,13 1,165 27,12 1,285 45,95
1,050 8,99 1,170 27,88 1,290 46,34
1,055 9,84 1,175 28,63 1,295 46,72 ♦
1,060 . 10,68 1,180 29,38 1,300 47*49
1,065 II, 51 1,185 30,13 1,305 48,26:
1,070 < "12,33 1,190 30,88 1,310 .. 49,07
-1,075’ ? ’ 13,15 1,195 31,62 1,315 .49,89
1,080 13,95 .1,200 32,36 1,320 50,71
’ 1,085 14,74 1,205 33,09 1,325 51,531
1,090 15,53 1,210 33,82 1,330 52,37 1'
.1,095 16,32 1,215 34,55 1,335 53,22:
1,100 17,11 1,220 35,28 1,340 54,07
1*105 .17,89 1,225 36,03 1,345 54,93 h
1,П0 18,67 1,230 36,78 1,350 55,79
1,115 19,45 1,235 37,53 1,355 56,66 г
- 92 -
Уд. вес при 15*| 4* Весовые проценты Удельный вес Весовые проценты Удельный вес Весовые» про “Я центы ||
1,360 ; 57,57 1,415 68,63 . 1370 ; 82,90. 1
-1,365 58,48 1,420 69,80 1,475. 84,45
1,370 59,39 . 1,425 70,98 1,480 86,05
1,375 60,30 1,430 72,17 1,485 87,70
1,380 61,27 1,435 73,39 1,490 89,60
1,385 ; 62,24 1,440 74,68 1,495 91,60
1,390 63^23 1,445 75,98 1,5® 94,09
1,395 64,25 1,450 77,28 1,505 96,39
1,400 . 65,30 1,455 78,60 1,510 98,10
1,405 66,40 1,460 . 79,98 1,515 99,07
1,410 67,50 1,465 81,42 1,520 99,67
Т а б л и ц a
1У
Удельные'-ве са водных растворов аммиака при 15’ 0 по
Удельный веа Проп,. NHt Удельный вес Проц. NHS
1,000 0,00 0,950 12,74
. 0,998 0,45 0,948 13,31
0,996 0,91 0,946 13,88
0,994 1,37 0,944 14,46
0,992 1,84 0,942 15,04
0,990 2,31 0,940 15,63
, 0,988 2,80 0,938 16,22
0,986 3,30 0,936 16,82
0,904 3,80 0,934 17,42
0,982 4,30 0,932 18,03
0,980 4,80 0,930 18,64
0,978 5,30 0,928 19,25
0,976 5,80 0,926 19,87
0,974 6,30 0,924 20,49
; 0,972 6,80 0,922 21,12
0,970 7,31 0,920 21,75
' 0,968 7,82 0,918 22,39
0,966 8,33 0,^16 23,03
0,964 8,84 0,914 23,68
0,962 9,35 0,912 24,33
0,960 9,91 0,910 24,99
0,958 10,47 0,908 25,65
0,956 11,03 0,906 26,31
0,954 П,60 0,904 26,98
0,952 12,17 0,902 27,65
Удельный ве с Проц, N Н5 Удельный ' вво Проц. NH,
0,900' 28,33 0,890 31,75;
- 0,898 29,01 0,888 32,50
0,896 29,69 0,886 33,^5 .
0,894 3OJ37 0,884 34,10
0,892 \ 31,05 0,882 34,95