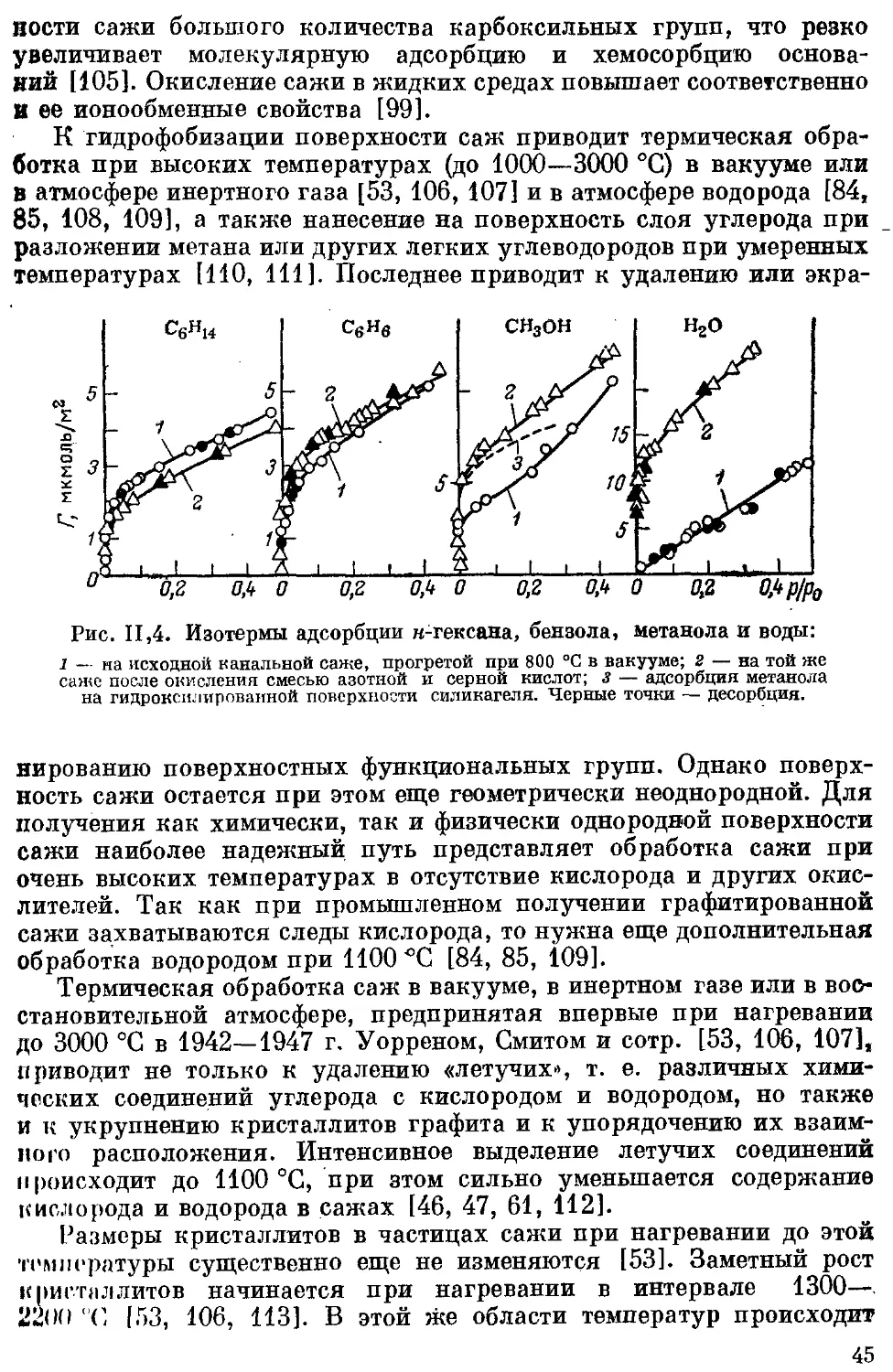
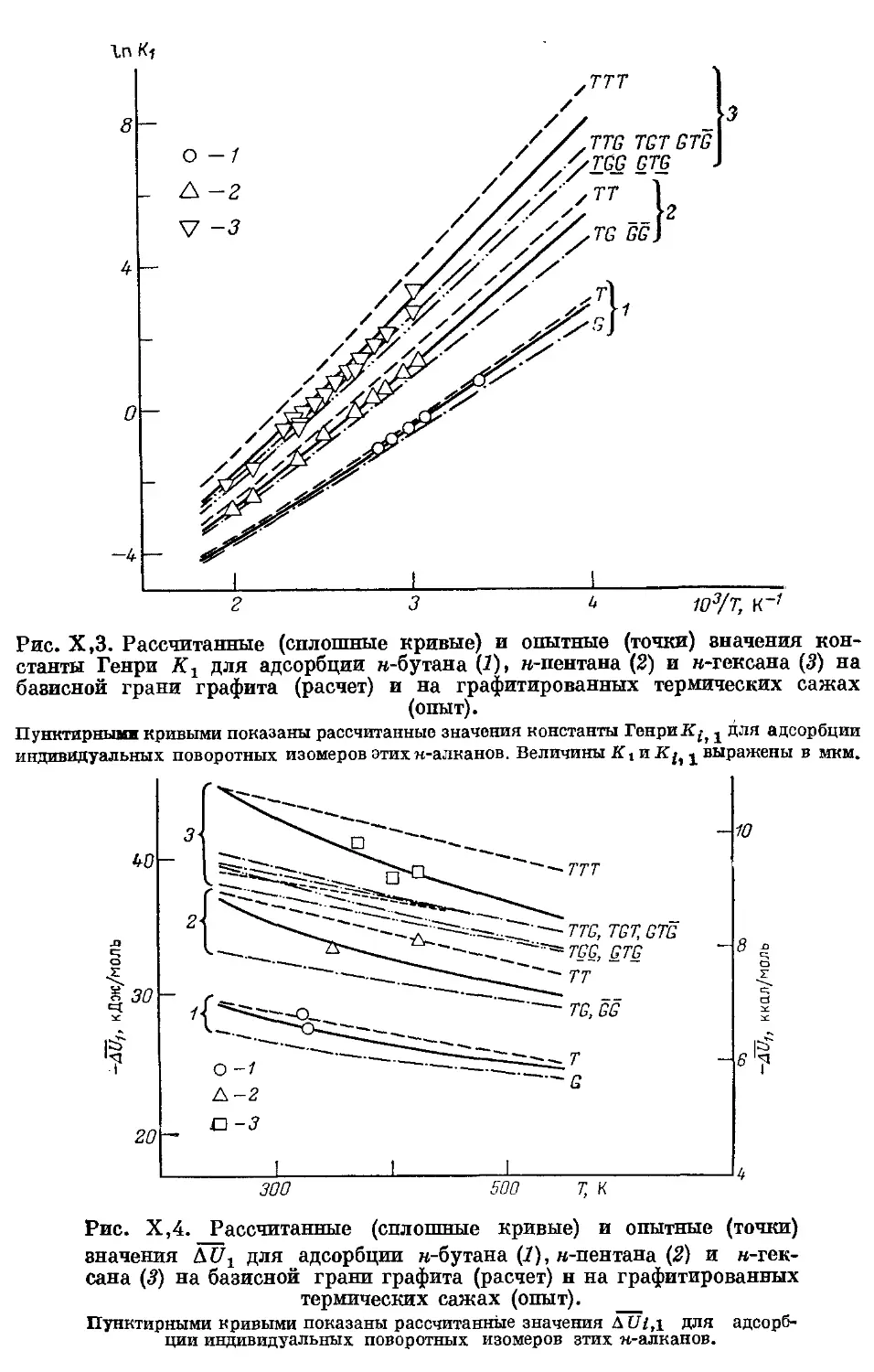
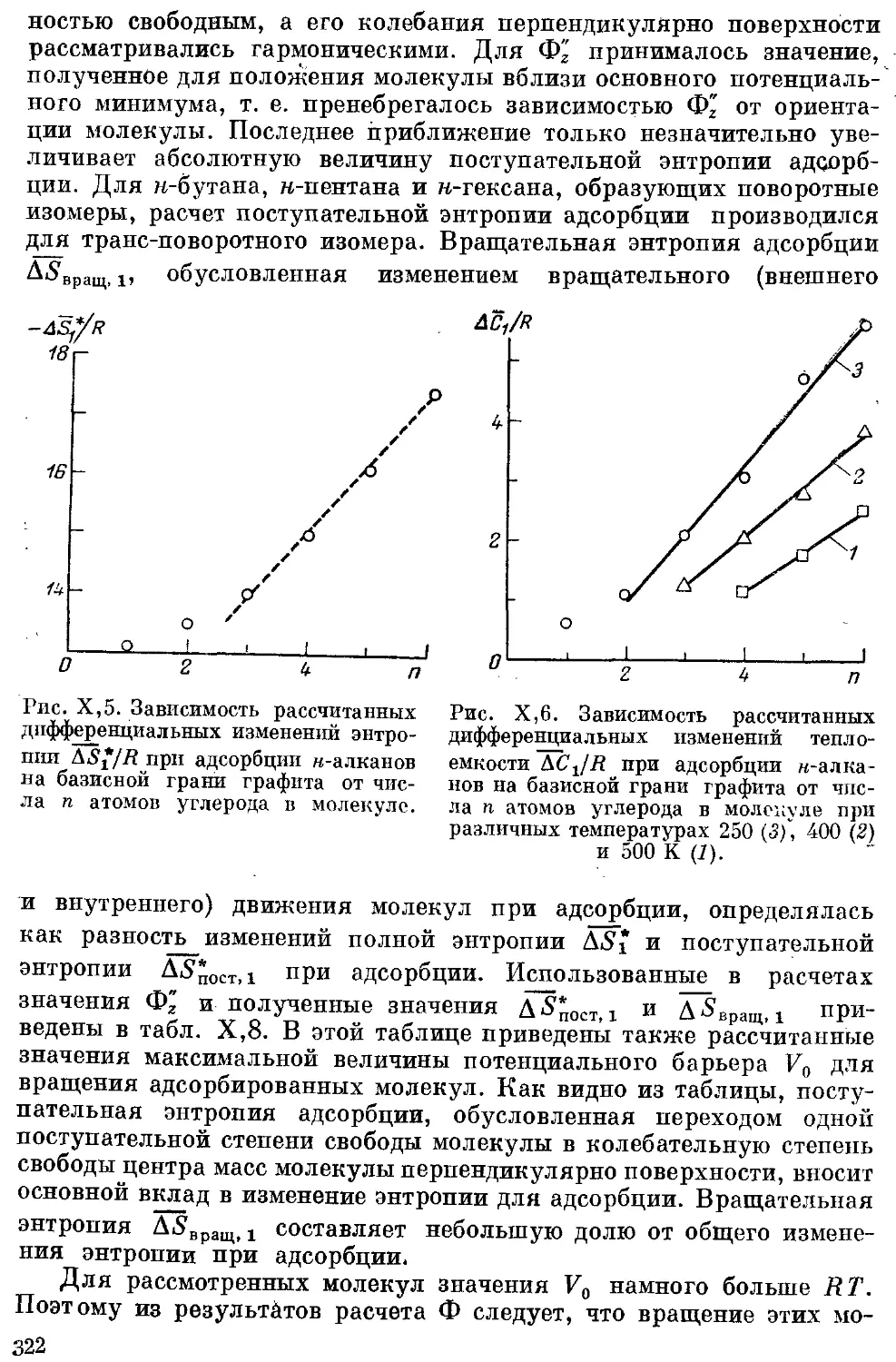
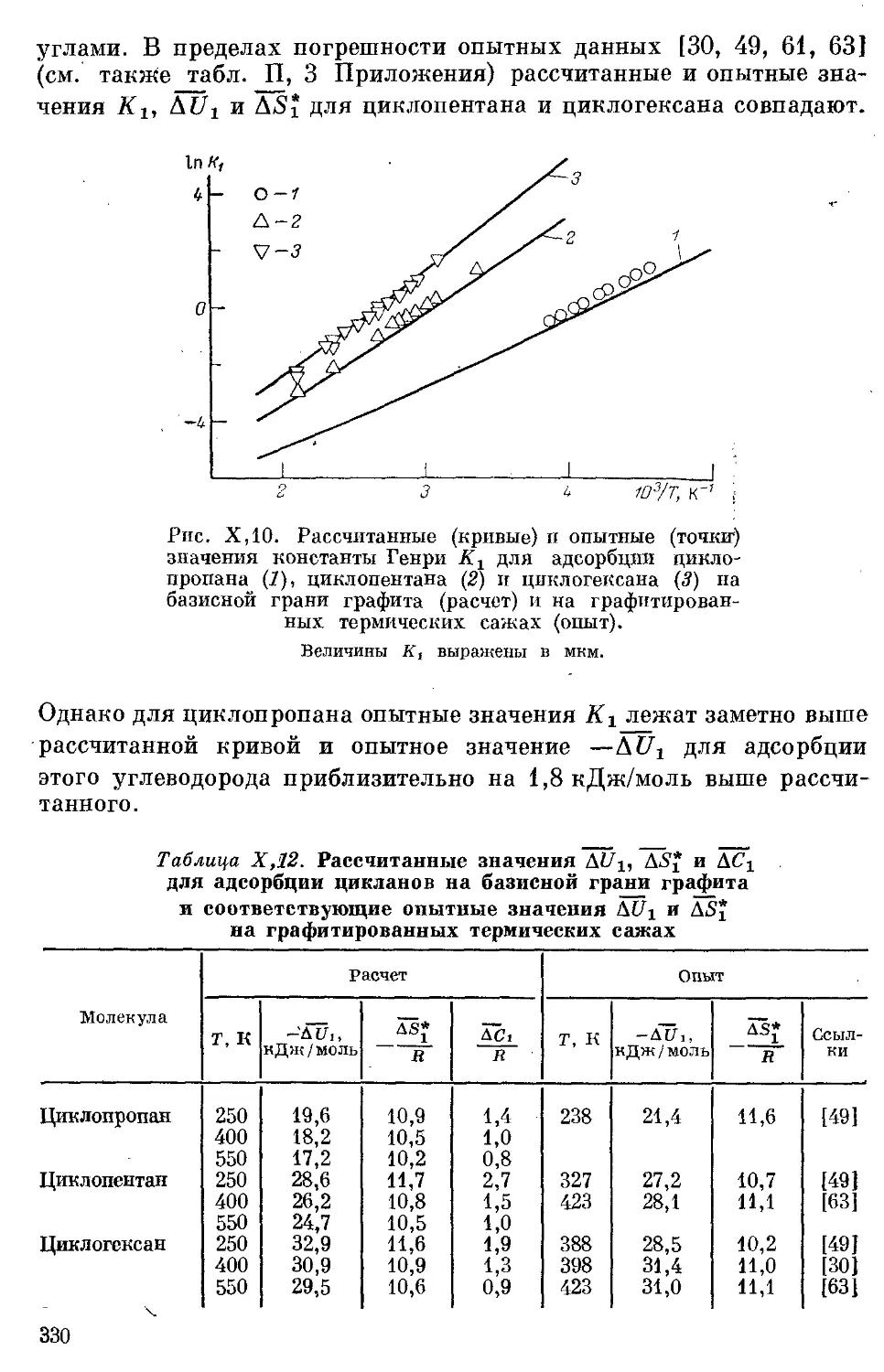
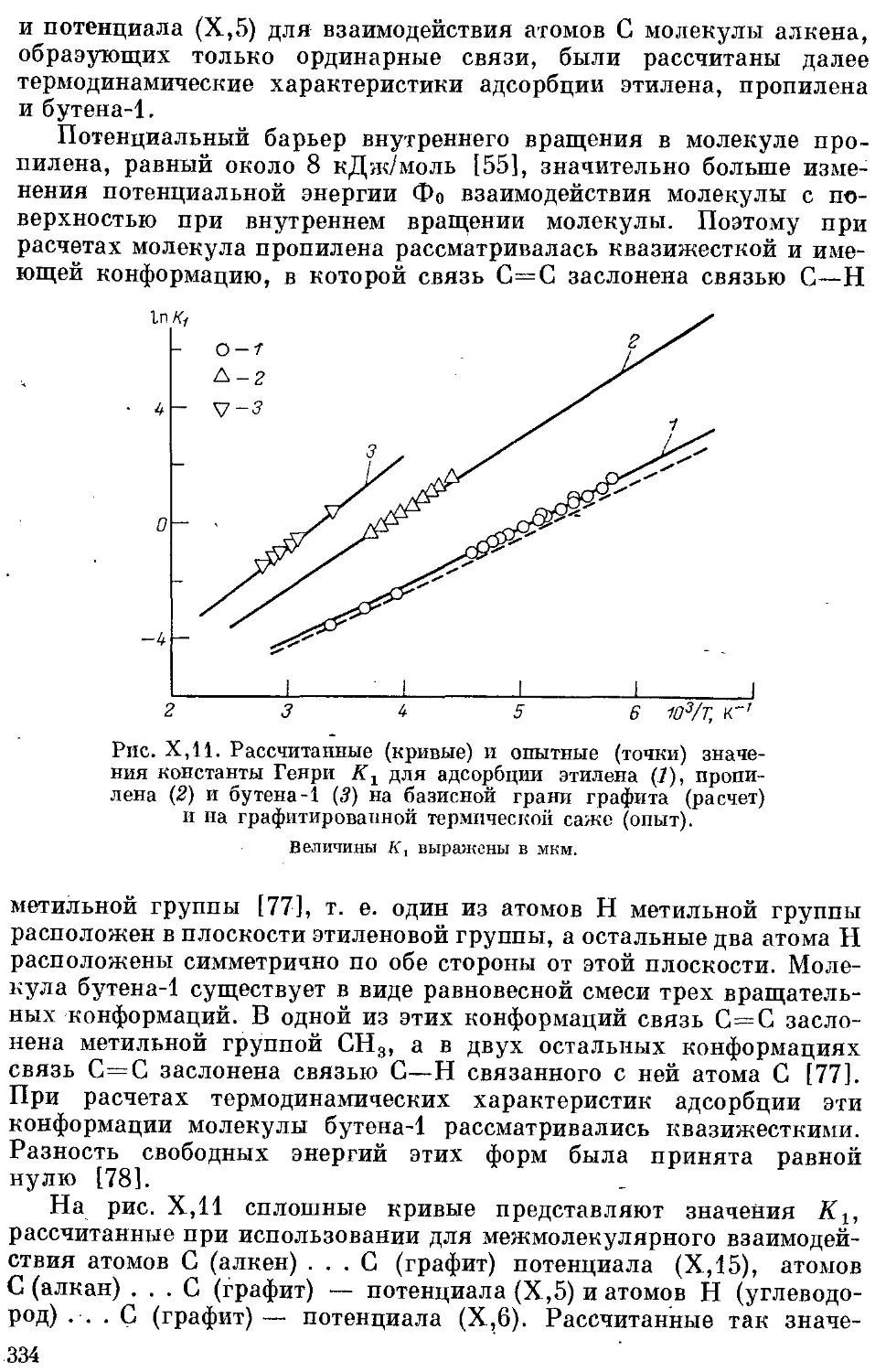
/
























































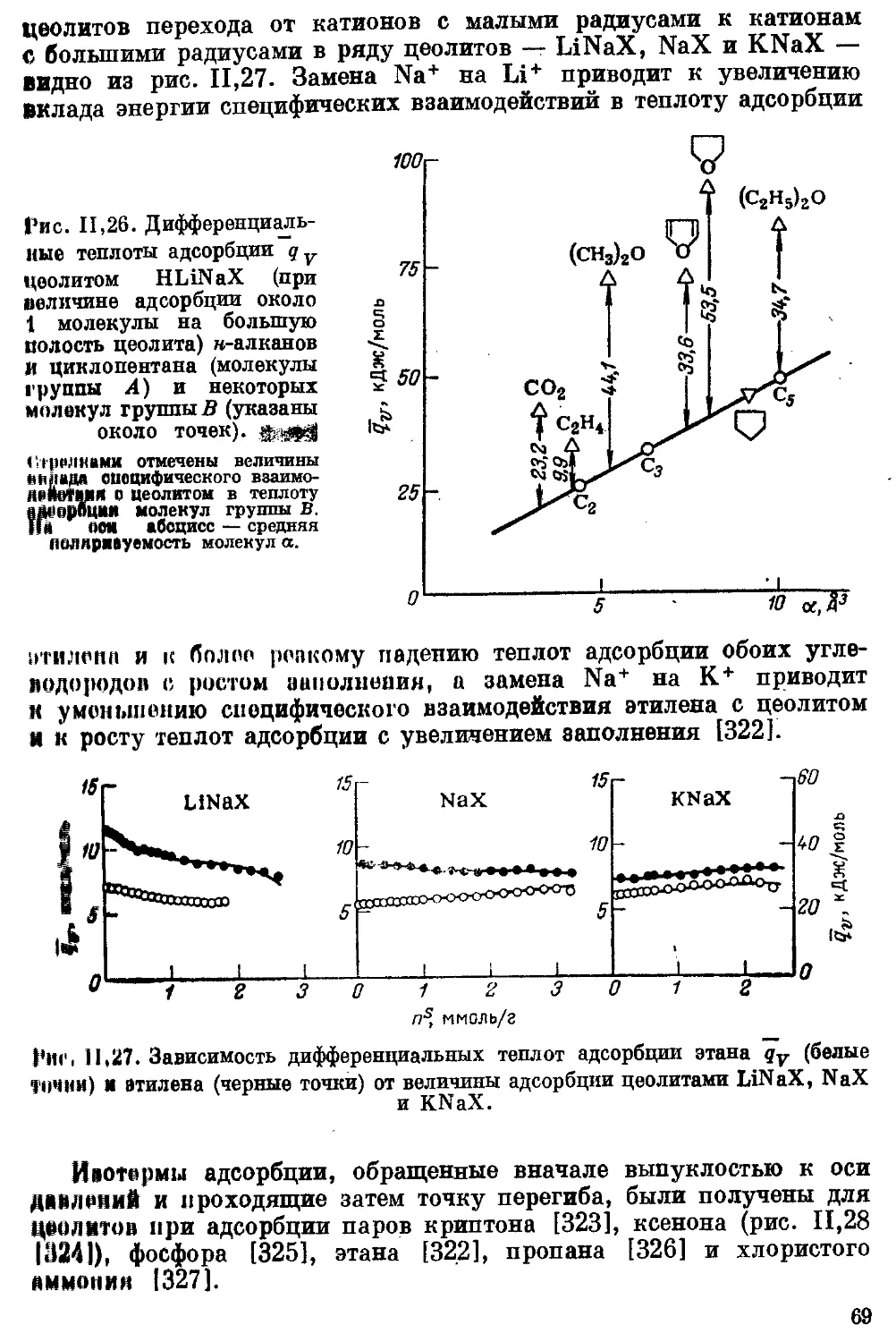



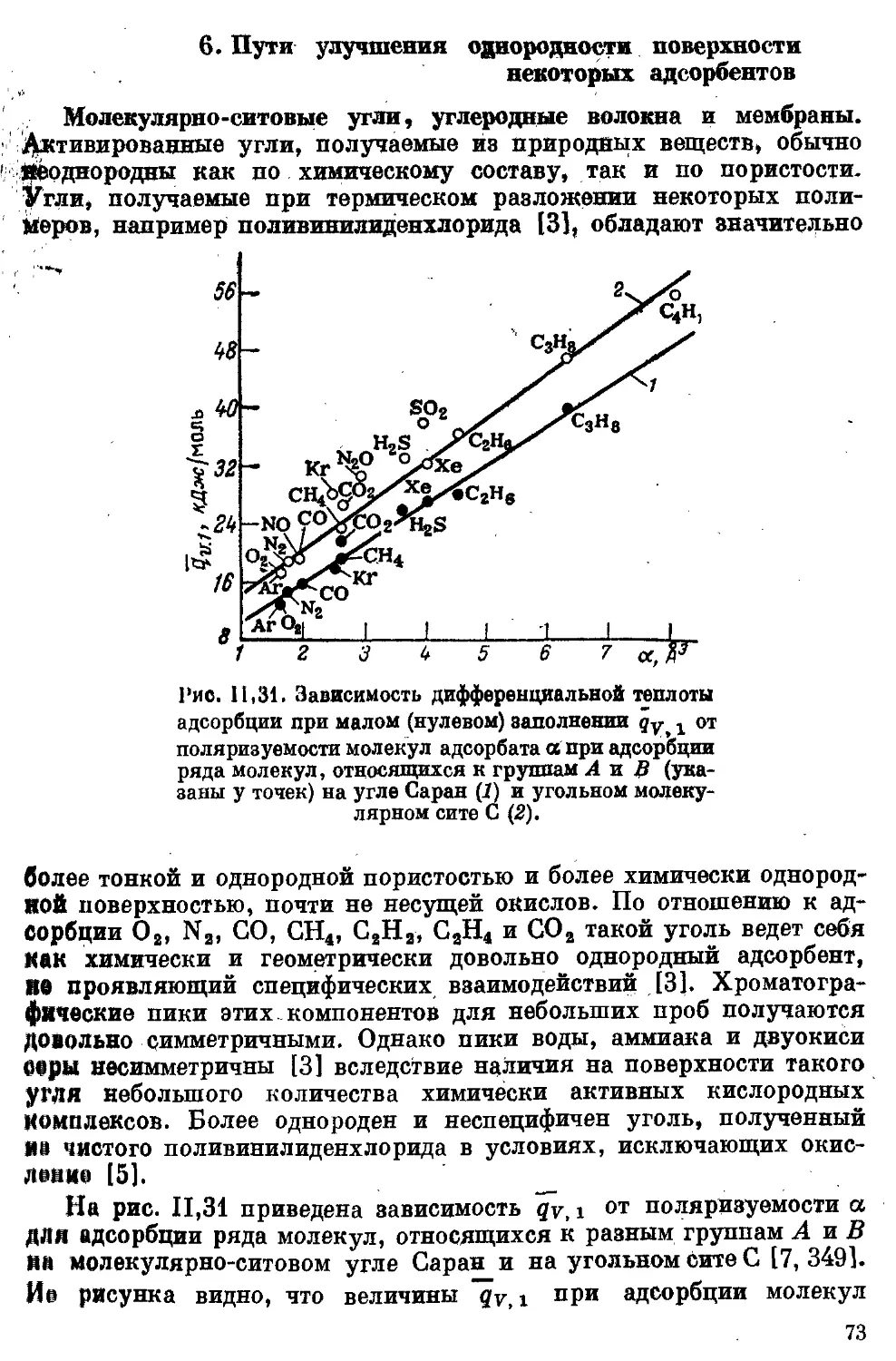
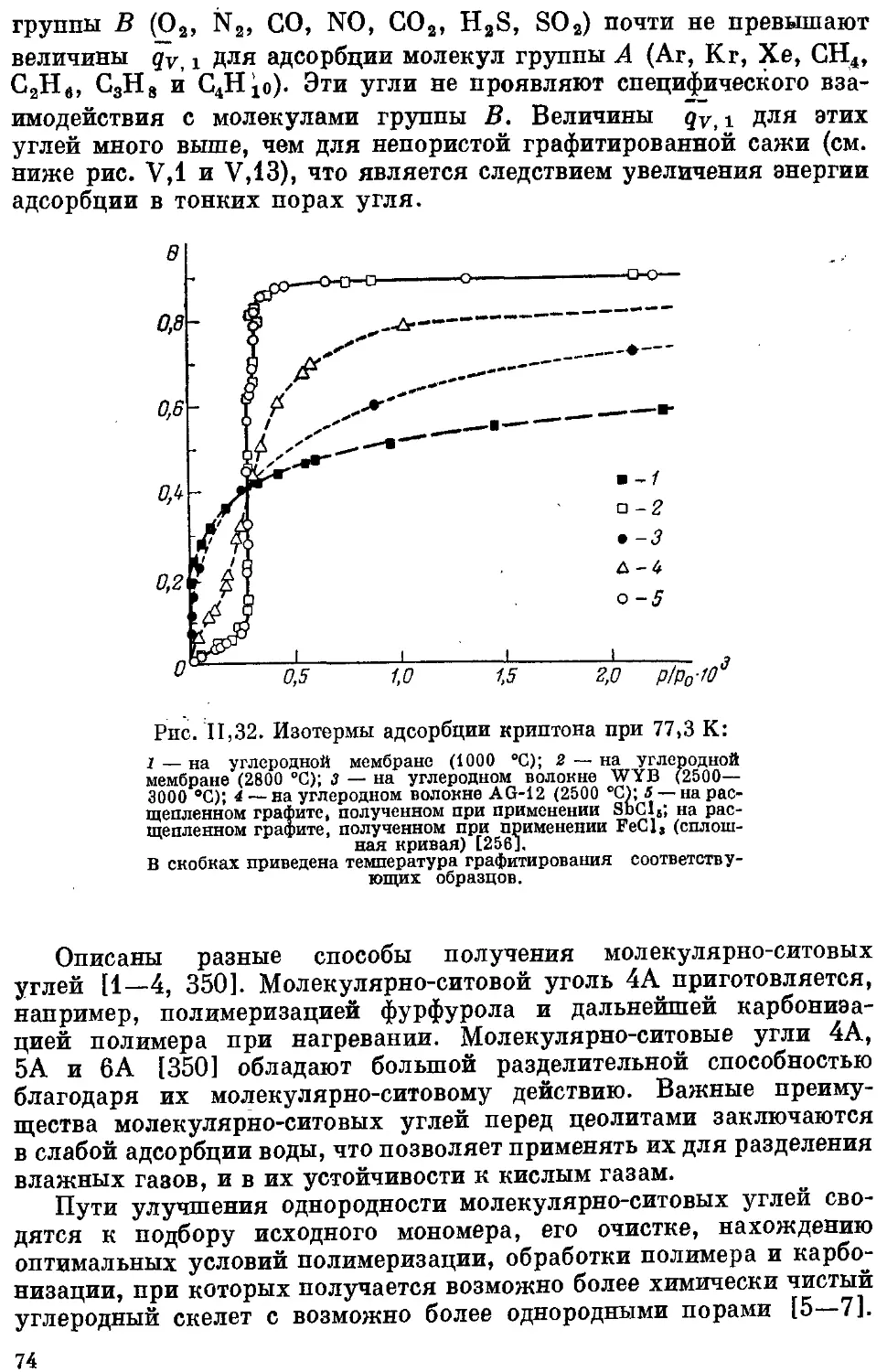


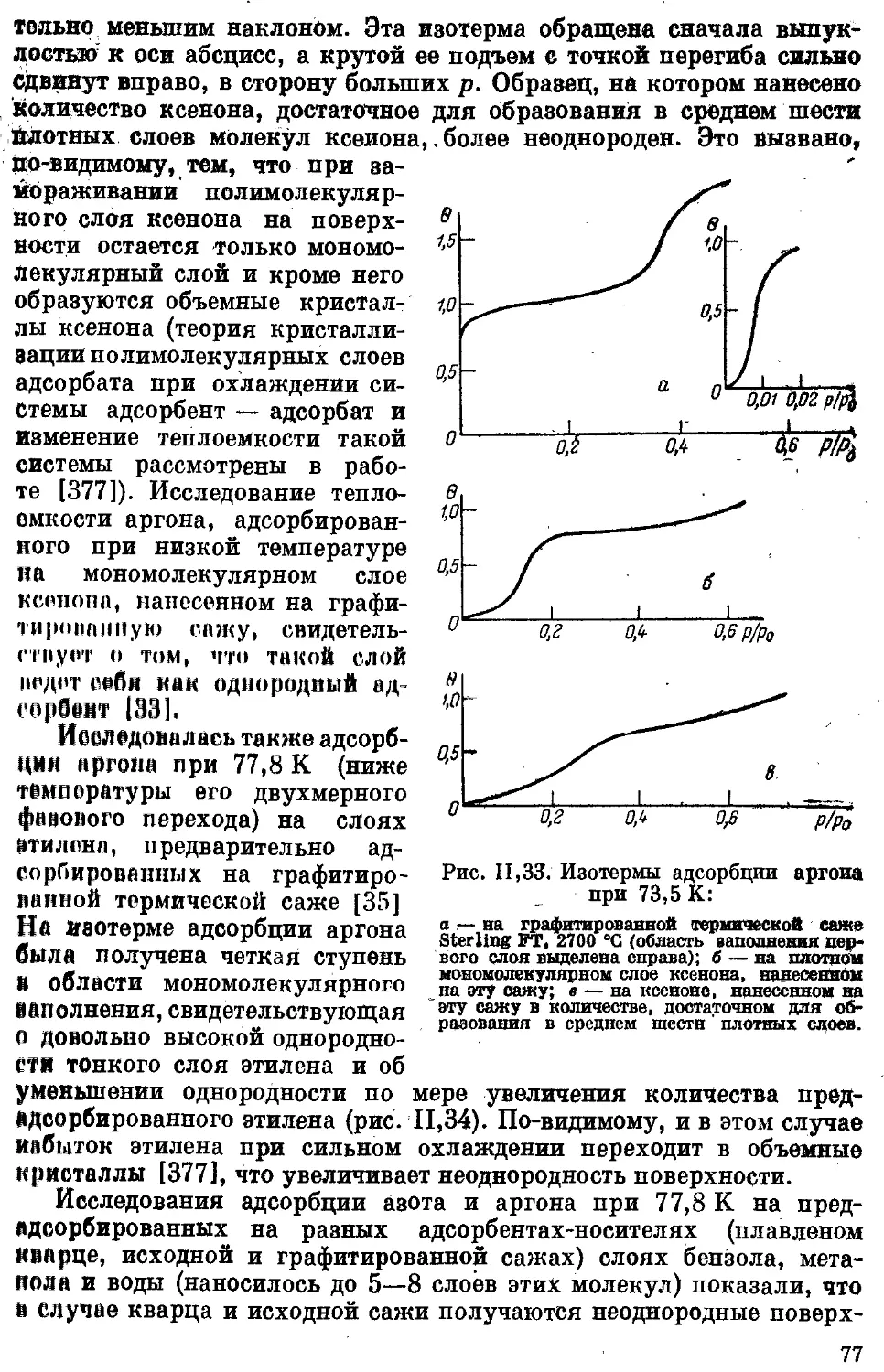






















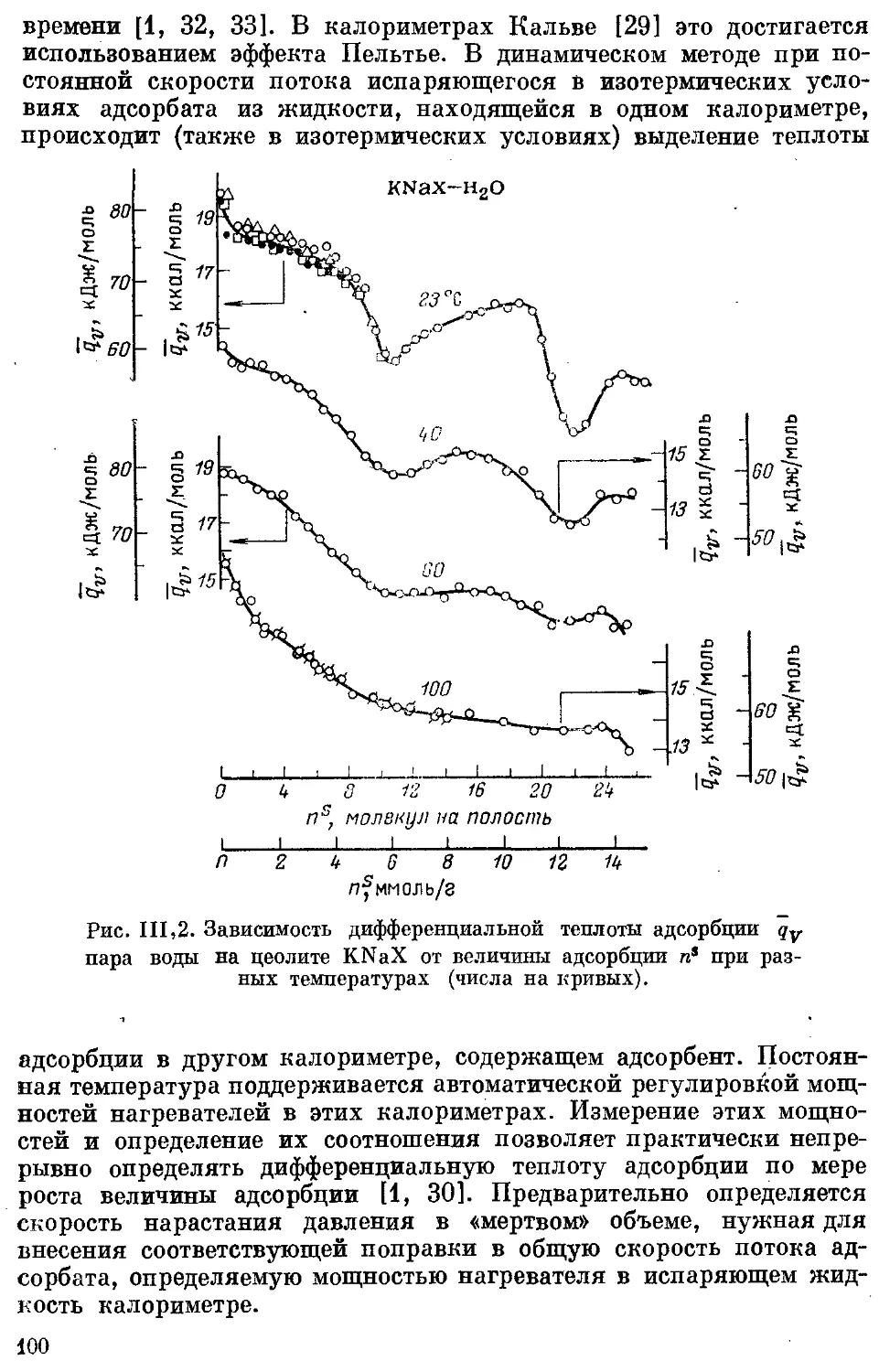
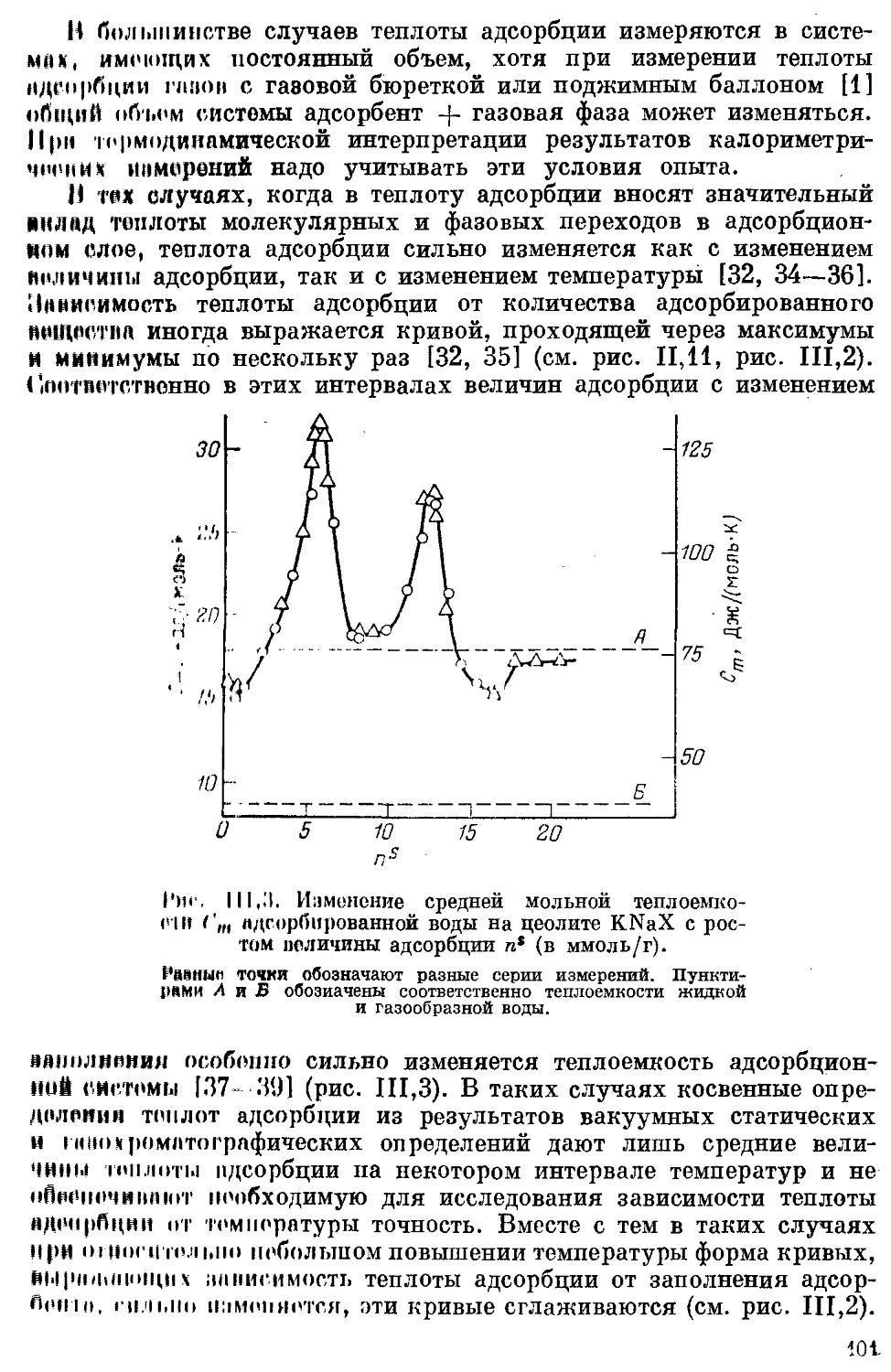





















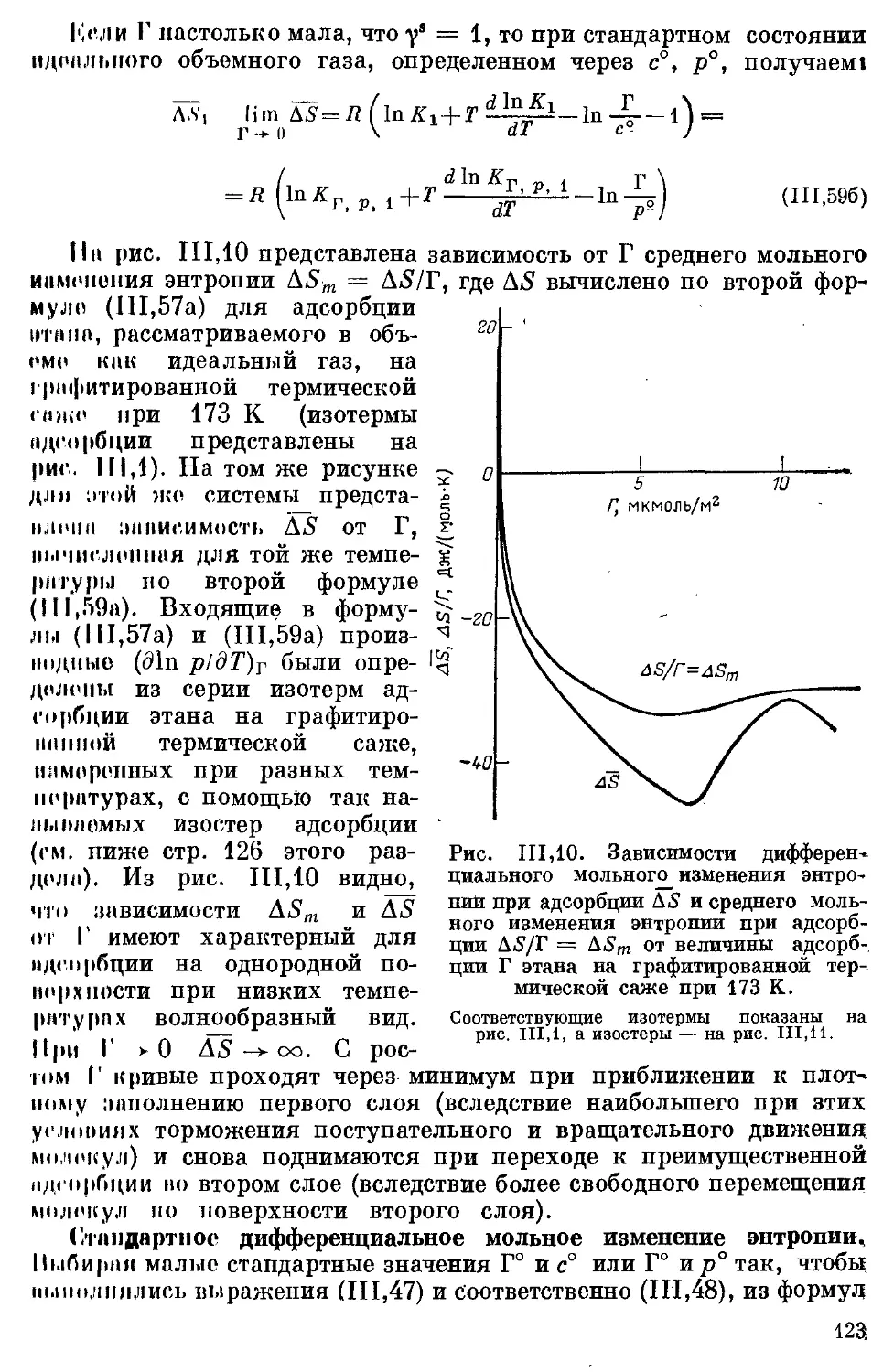


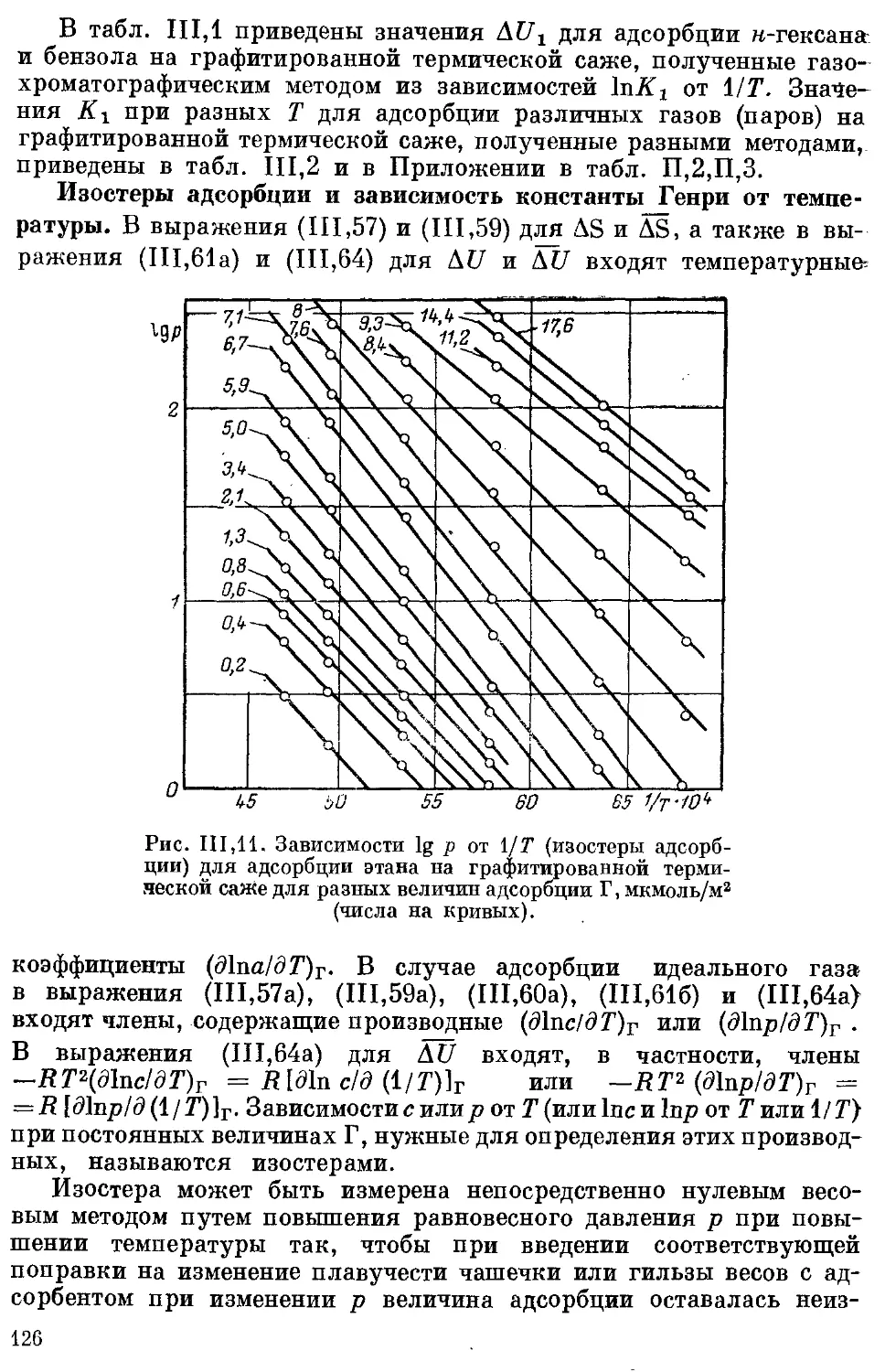































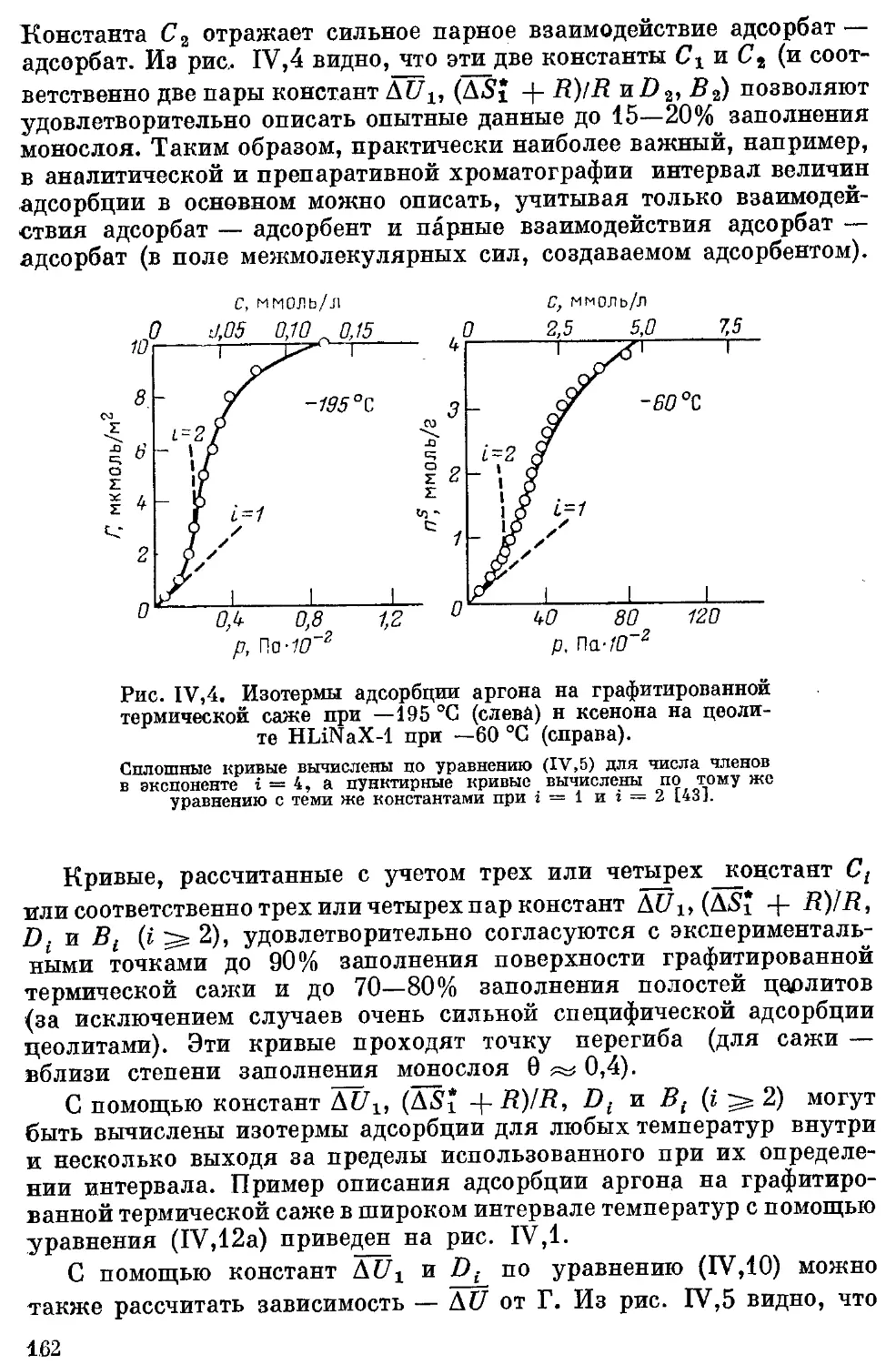

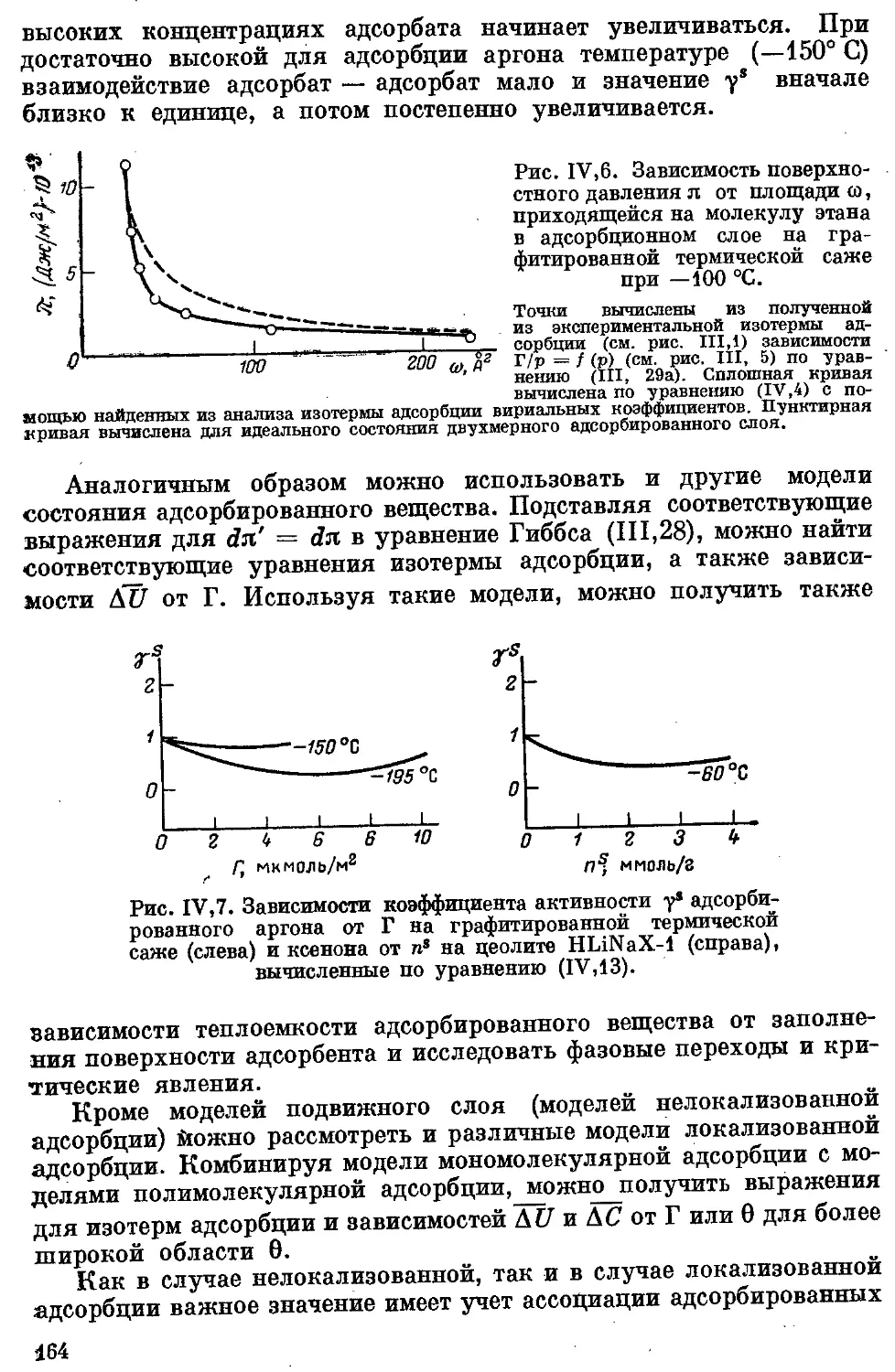







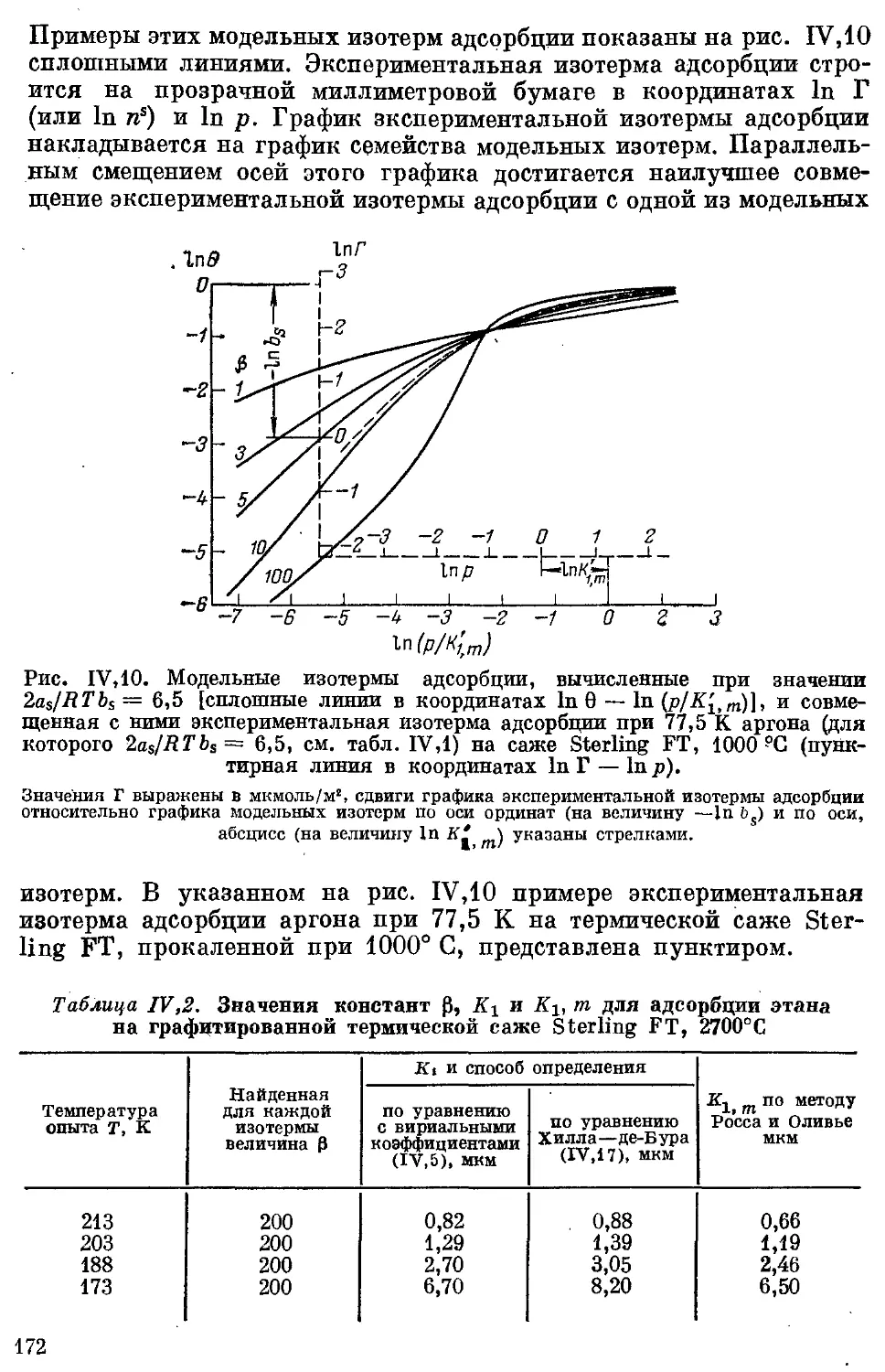

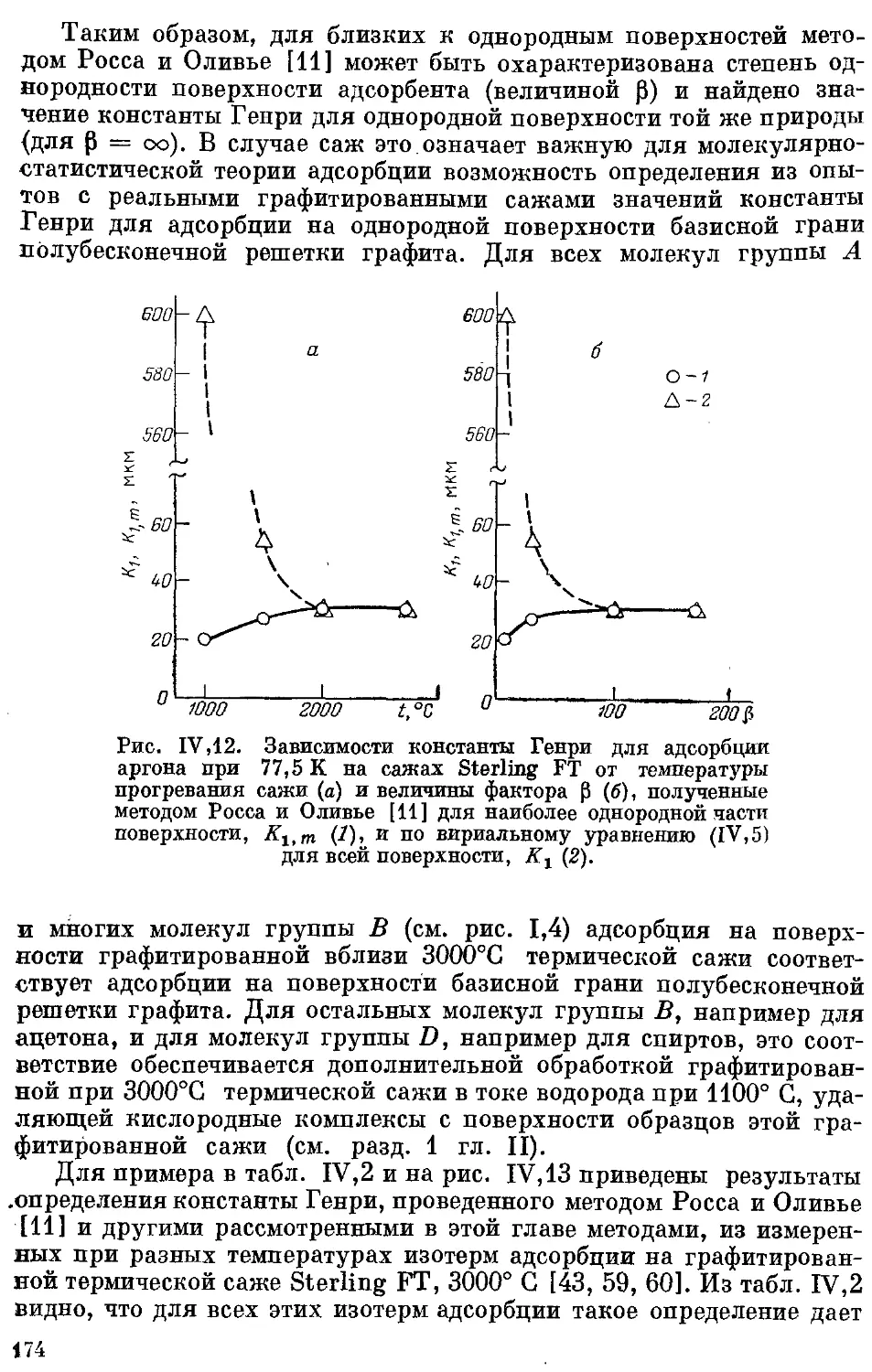

































































































































































































Текст
Н. Н А1Ч V III., А. В. КИСЕЛЕВ, Д. П. ПОШКУС
АДСОРБЦИЯ
ГАЗОВ
И ПАРОВ
НА ОДНОРОДНЫХ
ПОВЕРХНОСТЯХ
МОСКВА
„ХИМИЯ11 1976
Ь4|
А ’>2 5
УДК 541.183.5 : 54-13
А 18
Авгуль Н. BL, Киселев А. В., Пошкус Д. П.
Адсорбция газов и паров на однородных поверхностях.
М., «Химия», 1975.
384 с.; 62 табл.; 105 рис.; список литературы 970 ссылок.
В монографии описаны способы получения практически
важных адсорбентов с близкими к однородным поверхно-
стями, их адсорбционные свойства и применение в хромато-
графии. Рассмотрены общие уравнения термодинамики ад-
сорбции и уравнения, основанные на различных моделях
адсорбционного слоя. Приведены способы расчета термо-
динамических характеристик адсорбции из опытных данных
по газовой хроматографии, изотермам и теплотам адсорбции.
Изложена молекулярно-статистическая теория адсорбции
и теория межмолекулярных взаимодействий при адсорбции.
Рассмотрены результаты расчетов адсорбционных равновесий
для простых и сложных молекул на основе атом-атомных
потенциальных функций межмолекулярного взаимодействия.
Книга предназначена для научных работников, аспиран-
тов и студентов-химиков и физиков, изучающих межмолеку-
лярные взаимодействия и поверхностные явления. Она пред-
ставляет интерес для инженеров, работающих в различных
областях химической технологии, в которых используют
хроматографию, адсорбцию, катализ, применяют наполнители
и пигменты.
А 20503—195
050(01)—75
© Издательство «Химия», 1975 г.
СОДЕРЖАНИЕ
Предисловие ...................................................... *0
Г мм /. Значение адсорбентов с близкой к однородной поверхностью для
развития молекулярной теории адсорбции и практических при-
менений ..................................................... • 13
1« Представления 'об однородности поверхности..................... 13
0. Характер межмолекулярных взаимодействий при адсорбции и экспе-
риментальные термодинамические критерии однородности поверх-
ности ....... .................................................. 21
В» Роль соаднпин и iiayueinuT адсорбентов с близкой к однородной поверх-
ностью и равпитмн молекулярной теории адсорбции................. 31
4. Оснопиыо направления систематического экспериментального и теоре-
тического исследования адсорбции твердыми телами................ 34
Литература...................................................... 36
Глава II. Получение адсорбентов с близкой к однородной поверхностью 40
1. Графитированная сажа п расщепленный графит..................... 40
2. Нитрид бора................................................... 55
3. Металлы ....................................................... 56
4. Ионные адсорбенты с гранями одинаковых и разных индексов .... 61
Соли с кубической решеткой . ................................. 61
Соли со слоистой решеткой типа МХг............................ 64
Соли с комплексными анионами ................................. 67
5. Кристаллические окислы. Лед.................................... 70
6. Пути улучшения однородности поверхности некоторых адсорбентов . 73
Молекулярно-ситовые угли, углеродные волокна и мембраны .... 73
Пористые полимеры............................................. 75
Адсорбенты с нанесенными на поверхность модифицирующими слоями 76
Литература....................................................... 81
3
Глава III, Термодинамические характеристики адсорбции одиокомпонент-
иых газов и паров................................................... 93
1. Некоторые особенности определении термодинамических характери-
стик адсорбционных систем iui экспериментальных данных.............. 93
Статические измерения изотерм адсорбции ........................ 93
Газо-хроматографическое определение константы Генри и изотермы
адсорбции ..................... ............................... 97
Калориметрические методы определения теплот адсорбции и тепло-
емкости адсорбционных систем.................................... 99
2. Основные задачи термодинамики адсорбции...............“......... 103
Использование результатов исследования адсорбционных равновесии 103
Использование результатов калориметрических исследований теплот
адсорбции и теплоемкостей адсорбционных систем............... 104
3. Термодинамические условия равновесия адсорбционной системы . . . 105
Константа равновесия и уравнение изотермы адсорбции............ 105
Константа Генри................................................ 108
Коэффициент активности адсорбированного вещества ys............ Ill
Уравнение состояния адсорбированного вещества ,................ 111
4. Изменения при адсорбции поверхностных термодинамических функций 114
5. Изменения термодинамических функций при переходе поверхности
раздела из стандартного состояния адсорбент — вакуум и адсорбата
из стандартного или равновесного состояния объемного газа в равно-
весное или в стандартное состояние системы адсорбент — адсорбиро-
ванное вещество................................................. 11в
Выбор стандартных состоянии для поверхности адсорбента и объем-
ного газообразного адсорбата ............................... 116
Интегральное и дифференциальное изменение свободной энергии . . 116
Стандартное дифференциальное мольное изменение свободной энер-
гии ........................................................ 119
Дифференциальное мольное изменение свободной энергии адсорбата
при адсорбции в равновесных условиях........................ 120
Стандартное изменение химического потенциала адсорбата........ 120
Интегральное и дифференциальное изменение энтропии............ 121
Стандартное дифференциальное мольное изменение энтропии .... 123
Дифференциальное мольное изменение энтропии адсорбата при ад-
сорбции в равновесных условиях.....................I . . . . 124
Интегральное и дифференциальное изменение внутренней энергии . 124
Предельная величина дифференциального мольного изменения вну-
тренней энергии............................................. 125
Изостеры адсорбции и зависимость константы Генри от температуры 126
Дифференциальное мольное изменение внутренней энергии адсор-
бата при адсорбции в равновесных условиях................... 127
Интегральное и дифференциальное изменение теплоемкости .... 127
Дифференциальная мольная теплоемкость адсорбированного вещества 129
Предельная величина дифференциального изменения теплоемкости
при Г -> 0 ............................................. 129
4
Дифференциальное мольное изменение теплоемкости адсорбата при
адсорбции в равновесных условиях............................. 130
Примеры зависимости теплоемкости адсорбированного вещества от
величины адсорбции........................................... 130
Зависимость изменения внутренней энергии от температуры .... 131
li Изменений термодинамических функций в некоторых частных случаях
проведения адсорбционных опытов ................................ 132
Адсорбция газа, поступающего из резервуара с постоянным давле-
нием ...».................................................... 132
Адсорбция газа, поступающего из резервуара с постоянным объемом 134
Адсорбция пара, поступающего из резервуара с чистой жидкостью . 135
Теплота адсорбции . . .....................................- . 140
Случаи необходимости калориметрических измерений тепловых эф-
фектов адсорбции ..........................................., 140
Теплоты адсорбции газов и паров............................. 141
Изостерическая теплота адсорбции. Чистая теплота адсорбции пара 144
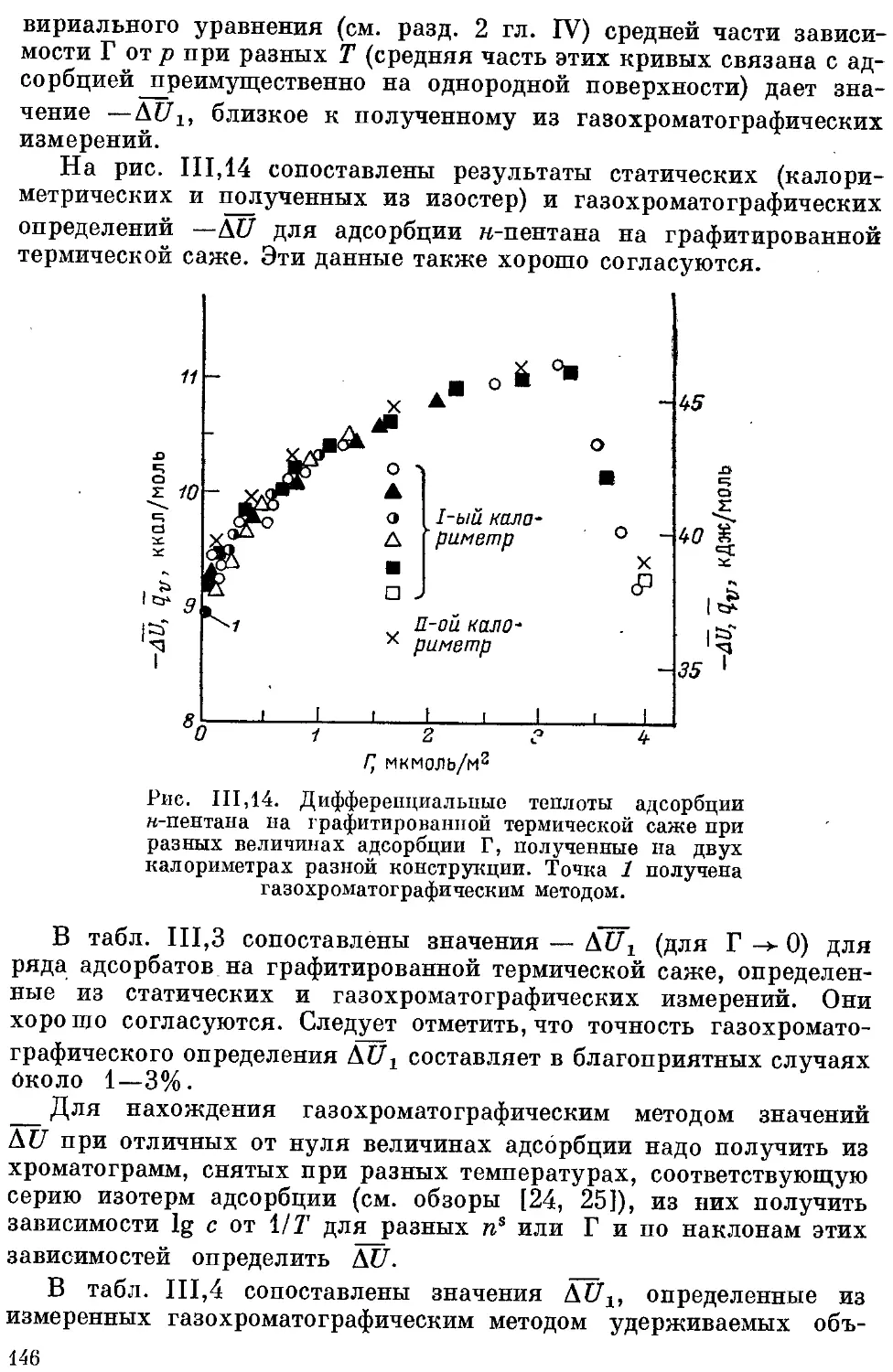
Сопоставление величин изменения внутренней энергии при адсорб-
ции, полученных разными методами............................. 145
О выборе вместо F и U других термодинамических функций для ха-
рактеристики адсорбционных процессов................ . . • 148
Литература....................................................... 148
Глава IV Термодинамическое описание адсорбции на однородной поверх-
ности и помощью некоторых моделей . .......................... • 152
1. Роль моделей в термодинамическом описании адсорбции.......... 152
В. Описание зависимости величины адсорбции от концентрации (давле-
ния) газа и температуры при использовании уравнения двухмерного
состояния адсорбата в вириальной форме......................... 155
I. Получение термодинамических характеристик адсорбции на однород-
ной поверхности из экспериментальных данных для реальной поверх-
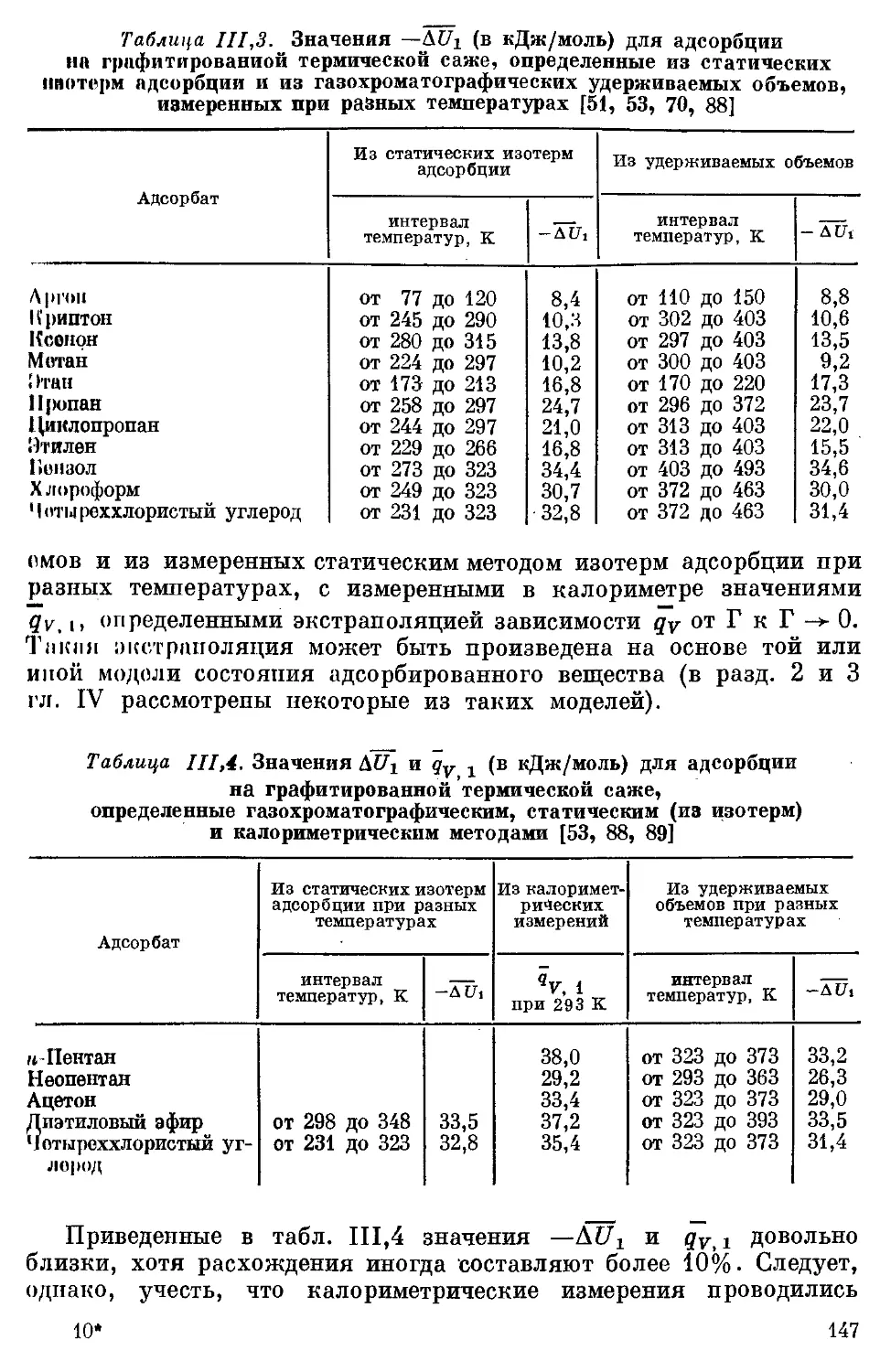
погтн .......................................................... 165
Литература....................................................... 177
Глава V. Термодинамические характеристики адсорбции различных ве-
ществ на графитированной термической саже при малом (нуле-
вом) заполнении поверхности, полученные из экспериментальных
данных .......................................................... 180
t. Нол учение термодинамических характеристик адсорбции из экспери-
ментальных данных.............................................. 180
2. Неорганические газы......................................... 183
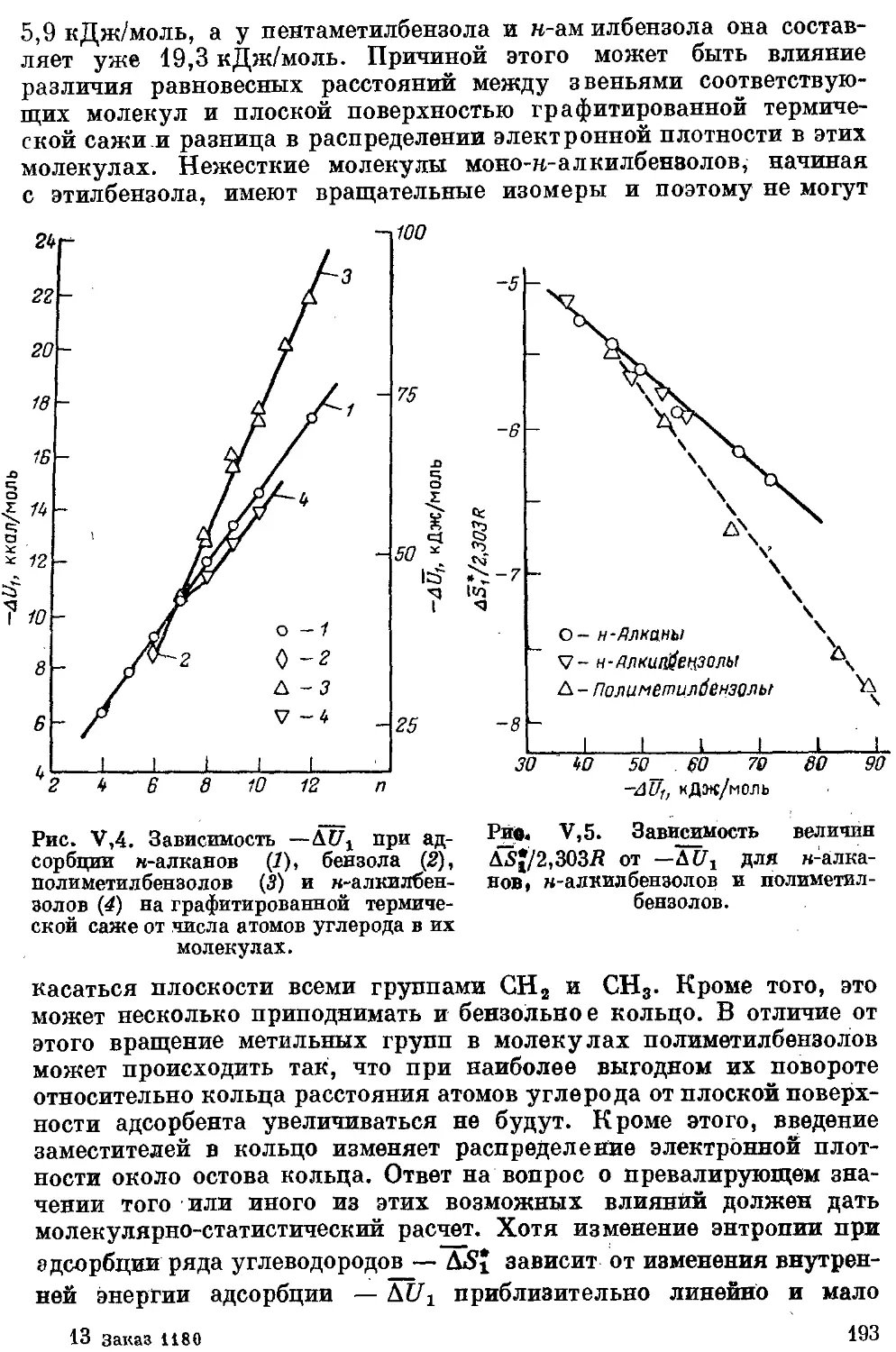
3. н-Алканы ..................................................... 185
4. Изоалканы ................................................... 186
5. Цикланы ...................................................... 186
*6, Алкены, алкадиены, циклены и‘ циклодиены..................... 189
5
7. Алкины ..................................................... 192
8. Бензол ц алкилбензолы .. .................................... 192
9. Полифенилы ............................................... 194
10. Ароматические углеводороды с конденсированными ядрами .... 194
11. Галогенпроизводные углеводородов .......................... 194
12. Простые эфиры........................................... . 198
13. м-Спирты ................•. ................................. 199
14. Терпены .................................................... 199
15. Дейтерозамещенные органические соединения................... 200
16. Азот- и серусодержащие производные углеводородов............. 201
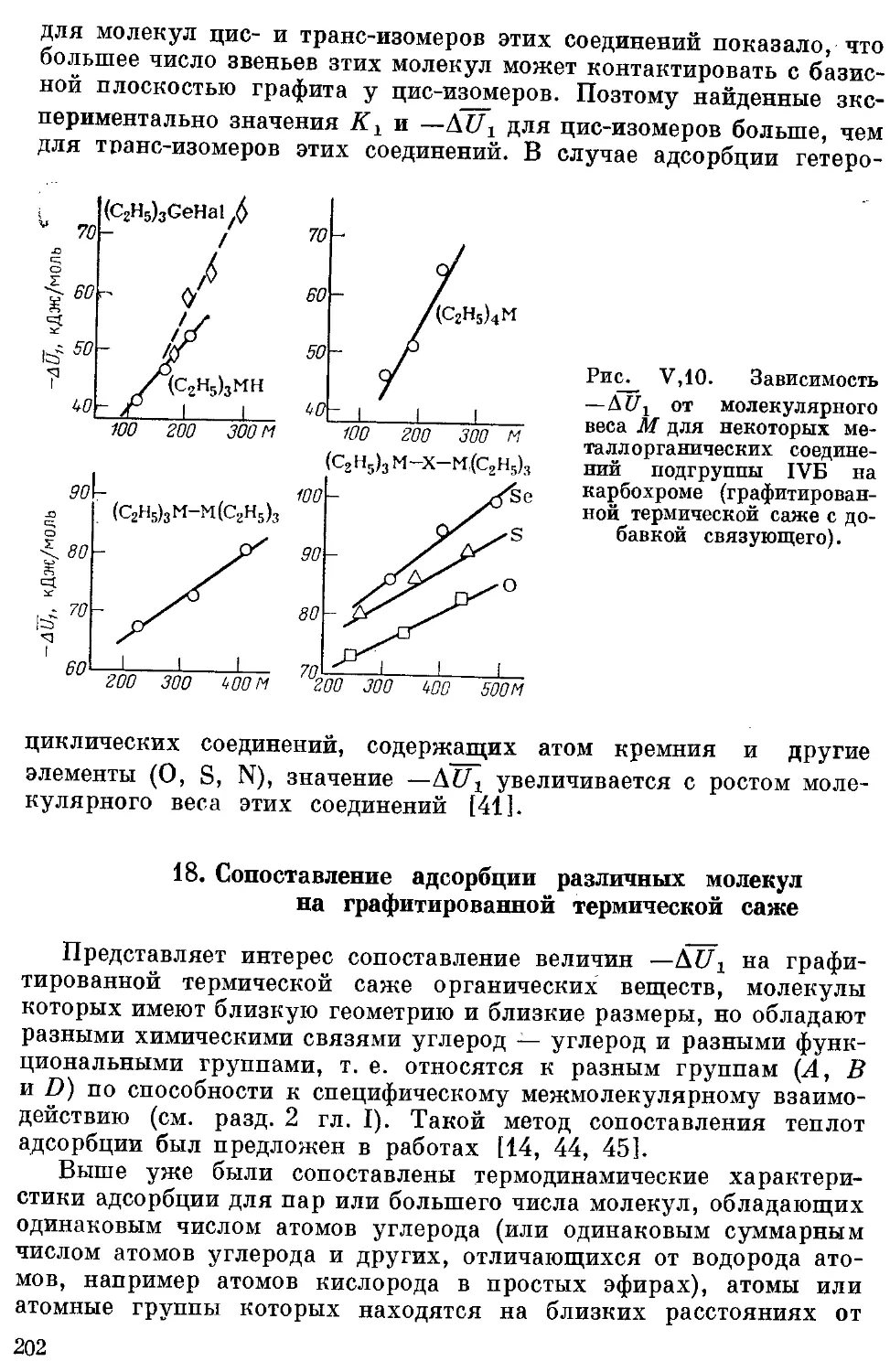
17, Элементорганическне соединения подгруппы IVB 201
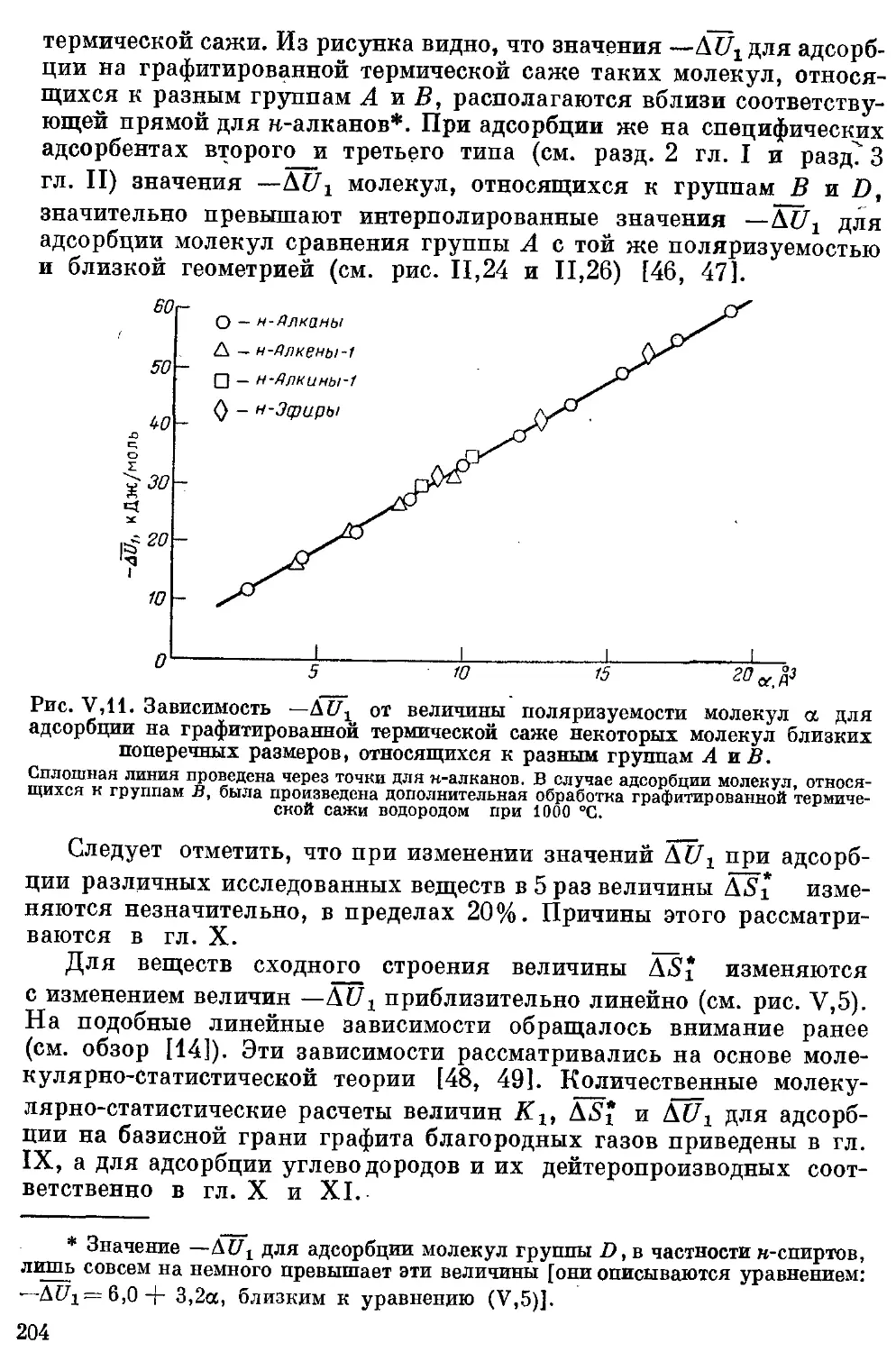
18. Сопоставление адсорбции различных молекул на графитированной
термической саже......................................... 202
Литература ............................................... » . . 205
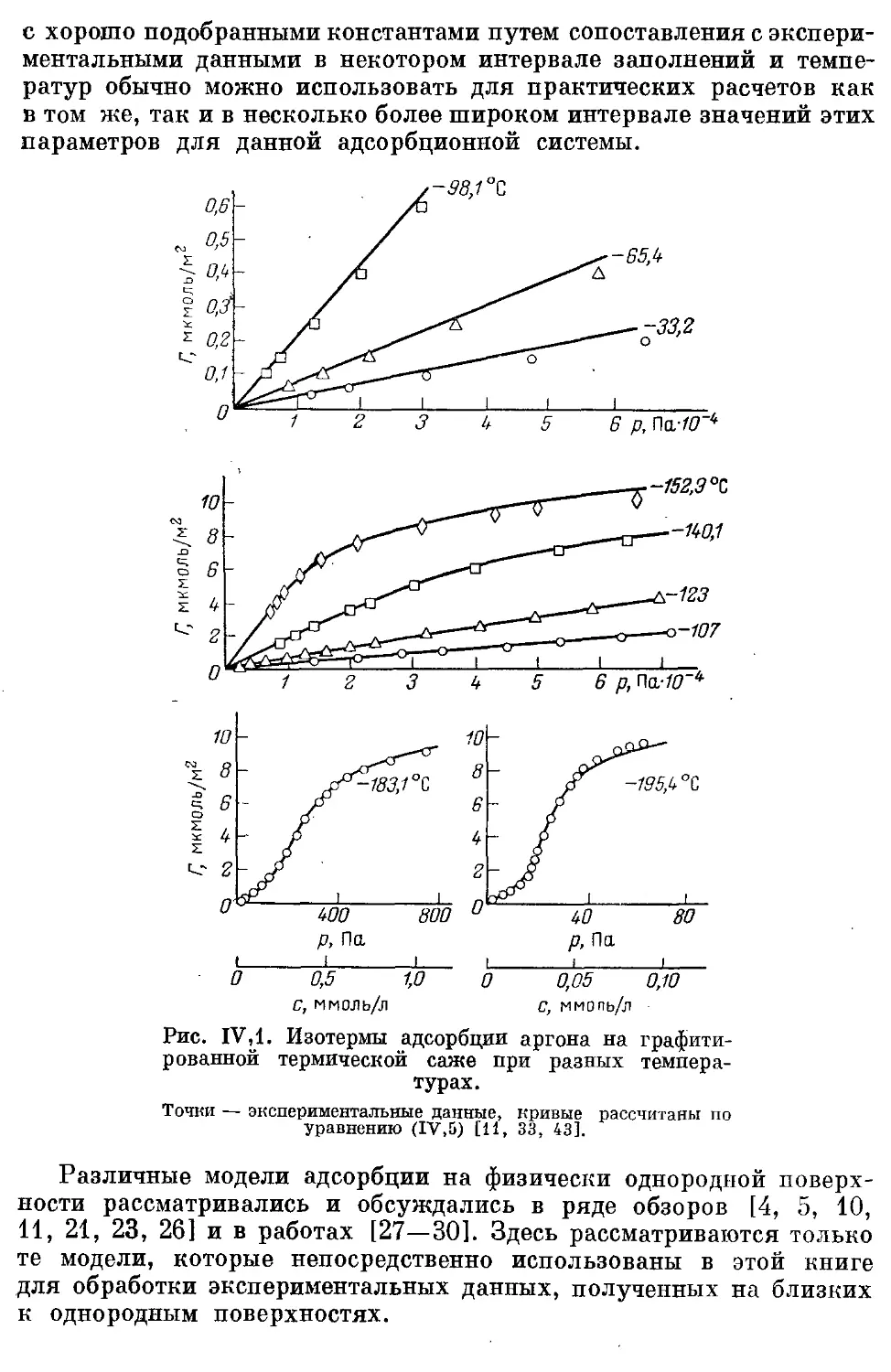
Глава VI. Статистические вириальпые выражении для термодинамических
характеристик адсорбции......................................... 207
1. Общие выражения.................................. , , * , . 207
2. Изотерма адсорбции однокомпонентного газа............... • • • 210
3. Уравнение состояния адсорбированного однокомпонентного газа * . 213
4. Изотерма адсорбции многокомпонентного газа.........'......... 214
5. Удерживаемый объем........................................... 217
6. Химический потенциал адсорбированного вещества............... 218
7. Изменение энтропии нри адсорбции............................. 219
8. Изменение внутренней энергии при адсорбции..............* . < 221
9. Изменение теплоемкости при адсорбции......................... 223
10. Частные случаи общих вириальных выражений.................... 224
Газ вдали от поверхности является идеальным.............. 224
Молекулы адсорбата взаимодействуют только с адсорбентом . , 225
Литература..................................................... 225
Глава VII. Статистические выражения для константы Генри и других тер-
модинамических характеристик адсорбции при нулевом запол-
нении поверхности................................................ 227
1. Общие выражения для константы Генри........................... 227
2. Приближенные выражения для нежестких молекул.................. 229
3. Выражения для молекул с поворотной изомерией.................. 232
4. Выражения для квазижестких молекул . ......................... 234
5. Выражения для разных моделей состояния адсорбированных квази-
жестких молекул................................................. 235
Свободное движение вдоль математически однородной поверхности . 235
Свободное движение вдоль математически однородной поверхности
и гармонические колебания перпендикулярно поверхности .... 235
6, Квантовые поправки ........................................... 237
Литература....................................................... 238
6
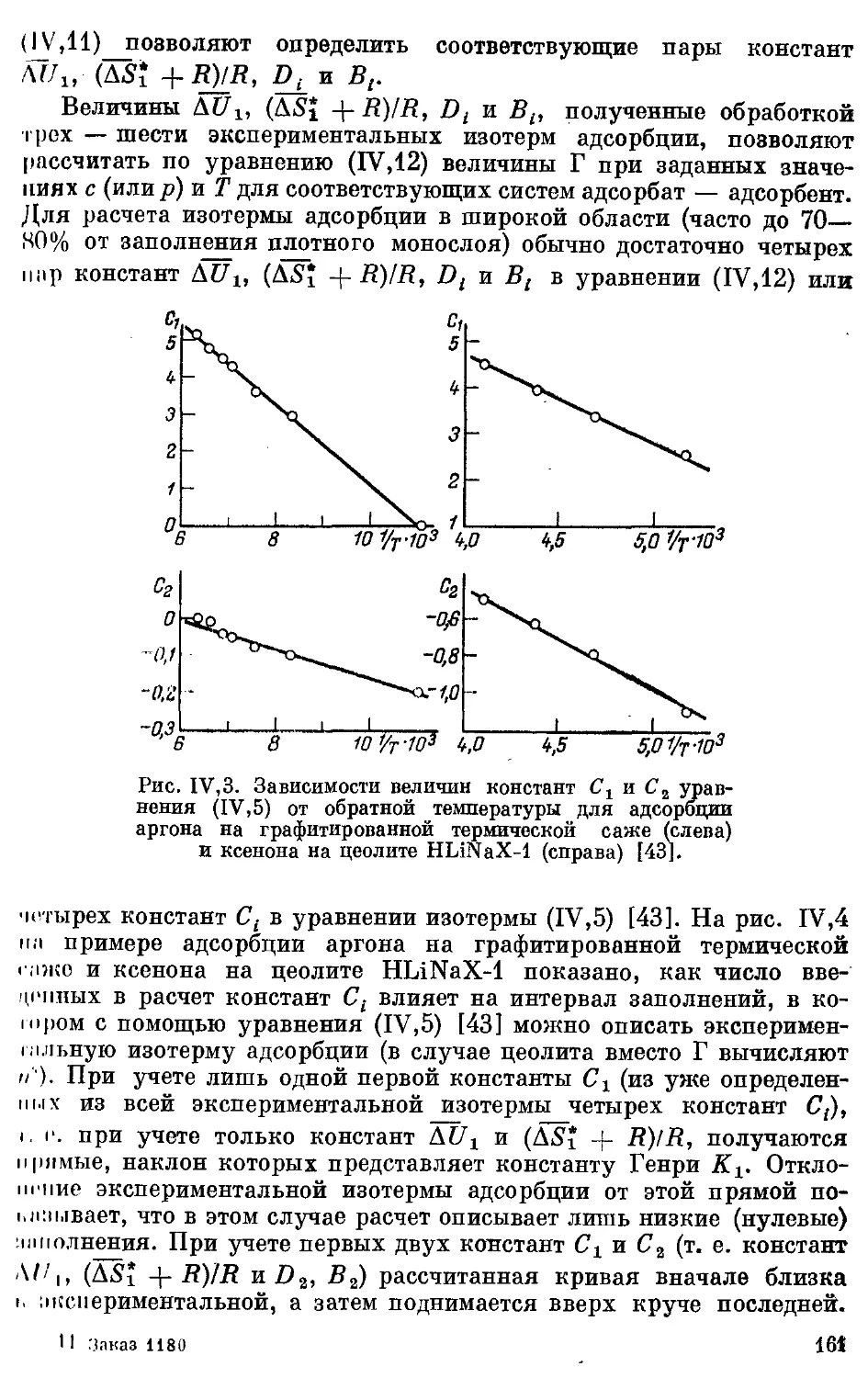
VIII. Межмолекулярные силы и потенциальная функция межмолеку-
лярного взаимодействия молекулы с адсорбентом .... 241
|f Общая характеристика проблемы................................. 241
Квантовомеханические теории взаимодействия молекулы с адсорбен-
том .......................................................... 248
I, Аддитивность потенциала межмолекулярного взаимодействия по по-
тенциалам взаимодействия пар силовых центров..................... 249
4i Выбор силовых центров в молекуле и их положения............... 254
Hi Выбор форк£ы атом-атомного потенциала межмолекулярного взаимо-
действия ...................................................... 255
Представление* Потенциала’в*вйде суммы слагаемых, выражающих
* вклады различных видов взаимодействия..................... 255
Обменная энергия первого порядка и^мен..................... 256
Энергия дисперсионного притяжения м^аи>сп................... 257
Обменная энергия второго порядка м^мен . . . .............. 257.
Форма атом-атомного потенциала межмолекулярного взаимодействия ч258
6. Оценка параметров атом-атомных потенциалов.................... 260
Константа .................................................. 260
Константа и отношение 264
Параметры потенциала отталкивания........................... 264
Prtwuonociioo расстояние при межмолекулярном взаимодействии двух
силовым центров.................,........................... 266
7. Суммирование атом-атомных потенциалов...................... . 268
Общее выражение............................................ 268
Расчет решеточных сумм...................................... 268
Расчет 2г-« пад базисной гранью графита и гранью (100) простой ку-
бической решетки............................................ 272
Аппроксимации зависимости Sr“M п £(z/d,m) от z.............. 275
Литература..................................................... 275
Глава IX. Расчет термодинамических характеристик адсорбции одноатом-
ных молекул на графите............................................ 284
1. Влияние формы атом-атомного потенциала межмолекулярного взаимо-
действия на значение константы Генри Ki........................ 284
2. Влияние приближений при суммировании атом-атомных потенциалов
на значения константы Генри К± .... *......................... 286
3. Гармоническое приближение .................................... 289
4. Оценка термодинамических характеристик адсорбции благородных
газов на базисной грани графита на основании свойств адсорбата и ад-
сорбента, взятых в отдельности .................................. 290
7
5. Определение параметра при использовании экспериментальных зна-
чений Л'1.............................. ...................... 297
6. Молекулярно-статистический метод определения удельной поверхности
графи тированной сажи .................... 299
Литература.............................. ..................... 303
ГлаваХ. Молекулярно-статистический расчет термодинамических харак-
теристик адсорбции углеводородов на*графите . . . ............. 305
1. Общие замечания о расчете термодинамических характеристик адсорб-
ции углеводородов...................<<...<»................ , 305
2. Алканы ................................................... 308
Оценка параметров атом-атомных потенциальныхТфункций межмоле-
кулярного взаимодействия атомов Н и С молекул алканов с атомами
С графита на основании свойств адсорбата и адсорбента, взятых
в отдельности................................................. 308
Термодинамические характеристики адсорбции м'етана, этана и про-
пана на базисной грани графита, полученные при использовании
приближенных атом-атомных потенциальных функций................. 310
Уточнение параметров атом-атомных потенциалов взаимодействия
атомов С (алкан) . . G (графит) и Н (алкан) ... С (графит) . . . 311
Расчет термодинамических характеристик адсорбции этана с учетом
внутреннего вращения молекул.................................... 315
н-Бутан, н-пентан и н-гексан.............................. 317
Расчеты термодинамических характеристик адсорбции н-алканов
при выборе в качестве силовых центров звеньев GHg и СЩ . . . 324
пзо-Алканы ............................................... 328
3. Цикланы .................................................... 329
4. Ненасыщенные и ароматические углеводороды. Зависимость меж-
молекулярного взаимодействия атомов углерода от их валентного со-
стояния ...................................................... 332
Преимущества использования адсорбции для определения зависи-
мости межмолекулярного взаимодействия атомов от их валентного
состояния ...................................................... 332
Алкены и алкины........................................... 333
Углеводороды с сопряженными связями....................... 338
Ароматические углеводороды................................ 340
5. Сопоставление потенциальных функций взаимодействия атомов С . . .С
и Н . . . С для различных проявлений межмолекулярного взаимодей-
ствия ......................................................... 345
Литература.................................................... 349
Глава XI. Изотопный эффект при адсорбции углеводородов на графите . 353
1. Влияние на адсорбцию водорода и углеводородов замещения атомов
водорода иа атомы дейтерия................................... 353
8
It Статистические выражения для термодинамических характеристик ад-
сорбционного разделения сложных изотопнозамещенных молекул . . 354
I. Молекулярно-статистический расчет изменений термодинамических
характеристик адсорбции углеводородов при замещении атомов Н на
атомы D..................................................... 358
Литература................................................... . . 362
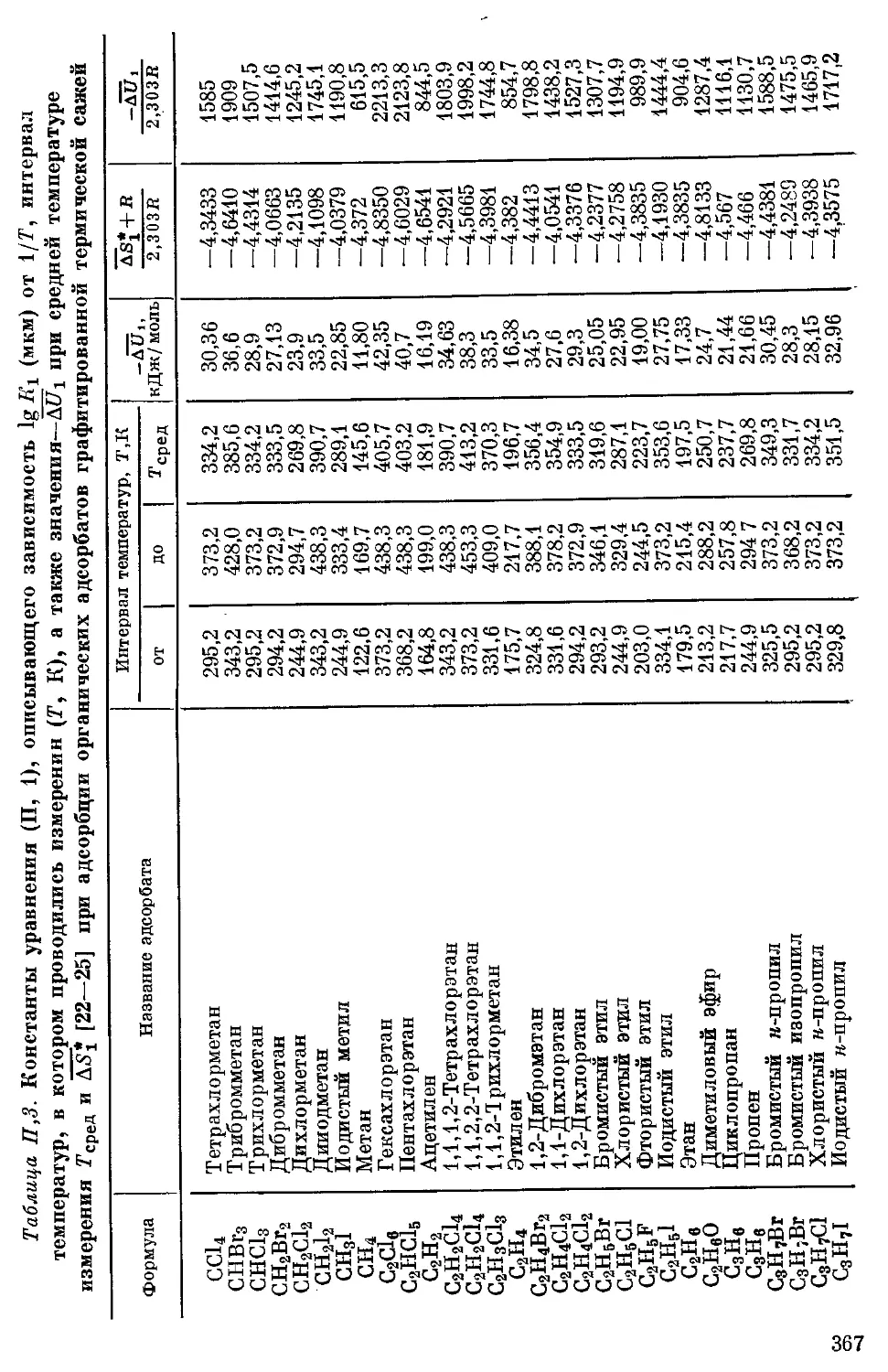
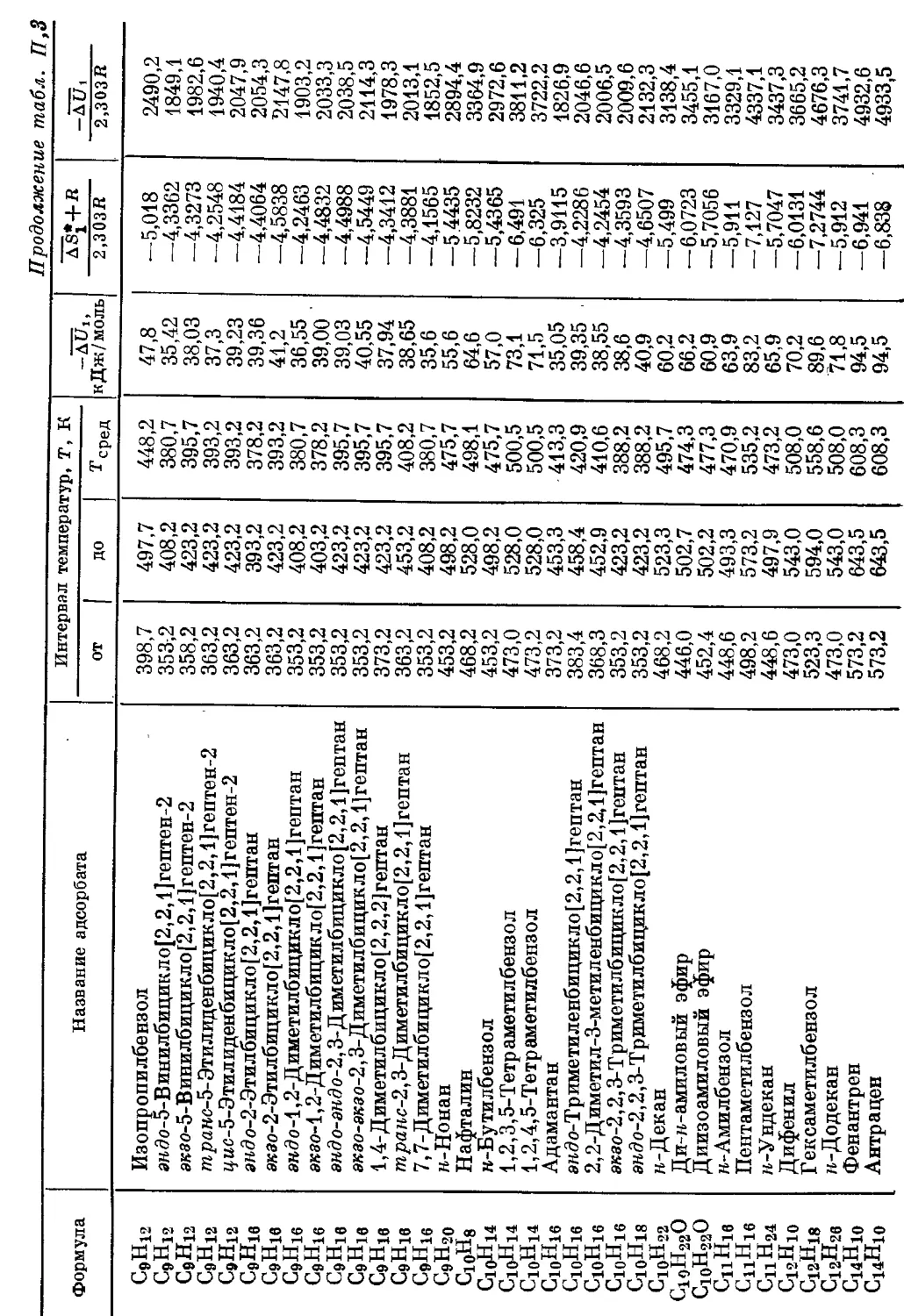
Приложение .................................................. 365
L Значения площади, приходящейся на молекулу в плотном мономоле-
куляриом слое при адсорбции на графитированной термической саже 365
I. Вйиисимости 1g К\ от температуры и значения Д5* и SUi при адсорб-
ции различных адсорбатов графитированной термической сажей . . 366
Литература...................................................... 371
Синеок некоторых обозначений.................................... 372
Предметный указатель ........................................... 378
ПРЕДИСЛОВИЕ
Предлагаемая читателям монография является четвертой книгой
серии «Адсорбция и химия поверхности». Ранее вышли три книги
этой серии: А. В. Киселев и Я. И. Яшин «Газо-адсорбционная
хроматография». М., «Наука», 1967 (исправленные и дополненные
издания этой книги вышли в Париже и в Нью-Йорке в 1969 г.);
А. В. Киселев и В. И. Лыгин «Инфракрасные спектры поверхно-
стных соединений и адсорбированных веществ». М., «Наука», 1972;
коллектив авторов, под ред. А. В. Киселева и В. П. Древинга «Экс-
периментальные методы в адсорбции и молекулярной хроматогра-
фии»,. М., изд. МГУ, 1973. Эти монографии посвящены только тем
разделам химии поверхности и адсорбции, в которых авторы имеют
длительный опыт работы (см. предисловие к серии в упомянутой
выше книге А. В. Киселева и В. И. Лыгина).
Название данной книги, указывающее на адсорбцию на одно-
родных поверхностях, может показаться несколько противоречи-
вым, так как нельзя получить идеально однородные поверхности
твердых ктел. Однако выполненные за последние 20 лет работы по
синтезу новых адсорбентов показали возможность получения по-
верхностей, близких к однородным. Влияние остаточной неодно-
родности поверхности на адсорбционные свойства оказалось или
соизмеримым с влиянием погрешностей экспериментальных методов
измерения термодинамических свойств адсорбционных систем, или
настолько малым, что его можно учесть при обработке эксперимен-
тальных результатов.
Создание адсорбентов с поверхностью, близкой к однородной,
не только имело практическое значение для молекулярной хромато-
графии, но и стимулировало развитие молекулярной теории адсорб-
ции и дало ей необходимое экспериментальное основание. Разра-
ботке одного из важнейших направлений этой теории — молекуляр*
но-статистической теории адсорбции — долгое время мешал разрыв
10
ИНаду предложенными в разных работах теоретическими моделями
1|аорбции, которые предполагают поверхность однородной, и экс-
ЦрШМентальными исследованиями, которые проводились на весьма
Однородных поверхностях, плохо воспроизводимых как по гео-
(Втрической структуре, так и по химическому составу. Только около
(О ДОТ назад создались благоприятные возможности для быстрого
раивмтия молекулярно-статистической теории адсорбции. Сюда отно-
•ИТОЛ, во-первых, создание близких к однородным непористых
пористых кристаллических адсорбентов, во-вторых, накопление
вдожпых и воспроизводимых значений термодинамических харак-
теристик адсорбции на таких адсорбентах и, в-третьих, использова-
электронных вычислительных машин. Это позволило выразить
Многие термодинамические свойства системы адсорбат — адсорбент
В виде физико-химических констант, характеризующих межмолеку-
ЛЯрныо взаимодействия адсорбат — адсорбент и адсорбат —
ВДеорбат в адсорбционных системах. Эти воспроизводимые физико-
Мнмичоские константы служат объектами молекулярно-статисти-
Чеоного расчета.
JI этой книге рассмотрена преимущественно бласть небольших
Вднолноний поверхности. Основное внимание в книге уделяется
рноталличоским нопопистым адсорбентам и среди них — графити-
рованной термической саже (базисной грани графита). Кристалли-
ЧМИНВ пористые адсорбенты — цеолиты привлекаются лишь для
Грнииоинн, так как их предполаг ется рассмотреть также в отдельной
Монографии.
И сякни с все возрастающим значением в адсорбционных сследо-
валиях измерений концентраций адсорбата в газовой фазе с омощью
детекторов, лримеппомых в хроматографии, приведенные^в этой
джиге термодинамические и молекулярно-статистические выражения
еаписалы как в терминах активности и мольной концентрации в газо-
вой фазе, так и в терминах давления в этой фазе, измеряемого
манометрами, применяемыми при исследованиях адсорбции статиче-
скими методами.
В книге рассмотрены величины адсорбции и поверхностных термо-
динамических функций, выраженные по Гиббсу. Молекулярно-ста-
тистические выражения для гиббсовых термодинамических харак-
теристик адсорбции получены с помощью большого канонического
ансамбля. Они даны в виде вириальных выражений для изотермы
адсорбции и происходящих при адсорбции изменений внутренней
энергии, энтропии и теплоемкости; Рассмотрены современное состо-
11
яние теории межмолекулярных взаимодействий и приближенное
выражение потенциальной функции межмолекулярного взаимодей-
ствия через парные атом-атомные потенциальные функции этого
взаимодействия, а также основы определения потенциальных функ-
ций межмолекулярного взаимодействия с адсорбентом благородных
газов, различных углеводородов и дейтероуглеводородов с учетом
валентного состояния входящих в них атомов углерода. Полученные
молекулярно-статистические выражения для термодинамических
характеристик адсорбции и выражения для потенциальных функций
межмолекулярного взаимодействия молекул с адсорбентом при-
менены для числового полуэмпирического молекулярно-статистиче-
ского расчета констант Генри и других термодинамических характе-
ристик адсорбции при малом (нулевом) заполнении базисной грани
полубесконечной решетки графита.
В приложении приведены справочные таблицы констант Генри,
дифференциальных мольных изменений внутренней энергии и энтро-
пии адсорбата при малой (пулевой) величине адсорбции чистых
веществ на графитированной термической саже.
Авторы будут признательны за указание ошибок и неясностей
и за пожелания, которые можно было бы. учесть при работе над
улучшением этой книги и над другими книгами этой серии.
Авторы благодарят Е. В. Калашникову и К. Д. Щербакову
за предоставление газохроматографических данных по адсорбции
на графитированной термической саже, а также В. Т. Авгуль,
А. П. Архипову, Л. Д. Воробьеву, Т. И. Горюнову, Т. С. Ки-
селеву и Р. А. Пошкене за помощь при подготовке рукописи.
А. В. Киселев
Глава I
ЗНАЧЕНИЕ АДСОРБЕНТОВ С БЛИЗКОЙ К ОДНОРОДНОЙ
ПОВЕРХНОСТЬЮ ДЛЯ РАЗВИТИЯ МОЛЕКУЛЯРНОЙ ТЕОРИИ
АДСОРБЦИИ И ПРАКТИЧЕСКИХ ПРИМЕНЕНИЙ
I. Представления об однородности поверхности
Получение и исследование адсорбентов с хорошо воспроизводи-
мыми гвойствами и с возможно более однородной поверхностью
в последнее десятилетие приобретает все большее значение как для
ронитин молекулярной теории адсорбции [1—34], так и для практи-
Чегиих применений в адсорбционной хроматографии [11, 18, 20,
2г», 2(1, 33 49]. Термодинамические адсорбционные свойства таких
ндсорбептон могут быть представлены в виде характеризующих
гигтвму адсорбат — адсорбент физико-химических констант [7, 11,
21, 24, 33, 44—49]. Только такие константы, неосложненные не-
шич|ро1Н1нодимостью строения поверхности адсорбента и влиянием
। илыюй и неконтролируемой ее неоднородности, могут быть исполь-
впйппы дли установления основных закономерностей проявления
йпчнмоленулнрных взаимодействий адсорбат — адсорбент и адсор-
Ант йдеорбвт в создаваемом адсорбентом поле межмолекулярных
сил, Иппольпун такие физико-химические константы, можно исследо-
вать потенциальные функции межмолекулярного взаимодействия
при адсорбции [10, 16, 22, 50, 51], а также исследовать некоторые
детали строения молекул [18, 33, 34, 40]. Кроме того, такие харак-
теристики адсорбционных систем позволяют идентифицировать не-
на1мчтныо вещества методом адсорбционной хроматографии [11,
33. 34 Г
Поверхность реальных адсорбентов — твердых тел в той или
иной степени неоднородна. Понятия однородности поверхности,
используемые в адсорбции, зависят от того, какие свойства самой
поверхности или системы поверхность — адсорбированная молекула
рассматриваются. В связи с этим выделяются несколько понятий
однородности поверхности: 1) химически однородная поверхность;
2) геометрически однородная поверхность; 3) физически однородная
поверхность и 4) математически или энергетически однородная
поверхность.
Одной из важнейших характеристик поверхности является ее
химический состав. В зависимости от способа получения и дальней-
шей обработки твердого тела химический состав его поверхности
может более или менее отличаться от его объемного состава. Под
химически однородной поверхностью обычно понимается поверх-
ность, химический состав которой одинаков на всех ее участках
1а
атомных или молекулярных размеров. К этим химически однородным
поверхностям приближаются поверхности многих кристаллических
и аморфных твердых тел, не содержащие посторонних примесей.
Например, к химически однородной близка поверхность чистых
окислов в предельно гидроксилированном состоянии [52, 53]. Хими-
чески однородные поверхности с гидроксильными группами исполь-
зуются, в частности, при изучении водородной связи при адсорбции.
Частично дегидроксилированные поверхности чистых окислов
или поверхности окислов, содержащие примесные центры, например
поверхность кремнезема с примесью алюминия, бора и других эле-
ментов, химически неоднородны [52, 54, 55]. Поверхность обрабо-
танной водородом графитированной термической сажи, состоящая
из атомов одного сорта, химически однородна [46—48]. В случае
кристаллов различных соединений можно говорить о химической
однородности определенной грани. В пределах же участка грани,
соответствующего элементарной ячейке или некоторой, ее части,
химический состав поверхности таких кристаллов может сильно
различаться. Однако такие участки на всей поверхности грани
повторяются строго периодически. Поэтому в целом грань идеаль-
ного кристалла можно рассматривать как химически однородную
поверхность. Следовательно, поверхность определенных граней чи-
стых ионных и молекулярных кристаллов с этой точки зрения также
химически однородна.
Под геометрически однородной поверхностью обычно понимается
поверхность постоянной кривизны, в частности, в адсорбции часто
рассматриваются такие модели геометрически однородной поверх-
ности, как плоская поверхность, поверхность цилиндра и шара.
Понятие геометрически однородной поверхности используется при
качественном или упрощенном количественном описании адсорбции
и капиллярной конденсации.
При исследовании межмолекулярных взаимодействий молекул
с адсорбентом для характеристики строения поверхности исполь-
зуются понятия физически и математически (энергетически) однород-
ной поверхности. Простейший случай физически однородной по-
верхности представляет одна бесконечная грань идеальной полу-
бесконечной решетки твердого тела. Такая физически однородная
поверхность является однородной и химически, и геометрически.
Основным свойством физически однородной поверхности является
периодическое изменение потенциальной энергии взаимодействия
молекулы с поверхностью при движении молекулы вдоль такой
поверхности. Это вызывается атомным строением твердого тела.
Изменение потенциальной энергии зависит как от строения поверх-
ности, так й от строения и размеров взаимодействующей с ней моле-
кулы. Отсюда следует, что понятие физической однородности не
сводится только к постоянству химического состава, т. е. оно предъ-
являет более жесткие требования к структуре поверхности, чем
понятие химической однородности, которому может соответствовать
поверхность аморфного вещества.
14
Придельный случай физически однородной поверхности пред-
М11ЛЯОТ математически или энергетически однородная поверхность.
Ирм ВТом пренебрегают периодическим изменением потенциальной
ВИВрс ИИ молекулы при ее движении вдоль поверхности и принимают*
ЧТО Потенциальная энергия молекулы при таком перемещении не
Такая упрощенная модель очень удобна для теорети-
4HHVX исследований адсорбции и поэтому широко используется
И теории адсорбции. Действительно, физически однородные поверх-
ности Часто п достаточно хорошем для многих задач приближении
МОЖНО рассматривать и как математически однородные.
Неоднородность поверхности кристаллических адсорбентов может
бьиь вывийна причинами двух родов. Во-первых, это наличие ребер*
PTVOooai роста, различных дислокаций, возможные фазовые и хими-
MfWKHO примеси у самих частиц. Такого рода неоднородность поверх-
ности твердых адсорбентов при выборе благоприятных условий
ОИнтНй и последующих обработок может быть значительно снижена.
Практически важно снизить такую неоднородность до такой сте-
пени, чтобы ее влиянием на адсорбционные свойства можно было бы
пренебречь.
Во-вторых, это выход на поверхность кристаллов граней разных
иидвиоов. С этой точки зрения физически однородна только такая
поверхность, которая образована лишь гранями одного индекса
нрнетАлли, нпиример, поверхность графитированной термической
ШНИ, пбрввоивпнАЯ лишь из базисной грани кристаллов графита,
ИЛИ поверхность кубического кристалла, образованная из грани
(100). Кристаллические адсорбенты, на поверхность которых выходят
епколько (часто однако немного, не более двух, трех) физически
однородных граней разных индексов, можно рассматривать как
емвсь равных адсорбентов с физически однородными поверхностями.
Мели раяличные грани значительно различаются по заселенности
атомами, ионами или молекулами, то межмолекулярные взаимо-
действии таких граней с адсорбирующимися молекулами сильна
ранличлютси по потенциальной энергии. Адсорбция на отдельных
гранях таких кристаллов при достаточно низких температурах
может происходить в основном по очереди на каждой из граней*
соответственно, при разных величинах давления пара адсорбата
в газовой фазе.
Преимущественно развитые грани кристаллов могут состоять
яа атомов одного сорта, например, из атомов углерода в случае
базисной грани слоистых кристаллов графита, образующей практи-
чески всю реальную поверхность графитированных термических
саж [6, 21] и почти всю поверхность листочков расщепленного гра-
фита [19]. Частицы графитированной термической сажи предста-
вляют собой полиэдры, ограненные базисными гранями, а листочки
расщепленного графита — тонкие пластинки с развитыми базис-
ными гранями с кристаллографическим индексом (0001). Эти адсор-
бенты обладают слоистой структурой. В случае базисной грани
графита повторяющимся участком является шестиугольник, обра-
15
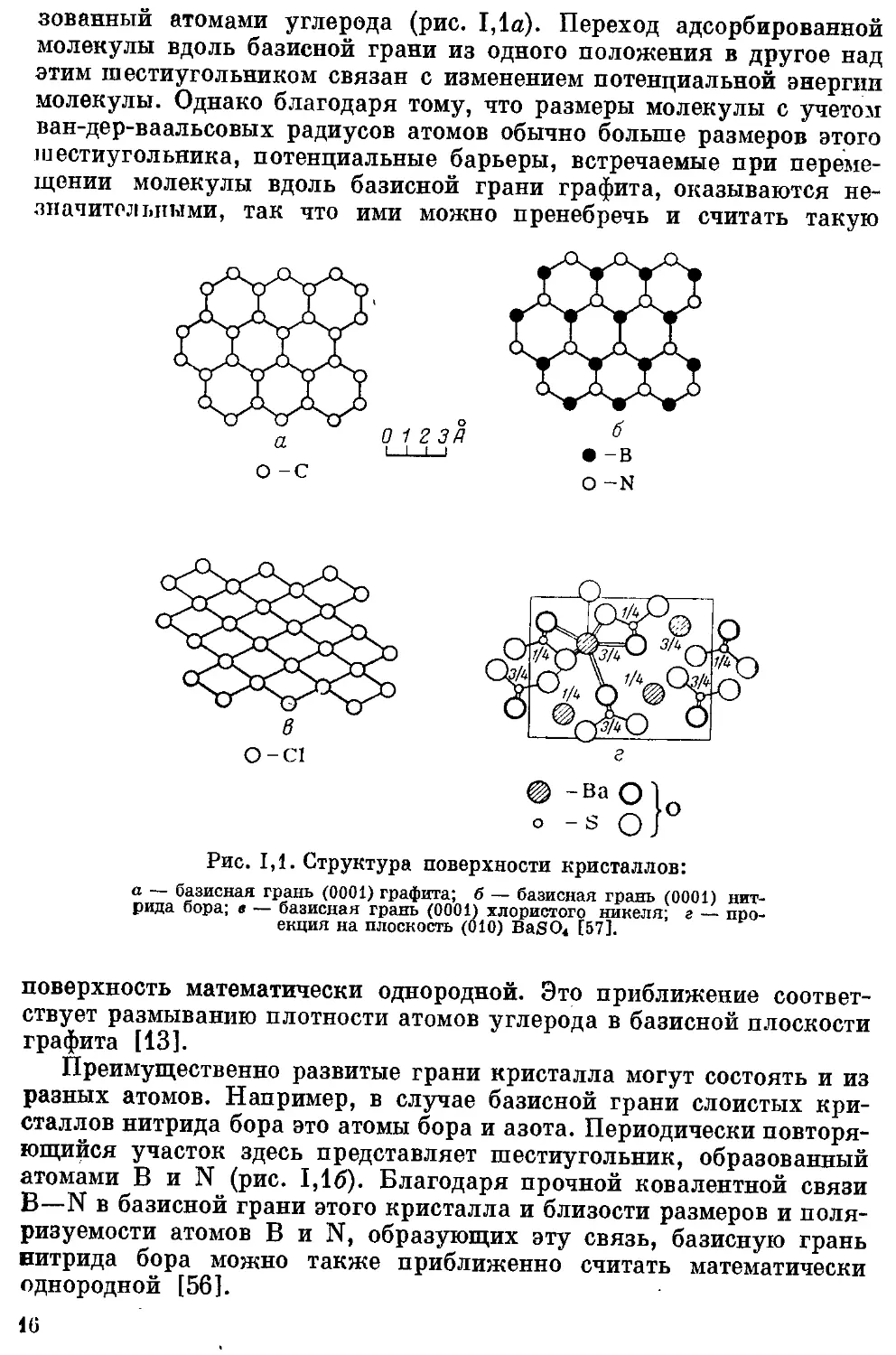
зованный атомами углерода (рис. 1,1а). Переход адсорбированной
молекулы вдоль базисной грани из одного положения в другое над
этим шестиугольником связан с изменением потенциальной энергии
молекулы. Однако благодаря тому, что размеры молекулы с учетом
ван-дер-ваальсовых радиусов атомов обычно больше размеров этого
шестиугольника, потенциальные барьеры, встречаемые при переме-
щении молекулы вдоль базисной грани графита, оказываются не-
значительными, так что ими можно пренебречь и считать такую
0 -ва О
° - s о
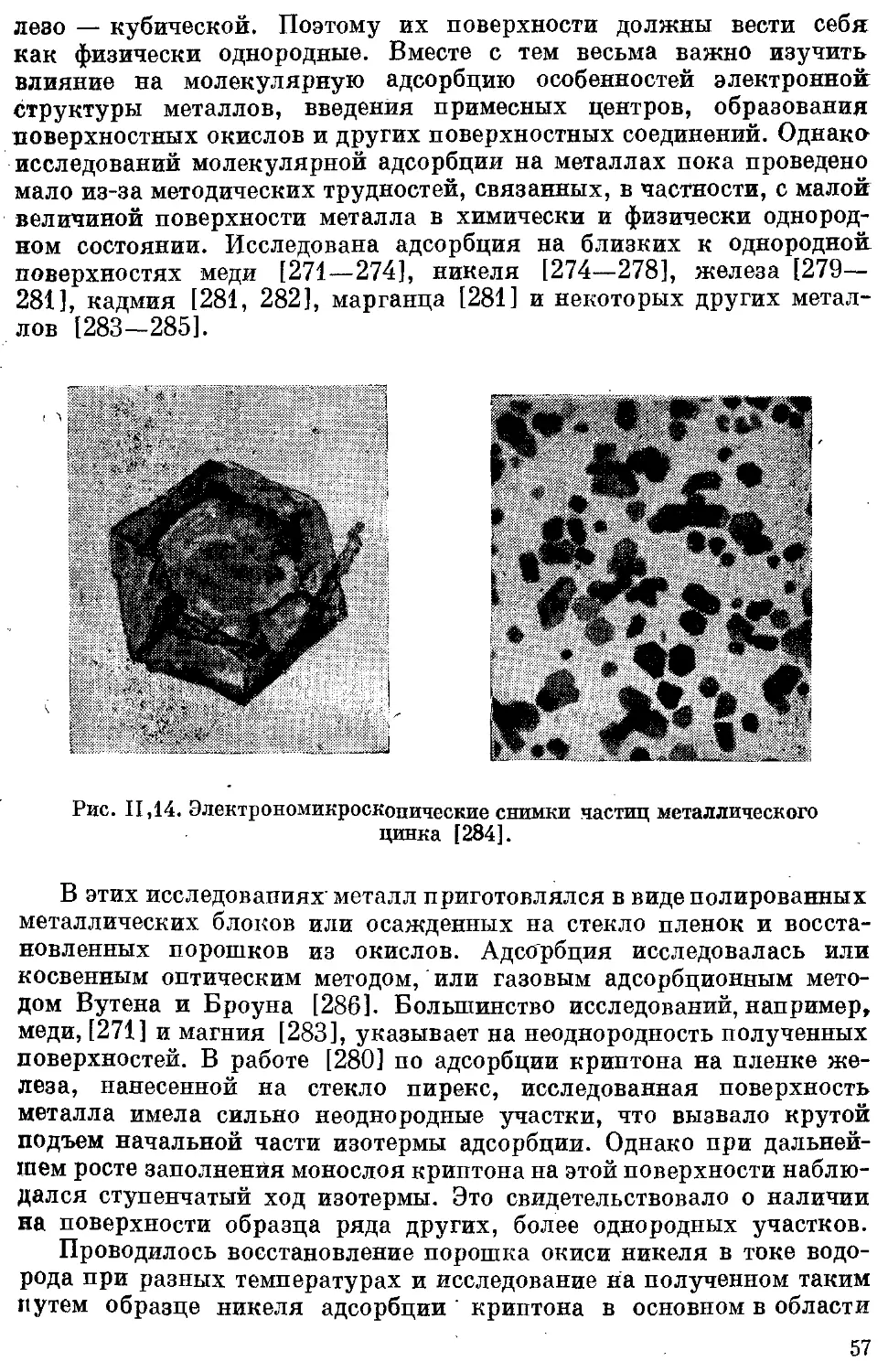
Рис. 1,1. Структура поверхности кристаллов:
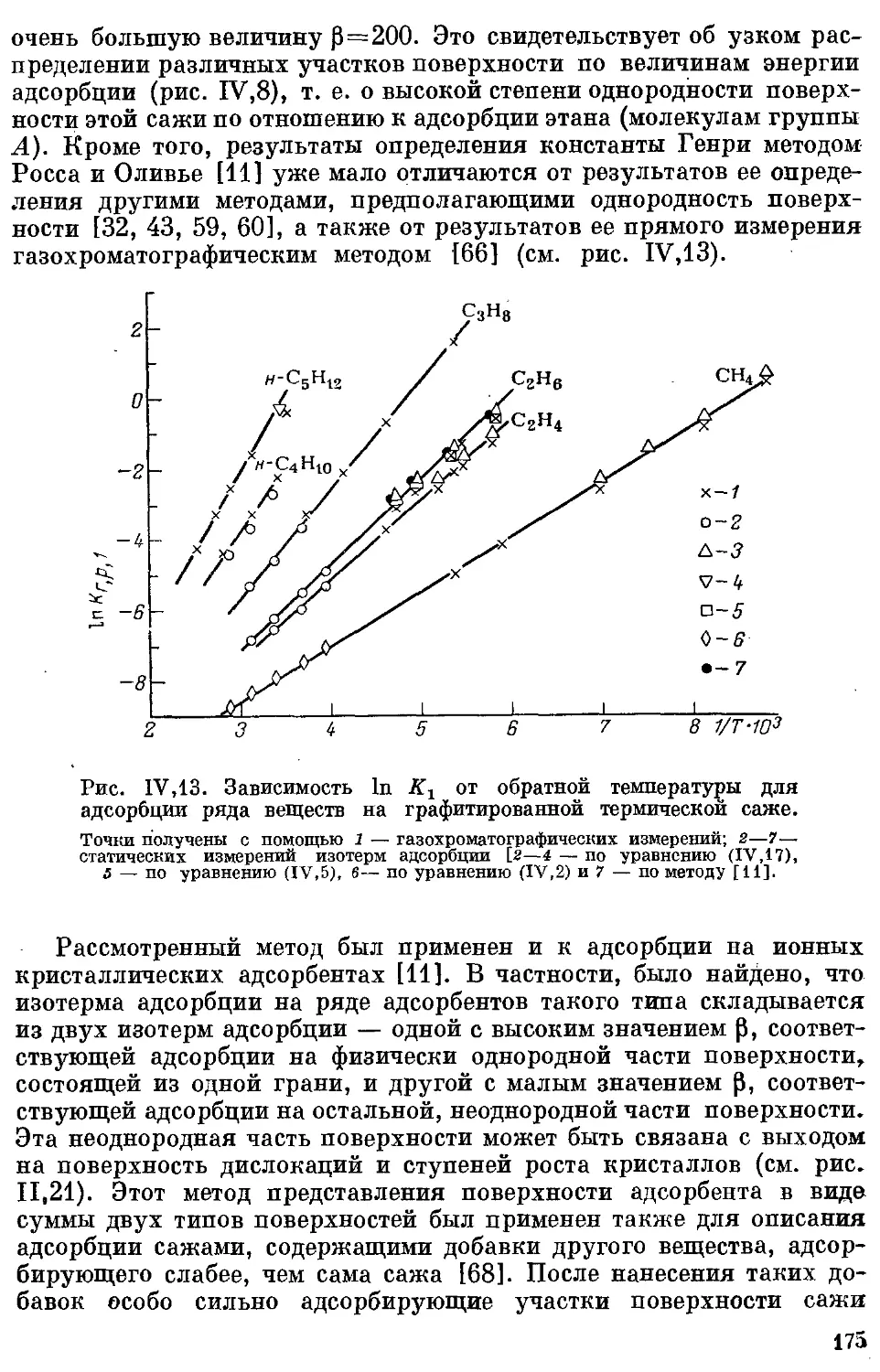
а — базисная грань (0001) графита; б — базисная грань (0001) нит-
рида бора; в — базисная грань (0001) хлористого никеля; г — про-
екция на плоскость (010) BaSO4 [57].
поверхность математически однородной. Это приближение соответ-
ствует размыванию плотности атомов углерода в базисной плоскости
графита [13].
Преимущественно развитые грани кристалла могут состоять и из
разных атомов. Например, в случае базисной грани слоистых кри-
сталлов нитрида бора это атомы бора и азота. Периодически повторя-
ющийся участок здесь представляет шестиугольник, образованный
атомами В и N (рис. 1,16). Благодаря прочной ковалентной связи
В—N в базисной грани этого кристалла и близости размеров и поля-
ризуемости атомов В и N, образующих эту связь, базисную грань
нитрида бора можно также приближенно считать математически
однородной [56].
16
Слоистой структурой обладают также многие ионные адсорбенты,
Иаример NiCl2, СоС12 и др. [58—61]. Поверхность базисной грани
К1С12 образована ионами хлора (рис. 1,1в). Под ними расположены
Юны никеля, противоположного знака и иной поляризуемости,
ЙОД которыми снова расположены ионы хлора. Ионы, образующие
Такие тройные слои, соединены сильными связями, но силы взаимо-
действия между тройными слоями значительно слабее. В базисной
грани хлористого никеля (0001) повторяющимся участком является
ромб, в вершинах которого расположены четыре иона хлора.
Однако у таких кристаллов иногда развиты также и грани, содер-
жащие и С1~.
II случае хорошо образованных кубических кристаллов, таких
|«ав кристаллы ряда металлов (Си, Fe), солей (NaCl) и окислов (MgO),
|к a iHinapxnoCTb, за исключением ребер и углов, может состоять
шльно ив граней (100), и поэтому она в основном построена одина-
шнш, Если зти кристаллы выращены и обработаны в условиях,
устраняющих или по крайней мере сильно снижающих образование
Вв поверхности граней ступеней роста и других дефектов, то поверх-
ность соответствующего адсорбента при достаточно больших раз-
мерах кристаллов, т. е. при небольшой удельной поверхности, при-
блишнотсн к физически однородной. При этих условиях влияние
тпних источников неоднородности, как ребра и углы кристаллов
И моста их контакта, невелико.
|||чц<*ст|ш с кубической решеткой могут кристаллизоваться и так,
чю парнду с гранями (100) будут образовываться также грани (111)
И (110).
Поверх кость ионных кристаллов с большими комплексными
инионами, например сульфатов щелочноземельных металлов, обра-
зуется большим числом граней разных индексов. Поэтому в целом
поверхность таких кристаллов физически неоднородна. На рис. 1,1а
маображона проекция на плоскость (010) BaSO4. Ионы бария и серы
расположены на расстояниях г/4 и 3/4 параметра кристаллической
р|чнетки пышо по оси с (перпендикулярной плоскости чертежа)
и плоскостях, параллельных плоскости чертежа. В этих парал-
лельных плоскостях также расположена часть ионов кислорода
(кружки, изображенные тонкой линией). Другая часть ионов кисло-
рода расположена на 1,18 А выше и ниже этих плоскостей (кружки,
изображенные жирной линией). Периодически повторяющиеся уча-
стки поверхности на каждой грани таких кристаллов с комплекс-
ными ионами значительно больше, чем у базисной грани графита
и нитрида бора или у граней (100) кристаллов типа NaCl. Потен-
циальные барьеры в этом случае имеют более сложную природу,
связанную с сильным изменением напряженности электростатиче-
ского поля, создаваемого ионами разных знаков. Даже физически
однородную поверхность отдельной грани в таких случаях уже
нельзя принимать за математически однородную, особенно по отно-
шению к адсорбции молекул, способных к сильному специфическому
взаимодействию с электростатическим полем, создаваемым у по-
2 Заказ 1180
17
верхности такой грани [18, 62, 63]. Это также относится к микропори-
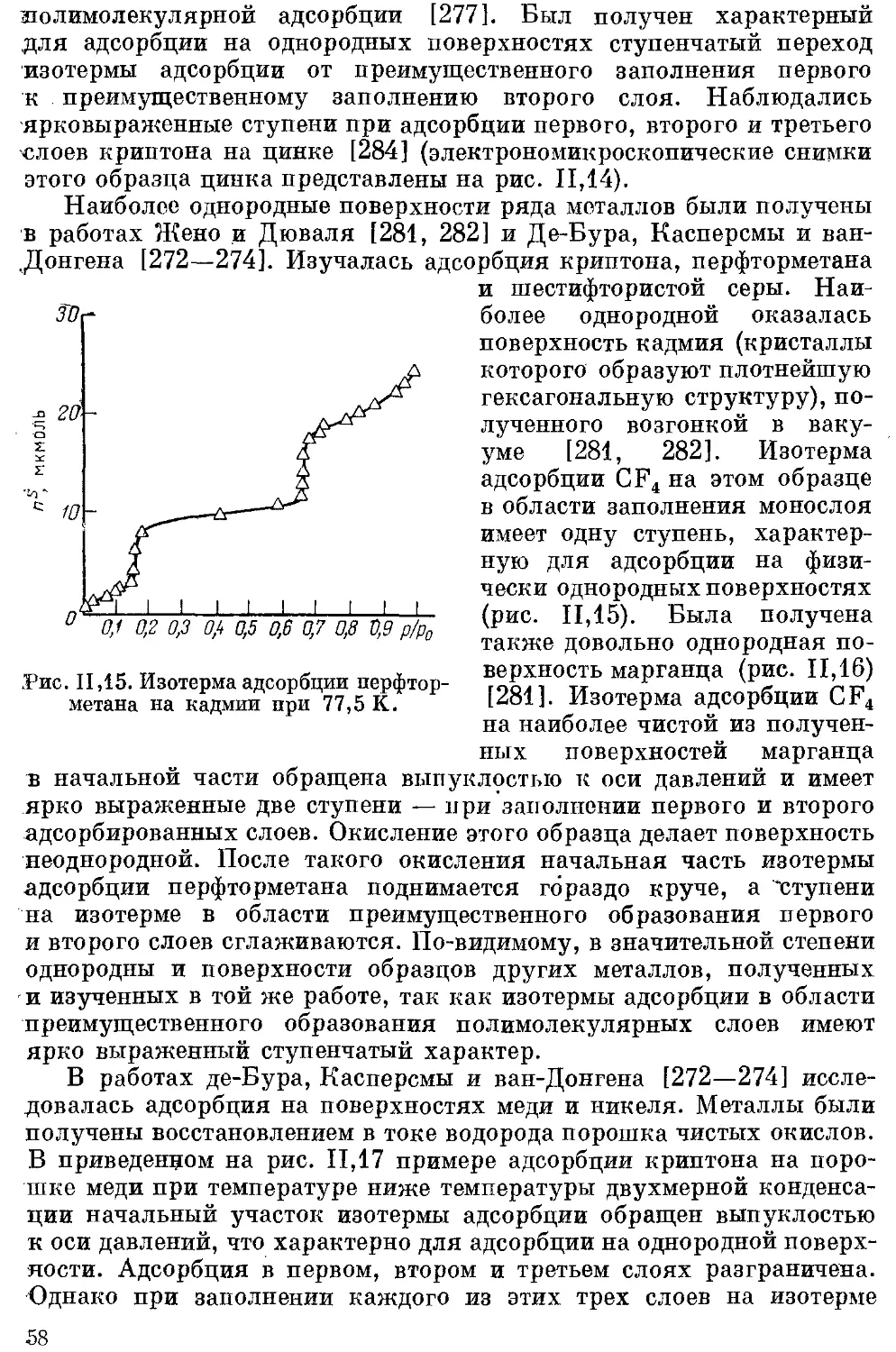
стым ионным кристаллам, например к цеолитам (рис. 1,2).
Каркас цеолитов построен из чередующихся алюминий- и крем-
ний-кислородных тетраэдров. Алюминий-кислородный тетраэдр
вследствие превышения координационного числа 4 над валентностью
алюминия 3 обладает единичным отрицательным зарядом, распре-
деленным по связям А1—О, и является поэтому комплексным ани-
оном большого размера. Отрицательный заряд алюминий-кислород-
ных тетраэдров нейтрализуется катионами обычно небольших раз-
меров. Число катионов значительно меньше числа ионов кислорода,
по которым распределен отрицательный заряд. Структурный элемент
решеток цеолитов типа А и X или Y представляет кубооктаэдр,
Рис. 1,2. Модель цеолитов типа А (а) и X или Y (б).
построенный из 24 алюминий- и кремний-кислородных тетраэдров,
причем ионы кислорода у смежных тетраэдров общие. На кубо-
октаэдр приходится 48 ионов кислорода. Он имеет шесть четырех-
членных и восемь шестичленных кислородных колец.
Различие в структуре алюмосиликатных скелетов цеолитов А
и X или Y заключается в разном пространственном расположении
кубооктаэдров. В цеолите типа А число алюминий- и кремний-кис-
лородных тетраэдров одинаково. Кубооктаэдры в этом цеолите
образуют простую кубическую решетку (рис. 1,2а). Каждый кубо-
октаэдр соединен с соседними шестью кубооктаэдрами через четырех-
членные кислородные кольца, образуя четырехгранные кислородные
призмы. Цеолиты X и Y имеют одинаковую кристаллическую ре-
шетку, но отличаются отношением Si : Al, которое изменяется
приблизительно от 1,1 для цеолита X до 2,5 для цеолита Y. Каждый
кубооктаэдр соединен с соседними четырьмя кубооктаэдрами через
шестичленные кислородные кольца, образуя не четырехгранные,
как у цеолита А, а шестигранные кислородные призмы (рис. 1,26).
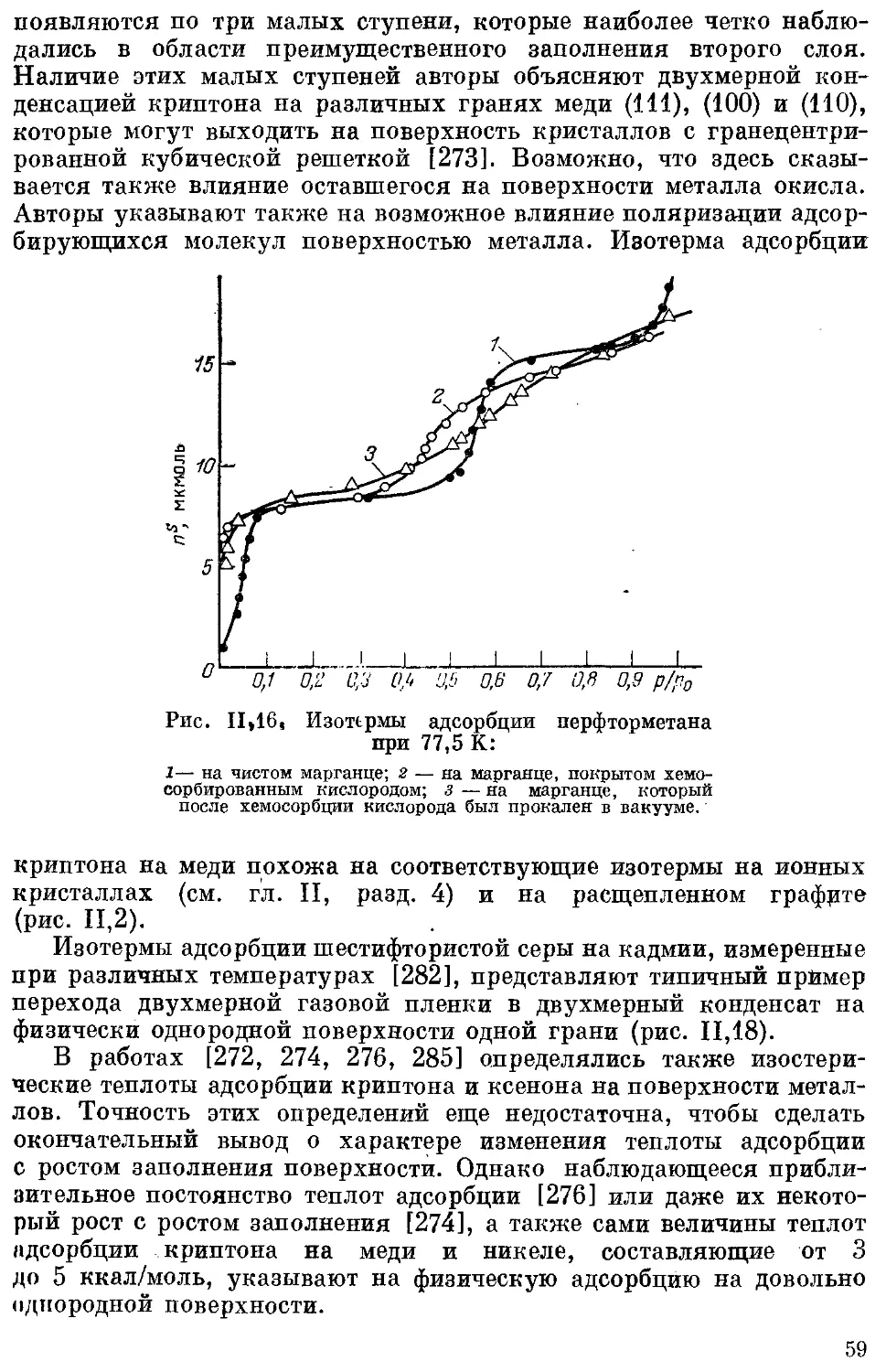
Из рис. 1,3 видно, что в большой полости цеолита NaA можно
выделить сравнительно небольшой повторяющийся сектор. Поэтому
18
•рюдическое изменение потенциала адсорбционных сил при пере-
ШЩгаии молекулы в каналах этого микропористого кристалла будет
Происходить не только от полости к полости, но и на более коротких
Расстояниях — от указанного на рис. 1,3 сектора к соседнему такому
МО сектору. Однако при перемещении молекулы вдоль разных уча-
ОТИОВ внутри такого сектора энергия ее взаимодействия с цеолитом
МОжет сильно изменяться (см. обзор [23]). В целом же микропори-
стый кристалл цеолита образует физически однородную поверхность
нахалов.
В случае микропористых кристаллов надо, однако, принимать
АО внимание не только величину поверхности, образованной ионами
Кислорода и обменными катионами, но также и ее кривизну.
Уис. 1,3. Элементарная ячейка цео-
лита типа А. Показан сектор с за-
штрихованным основанием, повторя-
ЮЩиЙся в большой полости цеоли-
та NaA.
Двойными кружками обозначены 8 мест,
ЙМИитых однозарядными катионами (около
ШШ5ТИ‘1леш1ых кислородных колец кубоок-
ТЛадрон, заштрихованных на схеме
рис. 1,2).
Простмми крутками обозначены 6 мест,
Ин IHH ИРМ Ч г ГП ГНГ |'НЧ1Ч*|(И ршчюложопы
4 ОЛН'НПрНППЫч hit IИОПН (ОКОЛО 1НМ‘ЬМИ~
4Ht<l|ttMN КИ1'.||О|ниП||4Х И11.1РЦ ОКОН МГ-
ищу 1НН1НПТНМИ, urn пмнн км щпптри X опи-
ны нн рис. 1,2).
z
На поверхности молекулярных кристаллов резко различаются
расстояния между соседними атомами, связанными химически
В одной молекуле, и расстояния между соседними атомами разных
Молекул. Однако поверхность отдельной грани таких кристаллов
можно рассматривать как физически однородную благодаря строгой
периодичности изменения потенциальной энергии при перемещении
адсорбированной молекулы вдоль такой поверхности. В этом случае
периодически повторяющиеся участки представляют собой площади,
•айимаемые молекулами на соответствующей грани кристалла. Эти
площади могут быть значительными, а периодичность может сильно
•ввисеть от направления перемещения адсорбированной молекулы
вдоль поверхности грани. Например, в случае кристаллов «-алканов
площади периодически повторяющихся участков часто составляют
десятки А2, причем движение вдоль оси одной вытянутой молекулы
н-алкана связано с периодичностью на расстоянии, определяемом;
длиной химической связи С—С, равной около 1,5 А, а при переходе
от одной молекулы w-алкана к другой периодичность определяется
ван-дер-ваальсовыми размерами групп СН2 соседних параллельно
расположенных молекул «-алканов, т. е. проявляется на рас-
2*
19
стоянии 4,9 А [64]. Поэтому приближение математически однородной
поверхности в этом случае или вообще нельзя использовать, или
можно использовать лишь при адсорбции молекул очень больших
размеров.
Поверхность некоторых специально приготовленных аморфных
адсорбентов, например пенористьтх, состоящих из глобул, и одно-
родпомакропористых глобулярной структуры, может быть довольно
однородной геометрически и химически. Так, химически чистый
аэросил, получаемый сжиганием SiCi4 в водородном пламени, с удель-
ной поверхностью ниже 100—200 м2/г состоит из отдельных шаро-
видных непористых частиц, причем у специально приготовленных
образцов размеры этих частиц очень близки, т. е. имеется узкое
распределение этих частиц по размерам [65]. Приготовленный из
такого аэросила однородномакропористый аэросилогель [66] (сило-
хром [67]) сохраняет довольно правильную глобулярную струк-
туру [68], в которой поры представляют повторяющиеся зазоры
между глобулами и также имеют узкое распределение по размерам.
Поэтому такие аэросилы и силохромы геометрически довольно
однородны, они не содержат в заметных количествах ультрапор [52]
п для них может быть использована глобулярная (в общем случае
корпускулярная) модель, соответствующая геометрически довольно
однородной поверхности [52, 68—80].
В предельно гидроксилированном состоянии поверхность этих
адсорбентов также и химически однородна, так как она не содержит
примесей, образующих сильно специфически адсорбирующие акцеп-
торные центры [52, 55, 81, 82]. В этом состоянии каждый поверх-
ностный атом кремния непористого или достаточно широкопористого
кремнезема удерживает в среднем одну гидроксильную группу [52,
83—85]. Эти гидроксильные группы определяют обратимую адсорб-
цию молекул органических оснований с образованием водородных
связей [86], а также обратимую адсорбцию воды (см. обзор [54],
а также [52,87—89]). Однако физически такая поверхность неодно-
родна, так как конденсированные кремневые кислоты образуют
полисилоксановые цепи и кольца разных размеров, по-разному
выходящие на поверхность, часто с разными зазорами между ними,
и поэтому нет строгой периодичности в расположении и ориентации
поверхностных гидроксильных групп. Тем не менее во многом адсорб-
ционные свойства таких химически чистых поверхностей сходны
с адсорбционными свойствами физически однородных поверх-
ностей.
Дегидроксилирование аэросилов и аэросилогелей вызывает воз-
никновение на их поверхности силоксановых групп, так что эта
поверхность, сохраняя геометрическую однородность и приблизи-
тельно ту же величину, становится химически неоднородной. Эта
неоднородность резко влияет на адсорбцию молекул, способных
к образованию водородных связей с силанольными группами. Однако
она мало влияет на адсорбцию не способных к образованию водород-
ной связи молекул [9, 11, 18, 52, 55].
20
Модифицирование неоднородной поверхности адсорбента нанесе-
ием небольшого количества органического вещества, адсорбиру-
ющегося в первую очередь на наиболее неоднородных местах поверх-
ности, или плотного монослоя, покрывающего всю поверхности
адсорбента-носителя, снижает потенциал адсорбционных сил осо-
бенно на наиболее неоднородных участках поверхности адсорбента-
носителя и делает поэтому модифицированную таким образом поверх-
ность более однородной. И хотя модифицирующие слои часто физи-
чески неоднородны, они могут вести себя как практически
однородные по отношению к адсорбции достаточно крупных моле-
кул [18, 43]. Модифицирование поверхности твердого тела плотными
монослоями молекул или макромолекул, содержащих соответству-
ющие функциональные группы, приводит к увеличению адсорбции
молекул, способных к специфическому межмолекулярному взаимо-
действию с этими группами [18, 36, 43].
& Характер межмолекулярных взаимодействий при адсорбции
и экспериментальные термодинамические критерии
однородности поверхности
Ив приведенного выше следует, что по отношению к адсорбции
станопь неоднородности поверхности надо рассматривать в зависи-
мости от того, какая молекула адсорбируется, в каком интервале
ННППЛН1ЧП1Й 1ннюрхпости и при какой температуре. В связи с этим
при ипучании инн рога о степени однородности поверхности твердых
ion ннжпоп вначпиио имеет характер аз а и мо действия адсорбат —
адсорбент, определяемый структурой как поверхности, так и моле-
кулы. Взаимодействие это может быть молекулярным (молекулярная
пли физическая адсорбция), когда адсорбированная молекула на
юрист своей химической индивидуальности, и химическим (хемо-
сорбция), когда между молекулой и поверхностью возникает хими-
ческни связь, в результате которой индивидуальность молекулы
1 l*p НОТСН.
Природа межмолекулярных взаимодействий едина и определяется
алектроппой структурой молекул. Однако для систематизации экс-
периментального материала межмолекулярное взаимодействие при
адсорбции, как и в других случаях проявления межмолекулярных
взаимодействий, удобно подразделить на неспецифическое и специ-
фическое. В зависимости от способности к этим видам межмолекуляр-
ных взаимодействий молекулы в свою очередь удобно подразделить,
на 4 группы: А, В, С й D [7, 9, 11]. К группе А относятся молекулы,
но способные к специфическому межмолекулярному взаимодей-
ствию. Это молекулы со сферически симметричными электронными
оболочками (атомы благородных газов) или о-связями (молекулы
насыщенных углеводородов). К группам В, С и D относятся моле-
кулы, способные к специфическому межмолекулярному взаимодей-
ствию. У молекул группы В электронная плотность локально сосре-
доточена на периферии отдельных связей или звеньев, например
21
л-связей или звеньев с неподеленными электронными парами в эфир-
ных, карбонильных, нитрильных, третичных аминных и т. п. функ-
циональных группах. Молекулы группы С имеют локально сосре-
доточенные на периферии положительные заряды, как в некоторых
металлоорганических соединениях. Молекулы группы D содержат
функциональные группы типа ОН, Nil и NHa. На соседних атомах О
и Н или N и Н этих групп сосредоточены как электронная плотность
(на атомах О или N), так и положительный заряд (на частично про-
тонизированных атомах Н тех же функциональных групп) [7, 9,
11, 36]. Молекулы группы В не образуют взаимных водородных
связей, но могут образовывать эти связи с молекулами группы D.
Молекулы же группы D образуют водородные связи как с молеку-
лами группы В, так и друг с другом.
Адсорбенты по той же классификации, т. е. в зависимости от
химического строения их поверхности, определяющего способность
к тому или иному виду межмолекулярных взаимодействий, делятся
на три типа. К первому типу относятся неспецйфические адсорбенты,
не несущие на своей поверхности нй ионов, ни каких-либо функци-
ональных групп, связей или центров с локально сосредоточенными
на периферии зарядами и не обладающие электронодонорными или
электроноакцепторными центрами. На таких адсорбентах любые
молекулы адсорбируются неспецифически. К адсорбентам этого
типа можно отнести графитированные сажи, в особенности графити-
рованную около 3000 °C термическую сажу, поверхность которой
состоит в основном из базисных граней графита. Кроме графитиро-
ванной сажи к неспецифическим адсорбентам относится чистый
нитрид бора, молекулярные кристаллы благородных газов и насы-
щенных углеводородов, а также пленки из таких углеводородов
и пористые углеводородные полимеры. Адсорбция на таких адсор-
бентах мало зависит от локального распределения в адсорбируемых
молекулах электронной плотности, в частности, от наличия л-связей
и неподеленных электронных пар. Различие в валентных состояниях
атомов углерода в таких адсорбентах, как, например, графит, с одной
стороны, и насыщенные углеводороды — с другой, сказывается на
адсорбции незначительно, хотя и может быть выявлено в некото-
рых системах (подробнее см. разд. 1 гл. II и рис. 11,12) [90, 91].
Адсорбенты второго типа — это специфические адсорбенты, на
поверхности которых сосредоточены положительные заряды, на-
пример гидроксильные группы с протонизированным в различной
степени водородом или катионы. Молекулы групп В и D адсорби-
руются на таких адсорбентах специфически, причем на гидрокси-
лированных поверхностях — с образованием водородных связей
с гидроксильными группами поверхности. При адсорбции на таких
адсорбентах всех молекул и особенно молекул групп В hD, имеющих
л-связи, квадрупольные или дипольные моменты, в энергию адсорб-
ции вносит вклад энергия электростатического взаимодействия
молекул адсорбата с электростатическим полем, создаваемым по-
верхностными полярными группами или поверхностными катионами.
22
|РТ0Т вклад относительно невелик, если поверхность ионных кри-
Ймллов образована гранями с одинаковой концентрацией череду-
ющихся положительных и отрицательных ионов равного заряда
Й близких размеров как в случае граней (100) кристаллов NaCl
ШЛИ MgO. Но этот вклад значителен для поверхностей адсорбентов,
Образованных катионами небольших размеров, особенно много-
Ирядными, и большими комплексными анионами. На поверхности
таких адсорбентов локально сосредоточены положительные заряды,
В то время как отрицательные заряды распределены по внутренним
Связям комплексных анионов.
Свойства адсорбентов второго типа проявляют, например, не-
которые грани непористых кристаллов сульфатов щелочноземельных
Металлов [18, 63], а также цеолиты. Выше было отмечено, что отри-
цательные заряды остова пористых кристаллов цеолитов (рис. 1,2)
распределены по многочисленным ионам кислорода. Положительный
Ж® заряды сосредоточены в обменных катионах. Электростатической
иоле в каналах цеолитов зависит от концентрации тетраэдров [А1О4]-1
В остове цеолита, от заряда и концентрации обменных катионов,
Причем главным образом тех катионов, которые расположены у по-
верхности каналов. Поэтому локальную (внутри повторяющихся
Секторов в полостях цеолита, см. рис. 1,3) неоднородность электро-
статического поля вблизи поверхности каналов цеолита можно
снизить, умешивая концентрацию алюминий-кислородных тетра-
iMipoii (шшримор, в случае цеолитов с решеткой фожазита, переходя
oi цполИтон типа X к цеолитам типа Y, имеющим большее отношении
MI/AI, чем у цеолитов типа X). Кроме того, можно уменьшить кон-
центрацию обменных катионов частичным дек абонированием, проис-
ходящим лри замене, например, катиона Na+ на NH4+ или Н3О+
(при длительной промывке водой) и последующем нагревании для
удаления NHS или Н2О. Это вызывает, однако, появление на по-
верхности различных кислотных центров [54]. По отношению к не-
мнщифичоски адсорбирующимся молекулам группы А декатиониро-
мннныо цеолиты ведут себя как адсорбенты со значительно более-
однородной поверхностью, поскольку энергия адсорбции этих моле-
кул на возникающих при декатионировании кислотных центрах
При невысоких температурах близка к энергии адсорбции на кисло-
родных центрах остова цеолита [23, 92—96].
Адсорбция цеолитами А и X происходит на наружных поверх-
ностях кристаллов и в больших полостях. Однако в эти полости
Проникают только такие молекулы, ван-дер-ваальсовы размеры
которых это допускают. Таким образом, цеолиты в зависимости от
химического строения и размера адсорбируемых молекул ведут себя,
как специфические адсорбенты П-го типа или как молекулярные-
сита (см. разд. 4 гл. II).
К адсорбентам третьего типа относятся специфические адсор-
бенты, на поверхности которых сосредоточены отрицательные за-
ряды. Такие адсорбенты можно получить, например, прямым син-
тезом пленок или пористых полимеров с поверхностными группами
23
-типа CN, СО и т. п. Такие адсорбенты можно также получить нане-
сением на поверхность адсорбента-носителя различных органи-
ческих оснований, молекулы которых относятся к группе В.
При этом в зависимости от содержания тех или иных связей
или групп в молекулах наносимых слоев на поверхности адсорбента
появляются непредельные или ароматические соединения,несущие
л-связи, или атомы О или N с неподеленными электронными парами
эфирных, карбонильных и карбоксильных групп, групп тре-
тичных аминов, групп—CN и т. п. К адсорбентам третьего типа
могут быть также отнесены базисные грани галогенидов двухвалент-
ных металлов, например NiCl2, СоС12 (см. рис. 1,1в), состоящие
из отрицательно заряженных ионов [27, 58—61]. Однако при оценке
специфичности всей поверхности таких адсорбентов надо принимать
во внимание структуру и протяженность остальных граней
кристалла.
При приготовлении твердых адсорбентов полностью устранить
геометрическую и химическую неоднородность поверхности не
удается. Однако при синтезе и обработке адсорбента эту неоднород-
ность можно сильно снизить и в благоприятных случаях сделать
настолько незначительной, что ее влиянием на адсорбцию при не
очень низких температурах можно пренебречь, особенно по отно-
шению к молекулам, неспособным к специфическим взаимодействиям
(т. е. к молекулам группы А). В настоящее время многие адсорбенты
всех трех типов могут быть получены и обработаны в таких усло-
виях, которые гарантируют достаточно низкую неоднородность
поверхности (в случае кристаллов — поверхности их граней). Это
позволяет успешно использовать такие адсорбенты для практических
целей разделения смесей в газовой и жидкостной адсорбционной
хроматографии и для получения термодинамических характеристик
адсорбции индивидуальных веществ в виде воспроизводимых
констант.
Для экспериментальной оценки степени однородности поверх-
ности адсорбентов применяются разные методы: термодинамические
{газохроматографический и вакуумный адсорбционный — методы
определения формы хроматографического пика и изотермы адсорб-
ции), калориметрический (определение зависимости теплоты адсорб-
ции от заполнения поверхности адсорбированными молекулами),
различные электронно-микроскопические методы (в частности,
метод декорирования), дифракция медленных электронов, спектро-
скопические методы, химические реакции с поверхностными соеди-
нениями, в частности, изотопный обмен [54, 97]. В соответствии
с содержанием этой книги ниже рассмотрены некоторые термодина-
мические методы такой оценки.
Используя чувствительные детекторы, например ионизационные,
в газохроматографических опытах можно применять весьма малые
пробы. Получение при таких весьма малых пробах симметричных
пиков для молекул, как неспособных, так и способных к специфи-
24
вескому взаимодействию (молекул групп А, В и /)), свидетель-
ствует об отсутствии сильно адсорбирующих примесных центров
И о практическом достижении предельного закона Генри для изо-
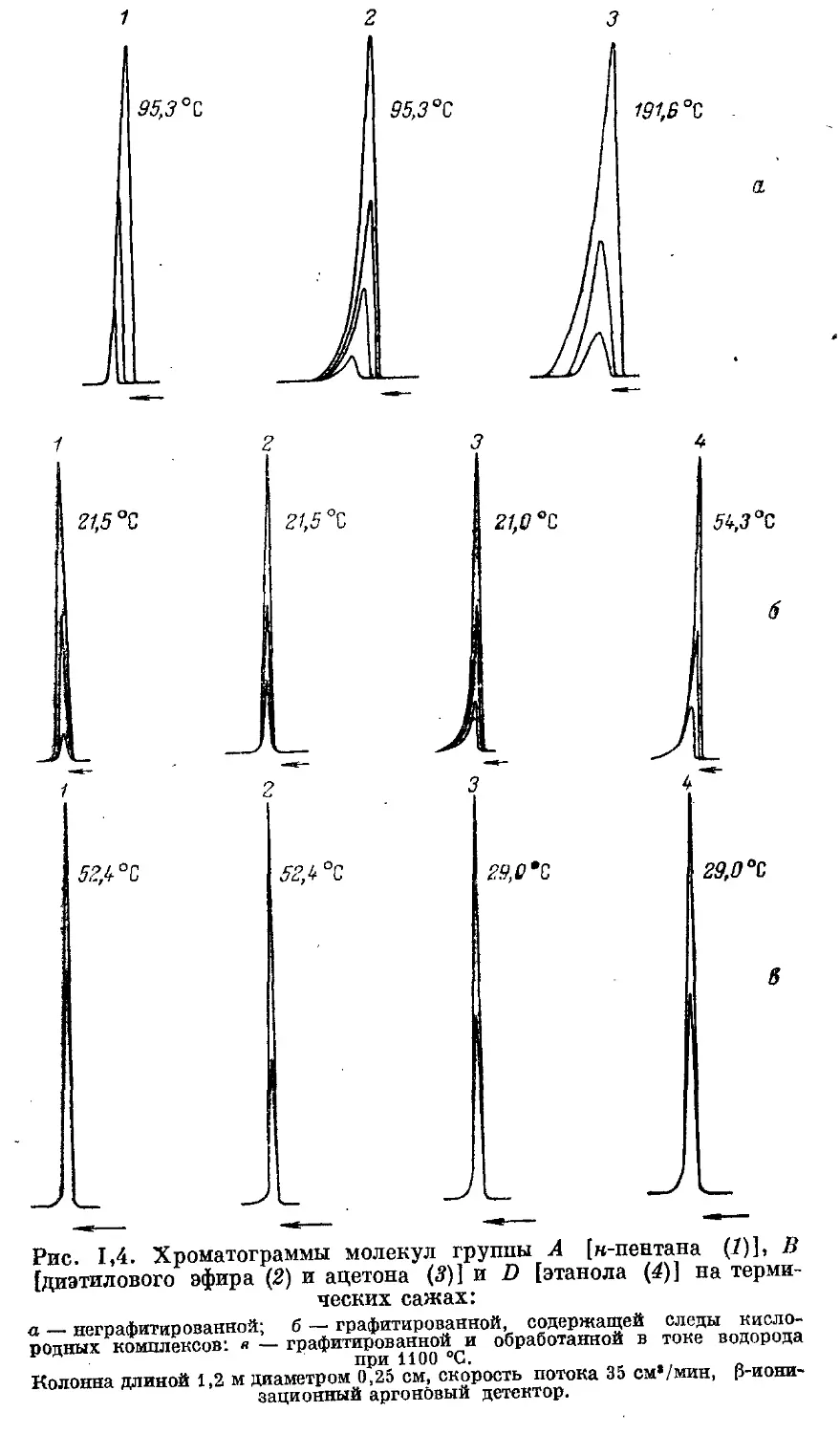
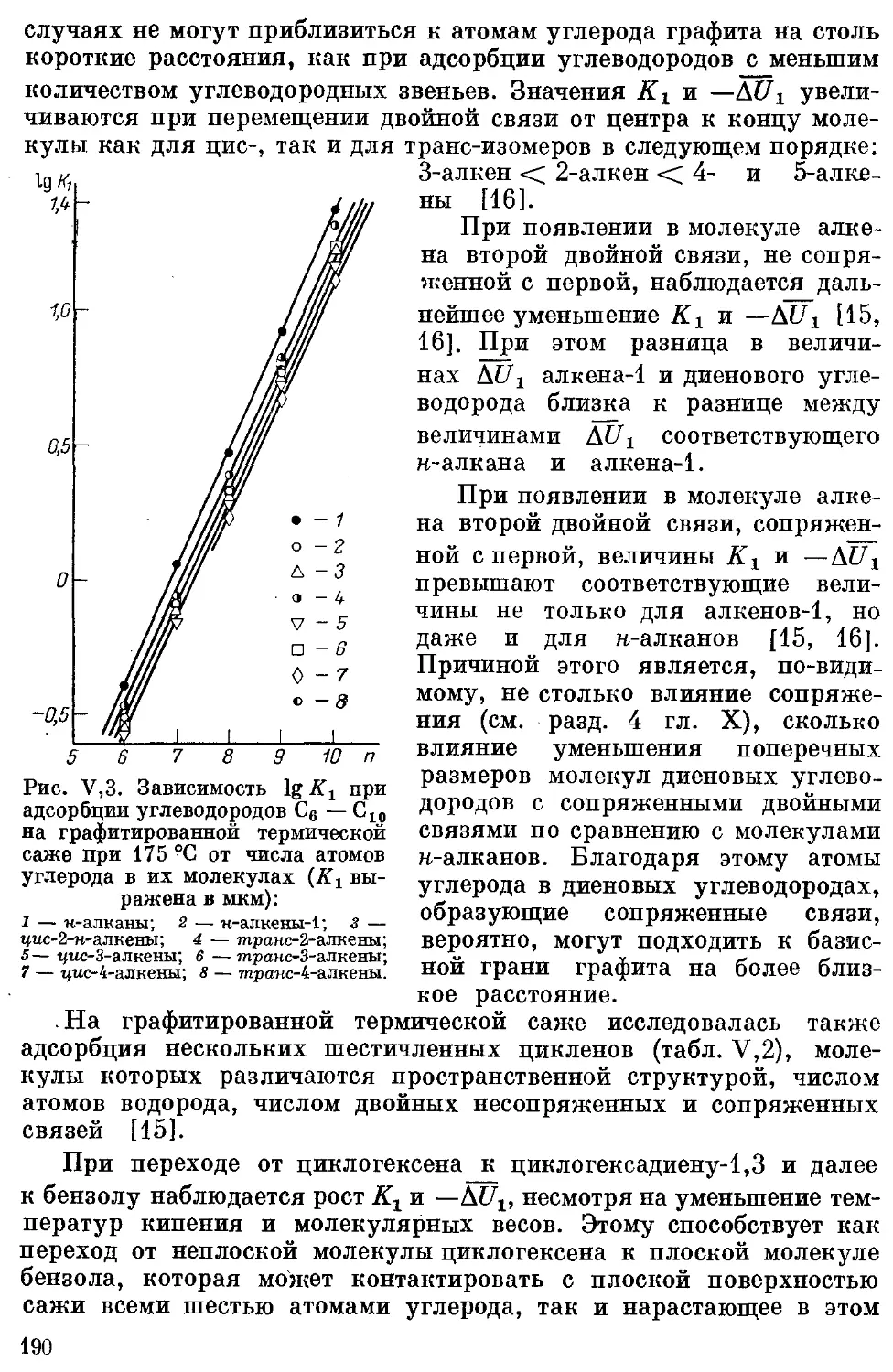
терм адсорбции всех этих адсорбатов [11]. На рис. 1,4 изображено
изменение формы хроматографических пиков для малых проб раз-
личных веществ при переходе от довольно неоднородной поверхности
неграфитированной термической сажи (рис. 1,4а) к весьма однород-
ной поверхности той же сажи после графитирования (рис. 1,46).
В случае графитированной термической сажи пики для углеводорода
н-пентана (молекулы группы Л) и диэтилового эфира (молекулы
группы В) становятся симметричными уже при обычной температуре.
В то же время хроматографические пики для молекул группы В
с ответвленной функциональной группой (ацетон) или молекул
группы D с концевой функциональной группой (спирт) остаются
несколько несимметричными. Это вызывается присутствием в обычной
графитированной саже следов кислородных комплексов, образу-
ющихся при соприкосновении этих саж с воздухом у дефектов базис-
ных граней графита или на стыках его кристаллов. Эти кислородные
комплексы можно удалить с поверхности графитированной сажи
обработкой водородом при 1100 °C [46—48]. Из рис. 1,4в видно,
что после такой обработки симметричные пики получены и для аце-
тона и спирта.
Однако сама по себе симметричность пиков при малых пробах,
представляя необходимое условие для того, чтобы считать поверх-
ность однородной, еще не является достаточным условием. Это вы-
текает из того, что при малых пробах адсорбата и высоких темпера-
турах колонны, особенно если этот адсорбат на данном адсорбенте
адсорбируется не очень сильно, можно практически приблизиться
к уравнению Генри для изотермы адсорбции, а следовательно, полу-
чить симметричные пики и в случае неоднородной поверхности.
Близкая к линейной изотерма адсорбции в области малых заполнений
может получиться и благодаря компенсации влияния неоднород-
ности поверхности противоположным влиянием взаимного притяже-
ния адсорбированных молекул. Однако сохранение симметричности
пиков и постоянства времени удерживания адсорбата в хроматогра-
фической колонне при изменении малых величин пробы для молекул,
относящихся к разным группам Л, В и D (т. е. близость начальной
части изотерм адсорбции всех адсорбатов к линейным), уже в зна-
чительной степени гарантирует высокую однородность всей изуча-
емой поверхности или составляющих ее основных частей (в случае
кристаллов — основных выходящих на поверхность граней).
Статические (вакуумные) и калориметрические методы измерения,
позволяющие выявлять форму изотермы адсорбции и вид зависимости
теплоты адсорбции от заполнения, также дают возможность судить
об однородности поверхности. При неспецифической адсорбции на
близкой к однородной поверхности, состоящей из кристаллической
грани одного индекса, получаются изотермы адсорбции, вначале
обращенные выпуклостью к оси давления в газовой фазе а затем
25
1
2
3
Рис. 1,4. Хроматограммы молекул группы А [w-пентана (7)], В
[диэтилового эфира (2) и ацетона (5)] и D [этанола (4)] на терми-
ческих сажах:
а — неграфитированной; б — графитированной, содержащей следы кисло-
родных комплексов: <? — графитированной и обработанной в токе водорода
при 1100 °C.
Колонна длиной 1,2 м диаметром 0,25 см, скорость потока 35 см*/мин, p-иони-
зационный аргоновый детектор.
Проходящие точку перегиба (рис. 1,5а). При достаточно низкой
Температуре изотермы адсорбции на такой поверхности с ростом
Наполнения образуют обычно одну ступень, т. е. практически вер-
тикальный подъем при постоянном давлении в газовой фазе р, вы-
виваемый сильной ассоциацией адсорбированных молекул друг
С другом в адсорбционном слое (например, при адсорбции спиртов
1.5. Изотермы адсорбции (а, б) и зависимости дифференциальной те-
H/HWW адсорбции qv от величины адсорбции (в, з) четыреххлористого углерода
ГМфктярованной термической сажей выше (а, в) и ниже (б, г) критической
температуры мономолекулярного слоя адсорбированного вещества (Г — вели-
чина адсорбции, р — равновесное давление в газовой фазе, 0 — степень запол-
нения поверхности).
Ий графитированной термической саже) или двухмерным фазовым пе-
реходом газ—конденсат в мономолекулярном слое. После такой
ступени эти изотермы обращаются выпуклостью к оси заполнения
Поверхности (рис. 1,56) [98, 99]. Благодаря проявлению притяжения
адсорбат — адсорбат теплота неспецифической адсорбции на одно-
родной поверхности, состоящей из одной кристаллографической
грани, растет в области заполнения поверхности от нулевого запол-
нения до 80—90% от емкости плотного мономолекулярного слоя
(рис. 1,5а). Дальнейшее падение теплоты адсорбции с ростом запол-
нения поверхности вызывается переходом к преимущественному
Заполнению второго и последующего слоев [98—100]. Из рис. 1,5г
видно, что теплота адсорбции с ростом заполнения в области двух-
мерного фазового перехода газ — конденсат быстро растет, а затем
остается постоянной до завершения этого фазового перехода.
27
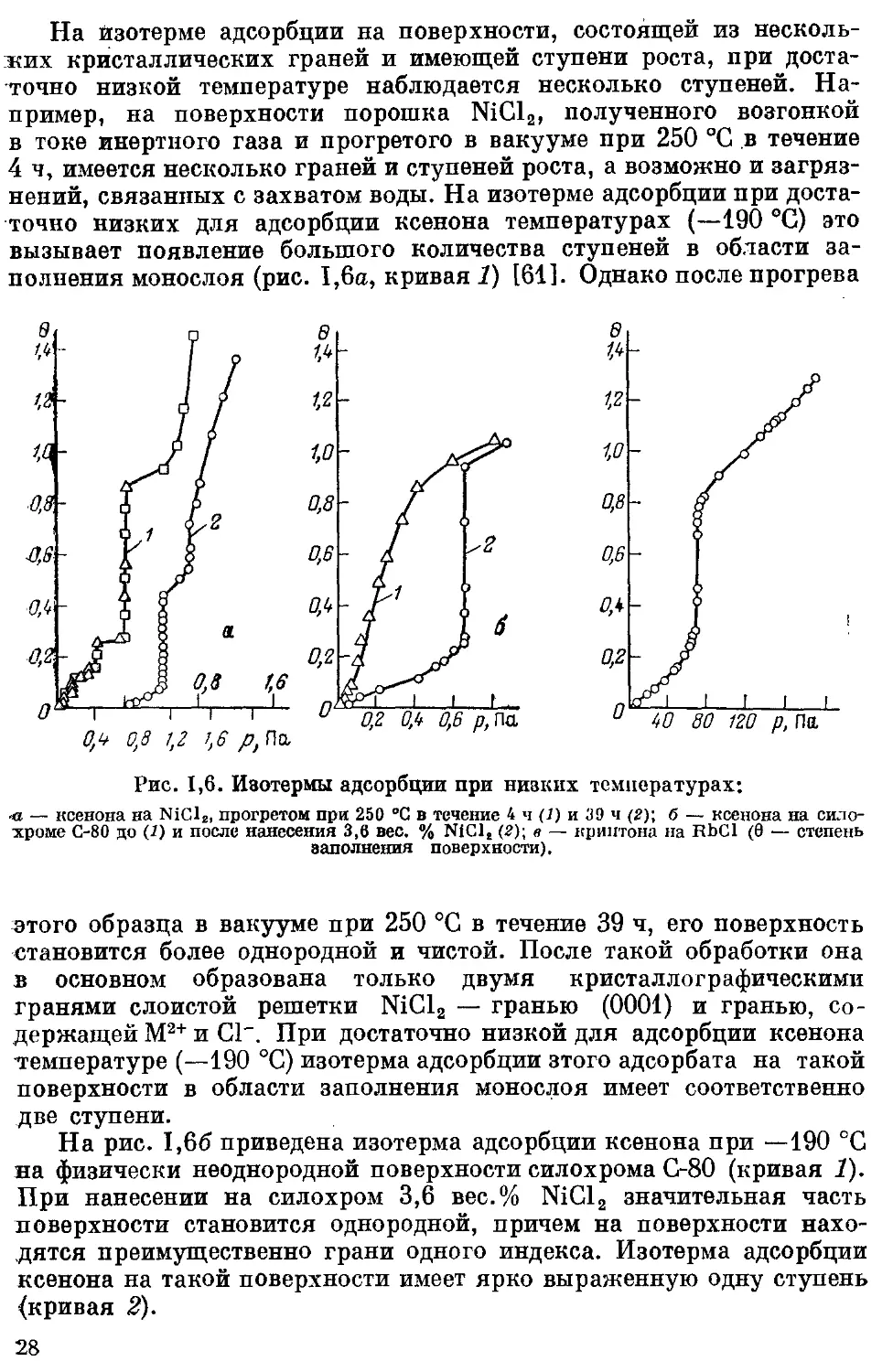
На изотерме адсорбции на поверхности, состоящей из несколь-
ких кристаллических граней и имеющей ступени роста, при доста-
точно низкой температуре наблюдается несколько ступеней. На-
пример, на поверхности порошка NiCl2, полученного возгонкой
в токе инертного газа и прогретого в вакууме при 250 °C в течение
4 ч, имеется несколько граней и ступеней роста, а возможно и загряз-
нений, связанных с захватом воды. На изотерме адсорбции при доста-
точно низких для адсорбции ксенона температурах (—190 °C) это
вызывает появление большого количества ступеней в области за-
полнения монослоя (рис. 1,6а, кривая 1) [61]. Однако после прогрева
Рис. 1,6. Изотермы адсорбции при низких температурах;
— ксенона на NiCl2, прогретом при 250 °C в течение 4 ч (]) и 39 ч (2)\ б — ксенона на сило-
хроме С-80 до (2) и после нанесения 3,6 вес. % NiCl8 (2); в — криптона на RbCl (0 — степень
заполнения поверхности).
итого образца в вакууме при 250 °C в течение 39 ч, его поверхность
становится более однородной и чистой. После такой обработки она
в основном образована только двумя кристаллографическими
гранями слоистой решетки NiCl2 — гранью (0001) и гранью, со-
держащей М2+ и С1". При достаточно низкой для адсорбции ксенона
температуре (—190 °C) изотерма адсорбции зтого адсорбата на такой
поверхности в области заполнения монослоя имеет соответственно
две ступени.
На рис. 1,66 приведена изотерма адсорбции ксенона при —190 °C
на физически неоднородной поверхности силохрома С-80 (кривая 1).
При нанесении на силохром 3,6 вес. % NiCl2 значительная часть
поверхности становится однородной, причем на поверхности нахо-
дятся преимущественно грани одного индекса. Изотерма адсорбции
ксенона на такой поверхности имеет ярко выраженную одну ступень
(кривая 2).
28
Из рис. I,6e видно, что в случае кубических кристаллов RbCl,
однородная поверхность которых образована гранями одного ин-
декса (100), изотерма адсорбции пара криптона при —195 °C в об-
ласти преимущественного заполнения первого слоя имеет только
Одну ступень (как и в случае графитированной термической сажи,
СМ. рис. 1,56) [101].
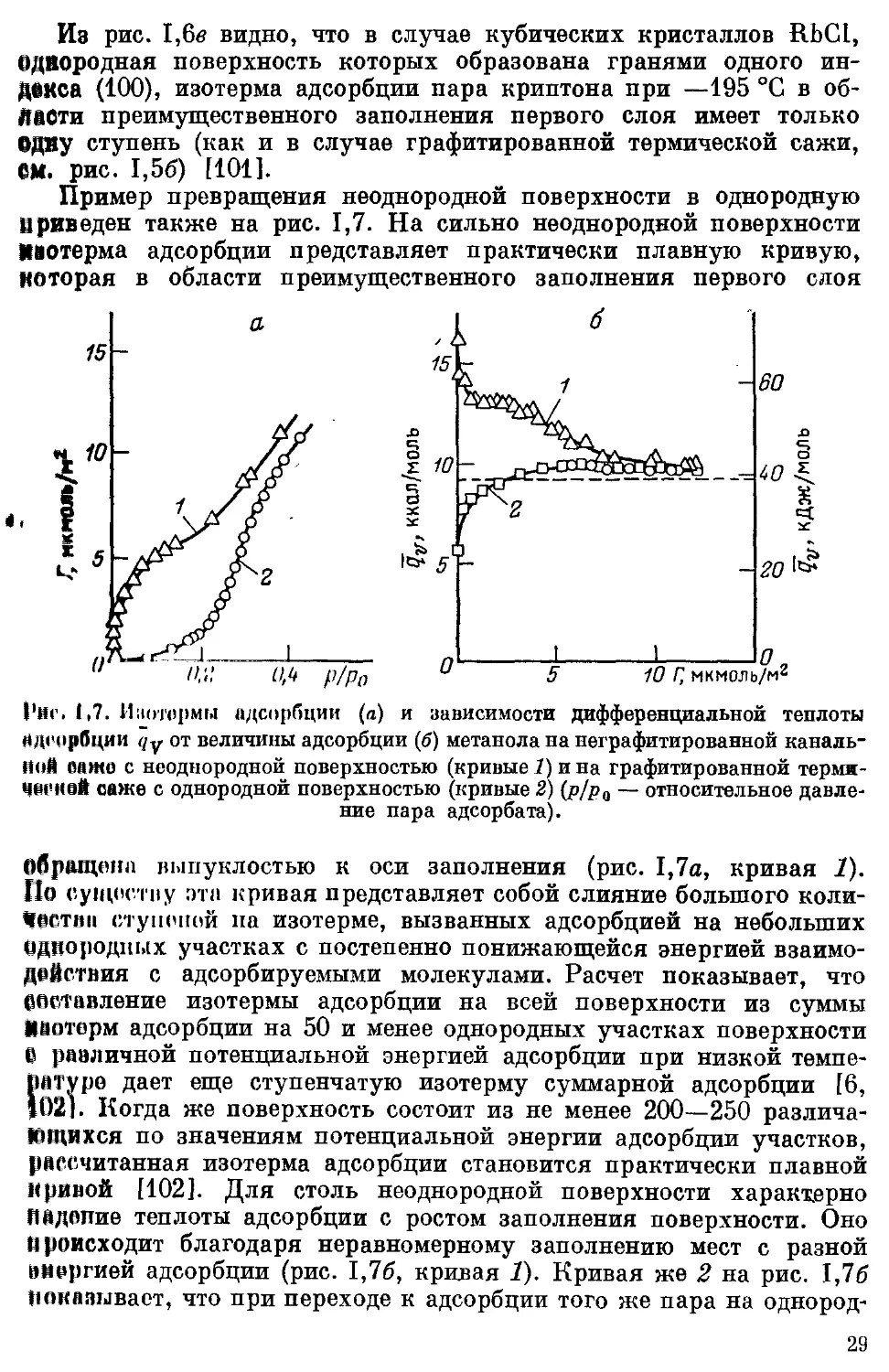
Пример превращения неоднородной поверхности в однородную
приведен также на рис. 1,7. На сильно неоднородной поверхности
Иаотерма адсорбции представляет практически плавную кривую,
которая в области преимущественного заполнения первого слоя
Риг. 1,7. Изотермы адсорбции (а) и зависимости дифференциальной теплоты
адсорбции г/у от величины адсорбции (б) метанола на неграфитированной каналь-
ной 6йжо с неоднородной поверхностью (кривые 1) и на графитированной термя-
саже с однородной поверхностью (кривые 2) (p/pQ — относительное давле-
ние пара адсорбата).
Обращена выпуклостью к оси заполнения (рис. 1,7а, кривая 2).
По существу эта кривая представляет собой слияние большого коли-
чества ступеней па изотерме, вызванных адсорбцией на небольших
Однородных участках с постепенно понижающейся энергией взаимо-
действия с адсорбируемыми молекулами. Расчет показывает, что
Составление изотермы адсорбции на всей поверхности из суммы
Цаотерм адсорбции на 50 и менее однородных участках поверхности
в различной потенциальной энергией адсорбции при низкой темпе-
ратуре дает еще ступенчатую изотерму суммарной адсорбции [6,
102]. Когда же поверхность состоит из не менее 200—250 различа-
ющихся по значениям потенциальной энергии адсорбции участков,
рассчитанная изотерма адсорбции становится практически плавной
кривой [102]. Для столь неоднородной поверхности характерно
Падение теплоты адсорбции с ростом заполнения поверхности. Оно
происходит благодаря неравномерному заполнению мест с разной
аморгией адсорбции (рис. 1,76, кривая 1). Кривая же 2 на рис. 1,76
показывает, что при переходе к адсорбции того же пара на однород-
29
нои поверхности при увеличении заполнения происходит рост те-
плоты адсорбции вследствие преимущественного проявления меж-
молекулярного притяжения адсорбат — адсорбат (см. также
рис. 1,5в).
Следует, однако, отметить, что уменьшение теплоты адсорбции
с ростом заполнения может происходить и на физически довольно
однородной поверхности. Так, в случае сильно декатионированного
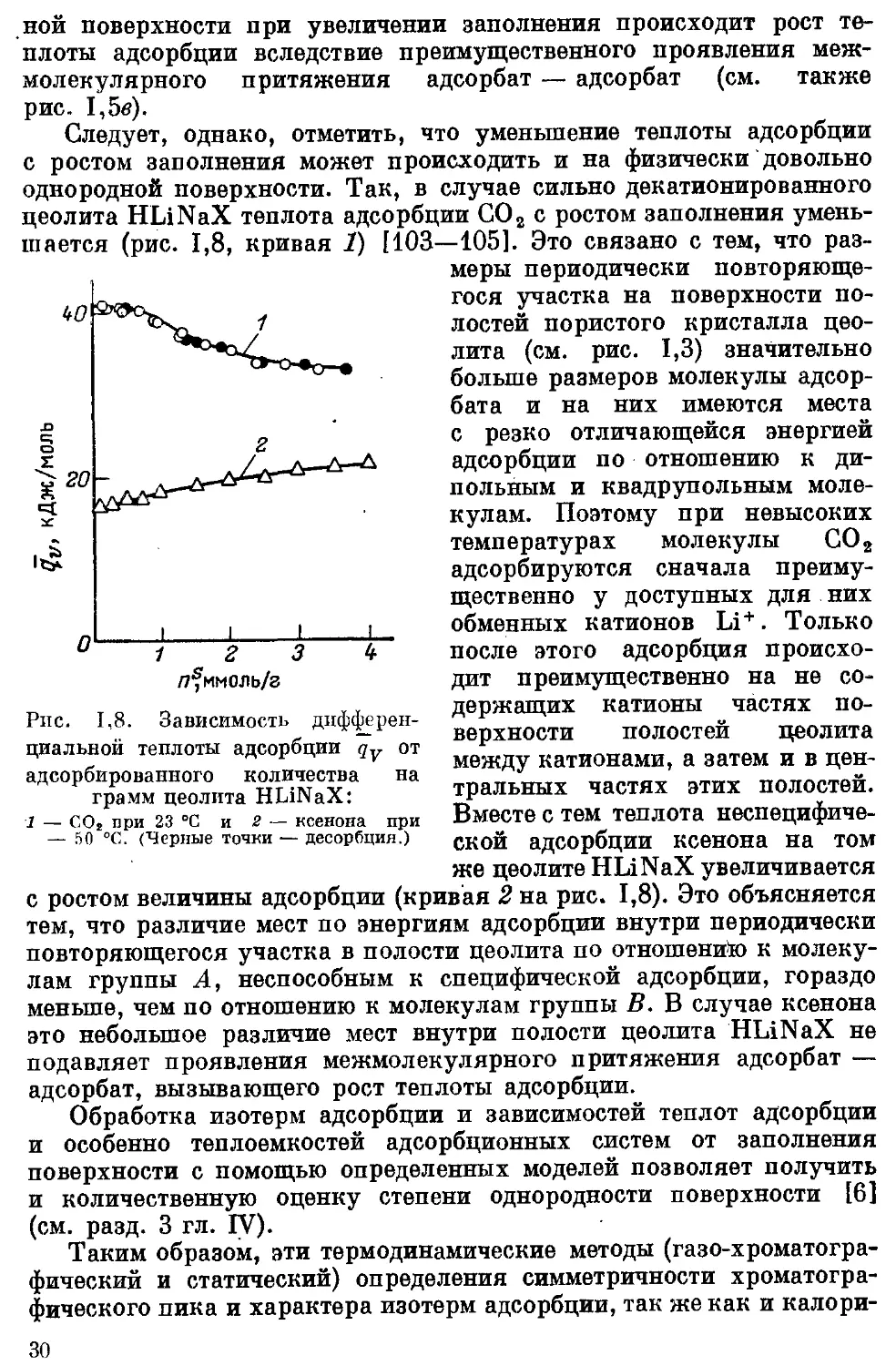
цеолита HLiNaX теплота адсорбции СО2 с ростом заполнения умень-
шается (рис. 1,8, кривая 1) [103—105]. Это связано с тем, что раз-
Рис. 1,8. Зависимость дифферен-
циальной теплоты адсорбции qv от
адсорбированного количества на
грамм цеолита HLiNaX:
1 — СО2 при 23 °C и 2 — ксенона при
— 50 °C. (Черные точки — десорбция.)
меры периодически повторяюще-
гося участка на поверхности по-
лостей пористого кристалла цео-
лита (см. рис. 1,3) значительно
больше размеров молекулы адсор-
бата и на них имеются места
с резко отличающейся энергией
адсорбции по отношению к ди-
польным и квадрупольным моле-
кулам. Поэтому при невысоких
температурах молекулы СО2
адсорбируются сначала преиму-
щественно у доступных для них
обменных катионов Li+. Только
после этого адсорбция происхо-
дит преимущественно на не со-
держащих катионы частях по-
верхности полостей цеолита
между катионами, а затем и в цен-
тральных частях этих полостей.
Вместе с тем теплота неспецифиче-
ской адсорбции ксенона на том
же цеолите HLiNaX увеличивается
с ростом величины адсорбции (кривая 2 на рис. 1,8). Это объясняется
тем, что различие мест по энергиям адсорбции внутри периодически
повторяющегося участка в полости цеолита по отношений) к молеку-
лам группы А, неспособным к специфической адсорбции, гораздо
меньше, чем по отношению к молекулам группы В. В случае ксенона
это небольшое различие мест внутри полости цеолита HLiNaX не
подавляет проявления межмолекулярного притяжения адсорбат —
адсорбат, вызывающего рост теплоты адсорбции.
Обработка изотерм адсорбции и зависимостей теплот адсорбции
и особенно теплоемкостей адсорбционных систем от заполнения
поверхности с помощью определенных моделей позволяет получить
и количественную оценку степени однородности поверхности [6]
(см. разд. 3 гл. IV).
Таким образом, эти термодинамические методы (газо-хроматогра-
фический и статический) определения симметричности хроматогра-
фического пика и характера изотерм адсорбции, так же как и калори-
30
Метрические методы измерения зависимости дифференциальной те-
ПЛОты адсорбции и теплоемкости адсорбционных систем от величины
Адсорбции (подробнее см. [106] и разд. 1 гл. III), особенно в их
совокупности, позволяют судить о степени однородности поверх-
ности адсорбентов. Информация об этом получается еще более полной
При привлечении упомянутых выше других методов исследования
Структуры поверхности, состава и свойств поверхностных соеди-
нений.
3. Роль создания и изучения адсорбентов с близкой
И однородной поверхностью в развитии молекулярной теории
адсорбции
Получение и исследование адсорбентов с поверхностями, близ-
кими к однородным, сыграло важную роль в развитии теории ад-
сорбции на поверхности твердых тел. Оно послужило необходимой
экспериментальной основой для разработки этой теории на моле-
кулярном уровне.
В развитии науки об адсорбции можно выделить два основных
этапа. К начальному этапу относится накопление и эмпирическая
обработка экспериментальных данных, полученных на адсорбентах
С Неопределенным химическим составом поверхности и неоднородной
пористостью, таких как активированные угли, получавшиеся из
Природных органических материалов, и многие ксерогели. На этом
Начальном этапе экспериментальные данные обрабатывались с по-
мощью различных эмпирических уравнений изотермы адсорбции
(от уравнения Фрейндлиха [107] до уравнения Дубинина и сотр.
1108—1101). Эмпирическое описание экспериментальных данных
оставляет однако неясным вопрос о физическом смысле констант,
Входящих в эти уравнения [6]. Остаются неясными также и вопросы
О ТОМ, применимы ли эти уравнения только к адсорбции в микропорах
ЯЛВ ж к адсорбции на поверхностях макропористых и непори-
ОТМХ [96, 111, 112] адсорбентов, а также вопросы об интервале за-
полнений, для описания которых эти уравнения оказываются при-
годными или непригодными. Чисто эмпирические уравнения не
отвечают на вопросы, связанные с природой адсорбции. Остается
поясным, почему один адсорбент адсорбирует одно вещество сильнее,
ЧОМ другое, а другой адсорбент, наоборот, адсорбирует это вещество
Слабее, чем другой? Как это связано качественно и количественно
О химией поверхности и структурой остова адсорбента и со строением
молекул адсорбата? Почему в одних случаях изотермы имеют, а в дру-
гих но имеют точки перегиба или разрывы? На такие вопросы может
дать ответ только молекулярная теория адсорбции.
Почти одновременно с зарождением эмпирического подхода к опи-
санию экспериментальных изотерм адсорбции и пересчету одних
ииотерм в другие [ИЗ—115] возникла и теория, основанная на
простейшей молекулярной модели адсорбционной системы: на модели
локализованной адсорбции на однородной поверхности не взаимо-
31
действующих друг с другом молекул (теория Лэнгмюра [116]).
Эта модель развивалась в последующих работах, в которых была
принята во внимание адсорбция во втором и последующих слоях
(вертикальная цепная ассоциация) [30, 115, 117, 118], парные вза-
имодействия адсорбат — адсорбат на соседних [119] и на более
удаленных друг от друга местах локализации [15], а также цепная
ассоциация локализованных молекул адсорбата как перпендику-
лярно поверхности, так и вдоль нее [24, 30, 106, 120].
Параллельно развивалась и другая молекулярная модель ад-
сорбционной системы — модель нелокализованной адсорбции
на однородной поверхности. Ван-дер-ваальсово взаимодействие моле-
кул ДРУГ с другом в прилегающем к поверхности двухмерном моно-
молекулярном слое было учтено в уравнении двухмерного состояния
этого слоя с помощью соответствующих вириальных коэффициен-
тов [10, 14, 16, 121—124] и в виде двухмерного аналога уравнения
состояния Ван-дер-Ваальса [1, 3, 6, 30, 120]. С помощью адсорб-
ционной формулы Гиббса [125] уравнения двухмерного состояния
преобразуются в соответствующие им уравнения изотермы адсорбции
(см. гл. IV). Адсорбция во втором и последующих слоях была учтена
в виде цепной ассоциации перпендикулярно поверхности [1, 30,
120]. Рассмотрена также модель нелокализованной адсорбции при
цепной ассоциации адсорбированных молекул вдоль поверхности
вместе с ван-дер-ваальсовым взаимодействием единичных молекул
и их ассоциатов друг с другом в первом слое и цепной ассоциацией
перпендикулярно поверхности [126].
Уравнения изотермы адсорбции, полученные на основании всех
этих приближенных молекулярных моделей для адсорбции на одно-
родной поверхности, содержат константу равновесия Генри, отра-
жающую взаимодействие адсорбат — адсорбент. Эти уравнения пере-
ходят в предельное уравнение Генри, когда концентрация адсорбата
в газовой фазе и величина адсорбции стремятся к нулю, т. е. они
имеют правильный нижний предел. Вместе с тем, в зависимости от
соотношения констант, связанных с межмолекулярными взаимодей-
ствиями адсорбат адсорбент и адсорбат — адсорбат, зти уравне-
ния объясняют и описывают изотермы адсорбции различной формы.
Сюда относятся изотермы, обращенные вначале выпуклостью как
к оси заполнения поверхности, так и к оси давления или концентра-
ции в газовой фазе и проходящие затем как одну, так и две точки
перегиба. Эти уравнения описывают также изотермы адсорбции
претерпевающие разрыв ниже критической температуры адсорбиро-
ванного вещества. Дифференцированием зтих уравнений по темпе-
ратуре получают соотношения, объясняющие и описывающие также
зависимости теплоты адсорбции и теплоемкости адсорбционной си-
стемы от заполнения поверхности [30, 106, 126].
Во всех указанных выше моделях адсорбционной системы поверх-
ность адсорбента рассматривается однородной. Поэтому сопоставле-
ния с опытом выведенных на основании этих моделей уравнений для
изотермы адсорбции и зависимости теплоты адсорбции и тепло-
32
•Мкости адсорбата от заполнения поверхности оказались возмож-
j йыми только после получения и изучения адсорбентов с достаточно
Однородной поверхностью. В первую очередь сюда относится полу-
^‘Мение графитированной термической сажи, поверхность которой
Аюразована в основном одной кристаллической гранью — базисной
Дранью графита. Сопоставления с опытом уравнений, основанных
Г НА некоторых из указанных моделей, рассмотрены в гл. IV.
От таких упрощенных моделей состояния адсорбированных моле-
кул свободно представление об адсорбированном веществе как о ре-
альном газе, находящемся в потенциальном поле межмолекулярных
Сил, создаваемом адсорбентом [10, 14, 16, 127]. В зтом случае при
; Яеболыпих заполнениях поверхности выражения для изотермы
адсорбции и для зависимостей от заполнения изменения энтропии
Я внутренней энергии адсорбции и теплоемкости адсорбированного
Вещества можно представить в вириальной форме, т. е. в виде сте-
ленного ряда. Константы первых членов зтих вириальных разложе-
ний представляют соответственно константу равновесия Генри,
Изменение внутренней знергии при адсорбции, а также изменение
Теплоемкости адсорбированного вещества при адсорбции при пре-
дельно малом (нулевом) заполнении, т. е. зти константы свя-
аапы только с межмолекулярным взаимодействием адсорбат — ад-
сорбент. Второй и последующие члены этих вириальных разложений
содержат константы, учитывающие парные, тройные и т. д. взаимо-
действия молекул адсорбата друг с другом при одновременном их
1вппмод<'йгтпии с адсорбентом. Молекулярная статистика дает фор-
мулы для вычисления зтих констант в вириальных разложениях
ИК основе соответствующих потенциальных функций межмолекуляр-
ЯОГО взаимодействия адсорбат — адсорбент и адсорбат — адсорбат
(Подробно зто рассмотрено в гл. VI и VII).
Втот расчет наиболее прост для адсорбции на однородной поверх-
ИОбТИ. Поэтому развитие молекулярной теории адсорбции в значи*
Юьной степени было связано с преодолением определенного рубежа
0 инжжримонтальной работе — с созданием адсорбентов с близкой
И однородной поверхностью и в первую очередь графитированных
f термических саж [128—130].
Ив зтом втором зтапе исследований адсорбции, т. е. при изучении
ЙДбОрбции па молекулярном уровне, графитированные сажи, осо-
ItNtto наиболее однородные из них термические графитированные
ипорпыо описанные Смитом и сотр. в 1953 году [130], сыграли
МОЛЬ важную роль, что и активированные угли на. первом этапе
ЯДООрбционпых исследований, т. е. при разработке эмпирических
ЯНпдоп описания экспериментальных данных, получаемых на адсор-
Йит с неопределенной структурой. По мере улучшения методов
ВННТава и обработки адсорбентов, приводящих к все лучшему воо
Нронаасдепию определенного химического состава и структуры
ЙйВбриногти, углубления всестороннего ее исследования и развития
МНлейумирной теории адсорбции все большее число адсорбентов
еганонип л объектами применения зтой теории. Объектами при-
। Н -4 ( |Н(|
33
менения этой теории могут быть не только адсорбенты с однородной
поверхностью, но также и адсорбенты с неоднородной поверхностью,
если строение этой поверхности хороню известно. В частности, она
может быть использована для исследования адсорбции на поверх-
ностях, состоящих из двух и более физически однородных граней
4. Основные направления систематического экспериментального
и теоретического исследования адсорбции твердыми телами
Изучение строения поверхностей твердых тел и их адсорбционных
свойств представляет один из наиболее важных и быстро развива-
ющихся разделов физической химии. Большое значение имеет плано-
мерность и всесторонность этого изучения. Относящиеся Сюда иссле-
дования должны, по-видимому, составлять цепь следующих
звеньев [23].
1. Получение хорошо воспроизводимых, достаточно химически
и геометрически однородных адсорбентов.
2. Исследование строения зтих поверхностей разными методами,
в частности, химическими, изотопнообменными, различными спе-
ктроскопическими, злектронномикроскопическими, различными
дифрактометрическими, а также контроль степени однородности
поверхностей адсорбционными термодинамическими методами — хро-
матографическими, вакуумными статическими и калориметрическими.
3. Экспериментальное исследование адсорбционных свойств зтих
поверхностей, особенно в области малых и средних заполнений
и в широком интервале температур, включая независимые измерения
как величин самой адсорбции, так и теплот адсорбции и теплоемко-
стей адсорбционных систем.
4. Разработка приближенных молекулярных моделей адсорбции,
упрощенно учитывающих межмолекулярные взаимодействия адсор-
бат — адсорбент и адсорбат — адсорбат, и вывод на основе этих
моделей уравнений для изотерм адсорбции и других термодинами-
ческих свойств адсорбционной системы.
5. Вывод статистических уравнений для термодинамических ха-
рактеристик адсорбционной системы, учитывающих строго меж-
молекулярные взаимодействия адсорбат — адсорбент и адсорбат —
адсорбат.
6. Разработка способов учета неоднородности поверхности адсор-
бентов и вывод соответствующих статистических уравнений.
7. Обработка с помощью полученных уравнений эксперименталь-
ных термодинамических данных и получение из них физико-хими-
ческих констант, характеризующих межмолекулярные взаимодей-
ствия в рассматриваемой системе, в частности, константы Генри,
второго вириального коэффициента, константы ассоциации и крити-
ческих параметров вещества в адсорбированном состоянии.
8. Исследование зависимостей зтих термодинамических констант
от строения поверхности и строения молекул адсорбата и от тем-
пературы.
34
9. Теоретические оценки потенциальных функций взаимодействия
молекула — адсорбент и молекула — молекула у поверхности ад-
сорбента и их использование в молекулярно-статистическом расчете
Термодинамических характеристик адсорбционной системы.
10. Корректировка этих потенциальных функций на основе наи-
более надежных зкспериментальных термодинамических и спектро-
скопических адсорбционных данных для молекул, типичных для
соответствующих классов адсорбируемых веществ (алканов, цикла-
нов, алкенов, алкинов, ароматических углеводородов, соответству-
ющих галогенпроизводных и т. д.).
И. Молекулярно-статистические расчеты термодинамических ха-
Вактеристик адсорбции на основе скорректированных потенциальных
ункций межмолекулярного взаимодействия как для эксперимен-
тально изученных, так и для неизученных случаев адсорбции раз-
личных молекул на разных адсорбентах.
При выполнении этой программы особенно важно комплексное
Применение различных химических и физических методов [52]
И, в частности, развитие разных спектроскопических методов иссле-
дования состояния адсорбированных молекул [54, 131, 132].
В последующих главах этой книги делается попытка система-
тического рассмотрения некоторых из этих вопросов как в общем
виде, так и специально для адсорбции на наиболее близких к одно-
родным и возможно более точно охарактеризованных поверхностях.
Многочисленные адсорбционные данные, полученные на пористых
X непористых адсорбентах с неопределенным строением скелета
И неизвестным химическим составом поверхности, в этой книге
не рассматриваются.
При термодинамической обработке результатов опытов в первой
Части книги (гл. III—V) основное внимание уделено адсорбции на
Особенно однородных поверхностях графитированных термических
важ. Термодинамические характеристики адсорбции выражены-коли-
чественно в виде соответствующих констант. Интерполированные
ревультаты измерений, отнесенные к единице поверхности, собраны
| таблицы и даны в Приложении.
Во второй части книги (гл. VI—XI) термодинамические харак-
теристики адсорбционного равновесия рассмотрены молекулярно-
ОТатистическим методом на основе соответствующих потенциальных
функций межмолекулярного взаимодействия адсорбат — адсорбент
И адсорбат — адсорбат. Рассмотрены также количественные молеку-
ДКрно-статистические расчеты термодинамических характеристик
едсорбции благородных газов, углеводородов разных классов и не-
которых дейтерозамещенных углеводородов при малом (нулевом)
1ДПолпепии базисной грани полубесконечной решетки графита,
Когда взаимодействием адсорбат — адсорбат можно пренебречь (прак-
тически это часто осуществляется в газовой хроматографии при
Малых дозах адсорбата).
Полученные при использовании опытных адсорбционных данных
выражения для атом-атомных потенциальных функций межмолеку-:
лярного взаимодействия, по-видимому, можно использовать не
только для расчетов адсорбции. Эти потенциальные функции имеют
и более общее значение, т. е. их можно использовать для расчетов
свойств реальных газов, молекулярных кристаллов, жидкостей
и других систем, в которых проявляются соответствующие меж-
молекулярные взаимодействия.
ЛИТЕРАТУРА
1. Hill Т. L. J. Chem. Phys., 1946, v. 14, р. 441—452.
2. HillT. L. Introduction to Statistical Thermodynamics. London, 1960.
508 p.
3. Де-Бур Я. X. Динамический характер адсорбции. М., Издатинлит,
1962. 290 с.
4. Young D. М., Crowell A. D. Physical Adsorption of Gases. London,
1962. 426 p.
5. Barter R. M. In: Non - Stoichiometric compounds. (Coll.). Ed. L. Mon-
delcom. New York — London, 1964. 674 p.
6. Ross S., Olivier J. On Physical Adsorption. New York, 1964. 401 p.
7. К и с e л e в А. В. ЖФХ, 1964, t. 38, c. 2753—2773; 1967, t. 41, c. 2470—
2504.
8. WolfeR., Sams J. B. J. Phys. Chem., 1965, v. 69, p. 1129—1135.
9. К i s e 1 e v A. V. Disc. Faraday Soc., 1965, v. 40, p. 205—231.
10. ПошкусД.П. ЖФХ, 1965, т. 39, с. 2962—2967.
И. Киселев А. В,, Яшин Я. И. Газо-адсорбционная хроматография.
М., «Наука», 1967. 256 с. Исправленные и дополненные издания: Gas-Adsorp-
tion Chromotography. New York, 1969. 254 p. La Chromatographie Gas-
Solid. Paris, 1969. 289 p.
12. Ricca F., P i s a n i C., G a г г о n e E. J. Chem. Phys., 1969, v. 51,
p. 4079—4091.
13. К p о у э л А. Д. В кн.: Межфазовая граница газ — твердое тело. Пер.
с англ. ред. Э. Флад. М., «Мир», 1970. 433 с.
14. Стил У. А. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ,
ред. Э. Флад. М., «Мир», 1970. 433 с.
15. X о н и г Д. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ,
ред. Э. Флад. М., «Мир», 1970. 433 с.
16. П ошку с Д. П. В кн.: Основные проблемы теории физической адсорбции
Под ред. М. М. Дубинина и В. В. Серпинского. М., «Наука», 1970. 475 с.
17. Бройер П., Лопаткин А. А. В кн.: Основные проблемы теории
физической адсорбции. Под ред. М. М. Дубинина и В. В. Серпинского.
М., «Наука», 1970. 475 с.
18. К i s е 1 е v А. V. J. Chromatog., 1970, v. 49, р. 84—129.
19. Thomy A., D u v а 1 X. J. Chim. phys., 1970, v. 67, р. 1101—1110.
20. Di С о г с i a A., L i b е г t i A. Trans. Faraday Soc., 1970, v. 66,
p. 967—975.
21. A v g и 1 N. N., К i s e 1 e v A. V. In: Chemistry and Physics of Carbon.
(ColL). Ed. P. L. Walker, New York, 1970, v. 6, 349 p.
22. PoshkusD. P., AfreimovichA. J. J. Chromatog., 1971, v. 58,
p. 55—59.
23.
24.
25.
26.
27.
CiselevA. V., Advances Chem. Ser., Molecular Sieves Zeolites II,
1971, v. 102, p. 37—68.
< is el e v A. V. In: Thermochimie, Collogues Internationaux du C. N. R. S.
№ 201, Marseille. (Coll.). 1972. 565 p.
Vid al-Ma d j ar C., Jacob L., GuiochonG. Bull. Soc.
chim. France, 1971, p. 3105—3109.
Vidal-Mad jar C., Gui oc h о n G. Bull. Soc. chim. France, 1971,
p. 3110—3118.
L a rh e r Y. J. Col. Interface Sci., 1971, v. 37, p. 836—848.
S6
16. Folman M., К о z i г о v s к i Y. J. Col. Interface Sci., 1972, v. 38,
p. 51-57.
20. D e Boer J. H. e. a. J. Col. Interface Sci., 1972, v. 38, p. 97—109.
|0. В e г e z i n G. I., К i s e 1 e v A. V. J. Col. Interface Sci., 1972, v. 38,
p. 227—233.
81. GenotB., DuvalX. J. Chim. phys., 1972, v. 69, p. 1238—1245.
82. Hagen D.E. J. Chem. Phys., 1972, v. 56, p. 5413—5416.
83. Киселев А. В. В кн.: Успехи хроматографии. К столетию со дня
рождения М. Цвета. М., «Наука», 1972. 295 с.
84. Киселев А. В. и др. Физико-химические применения газовой хромато-
графии. М., «Химия», 1973. 255 с.
85. Schneider W., BruderreckH., Halashl. Analyt. Chem.,
1964, v. 36, p. 1533—1540.
86. Kiselev A. V. In: Advances in Chromatography. (Coll.). Eds. J. C. Gid-
dings, R. A. Keller, v. 4, New York, 1967, 380 p.
87. Halash I., H e i n e E. In: Advances in Chromatography. (Coll.). Eds.
J. C. Giddings, R. A. Keller, v. 4, New York, 1967. 380 p.
88. Vidal-Madjar C., Gan ansi a J., GuiochonG. la: Gas
Chromatography 1970. (Coll.). Eds. R. Stock. S. G. Perri, London, 1971.
445 p.
89. KouznetsovA. V., Vidal-Madjar C., Guio ch on G. Bull.
Soc. chim. France, 1969, p. 1440—1448.
40. Киселев А. В., Щер бакова К. Д., Яшин Я. И. ЖСХ, 1969,
т. 10, с. 951—968.
41. Guio ch on G., PommierC. La Chromatographic en phase gazeuse
en chimie inorganique. Paris, 1971. 339 p.
42. JequirW., Robin J. Chromatographia, 1971, v. 4, p. 59—65.
43. Kiselev A. V., Kovaleva N. V., Nikitin Yu. S. J. Chroma-
tog., 1971, v. 58, p. 19—30.
44. Eisen O. G. e. a. Chromatographia, 1971, v. 4, p. 448—454.
45. К a 1 a s h n i k о v a E. V. e. a. Chromatographia, 1971, v. 4, p. 495—500.
46. Di CorciaA., Ceccioli P., Bruner F. J. Chromatog., 1971,
v. 62, p. 128—131.
47. Di CorciaA., Bruner F. Analyt. Chem., 1971, v. 43, p. 1634—1639.
48. В e z u s A. G. e. a. Chromatographia, 1974, v. 7, p. 246—250.
49. Kalashnikova E. V. e. a. Chromatographia, 1972, v. 5, p. 278—285.
50. К i s e 1 e v A. V., Poshkus D. P. J. Chem. Soc. Faraday Trans. I,
1975, v. 71.
51. Kiselev A. V., Poshkus D.P., Shcherbakova K.D. J.
Chromat. Sci., 1974, v. 12, № 12.
52. К i s e 1 e v A. V. Disc. Faraday Soc., 1971, v. 52, p. 14—32.
53. Atherton K., Newbold G., Hockey J. A. Disc. Faraday Soc.,
1971, v. 52, p. 33—45.
'54. Киселев А. В., Л ы г и н В. И. Инфракрасные спектры поверхност-
ных соединений и адсорбированных веществ. М., «Наука», 1972. 459 с.
55. Киселев А. В., Кузнецов Б. В., Никитин Ю. С. Кинетика
и катализ, 1970, т. 11, с. 500—512.
56. Grow ell A. D., С h a n g С. О. J. Chem. Phys., 1965, v. 43, р. 4364—
4368.
57. 3 е м а н И. Кристаллохимия. М., «Мир», 1969. 154 с.
58. LarherY., J. Chim. phys., 1968, v. 65, p. 974—976. These, Orsay,
1970. 142 p.
59. Белякова Л. Д., Киселев А. В., Калпа к ян А. М. Кол-
лоидн. ж., 1973, т. 35, с. 906—910.
60. V a n D • ng е n R. Н. Doctoral Disertation «Physical Adsorption on Ionic
Solids». Delit, 1972. 116 p.
61. Belyakova L. D., Kiselev A. V., Kalpakyan A. M. Chro-
matographia, 1974, v. 7, p. 14—18.
62. Belyakova L. D., Kiselev A. V., Soloyan G. A. Chroma-
tographia, 1970, v. 3, p. 254—259.
37
63. Б е л я к о в а Л. Д., К и с е л е в А. В., С о л о я и Г. А. ЖАХ, 1972.
т. 27, с. 1182—1187.
64. Кит ородский А. И. Молекулярные кристаллы. М., «Наукаэ,
65. В h a m b h a n i М. R. е. a. J. Col. Interface Sci., 1972, v. 38, p. 109—117.
66. Б e б p и с H. К., Киселев А. В., Никитин Ю. С. Коллоидн. ж.,
1967, т. 29, с. 326—332.
67. В е b г i s N. К. е. a. Chromatographia, 1971, v. 4, р. 93—97.
68. К и с е л е в А. В. ДАН СССР, 1954, т. 98, с. 431—434.
69. Carman Р. С., R а а 1 F. A. Proc. Roy. Soc., 1951, А209, р. 59—69.
70. Радушкевич Л. В. Изв. АН СССР, ОХН, 1952, с. 1008—1020.
71. Карнаухов А. П., Киселев А. В. ЖФХ, 1957, т. 31, с. 2635—
2643; 1960, т. 34, с. 2146—2155; 1970, т. 43, с. 2354—2360.
72. РадушкевичЛ. В., Колганов В. А. Коллоидн. ж., 1961, т. 23,
с. 86—94.
73. А р и с т о в Б. Г., К а р н а у х о в А. П., Киселев А. В. ЖФХ,
1962, т. 36, с. 2153—2161, с. 2486—2490, с. 2758—2763.
74. Wade W. Н. J. Phys. Chem., 1964, v. 68, р. 1029—1034; 1965, v. 69,
р. 322—326.
75. Venable R., W a d e W. H. J. Phys. Chem., 1965, v. 69, p. 1395—1401.
76. G i 11 e s p i e T., S e 11 i n e r i W. G. J. Col. Interface Sci., 1967, v. 24,
p. 199—202.
77. Kubota S., Higuchil. Nippon Kagaku Zasshi, 1967, v. 88, p. 43—48.
78. Нагиев M. Ф., Ибрагимов Ч. Ш., Гаджиева Э. А. ДАН
АзССР, 1967, т. 23, с. 24—27.
79. MassonG. J. Col. Interface Sci., 1971, v. 35, p. 279—287.
80. К a p н a у x о в А. П. «Кинетика и катализ», 1971, т. 12, с. 1025—1032,
1235—1241.
81. Ash S. G., Kiselev А. V., Kuznetsov B.V. Trans. Faraday
Soc., 1971, v. 67, p. 3118—3126.
82. БайгубековаТ. А., Киселев А. В., Никитин Ю. С. «Кине-
тика и катализ», 1972, т. 13, с. 755—761.
83. FripiatJ., UytterhoevenJ. J. Phys. Chem., 1962, v. 66,
p. 800—805.
84. А г з а м x о д ж a e в А. А. и др. Изв. АН СССР. Сер. хим., 1969, с.
2111-2116.
85. Z h и г a v 1 е г L. Т., К i s е 1 е v А. V. In: Surface Area Determination.
(Coll.). Ed. D. H. Everett. London, 1970. 405 p.
86. C u r t о у s G. e. a. J. Col. Interface Sci., 1974, v. 48, p. 58—72.
87. ZettlemoyerA. C., Klier K. Disc. Faraday Soc., 1971, v. 52,
p. 46—49.
88. Киселев А. В., ЛыгинВ. И., ЩепалинК. Л. «Кинетика и
катализ», 1971, т. 12, с. 185—190.
89. Д а в ы д о в В. Я. и др. ЖФХ, 1973, т. 47, с. 809—811.
90. Киселев А. В., ХудяковВ.Л., ЯшинЯ.И. ЖФХ, 1973,
т. 47, с. 2125—2126.
91. GvozdovichT. N., Kiselev А. V., Yashin Ya. I. Chroma-
tographia, 1969, v. 2, p. 234—238; 1973, v. 6, p. 179—182.
92. Боса чек В. В кн.: Цеолиты, их синтез, свойства и применение. М.,
«Наука», 1965. 395 с.
93. Aristov В. G., BosacekV., Kiselev А. V. Trans. Faraday
Soc., 1967, v. 63, p. 2057—2067.
94. BarrerR.M., Whiteman J.L. J. Chem. Soc., A1967, p. 13—18.
95. Bezus A. G., Kiselev A. V., SedlacekZ., Pham Quang
D u Trans. Faraday Soc., 1971, v. 67, p. 468—482.
96. A v g u 1 N. N. e. a. J. Col. Interface Sci., 1973, v. 42, p. 486—495.
97. Экспериментальные методы в адсорбции и молекулярной хроматографии.
Под ред. А. В. Киселева и В. П. Древинга. М., Изд-во МГУ, 1973. 447 с.
98. М а с h i n W. D., R о s s S. Proc. Roy. Soc. (London), 1962, A265, p.455 —
462.
38
90. Б ер ез ин Г. И. и др. ЖФХ, 1969, т. 43, с. 223.
100. Белякова Л. Д., Киселев А. В., Ковалева Н. В. ЖФХ,
1968, т. 42, с. 2276—2283.
101. TakaishiT., М о h г i М. J. Chem. Soc. Faraday Trans. I, 1972,
v. 68, p. 1921-1934.
102. Jaycock M. J., WaldsaxJ.C. R. J. Col. Interface Sci., 1971,
v. 37, p. 462—468.
103. Aristov B. G., В о s a с e k V., Kiselev A. V. Trans. Faraday
Soc., 1967, v. 63, p. 2057—2067.
104. АвгульН. H. и др. ЖФХ, 1968, т. 42, с. 2682—2685.
105. Bezus A. G. е. a. J. Col. Interface Sci., 1973, v. 45, p. 386—395.
106. Berezin G. I. e. a. J. Col. Interface Sci., 1972, v. 38, p. 335—340.
107. Freundlich H. Kapillarchemie, Leipzig, 1923. 1225 s.
108. DubininM. M. In: Chemistry and Physics of Carbon. (Col.). Ed.
P. L. Walker, v. 2, New York, Dekker, 1966, 384 p.
109. Bering В. P., DubininM. M., Serpinsky V. V. J. , Col.
Interface Sci., 1966, v. 21, p. 378—3Q3.
110. D u b i n i n M. M., Astakhov A. V. Advances Chem. Ser., Molecular
Sieves II, 1971, v. 102, p. 69—87.
111. Хобсон Дж. В кн.: Межфазовая граница газ — твердое тело. Пер.
с англ. ред. Э. Флад. М., «Мир», 1970. 433 с.
112. F г е е m a n Е. М. е. a. Carbon, 1970, v. 8, р. 7—12.
113. Polanyi М. Verh. deut. phys. Ges., 1914, Bd. 16, s. 1012—1030.
114. P о 1 a n у i M. Z. Electrochem., 1920, Bd. 26, s. 370—374.
115. Брунауэр С. Адсорбция газов и паров. М., Издатинлит, 1948, 781 с.
116. Langmuir I. J. Am. Chem. Soc., 1918, v. 40, p. 1361—1403.
117. Brunauer S., Emmett P. H., T e 11 e г E. J. Am. Chem. Soc.,
1938, v. 60, p. 309—319.
118. Брунауэр С., Коупленд Л., КантроД. В кн.*. Межфазо-
вая граница газ— твердое тело. Пер. с англ. ред. Э. Флад. М.,«Мир»,
1970. 433 с.
119. Фаулер Р., ГуггенгеймЭ. А. Статистическая термодинамика.
М., Издатинлит, 1949. 612 с.
120. Би би Р. А. и др. ЖФХ, 1964, т. 38, с. 708—718.
121. Bradley R. S. Phil. Mag., 1931, v. 11, ser. 7, p. 690—696.
122. W i 1 k i n s T. J. Proc. Roy. Soc., 1938, A164, p. 496—509.
123. BarrerR. M., GibbonsR.M. Trans. Faraday Soc., 1963, v, 59,
p. 2875—2887.
124. Barker J. A., Everett D. H. Trans. Faraday Soc., 1962, v. 58,
p. 1608—1623.
125. Г и б б с Д ж. В. Термодинамические работы. М., Техтеоретиздат, 1950.
492 с.
126. Berezin G. I., Kiselev А. V. J. Col. Interface Sci., 1974, v. 46,
p. 203—211.
127. П о ш к у с Д. П. Тезисы докладов на 2-ом Всесоюзном симпозиуме по
межмолекулярным взаимодействиям и конформации молекул, 3—9 июня
1974г., Волгоград, 112 с.
128. Biscoe J., Warren В. Е. J. Appl. Phys., 1942, v. 13, р. 364—371.
129. В е е b е R. А. е. a. J. Am. Chem. Soc., 1947, v. 69, р. 95—101.
130. Polley М. Н., Schaeffer W. D., S m i t h W. R. J. Phys. Chem.,
1953, v. 57, p. 469—471.
131. Литтл Л. Дополн. главы Кисе ле в а А. В., Л ы г и н а В. И.
Инфракрасные спектры адсорбированных молекул. Пер. с англ., М.,
«Мир», 1969. 514 с.
132. Suzanne J.. Coulomb J. Р., Bienfait М. Surface Sci.,
1974, v. 44, p. 141-156.
Глава II
ПОЛУЧЕНИЕ АДСОРБЕНТОВ С БЛИЗКОЙ
К ОДНОРОДНОЙ ПОВЕРХНОСТЬЮ
1. Графитированная сажа и расщепленный графит
Помимо важной роли в развитии теории адсорбции (см, разд. 1
гл. I) графитированные термические сажи представляют также инте-
рес как эталонные углеродные непористые адсорбенты с однородной
поверхностью при изучении свойств термически необработанных
саж, графитов, коксов и активных углей. В частности, сопоставление
с графитированной термической сажей важно при изучении адсорб-
ционных свойств новых важных адсорбентов — неокисленных моле-
кулярно-ситовых углей [1—7]. В последнее время графитированная
термическая сажа приобрела важное значение в газовой хроматогра-
фии [8—16], в особенности как адсорбент для разделения структур-
ных и пространственных изомеров [9,10, 12, 17, 18] и других соедине-
ний, отличающихся геометрией молекул [10, 18], а также дейтери-
рованных [И, 19—22], фторированных [23, 24], хлорированных,
Хромированных и иодированных углеводородов и их производ-
ных [25] и ряда элементорганических соединений [26, 27]. Кроме
того, графитированные сажи применяются как носители слоев трудно-
летучих и высокомолекулярных веществ [28—31]. Графитированная
сажа с успехом применяется также как носитель однородных адсорб-
ционных слоев более высококипящего адсорбата (например, ксенона
или этилена) при изучении адсорбции на поверхности таких слоев
при низкой температуре более низкокипящего адсорбата
(аргона) [32—37].
Адсорбционные свойства углеродных адсорбентов — графитов,
саж, активных углей, углеродных волокон и мембран — обусло-
влены особенностями их строения: размерами кристаллитов углерода
в скелете адсорбента, структурой аморфного углерода, химическими
соединениями углерода с другими атомами (в основном с кислородом
и водородом [38—42]), а также степенью шероховатости поверх-
ности, наличием и структурой пор. Наиболее сильно развита пори-
стость у активных углей, получаемых из природных материалов [43,
44], и у так называемых молекулярно-ситовых углей, получаемых
термическим разложением синтетических полимеров. Размеры пор
молекулярно-ситовых углей довольно однородны и очень малы [1—4].
Сажи, не окисленные специально нагреванием на воздухе или
в кислороде [45] или обработкой в жидких окислителях [40, 46, 47],
непористы, хотя их поверхность шероховата.
40
Кристалл графита имеет слоистую структуру [48] (рис. 11,1).
Расстояния между атомами углерода в гексагональных кольцах
Слоя (в направлении оси а, см. также рис. 1,1а) и между слоями
(в направлении оси с) составляют соответственно 1,42 и 3,35 А. Два
соседних слоя атомов углерода в решетке графита смещены друг
Относительно друга на 1,42 А (рис. 11,1). Атомы углерода в слое
прочно соединены с тремя соседними о-связями и дополнитель-
ными л-связями, образованными делокализованными электро-
нами [49]. Слои связаны друг с другом ван-дер-ваальсовыми
силами [50].
Помимо химически неактивной и адсорбирующей неспецифически
базисной грани кристалл графита имеет призматические грани.
Благодаря обрыву на этих гранях
гексагональных сеток они химически
активны и обычно насыщены содер-
жащими водород и кислород функ-
циональными группами.
Исследованы адсорбционные свой-
ства листочков расщепленного гра-
фита со значительной удельной
поверхностью [51]. В тонких листоч-
ках графита сильно развиты базис-
ные грани, а влияние призматиче-
ских граней относительно невелико.
На рис. 11,2 приведены изотермы
адсорбции ксенона на таком рас-
щепленном графите [51]. Обраще-
Рис. 11,1. Структура графита.
ние начала изотермы выпуклостью к оси давления пара р ксено-
на и резкий подъем изотермы адсорбции в средней части сви-
детельствуют о значительной однородности этого образца. Аналогич-
ные кривые получены на графите [52]. Однако в области завершения
плотного монослоя наблюдается еще вторая небольшая ступень,
появление которой было объяснено кристаллизацией двухмерного
конденсата на поверхности адсорбента [51, 52]. Возможно, однако, что
вторая ступень вызывается адсорбцией на оставшихся призмати-
ческих гранях, расположенных по периметру листочков или частиц
графита.
Даже наиболее однородные термические сажи обладают удельной
поверхностью от 6 м2/г и более. Поэтому для них возможны газо-
зсроматографические измерения удерживаемого объема, газохромато-
графические и статические измерения изотерм адсорбции, а также
калориметрические измерения зависимостей дифференциальных те-
пл от адсорбции и теплоемкостей адсорбированных веществ от за-
полнения поверхности. В зависимости от способа получения раз-
личают канальную, ацетиленовую, печную, ламповую, форсуночную
и термическую сажи. Необработанные сажи состоят из изолирован-
ных или слипшихся сферических частиц различных размеров, поверх-
ность которых в той или иной степени шероховата.
41
Диаметры частиц некоторых саж указаны в табл. 11,1. Там же
приведены величины удельных поверхностей, определенные по
адсорбции пара азота при —195 °C и из электронных микрофото-
графий. Для канальных саж удельная поверхность, определенная
Рис. 11,2. Изотермы адсорбции ксенона на листочках рас-
щепленного графита при различных температурах (К):
1 — 101,0; 2 — 105,6; 3 — 108,8; 4 — 112,6; 5 — Ц4.8; 6 — 115,9;
7 — 116,6; 8 ~ 117,6; 9 — 118,1.
по адсорбции пара азота методом БЭТ [64], в 1,3—1,4 раза больше
определенной из микрофотографий, т. е. поверхность таких саж
шероховата (отношение определенных этими двумя методами удель-
ных поверхностей называется фактором шероховатости).
Таблица 11,1. Некоторые характеристики исходных необработанных саж
[53-63]
Сажа Р ентгенострук- турные характе- ристики кристал- литов, А. Диаметр частиц, А Удельная поверхность, м2/г Фактор шерохо- ватости
La Lq по ад- сорбции пара азота из элек- тронных микро- фотогра- фий
Канальная Spheron-6 17 12 400 110 75 1,43
Ацетиленовая 47 26 500 64 55 1,08
Печная Statex 550 40 50 0,8
Ламповая 1400 25 23 1,07
Термическая 28 16 1500 20,7 21,0 0,96
42
По изменению адсорбционных свойств при переходе от канальных
саж к термическим и особенно к термическим графитированным
сажам можно судить о приближении к адсорбционным свойствам
наиболее однородной поверхности. Канальные сажи высокодис-
персны и довольно неоднородны. Частицы обычной канальной сажи
содержат углеродные образования сетчатого строения (кристаллиты
небольшого размера) с линейными параметрами вдоль оси a, La
^17А и вдоль оси с, Lc 12 А [54]. Эти кристаллиты беспоря-
дочно расположены внутри частиц сажи вместе с включениями
аморфного углерода и высокомолекулярных углеводородов, как
в углях и коксах [65]. Структура частиц термических саж более
упорядочена. В частицах необработанных термических саж (см.
табл. 11,1) имеются кристаллиты графита довольно больших раз-
меров. Даже в необработанной при высоких температурах терми-
ческой саже наблюдается частичное упорядочение зтих кристаллитов.
Углеродные слои располагаются параллельно поверхности частиц
сажи [66].
Канальные сажи получаются в окислительной атмосфере, поэтому
они весьма неоднородны по химическому составу. По сравнению
с другими сажами они содержат наибольшее количество кислорода
и водорода (табл. 11,2) [45—47, 67—69].
Таблица 1Ц2. Элементный состав некоторых канальных и термических саж
[46, 47]
Сажа Темпера- тура об- работки в ваку- уме, °C Удельная поверх- ность, м*/г Элементный состав
С н о
Канальная Spheron-6 220 115 94,84 0,45 4,71
950 109 96,89 0,45 2,66
Канальная [46] 200 148 94,8 0,75 4,45
Термическая Sterling FT 800 16 99,71 0,25 0,04
^Термическая [46] 800 27 99,66 0,27 0,07
Водородные соединения представляют собой остатки продуктов
полимеризации углеводородов, применяемых для получения сажи,
а кислородные соединения образуются в процессе получения саж
в канальных печах. Водород и кислород находятся внутри частиц
сажи и на их поверхности как в виде химически связанных с остовом
частицы самой сажи функциональных групп, так и в виде адсорби-
рованных смолистых веществ. Различные химические методы ана-
лиза, а также методы инфракрасной спектроскопии и ЭПР поз-
воляют обнаружить на поверхности саж, особенно на поверхности
окисленных саж, разнообразные функциональные группы: гидр-
оксильные, карбонильные, альдегидные, карбоксильные, хинонные,
лактонные, перекисные и др. (см. обзор [39] и работы [41, 42, 70—
86]), а также свободные радикалы и неспаренные электроны (см.
обзор [39, 62]), а также работы [78, 87—96]). У неграфитированных
43
саж, как и у углей, проявляются характерный сигнал ЭПР и соот-
ветствующий кислородный эффект. Для неграфитированных саж
этот сигнал имеет сложную форму, особенно у сильно окисленных
саж [77, 94]. Природа этого сигнала еще не ясна. Его связывают
с парамагнитными центрами двухмерных бензоидных структур в са-
жах и с образованием новых парамагнитных центров при действии
окислителя [93].
Благодаря различиям в геометрической неоднородности поверх-
ности разных саж и наличию на ней активных функциональных групп
величины адсорбции на
Рис. 11,3. Изотермы адсорбции
пара метанола при 20 °C на
исходных сажах, прогретых
ири 800 ₽С в вакууме:
1 — канальной Spheron-б и 2 —
термической.
единице поверхности адсорбента и формы
изотерм адсорбции на сажах разного
происхождения резко различаются [47,
68, 97]. Это различие особенно велико
при адсорбции молекул, относящихся
к группам В и D (см. разд. 2 гл. I),
способных к специфическому взаимодей-
ствию с кислородными комплексами
на поверхности саж. Так, изотермы
адсорбции (рис. 11,3) на исходных ка-
нальной и термической сажах, до-
полнительно прогретых в вакууме
при 800° С, в области невысоких
заполнений поверхности резко разли-
чаются [68, 97].
Дополнительной обработкой мож-
но сильно изменить химический со-
став поверхности саж, а также их
адсорбционные и адгезионные свой-
ства в двух противоположных напра-
влениях, как в сторону гидрофили-
зации, так и в сторону гидрофобизации. Во многих работах 146,
47, 72, 83, 98—105] показано, что к гидрофилизации поверх-
ности приводит окисление в газовой и особенно в жидкой среде.
Окисление в газовой среде, на воздухе и в кислороде, при повышен-
ных температурах приводит к резкому увеличению удельной по-
верхности сажи за счет частичного выгорания углерода и образова-
ния пор. Однако концентрация окислов на единице образующейся
поверхности, по-видимому, существенно не увеличивается [100].
Непродолжительное окисление в таких жидких средах, как растворы
перекиси водорода, гипохлорита натрия, марганцевокислого калия,
азотной и серной кислот и в растворах других сильных окислителей,
наоборот, не изменяя существенно величину поверхности, приводит
к резкому увеличению поверхностной концентрации функциональ-
ных групп. Это значительно увеличивает адсорбцию на такой поверх-
ности молекул, относящихся к группам В и D [46, 47] (рис. П,4).
Такая сажа становится настолько гидрофильной, что диспергируется
в воде без внесения смачивателей [99, 100].
Окисление азотной кислотой приводит к образованию на поверх-
44
яости сажи большого количества карбоксильных групп, что резко
увеличивает молекулярную адсорбцию и хемосорбцию основа-
ний [105]. Окисление сажи в жидких средах повышает соответственно
и ее ионообменные свойства [99].
К гидрофобизации поверхности саж приводит термическая обра-
ботка при высоких температурах (до 1000—3000 °C) в вакууме или
В атмосфере инертного газа [53, 106, 107] и в атмосфере водорода [84,
85, 108, 109], а также нанесение на поверхность слоя углерода при
разложении метана или других легких углеводородов при умеренных
температурах [110, 111]. Последнее приводит к удалению или экра-
Рис. 11,4. Изотермы адсорбции н-гексана, бензола, метанола и воды:
1 — на исходной канальной саже, прогретой при 800 °C в вакууме; 2 — на той же
саже после окисления смесью азотной и серной кислот; з — адсорбция метанола
на гидроксилированной поверхности силикагеля. Черные точки — десорбция.
пированию поверхностных функциональных групп. Однако поверх-
ность сажи остается при этом еще геометрически неоднородной. Для
получения как химически, так и физически однородной поверхности
сажи наиболее надежный путь представляет обработка сажи при
очень высоких температурах в отсутствие кислорода и других окис-
лителей. Так как при промышленном получении графитированной
сажи захватываются следы кислорода, то нужна еще дополнительная
обработка водородом при 1100 [84, 85, 109].
Термическая обработка саж в вакууме, в инертном газе или в вос-
становительной атмосфере, предпринятая впервые при нагревании
до 3000 °C в 1942—1947 г. Уорреном, Смитом и сотр. [53, 106, 107],
приводит не только к удалению «летучих», т. е. различных хими-
ческих соединений углерода с кислородом и водородом, но также
и к укрупнению кристаллитов графита и к упорядочению их взаим-
ного расположения. Интенсивное выделение летучих соединений
происходит до 1100 °C, при этом сильно уменьшается содержание
кислорода и водорода в сажах [46, 47, 61, 112].
Размеры кристаллитов в частицах сажи при нагревании до этой
температуры существенно еще не изменяются [53]. Заметный рост
кристаллитов начинается при нагревании в интервале 1300—
220(1 ‘С [53, 106, ИЗ]. В этой же области температур происходит
45
резкое изменение характера сигнала ЭПР термической сажи [114].
Особенно значительный рост кристаллитов и упорядочение их рас-
положения происходит в интервале температур 2800—3200 °C [53,
61, 106].{Размеры кристаллитов в обработанных при этих темпера-
турах сажах увеличиваются, и в случае термических саж La дости-
гает 150 А (табл. 11,3).
Таблица 11,3. Некоторые характеристики графитированных саж [55]
Сажа Темпера- тура об- работки, °C Удельная поверх- ность, м2/г Средний диаметр частиц, А Пикно- метричес- кая плот- ность, г/см8 рентгено- структурные данные
с/2, А ьо. а
Термическая Thermax 2700 6,0 5000 2,1787 3,395 147
3000 2,1796 3,38 150
Термическая Sterling МТ 2700 7,6 4000 2,161. 3,42 136
3000 2,1646 3,40 136
Термическая Sterling FT 2700 12,0 1500 2,15% 3,42 136
3000 2,1633 3,39 136
Печная Regal 2700 30 650 2,0529 3,44 90
3000 2,053, 3,42 90
Графит 2,22 3,39
Рост и упорядочение расположения кристаллитов приводит
к тому, что частицы сажи из круглых (с шероховатой поверхностью)
превращаются в полиэдры (рис. 11,5) [8, 9, 55, 115—1171- Поверх-
ность этих полиэдров образована однородными базисными гранями
кристаллитов графита. При таком нагревании термических саж
наступает полная ориентация кристаллитов вдоль граней полиэдров,
у канальных саж частично еще остается неупорядоченная струк-
тура [55, 118]. Метод микродифракции электронов [119] подтвердил
наличие в графитированной термической саже кристаллических
блоков и их азимутальную ориентацию. Таким образом, обработан-
ная вблизи 3000 °C термическая сажа оказывается в наибольшей
степени графитированной.
При переходе от исходных к графитированным вблизи 3000 °C
сажам меняется вид спектров ЭПР и их парамагнитные параметры.
Из рис. И,6 видно, что при уменьшении удельной поверхности
графитированных саж, т. е. при переходе к графитированным тер-
мическим сажам, g-фактор увеличивается до некоторого предельного
вначения [92], характерного, по-видимому, для объема частицы.
После прокаливания вблизи 3000 °C параметры решетки кристал-
литов и плотность частиц графитированных термических саж стано-
вятся близкими к таковым для графита (см. табл. II,3).
46
Однако частицы графитированной термической сажи отличаются
от обломков кристаллов графита тем, что их поверхность образуется
практически только базисными гранями решетки графита к(см.
Рис. 11,5. Электронные микрофотографии исходной термиче-
ской сажи Sterling МТ (а) и той же сажи после графитирова-
ния при 3100 °C (б).
рис. 11,5). Стыки граней полиэдров, на которых нарушается пра-
вильная упаковка атомов углерода в базисных гранях, составляют
лишь очень малую часть общей однородной поверхности. Кроме того,
Рис. 11,6. Зависимость величины g-фактора от удельной по-
верхности s для разных саж:
2 — сажа, графитированная при 3000 °C; 2 — исходная необработан-
ная сажа.
Разные точки относятся к данным разных работ.
эти стыки, по-видимому, осуществляются постепенно изгибающимися
графитовыми сетками, в которых расположение атомов углерода
близко к таковому в базисной плоскости графита [66]. Таким обра-
зом, именно поверхность графитированных термических саж хими-
47
чески и физически очень однородна. В разломах же обычного графита
наряду с базисными гранями имеются и химически активные призма-
тические грани. Только листочки расщепленного графита [51,
52] (рис. 11,7) и графитированная при 3000 °C углеродная мембрана
(см. ниже рис. 11,32) по однородности поверхности приближаются
к графитированным термическим сажам.
Рис. 11,7. Изотермы адсорбции криптона при 77,8 К на
листочках расщепленного графита (сплошная линия),
на термической саже Sterling МТ, 3000 9С (здесь и
далее температура, поставленная после марки сажи,
обозначает температуру графитирования) (кружки)
и на природном графите (квадраты).
Так как адсорбционные свойства очень чувствительны к степени
однородности поверхности, рассмотрим изменение адсорбционных
свойств сажи, происходящее при графитировании, выбрав молекулы,
относящиеся к разным группам по их способности к специфическому
межмолекулярному взаимодействию (см. разд. 2 гл. 1). Влияние
постепенно увеличивающейся температуры обработки термической
сажи в вакууме на величину адсорбции [120] молекул группы А
(аргона) показано на рис. 11,8. При переходе от исходной сажи
к графитированной форма изотермы адсорбции пара аргона изме-
няется от плавной s-образной, характерной для полимолекул ирной
48
адсорбции пара на неоднородной поверхности, до волнообразной
на графитированной саже. Изотерма адсорбции на наиболее однород-
ной графитированной вблизи 3000 °C термической саже в области
разных интервалов величин равновесных давлений представлена
на рис. 11,9.
При увеличении температуры обработки сажи соответственно
меняется и зависимость теплоты адсорбции от заполнения
(рис. 11,10) [121]. Типичное для неоднородной поверхности умень-
шение теплоты адсорбции с ростом заполнения поверхности исходной
сажи сменяется ростом теплоты адсорбции на графитированной саже
по мере заполнения первого адсорбционного слоя. В этом случае
отчетливо проявляется переход от преимущественной адсорбции
в первом слое адсорбированных молекул к преимущественной адсорб-
ции во втором и последующих слоях. Этот переход вызывает резкое
падение теплоты адсорбции после максимума и приближение ее
к теплоте конденсации.
Еще более сильное изменение адсорбционных свойств в результате
графитирования сажи наблюдается при адсорбции молекул группы В
Рис. 11,8. Изотермы адсорбции аргона на термичес-
кой саже Sterling, 2700 ?С (называемой ранее Р-33)
при 77,8 К. Температуры обработки образцов сажи
и размеры кристаллов графита указаны у кривых.
и особенно группы Р, например, при адсорбции молекул мета-
нола [46, 97, 122, 123] (см. рис. 1,7) и воды [46, 47, 123, 124]. Изо-
термы адсорбции и кривые, выражающие зависимость теплоты ад-
сорбции от заполнения поверхности, резко изменяют свою форму
при переходе от канальной сажи, прогретой при 900 °C и еще содер-
жащей поверхностные окисли [97], к более однородной термической
саже, графитированной при 3000 °C, особенно после дополнительной
обработки ее водородом при 1100 °C [109]. В этих случаях при малых
заполнениях возможна адсорбция изолированных молекул воды
и метанола. В соответствии с низкой поляризуемостью этих молекул
4 Заказ 1180
49
1 — данные работы [126]; 2 — данные работы [127].
в
Рис. 11,10. Влияние температуры (указана на рис.) обработки
канальной сажи Spheron-б на зависимость измеренных в калори-
метре дифференциальных теплот адсорбции аргона от заполнения
поверхности 0 при 77,8 К.
Пунктирная кривая относится к исходной саже.
(см. разд. 13 и 18, гл. V) при адсорбции изолированных молекул
выделяется очень небольшая теплота адсорбции. Начальные теплоты
адсорбции воды и метанола на графитированной термической саже
значительно меньше теплот конденсации. При конденсации воды
и спирта основную роль играет ассоциация, происходящая с обра-
зованием взаимных водородных связей. Энергия ассоциации вносит
большой вклад в теплоту конденсации. По мере роста заполнения
поверхности молекулами воды или метанола при адсорбции между
ними также возникает сильное специфическое взаимодействие, при-
водящее к ассоциации с образованием взаимных водородных связей.
Энергия этих взаимных водородных связей при ассоциации адсор-
бат — адсорбат прибавляется к небольшой энергии взаимодействия
адсорбат — неспецифический адсорбент, и теплота адсорбции
быстро растет.
Обработанные вблизи 3000 °C канальные сажи благодаря не-
полной графитизации и небольшим размерам частиц обладают за-
метной остаточной неоднородностью поверхности. Различие в вели-
чинах адсорбции на обработанных вблизи 3000 °C канальных и тер-
мических сажах в средней области не превышает 5%. Однако при
самых малых заполнениях это различие больше. Изотерма адсорб-
ции на наиболее однородной поверхности графитированной терми-
ческой сажи в начальной части имеет наименьший наклон по срав-
нению с другими сортами саж, обработанных при той же тем-
пературе. Соответственно рост адсорбции в средней части
заполнения первого слоя на поверхности графитированной терми-
ческой сажи выражен более резко, и волнообразный характер всей
изотермы адсорбции (при заполнении первого и последующих
слоев) проявляется более отчетливо И25].
Изотермы адсорбции на разных графитированных термических
сажах очень близки между собой (см. рис. 11,9) [126, 127], и для
единицы такой поверхности адсорбционные данные могут рассматри-
ваться как физико-химические константы, характеризующие адсорб-
цию па базисной грани полубескопечного кристалла графита. Изо-
терма адсорбции аргона на графитированной термической саже,
изображенная на рис. 11,9, является типичной изотермой, харак-
терной для адсорбции на однородной поверхности. Соответствующая
ей зависимость теплоты адсорбции от заполнения поверхности при-
ведена на рис. 11,11 [128]. Обращение изотермы адсорбции при ма-
лых заполнениях выпуклостью к оси давлений свидетельствует
о высокой однородности поверхности и о четком проявлении при-
тяжения адсорбат — адсорбат (рис. И,9). Точка перегиба вблизи
О ~ 0,4 и дальнейшее обращение изотермы адсорбции вогнутостью
к оси давлений связано с постепенным заполнением монослоя. Сле-
дующие точки перегиба связаны с переходом от преимущественной
адсорбции в первом слое к преимущественной адсорбции во втором
слое и т. д. В соответствии с этим теплота адсорбции при постепенном
наполнении первого слоя растет (рис. П,10 и рис. 11,11). При пере-
ходе к преимущественному заполнению второго слоя теплота адсорб-
4*
51
ции резко падает. Далее опять наблюдается слабый рост теплоты
адсорбции при преимущественном заполнении второго слоя
(рис. 11,11). Такой волнообразный ход теплоты адсорбции, так же
как и соответствующая волнообразная форма изотермы адсорбции,
характерны для неспецифической адсорбции на однородной поверх-
ности в результате проявления взаимного притяжения молекул
вдоль поверхности в первом и втором слоях.
Сопоставление величин дифференциальных теплот адсорбции
при малом (нулевом) заполнении поверхности qvд для молекул
групп А и В [8, 9, 129, 130] с близкой геометрией и с близкими
поляризуемостями, но с различной электронной структурой (см.,
Рис. 11,11. Зависимость
дифференциальной теп-
лоты адсорбции аргона
на графитированной тер-
мической саже Sterling
FT, 2700 РС (Р-33) от
заполнения поверхности.
Вертикальными пункти-
рами отмечены степени
заполнения поверхности
0 = 1 И0 = 2.
например, теплоты адсорбции рядов н-алканов, алкенов и алкинов —
разд. 3,6 и 7 гл. V, а также разд. 2 и 4 гл. X), свидетельствует о том,
что адсорбция на однородной поверхности графитированной терми-
ческой сажи имеет в основном неспецифический характер (подробнее
этот вопрос рассмотрен в разд. 18 гл. V). Однако для газохромато-
графического исследования очень малых проб веществ с молекулами,
относящимися к группам В и D, содержащими выдвинутую группу
СО или, соответственно, гидроксильную группу, необходимо про-
водить дополнительную восстановительную обработку поверхности
графитированной термической сажи водородом при температурах
около 1100 °C [84, 85, 109]. Такая обработка необходима для уда-
ления остатков кислородных соединений на стыках базисных граней
графитированной термической сажи. Эти кислородные соединения
вызывают повышенную адсорбцию, в частности, увеличение
(удерживаемого объема адсорбированного вещества в газохромато-
графической колонне, отнесенного к единице поверхцости адсор-
бента) таких молекул при малых заполнениях (см. рис. 1,7). После
проведения обработки поверхности водородом величины удержива-
емых объемов Кз,1 в случае ацетона и пропанола заметно сни-
жаются (табл. 11,4).
52
Таблица II\4, Удерживаемые объемы при 50 °C
при нулевом заполнении поверхности графитированной
термической сажи Туд * (мкм) до и после обработки водородом
(при 1100 ?С) [109]
Адсорбат Уд, 1 обработки Уа 1 после обработки
н-Пентан 4,22 4,27
Диэтиловый эфир 1,92 1,94
Ацетон 0,95 0,57
Пропанол 1,87 0,73
Теплоты адсорбцииа молекул, относящихся к разным группам Ау
В и D, но имеющих блпизкие средние поляризуемости, на графитиро-
ванной термической ссаже, обработанной дополнительно водородом
при 1100 °C, близки (;табл. 11,5) [109].
Таким образом, графитированная термическая сажа (особенна
после дополнительной: обработки водородом при 1100 °C [84, 85, 109]}
относится к неспецифическим адсорбентам. В соответствии с этим
адсорбция на такой сзаже молекул самого разного химического стро-
ения происходит в ocibobhom неспецифически.
Можно предположить, что все же имеющее место слабое усиление
адсорбции на графитированной и обработанной водородом термиче-
ской саже молекул, относящихся к группам В и D (см. гл. VF
разд. 18), происходит вследствие особого валентного состояния угле-
рода в графитовой решетке. Взаимодействие молекулы адсорбата
с атомом углерода :графита, образующим сопряженные л-свяэи^
несколько больше взаимодействия-той же молекулы с атомом угле-
рода, образующим то.дько о-связи. Этот вопрос подробно рассматри-
вается в гл. X, здесь, же мы ограничимся примером адсорбции на
графитированной термической саже и на полимеризованном углеводо-
родном адсорбенте га&ов N2, О2 и Аг. Молекулы зтих газов обладают
близкими массами и поляризуемостями. При достаточно низких для
увеличения удерживания этих газов температурах смесь этих газон
в колонне с графитированной термической сажей разделяется и ком-
Таблица 11,5. Теплоты адсорбции разных адсорбатов с близкими значениями
средней поляризуемости а при нулевом заполнении
поверхности графитированной термической сажи
после обработки водородом (при 1100 °C) [109]
Адеорбат Группа, к которой относит- ся молекула адсорбата а, А8 9у,1, кДж/моль
и Пентан А 9,0 34,0
Диэтиловый эфир В 9,0 33,5
н Пунтел ь 8,7 36,0
5$
поненты смеси выходят из колонны в следующей последовательности:
Аг, О2 и N2 (рис. 11,12) [22]. Эта последовательность соответствует
росту квадрупольного момента молекулы и не соответствует росту
молекулярного веса (N2 < О2 < Аг). В то же время при разделении
на полимолекулярном углеводородном адсорбенте норапаке Q - s
(рис. 11,126) эти газы выходят из колонны в соответствии с ростом
молекулярного веса. Однако разница в энергиях адсорбции этих
газов на графитированной термической саже невелика. Вели-
чины qv, 1 для Аг, О2 и N2 составляют соответственно 8,80, 8,90
и 9,10 кДж/моль.
a n2 со 6
I -- ! - 1 .1 -.--J_____i L___________I_______L_
О Z Ь мъ'.н О Ь 8 мин
460% 420%
Рис. 11,12. Хроматограммы смеси газов:
а — на графитированной термической саже при програм-
мировании температуры; б — на норапаке Q-s при —83 °C,
Длина колонн 1,5 м, скорость газа-посителя гелия 20 см*/мин.
Остаточная небольшая неоднородность поверхности обычных
графитированных термических саж (дефекты и кислородные ком-
плексы на гранях и между гранями полиэдрических частиц, возмож-
ные дислокации на гранях и места контактов между разными части-
цами) может несколько исказить начальный ход изотерм адсорбции
и кривых, выражающих зависимость дифференциальной теплоты
адсорбции от заполнения. Поэтому для получения констант адсорб-
ционной системы, характеризующих межмолекулярные взаимодей-
ствия адсорбат — адсорбент и адсорбат — адсорбат на однородной
поверхности, особенно важно производить обработку эксперимен-
тальных данных с учетом влияния на эти данные остаточной неодно-
родности поверхности. Эти вопросы рассматриваются в гл. IV.
Различные партии графитированных термических саж получаются
с одинаковой химической и геометрической структурой поверхности.
Это дает возможность определять хорошо воспроизводимые величины,
характеризующие адсорбционные свойства единицы их поверхности.
Изучая эти адсорбционные свойства, можно найти физико-химические
константы, характеризующие адсорбцию на базисной грани полу-
бесконечного кристалла графита.
•54
Исследования адсорбции и теплот адсорбции на графитирован-
ных сажах как вакуумными статическими, так и газохроматографи-
ческими методами проводились в работах Смита и Биби с сотр. [53,.
107, 111, 120, 121, 131—141], Пирса с сотр. [112, 142—149], Крауэла
и Янга [150, 151], Хэлси с сотр. [32, 128, 152—157], Цетлемойера
с сотр. [158—160], Росса с сотр. [116, 126, 161, 165], Киселева
с сотр. [8-11, 14, 16-18, 22, 24-27, 47, 108, 109, 111, 117, 122,
123, 125, 129, 130, 136, 137, 166—216] и рядом других авторов [12г
13, 15, 19, 21, 23, 28-30, 33-37, 52, 54, 63, 84, 95, 115, 217-262].
Некоторые надежные интерполированные данные по величинам
адсорбции при разных давлениях в газовой фазе и разных темпера-
турах и по величинам теплот адсорбции при разных величинах
адсорбции на поверхности графитированных термических саж даны
в обзоре [117].
Адсорбция на алмазе значительно отличается от адсорбции на
других углеродных адсорбентах [238] вследствие особенностей
строения его решетки, Однако она недостаточно изучена.
2. Нитрид бора
Кристаллическая структура нитрида бора BN подобна слоистой
труктуре графита (рис. 1,1). В ней имеются плоские слои гекса-
гональных колец. В каждом кольце три вершины (через одну) заняты
атомами одного элемента, а остальные три вершины — атомами
другого элемента. В отличие от структуры графита у нитрида бора
кольца разных слоев расположены точно друг под другом. При этом
вдоль оси третьего порядка, перпендикулярной базисной границ
атомы бора и азота чередуются. Параметры решетки нитрида бора а
и с равны соответственно 2,50 и 6,66 А. Соседние атомы в одной-
плоскости находятся на расстоянии 1,45 А, а в соседних плоскостях
на расстоянии 3,33 А- Ван-дер-ваальсовы размеры атомов бора
и азота близки между собой.Нитрид бора не обладает такой высокой
электропроводностью, как графит, поэтому весьма важно сравнение
энергии адсорбцйи разных молекул на зтих двух адсорбентах.
Способы получения и физические свойства нитрида бора описаны
в работах [263—265]. Один из технических синтезов нитрида бора
заключается в сплавлении мочевины с борной кислотой в атмосфера
аммиака и в пиролизе смеси при 500—950 °C [265]. Получающийся
при этом нитрид бора имеет неупорядоченную структуру, которая
при обработке при температурах около 1800 °C превращается в сло-
истую решетку с упорядоченной гексагональной структурой. При
прогреве высокодисперсного образца нитрида бора при 2100 °C
удельная поверхность его уменьшается (от 120 до 94 м2/г по данным
работы [266]).
Поверхность получаемых образцов нитрида бора далеко не всегда
бывает однородной [266, 267]. Существенное значение имеет чистота
образца и стехиометрическое соотношение В : N, которое должно
возможно более точно равняться единице, так как избыток или
55=
недостаток бора приводит к химической неоднородности поверхности
и придает ей специфические адсорбционные свойства. Методика
получения образцов нитрида бора с однородной поверхностью не
описана, однако адсорбционные свойства промышленных образцов
нитрида бора с весьма однородной поверхностью, состоящей в основ-
ном из базисной грани (0001), исследованы [161, 257, 268]. Удельная
поверхность их составляет около 10 м2/г. Однородность поверхности
-Этих образцов приближается к однородности поверхности графи-
тированных термических саж [9, 126, 127].
Адсорбционные свойства нитрида бора изучены значительно
меньше адсорбционных свойств графитированных саж [161, 257,
265—269]. Изотерма адсорбции арго-
200 ~ на на образце нитрида бора с одно-
. родной поверхностью представлена
|150 ~ на рис. 11,13 [268]. Она близка по
f 100 __ f форме к изотерме адсорбции на
е 9 графитированной термической саже
50- 7 " (см. Рис- И»9). Аналогичные резуль-
J таты получены при адсорбции окиси
fllx ।___ь__L__ i । азота [257]. Исследование теплот
100 300 500 яПа адсорбции некоторых молекул, от-
jPhc. 11,13. Изотерма адсорбции носящихся к группам А и В [161,
аргона при—195 еС на нитриде 257, 266], показывает, что нитрид
бора. бора представляет столь же неспеци-
фический адсорбент [9, 129, 130],
как и графитированная термическая сажа (см. табл. 11,5). Сходство
изотерм адсорбции на нитриде бора и графитированной термической
*саже и неспецифичность обоих адсорбентов свидетельствуют о том,
что проводимость графитированной сажи не сказывается суще-
ственно на ее адсорбционных свойствах.
3. Металлы
Часто при адсорбции металлами таких реакционноспособных
газов, как водород, кислород, окись углерода и другие, происходит
как физическая адсорбция, так и хемосорбция, которая приводит
к образованию новых поверхностных соединений. В этом случае
адсорбированная молекула или продукты ее превращения локали-
зуются на поверхности с большой знергией связи с поверхностными
атомами металла [270], так что значительно более слабыми меж-
молекулярными взаимодействиями хемосорбированных молекул друг
с другом можно пренебречь. Однако в случае благородных газов,
особенно таких, как криптон и ксенон, и некоторых других хими-
чески инертных молекул, таких, например, как перфторметан,
наблюдается только молекулярная (физическая) адсорбция на по-
верхности металла. Исследование молекулярной адсорбции на чистой
поверхности металла представляет значительный интерес для раз-
вития молекулярной теории адсорбции. Большинство металлов
обладает простой кристаллической решеткой, например, медь и же-
«56
лезо — кубической. Поэтому их поверхности должны вести себя
как физически однородные. Вместе с тем весьма важно изучить
влияние на молекулярную адсорбцию особенностей электронной
структуры металлов, введения примесных центров, образования
поверхностных окислов и других поверхностных соединений. Однако
исследований молекулярной адсорбции на металлах пока проведено
мало из-за методических трудностей, связанных, в частности, с малой
величиной поверхности металла в химически и физически однород-
ном состоянии. Исследована адсорбция на близких к однородной
поверхностях меди [271—274], никеля [274—278], железа [279—
281], кадмия [281, 282], марганца [281] и некоторых других метал-
лов [283—285].
Рис. 11,14. Электрономикроскопические снимки частиц металлического
цинка [284].
В этих исследованиях'металл приготовлялся в виде полированных
металлических блоков или осажденных на стекло пленок и восста-
новленных порошков из окислов. Адсорбция исследовалась или
косвенным оптическим методом, или газовым адсорбционным мето-
дом Вутена и Броуна [286]. Большинство исследований, например,
меди, [271] и магния [283], указывает на неоднородность полученных
поверхностей. В работе [280] по адсорбции криптона на пленке же-
леза, нанесенной на стекло пирекс, исследованная поверхность
металла имела сильно неоднородные участки, что вызвало крутой
подъем начальной части изотермы адсорбции. Однако при дальней-
шем росте заполнения монослоя криптона на этой поверхности наблю-
дался ступенчатый ход изотермы. Это свидетельствовало о наличии
на поверхности образца ряда других, более однородных участков.
Проводилось восстановление порошка окиси никеля в токе водо-
рода при разных температурах и исследование на полученном таким
путем образце никеля адсорбции ‘ криптона в основном в области
57
Рис. 11,15. Изотерма адсорбции перфтор-
метана на кадмии при 77,5 К.
шолимолекулярной адсорбции [277]. Был получен характерный
для адсорбции на однородных поверхностях ступенчатый переход
изотермы адсорбции от преимущественного заполнения первого
к преимущественному заполнению второго слоя. Наблюдались
ярковыраженные ступени при адсорбции первого, второго и третьего
ч;лоев криптона на цинке [284] (электрономикроскопические снимки
этого образца цинка представлены на рис. 11,14).
Наиболее однородные поверхности ряда металлов были получены
в работах Жено и Дюваля [281, 282] и Де-Бура, Касперсмы и ван-
.Донгена [272—274]. Изучалась адсорбция криптона, перфторметана
и шестифтористой серы. Наи-
более однородной оказалась
поверхность кадмия (кристаллы
которого образуют плотнейшую
гексагональную структуру), по-
лученного возгонкой в ваку-
уме [281, 282]. Изотерма
адсорбции CF4 на этом образце
в области заполнения монослоя
имеет одну ступень, характер-
ную для адсорбции на физи-
чески однородных поверхностях
(рис. 11,15). Была получена
также довольно однородная по-
верхность марганца (рис. 11,16)
[281]. Изотерма адсорбции CF4
на наиболее чистой из получен-
ных поверхностей марганца
в начальной части обращена выпуклостью к оси давлений и имеет
ярко выраженные две ступени — при заполнении первого и второго
адсорбированных слоев. Окисление этого образца делает поверхность
неоднородной. После такого окисления начальная часть изотермы
адсорбции перфторметана поднимается гораздо круче, а "ступени
на изотерме в области преимущественного образования первого
и второго слоев сглаживаются. По-видимому, в значительной степени
однородны и поверхности образцов других металлов, полученных
и изученных в той же работе, так как изотермы адсорбции в области
преимущественного образования полимолекулярных слоев имеют
ярко выраженный ступенчатый характер.
В работах де-Бура, Касперсмы и ван-Донгена [272—274] иссле-
довалась адсорбция на поверхностях меди и никеля. Металлы были
получены восстановлением в токе водорода порошка чистых окислов.
В приведенном на рис. 11,17 примере адсорбции криптона на поро-
шке меди при температуре ниже температуры двухмерной конденса-
ции начальный участок изотермы адсорбции обращен выпуклостью
к оси давлений, что характерно для адсорбции на однородной поверх-
ности. Адсорбция в первом, втором и третьем слоях разграничена.
Однако при заполнении каждого из этих трех слоев на изотерме
58
появляются по три малых ступени, которые наиболее четко наблю-
дались в области преимущественного заполнения второго слоя.
Наличие этих малых ступеней авторы объясняют двухмерной кон-
денсацией криптона на различных гранях меди (111), (100) и (110),
которые могут выходить на поверхность кристаллов с гранецентри-
рованной кубической решеткой [273]. Возможно, что здесь сказы-
вается также влияние оставшегося на поверхности металла окисла.
Авторы указывают также на возможное влияние поляризации адсор-
бирующихся молекул поверхностью металла. Изотерма адсорбции
Рис. II»16j Изотермы адсорбции перфторметана
при 77,5 К:
1— на чистом марганце; 2 — на марганце, покрытом хемо-
сорбированным кислородом; 3 — на марганце, который
после хемосорбции кислорода был прокален в вакууме.
криптона на меди похожа на соответствующие изотермы на ионных
кристаллах (см. гл. II, разд. 4) и на расщепленном графцте
(рис. 11,2).
Изотермы адсорбции шестифтористой серы на кадмии, измеренные
при различных температурах [282], представляют типичный пример
перехода двухмерной газовой пленки в двухмерный конденсат на
физически однородной поверхности одной грани (рис. 11,18).
В работах [272, 274, 276, 285] определялись также изостери-
ческие теплоты адсорбции криптона и ксенона на поверхности метал-
лов. Точность этих определений еще недостаточна, чтобы сделать
окончательный вывод о характере изменения теплоты адсорбции
с ростом заполнения поверхности. Однако наблюдающееся прибли-
зительное постоянство теплот адсорбции [276] или даже их некото-
рый рост с ростом заполнения [274], а также сами величины теплот
адсорбции криптона на меди и никеле, составляющие от 3
до 5 ккал/моль, указывают на физическую адсорбцию на довольно
однородной поверхности.
59
Рис. 11,17. Изотерма
адсорбции криптона на
порошке меди при
77,5 К.
Рис. 11,18. Изотермы адсорбции шестифтористой серы на
образце кадмия при разных температурах (числа на кривых).
Таким образом, зти пока еще немногочисленные примеры ад-
сорбции паров на специально подготовленных поверхностях метал-
лов показывают, что, несмотря на особенности строения металлов
и наличие в них электронов проводимости в случае неспецифической
адсорбции, поверхности этих металлов ведут себя аналогично одно-
родным поверхностям других адсорбентов (графитированной тер-
мической сажи, некоторых ионных кристаллов). В дальнейшем
представляет большой интерес исследование на однородных поверх-
ностях металлов физической адсорбции молекул, способных к спе-
цифическому межмолекулярному взаимодействию без заметной хемо-
сорбции.
4. Ионные адсорбенты с гранями одинаковых
и разных индексов
Соли с кубической решеткой. Поверхности многих ионных кри-
сталлов в целом химически и физически неоднородны благодаря
наличию граней разных индексов. Однако даже и при адсорбции
на кристаллах с гранями одного индекса (например, грани (100)
кристаллов типа NaCl) благодаря наличию углов и ребер, а также
ступеней роста и дислокаций по мере заполнения монослоя при
низких температурах часто получаются изотермы адсорбции не
с одной, а со многими ступенями и с начальным участком, обращен-
ным выпуклостью к оси заполнения. Такие изотермы получены,
например, на хлористом калии и иодистом цезии [287], хлористом
натрии, хлористом калии и фтористом кальции [288, 289], хлори-
стом калии [290], фтористом литии [291], иодистом серебре (см.
обзор [130]), хлористом натрии и хлористом калии [292] и на фто-
ристом кальции [293]. Сведения о более ранних из этих работ при-
ведены в книгах Брунауэра [294], Ильина [295] и Янга и Крау-
эла [151].
В случае кубических кристаллов КС1, поверхность которых
образована в основном гранями (100), равномерно заселенными
ионами противоположных знаков, наблюдается рост теплоты ад-
сорбции [296]. Поверхность же ромбоэдрических кристаллов KCI
образована в основном гранями (111), одни из которых содержат
только ионы калия, а другие — только ионы хлора. В этом случае
теплота адсорбции с увеличением заполнения поверхности посте-
пенно уменьшается благодаря значительной неоднородности общей
поверхности. Поэтому адсорбционные свойства кристаллических
непористых адсорбентов сильно зависят от способа их пригото-
вления.
На рис. 11,19 представлены [116] изотермы адсорбции этана на
двух образцах хлористого натрия: в образце А присутствуют как
грани (100), так и грани (111), а в образце В преимущественно грани
(100) [116, 289]. Из рисунка видно, что образец А обладает более
неоднородной поверхностью, благодаря чему изотерма адсорбции
при малых заполнениях обращена выпуклостью к оси заполнения.
61
В области средних заполнений поверхности изотерма адсорбции
на этом образце круто поднимается, причем имеется точка перегиба.
Только после этого на изотерме обнаруживается ступень, соответ-
ствующая преимущественной адсорбции на грани одного индекса.
Поверхность образца В более однородна. Хотя в начальной чазти
изотермы адсорбции ца этом образце величины адсорбции, по-види-
мому, несколько завышены по сравнению с теми, которые можно
ожидать для адсорбции на гладкой грани (100), в области более
высоких заполнений поверхности имеется всего один вертикальный
участок. Крутой подъем в средней части изотермы адсорбции на этом
образце происходит приблизительно при том же давлении, что и у со-
ответствующей ступени изотермы адсорбции на образце А. Поэтому
эта ступень соответствует преимущественному заполнению грани
Рис. 11,19. Изотермы адсорбции этана при —183 °C
двумя образцами хлористого натрия:
А — при наличии граней двух индексов (100) и (111); В —
при наличии преимущественно граней (100).
В случае кристаллов имеется небольшое количество различных
мест на поверхности. Это грани разных индексов (обычно от двух
до шести), ребра между гранями, ступени роста, отдельные дисло-
кации. Различные места на поверхности кристалла ограничены
в основном немногими типами и поэтому здесь нет непрерывного
распределения энергии адсорбционных сил по местам адсорбции.
Вследствие этого число ступеней на изотерме адсорбции в пределах
преимущественного заполнения монослоя не должно быть очень
большим. При соблюдении надлежащих предосторожностей во время
синтеза и последующих обработок таких кристаллов число ступеней
на изотерме адсорбции должно находиться в соответствии с числом
выходящих на поверхность кристалла граней разных индексов [297,
298]. Если это условие не достигнуто, то число ступеней на изотерме
адсорбции должно находиться в соответствии с числом выходящих
на поверхность граней и с числом возможных ступеней роста, типич-
ных для данного кристалла и условий его получения.
62
Несмотря на эти трудности, удается получить кристаллы солей,
например бромистого натрия, с весьма однородной поверх-
ностью [299]. Примеры этого приведены в обзорах [116, 300]. Весьма
однородный образец бромистого натрия был получен возгонкой соли
в быстром токе очищенного инертного газа [301]. Изотерма адсррб-
ции криптона на этом образце имеет форму, близкую к ожидаемой
при этой температуре для однородной поверхности.
Получены образцы солей NaCl, KGlnRbGl с весьма однородной
поверхностью [302]. Высокодисперсные порошки были приготовлены
осаждением из охлажденного насыщенного раствора солей путем
быстрого вливания в него сухого этанола. Для удаления оставшейся
Рис. 11,20. Изотермы адсорбции криптона на хлори-
стом рубидии при разных температурах (числа на
кривых).
воды осадки многократно промывались этанолом и затем эфиром.
Полученные изотермы адсорбции криптона на образце RbGl при
нескольких температурах вблизи двухмерной критической темпе-
ратуры приведены на рис. 11,20. Эти изотермы имеют типичную
форму для адсорбции на физически однородной поверхности. Резкий
подъем изотермы адсорбции в области преимущественного заполнения
монослоя связан с двухмерной конденсацией на грани (100). Неболь-
шая выпуклость (по отношению к оси заполнений) в начальной части
изотерм адсорбции может быть связана с наличием небольшого
количества дислокаций, ступеней роста, других граней и контактов
между кристалликами.
Изучение адсорбции на кристаллах с простой кубической решет-
кой особенно важно для теории адсорбции, так как в этом случае
легко рассчитать создаваемое у поверхности адсорбента электро-
статическое поле [287, 295, 299, 303]. Простые кристаллы типа NaGl
представляют также и практический интерес для газовой хромато-
63
графин, например для разделения многоядерных ароматических
углеводородов [8]. У таких кристаллов все грани (100) одинаково
заселены катионами и анионами близких размеров. Поэтому такие
адсорбенты не обладают большой специфичностью адсорбционного
взаимодействия.
Соли со слоистой решеткой типа МХ2. К ионным адсорбентам
СО СЛОИСТОЙ структурой ОТНОСЯТСЯ хлориды, бромиды, ИОДИДЫ И СуЛЬ-
фиды многих металлов. Адсорбция изучалась на кристаллах NiCl2,
СоС12, CdCl3, CdBr2, Cdl2, CoBr2, РЫ2, MoS2. В этих солях слой
ионов металла располагается между двумя слоями из ионов хлора,
брома, иода или серы, в результате чего образуются тройные Слои.
Силы химической связи внутри тройных слоев значительно больше,
чем межмолекулярные силы, действующие между тройными слоями.
Эти структуры характеризуются координационными числами 6 и 3.
В структуре Cdl2 каждый третий слой анионов повторяет первый,
каждый четвертый слой, повторяет второй и т. д. К структуре Cdl2
относятся многие дииодиды, дибромиды и другие кристаллы зтого
типа.
В структуре CdCl2 тройные слои располагаются друг над другом
так, что только четвертый слой анионов повторяет первый, пятый
слой повторяет второй и т. д. Вследствие этого элементарная ячейка
CdCl2 ромбоэдрическая. Сюда относятся многие дихлориды. В отли-
чие от CdCl2 и Cdl2 координационный многогранник молибдена
в MoS2 представляет тригональную призму.
Поверхность базисной грани с кристаллографическим индексом
(0001) адсорбентов со слоистой решеткой типа МХ2 заселена ионами
одного типа, например ионами хлора. Наряду с гранью (0001) на
поверхность таких кристаллов выходят и другие грани, которые
содержат ионы М2+ и С1". Из рис. 1,6, где приведены изотермы
адсорбции ксенона на образцах NiCl2 [304], видно существенное
влияние недостаточно откачанной воды на форму изотермы. Изотерма
адсорбции имеет несколько ступеней благодаря выходу на поверх-
ность кристаллов нескольких граней. В случае же образца, из кото-
рого длительной откачкой удалена вода, в области преимуществен-
ного заполнения монослоя на всей поверхности адсорбента наблю-
дается двухступенчатая изотерма адсорбции. Такой вид изотермы
связан с адсорбцией на двух кристаллографических гранях этой
соли: базисной с индексом (0001), поверхность которой состоит
из плотно упакованных ионов хлора, и грани, содержащей М2+ и С1“.
Росс, Оливье и Хинчен [300] исследовали адсорбцию аргона
довольно неоднородным образцом бромистого кадмия (рис. 11,21).
Однако, пользуясь методом оценки степени неоднородности поверх-
ности (см. разд. 3 гл. IV), авторы разделили сложную изотерму
адсорбции аргона на этом образце бромистого кадмия на две: одну —
для адсорбции на неоднородной части поверхности (кривая 7) и дру-
гую — для адсорбции на ее однородной части (кривая 2) (см.
рис. 11,21). Таким образом, измеренная величина адсорбции была
представлена в виде суммы величины адсорбции на однородной
64
и неоднородной частях поверхности этого образца при том же давле-
нии в газовой фазе. Это важный пример выделения из сложной экс-
периментальной кривой, полученной на физически неоднородном
образце, изотермы адсорбции на соответствующей физически одно-
родной поверхности.
Исследование адсорбции благородных газов на кристаллах солей
слоистой структуры (CdCl2 и Cdl2) было проведено Ляре и Нардо-
ном [301, 305]. Образцы с весьма однородной поверхностью были
получены возгонкой солей в быстром потоке очищенного инертного
газа при 500—700 °C. Удельные поверхности полученных адсорбен-
тов составляли от 15 до 35 м2/г.
Рис. П,21. Изотерма адсорбции аргона на броми-
стом кадмии при —195 РС (точки, кривая 5).
Кривые 1 и 2 — рассчитаны соответственно для
адсорбции па неоднородной и однородной частях
поверхности этого образца.
Изотермы адсорбции криптона на образце NiCl2, как в области
преимущественно мономолекулярной, так и в области преимуще-
ственно полимолекулярной адсорбции, изображены на рис. 11,22
и 11,23 [301]. Эти изотермы начинаются участками, обращенными
выпуклостью к оси давления газа, и при дальнейшем росте запол-
нения проходят ярко выраженные ступени. Однако в этих работах
пе указана масса и удельная поверхность образца, поэтому нельзя
точно сказать, чему соответствует вторая ступень изотермы адсорб-
ции (рис. 11,23). Эта ступень наблюдается при довольно большой
величине pipa 0,4—0,5 и может поэтому соответствовать как
заполнению преимущественно мономолекулярного слоя на поверх-
ности грани с малой энергией адсорбции, так и заполнению преиму-
щественно второго слоя на поверхности грани с большой энергией
адсорбции. Изотермы адсорбции аргона и ксенона на образце CdCl3
имеют подобный вид. Аналогичные результаты были получены ван-
Донгеном [278].
Образцы подобных солей были получены также прямым осажде-
нием из водных растворов. Полученный так образец РЫ2 с удельной
поверхностью 0,3 м2/г был весьма однороден [301].
Адсорбенты со слоистой структурой типа МХ2 представляют
интерес как для теории адсорбции, так и для применений в газовой
5 Заказ 1180 65
хроматографии, так как на их поверхности расположены отрица-
тельно заряженные ионы галогенов и они ведут себя как специфиче-
ские адсорбенты третьего типа [8, 9, 129, 130]. Это было обнаружено
Рис. 11,22. Изотермы адсорбции криптона на кристал-
лах NiCl2 в области малых и средних заполнений по-
верхности при разных температурах (числа на кривых).
при газохроматографическом исследовании адсорбции молекул
групп А, В и D на таких адсорбентах [306].
Адсорбция на MoS2 изучалась в работах [267, 307—311]. Полу-
чение однородной поверхности MoS2 затруднительно в связи со
I I I I , I
го ьо 60 оо юо # по.
Рис. П,23. Изотермы адсорбции криптона на кри-
сталлах NiClg в области высоких заполнении по-
верхности при разных температурах (числа на
кривых).
способностью к окислению. Однако в работе [308] была получена
изотерма адсорбции н-гептана на MoS3, обращенная к оси давлений
выпуклостью, как и на графитированной термической саже.
66
Соли с комплексными анионами. Большей специфичностью меж-
молекулярного взаимодействия при адсорбции по сравнению с рас-
смотренными выше обладают кристаллы, образованные сравнительно
небольшими по размерам многозарядными катионами и большими
но размерам комплексными анионами. По приведенной в гл. I клас-
сификации такие адсорбенты относятся к специфическим адсорбентам
второго типа. Однако в этом случае поверхность кристалла в целом
может быть физически неоднородна. Например, в кристалле серно-
кислого бария кроме в основном встречающейся грани (001) воз-
можен выход на поверхность граней индексов (110), (102), (ОН)
и (104) [312]. Изотермы адсорбции и зависимость теплоты адсорбции
от заполнения поверхности такого адсорбента определяются стати-
стическим распределением молекул адсорбата по всем этим граням.
Это распределение зависит от различий по величине и специфичности
потенциалов адсорбционных сил на разных гранях, площади поверх-
ности этих граней и температуры [313—316]. Существенное увели-
чен ио однородности поверхности кристаллов малорастворимых
со лей, например сернокислого бария, достигается при их термо-
обработке, а также при предварительной промывке растворами
Hopiniio растворимых солей с одноименными ионами [317].
Танин сильно специфические адсорбенты применяются в газовой
хроматографии для разделения молекул, близких по размерам,
пннфи! урпции и многим физическим свойствам, но различающихся
лопальным распределением электронной плотности. На сульфате
Ларин, нанримвр, хорошо рапдалиется смесь изомеров ксилола,
причем парным иыходит н ксилол, патом м-ксилол, потом о-кси-
лол (Jltrt|; пики практически симметричны. На рис. 11,24 показана
uahiiciiMociii дифференциальной теплоты адсорбции насыщенных,
ненасыщенных и ароматических углеводородов для малой (нулевой)
пробы г/г7Т| от| числа атомов углерода в молекуле. Из этого рисунка
видно, что значения qv,i при адсорбции цикленов и ароматических
уь'пчюдороднп пн BaSO4 значительно выше значений qVil при ад-
сорбции н алканов и цикланов с тем же количеством атомов углерода
в молекуле. Это указывает на сильную специфичность адсорбции
циклонов и ароматических углеводородов на таком адсорбенте.
Теплоты. адсорбции ксилолов заметно различаются между собой
и соответствуют последовательности выхода пиков на хроматограмме.
Кроме рассмотренных непористых кристаллических ионных ад-
сорбентов весьма важны микропористые кристаллические ионные
адсорбенты, обладающие каркасной структурой, и среди них цеолиты
(гм рис. 1,2 и 1,3) [318, 319].
Поскольку специфическое взаимодействие с молекулами групп В
И 1> при небольших заполнениях полостей цеолита происходит
а веноинпм с катионами, то на адсорбцию таких молекул в сильной
। Г1Ч1ННИ влияют заряд, радиус, расположение и подвижность кати-
онов на поверхности (на ребрах) полости цеолита (рис. 11,25). С мо-
пшц 1пмп же группы А, такими, как благородные газы или насы-
67
щенные углеводороды, цеолиты взаимодействуют неспецифи-
чески. На рис. 11,26 [320] приведена зависимость дифференциальной
теплоты адсорбции разных молекул qv при величине адсорбции
около одной молекулы на большую полость цеолита HLiNaX от
। i 1. <—t—r
fOfl
Рис. 11,24. Зависимость дифферен-
циальной теплоты адсорбции qv г
для ряда углеводородов на сульфате
бария от числа атомов углерода
в молекуле п:
1 — н-алканов; 2 — циклогексена; 3 —
циклогексадиена-1,3; 4 — бензола; 5 —
толуола; 6 — этилбензола; 7 — п-ксилола;
8 — лс-ксилола; 9 — о-ксилола.
Рис. 11,25. Модель большой полости
цеолита X с расположенными в ней
различными катионами.
На рисунке изображены катионы, располо-
женные в местах Su — около шестичленных
кислородных колец алюминий* и кремний-
кислородных тетраэдров и на местах 8Ш —
около четырехчленных кислородных колец
алюминий* и кремний-кислородных тетраэд-
ров.
поляризуемости этих молекул, относящихся к группам А и В. Этот
график аналогичен представленному на рис. 11,24 и позволяет по
разности величин qy для адсорбции молекул групп В и А оценить
вклад энергии специфических межмолекулярных взаимодействий
молекул группы В с цеолитом в их общую теплоту адсорбции. Линия,
проведенная через точки, соответствующие величинам qv для «-ал-
канов, характеризует теплоту адсорбции цеолитом неспецифически
адсорбирующихся молекул группы А. Стрелками отмечены вклады
специфического взаимодействия с цеолитом в дифференциальную
теплоту адсорбции приведенных на рис. 11,26 молекул группы В [9,
129, 130, 321].
Уменьшения различий в напряженности электростатического
поля в разных точках над поверхностью остова цеолита данного
длина и с данным отношением Si : Al (чему в обычном состоянии соот-
ветствует определенное количество катионов) можно осуществлять
двумя путями. Во-первых, можно проводить в цеолите замену кати-
онов большого заряда и малого радиуса на однозарядные катионы
большого радиуса. Во-вторых, можно уменьшать концентрацию
катионов на поверхности путем частичного декатионирования цео-
лита (без заметных искажений структуры остова). При этом энергия
молекулярной (физической) адсорбции, как правило, уменьшается.
Влияние на величины дифференциальных теплот адсорбции
молекул этана и этилена и на зависимость qy от заполнения полостей
68
цеолитов перехода от катионов с малыми радиусами к катионам
с большими радиусами в ряду цеолитов — LiNaX, NaX и KNaX —
видно из рис. 11,27. Замена Na+ на Li* приводит к увеличению
вклада энергии специфических взаимодействий в теплоту адсорбции
Рис. 11,26. Дифференциаль-
ные теплоты адсорбции qv
цеолитом HLiNaX (при
величине адсорбции около
1 молекулы на большую
полость цеолита) н-алканов
И циклопентана (молекулы
группы А) и некоторых
молекул группы В (указаны
около точек).
<'гур л нами отмечены величины
йййада специфического взаимо-
АеЙеТМЯ о цеолитом в теплоту
адеороцки молекул группы в.
II й оои абсцисс — средняя
лоллриауемоеть молекул а.
отилрпп и к более резкому падению теплот адсорбции обоих угле-
водородов с ростом заполнения, а замена Na+ на К* приводит
к уменьшению специфического взаимодействия этилена с цеолитом
И к росту теплот адсорбции с увеличением заполнения [322].
Pin', 11,27. Зависимость дифференциальных теплот адсорбции этана qv (белые
Точки) в этилена (черные точки) от величины адсорбции цеолитами LiNaX, NaX
и KNaX.
Ивотормы адсорбции, обращенные вначале выпуклостью к оси
дввлоии* и проходящие затем точку перегиба, были получены для
цеолитов при адсорбции паров криптона [323], ксенона (рис. 11,28
10241), фосфора [325], этана [322], пропана [326] и хлористого
аммония [327].
Причинами локальной неоднородности цеолитов могут быть
химические и фазовые примеси.
Исследование адсорбции цеолитами, так же, как и непористыми
ионными кристаллами, наряду с практическим значением для хро-
Рис. 11,28. Изотермы адсорбции ксенона
цеолитом HLiNaX при разных темпера-
турах (числа на кривых).
матографии, молекулярно-
ситовых и адсорбционных
разделений смесей, осушки
и катализа (см. обзоры
[319, 328—330]) представляет
большой интерес для моле-
кулярной теории адсорбции,
поскольку в этом случае
можно изменять взаимодей-
ствия адсорбирующихся мо-
лекул с адсорбентом в
заранее заданных направле-
ниях [319, 331, 332]. Термо-
динамике и молекулярно-
статистической теории ад-
сорбции на цеолитах будет
посвящена отдельная моно-
графия этой серии. Спектро-
сорбции различных молекул
монографии А.*В- Киселева
скопические проявления ад-
цеолитами описаны в предыдущей
и В. И. Лыгина [333].
5» Кристаллические окислы. Лед
Указанные выше причины неоднородности поверхности ионных
и молекулярных кристаллов относятся также и к кристаллическим
окисным адсорбентам, таким, как окись магния, анатаэ, рутил,
кварц и др. В этом случае часто возникают дополнительные ослож-
нения из-за химической неоднородности поверхности, так как де-
гидроксилированные окислы легко хемосорбируют воду, в результате
чего на поверхности образуются гидроксилированные участки, кото-
рые при дальнейшей откачке перед опытами по адсорбции частично
снова дегидроксилируются. Очень большое значение в этих случаях
имеют примеси. В частности, примеси алюминия или бора на поверх-
ности кремнезема создают сильные кислотные центры, вызывающие
хемосорбцию многих органических оснований (см. обзоры [333—
335]).
Кристаллы окиси магния обладают простой кубической решет-
кой. У окиси магния, так же как у NaCl, поверхность образована
в основном гранями (100). Размеры ионов Mg2+ и О2- близки, а за-
ряды равны. Поэтому специфичность взаимодействия в случае ад-
сорбции на окиси магния невелика [11]. Однако благодаря некото-
рому количеству оставшихся после откачки гидроксильных групп
на поверхности окиси магния и другим ее неоднородностям, при
70
малых заполнениях наблюдались повышенные теплоты адсорб-
ции [336].
Поверхности окислов, содержащих гидроксильные группы, при
нагревании в той или иной степени дегидроксилируются и поэтому
они более неоднородны по отношению к адсорбции молекул групп В
и D, способных к образованию водородной связи с поверхностными
гидроксильными группами. По отношению к неспецифической ад-
сорбции молекул группы А они могут вести себя как значительно
более однородные [334, 335]. С этой точки зрения представляет
интерес изотерма адсорбции криптона на гидроокиси натрия. Обра-
зец гидроокиси натрия был получен окислением кислородом тонкой
пленки металлического натрия и по-
следующей обработкой паром во- 0,20г 2
ды [337]. Изотерма адсорбции на J
этом образце (рис. 11,29) вначале о,15- /
обращена выпуклостью к оси давле- §
ния газа, а затем проходит точку | дш - а
перегиба, что характерно для одно- г
родной поверхности. 'V д05 _
Поверхность аморфных окислов- ’ £
ксерогелей: силикагелей, алюмоге- J < i t i t I _ > i
лей, силикаалюмогелей, феррогелей °>2 0,6 0,8 р/р0
и других ксерогелей с высокой
удельной поверхностью — обычно Рис* И,29. Изотерма адсорбции
более неоднородна. Аморфные кРиптона на гидроокиси натрия
окислы образуются, как правило,
в процессе полимеризации, поэтому
им присуща неоднородность, связанная с упаковкой макромолекул
и взаимодействием между ними. Дегидратация и дегидроксили-
рование объема и поверхности аморфных окислов представляют
собой сложные и часто необратимые процессы, а степень дегидра-
тации и дегидроксилирования как раз и определяет свойства
окислов как специфических молекулярных адсорбентов. Кроме
этого, наличие примесей может сильно влиять на кислотные и основ-
ные свойства поверхности окислов, а следовательно, на их ионо-
обменные, хемосорбционные и молекулярно-адсорбционные (осо-
бенно при небольших заполнениях), а также на каталити-
ческие свойства [333—335, 338—343].
Химия поверхности аморфных окислов и адсорбция на них раз-
ных молекул частично рассмотрены в предыдущих книгах этой се-
рии [9, 333]. Аморфные объемно- и поверхностно-пористые окислы,
применяемые в газовой и жидкостной хроматографии, будут рас-
смотрены А. В. Киселевым и Ю. С. Никитиным в одной из следу-
ющих монографий этой серии.
Кристаллы льда образуют структуру, сходную со структурой
алмаза, но упаковка молекул воды в кристалле льда довольно рых-
лая благодаря тому, что в этом случае тетраэдрическая структура
образуется направленными водородными связями. При исследовании
71
адсорбции пара азота при —195 °C на кристаллах льда, пригото-
вленных быстрым охлаждением, а также на кристаллах двуокиси
углерода и метанола [344] были получены изотермы адсорбции,
характерные для неоднородной непористой поверхности. Образова-
ние кристаллов с неоднородной поверхностью в этой работе быдо,
по-видимому, связано со стремлением получить высокодисперсные
кристаллы с большой удельной поверхностью для облегчения изме-
рений адсорбции вакуумным статическим методом. Повышение тем-
пературы опыта от —195 °C до —50 и —30 °C в случае кристаллов
льда приводит к миграции молекул воды и уменьшению удельной
поверхности. Поверхность, по-видимому, становится при этом более
однородной [345].
Рис. 11,30. Изотермы адсорбции н-гексана на льду (а), на жидкой
воде (б) при различных температурах (числа на кривых).
Адсорбция н-алканов исследовалась как на охлажденных до —50
и —30 °C кристаллах льда [345], так и на жидкой воде [346, 347]
(рис. 11,30). Начальный ход изотерм адсорбции в обоих случаях
типичен для адсорбции на однородной поверхности с малой энергией
взаимодействия адсорбат — адсорбент. Были также рассчитаны те-
плоты адсорбции на льду [345], однако точность этих результатов
невелика.
Лед также представляет интерес как один из наиболее тонко-
пористых адсорбентов [48]. Структура льда является примером
соединений с водородными связями, а водородная связь является
направленной связью. Поэтому структура таких кристаллов часто
не подчиняется правилам плотнейших шаровых упаковок. Между
молекулами воды в структуре льда образуются правильные пустоты,
имеющие размер молекул воды. Значительная адсорбция СО2 льдом
объяснена проникновением молекул СО2 в эти пустоты с образова-
нием клатратных соединений [348].
72
6. Пути улучшения однородности поверхности
некоторых адсорбентов
Молекулярно-ситовые угли, углеродные волокна и мембраны,
^тивированные угли, получаемые из природных веществ, обычно
Неоднородны как по химическому составу, так и по пористости.
Угли, получаемые при термическом разложении некоторых поли-
меров, например поливинилиденхлорида (31, обладают значительно
Рис. 11,31. Зависимость дифференциальной теплоты
адсорбции при малом (нулевом) заполнении qv х от
поляризуемости молекул адсорбата а при адсорбции
ряда молекул, относящихся к группам А и £ (ука-
заны у точек) на угле Саран (I) и угольном молеку-
лярном сите С (2),
более тонкой и однородной пористостью и более химически однород-
ной поверхностью, почти не несущей окислов. По отношению к ад-
сорбции О2, Na, СО, СН4, С2На, С2Н4 и СОа такой уголь ведет себя
Как химически и геометрически довольно однородный адсорбент,
КО проявляющий специфических взаимодействий [31. Хроматогра-
фические пики этих, компонентов для небольших проб получаются
довольно симметричными. Однако пики воды, аммиака и двуокиси
серы несимметричны [3] вследствие наличия на поверхности такого
угля небольшого количества химически активных кислородных
Комплексов. Более однороден и неспецифичен уголь, полученный
Ив чистого поливинилиденхлорида в условиях, исключающих окис-
ленио [5].
На рис. 11,31 приведена зависимость qvti от поляризуемости а
для адсорбции ряда молекул, относящихся к разным группам А и В
Ям молекулярно-ситовом угле Саран и на угольном сите С [7,349].
Ио рисунка видно, что величины ~qVix при адсорбции молекул
73
группы В (02, N3, СО, NO, СО2, H2S, S02) почти не превышают
величины qv х для адсорбции молекул группы А (Аг, Кг, Хе, СН4,
С2Нб, С3Н8 и С4Що)« Эти угли не проявляют специфического вза-
имодействия с молекулами группы В. Величины qV} г для этих
углей много выше, чем для непористой графитированной сажи (см.
ниже рис. V,1 и V,13), что является следствием увеличения энергии
адсорбции в тонких порах угля.
Рис. П,32. Изотермы адсорбции криптона при 77,3 К:
1 — на углеродной мембране (1000 °C); 2 — на углеродной
мембране (2800 °C); 3 — на углеродном волокне WYB (2500—
3000 вС); 4— на углеродном волокне AG-12 (2500 °C); 5 — на рас-
щепленном графите, полученном при применении SbCl8; на рас-
щепленном графите, полученном при применении FeCl, (сплош-
ная кривая) [256].
В скобках приведена температура графитирования соответству-
ющих образцов.
Описаны разные способы получения молекулярно-ситовых
углей [1—4, 350]. Молекулярно-ситовой уголь 4А приготовляется,
например, полимеризацией фурфурола и дальнейшей карбониза-
цией полимера при нагревании. Молекулярно-ситовые угли 4А,
5А и 6А [350] обладают большой разделительной способностью
благодаря их молекулярно-ситовому действию. Важные преиму-
щества молекулярно-ситовых углей перед цеолитами заключаются
в слабой адсорбции воды, что позволяет применять их для разделения
влажных газов, и в их устойчивости к кислым газам.
Пути улучшения однородности молекулярно-ситовых углей сво-
дятся к подбору исходного мономера, его очистке, нахождению
оптимальных условий полимеризации, обработки полимера и карбо-
низации, при которых получается возможно более химически чистый
углеродный скелет с возможно более однородными порами [5—7].
74
Получены также волокнистые углеродные материалы — углерод-
ные волокна, получаемые из полиакрилонитрила и гидратцеллюлоз-
ного во*локна [351—353]. В процессе карбонизации и дальнейшего
графитирования (при температурах до 2000 °C) углеродное волокно
обогащается углеродом. Величина удельной поверхности при графи-
тировании уменьшается, часть тонких пор при этом закрывается.
^Благодаря значительному увеличению кристаллитов и удалению
окислов поверхность углеродного волокна становится более одно-
родной, менее подверженной окислению и термостойкой. Адсорбция
и хроматография на углеродных волокнах изучены еще мало (см.,
например, [256, 354—358]), однако исследование адсорбции пара
криптона на графитированных волокнах [256] показало, что‘по-
верхность некоторых из них приближается к однородной (рис. 11,32).
Особенно однородной поверхностью обладала углеродная мемб-
рана [256], полученная из природного графита следующим обра-
зом [359]. Природный графит превращался в окись графита обра-
боткой смесью азотной кислоты и хлората калия или смесью серной
кислоты, перманганата калия и небольшого количества нитрата
натрия. Полученная вязкая суспензия окиси графита восстанавли-
валась гидразином или иодистым водородом и подвергалась медлен-
ному нагреванию в атмосфере азота до 250 °C. При дальнейшем
постепенном нагревании до 1000 ®С образовывалась мембрана, содер-
жащая 99% углерода.
Нагрев этой мембраны до 2800 *С приводит к образованию очень
однородной поверхности. На рис. П,32 сопоставлены изотермы
адсорбции криптона на такой поверхности, на образцах расщеплен-
ного графита с однородной поверхностью и графитированных угле-
родных волокнах. Из рис. 11,32 видно, что по однородности поверх-
ности полученная углеродная мембрана приближается к наиболее
однородным образцам расщепленного графита [51, 52, 256] и графи-
тированной термической сажи [52].
Пористые полимеры. Пористые полимеры представляют собой
обширную группу адсорбентов и находят важные применения в газо-
вой и жидкостной хроматографии (см. работы и обзоры [360—370])
и для очистки влажной атмосферы от вредных газов [371—373].
Изменяя условия синтеза сополимера стирола с дивинилбензолом,
регулируя природу и количество растворителя, можно в некоторых
пределах регулировать структуру пор. Варьируя участвующие в син-
тезе мономеры и последующие химические обработки, можно полу-
чать на основе сополимеризации с дивинилбензолом как малоспе-
цифические адсорбенты (хромосорбы 101 и 102, порапаки Q, Р, Q-s9
полисорб-1), так и специфические адсорбенты третьего типа, то есть
содержащие те же функциональные группы, что и молекулы группы В
(хромосорб 104, порапаки S, N, Т, полисорбат, макропористые
аниониты) [362—365, 367—374].
При хроматографическом разделении компонентов воздуха на
норапаке Q-s (при —82 °C) наблюдается порядок выхода газов
ни колонны, соответствующий росту поляризуемости этих газов,
75
что характерно для неспецифически адсорбирующей углеводородной
поверхности (рис. П,12б [374]).
Большим недостатком многих пористых полимеров является
низкая термостойкость и сильное удерживание углеводородов. Угле-
водороды легко проникают внутрь таких адсорбентов в пространства
между макромолекулами. В меньшей степени это проявляется в слу-
чае полиакрилонитрила [375]. Высокой термостойкостью обладают
пористые полиарилаты [376]. Получение более жестких однородно-
макропористых структур и введение в синтез или применение при
прививках разнообразных органических и элементорганических
мономеров, вероятно, даст возможность иметь наборы довольно одно-
родных адсорбентов с разной специфичностью межмолекулярногр
взаимодействия с газами и жидкостями. Хроматограммы показы-
вают, что на многих уже полученных макропористых сополимерах
с разными функциональными группами пики молекул, относящихся
к группам А, В и D, при малых дозах адсорбата симметричны [374].
Одним из путей улучшения однородности поверхности полимеров
является рассматриваемое ниже нанесение полимера в виде тонкого
слоя на поверхность непористого или макропористого адсорбента-
носителя с жестким скелетом.
Адсорбенты с нанесенными на поверхность модифицирующими
слоями. Помимо прямого синтеза или термической обработки адсор-
бентов для получения близкой к однородной поверхности возможен
и другой путь — адсорбционное или химическое модифицирование
неоднородной поверхности адсорбента-носителя. Обычно такое моди-
фицирование приводит к уменьшению энергии адсорбции вследствие
экранирования адсорбента-носителя. Поэтому для применений, на-
пример в газовой хроматографии, удельная поверхность адсорбента-
носителя должна быть достаточно велика.
Пренслоу и Хэлси [32] применяли адсорбционное модифициро-
вание графитированной сажи нанесением на нее плотных слоев
молекул ксенона, который сначала адсорбировался из газовой фазы,
а потом сильно охлаждался вместе с адсорбентом-носителем. При
этом модификатор — ксенон (предадсорбированное вещество) нано-
сился на поверхность сажи в количестве, соответствовавшем от
одного до шести молекулярным слоям. Поскольку при таком моди-
фицировании концентрация силовых центров на поверхности резко
уменьшается (валентные расстояния между атомами углерода в ба-
зисной грани графита заменяются ван-дер-ваальсовыми рассто-
яниями между атомами ксенона в его мономолекулярном слое),
энергия неспецифического взаимодействия адсорбат — адсорбент
резко уменьшается. В соответствии с этим изотермы адсорбции пара
аргона на плотном мономолекулярном слое ксенона, нанесенном
на графитированную сажу, типичны для адсорбции на близкой
к однородной поверхности с малым адсорбционным потен-
циалом (рис. 11,33). По сравнению с изотермой адсорбции аргона
на исходной графитированной термической саже (см. также рис. П,9)
изотерма адсорбции аргона на плотном монослое обладает значи-
76
только меныпим наклоном. Эта изотерма обращена сначала выпук-
лостью к оси абсцисс, а крутой ее подъем с точкой перегиба сильно
сдвинут вправо, в сторону больших р. Образец, на котором нанесено
количество ксенона, достаточное для образования в среднем шести
Плотных слоев молекул ксенона,,более неоднороден. Это вызвано,
по-видимому, тем, что при за-
мораживании полимол окуляр-
ного слоя ксенона на поверх-
ности остается только мономо-
Лекулярный слой и кроме него
образуются объемные кристал-
лы ксенона (теория кристалли-
заций полимолекулярных слоев
адсорбата при охлаждении си-
стемы адсорбент — адсорбат и
изменение теплоемкости такой
системы рассмотрены в рабо-
те [377]). Исследование тепло-
емкости аргона, адсорбирован-
ного при низкой температуре
на мономолекулярном слое
ксенона, нанесенном на графи-
тиронлнпую сажу, свидетель-
ствует о том, что такой слой
цодот себя как однородный ад-
сорбент [33],
Исследовалась также адсорб-
ция аргона при 77,8 К (ниже
температуры его двухмерного
фазового перехода) на слоях
втилонл, предварительно ад-
сорбированных на графитиро-
ванной термической саже [35]
На изотерме адсорбции аргона
была получена четкая ступень
В области мономолекулярного
заполнения, свидетельствующая
Рис. 11,33. Изотермы адсорбции аргона
при 73,5 К:
а — на графитированной термической саже
Sterling FT, 2700 °C (область заполнения пер-
вого слоя выделена справа); б — на плотном
мономолекулярном слое ксенона, нанесенном
на эту сажу; в — на ксеноне, нанесенном на
эту сажу в количестве, достаточном для об-
„ разевания в среднем шести ' плотных слоев,
о довольно высокой однородно-
сти тонкого слоя этилена и об
уменьшении однородности по мере увеличения количества пред-
ВДсорбированного этилена (рис. 11,34). По-видимому, и в этом случае
избыток этилена при сильном охлаждении переходит в объемные
кристаллы [377], что увеличивает неоднородность поверхности.
Исследования адсорбции азота и аргона при 77,8 К на пред-
йдсорбированных на разных адсорбентах-носителях (плавленом
кварце, исходной и графитированной сажах) слоях бензола, мета-
кола и воды (наносилось до 5—8 слоев этих молекул) показали, что
в случае кварца и исходной сажи получаются неоднородные поверх-
77
пости [378, 379]. В этих случаях глубокое охлаждение толстых
адсорбционных слоев приводит к образованию объемных кристаллов,
а не к замерзанию слоя целиком [377]. В случае же предадсорбции
на графитированной саже метанола в количестве 1,5—2 слоев полу-
чалась поверхность, близкая к однородной. При предадсорбции
воды на графитированной саже в количестве, достаточном для обра-
зования нескольких слоев, плотного замороженного слоя не полу-
чалось [379]. Это также вызывается объемной кристаллизацией
нанесенной воды [377].
Рис. 11,34. Изотермы адсорбции аргона при 77,8 К на
слоях этилена, предварительно нанесенных на графи-
тированную термическую сажу Sterling FT, 2700 ?С при
количестве слоев этилена:
1 — 2; я — 6; J — 26.
Большой интерес представляет исследование адсорбции на слоях
плоских молекул или на слоях соответствующих молекулярных
кристаллов, нанесенных на адсорбент-носитель. На поверхности
таких кристаллов резко различаются расстояния между атомами,
связанными химически в одной молекуле, и расстояния между ато-
мами разных молекул. В работах Гьошона, Видаль-Мадьяр, Куз-
нецова и Франкена [29, 30, 380—383] изучалась газовая хромато-
графия на слоях плоских молекул бензофенона, антрахинона, фта-
лоцианина и фталоцианинов комплексообразующих металлов, нане-
сенных на графитированную термическую сажу. Слои бензофенона
и антрахинона приготовлялись путем адсорбции из растворов с по-
следующим выпариванием растворителя и образованием некоторого
избытка кристаллов органического вещества сверх адсорбирован-
ного плотного монослоя на поверхности графитированной сажи.
Фталоцианин и фталоцианины большинства металлов, кроме фтало-
цианина лития, плохо растворимы. Поэтому вначале растворялся
в спирте фталоцианин лития, а затем этот раствор подвергался
78
действию серной кислоты и в этот момент добавлялась сажа. В ре-
зультате начинал осаждаться фталоцианин, образуя, по-видимому,
на поверхности графитированной сажи плотный монослой плоско-
ориентированных адсорбированных молекул и некоторый избыток
объемных кристаллов. Действительно, фталоцианины наносились
на графитированную сажу в количестве 5 и 10 вес. %, что при равно-
мерном покрытии поверхности сажи соответствовало бы приблизи-
тельно 10—20 плотным молекулярным слоям. Поэтому в этих слу-
чаях на поверхности графитированной термической сажи кроме
Рис. 11,35. Схематическое изображение плоского слоя
молекул фталоцианина, нанесенных на графитирован-
ную сажу.
адсорбированных плоских молекул фталоцианинов образовывались
также и объемные кристаллики избытка фталоцианинов. Поверх-
ность самих молекулярных кристаллов фталоцианинов в их а- и р-мо-
дификациях довольно неоднородна [384]. Ориентирующее действие
адсорбента-носителя позволяет получить более однородные поверх-
ности плотных монослоев.
Нанесение на поверхность графитированной термической сажи,
обладающей высокой концентрацией силовых центров (атомов угле-
рода базисной грани графита), слоя фталоцианина (рис. 11,35) резко
уменьшает среднюю концентрацию силовых центров на поверхности.
Это приводит к снижению энергии адсорбции. Так, теплота адсорб-
ции диэтилового эфира на графитированной термической саже при
милом заполнении поверхности составляет 37 кДж/моль, а на по-
верхности нанесенного фталоцианина — лишь 29 кДж/моль [291.
Адсорбция на монослоях фталоцианина и на его кристаллах
представляет также интерес благодаря наличию в молекулах фтало-
цианина разных функциональных групп, в частности, групп — N=:
и Nil—. Эти группы находятся в сопряжении, что способствует
79
более равномерному распределению электронной плотности во фтало-
цианиновом кольце. При плоском расположении молекул фтало-
цианина на адсорбенте-носителе потенциал адсорбционных сил при
перемещении вдоль такой поверхности изменяется определенным
образом над молекулой фталоцианина и от молекулы к молекуле.
Таким образом, поверхность такого слоя молекул фталоцианина
должна быть геометрически, химически и физически однородной,
хотя ее нельзя считать математически однородной. Специфичность
такого адсорбента можно изменять, используя фталоцианины разных
металлов — меди, цинка и кобальта [380—382]. Что касается объем-
ных кристаллов фталоцианина, то грани, образованные в основном
звеньями =СН— и — N=, обладают, по-видимому, значительно
более слабой энергией адсорбции по сравнению с гранями, содержа-
щими атомы металла, так как способные к специфической адсорбции
центры — N= лежат на некоторой глубине мёжду углеводородными
группами и, вероятно, трудно доступны для многих молекул [384].
Кроме практического значения для газовой хроматографии изуче-
ние адсорбции на фталоцианинах представляет интерес для молеку-
лярной теории адсорбции, так как для этих кристаллов возможен
расчет потенциальных функций адсорбированных молекул [382].
Для адсорбционного модифицирования применяются также моно-
слои полимеров [31, 385]. Во многих случаях адсорбированные на
адсорбенте-носителе монослои полимеров проявляют значительно
большую однородность по отношению к различным молекулам, чем
сами пористые полимеры. Это относится, например, к полиарилату,
нанесенному тонким слоем на силохром [386].
Помимо нанесения модифицирующих поверхность плотных слоев
адсорбированных веществ уменьшить неоднородность поверхности
адсорбента можно нанесением значительно меньших количеств (не-
сколько % от количества, нужного для заполнения плотного моно-
слоя) таких сильно адсорбирующихся веществ, которые сами слабо
адсорбируют молекулы газов и паров других веществ. Эти пред-
адсорбированные вещества экранируют наиболее неоднородные части
поверхности данного адсорбента. Так можно улучшить однородность
поверхности гранул из графитированных саж нанесением на эти
сажи небольших количеств полимера [387]. Благодаря тому что
энергия взаимодействия адсорбирующихся молекул с поверхностью
таких добавок значительно ниже знергии взаимодействия с поверх-
ностью собственно графитированной сажи, адсорбция на таком
адсорбенте происходит в основном на оставшейся свободной наиболее
однородной части поверхности сажи.
Улучшения химической однородности поверхности и механиче-
ской прочности гранул из графитированной сажи можно добиться
скреплением частиц сажи углеродом при пиролизе бензола [388].
Создание довольно однородной поверхности модификатора на
адсорбенте-носителе можно осуществить также расплавлением пред-
варительно осажденного на адсорбент-носитель вещества, например,
органического вещества [389] или соли [390, 391].
80
Значительного улучшения однородности поверхности адсорбентов
иногда можно достичь ее химическим модифицированием. При этом
можно использовать блокировку только особо активных центров,
Йанример сильных акцепторных центров, применяя соответству-
ющие органические основания 1392]. Можно произвести и более полное
химическое модифицирование поверхности адсорбента, используя
реакций с находящимися на ней ионами [393] или функциональными
Группами [306, 394, 3951, и получить довольно однородные адсор-
бенты с различной специфичностью [396].
ЛИТЕРАТУРА
1. Е v е г е tt D. Н.; Decay Н. R., F е n d 1 е у J. A. In: The Structure
and Properties of Porous Materials. Eds. D. H. Everett, F. Stone. London.*
1Й58. 389 p.
2. Marsh H., Wynne-Jones W. F. K. Carbon, 1964, v. 1, p.
269-280.
3. ГвоздовичТ. H., Киселев А. В., Яшин Я. И. Нефтехимия,
1968, т. 8, с. 476-480.
4. М a h a j а п О. Р., W а 1 k е г Р. L. J. Col. Interface Sci., 1969, v. 29,
p. 129—137.
5. Kaiser R. Chromatographia, 1969, v. 2, p. 453—461; 1970, v. 3, p. 38—43.
6. В о u c he r E. A., Everett D. H. Trans, Faraday Soc., 1971, v. 67,
p. 2720-2725.
7. Д олова И. А., Киселев А. В., Яшин Я. И. ЖФХ, 1974, т. 48,
с. 1285-1288.
8. К i а е 1 е v А. V.; Н а 1 a s h I., Н е i n е Е. In: Advances in Chroma-
tography. Eds. J. C. Giddings, R. A. Keller, New York, 1967, v. 4, 380 p.
9. К и с e л e в А. В,, Я ш и и Я. И. Газоадсорбционная хроматография.
М., «Наука», 1967. 256 с. Исправленные и дополненные издания: La Chro-
matography Gas — Solid, Paris, 1969, 289 p. Gas — Adsorption Chromatog-
raphy. New York, 1969. 254 p.
10. К исе л ев А. В,, Щербакова К. Д., Яшин Я. И. ЖСХ, 1969,
т. 10, с. 951—968.
11. К i s е 1 е v А. V. J. Chromatog., 1970, v. 49, р. 84—129.
12. V i d а 1 - М a d j а г С.» GanansiaJ., GuiochonG. In: Gas
Chromatography 1970. Eds. R. Stock, S. G. Perri, London, 1971, 445 p.
13. GuiochonG., PommierC. La Chromatographic en phase gazeuse
en chimie inorganique. Paris, 1971. 339 p.
14. К и с e л e в А. В. В ки.: «Успехи хроматографии. К столетию со дня
Рождения М. С. Цвета», «Наука», 1972. 295 с.
И С о г с i a A., S a m р е г i R. J. Phys. Chem., 1973, v. 77,1301 — 1306.
16. Кис ел е в А. В. и др. Физико-химические применения газовой хромато-
графии. М., «Химия», 1973. 255 с.
(17. Engewald V. е. a. Chromatographia, 1974, v. 7, р. 229—235.
18. К а 1 a s h n i k о v a E. V. e. a. Chromatographia, 1972, v. 5, s. 278—285.
19. Bruner F., C a r t о n i G. P. Analyt. Chem., 1964, v. 36, p. 1522—1525.
20. Киселев А. В., Пошкус Д. П. ЖФХ, 1969, т. 43, с. 285—291.
21. Di Corcia A., Liberti A. Trans. Faraday Soc., 1970, v. 66, p.967—
975.
22. Киселев А. В., Худяков В. Л., Я ш и н Я. И. ЖФХ, 1973,
т. 47, с. 2125-2126; 1974, т. 48, с. 448—450.
23. J е q u i г W., R о b i n J. Chromatographia, 1971, v. 4, р. 59—65.
24. Д о л о в а И. А., К и с е л е в А. В., Яшин Я, И. ЖСХ, 1972, т. 13,
с. 162-165.
25. 3 а п р о м е т о в А. Ю. и др., ЖФХ, 1972, т. 46, с. 1230—1233.
26. В я з а я к и и Н. С. и др., Изв. АН СССР. Сер. хим., 1969, с. 186—189.
6 Заказ 1180
81
27. Bortnikov G. N. е. a. Chromatographia, 1971, v. 4, р. 14—18.
28. Vidal-Madjar С., Guiochon G. Bull. Soc. chim. France, 1966,
p. 1096—1100.
29. Vidal-Madjar G., Guiochon G. Separation Sci., 1967, v. 2,
p. 155-158.
30. KouznetsovA. V., Vidal-Madjar G., Guiochon G. Bull.
Soc. chim. France, 1969, p. 1440—1448.
31. Kiselev A. V., Kovaleva N. V., Nikitin Yu. S. J. Chro-
matog., 1971, v. 58, p. 19—30.
32. Prenzlow C. F., Halsey G. D. J. Phys. Chem., 1957, v. 61,
p. 1158-1165.
33. В a r n e s M., Steele W. A. J. Chem. Phys., 1966, v. 45, p. 461—465.
34. S t e e 1 e W. A., Karl R. J. Col. Interface Sci., 1968, v, 28, p. 397—
402.
35. P r e n z 1 о w C. F., Bread H. R., Brundage R. S. J. Phys.
Chem., 1969, v. 73, p. 969—978.
36. R i с c a F., Gar rone E. Trans. Faraday Soc., 1970, v. 66, p. 959—
966.
37. P r e n z 1 о w C. F. J. Col. Interface Sci., 1971, v. 37, p. 849—859.
38. Schilov N. A., S c h a t u n о v s к а у a E. G., Tschmu-
t о v К. V. Z. phys. Chem. A. 1930, Bd. 148, s. 233—236; 1930, Bd. 150,
s. 31—36.
39. D о n n e t J. B. Les Carbones. Chapitre XXIV. Paris, 1965. 912 p.
40. В о e h m H. P., V о 11 M. Carbon, 1970, v. 8, p. 227—240.
41. Puri B. In: Chemistry and Physics of Carbon. Ed. P. L. Walker. New
York, 1970. v. 6. 349 p.
42. Puri B. R., Sharma S. K. J. Ind. Chem. Soc., 1971, v. 48, p. 629—
635.
43. Д у б и и и и М. М. ЖФХ, 1965, т. 39, с. 1305—1317.
44. D u b i n i n М. М. In: Chemistry and Physics of Carbon. Ed. P. L. Walker,
New York 1966. v. 2. 384 p.
45. Кельцев В. В., Те снер П. А. Сажа — свойства, производство
и применение. Гостоптехиздат, 1952. 172 с.
46. Киселев А. В., Ковалева Н. В., Королев А. Я., Коллоидн.
ж., 1961, т. 23, с. 582—591.
47. К i s е 1 е v А. V. Rev. Gen. Caoutchouc, 1964, v. 41, p. 377—393.
48. Б о к и й Г. Б. Введение в кристаллохимию. М., Изд-во МГУ, 1954.
490 с.
49. К р е б с Г. Основы кристаллохимии неорганических соединений. М.,
«Мир», 1971. 304 с.
50. Giri f al с о L. A., Lad R. A., J. Chem. Phys., 1956, v. 25, р. 693—
697.
51. Duval X., Thomy A. G. R. Acad. Sci. Paris, 1964, v. 259, p. 4007—
4009; 1969, v. 268, p. 1416—1419; J. Chim. phys., 1969, v. 66, p. 1966—
1973; 1970, v. 67, p. 1101—1110.
52. Lar her Y. J. Chem. Soc. Faraday Trans. I, 1974, v. 70, p. 320—
329.
53. В e e b e R. A. e. a. J. Am. Chem. Soc., 1947, v. 69, p. 95—101.
54. A n d e r s о n R. B., Emmett P. H. J. App. Physics, 1948, v. 19,
p. 367—373.
55. Rappeneau J., Fillatre A. F., Yvare M. Rev. Gen. Caout-
chouc, 1964, v. 41, p. 394—402.
56. L у о n F., S troy W. S. Paint Manufacture, 1966, v. 36, № 5, p. 35—41.
57. D о n n e t J. B., Schultz J., Eckhardt E. Carbon, 1968,
v. 6, p. 781—788.
58. M e d a 1 i a A. J., Heckman F. A. Carbon, 1969, v. 7, p. 567—582.
59. Печковская К. А. Сажа как усилитель каучука. М., «Химия»,
1968. 215 с.
60. Т е с н е р П. А., Полякова М. М. ДАН СССР, 1953, т. 93, с. 855—
858.
82
61. Houska С. K., Warren A. E. J. Appl. Phys., 1954, v. 25, p. 1503—
1509.
62. J. B. Donnet. Les Carbones, Ch. XXII, XXIII, Paris, 1965, v. 2,
912 p.
63. С. Д ж. Грегг, К. С. Синг. Адсорбция, поверхность й пористость.
М., «Мир», 1970. 407 с.
64. Brunauer S., Emmett Р. Н., Teller Е. J. Am. Chem. Soc.,
1938, v. 60, р. 309—319.
65. У с е н б а е в К. Исследование структуры и свойств переходных форм
углерода. Фрунзе, «Мектеп», 1970. 150 с.
66. D о n n е t J. В., В ouland J. С. Rev. Gen. Caoutchouc, 1964, v. 41,
p. 405-412.
67. Clark G. L., Eckert A. C., But on R. L. Ind. Eng. Chem.,
1949, v. 41, p. 201—208.
68. Киселев А. В., Ковалева H. В. Изв. АН СССР, OXH, 1959,
c. 989-998.
69. Given P. H., Hill L. W., Carbon, 1969, v. 7, p. 649-658.
70. V i 11 a r d D. S. J. Am. Chem. Soc., 1948, v. 70, p. 3655—3659.
71. Studebaker M. L. e. a. Ind. Eng. Chem., 1956, v. 48, p. 162—166.
72. G a r t e n V. A., Weiss D. E., Willis J. B., Austral. J. Chem.,
1957, v. 10, p. 295—308.
73. Hallum J. V., Drushel H. V. J. Phys. Chem., 1958, v. 62, p. 110—
117.
74. Лыгин В. И. и др. Коллоидн. ж., 1960, т. 22, с. 334—339.
75. Л ежнев Н. Н. и др. Кауч, и рез., 1961, т. И, с. 21—25.
76. Studebaker М. L. In: Proc, of the Fifth Conference on Carbon, 1961.
New York, 1963, v. 2, 673 p.
77. D о n n e t J. B. e. a. J. Chim. phys., 1963, v. 60, p. 426—432.
73, К и co л on А. В., Козлов Ю. A., Лыгин В. И. ЖФХ, 1965,
т. 31», с. 1256 -1263, 2773-2778.
70, Н ooh hi Н. Р. Angow. Chem., 1966, Bd. 78, s. 617—628.
HO. Coltharp M. T., Hackorman N. J. Phys. Chem., 1968, v. 72,
p, 1171-1177.
81, Mattson J. S., Mark H. B. J. Col. Interface Sci., 1969, v. 31,
p. 131-144.
82, Bansal R. C., Vastola F. J., Walker P. L. Carbon, 1970,
v. 8, p. 443—446.
83. Donnet J. B., Ehrburger P. Carbon, 1970, v. 8, p. 697—706.
84. Di Corcia A., Ceccioli P., Bruner F. J. Chromatog., 1971,
v. 62, p. 128—131.
85. D i Corcia A., Bruner F. Analyt. Chem., 1971, v. 43, p. 1634—
1639.
86. Donnet J. B., Ehrburger P., Voet A. Carbon, 1972, v. 10,
p. 737-746.
87. С о 11 i n s R. L., Bell M. D., Kraus G. Rub. Chem. Techn.,
1960, v. 33, p. 993—998.
88. Spackman J. W. Chem. Ind., 1961, v. 38, p. 1532—1536.
89. D onnet J. B., Henrich G., Riess G. Rev. Gen. Caoutchouc,
1961, v. 38, p. 1803—1811.
90. Riess G., Donnet J. B. Rev. Gen. Caoutchouc, 1964, v. 41, 413—
418.
01, Charlier A., Danan H., Taglang P. Rev. Gen. Caoutchouc,
1964, v. 41, p. 423-428.
92. Киселев А. В., Козлов Г. А., Лыгин В. И. КолЛоидн.
ж., 1964, т. 26, с. 651—653.
93. Deschler М. F., Charlier A. J. Chim. phys., 1967, v. 64, р. 1064—
1065.
04. Charlier М. F. е. a. Carbon, 1970, v. 8, р. 692—694.
95. Marchand A., Amiell J. Carbon, 1970, v. 8, p. 707—718.
96. Delhaes P., Carmona F. Carbon, 1972, v. 10, p. 677—690.
83
97. А в г у л ь Н. Н. и Др. ДАН СССР, 1953, т. 92, с. 1185—1188.
98. Королев А. Я., Малова Т. Н., Романова В. И. «Поли-
графическое производство», 1957, т. 7, с. 25—27.
99. Кисе л ев А. В. и др. ДАН СССР, 1959, т. 124, с. 617—620.
100. Киселев А. В. и др. Коллоидн. ж., 1962, т. 24, с. 195—200.
101. Smith W. R., Polley М. Н. J. Phys. Chem., 1956, v, 60, р. §89—
102. Boehm Н. Р., Diel Е., Heck W. Rev. Gen. Caoutchouc, 1964,
v. 41, p. 461—468,
103. Donnet J. B., Lahaye J. Rev. Gen. Caoutchouc, 1964, v. 41,
p. 469-474.
104. H о n a k E. R., M а г t i n e г P. Farbe, Lack, 1970, v. 76, p. 362—365.
105. Киселев А. В., Ковалева H. В., X опина В. В. ЖФХ,
1965, т. 39, с. 2982—2985.
106. Biscoe J., Warren В. Е. J. Appl. Phys., 1942, v. 13, р. 364—371.
107. Beebe R. А. е. a. J. Am. Chem. Soc., 1947, v. 69, p. 2294—2299.
108. А в г у л ь Н. Н. и др. ЖФХ, 1956, т. 30, с. 2106—2121.
109. Bezus A. G. е. a. Chromatographia, 1974, v. 7, р. 246—250.
110. Теснер П. А., Рафалькес И. С. Труды Всесоюзнонауч.-исслед.
Ин-та природных газов, М., 1953. 180 с.
111. Б а р м а к о в а Т. А., Киселев А. В., Ковалева Н. В.
Коллоидн. ж., 1974, т. 36, с. 133—136.
112. Pierce С., Smith R. N. J. Phys. Chem., 1954, v. 58, p. 298—302.
113. Marsh P. A. e. a. Carbon, 1971, v. 9, p. 797—805.
114. Касаточкин В. И., Недошивин Ю. Н. ЖФХ, 1965, т. 39,
с. 684-687.
115. Graham D., К а у W. S. J. Col. Sci., 1961, v. 16, р. 182—185.
116. Ross S., Olivier J. P. On Physical Adsorption. New York, 1964.
401 p.
117. A v g u 1 N. N., Kiselev A. V. In: Chemistry and Physics of Car-
bon. Ed. P. L. Walker, New York, 1970, v. 6. 349 p.
118. Касаточкин В. И., Каверов А. П. ДАН СССР, 1958, т. 120,
с. 1007—1010.
119. Попов Н. М., Касаточкин В. И., Лукьянович В. М.
ДАН СССР, 1960, т. 131, с. 609-612.
120. Polley М. Н., Schaeffer W. D., Smith W. R. J. Phys.
Chem., 1953, v. 57, p. 469—471.
121. Beebe R. A., Young D. M. J. Phys. Chem., 1954, v. 58, p. 93—96.
122. Авт ул ь H. H. и др. Изв. АН СССР, ОХН, 1961, с. 205-214.
123. Belyakova L. D., Kiselev A. V.^ Kovaleva N. V. Bulk
Soc. chim France, 1967, p. 285—290.
124. Ав гуль H. H. и др. ДАН СССР, 1953, т. 92, с. 105—108.
125. Авгуль Н. Н., К и с е л е в А. В., Лыгина И. А. Коллоидн. ж.,
1961, т. 23, с.’ 369—375.
126. Ross S., Winkler W. J. Col. Sci., 1955, v. 10, p. 319—329, 330-
337.
127. Аристов Б. Г., Киселев А. В. Коллоидн. ж., 1967, т. 29,
с. 631-637.
128. Sams J. Р., Constabaris G., Halsey G. D. J. Phys. Chem.,
1962, v. 66, p. 2154-2158.
129. Kiselev A. V. Disc. Faraday Soc., 1965, v. 40, p. 205—224.
130. Киселев А. В. ЖФХ, 1967, t. 41, c. 2470—2504.
131. А щ b e r g С. H,, Spencer W. B., Beebe R. A. Can. J. Chem.,
1955, v. 33, p. 305—311.; J. Phys. Chem. 1958, v. 62, p. 719—723.
132. Polley M. H., Schaeffer W. D., Smith W. R. J. Ащ.
Chem. Soc., 1951, v. 73, p. 2161—2165.
133. Millard B., Beebe R. A., Cynarsky J. J. Phys. Chem.,
1954, v. 58, p. 468—471.
134. Beebe R. A., Dell R. M. J. Phys. Chem., 1955, v. 59, p. 746—754,
754-762.
84
1'35. Beebe R. A., Holmes J. M. J. Phys. Chem., 1957, v. 61, p. 1684—
Fr 1686.
IM Б и б и P. А. и др. ЖФХ, 1964, т. 38, с. 708-718.
ЙМ Биби Р. А. и др. ЖФХ, 1964, т. 38, с. 939—946.
18&. Gale R. L., Beebe R. A. J. Phys. Chem., 1964, v. 68, р. 555-567.
Smith W. R., Ford D. G. J. Phys. Chem., 1965, v. 69, p. 3587—
fc<3592.
ЙИ0. Beebe R. A. e. a. J. Phys. Chem., 1966, v. 70, p. 1009—1016.
Kt. Beebe R. A., Gale R. L., Kleinstenber T. C. W. J.
Phys. Chem., 1966, v. 70, p. 4010—4014.
W. Pierce C., Smith R. N. J. Phys. Col. Chem., 1950, v. 54, p. 354—
W 364, 784—795, 795—803.
143. Smith R. N., Pierce C., Cordes H. J. Am. Chem. Soc., 1950r
<< v, 72, p. 5595—5597.
144. Pierce C. e. a. J. Am. Chem. Soc., 1951, v. 73, p. 4551—4557.
x1 45. M о о i J., Pierce C., Smith R. N. J. Phys. Col. Chem., 1953r
v. 57, p. 657—659.
146. Pierce C., Smith R. N. J. Am. Chem. Soc., 1953, v. 75, p. 846—
848.
147. Pierce C. J. Phys. Chem., 1959, v. 63, p. 1076-1079; 1968; v. 72r
p. 1955-1959; 1969, v. 73, p. 813-817.
148. Pierce C., Ewing B. J. Am. Chem. Soc., 1962, v. 84, p. 4070—
4075; J. Phys. Chem., 1964, v. 68, p. 2562-2568; 1967, v. 71, p. 3408—3413.
149. Davis B. W., Pierce C. J. Phys. Cliem., 1966, v. 70, p. 1051—
1058.
150. Crowell A. D., Young D. M. Trans. Faraday Soc., 1953, v. 49y
p. 1080—1085.
151. Young D. M., Crowell A. D. Physical Adsorption of Gases. Lon-
don, 1962. 426 p.
152. Steele W. A., Halsey G. D. J. Chem. Phys., 1954, v. 22, p. 979—
984.
153. Singleton J. H., Halsey G. D. J. Phys. Chem., 1954, v. 58„
p. 330-335, 1011-1017; Can. J. Chem., 1955, v. 33, p. 184—192.
154, fionstabaris G., Halsey G. D. J. Chem. Phys., 1957, v. 27,
p. 1433—1434.
ISt Constabaris G., Singleton J. H., Halsey G. D. J.
Phys. Chem., 1959, v. 63, p. 1350—1355.
165. Sams J. R., Constabaris G. D., Halsey G. D. J. Phys.
Chem., 1960, v. 64, 1689—1696; 1961, v. 65, p. 367—369.
<57. Contabaris G., Sams J. R., Halsey G. D., J. Chem. Phys.,
1962, v. 37, p. 915—915.
158. Young C. J. e. a. J. Phys. Chem., 1954, v. 58, p. 313—315, 887—890.
159. Chessick J. J., Young G. J., Zettlemoyer A. C. Trans.
Faraday Soc., 1954, v. 50, p. 587—592.
150. Bassett D. R., Boucher E. A., Zettlemoyer A. C.
J. Phys. Chem., 1967, v. 71, p. 2787-2790.
161. Ross S., P u 11 z W. W. J. Col. Sci., 1958, v. 13, p. 397—402.
102. Ross S., Olivier J. P. J. Phys. Chem., 1961, v. 65, p. 608-615.
103. Ross S., Sa el ens J. K., Olivieri. P. J. Phys. Chem., 1962
v. 66, p. 696—700.
154. Machin W. D., Ross S., Proc. Roy. Soc. (London), 1962, v. A265t
p. 455—462.
165. Ross S., Wiltshire I. J. J. Phys. Chem., 1966, v. 70, p. 2107—
2111.
100. Айгуль H. H. и др. Изв. АН СССР, ОХН, 1956, с. 1304—1311; 1957 у
с. 1021-1031.
107. Kiselev А. V. In: Proc. 2-nd Internet. Congress on Surface Activity.
Ed. E. Schullman, London. 1957, v. 2, 646 p.
158. А в г у л ь Н. Н. и др. Коллоидн. ж., 1958, т. 20, с. 298—304.
109. Киселев А. В. и др. Коллоидн. ж., 1958, т. 20, с. 444—455.
85
170. Авгуль Н. Н. и др. Изв. АН СССР, ОХН, 1959, с. 787—798.
171. Киселев А. В. ЖФХ, 1961, т. 35, с. 233—257.
172. Kiselev А. V. Quart. Rev., 1961, v. 15, р. 99—124.
173. В а си л ь е в а В. С. и др., ЖФХ, 1961, т. 35, с. 1889—1891.
174. Безус А. Г., Д р е в и и г В. П., Киселев А. В. Коллоидн. ж..
1961, т. 23, с. 389-397.
175. Ав гуль Н. Н., Киселев А. В., Лыгина И. А. Коллоидн.
ж. 1961, т. 23, с. 513—520.
176. Авгуль Н. Н., Киселев А. В., Лыгина И. А. Изв. АН СССР
ОХН, 1961, с. 2116-2125.
177. И с и р и к я н А. А., Киселев А. В. ЖФХ, 1962, т. 36, с. 1164—
1172, 1723-1730; 1963, т. 37, с. 1776-1785.
178. Киселев А. В. Коллоидн. ж., 1962, т. 24, с. 185—194.
179. Авгуль Н. Н., Киселев А. В., Лыгина И. А. Изв. АН
СССР, ОХН, 1962, с. 32—37.
180. Авгуль и др. Изв. АН СССР, ОХН, 1962, с. 769—777.
181. Kiselev А. V. In: Gas Chromatography 1962. Ed. M. van Swaay,
London 1963. 411 p.
182. Киселев А. В., Сердобов M. В. Коллоидн. ж., 1963, т. 25,
с. 543—548.
183. Avgul N. N., L у gin a I. A., Kiselev A. V. Trans. Faraday
Soc., 19'63, v. 59, p. 2113—2123.
184. Киселев А. В., Сердобов M. В. ЖФХ, 1963, t. 37, c. 2589—
2592.
185. Б e л я к о в а Л. Д., Киселев А. В., Ковалева Н. В.,
ДАН СССР, 1964, т. 157, с. 646—649; Analyt. Chem., 1964, v. 36, р. 1517—
1519.
186. Авгуль Н. Н., Лыгина И. А., Киселев А. В. ЖФХ,
1964, т. 38, с. 2055—2058.
187. Кисел ев А. В. и др. ЖФХ, 1964, т. 38, с. 161—169.
188. Безус А. Г., Киселев А. В., Древинг В. П. ЖФХ, 1964,
т. 38, с. 59—67, 947—954, 2924—2930.
189. Киселев А. В. ЖФХ, 1964, т. 38, с. 2753—2773.
190. Kiselev А. V. In: Gas Chromatorgaphy 1964. Ed. A. Goldup, Lon-
don, 1965. 350 p.
191. Киселев А. В., Ковалева H. В., Петрова P. С. Кол-
лоидн. ж., 1965, т. 27, с. 822—827.
192. Kiselev А. V., Y a s h i n Y a. I. In: Gas Chromatorgraphie — 1965.
Ed. G. H. Struppe, Berlin, 1965. 576 s.
193. Киселев А. В., Яшин Я. И. ЖФХ, 1966, т. 40, с. 429—433,
603-608.
194. Белякова Л. Д., Киселев А. В., Ковалева Н. В. ЖФХ,
1966, т. 40, с. 1494-1498; 1968, т. 42, с. 2276-2283.
195. Berezin G. I., Kiselev А. V. J. Col. Interface Sci., 1966, v. 22,
p. 161—*164.
196. Безус А. Г., Древинг В. П., Киселев А. В. ЖФХ, 1967,
т. 41, с. 2921—2927.
197. К ей б а л В. Л. и др. ЖФХ, 1967, т. 41, с. 2234—2239.
198. Киселев А. В., Не то в Г. М., Щербакова К. Д., ЖФХ,
1967, с. 1418-1425.
199. Киселев А. В., Мигунова И. А., Яшин Я. И. ЖФХ,
1968, т. 42, с. 1235—1239.
200. Kiselev А. V. J. Col. Interface Sci., 1968, v. 28, p. 430—442.
201. Авгуль H. H. и др. ЖФХ, 1968, т. 42, с. 1747—1753.
202. Onuska F. е. a. J. Chromatog., 1968, v. 34, р. 81—85.
203. Березин Г. И. и др. ЖФХ, 1969, т. 43, с. 224—224.
204. Березин Г. И. и др. ЖФХ, 1969, т. 43, с. 1601—1603, 2946—2947;
1970, т. 44, с. 523-524.
205. Бойкова А. С., Щербакова К. Д. «Нефтехимия», 1967, т. 7,
с. 451—457.
86
206. Киселев А. В. и др. ЖФХ, 1970, т. 44, с. 1272—1276.
207. Березин Г. И., Киселев А. В., Синицын В. А. ЖФХ,
1970, т. 44, с. 734—740.
208. Киселев А. В. и др. Коллоидн. ж., 1970, т. 32, с. 527—532.
209. Kalashnikova Е. V. е. a. Chromatographia, 1971, v. 4. s. 495—
500.
210. Березин Г. И. и др. ЖФХ, 1972, т. 46, с. 214-215.
211. Berezin G. I. е. a. J. Col. Interface Sci., 1972, v. 38, p. 335—340.
212. Kiselev A. V. In: Collogues internationoux du C. N. R. S., No. 201.
Thermochimie. Marseille, 1972. 565 p.
213. Калашникова E. В. Макогон A. M., Щербакова К.Д.
Тезисы доклада на 2-ом Всесоюзном симпозиуме по межмолекулярным
взаимодействиям и конформации молекул, 3—9 июня 1974 г. Волгоград,.
1974, 112 с.
214. Kalashnikova Е. V., Kiselev А. V., Shcherbakova
К. D. Chromatographia, 1974, v. 7, s. 22—25.
215. Eisen O.G. e. a. Chromatographia, 1971, v. 4, s. 448—454.
216. Батраков Г. H., Кисаров В. М., Киселев А. В. ЖФХ,
1974, т. 48, с. 1255—1257.
217. Joyner L. G., Emmett Р. Н. J. Am. Chem. Soc., 1948, v. 70,
p. 2353—2359.
218. C orrin M. L. J. Am. Chem. Soc., 1951, v. 73, p. 4061—4065.
219. Healey F. H., Yu Y. F., Chessick J. J. J Phys. Chem., 1955,
v. 59, p. 399—402.
220. Millard B. e. a. J. Phys. Chem., 1955, v, 59, p. 976—978.
221. Clark H. J. Phys. Chem., 1955, v. 59, p. 1068—1069.
222. Ross J. W., GoodR. J. J. Phys. Chem., 1956, v. 60, p. 1167-1171.
223. Groyson J., Aston J. G. J. Phys. Chem., 1957, v. 61, p. 610—61 <
224. Gi nlinm D. J. Phys. Chem. 1957, v. 61, p. 1310—1313; 1958, v. 62,
p, 1210 1211.
225. V а о о K. L.( Я i b о r t A, R. J. Phys. Chem,, 1959, v. 63, p. 1398—
1400; I960, v. 64, p. 061-963.
226, Y aria R., Sams J. R. J. Chem. Phys., 1962, v. 37, p. 571—576.
227. H a 1 a s z I., H о r v a t h C. Nature, 1963, v. 197, p. 71—73; Analyt.
Chem., 1964, v. 36, p. 1178-1186.
228. Schneider W., Bruderreck H., H a 1 a s h I. Analyt. Chem.,
1964, v. 36, p. 1533-1540.
229. Hsieh P. Y. J. Phys. Chem., 1964, v. 68, p. 1068—1071.
230. G r i f f i t h s D. W., Thomas W. J., Walker P. L. Carbon,
1964, v. 1, p. 515—524.
231. C h i r ns i d G. С., P о p e C. G. J. Phys. Chem., 1964, v. 68, p. 2377—
2381.
232. Myers A. L., P r a u s n i t z J. M. Trans Faraday Soc., 1965, v. 61,
p. 755-764.
233. Taylor G. L., Atkins J. H. J. Phys. Chem., 1966, v. 70, p. 1678—
1680.
234. Pierotti R. A., Smallwood R. E. J. Col. Interface Sci., 1966,.
v. 22, p. 469—481.
235. Bruner F., Cartoni G. P., Liberti A. Analyt. Chem., v. 38,
p. 298—299.
236. Hoory S, E., Prausnitz J. M. Trans. Faraday Soc., 1967, v. 63,
p. 455—460.
237. С о c h r a n e H. e. a. J. Col. Interface Sci., 1967, v. 24, p. 405—415.
238. Deitz V. R. J. Phys. Chem., 1967, v. 71, p. 830—837.
239. Goretti G. C., Liberti A., Nota G. J. Chromatog., 1968,
v. 34, p. 96—100; 1968, v. 38, p. 282—286.
240. Pierotti R. A. Chem. Phys. Letter, 1968, v. 2, p. 420—422.
241. V о e t A. J. Col. Interface Sci., 1969, v. 30, p. 264—269.
242. Mahajan О. P., Walker P. L. J. Col. Interface Sci., 1969, v. 31,
p. 79-84.
87
243. Wade W. H. J. Col. Interface Sci., 1969, v. 31, p. 111—115.
244. Davis B. W. J. Col. Interface. Sci, 1959, v. 31, p. 353—363.
245. Elkington P. A., C u r t h о у s G. J. Phys. Chem., 1969, v. 73,
p. 2321—2326.
246. Versino B., Geiss F. Chromatographia, 1969, v. 2,p. 354—359.
247. Холмс Дж. В кн.: «Межфазовая граница газ—твердое тело». Под ред.
Э. Флада. Пер. с аигл. М., «Мир», 1970. 433 с.
248. Di Corcia A. D., Bruner F. J. Chromatog., 1970, v. 49. р. 139—
145.
249. Di Corcia A. D., Fritz D., Bruner F. Analyt. Chem., 1970,
v. 42, p. 1500—1504; J. Chromatog., 1970, v. 53, p. 135—141.
250. Pare ja P., Amariglio H. J. Chim. phys., 1970, v. 67, p. 938—942.
251. Сычев Ю. H., Цирельников В. И. Вести. МГУ, химия II,
1970, с. 542—548.
252. Павлюченко Н. М. ЖФХ, 1970, т. 44, с. 271—273.
253. Brown С. Е., Hall Р. G. Trans. Faraday Soc., 1971, v. 67, p. 3558—
3564.
254. De-Boer H., Van Dongen В. H., Klaspersma J. H.
Surf. Sci., 1971, v. 28, p. 237—240.
255. Antoniou A. A., Scaife P. H., Peacock J. M. J. Chem.
Phys., 1971, v. 54, p. 5403—5412.
256. Thomy A., Matecki M.,Duval X. Carbon, 1971, v. 9, p. 587—592.
257. Thomy A., Duval X. C. R. Acad. Sc. Paris, 1972, v. C274, p. 15—18.
258. Vidal-Madjar C., Jacob L. Guiochon G. Bull. Soc.
Chim. France, 1971, p. 3105—3110.
259. Vidal-Madjar C., Guiochon G. J. Chim phys., 1972, v. 69,
p. 368—369.
260. Bruner F., Ce cel oli P., Di Corcia A. Analyt. Chem., 1972,
v. 44, p. 894-898.
261. Frycka J. J. Chromatog., 1972, v. 65, p. 432—434.
262. Пильт А., Ранг С., Эйзен О. Изв. АН Эст. ССР, Сер. хим.,
геолог., 1972, т. 21, с. 30—35.
263. Б регер А. X., Жданов Г. С. ДАН СССР, 1940, т. 28, с. 630—633.
264. Самсонов Г. В. и др. Бор, его соединения и сплавы. Киев, Изд-во
АН1УССР, 1960. 590 с.
265. Ниденцу'К., Д ajy сон Дж. Химия боразотных соединений. М.,
«Мир», 1968, 238 с.
266. Curthoys G., ElkingtonP. Б. J. Phys. Chem., 1967, v. 71.
p. 1477-1482.
267. Белякова Л. Д., Киселев А. В. Изв. АН СССР, ОХН,1966,
с. 638—642.
268. Winkler W. Thesis Ph. D., Rensselaer Polytechnic Institute. 1955,
cm. Ross S., Olivier J. P. On Physical Adsorption. New York, London, 1964,
p. 193.
269. Piero tti R. A. J. Phys. Chem., 1962, v. 66, p. 1810—1815.
270. Трепнелл Б. M. Хемосорбция. M., Издатинлит, 1955, 327 c.
271. Rnodin T. N. J. Am. Chem. Soc., 1950, v. 72, p. 4343—4348.
272. Van Dongen R. H., Kaspersma J. H., De Boer J. H.
Surface Sci., 1971, v. 28, p. 237—257.
273. De Boer J. H. e. a. J. Col. Interface Sci., 1972, v. 38, p. 97—100.
274. Kaspersma J. H. Thesis, Nijmegen, 1972. 132 p.
275. F о x P. G., К a t z M. J. J. Phys. Chem., 1961, v. 65, p. 1045-1047.
276. Baker B. G., Fox P. G. Trans. Faraday Soc., 1965, v. 61, p. 2001^
2012.
277. Trivin H., Bonn eta in L. C. R. Acad. Sc. Paris, 1968, v. 266,
p. 1488-1491; 1969, v. 268, p. 564-567.
278. Van Dongen R. H. Thesis, Delft, 1972, 116 p.
279. Haul R. A. W., Swartz E. R. Z. Elektrochem., 1957, Bd. 61,
s. 380—384.
280. Duval X., Schuller D. Z. Elektrochem., 1962, Bd. 66, s. 424—432.
88
281. G e n о t В., Duval X. C. R. Acad. Sc. Paris, 1967, v. 265, p. 285—287.
!82. G en о t B., Duval X. J. Chim. phys., 1972, v. 69, p. 1238—1245.
33- Young D. M., Anderson C. D. Can. J. Chem., 1965, v. 43, p. 309—
312
';j84. Larher Y., Maurice P. J. Chim. phys., 1968. v. 65, p. 669—672^
Maurice P. Etude de la condensation hidimensionnelle du krypton,
du xenon et.du methane sur la face de clivage du zinc, Novembre 1973,
< France, Saclay (These) 112 p. .
285. Steiger R. F. Surface Sci., 1969, v. 14, p. 279—304.
286. Wooten L. A., Brown C. J. Am. Chem. Soc., 1943, v. 65,p. 113—118..
287. Orr W. J. C. Proc. Roy. Soc., 1939, v. A173, p. 349—367.
288. R oss S., Clark H. J. Am. Chem. Soc., 1954, v. 76, p. 4291—4300- ’
280. Ross S., Winkler W. J. Am. Chem. Soc., 1954, v. 76, p. 2637—2640.
290. Hayakawa T. Bull. Chem. Soc. Japan, 1957, v. 30, p. 337—342;
343-349.
221. Yao Y. F. Y. J. Phys. Chem., 1965, v. 69, p. 2472—2475.
A p и с т о в Б. Г. и др. ЖФХ, 1968, т. 42, с. 984—987.
93. Davis В. W. J. Col. Interface Sci., 1969, v. 31, p. 353—363.
294. Бру нау эр С. Адсорбция газов и паров. М., Издатинлит, 1955. 781 с-
295. Ильин Б. В. Природа адсорбционных сил. М. JI. Гостехиздат, 1952-
124 с.
296. Young D. М. Trans. Faraday Soc., 1952, v. 48, p. 548—561; 1954, v. 50^
p. 838-841.
297. Даннинг В. В кн.: «Межфазовая граница газ—твердое тело». Под.
ред. Э. Флада. Пер. с англ. М., «Мнр», 1970. 433 с.
298. Jaycock М. J., Waldsax J. S. R. J. Col. Interface Sci., 1971,.
v. 37, p. 462-468.
299. Fischer В. B., Me Millan W. G. J. Chem. Phys., 1958, v. 28r
p. 549 -554, 555-561, 562-571.
300. Ц (j н m 8., 011 v lor J. P., H i n c h e n J. J. Advances in Che-
minify Ben, 1961, v. 33. p. 317—327; R о s s S., Hinchen J. J. Clean
Hurtacea. Ed. G. Goldginger, New York, 1970. 385 p.
301. L ar her Y, These, Orsay, 1970,142 p. J. Chim. phys., 1968, v. 65, p.
074—976; J. Phys. Chem., 1968, v. 72, p. 1847—1848; Le Vide, 1969, v.
139, P. 22—24; J. Col. Interface Sci., 1971, v. 37, p. 836-848.
802. T a k a i s h i T., Mohri M. J. Chem. Soc. Faraday Trans. I, 1972,.
v. 68, p. 1921—1934.
803. Takaishi T. J. Chem. Soc. Faraday Trans. I, 1972, v. 68, p. 801—813^
804. Белякова Л. Д., Калпакян A. M., Киселев A. B.
Коллоидн. ж. 1973, т. 35, с. 906—910.
305. Nardon Y., These, Nancy, 1972, 122 p.
306. Belyakova L. D., Kalpakyan A. M., Kiselev A. V.
Chromatographia, 1974, v. 7, p. 14—18.
807. Cannon P. J. Phys. Chem., 1960, v. 64, p. 1285—1289.
308, Ballou E. V., Ross S. J. Phys. Chem., 1953, v. 57, p. 653—657.
300. Ballou E. V. J. Am. Chem. Soc., 1954, v. 76, p. 1199—1200.
310. Johnston R. R. M., Moore P. J. M. J. Phys. Chem., 1964^.
v. 68, p. 3399-3406.
311. L о p e z G. J., В a г e а С. E., В a n a r e s M. M. A. An qui m_
Real soc. esp. fis. quim., 1964, v. 64, p. 699—704.
812. James R. W., Wood W. A. Proc. Roy. Soc., 1925, v. A109, p-
508-620.
313. А в г у л ь Н. Н. и др. Изв. АН СССР, ОХН, 1961, с. 1948—1954.
314. Белякова Л. Д., Киселев А. В. Изв. АН СССР, ОХН, 1962г
с. 1185-1189.
815. Machin W. D. Trans. Faraday Soc., 1969, v. 65, 529r-534.
816. Belyakova L. D., Kiselev A. V., Soloyan G. A. Chro-
ma tographia, 1970, v. 3, p. 254—259.
817. Белякова Л. Д., Киселев’А. В., Солоян Г. А. ЖАХ^
1972, т. 27, с. 1182-1187.
89*
318. Жданов С. П., Егорова Е.Н. Химия цеолитов. Л., «Наука»,
158 с.; Breck D. W. Zeolite Molecular Sieves. Structure, Chemistry
and Use. New York, 1974. 771 p.
319. Kiselev A. V. Molecular Sieves II, Advances in Chem. ser., 1970,
v. 102, p. 37-68.
320. Джигит О. M. и др. ЖФХ, 1969, т. 43, с. 968—969.
321. Barter R. М. J. Col. Interface Sci., 1966, v. 21, p. 415—434.
322. Bezus A. G. e. a. Trans. Faraday Soc., 1971, v. 67, p. 468—482.
323. Босачек В. В кн.: «Цеолиты, их свойства и применение», М., «Йаука»,
1965. 395 с.
324. Aristov В. G., В о s а с е k V., Kiselev А. V. Trans. Fara-
day Soc., 1967, v. 63, р. 2057—2067.
325. Barter R. M., Whiteman J. L. J. Chem. Soc., Set. A, 1967,
p. 13-18.
326. Джигит О. M. и др. ЖФХ, 1968, т. 42, с. 198—205.
327. Barter R. М., Kanellopoulos A. G. J. Chem. Soc. Ser. А,
1970, р. 775—782.
328. Топчиева К. В., Романовский Б. В. В кн.: «Современные
проблемы физической химии». Вып. 4. М., изд-во МГУ, 1970, 516 с.
329. Миначев X. М. и др. «Кинетика и катализ», 1972, т. 13, с. 1101—1112.
330. Гритчина Н. Д. и др. ЖФХ, 1975, т. 49.
331. Бройер П. и др. ЖФХ, 1968, т. 42, с. 2556-2562.
332. В е z u s A. G. е. a. J. Col'. Interface Sci., 1973, v. 45, p. 386—395.
333. Киселев А. В., Лыгин В. И. Инфракрасные спектры поверх-
ностных соединений и адсорбированных веществ. М., «Наука», 1972.459 с.
334. Киселев А. В., Кузнецов Б. В., Никитин Ю. С. «Ки-
нетика и катализ», 1970, т. 11, с. 500—512.
335. Kiselev А. V. Disc. Faraday Soc., 1971, v. 52, p. 14—32.
336. Anderson P. J., Horlock R. F. Trans. Faraday Soc., 1969,
v. 65, p. 251—256.
337. P i e г о 11 i R. A., Halsey G. D. J Phys. Chem., 1959, v. 63, p. 680—
686.
338. Day R. E., Kiselev A. V., Kusnetsov В. V. Trans. Fa-
raday Soc., 1969, v. 65, p. 1386—1393.
339. Ash S. C., Kiselev A. V., Kusnetsov В. V. Trans. Faraday
Soc., 1971, v. 67, p. 3118—3126.
340. Литл Л. Инфракрасные спектры адсорбированных молекул. М., «Мир»,
1969. 514 с. Г а л к и н Г. А., Киселев А. В., Лыгин В. И.
ЖФХ, 1969, т. 43, с. 1992—1998, 2309—2314; Давыдов В. Я. и др.
ЖФХ, 1973, т. 47, с. 809-811.
341. Киселев А. В., Лыгин В. И., Щепа л ин К. Л. «Кинетика
и катализ», 1971, т. 12, с. 185—190.
342. Leonard A. J. е. a. Disc. Faraday Soc., 1971, v. 52, p. 98—108.
343. Boehm H. P. Disc. Faraday Soc., 1971 v. 52, p. 264—275.
344. Adamson A., W., Dormant L. J. Am. Cnem. Soc., 1966, v. 88,
p. 2055—2057.
345. Orem M. W., Adamson A. W. J. Col. Interface Sci., 1969, v. 31,
p. 278—286.
346. Jones D., О 11 e w i 11 R. H. J. Chem. Soc., 1955, p. 4076—4088.
347. H a u x w e 11 F., Otte will R. H. J. Col. Interface Sci., 1968,
v. 28, p. 514-521,
348. Adamson A. W., Jones B. R. J. Col. Interface Sci., 1971, v. 37,
p. 831-835.
349. Белякова Л. Д. и др. ЖФХ, 1975, т. 49.
350. Walker Р. L., Austin L. G., Nandi S. P. In: Chemistry
and Physics of Carbon. Ed. P. L. Walker, New York, 1966, v. 2, 384 p.
351. Котина В. E. и др. Хим. волокна, 1969, № 6, с. 1—6.
352. Конкин А. А., Коннова Н. Ф. Журн. Всесоюзн. хим. о. им.
Д. И. Менделеева, 1972, т. 17, с. 632—636.
353. Walker Р. L. Carbon, 1972, v. 10, р. 369—382.
00
354. Burne G. A., Jeffries R. Chem. and Ind., 1971, p. 809—813.
355. Donnet J. B. , D auksch H. In: Int. Conf, on Carbon Fibres, their
Composites and Applications. London, 1971. 231 p.
356. Christner L. G., Walker P. L. In: 10th Conf, on Carbon Fibres,
Pennsylvania, 1971, 400 p.
357. Belinski C., Diot C., Keraly F. X. L. C. R. Acad. Sci.,
Paris, 1970, v. C271, p. 1025—1027.
358. Бовар А. И. и др. XTT, 1971, № 2, с. 149^153.
359. Maire J., Coles H., M a i 11 a r d P. Carbon, 1968, v. 6, p. 555—
560.
360. Zettlemoyer A. C., Chessick J. J., Holl alaugh C. M^
J. Phys. Chem., 1958, v. 62, p. 489—490.
361. Цилипоткина M. В. и др. Высокомол. соед., 1962, т. 4, с. 1844—
1848.
362. Н о 1 i s О. L. Analyt. Chem., 1966, v. 38, р. 309—316.
363. Т а г е р А. А. и др. Пласт, массы, 1966, т. 3, с. 23—28.
364. Гвоздович Т. Н. и др. «Нефтехимия», 1968, т. 8, с. 123—128.
365. Gvosdovich Т. N., Kiselev А. V., Yashin Ya. I. Chro-
matographia, 1969, v. 2, p. 234—238.
366. H a 11 P. G., Stoekli M. F. Trans. Faraday Soc., 1969,.v. 65, p.
3334—3340.
367. Mo сева Л. И., Ca ко дынский К. И. В кн.: «Газовая хрома-
тография». М. изд. НИИТЭХИМ, 1969. 215 с.
368. Сакодынский К. И., Панина Л. И. ЖАХ, 1972, т. 27, с.
с. 1024—1034.
369. Sakodirisky К. I., Panina L. I. J. Chromatog., 1971, v. 58r
p. 61-71.
370. Белякова Л. Д. и др. ДАН СССР, 1973, т. 210, с. 1117—1120.
371. II нм h Ida J., Nishimura М. J. Jap. Chem. Soc., 1971, v. 25,
p. 932-936.
372. R u c h о и н t о I и E., V a i d у a n a t h a n A. S., Youngquist
G. R. Chem. Eng. Sci., 1971, v. 26, p. 1305—1318.
373. Белякова Л. Д. и др. ДАН СССР, 1973, т. 213, с. 1311—1314.
374. Gvosdovich Т. N., Kiselev А. V., Yashin Y a. I. Chro-
matqgraphia, 1973, v. 6, р. 179—192.
375. Бебрис Н. К., Киселев А. В., Никитин Ю. С. «Нефте-
химия», 1969, т. 9, с. 631—637.
376. Киселев А. В., Ле Зунг, Никитин Ю. С. Коллоидн. ж.
1971, т. 33, с. 224—229.
377. Berezin G. I., Kiselev А. V., Kozlov A. A. J. Col. In-
terface Sci., 1973, v. 45, р. 190—196.
378. С а рахов А. И. и др. Изв. АН СССР, ОХН, 1961, с. 974—983.
379. Сар ахов А. И., Дубинин М. М., Березкина Ю. Ф.
Изв. АН СССР, ОХН, 1963, с. 1165-1175; 1968, с. 708-717; 1969, с. 2653—
2661.
380. Vidal-Madjar С., Guiochon G. In: Gas—Chromatographie
1968. Ed. H. G. Struppe, Berlin,. 1968. 450 s.
381. Vidal-Madjar C. These, Paris, 1969. 116 p.
382. Kouznetso v A. V., Guiochon G. J. Chim. phys., 1969, v. 66,.
p. 197—199.
383. Franken J. J., Vidal-Madjar C., Guiochon G. In:
Adv. in Chromatography 1971. Ed. A. Zlatkis, 1971, 254 p.
384. Д а в ы д о в В. Я., Киселев А. В., Силина Т. В. Коллоидн.
ж., 1972, т. 34, с. 30—35; 1974, т. 36, с. 359— 362 , 762—764.
ЗНГ» . В е л и к о в а Л. Д. и др. ЖФХ, 1968, т. 42, с. 177—181.
386. Киселев А. В., Ле 3 у н г , Н и к и т и н Ю. С. Коллоидн. ж.,
1972, т. 34, с. 64—67.
387. Киселев А. В., Ковалева Н. В., Торбина Л. А. В кн.:
«Гнлопня хроматография». Вып. 15, М., изд. НИИТЭХИМ, 1971, 88 с.
91
388. Б а р м а к о в а Т. В., Киселев А. В., Ковалева Н. В.
Коллоидн. ж., 1974, т. 36, с. 133—136.
389. Scott С. G., Phillips С. S. G. In: Gas—Chromatography 1964.
Ed. A. Goldup, London, 1965. 350 p.
390. Cado F. M., Yu vet R. S. In: Aspects Gas Chromatogr., Berlin,
1971,313 s.
391. Okamura J. P., Sawyer D. T. Analyt Chem., 1970. V. 43,
p. 1730—1735.
392. Джавадов С. П., Киселев А. В., Никитин Ю. С. «Ки-
нетика и катализ», 1967, т. 8, с. 238—243.
393. Taramasso М., Fuchs Р. J. Chromatogr. 1970, v, 49. р. 70—75.
394. Бабкин И. Ю., Киселев А. В., Королев А. Я. ДАН
СССР, 1961, т. 136, с. 373—376.
395. Борисенко И. В. и др. «Нефтехимия», 1966, т. 6, с. 776—781.
396. Eltekov Yu. A., Khokhlova Т. D., Kiselev А. V.
Chromatograpia, 1973, v. 6, р. 187—189.
Глава III
ТЕРМОДИНАМИЧЕСКИЕ ХАРАКТЕРИСТИКИ АДСОРБЦИИ
ОДНОКОМПОНЕНТНЫХ ГАЗОВ И ПАРОВ
1. Некоторые особенности определения
термодинамических характеристик адсорбционных систем
из экспериментальных данных
Для адсорбентов с близкой к однородной поверхностью произ-
ведены многочисленные измерения величин адсорбции и соответ-
ствующих величин давления или концентрации в газовой фазе при
постоянной (изотермы адсорбции) и при разных температурах.
Эти измерения производились как статическими, так и газохромато-
графическими методами. Значительно меньше сделано калоримет-
рических измерений (статических и динамических) теплот адсорбции.
Наконец, совсем немного сделано калориметрических измерений
теплоемкости адсорбционных систем. Однако именно все эти неза-
висимые измерения, вместе взятые, для одной и той же системы
адсорбат—адсорбент дают необходимую информацию о термодина-
мических свойствах адсорбционной системы. Вместе с тем перечис-
ленные методы измерений имеют свои особенности, которые необ-
ходимо учитывать как при оценке точности измеряемых величин,
так и при дальнейшей их обработке для получения термодинами-
ческих характеристик адсорбции, не зависящих от способа изме-
рений.
Статические измерения изотерм адсорбции. Изотермы равно-
весной адсорбции однокомпонентных газов и паров на твердых ад-
сорбентах измеряют обычно вакуумными статическими методами
(11. В этом случае адсорбент предварительно нагревают в вакууме
для очистки его поверхности от ранее адсорбированных веществ.
Температуру и продолжительность откачки, а также глубину вакуума
выбирают в зависимости от геометрической структуры, величины
и химического состава поверхности адсорбента. Химический состав
определяет термостойкость адсорбента и прочность связывания
поверхностью посторонних молекул. При изучении молекулярной
(физической) адсорбции следует избегать таких обработок адсор-
бента, которые приводят к возникновению химически активной по-
верхности по отношению к данному адсорбату.
Адсорбционные исследования проводятся обычно на поверхно-
стях, откачанных до давления 10" 3—10" 4 Па (10" б—10" 6 мм рт. ст).
При нагревании адсорбента такая откачка часто уже приводит к уда-
лению загрязнений с большей части его поверхности. При заданной
| '|убино вакуума степень очистки поверхности от физически адсор-
Лирнпанных на ней молекул зависит от природы этих молекул,
93
химии поверхности, температуры, продолжительности откачки, струк-
туры пор, а также от количества адсорбента и особенностей аппара-
туры. Неспецифически адсорбирующие поверхности (см. разд. 2
гл. I) легко освобождаются даже от сильно полярных молекул не-
больших размеров. Удаление специфически адсорбированных мо-
лекул, например молекул воды, связанных с поверхностью водород-
ными связями, требует более продолжительной откачки. За удале-
нием молекул воды с поверхности адсорбента и за ее адсорбцией
можно следить по изменению инфракрасного спектра в области ча-
стот колебаний, соответствующих составной полосе поглощения вален-
тных и деформационных колебаний молекул Н2О. Эта полоса (с мак-
симумом около 5300 см”1) не перекрывается с полосами поглощения
валентных колебаний поверхностных гидроксильных групп [2—7].
При более сильной специфической адсорбции молекул воды,
например, при их сильном электростатическом взаимодействии с одно-
и особенно многозарядными катионами солей, имеющих большие
комплексные анионы (см. разд. 4 гл. И), удаление адсорбированных
молекул воды с поверхности даже непористого адсорбента требует
длительной откачки при высоких температурах. Удаление воды
происходит еще медленнее в случае микропористых сильно специ-
фических адсорбентов, например, в случае цеолитов. Более полная
очистка цеолитов от адсорбированных молекул воды требует дли-
тельного прогрева в высоком вакууме [8] при температурах около
500 °C и выше (нагрев цеолита до столь высоких температур возможен
только в том случае, если при этих температурах не происходит
термодеструкция цеолитной структуры, что зависит от типа решетки
и состава цеолита). При выборе режима откачки и условий изуче-
ния адсорбции следует иметь в виду также возможность дополни-
тельных процессов проникновения молекул внутрь скелета адсор-
бента. Так, в случае цеолитов типа X при температурах выше 100—
110 °C наблюдается медленное поглощение молекул аммиака, ко-
торое связано, по-видимому, с проникновением этих молекул внутрь
образующих эти цеолиты кубооктаэдрических единиц через 6-ти
членные кислородные кольца [8]. Этот процесс требует преодоле-
ния значительных энергетических барьеров и поэтому не наблюдает-
ся при более низких температурах, при которых изотермы адсорб-
ции аммиака обратимы и адсорбционное равновесие на поверхности
больших полостей этих цеолитов наступает быстро. Для удаления
адсорбированных молекул из ультрапор и из других наиболее сильно
захватывающих данные молекулы мест часто требуется наряду
с нагреванием применение значительно более высокого вакуума —
до 10"8—10-10 Па (10“10—10"12 мм рт. ст.) [9—12]. Однако геомет-
рия и химия обнажаемых при такой обработке участков поверхности
твердого тела обычно отличаются от таковых для остальной более
однородной части поверхности.
Из статических методов измерения изотерм адсорбции газов
наибольшее распространение получили различные варианты так
называемых газовых объемных методов, связанных с применением
94
разного вида калиброванных газовых баллонов и газовых бюреток
II]. При этом измеряют давление адсорбата в газовой фазе. Вели-
чину адсорбции определяют по разности между числом молей ад-
сорбата в газовой фазе (в газовой бюретке) до адсорбции и числом
его молей в газовой фазе над адсорбентом (в так называемом «мертвом»
объеме [1]) после адсорбции.
Равновесное давление определяют обычно ртутными или мем-
бранными манометрами [1] и различными основанными на изме-
нении электрического сопротивления и других свойств электрон-
ными или другими подобными вакуумметрами, хотя часто точность
этих последних манометров и диапазон измеряемых давлений не
достаточны. Некоторые из наиболее точных установок для измере-
ния адсорбции газов и паров описаны в предыдущей книге [1] этой
серии и в работе [13]. Для определения очень малых величин ад-
сорбции можно использовать в качестве газовой бюретки капилляр
манометра Мак-Леода [ 1, 14].
Для исследования адсорбции пара вещества, находящегося
вблизи комнатной тенпературы в жидком состоянии, очень удобен,
а при не слишком малых величинах удельной поверхности адсор-
бента $ и достаточно точен, метод капиллярной жидкостной микро-
бюретки. Этим методом определяют величину адсорбции с помощью
измерения положения мениска жидкости в капилляре до и после
адсорбции. Иамопопие положения мениска пропорционально коли-
честву адсорбата, перешедшему из микробюретки в сосуд с адсор-
бентом И, 15, 1(|[.
Во всех этих случаях для определения величины адсорбции
необходимо измерепие количества адсорбата, остающегося в газовом
мертвом объеме. Его определяют обычно с помощью измерения
равновесного давления газа в этом объеме. По существу, надо оп-
ределить массу газа в мертвом объеме или его концентрацию.
Это можно сделать, в частности с помощью газового хроматографа
И]. Таким образом, обычными методами определяется разность
между введенным в ампулу с адсорбентом количеством адсорбата
и тем ого количеством, которое остается в мертвом объеме (вплоть
до поверхности адсорбента). Эта разность соответствует определению
гиббсовской величины адсорбции [17, 18].
Статические весовые методы измерения величин адсорбции при-
меняются обычно при изучении адсорбции паров веществ с неболь-
шим давлением насыщенного пара [1]. В этих случаях давление
адсорбата в газовой фазе мало и его нельзя точно измерить, т. е.
отсюда нельзя надежно определить величину адсорбции газовым
объемным методом. Весовой метод удобно применять также и при
адсорбции газов в тех случаях, когда надо следить за кинетикой
адсорбции или когда надо следить за изменением массы адсорбента
после откачки (происходящим в результате хемосорбции, терми-
ческой деструкции и т. п.) [19].
Кглн удельная поверхность адсорбента $ велика, для измерения
НИМ1Ч1ПНИН массы адсорбента в результате адсорбции или терми-
95
ческой деструкции используют весы Мак-Бэна и Бакра [20]
(см. [1]). Если величина s мала, то обычно используют коромысло-
вые крутильные весы с электромагнитной компенсацией изменения
массы адсорбента и автоматической записью [1, 21, 22].
Комбинация весового метода с методом газовой или жидкостной
бюретки позволяет в одном опыте контролировать глубину откачки
и термодеструкцию (например, дегидроксилирование) адсорбента
и измерять величину адсорбции газа или пара.
При измерении величины адсорбции весовцм методом равно-
весное давление часто регулируется и определяется с помощью
Рис. 111,1. Изотермы адсорбции пара этана на графи-
тированной термической саже при разных температу-
рах (числа на кривых). Черные точки — десорбция.
выбора температуры термостата криостата), в который погру-
жается сосуд с чистым жидким адсорбатом [1]. Предполагается, что
зависимость давления насыщенного пара жидкого адсорбата от тем-
пературы достаточно хорошо известна.
Вакуумными статическими методами обычно получают изотерму
адсорбции, т. е. зависимость величины гиббсовской адсорбции
(или полного количества адсорбированного вещества или степени
заполнения поверхности [1]) от равновесного давления или кон-
центрации адсорбата в газовой фазе.
Часто бывает нужно измерить изотермы адсорбции при несколь-
ких температурах в широком интервале температур. Удобный для
таких измерений интервал температур для данной системы адсор-
бат — адсорбент составляет обычно около 50 °C. На рис. Ill,1 при-
веден соответствующий пример для адсорбции этана на графити-
рованной термической саже [23]. За пределами этого интервала
при более низких температурах с ростом давления адсорбция рас-
тет очень быстро уже при малых давлениях, следовательно, точность
измерения этих давлений ртутным U-образным манометром в этой
области температур невелика. За пределами удобного для измере-
ний интервала при более высоких температурах величина адсорб-
ции растет с ростом давления очень медленно, т. е. приходится из-
96
мерять малые величины адсорбции, что снижает точность этих из-
мерений. При удачном выборе температуры погрешности, с кото-
рыми измеряют величины адсорбции и давления (или концентра-
ции) газа, можно сблизить. Если удельная поверхность со-
ставляет не менее 1 м2/г, то погрешности измерения величин
адсорбции и давления, начиная с 1000 Па (— 10 мм рт. ст.) могут быть
не больше 0,1%.
Газохроматографическое определение константы Генри и изо-
термы адсорбции. В методе газовой хроматографии [1, 24, 25] через
Заполняющий колонну адсорбент непрерывно пропускается поток
Газа-носителя, который обычно при температуре колонны на изучае-
мом адсорбенте практически не адсорбируется. Очистка поверхности
производится током этого инертного газа при повышенных темпера-
турах. Это приводит к несколько худшей очистке поверхности от
наиболее сильно адсорбированных примесей, чем в вакуумном ад-
сорбционном методе. Трудно удалить таким способом молекулы воды
и других полярных веществ с поверхности сильно специфических
адсорбентов [1, 24, 25]. Легче очищается поверхность неспецифи-
чегких адсорбентов. В этом случае, однако, предварительно адсор-
бированные молекулы могут остаться, по-видимому, только на наи-
более неоднородных местах поверхности. Основная, наиболее од-
нородная часть поверхности очищается от примесей. Таким обра-
1п>м, итог метод очистки поверхности имеет даже свои преимущества
нрн намерениях адсорбционных свойств однородных поверхностей,
особен пи и случае ппснецифичнских адсорбентов.
11ри фрон।алыюй хроматографии концентрация исследуемого
(нчцоства и потоке газа-носителя у входа в колонну поддерживается
настоянной и регистрируется нарастание его концентрации у выхода
ни колонны до величины, равной концентрации у входа в колонну.
Меняя постоянную концентрацию адсорбата в газе-носителе, полу-
чают различные выходные кривые, представляющие зависимости
нипцонтрпции адсорбата в выходящем из колонны газе от времени
пыкода. Анализ этих кривых позволяет в благоприятных случаях
(для ин слишком медленной адсорбции) получить ряд точек или
отровкоо для построения изотермы адсорбции. Существенно выбрать
оптимальные условия опыта — шаровидную форму, близкие раз-
меры и возможно более однородную упаковку зерен адсорбента (не
содержащих тонких пор), температуру колонны и скорость потока
гапа, чтобы в наибольшей степени приблизиться к равновесию в ко-
лонне и иметь возможность вычислить равновесную изотерму ад-
сорбции из выходных кривых.
При имульсной элюционной хроматографии адсорбат вводится
в ноток газа-носителя (элюента) у входа в колонну небольшими
порциями (пробами, дозами). У выхода из колонны получается кри-
вая в виде пика, выражающая нарастание и снижение концентра-
ции адсорбата в выходящем газе-носителе. Анализируя эту кривую,
можно рассчитать величины адсорбции и равновесного давления
для определенной точки на изотерме адсорбции или для отрезка
7 Заказ 1180 97.
изотермы до этой точки. Изменяя величину пробы, можно измерить
изотермы адсорбции в соответствующих интервалах величин адсорб-
ции [1, 24, 25]. Предполагается, что в колонне устанавливается
адсорбционное равновесие.
Измеряется при этом избыточное время удерживания в колонне
интересующего нас адсорбата по сравнению со временем удержи-
вания одновременно вошедшей в колонну порции газа-носителя
или другого, введенного одновременно с изучаемым адсорбатом прак-
тически не адсорбирующегося газа. Таким образом, этот метод поз-
воляет непосредственно определить избыточную, т. е. гиббсов-
скую [17, 18] величину адсорбции. Эту величину отражает про-
изведение измеренного избыточного времени удерживания адсор-
бата и скорости потока газа, приведенной к постоянному давлению
в колонне. Это произведение называют удерживаемым объемом
(подробнее см. разд. 3 этой гл.), его обычно относят или к единице
массы адсорбента в колонне Vm = VR/m (тп — масса всего адсор-
бента в колонне), или к единице его поверхности VА = VR/A (Л —
общая поверхность адсорбента в колонне). Вычисление изотерм
адсорбции из зависимости от величины пробы (количества ад-
сорбата, впускаемого в ток газа-носителя у входа в колонну) опи-
сано в книгах [1, 24, 25]. При этом очень важно обеспечить по-
стоянную температуру по всей длине колонны.
Благодаря применению ионизационных и других чувствитель-
ных детекторов газохроматографический метод обладает очень
высокой чувствительностью и поэтому позволяет работать с очень
малыми дозами адсорбата, т. е. при малых заполнениях поверхности.
Поэтому в импульсном элюционном методе особенно ценна возмож-
ность ввода в колонну очень малых доз адсорбата. Если поверхность
химически и геометрически близка к однородной, то при уменьше-
нии дозы даже специфически адсорбирующегося вещества пики
становятся симметричными и время удерживания перестает зави-
сеть от величины дозы (см. рис. 1,4) или же это время можно экстра-
полировать к нулевой пробе. Это позволяет прямо из эксперимента
получить удерживаемые объемы для нулевой пробы Vm, г или F a, i>
представляющие константы Генри [24, 25] [см. ниже выражения
(III,20) и (III,21)].
Методом элюционной газовой хроматографии можно исследо-
вать и более высокие заполнения поверхности, т. е. получить изо-
термы адсорбции из зависимости Vm или VA от величины дозы ад-
сорбата [24, 25]. Однако при переходе к большим величинам ад-
сорбции точность определения удерживаемого объема и соответ-
ствующей величины равновесной концентрации адсорбата в газо-
вой фазе уменьшается. Это связано как с возрастающими трудно-
стями обеспечения близких к равновесным условий работы колонны,
так и с необходимостью интегрирования пиков сложного профиля
[1, 24, 25].
Для обеспечения оптимальной скорости потока газа через запол-
ненную адсорбентом хроматографическую колонну создается не-
98
Коюрый нороннд давления газа в колонне от входа к выходу. Од-
пнно определяемые газохроматографическим методом удерживае-
мым объемы можно привести к нулевому перепаду давления газа
к колонне. 'Таким образом, определяемые из такого опыта величины
(пппгяни к открытой адсорбционной системе, находящейся при
inn 1ОН11ПОЙ температуре и постоянном давлении. Вместе с тем объем
riHihuhHonioft адсорбентом колонны также постоянен. Поскольку
гнадаииций давление в колонне газ-носитель при температурах газо-
Iроматографических опытов обычно практически не адсорбируется,
io при получении из газохроматографических данных термодинами-
ческим характеристик адсорбции это отличие условий хроматогра-
фического опыта от условий вакуумных статических измерений, про-
подимых при постоянной температуре и постоянном объеме системы,
не гущестноппо.
Рассмотрены погрешности определения теплот адсорбции из газо-
«роматосрафичоских измерений при разных температурах [26, 27].
Дли физико-химических исследований, в частности для исследований
Н'Норбции, специально предназначен газовый хроматограф серии
llaoi И |2Н|
11аден|Поо намерение величин адсорбции также возможно и дру-
I ИМ динамическим способом. Использовался, в частности, посто-
янный поток испаряющегося вещества из жидкого адсорбата, поме-
щенною по вспомогательный калориметр [29—31]. О количестве
адсорбата, перешедшего па адсорбент, судят по поглощенному при
нги Mcihipoiiiiii теплу. Зтот метод позволяет измерять адсорбирован-
ное количество с чуастнитольпостыо до 1()"7 г. В качестве источника
Нотоин адсорбата можно использовать и его десорбцию со вспомога-
1ильного адсорбента.
Калориметрические методы определения теплот адсорбции и те-
нлоемиогти адсорбционных систем. В тех случаях, когда равновесная
йннц(ш грпцпн адсорбата в газе с или его давление р малы и их нельзя
Иймнригь г нужной для определения температурного коэффициента
иди ityltiT точностью, нужны прямые калориметрические изме-
рения теплоты адсорбции. Измерения в калориметре необходимы
Й в твл случаях, когда дифференциальная теплота адсорбции сильно
иинаннптгн с изменением температуры [32] (рис. III.2).
Для измерения теплот адсорбции применяются в основном кало-
риметры, к которых производится компенсация теплового эффекта
при постоянной или близкой к постоянной температуре [1, 29, 30,
ИЛ| Достигнута столь высокая точность калориметрического изме-
рении теплоного эффекта, что погрешность определения средней
мнчышй и дифференциальной теплот адсорбции часто определяется
uni рншnor i ью измерения количества адсорбированного вещества.
Применяются как статические, так и динамические калоримет-
рические мы оды. В статическом методе при впуске порции газа
Пн йщпрЛшп пыделяющееся тепло компенсируют соответствующим
уменьшенном прирорыано подаваемой в калориметр мощности элек-
|ричегыпо hhi ронптелл, строго измеряемым и интегрируемым по
99
времени [1, 32, 33]. В калориметрах Кальве [29] это достигается
использованием эффекта Пельтье. В динамическом методе при по-
стоянной скорости потока испаряющегося в изотермических усло-
виях адсорбата из жидкости, находящейся в одном калориметре,
происходит (также в изотермических условиях) выделение теплоты
80
70
।
\^60
£ 80
о
X
с4 70
х
ns. молекул на полость
।____। ।____।____।_____।____। t
П 2 4 6 8 10 12 /4
л^ммоль/з
50
Рис. III,2. Зависимость дифференциальной теплоты адсорбции qv
пара воды на цеолите KNaX от величины адсорбции ns при раз-
ных температурах (числа на кривых).
адсорбции в другом калориметре, содержащем адсорбент. Постоян-
ная температура поддерживается автоматической регулировкой мощ-
ностей нагревателей в этих калориметрах. Измерение этих мощно-
стей и определение их соотношения позволяет практически непре-
рывно определять дифференциальную теплоту адсорбции по мере
роста величины адсорбции [1, 30]. Предварительно определяется
скорость нарастания давления в «мертвом» объеме, нужная для
внесения соответствующей поправки в общую скорость потока ад-
сорбата, определяемую мощностью нагревателя в испаряющем жид-
кость калориметре.
100
Н большинстве случаев теплоты адсорбции измеряются в систе-
мам, имеющих постоянный объем, хотя при измерении теплоты
адсорбции газов с газовой бюреткой или поджимным баллоном [1]
общий объем системы адсорбент газовая фаза может изменяться.
При термодинамической интерпретации результатов калориметри-
ческим измерений надо учитывать эти условия опыта.
Л тех случаях, когда в теплоту адсорбции вносят значительный
вклад теплоты молекулярных и фазовых переходов в адсорбцион-
ном слое, теплота адсорбции сильно изменяется как с изменением
Полинины адсорбции, так и с изменением температуры [32, 34—36].
!|||цисимость теплоты адсорбции от количества адсорбированного
пощостпа иногда выражается кривой, проходящей через максимумы
и минимумы по нескольку раз [32, 35] (см. рис. 11,11, рис. 111,2).
Соответственно в этих интервалах величин адсорбции с изменением
Риг, 111,3. Изменение средней мольной теплоемко-
НИ Cfft адсорбированной воды на цеолите KNaX с рос-
том величины адсорбции га* (в ммоль/г).
Ваннып точки обозначают разные серии измерений. Пункти-
рами Л и В обозначены соответственно теплоемкости жидкой
и газообразной воды.
ваиплжниия особенно сильно изменяется теплоемкость адсорбцион-
ной системы [37- 39] (рис. III,3). В таких случаях косвенные опре-
долшпп! теплот адсорбции из результатов вакуумных статических
и гнио хроматографических определений дают лишь средние вели-
чины тиллиты адсорбции па некотором интервале температур и не
нбеенечиаают необходимую для исследования зависимости теплоты
адсорбции от температуры точность. Вместе с тем в таких случаях
ори oi HiH ii гол апо небольшом повышении температуры форма кривых,
йырншнющнх ;и1нисимость теплоты адсорбции от заполнения адсор-
бент, ГНЛ1.НО изменяется, эти кривые сглаживаются (см. рис. III,2).
101
Поэтому здесь нужны, во-первых, непосредственные калориметри-
ческие измерения теплот адсорбции при разных температурах,
и, во-вторых, калориметрические измерения теплоемкости адсорб-
ционной системы также при разных температурах.
Измерения теплоемкости производятся при постоянном объеме
адсорбционной системы (адсорбент -|- газовая фаза). Часть адсор-
бата при нагревании такой системы десорбируется. В измеренную
теплоемкость вносится поправка на теплоту десорбции этой части
адсорбата [1]. Относящиеся сюда вопросы методики соответствую-
щих калориметрических измерений, результаты измерений и опре-
деления из них средней мольной и дифференциальной мольной теп-
лоемкости адсорбированного вещества подробно рассматриваются
в подготавливаемой к печати книге Березина и Киселева, про-
должающей эту серию монографий по химии поверхности и ад-
сорбции.
Калориметрические измерения тепловых эффектов необходимы
также в тех случаях, когда происходит хемосорбция. В этих случаях
выделение тепла происходит обычно медленно, поэтому особенно
большую роль здесь играет такое расположение адсорбента в кало-
риметрической ампуле, при котором обеспечивается по возможности
одновременная подача газа ко всем его частицам [40—42]. Это обес-
печивается, в частности, расположением мелких частиц адсорбента
тонким слоем на стенках ампулы [40, 41] или на надетых на перфо-
рированную трубку тарелочках [1], а также при напылении пленок
адсорбента, например металлических пленок, на стенки ампулы
[43-46].
В тех случаях, когда выделение теплового эффекта молекуляр-
ной адсорбции или хемосорбции затягивается, для получения рав-
новесной величины теплоты адсорбции особое значение имеет ис-
следование кинетики выделения тепла в калориметре, облегчающее
экстраполяцию [32].
Все эти особенности экспериментальных методов должны быть
учтены при оценке точности и обработке результатов адсорбцион-
ных измерений. Для дальнейшей обработки очень важно, в част-
ности, знать, при каких внешних условиях проводились измерения.
В частности, статические измерения изотерм адсорбции и калори-
метрические измерения теплот адсорбции обычно проводят в ваку-
умных установках с постоянным или мало изменяющимся объемом
и при постоянной температуре. Калориметрические измерения теп-
лоемкости адсорбционных систем проводят при их медленном нагре-
вании при постоянном объеме. Постоянство определенных термоди-
намических параметров адсорбционной системы при проведении
измерений очень важно, так как оно позволяет более надежно про-
изводить расчет воспроизводимых и сопоставимых термодинами-
ческих характеристик адсорбции, не зависящих от условий и спо-
соба измерений.
102
2. Основные задачи термодинамики адсорбции
При термодинамическом исследовании адсорбции надо рассмот-
реть следующие две задачи.
1. 11а хождение общих условий равновесия адсорбционной системы
и вытекающих из этих условий соотношений между обычно изме-
ряемыми равновесными термодинамическими величинами: вели-
чиной гиббсовской адсорбции п#, концентрацией с (или давлением р)
данного адсорбата в объемной газовой фазе и температурой Т.
Таким образом, в эту задачу входит нахождение зависимостей
ns = f(c, Т) (Ш,1)
НЛП
т) (III,1а)
Найди эти зависимости из экспериментальных данных или на основе
юоротичоских моделей, можно определить соответствующие кон-
г1ннгы адсорбционного равновесия К. Значения К удобно сопостав-
лен. дли разных систем адсорбат — адсорбент и вычислять с помо-
щью различных 1’(Ч»роги'к»ских моделей этих систем термодинами-
ческим и (гм гл IV) и молекул ирно-статистическими (см. гл. VI
и V11) методами. Па уравнений (111,1) или (III,1а) с помощью урав-
нен ил Гиббса 1171 можно найти коэффициенты активности адсор-
бированного вещества, а также получить его уравнение состояния.
2. Онредолепие интегральных и дифференциальных изменений
термодинамических функций адсорбционной системы (свободной
онергип Л', энтропии Л’, внутренней энергии U и теплоемкости С)
при переходе этой системы из некоторого начального состояния в не-
которое конечное состояние. Выбор начального и конечного состоя-
ний нроплнол<*н и определяется соображениями удобства сопостав-
лении получаемых таким путем термодинамических характеристик
(г о указанных пышс изменений термодинамических функций)
Irin разных адсорбционных систем. Поэтому следует выбрать опре-
деленные г।андаpiпые состоянии, одинаковые для всех адсорбцион-
ны h । иг Iом 11Н’рцос тело газ
Для на хождении пимеиеиий указанных выше термодинамических
функций адсорбционной системы в принципе достаточно решения
iiopinitt нндвчи, так как изменения F, S, U и С можно найти из урав-
нений (IIIJ) или (II 1,1а) а также из соответствующих констант
адсорбционного равновесия. Однако, поддерживая температуру no-
ri оннной, уравнения (III,1) или (III,1а) или константу равновесия Я
можно непосредственно использовать только для нахождения из-
менено)! Чтобы при той же температуре найти изменения S и U,
необходимо знать первые производные /, <р или К по температуре,
а зли нахождения изменений С — вторые производные /, <р или К
но температуре.
Использование результатов исследования адсорбционных равно-
весий, При дог га точно большой величине площади поверхности адсор-
бента .1 и соотпетстпугощем выборе температуры опытов обычно можно
измерить величины с (или р) и определить К с достаточной для
103
надежного определения изменения F точностью. Однако точность
измерения этих величин часто недостаточна для нахождения уже
первой производной функций/, <р или К по Т, особенно в тех случаях,
когда происходящее при адсорбции изменение U сильно зависит
от Т. Найти же с нужной для решения многих задач точностью вто-
рые и третьи производные/, ф или К по Т из результатов экспери-
ментальных исследований адсорбционных равновесий статическими
или хроматографическими методами пока затруднительно или даже
вообще невозможно из-за недостаточной точности этих исследова-
ний. Поэтому необходимы прямые калориметрические измерения
как тепловых эффектов адсорбции (теплот адсорбции), так и тепло-
емкостей (при разных температурах) адсорбционных систем.
Использование результатов калориметрических исследовании
теплот адсорбции и теплоемкостей адсорбционных систем. Резуль-
таты калориметрических измерений тепловых эффектов адсорбции
могут зависеть от условий протекания процесса адсорбции в экспе-
риментальной установке, а не только от выбранных начальных и ко-
нечных состояний системы адсорбат — адсорбент. Поэтому из ре-
зультатов таких калориметрических измерений надо найти термо-
динамические характеристики адсорбционной системы, не завися-
щие от условий проведения калориметрического опыта. Прежде
всего, сюда относится получение из результатов калориметрического
измерения теплоты адсорбции величины соответствующего изменения
внутренней энергии адсорбционной системы и получение из кало-
риметрических измерений при нагревании адсорбционной системы
собственно ее теплоемкости и ее изменения, происходящего при
адсорбции. Такая термодинамическая интерпретация результатов
калориметрических измерений часто встречает затруднения и тре-
бует рационального выбора условий проведения этих измерений
и учета их конкретных особенностей. При такой интерпретации
калориметрических измерений теплот адсорбции, соответствующих
переходу адсорбционной системы из некоторого начального состоя-
ния в конечное состояние равновесия или близкое к нему, надо,
в частности, исключить или учесть возможности совершения над
системой внешней работы или теплообмена вне калориметра.
Калориметрические измерения теплоемкости адсорбционных си-
стем связаны с их нагреванием и проводятся, исходя из начального
состояния системы газ — адсорбент, соответствующего термоди-
намическому равновесию или близкого к таковому. Однако при пе-
реходе в конечное состояние система может несколько отклониться
от равновесия. Кроме этого, вследствие нагревания, необходимого
при измерении теплоемкости адсорбционной системы, уменьшается
величина ns. Поэтому в измеряемую теплоемкость адсорбционной
системы вносит свой вклад теплота происходящего при нагревании
системы частичного перехода адсорбата из адсорбированного со-
стояния в газовую фазу. Величина этого вклада в измеряемую
теплоемкость зависит от конкретных условий опыта. Поэтому при
калориметрических исследованиях теплоемкости адсорбционных си-
104
г гем возникнет необходимость получения таких величин теплоемкости
адсорбционной системы, которые не зависят от конкретных условий
и роподопин этих измерений.
'Гоним образом, указанная выше вторая задача термодинами-
чески* исследований адсорбционных систем в той ее части, которая
относится к исследованию зависимостей изменений F, U, S и С
от температуры, связана с калориметрическими измерениями теп-
ловых аффектов и теплоемкостей.
II дальнейшем рассматриваются все эти задачи термодинамиче-
ского исследования адсорбционных систем. Так как настоящая
ИННгн носшпцопа адсорбции на поверхностях, приближающихся
н однородным, то размеры частиц твердого тела предполагаются
достаточно большими. Вопросы термодинамики малых объектов вы-
йодпт ид рамки этой книги. Эти вопросы рассмотрены в работах
(’нмпнчонко 1471 и Хилла [48].
3. Термодинамические условия равновесия
адсорбционной системы
Копетнптн равновесия и уравнение изотермы адсорбции. В соот-
ветствии с. надачами этой книги рассмотрим только молекулярную
(фииичоскую) адсорбцию. Примем, как это соответствует большинству
рассмотренных и гл. II практически важных случаев, что адсорбент
нредотапллот чистое (однокомпопоптпоо), нелетучее и нерастворяю-
ища адсорбат твердое тело. Примем также, что при адсорбции вели-
чина Я площади поверхности раздела твердое тело — газ не изме-
няется.
Рассмотрим систему, состоящую из твердой фазы — адсорбента
и ид пинтой фаны адсорбата. Вдали от поверхности раздела твер-
дое юле гни адсорбат находится а свободном от действия адсорб-
ЦИоннм* сип । аноном состонипп с концентрацией с и давлением р.
Объвй киситы Г поггоцпоН- Потому при постоянной температуре Т
йбЩиМ уелнйием ра пни нес и и и пл потея минимум свободной энергии.
Ий гнно условия следует, что химические потенциалы адсорбата
й COOTmiHHtt объемного num (т. о. вдали от поверхности раздела)
и в адсорбиронин ном состоянии ц при равновесии одинаковы
1171
|| (Ш,2)
Нпн1п имость р^ от активности адсорбата в объемном газе а можно
вырниить уравнением
Ina (III,3)
или
р* === (Г)4- RT In сув (III,За)
Н атом выражении ц* (Т) зависит от Т и не зависит от с, а коэф-
фициент активности у8 зависит от с, причем при с = 0, у8 = 1.
< 11 а лононие у8 от единицы характеризует межмолекулярное взаи-
Mojicftri Biie и газовой фазе. Выше указывалось, что газохроматогра-
105
фические детекторы можно прокалибровать по величинам с, поэтому
для объемного газа мы будем пользоваться величинами мольно-
объемной концентрации с и соответствующей активности а — cyg [50].
Рассмотрим теперь рациональное выражение для ц. Введенная
Гиббсом [17] величина адсорбции ns представляет собой в рассмат-
риваемом случае разность числа молей адсорбата п в объеме Vf
включающем область повышенной плотности вблизи поверхности
твердого тела, и числа его молей ng, находящегося в том же объеме V
в газообразном состоянии, свободном от влияния твердого тела:
п» = п—(Ш,4)
Гиббсовский избыток ns зависит от выбора положения разделяю-
щей поверхности, относительно которой определяется величина
ns [17]. В случае адсорбции твердыми телами в качестве разделяю-
щей поверхности целесообразно выбрать поверхность твердого тела.
Площадь этой поверхности А = ms, где т — масса адсорбента,
as — его удельная поверхность. Если величина s известна, то для
получения независимой от значения А величины адсорбции гиббсо-
вский избыток ns следует отнести к единице площади поверхности
адсорбента. При этом получается величина адсорбции по Гиббсу:
ns = r=4- (Ш.5)
Величину Г мы будем называть в дальнейшем просто величиной
адсобрции. Отметим, что под этим термином понимается именно
гиббсовский избыток, а не полное количество адсорбата в «адсорб-
ционном слое». Для определения полного количества адсорбата
в адсорбционном слое надо знать толщину этого слоя и распреде-
ление в нем концентрации адсорбата или сделать какие-либо допу-
щения об этом (см. гл. IV). Гиббсовские определения величин
адсорбции и других поверхностных термодинамических величин,
отнесенных к единице площади поверхности (индекс s внизу), —
свободной поверхностной энергии Fs , поверхностной энтропии Ssf
поверхностной внутренней энергии Us и поверхностной теплоем-
кости Cs свободны от каких-либо допущений о толщине адсорбцион-
ного слоя.
Для химического потенциала и активности адсорбированного
вещества можно использовать различные выражения (см., например,
[49]). Мы поступим здесь аналогично тому, как это делается
при определении активности реального газа, находящегося во внеш-
нем потенциальном поле, например в поле силы тяжести. В этом
случае химический потенциал газа р? на высоте h над некоторым
уровнем, для которого h = 0, представляется в виде суммы члена
рЛ (Т7), зависящего только от температуры [см. выражение (III, За)],
члена Mgh, зависящего от потенциальной энергии в поле силы тя-
жести (здесь М — мольная масса, a g — ускорение свободного па-
дения), и члена RTlnchyh, где cft—концентрация газа на высоте h,
a yh — его коэффициент активности, определяемый только межмо-
106
лииуллрпыми взаимодействиями в газе на высоте fe, т. е. =
11^(7') | /1/М | /ИЧгиЛу*. В соответствии с гиббсовским
определением величины адсорбции Г химический потенциал адсор-
б и ро а а и по го пегцества целесообразно представить в виде суммы
( ищу ющи х трех членов:
pEEpf (Г) + цв(Г)+ВГ In а» (Ш,6)
Норный член этой суммы, как и для объемного, т. е. свободного
от плияпия адсорбента газа [см. выражение (III,3)], отражает
спойстпа молекул адсорбата в отсутствие какого-либо межмолеку-
лнрпого взаимодействия. Второй член отражает взаимодействие
молекул адсорбата с адсорбентом, независящее от величины адсорб-
ции Г. Наконец, третий член отражает зависящее от Г взаимодей-
ствие адсорбированных молекул друг с другом и с адсорбентом.
Соответственно выражению (III,6) активность адсорбированного
шчцпства а представляет собой произведение активности а5,0 (ха-
рактеризующей взаимодействия адсорбат — адсорбент при Г ->0).
и а н । и иное гп ая (характеризующей взаимодействия адсорбат—ад-
сорбат при Г>0). Коэффициент активности адсорбированного
вещества, характеризующий взаимодействия адсорбат—адсорбат
при Г > О, можно представить
as
vs = — (111,7)
При Г 0 as Г и ys 1. Отклонение ys от единицы характе-
ризует межмолекулярное взаимодействие между молекулами адсор-
бированного вещества.
Подставляя выражения (III,3) и (III,6) в уравнение (III,2),
получаем:
|?(Г)4-7?Г1па5 — 7?Г1па (III,8)
ИЛИ
aS = jexp[-E-ip-]}a (Ш,9)
Пользуясь выражениями (III, За) и (III,7), получаем уравнение,
выражающее зависимость Г от с и Т*.
г = ^{ехр[-^Р-]}С = Хг С^с (ШЛО)
где независящая от Г и с величина
яг, с = ехР
Р5(П "1
НТ J
(III,И)
и роде гапляот константу адсорбционного равновесия. Индекс Г, с
у величины КГ|с указывает на то, что величина адсорбции выра-
жена по Гиббсу в единицах [моль/площадь поверхности], а концен-
трация адсорбата в объемном газе выражена в единицах [моль/объем].
'Гак как у" и безразмерные величины, то, выражая Г в моль/м2,
а г и моль/м3, получаем К?jC в м. В связи с тем, что числовые зна-
чении /»* г , имеют порядок 10-6 м, их удобнее выражать в мкм.
107
При постоянной Т уравнения (III,9) или (111,10) выражают
зависимость Г от с, т. е. представляют уравнения изотермы ад-
сорбции.
Константа Генри. При Г->0ис->0^->1 и у* -> 1, так что
независящая от Г и с величина
К
_rYs_
Г,с~
су
lim
Г, с->0
(111,12)
\ С J 1 , С, 1
Величина Кг, с, i ~ константа Генри, характеризующая взаимо-
действие адсорбат—адсорбент. Индекс 1 указывает на предельно
малые значения Г и с, т. е. на то, что константа Генри представляет
первый коэффициент в вириальном уравнении изотермы адсорбции
(см. разд. 2 гл. IV и разд. 2 гл. VI). Очевидно, что константа Генри,
выраженная через ns и с, имеет вид
К s = lim ( —<
n’c’1 ns с-хЛс' Г’ М
(Ш,13)
Так как при с —> 0 объемный газ становится идеальным, то,
введя в выражение (111,12) уравнение состояния идеального газа
р = cRT, получаем:
lim /XVflr lim (JL\ = RTK (111,14)
Г, с 0 \ С J г, р О \ Р J 1 * Р’ 1 л < р, 1 '
где Ку р х — константа Генри, выраженная через Г и р; К
Генри, выраженная через ns и р.
,р,1
— константа
Таким образом
з
*г, с, 1 — RTKr* £
(III,15)
Индекс 1 у величины Кг> 1? как и у величины Кг, с, i, указывает
на предельно низкие значения Г и р, а индексы с и, соответственно,
р — на то, какое свойство равновесного газа используется — его
мольно-объемная концентрация с или его давление р.
Если известно значение Кг, р, п причем Г выражена в мкмоль/м2,
ар — в мм рт. ст., то для того чтобы получить значение Кг, с, i с раз-
мерностью мкм, в формуле (111,15) надо использовать значение
R = 0,0624 мм рт. ст. «см3«мкмоль-1 «К-1, а если р выражено в пас-
калях (Па), то надо подставить, соответственно, значение R —
— 8,30 Па • см3 • мкмоль"1 • К-1.
Для определения константы Генри из экспериментальной изо-
термы адсорбции, измеренной статическим методом, надо привлечь
ту или иную модель состояния адсорбированного вещества, которая
позволяет выразить уравнение изотермы адсорбции в аналитической
форме, в частности в виде степенного ряда (вириальные разло-
жения по степеням Г или с), в котором первый член содержит только
константу Генри. Этот метод рассмотрен в разд. 1 гл. IV.
108
Константу Генри адсорбционного равновесия можно опреде-
лить в благоприятных случаях (при достаточно высоких темпера-
турах) и непосредственно. В последнее время приобрели большую
важность газохроматографические исследования адсорбции при ма-
лых дозах адсорбата, вводимых в практически неадсорбирующийся
на данном адсорбенте газ-носитель (см. разд. 1 этой главы и работы
И, 24, 25, 27, 28, 51—531). Измеряемые времена удерживания ад-
сорбата можно привести к нулевому перепаду давления газа в хро-
матографической колонне [25, 54]. Допущение о достижении рав-
новесия в колонне при достаточно высоких ее температурах оправ-
дывается, особенно для не очень больших энергий адсорбции, даже
в случае тонкопористых адсорбентов [24, 25, 55, 56]. При этом
измеряемую зависимость времени удерживания от концентрации
адсорбата в газе-носителе, т. е. выходную кривую при использовании
фронтальной хроматографии или растянутый край хроматографи-
ческого пика в элюционной хроматографии, можно пересчитать
в изотерму адсорбции [24, 25, 55, 56].
Разность времени удерживания в колонне адсорбирующегося
компонента и практически не адсорбирующегося газа-носителя, т. е.
исправленное время удерживания tc для концентрации адсорбата
в газе с на растянутом крае пика элюционной хроматограммы,
связано с наклоном изотермы адсорбции drfldc уравнением [1, 18,
24, 25, 56-58]:
(Ill,16)
tc —
А, Г
где т — масса адсорбента в колонне; w — объемная скорость потока газа,
приведенная к нулевому перепаду давления в колонне.
Величина tc зависит от условий опыта, т. е. от скорости потока
и?. Не зависит от w, а только от температуры колонны и концентра-
ции произведение tc на
VR^c^tcw-=m
(111,17)
называемое удерживаемым объемом. Величина Vr,c представляет
объем газа-носителя, прошедший через колонну с момента выхода
из колонны введенного одновременно с пробой адсорбата неадсор-
бирующегося газа до момента выхода из колонны адсорбата с кон-
центрацией с. Удельный удерживаемый объем, отнесенный к еди-
нице массы адсорбента в колонне, равен:
_tcw
Vm'c т L Ja, Т
(Ш,18)
а если известна удельная поверхность адсорбента $,то удерживае-
мый объем, отнесенный к единице поверхности адсорбента
tcw tcw ((9Г\
А ~~ ms \дс / а, Т
(Ш,19)
109
Из выражений (111,18) и (111,19) следует, что предельная вели-
чина удельного удерживаемого объема для нулевой дозы адсорбата
в соответствии с выражениями (111,12) и (111,13) представляет кон-
станту Генри для равновесия адсорбат—адсорбент:
lim
ns, с —*• О
(Ш,20)
Соответственно для единицы площади поверхности адсорбента
va i =
А’ Г, с-> 0
А.Г-^Г.сЛ
(1П,21)
Таким образом, газохроматографическим методом с высокочув-
ствительным детектором можно довольно быстро и надежно полу-
чать значения констант Генри для адсорбции на достаточно одно-
родной (для точной экстраполяции к нулевой дозе адсорбата)
поверхности адсорбента.
Таблица Ш,1. Значения Kv с 4 и AUi для адсорбции н-гексана
и бензола на графитированной термической саже,
определенные газохроматографическим методом в разных работах
Адсорбат КГ, с, 1 при 373 К, мкм — ДП1, КДЖ/МОЛЬ Литера- тура Адсорбат КГ, с, 1 при 373 К, мкм — АП1, кдж/моль Литера- тура
н-Гексан 38,0 [60] Бензол 35,1 [60]
44,0 [61] 1,81 [68]
3,10 41,5 [62] 1,70 38,0 [62]
3,20 40,5 [24, 63] 1,70 38,0 [24, 63]
3,13 41,5 [64] 1,94 36,4 [65]
3,18 39,2 [65] 41,0 [66]
43,5 [66] 2,11 38,0 [69]
3,29 40,0 [67] 2,33 36,5 [53]
3,44 38,6 [53] 1,84 37,7 [59]
2,70 37,3 [59]
В табл. Ill,1 сопоставлены значения VA1 = Кг с г для двух
углеводородов на графитированной термической саже. Результаты,
полученные разными авторами, в основном не отличаются между
собой более чем на 10%, причем значения VAtl, полученные для
очень малых доз адсорбата [53], наиболее надежны.
На рис. 111,4 сопоставлены наиболее точные значения
полученные из статических изотерм адсорбции [23, 70] (подробнее
см. разд. 2 и 3 гл. IV), со значениями VA>1, полученными из удер-
живаемых объемов [53]. Эти величины хорошо согласуются. Анало-
гичное согласие наблюдается также и в случае адсорбции других
веществ (см. рис. IV,13, IX,2, IX,4, Х,1, Х,7, Х,8 и Х,11).
110
.Чти результаты показывают, что в настоящее время газохро-
мнтографический метод, по крайней мере в случае адсорбции на таком
напористом и неспецифическом адсорбенте, как графитированная
термическая сажа, дает вполне надежные значения констант Генри
/G'.-.i- Что обстоятельство, а также возможность использовать
калиброванные по концентрации с детекторы и в статических изме-
рениях, что особенно удобно в случае газовых смесей, позволяют
и редпочесть для адсорбата в газовой фазе концентрацию с давлению р
и, соответственно, константу Генри Кг. с, i = VA,i константе Кг, р, i*
Поэтому в дальнейшем термодинамические характеристики адсорб-
ции даются в терминах концентрации объемного газа с (если адсор-
бируется газ идеальный) или его активности а. Так как в статиче-
ских методах исследования адсорбции
распространены измерения давления р,
то ниже даются также выражения для
термодинамических характеристик адсорб-
ции и через давление р, но только для
адсорбции идеального газа.
Коэффициент активности адсорбиро-
ванного вещества у\ Зная константу
Генри и изотерму адсорбции, можно ис-
следовать зависимость ys от Г. Из урав-
нений (111,10) и (111,12) получаем:
Рис. III,4. Зависимость
Хр с j от 1/Т для адсорбции
этана на графитированной
термической, саже. Точки 1
получены из газохромато-
графических измерений для
очень малых проб. Точки 2
и 3 получены обработкой
(см. разд. 2 и 3 главы IV)
изотерм адсорбции, измерен-
YS==KT, с,1 г (П1.22)
Если это идеальный газ, то = 1 и
Ч-%,1Т = КГ, р, 1-р (Ш’23)
Отношения с/Г или р/Г определяются ных статическим методом,
из изотермы адсорбции. Зависимость ys
от Г характеризует взаимодействия адсорбат—адсорбат на данном
адсорбенте.
На рис. III,5 приведены изотермы адсорбции бензола и этана
па графитированной термической саже, а на рис. III,6 — получен-
ные с помощью уравнения (111,23) зависимости ys от Г для адсорбции
этих веществ. Так как изотерма адсорбции бензола на графитиро-
ванной термической саже при данной температуре обращена к оси р
вогнутостью во всей начальной области Г, то ys > 1. Изотерма же
адсорбции этана благодаря сильному взаимодействию адсорбат—
адсорбат в начальной части обращена к оси р выпуклостью, поэтому
в области небольших Г значения ys < 1. Подробнее зависимость
от Г рассмотрена в разд. 2 гл. IV.
Уравнение состояния адсорбированного вещества- В случае
адсорбции на поверхности твердого тела нельзя непосредственно
111
измерить поверхностное давление адсорбированного вещества л
и найти таким образом зависимость л от Г, т. е. уравнение его со-
стояния. Однако это уравнение состояния можно определить из
Рис. III,5. Изотермы адсорбции этана (7) при
173 К и бензола (2) при 293 К на графитиро-
ванной термической саже.
экспериментальной изотермы адсорбции с помощью фундаменталь-
ного уравнения Гиббса [17]. При постоянной температуре в рас-
сматриваемом случае адсорбции одного компонента из объемной фазы
эта формула имеет вид:
Рис. III,6. Зависимости коэффициен-
та активности 7s от Г при адсорбции
этана (7) и бензола (2) на графити-
рованной саже.
— Га + Г (П-1,24)
Здесь Га — величина адсорбции
адсорбента (иногда ее называют
автоадсорбцией); ра — его хими-
ческий потенциал; величина о
имеет вид:
’-(ет)г„г
где Fs — поверхностная свободная энер-
гия, т. е. гиббсовский избыток вели-
чины свободной энергии системы ад-
сорбат-адсорбент у поверхности Л [17].
В случае поверхностей раздела
жидкость — газ и жидкость —
жидкость величина о представляет поверхностное натяжение и мо-
жет быть измерена непосредственно.
Как и при адсорбции нелетучих и нерастворимых монослоев
на поверхности жидкой воды, на адсорбированное вещество вдоль
поверхности действует давление
л-- о — о
(Ш,26)
<12
представляющее собой происшедшее в результате адсорбции изме-
нение а от аа для поверхности раздела твердое тело — вакуум
до а для поверхности раздела твердое тело — газ. Величину л по
аналогии с соответствующей величиной для нерастворимых и неле-
тучих монослоев на поверхности жидкости можно назвать поверх-
ностным давлением. Однако следует иметь в виду, что в рассматри-
ваемом случае поверхностное давление л создается лишь гиббсовским
избытком адсорбата Г, который может отличаться (особенно при
больших с) от полной поверхностной концентрации адсорбата, напри-
мер, от полной поверхностной концентрации двухмерного монослоя.
Подставляя в уравнение Гиббса (111,24) величину — do = йл
из выражения (111,26) и имея в виду, что при адсорбции на нераство-
ряющем адсорбат адсорбенте dpa = 0, получаем
dn=rd|i (Ш,27)
или, в соответствии с выражениями (III,2) и (III,3)
dn = P7T<Zlna (Ш,28)
Интегрируя эти уравнения, получаем, соответственно
(Ш,29)
II случае адсорбции идеального газа
л.- Н 7'5^ de
о
г
НТ I —dp
t) р
о
(III,29а)
!)ти формулы позволяют определить л из зависимости Г от с или р,
т, о. из изотермы адсорбции.
При с, р 0 подынтегральные функции в уравнениях (111,29)
и (111,29а) стремятся к предельным значениям [см. выражения (111,12)
и (III,14)] /Гр,,, । и, соответственно, Изуравнения (III,29а)
следует, что начальный (при Г —0) наклон кривой, выражающей
ннвисимость л от Г, имеет вид:
lim
г -к о VI /А, Г
lim
р -> о
т— ) lim
др JA, Гг ->
(111,30)
Па рис. II 1,7 показана вычисленная из экспериментальной изо-
термы адсорбции этана на графитированной термической саже при
173 К (см. рис. Ill,1) зависимость отношения Г/р от р. На рис. III,8
ориеодспа зависимость л от Г, вычисленная с помощью формулы
(111,29п) но приведенным на рис. 111,7 данным. Кривая, выражаю-
щая эту ипписимость, имеет точку перегиба. Она сильно отклоняется
от прямой
л-Л7Т (HI,31)
гоетпетствующой идеальному состоянию адсорбированного вещества.
в Нлкпл 11К0 ИЗ
Для получения уравнения состояния адсорбированного вещества
в аналитической форме надо привлечь ту или иную модель. В гл. IV
рассмотрены уравнения состояния в вириальной форме и в форме,
аналогичной уравнению Ван-дер-Ваальса. Подробно различные мо-
дели состояния адсорбированного вещества, в том числе модели,
учитывающие ассоциацию адсорби-
рованных молекул и критические
явления, рассматриваются в книге
Г. И. Березина и А. В. Киселева
Рис. III,8. Зависимости поверхно-
стного давления л = о? — о = —Ап,
изменений свободной энергии — AF
и внутренней энергии —&U от вели-
чины адсорбции Г этана на графити-
рованной термической саже при 173 К .
За стандартное состояние газа принято
р° = 760 мм рт. ст. Изотермы адсорбции
приведены на рис. III. 1.
Рис. III,7. Зависимость отношения Г/р
от р, полученная из изотермы адсорбции
этана на графитированной термической
саже при 173 К, показанной на
рис. III,1.
этой серии монографий по химии поверхности и адсорбции. Молеку-
лярно-статистический вывод уравнения состояния адсорбирован-
ного вещества дается ниже в разд. 3 гл. VI.
4. Изменения при адсорбции поверхностных термодинамических
функций
Поверхностная свободная энергия на границе твердое тело —
газ, отнесенная к единице площади поверхности раздела, равна [17]
Fs = "гГ'аЦа-р Г'п
(Ш,32)
Для поверхности раздела твердое тело — вакуум
+ (IIL33)
114
В случае нерастворяющего адсорбат адсорбента ра = р^. Изме-
нением автоадсорбции самого твердого тела в результате адсорбции
газа в рассматриваемом случае можно пренебречь. Поэтому изме-
нение' поверхностной свободной энергии в результате адсорбции
газа можно представить в виде:
AFS - Fs - F°s - о -о0 + Гр (111,34)
Учитывая выражения (111,26) и (111,29) и применяя формулу ин-
тегрирования по частям, получаем:
г
AFS-J р^Г (111,35)
о
Вводя сюда выражения (III,2) и (III,3) для р, получаем:
г
ДГз=Гр£(Т) + ЯТ f Inarfr (III,36)
. о
В это уравнение можно ввести константу Генри, используя соотно-
шении a --- Гу3/Ку, с, 1- Это дает:
г
A/'s Грг(Г)-ЯГГ1пК'г с 1+ЛТ J In (Гу5) dT (III,37)
о
Пи выражений (111,32) и (111,35) следует, что дифференциальное
мольное изменение поверхностной свободной энергии
AAV I1 (Ш.38)
где /’я дифференциальная мольная свободная энергия адсорбированного
шчце<тш1 (Л/Л, - Fs, так как в выражении (111,33) не зависит от Г).
Соответственно из выражения (111,36) следует, что
AFS р«(7’) | 7П’ 1п л (111,39)
н ил выражения (III,37), что
AFs = p«(T) — 7?TlnFr (111,40)
В случае адсорбции идеального газа у я = 1 и р == cRT, поэтому
ЛЛ = рг(Г) + ЛГ1пе-ц£(Г) — RT In ЯГ + ЯГ In р (111,39а)
Сели же и для адсорбированного вещества у* = 1 (малые Г), то
AFs=pfi (Г)-ЯГ In с, А+ЯГ In Г =
-р£(Г)~ RTlnRT-RTlnKY} р ЯГ In Г (III,40а)
Ни выражений (111,36)—(III,40а) видно, что помимо величин, харак-
теризующих адсорбцию на данном адсорбенте, т. е. величин Кг С>1
п Г, величины \FS и AFS через рй (Т) определяются свойствами
н* 115
самого адсорбата. Поэтому вычислить &FS и &FS из адсорбционных
измерений нельзя, можно лишь проследить sfa их изменениями с из-
менением Г. В выражение для изменения при адсорбции поверхно-
стной энтропии AS1., ™ —(dkFJdT)A, г войдет член, содержащий
рЛ (Г)+ dy8 (T)ldT, а в выражение для изменения поверхностной
внутренней энергии AC7S = &FS -|- TASs -— член, содержащий
d\L6(T)ldT. Наконец, в выражение для изменения поверхностной
теплоемкости ACS — (d\Us/dT)At г войдет член, содержащий
d2it8(T)/dT2. Таким образом, во все эти термодинамические харак-
теристики входят константы, относящиеся к адсорбату в объемном
газе при а = 1 [когда рЛ = pg (Г)].
Поэтому для получения термодинамических характеристик соб-
ственно адсорбции надо эти величины pg(T), d[i8 (T)/dT и d2[i8 (F)/dT2,
не связанные с адсорбцией, исключить. Это можно сделать,
выбрав начальное состояние адсорбата, отличным от равновесного
адсорбированного состояния.
5. Изменения термодинамических функций при переходе
поверхности раздела из стандартного состояния адсорбент —
вакуум и адсорбата из стандартного или равновесного
состояния объемного газа в равновесное или в стандартное
состояние системы адсорбент — адсорбированное вещество
Выбор стандартных состояний для поверхности адсорбента и объ-
емного газообразного адсорбата. В предыдущем разделе были полу-
чены выражения для изменения в результате адсорбции поверх-
ностной свободной энергии, причем в качестве стандартного началь-
ного состояния для поверхности раздела до адсорбции газа было
выбрано состояние поверхности раздела твердое тело — вакуум.
Введем теперь стандартное начальное состояние также и для сво-
бодного газообразного адсорбата до адсорбции. Обозначим относя-
щиеся к этому начальному состоянию величины концентрации, коэф-
фициента активности и давления соответственно через с°, у8, ° и р°.
Свободная энергия Г молей газообразного адсорбата в этом состоянии
Fg> ° = ГУ^°---Г °) (111,41)
где F^i ° — мольная свободная энергия; ц ° — химический потенциал; ° —
мольный объем газа при давлении р° и рассматриваемой температуре.
Интегральное и дифференциальное изменение свободной энергии.
Для получения интегрального изменения свободной энергии при
переходе адсорбата из выбранного выше начального состояния
адсорбент — вакуум в состояние равновесия адсорбат — адсорбент
надо из выражения (Ш,32) вычесть выражения (111,33) и (111,41)
или, что то же самое, вычесть выражение (111,41) иэ выражениями,35):
г
= — (F°s+Fg> °) = &Fs~Fg> o = ^Fs~-ГFgf f Дц^Г+Гр2Г£’ °
mJ' тп.
О
(П1,42)
116
где Ag = g — g° представляет изменение химического потенциала адсорбата
при его переходе из начального стандартного состояния объемного газа в состо-
яние равновесия с адсорбентом при величине адсорбции Г.
Пользуясь выражениями (III,2), (III,3), (III,7), (III,9), (111,11}
и (111,12), получаем
Др. = -ЯГ1п-^г= - RT (1п^с>1-1п^) (Ш,43>
При адсорбции идеального объемного газа в формулах (111,43}
надо а и а0 заменить на с и с°.
Если, кроме того, Г 0, то у® 1 и в этом случае
АИ1= -Нт(1пКт с j-ln-VU -RT (1пКг р
(Ш,43а>
Индекс 1 у величины Apt х указывает на условие Г -> 0.
Подставляя выражения (111,43) для Др в уравнение (111,42)».
в случае адсорбции неидеального объемного газа получаем:
(г \
Г In кт с !- Г In^dr °
’ ’ J О I
0 '
(III,44>
При адсорбции идеального объемного газа в формулах (111,44}
надо а/а° заменить на с/с° или plpQ, a p°F& ° на RT; при замене
в этом случае Kr,c,i на Kr,p,i надо а° — с° заменить на р°. Таким
образом, в случае адсорбции идеального газа
/г \ г г
AF^RT \ 1пуо ЙГ + Г = — RT Г (In Д-р, с> 4-1)- I ln-^-dr
\о / L о
- — RT
Г
Jr vs
о
(Ш,44а>
Среднее мольное изменение свободной энергии адсорбированного
вещества
AFm=~
(Ш,44б>
При адсорбции идеального газа
/ г \ / г \
\Fm = RT I 4г f 1п Л <*г + 1 1 = ЛГ Цг 1п <*г+! (П1,44в}.
I J. J С'" I \ k J I
\ О / \ О /
На рис. III,8 представлена зависимость —AF от Г, полученная
из изотермы адсорбции этана на графитированной термической
снже при 173 К (см. рис. Ш,1) по формуле (III,44а). Из рис. 111,8-
117
видно, что значения —&F и л = —До в области малых Г резко от-
личаются друг от друга.
Для дифференциального мольного изменения свободной энергии
при адсорбции газа из уравнения (111,42), учитывая выражение
ЦП,43), получаем:
.= ДИ+Р°Г& ° = RT In -^ + P9ve^ ° = - RT In кт +7?r ln£f°
(Ш,45)
Рис. Ill,9. Зависимости диффе-
ренциальных изменений свобод-
ной энергии —А/* и внутренней
энергии — А £7 от величины ад-
сорбции Г этана на графитирован-
ной термической саже при 173 К,
вычисленные из изотерм адсорб-
ции, приведенных на рис. Ill,1,
иизизостер рис. 111,11 (см. ниже).
Горизонтальный пунктир обозначает
теплоту конденсации L при 193 К.
Точка 1 рассчитана из статических
измерений по вириальному уравне-
нию (IV, 12).
При адсорбции идеального газа
kF = RT (in-^4
1) = RТ (in 1) = - RT (in KY c> , - In 1) =
= -Лг(1пКг p t_ln£f-l)
(III,45a)
На рис. Ill,9 представлен пример зависимости величины \F от Г,
вычисленной из изотермы адсорбции этана на графитированной
термической саже при 173 К (см. рис. Ill,1) по второй формуле
ЦП,45а).
Наиболее простые выражения для AF получаются для адсорбции
идеального газа при Г -+ 0:
AFX = limAT*’ — — RT (In Kr . 1
г-> о \ ’ ’ c~
- -RT (1пА-г> i~ln—-----------1
(111,456)
118
Стандартное дифференциальное мольное изменение свободной
энергии. Полученные выражения (111,44) — (III,44в) для &F и AFm if
выражения (111,45) — (111,456) для AF и А/\ содержат переменную ве-
личину Г. Сопоставлять адсорбционные системы удобно, приняв для Г
некоторое стандартное значение Г°. Подставляя величину Г° вместо Г
в формулы (111,45) и (III,45а), получаем выражения для стандарт-
ного дифференциального мольного изменения свободной энергии,
при адсорбции газа:
Д^° = RT (in ЯГ| с ! - In ГТ' + p°pg. 0 (III,46}
При с -> 0 и Г°О, когда можно пренебречь взаимодействиями
между молекулами адсорбата в объеме газа и в адсорбированном
состоянии
Л7; м°т1 -лг(1пкг>С)1-1пП—1)= -яг(1пкг>р,
(III,46а)
Для упрощения формулы (III,46а) целесообразно выбрать зна-
чения Г", с° и р° еще и так, чтобы при достаточно малых Г°, с°
и р° соблюдались условия:
^-=1 (Ш,47).
или
Г9
^-=1 (III,48)^
Получающуюся при таком выборе стандартных величин Г° и с°
величину стандартного дифференциального изменения свободной
энергии обозначим через AJ’r, Очевидно, что при Г°/с° = 1
Л?г, с, t = - нт (In t -1) (III,49).
Соответственно при Г°//?° = 1
А?г, р, 1 - ~ RT (In ! -1) (111,50)
Величины А/’г сд и Aff^j не равны друг другу вследствие-
того, что при Г%° = 1 соответствующая величина pQ отличается
от множителем RT, так как р° = cQRT. Поэтому, если стандарт-
ное состояние газообразного адсорбата выбрано так, что с — с°
и Г%° — 1, для того же стандартного состояния отношение
Г7р° 1. И наоборот, если стандартное состояние газа р = р°
выбрано так, что Г7р° = 1, то для этого стандартного состояния
Г7с° #= 1.
Для простоты записи величину Kr)C,i обозначим в дальнейшем
через а соответствующую величину AJ’r.c.i через AFJ.
Таким образом
(lntfi-1) (III,49а)
11Ф
Величины Кг и AFJ представляют термодинамические характе’
ристики взаимодействия адсорбат—адсорбент, так как при Г, с ->
-> 0 и Г°, с°->0 взаимодействиями адсорбат—адсорбат можно пре-
небречь (ys = 1 и у8 = 1). Примеры величин и AFJ для адсорб-
ции некоторых неорганических веществ разного строения на графи-
тированной термической саже при одной и той же температуре
приведены в табл. III,2.
Таблица 111,2. Термодинамические характеристики адсорбции
некоторых молекул на графитированной термической саже при 173 К.
Значения Кх получены из газохроматографических измерений [51],
а значения AF*, &S*/R и AZ71 рассчитаны соответственно по уравнениям
(Ш, 49 а), (III, 59 в) и (III, 64 б)
Адсор- бат Kf мкм дкг кДж моль ASJ -Д1Л, кДж моль Адсор- бат Kt мкм дг* кДж МОЛЬ R -Д1Л, кДж моль
R
Аг 0,0152 7,47 11,2 8,6 Кг 0,105 4,68 11,0 11,0
О2 0,0162 7,38 11,2 8,8 n2o 0,547 2,27 11,2 13,8
N2 0,0181 7,19 11,3 9,1 NOo 0,771 1,81 11,8 15,2
со 0,0305 6,46 10,9 9,2 Хе 1,44 0,94 10,9 14,7
Дифференциальное мольное изменение свободной энергии адсор-
бата при адсорбции в равновесных условиях. Обозначим эту
величину через kFer Очевидно [см. выражение (111,45)], что
°~F8l (111,51)
Подставляя сюда выражения (111,45) для АЛ1 и F8; ° — F8 =
= —А[х +pV^° ~p°V8i°f получаем
(III,51а)
При адсорбции идеального газа
"KFeq^RT (Ш,51б)
Эти выражения соответствуют равновесному переходу моля адсор-
бата из объемного идеального газа в адсорбированное состояние
при данном Г.
Стандартное изменение химического потенциала адсорбата. Под-
ставляя стандартную и достаточно малую (чтобы ys,° — 1) вели-
чину Г = Г° в уравнение (111,43) для адсорбции идеального газа,
получаем
f Г°\ / Г°\
= (^1п АГ с (^1п Аг Р( i-ln —J (III,52)
420
Выберем теперь стандартные малые значения Г° и с° или Г° и р°
так, чтобы выполнялись выражения (111,47) и (111,48). В этом случае
др,*, с t = - rt In Kr, Ct t (Ш,53)
и соответственно
Др,* i= — RT ln^r i (Ill,54)
Используя сокращенные обозначения Дрг.с, i = ДрГ и Кг,с, i =
= К19 получаем
Др* = — rt In Ki (III,53а)
Интегралвное и дифференциальное изменение энтропии. Диф-
ференцируя выражение (111,42) по температуре, получаем:
bS = ASs- rs£ ° = - f (^gVdr-rp° (III,55>
\ "* /Г О'-t
о
Из выражения (111,43) для Др, следует, что
При адсорбции идеального газа
Вводя выражения (111,56) в уравнение (111,55), получаем следую-
щие выражения для изменения энтропии в результате адсорбции Г
при той же активности а° в стандартном состоянии объемного газа
Д£=-Я
ln-^-dr + T
d In^i
1 ~dT
г \
Г IV I
rin^i-j In-—dr 1 + ЯТ
о '
dT
(111,57)
121
При адсорбции идеального газа
Среднее изменение энтропии на моль адсорбированного веще-
ства при сделанных допущениях относительно адсорбента равно:
Д5
(111,58)
Дифференциальное мольное изменение энтропии при адсорбции
газа в соответствии с выражениями (111,45), (111,55), (III,56) и (111,57)
имеет вид:
= « [i“ +т ¥ - т (i?)J -'’"тг2 <П1'59>
где Ss — дифференциальная мольная энтропия адсорбированного вещества.
При адсорбции идеального газа
Д5
d In Кр „ j f д In Vs \
= 2?[ln^r> р, i + T----------(~^Л)Г] (111,59a)
122
Кел и Г настолько мала, что у® = 1, то при стандартном состоянии
«доильного объемного газа, определенном через с°, р°, получаем!
A.S'> lim &S = R 1) =
Г -+• 0 \ al C J
/ din Ар ~ 4
= 7? (jnKr> р>1+Г------^1
(III,596)
На рис. 111,10 представлена зависимость от Г среднего мольного
«вменения энтропии \Sm = Д8/Г, где Д8 вычислено по второй фор-
муле (III,57а) для адсорбции
in n па, рассматриваемого в объ-
еме как идеальный газ, на
графитированной термической
гаже при 173 К (изотермы
адсорбции представлены на
рис. 111,1). На том же рисунке
дли этой лее системы предста-
нлепн зависимость Д8 от Г,
вычисленная для той же темпе-
ратуры по второй формуле
(III ,59а). Входящие в форму-
лы (III,57а) и (III,59а) произ-
водные (din р!дТ)у были опре-
делены из серии изотерм ад-
сорбции этана на графитиро-
ванной термической саже,
иимерепных при разных тем-
пературах, с помощью так на-
лынаомых изостер адсорбции
(см. ниже стр. 126 этого раз-
дела). Из рис. 111,10 видно,
что зависимости Д8т и Д8
от Г имеют характерный для
адсорбции на однородной по-
верхности при низких темпе-
ратурах волнообразный вид.
При Г >0 Д8 оо. С рос-
Рис. 111,10. Зависимости дифферен-
циального мольного изменения энтро-
пий при адсорбции Д5 и среднего моль-
ного изменения энтропии при адсорб-
ции Д8/Г = ЛЗт от величины адсорб-
ции Г этана на графитированной тер-
мической саже при 173 К.
Соответствующие изотермы показаны на
рис. 111,1, а изостеры — на рис. III,11.
гом I' кривые проходят через минимум при приближении к плот-
ному заполнению первого слоя (вследствие наибольшего при зтих
условиях торможения поступательного и вращательного движения
молекул) и снова поднимаются при переходе к преимущественной
адсорбции во втором слое (вследствие более свободного перемещения
молекул но поверхности второго слоя).
Стандартное дифференциальное мольное изменение энтропии^
Выбирал малые стандартные значения Г° и с° или Г° и р° так, чтобы
выполнялись выражения (111,47) и соответственно (III,48), из формуя
123,
111,596) получаем следующие выражения для стандартного диффе-
ренциального мольного изменения энтропии при адсорбции газа:
Д5г,с, 1 =bS* = R flnKi+T -'V1- 1) (111,59b)
1 \ al J
- -.у / d ] n К-p п j \
Д5Г, p, l = fl(hiKr> Pf l + ?---jf J (111,59г)
Дифференциальное мольное изменение энтропии адсорбата при
адсорбции в равновесных условиях. Обозначим эту величину
через Д5е?. Очевидно [см. выражение (111,59)], что
Д5е?=Л-< = Д5 + <’°-^ (111,60)
В случае адсорбции идеального газа, подставляя сюда выражения
(III,59а) для Д8 и — 8^ = Rin clc°, получаем
Л5„--Я [Г (У£Г)г + 1]_ _ ПТ (^)г-
rt['rrd\nK1 „
=7?L “i?—
-1 = RT
г J
d In К р р ।
dT
д In Ys
дТ
'Г.
(111,60а)
При Г -> 0 ys -> 1 и
&Seq, 1— R T
d In К\
dT
d In К р р ।
dT
(Ш,606)
1
Величина ASeq не равна величине —dAFeq!dT = — R [см. вы-
ражение (111,516)], потому что при адсорбции, как и при конденсации,
мы имеем дело с гетерогенной системой.
Интегральное и дифференциальное изменение внутренней энергии.
.Для получения интегрального изменения внутренней энергии при
адсорбции газа Ди = ДР + ТД8 надо воспользоваться выражени-
ями (111,42) для ДЕ и (111,55) для Д8
MJ=bUs~U$l
= J j(^)rdr+rz>
О О
тде
D== рО (vg,
U P * dT
(Ш,61)
(П1.62)
Подставляя сюда выражения (111,43) для Др, и (111,56) для (дДр/дТ)г,
получаем
г
Ц(^)г
^Г + Г/) = 7?Т2 Г
d In К\
~dT
(III,61а)
о
124
При адсорбции идеального газа D ~ 0 и
г г
ЛР= - № j (^)г ЙГ» j ЙГ+ГЛГ -
о о
= № Г
d In jfiTp р ।
dF
г
Кд In ys \
~~дТ~~/г
о
dr +ГЯТ
(III,616)
Если рассматривать столь малые Г, что можно принять ys = 1, то
/? 1 ri АГ 1 d In р tj 1
’ р’ ^ДГГ (111,61b)
d л CLJL
Среднее изменение внутренней энергии на моль адсорбированного
вещества при сделанных допущениях относительно адсорбента
Л£Лп = ^ (III,63)
Дифференциальное мольное изменение внутренней энергии при
адсорбции газа в рассматриваемых условиях получается из выраже-
ний (111,45) для AF и (111,59) для AS или дифференцированием вы-
ражения (III,61а) по Г:
At/ = At/s-E/fe °=US-U%1 “-Дц-Т ^)г+р = -ДТ2 + Д =
"d In К± f д In у® \
d7 к-dF” /Г.
(111,64)
где Us = Fs + TSs — дифференциальная мольная поверхностная внутренняя
энергия адсорбированного вещества; ° = + TS^ ° — мольная вну-
тренняя энергия адсорбата в объемном газе.
При адсорбции идеального газа D ~ 0 и из выражений (111,616)
следует, что
Предельная величина дифференциального мольного изменения
внутренней энергии. При Г -> 0, ys -> 1, поэтому
Лйг lim ДГ = tJJm, i = R Т2 = _R /*1п АХ = RT2 р> 1 J- R Т
Г""0 dT d(4-) dT
(Ш,64б)
где i — величина при Г -> 0.
125
В табл. 111,1 приведены значения Лиг для адсорбции w-гексана
и бензола на графитированной термической саже, полученные газо-
хроматографическим методом из зависимостей от 1/Т, Значе-
ния Кх при разных Т для адсорбции различных газов (паров) на
графитированной термической саже, полученные разными методами,
приведены в табл. III,2 и в Приложении в табл. П,2,П,3.
Изостеры адсорбции и зависимость константы Генри от темпе-
ратуры. В выражения (111,57) и (111,59) для AS и AS, а также в вы-
ражения (III,61а) и (111,64) для АС/ и АС/ входят температурные?
Рис. 111,11. Зависимости 1g р от 1/Т (изостеры адсорб-
ции) для адсорбции этана на графитированной терми-
ческой саже для разных величин адсорбции Г, мкмоль/м2
(числа на кривых).
коэффициенты (dlna/dT)r. В случае адсорбции идеального газа
в выражения (III,57а), (III,59а), (III,60а), (111,616) и (III,64а)
входят члены, содержащие производные (dlnc/dT)r или (dlnp/dT)r .
В выражения (III,64а) для АС/ входят, в частности, члены
~RT2(dlncldT)r = /?[6»1п с/д (1/Г)]г или —RT2 (д1пр/дТ)г =
— R [д1пр/д(1/Т)]г. Зависимости с илир от Г (или inc и 1пр от Т или ИТ)
при постоянных величинах Г, нужные для определения этих производ-
ных, называются изостерами.
Изостера может быть измерена непосредственно нулевым весо-
вым методом путем повышения равновесного давления р при повы-
шении температуры так, чтобы при введении соответствующей
поправки на изменение плавучести чашечки или гильзы весов с ад-
сорбентом при изменении р величина адсорбции оставалась неиз-
126
менной. Изостеры могут быть найдены также из серии Ьзотерм
адсорбции, измеренных при разных температурах. Пример таких
изотерм для адсорбции этана на графитированной термической саже
был приведен на рис. Ill,1. На рис. III,11 показаны определенные
из них изостеры — зависимости 1пр от ИТ для разных Г. Изостеры
во многих случаях довольно близки к линейным. Из выражения
(III,64а) видно, что наклон изостер изменяется с изменением Г
в соответствии с изменением A U.
___В выражения (111,57), (III,57а) для AS, (III,59) и (III,59а) для
AS, (111,596) для ASX, (Ш,59в) и (III,59г) для AS£ и (III,60а) и
(111,606) для AS^, а также в выражения (III,61а), (111,616) для
AU, (III,64), (III,64а) для АС7 и (111,646) для АС7Х входят члены,
содержащие температурный коэффициент 1пХг с х или In Хг,р, v
На рис. III,4 представлена зависимость In Хг> с, i от i/Т для рас-
смотренного выше случая адсорбции этана на графитированной
термической саже. Эта зависимость довольно близка к линейной.
Дифференциальное мольное изменение внутренней энергии адсор-
бата при адсорбции в равновесных условиях. Обозначим эту величину
через Лиег Очевидно [см. выражение (111,64)], что
Kueq^us-~vl = ku+u^ °~-и^ (III,65)
В случае адсорбции идеального газа Ufa ° = Ufa, так что
(Ш,65а)
где Д£/ выражается формулами (111,64а).
При Г 0, когда ys-> 1 получаем, в частности
kUeq, 1=~Кй1 (111,656)
где выражается формулами (111,646).
Интегральное и дифференциальное изменение теплоемкости.
Интегральное йзменение теплоемкости при адсорбции газа
Г в о
ЛС -_ (»?)г _ - 4 (^)г ir+
о
(111,66)
В соответствии с выражениями (111,56) производная
№
fd In Zd2 In а
\ дТ 7г+ 1 \ ^Г2’
Г dlnKx
= -2R \_~dr~
dlnys\ -1 ргЧпК! ZdMnfX -]
ОТ 7rJ L dT^ \ ат* 7rJ
(111,67)
Вводя эти выражения в уравнение (111,66) и обозначая для крат-
кости
№ ’ G
(111,68)
127
получаем:
kC^-RT
~dT
dY + T
---RT\2
— VG
d^Kx f Z^ln^\
0
(Ш,69)
В случае адсорбции идеального газа G = 0, поэтому
ДС = - RT
dV 4-7?Г —
о о
г dln^i
dT
= RT
d^lnKr C/^Iny^
dT* J \ dT* )v
о
d In Kr
dT~
p, 1
d2 In KY> p, A
Jf2
(III,69a)
= — RT
+ Т
Среднее изменение теплоемкости на моль адсорбированного вещества
АС
Ш — ТГ
(III,70)
Теплоемкость отличается от термодинамических функций F, щ S
(в рассматриваемом случае) и U тем, что для нее может быть получено
абсолютное значение. При сделанных допущениях относительно
инертности адсорбента средняя мольная теплоемкость адсорбирован-
ного вещества
128
Дифференциальное мольное изменение теплоемкости при адсорб-
ции газа в соответствии с выражениями (111,66) и (111,69) имеет вид
А — \г _с$’ ° — Се —С8* ° — __Т ( __G_
ЛЬ~\дГ )т~^5 CV,m — cs ('у,™— 1 \ qt% Jr
, „/d2lna\ "] „
— RT L2 ufJi-+ т ;rJ~G==
= RT{ 2
"din#! Zdlnys\ "1
_ dT \ QT /rj’1’
rd2 InKi
_ dT2
(111,72)
В случае адсорбции идеального газа в выражении (111,72) G = О,
поэтому
Л-С_„[2(^)г+Г(^)г]_
Дифференциальная мольная теплоемкость адсорбированного ве-
щества. В выражении (111,72) Cs представляет собой дифференциаль-
ную мольную гиббсовскую поверхностную теплоемкость. Таким
образом, из верхней строки выражения (111,72) следует, что диф-
ференциальная мольная поверхностная теплоемкость
(111,726)
Дифференциальная теплоемкость адсорбированного вещества Са
может быть получена из средней мольной его теплоемкости, т. е.
из выражения (111,71) по формуле, связывающей парциальные
и средние величины:
Cfl=Cm + r(^-)r = Cf.’/m+AC = Cs (111,73)
Предельная величина дифференциального изменения теплоем-
кости при Г -> 0. При Г -> 0 / -> 1, поэтому из формул (111,72)
следует, что
ДС1= ИтДС = ДСот,1 = 7?Т + Г =
Г -> 0 \ al al * /
(d In К р >n 1
2—
al
где Дб’/д,! — величина при Г -> 0.
9 Заказ 1180
d2ln^r n j
LT------L>
dT2
(111.72b)
129
Дифференциальное мольное изменение теплоемкости адсорбата
при адсорбции в равновесных условиях. Обозначим эту величину
через ДС^. Очевидно [см. выражение (111,72)], что
\С^С3~-С^ т = Хе + С^°т^С^т (111,74)
В случае адсорбции идеального газа Су\т = Cyiin, так что
АС^-АГ (III,74а)
где АС выражается формулами (III,72а).
При Г -> О, когда / -> 1, получаем, в частности,
АС^а^ДСг (111,746)
где АСа выражается формулами (III,72в).
Примеры зависимости теплоемкости адсорбированного вещества
от велйчины адсорбции. В уравнения (111,66), (111,67), (111,72),
(III,72а) и (III,72в) входят вторые производные по температуре
величин активности (концентрации) или давления адсорбата в объем-
ном газе или констант Генри и коэффициентов активности адсорби-
рованного вещества. Выше уже отмечались трудности определения
этих производных из статических или динамических измерений. Для
исследований же зависимости теплоемкости адсорбированного ве-
щества от температуры надо определять соответствующие третьи
производные по Т. Поэтому необходимы прямые калориметрические
измерения теплоемкости адсорбционных систем.
Измерения теплоемкости адсорбционных систем при разных п5
и Т подробно рассматриваются в одной из следующих монографий
этой серии, подготавливаемой Г. И. Березиным и А. В. Киселевым.
Поэтому здесь мы ограничимся только двумя примерами. На рис.
III,12а показана определенная из таких измерений зависимость
величины Ст [см. выражение (111,71)] от величины адсорбции Г
бензола на графитированной термической саже [38, 71, 72]. В этом
случае взаимодействия адсорбат — адсорбат малы, поэтому мольная
теплоемкость адсорбированного бензола при Г 0 меньше моль-
ной теплоемкости жидкого бензола, но больше мольной теплоемкости
газообразного бензола (см. разд. 4 гл. X). С ростом Г начинают про-
являться слабые взаимодействия адсорбат — адсорбат, в результате
чего мольная теплоемкость адсорбированного бензола постепенно
растет, а с переходом к преимущественно полимолекулярной ад-
сорбции приближается к мольной теплоемкости жидкого бензола С\п.
На рис. 111,126 представлен другой типичный случай — зависи-
мость от Г величины Ст этанола, молекулы которого на поверхности
графитированной термической сажи с ростом Г сильно ассоциируют
[38, 39, 73]. Благодаря этому в величину Ст большой вклад вносит
энергия, затрачиваемая на частичную диссоциацию (разрыв части
водородных связей этих ассоциатов), происходящую при нагревании
130
такой системы даже при постоянной Г. В результате этого с ростом Г
величина Ст вначале быстро растет, проходит через максимум и
только после этого начинает приближаться к мольной теплоемкости
жидкого этанола С1т.
Зависимость изменения внутренней энергии от температуры.
Зная зависимость &U или "KU от Г^при одной температуре и соответ-
ствующую зависимость ДС или ДС от Т и Г, можно определить ДС7
Рис. 111,12. Зависимости
средней мольной теплоем-
кости адсорбированного ве-
щества Ст от величины
адсорбции Г на графи-
ти рова иной термической
саже при 300 К:
а — для бензола; б — для эта-
нола.
Горизонтальные пунктиры ука-
зывают значение теплоемкости
соответствующих жидкостей,
вертикальные пунктиры - пере-
ход от преимущественного запол-
нения первого адсорбционного
слоя к преимущественному за-
полнению последующих слоев.
или \й для разных температур» Например, для*данного Г в интер-
вале температур от Т
' ___ - ш____
ДС7Т=Д£7Т1+ J Д€ dr (III,75)
Т1
Величина ДС при малых Г обычно не велика. Если для неболь-
шого интервала температур принять эту величину постоянной, то
в этом интервале
Д^т = м7Т1 + (г —гг) дс . (Ш,76)
В пределе при Г -х 0:
I* Tj + (T — 2’i)AC1 (111,76а)
Таким образом, по крайней мере при небольших Г величина —Kut
слабо и приблизительно линейно уменьшается с ростом температуры.
9* 131
При наличии фазовых и молекулярных переходов в адсорбиро-
ванном веществе (сюда относится, например, частичный разрыв
водородных связей) энергия, затрачиваемая на эти переходы при
нагревании адсорбционной системы при постоянной величине адсорб-
ции, включается в величину Ст (см. рис. III,3) [37—39, 71]. В этих
случаях Д?7 может очень сильно изменяться с изменением ns и Т.
Пример этого показан на , рис. III,2 [32, 34, 36] (в этом случае
—А £7 — qv — дифференциальной теплоте адсорбции, см. ниже
разд. 7 этой главы).
6. Изменения термодинамических функций в некоторых
частных случаях проведения адсорбционных опытов
В предыдущем разделе мы нашли изменения термодинамических
функций при рассмотренном переходе поверхности адсорбента с пло-
щадью А = 1 в стандартном состоянии адсорбент — вакуум и Г
молей чистого адсорбата в стандартном газообразном состоянии
при концентрации с° (давлении р°) в равновесное состояние адсор-
бент — Г молей адсорбированного вещества. Нас интересовало при
этом только количество Г молей адсорбата, перешедших в адсорби-
рованное состояние. Происходящие при этом изменения химического
потенциала Др. и интегральные изменения свободной энергии Д/\
энтропии Д.8, внутренней энергии А17 и теплоемкости АС выражаются
соответственно формулами (111,43), (111,44), (111,57), (ШДИа) и (111,69).
Мы нашли выражения для Д/\ Д5, Д<7 и АС в рассмотренном термо-
динамическом процессе, позволяющие вычислить их значения,
зная изотермы адсорбции (константы равновесия и коэффициенты
активности) при разных температурах и введя соответствующие
средние мольные величины для газообразного адсорбата в стан-
дартном состоянии °, S^°, Ufa ° и Cfy°m [см. выражения (111,42),
(111,55), (111,61) и (111,66)].
Рассмотрим теперь интегральные. изменения термодинамических
функций F, 5, U и С, происходящие в реальных процессах адсорбции,
проводимых при трех наиболее важных частных условиях подвода
адсорбата в сосуд с адсорбентом.
Адсорбция газа, (поступающего из резервуара с постоянным
давлением. Этот случай наиболее близок к рассмотренному в пре-
дыдущем разделе, так как выражение (111,41) предполагает, что
газообразный адсорбат в начальном состоянии находится при по-
стоянном давлении р°. Схема проведения опыта представлена на
рис. 111,13. Чистый адсорбент находится в подсистеме 7, а чистый
газообразный адсорбат в подсистемы Па (в поджимном баллоне,
в газовой бюретке [1]). Подсистемы I и Па соединены краном или
затвором. Давления газа в* подсистемах I и Па измеряются мано-
метрами, работающими без изменения объема газа [1] (например,
мембранными). Вся система 1-\-На находится в термостате, в част-
ности в изотермическом калориметре (см. разд. 7 этой главы).
132
При давлении р° средний мольный объем газа в подсистеме Па
составляет V^°, При сообщений подсистем I и Па из подсистемы
Па в подсистему I с адсорбевтои переводится п молей адсорбата
при сохранении в подсистеме Ih постоянного давления путем,
например, соответствующего сжатия сильфона или поднятия ртути
Рис. ШЛЗ. Схема установки,
позволяющей проводить адсорб-
ционный процесс в системе с по-
стоянным объемом:
1 — подсистема с адсорбентом; Па —
подсистема с газообразным адсорбатом
при постоянном давлении; Пб —
подсистема с газообразным адсорбатом
при постоянном объеме; Не — подсис-
тема с жидким адсорбатом; 1 — ад-
сорбент; 2 — кран или затвор, ведущий
к источникам адсорбата в подсис-
темах Па, Пб и Пв; 3 — подводящие
к этим подсистемам трубки; 4 — мем-
бранный манометр подсистемы I; 5 —
кран или затвор для откачки подси-
стемы /; в — газовая бюретка или
поджимной баллон с газом постоянного
давления; 7 — баллон с газом посто-
янного объема; 8— жидкостная микробюр(>ца1- » __________ ________*____________
мах Па и Пб; 10 — краны или затворы для откачки и впуска адсорбата в подсистемы Па,
Пб и Пв; 11 — кран или сильфон для поднятия ртути в газовой бюретке б (косой штрихов-
кой отмечено изменение объема газа при постоянном давлении в подсистеме Па).
У5
УЗО
yJO
VJO
*г-3
г
3-
3-
па
Пб
мембранные
манометры в
подсисте-
4
1
9
9
6
7
8
f
в газовой бюретке сосуда Па. При этом в подсистеме Па происходит
следующее изменение свободной энергии |см. уравнение (III,41)]
Д^„=-п(ц« (HI.77)
Из перешедших в подсистему / п молей адсорбата ns молей ад-
сорбируется, а п8 — п — п5 молей остается в равновесной газовой
фазе. Изменение свободной энергии подсистемы I составляет:
+ .lA/’gl ng(p-p^) (111,78)
где AAFS — изменение поверхностной свободной энергии; р — равновесное
давление; — средний мольный объем газа при давлении р.
Полное изменение F всей системы I 4- Па в рассматриваемом
процессе составляет
Па = Я АГ5 + ng (ц. pV 8)~п (р° - Z V% °) =
= ЛДЛ-п’(Дн+Р°^’) I ««(Дн + Ро^°-pVQ (Ш.79)
где Ар = р — р°.
Используя уравнения (111,35) и (III,42), получаем
ns
Д^, Па = J ДМ"* + "’Л* (Др+^Ч’ °-pV£) (111,80)
133
Для более точного измерения адсорбции газа по схеме 7, Па
«мертвый» объем газа над адсорбентом в подсистеме I стремятся
сделать небольшим. При достаточно большой величине площади
поверхности адсорбента А в этом случае п8 ns и из выражения
(111,80) следует, что
ns
11а f ^dn5+nsp°V^ ° (111,80a)
о
а для A = 1
—\ Afi + ° (111,806)
-Z1 <J
0
Таким образом, это выражение совпадает с выражением (111,42)
при условии ng< ws.
В случае адсорбции идеального газа
Па
А
Г
£ ДрйГ + ГЯГ
о
(III, 80в)
При условии п8 ns интегральные изменения энтропии, внут-
ренней энергии и теплоемкости при проведении процесса адсорбции
по схеме 7, 77а выражаются полученными в предыдущем разделе
формулами (111,57), (III,61а) и (111,69).
Адсорбция газа, поступающего из резервуара с постоянным
объемом. В этом случае используется подсистема 776 (см. рис. 111,13).
Постоянный объем Уп этой подсистемы 776 содержит вначале число
молей газообразного адсорбата п°, находящегося при давлении р°
(концентрации с°). В результате перевода в подсистему 7 с адсорбен-
том п молей адсорбата в подсистеме 776 остается п" ~ п° — п молей
при давлении р" (концентрации с"). Таким образом, при проведении
адсорбции газа по схеме 7, 776 химический потенциал газа, остающе-
гося в подсистеме 776 (в отличие от химического потенциала газа,
остающегося в подсистеме 77а) изменяется.
В соответствии с этим свободная энергия адсорбента в подсистеме
7 вместе со свободной энергией газообразного адсорбата в подсистеме
776 в начальном состоянии (без учета неизменной свободной энергии
самого адсорбента) составляет
^=Лае+п° (Ш,81)
В конечном состоянии системы 7, 776 свободная энергия равна
+ (111,82)
134
Учитывая, что п == ns + п#, и используя выражения (111,35) и
(Ш,42) для изменения свободной энергии в рассматриваемом про-
цессе адсорбции, получаем
ns
°4-пй(Дц—+
О
+ *'[(Н"-Н0)-/ Ot)'+P0V£*] (111,83)
При п& <^ns
ns
пб ™J Ан <** + »SP°V% ° + *" [(Н"-Н°)-р" (V^'+p^ °]
О
(III,83а
I [оследний член в этом выражении нужно знать только для составле-
ния уравнений материального баланса, нужных для расчета соответ-
ствующей точки на изотерме адсорбции ns = / (р) (см. гл. 4 в ру-
ководстве [1]). Изменение же свободной энергии при адсорбции п®
молей адсорбированного вещества по схеме I, Пб в расчете на еди-
ницу площади поверхности адсорбента составляет
—I АЦ (111,836)
о
Правая часть этого выражения совпадает с таковой для выражений
(111,806) и (111,42).
Адсорбция пар$9 поступающего из резервуара с чистой
жидкостью. При адсорбции паров изотермы адсорбции часто выра-
жаются в виде зависимостей ns или Г не от абсолютного давления
пара адсорбата р, а от относительного давления plp0. Поэтому при
изучении адсорбции паров представляют интерес также и изменения
термодинамических функций при выборе за стандартное состояние
адсорбата насыщенного пара или жидкого адсорбата. Такой выбор
соответствует, кроме того, обычным условиям экспериментального
исследования адсорбции паров, когда источником адсорбата служит
чистая жидкость в вакуумной микробюретке [1].
Схематически такая система представлена на рис. 111,13, Z, ZZe.
Чистый адсорбент находится в подсистеме Z, а чистый адсорбат —
в подсистеме Це (в жидкостной микробюретке) в виде жидкости и на-
сыщенного пара над ней. Из жидкого адсорбата предварительно
удаляются растворенные в нем газы. Давление газа в подсистеме I
измеряют манометром, работающим без изменения объема газа [1]
(например, мембранным). Таким образом, объем системы Z, Пв
не изменяется. При погружении подсистемы I и Пв в термостат та-
кая система в целом удовлетворяет условиям постоянства как V,
так и Т. В отличие от системы Z, Па внешние силы работы над си-
стемой Z, Пв не производят.
135
Примем за начальное состояние подсистемы I по-прежнему со-
стояние чистого адсорбента. Благодаря тому, что находящееся
в жидкостной микробюретке Це количество жидкого адсорбата
больше максимального его количества, переходящего в подсистему Z,
химический потенциал адсорбата в подсистеме Пв остается не-
изменным. Это сближает рассматриваемый случай адсорбции пара
с уже рассмотренным случаем адсорбции газа, начальное состояние
которого определялось постоянным значением р°.
Найдем изменение свободной энергии при адсорбции пара по
схеме 7, Пв. В этом случае в начальном состоянии в подсистеме Пв
свободная энергия адсорбата
Р (111,84)
гдеУЯ» °и F1» °—объемы газовой и жидкой фаз в подсистеме Пв\ р0 — давление
насыщенного пара; Р1 — гидростатическое давление жидкости; п° — общее
число молей адсорбата в микробюретке; р,0 — его химический потенциал.
Если пренебречь влиянием кривизны мениска жидкости на Р1,
то Р1 = pQ и
Рцв~ Ив Н- (III,84а)
гДе в “ Vg’9 + — объем всей микробюретки.
Делая те же допущения, что и ранее, для изменения свободной
энергии системы 7, Пв при адсорбции пара получаем
ns
+n-(Ap — pV^n) (111,85)
о
Соответственно при ng<^jis и в расчете на единицу площади по-
верхности адсорбента получаем отсюда
АЛЧ пв С
—(111,86)
о
Помимо различия в начальном состоянии адсорбата зти выраже-
ния для ДЕ/, цв1А отличаются от рассмотренных ранее выражений
для AFi,nalA^ полученных для адсорбции газа [см. выражение
(111,806)], отсутствием члена Гр0^’0. Это различие связано с тем,
что при подаче адсорбата из жидкостной микробюретки Пв объем
газа в ней практически не изменяется, в то время как при подаче
адсорбата из газового баллона над ним совершается внешняя работа,
равная Гр°У^’ °.
Введем в уравнение (111,86) выражения для Др. в соответствии
с формулами (111,43), заменив в них aG, с°, у° и р° на соответству-
ющие величины для насыщенного пара а0, с0, у0 и р0. Это дает
следующие выражения:
г
-Л^ -8^=ДГ Jln-^-dr (Ш,87)
о
136
В случае адсорбции идеального пара
AF Г Г
—= f in — dV = RT f 1п-^-йГ (III,87а)
A J c0 J Po
о о
где p/pQ — относительное давление пара в подсистеме Z.
При малых р/р0 значения AF/, nJA значительно превосходят
значения — Да = л (как и в случае адсорбции газа, см. рис. III,8),
однако по мере приближения р/р0 к единице значения AF/, цв/А
и Да сближаются (они становятся равными при p/pQ — 1). Такое
различие значений AF/> цв/А и Да было показано в работах [ 74 — 78].
Дифференциальное мольное изменение свободной энергии ад-
сорбционной системы Z, Це при n8 ns
— (d\Fj гт \
AZ, iie=RT\—(III,88)
*> *1в \ дп* /V, А,Т
В случае адсорбции идеального пара
= ЛТ1п~ (III,89)
При уменьшении Гис (или р) величина &FIt цв обращается
в минус бесконечность, а при больших Г, когда с е0, р -> р0,
опа обращается в нуль.
Следует рассмотреть также и изменения энтропии, внутренней
энергии и теплоемкости при адсорбции пара, потому что свойства
насыщенного пара зависят от температуры. Интегральное изменение
энтропии при адсорбции ns молей пара по рассматриваемой нами
схеме 7, Це при n8 < ns
„л
AS, Пв = - ---, (111,90)
•*’ 11в \ дТ /V, А, п*
Подставляя сюда выражение (111,86) для ДР^ цв9 получаем в рас-
чете на единицу площади поверхности
г
А5/’ 7/8 = - С f dr (111,91)
A J \ дТ Jv, А, Г
О
В случае адсорбции идеального пара, поскольку концентрация с0
и давление р0 насыщенного пара зависят от Т, в уравнение (111,91)
надо подставить выражения
Г, с ,T(dln.c\ dlnc0"]
Lla^ + 7A~Jr
= йГ1п-^+г(Ц^) (IIL92)
L Pq \ дТ Jr аТ J
Если пар не идеальный, то здесь и ниже надо с и с0 заменить соответ-
ственно на а и а0.
137
Подставляя выражение (111,92) в уравнение (111,91), получаем
Г Г г ч
^SI, He
LO О
' Г г
j lnpdr+Г j (
-О о
dr —‘Г/? ^IncQ-j-y
rdf — TR ^1пр0 + Г
din c0\___
dT. )~
11JLP05) (111,93)
dT J
В выражениях (111,93) последние члены представляют измене-
ние энтропии при испарении Г молей жидкости в подсистеме Пв.
Дифференциальное мольное изменение энтропии адсорбата в си-
стеме Z, Цв при адсорбции пара, если пё nS, составляет
= (d^si, Пв\ ___________/аДц\
11в \ дп* }vt А, Г \ dT Jvt А, Г
(III,94)
При адсорбции идеального пара
МцП'—R [ье + т(^)г]-/фпСо + Т^) =
= -Д [1пр + г(^)г]-Я^про+г£^) (III,95)
В выражениях (111,95) последние члены представляют мольную
энтропию испарения жидкости в подсистеме Пв.
Зависимости средней мольной величины AS/, 11в /ЛГ, найденной
по формуле (111,93), и величины Д5\ 11в , найденной по формуле
(111,95), от величины адсорбции Г для этана на графитированной
термической саже имеют характерный для адсорбции на однород-
ной поверхности волнообразный вид, такой же, как и зависимости
от F величин Д5/Г и AS (см. рис. 111,10), изменяется лишь поло-
жение нуля на оси ординат.
Интегральное изменение внутренней энергии при адсорбции
пара в системе /, Пв в случае, когда пё ns, в расчете на единицу
площади поверхности составляет
дМГ-( ЙГ (III,96)
A J J \ дТ /У, А, Г
О о
При адсорбции идеального пара
-о
-----ПТЪ (Ш,96а)
J \ дТ Jr dT
о
138
II ртом выражении величина
RT2^Po=.L (III,97)
dT
пред*'таил нот собой мольную скрытую теплоту испарения жидкости.
Проипнодение TL— теплота испарения Г молей жидкости в под-
системе Пв.
Дифференциальное мольное изменение внутренней энергии при
адсорбции пара по схеме I, IIв при п8 < ns составляет
(Ill,98)
В случае адсорбции идеального пара
Сравнение с выражением (III, 64а) для адсорбции идеального
гааа \U —RT2 (0 In р/дТ)? RT показывает, что при адсорбции
пара, испаряющегося из жидкости в подсистеме Пв, общее дифферен-
циальное мольное изменение внутренней энергии адсорбата в системе
Z, Пв _ _
\Ult 1Ie = \U—RT + L (111,99)
Предельная величина ДСТ/пв," при Г -> 0 может быть выражена
через температурные коэффициенты константы Генри подстановкой
в уравнения (III,98а) выражений (111,12) и (111,14):
Щ lie, 1 = Д У2("И KdT~-’-~ ~= RT2dla K^’-i + L (HI,ЮО)
* • \ di di / di
Зависимость — AUItIIe от Г для адсорбции этана на графитирован-
ной термической саже имеет такой же волнообразный вид, как и за-
висимость — ДС7газ от Г (см. рис. 1П,9), изменяется только поло-
жение нуля на оси ординат на величину L — RT.
Интегральное изменение теплоемкости адсорбционной системы
I, Пв, происходящее в результате адсорбции пара в том случае, когда
п8 ns, в расчете на единицу площади поверхности составляет
пв 7 Г /^2
A J \ № Jv,A,r
о
(111,101)
В случае адсорбции идеального пара в соответствии с выражениями
(III,96а) для AUmIe 1А получаем выражения, отличающиеся от
выражений (III,69а) членами
л т-п /2d In с о d2 In с о X / 2 d In d2 In Л -> g
~^RTr\~dT~^'T~dT^~)~~~RTr\~dT—
(111,102)
139
представляющими изменение теплоемкости адсорбата при переходе
из жидкости в насыщенный пар.
Дифференциальное мольное изменение теплоемкости в рассмат-
риваемом процессе адсорбции пара для случая ns ns
«.-( )v. А. т-^Т (тЙЧг. Л. Г <ПиЮ)
В случае адсорбции идеального пара
Sc,,n,—лг[г(-^)г+т(-^)г]-АСЙ-«-
—[2(тгЧг+7' (4?НГ]~ЛС”‘ (шда>
Предельная величина 1 для Г-> 0 получается подста-
новкой в уравнение (111,104) выражений (111,12) или (111,14):
^1. Пв, ! = ЯТ’ (2 + Т - ДС#в =
( dlnKr D г d2 1n/Cr . \ .
=яг + г---------------^дс5г<
(111,105)
Выше уже отмечалось, что надежное определение вторых про-
изводных (д2 In с/дТ2)г или (д2 In pldT2)? требует очень высокой
точности измерения всех трех величин ns (или Г), с (или р) и Г.
Это же относится к входящим в уравнение (111,105) производным
d2 In KJdT2 и d2 In Pt-JdT2, требующим очень точного опреде-
ления констант Генри (удерживаемых объемов) при разных Т. По-
этому и здесь нужны прямые калориметрические измерения тепло-
емкости адсорбционных систем.
7. Теплота адсорбции
Случаи необходимости калориметрических измерений тепловых
эффектов адсорбции. В формулы для изменения внутренней
энергии AZ7 при адсорбции газа или пара (см. разд. 5 и 6 этой главы)
входят изостерические температурные коэффициенты In с или In р.
Из рис. III,11 видно, однако, что изостеры близки к прямым. Это же
можно сказать и о зависимости In Кг от ИТ (см. рис. III,4). Отсюда
следует, что AZ7 в этих сучаях слабо зависит от температуры, особенно
в области малых заполнений однородной поверхности. Газохромато-
графические измерения удерживаемых объемов или статические
измерения изотерм адсорбции (см. разд. 1 этой главы) охватывают
довольно небольшой интервал температур, составляющий около
50—70 К (это видно из рис. 111,1). В этом случае зависимости In с
от ИТ при постоянной Г и зависимости In Кг от ИТ часто практи-
чески линейны. Если ЛЕ/ значительно превосходит RГ, то из формулы
(III,64а) следует, что и зависимости In р от ИТ при постоянной Г
(изостеры адсорбции) практически линейны. Это обычно облегчает
140
графическое нахождение производных [д In с/д (1/Т)]г или
Id In р/д (1/Г)]г и производных d In Kx/d(i/T) или d 1п7Гг>Р(1 id(l/T)
Из экспериментальных данных, относящихся к адсорбционному равно-
весию, хотя при соответствующей обработке наиболее надежных
данных обнаруживаются отклонения от линейных зависимостей
[26, 79].
Однако, как уже отмечалось выше, часто невозможно экспери-
ментально определить все эти величины ns (или Г) и с (или р), а
также Кг или К?, р> i при разных Т с той высокой точностью, которая
необходима для определения указанных выше производных. Например,
„этого нельзя сделать, когда значения р весьма малы [при достаточно
низких для данной системы температурах (см. рис. Ill,1)] или когда
AZ7 сильно изменяется с изменением температуры (см. рис. III,2).
В та,ких случаях необходимы прямые калориметрические изме-
рения теплового эффекта адсорбционного процесса и нахождение
величины ДС7 из этого теплового эффекта. Однако кроме чисто
методических трудностей, возникающих при самом калориметриче-
ском измерении тепловых эффектов адсорбционных процессов (свя-
занных главным образом с тем, что часто приходится измерять малые
и медленно выделяющиеся [32] тепловые эффекты), здесь возникают
также и трудности термодинамической интерпретации измеряемых
величин. Эти трудности вызываются тем, что не всегда можно пред-
ставить измеренный в калориметре тепловой эффект в виде рассмот-
ренного выше изменения тепловой функции состояния адсорбцион-
ной системы AZ7.
Теплоты адсорбции газов и паров. Рассмотренные в разд. 6 этой
главы схемы Z, Цб и 7, Цв процесса адсорбции (см. рис. 111,13)
предполагают постоянство объема системы. При этом условии тепло-
вой эффект процесса равен изменению внутренней энергии этой си-
стемы ДС7. Интегральное изменение внутренней энергии системы
при адсорбции пара по схеме Z, Цв выражается уравнением (111,96)
или приближенно уравнениями (III,96а). При их выводе мы пред-
полагали, что пар поступает в подсистему I с адсорбентом из под-
системы Цв без изменения давления пара над мениском жидкости
в микробюретке. На испарение перешедшего в подсистему I коли-
чества п = ns + п8 ns молей адсорбата была затрачена скрытая
теплота испарения nsL [или ГЬ в расчете на единицу площади
поверхности адсорбента в подсистеме I, см. выражения (III,96а)
и (111,97)]. Однако в рассматриваемом случае, т. е. при переходе
зтих ns молей адсорбата в подсистему 7, не производится какой-либо
работы внешними силами, так как при соединении подсистем I и Цв
пар расширяется в подсистему 7 самопроизвольно. В одном из опытов,
описанных Кальве [29], сосуд с адсорбентом, соответствующий
нашей подсистеме Z, и сосуд с жидким адсорбатом, соответствующий
нашей подсистеме Цв, помещались в один и тот же калориметр, в ко-
тором измерялась так называемая «чистая» теплота адсорбции, т. е.
разность между теплотой адсорбции пара и теплотой испарения
жидкости в соответствующих условиях. Если положительной счи-
141
тать выделяющуюся теплоту, то величина этой разности близка
к—AUIjIIe в формулах (111,96), (111,96а) и соответственно к—AfZj, цв
в формулах (111,98), (III,98а). В большинстве же случаев источник
пара адсорбата (например, жидкостная микробюретка на схеме
рис. 111,13, Пв) находится вне калориметра (см. руководство [1]).
Так как пар или газ входит в подсистему I из подсистемы Пв
или Пб, не производя работы против какой-либо внешней силы
(потому что система имеет жесткие стенки), все изменение внутрен-
ней энергии в подсистеме I с адсорбентом выделяется в окружающую
эту подсистему I среду в виде теплоты. В этих случаях измеренная
в окружающем подсистему I с адсорбентом изотермическом калори-
метре интегральная теплота при отсутствии теплообмена вне кало-
риметра
qVI=^~~^UI (111,106)
где — изменение внутренней энергии в подсистеме Z, представляющее
рассмотренную в разд. 5 этой главы величину &U.
В адсорбционной установке перехдд пара или газа в сосуд с ад-
сорбентом происходит обычно через узкие капилляры затворов
или тонкие щели кранов. С точки зрения внешней среды в установке
с жесткими стенками (см. схемы I, Пб и I, Пв на рис. 111,13) газ
или пар, проходя через эти отверстия, расширяется свободно, не
производя внешней работы. Однако при этом могут происходить
локальные динамические процессы трения, которые могут вызвать
некоторый локальный теплообмен вне калориметра, влияющий на
измеряемую величину ду,/. Кроме того, внутренняя энергия реаль-
ного газа зависит от концентрации, что может вызвать частичный
теплообмен уже в подсистеме IIб или IIв и поступление в подсистему I
потока газа с несколько измененной температурой. При свободной
расширении реального газа в подсистеме I также может произойти
теплообмен вне калориметра, потому что часто соединительные
трубки и часть газового объема подсистемы I до манометра находятся
вне калориметра. Процессы трения газа в капиллярах и кранах
и процессы расширения реального газа вне калориметра должны
приводить к тому, что измеренная в изотермическом калориметре
величина qV)i —Af7/. Поэтому в практической работе, чтобы
сблизить измеряемую величину qV} i с величиной соответствующего
изменения тепловой функции в подсистеме 7, т. е. с величиной
—AZ7i, надо следить, во-первых, за достижением в подсистеме /
адсорбционного и теплового равновесия и, во-вторых, по возмож-
ности устранить теплообмен вне калориметра. В случае медленных
процессов адсорбции особенно важно изучение термокинетики, ко-
торое позволяет экстраполировать измеряемые величины qv т ко
времени достижения адсорбционного и теплового равновесия [32].
Это позволяет сблизить находимые из калориметрических измерений
величины qVji или ffy,/, цб, Qv,i,iie с величинами соответству-
ющих изменений функции состояния адсорбционной системы —AZ7/
или AUit пб/ которые не зависят от условий проведения калориметри-
142
ческого опыта и для данной системы целиком определяются величи-
нами Т и Г (или пЧтп).
Таким образом, измеренная в калориметре в условиях постоян-
ства V и А теплота адсорбции qv при устранении теплообмена вне
калориметра и при обеспечении достижения равновесия или экстра-
поляции к равновесию имеет вполне определенный термодинамиче-
ский смысл величины —Af7 и может рассматриваться как физико-
химическая величина при сравнении свойств различных адсорбци-
онных систем при одних и тех же значениях Т и Г (или ns/m).
Кроме того, над газом, поступающим в подсистему I из под-
системы Па (например, из газовой бюретки), производится работа
сжатия внешней силой (поднимающей ртуть в бюретке, подробнее
см. руководство [1]). В разд. 6 этой главы показано, что при пё п3
интегральное изменение внутренней энергии &ицца1А выражается
в этом случае той же формулой (см. с. 134), что и полученная в разд. 5
формула (III,61а). При изотермическом сжатии газа работа сжатия
полностью (для идеального газа) или частично переходит в тепло,
и это учитывается в формулах (III,61а) и (111,806). Однако только
часть этого тепла достигает калориметра. Подробнее эти вопросы
могут быть рассмотрены только с учетом особенностей проведения
конкретного калориметрического опыта в данной конкретной уста-
новке. Такой анализ нужен для перехода от измеряемой в калори-
метре величины qv к величине —AUj.
Определенные затруднения связаны часто и с получением'из ре-
зультатов калориметрических измерений дифференциальных ве-
личин At/у или At7/(jj6. Обычно измеряют величины, входящие
в отношение
&qv dqv
"ДТ дГ~~ду
(111,107)
где ДГ — приращение величины адсорбции от Г до Г + ДГ; Дду — соответ-
ствующий этому ДГ тепловой эффект, зарегистрированный калориметром;
qv — истинная дифференциальная теплота адсорбции.
В случае медленных процессов адсорбции, впущенный в подси-
стему I адсорбат за время калориметрического измерения не успе-
вает равномерно распределиться на поверхности всех частиц адсор-
бента: ближайшие к потоку подводимого газа частицы адсорбируют
больше, чем более удаленные, особенно при больших энергиях ад-
сорбции, низких температурах и небольших (нулевых) р. Результаты
измерений при последовательных впусках адсорбата в подсистему I
в этих случаях представляют интегральные тепловые эффекты на
некоторой части адсорбента. Поэтому величина А ду/АГ только
в тех случаях достаточно близка к величине — AZ7, когда соблюдены
указанные выше условия: устранен теплообмен вне калориметра
и достигнуто адсорбционное и тепловое равновесие со всей поверх-
ностью адсорбента или произведена достаточно надежная экстра-
поляция термокинетических измерений, т. е. зависимости измеряе-
мой величины Аду от времени [32]. Кроме того, необходимо изме-
143
рять достаточно малые величины Aqv и ДГ с примерно одинаковой
точностью.
Если все эти условия соблюдены, то равенство [см. выражение
(1П,64)1 _
qv^-&U^--(Us-Uk °) (111,108)
при адсорбции идеального газа (когда U^° не зависит от выбора р°)
выполняется с достаточной точностью. Это имеет место при приме-
нении изотермических компенсационных калориметров [1].
Изостерическая теплота адсорбции. Чистая теплота адсорбции
пара. В соответствии с выражениями (III,64) и (111,108) дифферен-
циальная теплота адсорбции идеального газа
ду==_да=лт2(^^)г=№(Л^Р.)г_Дт (Ш.109)
В это уравнение входит величина RТ2 (д In р/дТ)^= —R 15 In р/д(1/Т)]г
обычно находимая из изостер, пример которых приведен на рис.
111,11. Поэтому эту величину обычно называют изостерической
теплотой адсорбции и обозначают через qsi. Из выражений (III,64а)
следует, что при адсорбции идеального газа
qst=RT^-~P-^F = ~SU + RT (111,110)
В соответствии с выражением (111,64) или (111,108) ДС7 = Us —
Так как Uffc ° RT — ° , где — мольйая энтальпия
идеального газа, то
= (III,110а)
(В это выражение входит E7S, а не Hs 180].) Из определения (111,110)
и выражения (111,108) следует, что
qst = qv+RT (Ш,111)
В случае адсорбции реального газа связь между q$t и qv сложнее,
поскольку внутренняя энергия реального газа'зависит от концент-
рации.
При адсорбции пара иногда пользуются термином «чистая теп-
лота адсорбции». Этому соответствует разность
qst-L= RT* )г (111,112)
Следует отметить, однако, что ни величина q3t, ни] величина qst —
— L не являются собственно тепловыми эффектами адсорбции, не-
посредственно измеряемыми в калориметре. В виде теплового эф-
фекта процесса адсорбции измеряется величина q^. Величина,
которую называют изостерической теплотой адсорбции, определяет-
ся из данных по адсорбционному равновесию [см. выражение
(111,110)], а не из калориметрических данных. Поэтому название
144
величины ЙТ2 (д In р/дТ)? «теплотой» адсорбции неправильно.
ЯтОт термин употребляется только по традиции и из-за удобства,
поЬкольку величина qsi непосредственно получается из изостер,
выраженных обычно близкими к линейным (при недостаточно точных
измерениях) зависимостями In р от ИТ (см. рис. 111,11). Введение
в термодинамику адсорбции величины q&t было связано с тем, что
при изучении адсорбции в вакуумных установках статическим ме-
тодом производились измерения в газовой фазе давления р (с по-
мощью различных манометров), а не концентрации с. Однако, исполь-
зуя детекторы, калиброванные по концентрации адсорбата в газо-
вой фазе, можно измерять в этом случае не р, а с, и определять
из таких измерений с помощью формулы (111,64) сразу изменение
соответствующей тепловой функции, т. е. изменение внутренней
энергии ДЕЛ Следует отметить также и то неудобство, что qsi =—
— KU 4“ RT вследствие влияния члена RT в некотором интервале
значений Г и Г может увеличиваться с ростом Т и проходить затем
через максимум. Из этого нельзя делать вывод о том, что —ЛЕЕ
проходит через максимум. Изменение —ЛЕЕ с ростом Т правильно
передает калориметрическая величина qy.
В литературе встречаются и другие термины для «теплоты»
адсорбции, отличающиеся друг от друга условиями, при которых
берутся соответствующие частные производные по температуре
[80—88]. Как и в случае эти величины не имеют непосредствен-
ного отношения к тепловому эффекту какого-либо адсорбционного
процесса.
Рассматривают также теплоты адсорбции, измеренные в калори-
метре, но в условиях, отличающихся от описанных выше изотерми-
ческих условий, например адиабатическую теплоту адсорбции.
Однако в последнее время теплоту адсорбции предпочитают изме-
рять в соответствии со схемами, представленными на рис. III,13,
в изотермических условиях компенсационными методами (см. руко-
водство [1] и разд. 1 этой главы). Поэтому теплоту адсорбции, изме-
ренную в адиабатическом калориметре, мы не рассматриваем.
Сопоставление величин изменения внутренней энергии при ад-
сорбции, полученных разными методами. Рассмотрим теперь резуль-
таты независимых определений величин' ДЕЕ как из исследований
адсорбционных равновесий статическими и динамическими мето-:
дами, так и из калориметрических измерений, теплот адсорбции.
На рис. III,9 сопоставлены величины —ДЕЕ для адсорбции этана
на графитированной термической саже, полученные из измерений
статическим методом изотерм адсорбции и из измеренных газохрома-
тографическим методом удерживаемых объемов при разных темпе-
ратурах. В случае статических измерений, проводимых в интервале
более низких температур, при малых величинах адсорбции заметно
влияние остаточной неоднородности поверхности, которое вызывает
в этой области Г повышение значения —ДЕЕ. Обработка с помощью
10 заказ 1180 145
вириального уравнения (см. разд. 2 гл. IV) средней части зависи-
мости Г от р при разных Т (средняя часть этих кривых связана с ад-
сорбцией преимущественно на однородной поверхности) дает зна-
чение — А?71, близкое к полученному из газохроматографических
измерений.
На рис. III,14 сопоставлены результаты статических (калори-
метрических и полученных из изостер) и газохроматографических
определений — AZ7 для адсорбции н-пентана на графитированной
термической саже. Эти данные также хорошо согласуются.
Рис. 111,14. Дифференциальные теплоты адсорбции
н-пентана на графитированной термической саже при
разных величинах адсорбции Г, полученные на двух
калориметрах разной конструкции. Точка 1 получена
газохроматографическим методом.
В табл. III,3 сопоставлены значения — \U1 (для Г-> 0) для
ряда адсорбатов на графитированной термической саже, определен-
ные из статических и газохроматографических измерений. Они
хорошо согласуются. Следует отметить, что точность газохромато-
графического определения составляет в благоприятных случаях
Около 1—3%.
Для нахождения газохроматографическим методом значений
AU при отличных от нуля величинах адсорбции надо получить из
хроматограмм, снятых при разных температурах, соответствующую
серию изотерм адсорбции (см. обзоры [24, 25]), из них получить
зависимости 1g с от i/T для разных ns или Г и по наклонам этих
зависимостей определить AU.
В табл. III,4 сопоставлены значения AUlt определенные из
измеренных газохроматографическим методом удерживаемых объ-
146
Таблица 111,3. Значения —ACTi (в кДж/моль) для адсорбции
на графитированной термической саже, определенные из статических
изотерм адсорбции и из газохроматографических удерживаемых объемов,
измеренных при разных температурах [51, 53, 70, 88]
Адсорбат Из статических изотерм адсорбции Из удерживаемых объемов
интервал температур, К -ДС/i интервал температур, К - ДП1
Аргон от 77 до 120 8,4 от 110 до 150 8,8
Криптон от 245 до 290 10,3 от 302 до 403 10,6
Ксенон от 280 до 315 13,8 от 297 до 403 13,5
Мотан от 224 до 297 10,2 от 300 до 403 9,2
I >тан от 173 до 213 16,8 от 170 до 220 17,3
Пропан от 258 до 297 24,7 от 296 до 372 23,7
Циклопропан от 244 до 297 21,0 от 313 до 403 22,0
Этилен от 229 до 266 16,8 от 313 до 403 15,5
Непзол от 273 до 323 34,4 от 403 до 493 34,6
Хлороформ от 249 до 323 30,7 от 372 до 463 30,0
Чотыреххлористый углерод от 231 до 323 32,8 от 372 до 463 31,4
омов и из измеренных статическим методом изотерм адсорбции при
разных температурах, с измеренными в калориметре значениями
и определенными экстраполяцией зависимости qv от Г к Г -> 0.
Таили экстраполяция может быть произведена на основе той или
иной модели состояния адсорбированного вещества (в разд. 2 и 3
гл. IV рассмотрены некоторые из таких моделей).
Таблица 111,4. Значения &U± и qv г (в кДж/моль) для адсорбции
на графитированной термической саже,
определенные газохроматографическим, статическим (из изотерм)
и калориметрическим методами [53, 88, 89]
Адсорбат Из статических изотерм адсорбции при разных температурах Из калоримет- рических измерений Из удерживаемых объемов при разных температурах
интервал температур, К -ДЕЛ V, 1 при 293 К интервал температур, К -Д171
и Пентан Неопентан Ацетон Пиэтиловый эфир Чотыреххлористый уг- лерод от 298 до 348 от 231 до 323 33,5 32,8 38,0 29,2 33,4 37,2 35,4 от 323 до 373 от 293 до 363 от 323 до 373 от 323 до 393 от 323 до 373 33,2 26,3 29,0 33,5 31,4
Приведенные в табл. III,4 значения —А£7± и qvi довольно
близки, хотя расхождения иногда составляют более 10%. Следует,
однако, учесть, что калориметрические измерения проводились
10*
147
при более низких температурах. Внесение поправок на убывание
— AZ7i с ростом температуры [см. выражение (111,75)] сближает вели-
чины qVil с величинами — вычисленными из газохроматогра-
фических определений удерживаемых объемов и из статических
определений семейства изотерм адсорбции, произведенных при более
высоких температурах.
О выборе вместо F и U jsjpyvm термодинамических функций для
характеристики адсорбционных процессов. Выше было отмечено,
что при проведении измерений изотерм, изостер и теплот адсорбции
в вакуумных установках с постоянным объемом (подсистемы Пб
и Пв) внешнее давление работы не производит^ В случае подсистемы
Па работа, производимая постоянным внешним давлением р°, учи-
тывается в величине ДЕЛ Давление газа внутри подсистемы I во всех
случаях изменяется, а объем этой системы остается постоянным.
Поэтому за рабочую и тепловую функции [17] для адсорбционной
системы мы выбрали соответственно свободную энергию Гельмгольца
F и внутреннюю энергию U. В случае физико-химических процессов,
осуществляемых при постоянном внешнем (гидростатическом) да-
влении Р во всей системе и переменном объеме системы, рабочей и
тепловой функциями системы являются соответственно свободная
энергия Гиббса G — F 4- PV и энтальпия Н = U 4- PV. Однако
по указанным выше причинам применение функций G и Н для опи-
сания адсорбционных опытов нецелесообразно. Также нецелесооб-
разно применение в этих случаях рабочей функции в форме F 4-
4-РУ 4-оЛ [90] и тепловой функции в форме U 4- PV -f-оЛ,
так как в адсорбционных опытах с твердыми телами о, во-первых,
изменяется, а, во-вторых, не измеряется. Поэтому применение по-
добных рабочих и тепловых функций для процессов адсорбции на
твердых телах может быть лишь формальным. В рассмотренных выше
случаях, когда в процессе адсорбции р и о изменяются, использова-
ние этих функций не упрощает записи термодинамических формул.
По этим причинам эти функции в этой главе не рассматриваются.
В разд. 1 гл. VI рассматривается функция Q = pV 4-лЛ, пред-
ставляющая сумму произведений обобщенных силовых и геометри-
ческих параметров системы газ — адсорбент, поскольку эта функ-
ция непосредственно связана с большой статистической суммой для
газа, взаимодействующего с поверхностью твердого тела.
ЛИТЕРА ТУРА
1. Экспериментальные методы в адсорбции и хроматографии. Под ред.
А. В. Киселева и В. П. Древинга. М., Изд-во МГУ, 1973, 447 с.
2. Киселев А. В., Лыгин В. И. Инфракрасные спектры поверх-
ностных соединений. М., «Наука», 1972, 459 с.
3. Kiselev А. V. Discuss. Faraday Soc., 1971, v. 52, p. 14—32.
4. Zettlemoy ет А. С., К 1 i e r K. Discuss. Faraday Soc., 1971, v. 52,
p. 46—49.
5. Д а в ы д о в В. Я. и др. ЖФХ, 1973, т. 47, с. 809—811.
6. Киселев А. В., Локуциевский В. А., Лыгин В. И.
Кинетика и катализ, 1972, т. 13, с. 1611—1613.
148
7. Киселев А. В., Лыгин В. И., Щепа л кн К. Л. Кинетика
и катализ, 1971, т. 12, с. 185—190.
В. Ж д а н о в С. П., X в о щ е в С. С. Кинетика и катализ, 1970, т. 11,
с. 1520—1526.
9. Франсуорт Г. В кн.: «Межфазовая граница газ—твердое тело». Под
ред. Э. Флада. Пер. с англ. М., «Мир», 1970, 433 с.
10. X о б с о н Дж. В кн.: «Межфазовая граница газ—твердое тело», Под
ред. Э. Флада. Пер. с англ. М., «Мир», 1970. 433 с.
И. Кошетов В. В., Мартинсон Е. Н., Шальников А. И.
Приборы и техника эксперимента, 1971, № 1, с. 180—182.
12. Robert. R. W., Vanderslice Т. A. Ultrahigh Vacuum and
Its Application. New York, 1963, 290 p.
13. С о n s t a b a г i s G., Singleton J. H., Halsey G. D., J.
Phys. Chem., 1959, v. 63, p. 1350—1355.
14. Woo t en L. A., Brown С. J. Am. Chem. Soc., 1943, v. 65,
p. 113—118.
15, Urquart A. K., Williams A. M. J. Text. Inst., 1924, v. 15,
p. 434-439.
16. Д p e в и н г В. П., Киселев А. В., ЭльтековЮ. А. ДАН,
1952, т. 86, с. 349-352.
17. Г и б б с Дж. Термодинамические работы. М., Гостехтеоретиздат, 1950,
492 с.
18. Г е р а с и м о в Я. И. и др. Курс физической химии. Под ред. Я. И. Ге-
расимова, Изд. 2-е. Том I, М., «Химия», 1969. 592 с.
19. Г а р к а в е н к о Л. Г. и др., ЖФХ, 1967, т. 41, с. 244—251.
20. М с Bain J. W., В a k г А. М. J. Am. Chem. Soc., 1926, v. 48, р. 690—
695.
21. Киселев А. В., МуттикГ. Г. Коллоидн. ж., 1957, т. 19, с. 562—
571.
22, С а р а х о в А. И. Весы в физико-химических исследованиях. М., «Наука»,
1968. 229 с.
23. Б е з у с А. Г., Д р е в и н г В. П., Киселев А. В. ЖФХ, 1964,
т. 38, с. 59—67.
24. К и с е л е в А. В., Яшин Я. И. Газоадсорбционная хроматография.
М., «Наука», 1967. 256 с. Исправленные и дополненные издания: Gas—
Adsorption Chromatography. New York, 1969, 254 p. La Chromatograph io
Gas — Solide. Paris, 1969, 289 p.
25. Киселев и др. Физико-химические применения газовой хромато-
графии. М., «Химия», 1972. 255 с.
26. В е г t u s h Т. I. е. a. Chromatographia, 1970, v. 3, р. 369—371.
27. В 1 u G., Jacob L., Guiochon G. J. Chromatog., 1971, v. 61,
p. 207—216.
28. Киселев А. В., Худяков В. Л., Я ш и и Я. И. ЖФХ, 1974,
т. 48, с. 757-759.
29. Кальве Э., П р а т А. Микрокалориметрия. М., Издатинлит, 1963.
477 с.
30. Б е р е з и н Г. И. и др., ЖФХ, 1969, т. 43, с. 223—223.
31. В е г е z i n G. I., S a g a t е 1 у a n R. Т. In: Thermochimie. Colleques
internationoux du C, N. R. S., No. 201. Marseille, 1972, 565 p.
32. C huykina V. K. e. a. Collect. Gzechoslow. Chem. Com., 1974, v. 39?
p. 1794—1796.
33. Кисел ев А. В., Муттик Г. Г. ЖФХ, 1961, т. 35, с. 2153—2155.
34. Березин Г. И. и др. ЖФХ, 1969, т. 43, с. 224—224.
35. D z h i g i t О. M. e. a. Trans. Faraday Soc., 1971, v. 67, p. 458—467.
36. S c h i r m e r W. e. a. Chem. Thechn., 1971, v. 23, s. 476—478.
37. В e r e z i n G. I., К i s e 1 e v A. V. I. Col. Interface Sci., 1972, v. 38,
p. 227—233.
38. В e r e z i n G. I. e. a. J. Col. Interface Sci., 1972, v. 38, p. 335—340.
39. Berezin G. I., Kiselev A. V. J. Col. Interface Sci., 1974, v. 46,
p. 203—211.
149
40. G a r n e r W. E., Veal F. J. J. Chem. Soc., 1935, p. 1436—1443.
41. Beebe R. A. In: Handb. d. Katalyse. 13d IV. Heterogene Katalyse I.
Herausgegeben von G. M. Schwab, Wien, 1943. 556 s.
42. BeeckO., Cole W. A., Wheeler A. Discuss. Faraday Soc., 1950,
v. 8, p. 314—321.
43. Трепнелл Б. Хемосорбция. M-, Издатинлит, 1958. 327 с.
44. К а в т а р а д з е Н. Н. В кн.: «Электронные явления в катализе и ад-
сорбции». М., Изд-во АН СССР, 1955. 255 с; ДАН 1957, т. 114, с. 822—825.
45. CernyS., PonecV., Catal Rev., 1968, v. 2, p. 249—255.
46. К 1 e m p e r e r D. F. In: Chemisorption and Reactions on Mettalic Films.
Ed. J. R. Anderson. London, New York 1971, v. 1, 555 p.
47. Семенченко В. К. Избранные главы теоретической физики. М.,
«Просвещение». 1966. 338 с.
48. Н i 11 Т. L. Thermodynamics of small systems, part 1, New York, Amster-
dam, Benjamin, 1963. 171 p.
49. Фаулер P., ГугенгеймЭ. Статистическая термодинамика. M.,
Издатинлит, 1949. 612 с.
50. В а г г е г R. М., R е е s L. V. Trans. Faraday Soc., 1961, v. 57, p. 999—
1007.
51. Киселев А. В., Худяков В. Л., Яшин Я. И. ЖФХ, 1973,
т. 47, с. 2125—2126.
52. Vidal-Madjar С., GanansiaJ., Guiochon G. In: Gas
Chromatography 1970. Ed. R. Stock, S. G. Perri, London, 1971. 445 p.
53. Kalashnikova E. V. e. a. Chromatographia, 1971, v. 4,
s. 495-500.
54. Кейлеманс А. Хроматография газов. M., Издатинлит, 1959. 320 с.
55. Glueckauf Е., Barker К. Н., KittG. Р. Discuss. Faraday
Soc., 1949, v. 7, р. 199—213.
56. Н ii b е г Н. F., Keulemans A. J. М. In: Gas Chromatography.
London, 1962. 420 p.
57. G r e e n e S. A., P u s t H. J. Phys. Chem., 1958, v. 62, p. 55—58.
58. L i t 11 e w о о d A. In: Gas Chromatography, Principles, Techniques and
Application. New York, London, 1962. 514 p.
59. PetitclercT., Guiochon G. Chromatographia, 1974, v. 7, p. 10—
13.
60. Ross S., Saelens J. K., Olivier J. P. J. Phys. Chem., 1962,
v. 66, p. 696—700.
61. C h i r n s i d e G. С., P о p e C. G. J. Phys. Chem., 1964, v. 68, p. 2377—
2379.
62. Б e л я к о в а Л. Д., Ковалева Н. В., Киселев А. В. ЖФХ,
1966, т. 40, с. 1494—1500.
63. Киселев А. В., Мигунова И. А., Яшин Я. И. ЖФХ, 1968,
т. 42, с. 1235—1239.
64. Бойкова А. С., Щербакова К. Д. Нефтехимия, 1967, т. 7,
с. 451—457.
65. ElkingtonP. A., CurthoysG. J. Phys. Chem., 1969, v. 73,
, p. 2321—2326.
66. Vers in о В., G e i s s F. Chromatographia, 1969, v. 2, p. 354—359.
67. E i s e n O. G. e. a. Chromatographia, 1971, v. 4, p. 448—454.
68. К и с e л e в А. В. и др. ЖФХ, 1964, т. 38, с. 161—169.
69. Кисел ев А. В. и др. ЖФХ, 1970, т. 44, с. 1272—1278.
70. A v g u 1 N. N. е. a. J. Col. Interface Sci., 1973, v. 42, p. 486—495.
71. В e r e z i n G. I., К i s e 1 e v A. V. J. Col. Interface Sci., 1966, v. 22,
p. 161—164.
72. Б e p e з и н Г. И., Киселев А. В., Синицын В. А. ЖФХ, 1967,
t. 41, c. 926—930.
73. Берез и нГ. И., Киселев А. В., КлешнинаИ. В. ЖФХ,
1969, т. 43, с. 2946—2947,
74. Киселев А. В. ДАН СССР, 1945, т. 49, с. 516—519.
75. К i s е 1 е v А. V. Acta Physicochimica USSR, 1945, v. 20, p. 947—968.
150
76. К и с е л е в А. В. Ученые записки НИИфизики МГУ (2), 1946, т. 86,
с. 116-146.
77. Киселев А. В. ЖФХ, 1946, т. 20, с. 239—256.
78. К и с е л е в А. В. Успехи химии. 1946, т. 15, с. 456—484.
79. Vidal-Madjar С., Guiochon G. Bull. Soc. Chim. phys. France,
1974.
80. Пейс E. В кн.: «Межфазовая граница газ — твердое тело». Под ред.
Э. Флада. Пер. с англ. М., «Мир», 1970. 433 с.
81. Huck el Е. Adsorption und Kapillarkondensation. Leipzig, 1928. 308 s.
82. E u c k e n A. Handbuch d. Experimentalphysik, 1929, Bd. 8, s. 657—690.
83. Hill T. L. J. Chem. Phys., 1949, v. 17, 520-535.
84. E v e r e t t D. H. Trans. Faraday Soc., 1950, v. 46, p. 453—459, 942—
956 957______969.
85. К i n g t о n G. L., A s t о n J. G. J. Am. Chem. Soc., 1951, v. 73, p. 1929—
1936.
86. Hill T. L. Trans. Faraday Soc., 1951, v. 54, p. 376—380.
87. Y о u n g D. M., Crowell A. D. Physical Adsorption of Gases. London,
1962. 426 p.
88. Ross S., О 1 i v i e г J. P. On Physical Adsorption. New York, London,
1964. kOl p.
89. A v g u 1 N. N., К i s e 1 e v A. V. In: Chemistry and Physics of Carbon.
Ed. P. L. Walker, New York, 1970, v. 6, 349 p.
90. Guggenhei m E. A., Ad am N. K, Proc. Roy. Soc., London, 1933,
ser. A v. 139, p. 218—230.
Глава IV
ТЕРМОДИНАМИЧЕСКОЕ ОПИСАНИЕ АДСОРБЦИИ
НА ОДНОРОДНОЙ ПОВЕРХНОСТИ
С ПОМОЩЬЮ НЕКОТОРЫХ МОДЕЛЕЙ
1. Роль моделей в термодинамическом
описании адсорбции
Общие уравнения термодинамики адсорбции, рассмотренные
в предыдущей главе, еще не позволяют получить уравнение состоя-
ния адсорбированного вещества или уравнения изотермы адсорб-
ции и зависимости от величины адсорбции Г дифференциальных
мольных изменений внутренней энергии А?7 и теплоемкости АС.
Они недостаточны также и для получения термодинамических ха-
рактеристик адсорбционной системы в виде констант, связанных
с межмолекулярными взаимодействиями адсорбат — адсорбент и
адсорбат — адсорбат. Строгое решение этих задач составляет пред-
мет молекулярно-статистической теории адсорбции, которая изла-
гается в VI и VII главах. Однако это строгое решение пока возможно
только для небольших заполнений поверхности.
Другой путь нахождения уравнения состояния адсорбированного
вещества, уравнения изотермы адсорбции и зависимостей А17 и АС
от Г представляет использование некоторых приближенных моделей
для адсорбированного вещества. Свойства этих моделей можно рас-
смотреть с помощью термодинамического или молекулярно-стати-
стического метода. Таким путем можно, например, найти уравнения,
приближенно описывающие изотермы адсорбции и зависимости
от Г величин АСУ и АС в довольно широкой области заполнения одно-
родной поверхности, включая переход к преимущественно полимоле-
кулярной адсорбции. При использовании только классического
термодинамического метода (т. е. уравнений, полученных в гл7 III)
связь входящих в эти уравнения констант с потенциальными функ-
циями межмолекулярного взаимодействия адсорбат — адсорбент и
адсорбат — адсорбат не раскрывается. Эти константы определяются
из опытных данных. Такое описание термодинамических свойств
адсорбционной системы во многих случаях, особенно при доста-
точно высоких заполнениях поверхности, оказывается единственно
возможным. Вместе с тем обработка экспериментальных данных
с помощью таких уравнений позволяет найти константы, отражающие
межмолекулярные взаимодейтвия адсорбат — адсорбент [кон-
станту Генри Кг и величины A5J, и (см. гл. III)] и адсор-
бат — адсорбат и исследовать их зависимость от природы адсорбата
и адсорбента и температуры. Полученные таким путем константы
152
нужны как для вычисления термодинамических свойств соответ-
ствующих адсорбционных систем при разных Г и Т (с помощью
содержащих эти константы приближенных уравнений), так и для
сопоставления с результатами расчета величин этих констант моле-
кулярно-статистическим методом.
Рассмотрение адсорбции молекул различного строения на одно-
родных поверхностях разной структуры обычно принято проводить
на основе моделей нелокализованной и локализованной адсорбции.
Однако при молекулярной (физической) адсорбции модель локали-
зованной адсорбции соответствует лишь предельному случаю состоя-
ния адсорбированного вещества при достаточно низких температурах
[1]. При молекулярной адсорбции при достаточно высоких темпера-
турах молекулы адсорбата всегда в той или иной степени способны
перемещаться вдоль поверхности. Поэтому в случае молекулярной
адсорбции при низких температурах и высоких потенциальных
барьерах для перемещения молекулы вдоль поверхности можно го-
ворить лишь о преимущественно локализованной адсорбции. О пре-
имущественно нелокализованной адсорбции можно говорить при
относительно высоких температурах и достаточно низких потенци-
альных барьерах для перемещения молекул вдоль поверхности.
С помощью различных моделей был получен ряд приближенных
уравнений изотерм как для мономолекулярной, так и для полимоле-
кулярной адсорбции из газовой фазы на однородной поверхности.
При этом использовались как модели локализованной адсорбции,
так и модели подвижного адсорбционного слоя [1—30]. Среди
них рассматривались модели, учитывающие различные взаимодей-
ствия адсорбат — адсорбат, в частности ассоциацию молекул ад-
сорбата. Взаимодействие адсорбат — адсорбат характеризовалось
обычно одной или несколькими константами, которые находились
из опытных данных. Применение этих приближенных уравнений:
для обработки экспериментальных изотерм адсорбции позволяет,
во-первых, определять константы Генри и, во-вторых, описывать
отклонения изотерм адсорбции от уравнения Генри не только при
малых заполнениях, но в благоприятных случаях и во всей об-
ласти преимущественного заполнения первого слоя до перехода
к преимущественно полимолекулярной адсорбции. Это же можно
сделать и для зависимостей от заполнения поверхности теплот ад-
сорбции [16, 18, 20, 27—30] и теплоемкостей адсорбционных систем:
[17-19, 27-30].
Определение из экспериментальных данных констант Генри,
теплот адсорбции при нулевом заполнении, ряда изотерм адсорбции
при разных температурах, зависимостей теплот адсорбции и тепло-
емкостей адсорбата от заполнения и сопоставление этих зависимостей
с получаемыми из расчета для разных моделей адсорбционных систем
дает важную информацию не только о влиянии межмолекулярных
взаимодействий адсорбат — адсорбент и адсорбат — адсорбат на
термодинамические характеристики адсорбционной системы, но и
о состоянии адсорбированного вещества. Помимо этого, уравнения
15&
с хорошо подобранными константами путем сопоставления с экспери-
ментальными данными в некотором интервале заполнений и темпе-
ратур обычно можно использовать для практических расчетов как
в том же, так и в несколько более широком интервале значений этих
параметров для данной адсорбционной системы.
с, ммоль/л
с, ммопь/л
Рис. IV, 1. Изотермы адсорбции аргона на графити-
рованной термической саже при разных темпера-
турах.
Точки — экспериментальные данные, кривые рассчитаны по
уравнению (IV,5) [11, 33, 43].
Различные модели адсорбции на физически однородной поверх-
ности рассматривались и обсуждались в ряде обзоров [4, 5, 10,
11, 21, 23, 26] и в работах [27—30]. Здесь рассматриваются только
те модели, которые непосредственно использованы в этой книге
для обработки экспериментальных данных, полученных на близких
к однородным поверхностях.
Любая правильная молекулярная модель адсорбции при Г О
дает уравнение Генри. В разд. 3 гл. III константа Генри была
введена как выражение предельного наклона изотермы адсорбции
при Г -> 0 [см. выражение (111,12) и (111,13)]. При достаточно вы-
соких температурах и низких концентрациях с в газовой фазе Г
остается приблизительно пропорциональной с в некотором интервале
малых значений Г, т. е. в этом интервале справедливы уравнения
[31]
Г && К±с или Г^Кг р ±р (IV,1)
В этом случае обычно говорят об «области Генри». Эта область имеет
практически важное значение, обеспечивая высокуиЗ симметричность
хроматографических пиков. Однако существование простой зави-
симости (IV,!) в конечном интервале величин Г связано либо с недо-
статочной точностью измерений Гис (или р) и недостаточно постоян-
ной Т, либо с тем, что влияние притяжения адсорбат — адсорбат
в некотором интервале величин Г > 0 практически полностью ском-
пенсировано влиянием неоднородности поверхности. Из рис. IV,1
видно, что изотермы адсорбции аргона на графитированной терми-
ческой саже близки к линейным при относительно высоких (для ад-
сорбции аргона) температурах и в области малых заполнений, мень-
ших 3—5% плотного монослоя (емкость монослоя составляет в этом
случае около 10 мкмоль/м2) [32, 33].
Уравнение (IV,1) приблизительно выполняется также и для
многих адсорбентов в целом с заведомо неоднородной поверхностью,
если эта поверхность состоит в основном из отдельных участков
(см., например, рис. 1,4 и 11,12), для каждого из которых имеется
своя константа Генри. Поскольку в газохроматографических опытах
при малых пробах имеют место лишь очень малые заполнения таких
участков поверхности, то и для всей поверхности в целом уравнение
Генри выполняется и хроматографический пик может оказаться
симметричным.
2. Описание зависимости величины адсорбции от концентрации
(давления) газа и температуры при использовании уравнения
fдвухмерного состояния адсорбата в вириальной форме
Уравнение в вириальной форме представляет наиболее общее
уравнение адсорбционного равновесия для небольших заполнений,
учитывающее межмолекулярные взаимодействия адсорбат — ад-
сорбент и адсорбат — адсорбат. Молекулярно-статистическая тео-
рия адсорбции, учитывающая эти взаимодействия при небольших
заполнениях поверхности, рассматривается в гл. VI и VII. Полу-
ченное этим путем вириальное уравнение изотермы адсорбции
имеет вид степенного ряда по степеням концентрации (или давления)
адсорбата в газовой фазе
. (IV,2)
155
или ряда по степеням Г:
с-КС1Г + Кс2Г2Н-- (IV,3)
Переход от концентрации с к давлению р может быть осуществлен
45 помощью уравнения состояния объемного газа.
Вириальные уравнения (IV,2) и (IV,3) не связаны с какой-либо
конкретной моделью адсорбционного слоя или его толщиной. Они
выражают связь гиббсовского избытка адсорбата Г, создаваемого
межмолекулярным взаимодействием молекул адсорбата с адсорбен-
том, с концентрацией адсорбата в объемном газе с.
Вириальное уравнение в частной форме можно получить на основе
модели двухмерного (мономолекулярного) адсорбционного слоя.
В этом случае считается, что адсорбированные в количестве Г мо-
лекулы образуют на поверхности адсорбента двухмерный реальный
газ. Для этого случая можно записать уравнение состояния такого
двухмерного (мономолекулярного) слоя в форме следующего вири-
ального разложения по степеням Г:
Я7 = кТГ + Ь2Г2 + Ь3Г3 + Ь4Г< + . . . (IV,4)
тде л' — поверхностное давление адсорбированного двухмерного слоя (моно-
слоя); к — константа Больцмана; д2, Ь3, &4 . . . — вириальные коэффициенты»
Здесь л' о° — п', где — поверхностное «натяжение» чистого
адсорбента, а о' — то же для адсорбента, покрытого монослоем
(для модели двухмерного слоя о' а и л' л, см. разд. 3 гл. III).
Объем газа V и площадь поверхности адсорбента А считаются по-
стоянными. Подставляя в адсорбционное уравнение Гиббса (111,28)
выражение для dn dnr, найденное из уравнения (IV,4), и инте-
грируя, получаем следующее уравнение изотермы мономолекуляр-
ной адсорбции идеального газа (а = с) с вириальными коэффициен-
тами в экспоненте:
с = Гехр(С1 + С2Г+С3Г2+. . .) (IV,5)
В уравнении (IV,5) константа интегрирования Сх = —In К19
где Кх — константа Генри [см. уравнение (IV,!)], константа С2 =
— 2ЬЯ, константа С3 = ЗЬ3/2, ^онстанта С4 — 4fc4/3. Уравнение
(IV,5) было выведено термодинамическим [1, 34—36] и статистиче-
ским [37] методами для адсорбции, а также методом термодинами-
ческой теории растворов (для адсорбции цеолитами, где вместо Г
использовали величину п& [38]). Если в вириальном разложении
(IV,5) ограничиться первыми двумя членами, то получается извест-
ное уравнение изотермы адсорбции Вильямса [39] — Генри [31].
Уравнение (IV,5) обычно хорошо описывает экспериментальные
изотермы адсорбции при малых и средних заполнениях поверхности.
При малых Г разложение экспоненциального выражения (IV,5)
156
в ряд Маклорена дает степенной ряд (IV,3)- Константы уравнений
IV,2), (IV,3) и (IV,5) связаны следующими формулами:
К1=~^—^~ехрС1
^c2 = ^2exPC’l (IV,6)
Кез = ^3^ ехР
Важную задачу представляет вычисление величин адсорбции
не только для разных концентраций адсорбата в газовой фазе при
постоянной температуре, но и при разных температурах. Уравнения
*с вириальными коэффициентами (IV,3) и (IV,5) можно использовать
и для этой цели [1]. Из уравнения (IV,5) вытекает, что
Г dine \ dC\ ( dC2 \ / dC3 \
\ дТ )г~~ dT '\dT ) dT )
ТГак как по формуле (III,64а) (3 In с/дТ)? = —&U!RT\ то из
жения (IV,7) следует, что
-“=ят’[-Й!-+(-зг)г+(-§-)Г!+- ]
Введя обозначения
Dt = ~RT^-^- (i=&2)
О1
получаем зависимость A U от Г в форме степенного ряда
(IV,7)
выра-
(IV,8)
(IV,9)
(IV,10)
В формуле (IV,10) величина \Ur = — RT* dCJdT ~RTzd In KjdT
[см. выражение (111,646)].
Коэффициенты ДС7b Z>2, D3 ... зависят только от природы
адсорбата и адсорбента и от температуры. Если пренебречь зависи-
мостью ДС7 от температуры, что обычно допустимо для небольшого
интервала температур в отсутствие молекулярных и фазовых пере-
ходов (см. гл. IX—XI, а также [17, 18]), то константы уравнений
(IV,5) и (IV, 10) связываются формулами [см. (III,59в)]
? I _ (Д^?+Д) । A^i
1-г НТ н нт
Ci = Bi +^r+(i^2)
til
(IV,11)
где = —(ASf 4" a Bi — константа интегрирования уравнений (IV,9).
Из уравнений (IV,5) и (IV,11) следует [1], что
е = Г ехр (51+5аг+58Г2+ . . .) хехр ( А?1+^Г-Н^Ч-• . . \ (J
' til 1
157
В уравнения (IV,5) и (IV,12) не входит емкость монослоя, которая
изменяется с температурой, и для многих адсорбентов представляет
трудноопределимую и часто имеющую лишь формальный характер
величину.
Применение уравнения (IV, 12) позволяет рассчитать из одной
экспериментальной обратимой изотермы адсорбции [см. уравнение
(IV,5)] и из определенной в калориметре при указанных в разделе 7
гл. III условиях зависимости дифференциальной теплоты адсорб-
ции qv = —AU от Г [см. уравнение (IV,10)] величины Г в широком
интервале температур и равновесных концентраций в газовой фазе.
Если независимых калориметрических определений qv для данной
системы адсорбат — адсорбент не сделано, то для определения кон-
стант — (Д57 + R)/R, AU1,Bi из уравнения (IV,12) необходимы
величины адсорбции, полученные при разных с и при разных (по
крайней мере при двух) температурах.
Уравнения (IV,5), (IV,10) и (IV,12) были применены для описа-
ния изотерм адсорбции и зависимостей AU или qst [см. выражение
(111,110)] от Г как для непористых адсорбентов (для графитирован-
ной термической сажи), так и для микропористых кристаллических
адсорбентов (цеолитов; в зтом случае вместо Г надо использовать ns).
Вириальные коэффициенты этих уравнений были определены для
адсорбции аргона, метана, этана и этилена на графитированной тер-
мической саже [43] и для адсорбции аргона, ксенона, этана, пропана,
этилена и СО2 на цеолите X с некоторыми щелочными и щелочно-
земельными катионами [40, 42, 43], а также для адсорбции низших
«-алканов цеолитом типа L [41].
Уравнения с вириальными коэффициентами описывают изотермы,
как с точкой перегиба в области заполнения монослоя, так и без нее.
Они также описывают зависимости теплоты адсорбции от заполне-
ния как при росте, так и при уменьшении теплоты адсорбции.
Уравнения (IV,5) и (IV,12) при Г -> 0 дают правильный нижний
предел — уравнение Генри. Однако они содержат несколько кон-
стант, связанных с парными, тройными и т. д. взаимодействиями
адсорбированных молекул друг с другом. До развития электронно-
вычислительной техники это создавало неудобства при практиче-
ском применении вириальных уравнений для обработки экспери-
ментальных данных с целью получения константы Генри и остальных
вириальных коэффициентов. Эти константы, однако, имеют четкий
физический смысл и по крайней мере первые две из них [в уравне-
ниях (IV,2) и (IV,5)] уже могут быть вычислены как из эксперимен-
тальных данных, так и с помощью молекулярно-статистической
теории.
Теоретические уравнения адсорбционного равновесия, получен-
ные с помощью молекулярной статистики (см. гл. VI и VII), содер-
жат несколько констант, связанных с геометрическими и силовыми
параметрами системы. Сюда относятся константы решетки, ван-дер-
ваальсовы радиусы, межъядерные расстояния и валентные углы
158
в молекуле, поляризуемости и магнитные восприимчивости силовых
центров молекулы и адсорбента, соответствующие квадрупольные
и дипольные моменты, характеристики локального распределения
электронной плотности и его изменения (например, при сильном
'Специфическом взаимодействии) и т. п., а также универсальные по-
стоянные.
В задачу молекулярно-статистической теории адсорбции входит
расчет констант Кх, ... в уравнении (IV,2) или констант С19
С... в уравнении (IV,5), а также связанных с этими константами
величин (ASJ + R)IR, В2, . . ., Atfi, Z)2, . . . в уравнении (IV,12) из
этих геометрических и силовых параметров. Свести число констант
в уравнении адсорбционного равновесия / (Г, с, Т) — 0 к двум или
трем при наличии достаточно сильного межмолекулярного взаимо-
действия адсорбат — адсорбат принципиально невозможно даже для
простейшего случая адсорбции на математически однородной по-
верхности.
В соответствии с этим в задачу рассматриваемой здесь термодина-
мической теории, связанной с определенной моделью мономолеку-
ляриого слоя [уравнения (IV,5) и (IV,10)], входит использование
этих уравнений для обработки экспериментальных данных и нахо-
ждения таких значений по крайней мере двух первых констант С г
и или копстант (А*$7 -{-/?)//?, В%, &Ц\ и D2, которые отражают
нааимодойствия адсорбат — адсорбент и соответственно парные вза-
имодействии адсорбат — адсорбат. Значения этих констант должны
зависеть только от свойств адсорбционной системы (структуры и фи-
зических свойств решетки адсорбента и молекулы адсорбата) и от
температуры. Находимые обработкой экспериментальных данных
значения констант Кх (илиС\) и МЦ практически не зависят от спо-
соба обработки этих данных. Определение остальных констант, учи-
тывающих взаимодействия адсорбат — адсорбат, затрудняется из-за
недостаточной точности экспериментальных данных. Однако практи-
чески ври использовании уравнения (IV,5) с числом членов не ме-
нее 3—G можно получить такие значения констант С\ и С2, которые
в пределах погрешности опытных данных и расчета методом наимень-
ших квадратов с помощью ЭВМ не зависят ни от числа членов, ни
от введенного в расчет интервала экспериментальных значений Г
(начинающегося с самых низких из охваченных измерениями запол-
нений и доходящего обычно до 50—75% от величины, соответству-
ющей заполнению плотного монослоя).
Примеры такого определения констант Сх и С2 из эксперимен-
тальных изотерм адсорбции приведены на рис. IV,2. Значения Сг
практически не зависят от величин интервалов Г или ns, введенных
и расчет, и от числа членов в экспоненте уравнения (IV,5), которое
менялось от трех до шести. Погрешность определения таким путем
величин составляет около 1—2%. Разброс значений С2 для раз-
ных величин обработанных интервалов изотерм адсорбции и разных
чисел членов ряда значительно больше, он составляет около 10—
159
15%. Для более надежного определения С2 точность измерений
на установках с газовыми бюретками еще недостаточна. По этой
причине величину С3 нельзя было определить таким же методом,
т. е. независимо от числа членов ряда и от интервала обрабатывае-
мой экспериментальной изотермы адсорбции. Поэтому можно опи-
санным выше методом пока определять только Сх и С3. Если для
практических целей кроме этого надо описать более широкую область
заполнений, то значения С3, С4 и т. д. следует определять из той же
I t I I -L
0 1 2 3 ь 5
Г, мкмоль/м2
£1---1----1----L
О 1 2 3
nst ммоль/г
i
4
С2 -123 °C
q-----а.....а--------о------и-
“45 °C
я—я-
-4
'ВО
/2345
Г, мкмоль/м2
о- 1
а~2
д-з
х-4
I I_____I___L
1 2 3 4
ns, ммоль/г
Рис. IV,2. Значение констант Сг и С2 уравнения (IV,5), рассчитанные для разных
интервалов величин Г аргона на графитированной термической саже (слева)
и ns ксенона на цеолите HLiNaX-1 (справа)» с учетом равного числа членов
ряда в уравнении (IV, 5):
I — 1 = 3; 2 — 1 = 4; 3 ~ г — 5; 4 ~ 1 = 6 [43].
Интервалы величин адсорбции вводились в расчет каждый раз от самой малой из измеренных
до указанной на рисунке измеренной величины адсорбции.
экспериментальной изотермы адсорбции, используя найденные рас-
смотренным выше методом значения констант и С2. При этом
полученные значения С3, С4, ... представляют лишь интерполя-
ционные коэффициенты, не имеющие ценности физико-химических
констант, как в случае описанного определения Сг и С2.
Примеры зависимости Сг и С2 от ИТ приведены на рис. IV,3.
Эти зависимости часто выглядят близкими к линейным. Это указы-
вает на то, что сделанное выше допущение о независимости AU
от Т в этих случаях невысоких заполнений и небольших интер-
валов температуры практически оправдывается. Однако этот вывод
также связан лишь с недостаточной точностью измерения изотерм
адсорбции при разных температурах.
Зависимости от НТ коэффициентов С3 и С4, полученных описан-
ным выше способом, имеют аналогичный вид. Поэтому уравнения
160
(IV,ll) позволяют определить соответствующие пары констант
/VTA, (AS? +Я)/7?, и
Величины ДС7\, (ASi JrR)/R, Dt и Bi9 полученные обработкой
трех — шести экспериментальных изотерм адсорбции, позволяют
рассчитать по уравнению (IV,12) величины Г при заданных значе-
ниях с (или р) и Т для соответствующих систем адсорбат — адсорбент.
Для расчета изотермы адсорбции в широкой области (часто до 70—
80% от заполнения плотного монослоя) обычно достаточно четырех
пар констант AUr, (Д££ -|-7?)/7?, Bz и Bt в уравнении (IV,12) или
Рис. IV,3. Зависимости величин констант С± и С2 урав-
нения (IV,5) от обратной температуры для адсорбции
аргона на графитированной термической саже (слева)
и ксенона на цеолите HLiNaX-1 (справа) [43].
четырех констант Ci в уравнении изотермы (IV,5) [43]. На рис. IV,4
на примере адсорбции аргона на графитированной термической
гаже и ксенона на цеолите HLiNaX-1 показано, как число вве-
прпных в расчет констант Ci влияет на интервал заполнений, в ко-
тором с помощью уравнения (IV,5) [43] можно описать эксперимен-
тальную изотерму адсорбции (в случае цеолита вместо Г вычисляют
//'). При учете лишь одной первой константы Сг (из уже определен-
ных из всей экспериментальной изотермы четырех констант С£),
г о. при учете только констант AUX и (AS* + B)/R, получаются
прямые, наклон которых представляет константу Генри Кх. Откло-
нение экспериментальной изотермы адсорбции от этой прямой по-
капывает, что в этом случае расчет описывает лишь низкие (нулевые)
‘пнюлнения. При учете первых двух констант Сх и С2 (т. е. констант
АН |, (AS{ + R)/R jiD^ B2) рассчитанная кривая вначале близка
к экспериментальной, а затем поднимается вверх круче последней.
11 Заказ 1180
161
Константа С2 отражает сильное парное взаимодействие адсорбат —
адсорбат. Из рис.. IV,4 видно, что эти две константы С\ и (и соот-
ветственно две пары констант АС71? (A5J + RVR иО2, В2) позволяют
удовлетворительно описать опытные данные до 15—20% заполнения
монослоя. Таким образом, практически наиболее важный, например,
в аналитической и препаративной хроматографии интервал величин
адсорбции в основном можно описать, учитывая только взаимодей-
ствия адсорбат — адсорбент и парные взаимодействия адсорбат —
адсорбат (в поле межмолекулярных сил, создаваемом адсорбентом).
Рис. IV,4. Изотермы адсорбции аргона на графитированной
термической саже при —195 °C (слева) н ксенона на цеоли-
те HLiNaX-1 при —60 °C (справа).
Сплошные кривые вычислены по уравнению (IV,5) для числа членов
в экспоненте г — к, а пунктирные кривые вычислены по тому же
уравнению с теми же константами при i — 1 и г = 2 [43].
Кривые, рассчитанные с учетом трех или четырех констант
или соответственно трех или четырех пар констант АС7Х, (ASJ + 7?)/7?,
и Bt (i 2), удовлетворительно согласуются с эксперименталь-
ными точками до 90% заполнения поверхности графитированной
термической сажи и до 70—80% заполнения полостей цеолитов
(за исключением случаев очень сильной специфической адсорбции
цеолитами). Эти кривые проходят точку перегиба (для сажи —
вблизи степени заполнения монослоя 0^0,4).
С помощью констант AU19 (AS* -j-7?)//?, и Bt (i 2) могут
быть вычислены изотермы адсорбции для любых температур внутри
и несколько выходя за пределы использованного при их определе-
нии интервала. Пример описания адсорбции аргона на графитиро-
ванной термической саже в широком интервале температур с помощью
уравнения (IV,12а) приведен на рис. IV,1.
С помощью констант AUX и Dt по уравнению (IV,10) можно
также рассчитать зависимость — AU от Г. Из рис. IV,5 видно, что
1.62
в определенном интервале Г вычисленная зависимость — АС7 от Г
удовлетворительно согласуется с точками, полученными из изостер,
построенных непосредственно по экспериментальным данным [43].
Зная константы уравнения изотермы адсорбции в вириальной
форме (IV, 5), определенные из экспериментальной изотермы адсорб-
ции, можно вычислить поверхностное давление л' мономолекуляр-
ного адсорбированного слоя по экспоненциальному уравнению
в вириальной форме (IV,4) для разных Г или разных величин пло-
щадей со, приходящихся на одну молекулу в адсорбированном моно-
Рис. IV, 5. Зависимости дифференциального моль-
ного изменения внутренней энергии при адсорб-
ции—ДС7 от величины адсорбции Г для аргона на
графитированной термической саже (слева) и от
величины адсорбции ns для ксенона на цеолите
HLiNaX-1 (справа) [43].
Кривые рассчитывались по уравнению (IV, 10), точки —
экспериментальные данные.
слое. Зависимость л' от со, рассчитанная по уравнению (IV,4) для
адсорбции этана на графитированной термической саже при —100° С,
показана на рис. IV,6. Величины л, изображенные точками, рассчи-
таны непосредственно из экспериментальных данных по уравнению
(III,29а). Величины л', вычисленные по уравнению (IV,4), хорошо
согласуются с величинами л, вычисленными непосредственно из
экспериментальных данных по уравнению (III,29а). На том же ри-
сунке приведена кривая для состояния идеального двухмерного
газа л'(о = кТ. Состояние адсорбированного этана при —100° С
сильно отличается от состояния идеального двухмерного газа. За счет
значительного притяжения адсорбат — адсорбат кривые для реаль-
ного состояния адсорбированного вещества идут значительно ниже
кривой для состояния идеального двухмерного газа.
Уравнение (IV,5) позволяет представить зависимость коэффи-
циента активности у5 адсорбата в адсорбционном слое [см. урав-
нение (111,23)] от величины адсорбции Г в виде ряда [43]:
у’ = ехр (С2Г + С3Г2 + С4ГЗ) (IV ,13)
Величины ys, рассчитанные по этому уравнению для некоторых
систем [43], представлены на рис. IV,7. Начиная от у3 = 1 при
Г = 0, для систем с сильным притяжением адсорбат — адсорбат
ys сначала уменьшается (до заполнения 0 0,4—0,5), а при более
11*
163
высоких концентрациях адсорбата начинает увеличиваться. При
достаточно высокой для адсорбции аргона температуре (—150° С)
взаимодействие адсорбат — адсорбат мало и значение у5 вначале
близко к единице, а потом постепенно увеличивается.
0
100
20Q
Рис. IV,6. Зависимость поверхно-
стного давления л от площади со,
приходящейся на молекулу этана
в адсорбционном слое на гра-
фитированной термической саже
при —100 °C.
Точки вычислены из полученной
из экспериментальной изотермы ад-
сорбции (см. рис. 111,1) зависимости
Г/р = / (р) (см. рис. III, 5) по урав-
нению (III, 29а). Сплошная кривая
вычислена по уравнению (IV,4) с по-
мощью найденных из анализа изотермы адсорбции вириальных коэффициентов. Пунктирная
кривая вычислена для идеального состояния двухмерного адсорбированного слоя.
Аналогичным образом можно использовать и другие модели
состояния адсорбированного вещества. Подставляя соответствующие
выражения для dn* = dst в уравнение Гиббса (111,28), можно найти
соответствующие уравнения изотермы адсорбции, а также зависи-
мости АС от Г. Используя такие модели, можно получить также
I 1-1______L
о 1 г з 4
л? ммоль/3
Рис. IV,7. Зависимости коэффициента активности у8 адсорби-
рованного аргона от Г на графитированной термической
саже (слева) и ксенона от п8 на цеолите HLiNaX-1 (справа)»
вычисленные по уравнению (IV,13).
зависимости теплоемкости адсорбированного вещества от заполне-
ния поверхности адсорбента и исследовать фазовые переходы и кри-
тические явления.
Кроме моделей подвижного слоя (моделей нелокализованной
адсорбции) йожно рассмотреть и различные модели локализованной
адсорбции. Комбинируя модели мономолекулярной адсорбции с мо-
делями полимолекулярной адсорбции, можно получить выражения
для изотерм адсорбции и зависимостей ACT” и АС от Г или 9 для более
широкой области 0.
Как в случае нелокализованной, так и в случае локализованной
адсорбции важное значение имеет учет ассоциации адсорбированных
464
молекул, особенно легко происходящей при адсорбции на поверх-
ности неспецифических адсорбентов молекул группы D, образу-
ющих взаимные водородные связи, а также молекул группы В,
обладающих значительными периферическими дипольными момен-
тами.
Эти важные вопросы выходят за пределы этой книги, в которой
в дальнейшем основное внимание уделяется небольшим заполнениям
поверхности. Подробно получение изотерм адсорбции и зависимостей
изменений внутренней энергии и теплоемкости от величины адсорб-
ции при использовании различных моделей уравнения двухмерного
состояния и моделей нелокализованной и локализованной моно-
и полимолекулярной адсорбции с учетом ассоциации адсорбат —
адсорбат рассматривается в подготавливаемой книге Г. И. Бере-
зина и А. В. Киселева, специально посвященной молекулярным
и фазовым переходам в адсорбционных системах и . теплоемкости
этих систем.
3. Получение термодинамических характеристик адсорбций
на однородной поверхности из экспериментальных данных
для реальной поверхности
В гл. II рассмотрены трудности получения твердых адсорбентов
с близкими к однородным поверхностями. Вместе с тем было пока-
зано, что, совершенствуя синтез адсорбентов и способы модифици-
рования их поверхности, можно в той или иной степени приблизиться
к химически и физически однородной поверхности заданной струк-
туры и состава.
Гранулы адсорбентов, обычно используемые в экспериментальных
исследованиях, представляют агрегаты или сростки частиц твер-
дого тела. На поверхности таких адсорбентов неизбежно остаются
геометрически и химически неоднородные участки. Относительное
число и влияние зтих неоднородных участков поверхности адсор-
бента можно значительно снизить, но нельзя устранить совершенно.
Для преодоления зтих затруднений возможны следующие пути.
Во-первых, нужна разработка такого метода анализа эксперимен-
тальных термодинамических данных, относящихся к не вполне
однородной поверхности, который позволил бы получить из этих
данных термодинамические характеристики для соответствующей
однородной поверхности. Например, такой метод анализа результа-
тов хроматографических и статических измерений на графитиро-
ванных сажах должен позволить найти термодинамические характе-
ристики (константы равновесия, теплоты и энтропии адсорбции,
теплоемкости) системы данный адсорбат — базисная грань полубес-
конечной решетки графита. Один из таких методов изложен в этом
разделе.
Во-вторых, нужно использовать комплекс прямых (не термодина-
мических) методов детального исследования реальной поверхности
применяемого в экспериментальных исследованиях адсорбента: хи-
165
мических и изотопнообменных методов определения химического
.состава поверхности (см. гл. II, а также работы [44—48]), различ-
ных дифрактометрических [49] и других кристаллографических
методов, спектроскопических методов определения концентрации
и состояния поверхностных соединений [50], электронно-микро-
скопических методов исследования поверхности (в частности, де-
корированной поверхности) с целью обнаружения и определения
характера ступеней роста и выхода дислокаций [51, 52] и других
химических и физических методов. Такое комплексное исследование
структуры именно той поверхности, к которой относятся эксперимен-
тальные адсорбционные термодинамические данные, помогает вы-
брать и уточнить модель поверхности твердого тела, принимаемую
для описания адсорбции на этой поверхности. Это позволяет ввести
экспериментально обоснованные функции распределения различных
мест поверхности по создаваемой ими энергии адсорбционного взаи-
модействия в молекулярно-статистические выражения для термоди-
намических характеристик адсорбции. Такое прямое физическое
обоснование функций распределения, учитывающих неоднородность
реальной поверхности адсорбента, позволит сделать более надеж-
ным анализ экспериментальных термодинамических данных по пер-
вому указанному выше пути — но пути расчета из эксперименталь-
ных термодинамических данных характеристик адсорбции на соот-
ветствующей однородной поверхности. Проверка правильности вы-
бора функции распределения при известных химическом составе
и структуре поверхности и объема адсорбента может быть сделана
молекулярно-статистическим путем с использованием найденных
для адсорбции на физически однородной поверхности атом-атомных
потенциальных функций межмолекулярпого взаимодействия. Ме-
тоды нахождения таких потенциалх>ных функций описаны в гл. X.
Работы по комплексному применению химических и физических
методов количественного изучения состава, структуры и состоя-
ния поверхностей параллельно изучению их адсорбционных свойств
еще только начинаются. Поэтому здесь будет рассмотрен только
первый путь, т. е. способ оценки термодинамических характеристик
системы адсорбат — физически однородная поверхность из экспери-
ментальных данных, полученных на поверхностях, по своему составу,
структуре и свойствам приближающихся к этой однородной поверх-
ности.
Недостатком ранних работ по адсорбции на неоднородных поверх-
ностях было отсутствие обоснованных независимо от адсорбционных
измерений физических моделей этих поверхностей и невозможность
сопоставления с соответствующими однородными поверхностями.
Обзор более ранних работ по адсорбции на неоднородных поверх-
ностях с учетом взаимодействия адсорбат — адсорбат дан Хонигом
[53, 54]. Здесь мы рассмотрим метод учета неоднородности реальной
поверхности для получения термодинамических характеристик ад-
сорбции на однородной поверхности той же природы, предложенный
Россом и Оливье [11, 55], как пример одной из первых попыток
166
i" niriHui этой проблемы с учетом взаимодействий адсорбат — ад-
* "|ц<иг( существенно влияющих на форму экспериментальных изо-
С I'M адсорбции.
Ji от метод представляет частный способ решения уравнения для
ыпигимости адсорбции на неоднородной поверхности от темпера-
»\ |<1.1 и концентрации (давления) адсорбата в газовой фазе. Поверх-
п'н и. при этом предполагается состоящей из однородных участков.
Нм ураинепие имеет следующий общий вид:
fe
е= J /(е)<р(р, е)йе (IV.14)
О
Здесь функция / (е) описывает распределение однородных участков
нонерхности по энергиям адсорбции на этих участках е, а функция
'I (/>, и) описывает изотерму адсорбции для участка поверхности
• энергией адсорбции е. При числовом решении уравнения (IV,14)
рш ематрипаомым методом [11] делается ряд допущений. Поверхность
шн ирбента рассматривается состоящей из некоторого конечного
числа однородных участков, каждый из которых характеризуется
онрндолоппой потенциальной энергией адсорбции, например f-тый
участок энергией Если рассматривается 50 таких участков [11],
in общаи величина заполнения поверхности
50
£-1
। ЛЛ/ - / (в/) Jf? — доли поверхности, приходящаяся па участок с энергией в/;
0( - ip (/», г,/) — заполнение этого участка, определяемое энергией 8/.
Допускается далее, что общий интервал значений ez ограничи-
iHiPim -20 кДж/моль.
Для описания состояния адсорбционной пленки на каждом
" а,породном участке была выбрана [11], в частности, модель двухмер-
ной пел окал изованной адсорбционной пленки па математически
однородной поверх пости адсорбента, состояние которой описывается
\р1шиоиием тина уравнения Вап-дер-Ваальса [7, 12]:
(я' + -^-) (fi> — bs) = kT (IV,16)
• и,' поверхностное давление монослоя; со — площадь поверхности, при-
*niniiiviiirii в среднем на одну молекулу; as и bs — константы притяжения и от-
ihtin.iiiiiii адсорбат — адсорбат (константа bs соответствует площади£сот,
причпдпщейся на молекулу адсорбата в плотном монослое).
II я этого уравнения состояния получается известное уравнение
монпмолекулярной нелокализованной адсорбции Хилла [6] — де-
Hvihi 17]:
fl>~exp ('i—e' ~Х26') (IV,17)
!н,ггь О' rfes — степень заполнения монослоя на однородной
нпнерхности адсорбента молекулами адсорбата; К[ — константа
167
Генри (выраженная через 0 и />, т. е. в единицах р), связанная с ра-
нее введенными константами КГ р1 и Kr>Cfl = Кг [см. выраже-
ние (111,15)] формулой
К> _ 1 _ kT ==s кТ (TV W
Х bsKT, р. 1 bSKT, с, 1 }
Константа же
отражает влияние на форму изотермы адсорбции взаимодействия
адсорбат — адсорбат. Так как as и fes принимаются постоянными,
то константа К2 обратно пропорциональна температуре.
Уравнение (IV,17) приближенно описывает изотермы адсорбции
на однородной поверхности графитированных саж аргона [56, 57],
криптона [56], ксенона [58], двуокиси углерода, шестифтористой
серы [13], метана, этана [59, 60], этилена [59], пропана, бутана
и изобутана [60], бензола [И], неопентана [61], дизтилового эфира
[62], четыреххлористого углерода [И, 61, 63], хлороформа и фтор-
хлорметана [11]. Поэтому его можно использовать и для прибли-
женного описания изотермы адсорбции на однородном участке z.
Для каждого z-го однородного участка поверхности надо различать
свое значение константы Генри K'lti уравнения (IV,17), определяе-
мое энергией е. взаимодействия адсорбат — адсорбент. Далее пред-
полагается [11J, что отношение констант двухмерного состояния as
и fes, учитывающее взаимодействие адсорбат — адсорбат, можно
вычислить из отношения соответствующих трехмерных констант <zy
и bv по уравнению [11]
д8 av
bt 2Ьу
(1V,2O)
причем
(IV.21)
Величины as и Ь$ для ряда адсорбатов приведены в табл 1
гл. VI книги Росса и Оливье [11]. В табл. IV,1 некоторые из вели-
чин К2, рассчитанных из указанных в этой книге значений as и bsi
сопоставлены с величинами К2, полученными обработкой экспери-
ментальных изотерм адсорбции с помощью уравнения (IV, 17).
Как правило, значение константы Я2, определенное из уравне-
ния (IV, 17), немного меньше значения, определенного из констант
av и bv. Расхождение это связано, вероятно, главным образом с тем,
что в рассматриваемой модели не принято во внимание влияние
адсорбента на взаимодействие адсорбат — адсорбат. Данные табл. IV,!
показывают тем не менее, что значения К2 [11], оцененные из объем-
ных свойств адсорбата с помощью формул (IV,19)—(IV,21), можно
считать приблизительно правильными.
Для описания адсорбции на однородных участках, составляющих
в целом неоднородную поверхность, можно использовать и другие
168
Таблица IV,1. Значения константы K$~2as/RTbs,
вычисленные из экспериментальных изотерм адсорбции
на графитированной термической саже с помощью уравнения (IV, 17)
и из объемных свойств адсорбата с помощью выражений (IV,19) — (IV,21)
Адсорбат Температура, °C Значения К2
из изотерм адсорбции из объемных свойств [11]
Аргон —195,7 5,5 [56, 57] 6,5
Криптон —183 6,2 [56] 7,1
Ксенон —120 5,8 6,7
Двуокись углерода —80 - 5,0 [13] 5,3
Этан —100 5,4 [59] 5,9
-85 4,8 [59] 5,2
—70 4,2 [59] 5,0
—60 3,8 [59] 4,8
—24,7 2,5 [60] 3,4
Четыреххлористый углерод -47,0 7,1 [63] 7,9
—29,8 6,7 [63] 7,4
20 5,5 [62] 6,1
уравнения изотермы адсорбции на однородной поверхности, доста-
точно полно учитывающие взаимодействия адсорбат — адсорбат [11К
Росс и Оливье [И] допустили, что
распределение участков поверхности
по величинам энергии адсорбции вы-
ражается функцией Гаусса:
Дбг^/(е) йе ——ехр [—₽ (м—вт)*] d&
П
(IV,22)
где Дб/ — доля поверхности с энергиями ад-
сорбции в интервале от до + Де; —
значение потенциальной энергии адсорбции
на данной поверхности, соответствующее
максимуму кривой распределения (рис. IV,8);
р — константа, обратно пропорциональная
дисперсии этой кривой и поэтому характери-
зующая неоднородность поверхности; п —
нормирующий фактор, соответствующий при-
нятому интервалу значений е/.
Можно избрать и другую функцию
распределения участков поверхности
по энергиям адсорбции, например
функцию Пуассона [64].
Для выбранной модели адсорбции
на _ неоднородной поверхности анали-
Рис. IV,8. Гауссовы кривые
распределения участков по-
верхности по величинам потен-
циальной энергии адсорбции
для разных значений р (числа
на кривых).
тическое решение уравнения (IV,22) невозможно [И, 55]. По-
этому уравнение (IV,22) решалось на вычислительной машине при
использовании конечных величин A6Z и Aez = 0,1 ккал/моль [11].
169
Описанным методом получают семейство модельных изотерм
адсорбции для набора разных значений констант К2 и К[г. и сопо-
ставляют с ними экспериментальную изотерму адсорбции. Модель-
ные изотермы адсорбции рассчитываются следующим образом.
Заполнение 0Z каждого j-ro участка поверхности с энергией адсорб-
ции е{. при давлении р в газовой фазе рассчитывается по уравнению
(IV,17). Величина 0 для всей поверхности определяется суммиро-
ванием по уравнению (IV,15). При этом константа Генри K[ti
определяется по приближенному уравнению
Ki,i = B?exp(-^r) (IV,23)
где В ° — энтропийный фактор, который принимается не зависящим от 8/ [И].
Энтропийный фактор рассчитывается по уравнению [11]
—1пт=^г+1п/,° <IV>24>
В этом уравнении Д5° — стандартное дифференциальное изменение
энтропии при переходе моля адсорбата из газа в его стандартном
состоянии р° в адсорбированное стандартное состояние 0°; Д£кнн —
изменение кинетической энергии адсорбированной молекулы при
переходе из газового в адсорбированное состояние. За стандартные
состояния выбраны р° ~ 760 мм рт. ст. и 0° = &s/4,087 [11, 65].
Принимается также, что Д£кнн = 72772.
Согласно принятой модели состояния адсорбированного вещества
[11] величина Д5° включает колебательную энтропию адсорбата
в адсорбированном состоянии и изменение энтропии, соответствующее
потере одной поступательной и одной вращательной степени свободы.
В расчетах принималось во внимание только изменение энтропии,
соответствующее потере поступательной степени свободы. Изменение
энтропии рассчитывалось по уравнению Закура — Тетроде
д5°, П0СТ=-1пМ + 1пГ + 2,31 (IV,25)
Константа К'1}ГП, соответствующая энергии ет в максимуме
кривой распределения, равна
X{fOT = B?exp (~^) (IV,23а)
Константы z для адсорбции на остальных участках поверхности
можно выразить через
*1, i = Kj,MexP [— (ez_em)] (IV,236)
Задаваясь величинами plK[tm, Р и К2 = 2 as/7?Tfes [7, И], для
каждого из 50 значений ez в рассматриваемом интервале рассчиты-
вают величины p!K[tl и 0р а затем суммарное 0 [(см. уравнение
(IV,15)]. Для р1К^т было взято 39 значений от 0,001 до 40, для р
взято 16 значений от 1 до 1000 и для К2 — 100 значений от 0,1 до 10.
170
Данные для построения рассчитанных модельных изотерм ад-
сорбции собраны в 100 таблицах [11]. Эти таблицы позволяют по-
строить серию модельных изотерм адсорбции практически для лю-
бого имеющего физический смысл значения К2 = 2aJRTs. На
рис. IV,9 представлены рассчитанные для 2ajRTbs — 6,5 изотермы
адсорбции, соответствующие кривым распределения участков по-
верхности по величинам энергии адсорбции, показанным на рис. IV,8.
Форма изотермы адсорбции меняется от кривой, обращенной вначале
выпуклостью к оси давления и проходящей затем точку перегиба,
характерной для поверхности с большой степенью однородности
Рис. IV,9. Полученное при значении 2as/RTbs = 6,5 се-
мейство модельных изотерм адсорбции, соответствующих
кривым распределения участков поверхности по величинам
потенциальной энергии адсорбции, представленным на \
рис. IV,8.
(большие Р), до кривой, обращенной повсюду вогнутостью к оси да-
вления. При наличии взаимодействия адсорбат — адсорбат такая
кривая характерна для весьма неоднородной поверхности (неболь-
шие Р).
Для исследуемого адсорбата, исходя из данных [И] (приведены
значения as и bs для 30 адсорбатов), рассчитывается величина
2aJRTbs, частично приведенная в табл. IV,1. Далее из упомянутых
выше 100 таблиц модельных изотерм выбирается та, которая соот-
ветствует этому значению 2aJRTbs. В этой таблице приведены
значения 0 и р/К'1т для разных значений р. Например, в таблице
для 2aJRTbs — 6,5 при р = 200 для значений 0, равных 0,0019;
0,0029; 0,0092; 0,0863; 0,1279; 0,2480; 0,3802; 0,6215; 0,7123; 0,7810
и 0,8814, значения р/К{ т равны соответственно 0,001; 0,002;
0,008; 0,050; 0,060; 0,080; 6,100; 0,170; 0,300; 0,800 и 40,000. Значе-
ния, приведенные в этой таблице, позволяют построить график се-
мейства модельных изотерм адсорбции, соответствующих значению
2as!RTbs = 6,5 для разных р, в координатах In 0 и In (р1К\^ т).
171
Примеры этих модельных изотерм адсорбции показаны на рис. IV,10
сплошными линиями. Экспериментальная изотерма адсорбции стро-
ится на прозрачной миллиметровой бумаге в координатах In Г
(или In ns) и In р. График экспериментальной изотермы адсорбции
накладывается на график семейства модельных изотерм. Параллель-
ным смещением осей этого графика достигается наилучшее совме-
щение экспериментальной изотермы адсорбции с одной из модельных
Рис. IV,10. Модельные изотермы адсорбции, вычисленные при значении
2as/RTbs = 6,5 [сплошные линии в координатах In 0 — In (р/К{>т)], и совме-
щенная с ними экспериментальная изотерма адсорбции при 77,5 К. аргона (для
которого 2aslRTbs=^ 6,5, см, табл. IV,1) на саже Sterling FT, 1000 РС (пунк-
тирная линия в координатах In Г — 1пр).
Значения Г выражены в мкмоль/м2, сдвиги графика экспериментальной изотермы адсорбции
относительно графика модельных изотерм по оси ординат (на величину —In bs) и по оси,
абсцисс (на величину In К* указаны стрелками.
изотерм. В указанном на рис. IV,10 примере экспериментальная
изотерма адсорбции аргона при 77,5 К на термической саже Ster-
ling FT, прокаленной при 1000° С, представлена пунктиром.
Таблица IV,2. Значения констант р, и т для адсорбции этана
на графитированной термической саже Sterling FT, 2700°С
Температура опыта Т, К Найденная для каждой изотермы величина 3 Kt и способ определения К1( m по методу Росса и Оливье мкм
по уравнению с вириальными коэффициентами (IV, 5), мкм по уравнению Хилла—де-Бура (IV,17), мкм
213 200 0,82 . 0,88 0,66
203 200 1,29 1,39 1,19
188 200 2,70 3,05 2,46
173 200 6,70 8,20 6,50
172
На этом рисунке
линии — соответ-
И Z/W G).
сплошные
Рис. IV,11. Изотермы адсорбции
аргона при 77,5 К на саже Sterling
FT, графитированной при разных
температурах (числа на кривых)»
Кривые вычислены по методу [И], точ-
ки — экспериментальные данные.
Необходимое для совмещения экспериментальной и модельной
изотерм адсорбции смещение осей двух этих графиков в направле-
нии оси ординат дает величину — In bs [или —In (fes/s)J, а смещение
в направлении оси абсцисс — величину In т. Наилучшим об-
разом совмещенная с экспериментальной изотермой адсорбции
модельная изотерма дает и соответствующий фактор р (см.
табл. IV,2 и IV,3), указывающий на степень однородности по-
верхности.
На рис. IV, 11 для примера представлены изотермы адсорбции
аргона на двух образцах термических саж с разной степенью графи-
тирования (обработанных при 10
точки — экспериментальные данн
ствующие модельные изотермы.
Из рис. IV,11 видно хорошее
отображение модельными изотер-
мами перехода от неоднородной
поверхности сажи, обработанной
при 1000° С (Р = 8) к весьма
однородной поверхности сажи,
обработанной при 2700°С (Р ~
= 170).
Приведенные на рис. IV,10 и
IV,И примеры показывают воз-
можность успешного применения
рассмотренного метода [И ] для
обработки и интерпретации экспе-
риментальных термодинамических
данных, полученных на адсорбен-
тах с разной степенью однородно-
сти. Весьма важно, что определен-
ная этим методом константа Генри
Кг>т, характеризующая взаимодействие адсорбата с адсорбентом,
начиная с температуры прокаливания термической сажи околи
1500°С, мало меняется при дальнейшем повышении температуры
прокаливания (рис. IV,12). В то же время значения констант Генри
К19 рассчитанные по уравнению с вириальными коэффициентами
(IV,5) для саж, прокаленных при более низких температурах, резка
изменяются с повышением температуры прокаливания. Однака
с ростом температуры графитирования сажи величина Кг стремится
к определенному предельному значению, совпадающему со значе-
нием К1} т, и практически достигает его уже при р = 100. Таким
образом, в этом случае рассмотренный метод [И] позволяет из изо-
терм адсорбции на не вполне графитированных сажах получить
значение константы Генри, характеризующее адсорбцию на пре^
дельно графитированной саже. Это предельное (для р = оо) значе-
ние резко отличается от значения константы Генри для изотермы
адсорбции, измеренной на неоднородной поверхности неграфитиро-
ванной сажи (см. рис. IV,И).
173
Таким образом, для близких к однородным поверхностей мето-
дом Росса и Оливье [И] может быть охарактеризована степень од-
нородности поверхности адсорбента (величиной 0) и найдено зна-
чение константы Генри для однородной поверхности той же природы
{для р = оо). В случае саж это.означает важную для молекулярно-
статистической теории адсорбции возможность определения из опы-
тов с реальными графитированными сажами значений константы
Генри для адсорбции на однородной поверхности базисной грани
нолубесконечной решетки графита. Для всех молекул группы А
Рис. IV,12. Зависимости константы Генри для адсорбции
аргона при 77,5 К на сажах Sterling FT от температуры
прогревания сажи (а) и величины фактора р (б), полученные
методом Росса и Оливье [11] для наиболее однородной части
поверхности, KlfTn (Z), и по вириальному уравнению (IV,5)
для всей поверхности, Кх (2).
и многих молекул группы В (см. рис. 1,4) адсорбция на поверх-
ности графитированной вблизи ЗООО°С термической сажи соответ-
ствует адсорбции на поверхности базисной грани полубесконечной
решетки графита. Для остальных молекул группы В, например для
ацетона, и для молекул группы Z), например для спиртов, это соот-
ветствие обеспечивается дополнительной обработкой графитирован-
ной при ЗООО°С термической сажи в токе водорода при 1100° С, уда-
ляющей кислородные комплексы с поверхности образцов этой гра-
фитированной сажи (см. разд. 1 гл. II).
Для примера в табл. IV,2 и на рис. IV, 13 приведены результаты
.определения константы Генри, проведенного методом Росса и Оливье
[11] и другими рассмотренными в этой главе методами, из измерен-
ных при разных температурах изотерм адсорбции на графитирован-
ной термической саже Sterling FT, 3000° С [43, 59, 60]. Из табл. IV,2
видно, что для всех этих изотерм адсорбции такое определение дает
174
очень большую величину £=200- Это свидетельствует об узком рас-
пределении различных участков поверхности по величинам энергии
адсорбции (рис. IV,8), т. е. о высокой степени однородности поверх-
ности этой сажи по отношению к адсорбции этана (молекулам группы
Л). Кроме того, результаты определения константы Генри методом
Росса и Оливье [11] уже мало отличаются от результатов ее опреде-
ления другими методами, предполагающими однородность поверх-
ности [32, 43, 59, 60], а также от результатов ее прямого измерения
газохроматографическим методом [66] (см. рис. IV,13).
Рис. IV,13. Зависимость In от обратной температуры для
адсорбции ряда веществ на графитированной термической саже.
Точки получены с помощью 1 — газохроматографических измерений; 2—1—
статических измерений изотерм адсорбции [2—4 — по уравнению (IV,17),
5 — по уравнению (IV,5), 6— по уравнению (IV,2) и 7 — по методу [11].
Рассмотренный метод был применен и к адсорбции на ионных
кристаллических адсорбентах [11]. В частности, было найдено, что
изотерма адсорбции на ряде адсорбентов такого типа складывается
из двух изотерм адсорбции — одной с высоким значением 0, соответ-
ствующей адсорбции на физически однородной части поверхности,
состоящей из одной грани, и другой с малым значением 0, соответ-
ствующей адсорбции на остальной, неоднородной части поверхности.
Эта неоднородная часть поверхности может быть связана с выходом
на поверхность дислокаций и ступеней роста кристаллов (см. рис.
11,21). Этот метод представления поверхности адсорбента в вида
суммы двух типов поверхностей был применен также для описания
адсорбции сажами, содержащими добавки другого вещества, адсор-
бирующего слабее, чем сама сажа [68]. После нанесения таких до-
бавок особо сильно адсорбирующие участки поверхности сажи
175
оказывались экранированными и степень однородности остальной
поверхности сажи увеличивалась.
В табл. IV,3 приведены примеры результатов аналогичных ана-
лизов экспериментальных изотерм адсорбции ксенона на некоторых
цеолитах [43, 69]. По отношению к неспецифической адсорбции
ксенона цеолиты ведут себя как довольно однородные адсорбенты
(значение 0 велико, особенно для системы Хе — HLiNaX-1).
По отношению же к специфически адсорбирующейся цеолитом дву-
окиси углерода этот метод позволяет сделать заключение лишь о
степени неоднородности цеолита.
Таблица IV,3. Значения констант 0, и т для адсорбции ксенона цеолитами
Адсорбционная система Температура опыта Т, К Найденная для каждой изотермы величина 3 Ki по уравнению с вириальными коэффициентами (IV, 5), мкм к1, т по ме- тоду Росса и Оливье, мкм
Хе —HLiNaX-1 [67] 193 1000 0,96 1,29
213 1000 0,44 0,48
228 1000 0,171 0,156
Хе —NaX [67] 213 70 1,02 0,80
228 70 0,48 0,40
243 70 0,27 0,24
Расчет рассмотренным методом [И] модельных изотерм адсорб-
ции для величин 2aJRTbs > 6,75, соответствующих температуре
ниже критической для двухмерного газа, дает при небольших 0
модельные изотермы адсорбции со многими небольшими ступенями
[70]. В связи с этим поверхность была разбита не на 50, а на 200
различающихся по величинам et- участков для больших и средних
значений 0 и на 250 таких участков для малых 0, соответствующих
более широкому распределению. При этом были получены кривые
без ступеней при 2aJRTbs >6,75. Следует, однако, отметить, что
сглаживание изотермы связано здесь лишь с практической неразли-
чимостью ступеней при построении кривых.
Метод Росса и Оливье [11] можно использовать лишь для таких
систем, для которых можно считать довольно правильными сделан-
ные этими авторами допущения. Тем не менее этот метод полезен
для обработки экспериментальных изотерм адсорбции на близких
к однородным реальных адсорбентах с целью оценки степени неод-
нородности поверхности и получения из этих экспериментальных
изотерм адсорбции констант, характеризующих адсорбционное рав-
новесие на соответствующих однородных поверхностях. По мере
развития комплексного применения новых химических и физиче-
ских методов количественного изучения состава, структуры и со-
стояния реальных поверхностей твердых тел можно будет в конкрет-
ных случаях уточнить многие из сделанных в этом методе допуще-
176
ний, в частности допущение о гауссовском характере распределения
участков поверхности по величинам потенциальной энергии адсорб-
ции. Часто выбором подходящих условий синтеза и обработки ад-
сорбента можно свести число физически однородных участков по-
верхности, сильно различающихся по их строению и потенциальной
энергии адсорбции, к очень небольшому числу (например, к двум
в случае кристаллов типа СоС12, см. рис. 1,6, разд. 2 гл. I и разд. 4
гл. II). В этом случае, определив тем или иным методом соотноше-
ние площадей соответствующих физически однородных частей по-
верхности и потенциальные кривые межмолекулярного взаимодей-
ствия молекул данного адсорбата с обоими типами этих однородных
частей поверхности или хотя бы оценив значения потенциальных
функций в минимуме потенциальных кривых (т. е. найдя значения
е1( т и s2( m)t можно вычислить изотерму и теплоту адсорбции данного
адсорбата на суммарной поверхности такого адсорбента. Таким
образом, можно моделировать ступенчатые изотермы, наблюдаемые
во многих случаях адсорбции на кристаллах с различающимися
по структуре гранями. Решение этой задачи для трех уровней
61,/п, б2, т и e3j/n предложено в работе [71].
ЛИТЕРАТУРА
1. Киселев А. В. ЖФХ, 1967, т. 41, с. 2470—2504.
2. L a n g m u i г I. J. Am. Chem. Soc., 1918, v. 40, p. 1361—1403.
3. BrunauerS. e. a. J. Am. Chem. Soc., 1938, v. 60, p. 309—319.
4. Фаулер P. X., Гуггенгейм E. А. Статистическая термодина-
мика. M., Издатинлит, 1949. 612 с.
5. БрунауэрС. Адсорбция газов и паров. М., Издатинлит, 1948. 781 с.
6. Н i 11 Т. J. Chem. Phys., 1946, v. 14, р. 441—452.
7. Де-Бур Дж. X. Динамический характер адсорбции. М., Издатинлит,
1962. 290 с.
8. Киселев А. В. ДАН СССР, 1957, т. 117, с. 1023—1026; Коллоидн.
ж., 1958, т. 20, с. 338—348.
9. Киселев А. В., П о ш к у с Д. П. Изв. АН СССР, ОХН, 1958,
с. 520-522.
10. Y о u n g D. М., Crowell A. D. Physical Adsorption of Gases, London,
1962. 426 p.
11. Ross S., Oliever J. P. On Physical Adsorption. New York, 1964.
401 p.
12. Г e p а с и м о в Я. И, и др. Курс физической химии. Под ред. Я. И. Ге-
расимова. Изд. 2-е. т. I, М., «Химия», 1969. 592 с.
13. Биби Р. А. и др. ЖФХ, 1964, т. 38, с. 708—718.
14. Sams J. R., Constab aris G., Halsey G. D. J. Phys. Chem.,
1960, v. 64, p. 1689—1696.
15. W о 1 f e R-, S ams J. B. J. Phys. Chem., 1965, v. 69, p. 1129—1135.
16. Б e p e з и н Г. И., К и с e л e в А. В. ЖФХ, 1966, т. 40, с. 1702—
1707.
17. Berezin G. I., Kiselev А. V. J. Col. Interface Sci., 1966, v. 22,
p. 161—164.
18. Б e p e з и н Г. И., Киселев А. В., ЖФХ, 1968, t. 42, c. 1987—
1991.
19. Березин Г. И., Киселев А. В., ЖФХ, 1969, т. 43, с. 1228—
1233.
20. Березин Г. И., Киселев А. В., СиницынВ. А., ЖФХ,
1967, т. 41, с. 926—930.
12 Заказ 1180
177
21. БрунауэрС., Коупленд Л., КантроД. В кн.: Межфазо-
вая граница газ—твердое тело. Пер. с англ. ред. Э. Флад. М., «Мир»,
1970. 433 с.
22. Стил У. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ,
ред. Э. Флад. М., «Мир», 1970. 433 с.
23. X о н и г Дж. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ,
ред. Э. Флад. М., «Мир», 1970. 433 с.
24. Р о сс С. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ,
ред. Э. Флад. М. «Мир», 1970. 433 с.
25. К и с е л е в А. В. В кн.: Межфазовая граница газ — твердое тело. Пер.
с англ. ред. Э. Флад. М., «Мир», 1970. 433 с.
26. А р и с т о в Б. Г. и др. В кн.: Основные проблемы теории физической
адсорбции. М., «Наука», 1970. 475 с.
27. В е г е z i n G. I., К i s е 1 е v А. V. J. Col. Interface Sci., 1972, v. 38,
p 227______233.
28. Berezin G.I. e. a. J. Col. Interface Sci., 1972, v. 38, p. 335—340.
29. Б e p e з и н Г. И., Киселев А. В., Синицын В. А., ЖФХ,
1970, т. 44, с. 734—740.
30. В е г е z i n G. I., К i s e 1 e v A. V. J. Col. Interface Sci., 1974, v. 46,
p. 203—211.
31. Henry D. C. Phyl. Mag., 1922, v. 44, ser. 6, p. 689—705.
32. Sams J. R., Constabaris G., H a 1 s e у G. D. J. Phys. Chem.,
1962, v. 66, p. 2154—2158.
33. Constabaris G., Sams J. R., H a 1 s e у G. D. J. Chem. Phys.,
1962, v. 37, p. 915—915.
34. Bradley R. S., Phil. Mag., 1931, v. 11, ser. 7, p. 690—696.
35. Everett D. H. Trans. Faraday Soc., 1950, v. 46, p. 957—969.
36. В a r k e r J. A., E veret t D. H. Trans. Faraday Soc., 1962, v. 58,
1608—1623.
37. W i 1 k i n s T. J. Proc. Roy. Soc., 1938, A164, p. 496—509.
38. В а г r e r R. M., Gibbons R. M. Trans. Faraday Soc., 1963, v. 59,
p. 2875—2887.
39. Williams A. M. Proc. Roy. Soc., Edinburgh, 1918, v. 38, p. 23—39;
Proc. Roy. Soc., 1919, A96, p. 287—297.
40. Ав г у л ь Н. Н. и др. ЖФХ, 1971, т. 45, с. 442—444.
41. В arrer R. М., Lee J. A. Surface Sci., 1968, v. 12, р. 341—353.
42. В е z u s A. G. е. a. Trans. Faraday Soc., 1971, v. 67, p. 468—482.
43. AvgulN. N. e. a. J. Col. Interface Sci., 1973, v. 42, p. 486—495.
44. Kiselev A. V. Rev. Gen. Caoutchouc, 1964, v. 41, p. 377—393.
45. Puri B. Chemistry and Physics of Carbon. (Coll.). Ed. P. L. Walker,
New York, v. 6, 1970. 349 p.
46. Агз а м x о джаев А. А., Журавлев Л. T., Киселев А. В.
Изв. АН СССР. Сер. хим. 1968, с. 1186—1191.
47. Zhuravlev L. Т., Kiselev А. V. Surface Area Determination.
' (Coll.). London, 1970. 405 p.
48. Kiselev A. V. Disc. Faraday Soc., 1971, v. 52, p. 14—32.
49. Фарнсуорт Г. В кн.: Межфазовая граница газ — твердое тело.
Пер. с англ. ред. Э. Флад. М., «Мир», 1970. 433 с.
50. К и с е л е в А. В., Л ы г и н В. И. Инфракрасные спектры поверхност-
ных соединений и адсорбированных веществ. М., «Наука», 1972. 459 с.
51. SernaJ., В г u L. Surface Sci., 1968, v. 12, р. 369—384.
52. Даннинг В. В кн.: Межфазовая граница газ — твердое тело. Пер.
с англ. ред. Э. Флад. М., «Мир», 1970. 433 с.
53. Honig J. М. Ann. N.Y. Acad. Sci., 1954, v. 58, p. 741—797.
54. X они г Дж. В кн.: Межфазовая граница газ — твердое тело. Пер.
с англ. ред. Э. Флад. М., «Мир», 1970. 433 с.
55. Ross S., Oliever J. Р. J. Phys. Chem., 1961, v. 65, p. 608—615.
56. Ross S., Winkler W. J. Col. Sci., 1955, v. 10, p. 319-329,330-335.
57. Аристов Б. Г., Киселев А. В. Коллоидн. ж., 1967, т. 29, с. 631»»
637.
178
58. Mahajan О. P., Walker P. L. J. Col. Interface Sci., 1969, v. 31,
p. 79—84.
59. Безус А. Г., Древинг В. П., Киселев А. В. ЖФХ, 1967,
t. 41, c. 2921—2927.
60. H о о г у S. E., Prausnitz J. M. Trans. Faraday Soc., 1967, v. 63,
p. 455—460.
61. Авгуль H. H. и др. Изв. АН СССР, ОХН, 1962, с. 769—777.
62. Авгуль Н. Н., Киселев А. В., Л ы г и н а И. А. Изв. АН СССР.
ОХН, 1961, с. 2116—2125.
63. М а с h i n W. D., Ross S. Proc. Roy. Soc. (London), 1962, A265,
p. 455—462.
64, Радушкевич Л. T. В кн.: Основные проблемы теории физической
адсорбции. Под ред. М. М. Дубинина и В. В. Серпинского. М., «Наука»,
1970. 475 с.
65. De В oer J, Н., KruyerS. Koninkle Ned. Akad. Wetenschp. Proc.,
1952, 55B, p. 451—463.; 1955, 58B, p. 61—72.
66. Kalashnikova E. V. e. a. Chromatographia, 1971, v. 4, p. 495—500.
67. Aristov B. G., BosacekV., Kiselev A. V. Trans. Faraday
Soc., 1967, v. 63, p. 2057—2067.
68. R oss S., Whiltshirel. J. J. Phys. Chem., 1966, v. 70, p. 2107—
2111.
69. Kiselev A. V. Molecular Sieves Zeolites — II, Adv. Chem. Ser., 1971,
v. 102, p. 37—68.
70. J aycock M. J., W a 1 d s a x J. С. R. J. Col. Interface Sci., 1971,
v. 37, p. 462—468.
71. К и с e л e в А. В., Лопаткин А. А., Л о у p ь e Б. И. Теорети-
ческая и экспериментальная химия, 1974, т. 10, с. 254—258.
Глава V
ТЕРМОДИНАМИЧЕСКИЕ ХАРАКТЕРИСТИКИ АДСОРБЦИИ
РАЗЛИЧНЫХ ВЕЩЕСТВ НА ГРАФИТИРОВАННОЙ
ТЕРМИЧЕСКОЙ САЖЕ ПРИ МАЛОМ (НУЛЕВОМ) ЗАПОЛНЕНИИ
ПОВЕРХНОСТИ, ПОЛУЧЕННЫЕ ИЗ ЭКСПЕРИМЕНТАЛЬНЫХ
ДАННЫХ
1. Получение термодинамических характеристик адсорбции
из экспериментальных данных
Получение из экспериментальных данных по адсорбционному
равновесию термодинамических характеристик адсорбции для ряда
молекул близкого и разного состава и строения необходимо как
для практических применений, так и для развития молекулярной
теории адсорбции и межмолекулярных взаимодействий вообще.
Во-первых, термодинамические характеристики являются опорными
для определения соответствующих величин для экспериментально
не изученных веществ, что, в частности, помогает идентифицировать
неизвестные вещества в адсорбционной хроматографии. Во-вторых,
эти данные нужны для определения атом-атомных потенциальных
функций межмолекулярного взаимодействия и теоретического рас-
чета термодинамических характеристик адсорбции на основании
структуры молекулы адсорбата и строения адсорбента (см. гл. X).
Наконец, в-третьих, эти данные нужны для решения обратных задач,
т. е. при известных атом-атомных потенциальных функциях меж-
молекулярного взаимодействия экспериментальные термодинамиче-
ские характеристики адсорбции позволяют сделать заключение
о структуре молекулы адсорбата (подробнее об этом см., например,
разд. 4 гл. X). В этой главе рассмотрены полученные из эксперимен-
тальных данных термодинамические характеристики адсорбции на
графитированной термической саже при малом (нулевом) заполнении
поверхности. Основная литература по экспериментальному исследо-
ванию адсорбции на графитированных термических сажах была
указана в разд. 1 гл. II. Поэтому здесь даются ссылки лишь на те
работы, в которых были получены наиболее точные данные, исполь-
зованные для определения термодинамических характеристик адсорб-
ции при нулевом заполнении поверхности.
Из рассмотренных в гл. III термодинамических характеристик
адсорбции наиболее точные значения могут быть получены для нуле-
вого заполнения пока только для величин Klt AS* и AU\ (значения
ACi определены только для немногих систем). Эти значения пред-
ставляют термодинамический эффект проявления межмолекулярных
взаимодействий адсорбат — адсорбент в наиболее простом случае —
в отсутствие взаимодейст вий адсорбат — адсорбат.
180
С целью получения таких термодинамических характеристик
адсорбции из экспериментальных данных были собраны, системати-
зированы и обработаны результаты газохроматографических и ста-
тических исследований около 150 систем: различные адсорбаты —
графитированная термическая сажа. Значения констант Генри,
полученные непосредственно газохроматографическим методом при
работе с очень малыми дозами, дающими симметричные пики, были
частично уже приведены в гл. II, III и IV (см., например, рис. III,4,
табл. IV,2 и рис. IV. 13). Из статических измерений непосредственна
получить значения константы Генри нельзя. Нужна экстраполяция
полученных изотерм адсорбции к нулевому заполнению. Поэтому
результаты статических измерений для отдельных адсорбционных
систем обрабатывались с помощью рассмотренных в разд. 2 гл. IV
вириальных уравнений в экспоненциальной форме и некоторых
других моделей и отсюда определялись значения константы Генри
для адсорбционного равновесия. Оба эти метода определения кон-
стант Генри для адсорбции на графитированной термической сажа
дали близкие результаты. Это позволило определить из эксперимен-
тальных данных значения констант Генри с нужной для дальнейших
сопоставлений точностью. В отдельных случаях наиболее точных
статических измерений были определены также вторые вириальныа
коэффициенты термодинамических характеристик адсорбции. Зна-
чения Кг при разных температурах для ряда адсорбционных систем
собраны в табл. П,2, П,3 Приложения. Константы Генри при-
ведены в виде приближенного-уравнения, выражающего их зависи-
мость от температуры
। л 1 А^ А
g 1 Т 2,303 Л 2,3037? Т
(V,l>
в котором Кх выражена в мкм. Энтропийная константа В = (Д3£ +
~ 7?)/2,3032? и связанная с дифференциальным мольным изменением1
внутренней энергии константа А — —Д £7х/2,3037? приняты в этом
уравнении не зависящими от температуры.
В гл. IX—XI полученные для нулевого заполнения значения
термодинамических характеристик адсорбции использованы для
полуэмпирического определения потенциальных функций межмоле-
кулярного взаимодействия и для сопоставления с результатами моле-
кулярно-статистических расчетов.
В этой главе приведены некоторые сопоставления полученных
термодинамических характеристик адсорбции К19 Д5* или (Д5\* +
4- R)/R и —ДС/\ на графитированной термической саже для раз-
ных адсорбатов и рассмотрены некоторые зависимости этих ха-
рактеристик от строения молекул и некоторых физических свойств^
этих адсорбатов.
Чтобы получить независимо от образца графитированной тер-
мической сажи (при соблюдении одинаковых условий его подготовки
181
к опытам) значения константы Генри, надо их отнести к единице
поверхности адсорбента А. Удельная поверхность s графитирован-
ных термических саж определялась методом Брунауэра, Эмметта
и Теллера (БЭТ) [1] по адсорбции пара азота при температуре его
кипения. Однако этот метод является приближенным. Допущения,
лежащие в основе модели БЭТ, и связанные с ними погрешности
определения величины s описаны в примечаниях к сборнику [2].
Поэтому, чтобы снизить погрешности определения величины s при
применении метода БЭТ, надо правильно выбрать адсорбат и темпе-
ратуру, учитывая, что модель БЭТ дает наименьшую погрешность
лишь для случая сильного взаимодействия адсорбат — адсорбент
и относительно слабого взаимодействия адсорбат — адсорбат (вза-
имодействия адсорбат — адсорбат в модели БЭТ не учитываются
вообще). Остальные допущения модели БЭТ в случае графитиро-
ванной сажи или оправдываются (однородность поверхности), или
не оказывают существенного влияния на определение емкости моно-
ч^лоя (допущение о локализации адсорбированных молекул на поверх-
ности адсорбента в первом слое, упрощенное представление о по-
строении второго и последующих слоев).
При подходящем выборе адсорбента и температуры измерения
изотермы адсорбции метод БЭТ позволяет из экспериментальной
изотермы адсорбции найти емкость плотного монослоя (если
концентрация адсорбата в газовой фазе невелика, то ат nsm,
где пД — соответствующая величина гиббсовской адсорбции). Вели-
чина удельной поверхности равна
s = amNАат ~ n»mNАат (V.2)
где Лд — число Авогадро'; — площадь, приходящаяся на молекулу в плот-
ном монослое.
Зная s для данного адсорбента и определив из изотермы
адсорбции на том же адсорбенте для разных адсорбатов (адсорбция
которых происходит по механизму, приближающемуся к модели
БЭТ), по уравнению (V,2) можно определить величины аут для этих
адсорбатов. Величина сот для одного и того же адсорбата непостоянна
для разных адсорбентов благодаря различному строению их поверх-
ности, вызывающему разную плотность упаковки молекул в моно-
слое. Поэтому для определения s выбирают стандартное значение
для эталонного адсорбата, изотерма адсорбции которого близка
к соответствующей модели БЭТ. В этой книге значения s определены
из изотерм адсорбции азота или аргона, измеренных при темпера-
туре кипения жидкого азота (равной 78 К), при стандартных значе-
ниях wwN2 = 16,2A2 и соответственно a>mAr = 13,8 А2 [3—5].
В случае графитированных термических саж этому практически
эквивалентно удобное определение s из изотермы адсорбции бензола,
измеренной при комнатной температуре, при значении а>тсвнв =
= 40 А2 [6]. Это значение а>тсвнв было определено при использова-
нии приведенного выше стандартного значения (i)wn2- Кроме того,
182
значения для молекул азота, аргона и бензола были определены
из ван-дер-ваальсовых размеров молекул при плотной упаковка
и плоской (бензол) ориентации на поверхности (см. табл. П, 1
Приложения). Для молекул азота эта величина gjwn2 = 16,2 А2
близка к полученной из определения удельной поверхности анатаза
независимым методом [7].
Величины были определены методом БЭТ для многих веществ
при адсорбции на графитированных сажах. Эти величины вмести
с величинами, определенными другими независимыми от адсорбци-
онных измерений методами (из ван-дер-ваальсовых размеров моле-
кул и из плотности жидкости при плоском расположении молекул
на поверхности) также приведены в табл. П, 1 Приложения.
Для быстрых определений s, особенно малых по величине, ши-
роко используются также изотермы адсорбции пара криптона,,
определенные при температуре кипения жидкого азота [8]. При этой
температуре давление пара криптона мало, так что можно применить
метод, разработанный Вутеном и Броуном [9], позволяющий опре-
делять весьма малые величины адсорбции, а следовательно, и весьма
малые величины s, благодаря использованию в качестве газовой
бюретки капилляра манометра Мак-Леода [8].
Независящее от допущений модели БЭТ определение удельной
поверхности графитированной термической сажи молекулярно-ста-
тистическим методом рассмотрено в разд. 6 гл. IX.
Для единицы поверхности графитированной термической сажи
были определены следующие термодинамические характеристики
адсорбции при нулевом заполнении: константы Генри К1} дифферен-
циальные изменения энтропии адсорбции AS* и внутренней энергии
адсорбции AZ71. Эти величины получены для следующих рядов
адсорбатов: благородных газов, некоторых неорганических газовг
гомологического ряда w-алканов, ряда изоалканов и цикланов,
некоторых w-алкенов и цикленов, алкадиенов и цикладиенов, к-ал-
кинов, бензола, мопоалкилбепзолов и полиметилбензолов, полифе-
нилов и ароматических углеводородов с конденсированными ядрами
н различных производных углеводородов, а именно галогенпроизвод-
ных, простых эфиров, w-спиртов, терпенов, дейтерированных углево-
дородов и их кислородсодержащих производных, азот- и серусодер-
жащих органических соединений и некоторых элементорганических
соединений. Системы, для которых уже сделаны молекулярно-ста-
тистические расчеты термодинамических характеристик адсорбции
при нулевом заполнении, подробно рассмотрены в гл. IX—XL
В этой главе эти системы рассмотрены лишь вкратце.
2. Неорганические газы
Термодинамические характеристики адсорбции благородных га-
зов при малых (нулевых) заполнениях были приведены в табл. 111,2.
п даны также в табл. П, 2 Приложения. Константа Геири Кг и
величина дифференциального изменения внутренней энергии адсорб-
18$
щии — AC/i благородных газов увеличиваются с ростом молекуляр-
ного веса и поляризуемости молекул, а изменение энтропии адсорб-
дии остается практически постоянным (подробнее см. гл. IX).
Была исследована также адсорбция азота, кислорода, окислов
азота, окиси углерода, двуокиси углерода и шестифтористой серы [4,
'6, 10, 12, 13] (см. табл. III,2 и табл. П, 2 Приложения).
На графитированной термической саже и других непористых
адсорбентах с небольшой удельной поверхностью при обычных
температурах газохроматографической колонны газы адсорбируются
ч;лабо, поэтому величины удерживаемых объемов в колонне VR в этом
случае недостаточны для четкого разделения компонентов газовой
Рис. V,l. Зависимость —Д£7г для адсорбции
неорганических газов при — 100°С на графи-
тированной термической саже от поляризуе-
мости молекул этих адсорбатов а.
смеси. Увеличить значения VR можно двумя путями. Во-первых,
можно увеличить удельную поверхность и сузить поры адсорбента.
В соответствии с этим газохроматографическое разделение легких
газов обычно ведется на тонкопористых адсорбентах с большой
удельной поверхностью и более высокой по сравнению с графитиро-
ванной сажей величиной —Во-вторых, увеличить значения VR
и Кг можно, понижая температуру [см. уравнение (V.1)]. Это позво-
ляет осуществлять хроматографическое разделение газов на непо-
ристых адсорбентах с небольшой удельной поверхностью. Киселев,
Худяков и Яшин использовали этот путь для разделения ряда газов
*на графитированной термической саже [10]. Газохроматографическая
колонна помещалась в криостат-хроматограф Цвет-И, модель 8 [11].
Из рис. V,1 видно, что величины —растут приблизительно
линейно с ростом поляризуемости молекул ряда газов а:
—= 1,0+4,6а кДж/моль (V,3)
В основном эти газы по мере роста их молекулярного веса лучше
удерживаются на графитированной термической саже. Однако для
труппы газов Аг, О2 и N2 порядок удерживания обратный (см.
рис. 11,12). Изменение порядка удерживания связано с наличием
л-связей в молекуле азота, электронная структура которой подобна
электронной структуре молекулы ацетилена (см. разд. 4 гл. X).
Наличие л-связей немного увеличивает межмолекулярное взаимо-
действие атома азота молекулы N2 с атомом углерода графита по-
184
добно увеличению межмолекулярного взаимодействия образующего*
л-связи атома углерода ацетилена с атомом углерода графита.
В значительно меньшей степени это относиться и к адсорбции О2 на
графитированной термической саже. При адсорбции же на углеводо-
родных адсорбентах, в которых атомы углерода связаны в основном
о-связями, эти три газа удерживаются в порядке роста М (см.
рис. 11,12).
Изменение энтропии при адсорбции этих газов практически оди-
наково (см. табл. III,2).
3. «-Алканы
Экспериментальные термодинамические характеристики адсорб-
ции «-алканов при разных температурах приведены на рис. Х,1—
Х,4 и в табл. Х,3, Х,8, Х,12, а также в табл. П. 3 Приложения.
Изменение In К± при 100°С и — \U1 с ростом числа атомов
углерода п в молекулах «-алканов показано на рис. V,2. Величины
Рис. V,2. Зависимость In Кг при 100 °C (а) и —Ai71 (б) для адсорбции w-алканов
на графитированной термической саже от числа атомов углерода в их молекулах.
In Кг и —MJ! для гомологического ряда «-алканов представляют
линейные функции числа атомов углерода в молекуле п. При отно-
сительно невысоких температурах эти молекулы, способные к вну-
треннему вращению, адсорбируются наиболее выгодно, т. е. преиму-
щественно в их транс-конформациях (см. гл. X). По мере удлинения
молекулы в гомологическом ряду «-алканов каждое новое добавля-
ющееся звено СН2 в этих молекулах вносит такой же вклад в вели-
чины In К± и —как и предыдущие звенья СН2, т. е. величины
In Кг и —аддитивны по звеньям СН2 [14—16]. Так, для «-алканов
C-i—Се __
— ДС71= 6,5 + 5,4п кДж/моль (V,4>
Следует, однако, иметь в виду, что -средняя температура газо-
хроматографических колонн (для интервала температур, при кото-
ром производилось определение величин —&UL для каждого угле-
водорода) с увеличением п от 1 до 6 увеличивается. В работе [15]
при увеличении п от 1 до 6 средняя температура колонны увеличи-
валась от 145 К до 400 К (в частности, измерения адсорбции метана
185*
проводились в интервале от 120 до 170 К, а для «-гексана — в интер-
вале 373—423 К). Так как —AC7j с ростом температуры несколько
уменьшается [17], представляет интерес сопоставление величин
—AC/i этого ряда углеводородов для одинаковой температуры.
Расчеты значений — A CZ1 для «-алканов Сг—Се при разных
Т проведены в разд. 2 гл. X. Здесь мы ограничимся сопоставлением
экспериментальных величин —&U19 приведенных к температуре
293 К. Экспериментальные данные по средней мольной теплоемкости
в адсорбированном состоянии на графитированной термической
-саже имеются лишь для бензола и «-гексана [17]. Уменьшение —-А[7г
вблизи комнатной температуры составляет в этих случаях около
Ю,1% на 1 К. Если принять такое уменьшение —At/j для всех рас-
сматриваемых углеводородов, то получается следующая приближен-
ная зависимость при 293 К:
— Д£7х^4,5+5,8п кДж/моль (V,4a)
Для адсорбции «-алканов на графитированной термической
саже наблюдается также линейная зависимость —At/j от средней
поляризуемости а адсорбирующихся молекул. Если а выражена
в А3, то
—MJ1 = 3,9 + 3,0а кДж/моль (V,5)
4. Изоалканы
Экспериментальные термодинамические характеристики адсорб-
ции изоалканов при разных температурах приведены на рис. X, 9,
в табл. Х,13 и в табл. П, 3 Приложения. По мере разветвления
молекул осуществляется контакт меньшего числа ее звеньев с пло-
ской поверхностью графитированной сажи, и величины К± и —/^U1
уменьшаются»
5. Цикланы
Экспериментальные термодинамические характеристики адсорб-
ции цикланов при разных температурах приведены на рис. Х,10,
в табл. Х,14 и в табл. П, 3 Приложения. При переходе от «-алканов
к цикланам с тем же числом атомов углерода в молекуле [15] про-
исходит уменьшение К± и |—АСТх в связи с тем, что, во-первых,
молекулы цикланов содержат меньшее число атомов водорода и,
во-вторых, они имеют почти всегда неплоское строение, так что одна
или несколько групп СН2 или других групп в случае замещенных
цикланов даже при энергетически наиболее выгодной ориентации
на базисной грани графита остаются приподнятыми над плоской
поверхностью адсорбента, и лишь плоская молекула циклопропана
ед может быть расположена параллельно поверхности базисной
грани графита. Число атомов водорода в молекуле циклопропана
486
на два меньше, чем в молекуле пропана, однако величина I—
циклопропана меньше величины —AUr пропана не настолько,
насколько следовало бы ожидать из-за уменьшения числа атомов
водорода в молекуле при переходе от пропана к циклопропану.
Это является следствием усиления межмолекулярного взаимодействия
циклопропана с графитом из-за значительных напряжений в трех-
членном цикле (см. разд. 3 гл. X).
Мостиковые бициклические углеводороды составляют основу
многих бициклических терпенов. Изучение адсорбционных свойств
этих соединений, в частности, замещенных бицикло [2,2,1] гептана
с одним или несколькими заместителями, осложняется большим
числом пространственных и структурных изомеров, разделении
и идентификация которых очень трудна.
Конфигурация молекулы бицикло [2,2,1 ]гептана (норборнана}
является единственно возможной (см. табл. V,l). В структуре нор-
борнана имеются как элементы шестичленного кольца, так и пяти-
членных колец. Циклопентановые кольца в норборнане имеют
гипертрофированную конформацию типа конверта с сильными иска-
жениями валентного угла между связями 1—7 и 4—7 у мостикового
углерода С7 (94°) [18]. Все это вызывает в молекуле норборнана
довольно сильное напряжение. Изомер триметиленнорборнана —
адамантан имеет очень высокую температуру плавления и его моле-
кула обладает уникально жестким, но свободным от напряжения
циклическим скелетом из атомов углерода. Она построена из трех
сконденсированных креслообразных циклогексановых колец (см.
табл. V,l). Длины всех связей С—С составляют 1,54 А, а все углы
равны 109,5°.
Термодинамические характеристики адсорбции некоторых про-
изводных норборнана и адамантана приведены в табл. V,1 и в табл.
П, 3 Приложения [19]. Увеличение значений К± и %ля изо-
меров определяется в основном характером наиболее выгодного
расположения молекул различного геометрического строения на
плоской поверхности графитированной термической сажи. Так,,
введение одного заместителя в положение 1 при адсорбции на пло-
скости оказывается менее энергетически выгодным, чем введение
заместителя в положение 2.
Для диметилбицикло [2,2,Цгептанов характерны более низкие
значения константы Генри при адсорбции молекул с двумя метиль-
ными группами у одного углеродного атома по сравнению с моле-
кулами, содержащими заместители у двух разных атомов углерода.
Минимальное значение К± получено для 7,7-диметилбицикло [2,2,1J
гептана. Все моно- и дизамещенные производные бицикло [2,2,1] геп-
тана дают меньшие Кг и —у эндо-изомеров, чем у экзо-изомеров.
Различие этих величин позволяет с успехом разделять эндо- и экзо-
изомеры углеводородов ряда бицикло [2,2,1] гептана на графитиро-
ванной термической саже [19]. Кроме того, газохроматографические1
исследования таких углеводородов можно использовать не только
187
Таблица V,7. Величины —Д^1 и структурные формулы
бицикло [2,2,1]гептана и его производных
Соединение Структурная формула -AUl, кДж/моль
-анс?о-5-Метилбицикло[2,2,1]гептен-2 32,6 '
акао-5-Метилбицикло [2,2,1]гептен-2 34,7
4-Метилбицикло[2,2,1]гептан 32,6
-внс?о-2-Метилбицикло[2,2, 1 ]гептан 32,6
лкао-2-Мети лбицик л о [ 2,2,1J гептан 35,2
2-Мети леибицик ло [2,2,1 ]гептан 33,9
7,7-Диметил бицикл о [2,2,1 ] гептан 35,6
-андо-5-Винп л бицик л о[ 2,2,1 ] гептен-2 35,6
лкао-5-Винилбицик ло [2,2,1 ]гептен-2 38,5
лндоА ,2-Диметилбицик ло [2,2,1] гептан 36,8
якзот i, 2-Диметилбицикл о [ 2,2,1] гептан 39,4
лгра«с-5-Этилиденбицикло[2,2,Цгептен-2 37,7
^ис-5-Этилиденбицикло[2,2,1]гептен-2 39,8
4 ,4-Диметилбицикл о [2,2,1 ]гепт ан 38,5
гид о-эндо-2,3-Диметилбицикл о [2,2,1] гептан 38,9
jn ра нс-2,3-Диметилбицикл о [2,2,1 ] гептан 38,9
^као-акао-2,3-Диметилбицикло[2,2,1]гептан 41,0
.акзо-2,2,3-Т римети лбицик л о [2,2,1] гептан 38,9
-вндо-2,2,3-Триметилбицикло[2,2,1]гептан 41,0
2,2-Диметил-3-метиленбицикл о [ 2,2,1 ]гептан 40,2
андо-2-Этилбицикло[2,2,1]гептан 39,8
^«зо-2-Этил бицикл о [2,2,1] гептан 41,4
Адамантан 39,8
для аналитических целей, но и для установления структуры того
или иного изомера по порядку выхода из колонны с графитирован-
ной термической сажей и для определения соотношения эндо- и экзо-
изомеров в исследуемых смесях.
6. Алкены, алкадиены, циклены и цикло диены
Экспериментальные термодинамические характеристики адсорб-
ции алкенов и алкадиенов Ci—Св при разных температурах приве-
дены на рис. Х,11 и Х,13, а также в табл. Х,15, Х,17 и табл. П,3
Приложения.
При переходе от ряда н-алканов к ряду н-алкенов-1 происходит
уменьшение величин Кг и — [15, 16] вследствие того, что влия-
ние уменьшения числа атомов водорода в молекуле превалирует над
влиянием увеличения взаимодействия с графитом атомов углерода
молекулы, находящихся у двойной связи (подробнее см. разд. 4
гл. X).
Как и при адсорбции н-алканов, для н-алкенов-1 наблюдается
линейная зависимость от числа атомов углерода в молекуле п
и от средней поляризуемости молекулы а. Для н-алкенов-1 С2—С5
в интервале температур хроматографической колонны с графитиро-
ванной термической сажей от 298 до 373 К были найдены выраже-
ния [15]
—AL/j — 5,7 +5,Зп кДж/моль (V,6)
И
—AtZj—3,5 + 2,9с% кДж/моль (У,7)
Зависимость 1g Кг при 373 К от п для ряда алкенов-1 С2—С6
выражается уравнением, в котором значения Кг выражены в мкм:
1g К i00°c = -3,16 + 0,615л (V,8)
Для алкенов-1 С6—С10 в интервале температур от 373 до 473 К
было получено уравнение [16]:
—ДL71 = 5,6+5,2n кДж/моль (V,9)
Перемещение двойной связи в алкенах из положения 1 в поло-
жение 2 вызывает увеличение Кг и соответственно —^.U1 (рис. V,3),
причем для транс-изомера в большей степени, чем для цис-изомера.
Транс-изомеры олефинов располагаются на поверхности графити-
рованной термической сажи энергетически более выгодно по сравне-
нию с цис-изомерами. Это позволяет использовать графитированную
термическую сажу для их разделения [15, 16]. Для пространствен-
ных изомеров бутена-2 получены значения Ки превышающие значе-
ния Кг для к-бутана при той же температуре (причины этого рас-
смотрены в разд. 4 гл. X).
Для алкенов же Cft—C10 (см. рис. V,3) величины Кг и —AC/j
остаются более низкими, чем при адсорбции н-алканов, по-види-
мому, вследствие того, что атомы углерода у двойной связи в этих
189
случаях не могут приблизиться к атомам углерода графита на столь
короткие расстояния, как при адсорбции углеводородов с меньшим
количеством углеводородных звеньев. Значения Кг и —ACZj увели-
чиваются при перемещении двойной связи от центра к концу моле-
кулы как для цис-, так и для
Рис. V,3. Зависимость 1g при
адсорбции углеводородов С6 — С10
на графитированной термической
саже при 175 9С от числа атомов
углерода в их молекулах вы-
ражена в мкм):
1 — н-алканы; 2 — н-алкены-1; 3 —
tjuc-2-н-алкены; 4 — транс-2-ъл¥&кы:,
5— t^uc-3-алкены; 6 — транс-З-алкены;
7 — t^uc-4-алкены; 8 — тпранс-4-алкены.
транс-изомеров в следующем порядке:
3-алкен <с 2-алкен <С 4- и 5-алке-
ны [16].
При появлении в молекуле алке-
на второй двойной связи, не сопря-
женной с первой, наблюдается даль-
нейшее уменьшение Кг и —AC/j [15,
16]. При этом разница в величи-
нах \U1 алкена-1 и диенового угле-
водорода близка к разнице между
величинами ^U1 соответствующего
н-алкана и алкена-1.
При появлении в молекуле алке-
на второй двойной связи, сопряжен-
ной с первой, величины Кх и —At/x
превышают соответствующие вели-
чины не только для алкенов-1, но
даже и для н-алканов [15, 16].
Причиной этого является, по-види-
мому, не столько влияние сопряже-
ния (см. разд. 4 гл. X), сколько
влияние уменьшения поперечных
размеров молекул диеновых углево-
дородов с сопряженными двойными
связями по сравнению с молекулами
н-алканов. Благодаря этому атомы
углерода в диеновых углеводородах,
образующие сопряженные связи,
вероятно, могут подходить к базис-
ной грани графита на более близ-
кое расстояние.
На графитированной термической саже исследовалась также
адсорбция нескольких шестичленных цикленов (табл. V,2), моле-
кулы которых различаются пространственной структурой, числом
атомов водорода, числом двойных несопряженных и сопряженных
связей [15].
При переходе от циклогексена к циклогексадиену-1,3 и далее
к бензолу наблюдается рост Кг и —AC71S несмотря на уменьшение тем-
ператур кипения и молекулярных весов. Этому способствует как
переход от неплоской молекулы циклогексена к плоской молекуле
бензола, которая может контактировать с плоской поверхностью
сажи всеми шестью атомами углерода, так и нарастающее в этом
190
Таблица V,2. Сопоставление термодинамических характеристик
адсорбции шестичленных цикленов на графитированной термической саже
с соответствующими характеристиками для циклогексана и бензола (373 К)
Адсорбат Ki, мкм - (Д8*+й)/2,303й -ДП1, кДж/моль
Циклогексан 0,43 4,0155 .28,6
Циклогексен 1,38 4,5075 33,3
Циклогексадиен-1,3 1,48 4,580 34,0
Бензол 2,37 4,690 36,2
ряду число участвующих в образовании л-связей атомов углерода
молекулы, контактирующих с плоской поверхностью графитирован-
ной термической сажи.
Молекулярно-статистический расчет приведенных в табл. V,2
величин дан в гл. X.
В случае адсорбции бицикло [2,2,1] гептенов и замещенных би-
цикло [2,2,1]гептенов, имеющих заместители с двойной связью
(см. табл. V, 1 и табл. П, 3 Приложения), величины Кг и — At/j
так же, как и для предельных бицикло [2,2,1 ]гептанов, определяются
различиями в возможных наиболее выгодных расположениях моле-
кул на поверхности (при наибольшем числе контактирующих с ней
звеньев молекулы). При замене метильной группы на метиленовую
в положении 2, т. е. при уменьшении числа атомов водорода в про-
изводных бицикло [2,2,1]гептана, величины Кг и —умень-
шаются. У непредельных производных бицикло [2,2,1]гептана так
же, как и у предельных, в случае адсорбции эндо-изомеров моно- и
дизамещенных производных наблюдаются меньшие величины Кх
и —Д?71, чем при адсорбции экзо-изомеров.
Газоадсорбционная хроматография на графитированной терми-
ческой саже позволяет разделить и идентифицировать стереоизомеры,
получающиеся, например, при термическом разложении при 500 °C
циклододекатриенов-1,5,9 [20]. В этом случае в основном полу-
чаются 4 возможных стереоизомера 1,2,4-тривинилциклогексана
12 3 4
1г, 2f, 4/ 1л 2/\ 4с 1л 2с, 4* 1г, 2с, 4с
Выделены стереоизомеры 1 и 2. Структурные же конфигурации
стереоизомеров 3 и 4 были установлены при помощи газоадсорбцион-
ной хроматографии на графитированной термической саже [20].
Найдена следующая последовательность величин Кг исследуемых
изомеров при адсорбции на плоской поверхности графитированной
191
термической сажи: 2 < 4 < 3 < 1. Введение двойной связи в конец
углеродной цепи молекулы приводит к уменьшению времени удер-
живания.
7. Алкины
Экспериментальные термодинамические характеристики адсорб-
ции алкинов при разных температурах приведены на рис. Х,12,
в табл. Х,16 и П, 3 Приложения. Появление в молекуле углеводо-
рода тройной связи вызывает еще большее (по сравнению с влия-
нием двойной связи) уменьшение Кг и —ДЕ7\ [15]. Здесь превалирует
влияние уменьшения числа атомов водорода в молекуле алкина,
так как изменение валентного состояния атома углерода, образу-
ющего тройную связь, увеличивает энергию межмолекулярного
взаимодействия этого атома углерода с атомами углерода графита
(см. разд. 4 гл. X). Зависимости величины —AC/j от п и а для 1-н-ал-
кинов С2—Се выражаются уравнениями [15]:
—— 5,4+ 5,On кДж/моль (V,10)
—AZ7x = 3,6+3,0а кДж/моль (V,ll)
8. Бензол и алкилбензолы
При переходе от н-гексана к бензолу величины поляризуемости
молекулы а и величины Кх и —ДС7х при адсорбции на графитирован-
ной термической саже уменьшаются (рис. V,4). Здесь превалирует
влияние резкого уменьшения числа атомов водорода в молекуле
бензола по сравнению с молекулой гексана.
Было рассмотрено два ряда производных бензола: моно-н-алкил-
бензолы и полиметилбензолы [21]. Дифференциальное изменение
внутренней энергии адсорбции — \иг толуола превышает —ДСТд
бензола на 8,2 кДж/моль (см. рис. V,4 и табл. П, 3 Приложения)..
Далее при переходе от толуола к этилбензолу —ДСТ^ возрастает
всего лишь на 3,5 кДж/моль, в то время как переход от толуола
к ксилолам вызывает значительно больший рост величины ——
на 9,4 кДж/моль. Это показывает, что замещение в «кольцо» не равно-
ценно удлинению боковой цепи при адсорбции этих изомеров.
Из рис. V,4 видно, что дифференциальное изменение внутренней
энергии —ДС7 при адсорбции и всех остальных полиметилбензолов
значительно превосходит —ДЕТ* изомерных им моно-н-алкилбензолов.
Из рис. V,4 также видно, что в случае моно-н-алкилбензолов
наблюдается линейная зависимость —ДПх от числа атомов угле-
рода в молекуле, причем линия для моно-н-алкилбензолов парал-
лельна линии для н-алканов.
Для полиметилбензолов С7—С12 значения —&U\ расположены
вблизи линии, имеющей значительно больший наклон (см. рис. V,4).
Разница величин —ДС7х у о-ксилола и этилбензола составляет
192
5,9 кДж/моль, а у пентаметилбензола и н-ам илбензола она состав-
ляет уже 19,3 кДж/моль. Причиной этого может быть влияние
различия равновесных расстояний между звеньями соответствую-
щих молекул и плоской поверхностью графитированной термиче-
ской сажи и разница в распределении электронной плотности в этих
молекулах. Нежесткие молекулы моно-н-алкилбензолов, начиная
с этилбензола, имеют вращательные изомеры и поэтому не могут
—AUi, ккал/моль
Рис. V,4. Зависимость — Д/7г при ад-
сорбции «-алканов (7), бензола (2),
полиметилбензолов (5) и «-алкилбен-
золов (4) на графитированной термиче-
ской саже от числа атомов углерода в их
молекулах.
Рио. V,5. Зависимость величин
Д5|/2,303Л от — ДС?! для «-алка-
нов» н-алкилбензолов и полиметил-
бензолов.
касаться плоскости всеми группами СН2 и СН3. Кроме того, это
может несколько приподнимать и бензольное кольцо. В отличие от
этого вращение метильных групп в молекулах полиметилбензолов
может происходить так, что при наиболее выгодном их повороте
относительно кольца расстояния атомов углерода от плоской поверх-
ности адсорбента увеличиваться не будут. Кроме этого, введение
заместителей в кольцо изменяет распределение электронной плот-
ности около остова кольца. Ответ на вопрос о превалирующем зна-
чении того или иного из этих возможных влияний должен дать
молекулярно-статистический расчет. Хотя изменение энтропии при
адсорбции ряда углеводородов — Д5| зависит от изменения внутрен-
ней энергии адсорбции — \Ur приблизительно линейно и мало
13 заказ 1180
193
отличается для разных адсорбатов, в случае адсорбции полиметил-
бензолов — A5J заметно больше, чем в случае н-алканов и н-алкил-
бензолов (рис. V,5).
9. Полифенилы
Молекулы дифенила в его кристаллах являются плоскими.
В газообразном состоянии молекулы дифенила могут образовывать
конформеры благодаря вращению вокруг связи между ароматиче-
скими кольцами. Исследование газообразного дифенила методом
дифракции электронов показало, что существует наиболее выгод-
ная конформация, когда угол между плоскостями колец составляет
41,6° [22].
Термодинамические характеристики адсорбции дифенила при-
ведены в табл. Х,18 и в табл. П, 3 Приложения. Величина — AC/j
для адсорбции дифенила составляет 70,2 кДж/моль [23]. Она на
4,2 кДж/моль меньше удвоенного значения —AC/j для бензола при
той же температуре. Это указывает на сохранение некоторого угла
между кольцами в адсорбированных молекулах дифенила при адсор-
бции на плоской поверхности графитированной термической сажи.
Молекулярно-статистический расчет этого угла приведен в разд. 4
гл. X.
Для терфенилов величины Kt имеют следующую последователь-
ность: орто-< мета-<С пара-, что соответствует уменьшающейся
степени отклонения от копланарности трех циклов в молекулах
этих углеводородов [24, 25].
10. Ароматические углеводороды
с конденсированными ядрами
Экспериментальные термодинамические характеристики адсорб-
ции нафталина, антрацена и фенантрена на графитированной терми-
ческой саже при разных температурах приведены на рис. Х,14
и в табл. Х,18 и П,3 Приложения. Для антрацена и фенантрена
значения — практически одинаковы при заметной разнице
значений Кг. Отношение Klt антрацен/Х1, фенантрен = I,23 [21].
Энтропийные характеристики адсорбции этих молекул различаются.
11. Галогенпроизводные углеводородов
Термодинамические характеристики адсорбции на графитиро-
ванной термической саже первичных моногалогенпроизводных угле-
водородов приведены в табл. П,3 Приложения [26].
Для адсорбции всех первичных моногалоген-н-алкилов незави-
симо от замещающего галогена величины Кх и —А?7\ больше Кг
и —\U1 для адсорбции соответствующих н-алканов. На рис. V,6
представлены зависимости —AC/j для F-, CI-, Вг- и 1-н-алкилов
от числа атомов углерода в молекуле п. Для сравнения представлены
данные для н-алканов. Для первичных моногалогенпроизводных
194
н-алкилов зависимости —A?7i от п = 1 до п = 6 линейны, причем
эти линии и соответствующая линия для н-алканов приблизительно
параллельны. Таким образом, имеется постоянная разница в зна-
чениях дифференциального изменения внутренней энергии адсорб-
ции Д (—Д£/\) между первичными моногалогенпроизводными н-алки-
лов и н-алканами. Величина Д (—ДС/i) составляет для фторпроиз-
водных 2,0 кДж/моль, для хлорпроизводных 6,0 кДж/моль, для бром-
производных 8,5 кДж/моль и для иодпроизводных 10,5 кДж/моль.
Рис. V,6. Зависимость —при ад-
сорбции н-алканов (1), F-н-алкилов (2),
С1-н-алкилов (3), Вг-н-алкилов (4), 1-н-
алкилов (5) на графитированной тер-
мической саже от числа атомов углеро-
да в молекулах этих веществ.
Зависимости —первичных
моногалогенпроизводных н-ал-
килов от средней поляризу-
емости их молекул а также
линейны [26].
0,9 1,0 V 1,2 1,3 Д
Рис. V.7. Зависимость — при
адсорбции на графитированной тер-
мической саже первичных моногало-
геналкилов и моногалогенбензолов
от величины ковалентного радиуса
галогена г:
1 — галогенметилы; 2 — галогенэтилы:
3 — галогенпропилы; 4 — галогенбензолы.
Из рис. V,7 видна линейная зависимость —ДСТ^ первичных
моногалогеналкилов от ковалентного радиуса галогена г [26, 27].
Для фторпроизводных н-алкилов, имеющих наименьший радиус
атома галогена, величины Кх и —ДС^ являются наименьшими по
сравнению с соответствующими величинами для хлор-, бром- и иод-
производных. По-видимому, влияние увеличения радиуса гало-
гена перекрывается влиянием роста его поляризуемости, что увели-
чивает энергию дисперсионного взаимодействия. Перемещение
галогена по нормальной углеродной цепи из положения 1 в положе-
ние 2 при адсорбции вторичных моногалогензамещенных н-алкилов
С3—С6 [27] вызывает уменьшение соответствующих значений Кх
и — Для —ДС71 оно составляет около 2,0 кДж/моль (см. табл,
и П,3 Приложения). Для наиболее подробно изученных вторичных
13*
195
бромистых н-алкилов — AUt также линейно зависит от числа атомов
углерода в молекуле п [27]. Перемещение галогена из положения 1
в положение 2 из-за большого радиуса галогена отодвигает ближай-
шие к атому галогена атомы углерода цепи от поверхности, что умень-
шает энергию дисперсионного взаимодействия при наиболее выгод-
ной ориентации этих молекул.
В случае адсорбции моногалогенпроизводных бензола на графи-
тированной термической саже наблюдается увеличение Кг и —ACZj
при переходе от бензола к его галогенпроизводным для всех
галогенов [26] (см. табл. П,3 Приложения). Как и в случае
первичных моногалогенпроизводных алкилов здесь наблюдается
линейная зависимость теплот адсорбции моногалогенпроизводных
бензола от ковалентного радиуса галогена (см.рис. V,7).
Изучалась также адсорбция CI-, Вг- и 1-полигалогенметанов,
а также адсорбция GI- и Вг-полигалогенэтанов [26] (см. табл. П,3
Приложения). Замена одного атома водорода в молекулах этана
на хлор, бром и иод вызывает рост Кг и —А?/!. Для галогенметиле-
нов, содержащих два атома галогена в молекуле, вклады в величину
—AUt приходящегося на каждый из этих двух атомов галогена,
представляют удвоенные значения вкладов в — AL/\ от соответству-
ющего атома галогена в молекулах моногалогеналкилов (при пере-
ходе к ним от н-алканов). Увеличение —At7i при переходе от метана
к хлороформу и бромоформу составляет 17,1 и 24,8 кДж/моль соот-
ветственно, т. е. приблизительно в три раза больше увеличения
—&иг при переходе от н-алканов к моногалогенпроизводным н-ал-
килам. Это показывает, что молекулы СНС13 и СНВг3 адсорбируются
на поверхности графитированной термической сажи преимуще-
ственно всеми тремя атомами галогена. При переходе же от СНС13
к СС14 значение возрастает всего лишь на 1,2 кДж/моль.
Это связано с тем, что молекула СС14 может касаться поверхности
адсорбента только тремя атомами хлора, а четвертый атом хлора
настолько сильно приподнят над поверхностью, что вносит значи-
тельно меньший вклад в энергию взаимодействия с графитированной
термической сажей по сравнению с вкладом каждого из первых трех
атомов хлора.
В случае адсорбции дигалогенпроизводных этана вклад обоих
галогенов в дифференциальное изменение внутренней энергии адсорб-
ции приблизительно равен удвоенному вкладу галогена первичных
моногалогенпроизводных. Значения Кг и —AZ71 при адсорбции
1,1-С2Н4С12 и 1,2-С2Н4С12 практически равны [27]. В противополож-
ность молекулам полигалогенпроизводных этана, содержащим 2
атома галогена, для молекул полигалогенпроизводных этана, содер-
жащих 3 и более атомов галогена, более симметричные молекулы
адсорбируются сильнее, чем менее симметричные. Например, Кх и
—-А/?! для 1,1,2,2-тетрахлорэтана больше, чем для 1,1,1,2-тетрахлор-
этана. Один из атомов галогена в группе —СС13 не может касаться
196
поверхности одновременно с двумя другими, что снижает вклад
этого атома в Кг и для 1,1,1,2-тетрахлорэтана.
При переходе к перфторпроизводным углеводородам от нефтори-
рованных или не полностью фторированных соединений наблюдается
изменение последовательности величин Кг и —&Ur для фторпроиз-
водных н-алканов по сравнению с н-алканами и для фторпроизвод-
ных бензола по сравнению с бензолом. Для изученных перфтор-н-ал-
канов величины KL и меньше, чем для соответствующих
н-алканов (—меньше на 4 кДж/моль) [28, 29]. В этом случае
влияние увеличения среднего расстояния атомов углерода моле-
кулы от поверхности за счет большего радиуса фтора, чем водорода,
Рис. V,8. Зависимость — &U
от Г для адсорбции хлорд-
прена на графитированной
термической саже (из стати-
ческих измерении в инте-
рвале температур от 25
до 80 ?С).
Точка, обозначенная крестом,
получена из хроматографиче-
ских измерений. Вертикальным
пунктиром отмечена величина
0 == 1.
мкмоль/м2
перекрывает влияние большей поляризуемости атома фтора по
сравнению с атомом водорода. Переход же от бензола к перфтор-
бензолу вызывает увеличение Кг и —ДС/р В этом случае замена
атомов водорода на атомы фтора не вызывает удаления атомов угле-
рода плоской молекулы бензола от плоской поверхности адсорбента.
Наряду с этими влияниями различий в геометрии молекул и в от-
меченных свойствах атомов водорода и галогенов в молекулах угле-
водородов и их галогенпроизводных возможно также влияние раз-
личий в распределении электронной плотности в этих молекулах,
хотя последнее при адсорбции на графитированной термической
саже не может быть значительным.
Результаты экспериментального исследования адсорбции моно- и
полигалогенпроизводных углеводородов, помимо значения для иден-
тифицирования в аналитической газовой хроматографии, необхо-
димы для дальнейшего развития молекулярно-статистической теории
адсорбции и межмолекулярных взаимодействий. Эти результаты
можно использовать для полуэмпирического определения модели
атом-атомной потенциальной функции межмолекулярного взаимо-
действия атом галогейа (в соответствующих производных углеводо-
родов) — атом углерода в графите. Такое определение потенциаль-
ных функций межмолекулярного взаимодействия атом углерода
углеводорода — атом углерода графита и атом водорода углеводо-
рода — атом углерода графита сделано в гл. X.
Хлоропрен (2-хлорбутадиен-1,3) адсорбируется на графитирован-
ной термической саже вблизи комнатной температуры также молеку-
197
лярно (физическая адсорбция). Рост — АС/ хлоропрена с ростом
заполнения поверхности графитированной термической сажи (рис.
V,8) типичен для адсорбции на физически однородной поверхности
[30] (см. разд. 1 гл. II и рис. 11,11). Величина —А£7\, полученная
экстраполяцией кривой —АС/ = / (Г) к Г = 0, совпадает с вели-
чиной —Al/j, определенной газохроматографическим методом, и со-
ставляет около 34,7 кДж/моль. Таким образом, при переходе
от бутадиена (см. табл. П,3 Приложения) к хлоропрену величина
—АС^! возрастает на 6,3 кДж/моль, т. е. на ту же величину, что
и при переходе от н-бутанак н-бутилхлориду (см. рис. V,6 и табл. П,3
Приложения).
Выше 80° С изобары адсорбции хлоропрена проходят через
минимум. Это указывает на вторичный процесс, протекающий вслед
за молекулярной адсорбцией хлоропрена. При этих условиях на
поверхности графитированной термической сажи происходит поли-
меризация хлоропрена [30].
12. Простые эфиры
Простые эфиры представляют собой наиболее простые кислород-
содержащие производные углеводородов Геометрическое строение
их молекул близко
Рис. V,9. Зависимость —ДС7Х при
адсорбции на графитированной
термической саже н-алканов (7)
и ди-н-алкилэфиров (2) от числа
атомов углерода в их молекулах.
к геометрическому строению молекул соответ-
ствующих углеводородов. Ван-дер-
ваальсовский радиус кислорода мень-
ше такового для групп СН2 и СН3.
Взаимных водородных связей моле-
кулы простых эфиров (молекулы
группы В) не образуют.
Исследована адсорбция димети-
лового, диэтилового, ди-н-пропило-
вого, диизопропилового и ди-к-бу-
тилового эфиров [31—33] (см.
табл. П. 3 Приложения).
Величины К± и —&UX для про-
стых эфиров немного больше соот-
ветствующих величин для н-алка-
нов с тем же числом атомов угле-
рода в молекуле (рис. V,9) и не-
много меньше соответствующих
величин для «-алканов с числом
атомов углерода в молекуле, рав-
ным сумме чисел атомов углерода
и кислорода в молекуле эфира.
Например, для диметилового эфира зти величины меньше, чем для
пропана, но больше, чем для этана; для диэтилового эфира меньше,
чем для «-пентана, но больше, чем для «-бутана, и т. д. Различие
этих величин находится в соответствии с различиями в величинах
поляризуемостей атома кислорода в эфире и группы СН2 (поляризу-
198
емость эфирного атома кислорода меньше поляризуемости группы
СН2, см. ниже рис. V,ll). Свободные электронные пары атома кисло-
рода простого эфира и соответствующий дипольный момент не вно-
сят заметного вклада в энергию межмолекулярного взаимодействия
простых эфиров с графитированной термической сажей.
Изомеризация алкильных групп эфира (например, переход
от ди-н-пропилового эфира к диизопропиловому эфиру) уменьшает
величины Кг и —AU\ в результате удаления части звеньев моле-
кулы от плоской поверхности адсорбента..
Результаты исследования адсорбции эфиров можно использо-
вать для их идентификации и для определения атом-атомной потен-
циальной функции межмолекулярного взаимодействия атома кисло-
рода простого эфира с атомами углерода графита, подобно тому, как
это описано в гл. X для адсорбции различных углеводородов.
13. н-Спйрты
Молекулы спиртов (молекулы группы D) способны образовывать
взаимные водородные связи, что ведет к сильной их ассоциации на
поверхности неспецифического адсорбента. Газохроматографические
исследования адсорбции спиртов при малых заполнениях поверх-
ности графитированной термической сажи при температурах около
100—200° С [14, 34] показали, что величины —A.U\ при адсорбции
«-спиртов значительно меньше теплот их конденсации —L (см.
рис. 1,7). Разность — — (L—RT) близка к энергии водородной
связи. Отсюда ясно, что при малых заполнениях поверхности гра-
фитированной термической сажи спирт адсорбируется в виде изоли-
рованных (неассоциированных) молекул. Однако вследствие того,
что молекулы многих спиртов содержат концевую гидроксильную
группу, они способны специфически взаимодействовать с остаточ-
ными кислородными соединениями на поверхности сажи с образова-
нием и с ними водородной связи. Поэтому полученные ранее (14, 34]
значения —Аиг при адсорбции спиртов, по-видимому, несколько
завышены. Новые систематические определения следует провести на
поверхности графитированной термической сажи, дополнительно
обработанной водородом [31, 35] (см. рис. 1,4 и разд. 1 гл. II). Это
относится также и к адсорбции кетонов, кислот и сложных эфиров,
молекулы которых содержат выдвинутые на периферию карбо-
нильные и карбоксильные группы.
14. Терпены
Терпены представляют собой сложные моно- и бициклические
углеводороды и их производные. Молекулы незамещенных терпенов
содержат 10 атомов углерода. Углеводороды таких терпенов имеют
общую формулу С10Н1в. Их производные могут содержать двойные
связи, кетонную или гидроксильную группы. Константы Генри К±
и величины—при адсорбции изученных терпенов на графитиро-
199
ванной термической саже изменяются в основном в соответствии с гео-
метрией их молекул, независимо от наличия двойных связей или
функциональных групп [36]. Наибольшие значения Кг и —At/j
получены при адсорбции тех терпенов, молекулы которых наиболее
вытянуты и поэтому могут касаться плоской поверхности наиболь-
шим числом звеньев. По мере искривления молекулы и уменьшения
числа ее звеньев, могущих непосредственно касаться плоской по-
верхности, величины Кг и —&иг в ряду терпенов при адсорбции на
графитированной термической саже уменьшаются [14, 36]. Наибо-
лее точные данные получены для камфена [19].
15. Дейтерозамещенные органические соединения
Термодинамические характеристики адсорбции на графитиро-
ванной термической саже обычных и дейтерированных углеводоро-
дов и их производных несколько различаются (см., например, ра-
боты [37—39], а также рис. XI,1—XI,3). Это позволяет при под-
ходящих условиях разделять смеси таких изотопных молекул.
На графитированной саже при не слишком низких температурах
(подробнее см. гл. XI) дейтерозамещеннце углеводороды удержи-
ваются слабее обычных углеводородов. Различие адсорбции D-
и Н-углеводородов [37] и их производных [38] в этом случае в основ-
ном определяется отношением поляризуемости атомов D и Н в этих
соединениях: поляризуемость D-соединений несколько меньше поля-
ризуемости Н-соединений. Газохроматографическое разделение CD4,
СН4 и C2D4, CsH4 произведено на колонне с графитированной терми-
ческой сажей [39], помещенной для увеличения Кг в криостат [11].
Термодинамические характеристики адсорбции для этих веществ
приведены в табл. V,3 [39].
Таблица У,3. Термодинамические характеристики адсорбции
Н- и D-углеводородов на графитированной термической саже
Адсорбат Температу- ра, к Ki, мкм -ASJ/R -Д171 Д(-ДП1)
кДж/моль
СН4 127,4 3,72 9,92 12,20 0,23
gd4 127,4 3,44 9,30 11,97
С2Щ 173,9 4,41 11,15 16,87 0,12
c2d4 173,9 4,18 11,12 16,75
Найденные экспериментально последовательности величин К г
для изученных Н- и D-углеводородов, а также приблизительные
значения соответствующих разностей А (—-А??!) согласуются с пред-
сказаниями молекулярно-статистической теории адсорбции (см.
гл. XI).
200
16. Азот- и серусодержащие производные углеводородов
Исследовались лишь отдельные представители азотсодержащих
производных углеводородов (амины, нитрилы, пиридин) и серусо-
держащих производных углеводородов (тиофен, тиофан) 114, 32].
Величины Кг и —&иг при адсорбции этих соединений в основном
возрастают в порядке увеличения молекулярного веса. Однако
благодаря резкому влиянию на адсорбцию этих веществ остаточных
кислородных соединений на поверхности [31, 35] необходимо
произвести уточнения этих данных, проводя измерения адсорбции
на графитированной термической саже, обработанной в токе водо-
рода при 1000 °C.
17. Элементорганические соединения подгруппы IVB
На графитированной термической саже хорошо разделяются
различные элементорганические соединения, содержащие элементы
подгруппы IVB (кремний, германий, олово) [40—42]. Величины
—AtG при адсорбции некоторых элементорганических соединений
на графитированной термической саже и карбохроме (модифици-
рованной графитированной саже, сохраняющей однородную ее
поверхность [43]), приведены на рис. V,10.
Величины —AUT увеличиваются с ростом молекулярного веса
(рис. ¥,10) в рядах многих соединений различного типа:
(С2Н6)3МН, (С2Н6)4М, (С2Н6)3М-М(С2Н5)3, (С2Н5)3М— X-М(С2Н6)3,
(C2H5)3GeHal, где М — металл (Si, Ge, Sn); X — О, S, Se, Hal —
соответственно F, Cl, Br и I.
Сопоставление рядов (C2H5)3MH и (С2Н6)4М для разных М
позволяет определить вклад в —АС7г четвертой этильной группы,
который составляет около 4 кДж/моль для кремний- и германий-
содержащих соединений. Сопоставление рядов (С2Н5)3М—М(С2Н5)3
и (С2Н6)3М—X—М(С2Н6)3 для разных М и X позволяет определить
вклад в —АЦ\ атома X, т. е. О, S и Se. Вклад кислорода при измене-
нии М от кремния к олову при этом уменьшается, так как небольшой
по размерам атом кислорода, находящийся между двумя атомами
олова, имеющими значительно больший размер, отодвигается от
поверхности адсорбента. Вклад более крупного атома селена в —AC7j
в этом ряду молекул остается постоянным. В ряду (C2H5)3GeHal
—Диг увеличивается с ростом М особенно сильно.
Константа Генри для адсорбции на графитированной термиче-
ской саже стереоизомеров изученных элементорганических соедине-
ний зависит от геометрического строения их молекул и от их ориен-
тации относительно поверхности [40]. Транс-изомеры соединений
типа (СН3)3М'СН=СНМ"(СН3)3, где М' и М" обозначают пары Si,
Si, Si; Ge и Ge, Ge, благодаря большим размерам атомов М ориенти-
руются на плоской поверхности адсорбента менее выгодно, чем
цис-изомеры [40]. Сопоставление моделей Стюарта — Бриглеба
201
для молекул цис- и транс-изомеров этих соединений показало, что
большее число звеньев зтих молекул может контактировать с базис-
ной плоскостью графита у цис-изомеров. Поэтому найденные экс-
периментально значения К± и —для цис-изомеров больше, чем
для транс-изомеров этих соединений. В случае адсорбции гетеро-
Рис. V,10. Зависимость
—от молекулярного
веса М для некоторых ме-
таллорганических соедине-
ний подгруппы IVB на
карбохроме (графитирован-
ной термической саже с до-
бавкой связующего).
циклических соединений, содержащих атом кремния и другие
элементы (О, S, N), значение —&UX увеличивается с ростом моле-
кулярного веса этих соединений [41].
18. Сопоставление адсорбции различных молекул
на графитированной термической саже
Представляет интерес сопоставление величин —на графи-
тированной термической саже органических веществ, молекулы
которых имеют близкую геометрию и близкие размеры, но обладают
разными химическими связями углерод — углерод и разными функ-
циональными группами, т. е. относятся к разным группам (Л, В
и D) по способности к специфическому межмолекулярному взаимо-
действию (см. разд. 2 гл. I). Такой метод сопоставления теплот
адсорбции был предложен в работах [14, 44, 45].
Выше уже были сопоставлены термодинамические характери-
стики адсорбции для пар или большего числа молекул, обладающих
одинаковым числом атомов углерода (или одинаковым суммарным
числом атомов углерода и других, отличающихся от водорода ато-
мов, например атомов кислорода в простых эфирах), атомы или
атомные группы которых находятся на близких расстояниях от
202
плоской поверхности адсорбента при энергетически наиболее выгод-
ной ориентации (см. разд. 1 гл. II).
Сопоставление термодинамических характеристик адсорбции на
графитированной термической саже таких пар или более многочис-
ленных рядов молекул показывает, что значения —Аи± для таких
молекул группы J5, как простые эфиры, не превышают намного
значения —АСЛг соответствующих молекул сравнения — молекул
группы А, близких по геометрии и величинам общей поляризуе-
мости. Сопоставление соответствующих пар молекул группы D
и группы А (например, молекул «-бутанола и н-пентана с одинако-
выми числами атомов углерода в молекуле н-алкана и атомов угле-
рода и кислорода в молекуле н-спирта) показывает, что значения
—AU1 для адсорбции молекул группы D лишь незначительно пре-
вышают значения —AUr для адсорбции молекул сравнения группы А.
Однако и это небольшое превышение значения —для адсорбции
«-спиртов над значениями —At/j для адсорбции соответствующих
н-алканов может быть вызвано побочными причинами. Во-первых,
оставшиеся на поверхности графитированной термической сажи кис-
лородные комплексы увеличивают энергию’взаимодействия спирт —
адсорбент (см. разд. 2 гл. I и разд. 1 гл. II). Во-вторых, такие места
остаточной неоднородности поверхности, как ступени или трещины,
благодаря увеличению общей энергии адсорбции способствуют
сближению адсорбированных молекул спиртов и образованию между
ними взаимных водородных связей уже при малых заполнениях,
что также увеличивает получаемое из измерений значение —Аиг.
Дополнительная обработка графитированной термической сажи
водородом при 1100° С (см. разд. 1 гл. II) заметно уменьшает это
различие в значениях —AU\ молекул групп Л, В ъО.
Таким образом, в случае адсорбции сходных по геометрии моле-
кул на графитированной термической саже (особенно после дополни-
тельной обработки ее водородом) при одинаковом расстоянии звеньев
этих молекул от плоской поверхности адсорбента при наиболее вы-
годной ориентации этих молекул относительно поверхности значе-
ния —AU\ изменяются, в основном, следуя изменению числа и поля-
ризуемости этих звеньев.
Чтобы избежать затруднений, возникающих в тех случаях,
когда для молекулы, относящейся к группам В или Z), нельзя по-
добрать соответствующей молекулы сравнения группы А с той же
геометрией и поляризуемостью, можно использовать предложенную
в работе [46] графическую интерполяцию зависимости величины
—AUT молекул сравнения от средней их поляризуемости (см. разд. 3
гл. II).
На рис. V,ll приведены зависимости значения —Аиг от поля-
ризуемости а для адсорбции вытянутых молекул, которые могут
располагаться параллельно плоской поверхности графитированной
203
термической сажи. Из рисунка видно, что значения —AtT* для адсорб-
ции на графитированной термической саже таких молекул, относя-
щихся к разным группам А и В, располагаются вблизи соответству-
ющей прямой для н-алканов*. При адсорбции же на специфических
адсорбентах второго и третьего типа (см. разд. 2 гл. I и разд? 3
гл. II) значения —А£7\ молекул, относящихся к группам В и Z),
значительно превышают интерполированные значения —А£7\ для
адсорбции молекул сравнения группы А с той же поляризуемостью
и близкой геометрией (см. рис. 11,24 и 11,26) [46, 47].
л1_____________I____________I_____________I_____________1—
и 5 Ю 15 20
Рис. V,ll. Зависимость —А £7Х от величины поляризуемости молекул а для
адсорбции на графитированной термической саже некоторых молекул близких
поперечных размеров, относящихся к разным группам А и В.
Сплошная линия проведена через точки для н-алканов. В случае адсорбции молекул, относя-
щихся к группам В, была произведена дополнительная обработка графитированной термиче-
ской сажи водородом при 1000 °C.
Следует отметить, что при изменении значений AU\ при адсорб-
ции различных исследованных веществ в 5 раз величины ASX изме-
няются незначительно, в пределах 20%. Причины этого рассматри-
ваются в гл. X. __
Для веществ сходного строения величины A5J изменяются
с изменением величин —At/j приблизительно линейно (см. рис. V,5).
На подобные линейные зависимости обращалось внимание ранее
(см. обзор [14]). Эти зависимости рассматривались на основе моле-
кулярно-статистической теории [48, 49]. Количественные молеку-
лярно-статистические расчеты величин К19 ASJ и АС^х для адсорб-
ции на базисной грани графита благородных газов приведены в гл.
IX, а для адсорбции углеводородов и их дейтеропроизводных соот-
ветственно в гл. X и XI.
* Значение —AZ7X для адсорбции молекул группы D, в частности н-спиртов,
лишь совсем на немного превышает эти величины [они описываются уравнением:
—Д^7х=6,0+ 3,2а, близким к уравнению (V,5)J.
204
В дальнейшем необходимы экспериментальные определения при
разных температурах констант адсорбционного равновесия, измене-
ний внутренней энергии и энтропии адсорбции и теплоемкости
адсорбированного вещества на графитированной термической саже
как для еще неизученных углеводородов и их производных, так и для
летучих неорганических и злементоорганических веществ. При экс-
периментальном определении термодинамических характеристик ад-
сорбции молекул с активными функциональными группами особенно
важно удаление с поверхности графитированной термической сажи
следов кислородных соединений, которые могут дать завышенные
величины констант Генри (см. разд. 1 гл. II) или привести к поверх-
ностным химическим реакциям.
В этой главе мы рассмотрели экспериментальные величины термо-
динамических характеристик адсорбции только для малых (нуле-
вых) заполнений. Необходимы также весьма тщательные хромато-
графические, статические и калориметрические измерения на обра-
ботанной водородом поверхности графитированной термической
сажи изотерм адсорбции и зависимостей Д5, AJ7 и АС от Г. Резуль-
таты зтих измерений должны послужить основой для определения
вторых вириальных коэффициентов адсорбированных веществ из
экспериментальных данных и облегчить молекулярно-статистические
расчеты термодинамических зависимостей Г (с, Т) — О, AJ7 (Г, Т) —О
и АС (Г, 7) = 0.
ЛИТЕРАТУРА
1. BrunauerS., Emmett Р. Н., Teller E.J. Am. Chem. Soc., 1938,
v. 60, p. 309—319.
2. К и с e л e в А. В. В кн.: Межфазовая граница газ — твердое тело. Пер.
с англ. ред. Э. Флад. М., «Мир», 1970. 433 с.
3. БрунауэрС. Адсорбция газов и паров. М., Издатинлит, 1948, 781 с.
4. Аристов Б. Г., Киселев А. В. Коллоидн. ж., 1967, т. 29,
с. 631—637.
5. Кантро Д., БрунауэрС., Коупленд Л. В кн.: Межфазовая
граница газ — твердое тело. Пер. с англ. ред. Э. Флад, М.,«Мир», 1970.
433 с.
6. Avgul N. N., К i s е 1 е v А. V. In: Chemistry and Physics of Carbon.
(Coll.). Ed. P. H. Walker, New York, 1970, v. 6, 349 p.
7. Harkins W. D., Yura G. J. Am. Chem. Soc., 1944, v. 66, p. 1366—
1373.
8. Киселев А. В., ДревингВ. П. Общ. ред. Экспериментальные
методы в адсорбции и хроматографии, Изд-во МГУ, М., 1972. 447 с.
9. Wooten L. A., Brown Г. J. Am. Chem. Soc., 1943, v. 65, р. 113—
118.
10. К и с е л е в А. В., Худяков В. Л., Я ш и н Я. И. ЖФХ, 1973,
т. 47, с. 2125—2126.
11. Киселев А. В., Худяков В. Л., Я ш и н Я. И. ЖФХ, 1974,
т. 48, с. 757-759.
12. Ross S., Winkler W. J. Col. Sci., 1955, v. 10, p. 319—329.
13. Sams J. R., Constabaris G., H a 1 s e у G. D. J. Phys. Chem.
1962, v. 66, p. 2154-2158.
14. Киселев А. В., Я ш и н Я. И. Газо-адсорбционная хроматография.
М., «Наука», 1967. 256 с. Исправленные и дополненные издания: La Chro-
205
matographie Gas — Solid, Paris, Masson, 1969. 289 p.; Gas-Adsorption
Chromatography Plenum Press, N. Y., 1969. 254 p.
15. Kalashnikova E. V. e. a. Chromatographia, 1971, v. 4, p. 495—500.
16. E i s e n 0. G. e. a. Chromatographia, 1971, v. 4, p. 448—454; П и л ь т А.,
РангС., Эйзен О. Изв. АН Эст.ССР, 1972, с. 211—217.
17. Березин Г. И., Киселев А. В., Синицын В. А. ЖФХ,
1967, т. 41, с. 926—930.
18. Петров Ал. А. Химия нафтенов. М., «Наука», 1971. 388 с.
19. Kalashnikova Е. V. е. a. Chromatographia, 1972, v. 5, р. 278—285.
20. Е n g е w а 1 d W. е. a. Chromatographia, 1974, v. 7, р. 229—235.
21. К а 1 a s h n i к о v а Е. V., KiselevA.V., ShcherbakovaK. D.
Chromatographia, 1974, v. 7, p. 22—25.
22. Al menniingend A., В as tiansen O. Det. Kgl. Norske, Vid.
Selsk. S. К. R., 1958, N 4, p. 25—30.
23. Vidal — MadjarC., Guiochon G. Bull. Soc. chim. France,
1971, p. 3110—3118.
24. OnuskaF., JanakJ., Tesarik K., Kiselev A. V. J. Chro-
matog, 1968, v. 34, p. 81—85.
25. Киселев А. В., Щербакова К. Д., ЯшинЯ. И. ЖСХ, 1969,
т. 10, c. 951—968.
26. Запр ометов А, Ю. и др. ЖФХ, 1972, т. 46, с. 1230—1233.
27. Калашникова Е. В. Канд, дисс., М., МГУ, 1973. 152 с.
28. J е q u i г W., Robin J. Chromatographia, 1971, v. 4, p. 59—65.
29. Д олова И. А., Киселев А. В., Яшин Я. И. ЖСХ, 1972, т. 13,
с. 162—165.
30. Батраков Г. Н., К и с а р о в В. М., Киселев А. В. ЖФХ, 1974,
т. 48, с. 1255—1258.
31. Bezus A. G. е. a. Chromatographia, 1974, v. 7, р. 246—250.
32. Elkington Р. A.,Curthoys G. J. Phys .Chem., 1969, v. 73, p. 2321.
33. Калашникова E. В., Макагон A. M., Щербакова К. Д.
Тезисы докладов на 2-м Всесоюзном симпозиуме по межмолекулярным
взаимодействиям и конформации молекул. 2—10 июня 1947 г. Волгоград.
34. Belyakova L. D., Kiselev А. V., Kovaleva N. V. Bull. Soc.
chim. France, 1967, p. 285—290.
35. Di Corcia A., Fritz D., Bruner F. J. Chromatog., 1970, v. 53,
p. 135-141 .
36. Киселев А. В., Петов Г. M., Щербакова К.Д. ЖФХ, 1967,
т. 41 с. 1418—1425.
37. Constabaris G., Sams J. Н., Н а 1 s е у G. D. J. Phys. Chem., 1961,
v. 65, р. 367—369.
38. Bruner F., Ceccioli P., Corcia A. Analyt. Chem., 1972, v. 44,
p. 894—898.
39. К и с e л e в А. В., Худяков В. Л., ЯшинЯ. И. ЖФХ, 1974, т. 48,
с. 757—759.
40. Вязанкин Н. С. и др. Изв. АН СССР. Сер. хим., 1969, с. 186—189.
41. Bortnikov G. N. е. a. Chromatographia, 1971, v. 4, р. 14—18.
42. Б о р т н и к о в Г. Н. Канд, дис., М., МГУ, 1975.
43. Б а р м а к о в а Т. В., Киселев А. В., Ковалева Н. В.
Коллоидн. ж., 1974, т. 36, с. 133—136.
44. Kiselev А. V. Disc. Faraday Soc., 1965, v. 40, p. 205—225.
45. К и с e л e в А. В. ЖФХ, 1967, t. 41, c. 2470—2504.
46. В arr er R. M. J. Col. Interface Sci., 1966, v. 21, p. 415—434.
47. Киселев А. В. и др. Физико-химические применения газовой хро-
матографии. М., «Химия», 1973. 255 с.
48. Barker J. A., Everett D. Н. Trans. Faraday Soc., 1962, v. 58,
p. 1608—1623.
49. К и с e л e в А. В., П о ш к у с Д. П. ЖФХ, 1965, т. 39, с. 398—402.
Глава VI
СТАТИСТИЧЕСКИЕ ВИРИАЛЬНЫЕ ВЫРАЖЕНИЯ
ДЛЯ ТЕРМОДИНАМИЧЕСКИХ ХАРАКТЕРИСТИК АДСОРБЦИИ
1. Общие выражения
В статистической теории физической адсорбции твердое тело
(адсорбент) обычно рассматривается инертным, т. е. принимается,
что все его термодинамические свойства практически не изменяются
при адсорбции газа. Таким образом, все изменения термодинамиче-
ских свойств адсорбата и адсорбента при адсорбции приписываются
адсорбату. Это допущение справедливо для многочисленных практи-
чески важных случаев адсорбции на нелетучих нерастворяющих
адсорбат и ненабухающих адсорбентах. Твердое тело в этом прибли-
жении рассматривается только как источник внешнего потенциаль-
ного поля, постоянного во времени и не зависящего от температуры.
В этом случае статистическая механика адсорбции сводится к стати-
стической механике адсорбата во внешнем потенциальном поле.
Физически адсорбированное вещество при не очень низких темпе-
ратурах можно рассматривать как флюид (газ, жидкость) во внешнем
потенциальном поле. Формальная статистическая механика свойств
флюида во внешнем потенциальном поле развита довольно полно [1,
2]. Однако не решены проблемы приведения полученных общих
формальных уравнений к полезным для практических применений
теоретическим выражениям при любых заполнениях поверхности.
Практически полезные статистические выражения для адсорбирован-
ного вещества при сравнительно больших заполнениях выводятся
обычно для определенных моделей состояния (для приближенных
моделей потенциальных функций) адсорбированных молекул (см.,
например, работы [3, 4]).
В случае низких заполнений поверхности физически адсорби-
рованное вещество при не очень низких температурах представляет
в сущности газ во внешнем потенциальном поле. В последнем случае
статистическая механика адсорбированного вещества сводится к ста-
тистической механике реального разреженного газа во внешнем
потенциальном поле. В этом случае, как и в случае разреженного
объемного газа, статистическую сумму можно разложить в ряд
и получить вириальные статистические выражения, не связанные
с какими-либо моделями состояния адсорбированных молекул [1, 2,
4—28]. Коэффициенты первого (главного для малых заполнений
поверхности) члена этих выражений определяются потенциальной
функцией взаимодействия только одной молекулы с поверхностью
207
твердого тела. Коэффициенты остальных членов этих выражений
определяются потенциальными функциями взаимодействия двух,
трех и т. д. молекул адсорбата с поверхностью адсорбента и друг
с другом. Вириальные выражения оказались очень полезными при
исследовании потенциалов сил, действующих между молекулой
и твердым телом, а также между адсорбированными молекулами при
физической адсорбции на однородной поверхности твердого тела
Однако они практически применимы только при низких концентра-
циях газа вблизи и вдали от поверхности.
При выводе вириальных выражений для адсорбции использова-
лись как статистическая сумма канонического ансамбля [1, 2,
5—12], так и статистическая сумма большого канонического ансамбля
[1, 13—28]. При выводе вириальных выражений для адсорбции
методом канонического ансамбля, как правило, делаются допущения
о том, что адсорбирующийся газ подчиняется законам классической
механики и состоит из одноатомных молекул. Вириальные выраже-
ния для адсорбции любого газа сравнительно просто можно полу-
чить, используя метод большого канонического ансамбля.
Большим каноническим ансамблем называется система, обмени-
вающаяся с окружающей средой как энергией, так и частицами.
Общая теория большого канонического ансамбля была рассмотрена
в ряде монографий (см., например, [16, 22, 29]). Это более общий
и более мощный статистический метод, чем метод канонического
ансамбля, рассматривающий равновесные системы, обменивающиеся
с окружающей средой только энергией.
Свойства большого канонического ансамбля, а также и других
равновесных ансамблей (например, канонического), могут быть
выражены через большую статистическую сумму S и ее производ-
ные. S является функцией 7, геометрических параметров х и хими-
ческих потенциалов всех компонентов системы ц. Наиболее простым
образом с S связан термодинамический потенциал системы й,
представляющий характеристическую функцию тех же перемен-
ных [29]:
ЩГ, х, р) = /сГ InS (VI,1)
Этот термодинамический потенциал определяется равенством [29]:
а = (Vi,2)
r=l
где Хг — обобщенная сила, соответствующая r-му геометрическому параметру
системы хл; тп — число геометрических параметров системы; и — хими-
ческий потенциал и статистически среднее число молекул Zc-ro компонента в си-
стеме; х — число компонент; F — свободная энергия системы.
Так как полный дифференциал свободной энергии равен
[т х
dF = — SdT — Xrdxr + ^ pkdNk (VI,3)
r=l fc=l
208
(где S — интегральная энтропия системы), то полный дифферен-
циал потенциала Q можно представить выражением:
m х
d£i = S dT + 2 xr dzr± 2 (VI A)
r«=l Ы
Из этого уравнения следует, что
(VI,5)
\ dp-fe /т, х, U;
(где — химические потенциалы всех компонентов, за исключе-
нием Л-го компонента) и
где ц — химические потенциалы всех компонентов газа.
Кроме того, используя соотношение U = F -|- TS и выражения
(VI,2) и (VI,6), для внутренней энергии U системы получаем [291:
где Л — абсолютные активности всех компонентов газа.
Абсолютная активность Л-го компонента газа связана с его
химическим потенциалом (в пересчете на одну молекулу) соотно-
шением:
^=exp(-g-) (VI,8)
Геометрическими параметрами адсорбционной системы являются
объем газа V и площадь поверхности твердого тела А. Соответству-
ющие силы — зто давление в объеме газа р = (dQ/dV)Ti аи по-
верхностное давление адсорбированного вещества л = (5Й/Л4)Г) yiU.
Поэтому для газа, взаимодействующего с поверхностью твердого
тела, из уравнений (VI,!) и (VI,2) следует, что
+ 1п8 (VI,9)
где 3 — большая статистическая ^сумма для газа, взаимодействующего с по-
верхностью твердого тела.
Для статистического среднего чисуха молекул Л-го компонента
Nk в газе, взаимодействующем с поверхностью твердого тела, из
уравнений (VI,!) и (VI,5) получаем
Nk = kT f (VI,10)
\ JT. V, A,
14 Заказ 1180
209
Если газ не взаимодействует с поверхностью твердого тела (гипо-
тетическая система сравнения), то геометрическим параметром
системы является только V. В этом случае
Q& = pV = kT In
(VI,И)
где S 8 — большая статистическая сумма для газа, не взаимодействующего
с поверхностью твердого тела, т. е. обычного объемного газа.
Статистическое среднее число молекул Л-го компонента в таком
газе
N% = kT
din Ве \
'Т, V, иу
(VI,12)
При наличии взаимодействия с твердым телом Nk >2Vf при одних
и тех же значениях Г, V и
В разд. 3 гл. III отмечены преимущества определения величины
адсорбции по Гиббсу как избытка числа молекул в системе, образо-
вавшегося в результате взаимодействия газа с адсорбентом, по
сравнению с числом молекул в несодержащем адсорбент газе (системе
сравнения) с теми же Г, V и р. Согласно этому определению Гиббса,
число адсорбированных молекул Л-го компонента на инертном твер-
дом теле
(VI ,13)
где Nk — статистическое среднее число молекул к-го компонента газа, реально
находящееся в объеме V при взаимодействии газа с твердым телом; N$ — соот-
ветствующее число молекул в этом же объеме при предположении, что кон-
центрация к-го компонента в этом объеме газа остается той же вплоть до поверх-
ности твердого тела, т. е. в отсутствие адсорбции.
Вычитая уравнение (VI,12) из уравнения (VI,10), для изотермы
гиббсовской величины адсорбции fc-ro компонента на единице поверх-
ности твердого тела получаем
*1
А
Г*
кт gin(a/ag)
л L fyk
Tj V, A, Цу
(VI,14)
2. Изотерма адсорбции однокомпоиентного газа
Большая статистическая сумма для однокомпонентного газа,
взаимодействующего с поверхностью твердого тела [16, 17, 23],
равна
S(V, А, К, = + Qn(V, A, T)Kn (VI.15)
Л>0
где QN — каноническая статистическая сумма для N молекул адсорбата, вза-
имодействующих с поверхностью твердого тела.
210
Для газа, не взаимодействующего с поверхностью, т. е. для обыч-
ного объемного газа, большая статистическая сумма [16, 17, 23]
равна:
38(У, Л, П = ехр(-^Р) = 2 Q<^V’ T}^N (vl’16)
n>q
гДе1(?# — каноническая сумма для N молекул в объеме газа.
Как и в случае объемного газа, вместо абсолютной активности Л
удобно ввести активность а, которая при низких давлениях равна
концентрации газа вдали от поверхности с = N8/V [16, 21]:
(VI,17)
где Qi — каноническая статистическая сумма для одной молекулы газа.
Большие статистические суммы в этом случае принимают следу-
ющий вид [16, 21]:
^|- (VI ,19)
Здесь
Zjv=(w)N7V!9jv (VI,20)
В классической статистической механике величины ZN и Z% равны
соответствующим конфигурационным интегралам.
Разлагая In Н и In В8 в ряды Тэйлора по степеням активности а,
получаем [16, 22, 23]
In 3 = 2 (VI,22)
In Bg=2 Vbfal ' (VI,23)
где коэффициенты разложения bj и by даются выражениями:
2 1 = и т. д-
1 ! Vb8 = Z8 = V
2 ! V&f = Zf~ (zf)2 и т. д.
(VI,24)
(VI,25)
14*
211
Статистические средние числа молекул в объеме V в зтих двух слу-
чаях равны соответственно [16, 22, 23]:
у W’-2e)
Ne = a ( Sg ) = \vjbfa1 (VI,27)
\ да ' т, V *
Изотерму гиббсовской величины адсорбции Г, т. е. зависимость
Г от активности а адсорбата, получаем при этом в форме следующего
вириального разложения [16, 22, 23]:
Г =JL^L _ 2 ~ i (bi - bf) Kial (VI,28)
/>1 />1
где
*/=—(b/-bf) (VI,29)
Для / = 1Г= Кга = Кгс, где Кг ~ константа Генри Кг, с, i
[см. выражение (Ш.12)].
Вводя выражения (VI,20), (VI,21), (VI,24) и (VI,25) в уравнение
VI,29), для констант Кг и К2 получаем следующие выражения:
^=4-(z,-z5)-4
ei-ef
Cf/v
(VI,30)
"4 (z, -Zi+2И«) - 4 + 2VB‘.
(VI,31)
где В f — второй вириальный коэффициент объемного газа [16]:
= (VI,32)
Активность а можно выразить через концентрацию с или через
давление газа р вдали от поверхности с помощью выражений (VI,16),
(VI,23) и (VI,27). Из этих выражений следует, что [16]
С= 2 ibfa1' (VI ,33)
и
-^ = 2^' (VI,34)
/>1
Решая эти уравнения относительно а, получаем [16]:
a=c + 2Bfc2 + - . . (VI,35)
’и
•-тк+-в1(тг)!+--- <v,'36)
212
Активность а можно также выразить через Г. Решая выражение
(VI,28) относительно а, получаем
Г
*2Г2 ,
Kf -Г • • •
(VI,37)
Вводя выражения (VI,35) и (VI,36) в уравнение (VI,28), получаем
следующие уравнения для изотермы адсорбции:
с2 + . . .
r=K^^+*iBf) (тг)2+• • •
(VI, 38}
(VI,39}
Решая уравнения (VI,38) и (VI,39) относительно с и р!кТ соответ-
ственно получаем обратные ряды:
Г (K2+2JfxBf)r2 ,
Kf +
_Е_____Г___(я2+ ЯХВ|) г2
кТ - Кг -----(
(VI,40}
(VI,41}
3. Уравнение состояния адсорбированного
однокомпонентного газа
Вычитая уравнение (VI,11) из уравнения (VI,9) и разлагая In Е
и In Е е в ряд Тзйлора с помощью выражений (VI,22) и (VI,23) для
зависимости л от а, т. е. для уравнения состояния адсорбированного
вещества, получаем [17, 23]
(VI.42)
•ь 1 яшл J
/>1
Величина л в уравнении (VI,42) не связана с какой-либо моделью
состояния адсорбированного вещества. Она связана с Г, т. е. она
не связана с толщиной адсорбционного слоя. Вводя выражение
(VI,37) в уравнение (VI,42), получаем:
л Я2Г
ГкТ 1 2К*
(VI,43}
При низких температурах и небольших заполнениях поверх-
ности движение адсорбированных молекул ограничено главным обра-
зом областью, близкой к поверхности. Поэтому в случае плоской
поверхности адсорбированные молекулы приближенно можно рас-
сматривать как реальный двухмерный газ во внешнем потенциаль-
ном поле, т. е. можно пренебречь колебаниями адсорбированных
молекул перпендикулярно поверхности, которые налагаются на
движение адсорбированных молекул вдоль поверхности [1, 9, 10,
213
17, 21]. Вириальное уравнение состояния для двухмерного газа
имеет вид [9, 10, 11]:
^r=l + ^2Dr + C2Dr2+. . . (VI,44)
где л' — двухмерное давление; В2р и C2D — двухмерные второй и третий ви-
риальные коэффициенты.
Двухмерный второй вириальный коэффициент В2в дается выра-
жением:
52О=-4'КеХР(-------W’)~i]dA (VI,45)
гДе wi2 — потенциальная энергия взаимодействия двух молекул друг с другом.
Уравнение (VI,43) имеет форму вириального уравнения состояния
двухмерного газа (VI,44). Так как для модели двухмерного газа л'
принимается равным л, то, приравнивая коэффициенты при одинако-
вых степенях Г в уравнениях (VI,43) и (VI,44), для B2d получаем
формулу
= —(VI,46)
позволяющую найти B2D из констант Кг и К2 изотермы адсорбции.
4. Изотерма адсорбции многокомпонентного газа
Вириальные разложения для гиббсовской величины адсорбции
многокомпонентного газа методом большого канонического ансамбля
были получены в работах [15, 22, 24]. Для системы, состоящей из х
компонентов, Е дается выражением (см., например, [22]):
8{Т, х, Л)= J • • • £ Qn^i' • • • *хх (VI,47)
.ZV1=O Лх=0
где Q/V = Q (Л х, JVu . . 7VX) — каноническая статистическая сумма системы
при заданных числах 2V1? JV2, . . Nx молекул 1, 2, . . к компонентов в си-
стеме; х — геометрические параметры системы.
Как и в случае однокомпонентного газа, вместо абсолютных актив-
ностей \ компонентов смеси газов введем новые активности
л1? ..., ах, определяемые соотношениями:
ak = —y (VI ,48)
где ak — активность Zc-ro компонента; Q*k — каноническая статистическая
сумма для одной молекулы к-го компонента в£объеме газа; — абсолютная
активность Zc-ro компонента.
214
При давлении газа р-+0 активности а19..., ап приближаются
к концентрациям сх, сх соответствующих компонентов (ck =
Вводя выражение (VI,48) в уравнение (VI,47), S можно*
записать в виде:
s=2 ••• 2(vli49>
7Vi>0 ^>0 k=l
где
„ / V \ Nk
zn=QnY\ 7^-) Nk\ (VI,50>
Разлагая In S и In в ряд по степеням активностей а15 ак
и обозначая коэффициент при а™*, а™* через Vb ..., /пх),
получаем соответственно для газа, взаимодействующего с поверх-
ностью твердого тела, и для объемного газа [22]:
bS= 2 тх)а™ . . . (VI,51>
mt >0 mx>0
1пЗв = 2 • • 2 • • •• mz)«r (VI,52>
mi>0 mx>0
Оставим в рядах (VI,51) и (VI,52) только члены, учитывающие
взаимодействия молекул газа с поверхностью и парные взаимодей-
ствия между молекулами газа как вблизи, так и вдали от поверх-
ности, и введем обозначения:
— = mk+1~0, . . тпх = О) — Ь?>
b(ml = Q, . . ., 0, m*+i = 0, . .
mz+i = 0, . . wx=O) = bJftn (VI,53)»
и соответственно
rnk-1, m£+i = 0, . . тх~0)~Ь%№
bi(m1 = ei . . mk-i~0, = ^+i = 0, . . =
mz+i~0, . . /пх==0) = &2^^ (VI,54)»
В этом случае выражения (VI,51) и (VI,52) можно записать в виде::
lnS=2 Vb[kiak+'^l 2 Vb<thl)akal+. . . (VI,55)>
Й=1 k=xl>k
In 3g = 2 Vbi Mak + 2 2 + • • • (VI’56>
k=l k^ll>k
где
1! Vb<^ = Z^
2 J (VI,57)
Vb^h = Z^~Z^ztf) и т. д.
215
и
1 I Vbf (k)=--Z% (fe)= V
2! y&f = W-(zfW)2 (VI ,58)
Вводя выражения (VI,55) и (VI,56) соответственно в уравнений
{VI,10) и (VI,12), для среднего числа молекул А-го компонента в объ-
еме при взаимодействии молекул с поверхностью твердого тела
и в отсутствие такого взаимодействия получаем соответственно:
^ = а*(‘4г^")т V л , = уЬг>ай+'Уп7Ь<«)а^+. . . (VI,59)
\ к /1 f vj , а 2
J Z=1
z pg \ H
^f = a4—)„ v =Vak + \nVb^kl>akai+. . . (VI,60)
\ cfak ' 3’, V, a}
! 1=1
где n = 1, если I #= к и n = 2, если I = к.
Отсюда для зависимости гиббсовской величины адсорбции А-го
компонента от активностей компонентов газа получаем:
2L
П =+ + .. (VI,61)
z~i
где
V
= — (Ц*>-1) (VI, 62)
K(M>=^.^kl)_bg(kl^ (VI,63)
Активности alt ax могут быть выражены через концентрации
..., ех компонентов газа вдали от поверхности [18]. Решая уравне-
ние (VI,60) относительно ak и аг, получаем:
ak~ck +2 nBf <W)c*cz+- • • (VI,64)
1=1
XL
“l=ci + 21 nBl^ckci + . . . (VI,65)
fc=l
где —bf — второй] вириальный коэффициент А-го компонента
газа; В^к1^ ~ — второй вириальный коэффициент газа, зависящий
от свойств двух молекул А-го и Z-ro компонентов.
Вводя выражения (VI,64) и (VI,65) в уравнение (VI,61), получаем
следующее вириальное уравнение для изотермы адсорбции многоком-
216
понентного газа, выраженной в виде функции концентраций ком-
понентов:
г* = K^ck + 2 \К^‘> + <w>] ekCl + . . . (VI,66)
1=1
trq — константа Генри для адсорбции &-го компонента; +
+ nK^Bl(И)] — константы, учитывающие парные взаимодействия между
адсорбированными молекулами, между адсорбированной молекулой и молекулой
в газе, а также между молекулами в газе.
Если газ вдали от поверхности принять за идеальный, тоБ( = О,
и тогда
rk^K(1k>ck + ^iK(^ckci+. . . (VI,67)
1=1
где
K(ki>=.^Y3 ’ (VI,68)
А
5. Удерживаемый объем
Удерживаемый объем А-го компонента смеси газов FJj* опреде-
ляется по формуле {30, 31] (см. также формулу (111,17)]:
V$’ = ip/ft-V (VI,69)
где w — средняя объемная скорость газа при температуре колонны Т и среднем
давлении р внутри колонны вдали от поверхности; tk — неисправленное время
удерживания &-го компонента, т. е. отрезок времени между введением смеси
в колонну и-выходом /с-го компонента в другом ее конце; V — объем свободного
газа в колонне (мертвый объем), равный удерживаемому объему той же колонны
по отношению к неадсорбирующемуся газу.
Если газ вдали от поверхности адсорбента принять идеальным, та
цкт
w = ~---
Р
(VI,70>
Где у] __ скорость газа, выраженная числом молекул, проходящих в секунду
через сечение колонны.
Кроме того, если принять равновесную модель для газоадсорб-
ционной хроматографии, т. е. предположить, что при прохождении
газа через колонну устанавливается адсорбционно-десорбционное
равновесие, а также принять, что газ входит и выходит из колонны
с постоянной скоростью ц, причем перепад давления р внутри ко-
лонны близок к нулю, то время, необходимое для прохождения
через колонну &-го компонента газа, т. е. неисправленное время
удерживания А-го компонента, равно
= (VI,71)
где Nk — среднее число молекул &-го компонента во всей колонне; — моль-
ная доля Л-го компонента в выходящем из колонны газе$ — число молекул
fc-ro компонента, покидающих колонну каждую секунду.
247
Вводя выражения (VI,70) и (VI,71) в уравнение (VI,69), для
удерживаемого объема Л-го компонента, отнесенного к единице
поверхности адсорбента, получаем:
где pk ~ p%k — парциальное давление &-го компонента равновесного газа
внутри колонны вдали от поверхности адсорбента; А — величина поверхности
адсорбента в колонне.
При указанных допущениях газ внутри адсорбционной хромато-
графической колонны можно рассматривать как открытую равновес-
ную систему. Среднее число молекул А-го компонента во всей колонне
JVA дается выражением (VI,59). Вводя выражение (VI,59) в уравне-
ние (VI,72) и принимая во внимание, что для идеального газа ak =
= ck = PklkT и di = Ci = pilkT, получаем [24]:
(VI,73)
где константа дается выражением (VI,62), а константа — выраже-
нием (VI,68).
В уравнении (VI,73) первый член учитывает взаимодействия
отдельных молекул А-го компонента газа с поверхностью адсорбента,
а второй член учитывает парные взаимодействия между адсорбиро-
ванными молекулами многокомпонентного газа.
При предельно малых (нулевых) дозах вводимых в колонну
компонентов в соответствии с выражением (111,21) = Кл\
6. Химический потенциал адсорбированного вещества
Для химического потенциала р газа, взаимодействующего с по-
верхностью твердого тела, в функции величины адсорбции Г, из выра-
жений (VI,8), (VI,17) и (VI,37) получаем:
kT I № ) Kl ф
(VI,74)
Вместе с тем для химического потенциала pgt0 газа вдали от поверх-
ности твердого тела при фиксированной концентрации с° (давлении
J9’) из выражений (VI,8), (VI,17), (VI,35) и (VI,36) следует:
(VI,75)
или
-In ( P°V \ , В& I
kT !;Т ~
(VI,75а)
218
Вычитая (VI,75) и (VI,75а) из (VI,74), получаем вириальное выра-
жение для изменения химического потенциала газа Дц при переходе
моля адсорбата из объема газа при концентрации с9 (давлении р°)
в адсорбированное состояние при величине адсорбции Г:
ИЛИ
тг2г К1
к2г
кТ 1 * ’ ’
(VI,76)
(VI,76а)
7. Изменение энтропии при адсорбции
Интегральная энтропия 5 и термодинамический потенциал Q
системы связаны соотношением (VI,6). Вводя выражение (VI,!)
в уравнение (VI,6), получаем для энтропии газа, взаимодействующего
с поверхностью твердого тела
S
d In В \
дТ /у, а, р.
(VI,77)
Энтропия газа, не взаимодействующего с поверхностью твердого
тела
S^klntf + krf gIn5g )
X дТ / у, ц
(VI,78)
Вычитая (VI,78) из (VI,77), получаем следующее выражение для
интегральной энтропии адсорбированных молекул № = 5—S8>
отнесенной к единице поверхности твердого тела:
(VI,79)
Разлагая In Н и In в ряды Тэйлора по степеням активности а
с помощью выражений (VI,22) и (VI,23), выражая а через Г с по-
мощью выражения (VI,37) и используя обозначения (VI,17) и (VI,29),
для Ss получаем:
к
d In Ki ( д Qi \ , I ГК
Кг
KI
d (Кг/К!
dT
(VI, 80)
Дифференцируя это выражение по числу молей адсорбированного
вещества Г/Na (Аа — число Авогадро) при постоянных А и Т, для
219
дифференциальной мольной энтропии адсорбированных молекул 5s
получаем:
cs d In Kt / д In Q? \ / гу x
T=1" *>+r Vi" (-Jf) +
+[4r+r^lt]r+ -- ' <v,'8i>
Дифференциальное мольное изменение энтропии адсорбата при
переходе из объемного газа при фиксированной концентрации с°
{давлении pQ) в адсорбированное состояние при величине адсорбции
Г равно [см. выражение (111,59)]:
(VI,82)
где ° — средняя мольная энтропия объемного газа при концентрации с-
^давлении р°).
Разлагая в формуле (VI,78) In Е* в ряд Тэйлора по степеням а
о помощью выражения (VI,23), деля полученное выражение на число
молей Ng/NA в системе, т. е. на уравнение (VI,27), и выражая а
через с или р с помощью уравнений (VI,35) и (VI,36), получаем:
или
-.1 In Г PF- ) | TplnQf \ г Р , (VI 83 al
Вычитая уравнения (VI,83) при с ~ с° и (VI,83а) при р — р° из
уравнения (VI,81), для дифференциального мольного изменения
энтропии адсорбата А5 при его переходе из объема газа при концен-
трации с° (давлении р°) в адсорбированное состояние при величине,
адсорбции Г, получаем [28]:
^-.+ь (Я + »-]г +
/ a dB»\
+ V + . . . (VI,84)
1пК, I Т , i_f Р° \ , Г К2 d(Zf2/^)-l
—Ki+p—;+L^T+ —dr—Jr +
(vi’84a)
al KL
Вычитая уравнение (VI,83) или уравнение (VI,83a) из уравнения
(VI,81) и выражая с или р через Г соответственно с помощью выраже-
ний (VI,40) и (VI,41), получаем следующее выражение для дифферен-
220
циального мольного изменения энтропии адсорбата nprf адсорбции
в равновесных условиях:
Д^е<? 5s—
Я ” R--------^T~dT-------1 +
+ Л/ДГП +^-(r . (VI,85)
8. Изменение внутренней энергии при адс(/РбЧии
Внутренняя энергия U системы и термодинамический потенциал
п CmaI?? соотношением (VI, 7). Вводя выражение (VI,!) в уравне-
ние (VI,/), для внутренней энергии газа, взаимодействуй011^61'0 с по-
верхностью твердого тела, получаем:
u=kT2(^^-) (VI,86)
\ дТ ) А, у, л
Внутренняя энергия U8 газа в объеме, т. е газа не взаимодей-
ствующего с поверхностью твердого тела ’
Us = kT^(-^^-] (VI,87)
\ дТ Jv, К
Вычитая уравнение (VI,87) из уравнения (VI,86), для Ин/Т6ГРальн°й
внутренней энергии адсорбированных молекул U = i(u~U8)/A,
отнесенной к единице поверхности твердого тела, полуй*аем следу-
ющее выражение:
(VX.88,
Л Л JA, V, X
Разлагая In S и In S« в ряды Тэйлора по степеням а Iе помощью
выражении (VI,22) и (VI,23), выражая а через Г с помощ»ью уравне-
чае d7 И испольузя обозначения (VI,17) и (VI,29), дл/я Us полу-
(vi>8s)
Дифференцируя это выражение по числу молей адсорбгйРованного
вещества Г^А при постоянных А и Т, для дифференциальной моль-
ной внутренней энергии адсорбированного вещества U* получаем:
£_е^_^+(ЭДу+г^+ ...(И.ад
Разлагая в формуле (VI,87) In S* в ряд Тэйлора по степеням а
с помощью выражения (VI,23), деля полученное выражений6 на пиело
молей газа N8/NА в системе, т. е. на уравнение (VI,27), йи выражая
221
а через с или р с помощью уравнений (VI,35) и (VI,36), для средней
мольной внутренней энергии объемного газа получаем:
( д In Ql \ У dB%\
RT* \ дТ /у к dT
или
р In <?Q dBf
RT* \ дТ 'v dT
(VI,91)
(VI,91а)
Вычитая уравнения (VI,91) и (VI,91а) при с = cQ ир = р° из уравне-
ния (VI,90), для дифференциального мольного изменения внутрен-
ней энергии адсорбата при переходе из объема газа при стандартной
концентрации с° (стандартном давлении р°) в адсорбированное со-
стояние при величине адсорбции Г получаем [28]:
dinKr d(K2/Kj) dBf ,
RT% RT* dT dT dT
или
___din Кг d(K^fKj) , dBf p° .
RT2 dT dT ^dT' kTTt,t
Вычитая уравнение (VI,91) или (VI,91a) из уравнения (VI,90)
и выражая с или р через Г соответственно с помощью уравнений
(VI,40) и (VI,41), получаем следующее выражение для дифферен-
циального мольного изменения внутренней энергии адсорбата при
адсорбции в равновесных условиях:
i,ueq _ us—u$t dinKj , Г d(K2/Ki) 1 jbT] ,VT<m
ЯТ2 dT "TL dT ' Кг dT J Wi,yo>
Из изотерм адсорбции, измеренных при разных температурах
статическим методом, часто определяют так называемую изостери-
ческую теплоту адсорбции [32—34] [см. формулу (111,110)]:
(VI,94)
где Vm — мольный объем равновесного с адсорбентом газа.
Так как равновесные величины Г и р связаны друг с другом
уравнением (VI,41), то
рУ|1=Я7’[1+4к + • ’ •]==ЛГ(1+-Й^+ ' • •) (VI’95>
Вводя выражения (VI,93) и (VI,95) в выражение (VI,94), получаем
qst______din Кг .__1___d (K2/Kl)
RT2 ~~ dT Т L dT
1 ( dBl
Кг \ dT
SLY
т
Г+.. .
(VI,96)
222
9. Изменение теплоемкости при адсо{/®ции
Обычно измеряется теплоемкость адсорбционной си(Утемы’ а^е
ее изменение при адсорбции. Однако в молекулярной теории аДс0Р
ции проще рассматривать не сами термодинамические свойёЗва аДс0Р"
бированного вещества, а только изменения этих св<?иств ПРИ
адсорбции. Полные значения термодинамических свой^тв аДс0Р"
бированного вещества при желании могут быть получены, ;ПРи^авляя
эти изменения к свойствам адсорбата в объемной фазе. -
Дифференциальное мольное изменение теплоемкости аД£°Рйата
при переходе из объема газа при стандартной концентраци и с \стан"
дартном давлении р°) в адсорбированное состояние при^ величине
адсорбции Г [см. выражение (III, 726)]
дс = с — т
(VI,97)
где Cs = (dUs/dT)p — дифференциальная мольная теплоемкость адсорбиро-
ванных молекул при данной величине адсорбции Г; ^°т = (д^т
мольная теплоемкость объемного газа при с = с° (или р = р°) и^хпостоянном
объеме V.
Используя выражения (VI,90), (VI,91) и (VI,91а),
ДС dln^ , _ ^InA'r
—=2Т —dT~+ т ат*
d2(7C2/41L~
I r2TjL(*2/*j)_ , T,
L27 dT ‘ 1 dT*
9Т dBLri d*B*}
2T-dT~+T*4fi~)
(VI,98)
ИЛИ
= din Kt in г а (К2/К1) , d2 (я8/тЯ1'| г +
R 47 dT dT* ‘ L dT
dT*
r dT* / kT
(VI,98a)
Для дифференциального мольного изменения теплоемости аМс^рбата
при адсорбции в равновесных условиях получаем:
&Ceq Bv\m d In Kr d* In Kx ।
—R--------R------- 2T —~+ T dT* +
d^/K^ , dHKzIKl) , 1 / dBl , d2Bf V)~|r+. . .
dT dT* n Kr \ dT dT* '
(VI,99)
В полученных выражениях учитывается взаимодей^ствие как
между адсорбированными молекулами, так и между М(чолекУлами
в объеме газа, а также взаимодействие между адсорбирРованными
молекулами и молекулами в объеме газа.
223
10. Частные случаи общих вириальных выражений
Газ вдали от поверхности является идеальным. При не очень
высоких температурах плотность газа вблизи поверхности значи-
тельно больше его плотности вдали от поверхности. Если плотность
газа вдали от поверхности очень мала, то в хорошем приближении
его можно принять идеальным, т. е. можно пренебречь взаимодей-
ствием между молекулами в объеме газа, а также взаимодействием
молекул в объеме газа с адсорбированными молекулами. В послед-
нем случае вириальные коэффициенты объемного газа равны нулю
и выражения для изотермы адсорбции (VI,38), (VI,39), изменения
химического потенциала (VI,76), (VI,76а), дифференциальных
мольных изменений энтропии (VI,84), (VI,84а), (VI,85), внутренней
энергии (VI,92), (VI,92а), (VI,93), (VI,96) и теплоемкости адсорбата
при адсорбции (VI,98), (VI,98а), (VI,99) переходят соответственно
в следующие выражения:
или
Г кТ ( кТ ) + ’ ‘ ’
я2г
(VI,100)
(VI,100а)
(VI,101)
(VI,101а)
, или
Д5 В или = lnK1+T^^~i+ln (4)+ [4t+7 dT J Г+.. . (VI,102)
Л5 В = \пК1 + Т — ^Д-1 + ln f p° \ \ кТГ ) r d(K2/Kl) dT (VI,102a)
&Seq В dT — i+r- d(K2/KJ) dT Г+... (VI,103)
Хи &U eq d In^i , d(K2/Kf) -Г+.. . (VI,104)
RT* RT2 ” dT 1 dT
Qsf d In Ki ( 1 d (K2/K{) Г + . . . (VI,105)
ВТ* dT T dT
Д'С &Ceq R R = 2T .d ^2 _ dT 1 a И" 1 dT2 InKi , Г !T2 ‘L r+.. . ~2T + (VI,106)
224
Молекулы адсорбата взаимодействуют только с адсорбентом.
Если концентрация газа достаточно низка не только вдали от
поверхности, но и вблизи нее, то можно пренебречь взаимодействием
друг с другом также и адсорбированных молекул. В последнем случае
обращаются в нуль вириальные коэффициенты адсорбированного
газа К] (VI,29), за исключением первого коэффициента Кг (VI,30)
характеризующего взаимодействие молекул адсорбата с адсорбентом.
Выражения (VI, 100)—(VI, 106) в этом случае переходят в уже при-
веденные в гл. III простые выражения соответственно для изотермы
адсорбции (111,12), А(ч (111,43а). (III,596), A5e?,i (HI,606),
Af/x (111,646) и ACj (III,72в). При сделанном в разд. 5 гл. Ill выборе
стандартных состояний (Г%° = 1) формулы (VI,101) и (VI, 102)
переходят соответственно в формулы (III,53а) и (III,59в).
ЛИТЕРАТУРА
1. Steele W. A.,Ross М. J. Chem. Phys., 1960, v. 33, р. 464—470.
2. Hill Т. L., Sai to N. J. Chem. Phys., 1961, v. 34, p. 1543-1553.
3. Хилл T. Л. В кн.: Катализ. Вопросы теории и методы исследования. М.,
Издатинлит, 1955. 571 с.
4. Стилл У. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ,
ред. Э. Флад. М., «Мир», 1970. 433 с.
5. Wilkins F. J. Proc. Roy. Soc., 1938, A 164, p. 496—509.
6. Freeman M. P, H a 1 s e у G. D. J. Phys. Chem., 1955, v. 59, p. 181—
184.
7. Kwan T., Freeman M. P., Halsey G. D. J. Phys. Chem., 1955, v. 59,
p. 600-603.
8. Freeman M. P. J. Phys. Chem., 1958, v. 62, p. 723—728.
9. Barker!. A., Everett D. H. Trans. Faraday Soc., 1962, v. 58, p. 1608—
1623.
10. SamsJ. R., Cons tabaris G., Halsey G. D. J. Chem. Phys., 1962,
v. 36, p. 1334—1339.
1. Krizan J. E., Crowell A. D. J. Chem. Phys., 1964, v. 41, p. 1322—
1326.
12. Krizan J. E. J. Chem. Phys., 1965, v. 42, p. 2923—2927.
13. Ono S. J. Chem. Phys., 1950, v. 18, p. 397.
14. О n о S. Memoirs of the Faculty of Engineering, Kyushu University, 1950,
v. 12, p. 9-19.
15. О n о S. J. Phys. Soc. Japan, 1951, v. 6, p. 10—15.
16. X и л л T. Л. Статистическая механика. M., Издатинлит, 1960. 486 с.
17. Н i 11 Т. L. J. Phys. Chem., 1959, v. 63, р. 456—460.
18. Н i 11 Т. L. An Introduction to Statistical Thermodynamics, London, 1960.
508 p.
19. S t e e 1 e W. A., R о s s M. J. Chem. Phys., 1961, v. 35, p. 850—862.
20. S t e e 1 e W. A. Advanc. Chemistry Series, 1961, v. 33, p. 269—280.
21. H i 11 T. L., Greenschlag S. J. Chem. Phys., 1961, V. 34, p. 1538—
1543.
22. О н о С., К о н д о С. Молекулярная теория поверхностного натяжения
в жидкостях. М., Издатинлит, 1963. 292 с.
23. S a m s J. R. J. Chem. Phys., 1965, v. 43, p. 2243—2250.
24. П ошк у с Д. П. ЖФХ, 1965, t. 39, c. 2962—2967.
25. M a c Rury T. B„ Bird C., Sams J. R. Molec. Phys., 1969, v. 17,
p. 91—100. ‘
15 заказ 1180 225
26. П о шк у с Д. П. В кн.: Основные проблемы теории физической адсорбции
Под ред. М. М. Дубинина и В. В. Серпинского. М., «Наука», 1970. 475 с.
27. В akri М. М. Physica, 1971, v. 52, р. 16—20.
28. Kiselev А. V., Poshkus D. Р. J. Chem. Soc. Faraday Trans., 1975,
v. 71.
29. Фаулер P., Гуггенгейм Э. Статистическая термодинамика. М.,
Издатиндит, 1949. 612 с.
30. Н а п 1 a n J. F., F г е е m а п М. Р. Canad. J. Chem., 1959, v. 37, р. 1575—
1588.
31. Герасимов Я. И. и др. Курс физической химии. Т. I. М., «Химия»,
1969. 592 с.
32. П ей с Е. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ,
ред. Э. Флад. М., «Мир», 1970. 433 с.
33. К in g t о n G. L., A s t о n J. G. J. Am. Chem. Soc., 1951, v. 73, p. 1929—1936.
34. Hill T. L. Trans. Faraday Soc., 1951, v. 47, p. 376—380.
Глава VII
СТАТИСТИЧЕСКИЕ ВЫРАЖЕНИЯ ДЛЯ КОНСТАНТЫ ГЕНРИ
И ДРУГИХ ТЕРМОДИНАМИЧЕСКИХ ХАРАКТЕРИСТИК
АДСОРБЦИИ ПРИ НУЛЕВОМ ЗАПОЛНЕНИИ ПОВЕРХНОСТИ
1. Общие выражения для константы Генри
В классическом приближении явные статистические выражения
для константы Генри и других термодинамических характеристик
адсорбции при нулевом заполнении поверхности зависят от строения
молекулы. Такие выражения получены для адсорбции одноатомных
[1—10], двухатомных [10, 11] и некоторых простейших многоатом-
ных [11, 12] молекул, а также для адсорбции сложных многоатомных
молекул, в том числе для молекул, обладающих степенями свободы
внутреннего вращения [13—18].
Константа Генри Кх дается общим выражением (VI,30). В класси-
ческом приближении статистическая сумма молекулы, взаимодей-
ствующей с твердым телом, равна
1 Г / E4-V \
1^7777 \ 0ХР (---) dpi * * * dpsdgi . . . d<h (VII,1)
fl U \ KI /
где s — число степеней свободы; о — число симметрии; Е — кинетическая
энергия и V — потенциальная энергия молекулы; дир — ее обобщенные коор-
динаты и соответствующие обобщенные импульсы; h и к — константы Планка
и Больцмана.
Как следует из выражения (VI,30), константа определяется
отношением (J j и Qf. Вклады энергий движения электронов, электрон-
ного и ядорного спинов молекулы в общую ее энергию вообще незна-
чительны, а их изменения, вызываемые молекулярной адсорбцией,
пренебрежимо малы. Поэтому при составлении отношения Q} й Qf
в хорошем приближении эти вклады в Qr и Qf можно не включать.
15 этом приближении атом можно рассматривать как материальную
точку, а молекулу — как систему материальных точек. Так как
положение материальной точки определяется тремя величинами, то
положение молекулы, состоящей иэ п атомов, определяется s = Зп
величинами, например, Зп декартовыми координатами ядер всех п
атомов. Любые Зп величины, однозначно определяющие расположе-
ние в пространстве системы из п материальных точек, называются
обобщенными координатами этой системы.
В качество первых трех обобщенных координат обычно выби-
раются координаты, определяющие положение центра масс молекулы
в пространстве. Сокращенно эти три координаты обозначим через
<71|ОСТ. В качестве следующих двух (для линейных молекул) или трех
15* 227
(для нелинейных многоатомных молекул) обобщенных координат
обычно выбираются соответственно два или три угла, определяющие
ориентацию в пространстве молекулы в целом или ее жесткого остова
при фиксированных взаимных расстояниях между атомами, соответ-
ствующих минимуму потенциальной энергии изолированной моле-
кулы W. Эти обобщенные координаты обозначим через двнеш. вращ-
Если в молекуле отсутствует внутреннее вращение и перегруппи-
ровки (квазижесткая молекула), то остальные обобщенные коорди-
наты соответствуют колебательным степеням свободы молекулы, их
мы обозначим через дцолеб. Если в молекуле возможно внутреннее
вращение и число степеней свободы внутреннего вращения равно t,
то t координат соответствуют этим степеням свободы молекулы, их
мы обозначим через двнуТф вращ- Координаты внешнего и внутреннего
вращения вообще обозначим через двращ.
Обычно принимается, что потенциальная энергия молекулы V,
взаимодействующей с твердым телом, равна сумме потенциальной
энергии изолированной молекулы W, т. е. не взаимодействующей
с твердым телом и с другими молекулами газа, и потенциальной
энергии Ф взаимодействия молекулы с твердым телом, т. е. V = W-\~
+ Ф. С точностью до внутриатомных (электронных и ядерных)
степеней свободы W является функцией только дВНут.вращ и дколеб.
В общем случае Ф представляет функцию всех обобщенных коорди-
нат молекулы.
Кинетическую энергию молекулы Е можно представить в виде
квадратичной функции обобщенных скоростей [19, 20]
£ = (VII,2)
A, I
или обобщенных импульсов
E^^bkiPkPl (VI 1,3)
k, I
где gjfe, qi — к-ая и Z-ая обобщенные скорости; р*, pi — соответствующие об-
общенные импульсы; aki и bki — функции только обобщенных координат моле-
кулы.
Выражение (VIII,3) можно привести к диагональному виду
с помощью ортогонального линейного преобразования импуль-
сов [19]:
S
А=*1
где Dk — функция старых обобщенных координат д; р£— импульсы молекулы,
соответствующие новым обобщенным координатам д'.
Вводя выражение (VII,4) в выражение (VIIel), получаем:
л V2 £ \
<?i j ехР I---------——£7-------------I dP'i dP'^ (Vii,5)
228
так как якобиан преобразования импульсов в данном случае равен
единице. Интегрирование по импульсам дает [19]:
п 1 / 2л/сГ V/2 Г / 1Г+Ф \ л ,vn^
) J (Р1 ’ ' * Ps) 1 ехр V--лГГ/ dq (VII’6>
Произведение (Рг. . ,DS) = [Л]"1 ~ [В], где [Л] и [В] — детерми-
нанты матриц, составленных соответственно из коэффициентов выра-
жений (VII,2) и (VII,3). Произведение (Dx. . .Ds) и детерминанты
[Л ] и [В] в общем случае являются функциями обобщенных коорди-
нат д, и поэтому их нельзя выносить за знак интеграла.
Для молекулы в объеме газа Ф = 0 и статистическая сумма
/иг 1 / Y/2 Г Г / И7 \ , /VTT -74
J [Л]/’ехр(—^dq (VII,7)
Вводя (VII,6) и (VII,7) в (VI,30) и принимая, что число симметрии
молекулы о не изменяется при ее межмолекулярном (не химическом)
взаимодействии с поверхностью, получаем следующее выражение
для константы Генри;
__V_ J И]1/гехр(—И7?сГ)[ехр(—Ф//сГ) —1] dq
А |[Л],/!ехр(—W/kT)dq
Расчет константы по этой формуле в общем встречает значитель-
ные трудности главным образом из-за сложности расчета детерми-
нанта [Л] для сложных молекул. Чтобы получить практически полез-
ные формулы для константы К19 приходится вводить некоторые
дополнительные приближения.
2. Приближенные выражения для нежестких молекул
Кинетическую энергию Е молекулы всегда можно представить
в виде суммы кинетической энергии поступательного движения центра
масс молекулы £пост и кинетической энергии движения отдельных
атомов молекулы относительно ее центра масс 2?отн, т- е- можно
записать [21—23]:
Е = £пост~|“ ^отн (VII,9)
п ри чем
Рх + Ру + Р*
(VII,10)
где Рхч Руч Pz — компоненты импульса центра масс молекулы, соответствующие
ого декартовым координатам л, yt z (сокращенно эти координаты обозначим
мороз т); М — масса молекулы»
Для случая малых колебаний, когда изменение расстояний между
атомами молекулы мало по сравнению с этими расстояниями, т. е.
когда можно пренебречь взаимодействием колебаний и вращений
молекулы, величину £отн можно представить в виде суммы кинети-
ческой энергии вращательного движения молекулы (внешнего и вну-
229
треннего) 2?вращ и кинетической энергии колебательного движения
-Е’колеб» т* е* в этом случае можно записать:
^отн — #вращ“Ь ^колеб (VI 1,11)
Кроме того, можно предположить, что W и Ф можно представить
в виде суммы двух слагаемых:
1V=W (#внут. вращ) ДИ7 (<?колеб) (VII,12)
Ф=Ф(?пост» З'врадОпНДФ (?колеб) (VII,13)
где W (двнуг. вращ) и Ф (<?пост1 <?вращ) — потенциальные энергии изолированной
молекулы и ее межмолекулярного взаимодействия с твердым телом при кон-
фигурации, соответствующей минимуму функции W} ДИ7 и ДФ — изменения
соответствующих потенциальных энергий при отклонениях атомов от их равно-
весных положений.
В этом случае можно представить в виде произведения стати-
стической суммы для поступательного и вращательного движения
(?пост. вращ и статистической суммы для колебательного движе-
ния Сколеб) Т. е.
С?1= (?пост, вращФколеб (VII,14)
где
п 1 / 2лМкТ \’Л / 2пкТ \®/2
(/пост, вращ- — ----J h2 ) Х
Ч/ С Г Л Г ИЧ^внут. вращ) + Ф (?ПОСТ., ?вращ)“| Jn
/\ I L-^вращ] ЭХр I импост ОДвращ
(VII,15)
Здесь со — число вращательных степеней свободы; [4Еращ] —детерминант
матрицы, составленной из коэффициентов квадратичного выражения (VII,2)
для кинетической энергии вращательного движения молекулы.
Для молекулы в объеме газа Ф = 0 и ее статистическая сумма
в этом приближении имеет вид:
V ( 2пМкТ \’/я / 2якТ \®/2 е
<21 = —(—fe2 —) <?“лебХ
X J МвращГ7’ ехр [- ^?™у-вращ) J (VII.16)
где Сколеб — колебательная статистическая сумма молекулы в отсутствие
внешнего потенциального поля.
Вводя выражения (VII,14)—(VII,16) в уравнение (VI,30) и при-
нимая, что о и (^олеб ПРИ переходе от газового в адсорбированное
состояние при молекулярной (физической) адсорбции практически
не изменяются, получаем:
j J [4вращ] ехр (— W/kT) {exp [ — Ф (#пост» £вращ)/&Т] 1} d^nocT <2#Вращ
J [4вращ] exp ( W/кТ} 4/<?вращ
(VII,17)
Детерминант Мвращ] в общем зависит от вращательных координат
молекулы и вынести его за знак интеграла нельзя.
230
Если молекула состоит из жесткого остова и прикрепленных
к нему t волчков и если в качестве обобщенных вращательных коор-
динат выбраны три эйлеровых угла ft, <р, ф (сокращенно эти коорди-
наты обозначим через 0), определяющие ориентацию трех главных
центральных осей остова по отношению к неподвижной системе
координат, а также углы ах, . . ., а/ (сокращенно эти координаты
обозначим через а), описывающие повороты волчков вокруг осей
внутреннего вращения, то для вращательных степеней свободы [19]
[-^вращ] = [5Al] sin2 'О' (VII,18)
Здесь [$£/] — детерминант матрицы, составленной из коэффициентов
квадратичной функции вращательной кинетической энергии
&вращ “"2“ ^Skilkh (VII,19)
А, I
где Ik и 1[ означает одну из величин Q2, Q3, ах, а2> . • а/, причем Q2,
Q3 — проекции угловой скорости вращения остова, а ах, а2, . . ., а/ — произ-
водные углов ап а2, . . а/ по времени. Детерминант не зависит от углов
Эйлера 0, он является функцией только углов внутреннего вращения а.
Вводя (VII,18) в (VII,17), получаем
к Г ехр ( — W/кТ) [exp (— )~«1] sin ft dxdti da
1 8л2Л |'[ад]м/йехр(~WjkT)da
(VII,20)
. Если волчки симметричны (например, представляют группы СН3),
то детерминант [s^] является постоянной величиной [19]. В этом
случае
% J ехр (— W/кТ [exp (— Ф/кТ) -1] sin ft dr dQ da
1 8nM Jexp ( — Hz//c7’) da (VII,21)
Последнее выражение было использовано при расчетах Кх и других
термодинамических характеристик адсорбции этана с учетом вну-
треннего вращения его молекул [16]. Вводя выражение (VII,21)
в уравнения (111,646), (III,59в), (III,72в), для ДЕ7Х, и полу-
чаем соответственно [16]:
^2
RT #1“^
In К _L*^2 ^2 4
AGX /S3 7?з\ /. Т?2/2*^2 ^3
я hJ sj
(VII,22)
(VII, 23)
(VII,24)
231
Здесь
w
кТ
Ф
кТ
sin# drdB da
(VII,25)
Si= f ( ^-W^} sin'» dx dQ da, (1 = 2,3) (VII,26)
J \ nl / \ Ki /
w \
^)da, (Z-l, 2, 3)
(VII,27)
3. Выражения для молекул с поворотной изомерией
При внутреннем вращении W проходит через максимумы и мини-
мумы. Если эти изменения W значительно больше RT, то молекула
будет в основном принимать такие конформации, которые соответ-
ствуют минимумам W с относительно редкими переходами из конфор-
мации, соответствующей одному минимуму W, в конформацию,
соответствующую другому минимуму W. В этом случае при внутрен-
нем вращении образуется несколько относительно устойчивых кон-
формаций молекулы — поворотных изомеров. Поворотная изомерия
имеет место для многих молекул [24—26], например для молекул
н-алканов, начиная с н-бутана.
Для молекул с поворотной изомерией расчет детерминанта [-4вращ]
весьма сложен [19, 27, 28]. Кроме того, о потенциальной функции W
для таких молекул имеются лишь ограниченные экспериментальные
сведения [24, 26, 29, 30] и существуют только грубые полуэмпириче-
ские методы ее расчетов [26, 31]. Поэтому расчеты Кг для таких
молекул по формуле (VI 1,17) встречают значительные трудности.
Ввиду того, что внутреннее вращение при поворотной изомерии
носит характер крутильных колебаний внутри потенциальных ям
с довольно редкими перескоками между потенциальными ямами для
различных поворотных изомеров, при расчетах как и при расче-
тах QI [25, 32—35], газ, состоящий из таких молекул с поворотной
изомерией, приближенно можно рассматривать как смесь поворотных
изомеров, находящихся в равновесии друг с другом. В соответствии
с этим в классических выражениях для и Q& полные пределы
изменения углов внутреннего вращения а* (от 0 до 2л)
можно разбить по пределам изменения этих углов, соответствующим
отдельным поворотным изомерам. В зтом случае
(VII,28)
i=l
m
<21= 2 «i (VI 1,29)
i-1
где gf и qi — статистические суммы i-го поворотного изомера в объеме газа
и соответственно взаимодействующего с поверхностью твердого тела; тп — число
поворотных изомеров рассматриваемой молекулы.
232
Вводя выражения (VII,28) и (VII,29) в выражение (VI,30), полу-
чаем:
tfi=2^£i,i (VII,30)
i=l
где Kit 1 — константа Генри для адсорбции f-го поворотного изомера.
(VII,31)
\ /
а мольная доля i-ro изомера в объеме равновесного газа [25, 351
равна
-ь (VII,32)
1 Ng Qgi
Здесь N8 — общее число молекул в объеме V вдали от поверхности"; TVf —
число молекул, имеющих конформацию /-го поворотного изомера, в том же
объеме.
Приближение (VII, 30) было использовано при расчетах К± и дру-
гих термодинамических характеристик адсорбции для н-бутана,
н-пентана [17], н-гексана [36] и других углеводородов с поворотной
изомерией [37, 38]. Вводя выражение (VII,30) в уравнения (111,646),
(III,59в), (III,72в), получаем [17]
7П т
дг7?= (VII,33)
г=1 г=1
__ т ______* т , . । _
(VII>34)
i=l г=1
А£1 = У ха I У ха 2Т п ,
R А г R 1 Zj ’ \ dT h № Л
г-1 г«1
V4 ( ®х<1 A d 1П (К/\
+2(тА^2—(VIU5>
г-1
где соответствующие термодинамические характеристики адсорбции
i-го поворотного изомера
KUi^RT^^^ (VII,36)
см
Ц^- = 1п КЛ1+т‘Ц^Ь1-1 (VII,37)
£1 (М
^-= 2Т d }~l—+ Т* -2 1 (VII,38)
ti ai alл
233
а средняя мольная доля i-ro изомера в адсорбированном состоянии
xa = Kl^x8i _Г;
г т р
h=l
(VII,39)
причем — величина адсорбции i-ro изомера; Г — общая величина
адсорбции данного адсорбата.
Мольные доли xf могут быть оценены по формуле [25]
(V.I.40)
1+ 2 ехр(—ДТ^/ЯГ)
1=2
где Д-Fz — разность свободных энергий в газе для i-ro вращательного^изомера
и наиболее устойчивого из этих изомеров (транс-изомера).
4. Выражения для квазижестких молекул
Квазижесткими называются многоатомные молекулы, не способ-
ные к внутренним вращениям и внутренним перегруппировкам.
Для таких молекул в выражении (VII,21) W = 0, а детерминант
[sfti] является константой. В случае адсорбции трехмерной квази-
жесткой молекулы выражение (VII,21) сводится к формуле:
*1 = 8?iM[eXP(
кт) J
sin О dx dy dz dft dq> dip
(VII,41)
Ориентация в пространстве линейных молекул определяется только
двумя углами й и ср. В этом случае
f[eXP (
sin Ф dx dy dz dft d<p
(VI 1,42)
Наконец, для адсорбции одноатомных молекул
dx dy dz
(VII,43)
(VII,44)
(VII,45)
(VII,46)
Для квазижестких молекул интеграл (VII,27) 7?г = 1, а инте-
гралы Т?2 = 1?3 = 0. Выражения (VII,22)—(VII,24) для термодина-
мических характеристик адсорбции квазижесткой молекулы прини-
мают поэтому следующий простой вид:
Д£1
RT 51
ДС1 5з Si
R “ 5i S2
234
где
Sl==S(^)1 lexp d9’ (Z==2,3) (Vll/8)
Здесь dq — соответствующий элемент фазового пространства, т , в.
для трехмерных молекул dq — sin ftdxdydzd'&dqdty, для линщн ых
молекул dq = sin ftdxdydzdftdq и для одноатомных молекул jg =
= dxdydz.
5. Выражения для разных моделей состояния
адсорбированных квазижестких молекул
Свободное движение вдоль математически однородной поверх-
ности. Модели полностью свободного движения адсорбированных
молекул вдоль поверхности, т. е. модели нелокализованной а^со^рб-
ции на математически однородной поверхности (см. гл. I), соответ-
ствует допущение, что потенциальная функция Ф не зависит от^^ор-
цинат х и у центра масс молекулы и угла <р, определяющего вра^е^ие
молекулы вокруг оси, перпендикулярной поверхности. Эта Нодуль
была широко использована при статистическом анализе опцтн1ых
данных по адсорбции на графитированной саже (на базисной ф^ни
графита) при нулевых [4, 13—18, 39—51] и малых [5, 47, 52—J65]
заполнениях поверхности. Физическим обоснованием этой лод^ли
является слабая зависимость от координат х и у потенциальных
энергий Ф межмолекулярного взаимодействия одноатомных мо^НУЛ
с базисной гранью графита, полученных путем суммирования цотг-ен-
циальных функций <р межмолекулярного взаимодействия молеКу-*лы
адсорбата с атомом углерода графита по всем атомам углерода ц0>лу-
бесконечной решетки графита [66—70].
При допущении, что функция Ф не зависит от координат у
и (р, для адсорбции трехмерной, линейной и одноатомной метнул
получаем соответственно:
A'i = J |ехр — -Ф — 1| s*n dz
Ki = -yJ|exp — Ф J -1} sin ft dz d$
K1 = J{exp[-^]-l}dz
fru *’49)
fruJ*51)
В этом приближении в выражениях (VII,47) и (VII,48) dq$ “
sin ftdzdftdty для трехмерных, dq = sin ftdzd'ft для лшей^ых
и dq --- dz для одноатомных молекул.
Свободное движение вдоль математически однородной поверх-
ности и гармонические колебания перпендикулярно повер^ое^™*
Этой модели состояния адсорбированных молекул соответсгв-8ует
235
допущение о том, что потенциальная энергия Ф не зависит от коор-
динат х, у и ср молекулы, а зависимость Ф от z дается разложением
Ф в ряд Тэйлора по степеням z — zot в котором учтены только
первые два члена:
ф=Фо+(2 - 2о)а (VII,52;
Здесь Фо и Ф2 представляют значения Ф и ее второй производной по
z при равновесном расстоянии zo- Значения Фо, Ф« и zo зависят только
от ориентации молекулы, т. е. от углов А и Это приближение часто
применяется при статистическом анализе опытных данных по адсорб-
ции на графите [42—44, 48—50]. В работах [14—18] это приближе-
ние было использовано при расчетах термодинамических характери-
стик адсорбции для ряда квазижестких молекул, а также для инди-
видуальных поворотных изомеров.
Вводя выражение (VII,52) в уравнения (VII,49)—(VII,51), пре-
небрегая единицей в подынтегральных функциях и производя анали-
тическое интегрирование по z (в пределах от —оо до -|-оо), получаем
соответственно для адсорбции трехмерной, линейной и одноатомной
молекул:
J к~ФГ/ ехр\“ £yjsinfldfldi|j (VII,53)
„ 1 P (2пкТ V/г / Фо\ . ала, ,,,тт
(-фг) exp(-^sinOdO (VII.54)
= / (VII 551
кт) \2лМкт) \hvj кТ)
где частота гармонических колебаний центра масс молекулы перпен-
дикулярна поверхности
1 /Ф^Ч'/г
Vz~ 2п\М )
(¥11,56)
Конфигурационные интегралы St (VII,47) и (VII,48) в этом случае
выражаются формулами: 51= Рх (VII,57) Sa=P2+vX (VII,58)
(¥11,59)
где соответствующие интегралы Pt (7 = 1, 2, 3) для трехмерных
молекул
КФоУ’1 / 2якТ\Чъ / Фо\ . алал лптйл\
ЛГ/ \“фГ/ ехр \ kf) smfldfl^ (VII,60)
для линейных молекул
„ (* /ФоМ"1 /* ( Ф0\ • а лА
Pl = \ ( —2 •) ( ) ехр ( — ) sin й dfl
J \кТ/ кФ*/ Д '
(VII,61)
236
и для одноатомных молекул
(VII,62)
Вводя выражения (VII,55), (VII,57)—(VII,59), (VII,62) в уравне-
ния (VII,44)—(VII,46), получаем следующие выражения для термо-
динамических характеристик адсорбции одноатомных молекул при
нулевом заполнении математически однородной поверхности:
Д^ = ЛГЛФо + ^
л =1П\ ф; / 2
ДС1_/1
Я “ 2
(VII,63)
(VI 1,64)
(VII,65)
6. Квантовые поправки
Если адсорбционная система отклоняется от классической, но
не сильно, то к классическим статистическим выражениям для кон-
станты Генри Кг и других термодинамических характеристик адсорб-
ции необходимо еще добавить соответствующие квантовые поправки.
Эти поправки можно получить, заменяя множитель Больцмана в клас-
сических выражениях суммой Слетера [45, 71, 72] или используя
приближение Питцера и Гвина [73]. Согласно последнему приближе-
нию статистическую сумму адсорбированной молекулы можно запи-
сать в виде [10, 11, 73, 74]:
п п Е@гарм. осц. квант
V = ¥КласН=)---------л
VrapM. осц. класз
где] <2клас — статистическая сумма адсорбированной молекулы в классическом
приближении; фгарм, осц. квант и фгарм, осц, клас статистические суммы для
квантуемых степеней свободы адсорбированной молекулы в квантовом и клас-
сическом приближениях, рассчитанные с использованием гармонического при-
ближения (VII,52).
При справедливости приближения (VII,66)
(VII,66)
К1=Я1»клас* *• (VII,67)
1 ДО ^1, клас — константа Генри в классическом приближении; v** *• квантово-
механический фактор [74], выражаемый формулой
v *• — @гаРм- °сц> «вантП hvr « ехр (—hvr/2kT) ™
(?гарм, осц« клас kT 1 — ехр ( JiVr/kT}
Здесь — частота гармонических колебаний для r-ой степени сво-
боды движения адсорбированной молекулы
1
Vr=^\—r)
(VII,69)
237
где Ф£ — вторая производная потенциальной энергии Ф молекулы по отноше-
нию к r-ой координате молекулы вблизи потенциального минимума; величина 1Г
равна массе молекулы в случае трансляционных или колебательных степеней
свободы движения центра масс адсорбированной молекулы перпендикулярно
и вдоль поверхности адсорбента и соответствующему центральному моменту
инерции молекулы в случае вращательных степеней свободы.
При справедливости приближения (VII,66) квантовомеханически^
поправки AC7f*, A5J* и ACJ* соответственно для AZ7X, А££ и
даются выражениями [75]:
Л гт** №dlnv**
=--------dT---
(VII,70)
Д5?* = R fin v**+ Т d
\ аТ
(VII,71)
(VII,72)
\ их Щ А
Если система сильно отклоняется от классической, то Кг необходимо
вычислить по квантовостатистической формуле с учетом энергети-
ческих уровней адсорбированной молекулы (см., например, [76, 77]).
В случае адсорбции одноатомных молекул при предположении, что
колебания молекулы перпендикулярно поверхности являются гармо-
ническими, а движение вдоль поверхности свободным [8, 42, 44, 49]
(VII,73)
(VII, 74)
--- RT NAhv г /j>v \
Д£/1 = ЛГаФ0-^- + -^- + ЛГа^ |_ехр (g)-l
*£.= 1П ( hZ Х/г 3 -I- hv!kT In Tl-exp ГЗД
R \2nMkT) 2 ' exp (/iv/Zc?)—1 L \кТ)_
_ (VII,75)
= _ _L J- (fev/fer)a exp (fov/feT1)
R 2 ' [exp(Jiv//cr)-l]2 kvn,/o)
При T oo выражения (VII,73)—(VII,76) переходят в выражения
(VII,55), (VII,63)—(VII,65).
Практически, квантовые расчеты константы Кг или квантовые
поправки к классическим значениям А\>клас необходимы только
для адсорбции нескольких легких молекул при низких температурах.
Классическая статистическая механика в хорошем приближении
описывает большинство систем с физической адсорбцией в области
температур, в которой обычно производятся измерения.
ЛИТЕРАТУРА
1. Steele W. A.,Ross М. J. Chem. Phys., I960, v. 33, р. 464—470.
2. С т и л У. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ,
ред. Э. Флад. М., «Мир», 1970. 433 с.
3. Freeman М. Р., Halsey G. D. J. Phys. Chem., 1955, v. 59, p. 181 —
184.
233
4. Freeman M. P. J. Phys. Chem., 1958, v. 62, p. 723—728.
5. BarkerJ. A.,Everett D. H. Trans. Faraday Soc., 1962, v. 58, p. 16Q8—
1628.
6. Хи л л T. Л. Статистическая механика M., Издатинлит, 1960. 486 с.
7. S t е е 1 е W. A., Ross М. J. Chem. Phys., 1961, v. 35, р. 850—862.
8. St е е 1 е W. A. Advanc. Chem. Ser., 1961, v. 33, p. 269—280.
9. H i 11 T. L. J. Chem. Phys., 1946, v. 14, p. 44t—453.
10. H i 11 T. L. J. Chem. Phys., 1948, v. 16, p. 181—189.
11. D r en an J. W., H ill T. L. J. Chem. Phys., 1949, v. 17, p. 775—781.
12. В a r r e r R. M., S tuart W. J. J. Chem. Soc., 1956, p. 3307—3311.
13. П о ш к у с Д. П. ЖФХ, 1965, т. 39, с. 2962—2967.
14. ПошкусД. П., Афреймовия А. Я. ЖФХ, 1968, т. 42, с. 1201—*
1205.
15. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1968, т. 42, с. 2546—2552.
16. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1968, т. 42, с. 2553—2555.
17. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1970, т. 44, с. 981—986.
18. П о ш к у с Д. П. В кн.: Основные проблемы теории физической адсорбции.
Под ред. М. М. Дубинина и В. В. Серпинского. М., «Наука», 1970. 475 с.
19. Г о д н е в И. Н. Вычисление термодинамических функций по молекуляр-
ным данным. М., Гостехтеоретиздат, 1956. 419 в
20. Л андауЛ. Д., ЛифшицЕ. М. Механика. М., Физматгиз, 1958, 206 с.
21. Лойцянский Л. Г., Лурье А. И. Курс теоретической механики.
М., Гостехиздат, 1955, I ч. 380 с.; II я. 595 с.
22. Бухгольц Н. Н. Основной курс теоретической механики. М., «Наука»,
1972. I ч. 467 с.; II я. 329 с.
23. Б л о х и н ц е в Д. И. Основы квантовой механики. М., Гостехиздат, 1949.
588 с.
24. М и д з у с и м а С. Строение молекул и внутреннее вращение М., Издатинлит,
1957. 263 с.
25. Волькенштейн М. В. Конфигурационная статистика полимерных
цепей. М.—Л., Изд-во АН СССР, 1959. 466 с.
26. Б и р ш т е й н Т. М., Птицын О. Б. Конформации макромолекул. М.,
«Наука», 1964. 391 с.
27. Р i t z е г К. S. J. Chem. Phys., 1946, v. 14, р. 239—243.
28. Kilpatrick J. E., P i t z e г К. S. J. Chem. Phys., 1949, v. 17, p. 1064—*
1075.
29. В a r t e 11 L. S., К о h 1 D. A. J. Chem. Phys., 1963, v. 39, p. 3097—3105.
30. S h e p p a r d N., S z a s z G. J. J. Chem. Phys., 1949, v. 17, p. 86—97.
31. Scott R. A., Scheraga H. A. J. Chem. Phys., 1966, v. 42, p. 2209—*
2215.
32. Волькенштейн M. В. ДАН СССР, 1945, т. 49, с. 111—115 ; 1951, т. 78,
с. 879—882.
33. Волькенштейн М. В. ЖФХ, 1952, т. 26, с. 1072—1083.
34. Г о д н е в И. Н., Морозов В. ЖФХ, 1947, т. 21, с. 799—809.
35. Hill Т. L. J. Chem. Phys., 1961, v. 35, р. 303—305.
36. Poshkus D. P., Afreimovich A. Ya. J. Chromatog., 1971, v. 58,
p. 55—59.
37. Kiselev A. V., Poshkus D. P. J. Chem. Soc. Faraday Trans. II,
1975, v. 71.
38. Kiselev A. V., P о s h k u s D. P., J. Chem. Soc. Faraday Trans. II,
19 75, v. 75.
39. Constabaris G.,Halsey G. D. J. Phys. Chem., 1957, v. 27, p. 1433—
1434.
40. S a m s J. R., Constabaris G., H a 1 s e у G. D. J. Phys. Chem., 1960,
v. 64, p. 1689—1696.
41. Constabaris G., S a m s J. R., H a 1 s e у G. D. J. Phys. Chem., 1961,
v. 65, p. 367—369.
239
42. Ross S., Olivieri. P. Advanc. Chem. Ser., 1961, v. 33, p. 301—308.
43. R oss S., Oli vier J. P. J. Phys. Chem., 1961, v. 65, p. 608—615
44. О 1 i v i e г J. P., R о s s S. Proc. Roy. Soc., 1962, A 265, p. 447—454.
45. Y a r i s R., Sams J. R. J. Chem. Phys., 1962, v. 37, p. 571—576.
46. Sams J. R„ Yaris R. J. Phys. Chem., 1963, v. 67, p. 1931—1933.
47. W о If e R., S a m s J. R. J. Phys. Chem., 1965, v. 69, p. 1129—1135.
48. Myers A. L.,P rausn itz J. M. Trans. Faraday Soc., 1965, v. 61, p. 755—
764.
49. HooryS. E.,PrausnitzJ.M. Trans. Faraday Soc., 1967, v. 63, p/455—
460.
50. Pierotti R. A. Chem. Phys. Letters, 1968, v. 2, p. 420—422.
51. Mac Rury T. В. J. Chem. Phys., 1968, v. 49, p. 1543—1545.
52. S a m s J. R., ConstabarisG., Halsey G. D. J. Chem. Phys., 1962,
v. 36, p. 1334-1339.
53. Krizan J. E., Crowell A. D. J. Chem. Phys., 1964, v. 41, p. 1322—
1326.
54. Krizan J. E. J. Chem. Phys., 1965, v. 42, p. 2923—2927.
55. Sams J. R. J. Chem. Phys., 1965, v. 43, p. 2243—2250.
56. Mac Rury T. B., Bird C., Sams J. R. Molec. Phys., 1969, v. 17,
p. 91-100.
57. J ohnsonJ. D.,Klein M. L. Trans. Faraday Soc., 1964, v. 60, p. 1964—
1972.
58. F r e e m a n M. P. J. Phys. Chem., 1958, v. 62, p. 729—732.
59. Sams J. R. J. Chem. Phys., 1962, v. 37, p. 1883—1884.
60. S a m s J. R. Molec. Phys., 1965, v. 9, p. 17—27.
61. Sams J. R. Molec. Phys., 1965, v. 9, p. 195—196.
62. Everett D. H. Disc. Faraday Soc., 1965, v. 40, p. 177—187.
63. W о If e R., S a ms J. R. J. Chem. Phys., 1966, v. 44, p. 2181—2188.
64. J ohnson J. D., К lei n M. L. Trans. Faraday Soc., 1967, v. 63, p. 1269—
1273.
65. Mac Rury T. B., Sams J. R. J. Chem. Phys., 1969, v. 51, p. 1302—
1310.
66. Crowell A. D.,YoungD.M. Trans. Faraday Soc., 1953, v. 49, p. 1080—
1085.
67. С г о w e 11 A. D. J. Chem. Phys., 1954, v. 22, p. 1397—1399.
68. P ас e E. L. J. Chem. Phys., 1957, v. 27, p. 1341—1346.
69. P a с e E. L. Proc. 4th Conf. Carbon, Buffalo, New York, 1960. 780 p.
70. Steele W. A.,KarlR. J. Col. Interface Sci., 1968, v. 28, p. 397—402.
71. Ги ршфельдер Дж., Кертис Ч., Берд Р. Молекулярная теория
газо# и жидкостей. М., Издатинлит, 1961. 929 с.
72. F г е eman М. Р. J. Phys. Chem., 1960, v. 64. р. 32—37.
73. Р it z er К. S., Gwinn W. D. I. Chem. Phys., 1942, v. 10, p. 428—440.
74. P о shkus D. P. Disc. Faraday Soc., 1965, v. 40, p. 195—204-
75. К и с e л e в А. В., П о ш к у с Д. П. ЖФХ, 1965, т. 39, с. 398—402.
76. W hite D., Lassettre Е. N. J. Chem. Phys., 1960, v. 32, p. 72—84. -
77. R icca F., Pisani C., Garrone В. J. Chem. Phys., 1969, v. 51, p. -
4079-4091.
Глава VIII
МЕЖМОЛЕКУЛЯРНЫЕ СИЛЫ И ПОТЕНЦИАЛЬНАЯ ФУНКЦИЯ
МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ МОЛЕКУЛЫ
С АДСОРБЕНТОМ
1. Общая характеристика проблемы
Как следует из молекулярно-статистических выражений, приве-
денных в предыдущих главах, термодинамические характеристики
адсорбции и газовой хроматографии при предельно низком (нулевом)
заполнении поверхности определяются потенциальной функцией Ф
взаимодействия молекулы с адсорбентом. Поэтому для интерпрета-
ции экспериментальных адсорбционных и хроматографических дан-
ных на молекулярном уровне необходимо определение потенциальной
функции Ф. Зная эту функцию, в принципе, можно рассчитать все
интересующие нас равновесные свойства адсорбционной системы при
нулевом заполнении поверхности. Поэтому разработка методов опре-
деления Ф составляет также важный этап развития методов предска-
зания (вычисления) свойств интересующих нас адсорбционных систем.
Взаимодействие молекулы с твердым телом при молекулярной
(физической) адсорбции представляет частный случай проявления
межмолекулярных (ван-дер-ваальсовых) сил. В случае физической
адсорбции основными силами взаимодействия молекулы с твердым
телом являются те же дисперсионные, индукционные и ориентацион-
ные силы притяжения и силы отталкивания, что и при взаимодей-
ствии молекул в газе, жидкости и молекулярных кристаллах. По-
этому основные принципы, разработанные для определения потен-
циальной функции межмолекулярного взаимодействия, применимы
и при определении Ф.
Потенциальная функция межмолекулярного взаимодействия в ос-
новном определяется двумя путями: 1) прямыми квантовомеханиче-
скими расчетами и 2) из изучения макроскопических свойств, зави-
сящих от межмолекулярных сил.
Хотя природа межмолекулярных взаимодействий ясна, количе-
ственные расчеты их энергии представляют значительные трудности.
Квантовомеханические расчеты на основании фундаментальных
констант и уравнения Шредингера пока возможны практически
только для простейших систем, содержащих лишь немного электро-
нов. Этим путем получены хорошие результаты при расстояниях,
близких к равновесному, только для взаимодействия двух атомов Н
[1]. Поэтому для определения Ф первый путь пока практически не-
применим.
16 Заказ 1180 241
Большая часть количественной информации о потенциале сил меж-
молекулярного взаимодействия получена путем анализа макроскопи-
ческих данных. Для определения потенциала межмолекулярных сил
практически пригодны только те зависящие от этих сил макроскопи-
ческие свойства, которые удовлетворяют следующим требованиям:
1) существует строгая статистикомеханическая теория, связывающая
межмолекулярные силы и макроскопические свойства; 2) связь между
макроскопическими свойствами и потенциалом межмолекулярных
сил не слишком сложна; 3) макроскопические свойства измеряются
с достаточной точностью [1]. Этим требованиям удовлетворяют опыт-
ные данные по адсорбции из газовой фазы на адсорбентах с однород-
ной поверхностью при низких ее заполнениях. Поэтому для опреде-
ления Ф, в принципе, могут быть использованы макроскопические
свойства адсорбционных систем.
Основными экспериментальными термодинамическими данными,
получаемыми независимыми измерениями, на основании которых
можно определить Ф, являются: а) изотерма адсорбции; б) удержи-
ваемый объем; в) теплота адсорбции; г) теплоемкость адсорбционной
системы. Из этих экспериментальных величин в благоприятных
условиях, т. е. при подходящем выборе температуры, часто наиболее
точно измеряются удерживаемые объемы при малой (нулевой) вели-
чине пробы, т. е. константа Генри и изотермы адсорбции. Связь
между Ф и экспериментально измеряемыми термодинамическими
характеристиками адсорбции наиболее проста в случае константы
Генри. Поэтому определение Ф удобнее всего производить, исполь-
зуя удерживаемые объемы и изотермы адсорбции. Остальные экспе-
риментальные величины могут служить для проверки справедливости
определенной из изотермы адсорбции потенциальной функции Ф.
Однако однозначно определить потенциал сил межмолекулярного
взаимодействия из макроскопических свойств системы нельзя прак-
тически [1]. Поэтому при анализе макроскопических свойств, в общем,
необходимо прибегать к математическим моделям этого потенциала,
а их параметры определять, сравнивая рассчитанные макроскопи-
ческие свойства с экспериментом.
При некоторых приближениях можно получить упрощенную
теорию межмолекулярных сил, которая хотя и не является количе-
ственно точной, но дает общую форму потенциала межмолекуляр-
ного взаимодействия. Поэтому при выборе математической модели
потенциала межмолекулярного взаимодействия необходимо исполь-
зовать общие теоретические соображения.
Термодинамические характеристики адсорбции определяются по-
тенциальной функцией Ф вблизи ее минимума. При таких расстоя-
ниях потенциальная энергия Ф межмолекулярного взаимодействия
молекулы с поверхностью твердого тела зависит не только от расстоя-
ния центра масс молекулы от поверхности, но из-за атомного строе-
ния твердого тела зависит также и от положения центра масс моле-
кулы над поверхностью (над физически однородной поверхностью,
см. разд. 1 гл. I). Чтобы учесть эту последнюю зависимость при
242
выборе модели потенциальной функции (потенциала) Ф, как правило,
принимается, что этот потенциал равен сумме потенциалов ф меж-
молекулярного взаимодействия молекулы с силовыми центрами
(атомами, группами атомов, ионами) твердого тела, т. е.
0=2^ (VIII,1)
В случае адсорбции сложных молекул потенциал Ф зависит не только
от положения центра масс молекулы, но также и от ориентации моле-
кулы по отношению к поверхности. Для правильного молекулярно-
статистического описания адсорбции сложных молекул необходимо
учесть обе эти зависимости [2—15] (см. гл. VII).
Необходимость учета зависимости потенциала Ф межмолекуляр-
ного взаимодействия молекулы с адсорбентом от ориентации молекулы
по отношению к поверхности создает значительные трудности при
выборе модели потенциала. Вместе с тем сильная зависимость Ф от
ориентации молекулы может быть использована для эмпирического
изучения зависимости межмолекулярных взаимодействий от ориен-
тации.
Квантовомеханические расчеты энергии межмолекулярного взаи-
модействия с учетом зависимости этой энергии от ориентации взаимо-
действующих систем проводились только для простейших случаев:
для взаимодействия атома с двухатомной молекулой [16—19] и для
взаимодействия двух двухатомных молекул [18—26]. Для учета
анизотропии дисперсионного взаимодействия некоторые сложные
молекулы рассматривались как асимметрические трехмерные осцилля-
торы [27—31]. Выбор формы потенциала Ф для взаимодействия слож-
ных молекул с адсорбентом на основании таких расчетов, по-види-
мому, пока практически невозможен. Вместе с тем определение зави-
симости потенциальной энергии Ф межмолекулярного взаимодей-
ствия сложной молекулы с адсорбентом от ее ориентации у поверх-
ности на основании экспериментальных адсорбционных данных
также практически невозможно из-за недостаточной чувствительности
термодинамических характеристик адсорбции к модели этой зависи-
мости. Кроме того, такие эмпирические определения формы потен-
циала Ф необходимо было бы проводить для каждой интересующей
нас системы в отдельности. Вместе с тем модели потенциалов, учиты-
вающие зависимость межмолекулярного взаимодействия от ориента-
ции молекул, применяемые при расчетах свойств разреженных газов,
по-видимому, не годятся для расчетов свойств адсорбционных систем,
так как адсорбционные свойства более чувствительны к геометри-
ческому строению молекулы, чем свойства объемных газов.
Потенциальную функцию Ф межмолекулярного взаимодействия
сложной молекулы с поверхностью адсорбента, учитывающую зависи-
мость энергии взаимодействия от ориентации молекулы, в первом
приближении, по-видимому, можно получить, представляя Ф в виде
суммы потенциальных функций ф межмолекулярного взаимодей-
16* 243
ствия силовых центров молекулы (атомов, атомных групп или валент-
ных связей) с силовыми центрами твердого тела, т. е. считая, что
ф=22ф (vin,2>
Для потенциала ф взаимодействия силовых центров молекулы и твер-
дого тела обычно принимается та же модель, что и для взаимодей-
ствия двух простых молекул в газе. При современном состоянии
теории межмолекулярных сил использование приближения (VIII,2),
по-видимому, дает пока единственную возможность какого-либо
прогресса при определений потенциальных функций Ф взаимодей-
ствия сложных молекул с учетом зависимости Ф от их ориентации.
Отдельные классы сложных молекул, в частности, углеводороды,
состоят только иэ немногих фрагментов, которые удобно принять за
силовые центры межмолекулярного взаимодействия. Молекулы од-
ного класса различаются числом этих силовых центров, их химиче-
ским (валентным) состоянием и их пространственным расположением.
Используя экспериментальные адсорбционные данные для сравни-
тельно немногих молекул, в принципе, можно определять потенциалы
Ф межмолекулярного взаимодействия для всех интересующих нас
пар силовых центров. Полученные так потенциалы ф далее могут
быть использованы для определения потенциальных функций Ф
взаимодействия любых других молекул, состоящих из тех же сило-
вых центров. Поэтому таким путем можно произвести расчет адсорб-
ционных свойств для таких систем, для которых нет эксперимен-
тальных данных, или таких характеристик адсорбции, измерения
которых представляют большие трудности (сюда относятся, например,
теплоемкость адсорбированных молекул при нулевом и низких за-
полнениях поверхности, величины адсорбции и равновесного давле-
ния при слишком высоких или слишком низких температурах для
непосредственного измерения, а также медленно выделяющиеся теп-
лоты адсорбции).
Используя приближение (VIII,2), термодинамические характери-
стики адсорбции при низких заполнениях поверхности можно свя-
зать с химическим и геометрическим строением молекулы адсорбата
и решетки адсорбента и, следовательно, произвести обоснованное
сопоставление адсорбционных свойств молекул, состоящих из тех же
или разных силовых центров, выявить влияние особенностей геоме-
трической и электронной структуры молекулы на ее адсорбцию. Нако-
нец, такие расчеты могут быть использованы для определения или
уточнения конформации молекулы на основании адсорбционных
и хроматографических данных [32] или для определения ее измене-
ния при адсорбции. Поэтому применение и развитие вышеуказанного
подхода в молекулярной теории адсорбции представляет значитель-
ный интерес как для самой теории адсорбции, так и для теории меж-
молекулярных взаимодействий вообще.
Значительные трудности представляет определение параметров
для выбранной модели межмолекулярного потенциала. Если потен-
244
циал содержит только несколько (один, два) параметров, то их
можно определить, сравнивая рассчитанные макроскопические свой-
ства с экспериментом. Полученные таким путем потенциальные функ-
ции далее могут быть использованы для предсказания (вычисления)
других свойств тех же систем, для которых они были определены.
Однако такой чисто эмпирический способ определения параметров
потенциальной функции имеет некоторые недостатки. Во-первых,
согласно этому способу определение параметров необходимо произ-
водить для каждой интересующей нас системы в отдельности. Во-вто-
рых, эмпирическое определение параметров потенциала взаимодей-
ствия в случае сложных систем встречает значительные трудности
из-за большого числа требующих учета параметров [33]. Поэтому
при использовании опытных адсорбционных данных параметры по-
тенциала Ф были определены только в случае адсорбции одноатом-
ных или квазиодноатомных молекул [34—44].
Приближенная теория межмолекулярных сил дает правила ком-
бинирования для входящих в потенциалы взаимодействия параметров
сил притяжения и сил отталкивания [1, 45—51]. С помощью этих
правил комбинирования параметры потенциала взаимодействия раз-
ных силовых центров могут быть оценены из параметров потенциалов
взаимодействия одинаковых силовых центров. Поэтому параметры
потенциальной функции Ф могут быть оценены с по-
мощью таких правил комбинирования независимо от эксперименталь-
ных адсорбционных данных при использовании параметров потен-
циальных функций межмолекулярного взаимодействия силовых цен-
тров адсорбата и силовых центров адсорбента, взятых в отдельности
[52]. Этим путем были получены потенциалы Ф взаимодействия
некоторых одноатомных и квазиодноатомных молекул с решетками
графита [45, 52—58], нитрида бора [59] и инертных газов [60—65].
Однако правила комбинирования дают только приближенные значе-
ния этих параметров [45]. Кроме того, для применения этого способа
сначала надо определить параметры потенциалов межмолекулярного
взаимодействия силовых центров адсорбата между собой и потенциа-
лов межмолекулярного взаимодействия силовых центров адсорбента
между собой, что само по себе часто затруднительно. Поэтому прак-
тическое применение этого способа, в общем, встречает значительные
трудности, а точность определенных этим способом параметров недо-
статочна для использования найденной таким способом функции Ф
для статистических расчетов термодинамических характеристик
адсорбции.
При достаточно больших расстояниях между взаимодейству-
ющими атомными системами энергию межмолекулярного притяжения
(энергию ориентационного, индукционного и дисперсионного притя-
жения) можно связать с экспериментально измеряемыми свойствами
индивидуальных атомных систем, такими как электрические моменты,
поляризуемости, потенциалы ионизации. Поэтому параметры сил
притяжения можно оценить полуэмпирическим путем, вводя в соот-
ветствующие квантовомеханические формулы необходимые экспери-
245
ментально определенные свойства этих систем. Этот способ определе-
ния параметров сил .притяжения использовался/особенно широко
при оценках параметров потенциальных функций Ф межмолекуляр-
ного взаимодействия молекул с графитом [7, 8, 38, 66—94], нитридом
бора [95, 96], ионными кристаллами [97—110], цеолитами [111—130]
и другими твердыми телами [131—135]. Этим способом были оценены
параметры потенциалов Ф взаимодействия как простых [7, 8, 38, 66,-
69, 72, 74-79, 86-90, 95-97, 99-104,107-117, 119, 120, 123-127],
так и сложных [70, 71, 73, 75, 77, 80—85, 91—94, 96, 98, 105, 106,
118, 121, 122, 128—130, 134] молекул. Параметры сил отталкивания
при этом обычно оценивались при использовании условия равно-
весия сил притяжения и сил отталкивания при равновесном рассто-
янии и разные эмпирические или полуэмпирические правила
комбинирования равновесных расстояний и параметров сил оттал-
кивания.
Последний способ определения параметров, как и предыдущий,
позволяет оценить параметры потенциалов Ф независимым от экспе-
риментальных адсорбционных данных путем. Кроме того, примене-
ние последнего способа определения параметров проще первых двух
способов. Такой способ оценки Ф сравнительно легко применим
к любым системам адсорбат — адсорбент. Оцененные этим способом
значения потенциальной энергии Ф в минимуме обычно получаются
близкими к опытным значениям теплот адсорбции. Однако функции
Ф, полученные этим способом, недостаточно точны для проведения
более глубоких исследований термодинамических свойств разных
адсорбционных систем. Для получения более точных потенциальных
функций межмолекулярного взаимодействия простых и сложных
молекул с адсорбентом необходимо использовать экспериментальные
адсорбционные данные. При этом значения параметров потенциаль-
ной функции Ф, оцененные последними двумя способами, т. е.
независимо от адсорбционных данных, могут быть использованы
в качестве первого приближения и далее уточнены методом проб
и ошибок при сравнении с термодинамическими свойствами соответ-
ствующих адсорбционных систем. В работах Киселева, Пошкуса
и Афреймовича [12, 13, 136—139] был применен этот способ расчета
и предложен простой способ уточнения главного параметра сил при-
тяжения путем использования экспериментальных значений кон-
станты Генри определенных при разных температурах.
Использование в молекулярно-статистических расчетах полуэмпи-
рических и затем исправленных по экспериментальным данным по-
тенциальных функций Ф в настоящее время представляет наиболее
надежный путь, связывающий молекулярные параметры адсорбата
и адсорбента с измеряемыми термодинамическими свойствами адсорб-
ционных систем.
При расчетах потенциальной энергии Ф взаимодействия молекулы
с базисной гранью графита атомная решетка графита обычно рассма-
тривается как диэлектрик. В случае адсорбции неполярных молекул
принимается, что взаимодействие молекулы с графитом обусловлено
246
только дисперсионными силами притяжения и силами отталкивания
[7, 8, 38, 52—54, 56, 57, 66—79, 82, 83, 85-91, 93, 94]. В случае ад-
сорбции молекул, обладающих электрическими моментами (диполи,
квадруполи), к этим силам добавляются еще силы индукционного
взаимодействия [55, 58, 80, 81, 84, 92]. Фактически же графит содер-
жит электроны проводимости [140]. Поэтому, в принципе, необхо-
димо было бы учесть влияние этих электронов на межмолекулярное
взаимодействие [141, 142]. Однако вклад электронов проводимости
графита в энергию межмолекулярного взаимодействия, по-видимому,
мал. На это указывает, например, близость экспериментальных
и рассчитанных, пренебрегая вкладом подвижных электронов гра-
фита, теплот адсорбции на графите и на нитриде бора, несмотря на
сильное различие электрических свойств этих адсорбентов [52, 53,
59, 95, 142].
При расчетах потенциальных энергий Ф взаимодействия молекул
с базисной гранью графита принимается также, что напряжение
электростатического поля над поверхностью графита равно нулю
или близко к нулю. С этим предположением согласуется наблюда-
емая близость экспериментальных значений теплот адсорбции на
графитированных термических сажах при низком (нулевом) заполне-
нии поверхности для веществ с близкой энергией дисперсионного
взаимодействия, но с различным электронным строением, например
для аргона (не обладает постоянными электрическими моментами)
и азота (обладает значительным квадрупольным моментом), для
к-пентана (электрические моменты близки к нулю) и диэтилового
эфира (обладает значительным периферическим дипольным момен-
том) [143—145].
Экспериментальные определения теплот адсорбции ряда других
молекул с большими периферическими дипольными моментами при
малых заполнениях поверхности графитированной термической сажи
дали величины, близкие к теплотам адсорбции алканов, соответству-
ющих по геометрии и общей поляризуемости молекул [143—145].
Вместе с тем экспериментальные значения теплот адсорбции этих же
пар веществ на специфических адсорбентах (гидроксилированная
поверхность кремнезема, катионированные цеолиты) значительно
различаются: теплоты адсорбции молекул, обладающих электриче-
скими моментами, значительно выше теплот адсорбции соответству-
ющих неполярных молекул [135, 143—145].
Как экспериментальное доказательство наличия над поверхностью
графита сильного быстро убывающего электростатического поля при-
водилось ослабление межмолекулярного взаимодействия двух моле-
кул при адсорбции на графитированных сажах [146]. Однако ослабле-
ние межмолекулярного взаимодействия между адсорбированными
молекулами может быть также обусловлено эффектом трех тел при
адсорбции [146—150]. Поэтому наличие сильного электростатиче-
ского поля над базисной гранью графита экспериментально одно-
значно еще не доказано. Вместе с тем, из указанных выше экспери-
ментальных данных следует, что если и существует электростатиче-
247
ское иоле над базисной гранью графита, то его влияние на адсорбцион-
ное взаимодействие самых разных по строению молекул относи-
тельно слабо.
2. Квантовомеханические теории взаимодействия
молекулы с адсорбентом
Выражение для энергии ван-дер-ваальсового взаимодействия
молекулы с поверхностью твердого тела при больших расстояниях ъ
молекулы от поверхности можно получить как частный случай из
общих выражений макроскопической теории ван-дер-ваальсойого
взаимодействия двух тел, принимая, что одно из тел является сильно
разреженным [151, 152]. При ао 2 Хо (где ао — атомный радиус,
а Хо — длина волн, характерных для спектров поглощения молекулы
и твердого тела) из общих выражений макроскопической теории
ван-дер-ваальсовых взаимодействий вытекает следующее выражение
для потенциальной энергии ван-дер-ваальсового взаимодействия
молекулы с твердым телом [152]:
СО ->
ф = —— 'S’ ( g»ola. Г,£(гй))—1~1 j (viп 3)
Я*о + <*)2 Le(i'<o) + 1 J Ф 1 }
п о
где — разность энергий молекулы в основном и n-ом возбужденном состо-
яниях; q = —ri — оператор электрического диполя; 8 (гео) — диэлектри-
ческая проницаемость твердого тела, выраженная как функция мнимой ча-
стоты Кд.
Для потенциальной энергии ван-дер-ваальсового взаимодействия
молекулы с полубесконечной изотропной диэлектрической средой
при ао <С 2 Хо методом электрических изображений Мак-Лечлен
[148] получил выражение
. со
Ф = -7-Ч( «(ад) (VIII,4)
4nz3J v Le(f(o) + 1 J 7
о
где a (io) — электрическая поляризуемость молекулы, выраженная как функ-
ция мнимой частоты г со.
Выражение (VIII,4), полученное при помощи макроскопической
модели, можно получить также при помощи микроскопической мо-
дели, принимая, что молекула адсорбата и атомы, составляющие
твердое тело, состоят из изотропных гармонических осцилляторов
[153, 154].
Оцененная по формуле (VIII,4) потенциальная энергия взаимо-
действия атомов благородных газов с графитом заметно меньше опре-
деленной эмпирически потенциальной энергии взаимодействия этих
атомов с графитированной сажей [90].
Проблема ван-дер-ваальсового взаимодействия молекулы с по-
верхностью твердого тела рассматривалась также с использованием
248
квантовомеханической теории возмущений. Применяя теорию возму-
щений второго порядка, для энергии Ф притяжения между сфериче-
ской неполярной молекулой и плоской поверхностью любого полу-
бесконечного твердого тела Синаноглу и Питцер [147] получили
выражение:
оо
ф=-4«11<о|л|о>р-4сч^2|<о|;1,т>|2 <VIII,5)
ту=0
где ах — поляризуемость молекулы; и — средние энергии возбуждения
молекулы и твердого тела; — мгновенное электростатическое поле, создава-
емое всеми электронами и ядрами твердого тела в центре молекулы адсорбата;
т — обозначает различные энергетические уровни твердого тела; первый член
равен энергии поляризации молекулы электростатическим полем над поверх-
ностью; второй член соответствует энергии дисперсионного взаимодействия.
При выводе всех этих выражений для энергии взаимодействия
молекулы с поверхностью твердого тела принимается, что расстоя-
ние молекулы от поверхности значительно больше атомных размеров.
Поэтому в этих теориях фактически учитываются только силы притя-
жения ван-дер-ваальсового типа; силы отталкивания в них не учиты-
ваются. Кроме того, в этих теориях не учитывается атомная струк-
тура твердого тела: твердое тело рассматривается как однородная
непрерывная среда с математически плоской поверхностью. Однако,
как отмечалось выше, адсорбция определяется главным образом по-
тенциальной энергией Ф взаимодействия молекулы с поверхностью
вблизи потенциального минимума. При таких расстояниях необхо-
димо учесть атомное строение поверхности твердого тела и силы
отталкивания. Поэтому указанные выше теории не могут быть ис-
пользованы для определения потенциальной энергии Ф вблизи
потенциального минимума. При выборе формы потенциала Ф и оценке
его параметров вблизи потенциального минимума обычно исполь-
зуются результаты приближенных квантовомеханических теорий
ван-дер-ваальсового взаимодействия двух молекул вблизи равновес-
ного расстояния.
3. Аддитивность потенциала межмолекулярного
взаимодействия по потенциалам взаимодействия пар силовых
центров
Аддитивность потенциала межмолекулярного взаимодействия мно-
гоатомных систем по потенциалам взаимодействия пар силовых
центров этих систем, т. е. справедливость приближения (VIII,2),
широко исследовалась в случае взаимодействия двух молекул.
Рассматривая межмолекулярное взаимодействие многоатомных моле-
кул, необходимо различать молекулы, в которых ответственные за
межмолекулярное взаимодействие внешние электроны практически
локализованы на образующих молекулы атомах и на их валентных
связях, и такие молекулы, в которых имеются нелокализованные
электроны, распределенные по многим атомам. Примеры молекул
249
первого типа представляют молекулы насыщенных углеводородов,
электроны которых, образующие о-связи, ведут себя так, как если бы
они были почти полностью локализованными [155, 156]. Примеры
молекул второго типа представляют молекулы ненасыщенных и аро-
матических углеводородов с сопряженными л-связями. Образующие
эти связи электроны практически нелокализованы, т. е. практически
свободно движутся вдоль сопряженных связей.
Исследования аддитивности энергии межмолекулярного взаимо-
действия двух молекул с локализованными электронами по энергиям
парного взаимодействия их силовых центров производились в первом
и втором порядках теории возмущений. Потенциальная энергия взаи-
модействия двух молекул в первом порядке теории возмущений
содержит в себе энергию электростатического взаимодействия и об-
менную энергию первого порядка, обусловливающую энергию оттал-
кивания. Электростатические силы взаимодействия двух молекул
является локально аддитивными, т. е. электростатическую энергию
можно записать в виде двукратного интеграла по элементам объема
в одной молекуле и по элементам объема в другой молекуле [157].
При взаимодействии неполярных молекул электростатические силы
равны нулю и единственными не равными нулю силами в первом по-
рядке теории возмущений являются обменные силы первого порядка.
Встречающиеся в большинстве проблем межмолекулярного взаи-
модействия (в том числе и при физической адсорбции) межъядерные
расстояния соответствуют очень небольшим перекрываниям электрон-
ных облаков взаимодействующих систем [24]. Условия аддитивности
обменной энергии первого порядка Ио^мен межмолекулярного взаимо-
действия многоатомных неполярных молекул при небольших пере-
крываниях рассматривались с помощью теоремы Хельмана — Фей-
мана [24]. В том случае, когда электронные плотности взаимодей-
ствующих молекул хорошо описываются локализованными на атомах
орбиталями, энергия и$Мен аддитивна по энергиям взаимодействия
пар атомов. Если же электронная плотность рассматриваемых моле-
кул описывается молекулярными орбиталями, охватывающими всю
молекулу, то энергия «оУмен должна быть локально неаддитивной [24].
Таким образом, общий теоретический анализ сил отталкивания
указывает на то, что во многих случаях их энергия приближенно
может быть представлена в виде суммы энергий соответствующих
атом-атомных взаимодействий.
Числовые квантовомеханические расчеты энергии отталкивания
НоУмен производились главным образом для взаимодействия простей-
ших молекул: Не. . ,Н2 [16, 17], Н2. - -Н2 [20—22, 24]. Энергия
Ноумен в этих случаях оказалась неаддитивной по атомам молекулы
Н2. Эти расчеты указывают на более слабую зависимость потенциала
сил отталкивания от взаимной ориентации молекул, чем это следует
из атом-атомного приближения. Однако если учесть небольшое
(около 0,1 А) смещение центра сил отталкивания валентно связан-
ного атома водорода от его ядра, то потенциалы отталкивания атома
Не и молекулы Н2 или двух молекул Н2 в хорошем приближении
250
можно представить в виде суммы потенциалов сил отталкивания
валентно не связанных атомов [158]. Смещение центра сил отталки-
вания для более тяжелых атомов значительно меньше, чем для атома
водорода, и им можно пренебречь [158].
При достаточно больших расстояниях, когда электронные обо-
лочки взаимодействующих молекул существенно не перекрываются,
потенциальную энергию z?2) взаимодействия двух молекул во вто-
ром порядке теории возмущений можно представить в виде суммы
поляризационной и&ляр и дисперсионной и$сп энергий взаимодействия
[157, 159].
Общее рассмотрение потенциальной энергии п<2) взаимодействия
двух неперекрывающихся многоатомных молекул показало, что
энергии поляризационных и дисперсионных сил локально аддитивны.
Это значит, что эти энергии могут быть представлены в виде дву-
кратных сумм по локализованным областям в одной молекуле и лока-
лизованным областям в другой в том случае, если корреляция элек-
тронов в пределах каждой молекулы убывает достаточно быстро
с расстоянием [157].
Полуколичественное исследование локальной аддитивности энер-
гии Идисп для взаимодействия двух многоатомных насыщенных моле-
кул производилось при использовании модели Друде, т. е. молекулу
представляли в виде группы взаимодействующих друг с другом изо-
тропных трехмерный гармонических осцилляторов, заменяющих
силовые центры (атомы или атомные группы) молекулы [160—162].
Если взаимодействие между осцилляторами внутри молекулы силь-
ное, то отклонения от аддитивности диполь-дипольного дисперсион-
ного взаимодействия могут быть значительными, т. е. потенциальная
энергия межмолекулярного взаимодействия осцилляторов, входящих
в молекулы, значительно отличается от энергии взаимодействия
тех же изолированных осцилляторов [160, 161]. Знак и величина
отклонения от аддитивности зависят от расположения силовых цен-
тров внутри молекулы и от относительной ориентации молекул
[160, 162]. Однако если вместо значений поляризуемостей изолиро-
ванных силовых центров молекулы а использовать значения средних
поляризуемостей а этих силовых центров в рассматриваемых моле-
кулах, т. е. если учесть изменения а силовых центров молекулы
из-за взаимодействия внутри молекулы, то энергия дисперсионного
взаимодействия двух сложных молекул оказывается в хорошем при-
бл иж ении аддитивной [161].
Таким образом, теоретические исследования указывают на то,
что для двух многоатомных систем, в которых взаимное влияние
атомов убывает достаточно быстро с расстоянием от рассматриваемого
атома и электронные плотности хорошо описываются локализован-
ными орбиталями, энергия межмолекулярного взаимодействия при-
ближенно должна быть аддитивной по энергиям межмолекулярного
взаимодействия пар валентных связей, пар атомов или пар атомных
групп этих систем. На это свойство межмолекулярного взаимодей-
ствия указывает также аддитивность теплот сублимации кристалли-
251
ческих углеводородов по фрагментам молекул (см., например, [163]).
Потенциальную энергию взаимодействия таких молекул можно рас-
считать, используя приближение (VIII,2).
Исследования взаимодействия молекул с нелокализованными
электронами производились главным образом во втором порядке
теории возмущений. В случае взаимодействия ненасыщенных угле-
водородов в соответствии с двумя типами электронов, образующих
а- и л-связи, общую энергию дисперсионного взаимодействия ндисп
можно представить в виде суммы трех вкладов [29, 164—167]:
идисп ~ иДисп 4"*мдисп 4“идисп (VIII,6)
где иД^сп, м^неп и мДисп представляют собой доли общей энергии дисперсионного
взаимодействия, обусловленные соответственно взаимодействием а-электронов,
я-электронов и ст- и л-электронов разных молекул.
Энергия Мд’исп в случае ненасыщенных молекул может быть рас-
считана таким же путем, как и в случае насыщенных молекул, содер-
жащих только а-электроны, локализованные на атомах или связях
атомных размеров. В частности, она может быть представлена в виде
суммы взаимодействия отдельных а-связей [27, 29, 164, 165].
В молекулах углеводородов с сопряженными двойными связями,
как уже указывалось выше, л-электроны практически нелокализо-
ваны, т. е. они свободно движутся вдоль сопряженных двойных
связей. Корреляция л-электронов в пределах такой молекулы, по-
видимому, убывает медленно [157]. По этой причине молекулу с со-
пряженными связями, в принципе, нельзя представить как набор
небольших существенно не взаимодействующих фрагментов молекулы
и энергию дисперсионного взаимодействия таких молекул нельзя
рассматривать аддитивной по ее отдельным фрагментам [27, 29, 157,
165, 167, 168].
Согласно теоретическим исследованиям [29, 165] относительный
(на один электрон) вклад л-электронов сопряженных двойных связей
в поляризуемость и в общую энергию дисперсионного взаимодействия
таких молекул больше, чем а-электронов, из-за большей подвижности
л-электронов таких связей. Так, вклад л-электронов в среднюю поля-
ризуемость молекулы бензола составляет 40% [29], тогда как доля
л-электронов в общем числе валентных электронов молекулы бен-
зола составляет 20%. Следовательно, относительный (на один элек-
трон) вклад л-электронов в среднюю поляризуемость молекулы бен-
зола приблизительно в 3 раза больше, чем а-электронов. Однако число
л-электронов в полиенах и ароматических углеводородах по крайней
мере в 4 раза меньше числа а-электронов, что значительно снижает
роль л-электронов в общей энергии дисперсионного взаимодействия
молекул с сопряженными связями и роль неаддитивности
Оцененный теоретически вклад л-электронов в энергию дисперсион-
ного взаимодействия молекул бензола в кристаллической решетке
составляет только около 40% от общей энергии дисперсионного
взаимодействия [29].
252
Доля йдисп от общей энергии кдисп зависит от числа сопряженных
двойных связей в молекуле [164, 168] и расстояния между молеку-
лами [164, 165]: с увеличением числа сопряженных двойных связей
и расстояния между молекулами доля растет. Однако и при этом
доля wjlcn не должна быть особенно большой [165]. Энергия мДисп
мало изучена [29, 164, 167].
Таким образом, согласно теоретическим исследованиям энергия
взаимодействия молекул ненасыщенных и ароматических углеводо-
родов с сопряженными л-связями неаддитивна по энергиям взаимо-
действия пар фрагментов таких молекул главным образом из-за
неаддитивности энергии взаимодействия делокализованных л-элек-
тронов. Однако отклонения от аддитивности энергии взаимодействия
таких молекул, по^видимому, невелики из-за сравнительно неболь-
шого вклада энергии взаимодействия л-электронов в общую энергию
вз аимодействия.
Средняя поляризуемость многоядерных ароматических молекул
с точностью около 5% может быть представлена в виде суммы сред-
них поляризуемостей их атомных групп [169]. Это является веским
указанием на возможность представления энергии дисперсионного
взаимодействия ароматических молекул в виде суммы потенциалов
межмолекулярного взаимодействия атомных групп. В соответствии
с этим эмпирически было установлено, что допущение о парной
аддитивности потенциала взаимодействия ароматических молекул
по валентно несвязанным атомам выполняется удовлетворительно,
причем эмпирические потенциалы взаимодействия валентно несвя-
занных атомов С и Н ароматических молекул практически совпадают
с соответствующими потенциалами для взаимодействия парафиновых
углеводородов [170—174].
Таким образом, энергию взаимодействия молекул ненасыщенных
углеводородов с сопряженными двойными связями и молекул арома-
тических углеводородов в первом приближении, по-видимому, также
можно рассматривать аддитивной по атомам или атомным группам
таких молекул.
В пользу такой аддитивности потенциальной энергии Ф взаимо-
действия молекулы с твердым телом говорит линейная зависимость
величины ACZjl при адсорбции н-алканов, н-алкенов и углеводородов
других гомологических рядов от числа атомов углерода в молекуле
[135, 145, 175-181].
Применимость приближения (VIII,1) к энергии взаимодействия
неполярной молекулы с поверхностью диэлектрика при больших
расстояниях молекулы от поверхности рассматривалось теорети-
чески [147, 153, 154].
Приближение (VIII,2) для потенциальной энергии ван-дер-вааль-
сового взаимодействия сложных систем с успехом применялось при
расчетах свойств молекулярных кристаллов (теплот сублимации,
параметров элементарных ячеек) [170—174, 182—217] и в конформа-
ционном анализе [218—251]. Это приближение также с успехом
253
было применено и при оценке потенциальных функций Ф взаимодей-
ствия многоатомных молекул с поверхностью [70, 71, 73—75, 77,
80-85, 91-94, 96, 105, 106, 118, 121, 122, 129, 130, 133, 139].
4. Выбор силовых центров в молекуле и их положения
Дисперсионные силы обусловлены взаимодействием тех же осцил-
ляторов, которые обусловливают дисперсию молекулы. Поэтому
проблема выбора силовых центров дисперсионного притяжения тесно
связана с проблемой расчета поляризуемости сложной молекулы
с помощью поляризуемостей ее фрагментов. Для этого пользуются
как поляризуемостями атомов или атомных групп, так и поляризу-
емостями валентных связей. В соответствии с этим в качестве силовых
центров дисперсионного взаимодействия многоатомных систем можно
также принять атомы или группы атомов и валентные связи.
Внешняя электронная оболочка молекулы, ответственная за меж-
молекулярное взаимодействие, формируется валентными связями.
Поэтому в качестве силовых центров сложных молекул, по-видимому,
следовало бы принять именно валентные связи. Однако потенциаль-
ная энергия межмолекулярного взаимодействия валентных связей
сильно зависит не только от расстояния между ними, но и от напра-
вления радиуса вектора по отношению к соответствующим валентным
связям [27, 29]. Были предложены приближенные методы учета по-
следней зависимости для диполь-дипольного дисперсионного взаимо-
действия валентных связей [27, 29]. Однако учет этой зависимости
при полуэмпирических расчетах энергии межмолекулярного взаимо-
действия встречает значительные трудности. Определенные трудности
вызывает также выбор положения силового центра валентной связи.
Валентные связи в качестве силовых центров выбирались лишь
в немногих работах при расчетах энергий молекулярных кристал-
лов [198, 200]. При этом пренебрегалось зависимостью межмолеку-
лярного взаимодействия валентных связей от их взаимной ориента-
ции, а силовые центры помещались в середине валентных связей.
При определениях потенциальной функции Ф взаимодействия моле-
кулы с твердым телом валентные связи в качестве силовых центров,
по-видимому, не рассматривались. За силовые центры сложной
молекулы при адсорбции выбирались атомы или группы атомов
(см. гл. X и XI). За силовые центры твердого тела при определениях
потенциала Ф, как правило, принимаются атомы или ионы.
Выбор атомов в качестве силовых центров молекулы при рассмо-
трении межмолекулярного взаимодействия (атом-атомное приближе-
ние) имеет ряд преимуществ перед выбором в качестве силовых цен-
тров валентных связей и атомных групп. Входящие в молекулу атомы
в гораздо большей степени, чем валентные связи или атомные группы,
можно рассматривать изотропными. Это позволяет принять, что
атом-атомные потенциалы межмолекулярных взаимодействий зави-
сят только от расстояний между центрами атомов [27, 204, 252, 253].
Чтобы силовые центры можно было рассматривать точечными, их
254
линейные размеры должны быть малыми по сравнению с расстоя-
ниями между ними. Это требование для атомов всегда выполняется
более строго, чем для атомных групп. Кроме того, атом-атомное
приближение позволяет ограничиться для расчета энергии межмоле-
кулярного взаимодействия большого количества веществ лишь очень
небольшим числом полуэмпирических атом-атомных потенциальных
функций. Например, при расчете Ф для адсорбции всех насыщенных
углеводородов на поверхности атомной решетки практически доста-
точно подобрать только две потенциальные функции (р для межмоле-
кулярного взаимодействия атомов G и Н молекулы с атомами адсор-
бента (подробнее см. гл. X).
Положения силовых центров атомов обычно выбираются в ядрах
атомов [173, 182, 198]. Однако рассеивание рентгеновских лучей
и теоретические расчеты указывают на то, что центры распределения
электронной плотности в валентно связанных атомах Н не совпадают
с положением их ядер [158]. Поэтому в некоторых работах [170,
171, 254] положение силового центра валентно связанного атома
водорода принималось-на 0,07—0,1 А смещенным в сторону связи.
Однако сумма атом-атомных потенциальных функций межмолекуляр-
ного взаимодействия, зависящих только от межатомных расстояний,
по-видимому, не передает точно зависимость энергии межмолекуляр-
ного взаимодействия молекул углеводородов от их ориентации [197].
Смещение положения силового центра атома Н в сторону атома С
не позволяет значительно исправить эту погрешность [197].
Использование атомных групп в качестве силовых центров значи-
тельно снижает трудоемкость расчетов энергии межмолекулярного
взаимодействия. Однако, как указывалось выше, это приближение
имеет ряд недостатков по сравнению с первыми двумя рассмотренными
приближениями, т. е. межмолекулярным взаимодействием валент-
ных связей и межмолекулярным взаимодействием атомов.
5. Выбор формы атом-атомного потенциала
межмолекулярного взаимодействия
Форма потенциала взаимодействия двух силовых центров (ато-
мов), входящих в молекулы, обычно принимается такой же, как
и форма потенциала взаимодействия двух изолированных атомов
(одноатомных молекул). Приближенная форма последнего потен-
циала может быть выбрана на основании приближенной теории
межмолекулярного взаимодействия.
Представление потенциала в виде суммы слагаемых, выража-
ющих вклады различных видов взаимодействия. Энергия межмоле-
кулярных сил обычно рассчитывается с помощью квантовомеханиче-
ской теории возмущений. Согласно этой теории потенциальную энер-
гию и притяжения двух молекул на расстояниях, при которых элек-
тронные оболочки молекул не перекрываются, можно представить
в виде суммы трех слагаемых [157, 159]:
и = мориент + миндукц 4“ мдисп (VIII,7)
255
где иорнент? ыиндукц и «дисп — потенциальные энергии ориентационного, ин-
дукционного и дисперсионного взаимодействии.
Энергию взаимодействия двух молекул в области небольшого
перекрывания их электронных оболочек с учетом обменных эффектов
приближенно можно представить в виде суммы [255—261]:
и — ыОрИент -|- мй(мен 4" мполяр + мсймен
(VIII,8)
где ы^ен — энергия электронного обмена первого порядка; WnoJiap — энер-
гия поляризации второго порядка, содержащая энергию индукционного и дис-
персионного взаимодействия; — обменная энергия второго порядка
(обменная поляризационная энергия).
В случае взаимодействия неполярных систем ориентационная
и индукционная энергии равны нулю и энергия межмолекулярного
взаимодействия двух молекул
м= ы1Илен4“ мдисп 4" ио^мен (VIII,9)
Обменная энергия первого порядка Подмен- Физическая природа
обменных сил достаточно ясна. Однако расчеты их энергии на основа-
нии фундаментальных констант представляют значительные труд-
ности и проведены только для простейших систем. Общей теоретиче-
ской информации об этих силах получено еще мало. Обменные силы
зависят от довольно тонких деталей распределения заряда во взаи-
модействующих молекулах, и, следовательно, нет оснований надеяться
на то, что их можно связать с другими простыми молекулярными
константами, подобно тому, как это можно сделать для дальнодей-
ствующих сил (ориентационных, индукционных и дисперсионных).
Для атомов с заполненными электронными оболочками обусловли-
вающая отталкивание обменная энергия первого порядка иймен
в общем равна сумме членов Р (г) е~аг, где Р (г) — относительно
медленно изменяющаяся функция межатомного расстояния г. По-
этому можно ожидать, что наиболее простую модельную функцию
для этой энергии представляет потенциал [1]
»8&еп==Вг-<1г (VIII,10)
где В и q — константы.
G помощью этой простой функции часто довольно точно можно
представить результаты квантовомеханических расчетов ^энергии
иймеиВ довольно широкой области г [51, 158, 262—266].
Для упрощения расчетов энергия и$мен часто аппроксимируется
степенной функцией
^меи = Ьг-п (VIII,И)
где В ия — константы.
256
Однако эта функция не имеет теоретического обоснования.
Таким образом, квантовая механика указывает на то, что наибо-
лее простой формой потенциальной энергии межмолекулярного оттал-
кивания должна быть экспоненциальная функция расстояния.
При определениях потенциальной функции Ф для потенциала сил
отталкивания между двумя силовыми центрами принимались как
экспоненциальная, так и степенная функции.
Энергия дисперсионного притяжения Ыдисп- Используя мульти-
польное разложение, энергию дисперсионного взаимодействия Ндисп
при отсутствии перекрывания электронных облаков взаимодейству-
ющих молекул можно представить в виде ряда [267]:
4исп--C1r-6-C2r“8-C3r-io4_ , . . (УШ,12)
где Clt С21 С3 представляют соответственно константы диполь-дипольного,
диполь-квадрупольного и суммарного квадруполь-квадрупольного и диполь-
октупольного взаимодействий.
Этот ряд сходится асимптотически [268, 269]. Погрешность, по-
лучающаяся при представлении энергии в виде ряда (VIII,12),
близка к последнему сохраненному члену ряда [268].
Мультипольное разложение может быть использовано также
и при небольших перекрываниях электронных облаков [270—273].
При расстояниях, близких к равновесному, необходимо учесть пер-
вые два члена ряда (VIII,12) и можно пренебречь третьим членом
этого ряда [267, 270, 274].
Таким образом, энергию дисперсионных сил притяжения при
больших и средних расстояниях г между взаимодействующими моле-
кулами можно аппроксимировать степенным рядом (VIII,12).
При выборе формы потенциальной функции Ф потенциальная
функция дисперсионного притяжения между двумя силовыми цен-
трами представлялась или в виде одного главного члена разложения
энергии дисперсионного взаимодействия в степенной ряд, или учиты-
валось несколько (2 или 3) первых членов этого разложения.
Обменная энергия второго порядка Ыобмен. Форма обменной
энергии второго порядка похожа на форму обменной энергии пер-
вого порядка [255, 256]. Она, как и обменная энергия первого
порядка, аппроксимируется простой экспоненциальной функцией
[256, 275]
= (УШ,13)
где А и а — константы.
Однако знак обменной энергии второго порядка тот же, что
и энергии дисперсионного притяжения второго порядка. Относи-
тельный вклад этого члена наибольший при промежуточных расстоя-
ниях, т. е. вблизи потенциального минимума. В области преоблада-
ния межмолекулярного притяжения вклад этого члена, по-видимому,
мал [255, 256, 276].
При выборе формы межатомного потенциала вкладам обменной
энергии второго порядка обычно пренебрегают, и обменная энергия
17 Заказ 1180
257
«обмен первого порядка складывается с энергией дисперсионного
взаимодействия u$cn второго порядка.
Форма атом-атомного потенциала межмолекулярного взаимодей-
ствия. Принимая для потенциала сил отталкивания экспоненциаль-
ную (VIII,10) или степенную (VIII,11) функцию и учитывая один,
два или более членов степенного ряда (VIII,12) для энергии диспер-
сионного притяжения, можно получить ряд моделей потенциала м^ж-
молекулярного взаимодействия. Для описания межмолекулярного
взаимодействия двух силовых центров при адсорбции были использо-
ваны главным образом следующие модели: 1) потенциал Леннард-
Джонса (6,12) [35-38, 40, 42-44, 52, 54-65, 67-74, 76, 78, 79,
89, 91, 92, 95, 111-115, 116, 120, 129-134]:
(р - — Сг-ь + Вг-кь (VIII,14)
2) потенциал Бакингема (6, ехр) [53, 89, 99, 101, 103, 104, 107]
Ф = — Cr-ь^в ехр ( — дг) (VIII,15)
3) потенциал Бакингема — Корнера (6,8, ехр) [7, 8, 80—88, 94,
136-139, 277]
ф^__ Схг-6 —C2r-8_|-5exp (~gr) — — Cjr-6 f 1 ^Bexp ( — gr)
(VIII,16)
где и С2 — параметры сил притяжения; В и q — параметры сил отталки-
вания.
Потенциал (VIII,16) пригоден вблизи потенциального минимума
и в сторону более высоких расстояний. Использовались и другие
формы потенциальной функции [38, 75, 77, 94, 96, 105, 119].
Потенциалы (VIII,14)—(VIII,16) можно записать в виде
Ф-ер(о) (VIII,17)
где е — энергия в минимуме при г = г0, о = г/г0; о (о) — функция формы.
В случае потенциала Леннард-Джонса (6,12)
и (о) =о~12 —2о-в (VIII,18)
в случае потенциала Бакингема (6, ехр)
v (о)— в ехр а (1 — а) — (VIII,19)
и в случае потенциала Бакингема — Корнера (6,8, ехр)
v (а)-[а (1+ ₽) -(6 + 8₽)J-i [(6 + 8(3) ехр а (1-о)-ао-е (1+₽О"2)]
(VIII,20)
где а = qrQ и £ = CJC^.
Потенциалы в форме (VIII,17), т. е. формулы (VIII, 18)—(VIII,20)
более удобны, когда их параметры определяются чисто эмпирически.
Если для оценки параметров Ct и С2 использовать квантовомехани-
258
ческие формулы, то более удобны первоначальные формы потенциа-
лов (VIII,14)-(VIII,16).
Из рассмотренных выше потенциалов теоретически наиболее обос-
нован потенциал (VIII,16) Бакингема— Корнера (6,8, ехр), в котором
для сил отталкивания принято экспоненциальное выражение, а для
сил дисперсионного притяжения учтены первые два члена мульти-
польного разложения знергии этих сил. В потенциале Бакингема
(6, ехр) учтен только один первый член этого разложения, а в потен-
циале Леннард-Джонса (6,12) и аналогичных ему потенциалах (6,п)
(где п — показатель степени в члене 5г"п), кроме того, для сил
отталкивания вместо экспоненциального члена эмпирически принят
степенной.
Потенциал (6,8, ехр) является хорошей аппроксимацией теоре-
тически рассчитанного потенциала межмолекулярного взаимодей-
ствия двух атомов Не в области потенциального минимума (2—4 А)
[276]. Он также лучше, чем потенциалы (6, п) и (6, ехр), описывает
второй вириальный коэффициент, коэффициент Джоуля — Томсона
и свойства кристаллических решеток благородных газов [278—280].
При записи потенциала (6,8, ехр), как и при записи других потен-
циалов межмолекулярного взаимодействия, не учитывается член
обменной энергии второго порядка, а для энергии дисперсионного
взаимодействия используется мультипольное разложение. Оба эти
приближения при расстояниях, близких к равновесному, могут быть
грубыми. Поэтому потенциал (6,8, ехр) в действительности предста-
вляет собой только удобную математическую модель, имеющую неко-
торое теоретическое обоснование. Погрешности, сделанные при выборе
модели потенциала межмолекулярного взаимодействия, поглощаются
числовыми значениями подбираемых констант [281—283].
Потенциалы (VIII,14)—(VIII,16) дают зависимость энергии взаи-
модействия ф двух силовых центров только от расстояния между
ними. Силовые центры молекул и твердых тел являются анизотроп-
ными. Энергия дисперсионного взаимодействия анизотропных сило-
вых центров зависит не только от расстояния между ними, но и от
взаимной ориентации их эллипсоидов поляризуемости [27, 284].
Решетка графита, например, обладает сильной анизотропией поля-
ризуемости [285]. Поэтомупотенциал дисперсионного взаимодействия
силового центра молекулы с атомом углерода графита должен сильно
зависеть от взаимной ориентации их эллипсоидов поляризуемости
[286—288]. Эту зависимость потенциала взаимодействия двух сило-
вых центров необходимо учесть при объяснении различия потенциала
Ф взаимодействия молекулы адсорбата с базисной и призматической
гранями решетки графита [286—288]. Были проведены расчеты энер-
гии Ф взаимодействия атомов инертных газов и СО2 с базисной и приз-
матической гранью графита, учитывая эффект анизотропии атом-
атомного потенциала [286, 287]. Однако потенциал Фдисп дисперсион-
ного взаимодействия силового центра молекулы с базисной гранью
графита, полученный на основании потенциала дисперсионного взаи-
модействия силового центра молекулы с атомом углерода графита
17'
259
с учетом анизотропии взаимодействия двух силовых центров, имеет
практически ту же форму, что и потенциал Фдисп, полученный без
учета эффекта анизотропии взаимодействия этих силовых центров
[286-288].
6. Оценка параметров атом-атомных потенциалов
Как было указано в первом разделе этой главы, параметры сил
дисперсионного притяжения атом-атомных потенциалов межмолеку-
лярного взаимодействия при адсорбции можно оценить, используя
квантовомеханические формулы. Кроме того, параметры сил притя-
жения и сил отталкивания атом-атомных потенциалов межмолеку-
лярного взаимодействия при адсорбции можно оценить с помощью
правил комбинирования на основе соответствующих параметров
атом-атомных потенциалов межмолекулярного взаимодействия моле-
кул адсорбата друг с другом и силовых центров твердого тела друг
с другом внутри кристаллической решетки.
Константа Сх. Используя теорию возмущений второго порядка,
Лондон [1, 66, 289] получил следующую строгую формулу для кон-
станты (\ диполь-дицольного дисперсионного взаимодействия двух
сферически симметричных атомов а и Ъ в основных состояниях:
С1
____у у
2 m2
771=1 71=1
(VIII,21)
где — сила осциллятора дипольного перехода атома а из основного состояния
с энергией Ag в тп-ое состояние с энергией со^ ~ Е^ — Ag — соответству-
ющая частота возбуждения; е — заряд; те — масса электрона; $ и ео£* — соот-
ветствующие величины для атома Ь.
Значения сил осцилляторов / атомов и молекул, в принципе, можно
получить, решая уравнение Шредингера. Однако такие расчеты пока
возможны только для простейших систем. Силы осцилляторов /
входят в аналогичные теоретические выражения для таких свойств
атомов (или молекул), как показатель преломления и поляризуемость,
которые измеряются экспериментально. Кроме того, значения /
должны удовлетворять определенным «правилам сумм». Эти правила
сумм можно получить из экспериментальных данных, таких, как
показатель преломления, константа Вердета. На основании этих
экспериментальных данных можно составить наборы значений /
и провести расчеты значений константы Сг [1, 255, 290—294]. Зна-
чения Сг можно также рассчитывать прямо из правил сумм [294,
295] или из показателя преломления [294, 296]. Этими способами
были рассчитаны значения константы Сг для ряда пар атомов и про-
стых молекул.
Формулу (VIII,21) можно также записать в виде [294, 297—299]:
J5
л
(гео) аь (ico) da
(VIII,22)
где аа (ieo) и (гео) — динамические поляризуемости атомов а и & при мнимых
частотах ieo.
260
Динамические поляризуемости a (ico) могут быть рассчитаны тео-
ретически для простейших систем или оценены при использовании
тех же зкспериментальных данных, как и при оценке распределения
сил осцилляторов [282, 295, 296, 298, 300]. Относительная погреш-
ность рассчитанных по формулам (VIII,21) и (VIII,22) полуэмпириче-
ским методом значений Ci меньше 10% [282, 294, 300—302].
Таким образом, квантовомеханическая теория межмолекулярных
сил дает формулы для строгого расчета значений константы Cj
диполь-дипольного дисперсионного взаимодействия. Однако расчеты
значений Сг по этим формулам встречают значительные трудности
из-за отсутствия необходимых данных о взаимодействующих силовых
центрах. Кроме того, строго эти формулы применимы только к взаи-
модействию сферически симметричных силовых центров.
С помощью некоторых упрощающих допущений можно получить
ряд приближенных формул для константы С19 содержащих такие
экспериментально измеряемые величины, как статическая поляризу-
емость, диамагнитная восприимчивость, потенциал ионизации, число
электронов во внешней электронной оболочке. Эти формулы могут
быть использованы для оценки значений Clf если нет достаточных
данных для расчетов значений Сх по строгим формулам (VIII,21)
и (VIII,22). Кроме того, расчеты по таким формулам значительно
проще, чем по строгим формулам.
Для молекул с одночленной дисперсионной формулой выражение
(VIII,21) для константы Сг диполь-дипольного дисперсионного взаи-
модействия можно записать в виде [66, 252, 267, 303, 304]:
(VIII,23)
где а — статическая поляризуемость; Д — средняя, или эффективная, энергия
возбуждения, т. е. характеристическая энергия в одночленной дисперсионной
формуле.
Величины А часто близки к потенциалу ионизации атомов I.
Позтому в формулу (VIII,23) вместо А часто вводится /.
Слэтер и Кирквуд [252, 267, 305], используя вариационный метод
и пренебрегая вкладом внутренних электронных оболочек, для взаи-
модействия сферически симметричных молекул с заполненными обо-
лочками получили формулу:
qag»
с _ ЗеЙ______________________________
1 2тне/2 (ae/^e)1/2 + (ab/-Vb)1/2
(VIII,24)
где Na и Nf, — числа электронов в валентных или внешних электронных обо-
лочках атомов а и Ь.
Выражения (VIII,23) и (VIII,24) идентичны, если принять, что
А = (N/a)'/s [23, 301, 306]. Кирквуд и Мюллер [23, 198, 252, 267,
303, 307] получили формулу:
С1 = -6отес2-7—т-\а?7--г-v (VIII,25)
где с — скорость света; % — диамагнитная восприимчивость.
261
Формулы Кирквуда — Мюллера (VIII,25) и Лондона (VIII,23)
эквивалентны при условии [198, 303]:
V
Д = — — (VIII,26)
ос
Используя теорию возмущений второго порядка и учитывая кор-
реляцию электронов, Салем [23] для сферически симметричных систем
получил выражение:
где
<(X^2>=2<T?>+S.<^> =
\\ i / / i vty
а <*;
Выражение (VIII,28) представляет квантовомеханическое среднее
координат всех электронов системы. Если в (VIII,28) пренебречь
вторым правым членом, учитывающим корреляцию электронов, то
формула Салема (VIII,27) переходит в формулу Кирквуда— Мюл-
лера (VIII,25). В общем, По этой причине
формула Кирквуда — Мюллера (VIII,25) должна давать завышенные
значения Сх.
В таблице VIII,! сопоставлены значения С\, полученные с по-
мощью приближенных формул (VIII,23)—(VIII,25), со значениями
полученными с помощью точной формулы (VIII,21). Сравнение
показывает, что приближенная формула Лондона (VIII,23) дает
несколько заниженные результаты, формула Слэтера — Кирквуда
(VIII,24) дает результаты, близкие к точным, а формула Кирквуда —
Мюллера (VIII,25) дает значительно завышенные результаты. Хотя
формулы (VIII,23)—(VIII,25) являются приближенными, они дают
полезные соотношения между значениями и экспериментально
измеряемыми свойствами атомов, т. е. они удовлетворительно пере-
дают функциональную зависимость значений константы Сг от природы
взаимодействующих атомов [252].
В случае взаимодействия анизотропных силовых центров кон-
станта диполь-дипольного дисперсионного взаимодействия зави-
сит от взаимной ориентации этих силовых центров. Было получено
приближенное выражение для константы Сj взаимодействия анизо-
тропных силовых центров с осевой симметрией [23, 27, 29, 284].
При определениях потенциальной функции Ф для оценки кон-
станты Сг в основном применялись приближенные формулы Лондона
(VIII,23) [66, 67, 70, 97, 114, 115], Слэтера — Кирквуда (VIII,24)
[131, 132] и Кирквуда — Мюллера (VIII,25) [7, 8, 38, 67, 68—89,
262
Таблица Значения Ci*1057 (Дж • мв • моль-*),
рассчитанные по строгой формуле Лондона и по приближенным формулам
Лондона, Слэтера— Кирквуда и Кирквуда — Мюллера,
а также величины отношения R приближенных и точных значений Cj
Атом По стро- гой фор- муле Лондона [294] (VIII, 21) По приближенной формуле Лондона (VIII, 23) По формуле Слэтера - Кирквуда (VIII,24) По формуле Кирквуда - Мюл- лера (VIII, 25)
Ci [252] н Ci [252] н Ci [252] н
Не 84,9 78,2 0,92 103 1,21 100 1,18
Ne 361 249 0,69 470 1,30 771 2,13
Аг 3 730 3 030 0,81 * 3 850 1,03 7 770 2,08
Кг 7 470 6 200 0,83 7 230 0,97 17 050 2,28
Хе 15 540 14 150 0,91 14 880 0,96 42 340 2,73
91—96, 99, 101, 103-107, ИЗ, 119, 120, 125, 129, 130, 137—139].
В случае адсорбции на графите для оценки константы Сх чаще всего
использовалась формула Кирквуда — Мюллера (VIII,25) [7, 8, 38,
67-89, 91-94, 137-139].
Как указывалось выше, значения С1аЬ для взаимодействия двух
разных атомов можно оценить из значений С1аа и Схьъ для взаимо-
действия одинаковых атомов с помощью правила комбинирования
[1, 45]:
Clab = (ClaaClbb)'/2 (VIII,29)
При точных значениях С1аа и С1ЬЬ зто правило комбинирования дает
верхний предел значения С1аЬ [47, 48], т. е.
Ctrf^iaaClbb)4' (VIII,30)
Более точные результаты можно получить с помощью формулы Слэ-
тера — Кирквуда (VIII,24), вводя в нее7Уа и7Уьв качестве подбира-
емых констант, рассчитанных из известных значений С1аа, Сlbb,
а(! и аь [4(5], или используя эмпирическую формулу [49]
а («ь/сха) С1аа+(оса/ссь) Схьь
Для оценки константы С i потенциальной функции Ф использова-
лось правило комбинирования (VIII,29) [52, 53, 55—59]. При этом
значения Сх для межмолекулярного взаимодействия атомов одного
сорта определялись из свойств адсорбата и адсорбента, взятых
в отдельности.
В случае сложных систем применение формулы Кирквуда — Мюл-
лера (VIII,25) проще [303]: в эту формулу входят а и %, которые для
сложных систем приблизительно аддитивно складываются из поля-
ризуемостей и диамагнитных восприимчивостей атомов. Кроме того,
оцененные с помощью этой формулы значения константы близки
к значениям оцененным с помощью правила комбинирования
(VIII,29), и приводят к рассчитанным энергиям адсорбции, близким
263
к измеренным теплотам адсорбции. Использование формул Лондона
(VIII,23) и Слэтера — Кирквуда (VIII,24) в случае сложных систем
представляет значительные трудности, так как приписывание отдель-
ным частям (атомам) молекулы и твердого тела эффективных энергий
возбуждения или соответствующих чисел электронов довольно про-
извольно.
Константа С2 и отношение С21(\. Для взаимодействия сфери-
чески симметричных силовых центров с одночленной дисперсионной
формулой константа С2 диполь-квадрупольного дисперсионного
взаимодействия может быть оценена по формуле [308]:
где va и vb — характеристические частоты в одночленных дисперсионных фор-
мулах взаимодействующих силовых центров а и Ь.
Если в формуле (VIII,32) выразить v через а и % с помощью соот-
ношения (VIII,26), то для константы С2 получается следующее удоб-
ное для расчетов выражение [105]:
С2
__ Г 1 1
32л2/пб а / \ п f ^а/^д \ 1 л
_ \ ) \ <*Ь/ХЬ / ‘
(VIII,33)
Экспериментальное определение константы С2 встречает большие
трудности, так как изменение значения этой константы практически
полностью можно компенсировать изменением значений остальных
параметров [278—280]. Поэтому при определении атом-атомного
потенциала межмолекулярного взаимодействия на основании макро-
скопических свойств значение константы С2 обычно оценивается
с помощью приближенных квантовомеханических формул [278—280,
283].
При переходе, например, от атома Не к атому Хе, значения кон-
стант и С2 изменяются приблизительно на два порядка, однако
отношение этих констант изменяется в сравнительно узких пределах
[267] (см. также гл. IX). Поэтому в качестве второго параметра сил
притяжения межатомного потенциала удобнее брать не саму кон-
станту С2, а отношение С21СГ [278—280, 283]. Точность значения от-
ношения С21Си оцененного с помощью приближенных квантовомеха-
нических формул, может быть значительно выше точности значений
Сх и С2 в отдельности [267].
Значение константы С2 при оценках потенциальных функций Ф
оценивалось с помощью формулы (VIII,33) [4—8, 38, 75, 77, 80—86,
88, 96, 105, 106, 137-139].
Параметры потенциала отталкивания. Как уже отмечалось выше,
энергию сил межмолекулярного отталкивания невозможно предста-
вить с помощью формул, в которые входили бы параметры невозму-
щенных молекул, т. е. квантовомеханическая теория пока не может
быть использована для оценки параметров потенциала сил отталки-
вания. Поэтому эти параметры приходится определять эмпирически.
264
Однако приближенные расчеты потенциала сил межатомного оттал-
кивания указывают на следующие правила комбинирования пара-
метров q и В в формуле (VIII,10) [1, 50]:
--------2------
(VIII,34)
Bab = (BaaBbb)^ (VIII,35)
Объединяя эти правила комбинирования для параметров q и В,
получаем:
wott, ab~ (мотт» аа^отт» bb) г (VIII,36)
Эти правила комбинирования связывают параметры qab и ВаЪ фор-
мулы (VIII,10) для взаимодействия атомов разных видов с парамет-
рами qaa, qbb, Ваа и Вьь для взаимодействия атомов одного вида.
Хотя приближенные теории, указывающие на эти правила комбини-
рования, слишком грубы для того, чтобы дать абсолютные значения
этих параметров с какой-либо точностью, правила комбинирования
(VIII,34)—(VIII,36) дают хорошие результаты [51].
Константу сил отталкивания В в формулах (VIII,10) и (VIII,11)
можно выразить через равновесное расстояние го и остальные пара-
метры атом-атомного потенциала межмолекулярного взаимодействия,
используя условие равновесия всех составляющих сил притяжения
и сил отталкивания при г = г0
(VIII,37)
В случае межмолекулярного взаимодействия атома с адсорбентом
константа В также может быть выражена через равновесное расстоя-
ние zQ взаимодействия атома с поверхностью, используя условие
равновесия:
(—Ц=о (VIII’38)
где Ф — потенциальная энергия межмолекулярного взаимодействия атома
с твердым телом, зависящая от расстояния z атома от поверхности твердого тела
и рассчитанная на основании ф при использовании приближения (VIII,1).
При определениях потенциальной функции Ф параметр q формулы
(VIII,10) оценивался с помощью правила конбинирования (VIII,34)
на основании значений q, определенных эмпирически для взаимодей-
ствия силовых центров адсорбата и адсорбента, взятых в отдельности
[53, 75, 80—85, 104], или принимался равным некоторому среднему
значению [89, 94]. Чисто эмпирический параметр п формулы (VIII,11),
как правило, принимался равным 12. Параметр В формулы (VIII,10)
оценивался с помощью правила комбинирования (VIII,35) [53, 99,
104, 107] или выражался через параметры сил притяжения и равно-
весное расстояние г0, используя условие равновесия (VIII,37) [67,
265
68, 70—74, 78, 79, 55—59, 95], или равновесное расстояние z0, исполь-
зуя условие равновесия (VIII,38) [38, 69, 76, 80—85, 89, 92, 93].
Равновесное расстояние при межмолекулярном взаимодействии
двух силовых центров. Равновесное расстояние г0 межмолекулярного
взаимодействия двух атомов, не входящих в состав сложной моле-
кулы (например, атомов инертных газов), обычно определяется как,
один из основных параметров потенциала межмолекулярного взаимо-
действия на основании свойств объемного газа и кристаллической
решетки.
Равновесные расстояния го межмолекулярного взаимодействия
атомов, входящих в состав сложной молекулы (например, атомов С
и Н молекул, углеводородов), обычно оцениваются на основании
кристаллохимических данных. Для равновесного расстояния г0> с ... с
межмолекулярного взаимодействия двух атомов С на основании
свойств решетки графита было получено значение 3,81—3,83 А
[184, 309]. Близкие к этому значению rQj с... с принимались в ряде
работ [172, 173, 184, 186, 205, 212] при расчетах межмолекулярных
и внутримолекулярных взаимодействий.
В кристаллических решетках углеводородов расстояние наиболь-
шего сближения атомов водорода соседних молекул равно 2,4—
2,6 А [310]. Часто это значение принимается равным равновесному
расстоянию Го,н....н межмолекулярного взаимодействия двух
атомов Н. Однако расчеты кристаллических решеток углеводородов
на основании атом-атомных потенциальных функций межмолекуляр-
ного взаимодействия С и Н показывают [172, 186, 228], что расстоя-
ние наибольшего сближения атомов Н соседних молекул в решетке
приблизительно на 0,3 А меньше значения равновесного расстояния
^о,н...н, принятого в расчетах потенциала межмолекулярного
взаимодействия двух атомов Н. Это обусловлено главным образом
тем, что расстояния между атомами сложных молекул в кристалли-
ческой решетке определяются минимумом потенциальной энергии
межмолекулярного взаимодействия всех силовых центров рассматри-
ваемой молекулы со всеми силовыми центрами остальных молекул,
а не потенциальным минимумом межмолекулярного взаимодействия
только наружных атомов Н. Таким образом, расстояние наибольшего
сближения атомов Н в молекулярных кристаллах не равно значению
г0>н. ..н для потенциальной функции межмолекулярного взаимо-
действия этих двух изолированных атомов Н. Чтобы из атом-атомных
потенциальных функций межмолекулярного взаимодействия полу-
чить расстояние наибольшего сближения атомов Н в кристалличе-
ской решетке н-гексана, равное экспериментально наблюдаемому,
для равновесного расстояния Го,н...н взаимодействия двух
атомов Н необходимо принять значение, равное 2,8—3,2 А [228, 229].
Необходимость введения более высокого, чем 2,4—2,6 А, значения
для Го, н ... н было отмечено и в других работах [173, 227].
Равновесное расстояние гои ... / межмолекулярного взаимодей-
ствия двух разных силовых центров (атомов) I и /, как правило, при-
нимается равным среднему арифметическому значению равновесных
266
взаимодеи-
(VIII,39)
расстояний r0( z z и r0> /,,, / межмолекулярного
ствия силовых центров г. . Л и /. . .у, т. е.
. Г0, t . . г+г0, Л . , j
О, г. . • j 2
Это правило комбинирования строго справедливо только для модели
твердых сфер. В случае реальных силовых центров оно не имеет
теоретического обоснования и его надо рассматривать лишь как
эмпирическое. Недавно было предложено другое эмпирическое пра-
вило комбинирования [311, 312]:
Г0, г. . . > = (Г0, L . . г ‘ Г0, /. . . jP2 (VIII,40)
Для учета зависимости равновесного расстояния го между одно-
валентными атомами от углов ipi и ф2 между межатомным вектором и
валентными связями была предложена формула [213]:
г0 = 4- [(е sin2 cos2i|?1)-1/t + (е sin2i|?2 + cos2 (VIII,41)
где а — равновесное расстояние вдоль связи; а/е — равновесное расстояние
поперек связи.
Однако эта зависимость, по-видимому, имеет небольшое влияние
на результаты расчета межмолекулярной энергии, так как во время
суммирования атом-атомных потенциалов происходит усреднение
различных направлений межатомных векторов по отношению к ва-
лентным связям [204].
При оценках потенциальной функции Ф равновесное расстояние
го межмолекулярного взаимодействия атома адсорбата с атомом ад-
сорбента оценивалось с помощью правила комбинирования (VIII,39)
[52, 54—58, 70—74]. Для равновесных расстояний r0> z t иг0( j . j
межмолекулярного взаимодействия атомов адсорбата и силовых
центров адсорбента принимались значения, определенные из
свойств адсорбата и адсорбента, взятых в отдельности.
При оценке константы В сил отталкивания с помощью условия
равновесия (VIII,38) равновесное расстояние zQ межмолекулярного
взаимодействия атома адсорбата с базисной гранью решетки графита
принималось равным сумме эффективного ван-дер-ваальсового ра-
диуса г* атома и половины межплоскостного расстояния d/2 =
= 1,68 А решетки графита [38, 75—77, 80—84, 89, 92]:
zo = -4-H-r* (VIII,42)
Для инертных газов г* принимался равным половине равновесного
расстояния го межмолекулярного взаимодействия двух атомов соот-
ветствующего инертного газа. Значение выбиралось на основании
свойств реального газа и отсюда получалось значение г* и z0. Однако
такой выбор значений z0 является довольно произвольным (см. ниже
разд. 2, гл. IX).
267
7. Суммирование атом-атомных потенциалов
Общее выражение. Чтобы рассчитать потенциальную энергию Ф
согласно аддитивной схеме (VIII,2), необходимо провести суммирова-
ние потенциалов <pz }- межмолекулярного взаимодействия ато-
мов i молекулы с атомами j твердого тела. В случае межмолекуляр-
ного взаимодействия молекулы с твердым телом, состоящим из атомов -
одного сорта, например, с графитом, вводя потенциал (6,8, ехр)
(VIII,16) в выражение (VIII,2), получаем:
ф=2 2 фо=2 ф/ (viii,43)
i j i
Здесь
ф/=2 ча=- ci'7 2 rvs~Ciii 2 r^+ Bi> S e”’z/r‘7 (VIII-44>
3 3 j j
где Ф/ — потенциальная энергия межмолекулярного взаимодействия г-го атома
молекулы со всеми атомами у твердого тела; г/у — расстояние между г-ым ато-
мом молекулы и у-ым атомом твердого тела.
Как видно из выражения (VIII,44), для расчета потенциальной
энергии Ф межмолекулярного взаимодействия молекулы с твердым
телом на основании атом-атомных потенциалов межмолекулярного
взаимодействия <pzy необходимо определить значения сумм 2Г7/
(п = 6,8) и 2 ехР (—’4nrij) в зависимости от положения рассматрива-
емой точки над поверхностью твердого тела.
Расчет решеточных сумм. Для расчета степенных сумм
2 необходимо учесть большое число слагаемых г~ц [309, 313],
что связано со значительным объемом вычислений. Было предложено
числовое суммирование вкладов отдельных атомов твердого тела
заменить на аналитическое интегрирование, принимая непрерывное
однородное распределение вещества твердого тела во всем его объеме
[66, 314, 315]. В этом приближении
V r~i? =-----—------— (VI 11,45)
А (п —2) (п —3) z(n-3) ?
3
где v — число атомов в 1 см3 твердого тела; z — расстояние рассматриваемой
точки от поверхности твердого тела.
Это приближение значительно упрощает расчет степенных сумм
2 г~п, однако оно дает для этих сумм сильно завышенные значения
[316].
Для более точного расчета сумм^Г" п и2 е~ qr наД базисной гранью
графита Крауэлл [69, 76] предложил принять непрерывное однород-
ное распределение вещества графита только внутри атомных базис-
ных плоскостей решетки графита и учесть дискретное распределение
вещества перпендикулярно этой грани. Это допущение приводит
268
к следующему приближенному выражению для степенных решеточ-
ных .сумм:
= "-2) <V,I,'4e>
3
здесь £ — обобщенная зета-функция Римана
со
ft=0
где d — межплоскостное расстояние; х = 0,382 А’2 — число атомов углерода,
приходящееся на 1 см2 базисной плоскости графита; к + 1 — порядковый
номер этой плоскости.
Принимая потенциал Леннард-Джонса (6,12) для энергии межмо-
лекулярного взаимодействия силового центра молекулы с атомом С
графита, используя приближение Крауэлла (УШ,46) и учитывая
вклад только наружной базисной плоскости решетки графита в2г”12
для потенциальной энергии Ф межмолекулярного взаимодействия
силового центра молекулы с базисной гранью графита, получили
[52, 76, 315] выражение:
4)+тЖ1Г’=
—-(т 4) - т (^гУ ((VIII’48>
Если для энергии межмолекулярного взаимодействия силового
центра молекулы с атомом С графита принять потенциал Бакингема —
Корнера (6,8 ехр), то при аналогичных приближениях (т. е. учитывая
вклад в энергию отталкивания только от наружной базисной грани)
для Ф получаем [8, 53, 88, 315]:
Ф 4)-^S(-T’ б)+^(<7-Н)е-’2 (VIII,49)
Допущение о непрерывном однородном распределении вещества
в базисных плоскостях решетки графита дает значения сумм 2 г~п
и £ехр близкие к значениям, полученным при числовом сум-
мировании по очень большому числу атомов углерода решетки гра-
фита [69, 309]. Это допущение было использовано в ряде работ [38, 45,
52—55, 57, 58, 69, 76, 137—139] при расчетах Ф для адсорбции на
базисной грани графита. Однако это приближение не позволяет оце-
нить величину потенциального барьера Fo для движения адсорбиро-
ванных молекул вдоль поверхности. Чтобы получить точные значе-
ния сумм2^"Л) необходимые для оценки Fo, производились трудоем-
кие числовые суммирования слагаемых г~п по большому числу N
ближайших атомов [67, 68, 75, 77]. Такие расчеты производились
для ряда значений z при немногих положениях рассматриваемой точки
над поверхностью. Вклад более отдаленных атомов определялся
269
интегрированием по остальному объему, предполагая непрерывное
равномерное распределение вещества в этом остальном объеме.
Однако даже при N = 100—200 этот способ расчета сумм дает зна-
чения, отличные от точных значений [309] на 0,5—2%. Рассматри-
вались аналитические методы учета зависимости решеточных сумм
от перемещения точки вдоль грани кристалла [317—319]. В работе
[319] использовалось одно из интегральных представлений гаммц,-
функции Эйлера и разложение решеточной суммы S г~ " (п > 3)
для простой решетки Бравэ (элементарной ячейкой которой Является
прямоугольный параллелепипед) в двойной ряд Фурье, который быс-
тро сходится. Полученные в этой работе разложения могут быть при-
менены и к другим, более сложным решеткам, которые могут быть
представлены как суперпозиции простых решеток Бравэ. В этой
работе показано, что такие разложения могут быть применены и к ре-
шетке графита, так как она может быть представлена как суперпо-
зиция 8 тождественных решеток Бравэ, отличающихся друг от друга
только сдвигом на некоторый вектор. Однако практическое примене-
ние этих рядов осложняется некоторыми трудностями расчетов коэф-
фициентов рядов Фурье. К настоящему времени эти коэффициенты
рассчитаны только для простой кубической решетки Бравэ [320].
Значения 2 6 наД базисной гранью слоистой решетки графита
были получены и прямым числовым суммированием по очень боль-
шому числу N ближайших атомов углерода, лежащих в I атомных
плоскостях — графитовых слоях (к — 1, 2, . . Z; к — номер плос-
кости) [309]. Вклад более отдаленных атомов, лежащих в Z и в следу-
ющих L — Z плоскостях (Z < к L), определялся соответственно
интегрированием внутри неучтенных площадей первых Z плоскостей
и внутри L — I следующих плоскостей, принимая непрерывное
равномерное распределение атомов внутри этих плоскостей. Вклад
атомов, лежащих в еще более отдаленных от поверхности плоскостях
(L < к <Z оо), определялся интегрированием по остальному объему
решетки, принимая непрерывное равномерное распределение атомов
в этой ее части.
Описанный выше способ расчета2^-л применим не только к слои-
стым решеткам, но также и к другим кристаллическим решеткам,
в которых могут быть выделены атомные плоскости, параллельные
рассматриваемой грани [316]. Кроме того, такой способ учета вклада
более отдаленных атомов, сохраняя большую точность рассчитанных
значений2 т*-п> позволяет значительно уменьшить число атомов 7V,
по которым необходимо производить прямое числовое суммирование
1316].
Если zk = z± + zlk — расстояние от рассматриваемой точки над
гранью до &-той атомной плоскости (zr — расстояние от рассматрива-
емой точки до первой плоскости; zlk — расстояние между первой
и &-ой плоскостями); — проекция на Л-тую плоскость (rz-A — г}
для атомов, лежащих в Л-той плоскости); хй — среднее число атомов
рассматриваемого сорта на 1 см2 &-той плоскости; Rk = (ЛГй/лхй) /г —
270
радиус круга, соответствующего переходу от суммирования к инте-
грированию в Л-той плоскости; Nk — число атомов Л-той плоскости,
по которым произведено числовое суммирование (N = 4-
4- . . . +ATfc + . . . 4-ATz);v —среднее число атомов рассматриваемого
вида в 1 см3 твердого тела, и если интегрирование по объему произ-
водится при z > Ч2 (zL + ZL+1) = Zy, то
I Г Nk 1
V Х?С? , п/2 , / Nk I _з\2—п/2 ,
ri =2 2( +
i fc-i L j
+4^-2 Kkz™+ z^n (vni’50>
k=l+l
Преимущества формулы (VIII,50) для расчета сумм ]£г"л пока-
заны на примере двух кристаллических решеток: базисной грани
графита и грани (100) простой кубической решетки, состоящей из
атомов одного сорта. Наиболее точные значения для этих решеток
были получены в работах [309, 313]. Для них расстояния d между
атомными плоскостями постоянны, поэтому zk = z± 4- (& — 1) и
= Zi 4- (L — l/2)d.
Для выбора N и I при помощи* формулы (VIII,50) при I — L — 1
и v — 0 были рассчитаны [316] значения ав2г"в над одной двух-
мерной гексагональной сеткой атомов углерода решетки графита
и над двухмерной квадратной сеткой атомов простой кубической
решетки в зависимости от 7V\ при разных значениях ^z-Ja (а — кон-
станта сетки). Результаты расчетов приведены в табл. VIII,2^ 3.
В табл. VIII,2 также приведены соответствующие значения
полученные в работе [309] прямым суммированием.
Из табл. VIII,2 и VIII,3 видно, что рассчитанные по формуле
(VIII,50) значения 2г"в наД одной атомной плоскостью с ростом
числа Ni быстро приближаются к точным значениям^ т*-6 , и рас-
хождение не более 0,2% наблюдается при V i 36 в случае базис-
ной плоскости графита и при N 24 в случае плоскости (100) про-
стой кубической решетки. При учете того, что для базисной грани
графита aid = 0,42 [для грани (100) простой кубической решетки
aid = 1], получаем из табл. VIII,2 и VIII,3, что при z-Jd 1,5
точные значения по атомам одной плоскости близки к рассчи-
танным по формуле (VIII,50) при = 0, и, следовательно, при
zx Id 5- 0,5 для обеих граней можно принять I = 1.
Для выбора L с помощью формулы (VIII,50) при N ™ 0, I — 0
были рассчитаны [316] значения (2<?4/лх) над кристалличе-
ской решеткой с независящими от к величинами d и х (v = x/d)
в функции L при разных значениях z-Jd. Результаты расчета при-
ведены в табл. VIII,4, из которой видно, что с точностью 0,2% можно
принять L = 3.
271
Таблица VIII,2. Значения сумм ав2г'в над одной бесконечной
базисной плоскостью графита для двух положений:
над центром шестиугольника из атомов углерода (положение h)
и иад атомом углерода (положение с)
Поло- жение Ni Zi/a—1 Zi/a==2 zt/a= 3
ав 2 Г-8 Погреш- ность, % 102ав Погреш- ность, % 102ав2 г"в Погреш- ность, о/ .0
0 1,2092 31,3 7,558 1,3 1,493 0,0
6 0,8498 2,3 7,679 2,8 1,517 и
12 0,8320 0,2 7,477 0,2 1,493 0,1
24 0,8316 0,1 7,497 0,5 1,504 0,8
h 36 0,8306 0,0 7,457 0,1 1,491 0,1
54 0,8308 0,0 7,466 0,1 1,495 0,2
72 0,8306 0,0 7,459 0,0 1,491 0,1
более 2000 [309] 0,8307 0,0 7,461 0,0 1,492 0,0
0 1,209 25,7 7,558 0,7 1,493 0,0
6 1,515 0,3 7,539 0,9 1,485 0,5
13 1,523 0,2 7,673 0,9 1,506 0,9
25 1,520 0,0 7,610 0,0 1,494 0,1
с 37 1,520 0,0 7,615 0,1 1,496 0,2
58 1,520 0,0 7,608 0,0 1,494 0,1
73 1,520 0,0 7,608 0,0 1,494 0,1
более 2000 [309] 1,520 0,0 7,607 0,0 1,493 0,0
При п > 6 вклады г,п убывают быстрее, чем вклады гД и зна-
чения параметров N, I и L будут не больше, чем в случае п — 6.
Расчет 2 г~е над базисной гранью графита и гранью (100) простой
кубической решетки. Результаты расчета ав2 г~0 по формуле (VIII,50)
при разных Nlt L и zja над базисной гранью графита и гранью (100)
простой кубической решетки для двух положений приведены в табл.
VIII,5. В этой таблице приведены также соответствующие наиболее
точные значения полученные прямым суммированием г-6
по весьма‘большому числу атомов решетки [309, 313].
Из табл. VIII,5 видно, что рассчитанные в работе [316] по формуле
(VIII,50) при I — 1 и L = 3 значения наД обеими решетками
при сравнительно небольших N ~ _ZV\ практически совпадают со
значениями ав2 г"6, полученными весьма трудоемкими расчетами
[309, 313]. При учете того, что для базисной грани графита aid —
= 0,42 из данных табл. VIII,5 получаем также, что допущение о не-
прерывном однородном распределении атомов в атомных плоскостях
(четвертый способ расчета) при z-Jd 1 с успехом может быть исполь-
зовано не только в случае слоистых решеток [69], но и в случае
других решеток, например кубических (сравните результаты, полу-
272
Таблица VIII,3. Значения сумм а6 г~6 наД одной бесконечной
атомной плоскостью (100) простой кубической решетки для двух положений:
над центром четырехугольника (положение h) и над атомом (положение с)
Поло- жение Ni Zi/ae0,5 Zi/a==l z*/a==l,5
а9 2 г"й Погреш- ность, % а9 2 г~* Погреш- ность, % а9 2 Погреш- ность, %
0 25,133 60,4 1,571 8,6 0,3103 0,7
4 10,159 2,0 1,489 3,6 0,3189 0,2
12 9,961 0,1 1,439 0,2 0,3096 0,5
16 9,958 0,0 1,438 0,1 0,3091 0,3
h 24 9,955 0,0 1,436 0,0 0,3080 0,1
32 9,956 0,0 1,437 0,1 0,3085 0,1
44 9,955 0,0 1,436 0,0 0,3081 0,0
76 9,955 0,0 1,436 0,0 0,3082 0,0
148 9,955 0,0 1,436 0,0 0,3082 0,0
0 25,13 164,7 1,571 10,9 0,3103 0,7
5 66,51 0,0 1,734 5,2 0,3108 0,6
9 66,56 0,0 1,753 5,7 0,3165 1,2
13 66,54 0,0 1,740 0,2 0,3113 0,4
с 21 66,54 0,0 1,744 0,1 0,3135 0,3
25 66,54 0,0 1,742 0,1 0,3126 0,0
37 66,54 0,0 1,752 0,1 0,3127 0,0
69 66,54 0,0 1,743 0,0 0,3126 0,0
149 66,54 0,0 1,743 0,0 0,3126 0,0
Таблица VIII,4. Результаты расчета (2<№/лк) 2 г~6
по формуле (VIII,50) при #=0
L Zi/d=l Zi/d=3 Zi/d==5
(2d*/nx) 2 г-* Погреш- ность, 0/ /0 (2d4/nx) 2 Погреш- ность, % (2d4/лх) 2 г~9 Погреш- ность, %
1 1,099 1,5 2,012 1,5 3,604 0,9
2 1,084 0,2 1,991 0,5 3,585 0,4
3 1,083 0,1 1,986 0,2 3,578 0,2
4 1,082 0,0 1,984 0,1 3,575 0,1
сю 1,082 0,0 1,982 0,0 3,571 0,0
ченные первым и четвертым способами). Вместе с тем, интегрирова-
ние по объему (пятый способ расчета) даже при z-Jd =1,5 дает зна-
чительно завышенные значения.
Таким образом, использованный в работах [69, 309] для слоистой
решетки графита способ расчета степенных сумм 2г~в применим
и к решеткам других типов. Примененный для графита [309] и обоб-
18 Заказ 1180
273
Таблица УПЦб. Значения а8 г~8 над базисной гранью графита
и над гранью (100) простой кубической решетки
Метод расчета Положение над гранью а® Для графита при a® Для кубический решетки при
Zi/a = l Zi/a«2 Z!/a=3 z1/a=0,5 Zi/a=l Zi/a—1,5
Числовое суммирование и интегрирование h более 2000 [309] 0,8418 0,07888 0,01696 около 500 [313] 10,32 1,565 0,3664
с более 2000 [309] 1,530 0,08034 0,01697 около 500 [313] 66,91 1,871 0,3708
По формуле (V111, 50) при 1— 1, L — 3 h 36 0,8417 0,07885 0,01695 24 10,33 1,566 0,3669
с 37 1,531 0,08043 0,01700 25 66,91 1,872 0,3715
По формуле (VIII, 50) при 1~ 0, £ = 3 (ин- тегрирование по плос- костям и по осталь-, ному объему) Не зависит от положе- ния 0 1,220 0,07985 0,01697 0 25,50 1,701 0,3691
По формуле (VIII, 50) при Z = 0, 1/=оо (ин- тегрирование по плос- костям) То же 0 1,220 0,07983 0,01696 0 25,50 1,700 0,3689
По формуле (VIII, 50) при 2 = 0, £ = 0 (ин- тегрирование по объ- ему) То же 0 0,31088 0,02836 0 — 4,189 0,5236
щенный на другие решетки [316] способ учета вклада более отдален-
ных атомов позволяет с удовлетворительной точностью (—0,2%)
рассчитать значения сумм 2Г"6’ производя числовое суммирование
лишь по сравнительно небольшому числу ближайших атомов.
Ашпфоксимации зависимости и £ (z/d, *и) от 2- Зависимости
значений 2г"в от 2 для разных положений над решеткой удовлетво-
рительно выражаются степенными функциями pnz~Qn, где рп и qn —
константы, которые находятся расчетом этих сумм для ряда значе-
ний z [75, 77, 105].
Из приведенных в табл. VIII,4 и VIII,5 результатов следует,
что суммирование вкладов отдельных плоскостей кристаллической
решетки может быть заменено суммированием вкладов только не-
скольких первых плоскостей и интегрированием по остальному объему
решетки. Отсюда вытекает, что значения обобщенной ^-функции
Римана можно с удовлетворительной точностью рассчитать по фор-
муле
00 р
t(-b ")-2(-з-+‘Г~2(-г+‘)”+
fc=o fc=0
(Z \1—т
— + -Р + 0.5)
—------—----- (VIII,51)
' 7П — 1
где р 2.
ЛИТЕРА ТУРА
1. MasonE. A., Monchick L. Advan. Chem. Phys., 1967, v. 12, p. 330—
387.
2. Hill T. L. J. Chem. Phys., 1948, v. 16, p. 181—189.
3. D r en an J. W., H ill T. L. J. Chem. Phys., 1949, v. 17, p. 775-781.
4. К и с e л ев А. В., П ошку с Д.П. ДАН СССР, 1961, т. 139, с. 1145—1148.
5. К и с е л е в А. В., П о ш к у с Д. П. ЖФХ, 1963, т. 37, с. 608-614;
Trans. Faraday Soc., 1963, v. 59, p. 428—436.
6. К и с ел е в А. В., П о шк у с Д. П. ЖФХ, 1963, т. 37, с. 1504-1509;
Trans. Faraday Soc., 1965, v. 59, p. 1438—1443.
7. К и с е л е в А. В., П о ш к у с Д. П. ЖФХ, 1965, т. 39, с. 398—402.
8. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1965, т. 39, с. 1190—1197.
9. Myers A. L., Prausnitz J. М. Trans. Faraday Soc., 1965, v. 61,
p. 755-764.
10. Hoory S. E., Prausnitz J. M. Trans. Faraday Soc., 1967, v. 63,
p. 455-460.
11. Pierotti R. A. Chem. Physics Letters, 1968, v. 2, p. 420—422.
12. Пошкус Д. П., Афреймович А. Я. ЖФХ, 1968, т. 42, с. 1201 —
1205.
13. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1968, т. 42, с. 2546—2552.
14. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1968, т. 42, с. 2553—2555.
15. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1970, т. 44, с. 981—986.
16. R oberts С. S. Phys. Rev., 1963, v. 131, р. 203—209.
275
17. К raus M., M ies F. H. J. Chem. Phys., 1965, v. 42, p. 2703—2708.
18. Vand ersli ce J. T., Mason E. J. Chem. Phys., 1960, v. 33, p. 492—
494.
19. V i c t о r G. A., D a 1g a r n о A. J. Chem. Phys., 1970, v. 53, p. 1316—1317.
20. D e В о er J. Physica, 1942, v. 9, p. 363—382.
21. E v e 11 A. A., M a r g e n a u H. Phys. Rev., v. 1953, v. 90, p. 1021—1023;
J. Chem. Phys., 1953, v. 21, p. 958—959.
22. Mason E. A., Hirscbfelder J. О. J. Chem. Phys., 1957, v. 26,
p. 756—766.
23. Salem L. Molec. Phys., 1960, v. 3, p. 441—452.
24. Salem L. Proc. Roy. Soc., 1961, v. A264, p. 379—391.
25. Magnasco V., Muss о G. F. J. Chem. Phys., 1967, v. 47, p. 4617—
4628, 4629—4641.
26. Tapia O.,BessisG.,BratozS. C. R. Acad. Sci. Paris, 1969, v. B268,
813—816.
27. London F. J. Phys. Chem., 1942, v. 46, p. 305—316.
28. Salem L. Molec. Phys., 1960, v. 3, p. 441—452.
29. Banerjee K., Salem L. Molec. Physica, 1966, v. 11, p. 405—420.
30. CraigD. P. e. a. Proc. Roy. Soc. (London), 1965, v. A 286, p. 98—116.
31. С r a i g D. P. e. a. Disc. Faraday Soc., 1965, v. 40, p. 110—116.
32. Киселев А. В., Щербакова К. Д., Яшин Я. И. ЖСХ, 1969,
т. 10, с. 951—968.
33. М а с R и г у Т. В., Sams I. R. Molec. Physics, 1971, v. 20, p. 49—55.
34. Constabaris G.,Halsey G. D.J. Chem. Phys., 1957, v. 27, p. 1433—
1434.
35. Freeman M. P. J. Phys. Chem., 1958, v. 62, p. 723—728.
36. Sams J. R., Constabaris G., Halsey G. D. J. Phys. Chem., 1960,
v. 64, p. 1689—1696.
37. ConstabarisG.,Sams J. R., Halsey G. D. J. Phys. Chem., 1961,
v. 65, p. 367—369.
38. Ross S.,Olivieri. P. Advan. Chem. Series. 1961. v. 33, p. 301—308,
309-316.
39. Ross S., Olivier J. P. J. Phys. Chem., 1961, v. 65, p. 608—615.
40. YarisR.,SamsJ. R. J. Chem. Phys., 1962, v. 37, p. 571—576.
41. Olivier J. P., R о s s S. Proc. Roy. Soc., 1962, v. A265, p. 447—454.
42. Sams J. R., Y a r i s R. J. Phys. Chem., 1963, v. 67, p. 1931—1933.
43. WolfeR., Sams J. R. J. Phys. Chem., 1965, v. 69, p. 1129—1135.
44. Mac R u г у T. B. J. Chem. Phys., 1968, v. 49, p. 1543—1545.
45. Sams J. R. Trans. Faraday Soc., 1964, v. 60, p. 149—156.
46. Wilson J. N. J. Chem. Phys., 1965, v. 43, p. 2564—2565.
47. T ang К. T. J. Chem. Phys., 1968, v. 49, p. 4727—4728.
48. W e i n h о 1 d E. J. Phys. (Atom. Molec. Phys.), 1969, v. В 2, p. 517-520.
49. Kramer H. L., Herschbach D. R. J. Chem. Phys., 1970, v. 53,
p. 2792—2800.
50. Z e n e r C. Phys. Rev., 1931, v. 37, p. 556—569.
51. Abrahamson A. A. Phys. Rev., 1964, v. 133, p. A 990—A 1004.
52. Crowell A. D., Steele R. B. J. Chem. Phys., 1961, v. 34, p. 1347—
1349.
53. Crowell A. D., Chang С. O. J. Chem. Phys., 1963, v. 38, p. 2584-
2586.
54. HellemansR., Van Itterbeck A., Van DaelW. Physica,
1967, v. 34, p. 429—437.
55. С г о w e 11 A. D. J. Chem. Phys., 1968, v. 49, p. 892—895.
56. S t e e 1 e W. A., К a rl R. J. Col. Interface Sci., 1968, v. 28, p. 397—402.
57. Brown J. S. Surface Sci., 1970, v. 19, p. 259—268.
58. Crowell A. D. Surface Sci., 1971, v. 24, p. 651—653.
59. Crowell A. D., Chang С. O. J. Chem. Phys., 1965, v. 43, p. 4364—
4367.
60. S t e e 1 e W. A. Advan. Chem. Series, 1961, v. 33, p. 269—280.
61. R о s s M., Steele W. A. J. Chem. Phys., 1961, v. 33, p. 862-871.
276
<>2. S t e e 1 e W. A., KebbekusE. R. J. Chem. Phys., 1965, v. 43, 292—304.
63. Barnes M. W., Steele W. A. J. Chem. Phys., 1968, v. 49, p. 2128—
2135.
64. Ricca F., P i s a n i C., Garrone E. J. Chem. Phys., 1969, v. 51,
p. 4079—4094.
65. Ricca F., Garrone Б. Trans. Faraday Soc., 1970, v. 66, p. 959—966.
66. L о n d о n F. Z. Phys. Chem., 1930, Bd. Bll, s. 222—251.
67. В а г r e r R. M. Proc. Roy. Soc,, 1937, v. A161, p. 476—493.
68. С г о w e 11 A. D., Young D. M. Trans. Faraday Soc., 1953,v. 49,
p. 1080—1085.
69. Crowell A. D. J. Chem. Phys., 1954, v. 22, p. 1397—1399.
70. А в г у л ь Н. Н. и др. ЖФХ, 1956, т. 30, с. 2106—2121.
71. А в г у л ь Н. Н. и др. Изв. АН СССР, ОХН, 1956, с. 1304—1311.
72. Расе Е. L. J. Chem. Phys., 1957, у. 27, р. 1341—1346.
73. АвгульН. Н., Киселев А. В. ДАН СССР, 1957, т. 112,
с. 673—676.
74. А в гуль Н. Н., К ис ел ев, А. В. Изв. АН СССР, ОХН, 1957,
с. 230—232.
75. А в г у л ь Н. Н. и др. Изв. АН СССР, ОХН, 1957, с. 1314—1327.
76. С г о w е 11 A. D. J. Chem. Phys., 1957, v. 26, р. 1407—1408.
77. А в г у л ь Н. Н. и др. Изв. АН СССР, ОХН, 1959, с. 1196—1206.
78. Р а с е Е. L., Siebert A. R. J. Phys. Chem., 1959, v. 63, р. 1398—1400.
79. Р а с е Е. L. Proc. 4th Conf. Carbon, Buffalo, 1960, 778 p.
80. А в г у л ь Н. Н., Киселев А. В., Л ы г и н а И. А. Изв. АН СССР,
ОХН, 1961, с. 1395—1403.
81. А в г у л ь Н. Н., Киселев А. В., Л ы г и н а И. А. Изв. АН СССР,
ОХН, 1961, с. 1404—1411.
82. Б е з у с А. Г., Д р е в и н г В. П., К и с е л е в А. В. Коллоидн. ж.,
1961, т. 23, с. 389—398.
83. А в г у л ь Н. Н., Киселев А. В., Л ыгина И. А. Изв. АН СССР,
ОХН, 1962, с. 1346-1353.
84. А в г у л ь Н. Н., К и с е л е в А. В., Л ы г и и а И. А. Изв. АН СССР,
ОХН, 1962, с. 32—37.
85. A v g u 1 N. N., L у g i n a I. A., Kiselev А. V. Trans. Faraday
Soc., 1963, v. 59, p. 2113—2128.
86. К и с e л e в А. В., П о ш к у с Д. П., Афреймович А. Я. ЖФХ,
1964, т. 38, с. 1514—1522.
87. Пошкус Д. П. ЖФХ, 1965, т. 39, с. 2962—2967.
88. Р о s h k u s D. Р. Disc. Faraday Soc., 1965, v. 40, p. 195—204.
89. Рубежный Ю. Г. ЖФХ, 1966, т. 40, с. 2190—2192.
90. Johnson J. D. J. Phys. Chem., 1968, v. 72, p. 3697—3698.
91. Kuznetsov A. V., Guiochon G. J. Chim. phys., 1969, v. 66,
p. 257—261.
92. К и с e л e в А. В. и др. ЖФХ, 1970, т. 44, с. 1272—1278.
93. Prenzlow С. F., В е а г d Н. R., Brundage R. S. J. Phys. Chem.,
1969, v. 73, р. 969—978.
94. Vidal-Madjar С., Jacob L., Guiochon G. Bull. Soc. chim.
France, 1971, p. 3105—3109.
95. P i e г о 11 i R. A., Petricciani J. C. J. Phys. Chem., 1960, v. 64,
p. 1596—1597.
96. Gurthoys G., Elkington P. E. J. Phys. Chem., 1967, v. 71,
p. 1477—1483.
97. Lenel F. V. Z. Phys. Chem., 1933, Bd. В 23, s. 379—399.
98. D e Boer J. H., Custers J. F. H. Z. Phys. Chem., 1934, Bd. В 25,
s. 225—237:
99. Orr W. J. C. Trans. Faraday Soc., 1939, v. 35, p. 1247—1265.
100. Ильин Б. В. Природа адсорбционных сил. М., Гостехтеоретиздат,
1952. 124 с.
101. Young D. М. Trans. Faraday Soc., 1951, v. 47, p. 1228—1233.
102. Drain L, E. Trans. Faraday Soc., 1953, v. 49, p. 650—654.
277
103. H ay aka wa T. Bull. Chem. Soc. Japan, 1957, v. 30, p. 236—243, 343—
349.
104. F i s h e г В. В., Me M i 11 a n M. G. J. Chem. Phys., 1958, v. 28, p. 562—571.
105. Киселев А. В., Пошкус Д. П. ЖФХ, 1958, т. 32, с. 2824—2833.
106. Пошкус Д. П., Кисел ев А. В. ЖФХ, 1960, т. 34, с. 2640—2645.
107. Anderson Р. J., Н о г 1 о с k R. F. Trans. Faraday Soc., 1969, v. 65,
p. 251—258.
108. Kozirovski Y., Folman M. Trans. Faraday Soc., 1966, v. 62,
p. 808—820.
109. Киселев А. В., Лопаткин А. А., Разумова E.P. ЖФХ,
1969, t. 43, c. 1795—1801.
110. Phillips M. A. K., Seymour R. S. Chem. Phys. Letters, 1970,
v. 6, p. 553—554.
111. Kington G. L., Mac L e о d A. C. Trans. Faraday Soc., 1959, v. 55,
p. 1799-1814.
112. Cannon P. Advan. Chem. Series, 1961, v. 33, p. 422—132.
ИЗ. В a r r e r R. M., Wasilewski S. Trans. Faraday Soc., 1961, v. 57,
p. 1140-1152.
114. Barter R. M., Gibbons R. M. Trans. Faraday Soc., 1963, v. 59,
p. 2569-2581, 2875-2887; 1965, v. 61, p. 948-961.
115. Rees L. V. C., W i 11 i a m s C. J. Trans. Faraday Soc., 1964, v. 60,
p. 1973—1984.
116. Бройер П. и др. ДАН СССР, 1965, т. 161, с. 853—856.
117. Бройер П., Киселев А. В., Лопаткин А. А. ДАН СССР,
1966, т. 171, с. 1126—1129.
118. Fiedler К., Spangenberg Н., Schirmer W. Monatsber.
Deutsch Acad, Wiss. Berlin, 1967, Bd. 9, s. 516—527.
119. Бройер П. и др. ЖФХ, 1968, т. 42, с. 2556—2562.
120. Kiselev А. V., L о р a t k i n A. A., In: Molecular Sieves. Ed. Soc.
Chem. Ind., London, 1968. 339 p.
121. Schirmer W. e. a. Chem. Techn., 1970, v. 22, p. 405—409.
122. Peinze T. e. a. Monatsber. Deutsch. Akad. Wiss., Berlin, 1970, Bd. 12,
s. 867-876.
123. Бройер П., Лопаткин А. А. В кн.: «Основные проблемы теории
физической адсорбции». Под ред. М. М. Дубинина и В. В. Серпинского.
М., «Наука», 1970, 475 с.
124. Kiselev А. V. Molecular Sieve Zeolites-II, Adv. Chem. Ser., 1971,
v. 102, p. 37—68.
125. Brauer P., Lopatkin A. A., Stepanez G. Ph. Molecular
Sieve Zeolites-II, Adv. Chem. Ser., 1971, v. 102, p. 97—104.
126. В arrer R. M., Cram P. J. Molecular Sieve Zeolites-II. Adv. Chem.
Ser., 1971, v. 102, p. 105-111.
127. Sargent R. W. H., Whitford C. J. Molecular Sieve Zeolites-II,
Adv. Chem. Ser., 1971, v. 102, p. 144—151.
128. Spangenberg H. J. e. a. Z. Phys. Chem., Leipzig, 1971, Bd. 248, s.
49—64.
129. Kiselev A. V., Lopatkin A. A., Ryaboukhina L. G. Bull.
Soc. chim. France, 1972, p. 1324—1329.
130. Bezus A. G. e. a. J. Col. Interface Sci., 1973, v. 45, p. 386—395.
131. Rosenberg A. J. J. Phys. Chem., 1958, v. 62, p. 1112—1119.
132. Green M., Seiwatz R. J. Chem. Phys., 1961, v. 35, p. 915—921.
133. Бабкин И. Ю., Кисел ев А. В. ДАН СССР, 1959, т. 129, с. 357—
360.
134. Kouznetso v А. V., Vidal-Madjar С., Guiochon G.
Bull. Soc. chim. France, 1969, p. 1440—1448.
135. Kiselev A. V. J. Chromatog., 1970, v. 49, p. 84—129.
136. P о s h k u s D. P. In: Gas —Chromatographie, Berlin 1968, Akademie —Ver-
lag, Berlin, 1968. 500 s.
137. Пошкус Д. П. В кн.: «Основные проблемы теории физической адсорб-
ции». Под ред. М. М. Дубинина и В. В. Серпинского. М., «Наука», 1970,
475 с.
278
138. Poshkus D. P., Af reimo vich A. Ya. J. Chromatog., 1971, v. 58,
p. 55—59.
139. Kiselev A. V., Poshkus D. P. J. Chem. Soc. Faraday Trans I,
1974, v. 70, p. 000.
140. Уббелоде A. P., Льюис Ф. А. Графит и его кристаллические
соединения, М., «Мир», 1965. 256 с.
141. Де Б у р Я. X. В кн.: Катализ. Некоторые вопросы теории и технологии
органических реакции. Под ред. А. А. Баландина и А. М. Рубинштейна.
М., Издатинлит, 1959, 367 с.
142. Pierotti R. A. J. Phys. Chem., 1962, v. 66, р. 1810—1815.
143. Kiselev А. V. Disc. Faraday Soc., 1965, v. 40, p. 205—218.
144. Киселев А. В. ЖФХ, 1967, т. 41, с. 2470-2504.
145. Киселев А. В., Яшин Я. И. Газоадсорбционная хроматография,
М., «Наука», 1967. 256 с. Исправленные и дополненные издания: Kiselev
А. V., Yashin Ya. I. Gas—Adsorption Chromatography, New York, Plenum
Press, 1969. 254 p.; La Chromatographie Gas—Solide., Paris, Masson. 1969.
289 p.
146. Everett D. H. Disc. Faraday Soc., 1965, v. 40, p. 177—187.
147. S i n a n о g 1 u О., P i t z e r K. S. J. Chem. Phys., 1960, v. 32, p. 1279—
1288.
148. Me Lachlan A. D. Molec. Phys., 1964, v. 7, p. 381—388.
149. Wolfe R., Sams J. R. J. Chem. Phys., 1966, v. 44, p. 2181-2188.
150. Mac Rury T. B., Linder B. J. Chem. Phys., 1971, v. 54, p. 2056—
2066.
151. Д з и а л о ш и н с к и й И. E., Лифшиц E. M., Питаевский
Л. П. Успехи физ. наук, 1961, т. 73, с. 381—422; Advances in Physica,
1961, v. 10, р. 165-209.
152. Mavroyanis С. Molec. Phys., 1963, v. 6, p. 593—600.
153. R e n n e M. J., N i j b о e r B. R. A. Chem. Phys. Letters, 1967, v. 1,
p. 317-320.
154. Nijboer B. R. A., R e n n e M. J. Chem. Phys. Letters, 1968, v. 2,
p. 35-38.
155. S k i n n e r H. A., Pilcher G. Quart. Rev. (London), 1963, v. 17,
p. 264—288.
156. Kalb A. J., C h u n g A. L. H., Allen Th. L. J. Am. Chem. Soc.,
1966, v. 88, p. 2938-2942.
157. Longuet-Higgins H. C., Salem L. Proc. Roy. Soc., 1961,
v. A 259, p. 433-441.
158. W i 1 1 i a m s D. E. J. Chem. Phys., 1965, v. 43, p. 4424—4426.
159. Longuet-Higgins H. C. Proc. Roy. Soc., 1956, v. A 235, p. 537—
543.
160. Sparnaay M. J. Physica, 1959, v. 25, p. 217—231.
161. Zwanzig R. J. Chem. Phys., 1963, v. 39, p. 2251—2258.
162. Yorke E. D., Rocco A. G. J. Chem. Phys., 1970, v. 53, p. 764—767;
1971, v. 55, p. 1900-1903.
163. Шимонаев Г. С. ЖФХ, 1967, т. 41, с. 365—368.
164. Haugh E.F., HirschfelderJ. О. J. Chem. Phys., 1955, v. 23,
p. 1778—1796.
165. Sternlicht H. J. Chem. Phys., 1964, v. 40, p. 1175—1188.
166. Rein R., Claverie P., Pollak M. J. Quant. Chem., 1968, v. 2,
p. 129—144.
167. Claverie P., R e i n R. J. Quant. Chem., 1969, v. 3, p. 537—551.
168. Coulson C. A., D a v i e s P. L. Trans. Faraday Soc., 1952, v. 48,
p. 777—789.
169. Schuyer J., Blom L., van Kreveln D.W. Trans. Faraday
Soc., 1953, v. 49, p. 1391—1401.
170. Williams D. E. J. Chem. Phys., 1966, v. 45, p. 3770-3778.
171. Williams D. E. J. Chem. Phys., 1967, v. 47, p. 4680—4684.
172. Китайгородский А. И., Мирская К. В. Кристаллография,
1961, т. 6, с. 507—514.
279
173. Китайгородский А. И., Мирская К. В. Кристаллография,
1964, т. 9, с. 174—181.
174. Kitaigorodsky A. L, Mirskaya К. V. Molec. Crystals and
Liquid Crystals, 1970, v. 6, p. 339—350.
175. Кейбал В. Л. и др. ЖФХ, 1967, т. 41, с. 2234—2239.
176. Киселев А. В., Мигунова И. А., Яшин Я. И. ЖФХ, 1968,
т. 42, с. 1235-1239.
177. Киселев А. В. и др. ЖФХ, 1970, т. 44, с. 1272—1278.
178. Kalashnikova E.V. е. a. Chromatographia, 1971, v. 4, р. 495—
500.
179. Kiselev А. V. Dans: Colloques Internationaux du C. N. R. S., No 201,
Thermochimie. Marseille, 1972, 565 p.
180. Kalashnikova E.V. e. a. Chromatographia, 1974, v. 7, s. 22—25.
181. Киселев А. В. В кн.: Успехи хроматографии. К столетию со дня
рождения М. С. Цвета, М., «Наука», 1972, 295 с.
182. Muller A. Proc. Roy. Soc., 1941, v. А 178, p. 227—241.
183. Hill T. L. J. Chem. Phys., 1948, v. 16, p. 399—404.
184. Crowell A. D. J. Chem. Phys., 1958, v. 29, p. 446—447.
185. Борисова H. П., Волькенштейн M. В. ЖСХ, 1961, т. 2,
с. 346—349.
186. К и т а й г о p о д с к и й А. И. ДАН СССР, 1961, т. 137, с. 116—119.
187. Kitaigorodsky A. I. Tetrahedron, 1961, v. 14, р. 230—236.
188. Tasumi М., Shimanouchi Т. J. Chem. Phys., 1965, v. 43,
p. 1245-1258.
189. Harada I., Shimanouchi T. J. Chem. Phys., 1966, v. 44, p. 2016—
2028.
190. Hermsen R. W., Prausnitz J. M. Chem. Eng. Sci.,1966, v. 21
p. 791-802.
191. Kitaigorodsky A. I. J. Chim. phys., 1966, v. 63, p. 9—16.
192. Harada I.,Simanouchi T. J. Chem. Phys., 1967, v. 46, p. 2708—
2714.
193. De Coen J. L. e. a. Nature, 1967, v. 216, p. 910-913.
194. Китайгородский А. И., Мирская К. В., Товбис А.Б.
Кристаллография, 1968, т. 13, с, 225—231.
195. М о m а п у F. A., Vanderkooi G., Scheraga Н. A. Proc. Nat.
Acad. Sci., USA, 1968, v. 61, p. 429-436.
196. Snook I. K., S p u r 1 i n g T. H. Austral J. Chem., 1970, v. 23, p. 819 —
823.
197. Warshel A., Lifson S. J. Chem. Phys., 1970, v. 53, p. 582—594.
198. Muller A. Proc. Roy. Soc., 1936, v. A 154, p. 624—639.
199. Давыдова M. А., Стручков Ю. T. ЖСХ, 1962, t. 3, c. 184—199.
200. Salem L. J. Chem. Phys., 1962, v. 37, p. 2100-2113.
201. Kim el S., R о n A., Horing D. F. J. Chem. Phys., 1964, v. 40,
p. 3351—3356.
202. Chapman D., Whittington S. G. Trans. Faraday Soc., 1964,
v. 60, p. 1369-1377.
203. Китайгородский А. И., Корешков Б. Д., Кул ь-
кин А. Г. Физ. тв. тела, 1965, т. 7, с. 643—645.
204. К i t a i g о г о d s к у A. I. Acta cryst., 1965, v. 18, р. 585—590.
205. Давыдова М. А.,Стручков Б.Т. ЖСХ, 1965, т. 6, с. 113—
122.
206. Мс К i n 1 е у М. D., Reed Т. М. J. Chem. Phys., 1965, v. 42, р. 3891 —
3899.
207. Craig D. Р. е. a. Proc. Roy. Soc. (London), 1965, v. A 286, p. 98—
116.
208. Craig D. P. e. a. Disc. Faraday Soc., 1965, v. 40, p. 110—116.
209. Зоркий П. M. и др. ЖСХ, 1966, т. 7, с. 914-916.
210. Зоркий П. М. и др. ЖСХ, 1967. т. 8, с. 312—316.
211. Giglio Е., Liguori А. М. Acta cryst, 1967, v. 22, р. 437—438.
212. П о л т е в В. И., Сухоруков Б. И. ЖСХ, 1968, т. 9, с. 298—304.
280
2!3. Китайгородский А. И., Высокомол. соед., 1968, т. А 10,
с. 2669-2671.
214. С lure D. W. J. Chem. Phys., 1968, v. 49, p. 1830—1839.
215. Giglio E. Nature (Engl.), 1969, v. 222, p. 339—341.
216. Мирская К. В., Козлова И. Е. Кристаллография, 1969, т. 14,
с. 412—416.
217. Mantione М. J. Theoret. chim. Acta (BerL), 1969, v. 15, p. 141—148.
218. Bartell L. S. J. Chem. Phys., 1960, v. 32, p. 827—831.
219. Cullough R. L., Me Mahon P. E. Trans. Faraday Soc., 1964, v. 60,
p. 2089—2096.
220. CignittiM.,Allen Th. L. J. Chem. Phys., 1965, v. 43, p. 4472—4478.
221. Дашевский В. Г. ЖСХ, 1965, т. 6, с. 888—897.
222. Scott В. A., S с h е г a g а Н. A. J. Chem. Phys., 1966, v. 44, р. 3054—
3069.
223. Scott R. A., S c h e r a g a H. A. J. Chem. Phys., 1966, v. 45, p. 2091 —
2101.
224. Abe A., J ernigan R. L., Flory P. J. J. Am. Chem. Soc., 1966,
v. 88, p. 631-639.
225. Дашевский В. Г. ЖСХ, 1966, т. 7, с. 93—102.
226. Дашевский В. Г.,Китайгородский А. И. Теорет. и экспер.
хим., 1967, т. 3, с. 43—57.
227. Hendrickson I.B. J. Am. Chem. Soc., 1967, v. 89, p. 7036—7043.
228. Allinger N. L. e. a. J. Am. Chem. Soc., 1967, v. 89, p. 4345—4357.
229. Allinger N. L. e. a. J. Am. Chem. Soc., 1968, v. 90, p. 1199—1210.
230. Дашевский В. Г. ЖСХ, 1963, т. 4, с. 637-640; 1968, т. 9, с. 289—
297; 1970, т. 11, с. 489—501.
231. Kitaigorodsky A. I., Dashevsky V. G. Tetrahedron, 1968,
v. 24, р. 5917—5928.
232. L i f s о n S., W a г s h e 1 A. .J. Chem. Phys., 1968 v. 49, p. 5116—5129.
233. Mark J. E. J. Phys. Chem, 1968, v. 72, p. 2941-2946.
234. Williams J.E. e. a. Annual Review of Physical Chemistry, 1968,
v. 19, p. 531-558.
235. Hill T. L. J. Chem. Phys., 1946, v. 14, p. 465.
236. Hill T. L. J. Chem. Phys., 1948 v. 16, p. 938—949.
237. Pitzer K. S., C a t a 1 a n о E. J. Am. Chem. Soc., 1956, v. 78,
p. 4844—4846.
238. Борисова H. П., Волькенштейн M. В. ЖСХ, 1961, т. 2,
с. 469—475.
239. Борисова Н. П., БирштейнТ. М. Высокомол. соед., 1963, т. 5,
с. 279—286.
240. С ou Iso п С. А., II a i g h С. W. Tetrahedron, 1963, v. 19, p. 527—544.
241 .Cignitti M., Allen T. L. J. Phys. Chem., 1964, v. 68, p. 1292—
1297.
242. Scott R. A., S c h e r a g a H. A. J. Chem. Phys., 1965, v. 42, p. 2209—
2215.
243. S о v e г s 0. J., К a p p 1 u s M. J. Chem. Phys., 1966, v. 44, p. 3033—
3037.
244. Jacob F. J., Thompson H. B., Bartell L. S. J. Chem.
Phys., 1967, v. 47, p. 3736-3753.
245. Грушецкий К. M., Бесценный А. Ф. ЖСХ, 1967, т. 8,
с. 325-331, 332—337.
246. Miller К., Murrell J. N. Trans. Faraday Soc., 1967, v. 63, p. 806—
811.
247. Китайгородский А. И., Дашевский В. Г. Теорет. и экс-
пер. хим., 1967, т. 3, с. 35—42.
248. Грушецкий К. М. ЖСХ, 1968, т. 9, с. 870—874.
249. Вилков Л. В., Садова Н. И., Тарасенко Н. А. ЖСХ,
1969, т. 10, с. 403—406.
250. Дашевский В. Г., Наумов В. А., Зарипов Н. М. ЖСХ,
1970, т. 11, с. 746-752.
281
251. И л и е л Э. и др. Конформационный анализ. М., «Мир», 1969, 592 с.
252. Pitzer К. Advanc. Chem. Phys., 1959, v. 2, p. 59—83.
253. Китайгородский А. И., Мирская К. В. В кн.: «Основные
проблемы теории физической адсорбции». Под ред. М. М. Дубинина
и В. В. Серпинского. М., «Наука», 1970, 475 с.
254. . Kuan Т. S., W а г s h е 1 A., Schnepp О. J. Chem. Phys., 1970,
v. 52, р. 3012—3020.
255. Margenau Н. Phys. Rev., 1939, v. 56, р. 1000—1008.
256. Murrell J. N., Randic M., Willi am s D, R. Proc. Roy.
Soc. (London), 1965, v. A 284, p. 566—581.
257. Salem L. Disc. Faraday Soc., 1965, v. 40, p. 150—163.
258. Musher J. I., Salem L. J. Chem. Phys., 1966, v. 44, p. 2943-2946.
259. Murrell J. N., Shaw G. J. Chem. Phys., 1967, v. 46, p. 1768—
1772.
260. M u г г e 1 1 J. N., Shaw G. Molec. Phys., 1967, v. 12, p. 475—480.
261. Van der Avoird A. J. Chem. Phys., 1967, v. 47, p. 3649—3653.
262. Slater J. C., Kirkwood J. G. Phys. Rev., 1931, v. 37, p. 682—
697.
263. Abrahamson A. A. Phys. Rev., 1963, v. 130, p. 693—707.
264. Matcha R. L., Nesbet R. K. Phys. Rev., 1967, v. 160, p. 72—75.
265. Kestner N. R., Sinanoglu O. J. Chem. Phys., 1966, v. 45,
p. 194-207.
266. Abrahamson A. A. Phys. Rev., 1969, v. 178, p. 76—79.
267. Margenau H. Rev. Mod. Phys., 1939, v. 11, p. 1—35.
268. Dalgarno A., Lewis J. T. Proc. Phys. Soc., 1956, v. 69,
p. 57—64.
269. Dalgarno A., Lynn N. Proc. Phys. Soc., 1957, v. A 70, p. 223—225.
270. Page С. H. Phys. Rev., 1938, v. 53, p. 426—430.
271. Dabler J. S., Hirschfelder J. O. J. Chem. Phys., 1956,
v. 25, p. 986—1005.
272. Murrell J. N., Shaw G. J. Phys. Chem., 1968, v. 49, p. 4731—
4733.
273. Kreek H., Meath W. J. J. Chem. Phys., 1969, v. 50, p. 2289—2302.
274. Margenau H. Phys. Rev., 1931, v. 38, p. 747—756.
275. Kim D. I. Z. Phys., 1962, Bd. 166, s. 359-369.
276. Murrell J. N., Shaw G. Molec. Phys., 1968, v. 15, p. 325—328.
277. Buckingham R. A., Corner J. Proc. Roy. Soc. (London), 1947,
v. A 189, p. 118—129.
278. Corner J. Trans. Faraday Soc. 1948, v. 44, p. 914—927.
279. В a r u a A. K. J. Chem. Phys., 1959, v. 31, p. 957—960.
280. В а г u a A. K. Indian J. Phys., 1960, v. 34, p. 76—84.
281. Rossi J. C., D a n о n F. Disc. Faraday Soc., 1965, v. 40, p. 97—109.
282. Gordon R. G. J. Chem. Phys., 1968, v. 48, p. 3929—3934.
283. Barker J. A., Pompe A. Aust. J. Chem., 1968, v. 21, p. 1683—
1694.
284. De Boer J. H., Heller G. Physica, 1937, v. 4, p. 1045—1057.
285. Lippincott E. R., S t u t m a n J. M. J. Phys. Chem., 1964, v. 68,
p. 2926-2940.
286. Meyer E. F., Deitz V. R. J. Phys. Chem., 1967, v. 71, p. 1521 —
1523.
287. M e у e r E. F. J. Phys. Chem., 1967, v. 71, p. 4416—4421.
288. Mey er E. F. J. Chem. Phys., 1968, v. 48, p. 5284—5285.
289. Dalgarno A. Advanc. Chem. Phys., 1967, v. 12, p. 143—166.
290. Dalgarno A., Lynn N. Proc. Phys. Soc. (London), 1957, v. A 70,
p. 802—808.
291. Dalgarno A., Kingston A. E. Proc. Phys. Soc. (London). 1961,
v. 78, p. 607-609.
292. Barker J. A., Leonard P. J. Phys. Letters, 1964, v. 13, p. 127—
128.
293. Kingston A. E. Phys. Rev., 1964, v. 135, p. A 1018—A 1019.
282
294. Dalgarno A. Advanc. Chem. Phys., 1967, v. 12, p. 143—166.
295. Davison W. D. J. Phys. (Proc. Phys. Soc.), 1968, v. В 1, p. 597—604.
296. Dalgarno A., Morrison I. H., Pengally R. M. Intern.
J. Quantum Chem., 1967. v. 1, p. 161—167.
297. Casimir H. B. G., Polder D. Phys. Rev., 1948, v. 73, p. 360—372.
298. Mavroyanis C., Stephen M. J. Mol. Phys., 1962, v. 5, p. 629—
638.
299. Me Lachlan A. D. Proc. Roy. Soc., 1963, v. A271, p. 387—401.
300. Langhoff P. W., Karplus M. J. Chem. Phys., 1970, v. 53,
p. 233—250.
301. Kramer H. L. J. Chem. Phys., 1970, v. 53, p. 2783—2791.
302. Starksch all G., Gordon R. G. J. Chem. Phys., 1971, v. 54,
p. 663—673.
303. Лондон Ф. Успехи физ. наук, 1937, т. 17, с. 421—446.
304. Hornig J. F., Hirschfelder J. О. J. Chem. Phys., 1952, v. 20,
p. 1812.
305. Slater J. C., Kirkwood J. G. Phys. Rev. 1931, v. 37, p. 682—
697.
306. Kramer H. L., Herschbach D. R. J. Chem. Phys., 1970, v. 53,
p. 2792—2800.
307. Kirkwood J. G. Phys. Z., 1932, Bd. 33, s. 57—60.
308. Heller R. J. Chem. Phys., 1941, v. 9, p. 154—163.
309. Girifalco L. A., Lad R. A. J. Chem. Phys., 1956, v. 25, p. 693—
697.
310. Pauling L. The Nature of the Chemical Bond. New York, 1960. 3 ed.
450 p.
311. Good R. J., H о p e С. J. J. Chem. Phys., 1970, v. 53, p. 540—543.
312. Good R. J., Hope C. J. J. Chem. Phys., 1971, v. 55, p. 111—116.
313. Steele W. A., Ross M. J. Chem. Phys., 1961, v.£35, p. 850—862.
314. X ц л л T. В кн.: «Катализ. Вопросы теории и методы исследования».
Издатинлит, М., 1955. 571 с.
315. Кроуэлл А. В кн.: «Межфазовая граница газ — твердое тело». Под
ред. Э. Флада. М., «Мир», пер. с англ, яз., 1970. 433 с.
316. Пошкус Д. П., Афреймович А. Я. ЖФХ, 1966, т. 40, с. 2185—
2189
317. Hove J., Krumhansl J. A. Phys.Rev., 1953, v. 92, р. 569—572.
318. Fumi F. G., Tosi M. P. Phys. Rev., 1960, v. 117, p. 1466—1468.
319. Степанец Г. Ф., Лопаткин А. А. ЖФХ, 1967, т. 41, с. 2746—
2572.
320. Степанец Г. Ф. Кандидат, дисс. М., МГУ, 1968, 101 с.
Глава IX
РАСЧЕТ ТЕРМОДИНАМИЧЕСКИХ ХАРАКТЕРИСТИК АДСОРБЦИИ
ОДНОАТОМНЫХ МОЛЕКУЛ НА ГРАФИТЕ
1. Влияние формы атом-атомного потенциала межмолекулярного
взаимодействия на значение константы Генри Кх
Расчеты константы Генри Кг для адсорбции одноатомных молекул
на базисной грани полубесконечной решетки графита производились
при использовании разных форм для атом-атомного потенциала (р:
Сюзерленда (6, оо) fl], Леннард-Джонса (6, 12) [1—5], Бакингема
(6, ехр) [5] и Бакингема — Корнера (6, 8, ехр) [5]. Эти атом-атомные
потенциалы различаются как формой члена, аппроксимирующего
потенциал сил отталкивания, так и числом членов,описывающих
потенциал сил дисперсионного притяжения. Были сопоставлены
рассчитанные значения константы при фиксированных значе-
ниях параметров &!кТ и г0 потенциалов (р взаимодействия атома благо-
родного газа с атомом углерода графита в форме (VIII,17) [5] с рас-
считанными значениями Кх при фиксированных значениях парамет-
ров Ф$!кТ и zQ для соответствующих потенциальных функций Ф
взаимодействия одноатомной молекулы с базисной гранью полу-
бесконечной решетки графита в форме [1, 5]:
Ф=Фо/(—) (IX,1)
где Фо — энергия в минимуме при z = z0; / (z/z0) — функция формы.
В табл. IX.1 сопоставлены значения Ф$!кТ и Кг1г$ для
адсорбции одноатомных молекул на базисной грани графита, рассчи-
танные при использовании атом-атомных потенциалов Леннард-
Джонса (VIII,14), Бакингема (VIII,15) и Бакингема — Корнера
(VIII,16) при фиксированных значениях s/кТ и r0 = d [5]. Сумми-
рование потенциалов <р по атомам углерода решетки графита произ-
водилось при использовании приближения Крауэлла (VIII,46).
Суммирование потенциала сил отталкивания производилось только
по атомам С наружной базисной плоскости. Соответствующие по-
тенциальные функции Ф даются выражениями (VIII,48) и (VIII,49).
Значения полученные при использовании потенциала Леннард-
Джонса (6, 12), зависят лишь от значения &1кТ. Значения Krir^
полученные при использовании потенциала Бакингема (6, ехр),
зависят уже от трех параметров: &1кТ, г0 и q, а значения Кг/г^
полученные при использовании потенциала Бакингема — Корнера
(6, 8, ехр), зависят от четырех параметров: &1кТ, r0 , q nC2/Cv
284
Таблица IXtla Значения z0/r0, Ф0/кТ и для адсорбции
одноатомных молекул на базисной грани графита, рассчитанные
при использовании атом-атомных потенциалов Леннард-Джонса (6,12),
Бакингема (6, ехр) и Бакингема — Корнера (6,8,ехр)
при фиксированных значениях параметров г/кТ и гo = d
Рассчитанные величины Принятые значения e/feT Использованные потенциалы
(6,12) (6, ехр) при 9=36 нм-1 (6,8, ехр)прид=36 нм-1, C2/Ci = 0,2d2
zo/ro 0,887 0,871 0,872
-Фь/kT 0,1 1,42 1,48 1,44
0,2 2,84 2,95 2,87
0,5 7,10 7,39 7,17
1,0 14,2 14,8 14,3
К1/г0 0,1 0,524 0,719 0,620
0,2 3,97 4,86 4,34
0,5 187 268 216
1,0 1,48 ПО5 2,87*105 1,86-105
Параметр q был принят равным 36 нм-1, а параметр C2/Ci — равным
0,2 d2.
Как видно из табл. IX,1, при фиксированных значениях &!кТ
и г0 величины и0 и Фо слабо зависят от формы атом-атомного потен-
циала. Однако Кг заметно зависит от формы этого потенциала.
Поэтому при расчетах термодинамических характеристик адсорбции
необходимо использовать теоретически наиболее оправданную форму
для атом-атомного потенциала.
В табл. IX,2 сопоставлены значения Хх/годля адсорбции одно-
атомных молекул на базисной грани графита, рассчитанные при
использовании атом-атомных потенциалов (6, 12), (6, ехр) и (6, 8,
ехр) при фиксированных значениях параметров Ф0/АГ и z0 = d
потенциала Ф в форме (IX,1).
Таблица IXt2, Значения K^/zq для адсорбции одноатомных молекул
на базисной грани графита, рассчитанные при использовании
атом-атомных потенциалов Леннард-Джонса (6, 12),
Бакингема (6, ехр) и Бакингема — Корнера (6, 8, ехр) при фиксированных
значениях параметров Ф^/кТ и zQ = d
Принятые значения -Фо/ЬТ Значения Ki/z0 при использо- вании потенциалов Принятые значения -Фо/ЬТ Значения Ki/z9 при использо- вании потенциалов
(6,12) (6, ехр) при 9=36 нм”1 (6, 8, ехр) при 9 = 36 нм-1, C2/Ci = 0,2 d2 (6, 12) (6, ехр) при 9=36 нм-1 (6, 8, ехр) при 9 = 36 нм”1, Cs/Ci-=0,2 d2
1,6 0,902 0,915 0,872 6,4 ИЗ 115 113
2,8 4,34 4,37 4,26 7,6 335 342 335
4,0 13,4 13,5 13,2 8,8 1013 1035 1014
5,2 38,8 39,3 38,5 10,0 3106 3175 3115
285
Значения K-jz^ полученные при использовании потенциала (6,
12), зависят лишь от значения параметра Ф^/кТ. Значения K^z^
полученные при использовании потенциала (6, ехр), зависят от трех
параметров: Ф^/кТ^ z0 и а значения Ki/z0, полученные при ис-
пользовании потенциала (6, 8, ехр), зависят от четырех параметров:
Ф0/кТ, zQ, q и С2/Сг. Параметры q и С2/Сг были приняты соответ-
ственно равными 36 нм-1 и 0,2 d2. Как видно из табл. IX,2, зна-
чения Kjzq различаются менее чем на 5% для рассмотренных трех
атом-атомных потенциалов. Таким образом, при заданном способе
суммирования потенциала ф и фиксированных значениях парамет-
ров Ф^кТ и z0 потенциала Ф значения Кх мало чувствительны к при-
нятой форме для атом-атомного потенциала ф. Они определяются
главным образом значениями параметров Ф^кТ и zQ.
2. Влияние приближений при суммировании атом-атомных
потенциалов на значения константы Генри Кг
Расчеты константы Генри Кг для адсорбции одноатомных молекул
на базисной грани графита производились на основании потенциаль-
ных функций Ф, полученных при использовании различных при-
ближений при суммировании одного и того же атом-атомного потен-
циала ф по атомам углерода решетки графита: при интегрировании
атом-атомного потенциала по всему объему [1—3] или по одной
наружной атомной плоскости [1, 3] полубесконечного твердого тела,
при интегрировании по объему твердого тела только потенциала
сил притяжения атом-атомного потенциала [1, 3, 4], при исполь-
зовании приближения Крауэлла (VIII,46) [5], а также при учете
атомного строения наружной атомной плоскости графита [5]. Рассчи-
танные значения константы Кг при фиксированных значениях пара-
метров &1кТ и г0 потенциала ф в форме (VIII,17) [5] сопоставлялись
со значениями Кг при фиксированных значениях параметров Ф^/кТ
и z0 потенциала Ф в форме (IX,1) [1, 5].
В табл. IX,3 приведены значения zjr^ Ф^кТ и KxlrQ для ад-
сорбции одноатомных молекул на базисной грани графита при
фиксированных значениях параметров &1кТ и rQ = d, рассчитанные
при использовании следующих приближений . при суммировании
атом-атомного потенциала Леннард-Джонса (VIII,14) по атомам
углерода графита.
1. При использовании приближения (VIII,50), учитывающего
атомное строение решетки графита. При этом было принято N = 36,
I __ 1, L = 3.
2. При использовании приближения Крауэлла (VIII,46). В этом
случае
ф-Е^[-5-(т)'с(т- 4)]
3. При использовании приближения Крауэлла (VIII,46) и учете
вклада в потенциал сил отталкивания только наружной атомной
286
Таблица IXt3. Значения z0/r0, Ф$/кТ и KjJrQ для адсорбции
одноатомных молекул на базисной грани графита, рассчитанные при разных
приближениях при суммировании по атомам углерода графита
атом-атомного потенциала (6,12) при фиксированных значениях
параметров &/кТ и rQ~d. В случае приближения 1 значения zq/pq
и Ф^/кТ приведены для двух положений молекулы относительно решетки:
над атомом С графита(г) и над центром шестиугольника
из атомов С графита (h)
Прибли- жение Zq/Tq —Фо/kT при значениях г/kT, равных Ki/tq при значениях e/kT, равных
0,1 0,2 0,5 1,0 0,1 0,2 0,5 1,0
1 0,896 (с) 1,39 (с) 2,77 (с) 6,94 (с) 13,9 (с)
0,865 (h) 1,50(Л) 3,00 (Ь) 7,49 (М 15,0(/г) — 4,0 190 1,58-105
2 0.881 1,42 2,84 7,10 14,2 0,523 3,97 187 1,47-105
3 0,887 1,42 2,84 7,10 14,2 0,524 3,97 187 1,48-105
4 0,891 1,28 2,57 6,41 12,8 0,282 2,82 98,3 3,92 • 105
5 0,765 0,669 1,34 3,35 6,69 -0,100 0,644 7,10 1,35-102
плоскости графита. Потенциальная функция Ф в этом случае дается
выражением (VIII,48).
4. При интегрировании атом-атомного потенциала по одной на-
ружной базисной плоскости графита, принимая равномерное распре-
деление плотности атомов С в этой плоскости. В этом приближении
и-т <«>
U \ <6 / |
где zQ — и Фо = —(6/5)/2) nxsrg.
5. При интегрировании атом-атомного потенциала по всему
объему полубесконечной решетки графита, принимая равномерное
распределение атомов С (приближение Лондона). В этом приближе-
нии
»и* m1 - (--н
где z0 = г0/у^5“ и Фо = — (10/9) Eimg.
В случае приближений 4 и 5 значения зависят только от
значения параметра г!кТ. В случае приближений 1—3 значения KrlrQ
зависят еще от значения параметра г0, однако эта зависимость слабая.
Как видно из табл. IX,3, значения z0, Фо и К19 полученные
при пренебрежении атомным строением базисных плоскостей графита,
но с учетом слоистого строения его решетки (приближения 2 и 3),
близки к значениям этих величин, полученным при учете атомного
строения этих плоскостей графита (приближение 1). Значения Фо
и Ki полученные при использовании более грубого приближения 4,
в котором учитывают вклад в потенциальную функцию Ф лишь
наружной базисной плоскости графита и пренебрегают атомным
строением этой плоскости, несколько ниже значений этих величин,
287
полученных при использовании приближений 1—3. Значения z0,
Фо и Ки полученные при использовании приближения Лондона
(приближение 5), предполагающего непрерывное распределение
вещества во всей решетке графита, т. е. пренебрегающего не только
атомным строением базисных плоскостей графита, но и слоистым
строением его решетки, значительно ниже значений этих величин,
полученных при использовании всех других рассмотренных выше
приближений. Значения z0 и Фо, полученные при использовании
приближения 1, зависят от положения атома над базисной гранью
графита. Однако эта зависимость слабая и она мало сказывается на
значениях Кг.
Таким образом, пренебрежение атомным строением базисных
плоскостей графита (приближение Крауэлла) при суммировании
атом-атомного потенциала межмолекулярного взаимодействия слабо
сказывается на рассчитанных значениях константы Кг для адсорб-
ции одноатомных молекул на базисной грани графита. Пренебре-
жение же слоистым строением графита (приближение Лондона)
приводит к сильно заниженным значениям этой константы. Поэтому
суммирование атом-атомных потенциалов необходимо производить
с учетом слоистого строения решетки графита.
Таблица IX,4. Значения KyJzQ для адсорбции одноатомных молекул
на базисной грани графита, рассчитанные при разных приближениях
при суммировании атом-атомного потенциала Леннард-Джонса (6,12)
по атомам углерода графита при фиксированных значениях
параметров Фъ/kT и z0
•|b в]*: 1 Значения Ki/z0 при использовании
приближения 2 приближения 4 приближения 5
zo = 0,9d z0 = d z0= 1 ,ld
1,6 0,890 0,902 0,911 0,797 1,37
2,8 4,31 4,34 4,37 4,08 5,59
4,0 13,3 13,4 13,5 12,8 16,6
5,2 38,6 38,8 39,0 37,3 47,2
6,4 112 ИЗ 114 109 136
7,6 334 335 337 324 402
8,8 1008 1013 1017 980 1211
10,0 3090 3106 3121 ЗОИ 3707
В табл. IX,4 приведены значения K-JzQ для адсорбции одноатом-
ных молекул на базисной грани графита при фиксированных значе-
ниях параметров Ф$!кТ и zQ потенциала Ф, рассчитанные при исполь-
зовании атом-атомного потенциала (6, 12) и вышеуказанных при-
ближений 2, 4 и 5 при суммировании этого потенциала. В случае
приближений 4 и 5 значения K-JzQ зависят только от значения
ф^кТ. В случае же приближения 2 значения K^lz^ зависят еще
от значения z0, однако последняя зависимость сравнительно слабая.
Как видно из табл. IX,4, значения Кг, полученные при использо-
вании приближений 2 и 4, учитывающих слоистое строение графита,
288
сравнительно близки. Вместе с тем, значения Klf полученные при
использовании приближения 5, пренебрегающего слоистым строе-
нием графита, на 20—50% больше значений К19 полученных при
использовании приближения 2. Таким образом, пренебрежение
слоистым строением решетки графита при суммировании атом-
атомного потенциала сильно влияет на рассчитанные значения Кг
как при фиксированных значениях параметров zlkT и г0 потенциала
ср, так и при фиксированных значениях параметров <&QlkT и z0 по-
тенциала Ф. Поэтому слоистое строение графита необходимо учиты-
вать как при определении потенциала Ф на основании потенциала q>
(при суммировании этого потенциала), так и при определении потен-
циала Ф на основании опытных адсорбционных данных, чтобы пра-
вильно выбрать форму этого потенциала.
3. Гармоническое приближение
При расчетах термодинамических характеристик адсорбции бла-
городных газов во многих работах [6—20] колебание центра масс
адсорбированной молекулы перпендикулярно поверхности рас-
Рис. IX,1. Отношения рассчитанных значений и Х1ггарм. осцил прн фикси-
рованных значениях и z0= d для адсорбции одноатомных молекул на
базисной грани графита.
Кривая J получена при использовании потенциала (IX,2); кривая 2 — при использовании
потенциала (VIII,49) при q =36 нм”1 и Ci/C, = 0; кривая з — при использовании потенциала
(VIII.49) при q = 36 нм*1 и Ct/Ct = 0,2d*; кривая 4 — при использовании потенциала
(IX,4) и кривая J — при использовании потенциал^ (IX,3).
сматривалось как гармоническое. Этому приближению соответствует
разложение Ф в ряд Тэйлора по степеням z — z& и учет в этом ряду
только первых двух членов, т. е.
Ф = Фо+-тг-(«~ *o)2 (IX.5)
где — вторая производная функции Ф по z при z == z0 (см. стр. 236).
В случае потенциалов (IX,3) и (IX,4)
ф;=—dx,6)
z0
где Р — константа, зависящая от формы потенциала.
19 заказ И 80 289
В случае потенциала (IX,3) Р = 40, а в случае потенциала
(IX,4) Р = 27. В случае потенциала (IX,2) параметр Р зависит
от значения z0, а в случае потенциала (VIII,49) — от значений z09
CZIC1 и q.
Константа Генри для адсорбции одноатомных молекул в гармо-
ническом приближении -ЙГ1, гарм. осцил ПРИ предположении, что
Фо и Фх не зависят от положения атома над поверхностью, т. е.
от координат х и у атома, дается выражением (VII,55). На рис. IX,1
приведены отношения констант Генри гарм>осцил при фикси-
рованных значениях Ф0/&Г и z0 — d, рассчитанные при использо-
вании потенциалов (VIII,49), (IX,2)—(IX,4). Эти отношения при
Фо/&7 >3 лежат в пределах от 1,1 до 1,25, т. е. погрешность в рас-
считанных значениях/^, вызываемая гармоническим приближением,
не превышает 25%.
4. Оценка термодинамических характеристик адсорбции
благородных газов на базисной грани графита на основании
свойств адсорбата и адсорбента <
взятых в отдельности
Как было сказано в гл. VIII, параметры атом-атомных потен-
циальных функций взаимодействия атомов адсорбата с атомами
адсорбента можно оценить на основании свойств адсорбата и адсор-
бента, взятых в отдельности. Их можно оценить при использовании
квантовомеханических формул для параметров сил дисперсионного
притяжения и правил комбинирования для параметров сил оттал-
кивания или при использовании одних только правил комбинирова-
ния параметров сил притяжения и сил отталкивания. Рассмотрим
результаты расчета термодинамических характеристик адсорбции
благородных газов на базисной грани графита, получающихся при
таком способе оценки параметров потенциальной функции взаимо-
действия атомов благородных газов с атомом С графита [8, 12—16,
21].
Для потенциальной функции ф взаимодействия атома благород-
ного газа с атомом углерода графита в этих работах был принят
потенциал Бакингема — Корнера (VIII,16). Параметры Сг и С2
сил притяжения этого потенциала были оценены с помощью формулы
Кирквуда — Мюллера (VIII,25) для С\ и аналогичной ей формулы
(VIII,33) для С2. Использованные при этом значения поляризуемости
а и диамагнитной восприимчивости % атомов благородных газов и
углерода приведены ниже [25, 26]:
Атом . . Ne Аг Кг Хе С
а - 1О30, мз . . • , . 0,398 1.63 2,48 4,01 0,937
—% • 10зв, м3 . , , . 11,95 32,20 46,50 71,40 10,54
Полученные значения параметров ^1» С-а И С21С1 приведены
в табл. IX,5.
*290
Таблица 1Х,5. Значения параметров С2 и С^/С^ потенциала
Бакингема — Корнера (VIII, 16) для взаимодействия атомов
благородных газов с атомом С графита
Атом С1*108Т, кДж«мв»моль”1 С2.10”, кДж «м8, моль-1 (Сг/С1)*102% м3 Значения Ci взяты из второго столбца, т. е. по формуле (VIII,25)
По фор- муле (VIII.25) По формуле (VIII,29)
Ne 0,90 0,92 [27, 28] 0,86 [29] 1,12 1,24
Аг 3,24 3,10 [27, 28] 2,93 [29] 3,01 [19] 4,35 1,34
Кг 4,86 4,06 [27—29] 6,57 1,35
Хе 7,66 6,57 [27] 6,32 [29] 10,60 1,38
Приведенные в таблице значения параметра Сг близки к значе-
ниям, оцененным аналогичным способом в работах [22—24]. В табл.
1Х,5 приведены также значения С\, полученные при использовании
правила комбинирования (VIII,29). Для Ne эти значения практически
совпадают со значением, оцененным с помощью формулы Кирквуда —
Мюллера. Для Аг они на 5—10%, а для Кг и Хе на 14—18% меньше
значений, оцененных с помощью формулы Кирквуда — Мюллера.
Параметр q для межмолекулярного взаимодействия атомов бла-
городных газов с атомом G графита был оценен при помощи правила
комбинирования (VIII,34). Для взаимодействия атомов G графита
было принято значение q = 36 нм"1. Это значение получено при
использовании свойств решетки графита [30]. Значения параметра q
для взаимодействия атомов благородных газов друг с другом опре-
делены в ряде работ [31—36]. Принятые значения параметра q для
взаимодействия атомов благородных газов друг с другом и получен-
ные значения q для взаимодействия атомов благородных газов с ато-
мом С графита приведены ниже:
Атом ....................... Ne Аг Кг
^...6 нм-i ....... . 42 [31] 34 [31] 29 [33]
qi с нм-1 ........................ 38 36 32
х • • • ъ,
Хе
29 [33]
32
Параметр В был выражен через остальные параметры и равно-
весное расстояние г0 взаимодействия атома благородного газа и атома
С графита с помощью выражения, полученного из условия равнове-
сия (VIII,37):
„__ 661 exp(gr0) / 4 Съ
Г? к Ф 3 (\г*
расстояние Го,/...с взаимодействия атома бла-
с атомом G графита было определено как среднее
значений равновесных расстояний межмолекуляр-
(IX,7)
Равновесное
городного газа
а рифметическое
пого взаимодействия атомов благородных газов it £ и меж-
молекулярного взаимодействия атомов G графита rOj с... с* Для
19*
291
равновесного расстояния г0,с.,.с было принято значение 0,382 нм,
полученное из свойств решетки графита [30, 37]. Для r0> £ 1£ были
приняты значения, полученные из свойств благородных газов [31,
33]. Принятые в расчетах значения г0, /... / и ro, t... с и оценен-
ные с помощью выражения (IX,7) значения параметра В приведены
ниже:
Атом . .... Ne Аг Кг Хе
г0, /.../, нм .... . - 0,316 [31] 0,387 [31] 0,408 [33] 0,445 [33]
Го» < • • С’ КМ ; .... 0,349 0,385 0,395 0,414
В • 10"Б, кДж * моль-1 . * . ♦ 1,72 4,56 2,29 4,78
Приведенные значения r0, i... с практически совпадают со зна-
чениями, полученными аналогичным способом в работах [19, 27—29].
Рис. IX,2. Рассчитанные (кривые) при использовании потенциала (VIII,49)
и опытные (точки) значения константы Генри Кг для адсорбции Ne (I), Аг (2),
Кг (3) и Хе (4) на базисной грани графита (расчёт) и на Графитированных тер-
мических сажах (опыт).
Способы расчета сплошных и пунктирных кривых указаны в тексте. Величины Kt выражены
в мкм. Белые точки измерены статическим методом [1,40—44], черные точки получены
газохроматографическим методом В. Л. Худяковым и Е. В. Калашниковой. / £
При суммировании атом-атомных потенциальных функций ф
(6, 8, ехр) по атомам G графита было использовано приближение
Крауэлла (VIII,46), т. е. суммирование функций ф по атомам С,
лежащим в той же базисной плоскости графита, заменялось инте-
грированием. Учитывался вклад в потенциал сил отталкивания
лишь одной наружной базисной плоскости графита. В этом прибли-
жении потенциальная энергия Ф межмолекулярного взаимодействия
атома благородного газа с базисной гранью графита дается выра-
2Й2
жением (VIII,49). Зета-функция Римана (zld, п) вычислялась по
приближенной формуле (VIII,51).
Расчеты термодинамических характеристик адсорбции благо-
родных газов на базисной грани графита в классическом приближе-
нии производились при использовании статистических выражений
(VII,44)—(VII,48), (VII,51). Квантовомеханические поправки оце-
нивались согласно приближению Птицера — Гвина (VII,66) по
формулам (VII,67)—(VII,72). На рис. IX,2 и IX,3 рассчитанные
зависимости логарифма константы Генри In Кг и изменения при ад-
сорбции внутренней энергии AUi сопоставлены с эксперименталь-
ными значениями, полученными в разных работах [1, 9, 38—44].
Сплошные кривые на этих рисунках рассчитаны при использовании
параметров атом-атомных потенциальных функций межмолекуляр-
ного взаимодействия, оцененных из свойств благородных газов и гра-
Рис. IX,3. Рассчитанные (кривые)
при использовании потенциала
(VIII,49) и опытные (точки) зна- £
чения AU1 для Ne (I), Аг (2), 1П
Кг (3) и Хе (4) на базисной грани Д и
графита (расчет) и на графитиро-
ванных термических сажах (опыт),
Способы расчета сплошных и пунктир- 1
ных кривых указаны в тексте.
С
фита, взятых в отдельности. Эти кривые лежат заметно выше соот-
ветствующих экспериментальных значений. Таким образом, термо-
динамические характеристики адсорбции благородных газов на ба-
зисной грани графита, оцененные указанным выше путем на основа-
нии свойств адсорбата и адсорбента, взятых в отдельности, заметно
отличаются от соответствующих экспериментальных значений. Рас-
смотрим возможные причины этого расхождения.
Основными факторами, определяющими рассчитанные значения
Xi и ДС71, являются форма и значения параметров атом-атомных
потенциалов ф, а также приближения, применяемые при суммиро-
вании этих потенциалов по атомам твердого тела. Как было показано
в разд. 1 этой главы, форма потенциала ф сравнительно слабо ска-
зывается на рассчитанных значениях К19 а следовательно, и на зна-
чениях Af/i. Суммирование потенциала ф производилось при исполь-
зовании хорошего приближения Крауэлла (см. разд. 2 этой главы).
Поэтому полученное выше расхождение между рассчитанными и опыт-
ными значениями Кг и ДС7Х может быть вызвано главным образом
погрешностями в значениях параметров потенциала ф.
293
Основными параметрами потенциала Бакингема — Корнера
(VIII,16) являются С\ и В, Параметр В выражался через остальные
параметры потенциала ф и равновесное расстояние г0 межмолеку-
лярного взаимодействия рассматриваемой пары атомов. В этом слу-
чае основными параметрами потенциальной функции ф являются
Cj иг0. Их значения изменяются в широких пределах при переходе
от одной пары атомов к другой (см. табл. IX,5 и данные на стр. JJ92).
При их оценке возможны значительные погрешности, которые сильно
сказываются на рассчитанных значениях константы Генри К± (см.
табл. IX,6).
Таблица IX ,6, Значения Фо/АТ, z0 и In Хг для адсорбции
одноатомных молекул на базисной грани графита,
рассчитанные при разных значениях параметров ? и го
атом-атомного потенциала Бакингема — Корнера (VIII,16)
(Ci/feD-lO®7, м* (С2/С1)- 1020, м* д, нм-1 гв, нм z0, нм Фо/feT In (Ki в мкм)
1 1,3 36 0,38 0,335 3,53 —5,84
2 1,3 36 0,38 0,335 7,06 —2,67
5 1,3 36 0,38 0,335 17,66 7,38
10 1,3 36 0,38 0,335 35,31 24,66
5 1,3 36 0,35 0,306 23,63 13,17
5 1,3 36 0,41 0,364 13,44 3,35
5 1,2 36 0,38 0,335 17,59 7,32
5 1,4 36 0,38 0,335 17,72 7,44
5 1,3 40 0,38 0,339 18,12 7,76
5 1,3 32 0,38 0,331 17,10 6,92
Значения же параметров С21СГ и q изменяются в сравнительно
узких пределах при переходе от одной пары атомов к другой (см.
табл. IX,5 и данные на стр. 291). Кроме того, погрешности в их оценке
слабо сказываются на рассчитанных значениях константы Генри Кг
(см. табл. IX,6). Поэтому полученное выше значительное расхожде-
ние рассчитанных и опытных значений Кг и Atfj для адсорбции
благородных газов на графите может быть вызвано в первую оче-
редь погрешностями значений параметров Сг и г0. Как известно,
формула Кирквуда — Мюллера дает завышенные значения для
С} (см. разд. 6 гл. VIII). Поэтому это расхождение рассчитанных
и опытных значений Кг и &Ui может быть вызвано главным образом
погрешностями в значениях Сг.
В работах [8, 10—16, 21] было получено удовлетворительное
согласие с опытом термодинамических характеристик адсорбции
благородных газов на графите, рассчитанных на основании свойств
адсорбата и адсорбента, взятых в отдельности. При этом параметр
сил притяжения как и выше, оценивался по формуле Кирквуда —
294
Мюллера. Однако параметр сил отталкивания В оценивался в этих
работах по формуле:
причем равновесное расстояние z0 оценивалось по формуле (VIII,42),
при этом эффективный ван-дер-ваальсов радиус г* атомов благород-
ных газов принимался равным половине равновесного расстояния
для взаимодействия пары атомов благородного газа
Рис. IX,4. Значения константы Генри Кг (в мкм) для адсорб-
ции Ne (7), Аг (2), Кг {3) и Хе (4) на базисной грани графита
(рассчитанные кривые) и на графитированных термических
сажах (опытные точки)
Расчет проведен при оценке параметра сил отталкивания В потенциала
(VIII,49) по формулам (IX,8) и (VIII,42)»
Кривые зависимости In Кг от 1/Т и ДС^от Т для адсорбции благо-
родных газов на базисной грани графита, рассчитанные указанным
выше путем при использовании приведенных в табл. IX,5 значений
иС2, а также приведенных выше (см. стр. 291 и 292) значений q и r0( z .. z
на рис. IX,4 и IX,5 сопоставлены с соответствующими опытными
значениями [1, 9, 38—44]. Рассчитанные и опытные значения In Кг
и Д{7Х для Аг, Кг и Хе, а также значения ДС^ для Ne, близки. Однако
близость рассчитанных и опытных значений в этом случае, по ви-
димому, получается из-за того, что погрешности в рассчитанных
значениях Кг и Д27х, вызванные завышенными значениями пара-
метра С\, компенсируются погрешностями, вызванными выбором
также завышенных значений z0.
Ниже сопоставлены значения z0 для взаимодействия атомов
благородных газов с базисной гранью графита, оцененные по формуле
295
(VIII,42) и при использовании атом-атомного потенциала Бакинге-
ма — Корнера (VIII,16) для взаимодействия атома i благородного
газа и атома С графита:
АТОМ Ne Аг Кг Хе
Zq, НМ по формуле (VIII, 42) . . 0,326 0,362 0,370 0,391
при использовании атом- атомного потенциал а (VIII, 16) ....... 0,308 0,340 0,346 0,364
При оценках этих значений z0 были использованы приведенные
выше (см. стр. 292) значения j и а также приведен-
ные в табл. IX,5 на стр. 291 значения параметров С2/Сх и q. Зна-
чения z0, полученные при использовании других атом-атомных
Рис. IX,5. Значения Atfj для ад-
сорбции Ne (7), Аг (2), Кг (3) и
Хе (4) на базисной грани графита
(рассчитанные кривые) и на гра-
фитированных термических сажах
(опытные точки).
Расчет проведен при опенке параметра
сил отталкивания В потенциала
(VIII,49) по формулам (IX, 8) и
(VIII,42).
потенциальных функций [27, 29], близки к приведенным выше
данным (см. стр. 296). Как видно из этих данных, значения zd, оценен-
ные по формуле (VIII,42), приблизительно на 0,02—0,03 нм больше
значений z0, оцененных при использовании атом-атомной потенци-
альной функции (VIII,16). Второй способ оценки значений z0, по-
видимому, более последователен, чем первый. Допущение о том,
что эффективный ван-дер-ваальсов радиус г* атома при его взаимо-
действии с поверхностью твердого тела равен половине равновесного
расстояния для взаимодействия между двумя атомами в газе rou г/2,
необосновано. Поэтому значения z0, оцененные по формуле (VIII,42),
по-видимому, на 0,02—0,03 нм завышены. По этой причине рассчи-
танные значения Кх и должны получаться сильно заниженными,
что, по-видимому, и компенсирует погрешности в значениях Кг
и ДС/1, вызванные выбором завышенных значений для параметра
С\, получаемых из формулы Кирквуда — Мюллера. Поэтому, чтобы
получить физически более обоснованные значения параметров атом-
атомных потенциальных функций, параметр сил отталкивания В
лучше определять при использовании формулы (IX,7), а не (IX,8).
Параметр же С\ следует определять при использовании опытных
адсорбционных данных.
296
5. Определение параметра С\ при использовании
экспериментальных значений Kt
Если предположить, что оцененные указанным выше способом
значения параметров г©, z...c, CJC-i и q близки к истинным, то
Ф = Рф* (IX,9)
и
где ф и Ф — соответствующие потенциальные функции, описывающие опытные
адсорбционные данные; ф* и Ф* — потенциальные функции, содержащие пара-
метр оцененный каким-либо приближенным способом, независящим от
экспериментальных адсорбционных данных.
В этом уравнении коэффициент 0 равен:
₽=y|-
где Сг и CJ — значения этого параметра, соответственно удовлетворяющие
опытным адсорбционным данным и оцененные независимым от опытных адсорб-
ционных данных способом.
Значение 0 можно определить, сравнивая рассчитанные при
использовании потенциальной функции Ф* и опытные значения Kv
Это удобно сделать на графике приблизительно линейной зави-
симости In Кг от ЦТ. В этом случае значение 0 равно значению
коэффициента, на который надо разделить абсциссы рассчитанной
кривой зависимости In Кг от 1/Т, чтобы получить ее совпадение
с опытными значениями In К}. Ниже приведены определенные ука-
занным выше способом значения коэффициента 0 и параметра Сг
для адсорбции благородных газов на графите, удовлетворяющие
опытным значениям Кг:
Атом .......................
0...........................
Ci’ 1057, кДж • мб • моль“1 . .
Ne Аг Кг Хе
0,70 0,81 0,79 0,79
0,63 2,63 3,84 6,05
При этом для параметров 7 и г0 были приняты значения,
приведенные выше (см. табл. IX,5 и стр. 291 и 292). Рассчитанные
ранее (см. стр. 293) при использовании потенциальной функции Ф*
кривые зависимости In Кг от 1/Т, абсциссы которых разделены на
соответствующие значения коэффициента |3, на рис. IX,2 показаны
пунктирными линиями.
Как указывалось выше, формула Кирквуда — Мюллера дает
завышенные значения (\. В соответствии с этим 0 < 1. Кроме того,
значения 0 для адсорбции Аг, Кг и Хе практически совпадают, и
только для Ne 0 несколько меньше, чем для остальных атомов, од-
нако для Ne экспериментальные значения Кг менее надежны.'Зна-
чения параметра С\, удовлетворяющие опытным значениям
ла 20—30% меньше значений этого параметра, оцененных по формуле
297
Кирквуда — Мюллера, и на 6—30% меньше значений этого пара-
метра, оцененных при использовании правила комбинирования
(VIII,29).
Таким образом, атом-атомные потенциальные функции межмоле-
кулярного взаимодействия атомов благородных газов с атомом С
графита, описывающие экспериментальные значения Кг для адсорб-
ции этих газов на графитированных термических сажах, даются
выражениями:
<pNe... G (графит) = “ 0,632 • 10^-6-0,784 - 10’5г-8 +1,20 • 105 ехр (-38г) (IX,12)
Фаг. .. G (графит) = “2‘63-10^-6-3,52 . Ю-5г-8 + 3,70 • 105 ехр (-36r) (IX,13)
Фкг... с (графит) = “3>84 ’ Ю“3г"6 —5,19 • 10-5г-8 +1,81 -105 еХр (-32r) (IX,14)
ФХе... С (графит) = -6’05 * 10^-6-8,37 • 10-5г-8 + 3,78 • 105 еХр (-32г) (IX,15)
Здесь ф в кДж/моль, аг — в нм. Эти функции ф можно использовать
для расчета Кг и других термодинамических характеристик адсорб-
ции благородных газов на графитированных термических сажах.
Значения рассчитанные при использовании этих функций ф,
на рис. IX,3 показаны пунктирными линиями. Значения и0, Фо,
А51/7? и &CJR, рассчитанные при использовании этих же атом-
атомных потенциальных функций, приведены в табл. IX,7.
Таблица IX,7. Значения Фо, z0, Д5? и A^i Для адсорбции
благородных газов на базисной грани графита, рассчитанные
при использовании значений Clf удовлетворяющих опытным
значениям Кг для адсорбции этих газов на графитированных
термических сажах, и экспериментальные значения Д5?
(Кг в мкм) для адсорбции благородных газов
на графитированных термических сажах
Атом Расчет Опыт
Фо, ' кДж/моль z0, нм Г, К R АС, R Т, К AS? Я
Ne 3,10 0,308 50 10,3 0,7
75 10,1 79 10,3 [1]
100 10,2
Аг 8,88 0,340 75 10,7 0,7 78 10,9 40)
100 10,4 0,7 78 11,6 42]
200 10,0 0,2 175 10,3 1]
Кг 11,5 0,346 75 10,7 0,6
100 10,5 0,7 90 12,8 [41]
200 10,0 0,6 187 10,4 [45]
300 9,9 280 9,9 [1]
Хе 15,4 0,364 100 10,7 0,6
200 10,2 0,7 173 12,4 [44]
300 9,9 0,6 226 9,8 [45]
400 9,9 295 10,2 [1]
298
В той же таблице приведены экспериментальные значения ASi/R,
полученные на основании экспериментальных значений Къ опре-
деленных при разных температурах по формуле (111,60) [1, 40—42,
44, 45]. В пределах погрешности опытных значений, рассчитанные
и опытные значения &.U1 и A5J/7? совпадают.
Для определения потенциальной функции Ф и сопоставления
результатов теоретических расчетов с опытом необходимо знать
опытные значения константы Генри Klt отнесенной к единице по-
верхности адсорбента. Такие значения Кг были получены путем
деления константы Генри, отнесенной к единице массы адсорбента
Kns c>i, на удельную поверхность адсорбентам. Значением обычно
определяется приближенным методом БЭТ. Погрешности в м вызы-
вают соответствующие погрешности в Кг. Поэтому потенциальные
функции ф взаимодействия атомов благородных газов с атомом угле-
рода графита, полученные выше при использовании эксперименталь-
ных значений Кг, содержат погрешности определения s. От погреш-
ностей величины м свободны экспериментальные значения A?7i.
Поэтому удовлетворительное согласие рассчитанных и опытных зна-
чений \U1 указывает и на то, что значения м, использованные при
расчетах значений Кг для графитированных термических саж,
близки к истинным.
6. Молекулярно-статистический метод определения
удельной поверхности графитированной сажи
Существующие адсорбционные методы определения удельной
поверхности м твердых тел имеют ряд недостатков. Например, опре-
деленная методом БЭТ емкость монослоя сильжо зависит от темпе-
ратуры. Значительные трудности представляет выбор истинной
площади, занимаемой молекулой адсорбата в плотном монослое.
Поэтому представляет интерес поиск новых, более надежных мето-
дов определения $ из адсорбционных данных.
Был предложен молекулярно-статистический метод определения
удельной поверхности з твердого тела из экспериментальных зна-
чений константы Генри Knscl = AK1( = msK1) для адсорбции
газов на этой поверхности [1, 46]. Согласно этому методу, сопоста-
вляя экспериментальные значения константы Генри sKx в функции
Т с рассчитанными значениями K^Izq в функции Ф0/7?Т, определяют
значения Фо и sz^ [1, 2, 5, 46]. Значение 5 получают из произведения
szQ, оценивая каким-либо способом значение z0. Этот метод был
применен для определения s графитированной термической сажи
Р-33 (Sterling FT, 2700) [1, 2, 5] (в дальнейшем принято пь = 1).
В табл. IX,8 приведены значения Фо и зи0, определенные ука-
занным выше методом для адсорбции Аг, Кг и Хе на графитирован-
ной термической саже Р-33 (Sterling FT, 2700) [1, 5].
299
Таблица 1Х,8, Значения Фо и sz0 для межмолекулярного
взаимодействия Аг, Кг и Хе с базисной гранью графита, определенные
из экспериментальных значений Kns, с, х — Кг8 для адсорбции этих газов
на графитированной термической саже Р-33 (Sterling FT, 2700) Ji]
при использовании разных потенциалов Ф
Формулы, выражающие потенциал Ф Ar Кг Хе
-Фо> кДж «моль-1 Л со о I I —Фо, ! кДж «моль-1 1 и" СО о —Фо, кДж «моль-1 см’ /г
(IX,4) 9,21 2,87 12,1 2,89 16,0 3,15
(IX,3) 9,13 3,79 11,9 3,96 15,9 4,11
(IX,2) при z0 = d (VIII,49) при значениях параметров: 9,17 3,60 12,0 3,70 15,9 3,90
a) = 0,2; z0 = d; q — 36 нм*1 9,17 3,61 12,0 3,77 15,9 3,98
6) CJC-^d2 =0; z0 = d\ q = 36 нм"1 9,13 3,58 12,0 3,73 15,9 3,90
в) CgJCxd^ — 0,2; z0 = l,ld; q = 36 нм"1 9,17 3,81 12,0 3,97 15,9 4,19
г) = 0,2; z0 — d\ q — 42 нм"1 9,17 3,99 12,0 4,16 15,9 4,39
При расчетах значений KJzq для функции Ф принимались по-
тенциалы (IX,4) [1], (VIII,49), (IX,2) и (IX,3) [5]. В случае потен-
циала (IX,2) значения Фл и яз0 определялись при использовании
рассчитанных значений K-Jz^ при z0 = d, а в случае потенциала
(VIII,49) — при использовании рассчитанных значений KJzQ при
z0 — d и 1,1 d, CJCxd2 = 0 и 0,2 и q = 36 и 42 нм*1. Как видно
из табл. IX,8, значения Фо слабо зависят от принятой модели для
функции Ф и параметров z0, С21С1 и q в случае потенциалов (VIII,49)
и (IX,2).
Значения sz3 зависят значительно сильнее, чем значения Фо,
от модели функции Ф и ее параметров z0, С21Сг и q, использованных
при расчетахзначений K-JZq. Значения^, полученные при исполь-
зовании потенциала (VIII,49) при z0 = d, q = 36 нм-1, CJC^d2 = 0
и CJC^d2 = 0,2 и потенциала (IX,2) при z0 = d, различаются лишь
на 1—2%. Однако значения sz0, полученные при использовании
потенциала (VIII,49), заметно зависят от принятых значений пара-
метров zQ и q: при изменении z0 от d до l,ld, т. е. при изменении z0
в пределах возможных значений z0 для рассмотренных молекул
(см. стр. 296), значения szQ изменяются приблизительно на 5%,
при изменении q от 36 до 42 нм"1 — приблизительно на 10%. По-
этому при более точном определении значений $z0 при использовании
потенциала (VIII,49) необходимо выбрать для каждого благород-
ного газа соответствующие значения параметров z0 и q. При исполь-
зовании потенциала (IX,3), учитывающего вклад в Ф только одной
300
наружной атомной плоскости графита, значения szd получаются на
5—8% выше, а при использовании потенциала (IX,4), не учитыва-
ющего слоистого строения графита, значения szQ получаются при-
близительно на 20% ниже, чем при использовании потенциала
(IX,2). Наиболее надежные значения szQ, по-видимому, получаются
при использовании потенциалов (VIII,49) и (IX,2), учитывающих
слоистое строение графита.
Было предложено оценивать значения z0 из значений Фо [1, 46].
В случае потенциала (IX,4)
z0=
vjiCi у/з
9ФТ/
(1Х116>
а в случае потенциала (IX,3)
1ОФо /
(IX,17)
Для оценки Ci были использованы формулы Кирквуда — Мюллера,
Кирквуда — Слэтера, Лондона, Бардина и Маргенау — Полларда
[1, 5, 46]. Ниже приведены значения z0, полученные по формулам
(IX,16) и (IX,17) при использовании приведенных в табл. IX,8
значений Фо и оцененных по формуле Кирквуда — Мюллера зна-
чений Сг (приведенных в табл. IX,5):
Атом................................
z0, нм
по формуле (IX, 16)...........
но формуле (IX, 17) ............
Аг Кг Хе
0,241 0,251 0,268
0,337 0,350 0,366
Как видно из приведенных данных, значения zQ сильно зависят
от принятой модели для функции Ф. Значения z0, полученные по
формуле (IX,16), значительно меньше полученных по формуле (IX,17).
Значения z0, полученные при использовании формулы (IX,17),
близки к приведенным выше значениям и0 (см. стр. 296), оцененным
при использовании атом-атомного потенциала (VIII,16).
Формула (IX,16) получена при использовании приближения Лон-
дона, которое приводит к сильно завышенным значениям решеточ-
ных сумм (см. табл. VII 1,5) и к сильно заниженным значениям Фо
при фиксированных значениях параметров атом-атомных потенци-
альных функций (см. табл. IX,3). Формула же (IX,17) получена
при использовании приближения Краузлла (VIII,46), которое при-
водит к значениям решеточных сумм и Фо, близким к значениям,
полученным при числовом суммировании (см. таблДЧП,5 и IX,3). По-
этому формула (IX,17) является более строгой, чем формула (IX,16),
и значения zQi полученные при использовании формулы (IX,17),
должны быть ближе к истинным, чем значения z0, полученные при
использовании формулы (IX,16).
В случае справедливости потенциала (VIII,49) связь между зна-
чениями Zq, Фо, С19 CJC-l и q сложна и определение z0 из значений
Фо в этом случае представляет значительные трудности.
301
Полученные из Фо значения z0 сильно зависят от использованной
формулы для оценки Сг [1]. Формулы Лондона, Кирквуда — Слэ-
тера, Бардина и Маргенау—Полларда дают более низкие значения
для чем формула Кирквуда — Мюллера [1, 47]. Поэтому зна-
чения г0, полученные при использовании этих формул для оценки
получаются более низкими, чем при использовании формулы
Кирквуда — Мюллера.
Было предложено также несколько способов определения z0
из экспериментальных значений Кг для адсорбции легких газов
при низких температурах, используя квантово-статистические эф-
фекты при адсорбции: квантово-статистические поправки для клас-
сических значений Кг [481, разность квантово-статистических эф-
фектов масс для изотопных молекул с одинаковыми Ф [49] или коэф-
фициент их разделения [50]. Значения z0 для адсорбции на графити-
рованных термических сажах были определены только последним
из этих трех способов [50]. Результаты сильно зависят от принятой
модели для функции Ф, и их точность, по-видимому, очень низка.
Таким образом, из рассмотренных выше способов оценки zQ
наиболее надежные значения получаются при использовании атом-
атомных потенциалов. Ниже приведены значения s* графитирован-
ной сажи Р-33 (Sterling FT, 2700), полученные из приведенных
в табл. IX,8 наиболее надежных значений [определенных при
использовании потенциалов (IX,2) и (VIII,49)] и из приведенных
выше (см. стр. 296) наиболее надежных значений z0 [определенных
при использовании атом-атомной потенциальной функции (VIII,16)]:
Потенциал • Значения $ (м2/г) для сажи Р-33 при адсорбции Аг (IX,2) 10,6 (VIII,49), а) ♦ 10,6 (VIII,49), б) 10,5 (VIII,49), в) 11,2
Кг 10,6 10,8 10,7 11,4
Хе ... . 10,7 10,9 10,7 11,4
* Обозначения те же, что и в табп. IX,8.
Эти значения s^ll м2/г практически не зависят от природы
адсорбата и лежат в пределах погрешности величины s = 12,5 ±
± 2 м2/г, определенной для этой сажи методом БЭТ [39, 43, 51, 52].
Таким образом, определенные молекулярно-статистическим ме-
тодом значения $, в отличие от значений $, определенных методом
БЭТ, не зависят от выбора площади, занимаемой молекулой на
данной поверхности. Однако полученные этим методом значения s
заметно зависят от принятой модели для функции Ф и способа оценки
z0. Поэтому при применении молекулярно-статистического метода
определения s надо использовать наиболее точную модель для функ-
ции Ф, учитывающую атомное строение твердого тела, и наиболее
надежные значения z0.
302
ЛИТЕРА ТУРА
1. Sams J. R., Constabaris G., Halsey G. D. J. Phys. Chem.*
1960, v. 64, p. 1689—1696.
2. Freeman M. P. J. Phys. Chem., 1958, v. 62, p. 723—728.
3. Yaris R., Sams J. R. J. Chem. Phys., 1962, v. 37, p. 571—576.
4. Wolfe R., Sams J. R. J. Phys. Chem., 1965, v. 69, p. 1129—1135.
5. К i sele v A. V., Poshkus D. P. J. Chem. Soc. Faraday Trans. IL
1975, v. 71.
6. С т и л л У. В кн.: Межфазовая граница газ — твердое тело. Пер. с англ.
Ред. Э. Флад. М., «Мир», 1970. 433 с.
7. Ross S., Olivier О. J. Advanc. Chem. Series, 1961, v. 33, p. 301—308.
8. Пошкус Д. П., Киселев А. В. ЖФХ, 1962, т. 36, с. 1735—1742.
9. R о s s S., S а е 1 е n s J. F., Olivier J. Р. J. Phys. Chem., 1962*
v. 66, p. 696—700.
10. К и с e л e в А. В., Пошкус Д. П. ЖФХ, 1963, т. 37, с. 770—777»
11. К и с е л е в А. В., Пошкус Д. П. ЖФХ, 1963, т. 37, с. 1504—1509.
12. Kiselev А. V., Poshkus D. Р. Trans. Faraday Soc., 1963, v. 59*
p. 176-19..
13. Kiselev A. V., Poshkus D. P. Trans. Faraday Soc., 1963, v. 59*
p. 1438—1443.
14. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ*
1964, т. 38, с. 1514—1522.
15. П о ш к у с Д. П. ЖФХ, 1965, т. 39, с. 2962—2967.
16. Р о s h k u s D. Р. Disc. Faraday Soc., 1965, v. 40, p. 195—204.
17. M у e r s A. L., P г a u s n i t z J. M. Trans. Faraday Soc., 1965, v. 61*
p. 755-764.
18. V a n D a e 1 W. Disc. Faraday Soc., 1965, v. 40, p. 225.
19. Hellemans R., Van Itterbeek A., Van Dael W. Physica*
1967, v. 34, p. 429—437.
20. Barnes M. W., Steele W. A. J. Chem. Phys., 1968, v. 49, p. 2128-^
2135.
21. Киселев А. В., Пошкус Д. П. ДАН СССР, 1960, т. 132, c. 876—*
879.
22. Ross S., Olivier J. P. Advanc. Chem. Series, 1961, v. 33, p. 309—316»
23. С г о w e 1 1 A. D., Young D. M. Trans. Faraday Soc., 1953, v. 49*
p. 1080—1085.
24, Pace E. L. J. Chem. Phys., 1957, v. 27, p. 1341—1346.
25. L andol t - В 6 ms t e in. Tabellen, Bd. I. Teil 4, 6 Auflage.
26. Barr er R. M. Proc. Roy. Soc. (London), 1937, v. A 161, p. 476—493.
27. Crowell A. D., Steele R. B. J. Chem. Phys., 1961, v. 34, p. 1347—*
1349.
28. Steele W. A., Karl R. J. Col. Interface Sci., 1968, v. 28, p. 397—402.
29. С г о w e 11 A. D., Chang С. O. J. Chem. Phys., 1963, v. 38, p. 2584^
2586.
30. С г о w e 1 1 A. D. J. Chem. Phys., 1958, v. 29, p. 446—447.
31. Corner J. Trans. Faraday Soc., 1948, v. 44, p. 914—927.
32. M a s о n E. A., Rice W. E. J. Chem. Phys., 1954, v. 22, p. 843—851»
33. В а г u a A. K. J. Chem. Phys., 1959, v. 31, p. 957—960.
34. В a r u a A. K. Indian J. Phys., 1960, v. 34, p. 76^-84.
35. В a rker J. A., P о m p e A. Austr. J. Chem., 1968, v. 21, p. 1683—1694»
36. Murrell J. N., Shaw G. Molec. Phys., 1968, v. 15, p. 325—328.
37. G i r i f a 1 с о A. L., Lad R. A. J. Chem. Phys,, 1956, v. 25, p. 693—
697.
38. G г e у s о n J., Aston J. G. J. Phys. Chem., 1957, v. 61, p. 610—616»
39. S a m s J. R., Constabaris G., Halsey G. D. J. Phys. Chem.*
1962, v. 66, p. 2154—2158.
40. R о s s S., Winkler W. J. Col. Sci., 1955, v. 10, p. 319-329.
41. Ross S., Winkler W. J. Col. Sci., 1955, v. 10, p. 330—337.
42. R о s s S., Olivier J. P. J. Phys. Chem., 1961, v. 65, p. 608—615»
303
43. Аристов Б. Г., Киселев А. В. Коллоидн. ж., 1967, т. 29, с. 631—
. 637.
44. Cochrane Н. е. a. J. Col, Interface Sci., 1967, v. 24, p. 405—415.
45. К и с e л e в А. В., Худяков В. Л., Яшин Я. И. ЖФХ, 1973,
т. 47, с. 2125—2126.
46. Steele W. A., Halsey G. D. J. Chem. Phys., 1954, v. 22, p. 979—
984.
47. Pitzer K. S. Advanc. Chem. Phys., 1959, v. 2, p. 59—83.
48. Yang K., Gant P. L., С о о p e r D. E. J. Phys. Chem., 1965, v. 69,
p. 1768—1770.
49. F r e e m a n M. P. J. Phys. Chem., 1960, v. 64, p. 32—37.
50. C a s a n о v a G. e. a. Disc. Faraday Soc., 1965, v. 40, p. 188—194.
51. Polley M. H., Schaeffer W. D., Smith W. R. J. Phys.
Chem., 1953, v. 57, p. 469—471.
52. E v e r e 11 D. H. In: Surface area determination. Butterworths. London,
1970. 405 p.
Глава X
МОЛЕКУЛЯРНО-СТАТИСТИЧЕСКИЙ РАСЧЕТ
ТЕРМОДИНАМИЧЕСКИХ ХАРАКТЕРИСТИК АДСОРБЦИИ
УГЛЕВОДОРОДОВ НА ГРАФИТЕ
1. Общие замечания о расчете термодинамических
характеристик адсорбции углеводородов
При взаимодействии многоатомных молекул с твердым телом
происходит торможение как поступательного движения центра масс
молекулы, так и ее вращательного движения. Для правильного
молекулярно-статистического описания адсорбции таких молекул,
как отмечалось в разд. I гл. VIII, надо учесть изменения обоих
этих видов движения молекулы при адсорбции. Молекулярно-ста-
тистические расчеты термодинамических характеристик адсорбции
углеводородов на графите с учетом изменения поступательного
и вращательного движения молекул адсорбата при адсорбции произ-
водились в ряде работ [1—18].
Константа Генри Кг и изменение внутренней энергии адсорбата
при адсорбции определяются главным образом потенциальной
энергией взаимодействия молекулы с адсорбентом вблизи главного
потенциального минимума Фо. Поэтому эти термодинамические
характеристики адсорбции удобны для исследования межмолекуляр-
ного взаимодействия при адсорбции. Расчеты Кг и для адсорб-
ции углеводородов на графите производились в работах [2—4,
7—14, 16—18]. Изменения энтропии AS* и теплоемкости адсор-
бата при адсорбции определяются только зависимостью потенциаль-
ной энергии Ф взаимодействия молекулы адсорбата с адсорбентом
от координат поступательного и вращательного движения моле-
кулы, но не зависят от абсолютной величины потенциальной энер-
гии Ф. Поэтому эти термодинамические характеристики адсорбции
удобны для изучения зависимости Ф от положения молекулы: поло-
жения центра масс и ориентации молекулы по отношению к поверх-
ности адсорбента. Эта зависимость определяет состояние адсорби-
рованных молекул при нулевом заполнении поверхности, т. е. ха-
рактер их поступательного и вращательного движения. Расчеты
AS? и АС\ для адсорбции углеводородов на графите производились
в работах [1, 3—6, 10, 11, 13, 16—18].
На основе приближения (VIII,2) Киселевым, Пошкусоми Афрей-
мовичем [2—11,13,14,17,18] были проведены расчеты Ф для взаимо-
действия молекул ряда углеводородов разных классов (алканов,
цикланов, алкенов, алкинов, полиенов с сопряженными связями,
ароматических углеводородов) с базисной гранью графита. В каче-
20 Заказ 1180 305
стве силовых центров молекул выбирались атомы С и Н [9—11,
14, 17, 18]. Расчеты для н-алканов и бензола проводились также
при выборе в качестве силовых центров молекулы звеньев СН3,
СН2 и СНаром [2—10, 13]. Параметры потенциальных функций
взаимодействия атомов Си Н и звеньев СН3иСН2 молекул углево-
дородов с атомами С графита сначала оценивались с помощью при-
ближенных квантовомеханических формул и правил комбинирова-
ния на основании свойств адсорбента и адсорбата, взятых в отдель-
ности [2—10]. Далее эти параметры уточнялись при использовании
экспериментальных данных по адсорбции нескольких молекул рас-
сматриваемого класса [9, 10, 17, 18]. Для межмолекулярного взаи-
модействия с атомами С графита атомов С молекулы, находящихся
в разных валентных состояниях, были введены разные атом-атомные
потенциальные функции [18]. На основании потенциальных функ-
ций Ф были рассчитаны константы Генри Кг или равные им удержи-
ваемые объемы V^,! [2—4, 7—9, И, 13, 14, 17, 18], изостерические
теплоты адсорбции’ qshl [3, 4, 8, 10, 11, 13, 17, 18], дифференциаль-
ные мольные изменения энтропии [3, 4, 10, 11, 13, 17, 18] и
теплоемкости [5, 6, 10, 13, 17, 18] адсорбата при адсорбции
углеводородов указанных выше классов на базисной грани графита
при нулевом заполнении поверхности. Результаты расчета были
сопоставлены с соответствующими опытными значениями, получен-
ными в разных работах. Таким образом были проведены исследова-
ния зависимости межмолекулярного взаимодействия углеводородов
с графитированными термическими сажами от химического состава,
пространственной структуры и конформации молекулы, а также от
валентного состояния атомов углерода и сопряжения двойных свя-
зей в молекуле углеводорода.
В работах [8—11, 13, 14, 17, 18] суммирование атом-атомных
или группа-атомных потенциальных функций межмолекулярного
взаимодействия ср по атомам решетки графита производилось с ис-
пользованием приближения Крауэлла (VIII,46), т. е. суммирование
по атомам углерода, лежащим в одной плоскости решетки графита,
заменялось интегрированием. Получающаяся при этом обобщенная
зета-функция Римана оценивалась с помощью приближенной фор-
мулы (VIII,51). Потенциальная функция Ф, взаимодействия i-ro
атома или группы атомов молекулы с базисной гранью графита,
полученная при использовании приближения Крауэлла, зависит
только от расстояния zi этого атома или группы атомов от базисной
грани графита (от плоскости, проходящей через центры наружных
атомов С базисной грани графита). Поэтому потенциальная функция Ф
взаимодействия всей молекулы со всей решеткой адсорбента, полу-
ченная суммированием потенциальных функций Ф^ по атомам или
звеньям молекулы, не зависит от декартовых координат центра масс
молекулы х и у, изменяющихся при движении этого центра масс
вдоль базисной грани графита, и от одного из углов Эйлера, изменя-
ющегося при вращении молекулы вокруг оси, перпендикулярной
этой грани.
306
При расчетах конфигурационных интегралов для зависимости Ф
от z в работах [2—14, 16—18] использовалось приближение гармо-
нического осциллятора (VII,52). Это приближение значительно
сокращает расчет этих интегралов. Вместе с тем оно вносит лишь
небольшие погрешности в результаты расчета (см. следующий раз-
дел этой главы).
При указанных приближениях, т. е. при допущении о гармони-
ческом колебании центра масс молекулы перпендикулярно поверх-
ности, свободном поступательном движении центра масс молекулы
параллельно поверхности и свободном вращении вокруг перпенди-
кулярной ей оси, термодинамические характеристики адсорбции
квазижестких молекул даются статистическими выражениями
(VII,44)—(VII,46), (VII,53), (VII,57)-(VII,60), а в случае адсорб-
ции молекул, образующих поворотные изомеры при внутреннем
вращении — статистическими выражениями (VII,30)—(VII,40) и
(VII,53).
Расчет потенциальных функций и конфигурационных интегра-
лов представляет весьма трудоемкую работу, особенно в случае
сложных молекул, состоящих из большого числа силовых центров.
Поэтому в работах [9—11, 13, 14, 17, 18] расчеты этих величин
производились на электронной вычислительной машине по специ-
ально составленной программе [19].
В работах [20—31] при использовании приближения (VIII,2)
производились расчеты потенциальных функций Ф взаимодействия
молекул углеводородов с базисной гранью графита. Оцененные
значения потенциальной энергии Фо вблизи наиболее глубокого
потенциального минимума сопоставлялись с опытными значениями
дифференциальных теплот адсорбции, экстраполированными к нуле-
вому заполнению поверхности. Однако значение потенциальной
энергии Фо с точностью до нулевой энергии адсорбированных мо-
лекул равно теплоте адсорбции только при ОК. Вместе с тем экспе-
риментальные значения теплот адсорбции обычно получаются для
температур значительно выше ОК. Так как при адсорбции проис-
ходит изменение теплоемкости адсорбата [32—38], то эксперимен-
тально полученные значения теплоты адсорбции должны отличать-
ся от теплот адсорбции при ОК. Поэтому сопоставление рассчи-
танных значений Фо с измеренными значениями теплот адсорбции
при температуре опыта не является строгим. Для сопоставления
оцененных каким-либо способом потенциальных функций Ф с опы-
том необходимо на основании этих функций Ф рассчитать термо-
динамические характеристики адсорбции при разных темпера-
турах. Только последние величины могут быть использованы для
сопоставления с опытом.
В работах [2—10, 20—31] параметры потенциальных функций ср
оценивались с помощью приближенных квантовомеханических фор-
мул и правил комбинирования на основании свойств адсорбата и
адсорбента, взятых в отдельности. Полученные таким путем потен-
циальные функции Ф недостаточно точны для решения ряда задач
20*
307
молекулярной теории адсорбции. Для получения более точных по-
тенциальных функций Ф необходимо использовать эксперименталь^
ные данные по адсорбции.
2. Алканы
Оценка параметров атом-атомных потенциальных функций меж-
молекулярного взаимодействия атомов Н и С молекул алканов с ато-
мами С графита на основании свойств адсорбата и адсорбента, взятых
в отдельности. В общем случае атом-атомные потенциальные функ-
ции характеризуют межмолекулярное взаимодействие атомов, ва-
лентно связанных с другими атомами в данной молекуле и в данном
твердом теле. Поэтому они, в принципе, должны зависеть не только
от природы взаимодействующих атомов и их валентного состояния,
но также и от природы соседних атомов, связанных с ними валентно.
В соответствии с этим в алканах следует различать 5 типов атомов С:
1) атом С, соединенный с четырьмя атомами Н;
2) атом С, соединенный с тремя атомами Н и одним атомом С;
3) атом С, соединенный с двумя атомами Н и двумя атомами С;
4) атом С, соединенный с одним атомом Н и тремя атомами С;
5) атом С, соединенный с четырьмя атомами С.
Обычно вклад атомов С типов 4) и 5) в общую энергию взаимо-
действия Ф меньше, чем атомов С типов 1) — 3), так как в энергети-
чески наиболее выгодных положениях молекулы атомы С типов 4)
и 5) более удалены от поверхности, чем атомы С типов 1)—3). По-
этому различие в атом-атомных потенциалах межмолекулярного
взаимодействия для атомов С типов 4), 5) и для атомов С типов 1)—3)
должно сказаться на термодинамических характеристиках адсорб-
ции слабее, чем различие в атом-атомных потенциалах этого взаимо-
действия для атомов С типов 1), 2) и 3).
Строго говоря, атом-атомные потенциальные функции межмоле-
кулярного взаимодействия должны зависеть также от природы и
расположения более удаленных, непосредственно валентно несвя-
занных атомов молекулы, так как электронные оболочки непосред-
ственно валентно несвязанных атомов в молекуле не являются неза-
висимыми. Однако в молекулах с несопряженными связями взаимо-
действие электронных оболочек валентно несвязанных атомов быстро
убывает с ростом расстояния от рассматриваемого атома [39]. По-
этому в случае таких молекул можно принять, что атом-атомные
потенциальные функции межмолекулярного взаимодействия прак-
тически не зависят от природы и расположения валентно несвязан-
ных атомов такой молекулы.
При оценках и уточнении параметров атом-атомных потенциаль-
ных функций для взаимодействия атомов С и Н молекул алканов
с атомами С решетки графита и при расчетах термодинамических
характеристик адсорбции алканов различиями в атом-атомных функ-
циях, вызываемыми различиями природы и числа валентно с ними
связанных атомов и различиями природы и расположения валентно
с ними не связанных атомов, пренебрегали [9—11, 14,17]. В этом
308
приближении различие адсорбционных свойств алканов сводится
только к различиям химического состава и геометрии адсорби-
руемых молекул.
Для энергии межмолекулярного взаимодействия с атомами С
графита атомов С и Н молекул углеводородов был принят теорети-
чески наиболее обоснованный потенциал Бакингема — Корнера
(VIII,16) [9—11, 14, 17]. В этом случае потенциальная функция Ф;
взаимодействия t-го атома молекулы с базисной гранью графита^
полученная при использовании приближения Крауэлла (VIII,46),
дается выражением (VIII,49). Параметры Сг, С21Си В и q этого
потенциала для взаимодействия с атомами С графита атома С и Н
молекулы сначала были оценены на основании только физико-хи-
мических свойств молекул и адсорбента, взятых в отдельности
[9, 10]. Параметры сил притяжения Сг и С2 были оценены по при-
ближенной квантовомеханической формуле Кирквуда — Мюллера
(VIII,25) для Ci и по аналогичной ей формуле (VIII,33) для С2. При-
нятые в расчетах значения поляризуемости а и диамагнитной вос-
приимчивости % для атомов Н и С молекул алканов и атомов С гра-
нита и полученные значения С19 С2 и С2/Сг приведены в табл. Х,1.
Таблица Xtl. Принятые значения поляризуемости а и диамагнитной
восприимчивости % для атомов С и Н молекул алканов и атомов С графита,
и рассчитанные по формулам (VIII,25) и (VIII,33) значения параметров сил
притяжения Clf С2 и атом-атомного потенциала (VIII,16)
для взаимодействия атомов С и Н молекул алкана с атомом С
графита [10]
Атом а.10”, м8 - %* 1088, м8 Ci- 1057, кДж »М8» МОЛЬ-1 С2-10”7, кДж »м8» моль-1 (С2/С1)-10гЧ м1
С (алкан) 0,96 12,3 1,590 2,470 1,55
Н (алкан) 0,43 3,7 0,574 1,090 1,90
С (графит) 0,937 10,54 — — —
Параметр сил отталкивания q был принят одинаковым для взаи-
модействия обеих пар атомов (С . . -С и Н . . . С) и равным 36 нм"1
[9,-11, 14, 17]. Это значение для взаимодействия пары атомов С
графита было получено эмпирически из свойств решетки графита [40].
Параметр сил отталкивания В в работе [17] выражался через
остальные параметры и равновесное расстояние г0 для взаимодей-
ствия рассматриваемой пары атомов, используя условие равно-
весия (VIII,37), т. е. оценивался по формуле (IX,7). Для равновес-
ного расстояния г0 ван-дер-ваальсового взаимодействия пары ато-
мов С . . .С было принято значение 0,382 нм, полученное из свойств
решетки графита [40, 41]. Равновесное расстояние г0 для взаимо-
действия атомов С . . .Н определялось как среднее арифметическое
значений г0 для взаимодействия атомов Н . . .Н и С ... С. При
этом для взаимодействия атомов Н . . .Н было принято значение
30&
rQ — 0,30 нм, полученное из свойств кристаллических углеводо-
родов (структуры и энергии решетки) [42, 43]. Принятые значения
г0 и полученные по формуле (IX,7) значения параметра В для взаи-
модействия пар атомов С. . .С и Н . , .С приведены ниже:
г0, нм...............
В-10*5, кДж-моль-1
Взаимодействующие ато-
мы ....................С (алкан) ... С (гра-
фит)
0,382
2,18
Н (алкан) ... С (гра- -
фит)
0,341
0,42
В работах [9—11, 14] параметр сил отталкивания В для взаимо-
действия атомов С . . .С и Н . . .С выражался через остальные па-
раметры и равновесное расстояние z0 межмолекулярного взаимо-
действия рассматриваемого атома со всей решеткой графита по
Рис. Х,1. Рассчитанные (кривые) и опытные (точки) значе-
ния константы Генри К± для адсорбции метана (I), этана (2)
и пропана (3) на базисной грани графита (расчет) и графити-
рованных термических сажах (опыт).
Способы расчета сплошных и пунктирных (см. стр. 313) кривых указа-
ны в тексте.
^формуле (IX,8). Равновесное расстояние z0 принималось равным
сумме половины межплоскостного расстояния решетки графита
dl2 = 0,168 нм и эффективного ван-дер-ваальсового радиуса г*
атома. Для атома Н принималось г* = 0,13 нм, а для атома С г * =
= 0,17 нм. Оцененные по формулам (IX,7) и (IX,8) значения пара-
метра В для взаимодействия атомов С . . . С и Н . . .С практически
совпадают. Однако оценка параметра В по формуле (IX,7) более
обоснована.
Термодинамические характеристики адсорбции метана, этана
и пропана на базисной грани графита, полученные при использо-
вании приближенных атом-атомных потенциальных функций. Зна-
чения In дяя адсорбции метана, этана и пропана на базисной
310
i рани графита, рассчитанные на основании приближенных атом-
атомных потенциальных функций, полученных описанным выше-
путем, на рис. Х,1 сопоставлены с соответствующими опытными
значениями для адсорбции этих углеводородов на графитированной
комической саже [44—49]. На рис. Х,2 такое сопоставление сде-
лано для рассчитанных и опытных величин AU\.
В молекулах этана и пропана внутреннее вращение вокруг
связей С—С сильно заторможено [50]. Внутреннее вращение этих
молекул при адсорбции изменяется слабо, и это изменение вносит
лишь незначительный вклад в тер-
модинамические характеристики ад-
сорбции (см. ниже разд. 4 зтой гла-
вы). Поэтому молекулы этана и про-
пана, как и молекула метана, при
расчетах изменений их термодинами-
ческих свойств при адсорбции [9,10]
рассматривались квазижесткими и
обладающими конформациями, со-
ответствующими положениям мини-
мумов потенциальных энергий этих
молекул в объеме газа.
Опытные значения Кг определя-
лись из изотерм адсорбции, изме-
ренных статическим методом [44—
48], и из удерживаемых объемов,
полученных методом газоадсорб-
ционной хроматографии [49] (см.
гл. Ill, IV и Приложение). Опыт-
ные значения были определе-
ны из графиков зависимости In Кг
от 1/Т, полученных из эксперимен-
тальных данных [44, 45, 47—49]
пой части кривой зависимости qst от
Рис. Х,2. Рассчитанные (кривые>
и опытные (точки) значения
для адсорбции метана (1), этана
(2) и пропана (5) на базисной гра-
ни графита (расчет) и графити-
рованных термических сажах
(опыт).
Способы расчета сплошных и пунк-
тирных (см. стр. 313) кривых указан-
ны в тексте.
или экстраполяцией линей-
Г к Г = 0 [46] и переходом
от qst,i к &Ult ___
Рассчитанные зависимости In Кг от 1/77 (см. рис. Х,1) и
от Т (см. рис. Х,2) лежат заметно выше соответствующих экспери-
ментальных значений. Основной причиной этого расхождения ре-
зультатов расчета с опытом, по-видимому, являются, как и в рас-
мотренном в разд. 4 гл. IX случае адсорбции благородных газов,
неточности параметров атом-атомных потенциальных функций меж-
молекулярного взаимодействия С (алкан). . . С(графит) иН (алкан). ..
С (графит), оцененных на основании свойств адсорбата и адсорбента,
взятых в отдельности.
Уточнение параметров атом-атомных потенциалов взаимодей-
ствия атомов С (алкан) ... С (графит) и Н (алкан) ... С (графит).
В работах [9, 10, 17] было принято, что оцененные выше на основа-
нии свойств адсорбата и адсорбента, взятых в отдельности, значения
311.
параметров CJC^ q и r0 потенциальных функций межмолекуляр-
ного взаимодействия атомов G ... С и Н . . . С близки к точным.
Кроме того, было принято, что отношение параметров Ci, с... с
и н ... с Для взаимодействия этих пар атомов дается формулой
Кирквуда Мюллера, т. е.
г гкм
°1, С. . . С с1, С. . . с zv п
С< и г ” гкм ' ’ ;
1, H. . . С bit н. . . С
где . си > с — значения параметра С± для взаимодействия ато-
мов С ... С и Н ... С, оцененные по формуле Кирквуда — Мюллера.
При справедливости этих приближений искомые потенциальные
функции ф и Ф = связаны с приближенными потенциальными
функциями ф* и Ф* = 22<р*, у которых параметры Су оценены
по формуле Кирквуда — Мюллера, соотношениями [см. выражения
< IX,9)-(IX,11)]:
ф=Рф‘ (Х,2)
я
ф=22ф=₽22ф*=₽ф* (х,з)
где коэффициент
P=-f^- = -fkg- 'С (Х-4)
Ч. с... с Ч, н... с
Здесь С х G q и Су н с—значения этих параметров, удовлетворя-
ющие экспериментальным адсорбционным данным.
Значение р можно определить, сопоставляя рассчитанные на
основании Ф* и опытные значения константы Генри Ку. Как и в слу-
чае адсорбции благородных газов (см. разд. 5 гл. IX), для этого
удобно использовать графический способ. Значение р равно зна-
чению коэффициента, на который надо разделить абсциссы рассчи-
танной кривой зависимости In от 1/77, чтобы получить ее совпа-
дение с экспериментальными значениями In Ку.
Определенные таким способом значения р для адсорбции этана
и пропана близки. В среднем р = 0,87. Таким образом, необходимое
уточнение Су составляет всего 13%. Как известно, формула Кирк-
вуда — Мюллера дает завышенные значения Су [51, 52]. Поэтому
полученное значение р < 1.
Для адсорбции метана значение р, полученное при использова-
нии определенных при высоких температурах опытных значений Ку
[44] близко к значению р, полученному для этана и пропана. Однако
-значение р, полученное при использовании опытных значений Ку,
определенных при более низких температурах [46, 49], заметно
€олыпе 0,87. Это расхождение может быть вызвано квантовостати-
стическими эффектами, проявляющимися при более низких темпе-
ратурах. Однако возможны и другие причины этого расхождения,
прежде всего отличающееся валентное состояние атома С в метане
312
(отсутствие связи с другим атомом углерода). Для суждения об этол®
необходимо уточнение опытных данных для адсорбции метана в ши-
роком интервале температур.
Рассчитанные на основании Ф* зависимости In Кг от 1/Т для
адсорбции метана, этана и пропана, абсциссы* которых разделены
на коэффициент р = 0,87, на рис. Х,1 показаны пунктирными ли-
ниями.
Полученные так уточненные атом-атомные потенциальные функ-
ции ср межмолекулярного взаимодействия атомов С и Н молекул
алканов с атомами С решетки графита даются выражениями:
Фс (алкан). .. С (графит)= 1,386* 10 ®г 6 2,148-10 5г 8~F
+1,89 • 105 ехр (—35,7г) кДж-моль’1 (Х,5>
Фн (алкай)... С (графит)= 0,498-10 8г 6— 0,950-10 5г 8 +
4-3,60-104 ехр (—35,7г) кДж-моль-1 (Х,6>
Расстояние г в этих формулах выражено в нм.
Потенциальные функции Ф взаимодействия атомов С и Н со всей
решеткой графита, рассчитанные на основании уточненных атом-
атомных потенциальных функций (Х,5) и (Х,6) путем их суммиро-
вания по атомам С решетки графита, имеют минимумы соответственно
при 0,338 и 0,296 нм. Если принять, что равновесное расстояние
z0 силового центра молекулы (атома Н или атома С молекулы угле-
водорода) от базисной грани графита равно сумме половины меж-
плоскостного расстояния решетки графита и эффективного радиуса
силового центра [см. формулу (VIII,42)], то для эффективных радиу-
сов атомов С и Н получаем соответственно 0,170 и 0,128 нм. Эти
значения приблизительно на 0,02 нм меньше значений ван-дер-вааль-
совых радиусов атомов С и Н при взаимодействии этих атомов не
с кристаллом графита, но с изолированным атомом С и соответственна
Н (см. данные на стр. 310). ___
На рис. Х,2 пунктирными кривыми показаны зависимости AZ7i
от Т для адсорбции метана, этана и пропана, полученные при исполь-
зовании уточненных атом-атомных потенциальных функций (Х,5)
п (Х,6). Они близки к соответствующим опытным значениям.
В табл. Х,2 приведены рассчитанные при использовании уточнен-
ных атом-атомных потенциальных функций (Х,5) и (Х,6) значения
Д51 и ACi для адсорбции метана, этана и пропана на базисной грани
графита [10]. В табл. X,2 также приведены экспериментальные зна-
чения ASi, определенные на основании экспериментальных зна-
чений Кг и AZ/i [49] (см. также табл. П,128 Приложения).
Рассчитанные и экспериментальные значения А5* близки друг
к другу. Это указывает на то, что полученные потенциальные функ-
ции Ф для метана, этана и пропана приблизительно правильно пере-
дают зависимость энергии межмолекулярного взаимодействия этих
молекул с базисной гранью графита от положения молекулы по от-
ношению к ее поверхности.
313
Таблица Х,2, Рассчитанные значения и ДС1!
для адсорбции метана, этана и пропана на базисной грани графита
и опытные значения Д5* (7^1 в мкм) для адеорбции этих молекул
на графитированной термической саже
Молекула Расчет Опыт
Г, К -Д8?/й ДС1/Я Г, К -Д81/2?
Метан 150 10,9 1,0 149 11,1
250 10,3 0,7
Этан 150 11,4 1,5
200 11,1 1,4 199 11,2
250 10,7 1,2
Пропан 150 12,5 2,6
250 11,2 1,9 253 11,5
В случае адсорбции сложных квазижестких молекул при пред-
положении, что движение вдоль поверхности и вращение молекул
в адсорбированном состоянии не заторможены, а колебания центра
масс молекулы перпендикулярно поверхности являются гармони-
ческими, как и в случае адсорбции одноатомных молекул,
не зависит от температуры и равно 0,5 R. Для этана и пропана рас-
считанные значения &Сг в несколько раз больше 0,5 R. Это показы-
вает, что изменение теплоемкости этих молекул при адсорбции обу-
словлено главным образом торможением вращения молекул при
адсорбции. Следовательно, для изучения зависимости Ф от ориен-
тации молекулы удобнее всего использовать изменение теплоем-
кости при адсорбции. Однако экспериментально такое изучение
в области малых Г сделано пока лишь для бензола (см. разд. 3
этой главы).
Как указывалось выше, расчеты термодинамических характери-
стик адсорбции метана, этана и пропана производились при исполь-
зовании гармонического приближения для зависимости Ф от z.
Термодинамические характеристики адсорбции этих углеводородов
были рассчитаны также и без использования гармонического при-
ближения [10, 19]. В табл. Х,3 сопоставлены результаты расчета
величин In К г и полученные при числовом интегрировании
конфигурационных интегралов по z и при использовании гармони-
ческого приближения для зависимости Ф от z. Из таблицы видно,
что погрешность, вносимая этим приближением в значения In
(при выражении Кi в мкм), меньше 0,2, а для Д Ur не превышает 5%.
Значения Л5* и ДС^, полученные при использовании гармониче-
ского приближения, только на 0,2 R — 0,3 R меньше значений,
полученных при численном интегрировании без этого допущения.
Поэтому расчеты для других молекул производились только при
использовании гармонического приближения для зависимости Ф от z.
314
Расчет термодинамических характеристик адсорбции этана с уче-
том внутреннего вращения молекул. При расчетах термодинами-
ческих характеристик адсорбции (см. стр. 310) молекулы этана
п пропана рассматривались квазижесткими и имеющими в адсор-
бированном состоянии те же конфигурации, что и в объеме газа.
Однако в молекулах этана и пропана происходит заторможенное
внутреннее вращение вокруг связей С—С, которое при адсорбции
может несколько изменяться. Чтобы определить вклад изменения
этого внутреннего вращения при адсорбции в термодинамические
характеристики, был произведен расчет термодинамических харак-
теристик адсорбции этана на графите с учетом внутреннего вращения
молекулы [11].
В классическом приближении термодинамические характери-
стики адсорбции этана с учетом внутреннего вращения молекулы
даются выражения (VII,21)—(VII,27). Расчеты производились при
использовании неуточненных потенциальных функций ср и гармо-
нического приближения для зависимости Ф от z. Для потенциальной
энергии W изолированной молекулы этана было принято выраже-
ние [53]:
W = -И»(1-cos па) (Х,7>
где Жо — 11,5 кДж/моль — потенциальный барьер для внутреннего вращения;
п — 3 — число потенциальных минимумов для внутреннего вращения молекулы
этана.
При справедливости выражения (Х,7) интегралы 7?z сводятся
к функции Бесселя.
В табл. Х,4 результаты расчета термодинамических характери-
стик сопоставлены с соответствующими значениями, полученными
для модели молекулы этана, как квазижесткой с транс-расположе-
нием атомов Н.
Из этой таблицы видно, что термодинамические характеристики
адсорбции этана на базисной грани графита, рассчитанные с учетом
внутреннего вращения молекулы и для модели квазижесткой моле-
кулы, близки. При одинаковой погрешности рассчитанных значений
конфигурационных интегралов значения АСг получаются со зна-
чительно большей погрешностью, чем остальные термодинамические
величины, причем эта погрешность больше при более низких темпе-
ратурах. Поэтому расхождение в значениях АС^, рассчитанных
при 150 К для модели квазижесткой молекулы и с учетом внутрен-
него вращения, вызвано, по-видимому, погрешностями расчета.
Для взаимодействия молекулы этана с базисной гранью графита
энергетически наиболее выгодно цис-расположение атомов Н в мо-
лекуле, Рассчитанное абсолютное значение Фо для молекулы этана
при цис-расположении атомов Н приблизительно на 2,5 кДж/моль
больше, чем при транс-расположении. В объеме же газа энергети-
чески более выгодно транс-расположение атомов Н в молекуле.
Поэтому суммарная величина потенциального барьера для внут-
315
Таблица Х,8, Рассчитанные значения In Ki и AUj. для адеорбции
метана, этана и пропана, полученные числовым интегрированием по z
и при использовании гармонического приближения для зависимости Ф от z
Молекула г, к InKi (К, в мкм) —ДПь кДж «моль’1
Числовое интегрирование Гармоническое приближение Числовое интегрирование Гармоническое приближений
Метан 100 4,74 4,69 12,4 * 12,4
200 —2,46 —2,58 11,3 11,6
300 —4,66 —4,85 . 10,6 11,1
Этан 150 3,83 3,75 17,7 17,6
200 0,35 0,26 17,0 17,0
250 —1,65 —1,76 16,3 16,6
Пропан 200 3,24 3,28 23,1 23,5
300 —1,22 —1,28 21,3 21,8
400 —3,29 -3,41 19,9 20,7
Таблица Х,4. Термодинамические характеристики адсорбции этана
на графите, вычисленные с учетом внутреннего вращения молекулы
и для модели квазижесткой молекулы
Термодинамическая характеристика 150К 250К
С учетом внутреннего вращения Для модели квазижесткой молекулы С учетом внутреннего вращения Для модели квазижест- кой молекулы
In Ki (Кг в мкм) 5,93 5,88 —0,52 -0,56
AtZx, кДж/моль —20,5 —20,5 -19,4 —19,4
ASJ/Я -11,6 —11,6 -10,8 —10,8
ACJR (1,7) 1,3 1,2 1,2
реннего вращения молекулы этана вокруг связи С—С в адсорбиро-
ванном состоянии на 2,5 кДж/моль меньше, чем в газе. Однако
это уменьшение значительно меньше величины самого потенциаль-
ного барьера внутреннего вращения для молекулы в объеме газа
0УО = 11,5 кДж/моль). Поэтому при адсорбции на поверхности
графита внутреннее вращение молекулы этана может измениться лишь
очень слабо и остается существенно заторможенным. Основной кон-
фигурации молекулы этана на поверхности, как и в газе, соответствует
транс-расположение атомов Н. Небольшое изменение внутреннего
вращения молекулы этана при ее переходе из объема газа в адсорби-
рованное состояние вносит лишь незначительный вклад в термодина-
мические характеристики адсорбции этой молекулы на графите.
Поэтому при расчетах изменений термодинамических свойств ад-
сорбата при адсорбции молекулу этана в хорошем приближении
можно рассматривать квазижесткой.
Обобщая эти результаты, можно утверждать, что при расчетах
термодинамических характеристик адсорбции молекулу адсорбата
316
можно приближенно рассматривать квазижесткой, если эта моле-
кула при внутреннем вращении не образует поворотных изомеров
и если потенциальный барьер для внутреннего вращения значительно
больше вызываемого этим вращением изменения потенциальной
энергии Ф взаимодействия молекулы с поверхностью. Этим требо-
ваниям удовлетворяет адсорбция на графите ближайшего к этану
гомолога — пропана. Поэтому при расчетах термодинамических
характеристик адсорбции молекулу пропана также можно рассмат-
ривать квазижесткой. По-видимому, это приближение применимо
ко всем молекулам углеводородов, состоящим из остова и прикре-
пленных к нему симметричных волчков, для которых барьеры внут-
реннего вращения того же порядка, как и в молекуле этана.
н-Бутан*, к-пентан и н-гексан. Молекулы н-алканов, начиная
с w-бутана, при внутреннем вращении вокруг связей С—С образуют
поворотные изомеры [54, 55]. У н-бутана при внутреннем вращении
вокруг центральной связи С—С возможны три поворотных изомера:
один транс-изомер Т и два энергетически одинаковых гош-изомера G
(правый и левый). У м-гептана поворотные изомеры образуются при
вращении относительно двух связей С—С и всего их девять: один
изомер ТТ, четыре TG, два GG и два GG (здесь использованы обозна-
чения Мидзусимы [54]). Общее число поворотных изомеров у я-алка-
нов равно 3”“3, где п — число атомов С в молекуле.
При расчетах термодинамических характеристик адсорбции н-бу-
тан, м-пентан и м-гексан рассматривались как смеси их поворотных
изомеров, находящихся в равновесии друг с другом [13, 14, 17].
В этом приближении термодинамические характеристики адсорбции
даются статистическими выражениями (VII,30)—(VII,40). Индиви-
дуальные же поворотные изомеры рассматривались квазижесткими.
Величины 1} Д5*,1 и ДСЬ1 для индивидуальных поворот-
ных изомеров вычислялись с помощью выражений (VII,44)-— (VII,46),
(VII,53), (VII,56)—(VII,60), т. е. пренебрегалось изменением коле-
бательной статистической суммы поворотного изомера при адсорб-
ции, а колебания центров масс изомеров перпендикулярно поверх-
ности рассматривались гармоническими.
При расчетах мольных долей изомеров в газе х% по формуле
(VII,40) значения .разностей свободных энергий ДГ£. гош-изомеров
и транс-изомера оценивались по формуле [56, 57:]
SFi^gib (Х,8)
где gi — число гош-положений в г-ом изомере; Ъ = 2,5 кДж/моль [56—59] —
средняя разность свободных энергий изомеров, различающихся одним гош-
поворотом.
В случае гош-положений разных знаков около соседних связей
С—С (например, для изомера GG м-пентана) значения ДГ* прини-
мались бесконечными [57, 59], т. е. такие изомеры принимались
стерически исключенными.
Результаты расчета термодинамических характеристик адсорб-
ции для индивидуальных поворотных изомеров м-бутана, н-пентана
317
и «-гексана, полученные при использовании уточненных атом-
атомных потенциальных функций межмолекулярного взаимодей-
ствия (Х,5) и (Х,6), приведены в табл. Х,5 и Х,6. В этих таблицах
также приведены мольные доли поворотных изомеров в объеме
газа xf и в адсорбированном состоянии х?, рассчитанные по фор-
мулам (VII,39), (VII,40) и (Х,8).
Таблица Х,5, Рассчитанные термодинамические характеристики
адсорбции энергетически различных поворотных изомеров «-бутана
и н-пентана на базисной грани графита и мольные доли этих изомеров
в объеме газа и в адсорбированном состоянии xf
(константа Генри К{, выражена в мкм, &Uit 1 — в кДж/моль)
Молекула и ее поворотные изомеры Число изо- меров т, к 1 1 1 R R xf
«-Бутан Т 1 250 3,08 29,6 12,1 2,5 0,63 0,76
400 —2,04 27,0 11,2 1,8 0,52 0,58
550 -4,18 25,0 10,7 1,4 0,46 0,50
G 2 250 2,44 27,3 11,6 2,3 0,37 0,24
400 -2,30 25,1 10,9 1,4 0,48 0,42
500 —4,31 23,9 10,6 0,7 0,54 0,50
«-Пентан
ТТ 1 250 5,92 37,5 13,1 2,9 0,42 0,70
400 —0,59 34,2 11,9 2,5 0,30 0,41
550 —3,28 31,4 11,2 1,9 0,25 0,30
TG 4 250 4,76 33,1 12,1 1,9 0,50 0,26
400 —1,04 30,9 11,3 1,7 0,57 0,48
550 —3,49 29,0 10,9 1,3 0,58 0,55
GG 2 250 4,73 33,2 12,2 2,2 0,08 0,04
400 —1,06 30,6 11,3 1,9 0,13 0,11
550 —3,50 28,8 10,8 1,2 0,17 0,16
Как видно из табл. Х,5 и Х,6, термодинамические характери-
стики адсорбции гош-поворотных изомеров этих молекул заметно
отличаются от таковых для транс-изомера, причем это различие
растет в ряду «-бутан, «-пентан и «-гексан. Значения Кц х и —ДС7Ь *
для гош-изомеров всегда меньше, чем для транс-изомера, который
может расположиться на поверхности плоско, т. е. энергетически
наиболее выгодным образом. Термодинамические характеристики
адсорбции для всех гош-изомеров сравнительно близки. Мольные
доли транс-поворотных изомеров в адсорбированном состоянии
значительно больше, чем в объеме газа, причем с понижением тем-
пературы они увеличиваются. Однако даже при самых низких рас-
смотренных температурах в адсорбированном состоянии еще содер-
жится значительная доля молекул в гош-конформациях.
На рис. Х,3 и Х,4 рассчитанные значения 1иДС76 г для от-
дельных поворотных изомеров, а также статистические средние
значения и ДС7Х для адсорбции «-бутана, «-пентана и «-гексана
318
tn Ki
Рис. X,3. Рассчитанные (сплошные кривые) и опытные (точки) значения кон-
станты Генри Кг для адсорбции «-бутана (7), к-пентана (2) и «-гексана (5) на
базисной грани графита (расчет) и на графитированных термических сажах
(опыт).
Пунктирными кривыми показаны рассчитанные значения константы Генрих^ * для адсорбции
индивидуальных поворотных изомеров этих н-алканов. Величины Кх и Кц выражены в мкм.
Рис. Х,4. Рассчитанные (сплошные кривые) и опытные (точки)
значения для адсорбции w-бутана (I), «-пентана (2) и «-гек-
сана (2) на базисной грани графита (расчет) н на графитированных
термических сажах (опыт).
Пунктирными кривыми показаны рассчитанные значения ДШд для адсорб-
ции индивидуальных поворотных изомеров этих н-алканов.
Таблица Х,6. Рассчитанные термодинамические характеристики
адсорбции энергетически различных поворотных изомеров н-гексана
на базисной грани графита и мольные доли этих изомеров
в объеме газа ж? и в адсорбированном состоянии
(константа Генри Ki, i вкГражена в мкм, Д€71 — в кДж/моль)
Поворот- ные изомеры Число изомеров т, к In К,., , -ди<, 1 1 R дс/, 1 хд Х1 хг
R
ттт 1 250 9,09 45,2 13,6 2,8 0,27 0,72
400 1,18 42,0 12,5 2,4 0,17 0,35
550 —2,15 39,1 11,6 2,3 0,13 0,21
TTG 4 250 7,27 40,4 13,1 2,7 0,32 0,14
400 0,23 37,2 12,0 2,4 0,31 0,25
550 —2,72 34,5 11,3 1,9 0,30 0,27
TGT 2 250 7,26 39,6 12,8 2,5 0,16 0,07
400 0,31 37,0 11,8 2,0 0,16 0,14
550 —2,63 34,7 11,3 1,8 0,15 0,15
TGG 4 250 6,86 39,4 13,1 2,9 0,10 0,03
400 0,01 36,0 12,8 2,5 0,15 0,10
550 —2,83 33,2 11,1 1,9 0,17 0,14
GTG 2 250 6,68 38,1 12,6 2,3 0,05 0,01
400 0,01 35,5 11,7 2,0 0,07 0,05
550 -2,80 33,2 11,1 1,6 0,09 0,07
GTG 4 250 7,17 39,1 12,6 1,8 0,10 0,04
400 0,29 36,8 11,8 1,9 0,15 0,13
550 —2,64 34,5 11,2 1,8 0,17 0,17
на базисной грани графита сопоставлены с соответствующими опыт-
ными значениями на графитированных термических сажах [49,
60—63]. Рассчитанные зависимости In Кх от 1/Г и от Т для всех
трех углеводородов находятся в хорошем согласии с опытом.
Статистические средние значения Кх и лежат между соот-
ветствующими значениями для транс- и гош-изомеров. При пони-
жении температуры они приближаются к соответствующим значе-
ниям для транс-изомера, а при повышении температуры — к зна-
чениям для гош-изомеров. Такое изменение значений Кх и
определяется главным образом изменением мольных долей поворот-
ных изомеров в адсорбированном состоянии с изменением темпера-
туры. Для правильного молекулярно-статистического описания
адсорбции молекул н-алканов, начиная с н-бутана, необходимо учесть
способность этих молекул к внутреннему вращению с образо-
ванием поворотных изомеров.
Рассчитанные значения и ДС\ для адсорбции н-бутана,
н-пентана и н-гексана на базисной грани графита приведены в табл.
Х,7. В этой таблице приведены также экспериментальные значения
AiSJ, определенные на основании экспериментальных значений Кг
и [49] (см. также табл. П,3 Приложения).
320
Таблица Х,7. Рассчитанные значения AS* и ACi
для адсорбции «-бутана, «-пентана и н-гексана на базисной грани графита
и опытные значения AS* для адсорбции этих молекул
на графитированной термической саже
Молекула Расчет Опыт
Т, К -дзГ/н ДС1/В Т, К -Д8?/Н
«-Бутан 250 12,2 3,0 325 11,5
400 11,1 1,9
550 10,6 1,2
«-Пентан 250 13,3 4,7
400 11,7 2,8 349 11,9
550 11,0 1,8
«-Гексан 250 14,6 5,6
400 12,4 3,8 398 12,1
550 11,4 2,5
Рассчитанные и экспериментальные значения A5J близки друг
к другу. Следовательно, полученные потенциальные функции Ф
для н-бутана, н-пентана и н-гексана приблизительно правильно
передают зависимость потенциальной энергии межмолекулярного
взаимодействия этих молекул с базисной гранью графита от поло-
жения молекулы по отношению к поверхности адсорбента.
Рассчитанные значения АС*! для адсорбции н-бутана, н-пентана
и н-гексана во много раз больше 0,5 R. Поэтому изменение тепло-
емкости этих углеводородов при адсорбции обусловлено в основном
торможением вращения этих молекул при адсорбции и изменением
равновесия между поворотными изомерами молекулы при адсорбции.
С ростом температуры рассчитанные значения ДСХ быстро умень-
шаются.
На рис. Х,5 и Х,6 приведены рассчитанные значения A5J и ACj
для н-алканов в функции числа п атомов углерода в молекуле. На-
чиная с пропана наблюдается приблизительно линейная зависи-
мость A5J от и. Величина ДС^ быстро растет с ростом п и при п 2
эта зависимость также приблизительно линейна.
Изменение энтропии при адсорбции обусловлено в основном
изменением поступательного и вращательного движения молекул
при адсорбции. Строго этих вкладов разделить нельзя, так как
поступательное и вращательное движения адсорбированных молекул
взаимосвязаны. Чтобы оценить вклады, которые вносят отдельно
поступательные и вращательные степени свободы молекулы в энтро-
пию адсорбции, была рассчитана поступательная энтропия адсорб-
ции Д*$*ост,1 w-алканов, обусловленная изменением только посту-
пательного движения молекул при адсорбции [17]. При расчетах
А5*0СТ)1, как и ПРИ расчетах полной энтропии адсорбции A5J,
движение центра масс молекул вдоль поверхности принималось пол-
21 Заказ 1180 321
ностью свободным, а его колебания перпендикулярно поверхности
рассматривались гармоническими. Для принималось значение,
полученное для положения молекулы вблизи основного потенциаль-
ного минимума, т. е. пренебрегалось зависимостью Ф? от ориента-
ции молекулы. Последнее приближение только незначительно уве-
личивает абсолютную величину поступательной энтропии адсорб-
ции. Для н-бутана, м-пентана и м-гексана, образующих поворотные
изомеры, расчет поступательной энтропии адсорбции производился
для транс-поворотного изомера. Вращательная энтропия адсорбции
Д5Вращ, 1, обусловленная изменением вращательного (внешнего
Рис. Х,5. Зависимость рассчитанных
дифференциальных изменений энтро-
пии &S*/R при адсорбции м-алканов
на базисной грани графита от чис-
ла п атомов углерода в молекуле.
Рис. Х,6. Зависимость рассчитанных
дифференциальных изменений тепло-
емкости &CJR при адсорбции н-алка-
нов на базисной грани графита от чис-
ла и. атомов углерода в молекуле при
различных температурах 250 (5)\ 400 (2)
и 500 К (7).
и внутреннего) движения молекул при адсорбции, определялась
как разность изменений полной энтропии Д5* и поступательной
энтропии Д5*ост>1 при адсорбции. Использованные в расчетах
значения Ф"г и полученные значения Д5*0СТ)1 и Д5вращ>1 при-
ведены в табл. Х,8. В этой таблице приведены также рассчитанные
значения максимальной величины потенциального барьера Уо для
вращения адсорбированных молекул. Как видно из таблицы, посту-
пательная энтропия адсорбции, обусловленная переходом одной
поступательной степени свободы молекулы в колебательную степень
свободы центра масс молекулы перпендикулярно поверхности, вносит
основной вклад в изменение энтропии для адсорбции. Вращательная
энтропия Д5враЩ(1 составляет небольшую долю от общего измене-
ния энтропии при адсорбции*
Для рассмотренных молекул значения Уо намного больше RT.
Поэтому из результатов расчета Ф следует, что вращение этих мо-
322
ленул в адсорбированном состоянии сильно заторможено. Однако
вращательная энтропия адсорбции для этих молекул составляет
только несколько энтропийных единиц. Поэтому о степени затормо-
женности вращения адсорбированных молекул трудно однозначно
судить на основании экспериментальных значений изменений энтро-
пии при адсорбции. Для определения состояйия адсорбированных
молекул необходимо определить Ф.
Таблица ХЛ8. Рассчитанные значения Ф^, Уо? Лапост, i
и АЗвращ, 1 Для адсорбции «-алканов на базисной грани графита
Молекула Фг-10—23, Дж / (моль «м2) Ио, кДж/моль т, к Допост, 1 В ДЗвращ, 1 R
Метан 46,9 4,2 150 10,6 0,2
250 10,3 0,0
Этан 64,5 5,9 150 10,8 0,6
250 10,5 0,2
Пропан 87,1 10,5 150 11,0 1,5
250 10,6 0,6
«-Бутан 111 18,8 250 10,7 1,5
400 10,6 0,5
«-Пентан 136 26,0 t 250 10,8 2,5
400 10,6 1,1
«-Гексан 161 33,5 250 10,9 3,7
400 10,8 1,6
550 10,6 0,6
Рассчитанные изменения теплоемкости при адсорбции н-алканов
значительно отличаются от нуля. Согласно законам термодинамики
теплота адсорбции таких веществ должна существенно зависеть от
температуры. Экспериментальное исследование этой зависимости
встречает значительные трудности, так как теплота адсорбции обычно
определяется в сравнительно узком интервале температур и часто
со значительной погрешностью (до 5%).
В табл. Х,9 приведены рассчитанные значения потенциальной
энергии Фо взаимодействия молекул м-алканов с базисной гранью
графита в основном потенциальном минимуме, изменения внутренней
энергии при адсорбции —^U1 и разности Фо —AtTY В случае
молекул в-алканов С4—CQ, образующих поворотные изомеры, в та-
блице приведены значения Фо для транс-поворотных изомеров.
Значения Фо с точностью до нулевой энергии адсорбированных мо-
лекул равны — At/i лишь при О К. Как видно из таблицы, значения
АС7Х близки к значениям Фо только для наиболее легких молекул
и при низких температурах. Для более тяжелых молекул при вы-
соких температурах значения АС7\ на 5—15 кДж/моль (10—30%)
меньше значений Фо, и эта разность быстро растет с ростом темпера-
21* 323
туры. Поэтому сопоставление рассчитанных значений Фо с опытными
значениями ДС7П полученными при Т ОК, как это часто делается,
не строго.
Таблица Рассчитанные значения Фо, и Фо—
для адсорбции «-алканов на базисной грани графита
Молекула —Фо» кДж /моль т, к -ДП1, кДж/моль - (фо-Д<71)
кДж/моль % ОТ Фо
Метан 13,9 100 12,4 1,5 11
200 11,6 2,3 17
Этан 19,8 150 17,6 2,2 И
250 16,6 3,2 16
Пропан 27,0 150 24,2 2,8 10
250 23,1 3,9 14
м-Бутан 34,5 250 29,3 5,2 15
400 26,3 8,2 24
м-Пентан 42,2 250 37,0 5,2 12
400 32,5 9,7 23
«-Гексан 49,9 250 45,2 4,7 9
400 39,3 10,6 21
550 35,5 14,4 29
Расчеты термодинамических характеристик адсорбции н-алканов
при выборе в качестве силовых центров звеньев СН3 и СН2. Расчет
термодинамических характеристик адсорбции метана, этана, про-
пана, н-бутана и н-пентана на базисной грани графита производился
также при рассмотрении в качестве силовых центров молекулы СН4
в целом [9, 10, 64, 65] и звеньев СН3 и СН2 для остальных молекул
[9, 10, 13]. Расчеты Ф и на ее основании расчеты термодинамиче-
ских характеристик адсорбции в работах [9, 10, 13] производились
путем, аналогичным использованному при выборе атомов в каче-
стве силовых центров молекулы. В этих работах для энергии меж-
молекулярного взаимодействия звеньев СН3 и СН2 молекулы адсор-
бата с атомом С графита был принят потенциал Бакингема — Кор-
нера (VIII,16). Параметры сил притяжения Сг и С2 этого потенциала
оценивались с помощью формулы Кирквуда — Мюллера (VIII,25)
и аналогичной ей формулы (VIII,33). Параметр сил отталкивания
в экспоненте q принимался одинаковым для взаимодействий СН4 . . .
С, СН3 ... С и СН2 ... С и равным 36 нм-1. Предэкспоненциальный
параметр сил отталкивания В для взаимодействия этих пар силовых
центров оценивался при условии равенства сил притяжения и сил
отталкивания между силовым центром молекулы и всей решеткой
графита при соответствующем равновесном расстоянии z0, т. е. при
использовании уравнения (VIII,38). При этом равновесное расстоя-
ние z0 принималось равным сумме эффективного ван-дер-ваальсового
радиуса г* силового центра молекулы и половины межплоскостного
324
расстояния решетки графита. Ван-дер-ваальсовы радиусы г* мо-
лекулы СН4 и звеньев СН3 и СН2 принимались равными 0,2 нм.
Принятые в расчетах значения поляризуемости а и диамагнитной
восприимчивости % для молекулы СН4 и звеньев СН3 и СН2, и полу-
ченные значения параметров Сг, С2 и В для взаимодействия этих
силовых центров с атомом С графита приведены ниже:
Силовой центр..........
а • Ю3®, м3............
—% * 10зв, и3..........
С1 • 1057, кДж • м6 * моль-1
С2 • 1077, кДж • м8 - моль-1
В-10"5, кДж-моль-1 . .
сн4 СН3 СН2
2,68 2,26 1,83
27,1 23,8 18,9
3,94 3,39 2,72
6,95 5,82 4,73
9,46 8,12 6,53
На рис. Х,7 рассчитанные так зависимости In К.х от ИТ для ад-
сорбции метана, этана и пропана на базисной грани графита сопо-
ставлены с соответствующими опытными значениями In#! для
Рис. Х,7. Рассчитанные (кри-
вые) и опытные (точки) значе-
ния константы Генри Кг для
адсорбции метана (Z), этана (2)
и пропана (3) на базисной
грани графита (расчет) и на
графитированных термических
сажах (опыт).
Сплошные кривые получены при
использовании приближенных потен-
циальных функций межмолекуляр-
ного взаимодействия силовых
центров СН< . . .С, СН8 . . .С и
СН# . . .С. Пунктирная кривая 1
получена при делении абсцисс
сплошной кривой 1 на коэффициент
р = i,Q9t а пунктирные кривые 2
и 3 получены при делении абсцисс
сплошных кривых 2 и 3 на коэффи-
циент Р « 1,05. Величины Кх выра-
жены в мкм.
адсорбции этих веществ на графитированных термических сажах
[44—49]. Рассчитанные кривые лежат заметно ниже соответствующих
экспериментальных значений. В этом случае, как и в случае выбора
атомов С и Н в качестве силовых центров молекулы, можно предпо-
ложить, что основной причиной расхождения рассчитанных и опыт-
ных значений Кх является погрешность в значениях параметров Сг
для межмолекулярных взаимодействий силовых центров СН4, СН3
и СН2 с атомом С графита. Было принято, что отношение парамет-
ров С\ для межмолекулярного взаимодействия этих силовых цент-
ров дается формулой Кирквуда Мюллера, т. е.
gi, сн8... с
е1, сн2. .. с
гкм
с1, сна... с
г.КМ
с1, сн,... с
(Х,9)
где — значения параметра Clt оцененные по формуле Кирквуда — Мюл-
лера.
325
В этом случае искомые уточненные потенциальные функци
взаимодействия силовых центров молекулы с атомом С графита i
и со всей решеткой графита Ф = XX Ф связаны с приближенным;
потенциальными функциями и Ф* = у которых пара
метры Сг оценены по формуле Кирквуда — Мюллера (или по какой
либо другой приближенной формуле, например по формуле Слэте
ра — Кирквуда), теми же соотношениями (Х,2) и (Х,3), как и в слу
чае выбора в качестве силовых центров молекулы атомов С и Н
Значение р в данном случае определяется соотношениями, анало
гичными соотношениям (Х,4), т. е,
о С1,сн3. ,.с _ С1,снг...с ,Y ,п
:---77*--------- (Х,10
Gl, СН3. . . С °1, сн2... с
где — значения параметра оцененные каким-либо приближенным спо
собом. В работах [9, 10] значение С* оценивалось с помощью формулы Кирк
вуда — Мюллера.
Значение р можно определить таким же способом, как и в случае
выбора атомов в качестве силовых центров молекулы, сопоставляв
рассчитанные на основании приближенной потенциальной функции
Ф* значения константы Кх с соответствующими опытными значе-
ниями (см. разд. 2 этой главы). Для метана р — 1,09. Для этана
и пропана значения р близки и в среднем р 1,05. Рассчитанные
выше кривые зависимости In КА от 1/7’, у которых значения 1/Т
разделены на р = 1,09 для метана и на р = 1,05 для этана и про-
пана, на рис. Х,7 изображены пунктирными кривыми.
Уточненные таким образом потенциальные функции межмоле-
кулярного взаимодействия СН3 ... С (графит) и СН2 ... С (графит)
даются выражениями:
<Рсн8. ..С(графит)^-3^6-10-3г'е-“641’10"5г-8+
+ 8,54 • Ю5 ехр (—35,7г) кДж/моль (Х,11)
Фсна. . . С (графит) = -2,86 . Ю-Зг-6-4,97.10-5Г-8 +
+ 6,87 • 105 ехр (—35,7г) кДж/моль (Х,12)
С помощью этих уточненных потенциальных функций были рас-
считаны другие термодинамические характеристики адсорбции этана
и пропана [10], а также термодинамические характеристики адсорб-
ции н-бутана и н-пентана [13]. При расчетах н-бутан и н-пентан рас-
сматривались, как и выше, как равновесные смеси их поворотных
изомеров. Из рис. Х,8 видно, что результаты расчета In К± для
н-бутана и н-пентана хорошо согласуются с соответствующими опыт-
ными значениями [48, 49, 60, 62, 63] (см. также табл. П,3 Прило-
жения).
Результаты расчета на основе потенциальных функций (X,11) и
(Х,12) величин ДС71, Д5Г и &Сг для адсорбции этана, пропана,
н-бутана и н-пентана приведены в табл. Х,10. В таблице приведены
также соответствующие экспериментальные ^значения ДСТД и Д5*.
326
Рассчитанные и экспериментальные значения AC/i и AS£ хорошо
согласуются для всех рассмотренных молекул.
Таким образом, при определениях потенциальных функций Ф
п расчетах на основании этих функций термодинамических харак-
теристик адсорбции м-алканов в качестве силовых центров молекулы,
начиная с этана, можно рассматривать группы СН3 и СН2. Полу-
ченные при использовании опытных данных для адсорбции этана
и пропана потенциальные функции (Х,11) и Х,12) межмолекулярного
Рис. Х,8. Рассчитанные (кривые) при ис-
пользовании уточненных потенциальных
функций межмолекулярного взаимодейст-
вия силовых центров СН3 ... С и
СН2 ... С и опытные (точки) значения
константы Генри Кг для адсорбции н-бу-
тана (7) и н-пентана (2) на базисной
грани графита (расчет) и на графитирован-
ных термических сажах (опыт).
Величины выражены в мкм.
взаимодействия групп СН3 и СН2 с атомом С графита дают хорошие
результаты и для более высоких членов ряда м-алканов. По-види-
мому, они могут быть использованы для расчетов потенциальной
функции Ф и на ее основании — термодинамических характеристик
адсорбции всех к-алкапов при низких заполнениях поверхности
графитированных термических саж. Однако для проведения анало-
гичных расчетов для других углеводородов, например мзо-алканов,
Таблица Х,10. Рассчитанные значения AC/j, А5* и АС\ для адсорбции
н-алканов на базисной грани графита и соответствующие опытные
значения At/X и A5J на графитированных термических сажах
Молекула Расчет Опыт
Т, К -ди1( кДж/моль _ AS* R ДС1 R Т, К -ДП1, кДж/моль _дй£ R Лите- рату- ра
Этан 157,5 17,2 11,2 1,4 193 16,3 10,9 [45]
210 16,7 11,0 1,1 196 17,2 11,2 [49]
262,5 16,2 10,9 1,0 288 16,7 11,0 [48|
Пропан 210 22,9 11,6 1,8 248 22,6 11,6 [49]
367,5 20,9 11,0 1,3 288 22,6 11,5 [48|
н-Бутан 300 20,5 10,7 [47|
262,5 29,4 12,2 2,6 327 27,6 11,5 [49]
472,5 25,7 11,2 1,6 323 28,9 12,2 [48|
н-Пентан 262,5 37,1 13,1 3,1 349 33,5 11,9 [49]
472,5 32,2 11,7 2,3 423 33,9 12,1 [63|
423 33,9 12,2 [62]
327
необходимо еще ввести потенциальные функции для межмолекуляр-
ного взаимодействия групп С Н и четвертичного атома С молекул
изо-алканов с атомами С графита, в то время как для расчетов при
использовании в качестве силовых центров атомов С и Н молекулы
в случае адсорбции изо-алканов можно обойтись теми же двумя
атом-атомными потенциальными функциями (Х,5) и (Х,6),-как
и в уже рассмотренном случае адсорбции н-алканов. Поэтому при
исследованиях термодинамических характеристик адсорбции других
классов углеводородов в качестве силовых центров рассматривались
только атомы С и Н молекулы [14, 17, 18].
Рис. Х,9. Рассчитанные (кривые)
и опытные (точки) значения констан-
ты Генри для адсорбции изобу-
тана (7), неопентана (2), изопента-
на (3) и «-пентана (4) на базисной
грани графита (расчет) и на графи-
тированной термической саже (опыт).
Величины Ki выражены в мкм.
изо-Алканы. На рис. Х,9 и в табл. Х,11 приведены термодина-
мические характеристики адсорбции структурных изомеров н-бутана
и н-пентана, а именно, изобутана, изопентана и неопентана, рассчи-
танные при использовании тех же атом-атомных потенциальных
функций межмолекулярного взаимодействия (Х,5) и (Х,6), что и для
адсорбции н-алканов [14, 17].
На рис. Х,9 и в табл. Х,11 также’приведены соответствующие
экспериментальные значения и AS* [49, 63] (см. также
табл. П, 3 Приложения). Было принято, что в молекулах изобу-
тана, изопентана и неопентана все валентные углы тетраэдрические,
а углы внутреннего вращения соответствуют транс-расположению
соседних связей С—Н. Для молекул изопентана реализуется, по-
видимому, только энергетически наиболее выгодная транс-конфор-
мация [57, 66, 67]. Поэтому расчеты для изопентана производились
только для этой конформации. Рассчитанные и опытные значения
хорошо согласуются. Для сравнения на рис. Х,9 приведены также
рассчитанные и опытные значения In Кг для н-пентана. Как видно
из этого рисунка, рассчитанные значения In К± для структурных
изомеров молекулы пентана (н-пентана, изопентана и неопентана)
имеют ту же последовательность, что и соответствующие экспери-
ментальные значения.
328
Таким образом, принятая в работах [9, 10, 14, 17] система при-
ближений позволяет хорошо описать зависимость адсорбционных
свойств алканов на базисной грани графита от геометрического
строения молекулы. Весьма важно то, что это может быть исполь-
зовано и для решения обратной задачи — для определения геометри-
ческого строения молекулы адсорбата на основании ее эмпирической
химической формулы, опытных (газохроматографических [68] или
статических) адсорбционных данных на графитированных терми-
ческих сажах и результатов молекулярно-статистического расчета
гермодинамических характеристик адсорбции для разных возможных
моделей геометрического строения молекулы.
Таблица Х,11. Рассчитанные значения At719 A5J и ACi
для адсорбции изоалканов на базисной грани графита и соответствующие
опытные значения AZ71 и А5* на графитированной термической саже
Молекула Расчет Опыт
Т, К -Д171, кДж/моль „ AS* R ДС1 н Т, К -Д171, КДж/МОЛЬ _ &s* R Ссылки
Изобутан 250 26,1 11,4 1,6 327 26,8 11,5 [49].
400 24,4 10,8 1,2
550 23,0 10,4 1,0
Изопентан 250 32,7 12,1 2,1 349 30,6 11,5 [49]
400 30,3 11,3 1,8 423 31,4 11,7 [63]
550 28,3 10,8 1,4
Неопентан 250 27,6 11,1 1,4 329 26,0 10,7 [49]
375 26,4 10,6 1,0
500 25,5 10,5 0,7
Удовлетворительное согласие результатов расчета с ОПЫТОМ
для w-алканов и изоалканов, полученное при использовании одних
и тех же атом-атомных потенциальных функций (Х,5) и (Х,6), ука-
зывает на то, что атом-атомная потенциальная функция межмолеку-
лярного взаимодействия атомов С алканов с атомом С графита в пре-
делах погрешности экспериментальных хроматографических и ад-
сорбционных данных не зависит от природы атомов, связанных
валентно с рассматриваемым атомом С молекулы алкана.
3. Цикланы
На рис. Х,10 и в табл. Х,12 приведены результаты расчета термо-
динамических характеристик адсорбции на базисной грани графита
для трех цикланов: циклопропана, циклопентана и цикло-
гексана [17].
При расчетах были использованы те же атом-атомные потен-
циальные функции межмолекулярного взаимодействия (Х,5) и (Х,6),
что и в рассмотренных выше случаях адсорбции алканов. Для цикло-
пентана была принята конформация формы конверта [69]. Для
циклогексана была принята конформация кресла с тетраэдрическими
329
углами. В пределах погрешности опытных данных [30, 49, 61, 63J
(см. также табл. П, 3 Приложения) рассчитанные и опытные зна-
чения К19 &иг и для циклопентана и циклогексана совпадают.
Рис. Х,10. Рассчитанные (кривые) п опытные (точки)
значения константы Генри К± для адсорбции цикло-
пропана (7), циклопентана (2) и циклогексана (3) на
базисной грани графита (расчет) и на графитирован-
ных термических сажах (опыт).
Величины Kt выражены в мкм.
Однако для циклопропана опытные значения Кг лежат заметно выше
рассчитанной кривой и опытное значение —&иг для адсорбции
этого углеводорода приблизительно на 1,8 кДж/моль выше рассчи-
танного.
Таблица Х,12. Рассчитанные значения At^i, и ACj
для адсорбции цикланов на базисной грани графита
и соответствующие опытные значения AU± и AS*
на графитированных термических сажах
Молекула Расчет Опыт
Т, К -ДЕЛ, кДж/моль Д8* в ДС1 в т, К -ДЕЛ, кДж/моль дз* Ссыл- ки
Циклопропан 250 19,6 10,9 1,4 238 21,4 11,6 [49]
400 18,2 10,5 1,0
550 17,2 10,2 0,8
Циклопентан 250 28,6 11,7 2,7 327 27,2 10,7 [491
400 26,2 10,8 1,5 423 28,1 11,1 [631
550 24,7 10,5 1,0
Циклогексан 250 32,9 11,6 1,9 388 28,5 10,2 [49J
400 30,9 10,9 1,3 398 31,4 11,0 [30J
550 29,5 10,6 0,9 423 31,0 11,1 [631
330
У молекулы циклопропана валентные углы С—С—С сильно
отклоняются от тетраэдрических. Это заметно сказывается на физико-
химических свойствах циклопропана [39]. Поэтому на основании
полученного расхождения рассчитанных и опытных значений Кг
и для циклопропана можно предположить, что атом-атомная
потенциальая функция межмолекулярного взаимодействия атомов С
циклопропана с атомами С графита отличается от соответствующей
атом-атомной потенциальной функции для рассмотренных выше
алканов. Однако это различие невелико. Если принять, что взаимо-
действие атомов Н молекулы циклопропана с атомами С графита
описывается той же потенциальной функцией (Х,6), что и взаимо-
действие атомов Н алканов, то получается, что энергия межмолеку-
лярного взаимодействия атомов С молекулы циклопропана с ато-
мами С графита только на 6% больше энергии соответствующего
взаимодействия атомов С алканов. В разд. 4 этой главы показано,
что примерно на столько же отличается атом-атомная потенциальная
функция межмолёкулярного взаимодействия с атомом С графита
атома С молекулы, образующего двойную или ароматическую
связь.
Напряжение валентных углов в молекулах циклопентана и цикло-
гексана сравнительно небольшое. Полученное согласие рассчитанных
и опытных значений Кг и для этих углеводородов показывает,
что взаимодействие с графитом слабо напряженных молекул цикла-
нов описывается теми же атом-атомпыми потенциальными функциями
межмолекулярного взаимодействия, что и взаимодействие алканов.
Адсорбционные свойства таких цикланов, как и алканов, опре-
деляются главным образом составом и геометрическим строением
их молекул.
Таким образом, проведенные расчеты термодинамических харак-
теристик адсорбции алканов и цикланов [9, 10, 14, 171 показали, что
атом-атомное приближение (VIII,2) для потенциальной функции Ф
межмолекулярного взаимодействия молекул насыщенных углеводо-
родов с поверхностью графитированных термических саж правильно
передает зависимость потенциала межмолекулярного взаимодействия
при адсорбции Ф от химического состава и геометрического строения
молекул этих углеводородов; а также зависимость Ф от ориентации
молекулы над поверхностью. Термодинамические характеристики
адсорбции насыщенных углеводородов при нулевом (малом) запол-
нении поверхности можно рассчитать в хорошем согласии с опытом
молекулярно-статистическим методом, исходя из химического и гео-
метрического строения молекулы углеводорода и поверхности твер-
дого тела. В случае адсорбции на базисной грани графита (на графи-
тированных термических сажах) для этого надо использовать полу-
эмпирические атом-атомные потенциальные функции межмолекуляр-
ного взаимодействия (Х,5) и (Х,6).
331
4. Ненасыщенные и ароматические углеводороды.
Зависимость межмолекулярного взаимодействия атомов
углерода от их валентного состояния
Преимущества использования адсорбции для определения зави-
симости межмолекулярного взаимодействия атомов от их валентного
состояния. Атомные инкременты многих свойств молекулы зависят"
от валентного состояния входящих в нее атомов. Поэтому можно
ожидать, что и потенциальные функции межмолекулярного взаимо-
действия атомов в принятом атом-атомном приближении зависят
не только от природы взаимодействующих атомов, но и от их валент-
ного состояния.
Свойства кристаллических углеводородов определяются меж-
молекулярным взаимодействием трех типов пар атомов: атомов
Н . . . Н, Н . . . С и С ... С, причем главным образом ван-дер-
ваальсовым взаимодействием периферических атомов Н . . . Н
и Н ... С. Взаимодействие между атомами С ... С при этом играет
второстепенную роль [70—72]. Поэтому на основании свойств кри-
сталлических углеводородов не было установлено различие в потен-
циальных функциях межмолекулярного взаимодействия атомов С . . .С
насыщенных и ароматических углеводородов [71, 73—75]. Потен-
циальная же энергия межмолекулярпого взаимодействия молекулы
углеводорода с базисной гранью графита определяется межмолеку-
лярным взаимодействием только двух типов пар атомов Н . . . С
и С ... С. Поэтому определение различия в потенциальных функ-
циях межмолекулярного взаимодействия атомов С ... С, находя-
щихся в разных валентных состояниях, на основании эксперимен-
тальных адсорбционных данных, полученных на графитированной
термической саже, в принципе, проще и более однозначно, чем на
основании свойств кристаллических углеводородов.
В соответствии с тремя главными валентными состояниями ато-
мов С в молекулах углеводородов, не имеющих сопряженных связей,
для описания их взаимодействия с атомами С графита надо ввести
следующие три атом-атомные потенциальные функции:
1) <Рс (алкаю ... с (графит) “ для взаимодействия атома С моле-
кулы алкана с атомом С графита;
2) фс(алкен>. .. С(графит) — Для взаимодействия атома С, обра-
зующего двойную связь в молекуле алкена, с атомом С графита;
3) <Рс (алкин;... с (графит; — Для взаимодействия атома С, обра-
зующего тройную связь в молекуле алкина, с атомом С графита.
Потенциальная функция межмолекулярного взаимодействия ато-
мов G (алкан) ... С (графит) дается выражением (Х,5). В работе [18]
аналогичным путем, т. е. при использовании опытных значений
константы Генри для адсорбции ненасыщенных углеводородов на
графитированных термических сажах, были определены потенциаль-
ные функции межмолекулярного взаимодействия атомов С (ал-
кен) ... С (графит) и С (алкин) ... С (графит). Исследовалось
также влияние сопряжения связей молекул ненасыщенных и арома-
332
гических углеводородов на их межмолекулярное взаимодействие
с графитом [18].
Алкены и алкины. Атом-атомные потенциалы межмолекулярного
взаимодействия фс (алкен) ... С (графит) И фс (алкин) * * * С (Графит)
были определены [18] при использовании экспериментальных зна-
чений константы Генри Кл для адсорбции алкенов и алкинов на
графитированных термических сажах [48, 49, 76]. При этом были
сделаны следующие приближения и допущения.
Обычно принимается, что поляризуемость и другие свойства
атома Н в молекулах углеводорода, определяющие его способность
к неспецифическому межмолекулярному притяжению, практически
не зависят от валентного состояния связанного с ним атома С [39].
ПОЭТОМУ При Определении ПОТеНЦИаЛОВ фС (аЛкен) ... С (графит)
и фс (алкии)... с (графит? неспецифического межмолекулярного
взаимодействия было принято, что межмолекулярное взаимодей-
ствие атомов Н . . . С (графит) не зависит от валентного состояния
атома С, с которым связан рассматриваемый атом Н молекулы, и вы-
ражается тем же атом-атомным потенциалом (Х,6), что и для адсорб-
ции на графите алканов и ненапряженных цикланов. Кроме того,
было принято, что от валентного состояния атома С молекулы не
зависят значения параметров С21СГ, го и q потенциала межмолеку-
лярного взаимодействия атомов С ... С. Для этих параметров
были приняты те же значения, что и в случае взаимодействия ато-
мов С (алкан) ... С (графит), а именно: CJC-i = 1,55* 10"20 м2,
/ о = 0,382 нм и q ~ 35,7 нм-1.
При этих приближениях и допущениях от валентного состояния
атомов С молекул углеводородов зависят только значения пара-
метров Cj и В потенциала межмолекулярного взаимодействия ато-
мов С ... С. Так как согласно условию равновесия параметр В
пропорционален параметру Съ то
Фс (алкен). . . С (графит) “^алкенФс (алкан). . . С (графит) (Хт13)
Фс (алкин). . . С (графит) ^алкинФс (алкан). . . С (графит) (Х,14)
где 6 — параметр, зависящий от валентного состояния атома С молекулы.
Следовательно, чтобы определить потенциалы взаимодействия
атомов С (алкен) ... С (графит) и С (алкин) ... С (графит) при
указанных выше допущениях и приближениях на основании опыт-
ных адсорбционных данных, надо определить только соответствующие
значения параметра 6.
Используя опытные значения для этилена [48, 49, 76], мето-
дом проб и ошибок было получено балкен — 1,07. Таким образом
Фс (алкен). . . С (графит)—М8 • 10^-6-2,30 > 10^ +
+ 2,026 • 105 ехр (—35,7г) кДж/моль (Х,15)
При использовании потенциала (Х,15) для взаимодействия с атомом С
графита атомов С молекулы алкена, образующих двойную связь,
333
и потенциала (Х,5) для взаимодействия атомов С молекулы алкена,
образующих только ординарные связи, были рассчитаны далее
термодинамические характеристики адсорбции этилена, пропилена
и бутена-1.
Потенциальный барьер внутреннего вращения в молекуле про-
пилена, равный около 8 кДж/моль [55], значительно больше изме-
нения потенциальной энергии Фо взаимодействия молекулы с по-
верхностью при внутреннем вращении молекулы. Поэтому при
расчетах молекула пропилена рассматривалась квазижесткой и име-
ющей конформацию, в которой связь С==С заслонена связью С—Н
Рис. Х,11. Рассчитанные (кривые) и опытные (точки) значе-
ния константы Генри Кх для адсорбции этилена (/), пропи-
лена (2) и бутена-1 (3) на базисной грани графита (расчет)
и на графитированной термической саже (опыт).
Величины Kj выражены в мкм.
метильной группы [77], т. е. один из атомов Н метильной группы
расположен в плоскости этиленовой группы, а остальные два атома Н
расположены симметрично по обе стороны от этой плоскости. Моле-
кула бутена-1 существует в виде равновесной смеси трех вращатель-
ных конформаций. В одной из этих конформаций связь С—С засло-
нена метильной группой СН3, а в двух остальных конформациях
связь С=С заслонена связью С—Н связанного с ней атома С [77].
При расчетах термодинамических характеристик адсорбции эти
конформации молекулы бутена-1 рассматривались квазижесткими.
Разность свободных энергий этих форм была принята равной
нулю [78].
На рис. Х,11 сплошные кривые представляют значения Кг,
рассчитанные при использовании для межмолекулярного взаимодей-
ствия атомов С (алкен) ... С (графит) потенциала (Х,15), атомов
G (алкан) ... С (графит) — потенциала (Х,5) и атомов Н (углеводо-
род) . . . С (графит)— потенциала (Х,6). Рассчитанные так значе-
334
пня Ку для всех рассмотренных молекул хорошо согласуются с соот-
ветствующими опытными значениями [48, 49, 76, 79] (см. также
табл. П, 3 Приложения). Пунктирной кривой на рис. Х,11 показаны
значения Ку для этилена, рассчитанные при использовании тех же
атом-атомных потенциалов (Х,5) и (Х,6), что и для адсорбции алка-
нов. Эти значения Ку заметно ниже соответствующих эксперимен-
тальных значений. Таким образом, установленное различие в атом-
атомных фуНКЦИЯХ фС (алкен)) ... С (графит) И ФС (алкан) ... С (графит)
лежит за пределами погрешности экспериментальных адсорбцион-
ных данных.
В табл. Х,13 приведены рассчитанные и опытные значения &Uy
и A5J, а также рассчитанные значения ДСХ для адсорбции этилена,
пропилена и бутена-1. Рассчитанные и опытные значения Д£7Х и Д51
близки.
Таблица Х,13. Рассчитанные значения AZ7X, Д5* и ДСХ
для адсорбции алкенов на базисной грани графита и опытные
значения \Uy и на графитированных термических сажах
Молекула Расчет Опыт
Т, К -ДЕЛ, кДж/моль Д8* ДС1 R Т, К -ДЕЛ, КДж/моль де* R~ Литера- тура
R
Этилен 150 17,5 11,9 2,1 183 16,7 11,3 [76]
250 16,0 10,9 1,5
350 14,9 10,5 1,1 200 16,3 11,1 [49]
276 16,3 11,0 [48]
Пропилен 150 25,2 13,1 2,2
250 23,2 11,8 2,5 270 21,8 11,3 [79]
400 20,6 10,8 1,7
Бутен-1 250 28,3 12,2 2,7
400 25,1 11,1 1,9 327 26,8 11,3 [49]
550 23,1 10,6 1,3
Используя опытные значения Ку для ацетилена [49], таким же
путем было получено значение баЛкии 1?27 [18]. Таким образом
Фс (алкин). .. С (графит) ~ 1,76-10 Зг 6 2,73-10 5г 8 +
+ 2,40 105 ехр (—35,7г) кДж/моль (Х,16)
При использовании потенциала (Х,16) для взаимодействия с ато-
мом С графита атомов С молекулы алкина, образующих тройную
связь, и потенциала (Х,5) для взаимодействия атомов С молекулы
алкина, образующих только ординарные связи, были рассчитаны
термодинамические характеристики адсорбции ацетилена, пропина,
бутина-1, бутина-2 и пентина-1.
Внутреннее вращение в алкинах вокруг связи С—С, соседней
с тройной связью СааС, практически свободно [55]. При расчетах
335
это вращение было принято полностью свободным. Этильная группа
в молекуле бутина-1 рассматривалась квазижесткой со скрещенным
расположением атомов Н в соседних метильной и метиленовых груп-
пах. При расчетах для пентина-1 было принято во внимание образо-
вание трех поворотных изомеров (одного транс-изомера и двух гош-
изомеров) при внутреннем вращении вокруг центральной связи С—С
пропильной группы молекулы. Для разности свободных энергий
гош- и транс-поворотных изомеров была принята та же величина,
что и для разности свободных энергий поворотных изомеров н-бутана,
т. е. 2,5 кДж/моль.
Рис. Х,12. Рассчитанные (кривые) и опытные (точки) значе-
ния константы Генри Ку для адсорбции ацетилена (7),
пропина (2), бутина-1 (3), бутина-2 (4) и пептина-1 (5) на
базисной грани графита (расчет) и на графитированной
термической саже (опыт).
Величины К, выражены в мкм.
На рис. Х,12 сплошные кривые представляют значения К19
рассчитанные при использовании для межмолекулярного взаимодей-
ствия атомов С (алкин) . . .~G (графит) потенциала (Х,16) и атомов
G (алкан) . . . G (графит) — потенциала (X,6). Рассчитанные значе-
ния Ку для алкинов практически совпадают с имеющимися опыт-
ными значениями для ацетилена и пентина-1 [49]. Пунктирной
кривой на рис. Х,12 показаны значения Ку для ацетилена, рассчи-
танные при использовании тех же атом-атомных потенциалов, что и
для алканов. Они лежат значительно ниже соответствующих экспери-
ментальных значений, так что установленное различие в атом-атомных
ПОТеНЦИаЛЬНЫХ функциях фс (алкин) ... С (графит) ® фС (алкай) . . . G (Графит)
выходит далеко за пределы погрешностей эксперимента. Рассчитанные
и опытные значения AUy и AS*, а также рассчитанные значения АС у
для рассмотренных алкинов приведены в табл. Х,14. Рассчитанные
и опытные значения АС71 и ASJ близки.
336
Таблица Х,14. Рассчитанные значения AZ71, ДЛ1* и A6\
для адсорбции алкинов на базисной грани графита
Молекула Расчет
Т, К -4(71, кДж/моль 4S* 4Ci R
Ацетилен * 100 16,0 11,8 1,2
200 15,0 11,0 1,1
300 14,2 10,5 1,0
Пропин 150 23,6 12,6 1,8
250 22,1 11,6 1,8
400 20,0 10,8 1,5
Бутин-1 200 27,8 12,6 2,7
300 25,7 11,6 2,2
450 24,2 11,0 1,8
Бутин-2 200 30,9 13,2 — ।
350 28,1 11,9 2,1
450 26,3 11,3 2,1
Пентин-1 ** 250 34,4 13,0 —
400 30,4 11,6 2,7
550 27,6 10,9 1,6
* Для адсорбции ацетилена на графитированной термической саже при 182 К найдены
следующие опытные значения: — 4(71 = 15,9 кДж/моль; —Д8*/Й==11,8 [49].
** Для адсорбции пептина-1 при 356 К найдены следующие опытные значения?
—4(71 = 29,7 кдж/моль: -4SJ/R = 11,1 [49].
Таким образом, полученные на основании опытных данных для
адсорбции алканов, цикланов, алкенов и алкинов на графитирован-
ных термических сажах атом-атомные потенциальные функции
межмолекулярного взаимодействия атомов С молекул этих угле-
водородов с атомами С графита зависят от валентного состояния
атома С молекулы углеводорода. Как и в случае насыщенных угле-
водородов, атом-атомное приближение для потенциальной энергии
взаимодействия молекул углеводородов с поверхностью графита
хорошо оправдывается в случае адсорбции ненасыщенных углеводо-
родов, если учесть зависимость атом-атомных потенциальных функ-
ций межмолекулярного взаимодействия атомов С . . . С от их валент-
ного состояния.
В соответствии с двумя типами электронов, образующих хими-
ческие связи в молекулах ненасыщенных углеводородов, энергию
дисперсионного взаимодействия двух молекул ненасыщенных угле-
водородов можно представить в виде суммы энергии дисперсионного
взаимодействия а-электронов, а- и л-электронов и л-электронов [80—
86]. Выше установленное более сильное взаимодействие с атомами С
графита атомов С молекулы углеводородов, образующих двойные
и тройные связи, чем атомов С, образующих только ординарные
связи, может быть вызвано тем, что относительный (на один элек-
22 заказ 1180
337
трон) вклад дисперсионного взаимодействия л-электронов молекулы
с атомами С графита несколько больше относительного вклада а-элек-
тронов молекулы. Как следует из выражений (Х,5), (Х,15) и (Х,16)т
относительные значения константы (\ для взаимодействия с ато-
мами С графита атомов С молекул углеводородов, образующих
только ординарные, только одну двойную и только одну тройную
связи, равны около 1 : 1,07 : 1,27. Согласно формуле Кирквуда —
Мюллера, эти относительные значения должны быть близкими
к относительным значениям поляризуемостей соответствующих ато-
мов С. Они равны около 1 : 1,6 : 1,9. Таким образом, формула Кирк-
вуда — Мюллера качественно правильно передает найденную зави-
симость константы (\ от валентного состояния атома С. Однако
рассчитанные по ней относительные значения константы Сх полу-
чаются значительно большими полученных описанным выше путем
при использовании экспериментальных адсорбционных и газохрома-
тографических данных.
Молекулы этилена и ацетилена, содержащие л-электроны, имеют
значительные квадрупольные моменты [87, 88]. Предполагалось,
что л-электроны решетки графита могут создавать вблизи поверх-
ности базисной грани графита сравнительно сильное, но быстро
убывающее с расстоянием электростатическое поле [89]. Поэтому
можно было бы предположить, что найденное указанным выше путем
более сильное взаимодействие с атомами С графита атомов С молекул
углеводородов, образующих двойные и тройные связи, чем атомов С,
образующих только ординарные связи, частично или полностью
обусловлено тем, что в общую энергию взаимодействия ненасыщен-
ных углеводородов с графитом заметный вклад вносит электростати-
ческое взаимодействие квадрупольного момента молекулы ненасы-
щенных углеводородов с электростатическим полем базисной грани
графита. Однако экспериментально установлено, что теплоты ад-
сорбции на графитированных термических сажах даже молекул
с большими периферическими дипольными моментами близки к те-
плотам адсорбции неполярных молекул с близкой средней поляри-
зуемостью и геометрической структурой [90, 91]. Например, теплота
адсорбции на графитированной термической саже диэтилового эфира
не превышает теплоту адсорбции н-пентана (см. гл. V). Таким обра-
зом, в случае адсорбции на графитированных термических сажах
энергия электростатического взаимодействия может вносить лишь
незначительный вклад в общую энергию адсорбции и электростати-
ческое взаимодействие молекул ненасыщенных углеводородов с гра-
фитом не может быть сильным.
Углеводороды с сопряженными связями. В случае ненасыщенных
углеводородов с сопряженными связями л-электроны делокализо-
ваны по всей длине этих связей. Согласно квантовомеханическим
исследованиям [80—82, 92, 93], межмолекулярное взаимодействие
таких углеводородов обладает специфическими закономерностями,
отличными от закономерностей межмолекулярного взаимодействия
углеводородов без сопряженных связей. Взаимное влияние атомов
338
или групп атомов в молекулах с сопряженными связями убывает
медленно и простирается по всей длине сопряженных связей. По-
этому энергия межмолекулярного взаимодействия таких молекул
не аддитивна по энергиям взаимодействия входящих в них пар атомов
или атомных групп (см. разд. 3 гл. VIII).
Базисная плоскость решетки графита представляет в сущности
большую молекулу, состоящую из очень большого числа конден-
сированных ароматических ядер. В этой плоскости л-электроны
делокализованы.Поэтому адсорбция молекул с сопряженными свя-
зями на базисной грани графита представляет удобный случай для
изучения межмолекулярного взаимодействия таких молекул. Это
представляет интерес как для изучения влияния подвижных л-элек-
тронов графита на межмолекулярное взаимодействие при адсорбции,
так и для выяснения закономерностей межмолекулярного взаимо-
действия углеводородов с сопряженными связями.
Полуэмпирические исследования влияния сопряжения связей
в молекуле на ее взаимодействие с базисной гранью графита могут
быть проведены двумя способами.
Во-первых, можно сравнить экспериментальные значения термо-
динамических характеристик адсорбции углеводорода с сопряжен-
ными связями на графитированных термических сажах со значе-
ниями, рассчитанными без учета влияния сопряжения на межмоле-
кулярное взаимодействие. Так, влияние сопряжения двух двойных
связей в молекуле бутадиена-1,3 на ее взаимодействие с графитом
можно оценить, сравнивая полученные из опыта адсорбционные
свойства бутадиена с адсорбционными свойствами этого углеводо-
рода, рассчитанными на основании атом-атомных потенциальных
функций, найденных для алканов [уравнение (Х,5)] и алкенов
с несопряженными связями [уравнение (X ,15) ].
Во-вторых, можно сравнить термодинамические характеристики
адсорбции на графитированных термических сажах углеводородов
с сопряженными связями и изомерных им соединений без сопряжен-
ных связей, например, можно сравнить термодинамические харак-
теристики адсорбции пентадиена-1,3 (молекулы с сопряженными
связями) и пентадиена-1,4 (изомерной молекулы с несопряжеиными
связями).
Исследование влияния сопряжения двойных связей на адсорбцию
на графитированных термических сажах проводилось первым спосо-
бом [18]. Были рассчитаны термодинамические характеристики
адсорбции бутадиена-1,3, цис- и тпранс-пентадиенов-1,3 и гекса-
триена-1,3,5 при использовании для взаимодействия атомов С ... С
потенциальных функций (Х,5) и (Х,15), определенных для взаимо-
действия с атомом С графита атомов С молекул алканов и алкенов
без сопряженных связей.
При расчетах для всех сопряженных диенов и гексатриена было
принято плоское транс-расположение двойных связей [77, 94].
Было принято также, что в пентадиенах, как и в пропилене, один
атом Н группы СН3 располагается в плоскости сопряженных двой-
22* 339
чения константы Генри Кг для
адсорбции бутадиена-1,3 (7), цис-
пентадиена-1,3 (2), транс-пента-
диена-1,3 (3) и гексатриена (4)
на базисной грани графита (рас-
чет) и на графитированной тер-
мической саже (опыт).
Величины Кх выражены в мкм.
ных связей, а остальные два атома Н расположены симметрично
по обе стороны этой плоскости. На рис. Х,13 и в табл. Х,15 резуль-
таты расчета сопоставлены с опытными данными [49] (см. также
табл. П, 3 Приложения).
Для молекул с двумя сопряженными связями экспериментальные
значения лежат лишь незначительно выше значений Klt рассчи-
танных без учета эффекта сопряжения двойных связей. Различия
в рассчитанных и опытных значениях AZ7X для этих соединений
находятся в пределах погрешности опытных данных. В согласии
с опытом, рассчитанные значения Кх и —At/X для znpawc-пентадиена
больше, чем для г{ис-пентадиена.
Согласно квантовомеханическим
исследованиям [80—82], влияние
сопряжения связей на межмолеку-
лярное взаимодействие должно ра-
сти с ростом числа сопряженных
связей. Поэтому, чтобы получить
более однозначный вывод о влиянии
сопряжения двойных связей моле-
кулы на ее взаимодействие с по-
верхностью графита, следует сопо-
ставить результаты аналогичных
расчетов с результатами опытов
для углеводородов с большим чис-
лом сопряженных двойных связей.
Однако опытных данных по адсорб-
ции таких соединений, по-видимому,
еще нет.
Таким образом, сопряжение двух
двойных связей в молекуле углево-
дорода при учете соответствующего
изменения геометрии молекулы слабо
сказывается на ее взаимодействии
с графитом. Поэтому энергию меж-
молекулярного взаимодействия таких
молекул, как и молекул с несопря-
приближенно рассматривать аддитив-
ной по энергиям взаимодействия пар атомов или атомных групп
этих молекул.
Ароматические углеводороды. В ароматических углеводородах
атомы углерода находятся в особом валентном состоянии. Взаимное
женными связями, можно
влияние атомов в ароматических соединениях, как и в ненасыщенных
углеводородах с сопряженными связями, передается по всей системе
сопряженных связей. Однако в отличие от линейных углеводородов
с сопряженными связями к ряду свойств ароматических соединений
(энтальпия образования, рефракция) применима аддитивная
схема [39, 95]. Эта схема применима и к энергии межмолекулярного
взаимодействия ароматических молекул друг с другом [71, 73, 74,
340
Таблица Х,15- Рассчитанные значения ДСЛь Д5£ и ДС^
для адсорбции углеводородов с сопряженными двойными связями
на базисной грани графита и опытные значения ДUi и Д5*
на графитированной термической саже [49]
Расчет Опыт
Молекула Т, К -ДС71, -Дб* Д(71 Т, К -Д/71, Дб*
кДж/моль R R кДж/моль R
Бутадиен-1,3 250 30,9 12,7 2,4
400 27,7 11,6 2,5 327 28,5 11,6
550 24,9 10,9 2,0
ц ис-Пентадиен-1,3 250 37,9 13,5 2,6
400 34,3 12,2 3,0 344 35,6 12,3
550 30,8 11,3 2,5
лграно-Пентадиен-! ,3 250 38,1 13,3 2,0
400 35,1 12,3 2,7 344 36,4 12,3
550 31,9 11,5 2,5
Гексатриен 250 46,1 14,0 2,0
400 43,4 13,0 2,6
550 39,9 12,2 2,9
96]. Энергия взаимодействия л-электронов ароматических углеводо-
родов с л-электропами базисной грани графита, рассчитанная мето-
дами теории молекулярных орбит [97, 98], приблизительно линейно
зависит от числа атомов С в молекуле. Поэтому можно ожидать,
что атом-атомное приближение (VIII,2) применимо и к энергии
межмолекулярного взаимодействия ароматических углеводородов
с базисной гранью графита.
В работе [18] было принято, что к энергии межмолекулярного
взаимодействия ароматических углеводородов с базисной гранью
графита применимо атом-атомное приближение (VIII,2). При этом
допущении использование опытных значений Кг для адсорбции
бензола на графитированных термических сажах [12, 30, 49, 63]
привело к следующему выражению для потенциальной функции
межмолекулярного взаимодействия атома С молекулы ароматического
углеводорода с атомом С графита:
ФС (аромат.). . . С (графит) = “М8 • 10^-6-2,30 • 10^-8 +
-4 2,03 • 10б ехр (—35,7г) кДж/моль (Х,17)
При использовании атом-атомных потенциальных функций (Х,6)
и (Х,17) были рассчитаны термодинамические характеристики ад-
сорбции на базисной грани графита бензола, нафталина, антрацена,
фенантрена и дифенила. Расчеты для дифенила проводились с учетом
заторможенного внутреннего вращения его молекул при исполь-
зовании полученной в работе [99] потенциальной функции Ж. На
рис. Х,14 и в табл. Х,16 результаты расчета сопоставлены с соответ-
ствующими опытными данными [12, 16, 30, 49, 61, 63, 79].
341
Рассчитанные и экспериментальные значения для всех рас-
смотренных ароматических углеводородов удовлетворительно согла-
суются, причем рассчитанные, как и экспериментальные, значения
константы Кх для фенантрена меньше, чем для антрацена. Однако
Рис. Х,14. Рассчитанные (кри-
вые) и опытные (точки) зна-
чения константы Генри Кг
для адсорбции бензола (7),
нафталина (2), антрацена (5),
фенантрена (4) и дифенила (5)
на базисной грани графита
(расчет) и на. графитирован-
ных термических сажах (опыт).
Пунктирная кривая для бензола
получена при использовании для
взаимодействия атомов С(аромат). . .
С(графит) потенциала (Х,5), сплош-
ные кривые получены при исполь-
зовании потенциала (Х.17) для
взаимодействия этих атомов. Вели-
чины выражены в мкм.
Таблица Х,16. Рассчитанные значения и ДСХ
для адсорбции ароматических углеводородов на базисной грани
графита и соответствующие опытные значения на графитированных
термических сажах
Молекула Расчет Опыт
Т, К -ДС71, кДж/моль -Д5* я SCi R т, к —AC7t, кДж/моль -AS* я Литера- тура
Бензол 250 41,0 13,7 2,3 391 36,0 11,8 [49]
350 38,9 12,9 2,8 393 37,7 12,6 [16]
450 36,8 12,2 3,1 398 37,7 12,3 [30]
Нафталин 423 36,0 11,9 [631
250 66,6 15,2 1,8 400 69,1 14,1 [30, 62]
400 64,1 14,3 2,2 498 64,5 14,5 [79]
550 61,1 13,5 2,9 553 62',0 ' 14,3 [16]
Антрацен 400 89,6 15,3 1,9
550 87,1 14,7 2,2 604 94,6 16,8 [79]
700 84,2 14,0 2,7 713 87,5 15,2 [16]
850 80,4 13,4 з,з
Фенантрен 400 89,6 15,3 1,9
550 87,1 14,7 2,2 604 94,6 17,0 [79]
700 84,2 14,1 2,7 713 87,1 15,2 [16]
850 80,4 13,5 3,4
Дифенил 250 72,4 15,3
400 71,2 14,9
550 67,8 14,0 503 70,3 14,8 [79]
342
отношение рассчитанных значений Кг для антрацена и фенантрена
(равное около 1,06) несколько меньше, чем соответствующее отноше-
ние экспериментальных значений Кг (равное около 1,25 116, 79I).
В пределах погрешности экспериментальных значений согласуются
также рассчитанные и опытные значения А£7Х и А5£.
Средняя мольная теплоемкость адсорбированного на графитиро-
ванной термической саже бензола Ст при 307 К была измерена
в работе [35]. В области мономолекулярной адсорбции опытные
значения слабо зависят от Г и позволяют довольно точно опре-
делить экспериментальное значение Са, г Оно равно около
100 Дж/(моль*К). Средняя мольная теплоемкость газообразного
бензола при 307 К Cy,w — 77,5 Дж/(моль*К) [100]. Отсюда экспе-
риментальное значение АСХ — 22,5 Дж/(моль* К). Оно практически
совпадает с рассчитанным при соответствующей температуре
[21,7 Дж/(мойь-К)].
Полученные результаты показывают, что атом-атомное при-
ближение удовлетворительно описывает межмолекулярное
взаимодействие с графитом также и ароматических углеводо-
родов.
Потенциальные функции межмолекулярного взаимодействия
с атомами С графита атомов С ароматических углеводородов (Х,17)
и атомов С алкенов у двойной связи (Х,15) практически совпадают.
Следовательно, каких-либо сильных специфических взаимодействий
ароматических углеводородов с базисной гранью графита, которые
вызывались бы близостью их структур [101] и наличием в них по-
движных электронов, не наблюдается.
В конденсированных ароматических соединениях можно выде-
лить несколько типов связей С—С [39]. Для учета этих особенностей
конденсированных ароматических соединений следовало бы ввести
несколько атом-атомных потенциальных функций. Однако погреш-
ности экспериментальных данных и расчета пока не позволяют сде-
лать такого уточнения. Вместе с тем из полученных результатов
следует, что различие между этими функциями небольшое и в при-
ближенных расчетах им можно пренебречь.
В работах [15, 16] молекулярно-статистическим методом были
рассчитаны термодинамические характеристики адсорбции бензола,
нафталина, антрацена и фенантрена на базисной грани графита.
При расчетах Ф в качестве силовых центров принимались атомы G
и группы атомов СН молекул ароматических углеводородов и атомы С
графита, причем потенциальные функции ф взаимодействия с ато-
мом С графита атома С и группы атомов СН молекулы принимались
одинаковыми. Параметры функций ф оценивались исходя из свойств
адсорбата и адсорбента, взятых в отдельности. Расчеты термодина-
мических характеристик адсорбции производились при использо-
вании разных приближений (моделей) для зависимости Ф от поло-
жения молекулы (от координат z, $ и ф). Различие эксперименталь-
ных значений для антрацена и фенантрена лучше всего описывает
343
следующее приближение (модель D [16]) для зависимости Ф от
положения молекулы:
ф? Fn
Ф = Фо-|---—(z — z0)2H-5— sin2$ + -^-(l—cosoip) (Х,18)
где Фо, Фа° и г0 — значения Ф, второй производной Ф по z и величины z в наи-
более глубоком потенциальном минимуме молекулы; Ф^°— минимальное зна-
чение второй производной Ф по sin Ф; Vo — величина потенциального барьера
для вращения молекулы вокруг оси с наибольшим моментом инерции (для изме-
нения ф) при параллельной ее ориентации по отношению к поверхности (ф =
= л/2); а — число симметрии вращения молекулы вокруг этой оси.
Таким образом, в этой модели предполагается, что адсорбирован-
ные молекулы антрацена и фенантрена свободно движутся вдоль
поверхности адсорбента и свободно вращаются вокруг оси, перпен-
дикулярной к поверхности, совершают гармонические колебания
перпендикулярно поверхности, гармонические крутильные колеба-
ния вокруг одной фиксированной оси, параллельной поверхности,
а зависимость Ф от ф дается последним членом выражения (Х,18).
В этом приближении
1/2
(Х,19)
Рассчитанные при использовании этой модели значения для
нафталина, антрацена и фенантрена, а также значение отношения Ki
для антрацена и фенантрена близки к соответствующим эксперимен-
тальным значениям, полученным в работе [16].
Рассчитанные значения Фо и Ф^0 для антрацена и фенантрена
одинаковы, различаются только значения Ф^° и "Ио. Поэтому при
справедливости указанной выше модели
К* Ф^0’ф
К® ~ Ф<Л А
in
(Х,20)
где верхние индексы АиФ показывают, что эти величины относятся
соответственно к антрацену и фенантрену. В работе [16] было полу-
чено, что Ф^°*ф = 464, Ф^°’Л = 333, Уф = 23, УА = 27 кДж/моль.
Вводя эти значения в (Х,20), получаем, что К^/К^ = 1,28. Основ-
ной вклад в это отношение вносит первый множитель Ф^,0’ Ф/Ф^°>А —
~ 1,39. Второй множитель (УФ/УА) = 0,92, т. е. действует в об-
ратную сторону, чем первый. Однако в этой модели делаются два
необоснованных предположения. Во-первых, в этой модели пред-
полагается, что гармонические крутильные колебания адсорбиро-
ванной молекулы происходят вокруг фиксированной оси, параллель-
ной поверхности. В действительности положение этой оси должно
изменяться. Учет этого изменения значительно снижает значение
отношения K^/Kf [16]. Во-вторых, в этой модели предполагается,
что потенциал торможения вращения молекулы вокруг оси с наи-
большим моментом инерции (зависимость Ф от угла ф) не зависит
344
от ориентации молекулы (от угла й) и равен потенциалу торможения
при параллельной ориентации этой же оси к поверхности адсорбента
(при О' = л/2). Однако в действительности зависимости Ф от if и #
неразделимы. При предположении, что Ф не зависит от ф, Ф также
не должна зависеть и от ф при О -> 0. Кроме того, это предположение
не является необходимым для описания различия значений для
антрацена и фенантрена, так как последнее предположение изменяет
отношение К^/К® в противоположную сторону, чем первое пред-
положение.
Таким образом, в работах [15, 16] различие значений для
антрацена и фенантрена объясняется только различными зависи-
мостями Ф от ориентации молекул антрацена и фенантрена,вызван-
ными различием геометрических структур этих молекул. Значе-
ния Фо для этих молекул рассматриваются одинаковыми. Однако
в действительности они могут быть различными из-за различия
электронных структур молекул антрацена и фенантрена. На это
указывают квантовомеханические расчеты энергии взаимодействия
этих молекул с базисной плоскостью графита [97, 98].
5. Сопоставление потенциальных функций взаимодействия
атомов С ... С и Н . . . С для различных проявлений
межмолекулярного взаимодействия
Атом-атомные потенциальные функции межмолекулярного
взаимодействия в первом приближении, по-видимому, универсальны,
т. е. они практически одинаковы для всех проявлений межмолеку-
лярного взаимодействия, т. е. для взаимодействий молекул в объеме
реальных газов, жидкостей и молекулярных растворов, для взаимо-
действий с поверхностью твердого тела при адсорбции, для взаимо-
действий в молекулярных кристаллах, при внутримолекулярных
взаимодействиях непосредственно валентно не связанных атомов-
и в других подобных случаях.
Потенциальные функции межмолекулярного взаимодействия ато-
мов С ... С и С ... Н, а также атомов Н . . . Н определялись как
на основании физико-химических свойств молекулярных систем,
зависящих от межмолекулярных взаимодействий (теплот сублима-
ции, параметров элементарных ячеек, коэффициентов упругости
кристаллических углеводородов и решетки графита, вторых вири-
альных коэффициентов газов, констант Генри для адсорбции), так
и на основании физико-химических свойств, зависящих от внутри-
молекулярных взаимодействий непосредственно валентно не свя-
занных атомов (энтальпий образования и изомеризации, геометрии
напряженных молекул, разностей энергий поворотных изомеров).
Разные экспериментальные данные дают информацию о разных
частях атом-атомных потенциальных функций. Физико-химические
свойства, зависящие от межмолекулярных взаимодействий, содержат
информацию главным образом о потенциалах взаимодействия атомов
при расстояниях, близких к равновесным при таком взаимодействии.
345
Физико-химические свойства, зависящие от внутримолекулярных
взаимодействий, дают информацию главным образом только об
отталкивательной части потенциала межмолекулярного взаимодей-
ствия атомов водорода [102].
Атом-атомные потенциальные функции зависят от валентного
состояния взаимодействующих атомов. При использовании адсорб->
ционных данных выше были получены потенциальные функции
^(Х,5), (Х,6), (Х,15)—(Х, 17) межмолекулярного взаимодействия с ато-
мом С графита атомов Н и атомов С в разных валентных состояниях
молекул углеводородов. На основании этих потенциальных функций
можно оценить потенциальные функции взаимодействия атомов
С...СиН...Св других валентных состояниях. В частности,
для потенциальных функций взаимодействия атомов С (алкан) ... С
(алкан) и Н . . . С (алкан) на основании потенциальных функций
(Х,5), (Х,6), (Х,15)-(Х,17), получаем:
Фс (алкан). . . С (алкан) = 0’^ Фс (алкан). . . С (графит) (Х,21)
И
Фн. . . С (алкан)= Фн. . . С (графит) (Х,22)
На рис. Х,15 сопоставлены потенциальные функции взаимодей-
ствия атомов С ... С, определенные при использовании физико-
химических свойств различных молекулярных систем, зависящих
от межмолекулярных взаимодействий атомов С ... С, С . . . Н
и Н . . . Н. Кривые 7, 2 и 3 получены Вильямсом [74, 75] из струк-
туры и теплот сублимации кристаллических углеводородов: кри-
вая 1 — из свойств алифатических углеводородов, кривая 2 —из
свойств ароматических углеводородов, кривая 3 — из свойств алифа-
тических и ароматических углеводородов. Кривая 4 получена Китай-
городским и Мирской [71, 103] из кристаллической структуры,
сжимаемости кристаллов и теплоты сублимации ряда ароматических
и алифатических соединений. Кривая 5 получена Крауэллом [40]
из межплоскостного расстояния и сжимаемости решетки графита.
Кривая 6 получена в работе [104] при использовании эксперимен-
тальных значений второго вириального коэффициента газообразного
метана. Кривая 7 получена Киселевым, Пошкусом и Афреймо-
вичем [9, 10] для взаимодействия атомов С (алкан) ... С (графит)
при использовании экспериментальных значений константы Генри
для адсорбции этана и пропана на графитированных термических
сажах, а кривая 8 получена для взаимодействия атовгов С (алкан)
... С (алкан) согласно соотношению (Х,21). Как видно из рис. Х,15,
полученные в разных работах при использовании разных физико-хи-
мических свойств различных молекулярных систем потенциальные
кривые межмолекулярного взаимодействия атомов С ... С заметно
различаются. Полученные Киселевым, Пошкусом и Афреймовичем [9,
10] кривые 7 и 8 близки к кривой 5, полученной Крауэлом из свойств
решетки графита, и к кривой 4, полученной Китайгородским и Мир-
ской из свойств кристаллических углеводородов. Вместе с тем,
кривые 7, 2 и 5, полученные Вильямсом также из свойств кристал-
346
лических углеводородов, имеют заметно более глубокие потенциаль-
ные минимумы, чем остальные потенциальные кривые приприблизи-
। ельно тех же равновесных расстояниях, а полученная в работе [104]
из свойств газообразного метана кривая 6, кроме того, смещена
в сторону меньших расстояний.
Рис. Х,15. Потенциальные кривые межмолекулярного взаимодействия ато-
мов С ... С, полученные из физико-химических свойств различных сис-
тем, зависящих от межмрлекулярного взаимодействия (указаны в тексте).
На рис. Х,16 сопоставлены потенциальные функции межмолеку-
лярного взаимодействия атомов Н . . . С. Кривые 7, 2 и 3 получены
Вильямсом [74, 75], кривая 4 — Китайгородским и Мирской [71,
103], кривая 5 — в работе [104] и кривые 6 и 7 — Киселевым, Пош-
кусом и Афреймовичем [9, 10, 17, 18] для взаимодействия атомов
Н . . . С (графит) и Н ... С (алкан). Указанные кривые получены
при использовании тех же физико-химических свойств, что и соот-
ветствующие потенциальные кривые межмолекулярного взаимо-
действия атомов С ... С, представленные на рис. Х,15. Как видно
из рис. Х,16, определенные в разных работах потенциальные кривые
взаимодействия атомов Н . . . С также заметно различаются. Полу-
347
ленные из изучения адсорбции кривые 6 и 7 ближе всего к кривым 1
и 5, определенным Вильямсом. Полученная Китайгородским и Мир-
ской кривая 4, а также кривая 5, полученная в работе [104], имеют
-значительно более глубокие потенциальные минимумы, чем осталь-
ные кривые. Глубина потенциального минимума у кривой 4 при-
близительно такая же, как и у полученной теми же авторами потен-
циальной кривой для взаимодействия атомов С ... С (см. рис. Х,15,
кривая 4), что мало вероятно.
Рис. Х,16. Потенциальные кривые межмолекулярного взаимодей-
ствия атомов Н . . .С, полученные из физико-химических свойств
различных систем, зависящих от межмолекулярного взаимодей-
ствия (указаны в тексте).
Наблюдаемое значительное различие в атом-атомных потенци-
альных функциях межмолекулярного взаимодействия не может быть
обусловлено различием условий проявления межмолекулярных сил,
так как наибольшие различия наблюдаются для кривых, полученных
на основании одних и тех же физико-химических свойств — свойств
молекулярных кристаллов (кривые 1—4). Как следует из выра-
жений (Х,5), (Х,6), (Х,15)-(Х,17), (Х,21), (Х,22) различие в ва-
лентных состояниях атомов С также не может быть главной причиной
наблюдаемого значительного различия в представленных на
рис. Х,15 и Х,16 атом-атомных потенциальных функциях. Основ-
ными причинами наблюдаемого различия в этих потенциальных
348
функциях, выбранных на основании разных физико-химических
данных, по-видимому, являются большие погрешности в самом
определении этих функций.
В случае кристаллических «углеводородов происходит одновре-
менное межмолекулярное взаимодействие трех типов пар атомов:
С ... С, Н . . . С и Н . . . Н. Поэтому на основании физико-хими-
ческих свойств углеводородов приходится одновременно определять
параметры для всех трех таких потенциальных функций. Ввиду
этого на основании физико-химических свойств углеводородов чаще
всего определяют значения только нескольких из этих параметров.
Значения остальных параметров оценивают, используя приближен-
ные квантовомеханические формулы для константы диполь-диполь-
пого притяжения, полуэмпирические правила комбинирования, эм-
пирически установленные зависимости параметров от свойств
взаимодействующих атомов и т. д. Поэтому точность определения
каждой из этих трех отдельных потенциальных функций межмолеку-
лярного взаимодействия атомов С...С, Н...СиН...Н при
использовании физико-химических свойств кристаллических угле-
водородов получается весьма низкой — значительно ниже точности
аналогичного определения эмпирических потенциальных функций
взаимодействия одноатомных молекул [74].
Межплоскостное расстояние и сжимаемость решетки графита
определяются ван-дер-ваальсовым взаимодействием только одного
сорта атомов — атомов углерода в одном и том же валентном состо-
янии. Ввиду этого па основании свойств решетки графита были
определены значения всех параметров потенциальной функции
взаимодействия атомов С (графит) ... С (графит) [40]. Поэтому
последнюю функцию (см. рис. Х,15, кривая 5), по-видимому, можно
рассматривать как определенную наиболее точно для этого валент-
ного состояния атомов С.
Взаимодействие молекул углеводородов с поверхностью графита
при адсорбции определяется взаимодействием двух типов пар атомов,
а именно, атомов С ... С и Н ... С. Поэтому определение потен-
циальных функций взаимодействия атомов С. ..С и Н...С на
основании адсорбционных данных с этой точки зрения проще и более
однозначно, чем на основании свойств кристаллических углеводо-
родов. Таким образом, потенциальную функцию межмолекулярного
взаимодействия атомовН (углеводород). . . С (графит) (см. рис. Х,16,
кривая 6), полученную на основании адсорбционных данных при
использовании потенциальной функции межмолекулярного взаимо-
действия атомов С (алкан) ... С (графит), определенной с учетом
потенциальной функции С (графит) ... С (графит), по-видимому,
можно рассматривать как наиболее точную.
ЛИТЕРА ТУРА
1. Drenan J. W., Hill Т. L. J. Chem. Phys., 1949, v. 17, р. 775—781.
2. Киселев А. В., П о ш к у с Д. П. ДАН СССР, 1961, т. 139, с. 1145—
1148.
3. К и с е л е в А. В., П о ш к у с Д. П. ЖФХ, 1963, т. 37, с. 608—614.
4. Kiselev А. V., Poshkus D. Р. Trans. Faraday Soc., 1963, v. 59.
p. 428-436.
5. Киселев А. В., П о ш к у с Д. П. ЖФХ, 1963, т. 37, с. 1504—1509.
6. Kiselev А. V., Poshkus D. Р. Trans. Faraday Soc., 1963, v. 59,
p. 1438—1443.
7. П ошкуе Д. П. ЖФХ, 1965, т. 39, с. 2962—2967.
8. Poshkus D. Р. Disc. Faraday Soc., 1965, v. 40, p. 195—204.
9. П о ш к у с Д. П., Афреймович А. Я. ЖФХ, 1968, т. 42, с. 1201 —
1205.
10. Киселев А. В., П ошкуе Д. П., Афреймов.ич А. Я. ЖФХ,
1968, т. 42, с. 2546-2552.
11. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1968, т. 42, с. 2553—2555.
12. Pierotti R. A. Chem. Phys. Letters, 1968, v, 2, p. 420—422,
13. Киселев А. В., Пошкус Д. П., Афреймович А. Я. ЖФХ,
1970, т. 44, с. 981—986.
14. Р о s h k u s D. Р., Afreimo vich A. J. J. Chromatog., 1971, v. 58,
р. 55—59.
15. Vidal-Madjar С., Jacob L., Guiochoh G. Bull. Soc. chim.
France, 1971, p. 3105—3109.
16. Vidal-Madjar C., Guiochon G. Bull. Soc. chim. France, 1971,
p. 3110—3118.
17. Kiselev A. V., Poshkus D. P. J. Chem. Soc. Faraday Trans. II
1975, v. 71.
18. Kiselev A. V., Poshkus D. P. J. Chem. Soc. Faraday Trans. II
1975, v. 71.
49 ., П ошкуе Д. П. Докт. дисс., Москва, 1972. 387 с.
20. Авгуль Н. Н. и др. ЖФХ, 1956, т. 30, с. 2106—2121.
21. Авгуль Н. Н. и др. Изв. АН СССР, ОХН, 1956, с. 1304-1311.
22. А в г у л ь Н. Н., Киселев А. В. ДАН СССР, 1957, т. 112, с, 673—
676.
23. А в г у л ь Н. Н. и др. Изв. АН
24. А в г у л ь Н. Н. и др. Изв. АН
25. А в г у л ь Н. Н., Киселев
СССР, ОХН, 1961, с. 1395—1403.
26. Авгуль Н. Н., Киселев
СССР, ОХН, 1961, с. 1404—1411.
27. Безус А. Г., Древинг В.
1961, т. 23, с. 389—398.
28. А в г у л ь Н. Н., Киселев
СССР, ОХН, 1962, с. 1346-1353.
29. Kouznetsov А. V., Guio
р. 257-261.
СССР, ОХН, 1957, с. 1314—1327.
СССР, ОХН, 1959, с. 1196—1206.
А. В., Лыгина И. А. Изв. АН
А. В., Лыгина И. А. Изв. АН
П., Киселев А. В. Коллоидн. ж,
А. В., Лыгина И. А. Изв. АН
h о n G. J. Chim. Phys., 1969, v. 66,
30. Киселев А. В. и др. ЖФХ, 1970, т. 44, с. 1272—1278.
34. Kalashnikova Е. V. е. a. Chromatographia, 1972, v. 5, р. 278—285.
32. Березин Г. И., Киселев А. В., Козлов А. А. ЖФХ, 1967,
т. 41, с. 1426—1430.
33. Б е р е з и н Г. И. и др. ЖФХ, 1969, т. 43, с. 1601—1603.
34. Б е р е з и н Г. И. и др. ЖФХ, 1970, т. 44, с. 523—524.
35. Березин Г. И., Киселев А. В., Синицын В. А. ЖФХ,
1970, т. 44, с. 734—740.
36. В е г t u s h Т. I. е. a. Chromatographia, 1970, v. 3, р. 369—371.
350
37. Пейс Е. В кн.: Межфазовая граница газ — твердое тело. Под ред.
Э. Флада, М., «Мир», 1970. 433 с.
(8. Berezin G. I. е. a. J. Col. Interface Sci., 1972, v. 38, p. 335—340.
39. Волькенштейн M. В. Строение и физические свойства молекул.
М. — Л., Изд-во АН СССР, 1955. 638 с.
10. Crowell A. D. J. Chem. Phys., 1958, v. 29, p. 446—447.
11. Girifalco L. A., Lad R. A. J. Chem. Phys., 1956, v. 25, p. 693—697.
12. A 1 1 i n g e r N. L. e. a. J. Am. Chem. Soc., 1967, v. 89, p. 4345—4357.
13. A 1 1 i n g e r N. L. e. a. J. Am. Chem. Soc., 1968, v. 90, p. 1199—1210.
14. Constabaris G., Sams J. R., H a 1 s e у G. D. J. Phys. Chem.,
1961, v. 65, p. 367—369.
15. Б e 3 у с А. Г., Д p e в и н г В. П., Киселев А. В. ЖФХ, 1964,
т. 38, с. 59—67; 1967, т. 41f с. 2921—2927.
16. Б е з у с А. Г., Д р е в и н г В. П., Киселев А. В. ЖФХ, 1964,
т. 38, с. 2924—2930.
17. Myers A.L., Prausnitz J.M. Trans Faraday Soc., 1965, v. 61,
p. 755-764.
48. H о о г у S. E., Prausnitz J.M. Trans. Faraday Soc., 1967, v. 63,
p. 455—500.
49. Kalashnikova E. V. e. a. Chromatographia, 1971, v. 4, p. 405—500.
50. Бирштейн T. M., Птицын О. Б. Конформации макромолекул. М.,
«Наукам, 1964. 391 с.
51. Pitzer К. S. Advanc. Chem. Phys., 1959, v. 2, p. 59—83.
52. S a 1 e tn L. Molec. Phys., 1960, v. 3, p. 441—452.
53. Pitzer K. S. J. Chem. Phys., 1937, v. 5, p. 469—472.
54. М ид зусима С. Строение молекул и внутреннее вращение. М., Издат-
инлит, 1957. 263 с.
55. Волькенштейн М. В. Конфигурационная статистика полимерных
цепей. М.—Л., Изд-во АН СССР, 1959. 466 с.
56. В а г t e l 1 L. S., К о h 1 D. A. J. Chem. Phys., 1963, v. 39, p. 3097-3105,
57. Pitzer K. S. J. Chem. Phys., 1940, v. 8, p. 711—720.
58. Sheppard N., S zasz S. J. J. Chem. Phys., 1949, v. 17, p. 86—93.
59. Person W. B., Pimentel G. C. J. Am. Chem. Soc., 1953, v. 75,
p. 532—538.
60. Бойкова А. С., Щербакова К. Д. «Нефтехимия», 1967, т. 7,
с. 451-457.
61. Киселев А.В., Петов Г. М., Щербакова К. Д. ЖФХ, 1967,
т. 41, с. 1418—1425.
62. Киселев А. В., Мигунова И. А., Яшин Я. И. ЖФХ, 1968,
т. 42 с. 1235—1239.
63. Е 1 k i n g t о n Р. A., Curthoys G. J. Phys. Chem., 1969, v. 73,
p. 2321—2326.
64. А в г у л ь Н. Н., Киселев А. В., Л ы г и н а И. А. Изв. АН СССР,
ОХН, 1962, с. 1353—1357.
65. A v g u 1 N. N., Kiselev А. V., L у g i n a I. A. Trans Faraday Soc.,
1963, v. 59, р. 2113—2128.
66. S z a s z G. J., Sheppard N. J. Chem. Phys., 1949, v. 17, p. 93—97.
67. Пентин Ю. А. В кн.: Современные проблемы физической химии. Под
ред. Я. И. Герасимова и П. А. Акишина. Т. I, М., Изд-во МГУ, 1968. 191 с.
68. К и с е л е в А. В., Щ ер б а ко в а К. Д., Я шин Я. И. ЖСХ, 1969,
т. 10, с. 951—968.
69. Pitzer К. S., D о n a t h W. Е. J. Am. Chem. Soc., 1959, v. 81, p. 3213—
3218.
70. Muller A. Proc. Roy. Soc., 1936, v. A 154, p. 624—639.
71. Китайгородский А. И., Мирская К. В., Товбис А. Б.
Кристаллография, 1968, т. 13, с. 225—231.
72. S п о о k I. К., S р u г 1 i n g Т. Н. Austr. J. Chem., 1970, v. 23, р. 819—
823.
73. Китайгородский А. И., Мирская К. В. «Кристаллография»,
1964, т. 9, с. 174—181.
351
74. W i 11 i a m s D. E. J. Chem. Phys., 1966, v. 45, p. 3770—3778.
75. W i 11 i a m s D. E. J. Chem. Phys., 1967, v. 47, p. 4680—4684.
76. Безус А. Г., П ревинг В. П.,Киселев А. В. ЖФХ, 1964, т, 38,
с. 947—954; 1967, т. 41, с. 2921—2927.
77. И л и е л Э. и др. Конформационный анализ. М., «Мир», 1969, 592 с.
78, В othner-By A. A., Naar-Colin С., Gunter Н. J. Am. Chem.
Soc., 1962, v. 84, р. 2748—2751.
79. К а л ашнико в а Е. В. Канд. дисс. Москва, 1973, 152 с.
80. Coulson С. A.,Davies Р. L. Trans. Faraday Soc., 1952, v. 48, p. 777—
789
81. Haugh F.,Hirschfelder J. O. J. Chem. Phys., 1955, v. 23, p. 1778—
1796.
82. S t e r n 1 i c h t H. J. Chem. Phys., 1964, v. 40, p. 1175—1188.
83. Banerjee K., Salem L. Molec. Phys., 1966, v. 11, p. 405—420.
84. Schweig A. Int. J. Quantum., Chem., 1969, v. 3, p. 823—849.
85. С 1 a v e r i e B., R ein R. Int. J. Quantum. Chem., 1969, v. 3, p. 537—551*
86. В a s i 1 e v s k у M. V. Theoret. Chim. Acta (Berl.), 1969, v. 13, p. 409—420.
87. Krishna j i, Prakash V. Revs Mod. Phys., 1966, v. 38, p. 690—709.
88. King A. D. J. Chem. Phys., 1969, v. 51, p. 1262—1264.
89. Everett D. H. Disc. Faraday, Soc., 1965, v. 40, p. 177—187.
90. Киселев А. В. ЖФХ, 1964, т. 38, с. 2753—2773.
91. К i s е 1 е v А .V. Disc. Faraday Soc., 1965, v. 40, p. 205—218e
92. London F. J. Phys. Chem., 1942, v. 46, p. 305—316.
93, Lo ngue t-H iggins H. C., Salem L. Proc. Roy. SoCe (London).
1961, v. A 259, p. 433—441.
94. В и л к о в Л. В., С а д о в а Н. И., Тарасенко Н. А. ЖСХ, 1969,
т. 10, с. 403—406.
95. Т а тев ск ий В. М. Классическая теория строения молекул и квантовая
механика. М., «Химия», 1973. 516 с.
96. М и р с к а я К. В., Козлова И. Е., «Кристаллография», 1969, т. 14,
97. Tyutylkov N., Shopov D., D у an ко v S. Comptes rendus de
L’Academie bulgare des Sciences, 1968, v. 21, p. 1077—1079.
98. Tyutylkov N., Vodenicharov R. Comptes rendus de L’Academie
bulgare des Sciences, 1969, v. 22, p. 451—453.
99. Dewar M. I. S., H a r g e t A. I. Proc. Roy. Soc. (London), 1970, v. 315,
p. 431—442.
100. Карапетьянц M. X. Химическая термодинамика. M., Госхимиздат,
1953. 611 с.
401. Groszek A. J. Disc. Faraday Soc., 1965, v. 40, p. 230.
102. Williams J.E., Stang P. J., Schleyer P. v о n R., Annual '
Rewiew of Physical Chemistry, 1968, v. 19, p. 531—558.
103. Kitaigorodsky A. I. J. Chim. Phys., 1966, v. 63, p. 9—14.
104. De Coen J. L. e. a. Nature, 1967, v. 216, p. 910—913.
Глава XI
ИЗОТОПНЫЙ ЭФФЕКТ ПРИ АДСОРБЦИИ УГЛЕВОДОРОДОВ
НА ГРАФИТЕ
1. Влияние на адсорбцию водорода и углеводородов
замещения атомов водорода на атомы дейтерия
Адсорбционные свойства дейтерированных и обычных форм водо-
рода и углеводородов несколько различаются [1—44]. В зависимости
от общей массы и строения молекулы, природы поверхности и тем-
пературы опыта дейтерированные формы молекулы могут адсорби-
роваться как сильнее [1—26, 29, 32], так и слабее [26—28, 31, 33—
44], чем соответствующие обычные формы. Более тяжелые изотопы
молекулы водорода во всех изученных случаях адсорбируются
сильнее, чем молекула Н2 [1—26]. Вместе с тем, все изученные дей-
тероуглеводороды на графитированной саже адсорбируются слабее,
чем соответствующие обычные углеводороды [26, 34, 36, 38—41,
43, 44]. Однако на сильно полярных адсорбентах некоторые дейтеро-
углеводороды адсорбируются сильнее, чем их водородные формы
[29—32]. Кроме того, в некоторых случаях при изменении темпера-
туры наблюдается инверсия изотопного эффекта, т. е. изменение
последовательности величин адсорбции изотопных молекул угле-
водородов [31].
Адсорбционные свойства изотопных молекул близки, и для их
полного разделения адсорбционными методами необходимо пра-
вильно выбрать адсорбент и условия разделения. Поэтому развитие
молекулярной теории изотопных эффектов при адсорбции пред-
ставляет не только теоретический, но и практический интерес.
Изменение адсорбции водорода и углеводородов при замещении
атомов Н на атомы D, по-видимому, обусловлено несколькими при-
чинами. Средняя длина валентной связи С—D меньше средней длины
связи С—Н из-за нулевых межатомных колебаний и ангармонич-
ности межатомного потенциала. По этой причине поляризуемость
дейтерозамещенной молекулы несколько меньше поляризуемости
обычной молекулы. Так как дисперсионные силы прямо пропорци-
ональны поляризуемости взаимодействующих частиц, то потен-
циальная энергия дисперсионного взаимодействия дейтерированной
молекулы с твердым телом должна быть несколько меньше потен-
циальной энергии дисперсионного взаимодействия соответствующей
водородной формы молекулы (квантовомеханический изотопный эф-
фект) [38, 45—49]. По этой причине адсорбция дейтерозамещенных
молекул должна быть меньше адсорбции соответствующих обычных
23 заказ 1180 353
молекул. В случае адсорбции углеводородов на графите этот эффект,
по-видимому, является основным 138, 45, 48, 49],
С другой стороны, энергетические уровни колебания центра масс
и вращения адсорбированной молекулы с более тяжелым изотопом
лежат ниже и ближе друг к другу, чем для адсорбированной моле-
кулы с более легким изотопом. По этой причине содержащие D
молекулы должны адсорбироваться сильнее своих более легких
аналогов, содержащих Н (квантовостатистический изотопный эф-
фект) [13, 21, 38, 45, 50—55]. Однако квантовостатистический изо-
топный эффект быстро убывает с ростом массы и момента инерции
молекулы, а также температуры опыта. Он имеет существенное
значение только для наиболее легких молекул при низких темпе-
ратурах. В случае адсорбции изотопных молекул водорода квантово-
статистический эффект является основным [13, 21, 50—53].
Наконец, из-за различия масс колеблющихся атомов в изотопных
молекулах происходящие при адсорбции изменения частот вну-
тренних колебаний молекул должны несколько различаться. Это
также должно вызвать некоторое различие в адсорбции изотопных
молекул (эффект нулевых энергий) [33, 54], причем в зависимости
от знака изменения силовых констант внутренних колебаний моле-
кулы при адсорбции эффект нулевых энергий может как увеличивать,
так и уменьшать адсорбцию дейтерированных молекул по сравнению
с обычными. С ростом температуры этот эффект должен убывать
значительно медленнее, чем квантовостатистический эффект. Кроме
того, его вклад должен расти с ростом числа замещенных атомов.
Поэтому эффект нулевых энергий может стать существенным для
углеводородов с большим числом замещенных атомов Н на
атомы D [33, 54, 56].
Из указанных выше трех возможных причин изотопного эффекта
при адсорбции довольно подробно исследован только квантово-
статистический эффект [13, 21, 38, 45, 50—55]. Однако при раз-
делении дейтерозамещенных углеводородов на графите вклад этого
эффекта, по-видимому, не является существенным [38, 45, 47—49].
Квантовомеханический эффект и эффект нулевых энергий при раз-
делении изотопнозамещенных углеводородов изучены мало [38,
45-49, 54].
2. Статистические выражения для термодинамических
характеристик адсорбционного разделения сложных
изотопнозамещенных молекул
Разделение изотопов часто осуществляется газохроматографи-
ческим методом. При предположении, что в хроматографической
колонне достигается равновесие и заполнение поверхности мало
(близко к нулю), коэффициент разделения изотопов равен:
izH
(XI,1)
Л1
354
где Kf и Л'Р — константы Генри, относящиеся соответственно к водородной
и дейтерированной формам рассматриваемой молекулы.
Статистическое выражение для коэффициента разделения w
сложных молекул при адсорбции было получено из статистического
выражения для изотопного эффекта в конденсированных системах
при использовании гармонического приближения для внутренних
и внешних степеней свободы молекулы [54]. Оно учитывает квантово-
статистический эффект и эффект нулевой энергии, но не учитывает
квантовомеханического изотопного эффекта.
Статистические выражения для учитывающие квантовомеха-
лический и квантовостатистический изотопные эффекты при адсорб-
ции, были получены для двух упрощенных моделей состояния адсор-
бированных молекул [38].
Статистические выражения для w и других термодинамических
характеристик адсорбционного разделения сложных изотопных моле-
кул при нулевом заполнении поверхности, учитывающие различие
потенциальных функций Ф взаимодействия этих молекул с адсор-
бентом, т. е. квантовомеханический изотопный эффект, были полу-
чены в работах [48, 49]. При выводе этих выражений были исполь-
зованы приведенные в гл. VII статистические выражения для кон-
станты Генри К г и других термодинамических характеристик
адсорбции сложных молекул.
Константа Генри Кг дается общим выражением (VI,30). При
допущении, что энергия внутренних колебаний изотопных молекул
отделима от остальной энергии и что к внешним степеням свободы
этих молекул применимы законы классической механики (т. е. при
пренебрежении квантовостатистическим эффектом), для w можно
записать выражение:
н /ngH
колеб' ^колеб
Q
w
^колеб/ ^колеб
(XI,2)
5?
Здесь <?колеб и <?колеб — колебательные статистические суммы изотопных
молекул, соответственно, в адсорбированном состоянии и в объеме газа; —
конфигурационные интегралы для изотопных молекул в адсорбированном со-
стоянии. В случае квазижестких молекул дается выражением (VII,47).
Отношение (<??оЛеб/С|<йеб)/(<22олеб/<21олеб) Дает вклад [эффекта
нулевой энергии в коэффициент разделения изотопных молекул,
а отношение Sp/Sf* дает вклад квантовомеханического эффекта.
В гармоническом приближении
<?колеб/колеб
<?колеб/ <?колеб
межатомные
колебания
ехр [— (uD — uH)s/2]
ехр [— (wD — wH)g/2]
1 —ехр (—u?) / 1 —ехр (-uf)]
’!-ехР / 1_ехр
(XI,3)
23*
355
Здесь и — hvlkT, где v — частоты гармонических межатомных
колебаний молекулы. Величины с нижними индексами s и g отно-
сятся соответственно к молекуле в адсорбированном состоянии
и в газовой фазе.
В случае наиболее легких молекул углеводородов (например,
метана) возможен небольшой вклад квантовостатистического эффекта
в термодинамические характеристики адсорбционного разделения
изотопных молекул. Для учета этого эффекта можно использовать
приближение Питцера и Гвина [57 , 58]. Согласно этому приближе-
нию, вклад квантовостатистического эффекта в коэффициент w
дается множителем [48, 49, 54]:
(Х..4>
uj? ехр (— uJ?/2) 1 — ехр (— и®)
Здесь иг = hvrlkT, где vr — частота гармонических колебаний
молекулы вблизи потенциального минимума для r-ой внешней сте-
пени свободы движения. Для колебания центра масс молекулы пер-
пендикулярно поверхности vz = (1/2л) (Ф^/М)^*, где — вторая
производная Ф по z вблизи минимума, М — масса молекулы. Ча-
стоты гармонических колебаний vr ~ (1/2л) (ФД/Г), где — вторые
производные Ф по углам вращения молекулы вокруг главных осей,
параллельных поверхности, Iг — соответствующие моменты инерции.
Приближение Питцера и Гвина, в сущности, является допущением
о том, что степени свободы движения, для которых вычисляются
квантовостатистические поправки, являются гармоническими коле-
баниями. В этом случае оно дает точные выражения для квантово-
статистического изотопного эффекта. Погрешность этого приближе-
ния будет тем больше, чем больше рассматриваемые степени свободы
отклоняются от гармонических колебаний. Если рассматриваемую
степень свободы движения нельзя в приемлемом приближении счи-
тать гармоническим колебанием, то для расчета квантовых поправок
необходимо воспользоваться суммой Слэтера [8, 45, 59], которая
позволяет учесть действительный вид потенциальной функции Ф.
Однако последний путь значительно сложнее первого.
При и <С 1, разлагая In w* в ряд Тейлора по степеням и и пре-
небрегая членами высших порядков, получаем [38, 54]:
г
Для других термодинамических характеристик адсорбционного
разделения сложных изотопных молекул, обусловленных только
квантовомеханическим эффектом, получаем:
/ оН ~d \
-(д^Н-д^-яг * (XI,6)
X *4 /
356
AS*H-A5*D = fl
I 5F
Mtf
ДСр-ДСР = Л
£?
5?
(XI,7)
(XI.8)
s* s? 1
a a
W 7°.
где конфигурационные интегралы Sf (I — 2, 3) в случае квазижестких
молекул даются выражением (VII,48).
Если потенциальная функция Ф практически не зависит от коор-
динат движения центра масс молекулы параллельно поверхности
(координат х и у) и от координат вращения молекулы вокруг оси,
перпендикулярной поверхности (от угла ср), а также если колебания
центра масс молекулы перпендикулярно поверхности можно считать
гармоническими [т. е. зависимость Ф от г дается выражением
(VII,52)], то конфигурационные интегралы St выражаются форму-
лами (VII,57)—(VII,62). Кроме того, если предположить, что
не зависит от ориентации молекулы над поверхностью адсорбента
и равна значению Ф^ для энергетически наиболее выгодной ориента-
ции (в этом приближении вращательные степени свободы молекулы
отделимы от колебательных степеней свободы центра масс) и что
потенциальные барьеры для вращения адсорбированных изотопных
молекул практически одинаковы, то статистические выражения
(XI,2), (XI,6)—(XI,8) для изотопного эффекта при адсорбции, обус-
ловленного различием потенциальных функций Ф изотопных моле-
кул, принимают вид:
In w — —
кТ
(XI,9)
-(ДС/Н-ДЕ/°)=-^(фН-ф5)) (XI,10)
ДСН-дСр = 0
(XI,И)
(XI,12)
где Фо — значение Ф в потенциальном минимуме для энергетически наиболее
выгодной ориентации молекулы.
Сделанные выше допущения приблизительно справедливы, на-
пример, в случае адсорбции на базисной грани графита.
Статистические выражения для разностей изменений внутренних
энергий, энтропий и теплоемкостей при адсорбции изотопных моле-
кул, обусловленных квантовостатистическим эффектом и эффектом
нулевой энергии, можно получить, вводя статистические выражения
357
для соответствующих множителей коэффициента w
ния [46]:
- (т - дрр)=_
\ 1 1 / dT
ТБ*Н Г7Г*В п /1 , Т dlUW \
^S±a -bS^^R Inw-i-----—---)
\ di /
д7Н r/vD n / 2T d In w П In w \
ДСг -ДСГ = R dT +—?2 )
в соотноше-
(XI,13)
(XI ,14)
(XI,15)
3. Молекулярно-статистический расчет изменений
термодинамических характеристик адсорбции углеводородов
при замещении атомов Н на атомы D
Исходя из физико-химических свойств углеводородов и дейтеро-
углеводородов, а также свойств решетки графита, в работах [48, 49]
были рассчитаны коэффициенты разделения w, т. е. отношения кон-
стант Генри К± (удерживаемых объемов Fs, J и разности
—(АС/н — АС7?) для ряда пар изотопных молекул (СН4 — GD4,
С2Не — G2De, С3Н8 — C3D8, CeHe GeDe, С2Н4 — G2D4
и СеН12 — GeD12) на базисной грани в функции температуры. В этих
расчетах были учтены квантовомеханический и квантовостатисти-
ческий изотопные эффекты. При оценках потенциальных функций Ф
взаимодействия изотопных молекул с графитом в качестве силовых
центров графита рассматривались атомы G, а в качестве силовых
центров молекулы рассматривались атомные группы [48] или
атомы [49]. Атом-атомное приближение, по-видимому, лучше учи-
тывает особенности пространственного строения рассматриваемых
молекул. Поэтому ниже рассмотрены результаты расчетов, полу-
ченные при использовании только атом-атомного приближения.
При взаимодействии неполярной молекулы с неполярным твер-
дым телом, в том числе с графитированной термической сажей,
основными силами притяжения являются дисперсионные. В этом
случае Ф определяется главным образом химическим составом и про-
странственным строением молекулы, а также электромагнитными
свойствами (поляризуемостью, диамагнитной восприимчивостью)
и ван-дер-ваальсовыми радиусами силовых центров молекулы.
В дейтероуглеводородах средняя длина связи G—D на 0,004—
0,005 А меньше длины связи С—Н [60—64]. Поляризуемость дейте-
роуглеводородов на 0,5—1,5% меньше поляризуемости соответству-
ющих углеводородов [61, 65—69].
Ван-дер-ваальсовы радиусы атомов Н и D в молекулах, по-види-
мому, практически одинаковы [45, 59, 70, 71]. Поэтому различие
потенциальных функций Ф взаимодействия дейтероуглеводородов
и обычных углеводородов с графитом может быть вызвано главным
образом различием длин связей С—D и G—Н и различием электро-
магнитных свойств силовых центров изотопных молекул. При расче-
тах Ф для дейтероуглеводородов и обычных углеводородов были при-
358
пяты во внимание оба эти различия изотопных молекул. Для вза-
имодействия атомов С и Н молекул углеводородов с атомами С гра-
фита были использованы приведенные в гл. X полуэмпирические
потенциальные функции (Х,5) и (Х,6).
Согласно формуле Кирквуда — Мюллера (VIII,25) отношение
констант Сх диполь-дипольного дисперсионного взаимодействия ато-
мов D и Н молекулы адсорбата с атомами С графита выражается
так:
_ __ Л , анДн~~арДр
Y ан авЛр + асЛс
(XI,16)
где ctjp aD, ас — поляризуемости; %с — диамагнитные восприимчи-
вости соответственно атомов Н, D и С.
Если предположить, что отношение констант диполь-квадру-
польного взаимодействия С^/С^ равно отношению С^/С^, а осталь-
ные параметры (положение потенциального минимума го и кон-
станта q) для атом-атомных потенциалов межмолекулярного взаимо-
действия атомов Н ... С и D ... С одинаковы, то
<Pd. .,c = Y<Ph...c <XI’17)
Кроме того, если предположить, что уменьшение а молекул
углеводородов при замещении атомов Н на атомы D обусловлено
меньшей поляризуемостью атомов D, чем атомов Н, то на основании
опытных значений а для пар изотопных молекул СН4 — CD4 [61],
СеН12 — CeD12 166, 67] и C6He — CeDe [66—68] получается, что
ах/ссн 0,98. Диамагнитная восприимчивость % дейтерированных
молекул, по-видимому, не измерялась. Поэтому принималось, что
при изотопном замещении остаются неизменными или отношение
а/% [45], или значение % [72] молекулы. Согласно выражению [XI,16)
получаем в этих двух случаях соответственно у = 0,98 и у = 0,99.
При расчетах Ф для дейтероуглеводородов было принято среднее
из двух указанных выше значений у, т. е. у = 0,985. Кроме того,
было принято, что положение силового центра атома D в молекуле
CD4 смещено на 0,004 А, а в остальных молекулах — на 0,005 А
в сторону атома С по сравнению с положением силового центра
атома Н.
Расчеты изменений термодинамических характеристик адсорбции
углеводородов при замещении атомов Н на атомы D, обусловленных
только квантовомеханическим изотопным эффектом, производились
при использовании гармонического приближения (VII,52). Вклады
квантовостатистического эффекта в эти изменения вычислялись
только для колебаний центров масс адсорбированных молекул пер-
пендикулярно поверхности при использовании выражения (XI,4).
На рис. XI,1 и XI,2 результаты расчетов In и? и ДС7}1 — ДС7Р
сопоставлены с опытными данными [38]. Рассчитанные кривые в об-
щем близки к опытным данным. Однако в отличие от эксперимен-
359
тальных данных рассчитанные значения In и? и А С/р — А С/р для
системы С2Н4 — C2D4 больше, чем для системы С2Нв — C2De, а для
системы СеНв — GeDe больше, чем для системы СвН12 — G6D12.
Рис. XI,1. Рассчитанные (кривые) и опытные (точки)
значения In w для адсорбции пар изотопных молекул
CH4-GD4 (7), G2H4-C2D4 (2), С2Н6—G2De (3),
CeHe — GeD6 (4) и CeH12 — CeD12 (5) на базисной гра-
ни графита (расчет) и на графитированных термиче-
ских сажах (опыт).
Это расхождение результатов расчета с опытом может быть вызвано
рядом причин. В частности, оно может быть обусловлено неучтенным
при расчетах эффектом нулевых энергий. Из-за большего числа
Рис. XI,2. Рассчитанные (кривые) и опытные (точки)
значения ДС7р — At/P для адсорбции пар изотоп-
ных молекул СН4 — GD4 (7), G2H4 — C2D4 (2),
G2H^ C2Dg (3), GeHe— G6D6 (4) и GeH12 — GeDl2 (5)
на оазисной грани графита (расчет) и на графитирован-
ных термических сажах (опыт).
замещенных атомов Н в C2De, чем в G2D4, и в GeD12, чем в CeDe,
а также из-за различия пространственного строения этих молекул
вклад эффекта нулевых энергий для системы С2Нв — G2De и СвН12—
GeD12 может быть больше, чем для систем С2Н4 — C2D4 и С6Не —
GeDe.
360
Расхождение результатов расчета с опытом может быть вызвано
также погрешностями оценки различия положений силовых центров
атомов Н и D. Изменение различия положений силовых центров
атомов Н и D практически не изменяет результатов расчета для пар
плоских молекул С2Н4 — C2D4 и СвНв — CeDe, но сравнительно
сильно изменяет результаты расчета для остальных пар молекул.
Гак, если предположить, что положения силовых центров атома D
и атома Н совпадают и различие в Ф для изотопных молекул обусло-
влено только различием в фн... с и Фо .. .с? т0 рассчитанные
Рис. XI,3. Рассчитанные (кривые) и опытные (точки)
значения In w для адсорбции пар изотопных молекул
CH4-CD4 (7), C2H4-C2D4 (2), С2Не — C2De (5),
CeHe — CeDe(4), CeH12 — CeD12 (5) на базисной грани
графита (расчет) и на графитированных термических
сажах (опыт).
Расчеты [проводились при предположении, что положения
силовых центров атомов D и Н совпадают.
кривые, выражающие зависимость In w от ИТ (рис. XI,3), распо-
лагаются в той же последовательности, что и экспериментальные
значения.
Результаты расчета весьма чувствительны к значению коэффи-
циента у. Так, изменение у на 0,005 изменяет значения In w и
—(Д17*1 — Д??Р) на 30—50%. Поэтому указанное выше расхожде-
ние рассчитанных и опытных значений In w и — AZ/p) может
быть также вызвано небольшими изменениями у при переходе от
одной пары изотопных молекул к другой.
Вклад квантовостатистического изотопного эффекта в различие
термодинамических характеристик адсорбции дейтероуглеводородов
и обычных углеводородов, обусловленный колебанием центров масс
изотопных молекул перпендикулярно поверхности, невелик. Для
пары изотопных молекул СН4 — CD4 при 100 К вклад этого эффекта
в In w составляет около —0,07, а в — (Д<7? — Д<7^) — 25 Дж/моль.
К этим значениям квантовостатистического эффекта надо было бы
еще добавить вклады квантовостатистического эффекта, обусловлен-
ные заторможенным вращением адсорбированных изотопных
361
молекул вокруг осей, параллельных поверхности. Учет зтих вкладов
несколько понизил бы абсолютные значения рассчитанных термо-
динамических величин для СН4 — GDV Для более тяжелых угле-
водородов при рассмотренных температурах квантовостатистические
изотопные эффекты незначительны и ими можно пренебречь.
Расчет вклада эффекта нулевых энергий в различие термодина-
мических характеристик адсорбции на графите дейтероуглеводородов,
и соответствующих обычных углеводородов, по-видимому, пока не-
возможен из-за отсутствия опытных данных по изменению частот
внутримолекулярных колебаний этих молекул при адсорбции на
графите [38].
Погрешности полуэмпирического расчета абсолютных значений
термодинамических характеристик адсорбции молекул, по-види-
мому, больше, чем различие термодинамических характеристик
адсорбции изотопных молекул. Однако при расчете коэффициента
разделения или разностей других термодинамических характеристик
адсорбции для изотопных молекул многие погрешности расчета
элиминируются, что значительно повышает точность молекулярно-
статистического расчета относительных величин и соответствующих
разностей по сравнению с точностью расчета абсолютных значений.
Таким образом, проведенные расчеты показывают, что главной
причиной различной адсорбируемости дейтероуглеводородов и обыч-
ных углеводородов на графитированной термической саже, а также
и на других неспецифических адсорбентах, является квантовомеха-
нический изотопный эффект, т. е. различие потенциальных функ-
ций Ф взаимодействия изотопных молекул с поверхностью. При
большом числе замещенных атомов Н на D в молекуле углеводорода
заметный вклад в изотопный эффект может, по-видимому, внести
также эффект нулевых энергий. Вклад квантовостатистического
изотопного эффекта для углеводородов невелик, и он заметен только
для наиболее легких молекул при низких температурах.
ЛИТЕРА ТУРА
1. Moore W. R., Ward Н. R. J. Am. Chem. Soc., 1958, v. 80, p. 2909—
2910.
2. G a n t P. L., Yang K. Science, 1959, v. 129, p. 1548—1549.
3. Moore W. R., Ward H. R. J. Phys. Chem., 1960, v. 64, p. 832.
4. S m i t h H. A., Hunt P. P. J. Phys. Chem., 1960, v. 64, p. 383—384.
5. Van Hook W. A., Emmett P. H. J. Phys. Chem., 1960, v. 64, p. 673—
675.
6. P a с e E. L., S i e b e r t A. R. J. Phys. Chem., 1960, v. 64, p. 961—963.
7. Basmad jian D. Canad. J. Chem., 1960, v. 38, p. 141—148.
8. F r e e m a n M. P. J. Phys. Chem., 1960, v. 64, p. 32—37.
9. Furuyama S., Kwan T. J. Phys. Chem., 1961, v. 65, p. 190—191.
10. H u n t P. P„ Smith H. A. J. Phys. Chem., 1961, v. 65, p. 87—89.
И. К i n g J. J. Phys. Chem., 1963, v. 67, p. 1397.
12. Carter E. H., Smith H. A. J. Phys. Chem., 1963, v. 67, p. 1512—1516.
13. H a u b a c h W. J., W h i t e D. J. Chim. phys., 1963, v. 60, p. 97—99.
14. W e s t D. L., M arston A. L. J. Am. Chem. Soc., 1964, v. 86, p. 4731.
15. К ачурихин В. E., Зел ьвенский Я. Д. ЖФХ, 1964, т. 38,
с. 2594—2603.
362
16. Панченков Г. М., Толмачев А. М., Зотова Т. В. ЖФХ,
1964, т. 38, с. 1361—1365.
17. Парбузин В. С., Панченков Г. М. ДАН СССР, 1965, т. 164,
с. 856—859.
18. Толмачев А. М., Зотова Т. В., Елисеева Н. М. ЖФХ, 1965,
т. 39, с. 1021—1025.
19. Парбузин В. С. и др. ЖФХ, 1967, т. 41, с. 193—194.
20. С о n t i М. L., L е s i m р 1 е M. J. Chromatog., 1967, v. 29, p. 32—43.
21. Haubach W. J. e. a. J. Phys. Chem., 1967, v. 71, p. 1398—1402.
22. J о n e s P. M., Huthcheson C. G. Nature (Engl.), 1967, v. 213,
p. 490—491.
23. Б л а г о й Ю. П., 3 и м о г л я д Б. Н., Ж у н ь Г. Г. ДАН СССР, 1968,
т. 181, с. 630—633.
24. К о ч у р и х и и В. Е., Зельвенский Я. Д. ЖФХ, 1969, т. 43,
с. 206-210.
25. G a n t Р. L. е. a. J. Phys. Chem., 1970, v. 74, р. 1985—1991.
26. Constabaris G., Sams J. R., Halsey G. D. J. Phys. Chem.,
1961, v. 65, p. 367—369.
27. G a n t P. L., Yang K. J. Am. Chem. Soc., 1964, v. 86, p. 5063—5064.
28. R о о t J. W., Lee E. K., Rowland F. S. Science, 1964, v. 143,
p. 676-678.
29. В r u n e r F., C arton i G. P. J. Chromatog., 1965, v. 18, p. 390—391.
30. С акодынский К. И., К a p т о н и Дж. Р., Пела А. ЖФХ, 1966,
т. 40, с. 2887—2888.
31. Bruner F., С а г t о n i G. Р., Liberti A. Analyt. Chem., 1966,
v. 38, р. 298—303.
32. С а г t о n i G. Р., L i b е г t i А., Р е 1 a A. Analyt. Chem., 1967, v. 39,
р. 1618-1622.
33. Р h i 11 i р s J. P., Van Hook W. A. J. Phys. Chem., 1967, v. 71,
p. 3276—3279.
34. G о r e 11 i G. C., L i b e r t i A., N о t a G. J. Chromatog., 1968, v. 34,
p. 96—100.
35. P о s s a n z i n i M. e. a. J. Chromatog., 1968, v. 38, p. 492—497.
36. Liberti A., N о t a G., G о r e 11 i G. C. J. Chromatog., 1968, v. 38,
p. 282—286.
37. Czubryt J. J.,Gess ar H. D.J. Gas Chromatog., 1968, v. 6, p. 41—42.
38. Di Corcia A., Liberti A. Trans Faraday Soc., 1970, v. 66, p. 967—
975.
39. Pare j a P., A m a r i g 1 i о H. J. Chim. phys., 1970, v. 67, p. 938—942.
40. Di Corcia A., Bruner F. J. Chromatog., 1970, v. 49, p. 139—145.
41. Di Corcia A., Fritz D., Bruner F. J. Chromatog., 1970, v. 53,
p. 135—141.
42. Gunter B. D.,Gleason J. D. J. Chromatog. Sci., 1971, v. 9, p. 191 —
192.
43. Bruner F., Ciccioli P., D i Corcia A. Analyt. Chem., 1972,
v. 44, p. 894—898.
44. К и с e л e в А. В., Худяков В. Л., Яшин Я. И. ЖФХ, 1974, т. 48,
с. 448—450.
45. Y а г i s R., Sams J. R. J. Chem. Phys., 1962, v. 37, p. 571—576.
46. Киселев А. В., Пошкус Д. П. ЖФХ, 1965, т. 39, с. 398—402.
47. М а с Rury Т. В. J. Chem. Phys., 1968, v. 49, р. 1543—1545.
48. Киселев А. В., Пошкус Д. П. ЖФХ, 1969, т. 43, с. 285—291.
49. Р о s h k u s D. Р. J. Chromatog., 1970, v. 49, р. 146—152.
50. W h i t e D., L a s s e t r e E. N. J. Chem. Phys., 1960, v. 32, p. 72—86.
51. К atorski A.,Eberhart J. G., White D.J. Chem. Phys., 1961,
v. 34, p. 2189—2190.
52. К a t о г s к i A., White D.J. Chem. Phys., 1964, v. 40, p. 3183—3194.
53. F и г и у a m a S., E b a r a N. Bull. Chem. Soc. Japan, 1965, v. 38, p. 1705—
1708
54. V a n Hook W. A. J. Phys. Chem., 1967, v. 71, p. 3270—3275.
363
55. Freeman M. P.,H agy ard M. J. J. Chem. Phys., 1969. v. 49, p. 4020—
4031.
56. Grigor A. F., S t e e 1 e W. A. J. Chem. Phys., 1968, v. 48, p. 1038—1046.
57. P i t z e r K. S., Gwinn W. D. J. Chem. Phys., 1942, v. 10, p. 428—440.
58. H i 1 1 T. L. J. Chem. Phys., 1948, v. 16, p. 184—189.
59. Гиршфельдер Дж., Кертис Ч., Берд Р. Молекулярная теория
газов и жидкостей. М., Издатинлит, 1961, 929 с.
60. Laurie V. W., Herschbach D. R. J. Chem. Phys., 1962, v. 37,
p. 1687—1693.
61. В e 1 1 R. P. Trans Faraday Soc., 1942, v. 38. p. 422—429.
62. Bartell L. S., Kuchitsu K., De N e u i R. J. J. Chem. Phys.,
1961, v. 35, p. 1211—1218.
63. Kuchitsu K., Bartell L. S. J. Chem. Phys., 1962, v. 36, p. 2470—
2481.
64. Bartell L. S.,Higginbotham H.K.J. Chem. Phys., 1965, v. 42,
p. 851-856.
65. Terhune R. W., Peters C. W. J. Mol. Spectroscopy, 1959, v. 3,
p. 138-147.
66. D a v i s R. T., Scbiessler R. W. J. Am. Chem. Soc., 1953, v. 75,
p. 2763—2764.
67. Dixon J. A., Schiessler R. W. J. Am. Chem. Soc., 1954, v. 76,
p. 2197—2199.
68. I n g о 1 d С. K., Raisin C. G., Wilson C. L. J. Chem. Soc.. 1936,
p. 915—921.
69. К n a a p H. F. P. e. a. Disc. Faraday Soc., 1965, v. 40, p. 135—139.
70. Michels A., De Graaff W., Ten Seklam C. A. Physica, 1960,
v. 26, p. 393—408.
71. К n a a p H. F. P., В e e n a k k e r J. J. M. Physica, 1961, v. 27, p. 523—
530.
72. Wolfe R., Sams J. R. J. Chem. Phys., 1966, v. 44, p. 2181—2188.
ПРИЛОЖЕНИЕ
1. Значения площади, приходящейся на молекулу в плотном
мономолекулярном слое при адсорбции на графитированной
термической саже
Таблица П,1- Значения площади приходящейся на молекулу
в плотном монослое на поверхности графитированной термической
сажи, определенные методом Брунауера, Эмметта и Теллера [1]
(принимая для адсорбции пара азота при 78 К = 16,2 А2),
из ван-дер-ваальсовых размеров молекул, из плотности жидкости
при плоском расположении молекул иа поверхности
и по уравнению (IV, 21) [2] (в А2)
Адсорбат Темпера- тура, Т, К По мето- ду БЭТ Из вандер- ваальсовых размеров молекулы Из плотности жидкости при плоской ориентации молекулы По урав- нению (IV, 21 >
Аргон [2, 3] 78 13,8 13,8 13,6
Криптон [4] 90 15,2 15,7
Ксенон 18,5
Азот 78 16,2 15,5
Двуокись углерода 15] 195 20 17,1 16,4
Двуокись серы [6, 7] 240 — 25 — 19,8
Бензол [8, 9] 293 40 41 39,7 32,7
Метан [10, 11] 113 18,1 16,0 16,4 16,4
Этан [10, И] 173 22,7 20,4 22,2
Этилен [10] 173 22,6 19,5 , 21,7 20,0
Бутан [12] 303 40,8 40
«-Гексан [12] 293 51 52
—СНа— в и-алканах [12] 293 5,5
Неопентан [13] 293 41 36
Циклопентан [14] 293 38,5 39
Метилциклогексан [15] 293 52 51
Диэтиловый эфир [16] 293 42 46
Пропанол [17] 293 28,2 29
Бутанол [17] 293 35,3 36 38
Четыреххлористый угле- 293 37 34 35,9
род [13, 18, 19]
Перфторметилциклогек- 293 70 69
сан [15]
365
2. Зависимости 1g К± от температуры и значения Д5? и AUi при
адсорбции различных адсорбатов графитированной термической сажей
Величины констант Генри Кг в определенном интервале темпе-
ратур описываются приближенной зависимостью.
-7? [ / 1 - ^* + 7? Mh 1____________
g 1 тЛ т 2,3032? 2,3037? Т
Д>$ 14'- 7? Д£71 1 , тт . . ,
19,18 19Д8 Т (Дж/моль) (ПЛ)
Величина К± выражается в мкм и представляет собой констан-
ту Генри Кг, с, 1, связанную с величиной Лт, р, i уравнением (III, 15).
Из величины А может быть рассчитано дифференциальное измене-
ние внутренней энергии адсорбции при нулевом заполнении по-
верхности по уравнению
_д^ = 19,18Л (Дж/моль) = 4,576/1 (кал/моль) (П,2)
Таблица П,2. Константы уравнения (П, 1), описывающего
зависимость lg Ki (мкм) от 1/7’, интервал температур,
в котором проводились измерения (7, К), а также значения
—Д771 при средней температуре измерения Тсред [4, 5, 20, 21]
и Д5* при адсорбции неорганических адсорбатов графити-
рованной термической сажей
Адсорбат Интервал температур, Т, К -ДП1, кДж/моль ASj+H 2,3032? -дел
от до Гсред 2,303Н
Аг 127,4 153,5 140,4 8,91 —4,1914 465,6
СО 118,0 148,1 133,0 9,03 —3,9372 470,7
со2 193,2 303,2 238,2 15,2 —4,784 792
Кг 164,0 209,5 186,7 11,1 —4,0860 580,0
no2 191,0 226,4 208,7 15,0 —4,383 783,2
n20 210,1 250,6 230,3 14,18 —4,1816 740,6
Хе 201,5 251,4 226,5 13,71 —3,8081 715,8
366
Таблица П,3. Константы уравнения (П, 1), описывающего зависимость 1g Ку (мкм) от 1/Г, интервал
температур, в котором проводились измерении (Т, К), а также значения—AUy при средней температуре
измерения ГСреД и AS* [22—25] при адсорбции органических адсорбатов графитированной термической сажей
Интервал температур, Т,К AS* 4-В -&U1
от до тсред кДж/моль 2,303Я 2,303В
СС14 Тетрахлорметан 295,2 373,2 334,2 30,36 —4,3433 1585
СНВгз Трибромметан 343,2 428,0 385,6 36,6 -4,6410 1909
СНС13 Трихлорметан 295,2 373,2 334,2 28,9 -4,4314 1507,5
СН2Вг2 Дибромметан 294,2 372,9 333,5 27,13 -4,0663 1414,6
СН2С12 Дихлорметан 244,9 294,7 269,8 23,9 -4,2135 1245,2
СН212 Дииодметан 343,2 438,3 390,7 33,5 —4,1098 1745,1
СН31 Йодистый метил 244,9 333,4 289,1 22,85 -4,0379 1190,8
сщ Метан 122,6 169,7 145,6 11,80 -4,372 615,5
с2с1в Гексахлорэтан 373,2 438,3 405,7 42,35 —4,8350 2213,3
С2НС16 Пентахлорэтан 368,2 438,3 403,2 40,7 -4,6029 2123,8
С2Н2 Ацетилен 164,8 199,0 181,9 16,19 —4,6541 844,5
С2Н2С14 1,1,1,2-Тетрахлорэтан 343,2 438,3 390,7 34,63 —4,2921 1803,9
G2H2G14 1,1,2,2-Тетрахлорэтан 373,2 453,3 413,2 38,3 -4,5665 1998,2
С2Н3С13 1,1,2-1 рихл орметан 331,6 409,0 370,3 33,5 —4,3981 1744,8
С2Н4 Этилен 175,7 217,7 196,7 16,38 -4,382 854,7
С2Н4Вг2 1,2-Дибромэтан 324,8 388,1 356,4 34,5 -4,4413 1798,8
С2Н4С12 1,1-Дихлорэтан 331,6 378,2 354,9 27,6 -4,0541 1438,2
С2Н4С12 1,2-Дихлорэтан 294,2 372,9 333,5 29,3 —4,3376 1527,3
С2Н5Вг Бромистый этил 293,2 346,1 319,6 25,05 -4,2377 1307,7
С2Н5С1 Хлористый этцл 244,9 329,4 287,1 22,95 -4,2758 1194,9
c2h6f фтористый этил 203,0 244,5 223,7 19,00 -4,3835 989,9
с2н51 Йодистый этил 334,1 373,2 353,6 27,75 —4,1930 1444,4
С2Нв Этан 179,5 215,4 197,5 17,33 —4,3835 904,6
с2нво Диметиловый эфир 213,2 288,2 250,7 24,7 -4,8133 1287,4
с3нв Циклопропан 217,7 257,8 237,7 21,44 -4,567 1116,1
с3нв Пропен 244,9 294 7 269,8 21,66 -4,466 1130,7
СЗН7ВГ Бромистый н-пропил 325,5 373,2 349,3 30,45 —4,4381 1588,5
Сзн^вг Бромистый изопропил 295,2 368,2 331,7 28,3 —4,2489 1475,5
С3Н7С1 Хлористый к-пропил 295,2 373,2 334,2 28,15 -4,3938 1465,9
“ с3н71 Йодистый н-пропил 329,8 373,2 351,5 32,96 —4,3575 1717,2
388
Продолжение табл. П,3
Формула Название адсорбата Интервал температур, Т, К —ДЕЛ, кДж/ моль Д8* + В 2,303В -Д171
от ДО тсред 2,303В
C3H7I Йодистый изопропил 324,8 388,1 356,4 31,1 -4,2348 1620,6
С3н8 Пропан 218,9 269,2 244,0 23,0 -4,6355 1199,8
с4нв Бутадиен-1,3 295,7 356,0 325,8 28,5 —4,599 1484,6
с4н8 Бутен-1 295,7 358,2 326,9 26,4 —4,4648 1375,8
с4н8 1{ис-Бутен-2 295,5 358,2 326,8 26,76 —4,377 1393,6
с4н8 т рд«с-Бутен-2 295,5 358,2 326,8 27,82 —4,459 1450,1 -
C4HgO Этилвиниловый эфир 325,4 373,2 349,3 32,16 -4,7502 1675,7
С4Н9ВГ Бромистый «-бутил 336,6 383,0 359,8 35,75 —4,5830 1862,2
С4Н9ВГ Бромистый втор-бутил 331,6 408,4 370,0 33,25 -4,3452 1732,0
С4Н9ВГ Бромистый изобутил 338,2 398,1 368,2 33,28 -4,3813 1736,1
С4Н9С1 Хлористый «-бутил 325,6 373,2 349,4 33,82 —4,6091 1762,7
С4Н9С1 Хлористый втор-бутил 331,6 393,5 362,6 31,2 -4,3303 1624,9
С4Н9С1 Хлористый втор-изобутил 294,2 373,2 333,7 27,2 -4,1104 1418,5
c4h9f Фтористый «-бутил 294,2 373,2 333,7 29,8 —4,8360 1552,3
С4Н91 Йодистый «-бутил 362,8 428,6 395,7 38,3 —4,5423 1997,5
С4Н91 Йодистый изобутил 358,1 423,6 390,8 36,0 -4,3612 1878,5
С4Н10 «-Бутан 295,7 356,0 ' 325,8 27,5 -4,5136 1433,4
С4Н10 Изобутан 295,7 356,0 325,8 26,4 —4,5586 1378,0
С4Н10О Диэтиловый эфир 325,4 423,2 374,3 31,64 -4,6707 1645,7
свн8 Пентин-1 328,4 383,2 355,8 29,5 —4,3987 1538,5
свн8 ц «с-Пентадиен-1,3 329,2 373,2 351,2 37,63 —4,9224 1963,4
Свн8 тра «с-Пент адиен-1,3 329,2 373,2 351,2 36,45 —4,9184 1898,3
сБн8 Пентадиен-1,4 295,0 382,9 339,0 28,9 -4,314 1509,4
Свн1о Пентен-1 328,2 383,0 355,6 31,35 -4,5678 1631,4
Свн1о ^«с-пентен-2 325,3 363,9 344,6 31,85 -4,6248 1657,8
Свн1о трд«с-Пентен-2 325,3 363,9 344,6 32,8 —4,7068 1710,8
Свн1о Циклопентан 295,5 358,2 326,8 26,93 -4,217 1400,9
СБНцВг Бромистый «-пентил 373,2 438,4 405,8 41,0 -4,7036 2135,0
СБНцВг Бромистый втор-пентил 358,1 438,4 398,2 39,0 -4,642 2030,6
СвНцВг Бромистый изопентил 353,3 433,5 393,4 38,2 -4,5086 1990,0
СВНПС1 Хлористый изопентил 353,3 433,5 393,4 36,2 -4,5034 1887,0
С5Н12 «-Пентан 329,2 373,2 351,2 33,5 -4,7476 1742,5
Свн1г Изопентан 325,2 373,2 349,2 30,8 -4,548 1604,7
tc c6H12 Неопентан 294,7 363,9 329,3 26,0 “4,190 1353,9
CeH6Br Бррмбензол 413,0 463,4 438,2 45,0 -4,7445 2341,1
CO 9» CeH6Cl Хлорбензол 403,1 458,2 430,6 43,8 “4,8627 2287,0
55 Ю CeH6F Фторбензол 358,0 428 4 393,2 38,1 “4,7663 1987,2
CO CeH6I Иодбензол 413,4 483,8 448,6 47,3 “4,6979 2464,4
CeHe Бензол 343,2 438,2 390,7 36,2 “4,690 1888,9
oo ceH8 Циклогексадиен-1,3 343,2 413,2 378,2 34,0 “4,580 1773,6
СвНю Гексин-1 348,2 423,2 385,7 35,0 “4,599 1827,3
СвНю Гексадиен-1,5 328,3 403,2 365,8 34,6 “4,5345 1805,1
СвНю Циклогексен 343,2 433,2 388,2 33,3 “4,5075 1734,8
CeHi0O Ди-н-аллиловый эфир 373,2 437,9 405,5 41,7 “5,0692 2174,2
CeHia Метилциклопентан 343,2 433,2 388,2 31,3 “4,3045 1632,7
CflHi2 Циклогексан 343,2 433,2 388,2 28,6 “4,0155 1489,6
CeHiaBr Бромистый «-гексил 403,1 463,4 433,2 46,3 “4,8705 2412,2
CflHigBr Бромистый ятпор-гексил 373,2 438,4 405,9 45,4 “4,9407 2364,0
CeHi3Cl Хлористый н-гексил 373,2 453,0 413,1 44,6 —4,9431 2328,8
CgHigl Йодистый н-гексил 373,2 488,0 430,6 48,9 “4,8637 2547,2
CeHu н-Гексан 348,2 453,2 400,7 38,4 —4,816 2003,5
CeHuO Ди-н-пропиловый эфир 373,2 437,2 405,2 41,6 “4,9913 2168,7
С8Н14О Диизопропиловый эфир 393,2 458,2 425,7 37,0 —4,6792 1928,7
C7H9 Толуол 388,2 476,2 432,2 44,4 “5,042 2319,7
C7H14 Метилциклогексан 363,2 463,2 413,2 35,8 —4,407 1862*6
C7Hie н-Гептан 423,2 473,2 448,2 44,0 —4,995 AW** f V 2292 3
GgHio Этилбензол 383,2 476,2 429,7 47,9 —5,175 2498 4
C8H10 Метаксилол 393,3 492,4 442,8 53,7 —5,523 2795,8
СвНю Орто-, нара-ксилолы 393,2 492,4 442,8 53,8 —5,523 2803 1
GeHia 2-Метиленбицикло[2,2,1]гептан 350,2 403,3 376,7 33,65 —4,1894 1756,2
C8H13 ан£о-5-Метилбицикло[2,2,1]гептен-2 353,2 403,3 378,2 32,50 —4,1296 1693Д
C8H12 аяао-5-Метилбицикло[2,2,1]гептен-2 353,2 403,3 378,2 34,50 —4,3018 1799^3
C8HU ан5о-2-Метилбицикло[2,2,1]гептан 348,2 408,5 378,3 32,74 —4,0562 1704*6
c8H14 а«зо-2-Метилбицикл о[2,2,1 ]гептан 348,2 403,2 375,7 35,03 —4,3085 1829*5
C8H14 1 -Метилбицикл о [2,2,1 ]ге птан 353,2 408,2 380,7 32,02 —4,0063 1669,4
C8Hi8 н-Октан 413,2 486,2 449,7 49,4 —5,1655 2576,3
G8H18O Ди-н-бутиловый эфир * 423,2 498,2 460,7 53,35 —5,4330 2777,9
C8H18O Диизобутиловый эфир 423,2 488,4 455,8 50,25 —5,2633 2620*2
G9H12 1,2,3-Триметилбензол 453,2 496,2 474,7 66,05 —6,257 3442,8
C9H12 1,2,4-Триметилбензо л 453,2 496,2 474,7 . 65,6 —6,252 3422*8
co G9H12 1,2,5-Триметилбензол 453,2 496,2 474,7 65,0 —6,257 3385 4
05 c©> C9H12 «-Пропилбензол 398,7 497,7 448,2 53,1 -5,378 2767,8
Продолжение табл. П,3
Формула Название адсорбата Интервал температур, Т, К —ДЕЛ, кДж/моль Д8*4-Н 2,303Н —АЕ71
от ДО т сред 2,ЗОЗН
С9Н13 Изопропилбензол 398,7 497,7 448,2 47,8 —5,018 2490,2
С9н12 ан5о-5-Винилбицикло{2,2,1]гептен-2 353,2 408,2 380,7 35,42 "4,3362 1849,1
С9Н12 акао-5-Винилбицикл о [2,2,1 ] гептен-2 358,2 423,2 395,7 38,03 "4,3273 1982,6
С9Н12 тракс-5-Этилиденбицикло[2,2,1]гептен-2 363,2 423,2 393,2 37,3 "4,2548 1940,4
С9Н12 !{ис-5-Этилиденбицикло[2,2,1]гептен-2 363,2 423,2 393,2 39,23 "4,4184 2047,9
С9н1в ан5о-2-Этилбицикло[2,2,1]гептан 363,2 393,2 378,2 39,36 "4,4064 2054,3
С9н1в екэо-2-Этилбицикло[2,2,1]гептан 363,2 423,2 393,2 41,2 -4,5838 2147,8
С9Н16 а«5о-1,2-Диметилбицнкло[2,2,1]гептан 353,2 408,2 380,7 36,55 "4,2463 1903,2
с9н16 акао-1,2-Диметилбицикло[2,2,1]гептан 353,2 403,2 378,2 39,00 -4,4832 2033,3
СдНгв зндо-дндо-2,3-Диметилбицпкло [2,2,1 ]гептан 353,2 423,2 395,7 39,03 —4,4988 2038,5
с9н1в 8кзо-8кзо-2,3-Диметил бицикл о [2,2,1]гептан 353,2 423,2 395,7 40,55 —4,5449 2114,3
С9н1в 1,4-Диметилбицикло[2,2,2]гептан 373,2 423,2 395,7 37,94 -4,3412 1978,3
С9Н1в транс-2,3-Диметилбицикло[2,2,1]гептан 363,2 453,2 408,2 38,65 -4,3881 2013,1
С9Н16 7,7-Диметилбицикло[2,2,1]гептан 353,2 408,2 380,7 35,6 -4,1565 1852,5
с9н30 н-Нонан 453,2 498,2 475,7 55,6 -5 4435 2894,4
СюН8 Нафталин 468,2 528,0 498,1 64,6 -5,8232 3364,9
С10Щ4 н-Бутилбензол 453,2 498,2 475,7 57,0 —5,4365 2972,6
СюН14 1,2,3,5-Тетр аметил бензо л 473,0 528,0 500,5 73,1 —6,491 3811,2
СюН14 1,2,4,5-Тетраметилбензол 473,2 528,0 500,5 71,5 -6,325 3722,2
C10H16 Адамантан 373,2 453,3 413,3 35,05 —3,9115 1826,9
QloH16 ан5о-Триметиленбицикло[2,2,1]гептан 383,4 458,4 420,9 39,35 -4,2286 2046,6
С19н16 2,2-Д иметил-3-мети л енбицик л о [ 2,2,1 ]гептан 368,3 452,9 410,6 38,55 —4,2454 2006,5
C10H16 акзо-2,2,З^Триметилбицикло[2,2,1]гептан 353,2 423,2 388,2 38,6 —4,3593 2009,6
C10H18 вндо-2,2,3-Триметилбицик ло[2,2,1] гептан 353,2 423,2 388,2 40,9 -4,6507 2132,3
^10^22 н- Декан 468,2 523,3 495,7 60,2 -5,499 3138,4
СюЩгО Ди-н-амиловый эфир 446,0 502,7 474,3 66,2 —6,0723 3455,1
С10Н22О Диизоамиловый эфир 452,4 502,2 477,3 60,9 -5,7056 3167,0
QtiHie н-Амилбензол 448,6 493,3 470,9 63,9 -5,911 3329,1
СцН16 Пентаметилбензол 498,2 573,2 535,2 83,2 —7,127 4337,1
СцНг4 н-Ундекан 448,6 497,9 473,2 65,9 -5,7047 3437,3
С12Н10 Дифенил 473,0 543,0 508,0 70,2 -6,0131 3665,2
С12Н18 Гексаметилбензол 523,3 594,0 558,6 89,6 -7,2744 4676,3
С12Н26 н-Додекан 473,0 543,0 508,0 71,8 —5,912 3741,7
С14Щ0 Фенантрен 573,2 643,5 608,3 94,5 —6,941 4932,6
С14Н10 Антрацен 573,2 643,5 608,3 94,5 -6,838 4933,5
ЛИТЕРА ТУРА
1. Brunauer S., Emmett P. H., Teller E. J. Am. Chem. Soc.,
1938, v. 60, p. 309—319.
2. R о s s S., Winkler W. J. Co]. Sci., 1955, v. 10, p. 319—329.
3. Аристов Б. Г., К и с e л e в А. В. Коллоидн. ж., 1967, т. 29, с. 631 —
637.
4. R oss S., W 1 п к 1 е г W. J. Col. Sci., 1955, v. 10, р. 330—337.
5. Б и б и Р. А. и др. ЖФХ, 1964, т. 38, с. 708—718.
6. К и с е л е в А. В. и др. Коллоидн. ж., 1958, т. 20, с. 444—455.
7. А в г у л ь Н. Н., Киселев А. В., Лыгина И. А. Изв. АН СССР,
ОХН, 1961, с. 1395.
8. А в г у л ь Н. Н. и др. Изв. АН СССР, ОХН, 1956, с. 1304—1311.
9. А в г у л ь Н. Н., Киселев А. В., Л ы г и н а И. А. Изв. АН СССР,
ОХН, 1962, с. 32-37.
10. Б е з у с А. Г., Д р е в и н г В. П., Киселев А. В. ЖФХ, 1964, т. 38,
с. 59—67; 947—954, 2924—2930.
11. Ноогу S. Е., Prausnitz J.M. Trans. Faraday Soc., 1967, v. 63,
p. 455—460.
12. Авгуль H. H. и др. Изв. АН СССР, ОХН, 1957, с. 1021—1031.
13. А в г у л ь Н. Н. и др. Изв. АН СССР, ОХН, 1962, с. 769—777.
14. Авгуль Н. Н. и др. Изв. АН СССР, ОХН, 1959, с. 787—796.
15. К и с е л е в А. В., С е р д о б о в М. В. ЖФХ, 1963, т. 37, с. 2589—2592.
16. А в г у л ь Н. Н., Киселев А. В., Лыгина И. А. Изв. АН СССР,
ОХН, 1961, с. 2116—2125.
17. А в г у л ь Н. Н., Киселев А. В., Лыгина И. А. Изв. АН СССР,
ОХН, 1961, с. 1404—1411.
18. Machin W. D., Ross S. Proc. Roy. Soc. (London). 1962, v. A 265,
p. 455-462.
19. D a v i s B. W., Pierce C. J. Phys. Chem., 1966, v. 70, p. 1051—1058.
20. Sams J. R., Constabaris G., Halsey G. D. J. Phys. Chem.,
1962, v. 66, p. 2154—2158.
21. Киселев А. В., X у д я к о в В. Л., Яшин Я. И. ЖФХ, 1973, т. 47,
с. 2125-2126.
22. К а 1 a s h n i к о v а Е. V. е. a. Chromatographia, 1971, v. 4, р. 495—500.
23. К а 1 a s h n i к о v а Е. V. е. a. Chromatographia, 1972, v. 5, р. 278—285.
24. 3 апрометов А. Ю. и др. ЖФХ, 1972, т. 46. с. 1230—1233.
25. Kalashnikova Е. V., Kiselev А. V., Shcherbakova K.D.
Chromatographia, 1974, v. 7, р. 22—25.
СПИСОК НЕКОТОРЫХ ОБОЗНАЧЕНИЙ
А — площадь поверхности твердого тела
а — активность однокомпонентного объемного газа в состо-
янии равновесия с адсорбированным веществом
а3 — активность адсорбированного вещества
as° (Т) — активность адсорбированного вещества цри Г О
as — константа притяжения уравнения состояния Ван-дер-
Ваальса для двухмерного газа
В — предэкспоненциальный множитель в выражении для по-
тенциальной энергии отталкивания
В% — второй вириальный коэффициент объемного газа
В 2D — второй вириальный коэффициент двухмерного адсорби-
рованного газа
bs — константа отталкивания уравнения состояния Ван-дер-
Ваальса для двухмерного газа
С — теплоемкость
Cs — поверхностная теплоемкость, отнесенная к единице пло-
щади разделяющей поверхности
С^ т — мольная теплоемкость объемного газа при постоянном
объеме, находящегося в равновесии с адсорбированным
веществом
С3^ т — мольная теплоемкость объемного газа при постоянном
объеме в стандартном состоянии
АС — изменение теплоемкости адсорбционной системы (или ад-
сорбата) при адсорбции газа
— средняя мольная теплоемкость адсорбированного вещества
АС — дифференциальное мольное изменение теплоемкости адсор-
бата при переходе из объема газа в адсорбированное со-
___ стояние
ACeq — дифференциальное мольное изменение теплоемкости адсор-
____ бата при адсорбции в равновесных условиях
АС$ — дифференциальное мольное изменение поверхностной тепло-
емкости
Cs Са — дифференциальная мольная теплоемкость адсорбирован-
_____ ного вещества
ACi — величина АС при Г -> О
С т х — величина Ст при Г -> О
СцС2, С$, . . . Ci — коэффициенты экспоненциального уравнения изотермы ад-
сорбции в вириальной форме
С, С± — константа диполь-дипольного дисперсионного взаимодей-
ствия
С 2 — константа диполь-квадрупольного дисперсионного вза-
имодействия
С\ — значение константы ди'поль-дипольного дисперсионного
притяжения С^ оцененное с помощью квантовомехани-
ческой формулы
с — равновесная концентрация адсорбата в газовой фазе
с* — стандартная (начальная) концентрация адсорбата в газовой
фазе
372
cq — концентрация насыщенного пара
d — межплоскостное расстояние в решетке графита
Е — кинетическая энергия молекулы
F — свободная энергия системы
F5 — свободная поверхностная энергия
F5 — свободная поверхностная энергия, отнесенная к единице
площади поверхности раздела твердое тело — газ
F$ — свободная поверхностная энергия ча единице площади
поверхности раздела твердое тело — вакуум
— свободная энергия газа в стандартном состоянии
F^l ° — мольная свободная энергия газа в стандартном состоянии
A Fs — изменение свободной поверхностной энергии при адсорбции
на единице поверхности раздела
Fs — дифференциальная мольная свободная поверхностная энер-
__ гия
AFS — дифференциальное мольное изменение поверхностной
свободной энергии
AF — изменение свободной энергии при адсорбции газа
AF — дифференциальное изменение свободной энергии при ад-
сорбции газа
\Feq — дифференциальное мольное изменение свободной энергии
адсорбата при адсорбции в равновесных условиях
АЛ9 — стандартное дифференциальное мольное изменение сво-
бодной энергии при адсорбции газа
Д/'р с г, &F* — стандартное дифференциальное мольное изменение сво-
бодной энергии при переходе адсорбата из состояния газа
при малой концентрации с° в адсорбированное состояние-
при малой (нулевой) величине адсорбции Г° в случае, когда
_ Г7с° = 1
Д/’р р t “ стандартное дифференциальное мольное изменение сво-
бодной энергии при переходе адсорбата из состояния газа
при малом давлении р°- в адсорбированное состояние при
малой (нулевой) величине адсорбции Г9 в случае, когда
Г°/р9 = 1
Нт, ° — мольная энтальпия газообразного адсорбата в стандартном
состоянии
Кр с г, Кг — константа Генри, выраженная через Гис
кл5 с г — константа Генри для всего адсорбента, выраженная через-
гиббсовскую адсорбцию ns на всем адсорбенте и мольную»
концентрацию с
Ку р г — константа Генри, выраженная через Г и давление газа р
KnS р г — константа Генри, выраженная через ns и р
К'ъ i — обратная величина константы Генри ]_ для г-го одно-
родного участка неоднородной поверхности
K'ljtn— обратная величина константы Генри для адсорбции на
неоднородной поверхности, соответствующая максимуму
кривой распределения участков поверхности по величинам
энергии адсорбции
М — молекулярный вес; мольная масса (масса 1 моль)
т — масса адсорбента
тп — число геометрических параметров системы
т — масса молекулы
— статистическое среднее число молекул в газе, не взаимо-
действующем с твердым телом
373;
— статистическое среднее число молекул к~го компонента
в объемном газе
TVfo — число адсорбированных молекул /с-го компонента согласно
определению Гиббса
ns — гиббсовский избыток числа молей адсорбированного ве-
щества
пё — число молей адсорбата в газовой фазе в равновесии с ад-
сорбентом
п — число атомов в молекуле
п — константа потенциальной энергии отталкивания
р — давление объемного газа вдали от поверхности, находя-
щегося в равновесии с адсорбированным веществом
р° — давление объемного газа в его стандартном состоянии
р0 — давление насыщенного пара
Qn — каноническая статистическая сумма для N молекул газа,
взаимодействующего с твердым телом
— каноническая статистическая сумма для N молекул газа,
вдали от поверхности твердого тела (обычного объемного
газа)
Qi — статистическая сумма одной молекулы, взаимодействующей
с твердым телом
Qi — статистическая сумма одной молекулы в объемном газе
(?колеб — статистическая сумма колебательного движения молекулы
в объеме газа
Фгарм. осц. квант — статистическая сумма для квантуемых степеней ^свободы
адсорбированной молекулы в квантовом приближении
<?гарм. осц. клас — статистическая сумма для квантуемых степеней свободы
адсорбированной молекулы в классическом приближении
qv j — интегральная теплота адсорбции, измеренная в калориметре
при постоянных Т, V и А
qv — дифференциальная мольная теплота адсорбции, измеренная
в калориметре при постоянных Т, V и А
q у 1 — величина qv при Г -> О
qst — изостерическая теплота адсорбции
q — обобщенные координаты
qi — статистическая сумма для г-го поворотного изомера моле-
кулы, взаимодействующего с твердым телом
— статистическая сумма для г-го поворотного изомера моле-
кулы в объеме газа
q — константа в экспоненте выражения для потенциальной
энергии отталкивания
R — универсальная газовая постоянная
г — расстояние между взаимодействующими силовыми цен-
трами (атомами, атомными группами)
го — равновесное расстояние для взаимодействия двух силовых
центров
г* — эффективный ван-дер-ваальсовый радиус силового центра
(атома)
S — энтропия системы
SS — интегральная энтропия объемного газа, не взаимодейству-
ющего с твердым телом
Ss — поверхностная энтропия
— поверхностная энтропия, отнесенная к единице площади
разделяющей поверхности
— дифференциальная мольная поверхностная энтропия
— средняя мольная энтропия объемного газа.
374
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоадсорбция 112
Адсорбент(ы)
ионные 61 сл.
неспецифические, первого типа 22
- носитель, адсорбционное моди-
фицирование поверхности
76
очистка поверхности 97
пути улучшения однородности
поверхности 73 сл.
я близкой к однородной поверх-
ностью получение 40 сл.
с нанесенными на поверхность
модифицирующими солями
76 сл.
специфические 22, 23
— второго типа 67
— третьего типа 67, 75
удаление воды с поверхности 94
химическое модифицирование по-
верхности 81
Адсорбционная система
геометрические параметры 209
термодинамические условия рав-
новесия 105 сл.
Адсорбционное равновесие, исполь-
зование результатов иссле-
дования 103
Адсорбционное разделение сложных
изотопнозайещенных моле-
кул, статистические выра-
жения для термодинамиче-
ских характеристик 354 сл.
Адсорбционный процесс в системе
с постоянным объемом, схе-
ма установки 133
Адсорбция
величина 106
водорода и углеводородов, влия-
ние замещения атомов во-
дорода на атомы дейтерия
на 353 сл.
газа, поступающего из резер-
вуара с постоянным давле-
нием 132
— — — — — — объемом 134
гиббсовая, определение 98
Адсорбция
дифференциальная теплота, за-
висимость от величины ад-
сорбции 27, 29
зависимость величины от концен-
трации газа и температуры
155 сл.
изостерическая теплота 222
локализованная 153
молекулярно-статистическая тео-
рия 152
мономолекулярная нелокализо-
ванная, уравнение Хил-
ла — де-Бура 167
на базисной грани полубеско-
нечного кристалла графита,
физико-химические кон-
станты 54
на графитированной термической
саже, значения площади,
приходящейся на молекулу
в плотном мономолекуляр-
ном слое при 365
на однородной поверхности, по-
лучение термодинамических
характеристик из экспери-
ментальных данных для ре-
альной поверхности 165 сл.
— — — термодинамическое
описание с помощью неко-
торых моделей 152 сл.
на слоях молекул 78
нелокализованная 153
основные задачи термодинамики
103 сл.
пара, поступающего из резер-
вуара с чистой жидкостью
135
различных молекул, сопоставле-
ние 202 сл.
роль моделей в термодинамиче-
ском описании 152
статические весовые методы из-
мерения 95
твердыми телами, основные на-
правления исследования
34 сл.
378
Адсорбция
тепловой эффект, необходимость
калориметрических изме-
рений 140
термодинамические характеристи-
ки
— — азотсодержащих произ-
водных углеводородов 201
— — алкадиенов 189, 339
— — н-алканов 185, 310,
315—318, 320, 324, 326
— — алкенов 189, 334, 335
— — алкинов 192, 337
— — ароматических углево-
дородов 192, 194, 341
— — вклад квантовостати-
стического изотопного эф-
фекта 361
— — галогенпропзводных
углеводородов 194 сл.
— — дейтерозамещенных
органич. соединений 200
— — изоалканов 186, 328
— — квазижесткой молекулы
234
— — квантовые поправки
237
— — молекул с поворотной
изомерией 232
— — молекулярно-статисти-
ческий расчет 305 сл.
— — на графитированной
термической саже 180 сл.
— — неорганических газов
183
— — одноатомных молекул
при нулевом заполнении
математически однородной
поверхности 237
— — однокомпонентных га-
зов и паров 93 сл.
— — оценка для благород-
ных газов 290
— — получение из экспери-
ментальных данных 180 сл.
— — простых эфиров 198
— — расчет 298
— — — для одноатомныхдио-
лекул 284 сл.
серусодержащих про-
изводных углеводородов
201
— — сопоставление 203
— — спиртов 199
— — статистические вириаль-
ные выражения 207 сл.
— — статистические выра-
жения 227 сл.
— — терпенов 199
Адсорбция
термодинамические характеристи-
ки
— — Н- и D-углеводородов
200
— — углеводородов, моле-
кулярно-статистический ра-
счет изменений при заме-
щении атомов Н на ато-
мы D 358 сл.
— — углеводородов с сопря-
женными связями 341
— — цикланов 186, 329
— — цикленов 189
— — циклодиенов 189
— — элементорганических со-
единений подгруппы IV Б
201
характер межмолекулярных взаи-
модействий 21 сл.
Активность адсорбированного веще-
ства 106, 107
Алканы, зависимость адсорбцион-
ных свойств от геометриче-
ского строения молекулы
329
Атом-атомные потенциальные функ-
ции межмолекулярного вза-
имодействия 298, 313
зависимость от валентного со-
стояния взаимодей ству-
ющих атомов 346
оценка параметров 308
сопоставление 345 сл.
Атом-атомный потенциал межмоле-
кулярного взаимодействия
влияние приближений при сум-
мировании на значения кон-
станты Генри 286 сл.
— формы на значение константы
Генри 284 сл.
выбор формы 255
оценка параметров 260
представление в виде суммы сла-
гаемых, выражающих вкла-
ды различных видов вза-
имодействия 255
суммирование 268
уточнение параметров 311
форма 258
Большой канонический ансамбль 208
Величина адсорбции по Гиббсу 210,
216
Внутренняя энергия
дифференциальная мольная ад-
сорбированных молекул 221
интегральная адсорбированных
молекул 221
системы 209
379*
Графит
изотопный эффект при адсорбции
углеводородов на 353 сл.
молекулярно-статистический рас-
чет термодинамических ха-
рактеристик адсорбции угле-
водородов на 305 сл.
оценка термодинамических ха-
рактеристик адсорбции бла-
городных газов на базисной
грани 290
.расчет термодинамических ха-
рактеристик адсорбции одно-
атомных молекул 284 сл..
расщепленный 40 сл.
структура 41
Диамагнитные восприимчивости ато-
мов в молекулах алканов
309, 325
Дифференциальная теплота адсорб-
ции
н-алканов 69
зависимости от величины адсорб-
ции 100
— — заполнения поверхности 52
— — поляризуемости молекул
адсорбата 73
— — температуры обработки
канальной сажи 50
— — числа атомов углерода
в молекуле 68
этана, зависимость от величины
адсорбции цеолитами 69
этилена, зависимость от адсорб-
ции цеолитами 69
Зависимость межмолекулярного вза-
- имодействия атомов угле-
рода от их валентного со-
стояния 332
Зета-функция Римана 269, 275, 306
Изменение внутренней энергии 147
адсорбции алканов 311, 319, 327
— — бензола 110
— — н-гексана 110
дифференциальное 124, 127
— для производных бицикло
[2,2,1 ]гептана 188
— зависимость от поляризуемос-
ти молекул адсорбатов 184
— мольное 139
— — зависимость от величины
адсорбции 163
— — предельная величина 125
— определение 183
— расчет 370
зависимость от молекулярного
веса металлорганических
соединений 202
— — величины адсорбции 114,
118, 164, 197
Изменение внутренней энергии
дифференциальное
— — поляризуемости молекул
204
— — температуры 131
— — числа атомов, углерода в мо-
лекуле адсорбата 193, 195,
198
интегральное 124, 134, 138
при адсорбции 221, 224, 323
— — благородных газов 293/ 296
— — галогенпроизводных угле-
водородов, зависимость от
величины ковалентного ра-
диуса галогена 195
— изотопных молекул 357
расчет 305
сопоставление величин, получен-
ных разнЬгми методами 145
Изменение свободной энергии 133,
135, 136
адсорбата дифференциальное
мольное 120
адсорбированного вещества, сред-
нее мольное 117
дифференциальное 116 сл.
— зависимость от величины ад-
сорбции 118
— мольное 137
— стандартное мольное 119
зависимость от величины адсорб-
ции 114
интегральное 116 сл.
Изменение теплоемкости
адсорбата, дифференциальное
мольное 130, 223, 306
дифференциальное 127, 129, 140
для адсорбции алканов 314, 321,
327
интегральное 127, 134, 139
при адсорбции 223, 224
— — изотопных молекул 357
расчет 305
Изменение энтропии
адсорбата, дифференциальное
мольное 124
для адсорбции алканов 314, 321,
327
дифференциальное 121 сл.
— мольное 138
— — зависимость от величины
адсорбции 123
— — расчет 306
— определение 183
интегральное 121 сл., 134, 137
при адсорбции 219, 224
— — изотопных молекул 357
расчет 305
среднее мольное, зависимость от
величины адсорбции 123
380
Изменение энтропии
стандартное дифференциальное
мольное 123
Изобары адсорбции хлоропрена 198
Изостера адсорбции 123, 126
Изостерическая теплота адсорбции
222, 306
Изотермы адсорбции 25, 27, 28, 29
аргона 49, 50, 56, 65, 77, 154,
162, 168, 173
бензола 45, 112, 168
бутана 168
воды 45
газохроматографическое опреде-
ление 97
w-гексана 45, 72
двуокиси углерода 168
диэтилового эфира 168
изобутана 168
криптона 48, 60, 63, 66, 71, 74,168
ксенона 41, 42, 70, 162, 168’
метана 168
метанола 44, 45
многокомпонентного газа 214
модельные 172
мономолекулярной, идеального
газа уравнение 156
неопентана 168
однокомпонентного газа 210
определение 242
перфторметана 58, 59
получение вакуумными статиче-
скими методами 96
пропана 168
статические измерения 93 сл.
уравнение 105, 108, 164, 224
— Вильямса — Генри 156
— вириальное 155
фторхлорметана 168
хлороформа 168
четыреххлористого углерода 168
шестифтористой серы 60, 168
этана 62, 96, 112, 168
этилена 168
Изотерма гиббсовской величины ад-
сорбции 210
Изотопный эффект
квантовомеханический 353
квантовостатистический 354
— вклад в различие термодина-
мических характеристик
адсорбции дейтеро-и обыч-
ных углеводородов 361
при адсорбции углеводородов на
графите 353 сл.
Калориметр Кальве 100
Квазижесткие молекулы 234
Квантовомеханические теории вза-
имодействия молекулы с
поверхностью 248
Квантовые поправки 237
Кинетическая энергия молекулы 228,
229
Константа
адсорбционного равновесия 103,
107
Генри 98, 108 сл., 155, 212
— влияние приближений при
суммировании атом-атом-
ных потенциалов на значе-
ние 286 сл.
— — формы атом-атомного по-
тенциала межмолекуляр-
ного взаимодействия на
значение 248 сл.
— газохроматографическое опре-
деление 97
— для адсорбции алканов 310,
319, 325, 327, 328
— — — алкинов 336
— — — ароматических углево-
дородов 342
— — — бензола 110
— — — н-гексана 110
— — — диметилбицикло [2,2,1 ]
гептанов 187
— — — линейной молекулы 235,
236
— — — одноатомной молекулы
235, 236, 290
— — — трехмерной молекулы
235, 236
— — — углеводородов с со-
пряженными связями 340
— зависимость логарифма от
обратной температуры
366 сл., 370
— — — температуры 111, 126,
181
— — — — прокаливания тер-
мической сажи 173, 174
— значения для адсорбции бла-
городных газов 292, 295
— квантовые поправки 237
— нежестких молекул 229
— общие выражения 227 сл.
— определение 153, 170, 181, 183,
242
— при адсорбции углеводородов,
зависимость от числа ато-
мов углерода 190
— расчет 298, 305, 306
— статистич еские выражения
227 сл.
— стереоизомеров элементо-
органических соединений,
зависимость от геометриче-
ского строения 201
диполь-квадрупольного дисперси-
онного взаимодействия 264
38
Константа
Генри
равновесия 105
Ci 156 сл. 161, 260
С2 156 сл., 161, 264
сил отталкивания 265, 267
Коэффициент
активности адсорбата, зависи-
мость от величины адсорб-
ции 163
— адсорбированного вещества
103, 107, 111
— — — зависимость от величи-
ны адсорбции 112
разделения изотопов 354
— — вклад квантовостатисти-
ческого эффекта 356
Кристаллические окислы, ад-
сорбция на 70 сл.
Лед, адсорбция на 70 сл.
Межмолекулярные силы и потен-
циальная функция меж-
молекулярного взаимодей-
ствия молекулы с поверх-
ностью 241 сл.
Мембрана(ы) 73 сл.
углеродная 75
Металлы, адсорбция на 56 сл.
Метод(ы)
большого канонического ансам-
бля 208, 214
Брунауэра, Эмметта и Теллера
(БЭТ) 182
весовой, измерения адсорбции 96
Вутена и Броуна 183
газовой хроматографии 97
калориметрические 99
капиллярной жидкостной микро-
бюретки 95
молекулярно-статистический 343
— — определения удельной по-
верхности графитирован-
ной сажи 299
Росса и Оливье 174
статический 94—96
учета неоднородности реальной
поверхности 166
электрических изображений 248
Модели (ль) состояния адсорбиро-
ванных квазижестких моле-
кул 235
Друде 251
Молекулярно-ситовые угли 73 сл.
Молекулярные сита 23
Нитрид бора, адсорбция на 55 сл.
«Область Генри» 155
Обменная энергия 256, 257
382
Обобщенные координаты системы
227
Отношение Cs/Ci 264
Параметр Сд, определение при ис-
пользовании эксперимен-
тальных значений констан-
ты Генри 297
сил отталкивания 291, 295
Поверхностная
внутренняя энергия 116
свободная энергия 114
теплоемкость 116
энтропия 116
Поверхностное давление 113, 163
зависимость от величины адсорб-
ции 114
— — площади 164
Поверхностные термодинамические
функции, изменения при
адсорбции 114 сл.
Поверхность
геометрически однородная 13, 14,
20
кристаллов, структура 16
математически или энергетически
однородная 13, 14
методы оценки степени однород-
ности 24
причины неоднородности 15
физически неоднородная 20
— однородная 13, 14
химически неоднородная 20
— однородная 13, 20
экспериментальные термодина-
мические критерии одно-
родности 21 сл.
Поляризуемости атомов в молекулах
алканов 309, 325
Пористые полимеры 75
Потенциал
Бакингема 258, 259, 284
Бакингема — Корнера 258, 259,
269, 284, 290, 309, 324
Леннард-Джонса 258, 259, 269,
284, 286
межмолекулярного взаимодей-
ствия, аддитивность по
потенциалам взаимодей-
ствия пар силовых центров
249
“ — атомов углерода, параметры
333
межмолекулярных сил, определе-
ние 242
отталкивания, параметры 264
Сюзерленда 284
Потенциальная функция
межмолекулярного взаимодей-
ствия 326
Потенциал
— — атома углерода молекулы
ароматического углеводо-
рода с атомом углерода
графита 341
— — атом-атомная 298, 313
— — молекул углеводородов с
базисной гранью графита,
расчет 305, 307
— — молекулы с поверхностью
241 сл.
— — пути определения 241
параметры, оценка 246
Потенциальная энергия
ван-дер-ваальсового взаимодей-
ствия молекулы с твердым
телом 248
взаимодействия двух молекул
251
— молекул н-алканов с базисной
гранью графита 323
межмолекулярного взаимодей-
ствия молекулы с твердым
телом, расчет 268, 269
притяжения двух молекул 255
Потенциальные кривые
межмолекулярного взаимодей-
ствия атомов водорода и
углерода 348
— — — углерода 347
Правила сумм 260
Приближение
гармоническое 289
Крауэлла 284, 286, 306
Лондона 287
Питцера и Гвина 237, 356
Равновесное расстояние при меж-
молекулярном взаимодей-
ствии двух силовых цен-
тров 266
Решеточные суммы, расчет 268
Сажа
ацетиленовая 41
гидрофилизация поверхности 44
гидрофобизация поверхности 45
графитированная 40 сл.
— молекулярно-статистический
метод определения удель-
ной поверхности 299
— термическая, значения пло-
щади, приходящейся на мо-
лекулу при адсорбции 365
— — сопоставление адсорбции
различных молекул 202 сл.
— — термодинамические харак-
теристики адсорбции 180 сл.
характеристики 46
диаметры частиц 42
Сажа
графитированная
—। термическая
зависимость g-фактора от удель-
ной поверхности 47
изменение адсорбционных свой-
ств 48
— химического состава 44
канальная 41, 43
ламповая 41
неграфитированная, си гнал
ЭПР 44
печная 41
термическая 41, 43
удельная поверхность 42
форсуночная 41
Свободная энергия, полный диф-
ференциал 208
Свободное движение вдоль матема-
тически однородной по-
верхности 235
Соли с комплексными анионами 67 сл.
с кубической решеткой 61 сл.
со слоистой решеткой 64 сл.
Степенные суммы
значения 272, 273, 274
расчет 272
Стереоизомеры, разделение и иден-
тификация 191
Сумма Слетера 237, 356
Теорема Хельмана — Феймана 250
Теория Лэнгмюра 32
Теплоемкость
адсорбированного вещества, диф-
ференциальная мольная
129
— — зависимость от величины
адсорбции 130, 131
— — — — заполнения поверх-
ности адсорбента 164
— — средняя мольная 128
адсорбционных систем, калори-
метрические методы опре-
деления 99
дифференциальная мольная по-
верхностная 129
зависимость от заполнения по-
верхности 153
средняя мольная, зависимость от
величины адсорбции 101
Теплота адсорбции 140 сл.
адсорбционных систем, использо-
вание результатов калори-
метрических исследований
104
газов и паров 141
дифференциальная 146
зависимость от заполнения по-
верхности 153
383
Теплота адсорбции
изостерическая 144
калориметрические методы опре-
деления 99
на графитированной термической
саже 53
пара, чистая 144
погрешности определения 99
Теплота испарения жидкости, моль-
ная скрытая 139
Термодинамические функции
выбор для характеристики ад-
сорбционных процессов 148
изменения 116 с л., 132 сл.
Термодинамические характеристики
адсорбционных систем, особен-
ности определения из экс-
периментальных данных
93 сл.
взаимодействия адсорбат — адсор-
бент 120
Термодинамический потенциал си-
стемы 208, 209
Углеродные волокна 73 сл.
Удерживаемый объем 98, 109, 217
на графитированной термической
саже 53
расчет 306
Уравнение
Генри 155
Дубинина 31
Закура — Тетроде 170
изотермы адсорбции 31 сл., 105,
108, 164, 224
— — Вильямса — Генри 156
— — вириальное 155
— мономолекулярной адсорбции
идеального газа 156
Уравнение
мономолекулярной нелокализо-
ванной адсорбции Хил-
ла — де-Бура 167
состояния адсорбированного ве-
щества 111
— — однокомпонентного газа
213 сл.
Фрейндлиха 31
Формула
Кирквуда — Мюллера 262, 312,
325, 338, 359
Лондона 262
Салема 262
Слэтера — Кирквуда 262
Функция
Гаусса 169
Пуассона 169
Хемосорбция 102
Химический потенциал
адсорбата, стандартное измене-
ние 120
адсорбированного вещества 106,218
изменение 224
X роматогр афия
газоадсорбционная 191
импульсная элюционная 97
фронтальная 97
Энергия взаимодействия, диспер-
сионная 251, 252,' 257
Энтропийный фактор, расчет 170
Энтропия
вращательная адсорбции 322
дифференциальная мольная ад-
сорбированных молекул 220
испарения жидкости, мольная 138
поступательная адсорбции н-ал-
канов 321
Эффект нулевых энергий 354
АВГУЛЬ НАТАЛЬЯ НИКОЛАЕВНА,
КИСЕЛЕВ АНДРЕЙ ВЛАДИМИРОВИЧ,
ПОШКУС ДИОНИЗАС ПРАНЦИШКАУС
АДСОРБЦИЯ ГАЗОВ И ПАРОВ
НА ОДНОРОДНЫХ ПОВЕРХНОСТЯХ
Редактор В. И. Щеголева
Технический редактор В. М, С кит ина
Художник В. М. К азинцев
Корректор Т. Е. И ль тер якова
Т-21156. Сдано в наб. 5/VIII 1974 г. Подл, в печ. 31/XII 1974 г.
Формат бумаги 60 X ЭО1/^. Бумага тип. № 2. Усл. печ. л. 24. Уч.-изд, л. 27,05.
Тираж 4600 экз. Зак. .№ 1180. Изд. № 435. Цена 2 р. 87 к.
Издательство «Химия», 107076, Москва, Стромынка, 13.
Ленинградская типография № 6 Союзполиграфпрома при Государственном комитете
Совета Министров СССР по делам издательств, полиграфии и книжной торговли.
196006. г. Ленинград, Московский пр., 91.