/

























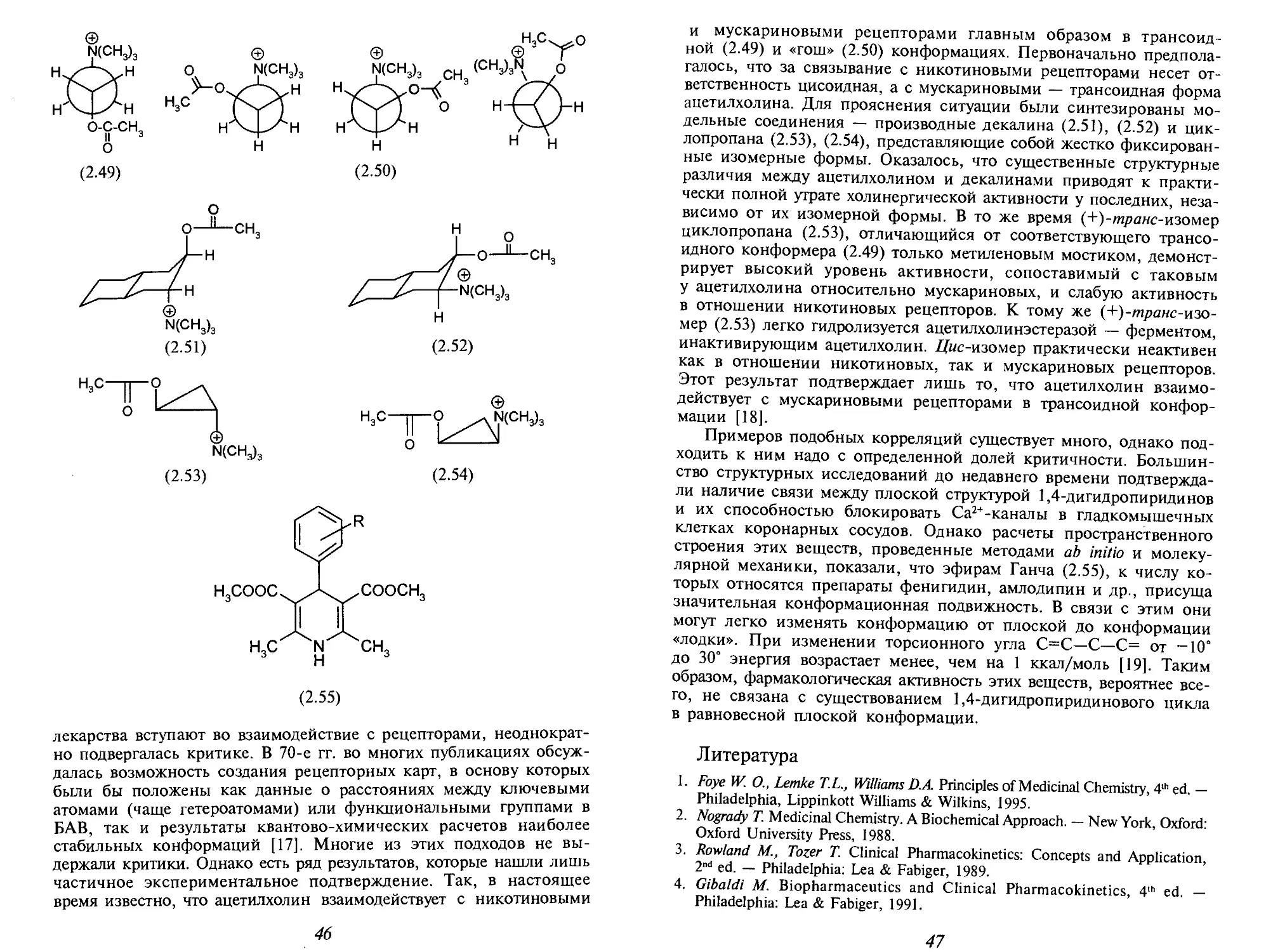




















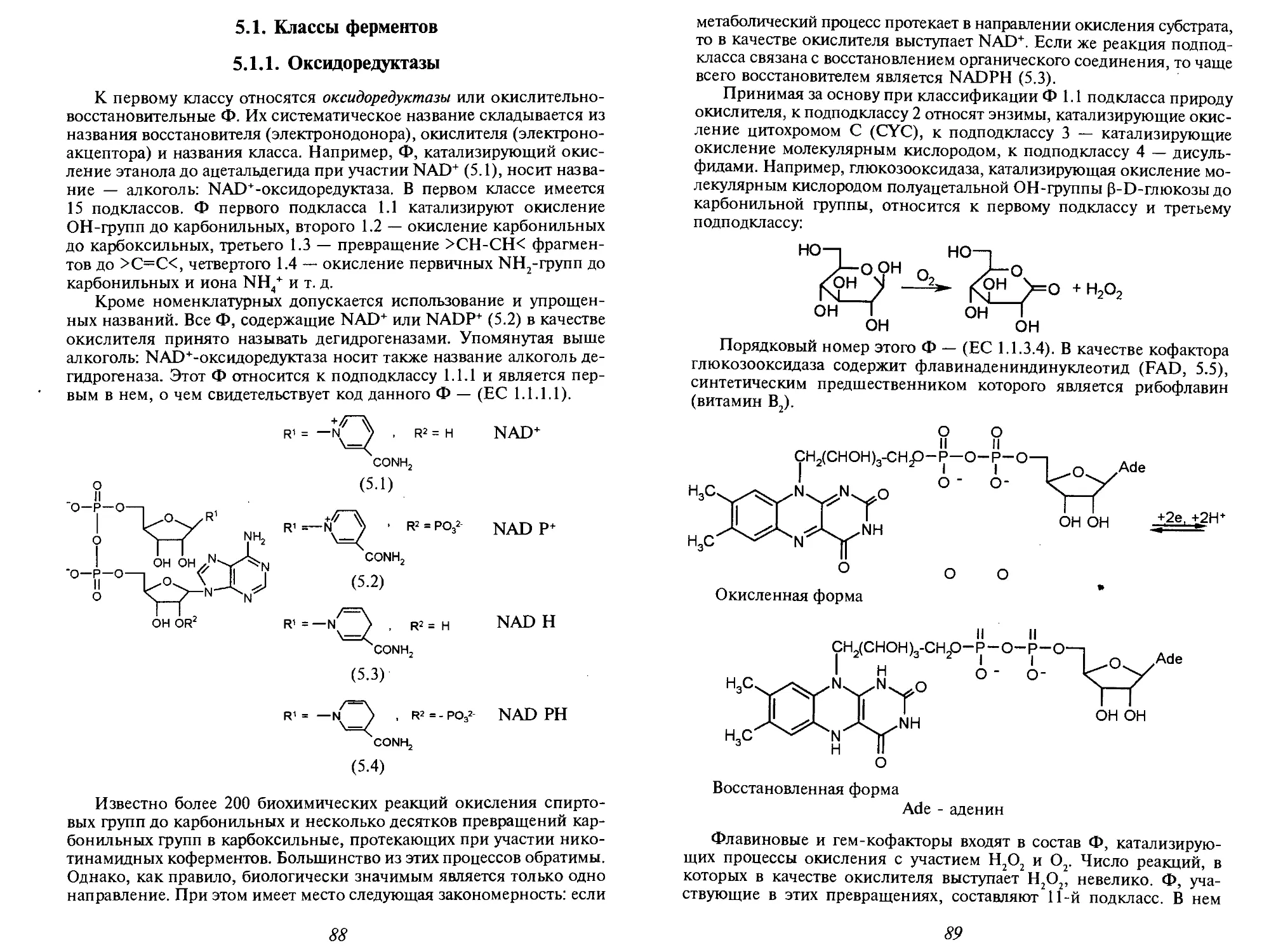

















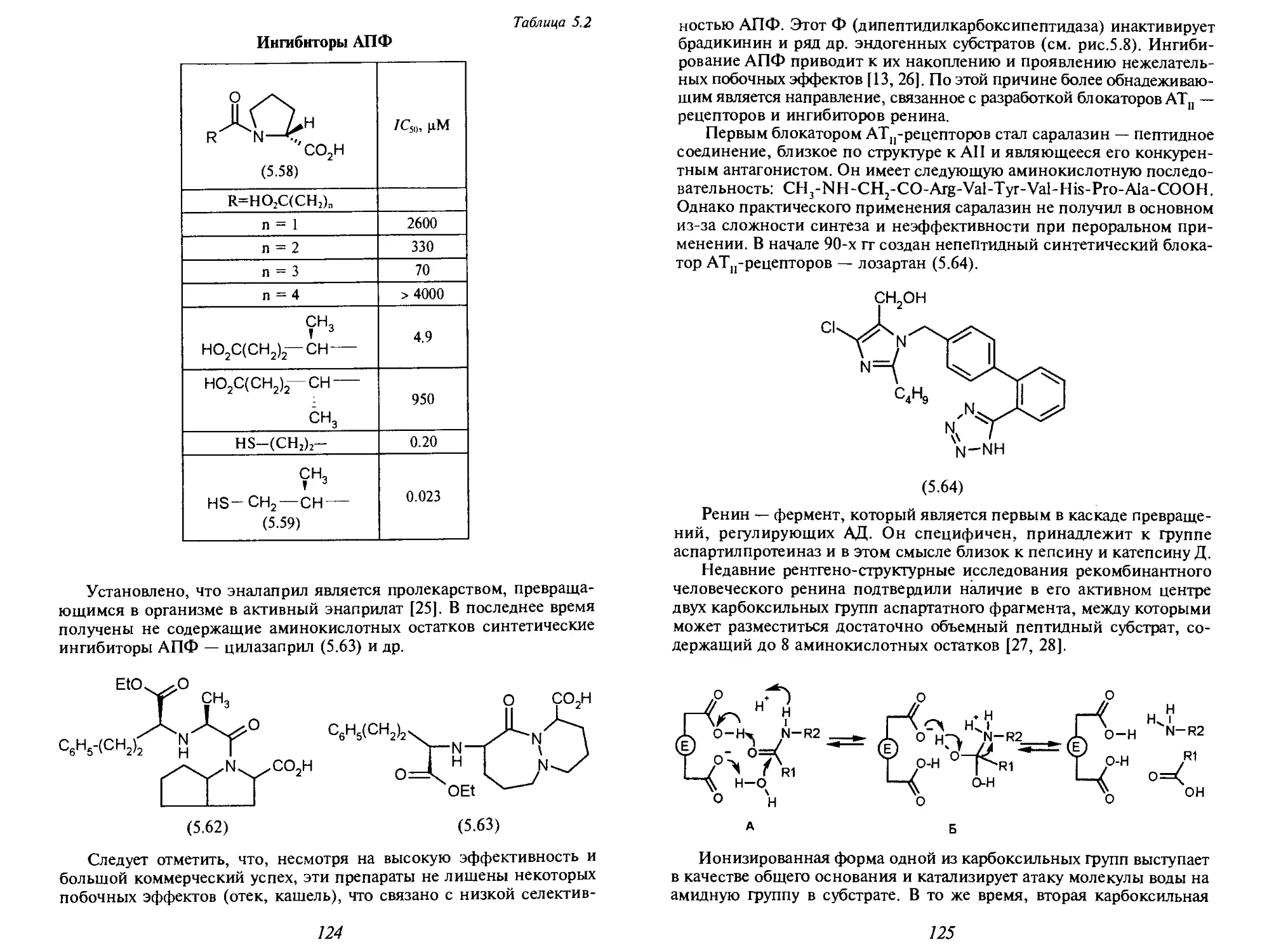






























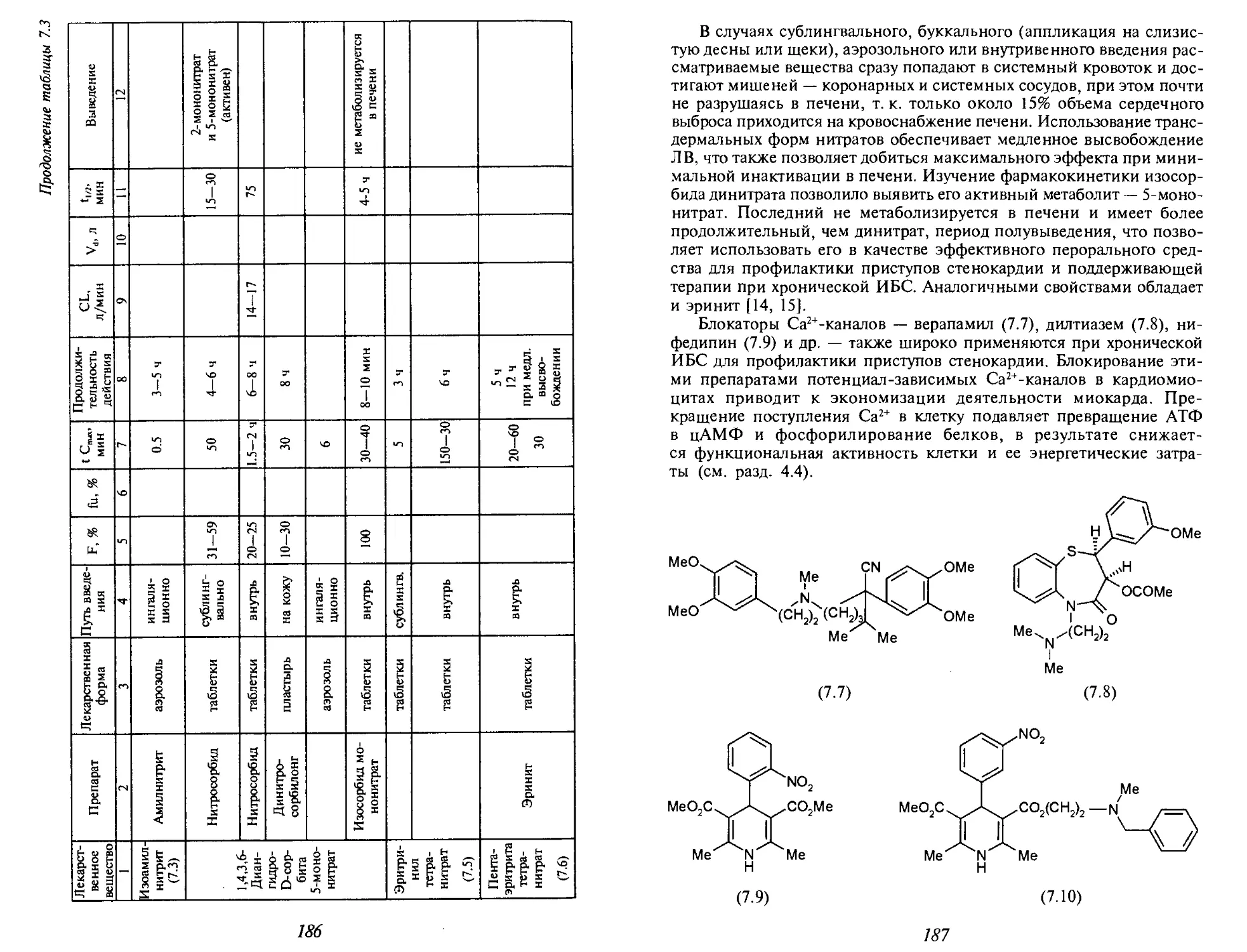


























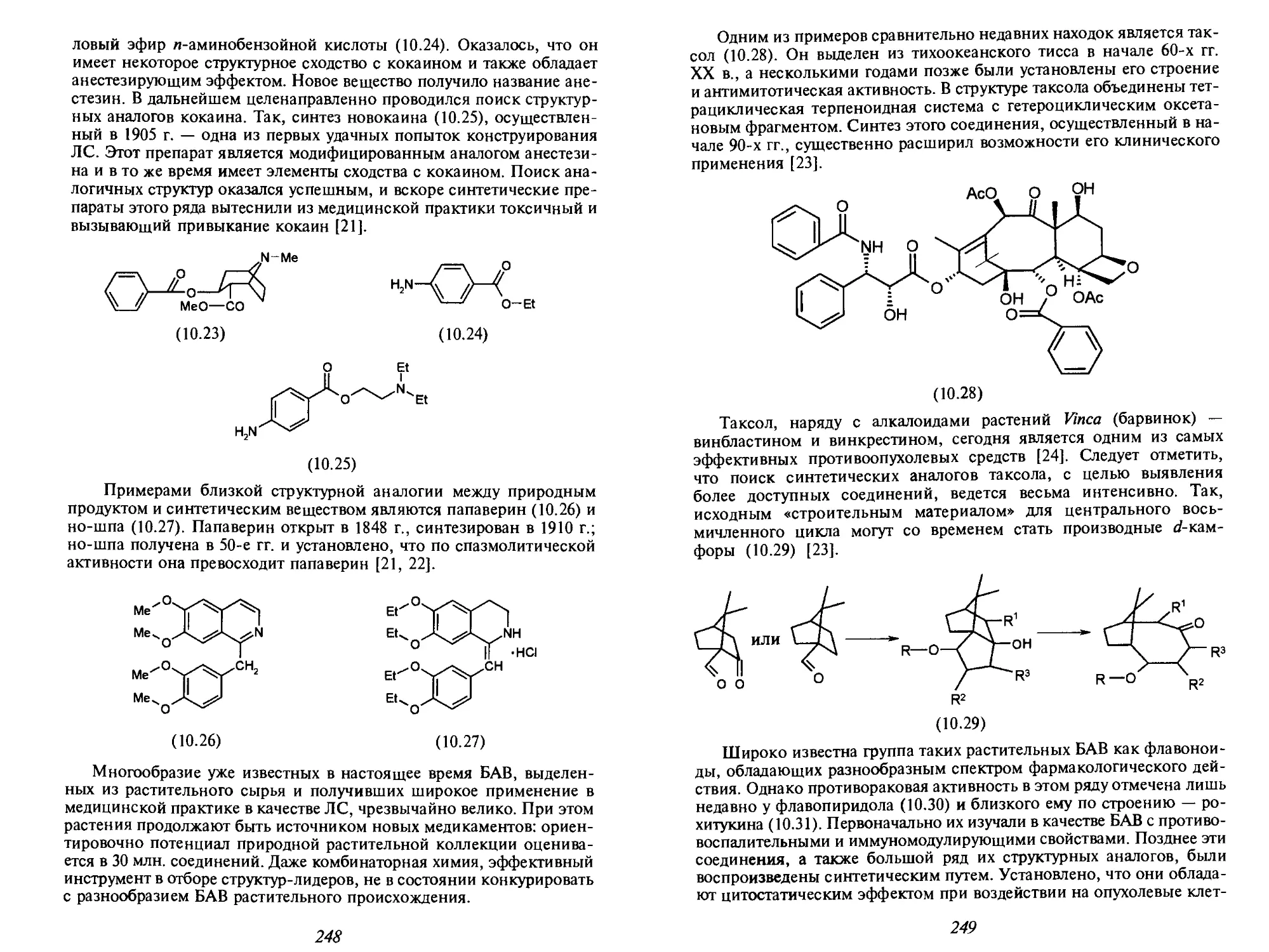



















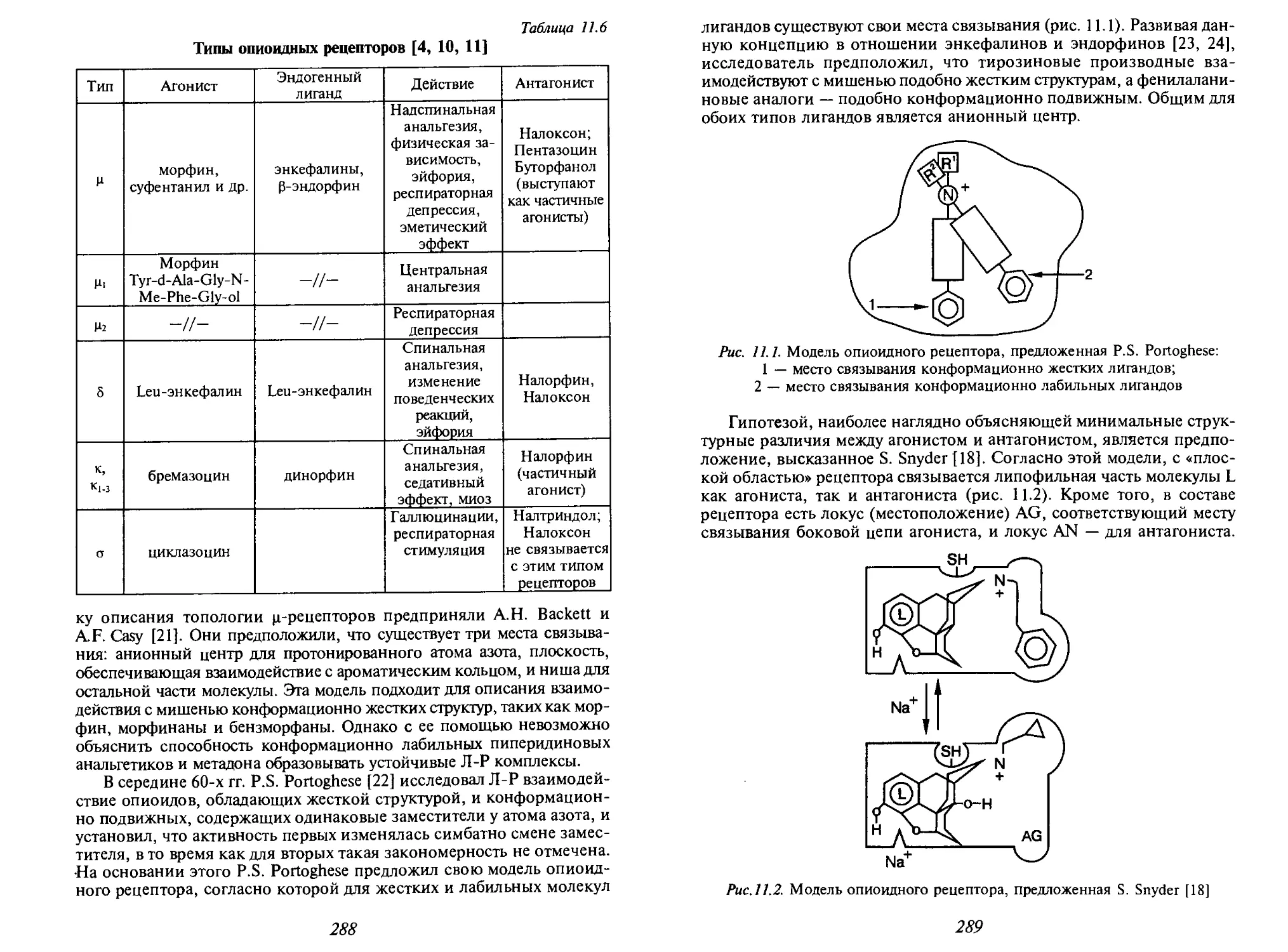















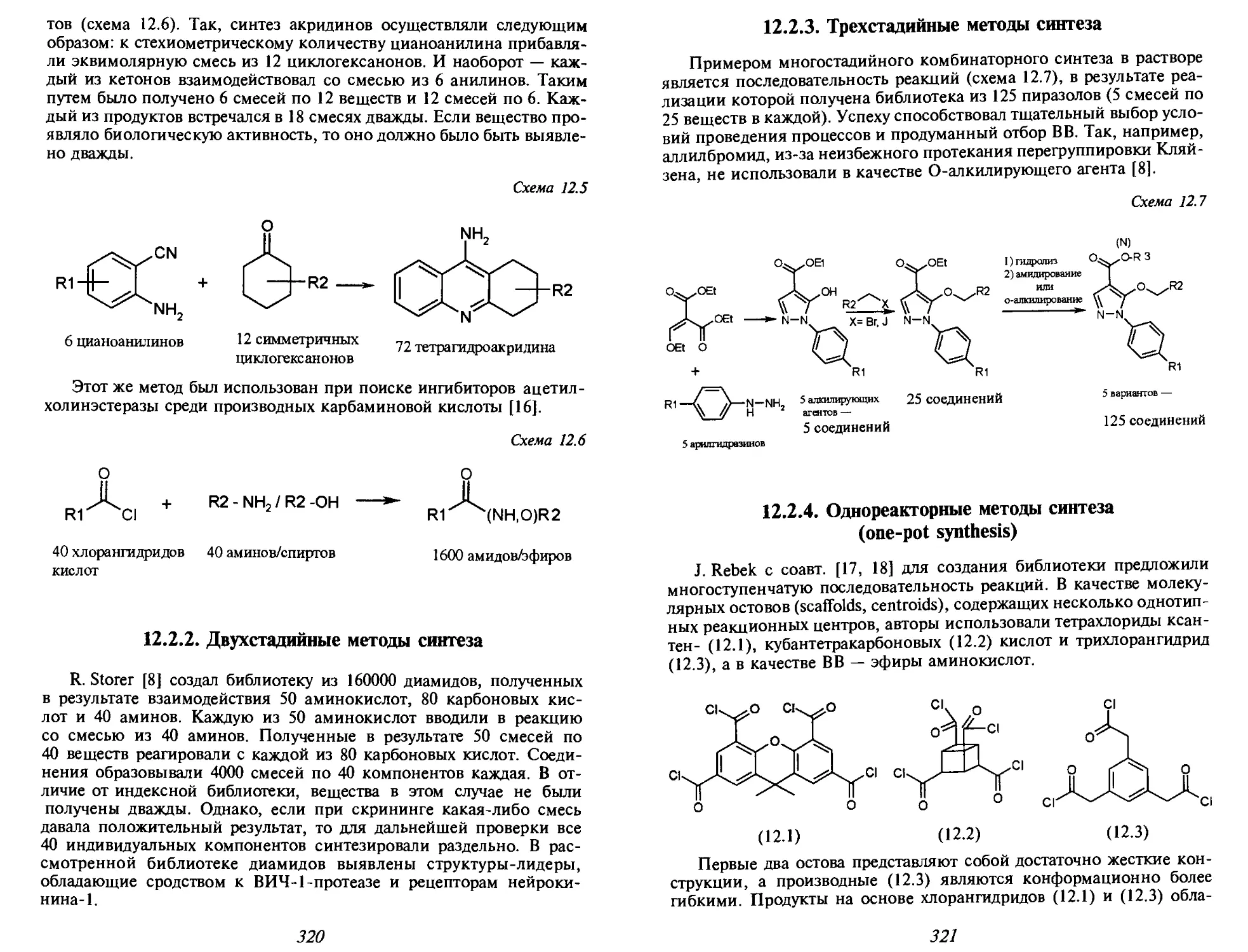








































































Автор: Иванов В.В. Орлов В.Д. Липсон В.В.
Теги: фармакология фармация токсикология медицина химия
ISBN: 966-03-3119-3
Год: 2005




Текст
МЕДИЦИНСКАЯ
ХИМИЯ
В учебнике кратко изложены основные понятия клеточной,
молекулярной биологии и биохимии (строение клетки
и биологических мембран, функции ферментов,
нуклеиновых кислот, вторичных посредников), необходимые
химикам для рассмотрения вопросов общей фармакологии
(фармакодинамики, фармакокинетики, метаболизма
лекарственных средств). Кроме того, в него включены
разделы, посвященные проблеме связи физико-химических
свойств биологически активных соединений с их
фармакологическим действием и методов количественной
оценки «структура-активность».
В книге проведен анализ современных подходов
в конструировании лекарственных веществ и в оценке
их специфических эффектов, таких как комбинаторный
синтез и тотальный скрининг, дана характеристика
этапов разработки новых лекарственных средств.
Учебник содержит обширный иллюстративный материал,
включающий химические формулы всех обсуждаемых
в тексте лекарственных веществ, а также таблицы
с данными о физико-химических, фармакологических
свойствах и фармакокинетических параметрах многих
препаратов.
Книга предназначена для исследователей, работающих
в области медицинской химии, студентов и аспирантов-
химиков, фармацевтов, специализирующихся в области
тонкого органического синтеза и создания новых лекарств.
^- - - #
1
\
*&4
^
<
ISBN 966-03-3119-3
'- ч , у
в. Д
в. в
в. в
Орлов
Липсои
Иванов
МЕДИЦИНСКАЯ
Учебник для спдентов
высших учеОньк
заведении
v
В. Д. Орлов, В. В. Липсон, В. В. Иванов
МЕДИЦИНСКАЯ
химия
Утверждено Министерством образования и науки Украины
как учебник для студентов химических специальностей
высших учебных заведений
Харьков
«Фолио»
2005
ББК 52.81
0-66
Утверждено Министерством образования и науки Украины
как учебник для студентов химических специальностей
высших учебных заведений
(Письмо № 14/18.2-15110 от 30.06.2005)
Рецензент:
академик НАН Украины,
директор Института органической химии НАН Украины
М. О. Лозинский.
Художник-оформлювач
А. С. Ленчик
© В. Д. Орлов, В. В. Липсон, В. В. Иванов, 2005
ISBN 966-03-31 19-3 © А. С. Ленчик, художественное оформление, 2005
ПРЕДИСЛОВИЕ
Настоящая книга предназначена для студентов и
аспирантов-химиков, специализирующихся в области тонкого органического
синтеза, а также для всех заинтересованных неспециалистов, желающих
познакомиться с основами медицинской химии — науки, предметом
исследований которой является поиск биологически активных веществ,
интерпретация механизма их действия на молекулярном уровне и
разработка на этой основе новых лекарственных средств.
Профессиональная подготовка химиков не предполагает знаний
основ биохимии, молекулярной и клеточной биологии,
фармакологии. В то же время, успех большинства проектов в области
конструирования лекарств (drug design) во многом зависит от того, насколько
продуктивно взаимодействие между представителями разных
специальностей, и в какой мере они готовы к такому сотрудничеству.
Одной из граней проблемы междисциплинарного взаимодействия
является адекватная формулировка фармакологической задачи на
структурном языке органической химии.
Свою скромную миссию авторы этой книги видят в том,
чтобы в какой-то мере восполнить у химиков недостаток в знаниях
по отдельным разделам биохимии и фармакологии, научить их
«говорить на одном языке» со специалистами в этих областях.
Основная цель состоит в формировании у читателей целостного
представления о процессе создания лекарств, начиная от момента
выдвижения идеи синтеза веществ определенного строения,
проведения скрининга и усовершенствования структуры, вплоть до
стадии клинических испытаний и организации производства. В этой
связи в книге рассмотрены темы, позволяющие получить
элементарные сведения о мишенях, фармакокинетике и метаболизме
лекарственных веществ в организме, о современных подходах в
конструировании лекарств и видах фармакологических испытаний новых
химических соединений, а также о методах количественной
оценки связи «структура — активность». По каждой из этих проблем
написана не одна монография, с которыми данная книга не
может конкурировать. Но в современной учебной литературе,
доступной студентам-химикам, эти вопросы освещены недостаточно. Книга
написана по материалам лекций, которые авторы на протяжении
ряда лет читают на химическом факультете Харьковского
национального университета им. В.Н. Каразина. Авторы ни в коей мере
не претендуют на исчерпывающую полноту изложения всех
проблем медицинской химии, и с благодарностью отнесутся к
пожеланиям и критическим замечаниям, которые возникнут у читателей
при анализе предложенного материала.
3
УСЛОВНЫЕ СОКРАЩЕНИЯ
АД — артериальное давление
АДФ — аденозиндифосфат
АНД — аналитический нормативный
документ
АТФ — аденозинтрифосфат
АМФ — аденозинмонофосфат
цАМФ — циклический АМФ
АПФ — ангиотензинпревращающий
фермент
БАВ — биологически активное
вещество
ВП — вторичный посредник
ВЭЖХ — высоко эффективная
жидкостная хроматография
ГАМК — у-аминомасляная кислота
ГДФ — гуанозиндифосфат
ГМФ — гуанозинмонофосфат
цГМФ — циклический ГМФ
ГЭБ — гематоэнцефалический барьер
/-ДОФА — /-диоксифенилаланин
ЗДМ — закон действия масс
ИФ — ингибитор фермента
ИБС — ишемическая болезнь сердца
ИНЗСД — инсулин-независимый
сахарный диабет
КОМТ — катехоламин-О-метилтран-
сфераза
ЛВ — лекарственное вещество
ЛС — лекарственное средство
ЛФ — лекарственная форма
НК — нуклеиновые кислоты
НПВС — нестероидные
противовоспалительные средства
ОНД — отраслевой нормативный
документ
ПАВ — поверхностно-активное
вещество
ПАУ — полициклические
ароматические углеводороды
ПК — пищеварительный канал
РСИ — рентгено-структурное
исследование
СД — сахарный диабет
СМ — сульфонилмочевина
ССС — сердечно-сосудистая система
ТСХ — тонкослойная хроматография
Ф — фермент
ФЫМТ — фенилэтаноламин-Ы-ме-
тилтрансфераза
ЧСС — частота сердечных
сокращений
ЦНС — центральная нервная система
ЭДТА — этилендиаминтетрауксусная
кислота
ЭР — эндоплазматический ретикулум
ЭПР — электронный парамагнитный
резонанс
ЯМР — ядерный магнитный резонанс
i
СТРОЕНИЕ КЛЕТКИ
Клетка — структурная и функциональная единица живого
организма, способная к самовоспроизведению. Различают два класса: про-
кариотические — клетки бактерий и сине-зеленых водорослей и
эукариотические — клетки некоторых одноклеточных организмов,
а также растений, животных и человека. Независимо от типа, клетка
окружена плазматической мембраной, характеризующейся
избирательной проницаемостью, благодаря которой обеспечивается защита от
чужеродных веществ и в то же время осуществляется обмен с
внешней средой.
Внутреннее пространство клетки — цитоплазма. В ней
располагаются органеллы клетки и протекают биохимические процессы,
составляющие основу ее метаболизма. Водная часть цитоплазмы — ци-
тозоль. В структуре прокариотических клеток (рис. 1.1) имеются: одна
хромосома — двуцепочечная молекула дезоксирибонуклеиновой
кислоты (ДНК), плазмиды — внехромосомные ДНК-содержащие
элементы, несущие лишь по нескольку генов. Некоторые из них кодируют
ферменты, благодаря которым клетка становится устойчивой к
антибиотикам [1].
Рибосомы — сложные органеллы, состоящие из двух субчастиц, в
составе которых имеются рибонуклеиновые кислоты (РНК),
обеспечивающие синтез белков, и сами белковые молекулы, главным
образом, ферменты. Скопления рибосом получили название полисом.
В цитоплазме бактерий имеются также гранулы, представляющие депо
питательных веществ. В цитозоле находятся неорганические ионы,
растворимые макромолекулы и их синтетические предшественники —
аминокислоты, пуриновые и пиримидиновые основания,
моносахариды.
Отличительной чертой прокариотических клеток, кроме отсутствия
ядра и митохондрий, является наличие плотной клеточной стенки,
находящейся снаружи от плазматической мембраны и выполняющей
функции защитного барьера. У бактерий клеточная стенка
образовала жесткой сетью из липидов, полисахаридов и белков (рис. 1.2),
которая предотвращает лизис клетки из-за осмотического шока.
В структурном отношении стенка бывает двух типов, в соответствии
с чем бактерии разделяют на грамположительные и грамотрицательные.
Первые обладают однослойной клеточной стенкой, толщина которой
5
\
Нить
Сине-зеленая водоросль
Рис. 1.1. Прокариотическая клетка [1]
составляет 20—80 нм при толщине плазматической мембраны 7.5 нм.
У грамотрицательных бактерий стенка двуслойная. Первый слой
(1 нм) — нелипидная мембрана — окружен вторым — липидной
мембраной (7.5 нм). При обработке грамположительных бактерий
гидролитическим ферментом — лизоцимом — стенка
«растворяется» и остаются клетки, имеющие только плазматическую
мембрану. Они получили название протопластов. Протопласты
чувствительны к осмотическому шоку. У грамотрицательных бактерий наружная
мембрана в присутствии лизоцима сохраняется, а образующиеся
клетки — сферопласты — несколько менее чувствительны к
осмотическому шоку. Материал клеточных стенок, который
утрачивается в обоих случаях, известен как пептидогликан (гликопептид или
мукопептид) [1, 2].
Плазматическая мембрана состоит из липидов, белков и
углеводов и выступает в качестве элемента, транспортирующего полезные
вещества внутрь клетки, а ненужные и вредные — наружу. Кроме
того, в ней происходит синтез и накопление аденозинтрифосфата
(АТФ) — основного энергетического вещества. Биосинтез клеточных
компонентов происходит в цитоплазме.
6
ГРАМОТРИЦАТЕЛЬНАЯ
ГРАМПОЛОЖИТЕЛЬНАЯ
E.coli S. aureus
Рис. 1.2. Клеточная стенка бактерий [1]
Процесс репродукции и передачи генетической информации у
бактерий чрезвычайно прост. Их клетки растут, пока не увеличатся по
размерам вдвое. Затем происходит деление на две одинаковые
дочерние клетки, каждая из которых содержит точную копию
родительской ДНК.
Устройство эукариотических клеток намного сложнее (рис. 1.3).
Из клеток такого типа состоят одноклеточные простейшие, грибы,
высшие растения и многоклеточные животные. Следует подчеркнуть,
что клетки из различных органов высших организмов могут
существенно отличаться по морфологии и функциям. Достаточно
упомянуть о лейкоцитах, эритроцитах, нервных клетках, клетках мышц,
почечных канальцев или островковых клетках поджелудочной
железы человека, чтобы обнаружить колоссальное разнообразие. По этой
причине на рис. 1.3 отражены лишь главные черты большинства
эукариотических клеток.
Для них характерно наличие окруженного оболочкой ядра со
сложной внутренней структурой. Ядерная оболочка состоит из двух
близко расположенных мембран, разделенных перинуклеарным
пространством. По всей поверхности мембраны равномерно
распределены ядерные поры, образованные белковыми гранулами. Через
поры осуществляется обмен веществами между ядром и
цитоплазмой. Внутри ядра расположено ядрышко. В нем содержатся РНК,
ДНК-белковые комплексы и осуществляется первый этап синтеза
рибосом.
В ядре распределен хроматин, состоящий из ДНК, РНК и
специфических белков. Перед делением (митозом) хроматин
собирается в определенное число гранул — хромосом. У человека в
диплоидных клетках число хромосом равно 46. После копирования
7
Триплеты микротрубочек
Окаймленная ямка
Комплекс Гольджи I Пиноцитозный пузырек
Ядерные
поры
ТОЛЬКО
ДЛ'Я
КЛЕТОК
ЭТОГО
ТИПА
ДЛЯ
ОБОИХ
ТИПОВ
КЛЕТОК
ТОЛЬКО
для
> КЛЕТОК
ЭТОГО
ТИПА
[растительная клетка
Хлоропласт
Фотоси нтези рующие
ламеллы
Рис. 1.3. Эукариотическая клетка [1]
8
(репликации) родительской ДНК и митоза хроматин вновь
распределяется по всему ядру.
«Энергетическими установками» клетки являются митохондрии.
Они образованы двумя мембранными системами — внешней
(гладкой) и внутренней (складчатой). Такие складки получили название
крист. Внутреннее пространство составляет гелеобразный матрикс,
содержащий рибосомы и митохондриальную ДНК. В митоходриях
реализуется ферментативное окисление питательных веществ до
углекислого газа и воды. Выделившаяся в результате окислительных
(катаболических) процессов энергия расходуется на синтез АТФ,
диффундирующего во все части клетки для выполнения необходимой
работы.
Цитоплазма эукариотических клеток располагает сложной
системой каналов, образованных мембранами и получивших название
эндоплазматинеского ретикулума (ЭР). Внутреннее пространство
каналов — цистерны. Они служат как для транспортировки веществ
из клетки во внешнюю среду, так и хранилищами питательных
веществ. Существует две разновидности ЭР — гладкая и
шероховатая. В шероховатом ЭР располагаются рибосомы и осуществляется
синтез белков. Их молекулы поступают внутрь цистерн и выходят
во внеклеточное пространство. На мембранах гладкого ЭР
протекает синтез липидов, а также гидролитическое расщепление
гликогена (гликогенолиз). Клеточные продукты из ЭР поступают в
комплекс Гольджи, представляющий собой скопление пузырьков,
окруженных мембранами. Здесь происходит модификация белков
(например, гликозилирование), предназначенных для секреции из
клетки. «Секреторные пузырьки», разрываясь, выбрасывают
содержимое во внеклеточную среду. Этот процесс носит название экзо-
цитоза. Другой тип пузырьков — лизосомы. Они содержат
гидролитические ферменты, основная функция которых — гидролиз
ненужных клетке белков, полисахаридов, липидов. Таким образом,
процессы синтеза и деградации соединений в клетке
пространственно разделены. Еще один тип «реакторов» представляют собой пе-
роксисомы. В них осуществляются ферментативные процессы,
связанные с образованием и распадом Н2Ог При этом остальная часть
клетки защищена от повреждающего воздействия перекисных
радикалов.
Своего рода «арматурой», поддерживающей определенное
внутреннее сопротивление в клетке и обеспечивающей ее целостность
при перемещении внутриклеточных структур и сокращении мышц,
служат нити из белковых молекул актина и миозина, получившие
название микрофиламентов.
Молекулы белков а- и р-тубулинов объединены в центриоли в
виде девяти триплетов микротрубочек и располагаются вблизи ядра.
Они выполняют транспортные функции и участвуют в процессах,
происходящих при делении клетки.
В цитоплазме располагаются образования, не имеющие мембран.
Это рибосомы, осуществляющие синтез белка, и гранулы гликоге-
9
на — запасные источники энергии, образованные молекулами
глюкозы. Здесь же содержатся белки и аминокислоты, нуклеотиды,
неорганические ионы, сахара.
Некоторые органеллы, такие как комплекс Гольджи, напрямую
связаны с поверхностью цитоплазматической мембраны; другие
(гладкий и шероховатый ЭР) непосредственно с ней не
контактируют. С внешней стороны цитоплазматической мембраны иногда
имеется еще и внешняя оболочка, через которую
осуществляются межклеточные контакты. Она способна принимать и
распознавать определенные сигналы, поступающие как от родственных
клеток при образовании тканевой структуры, так и от
биологических посредников — гормонов, нейромедиаторов. В состав
такой оболочки входят белки, полисахариды, липиды.
Распознающие участки — это, как правило, белковые образования или
полисахариды [2—4].
1.1. Краткие сведения
о строении биологических мембран
Мембрана отделяет цитоплазму клетки от окружающей среды, а
также служит барьером, ограничивающим функциональные единицы
органелл от других клеточных компонентов. Ее основной
структурный материал — липиды, белки и углеводы.
1.1.1. Мембранные липиды
Мембранные липиды — это, главным образом, фосфолипиды, у
которых одна или две жирнокислотные цепи эстерифицированы
глицерином или сфингозином.
Ацилглицериды — сложные эфиры глицерина и высших жирных
кислот (рис. 1.4).
Ацильные остатки могут существенно отличаться по длине цепи,
разветвленности и степени ненасыщенности. Из насыщенных
жирных кислот в состав липидов входят миристиновая, пальмитиновая,
стеариновая, арахиновая, бегеновая и лигноцериновая.
Ненасыщенные ацильные цепи присоединяются, как правило, ко второму
углеродному атому глицерина и содержат двойные связи у девятого атома
углерода или еще дальше к концу цепи. В месте нахождения двойной
связи реализуется г<мс-конфигурация. В составе мембранных липидов
встречаются ненасыщенные пальмитоолеиновая, олеиновая, лино-
левая, линоленовая и арахидоновая кислоты.
Присоединение к ацилглицериду полярной группы (например,
остатка фосфорной кислоты) придает веществу специфические
свойства. Липиды, содержащие такую полярную группу, называют амфи-
патическими. Эта группа может нести заряд или быть незаряженной.
10
О
н2с-о—"—(сн2)14—сн3 н2с-он
о
НС-0^-(СН2)14-СН3 НС-0-^(СН2)7-С=С-(СН2)7-СН3
о
о
Н2С-0^-(СН-2)1в-СНэ Н2С-0^-(СК2)14-СН.
Фосфолипиды
r=—Р-О —(СН2)2 —N+(CH3)3
О"
H.C-0-R п
| О ||
Н?—°~i)~(CH2)i~CH3 R = —Р—О—(СЮ, —N+H,
О
Н2С-0^(СН2)14—СН3 6"
О
н
R= —Р—О—СН9—С—COO
I- +l
О NH3
Рис. 1.4. Ацилглицериды
Наличие гидрофобного хвоста в виде остатков жирных кислот и
полярной головки позволяет фосфолипидам вступать как в гидрофильные,
так и в гидрофобные взаимодействия, что делает их идеальными
компонентами биологических мембран. Так, при избытке воды липиды
образуют мицеллы, в которых гидрофобные хвосты находятся внутри
сферической капли, а полярные головки расположены на ее
поверхности и контактируют с водой. Когда же в избытке находятся липиды,
наблюдается обратная картина: полярные головки обращены внутрь
сферы, включающей в себя и воду, а жирнокислотные цепи располагаются
снаружи. Такие «обращенные мицеллы» образуют эмульсию.
Часто к фосфатной группе присоединено азотсодержащее
основание. При этом головка имеет одновременно и положительный, и
отрицательный заряды, однако при физиологических значениях рН
является электронейтральной. Наиболее часто среди мембранообра-
зующих фосфолипидов встречаются производные холина (1.1), эта-
ноламина (1.2), серина (1.3) [4—6].
Неглицериновые липиды обнаружены во многих типах мембран
вместе с липидами, образованными на основе глицерина. Особенно
богаты ими нервные ткани, в частности, мозг. Существует три класса
неглицериновых липидов: цереброзиды, фосфосфинголипиды и ган-
глиозиды (рис. 1.5). В основе их структуры лежит сфингозин (1.4).
Последний имеет определенное сходство в строении с моно-
ацилглицерином, однако является аминоспиртом и содержит негид-
ролизуемую ненасыщенную углеводородную цепь.
11
н2с-он
СН—NH2
нс-£=с-(сн2)12—сн3
он
сфингозин (1.4)
ОН
R =
галактоцереброзид (1.6)
R=H
церамид (1.5)
R= —Р—О—(СН2)2 —N(CH3)3
0~
сфигномиелин (1.7)
I
NANA
R =р -1 Glc4-*-^— 1 Glc4-*-^-1 Gal Nac 3
t
P
1 Gal
ганглиозид (1.8)
H2C-OR 0
CH— N—u— (CH,)7—C=C—(CH2)7— CH,
| H 27 H H 27 3
не—c=c—(ск2)12—сн3
I H H 2'12 3
OH
R =
р-нейраминовая кислота (1.9)
Glc — Глюкоза
Gal — Галактоза
GalNac — Ацетилгалактозамин
NANA —Ацетилнейраминовая кислота
Рис. 1.5. Неглицериновые липиды
12
Церамид (1.5) — первичный сфинголипид — родоначальник всех
трех классов неглицериновых липидов; он является амидом
ненасыщенной жирной кислоты.
Цереброзиды (1.6) — гексозилированные церамиды. В качестве
моносахарида может выступать галактоза или глюкоза. Эти вещества
получили групповое название гликолипидов.
Фосфосфинголипиды — церамиды, эстерифицированные
фосфорной кислотой с азотсодержащим основанием. Они напоминают фос-
фоглицериды и являются амфипатическими. Типичный
представитель веществ этого класса — сфингомиелин (1.7).
Ганглиозиды (1.8) — церамиды, связанные гликозидной связью
Р-типа с коротким кислотным, как правило, разветвленным оли-
госахаридом. Носителем кислотных свойств является нейраминовая
кислота (1.9), присутствующая в качестве одной из ветвей саха-
ридной цепи. В тканях человека аминогруппа всегда ацетилиро-
вана [4, 6].
Простые липиды — это липиды, не содержащие остатков жирных
кислот. Их еще называют «неомыляемыми» липидами, поскольку они
не содержат эфирных связей. К этой группе принадлежат стероиды,
жирорастворимые витамины и терпены.
Стероиды — соединения, главным структурным фрагментом
которых является циклопентанопергидрофенантреновая система
(рис. 1.6). Так, в составе мембран грибов присутствует эргосте-
рин (1.10), а наиболее распространенным представителем стероидов
в животном мире является холестерин (1.11). Последний у человека —
предшественник всех стероидных гормонов и основная составная часть
желчных кислот. Размеры молекулы холестерина и присущие ему
слабые амфипатические свойства делают его распространенным
компонентом мембран многих животных клеток. Он составляет от 40 до
60% всех липидов эритроцитов млекопитающих [4, 5].
(1.10) (1.11)
Рис. 1.6. Представители группы простых липидов
Двойнойлипидный слой — структура, характерная для
плазматических мембран всех типов клеток. Толщина его колеблется в пределах
4—5 нм в зависимости от типов присутствующих в нем жирных
кислот. Липидные слои с внешней стороны образуют молекулы фосфа-
13
Рис. 1.7. Схематическое строение мембраны [1]
тидилхолина, а с внутренней — фосфатидилэтаноламина, фосфа-
тидилсерина или фосфатидилинозитола. Неполярные хвосты липид-
ных молекул обращены друг к другу, а полярные головки направлены
наружу. Таким образом формируются внутренняя и наружная
гидрофильные поверхности.
В составе мембраны имеются белки. Одна группа белков
способна пронизывать двойной липидный слой насквозь. Такие белки
получили название интегральных. Они связаны с липидами гидрофобным
взаимодействием и сохраняют нативную конформацию только в
комплексе с ними. Часто эти белки имеют углеводные фрагменты,
присоединенные к той части молекулы, которая выступает во
внеклеточную среду. Другие белки, связанные с мембраной менее прочно,
называются периферическими. Они, как правило, адсорбированы на
поверхности и удерживаются гидрофильными головками липидов за
счет электростатических сил. По своей структуре периферические
белки напоминают водорастворимые глобулярные белки.
В настоящее время общепризнанной является жидкомозаичная
модель структуры мембраны (рис. 1.7), предложенная в 1971 г. Г. Ни-
колсоном и С. Сингером [1, 5—7]. В соответствии с этой моделью
интегральные белки можно образно рассматривать как айсберги,
плавающие в липидном море. Пересекающие мембрану участки поли-
пептидной цепи часто образуют а-спирали. В такой структуре все
NH- и С=0-группы, за исключением находящихся на концах
спирали, образуют водородные связи. При этом обеспечивается оптимальное
14
для связывания с мембранными липидами расположение
гидрофобных боковых аминокислотных остатков.
Мембранные липиды, непосредственно примыкающие к белковым
молекулам, называются граничными. Их подвижность
значительно меньше, чем у основной массы липидов. В целом же
подвижность зависит от относительного содержания и типа
ненасыщенных жирных кислот, входящих в состав липидных молекул.
В присутствии ^-ненасыщенных кислот силы сцепления между
углеводородными цепями слабее, чем при наличии остатков только
насыщенных жирных кислот. В интервале температур 15—40 °С по
степени упорядоченности мембраны напоминают структуру жидких
кристаллов.
1.2. Перенос веществ через мембраны
Механизмы, посредством которых ионы или малые молекулы
проходят через плазматическую мембрану, принято подразделять на три
типа: диффузия, облегченная диффузия и активный транспорт.
Диффузия — это процесс, при котором частицы (молекулы или
ионы) переходят через мембрану из области с высокой
концентрацией в область с низкой концентрацией в результате броуновского
движения. Для незаряженных молекул скорость перехода описывается
законом Фика:
J = -Ddc/dx,
где /— поток вещества через мембрану, dc/dx — градиент
концентрации, D — коэффициент диффузии.
Скорость диффузии нейтральных молекул уменьшается с
увеличением их гидрофильное™.
Для ионов скорость перехода через мембрану описывается
модифицированным уравнением Фика:
/ = - Ddc/dx + Ady/dx,
где А — величина постоянная для данного иона, dy/dx —
трансмембранный градиент заряда.
Если клетка содержит полианионные протеины, а в окружающем
ее пространстве присутствуют ионы К+ и С1", то внутрь проникает
больше катионов К+, чем анионов С1\ В результате устанавливается
неодинаковое распределение ионов и возникает трансмембранный
электрохимический градиент [4, 6, 7].
Облегченная диффузия — процесс перемещения молекул по
градиенту концентрации при помощи белков-переносчиков,
локализованных в плазматической мембране. Он специфичен для определенных
молекул, осуществляется быстрее, чем обычная диффузия, достигает
насыщения. Специфические переносчики существуют для Сахаров,
аминокислот, пуриновых и пиримидиновых оснований, нуклеотидов,
глицерина и т.п. Избирательность обеспечивается наличием у пере-
15
носчика стереоспецифического места связывания. Например,
переносчики моносахаридов транспортируют только D-caxapa.
В клетке имеется конечное число переносчиков для каждого типа
частиц, и, когда все они задействованы, скорость переноса
становится максимальной. В отсутствие градиента концентрации
переносчики, продолжая работать, переносят молекулы или ионы внутрь и
наружу клетки с одинаковой скоростью, поэтому диффузия не
наблюдается [4—8].
Активный транспорт — это перенос частиц через
плазматическую мембрану против градиента концентрации. Он требует
притока энергии извне. Одна из наиболее изученных систем активного
транспорта — это Na+/K+-ATOa3a. С ее помощью поддерживаются
разные концентрации ионов Na+ и К+ внутри и снаружи клетки.
Обычно внутри преобладают ионы К+, а снаружи — Na+.
Поскольку ионы хотя и слабо, но все же диффундируют через мембрану,
их концентрации стремятся выравняться. Na+/K+-ATOa3a
осуществляет активный транспорт, что предотвращает выравнивание
концентраций указанных частиц. Перенос ионов кальция в
мышечные клетки обеспечивает Ca2+/Mg2+-ATOa3a. Системы активного
транспорта широко представлены в почечных канальцах. Здесь при
участии фермента глутамилтрансферазы осуществляется реаб-
сорбция аминокислот из мочи в виде у-глутамилпроизводных,
способных, в отличие от ионизированных аминокислот, проникать в
клетку, где происходит гидролиз на исходные глутатион и
аминокислоту [3, 5].
Еще один способ проникновения веществ в клетку — пиноци-
тоз. При этом небольшие области мембраны впячиваются, и
внутри клетки возникают пузырьки, несущие, например, внеклеточные
белки. Таким образом через мембрану проникают макромолекулы,
которые не могут пересечь этот барьер путем диффузии. Сходен с
пиноцитозом фагоцитоз: большие макромолекулярные комплексы
поступают в клетку или выделяются из нее в виде пузырьков,
окруженных липидными мембранами. Таким путем из клеток
поджелудочной железы секретируются гидролитические ферменты в
виде зимогеновых гранул, или высвобождается из нервных
окончаний в виде пузырьков медиатор центральной нервной системы
(ЦНС) — ацетилхолин. Если в клетку попадают чужеродные
белки, то в лизосомах они гидролизуются под действием протео-
литических ферментов. Бактерии и вирусы поступают в клетки также
путем фагоцитоза. В организме человека борьба с таким
вторжением осуществляется специальными клетками иммунной системы —
макрофагами. Они служат для разрушения проникших в организм
чужеродных белков до того, как последние успевают вызвать
повреждения клеток [1].
Литература
1. Рис Э., Стернберг М. От клеток к атомам. Иллюстрированное введение
в молекулярную биологию. — М.: Мир. — 1988.
16
2. Альберте Б., Брей Д., Льюис Ж. и др. Молекулярная биология клетки Т.1—
3. - М.: Мир. - 1994.
3. Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека. Т. 1—2. —
М.: Мир. - 1993.
4. Florence A.T., Attwood D. Physicochemical Principles of Pharmacy, 2 nd ed. —
New York: Chapman and Hall. - 1988.
5. Finean H.B., Coleman R.f Michell R.H. Membranes and their Cellular Function,
3rd ed.- Oxford, Blackwell Scientific Publications. — 1984.
6. Болдырев А.А. Биологические мембраны и транспорт ионов. — М.: Изд-во
Моск. ун-та. — 1985.
7. Foye W. О., Lemke T.L.f Williams D.A. Principles of Medicinal Chemistry, 4th
ed. — Philadelphia, Lippinkott Williams & Wilkins. — 1995.
8. Кантор Ч., Шиммел П. Биофизическая химия. Т. 1,2. — М.: Мир. —1984.
2
СВЯЗЬ ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ
БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
С ИХ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ
Одна из основных трудностей, с которой сталкиваются
специалисты в области конструирования лекарств, состоит в
понимании того, как физические, химические, молекулярные и атомные
свойства соединений способны влиять на характер их
взаимодействия с биологическими мишенями в организме. Далеко не всегда
фармакологическое действие веществ однозначно и прямо связано
с их химическими и физическими свойствами. В общем виде
биологическая активность (А) есть некая функция (/) структуры и/или
физико-химических параметров биологически активного вещества
(БАВ): A=f.
Схему поведения БАВ в организме можно представить в виде трех
составляющих (рис. 2.1): первая (1) — проникновение вещества к
месту действия (обычно путем пассивной диффузии, реже — с
помощью активного транспорта); вторая (2) — образование комплекса
лиганд (БАВ) — биологическая мишень (рецептор, фермент, ген);
третья (3) — индукция ответа на результат связывания.
Доза проникновение К Комплекс
С /\^/\^ | ^ | Л — Р >—> Ответ
(1) (2) (3)
Рис. 2.1. Схема поведения лекарственного вещества в организме:
С — концентрация (доза) БАВ, необходимая для достижения
определенного уровня биологического эффекта (ответа),
Л-Р — концентрация комплексов лиганд-рецептор,
К — константа устойчивости комплекса Л-Р
Вероятность (W) индукции ответа есть произведение
вероятностей этих трех составляющих и выражается формулой:
W ~ W1W2W3 >
А = kCW = kC-wiWiWi >
где С— концентрация (доза), к— коэффициент пропорциональности.
Рассматриваемое уравнение можно представить в
логарифмической форме. При этом активность принято выражать, как 1/С.
lg(l/C) = lgWl + \gWl + lg>v3 + ki
18
Для стадии образования комплекса Л-Р уравнение приобретает вид
где часть k^gwx описывает «транспортные» характеристики БАВ, а часть
k2\gK— устойчивость комплекса лиганд-рецептор [1].
2.1» Растворимость и липофильность
Известно, что взаимодействие между БАВ и клеточными и
субклеточными структурами, содержащими биологические мишени,
происходит в водной среде или в неводных слоях мембран, образованных
полярными или неполярными гидрофобными молекулами. В этой
связи чрезвычайно важными являются степень растворимости
исследуемого соединения и его липофильные свойства.
Теоретически нет абсолютно нерастворимых веществ: каждое
соединение способно в той или иной мере растворяться в водном или
липидном слоях. Растворимость БАВ определяется множеством
факторов: молекулярной структурой, способностью к ионизации,
размером молекул, стереохимическими особенностями, типом
кристаллической решетки и т. п. Классический пример корреляции между
фармакологическими свойствами и растворимостью демонстрирует
зависимость бактерицидной активности от растворимости
алифатических спиртов (рис. 2.2). В гомологическом ряду, начиная с
н-бутанола и заканчивая н-октанолом, бактерицидная активность в
отношении грамотрицательной Salmonella typhi возрастает с
увеличением молекулярной массы (Мм). Среди представленных
гомологов н-октанол оказывается наиболее эффективным, несмотря на
самую низкую растворимость в воде. В отношении более устойчивых
бактерий Staphylococcus aureus активны только н-бутанол и н-пента-
нол, т. к. высшие члены ряда из-за низкой растворимости не
достигают необходимой концентрации [2].
ig Стоке, Ю^М!
насыщение
S. aureus
В. typhosus
lg растворимости, 10
Рис. 2.2. Зависимость бактерицидной активности:
первичных алифатических спиртов от их водорастворимости
н-бутанол, 2 — н-пентанол, 3 — и-гексанол, 4 — н-гептанол, 5 — н-октанол
19
Важной характеристикой нерастворимых в воде веществ
является липофильность — один из факторов, определяющих
распределение в организме молекул БАВ. Показано, что возрастание липо-
фильности коррелирует с повышением биологической активности,
снижением водорастворимое™, ускорением метаболизма и
выведением, повышением скорости проникновения через кожу,
увеличением степени связывания с белками плазмы, ускорением
наступления пика активности и, в некоторых случаях, укорочением
длительности действия [2—5].
Соотношение растворимости вещества в органическом и
водном слоях носит название парциального коэффициента Р. Этот
коэффициент характеризует сродство молекулы к липидной фазе по
сравнению с водной и служит количественной характеристикой ли-
пофильности:
Гтчлтт/ч1 р п Г01Ш01октанол
Г DRUG] вода r^~^ fDRUG] октанол, Р = -Ц =Ц
L J L J [DRUG] вода
Бинарная система «-октанол — вода является удобной моделью
для оценки липофильности многих соединений. Так, С. Hansch
избрал «-октанол в качестве стандартного растворителя для
измерения IgP благодаря его формальному сходству с липидами:
длинная алкильная цепь и функциональная группа, проявляющая как
протонодонорные, так и акцепторные свойства. Кроме н-октанола,
для этих же целей используют н-бутанол (для Я-доноров),
хлороформ, гексан (для нейтральных веществ) и дипеларгонат пропи-
ленгликоля (для //-акцепторов). Удобным экспериментальным
подходом для определения величины IgP является метод
встряхивания. В качестве водной фазы используют обычно фосфатный
буфер с рН = 7.4. Кроме того, существует несколько
инструментальных методов, основанных на использовании
высокоэффективной жидкостной (ВЭЖХ) и обратимой тонкослойной
хроматографии (ТСХ) [1, 2, 5].
Если растворенное вещество склонно к диссоциации, то его
концентрация в неионизированной форме в октанольной фазе зависит от
рН в водной. Коэффициент распределения (IgD) при этом
рассматривают как эффективную липофильность соединения при заданном
значении рН, зависящую как от его липофильности в отсутствие
ионизации, так и от степени диссоциации. Указанное иллюстрируют
приведенные ниже уравнения.
Для кислот:
ГНА]
НА< >Н++А~ D = - =^L
^WKL
20
Для оснований:
Н*В.( >Н*+В D =
Для слабых кислот:
[в]
орг
[H+BL2o+Wh2o
lg(P/D-l) = pH-pKa
Липидный слой А| [недиссоциир. форма] не проникает
4
*
н2о
[недиссоциир. форма]
рК,
[диссоциир. форма]
Согласно рН-парциальной гипотезе только неионизованная
форма соединения способна проникать в неводную фазу. Если
допустить, что соединение может проявлять максимальную липофильность
ниже рА^ кислоты и выше рКа основания, то коэффициент Р должен
быть рН-зависимым (рис. 2.3).
\QD m
50% недис.
0%дис.
Р =
[НА]
орг
р =
[НА]
орг
lgP = 3, pK =2
а
Рис. 2.3. Связь между lg D и рН
Следует отметить, что связь между lg/) и рН будет иметь более
сложный вид, когда молекула содержит кислотные и основные
центры и возможно образование биполярных ионов. Любое амфотерное
вещество будет проявлять максимальную липофильность тогда, когда
полностью нейтрально (рис. 2.4). Ниже рКл1 и выше рКй2 преобладают
моноионизованные частицы.
Для экспериментального определения значения IgP необходимо
располагать веществом. Однако в большинстве случаев желательно
оценить этот показатель еще до того, как соединение будет синтезировано.
21
С целью предсказания значения lg-Рдля ряда замещенных бензола
С. Hansch предложил уравнение, аналогичное уравнению L.P. Hammett,
учитывающее электронные эффекты у замещенных бензойных кислот:
л.
= ig/> -ig/>
RH '
гдеях — параметр, характеризующий гидрофобные свойства
заместителя Xf PBYn Р.
^ж.. дд - парциальные коэффициенты пары исходного и
замещенного веществ соответственно.
Липидный слой
Н20
#
[ав+;
гав+;
Ч^
соо
рКа1 = 2.3
Рис. 2.4. Зависимость между lg D и рН для амфотерных веществ
Таким образом, пх характеризует изменения величины IgP, когда
атом Н заменен на X и пропорционален свободной энергии перехода
соединения из водной фазы в органическую (н-октанол) [5, 6].
Значения я приведены в табл. 2.1.
Значения параметра я
для наиболее распространенных заместителей
Таблица 2.1
Заместитель
ОН
F
ОСН3
СНз
С1
Вг
CF3
/-Bu
алифатич.
-1.16
-0.17
0.47
0.50
0.39
0.60
1.07
1.68
я
аромат ич.
-0.67
0.14
-0.02
0.52
0.71
0.86
1.16
1.68
Примечание. Знак «-» свидетельствует, что заместитель Xболее
гидрофилен, чем Н.
22
Используя значения п можно оценить изменения \gPxipw
введении в молекулу бензола группы CONH2 и атома CI:
lgP(C6H6) = 2.13
lgP(C6H5-CONH2) = 2.13 + (-1.49) = 0.64
lgP(jw-Cl-C6#4-CONH2) = 2.13 + (-1.49) + 0.71 = 1.35
Экспериментально полученное значение IgP для .ме/иа-хлорбенз-
амида составляет 1.51 [6]. Различия в величинах IgP, рассчитанной и
полученной экспериментально, существенны. Таким образом,
первоначальный подход С. Hansch потерпел неудачу, т. к. электронные,
стерические эффекты и водородные связи влияют на вклад
заместителя. Позднее С. Hansch, T. Fujita и др. использовали регрессионный
анализ для оценки экспериментальных данных \gPw предложили
новый параметр липофильности:
lg/, = S/
В этом уравнении уже учтены эффекты окружения и дан более
общий метод оценки значения IgP:
lgP(x,y9z) = fx+fy +fz +/(окруж.); fx =nx +/(Н-св.),
где х, у, z — заместители.
В конце 70-х гг С. Hansch и A. Leo создали алгоритм,
позволяющий подсчитать липофильность любой молекулы, используя метод
аддитивности. Они составили таблицы для ароматических и
алифатических заместителей, основываясь на большом фактическом
материале (табл. 2.2) [6]. Однако существует немало случаев, когда правило
аддитивности не соблюдается (взаимодействие между группами в
аминокислотах, передача электронных эффектов и т. п.). Для устранения
несогласованности для новых групп проводится параметризация или
вводятся поправки.
Таблица 2.2
Значения параметра пх, рассчитанные С. Hansch и A. Leo
<— Рост полярности и гидрофильности
CONH2 ОН NH2 N02 СООН Н С1 СН3 С6Н5
-2.11 -1.64 -1.54 -1.16 -1.11 0.23 0.06 0.88 1.92
-1.26 -0.44 -1.00 0.03 0.03 0.23 0.94 0.88 1.92
тс
алафатич.
аром.
Снижение полярности и гидрофильности
Таким образом, изменить липофильность соединения можно
введением функциональных групп, способных к диссоциации. Если же в
составе вещества уже имеются таковые, то желаемого результата можно
достичь, модифицируя рКл или включая в структуру молекулы
фрагменты с подходящим значением пх •
Все изложенное выше представляет физико-химические основы
понятия липофильности. Практическая же ценность величины пар-
23
циального коэффициента в том, что он помогает оценить
«транспортные» характеристики вещества, а, следовательно, и пути, по
которым БАВ достигает места действия. Так, если вещество
ионизировано, то оно не сможет преодолеть липидный слой и создать
значительную концентрацию в богатых липидами тканях и органах (мозг,
нервные клетки). В то же время, высоко липофильные вещества
будут задерживаться в липидных слоях клеточных мембран,
образовывать «депо» в жировой ткани, и также не смогут быстро достигать
цели. Величина IgP между —1 и +5 является оптимальной для
веществ, предназначенных для перорального применения: если
величина меньше —1, то вещество будет плохо всасываться из кишечника;
если же IgP >5, то соединение обладает значительной липофильно-
стью и способно надолго задерживаться в липидных слоях, что также
затрудняет абсорбцию [3—5]. Так, сукцинилсульфотиазол (2.1),
фталазол (2.2) и фтазин (2.3) в тонком кишечнике при рН ~ 8 будут
присутствовать в ионизованной форме в значительных количествах,
что практически исключает их проникновение в системное
кровообращение. Это и позволяет применять названные препараты в
качестве кишечных дезинфектантов.
\ /r~S02NH
(2.1)
N
7
ОН
S02NH
N.
7
(2.2)
N
Н
ОН
л >^S02NH
N-N
ОМе
(2.3)
Как отмечено выше, при количественной оценке активности
веществ величину \%Р принято коррелировать с \g(\/C). Связь между
этими двумя параметрами может носить как линейный:
\g{\lC) = kx\gP + k2
24
так и нелинейный (параболический) характер:
lg(l/C) = -^(lgP)2+^lgP + ^,
что наглядно иллюстрирует рис. 2.5.
|д(1/С) -
Р°
1дР
Рис. 2.5. Характер зависимости \gPoT Щ(\/С):
1) lg(l/C) = A:t lgi> + A:2; 2) lg(l/C) = -*, (\gPf +k2 \%Р + къ
Если для ряда БАВ сходного строения разброс в значениях IgP
невелик, то справедливо первое уравнение. Из него следует, что с
ростом липофильности увеличивается активность соединений,
т. к. возрастает вероятность их проникновения через мембраны и
достижения места действия — биологической мишени. Вместе с тем,
следует учитывать и тот факт, что, попадая в системное
кровообращение, эти вещества способны связываться с белками плазмы.
С ростом IgP эта способность также возрастает. Прочное
связывание уменьшает концентрацию свободного БАВ в крови, поэтому
рецептора достигает гораздо меньшее количество вещества, чем было
абсорбировано.
Значительный разброс в значениях IgP внутри одной и той же
группы структурно близких соединений приводит к параболическому
характеру зависимости их фармакологической активности от
липофильности. При малых значениях Р величина -£, {}%?)
пренебрежимо мала, и активность возрастает с изменением IgP почти линейно,
пока не будет достигнуто значение Р°, после которого дальнейший
рост lgP приведет к снижению эффекта [1, 6].
Необходимо подчеркнуть, что парциальный коэффициент
далеко не единственный фактор, оказывающий влияние на
биологическую активность веществ. Существует лишь небольшая группа
лекарственных средств (ЛС), для которых именно этот показатель
является определяющим. Так, величину парциального
коэффициента рационально использовать при объяснении фармакологических
эффектов так называемых «неспецифических лекарств»,
оказывающих действие на ЦНС — общих анестетиков, некоторых
снотворных, неспецифических дезинфектантов, действующих на
бактериальные мембраны и т. п. [1, 2].
В начале XX в. СЕ. Overton и Н.Н. Meyer предложили гипотезу,
объясняющую анестезию, индуцируемую веществами различной при-
25
роды. Было отмечено [2], что все нейтральные липидо-растворимые
вещества оказывают депремирующее действие на нейроны и эта
активность особенно заметна в клетках, богатых липидами. Эффект
возрастает с ростом парциального коэффициента, независимо от
структуры вещества. Значения Рдля некоторых веществ, обладающих
таким действием, приведены в табл. 2.3.
Таблица 2.3
Парциальные коэффициенты некоторых БАВ (октанол/Н20)
Вещество
Этанол
и-Бута нол
Валерамид
Бензамид
Салициламид
o-N02 анилин
Тимол
Р
0.10
0.65
0.30
2.50
5.90
14.0
950.0
С, моль/л
в водном слое
0.330
0.030
0.070
0.013
0.0033
0.0025
4.7-10"5
С, моль/л
в липидном слое
0.033
0.020
0.021
0.033
0.021
0.035
0.045
Как видно из приведенных данных, абсолютная концентрация
веществ, необходимая для достижения анестезии, различается
существенно. При этом содержание веществ в липидном слое, а,
следовательно, и в мембранах, находится в более узких пределах —
20—50 мМ для всех агентов. Таким образом, соединения разного
строения обладают сходным биологическим эффектом благодаря
сродству к органической фазе. В 1954 г. Mullins модифицировал
гипотезу СЕ. Overton — Н.Н. Meyer и предположил, что кроме
концентрации анестетика в мембране важен его объем,
выраженный как объемная фракция, вычисляемая как произведение
молярной фракции и парциального молярного объема. Это означает, что
анестетики расширяют мембрану и эффект возникает тогда, когда
достигается критическая величина, составляющая примерно 0.3—
0.5% от ее исходного объема. При этом также увеличивается
поверхность мембраны.
J. Ferguson, признавая гипотезу СЕ. Overton — Н.Н. Meyer,
утверждал, что анестезия наступает при определенной величине
термодинамической активности (а) вещества, которая представляет
собой не что иное, как величину относительного насыщения. Для
газов а - pt/ps , где р{ — это парциальное давление газа в воздухе,
a ps — давление газа в веществе. Для веществ, растворимых в
жидкостях, a = St/S0, где St — молярная концентрация растворенного
вещества, которая необходима для достижения эффекта, а£0 —
молярная растворимость вещества. Величина термодинамической
активности полезна для характеристики структурно специфических
и неспецифических агентов. Для последних величина а находится в
пределах 0.01—1.0, т.е. они активны в относительно высоких дозах,
26
и их биологическое действие не связано со строением, как это видно
из примера для анестетиков [1—5].
Большинство же фармакотерапевтических средств являются
структурно специфическими. Для них величина
термодинамической активности не превышает 0.001, и они действуют при очень
малых концентрациях. Эффект специфических лекарств
опосредован через селективные рецепторы, представляющие собой
макромолекулы липопротеинов или гликопротеинов. Плотность
рецепторов в мембране колеблется от 10 до 1000 на 1 мкм2. В то же
время, если активная концентрация неспецифического лекарства
составляет 2 мМ, это означает, что миллионы молекул такого
вещества помещаются на 1 мкм2 мембраны, и в этом случае
целесообразнее говорить не о рецепторе для такого агента, а о месте
его связывания.
Наиболее представительная группа неспецифических
лекарств - общие анестетики (рис 2.6). Они значительно отличаются
по структуре. К ним относятся как инертные газы, так и
некоторые стероиды.
Хе
ксенон (2.4)
N20
оксид азота(1) (2.5)
Л
циклопропан (2.6)
СНС1.
С2Н5С1
хлороформ (2.7) хлористый этил (2.8)
F3C-CHClBr
галотан (2.9)
CH30-CF2
СНС12 СНС1=СС13 С2Н5-0-С2Н5
метоксифлуран (2.10) трихлорэтилен (2.11) диэтиловый эфир (2.12)
S=<
н//° н /Я
N^V-C3H7
/\/-=С-С2Н5 0=<
Г\ I
сн.
СИ
о
н //
N-^C2H
тиопентал (2.13) метогекситал (2.14)
Барбитал
Ъг
с2н5
метилэтилглутаримид (2.16)
НО
СО-СН,
А1 -альфаксалон
27
со-сн2о-со
COONa
СО-СН,
предион (2.17)
альфаксалон (2.18)
со-сн2-он
CI
альфадалон (2.19)
с2н5
c2H5^Ny^0^Y"
О О-СН
сомбревин (2.20)
п-С3Н7
^v
NH-CH3 CI
°<^к
кетамин (2.21) мидазолам (2.22) этомидат( 2.23)
Et /=
itefo
N
Н
этоксадрол (2.24)
Рис. 2.6. Общие анестетики и их антагонисты
Термодинамическая активность Хе несколько ниже 0.01. Несмотря на то,
что это очень эффективный анестетик, он не нашел применения из-за
высокой стоимости.
Оксид азота (2.5) или «веселящий газ» — один из старейших
анестезирующих агентов. Английский химик Деви (1779 г.) обнаружил, что
вдыхание N20 может привести к потере сознания. Уэллс (1844 г.) в США
применил оксид азота для обезболивания при удалении зубов. В связи с тем, что
липофильность N20 ниже, чем у Хе, для достижения с его помощью нар-
28
коза необходимо, чтобы концентрация газа в дыхательной смеси была не
менее 70%. С липофильностью связана и скорость элиминирования
анестетика из организма. Оксид азота, благодаря высокому значению его
парциального давления в крови, элиминирует чрезвычайно быстро. Он
не создает глубокой анестезии, поэтому используется исключительно для
коротких операций. Преимуществом N20 является то, что он не
связывается в тканях и не образует метаболитов.
Углеводороды также обладают наркотическим эффектом, выраженность
которого возрастает в ряду метан — гептан, а затем снижается из-за
низкой растворимости в воде. Наркотическое действие возрастает с
увеличением кратности связей (этан<этилен<ацетилен). Замена атомов водорода
на полярные СООН- и ОН-группы снижает анестезирующий эффект, а
введение липофильных заместителей (CI, F) повышает активность.
Циклопропан (2.6) как мощный ингаляционный анестезирующий агент
используется с 1933 г. Однако он оказывает негативное действие на
сердце и взрывоопасен.
Галогенуглеводороды — хлороформ (2.7) (использовался с 1847 г.) и
хлористый этил (2.8) (использовался с 1894 г.) дольше всего применялись в
медицинской практике. В настоящее время они вытеснены менее
токсичными анестетиками — галотаном (2.9), метоксифлураном (2.10) и три-
хлорэтиленом (2.11).
В ряду галогенпроизводных наркотический эффект возрастает с
увеличением атомного объема: F<CKBr<I. Однако повышение реакционной
способности в этом же ряду делает невозможным медицинское применение
иодпроизводных. Известно также, что увеличение количества атомов хлора
повышает токсичность веществ. Хлороформ, например, оказывает
негативное действие на почки и печень. Комбинация же нескольких атомов
разных галогенов способствует росту анестезирующей активности.
Широкое распространение в современной анестезиологии получил гало-
тан (2.9), вошедший в медицинскую практику в 1956 г. Он обладает
мощным эффектом (в 1.5—2 раза превышает активность хлороформа и в 4
раза — диэтилового эфира), который наступает быстро (2—10 мин) без фазы
возбуждения и не сопровождается серьезными побочными эффектами.
Однако галотан нельзя использовать при заболеваниях печени,
гипотонии и аритмии, главным образом, из-за образования высокотоксичных
метаболитов (см. разд. 8.1, рис. 8.7).
Алифатические простые эфиры, подобно низшим алканам, оказывают
депремируюшее действие на ЦНС. Здесь справедливы те же корреляции
«структура — действие», что и в ряду углеводородов. Введение атомов
галогенов в состав молекул эфиров способствует сохранению активности
и снижает взрывоопасность.
Диэтиловый эфир (2.12) обеспечивает медленную индукцию наркоза,
медленный выход из него и глубокую анестезию. Обладает раздражающим
действием на слизистую дыхательных путей, взрывоопасен. В настоящее
время в хирургической практике не используется.
Среди анестетиков для внутривенного введения следует отметить
гетероциклические производные — барбитураты: тиОпентал (2.13) и метогекси-
тал (2.14). Первое соединение этого ряда, нашедшее применение в
медицине — барбитал (2.15), было получено Фишером и Мерингом в 1903 г.
29
Исследования связи «структура — действие» показали, что смещение
гипнотических свойств (а именно они определяют фармакологический
профиль этих веществ) в направлении наркотической активности
достигается заменой кислорода на серу во 2-ом положении пиримидинового кольца
и N-метилированием. Примерами таких структур как раз и являются
вышеупомянутые тиопентал и метогекситал. Тиопентал относится к
ультракоротким анестетикам, т. к. наступление эффекта и выключение
сознания наступают через несколько секунд после введения вещества. Выход
из этого состояния замедлен, поскольку тиопентал накапливается в
жировой ткани, тогда как ингаляционные анестетики выводятся из
организма через легкие. Липофильность метогекситала выше, чем у тиопен-
тала, т. к. в структуре имеется N—СН3 группа и непредельные связи в
боковой цепи. Наличие ненасыщенных фрагментов способствует
быстрому окислению и выведению метаболитов, поэтому метогекситал можно
использовать в широком диапазоне доз, что особенно важно в
хирургической практике. Способность барбитуратов к накоплению в жировой
ткани повышает риск передозировки. С фатальными последствиями
превышения доз этих препаратов столкнулись американские врачи во время
событий на военной базе Перл-Харбор в годы Второй мировой войны:
число летальных исходов вследствие применения барбитуратов для
общей анестезии превысило количество жертв от ранений.
Как правило, инъекционные анестетики комбинируют с ингаляционными,
что обусловлено снижением доз обоих компонентов. Существенным
недостатком внутривенных анестетиков является сложность контроля за
глубиной и продолжительностью наркоза. Следует отметить, что эффект
длительного действия барбитуратов опосредован через рецепторы у-аминомасляной
кислоты (ГАМК), а не только путем влияния на мембраны нейронов [9].
Неспецифическим антагонистом барбитуратов является производное глу-
таримида (2.16), обладающее аналептическим (стимулирующим ЦНС)
эффектом, которое и применяют при отравлениях этими веществами.
Селей в 1941 г. изучая фармакологические свойства стероидов, открыл
важную особенность — наряду с гормональным действием некоторые из
них проявили наркотический эффект. Первым препаратом, получившим
клиническое применение, был предион (2.17) — производное
прогестерона, лишенное гормонального действия. В настоящее время
используются альфаксалон (2.18) и альфадалон (2.19). Они активнее предиона в
13 раз. В рассматриваемых веществах За-ОН группа аксиальна, а Зр-эпи-
мер с экваториальной ОН группой неактивен. А16-Альфаксалон с
экваториальной боковой цепью у С—17 выступает как антагонист альфаксало-
на. К положительным качествам стероидных анестетиков следует отнести
отсутствие стадии возбуждения, достаточную мышечную релаксацию,
относительно быстрое пробуждение и низкую токсичность [2,8].
Кроме выше перечисленных, в медицинской практике в качестве общих
анестетиков применяются производные эвгенола (сомбревин, 2.20), цикло-
генксиламина (кетамин, 2.21), а также соединения из разных групп: мидазо-
лам (2.22), этомидат (2.23), этоксадрол (2.23). Наряду с наркотическим
действием они обладают гипнотическим или аналъгезирующим эффектами [8].
В чем же состоит механизм общей анестезии? Попытки объяснить связь
между разнообразными структурами анестетиков и их наркотическими
30
свойствами существуют со времени Овертона и Мейера, Фергюсона и Мю-
линса. Последний утверждал, что молекулы анестезирующих средств
сосредотачиваются в «свободном объеме» мембран нейронов [2]. Дальнейшее
развитие этой идеи нашло подтверждение в биофизических экспериментах
методами ядерного магнитного резонанса (ЯМР) и электронного
парамагнитного резонанса (ЭПР). Одна из гипотез состоит в том, что анестетики
расширяют и дестабилизируют мембраны, в результате чего возникает
флуидизация промежуточного бинарного слоя и превращение его из геля
в форму, подобную жидкокристаллической. Однако рентгеновские исследова-
ния и метод дифракции нейтронов этого не подтверждают. Предполагается \
также, что протеины, взаимодействуя с анестетиками, претерпевают '
конформационные изменения, что, в свою очередь, приводит к изменени- /
ям в мембранах нервных клеток и нарушает их функции: подавляется
передача сигналов через синапсы, т. к. нарушается выход нейротрансмиттеров *
через пресинаптические окончания нейронов [1, 2, 8].
2.2. Поверхностно-активные,
мембран-активные фармакологические агенты
и ион-проводящие антибиотики
Множество биохимических реакций протекает не только в
растворах, но и на поверхностях мембран, на границах раздела фаз.
Присутствующие в организме макромолекулы также обладают
развитой поверхностью. Так, 1 мл плазмы содержит белки,
поверхность которых составляет 100 м2. Мембраны играют важнейшую роль
в функционировании клеток, в том числе бактериальных и
грибковых. Поэтому любой агент, способный разрушить мембрану или
вмешаться в ее функции, является потенциальным бактерицидным
средством. Рассмотренное нами ранее бактерицидное действие
спиртов основано на повреждении мембран и приводит к быстрой
потере содержимого цитоплазмы бактериальной клетки. Фенол и
крезол обладают бактерицидным эффектом не только потому, что
денатурируют белки, но и действуют как поверхностно-активные
вещества (ПАВ), благодаря полярности и наличию гидроксильных
групп. Их активность повышается с ростом боковых алкильных
цепей, как это наблюдается в случае л-гексилрезорцинола (2.25).
Катионные ПАВ типа цетил-триметиламмоний хлорида (2.26) более
эффективны, чем анионные мыла. Неионные ПАВ, например,
тритон Х-100 — (полиэтиленгликоль)10-я-изооктилфениловый эфир —
обладают мягким эффектом, способствующим диспергированию
мембран. Он используется в качестве хирургического дезинфицирующего
средства. Хлоргексидин (2.27) — производное хлорфенилбигуаниди-
на — в этом смысле очень эффективен: в концентрациях 10—100 мкг/мл
он вызывает разрушение мембран и выход содержимого
цитоплазмы из бактериальных клеток.
31
CH3-(CH2)15-N+(CH3)3C1- CH3-(CH2)„-S03-Na+
(CH2)5-CH3
и-гексилрезорцинол катионное ПАВ
анионное ПАВ
(2.26)
CI
^JJ NH NH NH NH К^
CI
(2.27)
К числу мембраноактивных относятся и некоторые
противогрибковые средства. Отдельные виды Streptomyces продуцируют макроцик-
лические соединения, содержащие в своей структуре несколько
двойных связей. Примером может служить антибиотик амфотерицин В (2.28)
и близкий ему по структуре нистатин (2.29). Взаимодействуя с
молекулами стероидов, входящими в состав мембран микроорганизмов,
эти антибиотики объединяются по 5—10 молекул и образуют поры,
подобные ионным каналам, через которые ионы К+, сахара и белки
покидают клетку, тем самым приводя ее к гибели.
НООС
(2.28)
НООС
Азольные фунгицидные агенты кетоконазол (2.30), клотрима-
зол (2.31) и миконазол (2.32) также способствуют дестабилизации
мембран, но уже путем ингибирования синтеза эргостерона.
CI
^Ч
(2.31)
(2.32)
Еще одна группа веществ, интересная своими поверхностно-
активными свойствами, носит название ион-проводящих
антибиотиков или ионофоров. Некоторые бактерии продуцируют вещества,
которые, включаясь в липидный слой мембран, ускоряют
транспорт ионов, в частности, К+. Примером может служить
антибиотик валиномицин (2.33). Этот циклический пептидный лактон
состоит из трех молекул /-валина, D-cc-гидроксивалериановой
кислоты и /-лактата. Высоко поляризованные СО-группы
располагаются внутри кольца, в то время как алкилы оказываются вне его.
Таким образом, полярное внутреннее кольцо окружает ионы К+,
а снаружи возникает гидрофобный слой. Селективность к ионам
К+ по сравнению с ионами Na+ составляет 104:1, и ускорение
проводимости К+ через мембраны наблюдается при
концентрациях ниже 10~9 М.
Еще одним примером каналообразующего антибиотика является
грамицидин А — пептид, состоящий из 15 аминокислотных
остатков. Он индуцирует перенос через мембрану протонов, ионов
щелочных металлов, ионов таллия при концентрациях менее Ю-10 М.
Эти вещества не получили распространения в медицинской
практике, т.к. действуют на клетки млекопитающих так же, как и на
бактериальные, но они служат удобными моделями для изучения
явления транспорта через мембраны.
Синтетическими ионофорами являются краун-эфиры (2.34, 2.35)
и криптанты (2.36). Некоторые из них обладают интересными
фармакологическими свойствами, т.к. способны влиять на транспорт
33
L-Lac
D-Hyi\ о-*
(2.33)
С J
^—о о—1
YU
(2.34)
(2.35)
ГЛ
о
N
О О
v_y
(2.36)
ионов Са2+, что, в свою очередь, сказывается на коронарном
кровотоке диурезе, частоте сердечных сокращений (ЧСС). Многие из этих
веществ способны связывать радионуклиды. Однако их медицинское
применение ограничивает высокая токсичность.
2.3. Химическое связывание и биологическая активность
Связывание малых молекул лекарств с биологическими
субстратами может быть обратимым и необратимым. Обратимое
связывание подразделяют на несколько типов в зависимости от вида
связей обеспечивающих взаимодействие. В табл. 2.4 приведены типы
межмолекулярного связывания и соответствующие им значения
энергии. Л „
Дисперсионное или Ван-дер-Ваальсово взаимодействие существует
между всеми видами атомов, в том числе и инертных газов, что свя-
34
зано с возникновением мгновенных диполей, образуемых
электронной оболочкой и положительно заряженным ядром при сближении
атомов. Ван-дер-Ваальсовы силы резко ослабевают с увеличением
расстояния R между частицами и пропорциональны 1/R6. Несмотря на
то, что энергия дисперсионного связывания мала, при наличии
большого числа близко расположенных групп атомов она может
составить значительную величину. Так, сила притяжения между —СН2-
группами в липидных слоях мембран составляет 33 кДж/моль, что
приводит к упорядочению в расположении молекул. Введение в
такую углеводородную цепь двух двойных связей, имеющих
^-расположение, приводит к снижению энергии взаимодействия до 10—
12 кДж/моль. Наличие полярных липофильных заместителей,
напротив, повышает дисперсионное взаимодействие, что сказывается
на фармакологической активности. Как отмечалось ранее, галотан
(2.9) и метоксифлуран (2.10) более эффективные анестетики, чем
неполярные ксенон и циклопропан, т. к. они прочнее связываются с
липидами в нейронах.
Гидрофобный эффект — это тенденция неполярных групп к
взаимодействию друг с другом, препятствующая контакту последних с водой.
Таблица 2.4
Тип связывания и энергия взаимодействия
Тип связывания
Дисперсионное
Гидрофобное
Водородные связи
Перенос заряда
Диполь-дипольное
Ион-дипольное
Ионное
Ковалентное *
Пример
Хе...Хе
СбН6... CfiHg,
R-Alk...R-Alk
>С=О...Н-0-Н,
R—ОН ... О—R
1
Н
NC
NC
1 -*- н п
^ По*-'
CN
06"=C5+<-NR3
К+ Н20
NH4+ OOC
-С-С- , >С=С< , -S—S-
-ДЕ, кДж/моль
1.9
4.2
7-40
17
~5
171
685
346, 614, 250
* обратимое и необратимое.
Он играет ключевую роль в стабилизации конформации протеинов,
в транспорте липидов (посредством их переноса белками плазмы), в
проникновении веществ через мембраны клеток и во взаимодействии
молекул стероидов с рецепторами. Концепцию этого вида межмо-
35
лекулярного связывания впервые выдвинул Kauzman применительно
к химии белков. Он также объяснил низкую растворимость
углеводородов в воде неспособностью образовывать водородные связи.
Молекулы воды, располагаясь между углеводородными цепями,
претерпевают упорядочение, подобное тому, какое имеет место в
кристаллах льда, что термодинамически невыгодно из-за потери энтропии.
Физико-химические аспекты этого типа взаимодействия продолжают
обсуждаться [1, 2].
Широкое распространение в биологических системах
получило взаимодействие посредством водородных связей. Они
способствуют стабилизации структур за счет внутримолекулярного
взаимодействия (вторичная структура белков — сс-спираль и ДНК) и
обеспечивают .межмолекулярное связывание. Водородная связь
образуется за счет электростатического взаимодействия свободной
электронной пары атомов О, S или N с атомом водорода,
находящемся в составе ОН, NH и SH групп, и имеет линейную
направленность. Расстояние между атомами, образующими Н-связь,
составляет 2.5—2.7 А, а энергия связывания в апротонном
растворителе — 7—40 кДж/моль.
В белковых макромолекулах водородные связи возникают,
главным образом, между карбоксильными, гидроксильными,
карбонильными, амино, имино, амидными и тиольными группами. Самыми
сильными протоноакцепторами в таких системах являются карбокси-
лат-ионы, а донорами — ионы аммония.
Под переносом заряда понимают тип взаимодействия, который
осуществляется между электроноизбыточными соединениями
(донорами Р) и электронодефицитными (акцепторами А):
Р + А-к *РА?=±Р**А*~-К *Р+А'^ ^Р+ +А~
Среди органических соединений электроноизбыточность
присуща фурану, пирролу, тиофену, производным бензола с электронодо-
норными заместителями (—ОСН3, —NH2, —ОН), веществам с непо-
деленными электронными парами (амины, эфиры, карбонильные
соединения).
К числу электронодефицитных соединений относятся пурины,
пиримидины, производные бензола с акцепторными заместителями
(—N02, —S03H, —CN), тетрацианоэтилен и др.
Примером биологического эффекта, возникающего за счет
переноса заряда, является встраивание некоторых антибиотиков в
структуру ДНК (см. разд. 6.3) или связывание серотонина с АТФ. Энергия
такого взаимодействия определяется потенциалом ионизации донора
и энергией сродства к электрону акцептора и обычно не превышает
30 кДж/моль.
Если в молекуле существует частичное разделение зарядов, то
за счет этого она способна взаимодействовать с себе подобной (ди-
поль-дипольное взаимодействие) или с ионами (ион-дипольное
связывание). Энергия диполь-дипольного взаимодействия определяется
уравнением:
36
2^ cosB^
£ = ,
Ргг
где \ix и \i2 — дипольные моменты, 6 — углы между зарядами, Р —
диэлектрическая постоянная среды, г — расстояние между зарядами
в диполе.
Для ион-дипольного связывания уравнение имеет вид:
e\i cos 8
Е =
D(ssy
где е — заряд иона, d — длина диполя, г — расстояние между
зарядами иона и диполя.
Примером такого взаимодействия может служить образование гид-
ратной оболочки ионов.
Ионное взаимодействие возникает между противоположно
заряженными частицами. Его энергия описывается законом Кулона;
Dr2
где е} и е2— величины зарядов, Р — диэлектрическая постоянная
среды, г— расстояние между зарядами.
Значение энергии в этом случае обычно велико. Этот тип
связывания играет существенную роль во взаимодействии с мишенями БАВ,
содержащих способные к ионизации функциональные группы.
Ковалентные связи являются основными в органической химии,
однако они не вносят значительного вклада во взаимодействие
лекарственных веществ (ЛВ) в организме. Биологический эффект,
возникающий за счет ковалентного связывания, отмечен у ряда элемен-
тоорганических соединений, имеющих в составе атомы As, Sb, Bi.
Они необратимо связываются с атомами S, включенными в структуру
тиольных групп ферментов, которые присутствуют в клетках
микроорганизмов — возбудителей ряда заболеваний. За счет образования
ковалентных связей реализуется и цитостатический эффект
некоторых химиотерапевтических средств алкилирующего типа (рис. 2.7),
например метхлорэтамина (2.37).
о
CI-(CH2)2-N-(CH2)24(p j?
пи пи т Н2С—СН2—CI рп iSr NH
СН2-СНГС1 2/ 2 СН2-СН2—CI ?°4- <( \\ I
R-N _^R-jJ* i^R-f/ + dGDP CH2q N^n^NH;
г сн2—сн2 \LI/
ch2-ch2-ci
(2.37)
о=р-о-
Q
Рис. 2.7. Схема взаимодействия метхлорэтамина
с дезоксигуанилатдифосфатом (dGDP)
37
Антибиотики пенициллинового и цефалоспоринового ряда также
ковалентно связываются с транспептидазами и тем самым подавляют
синтез стеночных пептидов, входящих в структуру оболочки
бактериальной клетки (см. разд. 5.4).
2.4. Связь между фармакологической активностью,
электронными свойствами и константами ионизации
Существенную роль в способности соединений проникать к
биологической мишени и связываться с ней играют их кислотность и
основность. Неионизированные формы могут осуществлять
взаимодействие за счет дисперсионных сил, водородных связей и т.п., в то
время как ионизированные участвуют в передаче эффектов за счет
образования солевых мостов или ион-дипольного взаимодействия.
В составе молекул многих БАВ есть кислотные или основные
центры. Как правило, это слабые кислоты или основания, равновесие
между ионизированной и неионизированной формами которых
характеризуется константой Кл. Экспериментально Кй определяют,
используя методы потенциометрического титрования, кондуктомет-
рию, рН-метрию, ИК и ЯМР спектроскопию.
НОАс
MeNH;
Н +ОАс
К =
"н+~
ОАс
±MeNH+H+ К,
[НОАс]
[H+]-[MeNH2]
[MeNH;]
pH = -lg[H+]
рКа = рН + lg[HOAc] - lg[0Ac~ 1
трКа = рН + lg[MeNHjl- lg[MeNH2]
Впервые реальную попытку связать степень ионизации с
биологической активностью предпринял в середине XX в. A. Albert. Он
коррелировал выраженность биологического эффекта с константами
Гаммета для мета- и иора-замещенных бензойных кислот.
//
соон
//
соо + н
1 К*
lg1T
38
Если заместитель X — акцептор (Кх » Кн), то константа о
имеет положительный знак, если X — донор (Кх « Кн), то значение а
отрицательное.
Когда в молекуле несколько заместителей, связь между рК
и а-константой имеет следующий вид:
рКа = 4.20 + р£о
Однако для оценки активности орто-замещенных бензойных
кислот эти уравнения не подходят, т.к. существенное влияние на
константу кислотности наряду с электронными оказывают стерические
эффекты.
Для оценки активности веществ, не содержащих ароматических
заместителей, было предложено использовать уравнение Р. Тафта,
связывающее электронные эффекты с константами кислотного и
основного гидролиза алифатических эфиров:
X СН2-^- ^ X—СН2-^ + МеОН
О-Ме \он
О*
0,403[lg(^0L-lg(^0),
Электронодонорные группы снижают скорость гидролиза,
следовательно, о*— константа приобретает отрицательные значения.
Но электронные эффекты не являются единственным фактором,
сказывающимся на скорости рассматриваемого процесса. Не меньшее
значение имеет пространственное строение. Объемный заместитель
может препятствовать атаке на эфирную группу и тем самым снижать
скорость гидролиза. Для разделения электронных и стерических
эффектов и было предложено оценивать скорость процесса в кислой и
основной средах. В последнем случае имеют значение как стерические,
так и электронные факторы. При кислотном гидролизе только
стерические эффекты существенно сказываются на скорости процесса.
Таким образом, сравнив скорости реакции в различных условиях,
можно оценить электронные о и стерические Es факторы. В
результате связь фармакологической активности с липофильностью,
электронными эффектами заместителей и стерическими факторами,
зависящими от объема молекулы, можно выразить в виде следующего
уравнения:
lg (1/С) = k{ lg Р - к2 (lg Р)2 + k3a + *4 Я + к5
2.5. Стереохимические аспекты действия лекарств
Установить количественную связь между фармакологиче
ской активностью и пространственным строением молекул
гораздо сложнее, чем провести такую же оценку в отношении липо
39
фильности или электронных характеристик этих веществ. Р. Тафт
предложил использовать с этой целью упомянутый выше стериче-
ский фактор Е%. Другой способ оценки стерических эффектов
состоит в корреляции между активностью и молярной
рефракцией (MR), как мерой объема, который занимают атом или группа
атомов:
[п -\)М
MR = -Ц !—Л- = -2-71ЛГ а,
(n2+2)d 3 А
где Mw — молекулярная масса, d — плотность, п — индекс
рефракции (показатель преломления), а — электронная поляризуемость
молекулы, NA — число Авогадро.
Выражения (п2— 1) и (я2+2) показывают, насколько легко может
быть поляризована молекула, что особенно важно при наличии
заместителей, содержащих гс-электроны или неподеленные
электронные пары. В настоящее время MR, также как и IgP, входит в число
параметров (дескрипторов) молекулярной структуры, которые
используют для количественной оценки связи «химическое строение —
биологическая активность» (QSAR).
2.5.1. Оптическая изомерия
В связи с тем, что многие биологические макромолекулы,
образующие рецепторы, являются оптически активными, неудивительно, что
и лекарства, состоящие из смеси изомеров, обладают стереоспецифич-
ностью. Впервые это явление отметил Л. Пастер на примере дрожжей,
способных перерабатывать только один из изомеров винной кислоты.
Таким образом, определенное соответствие между асимметричным
лекарством и его асимметричным рецептором можно рассматривать как
один из критериев активности. Такое соответствие наиболее ярко
проявляется в случае высокоактивных и высокоспецифичных лекарств.
Оптические изомеры могут обладать различной физиологической
активностью. Согласно гипотезе Easson-Stedman, для связывания с
рецептором необходимо соответствие местам связывания трех
заместителей у асимметрического атома, следовательно, только один энан-
тиомер активен [1, 2]. Примером, иллюстрирующим данную
гипотезу, служит взаимодействие двух оптических изомеров адреналина (2.38)
и изадрина (2.39) с рецептором (рис. 2.8).
Из представленной схемы ясно, что (-)-адреналин и (-)-изадрин
связываются эффективнее.
Примером, стереоселективности взаимодействия фермент-субстрат
служит гидролитическое расщепление лактоилхолина (2.40) на хо-
лин и молочную кислоту под действием ацетилхолинэстеразы [10].
£(+)-Лактоилхолин гидролизуется значительно быстрее, чем Д-)-изо-
мер (рис. 2.9).
40
н3сч
(+>
N
Н
Н
ОН
н
н3с
н
он
н
N
н
он Т^-он
Оа r <j>^ /Oft к <^>f>
(-)
(2.38)
сн.
h3c^V
<+)
н
н
он
н
он
н3с
ск
N
н
н
н он |^он
(-)
/<5к R <J[y?/ /Оа r C£yp/
(2.39)
Рис. 2.8. Схема взаимодействия оптических изомеров
адреналина и изадрина с рецептором:
Л — анионный центр, R — рецептор, Р — плоская область
(2.40)
Рис. 2.9. Схема связывания лактоилхолина с ацетилхолинэстеразой:
А — активный центр фермента
Различия в активности оптических изомеров могут быть очень
существенными. Так, один из морфинанов — синтетический
анальгетик (-)-леворфанол — способен связываться с половиной морфино-
вых рецепторов в концентрации 10~9 М. Его оптический антипод —
(+)-декстрофан — практически неактивен как анальгетик [8]. Несмотря
на то, что энантиомеры демонстрируют различную степень
активности, они редко выступают антагонистами друг друга, т. к. различия
в их эффектах объясняются различиями в способности к связыванию.
Антагонист обычно сильнее связывается с рецептором, чем агонист,
а менее активный энантиомер не может вытеснить более активный с
рецептора [2]. Диастереомеры часто также активны в какой-либо
одной конфигурации. Однако это не всегда справедливо. Например,
41
в молекуле тиазолидиндионового производного троглитазона (2.41),
который по механизму действия является «усилителем» эффектов
инсулина, агонистом PPAR-рецепторов, есть два асимметрических
атома углерода, т.е. возможно наличие четырех изомеров. Все они
были выделены и исследованы. Ни один из них не имел преимуществ
перед рацемической смесью [11].
(2.41)
Часто разделение смеси или стереоселективный синтез одного из
возможных изомеров достаточно сложны. В этом можно убедиться на
примере репаглинида (2.42), схема синтеза которого представлена на
рис. 2.10. Этот антидиабетический препарат по механизму действия
является стимулятором секреции инсулина. Несмотря на то, что (+)-
изомер в два раза активнее рацемической смеси, в состав
лекарственной формы (ЛФ) все же входит рацемат [12].
Соотношение активностей эутомера (активного изомера) и дис-
томера (неактивного антипода) принято выражать соотношением:
EI = Igaffin^ - Xgaffin^^,
где affin — аффинность (сродство к рецептору).
Дистомер не всегда бывает «инертным» и может вносить свой вклад
в побочное действие препарата, т. к. «мешает» фармакологическому
эффекту эутомера или метаболизируется с образованием продуктов
с высокой токсичностью или нежелательной активностью.
Стереоспецифичность важна не только для процесса связывания,
но и для метаболизма вещества и скорости его выведения. Так,
метаболическое превращение диазепама (2.43) протекает стереоселектив-
но и приводит к образованию (5)-Ы-метилоксазепама (2.44).
Последний является фармакологически активным, и стереохимия имеет
решающее значение не только для проявления активности этого
метаболита, но и для скорости его выведения [2].
(2.43) (2.44)
42
сн,
о
9 (Г^Г^о'^сн,
НО' v ^ 'ОН
О У
о сн.
^\ —. н3с^Ч о [pV^OH О
" н
о
(АсО)20 / \ АсОН
NaHCO,/ \PhaP
|CCi4
TI(i-PrO)4
НзС-Ч О
СНЭ
НзС'Ч
Рис. 2.10. Схема синтеза репаглинида (2.42);
А — рацемический; Б — энантиоселективный
43
Канцерогенные свойства бензо[а]пирена (2.45) связаны со стерео-
селективностью его окисления в транс-диолы, которые затем
превращаются в эпоксиды (рис. 2.11). Соотношение между
аксиальным и экваториальным эпоксидом для (-)транс-изомера, составляет
9:1, а для (+)/я/?а//с-изомера, наоборот, — 1:22. Аксиальные
эпоксиды являются более эффективными электрофильными агентами,
атакующими нуклеотиды, в результате чего нарушается структура
ДНК [1, 2].
(2.45)
'он но
он он но*'
(+) транс {-) транс ОН
Рис. 2.11. Схема окисления бензо[а]пирена
Такая селективность метаболизма обусловлена существованием
селективных ферментных систем: оксидаз аминокислот, декарбокси-
лаз, дегидрогеназ. Метаболизм полициклических углеводородов, в том
числе и бензо[а]пирена, осуществляется при участии монооксигеназ
смешанного типа (цитохром Р450), также обладающих стереоспеци-
фичностью [13].
Метаболические превращения могут приводить к инверсии
конфигурации под действием изомераз, находящихся в кишечнике, как
это имеет место в случае ибупрофена (2.46). В табл. 2.5 приведено
экспериментально установленное соотношение изомеров
ибупрофена [14].
н сн
н он
(2.46)
Селективность имеет место и при взаимодействии БАВ с
асимметрическими центрами мембран. Если молекула, содержащая
асимметрический атом, должна проникнуть через богатую рецепторами
44
Таблица 2.5
Соотношение изомеров при метаболизме ибупрофена
№
образца
1
2
3
Соотношение изомеров
во вводимом образце, S/R
95 : 5
6 :94
50 : 50 (рацемат)
Соотношение изомеров
в экскретируемом образце, S/R
95 : 5
80 : 20
70 : 30
R (-) — менее активный изомер.
мембрану, то селективность в этом случае важна для биологического
ответа клетки на результат связывания Л-Р. Связывание же с белками
плазмы, напротив, характеризуется низкой стереоспецифичностью.
Исключение составляют оксазепам и другие бензодиазепины [15].
2.5.2. Геометрическая изомерия
Геометрические изомеры представляют собой обычно конформаци-
онно жесткие молекулы, часто в значительной степени различающиеся по
своим физико-химическим свойствам. В таком случае корреляцию
между пространственным строением и биологической активностью
вещества проводить достаточно сложно. Так, один из изомеров, при наличии
в структуре подходящих функциональных групп, может гораздо
эффективнее ионизоваться при физиологических значениях рН. Это
неизбежно скажется на абсорбции и распределении вещества в организме, что
может привести к значительным отличиям в биологической активности.
Примером такого типа различий между геометрическими изомерами
может служить диэтилстильбестрол (2.47) — аналог по действию
стероидного гормона эстрадиола (2.48). Транс-изомер диэтилстильбестрола в
14 раз активнее *<ис-изомера, потому что у транс-изомера расстояние
между ОН-группами такое же, как в и молекуле эстрадиола [16].
(2.47) транс- (2.47) цис- (2.48)
2.5.3. Биологическая активность конформеров
Вопросы зависимости действия БАВ от их конформационной
изомерии вызывают горячую дискуссию. Биофизическая концепция
0 существовании «привилегированной» конформации, в которой
45
©
М(СНз)3
нфн НзС
О-С-СН,
II 3
о
(2.49)
©
@НзСТ°
N(CH3)3 СН,(СНЛМ\ Р
(2.50)
(2.53)
н3соос
©
н,с—п—о^^снз),
(2.54)
сооск
(2.55)
лекарства вступают во взаимодействие с рецепторами,
неоднократно подвергалась критике. В 70-е гг. во многих публикациях
обсуждалась возможность создания рецепторных карт, в основу которых
были бы положены как данные о расстояниях между ключевыми
атомами (чаще гетероатомами) или функциональными группами в
БАВ так и результаты квантово-химических расчетов наиболее
стабильных конформаций [17]. Многие из этих подходов не
выдержали критики. Однако есть ряд результатов, которые нашли лишь
частичное экспериментальное подтверждение. Так, в настоящее
время известно, что ацетилхолин взаимодействует с никотиновыми
46
и мускариновыми рецепторами главным образом в трансоид-
ной (2.49) и «гош» (2.50) конформациях. Первоначально
предполагалось, что за связывание с никотиновыми рецепторами несет
ответственность цисоидная, а с мускариновыми — трансоидная форма
ацетилхолина. Для прояснения ситуации были синтезированы
модельные соединения — производные декалина (2.51), (2.52) и
циклопропана (2.53), (2.54), представляющие собой жестко
фиксированные изомерные формы. Оказалось, что существенные структурные
различия между ацетилхолином и декалинами приводят к
практически полной утрате холинергической активности у последних,
независимо от их изомерной формы. В то же время (+)-/я/?я//с-изомер
циклопропана (2.53), отличающийся от соответствующего трансо-
идного конформера (2.49) только метиленовым мостиком,
демонстрирует высокий уровень активности, сопоставимый с таковым
у ацетилхолина относительно мускариновых, и слабую активность
в отношении никотиновых рецепторов. К тому же {+)-транс-кзо-
мер (2.53) легко гидролизуется ацетилхолинэстеразой — ферментом,
инактивирующим ацетилхолин. Цис-изомер практически неактивен
как в отношении никотиновых, так и мускариновых рецепторов.
Этот результат подтверждает лишь то, что ацетилхолин
взаимодействует с мускариновыми рецепторами в трансоидной
конформаций [18].
Примеров подобных корреляций существует много, однако
подходить к ним надо с определенной долей критичности.
Большинство структурных исследований до недавнего времени
подтверждали наличие связи между плоской структурой 1,4-дигидропиридинов
и их способностью блокировать Са2+-каналы в гладкомышечных
клетках коронарных сосудов. Однако расчеты пространственного
строения этих веществ, проведенные методами ab initio и
молекулярной механики, показали, что эфирам Ганча (2.55), к числу
которых относятся препараты фенигидин, амлодипин и др., присуща
значительная конформационная подвижность. В связи с этим они
могут легко изменять конформацию от плоской до конформаций
«лодки». При изменении торсионного угла С~С—С—С= от —10°
до 30° энергия возрастает менее, чем на 1 ккал/моль [19]. Таким
образом, фармакологическая активность этих веществ, вероятнее
всего, не связана с существованием 1,4-дигидропиридинового цикла
в равновесной плоской конформаций.
Литература
1. Foye Ж О., Lemke T.L., Williams D.A Principles of Medicinal Chemistry, 4th ed. —
Philadelphia, Lippinkott Williams & Wilkins, 1995.
2. Nogrady T. Medicinal Chemistry. A Biochemical Approach. — New York, Oxford:
Oxford University Press, 1988.
3. Rowland M., Tozer T. Clinical Pharmacokinetics: Concepts and Application,
2«d ed __ Philadelphia: Lea & Fabiger, 1989.
4. Gibaldi M. Biopharmaceutics and Clinical Pharmacokinetics, 4th ed. —
Philadelphia: Lea & Fabiger, 1991.
47
5. Medicinal Chemistry: Principles and Practice. // Ed. F.D.King. — Cambridge,
the Royal Society of Chemistry, 1994.
6. Patrick G.L. An Introduction to Medicinal Chemisry. — Oxford: Oxford
University Press, 1995.
7. Ariens E.J., Soudijn W.y Timmermans P. Stereochemistry and Biological Activity
of Drugs. — Oxford: Blackwell Scientific, 1983.
8. Ковтуненко B.O. Лжарсыа засоби з д|ею на центральну нервову
систему. — Киш; 1рпшь: ВТФ «Перун», 1997.
9. Харкевич ДА. Фармакология. — М.: ГЭОТАР Медицина, 1999.
10. Auditore J.V., Rama Sastry B.V.// Arch. Biochem. - 1964. — Vol. 105. -
P. 506.
11. HorikoshiK Yoshioka T.y Kawasaki T.// Ann. Rep. Sankyo Res. Lab. - 1994. -
Vol. 46.-P. 1.
12. GraulA, CastanerJ.// Drugs Fut. - 1996. - Vol. 21, N7. - P. 694.
13. Jerina DM. Fifth Iiternational Symposium on Microsomes and Drug Oxidation.
/R.Sato, K.Kato Eds. // Japan Scientific Societies Press. — 1982. — P. 195.
14. Kaiser DG. et ai // J. Pharm.Sci. - 1976. - Vol. 65. - P. 269.
15. Muller W.E., Wollert К // Mol. Pharmacol. - 1975. - Vol. 11. - P. 52.
16. Walton E.} Browle G. // Nature. - 1943. - N 151. - P. 305.
17. Martin Y.C. Quantitative Drug Designs. — New York: Marcel Dekker, 1978.
18. Smissman E.E. et al. // J. Med. Chem. - 1966. - N9. - P. 458.
19. Shishkin O.V.I I J. Mol. Struct. - 1996. - N 385. - P. 209.
3
ВЗАИМОДЕЙСТВИЕ ЛЕКАРСТВО-РЕЦЕПТОР
(ОБЩИЕ СВЕДЕНИЯ)
Согласно теории J. Ferguson (см. разд. 2.1) в зависимости от вели- г
чины термодинамической активности а, ЛВ можно разделить на
структурно неспецифические (0.01<я<1) и структурно специфические
(я<0.001). Последние действуют в малых концентрациях и обычно
имеют определенные места связывания в организме — рецепторы (от
лат. receptio — восприятие). Клетки, как правило, содержат большое
количество различных мембранных, цитоплазматических и ядерных
рецепторов БАВ: ионные каналы, ферменты, нейромедиаторы,
транспортные системы и гены. Они необходимы организму для
нормального функционирования и адекватного реагирования на
изменения внешней и внутренней среды и, в конечном счете, для
поддержания гомеостаза. Наличие у живых существ органов,
воспринимающих внешние сигналы, было отмечено учеными еще в глубокой
древности. Однако понимание роли рецепторов как внутренней
системы «органов чувств», позволяющей организмам адаптироваться к
окружающей среде, стало складываться только в середине XIX в.
Впервые наличие специфических мест связывания БАВ установил Bernard
в опытах с кураре [1]. Несколько позже (в начале XX в.)
существование в клетках так называемых «рецепторных субстанций» выявил
J.N. Langley [2]. Изучая действие атропина и пилокарпина на
секрецию слюны, а также никотина на сокращение скелетных мышц,
исследователь обнаружил, что фармакологический эффект имеет место
только при аппликации этих алкалоидов на участки клеточной
поверхности с небольшой площадью. J.N. Langley также установил, что
кураре блокирует эффект никотина, т. е., в современном понимании,
выступает антагонистом рецепторов никотина (одного из подтипов
холинорецепторов). Н.Н. Dale в 1906 г. применил эту концепцию для
объяснения фармакологической активности алкалоидов спорыньи.
Однако основоположником рецепторной теории действия лекарств
следует признать P. Ehrlich (1907 г.). Он же впервые ввел термин
«рецептор». В результате изучения избирательности действия
синтетических красителей и мышьяксодержащих соединений на живые
клетки P. Ehrlich сформулировал основной постулат: «corpora non agun
nisi fixata» — «вещества не действуют, если не фиксируются». Это
обобщение стало теоретической базой химиотерапии [3]. P. Ehrlich
предполагал, что клетки содержат так называемые боковые ветви, или
рецепторы, в результате селективного взаимодействия с которыми ЛВ
49
индуцируют свой эффект. Согласно этой теории, лекарство имеет два
структурных фрагмента, один из которых — гаптофорная область —
соединяясь с рецептором, позволяет тем самым другому фрагменту —
эргофорной или токсофорной области — осуществлять биологический
ответ. Несмотря на то, что детали этой концепции были
пересмотрены и уточнены (в настоящее время эргофорный фрагмент
рассматривается не как лиганд, а как часть рецептора), многочисленные
исследования подтвердили правильность теории P. Ehrlich о
рецепторах как участках тканей, селективно связывающих ЛВ и
опосредующих реализацию их фармакологических эффектов [4—6].
Настоящая глава посвящена рассмотрению химической природы
рецепторов как макромолекул — мишеней Л В, в ней также даны общие
представления о кинетике взаимодействия лекарство — рецептор.
Общие и частные вопросы рецепторологии как одной из
важнейших концепций современной фармакологии изложены в большом
количестве монографий, обзоров и оригинальных статей [4—20]. Из
доступных отечественному читателю источников, наиболее подробно
теоретические основы рецепции рассмотрены в пособиях и
монографиях [4—8]. Во многих из вышеперечисленных работ даны определения
понятию «рецептор». В обобщенном виде его можно сформулировать
следующим образом: рецептором может быть любая
высокомолекулярная конформационно подвижная биоструктура, специфически
связывающая химическое соединение (лиганд, лекарство) на поверхности или внутри
клетки и трансформирующая полученную информацию в биологический
ответ. К таким макромолекулам относятся белки и нуклеиновые
кислоты. Специфическое взаимодействие лиганда с рецептором следует
отличать от неспецифического связывания эндогенных или экзогенных
БАВ с белками плазмы крови или мукополисахаридами
соединительной ткани. Белковые структуры такого типа получили название
«молчащих» рецепторов. В результате связывания с ними не реализуются
никакие эффекты. Плазменные белки количественно, а не качественно
влияют на проявление эффектов Л В [4—6, 9].
3.1. Семейства рецепторов и их химическая природа
Следует отметить, что до 60-х гг. XX в. представления о рецепторах
основывались лишь на косвенных данных. Изучалась, как правило,
зависимость величины биологического эффекта лекарства от его
структуры, дозы или концентрации в интактном (здоровом) организме и в
условиях применения антагонистов. На основании этих данных
оценивалось сродство ЛВ к тому или иному рецептору. При таком
подходе были выявлены и охарактеризованы различные виды
рецепторов, однако их химическая природа и механизм функционирования
оставались неизвестными. Только с появлением меченных
радиоактивными изотопами лигандов, а также благодаря развитию
физико-химических методов анализа макромолекул удалось в ряде случаев
выяснить, что же представляют собой рецепторы, и какую роль они
50
играют в трансформации молекулярного сигнала в физиологический
ответ клетки, ткани, органа. К началу 90-х гг. было выявлено более/
30 групп различных рецепторов [4—6, 8—11], отдельные из них
приведены в табл. 3.1. В последнее десятилетие, благодаря расшифровке \
генома человека, этот список значительно расширился и продолжает ?
пополняться [12, 14, 15].
Таблица 3.1
Некоторые виды рецепторов и их подтипы
Рецепторы
Аденозиновые рецепторы
агАдренорецелторы
ос2-Адренорецелторы
р-Адренорецепторы
Ангиотензиновые рецепторы
Брадикининовые рецепторы
ГАМК-рецепторы
Гистаминовые рецепторы
Дофаминовые рецепторы
Лейкотриеновые рецепторы
М (мускариновые)-холинорецепторы
Н(никотиновые)-холинорецепторы
Опиоидные рецепторы
Простаноидные рецепторы
Пуриновые рецепторы Р
Рецепторы возбуждающих
аминокислот (инотропные)
Рецепторы нейропептида Y
Рецепторы предсердного натрийуре-
тического пептида
Рецепторы, активирующие
пролиферацию пероксисом
Серотониновые рецепторы
Холецистокининовые рецепторы
Рецепторы белково-пептидных
гормонов
Рецепторы стероидных гормонов |
Подтипы
Аь Агл> А2в, Аз
С*[А, а1В> а1С
а2А> «2В, «2С
Pi, Pi, Рз
АТ„ АТ2
Bi, В2
GABAa, GABAb, GABAc
и,, н2, н3
Db D2, D3, D4) D5
LTB4, LTC4) LTD4
Mb M2, M3,M4
Мышечного типа, нейронального
типа
ц, 8, к, а
DP, FP, IP, ТР, ЕР„ ЕР2,ЕР3
Ргх, Ргу, Ргг, Ргт, Рги
NMDA, АМРА, каинатные
Y„Y2
ANPA, ANPB
PPARa, PPARp, PPARY
5-НТцд.р), 5-HT2(A.q, 5-HT3, 5-HT4,
5-НТ5(А.В), 5-НТ6, 5-НТ7
ССКА, ССКВ
Рецепторы инсулина, глюкагона,
пролактина, рецепторы тиреоид-
ных гормонов, лютеинизирую-
щего гормона, лактогенного
гормона, рилизинг-фактора, гонадо-
тропина
51
Во всем многообразии рецепторов выделяют 4 следующих типа
[4,6, 10, 11]:
• рецепторы, осуществляющие контроль за функцией ионных
каналов (н-холинорецепторы, САВАА-рецепторы, глутаматные
рецепторы);
• рецепторы, сопряженные с эффектором через систему G-протеи-
ны — вторичные посредники или G-протеины — ионные каналы
(рецепторы некоторых белковых гормонов и пептидов — ангио-
тензина, брадикинина, эндотелина и др.; биогенных аминов —
адреналина, дофамина, гистамина, серотонина; липидов — кана-
биноидов, простагландинов, лейкотриенов, тромбоксанов; нуклео-
зидов и нуклеотидов — аденозина, АТФ, АДФ и др., а также
ионов Са2+);
• рецепторы, осуществляющие прямой контроль за функцией эф-
фекторного фермента (они непосредственно связаны с тирозин-
киназой и регулируют фосфорилирование белков — рецепторы
инсулина и гормонов роста);
• рецепторы, контролирующие транскрипцию ДНК — растворимые
цитозольные или ядерные белки (рецепторы стероидных и тирео-
идных гормонов).
Первые три типа рецепторов принадлежат к числу мембранных, а
последний — к ядерным. Схематически семейства рецепторов
представлены на рис. 3.1.
ионы
,, 1
кл. мембрана
®4/
ИОНЫ
G О О
вторичн. фосфорилир.
посреди, белков
Рис. 3.1. Типы рецепторов:
А — агонист, Р — рецептор, Ф — фермент, G — G-белок
Большинство рецепторов представляют собой белки, точнее
липо- или гликопротеины, которые располагаются на цитоплаз-
матической мембране или мембранах органелл клетки. Они
могут пронизывать всю толщу мембраны (интегральные белки,
см. разд. 1.2) или находиться с ее внутренней стороны. Если лиганды
рецептора представляют собой гидрофильные молекулы, например,
нейромедиаторы, то связывающие участки рецептора должны
обязательно находиться с внешней стороны клетки. Экспериментально
доказано, что рецепторы таких молекул являются белками, прочно
связанными с мембранами, поэтому для их солюбилизации
необходимо использовать ПАВ. Рецепторы гидрофобных молекул слабо
52
связаны с мембранами, и образующиеся комплексы с лигандами
могут выходить из мембраны в цитоплазму.
В процессе эволюции образовались также рецепторы,
локализованные внутри клеток. Их лиганды, например, стероидные или ти-
реоидные гормоны, обладают способностью за счет своих
гидрофобных свойств проникать через мембраны путем пассивной
диффузии или активного транспорта. Структура и функции
внутренних рецепторов менее изучены, чем мембранных, поскольку
первые труднее выделить, не изменив при этом их первоначальной
структуры. Известно присутствие рецепторов различных лигандов
на рибосомах (к антибиотикам), в ядре (к стероидным и тиреоид-
ным гормонам, тиазолидиндионам, эссенциальным жирным
кислотам, фибратам), в комплексе Гольджи (к гормонам), в микросомах
(к гормонам). Биологический смысл такой внутриклеточной
рецепции заключается в более глубоком и продолжительном
изменении функций клетки в ответ на получение внешнего сигнала
[4, 6, 10, 11].
Белковые цепи и домены, образующие рецептор, имеют
определенную организацию. Установлено, что в механизме,
обеспечивающем закрытие ионного канала, ведущая роль принадлежит
н-ацетилхолинорецептору, состоящему из пяти белковых
субъединиц, каждая из которых имеет четыре трансмембранных домена
(рис. 3.2). Последние образованы пронизывающими всю мембрану
гидрофобными а-спиралями. Один из доменов — М2,
выстилающий канал с внутренней стороны, гидрофилен. В нем
преобладают ориентированные особым образом анионсодержащие
фрагменты аминокислот, что обеспечивает распознавание катионов. При
взаимодействии лиганда с таким рецептором, за счет изменений
в трансмембранном домене М2, происходит конформационная
перестройка белков, образующих стенки канала, и закрытие его
просвета [11, 13].
Рис. 3.2. Структура ионного канала
Представление о строении рецепторов, передача сигналов от
которых осуществляется с участием G-протеинов, можно составить по
рис. 3.3, из которого видно, что единственная полипептидная цепь
53
имеет семь трансмембранных а-спиралей, а находящаяся вне
мембраны цепь с терминальной ЫН2-группой, гликозилирована. Петли,
выходящие в цитоплазму клетки, различаются по количеству
аминокислотных остатков. Первая состоит из 10—20 аминокислот, вторая
содержит до 20 и третья — до 50 аминокислотных остатков. На
третьей петле расположены участки связывания с G-протеинами.
Оканчивается G-сопряженный рецептор цепью из 100 аминокислот и
содержит концевую карбоксильную группу, которая также обеспечивает
связывание с G-белками. Сведения о пространственном строении
рецепторов этого типа стали доступны лишь недавно. Благодаря рент-
гено-структурному исследованию (РСИ) бычьего родопсина была
впервые предложена трехмерная (3D) модель рецептора,
сопряженного с G-белками [12, 13].
Рис. 3.3. Схематическое изображение рецептора, связанного с G-белками:
G — домены, обеспечивающие связывание с G-протеинами
Рассматриваемые рецепторы принято относить к одному из трех
семейств — А, В или С [14]. К первому — самому обширному
семейству А — принадлежат так называемые родопсиноподобные
рецепторы или мишени биогенных аминов; ко второму — глюкаго-
ноподобные, а к третьему — глутаматные мишени. В настоящее
время в результате расшифровки генома человека выявлено
несколько сотен рецепторов, сопряженных с G-белками. Из них
только 30 являются мишенями широко распространенных ЛВ, а для
210 — идентифицированы лишь эндогенные лиганды. К этим
группам принадлежат рецепторы пептидов, биогенных аминов, нуклео-
зидов и лейкотриенов. Примерно 160 рецепторов, выявленных в
человеческом геноме, относят к так называемым
«рецепторам-сиротам», т. к. их лиганды и физиологические функции пока не
установлены [11, 14—17].
Обособленную группу образует семейство рецепторов, в передаче
сигналов от которых участвует тирозинкиназа (рис.3.4). К их числу
принадлежат инсулиновые рецепторы и рецепторы факторов роста.
Тирозиновый домен находится внутри клетки. Экстрацеллюлярная
54
часть содержит области, богатые цистеиновыми остатками, которые,
вероятно, и обеспечивают вторичную структуру, необходимую для мест
связывания лигандов [10, 11].
Характерной особенностью внутриклеточных рецепторов
стероидных гормонов является наличие домена, способного
связывать полипептидные цепи с лигандом за счет Zn-пальцев (рис. 3.5):
цистеиновые остатки окружают атомы Zn и обеспечивают связь
с ДНК [10, 15].
1 -
ноос ноон
Рис. ЗЛ Схематическое изображение рецептора инсулина:
цистеиновый домен; 2 — домен тирозинкиназы; 3 — мембрана
Рис. 3.5. Схема строения рецептора стероидных гормонов:
1 — Zn-цистеиновый комплекс; 2 — домен, связанный с ДНК; 3 — домен,
связывающийся с гормоном.
Взаимодействие лигандов с рецепторами осуществляется
благодаря образованию либо водородных и координационных связей, либо
комплексов с переносом заряда, ион-ионного, ион-дипольного или
дисперсионного взаимодействия (эти типы связывания рассмотрены
в разд. 2.2). Распространенные в органической химии ковалентные
связи в рецепции не столь важны. Необратимое ковалентное
связывание имеет место при взаимодействии некоторых БАВ и ЛВ с
ферментами и будет рассмотрено в разд. 5.
55
3.2. Кинетика взаимодействия лиганд—рецептор
При рассмотрении кинетических закономерностей взаимодействия
лиганд (лекарство) — рецептор (Л-Р) используется ряд специальных
терминов.
Агонист — вещество, в результате взаимодействия которого с
рецептором возникает ответ. Связывание агониста эффективно, и он
обладает внутренней активностью. Агонистом может быть как
эндогенный нейротрансмиттер или гормон, так и синтетическое БАВ. Если
при взаимодействии с рецептором такое вещество вызывает
максимальный эффект, его называют полным агонистом.
Частичный агонист — вещество, связывающееся с тем же
рецептором, что и полный агонист, но, несмотря на адекватную дозу, в
результате такого связывания возникает не максимально возможный
ответ, как в случае полного агониста.
Антагонист — ингибитор эффекта агониста, который в отсутствии
агониста связывается с рецептором, но при этом не индуцирует
ответа. Его связывание неэффективно, а внутренняя активность равна
нулю. Он может занимать как те же места, с которыми связывается
агонист, так и выступать в качестве аллостерического ингибитора. При
аллостерическом ингибировании антагонист, связываясь с рецептором,
нарушает его структуру, способную связываться с агонистом, т. е.
изменяет аффинность рецептора.
Аффинность — аффинитет (сродство), способность лиганда,
связываясь с рецептором, образовывать устойчивые комплексы Л-Р. Она
пропорциональна константе связывания Кд. Лиганды с низкой
аффинностью вызывают эффект, аналогичный высокоаффинным при
более высоких концентрациях. Антагонисты также характеризуются
аффинностью.
ДЦ50 — характеризует количество вещества, которое необходимо
для достижения полумаксимального эффекта или создания эффекта у
50% из участвующих в эксперименте животных. 1С50 — ингибирующая
концентрация антагониста, характеризующая его полумаксимальный
эффект.
Впервые попытку описать кинетику взаимодействия Л В с
рецептором, основываясь на данных фармакологического
эксперимента (влияние ацетилхолина на мышечные клетки), предпринял
A.J. Clark в 1926 г. [4, 5, 16]. Он отметил, что зависимость
биологического ответа клетки от количества введенного вещества
практически полностью соответствует зависимости максимальной
скорости реакции от концентрации субстрата, наблюдаемой при
изучении кинетики ферментативных процессов. A.J. Clark
предположил, что взаимодействие Л-Р есть обратимая бимолекулярная
реакция, в которой лекарство (D) и рецептор (R) аналогичны
субстратам и ферментам. Полумаксимальный биологический
эффект введенного ЛВ развивается при оккупации им 50%
рецепторов. Эта теория получила название простой оккупационной
теории. Однако, учитывая возможность связывания с рецепторами
56
антагонистов, которые не вызывают ответа, E.J. Ariens (1954 г.)
усовершенствовал концепцию, предложенную A.J. Clark, и создал
сложную оккупационную теорию. Обобщенные положения,
выдвинутые A.J. Clark и E.J. Ariens, носят в настоящее время название
«классическая теория действия лекарств».
Для математического описания взаимодействия Л-Р A.J. Clark
использовал закон действия масс (ЗДМ):
*,
[D] + [R>^[D-R]
к-1
Ldt j=^[p][R]-*-,[d-r] (3.1)
В условиях избыточной концентрации ЛВ по отношению к
количеству рецепторов [D] = [DJ, a [R0] = [R] + [DR] и решение этого
уравнения имеет следующий вид:
ID-R1 = ^^(1-exp[-(*iID»1 + *-)t])' (3.2)
где Rq и D0 — начальные концентрации рецептора и лиганда
соответственно, а КД = k xkx.
При достижении равновесия концентрация комплексов Л-Р не
изменяется, поэтому
d[D-R] _Q
dt
и уравнение 3.1 записывают следующим образом:
К , *-i _ [D][R]
д \ [D-R]' С3-3)
где Кж — константа диссоциации, кх и к_х — константы скорости
образования и распада комплекса D - R.
Классическая теория основана на 2-х постулатах:
• величина фармакологического эффекта (Е) прямо
пропорциональна концентрации комплексов Л-Р: Е = a[D - R];
• максимальный эффект имеет место при оккупации всех
рецепторов, т. е. когда [D - R] = RQ, Етш = aR0.
При объединении этих уравнений с учетом принятых выше
допущений выражение для величины фармакологического эффекта
согласно E.J. Ariens приобретает вид:
a[DJ[RJ E [DJ
f — 0 J l ОJ _ max l 0J
[DJ + K [D.] + K <3-4)
L 0J д l 0J д
57
Тогда с использованием обозначений, принятых при записи
уравнений 3.1 — 3.3, получаем зависимость:
[D-R] =
[D][R0]
[Dl + К
(3.5)
Следует отметить, что при проведении фармакологических
экспериментов определяют кажущуюся константу диссоциации
комплексов Л-Р (К), т. к. равновесная концентрация [D-R], как
правило, неизвестна. Величина, обратная Кх (т. е. константа
ассоциации), отражает аффинность лиганда к рецептору. Константу
пропорциональности а из уравнения 3.4 E.J. Aliens назвал
внутренней активностью [18], которая отражает величину эффекта
относительно количества комплексов Л-Р. Следовательно, для того,
чтобы БАВ оказывало действие, оно должно обладать как сродством
к рецептору, так и внутренней активностью, т. е.
взаимодействовать с рецепторами эффективно. Исходя из уравнения 3.4, Кп равна
ЕДЖ Чем больше величина константы диссоциации, тем меньше
аффинность рецептора к лиганду [4, 7, 18].
Согласно уравнению 3.4, зависимость эффекта is от дозы ЛВ
описывается гиперболой (изотерма сорбции Ленгмюра), соответствующей
процессу насыщения (рис.3.6). Именно такой вид довольно часто
имеют и экспериментальные кривые «доза-эффект».
Що = Ко
Рис.3.6. Зависимость величины эффекта от дозы ЛВ
При этом существует несколько способов представления
уравнений 3.4 или 3.5 в линейном виде [4, 7, 19]. Наиболее широкое
применение получили преобразования Скэтчарда 3.6 и
Лайнуивера—Берка 3.7.
J_[Ro]__L[D-R] = I£^l
К ° К [D]
Д Д L J
(3.6)
[*.]
[D]
(3.7)
58
Соответствующие им графики представлены на рис 3.7 и 3.8.
[DR]/[D],
R0 [D-R]
Рис. 3.7. График Скэтчарда
1 1 1,
Е [D-R] у
1/Е™*= [RJ,
г * *
г' -' '
V^
1
^
—*-
мк,
1/[D]
Рис. 3.8. График Лайнуивера—Берка:
1 — в отсутствие антагониста; 2 — в присутствии конкурентного антагониста
В координатах Скэтчарда тангенс угла наклона прямой,
отражающей зависимость отношения [D - R]/[D] от [D - R], равен — \/К.
Сама же прямая пересекает ось абсцисс в точке, равной начальной
концентрации рецепторов R^
График в двойных обратных координатах (1/[D — R], 1/[D])
(рис. 3.8, линия 1), отражающий взаимодействие агониста с
рецептором, представляет собой прямую, которая пересекает ось
ординат в точке 1/R0 = 1/Епт9 а ось абсцисс — в точке ~1/К .
Преобразование Лайнуивера—Берка используют для оценки конкуренции
нескольких лигандов за связывание с одним и тем же рецептором.
При конкурентном связывании число мест связывания не меняется,
зато изменяется аффинность. Указанное приводит к тому, что в
двойных обратных координатах точка пересечения графика (рис. 3.8,
линия 2) с осью ординат остается постоянной, а точка
пересечения с осью абсцисс изменяется.
Все рассмотренные нами модели взаимодействия Л-Р (кроме
линии 2 на рис. 3.8) относятся к случаю, когда с рецептором
связывается один тип лигандов. Это условие может быть соблюдено
только при исследованиях, проводимых in vitro. Однако in vivo в
микроокружении рецепторов одного типа, как правило, присутствует
достаточно большое количество различных лигандов, отличающих-
59
ся по степени сродства и специфичности к данному типу
мишеней. Признаком того, что взаимодействие Л-Р осуществляется по
более сложной схеме, чем один лиганд — один рецептор, является
отклонение экспериментально установленной зависимости доза
ЛВ — эффект от изотермы сорбции Ленгмюра или от прямой
линии в координатах Скэтчарда. Поэтому для определения модели ре-
цепторного связывания используют отличные от описанных
методы преобразования координат, например, преобразование Хилла 3.10.
В координатах Хилла принято анализировать так называемые
кооперативные взаимодействия Л-Р:
nD + R
±D R.
где: n — истинная степень кооперативности.
В условиях равновесия
d[DnR]
dt
= ^[D]n[R]-^,[DnR] = 0
(3.8)
1
[D R]
К, [R][D]r
И окончательно
lg
PR]
[RJ-[DR]
= n]g[D]-lgtf
(3.9)
(3.10)
График Хилла (рис. 3.9) представляет собой прямую линию,
тангенс угла наклона которой равен кажущейся кооперативности у.
Используя его, определяют наличие или отсутствие взаимодействия
рецепторов. Если у = 1, то взаимодействие Л с рецептором
некооперативно, если у >1, (у < п) — оно имеет положительную, а если у < 1,
(у ~ 1/п) — отрицательную кооперативность.
[Dn_R]
'9 Ка \д [D]
Рис. 3.9. График Хилла
Под неоперативностью понимают изменение сродства лиганда к
рецептору при его частичной оккупации. Точка пересечения изотер-
60
мы насыщения с осью абсцисс равна логарифму равновесной
константы диссоциации комплекса Л-Р. Анализ более сложных моделей можно
найти в изданиях [4, 7, 19].
В целом различают следующие виды взаимодействия нескольких
лигандов с одним типом рецепторов [7].
1. По конкуренции за места связывания:
• неконкурентное, когда связывание нескольких лигандов происходит
с различными местами связывания на одном и том же рецепторе;
D1+R< **' ±ЪХК D2+R<==^==±D2R
• конкурентное, когда различные лиганды связываются с одним и тем
же местом на рецепторе;
D,+R< h iD.R D+Rp^iDR
I *~ 1 2 k_2 2
• бесконкурентное, когда лиганд второго типа взаимодействует не
с рецептором, а с комплексами, образованными лигандом
первого типа с тем же рецептором.
d1+Rt-^d,R d2 + d,r^=e=±d2(d,r)
2. По изменению аффинности рецепторов:
• без изменения аффинности, когда связывание лигандов одного вида
не приводит к изменению аффинности рецептора к лигандам
другого вида;
• с изменением аффинности, когда связывание одного или
нескольких лигандов приводит к изменению сродства рецептора к
остальным лигандам.
3. По наличию взаимодействия между лигандами:
• без взаимодействия, когда концентрация двух и более видов
лигандов в окружении одного типа рецепторов изменяется только за
счет процессов Л-Р взаимодействия;
• с взаимодействием, когда концентрация лигандов изменяется за
счет протекания химической реакции между ними.
Особое значение при взаимодействии нескольких лигандов с
рецептором имеет случай, когда между лигандами наблюдается антагонизм.
Он проявляется в различиях реализации биологических эффектов в
ответ на связывание Л-Р. Механизм действия антагонистов имеет
разную природу. Так, различают:
• химический антагонизм, проявляющийся в развитии химической
реакции между агонистом и антагонистом. При этом
концентрация агониста уменьшается, и, следовательно, снижается
индуцируемый им эффект;
• функциональный антагонизм, имеющий место на уровне
передачи сигнала от рецептора в клетку. При этом агонист
взаимодействует с одной системой проведения сигнала, стимулируя клеточ-
61
ную функцию, а антагонист взаимодействует с другой системой,
угнетая эту функцию;
• физический антагонизм, проявляющийся при развитии
фармакологического ответа, например, когда действие одного
препарата нивелируется за счет эффектов другого. При этом они
взаимодействуют с различными органами и тканями (гипотетический
случай совместного применения ЛВ сосудосуживающего и
диуретического действия).
С точки зрения кинетики формально-математическое описание
взаимодействия агонистов и антагонистов с рецепторами в
большинстве случаев идентично. Поэтому ряд исследователей рассматривает
понятие антагонизма скорее как фармакологическое, а не как
биокинетическое [7].
В медицинской химии при поиске лигандов к рецепторам из
числа так называемых малых органических молекул с целью
создания на их основе новых ЛС чрезвычайно актуальна проблема
структурного подобия между агонистами и их конкурентными
антагонистами. В целом дать однозначный ответ на вопрос о том,
насколько должны быть структурно близки агонисты и
антагонисты, невозможно. Составить представление о таком подобии можно
на примере некоторых нейротрансмиттеров и их конкурентных
антагонистов (рис. ЗЛО) [9]. Примером чрезвычайно близкого
структурного родства могут служить молекулы агонистов и антагонистов
опиоидных рецепторов (см. разд. 10). Однако если поместить в этот
ряд эндогенные агонисты — эндорфины, имеющие пептидное
строение, то между ними и экзогенными лигандами названных
рецепторов практически нет ничего общего.
Подводя итог краткому рассмотрению математических моделей
комплексообразования Л-Р, следует подчеркнуть, что они служат
интерпретации результатов экспериментальных исследований,
проводимых, главным образом, радиорецепторным методом. Он
основан на определении количества связанного с рецепторами
меченного радиоактивной меткой лиганда по числу распадов. Основными
характеристиками рецепторов являются их концентрация и
аффинность - \/К. Исследование кинетики взаимоотношений Л-Р
позволяет определить вид рецептора, к которому данный лиганд имеет
наибольшее сродство. Группы родственных белков, расположенные
в разных тканях и имеющие некоторые различия в структуре и
аминокислотной последовательности, чувствительны к разным аго-
нистам. Так, для каждой из подгрупп адренорецепторов (а,, <х2, Рр
Р2, Р3) существуют собственные агонисты. Эти подгруппы (как,
впрочем, и все виды рецепторов, приведенные в табл. 3.1)
дифференцированы на основании значений 1/А^. Свойства рецепторов не
являются постоянными: со временем могут происходить изменения
их концентрации и аффинности. Принято считать, что изменение
аффинности обусловлено генетическими дефектами рецепторного
белка, а снижение концентрации рецепторов связано с
прижизненными повреждениями рецепторного аппарата [7].
62
63
3.3.Основные теории рецепции
Классическая теория действия ЛВ в настоящее время имеет больше
историческое значение. По мере накопления экспериментального материала было
выявлено немало фактов, не объяснимых с позиций этой теории. Одним из
таких фактов является несовпадение (иногда на несколько порядков)
кажущейся и истинной констант Ка [4, 7, 9, 19]. Данное несоответствие — результат
того, что максимальный эффект ЛВ часто наблюдается при оккупации лишь
небольшой части рецепторов. Это противоречит одному из положений
рассмотренной выше классической теории действия лекарств. Попытку
устранить это противоречие предпринял R.B. Stephenson в 1956 г. [20]. Он
модифицировал классическую теорию, предложив концепцию свободных рецепторов,
основанную на трех постулатах:
• максимальный эффект, вызываемый агонистом, имеет место при
оккупации лишь части рецепторов;
• величина эффекта есть неизвестная функция числа занятых рецепторов,
и она не прямо пропорциональна их числу, как следует из классической
теории;
• образование комплекса Л-Р приводит к возникновению стимула S,
величина которого прямо пропорциональна количеству оккупированных
рецепторов (Y): S= eY. Коэффициент е, согласно R.B. Stephenson, есть
эффективность, а эффект является неизвестной функцией стимула:
Е = т у=Ш=_ж_
К ' г [D] + К
t L J Д
Таким образом, R.B. Stephenson выделил линейную дозозависимую часть S= eY
и неизвестную, возможно нелинейную, дозонезависимую часть E=f(S).
Следовательно, из кривых доза — ответ нельзя непосредственно определить
величину KR как ЕД50, если е * а. Согласно рассматриваемой концепции
эффективность полных агонистов очень велика, хотя величина Y мала. В отличие от
внутренней активности, которая связывает величину эффекта с оккупацией
рецепторов, эффективность связывает величину стимула с оккупацией
рецепторов. Поэтому ЛВ с разной эффективностью вызывают различные
стимулы, но связь между стимулом и эффектом есть свойство ткани, а не ЛВ.
Вещество с низкой эффективностью способно индуцировать ответ, который
может быть меньше максимально возможного даже при оккупации всех
рецепторов. Это и есть эффект частичных агонистов. Они взаимодействуют с
теми же рецепторами, что и полные агонисты, но вследствие конкуренции за
связывание уменьшают их эффект (рис. 3.11), т.е. частичный агонист
действует как конкурентный антагонист в присутствии полного агониста [4, 18].
Однако нельзя не отметить, что данная теория не отвергает возможности для
некоторых агонистов вызывать максимальный ответ при оккупации всех
рецепторов (граничное условие, когда понятие агонист и частичный агонист
тождественны).
Как классическая теория действия ЛВ, так и концепция, предложенная R.B.
Stephenson, не дают ответа на вопрос, почему в некоторых случаях зависимость
доза — ответ имеет не гиперболическую, а сигмовидную или колоколообразную
64
/о '-max
Ю-7 Ю-6 D
Рис. 3.11. Кривые зависимости доза—ответ для:
1 — агониста, 2 — частичного агониста,
3 — агонист (концентрация const) + частичный агонист (концентрация растет)
форму. Необъяснимыми также остаются различия в эффектах агонистов и
антагонистов. Для разрешения этих противоречий были предложены две
теории — скоростная и аллостерическая.
Первую предложил в 1961 г. W.D. Paton [21]. Он считал, что величина
эффекта лекарства определяется скоростью его ассоциации с рецептором. Реакция
образования и распада комплекса Л-Р имеет следующий вид:
D + R<=b±D-R
Она характеризуется константами скоростей К+{ и К_{ соответственно. При
этом чем больше K+i, тем больше биологический ответ клетки. Если К {
велика, то вещество является полным агонистом, если имеет промежуточное
значение — частичным агонистом и, наконец, если мала — антагонистом.
Следовательно, согласно концепции W.D. Paton, антагонистом является
вещество, скорость взаимодействия которого с рецептором низка, но
образующийся комплекс стабилен. Агонист же связывается с рецептором с
высокой скоростью, однако образующийся комплекс является короткоживущим.
Скоростная теория объясняет наличие постоянного эффекта антагонистов и
снижение ответа при многократном воздействии агонистов. Ее характерной
чертой является то, что при использовании констант скоростей отпадает
необходимость вводить третью константу — а, не зависящую от природы
лиганда. Эта концепция может быть с успехом применена к веществам
нейромедиаторного типа действия, эффекты которых связаны с изменением
проницаемости плазматических мембран для ионов, а также в том случае,
когда агонисты и антагонисты взаимодействуют с одними и теми же
участками связывания, т. е. при прямой конкуренции между агонистами и
антагонистами. Однако скорость распада комплексов Л-Р для полных
агонистов не всегда высока. Например, для стероидных или белково-пептидных
гормонов, регулирующих метаболические процессы, скорость которых
исчисляется минутами и часами, характерны низкие значения К_{ [19]. Кроме
того, антагонист может взаимодействовать с участками, отличными от мест
связывания агониста, и изменять их конформацию, а, следовательно, и
эффект. В таком случае ближе к истине, по-видимому, аллостерическая теория
65
или теория 2-х состояний, предложенная в 1967 г. А.Т. Karlin [4, 9]. Суть ее
заключается в том, что постулируется существование рецепторов в двух кон-
формационных состояниях, называемых R (relaxed) и Т (tense). R-форма
соответствует активному состоянию, Т — неактивному. В отсутствие ЛВ обе
формы находятся в равновесии. Отношение [T]/[R]=ifL есть аллостерическая
константа. Агонист — D обладает сродством к R-, антагонист — I
(ингибитор) к Т-форме. Частичный агонист в одинаковой степени способен
взаимодействовать с обеими формами.
Т < L > R
+ 1 T\L К +D \*1 К
IT < > RD
Главное отличие аллостерической теории от рассмотренной выше
скоростной состоит в том, что антагонист необязательно должен иметь низкую
скорость диссоциации. Если такой лиганд взаимодействует с аллостерическим
участком, то неважно, как долго он с ним связан. Важно, чтобы его
концентрация была достаточно высока, и он индуцировал такое изменение кон-
формации рецептора, при котором последний не может взаимодействовать с
агонистом. Такое предположение оправдано, т.к. движение боковых цепей в
водно-белковой матрице происходит во времени, и в какой-то один момент
рецептор находится в конформации, комплементарной агонисту, а в другой —
соответствующей антагонисту. При связывании агонисты и антагонисты
«фиксируют» указанные конформации, в результате по аллостерическому
механизму изменяется вся конформация рецептора. Эта концепция хорошо
согласуется с тем фактом, что число оккупированных рецепторов не всегда
эквивалентно числу активированных.
В рамках аллостерической теории находит объяснение регулирующее
влияние ионов Na+ и Li+ на опиоидные рецепторы (см. также разд. 10), при
отсутствии которых эти мишени взаимодействуют, главным образом, с агонистами.
Однако при инкубировании рецепторов в среде, содержащей ионы Na+ в
концентрации < 5 мМ (значительно ниже физиологической нормы —150 мМ),
наблюдается преимущественное связывание рецепторов с молекулами
антагонистов. Следовательно, ионы Na+, согласно предложенной концепции,
сдвигают равновесие в сторону Т-формы. Присутствие этих ионов в организме в
указанной концентрации и обеспечивает повышенную аффинность опиоид-
ных рецепторов к антагонистам по сравнению с агонистами [9].
Следует отметить, что все рассмотренные выше теории во многом утратили
свое значение. На сегодняшний день накоплен экспериментальный
материал, свидетельствующий, что скорости взаимодействия агонистов и
антагонистов во многих случаях не отличаются друг от друга. Более того, скорость
ассоциации антагониста с рецептором может значительно превышать
скорость связывания агониста с тем же рецептором.
Главное противоречие теории 2-х состояний состоит в том, что число мест
связывания агонистов и антагонистов с биологическими мишенями
существенно не отличается друг от друга. Следует также принять во внимание то, что
один из основных постулатов классической оккупационной теории и ее
66
модификаций — взаимоотношения Л-Р есть простая бимолекулярная
реакция — не всегда обоснован. Все ЛВ — полифункциональные соединения, и
для каждой части молекулы лиганда может существовать комплементарный
участок рецептора.
В этом плане интересна предложенная A. De Lean в 1979 г. теория
связывания гибких полифункциональных лигандов со специфическими субучастками
рецепторов [22]. Основные положения этой теории состоят в следующем:
• субучасток представляет собой функциональную часть рецептора,
способную взаимодействовать с лигандом. Рецепторная область состоит из
нескольких таких субучастков. Лиганд содержит функциональные
группы, которые распознаются соответствующими специфическими
субучастками рецепторной области на поверхности клеток;
• взаимодействие функциональных групп лиганда с субучастками рецептора
должно происходить одновременно (для жестких молекул по типу ключ —
замок). В большинстве же случаев лиганд обладает гибкостью и может
находиться в нескольких конформациях как в свободном, так и в связанном
состоянии. В свою очередь, рецепторная область также обладает гибкостью
и характеризуется изменением конформации при связывании жесткого
лиганда;
• величина ответа на действие ЛВ определяется не оккупацией всех
рецепторов, а зависит от концентрации активных рецепторов, способных
стимулировать эффектор;
• с одним и тем же рецептором могут связываться несколько веществ, что
является результатом гибкости лигандов. При высокой концентрации
лигандов каждый из них мешает другим полностью связаться с
рецептором и тем самым вызвать активацию эффекторной системы;
• несмотря на то, что истинное число контактов лигандов с рецептором
может быть довольно велико, определяющее значение в комплексо-
образовании имеет только небольшое количество функциональных
областей.
Эту теорию можно рассмотреть на примере взаимодействия двухвалентного
лиганда с двумя субучастками рецептора (рис. 3.12). Когда число субучастков
равно 1, рецепторы представляют собой гомогенную систему,
подчиняющуюся ЗДМ. При этом существует только один связывающий массив —
активный пул. Данная модель не может объяснить феномен конкурентного
антагонизма и существование частичных агонистов.
При наличии двух субучастков у рецептора концентрация каждого пула Р.
есть функция концентрации свободных рецепторов R и концентрации
свободного лиганда F:
Р, =L1-R-F Р3 =Ц Ц R F
P2=L2.R.FP4=L,L4.R.F,
где L — константы.
Связывающие пулы разделены на 3 класса, содержащие 0, 1 или 2 лиганда.
Концентрация рецепторов в каждом из классов, соответственно числу
лигандов, может быть представлена следующими уравнениями:
67
Ч=Ро; C1=P|+P2+P3=K1RF; C2=P4=K2R.r
Рис. 3.12. Схема взаимодействия двухвалентного лиганда
с двумя субучастками рецептора
Макроскопические стехиометрические константы К, и К^ представляют
собой комбинации констант L:
К
Ч+Ц + Ц
ч>
K2=L,
Экспериментально можно определить только макроскопические константы.
Если только одна молекула способна связаться с рецепторной областью, то
класс С2 будет свободным и К^ = 0. Когда активен только пул Р3, то
концентрация активных рецепторов определяется как Р3 = L, L3R F. Отношение
макроскопической константы, характеризующей активный пул, к константам других
пулов данного класса KA/Kt (для С, класса) называют коэффициентом
внутренней активности агониста. Если КА/К, = 1, то лиганд — полный агонист, если
Кд/К, = 0, то лиганд — антагонист.
Согласно теории A. De Lean, наиболее стабилен активный, полностью
участвующий во взаимодействии пул Р3. При этом множественная
оккупация рецепторной области невозможна. В присутствии антагониста
наблюдается как увеличение величины ЕД50 для агониста, так и
уменьшение максимальной концентрации активного пула. Однофункциональный
неактивный лиганд может быть как конкурентным, так и
неконкурентным антагонистом.
В действительности рецептор может иметь значительно большее количество
специфических мест связывания (рис. 3.13). Так, более сильное взаимодействие
антагониста с рецептором может быть обусловлено наличием
дополнительного комплементарного именно ему субучастка.
Математическое описание модели множественных участков значительно
сложнее рассмотренного выше. Однако во многих случаях зависимость доза-
эффект достаточно хорошо подчиняется ЗДМ. Практически все
исследователи, изучающие кинетику лиганд-рецепторных отношений, поль-
зуютсяданным подходом. Не является исключением и теория, предложенная
68
|—| /~\ Агонист
\s~\_J Антагонист
Рис. 3.13. Схема взаимодействия двухвалентных лигандов
с рецептором, обладающим тремя субучастками,
один из которых специфичен для антагониста
E.J. Aliens в начале 80-х гг. XX в. [18], которая представляет собой попытку
объединить все перечисленные концепции. В обобщенном виде кинетика
взаимодействия Л-Р согласно E.J. Aliens может быть представлена
следующим образом:
D + Rr-^DRc h >DR'c Ь >D + R'( *4 >R* ** >R(reg),
k-\ k-2 k-3 k-4 k-S
где D — БАВ; R — рецептор в неактивном состоянии; DR* —
оккупированный рецептор в активном состоянии; R* — свободный рецептор в
активном состоянии; R(reg) -~ рецептор в не реагирующем (регенерирующем)
состоянии.
В соответствии с данной теорией, при взаимодействии ЛВ с рецептором
образуется неактивный комплекс, который затем обратимо может перейти
в активное состояние. При этом скорость активации Л-Р комплексов
антагонистов значительно уступает скорости ассоциации агонистов с
рецепторами, т. е для агонистов, как правило, константы скорости kl9k2nky
велики. В случае обратимой оккупации рецепторов без их активации, к2 по
сравнению с &, и к_{ очень мала (пример конкурентного антагонизма).
Внутренняя активность, а, следовательно, и величина ответа на результат
связывания ЛВ с рецепторами, определяется отношением к2/к_{,
отражающим переход занятых рецепторов в состояние DR* и/или R*. Таким
образом, фармакологический эффект пропорционален числу активных Л-Р
комплексов, а не суммарному числу оккупированных рецепторов. Переход
рецепторов в неактивное состояние R может быть обусловлен их
агрегацией и интернализацией (экзоцитозом). Однако это не всегда справедливо,
т.к. существует феномен потери чувствительности рецепторов, суть
которого заключается в снижении первоначального эффекта ЛВ при его
повторных воздействиях.
Теория потери чувствительности рецептора предложена в работах В. Katz и
A. Gero [9]. Она предполагает не только конформационное изменение
молекул рецептора при взаимодействии с Л В, но и увеличение свободной энергии
образовавшегося комплекса. Если в результате такого контакта не произошло
конформационных изменений или эти изменения недостаточны для
увеличения свободной энергии до определенного порогового значения, то ЛВ не
вызовет эффекта и будет выступать в качестве конкурентного антагониста.
Согласно этой концепции, потеря чувствительности рецептора происходит
вследствие перехода комплекса агонист — рецептор из состояния с высокой
69
энергией в состояние с более низкой. При этом независимо от того,
диссоциирует агонист или нет, рецептор находится в таком энергетическом
состоянии, которое не может обеспечить передачу молекулярного сигнала на
эффектор.
В целом действие ЛВ, согласно предложенной E.J. Aliens и общепризнанной
в настоящее время схеме, представляет собой сложный многоступенчатый
процесс, в котором ассоциация ЛВ с рецептором является лишь начальным
этапом реализации его биологической активности. В дальнейшем имеют
место последующие взаимодействия, в конечном счете, проявляющиеся в виде
фармакологического ответа на связывание ЛВ с мишенью. Рассмотренный
процесс представлен на рис. 3.14.
Лекарство / \ Начальн. ( Система А (Эффекторная \ эффект
Т JomMvn I Умения j -^система }
I -, \ усиления
J Стимул у
Рис.3.14. Схема индукции фармакологического эффекта ЛВ
Потеря чувствительности рецептора может быть связана не только с
изменением его конформации, но также обусловлена какими-либо другими
событиями, происходящими в промежутке между комплексообразованием
лекарства с рецептором и наблюдаемым эффектом [4].
На сегодняшний день, несмотря на большое количество различных
концепций и моделей, количественно описывающих лиганд-рецепторные
взаимоотношения, не существует единой теории, позволяющей учесть все
особенности такого взаимодействия.
Литература
1. Leake CD. A Historical Account of Pharmacology to the 20th Century.-
Springfield: Charles С Thomas, 1975.
2. Langley J.N.// J. Physiol. — 1905. - Vol. 33. - P. 374.
3. Collected Papers of Paul Ehrlich, Vol. Ill // Ed by F.Himmelweit. — London:
Pergamon, 1957.
4. Сергеев П.В., Шимановский Н.Л. Рецепторы физиологически активных
веществ. -~ М.: Медицина, 1987.
5. Галактионов С.Г., Голубович В.П., Шендерович М.Д., Ахрем А.А. Введение
в теорию рецепторов. -~ Минск, 1986.
6. Харкевич Д.А. Фармакология. -~ М.: ГЭОТАР Медицина, 1999.
7. Варфоломеев С.Д., Гуревич КГ Биокинетика: Практический курс. —
М: ФАИР-ПРЕСС, 1999.
8. Сергеев П.В., Галенко-Ярошевский П.А., Шимановский Н.Л. Очерки
биохимической фармакологии. — М.: Фармединфо, 1996.
9. The Receptors / R.D.O'Brien, Ed. — New York: Plenum Press, 1979.
10. Kenakin T. Pharmacologic Analysis of Drug -~ Receptor Interaction, 2nd ed. —
New York: Raven Press, 1993.
11. Kebabian /. W.f Neumeyer J.L. The RBI Handbook of Receptor Classification. —
Natik: MA, Research Biochemicals international, 1994.
12. Palczewski K., Kumasaka Г., Hon T. et al. // Science. - 2000. — Vol. 289. —
P. 739.
70
13. Teller B.C., Okada Т., Behnke C.A. et ai // Biochemistry. - 2001 - Vol 40 -
P. 7761.
14. Klabunde T. Hessler G. // Chem. Bio. Chem. — 2002. — Vol. 3. — P. 928
15. Kenakin T. P. Bond Я A., Bonner T.L // Pharm. Rev. — 1992. — Vol 44 — P 351
16. Venter J.C. et al. // Science — 2001. — Vol. 291. — P. 1304.
17. Wise A., Gearing K, ReesS.// Drug Discovery Today. — 2002. — Vol 7 -P 235
18. Ariens E.J. // J. Cardiovasc. Pharmacol. - 1983. - N 5. - S 8.
19. Варфоломеев С.Д., Зайцев СВ. Кинетические методы в биохимических
исследованиях. -~ М., 1982.
20. Stephenson R.P. // Br. J. Pharmacol. — 1956. — Vol. 11. — Р 379
21. Paton W.D.W. I/ Ргос. Roy. Soc, Ser. В. - 1961. - Vol. 154. - P. 21.
22. De Lean A. Munson P.J., Rodbard D. // Mol. Pharmacol. — 1979. — N 15. - P. 60
СИСТЕМЫ ПЕРЕДАЧ РЕЦЕПТОРНОГО СИГНАЛА
И ВТОРИЧНЫЕ ПОСРЕДНИКИ
Из рассмотрения процессов комплексообразования Л-Р следует,
что основным результатом связывания агонистов с рецепторами
является индуцирование ответа. Существуют два основных механизма
передачи сигнала от рецептора в клетку: с участием так называемых
«вторичных посредников» и без них. Термин «вторичные
посредники» (ВП) или «вторичные мессенджеры» был введен для того, чтобы
отличать передатчики сигналов, реализующие свои эффекты на пост-
рецепторном уровне внутри клетки, от БАВ, выступающих в качестве
лигандов рецепторов и участвующих в передаче информации между
разными клетками. Таким образом, первичные посредники — это в
общем случае лиганды (нейротрансмиттеры, гормоны, ЛВ, токсины
бактерий и др.). Через систему ВП осуществляется передача сигналов
в нервных клетках, клетках мышечной ткани. К числу ВП
принадлежат аденозин-31,51-циклический монофосфат (цАМФ, 4.1) и гу-
анозин-З'^^циклический монофосфат (цГМФ, 4.2), продукты
гидролиза мембранных фосфолипидов — диацилглицерин (ДГ, 4.3)
и инозитинтрифосфат (4.4), а также ионы Са2+ [1—7].
R1
О
А
О
(4.1)
сн2он
K.N
он
о
(4.2)
ОН
ОН
Y2
О
ро;
(4.3)
(4.4)
R1 — остаток арахидоновои кислоты
R2 — остаток стеариновой кислоты
72
В результате взаимодействия Л-Р концентрация ВП в клетке
меняется, при этом концентрация одних может возрастать, а
других — убывать. Отличительной чертой большинства сигнал-пере-
дающих систем является значительное усиление сигнала от
рецептора. При этом становится понятно, почему для достижения
максимального ответа совсем необязательно, чтобы все
рецепторы были заняты.
При передаче сигнала без участия ВП в результате
образования комплекса Л-Р активируется связанный с рецептором
специфический фосфорилирующий фермент — тирозинкиназа [5, 6, 8, 9].
По такому механизму запускается процесс утилизации глюкозы в
ответ на связывание инсулина с рецепторами. Известно, что инсу-
линовые рецепторы состоят из 2-х а- и Р-субъединиц (см. разд. 3.1,
рис. 3.4). С внешней стороны клеточной мембраны расположены
а-цепи, которые связываясь с инсулином, претерпевают конфор-
мационные изменения, отражающиеся на р-субъединицах в виде
усиления их протеинкиназной активности. При этом происходит
фосфорилирование молекул инсулинового рецептора, находящихся
с внутренней стороны мембраны (Р-цепей), и других белков.
Фосфорилирование может быть инициатором каскада реакций,
проявляющихся в виде различных эффектов инсулина.
Аналогичным образом через тирозинкиназу опосредован и механизм
действия факторов роста [2, 5, 9].
4.1. Аденилатциклазная система передачи сигнала
Первым из вторичных передатчиков сигнала от мембранных
рецепторов был открыт цАМФ. Этому событию предшествовал ряд
исследований. В начале 50-х гг. было установлено, что активирование
Р-адренергических рецепторов в клетках печени приводит к
изменению активности фермента глюкозо-6-фосфатазы. В 1956 г. Е. Krebs и
Е. Fisher выявили взаимосвязь между активацией указанного
фермента и содержанием АТФ в клетке. Выделение промежуточных
продуктов в каскаде превращений, приводящем к изменению
активности глюкозо-6-фосфатазы, позволило в 1958 г. Е. Sutherland и R. Rail
предположить, что ключевая роль в нем принадлежит аденин-нуклео-
тиду. В 1959 г. Д. Липкин с соавт. идентифицировали его как цАМФ
[5, 8, 6, 10]. Таким образом, цАМФ является посредником,
запускающим механизм гликогенолиза (высвобождения глюкозы из депо
гликогена) при активировании р-адренергических рецепторов в клетках
печени. Синтез данного ВП катализирует фермент аденилатциклаза
(АС). цАМФ является ВП и для ряда гормональных и нейро-
эндокринных эффектов. Причем некоторые агонисты вызывают рост
внутриклеточной концентрации цАМФ, другие же, напротив,
способствуют снижению его уровня. К группе стимуляторов относятся
агонисты р-адренергических, дофаминовых (Dj), гистаминовых (Н2)
рецепторов, а также рецепторов вазопрессина (V2), фолликуло-
73
стимулирующий и лютеинизирующий гормоны, глюкагон.
Ингибиторами являются агонисты а2-адренергических, дофаминовых (D2),
аденозиновых (\), ацетилхолиновых (мускариновых) рецепторов, а
также ангиотензин II и соматостатин. В 1971 г. Rodbell с соавт.
показали необходимость участия в процессе активирования АС, а,
следовательно, и синтеза цАМФ, гуанозинтрифосфата (ГТФ) [2, 5, 6].
Модель активирования системы АС представлена на рис. 4.1.
Каскад превращений, реализующийся в результате связывания
лиганда с рецептором, сопряженным с G-протеином, состоит из
нескольких стадий. На первом этапе, в результате связывания
агониста с рецептором, образуется активированный комплекс PR-A.
В зависимости от того, будет ли это процесс активирования или,
напротив, ингибирования синтеза цАМФ, PR-A комплекс
взаимодействует с соответствующим передающим белком — Gs- или
Gj-протеином, состоящим из трех субъединиц a J и у,
связанных с гуанозиндифосфатом (ГДФ). Вытеснение последнего ГТФ
приводит к активированию третичного комплекса PR-J1-GS. Далее
комплекс диссоциирует на неактивный рецептор Рт, лиганд Л,
Ру-су&ьединицу G-протеина и активированную субъединицу as-G-npo-
теина, связанную с ГТФ. Последняя, соединяясь с АС, про-
мотирует ее, что и приводит к синтезу цАМФ из АТФ.
Собственно АС и выполняет роль 'внутримембранного усилителя сигнала.
а5-Субъединица дезактивируется путем гидролиза ГТФ на ГДФ и
Р043_-ион, после чего as объединяется вновь с (Зу-субъединицами
G-протеина [5, 6, 8, 10].
' ^ J
Рис. 4.1. Схема аденилатциклазной системы передачи сигнала в клетке
в результате Л-Р взаимодействия [5]:
д _ агонист, Рт — свободный рецептор, PR — активированный рецептор,
G — белки, состоящие из а, (Зу — субъединиц, АС — аденилатциклаза
В случае взаимодействия антагониста с рецептором не
происходит изменения конформации рецептора, и последний не может
взаимодействовать с G-белком. Поэтому сигнал от Л-Р-комплекса
антагонистов с мембранными рецепторами не трансформируется во
внутриклеточный сигнал.
G-протеины представляют собой гетерогенную группу белков,
различающихся за счет a-субъединицы. Структура и функции их
74
окончательно не определены. В настоящее время достоверно
описаны пять групп G-протеинов [З, 8, 10—12]:
• Gs-6eAKu — содержат а5-субъединицу, являются
стимуляторами АС;
• Gx-6eAm — содержат агсубъединицу, являются ингибиторами АС,
активируют цАМФ-фосфодиэстеразу;
• Go-6ejiKU — приводят к угнетению потенциал-зависимых Са2+
каналов и стимуляции К+ каналов;
• G^-белки — активируют К+ каналы и ингибируют АС;
• G^-белки — активируют фосфолипазу С (PL-C).
Известно, что существует шесть типов аденилатциклаз,
большинство из которых активируются G-ag-субъединицами и ингибируются
G-ctj-субъединицами этих протеинов. Однако II и IV типы
аденилатциклаз промотируются GPY-Димерами [6, 7].
Рост внутриклеточной концентрации цАМФ приводит к
активации цАМФ-зависимых ферментов. Далее цАМФ-зависимые про-
теинкиназы фосфорилируют сериновые и треониновые остатки
целевых белков, некоторые из них, в свою очередь, являются
протеинкиназами. Примером типичного каскада, инициируемого
цАМФ, является высвобождение глюкозо-1-фосфата из гликогена
в печени (рис. 4.2).
Таким способом реализуются тканеспецифические и кина-
зоспецифические эффекты. Рассмотренный механизм
обеспечивает многократное усиление внутриклеточного сигнала, один
активированный рецептор может взаимодействовать с 10—100 G-npo-
теинами, а одна активная АС способна катализировать
образование 10—100 молекул цАМФ. Тем самым рецепторный сигнал
усиливается в W—W раз. Поскольку цАМФ не является конечной
целью передачи сигнала, и каскад превращений, связанный с
фосфорилированием белков, продолжается, то в конечном итоге
при реализации тканеспецифического эффекта, коэффициент
усиления достигает значений W—W. Как только цель передачи
сигнала от рецептора достигнута, клетка прерывает сигнальный
процесс. Это происходит благодаря участию ряда факторов,
таких как:
• удаление агониста из окружающей рецептор среды за счет его
разрушения внеклеточными ферментами;
• повышение константы скорости диссоциации Л-Р-комплекса
вследствие изменения конформации рецептора в процессе
передачи сигнала;
• изменение концентрации свободных и связанных рецепторов в
результате их интернализации (экзоцитоза);
• деградация комплекса а5-ГТФ;
• деградация цАМФ под действием фосфодиэстераз (последних
существует пять видов);
• дефосфорилирование активированных протеинов под действием
фосфопротеинфосфатаз;
75
А-Р,
ару
\
Неактивная
протеин-
фосфатаза
Активная
протеин-
фосфатаза
Gc, SPr
i
АТФ
•-Н
Неактивн ая ( Актив ная
АС I АС
(Активная \
АС Г
Активный
белок-
ингибитор
АМФ / фосфодиэстераза ц"
7
Р-Р
(
еактивный
белок-ингибитор
фосфатазы
Активная
А-киназа
Неактивная
А- киназа
i
1
Гликоген
Неактивная / Активная
гликоген- I гаикоген-
фосфорилаза \ фосфорилаза)
Глюкозо -1- фосфат
Гликолиз
Рис. 4.2. Схема проведения и усиления сигнала от рецепторов,
сопряженных с G-протеинами [З]
• потеря чувствительности в ряду взаимодействия рецептор —
G-протеин — АС при длительной стимуляции рецептора аго-
н истом.
Теоретически большинство из звеньев сложного каскада
передачи сигнала через аденилатциклазную систему может быть точкой
приложения действия БАВ. Однако пока такие примеры крайне
ограничены. Применительно к G-белкам известны только токсины,
которые с ними связываются. Так, с Gg-белком взаимодействует
токсин холерного вибриона, что приводит к росту концентрации цАМФ
в стенках кишечника. В результате развивается диарея, поскольку
цАМФ является активатором секреции жидкости в пищеварительном
канале (ПК). С Gj-белком связывается токсин палочки коклюша,
что приводит к бронхоспазму [5].
76
он ^f Т"с-сн
: ChL CHj h 2
Г Т бнТ
н3с сн^он
(4.5)
Некоторые БАВ оказывают влияние на ферменты, участвующие
в регуляции биосинтеза ВП. Дитерпен растительного
происхождения — форсколин (4.5) оказывает прямое стимулирующее
действие на АС [5, 13, 14]. Следует отметить, что это вещество
нашло применение в индийской народной медицине как средство,
оказывающее выраженное гипотензивное и бронходилататорное
действие [5].
4.2. цГМФ как вторичный посредник
Несмотря на то, что цГМФ был открыт практически
одновременно с цАМФ, его роль как ВП до конца не установлена. Тем не менее
известно, что цГМФ принимает участие в передаче сигналов в двух
важных системах, ответственных за регуляцию артериального
давления (АД). Одна из них обеспечивает нормализацию АД, регулируя
выведение жидкости из организма при участии натрийуретического
фактора, а другая — посредством вазорелаксации под действием
оксида азота N0 [6, 8, 15, 16].
Гуанилатциклаза, в отличие от АС, существует в двух формах:
1) специфическая (связана с мембраной); 2) растворимая
(находится в цитоплазме). Специфическая форма участвует в передаче
сигнала от натрийуретического фактора. При этом, в отличие от
механизма высвобождения цАМФ, в процессе активирования
синтеза цГМФ ГТФ и G-протеины не участвуют [15]. Растворимая
форма гуанилатциклазы активируется эндогенным N0,
производимым из /-аргинина с помощью двух форм
конститутивной NO-синтетазы, локализованных в сосудистом эндотелии и
мозге [16, 17]. Схема активирования гуанилатциклазы
представлена на рис. 4.3.
цГМФ играет важную роль в механизме расширения сосудов,
препятствует агрегации тромбоцитов. Увеличение концентрации цГМФ
внутри клетки приводит к активированию серии- и треонинпротеин-
киназ. Существуют две формы этих ферментов. Одна представляет
собой димер, каждая субъединица которого имеет собственное место
связывания с цГМФ. Другая форма является мономером, связанным
77
о
я
н
а
=;
s
X
cd
и
О
а
я
о-
о
CQ
Н
о
cd
О-
«
s
cd
CQ
О
о.
S
CQ
о.
78
с цитоплазматической мембраной. Она встречается только в клетках
кишечного эпителия.
О Мр
N
^^ сн3
(4.6)
Точное установление механизма передачи сигнала важно не
только для понимания причин возникновения тех или иных нарушений,
приводящих к развитию патологических процессов, но и для
определения возможных мест приложения фармакотерапевтических средств.
Так, вазорелаксация, происходящая под воздействием цГМФ, может
быть нарушена в результате гидролиза цГМФ в ГМФ под
действием фосфодиэстеразы-5 (ФДЭ-5). Применение ингибитора ФДЭ,
например, силденафила (виагра, 4.6), способствует увеличению
концентрации цГМФ у лиц со сниженной продукцией эндогенного N0,
и тем самым положительно сказывается на вазорелаксации и.на
мужской эрекции.
4.3. Продукты метаболизма фосфолипидов
как вторичные посредники
Еще один широко распространенный механизм передачи
сигнала основан на превращениях фосфатидининозитина, входящего
в состав клеточных мембран. Процесс схематически представлен на
рис. 4.4.
Первые два этапа аналогичны таковым для процесса
активирования АС. А далее сс-субъединица Оч-протеина, связанная с ГТФ,
активирует фосфолипазу С (фосфодиэстеразу, PL-C), что, в свою очередь,
приводит к гидролизу фосфатидининозитина (PI) на диацилглицерин
(DG) и инозитинтрифосфат (IP3). DG остается внутри мембраны и
промотирует протеинкиназу С (РК-С). Активирование этого фермента
может идти и прямым путем, без участия PI и DG, например, под
действием форболового эфира (4.7), являющегося стимулятором
роста опухолей [7, 8, 18, 19].
79
Прямое активирование
Рис. 4.4. Фосфатидининозитиновая система передачи сигнала
от рецептора в клетку [5]:
PL-C — фосфолипаза С, РК-С — протеинкиназа С, PI — фосфатидининозитин,
DG — диацилглицерин, СМ — кальмодулин
1Р3 диффундирует из мембраны в цитоплазму и способствует
высвобождению ионов Са2+. Последний прямо или в комбинации с каль-
модулином (СМ-Са2+) активирует протеинкиназу. Эта протеинкиназа,
также как и РК-С, фосфорилирует белки, инициируя тем самым
физиологический ответ на результат связывания агониста с рецептором.
К числу агонистов, активирующих PL-C, принадлежат ацетилхолин
(мускариновый), 5-гидрокситриптамин (5~НТ2), осгадренергические
агонисты, гистамин (Hj), аденозин (\), вазопрессин (Vj), брадики-
нин, ангиотензин II и некоторые экзогенные БАВ.
СН °
OHj? 3 п-й^^(СюН22\
но-н2с
(4.7)
Выявление причин активирования РК-С опухолевым
промотором (4.7), вероятно, способно пролить свет на один из возможных
механизмов онкогенеза, т.к. комбинированное воздействие форболо-
вого эфира и Са2+-ионофоров значительно ускоряет синтез белков
и приводит к неконтролируемому росту клеток [5, 20].
4.4. Ионы Са2+ как вторичные посредники
Ионы Са2+ являются важнейшим регулятором клеточного
метаболизма. Доказано, что деполяризация мембран частично связана с
транспортом значительных количеств ионов Са2+в клетку. Проникновение
80
секреция инсулина
Рис. 4.5. Схема передачи сигналов в р-клетках поджелудочной железы,
регулирующая секрецию инсулина:
G1 — глюкоза, GLUT-2 — транспортер глюкозы, G1-P — фосфорилированная
глюкоза, СМ — кальмодулин, РК-С — кальмодулинзависимая киназа,
SU — сульфонилмочевина, К+АТФ — АТФ-зависимый К+-канал,
VCa2+—вольтаж-зависимый Са2+-канал, К+Са2+ — кальций-зависимый К+-канал
Са2+ в цитоплазму осуществляется через вольтаж-зависимые ионные
каналы, а также из ЭР. Концентрация Са2+ в цитоплазме находится в
пределах 0.1—0.01 мкМ, в то время как в межклеточном пространстве
она достигает 1 мМ. Многие процессы, такие как сокращение
мускулатуры и др., инициируются, когда концентрация Са2+ в клетке
достигает 1 мкМ [1, 2, 5, 6].
Ионы Са2+ и многочисленные ассоциированные с ним
активирующие белки, из которых самый известный — кальмодулин, образуют
еще одну систему ВП [1, 5, 6, 21, 22]. Са2+в различных тканях, кроме
скелетных мышц и миокарда, связывается с кальмодулином. Этот белок
является мономерным глобулином, содержащим до 35 % аспартата и
глутамата, которые эффективно удерживают ионы Са2+. Кальмодулин
участвует в регуляции активности многих ферментов, таких как
аденилатциклазы, фосфодиэстеразы, триптофангидроксилазы, проста-
гландин 15-гидроксидегидрогеназы и др. Этот белок также оказывает
влияние на выход нейротрансмиттеров, эндо- и экзоцитоз, синтез ДНК,
а-адренергическую функцию, активацию конститутивной формы
NO-синтетазы и т.п. Существует большое количество препаратов,
связывающихся с кальмодулином (нейролептики, местные анестетики,
Са2+-антагонисты, противоопухолевые средства). Однако их
специфичность довольно низка, в связи с чем целесообразность применения
некоторых из них в настоящее время находится под вопросом [5, 6, 22].
Роль ионов Са2+в регуляции внутриклеточных процессов, а также
структура ионных каналов весьма показательно проиллюстрированы
на примере функционирования АТФ-зависимого К+-канала,
ответственного за секрецию инсулина из панкреатических Р-клеток.
Как отмечено в разд. 3.2, ионный канал представляет собой
просвет в цитоплазматической мембране, образованный пятисегмент-
ным протеином. Несколько сегментов могут быть связаны с аксо-
81
нами нервных клеток. Диаметр ионного канала соответствует
размеру иона без гидратной оболочки. Различают Na+-, K+-, Са2+-ка-
налы. Для распознавания катионов и удаления гидратной
оболочки на входе в канал существует «фильтр» в виде ионизированных
карбоксильных групп.
Панкреатические р-клетки содержат три вида каналов: два К+
и один Са2+ (рис. 4.5). Они функционируют следующим образом:
проникновение глюкозы в клетку и ее фосфорилирование
приводит к росту внутриклеточной концентрации АТФ, что инициирует
закрытие АТФ-чувствительного К+-канала и деполяризацию
клетки; в результате открывается вольтаж-зависимый Са2+-канал. Далее
ионы Са2+ связываются внутри клетки с кальмодулином, который
активирует РК-С и запускает механизм высвобождения инсулина
из везикул. Рост концентрации ионов Са2+ в цитоплазме приводит
к активированию К+-канала второго типа — Са2+-зависимого,
который функционирует как калиевый насос. Происходит
восстановление потенциала клетки, закрывается вольтаж-зависимый Са2+-ка-
нал, открывается К+АТФ-канал и, следовательно, прекращается
секреция инсулина [23—25].
Подобно глюкозе, однако независимо от уровня ее концентрации
в крови, действуют инсулиновые секретогены — производные суль-
фонилмочевины (СМ, табл. 4.1). Они связываются с
высокоаффинным рецептором, расположенным на внешней стороне цитоплазма-
тической мембраны рядом с К+АТФ-каналом. В результате такого
взаимодействия происходит конформационная перестройка белков,
образующих ионный канал, и он закрывается [25].
Из рис. 4.6 следует, что наибольшим сродством к рецептору
обладает глибенкламид (ГБ), наименьшим — толбутамид (ТБ) и карбута-
мид (КБ) [26]. В результате ГБ присуща наиболее выраженная
инсулиносекреторная активность.
-'дкд
4
6
8
10
- ig Ко.5
Рис.4.6. Зависимость активности препаратов ряда СМ
от прочности связывания с рецептором [27, 28]
Приведенные в табл. 4.1. препараты используются в медицинской
практике какгипогликемические средства для лечения
инсулин-независимого сахарного диабета (ИНЗСД). Самым новым из них является
82
Таблица 4.1
Антидиабетические препараты — агонисты рецепторов,
сопряженных с К+АТФ-каналами ^-клеток поджелудочной железы
Ях/Я jj
R1—S—N—я— N— R2
Н Н
R1
^-О-
Нзс4~)~
с|Ч^-
Нзс41)~
ОМе
~оч£
о
Карбутамид (КБ)
(4.8)
Толбутамид (ТБ)
(4.9)
Хлорпропамид
(4.10)
Гликлазид (ГК)
(4.11)
Глибенкламид (ГБ)
(4.12)
Глипизид (ГП)
(4.13)
Глимепирид (ГПр)
(4.14)
R2
-(СН2)з-СН3
-(СН2)з-СН3
-(СН2)2-СН3
N
>
Ч">
Ч">
-Д Н У~^г
глимепирид (ГПр). Он вошел в арсенал антидиабетических средств в
середине 90-х гг. ГПр образует с рецептором менее прочный
комплекс, чем ГБ и, следовательно, несколько уступает ему по выражен-
83
ности сахароснижающего эффекта. Однако этот препарат имеет и
существенные преимущества перед ГБ [28]. Дело в том, что К+АТФ-ка-
налы не являются исключительной принадлежностью Р-клеток
поджелудочной железы, а присутствуют также и в кардиомиоцитах,
клетках мозга, почек, гладкой мускулатуры, кровеносных сосудов. И эти
клетки, следовательно, тоже «откликаются» на действие СМ, но в
значительно меньшей степени. Взаимодействием с АТФ-зависимыми
К+-каналами в кардиомиоцитах и клетках сосудов ряд
исследователей объясняют негативные гемодинамические эффекты СМ [29, 30].
ГПр не взаимодействует с К+АТФ-каналами сердечно-сосудистой
системы (ССС). Поэтому он может применяться в течение длительного
времени без риска развития побочных эффектов со стороны органов
указанной системы [26].
Подводя итог рассмотрению систем передачи сигналов от
рецепторов в клетку, следует подчеркнуть, что они в большинстве
случаев универсальны, т. к. одни и те же ВП участвуют в передаче
сигналов от разных рецепторов. Связывая такой ВП в одном
месте, можно нарушить процессы передачи сигналов от других
рецепторов. Поэтому ВП являются наименее удачными мишенями при
конструировании лекарств. Более продуктивным является подбор
высокоаффинных лигандов к рецепторам или поиск ингибиторов
(активаторов) ферментов.
Литература
1. Альберте Б., Брей Д., Льюис Дэн:., Роберте К, Уотсон Дж. Молекулярная
биология клетки. — М.: Мир, 1994.
2. Сергеев П.В., Шимановский Н.Л. Рецепторы физиологически активных
веществ. — М.: Медицина, 1987.
3. Варфоломеев С.Д., Гуревич К.Г. Биокинетика: Практический курс. — М.:
ФАИР-ПРЕСС, 1999.
4. Сергеев Л.В., Галенко-Ярошевский Л.А., Шимановский Н.Л. Очерки
биохимической фармакологии. — М.: Фармединфо, 1996.
5. Nogrady Т. Medicinal Chemistry. A Biochemical Approach. — New York, Oxford:
Oxford University Press, 1988.
6. Medicinal Chemistry: Principles and Practice.// Ed. by F.D.King. — Cambridge,
the Royal Society of Chemistry, 1994.
7. Hokin L.E. // Annu. Rev. Biochem. - 1985. - Vol. 54. - P. 205.
8. Berridge MJ.// Sci. Am. - 1985. - Vol. 253, N 4. - P. 142.
9. Garrison J.C. //Annu. Rep. Med. Chem. — 1985. — Vol. 20. - P. 227.
10. Pfeuffer E.} Dreher R.M., Metzger H. et al // Proc. Natl. Acad. Sci. USA. —
1985. - Vol. 82. P. 3086.
11. SomlyoA.P, Somlyo A. V.// Acta Physiol. Scand. - 1998. -Vol. 64. - P. 437.
12. Klabunde T. Hessler G. // Chem. Bio. Chem. - 2002. - Vol. 3. - P. 928.
13. Seamon K.B. // Annu. Rep. Med. Chem. - 1984. - Vol. 19. — P. 293.
14. Cahn D.J., Melman A., Valcic. M. et al. / J.Urol. - 1996. - Vol. 155. - P. 1789.
15. Murad F. // J.Clin. Invest. - 1986. - Vol.78. - P. 1.
16. Salvemini D., Mollace V. // Drugs News Persp. - 1994. — Vol. 7. - P. 158.
17. Rosen M.G., Pei Tsai, SovitjPou// Chem. Rev. - 2002. - Vol. 102. - P. 1191.
18. Nishizuka Y./f Science. - 1986. - Vol. 233. - P. 305.
84
19. Majerus P.W., Conolly T.M., Deckmyn H. et al. // Science — 1986 —
Vol. 234. - P. 1519.
20. Parker P.J., Coussens L., Totty N. et al. // Science. — 1986. — Vol. 233. — P. 853.
21. Klee СВ., Crouch Т.Н., Richman P.G.// Ann. Rev. Biochem — 1980 —
Vol. 49. - P. 489.
22. West L.W./1 Fed. Proc. — 1982. - Vol. 41. — P. 2251.
23. Cook d.L., Hales C.N.// Nature. - 1984. - Vol. 311. - P. 271.
24. BoydA.E., AguilarB.L., Nelson D.A.// Am. J. Med. — 1990. — Vol 82(2A) —
P. 162.
25. Dorschner H.} Brekardin E., Uhde I. et al. // Mol. Pharmacol. — 1999. —
Vol. 55. - P. 1060.
26. Bijlstra P.J.// Diabetologia. — 1996. - Vol.39. - P. 1083.
27. Schmid-Antomarchi H. et al. // J. Biol. Chem. — 1987. — N 262. — P. 15840.
28. Scientific monograph Amaryl. — Frankfurt am Main: Hoechst Marion Roussel
1997.
29. Ни S., Wang S., Dunning B. // J Pharm. Exp. Ther. - 1999. - Vol. 291,
N 3. - P. 1372.
30. Jonas J.C, Garcia-Banado M.J., Angel I. et. al. // Eur. J.Pharmacol. — 1994 —
Vol. 264. - P. 81.
ФЕРМЕНТЫ
(ПРИНЦИПЫ ДЕЙСТВИЯ И РЕГУЛЯЦИЯ АКТИВНОСТИ).
ИНГИБИТОРЫ ФЕРМЕНТОВ В СОВРЕМЕННОМ АРСЕНАЛЕ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
Поиск регуляторов (главным образом, ингибиторов) активности
ферментов представляет на сегодняшний день одно из
магистральных направлений в конструировании лекарств. Примерно одна треть
современных препаратов обладает именно этим механизмом действия.
Ингибируя ферментно-катализируемые реакции, можно селективно
модулировать разнообразные биохимические и физиологические
процессы, а также прерывать главные метаболические пути, блокируя
образование ключевых или нежелательных метаболитов. Ингибиро-
вание ферментов комплементарно воздействию антагонистов на
рецепторы и в некоторых случаях используется для потенцирования
активности определенных разновидностей энзимов. Так,
манифестация биологического эффекта БАВ «X» (рис. 5.1) может быть
предотвращена посредством блокирования фермента, участвующего в его
биосинтезе. Такой же эффект достигается и при участии антагониста
рецептора R.
^^
qLt]
-*- Ответ
Рис. 5.1. Управление эффектом БАВ «X»:
субстрат, Е — фермент, I — ингибитор, R — рецептор, А — антагонист
Кроме того, связывание фермента, принимающего участие в
каскаде превращений при передаче сигнала от рецептора в клетку
(см. разд. 4.1), приводит к аналогичному результату — блокированию
ответа.
Однако, прежде чем рассматривать стратегию поиска
ингибиторов ферментов (ИФ), стоит хотя бы кратко коснуться особенностей
строения самих ферментов, их функций, причин специфичности и
необычайно высокой каталитической активности.
Ферменты (Ф) — протеины, содержащие хиральные
распознающие участки для специфических субстратов, и, в силу этого, являю-
86
щиеся высокоэффективными биокатализаторами, ускоряющими
химические реакции в 70ш—7016 раз. Слово «фермент» — производное от
латинского fermentum, т. е. «закваска», а часто употребляемый
синоним «энзим» произошел от греческого «эн зюме», что означает
«в дрожжах».
Энзимы могут быть однокомпонентными (простые белки),
двухкомпонентными и многокомпонентными; большинство из них —
глобулярные белки.
В составе Ф есть активный центр — особый участок в
молекуле белка, с которым связывается субстрат с образованием
фермент-субстратного комплекса (Е—S). В полипептидной
последовательности аминокислотные остатки, составляющие активный центр,
могут находиться далеко друг от друга, однако благодаря
специфичной для каждой белковой молекулы упаковке, эти фрагменты
пространственно сближены. Формирование комплекса Е—S
осуществляется, как правило, без ковалентного связывания, за счет
водородных связей, ион-дипольного, диполь-дипольного или ван-
дер-ваальсового взаимодействия. Однако известны случаи, когда
имеет место и образование ко валентных связей, в частности, при
функционировании Ф, принадлежащих к семейству сериновых иро-
теаз [1—4].
Во многих случаях в непосредственной близости к активному
центру Ф находится небелковый компонент. Последний получил
название кофермента (коэнзима) или кофактора, а белковая часть — апо-
фермента (ароЕ). В качестве коферментов могут выступать как
органические молекулы (главным образом витамины или их
производные), так и ионы Са2+, Zn2+, Mn2+, Se2+, Cu-f, Fe2f и др. В ряде
случаев кофакторы образуют с Ф прочные связи. При этом
небелковую часть Ф принято называть простетической группой. Далее по ходу
описания отдельных классов Ф будет дана характеристика и
важнейшим кофакторам.
В настоящее время зарегистрировано около 106 Ф, у
млекопитающих их примерно 2400. Согласно систематике,
предложенной Международным союзом биохимии (International Union of
Biochemistry, ШВ), Ф классифицируются не как индивидуальные
химические соединения, а по типу катализируемых п\ш реакции
Выделяют шесть классов Ф, каждый из которых подрайоне ;сн па
подклассы и более мелкие группы (подподклассы и отдельные Ф).
Для их обозначения используют четырехзначный код. В нем
первое число указывает на класс (1—6), второе — на подкласс (в
каждом классе разное число подклассов), третье число — подподкласс.
а четвертое — положение конкретного Ф в данном полподклассе.
Например, упомянутая в разд. 4.2 NO-синтетаза, согласно Enzyme
Classification (EC) имеет код (ЕС 1.14.13.39). Если механизм
действия Ф недостаточно изучен, его относят к общему подподклассу
99. Такая система позволяет классифицировать новые Ф, не
нарушая нумерации уже известных. Детально строение энзимов и их
функции рассмотрены в специальной ;:г.; .пуре fl Ь\,
87
5-1. Классы ферментов
5.1.1. Оксидоредуктазы
К первому классу относятся оксидоредуктазы или окислительно-
восстановительные Ф. Их систематическое название складывается из
названия восстановителя (электронодонора), окислителя (электроно-
акцептора) и названия класса. Например, Ф, катализирующий
окисление этанола до ацетальдегида при участии NAD+ (5.1), носит
название — алкоголь: НМ)+-оксидоредукгаза. В первом классе имеется
15 подклассов. Ф первого подкласса 1.1 катализируют окисление
ОН-групп до карбонильных, второго 1.2 — окисление карбонильных
до карбоксильных, третьего 1.3 — превращение >СН-СН<
фрагментов до >С=С<, четвертого 1.4 — окисление первичных NH2-rpynn до
карбонильных и иона NH4+ и т. д.
Кроме номенклатурных допускается использование и
упрощенных названий. Все Ф, содержащие NAD+ или NADP+ (5.2) в качестве
окислителя принято называть дегидрогеназами. Упомянутая выше
алкоголь: КАЕ>+-оксидоредуктаза носит также название алкоголь де-
гидрогеназа. Этот Ф относится к подподклассу 1.1.1 и является
первым в нем, о чем свидетельствует код данного Ф — (ЕС 1.1.1.1).
, R2 = н NAD+
CONH2
о (5.1,
о—р—о
*о—р—о
II
о
R1 ^v3 ' R2 = Р°з2 NAD р+
ОН OH/N-^N CONH^
о^ \. J J (5.2)
R1=~\=) ' R2=H NADH
CONH2
(5.3)
Ri = _r/~\ , R2 =. po32- NAD PH
CONH2
(5.4)
Известно более 200 биохимических реакций окисления
спиртовых групп до карбонильных и несколько десятков превращений
карбонильных групп в карбоксильные, протекающих при участии нико-
тинамидных коферментов. Большинство из этих процессов обратимы.
Однако, как правило, биологически значимым является только одно
направление. При этом имеет место следующая закономерность: если
88
метаболический процесс протекает в направлении окисления субстрата,
то в качестве окислителя выступает NAD+. Если же реакция подпод-
класса связана с восстановлением органического соединения, то чаще
всего восстановителем является NADPH (5.3).
Принимая за основу при классификации Ф 1.1 подкласса природу
окислителя, к подподклассу 2 относят энзимы, катализирующие
окисление цитохромом С (CYC), к подподклассу 3 — катализирующие
окисление молекулярным кислородом, к подподклассу 4 —
дисульфидами. Например, глюкозооксидаза, катализирующая окисление
молекулярным кислородом полуацетальной ОН-группы p-D-глюкозыдо
карбонильной группы, относится к первому подклассу и третьему
подподклассу:
НО—| НО—1
J—оон 0 J—Q
(& -^ ^>о+н2о2
он I он I
он он
Порядковый номер этого Ф — (ЕС 1.1.3.4). В качестве кофактора
глюкозооксидаза содержит флавинадениндинуклеотид (FAD, 5.5),
синтетическим предшественником которого является рибофлавин
(витамин В2).
н3с
о о
II II
сн2(снон)3-снр-р—о-р-о
Ade
ОН ОН
+2е, +2Н+
Окисленная форма
II
II
CH2(CHOH)3-CHp-P- О-Р-О
Ade
ОН ОН
Восстановленная форма
Ade - аденин
Флавиновые и гем-кофакторы входят в состав Ф,
катализирующих процессы окисления с участием Н202 и 02. Число реакций, в
которых в качестве окислителя выступает Н202, невелико. Ф,
участвующие в этих превращениях, составляют 11-й подкласс. В нем
89
представлены каталаза (ЕС 1.11.1.6) и пероксидаза (ЕС 1.11.1.7). Оба
Ф яачяются гемопротеидами.
Энзимы, катализирующие превращения, в которых окислителем
выступает молекулярный кислород, подразделяются на три группы:
оксидазы, монооксигеназы и диоксигеназы. Оксигеназы участвуют в
реакциях, в результате которых 02 восстанавливается до Н202 или
Н20. Примерами могут служить выше упомянутая глюкозооксидаза и
CYC-оксидаза. Последняя катализирует окисление ферроцитохрома
С до феррицитохрома:
4CYC(Fe2+) + 4IT +02 >4CYC(Fe3+) +2Н20
Монооксигеназы участвуют в процессах, в результате которых
один из атомов кислорода входит в состав субстрата, а второй
восстанавливается до Н20. Для подобного рода превращений
характерно участие двух доноров, один из которых является донором
электронов (чаще всего это NADPH), а второй — донором
протонов. Природа второго донора может быть различной. Суммарное
уравнение имеет вид:
RH + NADPH + Н+ + 02 > ROH + NADP+ + Н20
К монооксигеназам относится чрезвычайно важная группа
Ф—CYP450 (см. разд. 2.5 и 8.1). Эти энзимы являются одновременно
гемо- и флавопротеидами, причем флавиновые нуклеотиды
выступают в этом случае в качестве доноров протонов. Восстановление
связанного с белком флавинового фрагмента в CYP450 катализирует
NADPH: цитохром Р450-редуктаза и тем самым обеспечивает
цикличность работы этого семейства Ф.
Название CYP450 получили благодаря тому, что комплекс
оксида углерода СО с восстановленной формой гема в составе этих
Ф имеет максимум поглощения в видимой области спектра при
450 нм. Семейство CYP450 играет чрезвычайно важную роль в
метаболизме БАВ и других экзогенных органических веществ,
попадающих в организм извне и неспособных в нем ассимилировать
(см. разд. 8).
Диоксигеназы принимают участие в процессах, в результате
которых оба атома молекулы кислорода включаются в состав окисляемого
соединения, Например, окисление триптофана (5.6) до формилкин-
уренина (5.7). Это превращение катализирует триптофан 2,3-диокси-
геназа(ЕС 1.13.11.11).
О О NH3
^^ N 3 \"^ NH-CHO
н
(5.6) (5.7)
90
Чрезвычайно важная роль в защите организма на молекулярном и
клеточном уровнях от повреждающего действия сильного
окислителя — радикала Н02- принадлежит супероксиддисмутазе (ЕС 1.15.1.1).
Этот Ф является металлопротеидом и может содержать в качестве
кофактора ионы Zn2+, Mn2+, Cu2+, Fe2+. Реакция, катализируемая су-
пероксиддисмутазой, имеет следующий вид:
но2 + но
.н2о2+о2
5.1.2. Трансферазы
Трансферазы, катализирующие перенос различных групп с одной
молекулы на другую, образуют второй класс Ф. Эти превращения
можно представить следующим образом:
АВ + Х^
=±а + вх(ав, х*н2о, он )
Название Ф этого класса по систематической номенклатуре
составляется согласно схеме — АВ:Х В-трансфераза, например, серии
тетрагидрофолат гидроксиметилтрансфераза (ЕС 2.1.2.1). Разделение
на подклассы определяется природой переносимых групп.
К первому подклассу 2.1 относят энзимы, катализирующие
реакции переноса одноуглеродных остатков. Такого рода превращения
играют чрезвычайно важную роль в синтезе пиримидиновых и пури-
новых оснований. В качестве коферментов, переносящих одно-
углеродные фрагменты, в этих реакциях выступают тетрагидрофолат
(THF, 5.8) и S-аденозилметионин (5.9).
СОО
CONH-
СОО
У
(5.8)
сн.
>s
(СН2)/
V
NH
Ade
з ОН ОН
СОО
(5-9)
Присоединение к THF метильных и формальных групп может
происходить как по атому N(5), так и N(10) или приводить к
образованию одноуглеродного мостика между ними.
91
^НГ ^ II
^ 1\ н ^
^ 61 ti N \5
6
N ^ ю- Г
'и С N—
N(5)-CH,-THF N(5), N(10)-CH2-THF N(10)-HCO-THF
w
N(5), N(10)-HCO-THF N(5)-CHNH-THF
Перенос одноуглеродного фрагмента к THF — результат
взаимодействия последнего с серином. Этот процесс катализирует
упомянутая выше серии гидроксиметилтрансфераза (ЕС 2.1.2.1).
НО-СН2-СН-СОО~ +THF( ~>N(5), N(10)-CH2 -THF + +NH3CH2COO"+H20
I
+NH3
Дальнейшее превращение метиленового фрагмента в метиновый
катализирует метилен-THF дегидрогеназа (ЕС 1.5.1.5).
N(5), N(10)-CH2-THF + NADP*4==*N+(5), N(10)-CH - THF + NADPH
Восстановление метиленовой фуппы до N(5)-MeTnrc-THF
протекает при участии метилен-THF редуктазы (ЕС 1.5.1.20).
N(5), N(I0)-CH2-THF+NADP.H4=^=^N(5)-THF+NADP+
I
сн3
Формильное и метановое производные участвуют в каскаде
превращений, приводящем к образованию пуриновых оснований.
N(5)-Menui-THF алкилирует гомоцистеин, превращая его в метионин:
SCH
SH I
(СН2)2 + N(5)—THF »-((fH2>2+ + THF
I + I HC—NH-
НС—NH3 CH3 | 3
' - COO
COO
(5.10) (5.11)
Образовавшийся метионин далее используется в синтезе
протеинов и S-аденозилметионина (5.9):
(5.11) + АТФ—2^2_^(5.9) + РРР
92
Последний является главным метилирующим агентом в
клетках и участвует в десятках реакций, катализируемых метил-
трансферазами.
Энзимы, катализирующие перенос альдегидных и кетогрупп, образуют
подкласс 2.2. В нем представлены транскетолаза (ЕС 2.2.1.1) и транс-
альдолаза (ЕС 2.2.1.2), участвующие в синтезе моно- и полисахаридов.
Подкласс 2.3 ацетилтрансфераз объединяет Ф, которые
катализируют такие важные биохимические процессы как перенос ацильных
остатков. Коферментом, принимающим участие в большинстве
реакций энзиматического ацилирования, является так называемый
кофермент А (СоА, 5.12).
сн.
SH(CH2) — NHCO(CH2)2NHCO СН
ОН
о.
сн,—о—р—о—р—о
с к
о
о
2-
.Ade
03РО
ОН
(5.12)
Ацильные остатки связываются с СоАтиоэфирной связью,
поэтому указанный кофермент иногда обозначают и как CoA-SH. Перенос
ацильного остатка к CoA-SH осуществляется при окислительном де-
карбоксилировании а-кетокислот. Далее образовавшийся CoA-SCOR
ацилирует большое количество органических субстратов, например,
фосфат глицерина. Ацилпроизводные последнего являются
промежуточными продуктами в биосинтезе фосфолипидов и жиров:
СН0ОН
СНОН + CoA-SCOR
СН2ОР032"
CH.OCOR
I 2
СНОН + CoA-SH
сн2оро32"
CH-OCOR
I 2
СНОН
сн2оро;
+ CoA-SOR1
CH9OCOR
I
CHOCOR + COA-SH
ch2opo3
Эти реакции катализируют глицерофосфат ацилтрансфераза
(ЕС 2.3.1.15) и 1-глицерофосфат ацилтрансфераза (ЕС 2.3.1.51). Роль
СоА не ограничивается переносом ацильных групп. Многие
превращения производных жирных кислот, такие как их окислительная
деструкция, происходят не со свободными кислотами, а с их тиоэфира-
ми на основе СоА.
В четвертый подкласс 2.4 выделены гликозилтрансферазы,
катализирующие перенос гликозильных остатков. Эти Ф играют
ведущую роль в синтезе и первом этапе деструкции полисахаридов.
Очень важной группой Ф являются аминотрансферазы или
трансаминазы (2.6,1). Они катализируют реакции образования и
деградации аминокислот. Например, Ф аланинаминотрансфераза уча-
93
ствует в превращении пировиноградной кислоты (пирувата, 5.13)
в а-кетоглутаровую (а-кетоглутарат, 5.14):
СН,-СО -СОО" + ~ООССНХН,СНСОО~^=*~СН3-СН -СОО" + "ООССН2СН2СОСОО"
NH3 NH3
(5.13) (5.14)
Кофактором аминотрансфераз выступает пиридоксальфосфат
(производное витамина В6, 5.15).
сн2оро3
(5.15)
Пиридоксальфосфат образует основание Шиффа (5.16) с е-амино-
группой одного из остатков лизина (ароЕ). В присутствии
специфичной для данного Ф аминокислоты происходит вытеснение пири-
доксальфосфата с остатка лизина и образование нового основания
Шиффа (5.17), за которым следует изомеризация в азометиновое
производное пиридоксаминофосфата и ожетокислоты (5.18). Гидролиз
этого продукта сопровождается выделением а-кетокислоты и
формированием комплекса ароЕ-пиридоксаминофосфат (5.19).
H3C4/N. . NH ♦(сна)4-аро Е
NH3 НО^У^СНрРОз
\
"СН2ОРС^
N-(CH2)4-apoE R1-LC00-
(5.16) (5.17)
NHj^CH^ - ароЕ H3CYN^ -NH3 (CH^ - ароЕ
I 3 4 + Ri-COCOO
&- S^CHpPO^
4lH.+
(5.18) (5.19)
Присутствие в системе второй кетокислоты обеспечивает
продолжение описанного каскада превращений. Все реакции в этой цепи
являются обратимыми, и протекание их в обратном направлении
приводит к переаминированию с регенерацией азометина (5.16).
Один из самых больших подклассов трансфераз — 7-ой. К нему
относят Ф, катализирующие перенос остатков фосфорной кислоты.
94
В большинстве случаев донором фосфата является АТФ. Такие
энзимы принято называть киназами. Примером может служить аденил-
аткиназа (ЕС 2.7.4.3), катализирующая превращение АМФ в АДФ.
5.1.3. Гидролазы
Третий класс образуют гидролазы — Ф, катализирующие процессы
гидролитического расщепления различных связей. Энзимы,
обеспечивающие гидролиз эфиров карбоновых кислот, образуют первый
подкласс 3.1, гидролиз гликозидных связей — подкласс 3.2, гидролиз
простых эфиров и тиоэфиров — подкласс 3.3, гидролиз пептидных
связей — подкласс 3.4, гидролиз связей C-N, не относящихся к
пептидным, — подкласс 3.5 и т.д. Названия Ф образуются из названия
расщепляемого субстрата с добавлением окончания -аза. Ф, гидроли-
зующие внутренние пептидные связи в белках, называют протеазами,
а катализирующие отщепление N-концевой аминокислоты — амино-
пептидазами, отщепление С-концевой аминокислоты — карбокси-
пептидазами, гидролиз нуклеиновых кислот — нуклеазами.
Значительная часть гидролаз представлена пищеварительными ферментами
(амилазы, липазы, фосфолипазы, пепсин и т. п.), с помощью которых
поступающие с пищей полисахариды, жиры и протеины расщепляются
соответственно до моносахаридов, глицерина и жирных кислот,
пептидов и аминокислот. Последующая реутилизация образовавшихся
мономеров в ходе анаболических процессов, протекающих согласно
наследственным программам, приводит к построению новых биополимеров,
присущих данному организму.
Реакции гидролиза происходят не только в ПК, но и в таких
органеллах эукариотических клеток как лизосомы (см. разд. 1). Про-
теазы, располагающиеся в лизосомах, утилизируют макромолекулы,
которые выполнили свою биологическую функцию и становятся
ненужными на данном этапе жизнедеятельности клетки. Лизосомные
протеазы не способны проникать в цитоплазму через мембрану,
окружающую эти органеллы, к тому же они активны в слабокислой
среде и неактивны в нейтральной, характерной для цитоплазмы.
Таким образом, биополимеры остальных частей клетки защищены от
деструкции протеолитическими Ф лизосом.
Кроме утилизирующей роли следует подчеркнуть и такую
функцию протеаз, как процессинг белков и РНК. Процессингом называют
образование «зрелых» молекул белков из их синтетических
предшественников. Многие протеины, которые реализуют свою
активность вне пределов синтезирующей их клетки (некоторые Ф,
гормоны, иммуноглобулины), секретируются этими клетками в виде
пробелков. Предшественники Ф носят название зимогенов.
Последние имеют на N-конце первичной полипептидной цепи специальную
сигнальную последовательность, состоящую из 15—20 аминокислотных
остатков. После отщепления этой сигнальной последовательности с
помощью мембранных протеаз белок активируется не сразу, а по
достижении им определенной мишени. Например, синтетический
95
предшественник инсулина, секретируемый 0-клетками
поджелудочной железы в виде препроинсулина, после отщепления сигнальной
последовательности превращается в проинсулин. Проинсулин
самопроизвольно принимает пространственную структуру,
обеспечивающую сближение остатков цистеина и образование дисульфидных
мостиков. После этого из полипептидной цепи выделяется С-пептид,
и из проинсулина образуется инсулин.
Системы протеаз обеспечивают течение таких важных
физиологических процессов, как свертывание крови, регуляция АД,
расщепление чужеродных антигенов после их связывания с антителом
и др. Нарушение нормального функционирования какого-либо из
перечисленных процессов влечет за собой развитие серьезных
патологических состояний. В этой связи многие протеазы
являются мишенями для БАВ.
5.1.4. Лиазы
К четвертому классу Ф относятся лиазы. Они катализируют
процессы негидролитического расщепления субстрата с образованием
кратной связи (реже цикла), либо присоединения по такой связи.
Рассматриваемые Ф классифицируются по типу разрываемой связи:
Подкласс 4.1 образуют С-С-лиазы, подкласс 4.2 — С-О-лиазы,
подкласс 4.3 — C-N-лиазы и т. д. Систематическое название
складывается из названий расщепляемого субстрата, отщепляемого фрагмента
и названия класса — лиаза.
Ф подподкласса 4.1.1 катализируют отщепление С02 от карбоно-
вых кислот, поэтому они получили групповое название декарбокси-
лазы. Например, образующаяся при спиртовом брожении глюкозы
пировиноградная кислота декарбоксилируется под действием пиру-
ват декарбоксилазы с образованием уксусного альдегида:
сн3сосоо" —±i£—> сн3сно + со2
Кофактором этого Ф выступает тиаминпирофосфат (5.20).
О"
(5.20)
Ферментативным путем происходит декарбоксилирование ряда
аминокислот с образованием важных нейромедиаторов. Так, L-глу-
таминовая кислота под действием глутамат декарбоксилазы пре-
96
вращается в ГАМК. Другой Ф этой же группы — декарбоксила-
за ароматических аминокислот— катализирует декарбоксилирование
/-(3,4-диоксифенил)-аланина (L-ДОФА), превращая последний в нор-
адреналин. В большинстве случаев коферментом декарбоксилаз
аминокислот является описанный ранее пиридоксальмонофосфат (5.15).
Выделение С02 происходит через предварительное образование
оснований Шиффа так же, как это имеет место в протекающих при
участии трансфераз реакциях переаминирования. Направление
процесса — выделение С02 или же переаминирование — определяется
природой белка, т.е. ароЕ в структуре (5.16). Этот факт
свидетельствует о том, что именно ароЕ управляет работой кофермента.
Примером реакции, протекающей при участии лиазы и
приводящей к образованию цикла, является превращение АТФ в цАМФ (4.10).
Ade
*~ РР + (4.1)
ОН ОН
Процесс имеет место при передаче сигнала от рецептора в клетку
в результате образования Л-Р комплекса (см. разд. 4.1).
Катализатором в нем выступает АС (АТФ-пирофосфатлиаза циклизующая).
5.1.5. Изомеразы
Изомеразы, катализирующие процессы изомеризации, образуют
пятый класс Ф. Подкласс 5.1 составляют Ф, с помощью которых
происходит обращение конфигурации при асимметрическом атоме
углерода. Энзим катализирует обращение конфигурации в обоих
направлениях, что приводит к образованию рацемической смеси. Такие Ф
получили название рацемаз.
При наличии в молекуле нескольких хиральных центров обращение
конфигурации при одном из них приводит к образованию диастерео-
мерной пары. Ф, катализирующие такие процессы, называют эпиме-
разами. Они играют важную роль во взаимопревращениях углеводов.
Так, обратимое превращение рибулозо-5-фосфата в ксилулозо-
5-фосфат катализирует 0-рибулозо-5-фосфат-3-эпимераза:
СН2ОН СН2ОН
(=0 Р=0
снон ~*—*~ носн
I I
СНОН СНОН
CH2OPOf CH2OPOf
Подкласс 5.2 образуют Ф, катализирующие
цис-транс-изомеризацию. К подклассу 5.3 относятся энзимы, участвующие во
внутримолекулярных окислительно-восстановительных процессах. Например,
глюкозо-6-фосфат изомераза катализирует взаимопревращение глю-
rrr
К?
97
козо-6-фосфата и фруктозо-6-фосфата, что является одним из этапов
анаэробной деструкции глюкозы, гликогена и крахмала.
203Р-0—|
£^» ^ \у\он
он он
Внутримолекулярный перенос фрагментов, сопровождающий
деструкцию кислот с разветвленным углеродным скелетом,
происходит под действием Ф подкласса 5.4. Изомеризация метилмалоновой
кислоты в янтарную осуществляется при участи S-метилмалонил-СоА
мутазы. Коферментом в этом случае выступает кобаламин
(производное витамина В12), а субстратом — тиоэфир СоА и соответствующей
дикарбоновой кислоты:
CoAS - СОСНСОО" ( > CoAS - СОСН2СН2СОО"
Подводя итог краткой характеристике изомераз, следует отметить,
что не все биохимические процессы, завершающиеся изомеризацией
какого-либо субстрата, катализируют Ф этого класса. Например,
превращение лимонной кислоты в изолимонную осуществляется при
участии лиазы.
5.1,6. Лигазы (синтетазы)
Энзимы шестого класса — лигазы катализируют реакции
конденсации или присоединения, сопряженные с пиролизом одной из пирофос-
фатных связей в молекуле АТФ или ГТФ. Разделение на подклассы, как
и во всех описанных выше случаях, связано с типом образуемой связи.
К подклассу 6.1 принадлежат Ф, катализирующие образование связей
С-О, к подклассу 6.2 —образование связей C-S, к подклассу 6.3 —
связей C-N, к подклассу 6.4 — связей С-С и к подклассу 6.5 — связей Р-О.
Подкласс 6.1 представлен в первую очередь Ф, катализирующими
реакцию конденсации аминокислоты с ОН-группой З'-концевого аде-
нозина в транспортной РНК (тРНК). В результате таких реакций,
происходящих с каждой из входящих в состав белков 20
аминокислот, последние оказываются распределенными в строгом соответствии
с тРНК. Это имеет решающее значение при сборке белковых молекул
на рибосомах, где распознавание осуществляется не по
аминокислотным остаткам, а по связанным с ними тРНК.
Образование тиоэфиров карбоновых кислот с СоА катализируют
Ф подкласса 6.2. Примером служит окислительная деструкция
жирных кислот, начинающаяся с превращения:
RCOO" + СоА - SH + АТФ( )ГпА - SCOR + АМФ + РР
98
Реакцию катализирует ацилкофермент А-синтетаза (ЕС 6.2.1.3).
Реакции введения азотсодержащих групп в органические
соединения катализируют Ф подкласса 6.3. В частности, УТФ:аммиак лигаза
участвует в превращении уридин-5'-трифосфата в цитидиловый нуклеотид:
О
'NH АТФ; NH
J^ ^ И F + ЛДФ+НРО/
PPP-Rib ppp,^
Энзимы подкласса 6.4 катализируют процессы присоединения С02
к углеродному скелету. Так, при участии пируваткарбоксилазы
происходит превращение пирувата в оксалоацетат:
со2 + сн3сососг + атф< > ооссн2сосоо" + адф + нро*
Кофактором этого Ф выступает биотин (витамин Н, 5.21),
который связывается с ароЕ путем образования амидной связи
с £->Ш2-группой остатка лизина.
о
о
11 II
"J* у" МН.СНД-а^Е НмЛн СО, АТФ "°°^ А,Н + Адф + нро,
S^^tCHj^COO' S^(CH,)aCONH(CH2)4 - аро Е ^S^Sch2)4CONH(CH2)4 - аро Е
(5.21)
Биотин переносит С02, присоединяя его по имидному атому азота.
5.2. Коферменты
Большинство из рассмотренных выше ферментативно
катализируемых реакций протекают при участии коферментов, многие
из которых являются водорастворимыми витаминами. В организм
человека витамины или их синтетические предшественники
поступают с пищей. Еще одним источником этих жизненно важных
веществ является микрофлора кишечника. Современная
классификация коэнзимов основана на их химическом строении.
1. Коферменты алифатического ряда.
К ним относят глютатион — у-1—глютамил-Е-цистеинилгли-
цин (5.22) и липоевую кислоту (витамин N, 5.23).
НООС—|—(СН2)2—п—N-C-^-NH-CH2-COOH
NH2 О CH2-SH
Н I
С
(5.22)
99
/СН2-
н,с
/—(Ctt,)—СООН
(5.23)
Глютатион играет важную роль в
окислительно-восстановительных процессах, выступает кофактором Ф, катализирующих процессы
изомеризации кето-формы в енольную и т. п. Липоевая кислота —
фактор роста микроорганизмов, участвует также в процессах
окислительного декарбоксилирования а-кетокислот.
Синтетическим предшественником СоА является пантотеновая
кислота (витамин В3, 5.24).
СН- О
■ з о
но-сн.
NH(CH2)2—COOH
сн3он
(5.24)
2, Коферменты ароматического ряда.
К ним относят коэнзимы Qn — убихиноны общей формулы (5.25),
а также филлохинон (витамин К,, 5.26).
МеО
MeC^Y^
сн.
(СН2—С^СН^Н п-4-12
П
(5.25)
(CHJ;
'2'3
(СН2):
2'3
СН.
(СН2)3\ХН3
СН,
СН,
(5.26)
Убихиноны участвуют в переносе электронов на последних этапах
процесса дыхания. Витамин К — компонент карбоксилаз — служит
для образования протромбина в сложном каскаде биохимических
превращений, обеспечивающем формирование основных компонентов
системы свертывания крови.
100
3. Коферменты гетероциклического ряда.
Это самая обширная группа кофакторов. В ней представлены
упомянутые ранее никотинамидные производные — NADH и
NADPH. Их синтетическим предшественником является
никотиновая кислота — витамин РР или В5. К ряду пиридиновых ко-
энзимов принадлежит и пиридоксальфосфат (5.15), образующийся
путем ферментативного окисления и фосфорилирования из пири-
доксина (витамин В6, 5.27). Пиримидиновые производные
представлены тиаминпирофосфатом (5.20). Его синтетический
предшественник — тиамин (витамин Вр 5.28). Источником для синтеза
FAD (5.5) служит рибофлавин (витамин В2, 5.29). THF (5.8)
синтезируется в организме человека из фолиевой кислоты (5.30). Эта
кислота является витамином, который содержится, главным образом,
в зеленых частях растений. Отсюда и название от лат. folium —
лист. Коэнзимом одного из подклассов изомераз выступает коба-
ламин (витамин В12) — одна из самых сложных гетероциклических
систем, в основе которой лежит ядро коррина.
сн2он
сн2он
(5.27)
*V*\
Н3С N*" Н3С^Л
(5.28)
(СН2)2ОН
Н3С
Н3С
ЛЛмЛЛ
NH
N N О
I
СН (СНОН)3СН ОН
(5.29)
СООН
СООН
(5.30)
Для функционирования ряда Ф необходимы микроэлементы,
присутствующие в активном центре в виде ионов. Например, в состав
101
гемовых простетических групп входит Fe2+. Этот же элемент (о чем
свидетельствует само название) является неотъемлемой частью
железо-серного Ф. В простетической группе цитохромоксидазы,
ДНК-полимеразы, карбоангидразы присутствует медь. Ванадий —
кофактор нитратредуктазы, молибден — ксантиноксидазы, селен — глу-
татионпероксидазы, никель — уреазы.
Более подробно роль витаминов и микроэлементов в
ферментативном катализе освещена в литературе [3—7].
5.3. Принципы действия ферментов
Главные отличия Ф от синтетических катализаторов состоят в
достаточно высокой специфичности (способности энзима отличать
свой истинный субстрат от других родственных молекул) и
несравнимо большей эффективности. Для ряда Ф характерна также высокая
избирательность E-S взаимодействия и они катализируют реакции
только с одним субстратом, тогда как другие, менее специфичные,
участвуют в превращениях нескольких химически родственных
соединений. Примерами высоко селективных биокатализаторов служат:
энтеропептидаза, обеспечивающая расщепление связи Lys6-Ile7 в трип-
синогене, и ренин, катализирующий гидролиз связи Leu-Leu в анги-
отензиногене. Таким путем осуществляется превращение
синтетических предшественников трипсина и ангиотензина I в активные
формы Ф.
В качестве модели E-S-взаимодействия более 60 лет
просуществовала аналогия «замок — ключ», предложенная в конце XIX в. Fisher.
Суть ее в том, что к Ф (замку) подходит лишь свой субстрат (ключ).
В 1959 г. Koshland была высказана гипотеза «индуцированного
соответствия». Согласно этой общепризнанной в настоящее время
модели связывание Ф с соответствующим ему субстратом индуцирует в
белке небольшие конформационные изменения. В результате
каталитические группы Ф ориентируются таким образом, что становится
возможным превращение субстрата в продукт. Модель индуцированного
соответствия допускает также некоторую конформационную
перестройку в молекуле субстрата, которая получила название
напряжения. Гипотеза о возникновении конформационных изменений в Ф и
субстрате при их связывании позволяет объяснить тот факт, что
молекулы, очень близкие по строению к истинному субстрату, могут
связываться с Ф, но при этом не превращаются в продукт, т. е.
действуют как ингибиторы. В рамках аналогии «замок — ключ»
существование ИФ объяснить невозможно.
Основные принципы катализа наглядно иллюстрирует известная
зависимость свободной энергии G от координаты реакции (рис. 5.2).
Взаимодействие E-S подчиняется тем же закономерностям, какие
установлены для обратимых химических реакций.
Положение равновесия для обратимой реакции S^P не зависит
от присутствия в системе катализатора Е, а определяется величиной
102
G
i
о
t
У
1
1 1
*-
E+S E-S EA
Е+Р
1 2
Рис. 5.2. Зависимость свободной энергии G от координаты реакции:
1 — в отсутствие Ф; 2 — с участием Ф (Е)
AG0, которая, в свою очередь, равна разности свободных энергий S и
Р. В присутствии Ф (Е) свободная энергия исходных реагентов и
продуктов реакции не изменяется и, следовательно, не изменяется AG0.
Переходное состояние или активированный комплекс А — это
высокоэнергетическая промежуточная структура, которая образуется
во время реакции. Разность свободных энергий исходных реагентов и
переходного состояния — энергия активации AG*. Скорость реакции
зависит от величины AG*: чем она меньше, тем больше скорость.
Фермент увеличивает скорость реакции путем:
• понижения AG* за счет стабилизации А;
• увеличения энергии S при образовании комплекса E-S;
• поддержания микроокружения активного центра в состоянии,
отличном от такового в водной среде. В результате изменяется
(уменьшается) диэлектрическая проницаемость среды, что
способствует протеканию реакций с участием тех промежуточных
продуктов, которые нестабильны в водной среде;
• расположения реагирующих атомов в «правильной» ориентации и
на определенном расстоянии друг от друга так, чтобы обеспечить
оптимальное протекание процесса.
В целом скорость ферментативной реакции зависит от природы
реагирующих веществ (субстрата и Ф), их концентраций,
температуры (увеличивается до определенного предела, пока не начинается
денатурация белка), концентрации коферментов, рН, а также от
концентрации активаторов и ИФ.
5.4. Кинетика ферментативных реакций
(общие сведения)
Кинетические закономерности, описывающие E-S взаимодействия,
во многом сходны с рассмотренными ранее для Л-Р
взаимоотношений (см. разд. 3.2). Биокинетика получила развитие именно с изуче-
103
ния скоростей ферментативных реакций. Ко времени появления
первых теорий рецепции уже были описаны основные кинетические
схемы и модели ферментативного катализа. В 1913 г. Michaelis и Menten,
исследуя реакцию гидролиза сахарозы в присутствии дрожжевой ин-
вертазы в условиях значительного избытка субстрата по сравнению с
концентрацией Ф, показали, что зависимость начальной скорости
реакции от концентрации субстрата описывается гиперболической
функцией. Согласно схеме Михаэлиса—Ментен в процессе
каталитического превращения активный центр Ф с субстратом образуют
промежуточный комплекс E-S, внутримолекулярное превращение
которого приводит к образованию продукта Р с регенерацией активного
центра:
E + S< ** >E-S—^—>Е + Р,
*-i
где к{, кл — константы скорости образования и диссоциации E-S
комплекса; к2 — константа скорости мономолекулярного
превращения комплекса.
Согласно этой модели, образовавшийся комплекс E-S может либо
вновь диссоциировать на Е и S, либо превратиться в продукт Р и
свободный Ф. При этом предполагается, что реакция подчиняется
закономерностям первого кинетического порядка, т. е. продукт не
может обратно превращаться в субстрат. Это справедливо на ранних
стадиях реакции, когда концентрация продукта низка. При условии
избытка субстрата модель Михаэлиса—Ментен описывается
следующей системой уравнений:
-^-^ = *,ES0-(*1+*2)(E-S)
Vo=^ = ME-S) E0=E + (E-S)
где Е0 и SQ— начальные концентрации Ф и субстрата соответственно.
В условиях стационарности по промежуточному комплексу E-S,
когда d[E-S]/dt=0, уравнение начальной скорости имеет вид:
V ^EqSq)
к + к
где К = —■ получила название константы Михаэлиса.
К
Из уравнения 5.1 следует, что максимальная скорость реакции
достигается в том случае, когда все молекулы Ф связаны с субстратом.
V = *F
max 2 О
С учетом этого уравнение 5.1 можно представить в следующем виде:
v = v sn/(sn + *M)
max 0/ \ О М /
104
Константа Михаэлиса численно равна концентрации субстрата,
при которой скорость реакции составляет половину максимальной
величины.
Км — мера сродства данного субстрата к Ф (когда к2 « к _]).
Сродство, в свою очередь, отражает прочность связывания субстрата
с активным центром. График зависимости V от [S] (рис. 5.3)
представляет собой гиперболу.
Рис.5.3. Зависимость скорости реакции:
от начальной концентрации субстрата:
1 — модель Михаэлиса—Ментен; 2 — сигмовидная кривая
(положительная кооперативность)
Существуют альтернативные способы представления уравнения
Михаэлиса—Ментен — с помощью графика двойных обратных
координат (рис 5.4) и графика Скэтчарда (рис. 5.5). Аналогичные методы
были рассмотрены в разд. 3.2. для Л-Р взаимодействий. При
использовании двойных обратных координат величина, обратная скорости
реакции 1/V, связана с концентрацией субстрата S0 следующим
уравнением:
i/v = K/vmJ(i/s0) + i/v
1А/А
1ЛЛ
!аа_=Км^тах
1/К*
1/S
Рис. 5.4. Зависимость 1/V от 1/S0:
модель Михаэлиса—Ментен; 2 — положительная кооперативность
Прямая на графике (рис 5.4) пересекает ось ординат в точке,
соответствующей величине 1/Vmax, a tgct = К1Л/Утях.
105
В методе Скэтчарда уравнение 5.1 записывают в следующем виде:
VA=(1/^)Vmax-(1/^M)V
v/s0
Рис. 5.5. График Скэтчарда
Использование графического отображения преобразования
Скэтчарда позволяет определять значения Vraax и (—\/Кы).
В эксперименте довольно часто наблюдается не гиперболическая,
а сигмовидная зависимость скорости ферментативной реакции от
концентрации субстрата (рис. 5.2). Впервые она выявлена Хиллом
(1909 г.) при анализе зависимости степени связывания кислорода
гемоглобином от парциального давления 02. В качестве причины
отклонения полученных результатов от модели Михаэлиса было
предположено, что в молекуле гемоглобина существует несколько
центров связывания и процесс образования комплекса представляет собой
кооперативное взаимодействие нескольких частиц. Простейшим
объяснением механизма взаимного влияния этих частиц является
предположение о существовании молекул Ф в двух конформационных
состояниях, которые обладают разными свойствами в зависимости от наличия
или отсутствия субстрата на связывающем центре.
Сигмовидный характер зависимость «скорость — концентрация»
приобретает тогда, когда в системе имеется несколько центров
связывания. Существует ряд способов описания такого типа
взаимодействий Ф с субстратом, среди которых наиболее известными являются
симметричная модель Моно—Уаймена—Шанжё и последовательная
модель Кошланда—Немети—Филмера [8].
В симметричной модели предполагается, что каждый мультимер-
ный ферментный комплекс может существовать, по крайней мере, в
двух разных состояниях с неодинаковой четвертичной структурой Е
и Е1, находящихся в равновесии друг с другом. Е-Форма Ф имеет
высокое сродство к субстрату, а Е1 — низкое. Субстрат будет
предпочтительнее связываться с Е-конформерами, а взаимодействие его с
Е1 -формами приведет к дополнительному напряжению в последних,
что вызовет их одновременный переход в Е-конформацию. При
увеличении концентрации субстрата все больше молекул Ф будет
переходить из состояния Е' в состояние Е. Такой сдвиг равновесия в
присутствии субстрата представляет собой эффект положительной
кооперативности.
106
Согласно концепции последовательной модели предполагается, что
отдельные субъединицы мультимерной молекулы могут иметь разные
третичные структуры. Связывание субстрата с одной субъединицей
может вызывать изменение третичной структуры другой и, в
результате, увеличивать (положительная кооперативность) или уменьшать
(отрицательная кооперативность) их сродство к субстрату.
Аллостерическая регуляция ферментативной активности имеет
место тогда, когда небольшие молекулы (эффекторы) изменяют
скорость реакции при связывании с Ф не в области активного центра.
Так, действующий на Ф аллостерический ингибитор, описываемый
симметричной моделью, будет предпочтительно связываться и
стабилизировать его в Е1 состоянии. В результате наблюдается уменьшение
сродства Ф к своему субстрату (отрицательная кооперативность).
Простейшая кинетическая схема, приводящая к сигмовидной
зависимости скорости ферментативной реакции от концентрации
субстрата, имеет вид:
Е^=>Е*
E + S< К[ >E-S—^е + Р, Е' +S< *' >E'-S—^_>Е' +Р
E-S+S^==>E-S2—^E-S + P
E1 -S + S< h »E!-S—^E'-S+P,
где L — константа равновесия между двумя конформерами
(аллостерическая константа), Kv K2, AJ1, KJ — константы диссоциации
E-S комплексов, kv k2, &,1, k22— каталитические константы для форм
Е и Е1.
Подробное математическое описание этой аллостерической
модели приведено в пособии [8]. Аллостерические эффекты имеют
большое регуляторное значение, заключающееся в сведении к минимуму
изменений концентраций субстрата, необходимых для
существенного повышения или понижения активности Ф.
Предложенная Михаэлисом и Ментен схема ферментативного
катализа является очень упрощенной. В настоящее время
неизвестны Ф, которые действуют в прямом соответствии с этой схемой.
Большинство реакций протекает через множество стадий, на
каждой из которых образуется промежуточное соединение Ф с
субстратом. Но несмотря на то, что эта модель не соответствует на
молекулярном уровне ни одному из установленных на сегодняшний день
механизмов реакций, использование данного уравнения получило
широкое распространение. Причина в том, что модель Михаэлиса
отражает фундаментальную особенность ферментативных
процессов — образование на пути реакции лабильных промежуточных
соединений между субстратом и активным центром Ф. Наблюдаемые
отклонения от этой зависимости лишь свидетельствуют об
усложнении простейшей схемы.
107
5.5. Регуляция ферментативной активности.
Категории ингибиторов ферментов
Регуляция активности Ф может осуществляться различными
способами: ковалентной модификацией, активацией зимогена,
ингибированием, а также за счет кооперативных и аллостеричес-
ких эффектов. Ковалентной модификацией называется
присоединение к Ф или отщепление от него небольшого структурного
фрагмента, сопровождающееся значительным изменением активности
катализатора. Примером может служить инактивация гликогенсин-
тетазы после присоединения фосфатной группы к одному из сери-
новых остатков. Ф восстанавливает свою активность после
отщепления фосфата.
В поиске БАВ с целью создания новых ЛС особое значение
приобретает выбор стратегии конструирования ИФ [9]. Различают две
основные категории — обратимые и необратимые ИФ. Обратимые
обычно связываются с Ф посредством нековалентных или слабых ко-
валентных связей и могут быть удалены разбавлением, гелевой
фильтрацией или диализом. При этом каталитическая активность Ф
восстанавливается. Степень ингибирования при обратимом связывании
зависит от концентрации Ф и ингибитора. Необратимые ИФ
связываются с Ф ковалентно и не могут быть удалены указанными выше
способами, а степень ингибирования зависит от времени. Условность
этой классификации становится очевидной, если учесть, что
существуют мощные обратимые ИФ, связывающиеся нековалентно, но
чрезвычайно медленно диссоциирующие. В то же время такая
классификация полезна для описания многих систем Ф-ИФ.
5.5.1. Необратимые ингибиторы ферментов
Процесс необратимого ингибирования можно представить
следующим образом:
E + I >Е1.
При этом между активным центром Е и ингибитором I имеет
место реакция второго порядка с образованием неактивного
стабильного производного EI. Очевидно, что в присутствии ИФ
образование продукта Р в реакции, катализируемой Е,
приостанавливается:
E + S< >E-S >Е-Р >Е + Р.
Если первоначальная концентрация Е выше, чем I, то по
окончании связывания Ф с ИФ синтез Р возобновляется.
Ковалентное связывание Eel используется для получения
«меченых» Ф, например, алкилированием in vitro тиопротеаз с помощью
иодуксусной кислоты:
е - sh +1 - сн2 - со; > е - s - сн2 - со2" + н+ + г
108
Необратимым связыванием с ацетилхолинэстеразой объясняется
токсическое действие фосфорорганических соединений,
относящихся к категории боевых отравляющих веществ (зарин, зоман и др.)
или пестицидов. Эти соединения, фосфорилируя активный центр
указанного Ф, блокируют ферментативный гидролиз ацетилхолина в
местах соединения нейронов (синапсах). В результате прекращается
передача нервных импульсов в ЦНС животных и человека. Однако
необратимое связывание далеко не всегда сопряжено только с
токсическим эффектом.
Необратимые ИФ широко представлены в современном
арсенале ЛС [9]. Так, механизм действия антибиотиков пеницил-
линового и цефалоспоринового ряда основан на необратимом
связывании Ф с транспептидазой, трансгликозилазой и D-аланин-
карбоксикиназой. Эти Ф участвуют на заключительных этапах
синтеза гликопептидов, образующих стенки бактериальной
клетки. р-Лактамное кольцо как пенициллинов, так и цефалоспори-
нов, раскрываясь, ацилирует образующий Ф протеин, возле
активного центра [9—11].
Пенициллин — природное соединение, продуцируемое плесневым
грибом Penicillium notatum, был открыт в 1928 г. A. Fleming (Великобритания).
До открытия этого вещества р-лактамное кольцо как химическая
структура не было известно. Легкость гидролитического расщепления,
обусловленная нестабильностью напряженного четырехчленного цикла, не
позволила A. Fleming начать клиническое применение пенициллина, хотя
антибиотик проявил высокую эффективность при испытаниях in vitro.
Позднее, в годы Второй мировой войны, в США (сверхсекретный
англоамериканский проект) строение пенициллина было подтверждено
данными РСИ. Оказалось, что бициклическая структура образована двумя
аминокислотными остатками цистеина и валина:
Val
Полный синтез пенициллинов осуществлен в 1953 г. С тех пор получено
большое количество как синтетических, так и полусинтетических
производных этого ряда. Доступности последних в значительной мере
способствовало получение 6-аминопенициллиновой кислоты (5.31) в процессе
ферментации природных продуктов.
H2N
со2н
(5.31)
109
-HN
Cys I
О
со2н
Первые указания на возможный механизм действия пенициллина
появились в 1957 г., когда Laderberg обнаружил, что чувствительные к этому
антибиотику бактерии могут расти в его присутствии в том случае, если
находятся в форме протопластов, т. е. лишены клеточной стенки. В 1965 г.
Park и Strominger показали, что пенициллин препятствует образованию
сшивок на заключительном этапе синтеза клеточной стенки у бактерий [10].
Структуры наиболее распространенных препаратов пенициллино-
вого и цефалоспоринового ряда приведены в табл. 5.1. Все эти
вещества обладают широким спектром антибактериального действия,
главным образом, против грамположительных инфекций.
Самым существенным недостатком р-лактамных антибиотиков
остается развивающаяся к ним резистентность микроорганизмов.
Причина кроется в наличии у бактерий Ф — р-лактамаз (пени-
циллиназ), гидролизующих р-лактамное кольцо и превращающих
ЛВ в неактивную пенициллиновую кислоту. Грамположительные
бактерии экскретируют р-лактамазы в окружающую среду и
таким образом разрушают антибиотик до его проникновения в
бактериальную клетку. Грамотрицательные бактерии содержат
Р-лактамазы внутри клетки. Тем не менее, метод повышения устойчивости
антибиотиков пенициллинового ряда был найден, и он
представляет собой еще один пример необратимого ингибирования.
Ингибиторами пенициллиназ выступают сульфон (5.32) и клаву-
лановая кислота (5.33).
очЧ ,р н
=с-с-он
2
со2н со2н
(5.32) (5.33)
Механизм реакции с участием клавулановой кислоты можно представить
следующим образом:
C-CHL-OH
Н 2
Е-ОН
Ингибиторы р-лактамаз используются в комбинации с антибиотиками
для повышения их стабильности при создании ЛФ. Кроме того, ряд
веществ, в силу особенностей их строения, обладает повышенной
стабильностью относительно кислотного гидролиза, алкоголиза и действия ионов
Zn2+, Cu2+, Pb2"\ К их числу принадлежат феноксиметилпроизводное
пенициллина и большинство полусинтетических антибиотиков [10, 11].
110
Таблица 5.1
Антибиотики
пенициллинового и цефалоспоринового ряда
Производные ряда пенициллина
Н
R—n—N
о Ч] Г" VCH3
о^м^снз
со2н
Биосинтетические
R
F1
сбн—сн2—
бензил-
пенициллин
НО—^ у CHj
XIII
Н3С-(СН2)е—
К IV
-вн5— °— СН2
V
Полусинтетические
R
ОМе
ОМе
метициллин
X
W Л 1
\ %Хсн3
X=Y=H
оксациллин
1 2
с6н5—с—
6 5 н
ампициллин
Производные ряда цефалоспорина
Н
R—n—N Y о
8 Yr^l
X
R
НООС-С—(СН2)3
NH2
X
О-у-СНз
О
Y
Н
цефалоспорин С
N "
1 N —СН2 —
N-N
^sA^ch3
Н
цефазолин
№2
О || СИ3
о
н
клафоран
^^ ^СН2—
н3с
ОСН3
цефметазон
111
Более широким спектром бактерицидного действия и повышенной
стабильностью обладают цефалоспорины, открытые в 50-е гг. В основе их
химического строения лежит 7-аминоцефалоспориновая кислота.
Первый антибиотик этой группы — цефалоспорин С — был выделен из
культуры Cephalosporium acremonium. Соединения данного ряда также
являются необратимыми ингибиторами транспептидаз. Резистентные в отношении
3-лактамаз грачположительных бактерий, цефалоспорины подвержены
разрушению хромосомными лактамазами грамотрицательных
микроорганизмов. Для повышения их устойчивости, расширения спектра
антимикробного действия, улучшения фармакокинетических показателей
получены многочисленные полусинтетические аналоги цефалоспорина С.
Существует четыре поколения препаратов этого ряда. Приведенный в
табл. 5.1 клафоран относится к третьему, а цефметазон — к четвертому
из них [9—11].
Известные противоопухолевые средства — 5-фторурацил (5.34)
и фторафур (5.35) — сочетают в себе свойства антиметаболитов
(см. разд. 6) и необратимых ИФ. Эти соединения связываются с ти-
мидилат синтетазой, которая катализирует перенос одноуглеродного
остатка от метилентетрагидрофолата (см. разд. 5.1.2) к дезокеиури-
динмонофосфату, на пути превращения последнего в тимидинмоно-
нуклеотид. Угнетение синтеза тимидина тормозит быстрое деление
опухолевых клеток.
(5.34)
Первоначально предполагалось, что фторзамещенные урацила
выступают в качестве конкурентных ингибиторов тимидилат синтетазы.
н
H2N^ ^N_ ^N
мЛ^ич~)~л
соон
соон
Rib-P
Однако более поздние исследования показали, что эти соединения
мимикрируют переходное состояние E-S, ковалентно связываясь с Ф
и THF. В результате реакция не может завершиться природным путем
112
из-за необратимого связывания всех веществ, участвующих в данном
процессе [12]. Угнетение синтеза тимидина происходит до тех пор, пока
в системе присутствуют (5.34) или (5.35). Для возобновления
нормального течения биохимического процесса необходимо воспроизводство
новых порций Ф. К сожалению, нарушение продукции тимидина под
действием фторпроизводных урацила отмечается не только в
опухолевых, но и во всех быстро делящихся клетках (костный мозг, кишечный
эпителий). Этим объясняется большое количество побочных эффектов,
связанных с применением данных препаратов.
К категории необратимых ИФ, способных мимикрировать
переходное состояние E-S, относится и высоко аффинный ингибитор
аденозиндезаминазы — антибиотик коформицин (5.36). Он
препятствует основно катализируемому присоединению воды к аденози-
ну (5.37) на пути его превращения в инозин (5.38).
\^ОН уН2 H2N ОН ?Н
ноТсЛ ho^oJ ho^oJ но-^oJ
но он но он но он но он
(5.36) (5.37) (5.38)
Равновесие при взаимодействии (5.36) с Ф достигается очень
медленно в течение секунд или минут (вместо миллисекунд).
Образующийся комплекс E-I очень устойчив (Т 2= 40 ч), главным образом,
за счет перехода в состояние E-I*, близкое по структуре к E-S.
Превращения можно представить в виде следующей схемы:
E + S -3=^- E-S —*- Е + Р
E-I ^5=t E-I*
При значении А^, стремящемся к нулю, ингибирование становится
практически необратимым. Кроме того, E-I* не находится в состоянии
равновесия с субстратом, и он не может разрушить этот комплекс [13].
Рассматривая различные категории ИФ, невозможно обойти
вниманием такую обширную группу ЛС как нестероидные
противовоспалительные средства (НПВС). Механизм действия НПВС (сали-
цилаты, ибупрофен (2.46), вольтарен (5.39), индометацин (5.40),
напроксен (5.41), пироксикам (5.42), кетопрофен (5.43), мефенамо-
вая кислота (5.44), бугадион (5.45), анальгин (5.46) и др.) связан, главным
образом, с ингибированием простагландинсинтетазы (PGH-синтетаза).
PGH-синтетаза (циклооксигеназа) участвует на начальных этапах
превращения арахидоновой кислоты (5.47) в эндопероксид PGG2 и простаг-
113
MeO
Q-к-О
COOH
C02Na
(5.39)
(5.40)
MeO
он о
(5.41)
(5.43)
(5.44)
ceH5
(5.45)
н3с.
г
S
~
H3C N
C6H5
сн3
CH2S02Na
*o
(5.46)
ландин PGH2. Далее из PGH2 в результате реализации каскада энзимати-
ческих реакций образуется большое количество физиологических
посредников — простагландинов (рис. 5.6). Несмотря на то, что указанные
соединения подвергаются быстрой ферментативной деградации, их роль в
течении физиологических и патологических процессов в организме
чрезвычайно велика. Так, PGE2 стимулирует гладкую мускулатуру ПК и мио-
метрия, угнетает желудочную секрецию, способствует повышению
температуры тела, выступает в качестве медиатора боли, обладает вазо- и
бронходилататорным эффектами. PGF2 и PGD2, напротив, вызывают
бронхоспазм и вазоконстрикцию. Мощным вазоконстриктором является и тром-
боксан ТхА^ который также несет ответственность за тромбообразование
в сосудах. Его метаболит - ТхВ2 — неактивен. Простациклин PGI2 препят-
114
ствует агрегации тромбоцитов. Баланс между проста цикли ном и тромбокса-
ном играет важную роль в обеспечении гемостатического равновесия. PGI,
обладает вазодилататорным и гипотензивным эффектами.
Еще один вид посредников — лейкотриены (LT) — образуются из арахидо-
новой кислоты под действием липоксигеназы, глутатионтрансферазы и
др. Ф. LTB4 несет ответственность за проявление симптомов
воспалительного процесса: вызывает скопление лейкоцитов в очаге воспаления и
отек тканей. Повышенный уровень LTB4 отмечается при таких
заболеваниях как ревматоидный артрит, язвенный колит, бронхиальная астма. LTC,
и LTD4 вызывают бронхоспазм, обладают выраженным гипотензивным
эффектом. Таким образом, фармакологическое вмешательство в синтез
простагландинов влечет за собой большое количество как
положительных, так и отрицательных эффектов [14, 15].
Ингибирование PGH-синтетазы обусловливает проявление у НПВС
анальгетического, противовоспалительного, жаропонижающего
действия. В то же время снижение продукции простагландинов приводит
к побочным эффектам, связанным со стимуляцией эрозивно-язвен-
ных процессов в ПК. Опасным осложнением применения НПВС
является бронхоспазм: блокада синтеза простагландинов может вызвать
сдвиг метаболизма арахидоновой кислоты в сторону избыточной
продукции лейкотриенов, что влечет за собой отек тканей и спазм
гладкой мускулатуры бронхов [16].
Большинство НПВС относятся к категории обратимых
ингибиторов PGH-синтетазы. Необратимым ИФ является ацетилсалициловая
кислота (аспирин). Аспирин, связываясь с Ф, ацилирует гидроксиль-
ную группу в остатке серина, расположенном в локусе адсорбции
арахидоновой кислоты. Скорость инактивации Ф увеличивается с
ростом концентрации аспирина [8].
5.5.2. Обратимые ингибиторы ферментов
Существует два основных подтипа обратимых ИФ. Первый —
конкурентные ИФ (I). Молекула такого I близка по структуре к
молекуле субстрата, и Ф не различает их. В результате I конкурирует с
субстратом за место связывания с активным центром Ф.
E + S ^5=г E-S —»- Е-Р —*- Е + Р
Е-1
\т<е, +<g = [Тед — Q^ ♦ <ф
tl
115
116
В противоположность этому, неконкурентные ИФ (II)
связываются не с активным центром, а с каким-то другим участком Ф. Такой II
не снижает концентрацию комплексов E-S, а влияет на
эффективность превращения S в Р.
E + S
+11
E-S
Е-Р
и- «
+11
Е-
E-S
■*- Е + Р
^.<я»» ^
*s
it
°£^
S*^>
На практике классические неконкурентные ингибиторы довольно
редки, и чаще встречается смешанное ингибирование, когда и ИФ, и
субстрат связываются с Ф, но связывание одного влияет на другой.
Имеющая место природа ингибирования может быть установлена
посредством изучения кинетических свойств ферментной системы.
При этом удобно пользоваться преобразованием Лайнуивера — Бэр-
ка. Если к системе, содержащей Ф и субстрат, добавить
конкурентный или неконкурентный ИФ, график в двойных обратных
координатах будет изменяться в зависимости от типа ингибитора (рис. 5.7).
При этом I увеличивает Км реакции, но не влияет на Vmax.
Поскольку I конкурирует за активный центр Ф, его роль фактически
сводится к разбавлению субстрата. Поэтому для достижения
скорости реакции, равной половине V^, требуется большая концентрация
субстрата. В связи с тем, что нейтрализовать действие I можно путем
увеличения количества субстрата, Vmax не меняется. ИФ II понижает
Vmax, но не влияет на Км. Механизм ингибирования состоит в
снижении скорости, с которой субстрат в составе комплекса E-S
превращается в продукт, поэтому при этом типе ингибирования уменьшается
лишь величина Упах.
1/Vi,
1/V,i
1/К.
и
Рис, 5.7. Зависимость 1/V от 1/[S]
а — в присутствии I; б — в присутствии II
1/S
117
Классическим примером конкурентного ИФ, аналогичного
субстрату, является малоновая кислота, ингибирующая сукцинатдегидро-
геназу и тем самым препятствующая превращению янтарной кислоты
в фумаровую. Избыток малоновой кислоты полностью прерывает
образование фумаровой. Однако при добавлении янтарной кислоты
данная реакция снова идет в нужном направлении.
-ООС-(СН2) -СОО"
оос-сн = сн-соо
Рассмотренные ранее НПВС (вольтарен, индометацин, напрок-
сен, кетопрофен и др.) выступают конкурентами арахидоновой
кислоты на стадии ее ферментативного окисления в PGG2. Величина К;
изменяется в ряду НПВС от 10~4 М (бутадион, анальгин) до Ю-6 М
(индометацин, вольтарен) [8].
Антибактериальные препараты из группы сульфамидов —
стрептоцид (5.48), норсульфазол (5.49), сульфаметоксазол (5.50),
сульфадимезин (5.51) сульфадиметоксин (5.52), сульфален (5.53) и многие др. —
также принадлежат к категории субстратоподобных конкурентных ИФ.
Значение К. для них составляет ~10~9М.
R = H
(5.48)
D
(5.49)
(5.50)
R =
ОМе
Ч\ //'
ОМе
R =
N=
* $
МеО
(5.51)
(5.52)
(5.53)
Первым сульфамидным препаратом был Prontosil rubrum — красный
краситель (5.54), синтезированный и испытанный на бактериальных клетках
Donmagk в 1935 г. (Германия). Это открытие, наряду с открытием
пенициллина, положило начало современному направлению в борьбе с
инфекционными заболеваниями. Оказалось, что активным веществом
является не Prontosil rubrum, а л-аминобензолсульфонамид — стрептоцид (5.48),
образующийся при восстановлении красителя.
118
,SO,NH,
| | - H,N—({ V>— NH, + H,N
H„N
\ //
SO.NH.
(5.54)
(5.48)
Интересно отметить, что стрептоцид как химическое соединение был
получен еще в конце XIX в. Однако в то время никто не мог и предположить, что
это вещество обладает столь ценными фармакологическими свойствами.
Механизм действия сульфонамидов стал понятен к 1948 г., когда была
изучена структура и биосинтез фолиевой кислоты. Это была первая
теория, объясняющая действие ЛВ на молекулярном уровне. Несмотря на
то, что сульфамиды были открыты несколько позже пенициллина, они
гораздо быстрее получили широкое медицинское применение. Это
связано с их химической доступностью и стабильностью. На сегодняшний день
известно несколько тысяч соединений этого ряда, однако наибольшее
распространение получили примерно два десятка производных [16—181.
Сульфамиды конкурируют с природным субстратом — л-амино-
бензойной кислотой — за активный центр Ф — 7,8-дигидроптероат-
синтетазы. В клетках бактерий этот Ф катализирует реакцию
образования дигидрофолиата (DHF) из птеридинового производного,
л-аминобензойной и глутаминовой кислот.
сульфамиды
DHF
редуктаза
THF (5.8)
H,N N N
2 Н
DHF
Восстановление DHF в THF также протекает ферментативно при
участии дигидрофолатредуктазы. Этот Ф, в свою очередь, является
мишенью для ингибиторов — триметоприма (5.55) и хлоридина (5.56),
которые не принадлежат к ряду сульфамидов, однако используются в
комбинации с последними в таких препаратах, как бисептол (триме-
топрим и сульфаметоксазол).
NHj
МеО
МеО
(5.56)
119
Подавление продукции THF сразу на двух последовательных
этапах ее образования приводит к существенному повышению
антибактериальной активности. Роль THF в синтезе пиримидиновых и пури-
новых оснований была рассмотрена нами ранее (см. разд. 5.1.2). Тем
не менее, стоит еще раз подчеркнуть, что для человека фолиевая
кислота (5.30) является витамином и поступает в организм с пищей.
Бактерии же синтезируют ее самостоятельно рассмотренным выше
путем. При нарушении продукции THF микроорганизмы утрачивают
способность к воспроизводству собственных ДНК, что приводит к
подавлению их роста.
Выбирая игибирование Ф в качестве способа воздействия на
какой-либо физиологический или патологический процесс,
наряду с кинетическими показателями необходимо учитывать
множество других факторов, среди которых наиболее важными являются
видовая специфичность данного биокатализатора и селективность
ингибитора. В этой связи следует отметить, что дигидрофолат-
редуктазы обладают видовой специфичностью. Ф,
функционирующие в бактериальных клетках и клетках млекопитающих,
различаются, главным образом, структурой активного центра. Сродство
такого ингибитора как триметоприм к бактериальным Ф в 4-Ю4
раз превышает аналогичный показатель для млекопитающих [17].
Таким образом, в присутствии (5.55) или (5.56) ферментативное
восстановление DHF в THF подавляется исключительно в
клетках бактерий, а не в клетках организма человека. Завершая
рассмотрение механизма действия сульфамидных препаратов, важно
подчеркнуть и то, что, угнетая синтез «строительных блоков» ДНК,
они не нарушают при этом их структуры, следовательно, не
проявляют генотоксичных свойств.
Еще одним примером конкурентного ингибитора дигидрофолат-
редуктазы является метотрексат (5.57).
NH2
H2N
xXj
^Y^N' ^ "CCLH
I 2
Me
(5.57)
Сравнение структур DHF и (5.57) показывает, что это
родственные соединения, способные взаимодействовать с одинаковыми
фрагментами активного центра дигидрофолатредуктазы. Метотрексат
образует прочный комплекс с Ф как в бактериальных клетках, так и в
клетках млекопитающих. В связи с отсутствием видовой
селективности, такой ингибитор не может использоваться в качестве бактерио-
статика. Он применяется как противоопухолевое средство.
120
5.5.3. Регуляция активности мультиферментных систем
Обсуждая проблемы регуляции ферментативной активности, нельзя
не коснуться способов ингибирования в мультиферментных
системах, в которых субстрат претерпевает несколько последовательных
превращений, и каждая реакция катализируется своим Ф. Скорость
образования конечного продукта в таком каскаде определяется
скоростью самой медленно текущей - лимитирующей реакции. Ф,
катализирующий такую реакцию, называется регуляторным. В
мультиферментных системах на активность регуляторного Ф влияет
концентрация конечного продукта в каскаде превращений по
принципу отрицательной обратной связи;
t I
Такая система функционирует следующим образом: когда
концентрация продукта F превышает потребности клетки, наблюдается
ингибирование регуляторного Ф, в результате чего скорость процесса
падает, и концентрация вещества F приходит в соответствие с
потребностями биологической системы. При этом продукт F должен
быть либо структурно близок к субстрату регуляторного Ф ( на схеме
это субстрат А), т. е. действовать как конкурентный ингибитор, либо
связываться не с активным центром этого Ф, иными словами,
выступать в роли неконкурентного ингибитора.
Возможность ингибирования Ф продуктами его взаимодействия
с соответствующим субстратом чрезвычайно важна в плане
конструирования новых ЛС: если имеется информация о наличии
регуляторного Ф на какой-либо стадии метаболизма и есть
необходимость в поиске его ингибиторов, то такое исследование
целесообразно проводить в ряду структурных аналогов продуктов
ферментативной реакции. Один из наиболее ярких примеров такого
направления в конструировании лекарств — поиск ингибиторов
ангиотензин-превращающего Ф (АПФ), завершившийся созданием
новых антигипертензивных препаратов — каптоприла, эналапри-
ла и др.
Система ренин-ангиотензин-альдостерон является
многофакторно регулируемым каскадом ферментативно опосредованных
превращений, в котором гликопептид ангиотензиноген (ключевая
аминокислотная последовательность: Asp-Arg-Val-Tyr-Ile-His-Pro-
Phe-His-Leu-Leu-Val-Tyr-Ser-) превращается в ангиотензин I (AI:
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-), ангиотензин II (All: Asp-
Arg-Val-Tyr-Ile-His-Pro-Phe-) и ангиотензин III (AIII). Тем самым
обеспечивается главный механизм контроля АД млекопитающих
(рис. 5.8). Установление последовательности этих превращений
натолкнуло исследователей на мысль, что эффективно снижать АД
можно путем блокирования накопления АН, ингибируя ренин
или АПФ.
121
Ангиотензиноген (печень).
Ренин (почки, секретируется в кровь)
I i Кининоген
Ангиотензин I (декапептид) Проренин-*- Калликреин ^ I
*- Брадикинин
1С-
\
Ингибиторы АПФ Неактивные фрагменты
Ангиотензин II (октапептид)
I (ангиотензиназа
Ангиотензин III (гептапептид)
стимуляция секреции альдостерона "*■ AT II рецепторы
вазоконстрикция , рост АД
Рис. 5.8. Ренин-ангиотепзиновый каскад,
приводящий к вазоконстрикции и повышению АД [18]
Созданию препаратов, по механизму действия являющихся
ингибиторами АПФ, предшествовал ряд исследований.
В начале XX в. был обнаружен белок плазмы крови, вызывающий
повышение тонуса гладкой мускулатуры кровеносных сосудов — агиотензино-
ген. В 1934 г. установлено, что ангиотензиноген под действием фермента
ренина превращается в другой пептид — ангиотензин. К 1950 г. было
показано, что первой ступенью ферментативного расщепления ангиотен-
зиногена является декапептид AI, мало влияющий на сосудистое
сопротивление и АД. Под влиянием АПФ AI превращается в АН — октапептид,
обладающий сильной сосудосуживающей и прессорной активностью.
Изучение свойств АН позволило установить, что этот пептид связывается
со специфическими рецепторами АТП стенок кровеносных сосудов и
вызывает быстрое и длительное повышение АД. Кроме того, он стимулирует
секрецию альдостерона, а в больших дозах — и секрецию
антидиуретического гормона гипофиза, что сопровождается реабсорбцией ионов натрия
и воды и развитием гиперволемии. Таким образом, в результате
многолетних исследований стало очевидным, что ренин-ангиотензиновый
каскад играет важную роль в патогенезе гипертензии, что и явилось
теоретической основой для поиска препаратов, обеспечивающих новый подход
в лечении этого заболевания [18—24].
Принципиально существуют четыре возможности для снижения
АД путем воздействия на ренин-ангиотензиновый каскад:
• ингибирование образования ренина;
• ингибирование образования ангиотензиноген а;
• ингибирование образования или активности АПФ;
• блокада ангиотензиновых АТп-рецепторов.
До недавнего времени наиболее результативным было создание
ингибиторов АПФ. В настоящее время активно развивается поиск
блокаторов АТИ-рецепторов и ингибиторов ренина.
Первоначально поиск ингибиторов АПФ изобиловал случайными
находками по причине отсутствия сведений о структуре активного
центра этого Ф. Так, в 1960 г. было обнаружено, что способностью
ингибировать АПФ обладает яд бразильской змеи, из которого был
выделен пептид тепротид (pyroGlu-^-Pro-Arg-Pro-Glu-Ile-Pro-Pro-OH).
122
В 1971 г. осуществлен синтез этого пептида, но практического
применения это вещество не получило. В то же время стало понятно, что
направленный поиск субстратоподобных ингибиторов АПФ следует
вести среди пептидов близких по аминокислотному составу к тепро-
тиду или AI. С 1970 по 1974 гг. с этой целью было получено и
исследовано около 4000 соединений. Ключевую гипотезу в создании
АПФ выдвинули М. Ondetti и D. Cushman, предположив, что АПФ
принадлежит к числу металлопротеиназ и подобен карбоксипептида-
зе А [19, 20]. Ингибиторы последней были модифицированы и на их
основе получены вещества, способные связывать АПФ. В виду
отсутствия точных данных о строении активного центра Ф,
предположили, что для его эффективного блокирования необходимым условием
является наличие у ИФ групп, способных координировать ионы Zn2+
и тем самым препятствовать их каталитической функции в процессе
гидратации. Действительно, неспецифическими ингибиторами АПФ
являются ЭДТА, о-фенантролин, а,о>дипиридил, способные
образовывать устойчивые комплексы с ионами Zn2+. Объединение идеи
подбора субстратоподобных ингибиторов с гипотезой о хелатировании
ZnH послужило новым толчком в конструировании АПФ. Из
приведенных в табл. 5.2 данных можно составить представление о том, как
происходил отбор и модификация веществ с рассматриваемой
активностью.
В базовой структуре (5.58) наращивание боковой цепи от
концевой карбоксильной группы за счет метиленовых фрагментов
привело к росту ингибирующей активности, но при п=4 активность
резко снизилась. Следующий шаг был связан с изомеризацией цепи.
Введение в а-положение по отношению к амидной группе
метального радикала способствовало значительному росту активности.
Замена карбоксильной группы на тиольную привела к кап-
топрилу (5.59). В его структуре присутствует фрагмент /-пролина,
который входит в состав пептида тепротида, а также ангиотензи-
нов. Стереохимия этого фрагмента является решающим фактором
для проявления активности: замена его на d-пролин приводит к
снижению эффекта в 104 раз [21—24]. Вслед за каптоприлом был
создан целый ряд синтетических ингибиторов АПФ, в том числе
эналаприл (5.60), лизиноприл (5.61), рамиприл (5.62).
с6н5(сн2)2
с6н5(сн2)2
№
н н
N-
Т IN
1 Н
со2н
(CH2)4NH2
/N X02H
(5.60)
(5.61)
123
Ингибиторы АПФ
Таблица 5.2
со2н
(5.58)
R=H02C(CH2)n
п = 1
п = 2
п = 3
п = 4
СН,
! 3
Н02С(СН2)2—СН
Н02С(СН2)2—СН
сн3
HS-(CH2)2-
сн„
hs-ch2—сн—
(5.59)
/С50, цМ
2600
330
70
>4000
4.9
950
0.20
0.023
Установлено, что эналаприл является пролекарством,
превращающимся в организме в активный энаприлат [25]. В последнее время
получены не содержащие аминокислотных остатков синтетические
ингибиторы АПФ — цилазаприл (5.63) и др.
ЕЮ
CfiH5-(CH
X
СН,
2'2
ест
со2н
о со2н
С.Н«(СНА
(5.62)
OEt
(5.63)
Следует отметить, что, несмотря на высокую эффективность и
большой коммерческий успех, эти препараты не лишены некоторых
побочных эффектов (отек, кашель), что связано с низкой селектив-
124
ностью АПФ. Этот Ф (дипептидилкарбоксипептидаза) инактивирует
брадикинин и ряд др. эндогенных субстратов (см. рис.5.8). Ингиби-
рование АПФ приводит к их накоплению и проявлению
нежелательных побочных эффектов [13, 26]. По этой причине более
обнадеживающим является направление, связанное с разработкой блокаторов АТН —
рецепторов и ингибиторов ренина.
Первым блокатором АТп-рецепторов стал саралазин — пептидное
соединение, близкое по структуре к АН и являющееся его
конкурентным антагонистом. Он имеет следующую аминокислотную
последовательность: CH3-NH-CHrCO~Arg-Val-Tyr-Val-His-Pro-AIa-COOH.
Однако практического применения саралазин не получил в основном
из-за сложности синтеза и неэффективности при пероральном
применении. В начале 90-х гг создан непептидный синтетический блока-
тор АТп-рецепторов — лозартан (5.64).
СН2ОН
(5-64)
Ренин — фермент, который является первым в каскаде
превращений, регулирующих АД. Он специфичен, принадлежит к группе
аспартилпротеиназ и в этом смысле близок к пепсину и катепсину Д.
Недавние рентгено-структурные исследования рекомбинантного
человеческого ренина подтвердили наличие в его активном центре
двух карбоксильных групп аспартатного фрагмента, между которыми
может разместиться достаточно объемный пептидный субстрат,
содержащий до 8 аминокислотных остатков [27, 28].
Ионизированная форма одной из карбоксильных групп выступает
в качестве общего основания и катализирует атаку молекулы воды на
амидную группу в субстрате. В то же время, вторая карбоксильная
125
группа является протонодонором и участвует в образовании тетраэд-
рического промежуточного продукта. В отличие от АПФ, активный
центр ренина не содержит ионов цинка. Это означает, что
взаимодействие осуществляется за счет гидрофобных контактов фермента
с боковыми цепями аминокислотных остатков.
Наиболее привлекательным для специалистов является путь
поиска высоко аффинных химически стабильных ингибиторов,
которые воспроизводят геометрию и распределение электронной
плотности промежуточного состояния (Б). Это привело к созданию
миметиков гидроксиэтиленового переходного состояния (В),
включенных в структуру мошных ингибиторов ICI 219623 (5.65) и CGP
38560 А (5.66) [13, 27,28].
но' °нД^
переходное состояние (Б)
потенциальный миметик (В)
nh-c4h9 *
(5.65)
(5.66)
Однако эти вещества обладают низкой биодоступностью при
приеме внутрь и умеренной антигипертензивной активностью.
Исследования в этом направлении продолжаются.
Одним из сложных многостадийных процессов, включающий
большое количество энзиматических превращений является биосинтез
холестерина. Хорошо известна роль повышенного уровня холестерина в
крови в развитии атеросклероза. Ингибирование продукции
холестерина печенью с помощью подходящих ЛС снижает риск развития
серьезных сердечно-сосудистых заболеваний. Поэтому поиск БАВ,
обладающих выраженным гипохолестеринемическим действием, и
создание на их основе новых высоко эффективных ЛС весьма
актуальны.
Синтез холестерина осуществляется в организме в 26 стадий и
может быть предотвращен путем воздействия на Ф 3-гидрокси—
3-метилглутарил-СоА-редуктазу (HMG-CoA), который катализирует
126
превращение HMG-CoA в мевалоновую кислоту (5.67).
Ингибирование этого Ф позволяет подавить синтез холестерина на ранних этапах
и избежать возникновения и накопления токсичных продуктов на
более поздних стадиях.
НО
Me
S^^CO.H
НО,
Me
S^^CO.H
V
О HMG-CoA
ОН
редуктаза ^-^
SCoA (5.67)
\ У
холестерин
HMG-CoA\ /
^ Me /
N^ C02H
k,OH интермедиат
Н^
SCoA
В процессе поиска новых антибиотиков из культур Monascus ruber
и Aspergillus terreus, а также Penicillium citricum были выделены
мощные ингибиторы HMG-CoA — ловастатин и мевастатин [29], Этот
класс соединений, избирательно угнетающих синтез холестерина в
печени, получил общее название статинов. Определяющим для
структуры ингибиторов оказалось наличие ОН-группы у вторичного атома
углерода в 5-ом положении боковой цепи, содержащей
карбоксильную группу. Лактон (5.68) представляет собой пролекарство, которое
абсорбируется после перорального применения, проникает в печень
и эффективно ингибирует биосинтез холестерина. Сегодня самыми
известным препаратами этого ряда, полученными уже синтетическим
путем, являются симвастатин — зокор (5.69), правастатин — права-
хол (5.70) и флувастатин — лескол (5.71). Длительные клинические
исследования позволяют считать статины наиболее эффективными
современными гиполипидемическими средствами [30—32].
HCV7 ^\ -О ^
HCV:^. ^O
Me || :
ингибитор
(5.68)
127
(5.69)
HO./^.COjNa
ONa
(5.70) (5.71)
Подводя итог рассмотрению способов регуляции ферментативной
активности, следует подчеркнуть, что, имея возможность выбирать
различные пути, ведущие к определенной фармакологической цели,
важно определить, является ли применение ИФ наиболее
приемлемой стратегией для ее достижения. В связи с этим, перед тем как
приступить к конструированию каких-либо химических соединений —
потенциальных ингибиторов — желательно получить ответы на
следующие вопросы:
1. Что известно о структуре и механизме действия Ф?
2. Известна ли его первичная аминокислотная последовательность?
3. Установлена ли структура Ф с помощью РСИ или существует
только предполагаемая компьютерная модель, основанная на
аналогии с другими энзимами?
4. Обладает ли субстрат Ф биологической активностью, и не вызовет
ли рост его концентрации нежелательных побочных эффектов?
5. Обладаетли целевой Ф единственным основным субстратом
(абсолютная специфичность) или при ингибировании рассматриваемого
Ф будут нарушены другие биологические процессы, что имеет место
при групповой специфичности?
6. Существуют ли изоформы Ф, которые также могут быть
подавлены ингибитором?
7. В случае, если Ф — важный компонент бактериальной клетки,
является ли он уникальным, присущим только данному
биологическому виду или встречается в клетках хозяина?
К сожалению, перед началом реализации проекта по разработке
нового ЛС вряд ли существуют ответы на все эти вопросы. Однако
первым важнейшим шагом является понимание того, как действуют
Ф вообще и конкретный Ф в особенности. Возможно, изложенные
выше сведения в какой-то мере этому способствуют.
Литература
/. Диксон М„ УэббЭ. Ферменты. Т. 1-3. - М: Мир, 1982.
2. Страйер Л. Биохимия. Т. 1—3. — М.: Мир, 1984.
3. Ленинджер А. Основы биохимии. Т. 1—3. — М.: Мир, 1985.
4. Фёршт Э. Структура и механизмы действия ферментов. — М.: Мир, 1980.
5. Кретович В.Л. Введение в энзимологию. — М.: Наука, 1986.
6. Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека. Т. 1—2. —
М.: Мир, 1993.
128
7. Machlin I. J. Handbook of Vitamins , 2^ Ed. — New York, Marcel Dekker, 1990.
8. Варфоломеев С.Д., Гуревич. К.Г. Биокинетика: Практический курс — М ■
ФАИР-ПРЕСС, 1999.
9. Enzyme Inhibitors as Drugs // Ed. by Sandler M. — London: Macmillan, 1980.
10. Recent Progress in the Chemical Synthesis of Antibiotics // Ed. by Lukacs G.,
Ohno M. — New York: Springer, 1990.
11. Навашин СМ. Антибиотикотерапия на рубеже XX и XXI веков: Актовая
лекция / IV Рос. нац. конгресс «Человек и лекарство* // Антибиотики.
М.: Фармединфо, 1997.
12. Pratt W.B., Ruddon R. W. The Anticancer Drugs. — New York: Oxford University
Press, 1979.
13. Medicinal Chemistry: Principles and Practice // Ed. by F.D.King. — Cambridge:
The Royal Society of Chemistry, 1994.
14. Handbook of Cardiovascular and Anti-Inflammatory Agents // Ed. by
M.Verderame. — Boca Raton, FL: CRC Press, 1986.
15. Gooman and Gilman's The Pharmacological Basis of Therapeutics, 8Ih ed.
// d. by AGilman et al. — New York: Pergamon Press, 1990.
16. Харкевич ДА. Фармакология. — M.: ГЭОТАР Медицина, 1999.
17. Emerging Targets in Antibacterial and Antifungal Chemotherapy // Ed. by
J. Sutcliffe et al. — New York: Chepman and Hall, 1992.
18. Vogel H.G., Vogel W. Drug Discovery and Evaluation. — Heidelberg: Springer
Verlag, 1996.
19. OndettiM.A., Rubin В., Cushman D.W.// Science. — 1977. — Vol. 196. — P. 441
20. Ondetti M.A., Cushman D. W. // J. Med. Chem. - 1981. - Vol. 24. - P. 355.
21. Ondetti M.A., Cushman D.W.// CRC Crit. Rev. Biochem. - 1984. -Vol 16 -
P. 38.
22. Carini D.J., Duncia J.V. // Adv. Med. Chem. — 1993. — Vol. 2. — P. 153.
23. Carini D.J., Duncia J.V., Aldrich P.E. et al. // J. Med. Chem — 1991 —
Vol. 34. - P. 2525.
24. Alka Kurup, Rajni Garg, Carini D.J. et al. // Chem. Rev. — 2001. — Vol 101 —
P. 2727.
25. Ettmayer P., Amidon G.L., Clement B. et al. // J. Med. Chem — 2004 —
Vol. 47. - P. 2393
26. Goa K.L., Balfour J.A., Zuanetti G. // Drugs. — 1996. — Vol. 56. — P. 564.
27. Structure-based Ligand Design / Ed. by Gubemator K., Bohm H.J — Weinheinr
Wiley-VCH, 1998.
28. Structure-based Drug Design / Ed. by Veerapandian P. — New York" Marcel
Dakker, 1997.
29. Endo A. et al. // J. Antibiot. - 1976. - Vol. 29. — P. 1346.
30. The Lovastatin Study Grup IV//Am. J. Cardiol. — 1990. — Vol. 66. — P. 122B.
31. Преображенский Д. В., Сидоренко Б.А. // Кардиология. — 1995 — N° 9 —
С. 90.
32. Грацианский Н.Л. // Кардиология. — 1995. — № 5. — С. 76.
6
НУКЛЕИНОВЫЕ КИСЛОТЫ КАК МИШЕНИ
ДНЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
Специфическая активность ряда ЛВ, например, из группы
противоопухолевых, антибактериальных, противовирусных и некоторых др.
средств реализуется посредством связывания с нуклеиновыми
кислотами (НК) или Ф, участвующими в их синтезе. В этой связи для
успешного поиска новых ЛС чрезвычайно важны знания о строении,
биосинтезе и функциях НК.
6.1. Строение нуклеиновых кислот
НК — биополимеры, участвующие в хранении и передаче
генетической информации. Мономеры НК — нуклеотиды, состоящие из
азотистого основания, моносахарида и одной или нескольких фосфатных
групп (рис. 6.1). В составе НК все нуклеотиды являются
монофосфатами. Нуклеотид без фосфатной группы называется нуклеозидом. Сахар,
входящий в состав НК, представляет собой D-изомер и р-аномер ри-
бозы или 2-дезоксирибозы. Нуклеотиды, содержащие рибозу,
называются рибонуклеотидами и являются мономерами РНК, а нуклеотиды —
производные дезоксирибозы, являются дезоксирибонуклеотидами> и из
них состоит ДНК. Азотистые основания бывают двух типов: пурины —
аденин (6.1), гуанин (6.2) и пиримидины — цитозин (6.3), тимин (6.4),
урацил (6.5). В состав РНК и ДНК входят аденин, гуанин, цитозин;
урацил встречается только в РНК, а тимин только в ДНК.
Номенклатура нуклеотидов и нуклеозидов приведена в табл. 6.1.
Рибонуклеотид Дезоксирибонуклеотид
О—
I
-О—Р=0
I
О
5'СН- А, С, G, Т
он н
130
он он
Пиримидины
NH,
XX
СН.
и
(6.4)
Рис. 6.1. Структурные фрагменты НК
Номенклатура рибо- и дезоксирибонуклеозидов
Таблица 6.1
Основание
Аденин
Гуанин
Тимин
Урацил
Цитозин
Нуклеотид
Нуклеозид
Анденилат
(АМФ, AMP)
Дезоксиаденилат
(дАМФ, dAMP)
Гуанилат
(ГМФ, GMP)
Дезоксигуанилат
!(дГМФ, dGMP)
Тимидилат
(dTMP)
Уридилат
(УМФ, UMP)
Цитидилат
Дезоксицитидилат
(дЦМФ^СМР)
Трехбуквенное
обозначение
Аденозин
Дезоксиаденозин
Гуанозин
Дезоксигуа нозин
Тимидин
Уридин
Цитидин
Дезоксицитидин
Ado
dAdo
Guo
dGuo
dThd
Urd
Cyd
dCyd
Однобук-
венное
обозначение
dA
G
dG
dT
U
С
dC
В ряде случаев в НК присутствуют редко встречающиеся
минорные нуклеотиды, такие как дигидроуридин (6.6), 4-тиоуридин (6.7),
инозин (6.8) и др. Разнообразие их особенно велико у тРНК.
Минорные нуклеотиды образуются в результате химических превращений
оснований НК, происходящих уже после образования полимерной
цепи. Чрезвычайно распространены в РНК, и ДНК различные мети-
131
лированные производные: 5-метилуридин, 5-метилцитидин, l-N-ме-
тиладенозин, 2-1Ч-метилгуанозин. У РНК объектом метилирования
могут быть и 2'-гидроксигруппы остатков рибозы, что приводит к
образованию 2'-0-метилцитидина или 2'-0-метилгуанозина [1,2].
Рибонуклеотидные и дезоксирибонуклеотидные звенья
соединяются между собой с помощью фосфодиэфирных мостиков, связывающих
5'-гидроксильную группу одного нуклеотида с З'-гидроксильной груп-
пой следующего (рис. 6.2). Таким образом, регулярная основная цепь
образована фосфатными и рибозными остатками, а основания
присоединены к сахарам подобно тому, как присоединены боковые группы
в белках. Порядок следования оснований вдоль цепи называется
первичной структурой НК. Последовательность оснований принято читать
в направлении от 5'- к 3'- углеродному атому пентозы.
5' коней
°>
Г
HO'l
V
но<к
°$р
НО 1
О
Рис. 6.2.
°s
ОН(Н)
w
О ОН(Н)
W
^0 ОН РНК
3' конец
или (Н) -
Первичная структура
132
ДНК
НК
6.1.1. Структура ДНК
Модель структуры ДНК в виде двойной спирали была предложена
Уотсоном и Криком в 1953 г. на основании данных РСИ. Согласно этой
трехмерной модели, молекула ДНК состоит из двух противоположно
направленных полинуклеотидных цепей, которые относительно одной
и той же оси образуют правую спираль. Азотистые основания находятся
внутри двойной спирали, и их плоскости перпендикулярны основной
оси, а сахарофосфатные остатки экспонированы наружу (рис. 6.3).
Между основаниями образуются специфические Н-связи: аденин —
тимин (илиурацил), гуанин — цитозин, получившие название уотсон-
криковского спаривания. В результате более объемные пурины всегда
взаимодействуют с пиримидинами, имеющими меньшие размеры, что
обеспечивает оптимальную геометрию остова. Антипараллельные цепи
двойной спирали не являются идентичными ни по
последовательности оснований, ни по нуклеотидному составу, но они комплементарны
друг другу именно благодаря наличию специфического водородного
связывания между указанными выше основаниями.
Комплементарные пары оснований
расположены под прямым углом
А и G — пурины T и С — пиримидины .А^ Ось правой
двойной спирали
Рис. 6.3. Двойная спираль ДНК[1].
Комплементарность очень важна для копирования (репликации)
ДНК. Соотношения между числом различных оснований в ДНК,
выявленные Чарграффом с соавт. в 50-х гг., имели важное
значение для установления структуры ДНК: было показано, что число
адениновых остатков в основаниях цепи ДНК, независимо от
организма, равно числу тиминовых, а число гуаниновых — числу
133
цитозиновых. Эти равенства являются следствием избирательного
спаривания оснований [1, 2].
Геометрия двойной спирали такова, что соседние пары
оснований находятся друг от друга на расстоянии 0.34 нм и повернуты на 36°
вокруг оси спирали. Следовательно, на один виток спирали
приходится 10 пар оснований, и шаг спирали равен 3.4 нм. Диаметр
двойной спирали равен 20 нм и в ней образуются два желобка — большой
и малый. Это связано с тем, что сахарофосфатный остов расположен
дальше от оси спирали, чем азотистые основания.
Стабильность структуры ДНК обусловлена разными типами
взаимодействия, среди которых основными являются Н-связи между
основаниями и межплоскостное взаимодействие (стэкииг). Благодаря
последнему обеспечиваются не только выгодные ван-дер-ваальсовы контакты
между атомами, но и возникает дополнительная стабилизация
вследствие перекрывания р-орбиталей атомов параллельно расположенных
оснований. Стабилизации способствует также благоприятный
гидрофобный эффект, проявляющийся в защищенности малополярных
оснований от непосредственного контакта с водной средой. Напротив,
сахарофосфатный остов с его полярными и ионизированными группами
экспонирован, что также стабилизирует структуру.
Для ДНК известны три полиморфные формы: А, В и Z. Обычной
структурой является В-ДНК, в которой плоскости пар оснований
перпендикулярны оси двойной спирали (рис. 6.3). В А-ДНК
плоскости пар оснований повернуты примерно на 20° от нормали к оси
правой двойной спирали; на виток спирали здесь приходится 11 пар
оснований. Z-ДНК — это левая спираль с 12 парами оснований на виток;
плоскости оснований примерно перпендикулярны оси спирали. ДНК
в клетке обычно находится в В-форме, но отдельные ее участки могут
находиться в A, Z или даже в иной конформации [2].
6.1.2. Структура РНК
РНК бывают трех основных типов: информационная (матричная,
мРНК), транспортная (тРНК) и рибосомная (рРНК). В ядре клеток эука-
риот содержится РНК четвертого типа — гетерогенная ядерная (гяРНК).
мРНК является копией (транскриптом) соответствующей ДНК.
Этот транскрипт служит матрицей для синтеза белка. Каждые три
последовательных основания мРНК — кодон — детерминируют один
аминокислотный остаток. В одной эукариотической клетке более ста
различных молекул мРНК, каждая из которых кодирует одну или
несколько полипептидных цепей. Соответствующий кодон в мРНК
узнает тРНК и переносит нужную аминокислоту к растущей
полипептидной цепи. Аминокислотный остаток присоединяется к З'-кон-
цу молекулы тРНК. Для каждой аминокислоты имеется по крайней
мере одна тРНК. Узнавание кодона в мРНК осуществляется с
помощью трех последовательных оснований в тРНК, которые получили
название аитикодона. Между азотистыми основаниями кодона и ан-
134
тикодона образуются уотсон-криковские водородные связи при
условии, что полинуклеоТидные цепи антипараллельны (рис. 6.4).
тРНК 3'
5' Антикодон
мРНК 5' U—U С 3' Кодон
Рис. 6.4. Схема распознавания кодона в мРНК
Молекулы тРНК состоят примерно из 75 нуклеотидов и
начинаются с фосфорилированного 5'-конца. Первым основанием обычно
является гуанин. На З'-конце всегда присутствуют три основания —
ССА и концевая ОН-группа. В структуре тРНК, кроме обычных,
часто присутствуют минорные основания. Исходя из максимального
числа уотсон-криковских пар оснований при данной нуклеотидной
последовательности, структуру молекулы тРНК принято представлять
в виде «клеверного листа» (рис. 6.5).
Последовательность оснований
и структура клеверного листа
Водородные связи
уотсон-криковской
вторичной структуры
О-петля
Антикодоновая
ветвь
Антикодоновая
петля
Зчсонец
яд-— Акцепторный
Г 76 К0НвЦ
72 Акцепторная
ветвь
Т-ветвь
Т-петля
Вариабельная
Трехмерная структура
Т-ветвь
Т-петля 54 / 64
D-петпя
20
Вариабельная
петля
Водородная 44
связь,
не участвующая
в образовании
вторичной
структуры
5-конец
Звонец
Акцепторный конец
Акцепторная ветвь
Водородная I
саязь, 7мм
D-ветвь участвующая в образовании
вторичной структуры
Антикодоновая
ветвь
Антикодон
Антикодон
Антикодоновая петля
а б
Рис. 6.5. Структура тРНК [1]:
а — структура клеверного листа; б — трехмерная структура
Участки, в которых посредством Н-связей образовались пары
оснований, называются стеблями, а одно цеп очечные участки —
петлями. Все известные тРН К образуют «клеверный лист» с четырьмя
стеблями (акцепторным, D, антикодоновым и Т) и тремя петлями (D,
антикодоновой и Т). Некоторые тРНК имеют дополнительные петли
и/или стебли (например, вариабельная петля дрожжевой фенил-
аланиновой тРНК, рис. 6.5). Каждый стебель состоит из двух
антипараллельных цепей, образующих правую двойную спираль,
известную как А-форма РНК. Эта форма содержит 11 пар оснований на
виток, шаг спирали равен 3.1 нм. Плоскости оснований составляют
135
около 20° с нормалью к оси двойной спирали. А-форма РНК близка к
аналогичной форме ДНК. РНК не способна менять конформацию
и переходить в В-форму, что обусловлено наличием 2'-ОН группы в
рибозе, которой нет в дезоксирибозе. Пространственная (третичная)
структура молекулы тРНК напоминает по форме букву Г.
«Перекладину» этой буквы образует спираль из акцепторного и Т-стеблей, а
антикодоновый и D-стебли формируют «ножку». В каждой части
содержится примерно 10 пар оснований.
рРНК играют важную роль в структуре и биосинтетической
функции рибосом — органелл клетки, состоящих из двух субчастиц, на
которых происходит синтез белка. Каждая такая частица образована
молекулами протеинов и рРНК. Коэффициент седиментации рибосом
прокариот составляет примерно 70S, а для эукариот он равен 80S.
Своими собственными рибосомами обладают и митохондрии эукариоти-
ческих клеток. Их коэффициент седиментации равен 70S, и они во
многом сходны с рибосомами прокариот. 705-рибосома прокариот
состоит из 50S- и 30S-субчастиц, а SOS-рибосома эукариот — из 60S- и
405-субчастиц. Эти субчастицы, в свою очередь, могут диссоциировать
на составные части — белок и рРНК (рис. 6.6). Весовое соотношение
между ними для про- и эукариот составляет 2:1 и 1:1 соответственно.
Большинство рибосомных белков содержит в боковых цепях остатки
Arg+ и Lys+. Эти положительно заряженные группы взаимодействуют с
отрицательно заряженными фосфатными остатками молекул рРНК, за
счет чего стабилизируется комплекс протеин-НК. Для некоторых
молекул рРНК установлена нуклеотидная последовательность, в которой
обнаружены участки спаривания оснований, образующие вторичную
структуру, подобную той, что имеет место в стеблях молекулы тРНК [2].
недиссо циированная
риоосома
субчастицы
компоненты
20 нм
30 нм
23S-PHK + 5S-PHK
(1МДа) (ЗбкДа)
Прокариотичес кая
70S
(2.7 МДа)
30S
О МДа)
21 белок
23 им
35им
Эу кариотиче екая
80S
(4.3 МДа)
28S- РНК + 5,8 РНК + 5S- РНК
(1.5 МДа) (8 44кДа) (36 кДа)
около 45 белков
40S
18S- РНК
(700 кДа)
около 30 белков
Рис. 6.6. Структура рибосомы [1]
136
6.2. Биосинтез нуклеиновых кислот
(общие принципы)
Главными матричными процессами, присущими всем живым
организмам, являются репликация ДНК, транскрипция и трансляция.
Репликация — процесс, при котором информация,
закодированная в последовательности оснований молекулы родительской ДНК,
передается с максимальной точностью дочерней ДНК. При
полуконсервативной репликации дочерние клетки первого поколения
получают одну цепь ДНК от родителей, а вторая цепь является вновь
синтезированной. Процесс осуществляется при участии ДНК-поли-
мераз, которые относятся к классу трансфераз. Роль матрицы играют
разделенные цепи двунитевой материнской ДНК, а субстратами
являются дезоксирибонуклеозид-5'-трифосфаты.
Тринскрипция — процесс переноса генетической информации от
ДНК к РНК. Все виды РНК - мРНК, рРНК и тРНК -
синтезируются в соответствии с последовательностью оснований в ДНК,
служащей матрицей. Транскрибируется только одна, так называемая «+»-цепь
ДНК. Процесс протекает при участии РНК-полимераз. Субстратами
являются рибонуклеозид-5'-трифосфаты.
Процессы репликации и транскрипции у прокариот и эукариот
существенно различаются по скорости протекания и по отдельным
механизмам [3, 4, 5].
Трансляция — процесс декодирования мРНК, в результате которого
информация с языка последовательности оснований мРНК
переводится на язык аминокислотной последовательности белка.
Осуществляется трансляция на рибосомах, субстратами являются амино-
ацил-тРНК.
Матричный синтез складывается из трех основных этапов:
инициации, элонгации и терминации. Инициация — процесс, в котором
образуется первая связь между мономерными звеньями создаваемой
полимерной цепи. На матрице имеется специальный сигнал,
позволяющий Ф распознать кодирующий элемент, с которого начинается
информация о синтезируемой цепи биополимера. Для прекращения
роста цепи в определенном месте необходимо, чтобы присоединение
очередного мономера оказалось невозможным. Поскольку конец
продукта обычно не соответствует концу матрицы, на последней
закодирован специальный сигнал, обеспечивающий обрыв роста цепи —
терминацию. При нормальном развитии процесса на каждый акт
инициации и терминации в матричном синтезе приходится большое
число актов элонгации — соединений мономеров с растущей цепью.
Элонгация происходит на активном центре соответствующей полимеразы
НК или рибосомы, а непосредственными участниками этого
процесса являются концевая группа синтезируемого биополимера,
кодирующий элемент матрицы и очередная молекула мономера. Каждый
этап роста цепи начинается с отбора субстрата, который, вероятно,
происходит путем перебора всех альтернативных субстратов,
присутствующих в системе. При этом активный центр Ф должен обладать
137
сродством к имеющемуся у всех типов мономеров универсальному
фрагменту субстратов, в качестве которого могут выступать либо
остаток рибозы, либо дезоксирибозы или трифосфатный фрагмент. При
правильном протекании процесса каждый кодирующий элемент
должен участвовать в одном акте роста цепи и затем уступить свою роль
следующему за ним элементу. Чтобы сделать возможным следующий
акт элонгации, необходимо перемещение растущей цепи с
освобождением участка отбора мономера и одновременное перемещение
матрицы на один кодирующий элемент. Такое перемещение матрицы и
продукта носит название транслокации. Таким образом, каждый акт
элонгации складывается, по крайней мере, из трех этапов: отбора
мономера, химического превращения и транслокации. В
действительности же, элонгация является более сложным процессом, требующим
участия Ф и расходования энергии [4].
Матричный синтез ДНК, катализируемый ДНК-полимеразами,
выполняет две основные функции: репликацию ДНК — синтез
новых дочерних цепей и репарацию двунитевых ДНК, имеющих
разрывы в одной из цепей, образовавшихся в результате вырезания нукле-
азами поврежденных участков этой цепи. У прокариот и эукариот
существует три разновидности ДНК-полимераз. У прокариот
выделены полимеразы I, II и III типов, обозначаемые как poll, polll и polIII.
Последняя катализирует синтез растущей цепи, poll играет важную
роль в процессе созревания ДНК, функции polll изучены не
полностью. В эукариотических клетках в репликации хромосом участвует
ДНК-полимераза а, в репарации — ДНК-полимераза р, а у
разновидность является Ф, осуществляющим репликацию ДНК митохондрий.
Эти Ф, независимо от типа клеток, в которых происходит
репликация, присоединяют нуклеотид к ОН-группе на З'-конце одной из цепей
ДНК, которая растет в направлении 5'->3'. Поэтому говорят, что
данные Ф обладают 5'-»3'-полимеразной активностью. Помимо этого все
они проявляют способность деградировать ДНК, отщепляя нукле-
отиды в направлении 3'-»5', т. е. являются 3'-»5'-экзонуклеазами.
Обычно репликация начинается в строго определенных участках,
получивших название участков ori (от origin of replication), и от этих
участков распространяется в обе стороны. Участкам ori
предшествуют точки разветвления материнских цепей ДНК. Участок,
примыкающий к точке разветвления, получил название репликативной вилки
(рис. 6.7). В ходе синтеза репликативная вилка перемещается вдоль
молекулы, при этом расплетаются все новые участки родительской
ДНК до тех пор, пока вилка не дойдет до точки терминации.
Разделение цепей достигается с помощью специальных Ф — геликаз (топо-
изомераз). Энергия, необходимая для этого, высвобождается за счет
гидролиза АТФ. Геликазы перемещаются вдоль полинуклеотидных
цепей в двух направлениях.
Для начала синтеза ДНК необходима затравка — праймер. Роль
праймера выполняет короткая РНК (10—60 нуклеотидов). Она
синтезируется комплементарно определенному участку ДНК при участии
праймазы. После образования праймера в работу включается ДНК-
138
полимераза. В отличие от геликаз ДНК-полимеразы могут
перемещаться только от 3' к 5' концу матрицы. Поэтому элонгация
растущей цепи по мере раскручивания двунитевой материнской ДНК
может идти только вдоль одной цепи матрицы, той, относительно которой
вилка репликации движется от 3' к 5' концу. Непрерывно
синтезируемая цепь получила название ведущей. Синтез на запаздывающей цепи
также начинается с образования праймера и идет в направлении,
противоположном ведущей цепи — от вилки репликации.
Запаздывающая цепь синтезируется фрагментарно (в виде фрагментов
Оказаки), т. к. праймер образуется только тогда, когда вилка репликации
освободит тот участок матрицы, который имеет сродство к праймазе.
Лигирование (сшивание) фрагментов Оказаки с образованием
единой цепи носит название процесса созревания.
Рис. 6.7. Репликация ДНК [1]:
1 — репликативная вилка, 2 — ДНК-полимераза (pol I — созревание);
3 — ДНК-полимераза (pol HI — «корректорская правка»); 4—геликаза;
5—гираза (топоизомераза); 6—белки, дестабилизирующие двойную спираль
При созревании цепи РНК-затравка удаляется как с 5' конца
ведущей цепи, так и с 5' концов фрагментов Оказаки, а эти фрагменты
сшиваются друг с другом. Удаление затравки осуществляется при
участии 3'^5' экзонуклеазы. Этот же Ф вместо удаленной РНК
присоединяет дезоксинуклеотиды, используя свою 5'^3' полимеразную
активность. При этом в случае присоединения «неправильного» нук-
леотида осуществляется «корректорская правка» — удаление
оснований, образующих некомплементарные пары. Этот процесс
обеспечивает чрезвычайно высокую точность репликации, отвечающую одной
ошибке на 109 пар оснований. Еще один механизм удаления РНК-
фрагментов основан на присутствии в клетках особой рибонуклеазы,
получившей название РНКазы Н. Этот Ф специфичен к двунитевым
структурам, построенным из одной рибонуклеотидной и одной де-
зоксирибонуклеотидной цепи, причем он гидролизует первую из них.
139
РНКаза Н также способна удалять РНК-праймер с последующей
застройкой разрыва с помощью ДНК-полимеразы. На заключительных
этапах сборки фрагментов в нужном порядке действует ДНК-лигаза,
катализирующая образование фосфодиэфирной связи.
Раскручивание геликазами части двойной спирали ДНК в
хромосомах эукариот приводит к сверхспирализации остальной части
структуры, что неизбежно сказывается на скорости процесса репликации.
Сверхспирализации препятствуют ДНК-топоизомеразы.
Таким образом, в репликации ДНК, помимо ДНК-полимеразы,
принимает участие большой набор Ф: геликаза, праймаза, РНКаза Н,
ДНК-лигаза и топоизомераза. Этим перечень Ф и белков,
участвующих в матричном биосинтезе ДНК, далеко не исчерпывается. Однако
многие из участников этого процесса до настоящего времени
остаются мало изученными.
Матричный синтез РНК осуществляется с помощью
ДНК-зависимых РНК-полимераз, для которых характерна мультисубъединич-
ная структура. В строении этих Ф у прокариот и эукариот имеются
существенные различия. Так, в эукариотических клетках
представлены три вида РНК-полимераз. Их принято обозначать римскими
цифрами либо заглавными буквами латинского алфавита. РНК-полиме-
раза I (А) локализована в ядрышке, где она катализирует синтез
предшественников больших рибосомных РНК; РНК-полимераза II (В)
находится в нуклеоплазме и участвует в синтезе первичного
транскрипта мРНК; полимераза III (С) также локализована в
нуклеоплазме и катализирует синтез тРНК и 5S-pPHK.
Инициация транскрипции происходит на строго определенном
участке матрицы, расположенном после промотора. Последний
представляет собой участок молекулы ДНК, содержащий несколько
десятков пар оснований и находящийся вне пределов области,
программирующей синтез РНК. В свою очередь синтез РНК всегда начинается
с оснований А или G в «+»-цепи ДНК, а участок связывания Ф
расположен выше точки инициации примерно на 10 оснований.
Элонгация цепи РНК наступает после присоединения нескольких
нуклеозидов. В этот момент РНК-полимераза претерпевает
структурные изменения: у прокариот от Ф отделяется с-субъединица, а
оставшийся кор-фермент — комплекс (а2рр') субъединиц — катализирует
дальнейшее удлинение цепи РНК. Движущийся вдоль ДНК
кор-фермент действует подобно застежке-молнии и раскрывает двойную
спираль, которая замыкается позади Ф по мере того, как соответствующие
основания РНК спариваются с основаниями ДНК в «минус» — цепи.
Терминация цепи РНК происходит на специфических участках
ДНК — терминаторах. РНК чаще всего образуются в виде
предшественников, которые, уже без участия матричных систем,
превращаются в зрелые молекулы. Всю совокупность происходящих при
этом превращений как с РНК, так и с белками называют процес-
сингом. Например, молекулы мРНК образуются из больших по
размеру предшественников, называемых гетероядерной РНК
(гяРНК). Для образования зрелой мРНК из этих молекул фермен-
140
тативным путем удаляются лишние концевые олигонуклеотиды (по
5'- и З'-концам), а также имеет место «монтаж» (сплайсинг) —
удаление крупных вставочных последовательностей, соответствующих
интронам (некодирующим участкам) ДНК, с одновременным
воссоединением концов соседних экзонов (кодирующих участков) в
единую цепь. Наконец, многие РНК, в первую очередь тРНК,
подвергаются многочисленным химическим превращениям,
приводящим к образованию в составе зрелой молекулы широкого
спектра минорных компонентов, таких как (6.6) — (6.8).
Следует подчеркнуть, что Ф, участвующие в транскрипции,
являются мишенями для ряда БАВ, в частности антибиотиков и токсинов.
Например, токсин бледной поганки - а-аманитин - блокирует РНК-
полимеразу II эукариот, что приводит к прекращению синтеза новых
молекул мРНК и многих жизненно важных белков.
Трансляция — это процесс биосинтеза белков, который
осуществляется на рибосомах при участии мРНК, аминоацил-тРНК
и множества Ф. Полипептидная цепь белка растет в направлении от
N-конца к С-концу. Декодирование мРНК происходит
соответственно в направлении 5'->3'. Ф, катализирующий превращение сложных
эфиров (аминоацил-тРНК) в амиды (полипептиды) и
«вмонтированный» в рибосому, представляет собой пептидил-трансферазу.
Трансляция происходит при специфическом связывании кодона мРНК с
антикодоном тРНК. Этому предшествует активация тРНК —
превращение ее в аминоацил-тРНК. Этот процесс катализирует аминоацил-
тРНК-синтетаза. Связывание последней со своей и только своей
аминокислотой обеспечивается с очень высокой точностью: неправильная
активация тРНК приведет к тому, что в полипептидную цепь
включится не та аминокислота, которая именно в этом месте должна в ней
присутствовать. Для проверки правильности активации существует
механизм корректорской правки — при наличии ошибки в
связывании, соответствующая синтетаза автоматически деацилирует любую
неправильно активированную тРНК.
Синтез белка начинается с того момента, когда образуется
комплекс, состоящий из большой и малой субчастиц рибосом, мРНК и
двух молекул тРНК, одна из которых несет на З'-конце пептидный
фрагмент, а другая — аминоацильный. Участки рибосомы, на которых
связываются эти тРНК, соответственно называют Р-сайтом и А-сай-
том. Далее такая рибосома движется вдоль мРНК от 5' к 3' концу, что
сопровождается ростом полипептидной цепи. После того как синтез
полностью завершен, полипептидная цепь отходит от рибосомы,
которая в свою очередь отщепляется от мРНК и диссоциирует на
большую и малую субчастицы. В свою очередь эти субчастицы теперь
снова могут участвовать в синтезе новой молекулы белка.
Описанный механизм трансляции чрезвычайно упрощен. В
действительности на этапах узнавания кодона, образования пептидной
связи и транслокации участвует большое количество Ф и так
называемых факторов элонгации. Детально процессы матричного синтеза белка
и НК описаны во многих специальных источниках литературы [1—5].
141
6.3. Лекарственные средства,
влияющие на синтез нуклеиновых кислот и белков
6.3.1. Антибактериальные средства
Влияние на синтез НК и белков лежит в основе механизма
действия таких антибиотиков как рифамицины, аминогликозиды, тетра-
циклины, макролиды и хлорамфеникол, а также производные хино-
лонкарбоновых кислот.
Рифамицин В (6.9) — природный метаболит, выделенный из
культуры Streptomyces meditemanei. Он ингибирует ДНК-зависимую РНК-
полимеразу, связываясь с ее b-субъединицей как у грамположительных,
так и у грамотрицательных бактерий. Однако эффективность рифами-
цина В в отношении последних значительно ниже из-за его
неспособности проникать через клеточную стенку этих микроорганизмов.
он он н Ме
Ме1
C=N-N
Н
Невалентное связывание рифамицина В с мишенью
осуществляется за счет гидрофобного взаимодействия нафталинового фрагмента
с остатками ароматических аминокислот, входящих в состав РНК-
полимеразы. Указанный Ф принадлежит к числу металлопротеаз и
содержит два иона Zn2+ в активном центре. Гидроксильные группы,
расположенные у С-1 и С-8 атомов в структуре (6.9) хелатируют эти
ионы, тем самым обеспечивая более прочное связывание с мишенью,
а гидроксилы в положении С-21 и С-23 образуют водородные связи
с Ф. Селективность действия рифамицинов у прокариот и эукариот
обусловлена различиями в аминокислотной последовательности РНК-
полимеразы [6].
На основе продукта (6.9) получено большое количество
полусинтетических производных. Модификация структуры (6.9) позволила
выявить ключевые фрагменты, обеспечивающие высокую активность
соединений данного ряда. Это наличие свободных расположенных
в плоскости цикла гидроксильных групп у атомов С-1, С-8, С-21 и
С-23. Ацилирование по положению С-21 и С-23, восстановление двой-
142
ных связей в макроцикле и его раскрытие ведут к потере эффекта.
Напротив, замещение у атомов С-3 или С-4 позволяет получить
вещества, превосходящие (6.9) по активности. Вероятно, такие
производные обладают повышенной способностью к проникновению
через бактериальную стенку. Наибольшее распространение в медицине
среди полусинтетических аналогов рифамицина В получил рифампи-
цин (6.10). Он является одним из наиболее эффективных средств
против микобактерий туберкулеза и стафилококковых инфекций [7].
Ингибиторами синтеза белка на рибосомах являются
аминогликозиды (аминоциклитолы), тетрациклины, макролиды и
хлорамфеникол. Антибактериальное действие аминогликозидов и хлорамфе-
никола обусловлено их способностью избирательно взаимодействовать
с рибосомами прокариотических клеток. Селективность связывания
обеспечена различиями в белковых компонентах этих органелл у эука^
риот и прокариот. Аминогликозиды взаимодействуют с малой
субъединицей (30S), а хлорамфеникол — с большой (50S) субъединицей
вблизи пептидилтрансферазного центра рибосомы, подавляя тем
самым синтез белка у бактерий и не затрагивая биосинтез в клетках
высших организмов [8].
Стрептомицин (6.11), выделенный из Streptomycesgriseus в 1943 г.,
был вторым после пенициллина антибиотиком, эффективным в
отношении широкого спектра бактерий, главным образом,
грамотрицательных. В 50—60-е гг. он получил распространение как мощное
противотуберкулезное средство. В настоящее время применяется редко
из-за большого количества побочных эффектов, а также по причине
выявления в данном ряду новых более эффективных и менее
токсичных веществ. Однако стрептомицин остается в арсенале средств борьбы
с такими опасными инфекциями как бубонная чума и туляремия.
ОН
(6.11)
Гентамицин (6.12) и канамицины (6.13)-(6.16) являются
химически более стабильными структурами, чем стрептомицин. Они
устойчивы в кислой и основной средах, а также к действию высокой
температуры, что позволяет изготавливать на их основе широкий набор
препаратов.
143
Me—CHNH2 CH2NH2
\k°, H0^t
о
H2N A NH2 lf]0- NH
HO-> гЛ .„ , HO
NH, |^— NHR
о о он
\о-л Me H V
J Ч IVI
но^^1он о ,_он
NHCK
NK
ОН
(6.12)
Канамицин А (6.13) X = Y = ОН, R = Н
Канамицин В (6.14) X = OH, Y = NH2, R = Н
Торбамицин (6.15) X = R = Н, Y = NH2
Амикацин (6.16) X = Y = ОН, R = COCH(OH)-CH2-CH2-NH2 (L)
Причина развития резистентности к антибиотикам этого ряда
кроется в способности некоторых микроорганизмов N-ацетилировать,
О-фосфорилировать или О-аденилировать функциональные группы,
обеспечивающие связывание аминогликозидов с рибосомами. Решить
эту проблему в определенной степени удалось благодаря созданию
полусинтетических производных — торбамицина (6.15) и амикацина (6.16) [8].
Хлорамфеникол (левомицетин, 6.17) также является природным
продуктом и первоначально (1947 г.) был выделен из культуры Streptomyces
Venezuela. Однако простое строение этого вещества позволило довольно
быстро осуществить его синтез в промышленном масштабе [7].
НО н
СН2ОН
HN Н
\— СНС12
О
(6.17)
В структуре (6.17) есть два хиральных центра, но из четырех
возможных изомеров активен только R,R-xлopaмфeникoл. Существенными
для проявления этим веществом антибактериального действия, кроме
R,R-пpoпaндиoлoвoгo фрагмента, являются наличие 4-Ы02-группы в
фенильном кольце и незамещенных ОН-групп в боковой цепи. Хлор-
ацетамидный фрагмент влияет на эффект, но его замена на другую
электроноакцепторную группу существенным образом не
сказывается на активности. Хлорамфеникол нашел применение в лечении
144
кишечных инфекций, менингита, бруцеллеза и др. Рацемат левоми-
цетина используют наружно в виде синтомициновой эмульсии. Из-за
неблагоприятных эффектов в отношении кроветворения
хлорамфеникол относят к антибиотикам резерва и применяют в основном при
неэффективности других препаратов [9].
Тетрациклины по химическому строению являются высоко
функционализованными частично гидрированными нафтаценами
(табл. 6.2). Они проявляют амфотерные свойства, имеют изоэлектри-
ческую точку при рН ~ 5 и образуют трудно растворимые соли с Fe2+,
Al3+, Mg2+, Ca2+. Образование последних приводит к существенному
снижению активности тетрациклинов и накоплению их в виде
кальциевых солей в костной ткани при использовании в больших дозах в
течение длительного времени [10]. Первым в рассматриваемом ряду
в 1948 г. был открыт хлортетрациклин. Получают тетрациклины
ферментацией Streptomyces aureofaciens или Streptomyces rimosus и
модифицируют синтетическим путем.
Антибиотики тетрациклииового рада
R х v R1
н У(сн3)2
он
CONK
Таблица 6.2
Название Л В
Тетрациклин (6.18)
Хлортетрациклин (6.19)
Миноциклин (6.20)
Санциклин (6.21)
Окситетрациклин (6.22)
Доксициклин (6.23)
Метациклин (6.24)
R
Н
С1
N(CH3)2
Н
Н
н
н
Заместители
R1
Н
Н
Н
Н
ОН
ОН
ОН
X
ОН
ОН
Н
Н
ОН
-СН2
Н
Y
СН3
Н
Н
Н
СН3
—
СН3
Важным структурным фактором, определяющим наличие у этих
соединений антибактериальных свойств, является а-стереоориента-
ция диметиламиногруппы, расположенной у атома С-4. 4-Эпитетра-
циклин, который образуется в результате эпимеризации за счет
перехода в енольную форму в кислой среде, неактивен.
Тетрациклиновые производные, содержащие ОН-группу у С-6
атома, легко дегидратируются в слабо кислой среде, превращаясь в
неактивные ангидропроизводные. Этому способствует транс-атше-
рипланарная ориентация соседнего атома водорода. В отличие от
эпимеризации, данное явление наблюдается не только в растворах, но
и при длительном хранении этих веществ в ненадлежащих условиях.
145
N(CH3)2
ОН
CONK
CONR
Ангидротетрациклины имеют более глубокую, по сравнению с
исходными соединениями, окраску и обладают выраженными нефроток-
сическими свойствами. В этой связи наиболее безопасными из
приведенных в табл. 6.2 препаратов являются те, которые не способны
к дегидратации, например, метациклин и доксициклин.
Антибиотики тетрациклинового ряда по частоте применения в
медицинской практике уступают лишь пенициллинам, но, в отличие от
последних, их используют, главным образом, как пероральные средства
в терапии пневмоний разной этиологии, инфекций верхних
дыхательных путей, кишечника, мочеполовой системы, вызванных как
грамположительными, так и грамотрицательными бактериями. Бак-
териостатическое действие тетрациклинов обусловлено их
способностью взаимодействовать с 30S, а по некоторым данным и 50S
субъединицами в А-участках как прокариотических, так и эукариотических
рибосом. Этим они препятствуют отбору аминоацил-тРНК в А-участ-
ке и блокируют пептидный синтез. Причины, по которым избирательно
подавляется биосинтез именно у бактерий, окончательно не
установлены. Одно из предположений состоит в том, что тетрациклины
проникают через мембраны клеток микроорганизмов таким же путем, как
и пурины, т. е. бактерии ошибочно поглощают их как питательные
вещества. В результате в клетках прокариот эти вещества
накапливаются в токсических концентрациях гораздо быстрее, чем в клетках
млекопитающих. В то же время нельзя не отметить, что ингибирование
биосинтеза белка (антианаболический эффект) было отмечено у
человека при внутривенном введении тетрациклинов в больших дозах [7, 9].
Me
НО, N-Me2
Me ) S.
(6.25)
Макролиды (типичными представителями являются метаболит
Streptomyces erythreus — эритромицин (6.25) и его полусинтетические ана-
146
логи) по спектру антибактериального действия близки к пенициллинам.
По химическому строению они являются макроциклическими лактона-
ми. В отличие от пенициллинов эти вещества кислотоустойчивы,
благодаря чему макролиды, главным образом в виде солей различных
органических кислот или гидрохлоридов, применяют перорально [7, 11].
По механизму действия эти ЛВ являются ингибиторами
биосинтеза белка за счет блокирования транслокации аминоацил-тРНК на
50S субьединице рибосом. Эритромицин и его аналоги
используют для борьбы с инфекциями, устойчивыми к действию
пенициллинов. Однако быстро развивающаяся резистентность
микроорганизмов к этой группе антибиотиков ограничивает их применение [11].
Чрезвычайно важной группой современных антибактериальных-
средств являются производные хинолонкарбоновых кислот, которые
влияют на процессы транскрипции и репликации НК в
прокариотических клетках [12, 13]. К первому поколению ЛС названного
ряда относят налидиксовую (6.26), оксолиниевую (6.27) и пипемидие-
вую (6.28) кислоты.
(6.26) (6.27) (6.28)
Они получили распространение в середине 60-гг. в терапии
заболеваний, возбудителями которых являются грамотрицательные бактерии
(кишечная, дизентерийная, брюшнотифозная палочки). Но в
отношении грамположительных микроорганизмов (стафилококки,
стрептококки, пневмококки) хинолонкарбоновые кислоты оказались
неэффективными. В настоящее время их применяют, главным образом, в качестве
дезинфектантов мочевыводящих путей. Благодаря изучению широкого
круга аналогов (6.26)—(6.28) удалось установить, что наибольшей
активностью обладают соединения, содержащие атом фтора в
бензольном кольце бициклической хинолиноновой системы. Первым фторхино-
лоном, получившим широкое медицинское применение в середине
80-х гг., стал норфлоксацин (6.29). Вслед за ним появились пефлокса-
цин (6.30), офлоксацин (6.31), ципрофлоксацин (6.32) и ломефлокса-
цин (6.33).
О О
(6.32) (6.33)
По механизму действия препараты первого и второго
поколения являются ингибиторами топоизомераз (ДНК-гиразы и топо-
изомеразы IV) — Ф, поддерживающих сверхспиральную структуру
ДН К бактериальной клетки, без чего невозможно нормальное
осуществление процессов репликации и транскрипции. В грамот-
рицательных микроорганизмах первичной мишенью является ДНК-
гираза, вторичной — топоизомераза IV. Напротив, в грамполо-
жительных бактериях топоизомераза IV — первичная мишень.
Повышенная активность фторхинолонов по сравнению с
препаратами первого поколения объясняется их более высокой липофиль-
ностью и более выраженной способностью связываться с
соответствующими Ф [12, 13].
Фторхинолоны применяют при инфекциях мягких тканей и кожи,
дыхательных путей, мочевыводящей системы, ПК, туберкулезе и
многих др. заболеваниях [13, 14].
6.3.2. Противоопухолевые средства
Согласно современной классификации [9, 14]
противоопухолевые (антинеопластические, антибластомные) средства
подразделяются на:
• алкилирующие вещества — бис-(р-хлорэтил)амины, этиленами-
ны, алкилсульфонаты, нитрозомочевины, триазены;
• антиметаболиты — аналоги фолиевой кислоты, аналоги пуринов
и пиримидинов;
• вещества природного происхождения — алкалоиды, антибиотики;
• гормональные средства и их аналоги;
• ферменты;
• препараты разных химических групп — производные метилгидра-
зина, мочевины, антрацендионы, производные платины и др.
Среди вышеперечисленных средств непосредственное воздействие
на НК оказывают алкилирующие агенты, антибиотики,
производные платины, антрацендионы и акридины. Антиметаболиты —
аналоги пуринов и пиримидинов — нарушают синтез ДНК и РНК,
блокируя включение в их структуру соответствующих азотистых
оснований [15].
148
Вещества алкилирующего типа взаимодействуют с нуклеофиль-
ными центрами азотистых оснований ДНК, тем самым блокируя ее
репликацию. Сшивка двух цепей ДНК происходит по остаткам
гуанина (см. разд. 6.2). В результате нарушается способность клеток к
делению. Особенно выраженный цитостатический эффект проявляется
в отношении быстро пролиферирующих (размножающихся) клеток.
К группе алкилирующих средств принадлежат производные бис-(р-хлор-
этил)амина: сарколизин (6.34), эмбихин (6.35), циклофосфан (6.36),
допан (6.37), хлорбутин (6.38); этиленимины: тиофосфамид (6.39),
бензотэф (6.40); алкилсульфонат — миелосан (6.41) [15, 16].
(СН2)2С1
N.
\
(CHACI
(СН2)2С1
N —Ме-НС1
(СН2)2С1
(6.34)
С
\ /
P-N
' \ч \
0 0х
(СН2)2С1
•(СН2)2С1
(СН2)2С1
(СН2)2С1
(6.38)
(6.35)
н /Ме
Я-{ /ch2)2ci
о=< V\
N~^ (CH2)2CI
О
(6.37)
[>-Р-"3
N
(6.39)
Ph
Y
H П xi
N-P-N^j
I
N
IA
Me SO;
(CH2)3
(6.41)
S02Me
(6.40)
Главной мишенью N-нитрозаминов — ломустина (6.42) и кармус-
тина (6.43) — также является ДНК. Наличие в их структуре N-нитро-
зогруппы в а-положении по отношению к карбонильной группе в
значительной степени повышает электрофильные свойства последней.
Сшивка цепей НК происходит в результате взаимодействия
промежуточного алкилирующего агента с соседними нуклеофильными
компонентами ДНК.
149
NO
(6.42) (6-43)
Карбамоилирование
Алкилирование
Ингибирование репликации ДНК возможно и в результате
карбамоилирования под действием изоцианата [17]. Нитрозамины
способны выступать донорами N0, что также сказывается на
противоопухолевой активности этих веществ.
Свойства метилирующего агента проявляет дакарбазин (6.44).
В результате его микросомального окисления и дезалкилирования
образуется электрофильная частица CH3N2+, которая алкилирует
азотистые основания НК.
Н Me
(6.44)
К антиметаболитам — аналогам пуринов — относятся 6-меркап-
топурин (6.45) и тиогуанин (6.46), а также аналоги пиримидинов —
фторурацил (5.34), фторафур (5.35) и цитарабин (6.47).
NK
(6.45) (6.46) (6.47)
150
Механизм цитостатического действия этих веществ обусловлен
нарушением синтеза ДНК и РНК путем блокады включения в их
структуру соответствующих пуриновых и пиримидиновых оснований.
В разд. 5.4 уже были рассмотрены некоторые стороны механизма
действия фторурацила и его аналогов, в частности, их способность
необратимо связывать тимидилатсинтетазу. Однако, кроме ингибирования
названного Ф, эти соединения замещают истинные метаболиты в
структуре НК, что приводит к замедлению деления опухолевых
клеток. К сожалению, таким же образом эти препараты действуют и на
здоровые клетки, в особенности на быстро делящиеся клетки
костного мозга и кишечного эпителия [9].
Противоопухолевые антибиотики в большинстве случаев
являются природными соединениями и продуцируются различными видами
актиномицетов. Далеко не все такие вещества токсичны только для
опухолевых клеток, однако в связи с тем, что они, как правило,
действуют на механизм репликации, быстро делящиеся опухолевые клетки
поражаются ими в большей степени, чем обычные. Антибластомным
эффектом обладают митомицин С, блеомицин, доксорубицин, рубо-
мицин, дактиномицин, антрамицин.
Митомицин С (6.48) был выделен в середине 50-х гг. из культуры
Streptomyces verticillatus, а в начале 70-х гг. получен синтетическим
путем [18,19]. Этот антибиотик проявляет свойства алкилирующего
агента и одновременно является источником пероксидных радикалов, также
нарушающих структуру ДНК. Хиноидный фрагмент в молекуле (6.48)
in vivo претерпевает ферментативно катализируемое восстановление.
Кроме того, отмечается потеря ОСН3-группы. В результате в алкилирова-
нии ДНК принимает участие азиридиновый и уретановый фрагменты,
а гидрохиноновый является источником пероксидных радикалов [20].
Видовая чувствительность опухолей к этому антибиотику
объясняется, вероятно, различиями в способности клеток разных опухолей
репарировать поврежденную ДНК или нейтрализовать алкилирующие
агенты.
Блеомицины — группа антибиотиков, продуцируемых Streptomyces
verticillatus. По химическому строению это гликопептиды, содержа-
151
шие в структуре два тиазольных, имидазольный и аминопиримиди-
новый циклы. Эти вещества в виде комплекса с ионами Си2+ ингиби-
руют ДНК-лигазу, в результате чего в ДНК возникают разрывы [21].
Антрациклины — важнейший класс антибластомных средств,
обладающих политропным действием. Наиболее известными антрацик-
линовыми антибиотиками являются даунорубицин (6.49) и
доксорубицин (6.50).
R = CK
СН2ОН
(6.49)
(6.50)
ОН] '
NH2
Эти вещества встраиваются в ДНК за счет образования ионной
связи между аминогруппой гексозного фрагмента и фосфатным остатком в
НК, при этом плоская антрахиноновая система размещается между
парами оснований в двойной спирали. В результате такого встраивания
в двойную спираль — интеркаляции — снижается степень скрученности
ДНК и прекращается ее репликация. Доксорубицин ингибирует также
ДНК- и РНК-полимеразы. Кроме того, антрациклиновые антибиотики
активируют супероксидцисмутазу и способствуют продукции свободных
радикалов в клетках благодаря одно- и двухэлектронному
восстановлению в семихинон и гидрохинон соответственно. При окислении 02 эти
соединения образуют супероксид, перекись водорода и гидроксильный
радикал, которые вносят свой вклад в реализацию противоопухолевого
эффекта химиотерапевтических средств данного ряда [15, 20].
Среди производных антрацена выраженным антибластомным
действием обладают митоксантрон (6.51) и бисантрен (6.52).
(6.51)
(6.52)
Сходство этих структур с антрациклиновыми антибиотиками дает
основания предполагать, что их противоопухолевый эффект обуслов-
152
лен интеркаляцией ДНК. Однако в настоящее время это
предположение окончательно не доказано. Известно, что митоксантрон
связывается с гуаниновыми фрагментами в ДНК и обладает свойствами
индуктора свободных радикалов, повреждающих цепи НК [20].
Способность к интеркаляции лежит в основе антинеопластиче-
ского действия дактиномицина (актиномицина D, 6.53). Это
вещество открыто в культуре Streptomyces parvulus в середине 40-х гг. и вскоре
было получено синтетическим путем [22, 23]. В химической
структуре дактиномицина присутствуют феноксазин-3-оновый фрагмент и
две присоединенные к нему идентичные пентапептидные лактонные
группы. Известно, что благодаря наличию плоского фенокса-
зинонового фрагмента он способен встраиваться в структуру ДНК.
В отличие от рассмотренных выше случаев интеркаляции,
связывание дактиномицина обратимо. Однако образовавшийся комплекс
диссоциирует очень медленно, в результате оказываются блокированными
РНК-полимераза и биосинтез белка. Первоначально это вещество
испытывали в качестве антимикробного средства, однако из-за
высокой токсичности оно не получило клинического применения. В
середине 50-х гг. его с успехом стали применять для лечения
онкологических заболеваний. На сегодняшний день дактиномицин является
одним из наиболее широко изученных антибластомных средств.
.Sar -Sar
L-РгсГ ^L-Met-Val L-Pro ^L-Met-Val
II II
D-Va!^ /О D-Va!^ ^O
L-Тгео ^^L-Treo
Ю 0=
(6.53)
Антрамицин (6.54) также является продуктом природного
происхождения, однако имеет достаточно простую структуру пирро-
ло[1, 4]бенздиазепина, в составе которой есть высоко реакционно-
способный карбиноламинный фрагмент. Антрамицин ковалентно
связывается с гуанином и тем самым подавляет синтез НК [15, 20].
НО н ччОН
V N—< JH
ъу»'
CONH.
(6.54)
153
NH34 ,CI
Препараты платины — цисплатин (6.55), платин (6.56) и карбо-
платин (6.57) — появились в середине 70-х гг.
О
НО—NH2 с! Н2Ч Я-\ /\
,pt К А X/
NH'' <31 НО—NH, CI H2N о <s,
2 О
(6.55) (6.56) (6.57)
Главной мишенью для комплексов платины являются пуриновые
и пиримидиновые основания ДНК. При этом не затрагиваются ни
фосфорные, ни дезоксирибозные остатки. Препараты этой группы не
влияют на синтез РНК и белков и действуют одинаково эффективно
как на стадии активного синтеза ДНК в клетке, так и на стадии
митоза. Цитотоксическим эффектом обладают только цыс-производные
платины [9, 15, 20].
В заключении следует отметить, что все рассмотренные в данном
разделе ЛВ являются высоко токсичными соединениями. Их общий
недостаток — низкая избирательность действия. Антибластомные
средства алкилируюшего типа, антиметаболиты, интеркаляторы НК и др.
повреждают мишени не только в клетках, пораженных опухолевым
процессом, но и в нормально функционирующих, т.е. проявляют
свойства канцерогенов. В связи с этим поиск новых низкотоксичных и
высокоселективных средств чрезвычайно актуален. В настоящее
время в связи с такими достижениями в области молекулярной
генетики, как расшифровка генома человека, открываются перспективы для
выяснения тонких механизмов регуляции роста клеток. Так,
установлено, что одним из факторов, предотвращающим их неограниченный
рост, является специфическая генетическая программа,
определяющая конечное число делений, которое может претерпеть данная
клетка. По достижении этого числа делений клетки претерпевают
сложную систему деградации, которая получила название апаптоз.
Очевидно, нарушения в такой программе должны приводить к
неограниченному росту клеток, что свидетельствует о возникновении
опухолевого процесса. Понимание механизмов апаптоза способно
многое изменить в поиске антибластомных средств.
6.3.3. Противовирусные средства
Вирусы — неклеточные структуры, содержащие НК в белковой
оболочке — капсиде. Различают ДНК- и РНК-содержащие вирусы.
Для них известны четыре основных класса капсидов: спиральные,
икосаэдрические, сложные без оболочки, сложные с оболочкой.
В ДНК-содержащих вирусах генетический материал представлен либо
в одно-, либо в двухцепочечной ДНК, которая может быть как
линейной, так и кольцевой. Вирусная РНК также может состоять из
154
одной или двух цепей. Кроме того, вирусы классифицируют по
способности инфицировать про- и эукариотические клетки. Вирусы,
поражающие бактерии, носят название бактериофагов.
Процесс инфицирования, посредством которого рассматриваемые
неклеточные образования внедряются в клетку-хозяина и
«настраивают» ее метаболический аппарат на воспроизведение вирионов
(полностью сформированных вирусных частиц), протекает в несколько
этапов. Первый этап — адсорбция вируса на специфических рецепторных
участках клеточной поверхности, которые распознаются особыми
выступающими частями вириона и к которым он прочно
прикрепляется. Второй этап — проникновение вирусной ДНК или РНК в
клетку-хозяина — происходит у разных вирусов по-разному. Вирусы,
поражающие животных и человека, переносят НК в результате слияния
наружного слоя вириона с клеточной мембраной. При этом НК
попадает в клетку вместе с непосредственно прилегающими к ней белками,
последующее освобождение от них происходит ферментативным путем.
Транскрипция и репликация генетического материала вируса
осуществляются обычно с участием Ф клетки-хозяина. Сначала
вирусная ДНК копируется РНК-полимеразами клетки-хозяина, в
результате чего образуется мРНК, которая затем транслируется. При
этом транскрипция и репликация могут происходить как в ядре
(например, у вируса герпеса), так и в цитоплазме (например, у покс-
вирусов). Трансляция вирусной мРНК на рибосомах клетки-хозяина
приводит к образованию вирусных белков.
Сборка вируса и его компонентов обычно происходит спонтанно,
но может зависеть от участия вспомогательных белков. Вирусная ДНК
покрывается капсидом. Последний, в свою очередь, может
включаться в мембранную структуру, получаемую вирионом от клетки-хозяина.
РНК-содержащие вирусы, поражающие человека и животных,
могут быть как одно-, так и двухцепочечными. Одноцепочечные
вирусы принято разделять на два типа: с «плюс»-цепью и «минус»-це-
пью. У неклеточных структур первого типа РНК может
функционировать в клетке хозяина как мРНК, тогда как у структур второго типа
на «минус»-цепи должна сначала с помощью клеточных РНК-поли-
мераз образоваться «плюс»-цепь.
Примером вируса с «минуо-одноцепочечной РНК является вирус
гриппа, имеющий оболочку и спиральную сердцевиу. Основные белки
оболочки — гемагтлютинин и нейраминидаза. Сердцевина состоит из восьми
сегментов «минус»-РНК, которые в комплексе с белками образуют
спиралевидные структуры. Каждый сегмент кодирует один из белков вируса.
Инфекционный процесс начинается с прикрепления вируса к
поверхности клетки хозяина через гемагтлютинин. Происходит слияние оболочки
с клеточной мембраной, нуклеокапсид входит в клетку и кодируемая
вирусом РНК-зависимая РНК-полимераза синтезирует «плюс»-цепи мРНК
на вирусных «минуо-цепях. После этого на рибосомах клетки-хозяина
продуцируются вирусные белки. Репликация происходит в ядре, где с
помощью вирусной РНК-полимеразы (у вируса гриппа таких ферментов
три) образуются «минус»-цепи РНК. После того, как в ядро поступают
155
нуклеокапсидные белки, происходит сборка нуклеокапсида, который
затем проходит через цитоплазму, присоединяя при этом белки оболочки,
и покидает клетку. В процессе отпочкования вириона от плазматической
мембраны принимает участие нейраминидаза.
Совершенно иначе функционируют ретровирусы. Они являются «плюс»-
одноцепочечными структурами. Почти все ретровирусы онкогенны. Их
вирионы содержат РНК-зависимую ДНК-полимеразу (обратную транс-
криптазу), катализирующую обратную транскрипцию — синтез новых
молекул ДНК по программе, задаваемой вирусной РНК. С помощью этого
Ф в инфицированной клетке собирается единственная одноцепочечная
ДНК-копия вирусной РНК, которая при участии Ф хозяина превращается
в двуцепочечную ДНК. По ходу обратной транскрипции в качестве
промежуточных образований возникают гибридные ДНК-РНК-структуры, одна
цепь которых происходит из РНК вириона, а другая — из продукта
обратной транскрипции. Постепенно вирионная РНК в этом гибриде
разрушается, поскольку РНК-зависимой ДНК-полимеразе свойственна активность
РНКазы Н, т. е. способность катализировать гидролиз полирибонуклео-
тидной цепи в составе РНК-ДНК-гибрида. По мере разрушения РНК
синтезированная однонитевая ДНК становится матрицей для
формирования комплементарной ДНК-цепи. Полученная таким образом двухцепо-
чечная ДНК встраивается при участии специальных рекомбинационных
механизмов в ДНК хозяина, становясь частью хромосомы. В дальнейшем
новые молекулы вирусной РНК и белков, необходимые для образования
новых вирионов, производятся с участием общих систем транскрипции и
трансляции клеток хозяина. В результате заражения ретровирусом
меняется характер роста клеток: он становится быстрым и менее зависимым от
поступающих извне стимуляторов (таких как гормоны и факторы роста).
Основные группы вирусов приведены в табл. 6.3 и 6.4.
Таблица 6.3
ДНК-содержащие вирусы,
инфицирующие человека и животных [20, 24]
Группа
Аденовирус
Герпесвирус
Вид
Множество типов
Вирус простого герпеса
1 -го типа
Вирус простого герпеса
2-го типа
Вирус ветряной оспы
Вирус опоясывающего
герпеса
Вирус Эпщтейна-Барр
Цитомегаловирус
Заболевание
Респираторные
и глазные инфекции
Язвенный стоматит, герпесный
дерматит, экзема, конъюнктивит
Поражение гениталий, менингит
Ветряная оспа
Опоясывающий лишай
Гепатит, инфекционный моно-
нуклеоз, лимфома Буркетта,
карцинома и др.
Тератогенный эффект,
иммунодефицит, поражение
внутренних органов,
лимфоузлов, ЦНС
156
Продолжение таблицы 6.3
Группа
Герпесвирус
Паповирус
Поксвирус
Вид
Вирус герпеса человека
6-го типа
Вирус герпеса человека
7-го типа
Герпесвирус, связанный
с саркомой Капоши
Вирус папилом
Полиома вирус
SV40
Вирус оспы
Заболевание
Энцефалит, карцинома,
кофактор ВИЧ-инфекции
Синдром хронической усталости
Саркома Капоши
Папиломы человека и животных
Инфекции слюнных желез
Л ейко энцефалопатии
Оспа, коровья оспа
Таблица 6.4
РНК- содержащие вирусы,
поражающие человека и животных [20, 24]
Группа | Вид
Аренавирус
Ортомиковирусы
П арам иксовирусы
Пикорнавирусы
Реовирус
Ретровирусы
Рабдовирусы
Тогавирус
Вирус лимфоцитарого
хориоменингита
Вирус лихорадки Ласса
Вирусы гриппа А, В и С
Вирус парагриппа
Респираторный
сетчатый вирус
Вирус кори
Риновирусы
Энтеро вирусы
(коксаки А и В, эховирусы)
Ротавирус
Вирус Т-лимфоцитов
Вирусы иммунодефицита
человека 1-го типа
(HIV-1, HTLV-III/LAV)
Вирус бешенства
Вирус энцефалита
Вирус коревой краснухи
Арбовирус
Заболевание
Л имфоцитарный
хориоменингит,
геморрагические
лихорадки
Лихорадка Ласса
Грипп
Паротит, пневмония и
др.
Пневмония, бронхит
Корь
Респираторные
инфекции
Кищечные инфекции,
полиомиелит, менингит
Респираторные и
кишечные инфекции
Лейкемия, лимфома
и карцинома животных
Синдром
приобретенного
иммунодефицита
Бешенство, энцефалит
Менингоэнцефалит
Коревая краснуха,
энцефалит, гепатит
Гепатит, желтая
лихорадка и др.
157
Проблема создания эффективных и безопасных противовирусных
средств является одной из самых сложных в современной
медицинской химии и фармакологии. Сложность эта заключается в выявлении
веществ, способных избирательно подавлять репродукцию вирусов,
не затрагивая процессов жизнедеятельности клеток
организма-хозяина. Противовирусный препарат должен воздействовать на какой-либо
из этапов жизненного цикла вирусной инфекции:
• блокировать связывание вириона с определенными рецепторами
на поверхности клеток и последующее проникновение в клетку
путем дезинтеграции его оболочек;
• препятствовать транскрипции вирусного генома;
• ингибировать синтез специфических вирусных белков;
• подавлять активность вирус-специфических полимераз НК.
Строгая направленность действия — один из основных
критериев в отборе веществ с противовирусной активностью. При этом
биологическими мишенями являются вирусный геном,
вирус-специфичные ферменты (обратимые транскриптазы, протеазы, глюко-
зидазы). Вещества, тормозящие процесс дезинтеграции оболочки
вируса и перенос его генетического материала в клетку хозяина,
эффективны на начальных этапах развития инфекционного
процесса. К ним относят производные адамантана (ремантадин и др.),
которые применяют для профилактики и лечения начальной
стадии гриппа.
Агентами, влияющими на репликацию вирусных НК, являются
нуклеозиды и родственные им структуры. Так, галогенпроизводные
тимидина (6.58) эффективны в отношении ДНК-содержащих
вирусов. Наиболее известным препаратом этого ряда является идоксури-
дин (R = I), который под действием вирусной тимидинкиназы
превращается втрифосфат, ингибирующий клеточную ДНК-полимеразу.
Кроме того, галогенпроизводные тимидина выступают его
антиметаболитами. Встраиваясь в ДНК, они подавляют синтез мРНК и
вирусных белков [25, 26].
N
J R = I, F, Br, CF3 (6.58)
OH
Видарабин (6.59) получен из культуры Streptomyces antibiotycus.
Он нарушает репликацию вирусной ДНК и эффективен в
отношении вирусов герпеса различных типов. Основной недостаток ви-
дарабина — легкость дезаминирования в арабинозилгипоксантин
под действием аденозиндезаминазы. Период его полупревращения
158
составляет 1 ч. Для повышения стабильности вещества его
применяют совместно с ингибитором названного выше Ф —
соединением (6.60) [27, 28].
(СНА—Me
(6.60)
Цитарабин (6.61) используют чаще как антибластомное средство
для лечения лимфомы Буркетта. Он выступает конкурентом дезок-
сицитидина, ингибирует ДНК-полимеразу и редуктазу и тем самым
нарушает репликацию вирусной ДНК [29, 30]. Еще одним важным
представителем этого ряда ЛС является рибавирин (6.62),
обладающий широким спектром активности как в отношении ДНК-, так и
РНК-содержащих вирусов. In vivo рибавирин превращается в 5'-моно-
фосфат, являющийся ингибитором инозиндегидрогеназы. В
результате прекращается синтез гуаниловой кислоты, необходимой для
воспроизводства вирусных НК. Рибавирин эффективен в отношении
вирусов гриппа А и В, парагриппа, гепатита А, В и С, герпеса,
лихорадки Ласса [31, 32].
H2N
НО
о
. N
(6.61)
ОН ОН
(6.62)
Ациклические аналоги нуклеозидов, такие как ацикловир (6.63)
и ганцикловир (6.64), — высоко эффективные средства в
лечении герпеса. В клетках, пораженных вирусом, эти вещества под
действием вирус-специфической тимидинкиназы превращаются в
моно-, а затем в трифосфаты, которые тормозят синтез и
репликацию вирусной РНК. В здоровых клетках фосфорилирование
(6.63) и (6.64) не происходит, поэтому на репликацию клетки-
хозяина эти средства не влияют. Ганцикловир имеет ряд
преимуществ по сравнению с (6.63): обладает большей растворимостью
159
и действует на большее количество разновидностей герпеса (в том
числе и на цитомегаловирус) [33, 34].
0-Na+
ОН
Средством, блокирующим трансляцию мРНК и, как следствие,
синтез вирусных белков, является метисазон (6.65), проявляющий
наибольшую активность в отношении поксвирусов. В то же время
отмечена его эффективность и в отношении рино- и эховирусов,
вируса гриппа, парагриппа и др. [35].
(6.65)
В рамках важной проблемы поиска противовирусных средств
определенные особенности имеет отбор веществ с анти-ВИЧ
активностью. Несмотря на расшифровку генома ВИЧ-1 (состоит по крайней
мере из 9 генов) и выявления его специфического места связывания (так
называемый CD4 рецепторный белок) с клетками хозяина,
по-прежнему нерешенным остается вопрос о распознавании пораженных и не
пораженных вирусом клеток. Причина этого заключается в том, что
CD4 белок, кроме клеток иммунной системы (макрофагов, моноцитов
и т.п.), содержится также в клетках кишечного эпителия и многих др.
В этой связи возникает задача поиска веществ, которые были бы
активны только в пораженных вирусом клетках и не оказывали
токсического воздействия на здоровые ткани, что особенно актуально во время
длительного латентного периода (8—10 лет) до наступления
манифестной стадии заболевания. Не ясно также, каким образом по истечении
этого срока какая-либо инфекция активирует экспрессию генов ВИЧ-1
и как ее можно подавить [9, 20, 24].
Имеющиеся на сегодняшний день агенты, эффективные в
отношении вируса ВИЧ-1, разделяют на три группы:
• вещества — ИФ (обратимой транскриптазы, протеаз, глюкозидаз);
• вещества, препятствующие «раздеванию» оболочки вируса;
• вещества — ингибиторы экспрессии генов.
160
По химической структуре ингибиторы обратимой транскриптазы
ВИЧ-1 и ВИЧ-2 представляют собой модифицированные пентози-
Лированные пурины и пиримидины, выступающие в качестве
антиметаболитов, а также некоторые диазепиновые и пиперазиновые
производные, пиридоны. Первым соединением, получившим
распространение в качестве анти-ВИЧ средства, стал азидотимидин —
AZT (6.66), впервые полученный в 1964 г. [36]. Биологический эффект
AZT в отношении онковирусов, вызывающих лейкемию, установлен
в начале 70-х гг [37], но из-за высокой токсичности он не получил
распространения в качестве антибластомного средства, а в 1986 г. был
предложен в качестве анти-ВИЧ препарата [38, 39]. В настоящее
время круг нуклеозидных аналогов AZT расширился (6.67—6.74).
(6.66)
(6.67)
(6.68)
(6.69)
(6.70)
(6.71)
(6.72) (6.73) (6.74)
На основе исследования большого количества структур-аналогов
соединений (6.66) — (6.74) установлено, что высокую активность
обеспечивают:
161
• наличие 5-ОН группы;
• Н, N3, F в положении 3' или двойная связь в дезоксирибозном
фрагменте;
• наличие гетероатома, главным образом S, в фуранозном кольце;
• введение атома F в пуриновый или пиримидиновый фрагмент;
в то же время, наличие этого заместителя приводит к росту
токсичности [20, 24, 40].
В эксперименте противовирусную активность, значительно
превышающую AZT, проявили ациклические аналоги нуклеотидов —
соединения (6.75) - (6.77) [41].
NH
(6.75) (6.76)
(6.77)
• Специфическими ингибиторами ВИЧ-1-обратимой транскрипта-
зы являются соединения общего строения (6.78) и (6.79)- Эти
вещества проявляют активность в широком диапазоне концентраций:
для (6.78) ЕС50 ~ 0.06 - 1 мкМ, а для (6.79) ~ 0.0024 - 6.5 мкМ и ее
уровень существенным образом зависит от заместителей. В ряду (6.78)
наиболее активно соединение, у которого X = Y = СН3, а в ряду (6.79) —
соединение с X = Et, Y = Ph, Z = СН3 [42, 43].
О
X = Me, Et
Y = CH>OH,Me, Ph
Z = H, Me
(6-79)
Методами тотального скрининга из 600 различных структурных
классов были выявлены три циклические частично гидрированные
производные бензодиазепина (6.80), превосходящие по
выраженности и селективности действия AZT. Наличие в структуре (6.80) тио-
мочевинного фрагмента, заключенного в цикл, является
принципиальным для проявления этим веществом ингибирующей активности в
отношении ВИЧ-1-обратимой транскриптазы. Замена серы на
кислород приводит к снижению эффекта, а на селен, напротив,
способствует росту активности [44, 45].
s
(6.80) (6.81)
Еще одним представителем диазепинов, обладающим
выраженным анти-ВИЧ эффектом, является невирапин (6.81). Он активен и в
отношении штаммов вирусов, устойчивых к AZT. Наличие двух
симметрично расположенных пиридиновых циклов и NH-группы —
необходимое условие для сохранения высокого уровня активности [46, 47].
Среди ингибиторов протеаз представлены, главным образом, три-
и тетрапептиды, содержащие хинолиновые и частично
гидрированные инданоновые фрагменты (см. разд. 9) [48—50].
Ингибиторы экспрессии генов — олигонуклеотиды; ингибиторы
глюкозидазы — некоторые алкалоиды (кастаноспермин и его
производные). К группе веществ, препятствующих связыванию с CD4-pe-
цептором, относят некоторые соединения, существующие в форме
анионов, в частности, красители на основе симметрично
расположенных сульфонафталинов (голубой Эванса, трипановый голубой),
полимерные анионогенные соединения (поливинилсульфокислоты,
декстрансульфат) [24, 20].
В настоящее время поиск БАВ с анти-ВИЧ активностью
проводится с использованием технологий тотального скрининга, что по-
163
зволяет оценивать большие массивы соединений. В результате
выявлены несколько десятков веществ, активность которых нашла
подтверждение в исследованиях in vivo. Однако пока широкое
клиническое применение получили лишь несколько препаратов, главным
образом, из группы ингибиторов обратимой транскриптазы (AZT, неви-
рапин и некоторые др.).
Литература
1. Рис Э., Стернберг М. От клеток к атомам. Иллюстрированное введение
в молекулярную биологию. — М.: Мир, 1987.
2. Зенгер В. Принципы структурной организации нуклеиновых кислот. —
М.: Мир, 1988.
3. Стент Г, Кэлиндар Р. Молекулярная генетика. — М.: Мир, 1981.
4. KornbergA. DNA Replication. — San Francisco: W.H. Freeman, 1980.
5. Szekely M. From DNA to Proteins — the transfer of genetic information. —
London: Macmillan, 1980.
6. Arora S.K. /I J. Med. Chem. - 1985. - Vol. 28. - P. 1099.
7. Structure-Activity Relationship Among the Semisynthetic Antibiotics / Ed. by
D. Perlman. — New York: Academic Press, 1977.
8. The Aminoglycosides / Ed. by A. Whelton, H.C. Neu — New York: Marcel
Dekker, 1982.
9. Харкевич Д.А. Фармакология: Учебник. — 6-е изд., перераб. и доп. —М.:
ГЭОТАР Медицина, 1999.
10. Mitscher L.A. The Chemistry of Tetracycline Antibiotics. — New York: Marcel
Dekker, 1978.
11. Macrolide Antibiotics / Ed. by S. Omura. — New York: Academic Press, 1984.
12. Quinolone Antimicrobial Agents, 2nd ed. / Ed. by J.S. Wolfson, D.C. Hooper. —
Washington: D.C, Amer. Society of Microbiol., 1993.
13. Падейская Е.Н., Яковлев В.П. Антимикробные препараты группы фтор-
хинолонов в клинической практике. — М.: Логата, 1998.
14. Машковский М.Д. Лекарственные средства. Т. 1—2. — Изд. 13-е, новое. —
Харьков: Торсинг, 1998.
15. Chemistry of Antitumor Agents /Ed. by D.E.V. Wilman. — New York: Chapman
& Hall, 1990.
16. Hill D.L. A Review of Cyclophosphamide. — Springfield, IL: Charles С Thomas,
1975.
17. McCaleb G.S. et al. // J. Med. Chem. - 1963. - Vol. 6. - P. 669.
18. Webb J.S. et al. // J. Am. Chem. Soc. - 1962. - Vol. 84. - P. 3185.
19. Siuta GJ. et al. // J.Org. Chem. - 1979. - V. 39. - P. 3739.
20. Principles of Medicinal Chemistry 4,h ed. / Ed. by W.O. Foye, T.L. Lemke.
D.A. Williams. — Philadelphia: Lippincott Williams & Wilkms,l995.
21. Carter S. R. et al. Fundamental and Clinical Studies of Bleomycin. — Baltimore:
University Park Press, 1976.
22. Brockmann H. // Am. N. Y. Acad. Sci. - 1960. - Vol. 89. - P. 323.
23. MeienhoferJ. //J. Am. Chem. Soc. - 1970. - V. 92. - P. 3771.
24. Antiviral Agents and Viral Diseases of Man. 3rd ed. / Ed. by G.J.Galasso et al. —
New York: Raven Press, 1990.
25. Gold J.A. et al. // Am. N. Y. Acad. Sci. - 1965. - Vol. 130. - P. 209
26. Prusoff. W.H. II Pharmacol. Ter. - 1979. - Vol. 7. - P. 1.
27. Whitley R.J. etal. // N. Engl. J. Med. - 1977. - Vol. 297. - P. 283.
164
28. Whitley R.J. et al. // N. Engl. J. Med. - 1982. - Vol. 307. - P. 971.
29. Ward R.L.Stefens J.G 11 J.Virol. - 1975. - Vol. 15. - P. 71.
30. Nutter R.L. Rapp F. // Cancer Res. - 1973. - Vol. 33. - P. 166.
31. Ribavirin: a Broad Spectrum Antiviral Agent / Ed. by RASmith, W. Kirk-
patrick. — New York: Academic Press, 1980.
32. MolerF.W. etal. // N. Engl. J. Med. - 1991. - Vol. 325. - P. 1884.
33. Nilsen A.E. et al. // Lancet. - 1982. - N. 2. - P. 571.
34. Whitley R.J. et al. // N. Engl. J. Med. - 1986. - Vol. 314. - P. 144.
35. DoValle L.A. et al. // Lancet. - 1965. - N. 2. - P. 976.
36. Horowitz etal. // J. Org. Chem. - 1964. - Vol. 29. - P. 2076.
37. Ostertag W etal. // Proc. Natl. Acad. Sci-USA. - 1974. - Vol. 71. - P. 4980.
38. Mitsuya H et al Ц Proc. Natl. Acad. Sci-USA. - 1985. - Vol. 82. - P. 7096
39. Yarchoan R. et al. // Lancet. - 1986. - N 1. - P. 575.
40. BalzariniJ. etal. // Proc. Natl. Acad. Sci-USA. - 1989. - Vol. 86. - P. 332.
41. BalzariniJ. etal. // Proc. Natl. Acad. Sci-USA. - 1991. - Vol. 88. - P. 4961.
42. Camaraza M.-J. et. al. 11 J.Med. Chem. - 1992. - Vol. 35. - P. 2721.
43. Baba M. et al // Antiviral Res. - 1992. - Vol. 17. - P. 245.
44. White E.L. et al.// Antiviral Res. - 1991. - Vol. 16. - P. 257.
45. Parker K.A., Coburn C.A. // J. Org. Chem. - 1992. - Vol. 57. - P. 97.
46. Richman D. etal. //Antimicrob. Agents Chemther. — 1991. — Vol. 35. — P. 305.
47. Kohistaedt L.A. etal. // Science. - 1992. - Vol. 256. - P. 1783.
48. Ghosh A.K. Biker G, Schiltz G. // Synthesis . - 2001. - N. 15. - P. 2203.
49. De Clercq E. // Pure Appl.Chem. - 2001. - Vol. 73. - P. 55.
50. Liang Tong// Chem. Rev. - 2002. - Vol. 102. - P. 4609.
7
ФАРМАКОКИНЕТИКА
(ОСНОВНЫЕ ПОНЯТИЯ И МОДЕЛИ).
РОЛЬ ФАРМАКОКИНЕТИЧЕСКИХ ИССЛЕДОВАНИЙ
В СОЗДАНИИ НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
7.1. Основные понятия фармакокинетики
Из множества определений понятия «фармакокинетика»
наиболее точным, вероятно, является самое краткое: фармакокинетика —
это знание о перемещениях и превращениях ЛС в организме. В
иностранной литературе по фармакологии и медицинской химии при
рассмотрении превращений, происходящих с ЛС в организме, часто
используется заменяющая термин «фармакокинетика» аббревиатура
ADME от англ. absorption, distribution, metabolism, elimination.
Фармакокинетика тесно связана с фармакодинамикой, изучающей
специфическое действие ЛВ и представляющей набор сведений о том, каков
ответ организма на действие препарата [1—7].
Впервые термин «фармакокинетика» введен Ф. Достом в 1953 г. Однако
начало развития фармакокинетики как самостоятельного раздела общей
фармакологии связывают с возникновением учения о гистогематических
барьерах, предложенного Л.С. Штерн в 1918 г. Благодаря этому учению
сложилось представление о существовании в организме жидкостей,
относительно изолированных друг от друга. В современной фармакокинетике
исследуют перемещения и превращения ЛС в крови, лимфе, жидкости
анатомических полостей (ликворе, плевральной, перикардиальной и т. д.)
и межтканевой (иитерстициальной) жидкости. Попадая в какую-либо из
них, ЛВ имеет ограниченную возможность проникновения в другую. Первая
фармакокинетическая модель была предложена в 1919 г. В 30-е—50-е гг.
Т. Теорелл и Е. Крюгер-Тимер разработали более сложные, в том числе
и нелинейные модели [5].
Упрощенно путь ЛВ в организме можно представить в виде схемы
(рис. 7.1).
С «кинетической» точки зрения организм принято
рассматривать в виде совокупности сообщающихся отделов или камер.
Камера (compartment) представляет собой ограниченный не
изменяющийся во времени объем жидкости (ткани), в котором ЛВ
распределено равномерно. Молекулы ЛВ проникают в
различные отделы организма с разной скоростью. В кровь и
межтканевую жидкость большинство препаратов попадает быстро,
медленнее — в ткани и органы, имеющие хорошее кровоснабжение
(печень, легкие, почки, мышцы, мозг), и гораздо медленнее —
в кости, стекловидное тело, ногти, волосы.
166
выдыхаемый к
воздух /(Л I
легкие
почки
реабсорция
ликвор
ЦНС
связ.
с
белками
плазмы
фильтрация
экскреция
межклет.
Л-Р
связ.
жидкость
желудок
желчь
кишечник
экскреция
>ПК
Рис.7.1. Схема перемещений Л В в организме:
А — при введении внутрь; В — при внутривенном введении
Различают литеральные и парентеральные способы введения ЛС.
При энтеральном способе препарат, прежде чем попасть в системное
кровообращение, поступает в ПК. При парэнтеральном введении
всасывание происходит, минуя пищеварительную систему. К энтераль-
ным способам относятся пероральное (от лат. per os — через рот),
оральное или сублингвальное (под язык) и ректальное (от лат. per
rectum — через прямую кишку). Парэнтеральные пути введения —
трансдермальный, подкожный, внутримышечный, внутривенный, суб-
арахноидальный, ингаляционный и некоторые др. Вслед за
введением ЛС происходят абсорбция, распределение и выведение. Абсорбция —
процесс перемещения ЛВ от места введения к месту обнаружения.
Распределение — обратимое перемещение Л В от места абсорбции к
месту обнаружения. Выведение — необратимое перемещение ЛВ от
места обнаружения (метаболизм, почечная экскреция, желчная
экскреция, выделение через легкие, потоотделение, лактацию).
7.2. Физико-химические свойства лекарственных веществ
и их фармакокинетика
Все вещества, для того чтобы абсорбироваться, распределиться и
экскретироваться, должны пересечь множество барьеров, главным
образом, клеточных мембран. Пять основных механизмов
перенесения ЛВ через биологические мембраны (пассивная диффузия,
ускоренная диффузия, активный транспорт, пиноцитоз и фагоцитоз)
рассмотрены ранее (разд. 1Л.2).
Для ускоренной диффузии и активного транспорта характерно
участие соответствующих переносчиков. В связи с тем, что количе-
167
ство переносчиков ограничено, транспортные системы склонны к
насыщению. Транспортеры стерео- и структурно специфичны.
Падение концентрации ЛВ в месте его обнаружения с увеличением дозы
ЛС может быть обусловлено насыщением транспортной системы.
Проникновение с участием транспортеров возможно только для веществ,
имеющих строение, близкое к природным соединениям. Наиболее
общим способом проникновения большинства синтетических ЛС
является пассивная диффузия. Поскольку клеточная мембрана
представляет собой двойной липидный слой, вещество для
проникновения через мембрану должно растворяться в липидах. В мембранах
также имеются малые поры, через которые проникают вода и
растворенные в ней низкомолекулярные вещества. Количество таких пор
зависит от типа тканей. Наилучшим индикатором того, как будет
перемещаться вещество в организме, является величина его
парциального коэффициента Р (см. разд. 2).
Если ЛВ содержит способные к ионизации функциональные
группы, то, согласно ЗДМ, слабое основание, помещенное в основную
среду, будет существовать в недиссоциированнои форме; аналогичная
ситуация характерна и для слабой кислоты в сильнокислой среде. Не-
ионизированные вещества обладают большей способностью проникать
через двойной липидный слой (см. разд. 2). Таким образом, степень
ионизации является показателем, определяющим количество неионизи-
рованной формы, проникающей через мембраны. Между рН среды,
константой диссоциации и степенью ионизации существует взаимосвязь:
PH = p*a±lg(cV(l-a)),
где «+» — для кислот, «-» — для оснований
Следовательно, зная рКа, можно рассчитать степень ионизации
при заданном значении рН и определить относительную
концентрацию с каждой стороны мембраны:
С,
С2
С2
_ 1 + 10р"гр*.
июрН^л
1 + 10р*,-рН,
1 + 10p*,-pH2
Эти уравнения позволяют предсказать перемещения
исследуемого вещества in vivo.
7.3. Фармакокинетические модели
Для количественного описания процессов абсорбции,
распределения, метаболизма и выведения ЛВ из организма используются
специальные кинетические модели. Простейший вариант — линейные
модели, когда все формализуемые процессы описывают
кинетическими уравнениями первого порядка. Один из таких вариантов —
168
однокамерная модель, в которой весь организм представлен как
единое целое. На примере однокамерной модели удобно рассматривать
основные фармакокинетические показатели, такие как объем
распределения, клиренс, период полувыведения. Однако следует отметить, что
лежащее в основе этой модели предположение о гомогенности
распределения молекул ЛВ в организме является слишком упрощенным
(рис. 7.2). Кинетическое поведение ЛВ более адекватно описывается
с помощью двухкамерных или многокамерных моделей. Их
схематическое изображение представлено на рис. 7.3. Математическое
описание этих моделей приведено в изданиях [1, 2, 4—7].
w ^^ ©*®
Ik. \к к »гв
а б в
Рис. 7.2. Однокамерная фармакокинетическая модель
(однократное введение ЛВ):
а) внутривенное введение ЛВ, С—концентрация ЛВ, к,— константа скорости
элиминирования Л В; б) внесосудистое введение Л В, ка — константа скорости
абсорбции Л В из места введения А; в) внутривенное введение с учетом
образования одного метаболита, Сх — исходная концентрация ЛВ, Ст — концентрация
метаболита, rm — константа скорости метаболизма г. — константа скорости
элиминирования метаболита
he Ike U ^е *'•
а б в
Рис. 7.3. Многокамерные кинетические модели (однократное введение ЛВ):
а) двухкамерная модель, внутривенное введение ЛВ, Сх — центральная камера
(концентрация ЛВ в системном Кровообращении), С2— концентрация ЛВ в
периферических тканях, к12, Ц, — константы скорости проникновения и
элиминирования ЛВ в камеру С2; б) двухкамерная модель, внесосудистое введение ЛВ
из места А; в) трехкамерная модель, внутривенное введение С, — центральная
камера, Ст1 — концентрация первого метаболита в плазме, qm, сц —
константы скорости метаболизма и элиминирования первого метаболита, Ст2 —
концентрация второго метаболита в плазме, rm, г, — константы скорости
метаболизма и элиминирования второго метаболита
7.4. Абсорбция
Этот процесс характеризуется скоростью и количеством
поглощенного ЛВ и зависит от способа его введения.
Наиболее быстрым поступлением ЛВ в организм является
внутривенное введение. Этот путь используется исключительно для раство-
169
ров и обеспечивает легко предсказуемый уровень концентрации ЛВ в
крови. Таким способом вводят ЛС, обладающие коротким
периодом полувыведения и малой величиной терапевтического индекса
(ТИ = LD5l/ED50). Для пролекарств, которые в ПК должны
превращаться в активные формы, этот путь поступления в организм
непригоден. Вещества, которые не могут достичь ЦНС из-за неспособности
преодолеть гематоэнцефалический барьер (ГЭБ), также вводят не
внутривенно, а непосредственно в спинно-мозговую жидкость или
под твердые оболочки мозга (субарахноидально).
Внутримышечные инъекции менее надежны с точки зрения
скорости абсорбции, биодоступности и локальных эффектов. При этом
на'скорость всасывания оказывает влияние место введения ЛС. В
жировой ткани может образовываться депо ЛВ.
Самым распространенным и удобным для большинства
пациентов является пероральный способ введения лекарств. Поэтому чаще
всего обсуждается абсорбция из ПК. На процесс поглощения
вещества оказывает влияние:
• его водорастворимость;
• рН среды;
• рКл вещества;
• форма, площадь поверхности и тип кристаллической решетки;
• фармацевтическая форма (таблетки без покрытия и покрытые
оболочкой, капсулы, гранулы, драже и т. п.).
Желудок и пять отделов кишечника, составляющих гастроинтес-
тинальную систему, различаются анатомически, морфологически и
физиологически. Желудок выстлан гладким эпителием, среда в нем
кислая — рН находится в пределах 1—2.5. Теоретически это означает,
что вещества, содержащие кислотные функции, например,
ацетилсалициловая кислота (рЛГа= 3.5), абсорбируются, главным образом, из
желудка, т. к. кислая среда подавляет диссоциацию такого ЛВ.
Определить, какая часть его ионизированна при рН - 2, можно, используя
следующие уравнения:
lg(CI/Cua)=pH-pAi=2.0-3.5 = -1.5
С,/С =0.032
1/ Utl
При заданном значении рН только 3.2 % ацетилсалициловой
кислоты диссоциировано. Следовательно, неионизированная часть
препарата должна абсорбироваться в желудке. Но на практике не весь
введенный перорально аспирин попадает из желудка в системное
кровообращение. Не менее важны для всасывания площадь
поглощающей поверхности и время контакта с этой поверхностью. Так,
анатомически в желудке нет складок Kerckrin, villi и др., выстилающих
кишечник, благодаря чему поверхность последнего возрастает в
600 раз. Поэтому абсорбция слабых органических кислот относительно
низка в желудке и происходит главным образом в верхнем отделе
кишечника (двенадцатиперстной кишке), где значение рН = 5—6.
Неионизованные и липофильные соединения, напротив, легко
абсорбируются из желудка. Опорожнение желудка является относительно
170
быстрым процессом (Т|/2 = 20—60 мин). Можно ожидать, что вещества,
замедляющие высвобождение содержимого желудка (антимускари-
новые средства, Н2-антагонисты, опиоиды), снижают абсорбцию ЛВ,
в то время как препараты, промотирующие процесс опорожнения
(метоклопрамид), повышают всасывание. Пища также замедляет
поглощение веществ, т. к. замедляет опорожнение. Однако возможно и
обратное, если лимитирующей стадией процесса проникновения ЛВ
в ткани организма является его растворение [4, 6, 7].
Верхние отделы кишечника (двенадцатиперстная, тонкая и
подвздошная кишка) — наиболее важные места абсорбции. Они
обладают большой протяженностью и развитой поверхностью, рН
изменяется в пределах 5—7. Здесь имеется большое количество мест активного
транспорта и время пребывания веществ достаточно
продолжительное — от 4 до 6 ч.
Толстый кишечник имеет гораздо меньшую поверхность и
меньшее количество эффективных мест всасывания. В этом отделе ПК
наиболее щелочная среда: рН = 8—8.5. Здесь происходит распад
таблеток и капсул, покрытых кислотоустойчивой оболочкой. Такие ЛФ
изготовляют с целью защиты субстанций, склонных к деструкции под
воздействием кислой среды в желудке, например, р-лактамных
антибиотиков. В толстом кишечнике обычно абсорбируются и вещества,
образовавшиеся в тонком кишечнике из пролекарств.
Прямая кишка не имеет особого значения для проникновения
веществ, вводимых перорально. Однако здесь происходит всасывание
веществ, вводимых ректально (в виде суппозиториев, микроклизм и др.).
7.4.1. Количественные характеристики
процесса абсорбции
Большинство ЛС, используемых для приема внутрь,
представлены в виде твердых ЛФ (таблетки, капсулы, гранулы и т.п.), а не
растворов. Поэтому количество лимитирующих факторов,
препятствующих их усвоению, возрастает. Исследование эффективности
абсорбции из различных ЛФ приводит к понятию биодоступности.
Биодостунность в настоящее время определяют как
относительное количество ЛВ, которое в неизмененном виде достигает
системного кровообращения (степень биодоступности), и скорость, с
которой этот процесс происходит (скорость всасывания). Абсолютная
биодоступность имеет место тогда, когда вся введенная доза ЛВ
поступает в кровообращение и соответствует дозе, установленной при
внутривенном введении этого же вещества. В том случае, когда
невозможно осуществить внутривенное введение ЛС, проводят сравнение
между двумя разными пероральными формами, а полученное при этом
значение носит название относительной биодоступности [4, 6, 7].
Количественно биодоступность оценивают для одной дозы по
максимальному уровню концентрации ЛВ в крови или плазме,
времени наступления максимума концентрации и площади под кривой
171
зависимости «концентрация JIB — время». Изменение концентрации
ЛВ выражают уравнением:
dC/dt = -kcC
или в интегральном виде:
С = С0е
где С — концентрация в данный момент времени, С0 — введенная
доза, к, — константа скорости элиминирования, t — время.
Чаще используют логарифмическую форму:
ln(C0/C)=kct,
lnC = lnC0-kct
В графическом виде изменение концентрации ЛВ от времени
представлено на рис.7.4.
С, мг/мл '
С, мг/мл j
Рис. 7.4. Зависимость убывания концентрации Л В от времени:
а — внутривенное введение; б — пероральное введение
При этом график «а» соответствует простейшей однокамерной
модели (см. также рис. 7.2 а), а график «б» — однокамерной модели с
всасыванием (рис. 7.2 б). В фармакокинетических расчетах используют
значение AUC (area under the curve) — суммарной площади под кривой
концентрации ЛВ от момента его попадания в организм до полного
удаления из него. Математически AUC представляет собой интеграл:
AUC = jfCdt.
о
Во многих случаях рассчитывают не полную площадь под кривой,
а ее часть за определенный промежуток времени, как правило,
равный интервалам между двумя последовательными введениями ЛС.
Еще один важный параметр — среднее время удержания ЛВ в
организме MRT (mean residence time) — представляет собой отношение:
MRT = AUMC / AUC,
где AUMC (area under the moment curve) математически
выражается как интеграл от произведения времени на концентрацию
AUMC = jtCdt.
172
При внутривенном введении среднее время удержания ЛВ
обратно пропорционально константе скорости элиминирования:
MRT = l/ke.
Если вещество в значительном количестве в неизмененном виде
экскретируется с мочой, то эти данные также можно использовать
для оценки степени биодоступности. При этом, оперируя
параметрами AUC, абсолютную биодоступность определяют как:
раьс = ((AUC^ JAUC )■ (С/Сртг J) -100%,
■100%.
а относительную — как:
К, = ((AUCpcr и (test)/AUC„s os (stand)) • (Cpcr „ (standJ/C^ „ (test)))
AUC устанавливают интегрированием или любым методом
определения площади криволинейной трапеции.
В полулогарифмических координатах зависимость «концентрация
ЛВ — время», соответствующая внутривенному способу введения,
имеет линейный вид (рис. 7.5 а). При этом тангенс угла наклона
равен константе скорости элиминирования кс, а ордината точки
пересечения прямой с осью Y — логарифму начальной концентрации ЛВ.
Рис. 7.5. Изменение концентрации Л В во времени:
а — внутривенное введение; бив — пероральное введение
В случае однокамерной кинетической модели с всасыванием,
соответствующей пероральному способу введения ЛВ (рис. 7.5 б), 1пС0
находят как пересечение оси ординат с асимптотой к конечному
участку графика, а константы поступления ЛВ в камеру ка (см. также
рис. 7.2 б) и элиминирования кс по тангенсам углов наклона
асимптот, проведенных к начальным и конечным участкам графика в
координатах С/С0 от t [5].
Следует подчеркнуть, что абсорбция может быть не полной по
следующим причинам:
• нестабильность вещества (например, гидролиз р-лактамного кольца
антибиотиков пенициллинового ряда в кислой среде желудка);
• недостаточное время пребывания на абсорбирующей поверхности
кишечника;
• метаболизм под действием микрофлоры кишечника;
173
• метаболизм в кишечнике под действием пищеварительных Ф;
• первичный метаболизм в печени;
• биофармацевтические факторы.
Метаболические превращения ЛВ до поступления его в
системное кровообращение принято называть досистемным метаболизмом
или «йт-ра^-эффектом. Часть дозы, которая не подверглась досис-
темному метаболизму и достигла системной циркуляции,
определяется как произведение концентраций ЛВ, прошедших в
неизмененном виде через разные отделы ПК и печень:
F = F F. F ,
gui nit liver
где F ut, Fjnt, F!jvcr — доля ЛВ, попадающая в системный кровоток
после прохождения через желудок, верхние отделы кишечника и печень.
Из изложенного выше следует, что невозможно установить био-
доступность препарата, основываясь только на использовании меченного
радиоактивными изотопами ЛВ. Следует иметь в виду, что вещество
может быть абсорбировано на 100 %, а его биодоступность при этом
будет равна 0, т. к. ЛВ подверглось первичному метаболизму в печени.
При исследовании биодоступности очень важно использовать
рациональные ЛФ, т.к. роль биофармацевтического фактора также
чрезвычайно велика. Для абсорбции ЛВ важно, чтобы оно легко
растворялось. Скорость растворения, зависящую от площади
поверхности растворяемого вещества и вязкости среды, принято выражать как:
dC/dt = kS(Cs-C),
где S — площадь поверхности растворяемого вещества, Cs —
растворимость, С — концентрация в момент времени t, k — коэффициент,
пропорциональный вязкости среды.
Основным способом увеличения поверхности растворяемого
вещества является повышение степени его измельчения путем
уменьшения размеров частиц до 10 мм и менее — микронизация. Это
повышает скорость растворения, следовательно, и биодоступность.
Примером может служить гипогликемический препарат глибенкла-
мид (4.12), для которого степень выраженности фармакологического
эффекта существенным образом зависит от размера частиц,
образующих таблетку. Однако иногда результат оказывается
противоположным, т.к. микронизация способна вызвать агрегацию частиц, и в
растворе или в процессе таблетирования такие мелкие частицы могут
чрезвычайно плотно слипаться между собой. В отдельных случаях
повышенное измельчение ускоряет разложение вещества в ПК.
Использование аморфных порошков вместо кристаллических в
производстве твердых ЛФ положительно сказывается на их растворимости.
Часто желаемого результата можно достичь и путем перехода к
солевым формам ЛВ [4, 6].
Какие же вещества имеют наибольшие шансы на
удовлетворительный уровень биодоступности (содержание в крови не менее 40%
от общей величины введенной дозы)? В первую очередь, те
соединения, в структуре которых не более 5 протонодонорных и 10 протоно-
174
акцепторных групп, мол. масса которых не превышает 500, и \%Р
находится в пределах (-1)-(+5). Это так называемое мнемоническое
«правило 5» или принцип «лекарствоподобия» С.А. Lipinski [8]. Таким
образом, при пероральном способе введения оптимальными физико-
химическими характеристиками можно признать наличие у вещества
относительно небольшой мол. массы, умеренной липофильности
и водорастворимости, а также средней способности к образованию
Н-связей. Связь между мол. массой и биодоступностью некоторых
широко применяемых в медицинской практике препаратов наглядно
иллюстрирует рис. 7.6.
Кол-во, 11
ЛС -
4J
У .
1
fjl rfl rfl нп
Г 'I1 Ч1 Ч1 Ч' Ч' Ч1 ■!■ "i in m ^
1 2 3 4 5 6 7 8 9 10 11 Мол. масса
ЛС
Рис. 7.6 Зависимость мол. массы ЛВ и биодоступности [9]:
1—11 — группы ЛС с мол. массой в соответствующем интервале ед. массы
1. 100-149 4. 250-299 7. 400-449 10. 550-599
2. 150-199 5. 300-349 8. 450-499 11. 600-649
3. 200-249 6. 350-399 9. 500-549
Диаграмма составлена на основании данных, полученных для
286 препаратов, предназначенных для перорального применения. Ниже
уровня 40% расположено 118, выше — 168 ЛВ, т. е. 59% всех
исследованных препаратов. Мол. масса большинства из них находится в
пределах 250—400 ед. массы. Среди исследованных ЛВ низкой
биодоступностью обладают: оксациллин (33%), анаприлин (26%), спиронолактон
(25%), лизиноприл (25%), норфлоксацин (35%), пентоксифиллин (33%).
Средний уровень выявлен у вольтарена (54%), ранитидина (52%), ци-
метидина (62%), фуросемида (60%), ампициллина (62%), каптоприла
(65%). Высокая биодоступность присуща левомицетину (75—90 %),
индометацину (98%), а 100 % отмечена у фенобарбитала,
ацетилсалициловой кислоты, триметоприма, сульфаметоксазола, кофеина [9].
Рассматривая такие понятия как абсорбция и биодоступность,
следует еще раз подчеркнуть, что фармакологический профиль ЛВ
тесно связан с его фармакокинетическими показателями.
Следовательно, если специфическое действие перорального средства
реализуется за счет связывания с рецептором или блокирования какого-
либо Ф, находящихся во внутренних органах, ЦНС, крови и т. п., то
эффективность такого препарата во многом определяется его
биодоступностью. Однако существует ряд ЛВ, для которых эффективная
175
абсорбция из ПК нежелательна. Это кишечные дезинфектанты и
ингибиторы ряда пищеварительных Ф. К последним относятся
ингибиторы а-глюкозидазы — акарбоза и гастроинтестинальной липазы —
орлистат (7.1), местом действия которых является кишечник.
Н
OHC-N
н3с-(сн2)5 ^^(сн^
(7.1)
Абсорбция (7.1) из ПК чрезвычайно низка. При однократном
приеме орлистата внутрь в дозе 800 мг максимальная концентрация его в
плазме не превышает 5 нг/мл. Он не кумулирует при многократно
повторяющемся введении, при этом концентрация препарата в
неизмененном виде в плазме не превышает 1% от общей дозы. Такие
фармакокинетические свойства орлистата позволяют избежать
нежелательного влияния на активность печеночных липаз, которые
структурно близки к гастроинтестинальной липазе [10]. Таким образом,
слабое всасывание веществ, реализующих свои специфические
эффекты исключительно в ПК, является преимуществом.
7.5. Распределение лекарственных веществ
Биологические мишени, с которыми связываются ЛВ, находятся в
разных органах и тканях. Фармакологический эффект препарата зависит
не только от того, как быстро и в каком количестве абсорбируется
вещество, но и от того как, оно распределяется. Среди основных факторов,
влияющих на процесс распределения, находятся структура тканей, рН
среды и способность вещества связываться с макромолекулами.
Вещества с небольшой мол. массой и водорастворимые быстрее
всего проникают в клетки через водные поры. Наибольшее
количество пор содержат мембраны капилляров. Через стенки сосудов
лекарства поступают в экстраваскулярную жидкость. Клетки ЦНС
содержат очень малое количество водных пор и вещества должны
обладать значительной липофильностью, чтобы проникнуть в
мозговую ткань. Распределение препарата, при наличии в его структуре
кислотных или основных функциональных групп, также зависит от
соотношения ионизированной и неионизированной форм.
Различия в физиологических значениях рН в тканях влияют на
характер распределения ЛВ. Плазма несколько более основна, чем
ткани, поэтому и основные вещества в ней менее ионизированы.
Следовательно, они могут легко переходить в ткани, где при несколько
меньшем значении рН претерпевают протонирование, что препятствует
хт
176
их проникновению обратно в плазму. Противоположное справедливо
дл« слабых кислот. Таким образом, основные вещества стремятся
распределиться вне плазмы (в тканях) в большей степени, чем кислотные.
При оценке способности ЛВ связываться с макромолекулами
обычно наибольшее внимание уделяют связыванию с белками плазмы, т. к.
этот процесс легко поддается количественной оценке. Однако
связывание в тканях не менее важно. Ведь главный показатель
распределения — это соотношение между концентрацией ЛВ в тканях и в
плазме. Соединения кислотного характера связываются с альбумином,
присутствующим как в плазме, так и в тканях. Основные вещества
образуют связи с а-кислотными остатками гликопротеинов и липо-
протеинами. Скорость ассоциации и диссоциации для обратимого
связывания с белками очень высока, поэтому связанные и свободные
формы постоянно находятся в равновесии. Проникновение в клетки
и обратно в плазму возможно только для несвязанных веществ [1—4].
Количественной характеристикой распределения является его
объем. Под объемом распределения понимают соотношение между
концентрацией вещества в плазме (С0) и общим количеством
вещества в организме (С):
Часто используют и уравнение:
Vd=C/(AUCA2),
где Xz — тангенс угла наклона кривой «концентрация ЛВ — время» на
конечной стадии.
Таким образом, Vd — это воображаемый объем, в котором могли
бы равномерно распределиться молекулы ЛВ, если бы их
концентрация была такой же, как и в плазме. Обычно этот показатель не связан
с физиологическим объемом. Vd — математический показатель,
который имеет размерность объема. Объем плазмы у человека массой 70 кг
составляет - 3 л, крови ~ 5 л, внеклеточной жидкости без учета
плазмы ~ 12 л, а общий объем водной фазы около 42 л. Однако для
многих препаратов объем распределения значительно выше приведенных
значений. Например, для атропина Vd = 1600 л, дигоксина — 512.8 л,
в то же время для хлорпропамида Vd = 6.8 л. Высокие значения
рассматриваемого показателя свидетельствуют о предпочтительном
распределении препарата в тканях по сравнению с плазмой. Для тех
же ЛВ, которые задерживаются внутри сосудистого русла, Vd близок
к реальному объему крови [1,2, 4].
7.6. Выведение лекарственных веществ
Эффект ЛС снижается со временем за счет распределения ЛВ между
тканями, кумуляции и образования депо, например, для липофиль-
ных веществ в адипоцитах, а также выделения вещества в
неизмененном виде или экскреции его метаболитов.
177
Выведение состоит из метаболизма, почечной (ренальной) и
желчной (билиарной) экскреции, выведения через легкие,
потоотделение и лактацию. Метаболизм БАВ заслуживает специального рассмбт-
рения (см. разд. 8).
Процесс выделения ЛВ изучают как с помощью радиоактивной
метки, включенной в структуру исследуемого вещества, так и
специфическими методами, т. к. метаболиты тоже содержат метку.
Наиболее важными органами, через которые осуществляется
экскреция, являются почки и печень. Все низкомолекулярные
соединения, которые не связываются с протеинами с большой мол.
массой, фильтруются через почки. Количество вещества, выводимого
из организма с мочой, зависит от трех процессов в почках: клу-
бочковой фильтрации, канальцевой реабсорбции, а также каналь-
цевой секреции. Общая скорость клубочковой фильтрации
составляет приблизительно 125 мл/мин и обычно характеризуется
клиренсом креатинина. При этом удаляется только та часть
препарата, которая не связана с белками плазмы. Канальцевая секреция
включает активный транспорт, поэтому процесс не зависит от того,
связано вещество с протеинами или нет. Следует отметить, что
транспортные системы имеют различный механизм для кислот и
оснований. Однако внутри каждой группы этот процесс
неспецифический и наблюдается конкуренция между соединениями. Этот
факт часто используют для подавления быстрого выведения
некоторых лекарств, например антибиотиков цефалоспоринового ряда,
с целью увеличения времени их действия.
Очень важным фактором, относящимся к почечной экскреции,
является канальцевая реабсорбция, которая имеет место для
большинства ЛВ благодаря пассивной диффузии через нефроны.
Склонность веществ к реабсорбции зависит от их способности проходить
через мембраны, полярности и склонности к ионизации, а также от
рН мочи.
Полярные соединения, такие как р-лактамы, с трудом могут
реабсорбироваться и поэтому интенсивно выделяются через почки.
Если моча имеет кислую среду, основные вещества переходят в
ионизованную форму и не могут вновь абсорбироваться, следовательно,
их ренальная экскреция возрастает. Канальцевая реабсорбция
основных веществ наблюдается тогда, когда рН мочи > 8. Указанное
можно проиллюстрировать на примере никотина (рис. 7.7).
Таким образом, рН мочи имеет очень важное значения для
процесса выведения вещества. Обшая скорость ренальной экскреции
определяется как:
V =V +V -V
RE fill г seer reabs
Билиарная экскреция представляет собой комплексный процесс,
который не до конца изучен. Вещества, выводимые таким путем, сек-
ретируются в желчь против градиента концентрации и поэтому
требуют участия активных переносчиков. Факторы, влияющие на
уровень билиарной экскреции, включают полярность (наличие в структуре
178
^—1
—*-
t, min
Рис. 7.7. Зависимость концентрации никотина в плазме от времени
при различных значениях рН мочи: 1) рН> 7.5; 2) рН < 6.5 [11]
высокополярных групп облегчает выведение с желчью) и некоторые
другие структурные особенности. Одна из наиболее важных — размер
молекул. Установлено, что для веществ с мол. массой меньше 300
или, наоборот, с очень большой массой (белки) экскреция с желчью
чрезвычайно мала [12]. При мол. массе 300—500 существует видовая
специфичность (табл. 7.1).
Таблица 7.1
Видовая специфичность уровня билиарной экскреции веществ
с различной мол. массой
Вид
Крысы
Собаки
Морские свинки
Кролики
Приматы
Человек
Мол. масса
325
325
400
475
500
500
Выделение ЛВ через пот и с выдыхаемым воздухом для
препаратов, предназначенных для прерорального применения, не имеет
особого значения. Выделение же ЛВ с грудным молоком может
существенным образом сказаться на состоянии организма новорожденных,
поэтому эти исследования составляют интегральную часть
фармакологических испытаний новых ЛС.
7.6.1. Количественные характеристики
процесса выведения лекарственных веществ
Клиренс (очищение, коэффициент очищения) — один их
основных показателей, используемых в фармакокинетике. Он может быть
установлен только при выполнении специфических исследований.
Обычно клиренс рассчитывают при внутривенном введении
вещества, т. е. когда показатель биодоступности F = 1. Клиренс аддитивен
и складывается из ренального, билиарного и метаболического:
С, ng/ ml
100
10
179
CL = CL + CL., + CL
ren bil m
CLren =Ut/AUC ,
где Ut— количество вещества, экскретируемое почками в
неизмененном виде в единицу времени t, AUCt — площадь под кривой
зависимости концентрации в крови или плазме за тот же промежуток времени.
Зная ренальный и общий клиренсы, рассчитывают количество ЛВ,
экскретируемого с мочой в неизмененном виде:
f =(CLren/CL)-lOO%.
Эффективность удаления Л В тем или иным органом принято
описывать количественно с помощью такого показателя, как
экстракционное соотношение (ER). Этот параметр показывает, какая часть ЛВ
удаляется из системного кровообращения за одно прохождение
препарата через рассматриваемый орган:
Сгп / \ Cout
metab
ER = (C. -С )/С. ,
\ ia out // in'
где Cin — концентрация ЛВ в артериальной крови, Сош —
концентрация Л В в венозной крови.
Если орган не очень эффективен, то Сои1 - С in, ER - 0, и,
наоборот, эффективен, если Coul= 0, a ER = 1. Следовательно, ER может
находиться между значениями 0 (нет выведения) и 1 (полное
выведение). Предположим, что выделительным органом является печень.
При пероральном способе введения очень незначительное
количество ЛВ может избежать проникновения в печень, куда большинство
из них попадает через портальную вену при первичном всасывании.
Если при этом вещество подвергается метаболизму, то значение ER
приближается к 1 [12].
Величина ER связана с таким показателем как биодоступность
следующим соотношением:
F = 1 - ER, или F% = 100% - ER%,
где F — доля Л В, попадающая в системный кровоток после первого
прохождения через печень.
Вторым по значимости фактором, определяющим скорость
доставки препарата к элиминирующему органу, является скорость
кровотока — dQ/dt. Произведение величины ER и скорости кровотока
есть клирбнс:
CL = dQ/dtER.
При внутривенном введении ER->1, и клиренс приближается к
величине скорости кровотока в печени, почках и т. п. Клиренс крови
в печени позволяет установить, является ли испытуемое ЛВ объектом
первичного метаболизма при его внутривенном введении.
180
Если при клиническом использовании препарата предполагается
его многократное введение, и ответ организма (желательный —
терапевтический, нежелательный — токсический или др. побочный)
связан с уровнем концентрации этого вещества в плазме, то при
установленном значении клиренса можно легко определить число введений
в сутки.
Существует связь между клиренсом и такими фармакокинетиче-
скими показателями как Vd и к,, которая в рамках однокамерной
модели имеет следующий вид:
CL = C/AUC,
kc=CL/Vd.
В простейшем случае клиренс ЛВ — это суммарная скорость
элиминации всеми возможными способами из организма, деленная на
его концентрацию в плазме:
CL = кс/С.
В зависимости от того, какая концентрация рассматривается,
будет изменяться и название клиренса, т. е. клиренс из крови, клиренс
из плазмы и т. п.
Если клиренс, объем распределения и абсорбция изменяются от
количества вводимых доз, то кинетика носит название нелинейной.
Очевидно, что если клиренс используется для расчета режима доз, то
исследования должны выполняться таким образом, чтобы значение
этого параметра оставалось постоянным при изменении дозы (дозо-
зависимая кинетика) или времени (времязависимая кинетика).
Проверка дозозависимой кинетики обычно осуществляется путем
проверки того, изменяется ли в прямой пропорции AUC при увеличении
дозы (рис. 7.8). Времязависимую кинетику устанавливают путем
мониторинга устойчивого состояния уровня концентрации вещества
в плазме и проверки, как он изменяется со временем.
AUCA
Рис. 7.8. Виды кинетических кривых:
1 — линейная зависимость AUC от концентрации ЛВ; 2 — выделение с
насыщением; 3 — всасывание с насыщением при пероральном введении
или связывание с белками плазмы с насыщением при внутривенном введении
Период полувыведения (время полувыведения, t ) — еще один
широко используемый фармакокинетический показатель, который оп-
181
ределяют как время, необходимое для того, чтобы концентрация ЛВ
снизилась на 50%. И если процесс выведения строго экспоненциален,
то период полувыведения постоянен при любой концентрации.
Величину t1/2 легко установить, если зависимость 1пС от времени носит
линейный характер.
В рамках однокамерной модели справедливы следующие
соотношения:
-kct.»
С0/2 = Сое "2;
ke=ln2/t,/2=0,693/t1/2;
t1/2=0,693Vd/CL.
Период полувыведения является функцией объема
распределения и клиренса, поэтому не всегда отражает ожидаемые изменения
элиминации. К изменению t1/2 приводят как изменения в объеме
распределения и клиренсе, так и в обоих параметрах
одновременно. Исходя только из величины х причину этого установить
невозможно. Для выявления изменяющегося параметра вещество
вводят внутривенно в нескольких дозах. Если изменяется клиренс, то
уровень устойчивого состояния концентрации вещества в плазме
также изменяется. При изменении объема распределения уровень
устойчивого состояния в плазме остается постоянным.
Следовательно, период полувыведения Л В из организма только в том случае
реально отражает длительность элиминации, когда скорость
выведения имеет первый порядок.
В процессе конструирования новых ЛС, имея в распоряжении
основные фармакокинетические параметры, можно направленно
изменять структуру создаваемых веществ. Множество лидирующих
структур на этапе доклинических испытаний отсеиваются не
только из-за наличия у них нежелательных токсических свойств или
неудовлетворительного уровня специфического эффекта, но и из-
за низкого фармакокинетического профиля: слабой абсорбции,
высокой степени первичного метаболизма и неподходящего
времени полувыведения (очень длительного или, наоборот, короткого).
Для того, чтобы отклонить БАВ, обладающие неподходящими
параметрами на стадии доклинических исследований, необходимо
экстраполировать фармакокинетические характеристики, полученные
в эксперименте у животных, на человека. Это можно сделать,
приняв во внимание, что любой параметр (CL, Vd, t1/2) связан с
массой тела степенной функцией:
Par = к (масса тела)х
Функция (х) и коэффициент (к) специфичны как для каждого из
параметров, так и для каждого вещества. Например, х = 1 для Vd, для
CL х = 0.75, для t, 2 х = 0.25. На практике чаще всего строят
зависимость lg параметра от lg массы тела.
182
1.1. Фармакокинетика
некоторых групп лекарственных средств
Фармакокинетические исследования являются неотъемлемой
частью программы клинических испытаний кандидатов в ЛС.
Например, при изучении нового гипогликемического препарата глимепи-
рид (4.14) в клинике у 100 здоровых добровольцев было установлено,
что при внутривенном введении в дозе 1 мг Vd = 8.8 л, CL = 48 мл/мин.
Глимепирид имеет низкую растворимость — 0.03 мг/л при рН = 1.2 и
7.4 мг/л при рН = 7.4; Р(октанол/вода) = 100 или lg P~ 2 при рН = 7.0,
рЛГа = 6.2. Препарат легко связывается с альбумином плазмы,
количество не связанного вещества менее 1%. Биодоступность при перо-
ральном введении составляет 97%. Пик концентрации глимепирида в
плазме отмечается через 2—3 ч. Фармакокинетика препарата носит
линейный характер, т. е. разброс между значениями величины AUC
для разных доз незначителен и связан, главным образом, с низкой
растворимостью вещества. Различий в фармакокинетических
показателях между пациентами; страдающими ИНЗСД, и здоровыми
добровольцами не отмечено. Глимепирид не кумулирует при повторных
введениях. При однократной дозе 2—8 мг t1/2 для него составляет 5.2—
7.2 ч, а при повторном применении доз 4—8 мг этот показатель
увеличивается до 7.8—8.8 ч. Длительный период полувыведения
обосновывает целесообразность однократного приема препарата в течение
24 ч [13].
Фармакокинетические показатели четырех антидиабетических
препаратов (производных сульфонилмочевины) можно сравнить с
помощью данных, приведенных в табл. 7.2.
Таблица 7.2
Фармакокинетические показатели
некоторых антидиабетических препаратов [13]
Препарат
Глимепирид
(4.14)
Глипизид
(4.13)
Глибенкламид
(4-12)
Гликлазид
(4.11)
ftl, %
0.6
2.0
< 3
3-15
CL,
мл/мин
48
50
101
13
ХьЛ
9
10-12
8
15
tl/2, Ч
5-8
1-4
2-5,
при ИНЗСД
(8-13)
8-21
Выведение
2 метаб., 1 активн.,
40—50 % экскрет,
почками
неактивн. метаб.,
65—70 % экскрет.
почками
2 метаб., 1 активн.,
25—50 % экскрет,
почками
2 метаб., неактивн.,
60—70 % экскрет,
почками
fu, % — количество вещества, не связанного с белками плазмы.
Показательными для освещения роли фармакокинетических
исследований являются примеры из группы антиангинальных ЛС, к ко-
183
торым относятся нитраты и блокаторы Са2+-каналов, а также
представители из группы антиадренергических препаратов, в которой
будут рассмотрены р-адреноблокаторы. Эти средства используются в
лечении ишемической болезни сердца (ИБС). Препаратами первого
выбора для предупреждения и купирования приступов стенокардии
уже более ста лет остаются нитраты — нитроглицерин (7.2), изоамил-
нитрит (7.3), изосорбида динитрат (7.4), шосорбида мононитрат, эрит-
ринил тетранитрат (7.5), пентаэритрол тетранитрат (эринит, 7.6).
02NO
02NO H
н
ON02
(7.2)
.О
6no2
ON02 I
02NO—\ ^ON02
02NO ON02
(7.3)
02NO^r-
02NO—^ ^
-ON02
-ON02
(7.4) (7.5) (7.6)
Впервые антиангиналъный эффект был выявлен у изоамилнитрита в 1857 г.
Позже предположение о том, что нитроэфиры алифатических спиртов
должны обладать таким же фармакологическим действием, оправдалось,
и в арсенале сердечно-сосудистых средств появились все перечисленные
выше препараты. Несмотря на то, что они используются в медицинской
практике очень давно, механизм их действия стал понятен только после
того, как в середине 80-х гг. XX в. была установлена роль оксида азота —
N0 в контроле васкулярного тонуса (см. также разд. 4.2, рис. 4.3). Сейчас
полагают, что действие нитратов обусловлено, в первую очередь, тем, что
они являются донорами N0. За счет этого происходит расширение арте-
риол и снижается периферическое сосудистое сопротивление. Кроме того,
они экономизируют работу сердца, уменьшают потребность миокарда
в кислороде [14, 15].
По химическому строению соединения (7.2) — (7.6) являются эфи-
рами алифатических спиртов и азотной (изоамилнитрит — азотистой)
кислоты. Все они обладают высокой липофильностью, что определяет
их быстрое и практически полное всасывание через мембраны не только
слизистых оболочек полости рта и ПК, но также и при нанесении на
кожу. После приема внутрь и абсорбции большинство нитратов
претерпевает первичный метаболизм в печени под действием нитратредуктазы.
Поэтому для обеспечения терапевтического уровня концентрации в
системном кровообращении применяют специальные
пролонгированные ЛФ этих веществ. В табл. 7.3 приведены некоторые фармакоки-
нетические показатели нитратов, которые позволяют сравнивать
биодоступность, длительность действия и др. характеристики не
только различных веществ, но и разных ЛФ одного и того же ЛВ.
184
та и
К 3 Т
S n а
*£* 1
03 У ГО . - «
и 5 - g -
^ г
£&
о
5.
II
it
S
со
ё
>>
и
аз 3
н О.
f- =5 Й —
о а ч о
н И 4
sal
I
151
8.
с
т
1»!
о
-е-
е
и
Ш
В случаях сублингвального, буккального (аппликация на
слизистую десны или щеки), аэрозольного или внутривенного введения
рассматриваемые вещества сразу попадают в системный кровоток и
достигают мишеней — коронарных и системных сосудов, при этом почти
не разрушаясь в печени, т. к. только около 15% объема сердечного
выброса приходится на кровоснабжение печени. Использование транс-
дермальных форм нитратов обеспечивает медленное высвобождение
Л В, что также позволяет добиться максимального эффекта при
минимальной инактивации в печени. Изучение фармакокинетики изосор-
бида динитрата позволило выявить его активный метаболит — 5-моно-
нитрат. Последний не метаболизируется в печени и имеет более
продолжительный, чем динитрат, период полувыведения, что
позволяет использовать его в качестве эффективного перорального
средства для профилактики приступов стенокардии и поддерживающей
терапии при хронической ИБС. Аналогичными свойствами обладает
и эринит [14, 15].
Блокаторы Са2+-каналов — верапамил (7.7), дилтиазем (7.8), ни-
федипин (7.9) и др. — также широко применяются при хронической
ИБС для профилактики приступов стенокардии. Блокирование
этими препаратами потенциал-зависимых Са2+-каналов в кардиомио-
цитах приводит к экономизации деятельности миокарда.
Прекращение поступления Са2+ в клетку подавляет превращение АТФ
в цАМФ и фосфорилирование белков, в результате
снижается функциональная активность клетки и ее энергетические
затраты (см. разд. 4.4).
МеО
МеО
^ I* CN
^* Me
(СН2)2 (СН^
Me Me
ОМе
ОМе
ОМе
N-
I О
Me^N/(CH2)2
(7.7)
Me
(7.8)
Me
C02(CH2)2—N' /=
\ //
(7.9)
(7.10)
187
C02Me
Me' ~N' "Me
H
(7.11)
(7-12)
Me02C
CH2-0(CH2)2NH
(7ЛЗ) (7.14)
Препарат верапамил — производное фенил ал кил амина — предложен в
качестве коронарорасширяющего средства в 1962 г. Позже (1969 г.) было
установлено, что наряду с уменьшением сократительной активности
миокарда, верапамил проявляет свойства антагониста ионов Са2+. В 1973 г.
появился первый представитель из труппы 1,4-дигидропиридинов — нифе-
дипин, получивший вскоре широкое клиническое применение как анти-
ангинальное и антигипертензивное средство. Именно в этом ряду
химических соединений поиск антагонистов Саг+- каналов оказался наиболее
результативным. В настоящее время к нему относятся никардипин (7.10),
исрадипин (7.11), нитрендипин (7.12), амлодипин (7.13), фелодипин (7.14).
В 1977 г. появился препарат дилтиазем — представитель еще одной
химической труппы — бензтиазепинов [16—24].
Перечисленные ЛС имеют определенные различия в
проявлении специфических эффектов [16—22], которые обусловлены как
непосредственным коронарорасширяющим действием и умеренной
антиадренергической активностью (верапамил), так и связыванием
с разными группами рецепторов (L, T, N, R-типа),
расположенными в непосредственной близости с Са2+-каналами клеточных
мембран (дилтиазем, 1,4-дигидропиридины). Кроме того, ЛС этой
группы существенно различаются по фармакокинетическим
показателям. Так, из данных табл. 7.4 следует, что биодоступность
препаратов этой группы изменяется в широких пределах: даже в ряду
близких структурных аналогов — 1,4-дигидропиридинов — она
колеблется от 10—15% до 65—70%. Низкий уровень этого
показателя связан с досистемным метаболизмом, который претерпевают
ЛВ при первом прохождении через печень. Кроме того,
большинство из них обладает относительно коротким периодом
полувыведения. Поэтому до появления амлодипина (1988 г.), а также про-
188
189
локированных ЛФ нифедипина (прокардия XL, нифедипин СС),
приходилось прибегать к многократному приему этих ЛС в
течение суток [16—21, 23].
При введении верапамила и дилтиазема образуются активные
метаболиты, которые также вносят свой вклад в общий эффект
этих препаратов, что необходимо учитывать при их длительном
применении. На основе данных табл. 7.4 можно, в определенной
мере, составить представление о том, как осуществляется отбор
структур при создании новых ЛС — блокаторов Са2+-каналов:
наибольший интерес представляют те соединения, которые при
высоком уровне специфического эффекта не подвергаются
первичному метаболизму, а метаболиты не обладают активностью.
Важным критерием отбора является и длительный период
полувыведения.
Еще одна группа ЛС, которая заслуживает рассмотрения в
контексте обсуждаемых вопросов — р-адреноблокаторы. Эти
препараты также получили широкое применение в лечении ИБС. Их
специфическое действие обусловлено снижением
чувствительности миокарда к адренергическим импульсам, уменьшением его
энергетических затрат, потребности в кислороде и др. эффектами
[24—30]. Различают неизбирательные и избирательные (кардио-
селективные) р-адреноблокаторы. К первой группе относятся
пропранолол (7.15), окспренолол (7.16), пиндолол (7.17), надо-
лол (7.18), тимолол (7.19); ко второй — атенолол (7.20), метопро-
лол (7.21), талинолол (7.22), бетаксолол (7.23). Фармакокинети-
ческие характеристики некоторых из перечисленных ЛС приведены
в табл. 7.5.
N Me
Н -НС!
(7.15) (7.16)
,S-N
Л
Me
JU-Me
O'Y^N Me
N- ОН
О
(7.19)
(7.20)
Me
Me -. -. J<Me Me
О Т"^ N Me
N Me 1 ОН О T^ N Me
rl i Jh h
(7.21)
(7.22)
(7.23)
Появление р-адреноблокаторов явилось результатом направленного
конструирования ЛС, основанного на данных о физиологической роли р-ад-
ренорецепторов. Синтез проводился, исходя из структурного подобия
между вновь создаваемым веществом и эндогенным лигандом —
адреналином. Первым пригодным для клинического применения р-адренобло-
катором стал пропранолол (1964 г.). Неизбирательность пропранолола
приводит к проявлению как полезных терапевтических эффектов (снижение
АД и купирование аритмии), обусловленных блокадой р,-рецепторов, так
и нежелательных побочных явлений (спазм гладкой мускулатуры
бронхов и периферических сосудов, нарушение гликогенеза), связанных с
блокадой Рг-адренорецепторов. В связи с этим были предприняты попытки
синтеза новых более совершенных структурных аналогов этого препарата.
Селективными Р-адреноблокаторами, отвечающими требованиям
безвредности, оказались атенолол, метопролол и др.
Усовершенствование структур в рассматриваемом ряду
осуществлялось с учетом влияния фармако кинетических характеристик ЛС на
проявление специфических эффектов. Так, пропранолол при перо-
ральном введении почти полностью (>90% дозы) абсорбируется из
ПК. Липофильность и основные свойства обеспечивают быстрый
транспорт препарата через кишечный эпителий в кровь. Однако био-
191
доступность этого ЛС зависит от введенной дозы. Пропранолол
почти полностью метаболизируется в печени, причем в значительной
степени при первом прохождении, и около 20 его метаболитов
активны. Отмечается также насыщение микросомальных ферментных
систем печени, что существенным образом сказывается на
биодоступности препарата при длительном применении. Конъюгат пропранолола
с глюкуроновой кислотой обладает кумулятивным эффектом. При этом
уровень концентрации его в плазме в 4 раза превышает содержание
исходного пропранолола. Попадая с желчью в кишечник, конъюгат
расщепляется, а высвобождающееся ЛВ вновь абсорбируется, т.е.,
возникает кишечно-печеночная рециркуляция. В результате период
полувыведения конъюгата увеличивается до 12 ч. Этим частично
объясняется устойчивый гипотензивный эффект пропранолола при
двукратном приеме его в течение суток. Благодаря основным свойствам
как самого ЛВ, так и его метаболитов, экскреция его с мочой растет
по мере увеличения кислотности последней [25—27, 30].
Среди неселективных р-адреноблокаторов первичному метаболизму
в печени не подвергаются пиндолол и надолол. Метаболиты пиндоло-
ла — глюкурониды и сульфаты — неактивны. При длительном приеме
кумуляция этого препарата наблюдается только у лиц пожилого
возраста. Надолол обладает низкой биодоступностью из-за образования
стойких соединений с желчными кислотами. Из всех ЛС, приведенных в
табл. 7.5, этот препарат имеет самый большой период полувыведения,
что позволяет применять его один раз в сутки. Несомненным
преимуществом надолола является отсутствие активных метаболитов [30].
Кардиоселективный р-адреноблокатор — атенолол, по сравнению
со всеми остальными препаратами этой группы, характеризуется
более высокой водорастворимостью, поэтому он не проходит через ГЭБ
и в меньшей степени оказывает побочное действие на ЦНС. Это ЛВ
лишь в незначительном количестве подвергается метаболическим
превращениям в печени [30].
Метопролол абсорбируется из ПК практически полностью,
однако из-за интенсивного первичного метаболизма его биодоступность
составляет 50%. При длительном приеме препарата этот показатель
повышается благодаря насыщению Ф систем печени. При
недостаточности функции печени метопролол может накапливаться в
организме в значительных количествах [30].
Завершая рассмотрение основных параметров, характеризующих
перемещения ЛС в организме, следует подчеркнуть, что фармакоки-
нетика не просто количественно описывает процессы абсорбции,
распределения и выведения, но и позволяет оптимизировать структуры
новых ЛВ или усовершенствовать их ЛФ, например, в направлении
повышения биодоступности. Известно, что наиболее сложной
задачей в процессе конструирования новых ЛВ является не идентификация
высоко аффинных лигандов рецепторов или ингибиторов Ф, которые
можно выявить in vitro, а отбор структур с приемлемой
биодоступностью. В последнем случае не обойтись без фармакокинетических
исследований in vivo. Эти исследования позволяют:
193
• установить режим приема препарата с тем, чтобы исключить или
свести к минимуму негативные побочные эффекты;
• оптимизировать дозы ЛС, соответствующие особенностям
заболевания или возрастным группам;
• разработать рекомендации относительно нежелательного
взаимодействия с другими препаратами;
• создавать новые ЛС путем улучшения фармакокинетических
показателей уже известных препаратов;
• выявить вещества, подвергающиеся метаболическим превращениям
до достижения ими биологической мишени.
Литература
1. Соловьев В.Н., Фирсов А.А., Филов В.А. Фармакокинетика. — М.:
Медицина, 1980.
2. Холодов Л.Е. Яковлев В. П. Клиническая фармакокинетика. — М.:
Медицина, 1985.
3. Пиотровский В.К. // Фармакол. и токсикол. — 1991. — № 4. — С. 71.
4. Каркищенко Н.Н., Хоронько В.В., Сергеева С.А., Каркищенко В.Н.
Фармакокинетика. — Ростов н/Д: Феникс, 2001.
5. Варфоломеев С.Д., Гуревич КГ. Биокинетика: Практический курс. — М.:
ФАИР-ПРЕСС, 1999.
6. Gibaldi М. Biopharmaceutics and Clinical Pharmacokinetics. 4,h Ed. — Malvern,
Pensylvania: Lea & Febiger, 1991.
7. Rowland M., Tozer T.N. СМгйЫ Pharmacokinetics. Concepts and Applications'. —
Malvern, Pensylvania: Lea & Febiger, 1989.
8. Lipinski С A., Lombardo F, Domini B.W. et al. // Adv. Drug Delivery Rev. —
1997. - Vol. 23. - P. 3.
9. Fecik R.A., Frank K.E. et al. // Med. Res. Rev. - 1998. - Vol. 18. - P. 149.
10. Xenical (orlistat). Product Monograph. — Basel: F. Hoffmann-La Roch Ltd,
1998.
11. Filing H.P.A. Xenobiotic Metabolism and Disposition, The Design of Studies on
Novel Compounds. — Boca Raton, Florida: CRC Press Inc., 1989.
12. Drugs and the Liver: Clinical Applications. Pharmacokinetic Basis for Drug
Treatment // Ed. by L.Z.Benet et al. — New York: Raven Press, 1984.
13. Scientific monograph Amaryl. — Frankfurt am Main: Hoechst Marion Roussel,
1997.
14. Метелица В.И. // Кардиология. — 1994. — №1. — С. 64.
15. Метелица В.И., Давыдов А.Б, Препараты нитратов в кардиологии. — М.:
Медицина, 1989.
16. Андреев Н.А., Моисеев B.C. Антагонисты кальция в клинической
медицине. — М.: Фармединфо, 1995.
17. Маколкин В.И., Вихляев В.Д. // Клин. мед. — 1994. — №1. — С. 4.
18. Метелица В.Н. // Экспер. и клин, фармакол. — 1996. — №2. — С. 56.
19. Floyd D.M., Kimball S.D. etal./fi. Med. Chem. - 1992. - Vol. 35. - P. 756.
20. Faulkner IK, McGibney Detal. //Br. J. Clin. Pharmacol. - 1986. -Vol. 22. -
P. 21.
21. Kleinbloesem H., Van Marten J. et al // Clin. Pharmacol. Ther. — 1986. —
Vol. 40. -P. 21.
22. Edgar В., Regardh C.G. et al. // Clin. Pharmacol. Ther. - 1985. - Vol. 38. -
P. 205.
194
23. Soons P.A, Breimer D.D.//Br. J. Clin. Pharmacol. - 1991. - Vol. 32. - P. II.
24. Van de WaterbeemdH., Smith D.A. etal. f/i. Med. Chem. — 2001. — Vol. 44 —
P. 1313.
25. Южаков СД., Глушков Р.Г., Машковский М.Д. // Хим.-фарм. журн. —
1991.- № 5. -С 21.
26. Obach R.S. // Drug Metab. Dispos. - 1997. - Vol. 25. - P. 1359.
27. Manoury P.M., Binet J.L. et. al. // J. Med. Chem. - 1987. - Vol. 30. -
P. 1003.
28. Сидоренко Б.А., Преображенский Д.В. // Тер. Арх. — 1995. — № 12 —
С. 3-8.
29. Южаков СД., Машковский МД. //Хим.-фарм. журн. — 1995. — № 6. — С. 3.
30. Applied Pharmacokinetics: Principles of Therapeutic Drug Monitoring/ Ed. by
W.E.Evans et al. — Spokane, Applied Therapeutics, 1986.
8
МЕТАБОЛИЗМ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
(ОБЩИЕ СВЕДЕНИЯ)
Метаболизм БАВ — это химические изменения, происходящие с
данным веществом под воздействием биологической системы,
которые принципиальны для его удаления из этой системы.
Млекопитающие используют экзогенные (присутствующие во внешней среде)
вещества для синтеза биополимеров и медиаторов, обеспечивающих их
жизнедеятельность. Когда ксенобиотик — чужеродное вещество,
попавшее в организм, — не может в нем ассимилировать, оно должно
быть удалено из системы. Выведение ксенобиотиков, в том числе и
ЛВ, из организма может происходить (для водорастворимых веществ
с низкой мол. массой) прямо — без изменения их структуры через
почки и/или печень, и непрямо — посредством ряда ферментативно
катализируемых превращений с последующей экскрецией метаболитов
теми же путями или комбинацией этих процессов. Обычно в ходе
метаболизма образуются более растворимые соединения и,
следовательно, гораздо легче экскретируемые. Энзиматические превращения
ЛВ в большинстве случаев ведут к их детоксикации и потере
активности. Однако в результате окисления, восстановления, гидролиза или
других реакций могут образовываться новые продукты, обладающие
терапевтическим или токсическим эффектами. Этот процесс носит
название биоактивации. Таким образом, метаболическая судьба ЛС,
в свою очередь, влияет на их фармакодинамику и токсичность.
8.1. Метаболические пути и места
метаболических превращений ксенобиотиков
Метаболизм ксенобиотиков состоит из двух этапов — реакций I и II
фазы. Реакции I фазы приводят к образованию новых или
демаскированию уже имеющихся химических групп. Эти продукты либо легко
выводятся, либо становятся субстратами для реакций II фазы. Однако в ряде
случаев таким путем образуются и реакционноспособные, потенциально
токсичные вещества. Наиболее распространенными процессами I фазы
являются реакции окисления, восстановления и гидролиза (рис. 8.1).
К окислительным превращениям относятся гидроксилирование и
дезалкилирование. В последнем случае расщеплению связи С-гетеро-
атом предшествует гидроксилирование алкильнои группы, связанной
с этим гетероатомом.
196
(2.46) ибупрофен
Н ,0
H2N
Н ,0
N-
N
Ph
(8.1) нитразепам
(6.17) левомицетин
ОН Ph О ОН Ph ОН
0 0 ^ О' "О
(8.3) варфарин
со2н
со2н
H„N
хн
о
о^4^ — он
(8.4) ацетилсалициловая кислота
Et H>N
I
Et
<^s
C02H H2N'
Et
1
.N.
Et
(8.5) новокаинамид
Рис. 8.1. Реакции I фазы метаболизма некоторых ЛВ [1,2)
197
Восстановление нитро- и карбонильных групп приводит к
образованию аминов и спиртов соответственно. Гидролиз эфиров или
амидов завершается образованием спиртов, кислот и аминов.
Вторая фаза метаболизма включает реакции конъюгации,
отличающиеся от превращений I фазы тем, что они привносят новые
фрагменты в молекулу и являются, главным образом, реакциями присоединения.
Это процессы сульфирования, глюкуронирования, меркаптурирования,
ацилирования и др. (рис. 8.2). Их протеканию способствует образование
в ходе I фазы подходящих химических групп. Конъюгаты также
обладают повышенной растворимостью. Кроме того, О-, N- или S-алкили-
рованные производные более липофильны. Некоторые продукты II
фазы также могут вносить свой вклад в токсичность веществ.
Например, глюкурониды или сульфатные конъюгаты некоторых N-замещен-
ных гидроксиамидов обладают канцерогенными свойствами.
продукт I фазы метаболизма пропранолола (7.15)
°4 г-
ЧГ\ /Г""
со2н
о-4
I г
ОН
он
(8.6) парацетамол
НОХ
-О
*- нох
ю
им
(8.7) бензойная кислота
(8.5) новокаинамид
Рис. 8.2. Реакции II фазы метаболизма некоторых ЛВ [1, 2]
198
8.1.1. Реакции I фазы метаболизма,
катализируемые CYP450
Метаболизм происходит в основных тканях и органах — печени,
почках, кишечнике, крови/плазме, ЦНС, легких и др. Печень
является наиболее эффективным метаболизирующим органом,
обладающим большим потенциалом ферментативных реакций. Почки и
стенки кишечника — важные места для конъюгационных превращений.
Гидролиз эфиров и амидов может протекать так же хорошо, как в
крови/плазме, практически во всех других органах и тканях.
Внутри клетки ксенобиотики метаболизируются в ЭР
(микросомах), митохондриях и клеточном цитозоле (см. разд 1.1).
Большинство окислительных реакций осуществляется с участием оксидаз
смешанных функций, связанных с мембранами или с помощью CYP450.
В активном центре Ф этой группы находится гем. Все живые
существа от микробов до человека наделены гемсодержащими энзимами,
относящимися к суперсемейству CYP450, в состав которого входят
более 300 изоформ, способных катализировать около 60 типов
ферментативных реакций с громадным числом структур. Это
суперсемейство эволюционно очень древнее. Предполагают, что оно
существует в живой природе более 3.5 млрд лет [3].
RH
P450(Fe")
ROH у р^ P450(Fe'" RH)
P450[(ROH Fe"1)] Ye
P450[ROHFe'"]y P450(Fe" RH)
P450[(R-)(FelvOH)]
P450[(Fev=O)RH]
P450(Fje" RH)
P450(Fel(l RH)
P450(Fe[!l RHL^^r^n
ООН Нл
Рис. 8.3. Каталитический цикл CYP450 [4]
Основными функциями CYP450 являются участие в синтезе
регуляторов важнейших физиологических процессов, например,
стероидных гормонов, и метаболизме множества эндогенных веществ, таких как
желчные кислоты, простагландины, лейкотриены, биогенные амины,
а также ксенобиотиков. Изозимы CYP450 доминируют на I фазе
метаболических превращений. Следует подчеркнуть, что изозимами обычно
199
называют различающиеся аминокислотной последовательностью Ф,
которые действуют на один и тот же субстрат и катализируют
образование одних и тех же продуктов. Различные CYP450 либо воздействуют
на разные субстраты, либо разные CYP450 приводят к образованию
различных продуктов с одним и тем же субстратом. Поэтому термин
«изозимы» в данном случае не совсем корректен, хотя и является
широко употребляемым. На рис. 8.3 представлен механизм
каталитического превращения некоего субстрата RH под действием CYP450.
Основными окислительными процессами, которые катализирует
CYP450, являются [5]:
— ароматическое гидроксилирование
CH3-CO-NH-C6H5
(ОН']
CH3-CO-NH-C6H4-OH
(7.15) пропраналол
— алифатическое гидроксилирование
R-CH, ^R-CR-OH
^
н3с
so.
X.
"N NHC2H6
НОН2С
scwA
N NHC2H6
(4.9) толбутамид
дезаминирование
R-CH(NH2)-CH3 ^R-C(OH)(NH2)-CR -> R-CO-CH3 + NH4+
С К
NH.
С К
+ NH,
(8.8) амфетамин
— О-дезалкилирование
R-0-CH3i^R-0-CH2-OH-> R-OH + CH20
Et-0
\\ //
NHCOCK
НО
^ /
NHCOCK
(8.9) фенацетин
200
— N-дезаякилирование
Щ [R_N(CH2OH)CH3] -> R-NH-CH3+ СН20
R-N(CH3):
R_NH-CH3^[R-NH-CH2OH] -> R-NH2 + CH20
сн.
з acylCoA
R(-)
r=\ CH, acylCoA
(8.10) имипрамин
— N-окисление
(CH3)3-N-^ l(CH3)3~N+-OH] -> (CH3)3-NO + H+
— сульфокисление
R_S_R'i^a (R-S(OH)-R1 ] -> R-(S =0)-R' + H+
(8.12) хлорпромазин
Число изозимов, участвующих в катаболизме ксенобиотиков, видо-
специфично. Для метаболизма БАВ у человека важны три семейства
этих Ф: CYP1, CYP2, CYP3 [6].
Семейство 1. CYP1A играет ключевую роль в метаболизме двух
классов канцерогенных веществ: полициклических ароматических
углеводородов (ПАУ) и ариламинов. Наличие ПАУ в окружающей
среде — результат техногенного воздействия. Основным
источником загрязнения являются нефтеперерабатывающие и
коксохимические производства, тепловые электростанции, выхлопные газы
автомобилей, табачный дым. Ариламины попадают в организм
человека с мясными продуктами. В то же время CYP1A1 метаболизирует
проканцерогены в гидроксильные производные, не обладающие
мутагенным действием. Вопрос о том, как активировать
метаболические превращения именно в направлении образования
низкотоксичных продуктов, пока остается открытым.
201
Группа Ф CYP1A2 (также известная, как фенацетин-О-деэтилаза,
кофеиндеметилаза, aнтипиpин-N-дeмeтилaзa) метаболизирует арил-
амины, нитрозамины и ароматические углеводороды, включая
биоактивацию промутагенов и проканцерогенов, таких как афлатоксин В1.
Экспрессия CYP1A2 наблюдается в печени, кишечнике и желудке.
Энзимы активируются под влиянием ПАУ или курения (табл. 8.1).
Семейство 2. Примером видовой специфичности метаболизма
может служить подсемейство CYP2A, участвующее в гидроксилировании
тестостерона по 7а- и 15а-положениям в организме грызунов, но не в
организме человека. CYP2A6 вызывает гидроксилирование кумарина
(по положению 7 бицикла) и афлатоксина В1, биоактивацию нитроза-
минов и проканцерогенов ароматического ряда.
Группа изозимов CYP2D6 - одна из наиболее важных для
метаболизма более чем 30 ЛС (см. табл. 8.1).
CYP2E1 метаболизирует галогенпроизводные углеводородов,
включая пульмональные анестетики (хлороформ, галотан и др.), а также
органические вещества с низкой мол. массой (ацетон, ДМФА,
этанол, ацетонитрил, бензол). Гепато- и нефротоксичность
ингаляционных анестетиков объясняется образованием реакционноспособных
метаболитов — галогенангидридов соответствующих кислот (рис. 8.7),
которые ацилируют белки.
Экспрессия Ф рассматриваемой группы наблюдается в печени,
почках, кишечнике, легких и индуцируется этанолом, изониазидом и
др. веществами (табл. 8.2). Большинство из индукторов CYP2E1
выступают одновременно и в качестве субстратов. В связи с указанным,
это подсемейство Ф носит название микросомальной этанолокисли-
тельной системы, бензолгидроксилазы или анилингидроксилазы.
Энзимам CYP2E1 также присуща видовая специфичность. Удобной
моделью для человеческого CYP2E1 может служить аналогичная группа
Ф у крыс, однако в организме кроликов это подсемейство
отсутствует. Активация CYP2E1 имеет место при сахарном диабете, ожирении,
хроническом введении алкоголя [7, 8].
Семейство 3. Группа Ф CYP3A является наиболее
представительным подсемейством, эффекты которого реализуются в печени и
кишечнике. Однако только две формы — CYP3A4 и CYP3A5 —
охарактеризованы в настоящее время. Примерно две трети Ф печени
принадлежат именно к этому подсемейству. С их участием метаболи-
зируется большое количество широко распространенных ЛС
(см. табл. 8.1). Многие соединения, являясь индукторами CYP450,
выступают и в качестве субстратов. Известны также соединения'
ингибирующие эти Ф (табл. 8.2). Энзимы семейства ЗА связываются
преимущественно с липофильными веществами. Субстраты CYP3A
обладают обычно пониженной биодоступностью при пероральном
введении и могут претерпевать досистемный метаболизм под действием
кишечных форм Ф этой группы.
Механизмы окисления ксенобиотиков, происходящие при
участии CYP450, представлены на рис. 8.4—8.8.
202
CYP2D6
CYP2C18
CYP2C9,10
CYP2C8
CYP3A4
CYP1A2
CYPlAl
Лидокаин
Дебризохин
Каптоприл
Пропранолол
Метопролол
Тимолол
Амитриптилин
Имипрамин
Галоперидол
Метоксифенамин
Кодеин
Омепразол
Пропранолол
Диазепам
Имипрамин
Варфарин
Толбутамид
Ретинол
Напроксен
Ибупрофен
Диклофенак
Гексобарбитал
Тестостерон
Левомицетин
Варфарин
Толбутамид
Ретинол
Афлатоксин В1
Афлатоксин G1
6-Аминохризен
Фенобарбитал
Эстрадиол
Глюкокортикоиды
Рифампицин
Эритромицин
Фенацетин
Теофиллин
Кофеин
Имипрамин
Эстрадиол
Антипирин
Фторхинолоны
2-Нафтиламин
2-Аминофлуорен
2-Аминохинолин
Афлатоксин В1
Бенз[а]-
пирен
и др.
ПАУ
CYP2E1
CYP2E1
CYP2E1
CYP2E1
Теофиллин
ДМФА
Диэтиловый
эфир
Этанол
Глицерин
Анилин
Ацетон
Пиридин
Ацетонитрил
Энфлуран
Трихлорэтилен
Этилкарбомат
N-Нитрозоди-
метил анилин
Акрилонитрил
5 £ о х х §
и О О О О С
203
Таблица 8.2
Индукторы и ингибиторы некоторых изоформ CYP450 [7]
Индукторы
CYP1A
3-метилхолантрен, ПАУ, тетрахлор-
диоксин, табачный дым, р-нафто-
флавон, индол-3-карбинол
CYP2E
Этанол, диэтиловый эфир, бензол,
ДМСО, ацетон, изониазид, пира-
золы и др.
CYP3A
Фенобарбитал (CYP2B, CYP2C),
карбамазепин, дексаметазон и др.
глюкокортикоиды, рифампицин
и его аналоги, эритромицин и др,
макролиды, фенилбугазон, флуко-
назол и др. антифунгицидные «ко-
назолы»
CYP4A
Клофибрат и др. гиполипидеми-
ческие препараты
Ингибиторы
CYP2E
3-амино- 1,2,4-триазол, диэтилтио-
карбамат, диалл ил сульфид
CYP3A
Имидазол, триацетилолеандомицин,
17а-этинилэстрадиол, левомицетин,
(CYP2B, CYP2C), циметидин, фтор-
хинолоны (CYP1A2)
Н,СХ
сн
2 Fev = О
ОН
I
Fe1"
Felv ОН
Fe'vOH CH2
^-Fe^OH
,^Fe'"OH
У
®
/CHn
с к
Н+ Н2С
■2 Fe"'OH
Рис. 8.4. Предполагаемый механизм окисления алканов [9, 10]
8.1.2. Реакции I фазы метаболизма, катализируемые
FAD-содержащими ферментами
Альтернативный путь окисления азот- и серусодержащих
органических соединений, относящихся к так называемым «мягким» нуклео-
филам, осуществляется при участии флавин-монооксигеназы (FMO).
204
Fe^OH Fe»1
СН,ОН
Fev=0
л-комплекс ^
FeiiiO Fe'"0 Fe111 О
Рис. 8.5. Механизм окисления алкенов [IV
porph — порфирин
■N-CH,—
Fev=0 FewO
■*- -Д|—сн —
FelvO Fe»
CH-
-N-CH-
он
_____ нр^ (L_
"*" ~~ ~н н
Fe'vOH
Fe111
■N-CH,
Рис. 8.6 Механизм дезалкилирования и окисления [9, 10]
Fev=0
я-комплекс
S-Glut
FelV0
ст- комплекс
Рис 8 7. Механизм окисления ароматических углеводородов [9, 10]:
ЕрН О - эпоксид гидролаза; GlutSH - глугатион S-трансфераза
205
CHCI3 ^ НО-СС13
coci2 + nh2-Белок—
COCL + CI
Белок - NH - со - NH - Белок
coci2 + н2о
-* СО,+ 2HCI
е. в
СС14 - С14С ^ CLC + CI С1,С
+Н*
СНС13
:СС1,+ СГ
CI
Вг
CH3-0-FC
■в?
0
-CI
-»- F3C—CCIBr—ОН -^^- F3C
-CI
CI CI
—4— н —*- ch3-o-f2c —l— он
-CI
.0
Л CK-0-F2C
о V
NH - Белок
0
NH-Белок
■CH3-0-F2C
CI
н2о\
0
CI CI
ch3-o-f2c—со2н + cT
Рис 8.8. Схема окисления галогенуглеводородов [12—14]
Этот Ф содержит в активном центре FAD. Механизм окисления
субстрата S, катализируемого FMO, представляется следующим [15]:
^vNH H
н о II
HyO II
vo
I
H
FAD-ООН S-
H - -.
FAD
-S-O
Каталитический цикл FMO включает пять стадий (рис. 8.9). В
отличие от процессов, катализируемых изоформами CYP450, при
катализе FMO не наблюдается N- или S-дезалкилирования. В целом же
FMO и CYP450 взаимодействуют с одними и теми же субстратами
с образованием одинаковых продуктов.
Основными типами реакций, протекающих при участии FMO [15],
являются:
окисление третичных аминов (например, хлорпромазина,
атропина, имипрамина, никотина, диметиланилина)
R3—N+ ароЕ — FAD— ООН —- R3— N—- О + ароЕ — FAD —ОН
окисление вторичных аминов и гидроксиламинов (например, N-метил-
бензиламина, N-метиланилина).
R2— NH + ароЕ—FAD —ООН R2—NOH+ ароЕ—FAD —ОН
Ft,— NOH+ ароЕ —FAD —ООН ^ R2— N=0 +ароЕ— FAD—ОН
О"
206
FAD-OOH
а,-Л
FAD-H2
(NADP*)
J
W + NADPH
1
— FAD-^-
FAD-OH
Ко
FAD
(NADP+)
\
^NAC
Рис. 8.9. Каталитический цикл FMO [15]
окисление гидразинов
R—N—NH,+ apoE—FAD —ООН —- R—N —NH2 + apoE — FAD — OH
R R
окисление тиолов, дисульфидов и сульфидов
R—SH—^ R—SS— R R—S —R
R —SS—R
R—SS —R
I
О
О
I
R—S—R
О
Флавиновый кофактор обеспечивает каталитическую активность
моноаминооксидазы (МАО) и диаминооксидазы (DAO). Эти Ф
расположены в мембранах митохондрий и катализируют окисление моноаминов
в альдегиды. Последние в дальнейшем превращаются в соответствующие
кислоты или спирты при участии альдегидоксидаз или дегидрогеназ.
МАО обеспечивает реализацию важного механизма деградации
эндогенных катехоламинов и серотонина в нервных тканях [16].
Окисление метаболитов нуклеинового обмена — пуриновых
оснований — в мочевую кислоту осуществляется при участии ксанти-
ноксидазы. Этот Ф принадлежит к числу металлофлавопротеинов. ЛС,
являющиеся антиметаболитами пуринов (например, 6-меркаптопурин),
превращаются под действием ксантиноксидазы в меркаптуровую
кислоту [1, 5, 16].
SH
SH
n^vv
он
Окисление ксенобиотиков происходит не только под
действием энзимов, содержащих в качестве кофактора гем или FAD, но
207
и при участии Си2+ и NAD зависимых монооксигеназ и дегидро-
геназ например, дофамин р-монооксигеназы, алкогольдегидрогеназы
и др [1, 5].
8.1.3. Восстановительные процессы
В микросомах клеток печени содержатся Ф, способные
восстанавливать азо- и нитросоединения до аминопроизводных. К их числу
относятся NADPH-зависимая азоредуктаза и нитроредуктаза [1, 5].
Примерами продуктов восстановления N02-rpynnbi до NH2 являются
метаболиты левомицетина (6.17) и нитразепама (8.1).
Восстановление кетонов до гидроксипроизводных осуществляется
при участии кеторедуктазы, также содержащей в качестве кофактора
NADPH. Таким путем метаболизируются антагонист морфина налок-
сон, гидроморфан, даунорубицин и др. ЛС, содержащие кетогруппу.
8.1.4. Гидролиз
Метаболизм БАБ, являющихся по химическому строению эфира-
ми или амидами, происходит во всех органах и тканях при участии
карбоксиэстераз (ацетилхолинэстераза, арилкарбоксиэстераза и др.).
Скорость гидролиза во многом зависит от пространственного строения
субстрата. Расщепление стерически нагруженных эфиров, например,
атропина, протекает медленно. Поэтому 50% введенной дозы этого ЛВ
выводится из организма через почки в неизмененном виде. Амиды, как
правило, более устойчивы к гидролизу, чем эфиры. Это часто
используется в конструировании лекарств. Так, замена эфирной группы на
амидную при переходе от новокаина к новокаинамиду (8.4)
значительно повысила метаболическую стабильность последнего, что, в свою
очередь, позволило использовать (8.4) не как местноанестезирующее,
а как пероральное антиаритмическое средство [1].
8.1.5. Реакции II фазы метаболизма ксенобиотиков
Как отмечено выше, процессы II фазы метаболизма являются,
главным образом, реакциями присоединения. Образование конъюга-
тов характерно для липофильных веществ, легко абсорбирующихся и
плохо выводящихся с мочой. Только после образования продуктов
присоединения в их структуре появляются группы, способные к
ионизации, и, следовательно, повышающие водорастворимость (например,
сульфогруппа, остаток глюкуроновой кислоты или глицина). В
некоторых случаях, при наличии в исходной молекуле подходящей
функциональной группы, образование конъюгатов может
предшествовать реакциям I фазы (досистемный метаболизм) [17].
208
Обычно основные продукты II фазы — глюкурониды и
сульфаты — не обладают фармакологическим эффектом (т. к. не способны
в значительных количествах диффундировать в клетки) и обладают
низкой аффинностью к рецепторам.
Как уже неоднократно подчеркивалось, образование
конъюгатов — путь детоксикации ксенобиотиков. Тем не менее, в некоторых
случаях возможно образование реакционноспособных продуктов,
обладающих канцерогенными свойствами, вызывающих аллергические
реакции или оказывающих другие виды побочного действия на
различные органы и ткани организма [18].
Чем больше в молекуле ЛВ реакционноспособных
функциональных групп, тем большее количество метаболитов вообще и
конъюгатов в частности образуется. Одно и то же соединение может быть
субстратом для различных Ф, которые, в свою очередь, способны
конкурировать между собой за одни и те же функциональные
группы. Примером вещества, образующего большое количество
продуктов присоединения, является л-аминосалициловая кислота (8.13).
со2н
со2н
о£Г«-
он
-^Vco2h
ОН
он
N-CO„H
Образование конъюгатов с глюкуроновой кислотой. Это один из
важнейших способов удаления из организма липофильных веществ.
Продукты присоединения с глюкуроновой кислотой образуют фенолы,
спирты, карбоновые кислоты и амины, а из эндогенных субстратов —
стероиды, билирубин и тироксин.
Глюкурониды, как правило, хорошо растворимы и выводятся,
главным образом, через почки; те же конъюгаты, которые элиминируют с
желчью в кишечник, способны гидролизоваться под действием
присутствующей там р-глюкуронидазы. Таким способом исходное веще-
209
2 NAD
Gluc-1-P03+ УТФ ——- УТФ- Glue *-
УДФ -дегидрогеназа
С02Н С02Н
ство или его метаболит I фазы из ПК вновь попадают в системное
кровообращение. Примером является глюкуронид пропранолола [2].
Выше было отмечено, что конъюгаты этого типа гораздо менее
активны, чем их синтетические предшественники. Исключением
являются морфин—З-О- и — 6-О-глюкурониды. Первый выступает в
качестве антагониста опиоидных рецепторов. Он образуется в
результате досистемного метаболизма при пероральном введении
анальгетика и его концентрация в крови составляет - 20% от
концентрации морфина. Второй — 6-О-глюкуронид — присутствует в
крови в концентрации, в два раза превышающей содержание
морфина независимо от способа введения последнего, и проявляет более
выраженный анальгетический эффект, чем морфин. Таким
образом, эффект морфина является результатом взаимодействия как
самого ЛВ, так и двух его метаболитов с соответствующими
рецепторами [19].
Наименее стабильны из-за легкости гидролитического
расщепления при физиологических значениях рН — С'-О-ацилглюкурониды —
метаболиты алифатических и ароматических карбоновых кислот.
Такие конъюгаты могут выступать в роли переносчиков ацильных
остатков в реакциях переацилирования с участием эндогенных
белковых субстратов. В результате ацилированные протеины стимулируют
иммунный ответ в отношении исходных ЛС. Наибольшее количество
подобных иммунотоксических эффектов отмечено в ряду НПВС, т.к.
именно в этой группе ЛС широко представлены производные арил- и
гетерилалифатических кислот (ибупрофен, кетопрофен, сулиндак
и др.). Частота возникновения иммунотоксических реакций зависит
от стабильности глюкуронидного конъюгата, скорости миграции ациль-
ного остатка к эндогенным субстратам, концентрации и
стабильности образующихся антигенов [20].
Сульфатные конъюгаты. Сульфирование очень часто происходит
параллельно с глюкуронированием, в результате — эти процессы
конкурируют за один и тот же субстрат. При повышенных
концентрациях ксенобиотика преобладает глюкуронирование. Механизм
образования сульфатов представлен на рис. 8.10.
Из эндогенных веществ через сульфирование реализуется
метаболизм катехоламинов, желчных кислот, стероидных гормонов,
тироксина. Стероиды образуют сульфатные конъюгаты исключительно
в печени, а производные фенола — в печени, почках и кишечнике.
Сульфатные конъюгаты выводятся, главным образом, с мочой.
Исключение составляют стероиды, которые элиминируют с желчью [17].
210
л я 1)АТФ - сульфурилаза
S042" + АТФ У TJF »- 03SO-P
2)АФ8- фосфокиназа '
PAPS
сульфотрансфераза
Г
о
(8.5)
AOS- аденозинфосфосульфат
Рис. 8.10.
Среди сульфатов есть фармакологически активные метаболиты,
такие как дегидроэпиандростерона сульфат и морфина-6-сульфат.
Кроме того, сульфопроизводные могут вносить свой вклад в
токсичность. Сульфирование спиртов приводит к легко гидролизуемым
эфирам серной кислоты, а, следовательно, и к высоко реакционноспо-
собным электрофильным интермедиатам [18]. Способность метаболи-
зировать ксенобиотики посредством образования сульфатов зависит от
возраста: скорость их синтеза снижается в процессе старения организма.
Возможность образования сульфатов в кишечнике существенно
влияет на досистемный метаболизм некоторых Л В. За счет этого, например,
дезактивируются многие стероидные гормоны, а также а-метилдофа и
парацетамол. Данный факт следует учитывать при совместном приеме
нескольких препаратов, например, перорального контрацептива этиле-
нилэстрадиола и противовоспалительного средства парацетамола.
Аминокислотные конъюгаты. Образование продуктов
присоединения с глицином — важнейший путь выведения из организма
производных карбоновых кислот. Глицин образует легко ионизующиеся
водорастворимые конъюгаты с ароматическими, арилалифатическими
и гетероциклическими кислотами.
R-COOH + АТФ + СоА ацетилсинтетаза-^ R-CO S-CoA + АМФ
„ _ л лл1г» ни трансацетилаза
R-CO S-CoA + Ri —HN2 —£ ^ R_co_NHRi + CoA-SH
Синтез амидов на основе глицина конкурирует с образованием
глюкуронидов. Как и в случае сульфатов, при высокой концентрации
ксенобиотика преобладает синтез глюкуронидов [17].
211
Метаболизм алифатических кислот зависит от разветвленности их
углеводородной цепи. Жирные кислоты, входящие в состав липидов,
в большинстве случаев окисляются, не образуя конъюгатов.
Устойчивые к р-окислению арилалифатические и алифатические кислоты
образуют аминокислотные и/или глюкуронидные конъюгаты.
Следует отметить, что а-замещенные кислоты склонны к образованию глю-
куронидов в значительно большей степени, чем амидов. Метаболизм
производных бензойной и гетероциклических кислот осуществляется
исключительно по пути образования амидов с глицином [17].
Кроме глициновых конъюгатов, у человека и приматов возможен
синтез глутаматов, в частности, для арилуксусных кислот.
Интересно отметить, что при метаболизме производных 2-арил-
пропионовой кислоты (обширная группа НПВС — ибупрофен (2.46),
напроксен и др.) наблюдается рацемизация (см. также разд. 2.5.1) [21].
з acylCoA
=:\ у з гидролаза
^—* COSCoA
=\ СН, acylCoA
\ ^ у
%
рацемаза
СН
=\ ^пз гидролаза
СН,
COjH
В отличие от большинства метаболических превращений, при конъ-
югировании с аминокислотами образования токсичных продуктов до
настоящего времени не отмечено. Предполагают, что это путь деток-
сикации реакционноспособного ацил-СоА-тиоэфира [17].
Ацилирование. Это основной путь превращения первичных
ароматических и алифатических аминов, аминокислот, гидразидов и
сульфамидов, реализующийся при участии ацетил-СоА. Вторичные амины
не ацилируются. Процесс образования N-ацетилпроизводных
состоит в следующем:
Аг— NH,
Ar-N
СН,
Arv ^СОСН-
I
ОН
Аг^ ^ОСОСН,
. N 3
I _
Н
[Аг— NH+]
При ацилировании, в результате потери ацетоксигруппы,
образуется арилнитрениевый катион — электрофил, способный ковалентно
связываться с молекулами НК или белков, что может индуцировать
опухолевый процесс [22].
Продукты ацилирования часто обладают фармакологическим
эффектом, сопоставимым с исходным веществом, как это имеет
место в случае N-ацетилпрокаинамида.
В скорости протекания процесса ацилирования наблюдается
генетический полиморфизм. Различают быстрый и медленный
фенотипы. В случае быстрого фенотипа образование токсичных ацетоксиа-
212
риламинов ускорено, что обусловливает повышенный риск развития
рака крови и злокачественных новообразований печени [17].
Глутатионовые конъюгаты. Синтез меркаптуровой кислоты.
Мерка птуровая кислота является S-производным N-ацетилцистеина,
образующимся из глутатиона в ходе следующих превращений:
о сан
о
J
сан
i н i - - Н "
R—X + им I S-трансфераза HNS
HN- '—■ ^ к -г г -ъг -**- -NH
U LAJ2I-1 II Г
+ ..'. Н I £-тпанс<Ьепа-га HN. ^^
NH2
0 Glut - SH
, ^ ,CO,H
RS'
О СОН x^v^COjH "J T
JJ J ^S J ацетилаза hnn_/
глутаминтрансфераза RS'^^sf^N цисте инилшицнназа NH *. ]f
NH2 °
Образование конъюгата на первой стадии процесса — результат
нуклеофильной атаки глутатиона на электрофильный углерод,
связанный с легко уходящей группой (галогеном, алкилом, алкенилом,
арилом, алкилгалоидом, сульфо-, нитрогруппами либо легко
раскрывающимися малыми циклами — эпоксидами, р-лактонами) или
активированным р-углеродным атомом в а,р-ненасыщенных
карбонильных соединениях.
SR
Конъюгирование с эпоксидами обеспечивает защиту печени от
токсических эффектов последних. Их глутатионовые производные
неактивны [17].
Метилирование. Этот процесс имеет гораздо большее значение для
метаболизма эндогенных соединений (таких как катехоламины) чем
для превращений ксенобиотиков. Обычно продукты О-, S- или N-
метилирования обладают большей липофильностью и более
выраженным биологическим эффектом, чем их синтетические
предшественники. Образование метилирующего агента SAM осуществляется при
участии метионинаденозинтрансферазы (MeSAT). Далее от SAM
также ферментативным путем под действием метилтрансферазы (МеТ)
происходит перенос метильной группы на подходящий субстрат.
Различают О-, N- и S-MeT.
Катехол-О-метилтрансфераза (КОМТ) является Mg2+ зависимым Ф.
Субстраты для КОМТ — катехоламины (норадреналин, адреналин,
дофамин), L-ДОФА, а также эстрадиол и его 2- и 4-гидроксипроизвод-
ные. Фенилэтаноламин-1Ч-метилтрансфераза (ONMT) катализирует
213
coo
АТФ + ^S
MeSAT
NH,
п2гч
\ / + гшрофосфет + P;
OH OH
MeT SAM
SAM + RZH *■ RZ — CH3+ S аденозингомоцистеин
Z = O.NH,S
SAM
NHCH,
алкилирование аминогруппы в тех же субстратах. Однако этот Ф не
участвует в метилировании фенилэтиламинов. Их превращения
катализирует aMHH-N-метилтрансфераза (ANMT). Она играет важную роль
в механизме кишечно-печеночной рециркуляции N-деметилирован-
ных ЛВ, например, деметилированный имипрамин под действием
ANMT вновь метилируется и поступает в системное кровообращение [2].
S-Метилирование — важный этап детоксикации тиолов,
конкурирующий с их FAD-ферментативными превращениями. Таким
путем метаболизируются каптоприл, тиопурин, пенициламин, 6-про-
пил—2-тиоурацил [17].
8.2. Связь структуры и степени метаболизма
лекарственных веществ
Степень метаболизма зависит от двух факторов: водораствори-
мости ЛВ и доступности мест метаболизма. Например,
антидепрессант хлорпромазин (8.12), является высоколипофильным
соединением, поэтому, легко проникая во многие органы и ткани,
метаболизируется в них. Он претерпевает N-дезалкилирование,
ароматическое гидроксилирование, сульфо- и N-окисление.
Некоторые из продуктов I фазы метаболизма являются субстратами для
его II фазы, так, из фенольного метаболита образуется глюкуро-
нид. Все это приводит к появлению большого числа
метаболитов, которые экскретируются с мочой или с желчью. Количество
препарата, выводимого теми же путями в неизмененном виде,
минимально.
214
Образование большого количества метаболитов характерно и
для р-адреноблокатора пропранолола (7.15), который наряду с
ароматическим гидроксилированием О- и N-дезалкилируется, деза-
минируется и образует конъюгат с глюкуроновой кислотой.
Однако его близкий структурный аналог — атенолол (7.20),
обладающий большей водорастворимостью, выводится преимущественно
в неизмененном виде с мочой. Только незначительное
количество (< 5% дозы) экскретируется в виде гидроксиметаболита (см.
разд. 7.6).
Степень метаболических превращений многих Л В находится в
интервале между практически полным превращением и выделением
в неизмененном виде, т. е. экскретируется как само вещество, так и
его метаболиты. Внутри каждого ряда соединений степень
метаболизма и увеличение путей выведения возрастает с ростом липофиль-
ности (например, у Р-адреноблокаторов).
8.3. Факторы, влияющие на метаболизм лекарственных
веществ
Дозировка препарата. С увеличением концентрации Л В метаболи-
зирующая ферментная система способна насыщаться. Это может
привести к альтернативным путям превращения препарата, к
значительному росту концентрации как самого ЛВ, так и его активных или
токсичных метаболитов. Так, парацетамол (8.6) при применении его
в терапевтической дозе выводится из организма в виде легко удаляемых
сульфатного и глюкуронидного конъюгатов. При более высоких
дозах наблюдается насыщение ферментных систем печени, в результате
чего, наряду с названными метаболитами, образуются хиноидные
производные под воздействием CYP2E1, а на их основе — трудно
215
выводимые токсичные глютатионовые и белковые конъюгаты [23, 24].
Значительная передозировка приводит к гепато- и нефротоксическим
эффектам за счет накопления токсичных продуктов в печени и
почках. Сведения такого рода крайне важны для интерпретации фар-
макодинамических и токсикологических данных.
Путь введения препарата. При пероральном пути введения ЛС его
биодоступность во многом зависит от способности претерпевать до-
системный метаболизм («first-passe-эффект). Примером вещества с
чрезвычайно высоким «пг51-ра55»-эффектом является анальгетик меп-
тазинол (8.14). Он претерпевает досистемный метаболизм и
выводится в виде глюкуронидов. Этого можно избежать, если вещество
вводить ректально в виде суппозиториев. При этом ЛВ всасывается в той
части кишечника, из которой не попадает в портальную вену и
печень, что позволяет достичь большей концентрации в крови при
одинаковых дозах. При введении per os в дозе 50 мг максимальная
концентрация в крови составляет < 0.01 мкг/мл, при введении per rectum
в той же дозе С равна ~ 0.05—0.2 мкг/мл [22].
(8.14)
Видовая специфичность метаболизма. Существуют
количественные различия в I фазе метаболизма среди различных видов
млекопитающих. Эти различия часто трудно предсказать и интерпретировать.
Однако общий вывод таков: у человека метаболические
процессы, связанные с CYP450, протекают в 20 раз медленнее, чем у крыс,
и в шесть раз медленнее, чем у собак.
Ряд качественных отличий имеется и на II фазе метаболизма. Так,
у собак нет способности ацилировать ароматические амины и они
более чувствительны к фармакологическим и токсическим эффектам
препаратов, содержащих такие фрагменты; кошачьи лишены
способности образовывать глюкуроновые конъюгаты; и только приматы
образуют аминокислотные конъюгаты с глутамином. Существуют также
видовые различия в способности к экскреции: у человека вещества с
мол. массой до 500 выводятся через желчевыводящие пути, а у крыс
и собак это наблюдается в случае веществ с мол. массой до 350.
Половые различия. Отмечается разница в метаболических путях у
особей мужского и женского пола, что особенно характерно для
грызунов, в частности, для крыс. Так, самцы впадают в сон при
значительно больших концентрациях гексабарбитала, чем самки. Это обус-
216
ловлено половым диморфизмом в активности изозимов CYP450 у
данного вида животных, что, в свою очередь, проявляется в способности
самцов инактивировать названный препарат путем алифатического
гидроксилирования. У других биологических видов половые
различия в метаболизме разных ЛВ менее выражены.
Возрастные различия. Эта важная составляющая метаболизма ЛВ
лучше всего изучена у человека. В клинических испытаниях
препаратов обычно участвуют лица 18—45 лет. С возрастом печень и
другие органы обычно утрачивают способность к некоторым
метаболическим реакциям. В результате ЛВ, биодоступность которых
снижена за счет «first-pass^-эффекта, попадают в системное
кровообращение у лиц пожилого возраста в гораздо большей
концентрации, чем у молодых [25, 26]. Чтобы избежать побочных
эффектов, часто используют уменьшенные дозы ЛВ. Наглядным
примером возрастных различий в метаболизме служит имипра-
мин (8.10), для которого зарегистрированы следующие фармакокине-
тические показатели — у здоровых добровольцев младше 40 лет:
CL — 950 мл/мин, t1/2 —17 ч, Стах— 10—20 нг/мл; у лиц старше 70
лет: CL — 570 мл/мин, t — 30 ч, С^ — 40—45 нг/мл [2]. Подобные
различия в метаболизме отмечены также при пероральном
применении антагонистов Са2+-каналов — производных 1,4-дигидропи-
ридина, диазепама, пропранолола, верапамила, лабеталола, теофил-
лина, амитриптилина и некоторых др.
Проблема возрастных различий в фармакокинетике ЛС
приобретает особую актуальность в связи с увеличением
продолжительности жизни в развитых странах. На рубеже XXI в. около 20%
населения планеты составляют люди старше 65 лет. Они являются
потребителями ~ 25% всех назначаемых в мире ЛС, причем
большинство пожилых людей ежедневно принимают по несколько
препаратов, что также увеличивает риск проявления нежелательных
побочных эффектов [27—29].
Различия в фармакокинетике ЛС имеют место и в неонатальном
возрасте. Печень новорожденных неспособна метаболизировать
многие ЛВ. Например, невозможна конъюгация антибиотика левомице-
тина с глюкуроновои кислотой, что приводит к накоплению препарата
и проявлению токсического эффекта (в данном случае кардиоваску-
лярного), получившего название «синдром серых младенцев». Кроме
того, у детей в грудном возрасте имеется незрелость некоторых
систем почечных канальцев (что лимитирует процесс фильтрации) и
значительно снижен почечный кровоток — в почки поступает только
5% сердечного выброса по сравнению с 20—25% у взрослых. В
результате процессы экскреции ЛС у младенцев замедлены, что
приводит к повышению концентрации в крови как самих препаратов, так
и их метаболитов и развитию нежелательных эффектов [25—27, 30].
Заболевания. У пациентов с заболеваниями печени отмечаются
существенные изменения в кинетике тех препаратов, которые экск-
ретируются в виде метаболитов. Например, пропранолол и нифеди-
пин обладают повышенной биодоступностью у больных с нарушени-
217
ями функции печени из-за сниженного «first-pass»-3(j)(|)eKTa, кроме
того, увеличивается и период полувыведения этих ЛС из организма.
При заболеваниях печени может быть снижено превращение про-
лекарств в активные формы. Однако предсказать такие изменения
крайне сложно [2, 8, 31].
Взаимодействие лекарств. Одновременное использование
нескольких препаратов может приводить к изменению их
метаболизма. Происходит указанное из-за взаимодействия ЛВ с одними
и теми же ферментными системами. При этом в одних случаях
метаболизм замедляется, в других — ускоряется. Кроме того,
некоторые препараты (барбитураты, рифампицин, слиронолактон), а
также вещества, способные проникать в организм из окружающей
среды (галогенсодержащие инсектициды), являются индукторами
микросомальных ферментов печени, включая и оксигеназы (см.
табл. 8.2). Это повышает продукцию метаболитов и усиливает
побочное действие некоторых ЛВ за счет их активных
метаболитов. Механизм такого повышения активности Ф печени пока
недостаточно изучен [32—34].
Состав пищи. Существенное влияние на фармакокинетические
параметры и метаболизм ЛС оказывают количество потребляемой
пищи и ее состав [35, 36]. В частности, отмечено, что богатая перцем
чили «мексиканская диета» снижает абсорбцию ацетилсалициловой
кислоты в желудке. Связано это с тем, что капсаицин,
присутствующий в перце, стимулирует желудочную секрецию, что, в свою
очередь, приводит к увеличению объема жидкости в желудке и
повышению скорости высвобождения его содержимого.
Японская кухня также существенно отличается от традиционной
европейской. В ней преобладают углеводы, меньше белков и совсем
мало жиров. Более чем у 50% японцев старше 50 лет снижена
кислотность желудочного сока. Это неизбежно сказывается на
биодоступности ЛВ, всасывание которых зависит от рН в желудке, например,
метронидазола [35].
Ингибирование отдельных изоформ CYP450 некоторыми
природными соединениями, присутствующими в пище, приводит к
изменению метаболических путей ряда ЛВ. Установлено, что один из
компонентов сока грейпфрутов — фуранокумарин бергамоттин —
необратимо связывает CYP3A4. В результате биодоступность таких
препаратов как фелодипин (антагонист Са2+-каналов), диазепам, ми-
дазолам, карбамазепин (анксиолитики), ловастатин, симвастатин
(ингибиторы HMG-CoA) и некоторых др. повышается на 50% за счет
снижения их досистемного метаболизма. При этом отмечен ряд
побочных эффектов, обусловленных увеличением концентрации ЛВ
в крови и тканях [35].
Генетические факторы. Метаболическая «судьба» препарата во
многом зависит от экспрессии изозимной системы CYP450. У 10%
населения отмечена неспособность организма гидроксилировать
отдельные виды веществ Ф семейства CYP2D6, что приводит к
большому количеству побочных эффектов. Первоначально генетический по-
218
диморфизм был выявлен в отношении такого ЛС, как дебризохин.
Позже удалось установить, что у лиц со сниженным метаболизмом
дебризохина наблюдается такой же эффект в отношении более чем
20 других препаратов, в том числе р-адреноблокаторов,
антидепрессантов, антиаритмических средств, опиоидов и др. На основании этих
исследований были классифицированы медленный и быстрый
фенотипы [37—39].
Существуют генетические различия и в немикросомальных
формах метаболизма. Впервые это было установлено на примере
N-ацетилирования изониазида и связано с активностью N-ацетил-
трансферазы. Аналогичное явление наблюдается и при метаболизме
далсона. Низкий уровень N-ацетилтрансферазы отмечен у 60%
шведов, в то время как у эскимосов Канады, напротив, уровень
этого Ф высок [39].
8.4. Методы изучения метаболизма
Метаболические превращения препаратов изучают с помощью
нескольких методов:
• измерение общей радиоактивности с включением радиоактивной
метки (,4С, 3Н, 35S);
• экстракция и хроматографическое разделение (ТСХ, ВЭЖХ
и ВЭГХ);
• специфические аналитические методы, разработанные
специально для данного препарата и его возможных метаболитов;
• ЯМР и масс-спектрометрия.
Использование препаратов с радиоактивной меткой дает
возможность оценить все количество связанных с ним материалов в
комплексной смеси эндогенных веществ в биологических образцах.
Комбинация экстракционных и хроматографических методов позволяет
определить степень метаболизма. Таким путем метаболиты можно
накопить, выделить, очистить и установить их строение спектральными
методами. Прямой ЯМР-анализ образцов биологических жидкостей
(плазмы, мочи и др.) позволяет выявить минимальное количество ЛВ
или его метаболитов. Для концентрирования образцов используют
метод замораживания и высушивания с последующим растворением
в D20.
Метаболизм можно исследовать in vitro и in vivo. Для оценки
потенциальной судьбы соединений на стадии раннего отбора
кандидатов в ЛС используют печень экспериментальных животных, например,
культуру гепатоцитов, микросомы. Таким способом выявляют
метаболиты разных, но близких по структуре веществ с тем, чтобы определить
возможный характер их превращений в организме человека.
Специфические аналитические методы, разработанные для
препарата и его метаболитов, позволяют измерить их концентрацию в
биологических жидкостях и, следовательно, исследовать фармакоки-
нетику ЛВ.
219
8.5. Роль сведений о метаболизме
в конструировании лекарственных средств
Сведения о метаболизме ЛС оказывают существенную помощь в
его разработке. Так, низкая биодоступность, обусловленная низкой
абсорбцией или интенсивным «first-pass^-эффектом in vivo, может
приводить к низкой активности вещества, демонстрирующего
высокую активность in vitro. Например, большинство антибиотиков цефа-
лоспоринового ряда являются полярными соединениями с низким
значением \gP при физиологических значениях рН и, следовательно,
слабо абсорбирующимися из ПК. Эта проблема может быть
разрешена путем формирования на основе этих веществ подходящего эфира,
являющегося пролекарством. Последнее лучше абсорбируется и, гид-
ролизуясь в ходе первичного метаболизма в кишечнике или печени,
высвобождает активный компонент — собственно ЛВ. Если быстрый
метаболизм ограничивает фармакодинамику соединения, модификация
структуры должна проводиться в направлении снижения этого
эффекта. Такой подход был реализован в дизайне антагонистов р-адре-
норецепторов при модификации метопролола (7.21) в бетаксолол (7.23).
Для отбора более стабильного соединения использованы результаты
изучения метаболизма, полученные in vitro на гомогенатах печени и
микросомах экспериментальных животных [40].
Литература
1. filing H. P. A. Xenobiotic Metabolism and Disposition, The Design of Studies on
Novel Compounds. — Boca Raton, Florida: CRC Press Inc., 1989.
2. Obach R.S. // Drug Metab. Dispos. - 1997. - Vol. 25. - P. 1359.
3. Nelson DR., Kamataki T. etal. // DNA Cell. Biol. - 1993. - Vol. 12, № 1. - P. 1.
4. White R.E. // Pharmacol. Ther. - 1991. - Vol. 49. - P. 21.
5. Drugs and the Liver: Clinical Applications. Pharmacokinetic Basis for Drug
Treatment / Ed. by L.Z. Benet. — New York: Raven Press, 1984.
6. Wighton S.A., Stevens J.C. // Crit. Rev. Toxicol. - 1992. - Vol. 22. - P. 1.
7. Parkinson A. Reference Guide to Substrates, Inhibitors and Inducers of the
Major Human Liver Cytochrome P—450 Enzymes Involved in Xenobiotic
Transformation. — Kansas City: Xenotech LLC, 1996.
8. Ковалев Я.Е., Румянцева Е.И. // Хим.-фарм. журн. - 1999. - № 3. - С. 3.
9. Guengerich F.P., MacDonald T.L. // Accts. Chem. Res. - 1984. - Vol. 17. - P. 9.
10. Ortiz de Montellano PR. // Trends Pharmacol. Sci. — 1989. — Vol. 10. —
P. 354.
11. Guengerich F.P. // Crit. Rev. Biochem. Mol. Biol. - 1990. - Vol. 25. - P. 97.
12. ConneyA.H. etal. // Clin. Pharmacol. Ther. - 1977. - Vol. 22 - P. 707.
13. Thummel K.E. et al. // Drug Metab. Dispos. - 1993. - Vol. 21. - P. 350.
14. Elliot R.H., Strunun L. // Br. J. Anaesth. - 1993. - Vol. 70. - P. 339.
15. ZeigelD.M. // Trends Pharmacol. Sci. - 1990. - Vol. 11. - P. 321.
16. Hollenberg P.E. // FASEB J. - 1992. - Vol. 6. - P. 686.
17. Conjugation Reactions in Drug Metabolism: An Integrated Approach. / Ed. by
G.J. Mulder. — New York: Taylor and Francis, 1990.
18. Guengerich F.P. // Pharmacol. Ther. - 1992. - Vol. 54. - P. 17.
19. Mulder G.J. // Trends Pharmacol. Sci. - 1992. — Vol. 13. - P. 302.
220
20. Spahn-Langguth H., Benet L.Z. // Drug Metab. Rev. - 1992. - Vol. 24. - P. 5.
21. Tracy T.S. etal. // Drug Metab. Dispos. - 1993. - Vol. 21. - P. 114.
22. Bioactivation of Foreign Compounds // Ed. by M. Anders. — New York:
Academic Press, 1985.
23. Mitchell J.R., Jollow D.J, Potter W.Z. et al. // J. Pharmacol. Exp.Ther. -
1973. -Vol. 187.- P. 185.
24. Jollow D.J., Mitchell JR., Potter W.Z. et al. // J. Pharmacol. Exp.Ther. -
1973. -Vol. 187. - P. 195.
25. Durnas С etal. // Clin. Pharmacokinet. - 1990. - Vol. 19. - P. 359.
26. Woodhouse K., Wynne H.A. // Drugs & Aging. - 1992. - Vol. 2. - P. 243.
27. Гуськова T.A. // Хим.-фарм. журн. - 2001. - № 10. - С. 3.
28. Tsipis G, Krummen К, Sigel L.// J. Toxicol. Clin. Toxicol. — 1996. -Vol. 35. -
P. 632.
29. Roy M., Blanchon M.A., Beyens МЛ etal. // Rev. Geriatr. - 1999. - Vol. 24. -
P. 347.
30. Hall J, Kauffman R., Wasserman G. // J. Toxicol. Clin. Toxicol. - 1999. -
Vol. 37. - P. 629.
31. Kleinbloesem H., Van Harten J et al. // Clin. Pharmacol. Ther. - 1986. -
Vol. 40. - P. 21
32. Strain J.J., Caleind G. et al. // Psychosomatics. - 1997. - Vol. 38. - P. 201
33. D'Arcy И J. Pharm. Belg. - 1998. - Vol. 53. - P. 352.
34. Strolm Benedetti M.} Bani M. // Drug Metab. Rev. - 1999. - Vol. 31. - P. 665.
35. Abstracts of the Joint Meeting of: VII World Conference of Clinical Pharmacology
and Therapeutics. (15—20 July 2000, Florence) in association with British Journal
of Clinical Pharmacology. — London: Blackwell Science Ltd., 2000.
36. Shayam Bala Lall et. al. Ц Indian. J. Exp. Biol. - 1999. - Vol. 37. - P. 109.
37. Myer U.A. etal. // Pharmacol.. Ther. - 1990. - Vol. 46. - P. 297.
38. Eichelbaum M., Gross A.S. // Pharmacol.. Ther. — 1990. - Vol. 46. - P. 377.
39. Daly A.K. et. al. .// Pharmacol.. Ther. - 1993. - Vol. 57. - P. 129.
40. Van de Waterbeemd H., Smith D.A. et al. // J. Med. Chem. — 2001. -
Vol.44. - P. 1313
9
ВЫБОР СТРАТЕГИИ ИССЛЕДОВАНИЙ
ПРИ СОЗДАНИИ НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
И КРИТЕРИИ ОЦЕНКИ КАЧЕСТВА СТРУКТУРЫ-ЛИДЕРА
В современном арсенале ЛС, несмотря на его постоянное
обновление, существует немало препаратов, которые применяются в
медицинской практике еще с конца XIX в. До сих пор не
утратили своего значения нитроглицерин, анестезин, аспирин и др. В то
же время не прекратился поиск веществ с вазодилататорными,
анальгетическими, противовоспалительными и др. свойствами. Кроме
того, существует немало заболеваний, для которых пока еще нет
удовлетворительной фармакотерапии. Задача обеспечения
потребностей здравоохранения в новых ЛС, обладающих значительными
преимуществами перед уже существующими препаратами или
открывающими неизвестные ранее пути коррекции опасных
патологий, не теряет актуальности.
Преимущества вновь создаваемых ЛС могут состоять в
увеличении селективности воздействия на определенный биологический
механизм, повышении активности, снижении токсичности.
У любого начинания в области поиска новых ЛС есть несколько
аспектов: научный — создать новое вещество или ЛФ с заранее
заданными свойствами; социальный — способствовать улучшению
здоровья населения, повышению качества жизни, снижению инвалиди-
зации или смертности; коммерческий — выпустить на рынок новый
продукт. Поэтому, прежде чем какой-либо проект получит развитие,
он должен пройти всестороннюю оценку. В расчет принимаются:
медицинская потребность, коммерческие возможности, уровень
имеющейся на сегодняшний день терапии, сравнительная активность
предполагаемого средства.
Как правило, наиболее успешны проекты, связанные с
реализацией новых подходов в лечении тех или иных заболеваний.
Например, лечение кандидозов еще недавно осуществлялось только
с помощью наружных средств, большинство из которых высоко
токсичны. Принципиально новый подход возник с открытием
группы препаратов, пригодных для перорального применения и
получивших общее название коназолы (см. также разд. 2.2). Флуко-
назол (9.1) эффективен против двух видов грибов Candida и
Cryptococcus neoformans. Практически одновременно с ним
разработчики предложили еще одно средство — вориконазол (9.2),
подавляющий рост Aspergillus [1—3]. Такая продуктивность
объясняется удачным выбором лидирующей структуры UK—51060, лег-
222
кость модификация которой обеспечила доступ к большому
разнообразию аналогов (9.1).
(9.1) (9.2)
На стадии формирования проекта необходимо принять во
внимание и вероятность успешного прохождения предлагаемым
препаратом стадии клинических испытаний. Несмотря на то, что
такая оценка обычно проводится клиницистами, специалист в области
медицинской химии должен сознавать, что демонстрации
эффективности нового средства и его безопасности может оказаться
недостаточно для выявления преимуществ данного ЛС перед уже
существующими препаратами. Преимущества достаточно трудно
доказать в ситуации, когда уже сложившийся уровень терапии
обеспечивает удовлетворительные результаты, или когда требуются
особенно длительные курсы лечения, или когда проблематично
привлечь новых добровольцев к испытаниям. Например, клинически
высокая эффективность нового антибиотика для перорального
применения достоверно доказана и можно начать его производство,
однако выявление преимуществ перед уже имеющимися средствами
требует больших затрат. Следовательно, более выгоден уже
сложившийся подход в терапии.
Если же имеющиеся подходы обеспечивают эффективность
лечения, а назначаемые препараты хорошо переносятся пациентами, но
имеют недостатки, связанные с неудобной дозировкой (необходимость
многократного применения в течение суток), то целесообразно
продолжать поиски новых ЛВ в этом же классе соединений и пытаться
усовершенствовать фармакокинетические параметры, вероятно,
путем создания пролекарства или новой ЛФ. Так возникают новые
поколения препаратов одного ряда. Например, антагонист Са2+-кана-
лов — производное 1,4-дигидропиридина первого поколения
нифедипин (7.9) — требует частого применения *( 1—2 табл 3 раза в
день) и обладает рядом побочных эффектов. Препарат второго
поколения амлодипин (7.13) предназначен для однократного применения
в течение суток и лишен негативных эффектов, характерных для
средств первого поколения.
223
Таким образом, перед началом разработки нового проекта часто
возникает вопрос, стоит ли усовершенствовать существующий класс
препаратов или необходимо искать принципиально новый подход в
терапии. В первом случае исходная лидирующая структура известна.
Это, обычно, наиболее эффективное в данной группе средство.
Усовершенствуя его, осуществляют дизайн новых аналогов. Во втором
случае лидер, как правило, отсутствует. Как же осуществить его
выбор? Прежде всего, следует подчеркнуть, хотя это и очевидно, что
последовательность скрининга должна отражать свойства, которые
являются целевыми. Иными словами, проводить отбор БАВ нужно
по тем видам активности, которые должны присутствовать у
конечного продукта.
Чем больше сведений о механизме возникновения и развития
заболевания, для терапии которого предназначено разрабатываемое
средство, тем эффективнее работа по отбору веществ, тем
«придельней» используемые методики скрининга. В свою очередь, чем шире
набор веществ, предоставленных для исследования, тем выше
вероятность нахождения в дальнейшем достойного лидера.
Первый этап скрининга направлен на отбор соединений,
заслуживающих дальнейшего изучения, и исключение структур, не
обладающих выраженной активностью. Следовательно, выраженность
эффекта и селективность являются первыми критериями отбора.
Скрининг, проведенный in vitro, в большинстве случаев
гарантирует, что отобранные соединения действуют согласно
установленному механизму (связываются с рецептором или выступают в
качестве ИФ) и обладают активностью, превышающей минимальный
граничный уровень. Если вещества демонстрируют высокий
уровень селективности действия, то их активность мало зависит от
возмущающих факторов окружающей среды и значительно отличает
их от остальных соединений ряда. Для соединений с низкой
селективностью достоверность различий в выраженности
проявляемого эффекта может быть спорна, а результаты испытаний таких
веществ, полученные в разные дни, трудно сравнимы. В подобных
случаях возникает необходимость в проведении дополнительных
тестов in vitro. Важно, что первоначальное отсеивание
осуществляется до того, как приступают к изучению in vivo. Исследования,
проводимые на животных с моделями заболеваний,
воспроизводящими основные механизмы патологии у человека, призваны
подтвердить или опровергнуть эффективность веществ-«хитов»,
отобранных на первом этапе. При этом также полезно иметь большое
разнообразие структур-аналогов для эффективного поиска лидера.
Его целесообразно проверить на нескольких моделях. Обычно
такие исследования помогают выяснить, является ли наблюдаемый
эффект дозозависимым или нет, установить длительность действия,
оценить отдельные фармакокинетические параметры, токсичность.
Если же нет возможности перейти к более совершенным моделям,
то на клинические испытания передают лидер, выявленный в ходе
многоступенчатого отбора.
224
Задача специалистов в области медицинской химии состоит в
выявлении тех структурных особенностей, которые вносят
основной вклад в профиль действия потенциального препарата. При этом
полезными оказываются разнообразные методы компьютерного
моделирования для распознавания областей максимального
связывания, таких как места образования водородных связей, вносящие
главный вклад в селективность, или выявление гидрофобных
фрагментов, определяющих выраженность эффекта и т. п. Оптимизация
параметров модельных структур может привести к формулировке
новых целей синтеза, к получению этих веществ и к новой
последовательности биологических испытаний, направленной на отбор уже
следующего лидера. То вещество, которое будет обладать
достоверными преимуществами перед своими предшественниками, и станет
очередным лидером.
Рассмотренная последовательность отбора представляет собой
эволюционный подход к поиску лидирующей структуры. Однако
не исключена ситуация, когда в начале реализации некоего
проекта отсутствуют четкие представления о стартовых химических
структурах. В этом случае нет иного выхода, кроме тестирования
на наличие определенного вида активности максимально
возможного количества соединений из самых разных классов органических
веществ. По мнению некоторых специалистов, в конце 80-х гг.
такой тотальный скрининг в определенной мере потеснил
рациональные методы конструирования лекарств. Произошло это
благодаря изменениям в технологии самого скрининга: появилась
возможность проверять большие массивы соединений, используя
компактные тест-системы. Сегодня успехи фармакологии и
молекулярной биологии позволяют более точно формулировать
фармакологические цели. Исследователи получили доступ к
индивидуальным рецепторам и Ф, благодаря чему возросла скорость выявления
структур-«хитов», на основе которых осуществляется уже
рациональный отбор лидеров.
Оба подхода в поиске лидера, как эволюционный, так и
эмпирический, можно проиллюстрировать на примере
конструирования анти-ВИЧ препаратов [4—8]. Расшифровка последовательности
нуклеотидов в геноме ВИЧ позволила предложить структуру ВИЧ-
1-протеазы — Ф, участвующего в репликации вируса. Оказалось,
что данная протеаза состоит из двух доменов с последовательностью
в 99 аминокислотных остатков и родственна аспартилпротеазе —
ренину (см. разд. 5.4.3). В 1989 г. появилась 3D модель ВИЧ-1-про-
теазы, и это имело решающее значение для активного поиска ее
ингибиторов [9]. Многие исследования были весьма успешны в
создании потенциальных ингибиторов ренина: разработанные в
результате подходы легли в основу конструирования субстратоподобных
ингибиторов ВИЧ-1-протеазы. Общим для всех соединений стало
наличие гидроксиэтиленового фрагмента, мимикрирующего переходное
состояние Ф — субстрат. Именно этот фрагмент является
ключевым в молекулах ингибиторов ренина (5.65) и (5.66) [10]. Несмотря
225
на то, что синтез проводился в разных исследовательских группах,
большинство полученных соединений оказались очень близкими по
структуре, потому что во всех случаях использовались аналогичные
принципы дизайна. Возникли проблемы с патентной защитой
новых веществ. Более успешными в поиске ингибиторов ВИЧ-1 про-
теазы оказались те исследователи, которые синтезировали пептидо-
миметики с малым числом амидных связей. Одной из лидирующих
структур является L—685434. Важнейшим этапом в преобразовании
ее в индинавир (9.3) стало введение в боковую цепь протоно-
акцепторных пиперазинового и пиридинового фрагментов,
существенным образом повысивших растворимость и активность
конечного продукта.
L-685434
(9.3)
Кроме индинавира на клинические испытания были переданы еще
несколько кандидатов в Л С, и после их удачного завершения в 1995—
97 гг группу пероральных анти-ВИЧ средств пополнили секвина-
вир (9.4), нелфинавир (9.5) и ритонавир (9.6) [4, 7].
(9-4)
Стоит отметить наличие определенного структурного подобия
между соединениями (9.4) и (9.5). В данном случае такое сходство
вовсе не означает, что этот общий для обеих структур фрагмент
несет ответственность за проявляемую активность. Просто, как и в
описанном выше случае, разработчики исходили из одних и тех же
параметров 3D модели ВИЧ-1-протеазы и использовали
аналогичные приемы модификации лидеров с целью улучшения их фарма-
кокинетических показателей.
Следовательно, если конструирование БАВ осуществляется
рационально с привлечением широко известных данных об
особенностях их строения, важно найти прототипы, которые имели бы
менее «очевидные» структуры. Вероятно, имеет смысл обратиться
к более «сложной химии» с целью создания приоритетного ряда
соединений.
В этой связи Заслуживает внимания пример рационального
дизайна непептидных ингибиторов ВИЧ-1-протеазы. В данном случае
исследователи пошли по пути отбора соединений, способных
вытеснять молекулы воды с активного центра протеолитического Ф, и тем
самым препятствовать процессу гидролиза. Параметры, которым
должна удовлетворять такая структура, представлены на рис. 9.1 Они
также получены на основании 3D модели [5, 6].
8,2-12,0 А
R1
R2
3,5-6,5 А
Ч /
H-bond
donor/acceptor
Рид.,9.1. Геометрические параметры молекул
потенциальных ингибиторовВИЧ-1-протеазы [5]:
j R1, R2 — алкильные радикалы
Вещество (9.7) стало первым «хитом», подтвердившим
правильность гипотезы о необходимости удаления молекул воды с активного
центра Ф. В рассматриваемом случае место этих молекул заняли ме-
227
токсигруппы. Соединения (9.8)—(9.10) — промежуточные лидеры,
модификация структуры которых привела к кандидату в ЛС (9.11).
Значительному росту активности способствовало введение бензильных
заместителей; содержащих дополнительные центры образования
водородных связей. Для соединения (9.11) К, = 0.018 нМ.
R1
HN
О
Л
NH
R3\.
Л.
R*
R2
R1
ОН
R2
НО
ОН
R'= Я2-бензил (9.8) R'= Я2-нафтил-2-метил (9.9) (9.10)
(9.11)
Эмпирический подход к поиску структур с анти-ВИЧ
активностью, основанный на скрининге больших массивов веществ разного
строения, позволил выявить несколько соединений, которые по
механизму действия являются аллостерическими ингибиторами
ВИЧ-обратимой транскриптазы. Это структуры-лидеры U—85961,
E-BPU, R—82913 и L—697661. В эксперименте in vitrdona по
эффективности превосходили препараты сравнения — азид<Ьтимидин (6.66) и
невирапин (6.81) [11]. Их принадлежность к разным классам
органических соединений лишь подчеркивает возможность обнаружения
нового структурного типа при тотальном скрининге.
228
но
(6.66)
А
(6.81)
^.
N
I О
I
U-85961
NH-Et
д
Ч"
Et
S-Ph
"^Ph
E-BPU
R-82913
Немало примеров лидеров, отобранных эмпирическим путем,
можно позаимствовать из работ, посвященных ингибиторам ген-регуля-
торных протеинов ВИЧ. Роль этих протеинов установлена, но из-за
отсутствия данных, обосновывающих продуманный дизайн их
специфических антагонистов, лидирующая структура Ro 5—3335 была
выявлена методом тотального скрининга, а затем оптимизирована
в Ro 24-7429 [5].
Ro 5-3335
Ro 24-7429
Из изложенного выше ясно, что лидер — это соединение, которое
представляет особый интерес в плане дальнейшего продвижения его
как возможного кандидата в ЛС. Такое вещество должно
удовлетворять определенным требованиям. При его оценке принимают во
внимание следующие показатели:
• уровень активности и селективности действия;
• эффективность in vivo;
• возможность объяснения механизма действия, с тем чтобы
направить синтез в сторону оптимизации структуры;
• наличие в структуре групп, вызывающих токсический эффект;
• наличие или отсутствие сходства с конкурирующими веществами.
229
Одним из главных критериев оценки лидера как химического
соединения является возможность модификации его структуры прямым или
косвенным путем. Желательно также, чтобы отобранное вещество было
водорастворимым. При низкой растворимости показатель липофиль-
ности lg/*должен иметь значение в интервале от —1 до +5. Для перо-
ральных средств важно, чтобы мол. масса не превышала 500
(см. разд. 7.3.1). В структуре не должно быть реакционноспособных
групп, повышающих токсичность. Безусловными преимуществами
лидера являются простой способ синтеза и доступность сырья.
Исходные реагенты должны обеспечивать большое потенциальное
разнообразие заместителей в молекуле или возможность независимой
модификации боковых цепей.
Из анализа критериев оценки лидирующей структуры следует, что
выявить такое вещество, опираясь только на результаты скрининга,
невозможно. Необходимы физико-химические и токсикологические
данные. Один из наиболее спорных аспектов связан с тем, что
соединение «выглядит» токсичным. Под этим понимают структуры,
содержащие группы, которые входят в состав уже известных веществ с
высокой токсичностью. Это правило не является строгим, но в то же
время помогает определить фрагменты, присутствие которых
нежелательно именно по критериям токсичности. К числу таковых
относятся нитрофенильные, тиомочевинные и азогруппы. Безусловно,
можно привести большое количество примеров, когда наличие таких
фрагментов не приводило к повышению токсичности. Но все же, если
в отобранном соединении есть подобные группы, следует оценить его
токсичность до того, как приступить к углубленному изучению
специфического действия. При выборе лидера следует избегать веществ,
обладающих ацилирующими или алкилирующими (кроме
специальных целей) свойствами. Если же в распоряжении исследователей
имеется единственный доступный лидер, который содержит подобный
фрагмент, и целесообразно продолжить исследование этого вещества,
то необходимо избавиться от алкилирующих свойств, заменив эту
группу на биоизостерную.
Полезными в прогнозировании негативных видов активности,
безусловно, являются методы QSAR. Существует большое разнообразие
компьютерных программ, позволяющих с высокой вероятностью
предсказывать наличие или отсутствие у какого-либо соединения
канцерогенных, мутагенных, тератогенных свойств [12—14].
Проиллюстрировать перечисленные выше критерии оценки структур можно на
примере веществ (9.12—9.15).
п
Н
ХУГ
(9.12) (9.13)
230
NH2 /=N jT' j[
° U^cooh
Н Ph
(9.14)
Из четырех представленных лидеров, два вещества (9.12) и (9.14)
удовлетворяют структурным критериям. Они стабильны, и их
метаболические трансформации предсказуемы. Напротив, соединения
(9.13) и (9.15) не могут быть предложены в качестве лидеров,
поскольку амид (9.13) является алкилирующим агентом, а наличие
S03H группы в структуре (9.15) обусловливает выраженные
кислотные свойства этого вещества. При этом сложное стереохимическое
строение обеспечивает возможность эпимеризации, и каждый из
изомеров способен вносить свой вклад в биологическую активность
или токсичность.
При отборе лидера необходимо как можно раньше выявить те
структурные особенности, которые могут существенно сказаться на
фармакокинетических параметрах. Например, липофильные карбо-
новые кислоты с высокой мол. массой быстро экскретируются с
желчью и не могут служить отправной точкой для препарата, который
обладал бы длительным периодом полувыведения из организма. В
случае, когда никакие улучшения структуры лидера не привели к
желаемому результату и исчерпано реальное число близких структурных
аналогов, следует отказаться от такого лидера и обратиться к
альтернативным структурам.
Нет необходимости еще раз подчеркивать, что от удачного
выбора лидирующей структуры зависит успех проекта в целом. И чем
выше конкуренция в той области, в которой осуществляется
исследование, тем более сложные задачи стоят перед специалистами,
конструирующими БАВ. Примером может служить опыт двух
японских фирм Sankyo и Takeda, в течение трех десятилетий
занимающихся разработкой антидиабетических препаратов из ряда
производных тиазолидиндиона, специфическая активность которых
направлена на усиление действия эндогенного инсулина.
Преимущество этих препаратов перед уже существующими пероральными
антидиабетическими ЛС состоит в том, что с их помощью можно
осуществить принципиально новый подход к воздействию на
ключевой механизм возникновения инсулинорезистентности
(устойчивости организма к эффектам гормона), а именно на процесс
связывания инсулина с рецепторами в органах-мишенях (клетках
печени, гладкой мускулатуры, жировой ткани) [15—20]. В
настоящее время доказано, что тиазолидиндионы, выступая агонистами
ядерных PPAR-рецепторов у-подтипа, оказывают воздействие на
(9-15)
231
экспрессию генов, регулирующих синтез инсулиновых рецепторов
и транспортеров глюкозы [20—22].
Первыми к поиску веществ, обладающих гиполипидемическим
действием и эффективных при ИНЗСД, приступили в 1972 г.
специалисты фирмы Takeda. Ко времени начала проекта уже: 1) сложились
представления о специфическом связывании инсулина с
рецепторами; 2) получила развитие концепция устойчивости периферических
тканей к гормону в связи с нарушениями в инсулиновых рецепторах;
3) была установлена связь между сахарным диабетом (СД) и
ожирением; 4) разработаны экспериментальные модели инсулинорезистент-
ности у животных, пригодные для оценки эффективности средств
воздействия на нее.
В 1975 г. были предложены два соединения в качестве лидеров —
AL-294 и AL-321. В структуру последнего входит тиазолидиндионо-
вый фрагмент. Это вещество обладало выраженным антигиперглике-
мическим действием. Его модификация привела к созданию в 1978 г.
циглитазона (9.16) — первого препарата данного профиля [23].
Однако он не получил широкого распространения в клинической
практике из-за большого количества побочных эффектов. В начале 80-х гг.
был предложен более перспективный кандидат в ЛС — пиоглита-
зон (9.17) [24].
AL-294 AL-321
(9.16) (9.17)
Компания Sankyo начала разработку проекта по созданию
антигипергликемических препаратов в 1980 г. Поиск проводился в
ряду полиалкилфенолов. Успеху в значительной мере способствовал
накопленный ранее опыт в создании ингибиторов фотодеструкции
полимеров: стерически нагруженные фенолы, содержащие две алкиль-
ные группы в орто-попоженнн по отношению к фенольному гидро-
ксилу, являются эффективными фотостабилизаторами пластмасс.
Соединения такого строения, способные связывать свободные
радикалы, представляют интерес и в качестве потенциальных ЛС. На
основании большого экспериментального материала была показана связь
между деструктивной ролью кислородных радикалов и развитием
232
таких патологических процессов в организме человека, как ИБС,
атеросклероз, СД. Начался активный поиск антиоксидантных
средств [25, 26]. Строение и свойства природных антиоксидантов
(а-токоферола, аскорбиновой кислоты, р-каротина, глутатиона и су-
пероксиддисмутазы) были хорошо известны. Единственным же
синтетическим препаратом с антиоксидантными свойствами долгое
время оставался пробукол (9.18) [27].
У-
s s—v \—он
(9.18)
Первоначально в ходе осуществления проекта
синтезировали производные феноксипентанола (9.19), 1,3-бензоксатиола (9.20) и
2-хлор-З-фенилпропионовой кислоты (9.21), сочетающие гиполипи-
демические свойства с антиоксидантными [25, 26]. Проявлению
последних способствовал фрагмент стерически нагруженного фенола,
аналогичный таковому в молекуле а-токоферола (витамин Е).
(9.21)
Однако исследования этих лидирующих структур не получили
дальнейшего развития, поскольку у веществ (9.19) и (9.21) в эксперименте
на животных выявили гепатотоксический эффект, а бензоксатиол (9.20)
при высоких показателях антиоксидантной активности как гиполи-
233
hLN. .NK
ЛУ* H,NY
AcONa
>^NH мир
(9.22)
Rl = R2 = R3 = R4 = R5 = CH,
(9.23)
пидемический агент значительно уступал препарату сравнения про-
буколу [26]. К этому моменту получили распространение сведения об
антигипергликемических свойствах нового класса веществ — тиазо-
лидиндионов [18]. Возникло предположение, что при объединении в
одной молекуле тиазолидиндионового кольца с фрагментом а-токо-
ферола могут быть получены соединения, способные снижать
уровень глюкозы, триглицеридов и холестерина в крови и одновременно
препятствовать образованию продуктов ПОЛ. Эфиры 2-хлор-З-фенил-
пропионовой кислоты (9.21) как раз и открывали путь к
объединению в одной структуре указанных фрагментов. На их основе и был
синтезирован ряд веществ общей формулы (9.22), среди которых
выявлен троглитазон (9.23) [17, 18, 26].
Практически одновременно в 1982 г. фирма Takeda предложила
упомянутый выше препарат пиоглитазон. Клинические испытания
троглитазона в Японии проходили в 1987—93 гг., пиоглитазона — в
1993-96 гг., в Европе и США они начались в 1998—99 гг. [17,28,29].
Однако через несколько месяцев после начала испытаний в США
стали поступать данные о развитии у пациентов острой печеночной
недостаточности, связанной с приемом троглитазона [28—30].
Частота возникновения этого грозного осложнения по оценке амери-
234
канской Food and Drug Administration (FDA) составила 1 случай на
15154 чел в течение 8 мес. приема препарата. В марте 2001 г. FDA
запретила клиническое применение троглитазона [31].
Одновременно в США проходили испытания еще двух препаратов
тиазолидиндионового ряда — пиоглитазона и росиглитазона (9.24) [32, 33].
Случаев острой печеночной недостаточности, связанной с их применением,
не отмечено. Однако нарушение уровня активности некоторых Ф
печени наблюдалось у нескольких пациентов через 20—25 дн. после
начала приема росиглитазона [34, 35].
N N О ^^ ^
in, о
(9.24)
Структурные различия между названными средствами и трог-
литазоном состоят в строении боковой цепи. Гепатотоксичность
троглитазона связывают с наличием в его молекуле фрагмента а-то-
коферола [31], именно того, который так стремились ввести в
структуру препаратов этого ряда. Причина негативного эффекта
кроется в особенностях метаболизма производных фенола. При
высоких концентрациях этих веществ происходит насыщение
ферментных систем печени, в результате, наряду с легко удаляемыми
метаболитами (сульфатами и глюкуронидами), образуются
токсичные глютатионовые и белковые конъюгаты. Аналогичное явление
имеет место при передозировке витамина Е или парацетамола (см.
разд. 8.3) [36—38]. Троглитазон применяли в дозах 200—800 мг в
день. В среднем содержание а-токоферола в таком количестве
препарата превышает суточную дозу витамина Е, что может быть
причиной токсического эффекта. Однако негативные побочные
реакции отмечены и в случае росиглитазона, который не имеет
подобного структурного фрагмента. Возможно, гепатотоксичность
является общим свойством тиазолидиндионов. Ведь именно по
этой причине, как отмечено выше, в ходе доклинических
исследований были отклонены многие структуры-лидеры. На
сегодняшний день окончательный ответ на вопрос о причинах токсических
свойств рассматриваемых веществ не получен. Специалисты с
настороженностью подходят к производным тиазолидиндионового
ряда. Безусловно, эти вещества обладают мощным антигипер-
гликемическим эффектом, и потребность в препаратах такого рода
очень высока, в связи с чем интерес к ним огромен. Однако их
применение должно быть безопасным и дальнейшие испытания
покажут, какое из этих средств окажется наиболее эффективным
и наименее токсичным.
Для успешного развития данного направления в
конструировании антидиабетических ЛС есть два пути. Один из них связан с поис-
235
ком более активных веществ в ряду тиазолидиндионов, которые бы
обладали таким же эффектом, как и их предшественники, но в
значительно меньшей дозе. Примером может служить лидер нового
поколения (9.25). В эксперименте на животных с моделями ИНЗСД
показано, что его ЕД50 составляет 30 мг/кг [39].
(9.25)
Другой путь направлен на поиск агонистов PPARy-рецепторов
среди соединений нетиазолидиндионового ряда, т.е. на выявление
нового структурного типа. В этой связи особого внимания
заслуживают производные 1-тирозина — (9.26) и (9.27). Соединение (9.26) по
выраженности антигипергликемического эффекта в 100 раз
превышает троглитазон [40].
(9.26)
(9.27)
Указанные вещества получены посредством комбинаторного
синтеза, а их активность установлена в ходе тотального скрининга [40].
Этот пример является еще одним подтверждением эффективности
современных эмпирических методов отбора лидирующих структур.
Литература
1. Medicinal Chemistry: Principles and Practice // Ed. by F.D.King. — Harlow,
UK: Smith Kline Beecham Pharmaceuticals, 1994.
2. Emerging Targets in Antibacterial and Antifungal Chemotherapy // Ed. by
J.Sutcliffe et al. — New York: Chepman and Hall, 1992.
236
3. Principles of Medicinal Chemistry 4lh ed. // Ed. by W.O.Foye, T.L.Lemke,
D.A.Williams. — Philadelphia: Lippincott Williams &Wilkins, 1995.
4. Vacca J.P., Condra J.H. // Drug Discov. Today. - 1997. - Vol. 2. - P. 261-272.
5. De Lucca G.V., Erickson-Viitanen S., Lam P.Y.S. // Drug Discov. Today. —
1997. -Vol.2. - P. 6-18.
6. Lam P.Y.S, Jadhav P.K, Eyermann C.J. etal. //Science. — 1994. —Vol. 263. —
P. 380-384.
7. Kubinyi H. H Current Opin. in Drug Discov. Develop. - 1998. - Vol. 1, N 1. -
P. 4-15.
8. Roberts N.A. et al. // Science. - 1990. - Vol. 248. - P. 358.
9. Laskowski R.A., Hutchinson EG, Michie AD. //Trends Biochem Sci. — 1997. —
Vol. 22. - P. 488.
10. Bablne RE, Bender S.L. // Chem Rev. - 1997. - Vol. 97. - P. 1359.
11. Resent Advances in the Chemistry of Anti-infective Agents'. Special Publication
N 119. The Royal Society of Chemistry, Cambridge, 1993.
12. 3D QSAR in Drug Design. Vol.2. Ligand-protein Interaction and Molecular
Similarity // Ed. by H.Kibinyi, G. Folkers, Y.C. Martin. — Dordrecht: Kluwer
Academic Publishers, 1998.
13. Marrone T.J., Briggs J.M., McCammon J.A. // Annu. Rev. Pharmacol. Toxicol. —
1997. -Vol. 37. -P. 71.
14. www.ibmh.msk.su/PASS/
15. SatielA.R. Olefsky JM. // Diabetes. - 1996. - Vol. 45. - P. 1669.
16. Fulgencio J~P., Kohl C, Girard J. et al. // Diabetes. - 1996. - Vol. 45. -
P. 1556.
17. Spencer СМ., Markham A. // Drugs. - 1997. - Vol. 54. - P. 89.
18. Day С // Diabet Med. - 1999. - Vol. 16. - P. 179.
19. Липсон B.B. // Хим.-фарм. журн. — 1999. - № 7. - С. 13.
20. Nolte R.T., Wisely G.B., Westin S. et al // Nature. - 1998. - Vol. 395. -
P. 137.
21. Willson T.M., Brown P.J., Sternbach D.D. et al. // J. Med. Chem. - 2000. -
Vol. 43. - P. 527.
22. Tq/uri S.R. // Endocrinology. - 1996. - Vol. 137. - P. 4706.
23. Sohda Т., Mizuno K, Imamiyan E, et al. // Chem. Pharm. Bull. — 1982. —
Vol. 30. - P. 3580,
24. Campbell 1Ж // Drugs. - 2000. - Vol. 60. - P. 1017.
25. Horikoshi H., Yoshioka Т., Kawasaki T. // Ann. Rep. Sankyo Res. Lab. —
1994. -Vol. 46. - P. 1.
26. Fujita Т., Yoshioka T. In Medicinal Chemistry: Today and Tomorrow // Ed. by
M.Yamazaki — Tokyo: Sankyo. 1996. — P. 73.
27. HeelR.C. etal. // Drugs. -1978. - Vol. 15. - P. 409.
28. WatkinsP.B,, Whitcomb Я.И<//N. Engl. J. Med. -1998.-Vol. 338. - P. 916.
29. Herrine S.K, Choudhary С // Ann. Intern. Med. - 1999. - Vol. 130. - P. 163.
30. Vella A., De Groen P.C., Dinneen S.F.jj Ann. Intern. Med. — 1998. —
Vol. 129. - P. 1080.
31. Tolman K.G. // Int. J. Clin. Pract. - 2000. - Suppl. 113. - P. 29.
32. Smith U. К Int. J. Clin. Pract. - 2001. - Suppl. 121. - P. 13.
33. Hanefeld M. // Int. J. Clin. Pract. - 2001. - Suppl. 121. - P. 19.
34. Forman L.M., Simmons DA., Diamond R.H. // Ann. Intern. Med. — 2000. —
Vol. 132. - P. 118.
35. Al-Salman J., Arjomand H., Kemp D.G. et al. I I Ann. Intern. Med. — 2000. —
Vol. 132. - P. 121.
237
36. Mitchell JR., Jollow D.J., Potter W.Z. et al. // J. Pharmacol. Exp.Ther -
1973. — Vol. 187. — P. 185.
37. J^f-faMgf*^ J/^otter WZ- et Qi H J- Pharmacol. Exp.Ther. -
38. Elcock F.J., Lyon J.J., Hitchcock J. et al. // Diabetes - 1999 - Vol 48
(Suppl.l).-P. A95. 01- 48'
39. Lohray В., BhushanK,ReddyA.S.etal.//S. Med. Chem -1999 -Vol Ю
P. 2569 ' '"' vv"- ^-
40. Gampe R.T., Montana V.G., Lambert M.H. et al. // Molec Cell - 2000 -
N 5. — P. 545.
10
ИСТОЧНИКИ ПОИСКА
НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Источниками в создании новых ЛС служат:
• природные продукты;
• уже существующие в медицине препараты;
• физиологические посредники;
• синтетические органические соединения, выявленные в
результате скрининга.
Данная глава посвящена проблемам создания ЛС на основе БАВ,
относящихся к первым трем источникам. Вопросы, связанные с
отбором структур-лидеров из впервые синтезированных органических
соединений, будут рассмотрены в главе 12.
10.1. Природное сырье
как источник новых лекарственных средств
Природные продукты являются древнейшим источником БАВ.
Естественный отбор в ходе эволюции и борьба между видами создали
гигантский арсенал веществ, который активно используется
исследователями для отбора лидирующих структур. Например, открытие
пенициллина способствовало возникновению концепции поиска природных
антибиотиков и последующей модификации их структуры, исходя из
предположения микробиологов о том, что многие бактерии,
вызывающие инфекционные заболевания у человека, не выживают в почве
потому, что другие бактерии, обитающие в этой же среде, их
уничтожают. Интенсивные скрининговые программы по изучению почвенных
микроорганизмов позволили выявить большое количество
антибиотиков, на основе которых были созданы эффективные препараты:
стрептомицин (6.11), хлорамфеникол (6.17), хлортетрациклин (6.19),
эритромицин (6.25). Эти исследования продолжаются в настоящее время и
связаны они не только с поиском антибактериальных средств [1].
Примером недавней разработки служит препарат орлистат (7.1),
предложенный в качестве средства от ожирения (см. разд. 7.3) Действующим
веществом этого Л С является модифицированный природный продукт,
который первоначально был выделен из почвенных микроорганизмов
Streptomyces toxytricini и получил название липстатин. После выявления
239
у данного соединения способности ингибировать гастроинтестиналь-
ную липазу и установления строения, его структура была
модифицирована и синтетическим путем получено более стабильное производное —
тетрагидролипстатин (7.1). Последний, благодаря конкурентному
связыванию с кишечной липазой, препятствует расщеплению триглице-
ридов до моноглицеридов и свободных жирных кислот. В результате
организмом не усваивается до 30% жиров, поступающих с пищей [2].
Источником БАВ могут служить яды и токсины как белкового,
так и небелкового строения, используемые животными для защиты
или обездвижения своей добычи. Многие из этих природных
продуктов, например, тетродотоксин (10.1) рыбы puffer (фугу) или сакситок-
син (10.2) морских одноклеточных жгутиковых организмов,
блокирующие №+-каналы; карибдотоксин (из яда скорпиона), блокирующий
Са2+-зависимые К+-каналы; а-бунгаротоксин (яд змеи),
связывающийся с ацетилхолиновыми рецепторами, и батроксобин (из яда
гадюки), являющийся тромбино-подобным Ф, действуют в минимальных
концентрациях (~ 10"9 М).
H2N
Y
О
HN
+ NH<^N
(10.1) (10.2)
Общность физиологического действия тетродо- и сакситоксинов
определяется наличием в их структуре положительно заряженных гу-
анидиновых фрагментов. За счет этого они способны
взаимодействовать с кислотными остатками белковых молекул, формирующих
вход в ионный канал на внешней поверхности цитоплазматической
мембраны, и закупоривать его. Чрезвычайно высокая токсичность
рассматриваемых веществ (а это наиболее сильные из известных
небелковых ядов) не позволяет использовать их в фармакотерапевти-
ческих целях. Однако исключительное сродство нейротоксинов к
№+-каналам сделало их незаменимым инструментом в научных
исследованиях, позволив получить информацию о молекулярной
природе каналов и их локализации в нативных мембранах [3, 4].
Яды белковой природы также сыграли важную роль в изучении
механизмов функционирования рецепторов некоторых гормонов и Ф.
Недавно объектами исследований стали яды лягушек, пауков, губок.
Одним из наиболее богатых источников новых БАВ в настоящее
время служат морские организмы. Наиболее изучены среди них
водоросли и рыбы, наименее — морские бактерии. Большинство веществ,
извлеченных из морских микроорганизмов и обладающих
антибактериальными свойствами, весьма перспективны как антибиотики.
Органические вещества, выделенные из морских объектов, имеют ряд осо-
240
бенностей. Так, они отличаются высокой полярностью, наличием в
структуре большого числа атомов галогенов и гидроксильных групп,
например, соединения (10.3) — (10.5). Объясняется это тем, что
биосинтез таких веществ происходит в солевых растворах, в ряде случаев
при больших давлениях и низких температурах [5].
ОН О
Br. H,C CI
(10.5)
Новые БАВ, полученные из морских организмов, обладают не
только антибактериальным действием. Среди них обнаружены ИФ, вещества
с противоопухолевой, фунгицидной, противовирусной активностью.
Очень популярно направление, связанное с исследованием БАВ,
выделяемых цианобактериями. Цианобактерии — группа древних и
разнообразных фотосинтезирующих микроорганизмов, которые
выживают в различных, часто экстремальных, условиях окружающей
среды, что свидетельствует об их чрезвычайно высокой способности
к адаптации. Они могут существовать в неорганической среде,
используя для фотосинтеза солнечный свет и С02, и при этом
обеспечивают небывалое разнообразие высокоактивных метаболитов.
Наиболее характерным свойством этих метаболитов является способность
тормозить рост и развитие других организмов [6—8].
Биологическое разнообразие цианобактерии подстать
химическому разнообразию, которое присуще сегодняшнему состоянию процесса
поиска и создания лекарств. Производимые этими микроорганизмами
химические соединения, после установления их структуры, могут быть
исследованы в нативном состоянии или же модифицированы
методами органического синтеза в более мощные терапевтические агенты.
Следует отметить, что большинство подобных природных продуктов не
являются синтетически легкодоступными веществами, даже при
наличии самых современных методов конструирования органических
соединений. Число БАВ, впервые выделенных из цианобактерии, с каждым
годом возрастает в геометрической прогрессии. Виды активности с их
241
примерным долевым соотношением, выявленные у метаболитов этих
микроорганизмов, представлены на рис. 10.1 [7].
Рис. 10.1. Виды активности, присущие БАВ,
продуцируемым цианобактериями [7]:
1 — противоопухолевая; 2 — цитотоксическая; 3 — антибактериальная; 4 — фуи-
гицидная; 5 — противовирусная; 6 — др. виды активности; 7 — не исследованные
виды активности
Наиболее известны две группы веществ — криптофицины и курацины.
Исследование «структура-активность» 18 криптофицинов показало, что
среди них наиболее активны два: криптофицин-1 (10.6) и
соединение (10.7), полученное полусинтетическим путем. Криптофицин-1 (10.6)
был впервые выделен при отборе веществ, обладающих фунгицидной
активностью [9]. Однако из-за высокой токсичности он оказался
непригоден для использования по указанному назначению. Позднее при проверке
экстрактов из 1000 различных цианобактерий на разных типах
опухолевых клеток было установлено, что соединение (10.6), содержащееся в
Nostoc sp GSV224, является чрезвычайно эффективным цитостатиком [10]:
величина ДДХ для него in vitro составляет 3—5 ВДпг/мл. Таким образом,
(10.6) в 102—103 раз активнее, чем самые мощные из существующих ныне
противораковых средств — таксол и винбластин [11].
н,с ^сн, 3
(10.6)
CI
а
,сн,
н.сЧн, СНз
(10.7)
242
Курацины (10.8) — (10.10) выделены из Lyngbya majuscula (карибская
коллекция) в ходе реализации программы по получению новых липидных
компонент из цианобактерий [12]. Эти вещества оказались чрезвычайно
активными против лейкемии и др. злокачественных заболеваний:
величина ЕД50 — 9 нМ в отношении клеток лейкемии L-1210 и 0.2 мкМ — для
СА 46 клеток Burkitt lymphoma. Курацин А (10.8) был синтезирован путем
21 -стадийного синтеза с общим выходом 0.6% через год после открытия [13].
Исследованы 26 аналогов соединения (10.8) с модифицированной
боковой цепью, тиазолидиновым или циклопропановым кольцом и
установлено, что такие преобразования в указанных фрагментах приводят к
потере активности. Замена в боковой цепи метоксигруппы на метоксиме-
тильную также сопровождается снижением эффекта. Для обеспечения
Испытаний вещества (10.8) использовали микробиологический путь его
получения: из 640 л культуры извлекли 215 г сухого остатка, из которого
затем выделили 132 мг целевого продукта [14].
Н.С
(10.8)
(10.9)
Скрининговая программа по поиску ингибиторов протеаз среди
метаболитов цианобактерий была начата в Токийском университете. Главным
образом были изучены аэрогинозины А (10.11), (10.12) и В (10.13), (10.14) —
линейные пептиды, выделенные из Microcystis aeruginosa NIES 98.
Установлено, что ингибирующая активность соединений (10.11) и (10.13)
относительно тромбина, плазмина и трипсина практически одинакова:
величина ДДХ составляет 15, 10 и 0.9 мкМ соответственно для каждого Ф.
Аэрогинозин 298А (10.12) — мощный ингибитор тромбина и трипсина с
ДЦХ — 0.5 и 1.7 мкМ соответственно; в то же время он не влияет на
активность химотрипсина, эластазы, папаина [15, 16].
СН,
(10.10)
243
(10.12)
Цианобактерии примечательны и своей способностью продуцировать
циклические пептиды, такие как микроцистины и нодуларины. Микро-
пептины 478А (10.15) и В (10.16) из Microcystis aeruginosa NIES 478
селективно ингибируют плазмин (Щ50 — 0.11 и 0.45 мкМ соответственно для А
и В), не оказывая при этом воздействия на трипсин, тромбин, папаин,
химотрипсин и эластазу [17].
(10.13)
244
О Н
но он 0 ^^
ноДо
О" N
Н
(10.14)
Н
Y
NH
R= H
R = SO, H
(10.15)
(10.16)
Противовирусные агенты, выделенные из цианобактерии, принадлежат к
одной из двух категорий: пептидам или гликолипидам. Циановерин N —
пептид, содержащий 101 аминокислотный остаток, выделен в
национальном институте рака США из культуры цианобактерии Nostoc ellipsosporum
и воспроизведен путем кодирования ДНК-последовательности на
культуре E.coli. Тем самым было доказано его строение. Установлено, что в
концентрации 9 мкМ циановерин N нетоксичен для клеток хозяина, в то
же время в диапазоне концентраций 0.1—4.8 нМ он вызывает гибель
50% лабораторных образцов ВИЧ. Его Щ50 относительно штамма ВИЧ-1
изменяется в пределах 1.7—37 нМ. Циановерин N необратимо
связывается с гликопротеином оболочки вируса ВИЧ-1, что не дает возможности
последнему проникать в клетки хозяина. Еще одна важная особенность
245
циановерина — устойчивость к физико-химическим воздействиям, что
повышает вероятность успеха в разработке его как потенциального
средства против ВИЧ-1. Однако этот пептид неактивен против вируса
простого герпеса-1 и аденовируса-5 [18].
Сульфогликолипид (10.17) с анти-ВИЧ-активностью впервые выделен в
1989 г. в ряду гомологов из лабораторной культуры Lynbya lagerheimiiDN—
7—1 (гавайская коллекция). Близкие ему по структуре гликолипиды общей
формулы (10.18) составляют около 10% от состава органических веществ
цианобактериий. Они ингибируют ДНК-полимеразу ВИЧ-обратимой
транскриптазы в концентрациях <10 мкМ [19]. Разработка синтетических
подходов к получению гликолипидов осуществляется в связи с
необходимостью оптимизации их структуры. Соединение (10.19), полученное
синтетическим путем, обладает мощным противовирусным действием в
отношении одной из самых опасных разновидностей вируса герпеса
человека — вируса Эпштейна—Барр [20].
(10.17)
(10.18)
\ /
R2 R2
(10.19)
Поиск веществ с противовирусной активностью среди метаболитов циано-
бактерий находится на начальной стадии и имеет большое будущее. Воз-
246
росшая в последние годы в целом активность в исследовании указанных
микроорганизмов свидетельствует о том, что это исключительно
перспективный источник в поиске структур-лидеров для создания новых ЛС.
Народная медицина с давних пор использовала лекарственные
свойства растений. Свойства алкалоидов атропина и хиосцина из
растений семейства Solanaceae были известны античным грекам,
морфина — алкалоида из опийного мака — древним египтянам,
резерпина из раувольфии змеиной — жителям Индии, сердечные гликозиды
наперстянки — европейцам. Противовоспалительные свойства
природных продуктов — салицилатов, содержащихся в побегах ивы,
использовал Гиппократ. Сведения об этих средствах накапливались
эмпирически. Наши далекие предки, естественно, ничего не знали
ни о строении БАВ растительного происхождения, ни о механизмах
их действия. Однако с развитием органической химии уже в XIX и
начале XX вв все перечисленные выше алкалоиды были выделены,
установлено их строение, осуществлен синтез. Действующими
веществами растений являются не только алкалоиды и гликозиды, но и
такие химические соединения как терпены, сапонины, кумарины,
флавоноиды и др. Вскоре после открытия структуры многих
растительных БАБ начался интенсивный поиск их синтетических аналогов
более простого строения. Примером могут служить антихолинерги-
ческие препараты — спазмолитин (10.20) и метацин (10.21), имеющие
элементы структурного сходства с молекулой атропина (10.22) и аце-
тилхолина (2.48), что обеспечивает им эффективное связывание с
холинорецепторами. Первым препаратом данного ряда стал
спазмолитин, синтезированный в 1937 г. [21].
HCI
(10.20)
(10.21)
(10.22)
В 1898 г. было установлено строение природного алкалоида кока
ина (10.23), а годом позже открыто его местноанестезирующее лей
ствие. Вне связи с этими исследованиями в 1890 г. был получен эти
247
ловый эфир л-аминобензойной кислоты (10.24). Оказалось, что он
имеет некоторое структурное сходство с кокаином и также обладает
анестезирующим эффектом. Новое вещество получило название
анестезин. В дальнейшем целенаправленно проводился поиск
структурных аналогов кокаина. Так, синтез новокаина (10.25),
осуществленный в 1905 г. — одна из первых удачных попыток конструирования
ЛС. Этот препарат является модифицированным аналогом
анестезина и в то же время имеет элементы сходства с кокаином. Поиск
аналогичных структур оказался успешным, и вскоре синтетические
препараты этого ряда вытеснили из медицинской практики токсичный и
вызывающий привыкание кокаин [21].
N-Me
\-J MeO—CO N f О-В
(10.23) (10.24)
(10.25)
Примерами близкой структурной аналогии между природным
продуктом и синтетическим веществом являются папаверин (10.26) и
но-шпа (10.27). Папаверин открыт в 1848 г., синтезирован в 1910 г.;
но-шпа получена в 50-е гг. и установлено, что по спазмолитической
активности она превосходит папаверин [21, 22].
Me N
Mev J'
О
Me'°N
Me.QJ
f^r
и
'"'^S
„сн2
Е,Л
Е,Л
Е..0^
Лг
и
^
NH
|| -на
(10.26) (10.27)
Многообразие уже известных в настоящее время БАВ,
выделенных из растительного сырья и получивших широкое применение в
медицинской практике в качестве ЛС, чрезвычайно велико. При этом
растения продолжают быть источником новых медикаментов:
ориентировочно потенциал природной растительной коллекции
оценивается в 30 млн. соединений. Даже комбинаторная химия, эффективный
инструмент в отборе структур-лидеров, не в состоянии конкурировать
с разнообразием БАВ растительного происхождения.
248
Одним из примеров сравнительно недавних находок является так-
сол (10.28). Он выделен из тихоокеанского тисса в начале 60-х гг.
XX в., а несколькими годами позже были установлены его строение
и антимитотическая активность. В структуре таксола объединены тет-
рациклическая терпеноидная система с гетероциклическим оксета-
новым фрагментом. Синтез этого соединения, осуществленный в
начале 90-х гг., существенно расширил возможности его клинического
применения [23].
(10.28)
Таксол, наряду с алкалоидами растений Vinca (барвинок) —
винбластином и винкрестином, сегодня является одним из самых
эффективных противоопухолевых средств [24]. Следует отметить,
что поиск синтетических аналогов таксола, с целью выявления
более доступных соединений, ведется весьма интенсивно. Так,
исходным «строительным материалом» для центрального вось-
мичленного цикла могут со временем стать производные </-кам-
форы (10.29) [23].
л,„лиd^ —-R_0V4-roh -чГуR3
CI % у-* Яч,Ч
R2
(10.29)
Широко известна группа таких растительных БАВ как флавонои-
ды, обладающих разнообразным спектром фармакологического
действия. Однако противораковая активность в этом ряду отмечена лишь
недавно у флавопиридола (10.30) и близкого ему по строению — ро-
хитукина (10.31). Первоначально их изучали в качестве БАВ с
противовоспалительными и иммуномодулирующими свойствами. Позднее эти
соединения, а также большой ряд их структурных аналогов, были
воспроизведены синтетическим путем. Установлено, что они
обладают цитостатическим эффектом при воздействии на опухолевые клет-
249
ки in vitro Щ$0 для них < 0.1 мкМ. [25, 26]. В настоящее время флаво
пиридол находится на стадии доклинических испытаний как противо
опухолевое средство.
ОН О ОН О
(10.30) (10.31)
Среди флавоноидов, обладающих анксиолитическим действием,
опосредованным через центральные бенздиазепиновые рецепторы,
следует выделить хрисин (10.32) и церсилиол (10.33). Их активность
сравнима с диазепамом, но, в отличие от последнего, им не присущи
седативный и расслабляющий мускулатуру эффекты [27].
.ОН
ОН О
(10.32)
Оригинальный класс природных веществ артемизининов (10.34)—
(10.36) — пероксилактонов сесквитерпенового ряда с высокой анти-
малярийной активностью — обнаружен при изучении состава
эфирного масла полыни однолетней. С давних пор в традиционной
(10.34)
(10.35)
R=CONEt2
R=COC6H4C02Me
(10.36)
250
медицине Китая вытяжка из этого растения используется как
высокоэффективное антималярийное средство. Однолетняя полынь,
произрастающая в Средней Азии и Казахстане, содержит до 0.05% перокси-
да (10.34). Открытие лечебных свойств артемизининов стимулировало
работы по полному синтезу сесквитерпена (10.34) и его аналогов [6, 27].
Аскаридол (10.37), составляющий до 60% хеноподиевого масла,
получаемого перегонкой с водяным паром из растения Chenopodium
ambrosioides L., долгое время использовался в США в качестве
эффективного антигельминтного средства. Позднее было установлено, что
аскаридолу также присуща антималярийная активность [6, 27].
Примерами природных пероксидов с антипаразитарным
действием служат каниоян (10.38) и бисэпиканиоян (10.39), выделенные из
коры дерева Jatropha grossidentata, произрастающего в Парагвае.
Индейское население страны с давних пор использовало эту кору в
качестве антималярийного средства [6].
(10.37) (10.38) (10.39)
Несмотря на достигнутые успехи в получении БАВ из
растительного сырья, поиск новых лидеров ограничивается двумя основными
проблемами. Первая — сложность разделения экстрактов на индивидуальные
компоненты. Ряд фирм специализируется на технологиях разделения
смесей, содержащих компоненты в широком диапазоне концентраций.
Более серьезная проблема лежит в сфере общественных отношений и
связана с политикой. Большинство источников ценного растительного
сырья, пока еще недостаточно изученного, находится в развивающихся
странах. Выявление новых веществ и вывоз сырья часто осуществляются
без признания прав стран-хозяев, что приводит к хищническому
разграблению природных ресурсов. В 1992 г. ООН принята Конвенция о
биологическом разнообразии, призванная побудить правительства
развивающихся стран принять законы, охраняющие генофонд этих государств.
10.2. Официнальные лекарственные средства
как источник создания новых препаратов
Одним из источников для создания новых ЛС остаются
официнальные препараты. При этом некоторые из существующих ЛС
нуждаются в устранении определенных побочных эффектов, усо-
251
вершенствовании фармакокинетических параметров (повышении
абсорбции или длительности действия) и др. характеристик. Например,
антигистаминные средства, вошедшие в медицинскую практику в 40—
50-е гг. (пипольфен, димедрол), обладают выраженным седативным
действием, вызывают сонливость. Этот эффект в значительной мере
снижен у препаратов нового поколения (астемизол и др.).
Иногда появление новых средств становится возможным
благодаря «усовершенствованию» побочного эффекта известных ЛВ. Именно
по такому пути пошли в 50-х гг. создатели диуретических и гипо-
гликемических сульфамидных препаратов. При клиническом
применении сульфамидов было отмечено, что они вызывают щелочной
диурез у пациентов, принимающих большие дозы этих веществ.
Дальнейшие исследования показали, что происходит это из-за ингиби-
рования Ф карбоангидразы. Полученный результат использовали в
отборе новых структур, для которых данный эффект являлся
основным, что привело в 1957 г. к созданию хлортиазида (10.40). Тем
самым было положено начало новому направлению в фармакотерапии
артериальной гипертензии, быстро вытеснившему соединения ртути
из группы диуретических средств [21].
О
(10.40)
В 1942 г. французский врач М. Джанбон, изучая побочные
эффекты антибактериальных сульфамидных препаратов, обнаружил
симптомы гипогликемии. Позднее, в 1946 г., установили, что
сульфамиды способствуют снижению уровня глюкозы в крови благодаря
стимуляции секреции инсулина в поджелудочной железе. В 1955 г.
появились первые пероральные антидиабетические средства (см. разд.
4.4) — карбутамид (4.8) и толбутамид (4.9). На смену им пришли
препараты второго — глибенкламид (4.12), глипизид (4.13) и третьего —
глимепирид (4.14) поколений [28].
Длительное время механизм инсулиносекреторного действия этих
веществ оставался нераскрытым. Только в середине 80-х гг. выявлены
специфические рецепторы сульфонилмочевины — АТФ-зависимые
К+-каналы панкреатических р-клеток [29, 30]. Примерно в это же
время было показано, что вещество, представляющее
несульфамидную часть самого мощного гипогликемического препарата глибенк-
ламида, также обладает сахароснижающим действием [31]. Этот факт
стимулировал поиск несульфамидных инсулиновых секретогенов. На
сегодняшний день известен большой ряд веществ, не содержащих
сульфонилмочевинного фрагмента, но обладающих выраженным
гипогликемическим эффектом. В клинической практике в конце
90-х гг появилось два представителя этой группы — репаглинид
252
(см. рис. 2.10, разд. 2.5.1) и натеглинид (10.41). Механизм их
действия также опосредован через К+АТФ-каналы р-клеток
поджелудочной железы, однако, по сравнению с известными ранее перораль-
ными гипогликемическими препаратами, эффект наступает быстрее
(30 мин) и менее продолжителен (2—3 ч) [31, 32].
Т'
** tv °
О
(10.41)
Длительное клиническое применение сульфамидных
антибактериальных средств позволило выявить у них, кроме вышеупомянутых
побочных эффектов, слабо выраженную способность блокировать
рецепторы эндотелина (ЕТА-рецепторы), локализованные в
сосудистом эндотелии, и тем самым снижать АД. Так, для сульфотиазола
(норсульфазол, 5.49) /С50 в отношении ЕТА-рецепторов составляет
69 мкМ [33]. Замена тиазольного цикла на диметилизоксазольный,
реализованная в структуре (10.42), повысила активность в 100 раз, а
введение диметиламинонафталинового фрагмента привело к
получению BMS-182874 — высокоаффинного и селективного ЕТА-лиганда,
у которого отсутствует бактериостатический эффект. Это соединение
пригодно для перорального применения и оказывает
продолжительное гипотензивное действие. Дальнейшая оптимизация структур в
рассматриваемом ряду позволила выявить чрезвычайно мощные ЕТА-анта-
гонисты BMS-193884 и BMS-207940, которые, к тому же, обладают
100% биодоступностью и улучшают гемодинамику [34].
-KJ СНз СН I J Н СН,
H2N ^"^ СНз Ч^ омз
/С50 = 0.78 мкМ (10.42) BMS-182874 /С50 = 0.15 мкМ
В результате изучения действия, а затем и модификации
структуры уже упомянутого антигистаминного препарата пипольфен (10.43)
появились фентиазиновые нейролептические средства. В начале
50-х гг. предполагали, что пипольфен способен предотвращать
хирургический шок. Было доказано, что он частично блокирует вазодилата-
торное действие гистамина, однако обладает и необычным действием
на ЦНС. Исследование этого эффекта привело к получению
структурных аналогов пипольфена, которые оказывали депремирующее
253
BMS-193884 К, = 1.4 нМ MBMS-207940 К, = 0.010 нМ
действие на ЦНС, ошибочно принятое за способность снимать
хирургический шок. Наиболее активным веществом оказался
аминазин (10.44). В 1952 г. препарат был испытан у пациентов с
маниакальными состояниями. Полученные результаты позволили применять
это средство для купирования приступов шизофрении, что открыло
новую страницу в психиатрии [35].
(10.43) (10.44)
Среди современных примеров создания новых ЛС на основе
известных препаратов — направления, связанные с созданием анти-
эметических (противорвотных) препаратов и новой группы
гипотензивных средств — агонистов К+-каналов. В первом случае
начало было положено выявлением антиэметической активности у
метоклопрамида (10.45) — антагониста дофаминовых
рецепторов [36]. Изученные в данном аспекте более сильные дофаминовые
антагонисты оказались не настолько эффективны. Следовательно,
механизм антиэметического действия метоклопрамида опосредован
не через этот тип рецепторов. Выяснилось, что названный
препарат и его аналоги, содержащие тропановый фрагмент, являются
также антагонистами одного из подтипов 5-гидрокситриптаминовых
рецепторов (позднее этот тип серотониновых рецепторов
классифицировали как 5-НТ3). Дальнейшие исследования «структура-
активность» привели к получению 3,5-дихлорбензоата — MDL
72222, который превосходил по активности метоклопрамид в
40 раз. В результате замены бензольного кольца на индольное был
получен трописетрон (10.46). Наиболее выраженным эффектом в
рассмотренном ряду обладают гранисетрон (10.47) и ондансет-
рон (10.48) [37].
254
(10.45)
N-CH.
MDL 72222
N-CH,
N-CH.
(10.46)
(10.47)
N-CH,
(10.48)
Базовыми структурами в конструировании агонистов К+-кана-
лов кардиомиоцитов послужили (j-адреноблокаторы. Было
установлено, что при замыкании боковой цепи атенолола (7.20) в
бициклическую бензпирановую структуру (10.49) утрачивается ад-
ренергическая активность, но сохраняется гипотензивное
действие [38]. Исходя из этого синтезированы аналоги (10.49), для
повышения химической стабильности которых введены две метильные
группы в положение С(2). В ряду веществ (10.49) - (10.52)
наиболее активным «открывателем» К+-каналов является (-)-(3S,4R)-3HaH-
тиомер хромакалина (10.52).
(10.49)
он
255
NC' "^ Y OH
<y
(10.51) (10.52)
Большое количество фармацевтических продуктов появляется
благодаря незначительным изменениям в структуре существующих
препаратов. Это так называемые препараты «Ме-Тоо». Очень трудно
объяснить успех продуктов «Ме-Тоо», но некоторые из них становятся
препаратами первого выбора, вытесняя оригинальные средства или,
как минимум, приобретая широкое использование. Например,
антагонисты гистаминовых Н2-рецепторов коренным образом изменили
подходы к лечению язвы желудка. На короткое время самым продаваемым
и назначаемым препаратом данного профиля в мире стал циметидин
(10.53). Он появился на пять лет позже, чем ранитидин (10.54),
который в действительности значительно эффективнее и обладает
меньшим количеством побочных эффектов, в частности относительно
некоторых Ф печени. Однако через несколько лет ранитидин все же
вытеснил циметидин и занял первое место на рынке противоязвенных
средств [39]. Сегодня известны вещества, превосходящие по
выраженности антигистаминный эффект ранитидина.
HN^N
(10.53) (10.54)
Продукты «Ме-Тоо» обеспечивают главный путь, посредством
которого оптимизируются некоторые виды действия лекарств как по
селективности (устранение побочных эффектов), так и по
применению (пользуются особой популярностью у населения). Однако при
этом неоправданно увеличивается количество ЛС близкого профиля,
и необходимо много лет, чтобы клиницисты смогли выявить
наиболее приемлемые схемы фармакотерапии.
10.3. Физиологические посредники
как источник новых лекарственных средств
Первичные посредники — гормоны, простагландины, нейроме-
диаторы и др. — являются важным источником создания ЛС. В XX в.
изучение строения и физиологической роли эндогенных аминов (аце-
256
тилхолина, катехоламинов, серотонина, гистамина), нейропептидов,
глюкокортикоидов и т.п. привело к появлению большого количества
новых препаратов. Показательна в этом плане история создания ан-
тигистаминных средств.
Гистамин — 2-(имидазол—4-ил)этиламин — один из важнейших
эндогенных аминов, участвующий в регуляции как ряда
физиологических процессов (дыхание и пищеварение), так и в развитии многих
патологических состояний (аллергия, воспаление, болевой синдром,
ожог). Биосинтез гистамина в организме человека происходит путем
декарбоксилирования аминокислоты — гистидина. Этот амин
накапливается в тучных клетках и выбрасывается из них в кровяное русло в
ответ на соответствующий стимул, в качестве которого могут
выступать яды, токсины, протеолитические ферменты, пища, детергенты и
ряд др. химических веществ. Выброс гистамина регулируют ионы Са2+
и ГТФ. Эффекты гистамина опосредованы через три вида
рецепторов — Нр Н2 и Н3. Характеристика рецепторов приведена в табл. 10.1.
Впервые гистамин был выделен из продуктов гниения белков в 1876 г.
В 1910 г. Н.Н. Dale и P.P. Laidlaw обнаружили этот амин в спорынье,
синтезировали его и установили, что он вызывает сильное сокращение
гладкой мускулатуры, снижает АД [40, 41]. В 1919 г. было показано, что
гистамин при внутривенном введении животным вызывает шок. Интерес
к этому соединению значительно возрос после того, как в 1927 г. его
обнаружили в легких и других тканях животных. Изучение
биологической роли гистамина позволило рассматривать его как один из
медиаторов воспаления и аллергии. Первые антигистаминные препараты были
созданы именно для воздействия на эти патологические процессы.
Гипотеза о существовании специфических гистаминовых рецепторов впервые
подтверждена после того, как в 1933 г. обнаружили, что производное ди-
гидробензодиоксина — пипероксан (10.55) предотвращает развитие у
экспериментальных животных бронхоспазма, вызванного ингаляционным
введением гистамина. Позднее (1937 г.) A.M. Staub и D. Bovet с соавт. [42, 43]
установили, что производное триэтиламина (10.56) в несколько раз
превосходит эффект пипероксана.
ото сг~г
(10.55) (10.56)
Некоторое сходство в строении соединений (10.55) и (10.56)
стимулировало поиск среди их аналогов новых веществ — антагонистов гистамина,
которые в настояшее время относят к блокаторам Н,-рецепторов.
В середине 60-х гг. XX в. A.S.F. Ash и Н.О. Schield выдвинули гипотезу о
наличии еше одного класса рецепторов гистамина. Доказательством
справедливости этого предположения послужил в 1972 г. синтез J.W. Blak с
соавт. препарата буримамид, устранявшего те эффекты гистамина (в
частности стимуляцию желудочной секреции), которые не исчезали при
257
Таблица 10.1
Локализация рецепторов гнетами на и эффекты,
возникающие при их стимуляции0 [44 ]
Подтип
рецептора
н,
н2
Нз
Место локализации
Бронхи
Артерии:
мелкие
крупные
Сердце
Мочевой пузырь
Тонкий кишечник
цнс
Матка
Мелкие артерии
Сердце
Тканевые базофилы
и базофильные
лейкоциты
Мочевой пузырь
Подвздошная
и двенадцатиперстная
кишка
Париетальные
клетки желудка
ЦНС
Сердечно-сосудистая
система
Бронхиолы
Желудок
Поджелудочная
железа
ЦНС
Эффект
Сужение
Расслабление
Сокращение
Положительный
инотропный
Сокращение
Сокращение
Седативный,
тахикардия, гипертензия
Расслабление
Расслабление
Положительный
хроно- и инотропный
Регуляция
высвобождения гистамина по
механизму обратной связи
Расслабление
Расслабление
Стимуляция
секреции НС1
Ингибирование
электрической активности
нейронов коры
Снижение АД, общего
периферического
сопротивления сосудов
Релаксация
Снижение секреции
НО, гастрина
и соматостатина
Подавление
продукции а-амилазы
Регуляция синтеза
и секреции гистамина;
подавление электро-
ицдуцированных
судорог;
регуляция процессов
памяти и обучаемости
Способ передачи
сигнала2'
Сопряжен
с
фосфолипазой С
Сопряжен
с
аден ил атцикл азой
Стимулирует
потенциал-
зависимые
Са2+-каналы
_
Сопряжен
с
фосфолипазой С
—
—
'Данные получены па животных.
'Все подтипы рецепторов гистамина сопряжены с гуанилнуклеотид-зависи-
мыми G-протеинами.
258
применении Н(-антагонистов. Новый тип рецепторов получил
обозначение Н2[45, 46].
В 1983 г. J.M. Arrang и J.C. Schwartz впервые описали Н3-рецепторы,
локализующиеся на пресинаптических мембранах нервных окончаний ЦНС
и периферической нервной системы. Одна часть этих рецепторов
контролирует синтез и секрецию гистамина, а другая модулирует
высвобождение ацетилхолина, катехоламинов и серотонина [47].
Известно, что гистамин в зависимости от кислотности среды и
характера заместителей может существовать в таутомерных формах
NT-H и Nn-H (табл. 10.2).
Таблица 10.2
Иоииые и таутомериые формы гистамина [48]
Предполагают, что только в форме монокатиона этот амин
способен связываться с репепторами. В ряде исследований показано, что
таутомерия не имеет особого значения для связывания с Hj-подти-
пом, однако важна для активности Н2-подгруппы, представленной в
париетальных клетках желудка. Наиболее активна NT-H /иглг-форма
амина: замешенный по 4-му положению гистамин в этой форме
действует как протонирующий агент, что обеспечивает связывание
с Н2-рецепторами (табл. 10.3).
259
Таблица 10.3
Влияние заместителей в имидазольиом кольце гистамина
иа состояние таутомериого равновесия
и стимуляцию Н2-рецепторов [49]
Равновесие Nr-H-
и Ып~Н-форм
К =
[*-Н]
[n* - н]
%N*-H »>
н2-
агонистич.
активн.,%
- "VNH
ВН* +А*
Н
сн3
С1
Вг
N03
2.4
4.1
0.13
о.и
0.009
71
80
12
10
0.9
100
43
11
9
0.6
" Мольная фракция монокатиона.
Выявлена также определенная зависимость между конформацией
молекулы гистамина и ее активностью по отношению к определенному
подтипу рецепторов. По мнению L.B. Kier, возможны две
термодинамически устойчивые конформации этой молекулы: в конформации
I амин стимулирует преимущественно Н^рецепторы, в
конформации II — Н2-рецепторы [50].
H-N
Н Н
г
Н
НН-
N ^0,455 нм
I
,Н
H-N 'J-* Н
V^N 0,360 нм
Н
II
Агонисты, с помощью которых были дифференцированы
биологические мишени гистамина, приведены в табл. 10.4. Наибольшее
сродство к Нгрецепторам проявляют соединения (10.58), (10.59) и (10.60),
а к Н2-мишеням — соединения (10.57), (10.64) и (10.65). Наличие в
структуре БАВ имидазольного цикла не является необходимым
условием аффинности к Hj-рецепторам. При замене его на пиридиновый
или тиазольный фрагменты сродство к Н,-мйшеням сохраняется.
Вероятно, необходимым условием для связывания с Hj-подтипом
рецепторов является образования одной водородной связи, а для
эффективного взаимодействия с Н2-мишенями важно наличие в гетероцикле
двух атомов азота, которые могут образовывать одновременно две Н-свя-
зи [44, 48]. Наличие объемных алкильных групп в положении 4
молекулы гистамина ведет к снижению активности и частичному агонизму;
замещение по азоту в боковой цепи усиливает антагонистические
свойства. Указанное особенно наглядно при сравнении мест связывания аго-
ниста и антагониста (см. рис. ЗЛО, разд. 3).
260
Таблица 10.4
Н,- и Н2-агоиисты общей формулы: Het — СН2 — СН2 — X [48]
Соединение
(10.52)
(10.53)
(10.54)
(10.55)
(10.56 )
(10.57)
(10.58)
(10.59)
(10.60)
Заместители
Het
н
О
6
N—У
W
HN
H2N S(CH2)3N(CH3)2
н
Г н н Х">
Wn nh
X
NH2
NH2
N(CH3)2
NH2
NH2
NH2
NH2
—
—
Активность
относительно
гистамина
н,
0.23
16.5
44.0
12.7
5.6
<0.001
26.0
<0.0001
<0.001
н,
39.0
2.0
19.0
13.7
0.2
0.4
0.3
19.5
1680
261
Среди приведенных в табл. 10.4 агонистов димаприт (10.64)
является примером «нетрадиционного» фармакофора. Его изотиомо-
чевинный фрагмент (так же, как и имидазольный цикл гистамина)
содержит электронный секстет, кроме того, для димаприта
характерна таутомерия и он содержит группы, способные выступать как
донорами, так и акцепторами протонов. Вероятно, этим
объясняются его высокие селективность и аффинность по отношению к
Н2-рецепторам. Наиболее селективен в приведенном ряду импро-
мидин (10.65) [48].
Анализ зависимости активности аналогов гистамина от
структуры заместителей в имидазольном фрагменте позволил предложить
схему мест связывания рецепторов гистамина. Как Н,-, так и Н2-ре-
цепторы имеют центр, образующий водородную связь с
аммонийной группой этиламинной цепи. Вещества с липофильным
заместителем в положении 2 имидазольного кольца — агонисты Н,-, но
не Н2-рецепторов, а вещества с гидрофильным заместителем в этом
положении — агонисты Н2-, но не Н,-рецепторов. В отличие от
Н2-, Hj-мишени содержат липофильный диполь, который
принимает участие в образовании дополнительной связи с пятым
углеродным атомом гетероцикла [48].
Наименее изученный подтип — Н3-гистаминовые рецепторы. Они
локализуются на пресинаптических мембранах нервных окончаний
ЦНС и периферической нервной системы. Активация части из них
(так называемых ауторецепторов) вызывает снижение синтеза и
высвобождение гистамина, в то время как активация другой части (гете-
рорецепторов) модулирует высвобождение других нейромедиаторов
(ацетилхолина, дофамина, норадренапина и серотонина). В связи с тем,
что Н3-рецепторы выявлены сравнительно недавно, данные о местах
их локализации и эффектах получены пока только на животных [44,
48, 51].
Несмотря на то, что молекулярные механизмы, определяющие
специфический ответ при активации рассматриваемой группы
рецепторов, до конца пока неясны, имеются данные о том, что в клетках
желудка Н3-рецепторы сопряжены с фосфолипазой С, вызывают ее
торможение, что, в свою очередь, приводит к снижению секреции
НС1. В ответ на связывание агонистов с этими рецепторами в гладких
миоцитах сосудов наблюдается стимуляция потенциалзависимых Са2+-
каналов [52, 53].
Специфическими агонистами Н3-рецепторов являются: N-cc,
S-p-диметилгистамин (10.66), иметит (10.67), R-ct-метилгистамин (10.68)
и иммепип (10.69) [51].
Me
Н
N.
HN ^ Me HN^N Me
(10.66) (10.68)
262
HN
\^N
(10.69)
NH
В качестве фармакотерапевтических средств для воздействия на
гистаминергические процессы наибольший интерес, безусловно,
представляют антагонисты Hj-,H2- и Н3-рецепторов.
Большинство соединений, эффективно блокирующих активность
гистамина, опосредованную через Н^мишени, в химической
структуре содержит фрагмент:
Аг,
где Аг и Arj— арильные или гетерильные системы, одна из которых
может быть отделена от X метиленовой группой; X — (>С=, СН, N,
>C-N<).
По химическому строению блокаторы Н,-рецепторов
подразделяют на следующие группы:
— производные этилендиамина: мепирамин (10.70), супрастин (10.71)
и др.;
— производные этаноламина: димедрол (10.72), тавегил (10.73) и др.;
— производные пиперазина: цетиризин (зиртек, 10.74) и др.;
— производные алкиламинов: хлорфенирамин (10.75) и др.;
— производные фенотиазинов: пипольфен (10.43) и др.;
— производные пиперидина: терфенадин (трилудан, 10.76), астеми-
зол (гисманал, 10.77), лоратидин (кларитин, 10.78);
— тетрагидрокарболины: диазолин (10.79) и димебон (10.80) ;
— производные ципрогептадина: перитол (10.81);
— хинуклидины: фенкарол (10.82) и бикарфен (10.83).
(10.73)
(10.74)
263
(10.78)
(10.79)
Me
(10.80)
jO
OH
N Me
A?
О0-8*) (10.82) (10.83)
В настоящее время блокаторы Н,-гистаминовых рецепторов
принято подразделять на препараты первого и второго поколения. К
первому поколению относят димедрол, супрастин, пипольфен, диазолин,
фенкарол, перитол; ко второму принадлежат терфенадин, астемизол,
кларитин, зиртек, акривастин. Из-за большого количества побочных
эффектов, прежде всего связанных с воздействием на ПК и ЦНС (се-
дативный эффект, нарушение координации движений и зрения,
повышенная утомляемость, реже — эйфория, нервозность, потенцирование
действия алкоголя, снотворных, седативных и анальгетических ЛС), а
также наличием .м-холиноблокирующей активности, Н,-антагонисты
первого поколения применяются все реже [21, 22, 24, 44, 51].
264
Антигистаминные средства второго поколения разрабатывались по
принципу снижения нежелательного воздействия на ЦНС, ПК и
холинорецепторы. Усовершенствование их структуры вели в
направлении снижения аффинности к холинергическим и серотониновым
рецепторам. Не обладают седативным эффектом астемизол и
терфенадин. Их молекулы сильно полярны, не проникают через ГЭБ и,
следовательно, не связываются с перечисленными выше мишенями.
Однако им также присущ ряд побочных эффектов. Терфенадин и
астемизол при длительном применении способны вызывать
серьезные нарушения ССС и системы кроветворения [24, 44, 51].
Антагонисты Н2-рецепторов являются противоязвенными
средствами. Начало направлению усовершенствования структур Н2-антагони-
стов было положено открытием слабой антигистаминной активности
у частичного Н2-агониста — гуанилгистамина (10.84) [24, 51]. Замена
основной гуанидиновой группы на метилтиомочевинную привела к
созданию буримамида (10.85), который в дозе Ю-7 М проявляет
свойства конкурентного антагониста. Буримамид хотя и эффективен, но
токсичен и не пригоден для перорального применения, т. к. быстро
инактивируется в ПК. Повышению сродства к Н2-рецепторам за счет
стабилизации /леде-таутомерной формы и снижения рКл способствовало
введение 4-СН3 группы в имидазольное кольцо и электроноакцеп-
торного атома S в боковую цепь. Кроме того, снижение способности к
ионизации благоприятно сказалось на биодоступности веществ. Мети-
амид (10.86) в 10 раз активнее буримамида in vitro и в 5 раз in vivo [54,
55]. Однако наличие тиомочевинного фрагмента в его структуре
явилось причиной проявления нейтропении, нефро- и гепатотоксичнос-
ти. Решение проблемы было найдено при введении в гуанидиновый
фрагмент электроноакцепторных заместителей NO,- или CN-rpynn,
которые обеспечили удовлетворительный уровень рла. Так появились
упомянутые при обсуждении продуктов «Ме-Тоо» циметидин (10.53)
(Щх — 1-4 мкМ/кг) и ранитидин (10.54) (Щ50 — 0.6 мкМ/кг), а несколько
позже — фамотидин (10.87). Дальнейшее усовершенствование структур
Н2-антагонистов также проводилось в направлении повышения
аффинности к рецепторам и снижения количества побочных эффектов. Так,
препарат третьего поколения фамотидин по выраженности торможения
желудочной секреции в 20—150 раз превосходит циметидин и в 3—
20 раз ранитидин [24, 44, 51]. При этом фамотидин, в отличие от ци-
метидина, не проявляет антиандрогенного эффекта, не ингибирует
систему CYP450 печени и, следовательно, не влияет на биотрансформацию
других JIC, метаболизирующихся микросомальными Ф [44].
В настоящее время лидирующее место занимают препараты
четвертого — низатидин (10.88) и пятого — роксатидин (10.89)
поколений блокаторов Н2-рецепторов [51].
(10.84)
265
н н
(сн ^N-^N_CH'
HN I S
(10.85)
\^N
(10.86)
N^ ^NH?
V SO. 2
NH.
(10.87)
(10.88)
OTY°4,CH!,rV0
о
Л.
Me
(10.89)
Модуляторы Н3-рецепторов в клинической практике пока не
используются. Однако интенсивные исследования Н3-мишеней и
выявление их специфических лигандов позволяют определить область
применения таких средств. Доказано, что для блокаторов Н3-рецеп-
торов важнейшим структурным признаком является наличие 4(5)-за-
мещенного имидазольного кольца, а оптимальное расстояние между
этим кольцом и положительно заряженным атомом азота в а-амино-
группе составляет цепочка из пяти атомов углерода. Этим
структурным требованиям в полной мере удовлетворяет Н2-антагонист бури-
мамид (10.85). Действительно, оказалось, что он обладает в 100 раз
большей аффинностью к Н3-, чем к Н2-рецепторам. Однако из-за
наличия тиомочевинного фрагмента буримамид и его аналоги
неселективны. В настоящее время выявлено несколько высоко аффинных
266
антагонистов Н3-рецепторов [48]: тиоперамид (10.90), импентамин
(10.91), клобенпропит (10.92) и иодофенпропит (10.93). Первым в этом
ряду был выявлен тиоперамид, который можно рассматривать как
ригидный аналог буримамида, однако ему присущ иной тип
взаимодействия с мишенью. Это вещество является конкурентным Н3-анта-
гонистом. Установлено, что тиоперамид и клобенпропит способны
снижать судорожную восприимчивость головного мозга у
экспериментальных животных, и это может иметь значение при разработке
антиэпилептических ЛС [44].
\^N
(10.90)
HN
^
н
S^ ^N
NH
(10.92)
HN
^
(CH2),
/NH2
^
(10.91)
CI
HN
^
H
•(сн2)/ *Y
NH
(10.93)
Н3-блокаторы представляют интерес и как потенциальные ано-
рексигенные средства. Они увеличивают уровень высвобождения
гистамина в мозге и опосредованно влияют на центральные Hj-рецеп-
торы, при стимуляции которых происходит снижение аппетита.
В эксперименте показано, что назначение тиоперамида приводит
к уменьшению потребления пищи животными [53].
Имеются доказательства того, что гистаминергическая система
является одним из звеньев патофизиологического механизма
возникновения головокружения и тошноты при укачивании и болезни Ме-
ньера [57]. Антагонисты Н3-рецепторов показали свою эффективность
при данных расстройствах [58].
Еще одной областью применения Н3-блокаторов может стать
болезнь Альцгеймера. У пациентов с деменцией отмечается снижение
уровня гистамина в некоторых отделах мозга. Кроме того, изменения
267
в системе холинергической передачи сигналов в ЦНС
рассматриваются в качестве основного механизма нарушений процессов памяти,
а т. к. при блокаде Н3-рецепторов повышается высвобождение
эндогенного ацетилхолина, то это может указывать на целесообразность
поиска потенциальных средств лечения деменции среди Н3-антаго-
нистов [59, 60].
Современный арсенал антигистаминных средств возник
благодаря достижениям в изучении биологической роли гистамина и его
рецепторов. Несмотря на успехи в этой области, некоторые вопросы
по-прежнему остаются открытыми. В частности, еще не до конца
определена роль Н3-рецепторов в регуляции процессов в ЦНС и на
периферии. По этой причине пока отсутствуют ЛС, блокирующие
активность рецепторов данного типа. Не утратил актуальности и
поиск блокаторов Hj- и Н2-рецепторов, сочетающих высокую
активность с минимальными побочными эффектами. Следует подчеркнуть,
что подходы, реализованные в конструировании ЛС данного
фармакологического профиля, являются общими и могут быть
трансформированы на создание препаратов, модулирующих активность иных
физиологических посредников.
Литература
1. Burger's Medicinal Chemistry and Drug Discovery, Vol. 1 / Ed. by M.E. Wolff-
New York: John Wiley & Sons, 1995.
2. Xenical (orlistat). Product Monograph. — Basel: F. Hoffmann-La Roche Ltd,
1998.
3. Biomembrane Structure and Function, Topics in Molecular and Structural
Biology, Vol. 4 / Ed. by D. Chapman. — Weinheim: Verlag Chemie, 1984.
4. Болдырев А. А. Биологические мембраны и транспорт ионов. — М: Изд-
во. Моск. ун-та., 1985.
5. Стоник В.А. // Успехи химии. - 2001. - Т. 70. - С. 763.
6. Cragg G.M., Newman D.J., Snader KM. // J Nat Prod. - 1997. - Vol. 60. - P. 52.
7. Jaspars M., Lawton L.A. jj Current Opin. Drug Discov. Develop. — 1998. —
Vol. 1. - P. 77.
8. Burja A.M., Banagis В., Abou-Mansour E. et al. jj Tetrahedron. — 2001. —
Vol. 57. - P. 9347
9. Schwartz RE., Hirsch C.F., Sesin D.F. et ai jj J. Ind. Microbiol. - 1990. -
Vol. 5. - P. 113.
10. Patterson G.M.L., Baldwin C.L., Bolis CM. jj J. Phyc. — 1991. - Vol. 27. -
P. 530.
11. Panda D., Himes R.H., Moore RE. et.al. jj Biochemistry. — 1997. — Vol. 36. —
P. 12948.
12. Gerwick W. H., Proteau P.J., Nagle D.G. et al jj J. Org. Chem. - 1994. -
Vol. 59. - P. 1243.
13. White 3.D., Kim T.S., Nambu M. jj J. Arn. Chem. Soc. - 1995. - Vol. 117. -
P. 5612.
14. Rossi J. V.f Roberts M.A., Yoo H.D. et al. jj J. Appl. Phycol. - 1997. - Vol. 9. -
P. 195.
15. Sandler В., Murakami M, ClardyJ. jj i. Am. Chem. Soc. - 1998. - Vol. 120. -
P. 595.
268
16. Rios Steiner J.L., Murakami M., Tulinsky A. jj J. Am. Chem. Soc. — 1998. -
Vol. 120. - P. 597.
17. Moore RE. // J. Ind. Microbiol. - 1996. - Vol. 16. - P. 134.
18. Boyd M.R Gustafson K.R., McMahonJ.B.etal. //Antimic. Agents Chemother. -
1997. -Vol. 41. >— P. 1521.
19. ReshefV., MizrachiE, Maretzki T. et. al. //J. Nat. Prod. - 1997. - Vol. 60. -
P. 1251.
20. Colombo D., Scala A., Taino I. et al. jj Cancer Lett. -1998. - Vol. 123. - P. 83.
21. Машковский МЛ. Лекарства XX века. — М.:000 «Издательство Новая
Волна», 1998.
22. Машковский М.Д. Лекарственные средства. Т. 1—2. — Изд. 13-е, новое. —
Харьков: Торсинг, 1998,
23. Taxol, a molecular for all seasons // Chem. Commun. — 2001. — Vol. 4. —
P. 867.
24. Principles of Medicinal Chemistry / Ed. by W.O. Foye, T.L. Umke, D. A.
Williams„4'h ed. — Philadelphia, Baltimore, New York, London: Lippincott
Williams & Wilkins, 1995.
25. Naik R.G., Kattige S.I., Bhat S. V. et al. jj Tetrahedron. - 1988. - Vol. 44. -
P. 2081.
26. Sadlacek H. H., Czech J.t Naik R. et al. jj Inter. J. Oncology. - 1996. -
Vol. 9. - P. 1143.
27. Rotheim P. Plant-Derived Drugs: Products, Technologies and Application. -
Norwalk, Connecticut: Business Communications Co., 1998.
28. Henquin J.-C. jj Diabetologica. - 1992. - Vol. 35. - P. 907.
29. Rajan A.S// Diabetes Care. - 1984. - Vol. 13. - P. 349.
30. Cook D.L., Hales C.N. jj Nature. - Vol. 311. - P. 271.
31. GraulA., CastanerJ. jj Drugs Fut. - 1996. - Vol. 21, N 7. - P. 694.
32. Shinkai H., Nishikawa M.f Sato Y. et al. jj J. Med Chem. - 1989. - Vol. 32. -
P. 1436.
33. Stein P.D., HuntJ.T., Floyd DM. et al. //J. Med Chem. - 1994. - Vol. 37. -
P. 329.
34. Murugesan N., Gu Z., Spergel S. et al. jj J. Med Chem. - 2003. - Vol. 46. -
P. 125.
35. Laborit H.jj Presse Med. - 1954. - Vol. 62. - P. 359.
36. FozardJ.R, Mobarak All A.TM. jj Eur. J. Pharmacol. - 1981. - Vol. 49. -
P. 109.
37. BermudezJ., Fake C.S, Joiner GF.etal.//}. Med. Chem. - 1990.-Vol. 33. -
P. 1924.
38. Wermuth C.G. jj J. Med Chem. - 2004. - Vol. 47. - P. 1303.
39. Medicinal Chemistry; Principles and Practice / Ed. by F.D.King. — Harlow,
UK:Smith Kline Beecham Pharm., 1994.
40. Dale H.H., Laidlow P.P. // J. Physiol. (Lond.). - 1910. - Vol. 41. - P. 318.
41. Dale H.H., Laidlow P.P. jj). Physiol. (Lond.). - 1911. - Vol. 43. - P. 182.
42. Fourneau E., Bovet D// Arch. Int. Pharmacodyn. Ther. — 1933. — Vol. 46. —
P. 178.
43. Staub A.M. jj Ann. Inst. Pasteur. - 1939. - Vol. 63. - P. 400.
44. Спасов А.А., Черников В. М. jj Хим.-фарм. журнал. — 2000. — № 8. — С. 3.
45. Ash A.S., Shield И.О. // Brit. J. Pharmacol. - 1966. - Vol. 27. - P. 427.
46. Black J. W., Duncan W.A.M., Durant C.J. etal. jj Nature. - 1972. - Vol. 236. -
P. 385.
47. ArrangJ.M., GarbargM., Schwartz J. C.// Nature. - 1983. - Vol. 302. - P. 832.
269
48. Resent Advances in Receptor Chemistry / Ed. by F.Gualtieri, M.Gianella,
C.Melchiorre. -New York: Elsevier , 1979.
49. Drug Action at the Molecular Lavel / Ed. by G.C.K.Roberts. — Baltimor:
University Park Press, 1977.
50. KierL.B. //J. Med. Chem. - 1968. - Vol. 11. - P. 441.
51. Van der Gott H., Timmerman H. // Eur. J. Med. Chem. - 2000. - Vol. 35. -P. 5.
52. Cherift Y., Pigeon C, Le Romanser M. et al. // J. Biol. Chem. — 1992. —
Vol. 267. - P. 25315.
53. Oike M., Kitamura K.> Kuriyama H. // J. Phisiol. — 1992. — Vol. 448. — P. 133.
54. Hunghes M.J., Com LA. // Proc. Soc. Exp. Biol. (N.Y.). - 1975. - Vol. 148. -
P. 127.
55. Black J. W., Duncan W.A.M., Emmett J. С.Ц Agent Actions. — 1973. — Vol 3 —
P. 133.
56. Oohara A., Yoshimatsu H., Kurokawa M. et al. // J. Neurochem. — 1994. —
Vol. 63, N 1. - P. 677.
57. Schwaitz J.C, J.M. AtrangJ.M., M.Garbarg et. al. // Phisiol. Rev. — 1991. —
Vol. 71, N 1. - P. 1.
58. Barocelli E., Ballabeni V., Caretto A. et. al. // Agent Actions. — 1993. —
Vol. 38. - P. 158.
59. Bierer L.M., Haroutunian V., Gabriel S. et. al. // J. Neurochem. — 1995. —
Vol. 64, N 2. - P. 749
60. Mochizuki Т., Okakura- Mochizuki K., Horii A. et al. // J. Neurochem. —
1995. -Vol. 62, N 6. -P. 2275.
11
СОВРЕМЕННЫЕ МЕТОДЫ УСОВЕРШЕНСТВОВАНИЯ
СТРУКТУРЫ ЛИДЕРА
Процесс поиска кандидата в ЛС носит спирально-циклический
характер. Из БАВ, выявленных в ходе первичного скрининга,
отбирают промежуточный лидер. Это вещество, как правило, не может
сразу удовлетворять всем требованиям, предъявляемым к кандидату в
ЛС, а нуждается в дальнейшем усовершенствовании. Прежде всего,
речь идет об устранении имеющихся в нем недостатков, таких как
низкая специфичность, метаболическая и химическая нестабильность,
острая токсичность, Низкая биодоступность, неудовлетворительная
растворимость или даже отсутствие новизны структуры,
исключающее возможность его патентной защиты. В результате модификации
лидирующей структуры появляются новые соединения, которые
проходят аналогичный цикл и из них отбирается следующий лидер. Эта
процедура повторяется до тех пор, пока полученное вещество не
будет удовлетворять всем критериям, предъявляемым к кандидату в Л С.
В поиске и усовершенствовании структуры лидера существуют два
подхода: «рациональный» и «нерациональный». В данном случае это
не синонимы «плохой» и «хороший». Оба подхода одинаково широко
используются в современной медицинской химии и имеют свои
сферы применения. «Нерациональный» подход в отборе лидера связан с
синтезом и проверкой большого количества соединений. Еще в
начале 90-х гг. он рассматривался большинством специалистов как
интеллектуально непривлекательный. Однако с появлением тотального
скрининга (high-throughput screening) и комбинаторного синтеза отношение
к нерациональному подходу в поиске и отборе лидирующих структур
резко изменилось. «Рациональные» методы конструирования ЛВ
наиболее эффективны тогда, когда поиск проводят в ряду синтетически
трудно доступных соединений. Такие исследования, как правило,
предваряет внеэкспериментальный скрининг с использованием
компьютерных баз данных QSAR. Однако не стоит пренебрегать и тем, что
опытные практики называют «интуитивным» подходом в выборе
стратегии оптимизации структуры лидера.
При создании ЛВ, способных воздействовать на определенную
биологическую мишень, возможны четыре варианта состояния
начальной информации:
• нет сведений о строении мишени и нет данных о том, какие
соединения могут с ней взаимодействовать. В этом случае единственно
271
возможным будет подход, связанный с проверкой большого
количества соединений самого разнообразного строения. Это
«нерациональный» — эмпирический поиск, и успех его зависит от
возможностей скрининга и от количества «просеянных* веществ;
• нет сведений о строении мишени, но есть данные о том, какие
соединения могут выступать в качестве лигандов, связываясь с
таким рецептором. В этом случае обычно известны определенные
корреляции «структура — действие» и можно проводить более-
менее направленный поиск новых веществ или модифицировать
уже известные активные молекулы;
• есть данные о строении мишени, полученные, как правило, на
основе РСИ. Можно вести направленный подбор лигандов,
и в процессе такого подбора выявить структуру активного центра
этой мишени;
• известны и строение активного центра, и лигандов,
связывающихся с ним. При этом можно вести исключительно
направленный поиск структур. Во многих случаях речь обычно уже идет о
создании ЛС нового поколения и модификация их
осуществляется «рационально».
Существует три стандартных пути усовершенствования
лидирующих структур: биоизостерические перемещения, конформационные
ограничения и формирование пролекарств. В литературе наиболее
широко освещены методы биоизостерических перемещений [1, 2].
Методология конформационных ограничений раскрыта менее
подробно. По пути создания пролекарств специалисты обычно идут
тогда, когда необходимо усовершенствовать фармакокинетические
параметры, повысить биодоступность при приеме внутрь или повлиять
на способность вещества проникать через ГЭБ.
11.1. Биоизостерические перемещения
Изостерами называют заместители или группы, которые имеют
одинаковый размер или объем. Концепцию химического изостеризма
впервые выдвинул I. Langmuir в 1919 г. [3]. Он обратил внимание на
подобие физико-химических свойств атомов, групп, радикалов и
молекул со схожими электронными структурами. Согласно I. Langmuir,
сходство наиболее выражено у атомов, расположенных в
периодической системе в одной подгруппе друг под другом. Это объясняется
идентичностью внешних электронных оболочек, не слишком
большими различиями в размерах атомов и их массах. В меньшей степени
схожи между собой соседние атомы в горизонтальных рядах или
находящиеся в одной и той же подгруппе, но расположенные через три
ряда. Например, хлор и бром гораздо ближе друг к другу по
свойствам, чем хлор и сера или хлор и йод. В первом случае — из-за
различий в строении электронных оболочек, во втором — из-за
размеров атомов и массы. Решающим фактором схожести свойств
I. Langmuir считал подобие электронного строения. Его концепция
272
охватывала свойства элементов, неорганических молекул, ионов и
малых органических молекул, таких как диазометан и кетен.
Справедливость этих предположений подтверждают данные табл. 11.1.
Сравнительная характеристика
физико-химических свойств N20 и С02 [4]
Таблица 11.1
Физико-химический показатель
Мол. масса
Длины связей (А)
Плотность (при 20°С)
Вещество
N,0
44
N=N(re) 1.128
N-0(r.) 1.184
Мол, рефракция, (D линия при 16°С)
Диэлектрическая постоянная (при 0°С)
Растворимость в спирте (при 15°С)
148 • 10-
1.193
1.593
СО,
44
С=0(ге) 1.160
148* 10-
1.190
3.250
1.582
3.130
В 1925 г. H.G. Grimm сформулировал правило гидридного
замещения [5]. Согласно этому правилу вертикальные группы изостеров
образуются при замене атома в горизонтальном ряду периодической
системы на соседний слева атом с добавлением атома водорода или
гидрид-иона. Это правило иллюстрируют данные табл. 11.2.
Таблица 11.2
Группы изостеров,
образующиеся в результате гидридного замещения
Изостеры
И зо стеры
СН
Атомы элементов 2-го периода периодической системы
с In I о If I Ne
~СН I NH I ОН HF
NH
СН,
ОН3
NH,
СН,
I i .
Атомы элементов 3-го периода периодической системы
"si Г~Р | S | 01 I Аг
SiH
РН
SiH2
SH
РН2
SiH
НС1
SH2
РНз
SiH4
Следует отметить, что эта концепция не учитывала широко
используемые в настоящее время характеристики, которые вносят важный
вклад в физико-химические свойства веществ и влияют на
биологические эффекты. К ним относятся кислотность, основность,
электроотрицательность, угол между связями, размер и форма молекулярных
орбиталей, электронная плотность и др.
273
Концепция, предложенная I. Langmuir, изначально не имела
отношения к модификации БАВ, однако она создала теоретическую
основу для применения изостерического подхода в конструировании
лекарств. Впервые изостерическую замену этиленового фрагмента на
атом серы осуществил в 1916 г. О. Hinsberg [6]. Это инициировало
синтез новых органических соединений, в которых ароматические
кольца были заменены на тиофеновые, пиридиновые, пиррольные
или фурановые. Термин «биоизостеры» появился значительно позже.
В 1951 г. его впервые ввел H.L. Friedman [7]. Биоизостерами
называют группы одинаковые не столько по размерам или объему,
сколько имеющие аналогичные физико-химические свойства, благодаря
чему индуцирующие близкий фармакологический эффект. При
заменах такого рода не следует ожидать значительного роста активности,
однако могут произойти существенные изменения в селективности
связывания с мишенью, токсичности и метаболической стабильности
веществ.
Традиционно биоизостеры классифицируются на две группы:
классические изостеры, которые имеют приблизительно одинаковый
размер, форму, подобную электронную конфигурацию (табл. 11.3), и
неклассические (табл. 11.4), которые не имеют одинакового числа атомов
и не подчиняются правилам стерического и электронного подобия,
но несут ответственность за проявление аналогичных биологических
свойств [8].
Таблица 11.3
Классические изостеры, обладающие свойствами биоизостеров
Изостеры
Одновалентные
А: -СН3, -NH2, -ОН, -F, -С1;
Б: -CI, -PH2, -SH;
В: -Br, -i-Pr;
Г: -I, -t-Bu;
Трехвалентные
А:-СН=, -N=;
Б: -Р=, -As=;
Эквиваленты в цикле
A: -CH=CH-,-S-;
Б: -CH=,-N=;
В: -О-, -S-, -CHr, -NH-;
Двухвалентные
А: -СН2-; -NH-; -О-; -S-;
Б: -СОСН2-; -CONH-; -СОО-; -COS-;
Четырехвалентные
А: >С<; >Si<;
Б: =С=; =N+=;=P+=;
Циклические изостеры
бензол, тиофен;
бензол, пиридин;
тетрагидрофуран, тетрагидротиофен,
циклопентан, пирролидин
Следует подчеркнуть, что классические изостерические
перемещения «подобное-на-подобное», происходящие с сохранением числа
атомов, валентности, степени ненасыщенности и ароматичности, могут
рассматриваться как биоизостерические только тогда, когда
биологическая активность сохраняется. Неклассические биоизостеры способ-
274
Таблица 11.4
Неклассические биоизостеры
Группа
Биоизостеры
Карбонильная
группа —С=0
CN
CN
\ \
SO :
/ /
SO S02 —S02-N-
R
A
,У
CN
Карбоксильная
группа -COOH
PO(OH)NH2; S02NHR; S03H; CONHCN; CONHOH;
PO(OH)OEt;
^f° N_N
/)—ow
o^^K
AJ
OH
Сложноэфирная
группа
(-0=0)0-
N4/
\,—o
I»
NO-Me
A
Амидная группа
-CONH-
-CONMe-; -CSNH-; -CH2NH-; -NHCO-;
>C=C<; -CH2S- .
Гидроксил ьная
группа —ОН
-NHCOR; -NHS02R; -CH2OH; -NHCONH2;
-NHCN; -CH(CN)2
Полифенол
Галоген Hal
-CF3; -CN; NCN2-; -C(CN):
Тиомочевина
NHC(=S)NH2
NHC(=NCN)NH2; NHC(=CHN02)NH;
ствуют сохранению активности за счет таких характеристик как рКа,
электростатический потенциал, энергии высшей занятой и низшей
свободной молекулярных орбиталей, и др. Опираясь на
физико-химические параметры (табл. 11.5), можно осуществлять подбор
подходящего заместителя, что и используется при компьютерном
моделировании и оптимизации структур исходя из QSAR.
При этом важно учитывать, что в одних условиях два заместителя
могут выступать в качестве биоизостеров, а в других они уже
таковыми не являются. Так, если в связывании с биологической мишенью
ключевую роль играют диполь-дипольное, ион-дипольное или
ионное взаимодействия, находящиеся в прямой зависимости от
электронной природы заместителей, то определяющим параметром будет
а константа. Согласно данным табл. 11.5, для СОМе-группы биоизо-
стерна SOMe-группа. Если же фактором, существенно влияющим на
активность, выступает липофильность я, то для SOMe-группы био-
изостерной будет S02Me-rpynna.
Примером структур, полученных в результате
классического биоизостерического замещения, являются 6-меркаптопурин (6.45)
275
Физико-химические параметры заместителей [9]
Таблица 11.5
Параметр1
стр
<*т
Я
MR
-СОМе
0.50
0.38
-0.55
11.2
и
II
и
1
и
0.84
0.66
0.40
21.5
-SOMe
0.49
0.52
-1.58
13.7
О
СЛ
1
0.57
0.60
-1.63
13.5
%
X
Z
о
СЛ
1
0.72
0.46
-1.82
16.9
'и'
Z
о
и
1
0.36
0.35
-1.51
19.2
1в, стт — w- и л*- константы Гаммета; я
MR — молярная рефракция.
показатель липофильности Ханча;
тиогуанин (6.46). В результате замены —ОН на —SH в гипоксан-
тинах, последние были превращены в антиметаболиты, инги-
бирующие синтез ДНК. Замена Н на F в молекуле урацила, также
привела к появлению противоопухолевого антиметаболита 5-фто-
рурацила (5.34). Биоизостерны /и/?янс-диэтилстильбестрол (2.46)
и эстрадиол (2.47).
Неклассическое биоизостерическое перемещение имеет место
при замене карбоксамида на сульфамид в л-аминобензоиламиде —
синтетическом предшественнике тетрагидрофолиата (5.8), что
послужило отправной точкой в поиске новых сульфамидных
препаратов (11.1).
(11.1)
На основании многочисленных исследований по модификации
сульфамидов были установлены следующие корреляции «структура-
действие»:
• бензольное кольцо должно иметь заместители в положениях I и 4;
• NH2-rpynna должна находиться в пара-положении по
отношению к S02NH2 фрагменту и должна быть незамещенной или
высвобождаться in vivo в результате метаболической
трансформации;
• NH2-rpynna в составе сульфамидного фрагмента должна быть
монозамещенной; гетероароматические производные повышают
активность веществ этого ряда;
276
• сульфоновые, карбоксамидные или кетогруппы, замещающие
сульфамидный фрагмент, сохраняют активность, но на значительно
более низком уровне.
При выборе подхода к усовершенствованию структуры лидера
следует иметь в виду, что с изменением одной части
молекулы часто изменяются все свойства в целом. Изостеризм и био-
изостеризм не являются исключением. Например, простая замена
—СН — на —О— или — S— в ряду соединений изменяет размер,
форму, распределение электронной плотности, растворимость в
воде или липидах, рЛГа, метаболизм, способность к образованию
Н-связи, т. е. те свойства, которые могут неожиданно сказаться на
биологической активности. С другой стороны, изостерические и
биоизостерические замены способны дать чрезвычайно полезную
информацию относительно ключевых функциональных групп,
взаимодействующих с биологической мишенью. Например, если при
связывании с рецептором вторичная аминогруппа выступает в
качестве протоноакцептора, то замена ее на эфирную, кето- или
третичную аминогруппу сохранит активность. Однако, если
взаимодействие осуществляется за счет протонодонорного эффекта
NH-группы, эти изменения приведут к потере или
значительному снижению активности. Если же аминофуппа не участвует в
связывании с мишенью, то замена ее на —СН2—NH—, —СН2—S,
—СН=СН— и даже на — СН2—СНгфрагменты не должна
существенно сказаться на активности [I, 2].
11.2. Конформационные ограничения
Этот вид структурной перестройки молекулы направлен на
сохранение неизменными всех связывающихся с рецептором
функциональных групп, и в то же время стремится зафиксировать их
в «активной» конформации. Конформационные ограничения
являются очень мощным инструментом оптимизации структуры
лидера. Для достижения фиксированной конформации существуют
следующие пути:
• введение метильной группы, что стерически ограничивает
свободное вращение связи;
• использование внутримолекулярной Н-связи;
• введение ненасыщенной связи, что фиксирует относительное
положение геминальных и вицинальных заместителей;
• циклизация, что превращает заместители в эндоциклические или
оставляет их экзоциклическими.
В результате реализации конформационных ограничений можно
достичь:
• выделения из общей структуры «фармакофора»;
• повышения селективности в связывании с рецептором или Ф;
• повышения аффинности взаимодействия;
• выявления структуры антагониста;
277
• повышения химической (метаболической) стабильности;
• мимикрирования третичной структуры белка;
• выявления нового оригинального структурного типа [7].
Существует большое количество природных веществ, которые
обладают выраженным фармакологическим эффектом только в
строго фиксированной конформации. Один из ранних подходов
медицинской химии состоял в изучении способов упрощения структуры
таких соединений и на их основе создания новых систем, которые
сохраняли бы большинство свойств природных продуктов, но были
бы синтетически более доступны. Примером указанного являются
бензморфановые и пиперидиновые анальгетики, при создании
которых были широко использованы приемы, связанные с конфор-
мационной перестройкой молекул.
Морфин (11.2) — алкалоид, который получают из однолетнего
травянистого растения Papaver somniferum. Сок опийного мака
содержит около 25 алкалоидов, однако морфин является преобладающим
(~ 10%). Несмотря на то, что это вещество в индивидуальном виде
было выделено еще в 1806 г. В.А. Сертюрнером, его структура
окончательно доказана лишь в 1925 г. R. Robinson. Морфин имеет
Т-образную пространственную форму молекулы, в которой присутствуют пять
асимметрических атомов углерода. Природный алкалоид является
левовращающим изомером. Его антипод, полученный синтетическим
путем, не обладает анальгетическим эффектом. Для пиперидинового
кольца Е в молекуле морфина характерна конформация «кресла», а для
циклогексенового — «ванны».
N—Me
НО
N-Me
(11.2)
(11.2)
Полный синтез морфина, осуществленный в 1952 г. М. Гейтсом и
Ж. Чуди, безусловно принадлежит к числу значительных
синтетических достижений в органической химии в XX в. [10], однако он
достаточно сложен и не используется в промышленных целях.
Природными аналогами морфина являются кодеин (11.3) и теба-
ин (11.4). Кодеин в общей смеси алкалоидов содержится в очень
незначительных количествах — 0.2—2%, — и его получают из
морфина. В терапевтических дозах кодеин не обладает анальгетическим
эффектом, а используется как противокашлевое средство. Тебаин не
обладает наркотическими свойствами и является самым токсичным
из всех алкалоидов опия. Он используется в качестве сырья для
получения полусинтетических аналогов морфина.
278
(11.3) (11.4)
Первоначальные попытки выяснить, какие из фрагментов молекулы
несут ответственность за проявление анальгетических свойств,
сводились к модификации функциональных групп, и их не следует
рассматривать в качестве примера конформационных ограничений. В
большинстве случаев использовали изостерические замещения. Кратко
резюмируя результаты этих исследований, следует отметить, что:
• алкилирование фенольного гидроксила приводит к значительному
снижению анальгетического и повышению противокашлевого
эффекта (кодеин — метиловый эфир, этоморфин — этиловый эфир);
279
• ацилирование спиртового гидроксила (а-ацетилморфин) повышает
анальгетическую активность;
• ацилирование фенольного гидроксила снижает активность;
• ацилирование двух ОН-групп способствует как повышению
анальгетического эффекта, так и росту токсичности (героин);
• окисление ОН-группы в кольце С и снятие гидроксила у атома С-6
приводят к повышению анальгетического действия; при этом
(в отличие от морфина) кольцо С приобретает конформацию
«кресла» (дезоморфин). Последний случай можно рассматривать как
пример конформационной перестройки молекулы, приводящей
к росту активности [10].
Наиболее существенно на активности соединений этого ряда
сказывается замена метильной группы при атоме азота на ненасыщенный
алкил, что привело к созданию налорфина (11.5) — частичного
антагониста морфина. Чистым антагонистом является налоксон (11.6),
открытый в 1961 г. Это ЛС сегодня широко используется для выведения из
состояний, связанных с передозировкой наркотиков морфинового ряда.
"/N\^4
"/N\^4
(11.5)
(11-6)
Примером структурной перестройки морфина, приведшей к
фантастическому росту активности, является введение транс-вини-
леновой группы между атомами углерода С-6 и С-14 и объемных
радикалов при С-7. Полученное вещество — эторфин (11.7) или
соединение Бентли — синтезировали из тебаина следующим путем:
MeOv з
МеО
п-Рг
П-РГ
280
Эторфин в 5 • 103—10* раз активнее морфина. Однако его
аффинность к опиоидным рецепторам составляет только 20—30% от
аффинности морфина. Причина роста активности связана с высокой
липофильностью и способностью быстро проникать через ГЭБ.
Парциальный коэффициент эторфина — 1.4, в то время как для морфина
он составляет 1 • 10~4 в системе гептан-фосфатный буфер (рН=7.4).
Эторфин используется в ветеринарной практике для иммобилизации
крупных животных [10—12].
Бупренорфин (11.8) — производное с N-циклопропилметильным
заместителем и /w/je/w-бутильной группой вместо изо-пропильной. Он
является смешанным агонистом-антагонистом опиоидных
рецепторов и наиболее липофильным веществом среди продуктов
Бентли (Р=60). Бупренорфин в 100 раз активнее морфина как агонист
и в 4 раза превосходит налорфин как антагонист.
N—^
МеО ><
Me j ОН
t— Bu
(П.8)
В связи с тем, что полусинтетические производные морфина в
той или иной степени обладают его недостатками и их синтез не
всегда прост, дальнейший поиск эффективных обезболивающих
средств осуществлялся по пути упрощения структуры морфина. Так,
удаление кольца D привело к производным морфинана. (-)-Левор-
фанол (11.9) в 5—6 активнее морфина. (+)-Изомер — декстрорфан —
полностью утрачивает анальгетический эффект, т.к. не связывается
с рецепторами. Однако для этого соединения характерно наличие
выраженного противокашлевого действия. В медицинской
практике нашел применение его метиловый эфир — (+)-декстрометорфан [4,
10, 11]. Удаление фенольного гидроксила снижает активность до 20%
активности морфина. Перемещение этой группы в положение 2 или
4 в кольце А приводит к потере эффекта. Уменьшает активность
и превращение ОН-группы в СН30-группу. Замена метила у атома
азота на фенилэтильный радикал в феноморфане (11.10) в 20 раз
повышает активность по сравнению с исходным N-метилмор-
финаном. Введение метилцикропропильного или аллильного
заместителей приводит к антагонистам опиоидных рецепторов — цик-
лорфану (11.11) и левалорфану (11.12) [10, 11].
281
N—Me
(11.9)
(11.10)
(H.ll)
(11.12)
Наличие кольца С в молекуле морфинанов оказывает
незначительное влияние на уровень анальгетической активности. Раскрытие
этого кольца привело к бензоморфанам (11.13). Кольцо С может быть
обозначено двумя метильными группами в положении С-6 и С-11.
Исследование стереохимии этих метильных групп (при *<ис-располо-
жении) способствовало выявлению анальгетиков ряда метазоци-
на (11.14), самым мощным из которых является пентазоцин (11.15).
Этот препарат не вызывает привыкания, по эффективности он
близок морфину, однако не лишен негативного побочного эффекта всех
опиоидов — угнетения дыхательного центра. Его близкий
структурный аналог — циклазоцин (11.16), хотя и является эффективным
анальгетиком, не нашел применения в медицинской практике, т.к.
является мощным галлюциногеном [10—12]. В случае
транс-расположения СН3-групп у атомов С-6 и С-11 анальгетический эффект
сопровождается эффектом привыкания [10].
Me Me
(11.13)
(11.14)
282
nO<3
Me "H
(11.15) (11.16)
Бремазоцин (11.17) в 200 раз активнее морфина и не вызывает
привыкания и угнетения дыхания. Как и в рассмотренных выше
случаях, у производных бензоморфана высокий уровень
активности связан с присутствием фенольного гидроксила в положении
8 трициклической системы. Однако ключевым в проявлении аналь-
гетических свойств у соединений этого ряда является наличие
ароматического кольца. При его отсутствии активность
утрачивается [4, 10, 11].
ОН
(11.17)
Упрощение структуры бензоморфана привело к производным
пиперидина. Их можно рассматривать как аналоги морфина, у которых
сохранены А и D кольца. Производные петидиновой кислоты (11.18)
первоначально использовались как спазмолитики.
/—
H3C-N^~
СООН
Н3С^м^"
У
-J
—OEt
-Ph =
= &j
ЕЮ
N-"CH3
t
~^o
(11.18) (11.19)
Их анальгетическое действие было открыто случайно в
конце 40-х гг. XX в. Развитие нового направления в создании
синтетических анальгетиков неморфинового типа началось с
выявления определенной структурной аналогии рассматриваемых веществ
283
с производными морфина. Первым соединением этой группы
стал петидин (1J.19), получивший распространение в клинической
практике, несмотря на наличие привыкания. Позднее на основе
петидина был синтезирован достаточно представительный ряд
производных пиперидина, среди которых и широко известный
препарат промедол (11.20). Наличие в молекуле (11.20) двух
метальных групп у С-2 и С-5 атомов приближают его к структуре
морфина. Три хиральных центра обеспечивают наличие стереомер-
ных а-, р-, у-форм, различающихся уровнем анальгетической
активности. Наиболее эффективен ct-промедол, наименее — у. Метод
синтеза промедола из винилацетилена был разработан И.Н. Назаровым
с соавт. [10].
Me.
Me—N sV
Me
LPhLi
2. НэО+
О
MeO
Me +
0 О + ОМе
МеО
I— Me
Me
Me
Me—N
к
ОН
Me
■Ph
(ЕГСОШ
к
Me—N
OCOEt
-Ph
Me
Me
(11.20)
Сопоставление строения синтетических анальгетиков со
структурой морфина позволило выявить «опиоидный фармакофор» (ОФ).
Было сформулировано так называемое «морфиновое правило» [9—12],
выдвигающее три основных структурных требования к анальгетикам
такого типа:
• наличие в молекуле третичного атома азота;
• наличие фенильного кольца, соединенного с остальной частью
молекулы через зр3-гибридный атом углерода:
• отделение фенильного цикла от азота тремя углеродными атомами.
284
(ОФ)
Все рассмотренные выше соединения удовлетворяли этому
правилу. Однако среди пиперидиновых анальгетиков есть высоко
эффективные вещества, которым не присущи все структурные
признаки, постулируемые «морфиновым правилом». К таким веществам
принадлежат фентанил (11.21) и сулфентанил (11.22). Фентанил
(согласно разным источникам) в 50—400 раз активнее морфина,
благодаря большей, чем у последнего, способности проникать через ГЭБ
из-за высокой липофильности. По этой же причине сулфентанил
в 5000—6000 раз активнее морфина. Пик действия этих препаратов
наступает очень быстро, но он непродолжителен. Оба указанных ЛС
используются в хирургической практике в комбинации с
транквилизаторами [4, 10, 11].
К самостоятельной группе относят а,а-дифенил-у-аминокарбо-
нильные соединения. Их представители — метадон (11.23) и пропок-
сифен (11.24) — по структуре лишь отдаленно напоминают пипери-
диновые и морфиновые анальгетики. Уровень проявляемой метадоном
активности соответствует морфину, а при пероральном применении
даже превышает его; однако метадон обладает большей
длительностью действия и более токсичен. Метадон используется в ряде стран
для замены «тяжелых» наркотиков при лечении наркоманов.
Me
Ме^\ Me
N
NEt
(11.23)
(11.24)
285
С выявлением аналыетических свойств у веществ из ряда ари-
лалкиламинов «морфиновое правило» не только не было
опровергнуто, но, напротив, получило более широкую трактовку, т. к. до
появления метадона считалось, что наличие пиперидинового кольца
является необходимым условием для проявления анальгетического
эффекта [4, 9—12].
Арсенал наркотических анальгетиков не исчерпывается
рассмотренными выше группами. Частичное или полное соответствие «морфино-
вому правилу» можно отыскать в структурах трамадола (11.25), тили-
дина (11,26), цирамадола (11.27). Рассмотреть все структуры, которым
присуши эти свойства, не представляется возможным. Поиск
эффективных анальгетиков центрального действия продолжается. Наиболее
перспективны соединения, к которым не развивается привыкание [10].
(11.25) (11.26) (11.27)
Большое значение для понимания механизмов действия опиои-
дов имело обнаружение в экстрактах мозга животных пентапептидов,
обладающих свойствами наркотических анальгетиков. Произошло это
в середине 70-х гг. Работы проводили две группы исследователей под
руководством J. Hughes, L. Terenius [13, 14]. Выделенные пептиды
получили название энкефалинов от греческого «кефалос» — голова.
Различают Met-энкефалин — Tyr-Gly-Gly-Phe-Met и Leu-энкефа-
лин — Tyr-Gly-Gly-Phe-Leu. Met-энкефалин является частью
аминокислотной последовательности р-липотропина, а Leu-энкефалин —
динорфина, полипептидов гипофиза. Свойствами наркотических
анальгетиков обладают не только эти пентапептиды, но и более
длинные фрагменты р-липотропина, получившие название а-, р-, у-эндор-
финов. Наиболее эффективным анальгетиком является р-эндорфин,
который примерно в 50 раз активнее морфина. Модификация
структуры энкефалинов позволила выявить определенную взаимосвязь
«структура-активность» в этом ряду. Полипептидные анальгетики не
нашли медицинского применения, т. к. при пероральном введении
разрушаются в ПК под действием пептидаз, а при парентеральном —
не проникают через ГЭБ. Им присуши те же недостатки и побочные
эффекты, что и другим наркотическим анальгетикам природного
и синтетического происхождения [15].
Поиск веществ, устойчивых к гидролизу, привел к выявлению
циклических аналогов энкефалинов, полученных синтетическим
путем [16, 17], Соединение (11.28), содержащее в структуре два остатка
286
диметилцистеина (пеницилламина), обладает сродством к 5-типу опи-
оидных мишеней. Замена дисульфидного фрагмента на остаток
лейцина приводит к избирательному связыванию с ц-подтипом
рецепторов. Конструирование циклических пептидомиметиков,
подобных (11.28), является одним из примеров реализации метода конформа-
ционных ограничений, в результате которого удалось достичь
повышения метаболической стабильности веществ.
Опиоидные рецепторы описаны S. Snyder с соавт. в 1973 г. [18]
как стереоспецифические места связывания соответствующих лиган-
дов, образованные аллостерическими липопротеинами, активность
которых регулируется ионами Na+. В связи с тем, что концентрация
последних в ЦНС достаточно высока, рецепторы находятся в кон-
формации, обеспечивающей преимущественное связывание с
антагонистами (см. разд. 3,3). Ионы Li+ способны вытеснять Na+ с зоны
рецептора, а ионы К+ не оказывают влияния на активность. Сродство
агонистов к данному типу мишеней повышается в присутствии ионов
Cu2+, Mg2+, Mn2+, Ni2+, Меркаптогруппа, являющаяся важной частью
рецепторов данного типа, также оказывает существенное влияние на
их конформацию.
Опиоидные рецепторы сопряжены с Gj-белками. В результате
связывания агониста активируется а,-субъединица G-протеина —
ингибитор АС и промотор цАМФ-фосфодиэстеразы. В итоге снижается
концентрация цАМФ в клетке. Рассматриваемые мишени
располагаются на поверхности нейронов в головном и спинном мозге,
на стенках пищевода и мочевого пузыря. На сегодняшний день
известны ц-, к-, а-, 8-типы этих мишеней, которые, в свою очередь,
подразделяются на подтипы [4, 10, 11, 18—20]. Функции и лиганды для
каждого типа рецепторов приведены в табл. 11.6.
Наличие минимальных структурных различий между агонистами
и антагонистами, существование структур со смешанным эффектом —
частичных агонистов (налорфин, пентазоцин, буторфанол)
стимулировали поиск гипотез, объясняющих взаимодействие аналогов
морфина с рецепторами. В настоящее время известно несколько моделей
ц- и к-подтипов опиоидных мишеней [4, 18, 21—25]. Первыми попыт-
287
Таблица 11,6
Типы опиоидных рецепторов [4, 10, 11]
Тип
М
Mi
Из
5
к,
Ki-з
а
Агонист
морфин,
суфентанил и др.
Морфин
Tyr-d-Ala-Gly-N-
Me-Phe-Gly-ol
-II-
Leu-энкефалин
бремазоцин
циклазоцин
Эндогенный
лиганд
энкефалины,
р-эндорфин
-II-
-II-
Leu-энкефалин
динорфин
Действие
Надспинальная
анальгезия,
физическая
зависимость,
эйфория,
респираторная
депрессия,
эметический
эффект
Центральная
анальгезия
Респираторная
депрессия
Спинальная
анальгезия,
изменение
поведенческих
реакций,
эйфория
Спинальная
анальгезия,
седативный
эффект, миоз
Галлюцинации,
респираторная
стимуляция
Антагонист
Налоксон;
Пентазоцин
Бугорфанол
(выступают
как частичные
агонисты)
Налорфин,
Налоксон
Налорфин
(частичный
агонист)
Налтриндол;
Налоксон
не связывается
с этим типом
рецепторов
ку описания топологии ц-рецепторов предприняли А.Н. Backett и
A.F. Casy [21]. Они предположили, что существует три места
связывания: анионный центр для протонированного атома азота, плоскость,
обеспечивающая взаимодействие с ароматическим кольцом, и ниша для
остальной части молекулы. Эта модель подходит для описания
взаимодействия с мишенью конформационно жестких структур, таких как
морфин, морфинаны и бензморфаны. Однако с ее помощью невозможно
объяснить способность конформационно лабильных пиперидиновых
анальгетиков и метадона образовывать устойчивые Л-Р комплексы.
В середине 60-х rr. P.S. Portoghese [22] исследовал Л-Р
взаимодействие опиоидов, обладающих жесткой структурой, и
конформационно подвижных, содержащих одинаковые заместители у атома азота, и
установил, что активность первых изменялась симбатно смене
заместителя, в то время как для вторых такая закономерность не отмечена.
•На основании этого P.S. Portoghese предложил свою модель опиоид-
ного рецептора, согласно которой для жестких и лабильных молекул
288
лигандов существуют свои места связывания (рис. 11.1). Развивая
данную концепцию в отношении энкефалинов и эндорфинов [23, 24],
исследователь предположил, что тирозиновые производные
взаимодействуют с мишенью подобно жестким структурам, а фенилалани-
новые аналоги — подобно конформационно подвижным. Общим для
обоих типов лигандов является анионный центр.
Рис. 11.1. Модель опиоидного рецептора, предложенная P.S. Portoghese:
1 — место связывания конформационно жестких лигандов;
2 — место связывания конформационно лабильных лигандов
Гипотезой, наиболее наглядно объясняющей минимальные
структурные различия между агонистом и антагонистом, является
предположение, высказанное S. Snyder {18]. Согласно этой модели, с
«плоской областью» рецептора связывается липофильная часть молекулы L
как агониста, так и антагониста (рис. 11.2). Кроме того, в составе
рецептора есть локус (местоположение) AG, соответствующий месту
связывания боковой цепи агониста, и локус AN — для антагониста.
Рис.11.2. Модель опиоидного рецептора, предложенная S. Snyder [18]
289
Агонист — морфин — связывается с L-областью, слабо
взаимодействуя с AG- частью за счет N-CH3 группы. Мощный агонист —
циклазоцин (11.16) одинаково прочно связывается с L- и
AG-фрагментами рецептора. Слабый агонист метадон (11.23) не может
принять конформацию, необходимую для прочного связывания. Эта
модель хорошо объясняет эффект «аномально мощного* агониста —
фентанила (11.21). Частичные агонсты типа налорфина (11.5) могут
иметь как аксиальное, так и экваториальное расположение
заместителей у атома азота. У «чистого» антагониста налоксона (11.6) этот
заместитель всегда в экваториальном положении и изменять
конформацию ему не позволяет 14-ОН группа.
Представления о взаимодействии лигандов с опиоидным
рецептором изменялись по мере накопления новых данных. Было
установлено, что большое значение имеет ориентация электронной пары атома
азота для реакции переноса протона к N-атому, обеспечивающему
связывание с анионным центром рецептора и несущему
ответственность за проявление анальгетического эффекта. Исходя из этого,
предложен так называемый Zip-механизм взаимодействия. Согласно ему
контакт протонированной аминогруппы с анионным центром
вызывает дальнейшие конформационные изменения, обеспечивающие
сначала связывание ароматического фрагмента субстрата с
рецептором, а затем и той части молекулы, которая окружает третичный атом
азота [4, 10]. Эндорфины и энкефалины не имеют третичного атома
азота и при физиологических значениях рН представляют собой
биполярные ионы. Следовательно, для того, чтобы эти эндогенные
лиганды могли взаимодействовать с опиоидными рецепторами,
последние, кроме анионного центра и двух плоскостей для связывания
с агонистом и антагонистом, должны содержать еше один элемент.
В настоящее время в нативном виде выделены три типа опиоидных
рецепторов и предприняты попытки их клонирования [26—28].
Возможно, в ближайшие годы структура этих мишеней будет раскрыта.
Окончательное установление строения всех типов опиоидных рецепторов,
вероятно, позволит приблизиться к созданию «идеального» анальгетика.
Однако уже сейчас ясно, что искать такие вещества надо среди
частичных агонистов, обладающих мощным обезболивающим действием и не
вызывающих психической и физической зависимости [4, 10].
Анализ взаимодействия наркотических анальгетиков с
рецепторами позволяет подойти к понятию «фармакофор» с учетом
конформационного фактора. Фармакофор — это определенное
пространственное расположение функциональных групп в
молекуле, которое обеспечивает взаимодействие с биологической
мишенью. Существующие на сегодняшний день методы компьютерного
моделирования G-протеинов пока не настолько точны, чтобы быть
полезными для моделирования фармакофоров методами ab initio.
Поэтому при воссоздании подходящих фармакофоров
основываются на сведениях о третичной структуре белковых молекул,
образующих биологическую мишень. Наиболее информативным является
сопоставление данных о третичной структуре протеинов,
получению
ных как с .соответствующими лигандами, так и без них. Однако
сведения о пространственной структуре активных мест связывания
имеются далеко не для всех типов рецепторов. И это заставляет
специалистов в области медицинской химии искать другие пути.
Используя уточненные сведения о взаимосвязи «структура —
активность» для уже известных комплексов Л-Р, исследователи
переносят их на новые модели и на этом основании достраивают
полную картину взаимодействующих функциональных групп, варьируя
их расположение в пространстве. Наибольшие трудности
возникают тогда, когда частично перекрывающиеся данные соответствуют
разным, но очень гибким молекулам, при этом реконструируется
сразу несколько фармакофоров. Возникает необходимость
проверить каждый из вариантов с помощью синтеза пространственно
«ограниченных» молекул, которые имеют строго определенные
функциональные группы в потенциально «активных» конформаци-
ях. В случае положительного результата может быть выявлен
фармакофор.
Много проблем при усовершенствовании лидирующей структуры
связано с повышением селективности ее взаимодействия с тем или
иным Ф или рецептором. Последние могут содержать разные
функциональные группы, но иметь очень малые различия в
аминокислотной последовательности. Например, различие между 5-НТ!В и 5-HT|DB
триптаминовыми рецепторами заключается в одной аминокислоте.
Ясно, что малые молекулы с высокой конформационной
лабильностью, обычно способны взаимодействовать более чем с одним типом
белков, т.е. обладают низкой селективностью. Так, миансерин (11.29)
способен связываться с 5-НТ2-серотониновыми, с^-адренергически-
ми и Н,-гистаминовыми рецепторам. При определенных конфор-
мационных ограничениях, реализованных в структуре (11.30),
сродство к 5-НТ2- и а,-мишеням удалось сохранить, а к Ht- сродство было
утрачено [8].
Me Me
(11.29) (11.30)
Аналогичная селективность достигнута и в случае агонистов 5-НТ -
и 5-НТш-рецепторов. Установлено, что замена в гидрокситрипта-
мине (серотонине) 5-ОН группы на 5-карбоксиамидную (11.31)
приводит к более сильному взаимодействию с 1А-подтипом мишеней.
Замыкание этиламинового фрагмента в кольцо с образованием 3-ами-
291
но-6-карбоксиамидотетрагидрокарбазола (11.32) сопровождается
практически полной потерей 5-НТ]А-активности, но сохраняет сродство
к 1 D-рецепторам [29].
(11.31)
(11.32)
Повышения аффинности взаимодействия при реализации метода
конформационных ограничений можно ожидать по двум причинам:
• снижение неблагоприятного энтропийного фактора при
фиксированной конформации;
• стабилизации «активной» конформации.
Когда гибкая молекула связывается с рецептором, она теряет ряд
степеней свободы, приобретая неблагоприятные изменения
энтропии, которые снижают величину свободной энергии связывания. Этот
эффект особенно заметен для веществ, у которых вклад энтропийной
составляющей в общую энергию связывания высок, что имеет место
у соединений с разветвленной структурой и значительной липофиль-
ностью. Примером простой конформационной перестройки,
значительно повышающей сродство к рецептору, а, следовательно,
положительно сказывающейся на активности, может служить введение СН3
группы в а-положение молекулы ингибитора АПФ — каптоприла (5.59).
Для него отмечается повышение активности в 10 раз по сравнению с
неметилированным аналогом, и в 100 раз по сравнению с
оптическим антиподом (см. разд. 5.4.3) [30].
Введение двойной связи, снижающее гибкость углеродной цепи
или цикла, также существенно сказывается на активности. Так,
преднизалон (11.33) в 30 раз более активен, чем кортизол (11.34).
(11.33) (П.34)
Отмечены различия в активности между Е и Z изомерами; в
случае хлорпросиксена (11.35) Z-форма более активна.
292
\ /
Е< Z
(11.35)
За счет структурной перестройки молекулы можно добиться
существенной метаболической устойчивости лидера. Так, в качестве
специфической стратегии повышения стабильности амидной связи
пептидов, предназначенных для перорального применения,
используется циклизация. Этот подход основан на предположении, что
образование цикла предотвращает выравнивание пептида на активном
центре пептидазы, тем самым предохраняя структуру от разрушения
и сохраняя фармакологическую активность. Вообще циклизация —
широко распространенный способ, позволяющий избежать
нежелательных метаболических превращений отдельных функциональных
групп. Иллюстрацией могут служить как рассмотренный ранее
пример с циклическим аналогом энкефалина (11.28), так и
предотвращение быстрого окисления аминоэтильного фрагмента до
неактивной индолилуксусной кислоты в антимигрениевом препарате
суматриптане (11.36). Период полупревращения (11.36) в неактивный
метаболит составляет 2 ч. Включение третичной аминогруппы в пипе-
ридиновый цикл сохраняет активность, но значение t1/2 для (11.37)
увеличивается до 5 ч в связи с тем, что метаболические превращения
затрагивают 5-е положение этого цикла [8].
(11.36) (11.37)
Существует большое количество эндогенных лигандов
биологических мищеней, которые представляют собой белковые молекулы с
определенной третичной структурой. Однако связывание с
рецептором обеспечивает лишь небольшой участок такой молекулы.
Воспроизвести пространственное строение такого участка, используя
короткие пептиды с несколькими аминокислотными остатками, довольно
сложно, т. к. малые молекулы не образуют третичной структуры. Ее
нет и у органических соединений небелковой природы. В этом случае
необходимо построение фиксированной конформации с определен-
293
ной ориентацией боковых цепей и функциональных групп,
мимикрирующей третичную структуру белковой молекулы эндогенного
лиганда. Примером является гидроксиэтиленовый фрагмент,
включенный в структуру ингибиторов ВИЧ-1 протеазы (см. разд. 9).
11.3. Пролекарства
Многие соединения, обладающие мощным эффектом in vitro, при
проверке in vivo проявляют низкую активность, что может быть
следствием многих факторов, включая слабое всасывание, быстрый
метаболизм и/или выведение, медленное проникновение к месту действия.
Еще одним серьезным недостатком часто оказывается высокая
токсичность. Все это заставляет вести поиск структур, которые не
обладали бы вышеперечисленными отрицательными качествами. В
подобном случае полезным может оказаться создание пролекарства —
неактивного соединения, которые в результате биотрансформации в
организме превращается в активное, проникает к месту действия и
оказывает желаемый фармакологический эффект. В связи с тем, что
изначально такое вещество не обладает активностью, оно, в
большинстве случаев, не может быть выявлено при скрининге in vitro, т.е.
до тех пор, пока не будет реализован механизм его активации.
Впервые термин «Пролекарство» ввел в 1958 г. А. Альберт [31]. Однако
задолго до этого в 1886 г. попытка направленного конструирования такого
ЛВ была успешно осуществлена немецким биохимиком Ненцки,
который справедливо предположил, что при объединении в одной молекуле
остатка салициловой кислоты и фенола вновь образованное соединение
способно распадаться в организме на исходные компоненты и оказывать
противовоспалительное и антисептическое действие [32]. В результате был
синтезирован фенилсалицилат (салол). Пролекарствами, механизм
активации которых стал Понятен уже после того, как они получили
распространение в медицинской практике, являются уротропин, генерирующий
формальдегид в мочевыводящих путях, и Prontosil rubrum (5.54),
превращающийся в результате восстановления в стрептоцид (5.48).
Около 7% современных ЛС являются пролекарствами, например,
эналаприл (5.60), симвастатин (5.69), ацикловир (6.63) и др. В эту
группу не входят так называемые «ограниченные» пролекарства и
«мягкие» лекарства. Ограниченные пролекарства — это активные ЛВ,
метаболиты которых также вносят вклад в общий
фармакологический эффект (например, нитросорбид (7.4), пропранолол (7.15) и др.).
Они составляют ~ 4% нынешнего арсенала ЛС. Понятие о мягких
лекарствах было введено N. Bodor в 1984 г. для обозначения ЛВ,
которые после достижения места действия и реализации
фармакологического эффекта дезактивируются ферментативно контролируемым
путем, что позволяет избежать задержки в организме высоко
токсичных метаболитов [33].
Пролекарства классифицируют согласно двум основным
критериям: по химическому строению и по способу активации. Исходя из
294
структурных особенностей различают ЛВ, переносимые
специфическими носителями — линкерами, и биопрекурсоры. Переносимые
линкерами пролекарства обычно имеют некоторые подвижные группы,
аналогичные защитным группам, используемым в органическом
синтезе, которые, отщепляясь, высвобождают активное вещество. Этот
вид пролекарств, в свою очередь, подразделяется на системы,
состоящие из 2-х частей, т. е. из носителя и собственно ЛВ, и состоящие из
3-х частей, в которых носитель связан с ЛВ через промежуточное
звено. Примеры наиболее распространенных защитных групп,
которые нашли применение в конструировании пролекарств, приведены
в табл. 11.7.
Таблица 11.7.
Носители пролекарств [8]
х=
х=
-(О
-(С=
-(О
ЛВ-ОН -жро-ЛВ-ОХ"
=0)R; -(C=0)CH2NR2; -(C=0)CH2CH2COOR;
=0)CH2S03H; -(C=0)CH2CH2NR2
ЛВ-NH -шро-ЛВ-NX
-(OO)Ph
=0)R; -(C=0)CHRNH2; -(C=0)OPh; -CH2NH(C=0)Ar; =
=NAr; -CHR-0(C=0)R; -CH2- -(O0)CH2NR2;
>C(X)Y=
х=
-R;
ЛВ>С=0 ->про-ЛВ>С(Х)У
■- >C=NR; >C-NOR; >C(OR)2
ЛВ>С02Н -шро-ЛВ>С02Х
-CHR-0(C=0)R; -CHR-0(O0)NR2; -CH2(C=
-CHR-0(C=0)OR; -CH2CH2OH
=0)NR2;
-PO32-
^CHAr;
-CH2NR2
"X — носитель
Биопрекурсоры отличаются от рассмотренных выше систем
«линкер—ЛВ», тем, что в их структуре нет легко гидролизуемых связей.
Активация этих соединений происходит в результате образования
нового вещества — метаболита исходного. Типичными примерами
биопрекурсоров являются имипрамин (8.10) и L-ДОФА,
превращающиеся в организме в десметилимипрамин и дофамин соответственно.
Активация пролекарств может осуществляться как
ферментативным, так и неферментативным путем (спонтанная биоактивация). Оба
способа имеют свои преимущества и недостатки. Энзиматический путь
во многих случаях тканеспецифичен и достаточно легко
контролируется во времени. Недостатками его являются возможность
образования неожиданных метаболитов по причине генетического
полиморфизма и высокая вероятность взаимодействия ЛВ, в метаболизме
которых участвуют одни и те же группы Ф (см. разд. 8.3). При
спонтанной биоактивации пролекарств, генетический полиморфизм и
взаимодействие с другими ЛВ обычно не столь важны. Однако в этом
случае на первый план выходит проблема химической стабильности
вещества и необходимости установления места его превращения в
активное соединение. Следует отметить, что направленное конструи-
295
рование соединений, способных превращаться в активные ЛВ не-
энзиматическим путем — задача достаточно сложная, т.к. полностью
исключить влияние Ф на исследуемое вещество в организме
невозможно.
Как уже отмечено выше, к созданию пролекарства прибегают в тех
случаях, когда необходимо изменить фармакокинетические параметры
кандидата в ЛС: повысить биодоступность, повлиять на транспортные
характеристики или метаболическую стабильность. Оптимизировать эти
показатели можно за счет увеличения водорастворимости или,
наоборот, липофильности, предотвращения досистемного метаболизма или
путем избирательной доставки ЛВ к месту действия.
Способы повышения водорастворимости ЛВ. Для улучшения
данного показателя в структуру веществ вводят группы, способные к
ионизации (фосфатные, гемисукцинатные, аминоацильные, димети-
ламиноацетатные), или нейтральные ПЭГ фрагменты. Так, введение
фосфатного остатка в молекулу фентоина, обладающего противосу-
дорожным действием, позволило создать водорастворимое пролекар-
ство — фосфентоин (11.38), пригодное для парентерального
применения. Соединение (11.38) быстро гидролизуется в организме
НО ОН
о'Хо
к^
ы—<
О^^ у
U N
н
(?
{
UCeH5
сен5
(11.38) (11.39)
при участии фосфатаз с образованием активного 3-гидроксиметил-
5,5-дифенилгидантоина (фентоина) [34]. Еще одним примером
структурной перестройки, позволившей существенно повысить
растворимость, служит введение ацильных остатков в сульфамидные фрагменты
двух НПВС из группы диарилпроизводных азолов (11.39) и (11.40),
по механизму действия являющихся ингибиторами РОН-синтетазы-2.
Растворимость в воде натриевых солей (11.39) и (11.40)
составляет ~ 15 мг/мл, в то время как аналогичный показатель для не-
ацилированных сульфамидов не превышает 50 мкг/мл. Активные
ЛВ высвобождаются в результате первичного метаболизма про-
лекарств (11.39), (11.40) в ПК и печени, сопровождающегося
элиминированием ацильного остатка.
Повышению гидрофильности способствует введение в молекулу ЛВ
ПЭГ фрагмента. Для этих целей обычно используют ПЭГ с большой
мол. массой, чтобы избежать быстрого выведения ЛВ из организма.
Превращение таксола (10.28) в 2'-ПЭГ-таксол (11.41) позволило
296
^Г
Na+ p
NtO-
J^?
\r^N
сек
(11.40)
создать инъекционную ЛФ этого антибластомного средства. Гидролиз
эфира (11.41) протекает энзиматически. Следует подчеркнуть, что при
образовании 7'-ПЭГ-таксола подобной биоактивации не наблюдается.
Используя ПЭГ с различной мол. массой, можно регулировать
концентрацию ЛВ: при 2'-ПЭГ5000 растворимость (11.41) достигает 670 мг/мл,
при 2'-ПЭГ20000 - 200 мг/мл, при 2'-ПЭГ4000° - 125 мг/мл, в то время
у неэстерифицированного таксола она составляет всего 25 мкг/мл.
Acq о 9н
^v
(11.41)
Способы повышения липофильности ЛВ. Увеличение
липофильности способствует проникновению ЛВ через мембраны клеток ПК за
счет пассивной диффузии. В этом случае наиболее распространенной
тактикой усовершенствования структур является синтез эфиров.
Известно, что ампициллин (11.42) обладает умеренной (40—62%)
биодоступностью при пероральном способе введения [35, 36], а его эсте-
рифицированные производные — пивампициллин (11.43) и бакампи-
циллин (11.44) абсорбируются из кишечника на 98—99% [37]. Эти
пролекарства принадлежат к группе ЛВ, переносимых линкерами,
антимикробный эффект которых наблюдается только после
высвобождения ампициллина в сыворотке крови под воздействием эстераз.
Ph
NH2 s^J/ /°
(11.43)
297
о
Л /\
о
(11.44)
Пивалиновый (триметилацетатный) линкер, присутствующий в
структуре (11.43), является одним из наиболее популярных. Введение
двух таких остатков в молекулу адениннуклеотидного аналога — аде-
фовира, — обладающего выраженными противовирусными свойствами,
привело к пролекарству (11.45), которое в настоящее время проходит
клинические испытания в США как потенциальное пероральное ЛС,
эффективное в отношении гепатита В [38].
(11.45)
При подборе линкеров следует принимать во внимание
токсикологические характеристики продуктов, образующихся при их
распаде. Так, существенно повышает токсичность пролекарств
формальдегид, выделяющийся при гидролизе двойных эфиров, содержащих
метиленовый мостик. В этом смысле ацетальдегид как метаболит
более безопасен. Однако такой фрагмент вносит в структуру
дополнительный хиральный центр, а образовавшиеся в результате
модификации энантиомеры могут активироваться в организме с различной
скоростью. Недавно установлено, что и триметилуксусная кислота,
образующаяся в результате гидролиза пивалиновых эфиров, также
вносит в свой вклад в токсичность [39].
Формирование переносимых линкерами пролекарств — не
единственный способ повышения интестинальной абсорбции ЛВ. Часто
не менее эффективным оказывается конструирование
биопрекурсоров. Известно, что амидоксимы, гораздо менее основные, чем амиди-
ны, являются биопрекурсорами последних [40]. Данный подход
нашел отражение в модификации структуры антиагреганта мелагатрана
в двойное пролекарство — ксемилагатран (11.46). Восстановление ок-
298
сима в амидин происходит под действием редуктаз в ПК, почках,
печени, легких и др. органах и тканях. Ожидается, что
ксемилагатран, в случае успешного завершения клинических испытаний, станет
первым прямым ингибитором тромбина, пригодным для перорально-
го применения [41].
(11.46)
Способы повышения интестинальной абсорбции за счет активного
транспорта. С целью облегчения переноса ЛВ через биологические
барьеры широко используются специфические носители, для
которых существуют собственные транспортные системы. Олигопептиды
являются активными переносчиками ЛВ, в структуре которых
содержится остаток какой-либо аминокислоты, например, р-лактамных
антибиотиков, ингибитора АПФ эналаприла (5.60), валациклови-
ра (11.47) и многих др. [42]. Валацикловир — валиновый эфир ацик-
ловира (6.63) — обладает в пять раз большей биодоступностью при
введении per os, чем исходный препарат, благодаря активному
транспорту интестинальным дипептидом РЕРТ1 [43].
H.N
(11.47)
Активный перенос через мембраны способствует быстрому
всасыванию азидотимидина (6.66) и др. нуклеозидных аналогов.
Переносчики желчных кислот облегчают абсорбцию тироксиноподобных
соединений. Субстратами для транспортеров витаминов являются
метотрексат (5.57), а также производные, содержащие фрагмент
тиамина, никотиновой кислоты или ядро коррина (аналоги витамина
В12). Специфические доставщики органических катионов (холина,
дофамина и др.), анионов (метотрексата, правастатина) и транспортеры
299
глюкозы обеспечивают всасывание ЛВ, содержащих в структуре
соответствующие фрагменты. Несмотря на очевидные преимущества,
данный подход в конструировании пролекарств сопряжен и с
некоторыми трудностями. В частности, следует отметить, что транспортные
системы способны к насыщению, в результате чего не исключены
нарушения в фармакокинетике при одновременном приеме
нескольких ЛС и взаимодействие между ними [34].
Способы повышения метаболической и химической стабильности ЛВ.
Часто, с целью предотвращения быстрого досистемного метаболизма
ЛВ, прибегают к «маскировке» фармакофоров, формируя на их
основе фрагменты, претерпевающие более медленную биотрансформацию,
чем исходные. Например, превращение эффективного бронходилата-
тора тербуталина (11.48) в бамбутерол (11.49), за счет преобразования
фенольных групп в медленно гидролизующиеся неспецифическими
холинэстеразами карбаматные, позволило в три раза снизить
суточную дозу бронхорасширяющего средства [44].
Г н /
(11.49)
(11.48)
Кардиотоник докарпамин (11.50) представляет собой не что иное,
как пригодную для перорального применения транспортную форму
дофамина, высвобождающегося в результате гидролиза двойного про-
лекарства (11.50) в кишечнике и последующей метаболической
трансформации в печени [45].
У'
О
О.
V-o
г
И
HN
XT".
(11.50)
300
Методология конструирования пролекарств, распознаваемых
клетками-мишенями. Доставка ЛВ в клетки-мишени может происходить
путем пассивной диффузии, активного транспорта, биоактивации
специфическим Ф или в результате взаимодействия
антитела-носителя с клеточным антигеном. Проникновение путем пассивной
диффузии обеспечивают ПЭГ линкеры. Так, рассмотренный ранее 2'ПЭГ-
таксол, переносимый линкером с мол. массой > 50000, не только
обладает повышенной гидрофильностью, но и избирательно
накапливается в опухолевых клетках. Механизм этого явления
окончательно не выяснен [34].
Стратегия создания пролекарств, мишени которых расположены в
ЦНС, основана на следующих принципах: носитель должен
обеспечивать липофильность, достаточную для проникновения вещества в
нервную ткань, а затем превращаться in situ в более гидрофильную частицу,
не позволяющую ЛВ быстро элиминировать обратно в системное
кровообращение. Наиболее распространенным доставщиком такого типа
является 1Ч-метил-1,4-дигидропиридин-3-карбоновый эфир (11.51). Этот
фрагмент легко окисляется оксидоредуктазами как в плазме, так и в
ЦНС, с образованием четвертичного аммонийного метаболита (11.52).
Из плазмы неактивный метаболит элиминирует очень быстро. В
нервной же ткани, наоборот, высокополярное пролекарство (11.52)
задерживается гораздо дольше, и в результате ферментативного
гидролиза эфирной связи из него высвобождается активное ЛВ [34].
ЛВ
(11.51) (11.52)
В специальной литературе описано лишь несколько примеров
избирательной доставки ЛВ к мишеням за счет активного
транспорта [46]. Один из них — биопрекурсор дофамина — леводопа (L-ДОФА,
11.53), выступающий в качестве субстрата транспортеров
нейтральных аминокислот через ГЭБ. В ЦНС это пролекарство в результате
декарбоксилирования превращается в дофамин. С целью
предотвращения быстрой биоактивации леводопы в системном
кровообращении применяют комбинированную ЛФ, содержащую одновременно
и карбодофу — Ь-а-гидразино-а-метил-р-(3,4-диоксифенил)пропионо-
вую кислоту — ингибитор периферической ДОФА-декарбоксилазы,
не проникающий через ГЭБ.
Пролекарством, претерпевающим биоактивацию в результате
взаимодействия с тканеспецифичным Ф, является омепразол (11.54) —
представитель группы противоязвенных ЛС, угнетающих функцию
протонового насоса (Н+/К+-АТФазы) париетальных клеток желудка.
Препарат быстро всасывается, биодоступность составляет - 53%,
обладает слабыми основными свойствами и в нейтральной среде
неактивен. Однако в кислой среде канальцев париетальных клеток он
превращается в активный сульфенамид (11.55), который за счет
образования дисульфидных мостиков с цистеиновыми остатками
мембранной Н+/К+-АТФазы необратимо ингибирует ее. Высокая
избирательность действия омепразола обусловлена тем, что его биоактивация
происходит только в париетальных клетках, где есть необходимая для
этого среда. Процесс осуществляется достаточно быстро (2—4 мин), а
образовавшийся сульфенамид-катион не проникает в системный
кровоток. По этой причине концентрация омепразола в плазме не
коррелирует со степенью и продолжительностью подавления секреции
соляной кислоты в желудке [47, 48].
(11.54)
s"-*oh
(11.55)
Биоактивация специфическим Ф имеет место и в случае
противовирусного средства ацикловир (6.63). Его превращение в нуклеотид-
ный аналог, ингибирующий синтез вирусной ДНК, осуществляется
при участии фосфокиназы, кодируемой вирусным геномом. В
результате ацикловир и ряд др. структурно близких к нему антиметаболитов
нового поколения активируются только в инфицированных вирусом
клетках [49].
Проблема обеспечения селективной доставки антибластомных Л С
в опухолевые клетки является одной из самых актуальных и одновре-
302
менно самых сложных в конструировании этой группы химиотера-
певтических средств. Примеров ее успешного решения пока немного.
Один из них — тройное пролекарство на основе 5-фторурацила —
капецитабин (11.56). После абсорбции из ПК это вещество гидроли-
зуется и дезаминируется в печени при участии карбоксиэстеразы и
цитидиндезаминазы соответственно, и только в опухолевых
клетках под действием тимидинфосфорилазы происходит высвобождение
5-фторурацила [50].
n
N
л - ЧГТ
-он но' он
но
(11.56)
F
О
ХГг
^
NH _ HN NH
т
но' *он ° °
Высокоспецифичное распознавание пролекарством
клеток-мишеней может быть реализовано при взаимодействии антител-линкеров с
соответствующими антигенами, расположенными на поверхности
клеточной мембраны. Недавно в США после успешного завершения
клинических испытаний разрешено применение нового ЛС милотарг
(СМА—676) для лечения острого миелолейкоза. Это пролекарство
содержит рекомбинантный человеческий анти-СОЗЗ-линкер, связанан-
ный через N-атом лизинового остатка с мощным цитостатиком, —
?4-ацетил-у~калихемацином [51—53]. Указанный линкер распознается
только атипичными миелобластами, что позволяет осуществить
доставку ЛС в опухолевые клетки.
Энзим этическая активация пролекарства в пораженных опухолевым
процессом тканях может происходить и под действием экзогенного Ф.
Осуществляется это, как правило, в два этапа. Вначале в клетку
доставляют специфичный для данного пролекарства Ф, а уже затем в организм
вводят соответствующий ему субстрат — нетоксичное пролекарство,
которое подвергается биоактивации именно в той ткани или клетке, в
которой присутствует этот энзим. Данный метод позволяет достичь
высокой локальной концентрации ЛВ в клетках-мишенях.
Носителями?
ми (векторами доставки) экзогенных Ф выступают модифицированные
ДНК вирусов или бактерий, содержащие ген, кодирующий данный Ф,
химические доставщики генов, а также конъюгаты «моноклональное
антитело — Ф». Примером, подтверждающим перспективность такого
подхода в разработке антибластомных ЛС, является использование в
качестве экзогенного энзима, катализирующего превращение пролекар-
ства (11.57) в цитостатикалкилирующего типа 4-[бис-(2-хлорэтил)ами-
но]бензойную кислоту, бактериальной карбоксипептидазы G2 (CPG2).
Селективную доставку протеолитического Ф в опухолевые клетки
обеспечивает конъюгат «моноклональное антитело — CPG2». Важно
подчеркнуть, 4toCPG2 (EC 3.4.17.11) не встречается в клетках
млекопитающих. Это позволяет снизить риск развития нежелательных
побочных эффектов, обусловленных конкуренцией между близкими Ф за
один и тот же субстрат.
соон
(11.57)
Эффективность рассмотренного способа доставки ЛВ в клетки-
мишени нашла подтверждение и в эксперименте, и в ряде
клинических испытаний [54, 55]. Однако пока при разработке подобных
систем возникает гораздо больше проблем, чем существует способов из
разрешения.
Конструирование мягких лекарств. Как уже было отмечено выше,
мягкие лекарства являются активными аналогами известных ЛВ,
которые после реализации специфического эффекта, дезактивируются фер-
ментативно контролируемым путем, исключающим появление
нежелательных метаболитов. Это особенно важно для ЛС короткого действия,
применяемых в хирургии (эсмолол), анестезиологии (ремифентанил) и
кардиологии (ATI—2001) [56, 57]. Однако пока мягкие лекарства не
получили широкого распространения и используются, в основном, в
качестве средств, подходящих лишь для местного применения.
Методология создания Л В этого типа состоит в отборе базовых
структур среди неактивных метаболитов высокоэффективных ЛС и
модификации их в направлении «восстановления» активности, но не путем
возврата к исходной структуре. В большинстве случаев такие попытки имеют
ограниченный успех. Одними из немногих удачных примеров
являются местное противовоспалительное средство флукортин бутил (11.58) —
эфир неактивного метаболита фторкортолона (11.59) и лотепрендол
304
этабонат (11.60) — мягкий аналог гидрокортизона (11.61), нашедший
применение в офтальмологии [58, 59]. Стартовой структурой в
дизайне (11.60) стала неактивная кортиеновая кислота. Ее превращение в
эфир (11.60) привело к 20-ти кратному увеличению терапевтического
индекса (ТИ = ЛД^Щ^ по сравнению с гидрокортизоном.
(11.58)
(11.59)
,иОН
сн2он
(11.60) (11.61)
Определенные надежды в плане создания ЛС, эффективного в
лечении глаукомы, до недавнего времени связывали с адапроло-
лом (11.62) — мягким аналогом известного р-адреноблокатора мета-
пролола (7.21), полученным в результате модификации структуры его
основного метаболита. Однако на II стадии клинических
испытаний (1995 г.) дальнейшая разработка адапролола была прекращена [60].
Главными причинами неудач в конструировании мягких лекарств
являются их низкая, по сравнению с прототипами, специфическая
активность и метаболическая нестабильность.
?Н Н
(11.62)
В 1986 г. возникла идея объединить подходы в дизайне ЛВ,
связанные с про- и мягкими лекарствами. Цель данной концепции — обес-
305
печить вводимому в организм веществу предсказуемую энзиматическую
трансформацию в мягкое лекарство по месту действия, а затем
дезактивировать его, избежав высокотоксичных метаболитов. Примером
успешного воплощения этой идеи может служить использование для
цитостатика трифлуридина (11.63) в «транспортных» целях уже
рассмотренной ранее N-метилдигидропиридин — N-метилпиридиниевой ред-
окс-системы [61,62]. Этот препарат применяется в химиотерапии
злокачественных опухолей, локализованных в ЦНС. Соединение (11.63)
сильно полярно, поэтому не проникает через ГЭБ. Введение в
структуру остатков N-метилдигидропиридин-З-карбоновой итриметилуксус-
ной кислот обеспечивают проникновение вещества А в ЦНС (рис. 11.3).
Уже преодолев ГЭБ, оно окисляется в высоко полярное пиридиниевое
производное В, не способное элиминировать из ЦНС. Пиримидиний-
трифлуридин Г — мягкое пролекарство, из которого медленно
высвобождается активный трифлуридин (11.63). Частично гидролизованные
восстановленная Б и окисленная Г формы в сыворотке крови и
мозговой жидкости распределяются следующим образом:
— концентрация в плазме формы Б через 1 ч после внутривенного
введения — 7.9 мг/мл;
— концентрация в ЦНС формы Г через тот же промежуток времени
72 мг/г;
— концентрация в ЦНС формы Г через 4ч — 156 мг/г.
Me
Me l^
О Ni_u_
Me
ГЭБ .
X * Me
ИЛ
N—Me
Me)f ^й\
НО
N—Me
СГ N
1 N—N —
Me
НО
О' N
N—Me
(11.63)
Рис. 11.3. Транспортные формы трифлуридина [61]:
А — восстановленная форма; Б — частично гидролизованная форма;
В — окисленная форма; Г — пролекарство
306
Следует отметить, что, несмотря на определенные успехи,
практическая реализация подходов в усовершенствовании
структур-лидеров, связанных с созданием пролекарств, сопряжена с рядом
серьезных проблем. Многие специалисты скептически оценивают
возможности данной методологии из-за сложностей, возникающих
при подборе защитных групп, способных отщепляться от
основной структуры в нужном месте, с нужной скоростью, не
привнося при этом в организм токсичных промежуточных продуктов,
таких как формальдегид, алифатические кислоты и др. [34, 37].
Токсикологические критерии выступают на первый план, если
предполагается длительное применение препарата в больших
дозах. Идти по пути создания пролекарства целесообразно в том
случае, когда присутствие в структуре какого-либо из
высокоактивных фармакофоров (амидиновой, гуанидиновой, фенольной
группы) негативно сказывается на оральной биодоступности
потенциального кандидата в ЛС. Безусловно, модификация
такого фрагмента является одним из путей решения данной
проблемы. Однако, для того чтобы выбрать правильное направление
оптимизации структуры, необходимо установить точную причину
слабой абсорбции, будь то низкая растворимость в воде,
метаболическая нестабильность или др. факторы. Вряд ли стоит
заниматься синтезом веществ, способных проникать через все
биологические барьеры.
Литература
1. Thornber C.WЦ Chem. Soc. Rev. - 1979. - Vol. 8. - P. 536
2. Lipinski C.A. // Annu. Rep. Med. Chem. - 1986. - Vol. 21. - P. 283
3. LangmuirL // J. Am. Chem. Soc. - 1919. - Vol. 41. - P. 868.
4. Principles of Medicinal Chemistry, 4th ed. / Ed. by W.O. Foye, T.L. Lemke,
D. A. Williams — Philadelphia, Baltimore, New York, London: Lippincott
Williams & Wilkins, 1995.
5. Grimm H. G // Z. Elektrochemie. - 1925. - Vol. 31. - S. 474.
6. Hindsberg O. // J. Prakt. Chem. - 1916. - Vol. 93. - P. 302.
7. Friedman H.L. Influence of Isosteric Relacements Upon Biological Activity,
National Academy of Sciences // National Research Council Publication № 206,
Washington: D.C.U.S. Government Printing Office, 1951, P. 295.
8. Medicinal Chemistry: Principles and Practice / Ed. by F.D.King. — Harlow,
UK:Smith Kline Beecham Pharm., 1994.
9. Patrick G.L. An Introduction to Medicinal Chemistry. — New York: Oxford
University Press, 1995.
10. Ковтуненко B.O. JliKapcbici засоби з д]ею на центральну нервову систему.
Навч. поЫбник для студенев природн. фак. ун-т1в. — Кит; 1ртнь: ВТФ
«Перун», 1997.
11. Casey A.F., Parfitt R.T. Opioid Analgesics: Chemistry and Receptors. — New
York: Plenum Press, 1986.
12. Lenz G.R., Evans S.M., Walters D.E., Hopfinger A.J. Opiates. — Orlando:
Academic Press, 1986.
13. Hughes J. et al. 11 Nature. - 1975. - Vol. 258. - P. 577.
307
14. Terenius L. // Acta Phamacol. Toxicol. — 1973. — Vol. 32. — P. 317.
15. Громов Л.Л. Нейропептиды. — К.: Здоров'я, 1992.
16. Morley J.S. //Annu. Rev. Pharmacol. Toxicol. - 1980. - Vol. 20. - P. 81.
17. Chipkin R.E. // Drugs of the Future. - 1986. - Vol. 11. - P. 593
18. Snyder SH // Sci. Am. - 1977. - Vol. 236, N 3. - P. 44-56. -
19. James R. // Annu. Rep. Med. Chem. - 1986. - Vol. 21. - P. 21.
20. Ronsisvalle G., Afarrazzo A, Prezzavento O. et. al. // Pure Appl.Chem. — 2001. —
Vol. 73. -P. 1499.
21. Beckett AM., Cosy AF // J. Pharm. Pharmacol. - 1954. - Vol. 6. — P. 986.
22. Portoghese Ph. S. // J. Med. Chem. - 1965. - Vol. 8. - P. 609.
23. Portoghese Ph. S., Alreja B.D., Larson D.L. // J. Med. Chem. — 1981. -
Vol. 24. - P. 782.
24. Martin W.R. // Pharmacol. Rev. - 1984. - Vol. 35. - P. 283.
25. Portoghese Ph. S. // J. Med. Chem. - 2001. - Vol. 44. - P. 2259.
26. Evens C.J. etal. // Science. - 1992. - Vol. 258. - P. 1952.
27. Xie G-X, Miyajima A, Goldstein A // Proc. Natl.Acad.Sci. USA. - 1992. -
Vol. 89. -P. 4124.
28. Wang J.B. et al. // Proc. Natl. Acad. Sci. USA. - 1993. - Vol. 90. -
P. 10230.
29. KingF.D. etal. //J. Med. Chem. - 1993. - Vol. 36. - P. 1918.
30. Wyvratt M.J., Patchett A A. // Med. Res. Rev. - 1985. - Vol. 5. - P. 483.
31. Albert A. H Nature. - 1958. - № 182. - P. 421.
32. Машковский М.Д. Лекарства XX века. — M.: ООО «Издательство Новая
Волна*, 1998.
33. Bodor N. Kaminski J.J. Ц Annu. Rep. Med. Chem . - 1987. - Vol. 22. -
P. 303.
34. Ettmayer P. etal. // J. Med. Chem. - 2004. - Vol. 47. - P. 2393.
35. Харкевич ДА. Фармакология. — M.: ГЭОТАР Медицина, 1999.
36. Goodman & Gilman's The Pharmacological Basis of Therapeutics, 9lh Ed.
/ Eds. by J.G.Hardman, L.E.Limbird. - New York: McGraw-Hill, 1996.
37. Beaumont K. et al. // Curr. Drug. Metab. — 2003. — № 4. — P. 461.
38. Noble S, Goa K.L. 11 Drugs. - 1999. - Vol. 58. - P. 479.
39. Brass EP. // Pharmacol. Rev. - 2002. - Vol. 54. - P. 589.
40. Clement B. // Drug. Metab. Rev. - 2002. - Vol. 34. - P. 565.
41. Clement В., Lopian К. Ц Drug. Metab. Dispos. - 2003. - Vol. 31. - P. 645.
42. Swann P.W., Tukker J.J. //J. Pharm. Sci. - 1997. - Vol. 86. - P. 596.
43. Han H.K. et. al. // Pharm. Res. - 1998. - Vol. 15. - P. 1154.
44. Tunek A. etal. // Biochem. Pharmacol. — 1988. — Vol. 37. — P. 3867.
45. Yoshikawa M. etal. // Hypertens. Res. - 1999. - Vol. 57. - P. 653.
46. Bradley DA. //Adv. Drug Delivery Rev. - 1996. - Vol. 19. - P. 171.
47. Lindberg P. et al. // J. Med. Chem. - 1986. - Vol. 29. - P. 1327.
48. Senn-BilfigerJ. etal. //J. Org. Chem. - 1987. - Vol. 52. - P. 4582.
49. Richards DM. etal. // Drugs. - 1983. - Vol. 26. - P. 378.
50. Hwang J.J., Marshall J.L 11 Expert Opin. Pharmacother. — 2002. — Vol. 3. —
P. 733.
51. Giles F, Estey E, O'Brien S. Ц Cancer. - 2003. - Vol. 98. - P. 2095.
52. ffamann PR. etal. // Bioconjugate Chem. - 2002. -Vol. 13. - P. 47.
53. Sievers EL. // Expert Opin. Biol. Ther. - 2001. - Vol. 1. - P. 893.
54. Springer C.J. et al. I I J. Med. Chem. - 1990. - Vol. 33. - P. 677.
308
55. Napier M.P. et al. I I Clin. Cancer Res. - 2000. - Vol. 6. - P. 765.
56 Peacock J.E., Francis G. 11 Drugs Today. — 1997. - Vol. 33. - P. 611.
57. Befield P., Sokin EM. 11 Drugs. - 1987. - Vol. 33 - P. 392.
58. Herz-Huebner U., Taeuber U. // Arzneim.-Forsch. - 1977. - Vol. 27. - P. 2226.
59. Bodor N II Pharmazie. - 2001. - Vol. 56. - P. 67.
60. Standard NASDAQ Stock Reports; Analyst Report, August 7, 1995; Pharmos
Corp. — New York: Standard & Poors, 1995.
61. Nogrady T. Medicinal Chemistry. A Biochemical Approach, 2lh ed. — New
York: Oxford University Press, 1988.
62. Bundgaard H I I Adv. Drug Delivery Rev. - 1989. - Vol. 3. - P. 39.
12
КОМБИНАТОРНЫЙ СИНТЕЗ
И ЕГО РОЛЬ В ПОИСКЕ СТРУКТУР-ЛИДЕРОВ
Вплоть до последней четверти XX в. органическая химия
обеспечивала опережающие темпы в синтезе БАВ. Лимитирующей стадией
в поиске структур-лидеров долгое время оставался скрининг.
Быстрый прогресс в молекулярной биологии и генной инженерии принес
кардинальные изменения в процесс отбора потенциальных
кандидатов в ЛС. Благодаря достижениям в этих областях были установлены
на молекулярном уровне механизмы возникновения и развития
многих заболеваний. Появилась возможность выделять биологические
мишени и использовать их для проведения скрининга новых веществ
in vitro. Получил распространение high-throughput screening. При
таком подходе к оценке активности можно исследовать соединения в
следовых (Ю-9—10'12 М) количествах. К концу 80-х гг. скорость
скрининга стала опережать скорость синтеза новых веществ. Возникла
необходимость уже в high-throughput synthesis. Данная проблема
разрешилась с появлением комбинаторного синтеза.
В связи с тем, что комбинаторный синтез как направление в
органической химии возник сравнительно недавно, некоторые понятия,
принятые в этой области, еще не получили четких определений. Сегодня
термин «комбинаторная химия» применим к стратегии синтеза
больших массивов химических соединений и определяется как
совокупность высокотехнологичных методов, позволяющих получить в
короткий промежуток времени от нескольких сотен до десятков тысяч
веществ. Комбинаторный синтез характеризуется тем, что реакции с
участием большого числа однотипных реагентов — «строительных
блоков» (building blocks — ВВ) — происходят одновременно в смеси в
одном или параллельно в разных реакционных сосудах. Существует
и другая трактовка понятия «комбинаторный синтез», согласно
которой делать что-либо комбинаторно означает, что хотя бы на одной из
стадий процесса число реакционных сосудов, в которых получают
вещества, меньше, чем число синтезируемых веществ. Однако это
определение недостаточно конкретно, и с такой дефиницией
согласны не все специалисты, особенно сейчас, когда интерес к синтезу
смесей практически утрачен [1].
Одно из ключевых понятий комбинаторной химии — библиотека
соединений, т.е. большой массив веществ, подобранных по
определенному принципу. Основная функция такой библиотеки —
обеспечить максимальное разнообразие структур, которое во многом опре-
310
деляет успех в поиске лидера. В этом смысле лучший специалист в
области комбинаторного синтеза — природа. Самой обширной
природной библиотекой являются полипептиды. Менее обширен, но все
же значителен, массив алкалоидов, стероидов и др. Библиотека
веществ — понятие для медицинской химии не новое. Крупные
фармацевтические компании за многие годы работы накопили в своем
арсенале значительные коллекции соединений, которые можно
классифицировать как по структурным особенностям составляющих их
веществ, так и по фармакологическим свойствам соединений.
Однако у таких коллекций есть принципиальное отличие от
комбинаторных библиотек, состоящее в том, что последние создаются в
кратчайшие сроки за счет одновременно и параллельно происходящих реакций
с участием реагентов, отобранных специальным образом. Для того,
чтобы создавать такие обширные массивы веществ, необходимы
специальные условия проведения процессов: автоматизация и роботизация
операций взвешивания, загрузки реактивов, выдержки в
определенных условиях большого количества реакционных сосудов,
фильтрования и т. п. При этом сами реакционные сосуды приобретают формы,
несколько неожиданные для традиционного органического синтеза:
часто ими становятся ячейки пластикового планшета,
предназначенного для проведения high-throughput screening, или хроматографиче-
ские колонки.
Новые высокотехнологичные способы комбинаторного синтеза возникли
и совершенствовались одновременно и независимо в ряде лабораторий
мира. Многие специалисты разрабатывали методы, обеспечивающие
получение большого количества соединений в кратчайшие сроки. В этом
плане обозначились несколько подходов. Так, в 1983 г. в Германии R. Frank
синтезировал нуклеотиды, а позднее пептиды, используя в качестве места
проведения реакций диски из бумаги для хроматографии (spot synthesis
concept) [2]. В 1984 г. R.A. Houghten в США получил пептиды, применив
метод чайных пакетиков («tea bags») — контейнеров из полипропиленовой
сетки, содержащих гранулы из привитого полимера на основе
полистирола [3]. В 1986 г. М.Н. Geysen в Австралии предложил способ синтеза
пептидов с использованием фукционализированных полипропиленовых
«булавок» (pin technology), погруженных в микроцилиндры, содержащие
соответствующие активированные аминокислоты [4].
Приведенные примеры свидетельствуют, что первыми необходимость
усовершенствования способов синтеза ощутили специалисты,
занимающиеся пептидным синтезом, т. к получение больших белковых молекул —
длительный и трудоемкий процесс, связанный с наращиванием
аминокислотных последовательностей. Решению этой проблемы в
определенной степени способствовала идея R. Frank использовать подходящие
мембранные материалы для помещения на них молекул субстратов, которые
в дальнейшем наращивались с помощью реагентов одного типа. При этом
разнообразие целевых пептидов обеспечивалось за счет разнообразия
исходных субстратов. В итоге получалась смесь разных веществ, которые по
отдельности снимали с твердой основы химическим путем. Первые
синтезы были проведены с помощью целлюлозных дисков. Этот материал
оказался крайне прост в работе, т. к. позволял изготовлять диски любого
311
размера, которые удобно пропитывать, маркировать, размешать в
колоночном реакторе. Однако целлюлозе присущ один недостаток, который
не имеет большого значения в пептидном синтезе, но неприемлем,
например, в олигонуклеотидном, — наличие остаточных гидроксильных
групп, способных к эстерификации.
Иное оформление имеет метод капельного синтеза (spot-synthesis), в
котором первоначально в качестве носителя также использовали
целлюлозу. По этому методу на специальной мембранной поверхности
располагают пространственно изолированные места для одновременного
синтеза большого количества вешеств. Нанесенные в эти места малые
количества жидкости образуют круглые капли. Использование
растворителя с низкой летучестью, содержащего подходящий реагент,
позволяет из каждой такой капли создать открытый «реактор» и
осуществлять превращения с функциональными группами, закрепленными на
мембранной поверхности. По завершению всей цепи превращений с
помощью специальной процедуры (реакции, облучения или др.)
вещество снимают с носителя.
Решающими для развития комбинаторной химии явились достижения в
области твердофазного (solid phase) синтеза. Этот метод связан с
применением привитых модифицированных полимеров — смол (resins),
которые первоначально использовали в качестве основы для проведения все
тех же пептидных синтезов. Впервые такой синтез осуществил R.B. Merrifield
в 1962 г., исследуя способы получения брадикинина из отдельных
пептидных фрагментов. Перенесение данной технологии на синтез «малых»
органических молекул в начале 90-х гг. стало настоящей сенсацией.
Метод получил широкое распространение, о чем свидетельствует динамика
публикаций, посвященных этому вопросу: в 1971 г. всего 10 работ, в 1998 г.
уже - 600 [1].
Оригинальную технологию с использованием модифицированных
сополимеров в форме круглых гранул (beads), помешенных в аналогичные
чайным пакетикам сеточки из полипропилена, предложил R.A. Houghten. Этот
метод упростил многие операции в синтезе пептидов и позволил подойти
к автоматизации процесса в целом. Идея упростить технологию
получения полипептидной цепи возникла у R.A. Houghten еще во время работы
над дипломным проектом в 1971 г., когда нужно было синтезировать три-
пептид и охарактеризовать все промежуточные продукты. На выполнение
задания выделялось всего 6 недель. Позднее этот исследователь
синтезировал около 20 аналогов р-эндорфина, содержащих до 30 аминокислотных
остатков. Столь трудоемкая работа стимулировала поиск новых способов
синтеза. Интересно, что при первой же попытке проверить
предложенный метод «tea bags» в лаборатории, было изготовлено 550 пакетиков для
получения 550 различных пептидов. Указанное количество было выбрано
не случайно: во-первых, оно соответствовало номенклатуре каталога по
пептидам; во-вторых, примерно столько новых вешеств могли
синтезировать сотрудники R.A. Houghten за год, используя традиционные методы.
По окончании всех операций исследователи обнаружили исчезновение
поясняющих надписей, сделанных на пакетиках химическим карандашом.
Неразличимыми оказались 300 образцов. Это пример того, что при
наличии хорошей идеи «спешить надо медленно». Однако и 250 вешеств,
которые удалось выделить, оказалось вполне достаточно, чтобы
продемонстрировать преимущества данной технологии [3].
312
Вероятно, комбинаторный синтез не получил бы такого
распространения, если бы, в свою очередь, не получила подтверждения и развития
гипотеза относительно возможности проведения скрининга не среди
индивидуальных веществ, а используя смеси. Одним из авторов этой идеи
был все тот же R.A. Houghten. В 1984 г. он пришел к мысли, что смеси
веществ являются естественным окружением любой биологической
мишени, и если создать определенным образом смесь и провести
биологические испытания на предмет способности ее компонентов связываться с
мишенью, то в результате будет выявлено самое активное вещество,
несмотря на присутствие остальных соединений в смеси [5, 6, 13].
Требования, предъявляемые к соединениям, предназначенным для
скрининга, претерпели своего рода эволюцию. Основные этапы этой
эволюции четко прослеживаются во временном аспекте. Так, до 1984 г.
представления сводились к возможности исследования только
индивидуальных высокоочищенных соединений, а в период 1984—85 гг. уже
рассматривалась возможность скрининга индивидуальных неочищенных
пептидов, полученных параллельным синтезом, но не гетероциклических
соединений или каких-либо других традиционных продуктов
органического синтеза; в 1986—90 гг. вызвала интерес идея проведения
биологических испытаний смесей, однако твердой уверенности в
возможности ее практической реализации не было. В 1991 г. было показано, что
скрининг смесей реален, но только для пептидов, а итерационный
процесс разделения очень длителен; в период 1992—94 гг. получило
подтверждение предположение о возможности позиционного сканирования
смесей пептидов, пептоидов и пептидомиметиков, однако эта
процедура распознавания активных соединений в смесях считалась
неприемлемой для лекарствоподобных (drug-like) молекул. В 1995—96 гг.
установлено, что смеси пригодны и для испытаний гетероциклов, но это должны
быть маленькие смеси, обычно меньше 30 компонентов; в 1997 г.
получило подтверждение позиционное сканирование больших смесей
гетероциклических и других drug-like молекул, а в 1999—2001 гг. наиболее
результативным был снова признан скрининг больших библиотек
индивидуальных соединений, а не смесей [1, 6].
Первые работы, раскрывающие возможности использования
методологии комбинаторного синтеза применительно к малым
органическим молекулам, были опубликованы в 1992—93 гг. J.A. Ellman и
S.H. DeWitt с соавт. [1, 7]. Сегодня, когда еженедельно выходят
десятки статей, посвященных комбинаторной химии, и Американское
химическое общество издает специализированный журнал — «Journal
of the Combinatorial Chemistry», кажется невероятным, что всего лишь
несколько лет назад демонстрации ведущими фармацевтическими
компаниями технологий создания и проверки химических библиотек
встречали только скептицизм со стороны большинства специалистов, и
рассматривались скорее как курьез, а не как приемлемые для
промышленного использования. В настоящее время в программу
химических факультетов многих университетов включены специальные
курсы по комбинаторному синтезу. Первый такой курс был прочитан
в 1996 г. в университете Луисвилля (США) проф. AF. Spatola [1].
313
12.1. Стратегия конструирования и синтеза
химических библиотек
Суть комбинаторного подхода состоит в разработке методов
ускоренного синтеза веществ близкого строения. Основной принцип можно
проиллюстрировать на примере, представленном на схеме 12.1. Так,
если продукт АС образуется в результате взаимодействия реагентов А
и С, то одновременное использование 10 соединений А1../..А10 и
С1 С10 обеспечит получение уже не одного, а 100 разных веществ
однотипного строения.
Схема 12.1
А1 + 0=А1С\А1О А'С10
А2 + С2 = А2С\ А2С2 А2С10
А10 + С1 = А10С, А10С2 А10С
ю
Самая простая демонстрация такого подхода — одновременно
поместить в раствор несколько реагентов с целью получения всех
возможных продуктов, например, синтез 100 оснований Шиффа из
10 альдегидов и 10 аминов (схема 12.2).
Схема 12.2
R1 —^ + H2N-R2 *- R1 N
юН ю юо
Однако такой подход часто непродуктивен, т.к. по причине
различной реакционной способности первоначально взаимодействуют
самые активные реагенты, образуя объемные продукты. Остальные
же будут присутствовать в незначительных количествах, и такую
смесь трудно разделить. Тем не менее, этот метод используют, когда
скорости реакций примерно одинаковы. Именно таким способом
была создана библиотека из 97 000 соединений на основе реакций
тетрахлорида 9,9-диметилксантен-2,4,5,7-тетракарбоновой кислоты со
смесью аминов. Идентификация активных соединений из этой
библиотеки требовала повторного синтеза и их повторной
биологической проверки [8, 9].
Если перенести рассмотренный принцип на многостадийные
превращения, то при участии 10 реагентов 4-х типов А1 А10, В1 В10,
С1 С10 и D1 D10 всего в 4 стадии можно получить 10 000
соединений (схема 12.3).
Схема 12.3
I В1 В10 Ю С10 iD1 D10
А1 ... .А10 »- АПВП *■ АПВПСП »- AnBnCnDn
100 1000 10 000
314
Так, из 20 природных аминокислот можно получить 3 200 000
пентапептидов (205 = 3200000). Количество веществ на каждой стадии
будет определяться количеством реагентов, а общее количество
соединений в библиотеке еще и числом стадий [8].
Процесс создания библиотеки веществ в целом может
рассматриваться как итерационная процедура, когда одна библиотека
становится основой для создания новой, и состоит из следующих
этапов [9]:
• планирование библиотеки;
• отбор В В;
• химическая апробация синтеза в растворе или на твердом
носителе и контроль качества продукта;
• синтез библиотеки;
• биологический скрининг;
• идентификация активных соединений;
• подтверждение активности;
• интерпретация результатов.
При планировании библиотеки, в первую очередь, определяют
цель проведения скрининга. Если осуществляется подбор лигандов
к мишени, строение которой неизвестно, и нет сведений о
структуре соединений, способных с ней связываться, то это будет
«скрининг наугад» — random screening. Такой скрининг предполагает
проверку активности среди большого разнообразия структур и в
результате будет получена огромная библиотека или совокупность
нескольких более мелких, т. е. скрининг сведется к выявлению
активности в океане «неактивности». В противоположность этому,
целевые библиотеки содержат родственные структуры,
различающиеся только заместителями, и имеют гораздо меньший размер
по сравнению с массивами для broad или random screening. При
этом все члены библиотеки, как правило, обладают активностью,
и выбор сводится к идентификации наиболее аффинных среди них.
Способы конструирования библиотек этих двух типов различаются
количеством используемых ВВ, размером носителей, стратегией
декодирования (табл. 12.1).
Таблица 12.1
Характеристика библиотек химических соединений [9]
Библиотека для broad скрининг
Большой размер библиотеки
(от 10 до 100 тыс. соед.)
Широкое разнообразие структур
Нет специфической структурной
цели
Большое количество разнообразных ВВ
Неопределенное количество
комбинаций
Библиотека структур-аналогов
Небольшой размер библиотеки
(до 1000 соед.)
Однотипные структуры
Специфическая структурная цель
известна
Однотипные ВВ
Известно число комбинаций
315
Следующий важный этап в подготовке библиотеки — отбор ВВ.
Обычно исходят из того, что мол. масса веществ, образующих
массив, должна находиться в пределах 350—750 (см. разд. 7.3). Общее
количество соединений с массой < 750 можно оценить как 10200.
Отсюда очевидно, что потенциал комбинаторной химии офомен,
получить все эти соединения и проверить их активность на
сегодняшний день представляется маловероятным. И все же, если
предположить, что масса отобранных ВВ будет равна 150, то целевые
соединения можно будет «собрать» в 2-4 стадии из 2-5 фрагментов
(схема 12.4).
Схема 12.4
в с
При выборе ВВ путем ретросинтеза определяют их доступность и
реакционную способность, необходимость их синтезировать серийно
или с помощью параллельного синтеза. Большинство ВВ — это
амины, карбоновые кислоты, ароматические альдегиды и т.п. Удачным
является такой набор ВВ, который обеспечивает максимальное
разнообразие заместителей в целевых соединениях и все возможные
варианты их пространственного расположения. Учитывают также
физико-химические характеристики.
Теоретически количество соединений в библиотеке определяют
числом ВВ, участвующих в каждой стадии, и числом отдельных
стадий. Если ВВ = Ь, а стадий х, то общее количество веществ N=b\
Если число ВВ на каждой стадии различно (Ь, с, d в 3-х
стадийном синтезе), то N=bcd. Однако, как уже отмечалось, при
значительных различиях в реакционной способности участвующих в
синтезе ВВ, в первую очередь образуются продукты из наиболее
химически активных реагентов, и концентрация таких соединений
будет выше. Поэтому подбор ВВ осуществляют таким образом,
чтобы в результате образовались эквимолярные количества целевых
продуктов. При проведении синтеза в одном реакционном сосуде
(one pot synthesis) оптимальной для скрининга, по мнению
специалистов, является смесь из 20 веществ. В процессе
усовершенствования структуры лидера скрининг смесей вообще неприемлем. Для
таких целей создаются библиотеки индивидуальных соединений с
количеством < 1000 веществ [9, 10].
При разработке стратегии синтеза важным является определение
типа реакций, с помощью которых будут «собраны целевые
конструкции». В настоящее время в комбинаторной химии используется
316
довольно большой набор реакций. К ним относятся: циклоприсо-
единение [3+2], [2+2], [4+2]; конденсация; фоторасщепление;
окисление/восстановление; реакция Михаэля; нуклеофильное замещение;
восстановительное аминирование; алкилирование -N, S, О, С; метал-
лоорганический синтез; реакция Мицунобу; гидролиз; фосфорили-
рование; ацилирование -N,0, при чем число реакций постоянно
увеличивается.
Наиболее ресурсоемкой частью в создании библиотеки
является апробация получения соединений, которую обычно проводят с
помощью параллельных синтезов. В случае твердофазного синтеза
на этом этапе подбирают линкеры и проверяют адаптирован ность
полимерного носителя к выбранным ВВ, т. е. определяют характер
взаимодействия ВВ как с функциональными фуппами
модифицированного полимера, так и с аналогичными лигандами в растворе.
Кроме того, оптимизируют условия проведения реакций с учетом
свойств носителя и линкеров. На сегодняшний день твердофазный
синтез индивидуальных органических веществ так же хорошо
развит, как и синтез полипептидов. Схематически этот процесс
представлен на рис 12.1.
0w^| Лингёр~1-Д»-+^| Линкер ]—д—»-#w^| Линкер |—ДВ—*- +^[~Щшкер ( + АВ
Рис. 12.1. Твердофазный синтез малых молекул на полимерном носителе
• — полимерная гранула (resin bead), А и В — ВВ
Методы формирования многокомпонентных смесей при
твердофазном синтезе хорошо отработаны только для ограниченного
набора веществ, таких как амиды, пептиды и нуклеотиды. В случае
получения смесей малых органических молекул на твердом
носителе большое значение приобретают различия в реакционной
способности взаимодействующих реагентов и в скоростях протекающих
процессов.
Проблема различия в скоростях реакций нашла оригинальное
разрешение в методе расщепленного синтеза (split method),
позволяющем сформировать смесь из эквимолярных количеств веществ
[11—13]. Схема такого синтеза представлена на рис. 12.2. Так,
набор фанул твердого носителя разделяют на несколько равных
частей и помещают на них индивидуальные ВВ А1....А3, которые
ковалентно связываются с линкерами. После этого все фанулы
объединяют и вновь делят на 3 равные части, в результате чего
образуются 3 смеси, в которых связанные с линкерами ВВ присутствуют
в эквимолярных количествах. К каждой фуппе прибавляют
реагенты В1, В2 и В3 соответственно. При этом в реакционном сосуде
присутствует только один реагент, который взаимодействует с
закрепленными на носителе субсфатами. На каждой фануле формируется
индивидуальный продукт (one bead, one compound). Получают
3 фуппы веществ по 3 соединения в каждой. Перед прибавлением
317
ВВ С1....С3 процедуру смешивания гранул и последующего их
разделения на три равные части повторяют. Таким способом,
используя 9 ВВ, в три стадии можно получить 27 новых соединений. Для
раздельного синтеза этих веществ потребуется 81 стадия.
В1
А1В1
А1 В1
А1 В1
С1
-А1 В1 С1
-А2В1С1
-А3В1С1
-А1В2С1
-А2В2С1
-А3В1С1
-А1В3С1
-А2В3С1
-А3В3С1
В2
•—А1 В1
•—А1 В1
•—А1 В1
С2
#—А1В1С2
#—А2В1С2
#—А3В1С2
#—А1В2С2
#—А2В2С2
#—А3В2С2
#—А1В3С2
#—А2В3С2
•—АЭВЭС2
С3
■А3
-А1 В1
-А* В1
А1 В1
■А1В1С3
-А2В1С3
■А3В1С3
-А1В2С3
-А2В2С3
-А3В2С3
-А1В3С3
-А2В3С3
-А3В3С3
Рис. 12.2. Схема расщепленного синтеза
Синтез в растворах отличается от обычного органического
синтеза только аппаратурным оформлением и высокой степенью
роботизации. Это может быть как синтез индивидуальных веществ
(п соединений синтезируется в п реакционных сосудов), так и
смесей, когда число веществ намного больше, чем число реакторов, в
которых их одновременно получают. Стоит отметить, что
последний способ имеет, скорее, историческое значение и в настоящее
время мало используется.
Каждый из методов синтеза библиотек на твердом носителе и в
растворе имеет свои преимущества и недостатки. Сравнение этих
методов приведено в табл. 12.2.
318
Таблица 12.2
Сравнительная характеристика
жидкофазного и твердофазного синтеза
Синтез на твердом носителе
Синтез в растворе
Преимущества
Реагенты можно использовать в
избытке, не опасаясь впоследствии
проблем разделения. При этом
реакции идут до конца
Очистка продуктов осуществляется
промывкой гранул носителя
Простота автоматизации процесса
синтеза
Возможность проведения
расщепленного синтеза
Можно использовать любые
органические реакции
Не требуется адаптация реагентов к
новым условиям проведения процессов
Отсутствие дополнительных стадий,
связанных с присоединением к
линкеру и расщеплением линкера
Возможность получения
неограниченных количеств веществ
Недостатки
Нет точных данных о затратах
времени на проведение синтеза
Наличие дополнительных стадий по
связыванию с линкерами и их
расщеплению
Носители и линкеры ограничивают
использование широкого набора
реакций
Недостаточно развиты методы
контроля протекания процессов
Невозможно использовать реагенты
в избытке из-за сложностей очистки
целевых продуктов от примесей
исходных реагентов
Автоматизация процессов выделения
веществ и их очистки представляет
определенные сложности
12.2. Комбинаторный синтез в растворах
В этом направлении комбинаторного синтеза существует две
стратегии — синтез смесей и параллельный синтез индивидуальных
веществ. Последний, несмотря на то, что приводит к формированию
небольших коллекций соединений, имеет существенные преимущества
позволяет легко охарактеризовать полученные продукты (используя
традиционные методы установления строения органических веществ)
и проводить скрининг индивидуальных соединений. При синтезе
смесей возникают значительные трудности с идентификацией
продуктов.
12.2.1. Одностадийные методы синтеза
Примером формирования эквимолярных смесей в одну стадию
служат так называемые индексные библиотеки, созданные М.С. Pirrung
с соавт. [14, 15] из 72 тетрагидроакридинов (схема 12.5) и 54 карбама-
319
тов (схема 12.6). Так, синтез акридинов осуществляли следующим
образом: к стехиометрическому количеству цианоанилина
прибавляли эквимолярную смесь из 12 циклогексанонов. И наоборот —
каждый из кетонов взаимодействовал со смесью из 6 анилинов. Таким
путем было получено 6 смесей по 12 веществ и 12 смесей по 6.
Каждый из продуктов встречался в 18 смесях дважды. Если вещество
проявляло биологическую активность, то оно должно было быть
выявлено дважды.
Схема 12.5
6 цианоанилинов 12 симметричных 72 тетрагидроак идина
циклогексанонов
Этот же метод был использован при поиске ингибиторов ацетил-
холинэстеразы среди производных карбаминовой кислоты [16].
Схема 12.6
R1
О
А_ ♦
CI
R2-NH2/R2-OH
О
R1>^(NH,0)R2
40 хлорангидридов 40 аминов/спиртов
кислот
1600 амидов/эфиров
12.2.2. Двухстадийные методы синтеза
R. Storer [8] создал библиотеку из 160000 диамидов, полученных
в результате взаимодействия 50 аминокислот, 80 карбоновых
кислот и 40 аминов. Каждую из 50 аминокислот вводили в реакцию
со смесью из 40 аминов. Полученные в результате 50 смесей по
40 веществ реагировали с каждой из 80 карбоновых кислот.
Соединения образовывали 4000 смесей по 40 компонентов каждая. В
отличие от индексной библиотеки, вещества в этом случае не были
получены дважды. Однако, если при скрининге какая-либо смесь
давала положительный результат, то для дальнейшей проверки все
40 индивидуальных компонентов синтезировали раздельно. В
рассмотренной библиотеке диамидов выявлены структуры-лидеры,
обладающие сродством к ВИЧ-1-протеазе и рецепторам нейроки-
нина-1.
320
12.2.3. Трехстадийные методы синтеза
Примером многостадийного комбинаторного синтеза в растворе
является последовательность реакций (схема 12.7), в результате
реализации которой получена библиотека из 125 пиразолов (5 смесей по
25 веществ в каждой). Успеху способствовал тщательный выбор
условий проведения процессов и продуманный отбор ВВ. Так, например,
аллилбромид, из-за неизбежного протекания перегруппировки Кляй-
зена, не использовали в качестве О-алкилирующего агента [8].
Схема 12.7
O-^OEt
Et О
+
oet о
+
(N)
1) гидролиз 0^^,0-R 3
2) амидирование 1
О. .R2 и™ <AV'0^R2
о-алкилирование \V J
<v
N-N,
^
_ |4j|_| 5 алхилирующих
агдатов —
5 соединений
R1
5 вариантов —
125 соединений
5 арилгидразинов
12.2.4. Однореакторные методы синтеза
(one-pot synthesis)
J. Rebek с соавт. [17, 18] для создания библиотеки предложили
многоступенчатую последовательность реакций. В качестве
молекулярных остовов (scaffolds, centroids), содержащих несколько
однотипных реакционных центров, авторы использовали тетрахлориды ксан-
тен- (12.1), кубантетракарбоновых (12.2) кислот и трихлорангидрид
(12.3), а в качестве ВВ — эфиры аминокислот.
CI CI
(12.1) (12.2) (12.3)
Первые два остова представляют собой достаточно жесткие
конструкции, а производные (12.3) являются конформационно более
гибкими. Продукты на основе хлорангидридов (12.1) и (12.3) обла-
321
дают плоской симметрией, а производные (12.2) — сферической.
При этом реакционные центры во всех случаях расположены с
учетом минимального взаимного стерического влияния. В
результате реакции тетрахлорида ксантентетракарбоновой кислоты с 4, 7,
12 и 19 аминными компонентами теоретически образуется смесь
из 136, 1225, 10440 и 65341 веществ. Особый интерес вызывает
вопрос об абсолютно исчерпывающем синтезе всех возможных
продуктов реакций. Получить ответ на него удалось с помощью масс-
спектров. Однако в связи с тем, что масс-спектрометрический анализ
смеси из нескольких тысяч веществ — задача достаточно сложная,
было принято решение создать модельную библиотеку на основе
дихлорангидрида ксантендикарбоновой кислоты (12.4).
(12.4)
Теоретически на основе дихлорангидрида (12.4) с участием 8, 9 и
10 эфиров аминокислот можно получить 36, 45 и 55 диамидов. Масс-
спектрометрически в 36-компонентной системе были обнаружены
пики молекулярных ионов 85% ожидаемых продуктов, в 55-компо-
нентной — уже только 78%. Этот эксперимент позволил выявить
«проблемные» ВВ среди аминных компонентов. Так, если из набора ВВ
исключить производные аргинина, то в 55-компонетной смеси
идентифицируются 95% ожидаемых продуктов. Следовательно, для того,
чтобы большая часть предполагаемых соединений присутствовала в
синтезированном массиве веществ, необходимо тщательно отбирать
ВВ. В то же время анализ показывает, что 10% библиотеки составляют
побочные продукты.
Метод one-pot reaction позволяет получать очень большие
библиотеки веществ в виде смесей. Однако для этого пригоден
ограниченный набор реакций и ВВ. В настоящее время описанный метод
становится все менее популярным и постепенно утрачивает свое
значение.
12.2.5. Тандемные реакции
Особое место в комбинаторном синтезе занимают тандемные
реакции. При таком типе взаимодействия реакционная
последовательность берет начало после того, как смешаны все компоненты.
Отличие тандемных процессов от рассмотренных выше многокомпонентных
реакций состоит в реализации такого каскада превращений в смеси,
322
при котором продукты одной реакции становятся исходными
реагентами для другой. Самым простым примером тандемного превращения
служит синтез оснований Манниха. Пионером в разработке этого
направления в приложении к комбинаторному синтезу является I. Ugi.
Изучением многокомпонентных реакций этот исследователь занялся
еще в 1961 г., задолго до того, как возник комбинаторный синтез. Его
заинтересовала возможность получения большого количества
продуктов в смеси из 4 (в исключительных случаях более 4) реакционных
компонентов. I. Ugi с соавт. [19, 20] исследовали взаимодействие трех
аминокислот с тремя изонитрилами и тремя альдегидами в метаноле
(схема 12.8) и в результате получили библиотеку, теоретически
состоящую из 54 соединений (12.5), с учетом стереоизомеров. Масс-спектро-
метрически в ней было подтверждено наличие только 37 веществ.
При более тщательном подборе реагентов на основе этой же реакции
синтезированы три дочерние библиотеки по 18 веществ в каждой.
Таким образом, все 54 предсказанных вещества были получены, а
рассмотренная четырехкомпонентная конденсация получила
название реакции Ugi.
R3 CH,OH H
Схема 12.
R2 R3
R1_NC + R2^0 + h2N^COOH И Н
~ R1' V 'ГГ >Г ^Ме
(12.5)
Как и one-pot synthesis, метод тандемных реакций открывает
возможности для создания больших библиотек. Однако следует отметить, что
концентрации веществ в смеси, полученной по реакции Ugi, различны.
Многолетняя работа по совершенствованию этого метода позволила
авторам подобрать реагенты с одинаковой реакционной способностью
и, следовательно, получать продукты с удовлетворительным выходом.
В результате была создана библиотека производных пиридиндикар-
боновой кислоты (12.6), содержащая 8256 соединений (схема 12.9) [21].
Схема 12.9
НООС
2R1 —NC + 2R2 —СО + 2R3—NH2 +
R2 О О R2
НООС
О R3 Ч^ R3 °
(12.6)
323
12.2.6. Параллельный синтез
индивидуальных соединений
Учитывая недостатки комбинаторных библиотек, состоящих из смеси
веществ (присутствие трудно идентифицируемых примесей, разная
концентрация компонентов, сложности в распознавании активных
соединений), ряд исследователей развивают методы ускоренного синтеза
индивидуальных веществ. При этом также широко применяются
многокомпонентные реакции. Например, библиотека из 64 а-ацилоксиами-
дов (12.7) синтезирована путем комбинации 8 альдегидов, стольких же
карбоновых кислот и изонитрилов (схема 12.10). В каждой параллельной
реакции один эквивалент кислоты и изонитрила взаимодействовал с
двумя эквивалентами альдегида при комнатной температуре. Целевые
соединения выделяли путем испарения растворителя. Анализ продуктов
реакции методом ЯМР 'Н подтвердил, что степень превращения
исходного изонитрила — 80%. Образование стереоизомеров привело к
необходимости хроматографического разделения продуктов [22],
Схема 12.10
R1
О
X.
R2
О
л
он
о
Л.
R2' 'О О
I Н ll„0-Et
^k J*L .PC
- R1 ]|^ ^ O-Et
О
(12.7)
R3—СНО
+ ^^ .NC
HCI R1
R1-COOH
R2-NH2 cH3OH ?2 Q
Y т и
О R3
Схема 12.11
R2 О
О R3
R2 О
RK ^N
R4-Ofl " 'у
О
I
О R3 R4
R2 О
R4-SH R1
на
R3
'К
Y
О R3 R4
R4. Я5
R4 = R5 R-Г ^N^ ^R3
I
R2
(12.8)
324
Интересное развитие реакции Ugi в приложении к синтезу
индивидуальных веществ было предложено R.W. Armstrong и
ТА. Keating [23]. С помощью четырехкомпонентной конденсации
с участием разных кислот, аминов, альдегидов и одного цикло-
гексенизонитрила исследователи получили циклогексенамиды (12.8).
Последние, в свою очередь, стали источником четырех типов
структур благодаря участию в целом ряде реакций гидролиза, алкоголи-
за, (3+2)-циклоприсоединения и т.п. (схема 12.11).
12.2.7. Классические жидкофазные реакции
Наиболее очевидный способ комбинаторного синтеза
индивидуальных соединений — это параллельное проведение хорошо
известных реакций, например, конденсации Кневенагеля с участием
оксазолонов и альдегидов или раскрытия оксазолонового цикла нук-
леофилами (схема 12.12) [24, 25].
Схема 1212
R1
+ R2-CHO
R2
N—f-R3
R1—^ >=0
О
+ NuH
R2^ ^R3
Nu
О
Таблица 123
Соединения, используемые в параллельном синтезе
Субстраты
ROH
RSH
RNH2
RCONH2
RNHNH2
RMgX
RCOR
Реагенты
RCOCI
ROCOCI
R2NCOCi
RS02CI
RCN
RCH2X
RCHO
RCOOH
RCOR
Продукты
эфиры, (тио-)эфиры, спирты, альдоли, (тио-)
карбаматы, (тио-)карбонаты, амиды, амины,
тиосульфаты, уретаны, мочевины, гидразоны,
кетоны, сульфонамиды, сульфоны
Реакция Манниха также широко применяется для
проведения параллельного синтеза индивидуальных веществ в раство-
325
pax. С ее помощью создана библиотека 9000 аминофенолов исходя
из формальдегида, фенолов и вторичных аминов. Каждое вещество
было получено в количестве 20 мг, степень чистоты — 80% [8].
Основным требованием к реакциям, отбираемым для проведения
параллельного синтеза, является высокий (желательно
количественный) выход целевых продуктов. В табл. 12.3 приведен перечень ВВ,
которые обеспечивают удовлетворительные выходы продуктов
благодаря своей высокой реакционной способности.
12.2.8. Способы очистки реакционных растворов
от примесей
Часто для того, чтобы реакция прошла до конца, используют
избыток одного из реагентов, от которого по окончании процесса
необходимо избавиться. Для таких целей применяют реагенты-секвест-
ранты или «чистильщики». При этом существует два подхода. Первый
связан с разделением веществ между двумя несмешивающимися
слоями жидкости: в один слой переходят целевые соединения, в другом
остаются побочные продукты и непрореагировавшие исходные
реагенты. Второй подход представляет собой сочетание технологий
синтеза в растворах с твердофазным синтезом; «чистильщики»
закрепляют на полимерном носителе, добавление их в реакционную смесь
позволяет связать избыток исходных веществ или побочные
продукты, а после взаимодействия с примесями они легко удаляются из
раствора фильтрованием [26]. Например, полимеры, содержащие
аминогруппы, позволяют убрать из растворов электрофильные реагенты
(табл. 12.4). Так, доступные аминоэтилполистиролы эффективно
связывают альдегиды, имины, изоцианаты, изотиоцианаты, сульфохлориды,
хлорангидриды и ангидриды кислот. Для удаления из реакционной
смеси таких нуклеофилов, как тетра-бутиламмонийфторид (ТБАФ),
используют ионообменные смолы, содержащие сульфогруппы (ам-
берлит А-15) или их дикальциевые соли. Полимер с функцией мети-
лизоцианата эффективно удаляет из растворов избыток первичных и
вторичных аминов, а также гидразинов [27]. Амберлит IRA-900,
содержащий высоко основные четвертичные гидроксиаммонийные
группы, применяют для связывания слабых кислот — фенолов, гидрокси-
пиразолов и т.п. Для удаления побочных продуктов кислотного
характера, образующихся при ацилировании аминов, используют ани-
онообменный амберлит IRA-68 [26].
В тех случаях, когда минорные продукты реакции не содержат
ионогенных групп, очистку реакционной смеси проводят с
использованием высоко реакционноспособных бифункциональных реагентов
(sequestration-enabling-reagents), которые превращают трудно
удаляемые побочные продукты в легко секвестируемые. Так, при гидролизе
в реакциях с участием тиазолилизоцианата в качестве
нежелательного побочного продукта образуется аминотиазол (см. табл. 12.4).
Взаимодействием с фенилсульфонилизоцианатом его превращают в амино-
326
<и
£
2L
8.
о
ю
о
3
S
Г
Тип реакции
Секвестирующий реагент
Реагенты
аза-реакция
Дильса -Альдера
с*
I
21
i
2
образование (тио)мочевины,
ацилирование
ъ
xw
2
i
изоцианаты
сульфогалогениды
хлорангидриды
ангидриды
алкилгалогениды, альдегиды
многокомпонентные синтезы
тиазолидинонов
i
(П
■Z.T.
I
(О
и
О
1 + <о
а.
°<
см
а.
реакции снятия силильной
защитной группы
ТБАФ
синтез мочевин
синтез пиразолов
о
\\
о
Z
ft
нуклеофильное
присоединение
к тиазолил-2-изоцианатам
i „
1 - 1
О X Г
\ *={
1 wl
1,-
А
а.
St
О
\
А
327
тиазолил-Ы-сульфонилмочевину. Последняя легко удаляется
благодаря наличию в структуре NH-группы, обладающей слабыми
кислотными свойствами. Добавление в реакционную среду основной поли-
аминной смолы обеспечивает эффективное удаление как изоцианата,
так и мочевины. Примеры sequestration-enabling-reagents приведены в
табл. 12.5.
Использование закрепленных на полимерном носителе секвест-
рантов имеет ряд недостатков. В частности, взаимодействие с
примесями из реакционных растворов происходит в гетерогенной
среде. Кинетика протекания таких процессов трудно предсказуема,
поэтому очистка не всегда происходит эффективно. Попытка
обойти эти трудности привела к созданию растворимых секвестрантов
(tagged reagents). Такие реагенты должны взаимодействовать только
с побочными продуктами и быть инертны в условиях реакции. По
окончанию процесса и избыток tagged reagent, и связанный с ним
побочный продукт легко удаляются путем инкубирования
реакционной смеси с подходящей ионообменной смолой. Примеры tagged
reagent приведены в табл. 12.6.
12.2.9. Синтез
с использованием растворимого полимера
Попытка объединить преимущества метода проведения реакций в
растворах с возможностями твердофазного синтеза привела к
использованию в качестве носителя полиэтиленгликоля (ПЭГ) [8, 28]. Этот
полимер растворим в большинстве органических растворителей,
однако может быть легко осажден добавлением избытка диэтилового
эфира. Реакционная способность связанных с ПЭГ субстратов
изменяется в широких пределах, и реакции с участием таких реагентов
мало отличаются от обычных реакций в растворах (схема 12.13). При
этом процедура очистки целевых продуктов от исходных реагентов
существенно упрощается благодаря хорошей способности ПЭГ-про-
изводных к кристаллизации. Примером библиотеки, полученной
таким путем, является коллекция сульфамидов (12.9).
Н,СО -ПЭГ -ОН
SO.CI °
А,/-
Схема 12.13
R-NH,
г. JL /=\ т\-мп2
й НзС°-пэг-°^йЧ>^с| ^е
u ™ ^ ~Jx /=\ °.5 N Na0H /^K
Н3СО-ПЭГ-СК n / \_S02.NHR ^ h2N-^^— S02-NHR
0=N
О
(12.9)
328
S.
о
§
I
I
В
<и
3
о.
Г
ZX
■Ч
ZI
>°
XZ
чх 5
2
12
S
3
и
I
с*
| х Э
X
о
н
т
S
о
о.
ю
\=|
Z
ГЧ
X
У
т
xz
/ \
329
Методы, связанные с использованием растворимого носителя, не
могут составить конкуренцию твердофазному синтезу пептидных
макромолекул. Однако они весьма удобны для получения малых
органических молекул.
12.3. Комбинаторный синтез на твердом носителе
Твердофазный синтез, как уже отмечалось выше, возник в
начале 60-х гг., когда R.B. Merrifield [37] предложил новый метод
наращивания пептидных последовательностей. Основная идея
заключалась в том, что первый аминокислотный остаток в такой
последовательности должен быть связан с нерастворимым
полимером В то время иммобилизацию на твердом носителе
«строящейся* белковой молекулы осуществляли введением в полистирол хлор-
метильных групп, которые использовали для алкилирования
карбоксильной группы первого остатка аминокислоты (схема 12.14).
Каждый новый остаток вводили с помощью повторения двух
операций" а) снятия аминной защиты; б) связывания со следующим
защищенным по аминогруппе остатком кислоты под действием цикло-
гексилкарбодиимида.
Схема 12.14
О
>
НО
и
н
N-Y
На всех этапах реакционная система оставалась гетерогенной, а
растущий биополимер - связанным с нерастворимым носителем. Этот
метод позволил значительно снизить потери на стадиях выделения и
очистки. После создания необходимой последовательности
аминокислот, полученный пептид снимали с полимера с помощью
восстановительного расщепления, а затем очищали хроматографически.
Поскольку такой синтез проводят без очистки интермедиатов,
выходы на каждой стадии должны быть очень высоки. Иначе, если на
какой-либо стадии аминокислотный остаток не введен, в результате
будет получен пептид с нарушенной последовательностью. Так, при
синтезе цепи, содержащей 25 остатков с достоверностью введения
каждой единицы 99% требуемую аминокислотную последовательность
имеют только 75% конечных цепей. Можно предположить, что при
синтезе цепи из 100 остатков, если каждая последовательность имеет
хотя бы один дефект, выход «совершенного» пептида крайне низок.
Поэтому современные методы предполагают синтез отдельных
коротких цепей, а затем «сборку» их в более длинный полимер. Для
комбинаторных библиотек правильность последовательности не име-
331
ет такого значения, т.к. распознавание часто проводится после
скрининга, когда уже отобраны наиболее активные образцы.
В настоящее время существуют достаточно широкие
возможности для твердофазного синтеза малых молекул. Главное
достижение в развитии полимерных материалов для твердофазного синтеза
связано с именами Е. Bayer и W. Rapp, которые создали TentaGel™
(Rapp Polymer) — привитой на полистирольную основу
полимер ПЭГ (12.10). В середине 80-х гг. он нашел широкое
применение в пептидном синтезе. В настоящее время из этого материала
изготовляют мельчайшие гранулы (до 136 мкм в диаметре, 106
гранул/г), на которых и проводят «сборку» молекул органических
веществ из соответствующих В В [8, 9].
TentaGel™ (Rapp Polymer)
V-( у-[о-сн2-сн2]68-х
X = -NH2, -ОН, -SH, -СООН, -Вг
(12.10)
Полистирольная матрица является базовым полимером. ПЭГ-фраг-
мент выполняет роль линкера между матрицей и следующим
линкером, удерживающим на носителе «строящуюся» целевую молекулу.
Прогресс в этой области в значительной мере связан с появлением
большого разнообразия линкеров, пригодных для синтеза как
пептидных молекул, так и малых лекарствоподобных структур. Вид
конструкции, полученной в результате твердофазного синтеза,
представлен на рис. 12.3.
/ч н ^yyS
I I I I
Матрица пэг
из линкеп Фотолинкер Лиганд
полистирола р
Рис. 12.3. Лекарствоподобная молекула,
связанная с полимерным носителем
332
Me
Me
F
Кроме Rapp Polymer, в твердофазном синтезе нашли
применение такие смолы, как Affymax, Dowex 50W, Rink полимер
и многие др. [8, 9].
12.3.1. Линкеры
Направление в твердофазном синтезе, связанное с поиском
линкеров, возникло из химии защитных групп. Линкеры должны
удовлетворять следующим требованиям:
— быть доступными (простота синтеза и дешевизна);
— обеспечивать надежную связь с привитым полимером;
— легко расщепляться в мягких условиях, не затрагивая основную
молекулу;
— при расщеплении не допускать образования побочных продуктов
(«бесследные линкеры»);
— быть пригодными для синтеза с использованием самых разных ВВ
и для широкого набора реакций;
— обеспечивать возможность многократного использования.
Кроме того, желательно, чтобы линкеры имели структуру,
существенным образом не влияющую на спектральный мониторинг
реакций, происходящих на твердом носителе [29, 30].
Линкеры принято классифицировать по способу расщепления
связи, с помощью которой целевой продукт закрепляется на
полимерном носителе.
Кислотно-расщепляемые линкеры — наиболее распространенный
вид линкеров в пептидном синтезе. В настоящее время данный
вид линкеров нашел применение для закрепления на смоле
различных классов органических соединений: бенздиазепинов, ами-
нотиазолов, спиртов, амидов, производных гидроксамовой и кар-
баминовой кислот, мочевин, сульфонилмочевин, аминов, амидинов
и сульфамидов [29—31]. При планировании синтеза необходимо
учесть, что сильно кислая среда на какой-либо из стадий может
привести к преждевременному отщеплению еще «недостроенного»
вещества. Кроме того, если целевой продукт неустойчив в
кислой среде, то для его закрепления на носителе также нельзя
использовать кислотно-расщепляемый линкер. Долгое время самым
популярным был предложенный G.T. Wang и-оксибензиловый
линкер. J. Ellman для закрепления спиртов использовал тетрагидропи-
рановый линкер (схема 12.15), который в настоящее время
применяют в синтезе пуринов, тетразолов, производных гидроксамовой
кислоты [30]. Расщепление осуществляют воздействием трифтор-
уксусной кислоты (ТФУ).
333
Схема 12.15
ТФ5^
СН,С1,
Основно-расщепляемые линкеры приобрели распространение совсем
недавно. Общая стратегия их использования базируется на промоти-
руемом основаниями (3-элиминировании третичных аминов
(схема 12.16) [32].
Схема 12.16
О
R1 R2-CHO/NaBH(OAc)3
^ ~~~ ШГ 'О' ^ Ж
щ н
о
R3
Ч2 R2 R1
Сульфоновые линкеры получили широкое распространение с 1997—
98 гг. Группа — S02 более стабильна, чем большинство электроноакцеп-
торных групп, и способна выдержать действие агрессивной среды, в
частности, сильных кислот, нуклеофильных агентов (например,
реактивов Гриньяра) или гидридов металлов (схема 12.17). Еще одним
преимуществом линкеров данного типа является простота их
расщепления путем образования промежуточного тиоэфира. Причем, такой
эфир легко снова окислить в сульфон и тем самым немедленно
предотвратить нежелательное расщепление, если оно оказалось
преждевременным [33].
Схема 12.17
О-ч хО Rl.MnX R
^*
Rl-MgX
•^R^R CHa R' ^Rl
^*.^^ *- N N CF.
Me
02Si0-N- ^CF3 - r/
JJ4
Фото-расщепляемые линкеры (схема 12.18) позволяют
автоматизировать процесс синтеза. Однако при фотолизе могут быть
повреждены такие фрагменты молекул, которые содержат кето- или нитро-
группы. Поэтому перед стадией фоторасщепления они должны быть
надежно защищены [34].
Схема 12.18
.о
.F moc
^F moc
*^°
о о
Ph
II s-^s
*^°
F moc
Силиконовые линкеры (схема 12.19) используют для закрепления
карбоновых кислот, аминов, спиртов [35]. Они впервые описаны
J. Ellman в 1995 г. и с тех пор это направление активно развивается.
Большинство силиконовых линкеров содержат арилсилановый
фрагмент, который под действием кислот генерирует новую ароматическую
С—Н связь в месте прикрепления целевой молекулы.
Схема 12.19
ТБАФ^
или CsF НО
О
Л.
J. Ellman предложил также способ (схема 12.20), позволяющий
связывать ароматические соединения без предварительных процедур по-
стадийного наращивания линкера [29, 30, 35].
Схема 12.20
(CH2)3\srCI
Мех чМе
(С К
2'3 >
Me' 'Me
J^l X С(СН3
R2
R2
R3 R3
R1
335
Сохранить в продуктах реакции такие кислотно-расщепляемые
группы как ацетальные, позволяет использование аллилсилана
(схема 12.21); расщепление происходит в мягких условиях (3% ТФУ
в СН2С12) [29].
Схема 12.21
:si
Me' Me
Линкеры, расщепляемые с помощью соединений переходных
металлов, находят применение в синтезе некоторых циклических олефи-
нов, включая дигидропираны, пиколиновые эфиры и лактамы Фрей-
дингера (схема 12.22). В качестве расщепляющего агента используют
RuCl2(=CHPh)(P(C6Hn)3)2. В ряде синтезов применяют Pd-аллиловые
комплексы или соединения Rh (II) [36].
Схема 12.22
У3 О ^NH CH.
RuCI2(=CHPh)(P(CeHl1)3)
4-
Ph
Н
N Ov
^Y С(СНз):
О
О
Me
Хиральные линкеры. Линкеров указанного типа мало, поскольку
асимметрический синтез на твердом носителе пока не очень широко
развит. Примером хирального линкера для энантиоселективного метода
синтеза альдоля является производное тирозина (схема 12.23) [29].
Схема 12.23
1. nBu2BOTf
2. Ме2СНСН2СНО
3. Н202
Дальнейшее развитие химии линкеров, вероятно, будет идти как
по пути применения уже известных моделей в новых реакциях, так
и в направлении поиска оригинальных методов элиминирования це-
336
левых веществ. Наиболее перспективными являются бесследные
линкеры. К этой категории относятся рассмотренные выше сульфоновые
и силиконовые линкеры. Преимущество последних — в простоте
синтеза и расщепления. Особое внимание уделяется повышению
стабильности линкеров в связи с необходимостью широкого
использования в твердофазном синтезе активных нуклеофилов, таких как
металлоорганические реагенты (например, алкиллитий) и металло-
гидридов.
12.3.2.Синтез гетероциклов на полимерном носителе
Вероятно, solid phase синтез еще долго оставался бы методом,
специфически присущим химии белков, если бы пептиды
обладали подходящими фармакокинетическими свойствами. К сожалению,
их невозможно применять в качестве пероральных средств по
причине низкой биодоступности и легкости расщепления протеолити-
ческими Ф, но определенные структурные фрагменты пептидов
могут быть использованы в дизайне малых органических молекул.
Именно в направлении поиска ускоренных методов синтеза
последних и развивается сегодня химия solid phase. Первое сообщение
относительно возможности применения технологии твердофазного
синтеза для создания библиотеки малых молекул (бенздиазепинов)
было опубликовано в 1992 г. J. EUmann и его учеником В. Bunin [38].
Эта публикация вызвала большой интерес. Несколько позднее
стало известно и о работах S. DeWitt, также связанных с синтезом
бенздиазепинов [7, 39].
Выбор именно гетероциклических структур обусловлен тем, что
азотсодержащие гетероциклы представляют собой тот класс
соединений, в котором поиск новых лидеров в настоящее время наиболее
результативен. Большинство веществ этой группы удовлетворяют так
называемому «принципу лекарствоподобия» (см. разд. 7.3). В этом
классе много «привилегированных» структур, таких как бенздиазепи-
ны. С последними ассоциируется не только анксполитическая
активность и наличие анальгетических свойств, обусловленных селективным
связыванием с опиоидными рецепторами к-подтипа, но и
способность выступать в качестве ингибиторов гликопротеинов IIb/Ша, холе-
цистокининов А и В, а также фарнезилтрансферазы и обратимой транс -
криптазы, что важно для поиска анти-ВИЧ средств. Веществ со столь
широким спектром фармакологических свойств вне этого класса не
так уж много [10].
Классическая бенздиазепиновая библиотека J. Ellmann создана
параллельным синтезом с применением «solid phase pin» техники.
Авторы использовали три типа ВВ: аминобензофеноны, аминокислоты
и алкилирующие реагенты. Бенздиазепиновую структуру соединяли с
полимерным носителем через фенольный гидроксил (схема 12.24).
Гидроксигруппа сохранялась и после удаления смолы. Выход
соединений составлял 85—100%. *
337
Схема 12.24
ChLNHCOCH,^ ><^
2. Fmoc-amino
asid acil fluoride
F moc HN
О
Y"^NH О
ARM,
1. Piperidine
^
~>2. HOAc, NMP
^ ,ARM4
J °
2.TFA
Первоначально библиотека J. Ellmann содержала всего 9 веществ.
Позднее применение силиконовых линкеров позволило получать бен-
здиазепины, не содержащие ОН-группы (схема 12.25), и размеры
библиотеки расширились до 192, а затем и 1680 структур [41].
Схема 12.25
vA^
Ц^А0^(сн2)4
rr
NHBpoc
О
х
1.R1 CI
'S'*-- Sn(CH3)3 2. ТФУ/CHXI
Pd
Me Me
2"""2
NhL
R1
-vy-
3. R3-J, ДМФА
NHFmoc
Г iT - г Y
Me Me £ 2. ДМФА, пиперидин
1. 5%, HOAc. 65°C
„ Ph'
338
В группе S. De Witt для получения 1,4-бенздиазепин-2-онов
использовали закрепленные на полимерном носителе а-аминоэфиры,
2-аминобензофенонимины, полученные предварительно в растворе,
и алкилирующие агенты (схема 12.26). Процесс осуществляли
параллельным синтезом в созданном ими же аппарате (multipin Diversomer
apparatus), который представлял собой н'абор стеклянных
капилляров, заполненных 100 мг полимерных гранул с закрепленными на них
аминоэфирами. Время реакции подбирали, исходя из
предварительных экспериментов с участием наименее реакционноспособного
бензофенонимина. Таким методом были получены одновременно три
библиотеки, содержащие от 16 до 40 структурно близких дискретных
соединений. Общее количество синтезированных этими авторами
веществ равнялось 176. Выходы целевых соединений составляли 9—
63%. Чистота не превышала 60% (согласно данным ЯМР 'Н) [39].
Интересно, что в смеси из 40 веществ пять соединений имели тетра-
циклическое строение Б.
Схема 12.26
В настоящее время бенздиазепиновая библиотека содержит 11200
структур. На схеме 12.27 приведены примеры формирования бенздиа-
зепиндионов исходя из различных ВВ [8].
М.А. Gallop с соавт. [41] описали синтез пирролидинов —
потенциальных ингибиторов АПФ, состоящий в 1,3-биполярном
присоединении азометинилидов, полученных из иминов в основной
среде (схема 12.28). В качестве solid phase разработчики применили смолу
Апутах. Замещенные пирролидины ацилировали по атому азота. На
основе данной последовательности реакций методом расщепленного
синтеза получено 500 сульфанилацилпролинов — аналогов
ингибитора АПФ каптоприла (5.59). После четырех итерационных процедур
скрининга и синтеза той группы веществ, в которой были
обнаружены наиболее активные соединения, выявлен пирролидин (12.11). Это
вещество, подобно каптоприлу, содержит сульфгидрилизобутириль-
ный фрагмент. Установлено, что активность диастереомера (12.11) в
3 раза превышает эффект каптоприла, в то время как активность
другого изомера (12.12) значительно ниже [41].
339
Схема 12.27
•^°Y^J
NHj
,CI
CH£I2, ДМА 0 О NHR CHft. NaH2PQ4
/ H
О RN
l| RaH,NaN(TMC)2,TrQ
R*Me, CsjCC^, дмфа
r*°Y^7N
=^л //
=\ NH,
NH
[PdfP-Ph^cy, CuJ, N-Et3, ТГФ
2. ТФУ
-~ ч
OMe
0ц Jl^J NaBHfOAc),, 1%НОАс^ ДМФА Me°
OMe 2. R2
-ec
COOH
^4 ° NH,
O^ OMe 2
N^ „OLi
OMe
' jCty
2.R3-X
ТГФ/дмфа у^
*- MeO^^
1.R2«Br, Pd, R4 « B(OH)2
2. ТФУ/ДМС/HjO
R1 1.R2#Br, R4 = R2
R1 R1
0
Ar
R1 R2
ArCHO i
CH(OfXe)3
°YCI
R4
2. ТФУ.
Y^
Y
Схема 12.28
R2
N(Et),
•*"0^>^^ _R3_
AgN03, CH3CN
O,
N(Et)3 ТГФ
>-
<^R4
\ Ar
CRjCl;
HO
R1
R3
R2
MeO,C Me02C
C°2H ^W^N^CO,H ГГ^°" N ^""C02H
SH SH
Kf = 450pM Ki = 160pM Ki = 5 mkM
(5.59) (12.11) (12.12)
Примером тандемной реакции, проводимой однореакторным
способом с участием реагентов, закрепленных на смоле, может служить
синтез 4-тиазолидинонов (12.13) и 4-метатиазанонов (12.14),
иллюстрируемый схемой 12.29. В конденсации участвовали три компонента:
первичный амин, закрепленный на носителе Affymax, альдегид и тио-
уксусная или пропионовая кислоты (в зависимости от того, какой цикл
планировали получить). Библиотеки формировали методом
расщепленного синтеза. В массиве 3x125 соединений ряда 4-тиазолидинонов
выявлены ингибиторы циклооксигеназы-1 [42].
В твердофазном синтезе нашли применение и различные
варианты реакции Ugi (схема 12.30). В некоторых случаях, как это имело
место в синтезе библиотеки производных коричной кислоты (12.15), в
качестве аминного компонента в конденсации с альдегидами, изонит-
рилами и кислотой выступала аминогруппа Rink полимера,
одновременно являющаяся линкером. Полученные таким путем соединения
отличались достаточно высокой для комбинаторного синтеза чистотой —
90%. В указанном ряду выявлены эффективные ингибиторы тирозин-
фосфатазы, участвующей в гемопоэзе; 1С50 для наиболее активного
соединения (R1 = С6Н5, R2 = СН2-С6Н5) составляет 3,9 мкМ [43].
В комбинаторном синтезе получила щирокое распространение и
трехкомпонентная конденсация Бидженелли. На основе этой реакци
и описан твердофазный синтез дигидропиримидинов с участием р-оксо-
341
Схема 12.29
HS
ОН
R3
#а\0
NH
R1
О
Л
AT 'H
А
1. ТГФ
2.ТФУ
■*-но
R1 Аг
HS у ОН
R3
НО
1. ТГФ
2.ТФУ
1°х>
A^N S
R1 Аг
(12.14)
R3
эфиров, альдегидов и мочевины [8, 44]. Два варианта формирования
библиотек производных дигидропиримидина представлены на схеме
12,31. В первом случае на полимерном носителе закрепляли мочевину,
полученную исходя из у-аминомасляной кислоты. Затем в
реакционный сосуд помещали (3-оксоэфиры и альдегиды. Реакцию проводили в
кислой среде при слабом нагревании. Продукты (12.16) снимали с
носителя, используя ТФУ. Таким путем получены индивидуальные
вещества высокой степени чистоты (>95%) в количествах 10—20 мг (выход
67—98%). При втором способе тиопроизводные
дигидропиримидинов (12.17) синтезированы, исходя из закрепленных на твердом
носителе р-оксоэфиров. Оба ряда соединений представляют большой
интерес для скрининга, т, к. среди дигидропиримидинов обнаружены
блокаторы Са2+-каналов, вещества с противовирусной,
противоопухолевой, антигипертензивной активностью. Дигидропиримидиновый
остов также относится к числу «привилегированных» [10].
Схема 1230
соон
О ^^
о Г
II + R2NC + МеОН, СНС13
R1 Н
NH.
2. ТФУ, CH2CI2
R2
342
Схема 12.31
О О
о н «-АА.*.А °V"y°?
ОН
1.TTQ/HCI,t° С
2. ТФУ, CH2CI2
О О 1.R2-CHO
АЛ,
г. r2'sy""'**"
NH
3. НСООН
OR! R2
н (1216)
R2. ^y^R3
ОН R1
(12.17)
Схема 12.32
***\ \-SQ3Na
1. S02CI2, PhCH3, t°C
ТГФ
R1
NO.
% //
- Vfe
1. NaHSO?, H,Q, EtOH, t^C #^Y Link
R2
HN=< /
о—f
2.
Link
NH
CHO
A_NEt>
ВГ CHO
R1II HOAc.i-PrOH, t°C
N^4R2
H
K^ C02Et
(12.18)
343
Изящный пример применения ионообменной смолы Dowex
50W в качестве полимерного носителя для создания библиотеки,
состоящей из 125 производных N-фенилимидазолил акриловой
кислоты (схема 12.32), продемонстрировал В. Chenera с соавт. [45].
Для достижения полноты превращения авторы использовали
значительный избыток реагентов на каждой стадии. Полученные на
первом этапе л-нитробензилсульфонамиды, связанные с
полимерным носителем, взаимодействовали с иминоэфирами с
образованием N-фениламидинов, которые, в свою очередь, в реакции с
броммалональдегидом образовывали формилимидазолы.
Превращение последних в имидазолилакриловые кислоты достигалось
конденсацией Кневенагеля с моноэтилбензилмалонатами с
одновременной дегидратацией. По завершению синтеза целевые продукты
(12.18) снимали с носителя методом восстановительного аминиро-
вания, нагревая с гидроксиламин-О-сульфоновой кислотой в
присутствии NaOH.
Значительным достижением в технологии твердофазного
синтеза стал разработанный S. De Witt с соавт. метод снятия веществ с
носителя с одновременной циклизацией в целевые продукты. Он
был использован как в описанном ранее синтезе бенздиазепинов,
так и в разработанном этими же авторами способе получения ги-
дантоинов (схема 12.33) с потенциальной антиэпилептической
активностью [46]. Нагревание мочевин, полученных в результате
взаимодействия аминов с изоцианатами, с концентрированным раствором
НС1 приводит к гидролизу сложноэфирной связи, снятию вещества
с полимерной основы и одновременному замыканию имидазолин-
дионового цикла.
Схема 12.33
Acnh-
.R1 R4-NCO
R2 r3 R4 R3 О R1 О
В.А. Dressman с соавт. предложили иной вариант синтеза
библиотеки из 800 гидантоинов, исходя из 20 аминокислот и 80 первичных
аминов. Для закрепления аминокислот на полимере, содержащем гид-
роксиметиленовый фрагмент, использован карбаматный линкер
(схема 12.34). При этом снятие со смолы и циклизация осуществлялись
в основной среде [47].
Схема 12.34
ХХон ^н,ноВу^Х2>С%4^- RR23XV
N ][ ДМФА.ДСС W ^ N t°C,MeOH R3 ,N—^
R1 О R1 « R1
344
Интересен способ синтеза библиотек, когда на полимере
закрепляют блоки, являющиеся одновременно остовами будущих
молекул. Обычно такой остов содержит несколько
функциональных групп, и дальнейшие превращения осуществляют уже с их
участием. Примером является библиотека из 600 бисбензамидо-
фенолов [48], полученных в результате дериватизации 3-нитро-
4-аминофенола, закрепленного на карбоксилированной смоле
(схема 12.35).
Таким образом, твердофазный синтез широко используют не только
для создания библиотек различных гетероциклов, но и карбо- и
макроциклов. Это — бурно развивающаяся область, и рассмотреть все
типы реакций и варианты синтеза одних и тех же scaffolds с
использованием разнообразных ВВ в рамках настоящей книги не
представляется возможным.
Схема 12.35
NO,
*Ч~}~
СОгН
НО'
Nf-L
1.R1-COCI, Ру
DMAP, ДМФА
^~
О—^ Ъ— NH2 2SnCI2 2H20
XN '' ДМФА
~ск<й
X-R1
1.R2-Y-C1,CH2CI2
—*-
2. ТБАФ, ТГФ
3. NaOMe, МеОН ТГФ
=< X-R1
Y=CO, CONH.SOj
12.4. Методы установления структуры индивидуальных
компонентов библиотек
После проведения скрининга возникает необходимость в
установлении структуры активных веществ, Если библиотека создана
методом параллельного синтеза индивидуальных соединений, то проблем
с идентификацией структур не возникает. Достаточно легко
установить строение активных веществ и в том случае, когда скрининг
проводится среди веществ, закрепленных на твердом носителе:
полимерные гранулы могут иметь соответствующие метки, с помощью которых
соединения сортируют, после чего снимают с носителя, переводят в
раствор и устанавливают структуру адекватными
физико-химическими методами.
Наиболее сложная ситуация возникает, когда нужно установить
строение вещества, проявившего активность и находящегося при этом
в растворенном виде в смеси с другими продуктами. Существует не-
345
сколько способов распознавания веществ в смесях. Один из них
реализуется уже на стадии скрининга, когда биологическая мишень
связывается с самым аффинным компонентом и образует комплекс Л-Р
или ингибитор-Ф. Зная аминокислотный состав полипептидной цепи
мишени, можно методом масс-спектрометрии или ЯМР высокого
разрешения установить структуру лиганда. Однако чаще всего, для
упрощения процедуры распознавания, библиотеку формируют
специальным образом еще на стадии ее создания.
12.4.1. Индексные библиотеки
В синтезе индексной (ортогональной) библиотеки каждый
реагент Ап взаимодействует с эквимолярной смесью веществ В'-Вх и,
наоборот, каждое вещество Вт со смесью реагентов А'-Ау.
Схематически библиотеку, образованную 6 реагентами А и В, можно
представить в виде таблицы (рис. 12.4). Такой способ формирования массива
приводит к тому, что каждое вещество присутствует в нем дважды, а
каждая пара смесей имеет один общий компонент. Поэтому активное
соединение должно быть зарегистрировано дважды в процессе
скрининга. Например, если в эксперименте активными оказались две смеси
А'-А6В3 и А3В'-В6, то искомое вещество А3В3 [15].
АЬ6В'
А'-6В2
А1-6В3
А'"6В4
АЬ6В5
А'-6В6
А'В16
А2В'-6
А3ВЬ6
А3В3
А4ВЬ6
А5В1"6
A6Bi-6
Рис. 12.4. Принцип кодирования соединений
в ортогональной библиотеке [15]
12.4.2. Итерационная процедура распознавания веществ
(Deconvolution)
Этот метод идентификации активных компонентов смеси
получил распространение после работ A. Furka, M.H. Geysen и R.A. Houghten
[8, 13, 49, 50]. Указанные авторы описали способ выявления
определенных компонентов в смесях полипептидов, полученных
расщепленным синтезом. Данный метод нашел применение и при
распознавании активных соединений в библиотеках малых молекул. Сама
процедура выглядит следующим образом: четыре группы ВВ — А, В,
С и D — по 5 разновидностей в каждой могут образовать 625
соединений (рис. 12.5). В первом раунде создают 25 смесей по 25
соединений в каждой, в которых фрагменты А и В известны. Пусть наиболее
346
А1-5
31-5 54 = 625 соединений в библиотеке
5 смесей
А по 5 соед.
5 индивидуальных
соединений
Рис. 12.5. Схема итерационной процедуры распознавания веществ
в библиотеке
активной оказалась смесь с комбинацией А2В'. Исходя из этого, при
готавливают 5 смесей по 5 соединений в каждой, в которых А2В'
остаются неизменными и присутствуют во всех смесях и всех
соединениях. Заместитель С всюду известен — в первой смеси это С1, во
второй — С2 и т.д. Радикал D15 изменяется произвольно. Если на
этом этапе активной оказалась смесь А2В'С3, то в последнем раунде
синтезируют всего 5 индивидуальных веществ, варьируя заместитель
D, и выявляют активный продукт среди них. Тем самым
идентифицируют фрагмент D, несущий ответственность за проявляемую
активность (на рис. 12.5 это D5), и устанавливают структуру вещества
в целом.
Демонстрацией возможностей данного метода является
распознавание активных структур из библиотеки производных 1,4-дигид-
ропиридинов (схема 12.36). Для синтеза 300 веществ использовали
в качестве ВВ 10 альдегидов, 10 Р-кетоэфиров и ацетилацетон или
два других Р-кетоэфира. На твердом носителе закрепляли кетоэфи-
ры, превращая их в енаминопроизводные. Элиминировали целевые
соединения в кислой среде. Среди продуктов был и известный
блокатор Са2+-каналов — нифедипин (7.9). В результате скрининга
выявили два наиболее активных массива. Расшифровку структур
347
Схема 12.36
R1
ХА„
о о
R3" >Г 4R4
R2
■NH.
MS, DCM
R1
аа
.R2
V^ R4 = Alk, O-Alk
Py, Д
,R2 A
О f R4
Ar
TFA
R2.
HN R1 о R3
R4
i
R1
R3
10 альдегидов
0
r2.oA
R1*
I Ar|
]Tj[
N
H
0
R4
4R3
deconvolution
MeO'
t
tr
MeO
OMe
ACL 0591-3-3
10 Р-кетоэфиров
Ацетилацетоны
илн Р-кетоэфиры
провели с помощью итерационной процедуры и выявили
максимальную активность у нифедипина (/С50 = 18 нМ) и нового соединения
ACL0591-3-3 (/С50 = 14 нМ) [9].
Метод deconvolution имеет ряд недостатков: не всегда таким
способом можно выявить самое активное вещество, скрининг смесей
обеспечивает не столь надежный результат (по сравнению с
испытаниями индивидуальных веществ). К тому же, при таком методе
расшифровки библиотеки необходим дополнительный синтез подбиб-
лиотек.
12.4.3. Позиционное сканирование
R.A. Houghten был предложен неитерационный метод
позиционного сканирования [51]. Например, при наличии ВВ четырех типов
AM....DM (по 5 вариантов каждый) полную библиотеку, в отличие от
способа, рассмотренного выше, синтезируют в 4 этапа, каждый раз
по одной из 5 подбиблиотек по 125 соединений в каждой. Схема
идентификации активного соединения представлена на рис. 12.6.
Преимущество этого метода, по сравнению с итерационной
процедурой распознавания, состоит в том, что синтез библиотеки осуще-
348
Q1-5'
Д1-5
кг
Л
01-5
Заместители А
с1-5
Заместители В
Заместители С Заместители D
Д1-5 Д1-5
С
с1-5
ф-в- *-ф
Д1-5
_ В1"5
С1-5
AV5
D3—t^^-B1-5
С1"5
А1-5
X
С1-5
Рис. 2Z6. Схема позиционного сканирования
Целевое соединение A2B'C3D5
ствляется один раз. Однако, если выявлены несколько смесей с
примерно одинаковым уровнем активности, то каждое соединение,
входящее в их состав, должно быть синтезировано в индивидуальном
виде, и установлена его активность.
Значительно сложнее установить структуру веществ, образующихся
в ходе синтеза основной библиотеки в малых количествах. В этом
случае наиболее надежным оказывается метод масс-спектрометрии.
349
Для упрощения распознавания активных веществ при
твердофазном синтезегюльзуются молекулярными кодами на носителе (рис. 12.7).
Такая кодировка располагается со стороны, противоположной той, с
которой идет «сборка» целевого продукта. Кодировка может быть как
линейной, так и разветвленной [8].
А1
Соединение р*уф-^у Д1— дг — д3 Соединение -^фА*^ A2
^дз
Рис. 12.7. Способы расположения кодов на полимерном носителе [8]
Реагенты, образующие код, и условия реакций, в которые они
вступают, не должны оказывать воздействие на формирование
основной молекулы. Маркировку расшифровывают после снятия
продукта с полимерного носителя (схема 12.37). Кодировку
соединений в библиотеке осуществляют с помощью галогенарилокси-
алканолов [52].
0Yo
rNH .
о
/\ у Соединение
^N02
CI
'°^:
ho
■■ *■ Соединение XII
Се(МН4)2(МОз)6 Н0^па
CI'
Схема 12.37
CI
■
хх
В комбинаторном синтезе используют и нехимическое
кодирование, например, путем записи и считывания высокочастотных
сигналов [52, 53].
12.5. Перспективы дальнейшего развития
комбинаторного синтеза
Развитие комбинаторного синтеза идет по пути дальнейшей
автоматизации и миниатюризации процессов получения новых
соединений как в растворах, так и на твердом носителе: появляются новые
линкеры и секвестранты, расширяется набор реакций, пригодных для
осуществления твердофазных синтезов, совершенствуются методы
контроля состава реакционных смесей и способы разделения продук-
350
тов (экстракция с помощью реагентов, закрепленных на твердом
носителе, и препаративная ВЭЖХ).
Комбинаторный синтез открывает широчайшие возможности
перед исследователями, занимающимися поиском БАВ. Однако
создание больших массивов соединений сопряжено с определенными
трудностями. Одна из проблем связана с декодированием
библиотек. При этом наибольшие сложности возникают при
расшифровке массивов, превышающих 105 соединений. Даже если всего 1%
веществ в такой библиотеке проявит активность, то это составит
I03 структур. Возникает так называемая «ложная позитивность» —
при повторном скрининге среди соединений, близких по строению,
трудно выбрать самые активные, и, следовательно, необходимо
изменять критерии отбора. Кроме того, в больших библиотеках,
состоящих из смесей, невозможно быстро идентифицировать все
компоненты. Создается искаженное представление о том, что
большинство соединений неактивны, хотя они, вероятно, не были
получены в том количестве, чтобы их эффект можно было
зарегистрировать. Это усложняет процесс формирования новой библиотеки.
Время повсеместного увлечения чрезвычайно большими массивами
веществ (> 105), вероятно, прошло. Сейчас все большее число
исследователей склоняется к той точке зрения, что целесообразно
синтезировать ускоренными методами индивидуальные вещества, а
не создавать и испытывать смеси. Формируется два подхода к
созданию комбинаторных библиотек: первый направлен на поиск
лидера, второй — на оптимизацию его структуры. Этапы каждого
из указанных подходов отражены в табл. 12.7. Стоит еще раз
подчеркнуть, что и на этапе отбора лидера целесообразно
формировать библиотеку из индивидуальных соединений.
Таблица 12.7
Характеристика двух подходов
в создании библиотек соединений [8]
Поиск лидера
Обширная библиотека
Синтез разнообразных структур
Множество биологических мишеней
Твердофазный синтез
Формирование библиотеки методом
расщепленного синтеза ("one bead,
one compound")
Состав > 104 соединений
Количество < 1 мг каждого
соединения
Скрининг смесей с последующей
расшифровкой активных структур
Оптимизация лидера
Целевая библиотека
Соединения определенного строения
Одна биологическая мишень или
группа родственных мишеней
Твердофазный синтез/синтез
в растворе
Автоматизированный параллельный
синтез индивидуальных веществ
Состав << 104 соединений
Количество > 1 мг каждого
соединения
Оптимизированный скрининг
индивидуальных соединений
351
Литература
1. LeblM. И J. Comb. Chem. - 1999. - Vol. 1. - P. 3.
2. Frank R., Heikens W„ Heisterberg-Moutsis G.etal. //Nucl. Acid. Res. — 1983. -
Vol. 11. - P. 4365.
3. Geysen H.M. Meloen R.H. Barreling S.J. ff Proc. Natl. Acad. Sci. USA —
1984. -Vol. 81. - P. 3998
4. Houghten R.A. ff Proc. Natl. Acad. Sci. USA. - 1985. - Vol. 82. - P. 5131.
5. Pinilla C, Appel JR., Blondeelle S.E. ff Drug. Dev.Res. - 1994. - Vol. 33. -
P. 133.
6. Nejzi A., Ostresh J.M., Houghten RA. I I Chem. Rev. - 1997. - Vol. 97. -
P. 411.
7. DeWitt S.H, Kiely J.S., Stankovic C.J. et al I I Proc. Natl. Acad. Sci. USA. —
1993. -Vol.90. - P. 6909.
8. Balkenhohl F. et al. ff Angew. Chem. Int. Ed. Engl. - 1996. - Vol. 35. -
P. 2288.
9. Combinatorial Chemistry and Molecular Diversity in Drug Discovery / Ed. by
E.M. Gordon, J.F. Kerwin. - 1998, New York: Wiiey-Liss, Inc.
10. Fecik R.A., Frank K.E. et al. ff Med. Res. Rev. - 1998. - Vol. 18, N 3. -
P. 149.
11. Furka A., Sebesten F. et al. ff Int. J. Pept. Prot. Res. - 1991. - Vol. 37. -
P. 487.
12. Furka A. ff Drug. Dev. Res. - 1995. - Vol. 36. - P. 1.
13. Houghten R.A., Pinilla С etal. ff Nature. - 1991. - N 354. - P.84.
14. PirrungM.C, Chen J ff i. Am. Chem. Soc. - 1995. - Vol. 117. - P. 1240.
15. PirrungM.C, ChauJ. H-L., Chen J. //Chem.Biol. - 1995. - Vol. 2. - P. 621.
16. SmithP.W., LaiJ.Y.Q. etal. ffBioorg.Med.Chem. Lett. - 1994.-Vol.4. -P. 2821.
17. Pat. WO 95/19359 // Rebek J., Carell Jr.T., Wintner E.A
18. Carell Jr.T., Wintner E.A. et all I Angew. Chem. Int. Ed. Engl. - 1994. -
Vol. 33. - P. 2161.
19. Ugi I., Steinbruckner С ff Chem. Ber. - 1961. - Bd. 94. - S. 734.
20. Ugi I., DomlingA., Horl W. ff Endeavour. - 1994. - Vol. 18. - P. 115.
21. Ugi I., Gobel M. et al. ff Res. Chem. Intermed. - 1996. - Vol. 22. - P. 625.
22. Pat. WO 95/02566 // Armstrong R.W.
23. StrockerAM., Keating ТА. etal. //Tetrahedron Lett. - 1996. - Vol. 37. - P. 1149.
24. Pat. WO 94/01102 // Hogan J.C.
25. Peisach E. Casebier D. et ^/.//Science. - 1995. - Vol. 269. - P. 66.
26. Flynn D.L., Devraj R. V., Parlow J J. ff Curr. Opin. Drug Discov. & Develop. —
1998. -Vol. 1,N 1. - P. 41.
27. Parlow J.J., Flynn D.L.ff Tetrahedron. - 1998. - Vol. 54. - P. 4013.
28. Pat WO 95/16918 // Brenner S.
29. MorphyJR. ff Curr. Opin. Drug Discov. & Develop. - 1998. — Vol. 1, N 1. - P. 59.
30. BaskesJ.B. Ellman J.A.ff Curr. Opin. Chem.Biol. - 1997. - Vol. 1. - P. 86.
31. Fivush AM. Willson T.M.ff Tetrahedron Lett. - 1997. - Vol. 38. - P. 7151.
32. Kroll F.E.K., Morphy JR. ff Tetrahedron Lett. - 1997. - Vol. 38. - P. 8573.
33. Gayo L.M., Suto M.J. ff Tetrahedron Lett. - 1997. - Vol. 38. - P. 211.
34. Routledge A., Abell C, Balasubramanian S. ff Tetrahedron Lett. — 1997. —
Vol. 38. - P.1227.
35. Plunkett M.J., Ellman J.A. ff J. Org. Chem. - 1995. - Vol. 60. - P. 6006.
36. Piscopio A.D., Miller J.F, Koch K. ff Tetrahedron Lett. - 1997. - Vol. 38. -
P. 7143.
37. Merrifield R.B.ff Biochemistry. - 1964. - Vol. 3. - P. 1385.
352
38. Bunin B.A., Ellman J A. ffS. Am. Chem. Soc. - 1992. - Vol. 114. - P. 10997.
39. DeWitt S.H, Czarnik A. W. ff Ace. Chem. Res. - 1996. - Vol. 29. - P. 114.
40. Plunkett M.J., Ellman J.A. ff}. Am. Chem. Soc. - 1995. - Vol. 117. - P. 3306.
41. Pat. WO 95/35278 // Gallop M.A, Murphy M.M.
42. Pat. WO 96/00148 // Holmes C.P.
43. CaoX, Moran E.J etal. ff Bioorg. Med. Chem. Lett. - 1995. - Vol. 5. - P. 2953.
44. WipfP., Cunningham A. ff Tetrahedron Lett. - 1995. - Vol. 36. - P. 7819.
45. Pat. WO 95/16712 // Chenera В., Elliott J., More M.
46. Pat. WO 94/08711 // Cody D.R., DeWitt S.H. et al.
47. MeyrsH.V., Dilley G.J., Durgin T.L. <rfa/.//Mol.Diversity. - 1995. -Vol. 1. — P. 13.
48. Chabala J. С ff Curr. Opin. Biotechnol. - 1995. - Vol. 6. - P. 632.
49. Furka A. ff Drug Dev. Res. - 1995. - Vol. 36. - P. 1.
50. Geysen H.M., Rodda S.J. Mason T.J. ff Mol. Immunol . - 1986. - Vol. 23. -
P. 709.
51. Houghten R.A., Appel J.R., Blondelle S.E. etal ff BioTechniques. — 1992. —
Vol. 13.- P. 412.
52. Nicolaou K. C, Xiao X- Y, Parandoosh Z. et al. ff Angew. Chem. Int. Ed. Engl. —
1995.-Vol. 34.- P. 2289.
53. Moran E.J., Sashar S, Cargil J. F. et al, ff J. Am. Chem. Soc. — 1995. -
Vol. 117. - P. 1787.
54. Коваленко С.Н., Друшляк А.Г., Черных В.П. Основы комбинаторной
органической химии: Учебник для студ. высш. учеб. заведений. — Харьков:
Изд-во НфаУ:Золотые страницы, 2003.
13
БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ
НОВЫХ СОЕДИНЕНИЙ
Биологические испытания новых соединений проводят для того,
чтобы определить у них наличие и выраженность какой-либо
фармакологической активности, а для самого активного из
испытанных веществ устанавливают, обладает ли оно биологическим
эффектом in vivo в сочетании с желаемой длительностью действия при
использовании рационального пути введения. Скрининг, как метод
отбора веществ в зависимости от степени выраженности какого-
либо биологического эффекта, получил развитие в начале XX в.
Идея исследования синтетических органических веществ на
различных образцах биологических тканей была обоснована P. Ehrlich,
который установил, что синтетические красители при
избирательной абсорбции клетками могут убивать бактерии и паразитов без
оказания существенного негативного воздействия на организм
млекопитающих. В результате этих экспериментов в 1910 г. был
предложен сальварсан — соединение мышьяка для лечения сифилиса.
Несколько крупных химических компаний того времени,
производящих главным образом красители, использовали этот подход для
разработки собственных исследовательских программ по поиску
веществ, эффективных в борьбе с инфекционными заболеваниями,
и инициировали тем самым систематические биологические
испытания сотен химических соединений. Таким путем в 1931 г. Н. Do-
magk выявил антибактериальное действие сульфамидных
производных, использовавшихся в качестве полупродуктов в синтезе
красителей [1, 2].
Развитие представлений о рецепторах БАВ способствовало
проведению испытаний синтетических органических веществ на такие
виды активности как противосудорожная, анальгетическая,
гипотензивная, противовоспалительная, противоязвенная и др. Однако
существуют принципиальные различия между отбором соединений с
антибактериальными, противогрибковыми, антигельминтными,
противовирусными, антипротозойными свойствами и скринингом
веществ, фармакологический эффект которых направлен на
коррекцию метаболических нарушений. В первом случае вещества
должны быть губительными для патогенов, но безопасными для хозяина,
т.е. поиск сводится к различиям в межвидовой токсичности.
В противоположность этому причины метаболических нарушений,
354
приводящие, например, к СД, астме, раку и другим тяжелым
заболеваниям, пока недостаточно изучены. Отбор веществ для
коррекции перечисленных патологий заключается в поиске
селективности внутри того же бытия. При испытании новых соединений на
такие виды активности данные, полученные in vitro на
изолированных мишенях, могут существенным образом отличаться от
результатов исследований in vivo. В этом случае большую пользу
приносят модели заболеваний, специальным образом созданные у
животных. Однако, несмотря на то, что модели могут
обеспечивать адекватные симптомы заболевания, причины его
возникновения у человека и экспериментального животного различны.
Поэтому эффективное на стадии доклинических испытаний вещество
может оказаться неактивным при клинических исследованиях [2, 3].
Таким образом, для оценки фармакологической активности
какой-либо группы новых соединений необходимо реализовать
программу исследований, которая обычно состоит из нескольких этапов
испытаний in vitro и in vivo.
На этапе предварительного скрининга in vitro оценивают активность
испытуемых объектов и отсеивают те из них, которые не
представляют дальнейшего интереса. Эталоном активности служит
препарат сравнения — самое эффективное ЛС в данной фармакотера-
певтической группе. Если тестируемое соединение проявляет эффект,
превышающий эталонный уровень, то оно пригодно для испытаний
на следующей стадии. Целью первичного скрининга является
отбор веществ-«хитов», пригодных для дальнейших исследований.
Первоначальные тесты обычно носят качественный характер, и
критерием отбора служит уровень концентрации испытуемых соединений,
при котором они обнаруживают активность. На втором этапе
уровень активности оценивается уже количественно. Вещества с
высокими показателями предоставляются для специфических
исследований, которые также могут быть проведены in vitro. Однако в
этом случае, как правило, используют методики, отличающиеся от
первичных. Кроме того, круг исследуемых видов активности также
может быть расширен. Если вещество обладает подходящим
уровнем активности при первичной оценке и не проявляет других
видов действия, ограничивающих его дальнейшее исследование, оно
может быть передано на испытание in vivo.
Несмотря на то, что исследования in vitro дают достаточно много
информации о биологических свойствах изучаемых соединений, все
же удовлетворительное представление об их фармакологическом
профиле можно получить при комплексной проверке на все системы и
органы живого организма. Чаще всего исследования БАВ in vivo
начинают с анестезированных животных, изучают их влияние на
органы, с регистрацией количественных показателей. После этого
переходят к интактным (здоровым) животным, рефлексы которых носят
природный характер. Кроме того, обязательными являются
эксперименты на животных с моделями заболевания, для лечения или
профилактики которого планируют использовать в дальнейшем испыту-
355
емое вещество. Если Соединение проявило удовлетворительный
уровень эффекта in vivo, оно может быть передано на клинические
испытания первоначально на здоровых добровольцах, а затем на
пациентах с соответствующей патологией.
13.1. Исследования in vitro
Среди первичных тестов in vitro наиболее распространенными
являются исследования, связанные с изучением Л-Р взаимодействия,
измерением уровня вторичных посредников или оценкой
функционального ответа. Тестирование Л-Р взаимодействий может проводиться
как на всей ткани, так и на изолированных клетках, гомогенатах,
клеточных мембранах (культура ткани). Цель таких экспериментов
заключается, с одной стороны, в определении наличия или
отсутствия конкуренции между исследуемыми веществами и меченным
радиоактивной или флуоресцентной меткой уже известным лиган-
дом, а с другой — в выяснении того, какое из испытуемых веществ
обладает большей аффинностью. Исследования на образцах и на
культурах тканей имеют свои преимущества и недостатки. Преимущество
экспериментов на образце участка ткани заключается в его
доступности у животных. В то же время, участок этот состоит из разных
типов клеток, и нет уверенности в том, что связывание происходит
именно с интересующим видом клеток. Диспергированные клетки
получают из соответствующей ткани, однако доступность такого
препарата ограничена, в особенности если речь идет о человеческих
тканях. В этом смысле большими преимуществами обладает культура
тканей. Ее использование позволяет избежать привлечения большого
числа животных. Однако сама методика создания культуры тканей
трудоемка и может привести к некоторым трансформациям в самих
клетках. В настоящее время широко доступны клетки, полученные
методами генной инженерии. В них могут быть введены подходящие
рецепторы, однако это довольно сложный процесс, и нет гарантии,
что поведение рецептора в чужеродной мембране будет таким же, как
в нативной. Результатом этого вида исследований является
установление в изучаемом ряду веществ агонистов, частичных агонистов
или антагонистов рецепторов [4].
Активность агониста можно оценивать также по уровню
концентрации ВП (см. разд. 4). Наибольшее распространение получили
методы оценки эффективности агонистов, основанные на измерении
уровня цАМФ и инозитинтрифосфата. Эти исследования проводят в
тканях и в их гомогенатах, на культуре тканей и клонированных
клетках. При этом отслеживают рост концентрации посредника в
зависимости от роста концентрации лиганда. Кроме того, можно изучать
влияние концентрации потенциального антагониста на способность
агониста продуцировать все возрастающий уровень концентрации
посредника. При этом неважно, какую из клеточных функций
оценивать: проводимость, релаксацию или секрецию. В таких экспери-
356
ментах определяют выраженность эффекта, его специфичность,
а иногда и длительность действия [5, 6].
Количественную оценку активности агониста часто проводят на
препарате участка ткани, например, гладкой мышечной ткани как
наиболее доступном и простом. Для поддержания в «рабочем»
состоянии такой препарат помещают в изотонический раствор хлорида
натрия с температурой 37 °С, который насыщают смесью 95% 03и 5%
С02 при рН = 7.4. При размещении препарата учитывают
направление мышечных волокон, как это показано на рис. 13.1.
Н
Рис. 13.1. Препараты гладкой мышечной ткани:
а — сосуд; 6 ~ трахея
Абсолютную эффективность БАВ выражают в терминах
количества вещества, необходимого для того, чтобы вызвать
регистрируемый эффект. Обычно это ECSQ (Я£>50), т. е. концентрация,
необходимая для достижения полумаксимального ответа (см. также разд. 3).
Для определения £С50 строят график зависимости доза —
ответ (рис. 13.2). Проблема определения абсолютной эффективности
состоит в том, что на ответ часто влияет тип ткани: в различных
фрагментах тканей, содержащих одни и те же рецепторы, различия в
ЕС50 для одних и тех же агонистов могут быть весьма существенными.
Причина такого явления заключается в кооперировании рецепторов
с системой ВП и/или в плотности расположения рецепторов.
Поэтому наиболее приемлемой для количественной оценки активности
агониста является относительная эффективность, выраженная как
эквиэффективная концентрация (EEC). Сравнение проводят между
стандартным агонистом, одинаковым для всех испытуемых веществ
во всех экспериментах, и тестируемым веществом, что выражают как:
EEC = EC5Q test /EC5Q$t
Изолированный орган или участок ткани может быть погружен в
сосуд или размещен в камере (рис.13.3). Каждый из этих способов
имеет преимущества и недостатки. Основное преимущество первого
метода состоит в простоте размещения препарата, введения
испытуемого вещества и оценке его концентрации в окружающей жидкости.
357
С агониста
Рис. 13.2. Зависимость «доза — ответ» для тестируемого вещества (2)
и препарата сравнения (1)
Недостатком является удаление вещества из жидкости при обновлении
изотонического раствора NaCl. Этого можно избежать, если вводить
вещество одновременно с раствором, но тогда трудно контролировать
изменения концентрации. Еще одним недостатком является
накопление токсичных тканевых метаболитов в окружающей препарат
жидкости. Преимущество размещения участка ткани в камере состоит в
постоянном удалении из нее токсичных продуктов, поэтому такой
образец дольше сохраняет свои нативные свойства. Кроме того, нет
необходимости в замене раствора, т. к. его обновление происходит
непрерывно. Однако при этом способе трудно дозировать вещества
и оценивать их концентрацию.
9% NaCl
Рис. 13.3. Способы размещения образца ткани в экспериментах in vitro [4]
а) в сосуде; б) в камере
На изолированных участках гладкой мускулатуры могут быть
проведены измерения, связанные как с процессом сокращения, так и
расслабления. Из этих двух состояний обычно проще изучать
сокращение. В отсутствие внешнего стимула изолированный участок мы-
358
щечной ткани находится в состоянии релаксации, поэтому легко
оценить натяжение при добавлении исследуемого препарата.
Расслабление измерить сложнее, т. к. в начале нужно привести ткань в
состояние сокращения, а затем, добавив вещество, добиться снятия
спазма — релаксации. Сокращение индуцируют одним из двух
способов (рис. 13.4): тоническое сокращение вызывают однократным
добавлением спазмогенного вещества, а фазовое —
электростимуляцией нерва, находящегося внутри образца ткани. Это сокращение
происходит с регулярными интервалами и обеспечивает возможность
проверки БАВ, эффект которых направлен на расслабление мышц [5].
till
Рост
натяжения
\\\
ШШ
ш
спазмоген
.электростимуляция
Агонист
Рис. 13.4. Виды сокращений образца мышечной ткани:
а — тонические; 6 — фазовые
Существует два типа экспериментов на изолированных органах
или препаратах тканей: каскадный и параллельный. При
каскадном жидкость, омывающая первый перфузируемый орган, омывает
затем второй, третий и т.д. Каскад может состоять как из
нескольких видов тканевых препаратов, так и определенного числа
идентичных образцов. Этот тип исследований полезен в случае
испытаний соединений с коротким временем действия, например,
тромбоксана \ с периодом полупревращения 30 сек. Такое
соединение можно генерировать энзиматически во главе каскада и
исследовать практически одновременно на нескольких препаратах.
Первоначально данный тип испытаний применяли для
идентификации веществ из тканевых экстрактов: используя в каскаде
подходящие препараты, оценивали весь набор биологических
эффектов одновременно. Несмотря на это, в настоящее время существует
много новых подходов в оценке биологической активности веществ,
и каскадный метод используется редко.
Параллельное размещение нескольких препаратов, имеющее много
общего с экспериментами на отдельно взятом изолированном органе,
применяется гораздо шире. В этом случае образцы ткани находятся в
индивидуальных кюветах изолированно друг от друга. В омывающую
их жидкость добавляют агонист или антагонист и используют так долго,
как это необходимо. С помощью данного метода легко построить
концентрационные кривые, отметить момент наступления или
снижения эффекта и т. п. Таким путем можно оценить не только эффект
агониста, но и длительность его действия.
359
При проведении экспериментов по оценке сократительной
активности мышц следует учитывать такое явление как тахифилаксия,
состоящее в постепенном снижении интенсивности ответа ткани при
повторном введении агониста. Например, если ответ возникает через
определенные интервалы времени, то его интенсивность будет
прогрессирующе уменьшаться. Аналогично, если эксперимент для
полного агониста повторять несколько раз, то кривая доза-ответ будет
проявлять тенденцию к снижению (рис. 13.5). Чтобы рассматриваемое
явление не сказалось на оценке эффективности агониста,
необходимо увеличивать интервал между введениями БАВ или строить
единственную кривую для каждого образца отдельно [5].
Амплитуда
SSSSH
Рис. 13.5. Постепенное снижение амплитуды сокращений мышцы
при повторном введении агониста
Активность антагониста определяют как функциональную
способность БАВ препятствовать действию агониста. Для того, чтобы это
установить, необходимо измерить эффект агониста первоначально в
отсутствие, а затем в присутствии антагониста. Для количественной оценки
строят специальный график (рис. 13.6). При конкурентных взаимоотно-
шених агониста и антагониста по отношению к рецептору tgct= 1, а
показатель рАз — мера аффинности антагониста (см. также, разд. 3).
lg(CR-1)
РА,
igc
Рис. 13.6. Зависимость lg(CR— 1) от -lgC:
CR — концентрационное соотношение, С — конц. антагониста
Таким образом, даже весьма поверхностное рассмотрение методов
исследования in vitro позволяет понять, что биологические
испытания новых соединений — трудоемкий и дорогостоящий процесс.
360
13.2. Тотальный скрининг
В 80-е гг. с развитием технологий high-throughput screening время,
затрачиваемое на проведение доклинических исследований
кандидатов в ЛС, сократилось на 70—90%. Кроме того, снизились расходы на
биологические испытания, т. к. ранее только на скрининг приходилось
до 45% средств, предназначенных на разработку нового ЛС. В 90-е гг.
комбинаторный синтез расширил возможности органической химии
и позволил получать десятки и сотни тысяч соединений в очень
кроткий промежуток времени. Поэтому в настоящее время наибольший
интерес вызывает современная технология проведения биологических
испытаний, позволяющая оценивать до 105 соединений в день. Она
получила название тотального высокоскоростного скрининга, а
технология, обеспечивающая проверку свыше 105 веществ в день —
ультра-высокоскоростного скрининга [7].
В аппаратурном оформлении этих исследований повсеместное
распространение получили стандартные пластиковые планшеты с
96 (8x12) углублениями. Объем каждого такого углубления —
микроцилиндра составляет 100—300 мкл, в нем может содержаться ~ 109 М
вещества. Прогресс в этой области идет очень быстро. Менее 7 лет
назад был предложен планшет на 384 микроцилиндра с объемом
каждого 25—100 мкл (содержание вещества - 1010 М). Под этот
стандарт подходят прежние детекторные системы, позволяющие
считывать информацию, полученную в результате биологических
испытаний, системы для заполнения микроцилиндров веществами,
реагентами и т. п., а также средства для промывки таких
микрокювет. Однако практически одновременно с описанным появилось
новое поколение пластин с 864 и 1536 микрокюветами объемом
10—12 мкл. Компании DuPont и Merck предложили планшет на
96000 ячеек с объемом каждой микрокюветы 0.2 мкл. Заполнение
столь малых объемов вызывает определенные трудности. Для этих
целей разработаны новые системы промывки, инкубирования и
детектирования, что позволяет полностью автоматизировать процесс
скрининга. Универсальность рассматриваемых приспособлений для
проведения первичной оценки биологической активности новых
органических соединений в том, что такие же планшеты
применяются при создании комбинаторных библиотек. Для синтеза и
хранения веществ используют полипропиленовые планшеты,
устойчивые к действию растворителей, а для скрининга — полистирольные.
Их изготовляют прозрачными, полупрозрачными (с прозрачным
дном), прозрачными для излучения с определенной длиной волны,
а также с использованием специальных покрытий. Эти планшеты
пригодны для широкого круга биологических испытаний: на них
располагают испытуемые соединения (индивидуальные или смеси),
стандарты и контрольные образцы. Кроме того, планшеты можно
использовать как в скрининге веществ в одной и той же
концентрации, так и для определения дозозависимого эффекта для
одного вещества в разных концентрациях [8—12].
361
Уже более 20 лет для распознавания результатов биологических
испытаний на планшетах с 96 ячейками применяют спектрофотомет-
рический метод. В настоящее вре*1я не менее широкое
распространение для детектирования получили флуориметрические, сцинтилля-
ционные, люминесцентные датчики [13—15].
Сегодня тотальный высокоскоростной скрининг позволяет
оценивать многие виды биологической активности, изучать
взаимодействие Л-Р, Ф-ингибитор и др.
13.3. Скрининг комбинаторных библиотек
Создание «полезных» соединений не тождественно синтезу в
кратчайшие сроки максимально простым путем большого количества
новых веществ. «Полезные» библиотеки формируют с пониманием того,
как их будут исследовать. При этом важно не только то, какой класс
соединений получать и на какие виды активности их проверять, но и
какую из версий комбинаторного синтеза (жидкофазный,
твердофазный) выбрать, каков будет размер библиотеки, какие использовать
носители, линкеры, каким образом распознавать соединения, если
они находятся в смеси. Эти вопросы важны как для химиков, так и
для фармакологов. Поэтому скрининг комбинаторных библиотек
невозможен без тесного сотрудничества исследователей разных
специальностей и в значительной мере повышает продуктивность их
работы в поиске лидирующих структур.
Скрининг библиотек, полученных методами параллельного
синтеза, т.е. тех, которые состоят из индивидуальных соединений, ничем
не отличается от обычного высокоскоростного тестирования
больших массивов веществ, получаемых в крупных фармацевтических
компаниях. Преимущество состоит в том, что комбинаторные
библиотеки легко интегрировать в уже существующие схемы оценки
активности. Кроме того, эти исследования уже на самом начальном
этапе могут быть количественными, т.к. используют индивидуальные
соединения, а не смеси и, следовательно, о них можно получить
значительно больше достоверной информации [7].
К недостаткам библиотек, полученных методами параллельного
синтеза, следует отнести сравнительно малое количество соединений
по отношению к mix-split- или random synthesis-библиотекам (см.
разд. 12.4). Возможности высокоскоростного скрининга превышают
возможности, предоставляемые «параллельной» библиотекой.
Роботизация и миниатюризация технологии оценки активности в этом
случае способны лишь ускорить фармакологическое тестирование БАВ.
Важнейший аспект в оценке эффективности скрининга — способ
детектирования результатов испытаний. Примером исследования с
использованием сцинтилляционного метода распознавания активных
соединений в библиотеке является схема, представленная на рис. 13.7.
Если сцинтиллятор сцеплен с полимерным носителем, на который к
тому же привита белковая молекула (рецептор), то при связывании
362
такой мишени с меченым радиоактивной меткой лигандом возникает
эмиссия. Ее легко регистрировать с помощью соответствующего
датчика. Аналогичным образом могут быть устроены стенки или дно
микроцилиндра, в котором расположена мишень — энзим. Такую
кювету заполняют подходящим субстратом и добавляют исследуемое
вещество — потенциальный ингибитор данного Ф. Если ингибитор
препятствует взаимодействию Ф со своим субстратом, эмиссия
отсутствует. В качестве источников р-частиц используют 125J, 3H или 33Р [16].
о
^ Радиолиганд,
к~ связанный
с рецептором
эмиссия
*
ь>
о
отсутствует
связывание
отсутствует
эмиссия
Киназа
и ЭЗР АТФ,
белковый
субстрат
эмиссия
Тоже
+
ингибитор
эмиссия
отсутствует
б
Рис. 13.7. Принцип сцинтилляционного детектирования
комбинаторной библиотеки;
а — сцинтиллятор связан с полимерной гранулой;
6 — сцинтиллятор находится в дне микрокюветы
Скрининг соединений, иммобилизованных на полимерном носителе.
Методика оценки активности веществ, закрепленных на твердом
носителе, — on-bead-исследования — несколько отличается от
рассмотренной выше. Обычно для распознавания активных веществ,
иммобилизованных на полимерных гранулах, используют рецепторы, имеющие в
своем составе флуоресцентные или радиоактивные метки. Такие
рецепторы инкубируют с комбинаторной библиотекой. После интенсивной
промывки гранулы с закрепленными веществами и связанными с ними
рецепторами, отбирают методом флуоресцентной микроскопии или с
помощью другого флуоресцентно чувствительного детектора [7, 17,18].
В качестве флуоресцентной метки широкое распространение
получил GFT- флуоресцентный полипептид, впервые выделенный из
медуз Aequoria victoria. Главное преимущество GFT-полипептида
заключается в том, что это единственный белок, переходящий во
флуоресцирующее состояние самопроизвольно без участия Ф или
кофакторов. Он имеет относительно небольшую полипептидную цепь
363
(238 аминокислотных остатков). Флуорофорный фрагмент (13.1)
расположен внутри жесткой р-цилиндрической структуры, что
существенно снижает влияние на него окружающей среды [19—21].
(13.1)
Обычно с помощью GFT-пептида модифицируют гены,
экспрессию которых оценивают в ходе скрининга. Культуру клеток с
модифицированными генами инкубируют с библиотекой тестируемых
соединений и регистрируют интенсивность флуоресценции. Наличие
таковой свидетельствует о способности отдельных соединений из
испытуемого массива оказывать влияние на экспрессию генов
определенного типа, что и является критерием активности исследуемых
БАВ [21, 22].
Следует подчеркнуть, что on-bead-исследования в большинстве
случаев рассматривают как предварительные, позволяющие получить
качественный результат до того, как вещества будут сняты с носителя
и переведены в раствор. После этого обычно проводят повторные
испытания, результаты которых иногда не совпадают с данными
первичного скрининга. Последнее можно в определенной степени
объяснить тем, что в условиях гидрофобного окружения на носителе
соединения находится в конформации, отличной от той, в которой они
присутствуют в растворе. Кроме того, в процессе расщепления
линкеров не исключена возможность взаимодействия используемых для
этого реагентов с соединениями, входящими в состав библиотеки.
Эти недостатки ограничивают сферу применения on-bead-скрининга.
Off-bead-исследования. Вещества из mix-and-split библиотек
(см. разд. 12) можно исследовать как в закрепленном на матрице виде,
так и в виде соединений, переведенных с гранул в раствор или гель —
ofF-bead-исследования. Наиболее распространенным методом
скрининга указанных библиотек является гелевая диффузия [7, 18]. Этот
метод имеет много общего с зоной ингибирования в
микробиологическом скрининге. Обычно набор гранул с закрепленными на
364
них соединениями распределяют равномерно на тонком слое агаро-
зы, в котором и проводят биологические испытания. Соединения
снимают с носителя фоторасщеплением или парами кислоты.
Благодаря высокой вязкости агарозы, отдельно взятое вещество
диффундирует только в небольшой участок геля непосредственно под
гранулой, с которой оно было связано. Агарозу пропитывают субстратом,
подходящим для данного вида биологических испытаний.
Существующие методы детектирования позволяют установить место
нахождения активного соединения. Далее происходит расшифровка
закрепленных структур. В настоящее время этот метод представляет собой
очень мощный инструмент в отборе соединений из комбинаторных
библиотек.
13.4. Исследования на лабораторных животных
Исследования на анестезированных животных. Данный вид
испытаний БАВ позволяет изучать различные пути их введения, измерять
большое количество параметров, создавать при необходимости
большие экспериментальные группы. К недостаткам следует отнести
необходимость проведения исследований в условиях анестезии, когда
многие рефлекторные процессы подавлены, и невозможно оценить
поведенческие реакции. Кроме того, ЛС, вызывающие
продолжительную анестезию, могут повлиять на свойства испытуемых соединений.
В экспериментах на обездвиженных животных применимы те же
принципы, что и в исследованиях in vitro. Кривые «доза — ответ» для аго-
ниста строят при нескольких режимах введения, и эти эксперименты
повторяют до получения воспроизводимого результата*. Таким путем
определяют как абсолютную, так и относительную эффективность
агониста. Если используют стандартный агонист, то после
завершения испытаний может быть введен антагонист, и зависимость «доза -
ответ» оценивают уже в его присутствии. Это позволяет определить
выраженность эффекта антагониста in vivo. Изучаемое вещество
может быть введено как непосредственно в интересующий орган, так и
в системное кровообращение. При таком виде биологических
испытаний также можно столкнуться с явлением тахифилаксии, что
следует учитывать при планировании эксперимента и сводить к
минимуму возможные осложняющие факторы [23, 24].
Исследования на интактных животных или с моделями заболеваний.
На завершающей стадии доклинических испытаний
структуры-лидера или кандидата в ЛС проводят обычно исследования с
использованием интактных животных или животных с моделями
соответствующих заболеваний [25]. Эти исследования носят менее повреждающий
характер, приближены к клиническим условиям введения вещества,
могут продолжаться от нескольких минут до нескольких лет (в
течение всего периода жизни животного) и позволяют оценить
поведенческие реакции в динамике на одном и том же животном. На
интактных животных проверяют специфическое действие кандидата в ЛС
365
и проводят токсикологические исследования. Модели служат,
главным образом, для оценки специфических эффектов потенциального ЛС.
Модели заболеваний или отдельных симптомов каких-либо
патологических процессов разделяют на химически индуцированные
и генетически детерминированные. ^ первом случае патология
возникает при введении в организм животного определенного
химического вещества. Например, анальгетические свойства
ненаркотических веществ чаще всего изучают на модели «уксуснокислых
корчей» у крыс или мышей, противовоспалительные свойства БАВ
оценивают по способности вещества ингибировать отек, вызванный
подкожным введением экспериментальным животным каррагенина,
формалина или др. флогогена, антиоксидантную активность чаще
всего исследуют на модели однодневного токсического гепатита,
индуцированного пероральным введением 50% масляного раствора
СС14 [25—27] и т. д. Следует подчеркнуть, что рассмотренные
модели не воспроизводят ведущие звенья патологического процесса у
человека, а лишь отражают отдельные симптомы заболеваний. Они
пригодны в основном для первичного скрининга. На завершающих
этапах доклинических исследований целесообразно использовать
модели, максимально приближенные к особенностям патологии у
человека. Особенно остро вопрос соответствия механизма развития
заболевания у человека и животного стоит при моделировании
таких гетерогенных метаболических расстройств, как СД, ИБС,
бронхиальная астма, ревматоидный артрит и др. В этом случае наряду
с химически индуцированными моделями широко используют
линейных животных с генетически детерминированными
метаболическими нарушениями. Например, оценку антидиабетической
активности потенциальных ЛС, предназначенных для лечения
ИНЗСД, проводят на мышах линии КК, у которых отмечаются
такие патогенетические звенья СД, как гипергликемия, гиперинсу-
линемия, дислипидемия, или мышах линии db/db (гипергликемия,
гипоинсулинемия, дислипидемия), тучных крысах Zuker rat или fa/fa
(нормогликемия, гиперинсулинемия, дислипидемия). Ценность этих
моделей состоит в том, что они отражают гетерогенность
клинических форм ИНЗСД и, следовательно, обеспечивают практически
адекватную экстраполяцию полученных экспериментальных
результатов на человека [28, 29].
Проблема исследования БАВ на лабораторных животных
чрезвычайно многогранна, и отдельные ее стороны широко освещены в
специальной литературе [2, 5, 6, 23—25]. В последнее время вокруг
экспериментальных работ такого рода ведется большая полемика [30,
31]. Принимая во внимание этический аспект подобных
исследований, предлагается использовать альтернативные методы [32, 33].
Однако на завершающих этапах доклинических испытаний новых ЛС
замена экспериментов на животных альтернативными подходами
невозможна. Как уже было отмечено выше, экстраполяция на человека
результатов исследований, полученных на животных, представляет
определенные трудности, но переносить1на человека данные, получен-
366
ные только альтернативными методами, — еще более сложная задача.
Безусловно, при работе с экспериментальными животными
необходимо соблюдать определенные правила этического характера,
сформулированные в соответствующих документах [34]. Альтернативные
методы исследований можно с успехом применять при установлении
механизма специфического или токсического действия ЛС.
Значимость фармакологических испытаний на животных очень точно
выразил в свое время И.П. Павлов: «Чем полнее будет проделан опыт на
животных, тем менее часто больным придется быть в положении
опытного объекта со всеми печальными последствиями» [35].
Литература
1. Leake CD. A Historical Account of Pharmacology to the 20th Century. —
Springfield: Charles С Thomas, 1975.
2. Альберт А. Избирательная токсичность, Т. 1—2. — M.: Медицина, 1989.
3. Харкевич Д.А. Фармакология: Учебник. — 6-е изд., перераб. и доп. —М,:
ГЭОТАР Медицина, 1999.
4. Medicinal Chemistry: Principles and Practice / Ed. by F. D. King — Cambridge:
The Royal Society of Chemistry, 1994.
5. Daniel E.E., Paton DM. Methods in Pharmacology, Vol. 3. Smooth Muscle. —
London: Plenum Press, 1975.
6. Receptor-Ligand Interactions — A Practical Approach / Ed. by D. Rickwood,
B.D.Hanes. - Oxford: IRL Press, 1992.
7. Combinatorial Chemistry and Molecular Diversity in Drug Discovery / Ed. by
E.M. Gordon, J.F. Kerwin. - New York: Wiley-Liss, Inc, 1998.
8. Razvi E.S. // Drug & Market Development. - 1998. - Vol. 9. - P. 89.
9. Burbaum J. J.y Sigal N.H. // Curr. Opin. Chem. Biol. - 1997. - Vol. 1. - P. 72.
10. Knebel G // Am. Biotech Laboratory. — 1998. - Vol. 16. - P. 22.
11. Oldenburg K.R. et al. // J. Biomol, Screening. - 1998. - Vol. 3. - P. 55.
12. Kolb A J. Neumann K. // J. Biomol. Sceening. - 1997. - Vol. 2. - P. 103.
13. Lehel C. et al. // Anal. Biochem. - 1997. - Vol. 244. - P. 340.
14. Jolley M.E. H J. Biomol. Screening. - 1996. - Vol. 1. - P. 33.
15. Kolb J.M., Yamanaka G, Manly S.P. // J. Biomol. Screening. - 1996. -
Vol. 1. -P. 203.
16. Hill DC. II Curr. Opinion. Drug Discov. Develop. - 1998. - Vol. 1. - P. 92.
17. Gordon E.M. et al. 11 J. Med. Chem. - 1994. - Vol. 37. - P. 1385.
18. A Practical Guide in Combinatorial Chemistry / Ed. by A.W.Czarnik,
S. H. De Witt. - New York: John Wiley & Sons, 1997.
19. Misteli Т., Spector D.L. // Nature Biotech. - 1997. - Vol. 15. - P. 961.
20. Ormo M. etal. // Science. - 1996. - Vol. 273. - P. 1392.
21. ChiocchettiA. etal. Ц Biochem. Biophys. Acta. - 1997. - № 1352. - P. 193.
22. Finney N.S. 11 Curr. Opinion. Drug Discov. Develop. — 1998. — Vol. 1. —
P. 98.
23. Rang H.P., Dale M.M. Pharmacology. — Edinburgh: Churchill Livingstone,
1991.
24. Smith СМ., Reynard A.M. Textbook of Pharmacology. — Philadelphia: Saumders,
1991.
25. Доклгшчш дослщження лкарських засобш (методичш рекомендаш'О- —
К.: МОЗУкраши, 2001.
26. Катеуата А. // Drug. Res. - 1987. - Vol. 37, № 1. - P. 137.
367
27. Поздняков В Иванов Н. // Токсикол. новых промышленных химических
веществ. - 1979. - Вып.15. - С.87.
28. Portha В., Blondel О., Serradas P. et al. // Diabete & Metab - 1989 -
Vol. 15. — P. 61.
29. ZucherZM., Antoniades H.N. // Endocrinology. - 1972. - Vol. 90, N. 5. -
30. KnJnsha'nBA.H., Dammem S.J.P., West C. // Hum. and Exp. Toxicol.
1996. — Vol. 15, N. 5. — P. 452
31. Glomot R. И Sci. Et. Techn. Anim. Lab. - 1999. - Vol 24 N 1 - P 8
32. Clemedson C, Barile F.A., Ekwall B. et al. // AT LA - 1998 - Vol 26
Suppl.l. — P. 93. ■
33. Okamoto Y., Ryu A., Ohkoshi K. // ATLA - 1999. - Vol. 27, N 4 - P 639
34. Березовская И.В. 11 Токсикол. вестник. — 1999. — № 6. — С 2
35. Гуськова ТА. // Хим.-фарм. журн. — 2002. — № 10. — С. 3.
14
ЭТАПЫ СОЗДАНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Создание нового ЛС — комплексный процесс, требующий
значительных усилий по координации работы широкого круга
исследовательских групп в течение длительного промежутка времени. В
недавнем прошлом такие исследования продолжались 10—15 лет. На
современном этапе, благодаря глобальным изменениям в технологии
синтеза и скрининга новых соединений, время, затрачиваемое на
разработку нового ЛС, сократилось до 8—10 лет. Стоимость такой
разработки, в особенности на завершающих этапах при проведении
клинических испытаний с участием сотен пациентов, чрезвычайно высока.
По разным данным, в зависимости от фармакологического профиля
создаваемого ЛС, она составляет 350—500 млн $ [1]. Только одно из
10 веществ изучается на культурах тканей человека. В целом, из 104
соединений одно обладает всеми необходимыми свойствами,
позволяющими продвигать его в качестве кандидата в ЛС (рис. 14.1).
Относительные затраты на проведение каждого этапа разработки нового
препарата (в%) представлены на диаграмме (рис. 14.2).
1
10—15 лет а - 350—500 млн $
Материальные
затраты
Рис. 14.1. Материальные затраты, приходящиеся на разработку нового Л С
Наиболее распространенные причины отклонения кандидатов в ЛС
состоят в следующем [1]:
• низкая эффективность — 46%;
• неудовлетворительные показатели ADME — 7%;
• токсичность, выявленная на доклинической стадии — 17%;
• побочные эффекты, выявленные в клинике —16%;
• коммерческие причины — 7%;
• другие недостатки — 4%.
Время
369
Кпинич.
испыт.
30%
Др. исслед.
15% \
^-<lz /
10% / "
Разраб.
лек. формы
рг
/ 10%
Токсикол.
исслед.
Хим.
йработки 10%
-^Биоп. испыт.
~2) 15%
\ 10%
Фармакокин
исслед.
Рис. 14.2. Относительные затраты на каждом из этапов создания ЛС
Избежать отрицательного результата на завершающих стадиях
позволяет тщательная проработка проекта при выборе
направления исследования и высокие требования к отбору лидирующих
структур (см. разд. 9). Этапы, которые проходит новое химическое
вещество, прежде чем получит распространение в медицинской практике
как ЛС, представлены на рис. 14.3.
Химия
Выбор
способ*
Пилотно-технический гроект
Фарма колотя
Специфичвска»
активность
Клиника
Острая и
псдострая
токсичность
Хроническая токсичность
1 фиэа
II фаза
Шфиза
Анализ
результатов
Организация
Предоставление результатов о клинических исл.
Подача докуй.
Утверждение
Постмаркетинг
Фармтехиология
Фарма коки нетика
Разработка лек. форм и НТД на них
ADME у животных
Здоровые
добровольцы
Пациенты
Рис. 14.3. Этапы создания ЛС
Те виды деятельности, которые определяют основное время,
необходимое для разработки препарата вплоть до его регистрации,
называются «критическими» или на языке химической кинетики
«скорость определяющими». В интересах любой фармацевтической
компании в условиях экономического прессинга и жесткой
конкуренции свести к минимуму эти критические этапы. Кроме того, в
целях экономии времени многие исследования проводят параллельно,
например, клинические испытания совпадают по времени с
разработкой пилотных технологических схем производства.
370
Наиболее важными по степени риска являются выбор метода
синтеза, исследования безвредности, а также клинические испытания.
Главные этапы создания ЛС — доклинические и клинические
исследования. В большинстве стран принято проводить клинические
испытания в три стадии: I — на здоровых добровольцах, II —
эффективность и безопасность, подбор доз, III — фаза исследований,
охватывающая сотни пациентов. В Украине не проводят испытаний
новых ЛС с участием здоровых добровольцев.
14.1. Химические разработки
Скорость разработки препарата зависит от доступности ЛВ, что, в
свою очередь, определяется способом синтеза и стоимостью сырья,
из которого это вещество получают. Чистота вещества уже на этапе
изучения специфической активности и острой токсичности должна
удовлетворять имеющимся стандартам, чтобы можно было создать
подходящую ЛФ и, кроме того, обеспечить необходимое
экспериментальное обоснование для будущих клинических испытаний.
Для этого всесторонне изучают химические свойства целевого
продукта — ЛВ (субстанции), выбирают оптимальный путь синтеза
(наиболее эффективный, дешевый, безопасный, проходящий с
наименьшими потерями). Обязательным является выяснение состава
примесей в целевом продукте. Порой именно из-за неприемлемого
содержания в нем токсичных примесей приходится менять всю схему
получения ЛВ. Параллельно происходит разработка методов анализа
субстанции, позволяющих количественно охарактеризовать
содержание основного вещества, выявить полный состав примесей и
нормировать их. Главная цель аналитической аттестации — точное знание
состава продукта и его чистоты. В дальнейшем методики,
разработанные для оценки качества ЛВ, закладывают в аналитическую
нормативную документацию (АНД).
По мере продвижения кандидата в ЛС по этапам исследования
требуются все большие количества субстанции. Потребность в Л В
зависит от его активности, испытуемых доз, а также ЛФ. Например, при
изучении безвредности испытывают ЛВ и его ЛФ в терапевтической
и нескольких кратных ей дозах в течение длительного времени.
Пилотный проект по изготовлению ЛВ рассматривают как мини-
производство. Перед внедрением в промышленных масштабах этот
проект подвергают тщательному анализу, применяемые методики
оптимизируют. На сегодняшний день приемлемыми для
промышленного использования являются такие способы синтеза, которые
обеспечивают надлежащее качество и выход целевого продукта при
минимальном воздействии на окружающую среду.
Одним из основных условий производства ЛС и торговли ими на
мировом рынке является обеспечение высокого качества, в первую очередь, за
счет выполнения принципов и правил надлежащей производственной
371
практики (Good Manufacturing Practice — GMP). GMP — это система
мёр, направленная на обеспечение единых требований к организации,
контролю производства и оценке качества фармацевтической продукции.
Она впервые предложена Всемирной организацией здравоохранения (ВОЗ)
в 1967 г. С тех пор система GMP неоднократно пересматривалась,
уточнялась и расширялась. Последняя редакция правил по надлежащей
практике производства (НПП) переведена на русский язык и издана в 1995 г. [2].
Требования НПП представлены в трех частях.
Часть первая — «Управление качеством в промышленности
лекарственных средств: основные принципы и существенные составляющие» —
намечает общую концепцию обеспечения качества, а также принципиальные
компоненты или подсистемы НПП, которые объединяют ответственность
администрации, управление производством и контроль за ним.
Ответственность включает такие стороны производства как персонал,
помещения, гигиена, оборудование, материалы, документация (утверждения
процедур, технологий, испытаний и изменений к ним), работа по контракту,
рекламации, самоинспекция.
Часть вторая — «Надлежащая практика производства и контроля
качества» — обеспечивает указания по действиям, предпринимаемым
отдельно производственным персоналом и персоналом подразделения контроля
качества для внедрения общих принципов гарантий качества.
Часть третья — «Вспомогательные и дополнительные принципы» — в
настоящее время содержит два дополнительных руководящих принципа.
Один из них регламентирует организацию производства и контроль
качества стерильных фармацевтических препаратов, а второй устанавливает
основные требования к производству активных фармацевтических
ингредиентов, т.е. Л В или субстанций, для которых также принято название —
нерасфасованные конечные продукты. Но этот раздел НПП остается
незавершенным. Предполагается, что в дальнейшем будут разработаны
руководящие принципы, например, для биологических препаратов, матери-
лов для клинических испытаний и др.
В 1996 г. в Украине введен вдействие закон «Про л1карсью засоби»,
который регулирует правовые отношения, связанные с созданием,
регистрацией, производством, контролем качества и реализацией ЛС, определяет
права и обязанности предприятий, учреждений, организаций и граждан,
а также полномочия в этой сфере органов государственной
исполнительной власти и должностных лиц. В соответствии с этим законом
управление в сфере создания, производства, контроля качества, реализации ЛС
осуществляют Министерство здравоохранения (МЗ) Украины,
Государственный комитет Украины по медицинской и микробиологической
промышленности (Госкоммедбиопром) и специально уполномоченные ими
государственные органы.
Производство ЛС на территории Украины осуществляется на основании
лицензий. Основанием для выдачи лицензии является наличие
соответствующей материально-технической базы, квалифицированного
персонала, а также условий для контроля качества производимых ЛС. Общие
требования к материально-технической базе, производственному
контролю, а также технологическим регламентам устанавливает
Госкоммедбиопром с учетом государственных стандартов и международных норм
касательно производства ЛС.
МЗ Украины уполномочило заниматься вопросами регистрации ЛС
такие государственные органы как Фармакологический и Фармакопейный
372
комитеты. Фармакологический комитет — научно-экспертная
государственная организация, в компетенцию которой входят все вопросы,
связанные с экспертизой материалов доклинического и клинического изучения
ЛС, разработка нормативных документов, регламентирующих эти этапы
исследования препаратов, экспертиза образцов ЛС, направляемых на
клиническое изучение, выдача рекомендаций по регистрации ЛС и т. п.
Фармакопейный комитет — научно-экспертный орган, занимающийся
экспертизой и утверждением АНД на готовые ЛС, лекарственные и
вспомогательные вещества; этот комитет дает рекомендации к государственной
регистрации ЛС, принимает участие в аттестации производств и
лабораторий по контролю качества фармацевтических препаратов,
разрабатывает Государственную Фармакопею Украины и отраслевые нормативные
документы, регламентирующие требования к качеству ЛС и др.
Таким образом, в соответствии со стратегией ВОЗ в Украине созданы
и развиваются два звена системы обеспечения качества ЛС:
• система регистрации и лицензирования, которая предваряет
реализацию ЛС на рынке;
• система независимых испытаний ЛС на соответствие требованиям АНД
в отделах технического контроля (ОТК) предприятий и лабораториях
по контролю качества Л С, к которым имеют доступ все предприятия-
производители продукции фармацевтического назначения.
Следует подчеркнуть, что третье звено, связанное с обеспечением
гарантий качества ЛС за счет организации производства в соответствии с
правилами GMP, пока не получило должного развития. По этой причине
Украина в настоящее время не может присоединиться к системам
сертификации качества ЛС для международной торговли. В этой связи с 1997 г.
в Украине, как и в целом в СНГ, определен переходный период от
прежних норм и стандартов в области производства, контроля качества,
регистрации, лицензирования фармацевтической продукции к системе
правил GMP [3, 4]. На переходный период основными методическими
пособиями для стран СНГ по НПП являются законодательные
положения ЕС, руководства по GMP ЕС, ВОЗ и др. международных
организаций. Следует отметить, что принципы и правила GMP ЕС и ВОЗ
практически идентичны и отличаются, главным образом, порядком изложения [4].
В Украине согласно ОНД 09-001-98, основным документом,
отражающим технологию синтеза активного фармацевтического
ингредиента или переработки его в ЛФ является производственный
регламент [5]. Регламенты подразделяют на технологические и технические.
Технологический регламент — нормативный документ, в котором
приведены технологические методы, технические средства, нормы и
нормативы изготовления ЛС. Технологический регламент бывает двух
категорий — временный и промышленный. Согласно временному
регламенту выполняются лабораторные и опытно-промышленные
работы, изготовление опытных партий ЛС для проведения доклинических
и клинических исследований. Временный регламент является
документом на право получения разрешения к медицинскому
применению ЛС и утверждения временного АНД. Технологический
промышленный регламент разрешает серийное производство ЛС и является
основным документом регистрации препарата в Украине при поста-
373
новке его на производство. Срок действия такого регламента — пять
лет. Технологический промышленный регламент, независимо от
категории, включает разделы:
— характеристика готовой продукции;
— схемы производства и технологический процесс:
• схемы производства (химическая, технологическая);
• характеристика сырья, материалов, полупродуктов;
• описание стадий технологического процесса;
• материальный баланс;
— контроль производства;
— приложения: перечень технологических инструкций и перечень
форм протоколов производства.
Технический регламент — нормативный документ, в котором для
конкретного комплекса технологического оборудования изложены
условия, обеспечивающие выпуск полупродуктов или ЛС заданного
качества, условия эффективной и безопасной эксплуатации
оборудования и требования по охране окружающей среды. Технический
регламент включает следующие разделы:
— общая характеристика производства;
— аппаратурная схема, спецификация оборудования и контрольно-
измерительные приборы (КИП);
— эксплуатация технологического оборудования и КИП;
— общая схема контроля качества;
— безопасная эксплуатация производства и охрана окружающей
среды;
— общий перечень производственных инструкций;
— информационные материалы:
• приложение о техническом состоянии производства;
• информационные приложения о ЛС;
• протоколы валидации производства.
14.2. Разработка лекарственной формы
ЛФ — это расфасованный конечный продукт переработки Л В,
предназначенный для использования в медицинской практике. В
настоящее время существует широчайший набор ЛФ. Так, кроме
традиционных таблеток, мазей и растворов (инъекционные средства, капли)
применяют также гранулы, капсулы, трансдермальные
терапевтические системы, интраназальные, сублингвальные формы, аэрозоли,
суппозитории и т. п. Выбор ЛФ определяется профилем кандидата в ЛС.
Иногда для ускорения процесса исследований используют самую
простую ЛФ — раствор для перорального применения, которую изучают
на ранних этапах клинических испытаний, когда велик риск неудачи
(с целью экономии средств); на более поздних этапах изучают ту ЛФ,
которая будет выпущена на рынок.
Для создания ЛФ важны входящие в ее состав ингредиенты.
Например, при изготовлении инъекционных растворов, кроме ЛВ и ра-
374
створителя (обычно Н20), используют стабилизаторы (сульфит
натрия, ЭДТА), консерванты (этанол) и др. В производстве таблеток
применяют наполнители (сахара, крахмал), скользящие (стеарат
кальция или магния), связующие (поливинилпирролидон) и т.п.
вещества, способствующие формированию таблеточной массы. Выбор
вспомогательных веществ зависит как от технологических
характеристик субстанции, так и от профиля препарата. К технологическим
характеристикам относятся — угол откоса, насыпная масса, форма
кристаллов, прессуемость ЛВ и др. Эти показатели учитывают при
разработке технологии таблетирования.
При создании ЛФ принимают во внимание и фармакокинетические
свойства ЛВ. Так, для предотвращения негативного воздействия
кислой среды в желудке таблетки покрывают специальной оболочкой
или расфасовывают субстанцию в капсулы, обеспечивая тем самым
распадаемость ЛС в кишечнике. Используя подходящие наполнители,
можно изменить скорость всасывания ЛВ и за счет этого достичь
постоянной концентрации в крови в течение длительного
промежутка времени.
Производство ЛФ, также как и субстанции, осуществляется
согласно регламенту. Требования, предъявляемые к этому документу, и
основные разделы, представленные в нем, такие же, как и при
производстве субстанции. Показатели качества ЛС, так же как и ЛВ,
отражены в АНД. Цель этого документа — обеспечить идентификацию
активного фармацевтического ингредиента, представить основные
физико-химические характеристики, позволяющие оценить его
качество и качество ЛФ и стандартизовать их. В АНД на ЛВ входят
следующие показатели:
• описание;
• идентификация;
• растворимость;
• температура плавления;
• потеря в массе при высушивании;
• сульфатная зола;
• посторонние примеси;
• количественное определение;
• остаточные растворители.
Для ЛС, кроме перечисленных выше, приняты и специальные
характеристики, которые зависят от вида ЛФ. Например, для таблеток
используют такие показатели как средняя масса, растворимость и
прочность. Одной из наиболее важных характеристик ЛФ является ее
стабильность, которая определяет срок хранения препарата, обычно
не менее двух лет при хранении в естественных условиях. В течение
этого срока, через определенные промежутки времени (как правило
6 мес.) ЛФ, заложенную на хранение, анализируют по тестам АНД.
Требования, предъявляемые к АНД, отражены в Государственной
Фармакопее Украины [6]. Фармакопея — правовой документ, который
содержит общие требования к ЛС, фармакопейные статьи (монографии), а также
методики контроля качества ЛС. В Украине Государственная Фармако-
375
пея принята в 2001 г. Этот документ имеет законодательный характер. Ее
требования, предъявляемые к ЛС, являются обязательными для всех
предприятий и организаций Украины, независимо от форм собственности,
производящих, хранящих, контролирующих, реализующих и
использующих ЛС. Фармакопея содержит сведения о физических и
физико-химических методах анализа ЛС, способах идентификации, испытаниях на
допустимое содержание примесей, методах количественного определения,
биологических испытаниях (стерильность, пирогенность,
микробиологическая чистота и т. п.), фармако-технологи ческих испытаниях, а также
реактивах, используемых в фармацевтическом анализе. Кроме выше
перечисленного, в ней приведены монографии на отдельные ЛС и общие
статьи на ЛФ и субстанции. Основным отличием Государственной
Фармакопеи Украины от предыдущего нормативного документа —
Государственной Фармакопеи СССР XI издания — является то, что она
полностью гармонизирована с Европейской Фармакопеей, которая предполагает
изготовление ЛС согласно нормам GMP. В настоящее время в Украине
нет условий для перехода на обязательное исполнение этих требований.
Этот недостаток в какой-то мере приходится компенсировать за счет
повышения требований к качеству конечного продукта. Поэтому при
разработке Государственной Фармакопеи Украины соответствующие статьи
Европейской Фармакопеи были дополнены требованиями, которые
учитывают специфику фармацевтического производства в Украине.
14.3. Фармакологические испытания
Прежде чем кандидат в ЛС будет исследован в клинике, его
влияние на основные органы и системы организма оценивают в
эксперименте на животных. При этом обязательно рассматривают
влияние на сердечно-сосудистую, нервную, дыхательную, мочевы-
водящую, пищеварительную, кровеносную системы и печень.
Часто кандидат в ЛС исследуют на предмет взаимодействия с другими
медикаментами, которые, благодаря своему специфическому
действию или широкому употреблению, вероятно, могут с ним
конкурировать. Любой эффект антагонизма или синергизма должен быть
изучен для подготовки рекомендаций и предостережений
клиницистам. До начала клинических испытаний устанавливают также ЛС
для борьбы с возможными явлениями передозировки данного
средства, особенно если его терапевтическая доза невелика, что
свидетельствует о принадлежности испытуемого вещества к
сильнодействующим препаратам.
Следует отметить, что программа испытаний зависит от того,
является ли предлагаемое ЛС оригинальным или предполагаются
иной путь введения уже известного препарата, принципиально
новый подход к дозировке, применение по новому назначению и
т.п. Испытания проводятся и в связи с изменениями в технологии
синтеза субстанции или вида, состава и/или технологии
изготовления ЛФ. Наибольший объем исследований предполагается в случае
изучения новой субстанции. Он включает токсикологические, фар-
376
макологические, клинические испытания и фармакокинетические
исследования. Токсикологические испытания предполагают:
изучение острой токсичности на трех видах животных; подострой и
хронической токсичности; местнораздражающего, ульцерогенного и
кумулятивного действия; экспериментальное лечение отравлений при
передозировке; оценку аллергенности, иммунотоксичности, терато-
генности, гонадотоксичности, мутагенности и канцерогенности*;
выявления возможного развития лекарственной зависимости* (* — при
необходимости). Фармакологический блок исследований включает
общую фармакологию и оценку специфической активности по
нескольким критериям. Если предполагается регистрация уже
известного препарата, но с новым способом введения, то объем
испытаний сводится к изучению острой, подострой, хронической
токсичности и специфического действия. Любое изменение
технологии производства субстанции или ЛФ влечет за собой новый
набор биологических испытаний.
Особое внимание уделяется условиям проведения
токсикологических исследований. Они должны удовлетворять международным
требованиям по надлежащей лабораторной практике — Good Laboratory
Practice (GLP), которые регламентируют условия содержания
животных (рис. 14.4), требования к обслуживающему персоналу, ведению
документации, планированию эксперимента, контролю полученных
результатов и т. п [7, 8].
в
Помещение для
сжигания
отходов
Помещение для
контроля за
здоровьем
животных
Помещение для
контроля за
здоровьем
животных
Грязный
коридор •+
Душевая
Раздевалка
Помещение для
приёма животных
* Помещение для содержания животных
А: "Пред процедурная " зона
Б: "Процедурная" зона
В: "Постпроцедурная" зона
Рис. 14.4. Схема помещения для содержания животных,
участвующих в токсикологических исследованиях
377
Впервые эти правила были предложены в 1976 г. в США
управлением по контролю качества продуктов и медикаментов — US Food
and Drug Administration (FDA); в Японии эта система мер введена в
действие в 1983 г. Постепенно требования GLP были приняты и в
странах ЕС. В настоящее время, также как и нормы GMP, эта
система поэтапно внедряется в Украине. Цели установления правил GLP
следующие:
• обеспечение качества и согласованности представляемых данных;
• единообразие требований к лабораторным помещениям,
оборудованию и рабочему персоналу;
• составление и соблюдение подробной стандартной методики
экспериментальных работ — Standard Operating Procedure (SOP)
и порядка проведения испытаний (протокола);
• проверка испытаний в ходе их проведения путем создания
службы по качественной оценке испытаний — Quality Assurance Unit
(QAU);
• четкое распределение ответственности отдельных исполнителей
испытаний;
• точная регистрация данных;
• хранение всех данных испытаний, отчетов, препаратов;
• контроль за пригодностью учреждения к проведению испытаний.
Острая и хроническая токсичность. Токсикологические
исследования на животных проводят до того, как потенциальный препарат
будет испытан на людях, чтобы отклонить вещества с высокой
токсичностью, определить органы-мишени для токсического воздействия
и время наступления побочных эффектов. Это означает, что на
ранних стадиях клинических испытаний эти системы и органы будут
находиться под пристальным контролем. Очень важно установить,
является ли токсическое действие обратимым или нет, каков его
механизм и, следовательно, можно ли его предотвратить.
Токсикологические исследования проводят на 2—3-х видах
животных с учетом специфического эффекта препарата. Они
необходимы для подбора доз ЛС (размер, частота и курс приема), повторности
курсов и оценки продолжительности клинических испытаний в
будущем. Эти показатели являются основными для получения
разрешения на проведение испытаний с участием добровольцев.
Скандинавские страны, Великобритания, США, Австралия разработали
специальные рекомендации, регулирующие длительность воздействия
ЛС на человека в зависимости от продолжительности
токсикологических исследований.
Первоначально исследуют острую токсичность — вредное действие
препарата, проявляющееся после его однократного применения или
повторного введения через короткие (не более 6 ч.) интервалы в
течение суток. Целью эксперимента является определение переносимых,
токсических и летальных доз ЛВ и/или ЛС и причин гибели
животных. Исследования проводят на 2—3-х видах животных, один из
которых — не грызуны. При этом вводят все возрастающие дозы Л В/Л С
двумя путями, один из которых соответствует предполагаемому для
378
применения у человека. Эти исследования проводят на небольшом
количестве животных, но не менее 5—6 особей в группе. Таким
способом определяют ЛД50. Если из-за низкой токсичности вещества
определить ЛД50 не представляется возможным, то указывают
максимальную дозу, которая была введена. Общая продолжительность
наблюдения за животными при исследовании острой токсичности
составляет не менее 2-х недель, причем в первый день животные
находятся под непрерывным наблюдением. Это позволяет установить
максимально переносимые дозы для проведения тестов хронической
токсичности, определить разброс доз и идентифицировать органы-
мишени.
Основная цель изучения субхронической токсичности —
определить, как переносится ЛС при длительном (2—4 нед.) введении, при
том же способе введения, что и рекомендуемый для человека, на двух
видах животных (один вид — не грызуны). Обычно испытывают три
дозы: минимальную и максимальную суточные, минимальную
многократную — терапевтическую.
Общим требованием для испытаний нового ЛС в однократной
дозе на добровольцах является наличие результатов о
токсикологических исследованиях продолжительностью не менее 14 дн.
Токсикологические исследования, проведенные в течение 30 дн., позволяют
представить вещество для клинических исследований
продолжительностью 7—10 дн. Клинические испытания свыше 10 дн. требуют
предварительных токсикологических исследований в течение 90 дн.
Применение препарата более 6 мес. требует проверки его токсических
свойств в течение 12—18 мес. на двух видах животных, один из
которых — не грызуны.
Для выявления способности у кандидата в ЛС вызывать
новообразования у человека используется два типа тестов. Первый —
кратковременный тест in vitro на генотоксичность — обычно
проводится с использованием бактериальных клеток. Долгосрочный
эксперимент по проверке канцерогенных свойств продолжается в течение
двух лет на большом количестве лабораторных животных (мышах или
крысах).
Репродуктивная токсикология исследует влияние потенциального
препарата на фертильность, фетотоксичность, постнатальное
развитие. Перед проведением клинических испытаний с участием женщин
репродуктивного возраста необходимо изучить тератогенность на двух
видах животных. Для клинических испытаний на добровольцах
мужского пола данные о влиянии на репродукцию не требуются. Такие
сведения необходимы, когда препарат предназначен для длительного
применения у лиц репродуктивного возраста обоего пола.
Метаболизм и форма ко кинетика. Эти исследования проводят с
целью получения информации о превращениях ЛС в организме в
зависимости от вводимых доз и путях выведения метаболитов. Оценка
метаболизма требует измерения концентрации ЛВ и его метаболитов
в биологических жидкостях и тканях. Чаще всего для этого
используют ВЭЖХ или ГЖХ с детектированием УФ, флуориметрическими,
379
масс-спектрометрическими и др. способами. Проблема
детектирования стоит особенно остро, когда активность ЛВ очень высока и его
применяют в малых дозах, что приводит к присутствию его в тканях в
следовых количествах. В этом случае наиболее информативным
может оказаться радиоиммунологический метод. Однако при этом
возникает проблема специфичности, т.к. не только сам препарат, но и его
метаболиты могут содержать метку.
Информация о концентрации Л В и его метаболитов необходима для
подтверждения результатов токсикологических экспериментов и
подбора оптимальной дозы. При проведении фармакокинетических
исследований очень важно на кривой зависимости «концентрация — время»
выявить участок, который имеет линейный характер (см. также разд. 7).
Установление причин нелинейности, таких как метаболическое
насыщение и реабсорбция, способствует пониманию особенностей
токсических и фармакодинамических эффектов испытуемого ЛС.
14.4. Клинические испытания
При успешном завершении испытаний на животных проводятся
клинические испытания. Планирование этих исследований
осуществляют при участии разработчиков кандидата в ЛС (представителей
фармацевтической компании), клиницистов, комиссии по этике, а в таких
странах как США, Канада, страны ЕС, Россия и Украина вопрос
рассматривают специальные государственные учреждения. В США это FDA,
в Украине — Фармакологический комитет МЗ. Новый препарат
представляет спонсирующая компания (будущий производитель). В США
в течение 30 дней FDA обязана предоставить заключение
относительно безопасности клинических испытаний и, в случае положительной
оценки, разрешить их проведение. При отрицательной оценке требуются
дополнительные доказательства безопасности нового средства [7, 8].
В Украине для получения разрешения на проведение
клинических испытаний, кроме отчетов об изучении острой и хронической
токсичности, специфического действия, фармакокинетики и
программы клинических испытаний, необходимо представить в
Фармакологический комитет МЗ заключение о результатах рецензирования
технологической комиссией регламентов на изготовление субстанции и
ЛФ, технико-экономическое обоснование на их производство,
выписку из протокола об утверждении названия ЛС, проекты АНД,
сертификат контроля качества потенциального препарата и образцы Л С
в предлагаемой упаковке. При разработке программы клинических
испытаний важно четко обозначить профиль нового кандидата в ЛС
с тем, чтобы выявить его возможные преимущества, а в случае
неудачи — вовремя прекратить испытания. Основные задачи на этом этапе
состоят в следующем:
• определить безопасность и переносимость на людях;
• определить фармакокинетические показатели;
• установить фармакологический профиль.
380
Фармакологический комитет МЗ Украины осуществляет
экспертизу материалов доклинического исследования ЛС, анализ образцов
препарата, направляемого на клинические испытания, определение
клинических баз для таких испытаний, выдачу разрешения на их
проведение, утверждение инструкций и программы клинического
изучения, инструкций по применению, а также выдачу рекомендаций
касательно регистрации ЛС.
Ранние этапы клинических исследований дают возможность
уточнить уровень доз, выявить и контролировать побочные эффекты. При
этом необходимо получить максимум информации на минимальном
числе лиц и тем самым снизить риск для пациентов при дальнейших
исследованиях. Начинают испытания с однократной дозы, которую
постепенно увеличивают. Участвуют в этом небольшое количество
здоровых добровольцев, обычно молодых мужчин, состояние
которых постоянно контролируют. Таким путем подбирают максимально
переносимую дозу. Одна из причин привлечения здоровых молодых
людей состоит в необходимости выявления возможных побочных
реакций, связанных с применением потенциального препарата.
Оценка побочных реакций включает выявление повреждающего
действия изучаемого ЛС на ткани и органы, нарушающего их
функции вплоть до гибели клеток, и побочных явлений в виде
неожиданных и нежелательных эффектов, вызванных фармакологическим
действием ЛС в дозе, близкой к терапевтической. Женщин
репродуктивного возраста не привлекают к клиническим испытаниям, пока не
будут проведены для исследуемого вещества адекватные тесты на те-
ратогенность, эмбрио- и фетотоксичность. Клинические испытания
у мужчин в таких странах как США и Великобритания разрешено
начинать без данных о влиянии на репродукцию; в Японии же это
запрещено и исследования влияния вещества на фертильность
обязательно предваряют I стадию клинических испытаний.
Хотя все клинические исследования начинают со здоровых
добровольцев, изучение противораковых химиотерапевтических средств
и средств против ВИЧ проводят только с участием пациентов,
страдающих этими заболеваниями. В дополнение к данным о безопасности
на первой фазе клинических испытаний должны быть получены
фармакокинетические данные после однократного приема препарата и
последующего повторения доз. Определяют период полувыведения,
путь экскреции, возможность кумуляции.
Терапевтический эффект едва ли можно оценить в полной мере
на первой фазе, хотя все исследования проводят с учетом
специфического действия, принимают в расчет также и клиническую
«полезность». Путь введения ЛС должен быть тот же, что и при
токсикологических исследованиях. Начальную дозу для испытаний в клинике
выбирают, исходя из экспериментального материала. Приступать к
применению ЛС в нескольких дозах можно только после получения
надежных результатов при однократном введении. При
повторяющихся приемах в течение сут. временной интервал должен быть не
меньше, чем период полувыведения ЛВ из организма.
381
На П стадии кандидат в ЛС изучают у пациентов, которым
показаны средства данного профиля. Начинают эти испытания с
однократной минимальной дозы. При этом принимают во внимание все
аномалии в абсорбции, метаболизме, распределении, которые могут
возникнуть в связи с характером данной патологии. На более поздних
этапах II фазы исследования проводят с использованием placebo
(соответствующая ЛФ, которая не содержит активного фармацевтического
ингредиента) или препарата сравнения, если стоит вопрос о замене в
клинической практике одного ЛС на другое, более эффективное.
Наибольшее внимание уделяется органам-мишеням потенциального
препарата, а также печени и почкам, для того, чтобы
дифференцировать влияние данного ЛС от воздействия общего заболевания.
Третью фазу испытаний проводят с участием большого числа
пациентов. Эти исследования часто включают прямое сравнение с другими
терапевтическими средствами. Использование при этом placebo
зависит от вида заболевания и этических норм, принятых в стране. Так, в
Великобритании и большинстве стран Европы считается неэтичным
применять placebo тогда, когда больному нужна эффективная терапия;
в США требованиями FDA предусмотрено проведение хотя бы
одного хорошо спланированного исследования с placebo [7, 8].
Параллельно со второй и третьей фазами клинических испытаний
проводят специальные исследования, направленные на выяснение
взаимодействия кандидата в ЛС с препаратами, используемыми
влечении одновременно, или исследования с привлечением
специальных групп людей — пожилых или пациентов с нарушениями
функций печени или почек. Эти данные обеспечивают необходимую
информацию о возможных побочных эффектах при дальнейшем
клиническом применении нового ЛС.
В каждой стране существует правительственный орган,
рассматривающий результаты доклинических и клинических испытаний и
регулирующий выдачу разрешений на применение препарата.
Несмотря на различие критериев, общим для всех развитых стран является
однотипный подход: кандидат в ЛС должен быть подтвержденного
качества, безопасным и эффективным. Компании, желающие
продавать производимые ими препараты в другой стране, должны
провести проверку их безвредности и специфической активности на
животных по сокращенной программе испытаний, если предлагаемые
препараты широко известны, а также все необходимые этапы
клинических исследований в стране, покупающей эти препараты.
Литература
1. Combinatorial Chemistry and Molecular Diversity in Drug Discovery // Ed. by
E.M. Gordon, J.F. Kerwin. — New York: Wiley-Liss, Inc.,1998.
2. Надлежащая практика производства фармацевтических препаратов //
Комитет экспертов ВОЗ по спецификациям для фармацевтических
препаратов. 32-й доклад. Приложения!, Женева, Всемирная организация
здравоохранения. 1995. — С.18—116 — Серия технических докладов ВОЗ,
№ 823. —' М: Медицина, 1995.
382
3. Методические указания. Производство лекарственных средств.
Надлежащая практика и контроль качества МВ64У-1-97. — Киев: Госкоммед-
биопром Украины, 1997.
4. Надлежащая производственная практика лекарственных средств / Под
ред. Н.А. Ляпунова, В.А. Загория, В.П. Георгиевского, Е.П. Безутлой. —
К.: МОРИОН, 1999. - 896 с.
5. ГНД 09—001—98 Продукщя медично! та м1кроб1олопчно'1 промисловость
Регламенти виробництва лпсарських засоб1в. Зм1ст, порядок розробки,
узгодження та затвердження. — На замшу ОСТ 42У-2-92; Введ. 27.05.98. —
К.: Держком Укра'ши з медичноТ та яыкробюлопчноТ промисловосп, 1998.
6. Державна Фармакопея Украши / Державне пщприемство «Науково-екс-
пертний фармакопейний центр». — 1-е вид. — Харюв: Р1РЕГ, 2001.
7. Medicinal Chemistry: Principles and Practice / Ed. by F.D.King. — Harlow,
UK: Smith Юте Beecham Pharm., 1994.
8. Walker B.C. Walker S.R. Trends and Chenges in Drug Research and
Development. — London: Kluwer, 1987.
15
КОЛИЧЕСТВЕННЫЕ СООТНОШЕНИЯ
СТРУКТУРА - АКТИВНОСТЬ.
КРАТКИЙ ИСТОРИЧЕСКИЙ ОБЗОР,
ОСНОВНЫЕ МЕТОДОЛОГИЧЕСКИЕ ПОНЯТИЯ
Современные технологии конструирования ЛС ставят задачу
разработки методов надежного предсказания биологических видов
активности различных классов органических соединений. Эта
проблема имеет общее название количественное соотношение структура —
активность (Quantitative Structure — Activity Relationship, QSAR) и в
настоящее время является многопрофильной проблемой,
включающей в себя различные подходы не только в рамках медицинской
химии но и физики и математики.
История QSAR не имеет ясно очерченной даты начала. Первые
исследования связи структуры молекул с их активностью были проведены
в 60-х гг XIX века. Удивительно, что многие фундаментальные
закономерности QSAR, являющиеся в настоящее время базовыми при поиске
новых ЛС, были осознаны уже тогда. Следует упомянуть несколько
наиболее значительных исследований, положивших начало QSAR.
1863 год. A.F.A. Cros экспериментально исследуя (на
млекопитающих) токсичность спиртов, наблюдал ее увеличение в гомологическом
ряду, вплоть до некоторого максимального значения. Сама величина
токсичности связывалась с понижением растворимости спирта в воде.
1868 год. A. Crum Brown, T. Fraser, изучая биологические
эффекты алкалоидов (стрихнина, кодеина, морфина и т.д.) до и после
метилирования основного атома азота, пришли к естественному выводу о
связи физиологической активности исследуемой молекулы (у) и ее
структуры (х),
y = f(x)
1869 год. B.J. Richardson показал, что наркотическая активность
спиртов пропорциональна их молекулярному весу.
1893 год. С. Richet исследуя токсичность широкого класса
соединений (эфиров, спиртов, кетонов, альдегидов и др.) подтвердил
обратную пропорциональность токсичности и растворимости
соединений в воде.
1899, 1901 годы. Н.Н. Meyer, C.E. Overton предположили, что
токсичность органических соединений связана с их способностью
распределяться между водной и липоидной фазами. В качестве модели
такого распределения они предложили использовать систему
оливковое масло-вода.
384
1894 год. В работе Е. Fisher утверждалось, что энзимы и субстраты
должны соответствовать друг другу по принципу «ключ-замок».
1913 год. P. Ehrlich. «Вещество не действует, если не связано с
биологической субстанцией».
Следующий шаг был сделан в 30-х гг. XX века. Вслед за
известным уравнением Гаммета, связывающим реакционную способность
органических соединений с эмпирическими параметрами
заместителей, O.R.Hansen предложил так называемое «биологическое
уравнение Гаммета», которое имело формальный похожий вид:
lgVlgY^op.,
где lgY0 — некое численное выражение биоактивности незамещенной
молекулы, lgYs — биоактивность замешенной молекулы, а а и ps —
эмпирические параметры, характеризующие базовую молекулу и
заместитель соответственно. Однако наиболее важный шаг был сделан
в работах Хэнча, Фри и Вильсона лишь в начале 60-х гг. Ими, в
частности, было получено уравнение, в котором учитывались
наиболее важные факторы, описывающие биоактивность. Фактически
именно в этих работах и была реализована основа современных воззрений
на целенаправленную разработку новых ЛС.
Современный этап развития QSAR методологии характеризуется
широким внедрением математических методов с последующей
обработкой экспериментальных данных на электронно-вычислительных
машинах. Для того, чтобы найти соотношение между заданным
видом активности органических соединений и их структурой,
последнюю необходимо представить в численном виде (рис. 15.1).
с
в
о
й
с
т
в
о
структура
Рис. 15.1. Зависимость «структура-свойство» требует соответствующих
численных представлений
Численные характеристики, выражающие структурные
особенности молекулы, получили общее наименование «дескрипторы
молекулярной структуры» (или просто дескрипторы). В связи с этим главная
проблема QSAR — выбор такого (желательно минимального) набора
385
дескрипторов, который достаточен для описания заданного свойства.
Одним из источников дескрипторов являются методы квантовой
химии, с помощью которых можно проводить расчеты электронной
структуры и геометрии самых разнообразных химических систем вплоть
до моделирования эффектов среды и взаимодействия молекулы с
рецептором.
Для построения зависимости биоактивности от структуры
молекул используют широкий набор статистических методов,
позволяющих строить различные типы функциональных зависимостей. Здесь,
наряду с хорошо известным регрессионным анализом, применяются
методы, связанные с классификацией молекул по различным классам
активности. Особое значение имеют так называемые факторные
методы. Один из вариантов факторного анализа известен как метод
главных компонент — статистический подход, позволяющий
проанализировать структуру взаимосвязей элементов дескрипторного
набора и, что очень существенно, сжать его. Своеобразным гибридом
регрессионного анализа и метода главных компонент является частичный
метод наименьших квадратов (partial least squares, PLS),
использующийся в расчетах систем с большим количеством дескрипторов.
За последнее десятилетие появилось множество новых подходов.
Среди них так называемый трехмерный QSAR (3D-QSAR) — мощный
метод, предполагающий зависимость биоактивности от стерических
свойств молекул и их электростатических полей. Набирает
популярность метод искусственных нейронных сетей — алгоритм,
симулирующий функционирование нейронов. Он используется в проблеме
молекулярного распознавания и классификации. Генетические алгоритмы —
новый подход, позволяющий корректно обрабатывать данные,
которые содержат больше переменных (дескрипторов), чем объектов
(молекул).
Все эти методы наряду с проверенными известными подходами
составляют теоретическую базу для целенаправленного поиска новых Л С.
В целом методология QSAR позволяет заменить поиск
соотношений «структура — активность» анализом соотношений
«дескрипторы — активность» и на конечном этапе исследований получать
модельные функции вида:
y = f(x1,x2,...,xk),
где искомая биологическая активность (у) выражается через значения
к дескрипторов молекулярной структуры (х., i = 1, ... к). Подобные
уравнения фактически являются схемами для расчета самых
различных свойств органических соединений.
Область применения построенных моделей зависит как от их
качества с точки зрения статистики, так и от представительности базы
данных, использованной для их построения. В процессе
моделирования важно контролировать прогностическую способность
построенных моделей. Один из наиболее надежных подходов к решению этой
задачи состоит в разделении исходной базы данных на обучающую
выборку, использующуюся для построения моделей, и контрольную
386
выборку. Последняя необходима для независимого контроля
предсказательной способности моделей. Такой подход позволяет в
большинстве случаев избежать случайных корреляций.
Особую важность приобретает проблема отбора дескрипторов,
способных описать данный вид активности. Следует подчеркнуть, что
универсального набора дескрипторов для любых систем и любых
типов активности не существует. Уместно также напомнить принцип
Оккама: «Не приумножай сущностей более чем необходимо»,
поскольку чрезмерно «раздутый» дескрипторный набор увеличивает риск
случайных корреляций. В целом QSAR методология не предполагает
глубокого понимания сущности конкретных биохимических процессов
в организме, однако, имея какую-либо модель этих процессов,
можно существенно облегчить формулировку самого вида
количественного соотношения структура-активность.
Цель данного раздела состоит в кратком изложении основных
понятий и методологии QSAR, а также описании различных
систем молекулярных параметров и методов оценки биологической
активности на основе статистического анализа эмпирических
фармакологических данных. Для каждого из таких подходов предлагается
набор соответствующих иллюстраций. Все объекты, описанные в
этих примерах, и соответствующие данные об их биологической
активности взяты из оригинальных работ. Поскольку современная
QSAR литература является, как правило, англоязычной, для
многих распространенных понятий приведены также соответствующие
английские термины.
16
ДЕСКРИПТОРЫ МОЛЕКУЛЯРНОЙ СТРУКТУРЫ
Проблема расчета биологической активности требует
количественного описания структуры молекул в неких терминах. Такое
описание должно адекватно соответствовать наиболее типичным
особенностям исследуемой системы. Набор независимых параметров, которые
характеризуют электронные, структурные, геометрические или иные
особенности молекул, называют молекулярными дескрипторами. При
этом следует учитывать, что механизм взаимодействия химического
препарата и био-мишени (рецептора) обычно детально не известен
и, следовательно, строго говоря, не известно, какие именно
молекулярные параметры (дескрипторы) должны быть использованы в
предсказании того или иного вида активности. Это обстоятельство
приводит к необходимости использовать достаточно широкий набор
структурно-химических данных, каждый из которых может иметь
значение (или не играть никакой роли) в описании исследуемого
свойства. Следует отметить, что в настоящий момент насчитывается
несколько тысяч разнообразных молекулярных дескрипторов и их
полное перечисление может занять немало места. Ряд важнейших из
них, отображающих наиболее существенные аспекты молекулярной
структуры, описаны ниже.
16.1. Общая классификация дескрипторов
Молекулярные дескрипторы принято классифицировать в
соответствии с тем, на каком уровне они описывают структуру молекул.
• Обычно элементарными дескрипторами называют брутто-форму-
лу молекулы или ее атомный состав. Ясно, что такие величины
не характеризуют структуру молекулы, поэтому вряд ли могут
служить универсальными параметрами, описывающими
молекулярные свойства. Тем не менее, эти величины прекрасно
справляются с проблемой описания ряда свойств молекул простого строения.
Так, например, число углеродных атомов в насыщенных
углеводородах нормального строения (Nc) линейно связано с теплотой
сгорания (ДН298) и с высокой точностью описывается простым
уравнением;
388
ЛН"98 = 237.9 + 660.0 - Nc (кДж/моль).
• Электронные дескрипторы являются результатами квантовохими-
ческих расчетов и представляют собой набор индексов, так или
иначе характеризующих зарядовое распределение молекулы и ее
энергию.
• Топологические дескрипторы являются одними из наиболее
популярных. Обычно такие параметры базируются на исследовании
структурного графа молекулы.
• Геометрические дескрипторы — это параметры молекулы, которые
характеризуют ее геометрию и форму молекул. Среди таких
параметров эффективная площадь поверхности молекулы и
молекулярный объем. Обычно эти величины находятся с помощью
соответствующих аддитивных схем, построенных на данных о ван-
дер-ваальсовых радиусах молекул.
• Физико-химические дескрипторы — это группа дескрипторов,
которые описывают различные наблюдаемые физико-химические
параметры молекулярных систем. Эти величины могут быть
получены либо из эксперимента, либо вычислены с помощью какой-либо
эмпирической расчетной схемы. Важнейшими среди них
являются молекулярная рефракция (MR) и логарифм константы
распределения вещества в системе октанол-вода (липофильность, \%F).
• Естественными химическими дескрипторами являются
структурные элементы молекул, которые могут быть выделены различными
способами. К таким дескрипторам обычно относят не только
атомы периодической системы элементов, но и соответствующие
функциональные группы, атомы в различных гибридных состояниях
и т.д.
В следующих разделах описаны наиболее важные и
нетривиальные из молекулярных характеристик. Многие из них могут быть
вычислены с использованием популярных квантовохимических
пакетов программ Gaussian, GAMESS, HyperChem, в состав которых входит
набор полуэмпирических методов (CNDO, INDO, AMI, PM3),
используемых в расчетах QSAR. Программа HyperChem помимо
прочего позволяет оценить такие полезные величины как эффективная
площадь поверхности молекулы, молекулярный объем,
поляризуемость, рефракция. Существуют также специализированные
программы, ориентированные на расчет множества дескрипторов. Среди них
программа DRAGON, которая позволяет вычислять более тысячи (!)
всевозможных дескрипторов, включая топологические.
Немаловажным ее достоинством является то, что она свободно
распространяется (http://www.disat.unimib.it/chm).
16.2. Электронные дескрипторы
Множество электронных дескрипторов может быть получено
непосредственно в результате квантовохимических расчетов [1—3].
389
Электронная плотность (рг) характеризует вероятность
пребывания электронов в данной области молекулы (на данной атомной ор-
битали, г). В 71-электронном приближении рг может быть просто
выражена через соответствующие коэффициенты разложения:
р = Уп с
*г V i ir )
1
где п. — число электронов на i-той МО, cir — орбитальный
коэффициент разложения i-той молекулярной орбитали по r-той атомной
орбитали (атом г). С электронной плотностью связан также
частичный заряд на атоме (qr);
q =Z -p,
где Z — число электронов данного атома. Многие квантовохимиче-
ские дескрипторы получены из qr. Современные полуэмпирические
и неэмпирические методы используют так называемый Малликенов-
ский анализ атомных заселенностей для расчета зарядовых
распределений. Однако необходимо отметить, что подобные величины не
могут быть определены однозначно и, как следствие, имеется ряд
альтернативных методов определения частичного заряда на атомах.
В п-электронной теории определенную ценность представляет
также порядок связи, который характеризует степень л-электронного
связывания пары атомов. Эту величину для связи r-s можно вычислить
следующим образом:
Р =Уп.с.с.
i
Строго говоря, порядок связи однозначно определен лишь в
случае расчетов в я-электронном приближении, однако и во всевалент-
ных полуэмпирических методах квантовой химии имеются
аналогичные величины (индекс Вайнберга).
Индекс свободной валентности F. характеризует реакционную
способность углеродного атома г и согласно определению вычисляется
следующим образом:
F =N -N ,
г max r *
где N = уЗ — максимальная для атома углерода величина,
характеризующая способность к образованию л-связи, a Nr— сумма
порядков всех связей атома г:
Индекс свободной валентности является одной из величин, с
помощью которых характеризуют канцерогенную активность молекул.
Суперделокализуемость (сверхделокааизуемость) атома г (индекс
Фукуи) также является одной из характеристик реакционной
способности функциональной группы (атома). Обычно различают электрофиль-
ную — Se(r) и нуклеофильную — Sn(r) сверхделокализуемость атома г. Эти
390
величины выражаются через соответствующие коэффициенты
разложения и энергии вакантных (vac.) и занятых (осе.) орбиталей:
2
vac. С
S(r) = 2£
S(r) = 2£
^' '£(НОМО) +£(LUMO)'
е. U-A£(HOMO) +еаимог
где е,Н0М0) — энергия высшей заполненной молекулярной орбитали
(High Occupied МО), е номо — энергия нижней вакантной (Lowest
Unoccupied) МО. Se(r) и Sn(r) интерпретируются как индексы,
характеризующие способность к делокализации (стабилизации) электронов
в переходном комплексе при атаке электрофильным/нуклеофильным
реагентом. Суперделокализуемость как и индекс свободной
валентности также получила широкое распространение в связи с оценками
канцерогенной активности молекул.
Молекулярный дипольный момент (ц) является общей
характеристикой полярности системы и представляет собой вектор,
характеризующий разнесенность центров тяжести положительных и
отрицательных зарядов атомов молекулы. Значение этого дескриптора
может быть определено экспериментально или рассчитано с
использованием какого-либо квантовохимического метода. Однако
дипольный момент является лишь общей характеристикой молекулы и при
наличии значительных положительных и отрицательных зарядов
суммарный момент может оказаться достаточно мал. Поэтому наряду с
ц иногда в качестве дескриптора полярности используют значение
наибольшей разности зарядов между двумя атомами.
Важное значение в проблеме QSAR имеют индексы,
характеризующие поляризуемость молекулярной системы. Обычно различают
так называемую электрическую дипольную поляризуемость и набор ко-
улсоновских поляризуемостей п.
Электрическая дипольная поляризуемость (а) характеризует
изменение распределения электронной плотности молекулы при
помещении её в однородное электрическое поле. Эта величина может
быть получена из эксперимента по молекулярной рефракции или
вычислена как производная дипольного момента (Д) по напряженности
электрического поля (Е) согласно выражению:
М- = сс-£.
Обычно а используется в виде средней поляризуемости а :
а = —(а + а + а )
-J \ XX уу 2Z ) ,
или (реже) анизотропии поляризуемости:
ее =— (се -а |+(а -а )+(а -а \
2\хх уу/ \уу zz/' \ м хх }
391
Здесь ос , а , агг — компоненты тензора дипольной поляризуемости,
характеризующие отклик молекулярной системы на наличие
электрического поля, направленного вдоль соответствующей оси.
Коулсоновские поляризуемости (я) характеризуют изменение
электронных свойств атома или связи при изменении (возмущении)
параметров какого-либо атома системы. Известно несколько типов ко-
улсоновских поляризуемостей п.
1. Самополяризуемость атома (пг) характеризует изменение
электронной плотности атома при изменении
электроотрицательности (кулоновского интеграла) того же атома:
-. 2 2
Ор осе. vac. С. С
да i » е. -е
г 1 а
2. Сумма самополяризуемостей по всем атомам также может
служить хорошим дескриптором «подвижности» электронной
оболочки молекулы:
п = 5>г _
г
3. Атом-атомная поляризуемость (пк) характеризует изменение
электронной плотности на атоме (г) при изменении
электроотрицательности (кулоновского интеграла) другого атома (s):
иО осе. vac. С. С С. С
ir ar is as
= = t ?. ? —
sr K да
Кроме перечисленных известны также поляризуемости типа атом-
Эр Эр
связь (7t = ——) и связь-связь (7t = ——), которые, однако, ме-
r,st Эр ' V b.iu Эр » *
нее популярны "в проблеме QSAR. ш
Энергия системы является универсальным дескриптором в целом
ряде случаев. При квантовохимическом расчете обычно получают
полную энергию системы и оценивают энтальпию ее образования.
Определенный интерес представляет также энергия разрыва
химической связи:
ЕЬ=Е-Е\
где Е — полная энергия молекулярной системы, а Е — суммарная
энергия составных частей, на которые система распадается.
Энергия электронного перехода (X) в различных квантовохимичес-
ких подходах оценивается по-разному. В простейшем приближении
(например в методе Хюккеля) ее получают как разность
соответствующих молекулярных орбиталей (МО). Так, энергия длинноволнового
перехода вычисляется как разность энергий нижайшей вакантной
(е(шмо)) и верхней заполненной (е(НОМО)) МО:
А = Р - Р
c(LUMO) (HOMO)'
392
Эту же величину, после перенормировки, обычно называют
молекулярной жесткостью (ч\)\
Величина г\ является важным индексом, характеризующим
стабильность молекулярной системы, ее большое значение
соответствует относительно низкой активности в химических превращениях.
Концепция эта, однако, является достаточно грубой.
Отдельно взятые энергии молекулярных орбиталей, согласно
теореме Купманса, соответствуют потенциалу ионизации (IP) и сродству
к электрону (ЕА):
IP = - £
(HOMO) '
ЕА - Е<ШМО) •
Энергия делокализации, (ED) является фундаментальной
характеристикой сопряженной системы:
1
где второй член соответствует энергии системы изолированных
двойных связей данной молекулы.
Энергия протонирования (ДНН) — разность между полной
энергией протонированной и нейтральной формы.
Перечисленные выше квантовохимические индексы нашли
широкое распространение в проблеме QSAR. Известно множество
корреляционных функций, которые связывают наблюдаемый био- или
физико-химический эффект с параметрами электронной структуры
молекулы. В качестве примера приведем два уравнения. Так, из
данных о токсичности (\gLD5Q) серии из 20 нитрилов (п=20) было
получено следующее корреляционное уравнение [4]:
-lgID50=-1.69—+ 0.47, (п=20, г =0.87, а = 0.2)
ДЕ '
где а — средняя поляризуемость молекулы (А3), а ДЕ — ее энергия
протонирования (эВ), оцененная методами квантовой химии как
разность энергии нейтральной и протонированной формы. Величины,
характеризующие качество прогноза по этому уравнению (г —
коэффициент корреляции и а — стандартное отклонение) обсуждены в
следующем разделе. Во втором уравнении [5] описывается ингибиро-
вание ацетилхолинестеразы производными бензилпиперидина:
-Ig/C^ =2.21С4 -6.65ц + 1,18ц2 -1б2.9е(НОМО) -8.58е;н0М0).
(п=16,г * 0.939, а = 0.25).
В этом уравнении ц — дипольный момент (Дебай), С4 —
орбитальный коэффициент в высшей заполненной молекулярной орби-
тали, а е(НОМО) — ее энергия (эВ).
393
16.3. Топологические дескрипторы
Топологический подход к описанию молекулярной структуры
основан на анализе структурной формулы молекулы и вычислении
соответствующих индексов, которые остаются неизменными при
изменении нумерации атомов или связей (так называемые топологически^
инварианты) [6, 7, 8]. При этом обычно «легкие» (водородные) атомы
не учитываются в расчете. Рассматривается лишь каркас молекулы,
построенный из «тяжелых» атомов: С, N, О, S и т. д. Основная задача
топологических индексов связана с необходимостью описания
особенностей связывания атомов в молекулярной структуре. В
частности, именно топологические индексы призваны описать различия
в структуре изомеров.
Развитие топологического подхода тесно связано с
применением теории графов. Важнейшим понятием теории графов является
понятие топологической матрицы (или матрицы смежности)' (G). Это
матрица, размерность которой совпадает с количеством «тяжелых»
атомов. При этом номер строки (или столбца) соответствует номеру
атома. Если между парой соответствующих атомов имеется
химическая связь, то соответствующий матричный элемент матрицы
смежности полагается равным 1. Кроме топологической матрицы
используется также матрица расстояний GD. Каждый матричный элемент
gD(ij) равен минимальному числу ребер, соединяющих одну вершину
(Г) с другой (j). В качестве примера приведем соответствующие
матрицы G и GD для молекулы 2,3-диметилбутана (рис. 16.1):
G =
0
1
0
0
0
0
1
0
1
0
0
1
0
1
0
1
1
0
0
0
1
0
0
0
0
0
1
0
0
0
0
1
0
0
0
0
Go =
0
1
2
3
3
2
1
0
1
2
2
1
2
1
0
1
1
2
3
2
1
0
2
3
3
2
1
2
0
3
2
1
2
3
3
0
Рис. 16.1. Граф молекулы 2,3-диметилбутана
С использованием матриц графа (G и GD) можно получить ряд
топологических дескрипторов.
394
• Число путей в молекулярном графе определенной длины.
Число путей между вершинами длины «1» (Р,), т. е. число
химически связанных пар атомов, число путей длины «2» или индекс
Гордона-Скантлбери (Р2) и число путей длины «3» (Р3) получают
простым подсчетом количества единиц, двоек или троек в одном из
треугольников симметричной матрицы GD.
• Индекс Винера W равен сумме связей, существующих между
всеми парами «тяжелых» атомов в графе молекулы с п вершинами:
w=if;gD(ij).
• Индекс Рандича х(1) характеризует молекулярную связность:
(1) ^ / ч-1/2
где и и u — степени вершин графа, т. е. соответствующие количества
связей атомов i и j. Суммирование проводят по всем парам
связанных вершин (связанных атомов). Известны также обобщенные
индексы Рандича, где суммирование проводится по всем цепям
маршрутов длины к между вершинами i и j (между атомами i и j имеется
цепочка из к связей):
Х(к)= У (u...u...u)"1/2
• Индекс среднеквадратичных расстояний D2
2 W
D2 =-!
ур ,
Т 1
где Pj— число пар вершин в матрице GD, расстояние между
которыми (длина) равно i.
• Индексы загребской группы.
M,(G) = Y^,
M2(G) = J (uu)
(ij)
В первом выражении суммирование проводится по всем
вершинам, а во втором — по всем парам связанных атомов.
• Наибольшее собственное значение Атах матрицы смежности (G) как
топологический индекс был предложен в качестве меры
количества разветвлений в структуре графа.
Особенной группой топологических индексов являются так
называемые теоретико-информационные индексы, основанные на
использовании известной универсальной формулы Шеннона для оценки
неоднородности (информации) любой системы. Предположим, что
395
структура молекулы определенным образом разбита на подмножества/
Тогда, вычислив вероятность попадания того или иного элемента в
заданное подмножество, можно оценить информацию о
распределении всех элементов по подмножествам (формула Шеннона):
п. п,
Информация = -J—1-log —- (бит),
i П П
где а — количество элементов в подмножестве i, а п — суммарное
количество всех элементов системы, равное
В этом выражении логарифм берется по основанию 2, что
соответствует информации, выраженной в битах. На основе формулы
Шеннона введены следующие теоретико-информационные индексы.
• Неоднородность распределения расстояний между вершинами в
графе, ID, используется для характеристики разветвленности
молекулярной структуры. Здесь степень неоднородности структуры
оценивается на основе анализа распределения расстояний между
вершинами графа. Тогда ri= V. — количество связей длины «i», а
п — общее количество кратчайших расстояний между всеми
парами атомов структуры. Часто бывает удобно использовать общее
количество информации в пересчете на граф — TID:
TIo = »■'„•
• Информационное содержание графа относительно окрестностей к-го
порядка — /Ск, представляет собой информационное содержание
в расчете на одну вершину:
/Ск=-ХрЛо82р,;
где р] — вероятность того, что выбранная случайным образом
вершина графа (атом) попадет в i-тое подмножество, причем
вычисляется она с учетом окружения.
• Т/Су, — полное информационное содержание — мера сложности в
расчете на один граф.
Т/С = п-/С.
к к
• 5/Ск — структурное информационное содержание
S/Ck =/Ck/log2n.
• Z?/Ck — информационное содержание связывания
5/Ck=/Ck/log2Nb)
где Nb — полное число ребер (ковалентных связей) в молекулярном
графе.
396
• С/Су, — комплиментарное информационное содержание
CICk = log2 n - /С„ .
В качестве примера рассмотрим вычисление индексов ID и /С для
молекулы 2,2-диметилбутана (остальные информационные индексы
легко вычисляются из /С). Граф молекулы можно представить в виде:
Для вычисления индекса ID подсчитаем число путей между
вершинами длины «1», «2», «3» и т.д. Р,=5, Р2=7, Р3=3. Путей длины
более «3» в этом графе нет, поэтому
Р, +Р2 +Рз
n=r +v + г_ =5 + 7 + 3 = 15
И, следовательно, индекс ID равен
1о=-
^+^£+1^кЬ^(бт)-
В отличие от индекса ID индекс ICk вычисляется на основе
анализа вершин (атомов). Для 1С0 подсчитывается количество
неэквивалентных атомов без учета окружения. Поскольку в этой молекуле
все атомы одного сорта — атомы углерода, то все они эквивалентны
и, следовательно, относятся к одной группе.
Индекс /Cj вычисляется с учетом ближайших соседей. Анализ графа
показывает, что при таком рассмотрении можно выделить группы
атомов (см. табл. 16.1).
Группы атомов в молекуле 2,2-диметилбутаи
с учетом ближайшего окружения.
Таблица 16.1
Группа
Атомы группы
1.5,6,4
Число атомов
в группе
Следовательно, значение индекса /С, равно
'с,=
A loa А + 1 log 1 + 1 log2 ll = 1.252 (бИТ).
6 6 6 6 6 6,
397
Индекс /С, вычисляется с учетом ближайших и следующих за ними
соседей. Четыре группы эквивалентности атомов с учетом соседей
«второго порядка» приведены в табл. 16.2.
Таблица 16.2
Группы атомов в молекуле 2,2-диметилбутан
с учетом окружения второго порядка
Группа
I
2
3
4
Атомы группы
1,5,6
2
3
4
Число атомов
в группе
3
I
1
1
/с2=-
Ьб+М + ?^1+Ь{;
1.792 (бит).
В табл. 16.3 для сравнения приведены некоторые упомянутые
топологические индексы для трех изомеров гексана. Очевидно, что
многие из них позволяют различать изомеры.
Топологические индексы изомеров гексаиа
Таблица 16.3
Индекс
Nc
Pt
Р2
Рз
Ь
ICo
1С,
ic2
Г»
Хт
ЭГ
w
MiG
M2G
D2
(н-гексан)
6
5
4
3
2.149
0
0.918
1.585
2.914
1.707
0.957
35
18
16
7
(2,2-диметщ
■
[бутан)
6
5
7
3
1.506
0
1.252
1.792
2.561
2.914
1.061
28
24
22
4
■ ■ ■ /
(2-метилпентан)
6
5
5
3
1.909
0
1.459
2.252
2.770
2.183
0.866
32
20
18
5.6
398
Теперь рассмотрим более сложную систему — молекулу ментола,
в молекулярном графе которой (рис. 16.2) как и в предыдущих
случаях, атомы водорода не рассматриваем:
7
6
2
Г^^>
3
4
Y ^
Ю 8 9
. 16.2. Граф молекулы ментола
Нулевой порядок (1С0).
С учетом окрестностей нулевого порядка все десять углеродных
атомов эквивалентны. Следовательно, одиннадцать «тяжелых атомов»
системы могут быть сгруппированы в две группы. К первой
относятся все атомы углерода, а ко второй — кислород. Информационное
содержание в этом случае равно:
/С =-
^log^ + ^logi
10
11
10
11
11
= 0.439 (бит).
Т1С0= 4.829, 5/С0= 0.127, 5/С0= 0.127, С1Сп= 3.020.
Первый порядок (ICj). Рассмотрим окрестности первого порядка
(ближайшие соседи) для всех атомов.
С2
.с2
С1 С3 С;
Л
I 3 /Г4
/\ /l\
•С4
С3 С5 'О
/\ /\
С5 г СЮ
С8
'10
О
С4
Очевидно, что имеется 5 групп эквивалентности, а именно
С„ С9, С10, - 3 атома;
С2, С5, С8, - 3 атома;
С3, С6, С7, — 3 атома;
С4 — 1 атом;
О — 1 атом.
Используя эти данные, можно вычислить 1СХ:
399
77С, = 23.793, Siq = 0.625, BICX = 0.625, С/С, = 1.296.
Второй порядок (/С2). Окрестности второго порядка (учет соседей
через один атом) для всех эквивалентных атомов в первом порядке
выглядят следующим образом:
<?1
сГ\,
С;
\
/Ч
с2 с4
Ci С7 С5 О
с2
\.
\
с5
/К
*с7
\ / л /\
Ск С7 о С, г.. <
Итак, имеем следующие группы эквивалентности:
С,, С9, Ct0 — 3 атома;
С6, С7 — 2 атома;
а также шесть групп по одному атому.
/С
-2-log, -2-+ -2-log. 2. + 6- -J-log, -L I = 2.845 (бит)
И ^ 11 11 2 11 11 ^ 11
77C2= 31.295, S/C2= 0.822, Я/Сэ = 0.822, C/C2= 0.614.
Третий порядок (/С3). Рассматриваем все эквивалентные атомы во
втором порядке и выписываем окрестности через два соседних атома.
\,
/
с6
Хс5
/V
-С8
X
с6 с4
^\
Атомы Сд и С10 включаются в одну группу;
остальные группы по одному атому.
*Св.
>
400
1С = -f-2- log, -2- + 9 ■ -L log, -L | = 3.278 (бит)
3 1^11 ^11 11 ^llj
7YC3 = 36.058, ^/C3= 0.948, BIC^ 0.948, C/C3= 0.181.
Очевидно, что дальнейшее расширение окрестностей не
приведет к изменению информационного содержания, поскольку
атомы 9 и 10 топологически неразличимы по соображениям
симметрии.
Одним из самых популярных индексов в настоящее время
является так называемый электротопологический индекс [9], который
объединяет в себе как электронные, так и топологические особенности
молекулы. В формулировке этого индекса, прежде всего, вводится
понятие «внутренних» электронных состояний i-того атома (I.),
которые определяются как частное:
I.
(5;+1)
5
где 5. — кол-во с-связей С—С атома i, a 5V — общее количество
электронов, включая о, п и неподеленные пары (п):
8V = 5 + 71 + п.
Таким образом, 5; определяет локальный топологический индекс
атома — количество связей атома i в структурной формуле
молекулы, a 5V описывает особенности электронной структуры данного атома
в молекуле. Наиболее часто встречающиеся сорта атомов и
соответствующие индексы их электронных состояний приведены в табл. 16.4.
Таблица J6.4
Индексы «внутренних»
электронно-топологических состояний атомов
Сорт атома
^>c<Z
^:сн
сн2
>=
н3с
с
ип2
5.
1.25
1.333
1.500
1.667
'2.0
2.5
3.0
Сорт атома
О
NH 2
ОН
- и
NH
(5 +1)
I = —^ -
8
1
3.5
4
5
6
7
2.5
8
401
Итак, состояние атомов в графе отражается их внутренними
электронно-топологическими атрибутами. Влияние всех остальных
атомов на данный атом может быть учтено с помощью выражения:
I. -I.
J*1 Г.
где г„ — расстояние между i-тым и j-тым атомами выраженное в духе
теории графов как минимальное число химических связей в
маршруте, проходящем между двумя атомами i и j. Таким образом, каждый
атом в химическом графе, выражается как сумма двух компонент,
которые характеризуют внутреннее состояние (J) и соответствующее
молекулярное поле окружения (ДГ ):
S. = I. + Д1..
Множество корреляционных соотношений связывает структурные
и квантовохимические индексы молекул с наблюдаемыми
свойствами (в том числе и с физико-химическими) [9]. Так, теплота атомиза-
ции алканов (ДНа, ккал), может быть выражена через число атомов
углерода (N) и индекс Рандича (^0)):
ДНа = 283.33N - 6.321jc0) +115.72,
г = 0.9999 s = 0.960.
Температура кипения алканов Т(°С) также может быть
вычислена с помощью индекса Рандича:
Т(°С) = 57.85ха) -97.9.
Применение последнего уравнения для расчета Т(°С) изомеров
гексана демонстрирует удовлетворительное согласие с
экспериментальными величинами (табл. 16.5).
Таблица 16.5
Индекс Рандича и температура кипения изомеров гексана
(И
%
тсс)
тсо
эксп.
2-метилпентан
2.77
62.3
60.3
2,2 -дим етил бутан
2.56
50.2
49.7
2,3-диметил-бутан
2.64
54.8
58.0
н-гексан
2.91
70.4
68.7
В следующем уравнении [10] тот же индекс Рандича линейно
связан с минимальной блокирующей концентрацией анестетиков
(спирты, кетоны, эфиры, амины, фенолы) в мышечных и нервных клетках
(lg МВС).
IgMBC = -0.762xu>+3.55 (n = 36, г = 0.98, о * 0.39).
402
Вычисление топологических и информационных индексов
представляет собой относительно простую задачу ручного счета лишь для
малых систем. В тех же случаях, когда речь идет о крупных
молекулах, содержащих десятки атомов, такой расчет может оказаться
трудоемким и требует использования специализированных программ на
подобии упоминавшейся ранее программы DRAGON.
Следует подчеркнуть также то обстоятельство, что
топологические индексы не обладают определенным физическим смыслом и не
могут быть интерпретированы в терминах каких-либо наблюдаемых
физических величин. Эти индексы можно воспринимать лишь как
набор альтернативных описаний связности (топологии)
молекулярной структуры. Неоднозначность (в значительной степени
интуитивность) в определении понятия молекулярной связности и
определяет многообразие ее топологических характеристик.
16.4. Физико-химические дескрипторы
Как уже отмечалось выше, физико-химические дескрипторы, в
отличие от топологических, имеют вполне определенный
физический смысл и могут быть измерены с помощью соответствующего
эксперимента. В проблеме QSAR наиболее популярны два
дескриптора: молярная рефракция и липофильность.
Молярная рефракция (MR) — это величина, которая связывает
электронную поляризуемость вещества (а) с показателем преломления (п).
В области оптических частот она описывается известной формулой
Лорентца-Лоренца:
MR= V1 MW =±n NAa,
n2+2 d 3
где NA — число Авогадро, MW — молекулярный вес, ad — плотность.
Рефракция практически не зависит от температуры, плотности и
агрегатного состояния вещества, что делает ее чрезвычайно удобной для
построения различных корреляционных зависимостей, описывающих
связь молекулярной структуры со свойством. Важнейшей
особенностью рефракции является возможность представлять ее в виде суммы
отдельных вкладов атомов, групп атомов или химических связей. Так,
например, для насыщенных углеводородов с общей формулой СпН2п+2
рефракция может быть записана с высокой точностью в виде:
MR = n ■ MR(C) + (2n + 2) • MR(H),
где MR(C) и MR(H) — вклады (инкременты) от углерода и водорода
соответственно. MR является довольно многозначным параметром,
который описывает различные аспекты молекулярной системы.
Обычно с MR ассоциируется молярный объем, величина дисперсионных
сил и характеристика «подвижности» электронной оболочки
молекулы при действии внешнего возмущения.
403
На сегодняшний день построено множество корреляционных
соотношений, связывающих молекулярную рефракцию с
биологической активностью. В качестве примера приведем уравнение для
логарифма относительного сродства (Relative Binding Affinity, lgRBA)
16 = а замещенных эстрадиола (рис. 16.3) к соответствующим
рецепторам [11]:
lgRBA = -0.48MR + 2.08, г2 = 0.84,
где MR — парциальная рефракция заместителя X.
Рис. 16.3. 16-а замещенные эстрадиола
Липофильность или логарифм константы распределения в системе
1-октанол-вода;
С
lg/>=lg '-octa"ole =igC , -lgC
^ e p & J-octanote ° water
water
Эта величина является важнейшим молекулярным
дескриптором [9, 12], который моделирует распределение вещества между
водной и липиднои фазами (см. также разд. 2.1). Значимость ее была
осознана еще на заре QSAR. Соединения, для которых Р>\ (lg/^>0),
являются липофильными, в то время как соединения с Р< 1 (lg/*<0) —
гидрофильными. В целом липофильность отражает весь объем
эффектов взаимодействия вещества с растворителями и зависит (в
частности) от полярности растворенного вещества и возможного наличия
водородных связей.
Как уже отмечалось выше, в целом ряде случаев, приходится
изучать биологическую активность ряда соединений, которые
отличаются лишь заместителями в базовой структуре. В этом случае удобно
ввести понятие липофильности заместителя, которое характеризует
вклад в общую липофильность за счет заместителя. Исторически
первым подходом такого рода были константы липофильности
заместителей Хэнча—Фуджиты [13], которые можно получить из
соответствующего аналога уравнения Гаммета:
lg-L-igP-ig^p.^.
н
404
В этом уравнении величины Рх и Рн — коэффициенты
распределения Х-замещенной и незамещенной молекулы соответственно, а
ях — характеристика липофильности заместителя. При этом
предполагается, что величина р отражает характеристики растворителей и
для системы октанол-вода принимается равной единице. Величины
я для наиболее распространенных заместителей приведены в
табл. 16.6. Естественно, что данные для ароматических и
алифатических типов замещения различаются.
Таблица 16.6
Константы липофильности
распространенных заместителей (по Хэнчу)
в ароматическом и алифатическом радикалах
№
1
2
3
4
5
6
7
8
9
10
11
1?
М
14
И
16
Заместитель
-NH2
-I
-SCH3
-СОСН3
-CONH2
-СООСНз
-Br
-CN
-F
-CI
-COOH
-OCH3
-ос6н5
-N(CH3)2
-OH
-N02
Заместитель
в ароматическом кольце
*х=^с6н5х-^с6н6
-1.23
1.12
0.61
-0.55
-1.49
-0.01
0.86
-0.57
0.14
0.71
-0.28
-0.02
2.08
-0.18
-0.67
-0.28
Заместитель
в алифатическом фрагменте
-1.19
1.0
0.45
-0.71
-1.71
-0.27
0.60
-0.84
-0.17
0.39
-0.67
-0.47
1.61
-0.30
-1.16
-0.85
Суммарная липофильность полизамещенной системы выражается
через липофильности базовой молекулы (lgPH) и липофильности
заместителей (ях):
lg /> = lg Я. + f>
" X=I
где N — число заместителей в молекуле. Очевидно, что подобная
схема расчета \gP является довольно грубой, поскольку не
учитывает взаимодействие заместителей с остальными фрагментами
молекулы. Это обстоятельство стимулировало поиски более точных схем
описания липофильности, однако и величины ях, первоначально
предложенные Хэнчем и Фуджитой, все еще широко используются
405
в проблеме QSAR в тех случаях, когда исследуются эффекты
заместителей, но не требуется точное знание \%Р.
Параметры гидрофобности молекулярных фрагментов
Риккера [14] предполагают не только вклады в \%Р от различных
молекулярных фрагментов, но и также определенные так называемые
«факторы коррекции». В целом, липофильность модели Риккера
выражается с помощью обобщенной аддитивной схемы следующим
образом:
где fk — параметр гидрофобности молекулярного фрагмента к,
a Nk — число фрагментов типа к присутствующих в данной
молекуле. F.- факторы коррекции, которые призваны учитывать
изменение параметров гидрофобности за счет наличия в молекуле
определенных структурно-химических комбинаций (атом водорода в
полярной группе, арил-арил сопряжение и т.д.). На практике
факторы коррекции вычисляются с помощью специального
эмпирического уравнения [9]:
F = к.-0.219,
где к. принимает целочисленное значение, характеризующее j-тыЙ,
фактор, а величина 0.219 — так называемая «магическая константа».
Недостатком подхода Риккера является неоднозначность в выборе
молекулярных фрагментов, что, в свою очередь, ведет к
многообразию вычисляемых величин \%Р для различных способов разбиения
молекулы на фрагменты.
Одним из наиболее продуктивных и популярных подходов
является метод Лео—Хэнча, [12] в котором параметры гидрофобности
фрагмента выбираются с использованием так называемого
«конструктивистского подхода». В рамках этого метода точно
вычисляются величины липофильности для некоторых простых
соединений, имеющих обычно одну функциональную группу. \%Р для
изучаемых соединений находят с использованием аддитивной
схемы Риккера. При этом декомпозиция исследуемой молекулярной
структуры на фрагменты проводится с помощью простых и
однозначных правил и, таким образом, \gP вычисляется
однозначно. Факторы коррекции Fi в схеме Лео—Хэнча получены с
использованием соединений с более чем одним заместителем. Они
включают в себя эффекты, обусловленные наличием меж- и
внутримолекулярных водородных связей, электронные взаимодействия
в ароматических системах и т. д. Для успешного предсказания
величины \gP используют более 200 фрагментных констант и 14
факторов коррекции. Соответствующий программный комплекс
известен как Calculated \gP (CLOGP). В качестве примера
использования метода Лео—Хэнча приведем расчет CLOGP для молекулы
о-метилацетанилида (рис. 16.4.).
406
СНГ
СН3
Рис. 16.4. о-Метилацетанилид
Таблица 16.7
Расчет CLOGP для молекулы о-метилацетаннлида
Компонента липофильности
Фрагмент СО—NH
2 изолированных группы СН3
6 ароматических атомов С
10 атомов водорода связанных
с углеродом
1 цепочка связанных атомов
1 сопряжение
бензольное кольцо-цепочка
1 орто заместитель
Итого CLOGP
lgP получено экспериментально
Вклад
-1.510
0.39
0.78
2.270
-0.120
-0.150
-0.76
0.90
0.86
Еще одна популярный подход — модель Моригучи [9]
основывается на расчете площади молекулярной поверхности, доступной
растворителю (S) а также параметров, учитывающих эффекты полярных групп:
lgP = -1.06 + 1.9-S-£SK,
где SK— мера площади поверхности полярных групп, дающий
отрицательный (т. е. гидрофильный) вклад в суммарную величину \%Р.
Расчеты липофильности по методу Моригучи в научной литературе обычно
обозначаются как MLOGP.
Известно также множество других подходов к теоретической оценке
липофильности. Однако все они связаны либо с аддитивной схемой,
представляющей ^изучаемой системы как сумму фрагментных (или
атомных) вкладов, либо с корреляционным уравнением,
связывающим \%Р с молекулярными дескрипторами (наподобие уравнения
Моригучи).
Важным обстоятельством при вычислении и дальнейшем
использовании величин липофильности является тот факт, что ряд
практически важных соединений могут ионизироваться в растворе, при
этом значения рА"соединений могут значительно различаться. В этом
случае вместо \%Р принято использовать более корректную величину
407
lgD, которая отвечает распределению вещества между л-октанолом и
водным буфером с определенным рН. Действительно, если для
препарата (ХН) предполагается ионное равновесие в растворе (кислота):
ХН ^w Х- + Н+
то, очевидно, что его распределение между несмешивающимися
фазами, зависит от рК соединения и рН среды. В этом случае, вместо
\gP целесообразно использовать величину
lgD = lg/>-lg(l + 10pH-p*).
Если вещество ведет себя как основание:
X + Н+ «^w ХН*
то константа распределения принимает вид:
lgD=lg/>-lg(l + 10p*-pH).
Соответствующие выражения существуют также и для более
сложных равновесий. В качестве примера использования липофильнос-
тей запишем два уравнения (К. Хэнч, [15]). Первое связывает
летальную дозу спиртов (LDm) для кошек с \gP
lg—1— = 1.06 lg/> + 1.37.
LDm
Таким образом, увеличение липофильности спирта приводит к
увеличению токсичности, поскольку для достижения эффекта
(гибель животного) требуется меньшее количество вещества на единицу
массы.
Второе уравнение описывает изменение мембранного
потенциала моллюска Buccal ganglion под действием бензойных кислот.
Igi = 0.84 lgD +3.31.
V
В этом выражении С — концентрация бензойной кислоты
приводящая к увеличению мембранного потенциала на 20 мВ. В связи с
диссоциацией кислоты в водном растворе вместо \gPиспользуют lgD.
В настоящее время множество программных пакетов включают
расчет липофильности. Среди них особенно популярны ACDLabs и
упоминавшиеся ранее HyperChem и DRAGON.
Специализированный web-сайт (http://www.LogP.com) позволяет вычислить липофиль-
ность в онлайн-режиме. Информацию о программе CLOGP можно
получить на сайте (http://www.daylight.com/daycgi/clogp).
16.5. Химические дескрипторы. Фармакофоры
Описание молекулярных систем с использованием общих
физико-химических, квантовохимических и др. индексов является
довольно формальным, поскольку апеллирует к неким величинам,
408
которые довольно неоднозначно связаны с химической структурой.
Альтернативой такому описанию служит сугубо химический
подход, использующий элементы химической структуры в качестве
своеобразного алфавита для описания взаимоотношений «структура-
свойство». Этот подход базируется на некоторых тезисах (аксиомах),
выработанных на протяжении истории развития медицинской
химии исследований. Важнейшими среди таких аксиом являются
следующие утверждения:
1. Фармакологическое действие биологически активных соединений
определяется способностью взаимодействовать с биологической
мишенью — рецептором.
2. Во взаимодействии химических соединений с рецептором
принимает участие не вся молекула, а лишь ее часть (фармакофор).
Обычно такое взаимодействие ограничивается образованием
относительно слабых связей — ван-дер-ваальсовых,
электростатических, водородных.
3. Соединения, обладающие сходными фармакофорами, оказывают
сходное фармакологическое действие. Действие фармакофоров
может усиливаться или ослабляться другими функциональными
группами молекулы.
В этой связи возникает возможность сугубо химического
описания молекулярной структуры, с целью выявления
фармакофоров. Следует отметить, что разбиение молекул на фрагменты
может быть осуществлено различными способами. Один из вариантов
разбиения был предложен В.В.Авидоном в 1974 г. Это так
называемый метод фрагментарного описания химической структуры [16],
базисным элементом которого является понятие дескрипторных
центров (ДЦ). ДЦ это активные атомы и их группировки,
которые могут выступать реакционными центрами «слабого»
взаимодействия с рецептором. Обычно к ДЦ относят атомы или группы,
содержащие активные р- и d-электроны, электростатический
заряд и т. д. Таким образом, в качестве ДЦ принимаются все гетеро-
атомы: N, О, S, Р, галоиды, металлы, ароматические системы, а
также группировки с кратными связями.
Естественно, что различные атомы в различных валентных
(гибридных) состояниях обладают различными химическими
свойствами и поэтому могут быть отнесены к разным ДЦ. В целом, набор ДЦ
используется в специализированном языке — фрагментном коде
суперпозиции подструктур (ФКСП) Примеры ДЦ и соответствующие
кодовые обозначения приведены в табл. 16.8.
При этом собственно дескриптор молекулярной структуры
представляет собой более сложную структуру. Различают линейные и
циклические дескрипторы. Линейный дескриптор включает в себя два ДЦ
и цепочку атомов между ними. На языке ФКСП линейный
дескриптор описывается по следующей схеме:
ДЦ 1 Длина цепи ДЦ 2 Признак
\_ __ _ сопряжения
409
Таблица 16.8
Некоторые дескрипториые центры (ДЦ) н нх коды
дц
~N+-
-NH2, -NHR
~NR2
=NH
=NR
-OH
-OR
=0
Код
01
02
03
04
05
11
12
13
ДЦ
CHO (альдегид)
-О
-SH
-SR
CI, Br, I
F
-СН3, =СН2, ^СН
OX, -CR=X
Код
14
15
21
22
31
32
41
44
Таким образом, дескриптор кодируется семизначным числом.
Первые три позиции по две цифры. Последняя цифра — признак
сопряжения (1) или его отсутствие (0). Циклические дескрипторы
несут информацию о наличии различных циклов и количестве
электронов в системе сопряжения.
Геометрическая форма
циклической системы
Количество
я-электронов
в системе сопряжения
Расположение
гетероатомов
Две цифры отводятся на описание количества атомов в цикле и
количества электронов сопряженной системы. Структура
химического соединения на языке ФКСП описывается множеством
подструктур, которые появляются при разнообразном разбиении молекулы
на отдельные фрагменты. Следуя Розенблиту и Голиндеру [16]
опишем, в качестве примера, следующую молекулу:
сн
г\
В этой системе рассматривается 8 цепей линейных дескрипторов
(рис. 16.5) и один циклический дескриптор — бензольное кольцо,
которое кодируется как 6, 06 (шестичленный цикл и 6 р-электронов
в системе сопряжения).
Дальнейшее развитие идеи ФКСП связано с более точным
описанием структуры взаимосвязей дескрипторных центров молекулы.
Для этой цели предложено использовать матрицу геометрии
дескрипторных цепей (МГДЦ). Диагональные элементы МГДЦ — коды
дескрипторных центров, недиагональные — геометрические
расстояния (обычно в ангстремах).
410
N с=о
0201131
H2N—СН S-
0201220
■N С СН;
0202410
N с с с Nh2
0=с С С NH-
0203020
0203130
N С С С S-
о=с —с —с—s-
0203220
1303220
•Ph
2201330
Рис. 16.5. Пример линейных дескрипторов ФКСП и их кодировка
16.6. Индикаторные дескрипторы
Первые уравнения QSAR были структурно-зависимыми, в том
смысле, что ряды соединений отличались лишь природой
заместителей, в то время как положение замещения (например орто-, мета~,
или пара-) оставались неизменными. В дальнейшем, с накоплением
экспериментальных данных, исследователям приходилось иметь дело
с соединениями, которые существенно отличаются друг от друга в
структурном отношении. Например, методики комбинаторной химии
411
приводят к появлению тысяч соединений, имеющих множество
заместителей в различных положениях молекулы. Один из методов
решения этой проблемы связан с использованием так называемых
индикаторных дескрипторов (I). Такие дескрипторы применяют для
индикации наличия (или отсутствия) определенных
структурно-химических элементов в молекуле. Обычно I принимает значение нуль
или единица.
В этом разделе мы приведем два примера использования
индикаторных переменных. Первое уравнение получено классиком QSAR
К. Хэнчем [17]. Оно описывает константу связывания с рецептором
сульфонамидов общей формулы X - С6Н4 - S02NH2:
lg К = 1.55а + 0.64 lg P - 2.07 ■ I, - 3.28 • 12 + 6.94.
В этом уравнении а — константа заместителя, которая описывает
его электронные свойства (см. раздел 17.3). Индикаторные
дескрипторы принимают значение 1 для мета- (дескриптор 1{) и орто-
(дескриптор 12) замещенных. Во всех остальных случаях эти переменные
равны нулю. Таким образом, благодаря использованию
индикаторных дескрипторов, указанное уравнение описывает биологическую
активность широкого набора производных.
Второй пример (Kubinyi H. [18]) описывает глицинфениловые
эфиры как ингибиторы фермента папаина. В начале было получено
два независимых, но близких по форме уравнения. Для N-мезилгли-
цинфениловых:
\%К = 0.529 ■ MR + 0.379а + 1.877,
и для N-бензоилглицинфениловых эфиров
lg* = 0.771 ■ MR + 0.728а + 3.623.
С использованием индикаторных дескрипторов обобщенное
уравнение имеет вид:
\%К = 0.57 ■ MR + 0.56а -1.92 • I + 3.74.
При этом переменная I принимает значение 1 для мезил- и 0 для
бензоил- производных.
Литература
1. Karelson М., Lobanov V.S. // Chem. Rev. - 1996. - Vol. 96. - P. 1027.
2. Костриков P.P., Беспалов В.Я. Основы теоретической органической
химии. — Л.: Из-во Ленинградского университета, 1982.
3. Дьюар М. Теория молекулярных орбиталей в органической химии. —
М.:Мир, 1972.
4. Lewis D.F.V., Ioannides.C, Parke D.V. // Xenobiotica. — 1994.— Vol. 96.—
P. 401.
5. Cardoso M.G., Imura Y., et al // J.Med.Chem. - 1992. - Vol. 35. - P. 584.
6. Станкевич М.И., Станкевич И.В., Зефиров H. С. // Успехи химии. — 1988. —
Т. 57. - С.337.
412
7. Раевский О.А. Ц Успехи химии. — 1999. — Т. 68. — С 555.
8. Химические приложения топологии и теории графов / Под ред.
Р.Кинга.- М. Мир, 1987.
9. Todeschini R., Consonni К, Handbook of Molecular Descriptors. — NY, Toronto,
Wiley-VCH, Weinheim, 2000.
10. Kier L.B., Hall L.H., et al // J.Pharm. Sci. - 1975. - Vol. 64. - P. 1971.
11. Gao В., Katzenellenbogen LA., at. al. // Chem. Rev. - 1999. - Vol. 99. - P. 723.
12. Leo A.J. H Chem. Rev. - 1993. - Vol. 93, P. 1281.
13. Fujita Т., Iwasa J., Hansen C. 11 Joum. Amer.Chem.Soc. — 1964. — Vol. 86.—
P. 5175.
14. «cfeter^AorfM//J.Med. Chem.-Chimicatherapeutica.- 1979.—Vol 14,
P. 479.
15. Хэнч К. И Хим.-фарм. журнал. - 1980. - № 10. - С. 15.
16. Розенблит А.Б., Голиндер В.Е., Логико-комбинаторные методы в
конструировании лекарств. — Рига: Зинатне, 1983.
17. Hansen С, Мс Clarin J., etal. //Molecular Pharmacology. - 1985. - Vol. 27.—
P. 493.
18. Kubinyi H, In Burger's Medicinal Chemistry and Drug Discovery / Ed. M.E.Wolf,
Willey, NY, 1995.
17
РЕГРЕССИОННЫЕ МОДЕЛИ
БИОЛОГИЧЕСКОЙ АКТИВНОСТИ
ОРГАНИЧЕСКИХ МОЛЕКУЛ
Одним из наиболее распространенных статистических методов
описания биологической активности молекул является регрессионный
анализ. Этот метод связывает численное выражение биоактивности
(обычно — отрицательный логарифм количества ЛВ, приводящего к
заданному биоэффекту) и набор выбранных дескрипторов. При этом
предполагается, что введенное в биообъект вещество с некоторыми
потерями проходит к так называемому целевому рецептору — участку
организма (клетке), воздействие на который и определяет
биологическое действие. Сама функция, связывающая биоактивность (или
биоэффект) с дескрипторами, может быть как линейного, так и
нелинейного типа. Подбираемые параметры регрессии, определяющие
вклад того или иного дескриптора, обычно находят с помощью метода
наименьших квадратов (МНК). Подробное описание МНК содержится
во множестве учебников по статистике, поэтому мы ограничимся лишь
наиболее общим описанием МНК и важнейших параметров,
характеризующих полученное уравнение.
17.1. Регрессионный анализ
Простейшее уравнение регрессии имеет следующую форму:
У = ао+а,х,
и выражает линейную связь зависимой переменной (у) с независимой (х), а
величины а0 и гх определяют параметры этой линейной функции. В
методологии QSAR зависимая переменная (у) соответствует численно
выраженной биоактивности исследуемых систем, а переменная х —
это дескриптор, описывающий некий аспект молекулярной структуры.
В качестве простейшего примера подобного рода QSAR-уравнений
приведем выражение для острой токсичности а, р-ненасыщенных
альдегидов по отношению к фосфоресцентным бактериям Vibrio fischeri [l]:
lg—i— - 0.35 + 0.5 lg P,
LD5Q
которое связывает величину LD50c липофильностью (lg/7)
соединения. Данное уравнение было получено на основе изучения экспери-
414
ментальных данных по LD50k lg/'для семи альдегидов. Целью
регрессионного анализа, в этом случае, было нахождение коэффициентов
а0 = 0.35 и а, = 0.5. Графическая иллюстрация подобной задачи
показана на рис. 17.1.
YA
X
Рис. 17.1 Цель регрессии — минимизация отклонений
между наблюдаемыми и предсказанными значениями у
На этом графике наблюдаемые значения показаны в виде точек,
наклонная линия соответствует уравнению линейной регрессии, а
вертикальные отрезки, проходящие от точек до прямой —
отклонения действительных значений активности от рассчитанных.
Основная задача регрессии — найти такие значения коэффициентов
уравнения, чтобы суммарная величина отклонений была минимальной.
Одним из возможных (и наиболее распространенных) средств
решения задачи минимизации отклонений является метод наименьших
квадратов (МНК) [2—4]. Рассмотрим его для общего случая
множественной линейной регрессии, имеющей вид:
y = a0 + a1x1+a2x2+... + akxk.
Предполагая, что прогнозируемый биоэффект для i-той молекулы
(у) зависит от набора к дескрипторов, описывающих молекулярную
систему, соответствующее выражение для биоактивности одной (i-той)
молекулы может быть записано в следующем виде:
yi=a0 + a1x1.+a2x2i+... + akxk.,
где двухиндексная величина ху — является значением j-того
дескриптора для i-той молекулы. Искомые коэффициенты регрессии (а0, а,,
а2, К, \) в МНК находятся путем минимизации величины
A = i(yi-yi(e))2=i(a0 + a1xli+a2x2.+... + akxki-yi(e))2-
i i
суммы квадратов разностей биоактивностей, предсказанных из
уравнения (у;) и наблюдаемых в эксперименте (у^е)). В этом выражении
415
суммирование ведется по числу молекул (N), для которых известны
величины биоактивности (у(е)). Соответствующие уравнения для
коэффициентов а{ могут быть получены как система частных
производных Л по а., приравненных к нулю:
-24-=Д j = 0,...k.
Эа
j
Решение этой системы уравнений относительно величин а. и дает
искомое регрессионное уравнение.
Важнейшим вопросом регрессии является проверка качества
полученной функции, ее предсказательной силы. Для этой цели
используют ряд параметров, среди них стандартное отклонение (Standard
Deviation, SD):
Стандартное отклонение говорит о том, насколько хорошо
теоретические величины (у4), вычисленные с помощью уравнения
регрессии, соответствуют наблюдаемым величинам (у^е)). Величину SD
можно интерпретировать как среднюю точность расчета у; с
помощью уравнения регрессии.
Еще одним важным параметром является множественный
коэффициент корреляции (г) экспериментальных и теоретически
вычисленных с помощью уравнения регрессии величин биоактивности. Эта
величина может быть записана следующим образом:
1(У|(е)-У(с))(у|-у)
г- '———-
у£(У((е)-у(е))2£(У|-у)2 •
где а у и у(е) — средние значения теоретической и
экспериментальной биоактивности для данного набора соединений. Квадрат
коэффициента корреляции (г2) может приобретать значения от нуля до
единицы и, описывает ту долю изменения зависимой переменной (у),
которая может быть объяснена с помощью полученного уравнения
регрессии. С другой точки зрения, величина г выражает взаимную
согласованность (корреляцию) в изменении экспериментальных и
рассчитанных МНК данных. В литературе по статистике г2, обычно,
оценивают как:
«отличные» г2 > 0.99 ,
«хорошие» 0.98 < г2 < 0.99,
«удовлетворительные» 0.95 < г2 < 0.98,
«плохие» г2 < 0.9.
Следует отметить, однако, что такие оценки являются довольно
условными. В особенности это касается расчетов QSAR, которые
обычно опираются лишь на приближенные экспериментальные данные
416
о биологической активности и, поэтому, очень высокая точность
аппроксимации не имеет омыСла.
Для проверки значи**Ъсти корреляции используют ряд критериев.
Среди них популярен так называемый F-критерий:
F (N-k-1) г2
С помощью этой величины по специальным таблицам оценивют
вероятность того, что между биологическим эффектом и
дескрипторами имеется статистическая связь и, следовательно, уравнение
регрессии описывает эту связь.
Недостаток использования вышеописанных критериев оценки
прогностической способности уравнения регрессии состоит в том,
что проверка осуществляется на основе тех эмпирических данных, с
помощью которых и строилось данное уравнение. Для его
преодоления в методологии QSAR широко используется метод перекрестного
оценивания {cross-validation). В' этом методе все исходные данные по
биоактивности {исходная-выборка), делятся на так называемую
обучающую и контрольную выборки: Обучающая выборка используется для
получения искомого регрессионного уравнения. Затем, на основе
полученногоуравнения, дёЛается>расчет биоактивности для молекул,
которые вошли В контрольную выборку. Таким образом, именно на
основе данных для контрольной выборки делается вывод о
прогностической способности'регрессионной модели. Результатом такого
разделения является независимость шагов аппроксимации и
тестирования, что позволяет более корректно определить прогностическую
ценность уравнения. Теоретические величины биоактивности,
полученные для обучающей выборки, обычно называют рассчитанными, в
то время как данные для контрольной выборки называют
предсказанными. Одним из распространенных вариантов метода перекрестного
оценивания является метод скользящего контроля (в англоязычной
литературе известен как leave-one-out cross-validation, LOO, см.,
например, [5]). В этом методе из выборки с эмпирическими данными в
контрольную группу извлекается лишь одна молекулярная система.
Оставшаяся часть данных составляет обучающую выборку и на ее
основе находится уравнение регрессии. Затем делается прогноз
биоактивности для той единственной молекулы, которая была выведена
из обучающей выборки. На следующем этапе из обучающей выборки
удаляется другая система, в то время как первая возвращается. Новое
регрессионное уравнение используется для предсказания активности
уже другой системы. Процесс повторяется до тех пор, пока в
контрольной выборке не побывают все молекулярные системы исходной
выборки. На основе экспериментальных и предсказанных данных по
биоактивности вычисляют квадрат коэффициента корреляции (обычно
обозначают как q2), который и характеризует истинную
предсказательную силу уравнения. Таким образом, коэффициент корреляции,
вычисленный с помощью рассчитанных значений биоактивности (г2),
является мерой качества аппроксимации экспериментальных данных,
417
в то время как величина q2, вычисленная с использованием
предсказанных значений биоактивности, характеризует предсказательную силу
модели в целом. В методе скользящего контроля важное значение
имеет также параметр, являющийся аналогом стандартного
отклонения, который характеризует среднюю точность в предсказании
активности:
i(yi(e)-yl(prcd))2
° н -АР1—]
pred у j^
где у;(е) — экспериментальные, a y;(pred)- предсказанные значения
активностей для i-той молекулы.
В качестве примера использования указанных параметров
приведем уравнение, описывающее токсичность набора ароматических
соединений по отношению к реснитчатым Tetrahymena pyriformis [6]:
lg T^ = 0.603-IgP-0.33-ELUM0-1,
IGC50
(N = 239, r2 = 0.8, q2 = 0.796, a = 0.335, F = 476).
Величина ЮС50 — концентрация (мМ), ингибирующая рост
биологических объектов на 50%, ELUMO — энергия нижайшей вакантной
молекулярной орбитали (эВ). Величины в скобках говорят о том, что
QSAR уравнение было получено на основе анализа данных для 239
соединений, незначительные различия q2 и г2 свидетельствуют о
предсказательной силе уравнения.
В данном разделе приведены лишь наиболее общее описание
регрессионного анализа и МНК, поскольку оно имеется в ряде
учебников и монографий по статистике. Кроме того, этот метод и
соответствующие оценки значимости включают в себя множество
современных пакетов прикладных программ для персонального компьютера
(EXEL, CurveExpert, STATISTICA, ORIGIN). Существуют также
специализированные компьютерные программы, которые
предназначены для построения математических моделей QSAR.
17.2. Факторные модели в регрессионном анализе
Особенность проблематики QSAR заключается в том, что заранее
неизвестно сколько и какие именно дескрипторы необходимы для
описания заданного свойства. Поэтому зачастую возникает «соблазн»
выбрать очень широкий набор параметров, что приводит к
возникновению «случайных» корреляций. Часто возникает также ситуация, когда
число молекул сравнительно мало по сравнению с числом
дескрипторов. В этих случаях применяются так называемые факторные
методы [5, 7], которые основаны на изучении структуры корреляций
между параметрами предполагаемой регрессионной модели. К сожалению,
418
последовательное и строгое изложение теории факторного анализа
представляет собой довольно сложный материал, требующий
специальных знаний в области статистики. Поэтому в данном разделе мы
ограничились лишь наиболее общим его изложением, которого,
однако, на наш взгляд, вполне достаточно для понимания сущности
методологии.
Одним из распространенных факторных подходов является метод
анализа главных компонент (principal components analysis, PCA). В этом
методе анализируется структура корреляций между всеми
параметрами задачи с целью идентификации новых переменных, которые
суммируют информационное содержание широкого первичного дескрип-
торного набора. Источником такого сжатия исходных данных служит
тот факт, что многие дескрипторы, которые используются с целью
описания молекулярной структуры, могут в значительной степени
коррелировать друг с другом. Например, описанный в предыдущей
главе набор топологических (информационных) индексов 1СР 1С2и
т. д. характеризует тот же аспект молекулярной топологии, что и
известные индексы связности Рандича (х(1\ Х(1))- Поэтому очевидно, что
эти величины должны коррелировать друг с другом, т. е. есть
изменяться подобным образом при переходе от молекулы к молекуле.
Корреляция этих величин, в свою очередь, означает, что может быть
построена такая линейная функция, с помощью которой одну величину
можно было бы выразить через другую. Тем самым достигается
сжатие данных — вместо большого числа дескрипторов можно работать с
небольшим количеством величин, которые бы вбирали в себя
значительную часть информации, содержащейся в дескрипторах.
Простейший пример — корреляция двух молекулярных
параметров Xj и Xj (рис. 17.2).
Х2
X!
Рис. 17.2. Корреляция двух дескрипторов
Согласно этому рисунку, значения дескрипторов с некоторой
точностью лежат на прямой. Это означает, что вместо х, и ^ можно
419
ввести новую переменную, которая представляет собой линейную
комбинацию дескрипторов:
z = ах( +Ьх2.
Тогда задача линейной регрессии может быть трансформирована из
У-ао+а|х1+аЛ
в
у = а„ + a;z.
Такую скрытую (латентную) переменную (z) называют главной
компонентой {principal component, PC). В общем случае, когда речь идет о
наборе данных содержащих множество дескрипторов, приходится иметь
дело с несколькими главными компонентами, каждая из которых
вбирает в себя свою часть взаимных корреляций дескрипторов:
РС. =СПХ1+С12Х2+-+С1Л
PC2=c2ixi+c22x2+... + c2kxk
и т. д., где коэффициенты сц определяют вклад j-того дескриптора (х,)
в i-тую главную компоненту, а к — общее количество дескрипторов, с
помощью которых предполагается описывать данную задачу.
Обычно, для описания корреляций дескрипторного набора с данными по
биоактивности достаточно 1-3 главных компонент. В данном разделе
мы не описываем методики расчета PC, поскольку это очень
специальный вопрос. Отметим только, что множество пакетов прикладных
программ для персональных компьютеров содержит РСА как
составную часть факторного анализа. Среди них популярный пакет
STATISTICA.
Метод РСА дает ценную информацию, с помощью которой
можно сжать набор дескрипторов, выбрав наиболее важные компоненты
(дескрипторы) из разложения для PC. Однако существует и прямое
применение PC при построении уравнения регрессии. В этом методе
{principal component regression, PCR) уравнение регрессии приобретает
форму:
У = аРС. +а.РС,+... + а PC ,
112 2 р р >
где коэффициенты а. определяют вклад i-той главной компоненты в
исследуемую биоактивность (у), ар — количество отобранных
главных компонент. Наиболее последовательное развитие этого подхода
достигается в так называемом неполном методе наименьших
квадратов {partial leost squares, PLS, см. например [8]), в котором
учитывается структура совместной матрицы корреляций, включающей как
зависимую переменную (у), так и дескрипторный набор (х.). Уравнение
регрессии в случае PLS имеет вид: J
у = aZ +a,Z, +... + a Z ,
11 2 2 р р '
где латентные (скрытые) переменные Z., как и прежде, выражают
посредством линейных комбинаций дескрипторов:
420
22=С2.Х,+С22Х2 + - + С2Л ИТ'Д-
Отличие от PC, однако, заключается в том, что коэффициенты с.
в методе PLS выбирают таким образом, чтобы результирующая
функция (у) наиболее точно описывала изменение биоактивности от
молекулы к молекуле.
Методики типа PCR и PLS применяют обычно в случаях, когда
биоактивность систем не удается описать с помощью небольшого числа
дескрипторов. В таких задачах число молекулярных параметров может
значительно превышать количество объектов (молекул) и, как следствие
этого, стандартные методики регрессионного анализа неприменимы.
Например, активность по отношению к связыванию с рецептором для
32 различных хлорпроизводных дибензофурана (рис. 17.3) может быть
описана с помощью 188 различных дескрипторов [9]. Среди них
топологические, химические и геометрические параметры.
Рис, 17.3. Хлорпроизводные дибензофурана
Сопоставление качества прогноза для методов PCR и PLS
(табл. 17.1) говорит о том, что неполный метод наименьших
квадратов (PLS) обладает большей предсказательной силой,
характеризующейся значительной величиной q2 и относительно малой средней
ошибкой (opred).
Таблица 17.1
Параметры качества регрессии для методов PCR и PLS
2
q
pitd.
PCR
0.683
0.790
PLS
0.837
0.565
17.3. Эмпирические константы заместителей.
Уравнения Гаммета и Тафта
При построении регрессионных зависимостей «биоактивность —
структура» для рядов соединений, отличающихся только
заместителями, удобно использовать подходы с эмпирически подобранными
421
параметрами заместителей. Одним из таких подходов, который
базируется на термодинамическом принципе линейности свободных энергий,
является метод Гаммета [10]. Он применяется для описания я-элект-
ронных эффектов заместителей и основан на анализе констант
ионизации мета- и лара-замещенных бензойных кислот. Уравнение
Гаммета связывает свободную энергию Гиббса для данной реакционной
серии (AG) с аналогичной величиной, полученной для другой
(стандартной) реакционной серии (AG0), с тем же самым набором
варьируемых параметров (заместителей):
lgk-lgk = ! (AG-AGj,
0 2.303 RTV °'
где kf, и k — константы скорости реакции соответственно для
незамещенной и замещенной молекулы. Предполагая линейную связь
между AG - aG0 и параметрами заместителей, получаем уравнение Гаммета:
lgk-lgk0 = ро.
В этом уравнении параметр р — характеристика данной
реакционной серии, а а — величина, характеризующая заместитель. При
этом обычно различают константы заместителей, введенных в пара —
а и мета — ат положения бензольного кольца. Известны также
соответствующие величины для реакций в алифатическом ряду
(индукционные постоянные — а*).
Однако подчеркнем, что уравнение Гаммета описывает лищь
электронные эффекты взаимодействия заместителя с реакционным центром.
В тоже время известно много реакций, константы которых зависят
также от пространственных (стерических) особенностей
заместителей. В связи с этим при изучении кислотного гидролиза эфиров Таф-
том было получено выражение, по форме аналогичное уравнению
Гаммета:
lgk-lgk0 =SEs,
в котором Es — некоторая пространственная характеристика
заместителя, а 5 — мера чувствительности данной реакционной серии.
Параметры наиболее распространенных заместителей приведены в
табл. 17.2.
Эти величины, как характеристики электронных и стерических
эффектов заместителей, широко используются в качестве
дескрипторов регрессионных моделей биологической активности.
17.4. Аддитивная модель Фри—Вильсона
В 1964 году Фри и Вильсон [11] разработали математическую
модель, которая описывает биоэффекты от определенных
функциональных групп, находящихся в различных положениях базовой
422
Таблица 17.2
Эмпирические константы заместителей
Заместитель
-NH,
-N(CH3);
-ОСИ,
1-ос2н.
-СН3
-ан.
-0.660
-0.83
-0.268
-0.24
-0.170
-C3H7(i)
-С(СН3)з
-SCH3
н
-он.
-0.151
-0.126
-0.161
-0.211
0.115
0.1
-0.069
-0.07
1.450
1.366
0
-0.100
Es
0.97
0.86
0
-0.115
-0.197
-0.047
0
0.009
-С1
0.062
-Вг
-I
-СОСН3
-соон
0.227
0.232
-0.10
0.15
0.218
0.337
0.373
0.18
0.502
0.265
0.66
0.778
0.391
0.352
-0.300
0.600
3.08
2.92
-0.27
-0.56
-2.14
1.24
-0.90
0.49
2.78
2.36
0.376
0.355
0.56
0.710
1.65
2.1
3.6
3.9
0.18
0.06
-0.20
-0.75
-CN
-NQ2
молекулярной системы (рис. 17.4). При этом наличие или отсутствие
заместителя определенного типа в определенном положении
молекулы кодируется символами «1» или «0». В результате появляется
матрица Фри—Вильсона, которая, с одной стороны, описывает
молекулярную структуру, а, с другой стороны, коррелирует с биоактивностью
соответствующих замещенных.
Рис. 17.4. Схематическое представление молекулы по Фри—Вильсону —
базовая система и набор заместителей в различных положениях
Тогда, предполагая вклад заместителей аддитивным, уравнение для
биоактивности молекулы i (y;) можно записать следующим образом:
у. = У п. .а . +уп,
423
где аи— вклаД в общую активность заместителя j, находящегося в р-том
положении,!^— количество заместителей тиаа j в положении р
(обычно 0 или 1, редко — 2) для i-той молекулы, a y0 — биоактивность
незамещенной (базовой) молекулы. Численные значения
коэффициентов ай и у0 обычно находят с помощью процедуры МНК.
В качестве примера использования методики Фри-Вильсона
рассмотрим классический пример, описывающий антиадренергическую
активность К,К-диметил-2-бромфенетиламинов (рис. 17.5)
(экспериментальные данные взяты из [12]). Эти соединения блокируют
действие медиаторов, передающих возбуждение от постганглионарных
нервных узлов.
Вг СН3
Рис. J7.5. Замешенные ^>1-диметил-2-бромфенетиламинов
(X,Y = H, F, C1, Вг, I, СН3)
Соответствующая матрица Фри-Вильсона описывает 22
соединения, которые формируются пятью заместителями (F, C1, Вг, I,
СН3), введенными в мета- и пара- положения бензольного кольца
(табл. 17.3).
Антиадренергическая активность (lgVc) различных замещенных
выражаеься с помощью концентраций (С) приводящих к 50%
уменьшению соответствующего эффекта. Расчет коэффициентов а . и у
методом наименьших квадратов приводит к следующему
регрессионному уравнению:
lg^ = -0.301nm_F + 0.207nm_Q + 0.434nm_Br + 0.57911^ + 0.454пт_СНз
+0.340np_F + 0.768np_Q + 1.02011^ + 1.429n^ + 1-256пр_СНз + 7.821
(n = 22, r = 0.969, a = 0.194).
В указанном уравнении символы nm.F, nm.ci, nffl.Br, n^, nm.CH3 и np.F,
nP-cp nP-Br> nP-i» пр-снз соответствуют заместителям (фтор, хлор, бром,
йод или метальная группа) в мета- и пара- положениях. Таким
образом, величина п^ либо равна единице, если заместитель присутствует,
либо равна нулю — если в соответствующем положении его нет.
Простой анализ этого уравнения говорит о том, что наличие фтора в мета-
положении заметно снижает биологическую активность
(соответствующий коэффициент при nm.F отрицательная величина —0.301). В
тоже время йод в мета- положении бензольного кольца приводит к
существенному увеличению lg!/c.
424
Таблица 17.3
Матрица Фри-Вильсона и соответствующие антиадренергические
активности (IgVc) ^^диметил-2-бромфенетиламииов
Мета заместители
№
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
F
0
0
0
0
0
1
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
С1
0
0
0
0
0
0
1
0
0
0
1
0
0
1
0
0
1
0
0
0
0
0
Вг
0
0
0
0
0
0
0
1
0
0
0
1
0
0 '
1
0
0
1
0
0
1
0
I
0
0
0
0
0
0
0
0
1
0
0
0
0
0
0
0
0
0
0
0
0
0
сн3
0
0
0
0
0
0
0
0
0
1
0
0
1
0
0
1
0
0
1
1
0
0
Пара заместители
F
1
0
0
0
0
0
0
0
0
0
1
1
1
0
0
0
0
0
0
0
0.
0
С1
0
1
0
0
0
0
0
0
0
0
0
0
0
1
1
1
0
0
0
0
0
0
Вг
0
0
1
0
0
0
0
0
0
0
0
0
0
0
0
0
1
1
1
0
0
0
I
0
0
0
1
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
СН3
0
0
0
0
1
0
0
0
0
0
0
0
0
0
0
0
0
0
0
1
1
0
Активность
Ig'/c
8.16
8.68
8.89
9.25
9.30
7.52
8.16
8.30
8.40
8.46
8.19
8.57
8.82
8.89
8.92
8.96
9-00
9.35
9.22
9.30
9.52
7.46
Сопоставление рассчитанных и экспериментальных величин ан-
тиадренергической активности 1Ч,К-диметил-2-бромфенетиламинов
(рис. 17.6) демонстрирует вполне удовлетворительную корреляцию.
С этим выводом согласуются также величины коэффициента
корреляции (г = 0.959) и стандартного отклонения (а = 0.194).
Точности модели Фри-Вильсона вполне достаточно для целей
прогноза новых (не вошедших в табл. 17.3) соединений. Так
элементарный подсчет позволяет оценить lg !/c для дибромзамещенного
(пт-вг = * и пр-вг ~ *' а остальные величины п = 0):
lg J_ = + 0.434 + 1.020 + 7.821 = 9.275,
425
10
9,5
ф 9
т
о
со
.S 8,5
О
^5> 8
7,5
7
7,5 8 8,5 9
lg(1/C) эксперимент
9,5
10
Рис. 17.6. Линейная связь рассчитанных и экспериментальных величин
активности ^Ы-диметил-2-бромфенетиламинов
что характеризует структуру
как относительно активную.
17.5. Метод Хэнча. Значение липофильности
В методе Хэнча [13] рассматривается кинетическая модель,
согласно которой БАВ (С), взаимодействуя с биологической
мишенью (S), образует соответствующий комплекс (C:S) (см. также
раздел 3.3). В результате устанавливается равновесие с константой к:
С + S =^=
C:S
В соответствии с законами химической кинетики константа
равновесия выражается через отношение концентраций:
1: [C:S]
" [С] [S] '
426
Очевидно, что концентрация вещества С вблизи рецептора
пропорциональна его количеству, введенному в биообъект С0:
[С] = А.С0.
Поэтому концентрация С0 может служить мерой биологической
активности препарата (обычно рассматривается логарифм обратной
величины, lg!/c). Так, если заданный или стандартный биоэффект
(например замедление на 50% роста опухоли) наступает при очень
малых значениях С0, то препарат высокоактивен, и наоборот, —
большие значения С0 свидетельствуют о его малой активности.
Таким образом, в случае, когда С0 мало, величина lgl/c становится
большой, что соответствует высокой активности. Очевидно также,
что коэффициент пропорциональности А определенным образом
должен быть связан с параметрами, характеризующими
транспортировку ЛВ через клеточные мембраны к мишени. Предполагая так
называемую модель «случайных блужданий» (рис. 17.7), можно
ожидать, что ЛВ на своем пути приходится преодолевать ряд
водно-липидных слоев. В этом случае в качестве основного
параметра, описывающего продвижение ЛВ к рецептору, можно
использовать величину, которая характеризует распределение вещества
в различных фазах.
drug
receptor
Рис. 17.7. Модель случайных блужданий для транспорта лекарства
к рецептору. Пунктиром обозначены границы водно-липидных слоев
В настоящее время для этой цели принято использовать липо-
фильность — логарифм коэффициента распределения вещества в
смеси октанол-вода (\gP). Рассмотрим, каким же образом \%Р
связан с биоактивностью в рамках фармакокинетической модели.
Ответ на этот вопрос дает компьютерное моделирование
прохождения вещества через множество водно-липидных слоев. Численное
решение системы кинетических уравнений, описывающих такие
процессы, приводит к так называемому параболическому типу
зависимости lg— от \%Р [14]:
Со
lg-L~ a (lgp)2+blg?.
Со
Типичный график такой зависимости показан на рис. 17.8.
427
1д(1/с) и
igP
Рис. 17.8. Параболический тип зависимости активности lg('/c)
от липофильности ]gP
Очевидно, что при определенных значениях \gP достигается
максимум биологической активности препарата lg 1/C0 . Это
обстоятельство широко используется при разработке новых ЛС. Так» например,
анализ экспериментальных данных свидетельствует о том, что
соотношение lg.P~2 оптимально для того, чтобы ЛВ .преодолевало
ГЭБ (рис. 17.9).
1од1/С
нейротоксическая
активность
транспорт:
кровь-плацента
logP
Рис. 17.9. Нейротоксическая активность серии соединений
имеет максимум при IgP = 2. Перенос ЛВ через гемато-плацентарныЙ барьер
имеет менее отчетливый максимум в районе lgP = О
Иными словами, при данной величине липофильности
концентрация введенного вещества в ЦНС биообъекта максимальна. Это
означает, что поиск новых ЛС действующих на центральную нервную
систему целесообразно начинать с соединений имеющих lg P ~ 2.
428
Некоторые из таких эффективных ЛС, получивших широкое
клиническое применение, вместе с соответствующими значениями \gP
приведены в табл. 17.4.
Таблица 17.4
ЛВ, оказывающие депремирующее воздействие на ЦНС
и их липофильности
Препарат
Циклопропан
Дивиниловый эфир
Глутемид
Хлороформ
Хлорэтан
Этран
lg^
1.72
1.80
1.90
1.97
2.03
2.10
Препарат
Кетамин (кеталар)
М етоксифлуран
Трихлорэтилен
Галотан
Элениум
Диазепам
\%Р
2.18
2.21
2.29
2.30
2.44
2.82
Известны также оптимальные величины lg.P обеспечивающие
достижение других органов — мишеней (печени, почек и т. д.).
Параболическая зависимость активности от lgP, не является
единственной. Большое распространение в последнее время получила так
называемая билинейная модель (Кубиньи, [15]):
lgyc0 - a lgP + b lg(pP + l),
в которой числа а, Ь, и р — подгоночные параметры. При этом
величина р нелинейно входит в это уравнение, что существенно осложняет ее
оценку. Типичная билинейная зависимость представлена на рис 17.10.
1- »-»-в^_
lg(1/C) 1 / ^*""
_д4 1— ■-— | ■- ■ -г >р- i ■ 1 .
-20246
Рис. 17.10. Билинейная модель связи биоактивности и липофильности
Особенностью рассматриваемой модели является наличие двух
линейных участков вдали от максимума, что хорошо согласуется с
множеством экспериментальных данных.
Разумеется, не только величина липофильности оказывает
влияние на,эффективность воздействия вещества на организм. Очевидно,
что с химической точки зрения взаимодействие молекулы с мишенью
429
должно быть связано с параметрами электронной структуры
молекулы, такими как электронная плотность, дипольный момент, сверхде-
локализуемость и т. д. Не менее важны и особенности
пространственного строения молекулы БАВ. Если речь идет о ряде соединений,
различающихся набором заместителей, то ясно, что биологическое
действие может быть выражено (аналогично уравнению Гаммета)
через эмпирические константы заместителей, описанные ранее.
Таким образом, учитывая перечисленные выше факторы, можно
записать общее уравнение Хэнна (с параболической формой
зависимости lgl/C0 от \%Р), которое описывает биоактивность БАВ:
lgl/C0 =a (lgP)2+blg? + c £o, + d£E.+... + const.
В этом уравнении первые два члена характеризуют вклад липо-
фильности, третий член описывает электронные эффекты
заместителей, а четвертый член — стерические факторы. Кроме того,
уравнение может содержать ряд электронных индексов (заряды на
отдельных атомах, дипольный момент молекулы и т. д.)-
Варьируемые параметры (а, Ь, с, d,..., const) находят с помощью процедуры
МНК. Если вместо общей липофильности системы целесообразно
использовать только вклады от заместителей (тсх), то уравнение
приобретает вид:
lgl/C0 = а (Хя^ + ЬХяГ+с 5>i + dXEi+... +const.
Приведем несколько примеров нелинейных QSAR зависимостей.
Первое уравнение получено Хэнчем и описывает активность
производных нитрозомочевины (рис. 17.11) против L-1210 лейкемии
у мышей [16]:
lg J- = -0.0568 (lgP)2 + 0.0689\%Р + 4.527.
(N = 22, г = 0.922, s = 0.163).
X СН2 СН2 N С NH R
с I "
0=N О
Рис 17.11. Производные нитрозомочевины
Уравнение имеет пологий максимум в районе \%Р = 0.6 и
позволяет, используя лишь данные о липофильности, предсказывать
эффективность веществ с противораковым действием.
Другой наглядный пример использования QSAR — нелинейное
уравнение, описывающее фенантренкарбинолы (рис. 17.12) как
антималярийные агенты [16].
430
Рис. 17.12. Производные фенантренкарбинола
^^ = 0.29-дх+у+0.9-ах+у+0.11(яК]+71К2)--0.013(<|+<2) + 2.41.
(N = 212, г = 0.86, s = 0.32).
В этом уравнении С50 — молярная концентрация препарата,
необходимая для достижения полумаксимального эффекта. Очевидно, что
члены уравнения, включающие липофильности заместителей (я),
характеризуют способность БАВ достигать биологические мишени, в то
время как электронные параметры заместителей (а) —
взаимодействие с этой мишенью. В табл. 17.5 приведены данные по
биологической активности некоторых наиболее активных замещенных фе-
нантрена, которые вошли в обучающую и контрольную выборки.
Таблица 17.5
Антималярийная активность некоторых замещенных феиантрена
из обучающей и контрольной выборки
№
1
2
3
4
S
1
2
3
Заместители
в кольце
NR,R2
50
эксперимент
Активные системы из обучающей выборки
2,4-дихлор,
6 -трифторметил
2,3-дибром,
6 -трифторметил
3 -трифторметил,
5,7-дихлор
1,3,6-трибром
2,5,7-трихлор
2-пиперидил
N(C4H9)2
2-пиперидил
N(C4H9)2
2-пиперидил
4.34
4.36
4.35
4.35
4.29
Активные системы из контрольной выборки
1,2,3,4-тетрахлор,
6-трифторметил
2,4-д и(трифторметил),
7-хлор
2,4-ди(трифторметил),
6,7-дихлор
2-пиперидил
2-пиперидил
2-пиперидил
4.82
4.82
4.47
50
расчет
4.36
4.26
4.21
4.08
4.29
5.18
4.34
4.88
431
Эти данные свидетельствуют о том, что указанная регрессионная
модель хорошо справляется не только с задачей аппроксимации
исходных экспериментальных данных по *S /q t но и обладает
значительной предсказательной силой, что подтверждается величинами
активности для соединений, которые вошли в контрольную выборку.
Следующее уравнение в рамках билинейной модели описывает
ингибирование оксидазы аминами и спиртами при различных
значениях рН (Кубипьи).
lg J~ = 3.13lg />-3.379 lg(pP + l)- 3.507-1 - lg(l+ Юр*"рН) + 3.379,
К.
lgp = -1.781,
(N = 21, г = 0.999, a = 0.188).
В этом уравнении константа процесса сложным образом связана с
через константой распределения в системе октанол-вода (Р), а также рН
среды и рА'ионизации вводе. Индикаторная переменная I для аминов
принимает значение 0, адля спиртов I =1. Анализ этого уравнения
показал, что оптимум ингибирования достигается при lg/*= 2.45.
17.6. Метод сравнительного анализа молекулярных полей
Одним из наиболее значительных достижений QSAR в последнее
десятилетие является метод сравнительного анализа молекулярных
полей (Comparative Molecular Field Analysis, CoMFA, [17]). Метод
CoMFA основывается на том наблюдении, что взаимодействие БАВ с
мишенью определяется, первую очередь, межмолекулярными
эффектами, которые в основе своей нековалентны и в значительной степени
зависят от пространственных особенностей, формы молекул. Целью
CoMFA является изучение корреляций между трехмерными
характеристиками молекул и их биологической активностью. К трехмерным
молекулярным дескрипторам относятся стерические,
электростатические и др. пространственные параметры. Особенностью модели CoMFA
является то, что дескрипторы, описывающие молекулу, вычисляются
не в виде отдельных величин, характеризующих систему в целом, а в
виде трехмерной карты, описывающей заданное свойство в
пространстве. Таким образом, дескриптор соответствующий данному свойству,
представляет собой молекулярное поле охватывающее молекулу. Для
вычисления такого поля предполагается, что молекула находится в
трехмерной пространственной решетке (рис. 17.13).
Заданное свойство вычисляется в узлах этой пространственной
решетки как результат действия на данную точку пространства (узел)
со стороны структурных составляющих молекулы. Например, чтобы
определить электростатический потенциал, описывающий
пространственное распределение кулоновских сил вблизи молекулы, во все
узлы решетки необходимо последовательно поместить пробный за-
432
Рис. 17.13. Исследуемая молекула расположенная внутри
пространственной решетки
ряд. Суммарное действие на этот заряд со стороны всех атомов
молекулы и определяет значение электростатического потенциала в узле.
Стандартным пробным элементом CoMFA является углеродный атом
в 8р3-гибридном состоянии с эффективным радиусом 1.53А и зарядом
+ 1. Электростатический потенциал Е в узле j вычисляется с
помощью закона Кулона,
Е = V-^L
J tr.'
где qs — заряд на i-том атоме, а г. — расстояние между пробным
(единичным) зарядом и i-тым атомом молекулы. При вычислениях
геометрии и зарядовых распределений, как правило, используются
полуэмпирические методы квантовой химии (AMI, PM3, MNDO).
Для описания стерического молекулярного поля используется
известный потенциал Леннарда-Джонса (или 6-12 потенциал)
А..
и_
J 2
В
где А.
и В,-
параметры, которые зависят от ван-дер-Ваальсовых
радиусов соответствующих атомов.
В результате расчетов электростатических полей для каждой
молекулы обучающей выборки вычисляется огромное количество
(сотни или даже тысячи) параметров, соответствующих значениям
потенциалов в разных точках пространства. Таким образом, число
молекулярных дескрипторов значительно превышает число молекул.
В этом случае стандартные методики регрессионного анализа
оказываются неприменимы. Для построения уравнения регрессии CoMFA
433
используется метод PLS, описанный нами ранее. Общая форма
соответствующего уравнения остается той же:
Y = Ya.E.. +SbS. + const.
i y j и y j и
В этом выражении биологическая активность i-той молекулы (Y.)
связана со значениями электростатического и стерического
потенциала (E^Sjj) в различных точках пространства (индекс j).
Коэффициенты а. и Ь. определяют вклады пробы, расположенной в узле j по
отношению к электростатическому и стерическому полю соответственно.
Методу CoMFA идейно близок метод сравнительного анализа
индексов молекулярного подобия (Comparative Molecular Similarity
Indices Analysis, CoMSIA). Последний также оперирует с
трехмерными (пространственными) картами молекулярных полей. В методе
CoMSIA (как и в CoMFA) рассчитываются стерические и
электростатические поля. Кроме того, вычисляют поля, описывающие свойства
гидрофобности и способности к образованию водородной связи.
Однако, в отличие от CoMFA, вся информация об этих полях
концентрируется в специальном «поле подобия», которое имеет следующие
экспоненциальное представление:
W. =Wq.q. +Wv.v.+...
где qi — как и прежде заряд на атоме, a v.- ван-дер-Ваальсов радиус
атома. Величины We, Ws являются эмпирическими параметрами,
которые определяют вклад различных составляющих в поле подобия.
Благодаря использованию гауссовой формы при вычислении
расстояний ( "% ) метод CoMSIA порождает гладкую контурную карту поля,
с которой удобно работать и которая не имеет особых точек.
Наибольшее применение CoMFA и CoMSIA нашли в разработке
новых ЛС, для выявления тех структурных особенностей, которые
важны для эффективного связывания с соответствующей
биологической мишенью. Ряд коммерческих компьютерных программ
реализует эти алгоритмы. Среди них особую популярность приобрела
программа SYBYL [18].
Литература
1. Cronin М. T.D., Schultz Т. W. // Ecotoxicol. Envir. Safety. - 1998. - Vol. 39. -
P. 65.
2. Лоусон Ч., Хенсон P. Численное решение задач метода наименьших
квадратов. — М.: Наука, 1986.
3. Демиденко Е.З. Линейная и нелинейная регрессии. — М.: Финансы и
статистика, 1981, 302 с.
4. Статистические методы для ЭВМ / Под ред. К. Энслейна, Э. Релстона,
Г.С.Уилфа. - М: Наука, 1986.
5. Leach A.R., Gillet V.J. An introduction to chemometrics. — Dordrecht-Boston-
London Kluwer Academic Publishers, 2003.
434
6. Cronin M.T.D., Schultz ТЖ // Chem. Res. Toxicol. - 2001. - Vol. 14. -
P. 1284.
7. Факторный, дискриминантный и кластерный анализ. — М.: Финансы
и статистика, 1989.
8. Geladi P., Kowalski B.R. //Analytica Chimica Acta. - 1986. - Vol. 185.-
P. 1.
9. Basak S.C., Mills D., Hawkins DM. et al. // SAR QSAR Environ. Res. -
2002. -Vol. 13. - P. 649.
10. Днепровский А.С., Темников Т.Н. - Теоретические основы органической
химии. — Л.: Химия, 1991.
11. Free S.M. Wilson J.W.// Journ. of Med. Chem. - 1964. - Vol. 7. - P. 395.
12. Kubinyi H, Kehrhahn O.H. // Journ. of Med. Chem. - 1976. - Vol. 19. -
P. 1040.
13. Hansch С И Acc.Chem.Res. - 1969. - Vol. 2. - P. 232.
14. Hansch C, Clayton J.M. I I Journ. Pharm. Sci. - 1973. - Vol. 62. - P. I.
15. Kubinyi H. //Journ. Pharm. Sci. - 1978. - Vol. 67. - P. 262.
16. Хэнч К. II Хим.-фарм. журнал. - 1980. - № 10. - С. 15.
17. Cramer R.D., Patterson D.E., and Bunce J.D., J. Amer. Chem. Soc, 1988,
vol. 110, p. 5959-5967.
18. SYBYL/QSAR, Molecular Modeling Software, Tripos Inc., 1699 S.Hanley Road,
St Louis, MO 63944, USA.
18
СТАТИСТИЧЕСКИЕ МЕТОДЫ КЛАССИФИКАЦИИ
МОЛЕКУЛ ПО ИХ БИОЛОГИЧЕСКОЙ АКТИВНОСТИ
В связи с успехами органического синтеза, позволяющего
получать огромное количество новых структур, возрастает потребность
проведения систематического скрининга соединений с целью отбора
наиболее перспективных в плане их активности, дальнейшего
исследования и применения. Эта задача приводит к использованию
статистических методов классификации молекулярных структур по степени
выраженности их биологического действия. Наиболее
распространенной является классификация молекул на активные и неактивные. При
этом предполагается наличие определенной обучоющей выборки —
набора молекулярных систем, биологическая активность которых
известна. С помощью этой выборки проводится «настройка»
математической модели с целью выявления наиболее важных молекулярных
параметров, необходимых для описания заданного типа активности.
В данном разделе описаны три наиболее распространенных
подхода к классификации. Одним из таких подходов, опирающихся на
исследование связи элементов химической структуры молекулы с
биологической активностью, является так называемый
логико-комбинаторный метод. Достоинством его является возможность формулировки
характерных химических признаков для каждого типа
биологического действия.
Метод кластерного анализа относится к группе «обучения без
учителя». В основе этого метода лежит многопараметрическое
исследование заданного набора молекулярных структур посредством введения
метрических характеристик дескрипторного пространства.
К группе статистических подходов типа распознавания образов
относят дискриминантный анализ — метод позволяющий построить
классификационную функцию в явном виде как функцию от
молекулярных дескрипторов.
18.1. Логико-комбинаторный подход
Логико-комбинаторный подход (Розенблит, Голиндер, [1])
базируется на некоторых результатах формальной логики и статистики.
Рассмотрим наиболее важные в контексте этой проблемы
логические понятия.
436
Предположим, что нам предстоит задача исследования
закономерности «структура объекта»-»«явление или свойство». В этом
случае структура исследуемого объекта может быть описана с помощью
неких простых элементов (дескрипторов) А, В, С, D,.... Наличие
определенного свойства обозначим символом X, а его отсутствие X .
Предположим также, что не существует других факторов,
оказывающих воздействие на исследуемое свойство. Иными словами система
дескрипторов полна и не требует расширения. Тогда, в контексте
задачи «структура объекта»-»«свойство», можно определить несколько
формальных методов.
1. Сходство. Если два или больше исследуемых случаев имеют
общим одно обстоятельство (элемент структуры объекта), то это
обстоятельство и есть причина данного феномена. Схематически это
можно выразить следующим образом. Некоторая структура имеет
компоненты АВ и обладает свойством X: АВ-»Х. Другая структура
имеет компоненты АС и обладает тем же свойством X: АС—>Х.
Отсюда следует, что данное свойство связано с А: А—>Х,
АВ->Х
АС->Х
А->Х
2. Различие. Если объект для которого данное свойство X
проявляется, и объект, когда оно не проявляется (X), сходны во всех
компонентах структуры кроме одной, которая появляется только в
первом случае, то этот элемент структуры и есть причина данного
явления.
ABCD->X
BCD->X
А->Х
Эти два утверждения и являются основой для
логико-комбинаторного метода анализа биологической активности. Его основные
положения состоят в следующем:
1. Структура каждой молекулы \х описывается с помощью
специального вектора-описателя S . Этот вектор представляет собой
упорядоченное перечисление элементов молекулярной структуры и
позволяет однозначно охарактеризовать данную молекулу среди
других молекул обучающей выборки.
2. Соединения обучающей выборки классифицируются по типам
активности на Nk классов Ак, к - 1+Nk. В самом простом случае
предусматривается разбиение выборки на два класса (Nk = 2):
активные и неактивные (или слабоактивные).
3. Признаки активности fr(S) (или неактивности) определяются как
некоторые фрагменты молекулярной структуры, выраженные с
помощью вектора-описателя S . Присутствие этих элементов в
молекуле с высокой вероятностью указывает на то, что соединение
437
проявляет (или не проявляет) данную активность. Такие
потенциальные признаки находят путем сравнения пар молекул внутри
каждого класса с последующим выделением общего фрагмента:
f(S) = S (IS
4. Подсчитывается число появлений этого признака Lkr в каждом из
классов Nk, а также суммарное количество проявлений этого
признака во всех классах —L .
г
r k=i kr
5. Проводится статистическая оценка проявления этих признаков
для каждого класса. Поскольку точные значения вероятностей
того, что соединение, обладающее данным признаком f(S)
относится к классу \ неизвестны, для их оценки можно
воспользоваться методом Байеса. В этом методе вероятности
вычисляются так, что возможный ущерб от неверной оценки
минимизируется. В случае разбиения массива объектов из
обучающей выборки на два класса (активные — неактивные)
формула для вероятности отнесения к классу \ записывается
следующим образом:
p(Ajf) = bL±i
V k' '; L +2 •
г
При этом прогностическая ценность признака определяется как
и .РК'0
ь~ р(\) '
Величина прогностической ценности (Ukr) показывает, во
сколько раз увеличиваются шансы нахождения соединения с заданным
свойством в сравнении с методом простого перебора.
Проиллюстрируем этот метод следующим примером [1,2].
Предположим, что структура соединений различается только
заместителями R,, Rj, R3h R4. При этом соединения имеют различную
активность I (табл. 18.1).
•Здесь указан так называемый терапевтический индекс рп ~~
отношение токсичности к эффективной дозе. Величина I
характеризует эффективность данного препарата. Основным структурным
признаком в этой задаче для молекулы у. можно считать вектор S
содержащий информацию о заместителях: й
Так, для молекулы № 1 вектором-определителем является
s1=(H,ci,CHaa,Br)>
438
Таблица 18,1
Терапевтический индекс для (I) для различных замещенных
№
1
2
3
4
5
6
7
8
R,
н
a
н
С1
н
С1
н
a
R2
Cl
Br
QH5
Br
Br
QH5
Ci
QH5
R3
CH2C1
H
CH2C1
Cl
Cl
H
Cl
CH2C1
R4
Br
C2H4C1
C2H4C1
N02
N02
Br
N02
C2H4C1
LDn
6000
2500
4500
1500
1500
1200
80
150
ED50
300
100
320
100
800
450
50
40
I
20
25
14.06
15
1.88
2.67
1.6
3.75
а для молекулы № 2 —
S2S(Cl,Br,H,C2H4Cl).
Разделим весь набор по признаку I на два класса — активные
молекулы (№1—№4, 14.06 < I < 25 ) и неактивные (№5 —№8,
1.6 < I < 3.75 ).
Далее проводим отбор признаков специфичных для данного
класса. Например, векторы S, и S3 характеризуют структуры активных
молекул, а их пересечение Sj f|S3 может служить ключевым
признаком для классификации молекул:
1 вектор, Sr* (H, Cl, CH2C1, Br)
2 вектор, S3: (H, С6Н5, СН2С1, С2Н4С1)
Пересечение векторов, Sj П S3: (Н, 0, СН2С1, 0).
Здесь 0 — символ пустого множества. Признак S]f|S3
соответствует условию (R,=H)&(R3=CH2C1), где & — логическое «И». Таким
образом, сопоставляя структуры молекул №1 и №3, можно
предположить, что если Rj=H и R3=CH2C1, то молекула будет обладать данным
видом активности. Проводя последовательное попарное сравнение
структурных векторов S, получаем набор признаков биологической
активности и ее отсутствия (табл. 18.2).
Таблица 18.2
Признаки активности
Номер
признака
г
1
2
3
4
5
— ..._
Признак
f(S)
(R]=H)&(R3=CH2C1)
(R4=C2H4C1)
(R,=Cl)&(R2=Br)
(R,=H)&(R4=N02)
(R,=C1)&(R2=C6H5)
L,r
2
2
2
0
0
Ur
0
1
0
2
2
p(M0
0.75
0.60
0.75
0.25
0.25
р(мо
0.25
0.40
0.25
0.75
0.75
439
В таблице 18.2 кроме признаков приведены также числа их
появлений в классе активных молекул Llr и неактивных молекул Ц.
Соответствующие вероятности связи данного структурного
признака с активностью (?(\ I f)) или неактивностью (Р(А2 | f)) дают
оценку успешности классификации. Так, согласно этим данным,
наличие признаков 1 или 3 с вероятностью 0.75 позволяют отнести
неизвестную молекулу к классу активных систем. Признаки 4 и 5
с той же вероятностью позволяют отнести систему к неактивным
молекулам.
Используя таблицу признаков, можно оценить активности новых
соединений, которые не вошли в исходный обучающий набор,
описывающий терапевтический эффект (табл. 18.3).
Таблица 18.3
Оценка активности соединений не вошедших
в обучающую выборку
№
9
10
11
12
Ri
CI
CI
CI
н
R;
Br
QH5
CI
H
R3
Br
H
H
H
R4
Br
C2H4CI
H
C2H4C1
Активность
Активное (признак 3)
Неактивное (признак 5)
Отказ от прогноза - нет характерного
признака
Активное с малой вероятностью
(признак 2)
Очевидно, что соединения № 9 и № 10 уверенно
классифицируются как соответственно активное и неактивное.
Соединение № 11 на основании пяти перечисленных признаков не может
быть классифицировано, поскольку отсутствуют характерные
сочетания заместителей, позволяющие интерпретировать данную
систему как активную или не активную. Соединение № 12, с
относительно малой вероятностью (0.60), также может быть
отнесено к активным.
Очевидно, что эффективность описанного выше метода
существенно зависит от представительности обучающей выборки.
Например, для набора химических соединений, обладающих данным
видом активности, вообще может не оказаться похожих
структурных фрагментов. Тем не менее практическое применение такой
методики во многих случаях оказывается полезным. Некоторые из
полученных данных (Розенблит, Голиндер, Пирузян, [1—3])
приведены в табл. 18.4.
Величина Р(А( | f ) характеризует вероятность появления
указанного вида активности, если в молекуле присутствует данный
фрагмент (признак). Прогностическая ценность признака Ukr, как уже
отмечалось выще, характеризует эффективность его использования в
качестве структурно-химического критерия при разработке новых ЛС.
Конечно же, эти данные не следует интерпретировать таким образом,
что введение скажем группы СООН в некую базовую молекулу,
приведет к тому, что новое соединение непременно окажется эффек-
440
Таблица 18.4
Связь некоторых химических дескрипторов молекул
с активностью
Вид активности
Антибактериальная
Противоглистная
Противогрибковая
Противовоспалительная
Гипогликемическая
Гипотензивная
Симпатолитики
Антидепрессантная
П ротивосуд оржная
Спазмолитическая
Ингибиторы
холинестеразы
Антидепрессантная
Спазмолитическая
Р(М0
N02
0.34
0.06
0.11
СООН
0.15
NH2
NH
0.09
0.25
0.06
О
О С NRiRs
0,11
0,11
0.14
0.09
0- -о
2.46
О
5.69
uta
3.8
2.9
3.0
2.7
5.9
4.7
13.3
3.8
3.4
2.6
9.5
4.73
6.60
Р f Aj I f.) — вероятность проявления данного вида активности в
обучающей выборке, U^— прогностическая ценность дескриптора.
тивным противовоспалительным средством. Однако такие данные
позволяют ориентироваться (с некой долей вероятности) в
возможных видах активности вновь синтезированных структур.
441
Таблица 18.5
Примеры прогнозов связи структура-активность
Тип соединения
Бензотиадиазины
Пропиниламины
Сульфонамиды
Карбаноилпиперидины
Кетоны
Эфиры
Дофаминовые эфиры
Стероиды
Фенантрены
Сульфонамиды
Трифторметансульфонамиды
Фенилацетамиды
Пиридилметаны
Проназины
Ароматические и алифатические
кислоты
Хелатные комплексы с медью
Сульфоны
Вид активности
Гипотензивное действие
Ингибирование моноаминокеидазы
Ингибирование карбоангидразы
Ингибирование холинестеразы
Ингибирование холинестеразы
Гидролиз папаина
Гидролиз ариламидаз
Связывание
с прогистероновым рецептором
Антимикопл азматиче екая
Антим икоплазматическая
Гербицидная
Гипнотическая
Спазмолитическая
Нейролептическая
Противовоспалительная
Цитотоксичность
Антибактериальная
В современной QSAR литературе имеются также данные о
биологической активности рядов соединений (табл. 18.5) [4]. Такая
информация полезна при проведении целенаправленного поиска
соединений с заданным видом активности.
Немаловажным достоинством таких методик является также тот
факт, что в руках исследователей появляется возможность в
простых структурно-химических терминах описать широкий спектр
активностей.
18.2. Кластерный анализ
Кластерный анализ связи биоактивности и структуры
основывается на геометрическом представлении молекул в многомерном
пространстве дескрипторов. Иными словами, набор из N дескрипторов
(х;, i = 1, К, N), с помощью которых однозначно характеризуется
молекула, можно интерпретировать как совокупность координат
некой условной точки. Таким образом, молекула — это точка в
пространстве N измерений (дескрипторов). В качестве меры сходства (или
близости, подобия) между разными молекулами можно принять
расстояние R между соответствующими точками. Однако, поскольку дес-
крипторное пространство не является реальным, физическим, то та-
442
ковой метрикой могут служить самые разные функции,
удовлетворяющие аксиомам расстояния. Эти аксиомы для любых трех объектов
(молекул) Х;, X, Хк формулируются следующим образом:
1. R(X(, Х-)'> d для всех Х;, Х.} из данного набора молекул.
2. R(X;, Xj) = 0 тогда и только тогда, когда Х;= Xj т.е. когда речь идет об
одной и той же молекуле (неразличимость идентичных объектов).
3. R(X, X) = R(X, Xs) (симметрия).
4. R(x!, Xj) < R(X|, X^) + R(Xk, X) (неравенство треугольника).
Несколько наиболее популярных типов расстояний
удовлетворяющих указанным аксиомам, приведены в табл. 18.6.
Естественно предположить, что ряд молекул-точек, обладающих
близкими характеристиками (например, активные в отношении
данного свойства), будут находится на достаточно близком друг от друга
расстоянии. Неактивные же молекулы будут образовывать отдельную
группу. Такие группы принято называть кластерами^ а
соответствующий анализ данных, позволяющий разбивать совокупность объектов
на отдельные кластеры, кластерным анализом [5].
Метрические характеристики близости объектов
(расстояния)
Таблица 18.6
Название
Эвклидово расстояние
Линейное расстояние
Обобщенное расстояние
Минковского
Формула для вычисления
расстояния
R,(X1)Xi) =
Jtk-*J
R,(X1,xj) = x|xH-x1
R (X..X.)
Jt(x--xJ"
Таким образом, главная цель кластерного анализа — нахождение
групп (кластеров) схожих объектов. Распространенным способом
представления результатов кластерных методов является дендрограмма
(древовидная диаграмма), которая графически изображает
иерархическую структуру, порожденную объединением объектов в кластеры.
На рис. 18.1 показан пример иерархической дендрограммы
отображающей разбиение трех объектов (молекул) на два кластера. Структуры
А и В относятся к одному кластеру, тогда как структура С к другому.
А В С
Рис. 18.1. Пример иерархической дендрограммы
443
Различные варианты кластерного анализа программно
реализованы в множестве статистических пакетов прикладных программ,
например в STATIST! CA.
18.3. Дискриминантный анализ
Целью дискриминантного (или дискриминационного) анализа [5,
6] является вычисление функции (D), позволяющей
классифицировать системы по заранее заданным группам. Чаще всего
встречается необходимость классификации молекул на две группы
(активные — неактивные). При этом функция выбирается так, чтобы
ее значения максимально различались для типичных
представителей разных групп (активных — неактивных молекул). Наиболее
распространенный, линейный вариант дискриминантной
функции, строится как суперпозиция молекулярных дескрипторов (хр
Xj, х3,...), и значение D-функции для них выглядит
следующим образом:
D = k0+xI-k1+x2-k2+...
Искомые коэффициенты разложения D: Ц,, к,, 1^, К (не путайте
их с коэффициентами линейного регрессионного анализа!) можно
вычислить, максимизировав отношение:
max h(k) = -^— •
У W.k. k. ■
ft 1J ' J .
Величина k(k) построена с помощью так называемых
ковариационных матриц (Т и W), описывающих дисперсии. При этом
числитель характеризует разброс молекулярных параметров (дескрипторов)
между группами активности, а знаменатель — разброс параметров
внутри каждой из групп.
С геометрической точки зрения дискриминантный метод
близок к основам кластерного анализа. Каждая молекула, как и в
кластерном анализе, представляется в виде точки в условном
пространстве дескрипторов х. Тогда, при удачном выборе дескрип-
торного набора, молекулы, относящиеся к данной группе
активности, будут расположены достаточно «близко» друг к другу
(образуют кластер). Вычислив значение D-функции для молекулы с
неизвестной активностью, можно выяснить, к какому кластеру она
относится и, следовательно, оценить ее активность. Схематически
группы активных и неактивных молекул представлены на
рисунке (рис. 18.2).
Линия, разделяющая кластеры, соответствует значению функции
D = 0 . Из рисунка следует, что молекулу, помеченную символом z,
следует отнести к группе активных.
444
X, Active
D=0
щ • • . ^
Inactive
Рис. 18.2. Разделение молекул на активные и неактивные
в пространстве дескрипторов (х). Символом Z помечена молекула
с неизвестной активностью, жирная точка — центроид
Обычно D-функцию калибруют таким образом, чтобы ее
значения имели разный знак для активных и неактивных молекул.
Так, в качестве критерия активности удобно использовать
неравенство:
D(x1,x2,...)>0,
соответствующий критерий отсутствия активности имеет вид:
D(x,,x2,...)<0.
Таким образом, уравнение вида
D(x,,x2,...) = 0
определяет условную «плоскость», которая разделяет молекулы,
относящиеся к различным классам активности.
Обобщение линейной дискриминантной функции естественным
образом приводит к нелинейной зависимости вида:
Уравнение
D(x,,x2,...) = 0
в этом случае определяет более сложную поверхность в многомерном
пространстве дескрипторов.
Таким образом, основная задача дискриминантного анализа
состоит в нахождении наилучшего разделения молекул на группы
активности в том смысле, что различия между группами должны быть
максимальны, а внутри групп — минимальны.
Важным понятием дискриминантного анализа является понятие
центроида. Под центроидом понимают точку дескрипторного
пространства, имеющую значения дескрипторов, которые вычисляют как
средние по группе активности. Таким образом, в случае разбиения
молекул на два класса активности (активные или неактивные) можно
вычислить координаты двух центроидов. Каждую из них можно рас-
445
сматривать как наиболее типичную, чаще всего гипотетическую,
молекулу из данного класса активности. Значение дискриминантной
функции для центроида, в свою очередь, может служить опорным
при интерпретации расчетов D-функций реальных систем. На рис. 18.2
центроид схематически показан в виде жирной точки.
Качество дискриминации по группе активности а обычно
оценивается по отношению
где п — число молекул, верно отнесенных с помощью D-функции к
заданной группе, а па — суммарное число молекул в группе
активности. Для более уверенной интерпретации прогностической
способности дискриминантной функции, также как и в регрессионном
анализе, можно использовать метод скользящего контроля. В простейшем
варианте {leave-one-out cross validation, LOO) из обучающей выборки
поочередно удаляются молекулы, вычисляется дискриминантная
функция и делается прогноз активности для удаленной молекулы. Затем
на основе этих прогнозов вычисляется процент верно
классифицированных систем.
Целесообразность применения дискриминантного анализа в
проблеме прогноза биологической активности связана с тем, что в целом
ряде случаев экспериментальные данные носят чисто декларативный
характер. То есть в результате эксперимента регистрируется лишь факт
проявления активности либо ее отсутствие. В этих условиях
применение регрессионного анализа практически невозможно, поскольку
отсутствует количественная мера биологического действия
химического соединения.
Один из примеров возможного использования дискриминантного
анализа связан с исследованием канцерогенной активности. Канцероген-
ность — это свойство, характеризующее способность веществ вызывать
злокачественную опухоль в организме. Известно, что многие
химические соединения проявляют более или менее значительное
канцерогенное действие. В этом разделе мы проанализируем выраженную
канцерогенную активность конденсированных (полициклических)
углеводородов. Речь идет об ароматических углеводородах с четырьмя,
пятью или шестью аннеллированными (сочлененными) кольцами. Как
известно, многоядерные ароматические углеводороды могут быть ан-
неллированы линейно или ангулярно. В первом случае центры колец
лежат на одной прямой, а при ангулярном аннеллировании центры
бензольных колец могут быть соединены несколькими
пересекающимися прямыми. Типичным примером линейно аннеллированного
ароматического углеводорода является антрацен:
446
Изомер антрацена — фенантрен — пример ангулярно
аннеллированного конденсированного углеводорода
9 10
Многие из ангулярно сочлененных полициклических
углеводородов обладают значительной канцерогенностью. Среди них молекулы,
содержащие фрагмент пирена:
Примечательно, что сам пирен не обладает значительным
канцерогенным действием, но бенз[3,4]пирен — сильнейший
канцероген, а его изомер — бенз[1,2]пирен также не является
канцерогеном (рис. 18.3).
Рис. 18.3. А — бенз[3,4]пирен, В — бенз[1,2]пирен
Согласно концепции Пюльмана [7] канцерогенная активность
подобных систем зависит от электронных свойств и реакционной
способности так называемых К- и L- областей молекулы (рис. 18.4).
К-область соответствует связи 9—10 фенантрена, в то время как
L-область соответствует атомам 9—10 антрацена. Важность такого
разделения связана с тем, что в результате взаимодействия клеточного
рецептора (ферменты семейства СУР1А1) с К-областью образуется
ряд высокореакционных электрофильных соединений, нарушающих
447
структуру НК и белков, что приводит в конечном счете к проявлению
канцерогенного эффекта (см. также разд. 2.5.1 и 8.1.1).
Рис. 18.4. К- и L- области в молекуле бензантрацена
Конкурирующим фактором является взаимодействие ферментов
СУР1А1 с атомами рецептора с атомами L-области. В этом случае
образуются продукты окисления, которые легко выводятся из
организма и, не проявляют канцерогенного действия. Для описания
электронных свойств этих областей молекул предлагалось
множество квантовохимических индексов. Особенно эффективным
оказался индекс сверхделокализуемости [8], описанный нами ранее.
Этот параметр, как характеристика реакционной способности
соответствующих углеродных атомов, может быть использован в
качестве решающего дескриптора при описании канцерогенной
активности. В табл. 18.7 приведены максимальные значения электро-
Таблица 18.7
Суперделокализуемости К- и L- областей
и канцерогенная активность конденсированных углеводородов
№
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
Название
Трибензо[3,4;6,7;9,Ю]пирен
Трибензо[3,4;6,7;8,9]пирен
Дибензо[1,2;5,6]антрацен
Нафто[2,3;3,4]пирен
Дибензо[3,4;6,7]пирен
Дибензо[3,4;8,9]пирен
Бензо[3,4]пирен
Дибензо[ 1,2;3,4]пирен
Дибензо[3,4;9,Ю]пирен
Бензол
Нафталин
Антрацен
Тетрабензонафтацен
Бензо[ 1,2]нафтоцен
Дибензонафтоцен
Se(K)
2.003
2.I4I
2.061
2.196
2.033
2.175
2.140
2.114
2.206
1.667
1.867
1.995
1.815
2.139
2.028
S.(L)
1.993
2.100
2.292
2.724
2.004
2.094
2.046
2.065
2.035
1.667
1.989
2.626
2.398
2.863
2.772
Активность
+
+
+
+
+
+
+
+
+
-
_
-
-
-
-
D
+0.902
+2.394
+0.558
+0.765
+ 1.273
+2.886
+2.587
+2.156
+3.538
-2.490
-0.957
-1.629
-3.237
-0.551
-1.733
В последнем столбце указано значение дискриминантной функции.
448
фильных сверхделокализуемостей К- и L- областей некоторых
углеводородов вместе с экспериментальными данными о наличии (+)
или отсутствии (—) значительной канцерогенной активности.
Построенная дискриминантная функция, с помощью которой можно
разделить полициклические углеводороды на классы канцерогенов
и неканцерогенов, имеет следующий вид :
D = -19.07 + 13.78 Sc (К) - 3.82 Sc (L).
Положительное значение D>0 соответствует выраженной
канцерогенной активности углеводорода. Как очевидно из таблицы, знак
дискриминационных функций D правильно соотносятся с
активностью указанных соединений, что говорит о том, что функция обладает
значительной предсказательной силой.
Вычисленные значения D-функции для центроидов также могут
служить ориентиром для отнесения молекулы углеводорода к классам
активности. В данном примере эти величины равны
D(+) = +1.896
D(-) =-1.896,
здесь D(+) и D(-) соответствуют центроидам активных и неактивных
молекул соответственно.
Второй пример использования дискриминантных функций связан
с исследованием биологической активности замещенных эстрадио-
лов, которые относятся к классу стероидных гормонов (рис. 18.5).
Рис. 18.5. Производные эстрадиола
Биологическая активность этих соединений и их синтетических
аналогов, в том числе и нестероидного строения многие годы
находятся в центре внимания биохимиков и фармакологов. Этот интерес
стимулирует многочисленные попытки осмысления связи структуры
молекул с их активностью. Среди множества возможных подходов
QSAR к отношении эстрогенов можно выделить
многопараметрические методы Фри-Вильсона и Хэнча, опирающиеся на регрессионный
анализ данных. Применение их к анализу относительного сродства к
рецептору (Relative Binding Affinity, RBA) позволило получить ряд
корреляционных уравнений [9, 10], которые связывают численное вы-
449
ражение активности некоторых соединений с молекулярными
параметрами (рефракцией, константами Гаммета и т.д.). Для
исследования этих структур применяются также современные довольно
дорогостоящие подходы типа метода сравнения молекулярных полей
(CoMFA) из группы 3D-QSAR [11].
Следует отметить, однако что экспериментальная оценка
сродства лигандов к соответствующим рецепторам часто проводится с
использованием различных тканей и органов лабораторных
животных (мышей, крыс и др.), при разных температурах (0°С, 25°С,
37°С). Это приводит к тому, что методы регрессионного анализа
(основанные на уравнениях Фри-Вильсона и Хэнча), так же как
и подход CoMFA и аналогичные ему, опирающиеся на
методологию неполного метода наименьших квадратов, не могут быть
корректно применимы к большим массивам разнородных
экспериментальных данных, поскольку такие методы предполагают
единую шкалу биоактивности для всех БАВ. Указанная проблема в
значительной степени «снимается» при использовании дискрими-
нантных функций. Ниже приведен результат дискриминантного
анализ связи RBA с дескрипторами молекулярной структуры для
синтетических аналогов эстрадиола [12]. Экспериментальные
данные о биоактивности, а именно lg(RBA), были собраны в ряде
работ [9, 10]. При построении дискриминантной функции
использованы данные о lg(RBA) для более чем 140 соединений. Часть этих
данных приведена в табл. 18.8. Они численно отражают активность
производных относительно незамещенной молекулы, для которой
величина Ig(RBA) равна 2. Предположим, что соединения с lg(RBA) > 1.5
являются активными, а соединения с Ig(RBA) < 1.5 — неактивными
(или слабоактивными). До вычислении дескрипторов для всех
соединений проведена оптимизация геометрии полуэмпирическим
методом РМЗ. И в рамках этого же метода рассчитан набор
электронных дескрипторов. По результатам факторного анализа
наиболее важными дескрипторами оказались энергия верхней
занятой орбитали (£взм0> эВ) и поляризуемость молекулы (а, А3). Кроме
того, с использованием аддитивной схемы Моригучи были
вычислены значения \gP для соединений входящих в обучающую
выборку.
Подчеркнем, что перечисленный набор дескрипторов
характеризует всю молекулу в целом, а не отдельные заместители, как это
принято, например, в подходе Хэнча. Данное обстоятельство позволяет
получить решающую функцию, являющуюся в некоторой степени
универсальной, поскольку она не относится к какой-либо базовой
химической структуре и ее можно использовать для оценок lg(RBA)
широкого класса соединений.
По результатам проведенного анализа дискриминантная функция
для классификации молекул по указанным видам активности:
D, =+1.032-1.49енвм0-0.089а+ 1.41 lgP-0.17(lgP)2.
450
Таблица 18.8
Относительное сродство связывания с рецептором
для замещенных эстрадиола
R1
Et
Н
Н
Et
Et
Et
Et
Et
H
H
H
Et
OMe
H
H
R2
H
H
H
H
OH
H
OH
H
H
H
H
OH
H
OH
OH
R3
H
C^CH
H
C^CH
Me
Me
C^CH
C^CMe
Me
C^CMe
C6HS
H
HC=CMe
H
log(RBA)
2,60
2,05
2,00
1,94
1,93
1,92
1,90
1,82
1,76
1,67
1,48
1,45
1,42
1,32
с^сн 1,30
Rl
H
Et
Ome
OMe
H
OMe
H
OMe
OMe
H
OMe
OMe
OMe
OMe
OMe
R2
OH
OH
H
H
OH
H
OH
H
H
OH
OH
OH
OH
OH
OH
R3
C^CH
C^CMe
C^CMe
QH5
Me
C^CH
C6H5
H
Me
C^CMe
QH5
Me
H
C^CH
log(RBA)
1,30
1,26
1,26
1,85
1,15
1,15
0,90
0,90
0,78
0,70
0,70
0,60
0,0
0,0
C^CMe -0,10
Качество прогноза составляет 87%. Значения функции для
центроидов активных Dj(+) н неактивных D/-) молекул соответственно равны:
D,(+) = +0.401,
D,(-) = -0.502.
Как и прежде, положительное значение D соответствует активным
молекулам, а отрицательное значение — малоактивным.
Определенный интерес может представлять также и максимально упрощенная
функция разделения, использующая в качестве дискриминантного
параметра лишь величину липофильности:
D2 = -4.386 + 2.307 lg Р - 0.261(Ig P)2,
качество прогноза для которой также относительно велико и
составляет 78%.
Литература
1. Розенблит А.Б., Голиндер В.Е. Л о гико-комбинаторные методы в
конструировании лекарств. — Рига: Зинатне. 1983.
2. Баренбойм Г.М., Маленков А.Г. Биологически активные вещества. Новые
принципы поиска. — М.: Наука, 1986.
3. Пирузян Л.А., Маленков А.Г., Баренбойм Г.М. // Вестник АН СССР. —
1977. -С. 50.
4. ПирузянЛА, Маленков А.Г., Баренбойм Г.М.// Изв. АН СССР, сер. Биол. —
1977. - С. 325.
451
5. Факторный, дискриминантный и кластерный анализ. — М.: Финансы
и статистика, 1989. — 213 с.
6. Статистические методы для ЭВМ / Под ред. К. Энслейна, Э. Релстона,
Г.С. Уилфа. - М.: Наука, 1986.
7. Пюльман Б, Пюльман А. Квантовая биохимия. — М.; Мир, 1965.
8. Местечкин М.М., Сивякова Л.Н. Применение индексов реакционной
способности как показателей канцерогенной активности полициклических
углеводородов: препринт Академия наук Украинской CCPS Институт
теоретической физики. Киев, 1979.
9. Coluci V.R., Vendrana Я, Braga R.S. at ai // J. Chem. Inf., Сотр., Sci. -
2002. - Vol. 42. - P. 1479.
10. Gao H., Katzenellenbogen J.A, Garg R., Hansch С // Chem. Rev. - 1999. -
Vol. 99. - P. 723.
11. Yu S.J., Keenan SM, Тощ W., Welsh WJ. // Chem. Res., Toxicol. - 2002,
Vol. 15. -P. 1229.
12. Иванов В.В., Слета Л.А., Толстая А.А. // Журнал оргашчно'1 та фармацев-
тично! xiMii. — 2004. — Т.2, вип. 4(8). - С. 66-68.
СЛОВАРЬ ТЕРМИНОВ
Агонист. Вещество, в результате взаимодействия которого со
специфическим рецептором индуцируется цепь внутриклеточных
биохимических процессов.
Активный транспорт. Перенос растворенного вещества против
градиента концентрации, требующий энергетических затрат.
Аллергия. Приобретенная повышенная чувствительность организма к
веществам (аллергенам), обусловленная иммунологическими
механизмами.
Аллостерические ферменты. Регуляторные ферменты, активность
которых меняется при связывании с ними субстрата не в
каталитическом центре, а в другом участке.
Анаболизм. Процесс ассимиляции питательных веществ и
превращения их в компоненты клеток, требующий затрат энергии.
Антагонист. Вещество, связывающееся с рецептором, но не
вызывающее стимуляции внутриклеточного ответа.
Анальгезия. Состояние сниженного болевого восприятия, не
сопровождающееся отключением сознания.
Анестезия. Отсутствие чувствительности, наступающее в результате
медикаментозного угнетения (или заболевания) рецепции,
проведения импульсов или функционирования нервных центров.
Анальгетик. Средство, устраняющее чувство боли.
Анестетик. Вещество, обратимо угнетающее нервную функцию, что
сопровождается потерей болевых и/или других ощущений.
Анксиолитик. Вещество, устраняющее чувство страха (транквилизатор).
Антиангинальные средства. ЛС, увеличивающие приток крови к
миокарду или снижающие его потребность в кислороде.
Антибиотики. Вещества, продуцируемые различными видами
микроорганизмов и растений в окружающую среду. Выполняют
защитную функцию от других видов.
Антиген. Чужеродная для данного организма молекула, вызывающая
синтез специфического антитела у позвоночных.
Антипиретик. Вещество, снижающее лихорадку.
Антисептик. Вещество, обладающее обеззараживающим действием.
453
Биодоступность. Количественная характеристика неизмененного
вещества, поступившего в плазму крови, по отношению к исходной
дозе, введенного в организм ЛВ.
Брадикннин. Нонапептид, оказывающий выраженный
сосудорасширяющий эффект. Участвует в реакции организма на проходящий
в нем воспалительный процесс.
Вазодилатация. Расширение кровеносных сосудов.
Вазоконстрикция. Сужение кровеносных сосудов.
Вазорелаксация. Снижение тонуса сосудов.
Вирион. Полностью сформированная вирусная частица.
Генетический алгоритм (эволюционный алгоритм)*. Оптимизационный
алгоритм, симулирующий механизмы дарвиновской
эволюционной теории (случайные мутации и отбор) для нахождения
наилучшей дескрипторной модели, описывающей фармакохимические
данные.
Генетический код. Набор триплетов в ДНК, кодирующих
аминокислоты белков.
Ген. Единица наследственности, занимающая специфическое место
(локус) в хромосоме, способна к самовоспроизведению в
клеточном цикле, содержит информацию о последовательности
аминокислот зашифрованную в виде последовательности нуклеотидов.
Геном. Полный набор хромосом. Геном человека состоит из 23 пар
хромосом.
Гем. Железопорфириновая простетическая группа гемопротеинов.
Гиперволемия. Аномально повышенный объем крови в организме.
Гипергликемия. Повышенное содержание глюкозы в крови.
Гистоны. Основные белки, связанные с хромосомами эукариотических
клеток.
Гомеостаз. Физиологический процесс поддержания постоянства
внутренней среды организма, при котором различные параметры
организма (давление крови, температура тела, кислотно-щелочной
баланс и т.п.) поддерживаются в равновесии, несмотря на
изменения условий окружающей среды.
Деменция. Слабоумие — стойкое оскудение и упрощение
психической деятельности, характеризующееся ослаблением
познавательных процессов, обеднением эмоций и нарушением поведения.
Дескриптор*. Параметр, характеризующий некий структурный,
геометрический или физико-химический аспект молекулярной
системы.
Дискриминантный анализ*. Статистический метод нахождения
функций дескрипторного набора, с помощью которых можно
провести разделение молекулярных систем по различным классам
биологической активности.
454
Интеркаляция. Встраивание соединений между парами оснований ДНК.
Иитерлейкин 1. Монокин и полл и пептидный гормон,
вырабатываемый моноцитами; при действии на гипоталамус вызывает
повышение температуры тела, повышает катаболизм белков при
прямом действии на скелетные мышцы (лимфоциты активирующий
фактор, эндогенный пироген).
Интерлейкин 2. Комплекс факторов роста и дифференцировки Т-лим-
фоцитов.
Интрон (межгенная последовательность). Участок ДНК между двумя
экзонами; транскрибируется, но исчезает после созревания
матричной РНК и не кодирует белок.
Искусственная нейронная сеть*. Компьютерный алгоритм
симулирующий функционирование нейронов мозга. ИНС используется в
проблеме QSAR для задач связанных с распознаванием
биологически активных молекулярных систем.
Ишемия. Локальная анемия за счет механической закупорки (в
основном сужение артерий) или отсутствия кровоснабжения.
Капсид. Белковая оболочка вирусной частицы.
Катаболизм. Фаза метаболизма, включающая деградацию молекул
питательных веществ и сопровождающаяся выделением энергии.
Кинины. Биологически активные полипептиды, образующиеся в плазме
крови при наличии в организме повреждения или очага
воспаления. Повышают проницаемость сосудов, и расширение просвета
в них, снижают артериальное давление, сокращают гладкую
мускулатуру.
Кластерный анализ*. Метод группировки (кластеризации)
химических или фармакологических данных. Целью кластерного анализа
является выявление подобных молекулярных систем.
Количественные соотношения структура — активность (Quantitative
Structure-Activity Relationships, QSAR). Математические
соотношения связывающее параметры химической структуры молекул с их
фармакологической активностью. Методы QSAR обычно
включают в себя различные регрессионные подходы и методы
распознавания образов.
Липндемия (гиперлипидемия). Аномально высокая концентрация ли-
пидов в крови.
Методы распознавания образов (Pattern Recognition, PR)*. Группа
статистических методов, позволяющих идентифицировать различные
типы (классы) соединений по степени выраженности их
биологического эффекта (активные-неактивные-слабоактивные).
Пироген. Вещество, вызывающее повышение температуры тела.
Прокариоты. Простые одноклеточные организмы, содержащие одну
хромосому, не имеющие ядерной мембраны и окруженных
мембранами органелл.
455
Пролиферация. Рост за счет размножения клеток.
Промотор. Участок ДНК, с которым связывается РНК-полимераза,
инициируя транскрипцию.
Простагландииы. Класс жирорастворимых гормоноподобных регуля-
торных молекул, являющихся производными арахидоновой
кислоты и др. полиненасыщенных жирных кислот.
Постнатальный. Происходящий после родов.
Регрессионный анализ*. Метод построения зависимостей (линейных
или нелинейных) между набором дескрипторов и
биоактивностью молекул. В проблеме QSAR широко используется метод
наименьших квадратов (МНК) и т.н. неполный метод наименьших
квадратов (Partial Least Squares, PLS).
Ретровирусы. РНК-содержащие вирусы, в состав которых входит РНК-
зависимая ДНК-полимераза (обратная транскриптаза).
Рецептор. Белковая молекула на поверхности клетки или в цитоплазме,
или участок ДНК в ядре клетки, связывающиеся со
специфическими веществами — агонистами и антагонистами.
Сенсибилизация. Повышенная чувствительность организма к
воздействию какого-либо фактора окружающей или внутренней среды.
Симптом. Любой болезненный феномен или отклонение от
нормальной функции, возникающий или ощущаемый пациентом и
указывающий на болезнь.
Синдром. Относящаяся к болезни совокупность симптомов
патологического процесса.
Синергизм. Усиление действия ЛС при их совместном применении по
сравнению с суммарным действием.
Сплайсинг. С. генов, связывание одной молекулы ДНК с другой.
С. РНК. устранение интронов из мРНК-предшественника.
Сравнительный анализ молекулярных полей (Comparative Molecular Field
Analysis, CoMFA)*. Статистический метод нахождения
корреляций между биологической активностью набора молекулярных
систем и их трехмерными свойствами (электронными, стерическими
и др.).
Тахифилаксия. Снижение фармакологического эффекта ЛВ при его
длительном или периодическом повторном применении.
Тератоген. Вещество или фактор, при воздействии которых возникает
аномальное развитие плода.
Толерантность. Способность переносить или становиться менее
чувствительным к стимулу, особенно во время продолжительного
контакта с ним. Способность противостоять действию ЛВ или
токсина в больших дозах без проявления его повреждающего
эффекта.
Тонический. Находящийся в состоянии продолжительного
непрекращающегося действия; термин используется в основном
применительно к мышечным сокращениям.
456
Ульцерогенный. Вызывающий язву.
Фагоцит. Клетка, способная поглощать бактерии, простейших,
другие клетки и их остатки. К фагоцитам относятся лейкоциты и
макрофаги.
Фертильиость. Способность приносить потомство.
Фетотоксичиость. Отравление плода, могущее привести к смерти,
задержке развития или роста.
Фрн-Внльсона метод*. Регрессионная техника, выявляющая
корреляции между биологической активностью и наличием (или
отсутствием) определенных структурных элементов в молекуле.
Хроматин. Нитевидный комплекс ДНК, гистонов и др. белков,
образующих хромосомы эукариот.
Хэнча (Hansen) метод*. Построение регрессионного соотношения
между биологической активностью набора соединений и их физико-
химическими дескрипторами.
Цитозоль. Клеточная цитоплазма без митохондрий и эндоплазма-
тического ретикулума.
Цитоплазма. Вещество клетки за исключением ядра; состоит из
коллоидной протоплазмы, содержащей различные органеллы и
включения.
Цитохромы. Гем-содержащие белки-переносчики электронов
придыхании и фотосинтезе.
Широта терапевтического действия (LD50/EDS0). Диапазон между
минимальной терапевтической и минимальной токсической дозами.
Энтеральные способы введения препаратов. Способы, при которых ЛС,
прежде чем попасть в системный кровоток, поступает в
желудочно-кишечный тракт.
Экзои. Кодирующий фрагмент молекулы ДНК, копия которого в виде
информационной РНК транслируется на рибосомах.
Эукариоты. Организмы, клетки которых обладают окруженным
мембраной ядром с множественными хромосомами и
внутриклеточными органеллами.
Ядрышко. Структурная единица ядра эукариотических клеток,
участвующая в синтезе рРНК и образовании рибосом.
* Термин встречается в главах 15—18.
СОДЕРЖАНИЕ
Предисловие 3
1. СТРОЕНИЕ КЛЕТКИ 5
1.1. Краткие сведения
о строении биологических мембран 10
1.1.1 Мембранные липиды 10
1.2. Перенос веществ через мембраны 15
2. СВЯЗЬ ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ
БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
С ИХ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ 18
2.1. Растворимость и липофильность 19
2.2. Поверхностно-активные, мембран-активные
фармакологические агенты и и он-проводящие антибиотики . . 31
2.3. Химическое связывание и биологическая активность 34
2.4. Связь между фармакологической активностью,
электронными свойствами и константами ионизации 38
2.5. Стере охи мические аспекты действия лекарств 39
2.5.1. Оптическая изомерия 40
2.5.2. Геометрическая изомерия 45
2.5.3. Биологическая активность конформеров 45
3. ВЗАИМОДЕЙСТВИЕ ЛЕКАРСТВО-РЕЦЕПТОР
(ОБЩИЕ СВЕДЕНИЯ) 49
3.1. Семейства рецепторов и их химическая природа 50
3.2. Кинетика взаимодействия лиганд—рецептор 56
3.3. Основные теории рецепции 64
4. СИСТЕМЫ ПЕРЕДАЧ РЕЦЕПТОРНОГО СИГНАЛА
И ВТОРИЧНЫЕ ПОСРЕДНИКИ 72
4.1. Аденилатциклазная система передачи сигнала 73
4.2. цГМФ как вторичный посредник 77
4.3. Продукты метаболизма фосфолипидов
как вторичные посредники 79
4.4. Ионы Са2+ как вторичные посредники 80
5. ФЕРМЕНТЫ (ПРИНЦИПЫ ДЕЙСТВИЯ И РЕГУЛЯЦИЯ
АКТИВНОСТИ). ИНГИБИТОРЫ ФЕРМЕНТОВ
В СОВРЕМЕННОМ АРСЕНАЛЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ . . 86
5.1. Классы ферментов 88
5.1.1. Оксидоредуктазы 88
5.1.2 Трансферазы 91
458
5.1.3. Гидролазы 95
5.1.4. Лиазы 96
5.1.5. Изомеразы 97
5.1.6. Лигазы (синтетазы) , 98
5.2. Коферменты 99
5.3. Принципы действия ферментов 102
5.4. Кинетика ферментативных реакций (общие сведения) 103
5.5. Регуляция ферментативной активности. Категории
ингибиторов ферментов 108
5.5.1. Необратимые ингибиторы ферментов 108
5.5.2. Обратимые ингибиторы ферментов 115
5.5.3. Регуляция активности мультиферментных систем .... 121
НУКЛЕИНОВЫЕ КИСЛОТЫ КАК МИШЕНИ
ДЛЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ 130
6.1. Строение нуклеиновых кислот 130
6.1.1. Структура ДНК 133
6.1.2. Структура РНК 134
6.2. Биосинтез нуклеиновых кислот (общие принципы) 137
6.3. Лекарственные средства, влияющие на синтез нуклеиновых
кислот и белков 142
6.3.1. Антибактериальные средства 142
6.3.2. Противоопухолевые средства 148
6.3.3. Противовирусные средства 154
ФАРМАКОКИНЕТИКА (ОСНОВНЫЕ ПОНЯТИЯ И МОДЕЛИ).
РОЛЬ ФАРМАКОКИНЕТИЧЕСКИХ ИССЛЕДОВАНИЙ
В СОЗДАНИИ НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ 166
7.1. Основные понятия фармакокинетики 166
7.2. Физико-химические свойства лекарственных веществ
и их фармакокинетика 167
7.3. Фармакокинетические модели 168
7.4. Абсорбция 169
7.4.1. Количественные характеристики
процесса абсорбции 171
7.5. Распределение лекарственных веществ 176
7.6. Выведение лекарственных веществ 177
7.6.1. Количественные характеристики процесса выведения
лекарственных веществ 179
7.7. Фармакокинетика некоторых групп лекарственных средств . . 183
МЕТАБОЛИЗМ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
(ОБЩИЕ СВЕДЕНИЯ) 196
8.1. Метаболические пути и места метаболических превращений
ксенобиотиков 196
8.1.1. Реакции I фазы метаболизма, катализируемые CYP450 . 199
8.1.2. Реакции I фазы метаболизма, катализируемые
FAD-содержащим и ферментами 204
8.1.3. Восстановительные процессы 208
8.1.4. Гидролиз 208
8.1.5. Реакции II фазы метаболизма ксенобиотиков 208
8.2. Связь структуры и степени метаболизма лекарственных
веществ 214
459
8.3. Факторы, влияющие на метаболизм лекарственных веществ . 215
8.4. Методы изучения метаболизма 219
8.5. Роль сведений о метаболизме в конструировании
лекарственных средств 220
9. ВЫБОР СТРАТЕГИИ ИССЛЕДОВАНИЙ ПРИ СОЗДАНИИ
НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
И КРИТЕРИИ ОЦЕНКИ КАЧЕСТВА СТРУКТУРЫ-ЛИДЕРА ... 222
10. ИСТОЧНИКИ ПОИСКА НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ .. 239
10.1. Природное сырье как источник новых
лекарственных средств 239
10.2. Официнальные лекарственные средства как источник
создания новых препаратов 239
10.3. Физиологические посредники как источник новых
лекарственных средств 256
11. СОВРЕМЕННЫЕ МЕТОДЫ УСОВЕРШЕНСТВОВАНИЯ
СТРУКТУРЫ ЛИДЕРА 271
11.1. Биоизостерические перемещения 272
11.2. Кон форм ац ионные ограничения 277
11.3. Пролекарства 294
12. КОМБИНАТОРНЫЙ СИНТЕЗ
И ЕГО РОЛЬ В ПОИСКЕ СТРУКТУР-ЛИДЕРОВ 310
12.1. Стратегия конструирования и синтеза химических библиотек .. 314
12.2. Комбинаторный синтез в растворах 319
12.2.1. Одностадийные методы синтеза 319
12.2.2. Двухстадийные методы синтеза 320
12.2.3. Трехстадииные методы синтеза 321
12.2.4. Однореакторные методы синтеза (one-pot synthesis) ... 321
12.2.5. Тандемные реакции 322
12.2.6. Параллельный синтез индивидуальных соединений . . . 324
12.2.7. Классические жидкофазные реакции 325
12.2.8. Способы очистки реакционных растворов от примесей . 326
12.2.9. Синтез с использованием растворимого полимера .... 328
12.3. Комбинаторный синтез на твердом носителе 331
12.3.1. Линкеры 333
12.3.2. Синтез гетероциклов на полимерном носителе 337
12.4. Методы установления структуры индивидуальных
компонентов библиотек 345
12.4.1. Индексные библиотеки 346
12.4.2. Итерационная процедура распознавания веществ
(Deconvolution) 346
12.4.3. Позиционное сканирование 348
12.5. Перспективы дальнейшего развития
комбинаторного синтеза 350
13. БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ НОВЫХ СОЕДИНЕНИЙ . .-. 354
13.1. Исследования in vitro 356
13.2. Тотальный скрининг 361
13.3. Скрининг комбинаторных библиотек 362
13.4. Исследования на лабораторных животных 365
460
14. ЭТАПЫ СОЗДАНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ 369
14.1. Химические разработки 371
14.2. Разработка лекарственной формы 374
14.3. Фармакологические испытания 376
14.4. Клинические испытания 380
15. КОЛИЧЕСТВЕННЫЕ СООТНОШЕНИЯ СТРУКТУРА -
АКТИВНОСТЬ. КРАТКИЙ ИСТОРИЧЕСКИЙ ОБЗОР,
ОСНОВНЫЕ МЕТОДОЛОГИЧЕСКИЕ ПОНЯТИЯ 384
16. ДЕСКРИПТОРЫ МОЛЕКУЛЯРНОЙ СТРУКТУРЫ 388
16.1. Общая классификация дескрипторов 388
16.2. Электронные дескрипторы 389
16.3. Топологические дескрипторы 394
16.4. Физико-химические дескрипторы 403
16.5 Химические дескрипторы. Фармакофоры 408
16.6 Индикаторные дескрипторы 411
17. РЕГРЕССИОННЫЕ МОДЕЛИ БИОЛОГИЧЕСКОЙ
АКТИВНОСТИ ОРГАНИЧЕСКИХ МОЛЕКУЛ 414
17.1. Регрессионный анализ 414
17.2 Факторные модели в регрессионном анализе 418
17.3 Эмпирические константы заместителей.
Уравнения Гаммета и Тафта 421
17.4 Аддитивная модель Фри—Вильсона 422
17.5. Метод Хэнча. Значение липофильности 426
17.6 Метод сравнительного анализа молекулярных полей 432
18. СТАТИСТИЧЕСКИЕ МЕТОДЫ КЛАССИФИКАЦИИ
МОЛЕКУЛ ПО ИХ БИОЛОГИЧЕСКОЙ АКТИВНОСТИ 436
18.1. Логико-комбинаторный подход 436
18.2. Кластерный анализ 442
18.3 Дискриминантный анализ 444
Словарь терминов 453
Учебное издание
ОРЛОВ Валерий Дмитриевич
ЛИПСОН Виктория Викторовна
ИВАНОВ Владимир Венедиктович
МЕДИЦИНСКАЯ ХИМИЯ
Главный редактор Н. Е. Фомина
Ответственный за выпуск Л. В. Дмитриева
Художественный редактор Б. Ф. Бублик
Технический редактор А. С. Таран
Компьютерная верстка: Е. В. Подлесная
Корректор А. Я. Гопаченко
Подписано в печать 27.06.05. Формат 60x90 '/,6.
Бумага офсетная. Гарнитура Тип Тайме. Печать офсетиая.
Усл. печ. л. 29,0, Усл. кр.-отт. 29,5. Уч.-изд. л. 31,4.
Тираж 2 000 экз. Заказ № 5-\0.
ТОВ «Видавиицтво Фолю»
Свщоцтво про внесения суб'екта видавиичоУ справи
до Державного реестру видавщв, вигопвниюв
i розповсюджувач1в видавиичо'1 продукщ!
ДК№ 502 вш 21.06.2001 р.
ТОВ «Фолю»
Свщоцтво про внесения суб'екта видавиичоТ справи
до Державного реестру видавщв, вигот!Вник1в
i розповсюджувач1в видавиичоТ продукт!
ДК№ 683 вщ 21.11.2001 р.
6Ю57, XapKiB, вул. Доиець-Захаржевського, 6/8
Електроина адреса:
www.folio.com.ua
E-mail: realization@folio.com.ua
1нтернет магазин
www.bookpost .com .ua
Надруковано з готових позитивов
у ТОВ «ФОЛЮ-АРТ*
61143, м. XapKiB, вул. Велика кшьцева, 99
Свщоитво про реестрашю
ДК№2271 вщ 26.08.2005 р.
Орлов В. Д., Липсон В. В., Иванов В. В.
0-66 Медицинская химия / Худож.-оформитель А. С. Ленчик. —
Харьков: Фолио, 2005. — 461 с.
ISBN 966-03-3119-3.
В учебнике кратко изложены основные понятия клеточной,
молекулярной биологии и биохимии (строение клетки и биологических мембран,
функции ферментов, нуклеиновых кислот, вторичных посредников),
необходимые химикам для рассмотрения вопросов общей фармакологии (фар-
макодинамики, фармакокинетики, метаболизма лекарственных средств).
Кроме того, в него включены разделы, посвященные проблеме связи
физико-химических свойств биологически активных соединений с их
фармакологическим действием и методов количественной оценки
«структура—активность».
В книге проведен анализ современных подходов в конструировании
лекарственных веществ и в оценке их специфических эффектов, таких как
комбинаторный синтез и тотальный скрининг, дана характеристика этапов
разработки новых лекарственных средств.
Учебник содержит обширный иллюстративный материал, включающий
химические формулы всех обсуждаемых в тексте лекарственных вешеств, а
также таблицы с данными о физико-химических, фармакологических
свойствах и фармакокинетических параметрах многих препаратов.
Книга предназначена для исследователей, работающих в области
медицинской химии, студентов и аспирантов-химиков, фармацевтов,
специализирующихся в области тонкого органического синтеза и создания новых
лекарств.
ББК 52.81