/











































































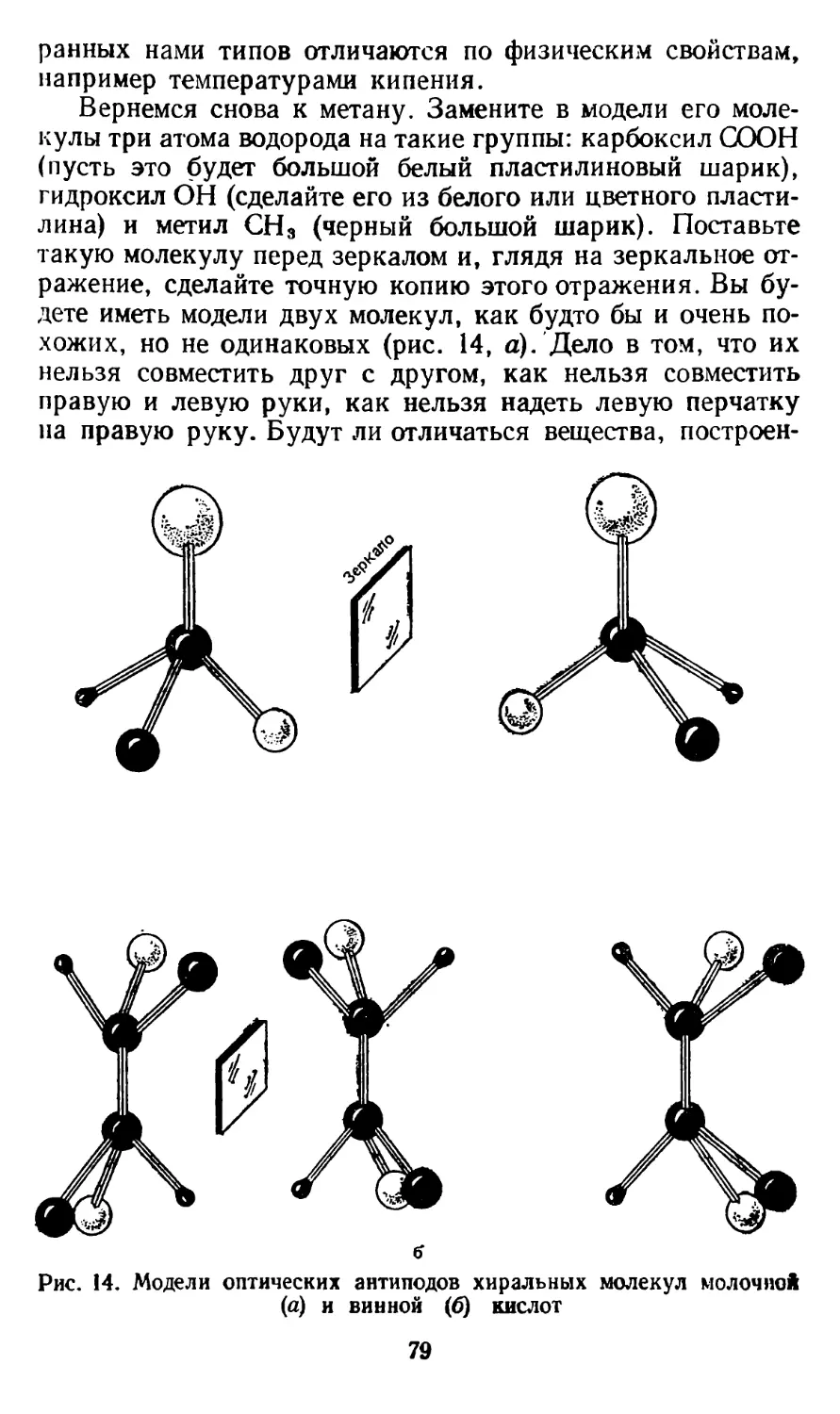


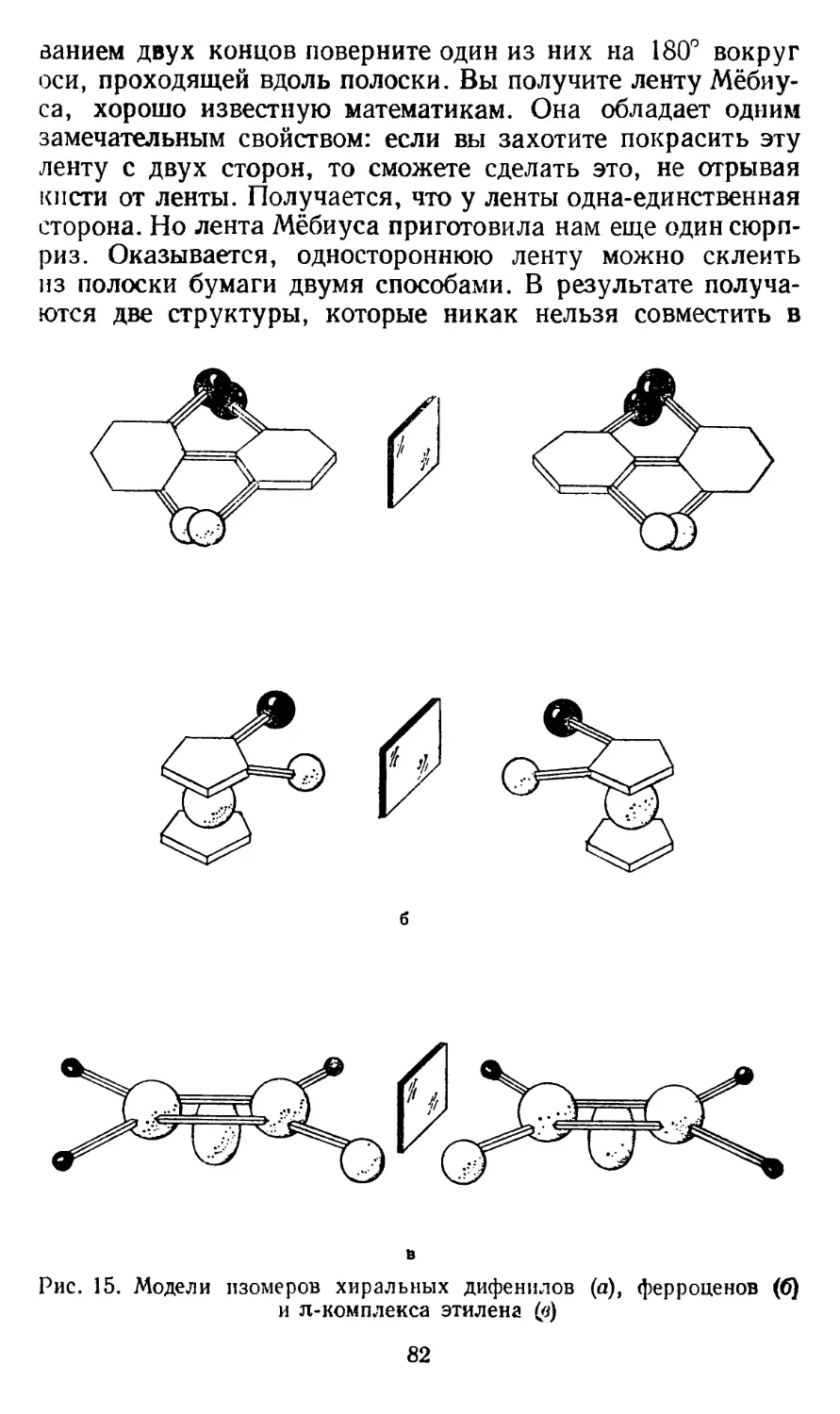































































Текст
ХИМИЯ&СЕХ
народный университет
естественнонаучный факультет
ИЗДАЕТСЯ С 1961 ГОДА
гв-шулышн
.кандидат химических наук
Основные понятия и простейшие опыты
ИЗДАТЕЛЬСТВО -ЗНАНИЕ»
МОСКВА 1987
ББК 24
Ш95
Автор: ШУЛЬПИН Георгий Борисович — кандидат химических наук, старший научный сотрудник Института химической физики АН СССР. Автор более 80 научных работ по вопросам металлоорганической химии, металлокомплексного катализа, активации насыщенных углеводородов. Им опубликованы монография «Химия комплексов со связями металл — углерод» (совместно с С. П. Губиным. Новосибирск, Наука, 1984), научно-популярные книги «Эта увлекательная химия» (М., Химия, 1984), «Мир необычных молекул: металлоорганические комплексы» (М., Наука, 1986), а также многочисленные статьи в журналах «Наука и жизнь», «Химия и жизнь», «Природа», «Химия в школе».
Рецензент: Хайрутдинов Р. Ф.— доктор химических наук.
Шульпин Г. Б.
Ш95 Химия для всех (Основные понятия и простейшие опыты).— М.: Знание, 1987.— 144 с.— (Нар. ун-т. Естественнонаучный фак.).
50 к. 80 000 экз.
Книга в популярной форме рассказывает о различных понятиях, методах химической науки. Так, например, читатель получит представление о полимерных материалах и квантовой химии, узнает о витаминах и о методах качественного и количественного анализа, познакомится с химией душистых веществ и катализом... Изложение основных понятий химии сопровождается описанием несложных опытов, большинство из которых читатель сможет провести дома, используя вещества и посуду, которые имеются в любом хозяйстве или продаются в аптеке и в магазинах.
Адресована самому широкому кругу читателей, интересующихся современной химией, слушателям народных университетов, старшеклассникам.
... 1801000000-005
Ш 073 (02)-87 24-87
ББК 24
Эта книга рассказывает об одной из важнейших наук — о современной химии. Начало же развития химии как науки можно отнести к середине XVIII в., когда был установлен закон сохранения массы вещества, когда рухнула теория, в основе которой лежало представление о несуществующем флогистоне. Следующие важнейшие на пути развития химии шаги были сделаны уже в XIX столетии. Это — введение представления об атомах, открытие Периодического закона элементов, создание теории строения органических соединений. За два века химия прошла огромный путь и превратилась в хорошо развитую науку, обладающую колоссальным запасом информации и основывающуюся на глубоких и четких теоретических посылках. В последние десятилетия стало очевидным, что целый ряд новых наук не может обойтись без привлечения понятий и методов химии. Здесь в первую очередь надо упомянуть медицину, молекулярную биологию.
Книга Г Б. Шульпина построена в виде отдельных глав-очерков. Каждая глава рассказывает о какой-то области химической науки, о ключевом понятии, об играющем важную роль веществе. Перефразируя пословицу, можно сказать, что для более глубокого понимания сути какого-либо научного понятия лучше поставить один опыт, чем сто раз об этом понятии прочитать. Характерная особенность предлагаемой вниманию читателя книги заключается в том, что рассказ об основах современной химии сопровождается описанием простейших опытов. Эти опыты легко провести в домашней химической лаборатории, а необходимые «реактивы» и оборудование читатель сможет найти в своем домашнем хозяйстве либо купит в аптеке, магазине хозяйственных или фотографических товаров. В качестве химической посуды автор чаще всего предлагает использовать обычные стаканы. Лишь несколько более сложных экспериментов потребуют особого оборудования и относительно редких химических реактивов. Эти опыты следует проводить в школьной лаборатории или в химиче
ском кружке. Повседневная деятельность ученого-химика включает в себя не только манипуляции с веществами, с колбами и пробирками. Полученные экспериментальные данные являются лишь основой для сложных подчас математических расчетов, после которых только и могут быть сделаны глубокие выводы. Наконец, современная химия имеет солидную теорию, основанную на положениях квантовой механики. Автор знакомит читателя с некоторыми принципами обработки экспериментальных данных, позволяющими в результате предсказывать свойства еще неполученных или неисследованных соединений. Проведя несложные расчеты, читатель сможет «убедиться» в том, что s-орбиталь имеет форму сферы, а р-орбиталь похожа на гантель. А изготовив из пластилина и проволоки модели орбиталей молекул, он поймет, почему возможны одни реакции и запрещены другие. Таким образом, главная задача автора — сделать активным усвоение материала об областях и проблемах химии, приобщить читателя к творчеству.
Книга не предназначена для чтения в автобусе или в постели перед сном. Читателю, принимающемуся за чтение, надлежит запастись некоторыми легкодоступными веществами, посудой. Впрочем, опыты можно и не делать, а лишь прочитать о них. Но вот карандаш и листок бумаги обязательно понадобятся: чтобы понять многие рассуждения, вам необходимо будет построить графики, нарисовать формулы, провести несложные математические преобразования.
Конечно, далеко не все разделы химической науки нашли отражение в книге. Одна из причин этого — небольшой объем издания. Другая заключается в том, что рассказ о некоторых областях химии было невозможно сопроводить описанием простых опытов. Например, радиационная химия связана с опасным для жизни излучением, а любые металлоорганические соединения — вещества малодоступные в быту. Тем не менее книга рассказывает о большинстве ветвей современной химии. Здесь органическая и неорганическая, физическая и коллоидная, биологическая и аналитическая химия, стереохимия. Специальные главы посвящены отдельным типам веществ — белкам, углеводам, красителям, лекарствам.
Автор этой книги, кандидат химических наук Г. Б. Шульпин, работает в Институте химической физики АН СССР, занимается химией органических, металлокомплексных и
металлоорганических соединений, гомогенным катализом, и многие темы, описываемые в книге, знакомы ему не только по литературе. В качестве примеров некоторых типов химических реакций автор берет самые последние достижения химии, предлагает опыты, описанные в оригинальных научных журналах последних лет.
Книга будет полезна всем, кто желает познакомиться с основными понятиями химической науки, и адресована в первую очередь слушателям народных университетов, преподавателям и школьникам старших классов.
Член-кор респондент АН СССР А. Е. ШИЛОВ
Что изучает современная химия? На первый взгляд может показаться, что ответить на этот вопрос очень легко. Действительно, химия — это наука о веществах и их взаимопревращениях. Однако попробуем проанализировать такое определение. Во-первых, о том, какие бывают вещества. Вот, например, гидроксид алюминия и основной карбонат меди. Эти вещества весьма интересны геологам, поскольку являются основой минералов боксита и малахита соответственно. Из первого минерала получают алюминий, второй — прекрасный поделочный камень, А такие вещества, как пенициллин или гемоглобин, интересуют, помимо химиков, также и медиков, и биологов. Далее: какие могут быть превращения веществ? Лед превращается в воду, гелий I переходит в сверхтекучий гелий II. Эти превращения веществ ни в коей мере не интересуют химика, Они — предмет забот физика, ведь не зря температуру плавления вещества относят к физическим свойствам.
А что же такое химические свойства? Это способность вещества вступать в реакцию с каким-то другим веществом. Химическая же реакция — это процесс превращения одних молекул в другие. При таких превращениях разрушаются только молекулы (которые составлены из атомов); сами же атомы не изменяются. Дело в том, что процессы взаимопревращений атомов исследуются уже не химией, а физикой, точнее, атомной и ядерной физикой. А как мы уже отмечали, превращения веществ, в которых не происходит разрушения ни атомов, ни молекул, также изучаются физикой. Вот и выходит, что физика как бы окружает химию с двух сторон— и «снизу» (атомный уровень), и «сверху» (надмолекулярный уровень).
Тут нужно сделать два замечания. Сами вещества в нереагирующем состоянии, их строение, структура все меньше и меньше интересуют современных химиков. Конечно, еще остались нерешенные вопросы в этой области, однако их решение входит уже в круг интересов физиков. И второе: существует немало пограничных между физикой и химией разделов, когда химические процессы изучаются с помощью физики, и наоборот — когда физическими, в общем-то,
явлениями занимаются химики. Например, на химический процесс, на элементарный акт взаимодействия двух или нескольких молекул можно посмотреть глазами физика. Вот такой раздел науки, занимающийся физикой химического превращения, называется химической физикой. С другой стороны, физические свойства сгустков многих молекул в растворе, называемых коллоидами, входят в компетенцию коллоидной химии, являющейся разделом физической химии.
И химическая физика, и физическая химия изучают свойства буквально всех типов веществ. Классификация разделов этих наук основывается или на методах стимулирования химических реакций (например, электрохимия, фотохимия, радиационная химия) или на способах изучения соединений и процессов (например, магнитная и оптическая спектроскопия, кинетические методы и т. д.).
Но существует и другая классификация разделов химии, которая принимает в расчет типы веществ, изучаемых данной областью. Все вещества делят на неорганические и органические. Органические соединения — это различные производные углеводородов, они обязательно содержат углерод. Неорганические соединения — это вещества, молекулы которых содержат любые другие элементы в любых сочетаниях. Поскольку атомы углерода обладают уникальной способностью соединяться в цепи, кольца и разные другие фигуры, и так, что одна молекула может состоять из сотни углеродных атомов, не удивительно, что веществ, содержащих углерод, известно во много раз больше, чем соединений неорганических. Органические соединения интересны тем, что они составляют основу живых организмов. Изучением веществ и процессов, происходящих в организмах, занимается биохимия. А в последние десятилетия возникла еще наука биоорганическая химия, которая смотрит на все реакции, протекающие в клетке, глазами химика-органика. В живом организме важнейшие функции выполняют ионы различных металлов, которые, связываясь с органическими молекулами, образуют биологические катализаторы (ферменты), переносчик кислорода — гемоглобин и другие нужные вещества. Исследованием таких соединений занимается очень молодая наука, именуемая био-неорганической химией. Известны и другие пограничные области химии, связывающие ее с биологией, с медициной, с сельским хозяйством,— это фармацевтическая, токсикологическая химия, агрохимия. Необходимо упомянуть еще
об одной очень важной области науки — о химии высокомолекулярных соединений, полимеров. Молекулы этих соединений — как органических, так и неорганических — составлены из большого и неопределенного числа одинаковых звеньев.
Мы очень кратко рассказали об одной из философских проблем химии — о предмете этой науки, о взаимоотношении химии с физикой. Надо заметить, что ученые до сих пор не пришли к общему мнению о том, что же считать предметом химии. Более подробно о философских вопросах современной химической науки можно прочитать, например, в книгах и статьях [1] (см. список литературы в конце книги).
Мы перечислили некоторые (далеко не все) разделы, области современной химии. О различных отраслях химии, о наиболее важных понятиях, веществах, методах этой науки — наш дальнейший рассказ.
Известно великое множество химических реакций, которые различаются и тем, какие вещества взаимодействуют, и в какой фазе протекает реакция (в газовой, в растворе или на поверхности раздела двух фаз), и тем, поглощается или выделяется тепло из реакционной массы. Но есть одно весьма важное свойство, которое делает одну реакцию непохожей на другую.
...Стрелок нажал на спусковой крючок, порох в патроне воспламенился, образовавшиеся газы вытолкнули пулю из ружья. Химическая реакция взаимодействия компонентов пороха протекла за считанные доли секунды. А вот другая реакция — превращения древесины, стволов умерших деревьев в каменный уголь. Этот химический процесс требует для своего завершения миллионов лет. Итак, реакции различаются скоростями. Но что такое скорость химического процесса?
Наверное, проще всего определить, что такое скорость движущегося тела. Это — частное от деления пути, пройденного телом, на время, за которое тело преодолело дан-
пый путь. Разумеется, при этом делении мы получили среднюю скорость, с которой тело двигалось весь отрезок пути. Для того чтобы определить скорость тела в данный очень короткий момент времени, нужно отрезок времени, за который измеряется скорость, делать все меньше и меньше, г. е. устремить его к нулю. В таком случае скорость будет математически выражаться производной от пути по времени.
Теперь представим себе такой случай: бригада каменщиков строит из кирпичей дом. Как определить скорость строительства? Один из вариантов — измерить скорость «исчезновения» кирпичей, припасенных для кладки. Разделив общее число кирпичей на время всего строительства, получим среднюю скорость. Но реальная скорость строительства постоянно менялась: сначала она была большой, потом рабочим пришлось носить кирпичи на более высокие этажи, и темп стал замедляться. Поэтому, чтобы вычислить скорость кладки в данный момент времени, нужно подсчитать количество кирпичей чуть-чуть раньше этого момента и чуть-чуть позже, разделить это количество на интервал времени от одного измерения до другого и затем интервал времени устремить к нулю.
Примерно так же поступают и химики, определяя скорость реакции. Только вместо кирпичей они имеют дело с молекулами, вступающими в реакцию. Однако пересчитывать молекулы в каждый момент времени — дело не очень удобное. Поэтому ученые измеряют концентрацию веществ, т. е. какое-то очень большое число молекул вещества в единице объема. Для того чтобы определить скорость реакции, химики берут концентрацию исходного вещества в какой-то начальный момент времени (пусть она равна сх) и затем измеряют концентрацию в момент (она оказывается равной с2). Разность концентраций с2—сх=кс делят на интервал времени t2—1Г=\1 и устремляют этот интервал к нулю. Скорость реакции W равна
U7 = — предел (при Дt 0) = —
Выражение dc/dt означает производную от концентрации по времени, а знак «минус» стоит перед выражением, чтобы сделать скорость величиной положительной (ведь с2 меньше сх).
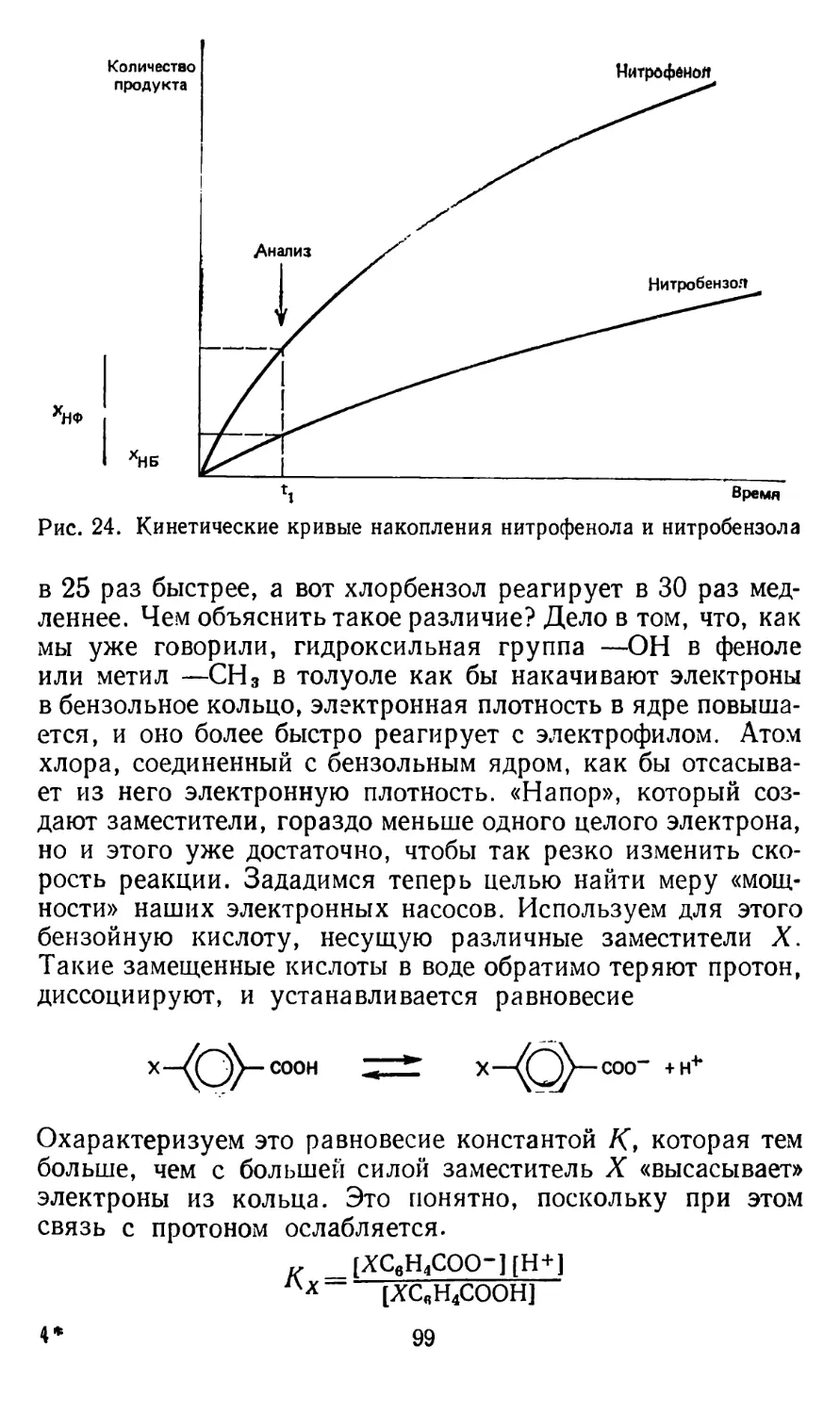
От чего зависит скорость реакции? Давайте проведем эксперимент. Известно, что при взаимодействии тиосульфата
натрия с уксусной кислотой происходит выделение коллоидной серы, которая выпадает из раствора в виде белого опалесцирующего осадка [2]. Уравнение реакции выглядит так:
Na2S2O3 + 2СН3СООН = Н2О + SO21 + S| + 2CH3COONa.
Растворите в стакане воды комнатной температуры 2 чайные ложки тиосульфата натрия (он называется еще гипосульфитом и широко применяется в фотографии в качестве закрепителя; его можно купить в любом магазине фототоваров). Теперь возьмите 4 стакана и отмерьте следующие количества нашего раствора: в первый стакан 2 чайные ложки, во второй — 4 ложки, в третий — 8 ложек и в четвертый стакан 16 ложек. Долейте в первые три стакана воды до объема, равного объему раствора в четвертом стакане. Вы получили 4 раствора разных концентраций. Концентрация во втором стакане в 2 раза больше, чем в первом, в третьем — в 4 раза больше, чем в первом, и в 2 раза больше, чем во втором. Быстро прилейте к первому раствору чайную ложку уксусной эссенции и заметьте время, которое пройдет с момента смешения растворов до момента появления мути. Только обязательно перемешивайте раствор чайной ложкой. Предположим, до появления белой мути прошло 90 с. Прибавьте последовательно по чайной ложке уксусной эссенции к остальным растворам и замерьте время появления мути. Предположим, оно оказалось для второго стакана 40 с, для третьего — 22 с и для четвертого — 12 с. Теперь на странице из тетради в клетку или на листке миллиметровой бумаги постройте график зависимости времени реакции от концентрации тиосульфата. По оси абсцисс отложите концентрацию (ее можно выразить в ложках начального раствора), а по оси ординат—время реакции до появления мути ts в секундах. Вы увидите, что кривая, проведенная через четыре точки, имеет вид гиперболы. Гипербола же — графическое выражение обратно пропорциональной зависимости. Поэтому если вы отложите по оси ординат не время а обратную ему величину, то экспериментальные точки лягут на прямую, проходящую через начало координат. Но ведь величина, обратная времени ts, пропорциональна скорости реакции в начальный момент UZ~l//y. Значит, скорость реакции тем больше, чем выше концентрация исходного вещества. В общем случае скорость реакции определяется таким уравнением:
Здесь k — коэффициент пропорциональности, не зависящий от концентраций реагентов и именуемый константой скорости реакции, сА, св — концентрации вступающих в реакцию веществ А и В, а и b — показатели степени при с, являющиеся одновременно коэффициентами, которые показывают, сколько молекул А или В вступают в реакцию.
Как видим, скорость реакции уменьшается по мере расходования реагентов. Вспомните наших каменщиков, которые по мере возведения стен строят их все медленнее. Впрочем, эта аналогия не очень глубокая. Более точно можно промоделировать обсуждаемое нами явление... игрой в биллиард. В первую очередь предположим, что химическая реакция происходит между двумя молекулами только тогда, когда молекулы столкнутся. Далее, представим себе, что мы имеем два типа молекул, которые изображаются белыми и черными шарами. Молекулы находятся в постоянном тепловом движении — будем катать шары по биллиардному столу в любой последовательности и в любых направлениях. Когда белый и черный шар столкнутся, снимем их со стола — реакция прошла, молекулы превратились в молекулы продуктов. А теперь представьте себе, что на столе находится 100 белых и 100 черных шаров. Разумеется, они будут часто сталкиваться, быстро удаляться со стола. Иная ситуация сложится, когда на столе останутся один белый и один черный шар: придется долго погонять их, прежде чем они столкнутся. Ясно, почему реакция идет очень медленно, когда концентрации реагентов малы.
Игра в биллиард подсказывает ответ и на такой вопрос: почему в уравнение для скорости реакции входит произведение концентраций, а к примеру, не их сумма. Мы уже говорили, что, для того чтобы прошла реакция, две реагирующие частицы должны столкнуться, т. е. одновременно оказаться водной точке пространства. Вероятность того, что в данной точке окажется молекула данного реагента, пропорциональна его концентрации. А вероятность того, что в данной точке окажутся сразу две молекулы, равна произведению вероятностей встретить здесь молекулы каждого реагента, т. е. пропорциональна произведению концентраций.
* Посмотрим, какие еще факторы оказывают влияние на скорость реакции. Положите в стакан две чайные ложки тиосульфата натрия, добавьте полстакана воды. Полученный раствор разлейте по две чайные ложки в 4 стакана и
долейте в них воды, чтобы объем растворов равнялся половине стакана. Прилейте к первому раствору, температура которого 20°С (измерьте ее термометром для ванн), чайную ложку уксусной эссенции и отмерьте время, через которое появится муть (перемешивайте раствор постоянно). Предположим, это время равно 75 с. Нагрейте раствор во втором стакане до 30°С и измерьте время появления мути. Вы увидите, что это время сократилось примерно в 2 раза (в нашем случае до 35 с). При 40°С муть появляется через 20 с, при 50°С — через 10 с.
Попытаемся математически обработать полученные результаты. Если построить график, откладывая по осям температуру и время реакции, получим некую кривую, смысл которой понять трудно. Но вот оказывается, что если по оси абсцисс откладывать величину, обратную абсолютной температуре (эту величину найдите как 1/7\ где 7'=273+ +/°С, если 1°С — температура в градусах Цельсия), а по оси ординат — натуральный логарифм времени реакции, то экспериментальные точки улягутся на прямую. (Для вычисления логарифмов воспользуйтесь таблицами или микрокалькулятором.) Математически нашу прямую можно описать уравнением:
In ts=b/T+a.
В этом выражении а и b — некие постоянные. Уравнение можно переписать и так:
ts=aleb/T,
где е — основание натурального логарифма, аг=еа.
Время реакции ts связано с константой скорости реакции k обратной пропорциональностью. Учитывая это обстоятельство, нетрудно прийти к выражению для k:
k=Ae-b/T.
Здесь А — новая постоянная данной реакции.
Очевидно, что взаимодействие между молекулами осуществляется только при их столкновениях. Однако опыты показывают, что не каждое столкновение приводит к реакции. Взаимодействие происходит только в том случае, когда энергия столкнувшихся молекул не меньше определенной для каждой реакции величины. Обозначим эту величину Еа. Зная это значение и абсолютную температуру реакционной смеси Т, можно вычислить число молекул п, которые способны при столкновении вступить в химическое взаимо-
действие. Эта величина определяется через экспоненту: n=Ne~Ea^Tt
где ft — общее число молекул; R — универсальная газовая постоянная. Отсюда уже недалеко до знаменитого уравнения Аррениуса, связывающего константу скорости реакции с температурой:
k=Pze-Ea/*T,
Где z — число столкновений молекул, вступающих в реакцию за секунду, а фактор Р (который всегда меньше единицы) показывает, что к взаимодействию приводят только столкновения молекул удобными, выгодными сторонами. Мы легко можем перейти к уравнению для константы скорости реакции, выведенному на основании экспериментальных наблюдений, если примем Рг—А и Ea/R = b. Если измерить константу скорости реакции при нескольких температурах, то из уравнения Аррениуса можно вычислить величину Еа, называемую энергией активации реакции. Графически Еа вычисляют таким образом. Откладывают по оси абсцисс величину 1/Т, по оси ординат — значения логарифма константы скорости реакции. Тангенс угла наклона полученной прямой равен EJR. Заметим, что энергия активации — очень важная характеристика реакции. Чем меньше энергия активации, тем легче протекает реакция.
До сих пор мы говорили о реакциях, которые проходят до конца, т. е. в которых за какое-то время из веществ А \\ В получаются целиком и полностью вещества D и Е (хотя, конечно, теоретически для полного завершения реакции требуется очень много времени). Но известны и такие случаи, когда соединения D и Е могут реагировать между собой, образуя вещества А и В. Тут мы имеем дело с обратимыми реакциями. Общее уравнение таких реакций:
aA+bB+±dD+eE.
Вот мы слили растворы веществ А и В, началась реакция, скорость которой По мере протекания ре-
акции концентрации веществ А и В уменьшаются, значит, реакция замедляется. Но, с другой стороны, нарастают концентрации веществ D и Е, следовательно, убыстряется реакция между ними, скорость которой UZ_1=/s_iC,£)6,e- Наконец, система приходит в равновесие, которое характеризуется константой К (в этом случае Величина К
равна отношению констант скоростей прямой и обратной реакций:
. d e
rs kf CD* Ce
k-i ca'^b
Слейте в стакане два разбавленных раствора хлорного железа и роданистого аммония. Отлейте полученный кроваво-красный раствор в три других стакана и прибавьте в первый стакан насыщенный раствор хлорного железа, во второй — насыщенный раствор роданистого аммония, в третий — насыщенный раствор хлористого аммония. Вы увидите, что в первых двух стаканах окраска растворов усилится (сравните с окраской четвертого контрольного стакана), в третьем стакане цвет побледнеет. Чем можно объяснить наблюдаемые изменения? Вот уравнение реакции:
FeCl3+3NH4SCN^Fe (SCN)3+3NH4C1.
Константа равновесия реакции (формулы в квадратных скобках обозначают концентрации с):
/f_[Fe (SCN)3][NH4C1]3 [FeCl3] [NH4SCN]3 ’
Поскольку К — величина постоянная, увеличивая концентрацию FeCl3 (в первом стакане), мы автоматически сдвигаем равновесие вправо, т. е. увеличиваем и концентрацию Fe(SCN)3. А интенсивность окраски и определяется концентрацией роданида железа. При добавлении хлорида аммония равновесие смещается влево, концентрация окрашенного вещества падает. Мы еще вернемся к этой реакции, позволяющей определять наличие ионов железа в растворе и их количество. Пока же подведем некоторые итоги. В этой главке рассказано о разделе науки, занимающемся изучением скоростей химических реакций и называемом химической кинетикой. О кинетике можно прочитать в учебниках по физической химии, в научно-популярных книгах [3].
Сначала — эксперимент. Смешайте на дне стакана или в пробирке этиловый спирт и уксусную кислоту (эссенцию) и нагрейте в кастрюле с кипящей водой. Известно, что
спирт и органическая кислота способны образовать сложный эфир, обладающий своеобразным фруктовым запахом. Однако вы не почувствуете запаха этилацетата. Теперь добавьте к смеси двух жидкостей каплю концентрированной серной кислоты и повторите нагревание. Скоро вы сможете ощутить приятный запах сложного эфира. В чем же заключается роль серной кислоты?
Она является катализатором процесса образования сложного эфира. Процесс идет и без катализатора, но очень-очень медленно. В современной науке катализаторами называют вещества, которые резко ускоряют химические реакции, в их ходе многократно вступая во взаимодействие с реагентами и промежуточными соединениями, но выходя из реакции в первоначальном виде. Это очень сложное, интересное и весьма важное явление — катализ.
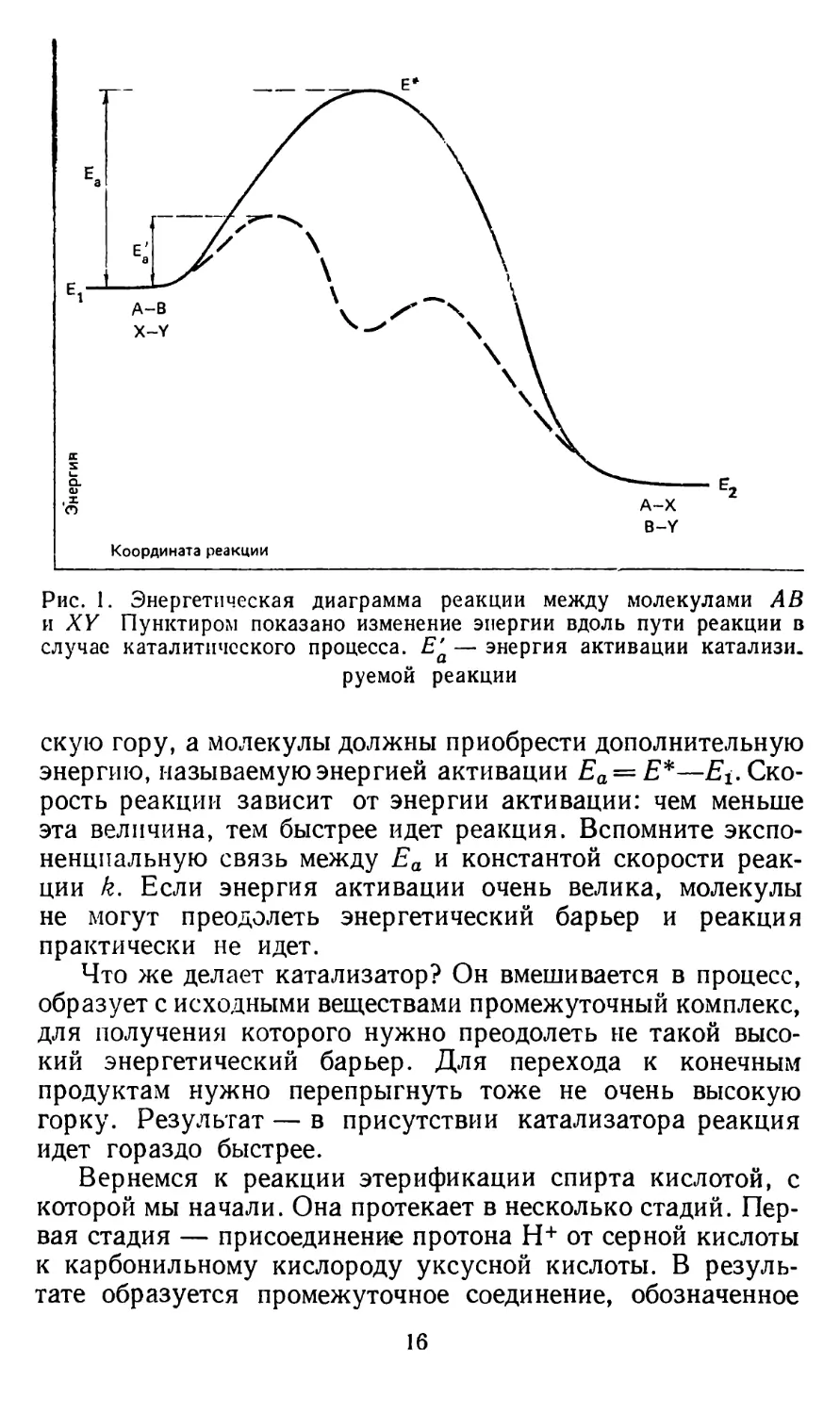
Как протекает химическая реакция? Рассмотрим такой пример: из вещества А—В (А и В — это части молекулы) и соединения X—Y при их взаимодействии образуются продукты А—X и В—Y Одним словом, молекулы как бы обмениваются частями. Построим график, в котором по оси абсцисс будем откладывать расстояния между атомами взаимодействующих молекул в каких-то условных единицах (это называется координатой реакции, ее путем). По оси ординат отложим энергию, которой обладают частицы. В левой части графика у нас отразится состояние нашей системы, когда расстояния А—В и X—Y малы, а расстояния А—X, В—Ху А—Y и В—Y велики (рис. 1). Это значит, что в системе присутствуют только молекулы А—В и X—Y Эти молекулы обладают запасом потенциальной энергии Ег. В правой части графика отразим состояние системы, состоящей из молекул А—X и В—Y Эти вещества характеризует энергия Е2. Пусть Е2 меньше Elt По этой причине состояние с Е2 выгоднее и вещества А—В и X—У, будучи смешанными, стремятся превратиться в А—X и B—Y
Пришло время сказать, что два плато с энергиями Ег и Е2 находятся по разные стороны от энергетического пика, характеризуемого энергией Е * Действительно, когда расстояния между атомами реагирующих молекул какие-то промежуточные, образуется так называемое переходное состояние, в котором одновременно связаны все четыре части Л, В, X и Y Вот этому переходному состоянию и соответствует максимальная энергия Е * Для того чтобы реакция прошла, система должна перевалить через энергетиче-
Рис. 1. Энергетическая диаграмма реакции между молекулами АВ и XY Пунктиром показано изменение энергии вдоль пути реакции в случае каталитического процесса. Е& — энергия активации катализи. руемой реакции
скую гору, а молекулы должны приобрести дополнительную энергию, называемую энергией активации Еа = Е*—^.Скорость реакции зависит от энергии активации: чем меньше эта величина, тем быстрее идет реакция. Вспомните экспоненциальную связь между Еа и константой скорости реакции k. Если энергия активации очень велика, молекулы не могут преодолеть энергетический барьер и реакция практически не идет.
Что же делает катализатор? Он вмешивается в процесс, образует с исходными веществами промежуточный комплекс, для получения которого нужно преодолеть не такой высокий энергетический барьер. Для перехода к конечным продуктам нужно перепрыгнуть тоже не очень высокую горку. Результат — в присутствии катализатора реакция идет гораздо быстрее.
Вернемся к реакции этерификации спирта кислотой, с которой мы начали. Она протекает в несколько стадий. Первая стадия — присоединение протона Н+ от серной кислоты к карбонильному кислороду уксусной кислоты. В результате образуется промежуточное соединение, обозначенное
ниже буквой А (все неустойчивые образования будем заключать в квадратные скобки). Соединение А присоединяет к центральному углеродному атому молекулу этилового спирта и переходит в вещество Б. Движущая сила такого превращения — взаимодействие положительного заряда на атоме углерода кислоты и свободной электронной пары на атоме кислорода спирта. Следующий этап — перескок протона на другой атом кислорода с образованием продукта В. После этого от В отщепляется молекула воды. Процесс заканчивается выбрасыванием протона от соединения Г и образованием молекулы этилацетата.
+н+ сн,-с-он 7 3 II о
+с2нБон
,СН3-С-ОН он-
.А
с2н5о(н)
СН3-С-ОН;
ЮН
Б
Как видим, протон, вступивший на первой стадии в водоворот химических превращений, на последнем этапе из него вернулся целым и невредимым. Поэтому-то мы можем говорить, что имеем дело с каталитической реакцией. Характерная особенность любой такой реакции: катализатора для ее осуществления требуется гораздо меньше, чем это следует из уравнения реакции, а точнее, из уравнения какой-либо стадии. Ведь протон, соединивший две молекулы кислоты и спирта, тут же принимается за сшивание другой пары молекул.
Различают два вида катализа. Если катализатор и реагирующие вещества участвуют в реакции, находясь в различных агрегатных состояниях (например, реакция газов, катализируемая твердым веществом), то говорят о гетерогенном катализе. Именно гетерогенный катализ участвует в весьма важных процессах превращения углеводородов нефти. В качестве катализаторов для таких реакций используют металлы или их окислы. Благодаря гетерогенным катализаторам удается в огромных масштабах из азота и водорода получать аммиак.
В качестве катализатора может выступать и жидкость. Смешайте на металлической пластинке порошки алюминия (он используется в качестве «серебряной» краски) и йода. Очень осторожно добавьте к смеси каплю воды, которая вызовет весьма бурную реакцию соединения алюминия и йода. Произойдет вспышка. Это пример катализируемого процесса образования из двух простых соединений более сложного. А теперь* проведите опыт по каталитическому разложению вещества. В пробирке к щепотке двуокиси марганца (этот черный порошок легко получить, смешав растворы перманганта калия, т. е. марганцовокислого калия, и хлорида двухвалентного марганца) прилейте осторожно раствор перекиси водорода. Происходит бурное разложение перекиси, выделяется кислород, который можно обнаружить, внеся в пробирку еле тлеющую лучинку — она загорится. L
Теперь проведем разложение перекиси водорода катализатором, который, как и реагент, находится в растворе (такой тип катализа называется гомогенным). Налейте в стакан светло-желтый раствор хромата калия в воде. Прибавьте к нему несколько капель перекиси водорода. Раствор становится фиолетовым (это окраска промежуточных в процессе соединений), и выделяются пузырьки кислорода. Реакция распада перекиси водорода ускоряется и ионами железа. Процесс это сложный, протекает в несколько стадий [3]. Первая стадия приводит к образованию очень реакционноспособного гидроксильного радикала, несущего один неспаренный электрон:
Fe2++H2O2->Fe3++OH~+OH*.
Этот радикал взаимодействует с другой молекулой перекиси, получается новый радикал:
ОН’+Н2О2->Н2О+НО2.
Радикал распадается на протон и анион-радикал кислорода:
НО2^Н++Ог-
Последний взаимодействует с ионом трехвалентного железа, восстанавливая его, а сам превращается в молекулу кислорода:
Fe3++O2-->Fe2++O2.
Сегодня ученые с большим интересом изучают весьма необычные каталитические процессы, в которых концентра-
Рис. 2. Кинетические кривые реакций: а — переход А в В при условии обратимости процесса; б — последовательное превращение А в В и В в С
ция одной из форм катализатора в ходе реакции то уменьшается, то увеличивается. Такие реакции стали известны сравнительно недавно. Впрочем, прежде чем начать разговор о столь необычных явлениях, нам будет полезно еще раз вернуться к самым, азам химической кинетики. Итак, химическая реакция превращения вещества А в вещество В. В начальный момент времени химик имеет чистое вещество Л, его 100%. Но вот он стал нагревать вещество, и оно начало переходить в продукт В. Если нарисовать график такого процесса, отложив по оси абсцисс время реакции, а по оси ординат — количество х (в процентах, но можно и концентрацию) вещества А и продукта В, то мы увидим две
кривые. Кривая, соответствующая содержанию Л, будет плавно убывать от 100% в начальный момент до 0% при бесконечно большом времени. Кривая для В будет как бы зеркальным отражением кривой Л — она будет возрастать от 0% при 1=0 до 100% при 1=оо. Количество вещества Л в любой момент времени t можно найти по формуле, включающей уже известную нам экспоненциальную зависимость:
xA=\00e~kt.
Здесь k — это константа скорости реакции.
Как мы уже говорили, реакция может идти и таким образом: по мере того как все больше молекул Л превращается в В, все интенсивнее начинает осуществляться обратный процесс — переход В в Л. В конце концов система через очень большое время приходит к равновесию. Например, если при таком равновесии в реакционной массе находится 75% вещества В и 25% вещества Л, кинетические кривые процесса имеют вид, показанный на рис. 2, а. Химики знают множество реакций, протекающих по более сложным механизмам. К примеру, вещество Л может переходить с какой-
то скоростью (определяемой константой &АВ) в вещество В, а вещество В, в свою очередь, превращается в соединение С (константа скорости kBC):
Проанализируем, как в этом случае будут выглядеть кинетические кривые накопления и расходования всех веществ. Для соединения А — все так же, как и в предыдущем случае. Ведь этому веществу «безразлично», что В затем переходит еще и в С. Но вот количество вещества В сначала возрастает, проходит через максимум и затем начинает падать (рис. 2, б). Почему так? Потому что, когда количество В мало, мала и скорость его превращения в С (ведь скорость тем больше, чем больше имеется данного вещества). Скорость превращения определяется наклоном кривой — чем больше в данный момент скорость, тем круче наклонена кривая относительно оси абсцисс. Далее. Естественно, пока мала убыль В, мало прибывает и вещества С. Затем положение меняется, скорость появления С начинает возрастать. Но так не может продолжаться бесконечно, ведь запас А и В ограничен. Значит, скорость образования С со временем начинает уменьшаться. В результате кинетическая кривая накопления С имеет вид буквы В. Нетрудно догадаться, что для каждого значения времени t сумма количеств всех веществ Л, В и С должна равняться 100%
Конечно, в разных химических реакциях, протекающих по схеме А—В—С, отдельные стадии могут иметь различающиеся значения констант kAB и kBC. В результате формы кривых содержания промежуточного вещества В для различных реакций будут отличаться. Но все они содержат лишь один-единственный максимум. А нельзя ли осуществить такую реакцию, чтобы максимумов концентрации какого-то вещества, образующегося на промежуточных стадиях реакции, было несколько, было много? Долгое время химики не знали таких реакций, больше того — многие думали, что их и вообще-то быть не может. Лишь в начале 50-х годов советский биохимик Б. П. Белоусов открыл первую реакцию, в ходе которой периодически и многократно изменяются концентрации промежуточных веществ. Некоторые из этих веществ являются катализаторами превращения исходного соединения в конечное.
Если растворить в воде (раствора должно быть 10 мл)
лимонную кислоту (2,0 г), сульфат церия (0,16 г), бромат калия (0,2 г) и серную кислоту (2 мл разбавленной в 3 раза водой концентрированной серной кислоты), то раствор время от времени (примерно через минуту-другую) меняет окраску от желтой до бесцветной [2, с. 60]. Дело тут в том, что периодически желтый сульфат четырехвалентного церия переходит в бесцветный ион трех валентного металла Се (III). Механизмы колебательных реакций были подробно исследованы советским физиком А. М. Жаботинским, и сегодня во всем мире такие процессы называются реакциями Белоусова — Жаботинского. Провести первую реакцию Белоусова — Жаботинского нетрудно даже в домашних условиях, но надо заметить, что бромат калия и особенно сульфат церия не принадлежат к общедоступным химическим реактивам. Проще осуществить другую реакцию такого типа, описанную недавно [4]. Из относительно труднодоступных реагентов здесь понадобится ацетат кобальта (другими словами, уксуснокислый кобальт) и бензальдегид, бензойный альдегид. Оба реактива можно найти в любом школьном кабинете химии.
Приготовьте несколько исходных растворов. Первый: растворите 0,3 г Со(ОСОСН3)2 -4Н2О в уксусной кислоте, разбавленной водой (к 40 мл ледяной, безводной кислоты прибавьте 10 мл воды, но можно взять 50 мл уксусной эссенции). Второй: растворите 0,03 г бромистого натрия (его можно заменить бромидом калия, продающимся в магазине фототоваров) в 10 мл 90-процентной уксусной кислоты (10 мл уксусной эссенции). Смешайте оба раствора в стакане и поместите стакан в кастрюлю с водой, которую нагревайте на электрической плигке до температуры около 70°С. Раствор надо непрерывно перемешивать. Эго можно делать рукой при помощи стеклянной палочки или даже чайной ложки, но лучше соорудить механическую мешалку, воспользовавшись для этого мотором от швейной машины или детской игрушки. Через раствор нужно пропускать ток воздуха. Для этого опустите в стакан стеклянную или резиновую трубку (можно купить в аптеке); воздух будет гнать через нее фен для сушки волос или пылесос. Теперь прилейте к вашему раствору 5 мл бензальдегида и, продолжая поддерживать температуру около 70°, интенсивно перемешивайте раствор и продувайте через него воздух. Цвет раствора через некоторое время (несколько секунд или минут) изменится от розового до черно-коричневого, но затем, по прошествии нескольких минут станет светло-розовым. За
тем окраска в очередной раз сменится на темно-бурую. Так может повторяться часами.
В ходе реакции происходит колебание концентрации иона трехвалентного кобальта — она то увеличивается (при этом раствор становится черным), то уменьшается за счет того, что Со (III) переходит в ион Со (II),— тогда раствор становится розовым. Кобальт в этой реакции выступает катализатором окисления бензальдегида кислородом воздуха. На первой стадии ион Со (II) окисляется кислородом до Со (III). Последний вступает во взаимодействие с бензальдегидом, в результате чего образуется снова Со (II) и бензоильный радикал:
Со(Ш)+СвН6СНО->Со(П)+СвН5СО+Н+.
Радикал реагирует с кислородом:
СвНБСО+О,->СсНБСОз
СвНБСОз+СвНБСНО->СсНБСО3Н+СвНБСО.
В результате получается надбензойная кислота СзН5СО3Н и новый радикал бензоила. Надбензойная кислота окисляет Со (II):
Со (Н)+СвН5СО3Н->Со (Ш)+СвНБСО2+ОН-.
Все эти процессы, комбинируясь во времени весьма сложным образом, и приводят к тому, что концентрация Со (III) и Со (II) постоян-io пульсирует.
О принципах катализа, о различных каталитических процессах рассказывают книги [3, 5].
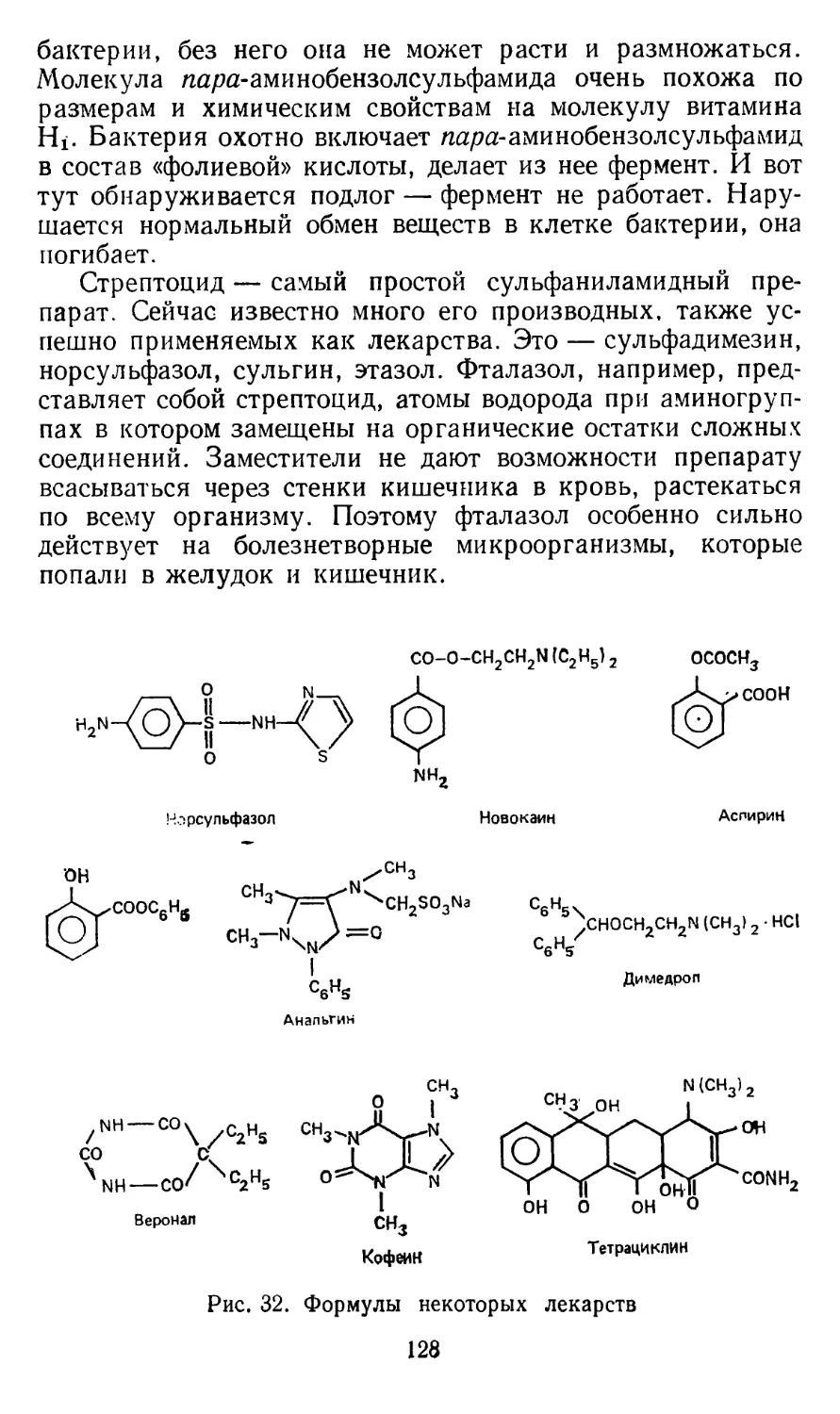
Часто по внешнему виду соединения совершенно невозможно определить, какие элементы в него входят. Но иногда бывает достаточно провести с веществом одну-две химические реакции, чтобы обнаружить присутствие в нем того или иного элемента. Методами качественного и количественного определения элементов в химических соединениях занимается особая наука — аналитическая химия. Конечно,
мы не сможем в домашней или школьной лаборатории провести все реакции, используемые химиком-аналитиком для установления состава вещества. Но некоторые химические превращения не требуют специального оборудования и труднодоступных реактивов. Давайте же посмотрим, по каким характерным реакциям узнают химики элементы [6, 7]. Будем читать «визитные карточки» элементов в соответствии с их «пропиской» в Периодической системе Д. И. Менделеева.
В первой ее группе располагается семейство щелочных металлов. Доказать химическим методом присутствие таких элементов в соединении — задача довольно трудная. Дело в том, что соединения щелочных металлов обычно не дают характерных реакций с окрашиванием растворов или выпадением осадков. Зато для определения этих элементов весьма удобны спектральные методы. Сделайте на конце нихромовой проволоки небольшую петельку. Смочите конец соляной кислотой и прокаливайте проволоку в пламени газовой горелки до тех пор, пока пламя не станет почти бесцветным. Теперь зачерпните петелькой несколько кристаллов поваренной соли и внесите проволоку в пламя. Яркое желтое окрашивание выдает присутствие натрия в соли.
Хорошо известно, что лучи света, окрашенные в какой-либо цвет, несут с собой определенную энергию. Например, энергия фиолетовых и синих лучей больше, чем энергия лучей красных или желтых. В солнечном спектре присутствуют лучи любой энергии, поэтому-то, если пропустить солнечный луч через стеклянную призму, он разложится в непрерывную радугу, в которой одни цвета непрерывно переходят в другие. Совсем иная картина получится, если пропустить через призму свет от пламени, окрашенного солью натрия. Вместо радуги в этом случае можно будет наблюдать лишь одну светящуюся желтую полоску, соответствующую лучам света с длиной волны 0,589 мкм. Дело в том, что под действием тепла пламени электрон в атоме натрия поднимается на более высокий энергетический уровень. Такое состояние, однако, атому невыгодно, электрон долго не задерживается на уровне с более высокой энергией и скоро возвращается на свое прежнее «место». Вот это «падение» электрона^ на более бедный энергией уровень, естественно, должно сопровождаться выделением энергии. Она выделяется в виде света, длина волны которого соответствует желтым лучам.
Спектры других щелочных металлов сложнее. Литий
окрашивает пламя в карминово-красный цвет; калий, рубидий и цезий — в различные оттенки лилового. Интересно заметить, что два последних элемента были впервые обнаружены именно спектральным методом и получили названия по характерным линиям в спектре (рубидус — темнокрасный, цезиус— небесно-голубой).
В одной группе со щелочными элементами располагаются такие не похожие на них медь, серебро и золото. Обнаружить эти металлы в химических соединениях не очень сложно. Соли двухвалентной меди обычно окрашены в синий цвет, при добавлении к ним нашатырного спирта образуются синефиолетовые аммиакаты. Можно провести и такую характерную реакцию. В пробирке к нескольким каплям раствора медного купороса добавьте 2—3 капли серной кислоты и несколько кристаллов тиосульфата натрия. При нагревании на пламени горелки выпадает бурый осадок сульфида одновалентной меди. Медь — металл неактивный и легко вытесняется из солей железом, цинком. Если в раствор соли меди опустить гвоздь, то на нем образуется красный налет вытесненной меди. Соли меди с галогенами окрашивают пламя в зеленый цвет. Серебро также дает характерные реакции — со щелочами соли серебра образуют бурый осадок окиси серебра, а с хлористым натрием — белый осадок хлорида серебра.
Доказать присутствие в веществе некоторых металлов второй группы — бериллия, магния, цинка, кадмия — не очень просто. Эти элементы не дают цветных солей, не окрашивают пламя. Зато кальций, стронций и барий можно легко отличить от других элементов. Их соли при добавлении серной кислоты выделяют белые осадки сульфатов, а при обработке раствором соды или поташа образуют белые осадки углекислых солей, растворимые в разбавленных кислотах. Мел — углекислый кальций — растворяется в кислотах, выделяя пузырьки углекислого газа. Все три элемента окрашивают пламя: кальций в кирпично-красный, стронций — в карминово-красный, барий — в желто-зеленый цвет. Обнаружить ртуть в растворе можно таким образом. Погрузите медную пластинку или монету, очищенную наждачной бумагой и азотной кислотой, в раствор соли ртути. Через несколько минут медь покроется блестящим налетом металлической ртути. Помните, что и сама ртуть и ее соли очень ядовиты!
Перейдем к представителям третьей группы. Бор. Докажем его присутствие в борной кислоте или буре. Смешайте
иа стекле или на блюдце несколько кристалликов вещества с чайной ложкой этилового спирта, прибавьте 2—3 капли концентрированной серной кислоты, снова перемешайте смесь и подожгите. Образовавшийся борно-этиловый эфир окрашивает пламя в зеленый цвет. Соли алюминия не дают каких-то очень характерных цветных реакций, но отличить металлический алюминий от других металлов нетрудно. Проделайте с ним несколько опытов. Поместите алюминиевую пластинку или проволоку в стакан с соляной кислотой. Выделяются пузырьки водорода. Теперь выньте пластинку, промойте ее водой и на короткое время опустите в стакан с концентрированной азотной кислотой, снова обмойте пластинку водой и погрузите в стакан с соляной кислотой. Теперь водород не выделяется. Дело в том, что концентрированная азотная кислота пассивирует алюминий. Второй опыт: налейте в стакан разбавленной серной кислоты, бросьте в нее кусочек алюминия и прилейте концентрированный раствор марганцовокислого калия. Фиолетовая окраска раствора быстро пропадет. И последний опыт. В пробирке к кусочку алюминия прилейте раствор щелочи и нагрейте его. Алюминий растворяется.
Самый интересный, самый важный представитель четвертой группы — углерод. Из неорганических производных углерода часто встречаются соли угольной кислоты — карбонаты. Их нетрудно отличить от других солей. Капните на кусочек мела уксусной кислоты — выделяются пузырьки углекислого газа. Чистый углерод выделяется из органических соединений при их горении или обугливании от сильного нагревания. Вы можете нагревать в консервной банке кусочек сахара до тех пор, пока он не обуглится. Выделить чистый углерод из органического соединения можно и не прибегая к нагреванию. Капните на кусочек сахара или полоску промокательной бумаги концентрированную серную кислоту — сахар и бумага чернеют. В быту мы широко используем различные соединения еще одного представителя четвертой группы, кремния,— это, например, обычное стекло. Из растворимых производных этого элемента наиболее доступен силикат натрия или калия, называемый также растворимым стеклом и применяющийся в качестве клея. При действии на раствор силиката разбавленных кислот (например, серной) выпадает осадок кремниевых кислот. Если к раствору силикатного клея прилить раствор хлорида бария, выпадает белый осадок силиката бария. Кремниевая кислота относится к слабым кислотам,
ее соли в водном растворе гидролизуются и поэтому имеют щелочную реакцию — силикатный клей скользкий на ощупь.
В пятой группе находятся два исключительно важных элемента — азот и фосфор. Чаще всего эти элементы входят в состав неорганических кислот. Обнаружить анион азотной кислоты можно двумя способами. В пробирке к нескольким каплям раствора азотнокислого калия или натрия (селитры) прилейте столько же концентрированной серной кислоты, бросьте небольшой кусочек меди и смесь нагрейте. Выделяется желтый газ — двуокись азота. На стекле к капле раствора селитры добавьте кристаллики железного купороса и каплю концентрированной серной кислоты. Вокруг кристаллика появляется бурое кольцо комплексной соли. Теперь проведите две реакции, характерные для аниона фосфорной кислоты. В одной пробирке прилейте к раствору какой-нибудь растворимой соли фосфорной кислоты раствор хлористого бария, в другой пробирке — азотнокислого серебра (ляписа). В первом случае выпадает белый осадок, растворимый в кислотах (кроме серной), во втором — образуется желтый осадок’фосфата серебра, который можно растворить в азотной кислоте.
Из представителей шестой группы остановимся лишь на сере: более известный элемент этой группы — кислород — содержится чуть ли не в любом веществе, но доказать его присутствие не так-то просто. Чаще всего сера в неорганических соединениях встречается в виде иона серной кислоты. Серная кислота и ее соли образуют с раствором хлористого бария и раствором азотнокислого серебра белые осадки. Сульфат бария не растворяется в кислотах, а сернокислое серебро растворимо в азотной кислоте.
Азотнокислое серебро — хороший реактив на соли кислот, образованных элементами седьмой группы — хлором, бромом и йодом. Если на стекле смешать капли растворов ляписа и поваренной соли, выпадает белый творожистый осадок, который не растворяется в кислотах, но растворим в нашатырном спирте. При этом образуется комплексная соль, которая под действием азотной кислоты разрушается, выделяя осадок хлористого серебра. С бромистым калием нитрат серебра дает желтоватый осадок, плохо растворимый в нашатырном спирте.
Соли металлов, расположенных в восьмой группе — железа, кобальта и никеля,— можно различить по внешнему виду. Двухвалентное железо обычно образует соединения,
окрашенные в зеленый цвет (например, железный купорос), трехвалентное железо дает желтые соли. Соединения кобальта чаще всего окрашены в розовый цвет, а никеля — в зеленый. Ион трехвалентного железа дает характерную реакцию с желтой кровяной солью (продается в магазинах фототоваров). При этом образуется синий осадок берлинской лазури. Докажите присутствие железа в гемоглобине крови. Для этого каплю крови выпарьте досуха на фарфоровой пластинке и сожгите, добавив 2—3 капли концентрированной азотной кислоты. Когда получите сухой порошок, охладите фарфоровую пластинку, соскоблите этот порошок на стекло, добавьте к нему несколько капель соляной кислоты и каплю раствора желтой кровяной соли. Читатель, конечно, помнит еще один характерный реактив иа железо — роданид аммония. Интенсивная кроваво-красная окраска роданистого железа позволяет использовать реакцию не только для качественного открытия железа, но и для определения его количества.
Мы подошли к разговору о количественном анализе. Представителям самых разных специальностей — медикам, пищевикам, гидрологам, геологам, металлургам — бывает необходимо знать не только качественный, но и количественный состав какого-то вещества, раствора, сплава. Действительно, от содержания углерода и марганца в стали, железа и кальция в воде, углеводов и белков в хлебе зависит качество и металла, и питьевой воды, и хлеба. Познакомиться с принципами количественного анализа нетрудно и в домашней лаборатории, используя вещества, которые почти всегда находятся под рукой. Однако совершенно необходимыми окажутся два прибора — весы для взвешивания и мензурки для измерения объемов растворов. Это оборудование можно приобрести в магазине фототоваров, но весы нетрудно изготовить и самому из проволоки, картона и ниток (в качестве гирек используйте медные монеты), а объемы измерять мерными стаканами, продающимися в хозяйственных магазинах, или бутылочками для детского питания. Очень полезными в вашей лаборатории окажутся бюретки. Их можно сделать самому из стеклянной трубки толщиной в палец. Разогрейте эту трубку на газовой горелке и оттяните так, чтобы ее конец был диаметром около 7 мм. Вложите в резиновую трубку диаметром примерно 7 мм и длиной 5—10 см стеклянную бусинку (она должна с трудом входить в трубку) и наденьте этот резиновый «хвостик» одним концом на узкий конец вашей стеклянной трубки —
бюретки, а другим — на толстый конец пипетки для глазных капель. Получившуюся бюретку надо испытать. Налейте в трубку воды — она не должна вытекать. Но если пальцами слегка надавить на резину вокруг шарика-бусинки, будет нетрудно откапать из бюретки нужное количество жидкости. Бюретку необходимо отградуировать. Для этого наливайте из мензурки по 5 мл воды и отмечайте на бюретке уровень жидкости (например, на полоске бумаги, наклеенной на бюретку). Разделите каждое расстояние между двумя отметинами на 5 равных частей и сделайте более мелкие отметки. Теперь вы можете отмерять объем раствора с точностью до полумиллилитра.
Вот задача, которую наиболее часто приходится решать при помощи количественного химического анализа. Предположим, нам нужно узнать, сколько щелочи содержится в фотографическом проявителе или в удобрении, или в стиральном порошке... Примем такой план действий: добавляем к раствору щелочи кислоту до полной нейтрализации. Зная, сколько пошло на это кислоты, можно рассчитать количество щелочи, используя уравнение Н++ОН_= =Н2О. Сначала нужно приготовить раствор кислоты известной концентрации. Прилейте осторожно 10 мл концентрированной серной кислоты к 1 л холодной воды. Вы получили раствор, который содержит 0,018 г H2SO4 в 1 мл, поскольку удельный вес кислоты 1,8 г/мл, и 10 мл весят 18 г. Надейте этот раствор в бюретку, а сами займитесь приготовлением раствора исследуемого вещества. Если щелочи много в этом веществе, естественно, его нужно отвесить поменьше или растворить в большем объеме воды. Предположим, вы растворили 10 г порошка, содержащего NaOH, в 1 л воды, затем отобрали 100 мл этого раствора в стакан. Добавьте к раствору 1—2 капли раствора фенолфталеина в спирте или одеколоне. (Фенолфталеин, называемый еще пургеном, продается в аптеке.) Щелочной раствор станет малинового цвета. При постоянном перемешивании стеклянной палочкой прибавьте по каплям к этому раствору раствор серной кислоты из бюретки. В тот момент, когда раствор вдруг станет бесцветным, прекратите прикапывание и определите объем раствора серной кислоты, пошедший на нейтрализацию. Пусть этот объем равен 7 мл. Это значит, что вы израсходовали 7x0,018=0,126 г H2SO4 на нейтрализацию. Уравнение реакции таково: 2NaOH+H2SO4= = Na2SO4+2H2O. Поскольку молекулярные массы равны 40 для NaOH и 98 для H2SO4, составляем пропорцию:
на нейтрализацию 2x40 г NaOH идет 98 г H2SO4, на нейтрализацию х г NaOH идет 0,126 г H2SO4.
Отсюда х=(80х0,126)/98=0,1 г. Итак, в 100мл щелочного раствора содержится 0,1 г NaOH, в 1 л вы имеете 10x0,1 = 1 г. Это же количество содержится и в 10 г вашего порошка, т. е. в нем 10% NaOH.
Титрование можно применять не только для проведения реакций нейтрализации. Растворите кусочек железного гвоздя в разбавленной серной кислоте. Получается раствор сернокислого железа или железного купороса. Концентрацию этого соединения в растворе определите следующим образом. Прикапывайте к нему из бюретки очень слабый раствор марганцовокислого калия в воде до тех пор, пока раствор не приобретет бледно-розовую окраску. Уравнение происходящего процесса таково: 10FeSO4+2KMnO4H-+8H2SO4=5Fe2(SO4)3+2MnSO4+K2SO4+8H2O. Ход расчета вам должен быть ясен.
Вообще подходов к решению задачи определения количественного содержания какого-либо химического компонента может быть очень много. Попробуем решить такую задачу: определить, сколько железа (III) содержится, например, в водопроводной воде. Вот тут обратимся еще раз к очень чувствительной реакции на железо (III) с роданид-ионом. Однако для количественного определения железа титрование в этом случае неприменимо: нет какого-то четкого перехода окрасок при равенстве концентраций Fe (III) и SCN-, и реакция, как мы помним, обратима. А что если сравнить цвет исследуемого раствора с цветом раствора, содержание железа в котором известно? Отвесьте определенное число граммов хлорного железа и растворите в определенном же объеме воды. Используя этот исходный раствор, приготовьте несколько растворов различной концентрации с очень небольшим содержанием железа. Для этого в стаканы налейте по 100 мл дистиллированной воды. Добавьте в стаканы из глазной пипетки по 1, 2, 3 и т. д. капель исходного раствора хлорида железа. Чтобы вычислить концентрации полученных растворов, вы должны узнать объем одной капли. С этой целью накапайте из той же пипетки в мензурку 20 (или 100) капель и определите объем. Капля имеет объем, в 20 (или 100) раз меньший. Итак, вы имеете серию растворов, для каждого из которых известна концентрация Fe (III). Добавьте в каждый стакан определенное количество раствора роданида аммония. При этом совсем не обязательно знать его концентрацию. Теперь
нужно только сравнить глубину окраски в стакане, содержащем водопроводную воду, с окраской стандартных растворов — и вы мгновенно определите, сколько железа содержится в водопроводной воде.
Если нужно получить небольшое количество водорода, лучший способ — подействовать на цинк кислотой. Налейте в стакан немного разбавленной серной или соляной кислоты и бросьте в нее несколько кусочков цинка. Металл начнет медленно растворяться в кислоте, покрываясь пузырьками водорода. Опустите на короткое время несколько кусочков цинка в раствор медного купороса и затем бросцте этот цинк в кислоту. Вы увидите, что он реагирует с кислотой быстрее, чем прежде [6, с. 147]. Как это объяснить? При растворении цинка положительно заряженные ионы водорода Н+ из кислоты получают от атомов цинка электроны и превращаются в нейтральные молекулы Н2. Сами же атомы цинка, лишившись двух электронов, превращаются в ионы Zn2t и переходят в раствор.
Но вот мы опустили кусочки цинка в раствор соли меди. Медь, как известно,— металл менее активный, нежели цинк. Поэтому электроны с цинка переходят на ионы меди, положительно заряженные ионы цинка поступают в раствор, а частицы металлической меди оседают на поверхности цинка. В результате получается цинк с вкраплениями частичек меди. Но в цинке свободных электронов больше, они легче отрываются от атомов. Поэтому цинк охотно передает свои электроны меди. А уже с меди электроны переходят на ионы водорода, превращая их в молекулы. Функции атомов цинка и меди различны: атомы цинка должны лишь переходить в раствор в виде ионов, атомы же меди должны только восстанавливать Н+ до Н2. В чистом цинке обе функции приходится выполнять атомам одного этого металла. Поверхность быстро покрывается молекулами водорода, растворение замедляется.
Итак, мы выяснили, что электроны охотно перетекают от меди к цинку. Но если разделить два этих металла и соединить их проводником, то электроны будут двигаться
по проводнику, т. е. по нему потечет электрический ток! Мы пришли к пониманию принципа действия химического источника тока. Мы дальше не будем подробно обсуждать его устройство. Проведем другие опыты, в которых воспользуемся готовым химическим источником тока — батарейкой для карманного фонаря.
Прикрепите к полюсам батарейки два изолированных медных провода длиной около 30 см каждый. Концы проводков намотайте в два-три витка на двухкопеечные монеты. В цепь последовательно подключите лампочку для карманного фонаря. Налейте в стакан примерно на одну треть чистой воды и опустите в нее электроды-монеты, только так, чтобы они не соприкасались. Лампочка не загорается, ничего не происходит. Замените чистую воду раствором сахара. Тот же эффект, точнее — отсутствие каких-либо эффектов. Теперь снова налейте в стакан чистой воды, погрузите в нее монеты и добавьте несколько капель серной кислоты. Лампочка загорается, а с обеих монет начинают подниматься пузырьки газов.
Следующий опыт. Налейте в стакан раствор поваренной соли, к которому добавьте две-три капли раствора фенолфталеина в спирте. Опустите в стакан электроды-монеты. Вы можете наблюдать очень красивую картину: на обеих монетах немедленно появляются пузырьки газов, а вокруг монеты, соединенной с отрицательным полюсом батареи (катодом), образуется малиновое облачко, постепенно распространяющееся навесь раствор. Лампочка горит. Как объяснить эти наблюдения? С отрицательного полюса батареи на катод поступают электроны, они скапливаются на монете, заряжают ее отрицательно. Положительный полюс батареи «высасывает» электроны с анода, заряжающегося положительно. Между монетами — слой чистой воды. А поскольку вода не проводит электрический ток, электроны не могут перетекать с катода на анод. Естественно, лампочка гореть не будет.
Заметим, что в воде есть кандидаты на роль переносчика электрических зарядов. Это — ионы Н+ и ОН-. Однако их концентрация очень мала в чистой воде, поэтому-то она и не проводит практически ток. Убедившись в плохой электропроводности воды, мы помещали электроды в раствор сахара. Здесь та же картина: молекулы этого вещества не распадаются в растворе на заряженные ионы. Но вот мы взяли раствор серной кислоты. Ситуация изменилась, раствор кислоты оказался хорошим проводником тока. Дело в
том, что молекулы кислоты в растворе распадаются на ионы водорода и отрицательно заряженные анионы. Здесь стоит подчеркнуть, что этот процесс происходит независимо от того, погружены или нет в раствор электроды, пропускают через него ток или не пропускают. К отрицательно заряженному катоду подходят положительно заряженные ионы Н+. Естественно, в растворе кислоты таких ионов много. На катоде протоны Н+ получают по электрону, образуются атомы водорода. Два атома дают молекулу Н2, молекулы образуют пузырьки газа. Итак, с катода электроны уходят, и батарейка вынуждена поставлять сюда все новые и новые порции отрицательных зарядов. Значит, по цепи пошел ток. Поэтому и загорается лампочка.
А что происходит на аноде? В упрощенном виде события здесь развертываются так. Ионы ОН“, которые, хотя их мало, все-таки присутствуют, отдают свои электроны аноду и превращаются в молекулы воды и кислорода. В итоге получается, что под действием электрического тока вода разлагается на составные элементы (рис. 3, а). Заметим, что в действительности электролиз раствора серной кислоты идет гораздо сложнее, образуются различные производные серы, перекиси водорода. Добавляя в воду любой электролит, т. е. вещество, распадающееся в воде на ионы, мы увеличиваем электропроводность воды. Можно добавлять и щелочи, В промышленности, например, воду разлагают электрическим током на водород и кислород, добавляя в нее едкий натр.
Какие же процессы вызывает электрический ток, проходя через раствор поваренной соли? Хлорид натрия распадается в растворе на ионы Na+ и С1~. Первые двигаются, естественно, к катоду, вторые — к аноду. На катоде ионы натрия, казалось бы, должны получать электроны и превращаться в металлический натрий. Но этого не происходит. Натрий — слишком активный металл и не может существовать рядом с водой. Поэтому электроны с катода переходят не на ионы Na+, а на ионы водорода Н+, которые всегда присутствуют в водном растворе. В результате на катоде выделяется водород (рис. 3, б). Вторая составная часть воды — ионы ОН~ Они накапливаются возле катода. Вот поэтому-то и краснеет раствор вокруг монеты-катода, если в нем присутствует фенолфталеин. Фенолфталеин, как известно,— хороший индикатор на щелочи.
На аноде скапливаются ионы хлора, отдают свои электроны аноду и переходят в молекулярный газообразный
a
Рис. 3. Схема электролиза; а — воды; б — раствора хлорида натрия
хлор. В растворе же остаются ионы Na+ и ОН-, т. е. составные части едкого натра. Не удивительно поэтому, что пропускание электрического тока через водный раствор поваренной соли — удобный промышленный способ получения сразу трех важнейших веществ: водорода, едкого натра и хлора.
Мы рассказали об азах важной ветви физической химии. Эта ветвь называется электрохимией и занимается исследованием химических реакций, происходящих под действием тока. Эту науку интересуют также и такие реакции, в результате которых возникает электрический ток. Не следует думать, что электрохимия оперирует только с неорганическими соединениями. Любые органические вещества представляют интерес для электрохимиков. В последние годы интенсивно развиваются электрохимические методы синтеза органических веществ. О некоторых сторонах электрохимии можно прочитать в популярных книгах [8].
Химические превращения обычно протекают либо с выделением, либо с поглощением энергии. В первом случае запас энергии, находящейся в продуктах реакции, меньше, чем в исходных веществах, во втором — наоборот. Как можно количественно оценить такие изменения в запасе энергии? Для этой цели вводится понятие свободной энергии, обозначаемой буквой G. Считается, что убыль этой величины AG для реакции, которая проходит при постоянных температуре и давлении, равна максимальной работе, которая может быть проведена за счет данной реакции. Изменение в свободной энергии ДО рассчитывается по такой формуле:
AG=SAG° (продукты) — SAG0 (реагенты).
Здесь AG° — изменение свободной энергии для каждого продукта или реагента при образовании его из элементов, происходящее при стандартных условиях, т. е. при 25°С и давлении в 1 атм. Знак 2 обозначает сумму этих величин по всем реагентам или продуктам. Стандартная свободная энергия любого элемента принимается равной нулю.
«> Из всего сказанного вытекает, что реакции, для которых Д6° является отрицательной величиной, идут с выделением энергии, что продукты обладают в этом случае меньшим запасом энергии, чем реагенты, и, следовательно, такие реакции идут самопроизвольно, подобно тому как самопроизвольно скатывается с горы камень. А теперь давайте посмотрим, каковы величины Д6° для реакций некоторых металлов с кислородом. Эти процессы приводят, естественно, к получению оксидов металлов. Изменения свободной энергии (в ккал на моль вещества), соответствующие образованию некоторых таких соединений, приведены в скобках: СиО (—31), NiO (—51), ZnO (—76), SnO2 (—124), MgO (—136), Fe2O3 (—177), Cr2O3 (—253), A12O3 (—378).
Что бросается в глаза, когда смотришь на эти числа? Все изменения свободной энергии отрицательны, а это значит, что окисел любого из приведенных здесь металлов устойчивее самого металла. При этом хорошо видно, что наиболее охотно оксид должен образовываться алюминием, а медь менее склонна к такому превращению. Итак, вывод, вытекающий из проведенного термодинамического анализа, такой: все металлы, за исключением очень немногих, в свободном состоянии не устойчивы, а должны в атмосфере, содержащей кислород, переходить в оксиды. Тем не менее все мы прекрасно знаем, что и стальные фермы мостов, и алюминиевые кружки, и медные провода не превращаются в груды окислов, а существуют очень долго. Почему? Продолжим наше сравнение свободного металла, находящегося в атмосфере кислорода, с поднятым в горы камнем. Да, он стремится оказаться у подножия горы, перейти в оксид. Но ведь далеко не все камни, находящиеся высоко в горах, катятся вниз. Причин тут несколько, одна из них — камень со всех сторон окружен горами. Чтобы скатиться, ему нужно преодолеть энергетический барьер, часто очень высокий. Ясно, что в таком состоянии камень может пролежать в горах миллионы лет, а металл столько же лет* остаться не окисленным.
И все же 1—1,5% всего металла, накопленного человеком, ежегодно теряется в результате процесса, называемого коррозией. Этот термин означает самопроизвольное разрушение металлов вследствие их взаимодействия с окружающей средой. При этом металл не обязательно превращается в окисел. Среди продуктов коррозии — и гидроксиды, и хлориды, и соли других кислот. Однако в любом случае металл переходит в окисленное состояние.
Провести опыт по корродированию железа в домашних условиях очень просто: оставьте кусочек железа во влажном месте, полейте его водой, и через несколько дней он покроется рыжим налетом оксида. В сухой атмосфере вам не удастся вызвать это превращение, не получите вы оксида из железа и в очень влажной, но не содержащей кислорода атмосфере. Значит, для ржавления необходимы и вода, и кислород. Железо при этом отдает два электрона: Fe=Fe24 +2е. Эти электроны восстанавливают кислород, образуя гидроксид-анионы: О2+2Н2О+4е=4ОН~ Катион железа реагирует с гидроксидом, давая гидроокись железа. Последняя постепенно теряет воду, переходя в оксид двухвалентного железа: Fe2++2OH~=Fe(OH)2->FeO+H2O. Гидроксид двухвалентного железа может окисляться кислородом до Fe(OH)3, который также распадается на воду и оксид железа (III). В результате всех этих процессов на поверхности металла возникает «слоеный пирог» из различных окислов.
Очень распространена коррозия другого вида — электрохимическая. Дело в том, что многие металлические конструкции постоянно находятся в соприкосновении с электролитами, т. е. с растворами солей, кислот, оснований, содержащих различные ионы. Чтобы понять суть электрохимической коррозии, вспомним устройство гальванического химического источника тока. Если в раствор электролита поместить железную (или цинковую) и медную пластинки и подключить их к гальванометру, то прибор покажет, что между пластинками возникает разность потенциалов. Меняя различные металлы в нашем элементе, мы будем получать различные значения разности потенциалов. В качестве одного из электродов можно рассматривать ионы водорода, переходящие в газообразный водород. Если потенциал такой системы принять за ноль, то по отношению к ней другие системы будут иметь такие разности потенциалов Е° в вольтах:
Na+-h?->Na
А13++Зе->А1
Zn2++2e->Zn Fe2++2e->Fe Cu2++2e->Cu Au3++3e->Au
—2,71
—1,68
—0,76
—0,47 +0,34 + 1,50
Видно, что наибольшие разности потенциалов можно получать в элементах, одним из электродов которых был бы натрий, алюминий, а другим — медь, золото. Легко ви
деть также, что железо по сравнению с медью должно окисляться легче. И если поместить железный гвоздь в раствор соли меди, то металлическая медь скоро покроет железо красным слоем. Происходящие при этом реакции можно суммировать:
Fe=Fe2++2e Е°=4-0,47 В
Cu2+-|-2e=Cu Е° =+0,34 В
Fe+Cu2+=Fe2++Cu Е°=+0,81 в
Известна формула, связывающая электродный потенциал Е° и свободную энергию окислительно-восстановительного электродного процесса:
AG°=— nFE\
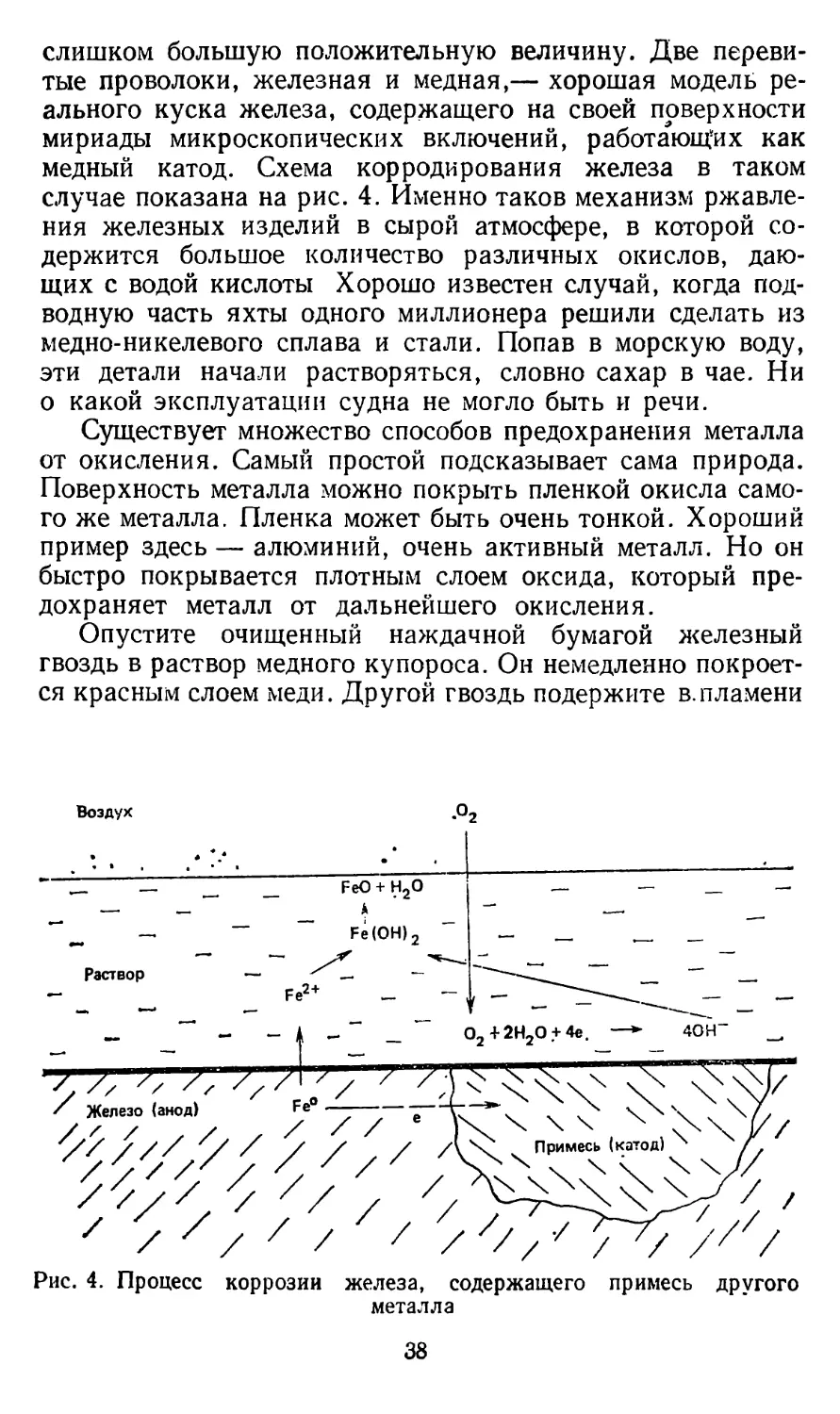
Здесь п — число молей переносимых в процессе электронов, a F— константа Фарадея, величина положительная. Для нашего железомедного элемента потенциал Е° положителен и, следовательно, значение AG0 отрицательно. Отсюда вытекает, что изображенный выше суммарным уравнением процесс термодинамически выгоден и идет с выделением энергии. Но какое все это имеет отношение к коррозии? Прежде чем ответить на этот вопрос, проведем один опыт. Растворите в стакане воды щепотку поваренной соли и добавьте в раствор немного красной кровяной соли (продается в магазинах фототоваров) и несколько капель спиртового раствора фенолфталеина. Теперь опустите в стакан связанные между собой железную и медную проволочки. Лучше их перевить, чтобы было много точек соприкосновения. Через несколько минут вокруг медной проволоки появится малиновое окрашивание, вокруг железной — синее. Что здесь происходит? Процесс похож на рассмотренную нами реакцию металлического железа и соли меди. Железо переходит в раствор в виде ионов Fe2+, которые дают с красной кровяной солью турнбулеву синь. В это же время на медной проволоке идет восстановление кислорода до гидроксид-анионов. При этом используются электроны, поставляемые по проволоке от железа. Наличие ионов ОН~ обнаруживается фенолфталеином. А теперь попробуйте провести аналогичный опыт, но без участия медной проволоки. Процесс окисления железа идет в этом случае гораздо медленнее.
Итак, железо под действием кислорода и в контакте с медью в растворе электролита окисляется, т. е. корродирует. Медь при этом не изменяется, поскольку ее потенциал имеет
слишком большую положительную величину. Две перевитые проволоки, железная и медная,— хорошая модель реального куска железа, содержащего на своей поверхности мириады микроскопических включений, работающих как медный катод. Схема корродирования железа в таком случае показана на рис. 4. Именно таков механизм ржавления железных изделий в сырой атмосфере, в которой содержится большое количество различных окислов, дающих с водой кислоты Хорошо известен случай, когда подводную часть яхты одного миллионера решили сделать из медно-никелевого сплава и стали. Попав в морскую воду, эти детали начали растворяться, словно сахар в чае. Ни о какой эксплуатации судна не могло быть и речи.
Существует множество способов предохранения металла от окисления. Самый простой подсказывает сама природа. Поверхность металла можно покрыть пленкой окисла самого же металла. Пленка может быть очень тонкой. Хороший пример здесь — алюминий, очень активный металл. Но он быстро покрывается плотным слоем оксида, который предохраняет металл от дальнейшего окисления.
Опустите очищенный наждачной бумагой железный гвоздь в раствор медного купороса. Он немедленно покроется красным слоем меди. Другой гвоздь подержите в.пламени
Рис. 4. Процесс коррозии железа, содержащего примесь другого металла
гйювой горелки примерно минуту, пока гвоздь не приобретет желтоватый цвет. Если такой гвоздь опустить в раствор купороса, медь выступит на нем несколько позже. Возьмите несколько других гвоздей и перед погружением в раствор медного купороса прокалите их в пламени горелки 2, 3 и 5 мин. После прокаливания гвозди приобретают красную, синюю или серо-зеленую окраску. Время, которое проходит до начала выделения меди на этих гвоздях, увеличивается в соответствии со временем прокаливания. Опыт объясняется просто — при прокаливании гвозди покрываются окисной пленкой, толщина и цвет которой зависят от времени прокаливания. Нанести на поверхность железа защитную пленку можно и другим способом — обработав его концентрированной серной или азотной кислотой. Очистите два гвоздя наждачной бумагой и один из них поместите на несколько минут в концентрированную азотную кислоту. Промойте оба гвоздя водой и опустите их в стакан с разбавленной серной кислотой. Вы увидите, что гвоздь, побывавший в азотной кислоте, не будет реагировать с серной кислотой, в то время как обычный гвоздь энергично выделяет из нее пузырьки водорода.
О коррозии и защите от нее см. [91.
G
Хорошо известно, что бумажный лист к пальцам не прилипает; но стоит смочить пальцы водой, как ситуация меняется. Этот «принцип», кстати, лежит в основе не очень хорошей привычки мусолить пальцы при чтении книг. Но почему же капелька воды вызывает такое разительное изменение? Обратимся сначала к молекулярному устройству жидкости. В глубине слоя воды, налитой, предположим, в стакан, каждая молекула окружена совершенно равномерно другими такими же молекулами воды. Поэтому во всех направлениях эта молекула испытывает одинаковые силы притяжения. Совсем иное положение с молекулами воды, находящимися на поверхности. Здесь снизу их притягивают такие же молекулы жидкости, а вот сверху над ними пролетают довольно редко попадающиеся молекулы газов,
входящих в состав воздуха. Ясно, что молекула воды на поверхности более сильно взаимодействует с водным слоем; на нее действуют силы, которые стремятся втянуть молекулу в объем. Предположим, что площадь водного поверхностного слоя в цилиндрическом стакане равна S. Наклоните немного стакан. Площадь поверхности воды увеличится на AS. Наклоняя стакан, вы заставили некоторые молекулы воды выйти из объема на поверхность, но поскольку вы действовали против сил, пытающихся втянуть молекулу с поверхности в объем, вы совершили некую работу. Обозначим эту работу по увеличению поверхности AW Если разделить эту величину на приращение поверхности, мы получим весьма важный параметр o=AW/AS, называемый поверхностным натяжением.
Увеличивая поверхность, вы повысили энергию системы. Но самопроизвольно система свою энергию повышать не станет, она всегда стремится ее уменьшить. Именно поэтому предоставленная сама себе капля воды принимает форму шара. Ведь шар имеет наименьшую площадь поверхности для тела данного объема. Капля ртути, помещенная на стекло, имеет форму несколько искаженного шара. А вот если вы капнете на чистое стекло воду, никаких капель не получится, вода равномерно растечется по стеклу. Почему такая разница? Дело в том, что атомы ртути гораздо сильнее притягиваются друг к другу, чем к молекулам, входящим в состав стекла. Молекулы же воды более склонны прилипать к частицам стекла. Взаимодействие воды и ртути со стеклом — это два крайних случая; здесь говорят о полном смачивании и несмачивании стекла. Но проведите по стеклу пальцем. Немного жира перейдет на стекло, и, если теперь на него поместить воду, она образует каплю, слегка размазанную по поверхности. Форму капли, а точнее, угол 0 между поверхностью твердого тела и касательной к поверхности капли, проведенной через точку соприкосновения поверхности капли с поверхностью «подложки», можно определить по формуле:
cosQ = отг-~ожт.
И ж г
Здесь Г — газ (например, воздух); Ж— жидкость; Т — твердое тело; о — поверхностное натяжение, существующее на границе между твердым телом и газом, жидкостью и твердым телом и между жидкостью и газом.
Поверхностное натяжение различных жидкостей нетруд
но, оценить в домашних условиях при помощи самодельного прибора. Соорудите весы из стеклянной или металлической палочки и нескольких ниток. Одну чашку можно сделать из алюминиевой жести или фольги, а вместо другой чашки используйте квадрат, согнутый из тонкой медной проволоки. Приведите коромысло весов в горизонтальное положение, нагрузив на чашку какие-нибудь гирьки, например изготовленные из кусочков пластилина. Теперь подставьте под квадрат из проволоки стакан с водой так, чтобы этот контур «прилип» к поверхности. Накладывая постепенно на чашку весов гирьки и отметив момент, когда квадрат оторвется от поверхности жидкости, мы в каких-то условных единицах измерим поверхностное натяжение. На практике величину о измеряют в эргах на см2. Для воды при 20°С о составляет 73 эрг/см2. Замените воду на спирт, и вы увидите, что поверхностное натяжение у этой жидкости гораздо меньше — 22 эрг/см2. У глицерина величина о, почти как у воды,— 62 эрг/см2.
Растворите в стакане воды небольшой кусочек мыла и определите поверхностное натяжение такого раствора. Вы увидите, что значение о после растворения мыла резко снизилось. Молекулы мыла состоят из двух частей, обладающих противоположными свойствами. Одна часть — натровое производное карбоксильной группы — COONa — прекрасно «растворяется» в воде, охотно притягивает к себе молекулы воды. Вторая часть — длинный хвост, состоящий из углеводородных звеньев, например —(CH2)i6CH3. Группы СН2 хвоста обладают явной тенденцией отталкивать молекулы воды. Когда молекула мыла попадает в воду, ее карбоксильная часть окружает себя молекулами воды, а вот хвост стремится выбраться из водного слоя. В результате молекула оказывается на поверхности, ее хвост лежит на самой границе вода — воздух, а карбоксильная головка утоплена в объем воды. Оказывается, что вся поверхность воды занята молекулами мыла, которое относится к поверхностно-активным веществам. Мыло понижает поверхностное натяжение воды.
Растворите в стакане воды кусочек мыла. Раствор получился мутноватый, что свидетельствует о том, что раствор этот коллоидный. Молекулы веществ, составляющих мыло, собраны в агрегаты, микроскопические капельки. Добавьте к такому раствору уксусной эссенции. Жидкость еще больше помутнеет, и через некоторое время из нее начнет всплывать белый маслянистый слой. Раствор при этом станет бо
лее светлым. Еще быстрее этот процесс пройдет, если вз'ять серную кислоту. Кислота вытеснит из стеарата натрйя, который составляет главную часть мыла, свободную стеариновую кислоту С17Нз5СООН. Эта кислота и образует сначала белый коллоидный раствор, затем коагулирует в более крупные капельки, которые затем отслаиваются в виде масла. Помимо стеариновой кислоты, в мыле присутствует олеиновая, интересная тем, что в ее длинной цепи содержится двойная связь СН3(СН2)7СН=СН (СН2)7СООН. Доказать присутствие двойной связи можно, взболтав в пробирке с выделенными из мыла кислотами несколько капель бромной воды. Бромная вода обесцветится, поскольку бром присоединяется по двойной связи. Стеариновая и олеиновая кислоты очень слабые; поэтому, попадая в воду, натровые соли этих кислот частично гидролизуются с образованием свободной кислоты и едкого натра. Именно это является причиной того, что растворы мыла имеют щелочную реакцию. Растворите кусочек мыла в небольшом количестве спирта или одеколона и добавьте в раствор крупинку фенолфталеина. Если теперь к раствору постепенно добавлять воду, жидкость по мере увеличения гидролиза будет приобретать все более интенсивный малиновый цвет.
Исследуем некоторые физико-химические свойства поверхностно-активных веществ. Соорудите остроумный прибор, называемый весами Лэнгмюра. Идея прибора такова (рис. 5). В прямоугольную ванну, заполненную до краев водой, помещается поверхностно-активное вещество, которое, естественно, растекается по поверхности воды. С одного края ванны имеется ограничитель, позволяющий изменять
Рис. 5. Идея конструкции весов Лэнгмюра
площадь ванны. На другом конце ванны размещается подвижный поршень, связанный с очень тонкой пружиной и стрелкой, которая отражает давление, оказываемое на поршень. Поместите на поверхность воды в ванне очень немного поверхностно-активного вещества. Это может быть смесь жирных кислот, выделенных из мыла (растворите эту смесь в эфире), или крупинка камфоры. Поверхностноактивное вещество мгновенно растечется по поверхности жидкости, а поскольку мы взяли его очень мало, толщина его слоя на поверхности жидкости будет составлять всего одну молекулу. Молекулы будут стремиться растечься по поверхности, но поршень мешает им это сделать. Поэтому на поршень будет оказываться давление, которое можно измерить по отклонению стрелки.
Будем теперь уменьшать площадь ванны, передвигая ограничитель. Если на графике откладывать по оси абсцисс площадь S, занимаемую поверхностно-активным веществом, а по оси ординат — давление Р, оказываемое на поршень, то на каком-то определенном участке А Б давление Р будет
шении поверхности их ванны S. Черные точки — группы СООН, зигзаги — углеводородные хвосты
расти пропорционально уменьшению площади S (рис. 6),. Произведение давления на площадь оказывается пропорциональным произведению универсальной газовой постоянной
на абсолютную температуру Т PS~RT. Но ведь точно так же записывается и уравнение состояния газа: PV=RT Здесь только вместо площади S стоит объем V. Значит, на участке А Б мы имеем дело как бы с двумерным газом. В этом состоянии молекулы вещества на поверхности находятся далеко друг от друга. Продолжаем уменьшать площадь S. В точке Б мы приходим к такой ситуации, когда вся поверхность занята молекулами поверхностно-активного вещества. Дальше при сжатии молекулам становится так тесно, что некоторые из них начинают поднимать хвосты над поверхностью воды. Участок Б В соответствует такому положению, когда отдельные островки из молекул с задранными хвостами — это как бы капли жидкости — плавают в море «распластанных» молекул. Уменьшая площадь S на этом участке, вы будете присоединять распластанные молекулы к каплям, число же распластанных молекул на единицу площади будет прежним. А это значит, что давление на поршень меняться не будет. Наконец, мы приходим в точку В, когда все молекулы плотно прижаты друг к другу и у всех хвосты торчат над поверхностью. Можно сказать, что мы получим некую двухмерную жидкость, а жидкости практически несжимаемы: небольшое уменьшение площади приводит к резкому повышению давления.
Зная количество нанесенного на поверхность вещества и площадь ванны, нетрудно рассчитать длину одной распластанной молекулы, вычислить диаметр хвоста молекулы.
Почему же мыло обладает такими удивительными моющими свойствами? Здесь несколько причин. Мы уже определили, что поверхностно-активные вещества резко, более чем в 2 раза, понижают поверхностное натяжение воды. А это значит, что теперь жидкость сможет проникнуть в самые мелкие поры на ткани, смочить все самые удаленные уголки, куда попали загрязнения. Это не все. Представим себе, что капелька жирового загрязнения оказалась на хлопковом волокне. Поместим такую ткань в чистую воду. Жировая капелька растечется по волокну. Мы уже знаем, что форма капли зависит от соотношения поверхностных натяжений на границах сред. Сверху каплю покроет слой воды, этим все и кончится. Теперь добавим к воде поверхностно-активное вещество. Его молекулы растекутся по поверхности воды и по поверхности жира. Жир, растекшийся
было по поверхности волокна, соберется в капельку. Внутрь капли будут смотреть хвосты молекул поверхностно-активного вещества, а головки вылезут из капли наружу и окружат себя молекулами воды. После этого капельки жира будут проявлять стремление оторваться от хлопка и перейти в воду. Поверхностно-активное вещество не дает каплям жира соединяться в большие капли, поэтому образуется устойчивая эмульсия. Эмульсию легко удалить водой — ткань очищается.
В том-то и красота науки, что возникающие задачи она решает обычно не прямо, так сказать, в лоб, а обходными, зачастую весьма изящными способами. Представьте себе, что вам надо измерить диаметр футбольного мяча или яблока. Ничего нет проще — приложите к мячу две дощечки и измерьте линейкой расстояние между ними. Но вот возникает такая задача — измерить диаметр частичек дорожной пыли или зубного порошка. Линейка тут вряд ли поможет. Оказывается, однако, что это можно сделать, взболтав порошок в воде и измерив скорость его осаждения. Конечно, здесь необходимы точные приборы: чем тоньше устроен прибор, тем точнее будет измерение. Но и в домашних условиях можно грубо прикинуть размеры частиц, соорудив относительно несложный аппарат. Дадим лишь идею прибора, предоставив читателю возможность проявить свои способности и конструктора и механика.
Основное в нашем эксперименте: определение массы осадка, поэтому главная часть прибора — весы. Можно использовать школьные лабораторные или аптекарские весы. Нам не потребуется определять абсолютную массу осадка в граммах, а только относительный вес. Поэтому в качестве гирь удобно использовать вырезанные из жести или фольги квадратики одинаковых размеров. Принцип опыта прост: погружаем одну из чашек наших весов во взвесь, суспензию порошка в воде, осадок постепенно откладывается на чашке, поэтому чашка опускается. Через определенные промежутки времени мы кладем на противоположную чашку столько
квадрагиков-гирек, сколько нужно для приведения коромысла в горизонтальное положение. Время и массу записываем в таблицу. Итак, взболтайте в воде щепотку-другую зубного порошка, залейте эту суспензию в стакан и погрузите в жидкость одну из чашек поближе ко дну. А пока суспензия будет оседать и вы сможете оценить, хватит ли вам времени и гирек для проведения опыта, займемся теоретическим анализом [10].
Известна формула, связывающая скорость оседания частицы суспензии с ее радиусом. Эта формула, вытекающая из закона Стокса, выглядит так:
где г — радиус частицы; т] — вязкость жидкости; &d — разность плотностей веществ частицы и жидкости; h — высота, с которой частица падает за время t\ g — ускорение силы тяжести; k—постоянная для данных системы и прибора величина. Положим, что все частицы в нашей взвеси имеют одинаковую массу и, следовательно, одинаковый радиус, и будем на графике откладывать по оси абсцисс время, а по оси ординат — массу осадка, скопившегося к данному времени на чашке весов. Такая зависимость будет
Рис. 7. Кривые оседания осадка, состоящего из одной или двух фракций (а) и осадка, представляющего собой смесь частиц любой массы (б)
выглядеть как отрезок прямой линии АВ, проходящей через начало координат (рис. 7, а). Действительно, в нулевой момент времени /=0 весь порошок находится во взвешенном состоянии. Предположим, что через 10 с на чашку осело 1000 частиц с массой 1000 условных единиц. Поскольку оседание происходит равномерно, через 20 с на чашке окажется 2000 частиц с условной массой 2000 единиц. Через 60 с на чашке будет масса 6000 единиц. После того как во время tB весь осадок окажется на чашке весов, масса осадка прибывать перестанет и прямая АВ перейдет в прямую ВС, параллельную оси абсцисс. Обратите внимание: время tB — это время, когда на чашку весов упали последние частицы. Очевидно, это те частицы, которые в начале опыта при t=0 находились на максимальной высоте от чашки, т. е. на уровне верхней границы жидкости. Таким образом, мы знаем высоту h от верхнего уровня жидкости до чашки весов (ее легко измерить линейкой) и время tB, за которое частица падает с этой высоты. Подставляя эти значения в формулу Стокса, мы можем вычислить радиус частиц.
Теперь рассмотрим более сложный случай — суспензия состоит из частиц двух сортов, различающихся радиусом и,
естественно, массой. Можно построить две прямые, соответствующие оседанию легких и тяжелых частиц. Предположим, что один тип частиц дает уже рассмотренную прямую АВ, а второй сорт состоит из более легких частиц. Ясно, что более легкие частицы оседают медленнее. Поэтому им должен соответствовать более пологий и более длинный отрезок AD. Как же изменяется масса осадка, когда в системе частицы двух сортов? Для того чтобы получить график такого изменения, нужно сложить ординаты двух кривых АВС и ADE. В результате получаем ломаную прямую AFGH. Точка F на этой линии соответствует времени t, когда полностью завершается оседание частиц более крупного сорта. В точке G отражено прекращение процесса оседания более легкой фракции. Если продолжить линию FG до пересечения с осью ординат с /=0, то отрезок MG М в окажется равным отрезку MdA. Но ведь MGA—это масса всего осадка, а М ;jA—масса легкой фракции. Значит, МВА — масса тяжелой фракции, MG Мв — масса легкой фракции. Что мы можем вычислить, исходя из этого графика? Закладывая времена /, соответствующие изломам, в формулу Стокса, нетрудно вычислить радиусы частиц первой и второй фракций, а соответствующие отрезки, отсекаемые на оси ординат, дадут нам относительные весовые количества этих фракций.
После такого чисто теоретического анализа обратимся к нашему эксперименту. Рассчитайте так, чтобы во время оседания зубного порошка сделать 5—6 взвешиваний, после чего весь осадок уляжется на чашку и дно и вес чашки станет постоянным. Учитывайте, что на первых стадиях все будет меняться быстрее, чем в конце эксперимента. Из общих соображений ясно, что зубной порошок состоит из частиц не одного строго заданного размера и даже не из двух или трех фракций. Состав частиц по радиусам меняется непрерывно в каких-то определенных пределах. Поэтому выбранные вами значения времени t в конечном счете будут определять границы каких-то условных фракций. Полученные результаты запишите в табличку, в которой будут фигурировать следующие величины: время в секундах, масса осадка — число гирек, масса осадка в процентах, масса данной фракции М в процентах от общей массы осадка, радиус частиц г в метрах, величина Ml&r. Вычисляя радиус, сначала удобно рассчитать константу k. Примем £г=9,8 м/с2, 11=0,001 н-с/м2, &d=d (мел)—d (вода)=2700—1000= = 1700 кг/м3, /х=0,1 м. Тогда /г=0,0002. Данные таблицы можно перенести на график. Линия оседания в этом случае
получится кривой (рис. 7, б). Это и понятно — ведь в смеси огромное количество плавно переходящих друг в друга фракций. Легко видеть, что кривая осаждения — как бы предельный случай ломаной линии при числе фаз, стремящемся к бесконечности. Но в первом приближении можно разделить кривую на 6 участков, отвечающих 6 фракциям.
Возьмем самый начальный участок кривой осаждения О—1. Будем считать, что он прямолинеен и отвечает осаждению самых крупных частиц. Осаждение этих частиц заканчивается в точке /, и содержание данной фракции составляет 5% Этот участок соответствует участку AM в на рис. 7, а и получается, если продолжить линию 1—2 до пересечения с осью ординат. Содержание фракции 1—2 мы получим, если продлим прямую 2—3 № пересечения с осью ординат и вычтем процент предыдущей фракции. Так можно вычислить относительное содержание всех выбранных нами фракций. Построим дифференциальную кривую осаждения. Для этого по оси абсцисс откладываем значения граничных радиусов частиц данной фракции, а по оси ординат величины /И/Аг, где М — относительное содержание фракции и Аг — интервал радиусов частиц фракции. Площадь каждого прямоугольника равна весовому содержанию фракции. Соединив средние точки оснований прямоугольников плавной линией, мы и получим дифференциальную картину распределения частиц по фракциям (рис. 8). Теперь хорошо видно, частицы какого размера преобладают в весьма пестрой смеси.
Мы рассказали об одной из задач так называемого седиментационного анализа. Это — раздел коллоидной химии, изучающий процесс осаждения, седиментации взвешенных в жидкостях частиц. Такие процессы осаждения играют важную роль в технике, в практике. Ведь со смесями приходится иметь дело каждому. Здесь и суспензии, т. е. взвеси твердых частиц в жидкостях, и эмульсии — это когда частицы жидкости диспергированы в нерастворяющей другой жидкости, и аэрозоли, т. е. туманы или дым — взвесь в газе жидкости или твердого тела. Краски, молоко — это все эмульсии, многие лекарства применяются в виде мазей-эмульсий.
Если посмотреть под микроскопом на взвесь тонкого порошка в воде, можно обнаружить очень любопытное явление. Маленькие частички суспензии находятся в непрерывном бесконечном движении. Этот эффект был впервые отк
рыт еще в 1828 г. английским ботаником Броуном. Как же объяснить броуновское движение? Представьте себе, что в воде плавает довольно большая частица. В каждый данный момент времени она атакуется молекулами воды, которые кишат словно пчелы в улье, сталкиваются между собой, со стенками частицы. Поскольку частица большая, она сталкивается одновременно с каждой стороны со многими тысячами и миллионами молекул. Ясно, что в такой ситуации, напоминающей положение, сложившееся в басне о лебеде, раке и щуке, частица двигаться не будет. Но вот мы уменьшили размеры частички, они стали в какой-то степени соизмеримы с размерами молекул. Оказывается, что теперь становятся возможными ситуации, когда в данный момент времени частица получает удар, предположим, 20 молекул воды с одной стороны и лишь 15 — с другой. Понятно, что результатом будет движение частички, причем движение совершенно беспорядочное и бесконечное. Такая неравномерность в распределении молекул воды — в данном объе-
Рис. 8. Дифференциальная кривая распределения частиц зубного порошка по фракциям
ме пространства находится несколько больше молекул, чем 'в соседнем,— называется флуктуацией. Флуктуации характерны для систем, состоящих из большого числа частиц, а даже в капле воды число молекул исчисляется миллиардами. Концентрации молекул, окружающих частицу с разных сторон, могут быть одинаковы. Но вот скорости движения молекул, соседствующих с частичкой с левой стороны, могут превышать скорости молекул, расположенных справа. Наконец, можно представить себе и промежуточную ситуацию: молекулы-соседки различаются и концентрацией и скоростью движения.
Как-то один нерадивый студент на экзамене на вопрос «Почему небо голубое?», подумав немного, ответил: «Потому что в нем море отражается». Такой ответ, разумеется, немало позабавил преподавателя. Как же объяснить голубизну неба? Известно, что свет, падающий на какую-то частичку, поглощается частицей, нагревает ее. Но немного света и рассеивается. Английским ученым Релеем было выведено уравнение для рассеяния света очень малыми частицами. Из этого уравнения следует, что интенсивность рассеянного света обратно пропорциональна длине волны падающего света в четвертой степени. В спектре видимого солнечного света наименьшую длину волны имеют фиолетовые, синие лучи. Поэтому-то они и рассеиваются гораздо сильнее, чем лучи желтые или красные. А раз так, небо выглядит голубым. Тут, разумеется, возникает вопрос: какие частицы рассеивают солнечный свет? Оказывается, в роли таких «частиц» выступают флуктуации в плотности воздуха. В огромной массе молекул газов атмосферы то и дело возникают небольшие области с повышенной или пониженной плотностью. Свет, падая на такие области, взаимодействует сними по-другому, нежели с основной массой, рассеивается сильнее.
Сначала мы говорили о довольно крупных частицах, какими являются зерна зубного порошка, потом перешли к молекулам. А что если рассмотреть нечто среднее, например частицы, размеры которых лишь в несколько раз превышают размеры молекул? Мы подошли к разговору об удивительнейших образованиях, называемых коллоидными растворами. Получить коллоидный раствор дома нетрудно, и сделать это можно несколькими способами. Растворите в спирте немного канифоли и теперь в стакане к чайной ложке такого раствора добавьте большой избыток воды. В другом опыте к кипящей воде добавьте хлорида трехвалентного
железа и полученный раствор кипятите еще несколько минут. В третьем опыте к очень разбавленному раствору желтой кровяной соли постепенно, помешивая, добавьте по каплям разбавленный раствор хлорида трехвалентного железа. Во всех случаях вы получили окрашенные в разные цвета коллоидные растворы [10, с. 271]. То, что это растворы коллоидные, а не истинные, легко показать. Осветите стаканы с этими растворами тонким лучом света сбоку. Вы увидите мутноватый конус, опалесценцию, что обусловлено рассеянием коллоидными частицами света.
В заключение отметим, что коллоидные системы распространены чрезвычайно широко. Знание свойств таких образований необходимо, например, и при очистке природных вод, и при добыче нефти [11].
Очень часто, для того чтобы провести взаимодействие двух веществ, необходимо их нагреть. Тепло — наиболее распространенный стимулятор химических превращений. Однако известны и другие виды энергии, например световая. Может ли свет вызывать химические реакции? Вспомните, как хорошо загорать в солнечный ветреный день, когда кожа совсем не ощущает солнечного тепла, и только свет вызывает образование в коже коричневого пигмента. Хорошо известен и обратный процесс — выцветание на ярком свету красителей ткани. Разрушение органических красителей под действием света — тоже химическое превращение. Итак, свет — отличный стимулятор химических реакций. Как же происходит взаимодействие света с веществом? Напомним сначала, что видимый свет — довольно узкий участок на шкале электромагнитных волн, он простирается от 4000 до 8000 А. Разные области этого участка воспринимаются глазом как свет определенного цвета. Так, волны с длиной около 4000 А или 400 нм — это фиолетовый свет; поток электромагнитных колебаний с длиной волны примерно 530 нм выглядит как зеленый свет, а красный свет имеет длину волны примерно 660 нм. Белый свет — это смесь всех лучей.
Свет можно рассматривать не только как волны, но и как поток частиц, называемых квантами или фотонами. Энергия Е, которую несут с собой кванты света, связана с частотой или длиной волны этого света таким соотношением:
E = hv = h^-, л
где h — постоянная Планка, равная 6,62 «10-27 эрг-с; v — частота света’ с-1; X — длина волны; с — скорость света. Отсюда видно, что чем меньше длина волны света, тем больше энергия, которую он несет. Поэтому фиолетовые и особенно ультрафиолетовые лучи обладают большей энергией, чем свет красный или желтый. Вспомним пример с загаром: можно быстро загореть под действием невидимых ультрафиолетовых лучей, а вот загореть у раскаленной печки, испускающей в основном красный и инфракрасный свет, еще никому не удавалось.
Еще в начале прошлого века был открыт первый закон фотохимии, т. е. науки, изучающей химические превращения под действием света. Этот закон, впрочем, достаточно очевиден: химические реакции вызываются только теми лучами, которые поглощаются веществом. Но вот обратное утверждение неверно: отнюдь не все поглощенные лучи вызывают химические превращения. Дело в том, что энергия этих лучей может затратиться на простой нагрев вещества. Другой закон фотохимии также легко понять: химическое действие света, как правило, пропорционально интенсивности света и времени его действия. Самый важный закон был сформулирован Эйнштейном, который установил, что каждый поглощенный квант света вызывает превращение лишь одной молекулы вещества. Количество квантов можно измерить, как и число молекул. Была введена даже специальная единица, равная одному молю квантов, т. е. 6,02 «1023. Эта единица называется эйнштейном. Используя вышеприведенную формулу, легко найти, что энергия одного поглощенного эйнштейна света с длиной волны 400 нм эквивалентна 71 ккал/моль. Энергия одного эйнштейна красных лучей с Х«800 нм эквивалентна 36 ккал/моль.
Вот квант света достиг молекулы. Что происходит? В любой молекуле связывание атомов осуществляется благодаря электронам. Эти электроны располагаются на нескольких уровнях (рис. 9). Рассмотрим некую молекулу с 4 электронами, способными взаимодействовать со светом, и таким же числом уровней энергии, на которых эти электро-
ны располагаются. В наиболее устойчивом состоянии молекулы электроны размещаются на двух нижних уровнях, по два на каждом. Обратите внимание: электроны обозначены стрелками, острия которых направлены в разные стороны. Это значит, что величины спинов электронов имеют противоположные знаки. Грубо говоря, это отражает то, что электроны вращаются в разные стороны относительно собственной оси. Падающий на молекулу квант света взаимодействует с электроном, передает ему свою энергию, и электрон как бы запрыгивает на более высокий энергетический уровень. При этом «прыжке» электрон может не менять направления вращения вокруг собственной оси, и тогда система приходит в так называемое синглетное состояние. В этом состоянии электроны хотя и находятся на разных уровнях, спины их спарены. В таком возбужденном состоянии молекула долго пребывать не может, электрон очень быстро, уже через 10-8 с, «соскальзывает» на нижний уровень. Этот процесс перехода электрона сопровождается выбросом кванта света, вещество как бы светится. Такое свечение называется флуоресценцией. Флуоресценцию нетрудно наблюдать. Погрузите в спирт лист зеленого растения. Когда раствор станет зеленым, профильтруйте его через вату или промокательную бумагу. Вынесите пробир-
Возбужденное синглетное состояние
Основное состояние
Возбужденно© триплетное состояние
Рис. 9. Взаимодействие света с молекулой вещества
йу или стакан такого раствора хлорофилла на свет. Вы увидите, что в отраженном свете (т. е. если смотреть на него сбоку) раствор флуоресцирует красноватым цветом.
Возможен и другой вариант: электрон при возбуждении не только переносится на более высокий уровень, но и в результате ряда превращений меняет ориентацию спина. Возникает частица в нестабильном триплетном состоянии. Продолжительность жизни такой частицы довольно большая, может достигать нескольких секунд. Но все-таки и здесь со временем электрон возвращается на более низкий уровень. Такой процесс сопровождается свечением, называемым фосфоресценцией. Если глазом флуоресценцию можно наблюдать, только пока на вещество падает свет, то фосфоресценция — это послесвечение, оно видно в течение нескольких секунд после того, как выключен источник света. Приведем рецепт фосфоресцирующей смеси, дающей зеленое свечение. Тщательно смешайте растертые компоненты: 10 г мела, 0,5 г сульфата натрия, 0,4 г буры, 3 г серы, 0,3 г сахара, 0,5 мл 5%-ного раствора нитрата висмута. Прокаливайте полученную смесь при температуре 800—900° С в течение 15 мин. И флуоресценция, и фосфоресценция, как правило, дают свет, обладающий меньшей энергией, нежели свет, падающий на вещество. Например, бензол, освещенный при температуре —200° С лучами с длиной волны 254 нм, флуоресцирует в ультрафиолетовой области при 290 нм, а фосфоресценция дает волны с длиной 340 нм.
До сих пор мы говорили о процессах, когда освещенная система со временем возвращается в исходное состояние. Но бывает и так, что возбуждения, вызванного квантом света, оказывается достаточно, чтобы произошел разрыв старых химических связей между атомами, чтобы завязались новые связи, одним словом, чтобы произошла химическая реакция. В этом случае перескок электрона на более высокий уровень — лишь первый этап коренной перестройки всей электронной системы. Налейте в пробирку немного концентрированного раствора перекиси водорода (пергидроль — опасное для кожи вещество) и поместите пробирку на яркий солнечный свет. Вы увидите пузырьки выделяющегося кислорода: 2H2O2+^v->2H2O+O2. Здесь символ hv обозначает квант света. Эта реакция начинается с образования радикалов гидроксила: Н2О2+/гу->2НО’.
Свет может расщеплять не только молекулу перекиси водорода. Квант света, упавший на кристалл бромистого серебра, который построен из чередующихся положитель
ных ионов серебра и отрицательных ионов брома, выбивает электрон из иона брома и «переселяет» его на ион серебра. В результате образуется атом металла и свободный галоген: 2AgBr+/iv->2Ag+Br2. Смоделировать этот процесс, лежащий в основе современной фотографии, нетрудно. В затемненной комнате прибавьте в стакане к раствору азотнокислого серебра (можно взять ляпис) раствор поваренной соли или бромистого калия. Выпавший осадок галогенида серебра аккуратно нанесите на промокательную бумагу тонким слоем и высушите в темном помещении. Еще лучше, если предварительно кашицу галогенида серебра смешать с набухшим в воде желатином. Теперь наложите на полученную «фотопластинку» трафарет из черной бумаги (можно вырезать любую фигуру) и поставьте «пластинку» на яркий свет. Через несколько минут снимите трафарет и осмотрите «пластинку» в затемненной комнате. Вы увидите, что в тех местах, куда попал свет, появилось темное окрашивание.
Надо заметить, что в отличие от человеческого глаза бромид, а тем более хлорид серебра чувствителен большей частью к ультрафиолетовым лучам. Поэтому в черно-белые фотоэмульсии, основу которых составляет галогенид серебра, вводят специальные сенсибилизаторы. Обычно это органические красители. Сенсибилизаторы отбирают.энергию всех падающих лучей и передают ее частицам галогенида серебра. Благодаря этому современная фотопленка по спектральной чувствительности подобна глазу (рис. 10). Зато на фотобумагу сенсибилизаторы не наносятся, такая эмульсия чувствительна только к синим и фиолетовым лучам. Поэтому с фотобумагой можно работать при красном свете.
Известно очень большое число самых различных химических реакций, происходящих под действием света. Здесь и разрушение молекул органических соединений, и присоединение различных веществ, например кислорода, и изомеризация. Наконец, известна одна реакция, которая протекает в зеленых растениях и имеет огромное значение. Это — фотосинтез. Именно благодаря фотосинтезу мы теперь можем жить на Земле, именно этот процесс позволяет превращать миллионы тонн углекислого газа атмосферы в необходимый живым организмам кислород. Общую схему фотосинтеза можно описать очень просто: CO2+H2O*-r/iv-> ->(СН2О)П+О2. Здесь (СН2О)П обозначает углевод, это может быть глюкоза, крахмал, клетчатка. Звездочка показывает, что кислород, выделяемый растением, берется не
из углекислого газа, а из воды, принимающей участие в процессе. Как ни просто выглядит это уравнение, реальный процесс фотосинтеза в растениях чрезвычайно сложен и во многом еще непонятен.
Процесс фотосинтеза можно разделить на два этапа. Первый этап протекает на свету, для осуществления химических реакций этой стадии требуется свет. Ключевая роль здесь принадлежит хлорофиллу, представляющему собой комплексное соединение, в котором атом магния заключен в кольцо из четырех азотсодержащих пиррольных циклов. Под действием кванта света молекула хлорофилла переходит в возбужденное состояние — электрон перескакивает на вышележащий энергетический уровень. После этого молекула может возвратиться в основное состояние, выбросив квант света. Флуоресценцию раствора хлорофилла вы наблюдали в ранее описанном опыте. В первой, световой, стадии фотосинтеза углекислый газ не участвует. В этот период под действием света при посредничестве хлорофилла происходит разложение воды. От воды отнимаются электроны, которые переходят к соединению с очень длинным названием — никотинамидадениндинуклеотидфосфату, сокращенно НАДФ. Это соединение переходит в свою восстановленную форму, обозначаемую условно НАДФ-Н. Уравне-
Рис. 10. Спектральная чувствительность галогенидов серебра, фотопленки и глаза человека
ние этой стадии: 2Н2О+2НАДФь4-/гт-^2НАДФ-Н+2Н4 -Н +О2. Одновременно идет и другой процесс — присоединение фосфатной группы к аденозин дифосфату, АДФ; образуется аденозинтрифосфат, АТФ. На этом световая стадия заканчивается. Что мы имеем в результате? Во-первых, мы получили хороший биологический восстановитель НАДФ-Н; во-вторых, мы зарядили энергией биохимический аккумулятор: из АДФ получили АТФ. Теперь уже без света в темновой стадии оба этих вещества будут использованы для восстановления двуокиси углерода в углевод. Уравнение темновой стадии: 6СО2+18АТФ-|-12НАДФ-Н4-12Н+-> ->CeH i 2Oe+18Фосфат+18 АДФ +12Н АДФ+.
Итак, продуктами фотосинтеза являются кислород и углеводы, например крахмал. Это нетрудно доказать. Веточку водного растения элодеи, широко распространенного в комнатных аквариумах, поместите в стакан с водой и накройте воронкой, на хвост которой помещена заполненная водой пробирка. Растение надо «покормить» углекислым газом, для этого желательно использовать газированную воду или бросить в стакан щепотку соды. Поставьте стакан на яркий свет. Постепенно пробирка заполнится газом. Поместите в пробирку тлеющую лучинку — она вспыхнет, подтверждая, что газ является кислородом. Второй опыт. Какое-либо комнатное растение, например примулу, поставьте на несколько дней в темную комнату. После этого поместите растение на подоконник, на яркий солнечный свет, а один из листочков прикройте с обеих сторон плотной черной бумагой, на которой вырезана какая-нибудь фигура. Через несколько часов или через день сорвите подопытный лист, поместите его в чашку с кипящей водой, затем в ста-жан с кипящим спиртом. Когда лист обесцветится, промойте его в холодной воде и положите в стакан с раствором йода и йодистого калия в воде. Йод дает с крахмалом синее окрашивание, которое будет заметно только в.тех местах, куда попал свет.
О биологическом фотосинтезе и его химическом моделировании можно прочитать в брошюрах и статьях [12].
В свое время на химическом факультете МГУ любили задавать абитуриентам такой вопрос: как получить из угля и любых неорганических веществ пикриновую кислоту, т. е. тринитрофенол? Вопрос этот хорош тем, что требует знания основных органических реакций, свойств веществ, а также способности, как в шахматах, видеть на несколько ходов вперед. Для нас вопрос интересен тем, что, проследив путь от угля до пикриновой кислоты, мы сможем узнать об одной из важных отраслей химии — органическом синтезе. Именно успехи органической синтетической химии привели к тому, что сегодня из угля, нефти и природного газа в промышленности получаются тысячи самых разнообразных веществ, среди которых лекарства, красители, средства защиты растений, душистые вещества.
Пикриновая кислота окрашена в ярко-желтый цвет и когда-то применялась как краситель для шелка и шерсти. Мы расскажем о различных путях синтеза этого соединения. Некоторые стадии синтеза вы сможете провести в своей лаборатории, но только некоторые, поскольку весь синтез потребовал бы специального оборудования. На самой первой стадии нам необходимо превратить углерод в какое-либо производное, из которого затем можно получить органическое вещество. Такой реакцией может стать взаимодействие угля и оксида кальция при повышенной температуре, в результате чего получается карбид кальция. Следующий «ход» нашей химической партии читателю, видимо, уже ясен: под действием воды карбид кальция выделяет ацетилен.
Получить ацетилен и исследовать некоторые его свойства можно с помощью простого прибора. Заткните пробирку резиновой или корковой пробкой с отверстием, через которое пропущена согнутая под прямым углом стеклянная трубка. Второй конец этой трубки входит в другую пробирку почти до дна. Поместите в первую пробирку маленький, не больше горошины, кусочек карбида кальция, капните на него несколько капель воды, быстро закройте пробирку пробкой с трубкой. Во вторую пробирку налейте слабый раствор марганцовокислого калия. Выделяющийся ацети
лен скоро обесцветит розовый раствор перманганата. Под действием перманганата ацетилен окисляется до щавелевой кислоты НООС—СООН, которая также взаимодействует с перманганатом, давая углекислый газ и воду. Перманганат при этом переходит в бесцветную соль двухвалентного марганца. Теперь повторите опыт, только во вторую пробирку налейте раствор ляписа, к которому добавлен нашатырный спирт — до растворения образующегося поначалу осадка. Пропустив через такой раствор ацетилен, вы получите желтый осадок ацетиленида серебра AgC=CAg. Только не высушивайте этот осадок — в сухом виде он легко взрывается при нагреве или ударе.
В 1922 г. советские химики Н. Д. Зелинский и Б. А. Казанский нашли, что, если пропускать ацетилен над активированным углем при температуре 500°С, он тримеризуется в бензол. Позднее было установлено, что такое превращение можно проводить и в гораздо более мягких условиях, используя катализаторы, которые представляют собой растворимые соединения различных металлов. Итак, мы «получили» бензол. В промышленности, правда, так не делают, ведь бензол содержится в нефти, его в больших количествах получают отщеплением водорода от насыщенных углеводородов, например от циклогексана.
От бензола к пикриновой кислоте ведут несколько путей (рис. И). Пойдем сначала по одному из них. Получим из бензола бензолсульфокислоту [13, с. 32]. Смешайте в пробирке несколько капель бензола и в 2 раза большее число капель концентрированной серной кислоты. Осторожно встряхивая пробирку, нагревайте раствор в кастрюле с кипящей водой. Через несколько минут, когда раствор станет однородным, вылейте содержимое пробирки в стакан с водой. Сульфокислота легко растворяется в воде и образует прозрачный раствор. Если твердую сульфокислоту сплавить со щелочью, образуется оксибензол, т. е. фенол. Мы уже близки к цели. Теперь фенол нужно пронитровать. Впрочем, если действовать разбавленной азотной кислотой, можно получить лишь мононитрофенол. Бросьте в пробирку несколько кристалликов фенола и добавьте несколько капель воды. В другой пробирке разбавьте концентрированную азотную кислоту водой в 2 раза и такой раствор очень осторожно по каплям приливайте к раствору фенола. Пробирку охлаждайте при этом в холодной воде. После окончания реакции вылейте содержимое пробирки в стакан с водой. Два изомера нитрофенола, орто- и пара-производные,
С + СаО
СаС2
Карбид кальция н2о
CH = СН Ацетилен
Cl2 (FeCI3)
Хлорбензол
HNO3
Рис. 11. Пути синтеза пикриновой кислоты
образуют нерастворимое масло с запахом горького миндаля. Если взять более концентрированную азотную кислоту, то в числе продуктов будут ди нитропроизводные фенола и нужная нам пикриновая кислота. Однако эта реакция не очень удобна — она протекает слишком бурно и дает много смолы.
Можно получить пикриновую кислоту и другим путем. Сначала бензол, нужно прохлорировать в присутствии хлорного железа как катализатора. Хлорбензол нитруют далее азотной кислотой. При этом получается тринитрохлорбензол, называемый также пикрилхлоридом. Замечательное свойство пикрилхлорида — большая активность атома хлора: под действием воды этот атом замещается на гидроксил. Еще один способ получения пикриновой кислоты: сначала синтезируют фенолдисульфокислоту. Проведите модельную реакцию в более мягких условиях и получите феносульфокислоту. Для этого в пробирку бросьте несколько кристаллов фенола и прибавьте каплю-другую серной кислоты. После нагревания в кипящей воде в течение нескольких минут вылейте реакционную массу из пробирки в стакан с водой. Образуется бесцветный раствор фенолсульфокислоты. Получающуюся в несколько иных условиях фенолдисульфокислоту нитруют азотной кислотой. При этом обе сульфогруппы заменяются на нитрогруппы, кроме того, в ядро входит е!це и третья нитрогруппа. В результате получается пикриновая кислота.
Наконец, тринитрофенол можно синтезировать и прямо из бензола, нитруя его азотной кислотой в присутствии солей ртути как катализатора. Читатель тут может воскликнуть: вот самый хороший способ! И зачем нужны другие более длинные пути? Дело, однако, в том, что самый короткий путь не всегда оказывается самым удобным в лабораторной или промышленной практике. Ведь очень большую роль играет то, с каким выходом образуется продукт; необходимо, чтобы не получалось много смолы, от которой трудно отделить продукт. Обычно приходится выбирать некий оптимальный вариант.
Мы рассказали о получении одного из желтых красителей, и путь к этому веществу довольно сложный. Более простым способом можно получить из того же фенола другую ярко-желтую краску, называемую аурином. Поместите в пробирку несколько кристаллов щавелевой кислоты и затем несколько большее количество фенола. Добавьте одну-две капли серной кислоты и осторожно нагрейте пробирку.
Вы увидите, как смесь постепенно приобретает желтый цвет. Реакционную массу охладите и залейте водой. Если к полученному желтому раствору добавить щелочь, он окрасится в оранжевый цвет. Проведенная вами реакция заключается в конденсации фенола с муравьиной кислотой, образующейся из щавелевой кислоты:
з
+ н — с
Итак, два красителя — тринитрофенол и аурин. А какие еще бывают красители, какими свойствами должно обладать органическое вещество, чтобы быть красителем? В первую очередь, естественно, краситель должен быть веществом окрашенным. Уже давно подмечено, что окраска вещества зависит от того, сколько в нем двойных сопряженных связей. Действительно, витамин А и каротин, содержащие несколько таких связей, окрашены в желтый цвет. То же относится и к конденсированным бензольным кольцам. Если нафталин и антрацен, содержащие два и три сцепленных кольца, бесцветны, то соединения, состоящие из пяти и шести бензольных ядер, имеют зеленый и синий цвет.
По одной из теорий цветности окрашенное соединение должно содержать так называемую хромофорную группировку, которой может быть, например, двойная углерод-углеродная С=С или азот-азотная N=N связь. Очень полезно присутствие в молекуле ауксохромной группы, которая углубляет цвет вещества. Пример ауксохрома — диметиламиногруппа— N(CH3)2. Придает цвет соединению и наличие в молекуле хиноидных фрагментов, подобных тому, что изображен в нижней части формулы аурина. Вот, к примеру, краситель метиловый оранжевый, очень широко применяющийся как индикатор на кислоты. В молекуле этого красителя присутствует хромофорная двойная связь между двумя атомами азота, ауксохромная ди метил-
аминогруппа, длинная цепь сопряжения двойных связей:
В этой формуле нет хиноидного фрагмента, но вещество можно изобразить и так:
Реально молекула имеет структуру среднюю между этими двумя. Но давайте прильем к раствору метилового оранжевого кислоту и присоединим тем самым протон к одному из атомов азота. В полученном соединении присутствует в явном виде хиноидный фрагмент да еще и целый положительный заряд:
А раз так, вещество должно иметь более глубокую окраску. Действительно, протонированная форма красителя имеет красный цвет. .Это свойство и позволяет использовать краситель в качестве индикатора на кислоты.
Органические соединения, применяющиеся как красители, относятся к разным классам. Очень широко используются так называемые азосоединения, о которых мы говорили (два атома азота, соединенных двойной связью, называются азогруппой). Получаются они из диазосоединений и фенолов или ароматических аминов. Диазосоединения, в свою очередь, можно синтезировать из производных анилина реакцией диазотирования и азосочетания [14, с. НО]. Для проведения этой реакции вам понадобятся два реактива — анилин и азотистокислый натрий, которые можно достать в любой химической лаборатории. Разбавьте в стакане концентрированную соляную кислоту водой в 3 раза. К 3—4 чайным ложкам такого раствора прилейте пол-ложки анилина и раствор охладите в кастрюле с толченым льдом. Пока раствор анилина охлаждается, приготовьте два других раствора: азотистокислого натрия и раствор йодистого калия и крахмала в воде. Последний раствор можно заменить таким — к разбавленной водой желтой йодной тинктуре
присыпьте аскорбиновой кислоты до обесцвечивания и затем добавьте немного крахмала. Теперь в стакане или пробирке при охлаждении льдом к раствору анилина (возьмите его несколько чайных ложек) по каплям прибавляйте раствор азотистокислого натрия. Во время прибавления смесь перемешивайте, встряхивая стакан. После прибавления нескольких капель берите пробу: каплю реакционного раствора смешайте на стекле с каплей йод-крахмального раствора. Как только будет появляться синее окрашивание, прекращайте прибавление раствора азотистокислого натрия. Вы получили в растворе хлористый фенилдиазоний.Вот химизм приведенных реакций. Из анилина и соляной кислоты образуется соль: СвНб—NH2+HCl->CeH6—NH3C1-. Из азотистокислого натрия и соляной кислоты получается свободная азотистая кислота: NaNO2+HCl->HNO2+NaCI. Соль анилина и азотистая кислота дают хлористый фенил-диазоний: CeH5 — NHSC1- + HNO2-^CeH5 — N=N-C1-+ +2Н2О. Избыток азотистой кислоты обнаруживается легко, поскольку эта кислота окисляет йодистый калий до свободного йода, дающего с крахмалом синее окрашивание.
Соли диазония — реакционноспособные соединения. Они легко реагируют с водой при повышенной температуре. Нагрейте в пробирке или в стакане немного полученного вами раствора хлористого фенилдиазония в кастрюле с кипящей водой. Вы увидите выделение пузырьков азота и почувствуете запах фенола: СвН5—N=N-СН+НбО->С6Н5— —OH+N2+HC1. В другой пробирке к раствору фенилдиазония прилейте раствор йодистого калия и смесь нагрейте в кипящей воде. После того как прекратится выделение пузырьков азота, на дне пробирки выделится тяжелая жидкость. Это йодбензол. Самое интересное свойство диазосое-динепий — способность замещать атом водорода в молекулах фенолов или анилина. При этом образуются азосоединения:
Проведите эту реакцию. В пробирке или в стакане прибавьте к фенолу раствор едкого натра и затем прилейте раствор хлористого фенилдиазония. Выпадает желтый осадок азосоединения. В этом красителе ауксохромом является гидрок
сильная группа. Эта группа очень полезна красителю еще и по другой причине: молекула благодаря ей прочно прикрепляется к волокну шерсти или шелка. Простейший краситель можно получить прямо на ткани. Для этого хлопчатобумажную ткань пропитывают щелочным раствором фенола и затем погружают ее в раствор соли диазония.
Мы видим, что анилин — необходимая составная часть азокрасителей. Но из анилина можно получать краски другого рода. Для этого его окисляют, образуется очень прочный черный краситель, так называемый черный анилин. Этот опыт можно провести в нескольких вариантах. В пробирке к нескольким каплям раствора анилина в разбавленной соляной кислоте добавьте раствор двухромовокислого калия или хлорной извести. Возникает темно-синее окрашивание. Взболтайте в пробирке смесь анилина с водой и прилейте к эмульсии раствор двухромовокислого калия в разбавленной серной кислоте. При нагревании раствора в горячей воде выпадает черный осадок. Если эмульсией анилина пропитать кусочек ткани и затем погрузить ее в раствор хромовой смеси, т. е. кислый раствор двухромовокислого калия, ткань окрасится в черный цвет.
Краситель еще одного типа можно получить из известных лекарств. Поместите в пробирку таблетку фталазола, щепотку резорцина (он используется в медицине как антисептик) и несколько капель концентрированной серной кислоты. Нагрейте пробирку осторожно на пламени горелки и дайте смеси охладиться. Добавьте к плаву раствор едкого натра и вылейте содержимое пробирки в стакан с водой. Вы имеете возможность наблюдать очень красивую желто-зеленую флуоресценцию. Для того чтобы такое свечение было еще заметно, достаточно в 40 000 000 частей воды растворить всего одну часть полученного вами красителя, называемого флуоресцеином. Это соединение используется для определения маршрутов подземных рек. В ручеек выливают немного красителя и затем в некоторых местах выхода подземных вод на поверхность наблюдают желто-зеленую флуоресценцию.
Мы провели несколько синтезов, но ни в одном из них не выделяли продукт в чистом виде. Существует много способов выделения веществ из реакционной смеси и их очистки. Кристаллические вещества, например, лучше всего очищать перекристаллизацией. Принцип этого метода вот в чем: примесей в веществе обычно довольно мало, и они не образуют насыщенного раствора даже при охлаждении.
Поэтому, если горячий насыщенный раствор какого-либо вещества охладить, выпадают кристаллы лишь этого вещества, а примеси остаются в растворе. Проведите синтез нитронафталина. На дне стакана к 1—2 чайным ложкам концентрированной азотной кислоты добавьте чайную ложку нафталина и погрузите стакан на 2—3 мин в сосуд с кипящей водой. Теперь вылейте содержимое в другой стакан, заполненный наполовину холодной водой. Получившийся а-нитронафталин выделяется в виде желтой массы. Продукт содержит примеси, для очистки перекристаллизуйте его. Во-первых, слейте воду и налейте в стакан с желтой массой этиловый спирт. Стакан нагрейте в сосуде с горячей водой и добавляйте спирт до тех пор, пока почти весь продукт не растворится. Теперь слейте горячий раствор в другой стакан. Через некоторое время по охлаждении раствора чистый нитронафталин выделится в виде желтых игольчатых кристаллов. Некоторые твердые вещества легко возгоняются, т. е. переходят в парообразное состояние, минуя жидкое. Конденсируясь снова в кристаллы, вещество избавляется от нелетучих примесей. Бросьте на дно стеклянной банки щепотку нафталина, закройте банку сверху металлической пластинкой. Теперь нагрейте снизу банку на газовой горелке или на свече. Кристаллы нафталина осядут на холодной металлической пластинке.
Об органическом синтезе см. популярные издания [15].
По значению для человека обоняние, конечно, не сравнится со зрением или слухом. Но насколько беднее была бы наша жизнь, насколько бледнее стал бы окружающий нас мир, если бы мы не воспринимали запахов. От чего же зависит запах вещества? Почему одни вещества пахнут приятно, другие — отвратительно, третьи вовсе не имеют запаха?
Всем хорошо знаком запах горького миндаля. Оказывается, этот аромат имеют сразу несколько различных веществ. При действии азотной кислоты на бензол образуется нитробензол, пахнущий миндалем. Так, может быть, сочетание бензольного кольца с нитрогруппой — это необходимое и
достаточное условие возникновения миндального аромата? Нет, оказывается, бензойный альдегид, в котором альдегидная группа связана с фенилом, имеет такой же запах. Сделаем такой вывод: сочетание бензольного кольца с различными группами приводит к тому, что вещество получает запах миндаля. И опять вывод неверный. Фенол, в котором бензольное кольцо связано с гидроксильной группой, пахнет отнюдь не миндалем. А с другой стороны, пропионитрил и синильная кислота, не содержащие бензольного кольца, имеют аромат миндаля. Вот два вещества:
сно сно
Они очень похожи и различаются только взаимным расположением групп ОН и ОСН3. Однако такое небольшое различие в строении приводит к драматическим изменениям в запахе соединений. Первое вещество называется ванилином и имеет всем хорошо известный аромат ванили. Второе соединение почти не пахнет. Ванилин является производным фенола, и это можно доказать. К раствору хлорного железа прилейте каплю-другую раствора ванилина в водке. Появляется характерное фиолетовое окрашивание.
Так можно ли найти хоть какие-нибудь связи между строением вещества и его запахом? В настоящее время нет четкой общепринятой теории запаха, уж больно сложна проблема (подробнее о теориях запаха и вкуса см. [16]). Существует воззрение, что любой запах можно разложить на отдельные элементы. Молекулы самых различных веществ, относящихся к одному и тому же типу запаха, должны иметь схожее пространственное строение. И тут уже неважно, какие атомы — углерода, кислорода, азота — находятся в данном месте молекулы. Главное, чтобы контуры молекул одного запаха были похожи. Дело в том, что предполагается, что при обонянии молекула вещества подходит к специальному рецептору и укладывается в особую щель. Щель эта имеет специфическую форму, соответствующую форме данной молекулы. Одним словом, принцип ключа и замка. На основе этой теории была проведена классификация запахов. Все запахи были сведены к семи типам: гнилостный, острый, эфирный, мятный, цветочный, мус
кусный, камфорный. Подавляющее большинство веществ, обладающих перечисленными здесь запахами, могут быть синтезированы в специальных хорошо оборудованных лабораториях или на заводе. Такие синтезы обычно сложны. Однако некоторые синтезы нетрудно провести и в домашней лаборатории.
Вещество, обладающее гнилостным запахом,— сероводород — всем известно. Этот запах имеет протухшее яйцо. Сероводород образуется из некоторых аминокислот белка при гниении яйца. Сероводород можно получить действием кислоты на сернистые соли металла. Газ этот весьма ядовит. Пример вещества, имеющего острый запах,— муравьиный альдегид. Раствор этого соединения в воде — формалин — доступен в быту. Другие альдегиды имеют более приятный, менее острый запах. Например, ацетальдегид СН3СНО пахнет прелыми яблоками. Это соединение получается при окислении этилового спирта. Смешайте в стакане чайную ложку серной кислоты с таким же объемом воды. Добавьте к полученной разбавленной кислоте раствор двухромовокислого калия в воде. Образовалась хромовая смесь. К ней осторожно добавьте несколько капель этилового спирта. Цвет раствора меняется от желтого до зеленого — появляется производное трехвалентного хрома. А этиловый спирт при этом окисляется в уксусный альдегид, который можно обнаружить по запаху прелых яблок. Если в таком соединении заменить метильную группу на этиленовый остаток, получим акролеин СН2=СН—СНО. Это соединение обладает резким неприятным запахом. Его можно получить из глицерина или из масла. Положите на сковороду щепотку кислого сернокислого калия, добавьте две капли подсолнечного масла и нагрейте на газовой горелке. Обнаружить выделяющийся акролеин легко по раздражающему слизистую оболочку запаху.
Запах простого диэтилового эфира всем хорошо знаком— это вещество применяется в медицине. Еще одно вещество напомнит вам своим запахом кабинет врача. Получите его из этилового спирта. Поместите на дно стакана 2—3 чайные ложки водки и несколько кристаллов йода (можно взять йодную тинктуру, разбавив ее водой). Теперь к раствору прилейте раствор едкого натра в воде так, чтобы окраска йода исчезла. Подогрейте стакан в кастрюле с теплой водой. Через некоторое время после охлаждения выпадут желтые кристаллы йодоформа СН13, который применяют в медицине как антисептик.
Вс® сложные эфиры, образованные органическими кислотами и спиртами, обладают приятным фруктовььм запахом. Их нетрудно получить, нагревая на водяной бане смесь органической кислоты и спирта в присутствии нескольких капель концентрированной серной кислоты. Вот какие запахи имеют некоторые сложные эфиры, получаемые: из изоамилового спирта и муравьиной кислоты — сливы, из бутилового спирта и уксусной кислоты — ананаса, изоамилового спирта и уксусной кислоты — груши, бензилового спирта и муравьиной кислоты — жасмина, фенилэтилового спирта и муравьиной кислоты — хризантемы.
Специфический запах природных продуктов обычно бывает обусловлен каким-то содержащимся в данном продукте веществом. Например, главная составная часть скипидара, придающая ему особый запах,— пинен, циклическое соединение, содержащее двойную связь. Наличие двойной связи легко подтвердить экспериментально. На дне стакана к нескольким каплям скипидара добавьте немного раствора перманганата калия, перемешайте. Исчезает розовая окраска, выпадает бурый осадок двуокиси марганца. В отличие от скипидара запах хлеба определяется очень сложной смесью разных веществ. Здесь и кетоны, и альдегиды, и органические кислоты. Но одно из основных веществ запаха хлеба — гетероциклический альдегид фурфурол. Его можно получить искусственно из сахаров, содержащих пять атомов углерода. А такие сахара получаются при действии кислоты на древесину. Насыпьте в пробирку немного древесных опилок и смочите их разбавленной соляной кислотой. Нагрейте смесь до легкого кипения. После охлаждения нейтрализуйте кислоту содой, добавьте воды и нагрейте. Фурфурол летит с водяным паром, и его можно обнаружить по характерному запаху.
Те, кто имеет некоторые навыки работы в химической лаборатории (например, в школьном кружке), могут провести более сложный синтез душистого вещества, смоделировав тем самым химическое производство. Попробуйте получить из нафталина вещества, обладающие запахом цветов акации и апельсинового дерева. В тонкостенном химическом стакане к двум чайным ложкам нафталина добавьте четверть стакана концентрированной серной кислоты. Нагревайте смесь при помешивании стеклянной палочкой на слабом пламени газовой горелки до получения однородной массы. Реакционную смесь охладите, добавьте к ней немного воды. Через некоторое время выпадут кристал
лы |3-нафталинсульфокислоты. Если они не выпадают, поставьте раствор в закрытой посуде в холодильник. Раствор слейте с кристаллов, к которым добавьте одну-две чайные ложки гранулированного едкого калия и такое же количество воды. Нагревайте эту смесь в химическом стакане на пламени горелки в течение нескольких минут. Образуется р-нафтол. Получившуюся серую массу осторожно вылейте в стакан, заполненный наполовину водой. Отфильтруйте раствор от осадка и прибавьте к раствору по каплям концентрированную серную кислоту. Выпавший осадок Р-нафтола отфильтруйте и высушите на фильтре на воздухе. Соскоблите этот осадок в пробирку, залейте его этиловым спиртом, прибавьте к смеси каплю концентрированной серной кислоты и нагревайте реакционную массу в сосуде с горячей водой до тех пор, пока спирт не испарится. В остатке вы получите этиловый эфир Р-нафтола — соединение со слабым приятным запахом, напоминающим запах цветов акации. Вот схема проведенных вами превращений:
Чтобы лучше почувствовать запах соединения, растворите вещество в эфире и смочите раствором кусочек промокательной бумаги. Если этиловый спирт заменить метиловым на последней стадии (метиловый спирт — очень ядовитое вещество), получается метиловый эфир Р-нафтола. Это вещество, называемое нерол и ном, имеет сильный запах цветов апельсинового дерева. Неролин широко применяется в парфюмерной промышленности, его добавляют в душистые мыла.
Проведите два опыта. Первый весьма прост. Прилейте к раствору азотнокислого серебра в воде каплю раствора поваренной соли. Немедленно выпадает белый осадок хлорида серебра. Второй опыт посложнее. Поместите в пробирку несколько капель концентрированной серной кислоты и
чрезвычайно осторожно прибавьте к ним по каплям такое же количество концентрированной азотной кислоты. Охладите полученную смесь в сосуде с холодной водой и прилейте в пробирку несколько капель бензола. Теперь заткните пробирку пробкой, в которую вставлена длинная стеклянная трубка. Нагревайте пробирку в сосуде с горячей водой — температура воды не должна быть выше 50°С. Через несколько минут осторожно слейте верхний слой в другую пробирку и добавьте к этой жидкости одну-две чайные ложки воды. Получившийся нитробензол — жидкость с запахом миндаля — тяжелее воды и опустится на дно пробирки.
Реакции, как видим, различаются весьма существенно. Если первая протекает за считанные доли секунды, при обычной температуре, то вторая требует нагревания в течение нескольких минут. Дело здесь вот в чем. В первой реакции встречаются два иона — серебра и хлора, и тут же образуется нерастворимое в воде соединение. Столь быстро протекают почти все реакции неорганических ионов. А теперь о второй реакции. Мы нитруем бензол азотной кислотой в присутствии серной кислоты. Когда мы смешиваем эти две кислоты, происходит химическая реакция, в результате которой образуется так называемый нитроний-катион NO^: HNO3+2H2SO4MH3O++2HSOz+NO2+.
В реакционном растворе присутствует еще и бензол. По шестиугольнику-его молекулы рассредоточено шесть л-элек-Уронов. Эти электроны взаимодействуют с нитроний-катио-ном, образуя так называемый л-комплекс, в котором группа NOJ одновременно «привязана» ко всем шести атомам углерода бензольного кольца (рис. 12).
Будем изображать ход реакции на энергетической диаграмме. На вертикальной оси диаграммы станем откладывать энергию реагирующей системы частиц. Очевидно, что если из одной точки диаграммы мы переходим в другую и при этом поднимаемся на более высокий энергетический уровень, то система при этом откуда-то приобретает энергию. Она может черпать ее, например, из запасов энергии хаотического движения частиц. Переход сверху вниз на энергетической диаграмме может совершаться спонтанно. Поэтому чем выше точка на диаграмме, тем неустойчивее соответствующее состояние системы. Из такого состояния система может выйти самопроизвольно.
На нашей диаграмме переход от одного местного, локального минимума энергии к соседнему соответствует стадии реакции. На протяжении же одной стадии движение
вдоль пути реакции можно понимать как взаимное перемещение реагирующих молекул, частиц или их частей. Вот самое начало реакции: сближаясь издалека, реагирующие частицы поначалу взаимно отталкиваются, и кривая на диаграмме идет вверх. В результате взаимодействия образуется относительно устойчивое образование — кривая снижается, возникает минимум. Именно так и изобразится на энергетической диаграмме образование л-комплекса бензола с нитроний-катионом. Их комбинация не очень устойчива и может легко распасться на исходные компоненты, что соответствует движению вспять на диаграмме. Но может произойти и другой процесс — перестройка молекулы и образование а-комплекса. При этом нитрониевая частица образует обычную ковалентную связь с одним из углеродных атомов бензольного кольца. Атом водорода, связанный с этим углеродом, немного выходит из плоскости кольца, положительный заряд с нитроний-катиона «соскальзывает» и размазывается по оставшимся пяти углеродным атомам. На энергетической диаграмме о-комплексу соответствует небольшая впадина «высоко в горах».
Следующая стадия реакции — отщепление протона Н+ от углеродного атома и образование л-комплекса с протоном. Протон в л-комплексе долго не задерживается, и скоро образуется нитробензол. Вот как сложно протекает простая на первый взгляд реакция замены атома водорода в бензоле на нитрогруппу. Эта реакция принадлежит к семейству
Координата реакции
Рис. 12. Энергетическая диаграмма реакции нитрования бензола нит» роний-катионом
хорошо известных в химии реакций электрофильного замещения. Здесь электрофильной, т. е. «любящей электроны», частицей выступает нитроний-катион, а бензол — вещество, поставляющее электроны. К реакциям электрофильного замещения относятся хлорирование, бромирование, сульфирование бензола и других ароматичесих соединений. Электрофильными частицами в таких реакциях могут выступать и ионы ртути, таллия, свинца.
В молекуле бензола все шесть атомов водорода равноценны, иная картина в случае толуола. Атакуя толуол, электрофильная частица оказывается перед выбором: она может заместить водород, соседний с метильной группой (это положение в бензольном ядре называется «орто»), она может вступить в противоположное от метильной группы пара-положение. Наконец, есть третья, мета-позиция:
сн3
Орто Орто
Мета Мете
Пара
В молекуле толуола два равноценных орто-положения, два мета-положения и лишь один пара-протон.
Метильная группа обладает способностью подавать, накачивать электронную плотность. Но вот эта дополнительная электронная плотность, поступающая в бензольное кольцо, распределяется в нем неравномерно. Чтобы определить места с наиболее высокой электронной плотностью, удобно нарисовать формулу бензола с традиционными, хотя и не отражающими реального положения, двойными связями. Вот как стрелками можно изобразить смещение электронного облака:
Здесь значком 6— (дельта минус) мы обозначили места с наибольшей! плотностью электронов. Видимо, именно к этим атомам, т. е. в орто- и пара-положения, и устремится нитрогруппа. Но это еще не все. Рассмотрим структуры возможных при замещении в разные положения о-комплек-сов. И условно нарисуем положительный заряд рассредоточенным не по всему кольцу, а сидящим на одном опре
деленном атоме. Вот о-комплекс при орто-замещении:
Обратите внимание: возникающий положительный заряд расположен рядом с метильной группой, которая несколько погашает этот «плюс». Такое погашение приводит к тому, что о-комплекс становится более устойчивым. Реакция идет охотнее. Посмотрим, что получится, если нитрогруппа входит в мета-положение:
сн3
В этом случае атом углерода, несущий заряд, отделен от метильной группы простой связью, и поэтому метил не может гасить положительный заряд. При замещении нитрогруппой пара-положения ситуация напоминает разобранную нами для случая орто-замещения (попробуйте показать это на формулах). Действительно, экспериментально установлено, что нитрование идет преимущественно в орто- и пара-положения. Если проанализировать смесь изомеров, получившихся при нитровании толуола, то окажется, что она на 60% состоит из орто-производного, 36% составляет пара-нитротолуол и только 4% приходится на долю метаизомера.
Мы разобрали последовательность превращений, которые происходят, прежде чем образуется нитропроизводное. Но откуда ученым известно, какие это превращения, какие промежуточные вещества образуются на пути реакции? Ведь мы имеем исходные вещества — бензол, азотную и серную кислоты, получаем конечное вещество — нитробензол. Каждое из них можно подержать в руках, взвесить, проанализировать. А что между начальным и конечным веществом? Ситуация напоминает «черный ящик», когда мы достоверно знаем лишь то, что в него кладут и что из него через какое-то время вынимают. А вот о том, что происходит в самом ящике, какие там разыгрываются превращения, какие возникают промежуточные соединения, часто приходится судить на основании косвенных данных. Представь
те себе, что вы проводите реакцию и по ее ходу измеряете концентрацию исходного и конечного вещества. Иногда оказывается, что через некоторое время после начала реакции исходное вещество израсходовалось в заметной степени, а конечного продукта образовалось еще очень мало. Значит, в реакционной смеси в этот момент времени находится некое промежуточное соединение.
Как же установить структуру такого соединения, концентрация которого зачастую очень невелика? Тут помогает физические методы исследования — ядерный магнитный резонанс, инфракрасная и электронная спектроскопия, электронный парамагнитный резонанс. Эти методы дают информацию о наличии в веществе тех или иных группировок, о порядке, в котором связаны атомы в молекулах. Впрочем, иногда промежуточные соединения можно обнаружить даже визуально. На стекле к капле концентрированной серной кислоты добавьте нафталин или фенол и затем крупинку нитрата натрия (можно использовать селитру или ляпис). Раствор окрасится в интенсивный желтый или зеленый цвет. Предполагают, что окрашенные вещества — это промежуточные в нитровании а-комплексы. Тут надо заметить, что недавно советские ученые установили, что подобные окрашенные соединения представляют собой продукт переноса одного электрона от ароматического соединения к иону NOf. В результате образуется так называемый ка-тион-радикал ароматического углеводорода и радикал NO2. Катион-радикал и радикал на следующей стадии соединяются, образуя уже известный нам о-комплекс. Как видим, механизм, включающий л- и a-комплексы, оказывается еще более сложным. Так что ученым предстоит большая работа, прежде чем станет в деталях известно, как же протекает столь давно открытая и, казалось бы, подробно изученная реакция, как нитрование.
Ученый-химик, прежде чем начинать растворять и фильтровать, растирать и упаривать, т. е. прежде чем начинать «истинно» химическую работу, частенько обращается к моделям молекул и на них проигрывает предстоящую реак
цию. Надо сказать, что иногда такие «игры» приносят больше совершенно неожиданных, великолепных открытий, чем чисто химический эксперимент. А сколько нетривиальных идей могут подсказать «химические игрушки»!
...Начало 50-х годов нашего века, Англия. Биолог Уотсон и физик Крик начинают работать над проблемой структуры дезоксирибонуклеиновой кислоты (ДНК), которая, как они предполагали, отвечает за передачу наследственных признаков организмов. Ученые решили воспользоваться подходом, который за несколько лет до этого позволил Полингу установить структуру белковых молекул. Подход этот заключался в том, что исследователи сделали из шариков, картона и проволоки модели отдельных частей молекулы ДНК и затем стали собирать из них целую нить. Делая заготовки этих частей, Уотсон и Крик часто советовались с химиками о форме того или иного гетероцикла, входящего в состав ДНК, о возможных направлениях тех или иных химических связей. Множество вариантов было перепробовано, много бессонных ночей проведено в раздумьях, прежде чем созрело простое, изящное решение: молекула ДНК состоит из двух нитей, переплетающихся в спираль, причем в центре вдоль спирали располагаются пары молекул-гетероциклов, а снаружи остов двух цепей образуется остатками фосфорной кислоты. Невозможно переоценить значения этого открытия века, сделанного, так сказать, «играючи».
Давайте и мы займемся моделированием молекул химических соединений. Для этого нам понадобится очень простое «оборудование»: коробка спичек, картон и несколько кусочков пластилина. В первую очередь изготовьте модель молекулы метана СН4. Для этого воткните в черный пластилиновый шарик размером с вишню, который будет изображать атом углерода, четыре спички так, чтобы головки спичек располагались по углам правильного тетраэдра. Форму тетраэдров имеют четырехгранные бумажные пакеты для молока. Головки спичек будут символизировать атомы водорода, а сами спички — связи углерод—водород. От метана нетрудно «перейти» к этану СН8СН8. Для этого сделайте вторую модель молекулы метана, но вместо одной спички углерод—водород воткните серной головкой в черный пластилиновый шарик одну из спичек первой модели. Теперь таким же образом присоедините к этану еще один метильный фрагмент —СН8. Вы получили модель молекулы пропана СН3СНаСН8.
Попробуем теперь сделать модель молекулы хлористого пропила. Для этого нужно в молекуле пропана заменить один из атомов водорода на атом хлора. Пусть атом хлора изображается большим белым шариком из пластилина. Достаточно воткнуть одну из спичек пропановой модели серной головкой в этот белый шарик. Но тут возникает вопрос: какую спичку воткнуть — крайнюю или соединенную со средним углеродом? Оказывается, заменяя на хлор водород при крайнем атоме углерода, вы придете к нормальному хлористому пропилу, кипящему при 45°С. Если же хлор будет соединен спичкой-связью со средним углеродом, получится модель хлористого изопропила, кипящего при 35°С (рис. 13,а). Два или несколько таких веществ, имеющих одинаковую общую формулу (в нашем случае С3Н7С1), но различающиеся порядком, в котором соединяются атомы друг с другом, называются изомерами. Изготовьте теперь модель молекулы этилена. Для этого соедините два углеродных шарика двумя спичками (двойная связь) и в каждый шарик воткните еще по две спички, лежащие в плоскости двойной связи. «Получите» производное этилена, несущее два атома хлора при разных углеродных атомах. Оказывается, и тут могут быть варианты: либо атомы хлора лежат по одну сторону от двойной связи (^uc-изомер), либо по разные стороны (/иран^-изомер) (рис. 13,6). Мы столкнулись с одной из разновидностей изомерии — геометрической цис-/ишнсизомерией. /{wc-изомер дихлорэтилена кипит при 60°С, /илляс-изомер — при 48°С. Как видим, изомеры разоб-
а Ь*
Рис. 13. Модели изомеров хлористого пропила (а) и дихлорэтилена (б)
ранных нами типов отличаются по физическим свойствам, например температурами кипения.
Вернемся снова к метану. Замените в модели его молекулы три атома водорода на такие группы: карбоксил СООН (пусть это будет большой белый пластилиновый шарик), гидроксил ОН (сделайте его из белого или цветного пластилина) и метил СН3 (черный большой шарик). Поставьте такую молекулу перед зеркалом и, глядя на зеркальное отражение, сделайте точную копию этого отражения. Вы будете иметь модели двух молекул, как будто бы и очень похожих, но не одинаковых (рис. 14, а). Дело в том, что их нельзя совместить друг с другом, как нельзя совместить правую и левую руки, как нельзя надеть левую перчатку на правую руку. Будут ли отличаться вещества, построен-
б
Рис. 14. Модели оптических антиподов хиральных молекул молочной (а) и винной (6) кислот
ные из таких левых и правых молекул? Будут, но только не температурами кипения и не большинством химических свойств. Однако есть свойство, которое их очень четко различает,— отношение к поляризованному свету. Свет — это поток электромагнитных волн. В обычном свете векторы электрического и магнитного полей смотрят перпендикулярно линии распространения луча, причем в любом направлении. Если же такой свет пропустить через специальные фильтры, то вектор магнитного (и электрического) поля в луче будет направлен вдоль линии, свет станет поляризованным. Пропустив свет, поляризованный в одной плоскости, через раствор левых или правых молекул, мы увидим, что плоскость поляризации повернется на какой-то угол. Самое замечательное, что для молекул одного типа она повернется влево, а для молекул-антиподов — на точно такой же угол вправо.
Вы держите в руках две разновидности молекул молочной кислоты СН3СН (ОН)СООН. Эти разновидности, относящиеся друг к другу как предмет и его зеркальное изображение, называются оптическими антиподами или энантиомерами. Свойство молекулы или любой геометрической фигуры не совпадать со своим отражением в зеркале называется хиральностью. Это слово происходит от греческого «хир», что значит «рука», ибо правая и левая руки — самое близкое нам всем воплощение хиральности. Для того чтобы молекула могла существовать в виде двух антиподов, необходимо, например, в ее составе иметь атом углерода, связанный с четырьмя разными заместителями (как в нашем случае, где заместители —Н, —СН8, —ОН и —СООН). Такой атом углерода называется асимметрическим или еще хиральным центром. Оптически активных соединений можно придумать великое множество, ведь мы можем присоединять к атому углерода любые заместители в любых сочетаниях. А что если в молекуле присутствуют два асимметрических атома углерода? Рассмотрим простейший случай, молекулу винной кислоты НООССН (ОН)СН (ОН)СООН. Повозитесь с моделями, и вы увидите, что для этого соединения можно придумать три варианта (рис. 14, б). Вещество, состоящее из молекул одного вида, вращает плоскость поляризации вправо, раствор молекул — зеркальных антиподов вращает влево. Наконец, возможен третий вариант, когда вещество не вращает плоскость поляризации. Это происходит потому, что одна половинка молекулы вращает плоскость вправо, а другая половинка — влево.
Оптическая изомерия — явление, чрезвычайно широко распространенное в природе. Все аминокислоты присутствуют в живых организмах лишь в виде одного энантиомера. Только при этом условии молекулы белков, построенные из аминокислот, могут образовывать правильную спираль. Сахар, глюкоза тоже существуют в виде оптических изомеров, причем в природе используется лишь один антипод. Это не случайно — ферменты, которые проводят химические реакции в клетке, четко различают относительное расположение заместителей вокруг асимметрических атомов углерода. Интересно, что наш язык иногда оказывается прекрасным прибором, позволяющим различать энантиомеры. Так, один из антиподов аспарагиновой кислоты имеет сладкий вкус, другой безвкусен. У аминокислоты изолейцина стереоизомеры различаются еще сильнее — один сладкий, другой горький.
Для появления оптической изомерии, оптической активности не обязательно наличие классического асимметрического атома углерода. Нужно, чтобы молекула лишь не совпадала со своим зеркальным отображением, т. е. была бы хиральной. Вырежьте из картона два шестиугольника. Это будут молекулы бензола. К трем соседним вершинам шестиугольников приклейте по спичке, на две крайние спички насадите белый и черный пластилиновые шарики, а средней спичкой соедините два таких замещенных кольца. Вы увидите, что соединить кольца можно двумя способами, что получаются два оптических антипода (рис. 15, а). Действительно, оптическая изомерия в таких дифенилах (белый шарик обозначает, например, группу —СООН, черный — нитрогруппу —NO2) была обнаружена еще в 1922 г. А вот совсем удивительные примеры, полученные в последние десятилетия. Сделайте из картона два правильных пятиугольника и соедините их в «бутерброд» пластилиновым шариком. Вы получили модель молекулы ферроцена. Пятиугольники— это углеводородные кольца С5Нб, шарик между ними — атом железа. «Заместите» два атома водорода при вершинах пятиугольников на черный и белый шарики. Вы увидите, что возможны два энантиомера (рис. 15, б). Сделайте молекулу акриловой кислоты: заместите в модели молекулы этилена один атом водорода на белый шарик, карбоксил —СООН. Прикрепите к двойной связи большой кусок пластилина, который будет символизировать группу Fe(CO)4. И опять возможны два энантиомера (рис. 15, в).
Склейте полоску бумаги в кольцо,*но только передсклеи-
ванием двух концов поверните один из них на 180° вокруг оси, проходящей вдоль полоски. Вы получите ленту Мёбиуса, хорошо известную математикам. Она обладает одним замечательным свойством: если вы захотите покрасить эту ленту с двух сторон, то сможете сделать это, не отрывая кисти от ленты. Получается, что у ленты одна-единственная сторона. Но лента Мёбиуса приготовила нам еще один сюрприз. Оказывается, одностороннюю ленту можно склеить из полоски бумаги двумя способами. В результате получаются две структуры, которые никак нельзя совместить в
в
Рис. 15. Модели изомеров хиральных дифенилов (а), ферроценов (б) и л-комплекса этилена (в)
пространстве, но которые относятся друг к другу как предмет к своему зеркальному отражению. Одним словом, лента Мёбиуса хиральна. Если перед склеиванием концов полоску повернуть на 360°, получатся структуры, которые также хиральны и обладают одним любопытным свойством (рис. 16 вверху). Чтобы это свойство обнаружить, воткните ножницы в середину полосок и разрежьте их вдоль оси. Получаются два переплетенных кольца, причем тоже хиральных (рис. 16 внизу).
Но какое отношение эти ленты имеют к химии? Представьте себе, что края наших лент — это цепочки углеродных атомов, а точнее, групп —СН2—. Время от времени на
Рис. 16. Хиральные ленты — модели молекул
своем протяжении эти цепочки соединяются между собой, например, атомами серы так, что получаются этакие замкнутые веревочные лесенки. Теперь уберем «ступеньки» лестницы, т. е. атомы серы. В результате получится молекула, представляющая собой два сцепленных кольца и называемая катенаном (catena — по-латыни «цепь»). Катенаны известны уже 20 лет, но получают их очень длинным и трудным путем. Идея же, которую мы здесь описали, до сих пор не реализована. Лишь совсем недавно удалось синтезировать молекулу, построенную в виде ленты Мёбиуса (см. [17]).
Разрезание молекул, построенных в виде лент,— это все-таки из области фантазий химиков. Но вот если очень внимательно присмотреться к моделям молекул хорошо известных соединений, то можно открыть новые, ранее неведомые свойства этих молекул. Возьмем тривиальнейшее соединение — глицерин. Его молекула устроена очень просто и «симметрично»:
н1 н н3
1 1 I яц
НО— с.—•&—с —он
L I 14
Н* ‘ОН н4
Кажется совершенно очевидным, что два атома водорода Н1 и Н2 у каждого из крайних углеродов ровно ничем не отличаются друг, от друга, равно как идентичны атомы Нх и Н3. Однако это совсем не так. Впрочем, займемся сначала более простыми примерами на моделях... более сложных соединений. Сделайте из картона несколько равносторонних треугольников. В середину треугольников воткните вертикально вязальные спицы или спички. Рассмотрим модель, в которой все вершины треугольника одинаковые. Будем считать, что они окрашены в белый цвет (структура А на рис. 17). Каждую вершину легко заменить любой из двух других, достаточно лишь повернуть треугольник вокруг спички на угол 60° Поэтому назовем такие взаимозаменяемые вращением вершины эквивалентными. Теперь покрасьте в треугольнике две вершины в черный цвет. Сделайте это для обеих сторон треугольника, в будущем это нам пригодится. В получившейся модели Б вы уже не сможете взаимозаме-нить вершины, помеченные номерами 1 и 2, никаким вращением. Однако мы получим на месте вершины 1 вершину 2, если поставим вертикально зеркало на белую вершину и на середину стороны, соединяющей 1 и 2. Значит, в этом случае вершины (и вообще все «места» в треугольнике)
вз а и моза меняются при отражении в зеркале. Назовем такие вершины энантиотопными (от греческих слов «энанти-ос» — находящийся напротив, и «топос» — место).
Мы рассмотрели два варианта: все три вершины одинаковые, две вершины одинаковые (в Б — черные) и одна — отличная от них. Возьмем третий вариант: все три вершины разные. Пусть это будут цвета белый, черный и серый. Но скомбинировать эти три цвета можно двумя способами, причем обе структуры относятся друг к другу как к зеркальному отражению. Поэтому такие структуры В и Г (см. рис. 17) можно назвать энантиомерными («мерос» — доля, часть).
Прежде чем заняться анализом более сложных построений, заметим, что все наши картонные игрушки имеют прототипы среди молекул реальных и хорошо известных соединений. Например, очевидно, что структура А — это модель молекулы хлороформа, а хиральные «игрушки» В и Г отражают определенные свойства жизненно важной аминокислоты аланина. Вершиной определенного цвета можно «зашифровать» не только атом, но и целую группу, например, метил —СН3. Хотя все молекулы имеют форму тетраэдра, они хорошо моделируются треугольником и вставленной в него спичкой, т. е. пирамидой.
Попробуем скомбинировать по две все наши конструкции в разных сочетаниях. Будем треугольники, как шашлык, нанизывать на вязальную спицу. Комбинации А с Б, В с Г в определенном смысле довольно малоинтересны. Во всех
Рис. 17. Модели и формулы молекул, позволяющие понять, что такое
энаитиотопия и энантиомерия
таких комбинациях можно представить себе, что А — это метильная группа. Независимо от соседа по спице вершины в треугольнике (атомы водорода в —СН3) будут эквивалентны. Проверьте это, меняя соседей треугольника с белыми вершинами и вращая треугольники на спице во всех направлениях. Никакого интересного влияния не будет оказывать и сам белый треугольник на соседей по спице. Мы с вами проанализировали такие, например, молекулы: СН3—СН8, СН3—СН2С1, СН3—CHBrCl. Следующий шаг — комбинируем структуру Б. Первый вариант: два треугольника Б. Получается структура Д, которая моделирует, например, молекулу дихлорэтана СН2С1—СН2С1 (рис. 18). Посмотрим, в каких отношениях находятся черные вершины. По условиям нашей игры можно безболезненно вращать треугольники на спице, что соответствует вращению в молекуле, скажем, дихлорэтана вокруг связи С—С. Если говорить о структуре Д, то здесь никак нельзя найти такого положения для зеркала, при котором обе черные вершины обоих треугольников оказались бы взаимозаменяемыми. Но поверните треугольники один относительно другого, и вы увидите, что к структуре Д' можно приставить зеркало, как мы сделали для Б. Значит, в Д черные вершины (/ и 2, а также I' и 2') в каждом треугольнике энантиотопны. Но зеркало можно поставить еще и перпендикулярно спице посередине треугольника. Отсюда следует, что имеются и другие пары, связанные энантиотопией: 1 и а также 2 и 2'
Переходим к более сложному варианту: комбинации Б+В=Е или Б+Г=Ж. Здесь, как ни крутите треугольники, вам не удастся «вставить» зеркало так, чтобы при этом вершины 1 и 2 взаимозаменялись. Интересно, что Е и Ж — энантиомеры, и в каждом из них есть взаимно соответствующие вершины, помеченные одинаковыми номерами. Назовем пару «одинаковых» вершин 1 и 2, не взаимозаменяемых ни при каких условиях, диастереотопными. По-гречески «диа» — через, между, приставка, означающая расхождение, «стереос» — пространственный, объемный. Последний вариант: комбинация двух хиральных структур, которых, по-видимому, должно быть три, В+В=3, Г~гГ= —И, Б+Г=Д. Структуры 3 и И хиральпы и связаны между собой зеркалом, это — энантиомеры. Структура К не хиральна. Такие неэнантиомерные пары, как К — 3 и К — И, называются диастереомерами.
Что касается всех этих моделей, то читателю, видимо, понятна их .связь с реальными молекулами. Например,
структуры 3, И и К могут служить моделями винной кислоты. А если взять не треугольники, а, к примеру, пятиугольники? Здесь можно найти много похожего. Структура Л (см. рис. 18) хиральна, больше того, белые вершины, обозначенные номерами 2 и 5 (а также 3 и 4), диастереотопны. Теперь в аналогичной игрушке М соедините два картонных пятиугольника мостом из мягкой проволоки, а на мост
Рис. 18. Комбинации моделей, соответствующие объединению различных фрагментов в одну молекулу
наденьте проволочную петлю с коротким и длинным хвостами. Такая петля должна свободно скользить по мосту. Эта модель позволит нам уяснить себе одну интересную особенность. Сдвиньте петлю поближе к одному из пятиугольников. Вы имеете хиральную структуру, в которой пары вершин 2—5 и 3—4 одного пятиугольника и 2'—5' и 3'—4' другого — диастереотопны. Но поместите петлю ровно посередине моста, на равном расстоянии от каждого пятиугольника. Модель перестанет быть хиральной (зеркало можно поставить в перпендикулярной спице плоскости, в которой лежит петля). Однако диастереотопия вершин сохранится.
У читателя наверняка накопились вопросы, на которые мы попытаемся ответить. Во-первых, какие реальные молекулы стоят за нашими построениями из пятиугольников? Прототипами структур Л и М можно считать производные ферроцена, о котором мы уже говорили. Вот формулы таких соединений (здесь Ф обозначает радикал фенил —С0Н5).
Несколько лет назад диастереотопия в этих молекулах была обнаружена экспериментально. Второй вопрос: как проявляется диастереотопия в свойствах веществ? Легко заметить, что каждый из четырех терминов — «энантиомерия», «энантиотопия», «диастереомерия» и «диастереотопия» — состоит из двух половинок, причем всего четыре варианта таких половинок. Оказывается, молекулы (в этом случае употребляется термин «мерия») или части молекулы, се отдельные атомы или группы (термин «топия») могут от-зюситься друг к другу как зеркальное отражение к предмету (термин «энантио»). Связанные таким отношением молекулы или их части обладают совершенно одинаковыми свойствами и отличаются лишь тогда, когда они реагируют с хиральными веществами. Ферменты организмов взаимодействуют исключительно с одним из энантиомеров или с одним из энантиотопных атомов. Диастереомеры — это совсем разные вещества, у которых и температуры кипения или плавления отличны, и реагируют они с другими веществами (любыми) по-разному. Точно так же разными физическими
свойствами и разной реакционной способностью обладают диастереотопные атомы или группы. Диастереомеры никак нельзя совместить друг с другом — ни вращением, ни отражением в зеркале. Это же относится и к диастереотопным частям молекулы. Часто бывает нелегко распознать диасте-реотопию. Чтобыэто сделать, применим такой тест. Заместим один из подозреваемой пары атомов молекулы на какой-нибудь другой, например водород на хлор. Если это была пара энантиотопных атомов, приходим к паре энантиомеров. Если же замену проводить среди диастереотопных атомов, получим один или другой диастереомер.
Вот теперь можно вернуться к глицерину. Проделайте такую операцию. «Замените» в этой молекуле атом Н1 или Н2 на С1, обозначьте всю правую группировку —СН3Н4ОН одной буквой /?. Если для полученной молекулы построить модели, то это будут «игрушки», помеченные буквами 3, И, Д'. Одним словом, вы придете к диастереомерам. А значит, атомы Н1 и Н2 в молекуле глицерина диастереотопны. А ведь это совсем было не очевидно!
Теперь ответим на главный вопрос: а зачем нужны все эти представления о диастереотопии, эти ленты — бумажные и из реальных атомов? Дело в том, что в природе почти все органические вещества построены из хиральных молекул. Возьмите, например, аминокислоты, входящие в состав белков. Только один из энантиомеров может быть использован при постройке белка. Строятся же белки ферментами, которые прекрасно разбираются в особенностях строения молекул. Знать эти особенности, по-видимому, необходимо и химику, производящему в колбе всевозможные перестроения молекул. И даже такие экзотические вещества, как ферроценовое производное типа Л, оказываются весьма эффективными компонентами катализаторов, с помощью которых производят только один нужный энантиомер аминокислоты в чистом виде. Что же касается перекрученных бумажных лет, то заметим лишь, что молекулы ДНК, представляющие собой две многократно перевитые цепи, иногда свертываются в кольца... Может быть, в решении и этой загадки помогут изготовленные из картона модели.
Мы рассказали о некоторых проблемах стереохимии. Более подробно об этой науке можно прочитать в популярных книгах и статьях [18].
Повседневная практика химика, конечно же, связана с колбами, пробирками, специфическими запахами. В течение долгого времени только эти «аксессуары» и сопровождали деятельность ученого-химика. Но вот уже последние полвека успешно развивается целая область химии, рабочими инструментами в которой являются лишь бумага да карандаш, а сравнительно недавно к ним прибавились еще и ЭВМ. Эта область — квантовая химия, возникшая на пересечении квантовой механики и химии. Квантовая химия оказывает огромное влияние на все другие разделы химической науки. Дело в том, что эта ветвь химии позволяет описывать химическую связь, структуру, электронное устройство молекул. И поэтому в наше время невозможно себе представить серьезное развитие ни одной из областей науки о превращениях молекул без привлечения методов квантовой теории.
В основе современных представлений о химической связи, объединяющей отдельные атомы в те или иные молекулы, лежит понятие об электроне — отрицательно заряженной частице, которая в тысячи раз легче атомного ядра. С некоторых пор (точнее, с 20-х годов нашего столетия) считают, что электрон — это не только частица, но одновременно еще и волна. Действительно, некоторые свойства электрона (например, дифракция) похожи на соответствующие свойства волн света, рентгеновских лучей. Попробуем смоделировать определенные свойства электрона. Для этого нам понадобятся... веревка или тазик с водой. Закрепите веревку с одного конца, а другой конец возьмите в руку и вызовите колебания в вертикальной плоскости. Колебания могут быть простыми, как показано на рис. 19, а. Но для этой же веревки можно получить и более сложную колебательную картину (рис. 19, б). Аналогичные колебания, называемые стоячими волнами, получаются и в тазу с водой, если колебания вызывать опущенной в воду ложкой. Можно ли математическим образом описать такую стоячую волну? Можно. Для этого введем функцию ф(х), которая равна амплитуде волны и зависит от аргумента х, т. е. расстояния вдоль волны от «стенки». Если мы обратимся к классической механике, то увидим, что задача описания колебаний
давным-давно решена, и, для того чтобы узнать вид нашей функции ф(х), необходимо решить дифференциальное уравнение второго порядка:
d-hHx) 4л* dx2 Х2 Ч> W
Здесь X — это длина волны; л — известное постоянное число.
Мы не будем разбирать, что такое дифференцирование, обозначаемое оператором d2!dx2. Скажем только, что этим оператором обозначают некую математическую операцию, процедуру, после осуществления которой с функцией ф(х) мы получаем какую-то другую функцию. Если мы узнаем, какой вид имеет функция ф(х), такая, что после подстановки ее в уравнение оно обратится в тождество, мы сможем сказать, что нашли математическое описание нашей системы. Иными словами, наша задача — отыскать функцию ф(х), обладающую следующим свойством: ее вторая производная функция равна самой функции ф(х), умноженной на —4л2/Х2, для любых значений х. Оказывается, таких функций может быть несколько, и «годятся» они лишь при некоторых определенных значениях К. Математически в нашем случае ф выражается синусом.
Рис. 19. Колебания веревки с образованием стоячих волн
Теперь сделаем следующий шаг. Представим себе, что ф — это функция, описывающая не волну на веревке, а волну, которая является электроном. Назовем ее волновой функцией. Поскольку электрон находится в трехмерном пространстве, придется сделать эту функцию зависящей от трех переменных координат: ф(х, у, z). Мы уже знаем, как записать уравнение для такой функции. По аналогии со случаем, когда волна возникала на веревке, подействуем на ф неким оператором Н, называемым оператором Гамильтона и включающим повторное дифференцирование. Продолжая нашу аналогию, приравняем получающуюся в результате функцию нашей функции ф, умноженной на значение энергии электрона Е. Действительно, энергия электрона связана с длиной «его» волны — чем меньше длина волны, тем больше энергия. Итак, мы получаем выражение H^=Ety, называемое уравнением Шредингера. Заметим, что в нем сокращать наф нельзя, поскольку если в правой части стоит произведение Е на ф, то в левой знак Н обозначает, какую манипуляцию следует провести с функцией ф.
Мы не будем решать уравнение Шредингера даже для простейшей системы — атома водорода, поскольку это решение требует основательного знакомства с теорией дифференциальных уравнений, а для более сложных атомов оно вообще точно не решается. Однако нам вполне достаточно будет обсуждения функций ф, получаемых при решении уравнения для системы протон плюс электрон. Но какой физический смысл имеет сама функция ф? Для стоячей волны на веревке одномерная функция ф — это амплитуда волны вдоль веревки. На рис. 19, б видно, что ф может принимать отрицательные значения, а в некоторых точках равняется нулю.
Примем теперь достаточно произвольное решение: волновая функция ф отражает месторасположение электрона в атоме. Но как это понять? Ведь если электрон — волна, он не может находиться в какой-то одной точке пространства; если это — частица, то ее координаты должны зависеть от времени, а время в уравнение Шредингера не входит. А что если «размазать» электрон по всему атому, превратить его в облако, а волновой функцией отражать степень «густоты», плотности этого облака? Тогда время нам не понадобится. Но вот неприятность — функция ф, по-видимому, может принимать и отрицательные значения, а что такое отрицательная плотность? Есть выход: пр и пи
сать плотность облака или, другими словами, вероятность встретить электрон квадрату гр2, значение которого всегда положительно.
Вот теперь, когда мы знаем, что гр — это некая функция трех координат х, у, г, квадрат которой равен плотности вероятности встретить электрон в точке с любыми заданными координатами yi, zi, но еще не знаем, как выглядит эта функция, мы можем задать ей некоторые свойства. Если какая-либо функция не будет удовлетворять этим требованиям, мы ее отбросим. Во-первых, гр должна быть равна нулю на очень большом расстоянии от ядра. Действительно, трудно представить себе какое-то облако бесконечных размеров. Во-вторых, поскольку электрон всегда существует и где-то да находится, общая вероятность его встретить должна равняться единице.
Дифференциальные уравнения обычно имеют много решений. На рис. 19, например, графически показаны два решения уравнения стоячей волны, соответствующие различным величинам X. Уравнение Шредингера в этом смысле исключения не представляет. Оказывается, ему удовлетворяют несколько функций и каждой соответствует свое конкретное значение энергии. Обозначим эти функции условными знаками по мере возрастания энергии: /s, 2s, 2р, 3s, Зр, 3d ... Мы не будем обсуждать, почему выбрали именно такие обозначения. Минимальному значению энергии, т. е. наиболее устойчивому состоянию атома водорода, соответствует функция 1s, которая математически выглядит так: е~г
4>(ls) = -^=. В этой формуле е — основание натураль-V л
ного логарифма, г — расстояние от ядра атома, выраженное в так называемых атомных единицах длины. Предоставим читателю, вооруженному микрокалькулятором и листком миллиметровой бумаги, всесторонне исследовать поведение этой функции, а также ее квадрата и, наконец, выражения r2hp(/s)]2. Поскольку г2 пропорционален площади сферы вокруг ядра, последняя функция является мерой вероятности того, что электрон находится на расстоянии г от ядра. Обратите внимание: в выражение для функции гр(Л) не входят в качестве аргументов координаты х, у, z. Единственная переменная здесь — радиус г. Значит, функция 1s должна совершенно равномерно «облегать» ядро. Говорят, что она имеет симметрию сферы. Сделайте из белого пластилина модель электронного облака, описываемого функцией гр (1s) и называемого орбиталью 1s. Чтобы показать распбло-
жение орбитали в пространстве, воткните в пластилиновый шарик три спички под прямыми углами. Эти спички будут символизировать оси х, у и г (рие. 20).
Орбиталь 2s (следующая по энергии) имеет несколько более сложное аналитическое выражение:
'*’(2s)== “ oW2 “ Г)е’Г/а
и эта функция ведет себя более сложным образом (постройте ее зависимость от г), но совершенно очевидно, что и она обладает симметрией сферы. Поэтому модель орбитали 1s может служить также моделью орбитали 2s.
Поднимаясь еще на одну ступеньку энергетической лестницы, приходим к функции ф(2р). И тут нас ждет сюрприз. Оказывается, в этом случае уравнению Шредингера удовлетворяют сразу три функции. Они все содержат общий член:
ге~г/2
4 К 2л’
умноженный на x/r, ylr или zlr. Соответственно этому второму сомножителю назовем функции ф(2рх), ty(2py) и ф(2рг). Первая составляющая функции зависит только от г и, подобно орбитали s, определяет лишь сферически симметричную часть функции ф и, так сказать, «погоды» не делает. Совсем иное дело, например, член х/r. Проанализируйте его поведение. Для этого удобнее перейти от обычных декартовых координат к сферическим, в которых координаты точки М задаются тремя параметрами: длиной вектора г, связывающего точку с началом координат, и углами $ и ф
Рис. 20. Модели орбиталей, изготовленные из пластилина
(рис. 21). Совершенно очевидно, что x=rsin^cos<p, y=rsinft simp и z=/'cosft. Учитывая эти соотношения, получаем:
ip(2pj^sin^cos(p,
a|?(2p(y)~sinftsimp,
4)(2pz)~cosft.
Мы получим функции, не зависящие от г. Постройте при помощи микрокалькулятора и транспортира на листке миллиметровой бумаги первую функцию. Для этого варьируйте значения углов ft и ф. Сначала составьте таблички для значений функции в плоскостях хг(ф=Ос), xr/(ft=90°) и /уг(<р=90°). Чем больше значений получите, тем точнее можно будет изобразить функции.
а Ф = 0° sin ft cos ср ф ft = 90° sin ft cos ср 0 ф —90° sin ft cos cp
0 0 0 1 0 0
45 0,7 45 0,7 45 0
90 1 180 — 1
Теперь постройте «разрезы» орбитали тремя плоскостями, вращая вектор вокруг начала координат на разные углы ft и <р и откладывая на луче отрезки, равные значениям функции (рис. 22). Итак, не зависящая от г (и определяющая форму орбитали) часть функции ty(2px) представляет собой два шара, центры которых лежат на оси х и которые соприкасаются в нулевой точке, где находится ядро атома. Обра-
тите внимание на очень важную особенность: в одном шарике значения функции sinft-coscp положительны, в другом — отрицательны. Это, конечно, не значит, что вектор, которым мы «вычертили» сферу, имеет отрицательную длину или что электрон в этой области находится с отрицательной вероятностью. Поскольку плотность вероятности встретить электрон равна ф2, он с одинаковой охотой размазывается по обе-
Рис. 21. Переход от декартовых координат к полгрным
им половинкам. Но все же не будем в дальнейшем забывать о знаках отдельных частей орбиталей. Постройте модель орбитали 2рх. Для этого соедините два пластилиновых шарика — белый (положительная половинка) и черный (отрицательная часть ф) — и воткните спички-оси (см. рис. 20).
У читателя должен возникнуть вопрос.5 а не забыли ли мы, строя модель орбитали, вторую часть функции ф, зависящую от г? Нет, не забыли, но помочь, как говорится, ничем не можем: нельзя в трехмерном пространстве скомбинировать обе составляющие. Но поскольку лишь второй член определяет форму и направление орбитали в пространстве, во многих случаях считают, что он и определяет всю орбиталь.
Предоставим возможность читателю самому определить форму и направление орбиталей 2ру и 2pz и после этого перейдем к устройству простейшей органической молекулы — метана, состоящей из центрального атома углерода, окруженного четверкой водородных атомов. Будем считать, что связь между двумя атомами осуществляется тогда, когда их орбитали накладываются друг на друга, перекрываются. У атома углерода четыре орбитали, пригодные для перекрывания,— 2s, 2рх, 2ру и 2pz. У каждого атома Н имеется орбиталь 1s. И вот тут нас поджидает одна трудность: как перекрыть четыре водородные орбитали с углеродными, да так, чтобы все атомы Н расположились на одинаковом удалении от С и друг от друга, если орбиталь 2s представляет собой сферу, а орбитали р — восьмерки, направленные вдоль осей х, у и г? Давайте пойдем на хитрость: положим, что орбитали могут как-то усредняться,образовывать некие гибриды. Как можно провести такую гибридизацию, показано на примере sp-гибридизации на рис. 23. Функции ф при такой процедуре складываются. Поскольку функция s положительна везде, а ру имеет и положительные и отрицательные стороны, в одной части пространства приходится
* = 0° = 90°
Рис. 22. Сечения функции гр (2рх)
<0 = 90°
z
Рис. 23. Гибридизация орбиталей s и ру
волновые функции вычитать. В результате и получаем вытянутое в одну сторону облако гибридной орбитали с большой положительной и маленькой отрицательной частями. Вот теперь нетрудно будет построить модель электронных облаков в молекуле метана. Четыре гибридные орбитали sp3 (поскольку они образованы s- и тремя р-орбиталями) направлены в вершины тетраэдра и перекрываются на концах со сферами /s-орбиталей водорода (см. рис. 20 справа).
Об электронном строении атомов и молекул можно прочитать в научно-полулярных изданиях [19].
Химия — наука экспериментальная. Как, впрочем, и физика, и геология, и генетика. И все-таки, если говорить о синтезе веществ, относительная роль эксперимента в химии превышает таковую для многих других наук. Это значит, что в химии все еще много экспериментальных наблюдений, которые не могут быть «поверены» теорией, что теория еще во многих случаях не может успешно предсказывать существование новых соединений и новых реакций. А как следствие — приходится надеяться на чистую случайность. В этом, конечно, есть своя прелесть. Однако ни в коем случае не следует думать, что синтетическая химия — наука совершенно «слепая» и что эксперименты химики ставят «на авось». Еще в прошлом веке появились основанные на обобщении многих наблюдений правила, позволяющие пред
сказывать, по какому пути пойдет реакция. Самое известное — правило Марковникова — гласит, что галогенводо-род, например НС1, реагирует с замещенным этиленом RCH=CH2 таким образом, что водород присоединяется к тому углероду, который несет больше атомов водорода, т. е. в нашем случае к СН2.
Сегодня химики знают немало методов и теорий, которые позволяют не только предсказывать, пойдет или не пойдет данная реакция, не только предугадывать продукты реакции, но и выдавать численные параметры, например, скоростей реакций. Хорошо известны и подробно изучены реакции электрофильного замещения в ароматических соединениях, в частности в производных бензола. Продукты таких реакций — весьма ценные вещества. Среди них аспирин, сульфамидные препараты, красители и т. д. Типичная реакция электрофильного замещения — нитрование. В главе «Желтая краска из черного угля» мы привели методику получения нитрофенола. Проведите аналогичный опыт с бензолом. Добавьте в пробирке к разбавленной азотной кислоте несколько капель бензола. Через несколько минут вылейте содержимое пробирки в стакан с водой и осторожно понюхайте. Вы не обнаружите запаха миндаля, характерного для нитробензола. Значит, реакция в этих условиях не прошла (однако бензол, как мы уже отмечали, реагирует с концентрированной азотной кислотой в присутствии серной). Итак, вывод очевиден: фенол реагирует в реакции электрофильного замещения гораздо быстрее бензола. Можно ли количественно сравнивать между собой фенол и бензол в реакции с азотной кислотой? Лучше всего это сделать таким образом: смешать бензол и фенол и добавить к ним немного азотной кислоты. Нитробензол и нитрофенол будут накапливаться во времени сначала быстро, потом все медленнее. Естественно, при этом нитробензола будет гораздо меньше. Если остановить реакцию в самом начале (время /ь рис. 24), когда накопление идет почти по прямой, и проанализировать, сколько образовалось нитробензола и нитрофенола, отношение их количеств х покажет отношение скоростей реакций:
цу__ 1Ф
А'Н Б
Таким образом можно определить соотношение скоростей нитрования для бензола и любого другого его производного. Например, по сравнению с бензолом толуол нитруется
Рис. 24. Кинетические кривые накопления нитрофенола и нитробензола
в 25 раз быстрее, а вот хлорбензол реагирует в 30 раз медленнее. Чем объяснить такое различие? Дело в том, что, как мы уже говорили, гидроксильная группа —ОН в феноле или метил —СНз в толуоле как бы накачивают электроны в бензольное кольцо, электронная плотность в ядре повышается, и оно более быстро реагирует с электрофилом. Атом хлора, соединенный с бензольным ядром, как бы отсасывает из него электронную плотность. «Напор», который создают заместители, гораздо меньше одного целого электрона, но и этого уже достаточно, чтобы так резко изменить скорость реакции. Зададимся теперь целью найти меру «мощности» наших электронных насосов. Используем для этого бензойную кислоту, несущую различные заместители X. Такие замещенные кислоты в воде обратимо теряют протон, диссоциируют, и устанавливается равновесие
Охарактеризуем это равновесие константой /<", которая тем больше, чем с большей силой заместитель X «высасывает» электроны из кольца. Это понятно, поскольку при этом связь с протоном ослабляется.
к _[ХСвН4СОО-][Н+]
Лх“ [ХСЯН4СООН]
Здесь выражения в скобках обозначают концентрации даш' ных частиц. Если взять логарифм отношения этой константы к константе диссоциации незамещенной бензойной кислоты (т. е. когда Х=Н), получим величину а, прекрасно характеризующую мощность электронного насоса X:
"-1еЙ-
Читатель легко сообразит, что о отрицательна для заместителей, подающих электроны, и положительна для «насосов» откачивающих. Оказывается, что логарифмы скоростей реакций электрофильного замещения линейно зависят от соответствующих величин о. Говорят, что в этом случае применимо уравнение Гаммета: IgVT =ро.
Давайте проанализируем одну недавно открытую реакцию. Если металлическую платину залить царской водкой, металл растворится и получится платинохлористоводородная кислота H2PtCl0. Эта кислота легко реагирует с замещенными бензолами при нагревании в растворе уксусной кислоты. Из бензола СвНбХ образуется производное H2[X'C6H4PtCl5]. Вот экспериментально полученные скорости реакций ароматических соединений с платинохлористоводородной кислотой. Рядом приведем значения а, соответствующие заместителям X:
X W а
ОСН3 8^5 —0,27
СН3 3 —0,17
Н 1 0
F 0,3 0,06
СООН 0,09 0,36
С1 0,08 0,37
Отложите на листке миллиметровой бумаги значения а и по оси ординат — соответствующие им величины lgW. Точки довольно неплохо ложатся на прямую. Проведите ее и из тангенса ее наклона определите величину р. Вы придете к такому уравнению Гаммета для нашей реакции lg W= =—За. Отрицательность величины р указывает на то, что реакция с платинохлористоводородной кислотой — типичное электрофильное замещение. Среди имеющихся у нас данных нет скорости реакции с фенолом, когда Х=ОН. Зная из справочников, что а для ОН равна —0,37, определите графически или по уравнению, во сколько раз быстрее бен
зола реагирует с платинохлористоводородной кислотой фенол.
...Еще более заманчиво научиться предсказывать химические превращения, не имея вообще никаких экспериментальных данных; только на основе теоретических соображений. В этом деле большую помощь оказывает квантовая химия, в частности один из ее разделов, называемый методом молекулярных орбиталей. Познакомимся с этим важнейшим методом современной химии, но предварительно запаситесь отрезками толстой медной проволоки и кусочками черного и белого пластилина.
Главное свойство атомных орбиталей — это способность к взаимному перекрыванию. В результате такого перекрывания образуется связь между атомами. Перекрываться облака могут двумя способами. Первый — когда ядра атомов и перекрывающиеся области лежат на одной оси. Второй — когда область перекрывания не пересекается этой осью и вы не можете беспрепятственно вращать все эти шарики (вылепленные, например, из пластилина) вокруг оси у (сделанной из проволоки). Первый тип связи обозначается буквой о, второй — л (рис. 25). Займемся теперь устройством молекулы этилена СН2=СН2. Два атома углерода, у каждого по четыре электрона. Один из четверки сидит на орбитали s, три другие — на р-орбиталях. Две р-орбитали гибриди-зуются с s-орбиталью. Образуются три гибридные sp2-op-битали. Две из них идут на связывание с двумя атомами водорода, а одна — перекрывается с такой же орбиталью углеродного партнера. Все эти перекрывания осуществляются по о-типу и образуют о-остов молекулы. Сделайте его из пластилина и проволоки (рис. 26). В дальнейшем нам о-остов не понадобится, и поэтому заменим его просто кусочком проволоки. Но прикрепим к ней оставшиеся две р2-орбитали. Только вот на что обратим внимание: пластилиновые шарики — это электронные облака, а облакам соот-
Рис. 25. Перекрывание из! оговленных из пластилина моделей орбита-лей по о- и л-типу
ветствуют волновые функции ф, значения которых имеют разные знаки для разных половинок р-орбитали. Будем положительную половинку изображать белым пластилином, отрицательную—черным. И вот оказывается, что две р2-орбитали можно расположить двумя разными манерами (см. рис. 26). Какой структуре отдать предпочтение и вообще — чем они отличаются? Станем перекрывание, комбинирование облаков отражать математически сложением соответствующих им функций ф. Таким образом, в результате перекрывания двух атомных орбиталей (АО) атомов А и В образуются молекулярные орбитали (МО) по закону: Фив=Фа+Фв- Что будет, если перекрываются части разных знаков? Логично предположить, что в этом случае (обозначим орбиталь звездочкой) фдВ=фд+ (—Фв)=Фа—Фв. Итак, для второго варианта имеем разность функций. Вычислим электронную плотность между атомами СА и Св в обоих случаях. Она равна квадрату МО: (фАВ)2= (фА+фв)2= =ф^+фв+2фдфв. Квадрат разности, как известно, будет: (Флв)2=(Фа“”Фв)2=Фд+ФЬ—2ФаФв- Значит, во втором случае в результате образования МО электронная плотность между атомами понижается, а для этилена она равна нулю, поскольку фА=Фв- Поэтому такая МО ф* называется разрыхляющей. Первая же МО весьма выгодна для образования связи и называется связывающей. Не удивительно, что ее энергия гораздо ниже, чем энергия каждой АО (рис. 27).
Рис. 26. Пластилиновые модели о-остова молекулы этилена и л-орби-талей
Поскольку на каждой орбитали (МО или АО) могут находиться два электрона (и не более), ясно, что в этилене оба л-электрона располагаются на более выгодной связывающей орбитали. На рисунке электроны изображены стрелками.
Теперь перейдем к более сложной системе — бутадиену. Изготовьте его о-остов из проволоки (сделайте четыре штуки). Распределите 16 пластилиновых моделей р-орбита-лей по всем углеродным атомам. Вы увидите, что сделать это можно четырьмя способами (рис. 28). Самая выгодная орбиталь — это фх, ее энергия минимальна (поэтому мы ее изобразили в самом низу), ведь здесь эффективно взаимодействуют, перекрываются по л-типу все четыре облака. Орбиталь ф2 — как бы комбинация двух невзаимодействующих этиленов. Третья орбиталь отражает молекулу с одной двойной связью, и, наконец, последняя орбиталь — наименее выгодная. Условно все структуры можно изобразить так:
Рис. 27. Энергии АО атомов А и В и образующихся из них МО ар и ар*
Рис. 28. Модели МО бутадиена
В бутадиене четыре л-электрона, все они размещаются на двух нижних связывающих орбиталях. Орбитали i|)3 и — разрыхляющие, они электронов не содержат. Здесь читатель захочет спросить: а зачем вести разговор о разрыхляющих орбиталях, если они электронов не несут, для связывания бесполезны, даже вредны? Во-первых, не во всех соединениях все разрыхляющие орбитали не заняты; во-вторых, при действии света на молекулу некоторые электроны соскакивают со связывающих и заселяют на какое-то время разрыхляющие орбитали. И в-третьих, вот правило, которое позволяет предсказывать возможность химической реакции. Это правило гласит, что реакция димеризации в цикл двух ненасыщенных углеводородов возможна в темноте, если при их взаимодействии наивысшая еще занятая электронами орбиталь одного реагента перекрывается с самой низкой свободной орбиталью другого по принципу «плюс с плюсом и минус с минусом». Проанализируем применимость этого правила. Попробуем присоединить молекулу этилена к бутадиену. Вот первый вариант перекрывания: самая низкая свободная орбиталь (это разрыхляющая орбиталь л*) с самой высокой занятой орбиталью бутадиена (это связывающая орбиталь г|)2). И второй вариант: перекрывание самой низкой свободной орбитали бутадиена (разрыхляющая 4>3) с занятой орбиталью этилена (связывающая л). Оба варианта изображены на рис. 29 слева. Но вот для димеризации этилена, как ни вертите модели, возможны только две комбинации, показанные на рис. 29 справа. Во втором случае никак не удается избежать «зебры» на месте стыковки орбиталей обеих молекул. Вывод такой: реакция бутадиена с этиленом возможна в темноте. Она протекает по схеме:
А вот димеризацию двух молекул этилена никак не удастся провести в темноте, но зато можно в одной из молекул све-
Рис. 29. Перекрывание орбиталей при циклодимеризации бутадиена с этиленом и двух молекул этилена
tdm забросить электрон на л*. Тогда становится возможным такое превращение:
□
Метод, о котором мы здесь дали некоторое представление, позволяет предсказывать возможность или невозможность многих реакций. За создание квантово-химической теории химических реакций несколько лет назад американский ученый Р. Хоффманн и японский специалист в квантовой химии К. Фукуи были удостоены Нобелевской премии.
Был век каменный, был век бронзовый, железный. Мы живем, безусловно, в век полимеров. Представить себе нашу сегодняшнюю жизнь без полимеров просто невозможно— без пластмасс, заменяющих дерево и металл, без волокон, используемых для изготовления тканей, канатов, без оргстекла и полиэтилена. Но что такое полимеры? Это не просто очень длинные, очень большие молекулы. Например, углеводород С80Н1в2 имеет молекулу весьма длинную, но к полимерам этот парафин не относят. К полимерным принадлежат такие вещества, молекулы которых состоят из повторяющихся звеньев мономеров, а число таких звеньев велико и неопределенно. Что значит неопределенно? Это значит, что в одной молекуле их может быть три тысячи, а в другой молекуле того же образца — три тысячи пятьсот, в третьей — две с половиной тысячи.
Полимеров сегодня известно великое множество. А в такой ситуации необходима какая-то классификация. Вот один из возможных вариантов. Грубо все полимеры можно разделить на три класса: полимеры природные (выделенные из природных продуктов), искусственные (т. е. полученные воздействием каких-то химических реагентов на природные полимеры) и, наконец, синтетические (полученные из мономеров на химических заводах). Внутри каждого из этих классов полимеры удобно подразделить в соответствии
с тем, из каких группировок построены полимерные цепи. Конечно же, и природные и синтетические полимеры могут быть примерно одинакового строения. Поэтому синтетические полимеры часто имеют свойства, похожие на свойства природных материалов. Можно расклассифицировать полимеры и по другим признакам, например, разделить их на волокна, пленки. Можно относить материалы к тому или иному классу в зависимости от устойчивости полимера, скажем, к нагреву.
Давайте последовательно рассмотрим наиболее известные полимеры, а в основу классификации положим их химическое строение. Наиболее просто устроены полимеры, относящиеся к классу так называемых карбоцепных соединений: в них цепь составлена только из атомов углерода. Атомы углерода могут быть соединены только с водородными или углеродными атомами. В этом случае имеем дело с полимерами, представляющими собой предельные углеводороды — полиэтилен и полипропилен. Вот уравнение реакции образования полиэтилена из этилена: /гСН2=СН2-> ->[—СН2—СН2—]п. Полиэтилен широко применяется в быту: из него делают прозрачную беловатую пленку, он идет на изготовление изоляционного материала для радиотехнических устройств, им пропитывают ткани, бумагу. Из полипропилена делают весьма прочное волокно. При обычной температуре эти материалы не растворяются ни в каких растворителях, но стоит поднять температуру до 80°, как они начнут набухать и затем растворяться в четыреххлористом углероде или толуоле. Полиэтилен легко отличить от других полимерных материалов. Внесите кусочек полиэтиленовой пленки в пламя газовой горелки. Полиэтилен расплавится, будет стекать каплями, затем загорится сначала голубоватым, потом желтым пламенем. При этом вы ощутите запах парафина. Это и не удивительно — полиэтилен и парафин имеют одинаковый состав.
Если в этилене один из атомов водорода заменить на фенильное кольцо, получим стирол, который легко полимеризуется в полистирол:
...—сн2----сн ---- сн2 ---сн -----сн2—...
Полистирол размягчается при нагревании уже до 80°; если к нему поднести пламя горелки или спички, кусочек
полистирола быстро воспламеняется и горит желтым, коптящим, светящимся пламенем, выделяя пары с характерным сладковатым запахом. Нагрейте в пробирке маленький кусочек полистирола на пламени горелки. Выделяются белые тяжелые пары с характерным запахом. Происходит деполимеризация, и образуется стирол. Полистирол применяют в качестве электроизоляционного материала, из него делают легкий пенопласт.
В длинной цепи полиэтилена некоторые атомы водорода можно заменить на атомы галогена, кислорода, азота и получить полимеры с новыми ценными свойствами. Но заменить водородные атомы непосредственно в полиэтилене — дело весьма трудное, если не невозможное вообще. Поступают другим образом — заменяют один или несколько водородов в этилене и затем полученный продукт полимеризуют. Вот самый простой вариант: замещаем в этилене один водород на хлор и винилхлорид подвергаем полимеризации.
2nCH2 = СНС1 —► / — СН2—СН — СН2 — СН — \ I I
\ Cl CI
В результате получаем поливинилхлорид, весьма широко применяемый как изолятор электрических проводов. Поливинилхлорид растворяется в ацетоне, хлороформе и этилацетате, еще лучше растворим в смеси ацетона с бензолом. Отличить поливинилхлорид от других полимеров нетрудно. Для этого используем то, что в состав этого материала входит хлор. Прокалите на газовой горелке медную проволоку, горячей еще проволокой коснитесь неизвестного вам полимерного материала и снова внесите проволоку в пламя. В присутствии хлора пламя окрасится в зеленый цвет. Значит, вы имеете дело с поливинилхлоридом или с сополимером винилхлорида, т. е. соединением, длинные молекулы которого содержат фрагменты поливинилхлорида и, например, поливинилацетата, полиакрилонитрила. В пламени поливинилхлорид сгорает с трудом, пламя имеет зеленоватый оттенок.
Очень ценен продукт полимеризации полностью фторированного этилена, политетрафторэтилен или тефлон: nCF2=CF2~>[—CF2—CF2—]п. Тефлон — белый, ни в чем не растворимый полимер, он не изменяется при охлаждении до —100° или нагревании до +250° Действие соляной, серной или азотной кислоты не приводит к разрушению тефло
на. Из тефлона делают электротехнические изделия, исполни зуют в радиотехнике, он идет на изготовление химически весьма стойких труб и насосов, получают из него и волокна. Отличить политетрафторэтилен нетрудно по его белому цвету, на ощупь он «жирный», словно мрамор.
Тефлон — продукт непрозрачный, на стекло мало похож. Полиэтилен пропускает лучи света, и если приложить полиэтиленовую пленку к листу с напечатанным текстом, легко можно будет прочитать текст. И все-таки на стекло полиэтилен тоже не очень-то похож: на большом расстоянии пленка из этого материала выглядит мутной, и через нее ничего толком не разглядишь. Но вот если в полиэтиленовой цепи заменить в каждом втором углеродном атоме один водород на метил, а второй — на сложноэфирную группу —СООСНз, получим весьма прозрачный полимер — полиметилметакрилат:
сн3 сн3
I I
... — сн2-------- с ------сн2 ------ с ------. . .
I I
соосн3 соосн3
Полиметилметакрилат — всем хорошо знакомое органическое стекло. Этот полимер легко растворяется в ацетоне, хлороформе, ’эти лацетате.
В углеводородную цепь полимерной молекулы можно включать бензольные кольца. Прилейте в пробирке или на дне стакана к чайной ложке кристаллического фенола столько же по объему раствора формальдегида в воде (40%-ного формалина). Перемешайте палочкой и добавьте к смеси несколько капель концентрированной соляной кислоты. Сразу же погрузите пробирку в холодную воду. Через несколько секунд погрузите в пробирку деревянную или стеклянную палочку и перенесите прилипший комок вязкой массы в другую пробирку со спиртом. Образовавшийся полимер растворяется в спирте. Что же это за полимер? Под действием кислоты формальдегид СН2О замещает в феноле орто-атомы водорода:
Выньте пробирку из холодной воды и перенесите в сосуд с кипящей водой. Через несколько минут ваш полимер станет твердым, вам придется разбить пробирку, чтобы вынуть кусок смолы. Попробуйте растворить его в спирте — он не растворится. Что же произошло? Реакция поликонденсации фенола с формальдегидом пошла дальше, молекулы формальдегида сшили между собой длинные нити резола, и получилась пространственная сетка резита:
Резит
Теперь молекулы растворителя не могут оторвать одну нить молекулы полимера от другой, поэтому-то он и не переходит в раствор. Итак, вы получили фенолоформальдегидную смолу, которая весьма широко применяется для изготовления электроизоляционных материалов, пластмасс, пластиков, из нее делают пуговицы и многие другие вещи.
Теперь перейдем к гетеронепным полимерам, нити которых, помимо углеродных атомов, включают атомы азота, кислорода и других элементов. Вот три гетероцепных полимера, из которых изготавливают волокна:
о и [-С-СН2СН2СН2СН2 СН2 -NH-] Капрон
О О
II II
[-C-CH2CH2CH2CH2-C-NH- (сн2) 6—NH— ] п
Найлон
О О
и II
[—о-с—с6н4—с-о—сн2сн2—] п
Лавсан
Первые два полимера имеют в своей основе структуру амида, для которого характерно наличие группы —CONH—, лавсан — это сложный эфир. Внесите в пламя газовой горелки кусочек ткани из полиамидного волокна. Нити расплавятся и потекут отдельными каплями. Обратите внимание на ха
рактерный очень неприятный запах. Через некоторое время от ткани останется коричневато-черная твердая масса. По-лиаглидное волокно растворяется в ледяной уксусной кислоте при нагревании. Полиэфирное волокно в пламени горелки горит медленно желтым пламенем с коричневыми парами и копотью. В отличие от полиамидного волокна лавсан не растворяется в кипящей концентрированной соляной кислоте, однако растворим в концентрированной азотной кислоте при кипячении. По этим признакам можно распознать тип волокна.
К гетероцепным полимерам относятся и природные волокна — шерсть, шелк, лен, хлопок. Шерсть и шелк состоят из белков, а белок, как известно, составлен из аминокислот. Таким образом, шерсть и шелк — это полиамидные волокна. В состав шерсти входит белок кератин, содержащий довольно много серы. Белки, образующие шелк, серы практически не содержат. Поэтому шелк нетрудно отличить от шерсти по запаху, если внести испытуемое волокно в пламя газовой горелки. Шерсть горит с более резко выраженным неприятным запахом паленых волос. Опустите белую шерстяную нитку в пробиркус концентрированной азотной кислотой и осторожно подогрейте пробирку. Шерсть окрасится в яркий желтый цвет. Это характерная реакция на белок. Промойте нитку водой и опустите в концентрированный водный раствор аммиака. Шерсть окрасится в оранжевый цвет. Лен и хлопок, как и бумага, состоят из целлюлозы. Поэтому сгорают они с запахом горелой бумаги. Целлюлоза представляет собой полисахарид, повторяющимся многократно фрагментом в ней является шестичленное кольцо, включающее один кислородный атом. Природный полисахарид — хлопковую вату — можно химически обработать, модифицировать и получить искусственные продукты. Для этого сначала приготовьте нитрующую смесь: в стакане, погруженном в сосуд с холодной водой, к концентрированной азотной кислоте очень осторожно прибавьте немного концентрированной серной кислоты. Погрузите в эту смесь клочок хлопковой ваты величиной с грецкий орех на 2—3 минуты (не больше!). Зацепите кусок ваты стеклянной палочкой и поместите его под струю водопроводной воды. Через несколько минут отожмите вату, расстелите ее на листе промокательной бумаги и высушите на воздухе. Вы получили динитрат целлюлозы, заменив в гидроксильных группах полимерной молекулы целлюлозы атомы водорода на нитрогруппы. После того как динитрат целлюлозы высохнет, раство
рите его в смеси эфира и спирта (примерно в соотношении 2 1). Вы получите вязкий раствор, который называется коллодием и используется для герметизации пробок на склянках с различными жидкостями. Удобно использовать коллодий и для заклеивания мелких ран на коже. В другом опыте к раствору камфоры в спирте (можно использовать камфорный спирт) прибавьте понемногу динитрат целлюлозы, смоченный спиртом. Полученную массу тщательно перемешайте и ровным слоем намажьте на металлический лист. Через некоторое время спирт испарится, оставив пленку материала, называемого целлулоидом. Нитраты целлюлозы применяются для изготовления пленки, лаков, пластмасс. Вместо азотной кислоты можно использовать уксусную; в этом случае получают ацетаты целлюлозы, которые идут на изготовление негорючей кинопленки, ацетатного волокна.
О полимерах можно прочитать в популярных книгах [20].
Попробуйте пальцами согнуть или растянуть железный гвоздь. Ясно, что такой эксперимент обречен на неудачу. А теперь повторите этот опыт с полоской резины. Какая разница! Действительно, для того чтобы растянуть полоску резины на одну сотую ее длины, требуется приложить усилие в 100 000 раз меньшее, чем для такого же растяжения стального гвоздя. Да не только в силе разница. Полоску резины можно увеличить в длине раз в 10, и она не порвется. Эта способность к растяжению в 1000 раз превосходит растяжимость нормальных твердых тел. Что придает резине такие удивительные свойства? Конечно же, причины нужно искать в строении вещества, в форме молекул. Резиновыми изделиями люди пользоваться начали давно. Но вот как это ни удивительно, теория «растяжимости» резины была создана лишь в 1932 г. швейцарцем Мейером.
Основу резины составляет каучук. Это вещество состоит из длинных полимерных молекул. Некоторые атомы углерода в такой молекуле соединены двойными связями. Каждая молекула каучука состоит из нескольких тысяч звеньев, и молекулярная масса вещества доходит до сотен
тысяч. Какова же длина полимерной молекулы? Если вытянуть молекулу в нить, то длина ее будет порядка микрона. Вы можете изготовить «модель» такой молекулы. Возьмите шелковую нитку длиной полметра — она будет передавать соотношение толщины молекулы к ее длине. Вот мы сказали: «если вытянуть молекулу» Дело в том, что в действительности в твердом теле или в растворе молекулы многих полимеров располагаются в виде зигзагообразных кривых. Бросьте нашу модель молекулы — нитку — на поверхность воды. Она примет форму причудливой кривой. Форму молекулы можно предсказать теоретически. Проделайте такой опыт, напоминающий игру в жмурки. Завяжите вашему товарищу глаза, раскрутите его и попросите сделать один шаг. Нанесите на бумагу направление его движения. Теперь снова раскрутите его, и снова — один шаг. Так вы получите ломаную кривую — математическую модель полимерной молекулы. Чем больше ваш товарищ сделает шагов, тем точнее вы приблизитесь к форме какой-то среднестатистической молекулы. Оказывается, размеры такого клубка можно рассчитать. Правда, нужно заметить, что в действительности молекулы располагаются в трехмерном пространстве, мы же «работаем» на плоскости, но это не меняет принципов анализа. Расстояние между концами молекулы г, т. е. между началом и концом пути вашего товарища, выражается формулой
r=lV п,
где I — длина его шага, п — число шагов. Отсюда, конечно, не следует, что если вы возьмете рулетку и измерите расстояние г в метрах, то оно точно совпадет с расстоянием, вычисленным по формуле. Но если вы будете повторять игру много раз, то чем больше будете играть, тем больше будет г приближаться к вычисленному по формуле. Поэтому г называется среднестатистическим. Длина же молекулы, т. е. весь путь вашего товарища, равен: h=ln. Соотношение hl г будет показывать степень «свернутости» молекулы или степень «бессмысленности» блужданий вашего товарища. Оно равно:/г/г=И^, т. е. чем длиннее молекула, тем сильнее она свернута.
Читатель может высказать догадку: значит, причина эластичности каучука в том, что когда мы его тянем, то распрямляем свернутые молекулы. Это действительно так, однако здесь есть одно «но». Давайте смоделируем такой процесс.
Положите на воду несколько не соприкасающихся друг другом ниток. Потяните две нитки в разные стороны. Когда вы прекратите «растяжение», назад нитки не вернутся, обратимой эластичности вы не получите. Мы смоделировали, таким образом, не резину, а мягкий пластичный парафин. А теперь сделайте вот что. Возьмите девять ниток и свяжите их узлами так, чтобы получилась правильная сетка. Положите эту сетку на поверхность воды и осторожно, чтобы нити не слипались, потяните за один конец, придерживая другой. Отпустите нити — сетка постепенно в р-нется в первоначальное положение. Вот мы и получили модель резины. Роль узлов, скрепляющих отдельные молекулы каучука в резине, выполняют мостики из атомов серы. Вводятся эти мостики при вулканизации каучука — обработки его серой при повышенной температуре.
При таком объяснении возникает несколько вопросов.
Вопрос первый. Что заставляет растянутую сетку, после того как убрали растягивающее усилие, возвращаться в исходное состояние? Дело в том, что молекулы обладают кинетической энергией, они всегда в движении. В резине сшивки не дают возможности огромным молекулам под действием тепла двигаться одна относительно другой. Но тепловое движение «расталкивает» отдельные части молекул, скручивает длинные молекулы. Вот это-то тепловое движение и возвращает сетку в исходное положение. Если растянуть кусок резины, а потом заморозить до очень низкой температуры, молекулы потеряют изрядную долю кинетической энергии, растянутая резина не сможет вернуться в исходное состояние. Но нагрейте ее до комнатной температуры — она снова сожмется. Есть у резины удивительное на первый взгляд свойство. Известно, что все «нормальные» тела и жидкости при нагревании расширяются. Резина, наоборот, сжимается. Проведите такой опыт. Вбейте в палку два гвоздика и соедините их металлической пружинкой и привязанной к ней резинкой. Если погрузить резину в горячую воду, она сожмется, что можно видеть по растяжению пружинки. Дело в том, что, увеличивая температуру, мы увеличиваем кинетическую энергию молекул, сетка стремится сжаться.
Вопрос второй. В каучуке нет поперечных сшивок серными мостиками, отдельные молекулы не связаны друг о другом. Тем не менее каучук тоже обратимо растягивается
(хотя он похож и на парафин). Почему? Оказывается, в каучуке тоже есть узлы, соединяющие длинные молекулы. Они возникают из-за того, что такие молекулы перепутываются, переплетаются друг с другом, как отдельные волокна в куске ваты. Однако отсутствие настоящих прочных узлов, связывающих молекулы, приводит к тому, что каучук менее эластичен, чем резина. По той же причине каучук растворяется в бензине, а резина только набухает (проверьте это).
Вопрос третий. Почему многие полимерные материалы, например полиэтилен, растягиваются необратимо, т. е. не сжимаются, после того как исчезает растягивающая сила? Дело здесь в том, что в отличие от каучука молекулы полиэтилена укладываются аккуратно в кристаллы. Такие молекулы могут только скользить друг относительно друга.
Мы говорили о молекулярной массе полимеров, о том, что ученые знают значения молекулярной массы полимерных материалов. Мы говорили о том, что реальная протяженность молекулы меньше, чем ее длина в вытянутом состоянии. Как ученые узнают такие вещи? Поскольку даже большую, длинную молекулу полимера не разглядишь в.микроскоп, применяют косвенные методы. Самый простой способ определения молекулярной массы вещества — измерение повышения температуры кипения раствора. Растворите в воде поваренную соль, налейте раствор в колбу и нагрейте до кипения. В тех же условиях нагрейте такой же объем чистой воды. Вы увидите, что раствор закипит позже, чем вода. Раствор соли замерзает при более низкой температуре, чем вода. В этом можно убедиться, поставив две жидкости в холодильник. Разница в температурах кипения или замерзания раствора и чистого растворителя зависит от концентрации растворенного вещества и от его молекулярной массы.
А как узнать о форме молекул в растворе? Тут помогает иЗхМерение вязкости раствора. Чем длиннее молекулы, чем более они вытянуты в цепь, тем им легче перепутаться между собой, тем большее сопротивление они оказывают «текущим» молекулам воды. Вот такая грубая аналогия: через коническую воронку гораздо легче просыпать речной песок, состоящий из мелких частичек, чем протянуть кусок ваты, составленный длинными перепутанными волокнами.
И в заключение этой главы приведем одну идею из области научной фантастики. Принцип эластичности резины можно изобразить так:
А теперь представьте себе, что нам удалось сделать вещество, молекулы которого состоят из множества продетых друг через друга колец. Два или три кольца соединять в цепь ученые уже научились. Но вот если молекула состоит из множества — сотен, тысяч — колец... Схема растяжения будет выглядеть так:
Совершенно очевидно, что растяжимость такого полимера должна в тысячи раз превышать растяжимость обычной резины.
Человеческий организм в определенном смысле слова можно сравнить с двигателем внутреннего сгорания, ведь он преобразует химическую энергию веществ, поступающих с пищей, в движение и тепло. Пища — это белки, углеводы, жиры, витамины, соли, вода. Все эти компоненты жизненно важны, необходимы, у каждого своя функция в организме. «Горючим» для человека и животных являются углеводы. Эти соединения содержат как бы несколько молекул воды и атомов углерода, за что и получили свое название. Например, формула наиболее известного и наиболее важного углевода глюкозы CeHi2Oe или Св(Н2О)в. Как же построена молекула глюкозы? Это одновременно и спирт, содержащий пять гидроксильных групп ОН, и альдегид (с группой СНО). Молекула глюкозы может существовать
в виде трех форм — двух циклических и одной разомкнутой,— находящихся в постоянном равновесии. Наличие групп —ОН и —СНО в глюкозе легко доказать такими опытами. Налейте на дно стакана несколько капель водного раствора глюкозы и прибавьте 2—3 капли раствора едкого натра. Теперь добавляйте по каплям раствор сернокислой меди, пока не образуется неисчезающая муть. Стакан нагрейте в кастрюле с горячей водой. Появляется желтое окрашивание и затем выпадает осадок. Эта реакция заключается в окислении альдегидной группы глюкозы в кислотную —СООН. Окислитель — гидроксид двухвалентной меди — восстанавливается при этом в оксид одновалентной меди Сп2О, осадок желтого цвета.
Другой аналогичный опыт. В нескольких каплях воды растворите небольшой кусочек ляписного карандаша (азотнокислое серебро), добавьте к нему нашатырного спирта до растворения осадка и затем немного глюкозы. При нагревании стакана на водяной бане серебро восстанавливается и выпадает в виде черного осадка. Это были опыты, подтверждающие наличие альдегидной группы. Теперь проведем реакции, в которых будут участвовать гидроксилы. На дно стакана или стекло поместите несколько капель раствора сернокислой меди, добавьте раствор едкого натра. К полученному осадку гидроксида меди прикапайте раствор глюкозы. Образуется соединение меди по кислородам гидроксильных групп глюкозы, в результате чего раствор окрашивается в синий цвет.
Глюкоза — весьма распространенное в природе соединение, но очень часто она встречается в связанном состоянии. Расскажем о наиболее известных производных глюкозы. Молекула обычного сахара (сахарозы) состоит из двух частей — глюкозы и фруктозы. Но сцеплены они так, что альдегидная группа глюкозы не способна восстанавливать гидроксид меди (проверьте это). Однако гидроксильные группы легко обнаруживают себя (проведите соответствующую реакцию). Сахар может распадаться на составные части — глюкозу и фруктозу — при кипячении с кислотой. Поместите в пробирку немного раствора сахара и несколько капель разбавленной серной кислоты. После 2—3-минутного кипячения и нейтрализации раствором щелочи вы сможете обнаружить в растворе глюкозу. Сахар — это димер, молекула которого состоит из двух единиц простейших углеводов. Но глюкоза может образовывать и длинные полимерные цепи. В зависимости от способа соединения звеньев по
лучается либо крахмал, либо клетчатка. Капните в раствор крахмала (крахмальный клейстер) йодной тинктуры. Синее окрашивание обусловлено тем, что молекула крахмала, представляющая собой длинный полый цилиндр, впитывает в себя йодные двухатомные молекулы, образуется так называемое соединение включения. На дне стакана к раствору сернокислой меди добавьте большое количество нашатырного спирта. В полученный раствор бросьте кусочек ваты. Через некоторое время он растворится. Если теперь в стакан добавить разбавленной соляной кислоты, клетчатка выделится из раствора обратно. И крахмал и клетчатка при нагревании с растворами кислот распадаются на отдельные глюкозные молекулы.
Итак, пища — это источник энергии. Как же это «топливо» сгорает, как содержащаяся в нем энергия используется? Вот мы за завтраком положили в рот кусочки хлеба и вареного картофеля. В хлебе содержится около 50% крахмала, в картофеле его примерно 20% . Мы съели рис или рисовую кашу. В рисе 80% крахмала. В завершение мы выпили стакан чая с сахаром. Следовательно, позавтракав, мы ввели в организм порцию углеводов. Уже во рту пища начинает перерабатываться. На крахмал, клетчатку, сахар действуют органические катализаторы — ферменты. Они расщепляют сложные сахариды на глюкозные звенья. То, для чего кислоте требуется кипячение и много минут времени, ферменты делают за считанные секунды. Каждая цепь крахмала или клетчатки распадается на много тысяч молекул глюкозы. Расщепление полимеров завершается в желудке под действием кислого сока. Через стенки кишок глюкоза всасывается в кровь и разносится ею по всему организму. Благодаря крови каждая клетка регулярно получает свою порцию глюкозы. Но клетки расходуют глюкозу неравномерно, ведь во время тяжелой работы энергии требуется больше, чем во время отдыха. Если в крови находится глюкозы больше, чем это требуется в данный момент клеткам, избыток сахара поступает с кровью в печень, где он снова полимеризуется в длинные молекулы животного крахмала гликогена. Но как кровь «узнает», что избыточную глюкозу нужно откладывать впрок? Для этого существует специальный гормон инсулин, производящийся поджелудочной железой. Если клеткам требуется дополнительная энергия, гликоген расщепляется и образующаяся глюкоза под1рсится кровью к клеткам.
Но вот молекула глюкозы проникла в клетку. Что о ней
происходит дальше? Превращения эти сложны, многостадийны, но сейчас ученым в общих чертах известны. В этих превращениях участвуют специальные ферменты и аккумулятор энергии — аденозинтрифосфат (АТФ). Это довольно сложное соединение, которое, отщепляя одну фосфатную группу, переходит в аденозинди фосфат (АДФ). При этом и выделяется энергия, необходимая клетке. На первой стадии молекула глюкозы реагирует с ферментом глюкокиназой и молекулой АТФ. Последняя при этом отдает свою энергию и превращается в АДФ. Дальше начинается целая цепь превращений, в которых молекула глюкозы дробится на более мелкие фрагменты. В результате всех этих процессов образуются молекулы АТФ, т. е. клетка получает энергию. Промежуточное вещество в превращениях глюкозы — пировиноградная кислота СНзСОСООН. Это соединение вовлекается в водоворот новых реакций и, реагируя с кислородом (который в клетку доставляется гемоглобином крови), превращается последовательно в лимонную, щавелево-янтарную, янтарную, яблочную кислоты и, наконец, в углекислый газ и воду, которые выносятся из клетки кровью. Весь процесс окисления глюкозы кислородом при дыхании включает 22 последовательные химические реакции и требует участия двух десятков ферментов. За одну «сгоревшую» молекулу глюкозы клетка получает 38 молекул АТФ, полученных из АДФ. КПД работы клетки равен 45%.
Куда идет вся эта энергия? В первую очередь на поддержание нужной температуры тела. Ведь многие процессы в клетке не могут происходить при пониженной температуре. Любой процесс, происходящий в клетке, требует энергии. Эта энергия высвобождается при переходе АТФАДФ. Например, известно, что специальный белок мышцы — миозин — способен расщеплять молекулу АТФ. В результате такого химического взаимодействия молекула белка укорачивается, мышца сокращается. Таков механизм превращения химической энергии углеводов пищи в движение мышцы.
Промежуточно образующаяся в окислительном процессе пировиноградная кислота, если кислорода много, окисляется полностью до углекислого газа и воды. В отсутствие же кислорода под действием специальных веществ это соединение восстанавливается в молочную кислоту СН3СН (ОН)СООН. Не удивительно, что у спортсменов, только что испытавших большую физическую нагрузку, наблюдается повышенное содержание молочной кислоты в мышцах: кислорода не хватает для окисления всей глюкозы,
а энергии требуется много. Наконец, при действии на глюкозу грибковых микроорганизмов — дрожжей, она переходит, как и при обычном дыхании, в пировиноградную кислоту, которая затем при отщеплении углекислого газа дает уксусный альдегид СН3СНО. Из него фермент дегидрогеназа этанола и особое восстанавливающее вещество «получают» этиловый спирт (рис. 30).
Углеводы — не единственное соединение, обеспечивающее наш организм энергией. Второй вид «топлива» живых существ — жиры. Это сложные эфиры органических кислот и трехатомного спирта глицерина. Они могут откладываться в тканях, выполняя те же функции, что и гликоген. Под действием щелочей жиры распадаются на глицерин и соли органических кислот, которые используются в качестве мыла. На дне стакана к нескольким каплям растительного масла прилейте немного спирта и бросьте кусочек едкого натра. Нагревайте смесь в сосуде с горячей водой, пока она не станет однородной. Вы получили раствор мыла. Если теперь к этому раствору добавить разбавленную серную кислоту, то на поверхности жидкости появятся капли органических кислот. В состав растительных масел входят глицериды ненасыщенных кислот. Это легко доказать: марганцовокислый калий реагирует с двойной связью, его раствор обесцвечивается. К нескольким каплям подсолнечного масла на дне стакана прибавьте немного раствора стиральной соды и несколько капель розового раствора марганцово-
кислого калия. При перемешивании смеси розовая окраска исчезает. Еще одна составная часть пищи — белки. Они используются для построения тела организма, для создания своих собственных белковых веществ. Но если организму не хватает энергии, он начинает сжигать аминокислоты белков. Это можно сравнить с тем случаем, когда в печке жгут ценные породы дерева. Конечно, топить можно и орехом и буком, но все-таки рациональнее делать из них красивую прочную мебель.
Белковых веществ известно огромное количество. Но все они состоят из похожих друг на друга составных частей аминокислот. В молекуле аминокислоты уживаются две противоположные по свойствам группы: кислая карбоксильная и основная аминная. Благодаря этим группам можно выстраивать длинные цепи, соединяя аминокислотные фрагменты: Н 2N—СН—СООН+Н—NH—СН— СООН-Ж 2N—
I I
—СН—СО—NH—СН—СООН+Н2О. Обычно молекула I I
белка состоит из десятков аминокислотных фрагментов.
Отделите яичный белок от желтка, хорошо взбейте его и разбавьте примерно ' десятикратным количеством воды. Теперь профильтруйте раствор через двойной слой марли. В растворе вы имеете яичный белок альбумин, в осадке белок глобулин. Глобулин можно растворить, добавляя раствор поваренной соли. К раствору альбумина (возьмите несколько капель на дне стакана или на стекле) прибавьте раствор щелочи и каплю-другую слабого раствора сернокислой меди. Появляется фиолетовое окрашивание. Дело в том, что атомы кислорода и азота пептидных связей белка образуют комплекс с медью.
Последовательность чередования аминокислот в длинной цепи молекулы белка называется ее первичной структурой. Сейчас ученые умеют определять первичные структуры очень сложных белковых молекул, но работа эта очень кропотливая и длительная. Для установления первичной структуры белка его молекулу расстригают специальными веществами на отдельные фрагменты, состоящие из нескольких аминокислот. Определить структуру таких кусочков несложно. А дальше составляют специальные карты, по которым воспроизводят структуру всей молекулы белка. Научились химики и синтезировать белок искусственно. Для этого цепь постепенно наращивают на одну аминокислоту, пока не получат всю молекулу.
Атомы водорода аминогрупп белковой цепи могут завязывать так называемые водородные связи с кислородами карбонильных групп С=О. Каждый водород NH-группы выбирает себе СО-группу из четвертого аминокислотного остатка. В результате вся длинная цепь аминокислот укладывается в правильную спираль. Каждую аминокислоту можно записать формулой H2NCH (R)COOH. Здесь группы —R, или радикалы,— это водород, метил или более сложные образования, например —CH2CH2CH2CH2NH2. Собственно этими группами аминокислоты и различаются. Когда белковая цепь укладывается в спираль, радикалы оказываются торчащими в разные стороны от этого спирального цилиндра. Спираль называется вторичной структурой белка.
Группы —R в пределах одной длинной белковой молекулы также могут взаимодействовать друг с другом в определенных сочетаниях, притягиваться одна к другой. А это значит, что спиралевидная молекула укладывается еще и в клубок. Но клубок получается не «какой попало», а по строго заданному плану (здесь можно привести сравнение с укладыванием в магазине мужских сорочек). Становится ясно, зачем белкам эти группы —R и почему мы получим совсем другой белок, заменив лишь в одном месте лишь одну группу —R. Свернутая в клубок молекула дает третичную структуру. Но это еще не все. Несколько клубков белковых молекул могут соединиться вместе, образовав четвертичную структуру. Именно так обстоит дело в случае молекулы гемоглобина. Определение третичной и четвертичной структуры белка — дело трудное. Для этого используют «просвечивание» кристаллов белка рентгеновскими лучами.
Все четыре структуры белка можно грубо промоделировать следующим образом. Возьмите гладкую длинную проволоку — это первичная структура белковой молекулы. Теперь изогните эту проволоку, как спираль для электроплитки,— вы получили модель вторичной структуры. Скомкайте спираль в спутанный клубок — это третичная структура. Соедините вместе несколько таких клубков, и вы синтезируете белок в естественном виде. Действительно, только молекула, обладающая третичной и четвертичной структурой, может «работать» в организме. Однако эту структуру легко нарушить. Соответствующий эксперимент проводил каждый, когда варил вкрутую куриные яйца. Что же происходите прозрачным, жидким, растворимым в воде белком куриного яйца? Оказывается, при нагревании рвутся водородные связи между группами —R, удерживающими белковую.
молекулу в форме компактного клубка. Третичная структура нарушается. Белок, молекулы которого представляют собой длинные перепутанные нити, конечно, не станет растворяться. Денатурацию белка, т. е. разрушение третичной структуры, можно провести и другими способами. Прибавьте к водному раствору яичного белка спирт или ацетон — раствор помутнеет. Денатурация еще не произошла, просто белок выпал в осадок. Действительно, достаточно добавить воды, и он снова перейдет в раствор. Но вот если этот осадок постоит некоторое время, третичная структура будет потеряна навсегда.
Поступившие с пищей белки в желудке расщепляются на отдельные небольшие фрагменты, которые в кишечнике разрушаются до отдельных аминокислот. Отсюда эти соединения всасываются кровью, которая разносит их по всем клеткам организма. В клетках аминокислоты снова собираются в белки, но уже другие, нужные именно этим клеткам. Синтез белка в клетке требует затрат энергии — и здесь вступают в работу молекулы-аккумуляторы АТФ, получившие энергию за счет окисления глюкозы. Некоторые аминокислоты, например валин, лейцин, лизин, обязательно должны содержаться в пище. Это так называемые незаменимые аминокислоты. Ежедневно их должно поступать с пищей около 30 г. Другие же аминокислоты — аланин, аспарагин,’ глутамин — не обязательно должны присутствовать в еде, в крайнем случае организм сможет их сам синтезировать.
Мы рассказали о химии пищи и попутно затронули некоторые вопросы биохимии. О биохимии, о том, какие опыты можно провести с белками, углеводами и другими веществами живых организмов, см. книги [21].
Сегодня все знают, что отсутствие в пище витаминов приводит к тяжелым заболеваниям, что жизнедеятельность организма без них невозможна. Но что это за соединения, сколько их, какую роль они выполняют в организме? История изучения витаминов началась в конце прошлого
века. Тогда уже было известно, что необходимые компоненты любой пищи — белки, жиры, углеводы, минеральные соли и вода. Без них организм не может нормально развиваться и погибает. В 1881 г. русский ученый Н. И. Лунин провел такой опыт. Он приготовил «искусственное молоко», т. е. смесь очищенных белков, жиров, углеводов и минеральных солей в той же пропорции, что и в натуральном молоке. Таким «молоком» стали кормить подопытных мышей. Через некоторое время все животные погибли. Стало ясно: в естественной пище содержатся какие-то незаменимые вещества, совершенно необходимые животному и человеческому организму. В 1911 г. польский биохимик К. Функ назвал эти незаменимые добавки «витаминами», т. е. «аминами жизни». В дальнейшем, впрочем, было показано, что многие витамины не имеют никакого отношения к аминам, но название осталось.
Сегодня насчитывается несколько десятков различных витаминов, известна их химическая структура, почти все они производятся в промышленности синтетически. В организме витамины входят как составные части в молекулы ферментов. Животный организм сам производить витамины не может, поэтому они обязательно должны поступать с пищей. Расскажем о некоторых витаминных веществах. Почти все они обозначаются латинскими буквами. Будем же следовать алфавиту.
При недостатке в пище витамина А нарушается обмен веществ, выпадают волосы, человек худеет. Но главное, в чем проявляется недостаток этого витамина, связано со зрением — наблюдается размягчение роговицы и сухость глаз, потеря способности видеть в сумерках (куриная слепота). Молекула витамина А, называемого еще ретинолом, состоит из длинной цепи углеродных атомов, соединенных попеременно простыми и двойными связями. Цепь эта оканчивается гидроксильной группой. Ретинол — светло-желтая вязкая масса, нерастворимая в воде, но хорошо растворяется в жирах. Производное витамина А ретинен (в его молекуле группа —СН2ОН заменена альдегидной группой —СНО) находится в сетчатке глаза. В палочках сетчатки он соединяется с белком опсином и образует зрительный пигмент родопсин. Когда свет попадает на палочку глаза, родопсин распадается на белок опсин и цис-форму ретинена. Цис-форма тут же переходит в транс-изомер, что сопровождается передачей нервного импульса в мозг (рис. 31). Импульс послан в мозг, человек увидел предмет, те
перь транс-ретинену снова предстоит перейти в цис-форму. Этот процесс можно сравнить с перемоткой фотопленки на один кадр, когда перед объективом встает незасвеченный еще кусок пленки. В глазу такая «перемотка» осуществляется в темноте по схеме: транс-ретинен под действием фермента и биологического восстановителя восстанавливается в транс-ретинол (т. е. витамин А), который специфическим ферментом изомеразой переводится в цис-ретинол. Это соединение далее окисляется в цис-ретинен. Глаз готов к «фотографированию» следующего кадра.
Витамин А содержится в рыбьем жире. Это можно доказать таким образом. На дне стакана или стекле к капле свежего рыбьего жира добавьте 5 капель хлороформа и каплю концентрированной серной кислоты. Раствор окрашивается в красный цвет. В природных продуктах широко распространен каротин. Молекула этого соединения состоит как бы из двух половинок — молекул витамина А. В ор-
Транс-ретинен
•Цис-ретинен
Цис-ретинол
Фермент, восстановитель
Фермент
Транс-ретинол (витамин А)
Рис. 31. Упрощенная схема превращений производных витамина А при восприятии света глазом. Черные точки — метильные группы
ганизме каротин превращается в ретинол. Разотрите мякоть шиповника с речным песком и прибавьте к ней в стакане или пробирке несколько капель хлороформа. Каротин переходит в хлороформ. Слейте этот раствор в другой стакан и прибавьте к нему несколько капель концентрированной серной кислоты. Верхний хлороформный слой окрашивается в зеленый, затем в синий цвет.
В шиповнике содержится также наиболее известный витамин С, аскорбиновая кислота. Это вещество представляет собой бесцветные кристаллы, хорошо растворимые в воде, оно довольно неустойчиво, легко разрушается при кипячении водных растворов. Витамин С — прекрасный восстановитель, именно на этом свойстве и основано его участие в биологических процессах. Он принимает участие в синтезе гормонов, предохраняет важное биологическое соединение — адреналин от окисления. Аскорбиновая кислота входит в состав некоторых ферментов. Витамин С является средством против цинги, его полезно применять при усиленной физической и умственной работе. На восстановительных свойствах аскорбиновой кислоты основаны многие реакции. Приготовьте вытяжку из шиповника, обогащенную витамином С. Для этого разотрите мякоть шиповника с водой и чистым речным песком. Полученную жидкость отфильтруйте через вату или промокательную бумагу и используйте в следующих опытах. В стакане к нескольким каплям раствора йода прибавьте вытяжку из шиповника. Раствор йода обесцвечивается — йод восстанавливается, а аскорбиновая кислота окисляется в дегидроаскорбиновую. В пробирке к очень разбавленному раствору красителя метиленового синего прибавьте каплю-другую раствора соды и несколько капель шиповниковой вытяжки. При нагревании краситель обесцвечивается. В пробирке или в стакане к капле раствора красной кровяной соли добавьте каплю вытяжки из шиповника. Выпадает синий осадок берлинской лазури. Разумеется, все эти опыты можно провести с раствором чистой аскорбиновой кислоты.
Рыбий жир, о котором мы уже говорили,— продукт весьма полезный. В нем, помимо витамина А, содержится другой ценный витамин — D, иначе называемый кальциферолом. Витамин D регулирует перенос кальция и фосфатов в организме. Его недостаток вызывает у детей рахит. Поместите в пробирку по нескольку капель рыбьего жира, хлороформа, анилина и каплю концентрированной соляной кислоты. При нагревании на пламени жидкость окрашивается в крас
ный цвет, обнаруживая присутствие витамина D. При встряхивании масляного раствора витамина D с хлороформом и концентрированной серной кислотой появляется красное окрашивание.
В 1936 г. венгерский биохимик Сент-Дьердьи сделал вывод, что на проницаемость кровеносных сосудов, с которой связана способность к кроветечению, оказывает влияние особое вещество — фактор проницаемости, или витамин Р Сейчас известно, что к веществам, оказывающим подобное влияние, относятся рутин, гесперидии, катехины и др. Рутин содержит фенольные гидроксилы, а значит, дает характерное окрашивание с хлорным железом. Смешайте в пробирке или на дне стакана насыщенный раствор рутина (продается в аптеке) с несколькими каплями раствора хлорного железа. Жидкость окрашивается в зеленый цвет. Рутин может восстанавливаться. К нескольким каплям раствора витамина Р добавьте кусочек цинка и две-три капли концентрированной соляной кислоты. Жидкость приобретает красный цвет. Катехины, составляющие витамин Р, содержатся в чае. Бросьте на дно стакана щепотку чая и залейте его спиртом. Через некоторое время раствор окрасится в желтый цвет. Слейте жидкость в другой стакан и прилейте к ней спиртовой раствор хлорного железа. Поя вл я-ется^ зеленоватое окрашивание.
Витамин РР не имеет ничего общего с витамином Р. Противопелларгический витамин РР по химической природе своей представляет амид никотиновой кислоты. Но не следует думать исходя из названия, что этот витамин — яд, подобный никотину. Однако от никотина нетрудно получить никотиновую кислоту, а от нее перейти к витамину РР.
Никотин, Никотиновая кислота Никотинамид
(Витамин РР)
Растворите в пробирке при нагревании немного никотиновой кислоты в нескольких каплях разбавленной уксусной кислоты. Нагрейте раствор до кипения и добавьте к нему раствор уксуснокислой меди. Жидкость мутнеет, окрашивается в голубой цвет, через некоторое время из нее выпадает синий осадок. В организме никотиновая кислота выполняет
исключительно важную роль. Амид никотиновой кислоты соединяется в клетках с сахаром, рибозой, фосфорной и адениловой кислотами и входит в таком комплексе в состав ферментов дегидрогеназ. Один из таких ферментов участвует в процессе восприятия света палочками глазной сетчатки. Восстановители, переводящие транс-ретинен в трансретинол, так называемые никотинамидадениндинуклеотид (НАД) и никотинамидадениндинуклеотидфосфат (НАДФ), содержат витамин РР.
О химии витаминов см. [22].
Значительную долю всех болезней человека вызывают самые разнообразные микроорганизмы, внедряющиеся в живые ткани и нарушающие нормальные химические процессы, в них происходящие. Как бороться с такими микробами? Один из способов — «отравить» микроорганизмы, введя в них какие-то химические соединения.
...С начала нашего века применяли так называемый napa-аминобензолсульфамид в качестве полупродукта в производстве красителей. Но вот как-то заметили, что это вещество губительно действует на микроорганизмы, вызывающие некоторые болезни. После многочисленных испытаний /шра-аминобензолсульфамид стал лекарством и получил новое имя «стрептоцид». Нагрейте на металлической пластинке таблетку стрептоцида. Образуется фиолетовый расплав, пахнущий аммиаком, что доказывает наличие в веществе аминогруппы. Теперь в пробирке прокипятите осторожно таблетку стрептоцида с концентрированной азотной кислотой, разбавьте раствор водой и добавьте к нему раствор хлористого бария. Выпадающий белый осадок доказывает присутствие сульфогруппы — SO2—. Почему же стрептоцид приводит к гибели микроорганизмов? Оказывается, бактериям для нормальной жизнедеятельности необходима /гара-аминобензойная кислота, называемая еще витамином Hi. /7ара-аминобензойная кислота входит далее в состав фолиевой кислоты, из которой бактерия изготавливает нужные ей ферменты. Витамин Hi — это фактор роста
бактерии, без него она не может расти и размножаться. Молекула /гара-аминобензолсульфамида очень похожа по размерам и химическим свойствам на молекулу витамина Hi. Бактерия охотно включает /шра-аминобензолсульфамид в состав «фолиевой» кислоты, делает из нее фермент. И вот тут обнаруживается подлог — фермент не работает. Нарушается нормальный обмен веществ в клетке бактерии, она погибает.
Стрептоцид — самый простой сульфаниламидный препарат. Сейчас известно много его производных, также успешно применяемых как лекарства. Это — сульфадимезин, норсульфазол, сульгин, этазол. Фталазол, например, представляет собой стрептоцид, атомы водорода при аминогруппах в котором замещены на органические остатки сложных соединений. Заместители не дают возможности препарату всасываться через стенки кишечника в кровь, растекаться по всему организму. Поэтому фталазол особенно сильно действует на болезнетворные микроорганизмы, которые попали в желудок и кишечник.
со-о-сн2сн2шс2н5)2
Аспирин
Норсульфазол Новокаин
/снз
CH3’^=f*NxcH2S°3Na
I c6hs
Анальгин
,CH0CH2CH2N (CH3) 2 • HCI
C6H/
Димедрол
Рис. 32. Формулы некоторых лекарств
Нагрейте в пробирке таблетку норсульфазола — образуется темно-бурый сплав, пахнущий сероводородом. Сероводород получается из серы пятичленного цикла, входящего в состав молекулы норсульфазола. Теперь возьмите три пробирки и прибавьте в них к раствору едкого натра норсульфазол, этазол и сульфадимезин. Если теперь в пробирки добавить растзор сернокислой меди, выпадут осадки: в первой пробирке грязно-фиолетового цвета, во второй — зеленого, через некоторое время чернеющего, в третьей — желто-зеленый коричневеющий осадок.
В медицине применяются и соединения настоящей пара-аминобензойной кислоты. Если в этом веществе водород карбоксильной группы заменить на радикал этил, получим анестезин, заменяя водород на более сложный радикал — CH2CH2N (С2Н5)2, приходим к новокаину. Оба соединения используются как местноанестезирующие, обезболивающие средства. В чем-то похожа на молекулу пара-аминобензойной кислоты салициловая кислота. Это бензол, в котором два соседних водорода заменены на карбоксил и гидроксил. Салициловая кислота — очень важное для медицины вещество. Сама она применяется как бактерицидное средство, при суставном ревматизме. Натриевая соль ее — болеутоляющее, жаропонижающее, противовоспалительное и противоревматическое средство. Метиловый эфир ее (вместо —СООН в нем —СООСНз) применяется в мазях как противовоспалительное, болеутоляющее и противоревматическое средство. Ацетилсалициловая кислота (вместо группы —ОН в ней присутствует группа —ОСОСН3) называется еще аспирином и хорошо известна. Фениловый эфир салициловой кислоты (вместо —СООН группы —СООСбНБ), салол,— хороший антисептик, дезинфицирующее средство. Натриевая соль napa-аминосалициловой кислоты (это соединение содержит еще дополнительно аминогруппу в бензольном ядре) — противотуберкулезный препарат.
Все фенолы дают с раствором хлорного железа фиолетовое окрашивание. Прибавьте к раствору салола в спирте каплю раствора хлорного железа. А теперь тот же опыт проделайте с аспирином. Окрашивание не появляется. Дело в том, что в молекуле ацетилсалициловой кислоты нет свободной гидроксильной группы, водород в ней заблокирован ацетилом. Но из аспирина легко получить салициловую и уксусную кислоты — прокипятите в пробирке таблетку аспирина с раствором едкого натра. После охлаждения подкислите раствор разбавленной серной кис
лотой и добавьте к получившемуся осадку салициловой кислоты каплю раствора хлорного железа — появляется фиолетовое окрашивание.
При лихорадке применяют эффективный препарат, называемый фенацетином. Молекула этого соединения состоит из бензольного кольца, связанного с группа”-!—NHCOCH3 и —ОС2Н5. С фенацетином можно провес: ", акие опыты. Бросьте в пробирку с разбавленной азотной кислотой маленький кусочек таблетки фенацетина. Появляется желтое окрашивание, а через некоторое время выпадает желтый осадок продукта нитрования. Прокипятите в пробирке разбавленную соляную кислоту с кусочком таблетки фенацетина, после охлаждения добавьте к раствору каплю раствора двухромовокислого калия. Появляется фиолетовое окрашивание, постепенно раствор становится красным.
Другой класс жаропонижающих и болеутоляющих средств — производные так называемого пиразолона. Сюда относятся антипирин, амидопирин (пирамидон) и анальгин. Они имеют схожие формулы. Антипирин с раствором хлорного железа дает красный цвет, а с подкисленным раствором азотистокислого натрия образуется нитрозопроизводное красивого зеленого цвета. В отличие от антипирина амидопирин образует с хлорным железом раствор синего цвета, выделяющий коричневый осадок. Если же такой раствор подкислить соляной кислотой, то появляется фиолетовая окраска. Такой же цвет приобретает раствор амидопирина при действии на него азотнокислого серебра.
В качестве снотворных средств используются производные барбитуровой кислоты — веронал (барбитал), фенобарбитал (люминал), барбамил, нембутал. Прилейте к раствору барбитала едкой щелочи и раствор медного купороса. Образуется синее окрашивание, а вскоре выпадает красноватый осадок. В состав барбамила входит натрий. Это легко доказать, внося немного вещества в пламя горелки — оно окрашивается в яркий желтый цвет.
В заключение несколько слов об антибиотиках. Эти вещества вырабатываются некоторыми микроорганизмами и способны губительно действовать на возбудителей целого ряда заболеваний. Пенициллин, например, не дает болезнетворной бактерии усваивать из пищи необходимые аминокислоты, бактерия лишается возможности строить оболочку клетки. Террамицин нарушает процесс фосфорилирования в бактерии, в результате которого та получает энергию. В присутствии тетрациклина или стрептомицина
бактерия не может синтезировать отдельные белки. Некоторые антибиотики дают характерные химические реакции. К нескольким каплям водного раствора стрептомицина добавьте каплю раствора едкого натра и смесь кипятите несколько секунд. К жидкости добавьте раствор соляной кислоты и затем хлорного железа. Появляется красно-фиолетовое окрашивание. Добавьте спиртовой раствор хлорного железа к раствору террамицина — раствор приобретает коричневый цвет. Если несколько кристалликов террамицина смешать с каплей концентрированной серной кислоты, появляется красное окрашивание. Растворите таблетку левомицетина в растворе едкого натра при нагревании. Возникает желтое, а затем красное окрашивание.
О лекарствах можно прочитать в книгах 122, 231.
Человеческий организм часто сравнивают в определенном смысле с огромным химическим комбинатом, выпускающим великое множество самых различных веществ. Одни химические соединения (например, белки) используются для создания тела организма, другие (углеводы) идут на снабжение энергией, третьи (например, гормоны) являются регуляторами, при помощи которых одни органы оказывают влияние на деятельность других. В нормальном здоровом организме выпуск «химической продукции» четко отлажен, каждый орган строго выполняет «план» производства своих соединений. И нужно сказать, что в данном случае не только недовыполнение, но и перевыполнение «плана» весьма нежелательно, ведь избыточное количество одного из веществ может привести к заболеванию, а то и к смерти.
Любой живой организм — система незамкнутая и постоянно обменивается веществами с окружающей средой. Одни вещества организм поглощает, перерабатывает, использует; другие — ненужные — выделяет. И можно только восхищаться тончайшей сбалансированностью всех сложнейших процессов, протекающих в организме. Но, оказывается, такую сбалансированность легко нарушить, для этого достаточно ввести некоторые химические соединения. Такие вещества в обыденной жизни называются ядами, и
их, к сожалению, известно очень много. Как же действуют яды? Почему одни вещества почти не влияют на работу организма, а введение очень небольших количеств других соединений вызывает тяжелейшие последствия? Чтобы полностью ответить на эти вопросы, нужно глубоко изучить функции самых различных соединений, вырабатываемых многими органами, понять механизм работы органов. Сегодня в общих чертах ученым известны механизмы действия ядов, но детали во многих случаях еще совершенно непонятны.
Рассмотрим действие некоторых ядовитых веществ, с которыми довольно часто приходится иметь дело в быту и при проведении простейших химических опытов. Все ядовитые вещества удобно разделить на неорганические и органические (хотя какой-либо «научности» в такой классификации нет).
Неорганические вещества, оказывающие вредное воздействие на человеческий организм, могут находиться в любом агрегатном состоянии — здесь и газы, и жидкости, и твердые соединения. Пожалуй, наиболее хорошо известна причина ядовитого влияния окиси углерода, называемой в последнее время моноксидом углерода, а в быту — угарным газом. Дело в том, что моноксид углерода очень легко образует так называемое комплексное соединение с ионом железа, входящим в состав гемоглобина. Присоединяясь к иону железа, моноксид углерода блокирует его, и человек как бы лишается гемоглобина. Транспортировка кислорода кровью становится невозможной, клетки перестают получать кислород. Моноксид углерода образуется при неполном сгорании углерода, поэтому отравления этим газом часто случаются в домах с печным отоплением. Предельно допустимая концентрация оксида углерода составляет 0,02 мг СО в литре воздуха. Разумеется, степень отравления зависит не только от концентрации оксида углерода, но и от времени вдыхания. Признаки отравления угарным газом — головокружение, головная боль, потеря сознания. При отравлении необходимо интенсивно вдыхать свежий воздух. Хотя СО прочнее связывается с железом гемоглобина, чем О2, кислорода все-таки при таком вдыхании попадает в легкие больше, и постепенно весь моноксид углерода будет вытеснен кислородом.
Мы рассмотрели действие оксида типичного неметалла— углерода. Среди оксидов металлов известны вещества совершенно нетоксичные (например, окись алюминия), но
бывают и сильные яды. Для того чтобы как-то количественно оценивать токсичность вещества, вводится понятие дозы, при которой за 14 дней гибнет половина всех взятых для испытания животных. Поскольку для животных и человека доза зависит/ от веса тела, делают перерасчет на килограмм веса животного. Если такая доза, обозначаемая DL^, меньше 15 мг яда на килограмм веса животного, вещество относится к чрезвычайно токсичным. О соединениях, для которых Z)L5o больше 1500 мг/кг, говорят как о малотоксичных. К токсичным оксидам относятся окислы ртути, таллия, свинца. Вообще чем тяжелее металл, тем токсичнее его соединения. Здесь есть два исключения: производные легких бериллия и меди также очень ядовиты. Обычно соли металла более ядовиты, чем оксиды.
Один из самых ядовитых металлов — ртуть. Весьма токсичны пары ртути. Поэтому, если вы разбили ртутный термометр, всю ртуть нужно тщательно собрать с помощью очищенной медной пластинки (капельки ртути прилипают к поверхности меди),: а остаток ртути уничтожить при помощи раствора хлорного железа. При этом металл переводится в соль. Соли ртути уже нелетучи, но они также обладают сильным токсическим действием, попадая в организм, например через рот. Тем не менее они широко применяются в лабораторной практике и даже в медицине. Как различить соли ртути? Существует несколько реакций, характерных именно для этого металла. Если подействовать щелочью на соль одновалентной ртути, образуется черно-бурый осадок закиси ртути. Из раствора соли двухвалентной ртути в этом случае осаждается желтый оксид ртути. К раствору соли двухвалентной ртути добавьте раствор йодистого калия. Выпадает красный осадок йодида ртути. Если добавить избыток раствора йодида калия, образуется раствор бесцветной комплексной соли.
Не следует думать, что соли ртути обладают какой-то феноменальной ядовитостью, и, только подержав в руках пробирку с соединением ртути, можно отравиться. Соли ртути применяются в нашей практике для медицинских целей. Так, однохлористая ртуть, коломель, используется иногда как слабительное и желчегонное средство (высшая допустимая доза 0,6 г), а очень разбавленные растворы двухлористой ртути, сулемы, применяются для дезинфекции (в организм должно попадать не более 0,02 г сулемы). Поэтому если химик работает осторожно и аккуратно, не допускает занесения солей ртути в рот, тщательно моет
химическую посуду и руки, то отравления исключены. При неаккуратной работе с соединениями ртути появляется металлический вкус во рту, боли в животе, понос, рвота. Если человек отравился солью ртути, ему следует пить молоко, промывать кишечник взвесью угля в воде, полоскать рот раствором перманганата калия.'Полезно выпить горячего чая, принять горячую ванну. Ни в коем случае нельзя употреблять внутрь поваренную соль и соленые продукты.
Соли меди менее ядовиты, однако при приеме их внутрь могут возникнуть тяжелые отравления. Различить производные меди довольно легко — чаще всего эти соединения синего или зеленого цвета. Железный предмет при действии на него раствора соли меди быстро покрывается слоем меди. Если к раствору соли двухвалентной меди добавить раствор йодистого калия, происходит выделение йода (медь восстанавливается до одновалентной), который с крахмалом дает характерную синюю окраску. Сульфат меди применяют в медицине как рвотное, антисептическое, прижигающее средство. Однократный прием сернокислой меди не должен превышать для взрослого человека 0,5 г.
Самый известный яд образуют производные мышьяка, который скорее относится к неметаллам. Отравление мышьяком может случиться и в быту, поскольку его соединения используются в медицине и для борьбы с грызунами. При острой отравлении мышьяком проявляется упадок сил, мышечная слабость. При больших дозах наступает паралич и смерть. Лечение острого отравления заключается в промывании желудка, нужно вызвать рвоту. Полезно пить взвесь в воде жженой магнезии.
Свинец — тоже весьма ядовитый металл, но в отличие от отравлений мышьяком чаще наблюдаются хронические отравления соединениями этого элемента. Первые симптомы хронического отравления людей, имеющих дело со сплавами свинца, например, в типографиях,— появление серой каймы на деснах и боли в животе. Напомним, что все мы также соприкасаемся с производными свинца, поскольку тетраэтилсвинец добавляют в автомобильный бензин. При остром отравлении соединениями свинца, которое сопровождается нарушением функционирования пищеварительного тракта, нужно принимать разбавленный раствор серной кислоты. Свинец при этом переходит в труднорастворимую соль. Токсическое действие производных тяжелых металлов связано с тем, что, попадая в организм, ионы этих элементов образуют прочные комплексы с белками. А раз так,
белки (среди которых и ферменты, и гормоны, и другие жизненно важные соединения) уже не могут выполнять свои функции. Нарушается идеальная сбалансированность всех процессов, происходящих в клетке. Известно, что пятивалентный ванадий, влияет на биосинтез холестерина и нарушает обмен аминокислот, в состав которых входит сера. Шестивалентный хром проникает в эритроциты крови и разрушает их.
В организме ионы металлов в виде комплексов с белками переносятся кровью, причем несколько задерживаются в тех органах, где присутствуют соединения или группировки, способные образовывать с ионами наиболее прочные связи. Интересно, что каждый металл имеет «любимые» органы. Так, в костной ткани накапливаются свинец, бериллий, барий, торий. Ртуть собирается в почках, мышьяк оседает в щитовидной железе, хром задерживается в поджелудочной железе, кадмий и цинк — в семенниках. Подмечено, что металлы оседают именно в тех органах, в которых содержание этих микроэлементов в нормальном состоянии повышено. Например, в здоровом организме процент ртути всегда больше в почках, свинца — в костях.
Токсичность элемента сильно зависит от валентного состояния, в котором он вводится в организм. Например, соединения трехвалентного мышьяка в 10 раз более токсичны, чем производные мышьяка пятивалентного. К сожалению, в организме пятивалентный мышьяк восстанавливается в более токсичное производное. А вот для хрома и ванадия соединения металлов в более высокой степени окисления токсичны, низковалентные же металлы не ядовиты. Поэтому при отравлении соединениями хрома, например двухромовокислым калием (химики широко используют его в хромовой смеси для мытья посуды), рекомендуется принимать аскорбиновую кислоту. Что при этом происходит, позволяет понять следующий опыт. К раствору бихромата калия в воде добавьте каплю серной кислоты и щепотку или таблетку витамина С. Желтый цвет бихромата калия перейдет в зеленый — аскорбиновая кислота восстановила бихромат в производное нетоксичного трехвалентного хрома.
Среди органических веществ ядовитых, к сожалению, ничуть не меньше, чем среди соединений, принадлежащих к неорганическому миру. Здесь и многие алкалоиды, выделяемые из растений (например, стрихнин, бруцин), и яды белковой природы (токсины ядовитых змей, скорпионов).
Токсичны почти все органические растворители, широко применяемые не только в химической лаборатории, но и в быту. В класс хлор содержащих растворителей входят хорошо известные четыреххлористый углерод (применяется для выведения жирных пятен, для тушения огня), хлороформ (все еще продолжает использоваться в медицине), винилхлорид (мономер для получения ценного полимерного материала), дихлорэтан (используется для склейки пластмасс). Все эти вещества вызывают при вдыхании нарушение сердечного ритма, жировое перерождение, цирроз и атрофию печени. Нарушается обмен веществ. Предполагают, что, попадая в организм, хлорсодержащие растворители реагируют с некоторыми веществами таким образом, что происходит разрыв связи углерод — хлор. При этом из четыреххлористого углерода образуются два радикала — С1 и СС13. Эти радикалы взаимодействуют с ненасыщенными жирными кислотами, образуя новые радикалы, которые далее реагируют с кислородом. В результате получаются производные перекиси водорода. Эти производные, в свою очередь, вызывают разрушение жирных кислот, т. е., в частности, распад мембран клеток. Вредное действие хлороформа усугубляется еще и тем, что при действии света из него образуется фосген, представляющий собой хлоран-гидрид угольной кислоты СОС12. Как и любой хлорангид-рид, фосген — соединение химически весьма активное, легко взаимодействует с белковыми и другими молекулами клетки.
Бензол — ароматический углеводород, используемый и как растворитель, и как полупродукт в органическом синтезе — вещество очень токсичное. Если в течение долгого времени неаккуратно работать с этой жидкостью, наблюдается снижение числа эритроцитов в крови, уменьшается содержание гемоглобина, происходят значительные изменения в нервной системе, нарушается деятельность сердечно-сосудистой системы. Первая помощь при отравлении бензолом — выпить взвесь активированного угля в воде, после чего вызвать рвоту. По сравнению с бензолом смесь насыщенных углеводородов — бензин — обладает меньшей токсичностью. Тем не менее при длительном контакте с бензином у человека наблюдаются изменения в нервной системе. Сильное отрицательное действие оказывает бензин на половую систему женщин и на развитие плода при беременности.
Теперь немного отвлечемся и рассмотрим, как устроены
нервные клетки и как нервное возбуждение передается по этим клеткам. Еще в XVIII в. итальянец Гальвани установил, что мышцы сокращаются под действием электричества, которое течет по нервам. Позднее немецкий физик Гельмгольц измерил скорость распространения нервных импульсов по нерву. Для лягушки эта скорость равна 30 м в секунду — скорость слишком маленькая для того, чтобы подтвердилась гипотеза, будто нервное возбуждение представляет собой лишь электрический ток. Необходимо было предположить, что в этом процессе участвуют и какие-то химические реакции.
Живая клетка, подобно радиотехническому конденсатору, несет разность потенциалов. Если два маленьких элект-родика подключить к клетке с внешней и внутренней сторон, то соединенный с электродами гальванометр покажет разность потенциалов, называемую потенциалом покоя. При любом раздражении нерва разность потенциалов быстро изменяется и передается по нерву. Но нервы имеют конечную длину, а между нервными клетками имеются «стыки». Как же перескакивает электрический импульс с нерва на нерв? Вот здесь-то и участвуют химические посредники.
Стык двух нервных клеток называется синапсом. Вот первый импульс, так называемый потенциал действия, дошел по первой (пресинаптической) клетке до места соприкосновения ее со второй (постсинаптической) нервной клеткой. Из специальных пузырьков в пресинаптической клетке выделяется особое вещество — ацетилхолин,— поступающее в синаптическую щель. Ацетилхолин действует на постсинаптическую клетку и вызывает в ней новый нервный им-' пульс, который распространяется дальше. Аналогичный процесс происходит и в месте стыка нервной клетки с мышечной тканью. Ацетилхолин, выделяющийся из нервной клетки, вызывает изменение разности потенциалов в клетках мышцы, возникает импульс, мышца сокращается. Ацетилхолин не должен непрерывно стимулировать нервный импульс в мышце или постсинаптической клетке. Сделавший свое дело химический посредник обязан немедленно уйти со сцены. Убрать его должен специальный фермент — ацетил холинэстераза. Этот фермент гидролизует ацетилхолин до холина, который биологической активностью не обладает.
Любое нарушение в прекрасно отлаженном механизме передачи нервного импульса сказывается весьма драматически. А такие нарушения могут вызываться многими хими
ческими соединениями. Например, яд кураре блокирует центры нервных клеток, чувствительные к ацетилхолину. Поэтому введение в организм экстракта растения кураре вызывает мгновенную парализацию (индейцы обмакивали в яд кураре концы своих смертоносных стрел). Надо заметить, что не всякий нервный импульс передается через синаптическую связку, проходят только достаточно сильные возбуждения. Но вот алкалоид стрихнин (это яд, выделяемый из растений) снижает сопротивление синапса, так что даже очень слабый раздражитель начинает вызывать судороги мышц.
Известны и вещества, действующие на фермент холинэстеразу, и их, к сожалению, довольно много. Наиболее активно дезактивируют холинэстеразу фосфорорганические соединения, т. е. вещества, содержащие в молекуле связанный с углеродом атом фосфора. К таким фосфорорганическим производным относятся хорошо известные отравляющие вещества зарин, зоман, табун и знаменитые пестициды тиофос, хлорофос, меркаптофос. Надо сказать, что пестициды гораздо сильнее действуют на насекомых, чем на теплокровных животных, но все-таки эти вещества ядовиты и для человека. Особенно ядовит тиофос, а хлорофос обладает средней токсичностью, бромофос же совсем мало ядовит.
Прекратить действие холинэстеразы введением ядовитого вещества — это то же самое, что ввести в организм избыток ацетилхолина. Ацетилхолин же вызывает сильнейшее биологическое воздействие — понижает кровяное давление, вызывает судороги.
С относительно нетоксичным хлорофосом можно провести такую характерную реакцию. Растворите несколько крупинок хлорофоса в чайной ложке спирта, добавьте чайную ложку ацетона и половину чайной ложки спиртового раствора едкого натра. Через несколько минут появится розовое окрашивание, затем оно перейдет в малиновое и, наконец, в оранжевое.
Ученые, поставив множество экспериментов на животных, нашли химические соединения, нейтрализующие действие различных ядовитых веществ. Например, сегодня известен целый ряд препаратов, которые могут восстанавливать, как говорят, реактивировать холинэстеразу, блокированную фосфорорганическим отравляющим веществом. Такие препараты называются антидотами и вводятся в пораженный организм. Но надо подчеркнуть, что и сам отрав
ленный организм пытается бороться с непрошенным пришельцем. Он использует многие биологически активные соединения, ферменты, кислород для очень сложных химических превращений, в результате которых ядовитое вещество превращается в менее ядовитые продукты и выводится из организма. Любое ядовитое вещество выводится из организма по экспоненциальному закону, т. е. наиболее активно выходит основная масса в первые минуты и часы, а выведение остатков растягивается на долгие дни и недели. К сожалению, иногда бывает, что умеренно ядовитое вещество в организме претерпевает такие превращения, что переходит в гораздо более токсичное соединение. Примером здесь может послужить окисление тиофоса в сильно ядовитый фос-факол. Поэтому организму удается справиться далеко не со всеми ядовитыми пришельцами.
В заключение напомним: обращаться с применяемыми в быту ядовитыми веществами нужно чрезвычайно осторожно. Все банки и коробки должны быть подписаны, снабжены надписью «ЯД» и спрятаны в такие места, откуда их не могут достать дети. После работы с ядом необходимо тщательно вымыть посуду и руки. Ну а если вы, несмотря на все предосторожности, почувствовали симптомы отравления?
В первую очередь нужно вызвать врача. А до его прихода необходимо лечь на спину и не совершать лишних движений. Если нет рвоты, нужно ее вызвать. Для этого пострадавшему дают выпить больше пяти стаканов чистой или слегка соленой воды и надавливают пальцами на основание языка. У потерявшего сознание человека вызывать рвоту нельзя. Полезно выпить несколько столовых ложек кашицы из смешанного с водой толченого активированного угля.
О ядах, противоядиях, о механизме их действия см. [24].
1. Кузнецов В. И. Тенденции развития химии.— М.: Знание, 1976; Добротин Р. Б. Философские проблемы современной химии.— Л.: Знание, 1977; В я з о в к и н В. С. Материалистическая философия и химия.— М.: Мысль, 1980; Кедров Б. М., Волков В. А. О предмете химии// Химия и жизнь, 1982, № 10; В а-с и л ь е в а Т. С., О р л о в В. В. Химическая форма материи.— Пермь, 1983.
2. И в а н о в а М. А., К о н о н о в а М. А. Химический демонстрационный эксперимент.— М.: Высшая школа, 1984.
3. Зайков Г. Е., Крицман В. А. Химическая кинетика.— М.: Знание, 1980; Пурмаль А. П., Слободецкая Е. М., Травин С. О. Как превращаются вещества.— М.: Наука, 1984.
4. Jensen J. Н. A New Type of Oscillating Reaction: Air Oxidation of Benzaldehyde// Journal Amer. Chem. Soc., 1983, vol. 105, p. 2639.
5. А ф а н а с ь e в В. А., Зайков Г. E. Катализ в химических реакциях.— М.: Знание, 1977; Они же. В мире катализа.— М.: Наука, 1977; Боресков Г. К. Некоторые проблемы катализа.— М.: Знание, 1981; Грязнов В. М., Смирнов В. С. Селективность катализаторов.— М.: Знание, 1983; Шилов А. Е. Металлокомплексный катализ и его место в науке о катализе// Природа, 1979, № 10.
6. Васильева 3. Г., Грановская А. А., Таперов а А. А. Лабораторные работы по общей и неорганической химии.— М.: Химия, 1979.
7. Б а б к о в А. В., Горшкова Г. Н., Кононов А. М. Практикум по общей химии с элементами количественного анализа.— М.: Высшая школа, 1978.
8. С л о в е ц к и й В. И. Электросинтез в органической химии.— М.: Знание, 1981; Чирков Ю. Г. Любимое дитя электрохимии.— М.: Знание, 1985.
9. И в а н о в Е. С., Иванов С. С. Коррозия и защита металлов.— М.: Знание, 1978.
10. Д у л и ц к а я Р. А., Фельдман Р. И, Практикум по физической и коллоидной химии.— М.: Высшая школа, 1978.
11. Бабенков Е. Д. Воду очищают коагулянты.— ДК: Знание, 1983; Фукс Г. И. Коллоидная химия нефти и нефтепродуктов.— М.: Знание, 1984.
12. Я с а й т и с А. А. От солнечного луча до нашего сознания.— М.: Знание, 1979; Комиссаров Г. Г Химия и физика фотосинтеза.— М.: Знание, 1980.
13. Аверина А. В., Снегирева А. Я. Лабораторный практикум по органической химии.— М.: Высшая школа, 1975.
14. Лабораторные работы по органической химии. Под ред. О. Ф. Гинзбурга, А. А. Петров а.— М.: Высшая школа, 1982.
15. К о ч е т к о в Н. К., Кондратьева Г. В. Тонкий органический синтез.— М.: Знание, 1984; Шульпин Г. Б. Эта увлекательная химия.— М.: Химия, 1984.
16. Ш у л ь п и н Г. Б. Загадки запаха // Наука и жизнь, 1978, № 1. Ш у л ь п и н Г. Б. Ученые спорят о вкусах И Наука и жизнь, 1979, № 10.
17. С о к о л о в В. И. Молекула — лента Мёбиуса // Химия и жизнь, 1983, № 3.
18. С о к о л о в В. И. Химическая топология.— М.: Знание, 1981; Дмитриев И. С. Молекулы без химических связей.— Л.: Химия, 1980; Ш у л ь п и н Г. Б. Математика без чисел И Химия и жизнь, 1983, №3; Ш у л ь п и н Г. Б. Стереохимия на пальцах // Химия и жизнь, 1985, № 6.
19. С а л е м Л. Чудесная молекула.— М.: Мир, 1983; Дмитриев И. С. Электрон глазами химика.— Л.: Химия, 1986.
20. Чертков И. Н. Эксперимент по полимерам в средней школе.— М.: Просвещение, 1980; Эскин В. Е. Мир невидимых великанов.— М.: Наука, 1976; Копылов В. В. В мире полимеров.— М.: Знание, 1983.
21. Филиппович Ю. Б. Биохимия белка и нуклеиновых кислот.— М.: Просвещение, 1978; Филиппович Ю. Б., Егорова?. А., Севастьянова Г. А. Практикум по общей биохимии. — М.: Просвещение, 1982.
22. С е н о в П. Л. Фармацевтическая химия.— М.: Медицина,
1971.
23. М а к а р о в К. А. Химия и медицина.— М.: Просвещение, 1981; Сафонова Т. С. Пути развития химии лекарственных веществ.— М.: Знание, 1978; Крылов Ю. Ф., Смирнов П. А. Удивительный мир лекарств.— М.: Знание, 1985.
24. Оксенгендлер Г. И. Яды и противоядия.— Л.: Наука, 1982; К и б я к о в А. В., Сахаров Д. А. Рассказы о медиаторах. — М.: Знание, 1978.
СОДЕРЖАНИЕ
Предисловие . . 3
Химия — наука о превращениях молекул 6
Скорость химического превращения 8
О катализе . . ... 14
Сколько железа в водопроводной воде? 22
Химические реакции и электрический ток 30
Ржавчина с точки зрения химика . 34
Вещество на поверхности, или химия стирки 39
Почему небо голубое? 45
Химия и свет 52
Желтая краска из черного угля 59
Духи из... нафталина 67
«Черный ящик» химического превращения 71
Химические головоломки и парадоксы 76
Электронные облака из пластилина 90
Теория предсказывает 97
Какие бывают полимеры . . 105
Почему резину можно тянуть? 111
Наша пища 115
Витамины глазами химика 122
Опыты с лекарствами 127
Почему яды ядовиты? 131
Литература 140
Георгий Борисович ШУЛЬПИН
ХИА4ИЯ ДЛЯ ВСЕХ (Основные понятия и простейшие опыты)
Главный отраслевой редактор Л. Нелюбов
Редактор Н. Феоктистова
Мл. редактор Н. Карячкина
Худож. редактор М. Бабичева
Техн, редактор Н. Лбова Корректор Н. Мелешкина
ИБ № 8486
Сдано в набор 30.05.86. Подписано к печати 02.12.86. А07907. Формат бумаги 84Х 1081/32. Бумага тлфдруч. Гарнитура литературная. Печать высокая. Усл. печ. л. 7,56. Усл. кр.-отт. 7,88. Уч.-изд. л. 7,72. Тираж 80 000 экз. Заказ № 2675. Цена 50 коп. Издательство «Знание». 101835, ГСП, Москва, Центр, проезд Серова, д. 4. Индекс заказа 876702. Отпечатано с матриц МПО 1-ой Образцовой типографии им. А. А. Жданова на полиграфкомбинате ЦК ЛКСМУ «Молодь». 252119, Киев-119, Пархоменко. 38- il. Заказ 7—32.
50 к.