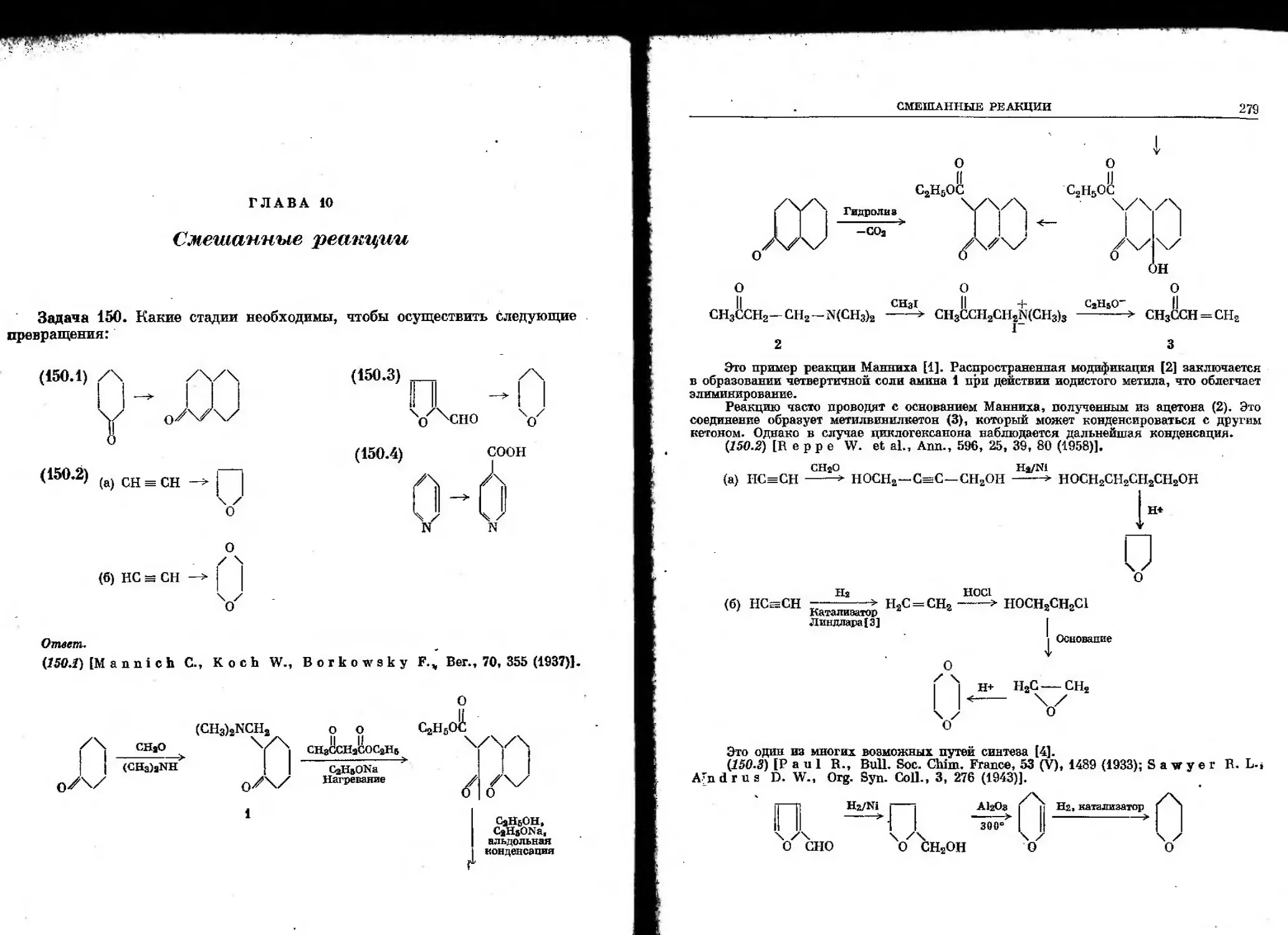
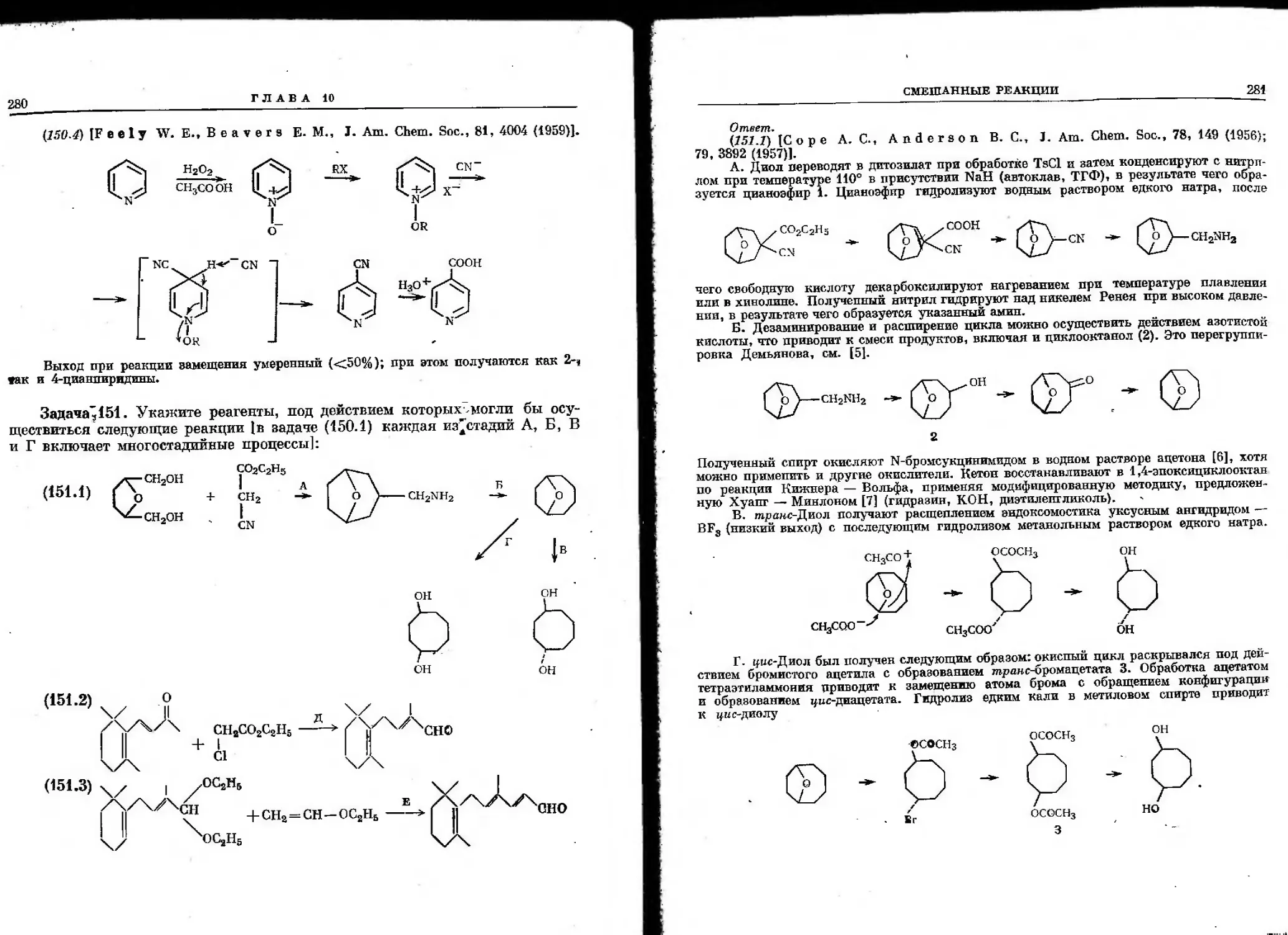
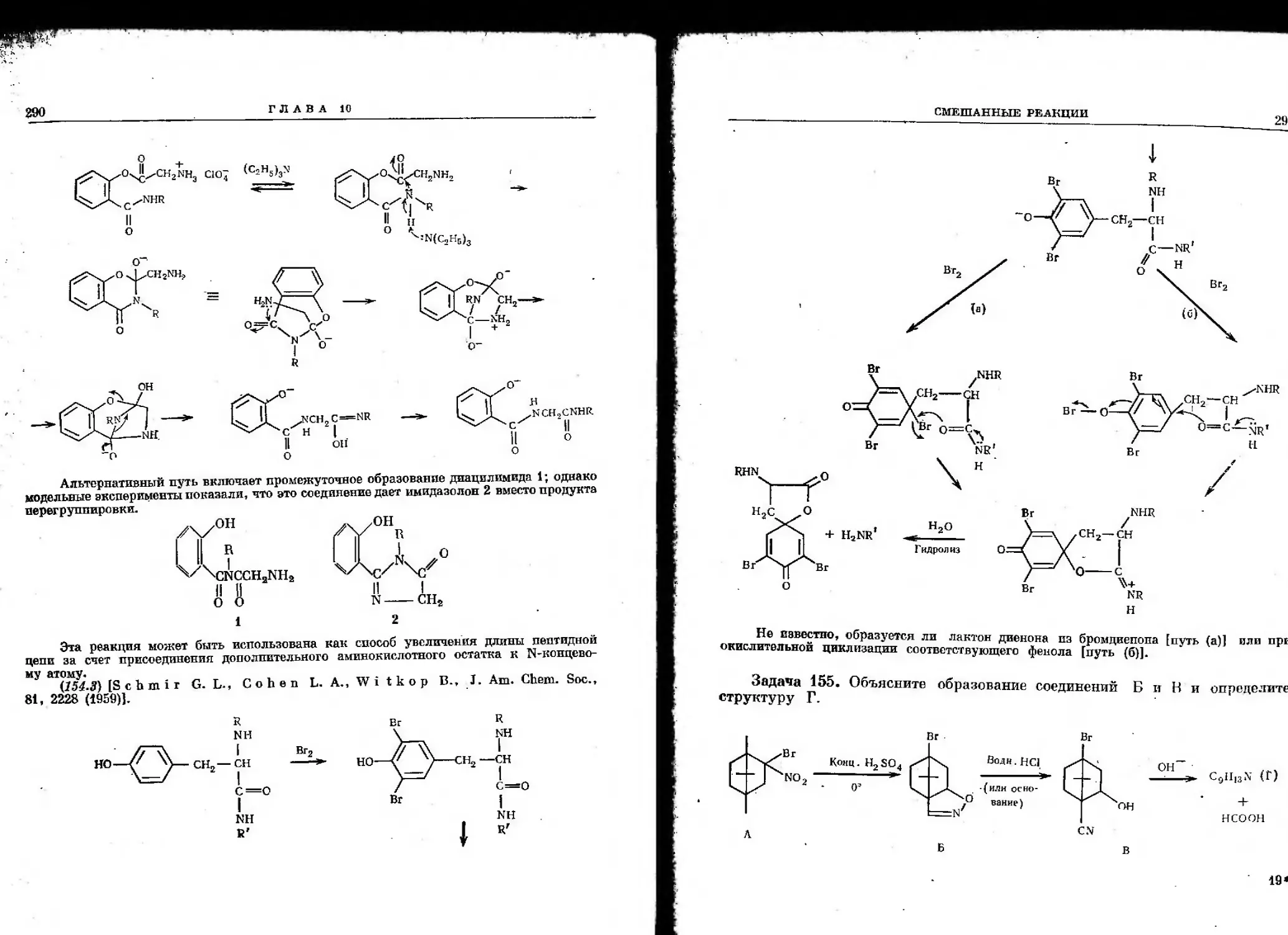
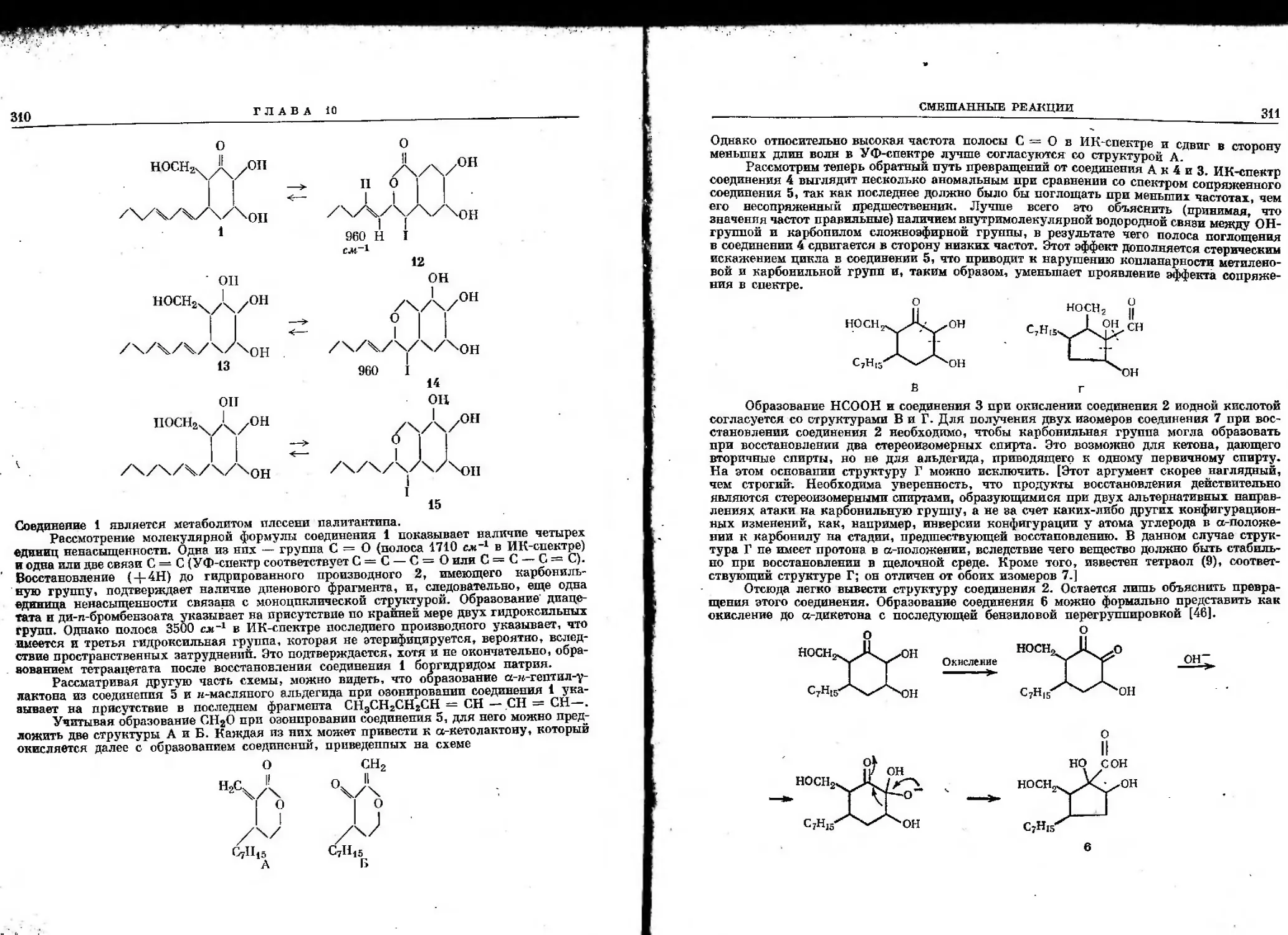
/
















































































































































Автор: Гото Т. Хирата И. Стоут Г.
Теги: химия органическая химия




Текст
Общая литература
1. Беллами Л., Инфракрасные спектры сложных молекул, ИЛ, Мм 1963.
2. Илиел Э., Стереохимия соединений углерода, изд-во «Мир», М., 1965.
3. Gould E. S., Mechanism and Structure in Organic Chemistry, Holt
N.Y., 1959.
4. Hine J.t Physical Organic Chemistry, 2nd ed., McGraw-Hill, N.Y., 1962.
5. Jactman L. M.r Application of Nuclear Magnetic Resonance
Spectroscopy in Organic Chemistry, Pergamon Press, London, 1959.
, 6. Royals E., Advanced Organic Chemistry, Prentice-Hall, N.Y., 1954.
7. Наканиси К., Инфракрасные спектры и строение органических
соединении, изд-во «Мир», М., 1965.
8. Ингольд К. К., Механизм реакций и строение органических
соединений, ИЛ, М., 1959.
9. Ионин Б. И., Ершов Б. А., ЯМР-спектроскопия в органической
химии, изд-во «Химия», Л., 1967.
10. Терентьев А. П., Потапов В. М., Основы стереохимии, Госхим-
издат, М.—Л., 1964.
ГЛАВА 1
Общие проблемы
Монографии, приведенные в списке общей литературы в начале книги,
следует рекомендовать при решении всех задач данной книги. Задачи
настоящей главы требуют знания основ, которые исчерпывающе изложены,
например, в работах Хайна и Гулда. Знание этих основ совершенно необходимо
для каждого химика-органика. Кроме того, можно рекомендовать ряд
монографий, посвященных тем или иным разделам данной главы [1—7].
Задача 1. Расположите перечисленные ниже группы (R = алкил}
в порядке возрастания величины указанного эффекта.
О
(1.1) —/-Эффект — SO", — SR, — SR, — S02R
R R
(1.2) +Ж-Эффект — NCOR, —NCR, — NCH2R, —NCR
I II I II
R NR R +NR2
(1.3) — jtf-Эффект — CONR2, —C —NR2, — C —NR2
II II
NR ■ +NR2
Ответ (Ингольд К. К., Механизм реакций и строение органических
соединений, ИЛ, М., 1959, стр. 52—77).
Система обозначений эффектов такая же, как у Ингольда. Хотя эта система не
слишком широко используется в США, она часто применяется в мировой литературе.
Символом / обозначают индуктивный эффект, а М — мезомерный или резонансный эффект *
в основном состоянии молекулы. Знак плюс указывает на электронодонорпое, а знак
минус — на электроноакцепторное действие.
о- о- о-
! I I
(1.1) — SR< — S — R< — S2+ — 0-<—S2+—R
I I
o- o-
Электроноакцепторный индуктивный эффект возрастает с увеличением формального
положительного заряда серы. Однако целый отрицательный заряд группы —SOjf умень-
* В советской литературе его также называют эффектом сопряжения.— Прим- перев.
10
ГЛАВА 1
шает этот эффект по сравнению с сульфопом —S02H.
(2.2) — N — С—R< — N — C-R<—N — C — И< — N— CH2R
f II J II I li I
R +NR2 R О R NR R
Электронодонорная способность атома азота с участием резонансной структуры А
возрастает с уменьшением вклада резонансной структуры Б. Если X — сильный акцептор
jL.f!_c_ ** =n—С *-» N=C —
fi X
А
I I
Б
электронов, то главный вклад обусловливает структура Б; в этом случае вклад
структуры А имеет меньшее значение. Электроноакцепторное действие (—М) будет изменяться
в ряду
= NR> = 0> = NR>H2.
(1.3) NR О NRa
If II II
—CNR2<-C-NR2<-C-NR2
Вклад резонансной формы А будет возрастать по мере увеличения электроноакцеп-
торной способности атома или группы X в ряду
О
:х:
= N-< = 0< = N^ l^LC-NR, *-Y=C_
\
NR,
Задача 2. Укажите, какое соединение в каждой из перечисленных ниже
дар имеет больший дипольный момент.
(2.1) м- и -И-Хлорнитробензолы.
(2.2) 3,5- и 3,6-Диметилнитробензолы.
(2-3)
CN
I
CN
СОСНд
I
/\
ч/
I
СОСН3
^=л ноос
s?— соон
(2.5)
С]
СН3
=v (2.6) ^ N
ci
СН;
/
CflCl
H5C
;CHci
ОБЩИЕ ПРОБЛЕМЫ
11
Ответ (Gould E. S., Mechanism and Structure in Organic Chemistry, Holt, N.Y.,
1959, Chap. 3; S ш у t h C. P., Dielectric Behavior and Structure, McGraw-Hill, N.Y., 1955,'
Chap. 8—11; см. также [8]).
Результирующий дипольный момент молекулы часто можно рассматривать как
векторную сумму индивидуальных моментов различных связей. Этот метод непригоден,
если между связями имеется взаимодействие, в результате которого изменяется
распределение электронов.'Однако его можно использовать для приближенного рассмотрения.
(2.1)
С1
Отсюда А>Б
Найдено: A = 3,69D
B = 2,78D
( *-
результирующий дипольный
момент)
CI
NOn
(2.2)
н,с
' }>
- U
Отсюда А>Б
(——-—■*-
результирующий дипольный
момент)
Дипольный момент связей СН3 — С направлен к циклу, тогда как связи С — NOj
направлены от цикла. В соединении Б вклады двух метильных групп взаимно
погашаются, но в соединении А они приводят к некоторому увеличению дипольного момента
молекулы.
12
ГЛАВА 1
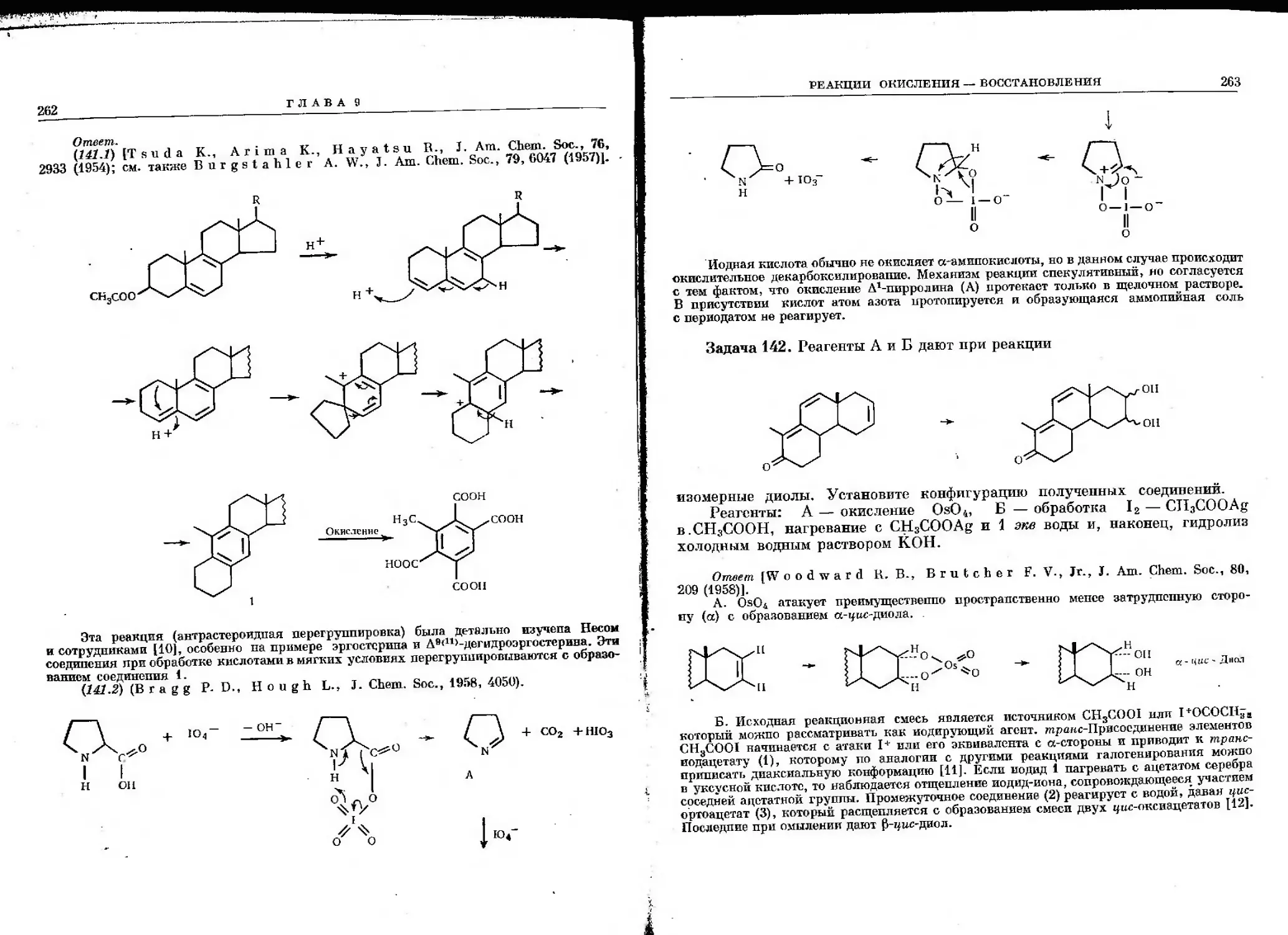
(2.3)
И
Ра зул ь тн рующий дипольны ft
момент =0
Отсюда Б > А
o^S сн
o>s ^сн3
^г/
в 4^
Результирующий
дипольный момент
в ^
Результирующий
дипольный момен,т=0
Дипольный момент ацетильной группы ориентирован приблизительно вдоль кар-
Т — -"^
бонила z> С = О. В одной из конформаций (В) моменты обеих групп погашаются, но в
другой (Б) получается суммарный дипольный момент. В растворе, где присутствуют обе
конформаций, мы будем наблюдать некоторый дипольпый момент. Так как циангруппы
в соединении А являются линейными, то результирующий дипольный момент постоянно
равен нулю.
(2А)
соон
со он
Результирующий
дипольный момент
НООС
НООС
Отсюда
Л>В
Результнру ющий
дипольный момент
Поскольку сам азулен имеет дипольный момент, то при определении суммарного
момента его производных необходимо учитывать вклад резонансных структур этого
ароматического соединения (сравните мезомерную структуру Б).
ОБЩИЕ ПРОБЛЕМЫ
13
(2.5)
Н
С1
А>Б
В соединении А связи С — О и С — С1 приблизительно направлены в одну и ту же
сторону, тогда как в соединении Б — в противоположные стороны.
(2.6) Циклопропан обладает промежуточными свойствами между свойствами о л ефинов
и предельных углеводородов. В частности, циклопропильная группа является более
электроотрицательной, чем алкильная, и облапает меньшей тенденцией отдавать электроны.
Этот эффект может быть обусловлен либо сопряжением, увеличивающим электронную
плотность в цикле {—М-эффект), либо увеличением «-характера связей, идущих от
трехчленного цикла. Во всяком случае, точное значение дипольного момента циклопропил-
хлорида (1,76D) ближе к дипольному моменту хлористого винила (1.44D), чем изопропил-
хлорида (2,15D).
Задача 3. Укажите, какое соединение в каждой из приведенных ниже
пар будет иметь большую теплоту гидрирования.
(3.1) Пентадиен-1,4 и пентадисн-1,3.
(3.2) транс- и ^ис-4,4-Диметилпентепы-2.
(3.3) Бутен-1 и транс-бу1ея-2.
(3.4) Этилиденциклогексан и 1-этилциклогексен-1.
(3.5) Бицикло-[2,1,0]-пептан и циклопентен.
Ответ (Тернер Р. Б., в книге «Теоретическая органическая химия», ИЛ, Мм
1963, стр. 89; см. также [9]).
Относительные теплоты образования двух изомерных ненасыщенных соединении
можно сравнивать лишь в том случае, если при гидрировании они дают одип и тот же
продукт и измерена теплота гидрирования. Менее стабильный изомер будет иметь
большую теплоту гидрирования, так как при этом он выделяет больше внутренней энергии.
Так как процесс происходит с выделением тепла, теплота гидрирования имеет
отрицательный знак.
(ЗЛ) СНа = СН — СН2—СН = СН2 Mi = — 60,8 ккал/молъ
СН2 = СН — СН=СН —СНз Д#=— 54,1 ккал/моль
Сопряженный диен стабилизирован вследствие резонанса, ипоэтому его гидрирование
протекает с большим трудом, чем гидрирование несопряженного изомера.
Пегзтаднен-М +2Н2
—60,8 ккад/моль
1'
Пеятадиен -1,3 + 2Н2
-54,1 икал /модь
|Г
н - Пентан
14
ГЛАВА 1
(3.2) [Turner R. В., Net tie ton D. E., Jr., P e г 1 m a n M., J. Am. Chem.
Soc, 80, 1430 (1958)].
Hx /C<CH3)3
С — С Д# = — 26,5 ккал/моль
СВзХ ХН
Н\ /Н
С = С Д# = —30,8 ккал/молъ
т/ чс(сн3)з
фис-Алкены менее стабильны, чем их транс-изомсры вследствие большего стериче-
ского напряжения, обусловленного отталкиванием двух объемистых групп, находящихся
по одну сторону от двойной связи. Разность в энергиях цис- и транс-изомеров изменяется
от приблизительно 1 ккал/молъ для бутена-2 и до 9,3 ккал/моль для 1,2-ди-трет-бутил-
этилена.
(3.3) {Kistiakowsky G. В., Ruhoff J- R., Smith H. A„Vau-
g h a n W. E., J. Am. Chem. Soc., 57, 876 (1935)].
CH3CH3CH = CH2 AH = — 30,3 ккал/молъ
С = С Д# = —27,6 ккал/моль
За исключением особых случаев, где большую роль играет напряжение молекулы,
стабильность олефшюв возрастает и теплота гидрирования уменьшается с возрастанием
степени вамещенности двойной связи. Имеются разногласия о причинах этого эффекта
для адкильных заместителей, но сам эффект, несомненно, реален.
(3.4) iTurner R. В.f Garner R. H., J- Am. Chem. Soc, 80, 1424 (1958)].
HC—CHS
Л
\/
CH2CH3
I
U
ДЯ = —26,3 ккал/моль
ДЯ= —25,1 ккал/моль
Из данных по тешштам гидрирования, а также из экспериментов по равновесной
изомеризации совершенно ясно, что двойная связь в шестичленном цикле (эндоцикличе-
ская) энергетически более выгодна, чем связь, выходящая из цикла (экзоциклическая).
Возможно, это обусловлено стерйческими причинами, так как наличие двойной связи
в цикле приводит к уменьшению числа неблагоприятных аксиально-аксиальных
взаимодействий атомов водорода. Этим, по-видимому, можно объяснить большее содержание
енольной формы в циклогексанонах по сравнению с алициклическими кетонами.
ОБЩИЕ ПРОБЛЕМЫ
15
{3.5)
ДН = — 55,1 ккал/моль
\
ДН ~- — 25,7 ккал /моль
Напряжение в бициклопентане очень большое, и оно отражается на теплоте
гидрирования. Отметил для сравнения, что величина Д#, вычисленная для циклопропана
(—37,6 ккал/молъ), значительно больше, чем для пропилена (—30,1 ккал/моль').
Задача 4. Определите направление дипольного момента родоначального
соединения и обоснуйте ваше решение, исходя из приведенных ниже
значений дипольных моментов производных.
U, Х- Н 3.7ID
1б, Х= Br 5.07D
NCH,
2а, Х=Н 1.24D
2б, Х = Вг 2,520
Ответ [Brasftn W. R., Holmquist Н. Е., В е n s о n R. E., J. Am. Chem.
Soc, 83, 3125 (1961)J.
Д идо л ьный момент свяаи Саром — Вг равен приблизительно 1,5D, причем атом брома
является отрицательным концом диполя. Отсюда следует, что замещение атомом брома
приводит к уменьшению результирующего дипольного момента трополона (1), но
увеличивает его у азотистого аналога (2). Таким образом, эти ненасыщенные соединения имеют
противоположно ориентированные дипольные моменты-
Тенденция семичленного ненасыщенного цикла превращаться в 6я-электронную
систему тропилия, имеющую положительный заряд, хорошо известна; см., например,
азу лен (3) и трополон (1). Аминосоединение (2), очевидно, не обладает такой тенденцией.
Вместо этого наблюдается перенос электронов от атомов азота к циклу. Это паводит
сн,
он
о-
N
I
сн.
3, 1.0D
16
ГЛАВА 1
на мысль, что перенос электронов должен включать участие резонансной формы,
изображенной на схеме, с Ю^электронпой структурой. Эту молекулу можно также Гедс^нть
как нолиен с изогнутой цепью (4), в котором дипольный момент возникав* в результате
переноса электрона от протона к сопряженной системе. результате
Нн
Задача 5. Какие из приведенных ниже соединений можно классифищг-
ровать как ароматические системы?
(5.1) Катион 1,2,3-трифенил- (5.6) Азулен.
циклопропенилия. (5.7) Сидноа.
(5.2) Дианион циклооктатет-
раена.
(5.8)
(5.3) Циклобутадиен. /д 9)
(5.4) Циклогептатриен-2,4,6-
карбоновая-1 кислота.
(5.5) и-Бензохинон.
Ответ (Gould E. S., Mechanism and Structure in Organic Chemistry, Holt, N-Y,
1959, pp. 412 ff; К р е й г Д., в книге «Небензоидные ароматические соединения», под
ред. Гинсбурга, ИЛ, М., 1963; см. также [10]).
Фундаментальной проблемой в данном вопросе является определение понятия
«ароматичность». По-видимому, лучшее определение следующее: ароматическим является
то соединение, которое значительно стабильнее, чем можно было бы ожидать для
гипотетической модельной молекулы, имеющей ту же структуру и фиксированное распределение
электронов, отвечающее нормальным простым и двойным связям. Разность в энергиях
между реальной молекулой и модельной называется «энергией делокализации».
Экспериментальным путем установлено, что ароматические соединения менее реакционноспо-
■собны, чем можно было бы ожидать на основании модели с фиксированными связями.
Ароматические соединения чаще вступают в реакции замещения (с сохранением ароматиче-
ОБЩИЕ ПРОБЛЕМЫ
17
ской системы) ^чем в реакции присоединения (в результате которых ароматическая система
разрушается). Современный критерий ароматичности соединения можно получить из
данных спектров ЯМР, а именно из данных по кольцевым токам, отражающим делокализацию
электронов в циклических ненасыщенных системах в присутствии магнитного поля[151.
Согласно правилу Хюккеля, основанному на простых квантовомеханических
расчетах, ароматические соединения должны обладать (An + 2) я-электронами в плоской,
полностью сопряженной циклической системе. Имеется много подводных камней в общем
приложении этого правила (особенно в отношении требования нланарности), тем не менее
оно часто оказывается хорошей путеводной нитью.
{5.1) [Breslow R., Y u a n С, J. Am. Chem. Soc, 80, 5994 (1958)].
с6н5
&К;.
CRHC
ХвН
СяНс
,с*н
6ПБ
с6п5
с6н6
Правило Хюккеля при п = 0 выполняется, и соединение ароматическое.
(5.2) [К a t z Т. J., J. Am. Chem. Soc, 82, 3784 (1960)1.
©--d-o -•■•■
Относительно легкое образование этого дианиона и наличие кольцевого тока
свидетельствуют о его ароматическом характере. Как 10л-электронная система, этот аниоп
удовлетворяет правилу Хюккеля с п = 2. Необходимо заметить, что незаряженный
предшественник этого дианиона — циклооктатетрасп — непланарный и не обладает
ароматическими свойствами. Это обусловлено присутствием только 8л-электронов (4п) в этом
ненасыщенном соединении.
(5.3) [Б е и к е р В., М а к-0 м и" Дж., в книге «Небензоидные ароматические
соединения», под ред. Гинсбурга, ИЛ, М:, 1963; см. также [11]).
-К-
Хотя для циклобутадиена можно написать простую бензолоподобную сопряженную
структуру, она не удовлетворяет правилу Хюккеля, вследствие чего этот углеводород
чрезвычайно нестабилен. И расчеты, и физико-химические измерения показывают, что
это соединение имеет форму скорее прямоугольника, чем квадрата, и, следовательно,
резонанс не имеет места.
(5.4)
сооп
соон
• Эта молекула не имеет замкнутой сопряженной системы и поэтому пе обладает арома
тическими свойствами. Потеря гидрид-иона Н~ приводит к катиону А, который содержит
2-374
18
ГЛ АВ А 1
бя-электронную систему тропилия и поэтому ароматичен.
О
Хотя п-бенвохинон имеет значительную энергию делокализации (16 ккал/моль), она
много меньше, чем у бензола, и молекулу хинона, вероятно, лучше всего рассматривать
как комбинацию фрагментов а, fl-непредельного кетона* имеющих относительно слабое
дополнительное взаимодействие в основном состоянии.
{5.6) (Хейльброннер И., в книге «Небензоидные ароматические
соединения» под ред. Гинсбурга, ИЛ, М-, 1963).
«т.д.
Авулен — ароматический углеводород, во многом сходный с нафталином, хот»
в менее стабильный. Он имеет 10л-электронную систему, которую можно изобразить как
комбинацию тропилий-катиона и циклопептадиенил-аниона (А) или, более грубо, как
простой сопряженный циклодекапентаен с одной внутрициклической связью.
{5,7) [Baker W.,OIlis W. D., Quart. Rev., 11,15 (1957) J.
хн=с-о- ,сн=с—o- . ,ch—c—o-
!N —О ^N = 0+ \N—0+
R—N
в т. д.
CH—С— О-
"N—О
Сидноны [12] относятся к классу «мезо-ионных соединений», которые нельзя
удовлетворительно изобразить формулой с простыми валентными связями, не включающей
разделенных зарядов. Они могут быть представлены как гибрид большого числа диполяр-
ных и тетралолярных структур. Их можно рассматривать как ароматические соединения,
так как пятичленный цикл содержит шесть я-электронов.
{5.8) [Sondheimer F., Wolovsky R.,J. Am. Chem. Soc, 84, 260 (1962)].
Если ацетиленовую связь рассматривать как электронную структуру, дающую
2л-электрона в сопряженную систему (другая л-связь расположена в плоскости
молекулы и не принимает участия в сопряжении), то этот тридегидро-[18]-анпулен
удовлетворяет правилу X юкке л я при п = 4. Для этого соединения показано наличие кольцевого
тока, его УФ-спектр похож на спектры ароматических соединений; крйме того, он болег
стабилен, чем его ациклические аналоги.
ОБЩИЕ ПРОБЛЕМЫ
19
к Частичное гидрирование ацетиленовых связей [13] приводит к [18]-аннулену (А)^
i't- который также обладает ароматическими свойствами, хотя и не очень стабилен.
■ (5.9) [Wind g ass en R. j.f Saunden W. H., Jr., Boekelheide V,
I. Am. Chem. Soc, 81, 1459 (1959)].
I
Это циклическое соединение, как и можно было ожидать, обладает заметными аро-
,матнческими свойствами, поскольку оно плоское и имеет на периферии Юя-алектронов.
!Т. е. удовлетворяет правилу Хюккеля. Такое представление — очень упрощенное. Более
точные расчеты показывают, что большой выигрыш резонансной энергии обусловлен
взаимодействием электронов атома азота с периферийной электронной системой, что
подтверждается малой основностью этого соединения.
Задача 6. Для каждого из перечисленных ниже соединений установите,
какая ив структур, приведенных в скобках, дает наибольший вклад в
резонансный гибрид данной молекулы. Обоснуйте ваш выбор.
(6Л) CH8CN(CH3— C = N, CH3C=N)
(6.3) СН3-СОСН = С-С2НЕ
(6.4)
'СН3—С^СН
■I
он ■
I
он
>-С1+)
сос2н6, сн3 - с=сн - ссан5<
о-
Ан )
Ответ (о выборе резонансных структур см. W h e I a n d G. W., Advanced Organic
i^TT1?; 3Да ed^XU^'N"Y"'4960' РР* 112-125; HineJ, Physical Organic Chemistry,
2nd ed., McGraw-Hill, N.Y., 1962). .
(6.1) Необходимо рассмотреть локализацию неподеленной электронной павы в двуз
структурах . *
СН3—C = N+ CH3—C = N^
2*
20
ГЛАВА 1
Б каждой из них один атом имеет октет, а другой — секстет, электронов. Хотя
ни одна из этих структур не является удовлетворительной, тем не менее структура с
локализацией отрицательного заряда на более электроотрицательном атоме азота и
положительного заряда на атоме углерода (Б) более приемлема, чем структура А.
{6.2)
= С1+
\=
Б
ci:+
В структуре А атом хлора, несмотря на его положительный заряд, имеет
заполненную внешнюю электронную оболочку (восемь электронов). Поэтому такое строение
энергетически более предпочтительна, чем структура Б, где атом хлора имеет только шесть
злектронов. (Это общее явление; сравните различия между очень стабильными частицами
rSh 2 и высокореакционноспособпыми RO+.)
(6-3) СН3
ОН О
-с=сн-с-
о-
он
сан5
сн3-с=сн-с-с2н5
Б
Ив этих двух структур только Б является резонансной структурой. Соединение А
представляет собой таутомер первоначальной структуры, т. е. химически отличную
частицу, образовавшуюся в результате переноса протона.
(6.4)
А
N
Структура А содержит атом азота с пятью связями, т. е. с десятью электронами.
У элементов второго периода периодической системы отсутствуют дополнительные низко-
лежащие орбитали, которые необходимы для образования такой связи. Поэтому такая
tструктура не вносит вклада в резонансный гибрид. Структура Б, напротив, является
одной из двух возможных структур, которые вносят наибольший вклад в резонансный
гибрид основного состояния алифатических диазосоединений; вторая структура
приведена в задаче*
Задача 7. Напишите резонансные структуры для следующих
соединений:
О
О
(7Л) r JLo-JLn'
(7.2) |Р)
Ответ.
(7.1)
S
0 о
« \
'<- — О— С— R'
(7-3) СН3=СНСОО-
(7.4) CH2 = CHCHR
R—C = 0 — C—R' ■
:ОГ
М~ С— 0=С —R'
ОБЩИЕ ПРОБЛЕМЫ 21
"+
Поскольку сера находится в третьем периоде периодической системы, она может
использовать Зй-орбитали для расширения октета. Поэтому необходимо учитывать допол-
яительные структуры А и Б.
(7.3) сн^сн-с^9-1 ~ сн2^сн—с^ ^снг-сн=с^-:*_
Первые две структуры вносят равный вклад, а третья — менее важна.
(7.4) СН2 = СН —С —R «-* СН2 —CH = CR
н н
Если И=алкил, то первая структура будет более важной, чем вторая, так как элек-
тронодонорный эффект группы R стабилизирует вторичный ион по сравнению с первичным.
Задача 8. Расположите следующие соединения в порядке увеличения
V%a (уменьшения кислотности):
(8.1) CeH5OH, CH3COOH, CH3S02CH2COOH, СНзСНаОН, п-СНзСеЩОН,
(СН3)3ССООН, (СвН5)3СН.
(8.2) Сопряженные кислоты следующих соединений: CH3NH2, NH2NH2, NH20H,
C(iH6NH2t CH3CN, (CH3)2NH, NH3, NH2-C-NH2, H2N-^_ ^-N02, ^ ^N
NH
N02 CN N02 '■
(8.3)
\ J\ СНз\А/СПз У\ СНз\У\/СНз
л/ ч/ л/
II I I I
OH OH OH OH OH
Ответ (Brown H.C.McDaniel D. H.,Hafligei О., в книге
«Determination of Organic Structures by Physical Methods», Vol. 1, В r a u d e E. A., N а с h о d F. C,
eda.T Academic Press, N.Y., 1955, pp. 567—662; Hine J., Physical Organic Chemistry,
2nd ed., McGraw-Hill, N.Y., 1962, pp. 5.8—65; см. также [7]).
22
Г Л А В А 1
Кислотность соединения определяется относительной стабильностью кислотной
и основной форм. Факторы, под действием которых одна из форм стабилизируется в
большей степени, чем другая, вызывают смещение равновесия между ними. В общем, электро-
яоакцепторные заместители имеют тенденцию дестабилизировать кислоты и
стабилизировать сопряженные им основания, тогда как электронодопорные группы обладают
противоположным действием.
(8.1) CH3S02CHaCOOH (ptfe 2,36), СН3СООН (рКа 4,76), (СН3)3ССООН (рКа 5,05),
СвН5ОН (рКа 9,95), п-СНзСвНйОН (рКа 10,19), Ш3СНаОН фКа 15,8), (СдазСН (pKa~25)
Эти соединения можно разделить на три группы. Во-первых, карбоновые кислоты —
наиболее кислые соединения вследствие стабилизации иона основания (карбоксилат-иона)
в результате резонанса, при котором заряд распределяется между двумя атомами
кислорода.
*0 _*н-
\он *^Г \0_ ** -0.
фи- чО~ ^0
Эдектроноакцёпторяый индуктивный эффект сульфонной группы дестабилизирует
кислотную форму и стабилизирует анион. Оба аффекта приводят к увеличению кислотности.
- С другой стороны, электронодонорные алкнльные заместители обладают
противоположным влиянием на равновесие и уменьшают кислотность.
В случае фенолов ароматический цикл не является таким эффективным акцептором
электронов, как карбонильная группа, и, следовательно, не обладает сильным
дестабилизирующим аффектом но отношению к соединенной с ним гидроксильной группе. Далее,
а резонансных структурах фенолят-иона заряд распределяется по атомам углерода,
которые принимают его менее эффективно, чем более электроотрицательный атом кислорода
в ионе карбоксила. Суммарный эффект делает фенольный гидроксил менее кислым, чем
карбоксильный гидроксил. Алкнльные заместители в ароматическом цикле фенола jeme
а большей степени уменьшают кислотность.
Кислотность двух последних соединений определяется главным образом
стабильностью их ионов и показывает, что заряд легче локализуется на атоме кислорода, чем
на большом числе атомов углерода.
{8.2) СН3Сэ=Ш (pKa<0), NOa-^ ~\—№1я(ъК„А-СЬ r.ti t
*-' Wo 1.0), CeH5NH3 (vKa 4,6),
_ «Л ад. H3N0H «. e,0), ^
СЩт «■ ** «Л (Р*. ю.8). NH„fim, ,„„ .л
Ю,8), NHaCNH2 (рКа 14)
Влия "2
""Рядок кисло*™™ —» -*-—- *£^2£kdZZ&%££
-aBaasaraaeitase
ОБЩИЕ ПРОБЛЕМЫ
a
23
жение нитрогруппы, обладающей электроноакцепторным индуктивным эффектом и
дестабилизирующей тем самым положительно заряженный аммониевый ион (кислоту), кроме
того, приводит к стабилизации основания за счет дополнительных резонансных структур,
в которых участвует пара электронов основания.
Основность простых аминов определяется электроноакцепторным (О > N ;> Н)
яли электронодонорньш (С > Н) эффектом заместителей. Это приводит соответственно
к уменьшению или увеличению стабильности положительно заряженной кислоты и,
следовательно, к возрастанию или уменьшению ее кислотности.
Ион гуанидиния особенно стабилен вследствие того, что в этом случае существует
возможность для эквивалентного распределения заряда на трех атомах азота.
HaN = C—NH2 *-»- H2N —C = NH2
(«■■»)
H2N-C-NHj,
II
+rffl2
{VKa 7,16),
NH5
(pKa 7,95),
I
NO,
CH^y^-XHa
(VKa 8,25),
OH
(ptfa 9,95),
(pKat 10,18).
Кислотность замещенных фенолов определяется главным образом влиянием
заместителя на распределение заряда в цикле (формулы 1 —3)
ОН
I
\
О
]|
■-<
2
Введение электроноакцепторной группы в пара-положение увеличивает
перераспределение заряда, стабилизирует ион и увеличивает кислотность. Так как группа N02 —
более сильный акцептор электронов, чем CN, то и влияние нитрогруппы больше. Однако
это в значительной степени зависит от вклада дополнительной резонансной структуры (4).
Если две метнльные группы находятся в орmo-положении к нитрогруппе, то стерические
препятствия уменьшают вклад структуры 4 и кислотность понижается. Однако она все
еще больше, чем для незамещенного фенола, вследствие как остаточного резонансного
взаимодействия, так и прямого индуктивного эффекта нитрогруппы.
Электронодонорные заместители, такие, как метальные группы, вызывают
увеличение электронной плотности в цикле и уменьшение стабильности иона.
и
ГЛАВА
Задача 9. Расположите следующие соединения в порядке уменьшения
основности:
0$> ф q-
Ответ [Wepster В. M.f Rec. trav. chim., 7i, 1171 (1952)].
О
(p/Cft=3,35J
(pKh=6,21)
fp*Q,=8,80J
Фенильная группа является акцептором электронов за счет как сопряжения, так
и индуктивного эффекта. В соединении А проявляется действие только индуктивного
эффекта, поскольку электронная пара атома азота находится в плоскости бензольного
цикла и не может взаимодействовать с л-элсктронной системой. В соединении В фенильная
группа уменьшает электронную плотность па атоме азота за счет как индуктивного
эффекта, так и участия резонансной структуры, например Г.
одти^Гастм^ыми^' КаКИе т првведенных ниже «-единений являются
(МЛ)
Н-
ИО-
соон
он
н
со/ж
А
соон
он
он
соон
Б
(10.2)
ос
I
NH
со
ОС^ "^NH
НГ^СО
н 'сн,
ОБЩИЕ ПРОБЛЕМЫ
25.
(10.3)
н-
но-
но-
соон
-он
-н
-н
соон
А
соон
-он
-он
-он
соон
Б
н-
но-
н-
соон
■он
-н
■он
соон
в
ж.
йг
(10.4)
СпНе
ноос
соон ноос
с,не
ноос
соон
соон
Ответ (И л и е л Э., Стереохимия соединений углерода, изд-во «Мир», М., 1965,
гл. 2 и 3; М i s 1 о w К., Introduction to Stereochemistry, Benjamin, N.Y., 1965; см. также
w eJ? с ?Л ,ь е в А. П., Потапов В. М., Основы стереохимии, Госхпмпздат,
*1--Л., 1964).
Основным условием для того, чтобы соединение могло быть расщеплено на
оптически активные формы, является нсидентичность молекулы ее зеркальному изображению
■или, в более общем виде, ее неспособность превратиться в зеркальное изображение при
каком-либо процессе, который в других условиях энергетически возможен. Работу
по определению взаимопревращаемости очень часто, но не всегда, можно упростить путем
поиска возможных (даже если они неправдоподобны) конформаций, которые имеют
плоскость или центр симметрии. Если такая конформация существует, то она идентична ее
зеркальному изображению и пе может существовать в оптически активной форме. Если
эта конформация имеет относительно высокую энергию, то расщепление соединения
на антиподы возможно; однако в этом случае будет протекать медленная рацемизация.
По мере того как отдельные молекулы будут достигать симметричной формы.
{10.1)
СООН
I
И»— С-«ОН
I
НО^^С—^Н
I
СООН
НООС
СООН
HOi
-с^-н н-^с
.зеркальная i
плоскость Г
-С—«ОН Н^-С-
iOH
iOH
НООС
СООН
НООС
i
НО^—С -^Н
I
НО»-С -—Н
i
НООС
А'
Б'
руктура А не иДентична ее зеркальному изображению А' и не может превра-
трлмтЯ В Н6ГО" Стаура Б идентична Б' (вращение в плоскости бумаги), и, следова-
ельно, зеркальное изображение и исходная структура идентичны.
26
ГЛАВА 1
\10.2)
сн3 ■ сн, н н
ch3 , сн3 сн3 °J
Структура А имеет центр симметрии (отмеченный точкой), и можно показать, что
-она идентична зеркальному изображению А' (путем вращения вокруг горизонтальной оси
а плоскости бумаги). Вследствие цыоконфигурации метильных групп структура Б не имеет
центра симметрии и не может превратиться при каком-либо вращении (или путем
инверсии кресло — кресло) в структуру Б'.
(10.3) Как и в случае {10.1, Б), соединения Б и В имеют плоскость симметрии
а оптически неактивны- Б этих случаях плоскость проходит через центральный атом
углерода; он, таким образом, является псевдосимметрическим. Структура А не имеет
- плоскости симметрии и оптически активна.
(10.4) Эта задача аналогична задаче (10.S). Соединения А и Б имеют плоскость сим-
-метрйи, а соединение Б не имеет.
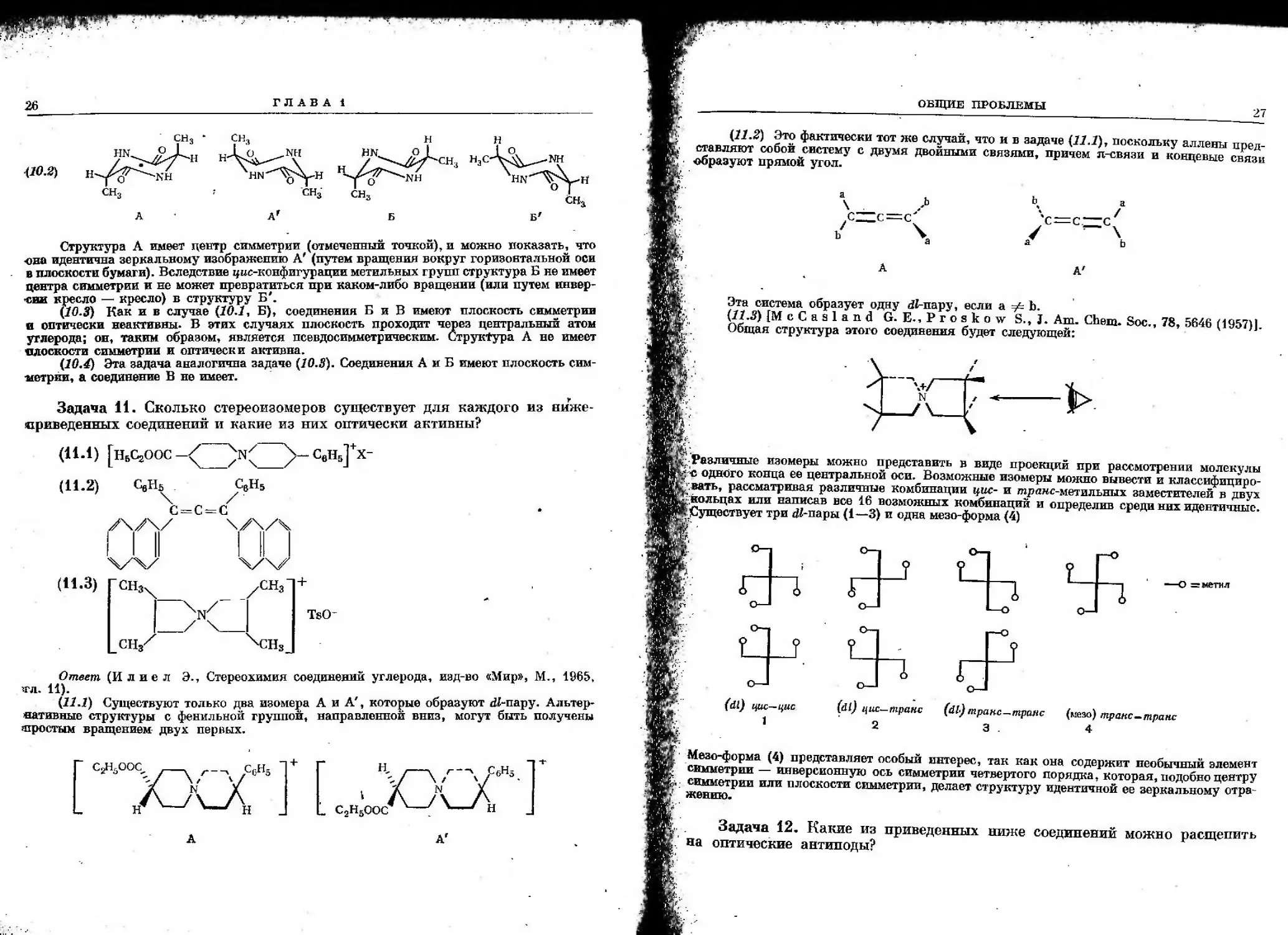
Задача 11. Сколько стереоизомеров существует для каждого из виже-
сриведенных соединений и какие из них оптически активны?
(11-1) [щсзоос-/"
(11.2)
вНБ]+
х-
CflHs . С6Н5
С = С = С
У\/\/ \/\/Ч
;н.З)
Гсн3
\
СНя^
уСН3
^CHS
+
TsC-
Ответ (И л и е л Э., Стереохимия соединений углерода, изд-во «Мир», М., 1965,
1ГЛ. 11).
(11.1) Существуют только два изомера А и А', которые образуют dJ-пару.
Альтернативные структуры с фенильной группой, направленной вниз, могут быть получены
^простым вращением двух первых.
ед/юс, — г_^ с0н5
Л-Л-А
\/—\Г\Р^
{„uCM,
А А'
ОБЩИЕ ПРОБЛЕМЫ
27
(11.2) Это фактически тот же случай, что и в задаче (11.1), поскольку аллены плел
С ЛВТЛКЯ ПНПИТТТЛИТТ (чгаътгп титл.. п_^.п. . _ „ ^f^U
ставляют собой систему
образуют прямой угол.
с двумя двойными связями, причем я-связи и концевые связи
,CZZc=c'
V
/
х:=с:
А'
Эта система образует одну <Й-пару, если а Ф Ь.
(11.3) [McCasland G. E.,Proskow S., J. Am. Chem. Soc, 78, 5646 (1957)].
Общая структура этого соединения будет следующей:
■Различные изомеры можно представить в виде проекций при рассмотрении молекулы
NS одного конца ее центральной оси. Возможные изомеры можно вывести и классифициро-
\вать, рассматривая различные комбинации цис- и m/хшс-метильных заместителей в двух
■кольцах или написав все 16 возможных комбинаций и определив среди них идентичные.
(Существует три $-пары (1—3) и одна мезо-форма (4)
if" -
1 Г
oj
3 I
О—|
.? t
1 i"
(dt)
1
т
(dt) цис-транс (и) транс-транс (ме3о) транс-трапе
2 3 . 4
cl^l™™ ( 2„S f ВЛЯвТ ОС°быИ интеРес> так ка* «на содержит необычный элемент
cSZSS -"н^РСиониую ось симметрии четвертого порядка, которая, подобно центру
ж^никТ плоскости симметрии, делает структуру идентичной ее зеркальному отра
Задача 12. Какие из приведенных ниже соединений можно расщепить
на оптические антиподы?
28
ГЛАВА 1
(12.1)
ноос соон
(12.2) НООС ОСН3
(12.5)
(12.3)
НоС
н„с
ff\
CHg
О V-CH2
соон
(12.6)
(12.7)
С0СН2СН2С00Н
(12.4)
н2соон
соон
—(ch2)jo —
Ответ (И л и е л Э., Стереохимия соединений углерода, шд-во «Мир», М„ 1965).
Ни одно из этих соединений не содержит асимметрического атома углерода,
вследствие чего возможная оптическая активность должна быть обусловлена общей
асимметрией молекулы, которая в этом случае не идентична своему зеркальному изображению.
(12.1) [A d a m a R-, Y u a n "H. С, Chem. Rev., 12, 261 (1933)].
Дифенилы, имеющие объемистые заместители в 2,2 ,6,6 -положениях, не могут
принять пленарную симметричную конфигурацию. Б действительности кольца располо
жены приблизительно в перпендикулярных плоскостях, и, как и в случае алленов,
возможны два оптических изомера. Б некоторых случаях может происходить
самопроизвольная рацемизация, если величина заместителей такова, что может наступить медленно
устанавливающееся равновесие за счет прохождения вращательного барьера
NO.,
0,N
СООН
НООС СООН
НООС NO
(12.2) Этот случай аналогичен вышерассмотренпому, так как свободное вращение
также затруднено; однако поскольку два заместителя с одном кольце идентичны друг другу,
то соединение совпадает^» своим зеркальным изображением и, следовательно, оптически
пе активно.
ОБЩИЕ ПРОБЛЕМЫ
29
(12.3) (L e s s 1 i e M. S., Т и г п е г Б. Е., J. Chem. Soc, 1933, 1588).
Триметиларсониевая группа настолько велика по объему, что она обусловливает
достаточно большой барьер вращения даже при прохождении мимо атома водорода.
Поэтому это соединение можно расщепить на антиподы, хотя оно быстро рацемизуется
в растворе.
(12.4) iLuttringhaus A„ Gralheer Н., Ann., 557, 112 (1945); L u t-
tringhaua A.,Eyring G., Ann., 604, 111 (1957)].
Б этом случае вопрос заключается в том, способно ли бензольное кольцо одной своей
стороной проходить при вращении внутри кольца из метиленовых звеньев или пет;
в последнем случае возможно существование антиподов.
(сн2)п
СООН
СООН
Г X
<сн2)п
НООС—«
и
(сн2)п
Если п = 10, то вращение свободное и соединение не расщепляется; если п = 9, то
соединение расщепляется на антиподы, но они рацемизуются при нагревании. Если п = 8,
рацемизации не происходит.
(12.5) [Cram D. J., A 11 i n g e г N. L., J. Am. Chem. Soc, 77, 6289 (1955)].
Расщепляется. Близкое расположение двух бепзольных колец делает невозможным
вращение, в результате которого один антипод переходит в другой.
(12.6) [Newman М- &., Н u s s е у A. S., I. Am. Chem. Soc, 69, 3023 (1947)].
Две метильные группы в положениях 4 и 5 перекрываются, поэтому они вынуждены
выйти из плоскости, в которой находится эта циклическая система. В результате
существуют две зеркальные формы А и А'. Расщепление возможно, но соединение очень легко
рацемизуется.
нйс
СН2СООН
СН2СООН
Л'
(12.7) [Adams R., Danker t L. J., J. Am. Chem. Soc, 62, 2191 (1940)].
Свободное вращение вокруг центральной связи С — N ограничено стерическими
препятствиями, создаваемыми о/»пго-метильньтми группами и заместителями при азоте;
это соединение можно расщепить па антиподы. Бромирование амида 1 дает дибромид 2,
30 Г Л АВ А 1
который оптически неактивен, так как он симметричен но отношению к вращению вокру г
связи С — N. Если бы оптическая активность была обусловлена асимметрическим атомом
азота, она должна была бы проявляться и в соединении 2.
нзС/ ссн2сн2соон н.с <:сн2снйсоон
няс JL .сн3 н3с
Ег0
Задача 13. Расположите следующие соединения в порядке возрастания
содержания енольной формы:
(13.1) Ацетон. (13.4) Ацетилацетон.
(13.2) Ацетоуксусный эфир. (13.5) Циклопентанон.
(13.3) Диэтиловый эфир мало- (13.6) Диацетил.
новой кислоты. (13.7) Циклопентандион-1,2¥
Ответ (Gould E. S., Mechanism and Structure in Organic Chemistry, Holt, N.W,
4959, pp. 376—380; Хэммонд Г.,в книге «Пространственные эффекты в органической
химии», под ред. М. Ньюмена, ИЛ, М., 4959, стр. 431; Wheland G. W.t Advanced
Organic Chemistry, 3rd ed., Wiley, N.Y., 1960, pp. 681—702).
(13.1) (2,bA0~*%)<(13.5) (4,8.10-s%) ~ (13.6) (5,6 A0^%)< (18.3) (0,1%)<
< (13.2) (7,5%) < (13.4) (80%) < (13.7) (100%).
Содержание енольной формы в соединениях с изолированной карбонильной группой
(как, например, ацетон) очень низкое. Введение карбонильной группы в пяти- или шести-
членный цикл заметно увеличивает, енолизацию (для циклогексанона содержание енольной4
формы составлет 2-10~8%), но она все еще очень мала. Причина этого эффекта сложна;
однако, возможно, он обусловлеп уменьшением отталкивания несвязанных атомов
водорода при образовании второго трйгонального атома в цикле (сравните задачу 3.4 об
относительной стабильности зкзо- и эндоциклических олефинов) [14].
Ациклические а-дикетоны, такие, как диацетил (13,6), обычно имеют трансоидную
конформацию и обладают относительно малой тенденцией к енолизации. В циклических
а-дикетонах диполи двух карбонильных групп расположены более или менее копланарно-
и параллельно. Отталкивание между эффективными зарядами дестабилизирует кето-форму
и сильно увеличивает содержание енола.
$-
[13.^) *~
СНд °+ "С—СН3
О
(1Э.7)
ОБЩИЕ ПРОБЛЕМЫ
31
Наиболее часто енольные формы встречаются у Р-дикарбонильных соединений.
Прибливительно можно считать, что содержание енола возрастает при следующем
изменении карбонильной группы:
О
/ ,
-С—OR'
О
II
R—С—R'
О
Я
R—СН
Так
сн/
СООС2Н5
^СООС2Н5
(13.3)
/
СООСаН5
у
СОСНз
<С СНгх <! СН2ч
хсосн3 нюсн3
(13.2)
(13.4)
I 5.
12,
13,
ЛИТЕГАТУГА
Hermans P. H., Introduction to Theoretical Organic Chemistry, Elsevier,
Amsterdam, 1954. Относительно старая и малоизвестная книга, которая, однако, содержит
много интересного материала по теоретическим основам различных химических
свойств."
Wheland G. W-, Advanced Organic Chemistry, 3rd ed., Wiley, N.Y., 1960.
Особенно следует рекомендовать при изучении теории резонанса и таутомерии.
Илиел Э., Стереохимия соединений углерода, изд-во «Мир», М., 1965. Обзор,
, охватывающий очень обширный материал.
Mislow К., Introduction to Stereochemistry, Benjamin, N.Y., 1965. Очень краткое,
но исчерпывающее руководство по симметрии и связанным с ней проблемам в
органической химии.
Brown Н. С, М с D a n i el D. H., Hafliger О., in «Determination of
Organic Structures by Physical Methods» (E. A. Braude, F. С Nachod, "eds.)t Academic
Press, N, Y., 1955, p. 567—662. Исчерпывающий обвор по константам диссоциации
и их применению.
.Б е к к е р Г., Введение в электронную теорию органических реакций, изд-во «Мир»,
М., 1965.
.Кинг Дж. Ф., в сборнике «Установление структуры органических соединений
физическими и химическими методами», изд-во «Химия», М-, 1967, том 1, стр. 367.
:.М и н к и н В. М-, Осипов' О. А., Жданов Ю. А., Дипольные моменты в
органической химии, изд-во «Химия», Л., 1968, стр. 164—209.
.Зефиров Н. С, С о к о л о в В. И., Усп. хим., 36, 243 (1967).
.Вольпин М. Е., Усп. хим., 29, 298 (1960).
•Р е 11 i t R. et al., J. Am. Chem. Soc., 87, 3253 (1965); Chem. Eng. News, 43, Л» 34,
38 (1965).
Stewart F. H., Chem. Rev., 64, 128 (1964).
Sondheimer F., Wolovsky R., Amiel Y., J. Am. Chem. Soc, 84, 274
(1962).
Turner R. В., Garner R. H., J. Am. Chem. Soc., 80, 1424 (1958).
Поп л Дж., HI ней дер В., Бернстейн Г., Спектроскопия ядерного
магнитного резонанса высокого разрешения, ИЛ, М., 1962.
Дополнения переводчика.
ГЛАВА 2
Задачи по спектроскопии
Задачи, приведенные в настоящей главе, касаются применения
различных физических методов для изучения природы и структуры органических
молекул. Литература, посвященная этому вопросу, довольно обширна:
общие обзоры спектральных методов [1—5], ультрафиолетовая (УФ) и
видимая -спектроскопия [5—7], инфракрасная спектроскопия (ИК) [5, 8, 9],
ядерный магнитный резонанс (ЯМР) [10—14], дисперсия оптического
вращения (ДОВ) и круговой дихроизм (КД) [15—17], масс-спектроскопия [18—21].
Задача 14. Вычислите Я°Й£Н (УФ) Для следующих соединений:
НО |
(ил) /\/\ (14-2) А/ч. <14-3)
/\/\/
О
I
<14'.4) /\
(14.5)
\/-\,
/\/\
(14.6)
Л/
I
О
/\
п
/\
•(14.7)-
/\
/\/\
/\Г
лул/
ч/\ /\/
Ответ (обсуждение и таблицы вкладов заместителей см. S с о 11 A. I., Interpretation
of the Ultraviolet Spectra of Natural Products, Pergamon, London, 1964, Chap. 2; см.
также [5]).
Максимум в УФ-спектре простых алпциклических сопряженных полиенов и енонов
может быть предсказан с неожиданно высокой точностью при использовании значения
максимума системы, лежащей в их основе, и аддитивных констант, отражающих
влияние заместителей.
{14.1) 215 (а, В-непре дельный кстон) +12 (В-заместитель) = 227 им. Найдено:
225 нм [22] и 230 нм [6].
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
33
(14.2) 215 +12 (р-заместитель) + 10 (а-заместитель) + 5 (экзоциклическая
двойная связь по отношению к кольцу циклогексана) = 242 нм. Найдено: 241 и 243 нм [221
(14.3) 215 + 2-12 (две В-группы) + 35 (а-ОН) = 274 нм. Найдено: 268 нм.
(14.4) 253 (гомоаннулярный диен) + 4-5 (2 заместителя и 2 остатка цикла) =
= 273 нм. Найдено: 262 нм.
(14.5) 214 (гетероаннулярный диен) + 4-5 (4 заместителя и остаток цикла) + 5
(одна экзоциклическая двойная связь) = 239 нм. Найдено: 238 нм.
(14.6) 253 (гомоаннулярный диен в центре) + 2-30 (двойные связи, увеличивающие
цепь сопряжения) + 5-5 (заместители) + 3*5 (экзоциклические двойные связи) = 353 нм.
Найдено: 355 нм [6] (для 3-СОСН3-производного; вклад группы 0C0CH3 я» 0).
(14.7). 215 + 30 (сопряженная двойная связь) + 2-18 (<у,б-остатки цикла) +5
(экзоциклическая двойная связь) = 286 нм. Найдено: 290 нм [6].
Задача 15. Объясните, почему замещение группы СН2 в положении X
соединения А вызывает сдвиг максимума поглощения в УФ-спектре в сто-
. рону коротких длин волн.
Ьмакс (95%-ный С2Н50Н), нм
(1) Х=—СН2— 238
I | X (2) X = \N—СН3 232,5
(3) х= >N< CIOj 229
А Х ХСН3
Ответ [Kosowei Б. М., R e m у D. С, Tetrahedron, 5, 281 (1959)].
Наблюдаемое поглощение обусловлено п -*- л.*-переходом в сс,р-непредельном кето-
не [7]. Очень грубо такой переход можно представить как А -»- Б; во всяком случае,
разделение зарядов в возбужденном состоянии больше, чем в основном*
I X _^1 ||Х
I I I
о -о +
А Б
Поэтому вклад такого диполярного соединения в возбужденное состояние
уменьшается в присутствии близкорасположенного положительного заряда, как в
соединении (3), что приводит к возрастанию энергии возбужденного состояния и сдвигу длины
волны электромагнитного колебания, возбуждающего этот переход, в сторону более
коротких длин-волн. Случай соединения (2) менее ясен. Однако, возможно, и здесь имеет
место взаимодействие диполя группы R3N;, положительный конец которого ближе, чем
отрицательный, к заряду, находящемуся в голове моста структуры Б.
Задача 16. Обычно Д*-3-кетостероиды имеют очень интенсивный
максимум в УФ-спектре в области 240—242 нм. В табл. 2-1 приведены величины
сдвигов этого максимума под влиянием заместителя в положении 6.
Объясните наблюдаемые сдвиги.
3—374
34
ГЛ АВ А 2
oJ^-^Y^
X
F
.С1
Вг
I
СДВИГ, НМ
«-заместитель
От —4 до —5
От —4 до —5
От —3 до —4
^-заместитель
От —5 ДО —8
От 0 до —1
От +6 до +8
От +11 до +13
Ответ [Ringold Н. J., Bowers A., Experientia, 17, 65 (1961)].
Высокоинтенсивпая полоса поглощения а,р-непредельных кетонов обусловлена
п~*> я* (V -*- г^-переходом, который грубо можно представить как
Энергия, необходимая для такого возбуждения, зависит от электронодонорного или
электроноакцепторного влияния заместителя па диполь в структуре А.
Для атомов галогенов в положении 6 первым эффектом, которого можно ожидать,
является индуктивный эффект: электроотрицательный заместитель в положении 6
приводят к возникновению частичного положительного заряда на Св, который
дестабилизирует структуру А. В результате этого наблюдается сдвиг в коротковолновую область
(гипсохромный сдвиг).
hv
-о^Л^У
Этот эффект проявляется для фтора в 6сс-п 66-положениях и для хлора и брома в 6а-поло-
жении. Очевидно, для С1, Вг и I в 6р-положении имеется другой тип взаимодействия,
так как сдвиг наблюдается в сторону длинных волн (батохромный).
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
35
Это приводит к выводу, что в этих случаях атом галогена вследствие
электронодонорного эффекта может стабилизировать положительный конец диполя в структуре А.
Это может быть осуществлено за счет мостиковой промежуточной структуры,
аналогичной той, которая была предложена для галогенирования олефинов [23] (заметим, однако,
hv
что в соответствии с принципом Франка — Кондона [7] атом галогепа не успевает
переместиться за время возбуждения; поэтому геометрия совершенно различная). Как и
следовало ожидать, величина этого эффекта возрастает с увеличением размера атома
галогена, его поляризуемости и его электроподонорной способности.
Различие между 6- и а-ориентациями связано с геометрией системы, В первом
случае возможно перекрывание орбиталей атома галогена и я-системы; однако для а-ориента-
ции это невозможно (сравните с преимущественным диаксиальным присоединением
галогенов к циклическим системам).
Аналогичное различие во влиянии атомов галогена, занимающих аксиальное
и экваториальное положения, наблюдается в случае п -»- л*-перехода как насыщенныхt
так и ненасыщенных кетонов [6].
Задача 17. Объясните, почему соединение, изображенное ниже и
имеющее г/шжонфигурацию (сочленения циклов), растворяется в сильных кисло-
'■ тах с образованием темно-желтого раствора (л.макс в 60%-ной H2S04
386, 427 нм), в то время как mjoflKC-изомер (сочленение циклов) не имеет
.сильных полос поглощения выше 300 нм в 60% -ной H2S04.
Ответ [Leal G.* P e 11 i t R., J. Am. Chem. Soc, 81, 3160 (1959)].
При растворении в сильных кислотах цие-соединение образует нсклассический
кдрбониевый ион (А), обладающий характеристичным УФ-спектром. Для
транс-соединения образование такого неклассического иопа невозможно. Так как рассредоточение
3*
36
ГЛАВА 2
заряда за счет некласспческого взаимодействия можно рассматривать как
стабилизирующий эффект, то цис-изомер образует карбониевый иоп в таких условиях, которые являются
слишком мягкими для образования карбоппевого иоиа из т/ш»с-изомера.
Задача 18. Каким образом с помощью ИК-спектроскопии можно
отличить одно соединение от другого в каждой из приведенных ниже пар?
(18.1)
(18.2)
(18.3)
(18.4)
\/
II
О
А
СгН5
Н
/\
II
О
(18.5)
О
I
/
О а
О
О СООС2Н5
/СНз
хн
^-с/1
соос2н5
А
п
н
СНз
У
/
л
сн.
Б
Вг
(18.6)
/\
/
Вг
'\/ \/
О Q
СН^
А
он о
! В
I II °
V—
А
о о
он
о
он
А
(18.7)
о
и
х\/\
он
Б
О
\х и
7\/\
Л/ N
о
Б
Б
Ответ.
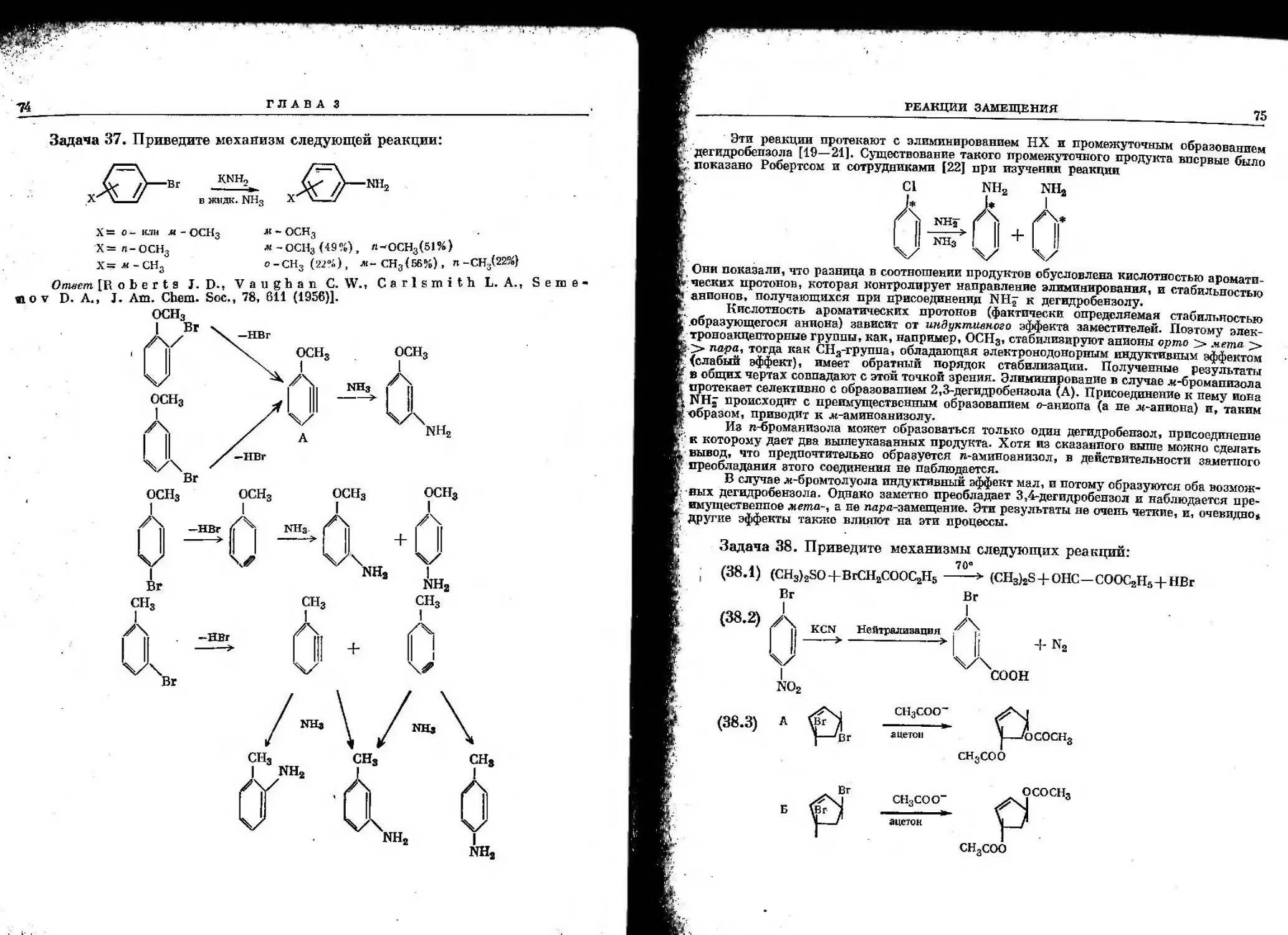
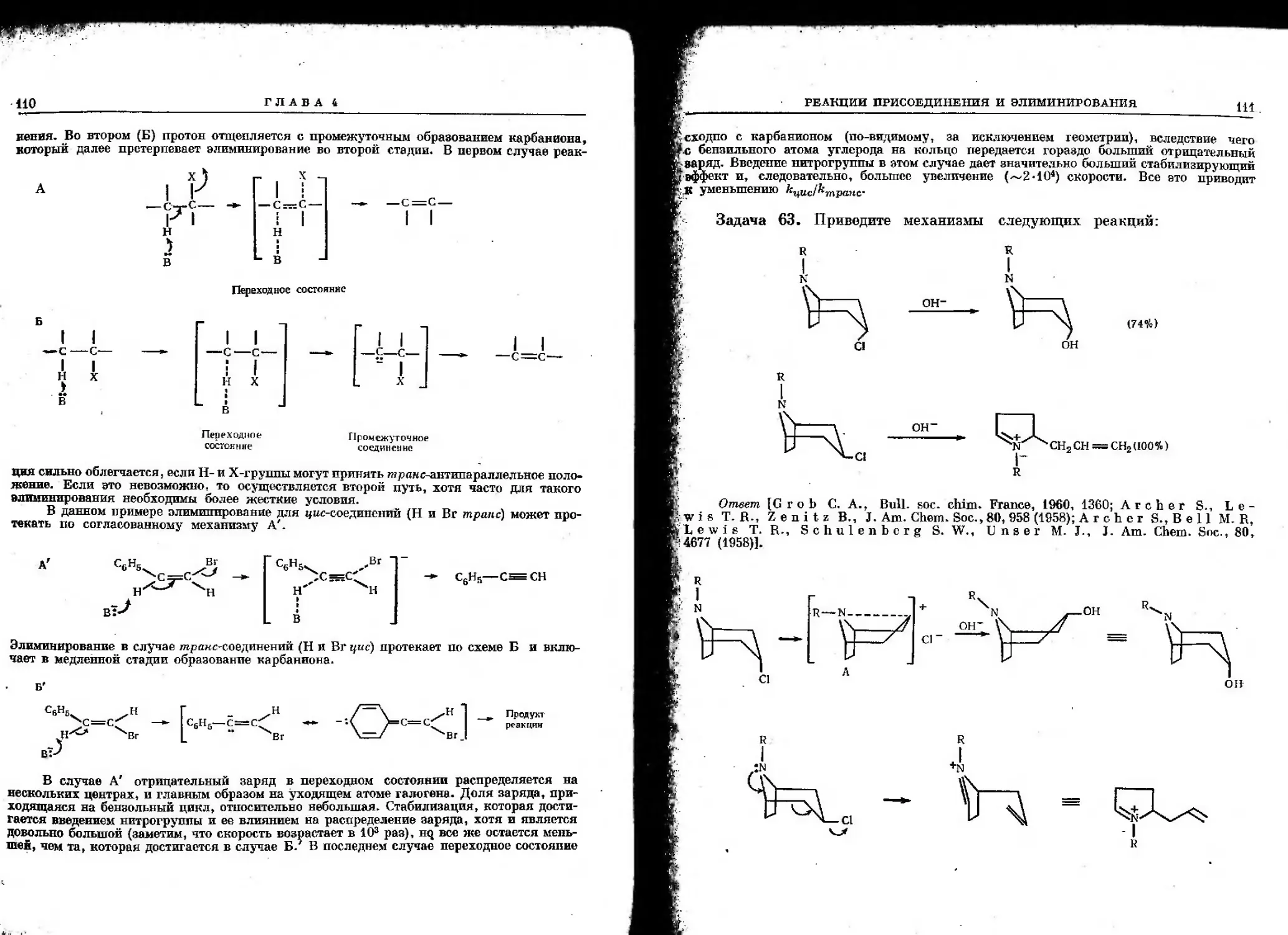
(18.1) (Наканиси К., Инфракрасные спектры и строение органических
соединений, изд-во «Мир», М„ 1965, стр. 51; Беллами Л., Инфракрасные спектры
сложных молекул, ИЛ, М-, 1963).
Частота поглощения циклопентанонов выше, чем циклогексанов:
vc=o: (А) 1745 елт*; (Б) 1715 см~1.
(18.2) (Наканиси К-, Инфракрасные спектры и строение органических
соединений, изд-во «Мир», М., 1965, стр. 30—31; Беллами Л., Инфракрасные спектры
сложных молекул, ИЛ. М-, 1963).
Имеются достаточно характеристические полосы поглощения С — Н-связеи для
цис- и трянс-дизамещенных алкенов.
(,„_„ (колебания в плоскости) *=С-Н Свнеплосностные колебания)
"С=С
(А) 1660 (ср) 1415 (ел)
(Б) 1675 (ел)
(с—сильная, ср—средняя, ел—слабая)
730—675 (ср)
965(c)
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
37
Валентные колебания рС = C<f обычно слабее в траис-олефине по сравнению с цис-та-
мером; однако интенсивность полосы в большей степени зависит от симметрии замещения
двойной связи, так что чем симметричнее система, тем слабее полоса поглощения. Так,
например, в ИК-спектре НгС = СН2 отсутствует полоса поглощения Vc=c B то время
как СН2 = СН — ОСН3 имеет очень сильное поглощение.
(18.3) [Беллами Л., Инфракрасные спектры сложных молекул, ИЛ, М., 1963;
Jones R.N"., Angell С. L., I to Т., Smith R. J. D-, Can. J. Chem., 37,
2007 (1959)].
Полоса поглощения группы С=Ов насыщенных -у-лактонах расположена около
1770 см'1. Если присутствует а,р-двойная связь (Б), то электронпая плотность
карбонильной группы уменьшается, т. е. уменьшается ее двоесвяванность. Полоса поглощения
смещается до ~1750 см~1.
1770 см-1
^о^^о хсг о: о о.
1780 - 1800 САГ1
1750 — 1760 елг1
С другой стороны, сложные эфиры енолов и фенолов (как и р\у-ненасыщенный лак-
тон А) имеют полосу поглощения группы С =0, сдвинутую в сторону более высоких
частот вследствие взаимодействия неподе ленной пары электронов атома кислорода с
двойной связью, что уменьшает его взаимодействие с карбонильной группой.
Интенсивность валентных колебаний С = С в соединении А больше, чем для Б.
На практике очень интенсивные полосы для а,р-ненасыщенных у~лактонов часто
находятся при ~1785 см'1. '
(18.4) (Duncanson L. A., Grove J- F., Zealley J., J- Chem. Soc,
1953, 1331).
Водородная,связь в соединении А сдвигает полосу поглощения карбонила в
сторону меньших частот за счет возрастания вклада структур с разделением: заряда. Так,
vc2o3; (А) 1738 сл_1: (Б) 1760 см-1-
(18-5) [Беллами Л., Инфракрасные спектры молекул
nard N- J., Gutowsky U.S., Middle ton W. J.,
J. Am. Chem. Soc, 74, 4070 (1952))
ИЛ, М., 1963; Leo
Petersen Б- М.
тения
В то время как соединение А является нормальным кетоэфиром и имеет полосы погло-
н как 1736 см'1 (сложноэфирная группа), так и 1718 см'1 (карбонильная группа),
38
ГЛАВА 2
соединение Б представляет собой смесь кетоэфира (1744 и 1718 см'1) и енольного эфира В,
так что можно наблюдать четыре полосы
1656 ел"
ОС2Н5
1618 сиг'
В
{18.6) IN i с к о п A., J. Am. Chem. Soc, 79, 243 (1957)].
В соединении А возможна слабая водородная связь между Вг и ОН. Она вызывает
небольшое смещение vo-н в сторону меньших частот и vc_o B область больших частот.
v0-h> cm~l vc_o, см-
3586 1078
3615 1051 (модель—циклогексанол)
(18.7) (Н е п Ь е s t Н. В., Woods G., J. Chem. Soc, 1952, 1150; В г а и -
d е Е- А., Т е ш m о п s С. J., J. Chem. Soc, 1955, 3766).
В соединении Б стерическое взаимодействие метилкетонной группы и метильных
заместителей цикла имеет тенденцию вывести ее из плоскости двойной связи. Это
приводит к уменьшению сопряжения и сдвигает vc=o в сторону высоких частот.
А 1689 (модельное соединение)
Б 1693
Задача 19. Объясните, почему для каждой пары изомеров полоса погло
щения карбонильной группы различная.
(19.1)
о
II
сн,со
Ф
он
Б, 1744 слг!
НО
но
о^ осн.
О' ОСНз
А, 1709 «Г'
Б, 1723 см-1
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
39
(19.3)
о
II
СН3ОС,
о^ ^осн3
1733 сл-1
' 1718 см-1
и
Б, 1658 см*1
Ответ. Положение полосы поглощения валентных колебаний карбонильной группы
сложных эфиров отражает характер ее двоесвязанности. Таким образом, ее положение
зависит от соотношения трех резонансных структур: 1, 2 и 3.
о :о:
-с—о— -*-*- ■—с — о—
+
:о:
^/'Структура 2 по сравнению с остальными менее благоприятна из-за электроноакцепторного
индуктивного эффекта эфирного атома кислорода. Если по соседству с карбонильной
группой находятся группы, способные подавать электроны, то стабильность структуры 2 уве-
■[■дпчивается. Водородная связь с карбонильной группой стабилизирует отрицательный
£$здряд атома кислорода и, таким образом, благоприятствует структурам 2 и 3. Водород-
ц£;.'ная связь с эфирным кислородом или сопряжение его неподеленной электронной пары
т.с другой системой (как, например, в сложных эфирах енолов) уменьшает вклад струк-
^ТУРЫ 3.
С увеличением вклада структур 2 и 3 уменьшается степень двоесвязанности карбо-
.йнльной группы и частота полосы поглощения понижается. Наоборот, заместители,
|&веблагоприятные для структур 2 и 3, вызывают увеличение частоты поглощения.
{19.1) (Henbest Н. В., L о v е 11 В. Г, J. Chem. Soc, 1957, 1965).
Соединение А имеет приблизительно нормальное значение поглощения карбониль-
Л\ вой группы при 1731 см'1; в соединении Б существует водородная связь
нх
^с'
:0:_
wiV
Она уменьшает вклад структуры 3» в которой электроны оттянуты от карбонильной
группы, вследствие чего наблюдается сдвиг в сторону более высоких частот.
{19.2) (Cole A. R. Н., М и 11 е г G. Т. A., J. Chem. Soc, 1959, 1224).
Этот пример также иллюстрирует влияние водородной связи, которая, однако,
образована карбонильным, а не эфирным кислородом ацетоксигруппы.
40
ГЛАВА 2
Водородная связь с карбонильной группой, стабилизирующая структуры 2 и 3
и понижающая частоту колебаний связи С =0, возможна в соединении А и отсутствует
в соединении Б из-за mpawc-диаксиального расположения заместителей.
(J9.S) [Wenkert Е., Jackson В. G.T J. Am. Chem. Soc, 81, 5601 (1959)].
Различие между этими двумя соединениями обусловлено тем, что они существуют
в различных таутомерных формах: соединение А представляет собой В-кетоэфир, а
соединение Б — енол.
1718 ел"1
/
осн,
о^ о
1733 слН |
СН
Полоса поглощения эфирной группы в соединении Б сильно сдвинута в область меньших
частот как из-за сопряжения, так и из-за водородной связи. Полоса поглощения кетоп-
ной группы, естественно, отсутствует.
Это наводит на мысль, что енольная форма соединения А, которую также можно
было бы ожидать, дестабилизирована стерическими препятствиями, возникающими при
взаимодействии 6<х-Н (экваториальный) и метоксигруппы (А').
сн3
А'
Задача 20. Установите строение продуктов ацилоиновой конденсации
от Ai до А4 (табл. 2-2). %
СООСаН5 СООС2Н5 Ацилоиновая конденсация
-* А
(СНа)
3\/
N
(СН2)3
R
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
41
Таблица 2-2
Соединение
Ai
А2
Аз
А4
R
СН3
с2н5
Ш(СНз)я
С(СПа)з
ИК, CAt-1
СС14
1661,3410
1671,3428
1691,3465
1698,3480
Перхлорат
в иуЗоле
3440
3425
3435
1706,3335
рк'а
66%-ный
ДМФА
10,6
10,2
6,9
6,4
ПаО
9,2
9,2
8,1
7,6
Ответ [Leonard N. J., О k i M., J. Am. Chem. Soc, 76, 3463 (1954)].
Общая структура продукта А следующая:
НО
но
ч
Gb
W
1
В соединениях такого типа имеется трансаннулярное взаимодействие между неподеленной
парой электронов атома азота и группой С = О [24], как это изображено в структуре Б.
Можно ожидать, что увеличение размера группы R будет вызывать
неблагоприятные стерические взаимодействия между этой группой и группировкой, находящейся через
цикл («^-напряжение») [25], что приведет к уменьшению N-*-- СО-взаимодействия.
Увеличение частоты поглощения группы С — О в ряду CHg -> mpem-C4H9
находится в согласии с увеличением характера двоесвязанности группы С = О, т. е. с
уменьшением трансаннулярного электронодонорного эффекта. В более поздних работах [26] было
показано, что можно разрешить две полосы, соответствующие двум типам молекул А и Б,
причем доля Б уменьшается с увеличением объема R.
В отличие от трансаннулярного взаимодействия в нейтральных соединениях твердые
перхлораты соединений Aj, А2 и А3 не показывают вообще поглощения карбонильной груи-
- пы и, следовательно, им должна быть приписана бициклическая структура В. С другой
стороны, А4-НС104 имеет полосу поглощения С = О, и для него характерна структура Г,
по крайней мере частично.
Уменьшение "значений рЯа при переходе от органического растворителя (66%-ный
ДИметилформамид) к воде характерно для соединений, в которых протон уходит от
группы —ОН (енолы, карбоновые кислоты) [27, 28], тогда как обратный случай наблюдается
+ 1
в соединениях, где протон уходит из фрагмента — NH [29]. Таким образом, в растворе
перхлораты А2 и Ai существуют в форме В, тогда как перхлораты А3 и А4 — в форме Г.
Это подтверждается наблюдением, что в D20 соли А3 и А4 имеют полосу поглощения
карбонильной группы, а соли Aj и А2 не имеют [26].
42
ГЛАВА 2
Задача 21,
(21.1) Установите структуры следующих продуктов:
/\
онс N сно
+ сн2:
ХООН пиридин, пиперидин,
N
\
соон
кипячение
ИК: 2660, 1674, 1632, 981, 811 смт* (нуйат)
i. HB/Pt в CHsCOOH
2. 88%-ная НСООН,
СНдО, нагревание
20 час
3. СгНьОН, бензол,
Н+, кипячение,
4. третп-С4Н9ОК, удаление Н20
ксилол, нагревание -^
C/iHioNO < С1бН29Гч04
Wl"19 5. Водн. Н30+, ie s» 4
3 нагревание *
ИК: 1664 см-1 (СС14)
УФ: 221 нм (6000)
(21.2) Установите конформацию соединения 3 (задача 21.1), учитывая
следующие значения дшгольных моментов:
Соединение 3 4,87
Циклогексанон 3,07
1-Метилпиперидин 0,8
1-Этилшшеридон-4 2,95
\
Ответ (Leonard N. J., Morrow D- F., Rogers M. Т., J- Am. Chem. Soc.,
79, 5476 (4957)].
(21
nur^iu^4 run
ОНСГ TST ^CHO
(0
HOOC^H H-^COOH
XX a XX 1
.но ост н ^соон Hoocr снй 4:oohJ
J(3)
(s)
OOCjHr,
(4)
cJheooc" ch3 ^cooc,hs
2
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
43
0
1 ИК: 2660, 1674: сс,6-непредельная кислота, —ОН и —С— [9J
1632: —С = С— [9]
Нч /
981: ;гС = С<Г тракс-алкеп, внеплоскостные колебания СН [9]
/ ХН
811: три атома водорода, соединенные с ароматическим циклом [9]
i -+■ 2 Стадия (2) представляет собой метилирование по Лейкарту [30].
3 Исключительно низкая частота поглощения карбонильной группы обусловлена
взаимодействием этой группы через цикл (трансаннулярное взаимодействие) с непо-
деленной парой электронов атома ааота. Поглощение в УФ-спектре, довольно
. неожиданное для насыщенного соединения, обусловлено, по-видимому, той же
причиной [31].
(21.2) Дипольные моменты циклогексанона и 1-метилпиперидина ориентированы
[['следующим образом:
9
^=^ !Л^
V Простейшее рассмотрение для дипольного момента 1-этилпиперидопа-4 будет
следующим:
С2Щ—N
1
7
Резул ьтир у ющий
момент
Это приводит к утверждению, что результирующий момепт должен Сыть меньше,
,чем для циклогексанона (для формы кресла вычислено 2,7 D).
Однако дипольныи момент соединения 3 больше 3,9 D — максимально возможного
значения, если бы дипольные моменты групп были независимы и расположены в одну
. линию. Поэтому наблюдаемый дипольныи момент может получиться, если имеет место
£ взаимодействие функциональных групп, которое вызывает еще большее разделение
зарядов. Вследствие этого преимущественной конформацией является следующая:
о-
находящаяся в согласии с ИК-спектром. Дипольныи момент соединения 3 предполагает
приблизительно 10%-ное разделение зарядов, хотя трудно интерпретировать это
взаимодействие при помощи теории резонанса, принимая определенный вклад структуры Б
в резонансный гибрид.
44
ГЛАВА 2
Задача 22. Установите структуры приведенных ниже соединений.
F
Н
Конц. H2S04 (СйН5)3М
С10НеС12 < C10H6F2C13 <
\.
СбН5
/
F Конц. H2S04
— —-?
С1
С1
cioh6ci2o
1
ИК: 1786 ел-1 УФ: 262 нм (21000)
УФ: 298 нм (25 500)
291 нм (25 300)
CioIIeF2Cla
УФ:~254кл (17 000)
п
\ /
N
Н
0
и
И К: 1786 см-1
УФ: 283 ил»
(24 700)
C10HeF20
10
HK:~1785cj№-i
УФ: 261 нм
(15 000)
н3о+
C20HaeN2F2 C1SH16NG1F2 <-
9
УФ: 309 нм (И 000)
Разб. Н3о+
^
2
Конц,
ОН-
Соль
iH2so4
(C15H17NOC1)2S04
3
ИК: 1786 см-i
УФ: 284 к* {> 15 300)
i. Нейтрализация
I 2. НаО
CI5H18N02C1
4
Раств. разб. Н+, ОН"
, C15H17N02
6
Раств. разб. Нч
Pd-c
ЦН5СН— СНаС-ОС2Н«
I I!
сно о
C|5H2iN02
5
Раств. разб. Н+, ОН'
/\
N
Н
2. На/Рб-С
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
45
Ответ [Jenny E. F., Druey J., J. Am. Chem. Soc, 82, 311 (I960)].
ci^^ ^o cu
QH,
-ci с6н.
/
CrH.
10
CEH.
•ci c6ns
-F
-CI
CI
CM
k"N\ Lf k^NN Lp -±k^
N*^4! CeH.
CI CM
GnS
CI
c"s ci
1
M
Щ'
о
ад
соон
CI
Y°\= »
ki
^—f
L W vci
з
CO OH
so.
Sf
Аномально высокая частота валентных колебаний группы С = О в соединении 1
подтверждает структуру цикл о бутанона ([9J, стр. 51). Превращспие А -»- 1 является
характерным для г<м1-дифторидов такого строения.
Реакция А -*■ 2 представляет собой SN 2'-замещение, приводящее к стабильному
винилгалогениду [32, 33J. Другие возможные структуры, такие, как
CI
>^
ОД»'
СкНт
л*-
46 Г Л А Е А 2
можно исключить, так как в первой присутствует очень реакционносиособный атом хлора,
а во второй должно наблюдаться взаимодействие неподеленной пары электронов атома
азота с системой стирола, что должно было бы приводить к заметному сдвигу максимума
в УФ-спектре в длинноволновую область (см. 9) [7].
Раскрытие цикла при переходе 3->4 протекает через промежуточное образований
кетена, изомерного нейтральной форме соединения 3.
.lb
V
<Г
Q.H-/
-ч
CI
zsn5^ Чл
<
ci
Н0О
Восстановление 6 -*■ 5* вероятно, связано с гядрогенолизом сложного аллилового-
эфира, протекающим либо перед гидрированием двойной срязи, либо одновременно с ним
Реакция А -*■ 7 представляет собой изомеризацию, которая наблюдается в
подобных системах в присутствии галогенид-ионов [32J. В данном случае она протекает по схеме
СеНг-
—CJ
С1
(с2н5)лй
(C2H5)3N,
С6Н5-
-F
XI
' а,
с6н5-
+ (C2H5)3N
XI
+ CI
Изменения в УФ-спектрах соединений А и 7, а также соединений 1 и 8 отражают эффект
замещения протона галогеном в хромофорной группировке.
Превращение 7 -»- 9 включает, очевидно, две последовательно протекающие
реакции SN2\ Приведенная структура подтверждается УФ-спектром, в котором имеется
сдвиг максимума поглощения, ожидаемый для структуры В-аминостирола [сравните с ЯмакС:
305 нм (14 700) для СвН5СН = СН ~ NCsH,0f. Альтернативная структура
скн
У
-F
-NC5H10
NC5H10
должна иметь УФ-спектр, аналогичный спектру соединения А.
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
47
Следует обратить также внимание на различие в УФ-спектрах соединений 1 и 3
имеющих последовательную цепь сопряжения, и соединения 10 с перекрестным сопряже-
L Задача 23. Установите структуры продуктов (1 — 6) следующих
превращений: *
NaOH Нагревание
Ci4Hi4Oe > С13Н,в05 > С12Н1в03
1 2 3-
(1. NaOHc
последующей
S (дегидрогенизация)
нейтрализацией
*$■ _ у_ ST V 2. CHaN2
§у C16H18N6 Ангидрид 2-окси- CI3Hi803
»';■ 6 нафталиндикар- ^
боновой-1,8 кислоты
Ha/Pd-C
п„ „ ■ Zn-Hg
9-Метил-цис-декалинкарбоповая-1 кислота < С13Н30О3
_ HCl
8 5
Соедкне- „„а
ние VKa
1
2
3
4
5
6
4,81
7,55
Дикарбоновая
кислота
Аманс нм
325(4,3)
[350 (4,3)J б
240(4,1)
[253 (4,0)] б
Нет сильного
поглощения
242(4,1)
Нет сильного
поглощения
325(4,3)
а В 80%-нога метилцеллоэольве.
В 0,01 н. едком натре.
в В хлороформе. -
ИК (нуйол), с.н-1
1748, 1709, 1699, проба
FcCIg положительная
1626, 1587
1761, 1715
1724«, 1680 В
1724, 1709
1742, 1724, 1692
с
48
ГЛАВА 2
Ответ [Gautochi F., Jeger 0., Pre log V., Woodward R. В.,
Helv. Chim. Acta, 37, 2280 (1954)1-
1798
&
1715
COOH
1-761
OH
\
CH3OOC
о осн3
[680
242 нм
сн3о.
HOOCi
CH3oc
Реакция 1 ->- 7 подтверждает общий скелет соединения 1, но не дает возможности
установить положение отщепившихся метальной и карбоксильной групп.
Превращение 5 ->■ 8, очевидно, является восстановлением карбонильной группы
по Клемменсену [34]. Эту кетонную группу разумно сопоставить с гидроксильной группой
соединения 7_ и, таким образом, установить ее положение в соединениях 2, 3, 6 или 7.
Изменение в УФ-спектре при переходе от соединения 4 к 5, так же как и сдвиг полосы
С = О в ИК-спектре (1680 ~> 1709), указывает, что соединение 4 представляет собой а, р-
ненасыщенный кетон ([9], стр. 51). Эта непасыщенность возникает при переходе 3 -»- 4
за счет р-элиминирования (сравните дегидратацию альдолсй основаниями) некоторых
групп [35]. Полоса при 1761 см"1 для соединения 3 подтверждает наличие группировки
V-лактона [9], которая и элиминируется. Отсюда наиболее вероятны следующие две
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
структуры:
49
ГТогда для соединения 4 структура А даст структуру А', а структура Б - Б'.
СНдООС
УФ вычислено:
215+2(12)+5=244к.«
СН3ООС
УФ вычислено:
215 + J2 = 227 «л
^Структура А' для соединения 4 дает гораздо лучшее согласие между вычисленным [36]
1ж.наблюдаемым УФ-снектром и, следовательно, является предпочтительной. Отсюда ясно,
?что карбоксильная группа соединения 4 не может быть ни одной из тех, которые имеются
^В соединении 7; поэтому скелет соединения 1 можно представить структурой В
Одна двойная связь
*^ZZ°лновый„ максимум в УФ-спектре, положительная проба с FeCl и титнование
^Гт2^Ти^яп^ГНаЯ «"««ЧИЧНиация ИК-снектра приведена в работе [9]
коу^пТожаыып^Т™™ опРеД^ить положение СООН-группы, и вполне №
vS^'^S^^f^^^ Пе^ПОЛ°ЖенИИ- °Д™ «РУ«Я* 1, приведенная
^m^^SS^^^^^J^^'^^ B результате гидролиза и декарбоксилиро-
ия ооразовавшеися р-кетокислоты, вытекает из сходства УФ-спектров соединений 2 и 4.
4—374
50
ГЛАВА 2
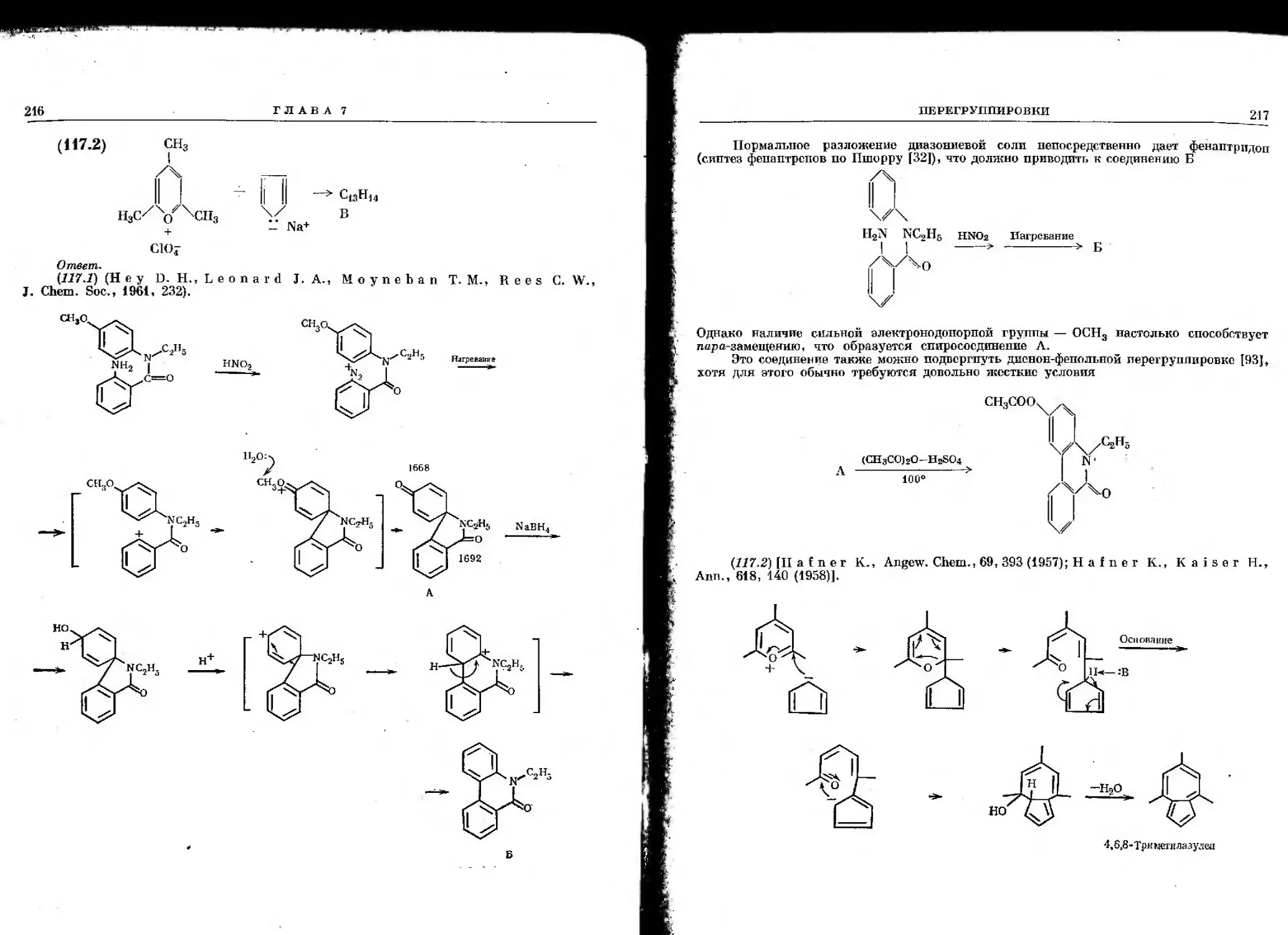
Задача 24. Установите структуры продуктов следующей реакции:
О.
НС — СдНд
II
ч/\/\
СН2С6Н5
о.
Pd/C(Ha)
0/\/\/
II
НС — СдНб
С24Н2403
Желтые
кристаллы
300е
ИК-спектр: ",
А (пуйол)
Б (нуйол)
В (СНС13)
Ответ [Leonard
НС
-СвНд
#э
*№
•> С24Н2в02 + С24Н2в02
А Б
Оранжевые Бесцветные
кристаллы кристаллы,
проба с
FeCla
положительная
+
\/\
О'
СПдСбНб
Желтые
кристаллы
LlAlHilJOa
Ca^Hae^a
В
1738 (плечо при 1755), 1605, 1400, 760, 699 смП
3300, 1697, 1657, 1605, 1587, 1502, 758, 700 елг*
3510, 1745, 1606, 1500, 703 см^
N. J-, L i 111 с J. С, J. Am. Chem. Soc, 80, 411 (1958)].
3310
HO
о
1745
C6H3
CKH,
Присутствие полос около 760 и 700 см~г в ИК-спектрах соединений А и Б
подтверждает наличие по крайней мере одной монозамещепной фенильной группы; однако это
не исключает возможности того, что другой цикл будет орто-дизамещенным [9].
Окраска соединения А указывает, что хромофорная группа в нем обусловливает
углубление окраски по сравнению с исходным соединением, песмотря на присоединение
водорода. Поэтому можно предположить, что неенолизующаяся система а-дикетона,
имеющаяся в исходном соединении, сохраняется, но карбонильные группы становятся
более копланарными и вследствие лучшего сопряжения окраска становится более
глубокой. Поскольку для многих а-дикетонов с параллельными карбонильными группами
(как, например, в пяти- и шестичленных циклах) наблюдается енолизация, то в данном
случае она должна подавляться. Это обусловлено либо отсутствием атомов водорода
в а-положении, либо наличием мостиковой бициклической системы, где енолизация
с образованием двойной связи в голове моста запрещена (правило Бредта [38]). Довольно
трудно представить себе структуру, не имеющую а-атомов водорода; однако
структура А удовлетворяет второму условию.
Альтернативную структуру Г па основании приведенных данных исключить нельзя.
ИК-спектр, хотя и подтверждает наличие сс-дикетопной группировки [9], также не повво-
ляет сделать выбора между этими структурами. В структуре Г мостик такого размера,
что енолизация представляется возможпой, хотя, вероятно, она отсутствует из-за
напряжения, все еще значительного.
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
51
Свойства соединения Б совпадают с ожидаемыми для енолизоватгой дикетонппй
^^™ССТаН°ВЛеНИе ЭТ0Г° ве1Ц-сства Д° соединения В, имеющего в ИК^ЖЕХое
же поглощение, как у насыщенного циклонентанона, позволяет сделать npemolJ™
о структурах соединений Б и В. *w*um© предположения
„ В°ССТановление енолизованных а-дикетонов алюмогидридом лития до кето-т™
в^идТсол^еноГа6010 Ш * °^Л« *»• ™ »Р*»ЙЛя ™ &££££
ОН
\У
О
LiAlH4
\/
о- м+
о
о- м+
,0- м+
ы
н+
L\X
\/
он
!ч/он
\/
о
/\
\/
/
он
СН,С6Н,
^ Задача 25. 2'-Дезоксиуридин (1) имеет спектр ЯМР (60 Мгц),
приведенный ниже. Какому протону этого нуклеозида соответствует каждый сигнал?
j=6,6
А
f
-79
Б В
I I
16 39
Г Д Е
I I I I
НО 120 145 165
Ж
I
250 (гц},
РполГот ТМС °; ббН30Л В КаЧеСТВе ВНеШНеГ0 ста«Д»Рта, -436 гц в слабой
4*
52
ГЛАВА 2
Ответ [J а г d e t z к у CD., J. Am. Chem. Soc, 83, 2919 (1961)].
Сигналы А и В следует приписать протонам при двойной связи урацила. Из
величин констант спин-спинового расщепления очевидно, что протон, которому отвечает
сигнал Жт находится в вицинальном положении к протонам, которым соответствуют
сигналы Б и Г. Поэтому отнесение сигпалов будет следующим:
А
Б
В
Г
4
Г
5
3'
Д
Е
Ж
4'
5'
2'
Задача 26- Установите структуры соединений А и Б.
СН,
СН3ч il u
чС = СНССЦз —>
/
СНз>
\1 V
^СНз + С12Н3оО + С^НгоО
сн/л
А
I
,с=о
ЯМР-спектры [в СС14? шкала т (м. д.)]
Б
А 9,11(ЗН) синглет
9,00(ЗН) синглет
8,80(ЗН) синглет
8,30(ЗН) синглет*
8,28(ЗН) синглет*
8Д9(ЗН) синглет*
5,71(Ш) синглет*
4,91(1Н) синглет*
9,12(6Н) синглет
9,04(6Н) синглет
8,08(ЗН) синглет*
7,85(2Н) синглет*
7,80(ЗН) синглет
*) Синглет имеет тонкое расщедишие
Ответ (Bacon N., В г е w i s S., Usher G. E., W a i g h t E- S-, J. Chem.
Soc, 1961, 2255).
H 4,91
7,fc5
9,04
CH, 8,19
CH3 8,08
0/ CH3 7,80
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
53
Отнесение сигпалов на приведенной выше схеме такое же, как и у авторов работы*
однако для некоторых групп сигналов нельзя сделать однозначного отнесения. В
частности, для группы
сн/
н
с-
сигналы метильных групп часто наблюдаются при 8,30 и 8,19 м. д., а сигнал винилъного
протона — при 4,75 м. д. [40]. Поэтому отнесение сигпалов в структуре А не является
однозначным. Тонкое расщепление некоторых пиков, по-видимому, обусловлено дальними
константами спин-спинового взаимодействия, особенно для винильного протона.
Задача 27. Установите структуры следующих соединений:
трет-С^ — СН = СН — C4H9-mpem
1. 30%-наяНяОя iG0° 1. 30%-вая НаОя
С^° «""i^£5 с*°н2зв --^ с10н31в -
* 13
2. Н20
ИК: 2500 см-1
2- Н20
ИК: 2500 см-1
С10Н22О2
4
15 Н
-«-50%-ный СС14
-*25%-ный СС14 2Н
TMC
6,13 6,73
8,68; 9,15 10,00
8,86
В СС14,ТМС в качестве внутреннего стан'дауга
БВ-йо^овай линия пика при ~9м.д. (возможно от J]is0_jj)
5,06
.М-
6,61 SB 6,65 9,14 TMC
В'СС14
54
ГЛ АВ А 2
Ответ (Logan Т. J., F 1 a u 11 Т. J., J. Am- Chem. Soc, 82, 3446 (I960)].
CH3 CH3
I I
сн3—с—сн=сн—с—сн3
I I
CH3 CH3
CH34 х^Нз
СНз CH3 \/
СНз-С-СН2-СН-С-СН3 -> СНг ^На CH»
сн3 вн2 СНз нв сн-с-сн3
1 3 СН3
1 ■ I
СНз СНз СНз CHS
II II
СНз—С —СН2 — СН — С—СН3 НО —СН2—С —СН3 —СН —С-СНз
111' ill
СН3 ОН СНз СН3 ОН СН3
2 4
Соединение 1 представляет собой продукт нормального присоединения ВН3 к оле-
фину; отклонение от обычного протекания реакции заключается в том, что из-эа
пространственных затруднений образуется моно замещенный боран, а не ди- или тризамещенный.
Окисление этого соединения и гидролиз приводят к спирту 2 [41]. Спектр ЯМР
подтверждает приписанную структуру:
6,73
8,68 Н
9,06 8,86 | 9,15
(СН3)3С - СН2 - С - С(СН3)3
I
он
6,13
Две группы метильных протонов неэквивалентны, вероятно, из-за того, что одна трет-
бутильная группа расположена ближе к атому кислорода, чем другая. Отнесение сигнала
при 9,15 м. д. более близко расиолощенным метильным группам сделано по аналогии со
спектром соединения 4 (см- ниже). Появление квадруплета при 6,73 м. д. показывает, что
два метиленовых протона неэквивалентны или вследствие затрудненности вращения около
центральной связи С—С, или вследствие асимметрии соседнего атома углерода * ([10],
стр. 99 и ел.).
Промежуточное соединение 3, которое не было выделено в чистом виде, возникает
при атаке бораном одного из насыщенных атомов углерода (атака сопровождается
дегидрированием) [43]. Соединение 4 получается далее при окислительном гидролизе. Альтер-
* Современное рассмотрение проблемы магнитной неэквивалентности («анизохрон-
ности») с точки зрения стереохимии, включающее введение понятий об энаптиотопных
и диастереотопных группах, проведено исчерпывающим образом в работе [42].—
Прим* перев.
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
55
нативными структурами для соединения 4 могут быть следующие:
СНз СН3
I I
сн2он сп3-с — сн—сн—сн—сн2он
СНя С Но
I 1
СНз-С— СН2СН—С
I I I
СНз ОН СН3
А
СН-
I I I
СНз ОН СНз
Б
а также аналогичные перегруппированные структуры и структура 1,2-диола В
СНз СНя
I I
СНз —С СН—СН — С-СН3
I I I I
СНз ОН ОН СН3
в
Однако структуры Б и В можно исключить на основании спектра ЯМР соединения 4t
который можно интерпретировать следующим образом:
т, м. д.
9,14
8,65
-СН3
-СН2
СН
I
ОН
не расщеплен, как можно было бы ожидать для соединения Б
в соединении В такой группы нет, расщепление не
соответствует структуре Б
£ 6,61 —СН—, —СН2ОН 3 протона, а не 2, как в структуре В
ОН
^.5,06 —ОН
Выбор между структурой А и 4 основывается па том, что простой синглет при 9,14 м. д.
подтверждает эквивалентность или «почти эквивалентность» метильных групп (спектр
1снят при 40 Мгц, и небольшая неэквивалентность может не разрешиться). В соединении 4
все СН3-группы находятся в р-положении к ОН-группе и поэтому их сигналы одинаковы.
В структуре А две метильные группы находятся в {1-положенйи к двум ОН-грутшам и три
метильные группы в ^-положении к одной ОН-группе, так что, по-видимому, следовало
бы ожидать расщепления сигнала, аналогичного наблюдаемому для соединения 2. Хотя
структура 4 и более вероятна, чем структура А, однако при отсутствии строгих аналогий
вряд ли ее можно считать окончательно доказанной.
Другим аргументом в пользу структуры 4 можно считать тенденцию к
преимущественному образованию пяти,- а не четырехчленного цикла. Пятичленный цикл образуется
дри переходе 1-*-3, тогда как структура А требует промежуточного образования
четырехчленного цикла.
Задача 28. Какое из двух соединений (А или Б) в каждой из
изображенных ниже пар может дать приведенный масс-спектр? Относительная
интенсивность пиков в задаче (28.3) дана в скобках. Часть спектра, стоящая после
символа X 50, должна быть увеличена в 50 раз.
56
ГЛАВА 2
(28.1) CH3CHa-CII—COOC2H5
NH2
L^L
30
об
CHS—CH — CH2COOC2H5
I
NH2
Б
74 102
m/e
(28.2) CH3CH2CH8CH3-CH-COOC2H5 \CHCH2-CH-COOC2H5
I CH3X I
NH2 NH2
А Б
-*- xSO
131
(28.3) ШууСН,
Nch3
\/\сн~снэ
\/\сн—cn3
I
CH3
A
I
CHg
Б
mjei 168(4,4), 139(38), 125(14), 97(18)
(28.1UB i e m a n n K., Seibl J., G a p p F., J- Am. Chem. Soc, 83, 3795
(1961)].
При фрагментации аминосоединепий обычно расщепляются связи в а-положении
к атому азота с образованием стабильного иона
Г -с^с-
4nh„
— с
J1
+nh2
• с —
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
57
Отсюда можно ожидать
СН3СН21—CH-'
i
NH2
102 58
А
СООС2Н5
н,с
—сн—
I I
NH2 J
116 44
сн2соос2н5
.Следовательно, наблюдаемый масс-спектр совпадает с ожидаемым для структуры А.
Остальные пики можно интерпретировать следующим образом:
СН3СН2СНСООС2Н5
сн2—-сн3—сн
. |-сн3сн2.
H2N^=CH— С^Г ^^=ССН2
NH2
58
—СН2===1СН2
-сн2—сн2
CH2=NH2
30
- H2N=CH-c4
.ОН
74
{28.2) [В i e m a n n К.,S e i bl J., G a p p F., J. Am. Chem. Soc, 83,3795 (1961)].
В этом случае основное направление расщепления, приводящее к пикам 86 и 102,
;,будет одинаковым для обоих соединений. Отличить их можно на основе различно
протекающей фрагментации боковой цепи. Так, для этих соединений можно ожидать
Н3С j— СН2{- СН2 J- СН2
\Ш I 130 N16.
-СНСООС-.Н.
102 NK2
СН,
I .
Hf,~CH — СН2-^-СНСООС2Н5
144 !ll6 |Ю2 N'H2
Отсутствие пика иона с mie 130 (пик иона с mie 127 обусловлен совсем другим процессом)
(Указывает на то, что отщепления этильной группы не происходит. Это в свою очередь
является доводом в пользу структуры Б. Дополнительное подтверждение можно
получить из рассмотрения относительной интенсивности пика иона М-15 с mie 144. Метил ьная
группа труднее отщепляется в случае высших гомологов, если только она не находится
в таком положении, которое особенно благоприятно для расщепления (например, в случае
сильно разветвленных углеводородов). Действительно, в масс-спектре соединения А
имеется пик иона с mie 130, а не 144.
{28.3) (Birch A. J., Grimshaw J., P e nf old A. R., Sheppard N-,
Spcake П. П., J. Chem. Soc, 1961, 2286).
58
Г Л АВ А 2
Эта вадача сходна с предыдущей. Можно предположить следующую фрагментацию:
/СН3 ,СН,
НзС
Действительно, в масс-спектре имеются пики, соответствующие отщеплению этиль-
ной и изопропильиой групп, но не метильной, так что структура Б является
предпочтительной. Присутствие иона с т/е 97, который образуется при перегруппировке либо иона
•с т/е 125 с отщеплением этилена (28), либо иона с т/е 139 с отщеплением пропилена (42),
подтверждает наличие двух боковых цепей с С2 и С3.
Задача 29. Природное соединение было синтезировано по
нижеприведенной схеме. Установите строение соединений i и 2 и объясните
происхождение основных пиков в масс-спектре соединения 2.
N-e-Карбобензоксилизин
+
а-Бромпропионовая кислота
ва(ОН)в
НВг
Нейтрализация
Нагревание
* C9H18N204
1
-> C13H2eN204
2
JVIacc-спектр соединения 2 следующий:
X -Ц JL J. ■■!■
30 84
\ diL uLi Jul
X5
156 184 201
274 (P)
C2H5OH
$
HC1
m/e
Ответ [Biemann K., Lioret C., AsselineauJ., LedererE.,
Polonsky J., Bull. Soc. Chim. Biol., 42, 979 (I960)].
О Бг
II I OH-
CeH5CH2OCNHCH2CH2CH2CH2CHCOOH-f CH3CHCOOH >
I
II НВг Нейтраливация
CeH5CH2OCNHCH2CH2CH2CH2CHCOOH —=-* **
I
NH
I
CH3-CH—COOH
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
59
—> H2NCH2CH2CH2CH2CHCOOH -> H3NCH2CH2CH2CH2CriCOOC2H5
I I
NH Nil
I ■ I
CH3CHCOOH CH3CHCOOC2H5
1 2
Масс-спектрометрия — очень удобный метод изучения аминокислот [особенно их
втиловых эфиров (£191, гл. 7; [21], гл. 26)], поскольку позволяет работать с очень малыми
количествами вещества. Фрагменты обычно хорошо идентифицируются и их можно
предсказать теоретически. Поэтому ценную информацию можно получить для соединений как
^с известной, так и с неизвестной структурой. Для данпой задачи процесс фрагментации
Судет протекать следующим образом:
н
НзКСНзСНзСН^К^СНСОгС^
-NH+
I
сн3снсо2с2н5
т/е 274
сн3снсо2с2н5
HjNC^CHgCHjCHjC
-•со2с2и5
20 Г
+ NH
I
сн3снсо2с2н5
н2к— снснгсн2сн2снг
NH
I
сн3снсо2с2н5
ex.
201
201
M NH
chco2c2hs
201 CH3
-NH,
О
I Перегруппировка
H2N=s=CH2
30
I
О
- NH2CHC02C2H5
CH,
CH,CH—С
164
I-
\,
CH,
О — CH,
H
84
CH, s=s CH,
О
CH3CHCOOH
156
^°личина™ ЭТИХ СТаДИЙ фрагментации аналогично наблюдаемым в масс-спектре само-
60
ГЛАВА 2
Задача 30.
(30.1) Можно ли на основании правила октантов различить приведенные
ниже соединения?
СН3 СН3
/\
/\
н
/\
0/\/1\/ 0/\/|\/
/\
н
(30.2) Изображенный ниже бромкетон имеет отрицательный эффект
Коттона. Какой из ротамеров (А или Б) является предпочтительным?
сн3соо
Ответ.
(30.1) Знак эффекта Коттона ([15], гл. 2) для оптически активных циклогексанонов
может быть предсказан на основе правила октантов ([15], гл. 13; [44]). В соответствии
с этим правилом (в несколько упрощенной форме; более детально см. [15], гл. 13 и [44])
необходимо ориентировать производное циклогексанона, как указано на схеме 1. При
этом каждый заместитель располагается в одном из восьми октантов, образующихся при
пересечении трех плоскостей
Обычно заместители слева от карбонильного атома кислорода отсутствуют; поэтому общую
картину можно упростить, рассматривая четыре наиболее важных октанта и используя
Р- условное изображение цикла.
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
61
5 и
3
1
6
£
В соответствии с этим правилом заместители в различных октантах обусловливают эффект
Коттона с тем знаком, который соответствует данному октанту, тогда как заместители,
находящиеся в плоскостях, не оказывают влияния. Если заместители влияют на знак
эффекта Коттона в противоположном направлении, то результирующий знак зависит от
! числа заместителей в каждом октанте, их природы и их расстояния от карбонильной
1 группы (что не дает возможности для точного предсказания).
' Для данной задачи
6
9
J
If)
СН,
*,
2
4
3
Ясно, что влияние атомов 4 и 9 уравновешивается и остающиеся «заместители»
'(6, 7 и 8) указывают на положительный знак эффекта Коттона. С другой стороны, для
| ifис-изомера возможны две конформации. В одной из них (А), аналогичной фрагменту
Астероидов с ifuc-сочленением циклов А и В [36], правило октантов указывает на отрица-
.тельный знак эффекта Коттона [44].
]0
f"3
оЕ
з '
2 ]
}
Г,
62
ГЛАВА 2
Для другой конформации, однако, можно предсказать сильный положительный эффект
Коттона
СИ?
Экспериментально было найдено, что i^uc-декалоны с такой абсолютной
конфигурацией обладают слабо отрицательным эффектом Коттона [15] в соответствии с их стеро-
идоподобной конфорыацией.
(30.2) (Джерасси К., Дисперсия оптического вращения, ИЛ, М., 1962).
Две возможные ротамерные конформации можно упрощенно изобразить как
Вг
-сн.
Вг
-?•
-СН,
А Б
Атом брома вследствие его большой электронной плотности и его близости к
карбонильной группе будет оказывать определяющее влияние на знак эффекта Коттона, и влияние
атомов углерода цикла можно просто не рассматривать. Используя правило октантов,
можно предсказать отрицательный знак эффекта Коттона для структуры А и
положительный для структуры Б. Поскольку экспериментально наблюдается отрицательный эффект
Коттона, то ротамерная конформация А более предпочтительна. Однако этот аргумент
скорее может рассматриваться как «довод в пользу», чем как «доказательство».
Задача 31. Объясните, почему кривая эффекта Коттона приведенного
ниже соединения значительно изменяется в присутствии следов соляной
кислоты, как показано на графике. Ш
н,с
в сн3он-на
Ответ {Herbst D-.Djerassi С, J. Am. Chem. Soc, 82, 4337 (I960)].
Знак эффекта Коттона можно предсказать на основании правила октантов, применяя
его к каждой из кетогрупп ([15], гл. 13; [44]). 6-Кетогруппа имеет 4С и 10 в (+)-октанте
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
63-
и 1С в (—)-октанте; 2-кетогруппа имеет 5С и 10 в (—)-октанте. Таким образом, 2-кето-
группа вносит большой вклад в отрицательный эффект Коттона, а 6-кетосистема —
несколько меньший вклад в положительный эффект Коттона. Суммарный результат,
поскольку обе группы находятся в одной молекуле, должен заключаться в слабом
отрицательном эффекте Коттона, что и наблюдается в действительности.
В смеси НС1 — СН8ОН возмоято частичное или полное превращение кетонных
групп в кетальпые, которые не показывают эффекта Коттона {[15], гл. 11). В этом случае
можно предвидеть, что менее пространственно затрудненпая 6-кетогруппа будет
подвергаться кетализации в большей степени, чем 2-кетогруппа. В результате кривая
дисперсии оптического вращения будет более отрицательна, как этого следовало бы ожидать
для одной 2-кетосистсмы.
Н,С
сн3
2
-е-
-СНЯ
СИ,
сн,
-СНЯ
Задача 32. Установите предпочтительную конформацию следующего
^сюединения:
8|-
■снч
>>
-12
В СНдОН
В октане
300 400 500
600
>.. нм
64
ГЛАВА 2
Ответ [Djerassi С, Geller L. E-, Eisenbraun E. J, J. Org. Chem-,
25, 1 (1960); Джерасси К., Дисперсия оптического вращения, ИЛ, М., 1962].
Две кресловидные конформации А и Б, по-видимому, более устойчивы, чем какие-
либо конформации ванны
Н С1 СН.
Знак эффекта Коттона на основании правила октантов будет ([15]* гл. 13; [44])
сн3
1 I J ■■ ■ ■
а
А Б
Умеренный + Большой-
Таким образом, преимущественной конформацией в метиловом спирте является А,
а в октане — Б. Подобный эффект для аналогичных а-галогенкетонов [45, 46] был
обнаружен также другими методами и объяснен на основе представлений об отталкивании Д|[по"
лей связей С — 0 и С — €1 (Вг), что приводит к предпочтительной аксиальной
конформации в неполярных растворителях. В полярных растворителях, обладающих большей
способностью к сольватации, отталкивание диполей уменьшается и в равновесной смеси
возрастает доля стерически более устойчивого экваториального копформера.
Несмотря на изменение кривой дисперсии оптического вращения, не следует
принимать, что конформации Б обязательно является преобладающей даже в октане.
Соответствующий л1ра»е-2-бром-5-метилциклогексанон, обладающий аналогичным конформа-
ционныы эффектом, имеет всегда отрицательный знак эффекта Коттона. Тем не менее
независимыми экспериментами было установлено [47], что содержание транс-диаксиальнои
формы (Б) в гептане составляет ~ 40% и уменьшается до 15% в метиловом спирте.
Однако даже при такой концентрации молекулярное вращение конформации- Б настолько
больше [СН3 и С1 находятся в (—)-октанте], чем конформации А [только СН3-группа
в (+)-октанте], что даже такая небольшая доля этой конформации Б определяет знак
кривой ДОВ. С другой стороны, если кривая становится положительной, как в
вышеприведенном случае, то это указывает па очень большое преобладание конформации А.
ЛМТЕГАТУ ТА
1- Silverstein R. M., Bassler G. С, Spectrometric Identification of Organic
Compounds, 2nd ed., Wiley, N. Y-, 1967. Охватывает ИК-, ЯМР-, УФ- и масс-спектро-
скопию. В книге не только обсуждаются методы, но приведены примеры для
тренировки, а также задачи, связанные с воспроизведением спектра.
2. Schwartz J., Physical Methods in Organic Chemistry, Holden-Day, San Francisco,
1965. Охватывает ИК-, УФ-, ЯМР- и масс-спектроскопию, дисперсию оптического
вращения и другие физические методы. Хорошая монография, знакомящая с
широким кругом современных методов.
/
ЗАДАЧИ ПО СПЕКТРОСКОПИИ
65
s»' 3. Dyer J. R-, Applications of Absorption Spectroscopy of Organic Compounds,
Prentice-Hall, Englewood Cliffs, N.J., 1965- Охватывает в основном ИК- и ЯМР-спектро-
скопию и частично УФ-спектроскопию. Хорошее пособие, включающее задачи с ис-
р. пользованием атих спектров.
I' 4. Mathieson D. W., Interpretation of Organic Spectra, Acad. Press, N.Y., 1965.
Охватывает ЯМР-, ИК- и масс-спектроскопию. Как и книга Сильверстейна и Бассле-
ра [1], данная монография включает реальные примеры, на обсуждении которых
и строится дискуссия. ЯМР-спектроскония рассмотрена более подробно; однако
|, отсутствует применение всех методов одновременно для изучения одного соединения.
с. 5*. У становление структуры органических соединений физико-химическими методами,
i под редакцией А. Вайсбергера, тома I и II, изд-во «Химия», М-, 1967.
.6. Scott A. I., Interpretation of the Ultraviolet Spectra of Natural Products,Macmillan,
J-, N. Y., 1964. Самый лучший и наиболее детальный обзор по интерпретации УФ-спект-
ч- ров сложных соединений.
fl. Jaffe H. H., OrchinM., Theory and Application of the Ultraviolet Spectroscopy,
\ Wiley, N.Y., 1962. Очень ясное изложение теоретических основ электронных спект-
t ров и их применения к различным системам.
'8. Беллами Л., Инфракрасные спектры сложных молекул, ИЛ, М-, 1963. Моногра-
% фия, в которой рассмотрены всевозможные корреляции для различных функциональ-
i ных групп.
j-9. Н а к а н и с и К., Инфракрасные спектры и строение органических соединений,
"i- изд-во «Мир», М., 1965. Имеются подробные таблицы полос поглощения различных
i групп, а также большое число задач по расшифровке реальных спектров. Читается
' легче, чем книга Беллами [8], но содержит меньшее число ссылок.
№. JackmanL. M-, Applications of Nuclear Magnetic Resonance Spectroscopy in
Organic Chemistry, Pergamon, London, 1959. Краткое, но полное пособие для
первоначального ознакомления с методом и его применением.
til. Бхакка П., Уильяме Д., Применение ЯМР в органической химии, изд-во
«Мир», М-, 1966. Приведены примеры использования ЯМР-спектроскопии для
решения структурных проблем. Хотя в основном использованы примеры из химии стеро-
ь.:. идов, большинство сведений имеет общее значение.
ЙЙ. П о п л Дж., Шнейдер В., Бернстейн Т., Спектры ядерного магнитного
| резонанса высокого разрешения, ИЛ, М., 1962. Более широкий по охвату и более
ТС глубокий в теоретическом отношении обзор, чем [10] и [11].
»1'3*.И онин Б.И., Ершов Б.А., ЯМР-спектроскопия в органической химии, изд-во
f «Химия», Л., 1967.
44*.Э мели Дж., Ф и н е й Дж., Сатклиф Л., Спектроскопия ядерного магнит-
4 ного резонанса высокого разрешения, изд-во «Мир», М-, том 1 (1968) и том 2 (1969).
£ Наиболее глубокий в теоретическом отношении обзор по сравнению со всеми
вышеприведенными. »
^5. Джерасси К., Дисперсия оптического вращения, ИЛ, М., 4962.
Д6. G г а Ь Ь ё P., Optical Rotatory Dispersion and Circular Dichroism in Organic Chemi-
l stry, Holden-Day, San Francisco, 1965.
17*.B еллюз Л., Легран М-, Грожан М., Оптический круговой дихроизм,
;,-: изд-во «Мир», М-, 1967.
if 18. Бейноп Дж., Масс-спектрометрия и ее применение в органической химии, изд-
що «Мир», М-, 1964. Детальный обзор техники этого метода, а также его применения.
/19. Biemann К., Mass Spectrometry, McGraw-Hill, N.Y., 1962. Охватывает рас-
& ■ смотрение оборудования, методов работы и интерпретацию масс-спектров.
|Г20. Будзикевич Г., Джерасси К., Уильяме Д., Интерпретация масс-
спектрой органических соединений, изд-во «Мир», М., 1966. Охватывает только
интерпретапию масс-спектров широкого круга соединений»
* Дополнения переводчика.
VG-374
66
ГЛАВА 2
21. Budzikewicz H., Djerassi С, Williams D.H., Structure Elucidation
of Natural Products by Mass Spectrometry, Vols. I and II, Holden-Day, San Francisco,
1964. Очень детальный обзор, в котором рассмотрены масс-спектры многих типов
природных соединений, и особенно алкалоидов.
22. Constants of Organic Compounds (ed. M. Kotake), Asakura Publishing Co., Tokyo, 1963.
23. G о u 1 d E. S., Mechanism and Structure in Organic Chemistry, Holt, N.Y., 1959.
24. Leonard N. J., Rec. Chem. Progr., 17, 243 (1956).
25- В г о w n H. C, Bartholomay H., Taylor M. D., J. Am. Chem. Soc,
66, 435 (1944).
26. Leonard N. J-, 0 k i M., Brader J., В о a z H.t J. Am. Chem. Soc, 77,
6237 (1955).
27. M i с h a e 1 i s L-, M i t z u t a n i M,Z. Physik. Chem. (Leipzig), 116, 135 (1925).
28. S p e a k m a n J. C, J. Chem. Soc, 1943, 270.
29. Mitzutani M., Z. Physik. Chem. (Leipzig), 116, 350 (1925).
,3.0. Moore M. L., Organic Reactions, Wiley, N.Y., vol. 5, 1949, p. 301.
-31. Leonard N. J., О k i M„ J, Am. Chem. Soc, 77, 6239 (1955).
32. С a ser i о М. С, S i m ю о n s H. E., Jr., Johnson A. E., R о Ь e r t s J. D.,
J. Am. Chem. Soc, 82, 3102 (1960).
S3. К i t a h a r a Y., С a s e r i о М. С, S с a r d i g 1 i a F.t R о b e r t s J. D., J. Am.
Chem. Soc, 82, 3106 (1960).
34. Мартин Э., Органические реакции, Издатинлит, М., 1948, том 1, стр. 194.
35. Alexander E. R-, Principles of Ionic Organic Reactions, Wiley, N.Y., 1950,
p. 180.
36. Ф и з е р Л., Физер М., Стероиды, изд-во «Мир», М., 1964.
37. М о г g a n К. J., В а г 11 г о р J. A., Quart. Rev., 12, 35 (1958).
38. F a w с е 11 F. S., Chem. Rev., 47, 219 (1950).
39. Trevoy L. W., Brown W. G., J. Am. Chem. Soc, 71, 1675 (1949).
40. Burling E. D., Jefferson A., Scheinmann F., Tetrahedron, 21, 2653
(1965).
41. Brown H. C, Hydroboration, Benjamin, N. Y., 1962.
42*.Мислоу К., Рабан М., в книге «Избранные проблемы стереохимии», под ред.
Н. Аллинжера и Е. Илиела, изд-во «Мир», М., 1970.
43. W i n t e r n i t z P. F., A.C.S. Abstracts of Papers, 135, 19М (1959).
44. Moffitt W., Woodward R. В., М о s с о wi t z А., К 1 у n e W.,
Djerassi C, J. Am. Chem. Soc, 83, 4013 (1961).
45. С or e у E. J., J. Am. Chem. Soc, 75, 2301 (1953).
46. Allinger N. L., Allinger J., J. Am. Chem. Soc, 80, 5476 (1958).
47. A 11 i n g e г N. L., A 11 i n g e r J., G e 11 e r L. E., Djerassi C, I. Org.
Chem., 25, 6 (1960).
ГЛАВА 3
Реакции замещения
Эти реакции описаны очень подробно во всех учебниках и пособиях,
! поскольку они составляют один из наиболее изученных разделов физической
органической химии. Имеется несколько более специализированных обзоров
(И-5].
Задача 33.
(33.1) Объясните, почему соединение с R = Н реагирует в 35 раз быст-
!рее, чем соединение с R = СН3.
R R
(33.2) Если нижеприведенные соединения обработать КНС03 в смеси
бензол — метанол — вода при 20° в течение 65 час, то соединение 1 гидро-
лизуется на 18%, а соединение 2 — на 70%. Объясните различие в
реакционной способности.
CKjCO
он
1 2
Ответ.
{33.1) [S р i t z е г W.CWheland G. W.. J. Am ГЬртп Чпг fi9 9оо* иа/с\\.
5*
68
ГЛАВА 3
Однако даже при наличии пространственных затруднений замещение протекает
гораздо легче, чем в самом бромбензоле. Это можно объяснить двумя способами, причем
оба, вероятно, приемлемы. Во-первых, взаимодействие между я-орбиталямп бензольного
кольца и нитрогруппы, необходимое для структуры Ai, мало изменяется при небольших
углах поворота (до 40°) вокруг соединяющей их связи. Поэтому часто даже очень большое
пространственное напряжение незначительно уменьшает резонансную стабилизацию.
Во-вторых, питрогруппа способна к стабилизации резонансной структуры А2 за счет
индуктивного эффекта, действующего через электроны а-связи, соединяющей нитрогруп-
пу с циклом. Этот эффект не зависит от поворота вокруг связи.
Br^ ^N— Brt tNr- Б г +к —
Н ^Н
R К
R R'
-*■ Продукт
реакции
{33.2) (Hcnbest Н. В., L о v е 11 В. J., J. Chcm. Soc, 1957, 1965).
Как правило [7, 8], экваториальные сложные эфиры гидролизуются легче, чем
аксиальные. Однако предлагаемый пример является исключением из-аа присутствия
5сс-гидроксильной группы.
Конформации^[7, 8] соединений 1 и 2 будут следующими:
сн,
сн-
н3с—с
II
о
н но
1
Н3<4 /°v Х-
В соединении 1 отсутствует взаимодействие между 5-ОН и ацетоксигруппой, вследствие
чего гидролиз этого эфира будет протекать со скоростью, обычной для экваториального
положения. (Очевидно, это упрощенное рассмотрение: в действительности и в этом случае
наблюдается небольшое увеличение скорости. Аналогичные результаты получены и для
Других соединений, в которых прямое взаимодействие между группами требует
значительных конформационпых изменений [9, 10].) Однако в соединении 2 гидроксильная группа
в положении 5 связана водородной связью с эфирным кислородом, как показано на схеме.
1акая связь вызывает уменьшение донорного влияния неподе лепной пары электронов
атома кислорода на карбонильную группу (форма В). Это уменьшает стабилизацию
Н3С
з^\.
:с:
Н,С
N;^
д.+
юг
РЕАКЦИИ ЗАМЕЩЕНИЯ
69
карбонильной группы в основном состоянии и увеличивает легкость атаки основаниями
(сравните ангидриды кислот, в которых вклад структуры Б уменьшен, так как существует
аналогичный вклад другой резонансной структуры, где стабилизируется другая
карбонильная группа) *.
Подтверждение этой точки зрения можно получить из ИК-спектров сложных эфиров
|Гс гидроксильной группой в положении 5 и без нее. Введение ОН-группы вызывает
увеличение частоты С = О-валентных колебаний (1733—1744 см'1) и понижение частоты
^слолшоэфирных колебаний С — О (1237—1224 см-1). Эти результаты указывают на воз-
врастание двоесвязанности группы С = О и уменьшение ее для —С — О — фрагмента [12],
'что можно предсказать на основании увеличения вклада структуры А (или уменьшения
^вклада структуры Б) в основное состояние сложного эфира.
Задача 34. Объясните, почему скорости сольволиза (SnI) уменьшаются
Ы порядке п — 5 ~ 7 ;> 6 ;> 4.
\ J^ ЯО^ный CsH5OH I I .ОН
(сн2>■ v &Г *■ (сн^, с<^
Ответ (Brown Н. С, В а г к о w в к i M., J. Am. Chem Soc, 74, 1894 (1952);
>$treit wiser A., Jr., Chem. Rev., 56, 666 (1956)].
Hr,0 f I J3H
f I S& Медленно |
(сн^ с Г^ (сн2)п_( ct-cH3 ~^ (4i c\rn
I I СНэ I I -H 1 I CH3
Медленной стадией в этой реакции сольволиза является ионизация галогенида с
образованием промежуточного карбониевого иона. Структура переходного состояния
неизвестна; однако обычно принимается, что она очень близка к структуре карбониевого иона.
ГТаким образом, дискуссия о возможном влиянии структурных изменений на стабильность
■ карбониевого иона имеет также непосредственное отношение и к переходному состоянию,
^предшествующему этому иопу.
Принимается, что влияние размера цикла на стабильность иона обусловлено
изменением величины внутреннего напряжения (I-напряжение) в различных циклических
системах [13]. Напряжение возникает или из-за отклонения величины углов между
связями от их нормального значения, или из-за отталкивания соседних атомов водорода.
При ионизации происходит переход тетраэдрического атома углерода (угол между связями
«^-гибридного атома 109°) в тригональный (sps —120°). В случае циклобутана, где угол
составляет около 90°, такой переход приводит к возрастанию напряжения, что затрудняет
'образование карбониевого иона и уменьшает скорость сольволиза.
Необходимо заметить, что в случае вторичных циклобутилгалогенидов важную роль
может играть дополнительный эффект, приводящий к возрастанию скорости сольволиза
(первого порядка) по сравнению со скоростями сольволиза производных циклопента-
■ на [14]. Этот эффект обусловлен неклассической делокализацией заряда очень
нестабильного вторичного иона, и он не наблюдается для более стабильных третичных ионов.
* Отметим, что медленной стадией при гидролизе сложных эфиров является
присоединение основания к карбонильной группе, вследствие чего дестабилизация основного
состояния будет приводить к увеличению скорости ([8], стр. 315 и ел.). Альтернативная
точка зрения, основанная на термодинамических измерениях модельных соединений,
высказана в работе [11].
70
ГЛАВА 3
тельноВмТлТи £S при ионизации относи-
существенно отличаются пГвеличи^^^
приводит к повышению энергии и понищени^?табш1^ти ^ аТСМ0В в°Д°Р°Да, что
U семичленных циклов по сравнению cSSSSSSS25S?U,TOr0 СОСТ0ЯПИя Д™ пяти-
лоте сгорания на одну CH2-rpynnvH15] T?SS SSST™ аНа (это Можно показать по теп-
ненных взаимодействий, уменьшается nL i?Zn»™P^6HHe' в03Еикак>щее за счет эасло-
тригоналышй атом. Это^риводит к ZlS^^Z^u^ &TOU& ^Ле^0^ в ^^^
сравнению с основным. оолыпей стабилизации переходного состояния по
реакц^йГ 35* КаКИе ПР°ДУКТЫ образуются в каждой из приведенных ниже
(35.i)
(35.2)
NaCN
\ /\ НгО*
О СН2С1
свн5-с-он -^
I
СН3
Оптически активный
(35.3)
(35.4)
-С*
I ОСН3
сн
/сн\
сн.
Ответ.
SOCI,
HNQ>
52, 1$<Щ]? П d в М* М" S с ° * 4 Е- W., J о h n s о п J. R.t j. Am. Chem. Soc,
РЕАКЦИИ ЗАМЕЩЕНИЯ
71
Реакция может протекать либо по механизму SN2' (как показано на схеме) пийп
iK SN1 через образование иона А *. Удовлетворительное объяснение того, noneMv
^получается именно продукт перегруппировки, отсутствует.
(35.2) (Cow й г у W. D., Hughes Е. D.,Ingold С. К., Master man Ч
с о 11 A. D., J. Chem. Soc, 1937, 1252). e г m a n ьм
H
cgh5—с—он
CH,
so a.
н н
ce%-с <^ps=о — с6н5— с — ci + so2
СП,
сн,
Эта классическая реакция SNi ([в], стр. 294 и ел.) протекает с сохранением как
роптическои активности, так и конфигурации.
"X}
o-^-s1^
SOCI,
HX,
H
'X
У/
I H,C
CI _^
л
H
CI
H
^™Эт° SN<'"Реакция ([6], стр, 296). Отметим, что связь С - CI образуется в
икс-положении к расщепляющейся связи С — О.
■ '5,4) [° п} а ,М** ,Gazz' СЫш" ItaL' 49> <1ГЬ *58 (1919); обсуждение и ссылки на
™^Y*oydoC"4d9l6,Ipap.^a0rff!,.PBD- nidd J- H- А-г-и.'яДлши». Butter
СН
н3с-- ^сн.
осн,
осн„
сн3
NpJ
-no;
3^ L«3
НдС ^СН:
4
-снСсн^з
сн.
«nmtJl Б Д^мяиФориамгда эта реакция протекает по механизму SN2 и приводит к
нормальному продукту _ нитрилу фурилуксусной кислоты [16].-Прим. пере*.
72
ГЛАВА 3
При обычном нитровании можно ожидать получения соединения 1 или 4, по ни
одно ий них не соответствует суммарной формуле полученного вещества. Промежуточно
образующееся соединение 1 подвергается дальнейшей атаке катионом нитрония. Однако
атака направляется не в положение 2 (с образованием соединения 4)', которое
пространственно затруднено, а в положение 4, которое пространствепно более доступпо и в то же
время активировано (так же как и положение 2). Промежуточно образующийся
комплекс 2 может превратиться либо обратно в соединение 1, либо с отщеплением изопропиль-
ной группы (в виде пропилена ) в соединение 3.
Можно привести также аналогичные реакции:
ОСН3
СН30 | ОСН3
\У\/
\
ОСН3
СН30 | ОСН3
I
СОСН3
он
Вг | Вг
I
NOa
ОН
Вг
Вг
Вг+
СООН Вг
Задача 36.
(36.1) Какой ив изомеров, А или Б, легче превращается в соединение В?
R R Вг К.
ОН"
ОН'
(36.2) Установите конфигурацию ацетильной группы в продуктах
следующей реакции:
R
сн3соон
TsO
РЕАКЦИИ ЗАМЕЩЕНИЯ
73
сн3со
Ответ.
(36.1) (Физер Л., Физер M.t Стероиды, иад-во «Мир», М., 1964).
Эта реакция относится к типу 8^2'-замещения ([6J, стр, 290—291); такие реакции
протекают наиболее легко при ^ис-расположении вступающей и уходящей групп.
Удобной, хотя и очень упрощенной, картиной этой реакции является представление о двух
согласованных процессах замещения, протекающих с тыльной стороны: первый процесс
связан с атакой олефина входящей группой, а второй заключается в атаке электронной
парой олефина уходящего атома галогена с противоположной стороны молекулы
(36.2) [Физер Л., Физер М., Стероиды, изд-во «Мир», М., 1964; Wins-
tein S., Adams R., J. Am. Chem. Soc, 70, 838 (1948)].
В этой мопомолркулярной реакции уход тозилатной группы ускоряется участием
«-электронов 5,6-двойной связи. Промежуточно образовавшийся ион А подвергается
далее атаке либо в положение 3, либо в положение 6 с образованием следующих
продуктов реакции Ц7, 18J:
СН3СОО'
TsO
Б обоих случаях заместитель приближается с верхней стороны молекулы. Атака
в положение 3 имеет сходство с S ^-замещением, в котором частичная связь между
атомами С8 и С5 играет роль уходящей группы. Атака в положение 6 напоминает обычное
присоединение к циклическим алкенам, которое протекает преимущественно с аксиаль-
Вой стороны.
74
ГЛАВ А 3
Задача 37. Приведите механизм следующей реакции:
£>-
вг J^E
В ЖИДК. NH3
Jz}-1*1*
X = о - или м - ОСН3 м ~ ОСН3
X=n-OCH3 л*-ОСН3(49%), «~OCH3(5l%)
Х=ж-СН3 о-СН3(Я%), л-СН3(Б6%), «-CHJ224
О^ШоЬвгН J.D.. VaughaiiCW., Carlsmith L. A., Seme
eiov D. A., J. Am. Chem. Soc, 78, 611 (1956)].
OCH3
-HBr
OCH, OCH3
РЕАКЦИИ ЗАМЕЩЕНИЯ
75
Эти реакции протекают с элиминированием НХ и промежуточным образованием
дегидробензола [19—21]. Существование такого промежуточного продукта впервые было
показано Робертсом и сотрудниками [22] при изучении реакции
С1 ГШЙ ГШз
Л/ Л/ "V
Они показали, что разница в соотношении продуктов обусловлена кислотностью
ароматических протонов, которая контролирует направление элиминирования, и стабильностью
анионов, получающихся при присоединении NH^ к дегидробензолу.
Кислотность ароматических протонов (фактически определяемая стабильностью
.образующегося аниона) зависит от индуктивного эффекта заместителей. Поэтому элек-
троноакцепторные группы, как, например, OCH3i стабилиаируют анионы орто ;> мета ;>
►> пара, тогда как СН3-группа, обладающая электронодонорным индуктивным эффектом
(слабый эффект), имеет обратный порядок стабилизации. Полученные результаты
; в общих чертах совпадают с этой точкой зрения. Элиминирование в случае л-бромапизола
протекает селективно с образованием 2,3-дегидробеязола (А). Присоединение к нему иона
NHj происходит с преимущественным образованием о-аниопа (а не jw-аниона) и, таким
^образом, приводит к jtt-аминоанизолу.
Из ?Н5романизода может образоваться только один дегидробензол, присоединение
: к которому дает два вышеуказанных продукта. Хотя из сказанного выше можно сделать
вывод, что предпочтительно образуется я-аминоанизол, в действительности заметного
•преобладания зтого соединения не наблюдается.
В случае ж-бромтолуола индуктивный эффект мал, и потому образуются оба
возможных дегидробензола. Однако заметно преобладает 3,4-дегидробензол и наблюдается пре-
■ имущественное мета-, а не пара-замещение. Эти результаты не очень четкие, и, очевидно*
другие эффекты также влияют на эти процессы.
Задача 38. Приведите механизмы следующих реакций;
70°
(38.1) (CH3)2SO+BrCH2COOC2H5
Бг
-> (CH3)2S + ОНС-СООС2Н5+ НВг
(38.2) А
KCN Нейтрализация
Вг
I
\/\
+ Na
соон
N02
(38.3)
а
СНяСОО-
р
ососн3
сн^соо
р
СН3СОО"
ацетон
Р
ососн3
сн,соо
76
Г Л А*В А 3
(38.1) [Hunsberger I. M., Tien J. M.T Chem. Ind. (London), 1959, 88).
н
г*.
(CH3)2S—ОГ CH2
Br
(CH3)2S
COOC2H5
i.0i?cH —- (ch3)2s + o—ch
соос2н5 соос2н5
Это удобный метод синтеза альдегидов. Аналогичную реакцию можно провести
с использованием тозилатов вместо галогенидов, что позволяет осуществить селективное
окисление первичных спиртов до альдегидов [23].
(38.2) [Koaenblum M., J. Am. Chem. Soc, 82, 3796 (I960)].
с=15й'н^Ъц
9*V
~<уЧ^о~
он
N-r-N
foH H<7CN
Br
coon
coo
H.O+
COOH
Это реакция Рихтера. Для нее было предложено большое числь механизмов, в том
числе и механизмы с промежуточным образованием дсгидробензола. Однако они не
согласуются с наблюдениями, что атомы азота отщепляются в виде N2 (получается не
в результате окисления NHS) и атом кислорода переходит от нитрогруппы к
карбоксилу 124). Модельное соединение А, предложенное в качестве промежуточного соединеният
было синтезировано специально; показано, что в присутствии основания оно также
претерпевает вышеуказанные превращения {25].
(38.3) [Saegebarth К. A., J, Org. Chem., 25, 2212 (I960)].
О
г ОССН,
4 :
сн,
' — с^СНз II
Р^о о A/ J °сснэ
РЕАКЦИИ ЗАМЕЩЕНИЯ
77
оссн-.
^jo-осснз^ ^ _
I
сн,
4
°4
оссн.
CHLCO
р
сн„
Применяемые условия благоприятны для протекания реакции Зщ2-замещепия
в аллильных системах, и замещение первого атома брома происходит именно таким путем.
Уход второго Вг~ включает участие соседней группы и протекает как S^2'-реакция (А)
или как более общий случай Б^-рсакции (Б) ([6], стр. 291). Различное направление,
по-видимому, обусловлено стереохимией промежуточных бромацетатов, так как SN2'-
реакция требует, чтобы атакующая и уходящая группы находились с одной стороны
плоскости молекулы [26], тогда как согласно Биомеханизму эти группы должны быть
расположены по разные стороны.
Задача 39. Приведите механизм следующей реакции:
ОСН3 ОСН3
1>СНСНаСНаСН2 OBs
,оА/
» нсоон Г
R > |
. . CEbo/N/V
СНз^г ' ■ • ^пз^
R
Выход 91%
Ответ [Н е с k R., W i n s t e i n S„ J. Am. Chem. Soc, 79, 3105 (1957)].
осп,
сн,о
CH.X>
OBs
осн,
ОСН,
ocrt
-HH
CH30'
Замещение группы —OBs в соединениях типа А может происходить в результате
как прямой атаки растворителем, приводящей к нормальным продуктам сольволиза, так
и атаки бензольным циклом, дающей промежуточное соединение типа Б. Оно может
перегруппироваться в соединение В, причем вторичный атом углерода будет более
подвижным, чем первичный. Последующее отщепление протона приводит к
вышеприведенному продукту реакции.
Важность замещения с участием арила определяется размером образующегося
цикла и влиянием заместителей в ароматическом кольце (например, —ОСН8) на
стабилизацию промежуточного иона Б. При этом выход тетралинов варьируется следующим
•образом:
Заместитель в ароматическом кольце Выход тетралина, %
2,4-Ди-ОСН3 91
4~OCHs 55
Незамещенный 20
78
ГЛАВА 3
Бели рассмотреть ряд соединений общей формулы Ar(CH2)nOBs, то соотношение между
длиной цепи и участием арила будет следующим:
п Участие
2 Очень важное
3 Исчезающе мало
4 От умеренного до значительного
5 Исчезающе мало
Бели п-= 2, то в системе наблюдается обычный «эффект соседней группы» [5J и ее
участие сопровождается сильным увеличением скорости отщепления иона OBs~.
Промежуточным соединением в этом случае, по-видимому, является соединение Г, в котором при
атаке растворителем раскрывается трехчленный цикл и образуется нормальный
продукт сольволиза.
Г/'
+
осн
. Задача 40. Установите структуру продуктов следующих реакций
[символ (А) на стрелке означает, что соединение обрабатывают сначала 2,4,6-три-
этилбензоилхлоридом и затем mpem-бутилатом калия]:
(40.1) \с/_у
^С —С^„ LiAID4 (A)
\ о н
V^V''' LiAlD* '" (A)
(40.2) V-V "А1Р4
Ответ [С и г t i n D. Y., К e 11 о m D. В., J. Am. Chem. Soc, 75, 6011 (1953)]»
N /°\ /°бНз свн^ ja с h л Сб1К sn
С—С. „Am_ B 54N^„ . Ч^Ч^ \с/
н^ с6н5 .с
H^^CbPs Н/^С6Н5 4
ЧчС—-С*' LiAIP,, НО Js?\ Эгерйфнкадня J jl
с*н
6nS
* А -н il .с6н5 И">Н* А / СбН5\С/°
Сб"5 С6Н* н^^с.Нз / ОДрч^о \ н^'
с«н.
5
,-а трет
Б
РЕАКЦИИ ЗАМЕЩЕНИЯ
79
Восстановительное раскрытие эпоксидного кольца под действием гидридов
металлов можно рассматривать как Бк2-реакцию с участием гидрид-иона. Так, транс-эиок-
сид (1) дает эритро-соединепие при атаке дейтерид-ионом со стороны, противоположной
? связи С — О; аналогичным образом ^ыс-эпоксид (2) дает тр«э-спирт.
Реакции элиминирования, катализируемые основаниями, требуют, чтобы две
отщепляющиеся группы (в данных случаях водород или дейтерий и этерифицировапная гидро-
ксильная группа) находились в траке-положении ([6], стр. 489). Каждый из продуктов
восстановления имеет две конформации (например, А и Б), в которых это условие
соблюдается. Элиминирование в одной конформации приводит к ^wc-алкену, а в другой —
к пгракс-алкену. В переходном состоянии, приводящем к тр аис-соединснию, возможно,
оба бензольных кольца будут расположены в плоскости образующейся двойной связи,
' что способствует резонансной стабилизации. С другой стороны, переходное состояние для
jfMc-алкена дестабилизируется вследствие стерического отталкивания объемистых фениль-
ных групп. По этой причине преимущественно образуется тр ан с-соединение и
элиминирование протекает в конформации, приводящей именно к пграис-алкену. Поэтому
,9 случае соединения 1 отщепляется дейтерий, тогда как для соединения 2 реакция
протвыкает в форме Б с отщеплением атома водорода.
Задача 41. Приведите механизм следующей реакции:
СН3 СНз
Кипячение
СвН5СОО—С—СН3 + СН3ОН ддч > СвН6СООСНз-г-СН3СОСНз + С6Н5СООН
<^Н3 61,9% ^ 22,6%
1V 60,7%
■Следует принять во внимание, что
СНз СНз
i Кипячение |
— ОН + СН3ОН — ~-^ СП3—С— ОСН3 отсутствует
7 дней
СНз СН3
Ответ [Cohen S. G., Schneider A., J. Am. Chem. Soc, 63, 3382 (1941)].
О СН3 г О СНз п СН3 Н О
"I н+ (| | сн3он | | ||
С—СНЧ —■> | СвН5СОИ + +С ~СН3 I > СН3С — О—СГГ3 + СвН5С — ОН
:СвН6СО
I
сня
i
Ня
I +
СН3
СН3
° енз сн, о
СвН6СОСН3+СН3С-ОСН3 ^^ СНз—С-ОСНз + СвНьСОН
СНя
н+
СНя
о
RC-f-OR' RCO-j-R'
1 2
€0
Г ЛА ВАЗ
Два фундаментально различных типа расщепления существуют для алкоголиза или
гидролиза сложных эфиров. Один включает расщепление связи ацил—кислород (1) (Адсг
и Влс-механизмыЬ а ВТ°Р°Й — связи алкил — кислород (2) (Адх,- и БАЬ"механИ8МЫ) I27»
28]-7Гля большинства сложных эфиров гидролиз протекает с расщеплением свяаи ацил —
кислород,- но приведенная реакция имеет другой механизм.
Нормальный механизм переэтерификации следующий:
О СН3 О СН3 СН3
II I I! I ? 1
СвНй-СО-С-СНз + СНзОН^СвНбСОСНз+НОС-СНз ~^ СН3ОССН3 + Н20
сы3 сн3 сн3 •
It
ЕйО
ф|
о
41
свн5сон+сн3он
Он объясняет все результаты, за исключением того, что второй эксперимент показывает
отсутствие реакции между метиловым и трem-бутиловым спиртами в используемых
условиях. Таким образом, метиловый эфир должен образоваться непосредственно из
первоначального сложного эфира.
Реакции третичных сложных эфиров, аналогичных вышеприведенному, протекают
по типу кислотного катализа даже в нейтральном растворе [29] и включают
промежуточное образование ионных частиц. С другой стороны, сольволиз аналогичных оптически
активных сложных эфиров сопровождается значительной инверсией [30], так что
остается неясным, является эта реакция моно- или бимолекулярной.
Задача 42. Приведите механизмы следующих реакций:
(42-1) .ОСОСНз /v /°ч гн
/Y CH3COONa /у \ /СНз
* I I С
| I СзЩОН | р / v
\/\0Ts \/\/ \ос2нЕ
(42.2) /N/OCH3 /Ч,Л
С1-
,,а
(42.4) ОСН3
н2о ^"з\
CH3CCH2OBs > ^СНСНО
СНз
СНз/
Bs = Br-^ ^-SOa-
РЕАКЦИИ ЗАМЕЩЕНИЯ
81
Ответ [Обзоры по участию соседних групп в сольволиве см. Capon В., Quart
Rev., 18, 45 (1964)1.
.:- (42.1) [W i ns t e i n S., Hess H. V., Buckles R. Б., J. Am. Chem. Soc,
Ш, 2796 (1942); Winstein S., Buckles R. E-, J. Am. Chem. Soc, 65, 613 (1943)].
О—С
:ос2н5
Н,С. ^ОСйНд
(\ (42.2) [Noyce D. S., Weingarten H. I., J. Am. Chem. Soc, 79, 3093 (1957)]
о
/^r
CO CI
CH,0
CI
o^v ^och.
н3со:—c-a ^o ^ "*<.•—*
С)
(42.3) (Berson J., в книге «Molecular Rearrangements», de Mayo P., ed., Vol. I,
tWiley-Interscience, N.Y., 1963, p. 185).
(42.4) [Winstein S., L i n d e g г e n C. R., Ingraham L. L., J. Am.
i$hem. Soc, 75, 155 (1953)].
СНз, СН3 Ш3
сн^-с-с4н — сн3.-с^с^н — сн3^с-с/ ■££* (снэ)2с-с-он2
' Cobs сн/ Чн н/ н
СНз1'
J
сн, осна
сщ ^н
о носн3
1-374
82
ГЛАВА 3
Альтернативный путь заключается в отщеплении протона от промежуточного
соединения 1 с образованием винилового эфира (2) и его последующем гидролизе. Однако в
действительности виниловый эфир (2) стабилен в условиях реакции.
Задача 43. Какой из изомеров (цис или транс) соединения А будет
претерпевать сольволиз с большей скоростью? Предскажите, как изменится
скорость сольволиза при введении нитрогруппы в пара-положение
бензольного цикла.
/\/
С1
80%-ный СаН5ОН
/\/
ОН
\/\
SCeHE
^/^SCeH
6П6
Ответ [Goering H. L-, House К. L., J. Am. Chem. Soc, 79, 6542 (1957)1
Существуют реакции, где два соединения различаются но реакционной способности,
поскольку в одном ив них сольволиз включает участие соседней группы [5], а в другом
из-за геометрии молекулы такое участие отсутствует. В случае 1,2-т/шкс-соединения
неиоделенная пара электронов атома серы может принимать участие в стабилизации
образующегося катионоидного центра в переходном состоянии Б и ускорять, таким образом,
отщепление аниона хлора. В ^ис-изомере отсутствие планарности связей С — С1 и С — S
препятствует такому взаимодействию и скорость сольволиза меньше, чем для цикло-
гексилхлорида.
6-
С1
С1
S6+
-^
—■*- Продукты
реакции
Электроноакцепторный эффект нитрогруппы, находящейся в пара-положении,
понижает электронную плотность на атоме серы и, следовательно, уменьшает ее участие
в реакции замещения. Поэтому скорость реакции для тракс-изомера при введении п-нит-
рогруппы уменьшается, хотя все же и остается гораздо большей, чем для ^ис-соедипе-
ния. Эффект замедления для цис-соединения очень мал, так как влияние нитрогруппы
не проявляется на значительном расстоянии от реакционного центра.
Задача 44. Объясните, почему гидролиз нижеприведенного хлорамина
в щелочной и в кислой средах приводит к изомерным спиртам*
РЕАКЦИИ ЗАМЕЩЕНИЯ
83
В щелочной среде
(C6H5CH2)2NCHCH3OH
(CeH5CH2)2NCH2CHCH3
В кислой среде
сн.
(CeH5CH2)2NCH2CHCH3
I
он
Ответ. В сильнокислой среде атом азота полностью протонирован, и его
неиоделенная пара электронов не участвует в гидролизе галогеппроизводного. Поэтому реакция
протекает нормально по схеме реакции замещения без участия соседней группы (А).
Если, однако, амин не связан в виде соли, то первой стадией гидролиза является
внутреннее замещение, приводящее к этилениммониевому иону В± ([6], стр. 570 и ел.).
В присутствии сильного основания (ОН-) он далее подвергается Эц}2-реакции с
раскрытием цикла у более реакционноспособного первичного атома углерода с образованием
продукта перегруппировки [31].
Дело, однако, осложняется тем, что промежуточное соединение Б, в почти
нейтральном растворе (НСО^) дает неперегруппированный продукт [32]. Объяснение,
предложенное в работе [2], состоит в следующем: предполагается, что переходное состояние при
атаке водой имеет больший положительный заряд на атоме углерода, чем при атаке ОН-,,
и поэтому реакция протекает по вторичпому атому углерода.
<г
А +
R9NH— СН2СНСНЭ
2 I
н2р.
R'2NH— Ш2СНСНз
*он0
н2о +
■ ** R2NHCHaCHCH3 + Н3СГ
он
R,№
сн2снсн, -»»
*С1
N
/ \
сн2—снсн3
он
:nr2
I
носн2—снен,
R,n:
сн2—снсн3
I
он
Задача 45. Объясните различия в соотношении продуктов (нитросое-
динсние : эфир азотистой кислоты), получающихся в результате двух
следующих реакций:
AgN02
«-С7Н151 > 8: 1
ъ эфире
6*
84
ГЛАВА 3
K-L.7Jti{5l > 5 : 2
в ацетонитриле
Ответ [Kornblum N., Smiley R. A., Blackwood R. К., Iff-
1 a nd D. C, J. Am. Chem. Soc, 77, 6269 (1955)].
Реакция нитрит-иона («амбидентного» аниона, способного реагировать либо по
азоту, либо по кислороду) с алкилгалогенидами протекает через промежуточные стадии,
прячемТпереходное состояние может иметь или SN1-, или SN2-xapaKTep (или оба). В
присутствий иона серебра, который облегчает уход галогена в виде аниона, имеется
тенденция к разрыву связи С—X перед образованием связи С- • • -N02. В результате в переходном
состоянии атом углерода имеет характер карбониевого иона» т. е. частичный
положительный заряд. Чем больше этот заряд, тем больше тенденция для ориентации
приближающегося нитрит-иона силами электростатического притяжения между положительно
заряженным атомом углерода и наиболее отрицательно заряженным атомом входящей группы
(обычно это наиболее электроотрицательный атом в сопряженной системе, имеющей
заряды). В данном случае такая ориентация должна приводить к образованию эфира
азотистой кислоты при атаке атома углерода атомом кислорода нитрит-иона.
Это подтверждается тем, что при отсутствия такого эффекта преобладает атака
наименее заряженного атома. Возможно, что вследствие меньшей сольватации
последнего при контакте реагирующих частиц необходимо удаление меньшего числа молекул
растворителя. Правильна эта точка зрения или нет» но, во всяком случае, результаты
согласуются с такой интерпретацией. В условиях» при которых следует ожидать
уменьшения ионного характера переходного состояния, атака по менее электроотрицательному
атому происходит в большей степени {для данного случая — это образование нитросое-
дцження).
В приведенном примере переходное состояние в ацетонитриле должно иметь
больший карбоний-ионный характер», чем в эфире, из-за того что ацетонитрил является более
полярным растворителем. Возрастание карбоний-ионного характера приводит к
увеличению выхода эфира азотистой кислоты.
Как и следует ожидать из вышеприведенной интерпретация, соотношение ннтросое-
динения и эфира азотистой кислоты должно сильно зависеть от структуры галогенида:
третичные галогеннды должны давать больше нитрита, чем вторичные, а последние —
больше, чем первичные. Равным образом замена нитрита серебра па нитрит натрия, в
котором ион натрия не катализирует уход галогенид-аниона, уменьшает вероятность
образования эфира азотистой кислоты.
Задача 46. Приведите механизмы следующих реакций:
(46.1) Вг О
CHS [|
' /\
Na+<CH3-C-T4 02)- | и
(46.2)
Н C(CH3)2N02
СНз \/
-NOz)-
\=/
ОтФет [Bersohn M., J. Am. Chem. Soc, 83, 2136 (1961)].
РЕАКЦИИ ЗАМЕЩЕНИЯ
85
(46.1)
(46.2)
Алкилировапие амбидентных ионов {способных реагировать двояко) приводит
к различным продуктам в зависимости от условий реакции. Как показал Корнблюм [33J,
реакции такого типа являются промежуточными между реакциями типа Swl и Sjj2 ([6],
.гл. 8). Поэтому очень важно, появляется ли более или менее выраженный
положительный заряд на атоме углерода, при котором осуществляется замещение, что в свою очередь
зависит от того, удаляется ли галогенид-анион вначале (SN1 как лимитирующая стадия)
или одновременно с вхождением атакующей группы (8^2 как лимитирующая стадия).
Экспериментальные результаты согласуются с представлением, что в реакциях с большим
«Sfjl-характером» направление атаки определяется электростатическим притяжением
между образовавшимся положительным зарядом на атоме углерода и наиболее
отрицательно заряженным атомом аниона (обычно наиболее электроотрицательный атом).
С другой стороны, для реакций с преобладающим «SN2-xapaKTepoM» имеется тенденция
к для атаки амбидентной системы по наименее электроотрицательному атому.
Направление алкилирования амбидентных ионов зависит также от природы
растворителя [34, 35] и физического состояния аниона [36].
В данном случае реакция с аллилгалогенидом обладает большой долей «БгД-харак-
стера» и, следовательно, атом углерода приобретает частичный заряд перед тем, как
образуется переходное состояние, несмотря на то что кинетика реакции, возможно, описывает-
[ уравнением второго порядка. Следовательно, реакция аниона 2-нитропропана
приводит к продукту О-алкилирования. Однако промежуточный продукт — эфир азотистой
кислоты — нестабилен [37] и перегруппировывается с образованием вышеуказанного
[ соединения.
Если такое направление реакции связано с появлением частичного положительного
"заряда в системе а лл ил бромида, то в случае стабильного тропилий-катиона оно должно-
наблюдаться в еще большей степени: здесь следует ожидать резкого преобладания О-ори-
!.ентации. Однако именно из-за большой стабильности тропилиевой системы
образующийся эфир азотистой кислоты находится в равновесии с исходными соединениями,
; вследствие чего в реакционной смеси накапливается более стабильный продукт С-алкиля-
Прования (в реакции осуществляется не кинетический, а термодинамический контроль).
86
Г Л АВ А 3
ЛИТЕГАТУГА
1. Streitwicser A., Jr., Solvolytic Displacement Reactions, McGraw-Hill, N.Y.,
1962. Классический обзор по реакциям замещения у насыщенного атома углерода.
2. Streitwieser A., Jr., Chem. Rev., 56, 571 (1956). Классический обзор по
реакциям замещения у насыщенного атома углерода.
3. Bunton С. A., Nucleophilic Substitution at a Saturated Carbon Atom, Elsevier,
Amsterdam, 1963.
4. Thornton E. R., Solvolysis Mechanisms, Ronald Press, N.Y-, 1964.
5- Капон Б., Усп. хим., 35, 1062 (1966). Обзор по эффекту участия соседних групп.
6. Gould E. S., Mechanism and Structure inorganic Chemistry, Holt, N.Y., 1959.
7. В а г t о n D., Experientia, 6, 316 (1950).
«.Barton D. H. R., С о о k s о n R. С, Quart. Revs., 10, 44 (1956).
9. Djerassi.C, Lippman A. E., J. Am. Chem. Soc, 77, 1825 (1955).
10. D j e г a s s i С, М i I 1 s J. S., J. Am. Chem. Soc, 80, 1236 (1958).
11. В r u i с e T. C, F И е Т. Н., Tetrahedron Letters, 1961, 263.
12. Jones R.N.,Sandorfy C, in «Techniques of Organic Chemistry» (A. Wessber-
ger ed.), Wiley-Interscience, N.Y., 1956, vol. IX, pp. 472 ff.
13. Brown H.C., Fletcher R.S., Johannesen R. В., J. Am. Chem. Soc,
73, 212 (1951).
W.Roberts J. D., С h a m b с r s V. C, J. Am. Chem. Soc, 73, 5037 (1951).
15. И л и е л Э., Стереохимия соединений углерода, изд-во «Мир», М., 1965.
16. Н о в и ц к и й К. Ю-, Г р е с л ь X., ЖОрХ, 1, 539 (1965).
17. W h i t m a n G. H., Proc. Chem. Soc, 1961, 422.
18. W i n s t e i n S., Kosower E. M., J. Am. Chem. Soc, 81, 4399 (1959).
19. Roberts J. D., Chem. Soc. (London) Spec. Publ., 12, 115 (1958).
20. Гуйсген Р.,в книге «. Химия металлоорганических соединений», под ред. Г. Цейс-
са, изд-во «Мир», М., 1964, стр. 55—114.
21. Н е а п е у Н-, Chem. Rev., 62, 81 (1962).
22. R о Ь е г t s J. D., S e m e n о w D. A., S i m m о n s H.E., Carlsmith L. A.,
J. Am. Chem. Soc, 78, 601 (1956).
23. Kornblum N., Jones W. J., Anderson G. J., J. Am. Chem. Soc, 81,
4113 (1959).
24. S a m u e 1 D., J. Chem. Soc, 1960, 1318.
25. 1 b n e - R a s a K. M., Koubek E-, J. Org. Chem., 28, 3240 (1963).
26. S t or k G., White W. N.. J. Am. Chem. Soc, 75, 4119 (1953); 78, 4607 (1956).
27. Ингольд К. К., Механизм реакций и строение органических соединений, ИЛ,
М., 1959.
28. Н i n e J., Physical Organic Chemistry, 2nd cd., McGraw-Hill, N.Y., 1962, Chap. 12.
39. D a v i e s A. G., К e n у о n J., Quart. Rev., 9, 203 (1955).
30. D о e r i n g W. von E., Zeiss H. N., J. Am. Chem. Soc, 65, 4733 (1953).
31. Ross S. D., J. Am. Chem. Soc, 69, 2982 (1947).
32. К e r w i n J. F., U 11 у о t G.E,Fuson R. S., Z i r k 1 e С L., J. Am. Chem.
Soc, 69, 2961 (1947).
33. Kornblum N., Smiley R. А., В 1 а с k w о о d R.K-, Iff land D. C,
J. Am. Chem. Soc, 77, 6269 (1955).
34. К о r n Ы u m N.. Berrigan P. J., L e N о b 1 e W. J., J. Am. Chem. Soc,
85, 1141 (1963).
35. Kornblum N.. Seltzer R-, H a b e r t i e 1 d P., J. Am. Chem. Soc. 85,
1148 (1963).
36. Kornblum N., Lurie A. P., J. Am. Chem. Soc, 81, 2705 (1959).
37. Mont a von M., L i n d 1 a r H., M а г Ь e t R., Ruegg R., Ryser G.,
S a n с у G., Zellar P., I s 1 e r O., Helv. Chim. Acta, 40, 1250 (1957).
ГЛАВА 4
Реакции присоединения и элиминирования
В дополнение к литературе, приведенной в начале книги, при решении
задач данной главы рекомендуются работы^ [1—3].
Задача 47. Дегидробромирование СНзСН2СВг(СНз)2 под действием
оснований приводит к смеси изомерных алкенов-1 и алкенов-2. Объясните,
почему выход алкена-1 возрастает; при использовании следующих
оснований:
Основание Выход алкена-i, %
Пиридин
2-Пиколин
2,6-Лутидин
25
30
45
Ответ [Brown H. С, Nakagawa M., J. Am. Chem. Soc, 78, 2192 (1956)].
Направление бимолекулярного элиминирования зависит от ряда различных
факторов, и не существует полной ясности относительно важности каждого из них [4—6]. Тем
не менее установлено, что иногда главным фактором является стерическое взаимодействие
различных групп в возможных переходных состояниях, что и определяет их
относительную стабильность и, следовательно, состав продуктов реакции.
Бимолекулярное дегидрогалогенирование обычно протекает с образованием
наиболее замещенного и, следовательно, наиболее стабильного олефина (правило Зайцева) [4].
В данном случае это будет алкен-2 (А). Однако если атом азота основания окружен метил ь-
ными группами, то их стерическое взаимодействие с группами, окружающими
отщепляющийся вторичный протон (.Р-напряжение [7—9]), делает переходное состояние,
приводящее к олефину А, менее стабильным. Поэтому большую роль начинает играть
альтернативное направление элиминирования, приводящее к алкену-1 (Б) (элиминирование по
Гофману) [4]. Это направление элиминирования, хотя и менее благоприятное с точки
зрения электронных факторов, становится относительно более важным, поскольку
отщепление первичного протона менее чувствительно к стерическим факторам.
СНзСНа С ■—'—v^ri 2
ОД3
88
ГЛ АВ А 4
Задача 48. Объясните, почему выход продукта 1,4-присоединения (Б)
возрастает с повышением температуры реакции.
Вгя
СН4=СН—СН-СНа > СН2—СН—СН = СН2 + СН2—СН = СН —СН2
Вг
Вг
А
Температура реакции.
Комнатная
100
Вг
Выход
А>Б
А<Б
Вг
Замечание. Преимущественное образование соединения Б
наблюдается также, если сначала провести реакцию при комнатной температуре
и затем нагреть смесь до 100° или просто оставить стоять на несколько дней.
Ошего [Farmer E. H., Lawrence CD., Thorpe J. F., J. Chem. Soc,
1928, 729; Hatch L. P., Gardner P. D., Gilbert R. E., J. Am. Chem. Soc,
81, 5943 (1959); Gould E. S., Mechanism and Structure in Organic Chemistry, Holt,
N.Y.f 1959, pp. 530 Й.].
Эта реакция является хорошо изученным случаем кинетического контроля
образования продуктов реакции. На то, что 1,4-дибромид (Б) более стабилен, чем 1,2-дибро-
мвд (А), как при 100°, так и ври комнатной температуре, указывает его преобладание
в равновесной смеси. С другой стороны, дибромид А образуется быстрее, по крайней мере
при комнатной температуре. Из этих данных невозможно установить, протекает ли
реакция при 100* непосредственно с образованием дибромида Б либо сначала образуется
дибромид А, который далее быстро изомеризуется в дибромид Б.
Бблыпая стабильность 1,4-дибромида (Б) обусловлена тем, что он имеет более
замещенную и, следовательно, более стабильную двойную связь, чем 1,2-дибромид
(А).'Быстрое образование дибромида А по сравнению с Б указывает, однако, что переходное
состояние, приводящее к нему, является более стабильным, чем приводящее к дибромиду Б,
Ход реакции может быть изображен схемой
Br-
Вг
Можно привести различные аргументы для объяснения преимущественного образования
дибромида А. Однако, поскольку детали строения активированного комплекса
неизвестны, ни один из них не может считаться достаточно строгим.
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ 89
Задача 49. Предскажите структуру основного ненасыщенного
соединения, получающегося в следующих реакциях элиминирования:
(49.1) /СЩОН
СаН6ОКа
H2SO4. 25°
>
-HsO
СНд/ ЧШ
(49.2) СН3
СН3СНаСН2ССН3
I
Вг
Вг
(49.3) *Ч^Цс -21*
L н" Чсн3 аце™
MQ А\ ,* ч Избыток Н30+
( ' <^7>-CIIBrCHsBr ~55йГ *
Ответ (Gould E. S., Mechanism and Structure in Organic Chemistry, Holt, N.Y.,
1959, Chap. 12; Химия алкенов, под ред. С. Патай, изд-во «Химия», Л., 1969).
{49.1) Дегидратация третичных спиртов, катализируемая кислотой, протекает
быстрее/чем первичных, и приводит к олефину с наиболее замещенной двойной связью
(правило Зайцева). Поэтому следует ожидать преимущественного образования олефина А
СН2ОН
. /
СНЯ
13
АГ
{49.2) Дегидрогалогенировапие, катализируемое основаниями, также протекает
\.ш соответствии с правилом Зайцева, по крайней мере для оснований с небольшими стери-
-■ческими требованиями*
£Н3СН2СН = ССН3
Шз
{49.3) Дегалогенирование такого типа требует (или по крайней мере сильно
облегчается) копланаряого антипараллельного расположения связей С—галоген.
0 н н
\sJf d 1 > С=С + ZnBr.
HiS н/ чсн3
Zn:-^ ^вс-Бутен-2
90
ГЛАВ A k
Вг Вг, 4 Вг
I l> N NH,
(49.4) Wj-™. * W=GH ~ c«HsCSCH -"
н н
H„N
_J
H,N^
cGH5csac"
|н3о +
с.,ен5с = сн
Так как группа —С=СН имеет кислый атом водорода и в условиях реакции
образует натриевую соль, то для выделения конечного продукта необходимо подкисление.
Задача 50. Объясните, почему в приведенных ниже реакциях
элиминирования выходы изомерных олефинов различны.
(50.1) сНз СН3
I | он-
яС — СНч — С—(
СНдС — CHg — С—СНз ~~^
СНз Вг
СНз СН3 СН3 СН3
> СН3ССН = ССН3 + СН3ССН2С = СН2
СН,
СНз - ^пз
18% 82%
(50.2)
C^H5ONa
0%
1О0-Й
CoH3OWa
С2Н5ОН
+■
78%
22%
(50.3) L/L
осбснч
-"^ s
20%
80%
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
91
Пиролиз
''''OCSCH,
70%
;ю %
Ответ (Gould E. S., Mechanism and Structure in Organic Chemistry, Holt, N.Y.,
1959, Chap. 12; Химия алкенов, под ред. С. Патай, изд-во «Химия», Л., 1969).
(50.1) [Brown Н- С, Moritani I., J. Am. Chem. Soc, 77, 3607 (1955)
и последующие работы].
СНз СНз
I [
СН3С — СНй — ССНз
СН3
L
Медленно
снй
СН
.С—СН<> —
СН3
СНз
I
ССН3
+
-н+
-н+
сня
сн.
СНз — С — CHj -— С = CH2
I
СНз
сня
/
сн.
СНз-С-СН=-С
I \
СНз СНз
Б
Это один из редких случаев реакции элиминирования, протекающей по
механизму Е1, которая не приводит к продукту, отвечающему правилу Зайцева. В данном
случае, .однако, олефин Б имеет в jfuc-полошении к метильной группе mpem-бутильную
и является сильно напряженным. Это напряжение проявляется и в переходном
состоянии, приводящем к олефину Б, что повышает его энергию настолько, что образование
олефина А" становится предпочтительным.
(50.2) (Hughes E. D., I n g о 1 d С. К., R о а е J. В., J. Chem. Soc, 1953, 3839).
Т^^И^^У
н 2
С1
С1
С*
с2н5о-
н
н
ос2н5
92
ГЛ АВ А 4
Экспериментальным путем было установлено, что эти реакции относятся к Е2-эли-
минированию, протекающему по второму порядку. Такому элиминированию сильно
способствует горякс-диаксиальное расположение, отщепляющихся групп. В случае
соединения А только один атом водорода, указанный/на схеме, может занять такое положение
по отношению к атому хлора, и элиминирование дает исключительно ментен-2. Вследствие
сильной нестабильности переходного состояния, обусловленной двумя аксиальными
заместителями, энергия активации больше, чем для элиминирования циклогексилхлори-
да; скорость элиминирования составляет около 0,01 по сравнению со скоростью
элиминирования циклогексилхлорида.
Во втором случае атом хлора находится в тракс-аксиальном положении по
отношению к двум атомам водорода. Такое соотношение двух продуктов часто наблюдается
для реакций элиминирования, подчиняющихся правилу Зайцева. В этом случае
отсутствует дополнительное пространственное затруднение в переходном состоянии, и скорость
реакции сравнима со скоростью элиминирования циклогексилхлорида.
(50.3)) [Н й с к е 1 W., Тар ре W., Legutke G., Ann., 543,Л91 (1940);
Gould Е. S., Mechanism and Structure in Organic Chemistry, Holt, N. Y-t 1959,
pp. 500-507).
SCH,
Пи ролнз
+ CH3SCSH
Пи роя из
SCH,
+ ch3scsh
Пиролитическое элиминирование ксантогенатов (реакция Чугаева [10, 11])
обычно приводит к алкену (алкевдш) в результате ^uc-опцепления. В соединении А имеется
только один атом водорода при С2, способный к такому элиминированию, вследствие чего
главным продуктом является ментен-2. С другой стороны, в соединении Б имеются два
атома водорода, удовлетворяющие этому требованию: при Сг и С4. В этом случае обычно
преимущественно образуется переходное состояние, приводящее к наиболее замещенной
двойной связи *, в ревультате чего отщепление от С4 наблюдается в большей степени,
чем от С2.
В то время как ментен-2, образующийся ив соединения Б, можно считать побочным
продуктом, получающимся при альтернативном направлении; ifuc-элимияирования, этого
нельая сказать о ментене-3, получающемся из соединения А. В качестве побочной
реакции при пиролизе обычно наблюдается трякс-эламинирование ([4], стр. 502—507). Оно
Иногда указывалось, что реакция Чугаева и родственные пиролитические
процессы подчиняются правилу Гофмана. Однако экспериментальные результаты [10, 11]
указывают, что конформационные эффекты и стабильность получающихся олефинов
усложняют вопрос настолько, что простое «правило» неприменимо.
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
93
протекает с гетеролитическим разрывом связи и образованием ионной пары, в которой
происходит движение ионов относительно друг друга, достаточное для того, чтобы анион
мог приблизиться к атому водорода, занимающему тракс-положение.
-iCSCH3
Задача 51. Объясните, почему дриведенные ниже реакции протекают
ПО-раЗНОМу. г л v aivi
I
/\|.
Пиридин,
комнатная
/V
\/ \/\ температура \/\/Ч
Н ° О 0 0
н
/\i
\/
\
н
/\/
СН2СО0Н
Пиридин,
кипячение 1
^ с обратным \^
О
холодильником
\ Ответ [Klein J., I. Am. Chem. Soc, 81, 3611 (1959)].
I Реакция соединения А представляет собой обычное mjoowc-элиминирование с отщеп-
'яением HI ([4], стр. 489 и ел.). Она протекает относительно легко, поскольку протон
отщемляется из «-положения по отношению к карбонильной группе и является более реакци-
няоепособным по отношению к основаниям (сравните с медленным образованием
вещества Б, см. ниже).
!>н. /
94 Г Л А Б А 4
Вторая реакция включает атаку основанием на атом иода с согласованным
замещением аниона кислоты
Изображенный продукт реакции получается с выходом только 22%. Основным продуктом*
является Б, который образуется при нормальной реакции дегидрогалогенирования (36%).
Альтернативный продукт Г не образуется, так как из-за г/ис-расположения иода и
протона, необходимого для такого отщепления, реакция будет протекать слишком медленно.
Хотя для реакции соединения Б известно несколько аналогий, она все же довольно
необычна. Особенно неясно, какие превращения происходят с атомом иода (формально он*
отщепляется в виде 1+). Можно было бы ожидать, что этот ион будет реагировать
с ионом 1~, который получается наряду с соединением В, с образованием 12. Однако автор-
не наблюдал выделения 12 при этой реакции.
Задача 52. Укажите структуры и конфигурации продуктов следующих
реакций:
№) <Си
TsOH
(С2Н5)20
(52.2) о
TsOH
(с2н5)2о
TsO'
НО'
tc&
(52.3)
Br,
СНдСООН
Вг «л*—-f^'^^^J
Вг Ч/А\^
Ответ (И л и е л Э., в книге «Пространственные эффекты в органической химии»;
под ред. С. Ньюмена, ИЛ, М., I960, стр. 65).
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
95
Эти реакции являются примером преимущественного т/?амс-диаксиального
раскрытия эпоксидного цикла и т/тнс-диаксиального присоединения брома по двойной связи.
(52.1)
Tso;
но --
+
TsO
H"i
н—=
он
{52.2)
ОН
НЧ1
TsO
(52.3)
Вг,
Вгу
-*- +™.
Rr -L-2
Вт
НИЩ
н—-?
Вт
'< Задача 53. Приведите механизмы каждой из приведенных ниже реакций
;'. декарбоксилирования.
(53.1)
н3о+
О
!— соон
(53.4)
Нагревание
N СООН
(53.2)
(53.3)
о о
„ И II
СН3ССН2СОН
он-
сн3
I 3
сн3с-с-соон
О СНя
(53.5) /х
N CHnCOOH
Нагревание
НвО 3
CeHeNHa
(53.6) н ОСН-,
АЛЛ w ■'vvs
■ II II j
Л/\/\/
/\
но соон
Ответ {Общее обсуждение механизмов декарбоксилирования см. Brown B. R.*
Quart. Rev., 5,131 (1951); Gould E. S., Mechanism and Structure in Organic Chemistry,
JjHolt, N.Y., 1959, pp. 346—353].
96
ГЛАВА 4
О
W) Высоси-* ЮКС^И " О
+ С0,
Ч* -Т-н
Такое декарбоксилирование имеет сходство с электрофильным ароматическим
замещением и включает присоединение протона с последующим элиминированием
карбоксильной группы из промежуточного соединения. Декарбоксилирование [12,13] и декарбонили-
рование (14] такого типа встречаются и для замещенных производных бензола.
(53.2) (Gould Е- S., Mechanism and Structure in Organic Chemistry, Holt, N.Y.(
1959, p. 346).
° ° f CI 5 °\ н S
oJcJoh ^ chJUS^c^"-~co> + си3с=сн2 ^i сн3ссн3
Декарбоксилирование по этой схеме протекает только в растворах, достаточно
щелочных, чтобы перевести ацетоуксусную кислоту в анион. Бели присутствует значительное
количество сопряженной кислоты, то она быстрее декарбоксилируется по циклическому
механизму
^c*^vL сн3с=сн2 + со2
н3с-^ ^сн^ ^о
Скорости цекарбоксилирования ацетоуксусной кислоты и ее аниона относятся как 53 : 1.
»Г3 Р Ч Г /СНз
{53.3) сн3с-с-с^°н + h2nc6h5— снаС^рД^ — сн3с=с^сн +Со2
сн3 НзС^ ^СНз 3
13
\ н2о
о
II
СН3ССН(СН3)2
Скорость декарбокешшрования р-кетскислот заметно «""^"S^iL^^K^
первичных аминов- Это ускорение не может быть обусловлено из^™?5?nfti2E£S
основания (см. предыдущий ответ). Данный механизм и был предложен для объяснения
возрастания скорости цекарбоксилирования.
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
97
(53.4) (Brown В. R., H a m m i с k D. L., J. Chem. Soc, 1949, 659).
N ^COOH
Показано, что анион хинальдиновой кислоты (Б) стабилен в тех условиях, когда
наблюдается декарбоксилирование свободной кислоты. В то же время метилированное
производное (В) разлагается очень легко. По этой причине приниыают, что пиколино-
вая кислота также декарбоксилируется через цвиттер-ион А.
\
N СОО-
+.N СОО-
I
СН3
В
(53.5) [D о е г i n g W. von E.* Pasternak V. Z.f J. Am. Chem. Soc, 72.
143 (1950)]. * ,JS»
CH„
A HVC=°
II /
ex - a
4- CO,
N^ ^CH,
о
^c<^_
Как и в предыдущем случае, свободная кислота легко декарбоксилируется, хотя ее
соли (где она выступает как кислота и как основание) стабильны. Механизм может
включать или образование цвиттер-иона Б, или согласованный процесс в кислоте А,
аналогичный декарбоксилированию ацетоуксусной кислоты (часть 2)
7—374
08
ГЛАВ А 4
(53.6) (Rigaudy J., Nedelic L-, Bull. soc. chim. France, i959, 648).
н ^OCH,
COOH
Хотя соединение Б при обработке кислотами также дает антранол, для этого
необходимы более жесткие условия, вследствие чего соединение Б не может рассматриваться
нак промежуточное при декарбоксилировании соединения А.
Задача 54. Объясните, почему скорость декарбоксилирования в
щелочной среде увеличивается в следующем ряду:
СБг3СООН > СС13СООН > CF3COOH.
Ответ [Auerbach I., Verhoek F, H., Henne A. L., J. Am- Chem.
Soc., 72, 299 (1950); Verhoek F. H., J. Am. Chem. Soc, 56, 571 (1934); d e G г о -
ote О., Bull. Soc. Chim. Beiges, 37, 225 (1928)1.
О
Медленно
CX3 —CO- > :СХз+С02 —>■ HCX3
Эти результаты не могут быть объяснены в рамках простой индуктивной
стабилизации СХз, так как индуктивный эффект уменьшается в ряду F ;> С1 ;> Вг. Как было
показано [151, этот ряд отражает большую способность брома и хлора к перераспределению
заряда в промежуточном ионе за счет или «резонанса без связи» (А), или расширения
внешней электронной оболочки до десяти электронов (Б). Вторая концепция
получила более широкое распространение.
Вг
Вг—с:~ ■**•
Вг
:Вг;
вг—с:
I
Вг
Вг
I
Вг—с:"
I
Вг
:вп
II
Вг—С
I
Вг
А
Б
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
99
U. Увели*ение скорости можно также объяснить увеличением стерического от™™™
*ания для кислот, содержащих более объемистые атомы галогена: такое^шстюан?т^™;
.напряжение должно уменьшаться при переходе к иону. пространственное
Ых3^ловиях? КаК°е И3 ДЕУХ Соединевнй легче Декарбоксилируется в дап-
Т55.1) сЦз
„ I
сн3ссн=снсоон
СН3
А
СН3
сн2=сн—с—соон
I
СН3
Б
Нагревание
при 250—300°
6П5
(55.2)
Вг СеН
н—с—с н
1 I
с6н5 соон
Вг СООН
н—с—с—н
L I
С6Н5 С6Н5
Слабое основание в С2П5ОН
t 131 (1951)]. * 1959»PP- 346-353; Brown B. R., Guart. Rev.,
% JB9(?4" °ld R'T-'ElffleraC"Dodscn B-M.. J. Am. Chen, Soc,
a$S£Sg£S5> ГханиГу! SSS^SS^' W™>t термическое
^Ненасыщенные кислоты n^fcSZSS^^^f^^*3"*^ С0СТ0ЯЕЕе 1-
Г перегруппироваться в б^невдсышенньп? ™SТ * аК КаК оии сначала Д°лж-
ушшровка невозможна .Р^pSS^S^S'^ 2™ А "" Uepe"
Ч
Н^^-^С—О
— с^ ^с-
+ CO,
*aSre%^^»^^^'u2iJ&
Вг
'*
&
//
о
:с-сь-
Вг
''с=сч4
+ со,
7*
100
ГЛАВА 4
В настоящем случае переходное состояние для элиминирования соединения А сходно
В г Вг
СОО~ ^Нд^ ХН
с<Р*'Т^п
CQO
Б
Н^
'с£*1
с цис-стильбеном, и две объемистые фенильные группы расположены ближе, чем в
исходном соединении. Поэтому первое переходное состояние менее благоприятно (щис-эф-
фект»), чем получающееся из соединения Б, и реакция протекает медленнее.
Задача 56. Приведите структуры соединений, получающихся в
результате следуюгцих реакций:
(56.1)
|.вгне
2-Н202
(56.3) С1-СНаСН = СН2
1. ВаЩ
>
2. ОН-
<56.2)/\ t
BgH«
2. НаОа
(56.4) f^H-^>
I Ди - втор - каоамнлборан
Ответ (Обзор по реакции гидроборирования см. Brown H. C.f Hydroboration,
Benjamin, N. Y.( 1962).
(56.1) [Brown H. C, Z w e i f e 1 G.t J. Am. Chem. Soc., 83, 2544 (1961)].
BeH,
■t-Ди — it триалкнлироваы-
иые продукты
н,о.
Реакция гидроборирования — окисления приводит к спиртам, которые можно
рассматривать как продукты цис-гидратации против правила Марковникова. Кроме того,
перегруппировки карбониевых ионов, часто наблюдающиеся при катализируемом кислотой
присоединении, в реакции гидроборирования встречаются очень редко (если вообще
встречаются).
В случае 6-линена цис-присоединение осуществляется с менее затрудненной сторо-
.ны; конфигурация получающегося продукта изображена на схеме.
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
103
(56.2) [S a g е Ъ а г t h К. A,, J. Org. Chem., 25, 2212 (I960)].
НО—S\— ОН
Подтверждено, что образование ^ш>диола может быть обусловлено образованием
\ промежуточного алкилборана А.
(56.3) [Hawthorne M.F.,Dupont J. A., J. Am. Chem. Soc, 80, 5830 (1958)].
OH
в H fr \ J он *
CI-CH2CH=CH2 JL^ («^CH^ChJ^B* -* /\ + C]" +H3B03
Эта реакция является общим методом синтеза циклопропанов.
(56.4) [Sondheimer F., Nussiem M., J. Org. Chem., 26, 630 (1961)].
НО,
Ди-втор-изоамплборан — пространственно более селективный агент, чем диборан.
'■В то время как присоединение диборана к 1,2-дизамещенным этиленам обычно приводит
Г к смеси приблизительно равных количеств двух возможных изомеров, использование
l ди-втор-изоамилборана приводит к преимущественному образованию пространственно
[менее затрудненного изомера.
£ Задача 57. Предскажите конфигурации соединений, получающихся
:в результате следующих реакций:
сн,
(57.1)
сн,
I. В2Н6
■■ -♦
2- H202
cV
он
(57.2)
(57.3)
н {
он
102
ГЛАВ А 4
вгогтгл:гк\^ъл^^.^^ьл^ ««. о-р. «
{57.1) СН3
А
Chem. Soc, 83, 2544'(1961)].
СН3 Н
\/ ОН
\/
\/
Реакция гидроборирования — окисления протекает как а/ш:-присоединение с менее
затрудненной стороны молекулы.
(57.2)
cb.-ctj.
ОН
з.трудя^ГпоХ'рпеяо 01тшению к »«™<У> является прсетраясиевно меиое
(ST.3) [Wecnter W. J., Chcm. Ind. (London), 1959, 294].
Из-за аксиальной метильной группы! расположенной на фронтальной стороне
молекулы* атака протекает с обратной стороны.
Задача 58. На основании относительных энергий переходных состояний
установите структуры соединений, образующихся в результате приведенных
ниже реакций.
(58 Л)
СН3СН=СН2
С12
нао
С3Н7ОС1
(58.2) /п + hi
(CH3)3N-CH = CH2 > [C5H13Nip
(58.3) НВг
С1СН = СН2 > С2Н4С1Бг
(58.4) СН=СНа
/\/
сн2о,
разб. HaSof^ C»Hl2°2
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
103
Ответ (Об электрофильном присоединении к олефинам см. Gould E. S.,
Mechanism and Structure in Organic Chemistry, Holt, N. Y., 1959, Chap. 13; H i ne J '
Г Physical Organic Chemistry, 2nd ed., McGraw-Hill, N. Y., 1962, Chap. 9).
(58.1)
сн3сн=сн2
A +
? CH3—CH —CH2C1
CI-C1
Cl-Cl
Б
н2о
Э
-H+
OH
CH3CHCH3C1
H3C—CH—CHa
T+ HjsO
CH3CHCH2 —{£> CH3CHCH2OH
CI CI
3 4
Возможны два направления. Путь А включает переходное состояние, сходное
с вторичным карбониевым ионом 1, который более стабилен, чем альтернативный ион 3,
являющийся первичным.
Проблема менее ясна, если рассматривать эту реакцию как протекающую через
«хлорониевый» промежуточный ион * 5, так как трудно предсказать, как может
раскрываться этот мостиковый ион. Существование таких ионов однозначно не доказано, хотя
эта концепция очень распространена. Продукты реакции можно предсказать на
основании рассмотрения открытых ионов.
(58.2) + ш
(CH3)3NCH = CH2 > (CH3)3N-CH3CH2
1
1-
HI
{CII3)3N-CH-CH3
3
(CH3)3N-CH2CH2I
2
I-
<CH3)3N-CHCH3
I
4
Хотя обычно вторичный карбониевый ион, такой, как 3, более стабилен, чем
первичный ион типа 1, в данном случае ион 3 дестабилизируется вследствие наличия зарядов
на соседних атомах. Поэтому продуктом реакции является соединение 2.
Хотя реакция так, как она написана, включает промежуточное образование ионов,
в настоящее время известны примеры молекулярного присоединения, и вполне вероятно,
что реакция протекает как согласованный процесс без образования собственно
первичного карбониевого иона. Также можно принять образование протонированного я-ком-
плекса (см. выше о «хлорониевом ионе»), который и раскрывается под действием аниона
с образованием продукта реакции.
* Возможность существования «галогенопиевых» ионов [17] доказана методом ЯМР
[18], хотя вопрос об их участии в реакциях присоединения остается открытым. Концепция
«галогенониевого» иона введена для объяснения наблюдаемого в большинстве случаев
транс-присоединения. В литературе рассмотрены различные факторы, влияющие на
соотношение открытого и циклического ионов [19—21] (или на «степень симметричности»
циклического иона) и, следовательно, на соотношение продуктов цис- и пграис-присоеди-
нения.— Прим. перев.
104
ГЛАВ А 4
(58.3)
:с1=сн—сн3
нвг ш *+ Вг
асн—сна —*■ :ci— сн— сн3 —
^НВг
асн,—сн.
Бг
Вг
I
аснсн3
2 •
С1СН3СН2Вг
4
Реакция аналогична предыдущей — вновь вопрос сводится к рассмотрению
устойчивости промежуточных ионов. Экспериментально было установлено, что главным
продуктом является соединение 2. Это указывает на дополнительную стабилизацию
промежуточного иона 1 в результате электроиодонорного ыезомерного эффекта атома хлора.
С«4> сн3=о+н*^сн2=5н^с+н2-он
он
СНаОН
%/
СНСН2СН3ОН /СНСНаСНаОН
+
CHjOH
СНСНо
I
сн2он
■ э
-н+
снснаон
I
снаон
НОСНСНз
I
СН.ОН
3
В кислых условиях формальдегид присоединяется к алкенам (реакция Прннса)
[20, 22]. В данном случае карбониевый ион бензильного типа (1) стабильнее первичного
иона 2, и реакция протекает по схеме с промежуточным образованием иона 1.
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
105
Альтернативный продукт 3 мог бы получиться по реакции электрофильного
ароматического замещения и гидратации алкена; однако энергия активации для замещения
'бензольного цикла намного больше, чем для присоединения по двойной связи стирола.
Задача 59. Объясните, почему 3,4тдвойная связь при пиролизе соедпце-
•ния А образуется быстрее, чем при пиролизе соединения Б.
s
II
CH,SCO
JC0
S
I!
CH3SCO
Ответ [O'Connor G. L., N а с е Н. R., J. Am. СЬепь Soc, 74, 5454 {1952)1.
Реакция Чугаева и другие аналогичные реакции элиминирования обычно
изображаются как протекающие по циклическому механизму с разрывом связей С — О
и С —Н. Частичный разрыв осуществляется в переходном состоянии, хотя и не
обязательно в равной степени. В результате образуется 3,4-двойная связь, которая для
соединения А будет сопряжена с уже имеющейся в молекуле 5,6-двойной связью. Такое сопрй-
\ жение стабилизирует переходное состояние, что находит свое отражение в уменьшении-
RS'
О'
И !
RS ^S
1
энергии активации. Действительно, разность Еа для соединений А и Е составляет
~i ккал/молъ. Если принять, что основные состояния эквивалентны и что эта разность
обусловлена исключительно частичным сопряжением в активированном комплексе
|<- (типа 1), получающемся из соединения А, то энергия сопряжения в этом случае составляет-
около одной четверти от наблюдаемой для бутадиена.
Задача 60- Установлено, что в присутствии ОН" в 50%-ном водном диок-
сане протекают следующие реакции:
о:
^so2c6h4ch3
OTs
^. ^S02C6H4CH3
^^-во2с5и4сщ
"OTs
106
ГЛАВА 4
•Обе реакции являются реакциями первого порядка и по ОН" и по тозилату,
причем кА/кв = 434.
Если ОН~ заменить буферным раствором (СН3)з^ и (СН3)зГШ+, то
скорость исчезновения тозилатов 1 и 2 будет увеличиваться по мере повышения
концентрации амина, даже если рН среды поддерживать постоянным.
Показано, что такое возрастание скорости происходит не за счет солевого эффекта.
Объясните, почему это происходит.
Ответ [Weinstock J., Pearson R. G., Bordwell F. G., J. Am.
<&em. Soc, 78, 3468, 3473 (1956)].
■:b
|О9^502с6н4сн3
OTs
*a
S02CeH4CH3
h^":B
r^^-so2c6H4c-H3 _*г ^4^so2cch4ch3
+ BH
aS02C6H4CH,
Два возможных механизма элиминирования приведены на схемах. Первый,
согласованный, имеет место для тех случаев, когда транс-уходящие группы и атомы углерода,
связанные с ними, можно расположить в одной плоскости ([5], гл. 12). Этот случай
приложим для соединения 1.
■ :в
""S02c.6h4ch,
Продукт-
реакции
а
OTs
Протекание ifuc-элиминпрования для соединения 2 вызывает удивление, так как
имеются атомы водорода, которые могут отщепляться по типу трднс-элиминирования.
Первоначально предполагали [23], что электропоакцепторный эффект сульфонильной
группы увеличивает кислотность протона, соединенного с ним, и реакция протекает через
образование аниона, который далее отщепляет тозилат-ион (механизм Б). Для такого
механизма схематически возможны два случая: в первом при к_х >■ к2 образуется
равновесная концентрация аниона; во втором при кг > k_t анион представляет собой
кратковременное промежуточное соединение после медленной стадии (kt). Отличие этого случая
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
107
от согласованного механизма довольно небольшое {по крайней мере эксперимента чмтгл
и их часто не разделяют. «щ^ьио;,
v^
А-1 > к.
ft_, < fo.
Эти два случая можно различить на основании данных по катализу амином.
Случай 1. Скорость исчезновения соединения 2 выражается как
отсюда с учетом равновесия
у = Ь2[3],
fc_,[3][BH+]=fc1[2][B]
_*i[2HB]
'[3] =
Случай 2.
k_t [BH+]
v-k ЛИЖ
V-k* JL,[BH+j
v=k±[2][B]
В случае 1 наблюдаемая скорость определяется отношением [В]/[ВН+], а не
концентрацией одного соединения В. Так как то же отношение определяет рН буфера, такое
поведение часто называют специфическим основным катализом. Однако в случае 2
отсутствует обратная реакция для аниона, и любое основание, которое имеется в реакционной
среде и которое может отщеплять протон, даст свой вклад в общую скорость реакции
независимо от концентрации его сопряженной кислоты. Это общий основной катализ.
Увеличение скорости с повышением концентрации амина, наблюдаемое для
соединения 1, находится в соответствии с согласованным механизмом. Однако такой эффект для
соединения 2 указывает также либо па согласованный механизм, либо па образование
аниона, соответствующего случаю 2 [24, 25].
Несмотря на то что реакция соединения 2 не включает образования стабильного
аниона, направление элиминирования, вероятно, все-таки определяется увеличением
кислотности из-за влияния сульфонильной группы. Вполне возможно, что переходное
состояние для укс-элиминирования включает в большей степени разрыв С — Н-связи,
чем С — OTs, в результате чего на атоме углерода цикла появляется частичный
отрицательный заряд. Сульфонильная группа облегчает этот процесс и тем самым стабилизирует
переходное состояние. Для того чтобы объяснить получепные результаты, необходимо
принять, что такая стабилизация может быть достаточно большой, чтобы перекрыть
энергию, благоприятствующую процессу трявс-отщепления. Большая скорость
элиминирования для соединения 1 указывает на те преимущества, которые получаются при
одновременном действии этих двух эффектов.
ь
108
ГЛАВА 4
Задача 61. Приведите приемлемый набор механизмов для отдельных
стадии следующих реакции: дельных
О
Нагревание f ^^
SO >
О
\х
00
с6н5
so
Нагревание
^-- Нм-рмицр. (^"Х^СНО
\/\
OS02CH3
/\/
\/
Нагревание
0SO2CH3
I.A^aSbtfcif«.Ul&(?9!5J!1 °" I-A^Chem.Scc.,76,121i(1954);Berti О..
С?
H.S=0 -*-
н
^ф^-СГ
н
o-^-s-^cr
и
о
о
н ^-v II
+ so.
\ | + so2
a
сен5
oso2ch3
С6Н5 О
<s
>н -,>5-°сна
-а
ссн
В1Ж5
а
сбН5
/у oso2ch3
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
109
Пиролитическое разложение а л кил сульфитов включает г етеро логический разрыв
связи С — О и промежуточное образование карбониевого иопа. В случае циклических
сульфитов (А, Б) продукты реакции сходны с продуктами, получающимися при пинако-
линовой перегруппировке днолов 1 и 2 ([4], стр. 601—607), имеющих такую же
ориентацию гидроксидов.
'ОН
сн,
сн.
-н2о
сн,
сн,
он
сн,
-н2о
сн.
сг
"ЧСН.
Аналогичным образом разложение ациклического сульфита В является гетеролити-
ческой реакцией, а не согласованным цис-элиминированием, как можно было бы ожидать
по аналогии с ксантогенатами и сложными эфирами ([4], стр. 500—507; I26J). Установлено,
что расщепление включает промежуточное образование ионной пары 3, после чего
сульфит-ион отщепляет протон с образованием более стабильного алкена. Аналогичные ионные
механизмы были предложены, чтобы объяснить появление небольших количеств продукта
тракс-влиминирования, который обычно всегда присутствует в других реакциях пиро-
литического элиминирования.
Задача 62. Объясните, почему для приведенных ниже реакций
элиминирования для цис- и трйнс-соединений отношение скоростей уменьшается,
если X = Н заменить X = N02.
X—/ Ч—CH^CIffir
NaOH
Х = Н:
X = N02:
цис
транс
цис
«ао-СаНтОН
к
3,00*10-3
1,4*10-8
3,71
транс 2,36-10-*
>— СнСН
к
цис/^транс
210000
16 000
Ответ [С г i s t о 1 В., Norris W. P., J. Am, Chem. Soc, 76, 3005 (1954)].
Для катализируемого основаниями бимолекулярного элиминирования были
предложены два механизма ([4], гл. 12). В первом (А) отщепление протона протекает
синхронно с элиминированием аниона без образования дискретного промежуточного лоеди-
110
ГЛАВА 4
нения. Во втором (Б) протон отщепляется с промежуточным образованием карбаниона,
который далее претерпевает элиминирование во второй стадии. В первом случае реак-
V
•стс-
н
X -,
— С=кС-
i I
н
I
- в
t I
Переходное состояние
1
1
н
в
I
1
X
,
г
1 1
i I
Н X
в
Г ~i
1 1
—с_-с—
Переходное Промежуточное
состояние
соединение
—с—с-
дня сильно облегчается, если Н- и X-группы могут принять трякс-антшгараллельное
положение. Если это невозможно, то осуществляется второй путь, хотя часто для такого
элиминирования необходимы более жесткие условия.
В данном примере элиминирование для цие-соединений (Н и Вг транс) может
протекать по согласованному механизму А'.
А'
СаНс
'V,
Вг
А>
Н^7*^
С6Н5ч.
н**
I
I
I
L в
.Вг
— С6Н!!—С=СН
Элиминирование в случае троне-соединений (Н и Вг цис) протекает по схеме Б и
включает в медленной стадии образование карбаниона.
Б'
Продукт
реакции
В случае А' отрицательный заряд в переходном состоянии распределяется на
нескольких центрах, и главным образом на уходящем атоме галогена. Доля заряда,
приходящаяся на бензольный цикл, относительно небольшая. Стабилизация, которая
достигается введением нитрогруппы и ее влиянием на распределение заряда, хотя и является
довольно большой (заметим, что скорость возрастает в 103 раз), щ все же остается
меньшей, чем та, которая достигается в случае Б/ В последнем случае переходное состояпие
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
111
Сходно с карбаниопом (по-видимому, за исключением геометрии), вследствие чего
\с бензильного атома углерода на кольцо передается гораздо больший отрицательный
;8аряд. Введение нитрогруппы в этом случае дает значительно больший стабилизирующий
■эффект и, следовательно, большее увеличение (~2*104) скорости. Все это приводит
;:К уменьшению кцис/ктранс.
Задача 63. Приведите механизмы следующих реакций:
R к
N
N
С!
R
1
1
он-
он-
U М (74%)
ОН
TJ ^СН,СН —СНа(100%)
1-
R
Ответ [G г о b С. A., Bull. soc. chim. France, I960, 1360; Archer S., Le-
Ivis T. R., Zenitz В., J. Am. Chem. Soc, 80, 958 (1958); А г с h e г S.,Bell M. R,
Lewis T. R., Schulenbcrg S. W., Unser M. J., J. Am. Chem. Soc, 80,
f 4677 (1958)].
N R —N
]a A
CI
OB
R
:n +n
112
ГЛАВА к
Обе эти реакции протекают со скоростями, которые па несколько порядков больше,
чем скорости соответствующей реакции циклогексилхлорида, так что влияние атома азота
сказывается как на увеличении скорости реакции (увеличение реакционной способности
галогена), так и на стереохимии получающихся продуктов.
Первая мономолекулярная реакция протекает за счет внутренней атаки атомом
азота с замещением галогена и образованием промежуточного соединения А. Размер
связи, образующей мостик в ионе А, а также распределение положительного заряда
между атомами азота и углерода не известны. Последующая атака ОН" приводит к
замещению атома азота и образованию продукта реакции. Для этих соединений известны
« другие аналогичные реакции.
Во второй реакции атом хлора в исходном соединении ориентирован так, что
прямое замещение его не может протекать с участием атома азота, как в предыдущем
примере. С другой стороны, геометрия этого соединеният акова, что четыре центра — CI, Са,
■Ср и Cv — копланарны, как и N, Ст, Ср. Это сильно облегчает электронные сдвиги,
показанные на схеме [сравните с более общим случаем трап с-диаксиального расположения
ори элиминировании ([4], стр. 489 и ел.)], которые приводят к фрагментативпому распаду
^циклической системы.
On
^ а* CJ
\^ ^Задача 64. Объясните, почему элиминирование BsO-группы по
механизму Е1 протекает для соединения А в 2,5 раза быстрее, чем для
соединения Б.
СН3
н*-с-^оснч
1
1
BsO"^-C-^H
сн3
А
трео
сн3
снэо^-с-*н
1
(сн2)2
1
BsO^-C-*H
СН3
Б
эритро
BsO=Br—(/ Vy_sCwo—
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
113
Ответ [W i п в t е i и S., А 11 г е А Е-, Heck R., G 1 i с k R., Tetrahedron,
3, 1 (1958)1.
rOBs
r>
сн3
+ О—СН,
'снч
Продукт
реакции
0В5
СН-
ОСН.,
Продукт
реакции
В обоих случаях стадией, определяющей скорость реакции, является
элиминирование брозилатной группы, сопровождающееся участием метоксильной группы и
приводящее к образованию ионов А' или Б'. Переходное состояние, из которого образуется ион
А/, несколько более стабильно, чем приводящее к иону Б', поскольку три метильные
группы находятся в трак с, троне-положении. С другой стороны, в ионе Б' группа
О — СН3 должна находиться в tfuc-положепии по отношению к одной из соседних групп
С — СН8. В результате этого стерического взаимодействия наблюдается небольшая
дестабилизация, которая и обусловливает различие в скоростях реакции.
Задача 65. Объясните, почему в результате следующих реакций
образуется главным образом один стерсоизомер олефина:
C2HEMgBr HaO OH- Nal, CHSCOOH
СН3С0СНС1СН3 * > СеН13ОС1 > >
6 а е ld CH8COONa
Рацемический
SnCla, РОС13
^
Пиридин
СНзч /СНз
\л fir
С2Н5
/
чс=с
\
н
8—374
114
ГЛАВА 4
Ответ (Cornforth J W г л » ^ f „ * L ^
J. Chem. Soc, 1959, 112). " Corntorth H. H., Mathew К. К.,
H,C'
CI
HO H
HsC2—jc—qC сн
н3с/ \i
P0C13, пиридин
SnCl„
OH
_[- cgHfiMgBr H >yj-yCH3':
hsc2A£LCH3
Kai
CI
}OH"
O
н5с2—с'—с— н
CH3COOH / \
CHaCOONa HaC CH3
HSC-2-- ^
-H
»CH,
HaC*" \+/ ^H3
1 ..
^-SnCIj
%
няс
CH3
+ I " + Sn
iV
Стереохимия образующегося алкена определяется направлением присоединения
реактива Гриньяра на первой стадии. Все последующие реакции требуют определенных
конформацнй (хотя это не достигается без напряжения) и являются стереоспецифичными.
Конформация А — одна из неопределенного числа возможных конформацнй и,
вероятно, не преобладающая, по крайней мере в полярных растворителях [27—291. Однако
конформер А является наиболее реакционноспособным из всех конформеров, поскольку
антипараллельное расположение связей С = О и С — CI приводит к легкой
поляризация карбонильной группы в переходном состоянии. Поэтому переходное состояние At
будет предпочтительным по сравнению с Б из-за меньшего дипольного отталкивания *.
о 6-
R---C—С
.а
в-
s:
Не вдаваясь в подробности, можно видеть, что полученный продукт реакции
совпадает с ожидаемым при атаке конформера А с наименее пространственно затрудненной
стороны, т. е. между двумя меньшими группами. В данном случае такими группами
являются Н и С1 (см. правило Крама) [31]. Последующие соединения получаются в результате
серии реакций /проис-замещения.
• Согласно последним данным ,30,, слтуацш, может ^ о6ратщ). _ ^ ^
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
115
Как и следовало ожидать, другой стереоиэомер этого алкена можно получить
аналогичным образом:
О С1
Li
CH3MgI
С2Н5С—CHCHS >
Н3СЧ Б
Х = С/
Хотя последовательность реакций приведена только для одного энантиомера
рацемической смеси, она должна быть аналогичной для другого с той разницей, что. конфигу-
■ рация будет обратной. Это не безразлично для случая реакций оптически: активных соеди-
, нений с симметричными реагентами. Так как последняя стадия включает потерю
асимметрических центров, все различия для этой пары исчезают и получается тот же* самый
продукт реакции.
Задача 66. Приведите механизмы следующих реакций:
(66.1) ceH5N^ /СъЩ CeH5v ^CeHs
#/\
I
Кон
/СеН5
он
Н30+
,/\
(66.2) свН- /CeHs
свн5/чо/\;освнв"
свн5
НбСеч у к >-С6Н5
■ С,
И 1/СеН5
о
|\ососн3 „Д»0^
он h5c/Vvo
Ответ [Yates P., Stout G. H., J. Am. Chem. Soc, 76, 5110 (1954)J.
(66.1) Предполагаемый механизм включает ретроальдольную реакцию.
с„нс
^С6Н, CsHG>ri^i +
•^о -
сг,Н5
С6Н5
С6Н5
сен5
С£Н,
ССН
сен,
б"5
8*
116
ГЛ А В А 4
(66*2) В отличие от гидроксильной группы в задаче (66.1) ацетоксигрулпа не может
вступать в ретроальдольную реакцию, и поэтому должна протекать альтернативная
реакция по схеме:
скН,.,
С6Н5
Wo
с6н
5 с6н5
с«н,
О-э—ССН,
Задача 67. Предложите механизм следующей реакции:
/VWh
+ ВгСН2СС6Н5 -^^ с19Н14о4 >
у\/у , /
CftHe
СаН5ОН
н2о
о'^о
Нереакциоино—
способен
по отношению
к КМпОа
Ответ [WidmanO., Ber., 51, 533 (1918); 52, 1652 (1919); Wawzonck S.
Що г г е а 1 С. Е., J. Am, Chem, Soc, 82, 439 (I960)].
Br—снссвн5
_r/CeH5
0=c
^CH,
| OH-
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
117
0=:C— СЯН<
£ 'IcAoAo
\w
Структура промежуточного продукта А была подтверждена независимым синтезом
(реакция диазоалканов с сопряженными двойными связями рассмотрена в работе [32]).
+ ' C6H5CCHN2
Последовательность стадий при гидролизе соединения А не установлена. Вавзонек
считал, что вначале протекает гидролиз лактона (перед расщеплением п;иклопропанового
кольца); однако он подтвердил, что декарбоксилирование протекает после расщепления
трехчленного цикла.
Задача 68. Приведите механизмы следующих реакций:
(68.1)
ВГз КОН
> _
^ О N0
>/\Лсоон
.ИЗ
ГЛАВА 4
(68.2) О
;
\;
/\
о
v\A
= 0 —
о
вгч П о
Вг» Пиридин Njf \K у \
* II I I Ч=о
>С0СН3
т:щ
ВГ2
н,о
Вг
D1 , ,»J
(68.4) £ДЗ)
Ответ. °
ОН"
н3о+
соон
(б*./) (Эльдерфильд£Р., Меиер В., в книге «Гетероциклические
соединения», под ред. Р. Эльдерфильда, т. 2, ИЛ, М., 1954, стр. 5).
ох *
он-
ш.
н,о'
соон
Ос
V
Л68>2) (Bart ogn D. H. R., de Mayo P., J. Chem. Soc, 1957, 150; В ar -
ton D- H. R., Bockman 0. C.f de Mayo P., J. Chem. Soc, 1960, 2263).
. Предполагают [33], что реакция протекает путем селективного бромировапия мети-
леновой группы, соседней с карбоншюм, несмотря на присутствие двойной связи. Так,
2Вг.,
Пиридин
-НВг
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
119
о }
Возможно» что иеханизы должен быть следующим!
*Вг—Вг ♦.
Повторение
Однако трудно установить причину малой реакционной способности двойной связи.
/ Разумной альтернативой может быть
I* Вг —В г
Pvl
у;Л о
э
о • н* II
Вг
1
И^;р,
.3) [Е a s t m a n R. H., Oken A., J. Am. Chem. Soc, 75, 1029 (1953)].
Вг>
Rr-
•Вг
120
ГЛАВА 4
co^J^^ST™ - б0ЛЬШ°ГО М В°-»0;™ "»»* ««нтезн. Другой 11уть
Вг"
-НВг
-Н^
Третий путь синтеза может включать раскрытие циклопропанового кольца пеоеп fin,
мированием а-положения в кетоне £33]. F^anUBuiu жильца перед оро-
(€8.4)
ОН
СУ0.-
7^—со
СОО"
о
/^-сс
соон
А
Реакция может включать согласованное отщепление брома и миграцию метите л ьпой
группы, как показано на схеме (возможно сочетание с декарбоксилированием),3£либо
протекать черев промежуточное образование карбена А.
Задача 69. Производное 1 не дает простого продукта элиминирования
при обработке коллидином, а вместо него образуется а;р-непредельный
альдегид (2), Объясните этот факт и напишите механизм такого превращения.
Кол лид ии
J
1(0°,6 час
TsO
OHC
^соесн3
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
121
Ответ [Thomas A. F., Hensler К., Miller J. M., Tetrahedron, 16,
264 (1961)].
Вследствие высокой степени замещенности в кольце А невозможно промежуточное
образование конформации ванны (В), которая необходима для легкого протекания
элиминирования при траиоантипараллельном расположении тозильпои группы и 2а-водо-
рода. Вместо этого происходит раскрытие цикла с последующей альдольпои конденсацией
и образованием соединения 2.
OTs
НО
но
со,снэ
Б
но |
TsCT
У^-
ПС
но
согсн3
НС
со2сн3
J3
I
со2сн;!
НС
II "cOjCHj
Стерическое отталкивание между Зр-тозильной и Юр-метильной группами может
быть достаточным, для того чтобы затруднить образование формы ванны даже в
отсутствие других заместителей. Так,
TsO
О
11 I
/лреда-С4Н90К У 1 |
:в
получается в качестве единственпого продукта [34].
Задача 70. Напишите механизм следующей реакции:
О ^СНпОН
сн3ссн2сн2соон
122
ГЛАВА 4
Ответ [В irk of ег L., В ее к man F., Ann., 620, 21 (1959)].
u ^ H2°+ H HO H
О CH2OH N OIj2
* л
A
1-й*
HjOJ HO
но^ /ГЛ о ~+ V-л. НСЧ . ■ но.
но ^ но но'Ч^о-' 3 но—t^ У—снз
о о
ft J^-сщ —- носсн2снассн3
НО— С л
I
он
Это один из предложенных механизмов [35], Все они включают промежуточное
образование соединения А. Имеются также веские доказательства промежуточного
образования соединения Б, но остальные стадии строго не доказаны. Однако если проводить
реакцию вметанольно-кислотпойереде, то можно выделить другие соединения, что
подтверждает приведенную схему [35а].
Г\-афн - О^н2ос„ч - сн3о4>-сн3 + ch.o-V^^och,
СН3ОН | Н +
о о Рснз
И JI ^£-й CI]3°^chLch2cch3
сн.о.ссн.сн.ссн, — сНз0/ »j
Задача 71, Напишите разумпьте структуры для изомеров А и Б, обра
зующихся в следующей реакции:
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
123
CHaOSOaCeH5
Кипячение
с пиридином CjiHte + CitHie
\ Б
в атмосфере
азота
"макс212 нм, Лиаксйоо нм,
log e 3,82, log e 3,0,
26%
Ответ [R о w e J. W-, Mel era A., Arigoni D-, Jeger С, Ruzic-
.к a L-, Helv. Chim. Acta, 40, 1 (1957)].
Карбониевый ион, получающийся при отщеплении бенаолсульфонатной группы
-от исходного соединения, относится к типу гомоаллильного [36] или неопентильного
катиона. Первая точка зрения подтверждается следующей реакцией:
\- тогда как вторая подтверждается протеканием перегруппировки Вагнера — Меервейна
<[4], стр. 584 и ел.).
ви^-н
Продукт первой реакции совпадает с А, так как наличие более коротковолнового
максимума в ИК-спектре согласуется с меньшей степенью сопряжения для винилцикло-
пропана, чем для диена-1,3. Приведенные данные не позволяют строго различить
структуры Bi и Б2, однако первая из них предпочтительнее, поскольку соединение Бй,
представляющее собой шестичленный гомоаннулярный диен, должно иметь максимум
поглощения около 270 нм [37]. Семичленный цикл в соединении Bi более гибкий, вследствие
чего он имеет большее сходство с ациклической структурой и поглощает в более коротко-
волновой области.
Задача 72. Укажите, какие продукты образуются в результате
следующих реакций:
(72.1) /х/ОН (72.2) /vOH
\/\ш
^■свн5
2
HNOa
HNOa
^СбН5 ►
\/\
**NHi
.124
ГЛАВА 4
Ответ [С и г t i n D. Y., S с h m и к 1 e r S., J. Am. Chem. Soc , 77 1105 fiqwi
Эти реакции включают диазотированис первичного амина и птелё™™»Jw«»2J"
рование азота из образовавшейся диазогругшьГ с одновре™ ым ^SZTLl^^
групп, находящихся к ней в трй„с-антипараллельпом положение Азо? отшепляе^я
настолько быстро, что направление перегруппировки определяется ™LS252Z5
конформациеи исходного амипа. f«wmuib±vh преимущественной:
<72J>
(уг.2)
он он
Н u U
он
СсНк
11
NHj
CfiHe
CeHs г с6н, с6н5
Оиесь
возможных продуктов,
реакции
Отсутствие миграции фенильной группы в случае (72.1) объясняют либо очень-
малой равновесной концентрацией реакционноспособной конформации исходного амина,,
либо очень большими стерическими затруднениями для миграции аксиальной
фенильной группы.
Задача 73. Напишите структуры продуктов следующих реакций:
(73.1) (С6Н5)2ССО0Н
СН
сн2
Вга
СН,
(73.2) CeH5NHCOCII2CH2Br
II
О
(СеН5)2СеН70яВг
Нейтральное
соединение
—НВг (нейтральный растворитель)
-> А
C6H5NHCOCIIsCHs>Br
(73.3) о
—НВг (основной растворитель)
-> Б
С]
-НВг в НаО
<=>-CNHCHaCH2Br » * C9HI0NO2Gl (A)
В CIIgOH
■* C10N12NO2CI (Б)
в СН3ОН/СН8СООК
C0H8NOC1 (B>
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
12;
Ответ.
(73Л) [С г a i i P. N.. Witt J. H., J- Am. Chem. Soc, 72, 4925 (1950)].
C£H
6"5
,о_н
C£H
CfiH,r—С—С
^s.
CH2—-C=CHi
6"S
CfiH5— С —С
\
■I- HBr
CH2_C~CH.,Bi
I
CH,
Присоединение брома по двойной связи сопровождается участием соседней
карбоксильной группы [38]. Структура 7-лактона для продукта данной реакции более вероятна,
*1вм альтернативная структура б-лактона, так как
./п «+/'""4 «-
г + (В>г — Вг
Вг + (
преимущественно атакует первичный, а не третичный атом углерода.
ИГ"" - " ------ -
% 0957)]
73.2) [Scott F. L.," G 1 i с k R. E^, W i n s t e i h S~, Experientia, 13, 183
О — СН,
о
\
О , CH„ —Br
Нейтральная j*
среда .. CeHeN=s=C^
О—CH2
Основание
C.:H,— N — C'
.O CH„Br
bO—CH„
о—с;
^6Н5
N~ CH,Br
I
•о—сн3
'о—снп
с6н5
N—CH2
^O—CH2
В нейтральных условиях атом кислорода более пуклеофилен, чем атом азота, тогда
как для анионов справедливо обратное. Так как в анионе заряд может находиться как
на атоме азота, так и на атоме кислорода, то эти результаты объяснить довольно трудно,
хотя экспериментальные факты не вызывают сомнений.
126
ГЛАВА к
(73.3) IHeine H. W.( J. Am. Chem. Soc, 79, 907 (1957)].
Ci
\=/ If-—^iH:
a
II CHaBr
О
н,о>
он
9 V_i/N
N—]
о—I
i
CI
-o-"^-r
CI
\=/ ^O—CH4
V—V H
3 = Б
О
fl
CH„CO
Jch3oh ci—^ 4—c^~]
* =B
-OCH,
2= A
ЕЦЗЯ- Первой стадией во всех растворителях является внутримолекулярное замещение,
приводящее к оксазолину и НВг {или их соли). В присутствии воды двойная связь
гидрагируется, и образовавшаяся ортокислота раскрывается с образованием соединения 2.
Возможно, что альтернативное раскрытие ортокислоты с образованием соединения 5
также происходит. Однако этот процесс обратимый, тогда как соединение 2 выделяется
в виде нереакциошюспособного бромгидрата.
-О-Г1!
осн
С1-
DH
з к,
f/ъ
+]
Аналогичное присоединение метилового спирта к соли оксазолина (1) дает
соединение 6. Поскольку соединения 1 и 6 находятся в равновесии, то это направление
заводит нас в тупик. Соединение 3 получается за счет совсем другой реакции:
Н
С1
\=J~~ °^^^z
В присутствии основания — ацетат-иона — соединение i просто отдает протсн
превращается в оксаволин h.
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
127
;.. Задача 74. Установите структуры нижеприведенных соединений.
Объясните, почему реакция А -*- Б протекает гораздо быстрее, чем А ->- Б', и
почему бромирование Б' затруднено.
С19Н1ВОВг <-
В
HI
Bra/CHsCOOH
-CCgHg
I
i. CeHeMgBr . Быстро
> [Промежуточный енол] , > L^riaoU
2. H80+ if.'
Л
о
if
Hv ^CCeH6
с
свн6/ \н
Медленно
i. CHs=CHCH=CHa
C2H6ONa
2. Hs/Pd
СаНьОН
Б'
Вга/CHsCOOH
Реакция не идет
Соединение Б' является изомером Б, а соединение А — енольной фор-
;мой (не выделена) обоих соединений Б и Б'.
Ответ [ZimmermanH. Ё., J. Org. Chem., 20, 549 (1955)].
Реакция Дильса — Альдера ([4], стр. 533; [39]) протекает как ^-присоединение,
# поэтому соединение Б' должно быть тровс-изомером, тогда как Б — менее стаоиль-
: вым t^wc-изомером.
Ч
О
нх ,сос6нв /ч/н ||
V .< >-— ссвн6 hi
о
II
свнб
Pd
fl ■
СС6Н5 + Г г-Н
\/\н СбН5 \/\н
Б' Б
128
ГЛАВА 4
Из рассмотрения' структур Б и Б' ясно, что реакция Гринъяра протекает как
1,4-присоединение по схеме [40]
СКН
б"5
СбН5
;С OMgBr
с—он
гу-*---- - ест- - а?
Промежуточный енолят А, получающийся в результате 1,4-присоединения, может
приводить к двум стереоизомерам Б и Б'. Образование соединения Б означает, что из
двух процессов, которые происходят при подкислении соединения А, быстрее протекает
реакция, приводящая к менее стабильному кетону В. Можно предложить два
объяснения этому паблюдению.
Первоначально Циммерман предположил, что направление реакции определяется
преимущественной атакой протоном енольной двойной связи с пространственно мспее
затрудненной стороны. Так, из двух возможных направлений для атаки донором
протона (переходные состояния! и 2) первое, которое приводит к кетону Б, будет более
благоприятно, поскольку атака НА протекает из менее пространственно затрудненной
экваториальной области.
НА
НА
СеН,
ОН
ССН
6П5
Однако в работе Джопсопа и Мальхотра [41, 42] было указано, что конформации
типа 1 и 2 будут иметь довольно значительное стерическое напряжение из-за
отталкивания экваториального заместителя в положении 2 и группы, соединенной с двойной связью.
Они привели доказательства, что в таких системах будет преобладать конформер 3 с
аксиальным положением заместителя. Поэтому для объяснения полученных
результатов необходимо принять быструю аксиальную атаку протоном (как ето обычно
наблюдается) копформера 3 с образованием кетона Б. Затем в равновесных условиях может
происходить медленная реакция путем либо экваториального протоиирования
конформации 3, либо аксиальной атаки конформации 2 с образованием стабильного кетона Б .
Таким образом, эти две группы авторов дали различные аргументы для объяснения
этих обратимых реакций. Однако они одинаково объяснили различную реакционную
способпость кетонов Б и Б' по отношению к брому, связывая ее с большей легкостью спо-
лизации кетона Б. Отмстим, что энергия активации енолизации кетона Б' гораздо
больше, чем кетона Б, так как основное состояние первого более стабильно, и
переходное состояние превращения Б' -»- А менее стабильно, чем Б -»- А (сравните скорости
•обратных реакций).
РЕАКЦИИ ПРИСОЕДИНЕНИЯ И ЭЛИМИНИРОВАНИЯ
129
1.
2.
3.
4.
5.
6.
7.
8*
9*
10.
11.
42.
лз.
16.
Й7<
С. К., Mandour A.M. M.,
,24.
}25.
Ж
27.
28.
29.
;30*
31.
32.
[33.
34.
ЛИТЕРА ТУГА
Bunnett J.F., Angew. Chem. Intern. Ed. Engl., 1, 225 (1962) {краткий, но очень
хороший обзор механизмов бимолекулярного элиминирования).
В anthorpe D. V., Elimination Reaction, Elsevier, Amsterdam, 1963 (общий
обзор реакций образования олефинов и их механизмов).
Химия алкенов, под ред. С. Патай, изд-во «Химия*, Л., 1969 (сборник статей,
прекрасно освещающий различные аспекты химии алкенов).
Gould E. S-, Mechanism and Structure in Organic Chemistry, Holt, N.Y., 1959
Chap- 12.
Dhr M.L, Hughes E. D., Ingold
M a r v G. A., J. Chem. Soc, 1948, 2093.
Brown H. C, Moritani I,, J. Am. Chem. Soc, 78, 2203 (1956).
Brown H. C, Record. Chem. Progr. Kresge-Hooker Sci. Lib., 14, 83 (1953).
.Г о л ь д ф а р б Я. Л., Беленький Л. И., Усп. хим., 29, 470 (I960).
.Йлиэл Э., Аллинджер Н., Энжиал С, Моррисон Г., Конформа-
ционный анализ, изд-во «Мир», М-, 1969, стр. 240.
D e P u у С. H.,King R. W-, Chem. Rev., 60, 431 (1960).
Н э с Г. Р., в сб. «Органические реакции», изд-во «Мир», М., 1965, сб. 12, стр. 71.
Schubert W. M., J- Am. Chem. Soc, 71, 2639 (1949).
Brown В. R., Hammick D. L., Scholefield A. J. В., J. Chem. Soc,
1950, 778.
Burkett H., Schubert W. M., Schultz F.rMurphy R. В.,
Talbot t R, J. Am. Chem. Soc, 81, 3923 (1959).
Glick R.-E., Chem. Ind. (London), 1955, 716.
Trumbull E. R., F i n n R. Т., I b n e - R a z а К. М., S a u e r s С. К., J.
Org. Chem., 27, 2339 (1962).
,Де л a Map П., Б о л т о п Р., Электрофильное присоединение к ненасыщенным
системам, изд-во «Мир», М., 1968.
.Olah et al., J. Am. Chem. Soc, 90, 947 (1968).
.Dewar M., Fahey R., Angew. Chem., 76, 330 (1964); J. Am.
2245, 2248 (1963).
.Smissman E., Schnettelbar R., Portoghese P.,
30, 797 (1965).
.Зефиров Н. С, Ш e x т м а н Н. М., К а р а х а н о в Р. А.,
(1967).
Arundalc E., M i k e s k a L. A., Chem. Rev., 51, 505 (1952).
В or dwell F. G., Kern R. J., J. Am. Chem. Soc, 77, 1141 (1955).
Weinstock J., Bernardi J. L., Pearson R. G., J. Am. Chem.
80, 4961 (1958).
H i n e J., Ramsey О. В., J. Am. Chem. Soc, 84, 973 (1962).
H i n e J., Physical Organic Chemistry, 2nd ed., McGraw-Hill, N.Y., 1962, pp. 515—
518
Bellamy L. J., Thomas L. Cv Williams R. L., J. Chem. Soc, 1956,
3704.
Bellamy L. J., Williams R. L-, J. Chem. Soc, 1957, 4294.
A 11 i n g e r N. L, Allinger J-, Tetrahedron, 2, 64 (1958).
-Karabatsos G. J.7Fenoglio D. J-, J. Am. Chem. Soc, 91, 1124 (1969).
Cram D. J., E 1 h a f e z F. A. A., J. Am. Chem. Soc, 74, 5828 (1952).
Эйстерт Б., в книге «Новые методы препаративной органической химии», ИЛ,
М., 1950, стр. 91.
Д е М а й о П., Терпеноиды, ИЛ, М., 1963.
Clayton R. D.t Henbest Н. В., S m i t h M., J. Chem. Soc, 1957, 1982,
Chem. Soc, 85,
J. Org, Chem.,
JKOpX, 3, 1925
Soc.
Дополнения переводчика.
! 8-374
130 ГЛАВА 4
35. Chemistry of Carbon Compounds, Vol. IVA, E. H. Rodd, ed., Elsevier, Amsterdam,
1957, p. 143.
35a. Birkofer L, Dutz R., Aim., 608, 17 (1957).
36. Канон Б„ Усп. хим., 35, 1062 (1966).
37. Физер Л., Ф и з е р М., Стероиды, изд-во «Мир», М., 1964.
38. V a n Tamelen E. Б., S h a m m a M., J. Am. Chem. Soc, 76, 2315 (1954).
39. A 1 d e г К., в кнвте «Newer Methods of Preparative Organic Chemistry, Interscience,
N.Y., 1948, pp. 381—511.
40. Kharasch M.S., Reinmuch 0., Grignard Reaction of Nonmetallic
Substances, Prentice-Hall, Englewood Cliffs, N.J., 1954, p. 196.
41. Johnson F., M a 1 h о t r a S. K., J. Am. Chem. Soc, 87, 5492 (1965).
42. Malhotra S. K-, Johnson F., J. Am. Chem. Soc, 87, 5493 (1965).
ГЛАВА 5
Смешанные реакции
Задача 75. Установите строение продуктов следующих реакций:
rNH,
CfH_,
Кр h2so4
NH,
<
100е
C16H]2N20
I 2
aN^/C°OH СЮ3| CH3COOH
N^ + CeH5COOH
Ответ [Smiitny E. J., Caserio M. C, Roberts J. D., J. Am. Chem.
Soc, 82; 1793 (I960)].
<
CRH
6"5
<
F
F
F
F
—
с6н5^
^
.
^
0
^NH2
4nh2
L С^Н;
Л
zo-
^
Л
ссн
6 "5
H
C6H3COOH
+
V^^
Первая реакция представляет собой специфический гидролиз фторированных цикло-
бутанов *.
* Эта реакция протекает к для полихлорированных соединений [1].— Прим* перее.
9*
132
ГЛАВА 5
Задача 76. Приведенное ниже соединение образует семикарбазон,
который при нагревании с кислотами превращается вновь в исходное соединение.
Предложите механизмы этих реакций.
\/
./
Ч<Л
Ответ [В и с h i G., Yang N. С, Chem. Ind. (London), 1955,357; Helv. Chim.
Acta, 38, 1338 (1955)].
Это соединение способно к таутомерному превращению по схеме
Вследствие различий в геометрии структуры А и Б не являются резонансными, хотя
энергия активации, необходимая для перехода от одной структуры к другой, очень мала. Как
и следовало ожидать, циклическая структура А более стабильна. Поэтому продукт
реакции имеет структуру А, хотя открытая форма, присутствующая в виде следов, тем не
менее может быть определена какой-либо реакцией (например, образованием семикарба-
'яона). Гидролиз ссмикарбазона приводит к кетону, который немедленно циклизуется
в пиран.
Согласованный механизм включает атаку семикарбазидом раскрывающегося
цикла. Хотя этот механизм и был предложен, он, по-видимому, не является необходимым
для данной реакции.
Аналогичный механизм можно лримепить для объяснения замечательно легкой
реакции распада 2,2-диметилхроменоп в довольно мягких основпых условиях:
ОН
| ^1 __ Прквдкты
i - распада
Задача 77. Приведите механизмы следующих реакций:
н»о"
<J^
СМЕШАННЫЕ РЕАКЦИИ
133
(77.2)
ососн3
(СН3СО)20
п- TsOH
СН-СОО
- <р>-
ососн.
сн
(77.3)
он-
HOQC-
соон
(77.1) (Aebi A., Barton D. H. R., Lindsey A. S., J« Chem. Soc.T
1953, 3124).
ЙЧ ы +
Cf*«
Oil
н2о:' +
Окись кариофилшенй
связи
Расщепление эпоксидной связи сопровождается трансаннулярньш образованием
. С — С. Напряжение.в четырехчленном цикле облегчает последний 1,2-сдвиг,
приводящий к образованию конечного продукта.
(77.2) (Barton D. H. R., В о с k m a n О. С, d е М а у о F., J. Chem. Soc.t
1960, 2263; Barton D. H. R., de M а у о Р., 3. Chem. Soc, 1957, 150).
OCOCH3
Пнретозин
ососн3
134
ГЛАВА 5
(77.3) [U 11m an E. F.? J. Am. Chem. Soc, 81, 5386 (1959)].
C,HtOC
II if II CHs
Br __^HsocvJL^r сгн5ос И
OH
CH,
HOOC^
CH,
Кислоте
Фей ста
Пиролиз И
-соон ** c^og
н,с-<40 р°
( ОН"
СНЧ
соон ■*- сан50СЧУг \ ^соон
сн,с-1-о-
I
он
В этом ряду также наблюдалась следующая реакция [2]:
О — О
НОС,
[] СН3
НОСч 1 ,Вг
ОН"
л
НзС^о^Чо ВД/У
Задача^78. Приведите механизмы следующих реакций:
(78.1) (] у _ HN°3 ^ Пиридин |т
о
(СН3СО>80
(78.2)
/\/
CeH7N05
нэо+
н н
онс/ \сно
Вг
СНСООСНз
Wo2
НС —СООСН3
Nal
СН300СII/
^^СНСООСН, h(!n(ch3»8 >/Y4COOCH
Вг
СООСНз
СМЕШАННЫЕ РЕАКЦИИ
135
Ответ.
(78.1) [Clause n-K aas N., Fakstorp J., Acta Chem. Scand., 1, 210
1947)1.
О -=c
11 "\
H H
1+
Ч0^М
■CV
*
\н<
Q
NO?
+ BH"
NO,
CH,COH CH,OX f==\ ^N0
нАоЛн
нго:л/=
+
'bo
снзСО H H
II
о
NO,
—*• сн3со о^н,
то +
н
Н /Н
_ о=с^ ^-с=о
':он2 н п
+ СН-СО0Н+ N02"
(75.2) [Blomquist А. Т., Meinwald Y. С, Bottomley С. G.,
Marti n P. W., Tetrahedron Letters, 1960 (24), 13].
со2сна
со2сн3
,сн
согснч
CHjOjjC
^v^>« __rr
С — Н Реакция
Дн лкса—Аль дера
кА
со2снэ
Ц^кс-i
со,сн<
со2сн3
Элиминирование с образованием промежуточного продукта А совершенно анаД°*£?Г
но более общей реакции превращения 1,2-дибромидов в алкены под действием I" 11<*Ь
стр. 494 и ел.).
136
ГЛАВ А 5
В некоторых случаях промежуточный о-хинодиметан циклизуется с образованием
производного бензциклобутена быстрее, чем протекает реакция димеризации, как, например,
f^Tf^ Nal Г^>1 Г
ЧА
СНЕгч
Nal
ДМФА
Оп
■Вг
Но даже в этом случае промежуточный продукт может быть зафиксирован
образованием аддукта при проведении реакции в присутствии активного диенофила [4].
Задача 79.
(79.1) Приведите механизм следующей реакции:
соос2н5
CH2N2
COOC^g
осн,
(79.2) Установите строение продукта реакции и приведите механизм
следующей реакции:
oJV°H *Ь
сэН10О5
А
тл гЯ~т^€т №ще& обсуждение реакций диазометана, приводящих к расширению цикла,
м " »£% т е Р т Р"» в книге «Новые методы препаративной органической химии», ИЛ,
М., 1950, стр. 91).
63, lisoiiJJoii п *z s с h А" С z а р р Е" Вег" 63, 566 (1930); А г n d * F" Ber-'
он— О ОВ~0 ОН...О
^1 ^^«ни^ VtV^n
-щ\
ОН-О
II
сос2н5
-N,
ос2н5
°-~CHfN2
ОН-0
II
сос2н5
CH2U=N
он—о
II
:-оед
/^пКтп?авило' Диазометан не метилирует хедатированную гидроксильную группу.
(79.2) [Yamada К., Hirata У., Bull. Soc. Chim. Japan, 31, 550 (1958)>
СМЕШАННЫЕ РЕАКЦИИ
137
О
\l -осн3 _ *
CH2N2> 0=(Т CHaN = N
о
Na—CH2^-0L^OCH
9/)/ч"осн
осн,
осн,
осн.
о о
Задача 80. Нижеприведенные реакции протекают с промежуточным-
образованием дегидробензола, что показано экспериментами с 14С (отмечен
звездочкой). Приведите последовательность реакций распада данного
соединения, па основании котордй можно было бы установить распределение 14С
в продукте реакции.
С1 * NH2 NH2
NaNH3
Л/
L\/J
Л
I II + II I
Ответ [R о Ъ е г t в 3-D., Semenov D. A., Simmons Н. Е., Jr., Car 1 -
smith L. A., J- Am. Chem. Soc, 78, 601 (1956)].
CI NH2 Щ OH OH
I* I I 1 I
*/4 HNOa
/\
HaO,
/\
H2
/\
KECr207
нагревание I 11 Ni 1'
f ^ NaN3, HC1 */4|
I (реакция I ми HN3, H2SO4
\y Шмидта) \/ "**a
*/2
COOH
*/4
Реакция Шмидта H2N
*/4 */4 */2
~i i-NH2-|-C02
~ 50%
активности
KMnOj
*v 60% актив-
НОСТИ
*/4 */4
0% ~ 50%
активности активности
HK3
HOOC COOH
v
/138 ГЛАВА Б
Предполагали, что распределение UC в анилине будет таким, как указано в
формуле А. Однако измерение радиоактивности продуктов расщепления отчетливо показало,
что такое предположение ошибочно.
Задача 81. Приведите механизмы следующих реакций:
(81.1) н . СОСНз
|+СН3С0С1 * С12Н180 - -»
\/':\У А нагревание
тт
н
н
(81.2)
ОН
250°
?
6 час
РзС\У\уЛУСРз
cf3
Я + ....
I \S р3с/^^\ср3
CF3
Ответ.
(£2J)(Baddeley G-, Heston В. С, Rosburn J. W,, J. Chem. Soc,
i960, 4713; 1961, 3828).
О
II
CRX-^CIAICL
It A
+ CH,CHO
00-00
>^н
снэс+
-н+ J| + h +
СМЕШАННЫЕ РЕАКЦИИ
139
If
н3с^.с^о
о—н
H,C
+■ нгО
н Wh+
Первой стадией является дегидрирование декалина в Д9-окталин, который затем
подвергается ацнлированию по Фриделю — Крафтсу- Высказано также предположение [5],
1 что первой стадией превращения окталина будет реакция с образованием
сня
который далее перегруппировывается в соединение А.
(81.2) [К г е s р а п С. G., McKusick В. С, Cairns Т. L., J. Am. Chem.
Soc, 83, 3428 (1961)].
О
^--rf-^
Р
„J - /О
I A
CF,
CF,C=CCF,
CF3 CF3
F,C
F,C
CF,
CF,
CF3C^CCF;
-H,
F,C
FaC
l^-^V^
CF,
CF.
Гексайторбутин-2 - очень активный диенофил [в], способный реагироватьдаже
с-авоматичНими соединениями. Такая реакция приводит к мостиковым структурам
5ипГ А Промежуточное образование соединения А в этой реакции показано выделением
(наряду с другими соединениями) продукта В.
РзС~~гГСРз
/о
Нагревание
ОС
+ нс=сн
140
ГЛАВА 5
Наблюдалось [7], что 1,5-присоединение к мостиковой системе типа А приводит
к соединениям, аналогичным по своей структуре соединению Б. Перегруппировка
приводит к менее напряженной структуре. Механизм перегруппировки неизвестен, но это
не удивительно, по крайней мере ретроспективно, если принять во внимание жесткие
условия проведения реакции. В последней стадии дегидрирование приводит к
стабильному ароматическому соединению.
Задача 82. Приведите механизмы следующих реакций:
(82.1) NH2 CN
N jj CeH6SOBCl, ^ jf -^
j || нагревание \
C6H/V4NH2 вшчп™ C6H!/VXNH2
(82.2) SH
АЛ/С°Н6 NC.N C6HB
N | у С1СН2СООН, водн. NaHCOs, j \\
| l шшнчсние j|
W4c6H5 h2n/V4c6ii5
Ответ.
(Ш)[Тау1ог Е. С, Jefford С. W., Cheng С. С, J. Am. Chem. Soc.
83, 1261 (1961)].
nh„ nh,2 ,:kh2
\ *
A2
CN ON 9N
,X„ -и* At.
И i ~* M I ^— II Г
-^5 N NH2 C^^N^SN^j CgH^ X N=C = N.H
Первая стадия не обязательно должна протекать через оксим А, так как тот же
самый продукт может получиться при атаке CeH5S02Cl по кислороду нитрозогруппы
с одновременным или последующим отщеплением протона от одной из аминогрупп.
СМЕШАННЫЕ РЕАКЦИИ
141
(82.2) IT a v 1 о г Б. С, Knopf R. J., С о gl i a no J. A., Barton 3. W.,
Pleiderer W., J. Am. Chem. Soc, 82, 6058 (I960)].
s~ sch2cooh
C«H5 NaHCQ3_ N'^N^^f^ clCHaCOOH П'^у'1*^*'*
CfiH5
I
OH-
SCH2COOH
SCH2C0011
"3LX2C
HCOOH +
H,N^N^CeH5
1
HO'
+ ~SCH9C00H
Легкость протекания реакции даже в таких очень мягких основных условиях
^ -существенно зависит от способности циклической системы стабилизировать промежутоя-
, но образующийся апион путем дслокализации заряда па электроотрицательных атомах
& и от наличия групп, которые могут необратимо отщепляться. Как было найдено, пири-
S мидиновый цикл, сконденсированный с другой циклической системой, которая помогает
i стабилизировать образующийся анион, может подвергаться расщеплению такого типа.
Задача 83. Приведите механизмы следующих реакций:
(83.1)
(83.2)
ел
6и5
СОСН3
н,о +
о ^о
С6Н5
и w
чО'
соон
сн.
Z—Л-с—сн.
1. CH3MgX
2- НВг
142 ГЛ АВ А 5
(снз)зс/С=С\н ^ «ялс-сна-свдсад,
СН3
- а..
АеВг . Вг
(83." ' " - '
с
Ответ.
(S3.J) [К о г t е F-, В и с h е 1 К. Н„ Angew. Chem., 71, 709 (1959)].
\У f* сн3
^/JK^o ссн5/Ч0/Ч0.-' с6н,Ло\о^нз0 +
хк. сн,
I
с=о
6П5
, , с=о
СсН,' ОН |
он
г-<СООН -но ^cooii I соон
ер
Приведенный в литературе альтернативный механизм [&] кажется менее вероятным.
(83.2) [J u I i а М., Julia S., Guegan R., С. R., 248., 820 (1959)].
n Br~N +H
Л Ч\ Л °MgX нвг f f0¥L V TS
)
CHgMgX
сн3 cH; CHi
Эта реакция может служить примером перегруппировки производных циклопро-
пилкарбинола в производные аллилкарбинола [9]. Образовавшийся галогенид можно
превратить в реактив Гриньяра и повторно использовать в данной реакции. Таким путем
можно построить цепь, состоящую из изопрсноидных единиц, хотя она всегда содержит
на один атом углерода (на конце цепи) больше, чем действительные терпены [10].
СМЕШАННЫЕ РЕАКЦИИ
14а
(88.3) [Puterbaugh W. Н., N е w m a n M. S,, J. Am. Chem. Soc, 81 1611
(1959)].
н,с^ ^с—с'
" н.С --сн,
С].
-CI
С1 СН,
няс^ ^сн:3н сн3
■-*■ нас
Н,С'
>,с—с—сен,
н I Г '
С1 СНЛ
, в
Jc,-
сн,
сн.
I
н,с— с—сн—сн— с— сн,
fill'
сн3С1 сн3 а
Было высказано предположение, что значительное стерическое экранирование
в ионе А препятствует сольватации и дестабилизирует его по сравнению с
перегруппированным ионом Б, в котором положительно .заряженный атом углерода более доступен.
(83.4) (Berson J. A., in «Molecular Rearrangements», de Mayo P., ed.t Vol. I,
Wiley-Interscience, N. Y., 1963, pp. 213 ff).
Br AgBr
BrAgEr
Задача 84. Приведите механизмы следующих аномальных реакции
Гриньяра:
(84.1) СНз
CHsMgl НвО I „ „
С6Н5СООС(С6Н5)3 ► > CeHsCOOH + СвНь-С-СвНв
I
СбН5
(84.2) /СН3
Г л С CeH5MgEr НйО
н3с -^ у— сооснсн2свн5 > =-
13 ^6П5
/у
-> н3с -^ \ Ч- соон+с6н6+сен,
(84.3)
6сн=снс6н6
СНз
CeH5CHaMgCI H2O СН2ОП
снго > > /\/
\*
144 Г Л А В А 5
(84.4)
С=0 m/w/rz-CjHgMgCl С=ГО
ОСН.
Ответ (Kharasch M. S., Reinmuth О., Grignard Reactions of Nonmetallic
■Substances, Prentice-Hall, Englewood Cliff, N. J., 1954; см. также [11—13]).
Строение реактивов Гриньяра очень сложное. Механизм их реакций с
карбонильной группой также очень сложеп, поскольку может включать различные сильносольвати-
рованные молекулярные комплексы. Реакция присоединения протекает через циклическое
шестичленное переходное состояние, включающее две молекулы этого реактива.
Аналогичный механизм с участием одной молекулы реактива Гриньяра можно принять для
часто наблюдаемого 1,4-присоединения к а ,(}-непрсдельным соединениям.
0Л у, -
Д1„Х
,„ __ -с-» л У
OMgX
К
xm/J*c^ хм^с^ но °^
Чс^с- ^с-с~ ^г/С-н
На практике строение продуктов реакции можно предсказать, условно принимая дта
реактивов Гриньяра строение R~MgX+.
препеммр™' S™pHe Н6 ДаЮТ ПР°ДУКТ0В' ожидаемых на основании такого простого
дК ™™; ПОД пазванием «аномальные реакции Гриньяра». Причины, oupe-
SSroSm^SSS6 РеаКЦИИ' МОГ^Т бЫ№ Различными' Е в том числе стерические
ных ^агентах иротекания нормальной реакции и особые электронные эффекты в
исходное ^ш1м№*н£а\? I £ R"' S \Р eJ s * е * п р- О- S h i v e r s J. С, J. Am. Chem.
*опШЫ?У19Л1 ? VrtaSCHhnMWS-l RVn,muth °" GrignMd factions of
nonmetallic Substances, Prentice-Hall, Englewood Cliff, N. J., 1954, pp. 567 ff]
Доказа^льгТт°^РеЛ11Ц«а М°ЖН° "P^*0™* Различные механизмы, но экспериментальные
^z^:^r:z^r:,z^сделать выбор между ™-' °«**^. ^
СМЕШАННЫЕ РЕАКЦИИ 145
^•о— с—сйн
/ \
И;А CGH5
3
в115
,OMgI
с6н3с^
I
+ с6н5с—cchs
C.H*
сн-
^S^w-1 ^
vo*-c—с6н5
СгНг
/
OMgCH3
;СН5СЦ
СН,Мп1
[ —ас6н5Ь
о
Второй путь имеет апалогии в случае пространственно затрудненных кислот
_/
сш о
* Н3С-
\=?
С—О —С4Н9-к
CeH6Mgl H20
/
СН,
■? Н3С —^ Л COOH+H-GiHgl,
\ня
~i3 хСНз
а также в реакциях алкилирования трифенилметилгалогенидов реактивами Гриньяра.
Тем не менее оба эти механизма основаны на стерических затруднениях при замещении
у четвертичного атома углерода как реакционного центра.
Альтерпативный механизм, сходный с предложенным для
трифенилметилгалогенидов, будет следующим:
о /г£х?
н^ /СбН5
«c^q>!_c--cGH5 —
\
C-1I,
сен5С^
OMgX^
О
с6н5
+ +c^-crhs C*W- н.с-с-свн.
\
С£Н,
поскольку Mg-соль, присутствующая в реактиве, способна реагировать как кислота Льюиса,
(84-2) (Kharasch M. S.t Reinmuth О., Grignard Reactions of Nonmetallic
Substances, Prentice-Hall, Englewood Cliff, N. J-, 1954).
A
Br
I
снг
// YS C —O* —/H -*- Mes—COMgBr +- СбН5СН=СНС6Н5 + C6H<
H,C
CH<
CRH
'6n5
CH,
csh3
■MB.
\s
// \N C^-X^ /^ • -*■ McsCOMgC6H5 + C6HsC-5-CHC6H5
сн.
II CEHC
s^l
(с.н,
Br
10—374
т
ГЛАВА 5
Как и в ответе (84.1), можно предложить два механизма без доказательства. Однако
второй из них подтверждается аналогиями.
(84.3) [S i e g е 1 S-, В о у е г W. M., Jay R. R., J. Am. Chem. Soc, 73, 3237
(1951); Kharasch M. S., Reinmuth 0., Grignard Reactions of Nonmetallic
Substances, Prentice-Hall, Englewood Cliff, N- J., 1954, pp. U33ff].
CH2 — MgCl
н—c=o
I
H
н—в -i
CH2—OMgCI
в
- a
CH-
CH2OMgCt
.. Это один из классических примеров аномальной реакции Гриньяра. Необходимо
ваметнть, что другие альдегиды (кроме формальдегида) реагируют по двум направлениям:
в кольцо и в боковую цепь.
CH2MgCl О
\\
■J* C1H3CH
\/
он
CH2CHCH3
V
он
уч/снЕснсн3
Ч/\
СНСНз
он
Механизм, вероятно, включает превращение промежуточного продукта А в новый
.CH3MgX
- 1: и
■CH2OMgX
aCH2,v
CH2OMgX
сн,
Me—X
/
- a
сн5—щ
I
о
сн
X
СМЕШАННЫЕ РЕАКЦИИ
147
;юньяроподобный реагент, хотя не ясно, почему атака в кольце отсутствует в случае
трмальдегида.
{84.4) [Fuson R. С, Friedlander W. S., J. Am. Chem. Soc, 75, 5410
953)].
Mgxa
К у I
^Ш^Ьоснз J^yQ-l-^-
У
OCH3
(CH3l3C^-MgCl
»
&^i
Mgx2
Задача 85. Объясните, почему реакции А и Б в каждом случае дают
различные продукты.
(85*1) _|_ Нагревание * jj 1
A CeHsCOCHNa+CeHsNHe + CeHbNHaBi -> |
4/N^Nr н
СНз СН3
| | Нагревание
Б CeHsCOCHNa + CeHsNH+CeHeNHaBr- *
f\ /СвН5 /\
(85.2)
/V
I
СНз
п
^4N^CfiH5
СНЭ
соон
К ,соон
Vi HBr- толуол
|R=HI
10*
*4В ГЛАВА 5
соон
н^^соон
толуол
(R = CH,)
(85.3) С3н6
I
А С6НБ—С—С0С1
I
сн.
н5с2 с2н5
(CH3)aCd C2HBLi
* * С6Н5—С - С - СН3
I I
СН3ОН
^п*. н5са с2н6
б ед-с-сош > > ед-А-А-сна
I I
сн> Н3С ОН
Соединения, полученные по реакции А и Б, являются диастерсомерами.
Ответ.
(85.1) [BladesCE., WildsA.L., J. Org. Chera,, 21, 1013 (1956)].
А О
H+ /*+ м„ II H
C„H,CCH— K=N * c6H5CCH2^N=N . ~N% C6HsC-CHj-SNCeHs
V. I H
CflHj, —NHj
shAn^V
CfiH
5NV.
-1Г
-HjOl C6H5NH2
Co"s-C-CH,NH2C6H5
СМЕШАННЫЕ РЕАКЦИИ
149
сн.
C6H5CCHN^N — — CeH5cCH2-^NC6Hc
H
CH,
Различие между этими реакциями можно объяснить следующим образом. В случае
анилина происходит однозначное превращение в иминопроизводное 1 с последующей цикли-
вацпей. Однако для алкиланилинов имин, аналогичный 1, не может образоваться. Поэто-
J. му для продукта присоединения амина по карбонильной группе равновесие будет менее
благоприятным, вследствие чего паблюдается альтернативная циклизация с
промежуточным образованием амина 2. Так как соединение 3 не может образовать имип, то оно цикли-
вуется как по вышеприведенной схеме, так и по схеме
+011,
сбн5— с—сн2—N:
ch3n: сн,
сн. ,
сн3
Н Г II
-C6H5NCH3 6 5
СО
сн,
Повышение "выхода 3-фенилиндола с увеличением размера N-алкильной группы
иожет уточнить выбор между этими схемами.
Эта реакция очень близка синтезу индолов по Бишлеру, в котором в качестве исход-
р ного соединения применяют не диазокетон, а а-галогенацетофеноп.
(85.2) IV a u g h a n W. R.t S с h о е n t h а 1 е г А. С, J. Аш. Chem. Soc, 79,
•5777 (1957)].
150
ГЛАВА 5
(At
R = H
CO ОН
Реакция А протекает как обычное присоединение. В неполярных растворителях,
как известно, переходное состояние включает несколько молекул галогеноводорода, и,
вероятно, присоединение брома протекает более или менее согласованно с протонированием.
Поэтому в реакции не наблюдается образования вторичного карбониевого иона. В
реакции Б образующийся третичный карбокатион может иметь достаточное время жизни, чтобы
произошла скелетная перегруппировка. Возможно также, что метильпая группа до
некоторой степени создает стерические препятствия приближению брома, что также приводит
к увеличению времени, необходимого для протекания перегруппировки.
(85.3) (G a u 1 t У., F е 1 k i n H., Bull. soc. chim. France, I960* 1342; И л и -
ел Э., Стереохимия соединений углерода, изд-во «Мир», М., 1965).
f2H5 о
с«н
c2hsv
6" 5
. , ^^— св-н5с — cch,s=
-^<:cH5)2cd J
Г2 5 гтз
сн3
СаНвС_сОС1
I
сн
I //
сбн5с—сс2н5
сн3
н,с
о"\С!Н5
Li +
СдНя
Li +
С2Н5
н.с
с,н.
С о Я,
СМЕШАННЫЕ РЕАКЦИИ 151
Если новый асимметрический центр образуется по соседству с уже имеющимся, то
конфигурация продукта в большей или меньшей степени определяется конфигурацией
' исходного вещества. Согласно правилу Крама Ц4], для присоединения по карбонильной
группе конфигурацию продукта можно предсказать, принимая ориентацию карбонила
между двумя наименьшими* заместителями (М, S) у асимметрического атома углерода
и принимая, что подходящий реагент (R) приближается со стороны наименьшей группы (S)
В данном примере можно принять, что фенил > этил > метил.
Задача 86.
(86.1) Приведите механизм следующей реакции (цифры в круглых
скобках обозначают полные атомные проценты 180 с точностью ±0,01/о):
С6Н5С-0-0-С-СеН5 + Р(СеН5)з
II II
о* о*
А(3,42%)
С6Н5С - О - С - СеН6-НСвН5)3Р ~>0
II 11
о о
Б(3,16%)
NH3
(0,20%)
CeH5CONHa-f CeH5COOH
(1,16%) - (1,99%)
(Заметим, что природная концентрация 180 составляет 0,2 ат.%.)^
(86.2) Используя приведенные выше данные, установите, какой атом
кислорода отщепляется PR3 в следующей реакции:
PR, NHs
Ч_10СНч > Ангидрид . >
/ ■ кислоты
02N-<f \-C—О-О-С-
О* JO
> OoN — <
/
(1,07%)
j-ICONHa
(1,12%)
(Заметим, что процент 180 в данном случае обозначает избыток по сравнению
с содержанием природного изотопа 180.)
Ответ. „ „ тт <■ * „. » v т. Am.
(86.1) [Greenbaum M. A., Denney D. В., Hoffman А. К., J. л
Chem. Soc, 78, 2563 (1956)].
152
ГЛ АВ А 5
r^JESST^%L18° (В атомных процентах) в исходной перекиси приведено на схеме А.
Содержание этого изотопа в нерекисной группе отвечает природному содержанию
0,20 0,20
С6Н6—С-О—0-С-С6Н5
о
1,51
О
1,51
Содержание ^О в фосфиноксиде совпадает с природным и указывает, что атом киглп-
рода отщепляется из перекисной группы. Кислород-18 (ЗД6 ат о™) Петидшде может
распределяться различными способами (Б, - Б4, см.таб л иду) Распределено в
продуктах аммонолиза в предположении, что атака* аммиаком протекает с равной вшяш
Таблица
Способ
распределения
Б,
Ба
Б3
Б4
Метка
О* О*
II II
-С—О-С-
0* О
1 »
—С—0*—С—
о* о*/2
-с-о*/2-с-
о*/з 0*/3
II 1)
-С-0—C-
*/3
Распределение
О1-48 о1-48
—с-о—с—
0,20
О1-48 о0'20
11 я
—с—о—с—
1,48
О1-48 О0'64
1 "
—с—о—с—
0,84
о1.05 о1,05
—с—о—с—
1,05
Содержание *Ю,
ат.%
кислота
1,68
2,32
2,00
2,10
l
амид
1,48
0,84
1,16
1,05
Очень хорошее совпадение между наблюдаемыми значениями и предсказанными для
случая Б3 указывает па то, что именно этот вариант реализуется в действительности.
Механизм, удовлетворяющий этим данным, можно представить схемой
с6н5-с
—&- о—с—сдн,—
*PR3
J6n5"
о о о-
II II I
r3p-o— сс6н5+ ссн5с—о" ++ с6н5с=о
С6Н5С~?~ССбН5 + К3Р-*0
СМЕШАННЫЕ РЕАКЦИИ
15а
Промежуточный продукт, вероятно, существует в виде ионной пары, поскольку
\использовался неполярпый растворитель. Оба атома кислорода бензоат-иопа
эквиваленты, что приводит к выравниванию распределения 1вО. Реакция замещения, приводящая
конечному продукту, возможно, протекает по уравнению
О
|]
с6н5со-
(0
1 +
-~~С — OPR3
1
С6Н5
0 0"л О О
II h>+ " и
-*" С6Н5С-° — С~ОРК3 -+ С6Н5СОСС6Н5
1 +
■ , С6Н5 +
ЙЯР—О"
{86.2) [D ев n e у D. В., Greenbaum M. A., J. Am. Chem. Soc.,79, 979 (1957)].
Эта несимметричная перекись имеет два неэквивалентных атома кислорода — аир,
которые могут подвергаться атаке фосфином. Такая атака приводит к ионным парам.
Я конечным продуктам, приведенным ниже
О
o2n—с6н4~ с —о—о—с—с6н4осн3
РЯу^ ^ PR,
^Х*« - Атака /Г - Атака
О
II +
0,2 N— С6Н4— С —OPR3
- -С-с6н4осн3
о
72
02N—С0Н4— С' -
0,
+. */2
О
R3P—ОС—С6Н4ОСН.
О О
II II
o2n—сен^—с—о— с—с6н4осн3
Jnh3
*/2
02N—С.6Н^— С — О— с — С,.!^ ОСИ
*/2
1 NH,
*/2
02N—С6Н4—CNHa
02N—C6H4—CKH,
Если первоначальная атака протекает по а-кислородному атому, то перераспределения
метки не должно происходить. Реакция по ^-кислородному атому должна протекать через
симметричный бензоат-ион, содержащий 180, что вызывает перераспределение метки
между карбонильным и ангидридным атомами кислорода. Наблюдаемое содержание 1еО
в амиде показывает, что метка сохраняется в карбонильной группе и, таким образом,
'осуществляется сс-атака.
.'*_" . 154
ГЛ ABA 5
Так как ионная пара, полученная при атаке по «-положению, термодинамически
менее стабильна (анион более слабой кислоты; положительный заряд находится рядом
с фрагментом! имеющим NOs-rpynny), чем ожидаемая при атаке по р-положению,
направление реакции должно определяться поляризацией исходной перекиси, а-Кислородный
атом более положителен, чем 6-атом кислорода, вследствие электроноакцепторного
эффекта нитрогруппы и поэтому является более реакционноспособным по отношению к фосфину.
Задача 87. При нагревании с ацетатом калия в этиловом спирте .при 60°
соединения 1, 2 и 3 циклизуются с образованием Д2-оксаэолина(4). цис-Изо-
меры 5, 6 и 7 подвергаются сольволизу с образованием смеси продуктов эли-
минирования и замещения.
OMs
H
HNCOCeH5
<ХХУсл
OMs
NHCOC6H5 тРеп- C4H9-
OMs
NHCOCrH
Gn5
R= h(i)
R = »i^m-C4H9 (2,3)
mpem-C^HjJ
OMs
HNCOC6H5
Ms = CH3S02—
Константы скорости первого (или псевдопервого) порядка будут
следующими (10%, сек'1):
1 252 5 0,69
2 6280 6 0,69
76 7 2,95
(87,1) Объясните относительную реакционную способность соединений
1, 2 и 3, а также ^iwr-соединений.
СМЕШАННЫЕ РЕАКЦИИ
155
(87.2) Обсудите на основании количественных данных относительную
'важность возможных промежуточных конформаций в реакции соединения 1
но сравнению с реакциями соединений 2 и 3.
Ответ [Sicher J., Tichy M., Sipos F., Pankova M., Coll. Czech
Chem- Commtin., 26, 2418 (1961); Proc. Chem. Soc, 1960,3841,
(57-?) Согласованный процесс замещения, приводящий к оксаэолину, требует транс-
.'-антипараллельного расположения реагирующих групп (А). Проблема заключается в
согласовании этого требования с ограничением, накладываемым стремлением объемистой
. mpem-бутильной группы занять экваториальное положение [15, 16].
OMs
СЙН,
трет-С^Щ
ОМ:
Ориентация типа А имеется в соединении 2, и вследствие стерического напряжения,
обусловленного двумя аксиальными группами, дестабилизирующими основное состояние,
реакция протекает с максимальной скоростью. С другой стороны, конформация Б для
соединения 3 является слишком напряженной, чтобы ее можно было рассматривать
в качестве промежуточной для данной реакции. Тем пе менее реакция протекает с
образованием продукта, возникающего по согласованному механизму и с такой скоростью,
которая по сравнению со скоростями цые-соединений указывает на значительное участие
соседней группы в переходном состоянии. Альтернативным предполжеыием является
допущение конформаций скрученной ванны (твист-конформация) (Б) [13,17], которая
позволяет реагирующим группам принять необходимую геометрию, но в то же время позволяет
«грет-бутильной группе занимать экваториальное, а не аксиальное положение.
трет-С^Ш^
трет-С^Щ
Н
/С6Н5
OMs
Родоначальное соединение 1 может реагировать или через трскс-диаксиальныи
конформер (Г), или через твист-конформацию (Б). Можно показать, что процесс протекает
по обоим 'направлениям (см, 87.2). В любом случае диэкваториальное основное состояние
соединения 1 стабильнее, чем диаксиальное для соединения 2, и реакция протекает
медленнее.
цыс-Сосдинения не могут принять конформаций А и при отсутствии участия
соседней группы сольволизуются медленнее по обычному механизму.
156
ГЛ А В А 5
{87.2). Конформационпо подвижное производное циклогексана 1 реагирует через
промежуточные конформации Г или Е, которые находятся в равновесии с диэкваториаль-
вой формой Д.
н—*Г || G 5 HN"
.сгн
6"Б
'НО
н
Продукт Про ду к т
реакции реакции
Наблюдаемая скорость v определяется выражением f 15¥ 16]
^ „ ЕЛ „ [Б]
^-W *Е~7дГ
Так как соединение Д значительно стабильнее, чем соединение Г или Е, то [Д] яй 1_
Поэтому [Г] ~ Кт [1] и [Е] ~ КЕ [I]. Поэтому
А: [1] = Аг^г [1J4-fcEiTE [1] или k^kTKr-\-kKKE.
В качестве очень грубого приближения можно принять, что скорость реакции через
образование твист-формы Е для соединения 1 будет равна скорости реакции соединения 3,
поскольку в обоих случаях происходит конверсия диэкваториальной конформации
в диаксиальную и можно приближенно считать, что ориентация mpem-бутпльной
группы не изменяется при образовании переходного состояния для соединения 3. Поэтому,
принимая скорость, наблюдаемую для соединения 3, равной КеК&, имеем
252*Ю-« = &гЛГг + 76.10
кгКг = П2- 10-е
^Г^г _ 172-10-е
*Е#Е 76-10-6
2,4.
Таким образом, реакция протекает несколько в большей степени через
производное, имеющее форму кресла, чем твист-форму, хотя оба эти пути имеют место.
ЛИТЕРА ТУРА
1*.С оборовский Л. 3., Якубович А. Я., Синтезы отравляющих веществ,
Оборонгнз, М., 1939, стр. 75.
2. F e i st F., Вег., 26, 747 (1893).
3. 6 о u I d E. S., Mechanism and Structure in Organic Chemistry, Holt, N.Y., 1959.
* Дополнения переводчика.
СМЕШАННЫЕ РЕАКЦИИ
157
4 Cava M P. D e a n a A.A., Muth К., J- Am. Chem. Soc, 81, 6458 (1959).
5 Ahmad M.S., BaddeLey G., Heston B. G., R о s Ь u г n J. W., Proc.
Chem. Soc, 1959, 395. . .
6. A d 1 e г К., in «Newer Methods of Preparative Organic Chemistry», Wiley-Interscience,
7 B*l o'm q u lit А. Т., M e i n w a 1 d Y. C, J. Am. Chem. Soc, 81, 667 (1959).
8. L а с е у R. N.. J. Chem. Soc, 1954, 822.
9 Roberts J.D,Mazur R. H., J- Am. Chem. Soc, 73, 2509 (1951).
i- 40. J u 1 i a M., Descoios C, Bull, soc chim. France, 1962, 1939.
; -11*.Синтетические методы в области металлоорганических соединений, цод ред. А. Н.
Несмеянова и К. А. Кочешкова, справочник но магнийорганическим соединениям,
том. 1-3, изд. АН СССР, М., 1950.
12*.Рупге Ф., Магнийорганические соединения, Л., 1937.
1 43*.R e u t о v О. A., Beletskaya I. P., Reaction Mechanisms of Organometallic
Compounds, North-Holland Puhl. Co., Amsterdam, 1968.
-14. Cram D. J., А Ь d Elhafez F. A., J. Am. Chem. Soc, 74, 5828 (1952).
15. winstein S., H о 1 n e s s N. J., J. Am. Chem. Soc, 77, 5562 (1955).
16. Eliel E. L.t J. Chem. Educ, 37, 126 (1960).
17. Илиел Э., Стереохимия соединений углерода, изд-во «Мир», М., 1965.
ГЛАВА 6
Реакции попденсации и родственные реакции
В данной главе приведены задачи, включающие реакции резонансно
стабилизированных карбанионов (или эквивалентных им частиц) с электро-
. фильным центром. При решении этих задач можно рекомендовать пособия
и монографии [1—31.
Задача 88. Предскажите положение, в которое будет вступать галоген»
при реакции каждого из приведенных соединений с 1 молем брома в
присутствии основания.
° s\ °
11 II II
СН3ССИ2СН3 С1СН2ССН3
: А уЧснз
о
Б
лака 0твет (G о u I d E. S., Mechanism and Structure in Organic Chemistry, Holt, N. Y.„
1959, pp. 372—384).
О О
BrCH3CCH2CH3 I CICH-C-CH3
A A A i
BK YNciIs Вг В
о
Б
Галогенирование кетонов, катализируемое основаниями, ебычпо протекает черев
медленное образование енолят-аниона путем отщепления сс-атома водорода с последующей
быстрой реакцией аниона с галогеном. Так,
О о- о
" _ Медленно
CH,CCHJ-OH~ w СН,С=СН2 - - > СНаССН,Вг
3 3 а г Быстро * z
В случае несимметричных молекул строение образующегося продукта будет
определяться стабильностью либо промежуточного аниона, либо переходного состояния,
приводящего к аниону. Поэтому замещение при галогенировании будет более благоприятно-
в РЯДУ первичный > вторичный > третичный, что находится в соответствии с изменением
стабильности анионов (отметим, что СН3 является донором электронов по сравнению с Н)-
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ
159
{'Замещение электронов таким акцептором, как атом галогена, способствует стабилизации
аниона и приводит к полигалогенированию одного атома углерода. Так, например,
йодоформенная реакция зависит от обоих этих эффектов. Первоначально иодируется
метильная группа, поскольку предпочтительно протекает замещение у первичного атома,
ja затем вновь иодируется тот же атом угяерода из-за стабилизации аниона уже
присутствующим атомом иода.
Задача 89. Объясните, почему для протекания бензоиновой конденсации
^необходим именно цианид-ион.
ОН О
СНО
X/
KCN
3>
ОН-
I
сн—с
\/
X/
Ответ (Gould Е. С, Mechanism and Structure in Organic Chemistry, Holt, N. Y.,
4959, pp. 394 ff.; А й д В., Бак И. С, в книге «Органические реакции», сб. 4, ИЛ, М.,
Й951, стр. 229).
с6н5 с=£о
-CN
н
с6н._с-о-
CN
ОН
— W£-c»
он
CfiH4C=C=N"
ОН
I н
ОН
I н
C6ns9 £С6Н5 -^ С6Н5С—С—С6Н5
CN О
А.
!Ь
i i
CN О1
V
с\ I
*CN ОН
}
о
' II
С6Н5ССНС6Н5
он
Вследствие способности CN-группы стабилизировать соседний отрицательный заряд
"путем резонансного участия циангидрин, образующийся в результате присоединения
HCN к альдегиду, может отщеплять протон с образованием аниона А. Аниоп А может
,далее присоединять вторую молекулу альдегида; эта стадия очень близка альдольпой
: конденсации.
В бензоиновую конденсацию обычно вступают альдегиды, не имеющие а-водородного
: атома, так как в противном случае CN"-hoh будет достаточно сильным основанием, чтобы
вызвать протекание альдольной конденсации.
160
ГЛАВА fl
Задача 90. Приведите подходящие методы селективного метилирования.
(90.1)
Ответ.
{90.1) [Woodward R. В., Patchett A. A., Barton D. H. R ,
Ives D. A. J., К el 1 e у R. В., J. Am. Chem. Soc, 76, 2852 (1954)].
(H9OK
H
{90.2) (Шмугакович Дж., в книге «Успехи органической химии», сб. 4, изд-во
«Мир», М., 1966; см. также [4]).
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ1РЕАКЦИИ 161
Задача 91. Приведите механизмы следующих реакций:
(91.1)
I. ОН
2. НаО
^о
(91.2)
.^и
*1 ОН"
о А л
НООС ч% .О,
° вУ О
(91.3)
он
!. ОН" НООС
2. Н30'
соон
СН2ОН
I
с=о
]
СНСН9СООН
сн
н3с nch3
Ответ.
(91.1) [Woodward R. В., В г u t s с h у F. J-, В а е г Н., 3. Am. Chem.
Soc, 70, 4216 (1948)]. •
fl—374
162
ГЛАВА 6
It
0
°^
f
л.
о о
"ООС
Саптоновая
"О каслогя
НООС
(91.2) [Woodward R. В., К"о vac h E. G., J. Am. Chem. Soc, 72,1009 (1950)].
Br
Er
ff J>T^% CO OH
НО ОС
Er
U£P
ooc
Это нример реакции Фаворского [5]; однако, поскольку исходное соединение не
содержит а-во дородных атомов, эта реакция пе мошет протекать через обычное
промежуточное обравование циклопропанона [6].
(91.3) [С о п г о у Н., J. Am. Chem. Soc, 79, 1726 (1957)].
ноос^ ^о
ooc -о
тюс^ч ..о.
н—он
±us
.-'он н+
"ООС--
он-
ДЬгадро-а - инкро^гоКсннайая
кжугота
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ
163
I
HOOCs ,Х°Н во* "OOC
V
V
он
он
>Л^соо-
/н/
о^^сн^он
HoJv
н
COO"
о
9 нс=о
НОч^СН ОН" CHOH
^^coo- ^ >>А^соо-
,соон
Задача 92. Приведите механизмы следующих реакций:
(92.1)
0>v ^Оч^ -CIL
сн3о
1. NaOH, CH3OIi СНз°
2. HgO"1"
сн.
(сн2)4снон
СН30 о
сн,о он
(92.2)
Cfc
КМп04 H30J
'СООН
С1"
''Д/СООР
соон
он
К Мп 04 Н30'
» ■■'■'■
MgS04
ЕдинствеЕшый продукт реакции
СООН
ОН
11*
164 Г Л А В А в
Ответ.
(02.J) (В г i с h A. J., Musgrave О. Cti Rickards R./W.s Smith H.,
J. Chem. Soc.t 1959, 3146).
сн3о
och3 о ^0H
сн3о
осна °
сн^о
OCH, О
сн3о
OH
(CH2)4CHCH3
OH
Это трансаннулярная конденсация Кляйзена, приводящая к производному
нафталина. Замыкание цикла такого типа совершенно аналогично (по крайней мере формально)
наблюдаемому при биосинтезе полифенолов из ацетата [7] {см. также [8], стр. 462).
(92.2) (Si mouse n J. L-, The Terpenes, Vol. II, Cambridge University Press,
Cambridge, 1949, pp. 280—289; де Майо П., Терпеноиды, ИЛ, М., 1963).
КМп04
at
КМпОа
сигон
^Aj^CHO
КМпО^
Qt
^JS ^COOH
он
он
он
"" сн=
он
"ОН
он
КМпО,
КМр04 {*\
£
соон
соон
Окисление перманганатом включает реакцию
Зе- + 2Н20+МпО* —*• Мц03 + 40H",
и реакционная смесь в отсутствие буфера становится сильнощелочной. В данном случае
образование кислоты 3 (и соответствующего гликоля 1, который также выделен) не
является неожиданным. Однако в присутствии основания альдегид 2 подвергается
перегруппировке с образованием кетола 4. Это, возможно, связано с уменьшением напряжения
ликла. Дальнейшее окисление кетола приводит к кислоте 5.
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ^
165
Прибавление сульфата магния связывает щелочь в виде нерастворимого Mg (ОН)я
| позволяет поддерживать нейтральную реакцию среды. В этих условиях
перегруппировка не наблюдается.
Задача 93. Приведите механизмы следующих реакций:
1)
Нагревание
СЙО
+ СН2(СООН)2
О "
CaH6ONa
93.2) CeH5-CH = CHCOOCaH5 + CH3CH(COOCaH5fo >
Нейтрализация
> СвН5-СН-СН(С02СяН5)2
I
сн3снсо2с2н5
Ответ,
{93.1) (В е г к о { i С. Е., С г о m Ь i e L-, Proc. Chem. Soc, 1959, 400),
соон
^^^у
Н + СНо
ч
соон
^
<глг
о--н
с—он
=4
соон
^ -
о*Чн
О о
166
ГЛАВА 6
Можно предложить и другие пути перехода от соединения А к конечному продукту;
однако в общих чертах они подобны вышеприведенному.
(93.2) [Simamura О., Inamoto N., Suehiro Т., Bull. Chem. Soc.
Japan, 27, 221 (1954)].
CH3CH(C02C2Hs>2
C2RsO-
c6h5ch=chco.,c2h5
CH3.C(CO;jC2Hs)2 -,
Реакция Михаэля
CfiH5CI-I —CHCOjCjHs
J •
сн3с—с^ос2н5
C02C2H*
It
+ C6HsCH—C—CO£C2Hs CfiH5CH — CHC02C2Hs
со2с2н5 со2сгн3
с6н6сн—chco2csh5
• cH3ct^_c—oc-Hs
I Гч
со2с2н5 o~
Продукт нормальной конденсации Михаэля (А) имеет очень реакционноспособный,
сильно основный карбанион, стабилизированный только одной соседней карбонильной
группой. При перегруппировке в соединение Б образуется менее основный карбанион,
имеющий две соседние стабилизирующие группы. Большая стабильность соединения Б
в сильноосновной реакционной среде приводит к тому, что реакция смещается в сторону
его образования.
Механизм такой «аномальной реакции Михаэля» был подтвержден экспериментами
с мечеными атомами.
Задача 94. Приведите механизмы следующих реакций:
(94.1)
ОНО R
I ! II I огн
кон
11+
ОН О R
1 I! I пги
Сплавление
Ho/N/VV'Xoh
+ (СН3)2СНСН2СООН + (СН3)2СГ1СН2СН2ОН
4-
но/^ЛЛ^он
(94.2)
СНдСОО'
с=о
i<W=nu& СП3ОК
. ^.
загадим як трубка
,соон
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ
167
Ответ.
(МЛ) [Yates P., Stout G, H., I. Am. Chem. Soc, 80, 1691 (1958)].
Элиминирование включает миграцию двойной связи боковой цепи в ™*РЯ**™°°
* ' тложе™к карбонильной группе (группам), получающейся при кетонизации Ф^™^
* г^др?кси4 (гидроксилов). Образовавшаяся сопряженная система гидратируется но схеме
™Ж™ Мп^чля (V91 сто 392 и ел.), и получающийся оксикетон расщепляется по типу
^&U^^V\£Si*L^ с образованием изовалериановогс»-J^gJ. Посио»-
^применяются жесткие щелочные условия, то альдегид Д*ЗД^Ч^^
допросом гидрид-иона и образованием соответствующих кислоты и спяРта- ^™^00eTS не
норционнрование (реакция Канниццаро) хорошо изв сстно^£«"*g^^Сальдо ля.
пгт7пяют в конденсацию, обычно, если это возможно, наблюдается °0Раз0ваЕ"„„„^ггтгя
O^TV^i^oo6vLo^mm обратима, и в жестких условиях может накапливаться
продукт диспропорционирования.
168
ГЛАВА в
^.ВД'Й'Й Bossard "•• »»«*!»« I-. i
eger 0., Helv.
'^он
с=ю
COO" =
COO"
COOH
ную J!££^.IS!EE3^Ja№- ретроальдольное расщепление и повтор-
Задача 95. Приведите механизмы следующих реакций:
(95.1) /J^"CH2C°OCH3 CHaONa^ JL^COOCHs
N—<
•соосн
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ
(95.2)
NaNHs °,v \° /
Ответ.
{95.1) [van Tamelen Е. Е., Н i I d a h I G. Т., J. Am. Chem. Soc, 78, 4405
(1956)].
CCH—COOCH3
COOCH3-
^"~OCH3
COOCHg
"А ^СООСНд
COOCH3
Реакция протекает через стадию циклизации по Дикману с образованием соедине-
вия А с последующей обратной конденсацией Михаэля. При этом снимается напряжение»
имеющееся в бициклической структуре А. Возможно, соединение Б образуется не через А,
т. е. не при раскрытии циклопропанового кольца; однако, поскольку было показано (10],
что бициклические соединения этого типа могут образоваться при конденсации Дикмана
я они неустойчивы по отношению к основаниям, этот механизм кажется вполне
удовлетворительным. Соединения А и Б легко взаимопревращаемы, и, возможно, что в
действительности в растворе существует таутомерное равновесие.
{95.2) [В псЫ G., Hansen J. Н., К nuts on D., Roller E., J.Am.
Chem. Soc, 80, 5517 (1959)J.
c^1"
170
Г Л А В А в
Хотя получены довольно убедительные доказательства общей структуры продукта
реакции, положение карбонильных групп [син-(Б) или «штои-расположение (Л)J
однозначно не установлено. Можно полагать, что (шиш-форма более вероятна*, так как переходное
соединение, приводящее к антu-структуре, имеет более благоприятную геометрию для
перекрывания, а также наибольшее разделение отрицательно заряженных атомов
кислорода. С другой стороны, нет оснований предполагать, что в реакции осуществляется
термодинамический, а не кинетический контроль, т. е. направление реакции определяется
равновесием, а не соотношением скоростей. Следует указать, что продукт реакции
стабилен в щелочных условиях, и наблюдается только ионизация а-атомов водорода {показано
' ебменом на дейтерий). Однако и в этом случае более благоприятпьш будет образование
лшйк-формы из-за минимального диполь-дипольного отталкиванй карбонильных групп.
Задача 96. Предложите механизм приведенной ниже реакции:
СН„0
сн,о
20%-шй КОН
снн5он
оснч
снао
сн3о
К=-сн2сн2м(снз)5
осня
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ ГЕАКЦИИ
17
Ответ [Bentley К. W., Dominguez J., RingeJ. Г., J. Org. Chcm
22, 418 (1957)].
CH,0
CH30
CH.0
OCH;
ch,o
CHoO
СЦ30
-HCOOH
-«
OCH,
CI]3o
H,C
ch,o
Углеродный скелет продукта реакции уже имеется в исходном соединении, и необ
ходимы только раскрытие кольца диклогексенона при гидратации и ретроальдольна)
реакция. Отщепление альдегидной группы, возможно, обусловлено способностью мето
ксилированного бензольного цикла стабилизировать частичный отрицательный заря,
в а-положении, который и появляется при разложении соединения А.
Задача 97. Приведите механизм следующей конденсации:
N02
-I
/\
• Л/ V
I
С!
CH2CN
I
кон/сн3оп
N—О
11 I
/\/\/v
I
С1
172
ГЛАВА 6
Ответ [Davis R. В., Р i z z i n i L. C, J. Org. Chem., 25, 1884 (1960)1-
ssAH-a*
"OR
"Q + OH
JIU^H^'OR
ОД*
он
jhc6h3 _0H.
CN
CI
6*
hV
CCfiH
Г OR
6"5
iC—C6H3
N—О
CN
CI
CN
CI
N О
C^H,
CRH,
6n5
Эта реакция не зависит от природы заместителей в цикле фенилацетонитрила, но
очень легко тормозится при изменениях в кольце нитробензола, как, например,
NC
NOa CH2CN
I I
+ I ■
\
CH-<
кон
CH3OH
OCH3
NO,
-J
Она также зависит и от природы растворителя
NO, CHjjCN
I I
NC
\
сн
-У
+
f
С1
КОН
Пиридин
I
N02
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ
173
Задача 98. Приведите механизмы следующих превращений:
о
II
OCQ.H
(98.1)
e"fi
тр ст.—С4НВ ОК
i ■ ■ »■»
трет— С4НдОН
СбН5С'
о
А
(снэ)2соон
(98.2)
/4/SNHCeH5
он-
V\
\/
N0.
О
и
S0H
N = NCeH5
?WiUY a t e s P , Anderson CD., J. Am- Chem. Soc, 80, 1264 (1958);
«5. 2937 (196^; CI a r k e R L , Hunter W.T., J. Am. Chem. Soc, 80, 5304 (1958)].
OC4H$- mpcm
C6H5^ ^°
•
*• iO
CCH
6n5
C„H
o^s
174
ГЛАВА 6
С6Н5С
CfiH
6П5
I
О -О
сен
н+
соон
с6н5
В согласии с приводимой схемой лактон А был выделен в работе Кларка и Хантера.
Яйтс и Андерсон покакали, что при действии щелочей он превращается в соединение Б *.
Аналогичная реакция
■осс6н5
II
О
трет - С4Н9ОК
0 Л
11 Л
СООН
.2). [Cava M. P.. Blake С. Е., J. Am. Chem. Soc., 78, 5444 (1956)].
"он
I
OZ
.. н
—N—СН-
o о он-
Ж.
цон
нс£/ хо~
Свойства циклопрошшкетонов см. в работе [И].— Прим. перев.
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ
175
ч0
I
N—К— С6П,
А
»он
ROH
aS—0"\ n -■ /v ^S—-ОН
^^-С6П3 kA^N-
-С-Н-
I
о-
он
он
I
s=o
>CRH
'6П5
Эта реакция рассматривается как внутримолекулярное окисление —
восстановление, поскольку пара-изомер (Б) не реагирует.
0»N —
\:
А_ s _ NHC6H5
-/ * •
Однако однозначпыс доказательства в пользу этого механизма отсутствуют, и можно
предположить также другие схемы. Так, промежуточное соединение А, образование
которого представляется очень вероятным; может перегруппировываться с
внутримолекулярным переносом кислорода и затем — с образованием конечного продукта
ОН
I.
N—СН
б" 5
- 00
N—ССН
б"5
S.
£l
I ^ПТ4
I
НО
он
vi+
0>-w
\
с«н
0"5
он
о
II
S—ОН
ос:
N=N—СЙН.
176
ГЛАВА 6
ffiw™H° ПреДЛ0ЖИТЬ и другие "У™'как- например, по аналогии с реакцией ФаВот>™«-
го 16]; однако этот механизм менее вероятен из-за стеринеского напряже^иГ Р
Задача 99. Установите структуры продуктов, образующихся в
результате следующих реакций; ±«»уль-
\ /
(99.1) /;
л
-N'
>+CH3CH2COC1^ic9H,A
(99.2) <^
(99.3) <^
X
2. Н+, НаО
^0 + 2СНзСН2СОС1 1^5. СДЛ
./
2. На0
ЧП _L 9H2COC1 l- (C2H6)j,N
-^ СН2С0С1 2- н2о ш 12 3
Ответ [обсуждение химии енаминов см- Шмушкович Дж., „ книге «Успмп
органической химиш, сб. 4над-во «Мир», М., 196вГ стр. 5; S t о Л Ъ* В r7z z ola-
га A., Landesman Н., Szmuszkovicz J Terrell р т д™ пь
Soc, 85, 207 (1962)]. «vicz j., lerrell R., J. Am. Chem.
mnBlffJ)TT^iUnig S'4?enzinE E., Lucke E., Ber., 90, 2833 (1957)- Ш м
Ушков ич Дж., в книге «Успехи органической химиш, сб. 4, изд-во «Мир»? м[Г, "Йв£
О
Ч^ с2н5
4
ч:г*(спн,)-
О
i»
н„о
о о
Лу^с2щ
Триэтиламин применяется для того, чтобы предотвратить связывание половины
исходного енамина хлористым водородом.
, {99.2) IH u n i g S., В е n z i n g E., Lucke E., Ber., 90, 2833 (1957)].
^**S^ 01*7" C~~C0Hr
н,о Г]^0""?
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ 177
Промежуточное соединение, получающееся приацилировании енамина, может
реагировать со второй молекулой хлорангидрида, образуя вышеприведенный продукт реакции.
{99.3) [Н ii n i g S-, Lucke E., Ber., 92, 652 (1959)].
Г } *а f Л 8
-на ^n:^ ох^с__0 4n/o—с
|н2о
о °~\
(57 Это превращение является внутримолекулярным вариантом реакции, описанной
в ответе (99.2).
'Задача 100. Дайте рациональное объяснение следующих превращении:
„О
(100.1) ^~~\-n' ] + 2СН2=С = 0 -> | ■ | ^
I
СНз
(100.2)
оо - tr - o6l
(100.3)
о«а--< - о-д
Ответ (общее обсуждение синтезов енаминов см- Шмушкович Дж., в книге
«Успехи органической химии», сб. 4, изд-во «Мир», М., 1966).
12-374
178
ГЛА В А в
(100.1) ГВегсЪ t old G. A., Harvey G. R., Wilson С. Е-, J. Org.
Chem., 26, 4776 (1961); Ш м ушко вич Дж., в книге «Успехи органической химии»,
сб. 4, изд-во «Мир», М.1 1966].
В - основание
(100.2) [Hiinig S., Benzin g~E., Н'и'Ь пег К., Вег.т 94,486 (1961); Шму-
ш к о в и ч Дж., в книге «Успехи органической химии», сб. 4, изд-во «Мир», М., 1966J.
РЕАКЦИИ КОНДЕНСАЦИИ И РОДСТВЕННЫЕ РЕАКЦИИ 179
(100.3) [S t о г k G., La n desman H. К., 3. Am- Chem. Soc, 78, 5129 (1956);
Щмушковпч Дж., в книге «Успехи органической химии», сб. 4, изд-во «Мир»,
М., 1966].
6
Ml
i
сн
н,о
но
сно
н^
О S,
сно
сно
о
О-^т
C*n=c'oN
-'а
_ | г-К]
+ он"
Вода, необходимая для протекания реакции в соответствии с этим механизмом,
может получаться in situ при получении самого енамина.
Хотя этот механизм и является спекулятивным, он указывает на то, что перенос
пирролидина, вероятно, протекает через стадию свободного амина. Таким образом, эта
реакция включает образование двух енаминов, которые дают продукты,
соответствующие всем четырем возможным комбинациям амипного и кетонного фрагментов [12].
ЛИТЕРА ТУТА
House Н. О., Modern Synthetic Reactions, Benjamin, N.Y., 1965, pp. 163—281
(обзор реакций конденсации, алкшшрования и ацилированин).
Шмушкович Дж.,в книге «Успехи органической химии», сб. 4, изд-во «Мир»,
М., 1966, стр. 5 {обзор химии енаминов).
Бидон Б., в книге «Установление структуры органических соединений
.физическими и химическими методами», изд-во «Химия», М., 1967, том 2, стр. 234
(обсуждение процессов, протекающих при сплавлении с щелочами в жестких условиях).
.В е й г а н д, Хильгетаг, Методы эксперимента в органической химии, изд-во
«Химия», М., 1968, стр. 481.
* Дополнения переводчика.
12*
180
ГЛАВА в
5. Кенде Э. С, в книге «Органические реакции», сб. II, изд-во «Мир», М., 1965,
стр. 267.
6. Loftfleld R. В., J. Am. Chem. Soc., 73, 4707 (1951).
7. Richards J. H.t Hendrickson J. В., The Biosynthesis of Steroids, Ter-
penes and Acetogenins, Benjamin, N.Y., 1964, Chap. 3.
8*.Д е М а и о П., Терпешшды, ИЛ, М., 1963.
9. Gould E. S-, Mechanism and Structure in Organic Chemistry, Holt, N.Y., 1959.
10. Farmer E. H., I n g о 1 d C.L.Thorpe J. F., J. Chem. Soc, 121, 128 (1922).
11*.Пространственные эффекты в органической химии, под ред. М. Ньюмена, ИЛ, М.,
1960, стр. 520.
12. Untch К. G., Doctoral Thesis, Columbia University, N. Y., 1959; Dissertation
Abstr., 20, 3962 (I960).
ГЛАВА 7
Перегруппировки
Рассмотрение перегруппировок имеется практически в каждом учебнике
по органической химии. Для более углубленного изучения можно
рекомендовать монографию Ц].
Задача 101. Приведите структуры продуктов следуюпщх реакций:
Конц.НвБОа
(101.1) (свн5)ас~— снсвн5 —_На0 >
ОН ОН
h2so4
(101.2) (CeH5)2C—С = СН > Изомер
ОН
(101.3) ГО^| S^ HCl-щщукт
(101.4)
CeHg
\
02N4 .. ,C = N
\/V/ i pci5 н2о
V\Br
OH ? *■ C7H4N303Br.CeII5
A
он-
>C7H3N203.CBH6
Б
(10?.?) [Collins С J., Quart. Rev., 14, 357 (1960); P о с k e r Y., в книге
«Molecular Rearrangements», de Mayo P., ed., Vol. lf Wilcy-Interscience, N. Y-, Wbd,
pp. 15 ff.].
182
ГЛ АЕ А 1
н сн5
н+ | Г6 5
(QHs^C—СНС6НГ *- //-«ч-_~- ~~ А
J6"5
он он
(СбН5)2с-с-с6н5 -Л*. (сен5)2с~с-н
он оД-н
н
, ! +
<^hsj2c—с—с6н5
он
(CgH5)3C—СН
л
о
(сбн5)2сн—сс6н5
Это пример пинаколиновой перегруппировки несимметричного соединения.
Третичный гидроксил отщепляется легче, поскольку дает более стабильный карбониевый ион.
Этот ион может далее перегруппировываться с миграцией либо фенила, либо водорода.
Наблюдаются оба эти направления- Установлено [2], что миграционная способность
обеих групп сильно зависит от условий реакции. Отношение А/Б варьируется от 7,3 в
концентрированной HZS04 до 0,04 в смеси НС1 — дяоксан.
(101.2) (do la Маге Р. В. D-, в книге «Molecular Rearrangements», de Mayo P.,
ed., Vol. 1, Wiley-Interscience, N. Y., 1963, p. 87).
H+ -H20
<CeH&)2C-C == CH " 1 (CeH5)2C-C = CH ~Zt(CeH5)2C — C = CH
OH
-HH
I
+ОШ
0
(CeH5)aC = CH-C-H *- (CeH5)2C = C=C/
H
HsO
-H^
HsO
+
-няо
V
H
хон2
-4 tzz^ {с6н5)2с-с=с:
X0H н+
Это пример перегруппировки Мейера — Шустера (превращение ацетиленовых
спиртов в ненасыщенные ,альдегиды или кстоны).
(101.S) (В о г s о n J. А., в книге «Molecular Rearrangements», de Mayo P., ed.,
Vol. 1, Wiley-Intcrscience, N. Y-, 1963, pp. 185 ff.).
«'Пнйен
Гидрохлорид пинепа
Борянлхлорид
•H
CI
ПЕРЕГРУПТШР ОВКИ
183
Если присоединение НО к ct-шгяену проводить при очень низкой температуре,
тщательно предохраняя смесь от попадания влаги, то удается получить гидрохлорид пинена.
Однако очень легко происходит перегруппировка с миграцией одной из углеродных
связей с образованием борнилхлорида. Миграция другой связи в данном случае
протекает в незначительной степени, но ее можно наблюдать при гидратации а-пинена и при
перегруппировке стереоизомерных спиртов, родственных гидрохлориду пинена [3].
(СНлСО)„0
fon
(СН3СО)20
а-Фенхилацетал
ОСОСН3
(сн3со)го
ОСОСНз
(сн3со)2о
\1 Борннлаце-
Вероятно, эти перегруппировки включают обычную стадию, очень похожую на
вальдеповское обращение, хотя не ясно, протекает ли такой сдвиг по согласованному
механизму.
(W1.4) [М е i s e n h e i m er J., Zimmermann P., von Kummer U.,
Ann., 446, 205(1926)1.
0,N
CfiH
6n5„
Br
02N ~" UNv^.T 02N £_
C=N—CBH5
OFT
ci-
ci
0»N.
X^n/C—KC^HS H2Q 02Nv^v.C—К—СйНд
Ы.
UOjN
CeH3
С—N
Ч^зЛ^о
вг
o<**
ад
6"5
с
^
-Br
184
Г Л ABA 7
лЯ P ™ SS*411" пР?ДОтавляет собой обычную перегруппировку оксима по Бекма-
*У ([11. стр. 48й и ел.; [4]), вторая - внутримолекулярное нуюгеофильпое замещение
ароматического галогена, активированного п-нитрогрушюй. Совокупность этих реакций
является одним из классических доказательств т/шке^тт^-миграции группы по
отношению к —ОН в перегруппировке Бекмана. /*•■*«
Задача 102. Установите строение продуктов следующих реакций:
(102.1) СНэ
^ тт *L л 10%-ный NaOH
CeH6N+— О- —— =► Изомер
I Нагревание г
СН2С6Н6
(102.2) ,ч/СОСН3
КаСоз
>
Пиридин
^чосс6нБ
о
Изомер
(102.3) S\ /V
' i ii ii i
CI&n13N
CHgONa
CeH6 NHC1
Ответ.
55,(5lf'(1922)]e.iSenheimer J"' Greeske н- Willmersdorf A., Ber.,
™з СН3
сн2
I
с6н5
Механизм этой реакции не установлеп, но весь процесс в целом нзоэлектронен с
перегруппировкой Стивенса ([1], стр. 378—382; [5], стр. 640), что позволяет предположить
существование аналогии между механизмами этих процессов. Эта реакция ограничивается
довольно узким кругом соединений, так как Мейзенгеймер [6] показал,, что необходимым
условием является наличие фенильных радикалов, связанных с атомом азота, а
мигрирующая группа должна быть либо бензилом, либо аллилом.
Для приведенного механизма основание не является необходимым; возможно, что
оно расходуется только на выделение окиси амина из соли, в виде которой она
получается и хранится.
ПЕРЕГРУППИРОВКИ
18ii
{102.2) (Baker W., J. Chem. Soc, 1933, 1381).
CH,
в:
о
CH,
„с—с6нг,
о
O/i
_ °
о о
qAA„
вн*
CH
_/с^сйн=
Хотя эта реакция иногда называется перегруппировкой Бекера — Венкатарамана,
^она по существу напоминает внутримолекулярную конденсацию Кляйзена.
[102.3) [Р i n с k L. A., H i 1 Ь е г t H. Б., 1. Am. Chem. Soc, 59, 8 (1937)].
сбн5 N—CI
Это пример перегруппировки Штиглица ([1), стр. 481), которая формально
аналогична перегруппировке Гофмана. Неизвестно, действительно ли в этой реакции
образуется питрен А в качестве промежуточного продукта или перегруппировка протекает
синхронно с уходом С1~.
Задача 103. Предскажите, какой продукт образуется в основном при
•бензиловой перегруппировке следующих соединений:
186
ГЛАВА 7
(103.2)
О О
СН30-<^>_|_Л_^>
(103.3) О
/\
/\
/\/\/
о
\/\/
Ответ [обзор бензиловых перегруппировок см. Selman S„ Eastham J.F
Quart. Rev., 14, 221 (I960)].
(i03.J) (Haworth R. D.T J e f f e r i e s P. R., J. Chem. Soc, 1951, 2067).
Сужение цикла в сильнощелочной среде — типичная реакция трополонов [7].
Н,с
ОН
н,с
он
н,с
но
НЯС
со он
"ОН
н,с
со он
ПЕРЕГРУППИРОВКИ
187
003.2) [Roberts J. D., Smith D. R-, Lee СfeT J- Am. Chem. Soc, 73,
«19 (1951)].
О О
сн
*-OS-43
ОН"
-5— СН
ОН"
сн.
•о^о
он
* I
ноос— с—с6н5
осн.
он
сн
jO-r -у-с—соон
ССН£
2
Иоп гндроксила может атаковать один из двух карбонилов и, таким образом,
привести к соединению 1 или 2. В действительности соединение 2 является главным продуктом
реакции, поскольку меченый карбопил менее реакц ион неспособен в реакциях
присоединения из-за э.чектронодопорного эффекта метоксильной группы. Одпако проблема
усложняется тем, что наблюдаемое распределение метки отражает не только различие в
константах равновесия для присоединения гидроксил-иопа, но и различие в скоростях
перегруппировки обоих промежуточных иродуктов [8]. Последняя в свою очередь зависпт
от относительной мигрирующей способности двух ароматических колец и в меньшей
степени от величины положительного заряда па двух различных карбонилах.
{103.3) tGoergian V., Kundu N-, Chem. Ind. (London), 1958, 1322; см. так-
же W e n d 1 e г N. L-, T a u b D., G г a b e г R. P., Tetrahedron, 7, 173 (1959)].
OH
188
ГЛАВА 7
НООС а
Эта реакция протекает стереоизбирательно с образованием 17 р-гидрокс ильной груп-
лы. Такая конфигурация соответствует преимущественной атаке гидроксил-ионом 17 а-,
а не 17-карбонильной группы. Можно предложить два объяснения такой селективности.
Во-первых, атака на 17-карбонил привела бы к промежуточному соединению Б, которое
нестабильно вследствие аксиально-аксиального взаимодействия О - и СН3-группы.
Поэтому реакция протекает по другому пути.
СН-
но
Альтернативное объяснение сводится к тому, что в щелочной среде такой а-дикетон
существует главным образом, если не исключительно, в виде аниона енола В. Тогда
только 17а-карбонильная группа доступна для атаки гидроксил-ионом. Когда ОН"
присоединится по 17а-карбоншту, то енолизации не происходит и протонирование атома
углерода приводит к соединению А. Затруднения, вызванного допущением необходимости
атаки гидроксил-ионом соединения, уже имеющего отрицательный заряд, можно
избежать, принимая, что реакция протекает с небольшим количеством енола, находящегося
в равновесии с анионом В.
Экспериментальных данных, позволяющих сделать выбор между этими возможными
механизмами, пока не получено; однако последний механизм более убедительно
обосновывает селективность атаки.
11 А
/Л
BF3
Задача 104. Установите структуры продуктов следующих реакций!
о
с,
С6Н5 С6Н5
(СбН5)зС3Н°2
(104.2)
&
Ч'
ссн
6Ж15
BF,
CfiH5CfiH7°2
ПЕРЕГРУППИРОВКИ jyc,
Ответ.
(104.1) [House H. О., U e i f D- J-, J. Am. Chem. Soc, 77, 6525 (1955)]
BHuCv /\ Я
си.
CRH]
e"6
BF,
~BF.
О £r3
II >SQ
:вн„с / \^H *-
4
сЕн3
C.H,
6"B
CeH«
о /I
И {яг
C0H3 С6Нг
I
о о
II II н
-с—с—с—сена
I
При действии кислот Льюиса, таких, как BF3, эпоксиды могут раскрываться с
перегруппировкой. Раскрытие цикла протекает таким образом, что промежуточно
образующийся карбониевый ион (частичный или полный, см- ниже) локализуется в положении,
где он будет обладать наибольшей стабильностью. В данном примере это будет атом
углерода, не соединенный с электроноакцепторным заместителем — ацильной
группой * 19].
Показано [10}, что эта перегруппировка протекает внутримолекулярно; в данном
случае [11] (но не во миогих других) она протекает по согласованному механизму.
(104.2) [House Н. О., W a s s о n R. L., J. Am. Chem. Soc, 78, 4394 (1956)].
"BF,
I 1 CGHS . —" I I C6H5
Pt-BF,
+
CH
CfiH,
СеН5
°rV° ♦
BF,
Предполагается, что эта реакция протекает с раскрытием эпоксидного цикла
и образованием более стабильного (бензильного) карбониевого попа, после чего
происходит миграция ацильной группы с образованием конечного продукта реакции [11].
В данной реакции наблюдается следующий порядок миграционной способности:
арил > ацил > алкил > Н [12].
О
* Предполагают, однако, что влияние —Ни —САг на соседний карбониевый ион
примерно одинаковое.
190
ГЛАВА 7
Задача 105. Приведите механизмы следующих реакций:
(105.1) Н3а
ХН-СН2С1 — > СНэСНСН2СНз+С1СН2СН2СН2СНз
_ / Нагревание I
н3с ^
(105.2)
СНз
I
СН3С-СН21
I
СН3
CHaCOOAg Н3Су /Н
С = С'
Н3С/ \СН3
о
\ /
- CH*CN
Вг2
\- CH2NH2
(105.3) О
ИС1 */2 у V I!
CeH6 — N—СОСН3 > С1—< >— NHCCH3
[ . N Х
С1*
* радиоактивная метка
Ответ (Molecular Hearragcments, de Mayo P., ed., Wilcy-Interscience. N. Y., 1963).
{105.1)
H3C.
H4C
;CHCH,C1 + A1C1.
н3с.
H3C'
:сн—сщ+ aici4'
н
— сн2—снсн,сн,
'2^"s
AIC1,
CICH2CH5CH,CH;
AICL
2^П2^Л2
С HgCH 2 CHn CH,
сн3снсн2сн3
CI
CH,
I
CH3C—СИ2Г + Ag
CH,
CH.
f—Vl
CELC—'CH,
CH,
сн3
л. + I -H+
+ Aglt -*- CH3 — C~CH2 >
CH,
H,C.
H,C
; C=CHCH
Как А1С13, так и Ag+ способны отщеплять гало ген ил-ион от алкилгалогенидов
с образованием карбопиевых ионов, которые далее могут подвергаться перегруппировке.
В обоих вышеприведенных случаях механизм дан, несомненно, очень упрощенно, и не
ясно, образуется ли настоящий карбопиенын иоп до перегруппировки или уход гало-
генида сопровождается перемещением метильных групп.
перегруппировки 191
(105.2) (S m I t h P. A. S., в книге «Molecular Rearragements de Mayo P., ed.,
Vol. i.f Wiley-Interscience, N. Y., 1963, pp. 457, 528 и ел.; G о u 1 d E. S., Mechanism and
Structure in Organic Chemistry, Holt, N.Y., 1959, p. 621).
O-
о
II
CH2CNH2
-o
О
II
CHjCNH
Br4
<o
CH9CNH
I
Br
OH
4Br
CH,^C
_n: —*- ( V- ch,—n—c—о
н,о
Оснт
-co2
сон
/ VcH2NH3
Это перегруппировка Гофмана. Мехапизм, приведенный здесь, включает
образование пптрепа А, хотя довольно вероятно, что отщепление брома и перегруппировка
протекают со гласи панно. Вследствие того, что нитрен, как п карбен, имеет шесть электронов,
.он обладает электроиоакцеитирнымп свопствами, несмотря па то, что он электроцентралей.
(105.3) (D e w а г М. J. S., в книге «Molecular Rearrangements*, de Mayo P., ed.,
Vol. 1, Wiley-lnterscience, N. Y., 1963, pp. 295, 309; G о u 1 d E. S., Mechanism and
Structure in Organic Chemistry, Holt, N. Y., 1959, pp. 650 ff.).
Ch:cCH*
i
OH
CI
CI*
Ароматическое
гали-еннрование
1
OH
I
CCH, + CI —СГ
о
II
NHCCH
Это перегруппировка Ортопа. Эта реакция пе является перегруппировкой в обычном
понимании, так как протекает межмолскулярпо. Источником С12 является N-хлорамид,
который реагирует с НС). Затем С12 независимо получается при реакции либо с образу то-
щимся ацетаиилидом, либо с какими-либо другими присутствующими молекулами.
Образование С12 доказывается тем, что с увеличением количества С1- доля меченого хлора,
который вступает в ядро, уменьшается. Это происходит в результате быстрого обмена
по схеме
С1-+С1-С1* ^ CI— C1 + C1*-,
и, таким образом, все хлорид-ионы и атомы хлора находятся в равновесии [13].
192
ГЛАВА 7
Существует большое число аналогичных «перегруппировок» общего типа:
А —X
АН
I
АХ
I
V
+
х
A=Nf О и т. д.
Х = галоген, N02, COR
которые включают (иногда это известно достоверно) разделение на фрагменты, которые
могут далее рекомбинировать. Сюда относятся следующие реакции:
а) Перегруппировка алкиланилинов ([1], стр. 310)
H2NR С1-
I
+NH3 С1-
Нагревание
К
б) Перегруппировка нитрозаминов ([1], стр. 310)
СН3ШО
HNCHa
нх
1«1>
NO
в) Перегруппировка эфиров фенолов (перегруппировка Фриса) ([1], стр. 318 и лс„;
О
OCR
AlCis
0=CR
ПЕРЕГРУППИРОВКИ
193
г) Перегруппировка простых эфиров фенолов ([1], стр. 313 и ел.)
OR ОН
I
Л Н3ро4 ^
Нагревание
д) Перегруппировка Якобсеяа ([1], стр. 299 и ел.; {15])
S03H R
H3S04
Нагревание
\у\-
\
so3n •
Задача 106. Приведите механизмы следующих реакций:
(106.1)
YY
ОСН2СН = СН2
Нагревание
CtijjOH — ^Н2
/\Л/0Н
Л/Ч/
* меченый углерод
(106.2)
+ он-
CeH5CCH2N(CH3)3
О
I!
С^НдССНСНз
I
N(CH3)2
Ответ (Molecular Rearrangements, de Mayo P., ed., Vol. 1 and 2, Wiley-Interscien-
ce, N. Y., 1963).
(106.1) (Rho ads S. J., в книге «Molecular Rearrangements», de Mayo P., ed.,
Wiley-Interscience, N. Y.t 1963, pp. 655, 660 ff.; Gould E. S., Mechanism and
Structure in Organic Chemistry, Holt, N. Y., 1959, pp. 644 ff.).
I
О
ч/\
н
\
о
/\/\/
\/\/
Епопизация
| ОН
4/V
Это перегруппировка Кляйзена. Она относится к числу перегруппировок,
включающих шестичленпое циклическое переходное состояние с участием л- и а-связей.
Механизм этой перегруппировки до сих пор точно не установлен, хотя, вероятно, формула А
13-374
194
ГЛАВА 7
наилучшим образом изображает переходное состояние, так как она обходит вопрос
направления движения электронов при образовании такого переходного состояния.
А
В данном случае показано, что n-электропная плотность двойной
1,2-связи"нафталина больше, чем 2,3-связи. Эта реакция приводит предпочтительно к а-замещенному
соединению.
(106.2) (Zimmerman Н. Е-, в книге «Molecular Rearrangements», de'Mayo P.,
ей.. Vol. 1, Wiley-Interscience, N. Y., 1963, pp. 345, 378 ff.; Gould E. S., Mechanism-
and Structure in Organic Chemistry, Holt, N. Y-, 1959, pp. 640 ff.).
CH, XV"* CH4
h" он" II?
-Т.. - Г-Н ГГНМ
C6HBCCH2NCH3 *~ CeH5CCHNCH3 —*- C6H5CCHN^ 3
CH
° сн3 о снч о
"Это перегруппировка Стивенса. Известно, что она является внутримолекулярной+
протекает с сохранением конфигурации мигрирующей группы и поэтому для нее
принимают согласованный механизм. Известны аналогичные перегруппировки, которые
включают промежуточное образование более неустойчивого аниона, чем в данном случае.
Однако они протекают в жестких условиях с более сильным основанием, как, например г
СН2СвН5 СН2СвН5
| NaNHa
CeH6CH2N(CH3)2 —^Г* C6HBCHN(CH3)2
Задача 107. Для реакции Фаворского были предложены два механизма:.
А и Б. Эксперимент с мечеными атомами показал, что в данном случае
реализуется механизм Б. Используя меченые атомы, предложите такой^экспе-
римент, чтобы можно было различить эти^два^механизма.
/Q OR -^ COOR
О" -
6
COOR
ПЕРЕГРУППИРОВКИ
195
Ответ [Loftfield R. В., J. Am. Chem. Soc, 73, 4707 (1951)].
&
OR"
О OR
\
OR-
Для реакции Фаворского был предложен ряд механизмов [16J, но большинство из них
требует, чтобы атом углерода, связанный с бромом, оставался в о-положении по
отношению к карбонилу (механизм А). Экспериментальные результаты, однако, полностью
соответствуют механизму Б.
COOR
Q
он-
Н"1
СООН
I
/\
LI
100%
вктввностн
СООН
|/0н
РС13
:
Вга'
О
СООН
1/вг
/\
U
CHaCOONa
CHsCOOH
KMn04
■ I
-> С02 +
ai
H2N~j |~NHa + C02
HNa HMe
50% 50%
активности активности
25%
активности
25%
активности
13*
196
ГЛАВА 7
Карбоксильная группа возникает из атома углерода кетонной группы
О
л.
U
С1
OR-
*COER
I
Другой предложенный механизм [17,18], объясняющий наблюдаемое распределение,
включает обмен галогена между а- и cc'-положеппями с последующей медленной
перегруппировкой но механизму А.
G1
О
II
ROH
\/
RO-
*/2/\*/2
Однако если реакцию остановить на стадии 50%-ного превращения и
проанализировать неизменный кетон, то перераспределения метки обнаружить не удается. Таким
-образом, этот эксперимент не согласуется с предположением о быстром равновесии гало-
ггенкетонов, предшествующем медленной стадии перегруппировки.
Механизм с промежуточным образованием циклопропанона объясняет главные
-особенности перегруппировки Фаворского; однако известны случаи, когда он заведомо
.неприменим (отсутствие а'-атомов водорода [19], наличие стерических затруднений [20]).
.'Поэтому для таких случаев принимается какой-либо другой механизм.
Задача 108. Предскажите структуры продуктов реакции Фаворского
.для нижеприведенных а-галогенкетонов:
(108.1)
о
(108.2)
Ij1\ /CGH3
Х/СНз
Ответ (К е и d e A. S.
М-* 1965, стр. 267).
в работе «Органические реакции», том 11, изд-во «Мир»,
ПЕРЕГРУППИРОВКИ
197
(108.1) [Stork G.t Borowitz I. J., J. Am. Chem. Soc, 82, 4307 (19G0)].
>Оснз
OH
tT'-cr
HOC CH
нос ch3
Общепринятый механизм реакции Фаворского включает образование
циклопропанона при внутримолекулярном замещении галогена [21]. Раскрытие трехчленного цикла при
атаке основанием дает наиболее стабильный анион (первичный ;> вторичный >
третичный).
{108,2) [W а 11 а с h О., Ann., 327, 125 (1903); 414, 233 (1918)].
ОН
ОН
4 Вт
($~
В данном случае согласованное отщепление Вг_ приводит к раскрытию циклопропа-
нонового цикла, причем легкость раскрытия цикла измепяется в порядке, обратном
приведенному выше ряду 4 изменения стабильности изолированных карбанионов.
Задача 109. Если пероксиэфир А, в котором карбонильный ато.м
кислорода мечен 180, подвергнуть серии превращений, показанных на схеме,
то весь 180 будет обнаружен в.бензиловом спирте. Перегруппировка А -*- Б
представляет собой гетеролитическую реакцию и ее скорость резко возрастает
при переходе от неполярного к полярному растворителю. Приведите
механизм этой перегруппировки. Введение какого заместителя в бензоатную
198
ГЛ ABA J
группу — ге-нитро- или п-метоксигруппы — вызовет большее увеличение
скорости?
0_0_С—CfiHg
СО
О—С—CrH
СН3СООН
гСО
6^5
LIAIHj
ОН
О
он
+ C6HSCH20H
Ответ, (Denney D. В., D e n n е у D. G., J. Am. Chem. Soc, 79, 4806 (1957)].
Реакция сходна с перегруппировкой Вагнера — Меервейна и может быть
представлена следующим образом ([1], стр. 1; [5], стр. 584 и ел.).
О—О—С—слН
CD - Cb
'C^CeH,
впе
О—с—CfiH
ens
о—с—сйнв
со
Изучение с конкурирующими ионами [22, 23] показало, что эта реакция не
включает стадии диссоциации на два кинетически независимых заряженных фрагмента. Вместо
втого протекает синхронная перегруппировка или образование тесной ионной пары.
Три возможные структуры были предложены для переходного состояния в этой
реакции (а, б и в):
ПЕРЕГРУППИРОВКИ
199
о—о—с—Сг,н
б"5
,о—с—свн6
*/2 о^' V/2
' 6-
Продукт реакции
Пунктирной линией обозначаются частичные ковалентные связи^или
электростатическое взаимодействие между ориентированными частичными зарядами. ^^^ л . ^
Распределение метки в продуктах реакции следующее:
о— с—ель
он
С9
*h
О */2
о—с—с=н,
СО
6 "5
?
6"3
О—С—СКИ
- со
LiAlH,
LiAIH4
L5A1H4
ОН
Ф он
*А он
+/2 0Н
*/2 ОН
+ с6н5сн2он
*/2
+ с6н5сн2он
+ с6н5сн2он
200
ГЛАВА 7
Наличие всего меченого кислорода в бензиловом спирте, получающемся при
восстановлении сложного эфира алюмогидридом лития, показывает, что реакция протекает
по схеме (а) и, таким образом, карбонильная группа сохраняет свою индивидуальность
при перегруппировке. Это может происходить либо при согласованной реакции, либо
при промежуточном образовании очень тесной ионной пары, в которой сложноэфирный
атом кислорода ближе к образующемуся карбопий-ионному центру, чем карбонильный
атом кислорода; при этом реакция протекает настолько быстро, что атомы кислород»
не имеют возможности поменяться местами.
Поскольку переходное состояние включает частичное или полное образование
аниона кислоты, то следует ожидать, что его стабилизация приведет к увеличению
скорости реакции. Это должно наблюдаться при введепии в бензольный цикл электроно-
акцвпторного заместителя, например N02-rpynnbi, что подтверждается экспериментально
124].
Задача 110. Приведите механизмы следующих реакций в ряду стероидов:
о
сосн3 И^-СЛО^
'.-ОН
кон
Qnteem (Wendler N. L., в книге «Molecular Rearrangements», de Mayo P., ed.,
Vol. 2, Wiley-Interscience, N.Y-, 1963, pp. 1114ff.).
ПЕРЕГРУППИРОВКИ 201
Эти ацилоиповыс перегруппировки взяты из большой серии сложных и неполностью
изученных реакций кетолов нормальных и D-гомостеройдов. Для соединения А механизм
реакции, катализируемой mpem-бутилатом алюминия, включает образование мостикового
комплекса за счет образования связи с обоими атомами кислорода (формула В)
снчс=о
AI(OR);
3 СН,
сн
(OR),
Миграция
связи (б)
спэ
"ОН
Ориентация карбонильной группы в этом соединении (под плоскость молекулы)
определяет ориентацию заместителей в продукте реакции.
С другой стороны, перегруппировка, катализируемая КОН, протекает через анион
соединения А, ъ котором О" и С = О имеют антипараллельную ориентацию вследствие
отталкивания двух атомов кислорода (Г)
СН*
Н3С
ОН
:0:~
Миграция
свиэн (а)
1,-СН3
Поэтому стереохимия получающегося продукта также определяется ориентацией групп
в промежуточном соединепии Г.
Однако этот механизм не объясняет, да и вообще пе ясно до сих пор, какие причины
вызывают миграцию связи (а) или (б). Было предложено большое число объяснений,
основанных как на электронных, так и на стерических факторах; однако ни один из них
не объясняет сколько-нибудь удовлетворительно все факты. Кроме того, установлено, что
существует равновесие различных продуктов перегруппировки и образование конечного
продукта может определяться как кинетическим, так и термодинамическим контролем.
Аналогично можно представить себе течение реакций в случае соединения Б
В этом случае при действии обоих реагентов мигрирует одна и та же связь (а)
он
202
ГЛАВА 7
Задача ill. Приведите механизм следующей реакции
ОАс £
ОН Н
/ ОН
Нейтрализация
ОАс Н
Ас=СОСН3
-Н
ОН Н
Ответ [Meinwald J., Wiley G. A., J. Am. Chem. Soc, 80t 3667 (1958) J.
OCOCH3
ОГГ
OCOCH,
OH
0H-»-
Нейтралп-
защщ
Продукт
реакции
Этот механизм согласуется с необычно легким раскрытием эпоксидного цикла при
катализе основаниями. Обычно экэо-норборнановые системы * довольно устойчивы
во отношению к нуклеофильной атаке в щелочных условиях.
Задача 112. Приведите механизмы следующих перегруппировок:
(112.1)
^N^4^
со2сн3
l.N2H4 2.HN02
3. Кипячение в С6Н5СН2ОН
^
<;
осн2свн5
NH
И родственные по геометрии системы.— Прим. перев.
ПЕРЕГРУППИРОВКИ
203
< 112.2)
Н"
(112.3)
Н+ CH3MgI H20 Se
310°
CI7H28°2
Ответ. , _ _ _„ ,.„,-„,,
(112.1) [D о е г i n g W. von. Б., "Goldstein М. J., Tetrahedron, 5, 53 (1959)].
со2сн3
о
II
CNHNH,
NH2NH2
^S^-^
„н/сн,о>
н=с=о
Реакция включает перегруппировку Курциуса ([1J, стр. 528—568), по последующая
перегруппировка изоцианата совершенно необычная. Вопрос о точном механизме этой
перегруппировки с образованием мостикового производного до сих пор не вполне ясен,
так как он осложняется неопределенностью конфигурации изоцианата. Если последний
имеет цис-конфигурациго (А), то легко представить согласованный механизм электронных
204
ГЛАВА 1
сдвигов. Однако если изоцианат имеет транс-конфигурацию (Б, на что указывают
некоторые данные), то требуется дискретное протекание стадий расщепления, вращения
и рекомбинации.
А Б
[112.2) [А г i g о n i D-, Viterbo R., Dunnenberger M., Jeger О.,
Ruzicka L-, Helv. Chim. Acta, 37, 2306 (1954)].
R
Хотя перегруппировка изображена постадийно, такие реакции часто рассматривают
как протекающие через синхронные 1,2-миграции трякс-диаксиально расположенных
групп ([1], стр. 813 и ел.). В данном случае реакция начинается атакой двойной связв
протоном с наименее пространственно затрудненной стороны. Важность ориентации
мигрирующих заместителей можно проследить при сравнении с перегруппировкой лаисстрина,
который имеет обратное сочленение циклов C/D.
Ланостсрин
ПЕРЕГРУППИРОВКИ
205
(112,3) [Вис hi G., Saar W. S., Escbenmoser A., Experientia, 13,
136 (1956)].
OH 1,2-Сдвиг
-H
+ HO
-H20
H+4
O.J
53-
HO
Se
Эта перегруппировка также включает серию 1,2-миграций горякс-дпаксиальных
струпп с последующим ацилировапием промежуточно образовавшегося алкена.
Задача 113. Приведите механизмы следующих реакций:
(113.1) /Ч
\1 IX
К они. H2S04
/X
Нагревание
/ч</\хюн
\
о
СООН
(113.2)
о
ССН3 HNOa | |
NOH
\Лч>
н80
\
206 ГЛАВ А 7
(113.3) О
Я
,ссн
Ч II ( +1 L
ч/ЧгАсн2снсс13 "^/V^cooh N/V^ch= снсоон
I H
OH
OH-
4N^ ^Cf^CHCClj
'4 ='4c
Метка находится
в атом положении
Ответ.
(Ш.1) [Meinwald J., Hwang H. С, Christensen D..Wolf A. P..
J. Am. Chem. Soc, 82, 483 (I960)].
о-Цякеновая
кислота
Героповая
кн слота
Эта перегруппировка очень необычна, и ее механизм долго оставался неясным-
Первоначально для нее принималась дальняя миграция метальной группы по
внутримолекулярному согласованному механизму. Однако изучение с помощью меченых атомов
[25] формально указывает на миграцию карбоксила (принимали внутримолекулярный
согласованный механизм [25, 26]). Выделение в этой реакции СО, а также введение 14С
в полученную героновую кислоту при добавлении к реакционной смеси иС позволили
предложить указанный механизм. Движущая сила этой реакции, возможно,
определяется стерическим напряжением из-за отталкивания 2,6-заместителей в а-циненовой кислоте.
Побочным продуктом реакции является лактон Б. Вышеприведенный механизм [27]
согласуется с образованием лактона, которое не требует протекания скелетной
перегруппировки и не включает обмена СООН-группы с добавлением иСО
ПЕРЕГРУППИРОВКИ
207-
Л ^ХХ -XX -XX
-^NrSxiaH ^о^Чхюн "о^соои ""^й^адсн
У 13.2) [L а п % i 1 о 11 i А. Е., Weiss М- J., 3. Org. Chem., 24, 1003 (1959)]~
+
J4 V.c—сн,
■^ СНЯ
i;
</S>
CKLCOOH +
Это общая реакция расщепления В-дикарбонильных соединений и замены ацильной
группы на оксимную [28]. В этом случае промежуточное соединение Б было выделено г
хотя не было приведено доказательств, что реакция включает внутримолекулярную
перегруппировку. Первоначальная трактовка этой реакции основывалась на предположении
о прямом гидролизе соединения А с образованием аниона оксима. Возможно, что она
является правильной для реакции, протекающей в более щелочных условиях (RONO, R О ")-
Очень похожая реакция термического разложения N-нитрозамидов в первой стадии
включает аналогичную перегруппировку [29]
N==0
U Нагре-
1 /■*■ ванне II
R—N-i-CCH, -*" R—N
N—О—ССН3
II
О
(113.3) [Dauben W. G. Vaughan С W.f Jr., J. Am. Chem. Soc, 75, 4651:
(1953)].
208
ГЛАВА 7
ОН
сн.
он
•ccl
он
—а
Н-,0
он
Kv^CH-CH^ca
он
он о
^/^•NV^vcOH Альдольмая\/*чм——г
II II КПНЛСНГЯПИО .. 1 _
он
от "L Jh
и * 3
-а-
-Г-Н2Ор-2НС!
]
ОН
Н ||
О
fl-H2o
конденсация
СООН
Ретроаль-
-*— Lо
расщепление \^^м
Н
^^N-^COH
н II
о
Выше приведен лучший из двух предложенных механизмов, которые согласуются
с наблюдаемым распределением меченого углерода. Прямые доказательства этого
механизма отсутствуют, хотя имеются аналогии для отдельных стадий этого процесса.
Неперегруппированный продукт может возникать при альтернативной циклизации
промежуточного соединения А
О
о
от он
-н2о
СООН
СООН
ПЕРЕГРУППИРОВКИ
209
Однако доказательства, что такой путь более вероятен по сравнению с прямым
гидролизом трихлорметильной группы и дегидратацией в присутствии основания (что должно
привести к тому же продукту реакции), конечно, отсутствуют.
Задача 114. Приведите механизмы следующих реакций:
(114.1)
сн3о
сн,о
NCH,
l.C6H5MgEr
2. Н20
снчо
NCH»
С fiH.
Тебаин
(114.2)
Тебаин
Разб. НС1 Нейтрализация
GHSNH /\/
ОН
H0~i(Y
cii3o-IN
N— CH,
Кгащ. HCI Нейтрализация
—■■ ■>■ >
.Морфин
Ответ.
(114.1) [Stork G., J. Am. Chem. Soc, 74, 768 (1952)].
Oi_
N—СЩ
CH3O
N—CH3
ХД1„0
CH30
CH30
14-374
210
ГЛАВА 7
сн,о
сн.о
Н20 Продукт
NCHg ■■ г* реакции
СЕНЧ
(114.В) (Stork G., в книге «The Alkaloids», Manske R.H.F., Holmes H. L., eds.,
Vol. II. Academic Press, N-Y-, 1952, p. 197).
сн,о
сн,о
N-CH,
сн3о CH
Г^7Н~СН3
N—СНЯ
Х-СНзН^О
сп3о
CH3o-
ТеСении
ПЕРЕГР УППИР О В КИ
211
{114.3) (Stork G., в книге «The Alkaloids*, Manske R. H. F.f Holmes H. L.,
eds., Vol. II, Academic Press, N. Y., 1952, p. 195).
-ТГ
но
Нейтрализации
Л по морф пи
Полностью аналогичная реакция тебаина приводит к морфотебаину (А).
Н0\/\/\
NCH£
W
но
\А/
Различие между реакциями в концентрированной и разбавленной кислотах
„определяется тем, что в первом случае азот находится исключительно в протонированнои
форме и не способен отдавать электроны, необходимые для превращений по схеме (114.^).
Задача 115. Приведите механизмы следующих перегруппировок:
14*
212
ГЛАВА 7
(115.1) о
(115.2)
сн
(СНзСО)йО
H2SO4
ОСОСН3
I
| У | (СН3СО)20 '
о
н3с/\ш3
^/V\ab
HsSOj
XII;
л/\/
* N о
сн3
СП3
!
^ о
Конц. HeSO* У\/ \У
NIICH3
4/+n 4conc6ii5
] I
О- СН3
(.MS../) [Arnold R.T., Buckley J. S., Jr., Richter J., J. Am. Chem. Soc,
69, 2322 (1947); Marvell E. N., G e i s z 1 e г А. О., J. Am. Chem. Soc, 74, 1259
(1952); Marvell E. N., Stephenson J. L.,J. Am. Chem. Soc, 77, 5177 (1955)],
Qoh
OCCH,
ПЕРЕГРУППИРОВКИ
213
Первая реакция представляет собой нормальную диенон-фепольпую
перегруппировку. Вторая является аномальным вариантом, что, вероятно, обусловлено высокой
энергией резонанспой формы В промежуточного иона, который должеп приводить к
нормальной перегруппировке, поскольку в этом ионе теряется энергия сопряжения бензольного
кольца.
О
ОССП3
чснч
Л/Ч/"'
в
+
{115.2) [И а Ь i Ь М. S., R e e s С. W., Chem. Ind. (London), 1959, 367].
он о
-н
t
II Т.Д.
СН
N1
„ Д> С=0
Н2°+К N1JCH,
0-*-Н
\и.Ук
он
О N.
сн,
Эта впутримолекулярная реакция напоминает электрофильное замещение,
происходящее в активированном фенольном цикле.
214
ГЛАВА 7
Задача 116. Приведите механизм следующей реакции:
О
уСН3
нсоон
/\/к
N ХХ
I
СН3
1> C10H19N Pd/C ,
Нагревание
Ответ [С е г v i n k a 0., Chora. Ind. (London), 1959, 4129].
OH
OH
3 HCOOH
-G
CH.
\
k
I ч jn_
CH, HCO
CH,
or
4-
те
CH,
CH,
+ /-OH
CH,
CH,
H —CO
CH3NH
NH <^J —-
-cO'
^ v^
л^снз II ^
■нс-те-
fCHq
Ч^уСНг
C10HigN
РЛ/С
*■
Нагревание
ch3 ch3
ПЕРЕГРУППИРОВКИ
215
Восстановление енамвнов муравьиной кислотой является общей реакцией (см.
реакция Лейкарта [30]). Ниже приведены результаты изучения соединения, меченного
дейтерием [31].
I
СИ,
HCOOD
сн,
D О
in
HCO
о*
сн.
сн.
СО-
псоон
сЬ
В первоначальном соединении аминогруппа, находящаяся в р-положении по отношению
к карбонилу, очень легко отщепляется, что дает возможность для протекания
дальнейших реакций.
Задача 117. Установите строение соединений, образующихся в
результате следующих реакций:
(117.1) . ОСН3
NCaH5
С1бН18^202
(1) 1Ш02. разб. HaS04, 0
(2) Н20, 70°
> Ct5Hl3NOa
ИК: 166S,
1692 см-i
Дает динитро-
фенилгидра-
эон
(1) NaBH4
(2) HCl, нагревание
\/\/СгНб
N
I I
/v/v0
C15H13N0
216
ГЛАВА 7
(Н7.2) СН3
I
~ || 1| -* с13н14
Изс/уЧпз VNa+ в
С10Г
UJ7.i) (Hey D. H., Leonard J. A., Moyneban Т. М-, R e e s C. W.,
J. Chem. Soc, 1961, 232).
CH.
NH2 |
HNO,
^
Нагревание
II,0:
NCj,-H5 NaBH4
О *
1692
NC„H.
NC2HS
ПЕРЕГРУППИРОВКИ
217
Нормальное разложение диазониевои соли непосредственно дает фенаптрпдоп
(синтез фепаптрспов по Пшорру [32]), что должно приводить к соединению Б
/V
II2N NC2H5 HNOa Нагревание
I 1 >
* Б
\/
Однако наличие сильной электронодопорпои группы — ОСН3 настолько способствует
пара-замещению, что образуется спирососдинение Л.
Это соединение также можно подвергнуть диснон-фепольиой перегруппировке [93],
хотя для этого обычно требуются довольно жесткие условия
СНзСОО
(CH3CO)sO~HaS04
Л »
100°
V4
(117.2) [IUfner К., Angew. Chem., 69, 393 (1957); Hafner К., Kaiser H.»
Ann., 618, 140 (1958)].
Основание
А й Й
11-«—=В
-НоО
4,6,8 - Три метила зулеа
218
ГЛАВА 7
Эта реакция аналогична синтезу азу ленов по Циглеру — Хафнеру, исходя
из N-алкилпрованных пиридинов [34—37].
Na+
О * Q-03
х- |
сн2с^н5
Азулен
(синего цвета)
Задача 118. Приведите механизмы следующих перегруппировок:
сн2соон
(118.1)
CHjjCO
Бг9
на,нао
сн2соон
сн„
сн.
(118.2)
сн.
Н3С N
он
он
сно
(118.3)
Н,С
лснз
* 'flit
н,о+
?н3
/сн2сн2он
НзО4-
-*
сн.
сн2сн3
&
сн3
он
сн2сн2он
Ответ.
(118.1) (Areas С. L., Bennett G. J. J. Chem. Soc, 1958, 3180).
ПЕРЕГРУППИРОВКИ
219
COOH
COOH
COOH
H*-:OH0
COOH
° Y 4-
"vSH's
COOH
COOH
-н„о
-HBr
Приведенный механизм расширен по сравнению с указанным в оригинальной работе.
Возможность расщепления пиноновой кислоты в кислой среде до лактона А была показана
в работе [38]. Хотя при бромировании соединения А в кислой среде и образуется 2,4-
диметилфенилуксусная кислота, скорость се образования слишком мала, чтобы можно
было рассматривать лактон А в качестве промежуточного продукта реакции.
^гн-
:ОН,
О ч ->-
H-^OHj
ОН
Циклизация соединения Б совершенно аналогична наблюдаемой для а- и fl-метил-
гептенонов и большого числа других родственных соединений [39]
/\
Н+
= 0
Медленно
0 Л I
н+
Быстро
/
ОН
н+
\)Н
/\/\
р-Гептенон и-Гсптенон
(118.2) [Meicwald J., Chapman 0. L., J. Am. Chem.Soc, 81, 5800 (1959)].
220
Г Л Л В А 7
N (СН3)2 ~
Н<Х I? „ Н ОН
и -н2о
NfCH^,
н2о
Вышеприведенный механизм согласуется с наблюдаемыми превращениями. Хотя
в общих чертах он, вероятно, правильный, тем не менее возмояшо, что порядок
отдельных стадий отличается от указанного на схеме. Так, возможно, что расщепление азотного
мостика происходит перед раскрытием эпоксидного цикла и образованием производного
циклопропана А. Кроме того, возможно, что в результате полного элиминирования азота
образуется соединение Б, которое далее претерпевает гидратацию и раскрытие
эпоксидного цикла.
№СН3)2
" ,J f-
н он
но
<^Q=o OJr ^=D
н
по
но
!
П .» Продукт
^У реакции
У -Тро полон
Для того чтобы принять последний механизм, необходимо допустить, что образование-
циклопропана протекает быстрее, чем альтернативное раскрытие эпоксидного кольца
с образованием у-трополона, поскольку было показано, что он стабилен в условиях
этой реакции.
(218.3) [Buchi G., Goldman I. M., J- Am. Chem. Soc, 79, 4741 (1957)]-
ПЕРЕГРУППИРОВКИ
221
H3C ч^0„Н2
'"/CHoCHsOH
'"/CHaCHjOH
''' CH,CI1,01[
CH
CH2CH3
Миграция двойной
__, ГЙЯЗН
— 11,0
CH
'CHg
CH3CH5OH
Дегидратация обоих третичных спиртов приводит к одному и тому же нсклассиче-
скому карбониевому иопу В. Вследствие напряжения оп далее перегруппировывается
в ион Г, который является плоским. Далее ион I1 превращается в конечный продукт.
Включение в схему неклассических ионов не обязательно; можно альтернативно
изобразить эту последовательность реакций и через образование классических ионов
СН3
СН3
?/'ОН9
'"гСН2СН2ОН
. —"'/снасн2он
222
ГЛАВА 7
Й
'СН2СН2ОН
сн.
XHs
"'/СН2СН2ОН
/y^CH2CH2OI-I
Продукты
реакция
Задача 119. Приведите механизм следующего превращения:
о
с со он
НООС СНз.
Нагревание
Pd/C
N^^O
Оптически
активный
Ответ f\V i е s л е г К., V а 1 е n t a Z., А у е г W. A., Fowler L. R.s
Francis J. E., ^ 87 (1958)].
COOil
НООС
сн.
соон
НООС
сн.
ПЕГЕГРУПШ1РОВКИ
223
сооп
НООС
НООС
соон
сн3
ноос сн3
нл
COOtI
НООС СН3
Предложенный механизм основан на двух реакциях (1 -> 2, 4 -э- 5) расщепления
циклобутанового кольца, обратных реакции Манниха [40]. Не имеет смысла писать
различные возможные таутомеры, поскольку реакцию проводят в присутствии катализаторов
дегидрирования. Отметим, что асимметрический центр, соседний с метильной группой,
но затрагивается во время этих превращений, что необходимо для сохранения оптической
активности получающегося продукта реакции.
Задача 120. Приведите возможные механизмы следующих реакций:
о
II
ХН2ССН2ОН
(120.1) Т^ЪСон
2н. H2S04-
ОН
(120.2)
Ответ.
(120.1) (Holker J.S.
Will i amso n W. R. N..
Пиридин-НС1
— *-
218°
E.,Robertson A.. Taylor J. H., Holker K. U.j
J. Chem. Soc, 1958, 2987).
224
ГЛАВА 7
НООС
сн2оц
Ппкротошл
Этот механизм отличается некоторыми деталями от предложенного в литературе,
но, по-видимому, более рационален.
Заметим, что в тех же условиях протекает реакция
он
Пикротоксинин
ПII к роти к сова я
кислота
ПЕРЕГРУППИРОВКИ
225
Различие обусловлено отсутствием слоншоэфирной аллильной группы.
(120.2) [С h i n n L.I.,Dodson R. M., J. Org. Chem., 24, 879 (1959)].
B:—H.
Кольцо В расщепляется по схеме винилогичной ретроальдольной конденсации
с последующим протеканием альдольнои конденсации после вращения кольца А
«округ 5,6-связи.
Задача 121. Объясните, почему при температуре установления
нижеприведенных равновесий общая конфигурация соединений и оптическая
активность сохраняются.
Ответ [Woodward R. В., Katz T. J., Tetrahedron, 5, 70 (1957)].
Эту перегруппировку нельзя объяснить полным ретродиеновым распадом с
образованием циклопентадиепа и оксициклопентадиена с последующей рекомбинацией этих
соединений. В этом случае наблюдалось бы образование равновесной смеси соединении
15—374
226
ГЛАВА 7
3 и 4 и потеря оптической активности. Поэтому циклопептадиеновые кольца должны
удерживаться вместе по крайней мере одной связью, чтобы сохранить стереоспецифичность.
Предполагается, что изомеризация включает самопроизвольное расщепление связи
(аб) в скелете дициклопентадиена (А) и образование новой связи между атомами (в) и (е).
На атом основании принимается, что и реакция Дильса — Альдера протекает в Две
стадии: сначала образуется связь (гд), что приводит к промежуточному соединению типа Б,
а затем соединение Б превращается в конечный продукт.
Однако было указано [41], что эта стереоспецифическая перегруппировка
эквивалентна перегруппировке Коупа ([1], стр. 684 и ел.) и не обязательно, чтобы такое же
переходное состояние было и в реакции Дильса — Альдера. Другие эксперименты [41,
42] показывают, что для аналогичных реакций, вероятно, имеется большой круг
возможных переходных состояний, в которых степень образования связей варьируется в
широких пределах.
Задача 122. Установите структуры следующих продуктов:
(122.1)
Пиридин
Нагревание
лчоНз
А
н3о+
пОл
Ci6Hio02
Желтый
Бесцветное,
один
активный водород
Б
Желтое
СгОз
ноос о
о
(122.2)
ноос
(CeH6)3C-CH2COAg ~> С21Н1502Вг
о
ПЕРЕГРУППИРОВКИ
227
Ответ.
(122.1) (Rigaudy J., Nedelec L., Bull. soc. chim. France, 1939, 6^3;^
Отсутствие окраски соединения А указывает, что а-дикетонная система не может
присутствовать в этой структуре, что приводит в свою очередь к необходимости принять
более предпочтительную мостиковую структуру А', находящуюся в равновесии с А-
(122.2) [Wilt J. W., О a t h о u d t D. D-, J. Org. Chcm., 23, 218 (1958);
Wilt J. W.t Finnerty J. L., J. Org. Chem., 26, 2173 (1961)].
15*
228 ГЛАВА 7
Вг,
•«^H^CCH^COAg —L
н
(С0Н5)2С—СН2С—О—Вг — - ^'- ?Чп/
0Vc=o
или
—Вг~ Ради
кал или ион
Вг О
Вг Вг О
| -II- иди НН
-НВг
Br, (CfiH,UC=CH
<ceH5}2tec—сос6И5 ^^- (с6н5.)2с-сн-^_о 1"е 52
с=о
Нормальная реакция брома с серебряной солью карбоновой кислоты (реакция
Хунсдиккера [43]) приводит к замещению СО ОН на Вг. Механизм реакции может быть
как ионным, так и радикальным. В данной задаче приведена аномальная реакция
Хунсдиккера. Аналогичная реакция протекает для данной кислоты при электролизе Г441
(реакция Кольбе [45]) r l '
О О
[[ Электролиз
(СвН5)3ССНаСОН —-^-^Г^ (СвН5)2ССН2С~ОСвН5
СН3ОН
о
'СНз
Для этой реакции обычно принимают радикальный механизм.
Задача 123. Установите структуры получающихся соединений и
приведите механизм, объясняющий образование соединения 1.
/\-~/
О
Л/\/\
+N СвН5
i-
CwH9NOa
HsO+ Cd(CeH6)a
> Изомер > C20Hl5O2N
ИК: 3500 cjH-i
1
NHaOH
на
ИК: 1642 cj«-i
NHaOH
HC1
Cd(CeHB)2
Оксим
ИК: 1637 cjw-i <-
2
COC1
N
ПЕРЕГРУППИРОВКИ
229
Ответ (Pink us J. L., Cohen Т., Sundaralingam M.,
Jeffrey G. A., Proc. Chem. Soc, 1960, 70).
Структуры соединений 1 и 3 устанавливаются на основании альтернативного
синтеза из хлорангиприда А. Образование третичного спирта при реакции с дифенилкадмием
довольно неожидапно и может быть объяснено использованием большого избытка этого
металлоорганического реактива, а также, возможно, и слишком жесткими условиями
проведения реакции. Первоначально предполагали осуществить встречный синтез
соединения 1 из А.
Структура соединения 1 была установлена рептгепоструктурным анализом до того,
как были получены химические доказательства.
Перегруппированная структура оксима 2 подтверждается невозможностью
обратного перехода соединения 1 в исходное вещество, в кислой среде, а также уменьшением
частоты связи С = N,-аналогичным для частоты связи С = О в соединении 1.
230
ГЛАВА 7
Задача 124. Установите структуры продуктов нижеприведенных реакций:
он
он
c9hJ2o3r
2
НТО,
1 моль,
■* chH-8o3«
Уф: 250км (Ю:0ОД)
ИК: 1730', j&9&a, -1
| СгО;
CgH1204R
4
CH2N^
C,uHn04K
5
ИЮ 1736, 3706 с.*-1
1;, СН3рН, CH3ONa
2. ОН", Н20, затем
нейтрализация
Ответ [Wiesner К., Bickelhaput F., В abin D, R., Gotz M.t
Tetrahedron, 9, 254 (I960)].
сн,о
сн~о-
но
он
7
СН30'
2
о ^соон
■7,30
3
сн3о-
СОО] [
4(R)
4
«/
)СПч
CILO
«
CH..O-
A
О -
II
ЧЮ
УФ-спектр соединения 3 указывает на присутствие фрагмента а, р-неиредельного кетона
пмГЛ^°ГЛ01^еНИС наол|°Дается ПРИ большей длине волны, чем для^'сопряженных цикло-
"витенонов (для циклопептенонов обычно наблюдается гипсохромный сдвиг на 8—10 нм
ПЕРЕГРУППИРОВКИ
231
по сравнению с замещенными циклогексенонами [46]), такие прецеденты известны. ИК-
спектр также больше согласуется со структурой пиклопентенона, чем циклогексенона [47].
Последовательность реакций 4 -+ 5 -> 3 включает конденсацию Дикмана с
образованием дикетона А, который раскрывается по схеме обратной реакции Дикмана с
образованием кетона Б. Очевидно, простая эфирная группа соединения 5 не может
элиминироваться хотя и находится в р-положепии к сложноэфирнои группировке, поскольку
такое элиминирование должно было бы привести к напряженной двойной связи в голове
моста (нарушение правила Бредта [48]). Однако в соединении Б отсутствует мостикова*
система, и элиминирование протекает легко.
Задача 125. Установите структуры и конфигурации соединений 3
7->10.
О
" СН3
О
о
Ч/\
\
сн3соо
С23Н3406
х\/.
/\/
ОН > 0а4НзвО88
.■ у. I "" пиридин * °
чон
ИК аналогичен 4
О
КУ
1
сн3он
трет-С4Н9ОК
торет-С4НеОН
CH8S02C1,
mpem-CeHgOK
/\
\/
'/
«ч
ОН
/\А/\
сн3
он
СН3СОО Н
|\/
mpem-C4H»OK
СгзНзйО*
I CH3SO2CU
пиридин
CH8S02C1, mpem-^4119""- •"*•"" "»-»-" г „ n q
С**1*0» ^^? С**Нз°°а5 яРы^ Сй1Нзо°5 ^"^с^о-н C2*Hse0fiS
кон
СНзОН
С21Н30О4
9
УФ: 279 нм (9450)
(СЯ3СО)20,
пиридин
СабНз^е
10
УФ: 241 нм (10750)
УФ: 282 нм (8300)
ИК: 1739, 1712, 1669,
1618 см-1
f I кон
i. Этерпфнкация i 1 н п
2. третп-С4Н9ОК I сНдОН
СООН
С21Нза05
ИК: 3571, 3448,
1730, 1712,
1370, 1250,
1176 сж-1
232
ГЛАВА 7
Ответ [Wendler N. L., Tetrahedron, 11, 213 (1960 ;WendlerNL Та-
u Ь D., I. Am- Chem. Soc, 82, 2836 (I960)]. L" U
chjcoo-
CH3C00
I
'СЩ02СН'з
о о о---но
В (3-OH)
4
CO OH
CH,
jfe
CH3
OH
ъ
в (3-OH)
Г i
10(3-OCOCH3)
о
б (3-OH)
Структура соединения 5 выводится из его соотношения с соединением 6.
Образование & ив мезилатов 3 и 4 можно представить схемой
'О— Н
"(^OS02CH3
Образование соединения 5 из 3 и 4 показывает, что мезилат 3 существует в форме
ванны> тогда как мезилат 4 — в форме кресла, хотя эти аргументы не однозначные.
4п*>п иК-спектр соединений 3 и 4 интерпретируется следующим образом: 3571, 3448(ОН)*
ПЗО и 1250(ОСОСН3), 4712(С = О) и 1370 и 1176 w-^RSOaO—).
ПЕРЕГРУППИРОВКИ
233
Превращение 1 -*■ 7 проще всего интерпретировать как миграцию метила,
катализируемую основанием, что приводит к конфигурации, показанной на схеме
"ОН
Прямые доказательства миграции метила отсутствуют; в других случаях подобные
перегруппировки протекают преимущественно со сдвигом связей цикла, а не миграцией
метила [49].
Последовательность превращений 7 -»- 9 приводит к структуре диосфенола
ОН'
н,с
он
сн.
D^C
9
Превращение мезилата 8 в дикетон 5 включает, во всяком случае формально,
перегруппировку метила, обратную наблюдаемой при переходе 1 -»- 7. Разумно, хотя и не
строго доказано, принять в качестве промежуточного соединения в этой серии реакций
соединение 3, которое далее нормально перегруппировывается с образованием дикетона 5.
Принимается, что различные обратимые миграции обусловлены тенденцией
системы к образованию конечных продуктов типа 9 и 5, которые в присутствии оснований
дают стабильные анионы.
♦Т JI Т Е Р Л Т У V А
1. Molecular Rearrangements, Vols. 1 and 2, de Mayo P., ed., Wiley-Interscience, N.Y.,
1963.
2. Collins С J., Am. Chem. Soc, 77, 5517 (1955).
3. В u г г о w s W. D., E a s t m a n R. H., J. Am. Chem. Soc, 81, 245 (1959).
4. Д о п а р у м а Л. Г., Хельдт В. 3,, в книге «Органические реакции», том 11,
изд-во «Мир», М., 1965, стр. 7.
Gould E.S., Mechanism and Structure in Organic Chemistry, Holt, N.Y., 1959.
Meisenheimer J., Ber., 52, 1667 (1919).
55, 1 (1955).
J. E., Quart. Rev.,
Wesson R. L., J.
Soc, 78, 2298 (1956).
J. Am. Chem. Soc
10
11
12
13
14
6
7. P a u s о n
8. S e 1 m a n
9. House H
House H
House H
House H
P. L-, Chem. Rev.
S., E a s t h а ш
. 0., R eif D. J.,
. O., J. Am. Chem.
R eif D. J.
14; 221 (1960).
Am. Chem. Soc, 79, 2490 (1957).
R e i i D. J., J. Am. Chem. Soc
Long F. A., Olson A. R., J. Am. Chem., Soc
Б л а т т
стр. 455.
79, 6491 (1957).
77, 6525 (1955).
58, 2214 (1936).
А. Г., в книге «Органические реакции», том 1, Госиниздат, М., 1948,
234
ГЛАВА 7
15*.С мит Л., в книге «Органические реакции», том 1, Госиниздат, М-, 1948, стр. 493.
16. К е н д е Э. С, в книге «Органические реакции», том 11, изд-во «Мир», М., 1965,
стр. 267.
17. R i с h а г d G., СИ., 200, 1944 (1935).
18. М с Р h e e W. D., К 1 i n g s b e r g E., J. Am. Chem. Soc, 66, 1132 (1944).
19. Tchoubar В., S а с k u г О., C.R., 208, 1020 (1939).
20. С о р е А. С, Graham E. S., J. Am. Chem. Soc, 73, 4702 (1951).
21.Loftfield R. В., J. Am. Chem. Soc, 73, 4707 (1951).
22. В art let t P. D., К ice J. L., J. Am. Chem. Soc, 75, 5591 (1953).
23. Gocring H., Olson A. C, J. Am. Chem. Soc, 75, 5853 (1953).
24. С г e g e e R., К a s p а г R., Ann., 560, 127 (1948).
25. Mernwald J., J. Am. Chem. Soc, 77, 1617 (1955).
26. Meinwald J., Cornwall С. С, J. Am. Chem. Soc, 77, 5991 (1955).
27. M e i n w a 1 d J„ H w a n g H. C., J. Am. Chem. Soc, 79, 2910 (1957).
28. To у стер О., в книге «Органические реакции», том 7, ИЛ, М., 1956, стр. 409.
29. StreitwieserA., Jr., S с h a e f f е г W. D., J. Am. Chem. Soc, 79, 2893 (1957).
30. M о ore M. L-, in Organic Reactions, Vol. 5, Wiley, N.Y., 1949, p. 301.
3i. Leonard N. J., Sauers R. R., J. Am. Chem. Soc, 79, 6210 (1957).
32. Д е Тар Д. Л. Ф., в книге «Органические реакции», том 9, ИЛ, М., стр. 529,
33. Woodward R. В., S i n g h Т., J. Am. Chem. Soc, 72, 494 (1950).
34. Ziegler K., Hafner K-, Angew. Chem., 67, 301 (1955).
35. Hafner K., Angew. Chem., 70, 419 (1958).
36. Reid D. H., Chem. Soc. (London), Spec Publ., 12, 69 (1958).
37. Келлер-Ширлейн Ч., Хейльброннер Е-, в книге «Небензоидпые
ароматические соединениям, ИЛ, М., 1963, стр. 279.
38. А г с u s С. L., В е n n e 11 G. J., J. Chem. Soc, 1955, 2627.
39. M e i n w a 1 d J., Y a n k e e I о v J. A., Jr., J. Am. Chem. Soc, 80, 5266 (1958).
40. Блик Ф.Ф.,в книге «Органические реакции», том1, Госшюиздат, М., 1948, стр. 399.
41. В е г s о n J. A., R e m a n i с k A., J. Am. Chem. Soc, 83, 4947 (1961).
42. S e 1 t z е г S., J. Am. Chem. Soc, 85, 1360 (1963); Goldstein M. J.,
Thayer G- L., Jr., J. Am. Chem. Soc, 87, 1925, 1933 (1965).
43. J о h n s о n R. G., Ingham R. K., Chem. Rev., 56, 219 (1956).
44. К о о g m a n E. S., Breederweld H., Rec. trav. Chim., 76, 297 (1957).
45. AllenM. J., Organic Electrode Processes, Chapman and Hall, London, 1958, pp. 95—
115.
46. F r e n с h H. S., J. Am. Chem. Soc, 74, 514 (1952).
47. Наканиси К., Инфракрасные спектры и строение органических соединений,
изд-во «Мир», М., 1965, стр. 51.
48. F a w с е 11 F. S., Chem. Rev., 47, 219 (1950).
49. Turner R. В., P e r e 1 m a n M-, Park К. Т., J. Am. Chem. Soc, 79, 1108
(1957).
* Дополнения переводчика.
ГЛАВА 8
Радикальные %i фотохимические реакции
В современных учебниках по органической химии радикальные реакции
обычно не рассматриваются, вследствие чего многие студенты довольно плохо
их знают. Однако хороший обзор этих реакций имеется в книгах Гол да [1]
и Хайна [2].
Фотохимия получила свое развитие главным образом в последнее
десятилетие, но всякие попытки обсудить ее в целом усложняются резким
несоответствием механизмов фотохимических реакций с механизмами обычно
наблюдаемых реакций. Значительные проблемы возникают при описании
фотовозбужденных состояний, которые иногда, с точки зрения большинства
химиков-органиков, являются просто бессмысленными. Очень часто
необходимо выбирать между простым описанием, которое сильно искажает
действительность, и более сложным, но менее наглядным.
Среди других пособий по радикальным реакциям можно
рекомендовать монографии 13, 41. Фотохимические реакции подробно рассмотрены
в работах [5—13].
Задача 126. Установите положение метки в продукте следующей
реакции:
])NBr (NBS)
.„. (СеН6СО)20Е
* = 14С
Монобромид
Ответ [обзор по химии N-бромсукцинимида см. Horner L., Wenkel-
m a n n E. H., Angew. Chem., 71, 349 (1959)].
Обычно N-бромсукцинщшд (NBS) реагирует с олефипами по цепному радикальному
механизму с образованием бромидов аллильного типа. В течение долгого времени
принималось, что переносчиком в этой цепной реакции является радикал сукцинимида. Однако
недавно было показано, что NBS реагирует с НВг с образованием Вг2 в постоянной и очень
малой концентрации, который и является истинным бронирующим агентом [1-4].
236
ГЛАВА 8
О
Br
hv
или инициатор
О О
. / \
>n-n/ |+бг2
V
о о
S /
[ yN~ Вг+НЕг • > \ \
О
Вг*+НВг+
yNH + Bre*.
Л
о
—с=с-с— -л
-с-с=с—
J I I J
н
Br
—С—C=G—
I I I
+
Br
вится^вгаален^™6 С^п?«Я молекУЛыприводит к тому, что ряд положений
становится эквивалентным, поскольку промежуточный радикал действительно «свободный»
и его образование и галогенирование протекают независимо ствительш> «св00оД*ыи»
Вг
/х.
\у'
i
\/
./К
\/
Вг *
\/
Вг
I*
/V
Вг
\/
rv
Делят?^^^^^ равновероятно, то метка будет распре-
Вг
\/
*
*/2
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
237
Заметим, что такое распределение получается при усреднении очень большого
числа молекул, большинство из которых немечено {действительная концентрация 14С очень
мала) и почти нет таких молекул, которые бы содержали больше одного меченого
атома углерода.
Наличие в исходном циклогексене метки в двух положениях объясняется
невозможностью различить два атома углерода на концах симметричной двойной связи
О ОН
I*
Л
-НаО
\>
Задача 127. Приведите механизмы следующих реакций:
С6Н5
н /-л т НзгРевание
сн3о2с
СОаСН3
Нагревание
сеН5
сн3о2с
скнк
СО^СН;
HN-
Нагревание /
■со2сн3
Н .С6Н,
6П5
со2сн3
н
с„н
6,J5
Ответ [Jones W. M,, J. Am. Chem. Soc., 80, 6687 {1958); 81, 5153 {1959)].
Общий механизм этой реакции следующий:
+ N.
Первая стадия — это медленное превращение пиразолина-2 в пиразолин-1 [45], а вторая,
быстрая стадия представляет собой стереоспецифическое отщепление N2 с образованием
циклопропана. Такое таутомерное превращение пиразолинов должно приводить к
наиболее стабильному пиразолину-1, т. е. к такому, в котором заместители при С3 и С4
находятся в траке-положении. Это стерео химическое расположение сохраняется и в
образующемся циклопропане
238
ГЛАВА Ё
/С6Н5
Щ X ' Н На гр евание
/ \/С02СН,, *-
со2сн3
Н
сн3о2с
соасн3
-н "н
со2сн3
СцЦ
«"■5
-СбН5
HN 6-Н N t.H
/ \/Н Нагревание // \^н Нагревание Зеркальное
•■С02СН3 ИЧ^""С02СН3 изображение А
С02СН,
1Г со3сн3
со2сн.
Н-.-реващи! я У^ Нагревание с Н,Оа С /\ со
6ДЖ5 *\1
2СН3
■сйн
6 "5
со2сн,3
н соаСН3
н
н
Задача 128. Приведите механизм следующего превращения:
н сн3
о=ссн.
ООН
HarpeeaFiHe
Ответ [Schmidt G. A., F i s h е г G. S., J. Am. Chem. Soc, 76, 5426 (1954)1
Предложен следующий механизм:
СН,
ООН
Гидроперекись
пинана
Нагревание /"^""""^^Ч
+ НО*
О .
RH
+ R
ч
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
239
Выход кетона возрастает в присутствии пинана, являющегося донором атомов
водорода.
Задача 129. Объясните, как влияют добавки ацетата натрия и серной
кислоты на следующую реакцию:
N0
Метиловый спирт, 25° Бензол — Анизол
С6Н5—N—СОСН3
+CH3COO-Na+
0,04 н. H£S04
25—30% 5—10%
40—45% 4%
10%
55—75%
Ответ [D е Tar D. F., J. Am. Chem. Soc, 73, 1443 (1951)].
C0H5-N
NT-
О
IT
CCH,
— с6н5-
■N CCH,
N—О
CH3CO- +N2 +C6H5-
C6Hf
C5H5OCH3
CH3OH
СЙН
+ N,
6"S
н+tl
о
CeH£-N=N-+ CH3CO
Медленной стадией реакции является перегруппировка N-нитрозоацетанялида
в диазоацетат А [16,17). Предполагается, что диазоацетат А может разлагаться с
образованием фенильного радикала, который далее отщепляет водород от растворителя с
образованием бензола. Существует также равновесие с ионизированной солью диазония Б,
которая разлагается медлеппее с образованием фенильного карбониевого иона, который
в свою очередь реагирует с растворителем с образованием анизола. Добавление кислоты
сдвигает равновесие в сторону Б за счет связывания образующегося ацетат-иона в
уксусную кислоту и, таким образом, увеличивает выход анизола по сравнению с бензолом.
Прибавление ацетат-иона повышает концентрацию А и вызывает противоположный эффект.
Задача 130. Приведите механизмы следующих реакций:
(130.1)
hv
240
ГЛАВА 8
(130.2)
oKAy^Y СгН5°н
о t^Q
CH3COOH
fly
Эфир
н3о
н2о \h..
Ответ.
{130.1) [Buchi G.t Goldman I. M., J. Am. Chem. Soc, 79, 4741 (1957)].
о h
вЬ~-&А -&r
***JZ^^" ТраКТуеТСЯ КаК Реальная, а стрелки обозначают одно-
140- СЬ°Л I?.'» *п Пт D4H' RV d ! Ч а I ° Р" S h a f i 4 M.t J. Chem. Soc, 1958,
14U, Chapman O. L., Englert L. F., J. Am. Chem. Soc, 85, 3028 (1963)].
Сантонин
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
241
Люмисантонин
н2о
НООС
Фото с а нто новая
кислота
Эта реакция изображена в терминах разделения зарядов (отвлекаясь от
действительной природы возбужденных состояний) вследствие большого сходства некоторых стадий
с более обычными карбоний-ионными перегруппировками [19J. Более вероятно, однако,
что промежуточно образуются радикалы, получающиеся ив фотовозбуждеиных трип
летных состояний.
Сложность перехода от сантонина к люмисантопину подтверждается тем, что, в то
время как люмисантонин (А) при обработке кислотой даст лактон изофотосантониновой
кислоты (Б), облучение сантонина в слабокислых условиях не приводит к продукту Б.
Таким образом, более вероятно промежуточное образование соединения 1, которое может
непосредственно превращаться в продукт Б.
Фотоизомеризация люмисантонина в эфире приводит к веществу В, которое
находится в фоторавновесии с кетеном 2 (доказательства неточности таких представлений
см. 119а]). Отсутствие воды более благоприятно для образования соединения В, но в
присутствии воды получается кислота Г.
он,
16-374
242 [ГЛАВА 8
Задача 131. Приведите механизмы следующих реакций:
(131.1)
\
N
hv
(131.2)
/\/
И' S
N
R' S
II \ /
\=С
R Я
hv
/'
\
R
СН3 СН3
свн5ссн3-нснз9=снснз * сен5
о
I
о-
сн<
сн,
Ответ.
(131.1) IK i г m s e W., Нагпег L., Ann., 614, 4 (1958); Kirmse W., Ancew
Chem., 71, 537 (1959)].
■■I ^>f^^
/iv
»- Ич
-N2 ^
с
f *.c
c*
I
R
C +
? ^
s
R. ^R'
С
II
с
II
s
/
Исходный 1,2,3-тиадиазол формально эквивалентен диазокетону, и первая стадия
точно такая же, как в фотокатализируемой перегруппировке Вольфа [20J
О О
II- + AV II
RCCHN = N > RCC: —> RC = C = 0
-N2 .
н н
(131.2) [В ц с h i G , I n m a n G. G., Lapinsky E. S., J. Am. Chcm. Soc*
76, 4327 (1954)].
II
О
C«H
hv
э-ч. /CH3
С*
I
О-
H3C^C /CH3
II '
CH3 СНз
I I
CgHg—С* wC—CHj
I I
О CHCH3
с6н5-
^
X
1
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
243
Получающиеся продукты соответствуют образовапию промежуточного наиболее
стабильного соединения 1 в результате присоединения фотовозбужденното триплстного
(бирадикального) состояния ацетофенона к алкену.
Задача 132. Предскажите строение продуктов следующих реакций:
(132.1) О
/\
/IV
(132.3) /^ О
+ C,H5OCN,
hv
(132.2)
(132.4)
о>
/IV
/ОСОСН3
'ОСОСНз
/IV
Ответ.
(132.1) [В 1 а с е t F. E-, Miller A., J. Am. Chem, Soc, 79, 4327 (1957); Sri-
nivasan R., J. Am. Chem. Soc, 81, 1546, 2601, 2604 (1959)].
Реакция приводит к большому числу продуктов, среди которых основными будут
сно
|| I7 /\ /\/Ч СО
\/ |_|
(132.2) [Cremer S., Srinivasan В., Tetrahedron Letters, 21, 24 (I960)].
Главными продуктами являются СО и гексадиен-1,5 (1); однако образуется также
и небольшое количество (5%) ожидаемого бидикло-[2,2,0]-гексана (2)
(132.3) [Н a f n е г К., К о п i g С, Angew. Chem., 75,89 (1963); U ojs ki W
M aricich T. J., M a 11 i n g 1 у Т. W., Jr., 3- Am. Chem. Soc, 85, 1200 (1963)].
Av
U3—COC2H5
N2 + N — COC2H5
Карбэтовдшнтрсн
О-
о
II
CQC2H5
О
II
N—COC2U5
(132.4) [Barton D. H. R., Helv. Chim. Acta, 42, 2604 (1959)].
Cn
_^OCOCH3
hv
^•ОСОСНз
^ ^ОСОСИз
ЮСОСНз
16*
244
ГЛАВА 8
Задача 133. Фотолиз органических нитритов, имеющих определенное
строение, может привести к внутримолекулярному обмену нитрозогруппы
и атома водорода, соединенного с б-атомом углерода. Эта реакция удобна
для избирательного введения кислородсодержащей функции к
неактивированному атому углерода.
ON H
о с
1 1
с с
\/
с
hv,-~NO
^
+ -NO
н
* \
о с
1 1
с с
\ /
с
н
0 -с
1 I
с с
\/
с
Продукты
реакции
(133.1) Фотолиз нитрита, полученного из к-октилового спирта, дает
оксим. Приведите структуру оксима.
CH3{CH2)eCH2ONO
i. hv в гексане
2. Нагревание
■*■ Оксим
(133.2) Фотолиз нитрита (1) дает три оксима — 2, 3 и 4. Оксим 2 при
обработке смесью нитрита натрия и уксусной кислоты приводит к
соединению 5. Установите строение оксима 2 и объясните образование
соединений 3, 4 и 5.
сн2ососн$
с=о
^^^KJ—J
hv
-NaN02
CH3CQOH
НО
сн2ососн3
с =о
N
НО''
3 син - форма
СН20СОСН3
1
II
N
чон
4,дкты-форш
СН2ОСОСНа
1
с=о
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ 245
(133.3) В ряде случаев при фотолизе нитритов получаются
промежуточные продукты, образование которых обусловлено иным течением реакции.
Ниже приводятся два таких случая. Объясните эти результаты.
ONO
(б)
hv
толуол
fiv
бензол
~rxr
Ответ [общий обзор см. Nussbaum A. L-, Robinson С. Н., Tetrahedron,
17, 35 (1962); механизм рассмотрен в статье Akhtar M., Pechet M. M., J. Am.
Chem. Soc.r 86, 265 (1964)].
(133.1) [Kabasakalian P., Townley E. R., J. Am. Chem. Soc, 84,
2711 (1962)].
C4H8
H
CH ONO
I I hv
HoC CH2
\/
с
н2
H
C4HS-CH -0
I 1
H2C CH2 + -NO
V
Hz
H
C4H8—С- HO
H2C CH2
\ /
с
н2
OH
/
N
It Нагревание
C4H8—CCHaCH2CH20H < Димер <-
C4H8
-NO
NO
i
CH OH
I I
H2C CH2
V
H2
Образование димера характерно для первичных и вторичных нитрозосо единении;
он может быть выделен из смеси, подвергаемой фотолизу, но при хранении или
нагревании перегруппировывается в изомерный оксим. ,Лпал\.
(183.2) [Barton D. H. R., Beaton J. M., J. Am. Chem. Soc, 82, 2641 (I960),
83, 4083 (1961)].
246
Г Л А В А 8
сигососн3
си£ососн,
I 21 И С I
HON = СИ
сн2ососн3
I
с=о
t шо2
t -NO
сн^ососн,
НО I л
I с=о
-СИ
ко
N
НО'||
к
Использование этой фотолитической реакции позволяет осуществить замечательный
четырехстадийный сиптез важного гормона альдостсропа (в виде ацетата) с выходом 15%,
считая на относительно доступный ацетат кортикостерона. Важность этого можно
оценить из следующего факта: природные источники настолько лимитируют получение
альдостерона, что структурный анализ его был проведен всего с 56 мв вещества [21].
{133.3) (a) [R e i m a n n Н., С а р о m a g g i A. S-, Strauss T-, 0 1 i
veto E. P., Barton D. H. R., I. Am. Chem. Soc, 83, 448 (1961)].
ONO.
V
HO
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
247
Перегруппировка изображена в виде последовательных стадий (А -*- Б -*- В), хотя
ее можно представить как согласованный процесс А->В. Возможно, перегруппировка
. происходит вследствие способности карбонильной группы стабилизировать электронное
состояние типа Б, в результате чего становится возможным энергетически выгодный
дуть превращений с образованием такого продукта, в котором будет отсутствовать
напряжение, имеющееся в исходпой стероидной системе.
(б) [Robinson С. Н., Gnoj О., Mitchell A., Wayne R., Town-
ley Е-, Kabasakalian P., Oliveto E. P., Barton D. H. R., I. Am.
Chem. Soc., 83, 1771 (1961)].
fiv
-NO
0
11 H n
он
I
N^ ^O
-R7
о _
I
N^ ^O
№f^
{?
-II н
pJ^N<Lo-
IXJ
Вышеуказанный механизм был предложен в соответствии с образованием гидроксамо-
бой кислоты. В некоторых случаях были выделены два изомера, вероятно отличающиеся
конфигурацией С13-метильной группы.
Однако общая теория, которая предсказывала бы, какой изомер получится в
каждом случае, отсутствует.
Задача 134. Установите структуру изомера.
О
\/ II
1 "^
/IV
АА
\/
о ч
+ Изомер
ЯМР-спектр изомера (шкала т, СС14, с — синглет, д — дублет, т —
триплет): 8,94(6Н, с), 8,3-8,7 (6Н, мультишшт), 7,96(311, с), 6,84{2Н, Д
(J = 7,1 гц), 5,49 (1Н,д) (/ = 2,6 гц), 5,04 (Ш, д) (/ = 2,6 гц), 4,67<Ш, т)
{/ = 7,1 гц).
248
ГЛАВА 8
Ответ [d e Mayo P., S t о t h е г s J. В., Yip R. W„ Can. J. Chern., 39,
3135 (1961)].
ЯМР-спектр подтверждает наличие следующих фрагментов [22]:
т=8,94
8,3-8,7
7,96
4,67 Н-С=;
5,49 5,04
(низкое значение константы спин-
спинового взаимодействия /
согласуется с первым фрагментом)
Структура А, имеющая циклопропановое кольцо, хотя и была предложена в
литературе [23], тем не менее не согласуется с данными спектра ЯМР, который однозначно
свидетельствует в пользу диеноновой структуры Б.
4,6 7
^СН3-<-7,96
J/e,«,5,t
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
249
Перенос водорода происходит во всех системах, имеющих следующую геометрию:
\А.
/\
hv
\у\
он
L/\
}
\/\.
Задача 135. Установите структуры продуктов следующих реакции:
СН3СОО'
СП
1
с =
х^
"Y
У^
^ J
\Х
с^н^Оз
}
=о
•н
/
hv / у Нюх
1
poct3t
гшрпдпп
ер
с2зН:и02
3
1 L.Os04
I2.HIO4
CtiIIuOa
2
N^LiAlH*
i
^ ТНадкнслота
СвН^Оа
4
| 1.0sO|
I2.HIO1
с22нЛ9о3
6
СН3%*
Для соединения 5 ИК-спектр: 2760, 1728, 1709, 1256 см'1; оно обладает
восстанавливающими свойствами; для соединения 6 ИК-спектр: 1778, 1735,
1245 см-К
Наряду с соединениями 1 и 2 при фотохимической реакции образуется
также соединение
/\1/\/\/
СН3СОО
Ответ [Б и с h s с h а с li е г P., Cereghetti M., Wehrli H-, Sliaf
£ п е г К., JegeiO., Helv. Chim. Acta, 42, 2122 (1959); С в г eg net11 М , W e
г 1 i H., S с h a f f n е г К., Jeger О., Helv. Chim. Acta, 43, 354 (19b0J].
250
ГЛАВА 8
-СНзСОО
170Q ^^та™вительныс свойства соединения 5 в сочетании с его ИК-спектром (2760,
отсоединений ,T??S££S™ ГРУППЫ -СН0' обР—йся нрИР неводе
I f
-СН = С~ > -СНО 0 = С-
Аналогичная последовательность реакций, сопровождающаяся отщеплением формальде-
СН2
гида при переходе 4-^6, позволяет установить наличие группировки — СН2 — С —
СНз
для соединения 4 и группировки - СН2 - С(ОН)- для соединения 1.
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
251
Поскольку эмпирическая формула требует для соединении 1,3 и 4 пентациклической
*TBvKTVDbi, то рассмотрение строения исходного вещества приводит к двум возможным
изошшам Конфигурация соединений 1 и 2 вытекает из реакции присоединения к их
ненасыщенным производным 4 и 6. Последние можно изобразить так:
f
\^s
R—СН2или О
Н*
М-1
и можнг ожидать, что реакция присоединения будет легче протекать с менее
пространственно затрудненной стороны (а) циклопентанового кольца.
^°
>
CH3MgI
.сн3
-он
с^
^
сн,
о-..
>
Нядквслота
LiAlH;
,ОН
—СН,
с^^> ^с^^Ъ
Обе реакции присоединения согласуются с этим предположением.
Предложенный механизм с обычными оговорками об изображении реакций с
возбужденным электронным состоянием выглядит следующим образом:
О
11
Н С
I
ней 1 Г4-" > I -> 1 к 2
•hi
Альтернативное направление, приводящее к побочному продукту, будет следующим:
нс3\
+. сн3ссна
252
ГЛ ABAS
Известно много аналогий для последней стадии как при фотохимических [241, так
я при масс-спектральных реакциях [25]. Равным образом было показано, что и другие
фотовозбужденные кетоны могут атаковать метиленовые группы (как bhvtdh- Г26 271
так и межмолекулярно [26]). l ' J'
Задача 136. Установите строение продуктов А и Б и приведите
механизм их образования (см. таблицу).
Спектральные данные*
Таблица 8.1
Спектр
ИК, cjtt-i
УФ, нм
ЯМР (т)
А
1689
226 (6400)
8,60 (ЗН, с)
6,95 (Ш)
6,45 (ЗН, с)
5,07 (1Н)
4,34(1Н,д,/=
2,55 (Ш, д,/ =
= 6,0
= 6,0
щ)
щ)
Б
1700
223 (9700)
7,94 (ЗН, с)
6,68 (Ш)
6,42 (Щ)
6,33(ЗН, с)
4,91 (Щ)
4,25 (1Н)
* с — синглет, д — д у Слет.
сэню03
СН,о Й
С!>Н|оО:
СН.,
hv
СН3ОН
СН,ОН
kv, Н20
— ■*
или
к.1° +
ft г- Н„0
Н..О'
СН,ОС
I Am°pfm [5 a u£.e ft,.?- G" Koch K> Chapman О. L., Smith S. L.,
J. Аш. Chem. Soc, 83, 1768 (1961); J. Am. Chem. Soc, 85, 2616 (1963)].
I
РАДИКАЛЬНЫЕ И ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
253
6,45
сн,с^
6,9
,07
■1,34
6,33
сн3о^
4,91
6,42
U-,
,У4
6.6К
СН,
СН3
8,60
А
Структуры А и Б могут быть выведены па основании анализа спектров ЯМР и
подтверждены данными гидролиза. Термическая перегруппировка вещества А до
соответствующего тропона является общей для соединений такого типа.
Образование этих соединений обусловлено глубокой фотоизомеризацией исходных
■соединений, протекающей по следующему механизму:
СНгО
В квадратных скобках изображено фотовозбужденное состояние. В соответствии
€, этпм механизмом было найдено, что
СН3о
СН,о.
hv
УФ-спектры отражают наличие л — п-взаимодействия, необходимого для пере-
групнировки и, вероятно, обусловленного геометрией этих циклических систем. Оба
вещества имеют максимум поглощения при большей длине волны, чем циклопентепоны,
возможно, вследствие увеличения сопряжения в возбужденном состоянии.
н2о:
о Г он' fo
сн.,0.
снэ
hv
или
о ону ^о он о
tYW Нзэ ~СНз°г 6 JHl°^
сн,
снд
сн,
Конечной стадией в последовательности этих реакций является гидратация эфира
енола (катализируемая светом или кислотой), протекающая по схеме ретроальдольной
конденсации или расщепления, обратного реакции Кляйзена.
254
ГЛАВА 8
ЛИТЕРА ТУРА
i. Gould E. S., Mechanism and Structure in Organic Chemistry, Holt, N.Y., 1959.
2. Hine J.f Physical Organic Chemistry, 2nd ed., McGraw-Hill, N.Y., 1962.
3. Walling C, Free Radicals Solution, Wiley, N.Y., 1957. Наиболее исчерпывающая;
монография по свободпорадикальным реакциям. Рассмотрены также процессы
полимеризации.
4. Sosnovsky GM Free Radical Reactions in Preparative Organic Chemistry, Ma-
cmillan, N.Y., 1964. Основное внимание уделено получению свободных радикалов.
Имеется очень полный список литературы.
5. Де Майо П., в книге «Успехи органической химии», том 2, изд-во «Мир», М.„
1964, стр. 368. Общее введение в фотохимию некоторых простых систем,
6- De Mayo P.,Reid S. Т., Quart. Rev., 15, 393 (1961). Фотохимические
перегруппировки.
7. Chapman О. L., в книге «Advances in Photochemistry», Vol. I, Wiley-Interscience,
N.Y., 1963, pp. 233—420. Рассмотрены в основном фотохимические перегруппировки.
Более специальным вопросам посвящены несколько томов этой серии.
8. Zimmerman H. E., Schuster D. I., J. Am. Chem. Soc, 84, 4527 (1962).
9. Z i m m e г m a n H.E., Wilson J. W-, J. Am. Chem. Soc, 86. 4036 (1964).
10. Hammond G. S., Saltiel J., Lamola A. A., Turro N. J., Brad-
shaw J. S., Cowan D. О,, С о u n s e 11 R.C.Vogt V., Da It on C, J.
Am. Chem. Soc, 86, 3197 (1964).
11. Турро Н., Молекулярная фотохимия, изд-во «Мир», М-, 1967. Общее введение-
в теорию и предмет органической фотохимии.
12. К an R. О-, Organic Photochemistry, McGraw-Hill, N.Y., I960. В этой работе
меньшее внимание, чем в работе [11], уделено вопросам теории. Рассматриваются в
основном практические аспекты фотохимических превращений.
13. К а л в е р т Дж., Питтс Дж., Фотохимия, изд-во «Мир», М-, 1968.
Исчерпывающий обзор, включающий вопросы теории, применение в органической химии и
экспериментальные методы.
14. House Н. О. Modern Synthetic Reactions, Benjamin, N.Y., 1965, pp. 159—161.
15. Jones W. M., J. Am. Chem. Soc, 82, 3136 (I960).
16. H u i s g e n R., Harold G., Ann., 562, 137 (1949).
17. H e у D. H., Stuart-Webb J., Williams G. H., J. Chem. Soc, 1952,
4657.
18. E у д з и к е в и ч Г., Д ж е р а с с и К-, У и л ь я м с Д., Интерпретация масс-
спектров органических соединений, изд-во «Мир», М., 1966.
19. В а г t о n D. H. R., d e MayoK., S h a f i q M. J. Chem. Soc, I960, 1.
19a. Zimmerman II. E., II a h n R. C, Morrison H., W a u i M. C, J. Am.
Chem. Soc, 87, 1138 (1965).
20. К i г m s e W., H a r n e r L.t Ann., 614, 4 (1958).
21. Ф и з е р Л., Ф и з е р М., Стероиды, изд-во «Мир», М., 1964.
22. Jackman L. M., Application of Nuclear Magnetic Resonance Spectroscopy in
Organic Chemistry, Pregamon London, 1959.
23. В u с h i G., J a n g NX., J. Am. Chem. Soc. 79, 2318 (1957).
23a. Наканиси К., Инфракрасные спектры и строение органических соединений,
изд-во «Мир», М., 1965.
24. М а г t i n Т. W., P i 11 s J. N.. J. Am. Chem. Soc, 77, 5465 (1955).
25. Biemann K-, Mass Spectrometry, McGraw-Hill, N.Y., 1962, p. 119.
26. Y a n g N. C, Yang D--D. H-, J. Am. Chem. Soc, 80, 2913 (1958).
27. В e r n а г d M-, Yang N. C, Proc. Chem. Soc, 1958, 302.
ГЛАВА 9
Реакции окисления—восстановления
Общая литература к главе см. работы J1—6].
Задача 137. Расположите приведенные ниже холестанолы в порядке
уменьшения легкости окисления хромовой кислотой.
но
но'
но
Ответ (Ф и з е р Л., Ф и з е р М., Стероиды, изд-во «Мир», М., 19е4)"
Скорость окисления изменяется в следующем порядке: 5>4>^>l>^- ljTdM"";
определяющая скорость окисления хромовой кислотой, заключается в разрыве связи
С — Н в а-положении к гидроксилу, который и подвергается дальнейшему окислению^j.
При этом атом кислорода изменяет свое положение. Изменение положения атома кисло
рода при переходе от спирта к кетону может уменьшать стерические взаимодействия
между аксиальным гидроксилом и соседней аксиальной группой, что делает иереходн
состояние относительно более стабильным и приводит к увеличению скорости Р^^11;1:
Б первом приближении, чем больше первоначальное напряжение (которое можно свяла№
с числом 1,3-диаксиальньгх взаимодействий), тем больше «стерическое ускорение» процве
са. Это наглядно демонстрируют данные таблицы.
256 ГЛАВА 9
Таблица 9-1
Соединение
5 (llp-OH)
4 (4В-0Н)
2 (2р-0Н)
1 (lcc-OH)
3 (За-ОН)
1:3-взаимодействие
ОН (акс)—
СН8 (акс)
2
1
1
0
0
ОН (акс)—Н (акс)
1
2
1
3
2
скорость
окисления Сг6+
60
35
20
13
3
Задача 138. Приведите механизмы следующих реакций окисления:
О О
1. os
(138.1) Н3С
н,с
\с-снсн2он 2.н2о
> Н2С = 0 + СН3ССНз + НСОН
(138.2)
Н5С2
с2н5
он
Zn
Н,0
Перегон ка
fпаром
н5с,ч
НО'
"СпНг
сно
Ответ \В a i 1 е у P. S., СЪеш. Rev., 58, 925 (1958)].
(13SJ) [Bailey P. S., Chem. Rev., 58, 947 (1958)].
Показано, что образование аномальных продуктов при реакции озонолиза происходит
яри перегруппировке соединений, имеющих фрагмент
С = С— С—G, где G = 0, N, S.
/ I
РЕАКЦИИ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
257
При этом получаются продукты перегруппировки [7], как, например:
НэС,
н.с
/
;с= снсн2он
о о
^C-i—CHCH2OH -*- 3 ^С=0 + / \
ЩС' Н3С^
о снсн2он
сн2=о + неон
ПС—О — СН90Н -*-
?С^>СН20Н
Во многих случаях, включая и вышеприведенный, возможно также и альтернативное
направление с образованием нормального озонида [8].
Н3С f^~ С \ щс От-О
;;с=о + сн—сн2он -*■ ^с^^
щс^ ^-^ -* + н3с" \v0
:онп
сн-^сн,-*Пон
н3с. «
-+ 3 N;=o + нсо-
н,С"
н5с=о + н,о^
Еще одна возможность связана с гидролизом образующегося озонида (особенно
в водном растворе) до нормальных продуктов, которые могут окисляться далее 02i 03
или Н20Е.
Возможно, что в некоторых случаях вероятны все эти направления и выбор между
ними определяется структурными особенностями соединения.
(138.2) (Nielsen А. Т., J. Am. Chem. Soc, 79, 2518 (1957)].
CnH
с2н5
С2Н5
но—н
СоИ
о<Ч^
VJ<CsH
Лг хне
Zn
*>
Н,0
н5с2
2115
Н^Ь
R
С„Н-
Т"2
сн
00 иг
Альдольная "b^SV.
II II кинденсация ^
+ с2н5ссн *>
Г)
17—374
258
ГЛАВА 9
Реакция сложнее, чем показано на схеме. Это вытекает из того, что 2-кетобутаналь
не выделен как таковой, а получается смесь НСООН, СН3СН2СООН и СН3СН2СНО-
Поэтому, если это дикарбонильнос соедипение действительно является промежуточным
продуктом, оно должно получаться на стадии окисления, а не восстановления.
Образование муравьиной и пропиоповой кислот можно объяснить схемой, приведенной в ответе
(138.1), но объяснить получение пропионового альдегида довольно трудно.
Задача 139. Приведите механизмы следующих реакций окисления:
о.
(139.1) 0
но
1.03(СНС13)
2.Н20
о
03
(139.2) || Ц —>с6н5ссно+с6н5соон
CeH/VXC6H5
+• сн3соон
Ответ (обзор реакций озонирования см. В a i 1 е у P. S., Ghcm. Rev., 58, 925 (1958)].
(139.1) {Barton D. H. R., d e Mayo P., Shafiq M., J. Chem. Soc.
1957, 929).
HO
o.
HO
н,о
Лактонизацня
yfi
о
ii
CH3COH
Если гидрировать неочищенный озонид, то получается трикетон (А). Выделение
этого соединения показывает, что превращение первоначальной карбонильной группы в
карбоксильную протекает не ири образовании озонида,апри его гидролизе. Механизм такого
аномального озонолиза отличен от механизма, приведенного в ответе (138.1).
РЕАКЦИИ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ 259
rv_
(139.2) [В a i 1 е у P. S., С о 1 о m Ь Н. Ом Jr., J. Am. Chem. Soc, 79, 4238 (1957)].
o3
XI
\СЛНЙ О CrH-
СвНэ'^Оз^ C6HS
н5-1Г1<СбП5..+
° :o7<>—§
CfiH,
или cfi
Hs-^ooi^c^,
O-rO
-VLJ
*-o5
о о
12%
При дальнейшем озонировании с выходом 14% получается фенилглиоксаль за счет
окисления дибензоилэтилена. Главный продукт (81%)— бензойная кислота —
образуется, по-видимому, при 1,2-, а не 1,4-атаке фуранового цикла.
Задача 140. Приведите механизмы следующих реакций:
О
II С6Н5
сеП5 „ „ ™ „ /\/
(140.1)
(140.2)
_/=
у СвН5С03Н . |Ч
свн
6П5
\/
сосн
снчсоо
CrHe
нсо3н
^_
COCHj
СП3СОО
Ответ.
(140.1) [Cauquil G-, Rouzard J., C.R., 237, 1720 (1953)].
17*
260
ГЛАВА 9
с6н5—с==о
( \ /Ссн5
Y=c
/ ^СйНк
с6н5
скн.
о~
/ ^сен5
1
■°^ ^С«НЯ
^с6н5
-1
он
ель
CfiHc
■н+
,2i,/c6HS
\
скн.
Принимается, что реакция надкислот с олефинами протекает через образование
я-комплекса типа А, причем олефин является донором электронов по отношению к ;0.
& данном случае реакция может протекать нормальным образом с образованием эпоксида Б,
который далее легко перегруппировывается под влиянием следов кислоты.
Перегруппировка может осуществляться и непосредственно в л-комплексе без образования
диоксида Б.
Интересно отметить, что перегруппировка такого типа может происходить и в
обратном направлении 19].
/
О
/ СвН5
IH]
ОН
/ сен5 рвг3
CgHs
CeII5
/
ч
CflHg
(140.2) [D j e г a s s i C, Mancera O., Stork G., Rozenkranz G.,
J. Am. Chem. Soc, 73, 4496 (1951)1.
РЕАКЦИИ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
261
сосн3
сн.соо
сосн3
нсо3н
СН3СОО
Перегруппировка Н
СОСН3
сн3соа
Эпоксидирование протекает с пространственно менее затрудненной а-стороны.
Задача 141. Приведите механизмы следующих реакций:
R
! СН3
/\
(141.4)
СН3СОО
\
/\/
/Ч
HN03
/V/"
100е, 30 час
НООС | СООЙ
/Ч/\
НООС СООН
NaIo4
(141.2)
П
N СООН
н
ню4
N Ъ
II
О
N
262
ГЛАВА
Ответ.
(141.1) IT s u d а К., А г i m а К., Н а у a t s u R-, J. Am. Chem. Soc, 76,
2933 (1954); см. также Burgstahler A. W.f J. Am. Chem. Soc, 79, 6047 (1957)1.
CH3cOO
H +
COOH
Окисление
H3C^ JL .COOH
coon
Эта реакция (антрастероидная перегруппировка) была детально изучена Несом
и сотрудниками [10], особенно па примере эргостсрипа и Дв*п>-дегидроэргостерина. Эти
соединения при обработке кислотами в мягких условиях перегруппировываются с
образованием соединения 1.
(141.2) (В г a g g P- D., Hough L., J. Chem. Soc, 1958, 4050).
I I
H OH
Ю
- - OH-
+ co2 +hio3
H
I
о о
|юг
PEАКЦИИ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
263
о.
Н
о?:
О— 1 — 0
о
Q-
■NJO*
Г I
О—] —О"
II
о
Йодная кислота обычно не окисляет ос-аминокислоты, но в данном случае происходит
окислительное декарбоксилировапие. Механизм реакции спекулятивный, но согласуется
с тем фактом, что окисление Д1-пирролина (А) протекает только в щелочном растворе.
В присутствии кислот атом азота протопируется и образующаяся аммонийная соль
с периодатом не реагирует.
Задача 142. Реагенты А и Б дают при реакции
изомерные диолы. Установите конфигурацию полученных соединений.
Реагенты: А — окисление Os04, Б — обработка I2 — CTI3COOAg
в.СН3СООН, нагревание с CH3COOAg и 1 эке воды и, наконец, гидролиз
холодным водным раствором КОН.
Ответ [Woodward R. В., В г u t с h e r F. V., Jr., J. Am. Chem. Soc, 80,
209 (1958)].
A. Os04 атакует преимущественно пространственно менее затрудненную
сторону (а) с образованием ос-уис-диода. .
"о.
-О'
II
:os'
г*4^ч\^*н
■*о
UkA
он
-— ОН
н
Днол
Е. Исходная реакционная смесь является источником CH3COOI или 1+ОСОСН^п
который можно рассматривать как иодирующий агент. транс-Присоединение элементов
СН3С001 начинается с атаки 1+ или его эквивалента с ос-стороны и приводит к транс-
иодацетату (1), которому по аналогии с другими реакциями галогенирования можно
приписать диаксиальную конформацию [11]. Если иодид 1 нагревать с ацетатом серебра
в уксусной кислоте, то наблюдается отщепление иодид-иона, сопровождающееся участием
соседней ацетатной группы. Промежуточное соединение (2) реагирует с водой, давая цис-
ортоацетат (3), который расщепляется с образованием смеси двух tfuc-оксиацетатов 112].
Последние при омылении дают ft-i^uc-дио л.
264
ГЛАВА 9
to - о ^ й$г.
2 3
ю:г -гсс
^сн3
44 он
кОН *^\^^0Н
р- цис - Диол
.ОН
Задача 143. Укажите, какие продукты образуются при окислении
нижеприведенных соединений нериодатом в заданных условиях.
(143.1) СНО
НСОН
I
Н3СОСН 5°, 1 моль
НСОН 30°, 3 моля
НСОН
I
СНйОН
(143.2) HOCHjjCHCOOH 1 моль
NHa
(143.3) С2Н5
О*. А аО 3 моля
\/
РЕАКЦИИ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ 265
Ответ (Общий обзор реакций иериодатного окисления см. Джексон Э. Л.,
в книге «Органические реакцию, том 2, ИЛ, М., 1950, стр. 362; Bobbitt J.M., в
книге «Advances in Carbohydrate Chemistry», Vol. 11, Academic Press, N.Y., 1956, p. 1).
Как йодная кислота, так и тетраацетат свинца являются специфичными
окислителями, которые применяются для расщепления связи С — С между атомами углерода,
соединенными с амино- или кислородсодержащими заместителями. Йодная кислота
расщепляет следующие связи:
— С—С—
I I
ОН ОН
-U-
NHOH
_С—С —
I I
Nil NH
I I
С—С-
ОНО
—С—С
оо
Тетраацетат свинца окисляет также сс-окси- и а-кетокислоты.
Эти реакции включают циклический комплекс типа 1 и протекают быстрее, если*
гидроксильные группы занимают цис-положение [2].
1
он
I
С —ОН
—с-
0 /0-
/ \<
— С—О
i, x=i, Pb
I
—c=o
—C^O
I
(143.1) [Huffman G. W., Lewis B.
bach D. R., J. Am. Chem. Soc, 77, 4346 (1955)].
A., Smith F., Spriesters
CHO
I
HCOH
I
CH30CH
I
HCOH
I
HCOH
I
CH2OH
ЙО
Ш04
>-
CH2OH J)
OCH
CHO
H2q
>
30°
HCOH
+
CHO
CH
|0—I— H
HCOOH + CH20
2HK>4
CHO
Исходный углевод в растворе существует в циклической форме. Первая реакция
окисления приводит к эфиру муравьиной кислоты, который стабилен на холоду и не
содержит свободных а-гликольных групп. С повышением температуры этот сложный
эфир гидролизуется и протекает дальнейшее окисление.
266
ГЛАВА 9
{143.2) [N i с о 1 с t В. Н.} S h i n n L. A., J. Chem. Soc, 61, 1615 (1939)].
; hio4 н2о
HOCH2^CHC0OH * HCHO -f HN ^ CHCOOH —* NH3 1-OCCOOH
I
H
Если реакционную смесь оставить приблизительно на депъ, то наблюдается
медленное образование глиоксиловой кислоты. Однако ее образование легко отличается от
быстрой реакции расщепления ашшоспирта.
(143.3) [Wolfrom M.L., В о Ъ Ь i 11 J. M., J. Am. Chem. Soc, 78, 2489 (1956)1.
6c
C2H5
о
НЮ/
НЮ 4
UIOz
^соон
Ч^^соон
+ С2Н5СООН
Окисление метиленовой или метиловой группы, активированной двумя а-карбопиль-
ными группами, при обработке йодной кислотой является общей реакцией. Циклогексан-
дпоп-1,3 реагирует с 4 молями периодата с образованием глутаровой кислоты и С03.
Принимается, что реакция иротекает через последовательное окисление енольных форм
с образованием неенолизующегося 1,2,3-трикетона, который затем реагирует с
расщеплением цикла.
НЮ4
ШСЬ
2НЮ4
,-СООН
-. ^
соон
+ со2
Задача 144. Приведите механизмы следующих реакций:
(144.1) ОС2Н5
У\ 1. LiAlH* /\
| 2. Н30+
0^\/\СООС8Н5 О^/^СНаОН
РЕАКЦИИ ОКИСЛЕНИЯ— ВОССТАНОВЛЕНИЯ
267
(144.2)
R\A/CR
он
о
Ha/Pd ^f >/
СН3ОН
o^Vxoh
XC =- CHCHo CHoCH = С
HO
УЛ/\
OH
/
(144.3)
H,C
\A/>
\сн.
CH2CH2CH
R и И' = алкильные групиы
\/
Восстановление
по Киншеру—Вольфу
> СН3(СН2)7СООН
Ответ-
{144.1) [van Tamelen E. E-, II i 1 d a h 1 G. Т., I. Am. Chem. Soc, 78,
4405 (1956)].
OC2H5
COOC2H5
LiAlH4
OC2H5
ЛХ
HOH^^^CH^H
H +
+ г н n+ О — H
CH2OH
CH2OH
CH2OH
Превращение эфиров енольных форм а-дикетонов в агр-пепредельные кетоны
является общей реакцией. Имеется, однако, одна реакция, в результате которой пропсходит
гидрогенолиз эфира епола [13]
ск,
OCgHl
L1AIH4
268
ГЛАВА 9
(m.2)[Riedl W., Nick I J., Ber., 89, 1839 (1956)].
C —R
„ >-сиснГ \нгсн-сГ" 3
V
CH,
>C^CHCII2 XCH2CH=C^ л
H2/Pd
CH2CH = C
^CH*
CH2CH2CH
'CH,
Этот механизм предложен в оригинальной работе. Оп основан на том, что в кислой
среде (СН8ОН — НС1) исходное соединение отщепляет две изопентенильныо группы
в виде изопрена, вероятно, по схеме
он
сн3 и,с
'но
tr-
■ *~ Другие продукты
Н,С"
СНз нчс'
с= снсн2
сн.
сн3
СН^СН-С-СНз -«—*- СН2-СН = С_С1Г3
сн*
сн2-сп_ с=сн3
--«а^ГИ переходным состоянием,
кулу, адсорбированную пакатадиза^ооГиnZL™™7™™5'* Д°Тпо В1™ать моле-
гетеролитичес/ой?) Н* в «.^2^x5^^^^^" &ТаКе <™молитической или
РЕАКЦИЯ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ
269
{144.3) [S t e 11 e r H-, D i e r i с k s W., Ber., 85, 61, 1061 (1952)].
XX
oil"
О
ОН
R(CH2)6COOH
I
о
II
сОн
со он
Это общеупотребительный метод синтеза для удлинения цепи на шесть атомов
углерода, так как исходное вещество А может быть получено алкилированием относительно
доступного циклогександиопа-1,3 (дигидрорезорцина)
R
Na+
RX-
\/
+ NaX
\У
Задача 145* Приведите механизмы следующей реакции:
Ег СН2ОН
LiAlH4
(145.1) ' 4
(145.2)
ОН
О
/ \ L1A1D4 НаО [
(с6н5)2с—снсен5 — > > (свн5)2сн-ссвн5
Aicia
Ответ.
(145.1) [Соре А. С, Graham E. S., М а г a h а 11 D. J., J. Am. Chem.
Soc, 76, 6159 (1954)].
Н—А1Нз
СНО
LiAlH4
СТ>
снэон
AEN03
аз
соон
270
ГЛАВА 9
Эта реакция формально близко напоминает перегруппировку Фаворского.
Действительно [14], при реакции с нитратом серебра исходное соединение легко образует
перегруппированную кислоту А. Однако обычный механизм с промежуточным
образованием циклопропанопа [15] в данном случае не применим, поскольку мостиковыс а-ато-
мы водорода не являются кислыми, вследствие чего предпочтение отдастся механизму типа
«гуш-нулл» («push-pull»).
(145.2) [Е 1 i el E. L-, Dclmonte D. W-, J. Am. Chem. Soc, 80,1744 (1958)].
A 1С].
a
/\
(C6H5)2C—CHC6H5
CMA1CI,
+ r
(C6H5}2—C —C —CGH5
1 H
(CgHs^CH— CC^Hj
2
OH
I
(C0H6)2C—C—C6H5
D H
+ L1AID4
t H+
OH
I
(CeH5)2CH—C —C6H5
Если бы промежуточно образующийся ион 1 непосредственно восстанавливался
LiAlD4> то продуктом реакции должен бы быть спирт 4. Поскольку в действительности
получается спирт 3, то перед стадией восстановления должна протекать
перегруппировка с образованием кетона 2 (или какого-нибудь эквивалентного ему комплекса).
Задача 146. Объясните, почему отношение 2(5- и 2а-спиртов,
образующихся при восстановлении приведенного ниже кетостероида, наибольшее
при каталитическом гидрировании, меньше при действии NaBH4 и
минимальное при действии ЫА1Н4.
НО'
:са ™сн
Каталнгнческое
гидрирование
NaBH4
UAIH4
-*-
Н
2а- спирт, %
0
16
37
Н
2р-спирт, %
100
7]
52
Ответ [Dauben VV. G.,Blanz E. J., Jr., 1 i u I.,Micheli R. A., J. Am.
Chem. Soc, 78, 3752 (1956)].
Влияние стерических факторов на направление восстановления карбонильной
группы может быть обобщено в виде двух эффектов: «контроля образующегося соединения»
(«product development control») и «стерического контроля подхода» («steric approach
control») [16, 17]. В первом случае реакция протекает с образованием продукта иосстанов-
РЕАКЦИИ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
271
ления, обладающего наиболее стабильной конфигурацией. Во втором случае реакция
определяется подходом восстанавливающего агента с пространственно наименее
затрудненной стороны и обычно приводит к нестабильному изомеру. Имеются случаи, где
действуют оба эти эффекта, но в общем первый справедлив для прострапственпо
незатрудненных, кетопов, тогда как роль второго эффекта становится более важной с увеличением
пространственных препятствий в кетоне.
В случае 2-кетостероидов более устойчивый изомер — экваториальный 2а-спирт —
образуется при аксиальном приближении восстанавливающего агента, тогда как более
легкий подход с экваториальной стороны приводит к 2р-спирту.
Менее, стабильный продукт
Более стабильный продукт
Одпим из важных факторов, определяющим направление присоединения, является
объем гидрида. Реагент, больший по объему, из-за больших сил отталкивания,
обусловленных стерическими взаимодействиями, будет подходить с менее пространственно
затрудненной стороны. NaBH4 в метиловом спирте обычно ведет себя как более объемистый
реагент, чем LiAlH4 в эфире, и выход нестабильного изомера при этом больше.
Каталитическое гидрирование, являющееся гетерогенной реакцией на
поверхности катализатора, нельзя прямо сравнивать и с восстановлением гидридами металлов;
однако очень часто каталитическое гидрирование приводит к таким изомерам, которые
образуются в том случае, если бы катализатор был очень объемистым реагентом.
Задача 147. Приведите механизм окисления, протекающего по схеме
Se02
СН3СОО
СНЧСОО
ш к о ч л Т С S о г i n e P. S., J. Chem. Soc, 1954, 1989; Brow n-
Omaem (M с К с a n L. U,,ь P " n g г. ,
lie G-, Spring F. S., J. Chem. Soc, 1956, 1949).
272
ГЛАВА В
Этот механизм [18] согласуется и с механизмами других аналогичных
перегруппировок. Образование промежуточного трикетона А было подтверждено при изучении
реакций аллильного окисления БеОг» которые сопровождаются перегруппировкой.
Движущей силой перегруппировки, по-видимому, является исчезновение неблагоприятного
диполь-дипольного отталкивания близко расположенных карбонильных групп.
Такой же лактоп получен при окислении стероидов Б и В Сг03 и, возможно, эта
реакции также протекают через образование трикетона А.
Задача 148. Установите строение продуктов реакций и приведите
механизмы их образования.
РЕАКЦИИ ОКИСЛЕНИЯ— ВОССТАНОВЛЕНИЯ
273
X О
I II
сн2— снсс6н5
СН2—СН СС6Н5
2п;-(сн3)2со
X О
сн3о~ |сн3он
СИН15О2Х
1
. [zn
СН3ОН
Za
СНаСООН
ciaHi8°2
3
ИК:~ 1690с*-1
Zn
(СНз)2СО
(выход.
низкий)
(СН3СОО)4РЬ
с18н16о2 »-
СбНб—C2II5OH
спнюо + с6н5сос2н5
ИК-346Сиб81с.«-1 ИК-1726 см'1
O^mlWasserman H. H., Gorbunoff M. J., J- Am* Chem fioc 80,
4568 (1958); Fuson R. С, Farlow M. W., J. Am. Chem. Soc, 56, 1593 (1934)].
■COC6H5
■COCcH5
CH2 —CH2CC^Hg
сн2—сн2сс6н5
II
о
18-374
274 г л а В А 9
СйН$
-ССвН5
I _t + с6н5сос2н5
4 НО о
Реакция А —э- 1 представляет собой дегидрогалогенирование, катализируемое
основанием. Возможный механизм следующий:
А-»-
)>
II
•CCflH3
Т"^еН5
о
II
-СС6Н5
V
■срёНб
о
сен5
X ЯС6Н5
V
с6н5_
с6н5
Очень вероятным представляется образование соединения Б. Тем но менее это
соединение не было получено при реакции сс-галогенкетонов; кроме того, его образование
несогласуется с вышеприведенной последовательностью реакций и особенно с переходом
4 -*- 5. Образование эпоксикетона формально аналогично синтезу глицидных эфиров
по Дарзану [191. Восстановление, приводящее к кетоспирту (4), может протекать через-
Zm.
ОН
С*Н
елд
-с«н,
'С- СбН5
C6HS
;с — с«н
е"5
АС—CfiH
■еп5
РЕАКЦИИ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ 275
промежуточное образование веществ В или Г. Из двух продуктов Д и Е соединение Е
должно легко расщепляться тетраацетатом свинца с образованием соединения 5 и этил-
бензоата. Кроме того» частота полосы поглощения С = О в соединении 4 по своему
значению ближе к частоте поглощения С = О изолированной бензоильной группы, чем
карбонила, сопряженного с дополнительной двойной связью [201 (как известно,
соединение Д имеет полосу поглощения при 1653 см'1).
Переход А -»- 2 напоминает реакцию Реформатского [21]
2п;
>х
О)
-СС6Нд
/
-СС6Нг
о
гг%
~ScceHj,
тогда как переход А -*- 3, возможно, представляет атаку цинка на оба атома галогена
и простое восстановление без образования С — С-связи.
Переход 1—2 протекает по схеме
.с6нБ
СС5Н5
OZn
тогда как переход 1 ->- 3 предполагает первоначальную атаку цинка по сс-положепию
«о отношению к карбонилу
с-н
сн1^
ссбн5
6П5
;СС6Н5
о-
Zn
Очевидно, реакция с цинком зависит главным образом от природы растворителя
и, возможно, от условий выделения. Промежуточные соединения, несомненно, имеют
более сложное строение, чем это изображено на схемах, поскольку реакции протекают
па поверхности цинка. Заметим, что одно и то же промежуточное соединение было
предложено и для перехода А -»- 2 и для 1-^3, хотя эти реакции приводят к различным
продуктам. Таким образом, предложенный механизм далек от совершенства.
Задача 149. При изучении гипотетической схемы биосинтеза- для
превращения цинхонамина (1) в цинхонин (2) 2-метилтриптофан (3) был гладко
превращен в 4-ацетилхинолин (4) в одну стадию (отметим, что требуется
2 же гипохлорита). Приведите механизм этого превращения.
18*
276
ГЛАВА 9
СпОТ
н
2NaOCl
50'
Ответ (van Tamelen Е- Е., Haarstad V. Б-, Tetrahedron Letters,
1961, 390).
CoC
-COOH
Mb
HO CI
-H20
о
II
С О—II
Л П HN-^CI^
н
— со,
-НС1
1ЬО
счо
-он
-н-
спо
н—on
он
Порядок различных стадий этого механизма может быть изменен, но ковочный
результат при этом не меняется.
РЕАКЦИИ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
277
ЛПТEFА ТУРА
1. House Н. О., Modern Synthetic Reactions, Benjamin, N.Y-, 1965, pp. 1—133.
Очень хороший обзор реакций окисления и восстановления всех типов.
2. Stewart R., Oxidation Mechanisms: Applications to Organic Chemistry, Benjamin,
N.Y., 1964. Введение в очень сложную область механизмов реакций окисления.
3. Гейлорд Н., Восстановление комплексными гидридами металлов, ИЛ, М.,
1959. Обстоятельный и подробный обзор старых реакций восстановления гидридами
металлов; в нем отсутствуют некоторые новые методы (см. [1]).
4. Мичович В., Михайлович М., Алюмогидрид лития, ИЛ, М., 1957.
Небольшой, но очень хороший обзор.
5. В a i 1 е у P. S., Chem. Rev., 58, 925 (1958). Обзор реакции озонолиза.
6. Поттс К- Т., в книге «Установление строения органических соединений
физическими и химическими методами», изд-во «Химия», М., 1967, том 2, стр. 425; Бент-
ли К., там же, том 2, стр. 341. Обзор способов окислительного расщепления
связей С — С и С = С.
7. Young W.G.,McKinnis А. С, W е Ь b I. D., R о Ь е г t s J. D.t J- Am.
Chem. Soc, 68, 293 (1946).
8. В u rt о n D. H.R., Seoane E., J. Chem. Soc, 1956, 4150.
9. L у 1 e R. E., M а г t i n N. В., Jr., Fielding H- L., J. Am. Chem. Soc, 75,
4089 (1953).
10. N e s W. R., Mosettig E., J. Am. Chem. Soc, 76, 3182 (1954).
11. Физер Л., Фпзер М., Стероиды, изд-во «Мир», М., 1964.
12. Gould E.S., Mechanism and Structure in Organic Chemistry, Holt, N.Y., 1959.
13. А г t h G. E., P о о s G. I., S а г e 11 L. H., J. Am. Chem. Soc, 77, 3834 (1955).
14. С о p e A. C, Graham E. S., J. Am. Chem. Soc, 73, 4702 (1951).
15. L о f t f i e 1 d R. R-, J. Am. Chem. Soc, 73, 4707 (1951).
16. D a u b e n W. G-, Г о r k e n G. J., N о у с e D. S-, J. Am. Chem. Soc, 78, 2579
(1956).
17*.И лиел Э., Аллинжер Н., Энжиал С, Моррисон Г., Конформа-
ционный анализ, изд-во «Мир», М., 1969, стр. 143.
18. Y a t e s P., Stent G. H.t J. Am. Chem. Soc, 76, 5110 (1954).
19- Ньюмен М. С, Мейджерлейн Б. Дж., в книге «Органические реакции»,
т. 5, ИЛ, М., 1951, стр. 319.
20. Наканиси К., Инфракрасные спектры и строение органических соединений,
нац-во «Мир», М., 1965.
21. Шрайнер Р., в книге «Органические реакции», том 1, Госиниздат, М-, 1948,
стр. 9.
* Дополнение переводчика.
ГЛАВА 10
Смешаиные реакции
Задача 150. Какие стадии необходимы, чтобы осуществить следующие
превращения:
(150.1) /\ /\/\
I I- Г1 I
Y о>\/\/
(150.2) (&) сн _ сн _^ р
\ /
О
(150.3) /\
VXCHO V
(150.4) СООН
\ /
N
N
(б) НС = СН
о
V
Ответ.
(150Л) [Mannich С, Koch W., Borkowsky F.^ Вег., 70, 355 (1937)1.
О
(CH3)2NCH2 о О С2Н6ОС
CHjO
(CH3)aNH
СНзССНаСОСгНб
C2H»0Na
Нагревание
О
СаН6ОН,
CaHjONa,
альдопьная
конденсация
СМЕШАННЫЕ РЕАКЦИИ
279
О
Гидролна
-СОа
О
С2Н50С
\/\/\
О
он
о
о
о
СНз1 ■+• С2Н50-
СН3ССН2—СН2—N(CH3)2 > CH3CCH2CH2N(CH3)3 > СН3ССН = СН2
I"
2 3
Это пример реакции Манниха [1]. Распространенная модификация [2] заключается
в образовании четвертичной соли амина 1 при действии йодистого метила, что облегчает
элиминирование.
Реакцию часто проводят с основанием Манниха, полученным из ацетона (2). Это
соединение образует метилвинилкетон (3), который может конденсироваться с другим
кетовом. Однако в случае никлогексанона наблюдается дальнейшая конденсация.
(150.2) [Reppe W. et al„ Ann., 596, 25, 39, 80 (1958)].
CH20 Hj/Ni
(a) IIC=CH > HOCH2—C=C—CHBOH * HOCH2CH2CH2CH2OH
i.
0
о
(б) НСееСН
HS
Каталиаатор
Линдлара[3]
HOC1
H2C = CH2 > H0CH2CH2C1
Основание
О
О
H+ H2C—CH2
— v
Это один из многих возможных путей синтеза [4].
(150.3) [Paul R., Bull. Soc. Chim. France, 53 (V), 1489 (1933); Sawyer R. L-i
A'ndrus D. W., Org. Syn. Coll., 3, 276 (1943)].
П
Нг/Ni
О CHO
О CH2OH
AI2O3
300°
" H2, катализатор
О
О
280 ГЛАВА 10
(150-4) IF eelyW. E.,Beavers E- M., J. Am. Chem. Soc, 8i, 4004 {1959)].
OH202 jJ^S RX S^ _ПГ_
CH.COOH IU;J 1+^X~
I
OR
N
OH
CN COOH
N
Выход при реакции замещения умеренный (<50%); при этом получаются как 2-?
»ак и 4-цианпиридины.
Задача, 151. Укажите реагенты, под действием которых7.могли бы
осуществиться следующие реакции (в задаче (150.1) каждая из^стадий А, Б, Б
и Г включает многостадийные процессы]:
(151.1) AJ
СН2ОН
со2с2н5
СН2ОН
+ сн2
CN
-** О ) CH2f
CH2NH2
4 ©
он
он
(151.2) О
/V4/\ снасо2с2н5 -^ Л/ЧУХ
+ I
сно
\/\
(151.3)
,осанй
/\/\/\(4 Е
™ +сн2=сн-осан5 —
Х/хА
он
он
У\
оно
СМЕШАННЫЕ РЕАКЦИИ 284
Ответ.
(251.1) [С о р е А. С, Anderson В. С, J. Am. Chem. Soc, 78, 149 (1956):
79, 3892 (1957)].
А. Диол переводят в дитозилат при обработке TsCl и затем конденсируют с
нитрилом при температуре 110° в присутствии NaH (автоклав, ТТФЬ в результате чего
образуется цианоэфир 1. Цианоэфир гидролизуют водным раствором едкого натра, после
®CC2HS - ®<Г - ®~ - ©-«
чего свободную кислоту декарбоксилируют нагреванием при температуре плавления
или в хинолине. Полученный нитрил гидрируют над никелем Ренея при высоком
давлении, в результате чего образуется указанный амин.
Б. Дезаминирование и расширение цикла можно осуществить действием азотистой
кислоты, что приводит к смеси продуктов, включая и циклооктанол (2). Это
перегруппировка Демьянова, см. [5].
Полученный спирт окисляют N-бромсукцинимидом в водном растворе ацетона [6], хотя
можно применить и другие окислители. Кетон восстанавливают в 1,4-эпоксициклооктан
по реакции Кижнера — Вольфа, применяя модифицированную методику,
предложенную Хуапг — Минлоном [7] (гидразин, КОН, диэтилепгликоль).
В. транс-Диол получают расщеплением ендоксомостика уксусным ангидридом —
BF3 (низкий выход) с последующим гидролизом метанольным раствором едкого натра.
сн,со+ осос% он
CH3CQO-^ СН3СОО'
Г. tyuc-Диол был получен следующим образом: окиспый цикл раскрывался под
действием бромистого ацетила с образованием транс-бромацетата 3. Обработка ацетатом
тетраэтиламмония приводит к замещению атома брома с обращением конфигурации
и образованием цис-дпацетата. Гидролиз едким кали в метиловом спирте приводит
к ifitc-диолу
■ососнз ?сосн*
ОН
©
282
ГЛАВА 10=-
(151 2) [I s 1 e r 0., H u b e г W., Ronco A., Koflei M., Helv. Chim.
Acta, 30, 1911 (1947)1-
COC.2Hs
Это реакция Дарзана 17]. Глицидный эфир 1 -получают при использовании в
качестве катализатора такого сильного основания, как пгрет-бутилат калия. Гидролиз водным
раствором щелочи сопровождается декарбоксилированием и раскрытием эпоксидного
цикла. Получившийся енол протонируется с образованием альдегида.
Эта серия реакций является первой стадией в промышленном синтезе витамина А.
Положение двойной связи в альдегиде 2 в настоящее время строго установлено [8].
(151.8) [I s 1 е г О., Lindlar H., Montavon М., Ruegg R., Zelb
i 8 г P., Helv. Chim. Acta, 39, 249 (1956)].
~BF3
+осгн5
CH
OC2H5
j (BF3:OC2tt5)'
CHO
H,Q
СМЕШАННЫЕ РЕАКЦИИ
283
В качестве катализаторов присоединения этилвинилового эфира к а, р-иепредельным
ацеталям применяются кислоты Льюиса, как, например, ZnCl2 или BF3- Реакцию
проводят при низкой температуре; в этих условиях исходное соединение дает сопряженный
ион 1 и далее промежуточный ацеталь 2, который стабилен и вступает в дальнейшие
реакции.
Это общий метод удлинения цепи, имеющей двойную связь, на два атома
углерода [9].
Задача 152. Приведите методы, позволяющие осуществить следующие
превращения;
(152.1) X
(152.2)
Ч/ \/
(Х=Вг, ОН, СООН, NOa)
СООН КН2
А Л\ Б
(152.3) RCOOH -
(152.4) 1
п-
А
> RCOCHg
1
1
/\
Ответ. (Очень хороший обзор синтетических методов имеется в работе
Fieser L. F., Fieser M., Advanced Organic Chemistry, Reinhold, N-У., 1961; ссылки
на экспериментальные работы приведены в работе Wagner R. В., Zoofc H. D.,
Synthetic Organic Chemistry, Wiley, N.Y, 1953.)
(152J) X = Br({10], стр. 114—117; [11], стр. 9)
MgBr
Br
I
Me
(С2НбЬО
ч
\s
НгО или
с2н5он
\
V
284
ГЛАВА 10
Х=ОН ([10], стр. 667—668; [11], стр. 8; [12])
ОН
ОН
НОР(ОС2ЕБ>^
(C2H6)sN
Перегонка с Zn-оылью
, >
о-
I
0-Р(ОС2Н5)а
| +
/Ч
\
Na
1^Н3
А
\i
Перегонка с цинковой пылью протекает в очень жестких условиях; эта реакция
неприменима для синтеза и ненадежна при выводе структуры. Выходы обычно очень
низкие. Восстановление гипофосфитом лимитируется масштабом реакции, но
потенциально опа более пригодна для синтеза.
Х=СООН ([10], стр. 667; [11], стр. 13)
СООН
NaOH-Ca(OE)a
э
Нагревание
X = N02 ([10], стр. 706—708, 723-726, 734—736; [11], стр. 14, 654, 772).
N02 NH2 Щ
/\
*/
sn
на
/Ч
HONO
Н3РО2
(152.2) (а) (В и л ь с о н Ч. В., в книге «Органические реакции», том 9, ИЛ, М.7
1959, стр. 445).
СООН
COOAg
+ Agr20
Вг2
CCL
4- AgBr + СО:
Это реакция Хунсдиккера.
(б) (Органические реакции, том 3, ИЛ, М., 1951, ст?. 243, 255, 322).
СМЕШАННЫЕ РЕАКЦИИ
2
I—
СООН
NaN^, H2SO4 (перегруппировка Шмидта)
coci
СОШг
NH,
Для мостиковых кислот использовались все эти методы- Реакция Шмидта наибод
легко выполнима при работе с небольшими количествами соединений, устойчивых в сш
нокислой среде. Однако азотистоводородлая кислота очень опасна при работе с больпш
количествами.
(152.3) (а) (Ш и р л и Д. А., в книге «Органические реакции», том 8, ИЛ, Ь
1954, стр. 44).
SOCl2 (CH3)2Cd
RCOOH > RCOC1 > RCOCH3
Можно также использовать реактив Гриньяра CH3MgI, но выходы в этом случ
значительно ниже (см., однако, [13]).
(б) (Г у т ше К. Д., в книге «Органические реакции», том 8, ИЛ., М., 1954, стр. 46'
RCOC1
-*- RCHO
CHaN,
Pd — BaS04
хино-лик-S
Р
R—C — CR, —K = N
I/' '
H
* -K2
О.
II
RCCH3
Первой стадией является восстановление по Роземунду [14].
(в) [Teg пег С, Acta Chem. Scand., 6, 782 (1952)].
0-1Л+ О
I н3о+ ||
R€00H + 2CH3Li —> R—С—0~Li+ > RCCH3
CH3
286
ГЛАВА 10
(252.1)
соон
COOR
COOR
Н,0"
сток
£
А/снз д
А: 03; Н2Ог [15] или КМп04, НЮ4 [16].
Б: CH2N2 или СгН5ОН, Н+, бенаол, кипячение с водоотделителем [17].
В: HOCHgCH2OH, TsOH, бензол, кипячение с водоотделителем [18].
Г: 1ЛАШ4; осторояшый гидролиз водным раствором NH4CI [19].
Д: TsCl, пиридин; LiAlHj [19].
Задача 153. Предложите методы, позволяющие осуществить следующие*
превращения:
(153.1) СООСНз СООСНз
1 /\/
" ' I !i \
(153.2)
(153.3)
II
О
(CHS)2N
7Г
Н
О
он
но
\/\У
о
сн3о
осн.
СМЕШАННЫЕ РЕАКЦИИ
287
Ответ.
(153.1) [Lloyd H. A., Horning E. С, J. Am. Chem. Boc, 76, 3651 (1954)].
СООСНо
I
HaNOH
II
О
СООСНз
[
II
NOH
СООСНд
\
Полифосфорная У\/ \
кислота, II \
нагревание ,. jj /
н X
СООСНз
I .
/\/—\
/\
СООСНз
J\/\
СООСН3
4/\NH2 С00Снз \X\n
н2
СН3ОН
—>
НС1
О
/\
СООСНд
У\/\
н
= 0
Эта реакция включает перегруппировку Бекмана, расщепление лактама и
циклизацию, приводящую К более стабильному пятичленному циклу.
(153.2) [Buzzetti F-, Wicki W, Kalvoda J., JegerO., Helv.
Chim. Acta, 42, 388 (1959); см. также Ruschig H, Pritach W., Schmidt-
Thome J., Haede W., Ber., 88, 883 (1955)].
fcH3)2N
CHo—N
I
CI
BrCN
-CH3Bi
rK-KJ**J
(с2н5)2о
I CH3ONa
•288
ГЛАВА 10
сн
лЛ^К^
IUO"
АД^
Первая стадия — это реакция Брауна 1201. «ОКП\1
<J5§.3) [Gates M„ J. Am. Chem. Soc, 72, 228 (1950)].
ОН о
RCC1
OH
HO
1 моль
* о
jJJo/VV
NaNOa
>
CH3COOH
NO
KH,
OH
о J J
RJi0/\/\/
0
H2
/\/v/
он
FeCi3
Pd/C
■> О
b6o/N/\/
OH
,0
О
Восстановление
кс0/х/\/-
S02
~> о
RCO
/\/\/
/x/\/
,он
K2COS
(CHs)aS04
OCH3
OCHs
0
II
RCO
OCH3
OH-
^2o
■OCHs
HNOs
HO
Восстановление FeCl3
. > >
OCH3
0/\/\/
Задача 154. Объясните следующие наблюдения.
(154.1) Ацетилирование оптически активного глициллейцина уксусным
ангидридом или кетеном дает оптически активный продукт, если
поддерживать рН около 10. Если поддерживать рН около 4, то получается рацеми-
СМЕШАННЫЕ РЕАКЦИИ
289
ческий продукт. Однако лейцилглицнн дает оптически активный продукт
в обоих случаях.
(154.2)
о
+
/\/
OCCH2NH3 ClOj
и
\:nhr
II
о
(C2H5)3N
CHCI3
S\/
ОН
CNHCHaCNHR
(154.3) Обработка полипептида, содержащего остаток тирозина, водным
раствором N-бромсукцинимида или бромом приводит к избирательному
расщеплению тирозил-пептидной связи
О
II .
RNHCHC Ч- NHR'
I
сн2
I
/\.
Y
он
Ответ.
(154.1) [С a h i 11 W. М., В u n t о n I. F.f J. Biol. Chem., 132, 161 (1940)].
R
R'
I
RCNHCHCOH
Jl II
о о
(СН3СО)20
N —
ОН- {очень быстро)
HgO+ (медленнее)
— Н
,R'
К-
R^V4o нЛЛон
Азлактон а-Оксиоксазол
Ацетилирующие агенты легко превращают а-ациламинокислоты в азлактоны [21, 22].
Вследствие легкого таутомерного перехода в резонансно стабилизированный оксааольный
цикл асимметрический центр по соседству с карбонильной группой быстро рацемизуется.
В глнциллейципе асимметрический атом углерода находится по соседству с
карбоксильной группой, и поэтому он легко рацемизуется. В лейцилглицине это не так, и рацемпза'
ция не происходит.
Различные результаты, полученные при проведении реакции в кислой и щелочной
средах, обусловлены быстрым гидролизом азлактона в щелочной среде, вследствие чего
он распадается раньше, чем произойдет рацемизация.
(154.2) [В г е п пе г М., Zimmermann 3. P., Wehrmuller J.,
Quit t P., Hartmann A., Schneider W., Beglinger V., Helv. Chim.
Acta, 40, 1496 (1957)].
19—374
290
ГЛАВА 10
ОС
о +
OvJl^CHjNHj СЮ7 (C=*W^
^NHR
Л
■CI-LNH,
t|XR
II
4:N(C,HS)
s;3
О vb-CHsNHj
RN CHS
C—N"H2
'o-
OC^r™ ■* GCc^cB!
on
„CNHR
II
О
Альтернативный путь включает промежуточное образование диацилимида 1; однако
модельные эксперименты показали, что это соединение дает имидазолон 2 вместо продукта
перегруппировки.
/S/
ОН
R
ч/он
й I ft—Jh,
Эта реакция может быть использована как способ увеличения длины пептидной
цепи за счет присоединения дополнительного аминокислотного остатка к
N-концевому атому.
(154.3) [Schmir G. L-, Cohen L. A., W i t k о р В., J. Am. Chem. Soc,
81, 2228 (1959)].
R
NH
I
сн„— сн
R
NH
Br,
-CH
c=o
I
NH
Br
но—P у—сн2—(
/ c=o
Br I
NH
I R'
СМЕШАННЫЕ РЕАКЦИИ
29
Br
Br
4\
CH,—CH
/KHR
NR'
RHN
H,C ^O
\"
Br
H
У
Br
+ HaNR'
h,o
Гидролиз
-PCI
/
CH
NHR
Br
С
NR
H
™„ ™ B8BeS™°' образуется ли лактон диенона из бромдвепопа [путь (а)] или npi
окислительной циклизации соответствующего фенола [путь (б)].
Задача 155. Объясните образование соединений Б и И и определите
структуру Г.
Вг
Вг
£fl
Конц
-"2so4 r^N
Водн.НС]
:N
(или осно
■ ванне)
он
он
C0II,3N <Г)
■*- ^9^13
+
нсоон
CN
19*
292
ГЛАВА 10
Ответ \v a n Tamelen E. E-, Brenner J. E, 3. Am. Chem. Soc, 79,
3839 (1957)].
frfi.
no2 и
-Вг
Вг
— IV
NO"1
Н,С
N0
О.
//
Г"СН
NT
Вг
-Нн
ОН*' ОН
он
с
111 >
N
КС
„н>о"
NC С
ОН
/fc
CN
CN
+ НСООИ
Г
СМЕШАННЫЕ РЕАКЦИИ
29;
Задача 156. Установите строение продуктов следующих реакций:
H,CL ПН
СНЭ
а-Пинен
NOC1 TpeT-C4HgONa
*■ C10H1BONC1 *- Ci0H]5ON
CgH]2ON2 ~« 1~~ C9H,302N
OH
4 3
ИК: 2151,1678 сж~1 HK:3205, 1724, J629 см~]
(h*. \ hv,
диоксин,
н2о
лииксан,
(CH3)2NH
i.socb
C9HH02 ■ *- C„H,9NO
2.(CH3)2NH
5 6
ИК: 3125,1695 с*-' ИК: 1635 см~1
LiAlH4
].КМп04рН~ CnH2lN
2-CH2N2" 7
3.LiAlH4
11.H20.
2. 120'
в вакууме
Os04
Cglile02 -* 2_ Cellu
9 8
ИК: 166], 870 сл-l
294
ГЛАВА 10
Ответ IM e i и w а 1 d J.f Gassman P. G., J. Am. Chem. Soc, 82, 2857 (I960)].
H3CwCH3
CH2
NOH
/lW A \№>2NH
3125 ex-1
L724 ся-1
NOH -*- 3205 CM
-1
1629 см
-1
+ 1635 слН
V «
Д CN{CH^
\
сн2Мснэ)2
H
CH2OH
oso4
/
^ 1661 CJH '
Реакции 1-^4 протекают нормально; переход 4 -»- 5 является фотохимической
перегруппировкой Вольфа, которая протекает через образование карбена,
перегруппировывающегося в кетен [23, 24]. Этот кетен (А) реагирует затем с водой или диметил-
ашгаом с образованием указанных продуктов реакции. Гидратация кетена протекает
СМЕШАННЫЕ РЕАКЦИИ
295
с наименее пространственно затрудненной стороны (со стороны незамещенного одноугле-
родного мостика) с образованием продукта, в котором карбоксильная группа направлена
вверх (р-конфигурация). Аналогичным образом атака протекает с пространственно
не затрудненной стороны и в других случаях (5->Б,8-»-9).
/IV
Задача 157. Установите строение следующих соединений:
НзС\/ч
сн2о <снзСО)аО
> CeH13NOe — —т* CuH^NOj
YNjh.
160°
Кипячение
ИК: 1735, 4635, 1595, 1565,
1480, 1230 ел"1
NaH
сн2<соаСаН5)«
CuHmNOs <
4
ИК: 1735, 1645 елг*
Нагревание
H2/Pt
<ci^oli c«№iN04
ИК: 1735, 1630, 1600,
1562, 1486 сж-1
1, Конц. НС1
2. СгНБОН, НС1
с/
о
c'oi
Ci3Ha5N02
К2СОа
Натронная иявесть,
-> ClsHa7N04 сухая перегонка
ИК: 3300, 1735 см-i
ИК: 3110, 1735, 1625,
1573, 1505, 875 см~1
СцНщГЮ <
8
ИК: 3110, 1505, 875 емт*
На
Pd
С16Н21
N0
ИК: 3110, 1645, 153& 1505,
875 см-1
Отпвет [К о t a k e M., Kawasaki I., Okamoto Т., К us u mo to S.,
К an е ко Т., Ann., 636, 158 (1960); Капеко Т., Kawasaki I., О к a m о -
t о -Т., Chem. Ind. (London), 1959, 1191].
wtx _ - "'XX
н,с
^71 /""
XHaOH
1635
■N ^СН3
N "CH
N С
>сн2
сн2он
2 1230
СНоОССН^
/
О -«-1735
Н„С
1645 с_оС2Н5
1735
1630
СН2 1735
I •"
сн2—сн(сос2н5)2
3300 J
.с—ос2н5
НзСт^
Lo' о\
1735
173Г
Н,С
1UC
О ч
СМЕШАННЫЕ РЕАКЦИИ
297
Полосы поглощения при 1600, 1560 и 1480 см'1 в соединениях 2 и 3 можно
отнести к пиридиновому циклу. Аналогично полосы поглощения при 3110 (СН), ~1580,
1505 (С = С) и 875 ем-1 в соединениях 6 и 7 можно отнести к фурановой системе [25[,
причем полоса ^^1580 сл~х появляется при наличии сопряжения.
Соединение 8 представляет собой смесь диастереомерных форм природного
алкалоида девоксипуфаридина. Хроматографией смеси было выделено (/-соединение [26J,
которое далее было расщеплено па антиподы с образованием природного алкалоида.
Задача 158. Установите структуры следующих продуктов:
НС10Ц
[C«H18N]+C10T
1
CHaN2
[CuHaoN]*C10j
2
C10H„N
/\
\/
ИК: 1665 CM-i
i. CH3Mgl,
эфир
2. НОЮ*
[C,iH2aN]+C[07
3
H2/Pt
ИК: нет полос при—3100
или 1665 елг1
сн3он,
кипячение
1. CICHsCH20H
2. Нейтрализация,
кипячение в эфире
3. HCIO*
[C12H34NOI+C10x [C13Ha4NO]+CIOj
4 5
ИК: 3125 сл-1
1. ВгСН8СН2СНаСН2Вг
2. НСЮ4
NHB LIAIH4
> C8H16N02 —> C8H17NO
6 7
СНзО-^СОСНд
II
О
СдНдеОд
Ответ [Leonard N. J., J a nn К., J. Am. Chem. Soc, 82, 6418 (1960)1.
298
ГЛАВА 10
сю4-
О
Q
сн3о' "сосн3
CH30~ ~CNH2
II
о
к.
сю4-
Q
Полоса поглощения при 1665 см'1 в ИК-спектре соединения 1 может быть
приписана группе \ С = Й/ 1251, хотя, вероятно, она не очень надежно отличается от полосы
СМЕШАННЫЕ РЕАКЦИИ
299
группы у, С = С — NH—. Отсутствие NH-валентпых колебаний при —3100 см-1 (см.
ниже) исключает, однако, вторую возможность.
Реакция 1 ~> 2 напоминает образование эпокспдов при реакции диазометана
с кетонами [Z7J,
Различие в направлении раскрытия цикла 2 -> 3 и 2 -»- 4, вероятно, связано с
различным протекапием, с одной стороны, сольволиза, при котором образующийся иоложи-
тельный заряд находится преимущественно у третичного центра (-»- 4), и, с другой
стороны, реакции на поверхности катализатора, который атакует молекулу с наименее
пространственно затрудненной стороны (-*- 3).
Связь N —- Н в солях третичных аминов, таких, как 4, по-разному поглощает
в ИК-спектре в зависимости от природы аниона. Для анионов С1~ ->■ Вг~ -* CIO fg51
«двиг составлял ~2600 -* 2800 -+ 3100 см'1. 4 J
Задача 159. Соединение А (рацемическая форма природного
соединения 1) получено серией реакций, приведенной на схеме. Установите
строение всех полученных соединений.
СН20(97%)
СН3С = СНгН- N»CHC02C2Hb
Си CuSOi
V
C7U±iBr02
I NaH, эфир
ч
СуН^оОг
ИК: 1745 (слабая), 888 еж*
I
LiAlH*
TsCl
СОаСаНв
Na+C-NHCHO
ЧС04СяН6
I
CvHeNOfCOgCaHsJs
NaOH
Избыток
HIOa
C7H11N02 (Природное соединение)
1
ИК: 888 см'1
Не/Ft
i. Оз, CHaCOOH
2. Н30+ .
C7H13N02
2
Нет полосы около
888 сл*-1
Ha/Pt
т
C7H15N02
3
Смесь трех
изомеров
сн2о
О
||
СН3С —СНз
С^тОдИ
4
СНзСООН
CylljiNOa
А
Ответ [синтез описан в работе Carbon J. Л., Martin W. В., S w e t t
L. R., J. Am. Chem. Soc, 80, 1002 (1958)].
сн3—с=сн2+n2chco2c2h5
I
Бг
н3с
Вг
V
C02C2Hs
н2с
С02С2Н5
Н2С—I I
СН2ОН
н2с=зг-у
CH,OTs
н,с
н,с
сн2о
сн2—снсоон
н,с
СН2СНСООН
I
кн2
*
СНчСНоСНСН,СНСООН
I I
СН3 NH2
+
сн3сн2сн2сн2сн2снсоон
кн2
+
н,с.
I
сн2—с—со2с2п5
I
| NHCHO
+
NHS
сн,снсоон
NH2
Гипоглниин
сн2о
+
нчс
\
сн,снсоон
I
NH2
гел г и ;снсн2сн2снсоон и
со2с2н6 н / \ II
1 * I Г.Н-Г.
о
If
CH-iCCHo
I
I
сн,снсоон
I
■
♦
о
СН3ССН3
+
нооссн2снсоон
NH2
4
СМЕШАННЫЕ РЕАКЦИИ
301
Природное соединение гипоглицин встречается в неспелых плодах деревьев,
произрастающих в Западной Индии, и представляет интерес из-за своей замечательной
способности уменьшать концентрацию сахара в крови. Его структура была установлепа
почти одновременно в шести лабораториях [28—33].
Синтез этого соединения сравнительно прост; можно сделать замечания только
относительно положения экзоциклической двойной связи. Она идентифицируется полосой
. /Н
поглощения при 888 см-1 {=СН2; 1745 см*1 Р^с.^=С ? [34J, 888 см-1 обертон?) и ана-
логией с кислотой Фейста (Б) [35]
ноос соон
ъ
Метиленциклопропановая структура более вероятна, чем метилциклопропеновая,
даже если последняя является сопряженной.
Озонирование гипоглицина протекает аномально: после гидролиза вместо
формальдегида в основном получаются продукты расщепления цикла. Однако окисление
избытком НЮ4 (малоизвестный, но потенциально интересный метод расщепления алкенов [36];
сравните реагент Лемье [16] КМп04 — Н104) дает очень хороший выход формальдегида.
Кислота Фейста при подобной обработке ведет себя аналогично [37, 38].
Различпые авторы [29, 30] расходятся по вопросу об относительных количествах
кислот, образующихся при гидрировании гипоглицина и имеющих открытую цепь.
Вероятно, при этом образуются все возможные продукты реакции.
Задача 160. Установите строение природного соединения 1 и его
производных.
H+ NaBH4 1н. NaOH f СН3СООИ
с8н14о3 < ^— ад^ -Комн тем^ ( с7н10о
5 i 4
ИК: 1757 см'1 ИК: 1795, 1765, 1715" емг1. УФ: 235 нм{\о?е 4,16)
Нейтральное (нет полос при 2730 и 1730 см~х) 300 «.«{loge 1,76)
соединение Иодоформная проба положительна ^
Проба с хлоридом железа(Ш) отрицательна
Тетранитрометановая проба отрицательна
4С—СН3; нет О — СН3
CHsCOONa/HaO
90-95°
СН3 СН3
I I
НООС —СН—СН-СООН
NaOI
rCNH4)BC03 f
CvHuN < C7H1202
6 2
AgaO
C7HJ2O3
3
302
ГЛАВА 10
Ответ [Ett linger L., Gaumann E., Hu ttor R., Ke Her-Schierlein W.,
Kradolfer F., Neipp L., Prelog V., Zahner H., Helv. Chim. Acta, 41, 216
(1958); Keller-Schierlein W., Mihailovic M. L., Prelo g V., Helv. Chim.
Acta, 41, 220 (1958)].
1715
CH, СШ 9H
СНя СНз 9
\
-СНСНз
сен.
0/\0/V0
1757
СНз
>с=о
1795
1765
Н_ЧС
ч /
СНз
I
УЧсн*
Н
6
СН3
не/
II
о
1730
сш
II
о
1715
СН<
СН3 СН3
I 1 С№
ноос^ Y
о
СНз СНд
НООС
соон
Последовательность реакций 2 -»- 3 ->■ диметилянтарная кислота ясна из схемы.
То, что соединение 2 является 1,4-дикарбонильпым соединением, подтверягдается
получением производного пиррола (6).
Переход 2-^4 представляет собой альдольпую конденсацию с последующей
миграцией двойпой связи
СНЯ
НС
II
о
J-
сн3
т
ОН"
н3с сн3
СИ-, СНа
w
О - V-J
LH° V
З^СНз
2,3-Положение двойной связи устанавливается из УФ-спектра, который
вычисляется [39, 401 следующим обраэом для соединения 4: 215+2 (12) + 10 — 10 = 239 нм, и для
изомера А 215+12 — 10=217 нм.
СМЕШАННЫЕ РЕАКЦИИ
303
* — н ova
(Заметим, что приблизительно 10 к.» следует вычесть, поскольку имеют дело с сиотрмпй
циклопентенона, а не циклогексенона [40].) А системой
ты, т ^Тил^В6 С°2 ПРИ ГИДР0ЛШе п°Дтверждает присутствие фрагмента 6-кетокпсло-
сн3 сн3
0=<J 7ЧгСНз
Н НООС о
НООС
^
СН,
соон
сн,
-ч_^сн3
В соответствии с данными гидролиза, приводящего к уксусной кислоте, и ИК-спек
тром (1765 и 1795 см-1) [411 можно предложить две структуры эфиролактонов Г и Д
няс сн
н 7 \
Л VCH)
о^о^ос
ОССНд
II
О
Так как соединение 1 обладает скорее кетонными, чем альдегидными свойствами, и так
как его ИК-спектр не согласуется с альдегидной структурой, то формула Г более вероятна.
Превращение соединения 1 в 5 можно рассматривать как гидролиз сложноафир-
ной и лактонной групп в слабощелочном растворе NaBH4, восстановление кетоппой
и альдегидной групп и повторную лактониаацию при подкислении.
Задача 161. Установите строение природного соединения 1 и его
производных:
С10Н14О2
1
Присоединяет 1 моль брома
НяОн! Т™*™*
<CHsCO)20 CHbNs]
С12Н1804 < С10Н|вО3 *■ СиН1803
5 2 3
Не присоединяет . 2С—СН3
брома ,
Иодоформная проба
отрицательна
Присоединяет NaHS03
Образует оксим
ИК: 1706, 3290 сл"1
Ha02(NaOH)
CHjOH, HjSOi,
"ч^ кипячение
CHsOH,
H1SO4
С12Нго03
4
Присоединяет 1 моль брома
304
Г Л ABA 10
NaOI
11
{CHsCO)20
CgHio03
12
ИК: 1852, 1776 cm~i
СвНиОз+НСООН
6
Иодоформная проба
положительна
PbQa/l5De
у
7
ИК: 1660, 1613 ел"»
УФ; 237 им (log e 4,24)
Гн3с
КМпО*
Hs/Pd
— СООН
СООН
СН3СООН
СеНиО
10
Дает дибензилиденовое
производное
\\. ОН"
1 2. СгОз
с8н,4о
8
СвН5СОООН
■* с8н14о2
9
1558 (1941)
Ответ [М с Е1 v a i n S. М., В г i g h t R. D., J о h ns о n P. R., J. Am. Chem. Soc, 63,
[1941); McEl vain S. M., Eisenbraun E. J., J. Am. Chem. Soc, 77,1599(1955)].
О
II
\/\
О
I If
"Y^OCOCHs
u
о
II
/х/сон
L/1/cho
/\/
о
II
СОСНз
3
сно
о
II
хон
А/-
и -
N:oh
/ч/
о
II
сон
и
f
о
-f-HCOOH
\/
ОСНя
I
4
СНОСНз
I
СМЕШАННЫЕ РЕАКЦИИ
305
о
/\/\
о
./
о
12
\
\/К
хсоон
-СООН
СП3СООН
\
/\
\sy
\
о
I
С = 0
I
СНя
/\
"Чш
/\
10
Соединение 1 выделено из кошачьей мяты и известно под названием непеталактон.
Анализ этой задачи, вероятно, проще всего начать с окисления соединения 7, Уксус-
пая][и а-метилтлутаровая кислоты могут получиться из трех возможных
фрагментов А, Б и В
сн
■СС
Из них только фрагмент В может легко подвергаться каталитическому
гидрированию по двойной связи С = С с образованием соединения 8, которое после окисления
по Байеру — Виллигеру 142] (8 ~> 9) может далее гидролизоваться с потерей двух
атомов углерода (9 ~> 10). Структуры А и Б при окислении должны приводить к лактонам,
и гидролиз должен протекать без отщепления атомов углерода. Положение метильноп
группы в соединении В (7) вытекает из образования соединением 10 дибспзилиденового
производного. Поэтому соединение 10 должно иметь частичную структуру •
О
Ц СвНбСНО
—сн2ссн2 он_ >
СвН6СН О НССвН5
(I II II
—С-С — С—
20—374
306
ГЛАВА 10
и, следовательно, для соединения 7 более вероятна структура В1, а не альтернативная
структура В 2
О
О
/
В1
/\/\ /Ч/Ч
В2
Структура соединения 6 подтверждается окислительным декарбоксилированием —
дегидрированием 6 -*- 7 (сравните окислительное бис-декарбоксилирование [43, 44]
Г -»- Д); это превращение устанавливает положение двойной связи в соединении 7.
/ч/соон
г
РЬОа
Декалин, кипячение
-> [ || + 2С02
Д
Эта структура подтверждается также окислением соединения 6 в 11 п образованием
при дегидратации соединения 11 продукта, ИК-спектр которого однозначно указывает
на структуру пятичленного циклического ангидрида [25].
При рассмотрении схемы с другого конца было показано, что соотношение
соединений 1 и 2 такое же, как между лактоном и оксикислотой. То, что это еноллактон,
подтверждается присутствием ненасыщенности в соединении 1 и альдегидными свойствами
соединения 2. Сопоставление этик наблюдений и структуры соединения 6 позволяет
сделать заключения о реакции 2-^6, хотя она протекает необычно. Поэтому были
проведены дополнительные реакции для подтверждения структуры 2.
Установление строения соединения 2 представляет определенную проблему. Для
объяснения реакции было постулировано равновесие трех структур Е, Ж и И
О
о
II
о
I
сно
\/\
/\/
о
(I
сон
он
снон
ж
и
Хотя две первые структуры довольно вероятны и можно ожидать, что вторая будет
преобладающей, трудно объяснить, почему должно существовать заметное количество споль-
ной формы- Возможно, что реакция 2 -»-1 представляет дегидратацию соединения Ж,
а не этерификацию И, как это было предложено в литературе. Реакцию 2->6 трудно
согласовать с какой-либо структурой. Принимается, что механизм реакции включает
присоединение НОО~ к альдегидной группе структуры Е с последующей
перегруппировкой (см. окисление ароматических оксиальдегидов по Дэкину [45]), либо атаку НО- на
анион енолята Е и дальнейшее окисление получившегося а-оксиальдегида.
СМЕШАННЫЕ РЕАКЦИИ
прш^х.162' УС1аНОЕИТС СТР>'«т>ы природного соединения 1 и ,
»-вк*адос. (СНаС0)г0
п jS шид. отри
°»/ ИК: 3333, 1715, 984, тельпа) V
956 c.w.-i
УФ: 232 нм (34000)
Оптически активно
Ди-л-бромбензоат •«-
ИК: 3500 и другие
полосы
«-Масляный альдегид
ЫА1Н4
Na/Hg
PtJH2
9
Hgl2
C14H28O4
7 2
(два изомера) ИК: 1706 см-i
(СНэСОО)4РЬ
шо4
Н104
СпН2604 -^ cllH2e05 *+ с14н2405
О
неон с13нио4
3
6
Кислое в-во
Реакция
Курциуса
10
NaBH4
11
Кислое в-во,
проба Фелинга
положительна
NaBH|
4
ИК: 1718 cjk-i
1. TsCI
2. Нагревание
в пиридине
снао**- с13п22о2 -ii£_ 3
5 32;§Й-СПзСООН
ИК: 1733, 1631 см-1
УФ: 212 нм (6000)
ИК: 1742 см-i ИК: 1780 елг*
■U
СцН2оОг
20"
308
ГЛАВА 10
NaBH4
или
Na/Hg
(СН3С0)2О __
Тстраацетат —„.„„„„. > Ci;H2404 ^.
^_- C14H2i04I
zn/CH3cooH 12
ИК: 960 cjw-i
12
Пиридин
13
Zn/CHaCOOH
± С14Н23041
14
ИК: 960 см-1
На
Pd
СцНзбОд ^_
16
ИК: 959 cjk-i
Zn/GH3COOH
_ С14Н25041
15
ИК: нет полос
около 960 см-1
Замечания:
(а) Переход, аналогичный 1 —►■ 12, не наблюдается для диацетата 1.
(б) Соединения 1, 2 и 12 дают изопропилиденовые производные при
обработке ацетоном и хлористым цинком.
(в) Частоты в ИК-спектрах приведены не полностью.
Ответ (В о w d е п К., L у t h g о е В., Marsden D. J. S., J» Chem. Soc,
1959, 1662).
HOC!
C,H
'/V^oh
HO H
носн-ч^соон носн2
C7H
XCOOH HOCH,\ ^,0
•$-■
7J'I5 H C7H15
3 10
I
C7H15
OH
CMgOH
HOCH.
"7nI5
/ 4
О 1733
сн2о
1631 II
coon
C7l[]5
с?н,5
OH
'JO
310
ГЛАВА 10
О
II
г\/\/У
/\/4/V/\/\on
1
" ой
Ч/\/*
он
и о
I I
/\/Ч/\/\/\он
960 H I
ел*-1
12
ОН
/\/4/V/\/\oh
13
Oil
IIOCH2x /\/0H
/\/\/4/\/\qh
Соединение 1 является метаболитом плесени палитантипа.
Рассмотрение молекулярной формулы соединения 1 показывает наличие четырех
единиц ненасыщенности. Одна из них — группа С = О (полоса 1710 см~х в ИК-спектре)
и одна или две связи С = С (УФ-спектр соответствует С = С — С = О или С = С — С — С).
Восстановление ( + 4Н) до гидрированного производного 2, имеющего
карбонильную группу, подтверждает наличие диенового фрагмента, и, следовательно, еще одна
единица ненасыщениости связана с моноцпклической структурой. Образование' диаце-
тата и ди-п-бромбензоата указывает на присутствие по крайней мере двух гидроксильных
групп. Однако полоса 3500 см-1 в ИК-спектре последнего производного указывает, что
имеется и третья гидроксильная группа, которая не этерифицируется, вероятно,
вследствие пространственных затруднений. Это подтверждается, хотя и не окончательно, обра-
аованием тетраацетата после восстановления соединения 1 боргидридом натрия.
Рассматривая другую часть схемы, можно видеть, что образование а-и-гептил-у-
лактона из соединепия 5 и н-масляного альдегида при озонировании соединения 1
указывает на присутствие в последнем фрагмента СН3СН2СНзСН = СН — СН — СН .
Учитывая образование СН20 при озонировании соединепия 5, для него можно пред^
ложить две структуры А и Б. Каждая из них может привести к а-кетолактону, который
окисляется далее с образованием соединений, прпведеппых на схеме
Н>С
О
I!
О
сн2
II
I О
с7п15
А
с7н15
г
;
СМЕШАННЫЕ РЕАКЦИИ
311
Однако относительно высокая частота полосы С = О в ИК-спектре и сдвиг в ctodohv
меньших длин волн в УФ-спектре лучше согласуются со структурой А
Рассмотрим теперь обратный путь превращений от соединения А к 4 и 3 ИК-спектп
соединения 4 выглядит несколько аномальным при сравнении со спектром сопряженного
соединения 5, так как последнее должно было бы поглощать при меньших частотах чем
его несопряженный предшественник. Лучше всего это объяснить (принимая' что
значения частот правильные) наличием внутримолекулярной водородной связи между ОН-
группой и карбопилом сложноэфирной группы, в результате чего полоса поглощения
в соединении 4 сдвигается в сторону низких частот. Этот эффект дополняется стерическим
искажением цикла в соединении 5, что приводит к нарушению копланарности метилено-
вой и карбонильной групп и, таким образом, уменьшает проявление эффекта
сопряжения в спектре.
носн
НОСН2
е7н[£
в г
Образование НСООН и соединения 3 при окислении соединения 2 йодной кислотой
согласуется со структурами В и Г. Для получения двух изомеров соединения 7 при
восстановлении соединения 2 необходимо, чтобы карбонильная группа могла образовать
при восстановлении два стереоизомерных спирта. Это возможно для кетона, дающего
вторичные спирты, но не для альдегида, приводящего к одному первичному спирту.
На этом основании структуру Г можно исключить. [Этот аргумент скорее наглядный,
чем строгий. Необходима уверенность, что продукты восстановления действительно
являются стереоизомерными спиртами, образующимися при двух альтернативных
направлениях атаки на карбонильную группу, а не за счет каких-либо других
конфигурационных изменений, как, например, инверсии конфигурации у атома углерода в
а-положении к карбонилу на стадии, предшествующей восстановлению. В данном случае
структура Г не имеет протона в а-положении, вследствие чего вещество должно быть
стабильно при восстановлении в щелочной среде. Кроме того, известен тетраол (9),
соответствующий структуре Г; он отличен от обоих изомеров 7.]
Отсюда легко вывести структуру соединения 2. Остается лишь объяснить
превращения этого соединения. Образование соединения 6 можно формально представить как
окисление до а-дикетона с последующей бензиловой перегруппировкой [46].
НОСН
с7н
Окисление
носна
с7н
носн
С 7^5'
312
ГЛАВА 10
Примененный реагент (реагент Девре [47]) отличается от обычных реактивов Тол-
ленса и Фелинга, содержащих окислы тяжелых металлов. Хотя последние обычно
рассматриваются как специфические реактивы на альдегидную группу, они также
окисляют и а-кетолы.
При окислении соединения 6 тетраацетатом свинца оно расщепляется па а-гли-
кольную группу, и группировку сс-оксикислоты с образованием соединения 3. Однако
НЮ4 с фрагментом а-окспкислоты реагирует медленно, и реакция останавливается на
стадии соединения 8 [48]. Строение лактона 11, образующегося после восстановления
вещества 8 NaBH4, подтверждается ИК-спектром.
Последняя серия реакций является обратимым оксииодированием одной из двойных
связей боковой цепи. Несомненно, что такое течение реакции определяется
благоприятным расположением заместителей в цикле. Отсутствие реакции с диацетатом
соединения 1 указывает на ацетилирование оксиметилъной группы. Инертность этого диацетата
по отношению к окислению периодатом приводит к структуре Д. Однако в этом случае
трудно объяснить, почему оставшаяся гидроксильная группа переакцшшноспособна,
хотя может ацетилироваться после восстановления карбонильной группы.
Альтернативной структурой диацетата может быть формула Е. Возможно, что в этом случае
отсутствие периодатного расщепления системы а-кетола обусловлено стерическимн причинами.
У н
CH3COOCH2v 1 ,0
/\/Ч/Ч/\/\0ц /\/V/V/\/\0cocii3
Д Е
ЛМТЕРАТУРА
1. Блик Ф. Ф., в книге «Органические реакции», том 1, Издатинлит, М-, 1948, стр. 399.
2. D u Feu Е- С, М с Q u i 11 i n F.J., Robinson R., J. Chem. Soc, 1937, 53.
3. L i n d 1 a r H., Helv. Chim- Acta, 35, 446 (1952).
4. Кремер Ч., Рохен Л., в книге «Гетероциклические соединения», под ред.
Эльдерфильда, том 6, ИЛ, М., 1960, стр. 5.
5. Смит П.А., Бо«р Д. Р., в книге «Органические реакции», том 11, изд-во «Мир»,
М., 1965, стр. 167.
6. Р 11 е г R., Chem. Rev., 63, 21 (1963).
7. Todd D., Organic Reactions, 4, 1948, p. 378.
8. Ньюмен М. С, Меджерлейн Б. Дж., в книге «Органические реакции»,
том 5, ИЛ, М., 1952, стр. 319.
9. С he е se m an G-W. Н., Н е i 1 Ь г о n I., J о n e s Е. R. H., S о n d h e i ш е г F.,
Weedon В. С. I., J. Chem. Soc, 1949, 1516.
10. Ислер О., Шустер И., в книге «Успехи органической химии», том 4, изд-во
«Мир», 1966, стр. 124.
11- Fieser L.F., Fiescr M-, Advanced Organic Chemistry, Reinhold, N-Y-, 1961.
12. Wagner R.B., Zook H.D., Synthetic Organic Chemistry, Wiley, N.Y., 1953-
13. Pelletiei S. W.? L о с k e D. M., J. Org. Chem.. 23, 131 (1958).
14. Cason J., Kraus K- W., J- Org. Chem., 26, 1768 (1961).
15. M о з е т т и г Э., М о з и н г о Р., в книге «Органические реакции», том 4, ИЛ,
М., 1951, стр. 337.
16. Long L., Jr., Chem. Rev., 27, 437 (1940).
17. L e m i e u x R. V., v о n RudlofI E., Can. J. Chem., 33, 1701, 1710 (1955).
18. W e у g a n d C, Organic Preparations, Wiley-Interscience, N.Y., 1954, p, 171.
CH3COOCH2 " .OCOCHg
СМЕШАННЫЕ РЕАКЦИИ
313
19. Keana J.W., Steroid Reactions, C. Djerassi, ed., Hold en-Day, San Francisco 19(53
Pp. 3.
20. Г с и л о р д П., Восстановление комплексными гидридами металлов, ИЛ, М., 1959
21. Хейгеман X. А., в книге «Органические реакции», том 7, ИЛ, М., 1956, стр. 260
22. Г р и н ш т е й н Дж., В и н и т ц М., Химия аминокислот и пептидов, изд-во «Мир»
М., 1965.
23. К а рте р Г. Е-, в книге «Органические реакции», том 3, ИЛ, М., 1951, стр. 190.
24. Molecular Rearrangements, de Mayo P., ed., Wiley-Interscience, N.Y., 1963, pp. 528ff.
25. G о u 1 d E. S., Mechanism and Structure in Organic Chemistry, Holt N.Y 1959
pp. 627ff. ' ' '
26. Наканиси К., Инфракрасные спектры и строение• органических соединений
изд-во «Мир», М., 1965-
27. К о t a k e M-, Kawasaki I., Matsutani S-, Kusumoto S., Kane-
k о Т., Bull. Chem. Soc. Japan, 35, 698 (1962).
28. Эйстерт Б., в книге «Новые методы препаративной органической химии», ИЛ
М.( 1950, стр. 91.
29. W i I k i о s о и В., Chem. Ind. (London), 1958, 17.
30. V о n Holt C, Leppla W., Angcw. Chem., 70, 25 (1958).
31. D e R a p p R. S., v о n Meter J. C, d e R e n z о E. С, М с К e r n s K. W.
Pi d ocks C.,B ell P. H., U П m a n E.F., S af ir S. R., F a n s h a w e W. J.'
D a v i s S. В., J. Am- Chem. Soc, 80, 1004 (1958).
32. E 11 i n g t о n E. V., II a s s а П C.H,,Plum m e.r J. R., Chem. Ind. (London)
1958, 329.
33. Anderson H.V., Johnson J. L., Nelson J. W., Olson E.C., Spee-
ter M- E., Vavra J. J., Chem- Ind- (London), 1958, 330.
34. Renner U., J о h 1 A., S t о 11 W- G., Helv. Chim. Acta, 41, 589 (1958).
35. G г а у so n J. Т., G г e e n 1 e e K. W., D erf er J. M., В о or d С. Е., J. Am.
Chem. Soc, 75, 3344 (1953).
36. Ettlinger M. G., Kennedy F., Chem. Ind. (London), 1956, 166.
37. С h a 11 e r j e e A., M a j u m d a r S. G.( Anal. Chem., 28, 878 (1966).
38. Goss F.R., Ingold C. K., T h о г p e J. F-, J. Chem. Soc, 123, 327 (1923).
39. LI о у d D., Chem. Ind. (London), 1956, 874.
40. Ф и з е р Л-, Ф и з е р М., Стероиды, изд-во «Мир», М., 1964.
41. French H. S., J. Am. Chem. Soc, 74, 514 (1952).
42. R a smu ssen R. S., Brat tain R. R., J. Am. Chem. Soc, 71, 1073 (1949).
43. Хассел Ч. X., в книге «Органические реакции», том 9, ИЛ, 1959, стр. 82.
44. D о е г i n g W. von Е., F а г Ь е г М., Sayigh A., J. Am. Chem. Soc, 74, 4370
(1952).
45. G г о b L. A. et al., Helv. Chim. Acta, 61, 1191 (1958); Зефиров Н. С, Ивано
в а Р. А., Ф и л а т о в а Р. С, Юрьев Ю. К., ЖОХ, 36, 1901 (1966).
46. D a k i n H. D., Am. Chem. J., 42, 474 (1909).
47. S e 1 m a n S-, Eastham J. F., Quart. Rev., 14, 221 (1961).
48. D о e u v r e J., Bull. Soc. Chim. France, 41, 1145 (1927).
49. Крнге Р., в книге «Новые методы препаративной органической химии»,' ИЛ, М-,
1950, стр. 139.
Указатель типов задач*
Задаче, требующие стереохимпческой и конформационной
интерпретации
10-12, 17, 19, 30-33, 35, 36, 40, 50-52, 55, 57, 60, 61, 64, 65, 69, 74, 85, 87,
110, 112, 119, 121, 125, 127, 135, 137, 142, 146, 154, 156, 162.
Задачи, требующие объяснения
' 33, 34, 44, 45, 47, 48, 50, 51, 54, 59, 60, 62, 64, 65, 69, 74, 85, 89, 121, 129,
133, 143, 146, 154.
Задачи, требующие интерпретации физико-химических
и химических данных
37, 43, 60, 62, 86, 87, 109.
Задачи, требующие интерпретации спектральных данных
Все задачи главы 2; 71, 117, 123—125, 134—136, 148, 156—162.
Задачи, в которых необходимо предсказать продукты реакции
■ . 20, 21, 24, 26, 27, 29, 35, 36, 40, 49, 52, 55—58, 71-73, 79, 88, 99, 101-104,
108, 117, 122, 126, 132-134, 142, 143, 155.
Установление механизмов реакции
38, 39, 41, 42, 46, 53, 61, 63, 66-68, 70, 76-79, 81-84, 91-98, 100, 105, 106,
110—116, 118-120, 127, 128, 130, 131, 138—141, 144, 145, 147, 149, 155.
Проведение синтеза или расщепления
80, 90, 107, 150-152.
Установление структуры большого числа соединений,
связанных взаимными превращениями
22, 23, 74, 75, 123—125, 135, 136, 148, 156, 157—162.
* Приведены ссылки на номера задач.
Предметный
Азулен 2, 4, 5
Алкилирование кетонов 90
Алкилсульфиты 61
Амбидентные ионы 45, 46
Аминокислоты 28, 29, 154
Анну лены 5
Ароматичность 5
Вигилера синтез индолов 85
Брауна реакция 153
N-Бром- и N-хлорсукцинпмиды 126, 151,
153, 154
Водородная связь 18, 19, 33, 79
В осста новл ение
алюмогидридом лития 20, 40, 109, 135,
144—146, 152, 156, 158, 159, 162
амальгамой натрия 162
боргидридом натрия 146, 160, 162
каталитическое гидрирование 3, 21 —
24, 74, 80, 144, 146, 150, 151, 157™
159, 161
по Кияшеру — Вольфу 144, 151
по Клемменсепу 23
муравьиной кислотой 21, 116
по Роземунду 152
сернистым ангидридом 153
а-Галогепкетоны 74, 88, 148
Гидроборирование 27, 56, 57
Гидролиз 33, 43, 44, 70, 83
Гриньяра реакция 65, 74, 83, S4, 114. 135,
152, 158
Дегидратация 49
Дегидробензол 37, 80
Дегидрогалогенирование 47, 49—51, 62,
68, 73
* Приведены ссылки на номера задач.
указатель
Дегидрирование 23, 81, 112, 119
2'-Дезоксиуридин 25
Декарбоксилирование 21, 23, 53—55, 161
Децевиновая кислота 23
Диазометан, расширение цикла 79
Диазосо единения 117, 129
Дигидро-а-пикротоксиновая кислота 91
Дильса — Альдера реакция 74, 81, 121
Дипольные моменты 2, 4, 21
Дисперсия оптического вращения 30—32
Енамины 90, 99, 100, 116, 158
Енолизация 13, 18, 19, 24, 74, 88, 103,125
Замещение ароматическое
нуклеофильное 33, 37, 38, 101
электрофильное 35, 39
Замещения реакции 22, 34—36, 38, 39, 41,
42, 45, 46, 63, 87, 151
Днеолъда символика 1
Индуктивный эффект 1, 8, 9, 16, 37, 54
Инфракрасные спектры 18—24, И 7, 123—
125, 135, 136, 156—160, 162
Канниццаро реакция 94
Кариофиллен 77
Кинетический (термодинамический)
контроль 46, 48, 74
Кислотность 8, 9, 20, 23
Кольбе электролиз 122
Коммовая кислота Е 69
Конденсация 21, 93, 97, 122, 157
альдольная 67, 69, 94,120,138,150,161
ацилоиновая 20
бензоиновая 89
Дарзана 148, 151
Кляйзена (Дикмана) 21, 92, 95, 98,
124, 125
316
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Манниха 119, 150
Михаэля 67, 91, 94, 95
— аномальная 93
ретроальдольная 66, 91, 94, 96, 120,
136, 138
Ланостерин 112
Левулиыовая кислота 70
Линдлафа катализатор 150
Люмисантонин 130
Масс-спектры 28, 29
Меченые атомы 80, 86, 105—107, 109, 113,
126
Морфии 1 4
Напряжение (В, I, F) 20, 34 41
Небензоидные ароматические соединения
2-5, 7, 46
Неклассические ионы 17, 36, 39
Неопикротоксин 120
Непеталактон 161
Нитрозамиды 129
Окисление
азотной кислотой 141
гипогалогенитами 149, 160, 161
двуокисью селена 147
', Девре реагентом 162
диметилсульфоксидом 38
иодом и ацетатом серебра 142
. " йодной кислотой и ее солями 124, 135,
141, 143, 159, 162
озоном 138, 139, 162
перекисями и надкислотами 135, 140,
156, 161, 162
перманганатом калия 80, 92, 107, 156,
161
перманганатом и йодной кислотой 159
тетраацетатом свинца 143, М8, 162
тетраокисью осмия 135, 142, 156
хлоридом желева(Ш) 153
хромовым ангидридом 75, 137, 161
Октантов правило 30—32
Оптическая активность 10—12,119,121,154
Палитантип 162
Перегруппировки 68, 69, 77, 81, 83, 85, 92,
104, 109, 111, 113-115, 117-120, 123,
128, 140, 147, 155
алкиланилинов 105
антрастероидов 141
ацилоинов 110, 124, 125
Бекмана 101, 153
бензиловая 103
Бекера — Венкатарамана 102
Вагнера — Меервейпа 42, 71, 101,
105, 112
Гофмана 105, 152
дезаминированпе 72, 151
диенон-фенольная 115
Кляизена 106
Коупа 121
Курциуса 112, 152, 162
Мейера — Шу стера 101
нитроааминов 105
Ортона 105
пинаколиновая 101'
Стивснса 102, 106
Фаворского 91, 107, 108, 145
фотохимические 130, 131, 156
Фриса 105
^-холестерина 36
Хунсдиккера 122, 152
Шмидта 80, 152
Штиглица 102
эфиров фенолов 105
Якобсепа 105
Перекиси 86, 109, 128
сс-Пинен 156
Пиразолины 127
Пиретозин 77
Пиримидины 82
Пиролиз 50, 59, 61
Иринса реакция 58
Присоединения реакции к алкенам 48, 52,
58, 68, 73, 83, 85, 93, 131, 161
Птеридины 82
Резонанс и резонансные эффекты 1—9, 18,
19, 33
Рихтера реакция 38
Сантонин 91, 130
Сидновы 5
Сольволиз 34, 36, 41, 43, 44
Соседних групп участие 36, 38, 39, 42—44,
63, 64, 71, 73, 87
Стереохимия 10—12
Стерический контроль присоединения 52
57, 65
Таутомерия 6, 76
Тебаин 114
Теплоты гидрирования 3
Термодинамический (кинетический) копт-
роль 46, 48, 74
Трансапнулярное взаимодействие 20, 21, 24
Трополоны 4, 103
Ультрафиолетовые спектры 14—17, 21, 22,
71, 124, 125, 136, 160, 162
Фейста кислота 77, 159
Фотолиз 132—136
Фотосантониновая кислота 130
Фриделя — Крафтса реакция 81
Фторциклобутаны 22, 75
Фуран 78
Фурфурол 70
о-Хинодиметан 78
УКАЗАТЕЛЬ
Циглера — Хафнера сиптез 117
Циклазины 5
Циклобутадиен 5
Циклооктатетраенил-дианиоп 5
Циклопронаны, получение 07, 71 77
127, 130, 133 ' '
Циклопропенилий-катиоп 5
Цинсповая кислота 1i3
Чугаева реакция 50, 59
Эифол 112
Элиминирование 23, 40, 59—61, 63—66 69
71, 72, 124
Эпоксиды 40, 52, 65, 77, 91, 104, 111, 118,
120, 140, 145, 148.
ЯМР-спектры 25—27, 134, 136
ОГЛАВЛЕНИЕ
Предисловие 5
Предисловие авторов 7
Общая литература 8
Глава 1. Общие проблемы 9
Литература 31
Глава 2. Задачи по спектроскопии 32
Литература 64
Глава 3. Реакции замещения 67
Литература 86
Глава 4. Реакции присоединения и элиминирования ... 87
Литература 129
Глава 5. Смешанные реакции 131
Литература 156
Глава 6. Реакции конденсации и родственные реакции . , . 158
Литература 179
Глава 7. Перегруппировки ■. ... 181
Литература 233
Глава 8. Радикальные и фотохимические реакции .... 235
Литература 254
Глава 9. Реакции окисления — восстановления 255
Литература 277
ОГЛАВЛЕНИЕ
Глава 10. Сметанные реакции 07я
Литература
Указатель типов задач
Предметный {указатель ... „,_