/














































































































































































































































































































































































































































































































































































































































































































Автор: Попова Т.Н. Артюхов В.Г. Жеребцов Н.А.
Теги: науки о земле геологические науки химия биология биохимия
ISBN: 5-7455-1183-4
Год: 2002



Текст
Н.А. Жеребцов, Т.Н. Попова,
В. Г. Артюхов
И
Допущено Министерством образования
Российской Федерации в качестве учебника
для студентов высших учебных заведений,
обучающихся по направлениям и специальностям
медико-биологического профиля
Издательство
■
Воронежского государственного
университета
2002
УДК 557.1
1
Жеребцов Н. А., Попова Т. Н., Артюхов В. Г. Биохимия: Учебник. —
Воронеж: Издательство Воронежского государственного университета,
2002. — 696 с.
18ВЫ 5-7455-1183-4
Учебник охватывает основные разделы биохимии: строение и
свойства биологических макромолекул, биоэнергетика и метаболизм клетки,
хранение, передача и реализация генетической информации. Большое
внимание уделено ряду прикладных аспектов биохимии: экологическим,
медицинским, фармацевтическим, а также некоторым вопросам
хранения и переработки сельскохозяйственного сырья.
Книга предназначена для студентов, аспирантов и преподавателей,
специализирующихся в области биохимии, молекулярной биологии,
медицины, фармации, технологии пищевых продуктов.
Рецензенты :
доктор биологических наук, академик РАН В. П. Скулачев
(профессор кафедры биохимии Московского государственного
университета),
доктор медицинских наук, профессор В. В. Алабовский
(заведующий кафедрой биохимии Воронежской государственной
медицинской академии)
Жеребцов Н. А., Попова Т. Н.,
Артюхов В. Г., 2002
18ВЫ 5-7455-1183-4 © Издательство Воронежского
государственного университета, 2002
ОГЛАВЛЕНИЕ
Предисловие 9
Введение 11
Часть I. Биомолекулы: структура, свойства, функции 17
Глава 1. Белки 19
1.1. Элементарный состав белков 19
1.2. Аминокислоты — строительные блоки белковой молекулы 20
1.3. Модифицированные аминокислоты 22
1.4. Кислотно-основные и электрохимические свойства
аминокислот 24
1.5. Специфические химические реакции аминокислот 26
1.6. Классификация аминокислот 29
1.7. Полипептидная теория химического строения белков 32
1.8. Биологические функции белков 35
1.9. Роль белков в питании 36
1.10. Физико-химические свойства белков 38
1.11. Форма белковых молекул 43
1.12. Способы свертывания и ассоциации полипептидных цепей 44
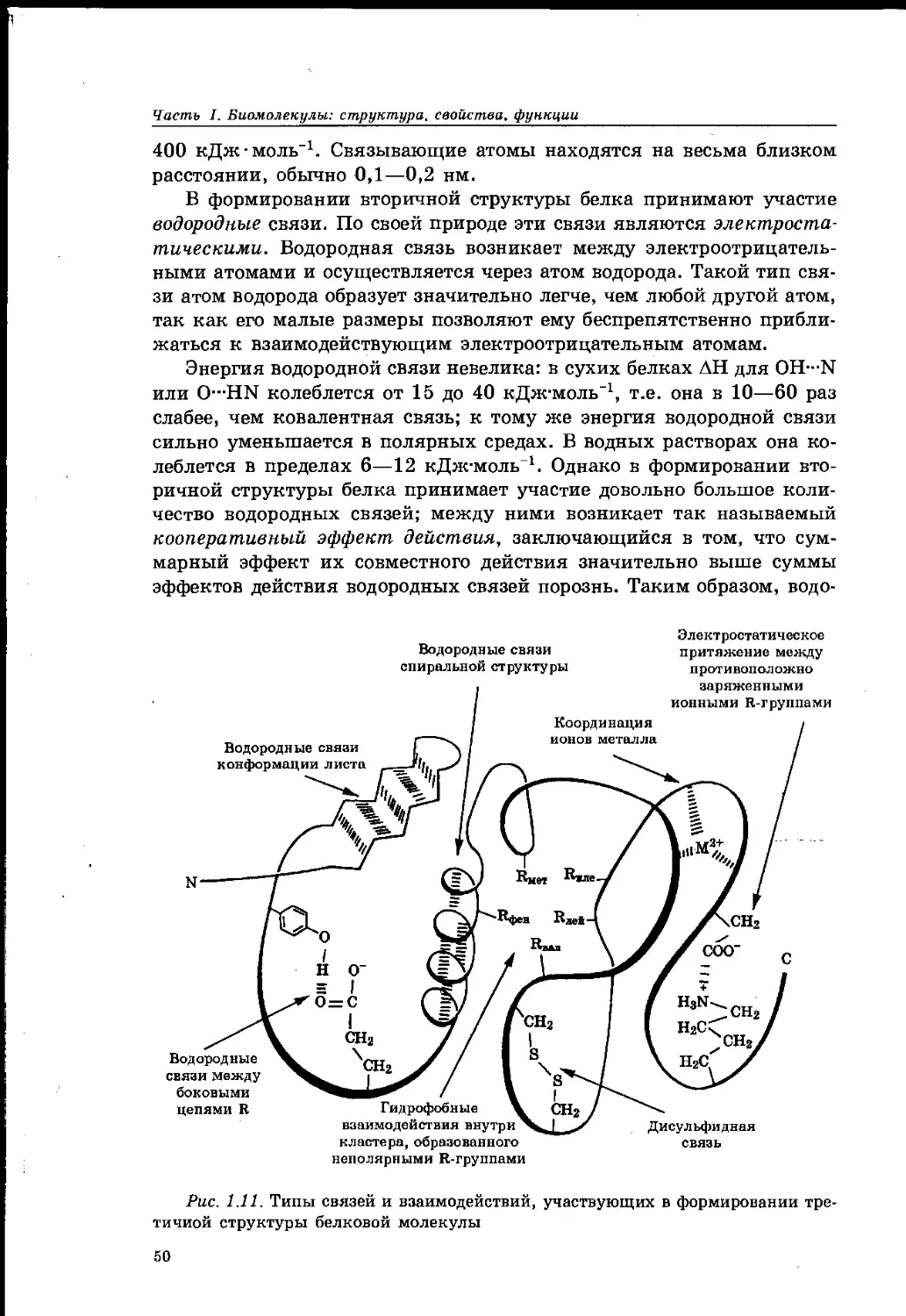
1.13. Природа и типы связей, участвующих в формировании
структуры белка 49
у
1.14. Электрохимические свойства белков 53
1.15. Гидрофильность белков 55
1.16. Осаждение белков 57
1.17. Денатурация белков 58
1.18. Исследование структуры белка. Цель, подходы и методы 60
1.19. Классификация белков 68
Краткое содержание главы 72
Хронология важнейших дат в исследовании белков 74
Литература 74
Вопросы и задачи 75
Глава 2. Нуклеиновые кислоты 76
2.1. Общая характеристика нуклеиновых кислот 76
2.2. Структура нуклеотидов 78
2.3. Первичная структура полинуклеотйдов 82
2.4. Вторичная и третичная структуры ДНК ♦ 83
2.5. Физико-химические свойства ДНК... 88
2.6. Биологические функции ДНК 89
2.7. Процессы метилирования ДНК 90
2.8. Структура и физико-химические свойства РНК 92
2.9. Типы РНК и их биологические функции 93
2.10. Комплексы нуклеиновых кислот и белков 96
Краткое содержание главы 101
Хронология важнейших дат в исследовании нуклеиновых
кислот 102
Литература 103
Вопросы и задачи..' 103
Глава 3 . Углеводы 104
3.1. Общая характеристика углеводов. Классификация 104
3.2. Моносахариды 105
3.3. Олигосахариды 114
3.4. Полисахариды 119
3.5. Гликопротеины 132
3.6. Подходы, приемы и методы в исследовании углеводов 132
3.7. Углеводы в пищевых продуктах 135
Краткое содержание главы 137
Хронология важнейших дат в исследовании структуры
углеводов 140
Литература 140
Вопросы и задачи 140
Глава 4. Липиды 142
4.1. Жирные кислоты 142
4.2. Простые липиды 146
4.3. Липиды — основные компоненты биологических мембран 150
4.4. Биологические мембраны 160
4.5. Методы исследования липидов 171
4.6. Липиды и их роль в питании человека 174
Краткое содержание главы , 177
Хронология важнейших дат в исследовании липидов 180
Литература 180
Вопросы и задачи 180
Глава 5. Витамины 181
5.1. Общая характеристика. Классификация и номенклатура 181
5.2. Жирорастворимые витамины. 183
5.3. Водорастворимые витамины 187
5.4. Витаминоподобные вещества 197
5.5. Количественное определение витаминов , 199
5.6. Витаминизация пищевых продуктов 201
Краткое содержание главы 203
Хронология важнейших дат в исследовании витаминов 205
Литература , 206
Вопросы и задачи ., 206
Глава 6. Вещества вторичного метаболизма растений 207
6.1. Терпены 207
6.2. Фенолы 212
6.3. Алкалоиды 220
6.4. Гликозиды 222
6.5. Фитогормоны 225
Краткое содержание главы 229
Хронология важнейших дат в исследовании веществ вторичного
происхождения 231
Литература 231
Вопросы 232
Часть II. Метаболизм веществ и энергии в клетке 233
Глава 7. Принципы биоэнергетики клетки 235
7.1. Биохимическая термодинамика 236
7.2. Роль адениловой системы в энергетике клетки 241
4
7.3. Термодинамическая шкала фосфоорганических соединений 243
7.4. Характеристика важнейших высокоэнергетических
соединений 245
7.5. Биохимические реакции сопряжения ..... 247
Краткое содержание главы 248
Хронология важнейших дат в исследовании биоэнергетики
клетки 250
Литература 250
Вопросы и задачи 251
Глава 8. Ферменты 252
8.1. Общая характеристика 252
8.2. Структура ферментов 254
8.3. О механизме ферментативного катализа 256
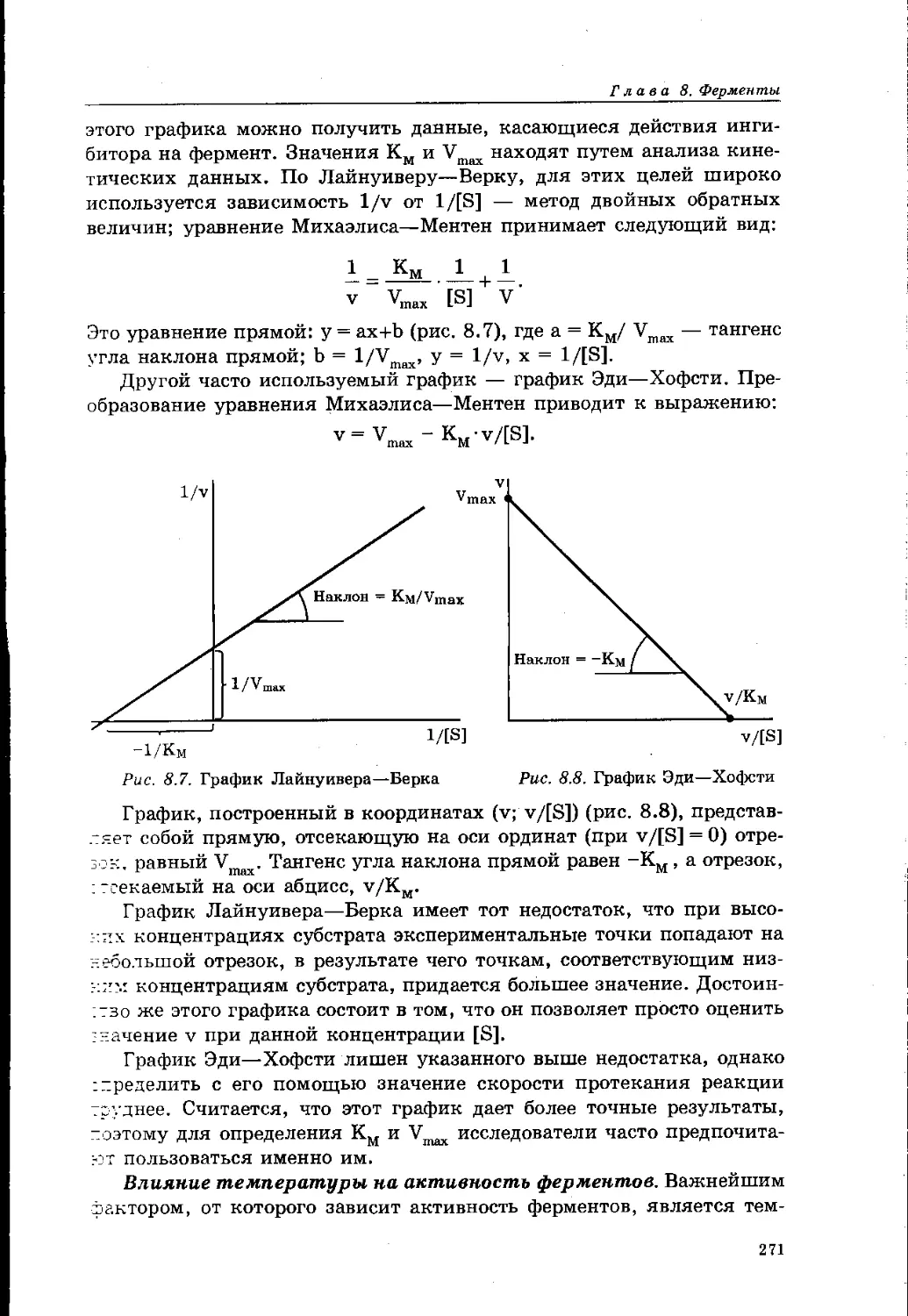
8.4. Кинетика ферментативных реакций 265
8.5. Номенклатура и классификация ферментов 279
8.6. Характеристика отдельных классов ферментов 281
8.7. Регуляция активности ферментов 294
8.8. Множественные молекулярные формы ферментов 299
8.9. Методы исследования ферментативного катализа 300
Краткое содержание главы 305
Хронология важнейших дат в исследовании ферментов , 307
Литература : 308
Вопросы и задачи .... 308
Глава 9. Катаболизм углеводов. Генерирование энергии
фосфатной связи 310
9.1. Гликолиз 310
9.2. Брожение 317
9.3. Биоэнергетика анаэробного разложения углеводов 323
9.4. Цикл трикарбоновых кислот 325
9.5. Биоэнергетика цикла трикарбоновых кислот 333
9.6. Регуляция превращения пирувата в ацетил-СоА
и цикла трикарбоновых кислот 333
9.7. Гексозомонофосфатный путь 335
9.8. Транспорт электронов и окислительное фосфорилирование 338
9.9. Транспортные системы внутренней митохондриальной
мембраны 359
9.10. Эффективность окисления глюкозы и контроль
окислительного фосфорилирования 363
9.11. Токсичные формы кислорода и их роль в апоптозе 366
9.12. Взаимосвязь процессов гликолиза и дыхания 374
9.13. Исследование процессов катаболизма углеводов 376
Краткое содержание главы 380
Хронология важнейших дат в исследовании катаболизма
углеводов . 383
Литература 384
Вопросы и задачи 384
Глава 10. Биосинтез углеводов 385
10.1. Глюконеогенез 385
10.2. Регуляция глюконеогенеза и гликолиза 387
10.3. Фотосинтез 389
10.4. Пигменты хлоропластов высших растений 392
5
10.5. Стадии фотосинтеза 394
10.6. Два типа фотосинтезирующих систем..... 396
10.7. Циклический и нециклический транспорт электронов 398
10.8. Темновая стадия фотосинтеза. Взаимопревращения
моносахаридов 405
10.9. Оксигеназная активность рубиско и ее снижение
при функционировании С4~пути... 414
10.10. Биосинтез олигосахаридов 420
10.11. Биосинтез полисахаридов 422
Краткое содержание главы 430
Хронология важнейших дат в исследовании анаболизма
углеводов 432
Литература . 433
Вопросы и задачи 433
Глава 11. Метаболизм липидов ♦ 434
11.1. Катаболизм ацилглицеролов 434
11.2. Глиоксилатный цикл 440
11.3. Энергетика окисления жирных кислот 442
11.4. Катаболизм фосфолипидов 444
11.5. Биосинтез жирных кислот 444
11.6. Регуляция биосинтеза жирных кислот 448
11.7. Биосинтез липидов 449
11.8. Исследования процессов метаболизма липидов 452
Краткое содержание главы 455
Хронология важнейших дат в исследовании метаболизма
липидов 457
Литература 457
Вопросы и задачи 457
Глава 12. Метаболизм аминокислот и нуклеотидов 459
12.1. Круговорот азота в природе 459
12.2. Азотфиксация 460
12.3. Нитрификация 463
12.4. Синтез аминокислот 464
12.5. Катаболизм аминокислот 469
12.6. Биосинтез нуклеотидов 177
12.7. Распад нуклеотидов 483
Краткое содержание главы 485
Хронология важнейших дат в исследовании азотсодержащих
соединений 487
Литература 487
Вопросы и упражнения 488
Г лава 13. Синтез ДНК, РНК и белков. Перенос генетической
информации 489
13.1. Репликация ДНК 490
13.2. Репарация генетических повреждений в ДНК 496
13.3. Рекомбинация ДНК 498
13.4. Генная инженерия. 502
13.5. Транскрипция 508
13.6. Обратная транскрипция... 513
13.7. Синтез белка (трансляция) 514
13.8. Адресный транспорт белков 522
6
13.9. Регуляция синтеза белка 523
13.10. Ингибиторы транскрипции и трансляции
и их биомедицинское значение 531
Краткое содержание главы 532
Хронология важнейших дат в исследовании синтеза
нуклеиновых кислот и белков ... 535
Литература 536
Вопросы и задачи 536
Глава 14. Молекулярные основы действия клеточных
медиаторов 537
14.1. Передача сигналов через клеточные мембраны с помощью
специфических рецепторов и эффекторных систем 537
14.2. Активация аденилатциклазного сигнального пути
с помощью гормонов . 538
14.3. Роль О-белков как сигнальных трансдукторов 540
14.4. Медиаторы инозитолфосфолипидного сигнального пути 543
14.5. Активация тирозинкиназных рецепторов под действием
ростовых факторов 545
14.6. Некоторые онкогенные продукты, участвующие
в сигнальных процессах 547
14.7. Гормоны — медиаторы на уровне целостного организма 549
14.8. Классификация и общие биологические признаки гормонов ... 551
14.9. Характеристика гормонов, продуцируемых различными
эндокринными системами 552
Краткое содержание главы 565
Хронология важнейших дат в исследовании молекулярных основ
действия клеточных медиаторов 566
Литература 567
Вопросы и упражнения 56 7
Часть III. Элементы прикладной биохимии 569
Глава 15. Некоторые аспекты экологической биохимии 571
15.1. Адаптация организмов к изменениям условий внешней
среды 572
15.2. Роль веществ вторичного происхождения в процессах
экологических взаимодействий ...: 574
15.3. Аллелопатические хемоэффекторы и токсины растений.
Механизмы их обезвреживания 579
15.4. Загрязнение окружающей среды. Мутации 584
15.5. Основные реакции биотрансформации ксенобиотиков 586
15.6. Некоторые прикладные аспекты экологической биохимии 588
Краткое содержание главы 589
Хронология важных дат в развитии экологической биохимии 591
Литература 591
Вопросы 592
Глава 16. Некоторые аспекты медицинской и фармацевтической
биохимии 593
16.1. Некоторые аспекты медицинской биохимии 593
16.1.1. Биохимические основы наследственных болезней 593
16.1.2. Элементы патологической биохимии питания 599
16.1.3. Биохимические механизмы, лежащие в основе
нервно-психических заболеваний человека 605
16.1.4. Некоторые аспекты патобиохимии онкологических
заболеваний 609
16.1.5. Патобиохимия нарушений функций сердца
при ишемии 612
16.2. Некоторые аспекты фармацевтической биохимии 614
16.2.1. Лекарственные и диагностические средства 614
16.2.2. Биотехнология лекарственных средств 617
16.2.3. Биохимические методы, применяемые для
мониторинга и контроля качества лекарственных средств 622
16.2.4. Некоторые аспекты метаболизма лекарств 628
Краткое содержание главы , 629
Хронология важнейших дат в развитии медицинской
и фармацевтической биохимии 631
Литература 632
Вопросы 633
Глава 17. Биохимические превращения важнейших пищевых
компонентов при созревании, хранении и переработке
сельскохозяйственного сырья 634
17.1. Биохимические превращения при отложении запасных
веществ в растениях ". 634
17.2. Биохимические превращения при хранении
сельскохозяйственного сырья 641
17.3. Ферменты в технологии пищевых продуктов 646
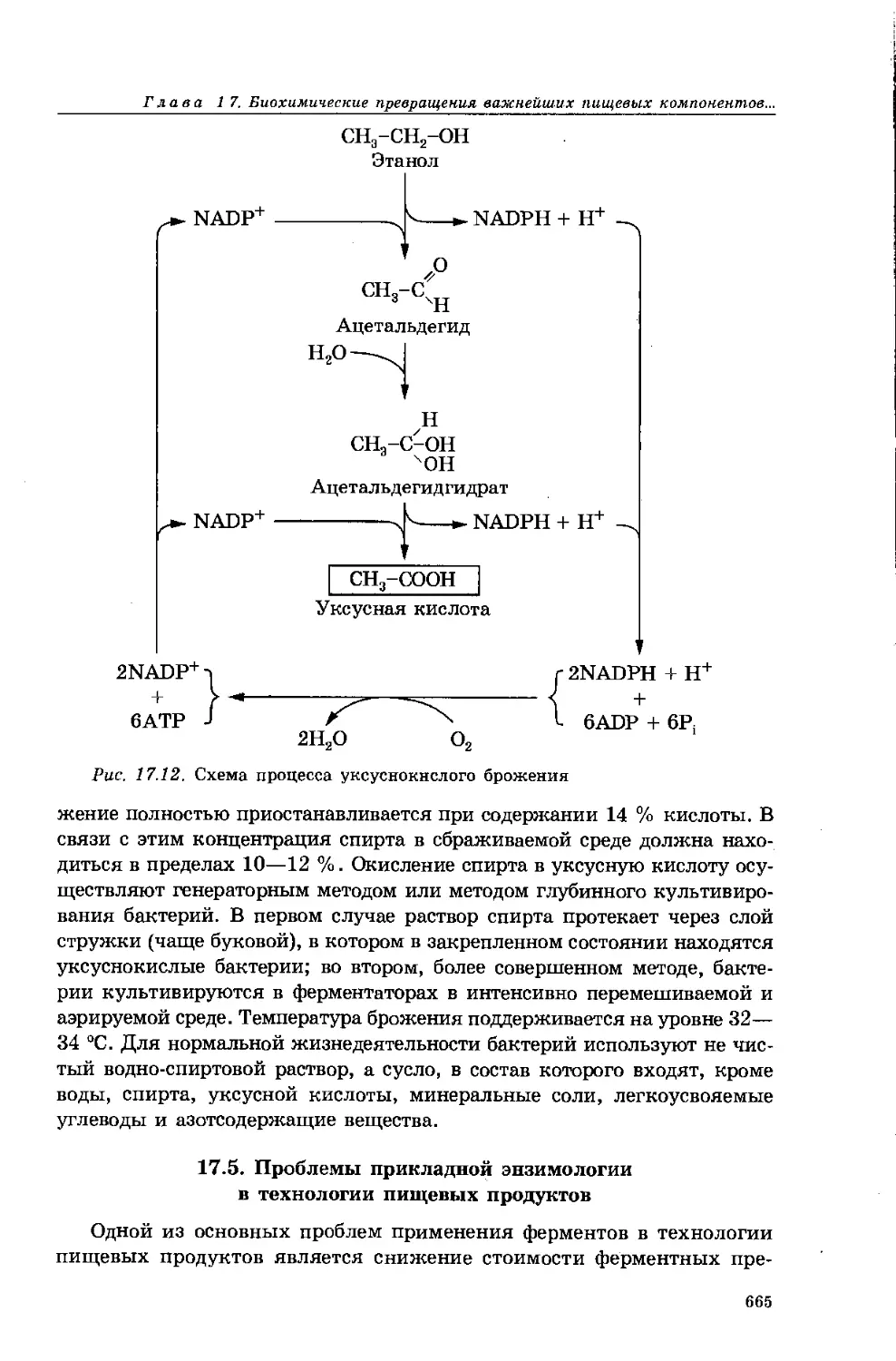
17.4. Процессы брожения и дыхания в технологии пищевых
продуктов 660
17.5. Проблемы прикладной энзимологии в технологии пищевых
продуктов 665
Краткое содержание главы 668
Хронология важнейших дат в исследовании биохимических
процессов при созревании, хранении и переработке
сельскохозяйственного сырья 671
Литература 671
Вопросы и задачи 672
Приложение. Ответы на вопросы и задачи 673
Предметный указатель 681
ПРЕДИСЛОВИЕ
Учебник "Биохимия" составлен на основе накопленного авторами
многолетнего опыта чтения лекций по общей биохимии и
молекулярной биологии. При написании книги авторы ставили перед собой цель
изложить материал так, чтобы студенты могли овладеть
биохимической терминологией и освоит^ общие концепции биохимии,
стремились творчески осмыслить эти концепции и понять молекулярную
логику жизнедеятельности.
Учебник состоит из трех частей:
Часть I "Биомолекулы: структура, свойства, функции"
(статическая биохимия) посвящена описанию структуры и функции белков,
нуклеиновых кислот, углеводов, липидов, витаминов.
Часть II "Метаболизм веществ и энергии в клетке" (динамическая
биохимия) включает вопросы биоэнергетики и биокатализа, анаболизма
и катаболизма углеводов и преобразования аккумулированной в них
энергии в энергию фосфатных связей. Рассматриваются метаболизм
нуклеиновых кислот, белков, жиров и сопряженные с ним
биоэнергетические процессы. В анаболизме нуклеиновых кислот и белков
особое внимание обращено на вопросы, связанные с важнейшей
проблемой молекулярной биологии — переносом генетической информации.
Ключевую роль в метаболизме клетки играют биологические
катализаторы — ферменты; в этой связи рассмотрены принципиальные
представления о кинетике, механизме действия, влиянии различных
физико-химических факторов на их активность и стабильность. При
освещении вопросов биоэнергетики особое внимание уделено такой
функции мембран, как аккумуляция энергии для синтеза АТР. В этой
связи рассмотрено функционирование нового класса ферментов —
мембранных протонных помп. В последнее время интенсивно
развиваются исследования процесса, обратного митозу, а именно разборки
клетки — апоптоза. Этот вопрос также нашел отражение в учебнике во
взаимосвязи с ролью активных форм кислорода.
Часть III "Элементы прикладной биохимии" рассматривает
прикладные вопросы биохимических процессов. Возрастание степени
угроз экологических катастроф на Земле побудило авторов включить ё
книгу некоторые аспекты экологической биохимии. При этом особое
внимание обращено на ту роль, которую играет биохимия в
предотвращении этих угроз. В последних главах учебника описано значение
биохимических процессов при хранении, переработке основных видов
сельскохозяйственного сырья. Здесь нашел отражение тот аспект
биохимии, который может быть важен в совершенствовании традицион-
9
ных биотехнологий и в организации новых технологий продуктов с
ценными биологическими характеристиками. Освещены также
важнейшие аспекты фармацевтической и клинической биохимии.
На протяжении всей книги авторы стремились дать физическую
интерпретацию биохимическим процессам. Глава по ферментативному
катализу включает математическое моделирование, позволяющее четко
представить механизм действия ферментов. В конце каждой главы
даются в историческом аспекте пути и подходы решения актуальных проблем
биохимии, краткое содержание главы. К каждой главе прилагаются
вопросы, задачи и упражнения; они позволят акцентировать внимание
студентов на основных вопросах разделов учебника. Авторы надеются, что
организация практических семинарских занятий по биохимии на основе
этих вопросов вызовет живой интерес у студентов к познанию этой
увлекательной области науки, привьет навыки вдумчивого аналитического
подхода к изучению учебного материала.
Авторы выражают глубокую благодарность директору Института
физико-химической биологии (Московский государственный
университет имени М. В. Ломоносова), академику РАН В. П. Скулачеву,
заведующему лабораторией структуры и функций мембран этого же'
Института, профессору Л. С. Ягужинскому и заведующему кафедрой
биохимии, профессору Воронежской государственной медицинской
академии В. В. Алабовскому за ценные критические замечания и труд
по рецензированию данной книги. Авторы весьма признательны
ассистенту кафедры аналитической и медицинской биохимии и
микробиологии Воронежского государственного университета Л. В. Матасовой
за добросовестную помощь в работе, зарубежному коллеге профессору
Университета Мадейры (Португалия) Мигелю Анжело Пинейру де Кар-
валью за помощь при подготовке некоторых разделов учебника.
Авторы искренне благодарны сотрудникам кафедры биофизики и
биотехнологии Воронежского государственного университета К. П. Пенско-
му, кафедры биохимии и микробиологии Воронежской
государственной технологической академии Л. В. Афанасьевой за большую
помощь при подготовке книги к изданию, сотрудникам и аспирантам
кафедры аналитической и медицинской биохимии и микробиологии
Воронежского государственного университета за участие в
оформлении рукописи, а также работникам Издательства Воронежского
государственного университета.
При подготовке книги авторы широко пользовались трудами
таких крупных ученых, как А. И. Опарин, В. Л. Кретович, В. П. Скула-
чев, А. Ленинджер, Л. Страйер, М. Диксон, Э. Уэбб, Т. Гудвин, Э. Мер-
сер, Д. Мецлер, Дж. Уотсон, Б. Альберте и др.
Авторы будут глубоко признательны читателям за добрые пожела-
ния и критические замечания, которые могут быть существенны для
переосмысления определенных аспектов биохимии или же подходов к
их изложению, что будет весьма полезно и необходимо для
последующего переиздания книги.
ВВЕДЕНИЕ
Биохимия (от греч. Ыоз — жизнь) — это наука о молекулярных
основах жизни. Она изучает химический состав организмов,
превращение веществ и энергии, которое осуществляется в процессе их
жизнедеятельности.
Необходимость изучения химического состава и строения клетки
и в целом живых организмов, а также химических превращений в
них возникла в далеком прошлом. Она была вызвана насущными
практическими задачами развития сельского хозяйства и
перерабатывающей его продукцию промышленности, медицины, а также логикой
развития самого естествознания. В настоящее время биохимия ставит
перед собой главную задачу — выяснить, каким образом неживые
молекулы, составляющие живую клетку, взаимодействуют друг с
другом и поддерживают живое состояние этой клетки.
Из определения предмета биохимии видно, что эта наука с
методологической точки зрения может быть подразделена на две части: на
статическую биохимию и динамическую биохимию. Статическая
биохимия изучает химический состав живых клеток и близка к
органической химии. Динамическая биохимия рассматривает
закономерности превращений веществ и энергии в этих клетках. По своему
характеру и основным установкам вторая часть близка к физиологии
клетки. Однако обе части биохимии неразрывно связаны между собой:
изучение механизма и путей превращения веществ невозможно без де-
г
тального глубокого знания свойств, специфических особенностей этих
веществ.
Возникнув на стыке органической химии и физиологии, биохимия
в то же время не стала неким механическим объединением этих
дисциплин. И хотя у нее очень много общего с органической химией
(особенно это относится к методам, применяемым для изучения
природных веществ), перед биохимией и органической химией стоят разные
задачи. В свете задач органической химии представляют интерес прежде
всего строение и свойства химических соединений (электронная
структура, порядок связи и механизм ее образования, изомерия, конформа-
ция), информацию о которых эта наука получает с помощью
специальных методов (структурный и стереохимический анализы, методы
молекулярных орбиталей, встречного синтеза, химической
модификации). Задача физиологии — изучить физиологическую сущность
биологических явлений. Главным же для биохимии является выяснение
взаимосвязи между структурой веществ и их функциями,
превращений веществ и энергии в живой клетке, регуляции и координации
11
I*
метаболических процессов, молекулярных механизмов переноса
генетической информации.
Статическая биохимия выявила характерную черту живых
клеток — их сложность и высокий уровень молекулярной организации,
переход от более простых компонентов клетки к более сложным.
Структурную организацию живой клетки можно представить в виде
следующей схемы (т — молекулярная масса в дальтонах, Да):
Неорганические вещества (т = 18—44)
(Н20, Ы2, С02, 02, Р, 3)
Мономеры (т — 50—250)
(нуклеотиды, аминокислоты, моносахариды, жирные кислоты,
глицерин)
Макромолекулы (т = 103—107)
(нуклеиновые кислоты, белки, полисахариды, липиды)
I
Сложные макромолекулы (т = 103—109)
(нуклепротеины, гликопротеины, липопротеины)
I
Комплексы (т = 106—1010)
(рибосомы, ядрышко, мембраны, сократительные системы)
Органеллы (т = 1011—1013)
(ядро, митохондрии, лизосомы)
Клетка (т - 1012—1015)
На первой ступени структурной организации стоят
низкомолекулярные предшественники клеточных компонентов: Ы2, С02, Н20, 02,
8, Р. Из этих веществ образуются биологические молекулы
(мономеры), которые в дальнейшем усложнении компонентов живой клетки
играют роль структурных единиц или строительных блоков.
Мономеры, соединяясь друг с другом, образуют макромолекулы или
биополимеры, имеющие большую молекулярную массу. Большая часть
макромолекул клетки относится к четырем основным типам соединений:
нуклеиновым кислотам, белкам, полисахаридам и ансамблям липид-
ных молекул. Молекулы данных биополимеров представляют собой
цепи, строительные блоки в которых соединены между собой
прочными ковалентными связями. Строительными блоками нуклеиновых
кислот являются нуклеотиды, в состав которых входят пять типов
оснований: аденин, гуанин, урацил, тимин, цитозин; строительными
блоками белков — 20 типов аминокислот, полисахаридов —
различные моносахариды. Хотя число строительных блоков в каждом типе
молекул невелико, но благодаря тому, что в цепях эти блоки
находятся в самых различных последовательностях, соотношениях и
сочетаниях, каждый тип макромолекул состоит из огромного числа различ-
12
ных по своим свойствам соединений. Так, из 20 аминокислот
образуются до 1012 различных белков, из 5 нуклеотидов — до 1010
разновидностей нуклеиновых кислот.
С точки зрения развития представлений о переходе неживой
материи в живую необходимо особо отметить, что макромолекулы
нуклеиновых кислот и белков несут в клетке информационную роль, так как
определенные последовательности их строительных блоков отражают
и специфические особенности клетки, и в целом генетическую
индивидуальность различных видов организмов. Полисахариды, напротив,
не являются информационными соединениями, поскольку они
состоят из одинаково повторяющихся мономеров. Именно в
информационных макромолекулах уже появляется способность к выполнению
биологических функций (способность к катализу, к самокопированию).
Макромолекулы способны соединяться друг с другом, образуя
сложные макромолекулы (например, нуклеопротеины, липопротеины, гли-
копротеины, гликолипиды и т. д.).
Взаимодействие макромолекул (простых и сложных) формирует
надмолекулярные структуры или комплексы (например, мембраны,
рибосомы, мультиферментные комплексы, метаболоны)*
Следующая ступень организации — клеточные органеллы
(митохондрия, ядро, хлоропласт, лизосома), которые отличаются уже
относительной автономностью в выполнении специфических функций,
определяющих существование клетки (например, митохондрии
участвуют в производстве энергии, лизосомы — в переваривании веществ).
Наконец, система органелл образует клетку.
Переход от простых биомолекул к сложным биологическим
структурам основывается на физико-химических принципах
самоорганизации. В основе самоорганизации лежат химические взаимодействия
между молекулами, входящими в состав живой материи. Ковалент-
ные связи обеспечивают все многообразие простых мономеров и
макромолекул. Укладка макромолекул в пространстве и организация над-
макромолекулярных структур, органелл в клетке осуществляются с
помощью остаточных валентных сил, водородных, ионных, ван-дер-
ваальсовых связей. Если ковалентные связи обусловливают прочность
и устойчивость биомолекул, то слабые, остаточные валентные силы
обеспечивают лабильность, динамичность биологических структур.
Структурная иерархия клетки явилась продуктом длительного
эволюционного развития живой материи на Земле.
Важнейшей задачей динамической биохимии является изучение
обмена веществ, или метаболизма клетки. Обмен веществ —- это
совокупность двух диаметрально противоположных, но гармонически
сочетающихся процессов — синтеза (анаболизма) и распада
(катаболизма) веществ. Метаболизм лежит в основе логических представлений о
развитии живой материи, подчеркивает огромнейшую разницу между
живой и неживой природой; обусловливает непрерывную связь
организма с внешней средой. В клетку поступают вещества среды, которые за-
13
тем превращаются ею в собственные, преемственные для нее соединения;
последние разрушаются и выводятся в окружающую среду.
Обмен веществ в клетке не отделим от обмена энергии. Синтез
веществ организма, сложность его структуры невозможны без затраты
химической энергии* Эту энергию организм черпает из окружающей
среды вместе с питательными веществами. Свободная энергия этих
веществ в клетке преобразуется в энергию химических связей веществ
самой клетки, которая при разложении этих веществ вновь
возвращается в окружающую среду в форме тепла или других бесполезных для
клетки форм энергии.
С точки зрения термодинамики живая клетка представляет собой
открытую систему, однако не в том смысле, что она находится в
равновесии с внешней средой, а в том, что через эту систему идет
постоянный поток веществ и энергии; для нее характерно неравновесное
стационарное состояние.
Термодинамическое равновесие {неживая замкнутая система) и
стационарное неравновесное состояние (живая клетка) сходны между
собой; как в том, так и в другом случае система сохраняет свои
свойства постоянными во времени. Коренное же отличие этих систем
заключается в том, что при равновесии вообще не происходит изменение
свободной энергии (АО = 0), а при стационарном состоянии оно совер-
шается непрерывно, но с постоянной скоростью (ЛО = сопзЪ).
Организмы извлекают из окружающей среды энергию: растения — в виде
квантов света, животные, микроорганизмы — в виде малоокисленных
соединений, которые окисляются в процессе дыхания* За счет этой
энергии они строят свою упорядоченность.
Другой характерной чертой обмена веществ в клетке является
тонкое регулирование скорости протекания отдельных химических
реакций. Живая клетка — это саморегулирующаяся система обмена
веществ. Накопление промежуточных продуктов (метаболитов) в
количествах, превышающих критический уровень, действует как сигнал,
который может вызвать уменьшение скорости реакций, приводящих
к образованию этих веществ.
Важную роль в регулировании процессов метаболизма играют
биологические катализаторы — ферменты. Регуляция клеточного
метаболизма может осуществляться либо путем активации или
подавления действия ферментов, либо за счет изменения скорости их
биосинтеза в клетке.
Пожалуй, самый уникальный признак живых организмов — это
самовоспроизведение или передача наследственной информации, что
не имеет места в неживой природе- Многообразие живого мира
определяется генетической программой, заложенной в нуклеиновых
кислотах. Вся генетическая программа хранится в дезоксирибонуклеино-
вой кислоте (ДНК). Особенностью ее строения является
потенциальная возможность самокопирования и, следовательно, передачи
наследственных признаков от одного поколения организмов к другому, В про-
14
цессе жизнедеятельности клетки информация, заложенная в ДНК,
реализуется через рибонуклеиновую кислоту (РНК) в структуре
соответствующего белка* У всех живых организмов информационная роль
нуклеиновых кислот выполняется с чрезвычайно высокой стабильностью.
Как самостоятельная область научных знаний биохимия начала
развиваться около 100 лет назад с осознанием учеными того факта,
что процессы, протекающие в живой клетке, могут быть объяснены с
позиций точных наук — химии и физики. Термин "биохимия" был
предложен К. Нейбергом в 1903 г. В течение последних 50 лет
биохимия преобразовалась в крупную область научного знания. Возникли
такие направления, как биохимия человека, биохимия животных,
биохимия растений, энзимология — наука о биологических
катализаторах-ферментах, генная инженерия, клиническая биохимия,
формируется экологическая биохимия. Следует отметить такие выдающиеся
достижения, как установление важного принципа молекулярной
биологии — принципа комплементарности, на основе которого была
установлена двухцепочечная спираль молекулы ДНК, расшифрован
генетический код, определена трехмерная структура значительного числа
белков, описаны важнейшие метаболические и энергетические пути в
клетке и на основе этого установлены общие принципы организации
живой материи. Оказалось, что столь различные организмы, как
бактерия и человек, имеют много общего на молекулярном уровне. В
синтезе макромолекул эти организмы используют одни и те же
строительные блоки. В принципе одинаковы (или по меньшей мере сходны)
и пути запасания, хранения энергии и ее использования в клеточном
метаболизме, передача генетической информации от ДНК к РНК и
далее к белкам.
Достижения биохимии и молекулярной биологии дали
значительный стимул для развития генной инженерии. Глубоко изучена роль
биологических мембран в биоэнергетике. В последние годы
интенсивно проводятся исследования механизмов запрограммированной
клеточной смерти, с помощью которых организм контролирует
инициацию и осуществление процесса гибели клетки. В настоящее время
очевидно, что, как это не парадоксально, смерть определенной части
клеток представляет собой существенную часть роста и развития
многих эукариотных организмов, включая и растения, и животных.
Клеточная смерть может быть компонентом ответа на биотический и
абиотический стрессы. В этой связи получило развитие новое научное
направление по изучению универсального биологического явления —
апоптоза, а также роли активных форм кислорода в этом процессе.
Показано, что развитие организма и его смерть находятся под
контролем регуляторных систем клетки.
Исключительно велико значение биохимии в совершенствовании
технологии пищевых продуктов, в развитии медицины,
фармацевтической промышленности. Различные заболевания человека
сопровождаются сдвигом в процессах обмена веществ, и важно выяснить при-
15
чины этих сдвигов и возможности их устранения. Диагностика и
лечение многих заболеваний оказались возможными только на основе
последних достижений биохимии, обеспечивающих понимание
молекулярных механизмов развития патологических процессов.
Итак, глубокое и всестороннее изучение обмена веществ является
одной из актуальных задач в познании сущности жизни. Однако это
изучение имеет и важное прикладное значение для медицины,
животноводства, растениеводства, биотехнологии, промышленной
переработки сельскохозяйственного сырья.
Цель возделывания сельскохозяйственных растений — получение
определенных химических соединений: белков, жиров, крахмала,
сахара, витаминов, которые используются в питании человека или
служат сырьем для перерабатывающей промышленности. Вместе с тем
чтобы управлять развитием и ростом растений и оказывать влияние
на накопление определенного вещества, надо глубоко изучить путь
его биосинтеза и сопряженные с ним процессы, иными словами,
нужно хорошо понимать тип обмена веществ данного растения. Так,
особого внимания заслуживает глубокое изучение и влияние различных
факторов на синтез и биологическое качество белков пшеницы, на
синтез сахарозы в сахарной свекле, крахмала в картофеле, жира — в
подсолнечнике. То же самое можно сказать и об увеличении
продуктивности животноводства.
Для решения научных проблем биохимии в России созданы
научные центры. В составе Российской Академии наук функционируют
институты биохимии, физико-химической биологии, биоорганической
химии, физиологии и биохимии микроорганизмов. Созданы кафедры
биохимии при биологических факультетах университетов, в
медицинских, технологических, сельскохозяйственных вузах. В них
развернуты исследования по таким важным проблемам, как взаимосвязь
между структурой биомолекул, надмолекулярных комплексов, клеточных
органелл и их функциями; расшифровка механизмов
ферментативного катализа и их роли в регуляции обмена веществ и энергии;
проблемы молекулярной генетики, переноса генетической информации,
генной инженерии, клеточной биоэнергетики. Все шире развертываются
исследования по медицинской биохимии, по актуальным вопросам
экологической биохимии.
Часть I
БИОМОЛЕКУЛЫ:
СТРУКТУРА, СВОЙСТВА,
ФУНКЦИИ
Глава 1. БЕЛКИ
Белки (протеины) — высокомолекулярные природные полимеры,
необходимая составная часть всех организмов.
Основная масса протоплазмы клетки представляет собой белки, на
их долю приходится около половины сухих веществ клетки. Велико
разнообразие белков: в одной клетке могут находиться сотни
различных видов этих макромолекул.
Белки играют первостепенную роль в структуре и функциях
клетки. К белкам относятся биологические катализаторы — ферменты,
занимающие центральное место в обмене веществ организма. Белки,
содержащиеся в мембранах клеток, способствуют транспорту веществ
внутрь клеток и наружу. Данный тип макромолекул участвует в
осуществлении и множества других биологических функций. Таким
образом, белки являются молекулярными инструментами, с помощью
которых реализуется генетическая информация. Это уникальные
соединения, обеспечивающие организацию и регуляцию обмена веществ
организма.
Белки имеют особое значение в питании человека и животных: они
выполняют пластическую роль в биосинтезе структурных элементов
организма. В организме человека почти нет резервов белков, поэтому они
являются совершенно незаменимыми в ежедневном питании. Белковое
голодание приводит к тяжелым расстройствам в организме. Длительное
безбелковое питание неизбежно приводит к смерти.
1.1. Элементарный состав белков
Средний элементарный состав большинства белков, %
Углерод 50—54
Азот 15—18
Кислород 20—23
Водород 6—8
Сера 0—2,5
Как видно, 99 % и более всей массы белков приходится на
углерод, азот, кислород, водород. Данные элементы наиболее подходят для
обеспечения биологических функций белков. Это объясняется
следующими свойствами данных элементов:
1. Все четыре элемента легко образуют ковалентные связи
посредством спаривания электронов.
2*
19
Часть I, Биомолекулы: структура, свойства, функции ' __
2. Они самые легкие среди всех элементов, а прочность ковалент-
ной связи обратно пропорциональна атомным массам элементов., т. е.
они дают сильные ковалентные связи.
3. Углерод, азот и кислород образуют и одинарные, и двойные
связи, благодаря чему они обусловливают разнообразие свойств белка.
4. В белках углерод образует каркасы разных форм и
конфигураций, дает ответвления, трехмерные структуры.
И, наконец, важная особенность углерода, обусловливающая его
незаменимость в живой материи: все основные жизненные функции
невозможны без наличия конъюгированных или резонансных
молекулярных систем с характерным для них явлением делокализации
электронов. Только углерод обладает этим уникальным свойством,
которое особенно ярко проявляется в биологических катализаторах —
ферментах, оно лежит в основе механизма их действия.
Именно из-за отсутствия этой особенности такой элемент, как
кремний, столь близкий к углероду по способности образовывать
ковалентные связи и давать бесчисленные множества каркасов
молекул, не может служить основой для структурных элементов живой
клетки*
Белки дают специфические цветные реакции. Так, биуретовая
реакция (реакция с сернокислой медью) указывает на наличие в
белке пептидных (кислотно-амидных) связей, играющих
важнейшую роль в формировании остова белковой молекулы. Ксантопро-
тейповая реакция (реакция с азотной кислотой) свидетельствует о
наличии в белке бензольных колец; а реакция Миллона — о
наличии в белке индольных и фенольных колец. Эти реакции убеждают
нас в том, что в белковой молекуле находятся самые разнообразные
по своим физико-химическим свойствам функциональные группы.
Именно они обеспечивают столь громадное разнообразие белков в
живой природе, столь различный характер их биологических
функций.
Содержание белка в объектах различного происхождения, %
Мышцы, сердце, печень 15—23
Семена зернобобовых (фасоль, соя, горох) 18—28
Семена хлебных злаков:
Рожь, ячмень 8—13
Пшеница 12—21
Стебли, листья растений 1,5—3,0
Овощи, фрукты 0,5—1,7
1.2. Аминокислоты — строительные блоки белковой молекулы
Строительными блоками (мономерами) белков являются
аминокислоты. Об этом свидетельствует тот факт, что при кислотном,
щелочном, ферментативном гидролизе белков в качестве его конечного
продукта образуются аминокислоты,
20
Глава 1. Белки
Всего в природе найдено около 300 аминокислот, однако в состав
белков входит лишь 20, получивших название белковых, или протеи-
ногенныХу аминокислот.
В виде очищенных препаратов белковые аминокислоты
представляют собой белые кристаллические вещества: сладкие, горькие или не
имеющие вкуса.
Белковые аминокислоты являются а-аминокислотами с
характерной общей структурной особенностью: наличием карбоксильной и амин-
ной групп, связанных с атомом углерода в а-положении:
Н
I
I
к-|-са—соон
\ I
N112
Часть этой структурной формулы (вправо от штриховой линии)
одинакова у всех аминокислот, а-остаток (радикал К) аминокислоты,
ее функциональная группа, которая у разных аминокислот
неодинакова по структуре, электрическому заряду и растворимости.
Благодаря специфическим особенностям радикалов каждая аминокислота
наделена индивидуальностью. Поэтому все группы К аминокислот, по
образному выражению А. Ленинджера, можно назвать алфавитом
"языка" белковой структуры. Соответствующие последовательность и
сочетание этих групп в молекуле белка определяют ее биологические
функции,
В зависимости от химических свойств К-групп аминокислоты
подразделяются на четыре основных класса, что принято называть
классификацией по полярности:
1) неполярные, или гидрофобные*, с алифатическими или
циклическими углеводородными группами;
2) полярные\ с сульфгидрильной, гидроксильной, амидной
группами;
3) отрицательно заряженные: с карбоксильной группой;
4) положительно заряженные: с аминогруппой, гуанидиновой,
имидазольной группами.
По своей стехиометрической конфигурации все аминокислоты, за
исключением глицина, имеют асимметрический атом углерода в
а-положении, с которым связаны 4 разные группы: К-группа,
1ЧН2-группа, СООН-группа и атом водорода. Таким образом,
аминокислоты обладают оптической активностью и являются либо
правовращающими (+), либо левовращающими (-).
В связи с тем, что валентные связи вокруг а-атома углерода имеют
тетраэдрическое расположение, 4 различных замещающих группы
могут размещаться в пространстве двумя разными способами, так что
молекула аминокислоты может существовать в виде стереоизомеров,
21
Часть I. Биомолекулы: структура, свойства, функции
в Ь-форме или В-форме. При отнесении аминокислот к В- или Ь-ряду
принято рассматривать абсолютную конфигурацию замещающих групп
вокруг асимметрического углеродного атома; при этом их сравнивают
с эталоном — простейшим трехуглеродным сахаром — глицеральде-
гидом:
соон соон
I I
СНз СН8
Ь-Алании О-Алании
2
СНО СНО
- 1
НОс=~ С -=>Н Нп=- С -^ОН
! I
сн2он сн2он
Ь-Глицеральдегид В-Глицеральдегид
Все белковые аминокислоты имеют Ь-форму. Живые клетки
обладают уникальной способностью синтезировать Ь-аминокислоты
благодаря высокой специфичности ферментных систем. В-аминокислоты в
настоящее время выделены из ряда источников в свободном
состоянии и в качестве компонентов других структур. В частности, В-амино-
кислоты обнаружены в структуре пептидных антибиотиков. Так, в
составе антибиотика бацитрацина Л9 синтезируемого штаммами
бактерии ВасШиз ИскепЬ^огтгз, обнаружена как Ь-, так и В-аспарагино-
вая кислота. В качестве другого примера можно назвать вещество
клеточной стенки бактерий 81арНу1ососсиз аигеиз пептид огликан,
содержащее О- и Ь-аланин,
1,3. Модифицированные аминокислоты
Помимо стандартных аминокислот в белках встречаются и
модифицированные аминокислоты. Они являются производными обычных
стандартных аминокислот. Как правило, химическая модификация
происходит после включения аминокислоты в состав белка*
Биологическое значение модифицированных нестандартных
аминокислот может быть продемонстрировано на примере у-карбокси-
глутаминовой кислоты:
НООС\? I //
сн-сн2~с~-с
НООС/ I ч пн
н
у-Карбоксиглутаминовая кислота
22
Глава 1. Белки
Наличие у-карбокеиглутаминовой кислоты, содержащей
дополнительную группу -СООН, обеспечивает оптимальное связывание ионов
Са2+ белком плазмы крови протромбином, что необходимо для его
активации и превращения в тромбин. Этот процесс сопряжен с
превращением другого белка плазмы — фибриногена в фибрин,
необходимый для образования кровяного сгустка и остановки кровотечения.
При недостатке витамина К происходит нарушение процесса карбо-
ксилирования глутаминовой кислоты в составе протромбина и, как
следствие, замедление свертывания крови.
Другие примеры соединений, содержащих модифицированные
аминокислоты, — трийодтиронин (ТЗ) и тетрайодтиронин (тироксин, или
Т4). Йодсодержащие аминокислоты синтезируются в щитовидной
железе из тирозина. ТЗ и Т4 — важнейшие гормоны, участвующие в
регуляции многих процессов жизнедеятельности:
ОН
сн2
I 2
н- с - ш„
I 2
СООН
Тироксин (Т4)
ОН
н-с-ш.
СООН
Трийодтиронин (ТЗ)
Примером модификации является окисление двух -8Н~групп цис-
теиновых остатков, что приводит к образованию аминокислоты цис-
тина, содержащей дисульфидный мостик. Дисульфидные мостики
играют важную роль в формировании структуры некоторых белков,
например иммуноглобулинов, инсулина. В таких белках цистеиновые
остатки молекулы цистина входят в состав двух разных
полипептидных цепей, которые благодаря дисульфидной связи оказываются
поперечно сшитыми между собой.
>ш2 кн2
^С-С -СН2-8-В-СН2- С -С ^
но I I он
н н
Цистин
Гидроксипролин и гидроксилизин — также примеры
модифицированных аминокислот. Они входят в состав коллагена — основного белка
соединительной ткани:
23
Часть I. Биомолекулы: структура, свойства, функции
НО-НС
СН.
2 О
нзС ^^сн—соон
н
4-Гидррксипролин
1Ш^-СН2— СН—СН2
ОН
-сн„-
КН
1 ^
с—с
I
н
\
он
5-Гидроксилизин
1.4. Кислотно-основные и электрохимические
свойства аминокислот
По Бренстеду, кислыми группами аминокислот являются
карбоксильная группа (-СООН —> -СОО" + Н+) и протонированная ос-амино-
группа (-ИНд" —» -МН2 + Н+); основные же группы — карбоксилат-ион
(-СОО- + Н+ -> -СООН) и а-аминогруппа (-МН2 + Н+ -> -Шф. Для
каждой аминокислоты имеется как минимум две константы
кислотной диссоциации: одна — для карбоксильной группы (рК колеблется
в пределах 1,8—5,5), другая — для протонированной а-аминогруппы
(рК в пределах 9,0—13,0). (Напомним, что рК соответствует значению
рН, при котором степень диссоциации составляет 50 %, т.е. в данной
точке соединение присутствует в виде эквимолярной смеси донорной
и акцепторной форм.) Находясь в непосредственной близости от
а-углеродного атома, карбоксильная и аминная группы резко влияют
друг на друга. Так, рК карбоксильной группы глицина — 2,34,
р-аланина — 3,6, тогда как рК уксусной кислоты — 4,75.
В растворе возможно существование четырех электрохимических
форм аминокислот:
Е —СН—СООН
1
Нейтральная
к—сн—соо
I
КН2
Отрицательно
заряженная
Е—СН —СООН
I
+
НН,
П о л ожител ьно
заряженная
к—сн—соо
I
+
1Ш8
Амфотерная
В водных растворах аминокислоты находятся в виде амфотерных
попову о чем свидетельствуют следующие факты:
1) они лучше растворимы в воде, чем в менее полярных
растворителях;
2) в водных растворах аминокислоты обладают высокими
диэлектрическими постоянными и большими дипольными моментами;
24
Глава 1 > Белки
3) при кристаллизации из нейтральных водных растворов
аминокислоты образуют прочные кристаллические решетки: кристаллы
аминокислот плавятся при достаточно высоких температурах (200—
300 °С), обычно с разложением. Объяснить это можно тем, что
прочность кристаллической решетки аминокислот обусловлена
электростатическими силами притяжения между противоположно
заряженными химическими группами расположенных рядом молекул.
На ионизацию аминокислот в водных растворах большое влияние
оказывает значение рН среды. В кислой среде высокая концентрация
протонов подавляет диссоциацию карбоксильных групп и
аминокислоты заряжаются положительно (переходя в форму катионов):
к~сн -соо" + н+*± к-сн-соон
В щелочной среде при избытке ОН"-ионов аминокислоты находятся в
виде анионов, так как диссоциируют протонированные аминогруппы:
к-сн -соо'+ <яГ*± к-сн -соо" + н^о
I + I
Поскольку в кислой среде аминокислоты заряжены
положительным электрическим зарядом, в щелочной — отрицательным,
величина заряда аминокислоты находится в тесной взаимосвязи с значением
рН среды. В растворе с рН 4—9 аминокислоты существуют в виде
амфотерных ионов. Состояние аминокислоты, когда ее суммарный
электрический заряд равен нулю, называется изоэлектрическим, а
значение рН, обусловливающее это состояние, называется изоэлект-
рическои точкой аминокислоты (ИЭТ, рН^. ИЭТ определяется как
среднее арифметическое двух величин рК;
рН, = 1/2 (рК/ + рК2')
Например, ИЭТ аланина равна
рНг = 1/2 (рКС00Н + рК^) = 1/2 (2,34 + 9,69) = 6,02.
Величина ИЭТ четко отражает кислотно-основные свойства
аминокислот; так, ИЭТ гидрофобных аминокислот близка к нейтральным
значениям рН (от 5,5 для фенилаланина до 6,3 для пролина), для кислых /
она имеет значения: для аспарагиновои кислоты — 2,8, для глутамино^
вой — 3,2; основные аминокислоты — гистидин, лизин и аргинин имеют
ИЭТ в щелочной зоне рН* В изоэлектрической точке аминокислоты
обладают минимумом растворимости, минимальной буферной емкостью и,
наоборот, вблизи рК каждой группы буферная емкость максимальна.
Как амфотерные электролиты аминокислоты имеют характерные
кривые титрования. На рис. 1.1 показаны кривые титрования,
отображающие последовательную ионизацию групп в основной аминокислоте
(лизине) и в кислой аминокислоте (аспарагиновои).
25
Часть I. Биомолекулы: структура, свойства, функции
О
СООН СОО"
I _ I
СНИН^ ОН" СНШ^ °Н
! ' » I
(СН2)3 Н+ (СН2)3
СН^Щ" СН2МН^
8
10
14 рН
Н
+
СОО" СОО"
I _ I
снын2 он снинг
(СН2)8 ' Ж* (СН2)з
I . I
СНгИН^ СН2МН2
рК^ 2,18 рК2= 8,95 рК3~ 10,5.3
„ СООН СОО"
г. | )
СНЫНз" ОН" СНШ^ 0Н|
сн2 ' н+ сн2 *а+
I I
СООН СООН
рК~2,01 рК2=3,80
СОО" СОО"
I I
снга^ °н" снмн2
сн2 ' н+ сн2
I I
СОО" СОО"
рК =9,93
3
Рис. 1.1. Кривые титрования лизина (кривая 1)
и аспарагиновой кислоты (кривая 2)
Поскольку при одном и
том же значении рН
каждая аминокислота несет
определенной величины
заряд, смесь аминокислот
можно разделить,
подвергнув ее воздействию
электрического поля. В этих
условиях аминокислоты
будут двигаться с
различными скоростями или даже в
разных направлениях. На
этом свойстве аминокислот
основано их разделение
методом электрофореза.
На амфотерных
свойствах аминокислот
основан их количественный
анализ методом
ионообменной хроматографии
(рис 1.2).
Хроматографическую
колонку заполняют
гранулами синтетической
смолы, которая содержит либо анионные (-) группы, (смола катионооб-
менная)* либо катионные (+) группы (смола анионообменная). На
колонку, например, с катионообменной смолой наносят кислый раствор
(рН около 3,0), содержащий смесь аминокислот; в таком растворе
аминокислоты находятся в виде катионов. Аминокислоты, имеющие
наибольший положительный заряд (лизин, аргинин, гистидин), наиболее
прочно ив первую очередь (в верхнем слое колонки) связываются со
смолой; аминокислоты, несущие наименьший положительный заряд
(глутаминовая и аспарагиновая кислоты), — с наименьшей
прочностью и в последнюю очередь (нижние зоны колонки). Другие
аминокислоты займут промежуточные зоны в адсорбционной колонке. При элю-
ции аминокислоты будут распределяться по фракциям в зависимости
от величины их заряда: в первых фракциях — аминокислоты с
наименьшей величиной заряда, в последних — с наибольшей. """
Следует отметить, что электрофорез и ионообменная
хроматография нашли широкое применение в исследовании аминокислотного
состава белков, их структуры и биологической оценке.
1.5. Специфические химические реакции аминокислот
Благодаря наличию карбоксильных и аминных групп аминокислоты
имеют свои специфические химические реакции, нашедшие применение
26
Глава 1. Белка
при разделении, идентификации и
количественном определении аминокислот. Реакции
по карбонильным группам аминокислот,
аминогруппа которых защищена, протекают
аналогично превращениям карбоновых кислот.
Аминокислоты легко образуют соли,
сложные эфиры, тиоэфиры; с формальдегидом дают
метильные или метиленовые производные*
При обработке эфиров аминокислот изоциа-
натами или изотиоцианатами образуются
производные мочевины и тиомочевины.
Аминокислоты способны реагировать
своими карбоксильными группами со
спиртами, образуя сложные эфиры:
К.-СН-СООН +Н0~Ко:
1 I
о
II
к -сн-с + нр.
I I
инл ок
2 *'"» ~"2
Сложные эфиры разных аминокислот со
спиртами имеют различные коэффициенты
летучести. Поэтому их легко можно
разделить путем фракционной перегонки в
вакууме. Этой реакцией широко пользовался
Э. Фишер при проведении аминокислотного
анализа белков.
В специфических реакциях аминокислот
особую роль играет реакционная способность
ос-аминогруппы* Аминокислоты могут
реагировать с азотистой кислотой, образуя при
этом гидроксикислоту и газообразный азот:
к-сн-соон + нжх
I
ъ- сн-соон +н2+н2о.
I
га2 он
Эта реакция получила название реакции
Ван-Слайка, В 20—40-х гг. XX в. она часто
использовалась для количественного
определения аминокислот по выделившемуся
газообразному азоту.
а-Аминогруппа аминокислот может
вступать в реакцию с формальдегидом:
^-СН-СООН-Ь 20 = С
1
ЫНо
/
\
н
н
кх-сн-соон,
нон2с
N
• \
+++
+++
++
Рис. 1.2. Ионообменная
хроматография. В методе
ионообменной
хроматографии разделяются
компоненты с различными зарядами.
В качестве элюента
используется раствор с
возрастающей ионной силой. Матрица
геля может нести либо
отрицательные (как это указано
на рисунке), либо
положительные группы. В случае
отрицательно заряженной
матрицы геля будут
адсорбироваться компоненты
образца, имеющие
положительный заряд, В случае
десорбции положительно
заряженные компоненты образца
будут обмениваться на ионы,
содержащиеся в элюенте.
При этом каждый компонент
образца будет десорбировать-
ся при определенной ионной
силе и непрерывно
выводиться из колонки
СН2ОН
27
Часть I. Биомолекулы: структура, свойства, функции
Реакция известна под названием реакции Зёренсона. Аминогруппа
блокируется гидроксиметиленовой группой и теряет свои основные
свойства, сохраняя кислотные. Она нашла применение при
количественном определении аминокислот по методу формольного
титрования аминокислот.
Для обнаружения, идентификации и количественного анализа
аминокислот широкое применение получила цветная реакция с нингидрином:
он
+ к-сн-соон
+ со0 + к- с
он
ын.
Нингидрин
О ""2
Аминокислота
ОН
ОН
^
О
\
Н
Альдегид
И + ЗЦр + Н+
О" °
Продукт с сине-фиолетовой
окраской (пролин дает с нингидрином
продукт с желтой окраской)
При рН 5,5 и нагревании с избытком нингидрина аминокислота
дегидрируется, декарбоксилируется с образованием С02, ЫН3 и альдегида,
а нингидрин превращается в восстановленный нингидрин, Нингидрин,
восстановленный нингидрин и аммиак затем конденсируются с
образованием окрашенного соединения, причем образуется пигмент
сине-фиолетового цвета, если аминокислота содержит свободную аминогруппу, и
желтый пигмент, если а-аминогруппа замещена, как, например, у про-
лина, гидроксипролина. Интенсивность окраски можно использовать для
колориметрического определения концентрации аминокислот, например,
в белковых гидролизатах после разделения аминокислот бумажной,
ионообменной хроматографией или электрофорезом. Нингидриновая реакция
обладает высокой чувствительностью и используется для обнаружения и
точного определения небольших количеств аминокислот.
При слабощелочных значениях рН и комнатной температуре
аминокислоты вступают в реакцию с 1-фтор-2,4-динитробензолом:
°2М
™2
сн
I
к
о^
сн-соон + нг
I
к
1-Фтор-2,4-ди- Аминокислота 1-Аминоацил-2,4-динитробензол
нитробензол
Динитробензольные производные аминокислот обладают высокой
устойчивостью к кислотному гидролизу. Эту реакцию используют для
идентификации аминокислот при исследовании аминокислотной после-
28
Глава 1. Белки
довательности пептидов. Ее успешно применил Сенгер при
определении аминокислотной последовательности гормона инсулина.
Аналогичная реакция с а-аминогруппой осуществляется с фенил-
изотиоцианатом:
I
=с=н-/^
К-СН-ОТ1Л+ 8=С ^тЧГ ч> ~+ N4! 7 + Н.О
'%
соон
Фенилизотиоцианат Фенилтиогидантоин
Эту реакцию впервые успешно использовал Эдман для
определения аминокислотной последовательности пептидов путем
постепенного отщепления концевых аминокислот кислотным гидролизом.
При нагревании растворы аминокислот и редуцирующих Сахаров
вступают в реакцию, известную под названием реакции Майяра, или
реакции меланоидинообразования. Реакция Майяра полностью не изучена,
протекает через ряд промежуточных стадий; Она сопровождается
разложением исходных соединений (подобно нингидриновой реакции) на
аммиак, альдегиды, фурфурол, оксимети л фурфурол. Продукты
разложения вновь вступают с исходными веществами (аминокислотой и
сахаром) в окислительно-восстановительные реакции, реакции конденсации
и полимеризации* В конечном счете образуются высокомолекулярные,
окрашенные в темно-коричневый цвет вещества, — меланоидины.
Меланоидины являются важными компонентами пищевых
продуктов. При кулинарной обработке пищевых продуктов, при тепловом
воздействии на сырье аминокислоты и редуцирующие сахара
образуют меланоидины, которые играют важную роль в формировании
вкуса, запаха, цвета пищевого продукта.
1.6- Классификация аминокислот
Характерные свойства отдельных аминокислот определяются
природой радикала К. В зависимости от строения этого радикала
аминокислоты подразделяют на алифатические (к ним принадлежит
большинство аминокислот), ароматические (фенилаланин и тирозин),
гетероциклические (гистидин, триптофан) и амингидрокислоты (про-
лин и гидроксипролин), у которых атом азота, стоящий при а-угле-
родном атоме, соединен с боковой цепью в пирролидиновое кольцо.
По числу аминных и карбоксильных групп аминокислоты делятся
на моноаминомонокарбоновые> содержащие по одной карбоксильной
и аминной группы; моноаминодикарбоновые (две карбоксильные и одна
аминная группы); диаминомонокарбоновые (две аминные и одна
карбоксильные группы).
По химическому составу входящих в радикал К замещающих групп
различают гидрооксикислоты (содержат спиртовую группу),
серосодержащие (в радикале находится атом серы).
29
Часть /• Биомолекулы: структура* свойства, функции
Аминокислоты с углеводородными радикалами придают белкам
гидрофобные свойства; если радикал содержит гидрофильные группы
(-ОН, -СООН, -8Н), белковая молекула приобретает гидрофильные
свойства.
Краткая характеристика важнейших физико-химических свойств
аминокислот представлена в табл. 1.1
Таблица 1.1
Химическое строение и физико-химические свойства аминокислот
Название
(тривиальное и
*
рациональное)
1
щенное
обозначение
2
Строение
химического
радикала К
3
Удельное
вращение
в водном
растворе
при 25 °С
4
Константы
кислотной
диссоциации
рк,
5
Рк2
6
рк3
7
иэт
8
Растворимость
при
25 °С,
г/100 г
воды
9
Глицин (а-аминоуксусная
к-та)
Алании (а-аминопропио-
новая к-та)
Валин (а-амино-|5-метил-
масляная к-та)
Лейцин (а-аминоизокал-
роновая к-та)
Изолейцин (а-ашгао-[5-ме-
тилвалериановая к-та)
Фенилаланин (а-амино-р-
фенилпропионовая к-та)
Тирозин (а-амино-р-гид-
роксифенилпропионовая
к-та
Аспарагиновая (а-амино~
янтарная к-та)
Глутаминовая (сс-амино-
глутаровая к-та)
Лизин (а, е-дианинокап-
роновая к-та)
Аргинин (а-амино-5-гуа-
нидинвалериановая к-та)
Серии (сс-амино-Р-гидро-
ксипропионовая к-та)
Моноаминомонокарбоновые кислоты
01у
Гли
А1а
Ала ***** +1,6 2,39 9,69
Н-
сн3-
_ _ 5,97 24,99
— 6,0 16,51
УаГ
Вал
Ьеи
Лей
Не
Иле
РЬе
Фен
Н8С-СН-
сн
з
+6,6 2,32 9,62 — 6,0 8,85
н3с-сн-сн2-
сн3
Н#С—СНг-СН—
СНз
^>-СН2-
-14,4 2,36 9,60 — 6,0 2,19
+16,3 2,26 9,62
57,0 1,83 9,13
5,9 4,117
3,5 2,965
Туг I |
Тир Н<Г^ -6,6 2,2 9,11 10,07 "5,7
Моноаминодикарбоновые кислоты
Аар ноос-сн -
Асп 2 +6,7 1,88 3,65 9,00 2,8
51и ноос-сн-сн-
Глу 2 2 +17,7 2,19 4,25 9,07 3,2
Диамин омонокарбоновые кислоты
Лиз ИНг-(СН2)4- +19>7 ^20 8>90 1() 8 д^
0,045
0,5
0,8
Аге
Арг
8ег
Сер
Н2Ы-С — Ш
II I .
N11 (СН2)3 +2,8
Гидрооксикислоты
2,18 9,09 13,2 10,9 —
Треонин (а-амино-р-гидро- Тпг
ксимасляная к-та) Тре
НО-СН2-
Н3С-СН-
-7,9 2,21 9,35 — 5,7 5,03
I
ОН
-33,9 2,15 9,12 — 5,6 20,50
30
1
|
2 1
3
Глава 1. Белки
Окончание табл. 1.1
1 4 [ 5 | 6 | 7 | 8 | 9
Тиоаминокислоты
Цистеин (а-амино-Р-тио- Суз но-гк _
пропионовая к-та) Цис ОМ* -20,0 1,71 8,33 10,78 5,0 —
Цистин (ди-ос-амино-р-тио- (Сув)2 _с„ _в «рд _
пропионовая к-та) (Цис)2 2 2 — 1,40 2,01 8,02 5,0 0,011
Метионин (а-амино-р~ МеЪ ртт _о_/гот ч _
метилтиомасляная к-та) Мет 3 22 -14,9 2,28 9,21 — 5,7 3,35
Гетероциклические аминокислоты
Триптофан (а-амино-Э- Тгр ' " "
индолилмасляная к-та) Три ^^^М' -68,8 2,38 9,30 — 5,9 1,14
-СН*-
Гистидин (а-ашгао-р-импа- Н1з
дазолияпропионовая к-та) Гис N^,N11 +59,8 1,78 5,97 8,97 7,6 4,29
Пролин (пирролидин-а- Рго
карбоновая к-та) Про
%
N •-^ООН -99,2 1,99 10,00 — 6,3 162,3
Н
ГИДРОКСИПРОЛИН* Д0
. ~„, г ак>-\ 1
(о-гадроксипирролидин- Нур I [ „
а-карбоновая к-та) Опр ^/-КЮОН _99 6 1&2 д65 _ 58 Зб п
Н_ __
* Иминокислоты; формулы представлены полностью. - ■
В состав белков, синтезируемыми живыми организмами, обычно
входят в разных сочетаниях охарактеризованные нами 20 типов
аминокислот. Однако биохимики из Франкфуртского университета
(Германия) обнаружили в "живом" белке еще одну, 21-ю аминокислоту —
ами нол имонную:
н
I
н2к-с-соон
I
но-с-соон
I
н-с-соон
I
н
Аминолимонная кислота
Эта аминокислота была найдена в белках рибонуклеопротеиновых
комплексов, выделенных из организмов самого различного
систематического положения: из тимуса теленка, из селезенки коровы и
человека, а также из некоторых бактерий, в том числе и кишечной
палочки.
Функция аминолимонной кислоты пока не выяснена, но,
по-видимому, как-то связана со свойством, отличающим эту аминокислоту от
всех других, входящих в состав белков, — с ее весьма высоким
отрицательным зарядом за счет ионизации карбоксильных групп.
31
Часть I. Биомолекулы: структура, свойства, функции
1.7. Полипептидная теория химического строения белков
После того как было выяснено, что аминокислоты являются стро-
ительными блоками белков (мономерами), встал вопрос о том, каким
образом они связаны в молекуле белка.
Представление о наличии в молекуле белка определенных типов
связей между аминокислотами дало изучение биуретовой реакции.
При добавлении слабых растворов сернокислой меди к биурету
КН2-СО-КН-СО-№12 появляется фиолетовое или
красно-фиолетовое окрашивание, обусловленное образованием комплексных
соединений меди с биуретом. Биуретовую реакцию дают все без
исключения белки.
На основании изучения этой реакции в 1888 г. А. Я, Данилевским
высказано предположение, что пептидная (кислотно-амидная) группа
н
—С—N— является основной связью в полипептидном каркасе белко-
II
О
вой молекулы.
Полипептидную теорию строения белка разработал и
экспериментально обосновал Э. Фишер. Работами, проведенными им в 1902—
1919 гг. (Нобелевская премия 1921 г.), доказано, что основным типом
связи в молекуле белка является пептидная связь.
Название пептидов складывается из названия аминокислот,
входящих в состав пептида. Причем те аминокислоты, которые
реагируют активированной карбоксильной группой, меняют окончание в
своем названии на "ил"; так, дипептид, состоящий из глицина и алани-
на, называется глицил-аланин (сокращенно 01у-А1а); трипептид,
содержащий глицин, аланин, лизин, называется глицил-аланил-лизин
(Сг1у-А1а-1л8). Концевая аминокислота, в которой карбоксильная
группа остается незатронутой в пептидах, не меняет своего названия.
Полипептидная теория удачно объясняла многие
физико-химические и биологические свойства белков. Она четко ответила на вопрос,
почему в природе существует огромное количество белков с их
самыми разнообразными физико-химическими свойствами и
биологическими функциями. Возьмем, к примеру, две аминокислоты: глицин и
аланин; из них можно получить дипептид глицил-аланин:
ад-сна-соон + ын^сн-соон -► кн2-сн2-со-кн-сн-соон + н2о
а также другой дипептид аланил-глицин:
СНз-СН-СООН + 1Ш2-СН<гСООН -> СН8-СН-СО-КЙ-СН2-СООН + Н20
ш2 *га2
Следует особо подчеркнуть, что данные изомеры имеют различные
физико-химические свойства, обусловленные последовательностью
аминокислот. Из трех аминокислот (глицина, аланина, лизина) мож-
32
Глава 1. Белки
но получить в результате различных сочетании аминокислот в их
последовательности следующие трипептиды:
1) 01у-А1а-Ыз, 3) А1а-01у-Ыз, 5) 1да-А1а-С1у,
2) 01у-Ыз-А1а, 4) А1а-1ла-С1у, 6) 1лз-С1у-А1а.
Как видно, различные сочетания трех аминокислот (1*2*3 = 6)
дают уже 6 трипептидов, имеющих также различные
физико-химические свойства.
Учитывая, что в химическом строении белков принимают участие
20 типов аминокислот, легко определить, сколько изомеров с
различными свойствами может из них образовываться.
Количество возможных изомеров
Количество аминокислот (20п, где п — число аминокислотных
звеньев в составе белковой молекулы)
2 2
3 б
4 24
5 120
10 3 628 800
15 330 767 438 000
20 243 290 200 817 664 000
Из 20 аминокислот можно получить астрономическое число
изомеров, равное примерно 2,4-1018, и все они имеют различные физико-
химические свойства. Но ведь полипептид, состоящий из 20
аминокислот, является коротким, с молекулярной массой около 2600.
Однако сочетание из 20 аминокислот не является предельным. При
синтезе белка многие из них в молекуле могут многократно
повторяться. В связи с этим возрастает и число полипептидных изомеров,
которое может возникнуть в результате различных сочетаний всех (в том
числе и многократно повторяющихся) аминокислот» Если взять,
например, белок с молекулярной массой 34 000, в котором 12
различных аминокислот представлены в различных соотношениях, то
получится 10300 различных изомеров. Если теперь допустить, что белок
построен из 20 различных аминокислот в разных соотношениях, то
число возможных последовательностей будет намного больше. Если
бы существовало только по одной молекуле каждого из этих
возможных изомеров таких белков, то общая масса всех этих молекул
значительно превышала бы массу Земли.
Эти простые расчеты позволяют дать ответ на вопрос, почему в
живой природе так многочислен и разнообразен класс белков. В
кишечной палочке, например, содержится 3000 различных белков, в
организме человека обнаружено около 5 млн белков, причем ни один
белок кишечной палочки не идентичен белкам человека. Поскольку
на нашей планете известно 1,2 млн видов организмов от бактерий до
человека, можно рассчитывать, что все виды, вместе взятые,
содержат приблизительно от 1010 до 1012 различных белков.
3. Заказ 3486
33
Часть I. Биомолекулы: структура, свойства, функции
Таким образом, 20 белковых аминокислот обладают практически
неограниченными возможностями в синтезе огромного количества
разнообразных белков» Они представляют собой благодатный
строительный материал в эволюционном развитии живой материи на
нашей планете. В то же время наличие в белках самых различных
химических группировок — аминных, карбоксильных, имидазольных,
бензольных, гидроксильных, сульфгидрильных, индольных
обусловливает также огромное разнообразие реакционных возможностей бел-
ковых молекул.
Необходимо отметить и то обстоятельство, что пептидная связь в
молекуле белка проявляет кето-енольную таутомеризацию:
н
I
— С—И— *± —С = N—
II I
О ОН
Кето-форма Енол-форма
Поскольку енольная форма пептидной связи химически более
активна, это еще более увеличивает реакционные возможности белковой
молекулы.
Кроме того, пептидная связь характеризуется следующими
свойствами:
1. Четыре атома пептидной связи лежат в одной плоскости,-т.е.
для пептидной связи характерна компланарность.
2. Атомы О и Н пептидной связи имеют трансориентацию.
3. Длина С-И-связи в пептидной связи, равная 0,13 нм, имеет про-
межуточное значение между длиной двойной ковалентнои связи
(0,12 нм) и одинарной ковалентнои связи (0,15 нм). Следовательно,
связь С-Ы имеет частично характер двойной связи (я-связи), из чего
следует, что вращение вокруг оси С-И затруднено.
Полипептидная цепь с различными функциональными
группировками может быть представлена следующим образом:
Цистеин
Валин ?Н
СНз—СН—СНд ^тт
I
N1*2 СО СН ГШ СО СН ИН СООН
\/ \/ \/ \/ \/ X/ К /
:н ин со сн *ш со сн
I
сн2
Алании . I
<г
СН3
Триптофан
(А1а-УаЬТгр-Суз-Ьеи)
НзС СН3
Лейцин
34
Глава 1. Белки
Аминокислотный остаток, находящийся на том конце цепи, где
имеется свободная аминогруппа, называется аминоконцевым, или
Ы-концевым, остатком, а остаток на другом конце, несущем
свободную карбоксильную группу, — карбоксиконцевым, или С-концевым,
остатком. Название пептида складывается из названия
аминокислот, начиная с 14-концевого остатка.
1,8. Биологические функции белков
Биологическая функция того или иного белка является
результатом интеграции его физико-химических характеристик. Поскольку
20 аминокислот дают огромное разнообразие белков с их различными
физико-химическими свойствами, естественно, и биологические
функции белков самые разнообразные. Та или иная белковая молекула
имеет строго определенный генетически закодированный
аминокислотный состав, специфические физико-химические свойства и строго
определенную специфическую биологическую функцию в живой
клетке, что обусловливает определенное целевое назначение белка и
строжайшую молекулярную экономию в обмене веществ организма.
Специфичность биологических функций белков явилась основой
создания в живой клетке стройной гармонии обмена веществ. Ни один
класс биологически активных веществ не наДелен такими
свойствами, ни одна биологическая функция не протекает без прямого или
косвенного участия белков. Отметим лишь одну функцию в
организме — генетическую, принадлежащую нуклеиновым кислотам,
структура которых приспособлена для хранения и воспроизведения
генетической программы.
Поскольку обмен веществ в организме представляет собой
совокупность огромного числа биохимических реакций и в этих
реакциях, а также в организации различных надмолекулярных структур
принимают участие белки, трудно дать исчерпывающее описание их
биологических функций. Отметим лишь главные из них:
1. Ферментативная (каталитическая) — без нее не протекает ни
одна биохимическая реакция в живой клетке. Ферменты — самый
разнообразный по своей специфике функционирования тип белков*
Синтез и распад веществ, их регуляция, перенос химических групп и
электронов от одного вещества к другому осуществляются с помощью
специфических белков. В настоящее время открыто около 3000
различных ферментов, каждый из которых служит катализатором
определенной химической реакции. На примере ферментов мы
убеждаемся, сколь многосторонни специфические биологические функции
белков и какие большие возможности заключены в модификациях
структурно-функциональных состояний биомолекул.
2. Регуляторная — обеспечение регуляции и интеграция
клеточного обмена веществ. К белкам, участвующим в регуляции клеточной
з*
35
Часть I. Биомолекулы: структура, свойства, функции
или физиологической активности, относятся многие гормоны.
Например, гормон инсулин регулирует углеводный, белковый, жировой
обмен. К регуляторным белкам относятся белки-репрессоры,
координирующие экспрессию генов хромосом и обеспечивающие тем самым
регуляцию биосинтеза необходимых белков,
3. Транспортная — связывание и транспорт веществ между
тканями и через мембраны. К таким белкам относятся липопротеины,
переносящие липиды между тканями, миоглобин — переносящий
кислород в мышечной ткани; гемоглобин — белок, связывающий
кислород воздуха и доставляющий его к периферическим тканям; пермеа-
зы — белки, связывающие глюкозу, аминокислоты и другие вещества
и переносящие их через мембраны внутрь клеток.
4. Механохимическая (сократительная) — функция белков,
обеспечивающая преобразование свободной химической энергии в
механическую работу. Сюда относятся белки мышечной ткани миозин и
актин.
5. Структурная — белки, участвующие в построении различных
мембран (плазматических, митохондриальных). Сюда относятся
также белки, обеспечивающие прочность опорных тканей: коллаген —
структурный элемент опорного каркаса костной ткани, хрящей,
сухожилий; кератин — структурная основа шерсти, волос, перьев, копыт,
рогов; эластин — белок, способный растягиваться в двух измерениях
и обеспечивающий свойства эластической ткани.
6. Защитная — эту функцию выполняют иммуноглобулины
(антитела), образующиеся у позвоночных в лимфоцитах. Они обладают
способностью обезвреживать бактерии, вирусы, чужеродные белки,
попавшие в организм извне. Фибриноген и тромбин — белки,
принимающие участие в свертывании крови и предохраняющие организм от
потери крови при случайных ранениях.
7. Резервная — использование белков как запасных материалов для
питания развивающихся клеток. Сюда относятся про л амины, глютели-
ны — белки хлебных культур; альбумин — яичный белок,
используемый для развития зародыша; казеин молока — белок, используемый для
кормления новорожденных млекопитающих. Резервные белки являются
важнейшими компонентами растительной и животной пищи.
1.9. Роль белков в питании
Среди многочисленных пищевых веществ белки играют наиболее
важную роль. Аминокислотный состав белков не только определяет
их биологическую функцию, но и является важным критерием их
биологической ценности как компонентов пищи. Огромный
экспериментальный материал, полученный при анализе гидролизатов
животных, растительных, микробных белков, свидетельствует о том, что
они имеют самый различный аминокислотный состав. В состав
многих белков входят не все 20 белковых аминокислот. Так, например,
36
Глава 1. Белки
важнейший белок кукурузы — зеин не содержит глицина, лизина,
триптофана; в желатине много глицина, но отсутствуют тирозин и
триптофан; в белках пшеницы — глиадине и глютелине — мало
лизина; протамины, находящиеся в молоках рыб, содержат до 85 %
аргинина, но в них отсутствуют циклические и кислые аминокислоты.
Наиболее часто в белках находят аланин, глицин, лейцин, серии.
Вводимый с пищей чужеродный белок подвергается в процессе
пищеварения ферментативному гидролизу до простых структурных
мономеров — аминокислот. Всасываясь из кишечника в кровь,
аминокислоты поступают в печень, где частично используются для
биосинтеза специфических белков, частично разносятся кровью в органы
и ткани в качестве структурного и иногда энергетического материала.
В организме человека обновление белков крови и печени происходит в
течение 5—7 дней.
Для биосинтеза собственных белков организму человека далеко не
безразличен аминокислотный состав белков пищевых продуктов. Он
является важным критерием их биологической оценки. С этой точки
зрения все белковые аминокислоты можно подразделить на три
группы; незаменимые, полузамеиимые, заменимые.
Незаменимые аминокислоты не могут синтезироваться
организмом человека и животных из других соединений, они обязательно
должны поступать вместе с пищей. Абсолютно незаменимых аминокислот
восемь: валин, лейцин, изолейцин, треонин, метионин, лизин, фёнй-
л аланин, триптофан.
Полузаменимые аминокислоты синтезируются в организме, но не
в достаточном количестве, поэтому частично должны поступать с
пищей. Такими аминокислотами являются аргинин, тирозин, гистидин;
к тому же в организме детей гистидин вообще не синтезируется*
Заменимые аминокислоты синтезируются в организме в
достаточном количестве из незаменимых аминокислот или других
соединений. Организм может обходиться без них долгое время, если, конеч-
I .Г ■ ■ » . ш ш
но, с пищевыми продуктами поступают вещества, из которых эти
аминокислоты могут быть синтезированы. К заменимым аминокислотам
относятся остальные, не названные выше аминокислоты, хотя некото-
рые из них можно отнести к условно заменимым. Так, тирозин
образуется в организме только из фенилаланина и при поступлении
последнего в недостаточном количестве может оказаться незаменимым.
Подобно этому цистеин и цистин могут образовываться из метионина, но
необходимы при недостатке этой аминокислоты.
Таким образом, биологическая ценность белков определяется в них
наличием незаменимых аминокислот и степенью их усвоения. Чем
ближе потребляемый белок по аминокислотному составу подходит к
составу белков данного организма, тем выше его биологическая
ценность* При отсутствии незаменимых аминокислот наряду с
нарушением биосинтеза белка наблюдаются еще и особые расстройства, завися-
37
Часть I, Биомолекулы: структура, свойства, функции
щие от специфического значения данной аминокислоты. Так,
недостаток триптофана вызывает заболевание человека пеллагрой (от итал.
рейе — шершавая, а§га — кожа); метионин, содержащийся в
достаточном количестве в молоке и молочных продуктах, играет большую
роль в обеспечении защитных функций печени, препятствует
накоплению жира и способствует его выведению из печени (предупреждает
жировое перерождение печени).
В зависимости от содержания незаменимых аминокислот белки
условно можно подразделить на полноценные и неполноценные. К
первым относятся белки всех молочных продуктов и животные белки,
ко вторым — растительные белки. Однако, используя различные
сочетания растительных белков, можно восполнить недостаток отдельных
незаменимых аминокислот в одних белках их наличием в других.
1.10. Физико-химические свойства белков
Молекулярная масса белков. В зависимости от длины цепи все
белковые вещества можно подразделить на пептиды (содержащие от
2 до 10 аминокислот), полипептиды (от 10 до 40 аминокислот) и
белки (свыше 40 аминокислот). Если принять среднюю молекулярную
массу аминокислоты за 100, то молекулярная масса пептидов
приближается к 1000, полипептидов — к 4000, а белков от 5 тыс. до
нескольких миллионов. Приведем примеры молекулярных масс некоторых
белков:
Белок Молекулярная масса
Инсулин 6000
Рибонуклеаза 12 700
Гордеин ячменя 27 500
Альбумин яйца 40 000
а-Амилаза ячменного солода 60 000
Уреаза 480 000
Глутаматдегидрогеназа 1 000 000
Существуют различные методы определения молекулярной массы
белков. Одни из них основаны на химическом определении тех
элементов или аминокислот, которые содержатся в белке в минимальном
количестве. Примером такого метода может служить определение
молекулярной массы гемоглобина по содержанию в нем железа. Гемоглобин
млекопитающих содержит 0,34 г железа в 100 г белка. Так как атомная
масса железа равна 56, то 0,34 г соответствует 0,34 ; 56 — 1 ; 165 г-экв.
Следовательно, количество белка на 1 г-атом железа (56 г) равно
165 -100 — 16 500. Эта величина, представляющая молекулярную массу
гемоглобина, долгое время считалась истинной. Физико-химические
методы дают молекулярную массу гемоглобина в растворе 68 000, что
соответствует частице, состоящей из четырех субъединиц.
Аналогичным путем минимальная молекулярная масса белка
может быть рассчитан^ по содержанию аминокислоты, которой в белках
38
Глава 1. Белки
минимальное количество. Другой прием заключается в
количественном определении концевых а-аминогрупп. Например, если 1 г белка
содержит 0,025 • 10~3 ммоль концевых а-аминогрупп, то
минимальная молекулярная масса равна 1 ; (0,025 • 10"3) «* 40 000.
При использовании физико-химических методов определения
молекулярной массы белков результат зависит не только от массы, но и
от электрического заряда и формы молекулы белка, особенно при
измерении скорости диффузии белка, скорости седиментации в
гравитационном поле. В этом случае хорошие результаты получаются только
для молекул, форма которых близка к сферической- Скорость
диффузии и седиментации вытянутых молекул существенно замедляется с
увеличением коэффициента трения; в концентрированных растворах
белков такие молекулы подвержены более интенсивным сцеплениям.
Наконец, при применении динамических методов на результате
сказывается гидратация частиц, обусловливающая увеличение
эффективных размеров молекул белков и замедляющая их движение в
растворителе- В связи с этим при определении молекулярных масс белков
предпочтительнее статические методы, когда белковый раствор
находится в состоянии равновесия, например, путем измерения
осмотического давления или градиента концентрации в гравитационном поле
ультрацентрифуги. Для сведения к нулю межмолекулярных сил
определение молекулярной массы белков можно проводить при
различных концентрациях белка с последующей экстраполяцией
тестируемых параметров к бесконечному разбавлению, т.е. к с -» 0 .
Ниже приводится описание используемых в настоящее время
методов определения молекулярных масс белков.
Осмометрический метод основан на зависимости
где к — осмотическое давление; с — молярная концентрация вещества в
растворе. По Вант-Гоффу, эта зависимость описывается уравнением
те « сКТ, или те = ^КТ,
ц М
где К — газовая постоянная, равная 0,08207 л * ат • град"1; Т —
абсолютная температура; св — весовая концентрация вещества; М — его
молекулярная масса.
Однако пропорциональность между лис наблюдается только для
очень разбавленных растворов и относится к ограниченной области
концентраций. Отклонение от поведения идеального раствора
описывается уравнением
М =
с.КТ
в
я-КсГ
где К — постоянная для данной исследуемой системы, учитывающая
объем, занимаемый самими молекулами белка.
39
Часть I. Биомолекулы: структура, свойства» функции
Уравнение можно преобразовать:
^^-м. л*н+Кв;-
М свКТ св М
При очень низких концентрациях белка с2 становится весьма ма-
лой величиной и членом Кс* можно пренебречь. Если построить
график зависимости я/св от св и экстраполировать кривую к точке св= О,
то отрезок, отсекаемый кривой на оси я, можно рассматривать как
осмотическое давление идеального раствора; эту величину можно
использовать для вычисления М по уравнению Вант-Гоффа.
Метод определения М по л привлекателен с теоретической точки
зрения. Однако возникают трудности в связи с амфотерной природой
белков. Эти трудности можно избежать, если проводить измерения
при значениях рН, соответствующих ИЭТ белка.
Метод седиментации основан на функции V = /(М), где V —
скорость седиментации белка с молекулярной массой М, возникающей
под действием центробежной силы. В этом методе используется
аналитическая ультрацентрифуга с устройством для наблюдения и
фоторегистрации скорости седиментации белков при очень высоких
частотах вращения (20 000—70 000 в мин). При движении молекул белка в
растворителе с постоянной скоростью на нее действуют две взаимно
уравновешенные силы: центробежная Рс и сила трения поверхности
молекулы белка в растворителе Гг* Если б — плотность белка, ар —
плотность растворителя, то центробежная сила будет описываться
уравнением
Кс-М(о2х(1"Р/5),
где М — масса одной молекулы белка; со — угловая скорость ротора
центрифуги, радиан-с"1; х — расстояние молекулы белка от оси
ротора; (1 - р/5) — плавучесть белка; член р/5 обычно заменяют
выражением рУ, где V — средний удельный объем белковых молекул,
находящийся в пределах 0,70—0,75 ем3/г.
Сопротивление трения растворителя описывается уравнением
где V — скорость движения молекулы; I — коэффициент трения,
равный КТД), где К — газовая постоянная, эрг-мольтрад"1; Т —
абсолютная температура; В — коэффициент диффузии (для белков он
колеблется в пределах^Ю6—ДО8 см2-с-1). В случае постоянной скорости
Гс - Гг и Мсо2х(1 - рУ) - V -КТД) и
™ у кт ™ БКТ
М = —т ; =- , ИЛИ М =
йГх В(1-рУ) ОЦ-рУ)
где 8 = у/ю2х — кинематический член, называемый коэффициентом
седиментации; его зцачение для белков лежит в пределах 1-Ю"18—
20'10"13. Обычно коэффициенты седиментации выражают в единицах,
40
Глава 1. Белки
называемых сведбергами (в честь Сведберга — изобретателя
ультрацентрифуги). Для их обозначения применяют символ 8 (1 8 = 1*10~13 с); так,
коэффициент седиментации 5 8 равен 5*10~13 с.
Для получения более точных значений 8 и В проводят измерения
при различных концентрациях белка и экстраполируют результаты к
бесконечному разбавлению.
С помощью этого метода можно получить ценную информацию не
только о молекулярной массе белка, но и о степени его гомогенности,
а также о составе исследуемой смеси, поскольку разные белки
осаждаются с различными скоростями.
Разновидностью определения молекулярных масс белков методом
седиментации является седиментация в градиенте плотности.
Существуют два варианта этого метода. В первом варианте в центрифужной
пробирке создают градиент плотности сахарозы (или другого вещества),
смешивая концентрированный раствор сахарозы с водой в достаточно
уменьшающихся соотношениях. Поверх градиента наслаивается раствор
белка. Константу седиментации белка определяют исходя из положения его
полосы в градиенте по отношению полос "белковых метчиков",
добавленных в ту же пробирку (т.е, белков, константы седиментации которых
известны). Положение отдельных полос определяют либо оптическим
методом, либо путем анализа фракций, отбираемых осторожным сифо-
нированием содержимого пробирки, либо через отверстие, проколотое в
ее дне. Во втором варианте исследуемый раствор замораживают в
пробирке из эластичной пластмассы и нарезают тонким слоем для анализа.
Метод гель-фильтрации (метод "молекулярных сит") основан на
том, что скорость прохождения молекул через колонку, заполненную
гелем, будет прямо пропорциональна его молекулярной массе.
Гранулы геля изготовлены из полимера со сшитой структурой, подобной
ситу, В качестве такого полимерного материала чаще всего
используется сшитый декстран (соединение представляет собой полисахарид)
или сшитый полиакрил амид. В водной среде полимерный материал
сорбирует воду и набухает, превращаясь в гелеподобные гранулы,
сохраняющие пористую структуру, причем размер пор такого
набухшего материала, определяется степенью сшивки полимера. Нанесенные
на колонку соединения (в виде раствора в подвижной фазе) начинают
взаимодействовать с гранулами геля, проникая в объем гранул через
поры (рис. 1.3), что замедляет движение этих соединений по колонке.
Молекулы меньших размеров легче проникают в объем гранул через
поры, что замедляет их движение. Молекулы с размерами,
большими, чем размеры пор, совсем не будут проникать в гранулы и в виде
раствора в подвижной фазе просто пройдут через колонку.
Результаты хроматографического разделения представляются
графически в виде зависимости измеряемого свойства (активность
фермента, поглощение в УФ-области) от объема элюата, вышедшего из
колонки (рис. 1.4). Пики на таком графике соответствуют выходу ин-
41
Часть /. Биомолекулы: структура, свойства, функции
о о о
та5
о
«и»*
&Аэо8
%оо0
ъ°с?
Рис. 1.3. Гель-фильтрация. Гель
действует подобно молекулярному ситу,
разделяя молекулы в зависимости от
молекулярной массы и размера. Матрица
представляет собой множество пористых
частиц, между которыми находится элю-
ент, Еели в верхнюю часть колонки
внести анализируемую смесь, то большие
молекулы не могут войти в поры частиц
геля и будут элюироваться первыми.
Молекулы меньшего размера, имеющие
доступ к порам, задерживаются на
некоторое время в геле и поэтому элюируются
после больших молекул в порядке
уменьшения молекулярной массы и размера
1еМ
1*М,
С6,А
•*Ч^ с
/а| \/в| \/бх] \/с | \
^е(А) Уе(В) У€(ВХ) У^С) V
Рис. 1.4. График калибровки
колонки с гелем. Через колонку пропускают
несколько белков (А, В, С) с известной
молекулярной массой и строят график,
откладывая по оси ординат логарифм
молекулярной массы (1§ М), по оси
абсцисс — величины элюционных
объемов Уе. Элюционные кривые белков А,
В, С строят или по сб (концентрации
белка, определяемой по оптической
плотности при 280 нм), или по А
(биологической активности белка).
Пользуясь графиком и элюционной кривой
белка В, находят экстраполяцией его
молекулярную массу (1&&1)
дивидуальных белков по зонам, отклоненным друг от друга из-за
разных размеров молекул. Каждый компонент разделяемой смеси
описывается объемом элюирования Уе> который равен объему элюата,
собранного до момента» соответствующего максимуму пика. Если при
разделении получается только один пик, то это указывает на чистоту
исходного препарата, нанесенного на колонку.
Неизвестную молекулярную массу белка можно определить, если
предварительно колонка с гелем проградуирована, т.е. на этой
колонке в тех же условиях определены V чистых белков с известной моле-
кулярной массой.
42
Глава 1. Белки
Метод гель-фильтрации прост в исполнении и в то же время
обладает большим преимуществом по сравнению с другими методами* Он
дает возможность определить молекулярную массу белка,
находящегося в смеси с другими белками, если этот белок обладает
биологической, например ферментативной, активностью. Вытяжки из
различных биологических объектов могут содержать большое количество
различных белков; тем не менее по этому методу можно определить
молекулярную массу одного белка, не выделяя его из вытяжки, а просто
пропуская экстракт через колонку и установив положение пика по
биологической активности данного белка. Присутствие других белков
не мешает определению, поскольку все белки движутся по колонке
независимо друг от друга.
1.11. Форма белковых молекул
По форме молекулы белки подразделяются на глобулярные и
фибриллярные. Молекулы первых свернуты в компактные глобулы
сферической или эллипсоидальной формы, молекулы вторых образуют
длинные волокна (фибриллы). К фибриллярным белкам относятся кера7
тин, коллаген, эластин (структурные белки), миозин, актин (белки,
выполняющие механохимические функции), фиброин тутового
шелкопряда, фибриноген крови (защитные белки).
Большинство белков имеют глобулярную форму (ферменты,
гормоны, антитела, запасные белки)- Следует отметить, что форма
глобулярных белков бывает самая разнообразная (эллипсоид вращения, диск,
вытянутая палочка). Судить о форме белковых молекул можно по так
называемому коэффициенту дисимметрии, представляющему собой
отношение большой оси глобулы Ь к малой оси В.
Коэффициент дисимметрии определяется исходя из данных
гидродинамических характеристик белков (например, из данных по вязкости,
двойному лучепреломлению в потоке, скорости седиментации). Однако
данные из гидродинамических характеристик дают весьма
приближенное, ограниченное представление о форме белковой глобулы.
Исчерпывающую информацию можно получить рентгенографическим методом.
Исследования показали, что белковые глобулы имеют весьма
сложную форму, рельеф их поверхности включает выступы, впадины, щели.
Это существенно осложняет определение коэффициента дисимметрии.
В то же время рельеф поверхности белковой глобулы играет важную
роль в формировании биологической функции белка.
Приведем примеры, позволяющие судить о разнообразии форм
белковых глобул:
Белок Коэффициент дисимметрии
Альбумин яйца 3
Уреаза 4
Глиадин пшеницы 11
Зеин кукурузы 20
43
1'
Часть I. Биомолекулы: структура, свойства, функции
Из этих данных следует, что нет резкой границы между
глобулярными и фибриллярными белками. Более того, между этими формами
белковых молекул возможен переход. Так, глобулярный актин при
внесении в раствор ионов К* превращается в фибриллярный, а при
удалении этих ионов актин вновь переходит в глобулярную форму.
1Д2, Способы свертывания и ассоциации полипептидных цепей
По К. Линденштрем-Лангу, можно выделить четыре уровня
структуры белка: первичный, вторичный, третичный, четвертичный. Эти
уровни означают соответственно линейную последовательность
аминокислот, упорядоченное строение главной цепи полипептида,
трехмерную структуру белковой глобулы и структуры белковых
агрегатов. На основании более детальных исследований глобулярных белков
можно добавить еще.два уровня: сверхвторичная структура,
характеризующая энергетически предпочтительные агрегаты вторичной
Л
структуры, и домены — части белковой глобулы, представляющие
собой достаточно обособленные глобулярные области.
Схема структурной организации молекулы белка представлена на
рис. 1.5.
Установлено, что организация вторичной, третичной и высших
структур не требует никакой специальной генетической информации. Она
полностью определяется аминокислотной последовательностью белковых
молекул. Поэтому между всеми уровнями имеется взаимосвязь, так что
структурные элементы более низких уровней определяют элементы
более высоких уровней. Совокупность этих уровней получила название кон-
формации белковой молекулы. Установление конформации белка
является важнейшей проблемой биохимии, основная цель которой — найти
взаимосвязь между структурой и функцией белка.
Перейдем к краткому описанию уровней структуры белковой
молекулы.
Первичная структура — это каркас белковой молекулы,
аминокислотные остатки которой соединены между собой в цепь пептидными
связями. Для каждого индивидуального белка последовательность
аминокислот в полипептидной цепи является уникальной. Она определяется
генетически и в свою
очередь определяет бо-
+ ♦ %. лее высокие уровни
Н}"1 1 г > Глобула белка организации данного
Домен белка. Замена даже од-
Сверхвторич ная структура НОГО аМИНОКИСЛОТНОГО
Вторичная структура остатка полипептидной
;^оошхдооооооосооосо& Первичная структура
**■■"** цепи, состоящей из со-
Рис. 1.5. Уровни структурной организации глобу- тен аминокислот,
молярных белков жет весьма существен-
44
Г лае а 1. Белки
но изменить свойства белка и даже полностью лишить его биологической
активности. Пептидная связь по своей химической природе является
ковалентной и придает высокую прочность первичной структуре
белковой молекулы. Являясь повторяющимся элементом полипептидной цепи
и имея специфические особенности структуры, пептидная связь влияет
не только на форму первичной структуры, но и на высшие уровни
организации полипептидной цепи (рис. 1.6).
В результате того, что пептидная связь может существовать в кето-
енольнои форме, вращение вокруг С-Ы-связи является запретным и
все атомы, входящие в пептидную группу, имеют планарную
трансконфигурацию. Дцс-конфигурация является энергетически менее
выгодной и встречается лишь в некоторых циклических пептидах.
В составе полипептидной цепи жесткие структурные элементы
(плоские пептидные группы) чередуются с относительно подвижными
участками (-СНК), которые способны вращаться вокруг связей.
Каждая пептидная группа может образовывать по две водородные
связи с другими группами, в том числе и пептидными. Исключением
являются пептидные группы, образованные с участием аминокислот
пролина или гидроксипролина, которые способны образовывать толь-
ко одну водородную связь. Полипептидная цепь на участке, где
находится пролин или гидроксипролин, легко изгибается, так как не
удерживается, как обычно, второй водородной связью.
Такие особенности строения пептидной группы влияют на укладку
полипептидной цепи в пространстве и прежде всего на формирование
вторичной структуры белка.
Вторичная структура
белков представляет собой способ
укладки полипептидной цепи в
упорядоченную форму за счет
системы водородных связей.
Различают две формы
вторичной структуры: спиральную,
возникающую в пределах одной
полипептидной цепи, и слоисто-
складчатую — между
смежными полипептидными цепями.
Хотя спиральная структура
в полипептидных цепях белков
обнаружена в виде отдельных
участков, она придает
молекуле белка достаточно высокую
прочность, обусловливает в ней
как ближний, так и дальний
порядок сил, участвующих в
создании водородных связей.
45
Ш! 110°
а
Рис. 1.6. Пептидная связь: а — струк
тура; б — фрагмент пептидной цепи
Часть I. Биомолекулы: структура, свойства, функции
На основании молекулярно-физических представлений и данных
рентгеноструктурного анализа о геометрических размерах атомов и
групп Л. Полингу и Р. Кори удалось точно предсказать структуру
спирали, названной ими а-спиралью (рис. 1,7).
а-Спираль учитывает все свойства пептидной связи, ее
конфигурация имеет винтовую симметрию. Витки спирали регулярны; все
аминокислотные остатки в остове спирали равнозначны независимо от
строения их боковых радикалов К, причем последние не участвуют в
образовании а-спирали. По Л. Полингу и Р. Кори, в одном витке
а-спирали находится 3,6 аминокислотных остатка, т.е. период
идентичности вдоль оси составляет 18 остатков или 5 витков; шаг одного
витка спирали — 0,54 нм, т.е. каждый аминокислотный остаток вдоль
оси занимает 0,54 : 3,6 = 0,15 нм, что и позволяет говорить о
равнозначности всех аминокислотных остатков в а-спирали. Диаметр ци-
Рис. 1,7. Модель а-спирали Полинга—Кори: --- — водородные связи в
структуре а-спирали
46
Глава 1. Белки
линдрической поверхности, на которой расположены все а-углерод-
ные атомы, равен 1 нм; длина одного периода 0,15 • 18 — 2,7 нм.
Спираль может быть описана последовательностью
Н5+* О6-
^-[СО-СН(К)-Ш2]3-С-
с 13 атомами в кольце (К — остатки аминокислот), где О—Н —
водородная связь.
Каждая пептидная группа образует водородную связь с четвертой
от нее по счету пептидной группой.
а-Спираль Полинга и Кори обеспечивает наименьшее напряжение
связей, минимальные размеры незанятого пространства вблизи оси и
минимальные размеры витка спирали. Предсказанная Полингом и Кори
а-спираль впервые была обнаружена в кристаллическом гемоглобине,
а позднее — почти во всех глобулярных белках.
Слоисто-складчатая структура известна также под названием
Р-структуры, или р-складчатого листа. Она имеет слегка изогнутую
конфигурацию у р-углеродного атома полипептидной цепи и
формируется с помощью межцепочечных водородных связей (рис. 1.8).
[5-Складчатые листы могут быть образованы параллельными
(К-концы направлены в одну сторону) и антипараллельными (Ы-концы
направлены в разные стороны) полипептидными цепями. Возможны
смешанные параллельные и антипараллельные р-складчатые листы.
Если смотреть вдоль полипептидного остова, видно, что боковые
аминокислотные радикалы ориентированы поочередно то в одну, то в
другую стороны средней плоскости, причем связи Са~Ср приблизительно
перпендикулярны плоскости.
/
н^
НСЕ
/
ос
\
ЙН"
ксн
со-
/
1Ш
\
НСЕ
/
ос
\
ын-
/
ЕСН
\
СО"
/
\
со-
ксн
\
ЫН"
•ос
НСЕ
-ны
\
СО"
/
ЕСН
\
ин-
/
•••ОС
\
НСЕ
/
••ни
\
/
-ны
ИСК
/
••ОС
\
нн-
ЕСН
СО-
/
.•ны
\
НСЕ
/
"ОС
\
*ш«
/
ЕСН
\
СО"
/
\
г
ЕСН
\
Г
•ос
НСЕ
-НЫ
\
СО
/
ЕСН
\
ин
/
•••ОС
\
НСЕ
/
••ны
\
Рис. 1.8. Модель Р-структуры полипептидной цепи
47
Часть I. Биомолекулы: структура, свойства, функции
Складчатые структуры наблюдаются очень часто. Они были
обнаружены во многих структурных белках (коллагене, кератине,
фиброине шелка). У трипсина, рибонуклеазы значительная часть
полипептидной цепи укладывается в слоистые {5-структуры. У
глобулярных белков эти структуры составляют 15 % и более. Возможны
переходы от ос-структур к {3-структурам и обратно вследствие
перестройки водородных связей между вытянутыми фрагментами
полипептидной цепи.
Совокупность а-спиралей и (5-структур является важным
критерием, по которому можно судить о степени упорядоченности структуры
белковой молекулы, стабильности белков при действии
физико-химических факторов сред.
Сверхвторичная структура (суперспираль) — это ансамбли
взаимодействующих между собой вторичных структур. Возникновение этих
ансамблей указывает на то, что они предпочтительны с точки зрения
либо кинетики процесса свертывания, либо выигрыша свободной
энергии в уже свернутом белке.
Суперспирализованная а-спираль встречается в фибриллярных
белках. На рис. 1.9 показана двухцепочечная а-спираль. Полипептидные
цепи скручены относительно друг друга, образуя суперспираль; обе
ос-спирали параллельны. Суперспира-
лизованные а-спирали обнаружены в
а-кератине, тропомиозине, парамиози-
не. Короткие участки такой
сверхвторичной структуры встречаются в гло-
Рис. 1.9. Скрученная двухцепо- булярных белках, содержащих а-спи-
чечная а-спираль рали, упакованные приблизительно
параллельно или антипараллельно.
Структурные домены довольно четко разграничены при рентгено-
структурном анализе на картах электронной плотности. Установлено,
что многие белки состоят из нескольких глобулярных областей,
довольно слабо связанных между собой. Четко определенные домены
были обнаружены в иммуноглобулинах, в химотрипсине. Часто доме-
ны расположены вдоль полипептидной цепи как жемчужины в
ожерелье. В них включены близкие по цепи аминокислотные остатки, что
дает основание рассматривать домены как участки полипептидной цепи,
которые свертываются независимо друг от друга. Вообще под
доменами принято понимать компактные автономные субобласти в составе
белка, характеризующиеся минимальным отношением поверхности к
объему, а также тем, что число функциональных связей в составе
домена значительно превышает таковое по сравнению с соседними
доменами. Как правило, домены выполняют определенные функции, в связи
с чем их называют функциональными доменами. Они часто состоят из
нескольких структурных доменов, представляющих собой
геометрически обособленные субобласти.
48
Глава 1. Белки
Третичная структура белковой молекулы — это специфическая
укладка упорядоченных (регулярных) и неупорядоченных (аморфных)
участков полипептидной цепи в компактное тело — глобулу
(рис. 1.10, а). В третичной структуре формируются биологические
функции белка.
Какова взаимосвязь между вторичной и третичной структурами
белковой молекулы? Вторичная структура нарушается в точках, где
находятся пролин и гидроксипролин, в них полипептидная цепь
изгибается в трехмерном пространстве. Третичная структура не
может существовать при разрушенной вторичной структуре,
деформации доменов. Между этими уровнями существуют тонко
сбалансированные взаимоотношения. Однако биологическая функция белка
зависит целиком от состояния третичной структуры. Ее
деформация или разрушение приводят к частичной или полной потере этой
функции.
Четвертичная структура белка представляет собой ассоциацию
нескольких полипептидных цепей с определенной конформацией в
стабильный агрегат, не имеющий ковалентных связей (рис. 1Д0, б).
Такой агрегат яляется структурной единицей со специфической
биохимической функцией: составляющие ее полипептидные цепи
называются субъединицами. Так, ое-амилаза ВасШиз зиЫШз состоит из 2
субъединиц, гемоглобин — из 4 субъединиц. Установлено, что в аминоки-
а
слотнои
последовательности каждой
субъединицы
содержится информация не
только о конформа-
ции самой
субъединицы, но и о
специфических
взаимодействиях этих
субъединиц, приводящих к
образованию устойчи- „ , ,. _ „ , л „ . _,ч
г * Рис. 1.10. Схема третичной (а) и четвертичной (б)
вых агрегатов. структур белка
1.13» Природа и типы связей,
участвующих в формировании структуры белка
В формировании рассмотренных выше уровней структурнбй
организации белков принимают участие самые различные типы связей и
взаимодействий (рис, 1.11).
Первичная структура представляет собой совокупность ковалент
пых (пептидных) связей. Они образуют энергетический остов
молекулы белка и действуют вдоль пептидной цепи. Ковалентные связи
характеризуются наибольшей электронной плотностью между двумя
связывающими атомами, поэтому энергия связи велика, порядка 140—
4. Заказ 3486 49
Часть I. Биомолекулы: структура, свойства» функции
400 кДж*моль~Ч Связывающие атомы находятся на весьма близком
расстоянии, обычно ОД—0,2 нм.
В формировании вторичной структуры белка принимают участие
водородные связи* По своей природе эти связи являются
электростатическими. Водородная связь возникает между
электроотрицательными атомами и осуществляется через атом водорода. Такой тип
связи атом водорода образует значительно легче, чем любой другой атом,
так как его малые размеры позволяют ему беспрепятственно
приближаться к взаимодействующим электроотрицательным атомам.
Энергия водородной связи невелика: в сухих белках АН для ОН—N
или О—НИ колеблется от 15 до 40 кДжмоль"1, т.е. она в 10—60 раз
слабее, чем ковалентная связь; к тому же энергия водородной связи
сильно уменьшается в полярных средах. В водных растворах она
колеблется в пределах 6—12 кДж-моль"1. Однако в формировании
вторичной структуры белка принимает участие довольно большое
количество водородных связей; между ними возникает так называемый
кооперативный эффект действия, заключающийся в том, что
суммарный эффект их совместного действия значительно выше суммы
эффектов действия водородных связей порознь. Таким образом, водо-
Водородные связи
спиральной структуры
Электростатическое
притяжение между
противоположно
заряженными
ионными В-группами
Водородные связи
конформации листа
Водородные
связи между
боковыми
цепями К
Гидрофобные
взаимодействия внутри
кластера» образованного
неполярными К-группами
Дисульфидная
связь
Рис. 1.11. Типы связей и взаимодействий, участвующих в формировании тре
тичвой структуры белковой молекулы
50
Глава 1. Белки
родные связи обеспечивают достаточную стабильность и
упорядоченность в структуре белковой молекулы.
I Третичная структура определяется свойствами боковых К-радика-
лов аминокислот и поэтому здесь принимают участие разные по своей
природе типы связей и взаимодействий (ковалентные,
электростатические, ван-дер-ваальсовы и гидрофобные взаимодействия).
К ковал ентным связям нужно отнести дисульфидные 8-8-связи. Они
обусловливают изменение направления полипептидной цепи, скрепляют
ее отдельные участки. Эти связи возникают между двумя молекулами
цистеина, находящимися на различных участках цепи, за счет отрыва
водорода от сульфгидрильных групп. Хотя под действием дисульфидных
связей и уменьшается степень упорядоченности вторичной структуры,
наличие этих связей укрепляет структуру оставшихся а-спиралей и р-
складок и делает ее более стабильной к воздействию таких
физико-химических факторов среды, как температура и рН.
К типам связей, носящих электростатический характер, относятся
ионные связи (солевые мостики). Солевой мостик СОО"*"КНд близок по
своей природе к водородной связи. Он возникает между К- и С-концевыми
аминокислотами, а также между карбоксильными группами кислых
аминокислот и аминогруппами основных аминокислот, которые в
растворе, как правило, находятся в ионизированном состоянии.
Ионные связи являются наиболее дальнодействующими. В водной
среде, имеющей высокую диэлектрическую проницаемость, эти связи
могут быть, подобно водородным связям, сильно ослаблены из-за
электростатического взаимодействия с диполями воды. Ориентировочно
АН ионной связи равна 40 Дж-моль"1. Однако электростатические
взаимодействия многих поликатионных и полианионных участков могут
создать довольно сильную стабилизацию третичной структуры,
особенно "обезвоженных" участков, вследствие скопления гидрофобных
К-остатков аминокислот.
В создании и стабилизации третичной структуры нередко важную
роль играют катионы металлов. В качестве оригинального примера
можно привести фермент а-амилазу, в стабилизации конформации
которой принимают участие ионы кальция. Связывание катионом
отдельных участков полипептидной цепи может быть также довольно
прочным, если мостик, образованный между металлом и группами,
специфически реагирующими с ним, находится в глубинных слоях
белка и, следовательно, экранирован от молекул воды.
В формировании третичной структуры белка принимают участие и
водородные связи. Они возникают, например, между ги дрок сильной
группой серина и имидазольным кольцом гистидина, между гидро-
ксильной группой тирозина и заряженной карбоксильной группой
аспарагиновой или глутаминовой аминокислот.
И, наконец, в формировании третичной структуры белковой
молекулы важную роль играют ван-дер-ваальсовы силы. Они ответственны
51
Часть I. Биомолекулы: структура, свойства, функции
за окончательное свертывание полипептидной цепи. В зависимости от
характера взаимодействия они подразделяются на индуцированные и
дисперсионные.
Индуцированные силы возникают в результате взаимодействий,
имеющих место при наведении диполя в какой-либо группировке
полем постоянного заряда другой группировки, при взаимодействии ионов
с индуцированным диполем или постоянного диполя с
индуцированным диполем.
Дисперсионные взаимодействия возникают между гидрофобными
неполярными К-радикалами аминокислот (лейцин, изолейцин, валин).
Энергия взаимопритяжения ван-дер- ваальсовых сил весьма невелика
и обратно пропорциональна шестой степени расстояния между
атомами; для атома Н она равна 0,075, для атома О — 0,21, для атома С — 1,8,
для атома N — 0,56 кДж-моль"1. Однако в молекуле белка число
взаимодействующих атомов велико и дисперсионные силы вносят
существенный вклад в формирование и стабилизацию третичной
структуры белка.
В тесной взаимосвязи с ван-дер-ваальсовыми силами связи
находятся гидрофобные взаимодействия.
При укладке в глобулу гибкая полипептидная цепь будет стремиться
принять более энергетически выгодную форму. В результате
образования между диполями воды водородных связей неполярные К-группы
будут вытесняться из сферы действия водной среды и контактировать
друг с другом. В свою очередь, этот контакт будет способствовать
развитию ван-дер-ваальсовых сил, В результате таких взаимодействий
водной среды и полипептидной цепи неполярные К-группы образуют
внутреннюю часть белковой глобулы (гидрофобное ядро), а ионные и
полярные К-группы располагаются снаружи этого ядра.
Хотя энергия гидрофобного взаимодействия невелика и
колеблется в пределах 1—6 кДж-моль"1, надо иметь в виду, что неполярные
группы в белках составляют от трети до половины всех остатков. Вполне
очевидно, что выигрыш свободной энергии при образовании
гидрофобного ядра может быть вполне достаточным, чтобы
стабилизировать белковую глобулу.
Итак, формирование структуры белковой молекулы,
определяющее ее биологические функции, представляет собой сложный процесс.
В нем принимает участие достаточно большое число групп,
разнообразных по своим физико-химическим свойствам, возникают
различные типы связей и взаимодействий, тесно связанных между собой. В
конечном счете в молекуле белка устанавливается тонкое равновесие
сил, соответствующее минимуму свободной энергии. В то же время
необходимо отметить, что поскольку аминокислотный состав белков
весьма разнообразен, то, безусловно, все типы связей и
взаимодействий у различных белков будут выражены по-разному: у одних в
совокупность взаимодействий основной вклад будут вносить электро-
52
Глава 1. Белки
статические силы, у других — гидрофобные, у третьих — ковалент-
ные связи. Каждый конкретный белок будет иметь свой особый путь
формирования структуры, который и предопределяет специфические
особенности его молекулы при воздействии на нее
физико-химических факторов среды.
Те же самые типы связей и взаимодействий, ответственных за
третичную структуру, принимают участие в формировании четвертичной
структуры белков.
1Л4* Электрохимические свойства белков
Амфотерная природа белков. Белки являются амфотерными
полиэлектролитами- Поскольку большая часть ионных и полярных К-групп
находится на поверхности белковой глобулы, то именно они
определяют амфотерные свойства и заряд белковой молекулы. Кислые
свойства белку придают аспарагиновая и глутаминовая аминокислоты,
диссоциация их карбоксильных групп является источником
отрицательных электрических зарядов на поверхности белковой молекулы*
Основные свойства белку придают лизин, аргинин, гистидин,
способные к протонированию и созданию на поверхности белковой
молекулы положительных зарядов. Определенный вклад в амфотерную
природу белковой молекулы вносят ее К- и С-концевые аминокислоты.
Слабая диссоциация 8Н~групп цистеина и ОН-групп тирозина весьма
несущественно влияет на амфотерность белков. В целом чем больше
кислых аминокислот содержится в белке, тем сильнее выражены его
кислотные свойства, тем выше суммарная плотность отрицательного
заряда. Чем больше основных аминокислот, тем ярче проявляются
основные свойства белка и выше плотность положительных зарядов в
его молекуле. Следует отметить, что величина рК К-радикалов
аминокислот колеблется в довольно широких пределах (табл. 1.2).
Широкий диапазон колебаний значений рК функциональных
групп связан с их топологией и взаимным индуктивным действием.
Рядом расположенные кислые и основные группы будут
способствовать снижению величины рК кислой группы, возрастанию рК
основной группы.
Таблица 1.2
Значения рК функциональных групп в белках
Функциональные группы
а-СООН
у-СООН
Имидазольная группа
8Н-группа цистеина
ОН-группа тирозина
се-НН2-группа
е-ЫН2-группа лизина
РК
1,5—3,0
2,0—4,0
5,5—7,5
8,0—9,0
9,0—10,0
10,0—11,0
11,0-12,5
53
Часть /. Биомолекулы: структура, свойства, функции
Амфотерная природа белков обусловливает определенную
буферную емкость их растворов.
Суммарный заряд белковой молекулы определяется соотношением
в ней кислых и основных радикалов К аминокислот, их величин рК и
рН среды. Если в белке кислые аминокислоты преобладают над
основными, то в целом молекула белка электроотрицательна, т*е. она
находится в форме полианиона, и наоборот, если преобладают основные
аминокислоты, — в форме поликатиона.
Влияние рН среды на электрохимические свойства белков. Амфо-
терный характер белков особенно ярко проявляется при изменении
величины рН белкового раствора. В кислой среде идет подавление
кислотной диссоциации карбоксильных групп и интенсивное прото-
нирование N112-, N11-, имидазольной групп: суммарный заряд
белковой молекулы будет положительным. В щелочной среде при избытке
ОНг-ионов будет наблюдаться обратная картина: интенсивная
диссоциация карбоксильных групп и депротонирование основных групп —
суммарный заряд белковой молекулы будет отрицательным. Подобно
аминокислотам, каждый белок может находиться в
газоэлектрическом состоянии, величина рН, обусловливающая это состояние, будет
являться изоэлектрической точкой (ИЭТ, р1) белка. В этой точке
белок не обладает подвижностью в электрическом поле, так как его
электрический заряд будет приблизительно равен нулю; белок имеет
наименьшую растворимость в воде; раствор белка обладает минимумом
устойчивости, минимумом осмотического давления.
ИЭТ каждого белка определяется соотношением кислых и основных
групп, величиной их рК: чем выше это соотношение и ниже величины
рК групп, тем ниже ИЭТ белка. У кислых белков ИЭТ ниже 7, у
нейтральных — около 7, а у основных — больше 7; при рН ниже ИЭТ белок
будет находиться в форме поликатиона, при рН выше ИЭТ — в форме
полианиона, в ИЭТ — в форме амфотерного полииона. ИЭТ большинства
белков животных, растений, микроорганизмов лежит в пределах 5,5—
6,0, а внутриклеточная величина рН находится при 7,0—7,2
(физиологическое значение рН). Следовательно, клеточные белки имеют в общем
отрицательный заряд, который уравновешивается неорганическими
катионами.
Электрофорез белков. Так как каждый белок в водных или
буферных растворах имеет свой определенной величины суммарный заряд,
то это свойство белков нашло широкое применение при их разделении
методом электрофореза. Электрофорез основан на передвижении
заряженной частицы в электрическом поле. Движение ее происходит в
жидкой среде, которая удерживается инертным твердым носителем,
например, полоской бумаги, гелевой пленкой из крахмала, полиакрил-
амида, декстрана, ацетата целлюлозы.
При постоянном приложении напряжения движение заряженной
молекулы белка определяется отношением заряда к ее размеру:
54
Глава 1. Белки
ц = ад/г),
где <3 — суммарный заряд белковой молекулы; г — радиус молекулы.
С увеличением этого соотношения подвижность молекулы растет.
Поскольку каждый белок имеет свою определенную величину <3/г,
скорость перемещения различных белков в электрическом поле будет
различной. Электрофорез белков нашел широкое применение для
определения их молекулярных масс.
1.15. Гидрофильность белков
Гидрофильность белков, т.е. способность белков связывать на
своей контактной поверхности воду, — одно из характерных
физико-химических свойств белков, имеющее исключительно большое значение
в организации биологических систем. С гидрофильностью связаны
такие процессы, как набухание и растворимость белков, их осаждение
и денатурация. Рассмотрим эти свойства более подробно.
Набухаемость и растворимость белков. Гидрофильность
представляет собой следствие действия электростатических сил притяжения,
развивающихся между ионными и полярными группами белковой
глобулы и диполями воды. Выше уже было отмечено, что белковая
глобула в принципе представляет собой структуру, состоящую из двух
зон: внутреннего гидрофобного ядра и внешней оболочки,
содержащей в основном ионные и полярные группы.
Гидратация ионных групп белка обусловлена ориентацией диполь-
ных молекул воды в электрическом поле иона, а гидратация
полярных групп белка — ориентацией молекул воды в результате
взаимодействия диполей и образования водородных связей.
В результате действия электростатических сил поверхность
белковой глобулы покрывается гидратной оболочкой. Первый слой
молекулы воды (мономолекулярный слой) довольно прочно адсорбирован на
поверхности, последующие слои гидратной оболочки, по мере того как
электростатические силы ослабевают, становятся все менее и менее
упорядоченными. Возникает ситуация, характерной особенностью
которой является медленный переход от твердой фазы (белковой
глобулы) к дисперсионной среде (воде); белковая глобула как бы
закреплена на "якорях", роль которых выполняют диполи воды. Поскольку
диэлектрическая проницаемость воды достаточно высокая, взаимосвязь
между белковой глобулой и водной средой достаточно стабильна.
При контакте сухого белка с водой он набухает, молекулы воды
проникают в белковую массу, гидратируют молекулы белка, разъединяя их.
Важную роль здесь играют не только электростатические силы, но и
силы осмоса. Дальнейшее поглощение воды приводит к растворению белка.
Поскольку аминокислотный состав белков различен, растворимость
белков колеблется в широких пределах и будет определяться
соотношением гидрофильных (ионных и полярных) и гидрофобных (непо-
55
Часть I. Биомолекулы: структура, свойства, функции
лярных) К-групп, спецификой их укладки в трехмерную структуру:
чем больше гидрофильных К-групп находится на поверхности
белковой глобулы, тем выше ее гидрофильность и растворимость белка;
поверхностные гидрофобные К-группы снижают растворимость.
Глобулярные белки обладают лучшей растворимостью, чем
фибриллярные. При достаточно большом количестве поверхностных
гидрофобных К-групп полное растворение белка не происходит и завершается
лишь ограниченным набуханием.
Явление набухания белков широко распространено в пищевой
технологии: оно, например, играет ведущую роль в образовании
пшеничного теста с присущими ему свойствами. Набухшие белки муки глиа-
дин и глютенин известны под названием клейковины. Они образуют в
тесте трехмерную губчато-сетчатую структурную основу,
обусловливают упругость, растяжимость, эластичность, пластичность теста.
Качество клейковины определяет в основном качество хлебобулочного
изделия (его вкус, форму, структуру мякиша).
Водные растворы белков, в сущности, можно отнести к истинным
молекулярным растворам. Однако высокая молекулярная масса
белков придает растворам коллоидный характер. Молекулы белков не
способны диффундировать через полупроницаемые мембраны — кол-
лодиевую пленку, обезжиренный рыбий пузырь, целлофан. На этом
явлении основаны очистка белков от низкомолекулярных примесей
методом диализа, очистка и концентрирование белков методом улът-
рафилътрации. При диализе целлофановый мешочек с раствором
белка помещают в сосуд с проточной водой. Внешние стенки мешочка
омываются водой. Низкомолекулярные вещества диффундируют
через мембрану и удаляются вместе с водой* При ультрафильтрации
мембрана действует как молекулярный фильтр.
Характерными признаками коллоидного характера белковых
растворов являются их опалесценция, блеск и способность рассеивать лучи
света (эффект Тиндаля).
Если через кювету с раствором низкомолекулярного вещества,
например ЫаС1, пропустить пучок света, то в кювете он не будет
обнаружен, раствор является "оптически пустым". Иная картина
наблюдается в кювете с раствором белка: при ее боковом освещении появляется
светящийся конус. Объясняется это вполне просто. Радиус ионов Ыа+
и С1" гораздо меньше длины волны видимого света и световые волны
будут "обтекать" эти ионы, не меняя своего первоначального
направления. В случае прохождения света через раствор, содержащий
белковые глобулы, радиус которых намного превышает длину волны света,
имеет место дифракция света: падая на белковую глобулу, свет будет
отражаться в различных направлениях (рассеиваться под
различными углами).
Светорассеивающая способность белков может быть использована
при определении концентрации белковых растворов методом нефело-
56
Глава 1. Белки
метрии, основанном на сравнении интенсивности светорассеивания
этих растворов и стандартного золя.
Согласно закону Рэлея, интенсивность светорассеяния
представляет собой функцию в четвертой степени радиуса рассеивающей
частицы, что может быть использовано для ориентировочного расчета
молекулярной массы белка по радиусу его глобулы:
М - 4/371г3рЫ,
где М — молекулярная масса белка; г — эквивалентный радиус
белковой глобулы; р — плотность белка; N = 6,024023 — число Авогадро.
Высокая молекулярная масса белков обусловливает их высокую
вязкость, причем особую роль играет форма белковой молекулы.
Растворы фибриллярных белков более вязкие, чем растворы глобулярных
белков (при приблизительно равных молекулярных массах).
Повышение концентрации белка, снижение температуры его
раствора, внесение в раствор соответствующих электролитов способствуют
резкому возрастанию межмолекулярных сил притяжения (сцепления)
и к резкому увеличению вязкости белкового раствора; раствор теряет
свою текучесть; происходит образование сеток (трехмерных каркасов),
внутри которых находятся захваченные (иммобилизованные)
молекулы воды. Система переходит в состояние геля (студня).
Студни используются в процессе приготовления всевозможных
заливных блюд. Производство кисломолочных продуктов (сметаны,
простокваши, кефира, кумыса), по сути говоря, представляет собой
перевод казеина молока из молекулярного состояния в состояние геля.
Желатина — продукт неполного гидролиза коллагена — широко
используется в пищевой промышленности для структурирования
пищевых продуктов, в микробиологических исследованиях при
приготовлении твердых питательных сред. Достаточно в эту среду внести 1 —
2 % желатины, чтобы превратить ее в состояние геля.
1.16. Осаждение белков
Гидрофильность белков является важнейшим фактором
устойчивости их растворов. Если в такой раствор внести какую-либо нейтральную
соль [(ЫН)2804, Ка2804, ЫаС1], то белки выпадают в осадок. Механизм
осаждения состоит в том, что анионы и катионы вносимой в раствор
белка соли, имеющие достаточно большие электростатические заряды,
интенсивно гидратируются диполями воды. Между гидрофильной
поверхностью белковой молекулы и ионами соли развивается конкуренция за
обладание молекулами воды. Более мощные электростатические силы
соли снимают гидратную оболочку с поверхности белковой молекулы.
Лишенные гидратных оболочек белковые молекулы при броуновском
движении сталкиваются между собой, агрегируются и, в конечном
счете, выпадают в осадок (рис. 1.12). При внесении нейтральной соли
снижается также диэлектрическая проницаемость воды.
57
Часть I. Биомолекулы: структура, свойства, функции
Рис. 1.12. Дегидратация и высаливание белков из водных растворов: / —
гидратированная белковая молекула; 2 — диполи воды; Зу 4 — ионы
нейтральной соли; 5 — агрегат белка; 6 — гидратированные ионы
Сильной гидратационной способностью обладают также некоторые
органические растворители (этанол, ацетон, пропанол, изопропанол); они
также осаждают белки из водных растворов. Осаждение белков является
обратимым процессом. Оно нашло широкое применение при их
фракционировании. В основу этой операции положены различия в размерах гид-
ратной оболочки: чем плотнее электрические заряды на поверхности
белковой молекулы, тем выше гидрофильность поверхностного слоя
молекулы, тем больше потребуется нейтральной соли для осаждения данного
белка* Таким образом, внося в раствор, содержащий смесь различных
белков, нейтральную соль порционно, но все в более возрастающем
количестве, белки можно разделить на отдельные фракции. На эффект
фракционирования существенное влияние оказывает величина рН: она
должна быть близка к изоэлектрической точке белка.
Выделение и фракционирование белков органическими
растворителями применяют или как самостоятельную операцию, или при
более глубокой очистке исследуемого белка после фракционирования
солью. Поскольку многие белки при контакте с органическими
растворителями подвергаются денатурации (см. ниже),
фракционирование необходимо проводить при низких температурах, близких к
температуре замерзания смеси растворителя с водой.
1.17. Денатурация белков
Нативная (естественная) конформация глобулярного белка,
обладая высокой лабильностью, подвержена глубоким изменениям под
58
Глава 1. Белки
действием жестких физико-химических факторов среды. Эти
изменения получили название денатурации белка. Денатурация является
следствием нарушения уникальной конформации (вторичной,
третичной, четвертичной структур) нативного белка с сохранением его
первичной структуры. Типичным примером денатурации является
свертывание яичного альбумина при варке яиц. При денатурации белок
изменяет свои физические, химические свойства и теряет
биологическую функцию. Полипептидная цепь при этом развертывается и
превращается в беспорядочный, хаотический клубок (рис. 1.13).
При денатурации белок из гидрофильного состояния переходит в
гидрофобное, в котором его молекулы могут оставаться в растворе
только при наличии какого-либо стабилизирующего фактора, например,
электрического заряда. В противном случае при броуновском
движении молекулы белка, сталкиваясь, агрегируются и в виде хлопьев
выпадают в осадок. При денатурации наблюдается изменение
оптической активности белков, увеличение реактивности химических групп,
ранее экранированных внутри глобулы; денатурированные белки дают
более интенсивные цветные реакции. На изменение расположения
пептидных цепей в ходе денатурации указывает увеличение двойного
лучепреломления в потоке, повышение вязкости; изоэлектрическая
точка денатурированного белка сдвигается в сторону более высоких
значений рН. В изоэлектрической точке денатурация протекает
медленнее и ускоряется в кислых и щелочных растворах.
Факторы, вызывающие денатурацию белка, можно подразделить
на физические и химические. К физическим факторам относятся
нагревание, механическое перемешивание, ультразвук,
ультрафиолетовое и ионизирующее излучения; к химическим — кислоты, щелочи,
соли тяжелых металлов, мочевина, таннин, ди- и трихлоруксусная
кислота, гуанидинхлорид, гуанидинсульфат, лаурилсульфат и др.
Тепловая денатурация является одним из характерных признаков
белков. Однако для различных белков она различна. Многие белки,
особенно животного происхождения, термолабильны: процесс их
денатурация начинается уже при 40 °С и быстро ускоряется с
повышением температуры. Однако
известны белки,
устойчивые к нагреванию,
например, ТРИПСИН," ХИМО- Ц /2^75РШЫ. Денатурация
трипсин, некоторые
белки мембраны. Особенно ^Г^йё^5^^ Ренатурация
устойчивы белки
термофильных
микроорганизмов. Следует отметить,
что компактно свернутые
пептидные цепи нативно- Рис }13 Денатурация й ренатурация моле-
го белка не разворачива- кулы белка
59
Часть I. Биомолекулы: структура, свойства, функции _
Л
ются до тех пор, пока в пространство между цепями не попадет вода.
Поэтому сухие белки (например, гемоглобин» яичный альбумин) более
устойчивы к тепловой денатурации, чем белки в растворе. По той же
причине большей устойчивостью обладают концентрированные
белковые растворы по сравнению с разбавленными.
Некоторые химические соединения оказывают на белки защитное
действие. Так, денатурация тормозится концентрированными
растворами глицерина, глюкозы и других сахаров, что связано, видимо, с их
адсорбцией на глобулах белков и образованием крупных
гидрофильных комплексов*
Процесс денатурации белков играет большую роль в технологии
пищевых продуктов при их тепловой обработке. При выпечке
хлебобулочных изделий примерно при 60—70 °С белки теста (клейковина)
денатурируют; этот процесс имеет большое значение в формировании
мякиша изделия. Обработка свекловичной стружки водой при
температуре 70—80 °С, осуществляемая при получении диффузионного сока,
вызывает денатурацию белков мембран и протоплазмы клеток
свекловичной стружки и резко повышает скорость диффузии сахарозы из
нее. Блашдировка плодов и овощей перед их консервированием
вызывает денатурацию (инактивацию) окислительных ферментов, ведет к
сохранению витамина С. Тепловая обработка пищевых продуктов
повышает их вкусовые качества, а также биологическую ценность,
поскольку Денатурированные белки обладают лучшей атакуемостью
пищеварительными ферментами.
1.18. Исследование структуры белка. Цель, подходы и методы
Исследование структуры белков является одним из актуальных
вопросов современной биохимии. Основная цель исследований —
установить взаимосвязь между структурой и функцией белка* Решение этой
задачи на современном этапе развития физики и химии белка
представляет определенные трудности. Чем выше молекулярная масса
белка, чем больше степень упорядоченности структуры, тем больше
осложнений в ее расшифровке* Пока удалось полностью установить кон-
формацию для немногих белков, молекулярная масса которых не
превышает 50 000 (инсулин, рибонуклеаза, лизоцим, химотрипсин, мио-
глобин, гемоглобин). По этим белкам имеются данные, позволяющие
найти взаимосвязь между структурой и функцией: четвертичная
структура гемоглобина и ее роль в транспорте кислорода, третичная
структура лизоцима, химотрипсина и ее роль в формировании активных
центров данных ферментов и обеспечении самого акта катализа.
В вопросе изучения конформации белков решающую роль играют
физико-химические методы, причем наиболее ценная информация
может быть получена в случае их комплексного использования.
Такой подход дает возможность получить либо взаимодополняющую
60
Глава 1. Белки
информацию, либо независимые экспериментальные критерии,
позволяющие развить более объективные представления о конформации
белковых молекул.
Выделение белков. В исследовании конформации белка
обязательным требованием является получение белка высокой степени
чистоты* Объект локализации исследуемого белка (орган, ткань, клетки)
содержат десятки, сотни других сопутствующих белков.
Предварительная подготовка биологического материала заключается в
механическом измельчении, гомогенизации взятого объекта, что позволяет
повысить эффективность извлечения искомого белка из объекта.
Белок переводят в растворимое состояние путем экстракции водой,
буферными растворами солей и детергентов, иногда неполярными
растворителями* Затем часто следует фракционное осаждение
нейтральными солями, обычно (>Ш4)2804, или органическими
растворителями. Важным условием является определение оптимальных значений
рН, ионной силы, температуры. Величина рН должна соответствовать
ИЭТ выделяемого белка. Для предотвращения его денатурации, как
правило, создают низкую ионную силу раствора, осаждение проводят
при пониженной температуре (около 4 °С), Следующей после
высаливания стадией очистки, как правило, является освобождение белка от
низкомолекулярных примесей; раствор подвергают диализу
(электродиализу), гель-фильтрации, ультрафильтрации.
Дальнейшую более тонкую очистку проводят по схемам,
специально разработанным для отдельных белков с учетом их
физико-химических и биологических свойств. Широко распространенными
методами являются кристаллизация и перекристаллизация исследуемого
белка, адсорбционная и ионообменная хроматография,
гель-фильтрация. Особенно эффективные методы — жидкостная хроматография
высокого разрешения и аффинная хроматография. Нет возможности
дать более подробное описание каждого метода, используемого для
глубокой очистки белка. Отметим лишь, что в основе ионообменной
хроматографии лежит различие в кислотно-основных свойствах
белков, адсорбционной хроматографии — различие белков в сродстве к
неполярному адсорбенту, гель-хроматографии — различие в
молекулярной массе белков, аффинной (лигандной) хроматографии —
специфическое связывание белков с лигандами. Амфотерная природа
белков, различие в знаке и величине заряда у них широко используются
для разделения белков электрофорезом в различных
поддерживающих средах (крахмал, агар, полиакриламид, ацетат целлюлозы, дек-
стран). Различие в ИЭТ применяется для разделения белков методом
ИЭТ-фокусирования в градиенте рН,
Степень очистки белка проверяют по так называемой его удельной
активности (его специфической биологической функции), т.е* по
активности, приходящейся на мкг, мг, г белка. Критерием
гомогенности идентифицированного белка является неизменяющаяся удельная
61
Часть I. Биомолекулы: структура, свойства, функции ^
активность при попытках дальнейшей очистки, одна полоса или
единственный пик при ультрацентрифугировании, электрофорезе,
хроматографии, Одноцепочечный белок должен быть гомогенным при 1М"-,
С-концевом анализе. Примеси сопутствующих белков определяют по
их специфической биологической функции.
Методы и приемы исследования первичной структуры белка.
Расшифровка первичной структуры белка — основа для получения
предварительной информации о более высоких уровнях структуры (вторичной,
третичной, четвертичной), для выяснения топографии функциональных
групп белка и построения модели его функционирования.
При определении аминокислотной последовательности белка прежде
всего разделяют его полипептидные цепи (если макромолекула
состоит из нескольких цепей), затем определяют аминокислотный состав
цепей, N4 С-концевые аминокислотные остатки, аминокислотные
последовательности. Полипептидные цепи подвергаются
специфическому расщеплению протеолитическими ферментами или химическими
реагентами. Смесь образовавшихся фрагментов разделяют и для
каждого из них определяют аминокислотный состав и аминокислотную
последовательность. При необходимости крупные фрагменты
дополнительно расщепляют каким-либо способом на более мелкие. Порядок
расположения фрагментов выясняют путем расщепления молекулы
белка по другим связям и анализа образующихся при этом
"перекрывающихся" фрагментов.
Анализ аминокислотного состава включает полный гидролиз
исследуемого белка или пептида и количественное определение всех
аминокислот в гидролизате. Для гидролиза обычно используют
5,7 ]Ч-раствор соляной кислоты, а при анализе содержания
триптофана, неустойчивого в кислой среде, — раствор щелочи; количественное
определение аминокислот в гидролизате проводят с помощью
аминокислотного анализатора. В большинстве таких приборов смесь
аминокислот разделяют на ионообменных колонках, детекцию
аминокислот осуществляют спектрофотометрически с использованием флуорес-
камина или о-фталевого диальдегида.
Наибольшее распространение для определения Ы-концевых
аминокислотных остатков находит дансильный метод. Его первая стадия —
присоединение дансилхлорида (1-диметиламинонафталин~5-сульфо-
хлорида) к непротонированной а*аминогруппе с образованием дансил-
пептида (Д НС-пептид а). Затем последний гидролизуют 5,7 И-раствором
соляной кислоты при 105 °С> в результате чего освобождается И-кон-
цевая а-ДНС-аминокислота, которая обладает интенсивной
флуоресценцией в УФ-области спектра*
Для определения С-концевых аминокислотных остатков чаще
всего используют гидролиз карбоксипептидазой, которая специфически
расщепляет пептидные связи, образованные С-концевыми остатками.
Поскольку после отщепления первой С-концевой аминокислоты фер-
62
_ Глава 1. Белки
мент атакует последующие пептидные связи, измерение скорости
отщепления отдельных аминокислот позволяет анализировать также и
С-концевую аминокислотную последовательность*
Среди ферментативных методов расщепления полипептидной
цепи на фрагменты широко используется трипсин, который гидро-
лизует связи, образованные карбоксильными группами основных
аминокислот •— лизина, аргинина; химотрипсин, который гидроли-
зует пептидные связи, образованные карбоксильными группами
ароматических аминокислот — тирозина, фенилаланина и
триптофана. С меньшей скоростью гидролизуются связи лейцина, метио-
нина, гистидина.
При выборе методов разделения пептидов используют
ионообменную хроматографию, электрофорез на бумаге или пластинках с
тонким слоем целлюлозы или силикагеля, жидкостную хроматографию
высокого разрешения*
При изучении последовательности аминокислот осуществляют их
химическое отщепление по методу П. Эдмана (с применением фенил-
изотиоцианата). Этот метод позволяет последовательно отщеплять
1Ч-концевые аминокислоты в виде фенилтиогидантоинов, которые
поглощают свет в УФ-области с максимумом поглощения при 265—•
270 нм. Идентификацию фенилтиогидантоинов осуществляют с
помощью тонкослойной хроматографии, жидкостной хроматографии
высокого разрешения, а также масс-спектроскопии; используют также
прием, сочетающий метод Эдмана с анализом И-концевых остатков
аминокислот в виде их ДНС-производных.
Для непосредственного анализа первичной структуры белков
используют секвенатор — прибор, осуществляющий последовательное
автоматическое отщепление Ы-концевых остатков аминокислот по
методу П. Эдмана.
Определение аминокислотного состава, 1Ч-, С-концевых
аминокислот, их последовательностей в пептидах позволяет расположить
аминокислоты в той последовательности, в которой они были в исходной
молекуле белка. Особенно важно найти те участки, в которых
пептиды второго частичного гидролиза (химотрипсином) перекрывают
пептиды первого частичного гидролиза (трипсином).
Методы и приемы исследования пространственной структуры
(конформации) белков. При исследовании вторичной, третичной и
четвертичной структур применяют два принципиальных подхода:
исследование в растворе и в кристаллическом состоянии.
Основным методом, позволяющим получить непосредственную
информацию о пространственном расположении атомов в молекуле
белка, является рентгеноструктурный кристаллографический анализ.
Метод основан на дифракции рентгеновских лучей электронами,
окружающими ядра атомов в кристалле белка, поскольку длина волны
рентгеновского луча соизмерима с межатомными расстояниями в кри-
63
Часть I, Биомолекулы: структура, свойства, функции
сталле белка (рис. 1.14). На фотопленке, помещенной за кристаллом,
рентгеновское излучение дает дифракционную картину, характерную
для третичной, вторичной структур белка. С использованием
информации, полученной с помощью рентгеновской диффракционной
картины подходящих белковых кристаллов, возможно создание карт
электронной плотности, отражающих расположение в пространстве атомов
в белках. Имея такие карты и зная аминокислотную
последовательность, можно определить трехмерную структуру белка,
Рентгеноструктурный анализ фибриллярных белков был впервые
предпринят в 1930-х гг. Выявление повторяющихся структурных
единиц в фибриллярных белках привело к предположению о
существовании сс-спирального и р-складчатого типов вторичной структуры
белков. Однако определение трехмерной структуры глобулярной
белковой молекулы, включающей тысячи атомов, было связано с
большими техническими затруднениями. Основной среди них была трудность
расчета атомных позиций, исходя из углов отклонения и интенсивно-
стей дифрагированных рентгеновских лучей. Не удивительно, что
развитие рентгеноструктурного кристаллографического анализа тесно
взаимосвязано с развитием компьютерной техники. В 1959 г. ученые
из лаборатории Дж. Кендрью получили кристаллы миоглобина,
подходящие для рентгеноструктурного кристаллографического анализа.
Дж. Кендрью в конечном итоге выяснил структуру этого
глобулярного белка с разрешением 0,2 нм. Определение структуры миоглобина
Дж. Кендрью и несколькими
годами позже гемоглобина его
коллегой — М. Перутцем обеспечило
первые представления о природе
трехмерной структуры глобулярных
белков. Труды этих ученых были
г
вознаграждены Нобелевской
премией в 1962 г. С тех пор с
помощью рентгеноструктурного
кристаллографического анализа была
определена структура многих
белков. Хотя анализ рентгеновских
дифракционных картин был
усовершенствован благодаря
техническим достижениям, включая
использование высокоскоростных
компьютеров, определение
структуры белков ограничивается
трудностью получения кристаллов
подходящего качества.
Рентгеноструктурный анализ применим только
для хорошо кристаллизующихся
Источник
рентгеновских
лучей
V
Пучок
рентгеновских
лучей
Кристалл белка
у^ч Дифрагированные
\ \л>ентгеновские лучи
Фотопленка
Рис. 1.14. Схема
рентгеноструктурного кристаллографического анализа.
Однонаправленный рентгеновский
поток падает на белковый кристалл,
который вызывает дифракцию лучей
64
Глава 1. Белки
белков. Однако кристаллизация белков — весьма трудоемкая задача.
Далеко не каждый белок удается получить в кристаллическом
состоянии. Теория кристаллизации белков с их трехмерной структурой,
дающей хотя бы в общих чертах представления о закономерностях этого
процесса, отсутствует. Нужны определенная интуиция, сочетание
малоэффективных операций. Необходимое условие кристаллизации —
сохранение нативной конформации белка, которая часто реализуется
лишь в условиях, приближенных к физиологическим. При этом
наряду с кристаллами нативного белка необходимо получать производные,
содержащие тяжелые атомы, которые давали бы кристаллические
структуры, подобные исходному белку. Введение атомов металлов в
виде изоморфных заместителей увеличивает разрешающую способность
рентгеновских установок.
В кристаллах макромолекул имеют место большие пространства
между соседними молекулами; эти пространства заполнены
растворителем (так называемым материнским ликвором). Присутствие
растворителя в кристаллах лимитирует разрешающую способность
дифракции. Однако высокое содержание растворителя дает и определенные
преимущества. Первое заключается в том, что нативная конформация
белка сохраняется в кристаллах; второе что кристаллы могут быть
насыщены материнским ликвором, содержащим лиганды, такие как
аналоги субстрата или ингибиторы, благодаря чему могут быть
определены структуры комплексов белка с данными лягандами.
Во многих случаях хорошие результаты получают, применяя
нейтронографию* Нейтроны имеют низкую энергию, в отличие от
рентгеновских лучей не разрушают кристаллы белка, в результате чего можно
получить полный набор дифракционных данных от одного кристалла,
С использованием этого метода удается обнаружить в структуре
белковой молекулы отдельные атомы водорода.
Однако с помощью этого метода можно исследовать лишь
кристаллы белков, в то время как сами белки обычно проявляют свои
функции в водной среде. В кристаллах белков рентгенографический метод
способен дать информацию только о статическом состоянии молекулы
белка. В живой клетке или вообще в любых биологических средах эта
молекула представляет собой динамическую систему, непрерывно
изменяющую в определенных пределах свою конформацию при
выполнении биологической функции. Но основная задача изучения
пространственной структуры белка — это определение биологической роли
тех или иных конформационных состояний (превращений) белковой
молекулы.
В отличие от рентгеноструктурной кристаллографии,
ядерно-магнитная резонансная (ЯМР) спектроскопия позволяет изучать белки в
растворах и, следовательно, не требует трудоемкого получения
кристаллов. ЯМР спектроскопия, основанная на абсорбции
электромагнитного излучения молекул в магнитных полях, позволяет определить
5, Заказ 3486
65
Часть I. Биомолекулы: структура, свойства, функции
" "" "~Г '1'1 ' " "■ I ' ' |1Т*~ |"и~[- г г 1~|— - " -| . I | I
состояние спинов определенных атомных ядер. При изучении
белковой структуры с помощью ЯМР-спектроскопии обычно определяют
спины водородных атомов. Специализированная ЯМР техника
позволяет регистрировать взаимодействия между водородными атомами,
близко расположенными друг к другу. С помощью этих результатов и
зная аминокислотную последовательность белка, можно определить
его конформацию.
Как рентгеноструктурная кристаллография, так и ЯМР
спектроскопия требуют больших количеств очищенного белка, и
высокоразрешающий структурный анализ белка может потребовать многие годы.
Когда трехмерные координаты атомов в макромолекулах определены,
то они обычно отправляются в банк данных, где доступны для других
исследователей. Такие банки данных обеспечивают помощь при
моделировании трехмерной структуры белков, которые гомологичны
белкам, трехмерная структура которых известна в деталях.
Трехмерная структура белков может быть изображена
несколькими способами* Каждое изображение имеет свои преимущества, но
стереоскопические виды молекул значительно улучшают представление
трехмерных параметров. Ленточные модели глобулярных белков
(рис. 1.15, а) позволяют дать трехмерное изображение извивающейся
полипептидной цепи. Ленточные модели наглядны для объяснения
действия сил, стабилизирующих белок. Расширенный вид,
показывающий индивидуальные атомы, изображает области активного центра
белков (рис. 1.15, б). Детальные модели этого типа могут быть
использованы для установления связи между структурой и химическим
механизмом функционирования белка. Пространственная модель
(рис. 1.15, в) отражает плотную, сильно упакованную структуру
глобулярных белков. Глобулярные белки действительно очень
компактны; участки полипептидных цепей свиты вместе таким образом, что
внутренняя область белка практически непроницаема даже для таких
небольших молекул, как вода.
Одной из интересных физико-химических характеристик белков
является их способность вращать плоскость поляризации света,
падающего на их образцы. Величина этого вращения представляет
собой сумму двух слагаемых: а) оптической активности, присущей
асимметрическому углероду остатков аминокислот (конфигурационной
оптической активности); б) оптической активности, обусловленной
расположением этих остатков относительно друг друга, т.е.
упорядоченности вторичной структуры (конформационной оптической
активности).
Поскольку конфигурационная оптическая активность мало зависит
от молекулярной массы белков, основную роль в изменении величины
угла вращения играет конформация полипептидной цепи. Конформаци-
онная оптическая активность изменяется с длиной световой волны. Это
изменение получило название дисперсии оптического вращения (ДОВ).
66
Исследование ДОВ — один из
наиболее важных методов изучения кон-
формации белков в их водных
растворах.
Измерение угла вращения плос-
кополяризованного света проводят на
спектрополяриметре. Сущность
метода заключается в следующем:
полипептидная цепь, находясь в виде
беспорядочного клубка, вращает
плоскость поляризации света влево;
ос-спираль и (3-структура — вправо.
Чем больше степень
упорядоченности, т.е. больше а- и (3-структур в
белке, тем больше правое вращение. С
помощью метода ДОВ были
обнаружены определенные корреляции
между характером первичной
структуры полипептида и типом его
вторичной структуры. В ряде
случаев обнаружена связь между
изменением степени
упорядоченности пространственной структуры
белков и их биологическими
функциями. Однако и метод ДОВ имеет
ограниченность. Это связано с тем,
что во многих белках а- и р-струк-
туры отнюдь не являются
доминирующими, основной вклад в
формирование пространственной
структуры этих белков вносят ван-дер-
ваальсовы силы, гидрофобные
взаимодействия.
Совокупность
экспериментальных данных по рентгеноструктурно-
му анализу и методу ДОВ может дать
более исчерпывающую информацию
о структуре и функциях белка.
В исследованиях вторичной
структуры используется также
ультрафиолетовая спектрофотомет-
рия. Метод основан на гипохром-
ном эффекте, т.е. понижении
поглощения света при длине волны
190 нм белком, имеющим ос-спира-
Глава 2 , Белки
в
Рис, 1.15. Структура белковой
молекулы сШТРазы из ЕвсНепсШа соИ: а —
ленточная модель; б — расширенный вид
активного центра; в — пространственная
модель (Ларссон Г. и др., 1996)
5*
67
Часть I. Биомолекулы: структура, свойства, функции
ли и р-структуры по сравнению с белком с разрушенными этими
структурами. По наличию гипохромного эффекта можно судить о степени
спирализации белка: чем он выраженнее, тем выше спирализация
белковой молекулы.
При исследовании доменов в молекуле белка рентгенографический
анализ является наиболее достоверным методом. С успехом может быть
использована и электронная микроскопия, В этом случае для
повышения контрастности (электронной плотности) белков применяют
специальные покрытия, компоненты которых адсорбируются
поверхностью белка. При облучении таких белков пучком электронов
происходит их поглощение контрастирующими веществами (на пленке они
темные), которые покрывают домены. Применяют также
ограниченный протеолиз, который проводят в мягких, не денатурирующих
условиях, с расщеплением пептидных связей между доменами. Таким
образом, получают информацию о доменной структуре белка*
Изложенные выше подходы в исследовании структуры белков хотя
и весьма трудоемки в экспериментальном исполнении, но все же
позволяют получить достаточно глубокую, исчерпывающую
информацию об их структуре и найти взаимосвязь между ней и биологической
функцией белка*
1.19. Классификация белков
В связи с огромным разнообразием белков, различием их
физических, химических свойств и биологических функций вопрос о
классификации и номенклатуре белков разработан далеко не полностью;
точнее, строго научная классификация белков пока отсутствует.
Систематизация и номенклатура белков, за исключением белков,
обладающих ферментативной активностью, не существуют. Название
белков дается по случайным признакам, учитывая преимущественно
источник выделения или какие-либо особенности химического
состава, конфигурации молекул.
В настоящее время наиболее удачной следует считать
классификацию по структурным признакам с определенным сочетанием харак-
терных физико-химических свойств белков. На основании такого
подхода класс белковых веществ подразделяется на две большие группы:
протеины (простые белки), в состав которых входят только остатки
аминокислот, и сложные белки — соединение простого белка с каким-
либо веществом небелковой природы — простетической группой
(например, гемом, углеводом, нуклеиновой кислотой и т.д.). Протеины и
сложные белки в свою очередь делятся на ряд подгрупп.
Протеины. К простым белкам относятся альбумины, глобулины,
проламины, глютелины, гистоны, протамины, протеноиды. В основу
классификации протеинов положены их растворимость в
специфическом растворителе и некоторые химические признаки (основность, кис-
68
_^ Глаба 1' Белки
лотность). По своим функциям протеины весьма разнообразны.
Некоторые из них играют роль запасных белков и служат питательными
веществами для растущих тканей; большая группа этих белков наделена
биологической активностью и включает в себя ферменты, гормоны,
антитела. Протеины — важнейшие компоненты пищевых продуктов.
Альбумины и глобулины. В растительных и животных клетках
всегда встречаются совместно, но отличаются по степени
растворимости. Альбумины — водорастворимые белки с высокой гидрофильное-
тью, выпадают из раствора при полном (100 %-ном) насыщении
сульфатом аммония. Имеют относительно небольшую молекулярную
массу (15 000 — 70 000), обладают кислыми свойствами (ИЭТ около 4,7)
из-за большого содержания глутаминовой кислоты.
Типичным представителем подгруппы альбуминов является белок
куриного яйца — овалъбумин, составляющий половину белков
куриного яйца. Легко кристаллизуется, содержит все незаменимые
аминокислоты.
В молочной сыворотке содержится лактальбумин; он получен в
кристаллическом виде, молекулярная масса 16 300. При нагревании
белка до 100 °С в течение 10 мин он сохраняет нативное состояние.
Небольшое количество альбуминов находится в зеленых частях
растений. В зародышах пшеничного зерна содержится лейкозин> в
семенах гороха — легумелин. Растительные альбумины представляют
собой смеси ряда белков, отличающихся друг от друга по способности
осаждаться при различных концентрациях нейтральной соли.
Глобулины не растворимы в дистиллированной воде, растворимы в
слабых солевых растворах. При извлечении глобулинов из различных
объектов обычно используют теплый 2—10 %-ный раствор хлорида
натрия. Это слабогидратированные белки, поэтому они осаждаются
уже из полунасыщенных растворов сульфата аммония. Отделение
глобулинов от альбуминов может быть проведено путем диализа
белкового раствора. По мере снижения концентрации соли в растворе
глобулины выпадают в осадок, а альбумины остаются в растворе. Многие
глобулины получены в кристаллическом состоянии.
Глобулины — слабокислые или нейтральные белки (ИЭТ лежит в
интервале рН 6,0—7,3), содержат меньше, чем альбумины, кислых
аминокислот. При электрофорезе происходит довольно четко
разделение этих белков, поскольку они обладают разной подвижностью в
электрическом поле. Глобулины сыворотки крови в электрическом поле
распределяются на фракции: а-, (3-, у-глобулины; фракция у-глобули-
нов представлена смесью различных иммуноглобулинов с
константами седиментации б 8 и 9,5 8.
Глобулины составляют большую часть белков многих семян,
особенно бобовых растений, масличных культур. Так, в семенах гороха и
чечевицы содержится легумин; фасоли — фазеолин; конопли — эдес-
тин; соевых бобов — глицинии.
69
Часть I. Биомолекулы: структура, свойства, функции ______
Жмых из подсолнечника, хлопчатника, льна, остающийся после
извлечения из семян масла, состоит в основном из глобулинов.
Проламины и глютелины — (запасные резервные) белки,
содержащиеся исключительно в растениях, особенно в семенах злаков
(пшеница, рожь, ячмень). Проламины растворимы в 60—70 %-ном этаноле.
При гидролизе проламины образуют большое количество пролина
(отсюда и название "проламины") и глутаминовой кислоты. Большим
содержанием пролина (неполярной аминокислоты) можно, видимо, объяснить
растворимость проламинов в этаноле. В проламинах мало лизина -—
незаменимой аминокислоты, на что обращено особое внимание в
селекционных работах на высоколизиновые сорта пшеницы. Наиболее
характерный проламин, содержащийся в эндосперме зерна пшеницы, — глиадин;
из зерна ячменя выделен гордеин; из овса — авенин.
Проламины не являются гомогенными белками и представляют
собой смеси различных белков со сходными свойствами, но различной
молекулярной массой. Глиадин был разделен на две фракции с ИЭТ
при рН 5 и 7 соответственно.
Глютелины находятся, как правило, совместно с проламинами, не
растворяются ни в солевых растворах, ни в спирте, но экстрагируются
из размолотого зерна гидроокисями щелочных металлов; особенно легко
переходят в раствор при действии 0,2 %-ных растворов щелочей. Они
также не гомогенные белки, а представляют собой смеси разных
белков со сходными свойствами.
Хороню исследованными глютелинами являются глютенин
пшеницы, орезенин риса.
В технологии хлебопечения при переработке пшеничной муки особо
важную роль играет комплекс двух белков — глютенина и глиадина, —
известный под названием клейковины. Клейковина, придающая тесту
характерную консистенцию, играет важную роль в формировании
качества хлеба. Она может быть выделена из муки в виде клейкой массы
путем постепенного отмывания зерен крахмала, отрубенистых частиц и
основной массы водорастворимых веществ муки из кусочка теста.
В зависимости от сорта пшеницы, почвенно-климатических и
агротехнических условий ее возделывания общее количество белка
может колебаться в пределах 10—25 %, две трети которых приходится
на долю глиадина и глютенина. Следует отметить, что в зародыше
семени пшеницы и клетках алейроновского слоя (слоя, прилегающего
к семенной оболочке) белка больше, чем в эндосперме; однако они не
способствуют образованию клейковины. Глиадин и глютенин
сосредоточены в эндосперме. Чем больше в процессе мукомолья удалено
зародышей и оболочек (отрубей), т.е. чем выше сортность муки, тем
больше в ней содержится глиадина и глютенина.
И все же чем больше в зерне (муке) белков, тем выше количество
отмываемой клейковины. Поэтому селекционеры, специалисты
мукомольной и хлебопекарной промышленности считают содержание бел-
70
Г лава 1. Белки
ков в зерне пшеницы одним из основных показателей хлебопекарных
достоинств муки*
Протамины и гистоны. Протамины — низкомолекулярные белки
(молекулярная масса 4000—12 000). Они обладают ярко
выраженными основными свойствами из-за большого содержания аргинина (до
двух третей от общего аминокислотного состава). ИЭТ протаминов
лежит в щелочной зоне рН (10,5—13,5), в клетках животных
организмов — это поликатионные белки. Помимо аргинина протамины
содержат небольшое количество моноаминокислот и пролина, в них
отсутствуют цистеин и цистин, а также незаменимые аминокислоты: мети-
онин, триптофан, фенил ал анин.
При нейтрализации их гидрохлоридов или сульфатов эквивалентом
щелочи образуется плотный маслянистый слой свободных протаминов .
В больших количествах протамины обнаружены в молоках и икре рыб.
Наиболее подробно изучены клупеин сельди, салъмин лососевых рыб.
Гистоны — тканевые белки (от греч. Ыз1о$ — ткань) животных
организмов с несколько меньшей основностью, чем протамины; ИЭТ
колеблется в пределах 8,5—11,0. Из растворов их осаждают
прибавлением аммиака* Молекулярная масса этих белков несколько выше,
чем у протаминов (11 000 — 24 000).
Гистоны находятся в ядрах клеток, содержат 20—30 % аргинина,
лизина, гистидина; в них мало триптофана или он вообще
отсутствует, не обнаружены цистеин и цистин. Много гистонов содержится в
зобной железе, эритроцитах; выделены из растительных тканей.
Основная функция гистонов — структурная и регуляторная.
Структурная функция состоит в том, что гистоны участвуют в стабилизации
пространственной структуры ДНК. Регуляторная функция
заключается в способности блокировать передачу генетической информации
от ДНК к РНК.
Протеноиды — подгруппа фибриллярных белков, содержащихся
в опорных тканях животных. Не растворимы ни в воде, ни в солевых
растворах, ни в разбавленных кислотах и щелочах, В силу
специфической структурной организации они плохо поддаются гидролизу про-
теолитическими (пищеварительными) ферментами.
Наиболее известными протеноидами являются коллаген
соединительной ткани, кератин волос, шерсти, перьев, копыт, эластин сухо-
жилий и связок, фиброин шелка.
Коллаген относится к самым распространенным белкам организма
животных (около одной трети массы всех белков). Он содержится в
тканях, обладающих высокой прочностью и малой растяжимостью
(кожа, хрящи, сухожилия, кости, зубы).
В коллагене треть аминокислотных остатков приходится на
глицин, а около четверти — на пролин и гидроксипролин. Из всех
незаменимых аминокислот коллаген не содержит триптофан, в малом
количестве имеет метионин, отсутствуют цистин и цистеин.
71
Часть I. Биомолекулы: структура, свойства, функции
Необычайным свойством молекулы коллагена является
разветвление пептидной цепи, обусловленное остатками лизина, образующими
точки ветвления на участках е-аминогрупп лизина. В соединительных
и опорных тканях коллаген находится в виде волокон, каждое
диаметром 20 мк.
В коллагеновом волокне полипептидные цели идут
преимущественно в продольном направлении параллельно или антипараллельно друг
другу.
Кератины (от греч. кегаз —- рог) являются характерными протено-
идами волос, шерсти, рогов, перьев, эпидермиса. Кератины
синтезируются в клетках эпидермиса. Высокая стабильность и
нерастворимость кератина обусловлены большим числом дисульфидных связей
между пептидными цепями. Кератины содержат большое количество
цистиновых остатков (5—12 %). Кроме цистина, кератины содержат в
достаточно большом количестве кислые и основные аминокислоты; в
небольших количествах в его состав входят все незаменимые
аминокислоты.
Подобно коллагену, молекулы кератина образуют комплексы. Длина
волокон зависит от содержания в них молекул воды. Увеличение
длины влажного волоса используется в гигрометре для определения
влажности воздуха. Кератиновые волокна под действием механических сил
растягиваются, обладают эластичностью.
Эластин — волокнистый структурный белок. Подобно коллагену,
он содержит много глицина и пролина, однако более устойчив к
действию протеолитических ферментов, кислот и оснований. Это,
видимо, связано с высоким содержанием неполярных аминокислотных
остатков. Эластин гидролизуется специфическим ферментом эластазой,
обнаруженной в поджелудочной железе и в некоторых бактериях.
Сложные белки. В зависимости от природы простетической группы
они делятся на гликопротеины, липопротеины, хромопротеины, фосфо-
протеины, металлопротеины, нуклеопротеины. Фактически многие из
этих белков представляют собой комплексы смешанных макромолекул.
Отметим лишь, что наряду с большими различиями указанных
протеинов у них есть общее свойство, а именно стабилизация белкового
компонента простетической группой. Видимо, большие простетические
группы протеинов занимают значительную часть поверхности глобулы
белка, препятствуя развертыванию полипептидных цепей и поддерживая
их нативную конформацикх Отрыв простетической группы, даже если
он осуществляется в мягких условиях, приводит к понижению
стабильности белкового компонента, а часто к его денатурации.
Краткое содержание главы
Белки — высокомолекулярные азотистые органические
соединения, состоящие на 99 % и более из химических элементов -— углеро-
72
Глава 1, Белки
да, кислорода, азота и водорода. Эти элементы способны к образованию
ковалентных связей, обусловливают прочность белковых структур, а
углерод с его способностью к созданию конъюгированных соединений
является основой возникновения в белке каталитических функций,
играющих важнейшую роль в обмене веществ живой клетки.
Структурными единицами (блоками) служат аминокислоты. В
состав белков входят 20 типов аминокислот, носящих название
белковых аминокислот. Эти аминокислоты построены по одному и тому же
принципу, относятся к а-аминокислотам, химические группы в
которых — радикалы — связаны с а-углеродным атомом. Радикалы
имеют различное химическое строение, являются "буквами"
молекулярного алфавита белков, их генетическими детерминантами.
Аминокислоты — амфотерные электролиты, каждая из них имеет
характерную изоэлектрическую точку. На электрохимических свойствах
аминокислот основано их разделение методами электрофореза,
ионообменной хроматографии, электрофокусирования. В результате наличия в
аминокислотах а-аминогруппы они обладают высокой реакционной
способностью. Специфические реакции аминокислот нашли широкое
применение в методах их количественного определения.
Определенная последовательность аминокислот в белках
обусловливает их физико-химические свойства и биологическую функцию.
20 типов белковых аминокислот при их разнообразном сочетании и
повторяемости в полипептидной цепи дают практически
неограниченные возможности для синтеза самых различных белков.
Белки в питании играют роль пластических веществ; как источник
химической энергии они выполняют второстепенную роль. Особое
значение в белках как важнейших компонентах пищи имеют незаменимые
аминокислоты; для человека необходимы 8 незаменимых аминокислот,
по наличию и количественному соотношению которых судят о
биологической ценности белков, входящих в пищевой продукт.
Аминокислоты, входящие в состав белка, придают ему
специфические электрохимические свойства, которые обусловливают их гид-
рофильность, растворимость и осаждаемость из растворов. На
различии электрохимических свойств белков основаны методы их
извлечения из разрушенных клеток, их выделение и очистка методами
осаждения нейтральными солями и органическими растворителями,
электрофореза, хроматографии.
Конформация белковой молекулы включает четыре уровня: пер-
вичную (определенная последовательность аминокислот), вторичную
(а-спирали и (3-складки), третичную (компактную глобулу) и
четвертичную (ассоциацию отдельных субъединиц) структуры. В
формировании конформации белков принимают участие различные типы
связей и взаимодействий: ковалентные, электростатические связи и
гидрофобные взаимодействия. Принципы, подходы и методы
расшифровки пространственной структуры обусловлены прежде всего первичной
73
Часть /. Биомолекулы: структура, свойства, функции __^__^^^_
структурой белка и расшифровкой типов связей, формирующих
пространственную структуру белка. Здесь используются физические и
химические методы, среди которых особое место занимают секвениро-
вание, рентгеноструктурный кристаллографический анализ и ядерно-
магнитная резонансная спектроскопия.
Хронология важнейших дат в исследовании белков
1838 г. — В. Мюльдер впервые провел систематические
исследования белков.
1870 г. — Н. Н. Любавин впервые высказал мысль, что белки
состоят из аминокислот.
1884 г. — А. Я, Данилевский получил белковоподобное вещество,
показал, что в основе структуры белка лежит пептидная связь.
1903—1920 гг. — Э. Фишер синтезировал из аминокислот
пептиды, развил полипептидную теорию структуры белков.
1910 г, — А. Коссель выдвинул идею о том, что белки построены в
основном из аминокислот.
1935 г. — А, Розе открыл треонин — последнюю из числа
незаменимых аминокислот. ^
1948 г. — А. Тизелиуеу присуждена Нобелевская премия за
разработку метода электрофореза в свободной фазе.
1951 г. — Л. Полинг и Р. Кори сформулировали теорию
формирования вторичной структуры белка — теорию спиралей. Нобелевская
премия 1952 г.
1952 г. — А. Мартин и Р. Синг. удостоены Нобелевской премии за
разработку метода хроматографии на бумаге.
1958 г. —• А. Сенгеру присуждена Нобелевская премия за
расшифровку структуры инсулина.
1962 г. — Дж. Кендрью и М. Перутц удостоены Нобелевской
премии за расшифровку третичной структуры миоглобина и
четвертичной структуры гемоглобина.
ЛИТЕРАТУРА
Албертс Б*, Брей Д., Льюис Дж., Рэфф М., Роберте К., Уотсон Дж.
Молекулярная биология клетки. — М.: Мир, 1994. — Т.1. — 516 с.
Артюхов В. /\, Башарина О. В., Вашанов Г. А, Наквасина М. А, Пупгинце-
ваО.В. Олигомерные белки: структурно-функциональные модификации и роль
субъединичных контактов. — Воронеж; Изд-во ВГУ, 1997. — 264 с.
Блюменфельд Л. А. Проблемы биологической физики. — М.: Наука, 1977. —
336 с.
Кретович В. Л. Биохимия растений. — М.: Высш. шк., 1986. — 503 с.
Ленинджер А, Основы биохимии. — М.: Мир, 1985. Т.1. — 366 с.
Марри Р., Греннер Д., Мейес Л„ Родуэлл В. Биохимия человека. — М.: Мир,
1993. Т. 1. — 381 с.
Остерман Л. А Методы исследования белков и нуклеиновых кислот. — М.:
Наука, 1981. — 285 с.
74
Глава 1. Белки
Проблема белка. Т. 3: Структурная организация белка / Е. М. Попов. — М.:
Наука, 1997- ~~ 604 с.
Степанов В, М* Молекулярная биология. Структура и функции белков. —
М.: Высш. шк„ 1996. — 335 с.
Страйер П. Биохимия. — Мл Мир, 1984. — Т, 1. — 228 с.
Шульц Г., Ширмер Р. Принципы структурной организации белков. — М,:
Наука, 1982. — 534 с.
ВОПРОСЫ И ЗАДАЧИ
1. Количественное определение белков;
9
а) На каких свойствах белков основаны методы их количественного
определения?
б) В 5 см3 раствора белка содержалось 0,01 г азота. Рассчитайте массовую
концентрацию белка в растворе.
2. Структура белков, пептидов:
а) Какие типы связей участвуют в формировании структуры белков?
б) Используя сокращенные обозначения для аминокислот, напишите все
возможные трипептиды, содержащие аланин, метионин, аспарагиновую кислоту.
в) Изобразите полную структуру следующих пептидов (ионизированные
группы изобразите в протонированном состоянии): 1) Ме1-01и~01у; 2) РЬе-Азр-ТЬг.
г) Биологическая оценка белков как компонентов пищи. Основным
критерием оценки белков в питании является наличие в них незаменимых аминокислот.
Дайте биологическую оценку следующим гексапептидам:
1) 01и-Азр-Рго-А1а-С18-С1у;
2) 01и-А8р-Рго-А1а-УаЬС1в;
3) 01и-Уа1-Рго-Тгр-А1а-С1у;
4) Уа1-Рго-1д8-Тгр-А1а-РЬе;
5) Уа1-Ьеи-Ме*-ТЬг-Тгр~Ь13.
3. Физико-химические свойства белков, пептидов, аминокислот
Растворимость полипептидов зависит от степени полярности группы,
особенно от числа ионизируемых групп: чем больше ионизируемых групп, тем выше
растворимость полипептида- Какой трипептид из указанной пары более
растворим в приведенных условиях:
А1а-Ьеи-Ме* или Азр-1л8-С1и при рН 7?
4. Разделение белков, пептидов, аминокислот на основе их
электрохимических свойств:
а) Электрофорез аминокислот на бумаге. Каплю раствора, содержащего смесь
лейцина, аспарагиновой кислоты, аргинина, нанесли на полоску бумаги,
предварительно смоченную буфером с рН 6, и к концам полоски приложили
электрическое напряжение. Какая аминокислота будет двигаться к аноду, какая — к
катоду, какая останется на стартовой точке или вблизи нее?
б) Полипептид (Ыз)1О-(Аг0)5 интенсивно сорбируется на колонке с катионо-
обменником при рН 3. Какова причина сорбции? Этот полипептид можно
удалить с колонки, пропуская буфер с рН 10. Объясните, почему?
г
Глава 2. НУКЛЕИНОВЫЕ КИСЛОТЫ
2-1. Общая характеристика нуклеиновых кислот
Термин нуклеиновые кислоты был предложен немецким химиком
Р* Альтманом в 1889 г* после того, как эти соединения были открыты
в 1868 г. швейцарским врачом Ф. Мишером. Он экстрактировал
клетки гнойного пневмококка разбавленной соляной кислотой в течение
нескольких недель и получил в остатке почти чистый ядерный
материал, назвав его нуклеином (от лат. писЫиз — ядро). По своим
свойствам нуклеин резко отличался от белков: он был кислым, не
содержал серы, было много фосфора. Нуклеин хорошо растворялся в
щелочах, но не растворялся в разбавленных кислотах.
Впоследствии из животных, растительных объектов и
микроорганизмов были выделены разные нуклеиновые кислоты. Их наилучшим
источником оказались клетки, имеющие большие ядра.
Химически нуклеиновые кислоты представляют собой
биополимеры, состоящие из мономерных звеньев — нуклеогпидов.
Каждый нуклеотид содержит три различных компонента:
азотистое (пуриновое или пиримидиновое) основание (N1$),
моносахарид пентозу (рибозу или дезоксирибозу) (КЬ), остаток фосфорной
кислоты (Р). Как показал специфический гидролиз (кислотный,
щелочной), а также гидролиз ферментами-нуклеазами, эти
компоненты соединены друг с другом в такой последовательности:
азотистое основание — пентоза — фосфат. Соседние нуклеотиды связаны
друг с другом посредством эфирной связи между моносахаридом и
фосфатом другого нуклеотида:
Фрагмент полинуклеотидной цепи
КВ N8 ИВ N6 N8 N8
Р—КЬ—Р—КЪ—Р—ЕЬ—Р—КЬ—Р—КЬ—Р—КЬ-
Остаток [ [
нуклеотида Фосфодиэфирная связь
Поскольку остаток пентозы и фосфат соединены эфирной связью,
то при образовании полинуклеотидной цепи связь КЪ-Р-КЪ
называется фосфодиэфирной.
Азотистые основания не участвуют в образовании никаких других
ковалентных связей, помимо связывающей их с остатками пентозы
76
Глава 2. Нуклеиновые кислоты
сахарофосфатной цепи. Именно последовательность азотистых
оснований в полинуклеотидной цепи определяет уникальную структуру
и специфическую функцию молекул нуклеиновых кислот.
Гидролиз нуклеиновых кислот, выделенных из ядер клеток,
показал, что они состоят из пуриновых (аденина, гуанина) и пиримидино-
вых (цитозина, тимина) оснований, 2-дезоксирибозы и фосфорной
кислоты. Эта нуклеиновая кислота была названа дезоксирибонуклеи-
новой кислотой (ДНК). Из дрожжей была получена другая по
химическому составу нуклеиновая кислота, содержащая вместо тимина
урацил и вместо дезоксирибозы рибозу. Ее назвали рибонуклеиновой
кислотой (РНК).
Биологическая функция нуклеиновых кислот оставалась
неизвестной в течение почти столетия. Только в 40-х гг. XX в. О. Т.
Звери, К. Мак-Леод и М. Мак-Карти установили, что эти биополимеры
ответственны за хранение, репликацию (воспроизведение),
транскрипцию (передачу) и трансляцию (воспроизведение на белок)
генетической (наследственной) информации. В 1953 г., когда Дж. Уот-
сон и Ф. Крик сообщили о расшифровке молекулярной структуры
ДНК, биохимия и вообще биология начала отсчет новой эры
познания живой материи.
Все нуклеиновые кислоты представляют собой
высокомолекулярные соединения, хотя их молекулярная масса сильно варьирует от
15 000 до 1 млрд и более.
Основная масса ДНК содержится в хромосомах клеток эукариот;
поскольку хромосомы сконцентрированы в ядрах клеток, то ДНК
часто называют ядерной нуклеиновой кислотой. РНК большей частью
содержится в цитоплазме клеток и называется цитоплазмической
нуклеиновой кислотой. Кроме того, ДНК присутствует в хлороплас-
тах растений, митохондриях.
Общее содержание ДНК и РНК в клетках зависит от
специфических функций клеток. В некоторых клетках, например спермиях,
содержание ДНК может составлять 60 % (в пересчете на сухую массу
клеток), в большинстве клеток — 1—10, а в мышцах — около 0,2 %.
Содержание РНК, как правило, в 5—10 раз больше, чем ДНК.
Соотношение РНК/ДНК в клетках тем выше, чем интенсивнее в них синтез
белка. Особое место занимают вирусы. У них в качестве генетического
материала может быть либо ДНК (ДНК-овые вирусы), либо РНК
(РНК-рвые вирусы),
В клетках бактерий, не имеющих ядра (прокариоты), молекула
ДНК (хромосома) находится в специальной зоне цитоплазмы — пук-
леоиде. Если она связана с клеточной мембраной бактерии, то ее
называют мезосомой. Фрагмент меньших размеров, локализуемый вне
хромосомной зоны, называется плазмидой. РНК, в отличие от ДНК,
распределена по клетке более равномерно. Уже это обстоятельство
свидетельствует, что функция РНК более многообразна и динамична.
77
Часть I. Биомолекулы: структура, свойства, функции
2.2. Структура нуклеотидов
Пуриновые основания имеют следующее строение:
1^1
Пурин
Ш<
8СН
9 /
н
>ну
сн
н
/
Аденин (А)
(б-аминопурин)
О
II
I II >н
Гуанин (О)
(2-амино- 6-ги д роксипурин)
Сам пурин не входит в состав нуклеотидов, а входят его произвол
ные — аденин (А) и гуанин (О).
Пиримидиновые азотистые основания имеют следующее строение
н
К'
^Чсн
а
нм
нсЧАхсн
Пиримидин
О
II
II
о^с\к/сн
н
Урацил (Ц)
(2,4-дигидрокси-
пиримидин)
N11
N
СН
н
Цитозин (С)
(2-гидрокси-
4-аминопиримидин)
НЫ'
О
II
С\С^СН3
о*с\^сн
н
Тимин (Т)
(б-метилурадил)
Пиримидин также не входит в состав нуклеотидов, а входят его
производные — урацил (V), тимин (Т), цитозин (С).
Молекулы пиримидинов имеют плоское кольцо; пуринов — почти
плоское с небольшой складкой. Эти основания плохо растворимы в
воде, существуют в таутомерных (кето-енольных) формах:
О
ны
^с
сн,
N
он
I
с
о
н
сн
но
Чк/
II
сн,
сн
Тимин (лактам)
Тимин (лактим)
Атом азота в их кольцах имеет слабо основный характер: значения
рК лежат в области рН 9—10; 6-аминогруппа аденина имеет рК 4,2;
2-аминогруппа гуанина — рК 3,2; следовательно, при рН 7,0 эти
группы не протонированы.
Наличие в пуриновых и пиримидиновых основаниях сопряженных
двойных связей обусловливает интенсивное поглощение ими света в
УФ-области спектра с максимумом при длине волны около 260 нм.
Из-за гетероциклической ароматической природы это свойство
широко используется для их количественного определения, а также
количественного определения нуклеозидов, нуклеотидов и нуклеиновых кис-
78
Глава 2. Нуклеиновые кислоты
лот. Свободные пуриновые и пиримидиновые основания легко
разделяются и идентифицируются методом тонкослойной хроматографии.
Пуриновые и пиримидиновые азотистые основания играют
важную роль в качестве стимуляторов роста микроорганизмов.
В составе ДНК и РНК встречаются более редкие азотистые
основания, например, 5~метилцитозин> 4-тиоурацил и дигидроурацил; они
получили название минорных оснований.
В состав ДНК входят р-В-2-дезоксирибоза, в состав РНК —
(З-В-рибоза. И в том, и в другом случае эти монозы являются пентозои
(пять углеродных атомов), различия касаются лишь второго
углеродного атома. В рибозе углерод-2 связан с ОН-группой, тогда как в дез-
оксирибозе на месте ОН-группы находится Н, отсюда префикс "дезок-
си". Буквы р и В отражают специфическую конфигурацию при
атомах С-1/ и С-4' фуранозного цикла:
З'Т Г 2'
ОН ОН
Р-П-рибоза
3'1 Г 2'
ОН Н
Р-В- 2'-дезоксирибоза
Нуклеозиды — соединения, в которых пуриновые или
пиримидиновые основания связаны с рибозой (рибонуклеозиды) или дезоксири-
бозой (дезоксирибонуклеозиды). Нуклеозиды относятся к Ы-гликози-
дам: атом С-1' рибозы или дезоксирибозы связан с N-9 пуринового
или N-1 пиримидинового основания:
кн
НОНоС /О
9-9
он он
Аденозин
О
НЖ" ^СН
0^Ь\Ы^
СН
бнон
Уридин
V
н,
N
^ч
нсЧк^с^]/
СН
НОН2С/Оч
ад
9т
онн
2' - Дезоксиаденозин
В состав ДНК и РНК входят следующие нуклеозиды:
ДНК
Аденин + дезоксирибоза = дезоксиаденозин.
Гуанин + дезоксирибоза = дезоксигуанозин.
Цитозин + дезоксирибоза = дезоксицитидин.
Тимйн + дезоксирибоза = дезокситимидин.
79
Часть I. Биомолекулы: структура, свойства, функции
РНК
Аденин + рибоза = аденозин.
Гуанин + рибоза = гуанозин.
Цитозин + рибоза = цитидин.
Урацил + рибоза = уридин.
Кроме вышеперечисленных главных нуклеозидов встречаются и
минорные нуклеозиды, из которых наиболее распространены дигид-
роуридин, псевдоуридин; в последнем отсутствует обычная Ы-глико-
зидная связь: в нем атом С-1/ рибозы соединен с атомом С-5 урацила.
Нуклеозиды лучше растворимы в воде, чем исходные азотистые
основания. Их легко можно разделить и идентифицировать методом
тонкослойной хроматографии. Они устойчивы к щелочам, но легко
гидролизуются кислотами, а также ферментом нуклеозидазой.
Нуклеотиды представляют собой нуклеозиды с присоединенной
эфирной связью к остатку рибозы или дезоксирибозы фосфатной
группой. В образовании связи участвует б'-углеродный атом пентозы. В
зависимости от строения пентозы все нуклеотиды можно разделить на
рибонуклеотиды и дезоксирибонуклеотиды:
N11, МН2
сн
сн
нсЧк/с'^^/ нсЧк/с^к/
НО-Р-0-СН2
I | }0
о \/
?Н 5'
но-р-о-сн2 л
I I уО
о
нЧс -сн
I I
он он
Аденозин- 5'- монофосфат 2'- Дезоксиад енозин - 5'-монофосфат
В зависимости от числа остатков фосфорной кислоты нуклеотиды
подразделяются на нуклеозид-5'-монофосфаты, нуклеозид-5'-дифосфаты
и нуклеозид-5'-трифосфаты:
О"
Аденозин-б'-монофосфат (АМР) о-р-о-
(адениловая кислота) о
О" О"
Аденозин-5'-дифосфат (АБР) О^р-О-р-О-
II II
о о
? ? ?
Аденозин-5'-трифосфат (АТР) О'-р-о-р-О-р-о-
О о о
В принципе нуклеозид может быть фосфорилирован до тетрафосфата.
80
Глава 2. Нуклеиновые кислоты
Ниже приводятся названия и сокращенные обозначения
нуклеотидов:
Названия Сокращенные обозначения
Рибонуклеотиды
Аденозинмоно-, ди-, трифосфат АМР, АВР, АТР
Гуанозинмоно-, ди-, трифосфат ОМР, ОВР, СгТР
Цитидинмоно-, ди-, трифосфат СМР, СВР, СТР
Уридинмоно-, ди-, трифосфат 1МР, ИВР, 1ТТР
Дезоксирибонуклеотиды
Дезоксоаденозинмоно-, ди-, трифосфат йАМР, <1АВР, <1АТР
Дезоксигуанозинмоно-, ди-, трифосфат <ЮМР, (ЮВР, сЮТР
Дезоксицитидинмоно-, ди-, трифосфат ЙСМР, сЮОР, сЮТР
Дезокситимидинмоно-, ди-, трифосфат йТМР, сШЭР, с!ТТР
Данная номенклатура нуклеотидов рассматривает их как фосфорные
эфиры. В то же время благодаря наличию кислотной фосфатной
группы удобно рассматривать ну к л еозид монофосфаты как кислотные
производные исходных нуклеозидов, например, адениловая, уридиловая,
гуанидиловая, цитидиловая кислоты.
Нуклеотиды — сильные кислоты, значения рК для двух
свободных ОН-групп остатка фосфорной кислоты соответствуют
приблизительно 1,0 и 6,2. Поэтому при рН 7,0 нуклеотиды находятся в
О"
форме К~р=0.
О"
Нуклеотиды интенсивно поглощают свет при 260 нм, легко
разделяются методом электрофореза и хроматографии на ионообменных
колонках. Они гидролизуются под действием нуклеаз.
Уникальны биохимические функции нуклеотидов, В качестве
основных можно отметить следующие:
1) являются строительными блоками нуклеиновых кислот (ДНК
и РНК); участвуют в молекулярных механизмах, с помощью
которых генетическая информация хранится, реплицируется и
транскрибируется;
2) выполняют важную роль в энергетическом (фосфорном) обмене,
в аккумулировании и переносе энергии;
3) служат агонами (коферментами и активными простетическими
группами) в окислительно-восстановительных ферментах;
4) играют важную роль в синтезе олиго- и полисахаридов, жиров.
Таким образом, нуклеотиды — универсальные биомолекулы,
играющие фундаментальную роль в обмене веществ и энергии живой
клетки.
6. Заказ 3486
81
Часть I. Биомолекулы: структура, свойства* функции
2,3. Первичная структура полинуклеотидов
Последовательность азотистых оснований. ДНК и РНК
представляют собой полинуклеотиды, имеющие три уровня структуры:
первичную, вторичную, третичную.
На рис. 2.1 показана первичная структура фрагмента полинук-
леотида.
Полинуклеотиды состоят из нуклеотидов, соединенных фосфорно-
эфирными связями с участием 3'- и 5'- углеродных атомов пентозных
остатков двух соседних нуклеотидов. Длинные полинуклеотидные цепи
содержат тысячи, миллионы нуклеотидных остатков. Фосфатные
группы в цепях обладают сильнокислыми свойствами и при рН 7,0
полностью ионизированы. Поэтому в живых клетках нуклеиновые кислоты
существуют в виде полианионов > Нуклеиновые кислоты плохо
растворимы в растворах кислот. Они экстрагируются из разрушенных
тканей и клеток растворами нейтральных солей или фенолом.
В соответствии с химическим строением существуют три группы
методов количественного определения нуклеиновых кислот: а) по
содержанию азотистых оснований; обычно пользуются определением
величины поглощения света в ультрафиолетовой части спектра, т.е.
спектрометрией; б) по цветным реакциям на рибозу и дезоксирибозу; в) по
количеству фосфора. Спектрофотометрические методы дают
удовлетворительные результаты для хорошо очищенных нуклеиновых
кислот. Методы второй группы специфичны к типу нуклеиновых кислот
и позволяют отличать ДНК от РНК.
Для отделения ДНК от РНК можно
использовать их различную растворимость
в №С1: ДНК не растворяется, РНК,
напротив, растворима. При этом применяются
ионообменная, адсорбционная, гельпрони-
кающая и аффинная хроматография,
электрофорез в полиакриламидном геле и
ультрацентрифугирование в градиенте
плотности (например, в градиенте плотности СзС1),
Подходы, пути и методы расшифровки
нуклеотидной последовательности в
нуклеиновых кислотах в определенной степени
аналогичны таковым в установлении
первичной структуры полипептидной цепи.
Однако здесь имеются и определенные
трудности, связанные с более высоким однооб-
! разием пол и нуклеотидной цепи,
ограничивающим разнообразие этих подходов и ме-
Рис. 2.1. Схематическое ^ - _ __
- , тодов. Важную роль играет ферментатив-
изображение фрагмента по- "^ *■ *- *- ^ г
линуклеотида ньш гидролиз межнуклеотидных связей,
О
1' 5'
Основание —- пентоза—ОР—О
3'
'О
ОН
1' 5'
Основание — пентоза—ОР^О
.3'
О
I
ОН
1' 5'
Основание — пентоза—ОР^О
.3'
О
ОН
82
Глава 2. Нуклеиновые кислоты
который осуществляется с помощью специфических эндонуклеаз. Этот
гидролиз позволяет получить ряд олигомерных фрагментов, которые
фракционируются, например, методом тонкослойной хроматографии
или электрофорезом в полиакрил амид ном геле, после чего они
полностью расщепляются с целью определения их нуклеотидного состава.
Для установления нуклеотидной последовательности фрагментов
используют экзонуклеазы. С их помощью осуществляется
последовательное отщепление нуклеотидов с одного конца полинуклеотидной цепи,
С помощью другой специфической зндонуклеазы осуществляется
новая фрагментация для определения нуклеотидной
последовательности. Полученные взаимно перекрывающиеся фрагменты позволяют
установить последовательность нуклеотидов в нуклеиновой кислоте.
2.4. Вторичная и третичная структуры ДНК
Растворы ДНК характеризуются аномальной (структурной)
вязкостью. В потоке обладают двойным лучепреломлением, что
объясняется удлиненной формой молекул ДНК.
В расшифровку структуры ДНК большой вклад внесли
исследования Э. Чаргаффа и его сотрудников (1945—1951 гг.). Для разделения
оснований, полученных при кислотном гидролизе ДНК, Э. Чаргафф
использовал метод хроматографии. Каждое из этих оснований было
определено спектрофотометрически. Он впервые определенно заявил,
что ДНК обладают выраженной видовой специфичностью. ДНК,
выделенные из различных источников, отличаются друг от друга по
соотношению входящих в их состав азотистых оснований. Э. Чаргафф
сформулировал закономерности состава ДНК, известные под
названием правил Чаргаффа. Независимо от происхождения ДНК эти
закономерности представляются следующим образом:
1) количество молекул аденина равно количеству молекул тимина
(А - Т);
2) количество молекул гуанина равно количеству молекул цитози-
на (С = С);
3) количество молекул пуриновых оснований равно количеству
молекул пиримидиновых оснований (А + О = Т + С);
4) количество оснований с 6-аминогруппами в цепях ДНК равно
количеству оснований с 6-гидроксигруппами {А + С = О + Т);
5) отношение (С+С)/(А+Т) резко отличается для разных видов ДНК,
но постоянно для клетки одного вида; это отношение называется
фактором специфичности.
Фактор специфичности одинаков для ДНК различных органов и
тканей одного организма и практически не отличается у разных видов
животных и растений в пределах одного класса. У высших растений и
животных его величина находится в пределах 0,55—0,93; у
бактерий — 0,35—2,73.
6*
83
Часть I, Биомолекулы: структура, свойства, функции
Правила Чаргаффа сыграли решающую роль в разработке проблем
молекулярной биологии. Именно они легли в основу открытия
строения ДНК, ее вторичной структуры.
Прежде чем приступить к рассмотрению этой структуры,
необходимо отметить следующее. Интерес к проблеме изучения структуры
ДНК возрос в связи с полной неясностью механизма воспроизведения
(репликации) ДНК, который отличается очень высокой степенью
точности. На основании уже полученных экспериментальных данных
предполагалось, что генетическая информация в живой клетке
зашифрована (т.е. записана с помощью определенного кода) в
специфической последовательности оснований ДНК. Однако было неясно, как
воспроизводится такая последовательность,
В 1953 г. Дж. Уотсон и Ф. Крик предложили модель структуры
ДНК. Она учитывала рентгеноструктурные данные Р. Франклин и
М. Уилкинса и "эквимолярность" оснований, открытую Э. Чаргаффом,
Модель Уотсона—Крика не только объяснила физико-химические
свойства ДНК, но и дала основание высказать предположение о
возможном механизме репликации ДНК. Согласно их выводу, молекула ДНК
должна быть двухцепочечной. Каждое основание одной цепи
"спарено" с лежащим в той же плоскости основанием второй полинуклео-
тидной цепи. Это спаривание специфично; поскольку количество
оснований с гидроксигруппами равно количеству оснований с
аминогруппами, то, по Уотсону и Крику, только определенные пары
оснований входят в структуру так, что могут образовывать друг с другом
водородные связи. Так как аденин содержит аминогруппу, а тимин —
гидроксигруппу, цитозин и гуанин — соответственно эти же группы и
поскольку они находятся в ДНК в эквимолекулярных количествах, то
разрешенными являются только пары А-Т и О-С:
0,28 нм
ы-н
о снз
н
1- О^Онм^
О
1,11 нм
-< >
(А:::Т)
о
0,29 нм , Н
——М/
Н-
# [щ °'30 нм
* * * *
1,08 нм
(ОНГС)
84
Глава 2. Нуклеиновые кислоты
Между А и Т образуются две водородные связи, между О и С —
три водородные связи.
Две полинуклеотидные цепи ДНК отличаются одна от другой как
последовательностью оснований, так и нуклеотидным составом. Однако
основания, стоящие в данном положении в одной цепи, определяют
природу основания в другой цепи. Например, если в одной цепи стоит аде-
нин, то напротив него в другой цепи будет располагаться тимин, и
наоборот. Если в одной цепи стоит гуанин, то в другой обязательно будет цито-
зин, и наоборот. Действительно, рентгеноструктурный анализ ДНК
показал, что пуриновые и пиримидиновые основания нуклеотидных
остатков ДНК лежат в одной плоскости, перпендикулярной продольной оси
молекулы, тогда как циклы дезоксирибозы находятся в плоскости,
почти перпендикулярной той, в которой лежат циклы оснований.
Явление, при котором последовательность оснований одной цепи
однозначно определяет последовательность оснований другой цепи,
получило название комплементарности. Таким образом, цепи моле-
кул ДНК комплементарны по отношению друг к другу (рис. 2.2).
На рис. 2.3 представлены двойная спираль молекулы ДНК и ее
модель* Сформулированный Уотсоном и Криком принцип
комплементарности явился универсальным принципом в биологии. Он дал
начало развитию новых научных направлений — молекулярной биологии,
молекулярной генетике, генной инженерии. Водородные связи
обеспечивают способ спаривания оснований, стабильность двухцепочеч-
ной системы. Основания плотно упакованы, причем расстояние
между центрами оснований, лежащих друг над другом, равно 0,34 нм. На
каждый полный виток двойной спирали приходится 10
нуклеотидных пар. Упакованные внутри двойной спирали основания гидрофоб-
ны и недоступны молекулам воды. Ионизированные фосфатные
группы и гидрофильные остатки дезоксирибозы находятся на поверхности
молекулы и контактируют с молекулами воды. Таким образом,
двойная спираль стабилизирована не только
водородными связями между
комплементарными основаниями, но и
гидрофобными взаимодействиями между
основаниями, расположенными вдоль
длинной оси молекулы ДНК. Из-за
высокой степени упорядоченности
макромолекул ДНК ее иногда называют
апериодическим одномерным кристаллом.
При рентгенографическом
исследовании головок сперматозоидов получается
такая же дифракционная картина, что
и для образцов ДНК, т.е. спираль Уот-
Цепь I
Цепь II
5'
3'
3'
\
5'
Рис. 2.2. Комплементарйость
цепей I и II (точками обозначены
сона—Крика наблюдается непосред- водородные связи между основа-
ственно в живых клетках.
ниями; цепи антипараллельны ?^)
85
Часть I. Биомолекулы: структура, свойства, функции
Рис. 2.3. Структура ДНК: а — двойная спираль молекулы ДНК (модель Уот-
сона—Крика): А — аденин; Т — тимин; С — гуанин; С — цитозин; В — дезокси-
рибоза; Р — фосфат; 3,4 нм — величина (высота) витка спирали; 1 нм — радиус
спирали; 0,34 нм — расстояние между нуклеотидами; стрелки указывают на-
првление витка спирали; б — модель молекулы ДНК
Модель строения ДНК в настоящее время является
общепризнанной. За расшифровку структуры ДНК Дж. Уотсону, Ф* Крику и М. Уил-
кинсу в 1962 г. была присуждена Нобелевская премия.
Третичная структура ДНК образуется в результате
дополнительного скручивания в пространстве двухцепочечнои молекулы. Она имеет
вид суперспирали или изогнутой (сломанной) двойной спирали.
В настоящее время описаны три формы структуры ДНК: А-, В- и
2-формы (рис. 2.4). Параметры модели Уотсона—Крика
соответствуют конформации ДНК в физиологических условиях (В-форме ДНК).
Однако при изменении условий среды двойная спираль может
принимать другие формы. Так» при уменьшении влажности (в препарате
образца для рентгеноструктурного анализа) ДНК переходит в А-фор-
му. Этот переход связан с изменением конформации в остатках дезок-
сирибозы, уменьшением расстояния между фосфатными группами са-
харофосфатного остова. Расстояние между парами нуклеотидов вдоль
оси спирали, равное 0,34 нм в уотсон-криковской модели,
уменьшается (примерно до 0,25 нм при 11 нуклеотидных остатках на один виток
86
Глава 2. Нуклеиновые кислоты
В-ДНК 2-ДНК
Рис. 2.4. В- и 2-формы структуры ДНК
спирали). Диаметр спирали увеличивается; изменяются ширина и
глубина бороздок; комплементарные пары азотистых оснований
образуют с осью спирали угол 20° и, главное, они смещаются к периферии
спирали. Поэтому двойная спираль похожа на пологую винтовую
лестницу и внутри нее возникает полость диаметром 0,40 нм.
Переход молекулы ДНК из В- в А-форму можно осуществить при
понижении активности воды в растворе (при внесении в него
органического растворителя, например, этанола). Существует мнение, что В-
форма представляет собой некую промежуточную форму двух или
большего числа конформаций. Одной из особенностей В-формы,
называемой В'-формой, является способность менять в молекуле ДНК
положение двух цепей на обратное. Более того, В-форма может
существовать в виде как правой, так и левой спирали.
Хотя более стабильными в А- и В-формах являются правозакру-
ченные спирали, существуют довольно устойчивые и левозакручен-
ные спирали ДНК. Одна из таких спиралей была получена в 1979 г.
А. Ричем. Из-за нерегулярного зигзагообразного изгиба еахарофосфат-
ного остова она была названа 2-формой (см. рис. 2.4). Повторяющаяся
единица в 2-форме ДНК включает две пары нуклеотидов, а не одну,
как в В- и А-формах. Вследствие этого линия, соединяющая фосфат-
87
Часть I. Биомолекулы: структура, свойства, функции _
ные группы, через каждые две пары нуклеотидов имеет излом и
принимает зигзагообразный вид. По сравнению с В-формой в левой 2-форме
резко изменен характер стэкинга оснований: сильные и слабые
межплоскостные взаимодействия также чередуются. 2-форма может
переходить в В-форму при снижении ионной силы раствора, добавлении
этанола. Однако вопрос о существовании 2-формы ДНК ж ишо и ее
биологической роли до конца не выяснен. Высказывается мнение, что
переход правозакрученнои формы в левозакрученную может служить
регуляторным сигналом, контролирующим экспрессию генов.
2.5, Физико-химические свойства ДНК
ДНК — довольно сильная многоосновная кислота, полностью
ионизированная при рН 4,0. Фосфатные группы расположены по
периферии. Они прочно связывают ионы Са2+ и М#2+, амины, гистоны —
положительно заряженные белки. Устойчивость комплементарных пар
оснований зависит от величины рН. Пары оснований наиболее
устойчивы в интервале рН 4,0—11,0. За его пределами двухцепочечная
спираль ДНК теряет устойчивость и раскручивается.
Молекулярная масса ДНК неодинакова и зависит от источника ее
получения. К тому же даже при самых тщательных и щадящих
процедурах выделения ДНК подвергается некоторой деградации.
Препараты, полученные современными методами из тканей животных и
растений, имеют молекулярную массу 6*106—10-Ю6. Однако
истинная молекулярная масса ДНК животных и растений, определенная по
вязкости и по длине молекул, значительно выше и достигает десятков
миллиардов.
У большинства вирусов ДНК представляет собой двойную спираль,
линейную или замкнутую в кольцо. У некоторых вирусов она
представляет собой одну полинуклеотидную цепь, замкнутую в кольцо и
имеющую сравнительно небольшую молекулярную массу — 2Ю6. ДНК
сравнительно легко деполимеризуется под действием некоторых химических
соединений, ультразвука, ионизирующей и ультрафиолетовой радиации.
Нагревание растворов ДНК до температур 70—80 °С, а также их подще-
лачивание вызывают денатурацию ДНК, заключающуюся в плавлении
двойной спирали (разрушение водородных связей и гидрофобных
взаимодействий), и расхождение полинуклеотидных цепей. Денатурация
сопровождается понижением вязкости раствора, повышением поглощения
в ультрафиолетовой области, увеличением отрицательного удельного
вращения плоскости поляризации света, увеличением плавучей плотности
образцов ДНК. Возрастание светопоглощения света при 260 нм
называется гипохромным эффектом; это важнейший критерий денатурации
ДНК, по которому можно контролировать этот процесс.
В отличие от многих глобулярных белков, денатурация которых
происходит постепенно в широком температурном интервале, натив-
ные ДНК денатурируют в узком интервале температур (-10 °С), поэто-
88
Глава 2. Нуклеиновые кислоты
му тепловую денатурацию часто называют плавлением. Температура
плавления тем выше, чем больше в молекуле ДНК ОС-пар; этот
показатель может использоваться для определения нуклеотидного состава
ДНК. Установлено, что температура плавления линейно связана с
составом ДНК: ее повышение на 1° соответствует 2,5 молярных % ОС-пар.
Гомогенные препараты ДНК характеризуются плавлением с резким
переходом спираль—клубок, тогда как гетерогенные препараты дают
сравнительно широкую зону плавления, что может служить мерой
гетерогенности ДНК. При быстром охлаждении после тепловой
денатурации ДНК не восстанавливает своих нативных свойств; однако при
медленном охлаждении полинуклеотидные цепи реассициируются по
принципу комплементарности, т.е. происходит ренатурация молекул
ДНК. Это продемонстрировано, в частности, на препаратах ДНК
пневмококка с помощью методов электронной микроскопии и
градиентного ультрацентрифугирования в СзСЬ
2.6. Биологические функции ДНК
Важнейшая биологическая функция ДНК — генетическая, т.е.
хранение и передача наследуемых признаков. В 1943 г. О. Т. Звери,
К. Мак-Леод и М. Мак-Карти из Рокфеллеровского института
обнаружили это впервые. Они экспериментально установили, что невирулентный
штамм бактерии Рпеитососсив может превратиться в вирулентный
простым добавлением ДНК, выделенной из вирулентных пневмококков.
Исследователи заключили, что ДНК может содержать генетическую
информацию* Работа О. Т. Эвери и его сотрудников признана выдающейся,
представляющей собой важную историческую веху в исследовании
генетической функции ДНК. Сейчас многочисленными экспериментами
установлено, что ДНК — основной компонент клеточных органелл-хромо-
сом. Трансформирующаяся ДНК включается ковалентно в ДНК
невирулентной клетки (клетки-реципиента) и, таким образом, реплицируется
вместе с хромосомой реципиента; свойство вирулентности наследуется. В
то же время возможность передачи генетической информации
бактериальным клеткам в результате введения РНК или белка не получила
экспериментального подтверждения.
На вопрос, почему наследуемые признаки копируются с
удивительной точностью, дает ответ принцип комплементарности. Модель ДНК,
разработанная Уотсоном и Криком, четко объясняет механизм
передачи информации. В связи с тем, что последовательность азотистых
оснований одной цепи однозначно определяет последовательность
оснований в другой цепи, репликация ДНК в клетке происходит в
результате расхождения двух полинуклеотидных цепей и последующего
синтеза двух новых (дочерних) цепей на старых (родительских) цепях
как на матрицах. В результате образуются две дочерние двухцепочеч-
ные молекулы ДНК, идентичные родительской молекуле ДНК,
содержащие по одной цепи из родительской молекулы. Такой механизм
89
Часть I. Биомолекулы: структура, свойства, функции
репликации был назван полуконсервативным (рис. 2.5). Он получил
блестящее подтверждение в экспериментах с клетками Е.соИ и
меченым азотом К15 , проведенных М. Мезелсоном и Ф. Сталем в 1957 г.
Полученные ими результаты точно согласуются с
полуконсервативным механизмом редупликации макромолекул ДНК.
Родительская ДНК
Цепь I
:: Цепь и
Репликация
I
Дочерние ДНК
Рис. 2.5. Репликация ДНК: образование двух дочерних молекул ДНК,
идентичных материнской молекуле ДНК
2.7. Процессы метилирования ДНК
Метилированные азотистые основания в ДНК обнаружены свыше
50 лет назад (К. В. НоЪсЫизз, 1948). ДНК прокариот содержит
модифицированные основания Н6-метиладенин и 5-метилцитозин, тогда как
ДНК высших эукариот — в основном 5-метилцитозин.
Метилирование остатков цитозина ДНК имеет место у бактерий, растений,
животных, в том числе млекопитающих (включая человека), но отсутствует
у дрожжей и нематод. Эта реакция осуществляется ферментативно в
первые минуты после репликации ДНК, т.е. пострепликативно.
В ДНК эукариот на долю 5-метилцитозина приходится около 5 %
от общего содержания цитозина. Степень метилирования ДНК
изменяется в процессе развития, а также зависит от возраста организма и
гормональных воздействий. Цитозин метилируется, если находится в
составе комплементарной последовательности р р . Особенно сильно
метилирована ДНК позвоночных, где модифицированы от 60 до 90 %
таких динуклеотидных последовательностей. Последовательности СО
сконцентрированы в отдельных районах генома, включающих
несколько сотен нуклеотидных пар. Число названных районов у
млекопитающих составляет около 30 000. Такие "островки" последовательностей
СО являются мишенями метилирования ДНК. "Рисунок" (профиль)
90
Г лава 2. Нуклеиновые кислоты
метилирования участков ДНК не одинаков в разных тканях и на
разных стадиях развития. Метилирование может сопровождать процесс
репликации ДНК, причем цитозин метилируется в дочерней нити ДНК
только в том случае, если он метилирован и в родительской:
тСО Метилаза
ос ■
тСО
ОСт.
Это обеспечивает наследование либо метилированного, либо деметилиро-
ванного состояния участка ДНК при последующих клеточных делениях.
Метилирование позволяет отличить "старую" цепь ДНК от "новой".
В течение нескольких минут после репликации, пока не
подействовала метилаза, новосинтезированная ДНК не метилирована, т. е.
отличается от матричной цепи (см. схему метилирования ДНК).
Метальная
группа
® ®
I
М
Репликация
М п
\
Метилаза
\
(м>
<=>
(м>
<м)
III
В данной схеме I — полностью метилированная ДНК; II —
полуметилированная ДНК; III — полностью метилированная ДНК.
Метилирование способствует не только репарации ДНК, но и может, напротив,
облегчать мутагенез, так как каждый метилированный цитозин
потенциально мутагенен. Эукариоты (позвоночные) используют метилирование
ДНК для целей дифференциального контроля генной активности. Роль
локального метилирования ДНК как фактора, контролирующего
активность генов, подтверждается прежде всего реактивацией некоторых
неактивных генов после частичного деметилирования.
Значение метилирования очень велико при определении так
называемого "родительского импринтинга" генов у млекопитающих. Известно,
что роль отцовских и материнских генов в развитии не одинакова. Сте-
91
Часть I. Биомолекулы: структура, свойства, функции
пень и характер экспрессии генов может зависеть от того, получены они
от отца или от матери. Выявлено, что метилированные гены
трансгенных мышей, полученные от матери, могут не экспрессироваться, тогда
как эти же гены, полученные от отца и деметилированные в процессе
гаметогенеза, способны к тканеспецифической экспрессии. Процессы
метилирования, несомненно, участвуют в инактивации одной из двух
Х-хромосом в клетках млекопитающих.
Итак, метилирование ДНК у эукариот, по-видимому, представляет
собой механизм регуляции экспрессии генов и клеточной дифферен-
цировки.
2.8. Структура и физико-химические свойства РНК
Рибонуклеиновые кислоты (РНК) — однонитевые молекулы,
поэтому в отличие от ДНК их вторичная и третичная структуры
нерегулярны. Нуклеотидная цепь РНК обладает гибкой структурой, ее
длина в зависимости от вида РНК может варьировать в очень широких
пределах — от нескольких десятков до десятков тысяч нуклеотидных
остатков; молекулярная масса РНК находится в пределах 104—106,
Последовательность нуклеотидных звеньев, соединенных фосфоди-
эфирной связью в неразветвленную полинуклеотидную цепь,
представляет собой первичную структуру РНК. Вторичная и третичная
структуры РНК, определяемые как пространственная конформация полинук-
леотидной цепи, формируются в основном за счет водородных связей и
гидрофобных взаимодействий между азотистыми основаниями. Если для
молекулы нативной ДНК характерна устойчивая спираль, то структура
РНК более многообразна и лабильна. Рентгеноструктурный анализ
показал, что отдельные участки полинуклеотидной цепи РНК» перегибаясь,
навиваются сами на себя с образованием внутриспиральных структур.
Стабилизация структур достигается за счет комплементарных
спариваний азотистых оснований антипараллельных участков цепи;
специфическими парами здесь являются А-ХТ, О—С и, реже, С-1Т,
Образование спиральных структур сопровождается гипохромным
эффектом — уменьшением оптической плотности образцов РНК при
260 нм. Разрушение этих структур происходит при понижении
ионной силы раствора РНК или при его нагревании до 60—70 °С; оно
также называется плавлением и объясняется структурным переходом
спираль — хаотический клубок, что сопровождается увеличением
оптической плотности раствора нуклеиновой кислоты.
Хотя РНК относится к однониточным полинуклеотидам, вместе с
тем в ее цепях имеются участки различной длины, состоящие из
комплементарных друг другу нуклеотидных последовательностей,
включающих от десятков до тысяч нуклеотидных остатков,
расположенных на небольшом удалении друг от друга. Благодаря этому в
молекуле РНК возникают как короткие, так и протяженные биспиральные
92
Глава 2. Нуклеиновые кислоты
участки, принадлежащие одной цепи; эти участки носят название
шпилек. Модель вторичной структуры РНК со шпилькообразными
элементами была создана в конце 50-х — начале 60-х гг. XX в. в
лабораториях А. С. Спирина (Россия) и П. Доти (США).
2.9. Типы РНК и их биологические функции
Клетки содержат три основных типа РНК: рибосомную — рРНК,
транспортную — тРНК и матричную (информационную) — мРНК.
Каждая из этих РНК выполняет специфическую роль в сложном
процессе биосинтеза белка, при котором последовательность аминокислот
однозначно определяется нуклеотидной последовательностью ДНК, что
можно представить в виде следующей схемы взаимосвязанных
процессов;
рРНК — •>- Рибосома
ДНК <^--»» мРНК т—9т- •^>- »- Полипептид
^ ТРНК —^ »■ Аминоацил-тРНК
1 Аминокислоты
Транскрипция Трансляция
В эукариотических клетках существуют также малые ядерные РНК
(мяРНК), являющиеся участниками процессинга РНК, и
гетерогенные ядерные РНК (гяРНК)> представляющие собой
предшественников мРНК. Кроме того, обнаружена так называемая антисмысловая
РНК, участвующая в регуляции процесса репликации ДНК.
В процессе транскрипции нуклеотидная последовательность локу-
са (место в хромосоме, где находится ген) в ДНК копируется в
молекулу РНК. Транскрибируются три вида генов. Транскрипты генов рРНК
используются в синтезе рибосом, нуклеотидная последовательность
мРНК переписывается в последовательность аминокислот при синтезе
полипептида на рибосоме, а транскрипты генов тРНК связываются с
аминокислотами, которые затем переносятся в рибосомный
синтезирующий центр в последовательности, зашифрованной в мРНК; этот
процесс называется трансляцией.
Дадим краткую характеристику каждого типа РНК.
Рибосомная РНК. Она входит в состав клеточных органелл —
рибосом. Биохимическая функция рРНК пока до конца не изучена.
Предполагается, что она выполняет роль молекулярного каркаса, на котором
крепятся участники процесса трансляции; рРНК имеет большую
молекулярную массу (до 2*106), характеризуется метаболической
стабильностью. На ее долю приходится до 85—90 % всех клеточных РНК. Степень
спирализованности молекул рРНК находится в пределах 70—80 %.
93
Часть I. Биомолекулы: структура, свойства, функции
Предполагается, что в белоксинтезирующей системе клетки
функция рРНК не исчерпывается ролью структурного компонента. У про-
кариотов обнаружено, что в рРНК имеются небольшие участки,
комплементарные участкам мРНК. Спаривание этих участков, видимо,
способствует первоначальному связыванию мРНК с рибосомой. Не
исключено, что некоторые участки рРНК играют определенную роль в
формировании пептидтрансферазного центра рибосомы,
ответственного за образование пептидных связей при синтезе белка.
Транспортные РНК. Это низкомолекулярные нуклеиновые
кислоты; молекулярная масса колеблется в пределах 23 000—30 000,
каждой из 20 белковых аминокислот соответствует, по крайней мере, одна
тРНК. Однако некоторым аминокислотам специфичны от 2 до 6 тРНК;
предполагается их общее количество около 60. Они составляют
примерно 15 % общего количества клеточных РНК. Многие тРНК
получены в гомогенном состоянии, некоторые — в кристаллическом виде.
Небольшая молекулярная масса, наличие достаточно большого
количества (до 10 %) минорных оснований, которые являются
прекрасными маркерами, существенно облегчают проблему определения нук-
леотидной последовательности тРНК. В 1965 г. Р. Холли и его
сотрудники установили полную нуклеотидную последовательность аланино-
вой тРНК дрожжей; в 1967 г* А, А. Баев и сотрудники установили
последовательность нуклеотидов валиновой тРНК дрожжей. А. Рич и
др. (1975—1977 гг.) провели полную расшифровку пространственной
структуры фенилаланиновой тРНК на основе рентгенограмм с
.разрешением до 0,4 нм.
Вторичная структура тРНК в плоском изображении имеет вид
клеверного листа (рис* 2.6). тРНК содержит 4 двухцепочечных спиральных
участка, 3 из которых являются "шпильками", несущими петли из не-
спаренных нуклеотидов; 3'- и б'-концы полинуклеотидной цепи
объединены в наиболее длинный спиральный участок, образованный
водородными связями между азотистыми основаниями и завершающийся неспа-
ренным тринуклеотидом ССА, Кроме четырех основных ветвей, более
длинные тРНК содержат короткую пятую, или дополнительную, ветвь.
Две из основных ветвей непосредственно обеспечивают функцию тРНК
как адалтора (между двадцатибуквенным кодом белков и
четырехбуквенным кодом нуклеиновых кислот). Антикодоновая ветвь имеет анти-
кодон, представляющий собой специфический триплет нуклеотидов,
комплементарный кодону мРНК и способный образовывать с ним пары
оснований. Акцепторная ветвь присоединяет специфическую аминокислоту
за счет образования эфирной связи между ее карбоксильной группой и
гидроксильной группой З'-концевого остатка аденина в тРНК, Две
другие главные ветви тРНК называются дигидроуридиловая ветвь и
ТЧ'С-ветвъ. Первая содержит необычный нуклеозид дигидроуридин, а
вторая — нуклеозиды псевдоуридин (Ч*) и риботимидин (Т), обычно не
присутствующие в составе РНК.
94
Глава 2. Нуклеиновые кислоты
б'-кояец
©
М1
е
^з
З'-конец
I II!!
С
I
С
I
А
Антикодон
а б
Рис. 2.6. Схемы структуры транспортной РНК (тРНК): а — структура типа
"клеверного листа"; 2, 2, 3, 4 — двухцепочечные фрагменты, образованные при
сворачивании цепи и стабилизированные водородными связями между
комплементарными парами оснований; I, II, III — периферические петли (нет
водородных связей); IV — минорная петля (имеет неодинаковые размеры в разных тРНК);
б — пространственная структура тРНК
Исследования структуры тРНК методом рентгеноструктурного
анализа показали, что их нативные молекулы имеют компактную форму;
отдельные двухспиральные "шпильки" клеверного листа
складываются в специфическую третичную структуру, которая является
близкой для всех тРНК.
После ферментативной этерификации свободной З^гидроксигруп-
пы концевого остатка адениловой кислоты в последовательности ССА
специфической в отношении тРНК аминокислотой образуется
активная форма, называемая аминоацил-тРНК. Остаток этой
аминокислоты переносится к концу растущей полипептидной цепи. Антикодон
обеспечивает специфичность взаимодействия тРНК с мРНК. Боковые
петли, видимо, играют важную роль в связывании тРНК с аминоа-
цил-тРНК-синтетазой и с комплексом рибосома—мРНК. Аддукты ами-
ноцил—тРНК располагаются в определенной последовательности,
связанной с последовательностью кодонов мРНК.
Матричная РНК составляет незначительную часть (3—10 %) всех
клеточных РНК; молекулярная масса колеблется в широких пределах
и доходит до 14*106. Она программирует синтез всех клеточных
белков цитоплазмы. Относительное содержание индивидуальной мРНК в
суммарном препарате РНК может составлять тысячные доли
процента. Первые экспериментальные доказательства существования мРНК
получили А. Н. Белозерский, А. С. Спирин и их сотрудники (1957—
1960 гг.). Они показали, что нуклеотидный состав общей РНК бакте-
95
Часть I. Биомолекулы: структура, свойства, функции
рий Е. соИ коррелирует с составом их ДНК, и пришли к заключению
о наличии по крайней мере двух типов РНК, один из которых
(большая фракция) имеет состав, не отражающий состава ДНК, а второй
(меньшая фракция) воспроизводит состав ДНК. В дальнейшем
выяснилось, что первая фракция — это рибосомная РНК, а вторая — мРНК.
Но это сделали в 1961 г. Ф. Гросс и сотр.
Если рРНК и тРНК метаболически устойчивы, то мРНК в
большинстве случаев, особенно у прокариот, является относительно ко-
роткоживущей. Ее нуклеотидный состав близок к составу ДНК,
выделенной из того же организма.
мРНК имеют отчетливо выраженную вторичную структуру; в
состав двухцепочечных участков включено до 75 % всех нуклеотидных
последовательностей мРНК. Значительная часть участков вторичной
структуры в мРНК идентифицирована "шпильками". Однако роль
участков вторичной структуры в реализации матричных функций пока
точно не установлена. Предполагается, что "шпильки" выполняют роль
специфических структур, обусловливающих узнавание определенных
участков рибосом при их связывании с мРНК.
Если рРНК и тРНК относятся к обслуживающему аппарату белок-
синтезирующей системы клетки, то мРНК является прямым
посредником между ДНК и белками, играет роль матрицы для синтеза
последних, поэтому считают, что она выполняет роль мессенджера (гпеззеп-
§ег КИА). Сама мРНК синтезируется в ядре клетки в процессе
транскрипции у в ходе которой нуклеотидная последовательность одной из
цепей хромосомной ДНК ферментативным путем "переписывается"
.(транскрибируется) с образованием предшественника пре-мРНК;
последняя имеет копии палиндромов ДНК, поэтому ее вторичная
структура содержит шпильки и линейные участки. При созревании пре-
мРНК шпильки отсекаются ферментами и образуется мРНК.
2.10* Комплексы нуклеиновых кислот и белков
Комплексы нуклеиновых кислот с белками являются основными
компонентами многих биоструктур, к важнейшим из которых
относятся рибосомы и вирусы.
Рибосомы — внутриклеточные органеллы, обеспечивающие синтез
белка* Впервые были описаны в 1955 г. Дж. Пелейдом; термин
"рибосомы" был предложен Р. Робертсом. Рибосомы прокариотических
клеток состоят на 60—65 % из рРНК и на 35—40 % из белка; в эукарио-
тических клетках их находится приблизительно одинаковое
количество. Рибосомы представляют собой плотные сферические
образования. В бактериальных клетках рибосомы находятся в цитоплазме в
свободной форме, причем на их долю приходится до 25 % общей
массы клетки. В одной клетке находится до 1500 рибосом* Масса одной
рибосомы — 2,840е, а диаметр ее — около 20 нм. В эукариотических
клетках рибосомы крупнее (диаметр — 25—30 нм).
96
Глава 2. Нуклеиновые кислоты
Рибосомы всех клеток
имеют идентичную структуру.
А. Тисьер и сотр, (1958 г.)
обнаружили, что каждая
рибосома диссоциирует на две
неравные субъединицы (рис. 2.7).
У прокариот — это 508- и
308-субъединицы; у эукари-
от — 608- и 408-субъединицы.
Контуры рибосомных
субъединиц (а) и модель
рибосомы прокариот (6),
построенные на основе данных
электронной микроскопии,
представлены на рис. 2.8.
Рибосомы устойчивы в
растворах с относительно
высокой концентрацией ионов
М§"2+, со снижением
концентрации этих ионов
диссоциируют на субъединицы. В
состав обеих субъединиц входит
много белков, в каждой из них
молекула рРНК служит
каркасом, на котором собираются ри-
босомные белки.
Сформировавшийся рибонуклеопротеино-
вый комплекс — так
называемый рибонуклеопротеино-
вый тяж — преобразуется в
сложную компактную
частицу — собственно рибосомную
субъединицу. Концепция тяжа
как основы структурной
организации рибосомы
разработана в 60-х гг. XX в. А. С.
Спириным.
В сборке комплекса рРНК—
белок важную роль играют
электростатические
взаимодействия. Об этом
свидетельствует тот факт, что
большинство рибосомных белков
имеет основный характер, что
обусловлено высоким содер-
233-РНК
56-РНК
508-субчастица
Примерно
34 белка
708-рибосома
168-РНК
ЗОВ-субчаетица ПрИмердо 21 белок
Рис. 2.7. Структура рибосомы прокариот
308
508
а
Рис. 2.8. Построенные по данным
электронной микроскопии контуры рибосомных
субчастиц (а): большая (508) и малая (308)
субчастицы, и модель рибосомы (б) в двух
различных проекциях; слева — в так
называемой перекрывающейся проекции (308-суб~
частица закрывает собой часть 508-субчас-
тицы); справа — в боковой проекции
(Васильеву В. Д. и др., 1986)
7. Заказ 3486
97
Часть I, Биомолекулы: структура, свойства, функции
жанием в них аргинина и лизина. Однако этот тип взаимодействий не
исчерпывает всех связей между рРНК и белками. Экспериментальные
данные по реконструированию субъединиц свидетельствуют о том, что
молекулы рРНК и белков в субъединицах взаимодействуют
упорядоченным образом. Однако основные принципы нуклеиново-белкового
"узнавания" в рибосомах пока недостаточно выяснены.
Вирусы (лат. иЬгиз — яд) — неклеточные формы жизни,
обладающие собственным геномом и способные к воспроизведению в клетках
более высокоорганизованных организмов. Для вирусов характерны
две формы существования: внеклеточная или покоящаяся (вирионы)
и внутриклеточная, вегетативная, размножающаяся. Вирусы
являются облигатными внутриклеточными паразитами, поскольку их
репродукция может происходить только внутриклеточно. Они
относятся либо к рибонуклеопротеинам, либо к дезоксирибонуклеопротеи-
нам; вызывают заболевание человека, животных, растений,
микроорганизмов.
Вирусы были открыты в 1892 г. Д. И. Ивановским при изучении
мозаичной болезни табака. В настоящее время изучено большое
количество вирусов, многие из них получены в кристаллическом виде.
Получены важные данные о структуре вирусов, химической природе их
инфекционности. Вирусы характеризуются сравнительно большой
вариабельностью размеров (от 15—18 до 3000—3500 нм). Большая часть
их обладает субмикроскопическими размерами и различима лишь в
электронном микроскопе.
Химическое строение вирусов относительно простое; с этой точки
зрения их можно подразделить на две группы: первая — типичные
нуклеопротеины; вторая —• наряду с нуклеопротеиновым комплексом
они содержат в своем составе липиды и углеводы в форме липо- и
гликопротеинов. В свою очередь, в пределах этих групп существуют
градации. Так, первую группу можно подразделить на две подгруппы,
различающиеся по степени сложности белкового компонента: в одной
подгруппе белок состоит из одного вида полипептидных цепей; в
другую подгруппу входят вирусы, белок которых образован несколькими
типами полипептидных цепей.
Форма вирусов — самая разнообразная: одни из них имеют
палочковидную форму, другие — шарообразную, третьи — форму полиэдра
(шестигранника, восьмигранника и т.д.). На рис. 2.9 представлены
частицы вируса табачной мозаики (а) и модель этого вируса (б).
Форма вируса является отражением геометрических особенностей
нуклеино-белкового комплекса.
Белок вируса выполняет двоякую функцию: во-первых, он
образует наружную оболочку — капсид, защищающую нуклеиновую
кислоту от различных внешних воздействий; во-вторых, этот белок
обеспечивает специфическую адсорбцию вируса на тех клетках живых
организмов, где может происходить его размножение.
98
Глава 2. Нуклеиновые кислоты
а б
Рис. 2.9. Вирус табачной мозаики: а — частицы вируса; б — модель отрезка
частицы вируса (каждый капсомер состоит из одной полипептидной цепи с
молекулярной массой 17 420)
Г. Шрамм я Г. Френкель-Конрат независимо друг от друга на
примере вируса табачной мозаики показали, что инфекционностью
обладает не белок, а нуклеиновая кислота, входящая в состав вируса.
Вирусы оказались очень ценным инструментом в
фундаментальных исследованиях в области молекулярной биологии и генной
инженерии. Их можно рассматривать как модельные системы, которые могут
служить для исследования сложных биологических процессов
дупликации генов и их экспрессии,
У вирусов заключенная в нуклеиновых кислотах информация
невелика, она достаточна для репликации нуклеиновых кислот и
синтеза одного или нескольких белков. Внутриклеточный паразитизм
вирусов обусловлен тем обстоятельством, что они в силу своей простоты
организации используют для своего воспроизведения клеточный
синтетический аппарат (мембраны, рибосомы), ферменты и энергогене-
рирующие системы. Связь между двумя формами существования
вируса вне клетки (покоящейся) и внутри клетки (вегетативной)
осуществляется через нуклеиновую кислоту, которая индуцирует в
зараженной клетке специфические синтезы и, в конечном счете,
формирование дочерних вирусных частиц.
С биологической точки зрения вирусы подразделяются на вирусы
человека, животных, растений, бактерий; вирусы бактерий получили
название бактериофаги.
Размножение вирусов в клетке-хозяине — сложный процесс.
Первым этапом является образование комплекса вирус—клетка, после чего
7*
99
Часть I. Биомолекулы: структура, свойства, функции
вирусная нуклеиновая кислота проникает в клетку-хозяина.
Проникновение может осуществляться двумя путями: либо по механизму,
близкому к пиноцитозу (полному поглощению вирусной частицы
клеткой), либо инъекцией нуклеиновой кислоты в клетку. Первый путь
чаще всего имеет место у вирусов человека, животных, растений,
второй — у бактериофагов.
В обоих случаях вирусная нуклеиновая кислота, проникнув в
клетку-хозяина, "овладевает" биохимическими механизмами этой клетки и
переключает их на синтез молекулярных компонентов вирусной
частицы. В случае РНК-содержащих вирусов рибосомы клетки-хозяина
предпочтительно связываются не с молекулами мРНК клетки-хозяина, а с
молекулами вирусной РНК- Эт последние начинают функционировать
в качестве матриц при синтезе белков оболочки, некоторых
дополнительных ферментов, необходимых для репликации самой вирусной РНК
и других структурных компонентов вируса. В ДНК-содержащих вирусах
молекулы вирусных ДНК служат матрицей для транскрипции
комплементарных молекул вирусной РНК, способных к интенсивному комп-
лексообразованию с рибосомами клетки-хозяина и, следовательно, к
синтезу вирусных белков и ферментов, необходимых для синтеза вирусной
ДНК- После репликации вирусной нуклеиновой кислоты и синтеза
вирусных белков в клетке начинается формирование вирусных частиц,
причем оно носит спонтанный характер, представляющий собой чисто
физико-химический процесс агрегации; он заранее запрограммирован в
нуклеиновой кислоте. Об этом свидетельствуют эксперименты: если в
раствор депротеинированной нуклеиновой кислоты вируса внести его
белки, то происходит самосборка вирусной частицы.
Заключительный этап вирусной инфекции — освобождение
дочерних вирусных частиц во внешнюю среду. Здесь происходит либо
лизис клетки, либо выход вирусных частиц без ее разрушения.
Следует отметить, что образование комплекса вирус—клетка —■
строго специфический процесс: каждый вирус имеет своего определенного
партнера клетку-хозяина. Например, каждый бактериофаг поражает
свою определенную бактериальную клетку. Бактериофаги ЕзсНепсЫа
соИ поражают только клетки этой бактерии. В ряде отраслей
микробиологической промышленности они являются опасными
возбудителями инфекции. Так, среди культур бактерий С1о81гШит асеЪоЪЫу-
Исит, используемых при производстве растворителей (ацетона» бута-
нолов, этанола) методом брожения, есть мезогенные, чувствительные
к специфическому бактериофагу. При попадании в сбраживаемую среду
бактериофаг быстро поражает бактериальные клетки, брожение
внезапно прекращается. Культура С1. асе^ЬШуИсигп, имеющая
нормальную физиологическую характеристику до прекращения брожения,
быстро теряет подвижность, мельчает и лизируется.
Строго специфично действие на человека вируса СПИДа (синдрома
приобретенного иммунного дефицита); болезнь, вызываемая этим ви-
100
Глава 2. Нуклеиновые кислоты
русом, опасна и приводит к летальному исходу. Вирус СПИДа состоит из
молекулы РНК и липопротеиновой оболочки. Именно такая природа
оболочки способствует образованию комплекса иммунной Т-клетки
лейкоцитов с вирусом. Проникая в эту клетку, вирус переключает ее синтез с
антител на синтез самих вирусных частиц с последующим разрушением
клетки; иммунитет ослабевает, развивается СПИД. Любая "дремлющая"
инфекция или новые вирусы, болезнетворные бактерии вызывают
соответствующий недуг или несколько болезней сразу. Чаще всего человек
заболевает пневмонией, саркомой, лейкозами, лимфомами.
Краткое содержание главы
Структурными блоками гигантских молекул нуклеиновых кислот
являются нуклеотиды. Каждый нуклеотид содержит три различных
компонента: азотистое (пуриновое или пиримидиновое) основание,
моносахарид пентозу (рибозу или дезоксирибозу), остаток фосфорной
кислоты. Эти компоненты соединены друг с другом в такой
последовательности: азотистое основание — пентоза — фосфат. Соседние
нуклеотиды соединены друг с другом посредством эфирной связи между
моносахаридом и фосфатом другого нуклеотид а.
Гидролиз нуклеиновых кислот, выделенных из ядер клеток,
показал, что они состоят из пуриновых и пиримидиновых оснований (аде-
нина, гуанина, цитозина, тимина), дезоксирибозы и фосфорной
кислоты. Эта кислота была названа дезоксирибонуклеиновой (ДНК). Из
дрожжей была получена другая нуклеиновая кислота, содержащая
вместо дезоксирибозы рибозу. Ее назвали рибонуклеиновой кислотой
(РНК)* В нее входят основания — аденин, урацил, цитозин и гуанин.
ДНК и РНК ответственны за хранение, репликацию
(воспроизведение), транскрипцию (перенос) и трансляцию (передачу) генетической
(наследственной) информации.
Уникальны биологические функции нуклеотидов. Помимо того, что
нуклеотиды входят в состав нуклеиновых кислот, они выполняют
важную функцию в энергетическом (фосфорном) обмене, в
аккумулировании химической энергии и ее переносе; служат активными простети-
ческими группами в окислительно-восстановительных ферментах,
играют важную роль в синтезе жиров, олиго- и полисахаридов.
На основе правил Чаргаффа и данных рентгеноструктурного
анализа, Дж. Уотсон и Ф. Крик предложили двухспиральную модель ДНК
и развили важную для биохимии концепцию комплементарности. Они
предложили три уровня структуры ДНК: первичную
(последовательность нуклеотидов), вторичную (структура двух правозакрученных
спиралей), третичную (дополнительное закручивание в пространстве
двухспиральной молекулы). В образовании вторичной и третичной
структур важную роль играют водородные связи, возникающие
между парами оснований аденин—тимин и гуанин—-цитозин, а также
гидрофобные взаимодействия между основаниями, направленные вдоль
101
Часть I. Биомолекулы: структура, свойства, функции
цепей молекулы ДНК. Разрушение этих связей нагреванием или под-
щелачиванием растворов вызывает денатурацию ДНК.
Точное копирование молекулы ДНК (ее репликацию) обеспечивает
так называемый полуконсервативный механизм, заключающийся в
расхождении двух цепей материнской ДНК и использовании их в
качестве матриц для синтеза двух новых (дочерних) цепей ДНК, Этот
механизм доказан экспериментально.
РНК — однонитевые молекулы, в отличие от ДНК; их вторичная и
третичная структуры нерегулярны. По своим биологическим
функциям РНК подразделяются на три типа: рибосомная — рРНК,
транспортная — тРНК и матричная — мРНК.
рРНК входит в состав клеточных органелл — рибосом. Данный
тип РНК участвует в формировании структуры рибосом, на которых
осуществляется синтез белка. тРНК выполняют функцию
переносчика аминокислот к месту синтеза белка. мРНК передает считанную ею
информацию с ДНК на синтезируемый белок, выполняет роль
матрицы при синтезе полипептидной цепи.
К комплексам нуклеиновых кислот и белков относятся рибосомы
и вирусы. Рибосома — фабрика синтеза белка. Она считывает
информацию с мРНК и транслирует ее на молекулу белка. Вирусы —
мельчайшие частицы, проявляющие свою жизнедеятельность в случае
проникновения в клетки организма человека, животных, растений и
бактерий и вызывающие их заболевание. Вирусы — важный инструмент
в решении проблем молекулярной биологии.
Хронология важнейших дат в исследовании нуклеиновых кислот
1868 г. — Ф. Мишер выделил из лейкоцитов нуклеин,
впоследствии оказавшийся ДНК.
1889 г. — Р. Альтман ввел в науку термин "нуклеиновая кислота".
1892 г. — Д. И. Ивановский открыл вирусы, что послужило
началом развития новой науки — вирусологии.
1943 г. — О. Т. Эвери, К. Мак-Леод и М. Мак-Карти показали, что
выделенная из пневмококков молекула ДНК обладает генетической
информацией (трансформирующей активностью).
1949 г. — Э. Чаргафф открыл и сформулировал уникальные
закономерности состава ДНК, вошедшие в науку под названием правил
Чаргаффа и позволившие расшифровать структуру ДНК.
1953 г. — Дж. Уотсон и Ф. Крик предложили трехмерную модель
структуры ДНК.
1957 г. — Г. Шрамм и Г. Френкель-Конрат выделили из вируса
табачной мозаики РНК и показали ее инфекционность.
1957 г. — М. Мезелсон и Ф. Сталь установили
полуконсервативный путь репликации ДНК.
1962 г. — Дж. Уотсон, Ф. Крик и М. Уилкинс удостоены Нобелев-
102
Глава 2. Нуклеиновые кислоты
ской премии за расшифровку структуры ДНК и ее возможной
репликации.
1965 г. — Р. Холли установил структуру аланиновой тРНК* В
1966 году он удостоен Нобелевской премии за разработку блочного
метода анализа последовательности нуклеотидов.
1968 г. — А. Корана и М. Ниренберг удостоены Нобелевской
премии за исследование генетического кода.
1968 г. — А. А. Баев и сотрудники установили последовательность
нуклеотидов в дрожжевой валиновой тРНК.
ЛИТЕРАТУРА
Биополимеры /Пер. с яп. Т. Оои и др.; Под ред. Ю. Иманиси. — М.: Мир,
1988. — 544 с.
Зенгер В. Принципы структурной организации нуклеиновых кислот / Пер. с
англ. — М*: Мир, 1987. — 584 с.
Киселев Л. Л.9 Фаворова О. О., Лаврик О. Я. Биосинтез белков от
аминокислот до аминоацил-тРНК. — М.: Наука, 1984. — 408 с.
Ленинджер А. Основы биохимии. ~ М,: Мир, 1985. — Т. 3. — С. 742—1055.
Молекулярная биология: структура и биосинтез нуклеиновых кислот:
Учебник / В. И. Агол, А. А. Богданов, В. А, Гвоздев и др.; Под ред. А. С. Спирина. —
М.: Высшая школа, 1990* — 352 с.
Остерман Л. А. Методы исследования белков и нуклеиновых кислот. — М.:
Наука, 1981. — 285 с.
Рекомбинантные молекулы: значение для науки и практики / Под ред. Р.
Бирса и Э. Бесита. — Мл Мир, 1980. — 624 с.
Страйер Я. Биохимия. — М.: Мир, 1985, Т. 3- — 396 с.
ВОПРОСЫ И ЗАДАЧИ
1. Какие основания входят в состав макромолекул ДНК и РНК?
2. Что такое нуклеозид, нуклеотид? Почему растворимость в воде от
основания к нуклеотиду возрастает?
3. Какие функции выполняют в клетке нуклеотиды?
4. Что такое комплементарность нитей ДНК? Какая форма связей
обусловливает ее? Объясните причину образования пар аденин—тимин, гуанин—цитозин.
5. Что такое репликация ДНК и каков ее механизм?
6. Молекула ДНК содержит 15 тыс. пар оснований. Рассчитайте длину
молекулы.
7. Согласно данным электронной микроскопии длина хромосомы Е. сой равна
1,2 мкм. Сколько пар комплементарных оснований присутствует в хромосоме?
Глава 3. УГЛЕВОДЫ
3.1. Общая характеристика углеводов. Классификация
Углеводы — это полигидроксиальдегиды и полигидроксикетоны с
общей формулой {СН20)п , наиболее распространенный на Земле класс
органических соединений, входящих в состав всех живых
организмов. В клетках животных тканей их содержится около 2 % сухой
массы, в клетках растений — 80 % и более.
Углеводы являются первичными продуктами фотосинтеза, в
круговороте веществ в природе они играют роль своеобразного моста
между неорганическими и органическими соединениями. Выделяемая при
биологическом окислении углеводов химическая энергия
используется в реакциях синтеза, активном переносе веществ через клеточные
мембраны, преобразуется в механическую работу. Промежуточные
продукты распада углеводов служат исходными веществами для
синтеза других соединений, необходимых живой клетке. На долю
углеводов приходится 60—70 % пищевого рациона* Они содержатся
преимущественно в растительных продуктах, являются основными
компонентами хлеба, круп, макарон, кондитерских изделий; служат
исходными веществами в бродильной промышленности (спирт, вино, пиво),
в производстве пищевых кислот — уксусной, молочной, лимонной;
служат важнейшими субстратами при микробиологических синтезах
ферментов, антибиотиков. Из животных продуктов углеводы
содержатся в молоке, В результате промышленной переработки
сельскохозяйственного сырья (кукурузы, картофеля, сахарной свеклы)
получают чистые продукты — сахар (сахарозу) и крахмал.
Углеводы явились первыми веществами, выделенными в чистом
виде, ранее других соединений были подвергнуты кислотному и
ферментативному гидролизу, сбраживанию; одними из первых были
синтезированы химическим путем. Большой вклад в изучение строения и
химии углеводов внесли А. М* Бутлеров, Э. Фишер, В. Н. Хеуорс.
Класс углеводов включает самые разнообразные соединения,
начиная от низкомолекулярных, содержащих три углеродных атома, и
кончая соединениями с длинными углеродными неразветвленными и
многократноразветвленными цепями и с большой молекулярной
массой, достигающей нескольких миллионов дальтон.
Классификация углеводов основана на их структуре и
физико-химических свойствах. Структурную классификацию этих веществ можно
представить в виде следующей схемы:
104
Глава 3. Углеводы
МОНОСАХАРИДЫ
(от 3 до 10 С-атомов)
УГЛЕВОДЫ
(САХАРИДЫ)
ОЛИГОСАХАРИДЫ
(от 2 до 10 остатков
моносахаридов)
>
ПОЛИСАХАРИДЫ
(свыше 10 моиосаха-
ридных звеньев)
Производные моносахаридов
Гомополисахариды Гетерополисахариды
Уроновые Аровые Гликозиды Фосфосахара
кислоты кислоты
Как видно из схемы, условно углеводы делят на три подкласса;
моносахариды* олиеосахариды, полисахариды.
По физико-химическим свойствам углеводы делятся на
нейтральные, содержащие только гидроксильные и карбонильные группы; ос-
новные> включающие кроме названных групп аминогруппу (аминоса-
хара); кислые* содержащие кроме гидроксильных и карбонильных
групп карбоксильные группы,
3*2. Моносахариды
Моносахариды — простые сахара с п = 3-^-10 углеродных атомов,
представляющие собой полигидроксиальдегиды или полигидроксике-
тоны. Они называются соответственно алъдозами или кетозами
(окончание оза означает принадлежность к углеводам, для обозначения
простых кетоз иногда используют окончание — у лоза). В зависимости
от числа углеродных атомов, содержащихся в молекуле,
моносахариды делятся на триозы, тетрозы> пентозы, гексозы> гептозы, окто-
зы, нонозы. Самыми простыми моносахаридами являются триозы —
глицеральдегид и дигидроксиацетон:
Н О
V
н-с-он
НоС-ОН
оо
н*с~он
Глицеральдегид
Н2ООН
Дигидроксиацетон
Наиболее широко распространены в природе гексозы и пентозы.
По расположению гидроксильной группы у асимметрического
углеродного атома, дальше всех отстоящего от карбонильной группы,
все моносахариды относятся к Б- или Ь-ряду:
105
Часть I. Биомолекулы: структура, свойства, функции
Н-ООН НО-С-Н
сн2-он сн2-он
Вряд Ь-ряд
Распространенные в природе моносахариды относятся к В-ряду:
Ал ьдоз ы Кетозы
Глицеральдегид (ЗС) Дигидроксиацетон (ЗС)
Эритроза (4С) Ксилулоза (5С)
Рибоза (5С) Рибулоза (5С)
Глюкоза (6С) Фруктоза (6С)
Галактоза (6С) Седгептулоза (7С)
Маныоза (6С) Октулоза (8С)
Ниже приведены наиболее важные в физиологическом отношении
гексозы; из альдоз — глюкоза, галактоза, манноза; из кетоз — фруктоза*
Нчс^о
н-с-он
НО-С-Н
н-с-он
н-с-он
сн2он
О-Глюкоза
Нчс^о
н-с-он
1
но-с-н
но-с-н
1
1
н-с-он
сн2он
Б-Галактоза
Нхс^о
1
но-с-н
но-с-н
1
н-с-он
1
!
н-с-он
сн2он
О-Манноза
сн2он
с=о
но-с-н
н-с-он
1
н-с-он
сн2он
О-Фруктоза
В-Глюкоза является основным компонентом виноградного сахара,
хорошо сбраживается дрожжами ЗассНагогпусез сегеи1в1ае и
используется в производстве виноградных вин. В чистом виде глюкоза
усваивается организмом лучше всех других углеводов. Ее получают при
кислотном или ферментативном гидролизе картофельного,
кукурузного крахмала. Глюкоза входит в состав фруктов, зеленых частей
растений, семян хлебных злаков, играет важную роль при сбраживании
пшеничного и ржаного теста в производстве хлебобулочных изделий.
О-Фруктоза содержится в растениях, в больших количествах в меде,
плодах, поэтому часто называется фруктовым или плодовым сахаром.
Из всех моносахаридов фруктоза — самый сладкий сахар, она в 1,5 раза
слаще сахарозы, в 2 раза — глюкозы. Она заслуживает особого
внимания при производстве кондитерских изделий для больных сахарным
диабетом. Существует ферментативный метод получения фруктозы из
глюкозы. Она также сбраживается дрожжами ЗассНагогпусез сегеигзгае.
В-Галактоза сбраживается дрожжами ЗассНагогпусез /га§1Из9
широко используется в производстве кисло-молочных продуктов. В боль-
106
Глава 3, Углеводы
ших количествах она присутствует в сульфитных щелоках,
образующихся в качестве побочных продуктов при обработке древесины
бисульфитом кальция в целлюлозной промышленности.
В-Манноза сбраживается дрожжами 5. сегетзгае несколько
медленнее, чем глюкоза и фруктоза. В больших количествах
обнаруживается в сульфитных щелоках, что делает их пригодными для многих
процессов ферментации (производство кормовых дрожжей, этанола).
Моносахариды представляют собой нелетучие, легко растворимые
в воде и других полярных растворителях соединения, что обусловлено
наличием в их молекулах большего числа гидроксильных групп.
Многие из них легко кристаллизуются, устойчивы при воздействии
разведенных кислот. В более жестких условиях при воздействии кислот из
пентоз образуется фурфурол, из гексоз — оксиметилфурфурол.
Моносахариды неустойчивы при воздействии на них щелочей: в их
молекулах происходит разрыв углерод-углеродных связей.
За исключением дигидроксиацетона, все простые сахара содержат
асимметрические углеродные атомы; общее их число равно числу
расположенных внутри углеродной цепи групп -С(Н)ОН, а число стерео-
изомеров равно 2П, где п — число асимметрических атомов углерода.
Простейшая альдоза глицеральдегид с п » 1 может существовать в
виде двух различных стереоизомеров:
СНО СНО
I
I !
сн2он сн2он
Ъ-Глицеральдегид В-Глицеральдегид
Альдогексоза С6Н1206 с п = 4 может быть представлена любым из
16 возможных стереоизомеров, 8 из которых относятся к В-ряду, а
остальные 8 — к Ь-ряду. Если два стереоизомера различаются
конфигурацией одного из асимметрических углеродных атомов, они
называются эпимерами; так, манноза является эпимером глюкозы по
второму углеродному атому, галактоза — эпимером глюкозы по четвертому
углеродному атому.
Поскольку моносахариды содержат асимметрические углеродные
атомы, то они обладают оптической активностью. Интенсивность
вращения плоскополяризованного луча для разных Сахаров является
различной; она характеризуется удельным вращением, [аЦ°, представляющим
собой угол, на который поворачивается плоскость поляризованного луча,
образованного пламенем натрия (фраунгоферова линия В, составляющая
589,3 нм) и проходящего через раствор сахара (1 г/см3) в трубке длиной
100 мм при 20 °С. Моносахариды обладают как правым (+), так и
левым (-) вращением; так, В-глюкоза имеет [аЦ° = +52,5°, а В-фруктоза —
[а]^° - -92,4°. Оптическая активность Сахаров нашла широкое примене-
107
Часть I. Биомолекулы: структура, свойства, функции
ние (особенно в медицине) при их количественном определении методом
поляриметрии.
В водном растворе моносахариды образуют равновесные системы,
состоящие из молекул с развернутой цепью и молекул с циклической
структурой. Циклические структуры отсутствуют у триоз, тетроз. Начиная с
пентоз, происходит самопроизвольная реакция внутримолекулярной
конденсации одной из гидроксильных групп и карбонильной группы
моносахарида с образованием цикла (рис. 3.1).
нС-°
-о
сн2он
Замыкание связи
С1-С5 и образование
циклического
соединения
н-с-он
I а
н-с-он
но-с-н
н-с-он
I *
н-с-""
н^°
н-с-он
о
сн2он
а-Б-Глюкоза
[а]2о0 = +18,7'
37%
НО-С-Н
Н-С-ОН
Н-С-ОН
сн»он
О-Глюкоза
<-
од %
НО-С-Н
Р|
Н-С-ОН
I о
НО-С-Н
Н-С-ОН
н-с--*'"
СН2ОН
р-Э-Глюкоза
[а]*0 = +112,2°
63 %
<^
при стоянии раствора глюкозы в течение
нескольких часов [<х]^° = +52,5°
Рис. 3*1. Схематическое представление образования циклической структуры
Б-глюкозы в ее а- и Р-аномерных формах и явления мутаротации глюкозы
(штриховая линия — связь за плоскостью кольца)
В случае альдогексоз возникает шестичленное кольцо
(формируется связь С1-С5)> кетогексозы образуют пятичленное кольцо (С2-С5,
соответственно). Углеродный атом карбонильной группы становится,
таким образом, новым центром асимметрии и называется аномерным
углеродным атомом, а структуру с гидроксильной группой,
расположенной справа от аномерного углеродного атома (С-ОН), называют
а-аномером9 слева (НО-С) — $-аномером. Реакция образования полу-
ацеталя обратима, а два аномера способны превращаться друг в друга,
проходя через стадию образования молекулы со свободной
карбонильной группой. В биохимических реакциях иногда участвуют лишь один
из изомеров, иногда — оба, а иногда необходимо наличие сахара со
свободной карбонильной группой.
По физико-химическим свойствам а- и р-аномеры существенно
отличаются друг от друга. Входя в олиго- и полисахариды в качестве
строительных блоков, они образуют совершенно различные углеводы:
так, ос-В-глюкоза является структурной единицей полисахарида амило-
108
Глава 3. Углеводы
зы, р-В-глюкоза — целлюлозы; оба полисахарида имеют резко отличные
физико-химические свойства, различные биологические функции в
живой клетке. Аномеры обладают различной оптической активностью; так,
а-В-глюкоза имеет [а]*0 = +18,7°, Р-В-глюкоза — [аЦ°- +112,2°. Оба
эти соединения были выделены в чистом виде. При растворении а- или
(3-изомера В-глюкозы в воде оптическое вращение раствора постепенно
меняется, достигая [ос]^° = +52,5°; в случае раствора р-В-глюкозы
величина этого показателя уменьшается, раствора а-В-глюкозы —
возрастает; это явление получило название мутаротации.
Представленные выше формулы не способны дать всесторонние
геометрические представления о полуацетальной структуре монозы. В 1928 г.
Хеуорс предложил проекционные формулы, более близко отражающие
реальные структуры веществ. Пятичленные и шестичленные структуры
изображаются в виде планарных циклических систем, гидроксильные
группы которых у каждого углеродного атома ориентированы либо вверх,
либо вниз от плоскости кольца; передний край этой плоскости имеет
утолщенные линии. Хотя и эти структуры не передают истинной
пространственной ориентации молекулы моносахарида, тем не менее они
широко используются как удобные для быстрой оценки относительной
ориентации гидроксильных групп в молекуле моносахарида. Из-за
внешних сходств шестичленных кольчатых структур альдогексоз с пираном
они получили название пиранозы; сходные со структурой пятичленного
кольца фурана кетогексозы, пентозы называются фуранозамш
с
1
Структура пиранозы
Пиран по Хеуорсу
I
о
1
Структура фуранозы
Фуран по Хеуорсу
Для упрощенного написания структур пиранозы или фуранозы часто
углеродные атомы в составе кольца опускают.
При написании формулы любого Б-моносахарида по Хеуорсу
следует придерживаться следующих правил: 1) все группы,
расположенные справа от углеродного остова в обычных формулах (формулах
Фишера), в формулах Хеуорса занимают положение под плоскостью
кольца; 2) группы слева — над плоскостью кольца; 3) концевую
группу — СН2ОН также помещают над плоскостью кольца. Для Б-моноса-
харидов, имеющих а-конфигурацию, гидроксильная группа при ано-
109
Часть I. Биомолекулы: структура, свойства, функции
мерном углероде ориентирована вниз, а для р-конфигурации -г- вверх.
Для иллюстрации приведем переход от простой формы Фишера к
проекции Хеуорса для а-Р-глюкопиранозы и р-О-фруктофуранозы:
Н^С-ОН
н-с-он
1
I
I
I
I
1
1
I
I
СН2ОН
СНсОН
но-с-н
3 I
о
Н-С-ОН !
но-р
о
он
но-с-н
»1 1
6
СНоОН
н-с-он :
Н-С *""
51
СНоОН
он
сн2он
3 1
он н
в
а-О-Глюкопираноза
р-Б-Фруктофураноза
В форме Хеуорса галактозу, маннозу, ксилозу, арабинозу можно
представить в виде таких структурных формул:
сн2он сн2он н н
но
но
н
он
он н
он но \_к он
Р-Ю-Галактопираноза а-Б-Маннопираноза а-Ь-Арабопираноза а-Б-Ксилопираноза
В принципе альдогексозы могут существовать в виде альдофура-
нозных форм. Однако альдопиранозные формы значительно
устойчивее, чем фуранозные; поэтому именно они и преобладают в растворах
альдогексоз.
Проекционные формулы Хеуорса могут создать неправильное
представление о пространственном строении молекул углеводов ~ будто
пиранозные и фуранозные кольца являются плоскими, что в
действительности не так. На самом деле пиранозное кольцо может принимать
две конфигурации — форму кресла и форму лодки:
сн2он
но
но
но
Кресло Лодка сс-Б-Глюкоза
а — аксиальное положение (под плоскостью или над плоскостью
листа);
э — экваториальное положение (в плоскости листа).
110
Глава 3. Углеводы
С энергетической точки зрения форма кресла более устойчива;
именно она преобладает в большей части природных моносахаридов.
Однако широкое распространение получили проекции Хеуорса; они
проще и лучше отображают химические свойства моносахаридов.
Присутствие в моносахаридах гидроксильных, альдегидных и ке-
тонных групп позволяет им вступать в реакции, характерные для
спиртов, альдегидов и кетонов. Кроме того, возможны реакции,
определяемые наличием циклических структур.
Модификация имеющихся групп или введение новых
заместителей в молекулу ведет к образованию производных моносахаридов. Одни
из них используются для синтеза полисахаридов, другие являются
промежуточными продуктами обмена клетки.
Как полиолы, моносахариды могут вступать в реакцию со
спиртами с образованием простых эфиров. Аномерный атом углерода
обладает наибольшей реакционной способностью, а образовавшаяся
структура полного ацеталя называется гликозидом, образовавшаяся связь —
гликозидной связью. Так, гидроксильная группа аномерного атома
углерода легко реагирует в кислой среде с метиловым спиртом, давая
О-метильные производные — метилгликозиды. В-глюкоза дает с ним
метил-а-В-гликозид и метил-р-В-гликозид. Метилирование остальных
гидроксильных групп моносахаридов требует более жестких условий,
например, обработки диметилсульфатом или метилиодидом в
присутствии оксида серебра*
СНоОН СН*ОН СН2ОСН«
о, н
. гтт п„ Кислая /Н \] СН^ |/Н
НО N К ОН НО N V ОСН3 СН30 Л) X оснз
он н он н
сс-В-Глнжоза Метиловый спирт а-В-Метилгликозид аЛ>5-Метилгликозид
Реакция метилирования нашла широкое применение при
изучении структуры моносахаридов.
Гликозидная связь при аномерном атоме углерода легко
образуется с другими спиртами, а также в результате реакции
моносахарида с гидроксильными группами других моносахаридов.
Продуктом такой реакции является дисахарид. Полисахариды
представляют собой цепи моносахаридов, соединенных гликозидными
связями, Гликозидная связь устойчива к действию оснований, но гидро-
лизуется при кипячении с кислотами с образованием свободного
моносахарида и свободного спирта. В то же время моносахариды
обладают высокой устойчивостью к кислотам. Кислотный гидролиз
олиго- и полисахаридов нашел широкое применение в химических
методах их количественного определения, а также в крахмало-па-
111
Часть /♦ Биомолекулы: структура, свойства, функции
точной и гидролизной промышленности при гидролизе крахмала и
целлюлозы.
Из производных Сахаров наиболее важное биологическое значение
имеют эфиры фосфорной кислоты и нуклеозиддифосфатсахара:
сн2он 0 0
Н0 У °ч О-Р-О-Р-О- рибозо-11
О" О
тЛ \П- '-
Г\ОН н/ г
н
н он
р-В-Тлюкозо-6-фосфат Уридиндифосфат-р-В-галактоза
(1ШР-галактоза)
В клетке эти производные легко образуются при ферментативных
реакциях. Широко распространены глюкозо-6-фосфат, рибозо-5-фос-
фат, фруктозо-6-фосфат. Возможно и образование дифосфатов,
например фруктозо-1,6-дифосфата. Биологическое значение сахарофосфатов
состоит в том, что они представляют собой метаболически активные
формы Сахаров, играющие важную роль в биоэнергетике живой
клетки. Фосфорилирование моносахаридов переводит их в мобильное ре-
акционноспособное состояние, а при их биологическом окислении
химическая энергия моносахарида аккумулируется в фосфатном
радикале.
Фосфорилирование выгодно клетке еще и потому, что клеточная
мембрана мало проницаема для фосфорных эфиров моносахаридов,
что очень важно для активного транспорта моносахаридов из внешней
среды внутрь клетки.
Что касается нуклеозиддифосфатов, то они играют важную роль в
клетке при взаимопревращении моносахаридов, а также в биосинтезе
олиго- и полисахаридов.
При химическом или биологическом окислении концевых групп
альдоз до карбоксильных образуются три различных производных
альдоз. Кислоты, образующиеся при окислении альдегидной группы,
называются алъдоновыми* Эта реакция успешно используется в
методах количественного определения моносахаридов с помощью таких
окислителей, как фелинговые реактивы, К3Ре(СК)б, йод. Если
окислению подвергается концевая группа -СН2ОН, то при этом образуются
уроновые кислоты. Среди них важную роль играют глюкуроновая,
маннуроновая, галактуроновая кислоты. Окисление двух концевых
групп до карбоксильной дает альдаровые кислоты — глюкаровую,
галактаровую, биологическая роль которых не установлена.
Альдаровые кислоты образуются при действии достаточно сильных
окислителей, таких как азотная кислота.
112
Глава 3. Углеводы
Продукты окисления В-глюкозы:
соон сно соон
н-с-он н-с-он н-с-он
но-с-н но-с-н но-с-н
I I I
н-с-он н-с-он н-с-он
н-с-он н-с-он н-с-он
сн2он соон соон
В-Глюконовая В-Глюкуроновая В-Глюкаровая
кислота кислота кислота
Карбонильная группа моносахаридов легко вступает в реакции
восстановления с образованием сахароспиртов. В клетках растений,
микроорганизмов это восстановление осуществляется при помощи
ферментов. Простейшим сахароепиртом является трехатомный спирт
глицерин , образующийся при восстановлении глицеральдегида. Глюкоза
при восстановлении дает шестиатомный сахароспирт сорбит,
галактоза — дульцит, манноза — маннит:
СН2ОН СН2ОН СН2ОН
н-с-он н-с-он но-с-н
но-с-н но-с-н но-с-н
сн2он н-с-он но-с-н н-с-он
н-с-он н-с-он н-с-он н-с-он
сн2он сн2он сн2он сн2он
Глицерин Сорбит Дульцит Маннит
Глицерин является важным компонентом липидов. Сорбит (глю-
цит) часто встречается в различных фруктах, ягодах. В ягодах
рябины его содержание достигает 7 %, заметное количество сорбита
обнаружено в плодах слив, яблок» вишен, абрикосов, персиков» Сорбит
используется в кондитерской промышленности для придания
изделиям сладкого вкуса. Дульцит (галактит) содержится во многих
растениях, выделяется на поверхности коры деревьев. Маннит также часто
выделяется на коре деревьев; в больших количествах содержится в
"манне", которая представляет собой высохшие выделения некоторых
видов ясеня; имеется также в водорослях, плодах (ананасе), овощах
(моркови, луке). Содержание маннита в бурых водорослях (морской
капусте) Дальнего Востока колеблется от 5 до 10 %. Эти водоросли
используются в качестве сырья при получении маннита.
8, Заказ 3486
113
Часть I. Биомолекулы: структура, свойства, функции
Из других производных моносахаридов следует отметить аминоса-
хара, содержащие аминогруппу вместо гидроксильной группы, и аце-
тиламиносахара> содержащие ацетиламиногруппу (Т^НСОСНд).
Наиболее распространены 2-амино-2-дезокси-0-глюкоза (В-глюкозамин),
2-амино-2-дезокси-В~галактоза (О-галактозамин) и соответствующие
И-ацетилированные производные (Ы-ацетил-В-глюкозамин и Ы-ацетил-
В-галактозамин):
ЫН2 Н гач>СНв
О
р-В-Галактозамин ]Ч-Ацетил-(3-В-Глк>козамин
Эти производные играют важную роль в синтезе полисахаридов
клеточных стенок, мембран бактерий.
3.3. Олигосахариды
Олигосахариды представляют собой углеводы, построенные из
небольшого количества моносахаридов (от греч. о1у#оз — немногий). Они
образуются из циклических форм моносахаридов, соединяемых О-гли-
козидными связями между полуацетальными или между полуацеталь-
ной и гидроксильными группами в реакциях с отщеплением воды.
Олигосахариды делят на ди-, три-, тетрасахариды и т.д. по числу
остатков входящих в них моносахаридов. Если молекула олигосахарида
построена из остатков одного и того же моносахарида, то такой
моносахарид называют гомоолигосахаридом; если же такая молекула
построена из остатков различных моносахаридов — гетероолигосаха-
ридом. В зависимости от того, свободна или занята карбонильная группа
в каком-либо остатке моносахарида, олигосахариды могут быть
редуцирующими и находиться в а- или (3-аномерных формах либо нереду-
цирующими с отсутствием этих форм. Олигосахариды различают
также и по типу связи между остатками моносахаридов. Если в
моносахаридах а- или р-формы легко превращаются одна в другую, то при
образовании гликозидной связи конфигурация относительно аномер-
ного атома углерода становится стабильной.
Олигосахариды имеют небольшую молекулярную массу, хорошо
растворимы в воде, легко кристаллизуются и, как правило, сладкие
на вкус, Будучи по своему строению гликозидами, они относительно
легко гидролизуются кислотами, ферментами; при действии щелочей
разрушаются с разрывом межуглеродных связей. В клетках и
биологических жидкостях они находятся как в свободном виде, так и в
114
Глава 3. Углеводы
20
[а]§> = +52,5° [а]*0 = -93,0°
составе смешанных углевод-белковых комплексов (гликопротеинов),
соединяясь с белками ковалентным способом»
Среди широко распространенных олигосахаридов и имеющих
важное значение как компоненты пищевых продуктов следует отметить
дисахариды: сахарозу, содержащуюся в растительном мире и уступа-
ющую по сладости только фруктозе; лактозу, содержащуюся в
молоке. Оба дисахарида — гетероолигосахариды.
Сахароза состоит из остатков а-В-глюкопиранозы и (З-В-фруктофу-
ранозы, соединенных а, р (1->2)-гликозидной связью (рис. 3*2).
Сахароза не обладает редуцирующей способностью. Она имеет
удельное вращение [сс]р° = +66,5°, величина которого не зависит от
концентрации сахарозы и используется при определении содержания сахарозы в
корнях сахарной свеклы с помощью поляриметра (сахариметра).
Сахароза хорошо растворима в воде. С повышением температуры растворимость
повышается с 64,4 % при 0 °С до 82,9 % при 100 °С, что широко
используется при ее выделении из растворов в кристаллическом виде.
При действии кислот и специфических ферментов происходит
гидролиз сахарозы с образованием эквимолекулярной смеси глюкозы и
фруктозы:
Сахароза + Н20 ^ ос-В-глюкоза + р-В-фруктоза
[аЦ° = +66,5°
В результате этой
реакции раствор будет иметь
[<хЦ° =-40,5°. При полном
гидролизе сахарозы ее
правовращающий раствор
становится левовращающим.
Такое явление называется
инверсией, а образующийся
сахар — инвертным
сахаром.
В наибольшем
количестве сахароза содержится в
сахарной свекле (14—20 %
на массу), в сахарном
тростнике (12—18 %), поэтому
ее часто называют
свекловичным или тростниковым
сахаром. Эти растения
являются исходным сырьем
для получения сахара в пи-
щевои промышленности, а ^ 3.2. Химическая структура (а) и про-
сама же сахароза важней- странственная модель (б) молекулы сахарозы
ший сахар для непосредст- (ос-Б-глюкопиранозил-р-Б-фруктофуранозида)
СН2ОН
I " п НОСН2 о
но М—/ о ^ \г сн2он
он
он
Сахароза
[сс-Б-Гл юкопиранози л- (1 —*■ 2 )-фруктофуранозид ]
а
8'
115
Часть I. Биомолекулы: структура, свойства, функции
венного использования в пищу, при выработке всевозможных
кондитерских изделий. В большом количестве (около 50 % на массу)
сахароза содержится в отходе сахарного производства — мелассе, которая
широко используется в качестве кормовой добавки скоту, в
бродильной промышленности для производства различных продуктов
методом ферментации.
Лактоза (молочный сахар) содержится в молоке; для растений
она является редким дисахаридом: содержится в каучуконосном
растении АсНгав зароШ и в пыльниках цветов ТогзуШа. Это второй,
наиболее важный с пищевой точки зрения сахар; в его состав входят
р-В-галактоза и а-О-глюкоза, связанные между собой через
р-1-углеродный атом галактозы и четвертый атом углерода глюкозы, т.е.
р(1-»4)-гликозидной связью:
но
сн2он
>~м
он
сн2он
\ )~ ^
\
он
Лактоза
[Р-В-галактопиранозил-(1->4)-а-В-глюкопираноза]
Поскольку аномерныи атом углерода остатка глюкозы не
участвует в образовании гликозидной связи, то лактоза представляет собой
редуцирующий сахар. Эта способность лактозы используется для
количественного определения ее в молоке и молочных продуктах. Она
существует в а- и р-форме. Обе формы различаются по растворимости
в воде, форме кристаллов и физиологическому действию.
а-Лактоза имеет [а]§° = +88°, р-лактоза — [а]^° - +34°, поэтому в
процессе установления равновесия между этими формами растворы
лактозы мутаротируют. Растворимость Р-лактозы выше, чем а-лактозы; с
повышением температуры растворимость увеличивается (с 10 % при 0 °С
до 59 % при 90 °С). Гликозидная связь в лактозе весьма прочная и для ее
расщепления в процессе гидролиза следует применять крепкие растворы
соляной или серной кислоты, высокую температуру.
Довольно трудно сравнивать сахара по степени сладости. Для
ориентировки могут служить данные Грютте: сахароза — 1,00;
фруктоза — 1,25; глюкоза — 0,70; мальтоза — 0,60; лактоза — 0,25.
Лактоза присутствует в молоке всех видов млекопитающих. Этот
дисахарид обеспечивает питательную ценность молока, является
важным компонентом питания для новорожденных, служит исходным
веществом для процесса брожения, используемого при получении
кисломолочных продуктов, и в то же время — основной причиной низкой
стойкости молока при хранении.
Лактоза находит применение в фармацевтической промышленное-
ти как хорошая основа при формировании таблеток. Здесь использу-
116
Глава 3. Углеводы
ется аморфная лактоза, образуемая обычно при быстром удалении влаги
из горячих растворов лактозы и представляющая собой
высококонцентрированный раствор а- и р-лактозы в соотношении 1,5:1. В виде
готового продукта лактозу получают из молочной сыворотки,
образующейся при получении творога и сыра.
К гомоолигосахаридам относятся дисахариды мальтоза, изомаль-
тоза, трегалоза, целлобиоза, гентибиоза. При их кислотном или
ферментативном гидролизе образуется глюкоза. Различие их
физико-химических и биологических свойств обусловлено природой гликозид-
ной связи.
Мальтоза обнаружена во многих растениях в небольших
количествах; накапливается в больших количествах в солоде — обычно в
семенах ячменя, проросших в определенных условиях. Поэтому
мальтозу часто называют солодовым сахаром. Он образуется в результате
гидролиза крахмала под действием амилаз. Мальтоза содержит два
остатка О-глюкопиранозы, соединенных между собой а~(1->4)-глико-
зидной связью:
сн2он сн2он
но Л4—г о л<—г он
он он
Мальтоза
[а-В-глюкопиранозил-(1—>4)-а*В-глюкопираноза]
У второго остатка глюкозы имеется аномерный атом углерода,
который может быть как в а-, так и р-форме (преобладает р-форма).
Мальтоза обладает редуцирующими свойствами, что используется при
ее количественном определении химическими методами. Мальтозная
патока, полученная из кукурузного или картофельного крахмала
ферментативным гидролизом, широко используется в кондитерской
промышленности.
Мальтоза кристаллизуется в виде игл с одной молекулой воды.
Она легко растворима в воде. Раствор обнаруживает мутаротацию, при
переходе а-формы в р-форму мутаротация усиливается до
окончательной величины [аЦ0 = +136°.
Изомальтоза отличается от мальтозы лишь тем, что вместо сх-(1—»4)-
имеет а-(1-™>6)-гликозидную связь. Она входит в состав амилопекти-
новой фракции крахмала и гликогена, возникает при
ферментативном гидролизе крахмала.
Трегалоза имеет такой же тип а-связи. Она называется также
грибным сахаром; содержится в грибах, сине-зеленых и красных водорослях.
В пекарских дрожжах содержание трегалозы достигает 18 % на сухую
массу; ее полное название — а-В-глюкопиранозил-(1-»1)-а-В-глю-
копиранозид; не восстанавливает фелингову жидкость.
117
Часть I, Биомолекулы: структура, свойства, функции
Из дисахаридов, имеющих гликозидную связь р-типа, следует
отметить целлобиозу и генгпибиозу. Полное название целлобиозы — [5-Б-глю-
копиранозил-(1—»4)ЛЭ-глюкопираноза, гентибиозы — р-В-глюкопирано-
зил-(1^6)-В-глюкопираноза. По этим названиям легко представить
структурные формулы данных дисахаридов. Оба дисахарида имеют аномер-
ный атом углерода, восстанавливают фелингову жидкость* Целлобиоза
не синтезируется в растениях и существует в них в свободном виде,
появляясь в результате ферментативного гидролиза целлюлозы; в достаточно
больших количествах содержится в соке некоторых деревьев. Гентибио-
за в свободном виде не встречается, она входит в состав многих гликози-
дов. По-видимому, она образуется в крахмало-паточной
промышленности при кислотном гидролизе крахмала вследствие вторичной реакции
конденсации глюкозы (реверсия глюкозы).
Три-у тетра- и пентасахариды были выделены из многих
растений. Они, как правило, содержатся в небольших количествах.
Исключение представляет рафиноза* молекула которой включает галактозу,
глюкозу, фруктозу:
сн2он сн2
сн2он
он
Глюкоза
Рафиноза
[В-галактопиранозил-(а1—>^6)-В-глюкопиранозил-(а1-|32)-фруктофуранозид]
ОН
Галактоза
ОН
Фруктоза
В значительных количествах рафиноза содержится в семенах
хлопчатника, в сахарной свекле. В свежеубранных корнях сахарной
свеклы, содержащих до 20 % сахарозы, рафиноза составляет от 0,2 до 1 %
при расчете на сахарозу. При хранении свеклы содержание рафинозы
возрастает. При производстве сахара из свеклы она накапливается в
больших количествах в мелассе.
Из водных растворов рафиноза кристаллизуется в виде длинных
игл с пятью молекулами воды; [ос]р° = +105,2°; она не восстанавливает
фелингову жидкость.
Рафинозу можно считать представителем семейства олигосахаридов,
основу структуры которых составляет сахароза плюс один остаток или
более О-галактозы. Так, тетрасахарид стахиоза содержит рафинозу плюс
галактозу; ее полное название: 0-галактопиранозил-(а1->б)-0-галакто-
пиранозил-(а1-^6)-0-глюкопиранозил-(а1^р2)-В-фруктофуранозид.
Тетрасахарид вербиоза — это стахиоза плюс галактоза. Из названий стахио-
зы и вербиозы нетрудно представить их структурные формулы. Оба оли-
госахарида содержатся в семенах бобовых: сое, вике, чечевице, люцерне.
Считают, что они представляют собой запасную форму О-галактозиль-
ных, В-фруктозильных остатков в растениях.
118
Глава 3. Углеводы
К гомоолигосахаридам относятся мальтотриоза — В-глюкопира-
нозил-(а1^4)-В-глюкопиранозил-(а1^4)-В-глюкопираноза, паноза —
В-глюкопиранозил-(а1-»6)-В-глюкопиранозил-(а1—>4)-В-глюкопирано-
за. Мальтотриоза является продуктом ферментативного гидролиза
крахмала.
3,4, Полисахариды
\
Полисахариды (долиолы, гликаны) — высокомолекулярные
углеводы, представляющие собой продукты конденсации моносахаридов,
содержащих от нескольких десятков до сотен тысяч моносахаридов,
соединенных гликозидными связями. Они могут быть как линейны-
ми, так и разветвленными; делятся на гомо- и гетерополисахариды в
зависимости от того, построены ли их молекулы из остатков
моносахаридов одного вида или из остатков различных моносахаридов.
Полисахариды не дают отчетливо оформленных кристаллов, не
растворяются в воде, набухая в ней. Они практически не обладают
восстанавливающими свойствами. Под действием кислот гидролизу-
ются до моносахаридов, а под действием щелочей деградируют*
Преобладающей моносахаридной единицей полисахаридов является В-глю-
коза. Распространены в природе также, но в меньшей степени,
полисахариды О-маннозы, О-фруктозы, В- и Ь-галактозы, В-ксилозы,
В-арабинозы.
Название гомополисахаридов складывается исходя из природы
мономерных единиц. Так, полисахариды, построенные из В-глюкоз-
ных единиц (крахмал, целлюлоза), называют глюканами;
полисахариды, построенные из маннозы, — маннанами.
Поскольку гетерополисахариды обычно состоят только из двух
различных мономеров, расположенных повторяющимся образом, они не
являются информационными молекулами. Полисахариды сильно
различаются по размеру; к тому же количество мономерных остатков в составе
одного и того же соединения может различаться для разных образцов.
Поэтому молекулярная масса полисахаридов лишь до некоторой степени
может характеризовать их физико-химические свойства.
В связи с биологической функцией полисахариды делятся на
резервные и структурные. Большинство резервных полисахаридов
(крахмал, гликоген, инулин) являются важнейшими компонентами
пищевых продуктов, выполняя в организме человека функцию источника
углерода и энергии. Структурные полисахариды (целлюлоза, гемицел-
люлоза) в клеточных стенках растений образуют протяженные цепи,
которые, в свою очередь, укладываются в прочные волокна или
пластины и служат своего рода каркасом в живом организме.
Крахмал — главный резервный полисахарид растений, запасается
во многих семенах, клубнях, корневищах и используется только
тогда, когда эти органы прорастают. В клубнях картофеля его
содержится около 20 %, кукурузе — 55—60 %, рисе — около 80 %, в муке и
119
Часть I. Биомолекулы: структура, свойства, функции
крупах — 70—80 % ■ Крахмал является основным компонентом
важнейших пищевых продуктов. В цитоплазме клеток он откладывается
в виде зерен, которые могут быть выделены из гомогенатов методом
дифференциального центрифугирования. Крахмальные зерна —
высокоорганизованные структуры, форма и размеры которых очень
разнообразны, но, как правило, характерны для каждого вида растения.
Форма зерен может быть сферической, яйцевидной, чечевицеобразной
или неправильной и размер их может колебаться от 1 до 150 мкм, в
среднем 30—50 мкм. Зерна содержат до 20 % воды (из которых 10 %
химически связаны с крахмалом) и состоят из ряда концентрических
слоев крахмала. Однако молекулярная архитектура этих слоев
остается неизвестной.
Рентгенографические исследования позволили установить, что
крахмальные зерна имеют микрокристаллическую структуру, хотя по
внешнему виду — это белый аморфный порошок. Обладая высокой
степенью организации, крахмальные зерна состоят из участков с
кристаллической структурой и участков, имеющих аморфное или гелеобраз-
ное строение. В определенных условиях (например, при повышении
температуры) кристаллические участки разрушаются, переводя зерна
целиком в аморфное состояние. Минеральные кислоты при
комнатной температуре быстро разрушают аморфную часть зерен крахмала,
а оставшаяся нерастворимая часть зерен приобретает отчетливо
выраженное кристаллическое строение. Наиболее крупные крахмальные
зерна — у картофеля, а самые мелкие — у риса и гречихи. Зерна
подразделяются на простые и сложные; простые зерна представляют
собой однородные образования (картофель, пшеница, рис); сложные
зерна — сочетание более мелких частиц (овес). Плотность воздушно-
сухого крахмала достаточна высокая и в среднем равна 1,5. Это
используется при его выделении на крахмало-паточных заводах из
крахмальных суспензий методами седиментации (отмучивания) или
центрифугирования. Водные растворы крахмала оптически активны, [аЦ°
колеблется от 180 до 220°.
Крахмал подобно белкам обладает гидрофильными свойствами.
Однако в холодной воде крахмальные зерна лишь набухают, но не
растворяются. Процесс набухания крахмала носит эндотермический
характер. Если водную суспензию крахмала нагревать, то
интенсивность набухания возрастает. Под действием тепловой энергии
разрушается структура крахмальных зерен, они увеличиваются в размерах
и при определенной температуре разрушаются; крахмальная
суспензия превращается в вязкий коллоидный раствор — крахмальный
клейстер. Температура клейстеризации крахмала не одинакова для
различных растений и находится в пределах 55—75 °С. При дальнейшем
повышении температуры вязкость крахмального клейстера
снижается; при снижении температуры до комнатной клейстер превращается
в упругий прочный гель с трехмерной структурой. Эти физико~хими~
120
Глава 3. Углеводы
ческие свойства крахмала играют важную роль при переработке крах-
малосодержащего сырья в пищевой промышленности. Так, в
технологии хлебопечения формирование мякиша хлеба связано с
образованием геля. От процесса старения этого геля при хранении хлеба в
основном зависит его черствение. При тепловой обработке крахмалосодер-
жащего сырья разрушение крахмального геля является важной
технологической операцией в крахмалопаточной, пивоваренной и
спиртовой промышленности.
Сырьем для промышленного получения крахмала служат зерна
кукурузы, пшеницы, риса, сорго, картофель. Из зерновых, богатых
белками, набухшие семена подвергаются сырому помолу с
последующим отмучиванием (отделением) крахмальных зерен от
сопутствующих веществ. После растирания клубней картофеля зерна крахмала
отмываются, отстаиваются из суспензии и перемалываются.
Характерным свойством крахмала является его способность
окрашиваться йодом в темно-синий цвет, причем эта окраска появляется в
присутствии следовых количеств крахмала в водной среде.
Крахмал на 96—98 % состоит из полисахаридов. В нем найдены в
небольшом количестве белки, высокомолекулярные жирные
кислоты, которые адсорбированы на крахмальных зернах, а также
минеральные кислоты — фосфорная и кремниевая.
Полисахаридная фракция крахмала состоит из двух компонентов:
амилозы и амилопектина.
Амилоза легко растворима в теплой воде и дает нестойкие раство-
ры со сравнительно низкой вязкостью. Длительное хранение раствора
амилозы (особенно на холоде) приводит к выпадению ее в осадок. Этот
процесс, заключающийся в формировании организованных агрегатов,
носит название ретроградации амилозы. Этим, отчасти, можно
объяснить процесс черствения хлеба при его хранении. При внесении в
раствор амилозы йода он окрашивается в синий цвет.
Амилопектин растворяется в воде при кипячении; особенно
интенсивно он переходит в раствор при температуре 120 °С. Растворы
амилопектина, вязкие и стойкие при хранении^ при действии на них йодом
окрашиваются в красно-фиолетовый цвет.
Молекула амилозы имеет линейную структуру, представляет
собой длинную цепочку из остатков а-В-глюкопиранозы, соединенных
а-(1^4)-гликозидными связями:
ОН 1 ОН 1
а-(1^4)-Гликозидная связь
• — восстанавливающий конец
121
Часть I. Биомолекулы: структура, свойства, функции
Один из концевых остатков ос-В-глюкопиранозы (правый на
рисунке) является редуцирующим*
Цепи амилозы полидисперсны, их молекулярная масса варьирует
от нескольких тысяч до 500 000 и более. Количество остатков
глюкозы в каждой цепи колеблется от 100 до нескольких тысяч. С помощью
метода дифракции рентгеновских лучей показано, что молекула
амилозы скручена в спиралевидную структуру диаметром 13 нм; в
каждый виток спирали входит б остатков глюкозы. Глубина и тип
окраски в характерной реакции с йодом зависят от длины цепи (степени
по л имериз ации):
Степень полимеризации Окраска
45 и выше Синяя
35—40 Пурпурная
20—30 Красная
12 и ниже Нет окраски
Эти переходы окраски отчетливо можно проследить при
кислотном или ферментативном гидролизе крахмала,
Амилопектин, в отличие от амилозы, имеет сильно разветвленную
(древовидную) структуру; в его молекулу входит до 50 000 а-О-глюко-
пиранозных остатков. Наряду с а-(1^4)-связями он имеет также
а-(1—»6)-гликозидные связи, представляющие собой точки ветвления
(рис. 3.3). Гликозидные ос-(1—»6)-связи составляют около 5 % от
общего количества связей, содержащихся в молекуле амилопектина.
Несмотря на ветвистое строение, молекула амилопектина имеет только
один восстанавливающий конец. В связи с ничтожной долей
восстанавливающих гликозидных концов, содержащихся в амилозе и ами-
лопектине, крахмал относят к нередуцирующим полисахаридам.
Красно-фиолетовая окраска амилопектина с йодом дает основание
считать, что ветви амилопектина скручены в спирали и в каждой из
них содержится не менее 25 глюкозных остатков. Во всяком случае,
структура амилопектина значительно сложнее, чем это изображено на
рис. 3.3: во-первых, число точек ветвления значительно больше и, во-
вторых, структура является трехмерной, с ветвями, идущими во всех
направлениях и придающими молекуле сферическую форму. Около
50—60 % гликозидных остатков молекулы амилопектина содержится
в наружных цепях.
В крахмале большинства растений на долю амилопектина
приходится 70—90 %, остальные 10—30 % составляет амилоза. Однако
содержание этих компонентов может изменяться в зависимости от сорта
растения, типа ткани, из которой он извлечен. Различается крахмал
из круглых и мозговых сортов гороха, из листьев и клубней
картофеля, из различных сортов кукурузы. Если содержание амилозы в
крахмале из зрелого картофеля составляет 22 %, то в молодых побегах —
46 %. Если в крахмале из зерна обычной кукурузы содержится
амилозы 20 %, то в крахмале восковидной кукурузы амилоза отсутствует
122
Глава 3. Углеводы
СН2ОН
СН2ОН
А он X Аон /
он
но
а-(1-»6)-Глико.
зидная связь
СН2ОН
СН,ОН
СН2ОН
СН
НО
* + щ
ОН
ОН
он
он
ос-(1^4)-Гликозидная связь
Рис. 3.3. Химическая структура молекулы амилопектина: • —
восстанавливающий конец; ® — невосстанавливающий конец; 00 — а-(1~»4)-гликозидная
связь; О© — а-(1—»6)-гликозидная связь
полностью. С другой стороны, выведены сорта кукурузы, крахмал
которых содержит около 80 % амилозы. Соотношение амилоза/амило-
пектин изменяется также во время созревания кукурузного зерна.
Гликоген — животный крахмал, сходен с амилопектином, также
представляет собой форму хранения углерода и энергии в клетках
человека и животных. Больше всего гликогена содержат клетки печени
и мышц. Молекула гликогена имеет более "упакованную" структуру,
сильно разветвлена; ветви короче, каждая содержит не более 15—
20 глюкозных колец; точки разветвления у него встречаются через
каждые 8—10 остатков. Молекулы гликогена пол и дисперсны, их
молекулярная масса гораздо выше, чем амилопектина, и колеблется в
пределах 107—109. Наряду с огромным количеством нередуцирующих
групп молекула гликогена, подобно амилопектину, имеет одну
редуцирующую группу. Под действием йода гликоген окрашивается в
красно-коричневый цвет.
Гликоген встречается и в растениях. Так, в кукурузе помимо
обычного крахмала (амилозы и амилопектина) находится фитогликоген,
который имеет более высокую степень ветвления, чем амилопектин;
около 10 % его гликозидных связей находится в виде а-(1^6)-связей,
тогда как в обычном амилопектине - 5%; встречается в грибах и дрож-
123
Часть I. Биомолекулы: структура, свойства, функции
жах. Контроль дрожжей на присутствие гликогена является важным
показателем в бродильной технологии, по которому судят о
физиологической активности дрожжей.
Гликоген может быть выделен из животных тканей при обработке
их гомогенатов горячим раствором КОН.
Фруктаны (полифруктозиды) широко распространены в
растениях. Как правило, они представляют собой низкомолекулярные
полимеры В-фруктофуранозы, которые играют роль запасных
питательных веществ, являются ценным углеводным компонентом пищевых
продуктов. Строение фруктанов в деталях еще не выяснено, но
известно, что большинство из этих полимеров имеет разветвленную
структуру. Лучше всего из растительных фруктанов изучен инулин. Он
обнаружен в клубнях топинамбура, корнях цикория (10—16 % массы
растений), в клубнях георгина, в корневищах кок-сагыза, одуванчика и
в некоторых водорослях.
Получают инулин из гомогенатов клубней топинамбура
экстрагированием водой с последующим вымораживанием экстракта или
осаждением спиртом. Инулин представляет собой белый гигроскопический
аморфный порошок, трудно растворимый в холодной воде, хорошо —
в горячей воде. При растворении дает вязкие коллоидные растворы
сладковатого вкуса.
Инулин легко гидролизуется под действием органических и
неорганических кислот, фермента инулазы с образованием О-фруктозы,
В-инулибиозы, В-глюкозы.
Молекула инулина состоит из остатков р-В-фруктофуранозы
(около 95 %),и сс-О-глюкопиранозы (около 5 %) и представляет собой цепь
из 35—40 фруктозных остатков, соединенных (3-(2—»1)-гликозидными
связями. Остаток молекулы а-В-глюкопиранозы присоединен к
редуцирующему концу полифруктозидной цепи так же, как и в молекуле
сахарозы (рис. 3.4).
Средняя молекулярная масса инулина, выделенного из различных
источников, колеблется в пределах 4000—6000. В растениях инулин
часто встречается вместе с рядом соединений, близких к нему по
структуре. Предполагается, что эти соединения служат
предшественниками для его синтеза или продуктами его ферментативного гидролиза
при прорастании клубней, корневищ.
Другим типом фруктанов является леван» у которого остатки
фруктозы соединены гликозидными (3-(2—>6)-связями. Леван, как и
инулин, также содержит а-В-глюкопиранозный остаток. Таким образом,
и инулин, и леван можно считать запасными полисахаридами —
производными сахарозы. Ни один конец полисахаридной цепи этих
фруктанов не является восстанавливающим, поскольку аномерные
углеродные атомы концевых моносахаридных остатков участвуют в
образовании гликозидной связи. Цепи у левана короче, чем у инулина;
обычно содержат 7—10 остатков Р-Б-фруктозы.
124
Глава 3. Углеводы
НО
НОСН2 .0
НОСН2 .0
ОН
а
> Сахароза
НОСН2 п
О .
11=304-40
"СН2 г,
СН2ОН
СН2ОН
НО
О --СНо л
ез
Г
сн2он
он
р.
СН2ОН
Рис. ЗА. Химическая структура молекул инулина (а) и левана (б)
Леваны обнаружены в листьях, стеблях и корнях однодольных
растений, среди которых наибольшее значение имеют злаковые- У этих
растений леваны функционируют главным образом как временный
запасной полисахарид. Их синтезируют также некоторые
непатогенные микроорганизмы — Вас. зиЫШз, Вас. пи8еп1епеи8у
развивающиеся в средах, содержащих сахарозу. Эти бактерии являются опасными
вредителями в технологии хлебопечения, вызывая "тягучую" или
"картофельную" болезнь хлеба.
Целлюлоза (клетчатка) — высокомолекулярный линейный
полисахарид, состоящий из остатков глюкозы, соединенных (3-(1—»4)-гли-
козидными связями (рис. 3.5). Число глюкозных остатков в молекуле
целлюлозы колеблется от 1400 до 14 000, что соответствует
молекулярной массе от 250 000 до 2 000 000. В растительных клетках
целлюлоза играет роль структурного компонента. Молекулы целлюлозы
отличаются высокой прочностью. Эта прочность, как полагают,
является результатом пространственного расположения гликозидных
остатков, обусловленного наличием р-(1—>4)-связей, а также водородных
связей между гидроксильными группами, находящимися у шестого и
второго углеродных атомов смежных остатков глюкозы.
р-Конфигурация гликозидных связей в молекуле целлюлозы
обусловливает сильно вытянутую структуру полимерной цепи.
Повторяющимся звеном в молекуле целлюлозы является остаток целлобиозы.
В растительных клеточных стенках молекулы целлюлозы связаны
друг с другом бок о бок, образуя структурные единицы, получившие
125
Часть I. Биомолекулы: структура, свойства, функции
СН^ОЫ ОН СН2ОН ОН
0Н А СН2ОН 1 0Н СН2ОН
Р-(1->4)-Гликозидная связь
Рис. 3.5. Химическая структура молекулы целлюлозы
название микрофибрилл. Каждая микрофибрилла состоит из пучка
молекул целлюлозы, расположенных по ее длине параллельно друг другу.
В поперечном разрезе микрофибрилла имеет овальную форму.
Молекулы целлюлозы, составляющие "ядро" микрофибриллы, расположены в
трехмерном пространстве так упорядоченно, что образуют
кристаллическую решетку. Вокруг кристаллического ядра находится область, в
которой молекулы целлюлозы хотя и расположены параллельно, но менее
упорядоченно; эта область называется паракристаллической. В паракри-
сталлическую область входят и другие полисахариды — чаще всего ге-
мицеллюлозы; обычно их меньше всего у поверхности "ядра", но по мере
удаления от него количество молекул гемицеллюлоз возрастает.
Предполагается, что именно смесь цепей целлюлозы и гемицеллюлоз не дает
молекулам целлюлозы паракристаллической области образовывать
кристаллическую решетку. Молекулы воды способны проникать в паракри-
сталлическую область, но не в кристаллическое "ядро" микрофибриллы.
Микрофибриллы в растительных стенках достигают в длину несколько
микрон. Относительно небольшое число молекул целлюлозы проходит
по всей длине микрофибриллы. Большая часть молекул целлюлозы
короче микрофибриллы. Поэтому по длине микрофибриллы встречаются
зоны, где одни молекулы целлюлозы оканчиваются, а другие
начинаются. В этих зонах кристаллическая структура нарушена.
Длина большой и малой осей поперечного среза микрофибриллы
всех высших растений составляет 8,5 и 4,5 нм соответственно. Длина
этих осей кристаллического "ядра" равна 5 и 3 нм. На поперечном
срезе "ядра" насчитывается примерно 50 молекул целлюлозы. На
площади поперечного среза всей микрофибриллы находится около 100 по-
лисахаридных молекул (целлюлозы и гемицеллюлоз).
Рентгеноструктурные исследования показали, что в полимерной
цепи остатки молекул глюкозы повернуты относительно друг друга на
180°, что делает возможным образование водородных связей между
ОН-группой при атоме С-3 одного глюкозного остатка и кислородом
пиранозного кольца следующего остатка глюкозы. Это препятствует
вращению расположенных рядом остатков глюкозы вокруг
соединяющей их гликозидной связи. В результате образуется похожая на ленту
молекула, в которой все пиранозные кольца в конфигурации кресла
лежат в одной плоскости.
126
Глава 3. Углеводы
Выполняя в растениях роль структурного, опорного материала,
целлюлоза широко распространена в природе. В некоторых растениях
она присутствует почти в чистом виде. Например, особенно богаты
целлюлозой волокна хлопка, где ее содержание составляет 95—98 %;
в лубяных волокнах льна, джута, рами — 60—65 %; древесные
породы содержат до 50 % целлюлозы на сухие вещества.
Целлюлоза не усваивается организмом человека. Она усваивается
травоядными животными, в желудочно-кишечном тракте которых
находится специфическая микрофлора, расщепляющая целлюлозу и
преобразующая ее в легко усвояемые животными соединения.
Кислотный гидролиз целлюлозы при температуре 170 °С приводит
к образованию глюкозы, которая используется для получения
кормовых дрожжей, этилового спирта.
Для получения самой целлюлозы из древесины чаще всего
применяют сульфитный метод, заключающийся в обработке целлюлозоео-
держащего сырья смесью бисульфита кальция или натрия и
сернистой кислоты при повышенных температурах. Такая технологическая
операция приводит к удалению лигнина, гемицеллюлоз и пектиновых
веществ. Целлюлоза широко используется в целлюлозно-бумажной
промышленности.
Целлюлоза является самым распространенным органическим
соединением на нашей планете; в ней заключено до половины общего
количества диоксида углерода, вовлекаемого в процесс фотосинтеза зелеными
растениями из атмосферы. Громадные ресурсы целлюлозосодержащего
сырья делают ее неисчерпаемым источником для получения
питательных веществ {пищевого сахара, белков, жиров, пищевых кислот), а
также биологически активных веществ в результате микробиологического
синтеза (ферментов, витаминов, лекарственных средств).
Дексгпраны. К ним относятся нейтральные гомополисахариды,
состоящие из остатков а-В-глюкозы, соединенных гликозидной связью
а-(1^б). Эти соединения образуют растворы чрезвычайно высокой
вязкости. Молекулы декстранов сильно разветвлены: наряду с основной
а-(1н>6)-евязыо имеют а-(1н>4)- и а-(1^3)-связи, являющиеся
точками ветвления (рис. 3.6).
Молекулярная масса декстранов исчисляется десятками миллионов;
величина [а]р° = +200°. Декстраны синтезируются слизеобразующими
бактериями. Они не входят в состав пищевых продуктов. У бактерий
ос~(1^6)-Гликозидная связь
СН2 л ф у^СН2 вуСН,
У<^о^
он
а
он он он он
|и^— а-(1->3)-Глюкозидная связь
Рис. 3.6. Химическая структура молекулы декстрана
127
1
Часть I. Биомолекулы: структура, свойства, функции
эти вещества играют роль резервных полисахаридов и выполняют
защитные функции, Декстраны откладываются на поверхности
бактериальной клетки, образуя защитный слой — капсулу, в десятки раз
превышающий размеры самой бактериальной клетки. Капсула
защищает бактерию от бактериофагов и от воздействия жестких физико-
химических факторов. Бактерии выдерживают температуру выше 85 °С,
при температуре 40 °С способны размножаться. На предприятиях
пищевой промышленности, связанных с производством и переработкой
сахарозы, они являются опасными вредителями, способны
развиваться в застойных зонах, образуя вязкую гелеобразную массу, известную
под названием "клек", осложняющую тралспортировку жидких
продуктов по коммуникациям и вызывающую прямые потери сахарозы
на сахарных заводах при получении сахара, в бродильных отраслях
промышленности, перерабатывающих мелассу. Декстрановое
брожение может быть причиной порчи молочных консервов, куда
слизистые бактерии попадают вместе с сахарозой.
С другой стороны, декстраны нашли практическое применение; из
них готовят сефадексы, широко используемые в
научно-исследовательских работах в качестве "молекулярных сит"» Продукты их неполного
гидролиза с молекулярной массой 70 000—90 000 применяются в
качестве заменителя плазмы крови.
Гемицеллюлозы — это сложная смесь полисахаридов, не
растворяющихся в воде, но растворимых в щелочных растворах.
Гемицеллюлозы всегда сопутствуют целлюлозе; в больших количествах
содержатся в соломе, семенах, отрубях, кукурузных початках, древесине.
В комплексе с целлюлозой выполняют структурную функцию.
Под действием кислот гемицеллюлозы гидролизуются легче, чем
целлюлоза. Продуктами гидролиза у различных гемицеллюлоз являются
манноза, галактоза, арабиноза, ксилоза. Гемицеллюлозы могут быть
подразделены на гексозаны (маннаны, галактаны) и пентозаны (арабаны,
ксиланы). Встречаются либо гомо-, либо гетеродолисахариды.
Галактаны — полисахариды, молекулы которых построены
преимущественно из остатков О- или Ь-галактозы; в состав галактанов
входят иногда остатки арабинозы. Одним из наиболее
распространенных соединений является галактан, построенный из остатков
В-галактозы, соединенных р-(1—>4)-гликозидными связями (рис. 3.7). В
молекуле содержится 100—400 остатков О-галактозы.
СН2ОН СН2ОН СН2ОН СН2ОН
Т^°у о у^ча/° у <у^° м\
он он ^ он он
Р-(1-»4)-Гликозидная связь
Рис. 3.7. Химическая структура молекулы галактана
128
Глава 3* Углеводы
К галактанам относится агароза — составная часть агара,
выделенная из красных водорослей. Она имеет линейное строение и
содержит остатки В-галактозы и 3,6-ангидро-Ь-галактозы, соединенных
поочередно Ь-(1—»4)- и а-(1^3)-гликозидными связями.
Агароза является основным компонентом агар-агара —
полисахарида, встречающегося в некоторых морских водорослях. В холодной
воде агар-агар не растворим, но переходит в растворимое состояние в
горячей воде; при охлаждении раствор агар-агара превращается в гель.
Это свойство агар-агара широко используется в кондитерской
промышленности при изготовлении желе, пастилы, мармелада, джемов, в
микробиологических лабораториях — для изготовления твердых
питательных сред. В хвойных деревьях, особенно в лиственнице,
содержится растворимый арабаногалактан, имеющий разветвленную
структуру, основой которой являются остатки В-галактозы, соединенные
Р-(1—»3) и Р-(1—>4)-связями. В небольшом количестве галактан
содержится в клубнях картофеля, в корнях сахарной свеклы, в дрожжах.
Маннаны представляют собой полимерные цепи остатков
маннозы, соединенных р-(1—»4)-связью с некоторыми р-(1^6)-разветвления-
ми; содержат от 200 до 400 остатков маннозы в молекуле. В виде
аморфных образований они занимают пространство между
фибриллами целлюлозы. Известны гетерополимеры маннозы (М) и
галактозы (Г) — галактоманнаны:
Г Г Г
I I I
МММ Г Г
м-м-м-м-м-м- м-м-м-м-м-м-
МММ Г
г г г
Являясь наполнителями клеточных стенок растений, маннаны
часто входят в состав резервных полисахаридов, играющих
определенную роль в энергетическом обмене растений.
Маннаны входят в состав некоторых водорослей, древесины
хвойных деревьев.
Арабаны — полимеры Ь-арабинозы, остатки которой соединены
ос-(1^5)-связью. Степень полимеризации колеблется в пределах 150—
200; [ос]^° = ~129°. К каждому остатку цепи через ос-(1-»3)-связь
присоединено ответвление из одного остатка моносахарида. В большом
количестве арабаны содержатся в плодах и овощах.
Ксиланы содержат остатки ксилозы, соединенные р-(1—»4)-связью;
степень полимеризации варьирует от 50 до 200 ксилозных остатков.
Эти полисахариды нередко частрхчно ацетилированы и содержат
боковые ответвления в виде остатков Ь-арабинозы.
9. Заказ 3486 129
ъ
Часть I. Биомолекулы: структура, свойства, функции -
Ксиланы в большом количестве (до 25 %) имеются в соломе, в
некоторых видах твердой древесины, кукурузных початках. Они
содержатся в ржаной и пшеничной муке, обладают свойствами к
неограниченному набуханию, в связи с чем играют важную роль в
формировании теста.
При продолжительном кипячении пентозанов с крепкой соляной
кислотой (12—14 %) образуется фурфурол:
сн— сн о
К II I
СН С —С
^о" I
н
Фурфурол
Таким путем фурфурол получают на гидролизных заводах.
Исходным сырьем для этого являются отходы сельскохозяйственного сырья
(хлопковая шелуха, подсолнечная лузга и т.д.). Сам фурфурол
широко используется в производстве полимеров.
Многие гемицеллюлозы наряду с гексозанами и пентозанами
содержат метилированные уроновые кислоты: либо метилглюкуроновую
кислоту (МГ) и ксилозу, либо метилгалактуроновую кислоту и арабинозу:
. ..Ксилоза~МГ—Ксилоза—МГ—Ксилоза...
Ксилоза Ксилоза
Ксилоза Ксилоза
I I
Ксилоза Ксилоза
Например, пшеничная солома содержит гемицеллюлозу, состоящую
из галактуроновой кислоты, арабинозы и ксилозы примерно в
соотношении 1:1:23.
Пектиновые вещества (полиурониды) — сложные эфиры полига-
лактуроновой кислоты и метилового спирта. Полиурониды,
состоящие главным образом из остатков галактуроновой кислоты,
соединены а-(1—»4)-гликозидной связью. В клеточных стенках растений,
образованных из целлюлозы, они вместе с гемицеллюлозами
выполняют структурные функции, являются цементирующим материалом этих
стенок, объединяют клетки в единое целое в том или ином органе
растений.
Различают три основные группы пектиновых веществ:
пектиновая кислота, пектин и протопектины.
Пектиновая кислота — это цепь, состоящая из остатков О-галак-
туроновой кислоты. Большинство пектиновых кислот содержит от 5
до 100 этих остатков. Молекула представляет собой растянутую цепь,
в которой имеется незначительная возможность свободного вращения
вокруг гликозидных связей.
130
Глава 3, Углеводы
Пектин — это пектиновая кислота, у которой ряд свободных
карбоксильных групп образуют сложные эфиры с метиловым спиртом
(рис. 3.8),
С—ОСН3
он * соон он с-осн3
а-(1-^4)-Гликозидная связь
Рис. 33* Химическая структура молекулы пектина
Пектин содержит 100—200 остатков В-галактуроновой кислоты.
Определить степень метоксилирования пектина затруднительно, так как
эфирные связи при экстракции легко разрываются. Однако установлено,
что пектин из различных растений имеет различную степень
метоксилирования и ни один из них полностью не метоксилирован. Метоксилиро-
вание влияет на устойчивость и растворимость полимера. В то время как
пектиновые кислоты относительно устойчивы, пектины быстро деполи-
меризуются в нейтральных и особенно щелочных средах. Экстракты
пектиновых веществ гетерогенны, с трудом поддаются анализу.
Растворимые в воде, особенно при нагревании, пектиновые вещества — аморфные
соединения, осаждаются этанолом, ацетоном; осадок имеет вид студня.
Они довольно устойчивы к кислотному гидролизу.
Получают пектиновые вещества из различных плодов, сахарной
свеклы и очищают многократным переосаждением. И пектиновая кис*
лота, и пектин образуют весьма вязкие коллоидные растворы.
Благодаря наличию свободных карбоксильных групп они несут высокий
отрицательный заряд и способны осаждаться ионами металлов. Эта
способность тем выше, чем ниже степень метоксилирования их
молекул. В кислой среде диссоциация карбоксильных групп пектиновой
кислоты и пектина подавляется, что способствует их агрегированию и
переходу в гелеобразное состояние. Особенно интенсивно этот процесс
идет при рН 3—4, в 60—80 %-ных растворах сахарозы. Присутствие
даже небольших количеств пектиновых веществ (0,5—1,0 %) ведет к
образованию твердого геля. Поэтому пектиновые вещества находят
широкое применение в кондитерской промышленности при
изготовлении желе, джема, мармелада, зефира и начинок для карамели.
Для всех нерастворимых пектиновых веществ существует общее
название протопектин. Протопектин легко расщепляется, переходя в
растворимую форму, поэтому его строение и состав в деталях не
известны. Препараты протопектина неизменно содержат целлюлозу, геми-
целлюлозы. Видимо, в состав протопектина входит пектин, ковалент-
но связанный с целлюлозой и гемицеллюлозами.
9*
131
Часть 7. Биомолекулы: структура, свойства, функции _^_
Пектиновые вещества широко распространены в качестве
важнейшего компонента клеточных стенок растений. В достаточно больших
количествах они содержатся в овощах и плодах. Так, в сахарной
свекле, картофеле их количество колеблется от 1 до 3 %; в яблоках,
грушах, сливах, цитрусовых — от 0,5 до 2,5 %♦ Большая часть
пектиновых веществ в них существует в форме протопектина.
Предполагается, что при созревании плодов и овощей часть протопектина
переходит в растворимую форму — плоды и овощи размягчаются. Пищевой
пектин получают из плодов (яблок, цитрусовых) и из свекловичного
жома.
Пектиновые вещества играют в пищевой промышленности и
отрицательную роль. В свеклосахарном производстве пектиновая кислота
и пектин из свекловичной стружки переходят в диффузионный сок, в
котором при его дальнейшей очистке с помощью известкового молока
[водной суспензии Са(ОН)2] образуются пектаты кальция, в
результате чего резко возрастает вязкость очищенного сока, что затрудняет его
фильтрацию.
3.5. Гликопротеины
Многие углеводы в виде линейных или разветвленных олигосаха-
ридов входят в состав белков в качестве простетических групп. Они
ковалентно связаны с определенными боковыми цепями
аминокислотных остатков, в больших количествах содержатся в клеточных
мембранах. Гликопротеины весьма разнообразны с точки зрения
химической структуры углеводных компонентов. Углеводный фрагмент
может составлять от 1 до 80 % от молекулярной массы гликопротеи-
на. Ковалентная связь возникает путем образования гликозидной
связи между углеводов и остатками серина, треонина, аспарагина
боковых полипептидных цепей белковой макромолекулы.
Моносахаридами, входящими в состав углеводных фрагментов, могут быть О-глю-
коза, В-галактоза, В-манноза.
Основная функция гликопротеинов — передача сигналов
узнавания клетки — клеткой, клетки — органеллой, клетки — молекулой,
молекулы — молекулой. Они выполняют важную функцию в
биоэнергетике клетки.
3.6. Подходы, приемы и методы в исследовании углеводов
При изучении химической природы углеводов очень часто трудно
получить их в чистом виде. Перегонка этих веществ невозможна, а
ряд других веществ, в частности минеральные соли, белки,
присутствующие в растениях, осложняют процесс получения чистых
препаратов углеводов. Здесь большую роль в прошлом сыграли приемы
введения в молекулу углевода различных органических радикалов,
например, метильного СН3- или ацетильного СН3СО~* Метилирование и
132
Глава 3, Углеводы
ацетилирование, проводимые в мягких условиях, позволяют получить
препараты метальных и ацетильных производных
высокомолекулярных полисахаридов большей чистоты, чем исходные вещества. Вместе
с тем введение метильных или ацетильных групп в молекулу
полисахарида существенно упрощает определение структуры входящих в его
состав моносахаридов. Метилированные производные моносахаридов
устойчивы в кислой среде. Исчерпывающее метилирование нашло
широкое применение при определении типа гликозидной связи в оли-
го- и полисахаридах, поскольку атомы углерода гликозидной связи не
участвуют в метилировании, но легко расщепляются при кислотном
или ферментативном гидролизе.
Весьма важный прием в изучении химической структуры
полисахаридов — их частичный кислотный или ферментативный гидролиз.
Так, с помощью кислотного гидролиза было показано, что
структурным блоком целлюлозы является целлобиоза, ферментативным же
гидролизом установлено, что основной структурный блок крахмала —
мальтоза. В то же время на примере крахмала и целлюлозы можно
видеть, какое решающее значение имеет тип связи. Оба полисахарида
состоят из одних и тех же звеньев В-глюкопиранозы, но в крахмале
они соединены а-(1^4)-связью, в целлюлозе — р-(1—>4)-связью.
Однако оба полимера существенно отличаются по своим
физико-химическим свойствам и биологическим функциям.
За последние 15—20 лет наши представления о молекулярной
массе многих полисахаридов, таких, как крахмал, гликоген,
существенно изменились. Показано, что величины молекулярных масс были
занижены на порядок, а в некоторых случаях на несколько порядков,
* ■ ш ш
Оказалось, что прочность гликозидных связей в молекуле
полисахаридов сильно переоценивалась и в результате при получении чистых
препаратов полисахаридов мы имели дело с продуктами
деполимеризации, образующимися при относительно жестких условиях
выделения. Применение щадящих методов (фенольная экстракция,
экстракция холодной водой, дифференциальное центрифугирование) дало
возможность получить препараты, более близкие к нативным.
В исследовании структуры полисахаридов большое значение имеет
метод рентгеноструктурного анализа. Так, с помощью этого метода
было установлено, что целлюлоза имеет участки с кристаллической и
аморфной структурой, что целлюлозные волокна представляют собой
пучки фибрилл, покрытые оболочкой из пектиновых веществ и геми-
це л л юл оз.
Чтобы выяснить, в какой форме находится моносахарид — фураноз-
ной или пиранозной, проводят его окислительную деградацию в кислом
растворе в присутствии солей йодной кислоты (перйодатов). Йодная
кислота растворяет моносахарид. Характер образующихся продуктов позво-
ляет определить тип кольца. Пиранозиды при окислении дают диальде-
гид и муравьиную кислоту, фуранозиды — лишь диальдегид.
133
Часть I- Биомолекулы: структура, свойства* функции
В исследованиях углеводного остатка клетки, химической
структуры углеводов большое значение имеют методы их определения. Они
подразделяются на методы качественной пробы и количественные
методы. Целью качественных проб очень часто является
идентификация или самого углевода, или олиго- и полисахаридов, установление
химической природы их мономеров. В последнем случае эта
возможность представляется только после гидролиза этих соединений.
Обязательным условием является удаление посторонних примесей,
особенно белков, пигментов, С этой целью используется соединения,
денатурирующие белки и специфические адсорбенты.
Большинство качественных проб на сахара основано на их
восстановительной способности. Следует особо подчеркнуть качественную
пробу на крахмал-йодную реакцию. Она широко используется в
биохимических научных исследованиях, поскольку является
специфической на крахмал.
Количественные методы определения Сахаров нашли широкое
применение в исследованиях биологических жидкостей,
сельскохозяйственного сырья и пищевых продуктов. Они весьма разнообразны.
Важными из них являются поляриметрический, титрометрический*
ферментативный.
Поляриметрический метод широко применяется при определении
содержания сахара в плодах и овощах, сахарной свекле, крахмала в
картофеле, семенах хлебных злаков, а также в исследовательских
работах по биохимии и биотехнологии. Титрометрический (редуктомет-
рический) метод основан на восстанавливающей способности Сахаров.
Путем определения количества окислителя, восстановленного
раствором сахара, можно вычислить концентрацию сахара. На этом, в
частности, основаны методы определения глюкозы в крови и моче при
диагностике сахарного диабета, при котором наблюдается
значительное возрастание концентрации глюкозы в крови и выведение избытка
ее с мочой. При определении Сахаров по методу Бертрана используют
реактивы Фелинга (щелочной раствор сегнетовой соли и раствор
сернокислой меди). При кипячении раствора сахара со смесью этих солей
в щелочной среде в результате окислительно-восстановительной
реакции сахар окисляется до соответствующей кислоты, а двухвалентная
медь восстанавливается до одновалентной и выпадает в осадок в виде
Си20, который количественно определяют методом перманганатомет-
рии. В качестве окислителя широко используется красная соль
К3Ге(СК)6. При кипячении раствора сахара с этой солью в щелочной
среде она восстанавливается в желтую кровяную соль К4Ре(СН)6. Этот
окислитель используется в методах Хагедорна—Иенсена, Иссекутца.
Избыток непрореагировавшей соли К3Ре(СК)6 определяют йодомет-
рически. Титрометрические методы имеют высокую точность, тем
не менее все они трудоемки, не являются специфическими для
отдельных Сахаров, С этой целью были осуществлены модификации
134
Глава 3. Углеводы
этих методов — переход с редуктометрических анализов на
колориметрические.
При осторожном нагревании моносахаридов с неорганическими
кислотами происходит их превращение в фурфурол, оксиметилфурфурол*
Последние в кислой среде могут давать окрашенные соединения с
различными циклическими веществами (а-нафтолом, антроном, фенолом).
В биохимических исследованиях при определении глюкозы, в
частности, достаточно широко используется антроновый метод, при котором
интенсивность окраски определяется спектрофотометрически.
Альдосахара при нагревании в слабокислой среде способны
конденсироваться с ароматическими аминами (анилином,
дифениламином, о-толуидином) и образовывать окрашенные соединения. Это
свойство можно использовать в методах их количественного определения
колориметрически.
Фруктозу можно обнаружить при помощи пробы с резорцином.
Резорциновый метод основан на превращении фруктозы в присутствии
концентрированной серной или соляной кислот в
оксиметилфурфурол, образующий с резорцином красно-коричневое окрашивание,
интенсивность которого пропорциональна концентрации фруктозы.
Лактозу и галактозу можно обнаружить по образованию слизевой
кислоты путем их окисления азотной кислотой. Слизевая кислота выпадает
из раствора в виде осадка. Сахарозу можно определить по кислотному
гидролизу и изменению величины угла вращения поляризованного
света, по восстанавливающей способности инвертного сахара.
Однако самой высокой специфичностью обладают методы,
основанные на использовании высокоочищенных препаратов ферментов.
Этот подход все шире применяется в настоящее время в
исследованиях углеводов.
Несмотря на разнообразие химических и ферментативных методов
обнаружения и количественного определения углеводов, ими нельзя
охарактеризовать весь состав Сахаров в исследуемой пробе. Это стало
возможным при использовании электрофоретического разделения борат-
ных комплексов Сахаров, различных видов хроматографии на бумаге, в
тонком слое силикагеля, колоночной и газожидкостной хроматографии.
Применяя различные системы растворителей и проявителей, можно с
высокой степенью достоверности определить качественный состав
Сахаров в пробе, измерить количество каждого сахара и выделить его
препаративно для дальнейших исследований. Наиболее перспективными и
точными методами идентификации и количественного определения
углеводов являются методы газовой и газожидкостной хроматографии.
3.7. Углеводы в пищевых продуктах
Основным источником углеводов в пище человека являются
продукты растениеводства. С биохимической точки зрения все углеводы
пищи можно подразделить на усвояемые организмом и неусвояемые.
135
Часть I. Биомолекулы: структура, свойства» функции
Неусвояемые углеводы не перевариваются в желудочно-кишечном
тракте, не всасываются в кишечнике, а если и всасываются, то не
вступают в метаболические процессы в организме. К неусвояемым
углеводам относятся в основном полисахариды: целлюлоза, гемицеллюлозы,
пектиновые вещества; олигосахариды, в частности рафиноза, а также
некоторые простые сахара. Пищеварительные секреты слюнной
железы, желудка и кишечника не выделяют ферментов, способных
расщеплять эти углеводы. Неусвояемые полисахариды образуют группу
балластных веществ, называемых пищевыми волокнами. Хотя в
технологии пищевых продуктов стремятся от них избавиться с целью
получения более "рафинированной" пищи, однако присутствие их в
пищевых продуктах необходимо, поскольку они играют
существенную роль в поддержании нормальной регуляции питания и
метаболизма ряда веществ, способствуют лучшему функционированию
(перистальтике) кишечника. Пищевые волокна нормализуют обмен
холестерина, способствуют удалению из организма вредных веществ
(токсинов), катионов тяжелых металлов, нормальному развитию
полезной кишечной микрофлоры.
Поскольку усвояемые углеводы являются основным источником
химической энергии в организме человека, потребность в них
колеблется в широких пределах. Человек при физическом труде средней
тяжести должен получать 350—400 г углеводов в сутки; при особо
тяжелых работах потребность в углеводах достигает 600 г; у лиц,
занятых в основном умственным трудом, потребность в углеводах
составляет 300—380 г. Углеводы должны покрывать половину
потребности организма в энергии. Такая доля потребления углеводов
достаточна для обеспечения глюкозой клеток головного мозга и
эритроцитов, для которых глюкоза является единственным источником
энергии. Недостаток углеводов в рационе вынуждает организм
расходовать для образования глюкозы аминокислоты, что может вызвать их
дефицит в организме.
Следует отметить, что в технологии пищевых продуктов их
тепловая обработка оказывает некоторое отрицательное воздействие;
особенно это касается восстанавливающих моно- и дисахаридов* Эти
сахара вступают в сахароаминную реакцию с аминокислотами,
особенно с незаменимыми аминокислотами — лизином и метионином,
увеличивая их дефицит в пищевых продуктах.
Все пищевые продукты, служащие источником усвояемых
углеводов, можно подразделить на три группы: хлеб и хлебопродукты,
сахаристые продукты, овощи и фрукты.
Основным углеводным компонентом хлеба и хлебопродуктов
является крахмал. Средний углеводный состав хлеба: 35—45 % крахмала,
небольшие количества моно-, дисахаридов (до 2—4 %) и целлюлозы и
гемицеллюлоз (1—5 %)• В крупах и макаронных изделиях крахмала
65—70 %, моно- и дисахаридов — 2—3 % на массу изделия. В сред-
136
_ Глава 3, Углеводы
нем за счет хлеба и хлебопродуктов обеспечивается ежедневное
поступление в организм человека 150—200 г углеводов.
Сахаристые продукты (сахар, патока, мед, варенья, джемы,
шоколад, продукты переработки фруктов и ягод с высоким содержанием
сахара — мармелад, пастила) более чем на 50 % состоят из моно- и
олигосахаридов на массу продукта*
Калорийность сахаристых продуктов весьма высока и находится в
пределах 1200—1600 кДж на 100 г продукта. Ввиду того, что в
современных условиях энергозатраты человека сравнительно невелики,
рекомендуется умеренное потребление сахаристых продуктов» При
систематическом злоупотреблении они способствуют развитию
тяжелых недугов (ожирение, гипертония). Установлено, что каждые
лишние 25 г сахара способствуют образованию 10 г жира. Продукты
обмена углеводов легко превращаются в холестерин, способствуют
возникновению сахарного диабета.
Замена в сахаристом продукте менее сладкого сахара на более
сладкий или тем более его полная замена на сладкое вещество неуглевод -
ной природы может существенно снизить калорийность продукта,
повысить его биологическую ценность. Ксилит, сорбит, фруктоза
ассимилируются клетками печени без участия гормона инсулина, что
позволяет использовать их в питании больных сахарным диабетом,
ксилит и сорбит — в питании людей, склонных к полноте. Ценным
источником фруктозы в сахаристых продуктах может явиться инулин,
который сравнительно легко расщепляется до этого моносахарида
кислотным или ферментативным гидролизом.
Овощи и фрукты используются в качестве пищевых продуктов и как
сырье в пищевой промышленности. В большинстве овощей содержание
углеводов — в среднем 8 %, из которых моно- и дисахаридов — около
5 %; в некоторых овощах содержится значительное количество
крахмала (в картофеле — 18 %, в бобовых — 30 %)♦ Глюкоза и фруктоза
содержатся во всех фруктах; в яблоках и грушах (семечковых плодах)
преобладает фруктоза, а в сливах, вишне, абрикосах — глюкоза и сахароза.
На долю усвояемых углеводов во фруктах приходится от б до 18 %.
Плоды и овощи содержат относительно большое количество балластных
углеводов (до 1,5 %) — целлюлозы, гемицеллюлоз, пектиновых веществ.
Краткое содержание главы
Углеводы — класс органических соединений, широко
распространенных на Земле. В клетках животных тканей их содержится около
2 %, в клетках растений — 80 % и более.
По структуре углеводы делятся на три подкласса —
моносахариды, олигосахариды, полисахариды.
Моносахариды содержат от 3 до 10 атомов углерода в молекуле.
Наиболее распространены в природе гексозы и пентозы О-ряда.
Важными гексозами являются глюкоза, галактоза, манноза, фруктоза.
137
Часть I. Биомолекулы: структура, свойства, функции
В метаболизме клетки особую роль играет глюкоза как источник
углерода и энергии. Моносахариды оптически активны, обладают как
правым, так и левым вращением плоскополяризованного луча света, В
водных растворах они образуют равновесные системы, состоящие из
молекул с развернутой цепью и молекул с циклической структурой,
имеющей а- или р-аномерный атом углерода. Аномерный атом
углерода обладает наибольшей реакционной способностью, может вступать
в реакцию с соединениями, содержащими ОН-группу,
Образовавшееся при этом соединение называется гликозидом, а возникшая связь —
гликозидной.
Олиго- и полисахариды представляют собой цепи моносахаридов,
соединенных гликозидными связями.
Олигосахариды делятся на ди-, три-, тетрасахариды и т.д. по
числу остатков входящих в их состав моносахаридов. В зависимости от
того, свободна или занята карбонильная группа в каком-либо остатке
моносахарида, олигосахариды могут быть либо редуцирующими в
а- или (5-аномерных формах, либо нередуцирующими с отсутствием
этих форм. В зависимости от типа гликозидной связи олигосахариды
бывают а- или (3-гликозидами.
Среди широко распространенных олигосахаридов и имеющих
значение как компоненты пищевых продуктов следует отметить гетеро-
сахариды: сахарозу, содержащуюся в растительном мире; лактозу,
содержащуюся в молоке.
Молекула сахарозы представляет собой а-В-глюкопиранозил-р-В-фрук-
тофуранозид с а1-(32-гликозидной связью. Сахароза является нереду-
цирующим сахаром, хорошо растворима в воде. При действии кислот
и специфических ферментов она гидролизуется, образуя
эквимолекулярную смесь глюкозы и фруктозы. Гидролиз сахарозы получил
название "инверсия", а продукты гидролиза называются инвертным
сахаром.
Лактоза [^-В-галактопиранозил-(1^4)*а(р)-В-глюкопираноза]
является редуцирующим сахаром. Она присутствует в молоке всех видов
млекопитающих.
К гомоолигосахаридам, структурным блоком которых является
В-глюкоза, относятся мальтоза, изомальтоза, трегалоза, целлобиоза,
гентибиоза.
Полисахариды представляют собой продукты конденсации
моносахаридов, содержащих от нескольких десятков до сотен тысяч
моносахаридов. Они могут быть линейными и разветвленными, делятся на
гомо- и гетерополисахариды. Преобладающей моносахаридной
единицей является В-глюкоза.
В связи с биологической функцией полисахариды делятся на
резервные и структурные. Резервные полисахариды (крахмал,
гликоген, инулин) являются важнейшими компонентами в питании
человека. Структурные полисахариды (целлюлоза, гемицеллюлоза, пекти-
138
Глава 3. Углеводы
новые вещества) в клеточных стенках растений образуют
протяженные цепи, которые, в свою очередь, укладываются в прочные волокна
и служат каркасом в растении.
Главным резервным полисахаридом растений является крахмал;
он запасается в семенах хлебных злаков, клубнях, корневищах.
Состоит из двух компонентов: амилозы и амилопектина. Молекула
амилозы имеет линейную структуру, представляет собой цепочку из
остатков а-В-глюкопиранозы, соединенных <х-{1-^4)-гликозидными
связями. Цепи амилозы поли дисперсны, их молекулярная масса
варьирует от нескольких тысяч до 500 000 и более.
Амилопектин, в отличие от амилозы, имеет сильно разветвленную
структуру, в его молекулу входит до 50 000 а-О-глюкопиранозных
остатков. Наряду с а-(1—>4)-связями он имеет также ос-{1—И>)-гликозид-
ные связи, представляющие собой точки ветвления цепей. В связи с
ничтожной долей восстанавливающих гликозидных концов,
содержащихся в амилозе и амилопектине, крахмал относят к нередуцирую-
щим полисахаридам*
В крахмале большинства растений на долю амилопектина
приходится 70—90 %, а остальные 10—30 % составляет амилоза. Близким по
структуре к амилопектину является животный крахмал — гликоген.
Больше всего он содержится в клетках печени и мышц. В отличие от
амилопектина, гликоген имеет более "упакованную" структуру,
молекулярная масса его гораздо выше и колеблется в пределах 107—109.
Фруктаны широко распространены в растениях. Они
представляют собой низкомолекулярные полимеры О-фруктофуранозы. Среди них
хорошо изучен инулин, содержащийся в клубнях топинамбура,
цикория. Он легко гидролизуется кислотным и ферментативным
способами. Молекула инулина состоит из остатков р-В-фруктофуранозы
(около 95 %) и а-В-глюкопиранозы (около 5 %) и представляет собой цепь
из 35—40 фруктозных остатков. Заслуживает внимания как
источник получения фруктозы.
Целлюлоза — высокомолекулярный линейный полисахарид,
состоящий из остатков В-глюкозы, соединенных Р-(1->4)-гликозидны-
ми связями. Длинные прямые цепи молекул целлюлозы легко
вступают в межмолекулярные взаимодействия с образованием
надмолекулярных структур. В клеточных стенках растений эти структуры
плотно "упакованы" в слои, которые дополнительно стабилизованы геми-
целлюлозами и пектиновыми веществами. Эти структурные
компоненты не усваиваются организмом человека. Они усваиваются
травоядными животными, в желудочно-кишечном тракте которых
находится специфическая микрофлора, расщепляющая эти компоненты
до легко усвояемых животными соединений.
К полисахаридам бактериального происхождения относится дек-
стран. В нем остатки а-В-глюкопиранозы соединены а-1,6-гликозид-
ными связями. Это ветвящийся полисахарид, который наряду с ос-
139
Часть I, Биомолекулы: структура, свойства, функции
новной ос-1,6-связыо имеет <х-(1—»4)- и а-(1—»3)-связи. В водных средах
декстран дает гелеобразную малоподвижную массу.
Гемицеллюлозы — это сложная смесь полисахаридов, не
растворяющихся в воде, но растворимых в щелочных растворах. В
зависимости от структурного блока, входящего в состав их молекулы,
гемицеллюлозы подразделяются на маннаны, галактаны, арабаны, ксиланы.
В растениях распространены также гетерогемицеллюлозы.
Углеводы — важнейшие компоненты пищевых продуктов. Они
являются основным источником углерода и энергии для организма
человека.
Хронология важнейших дат в исследовании структуры углеводов
1861 г, — А. М. Бутлеров создал теорию химического строения
органических веществ. Им впервые был получен формальдегид, а из
него — смесь синтетических сахароподобных веществ.
1902—1915 гг. — Э. Фишер определил структуру и
пространственную конфигурацию молекул Сахаров, осуществил синтез некоторых
из их производных.
1928 г. — В. Н. Хеуорс предложил проекционные формулы
моносахаридов, близко отражающие их реальные структуры.
ЛИТЕРАТУРА
Березов Г. Т., Коровкин Б. Ф. Биологическая химия: Учебник. — 3-е изд.,
перераб. и" доп. — М.: Медицина, 1998. — 704 с.
Лектины в исследовании белков и углеводов / Под ред. Ю. А. Овчинникова.
— М.: ВИНИТИ, 1985, — 288 с.
Степаненко Б. Н. Химия и биохимия углеводов (моносахариды). — М.:
Высшая школа, 1977. — 224 с.
Степаненко Б. Н* Химия и биохимия углеводов (полисахариды). — М.:
Высшая школа, 1978. — 256 с.
Степаненко Б. Н. Современные проблемы биохимии углеводов. — М.:
Наука, 1979. — 54 с.
Филиппович Ю* Б. Основы биохимии: Учебник. — 4-е изд., перераб* и доп. —•
М.: Изд-во "Агар", 1999. — 512 с.
ВОПРОСЫ И ЗАДАЧИ
1. Для каждого углевода выберите одно верное определение:
а) глюкоза является кетогексозой, дисахаридом, альдопентозой, кетопенто-
зой, альдогексозой;
б) фруктоза является кетогексозой, альдогексозой, кетопентозой,
альдопентозой, дисахаридом;
в) полисахарид, в состав которого входят остатки р-В-фруктозы, является
целлюлозой, декстраном, амилозой, инулином, пектином, леваном,
2. В результате кислотного гидролиза (инверсии) сахарозы получено
10,5 ммолей глюкозы. Рассчитайте количество сахарозы в исходном растворе.
3. При полном гидролизе крахмала, содержащегося в 10 г пшеничной муки,
образовано 7,6 г глюкозы. Определите крахмалистость пшеничной муки.
140
Г лава 3. Углеводы
4. Выберите из нижеследующих утверждений правильное:
а) моносахариды являются гидрофильными, гидрофобными веществами;
б) полисахариды, состоящие из остатков а-О-глюкозы: целлюлоза,
пектиновые вещества, леван, инулин, крахмал, гликоген.
5. Из 10 г гомогената картофеля был извлечен крахмал и подвергнут
кислотному гидролизу, при этом получено 1,8 г глюкозы. Рассчитайте крахмалистость
картофеля.
6. Из 200 г корнеплода сахарной свеклы извлечена сахароза и подвергнута
кислотному гидролизу. Химическим методом определено ОД г-моля инвертного
сахара. Рассчитайте сахаристость корнеплода сахарной свеклы.
Глава 4, ЛИПИДЫ
Липиды (от греч. Проз — жир) — разнородная группа природных
соединений, почти или полностью нерастворимых в воде, но
растворимых в органических растворителях (сероуглероде, хлороформе, эфире,
бензоле), дающих при гидролизе высокомолекулярные жирные
кислоты. Наряду с белками, нуклеиновыми кислотами и углеводами
липиды образуют четвертый класс соединений в живых клетках. Термин
липид относится не к какой-то определенной группе соединений,
обладающих общими, легко идентифицируемыми особенностями строения*
Он объединяет самые разнообразные по структуре вещества, общий
признак которых — наличие высокомолекулярных жирных кислот и
гидрофобность. Многие липиды, однако, содержат как минимум одну
полярную группу, которая может служить местом связывания с
другими компонентами. Липиды, содержащие как полярные, так и
неполярные группы, называются амфипатическими. Они обычно встречаются
в биологических мембранах и других поверхностях раздела между
водной средой и гидрофобными областями внутри клеток.
В организме липиды выполняют четыре основные функции: 1)
являются резервными соединениями, основной формой запасания
энергии и углерода; 2) служат формой, в которой транспортируется эта
энергия; 3) являются структурными компонентами мембран и 4)
несут защитную функцию в плодах, овощах, листьях растений, в
клеточных стенках бактерий.
По структуре липиды можно подразделить на три группы: 1)
простые липиды; 2) сложные липиды; 3) производные липидов.
К простым липидам относятся только эфиры жирных кислот и
спиртов. В состав сложных липидов входят помимо жирных кислот и
спиртов другие компоненты различного химического строения*
Производные липидов — это, в основном, жирорастворимые витамины и
их предшественники (см. гл. 5 "Витамины")*
4.1. Жирные кислоты
Более 100 различных жирных кислот идентифицированы в липи-
дах микроорганизмов, растений и животных. Их молекулы
представляют собой длинные углеводородные цепи с концевой карбоксильной
группой (рис. 4.1). Жирные кислоты отличаются друг от друга дли-
ной их углеводородного хвоста, степенью их ненасыщенности (т.е.
142
Глава 4. Липиди
числом двойных
углерод-углеродных связей в углеводородном
хвосте) и положением двойных
связей в цепях жирных кислот.
Природные жирные кислоты
весьма разнообразны, однако
имеют ряд общих черт. Они
представляют собой монокарбоновые
кислоты, содержащие линейные
углеводородные цепи, причем чаще с
четным числом атомов углерода
от 14 до 22. Гораздо реже
встречаются жирные кислоты с более
короткими цепями или с
нечетным числом атомов углерода.
Содержание ненасыщенных
жирных кислот в липидах, как
правило, выше, чем насыщенных.
Если в их молекулах
присутствуют две или большее число
двойных связей, почти всегда они
разделены метиленовои группой
-СН=СН-СН2-СН=СНК В
липидах высших организмов двойная
связь ненасыщенных жирных
кислот находится в большинстве
случаев между 9-м и 10-м
атомами углерода. Дополнительные
двойные связи обычно
встречаются на участке между 10-м
углеродным атомом и метильным
концом. Двойные связи почти всех
природных жирных кислот
имеют цис- конфигурацию.
Физические свойства
насыщенных и ненасыщенных
жирных кислот значительно
отличаются. Обычно насыщенные
жирные кислоты имеют твердую
воскообразную консистенцию при
комнатной температуре (22 °С),
тогда как ненасыщенные жирные
кислоты представляют собой
масла при данной температуре.
Ненасыщенные жирные кислоты
Жирная
кислота
Жирная
ацильная
группа
Углеводородный
хвост
;:
/
о=с
а СН2
/*
(ЗСН2
гт2
8СН2
*\
6 СН2
/•
СН2
7\
СН2
/»
сн2
сн2
сн2
соСНз
12
Рис. 4.1. Основная структура и
номенклатура жирных кислот.
Жирные кислоты содержат длинный
углеводородный хвост, связанный с
карбоксильной группой. Поскольку
рКа карбоксильной группы около
4,5—5,0, то жирные кислоты при
физиологических значениях рН
представляют собой анионы. Согласно
номенклатуре ГОРАС (Международный
союз фундаментальной и
прикладной химии) углерод карбоксильной
группы нумеруется СМ, а остальные
углеродные атомы нумеруются
последовательно, начиная с этой
позиции. По общей номенклатуре
углеродный атом, примыкающий к
углероду карбоксильной группы,
обозначается через а, остальные
углеродные атомы обозначаются р, у, 8, е и
т.д. Углеродный атом, наиболее
удаленный от углерода карбоксильной
группы, обозначается как ю-атом, не-
зависимо от длины хвоста. На
рисунке изображен лаурат — жирная
кислота, содержащая 12 углеродных
атомов и относящаяся к
насыщенным кислотам, поскольку не
содержит двойных углерод-углеродных
связей
143
Часть I. Биомолекулы: структура, свойства, функции ^
имеют более низкую температуру плавления, чем насыщенные.
Большинство нейтральных жиров, богатых ненасыщенными жирными
кислотами, остается в жидком состоянии до 5 °С и ниже. Ненасыщенные
жирные кислоты, имеющие только одну двойную связь называются
мононенасыщенными , а имеющие две и более — полиненасыщенными.
Длина углеводородной цепи жирной кислоты и степень
ненасыщенности влияют на точку плавления кислоты. Точки плавления для
ряда жирных кислот указаны в табл. 4.1. С увеличением длины
углеводородного хвоста точки плавления тоже повышаются. Ван-дер-вааль-
совы взаимодействия между соседними углеводородными хвостами
усиливаются по мере увеличения их длины, и больше энергии
требуется для разрыва ван-дер-ваальсовых связей при плавлении. В
качестве примера можно указать на такие жирные кислоты, как лаурат
(12:0), миристат (14:0) и пальмитат (16:0).
Насыщенные и ненасыщенные жирные кислоты сильно различаются
по своей структурной конфигурации- В насыщенных жирных кислотах
углеводородный хвост в принципе может принимать бесчисленное
множество конфигураций вследствие полной свободы вращения вокруг
одинарной связи; однако наиболее вероятной является вытянутая форма,
поскольку она энергетически наиболее выгодна- В ненасыщенных
кислотах наблюдается иная картина: невозможность вращения вокруг
двойной связи (или связей) обусловливает жесткий изгиб углеводородной цепи,
В природных жирных кислотах двойная связь находится в цис-конфигу-
рации. Она дает изгиб цепи под углом приблизительно 30°; в случае транс-
формы двойной связи конформация цепи мало отличается от конформа-
ции насыщенной цепи, Дис-форма менее стабильна, чем /яранс-форма-
Таблица 4.1
Некоторые представители жирных кислот,
входящих в состав липидов мембран
Число
С-атомов
12
14
16
18
20
24
16
18
18
18
20
Число
двойных
связей
0
0
0
0
0
0
1
1
2
3
4
Найме н ован ие
Лаурат
Миристат
Пальмитат
Стеарат
Арахинат
Лигноцерат
Пальмитоолеат
Олеат
Линолеат
Линоленат
Арахидонат
Точка
плавления, °С
44
52
63
70
75
84
-0,5
13
-9
-17
-49
Молекулярная формула
(анионная форма)
СН3(СН2)]0СОО-
СН3(СН2)12СОО-
СН3(СН2)14СОО-
СН3(СН2)16СОО-
СН3(СН2)18СОО-
СН3(СН2)22СОО-
СН3(СН2)5СН=СН(СН2)7СОО-
СН3(СН2)7СН=СН(СН2)7СОО"
СН3(СН2)4(СН=СНСН2)2(СН2)6СОО-
СН3(СН2ХСН=СНСН2)3(СН2)6СОО"
СН3(СН2)4(СН=СНСН2)4(СН2)2СОО-
144
Глава 4. Липиды
При высоких температурах и в присутствии определенных
катализаторов цис-форша. может переходить в транс-форту. В жирных кислотах с
несколькими двойными связями г^с-конформация придает
углеводородной цепи изогнутый и укороченный вид.
На рис. 4.2 представлены структуры стеарата (18:0), олеата (18:1) и
линолеата (18:3). Хотя стеарат показан в вытянутой конформации, его
насыщенный углеводородный хвост подвижен, так как каждая углерод-
углеродная связь свободно вращается. Присутствие цис-двойных связей
з олеате и линолеате вызывает изгиб углеводородного хвоста, так как в
этом случае вращение вокруг двойных связей затрудняется. Этот изгиб
-репятствует формированию упорядоченной структурной организации
между соседними молекулами, характерной для насыщенных жирных
:-:ислот, и, следовательно, ван-дер-ваальсовы взаимодействия между
углеводородными хвостами ненасыщенных кислот ослабляются. В
результате ^мс-ненасыщенные жирные кислоты имеют более низкую темпера-
гуру плавления, чем насыщенные. По мере увеличения степени
ненасыщенное™ липиды становятся более текучими. Следует подчеркнуть, что
стеарат (точка плавления 70 °С) имеет твердую консистенцию при
температуре тела, тогда как олеат (точка плавления 13 °С) и линолеат (точка
плавления 17 °С) представляют собою жидкости.
о°
/
о=с
\
2СН2
•?сн2
^сн2
$сн2
«ен2
/
?сн,
\'
зСН2
9СН2
V
/оСН2
/
/2СН2
/
мСЯз
\
«СНг
п СН2
\
«СН2 б в
/
*7СН2 Рис. 4.2. Структуры трех С18 жирных кислот: а — стеа-
,„)-,„ рат, насыщенная жирная кислота; б — олеат, мононенасы-
0°
/
о-с
2СН2
/
ЗСН2
\
'сн2
/
5СН2
\
«сн.
/ "
7СН2
\
«СНЧ
/
н-с
9^ 11
с-сн2
/10 \
н н2с-
12
13
-сн2
\
н2с-
14
/5
-сн2
\
н2с-
10
Олеат
17
-сн2
\
«СН3
Н-
н-
17
н2с-
7
Н3С/я
0°
/
о=с
\
2СН2
/
зСН2
\
*сн2
/
5СН2
\
«СН2
/
7СН2
и \
сн2 *сн2
12 /\ /
—С юС=С 9
, „ II 1 \
-с н н
\
/4СН2
«''
,/^"н
-ч
\
н
Линоленат
3
щенная жирная кислота; в — линоленат, полиненасыщен-
Стеарат Ная жирная кислота, Ццс-двойные связи вызывают изгиб
я хвостов ненасыщенных жирных кислот
10. Заказ 3486
145
Часть I. Биомолекулы: структура, свойства, функции
Полиненасыщенные жирные кислоты быстро окисляются на
открытом воздухе. Кислород реагирует с двойными связями с
образованием пероксидов и свободных радикалов (соединений, имеющих
неепаренный электрон и в этой связи весьма реакционноспособных).
Такие соединения могут повреждать другие липиды, белки и
нуклеиновые кислоты. Поэтому окисленные масла довольно токсичны.
Так, например, был описан синдром испанского токсичного масла,
после употребления которого некоторые люди в Испании умерли.
Это масло было окисленным вследствие несоблюдения
определенных правил торговли.
При всем многообразии жирных кислот в высших растениях
присутствует в основном насыщенная пальмитиновая кислота и две
ненасыщенные кислоты — олеиновая и линолевая. Насыщенная
стеариновая кислота в растениях почти не встречается, а кислоты от С20 до
С24 встречаются редко (за исключением эпидермиса листьев). У
бактерий полиненасыщенные жирные кислоты, как правило, отсутствуют.
Жирные кислоты с длинной цепью (содержащие 16 или 18 атомов
углерода) практически нерастворимы в воде, но их натриевые или
калиевые соли, называемые мылами, образуют в воде мицеллы,
стабилизируемые за счет гидрофобных взаимодействий. Мыла обладают
свойствами поверхностно-активных веществ — детергентов; рК
жирных кислот находится в интервале значений 4—5.
Природные жирные кислоты не поглощают свет ни в видимой, ни
в близкой ультрафиолетовой области: их молекулы поглощают
энергию света в области длин волн короче 230 нм.
Ненасыщенные жирные кислоты участвуют в реакциях
присоединения по двойным связям; к ним легко присоединяются йод или
хлор.
4.2. Простые липиды
Простые липиды — соединения, состоящие только из жирных
кислот и спиртов. Они делятся на две группы: 1) нейтральные ацилглице-
ролы и 2) воски.
Нейтральные ацилглицеролы — это сложные эфиры трехатомного
спирта глицерина и одной, двух или трех жирных кислот. В
зависимости от этого различают соответственно моноацилглицеролы, диацил-
глицеролы, триацилглицеролы; последние в природе встречаются чаще
всего. Простые ацилглицеролы не содержат ионных групп, являются
нейтральными липидами, относятся к Ь-ряду. Триацилглицеролы
имеют и другие названия: глицериды, триглицеролы, ацилглицерины
или нейтральные жиры. Таким образом, глицериды в зависимости от
числа ацильных остатков (КСО-) можно также делить на: моно-, ди- и
триглицериды. Среди природных нейтральных жиров преобладают
ненасыщенные триглицериды:
146
Глава 4, Липиды
о о о
II И II
сн2-о-с-к 0 сн2-о-с-к 0 сн2-о-с-к
( II I II I
НОСН К'-С-О-СН К'-С-О-СН п
I I I II
СН2ОН СН2ОН СНг-О-С-Е"
1-Ацил-Ь-глицерол 1,2-Ацил-Ь-глицерол Триацил-Ь-глицерол
Основная функция триацилглицеролов — запасание и хранение
энергии и углерода, поэтому они являются важнейшими
компонентами пищевых продуктов. Триацилглицеролы, которые имеют при
комнатной температуре твердую консистенцию, называют жирами,
жидкую — маслами. Ненасыщеные жирные кислоты имеют низкую
температуру плавления, являются основными компонентами масел, они
и обусловливают жидкое состояние последних. Жиры, как правило,
содержатся в животных тканях, масла — в плодах и семенах
растений. Среднее содержание масел в семенах и плодах важнейших
культурных растений следующее:
Культура
Подсолнечник
Хлопчатник
Соя
Лен
Арахис
Горчица
Кукуруза
Мак
Содержание, %
35
28
20
29
49
32
5
45
Семена этих культур используются в жировой промышленности
для получения пищевых масел.
Триацилглицеролы различаются природой и расположением трех
остатков жирных кислот, соединенных с глицерином сложноэфирной
связью. В зависимости от типа остатков жирных кислот, этерифици-
рующих гидроксильные группы глицерина, нейтральные жиры делятся
на простые и смешанные. Если во всех трех положениях стоят
остатки одной и той же жирной кислоты, то их относят к простым триацил-
глицеролам, название которых определяется названием
соответствующей жирной кислоты. Примерами простых триацилглицеролов могут
служить тристеарилглицерол, трипальмитилглицерол, триолеилгли-
церол, или проще: тристеарин, трипальмитин, триолеин.
Триацилглицеролы, содержащие остатки двух или трех разных
жирных кислот, называются смешанными триацилглицеролами.
Смешанные триацилглицеролы, содержащие остатки двух жирных
кислот Жг и Ж2, могут быть связаны с глицерином Г в виде шести
ю*
147
Часть I. Биомолекулы: структура» свойства, функции
разных молекулярных форм: ГЖ^Ж^ ГЖ^Ж,,, ГЖ^Ж^
ГЖ^р^Ж^ ТЖгЖ2Ж2 и ГЖ2Ж2ЖГ У триацилглицеролов,
содержащих остатки трех разных жирных кислот, число разных
молекулярных форм равно 18. Называются смешанные триацилглицеролы
просто, например: 1-пальмитоилдистеарилглицерол, 2-олеилдипаль-
митоилглицерол.
Большинство природных жиров представляют собой чрезвычайно
сложные смеси простых и смешанных триацилглицеролов* Однако здесь
имеет место так называемый закон максимальной разности, в
соответствии с которым в состав жиров и масел входят преимущественно
смешанные триацилглицеролы, в составе молекул которых находятся
три разные жирные кислоты. Молекулы, содержащие две разные по
природе кислоты, присутствуют в меньших количествах, а молекулы
триацилглицеролов, содержащие жирную кислоту только одного вида,
присутствуют в весьма небольшом количестве. Кроме того,
ненасыщенные жирные кислоты находятся преимущественно у второй гид-
роксильной группы глицерина.
В большинстве масел содержание насыщенных жирных кислот
невелико (10—15 %), однако в некоторых из них (хлопковое,
арахисовое) оно может достигать 20 %.
По способности высыхать на воздухе масла подразделяются на
высыхающие (льняное, конопляное), полувысыхающие (подсолнечное,
кукурузное, хлопковое, соевое, арахисовое), невысыхающие
(оливковое, миндальное, касторовое). Это свойство масел определяется их
жирнокислотным составом. Высыхающие масла содержат большое
количество линоленовой кислоты; в состав полувысыхающих масел
входит значительное количество линолевой и олеиновой кислот;
невысыхающие масла в основном представлены триацилглицеролами
олеиновой, рициноленолевой и насыщенных кислот.
Триацилглицеролы способны вступать во все химические реакции,
свойственные сложным эфирам. Наибольшее значение имеет реакция
омыления, в результате которой из триацилглицерола образуются
глицерин и жирные кислоты:
о
II
О СНд-О-С-К СН2ОН ЕСООК
II I КОН I
к-с-о-сн 0 ^ носн + ксоок
I I
СКрО-С-К' СН2ОН К'СООК
Триацилглицерол Глицерин Калиевые соли
жирных кислот
Жиры при сильном и продолжительном взбалтывании с водой
образуют эмульсии — дисперсные системы с жидкой дисперсной фазой
(жир) и жидкой дисперсионной средой (вода). Однако эти эмульсии
148
• Глава 4. Липиды
нестойки и быстро разделяются на два слоя — жир и воду. Жиры
плавают над водой, поскольку их плотность меньше плотности воды
(от 0,87 до 0,97).
Для получения стойких эмульсий жира в воде необходимо
присутствие третьего вещества — эмульгатора, легко адсорбирующегося на
поверхности раздела двух фаз. Все эмульгаторы имеют амфипатичес-
кую структуру. Молекула эмульгатора состоит из двух частей: из
углеводородной цепи (гидрофобный хвост) и какой-либо полярной или
ионной группы (гидрофильная головка). К таким соединениям
относятся мыла, белки, фосфатиды, соли желчных кислот, а также
некоторые синтетические поверхностно-активные вещества. При
взбалтывании жира с водой в присутствии эмульгатора, т.е. при дроблении
жира на капли, на последних появляется тончайшая пленка (которая
может быть и не сплошной), состоящая из адсорбированных молекул
эмульгатора, гидрофобный конец которых погружен в капельку жира
(масла), а гидрофильный — в водную фазу. Наличие на поверхности
каждой капли жира такой пленки, наружный слой которой состоит
из гидрофильной части, снижает поверхностное натяжение на
поверхности капли воды, создает непрерывный переход от капли жира к
водной фазе и препятствует слиянию капель друг с другом. Эмульсия
масла в воде приобретает свойство стабильности.
Эмульгирование имеет большое физиологическое значение при
всасывании и усвоении организмом жиров, а также при образовании
биологических мембран. Эмульсии широко используются в пищевой
промышленности, мыловарении, при изготовлении косметических средств,
в производстве красителей.
Воски. Это сложные эфиры высокомолекулярных жирных кислот
и одноатомных спиртов с длинной углеводородной цепью:
Воски являются твердыми соединениями с ярко выраженными
гидрофобными свойствами. Типичным примером восков является пчелиный
воск, основным компонентом которого является мирицилпальмитат:
О
СН3-(СН2)5Г0-С-(СН2)1ТСН8
Жирные кислоты в них содержат от 24 до 36 углеродных атомов
(четное число углеродных атомов), а высокомолекулярные спирты
имеют в углеводородной цепи 16—30 углеродных атомов. Часто в
состав восков входят в свободном состоянии эти же кислоты и спирты, а
также высокомолекулярные кетоны. В состав восков входят также
обычные жирные кислоты, содержащиеся в триацилглицеролах
(пальмитиновая, стеариновая, олеиновая).
149
Часть I. Виомолекулы: структура, свойства, функции
Основная функция природных восков — образование защитных
покрытий. Восковой налет на листьях, плодах растений уменьшает
потерю влаги й снижает возможность поражения микроорганизмами.
Нарушение этого налета приводит к быстрой порче плодов при их
хранении.
4.3. Липиды — основные компоненты биологических мембран
Многие липиды являются важнейшими компонентами
биологических мембран, окружающих протоплазму клеток и
содержащиеся в ней субклеточные структуры: ядро, митохондрии, пластиды,
мезосомы.
Фосфоглицериды — основные компоненты биологических
мембран. Хотя нейтральные жиры представляют собой наиболее
распространенный класс липидов у животных в расчете на массу, тем не менее они
не являются структурными компонентами биологических мембран,
потому что они — не амфипатические соединения и, следовательно, не
могут образовывать липидный бислой. Наиболее распространенные
липиды в мембранах — это фосфоглицериды, называемые также глицеро-
фосфолипидами, или фосфоацилглицеролами (рис. 4.3). В отличие от три-
ацилглицеролов, в фосфоацилглицеролах одна из первичных спиртовых
групп глицерина этерифицирована не жирной, а фосфорной кислотой.
Таким образом,
исходен
онр-сР
о
/2 31
Н2ОСН-СН2
но он
Глицерол-3-фосфат
О
1 2 3|
0 о
1 I
о~е оо
о° 1
У I Полярная
О-Р-сР У (гидрофильная)
I головка
Неполярный
У (гидрофобный)
хвост
(ВО (Кз)
Фосфатидат
б
Рис. 4,3. Фосфоглицериды: а — глицерол-3-фос-
фат; б — фосфатидат, содержащий глицерол-3-фос-
фат с двумя жирнокислотными адильными
группами (К^ и К2), этерифицирующими его гидроксиль-
ные группы при С-1 и С-2 атомах соответственно.
Фосфатидаты — простейший тип фосфоглицеридов.
Подобно другим фосфоглицеридам, фосфатидаты
имеют полярную головку и неполярный хвост
ным соединением для
фосфоацилглицеролов
является не глицерин, а
фосфатидная (глицеро-
фосфорная) кислота.
Она представляет собой
1,2-диацилглицериновый
остаток, этерифициро-
ванный в положении 3
фосфорной кислотой.
Большинство
фосфоацилглицеролов
содержит еще спиртовый
компонент Х-ОН, гидро-
ксильная группа
которого этерифицирована
фосфорной кислотой.
Простейшим представителем
фосфоглицеридов
является фосфатидат,
включающий две жирнокис-
лотных ацильных
группы, этерифицирующих
150
Г лава 4. Липиды
С-1 и С-2 атомы глицеролфосфата. Фосфатидаты обычно содержатся
только в небольших концентрациях как интермедиаты биосинтеза или
распада более сложных по структуре фосфоглицеридов, в которых
фосфатная группа связана как с глицеролом, так и с другим
компонентом, несущим ОН-группу. В табл. 4.2 показаны некоторые
представители глицерофосфолипидов.
Структуры трех наиболее распространенных мембранных липидов —
фосфатидилэтаноламина, фосфатидилсерина и фосфатидилхолина
показаны на рис. 4.4. Молекула фосфатидилхолина имеет типичную
амфипатическую структуру, В ней можно выделить два фрагмента:
полярную головку, образованную фосфорной кислотой и азотосодер-
Таблица 4.2
Некоторые заместители,
связанные с фосфатной группой фосфоглицеридов
О
*1 X — остаток полярной
(Щ)ЧАААА^-0^-Н О ГОЛ0ВКИ
О СН2-0-Р-0-Х
3 I _
о
Предшественник X в формулах (НО—
Вода
Холин
Этаноламин
Серии
Глицерол
-Н
-СН2СН2К(СН2)3
-сн2сн2ын8
ЙНз
/
-сн2- сн
СОО"
-СНд- СН-^СНй-
он
Фосфатидилглицерол
—СН^СН ~СН 2
лшо-Инозитол
он
Место . ,
связывания
он
-0-
1К.
н
X)
0
О СН2ОСК3
II 1
О К4СОСН
II 1
-р—о—сн2
1 _
0
н
ОН
Л>Н
2Г~
Н
ОН
■ ял
[5
он,
Н
1
н
л4
он
Название семейств
фосфоглицеридов
Фосфатидат
Фосфатидилхолин
Фосфатидилэтаноламин
Фосфатидилсерин
Фосфатидилглицерол
Дифосфатидилглицерол
(кардиолипин)
Фосфатидилинозитол
151
Часть I. Биомолекулы: структура, свойства, функции
.ОТ*
Этаноламин ^^^
. сн3
о
О-Р-0
I
о
1 2 3\
Н 2С~ СН—СН 2
1 I
0 о
1 I
о-с с=о
Серин
ЙН3
СН-СОО
I
СН.
+
Н3С-]*-СНз
Холин
СН-
сн2
о
ОЧР-0
I
о
1 2 3\
Н2С-СН-СН2
I I
о о
I I
о=с сю
сн2
о
0=Р-0
I
о
1 2 3|
Н2С~СН—СН2
I I
о о
I I
О-С ОО
Полярные
У (гидрофильные)
головки
Неполярные
У (гидрофобные)
хвосты
(В1)(Н2) (К!)(В2)
Фосфатидилэтанолашга Фосфатидилсерин
(Кх) (К,)
Фосфатидилхолин
а
в
Рис. 4А* Структурные изображения; а — фосфатидилэтаноламина; б — фос-
фатидилсерина; в — фосфатидилхолина* Поскольку каждый из этих липидов
может содержать многие комбинации жирнокислотных ацильных групп, то
название относится к семейству соединений, а не к единственной молекуле
жащим соединением — холином, и две углеводородные цепи —
гидрофобный хвост. Амфипатическую структуру имеют и другие фосфогли-
церолы: фосфатидилэтаноламин, фосфатидилсерин. Головка несет
электрический заряд. При физиологических значениях рН (-7,0)
фосфатная группа всегда несет отрицательный заряд, поскольку
величина рК этой группы лежит в пределах 1—2; Х-группы
фосфатидилхолина, фосфатидилэтаноламина при рН 7,0 несут положительный
заряд, так как величины рК для их аминогрупп равны соответственно
13 и 10. Таким образом, при рН 7,0 эти два фосфоглицерола
представляют собой биполярные ионы. Точно так же Х-группа фосфатидилсе-
рина содержит а-аминогруппу с рК 10 и карбоксильную группу с рК 3,0;
следовательно, при рН 7,0 молекула фосфатидилсерина имеет две
отрицательные и одну положительную группы. Этим и объясняется
высокая гидрофильность полярных головок фосфоглицеролов.
Семейство фосфоглицеридов содержит не только соединения,
представленные в табл. 4.2. Другие молекулы этого типа содержат такую
же полярную головную группу и различные цепочки жирных кислот.
Например, известно, что мембраны эритроцитов человека содержат по
меньшей мере 21 тип фосфатидилхолина. Эти типы отличаются друг
от друга жирнокислотными ацильными остатками, этерифицирующими
С-1 и С-2 атомы глицерола* В общем, в каждой позиции тип жирной
кислоты встраивается не наугад, — насыщенные жирные кислоты
обычно этерифицируют С-1, а ненасыщенные — С-2 атом в фосфоглицери-
дах. Исключением из этого правила является дипальмитоилфосфати-
152
Глава 4. Липиды
дилхолин (ДПФХ), в котором пальмитиновая кислота этерифицирует
как С-1, так и С-2 атомы.
Специфические позиции жирных кислот в фосфоглицеридах могут
быть определены с помощью ферментов - фосфолипазы Ах и фосфоли-
пазы А2, которые осуществляют специфический катализ гидролиза
связи между жирнокислотным остатком у С-1 или С-2 соответственно.
Высокие концентрации лизофосфоглицеридов > возникающих в
результате действия фосфолипазы А, могут разрушать клеточные
мембраны. Змеиный, пчелиный и осиный яды — это источники, богатые фос-
фолипазой А2. Введение змеиного яда в кровь может привести к
угрожающему жизни гемолизу, т.е. лизизу мембран эритроцитов.
Существуют также два других класса фосфолипаз, а именно фосфолипаза
С, которая катализирует гидролиз связи между глицеролом и
фосфатом, высвобождая диацилглицерол, и фосфолипаза В,
катализирующая образование фосфатидатов из глицеролфосфолипидов.
Следует обратить внимание, что фосфоглицериды представляют
собой амфипатические молекулы, имеющие полярную головку (с
ионизированным фосфатом и часто одну или две другие заряженные
группировки) и длинные, неполярные хвосты (рис. 4.5).
ДПФХ —• глицерофосфолипид, составляющий более 50 % секрета,
известного под названием легочный сурфактант. Легочный сурфактант
уменьшает натяженность поверхности альвеол, что позволяет кислороду
эффективно поступать из воздухоносных путей внутрь клеток. В
последнее время ДПФХ используют при синдроме недостаточности дыхания,
который может приводить к летальному исходу. Это состояние встреча-
ется у недоношенных детей, в чьих недоразвитых легких не может
образовываться достаточное количество сурфактанта. ДПФХ назначается
таким детям в качестве аэрозоля для
облегчения дыхания.
Фосфатидилинозитол (ФИ) является
предшественником более полярных липидов,
которые образуются за счет добавления
фосфатных групп к кольцу инозитола. К таким
производным относятся фосфатидилинозитол-4-
фосфат (ФИФ) и фосфатидилинозитол-4,5-
бисфосф&т (ФИФ2). Структуры ФИ, ФИФ и
ФИФ2 показаны на рис. 4.6. ФИФ2
представляет значительный интерес, так как он вовле,-
чен в сигнальные процессы и участвует в
передаче сигналов через мембраны. Например,
связывание нейромедиатора ацетилхолина с
его рецептором в нервных окончаниях
активирует фосфолипазу С, которая катализиру* » , ,
*ттЛ кои молекулы фосфоглицери-
ет распад ФИФ2 до диацилглицерола и ино- да. Показаны головка и два
зитол-1,4,5-/гграсфосфата. Диацилглицерол и неполярных хвоста липида
I
Полярная
(гидрофильная)
головка
Неполярный
/* (гидрофобный)
хвост
Рис. 4.5. Схематическое
изображение амфипатичес-
153
Часть I. Биомолекулы: структура, свойства, функции
О
СН2-0-С-К1
о
I
Н-0-С-Е2
°о-р-о-сн2 о
Фосфатидилиноаитол
(ФИ)
0РО3©
Фосфатидилинозитол-4-фосфат
(ФИФ)
?
О
Н2-0-Й-Е1
СН-О-С-Ко
о Л I *
н орор
орф
Фосфатидшшнози-
тол-4,5-^иофосфат
(ФИФ2)
Рис. 4.6. Структурные изображения фосфатидилинозитола (ФИ), фосфатидил-
инозитол-4-фосфата (ФИФ) и фосфатидилинозитол~4,5-б'#сфосфата (ФИФ2).
Дополнительные фосфатные группы, присоединенные к кольцу инозитола, делают
ФИФ и ФИФ2 высокополярными соединениями
■
инозитол-1,4,5-#грысфоефат действуют как вторичные медиаторы внутри
клетки, активируя протеинкиназу и вызывая высвобождение ионов Са2+,
таким образом индуцируя разнообразные метаболические ответы клетки
(см. гл. 14). Следует обратить внимание, что префикс трис используется
для обозначения трех фосфатных групп у трех разных углеродных
атомов, тогда как три указывает на три фосфата, последовательно
соединенные фосфоангидридными связями,
Кардиолипин является главным компонентом фосфолипидов в мито-
хондриальных мембранах, хотя его содержание в клетках — 2—5 %
общей массы липидов. Его название связано с тем обстоятельством, что
впервые он был выделен из сердца быка, но впоследствии был обнаружен
во многих тканях различных организмов. В связи с особенностями его
структуры кардиолипин иногда называют "двойным" фосфоглицеридом
(см. табл. 4.2).
Чистые фосфатидилглицеролы — плохо кристаллизующиеся,
белые, воскообразные вещества. На воздухе и свету они темнеют в
результате сложных химических превращений, связанных со
способностью их ненасыщенных компонентов образовывать под действием
атмосферного кислорода различного рода соединения перекисного типа.
Сфинголипиды — вторая группа липидов, входящих в состав
биологических мембран. Хотя фосфоглицериды составляют
основную часть липидов в большинстве мембранных систем, другие амфи-
патические липиды, называемые сфинголипидами, также
присутствуют в мембранах растений и животных. У млекопитающих они
особенно распространены в тканях центральной нервной системы.
Скелетом структуры сфинголипидов является сфингозин (трокс-4-сфин-
генин), неразветвленный С18 спирт с двойной связью в
траке-конфигурации между С-4 и С-5, аминогруппой при С-2 и гидроксильными
группами при С-1 и С-3 (рис. 4.7, а). Сфингозин имеет амфипатическую струк-
154
Глава 4. Липиды
туру, состоит из двух фрагментов — гидрофильной головки и
гидрофобного хвоста, а его производные сфингофосфатиды, содержащие остатки
жирной кислоты и фосфохолина, в еще более яркой форме проявляют
амфипатические свойства. Таким образом, по конформации
сфингофосфатиды весьма сходны с фосфоглицеридами. Сфингозины относятся к
аминоспиртам с длинной ненасыщенной цепью; они отличаются друг от
друга длиной углеводородной цепи.
Церамид содержит жирную кислоту, присоединеную к
аминогруппе при С-2 атоме сфингозина с помощью амидной связи (рис. 4.7, б).
Церамиды — метаболические предшественники всех сфинголипидов.
Тремя важнейшими семействами сфинголипидов являются сфингоми-
елины, цереброзиды и ганглиожды. Среди них только сфингомиелины
содержат фосфат, и, следовательно, могут классифицироваться как
сн3
®|
Н3С-М-СН3
сн2
сн2
о
н
%
• 3?н
НаС-СН-СН
ФЫН3 #СН
НС*
л
НаС-СН-СН
*с:
%
2 3
он
*сн
НС5
н2с-сн-сн
ын 'сн
<ш2
сн2
сн2
сн2
сн2
сн2
\н2
сн2
сн2
/вСН3
:н.
/ "
СН2
СН2
<5н2
сн2
сн2
СН2
сн2
^Н,
СН2
сн2
18
а
НС5
\
сн2
сн2
сн2
сн2
сн2
сн2
Сфянгозян
(шраяс-4-сфингвнин)
Церамид
Сфингомнелин
а б в
Рис. 4.7. Структурные изображения сфингозина, церамида и сфингомиелина;
а — сфингозин — длинноцепочечный спирт с аминогруппой при С-2 атоме,
служит в качестве скелета для сфинголипидов — второго типа молекул липидов,
обнаруженных в биомембранах; б — церамиды, имеющие длинноцепочечную
ацильную группу, присоединенную к аминогруппе сфингозина; в — сфингомие-
лин, имеющий фосфатную группу, присоединенную к гидроксильной группе при
С 1 атоме церамида, и холиновую группу, присоединенную к фосфату
155
Часть I. Биомолекулы: структура, свойства, функции
СН2ОН
НО
Огалактоза
(полярная головка)
к * * Н * * * *V ■ V ■ * ■ г \у ^ 2Ц * Р * # * ^ * ^А /ф 4, Э^4 ,
■■* * * 1ч'*^ч* ч*/* "* . * V* ^—* **» г «* г *
*^*^А* И>*/р</1 !.«У } ДО . А*
Ч*ТА* ■»> А*^А*^^^ /ЛЛ^1^ЧЛ*,( »
:^\ >/. У О }\ *&>$?* Я*^в&(* ■. ■!■. ■:.' I л^ и» » чУЛуО*
• *Г< "* ^ *ГГ * Л (Л1» . ... ... V '^ V . .ь^ Ч* Ч Г*1-» А*Г 4 * /Л^ *
. * 9* * * 9 * *а*Л %! 'уа*^ ■..■■._..■ Ч .-.:■ ' •л^вЛМ О*
: з« хдо;к&Е1з Я ♦« даИ1!ИЯ!
.-.'-■.. ■.-.■...■■. 11 .-...■.■.•■. ^ т^* ^, «, Л^ , ^ > ^+А* Г *Г
ч > * ..V* * ■ ■ ■, ' II , ' ■■ ■ ■■■■■■■■: ■ +■ ■
А..', *' **< * ■ .
кич«кг: О^О
^ т Ач>*7»>< > » ^Ь
* * *Ч ЧФ* < * * ^
■»** ***"**■ гш ч—Ж
А#4«}Г< г * &
*&Ш СНо
Сфингоаин
(выделен на темном
фоне)
:< гч«ъч ^
*У/ ^ АУ
фосфолипиды; цереброзиды и ганглиозиды содержат углеводородные
остатки и классифицируются как гликосфинголипиды.
Сфингомиелин содержит фосфохолин, присоединенный к С-1 гид-
роксильной группе церамида (рис. 4.7, в). Сфингомиелины
представляют собой сложные эфиры, состоящие из жирной кислоты, холина,
фосфорной кислоты и сфингозина. Следует обратить внимание на
сходство амфипатической молекулы сфингомиелина и молекулы фосфати-
дилхолина (см. рис. 4.4, в); обе являются цвиттерионами, содержащими
холин, фосфат и два длинных гидрофобных хвоста, Сфингомиелины
присутствуют в плазматических мембранах большинства клеток
млекопитающих и являются основными
компонентами миелиновых оболочек не-
, рвных клеток некоторых типов.
Цереброзиды — гликосфинголи-
пиды, содержащие один моносаха-
ридный остаток присоединенный
через р-гликозидную связь к С-1
атому церамида. Галактоцереброзиды,
известные также как галактозилце-
рамиды, представляют собой це-
рамиды, в которых единственный
р-галактозильный остаток
является полярной головной группой
(рис* 4.8). Галактоцереброзиды
распространены в тканях нервной
системы и составляют около 15 % ли-
пидов миелиновых оболочек.
Многие другие ткани млекопитающих
содержат глюкоцереброзиды, в
которых р-В-гликозильный остаток
связан с С-1 атомом церамида. В
некоторых таких гликосфинголипидах
линейная цепочка из трех
дополнительных моносахаридных остатков
присоединена к галактозильному ос-
татку галактоцереброзида или к гли-
козильному остатку гликоцеребрози-
да. Поскольку они содержат более
чем один моносахаридный остаток,
то такие гликосфинголипиды не
называются цереброзидами; они
имеют различные названия.
„ А 0 „ - ™ Среди гликолипидов особенно
Рис, 4.8, Галактоцереброзид* Жир- г
ная кислота в составе галактоцеребро- широко распространены галакто-
зидов содержит 24 атома углерода зилоийцилглицеролъы
Р2
п^А"^ Л
?Х(Хч??
то
:.:-■
Жирная кислота
с длинной цепью
156
Глава 4, Липиды
О
II
О СН2-ОС-К'
но
К"-СО-СН
СНоОН л
' О ?"СН2
он
Галактозилдиацилглицерол
Эти соединения содержатся в самых различных растительных
тканях. Они обнаружены в митохондриях, хлоропластах и локализованы
в мембранах; содержатся в водорослях, некоторых фотосинтезирую-
щих бактериях.
Ганглиозиды — наиболее сложные гликосфинголипиды, олигоса-
харидные цепи которых содержат Ы-ацетилнейраминовую (сиаловую)
кислоту, присоединенную к церамиду (рис. 4.9). Вследствие
присутствия карбоксильной группы сиаловой кислоты ганглиозиды
представляют собой соединения анионного характера. Остатки Сахаров
формируют большие полярные головки ганглиозидов.
Значительное разнообразие структурного строения ганглиозидов
является результатом различного состава и последовательности
остатков Сахаров; охарактеризовано более 60 видов ганглиозидов.
Ганглиозид Ош служит как рецептор холерного токсина,
продуцируемого бактерией УьЬпо сНоЫгае. Бактериальный токсин, проникая в
клетки, приводит к массивной потере жидкости, вызывая диарею и в
конечном итоге смерть в результате дегидратации. Холерный токсин
представляет собой белок, который специфически связывается с 0М1
на поверхности клетки. Оказавшись внутри клетки, токсин
стимулирует высвобождение больших количеств жидкости, постоянно
воздействуя на определенный сигнальный процесс (см, гл. 14). "М" в
сокращенных названиях Ом1 и СгМ2 означают "моносиало", указывая, что
эти липиды содержат только один остаток Ы-ацетилнейраминовой
кислоты в основе их структуры (Ом2 был вторым охарактеризованным
моносиалоганглиозидом, поэтому он и обозначается "2м).
Метаболизм ганглиозидов — сфера активных медицинских
исследований. Установлено, что состав мембранных гликосфинголипидов
может значительно изменяться при развитии злокачественных
опухолей. Известно также, что генетически обусловленные дефекты
метаболизма гангиозидов связаны с рядом заболеваний,
сопровождающихся слабоумием и часто заканчивающихся летальным исходом, таких
как болезнь Тея-Сакса и генерализованный ганглиозидоз.
Стероиды — третья группа липидов биологических мембран.
Стероиды представляют собой третью группу липидов, обычно
обнаруживаемых в мембранах эукариот. Стероиды и жирорастворимые
157
Часть I. Биомолекулы: структура, свойства, функции
сн2он
НзС НСОН
I I
0=С НСОН
— оч соон
сн2он
НзС НСОН
0=С НСОН
т*
Ганглиоаид Омг
Ь1-Ацети лнейрамн новая
кислота
НО
НС
, \
(СНг)16 (С(Н2)12
СНа СНз
Р-Б-Глюкоза
Р-Б-Галактоза
нэс ^о
Ы-Ацвтил-р-Б-галактозам ин
СН2ОН
НаС НСОН
I I
0=С НСОН
СН?ОН
но
Ганглиозид Ом
он
сн2он н2с—сн—сн
НА о. о' т1гн Ч?н
о-с нсч
Н (сн2)1в ^>
н он I сн
р-В-Глюкоза
12
3
На(Г ^О
р-В-Галактоза
ОН М-Ацетил-р-В-галак
тозамин
в
р-В-Галактоза
Рис. 4.9. Структура ганглиозидов: а — Ы-ацетилнейраминовая кислота —
компонент ганглиозидов. Представители ганглиозидов: б ~~ Оио; в — С
М2
'М1
витамины, наиболее широко классифицируемые как полипреноловые
соединения, —- это соединения, структура которых построена из пяти-
углеродных молекул изопрена (рис. 4Л0). Стероиды имеют
характерные циклические ядра, представляющие собой четыре соединенных
кольца: три шестиуглеродных кольца, обозначаемых А, В, С, и пяти-
углеродное кольцо (рис. 4Л0). Холестерол является одним из
примеров стероидов. Это важнейший компонент плазматических мембран
млекопитающих, но он редко встречается в растениях и никогда — в
прокариотах. Холестерол может быть назван более специфически сте-
ролом (стероидный спирт), потому что в его структуре имеется ОН-груп-
158
Глава 4. Липиды
сн3
Н2С=С-СН=СН2
Изопрен
20
22 24 27
23 2$\
12
18
26
НО
и/
19 А
*1Л
В
*ч^
13
С
14
\т
V
6
Холестерол
НО
ОН
Тестостерон
(стероидный гормон)
НО
Стигмастерол
(растительный стерол)
СОО° Nаф
ОН
Холат натрия
(желчная соль)
Рис. 4.10. Структурные изображения изопрена, являющегося основой
структурных единиц всех полипрениловых соединений, к которым относятся стероиды и
жирорастворимые витамины, и нескольких стероидов. Сопряженная кольцевая
система стероидов содержит четыре кольца (обозначаются буквами А, В, С и В)
па при С-3 атоме. Следует обратить внимание на наличие метильных
групп при С-10 и С-13 атомах и воеьмиуглеродную часть цепи,
присоединенную к С-17 атому.
Другие стероиды — это стеролы растений и грибов, которые
содержат по меньшей мере 27 углеродных атомов и, подобно холестеролу,
имеют гидроксильную группу при С-3 атоме; стероидные гормоны
млекопитающих (такие, например, как С18 эстрогены, С19 андрогены).
Структура этих стероидов отличается по длине углеводородной цепи,
присоединенной к С-17 атому сопряженной кольцевой системы,
количеством и расположением метильных групп, двойных связей, гидро-
ксильных групп и в некоторых случаях кетогрупп.
Несмотря на определенное отношение к развитию
сердечно-сосудистых заболеваний, холестерол играет существенную роль в
жизнедеятельности млекопитающих, Холестерол, синтезируемый в клетках
млекопитающих, — это не только компонент различных мембран, но
и предшественник стероидных гормонов и солей желчных кислот. К
стероидным гормонам относятся тестостерон (мужской половой
гормон), эстрадиол (один из женских гормонов), альдестерон
(образующийся в коре надпочечников и регулирующий водно-солевой баланс).
Холестерол намного более гидрофобен, чем глицерофосфолипиды и
сфинголипиды. Из его структуры (рис. 4,10) видно, что единственной
его полярной составляющей является гидроксильная группа при С-3
159
Часть I. Биомолекулы: структура, свойства, функции
атоме. В этой связи максимальная концентрация свободного
холестерола в воде — 10~8М. Кольцевая структура холестерола достаточно
жесткая, и его молекула обладает значительно меньшей
подвижностью, чем большинство других липидов. Поэтому наличие холестерола
определяет текучесть клеточных мембран млекопитающих.
При этерификации путем присоединения жирных кислот к С-3 гид-
роксильной группе холестерола образуются эфиры холестерола (рис. 4.11).
Поскольку 3-ацильная группа эфиров является неполярным
заместителем, то эфиры холестерола более гидрофобны, чем сам холестерол.
Эфиры холестерола обнаружены в липопротеинах крови. Они образуются
внутри клетки в качестве депо хранения холестерола.
о
(Н) ^^/\/\/\/^-с-о
Рис. 4.11. Эфир холестерола. Наличие жирнокислотной ацильной группы при
С-3 атоме делает эфиры холестерола более гидрофобными, чем холестерол
4-4. Биологические мембраны
Структурная основа биомембран — липидные бислои. С
химической точки зрения биомембраны представляют собой комплексы
липидных и белковых молекул. Обычно в состав мембран входит
около 40 % липидов и 60 % белков (по массе) и до 10 % углеводов (в
качестве компонентов гликолипидов и гликопротеинов). Однако
следует отметить, что состав биологических мембран не одинаков не только
для разных видов, но и для различных клеток многоклеточных
организмов. Соотношение белков и липидов в различных мембранах
варьирует от 1/5 белка + 4/5 липидов до 3/4 белка + 1/4 липидов.
Углеводы в форме гликолипидов и гликопротеинов могут составлять 0,5—10 %
всех веществ мембран. Типичная мембрана содержит фосфолипиды,
гликосфинголипиды и (в некоторых эукариотах) холестерол.
Липидные компоненты мембран имеют одну общую особенность: все
они являются амфипатическими соединениями, содержащими полярную
и неполярную части. Поэтому в водных растворах при определенных
условиях полярные липиды способны формировать мицеллы (рис. 4.12.).
Если любой фосфолипид поместить в систему масло—вода, то будет иметь
место ориентация гидрофильной головки в воду, гидрофобного хвоста в
масло, образуется слой фосфолипида, — "частокол" из его молекул. При
встряхивании фосфолипида в воде образуются мицеллы — агрегаты
молекул фосфолипида. Мицеллы содержат иногда тысячи фосфолипидных
молекул, так что масса такой частицы может быть довольно
значительно
Глава 4. Липиды
Во^а
Мицелла
а б
Рис, 4.12. Липидный слой (а) на границе раздела в системе масло—вода и
хпцелла фосфолипида (б) после интенсивного диспергирования липида в воде
ной. В этих структурах гидрофильные головки молекул липидов распо-
- о жены на поверхности частицы и взаимодействуют с окружающими
молекулами воды, а гидрофобные углеводородные хвосты находятся внут-
;:л. Триацилглицеролы сами по себе диспергируют плохо и мицелл не
б разуют, но они могут включаться в структуру мицелл фосфоглицеро-
тов, в результате чего образуются мицеллы смешанного типа. Фосфоли-
-иды могут растекаться по поверхности водного раствора в виде мономо-
"екулярного слоя, в котором углеводородные хвосты молекул обращены
з сторону сравнительно гидрофобной воздушной фазы, а гидрофильные
: о ловки направлены в сторону водной среды.
Однако т ишо липиды имеют тенденцию образовывать липидные
*ысл0и* В мицеллах углеводородные хвосты липидов не упакованы
достаточно хорошо, тогда как в бислоях они прекрасно подогнаны
хруг к другу (рис. 4.13).
Не все амфипатические липиды могут формировать бислои.
Например, холестерол хотя и является амфипатическим соединением, но сам
::о себе не может формировать би-
: лой, так как его полярная ОН-груп-
::а очень мала по отношению к
гидрофобной углеводородной кольцевой
системе. В биологических
мембранах холестерол и другие липиды, не
способные формировать бислои (на
их долю приходится около 30 % от
общего содержания липидов),
стабилизируются за счет окружения
другими липидами.
Липидный бислои обычно
имеет толщину около 5—7 нм.
Молекулы липидов внутри бислоя
ориентированы их гидрофобными
хвостами во внутреннюю часть, а их
гидрофильные головки
контактируют с водной средой. В результа-
11. Заказ 3486 161
Масло
Липидный
слой
Вода
_
Липидный
б лед о
Водная фаза
Окруженный ЛИ1ШДЛ1ЛМ бислоем
водный компартмеят
Рис. 4.23. Схематическое
изображение "среза" липосомы. Бислои состоит
из двух пластов. В каждом пласте
полярные головки амфипатических
липидов обращены в сторону водной среды,
а неполярные углеводородные хвосты —
вовнутрь и контактируют друг с
другом с помощью ван-дер-ваальсовых сил
Часть I. Биомолекулы: структура, свойства, функции
те образуется внутренний непрерывный углеводородный двойной слой,
а заряженные головки липидных молекул формируют по его
границам ионизированную поверхность. Формирование липидных бислоев
возможно благодаря действию тех же сил, которые стабилизируют
структуру глобулярных белков. Как рассматривалось в гл. 1,
полипептидные цепи в белковых молекулах уложены таким образом, что
гидрофильные полярные К-группы аминокислотных остатков
находятся на поверхности белковой глобулы, тогда как гидрофобные К-
группы формируют гидрофобное ядро внутри молекулы.
Бимолекулярные слои очень близки по своей структуре к природным
биологическим мембранам. Непрерывный слой углеводородов имеет низкую
диэлектрическую проницаемость и является плохим проводником
электричества, поэтому он характеризуется высокой электрической
емкостью и высоким сопротивлением. Все вариации в размерах, форме,
заряде полярных головок оказывают очень важное влияние на
структуру мицелл, слоев фосфоглицеролов и мембранных систем.
Поскольку липидные бислои могут легко изгибаться, то они могут,
сближаясь, формировать сферические структуры. Такие структуры,
называемые липосомами, можно легко получить в лабораторных условиях.
Липосомы могут быть использованы в качестве переносчиков
лекарственных препаратов к определенным тканям организма (см. гл. 17).
Структура и свойства биологических мембран. Мембраны —
это функционально активные поверхностные структуры клеток,
ограничивающие цитоплазму и внутриклеточные органеллы. Они
составляют обычно более половины всей массы клеток (в пересчете на сухие
вещества), что объясняется их высокой плотностью и широко
развитой поверхностью. Липидные бислои формируют основу
биологических мембран, включая плазматические мембраны и внутриклеточные
мембраны, и они определяют их многие физические свойства.
Липидные бислои могут существовать в отсутствие белков, но
белки являются важнейшими компонентами биологических мембран.
Мембранные белки непосредственно вовлечены в транспорт молекул
через липидный бислои, передачу сигналов внутрь клетки и
взаимодействие между плазматическими мембранами и цитоскелетом.
Толщина биологических мембран обычно б—10 нм вследствие наличия
белков, локализованных в бислое.
В 1972 г. С. Дж. Сингер и Г. Николсон предложили жидкостно-
мозаичную модель структуры биологических мембран. Согласно этой
модели мембрана состоит из бислоя амфипатических липидов, в
которых находятся глобулярные белки (рис* 4.14). Амфипатические
молекулы в бислое расположены так, что их гидрофобные хвосты
направлены друг к другу и частично перекрываются. Обе поверхности
бислоя составлены гидрофильными головками. Поэтому в водной
среде бислои стабилен, поскольку его поверхность легко ассоциирует с
водой.
162
Глава 4. Липиды
Олигосахаркдные цепи Окружающая клеточная среда
Рис. 4.14. Структура типичной эукариотической плазматической мембраны.
Липидный бислой формирует матрикс биологических мембран, а белки
(некоторые из них являются гликопротеинами) ассоциированы с ним различными
способами. Интегральные мембранные белки локализованы внутри бислоя и обычно
пронизывают оба пласта. Гидрофобные аминокислотные остатки
трансмембранных сегментов этих белков участвуют в ван-дер-ваальсовых взаимодействиях с
гидрофобными хвостами липидных молекул. Периферические мембранные
белки слабо ассоциированы с поверхностью мембраны через ионные взаимодействия
и водородные связи либо с полярными головками липидных молекул, либо с
интегральными мембранными белками. "Заякоренные" в липидах мембранные
белки связаны с мембраной за счет ковалентных связей с несколькими типами
липидных молекул
Мембраны содержат большое число различных глобулярных
белков с молекулярной массой от 25 000 до 230 000. Белки, заключенные
в бислое, можно подразделить на два типа — периферийные и
интегральные. Периферийные белки легко удаляются из мембраны
растворами с высокой ионной силой (например, 1 М МаС1); из этого следует,
что в бислое они удерживаются только или преимущественно
электростатическими силами. Напротив, интегральные белки глубоко
погружены в липидный бислой, причем одни из них погружены в бислой с
той или другой его стороны, но не пронизывают бислой насквозь;
другие — пронизывают бислой насквозь и, следовательно, вступают в
соприкосновение с водным раствором с обеих сторон бислоя; третьи —
полностью погружены в гидрофобную сердцевину бислоя. Та часть
поверхности интегральных белков, которая находится в гидрофобной
области бислоя, обладает гидрофобными свойствами, а остальная часть
поверхности, обращенная к водному окружению, — гидрофильными.
Мембранные белки "плавают" в липидном бислое. Интегральные
белки находятся внутри бислоя, контактируя с гидрофобным
окружением. Периферийные мембранные белки более слабо ассоциированы с
мембранной поверхностью.
11*
163
Часть I. Биомолекулы: структура, свойства, функции
В соответствии с жидкостно-мозаичной моделью мембрана
представляет собой динамичную структуру, в которой как белки, так и
липиды способны перемещаться внутри бислоя. Мембранные
структуры сформированы за счет слабых электростатических сил (полярных,
водородных связей) и гидрофобных взаимодействий. Ковалентные связи
в формировании этих структур играют второстепенную роль. Поэтому
ясно, что биологические мембраны — структуры динамичные;
молекулярные компоненты в них сохраняют довольно высокую
подвижность, однако они могут двигаться легко только в плоскости
мембраны. Поперечная диффузия происходит очень медленно. Она связана с
переходом полярной головки структурного компонента через
гидрофобную сердцевину бислоя. Белки, имеющие более высокую
молекулярную массу, движутся более медленно, чем липиды. Движение
белковых глобул может быть вращательным в пределах монослоя и даже
бислоя. Кроме того, сами белки способны претерпевать изменения своей
трехмерной конформации в составе мембраны, причем изменение кон-
формации одной молекулы, вероятно, может влиять на конформацию
молекул белков из ближайшего микроокружения.
Мембраны — это системы, соответствующие минимуму свободной
энергии и поэтому способные к самосборке. Жидкостно-мозаичная
модель мембраны легко объясняет различную гетерогенность в
вязкости ее отдельных участков. Низкие значения вязкости наблюдаются в
углеводородном слое липидов. Универсальным свойством
биологических мембран является их способность к структурным перестройкам,
вызываемым разнообразными экзогенными и эндогенными
факторами. Как правило, эти перестройки происходят без разрывов или
образования прочных ковалентных связей и сводятся к изменению
мономолекулярных и межмолекулярных взаимодействий. В различных
условиях эти переходы могут быть локальными или
распространенными на значительные участки мембраны. Структурные перестройки
сопряжены с большим числом важнейших функций мембран.
Хотя некоторые аспекты, касающиеся первоначально
предложенной жидкостно-мозаичной модели, со временем были пересмотрены и
добавлены новые характеристики, но в целом модель сохраняет свою
значимость в настоящее время.
Липидные бислои и мембраны — избирательно проницаемые
барьеры. Весьма существенно, что биологическая мембрана
физически отделяет живую клетку от окружающей среды. Но не менее важно,
что вода, кислород и остальные вещества, необходимые для роста,
способны входить в клетку и что продукты, синтезирующиеся
клеткой на экспорт (такие, как гормоны, некоторые пищеварительные
ферменты, токсины), так же, как и отходы жизнедеятельности (диоксид
углерода, мочевина), способны покидать ее. Гидрофобные или
небольшие незаряженные молекулы могут свободно диффундировать через
биологические мембраны, но в то же время липидные бислои пред-
164
Глава 4. Липиды
ставляют собой практически непроницаемый барьер для большинства
заряженных молекул.
Небольшие неполярные молекулы могут диффундировать через
мембрану со стороны с высокой их концентрацией на сторону с более
низкой концентрацией. Равновесие достигается, когда концентрации
с каждой стороны мембраны выравниваются. Скорость, с которой
вещества перемещаются с одной стороны мембраны на другую, зависит
от разницы концентраций, т.е. от градиента концентрации, между
двумя компартментами. Диффузия молекул по концентрационному
градиенту происходит самопроизвольно, так как приводит к
возрастанию энтропии и, следовательно, к уменьшению свободной энергии.
Движение какого-либо вещества через мембрану по закону обычной
диффузии, т.е. по градиенту концентрации этого вещества, иногда
называют пассивным транспортом.
Способность многих ионов и небольших молекул диффундировать
через липидные бислои оценивается их коэффициентами
проницаемости, варьирующими от 10~4 до 10"14 м-с"1. Величина коэффициента
проницаемости приблизительно соответствует растворимости веществ
в неполярных растворителях. Гидрофобные соединения
диффундируют быстро. Индол, например, проходит через липидный бислой менее
чем за 1 с. Полярные и заряженные молекулы пересекают бислои
намного медленнее, так как они должны оставлять молекулы воды в
окружающей их среде, пока они движутся через промежутки между
липидными молекулами. Этот процесс термодинамически невыгоден
для полярных молекул и особенно ионов. Например, для пересечения
мембраны глюкозой, представляющей собой полярную молекулу,
требуется время от минут до часов, тогда как для ионов натрия — дни и
недели. Исключением из общего правила является вода; ее молекулы
диффундируют через мембраны чрезвычайно быстро, достигая
равновесия за миллисекунды. Многие биологически важные молекулы
входят и выходят из клетки путем простой диффузии, включая
неполярные газы, такие как 02, С02, и гидрофобные молекулы, такие как
стероидные гормоны, жирорастворимые витамины, а также
некоторые лекарства.
Скорость диффузии ионов через мембраны значительно
усиливается ионофорами (переносчиками ионов). Многие из этих соединений
представляют собой токсины, синтезирующиеся грибами и
бактериями. Существует два типа ионофоров: мобильные переносчики и канал-
формирующие ионофорьь И те, и другие обеспечивают передвижение
ионов по концентрационному градиенту. В качестве примера можно
привести валиномицин, синтезируемый видом 81гер1отусе$. Валино-
мицин диффундирует через мембрану, перенося ион на другую
сторону, где высвобождается. Он обладает значительной ионоселективнос-
тью; К+ связывается в 1000 раз эффективнее, по сравнению с Ма+.
Неполярные участки молекулы ионофора контактируют с ацильными
165
Часть I. Биомолекулы: структура, свойства, функции
0,4 нм
цепями мембранных липидов, в результате чего входящий комплекс
"валиномицин—К+" становится жирорастворимым* Другой пример —
грамицидин Р, каналобразующий ионофор, синтезируемый
бактериями ВасШиз Ъгеи1з. Грамицидин О внедряется в мембраны, формируя
спиральную структуру, в результате чего возникает пора (рис. 4.15).
Одна молекула грамицидина О способна пронизывать только один пласт
бислоя; вся мембрана полностью пронизывается двумя спиральными
молекулами, формирующими димер за счет взаимодействия К-конце-
вых участков молекул.
Образующаяся пора имеет диаметр
около 0,4 нм и позволяет
быстро проникать моновалентным
катионам, таким как Н+, Ыа+
и К+. Поскольку каналформи-
рующим ионофорам, таким
как грамицидин В, не нужно
диффундировать через бислой
как мобильным переносчикам,
то скорость трансмембранной
диффузии ионов через грамици-
диновые поры в 104 раз выше по
сравнению с диффузией с
помощью валиномицина. Ионофоры,
такие как валиномицин и
грамицидин В, являются
сильными антибиотиками. Они
убивают другие клетки, нарушая ка-
тионные концентрационные
градиенты, имеющие значение
о
ьй1у 1
Ьеи ь
5^Уа1ь
ьЛ"а1
в^Тгр
Ьеи I,
ю"Тгрп
в^Тгр
ь Хеи
* Дгр
С 15
N11
сн2
сн2
он
а
Грамицидин О
Рис. 4.15. Грамицидин В: а — ковалент-
ная структура; б — димер грамицидина В,
образующий канал в мембране
для генерирования АТР.
Мембранные транспортные системы. Перенос небольших
молекул и ионов через мембраны обеспечивается тремя типами
интегральных мембранных белков: каналами и порами; пассивными
транспортерами и активными транспортерами.
Каналы и поры функционируют весьма сходно с ионофорами,
обеспечивая перенос ионов и небольших молекул по концентрационному
градиенту. Для этого процесса энергия не требуется, и он может
рассматриваться скорее как диффузия через белки, нежели чем через липидный
бислой. Однако этот процесс существенно превосходит по скорости
свободную диффузию, поэтому иногда его называют облегченной
диффузией. Некоторые каналы позволяют транспортироваться веществам
чрезвычайно быстро. Например, через ацетилхолин-рецепторный ионный
канал может за секунду проходить 107 ионов Ка*. Поры и каналы
представляют собой трансмембранные белки, внутри которых имеется
гидрофильный проход. Следует отметить, что термин "поры" используется,
166
Глава 4. Липиды
как правило, для бактерий, а "каналы" — для животных. Молекулы
веществ с соответствующими размером, зарядом и геометрической
конфигурацией могут быстро проходить через эти проходы, диффундируя в
направлении концентрационного градиента (рис. 4.16). Внешние
мембраны грамотрицательных бактерий богаты поринами, относящимися к
семейству белков, образующих поры и позволяющих ионам и
небольшим молекулам получать доступ к специфическим транспортерам ци-
топлазматической мембраны- Порины
только слабо селективны по отношению
к транспортируемым веществам и
обычно остаются постоянно открытыми.
Напротив, клеточные мембраны животных
содержат много каналобразующих белков,
которые высоко специфичны по
отношению к определенным ионам* Некоторые
из этих каналов открыты постоянно,
тогда как другие служат в качестве ворот,
открываясь и закрываясь в ответ на
определенные сигналы. Ионные каналы,
представляющие собой так называемые
"лигандзависимые ворота", открываются
в ответ на связывание специфических
сигнальных молекул. Другие каналы
открываются и закрываются только тогда,
когда меняются электрические свойства мембраны* Существуют также ка-
налы, восприимчивые к изменениям натяженности и тургорного
давления в мембране.
Активные и пассивные транспортеры, в отличие от каналов и пор,
специфически связывают и транспортируют вещества через
мембрану. Действие этих белков во многом сходно с ферментами, за
исключением того, что вместо катализа химического изменения субстрата
они перемещают его с одной стороны мембраны на другую* Вещества
могут транспортироваться против концентрационного градиента, и этот
процесс требует затрат энергии. Скорость транспортировки
активными или пассивными белковыми переносчиками соизмерима со
скоростью ферментативных реакций.
Транспортные белки обычно специфичны по отношению к
определенным молекулам или группе структурно сходных молекул. Транспортер,
как правило, стереохимически специфичен и транспортирует только
биологически важные стереоизомеры. В простейшем случае мембранные
транспортеры обеспечивают так называемый уыипорт, т.е* они
переносят только один тип веществ через мембрану (рис. 4.17, а). Многие
транспортеры обеспечивают котранспорт двух веществ, перенося их либо- в
одном направлении (в этом случае имеет место симпорт), либо в
различных направлениях (антипорт) (рис. 4.17 б, в). Некоторые транспортеры
Рис. 4.16 Мембранный
транспорт через поры или каналы.
Такие белки содержат
гидрофильный проход, который
позволяет молекулам и ионам с
подходящими размером, зарядом и
геометрической конфигурацией
проникать через мембрану в
каком-либо направлении
167
Часть I. Биомолекулы: структура, свойства, функции
электронеитральны, т.е. в результате их действия не происходит
изменения заряда по разные стороны мембраны. Транспорт с помощью
электрогенных транспортеров приводит к переносу зарядов через мембрану и
может, следовательно, существенно изменять мембранный потенциал
(разницу электрического потенциала на противоположных сторонах
мембраны). Все транспортные белки действуют как ворота. Транспортер
может принимать различную конформацию, так что связывающий центр
будет обращен наружу или вовнутрь в зависимости от того, как этого
требует конкретная ситуация. Находясь в конформации с обращенным
наружу связывающим центром, белок связывает специфическую
молекулу или ион и претерпевает конформационное изменение, в результате
чего связывающий центр оказывается на другой стороне мембраны.
Транспортируемая молекула высвобождается на внутренней стороне
мембраны, и транспортер возвращается в исходное состояние (рис. 4.18). Кон-
формационные изменения белка-транспортера часто вызываются
связыванием переносимой молекулы, подобно
индуцированному соответствию определенных
ферментов их субстратам. При активном
транспорте конформационные изменения
могут осуществляться за счет гидролиза АТР
или другого источника энергии.
В качестве примера пассивного
транспортера можно привести глкжозный
транспортер эритроцитов. Эритроциты
используют в основном глюкозу в качестве
источника энергии. В-глюкоза переносится
из крови (где ее концентрация около 5 мМ)
по концентрационному градиенту в
эритроциты путем пассивного транспорта с
помощью глюкозных транспортеров. Этот
транспортер представляет собой
мембранный гликопротеин, связывающий
глюкозу, когда он находится в конформации,
обеспечивающей расположение
связывающего центра на внешней поверхности
мембраны. Затем транспортер, испытывая
конформационные изменения, переносит
глюкозу через бислой. Глюкоза
высвобождается на цитоплазматической стороне, а
транспортер вновь переходит в исходную
конформацию. Глюкозные транспортеры,
подобные этому эритроцитарному белку,
присутствуют во всех клетках
млекопитающих, и их активность может тонко
регулироваться.
0
Рис. 4.17. Типы
мембранного транспорта: а — уни-
порт; б — симпорт; в — ан-
тидорт
168
Глава 4. Липиды
Активный транспорт требует затрат
энергии, и она может поставляться в
различных формах. Наиболее часто
используемый источник энергии — АТР.
Большая группа АТР-зависимых ионных
транспортеров —■ ионтранспортирующие
АТРазы, присутствующие во всех
организмах. Эти активные транспортеры,
включающие Ма+-К+-АТРазу и Са2+-АТРазу,
играют существенную роль в создании и
поддержании градиентов концентрации
ионов через плазматические мембраны и
мембраны внутриклеточных органелл.
Источником энергии для некоторых
активных транспортеров, например бактерио-
родопсина, является свет. Первичный
активный транспорт обеспечивается
прямыми непосредственными
источниками энергии, такими как АТР, свет или
электронный транспорт (см. гл. 9, 10),
Вторичный активный транспорт
возможен благодаря ионным
концентрационным градиентам.
Следует отметить, что в транспортный
процесс может быть вовлечен не один
компонент мембраны, а много больше. Зат-
раты энергии на активный транспорт
очень велики, например, у человека
составляют более трети всей энергии,
аккумулируемой в процессе метаболизма.
Функции биологических мембран.
Мембраны участвуют в формировании
клеточных структур, в которых
осуществляются
окислительно-восстановительные процессы, аккумулируется и
транспортируется энергия,
регулируется транспорт веществ. Но биологические
мембраны представляют собой не
просто инертные оболочки клеток и
субклеточных структур, они выполняют ряд
1
1
I
••
Рис. 4.18. Функционирование
транспортного белка.
Транспортные белки функционируют как
"ворота". Белок связывает свой
специфический субстрат и затем
испытывает конформационное
изменение, что позволяет молекуле или
иону высвободиться на другой
стороне мембраны. Котранспортеры
имеют специфические
связывающие центры для каждого типа
транспортируемых молекул
важнейших и разнообразных функций.
Участвуя в формировании клеточных структур, мембраны играют
роль механического барьера, ограничивающего эти структуры от
внешней среды. Являясь гибкими, лабильными, постоянно
обновляющимися образованиями, мембраны сохраняют в то же время прочность и
169
Часть I. Биомолекулы: структура, свойства, функции _^^
эластичность при деформациях. Хотя основную механическую нагрузку
(осмотическое и гидростатическое давление внутри клеток) у высших
растений несет клеточная стенка, построенная у растений в основном
из целлюлозы, гемицеллюлоз, пектина и белка экстенсина, а у
бактерий сложного полисахарида — муреина, клеточная мембрана играет
важную роль в регулировании механической нагрузки протоплазмы.
Внутриклеточные мембраны часто образуют сложные системы, такие
как внутренняя и наружная мембраны митохондрий, мембраны эн-
доплазматического ретикулума, ядра, лизосом. Суммарная поверхность
мембран настолько велика, что позволяет им нести большие
функциональные нагрузки.
В большинстве мембран содержатся ферменты, а также
ферментные комплексы и метаболоны, обеспечивающие важнейшие
метаболические процессы*
Как было рассмотрено выше, мембраны выполняют также
транспортную функцию. Транспортные системы, локализованные в
мембранах, обеспечивают перенос определенных соединений. И в то же
время мембраны выполняют разделительную функцию, регулируя
поток веществ внутрь клетки и из нее, что важно для сохранения
постоянства внутриклеточной среды. Характерной чертой природных
мембран является их избирательная проницаемость, благодаря чему
разные клеточные органеллы могут содержать разные химические
компоненты и выполнять различные функции. Избирательная
проницаемость основана на системах специфичности транспортных белков,
способных узнавать определенное соединение и проводить его через
мембрану. Транспортная функция мембран сопряжена с такими
важнейшими явлениями, как поддержание внутриклеточного гомеостаза,
запасание и трансформация энергии, с процессами метаболизма веществ.
На мембранах происходит формирование градиентов
электрического потенциала, что важно для передачи нервных импульсов, а также
для осуществления энерготрансформирующих процессов в клетке.
Мембраны являются теми клеточными структурами, благодаря которым
возможна трансформация осмотической и электрической форм
энергии в энергию макроэргической фосфатной связи АТР.
Трансформация и аккумулирование энергии протекают в специализированных
мембранах. Так, процессы фотосинтеза и дыхания осуществляются,
соответственно, в мембранах хлоропластов и митохондрий. Фотосинте-
зирующие мембраны трансформируют энергию света в энергию
восстановленных соединений — углеводов — основного химического
источника энергии для гетеротрофных организмов. При дыхании эта
энергия освобождается в мембранах митохондрий и используется в
процессах биосинтеза различных веществ.
В связи с наличием в мембранах специфических белковых
рецепторов клетки способны воспринимать внешние сигналы и в ответ на
них перестраивать свой обмен веществ.
170
Глава 4. Липиды
Важнейшей функцией мембран является также адгезивная
функция, благодаря которой клетки способны взаимодействовать друг с
другом, что существенно, в частности, при формировании тканей.
4.5. Методы исследования липидов
В недалеком прошлом биохимия липидов считалась неинтересной и
запутанной областью знаний. Однако с усовершенствованием методов
выделения, разделения и анализа липидов открылись возможности для
более глубоких исследований. Они оказались наиболее важными в
познании роли липидов в структурировании сложных биоорганических
систем, в частности мембран, и механизмах их функционирования.
Первым этапом исследования липидов является их выделение. Как
правило, липиды экстрагируют из свежей ткани в виде сложной
смеси. Для этой цели используются органические растворители или их
смеси, полностью или в значительной степени смешивающиеся с
водой, — такие, как этанол, ацетон или смесь хлороформ: метанол (2:1)
по объему. Смешиваемость растворителей с водой необходима потому,
что в свежей ткани содержится более 70 % воды.
Для разделения и идентификации ацилглицеролов, фосфатидил-
глицеролов, сфинголипидов и гликолипидов применяется метод
тонкослойной хроматографии. Суть его заключается в следующем. На
стеклянную пластинку размером приблизительно 10x10 см наносится
ровный слой густой водной пасты, изготовленной из мелкоизмельчен-
ного инертного адсорбента (силикагеля, целлюлозы) с добавлением
связывающего вещества (например, гипса). Пластинку подсушивают,
а затем прокаливают для удаления воды, после чего на стекле
остается тонкий однородный слой прочно связанного адсорбента. Нижний
край пластины погружают в кювету с растворителем и над
растворителем наносят на пластинку каплю смеси, подлежащую анализу; всю
систему помещают в закрытую камеру. Растворитель под действием
капиллярных сил поднимается по пластинке вверх, увлекая за собой
смесь липидов, которая по мере подъема растворителя распределяется
по дискретным зонам. Пластинку высушивают и, нанося подходящий
индикатор, определяют положение разделившихся компонентов.
Разделившиеся липидные компоненты собирают, соскабливая с
пластинки пятна с адсорбентом, и элюируют из них липиды.
Этот метод используется обычно для разделения ацилглицеролов в
очень малых количествах. Однако если слой адсорбента сделать
достаточно толстым, то можно проводить разделение и в более крупных
масштабах; применяются также методы двухмерной тонкослойной
хроматографии.
Поскольку липиды имеют низкую плотность (-0,95 г/см-3), а
белки — высокую («1,20 г/см"3), то липопротеины с разным
соотношением липида и белка могут быть разделены с помощью
ультрацентрифугирования.
171
Часть I. Биомолекулы: структура, свойства, функции
При исследовании компонентного состава липидов широко
используются щелочной, кислотный и ферментативный виды гидролиза. Что
касается ферментативного гидролиза, к нему мы вернемся в гл.
"Ферменты". При щелочном гидролизе используют следующий подход. Мягкий
щелочной гидролиз триацилглицеролов, фосфоглицеридов приводит к
отщеплению жирных кислот, но не затрагивает глицерофосфоспиртовый
остов исходной молекулы. При гидролизе фосфоглицеридов в сильно
щелочной среде отщепляются как обе жирные кислоты, так и спирт.
Поскольку связь между глицерином и фосфорной кислотой
сравнительно устойчива к щелочному гидролизу, еще одним продуктом гидролиза в
сильно щелочной среде является глицерол-3-фосфат. Это соединение
распадается при кислотном гидролизе.
Анализ смесей жирных кислот, полученных в результате гидролиза
липидов, наиболее четко и точно проводится методом газожидкостной
хроматографии. С целью перевода жирных кислот в летучие соединения
их превращают в метиловые эфиры жирных кислот. Эфиры
впрыскивают в нагретую до высокой температуры колонку, изготовленную в виде
полой спирали, внутренняя часть которой покрыта стационарной
жидкой фазой. В качестве жидкой фазы применяют высокотемпературную
парафиновую или силиконовую смазку. В качестве "носителя"
используется какой-либо инертный газ (азот, аргон). Метиловые эфиры жирных
кислот, введенные в нагретую колонку, вместе с газом-носителем, под
давлением движутся через колонку. Распределение основано на
различной растворимости эфиров в стационарной жидкой фазе. Поскольку
жидкая фаза в колонке находится при повышенной температуре,
адсорбированный на ней эфир вновь испаряется и поступает в подвижную газовую
фазу. Чем ниже растворимость эфира в жидкой фазе, тем быстрее он
покидает вместе с "носителем" колонку. Газовая смесь попадает в
специальное устройство (детектор) для обнаружения и количественного
определения отдельных соединений. Анализ основан на каком-либо
чувствительном физическом методе. В одном из этих методов регистрируется
изменение ионизационных свойств газовой смеси, вызванное действием
у-излучения. В результате получают набор отдельных пиков, каждый из
которых соответствует определенной жирной кислоте (рис. 4,19).
Анализ методом газожидкостной хроматографии выполняется
достаточно быстро. Метод имеет высокую разрешающую способность и
большую чувствительность (~10~12 моль).
В исследовании химических свойств жиров и масел существенное
значение имеют такие показатели, как кислотное, йодное числа,
число омыления.
Кислотное число — количество мг гидроксида калия,
необходимого для нейтрализации свободных жирных кислот, содержащихся в 1 г
жира (масла). Это весьма важный показатель свойств и состояния жира,
так как он может быстро возрастать при хранении жира и продуктов,
содержащих жир.
172
Глава 4. Липиды
СНзОН и
ЕСООН —*■* КСОСНз
Летучий метиловый эфир
6 9
Время, мин
Рис. 4.19. Типичный профиль разделения смеси летучих метиловых эфиров
жирных кислот методом газожидкостной хроматографии. Достигается очень
четкое разрешение (узкие зоны) для компонентов-гомологов
Йодное число — количество г йода, связываемое 100 г жира.
Поскольку йод присоединяется по месту двойных связей ненасыщенных
жирных кислот, йодное число определяет степень ненасыщенности
жиров; чем оно выше, тем более жидкую консистенцию имеет жир,
тем он легче и быстрее окисляется и более пригоден для изготовления
олифы, лаков, красок.
Число омыления — количество мг гидроксида калия, необходимое
для нейтрализации как свободных жирных кислот, так и связанных с
глицерином жирных кислот, получающихся при омылении
(гидролизе) 1 г жира (масла)*
В исследовании структуры и функций мембран используют их
препараты, выделяемые из тканей, отдельных клеток и клеточных
органоидов. Объектом исследования могут быть изолированные фрагменты
мембран, искусственные мембраны и реконструированные системы.
Выделение мембран из клеток осуществляют с помощью комбинации
различных приемов частичного разрушения структур клеток,
последующего фракционирования и концентрирования мембран клеток. Для
фрагментации мембран используют механическую гомогенизацию,
воздействие ультразвуком, чередование замораживания и оттаивания,
гидродинамический удар, осмотический шок, обработку детергентами и
ферментами. Выделяют и концентрируют клеточные органоиды или
фрагменты мембран с помощью центрифугирования или ультрафильтрации.
В исследовании структуры мембран, механизмов их
функционирования широко используют реконструированные и искусственные
мембранные системы; к последним относятся мономолекулярные липид-
173
Часть I. Биомолекулы: структура, свойства, функции
ные слои на поверхности вода—воздух, вода—ткань. Широко
применяется также метод замораживания — скалывания. Суть его
заключается в следующем. Клетки или фрагменты мембраны быстро
замораживают при температуре жидкого азота (-195,5 °С). Образец затем
скалывают, быстро оторвав его от ножа микротома. Скол проходит по
линии наименьшего сопротивления — по середине липидного бислоя,
который практически лишен воды, а прочность замороженного образ-
ца определяется главным образом содержанием воды, которая и
сосредоточена в гидрофильных поверхностных зонах мембраны* Если
после замораживания—-скалывания удалить лед сублимацией, то в
электронном микроскопе можно увидеть наружную поверхность
мембраны.
Объектом интенсивного исследования являются белки мембран.
Поверхностные белки обычно легко отделить от мембраны, получить в
очищенном виде и изучить их физико-химические свойства. Иное дело —
интегральные белки; при их выделении приходится обращаться к
помощи поверхностно-активных веществ, обрабатывать мембраны
ультразвуком с целью разрушения гидрофобных взаимодействий между
неполярными группами, находящимися на поверхности белковых глобул, и
контактирующими с ними углеводородными фрагментами липидов.
Сложность заключается в том, чтобы подобрать такие условия, которые были
бы достаточны для отделения белков от липидного матрикса, но не
вызывали денатурацию белков в процессе выделения.
4.6. Липиды и их роль в питании человека
Из липидов к основным пищевым веществам относятся триацил-
глицеролы. Они служат важным источником энергии, их можно
рассматривать как природный пищевой концентрат большой
энергетической ценности- При окислении 1 г жира выделяется около 40 кДж
энергии, т.е. примерно в 2 раза больше, чем при окислении того же
количества белков. В нормально сбалансированном пищевом рационе
соотношение количества белков, жиров, углеводов составляет 1:1:4.
Потребность человека в жире зависит от возраста, характера трудовой
деятельности, климатических условий. В пожилом возрасте, а также
при малой физической нагрузке и занятости умственным трудом
потребность в жире снижается; в холодном климате — повышается. В
среднем потребность в жире — 80—100 г в сутки.
Жиры являются растворителями витаминов А, В, Е и К, в связи с
чем обеспеченность организма этими витаминами в значительной
степени зависит от содержания жиров в пищевых продуктах, С жирами
поступают такие биологически активные вещества, как фосфатиды,
стеролы. Фосфатидилхолин является важным донором метальных
групп и относится к витаминоподобным соединениям.
Жиры оказывают существенное влияние на вкусовые качества пищи,
придавая ей при кулинарной обработке приятный вкус и аромат.
174
Глава 4. Липиды
Пищевые продукты, содержащие 70 % и более триацилглицеролов,
называются жировыми продуктами, — это выделенные и обработанные
резервные жиры. По происхождению жировые продукты
подразделяются на три группы: животные, растительные и искусственные.
К животным жировым продуктам относятся говяжий, бараний,
свиной жиры, получаемые вытапливанием из жировой и костной
тканей. Они характеризуются высоким содержанием насыщенных
жирных кислот и высокой температурой плавления (-40 °С), влаги в них
содержится не более 0,3 %, кислотное число — не более 2, йодное
число — 35—60.
К жировым продуктам растительного происхождения относятся
масла, на 95—97 % состоящие из триацилглицеролов и небольшого
количества сопутствующих веществ (фосфатиды, токоферолы, стеро-
лы, пигменты). Сопутствующие вещества повышают биологическую
ценность масел, стойкость при хранении, обусловливают их аромати-
ческие и вкусовые особенности, окраску. Йодное число масел лежит в
пределах 100—175, кислотное — от 1 до 3. Масла получают из семян
высокомасличных культурных растений и называют по их источнику
производства: подсолнечное, хлопковое, льняное, конопляное, соевое,
оливковое. Из измельченных семян масла либо экстрагируют каким-
либо органическим летучим растворителем с его последующим
удалением, либо извлекают прессованием. По степени очистки масла
подразделяются на сырые, нерафинированные, рафинированные,
очищенные. Сырые масла — это масла, подвергнутые только фильтрации. В
результате рафинирования обеспечиваются прозрачность масла,
отсутствие в нем отстоя (в основном фосфатидов), а также запаха и вкуса.
Рафинирование — обязательная технологическая операция при полу-
чении хлопкового масла. Это связано с тем, что в ядре семян
хлопчатника содержится соединение госсипол, которое переходит в масло,
придает ему бурый или почти черный цвет, является токсичным
соединением. При рафинировании госсипол полностью удаляется*
К нерафинированным относят масла, подвергнутые частичной
очистке: отстаиванию, фильтрации, гидратации (промывке водой) и
нейтрализации. Наиболее биологически полноценным является сырое
растительное масло, в котором полностью сохраняются фосфатиды, сте-
ролы и другие ценные компоненты.
В организме человека и животных легко синтезируются
насыщенные жирные кислоты, синтезируются также кислоты с одной двойной
связью (например, олеиновая). Ткани человека и ряда животных не
способны синтезировать полиненасыщенные кислоты: линолевую, ли-
ноленовую, арахидоновую. Их относят к категории незаменимых
жирных кислот. При длительном отсутствии их в пище у животных
наблюдается отставание в росте, выпадение волосяного покрова, пора-
жение кожи. Линолевая и линоленовая кислоты, содержащие
соответственно две и три двойные связи, а также родственные им полине-
175
Часть I, Биомолекулы: структура, свойства, функции
насыщенные жирные кислоты объединяют в группу под названием
витамин Г. Именно в связи с этим высокую биологическую ценность имеют
растительные масла. Так, в подсолнечном масле около 60 % всех
жирных кислот, а в хлопковом — около 50 % приходится на долю линолевой
кислоты. Высоким содержанием полиненасыщенных жирных кислот
отличаются льняное, конопляное, соевое, кукурузное масла.
Искусственные жировые продукты представляют собой жиры,
основой которых являются гидрогенизированный жир — саломас. Он
получается путем превращения растительного масла в продукт, обладающий
твердой консистенцией. Принцип такого превращения заключается в
воздействии водорода при повышенной температуре в присутствии
катализатора на ненасыщенные кислоты. В процессе присоединения
водорода последние превращаются в насыщенные жирные кислоты твердой
консистенции. Калорийность саломаса возрастает; однако теряются
ценные полиненасыщенные жирные кислоты, витамины, фосфатиды. При
производстве из саломаса маргарина его обогащают линолевой кислотой,
добавляя к нему ненасыщенные жирные кислоты (подсолнечное масло),
а также жирорастворимые витамины (см. гл. 5. "Витамины"). Изменяя
условия процесса гидрогенизации (температуру, парциальное давление
водорода, катализатор), можно получить саломас с различными
заданными физическими характеристиками.
Давая общую биологическую оценку жировым пищевым
продуктам, следует отметить, что она определяется наличием в них
жирорастворимых витаминов, полиненасыщенных жирных кислот, фосфати-
дов. Практически ни один из жиров, используемых в питании, не
является биологически полноценным во всех отношениях.
Биологическая полноценность жировых продуктов может быть обеспечена
только сочетанием различных жиров животного и растительного
происхождения. Оптимальные в биологическом отношении сочетания
создаются при включении в рацион 70—80 % животного и 20—30 %
растительного жира- Недостаток жиров в пище или постоянное
нарушение их оптимального соотношения приводит к различным
нарушениям обмена веществ и служит причиной многих заболеваний.
Растительные жировые продукты имеют высокую степень
ненасыщенности (большие йодные числа), поэтому весьма неустойчивы при
хранении. Они претерпевают определенные изменения, приобретают
неприятный вкус и запах, прогоркают. Процесс порчи жировых
продуктов может вызываться чисто химическими реакциями окисления,
связанными с действием воздуха, света, воды. Установлено, что
основную роль здесь играют процессы окисления ненасыщенных
жирных кислот кислородом с образованием пероксидов жирных кислот:
Г°1
К-С=С-К: + 02 -> Е-С-С-Й!
II II
н н н н
Пероксид
176
Глава 4. Липиды
Часто этому процессу предшествуют процесс гидролиза ацилгли-
церолов и переход жирных кислот в свободное состояние. В
результате дальнейшего разложения пероксидов получаются альдегиды,
придающие продукту неприятный запах и вкус.
Очень легко образуют пероксиды сложные липиды, содержащие
много ненасыщенных жирных кислот. Пероксиды, в свою очередь,
окисляют другие компоненты (белки, углеводы, витамины). Такое
разрушительное действие вызвано превращением пероксидов в перо-
ксид-радикалы — весьма реакционноспособные реагенты. Особенно
интенсивно идет переход пероксидов в пероксид-радикалы в
присутствии ионов металлов. Лучшими условиями хранения являются
герметическая упаковка в среде инертного газа (азота, диоксида
углерода) при температурах не выше 18 °С. Важную роль в повышении
стойкости жировых продуктов играют такие пищевые добавки, как
токоферолы, фосфатиды, аскорбиновая кислота. Каждое из этих
соединений способно выступать в роли антиоксид анта^ предохраняя
чувствительные биомолекулы липидов от окислительных реакций, а
продукты — от быстрой порчи.
Краткое содержание главы
Липиды.— это разнородная группа природных соединений,
полностью или почти полностью не растворимых в воде, но растворимых в
органических растворителях, дающих при гидролизе
высокомолекулярные жирные кислоты. Липиды, в отличие от белков, нуклеиновых
?: и слот и полисахаридов, не являются высокомолекулярными
соединениями, их структура весьма разнообразна, они имеют лишь один
:<бщий признак — гидрофобность. В организме липиды выполняют
четыре основные функции: 1) являются резервными соединениями,
основной формой запасания энергии и углерода; 2) служат формой, в
которой транспортируется эта энергия; 3) являются структурными
компонентами мембран; 4) несут защитную функцию в плодах,
овощах, листьях растений, в клеточных стенках бактерий. По структуре
липиды можно разделить на три группы: а) простые липиды — эфиры
жирных кислот и спиртов; б) сложные липиды, содержащие помимо
:кирных кислот и спиртов другие компоненты различной химической
природы; в) производные липидов — жирорастворимые витамины и
их предшественники.
Жирные кислоты представляют собой длинноцепочечные монокарбо-
новые кислоты. Большинство природных жирных кислот содержит от
12 до 20 углеродных атомов. Жирные кислоты, которые не содержат
двойных углерод-углеродных связей, относятся к насыщенным жирным
кислотам; те, которые содержат одну двойную углерод-углеродную связь,
классифицируются как мононенасыщенные, а те, которые содержат
более чем одну углерод-углеродную двойную связь, — как
полиненасыщенные. Насыщенные и ненасыщенные жирные кислоты сильно разли-
12. Заказ 3486 177
Часть I. Биомолекулы: структура, свойства, функции ^^^
чаются по своей структурной конфигурации. В насыщенных жирных
кислотах углеводородная цепь может принимать бесчисленное
множество конфигураций вследствие полной свободы вращения вокруг
одинарной связи; однако наиболее вероятной является вытянутая,
энергетически выгодная форма. В ненасыщенных жирных кислотах невозможность
вращения вокруг двойной связи обусловливает жесткий изгиб
углеводородной цепи под углом приблизительно 30°. Эта связь находится в ц
реформе. Большинство двойных связей в ненасыщенных жирных
кислотах имеют ццс-конфигурацию. Насыщенные и ненасыщенные жирные
кислоты входят в состав разнообразных липидов.
Простые липиды делятся на нейтральные ацилглицеролы и воски.
Нейтральные ацилглицеролы — это сложные эфиры трехатомного
спирта глицерина и одной, двух или трех жирных кислот. Основная
функция ацилглицеролов — запасание энергии и углерода, поэтому они
являются важнейшими компонентами пищевых продуктов. В
зависимости от физического состояния при комнатной температуре твердые
триацилглицеролы называют жирами, жидкие — маслами.
Основными компонентами в последних являются ненасыщенные жирные
кислоты — незаменимые компоненты пищи. Жиры, как правило,
содержатся в животных тканях, масла — в плодах и семенах растений.
Особенно высоко содержание масел (20—40 %) в семенах
подсолнечника, хлопчатника, сои, льна; они используются в пищевой
промышленности для получения пищевых масел.
Триацилглицеролы способны вступать во все химические реакции,
свойственные сложным эфирам. Наибольшее значение имеет реакция
гидролиза (омыления), ведущая к образованию глицерина и жирных
кислот.
Воски — сложные эфиры высокомолекулярных жирных кислот и
одноатомных спиртов с длинной углеводородной цепью. Это твердые
соединения с ярко выраженными гидрофобными свойствами.
Жирные кислоты в них содержат от 24 до 36 углеродных атомов, а
высокомолекулярные спирты — от 16 до 30 углеродных атомов. Часто эти
компоненты находятся в восках в свободном состоянии. Основная
функция природных восков — образование защитных покрытий на
листьях, плодах растений, которые уменьшают потерю влаги растением и
снижают возможность поражения микроорганизмами.
Фосфоглицериды — амфипатические липиды, являющиеся
основными компонентами биологических мембран. К ним относятся фосфа-
тидилхолин, фосфатидилэтаноламин, фоефатидилеерин, фосфатидил-
инозитол. Фосфатидшшнозитол-4,5-бисфосфат — производное фосфа-
тидилинозитола, играющее важную роль в сигнальных процессах. Кар-
диолипин является основным структурным компонентом мнтохонд-
риальных мембран.
К другим классам липидов, содержащихся в биомембранах,
относятся сфинголипиды и стероиды. Структурной основой сфинголипи-
178
Глава 4. Лапиды
дов является длинноцепочечный спирт сфингозин. Три важнейших
типа сфинголипидов — сфингомиелины, цереброзиды и ганглиозиды.
Холестерол — это стероид, являющийся важнейшим компонентом
мембран животных, он служит также как предшественник различных
гормонов.
Мембраны — это универсальная и доминирующая форма
структурной организации живой материи. Биологические мембраны формируют
внешнюю границу клетки и разделяют внутриклеточные компартменты.
Типичная мембрана содержит липиды и белки, а также небольшое
количество углеводов в составе гликосфинголипидов и гликолротеинов.
Наиболее удовлетворительной моделью мембраны признана жидкостно-мо-
заичная модель, согласно которой мембрана состоит из бислоя сложных
липидов, в которых находятся глобулярные белки. Липиды в бислое
расположены так, что их гидрофобные хвосты направлены друг к другу и
частично перекрываются* Обе поверхности бислоя составлены
гидрофильными головками, поэтому легко ассоциируются с водой* Большинство
интегральных белков пронизывают гидрофобную внутреннюю часть
бислоя, тогда как периферийные мембранные белки более свободно
ассоциированы на поверхности мембраны. Мембранные структуры организова-
ны за счет слабых электростатических сил и гидрофобных
взаимодействий; ковалентные связи здесь играют второстепенную роль. В связи с
этим биологические мембраны являются динамичными структурами, что
обусловливает их способность к различным перестройкам. Холестерол
воздействует на текучесть мембран клеток животных, нарушая
упаковку липидов в гелевой среде и уменьшая подвижность ацильных цепей в
липидной жидко-кристаллической фазе.
Липидный бислой представляет собой селективно избирательный
барьер, который непроницаем для большинства заряженных молекул,
но в то же время позволяет воде и гидрофобным молекулам свободно
диффундировать через него. Скорость диффузии ионов через
мембрану может быть значительно увеличена с помощью ионофоров.
Специфические транспортеры, а также белки, формирующие каналы и поры,
обеспечивают движение ионов и полярных молекул через мембраны.
Кана л образующие белки позволяют быстро диффундировать
большому количеству определенных ионов или небольших молекул через
центральный проход внутри них по концентрационному градиенту.
Транспортные белки связывают субстрат и переносят его через мембрану,
изменяя свою конформацию. Пассивные транспортеры перемещают
молекулы по концентрационному градиенту и не требуют затрат
энергии. Для функционирования активных транспортеров, переносящих
субстраты против концентрационного градиента, необходимы затраты
энергии. При первичном активном транспорте энергия поставляется
непосредственно за счет гидролиза АТР, энергии света или
электронного транспорта• Вторичный активный транспорт обеспечивается с
помощью ионных градиентов.
12*
179
Часть I. Биомолекулы: структура, свойства, функции
Функции мембран разнообразны и чрезвычайно важны. Они
участвуют в формировании клеточных структур, осуществляют окислительно-
восстановительные процессы, регулируют транспорт веществ.
Важнейшая функция биологических мембран — генерирование
биоэлектрического потенциала, аккумулирование и трансформация энергии. В
мембранах хлоропластов происходит трансформация энергии света в энергию
восстановительных соединений — углеводов — основного химического
источника энергии для организмов. При дыхании эта энергия
освобождается и аккумулируется благодаря молекулярным механизмам,
локализованным во внутренней митохондриальной мембране.
Хронология важнейших дат в исследованиях липидов
1854 г. — П. Э. Бертло установил, что глицерин является
трехатомным спиртом; синтезировал моно-, ди~, триацетаты.
1890 г. — П. Э. Бертло осуществил синтез жиров.
1905 г. — Ф. Кнооп открыл процесс окисления жирных кислот.
ЛИТЕРАТУРА
Антонов В. Ф,? Черныш А М., Пасечник В. И., Вознесенский С. А, Козлова Е. К.
Биофизика: Учебник. — М.: Гуманит. Изд. центр ВЛАДОС, 1999. — 288 с.
Аргпюхов В. Г., Наквасина М. А. Биологические мембраны: структурная
организация, функции, модификация физико-химическими агентами. — Воронеж:
Изд-во ВГУ, 2000. — 276 с.
Березов Т. Т.> Коровкин Б. Ф. Биологическая химия: Учебник. — 3-е изд.,
перераб, и доп. — М.: Медицина, 19,98- — 704 с.
Геннис Р. Биомембраны: Молекулярная структура и функции / Пер. с англ.
— М.: Мир, 1997, — 624 с.
Кретович В. Л. Биохимия растений. — М.: Высшая школа, 1986. — 503 с,
Ленинджер А* Основы биохимии, — М,: Мир, 1985. — Т. 1. — 365 с.
Строев И. М. Биологическая химия. — М.: Высшая школа, 1986. ~ С. 84—98.
Могап Ь. А, Зсптёеоиг К. С, НоНоп Н. К, ОсНв Д. 5„ Кашп </. I). ВюсЬепивЪгу.
— 2 Ей. — Ш1 Райегеоп РиЬ., Епе1еттооа СШв, 1994. — 1200 р.
ВОПРОСЫ И ЗАДАЧИ
1. Какой из двух триацилглицеролов имеет более высокую точку плавления:
триацилглицерол, содержащий стеариновую, пальмитиновую, олеиновую
кислоты, или триацилглицерол, содержащий олеиновую, линолевую, пальмитиновую
кислоты?
2. Изобразите структуру исходного липида, соответствующего составу
каждой из следующих смесей, полученных при полном гидролизе фосфолипида: а)
глицерин, пальмитиновая кислота, олеиновая кислота, неорганический фосфат; б)
глицерин, стеариновая кислота, неорганический фосфат, холин.
3. Рассчитайте молярное соотношение между липидами и белками в
биологической мембране, содержащей 56 % липидов и 44 % белков. Примите
среднюю молекулярную массу липидов 700, белков — 55 000.
4. В состав хлопкового масла входит а-стеародиолеин. Напишите
структурную формулу соединения.
Глава 5. Витамины
Глава 5. ВИТАМИНЫ
5.1- Общая характеристика. Классификация и номенклатура
Витамины (от лат. \)Иа — жизнь) — класс биологически
активных соединений, объединяемых по признаку строгой необходимости в
обмене веществ в организме человека и животных; это
низкомолекулярные пищевые вещества различной химической природы, причем
требуются они организму в ничтожно малых количествах (от
нескольких мкг до нескольких мг в сутки). Организм человека и животных
не синтезирует витамины или синтезирует их в небольшом
количестве. В отличие от других незаменимых факторов (незаменимых
аминокислот, полиненасыщенных жирных кислот), витамины не
являются пластическими веществами или источником энергии и
участвуют в обмене веществ не как субстраты биохимических реакций, а как
участники процессов биокатализа и регуляции этих реакций. Они
часто входят в состав коферментов, необходимых для
функционирования биологических катализаторов — ферментов.
Недостаток витаминов в организме приводит к развитию гипови-
таминозов) а полное их отсутствие — авитаминозов, к нарушению
обмена веществ и к тяжелым заболеваниям.
Открытие витаминов принадлежит нашему соотечественнику Н. И.
Лунину. Он установил, что в пищевых продуктах помимо белков, жиров,
углеводов, минеральных солей и воды для нормальной жизнедеятельности
необходимо также наличие еще неизвестных компонентов питания. Его
эксперименты показали, что белые мыши, получавшие в качестве пищи
цельное молоко, проявляли нормальную жизнедеятельность, но
погибали, когда их кормили смесью основных компонентов молока: казеина,
жира, лактозы, солей и воды. Эти данные были подтверждены С. А,
военным и Ф. Гопкинсом. В 1911—1912 гг. польский ученый К. Функ
выделил активное соединение из рисовых отрубей, предотвращающее
заболевание "бери-бери" (полиневрит). Это вещество, содерлеащее аминогруппу,
он назвал витамином (необходимым для жизни амином). В дальнейшем
этот термин был распространен и на другие вновь открытые
биологически активные соединения, хотя и не содержавшие аминогрупп.
В настоящее время известно около двух десятков витаминов. Они
подразделяются на две группы: жирорастворимые и водорастворимые
витамины. Первые — типичные гидрофобные соединения с ярко
выраженной циклической и ациклической углеводородной структурой. Все
они построены из строительных блоков изопренового типа. Подобно ли-
181
Часть I. Биомолекулы: структура, свойства, функции ^^
пидам, они растворимы в органических растворителях, их также
называют производными липидов. Вторые — вещества гидрофильные, самого
различного химического строения, растворимые в воде»
В растениях, микроорганизмах встречаются вещества, которые сами
по себе не проявляют биологической активности, но являются
предшественниками образования в организме человека или животных
витаминов. Они получили название провитаминов.
Наряду с витаминами, дефицит которых приводит к ярко
выраженной витаминной недостаточности, вызывает специфические
заболевания, имеются и другие биологически активные соединения не столь
сходного с витаминами характера. По своим функциям эти
соединения близки к другим незаменимым пищевым веществам
(полиненасыщенным жирным кислотам, незаменимым аминокислотам); они
получили название вигпаминоподобных веществ.
Ряд витаминов представлен не одним, а несколькими
соединениями со сходными биологическими активностями. Они называются ей-
тамерами. Для обозначения витаминов применяют термин "витамин"
с каким-либо буквенным обозначением (витамин А, витамин В,
витамин Е и т.д.). Для отдельных соединений, входящих в состав витаме-
ров, используется рациональное название, отражающее их
химическую природу, например ретиналь (альдегидр&я форма витамина А),
эргокальциферол (форма витамина О).
Гиповитаминозы, авитаминозы чаще всего возникают в связи с
низким содержанием витаминов в пище при однообразном,
несбалансированном питании, например, преобладании в продукте углеводного
компонента при низком содержании белков, жиров, при отсутствии
свежих овощей и фруктов. Иногда витаминная недостаточность
возникает из-за нарушения процессов пищеварения, каких-либо заболеваний.
Витаминная недостаточность часто возникает у жителей
слаборазвитых стран, при длительных морских путешествиях, экспедициях в
отдаленные районы, во время войн. В экономически развитых
странах гиповитаминозы связаны с использованием рафинированных
пищевых продуктов, с сезонными колебаниями содержания витаминов в
пище, неправильным режимом хранения продуктов, несовершенством
их технологической обработки. Витаминная недостаточность
возникает также тогда, когда в пищевых продуктах содержатся вещества,
подавляющие функции витаминов, — антивитамины. По механизму
действия эти вещества подразделяются на три группы: 1)
структурные аналоги витаминов — соединения, идентичные по химической
структуре с витаминами, но отличающиеся какой-либо одной
функциональной группой от витаминов; 2) ферменты, разрушающие
витамины; 3) соединения, дающие прочные комплексы с витаминами.
В экономически развитых странах в широких масштабах
осуществляется производство витаминов двумя методами синтеза —
химическим и микробиологическим. Полученные витамины используются
182
Глава 5. Витамины
в качестве добавок в пищевые продукты либо в качестве медицинских
препаратов.
5.2. Жирорастворимые витамины
Эта группа витаминов растворима в жирах и других органических
растворителях (хлороформе, петролейном эфире, бензоле). К ней
относятся витамины с буквенными обозначениями А, В, Е, К.
Витамин А. Этот витамин представляет собой группу витамеров:
ретинол, ретиналь, ретиноевая кислота, которые являются
производными Р-ионона:
СНз СНз 0
и
•сн=сн-с-сн3
СНз СНз Н
СНз
р-Ионон
СН3 Н
I 1
СНз
Сх /С<ч. /С^ ,/С.ч, /К
I
н
I
н
I
н
I
н
СНз
Витамин А1
^
СНзСЩ
СНз СНз
СНз {
Группа изопрена
р-Каротин
СН2ОН; ретиналь К = СНО; ретиноевая
Витамин Ах: ретинол К =
кислота К = СООН*
Позднее из печени пресноводных рыб был выделен витамин А2 (дегид-
роретинол), отличающийся от витамина Ах тем, что в (3-иононовом
кольце имеется одна дополнительная связь между С-3 и С-4 атомами углерода.
Очищенный витамин Аг образует крупные кристаллы
светло-желтого цвета. У всех соединений, кроме 11-^«е-ретиналя, находящегося
в сетчатке глаз, двойные связи в изопреновых группах находятся в
тяргшс-конфигурации.
Витамин А — витамин роста для детей. Первым признаком
гиповитаминоза А является ночная (куриная) слепота. Это связано с
недостаточным образованием в сетчатке глаза сложного белка родопсина,
в синтезе которого участвует данный витамин. Развитие
гиповитаминоза А проявляется сухостью роговой оболочки глаз, сухостью кожи и
волос, блеклостью и шелушением кожных покровов, склонностью к
появлению угрей, фурункулезу, повышенной утомляемостью.
Функциональные группы К витамина А придают ему различную
биологическую активность. Так, ретинол необходим для роста,
дифференциации и сохранения функций эпителиальных и костных тканей, а также
размножения (стимулирования образования спермы). Ретиналь играет
183
Часть I. Биомолекулы: структура, свойства, функции
важную роль в механизме зрения, образуя с белком опсином пигмент
родопсин, представляющий собой первичный рецептор света в
светочувствительных клетках, которые затем передают информацию клеткам
нервной системы. Ретиноевая кислота на порядок активнее ретинола в
клеточной дифференциации, но менее активна в процессе размножения.
Однако механизм действия витамина А на молекулярном уровне не
изучен. Имеются сведения о том, что биохимическая функция его связана с
транспортом Сахаров, с синтезом гликолипидов клеточных мембран.
Витамин Ах содержится исключительно в животной пище.
Особенно богат им жир печени морских животных и рыб (20—30 мг на 100 г);
в меньших количествах он содержится в молочных продуктах,
яичном желтке.
Хотя в растениях витамин А не содержится, в них широко
распространены каротины (а-, (5-, у-) — провитамины А. Богаты каротинами
морковь, шиповник, черешня, тыква, томаты, листовая зелень
(петрушка, шпинат), абрикосы, апельсины (1—10 мг на 100 г). Особенно широко
распространен (3-каротин. Он же обладает наибольшей биологической
активностью. В организме человека и животных в результате
окислительного расщепления из (З-каротина образуются две молекулы витамина А1,
Суточная потребность взрослого человека в витамине А — около
1—2 мг- При введении его в организм в дозах, существенно
превышающих физиологическую потребность, возможно развитие гипервита-
миноза; возникают головная боль, тошнота, шелушение кожи,
выпадение волос, повышенная раздражительность.
Витамин Ах устойчив в щелочных растворах, но разрушается в
кислой среде. Особенно быстро он разрушаются на свету, при
действии кислорода воздуха.
Выделяют витамин Ах из природных источников (морковь, плоды
шиповника) или синтезируют химическим путем из Р-ионона.
Витамин Ъ. Витамин О включает в себя группу витамеров —
кальциферолов, обладающих антирахитическим действием.
Витамин ^ содержится лишь в животных продуктах. Образуется он
из стеролов — одноатомных циклических спиртов, являющихся
производными циклопентанпергидрофенантрена. В растениях широко
распространен эргостерол, в животных тканях — 7-дегидрохолестерол. Эти
соединения играют роль провитамина В. При действии УФ-лучей в
результате пространственной перегруппировки они превращаются в витамин О:
Циклопентан- Стерол Витамин В
пергидрофенантрен
184
Глава 5. Витамины
При облучении эргостерол превращается в витамин Б2 (эргокаль-
циферол; К=СН-СН=СН-СН-СН-СНз), 7-дегидрохолестерол — в вита-
СНз СНз СНз
мин Б3: (холекальциферол; КЮН-СН2-СН2-СН2-СН-СН3).
СНз СНз
В больших количествах эргостерол содержится в дрожжах;
7-дегидрохолестерол — в коже человека. Эти витамеры — соединения бе-
лого цвета, чувствительные к действию света, кислороду воздуха,
особенно при нагревании. Недостаток витамина О приводит к
нарушению у человека кальциевого и фосфорного обмена; кости становятся
мягкими и пластичными, что приводит их к деформации; у детей
развивается рахит, особенно четко проявляющийся в искривлении ног и
деформации грудной клетки, а у взрослых — крошливость зубов, боли
в костях, хромота, утиная походка, вялость, утомляемость.
Содержание витамина В в продуктах питания невелико;
например, в печени быка — 0,01 мкг/г, в сливочном масле — 0,01 —
0,03 мкг/г; исключение составляет жир из печени трески и тунца, в
которых этого витамина содержится соответственно 1,25—8,75 мкг/г.
Добавляя значительные количества витамина В в рацион кормов
сельскохозяйственных животных, можно существенно повысить его
содержание в молочных продуктах и мясе. Как правило, в летний
период вследствие обильного воздействия УФ-лучей на животных эти
продукты богаче витамином В.
Потребность человека в витамине В составляет 10 мкг/сутки. При
достаточном и регулярном действии УФ-лучей организм человека
почти полностью обеспечивается витамином В за счет фотохимического
синтеза в коже. В дозах, существенно превышающих
физиологические потребности, витамин В высокотоксичен, вызывает кальцифика-
цию внутренних органов и тканей, что ведет к необратимому
нарушению их функций и в наиболее тяжелых случаях к летальному исходу.
В промышленности получают витамин В из дрожжей. Эргостерол
дрожжей извлекают экстракцией с последующим его облучением
УФ-светом с длиной волны 280—320 нм. В виде мелкоизмельченного
порошка облучают также и сами дрожжи. С этой целью используют
ртутные лампы высокого напряжения; облучение ведут на конвейерах в
форме каскадов. Облученные (витаминизированные) дрожжи имеют
существенное преимущество перед чистыми препаратами витамина В,
поскольку отличаются, с одной стороны, большой стабильностью препарата, с
другой — незначительной токсичностью, связанной с появлением
побочных продуктов при обработке УФ-лучами очищенного эргостерола. В
облученных дрожжах сопутствующие витамину В вещества (например, глу-
татион) действуют как антиоксиданты и стабилизаторы.
Витамин Е — антиоксидант; представляет собой группу витаме-
ров, известных под названием а-, р-, у-, Ь-токоферолов (от греч. 1осов —
потомство, рНегоз — несу), являющихся производными токола:
185
Часть I. Биомолекулы: структура, свойства, функции
*'^в
но
Ч/ сн2 с
СНз Н
К/
СНя С
сну,
СН2 С
Ч/\/\/ЧУ\/Ч/\
СН2 СНз СН2 СН2 СН2 СН2 СНз
К = К' - К" = Н
К = К' = К" - СН3
К' = Н; К ~ К" = СН3
К" - Н; К - К' = СН3
К' - К" = Н; К = СН,
Токол
а-Токоферол
Р-Токоферол
у-Токоферол
5-Токоферол
а-, Р-, у-, 6-Токоферолы представляют собой вязкие светло-желтые
жидкости. Их растворы в органических растворителях интенсивно
флуоресцируют (максимум возбуждения при длине волны 295 нм),
устойчивы в кислой среде. Под действием кислорода воздуха и других
окислителей превращаются в хиноны, разлагаются при облучении УФ-светом;
стабильны при нагревании в отсутствие кислорода воздуха.
Гиповитаминоз Е проявляется мышечной слабостью и гипотонией
вплоть до мышечной дистрофии; возникают бесплодие, некроз
печени, наклонность к самопроизвольным абортам. Наибольшей
биологической активностью обладает ос-токоферол; р-, у-, 6-токоферолы
составляют соответственно 25, 10 и 1 % от активности а-токоферола.
Витамин Е содержится главным образом в липопротеиновых
мембранах клеток и субклеточных органелл, благодаря
межмолекулярному взаимодействию с. ненасыщенными жирными кислотами. Его
биологическая активность проявляется в способности образовывать
устойчивые свободные радикалы в результате отделения атома водорода
от гидроксильной группы. Эти радикалы могут вступать во
взаимодействие со свободными радикалами, участвующими в образовании
органических пероксидов. Тем самым витамин Е предотвращает
окисление ненасыщенных липидов и предохраняет биологические
мембраны от разрушения. В качестве антиоксиданта он нашел широкое
применение как добавка в пищевые жировые продукты, предохраняя
их от преждевременной порчи.
В большом количестве витамин Е содержится в масле из
пшеничных зародышей (около 300 мг на 100 г), причем больше половины
приходится на долю а-токоферола. Из пищевых продуктов этим
витамином наиболее богаты растительные масла, особенно подсолнечное
(40—70 мг на 100 г), кукурузное (40—80 мг), хлопковое (50—100 мг),
соевое (40—80 мг), от одной трети до половины в которых содержится
а-токоферол. Из этих масел выделяют витамин Е в промышленности.
а-Токоферол синтезируется также химическим путем.
Потребность человека в витамине Е точно не установлена.
Рекомендуемая норма составляет около 10 мг в сутки.
186
Глава 5. Витамины
Витамин К . Витамин К принимает участие в свертывании крови.
В него входят две главные формы — витамин Кг и витамин К2. Они
представляют собой производные нафтохинона с углеводородными
цепями разной длины, структурной единицей которых является
группа изопрена:
О
сн3
(СН2~СН-С-СН2)П-Н
2-Метил- Группа изопрена
1,4-нафтохинон
Витамин К^ имеет п — 4; витамин К2 имеет группу витамеров от
п = 6 до п = 13 изопреновых единиц; химическим синтезом получен
витамин К3 с п = 0 (см. приведенную формулу).
Витамин К — вязкая желтая жидкость; неустойчив к действию
кислот, растворов щелочей, к кислороду и УФ-свету.
Недостаточность витамина К сопровождается кровотечением из
десен, носа, в желудочно-кишечном тракте, подкожными
кровоизлияниями. Установлено, что нарушение свертывания крови связано с
утратой способности организма в отсутствие витамина К синтезировать
специфический белок протромбин — необходимый предшественник
тромбина, участвующего в образовании кровяного сгустка.
Витамин К^ встречается преимущественно « зеленых частях
растений. Витамин К2 синтезируется микроорганизмами, он наиболее
активен.
Из пищевых продуктов витамином К богаты шпинат (0,04 мг/г
сухой массы), цветная и белокочанная капуста (0,01—0,03 мг/г),
томаты (0,004—0,006 мг/г).
Вследствие достаточно широкого распространения витамина К в
пищевых продуктах и синтеза его микрофлорой кишечника
недостаточность этого витамина у человека встречается относительно редко. В
лечебных целях применяют при кровоточивости, связанной с пониженным
содержанием в крови протромбина. Суточная потребность взрослого
человека в витамине К составляет ориентировочно 0,2—0,3 мг.
5.3. Водорастворимые витамины
К водорастворимым витаминам относятся витамины группы В,
аскорбиновая кислота, биотия. В группу витаминов В входят следующие
витамины: Вх, В2, РР, В6, пантотеновая кислота, Вс, В12, Витамины
группы В — соединения весьма различного химического строения,
имеющие разнообразные физико-химические характеристики. В от-
187
Часть I. Биомолекулы: структура, свойства, функции
личие от жирорастворимых витаминов биологические функции
витаминов группы В на молекулярном уровне хорошо изучены. За
исключением витамина В12 эта группа имеет один общий признак —
витамины содержатся в достаточно больших количествах в пшеничных,
рисовых отрубях и в дрожжах.
Витамин Вг (тиамин) является производным пиримидина и тиазола,
устойчив в форме катиона, чаще всего в виде хлоридгидрохлорида:
Г Н=С-КН2-НС1 П
I I +
Н8С-С С-СНг-К-С-СНз -
II I II II Ы
ы-сн нс с-сн2-сн2-он
Гиповитаминоз Вх приводит к нарушению центральной нервной
системы в виде психической подавленности, общего недомогания,
повышенной утомляемости, головной боли, бессонницы, ослабления
внимания. Значительный дефицит витамина Вх в организме ведет к
тяжелому заболеванию, которое называется "бери-бери". Оно
сопровождается полиневритом, параличами конечностей, сердцебиением,
потерей чувствительности периферической нервной системы.
Витамин Вх играет большую роль в регуляции углеводного,
жирового, минерального и водного обмена веществ. При недостатке этого
витамина окисление углеводов не доходит до конца. В тканях
накапливаются промежуточные продукты обмена — пировиноградная и
молочная, кислоты, в результате чего нарушаются процессы передачи
нервных импульсов.
Тиамин хорошо растворяется в воде (100 г на 100 см3). Свет,
влажность, действие кислорода воздуха, нагревание снижают устойчивость
витамина. Он довольно стабилен в кислой среде, легко разрушается в
нейтральных и щелочных средах. Тиамин был первым витамином,
полученным в очищенном виде польским ученым К. Функом. Именно
аминогруппа, содержащаяся в пиримидиновом кольце, явилась
причиной того, что тиамин был назван витамином, т.е. амином жизни.
Химический синтез тиамина в промышленных условиях
осуществляется реакцией конденсации производных пиримидина и тиазо-
ла. Биологический способ синтеза с использованием ферментов для
получения чистых препаратов тиамина не разработан. Помимо
дрожжей, рисовых и пшеничных отрубей тиамин содержится в
достаточном количестве во внутренних органах животных (сердце, печень,
почки). В полированном (очищенном) рисе он практически
отсутствует. Тиамин содержится в ржаном хлебе, гречневой и овсяной крупах.
Лучшим источником витамина являются цельные зерна злаков,
плоды бобовых растений, орехи, дрожжевые напитки, хлебный квас.
Суточная потребность взрослого человека в тиамине — 2 мг.
Однако потребность в нем возрастает при большом содержании в пище
углеводов, при заболеваниях кишечника, невритах и радикулитах.
188
Глава 5. Витамины
Антивитамином Вг является расщепляющий его фермент тиами-
наза, содержащийся в печени пресноводных рыб.
Витамин В2 (рибофлавин) состоит из флавинового красящего
вещества люминохрома и спирта рибитола:
Люминол
(7,8-диметил-изоаллоксазин)
О
а
N
сн2
I
н-с-он
н-с-он
н-с-он
н2сон
> Остаток рибитола
Рибофлавин (10-рибитол-
7,8-диметил-изоаллоксазин)
Гиповитаминоз В2 вызывает сухость и синюпгаость губ,
вертикальные трещины на губах, трещины и корочки в углах рта, сухой ярко-
красный язык. Гиповитаминоз снижает зрительную функцию,
приводит к нарушению сердечной деятельности; вызывает понижение
аппетита, похудание, слабость, апатию, головные боли, зуд, резь в глазах.
Этот витамин входит в состав ряда ферментов, участвующих в
синтезе белков, жиров, в обмене углеводов.
Наиболее важным источником рибофлавина являются цельное
молоко, простокваша, ацидофилин, кефир, сыр, печень, почки, сердце,
яичный желток, пекарские и пивные дрожжи; витамин устойчив при
кулинарной обработке продуктов. Его суточная потребность
составляет 2,5—3 мг.
В промышленности рибофлавин получают главным образом в
результате химического синтеза, а также микробиологическим синтезом.
Витамин В6 встречается в виде трех витамеров — пиридоксин, пи-
ридоксаль и пиридоксамин:
' он о нн2
нон2с
с—н
НОН2С
НОН,С
ОН
СН
3
Пиридоксин
Пиридоксаль
Пиридоксамин
В основе структуры витамина В6 лежит кольцо пиридина.
Гиповитаминоз В6 проявляется в задержке роста,
желудочно-кишечных расстройствах, потере аппетита, дерматите, возбудимости,
189
Часть I. Биомолекулы: структура, свойства, функции
конъюнктивите. Однако гиповитаминоз встречается редко, так как
витамин В6 синтезируется в кишечнике микрофлорой.
Витамин В6 входит в состав многих ферментов, участвующих в
обмене аминокислот, ненасыщенных жирных кислот, холестерола.
Указанные витамеры хорошо растворимы в воде, легко образуют
соли. Соли с НС1 стабильны при действии повышенных температур и
кислорода воздуха. В щелочных, нейтральных растворах они
разрушаются на свету.
Важным источником витамина В6 являются пищевые продукты:
мясо, печень, почки, мозги, икра трески, желток яиц, молоко,
хлебные злаки и зеленые овощи. В процессе кулинарной обработки
содержание витамина В6 снижается на 20—35 %, при копчении,
консервировании мяса — более чем на 50 %; его содержание в молоке (в
стеклянных бутылках на солнечном свету) за 2 ч может снизиться на 50 %
и более. Средняя суточная доза — 2—2,5 мг.
Витамин РР (никотиновая кислота, В5) и ее амид. В основе
структуры этого витамина лежит пиридиновое основание:
г<^^>
1
г<°
^ОН
\я'
N
^\
^Чт/
N
г/
х>га2
Никотиновая кислота Амид никотиновой кислоты
Никотиновая кислота (ниацин) и ее амид предупреждают
заболевание пеллагрой (поражение кожи на открытых местах тела,
психические расстройства, апатию, быструю утомляемость, сердцебиение,
снижение аппетита). Это белые кристаллические вещества; первое —
слабо (1,7 г/100 см3), второе — хорошо (100 г/100 см3) растворимы в
воде. Витамин РР устойчив в водных растворах к действию кислорода
воздуха, света, к повышенной температуре. Он устойчив при консер-
вировании и сушке пищевых продуктов. Биологическая активность
обеих форм витамина РР одинакова.
Амид никотиновой кислоты входит в состав коферментов,
участвующих в ферментативных реакциях, сопряженных с переносом водорода.
Для удовлетворения суточной потребности в витамине РР особенно
существенное значение имеют мясо, печень, почки; из растений —
арахис, грибы; им богаты пивные и пекарские дрожжи. В промышленном
масштабе никотиновую кислоту получают химическим синтезом.
Витамин В3 (пантотеновая кислота). В состав пантотеновой кислоты
входят остатки диметилгидроксимасляной кислоты и р-аланина.
СН3ОН
НО—СН2—С—СН—СО—ЗШ—СН2—СН2—СООН
СНз
Диметилгидрокси- р.Аланин
масляная кислота г
190
Глава 5. Витамины
Свободная пантотеновая кислота — нестабильное и чрезвычайно
гигроскопическое соединение. Она используется обычно в форме
солей кальция и натрия. Большое значение приобрел пантенол —
производное пантотеновой кислоты, имеющее в остатке р-аланина вместо
карбоксильной группы гидроксильную группу. Пантенол так же
биологически активен, как и пантотеновая кислота. Кальциевые и
натриевые соли пантотеновой кислоты хорошо растворимы в воде
(40 г/100 см3). Они устойчивы при действии кислорода воздуха и
света; нестабильны при нагревании, быстро гидролизуются, особенно в
кислой и щелочной средах.
Недостаток пантотеновой кислоты вызывает у животных
задержку роста, поражения кожи, нарушение деятельности нервной системы
и желудочно-кишечного тракта. Она является фактором роста
некоторых молочнокислых бактерий.
Пантотеновая кислота в природе в свободном виде находится в
редких случаях. Обычно она является составной частью кофермента А,
играющего важную роль в углеводном и жировом обмене.
Наиболее богаты пантотеновой кислотой следующие продукты:
печень, почки, яичный желток, молоко, рыба, горох, дрожжи, свежие
фрукты, капуста, картофель, В промышленных масштабах пантотено-
вую кислоту получают с помощью химического синтеза.
В процессе термической обработки пищевых продуктов потери
витамина могут достигать 25—50 %, в связи с чем может возникнуть
дефицит пантотеновой кислоты в организме человека. Суточная
потребность в ней составляет 10—12 мг.
Фолиевая кислота — это птероилглутаминовая кислота.
Фолиевая кислота содержит птериновое. кольцо —
гетероциклическую систему, состоящую из пиримидинового и пиразинового
оснований, и остатки д-аминобензойной и глутаминовой кислот.
Фолиевая кислота и фолаты ™ производные, содержащие от двух до семи
молекул глутаминовой кислбты, — получили название витамина Вс.
ОН
^сНз-^га-^3- сснчн-сн-сн2- сн^-соон
& соон
II
Птерин ^-Аминобензойная Глутаминовая
кислота кислота
Чистая фолиевая кислота имеет вид желтого мелкокристаллического
вещества без вкуса и запаха; очень плохо растворима в воде, но хорошо
растворима в слабощелочных растворах. На свету она разлагается*
Витамин Вс является важным фактором роста животных и
микроорганизмов. Гиповитаминоз Вс ведет к развитию анемии
(малокровия). Восстановленная форма фолиевой кислоты — тетрагидрофолие-
вая кислота — участвует в различных ферментативных реакциях, свя-
191
Часть I. Биомолекулы: структура, свойства, функции ^ ^_
занных с отщеплением и переносом одноуглеродных фрагментов.
Благодаря этому тетрагидрофолиевая кислота играет важную роль в
обмене ряда аминокислот — глицина, серина, гистидина, в синтезе ме-
тионина, пуриновых и пиримидиновых азотистых оснований.
Фолиевая кислота и ее производные широко распространены в
природе. Ими богаты зеленые овощи и фрукты, земляника; сравнительно
много ее в хлебе. Из животных продуктов исключительно богата ею
говяжья печень; мясо, яйца и молоко бедны фолиевой кислотой.
Фолиевая кислота и ее производные довольно неустойчивы,
поэтому легко могут разрушаться при технологической и кулинарной
обработке пищи. Особенно легко они разрушаются в овощах, при
длительной варке которых потери могут достигать 80—95 %.
Рекомендуемая норма потребления фолиевой кислоты взрослым
человеком составляет 200 мкг в сутки.
Витамин В12 (цианкобаламин) — сильный кроветворящий
фактор, широко используемый при злокачественной анемии, лучевой
болезни, заболеваниях печени.
Витамин В12 имеет весьма сложную структуру, и это —
единственный витамин, содержащий металл (кобальт). Основной частью
молекулы витамина является комплекс кобальта с азотистым
макроциклом — коррином. Ядро коррина содержит четыре восстановленных
пиррольных кольца А, В, С, Д, имеющих в р-положении метальные,
ацетамидные, пропионамидные заместители (рис. 5.1).
В циаикобаламине, обладающем биологической активностью, в
качестве лиганда кобальта функционирует цианогруппа. В коферментной
форме витамина В12 цианогруппа замещена на б'-дезоксиаденозильную
группу; эта форма витамина В12 называется 5'-дезоксиаденозилкобал-
амином. Ферменты, нуждающиеся в кобаламине (кофермент В12),
осуществляют реакции обмена между связанным с углеродом атомом
водорода и какой-либо группой, связанной с соседним атомом углерода (гидро-
ксильной, карбоксильной, алкильной или аминогруппой). Витамин В12 —
темно-красный кристаллический порошок, слабо растворимый в
воде (1,2 г/100 см3)» Сам он и его нейтральные и слабокислые растворы
относительно стабильны при действии повышенных температур и
кислорода воздуха, но неустойчивы при действии света или УФ-лучей, при
действии щелочей, сильных кислот и восстановителей.
Цианкобаламин участвует в переносе метильных групп, например,
при синтезе метионина, а также в процессах синтеза нуклеиновых
кислот и обмена тетрагидрофолиевой кислоты.
Витамин В12 синтезируется почти исключительно
микроорганизмами, особенно актиномицетами, сине-зелеными водорослями. Он
найден практически во всех животных тканях. Наиболее богаты им
печень, почки, яичный желток, сыр. На 100 г сырой массы продукта его
содержание достигает в мкг: говяжья печень — 50—130; говяжьи
почки — 20—50; печень трески — 40.
192
Глава 5. Витамины
Н2К-СО-СН2-СН2 СН3 СН3 ^СН2-СО-КН2
сн
\
;ч.
Н2К-СО-СН2^_/ "***Г N^"0" \
н3с- \
С А ||
Н3С
1У*\ С Я<тС
I в сн-сн2-сн2-со-ш2
Со+ СН
рн /*\ *
N А /СН,
С С
Н2Ы-СО-СН2-СН Б |
СО-СНг-СНг^СНдЧ^СНо ЧСН2-СН2-СО-КН2
(4
н
\са
I
сн2
I
н3с-сн
о
о
^
сн ||
о он
но-сн2 °
Рис, 5.1. Химическое строение витамина В12
Человек обеспечивает себя витамином В12 в результате потребления
продуктов питания животного происхождения и синтеза его
микрофлорой пищеварительного тракта; суточная потребность в нем — 1—2 мкг.
Травоядные животные снабжаются этим витамином
микроорганизмами пищеварительного тракта, в основном рубца. Обогащение
кормов витамином В12, обычно дефицитных по метионину, улучшает их
биологическую ценность, повышает продуктивность животноводства*
В промышленности витамин В12 получают исключительно
биологическим синтезом с использованием пропионовокислых и метанообразую-
щих бактерий. Большое количество витамина В12 образуется при
биологической очистке сточных вод, в составе так называемого активного ила9
который может служить источником выделения витамина В12.
Витамин С (аскорбиновая кислота). Отсутствие аскорбиновой
кислоты в пище вызывает заболевание — цингу. Все растения и
животные синтезируют аскорбиновую кислоту, за исключением человека,
обезьян и морской свинки. Внешне авитаминоз С проявляется в виде
синюшности десен, их кровоточивости, бледности и сухости кожи,
быстрой утомляемости при физических нагрузках, боли в мышцах; на
ногах и туловище появляются кровоизлияния.
Аскорбиновая кислота представляет собой у-лактон гексоновой
кислоты с енольнои формой структуры при С-2 и С-3 атомах; наиболее
активной является Ь-аскорбиновая кислота.
Аскорбиновая кислота представляет собой белый кристаллический
порошок кислого вкуса, хорошо растворимый в воде (более 20 %). Она
13. Заказ 3486
193
Часть I. Биомолекулы: структура, свойства, функции
стабильна в сухом виде в темноте. В водных растворах, особенно в
щелочной среде, эта кислота быстро окисляется обратимо до дегидро-
аскорбиновой кислоты, которая обладает такой же витаминной
активностью, что и аскорбиновая кислота. Дальнейшее окисление де-
гидроаскорбиновой кислоты ведет к необратимому разложению ее до
щавелевой и треоновой кислот. Окисление катализируется светом,
ионами железа, меди, серебра и ферментами растений — аскорбинок-
сидазой и полифенолоксидазой. Процесс окисления задерживается в
присутствии восстановителей, содержащих сульфгидрильные группы
(цистеин, глутатион). Большинство белков и аминокислот
задерживают окисление путем образования комплексов с аскорбиновой
кислотой, известных под названием аскорбиноген. Аскорбиновая кислота
легко восстанавливает окислители: бром, йод, 2,6-дихлорфенолиндо-
фенол. В пищевых продуктах она играет роль антиоксиданта >
о О
я и
С—1 -2Н С-
но-с Ъ ++ °=<г о
НО-С | ..отт 0=С
I
II у
ис. I
+2Н
Н-С ■ Н-С
I I
но-с-н но-с-н
I I
СН2ОН СН2ОН
Ь-Аскорбиновая Ь-Дегидроаскорбиновая
кислота кислота
Биологические функции аскорбиновой кислоты многообразны и
окончательно еще не расшифрованы* Она участвует в гидроксилиро-
вании пролина. Гидроксипролин является важнейшим строительным
блоком коллагена соединительной ткани* Авитаминоз С ведет к
нарушению синтеза коллагена и связанного с ним появления хрупкости
кровеносных сосудов, разрушению кровеносных капилляров.
Существенные изменения происходят в костях, зубы расшатываются и
выпадают; нарушается связь между хрящом и костью.
Аскорбиновая кислота участвует в окислении аминокислот
тирозина и фенилаланина; снижает потребность человека в витаминах В1,
В2, А, Е, фолиевой, пантотеновой кислот, что, видимо, связано с ее
антиоксидантными свойствами.
Аскорбиновая кислота широко распространена в природе. Она
содержится почти во всех овощах, фруктах, ягодах (табл. 5.1). Особенно
богаты аскорбиновой кислотой черная смородина, черноплодная
рябина, красный перец, петрушка, цитрусовые, белокочанная капуста.
Кладовой аскорбиновой кислоты являются плоды шиповника,
содержащие 1200 мг на 100 г сухих плодов.
Суточная потребность человека в аскорбиновой кислоте составляет
75 мг; она, по крайней мере, на порядок выше суточной потребности в
194
Г л ава 5, Витамины
Таблица 5.1
Содержание аскорбиновой кислоты
в овощах, фруктах, ягодах (мг/100 г)
Наименование овощей
Горошек зеленый
Кабачки
Капуста белокочанная
Капуста квашеная
Картофель
Лук зеленый
Огурцы
Перец зеленый сладкий
Перец красный
Редис
Редька
Томаты
Хрен
Щавель
Содержание
витамина С
25
10
40
25
20
25
15
125
250
50
20
35
160
60
Наименование фруктов,
ягод
Абрикосы
Апельсины
Брусника
Виноград
Вишня
Груши
Дыня
Земляника
Крыжовник
Лимоны
Мандарины
Малина
Смородина черная
Рябина черноплодная
Яблоки 1
Содержание
витамина С
10
50
15
5
15
5
20
60
40
50
30
25
250
200
20
других витаминах. Постоянным источником ее в нашей пище
являются картофель и капуста.
Исходным соединением для промышленного синтеза аскорбиновой
кислоты служит Б-глюкоза, которая в результате каталитического
гидрирования {Н2, N1) превращается в сорбит, Б-сорбит подвергается
биохимическому окислению под действием Асе1оЪасЬег виЪохуёапв с
образованием Ь-сорбозы. Последняя окисляется кислородом (в
присутствии платины) или КМп04 до 2-кетогулоновой кислоты, которая
при нагревании с разбавленными кислотами в результате
дегидратации образует аскорбиновую кислоту:
о
и
С-Н
н-с-он
но-с-н
н-с-он
н-с-он
сн2он
Б-Глюкоза
Н2, N1
СН2ОН
Н-С-ОН
** НО-С-Н
Н-С-ОН
н-с-он
сн2он
О-Сорбит
сн2он
но-с-н
н-с-он
но-с-н
сн2он
Ь-Сорбоза
КМпО.!
соон
I
с=о
но-с-н
н-с-он
но-с-н
Нагрев
-Н->0
0=С^
но-с
и
но-с
и
сн2он
2-Кето-Ь-гулоновая
кислота
н-с
но-с-н
сн2он
Ь- Аскорбиновая
кислота
13*
195
Часть I. Биомолекулы: структура, свойства, функции
При варке пищи может теряться до 50 % аскорбиновой кислоты.
Еще больше ее теряется при хранении готовой продукции. При сушке
и консервировании плодов и овощей аскорбиновая кислота также
легко разрушается. Разрушение ее происходит в результате окисления и
особенно сильно в присутствии аскорбиноксидазы и полифенолксида-
золы. Эти ферменты проявляют свое действие при очистке и
измельчении овощей, при лежании в нарезанном виде, при закладке их в
холодную воду, содержащую в достаточном количестве растворенный
кислород. Повышение температуры активизирует разрушительное
действие окислительных ферментов. Поэтому наиболее правильно варить
овощи, опуская их сразу в кипящую воду. Кипящая вода
практически не содержит растворенного кислорода, а ее высокая температура
ведет к быстрой денатурации (инактивации) ферментов. В консервной
промышленности с целью инактивации ферментов нарезанные
овощи, плоды предварительно бланшируют, т.е. обрабатывают кипящей
водой или острым паром.
Аскорбиновая кислота обладает достаточно высокой
устойчивостью в кислой среде (рН 4—6), поэтому с целью ее длительной
сохранности широко используется молочнокислое брожение, которое
является основным процессом при квашении капусты, солении огурцов и
томатов, мочке яблок, производстве кисломолочных продуктов.
Хороший метод сохранности аскорбиновой кислоты — замораживание
овощей и плодов, а также варка ягод и фруктов с сахаром,
Биотин (витамин Н) — циклическое производное мочевины с
кольцом тиофена; представляет собой белое кристаллическое соединение,
плохо растворимое в воде {примерно 20 мг/100 см3).
Остаток молекулы
мочевины
НС— СН
нгС \а^СН(СН2)4СООН
I ! и I
Кольцо Остаток
тиофена молекулы
валериановой
кислоты
Сухой кристаллический биотин при нагревании в присутствии
кислорода воздуха устойчив в слабокислых и слабощелочных растворах;
при действии УФ-света он постепенно разрушается.
Биотин является важным фактором роста для дрожжей и ряда
других микроорганизмов. Он частично присутствует в свободной
форме (овощи, фрукты, молоко, отруби), частично в связанной форме с
белком (органы животных, семена растений, дрожжи). Антивитами-
196
О
Н№
МН
Глава 5. Витамины
ном биотина является яичный белок авидин, образующий прочный
комплекс с биотином. Высокое содержание в пище сырого яичного
белка может привести к недостаточности биотина, признаком которой
является поражение кожи, ногтей, выпадение волос-
Биологическая функция биотина заключается в том, что он входит
в состав активной группы ферментов, участвующих в процессе кар-
боксилирования (присоединения С02) жирных кислот. У
микроорганизмов он принимает участие в биосинтезе аспарагиновой кислоты,
серина, треонина.
Потребность человека в биотине покрывается в основном за счет
биосинтеза его кишечной микрофлорой. Некоторая часть его
поступает с пищей. Богаты биотином цветная капуста, соя, горох, грибы,
яичный желток, печень, сердце, дрожжи. Суточная потребность человека
составляет 150—200 мкг.
Получают биотин с помощью химического синтеза.
5.4. Витаминоподобные вещества
Витаминоподобные соединения похожи по биологическим свойствам
на витамины, хотя их функционирование в качестве витаминов еще
не доказано и требуются они человеку в гораздо больших количествах,
чем витамины. К витаминоподобным соединениям относятся я-ами-
нобензойная кислота, инозит, оротовая и липоевая кислоты, метилме-
тионин, холин.
гс-Аминобензойная кислота (витамин Н1) — ростовой фактор для
ряда микроорганизмов, метаболический предшественник фолиевои
кислоты:
соон
п-Аминобензойная
кислота
Человек и животные не способны использовать тг-аминобензойную
кислоту для синтеза фолиевои кислоты; последнюю должны получать
с пищей. Поэтому д-аминобензойная кислота для человека и
животных не является витамином. Ее действие как фактора роста ряда
патогенных микроорганизмов подавляется сульфаниламидными
препаратами, выполняющими роль антивитаминов* На этом основано их
применение в качестве лекарственных средств.
д-Аминобензойная кислота представляет собой белое
кристаллическое соединение; не разрушается при кипячении в кислых и щелочных
средах. Она широко распространена в природе, содержится в пищевых
продуктах. Особенно ею богаты печень, почки, молоко, дрожжи, грибы.
197
*■
Часть I. Биомолекулы: структура, свойства, функции
Инозит (витамин В8) — шестиатомный циклический спирт. Из 8 сте-
реоизомеров биологической активностью обладает только один —мио
инозит:
Это белое кристаллическое вещество, стойкое к кислотам и
щелочам, не разрушается на свету. Он входит в состав миоинозитфосфати-
дов, содержащихся во всех тканях растений* При его недостатке
развиваются жировая и мышечная дистрофия, задержка роста. Миоино-
зит обнаружен во многих пищевых продуктах. Его содержание
(в мг/100 г): в пшеничной муке — 110; зеленом горошке — 150—250;
капусте, зеленом луке, томатах, моркови — 45—95; картофеле,
свекле, яблоках — 20—30; в курином мясе, свинине, яйцах — 30—50.
При варке пищи разрушается до 50 %. Биологическая роль миоино-
зита для человека мало исследована.
Оротовая кислота (витамин В13) — 2,4-диокси~пиримидин-6-кар-
боновая кислота — предшественник пиримидиновых оснований,
используемых для синтеза нуклеиновых кислот, способствует синтезу
белка:
СООН
Оротовая кислота
Оротовая кислота — белое кристаллическое вещество, обладает
кислотными свойствами, легко образует соли с металлами. Оротовая
кислота является фактором роста микроорганизмов. Истинным
витамином оротовая кислота не является, поскольку в организме человека и
животных она в достаточном количестве синтезируется из Ь-аспара-
гиновой кислоты и карбаматфосфата. Используется как стимулятор
роста детей. Норма для человека не установлена. Особо богаты ею
молоко, печень, дрожжи.
Липоевая кислота (витамин Ы) — представляет собой
циклический дисульфид с карбоксильной группой в боковой цепи; существует
в восстановленной и окисленной формах;
СООН +211 ^ ^ ^ /СООН
8 ~2Н 8Н 8Н
Она соединена с белком в форме амида.
198
Глава 5. Витамины
Липоевая кислота участвует в окислительном декарбоксилирова-
нии а-кетокислот — важной промежуточной реакции при дыхании.
Наиболее богаты ею молоко, мясные продукты, дрожжи. Потребность
человека в ней не установлена.
8-метилметионин (витамин V) играет роль противоязвенного
фактора (от лат. и1еиз — язва). По химическому строению является
метилированным производным метионина. Метилметионин — белое
кристаллическое соединение, слегка солоноватого вкуса.
Гнх ^
а I 8-СН9-СН9~СН~СООН
С1"
А
При нагревании, длительном хранении разлагается, не устойчив на
свету. В большом количестве содержится в белокочанной капусте (35—
85 мг на 100 г), спарже (100—160 мг), в петрушке, репе, перце,
моркови, томатах, луке. Метилметионин, как и метионин, является
активным донором метильных групп и тем самым способствует синтезу
в тканях организма холина, холинфосфатидов. Используется для
лечения язвенной болезни желудка и двенадцатиперстной кишки,
гастритов. Тем не менее необходимость метилметионина для человека и
животных не доказана, поэтому он не может быть отнесен к
витаминам или другим незаменимым пищевым веществам, а является вита-
миноподобным соединением.
5.5. Количественное определение витаминов
Количественное определение витаминов имеет специфическую
особенность. Во-первых, витамины представляют собой самые различные по
химической природе соединения, методы количественного определения
которых весьма разнообразны; во-вторых, в исследуемых объектах они,
как правило, присутствуют в ничтожно малых количествах, поэтому эти
методы должны обладать высокой точностью; в-третьих, исследуемые
объекты, к которым относятся пищевые продукты, имеют сложный
химический состав и возникает необходимость либо проводить глубокую
очистку витаминов, либо прибегать к строго специфическим методам
количественного определения витамина в данном объекте*
Все методы количественного определения витаминов можно
подразделить на три группы: химические, физико-химические и
биологические.
К химическим методам прибегают в том случае, когда витамин в
исследуемом объекте присутствует в достаточно большом количестве
и когда он обладает ярко выраженным специфическим химическим
свойством. Однако и в данном случае не исключается необходимость
основательной очистки витамина от сопутствующих примесей.
Классическим примером определения витаминов химическим
методом является аскорбиновая кислота. В пищевых продуктах (ово-
199
Часть /♦ Биомолекулы; структура, свойства, функции
щах, фруктах, ягодах) она, как правило, присутствует в
концентрациях, на порядок и более превышающих другие витамины, а с
химической точки зрения она имеет ярко выраженную способность к
окислению* Основные этапы определения аскорбиновой кислоты следующие:
1) получение материала; 2) его хранение; 3) экстрагирование
аскорбиновой кислоты из образца; 4) освобождение полученного экстракта от
примесей, мешающих определению аскорбиновой кислоты; 5)
определение количества аскорбиновой кислоты.
Аскорбиновая кислота легко разрушается, поэтому обеспечение
сохранности весьма существенно для любого метода определения ее
количества. Аскорбиновую кислоту необходимо предохранять от
солнечного света, аэрации, повышения температуры, от соприкосновения с
металлическими предметами, от высоких температур и повышения
рН среды. Хранение материала лучше осуществлять на холоде,
экстрагирование необходимо проводить при рН не более 4, при
предварительном связывании ионов металлов. Для экстрагирования
применяют растворы уксусной, трихлоруксусной, щавелевой и метафосфор-
ной кислот. Наиболее предпочтительна 5—6%-ная метафосфорная
кислота, хорошо стабилизирующая аскорбиновую кислоту,
осаждающая белки и инактивирующая в сырых растительных объектах
фермент аскорбиноксидазу. Освобождение от примесей, мешающих опре-
делению, проводят с помощью осаждения последних, а также с
использованием различных методов хроматографии (тонкослойной,
ионообменной).
Для количественного определения аскорбиновой кислоты в
биологических материалах используют 2,6-дихлорфенолиндофенол,
который, восстанавливаясь под действием аскорбиновой кислоты,
переходит в лейкоформу, что тестируется спектрофотометрически. При
исследовании продуктов, содержащих редуцирующие вещества,
которые вступают в реакцию с 2,6-дихлорфенолиндофенолом (сиропы,
компоты, сушеные овощи, фрукты и пр.), лучше всего обрабатывать
экстракт формальдегидом. При анализе объектов, содержащих пигменты,
часто применяют титрование в присутствии органических
растворителей (хлороформ, ксилан, изоамилацетат), экстрагирующих избыток
красителя. При определении аскорбиновой кислоты в окрашенных
фруктовых и ягодных соках применяют титрование
2,6-дихлорфенолиндофенолом, конечную точку которого определяют по изменению
окислительно-восстановительного потенциала — потенциометрически,
по появлению поляризационного тока — амперометрически.
Для определения дегидроаскорбиновой кислоты ее
восстанавливают до аскорбиновой кислоты с последующим титрованием
2,6-дихлорфенолиндофенолом. В качестве восстановителей применяют сульфгид-
рильные соединения (гомоцистеин, цистеин, 2,3-димеркаптопропанол).
Физико-химические методы в большинстве случаев основаны на
образовании из витаминов окрашенных соединений, концентрация
200
Глава 5. Витамины
которых определяется методами фотометрии. В основном эти методы
применяются при исследовании чистых препаратов. В простых по
составу средах, содержащих примеси, витамин отделяют методом
хроматографии. В пищевых продуктах витамины содержатся в
незначительных количествах, поэтому физико-химические методы в
большинстве случаев непригодны. Для количественного определения
витаминов используют поглощение в УФ-области спектра (витамины В2, В,
Е), газожидкостную хроматографию (пантотеновая кислота), флюоро-
метрические методы (фолиевая кислота, витамины В2, В12, К).
Биологические методы основаны на том, что при использовании
специально составленных рационов у животных вызывают витаминную
недостаточность и выявляют дозу витамина, устраняющую явление
авитаминоза. Проведение такого рода исследований трудоемко и длительно.
Однако оно позволяет количественно определить витамины в сложных
по составу объектах, в том числе и в пищевых продуктах.
Для количественного определения в пищевых продуктах
водорастворимых витаминов — Вх, В2, В6, В12, пантотеновой и фолиевой
кислот, а также биотина широко используют микробиологические
методы. Они основаны на том, что для некоторых микроорганизмов
витамины являются незаменимыми факторами роста. При этом в
пределах определенной концентрации витаминов наблюдается линейная
зависимость между скоростью роста и размножения микроорганизма
и содержанием витамина в культуральной среде.
При количественном определении витамина В12 широко
используют ферментативные методы. Они обладают высокой чувствительное-
тью и позволяют найти низкие концентрации этого витамина. В этих
методах используются ферменты диолдегидрогеназы, глицеролдегид-
рогеназу. В ходе реакций к избытку фермента добавляют кофермент-
ные формы витамина В12. Скорость реакции находится в
пропорциональной зависимости от количества добавленного витамина.
5.6. Витаминизация пищевых продуктов
Нормальный обмен веществ в организме немыслим без витаминов.
Организм должен получать их регулярно и в полном наборе. Еще
недавно эпидемии цинги, "бери-бери", пеллагры уносили десятки и
сотни тысяч жизней. Сегодня в экономически развитых странах
авитаминозы встречаются редко. Тем не менее, по данным Института
питания АМН России, гиповитаминозы — явление, широко
распространенное в нашей стране. Особенно часто витаминная недостаточность
проявляется весной, когда в пищевых продуктах в это время менее
всего витаминов. Она создает благоприятный фон для формирования
и развития заболеваний, особенно это относится к витаминам С и В1»
недостаток которых причиняет ущерб здоровью миллионов людей. В
качестве действенной меры предупреждения С-витаминной
недостаточности требуется не просто в больших дозах витамин С, а комплекс,
201
Часть I. Биомолекулы: структура, свойства, функции
состоящий из набора витаминов. Этот комплекс действует синергети-
чески, препятствуя развитию указанных выше заболеваний.
Витамин Вг связан с углеводным обменом* В настоящее время, когда
все шире распространяются малая физическая нагрузка и связанная с
ней избыточная масса тела, неврозы, ранний атеросклероз,
гипертония, устранение В1-витаминной недостаточности стало особо
актуальным, И как раз с этим витамином дело обстоит особенно сложно. С
одной стороны, возросло потребление легкоусвояемых углеводов
(сахар, кондитерские изделия), что требует увеличения потребности в
витамине Вг С другой стороны, источники потребления витамина В2
становятся более скудными, его содержание в пищевых продуктах
снижается-
Технический прогресс в пищевой промышленности, высокая
очистка продуктов привели к тому, что в конечном продукте остается все
меньше витамина Вг Как правило, этот витамин находится в тех
частях продукта, которые по нынешней технологии удаляются, наше
питание становится все более рафинированным, и все реже мы имеем
дело с природными продуктами. При изготовлении муки высших
сортов теряется с отрубями до 80—90 % содержащегося в исходном зерне
витамина Вг
Происходят потери витаминов при рафинировании и
гидрогенизации масел, термической обработке, консервировании продуктов.
Наибольшим изменениям в пище подвержено содержание аскорбиновой
кислоты, которая быстро разрушается, особенно при нагревании. При
обычной варке овощей теряется примерно от одной трети до половины
содержащейся аскорбиновой кислоты. Существенно снижается ее
содержание при хранении овощей и фруктов. В связи с этим опасность
развития гиповитаминоза С возникает чаще, чем других гиповитами-
нозов. Влияет на содержание аскорбиновой кислоты в овощах и
плодах и использование интенсивных агротехнических приемов. По
данным Японского национального института питания» содержание
аскорбиновой кислоты в высокопродуктивных сортах овощей и фруктов в
10—20 раз ниже, чем в дикорастущих плодах.
Повышение витаминной ценности пищевых продуктов — одна из
важнейших проблем пищевой промышленности; она может быть
решена их витаминизацией — внесением в пищевой продукт или пищу
витаминов. В нашей стране осуществляют витаминизацию продуктов
массового потребления: пшеничной муки высшего и первого сортов —
витаминами Вх и В2, РР; маргарина бутербродных сортов —
витамином А, молока — аскорбиновой кислотой.
Аскорбиновая кислота используется также в качестве антиокси-
данта при производстве пищевых жиров, фруктовых соков, для
предотвращения образования в мясных и колбасных изделиях
канцерогенных нитрозаминов из нитритов, добавляемых к этим продуктам
для сохранения природной окраски.
202
Глава 5. Витамины
Свойство растворимости витаминов определяет область их
применения. Аскорбиновая кислота растворима в воде, витамин Е — в жирах.
Поэтому витамин С используют для стабилизации напитков (пива, вина,
фруктов, соков), а витамин Е — для стабилизации жиров и масел.
Витамин С применяют для обогащения муки с незначительным содержанием
клейковины в качестве улучшителя хлебопекарных свойств.
Каротины используются в пищевой промышленности в качестве
безвредных красящих веществ, причем их витаминная активность
придает дополнительную биологическую ценность пищевому
продукту. Они применяются для окрашивания сыров и жиров.
Хорошим источником витаминов в хлебобулочных изделиях
являются дрожжи, используемые для брожения теста.
Особого внимания заслуживает витамин Е, содержащийся в
больших количествах в зародышах семян злаковых. В настоящее время
есть уже промышленные способы отделения зародышей от зерна на
мельницах; молотые зародыши можно потом добавлять в муку и это
практически не скажется на вкусе хлеба.
Краткое содержание главы
Витамины — класс биологически активных соединений,
объединяемых по признаку строгой необходимости в обмене веществ в
организме человека и животных; это низкомолекулярные пищевые
вещества различной химической природы, причем требуются они в
организме в ничтожно малых количествах. Витамины не являются
пластическими веществами или источниками энергии, они входят в состав
биологических катализаторов — ферментов, участвуя в механизмах
катализа и регуляции биохимических реакций.
В настоящее время известно около двух десятков витаминов. Они
подразделяются на две группы: жирорастворимые (витамины А, В, Е,
К) и водорастворимые (витамины группы В, витамин С, биотин).
Первые — типичные гидрофобные соединения с ярко выраженной
циклической и ациклической углеводородной структурой; в конечном счете
все они построены из строительных блоков изопренового типа.
Вторые — вещества гидрофильные; в отличие от жирорастворимых
витаминов, биохимические функции водорастворимых витаминов сейчас
достоверно установлены.
Ряд витаминов представлен не одним, а несколькими
соединениями со сходными биологическими активностями. Они называются ви-
тамерами.
В растениях, микроорганизмах встречаются вещества, которые сами
по себе не проявляют биологической активности, но являются пред-
шественниками образования витаминов. Они получили название
провитаминов.
Витамин А представляет собой группу витамеров — ретинол, рети-
наль, ретиноевую кислоту; это витамин роста детей. Первым призна-
203
Часть I. Биомолекулы: структура, свойства, функции
ком его недостатка является (ночная) куриная слепота, поражение
кожи. Содержится в животной пище; в растениях широко
распространены провитамины А — каротины а, Р, у.
Витамин В включает в себя группу витаминов — кальциферолов,
обладающих антирахитическим действием. Содержится лишь в живот-
ных продуктах. Особенно много его в жире, выделенном из печени
морских животных и рыб* В растениях и особенно в дрожжах встречается
эргостерол, который при действии УФ-лучей превращается в витамин
иг В подкожном жире человека, животных содержится 7-дегидрохоле-
стерол, который при действии УФ-лучей превращается в витамин ^^
Витамин Е — антиоксидант; представляет собой группу витамеров —
токоферолов а, Р, у, 8. Недостаток токоферолов ведет к нарушению
функционирования клеточных мембран в результате нарушения их
структуры под действием реакционно активных форм кислорода. В большом
количестве витамин Е содержится в масле из пшеничных зародышей; из
пищевых продуктов им богаты растительные масла.
Витамин К принимает участие в свертывании крови; в него входят
два витамера Кх и К2. Витамер Кг встречается преимущественно в
зеленых частях растений; витамер К2 синтезируется микроорганизмами.
К водорастворимым витаминам группы В относятся витамины Вь,
В2, РР, В6, пантотеновая кислота (В3), Вс, В12. Биологические
функции витаминов группы В на молекулярном уровне хорошо изучены.
За исключением витамина В12, эти витамины имеют один общий
признак — они содержатся в достаточно больших количествах в
пшеничных, рисовых отрубях и в дрожжах. Водорастворимые витамины
играют важную роль в регуляции белкового, углеводного, жирового
обмена веществ в организме; являются факторами роста детей, молодых
животных, птиц, микроорганизмов. Они входят в состав ферментов,
играющих ключевую роль в метаболизме веществ клетки.
В обмене веществ в организме человека особенно важна роль
витамина С, его недостаток ведет к тяжелому заболеванию — цинге. В
водных растворах аскорбиновая кислота неустойчива, быстро
окисляется до дегидроаскорбиновой кислоты, которая обладает такой же
витаминной активностью, что и аскорбиновая кислота. Дальнейшее
окисление дегидроаскорбиновой кислоты ведет к необратимому
разложению. Процесс разложения катализируется светом, ионами меди,
железа, цинка и ферментами аскорбиноксидазой и полифенолоксидазой.
Ингибиторами окисления являются восстановители, содержащие сульф-
гидрильные группы (цистеин, глутатион), белки, аминокислоты.
Биологические функции витамина С многообразны. Он переводит в гид-
роксоформу пролин, который является важнейшим строительным
блоком коллагена соединительной ткани. Витамин С содержится в
овощах, фруктах, ягодах. Особенно богаты им плоды шиповника, черная
смородина, черноплодная рябина, перец, петрушка, белокочанная
капуста, цитрусовые. При сушке и консервировании плодов и овощей
204
Глава 5. Витамины
витамин С легко разрушается, особенно интенсивно —> ферментами
оксидазами, которые проявляют свое действие при очистке и
измельчении плодов, овощей.
Биотин необходим для функционирования ферментов,
катализирующих реакции карбоксилирования. В этих ферментах биотин кова-
лентно присоединен к белку при помощи амидной связи, в
образовании которой участвует е-аминогруппа остатка лизина,
локализованного в активном центре фермента. Поэтому биотин выделяют из био-
тинсодер^ащих ферментов в виде биотиниллизинового остатка,
называемого биоцитиноМш
Близкими к витаминам являются витаминоподобные вещества
(я-аминобензойная кислота, инозит, оротовая и липоевая кислоты).
Эти соединения похожи на витамины, хотя их функционирование в
качестве витаминов еще не доказано. Требуются они человеку в
гораздо больших количествах, чем витамины.
Все методы количественного определения витаминов можно
подразделить на три группы: химические, физико-химические и
биологические.
Примером химических методов может служить количественное
определение аскорбиновой кислоты (путем ее окисления 2,6-дихлор^
фенолиндофенолом).
Физико-химические методы определения витаминов основаны на
образовании из них окрашенных соединений, концентрация которых
определяется методом фотометрии.
В биологических методах используют микроорганизмы, для
которых тот или иной витамин является незаменимым фактором роста.
С целью повышения биологической ценности пищевых продуктов
их обогащают витаминами. В пищевой продукт чаще всего вносят
витамины С и Вх, которые в большинстве случаев являются
дефицитом в питании человека.
Хронология важнейших дат в исследовании витаминов
1880 г. — Н. И. Лунин впервые предположил, что организм человека и
животного нуждается в биологически активных веществах, названных впоследствии
"витаминами",
1897—1906 гг. — К. Эйкман доказал, что заболевание "бери-бери"
обусловлено нехваткой каких-то веществ в пище и что введение в рацион водной
вытяжки из рисовых отрубей ведет к излечиванию болезни.
1906 г. — М. С- Цвет предложил хроматографический адсорбционный метод
разделения пигментов (каротинов, хлорофилла) из растений на колонке
карбоната кальция.
1914 г. — К. Функ выделил в кристаллическом виде вещество,
содержащееся в рисовых отрубях, и предложил термин "витамин" (витамин Вх).
1917 г. — Э. Мак-Коллум показал, что ксерофтальмия вызывается
недостатком витамина А.
1922 г. — Э. Мак-Коллум показал, что недостаток витамина О вызывает
рахит.
205
Часть I. Биомолекулы: структура, свойства, функции
1927 г. — А. Виндаус показал, что эргостерол является предшественником
витамина В2.
1928 г. — Л. Б. Эйлер выделил каротин и показал, что он обладает
активностью, характерной для витамина А.
1928 г, — К- Эйкману и Ф. Гопкинсу была присуждена Нобелевская премия
за открытие витаминов.
1928—1932 гг. — Э. Сент-Дьердьи, С. Глин-Кинг и У. Л. Во выделили из
лимонного сока аскорбиновую кислоту.
1935 г. — Р. Уильяме и его сотрудники установили структуру витамина Вг
1938 г. — Р. Куну присуждена Нобелевская премия за открытие и синтез
рибофлавина, идентичного ранее открытому витамину В2. Это открытие
послужило началом подтверждения важной роли витаминов в действии ферментов.
1951 г. — А. А. Шмидт осуществил промышленный синтез аскорбиновой
кислоты.
ЛИТЕРАТУРА
Берегов Т. Т., Коровник Б. Ф* Биологическая.химия: Учебник. — 3-е изд.,
перераб. и доп. — М.: Медицина, 1998. — 704 с.
Бышевский А П1.9 Терсенов О. А. Биохимия для врача: Учебник. —
Екатеринбург: Изд-во "Уральский рабочий", 1994. — 384 с.
Кнорре Д. Г., Мызина С. Д. Биологическая химия: Учебник. — 2-е изд.,
перераб. и доп. — М.: Высшая школа, 1998. — 479 с.
Ленинджер А. Основы биохимии: В 3-х т. —М.: Мир, 1985. — Т. 1. — 367 с.
Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека: В 2-х т. ^~
М.: Мир, 1993. — Т. 1. — 381 с.
МецлерД. Биохимия: В 3-х т. — М.: Мир, 1980. — Т. 1. — 487 с. —Т. 2. —
606 с.
ВОПРОСЫ И ЗАДАЧИ
1. Что такое авитаминозы, гиповитаминозы, гипервитаминозы?
2. Охарактеризуйте водорастворимые и жирорастворимые
витаминычпровитамины, витамеры, витаминоподобные вещества, антивитамины.
3. Изобразите структурную формулу ретинола. Исходя из специфической
структуры витамина, дайте ответ на вопрос: почему он не растворим в воде?
4. Расскажите о биологической роли витаминов В, Е, К. Какую роль
витамин Б играет в пищевых жировых продуктах?
5. Каковы биохимические функции витаминов В1? В2, РР, В6?
6. С чем связано возникновение цинги при С-авитаминозе? В синтезе какого
белка принимает участие аскорбиновая кислота?
7. С учетом специфических особенностей формул витамина Е и аскорбиновой
кислоты ответьте на вопрос, почему витамин Е растворим только в жирах и
других органических растворителях, витамин С — только в воде?
8- На основании структурной формулы аскорбиновой кислоты объясните,
почему она весьма неустойчива в присутствии кислорода воздуха, на свету?
9, Рассчитайте, сколько необходимо использовать в пищу картофеля,
капусты, яблок, смородины, чтобы обеспечить суточную потребность взрослого
человека в аскорбиновой кислоте?
Глава 6. ВЕЩЕСТВА ВТОРИЧНОГО
МЕТАБОЛИЗМА РАСТЕНИЙ
Наряду с основными компонентами живой клетки — белками,
нуклеиновыми кислотами, углеводами, липидами и витаминами в растениях
осуществляется биосинтез различных веществ, не входящих в состав
компонентов важнейших метаболических путей. Эти вещества получили
название веществ вторичного происхождения, В свое время эти
соединения рассматривались как вторичные растительные вещества, исходя, по-
видимому, из предположения, что они являются примером
биосинтетической "расточительности" растений, продуктом "тупикового" звена в
обмене ключевых предшественников. Однако исследования последних лет
показали, что многие вещества вторичного происхождения имеют
большое значение в метаболизме растений, хотя механизм их действия на
молекулярном уровне в большинстве случаев остается не выясненным,
В настоящее время эти вещества привлекают к себе внимание как
соединения, играющие важную роль в обеспечении экологических
взаимосвязей в экосистемах (гл. 15 "Некоторые аспекты экологической
биохимии"). Многие из них являются физиологически активными
соединениями — стимуляторами и ингибиторами роста, токсинами; играют важную
роль в процессах дыхания и фотосинтеза. Поэтому термин "вещества
вторичного происхождения" является в известной степени условным и
свидетельствует скорее всего о малой изученности этих соединений.
Некоторые из веществ вторичного происхождения имеют значение
в формировании цвета, запаха, вкуса плодов и овощей, нашли широ-
кое применение в медицине.
Вещества вторичного происхождения по химической природе
характеризуются огромным разнообразием. Особо важные из них можно
подразделить на следующие группы: 1) терпены; 2) растительные
фенолы; 3) гликозиды; 4) алкалоиды и 5) фитогормоны.
6.1. Терпены
Терпены — углеводороды алифатического или циклического ряда.
В основе их химического строения находится молекула
(^-соединения — изопрена:
сн3
Алифатические терпены тесно связаны взаимными переходами с
циклическими терпенами. В большинстве случаев эти вещества не.раст-
207
Часть I. Биомолекулы: структура, свойства, функции
воримы в воде, но растворимы в различных органических
растворителях. Они являются основными компонентами эфирных масел,
выделяемых железистыми волосками, чешуйками растений, а также смол,
находящихся в смоляных ходах растений; обусловливают
определенный аромат многих растений. Эфирные масла нашли широкое
применение в парфюмерной, мыловаренной, фармацевтической
промышленности, при изготовлении конфет и различных напитков; эфирные
масла семян кориандра и тмина используются в качестве ароматических
приправ в хлебопечении. Сами по себе алифатические терпены в
создании аромата эфирных масел играют незначительную роль. Более
важны в них и гораздо шире распространены кислородсодержащие
производные терпенов — альдегиды и спирты. В свою очередь спирты
могут находиться как в свободном виде, так и в виде гликозидов.
Терпены классифицируются по числу содержащихся в их
молекуле атомов углерода, которое кратно числу изопреновых единиц (С5-еди-
ниц): монотерпены С10; сесквитерпены С15; дитерпены С20; тригпер-
пены С30; тетратерпены С40 и политерпены с числом углеродных
атомов от 7,5-Ю3 до 3405.
Монотерпены можно подразделить на ациклические и циклогекса-
ноидные; последние в свою очередь подразделяются на моно-, би- и
трициклические соединения:
Ациклические Моноциклические' Бициклические Трициклические
Монотерпены широко распространены в растениях, обладают
сильным ароматом, содержатся в особых секреторных железах (например, у
мяты). Их можно выделить, подвергая растения перегонке с парами воды.
Наиболее широко распространены кислородные производные
монотерпенов — спирты линалоол, гераниол, получившие свои названия
от названия содержащего их растения:
н3с
Н3С
он
с=сн-сн2-сн2~с-сн=сн2
Лиыалоол
I
СН.
Н3С
нэс
н3с
Н3С
с-снч:н2-сн2«с=сн-сн2он
сн3
Гераниол
С-СН-СН2-СН2-СН-СН2-СН2ОН
сн3
Цитронеллол
208
Глава 6. Вещества вторичного метаболизма растений
Линалоол содержится в цветках ландыша, в апельсиновом и
кориандровом маслах; используется в парфюмерии. Это жидкость с
запахом ландыша. Гераниол выделен из цветков герани, из эфирного мас-
-а эвкалипта. Цитронеллол обладает запахом розы и содержится в
розовом, гераниевом и других маслах.
При окислении гераниола образуется альдегид — цитралъ (смесь
.*:вух изомеров — аир):
3 % С-СН»-СНо-СН«-С=СН-С=0
3 сн3 н
а-Цитраль
3 * с=сн-сн«-сн«-с-сн-с=о
н°с сщ к
Р-Цитраль
Цитраль содержится в померанцевом масле. Среди моноцикличес-
:-:нх монотерпенов широко распространен лимонен. Он содержится в
:-хипидаре, в маслах укропа и тмина. Из кислородных
моноциклических монотерпенов можно отметить ментол — вторичный спирт,
содержащийся до 70 % в эфирном масле перечной мяты, а также кар-
:-.)н — кетой, выделенный из эфирных масел укропа и тмина:
с
А
н,с
с
сн3
Лимонен
СН
СН
н,с
н,с
I
/с^
снон
СН,
СН
сн3
Ментол
3 ЧС, 2
н,с
/\
СН.
НС
ч
С-0
С
сн3
Карвон
К бициклическим монотерпенам относятся пинен и камфен, а так
же их кислородные производные — борнеол й камфора:
Н,С
СН
Ч
СН.
НС
НзС-С-СН
С
СН3
Пинен
Н,С
СН Н2С
СН
\ ,сн3
с-сна
СН.
СН
Камфен
нх
СН
ч
н,с-с-сн
,С—СН^ НзС
сн2
\
он
с
ён3
Борнеол
СН
НзС-С-СНд!
с=о
н2с
С
сн3
Камфора
14. Заказ 3485
209
Глава 6. Вещества вторичного метаболизма растений
мулу). Большинство стеролов имеют алифатическую боковую цепь изо-
пренильной природы:
Циклопентан
Пергидрофенантрен
Стеролы — провитамины группы В. Из растительных стеролов
широко распространен эреостерол9 содержащийся в дрожжах, грибах
и являющийся провитамином В2; в больших количествах в
подкожном жире человека и животных содержится холестерол —
провитамин В3 (см. гл. "Витамины"). К растительным стеролам принадлежат
сапогенины. В растениях сапогенины встречаются в виде гликозидов —
сапонинов, обладающих детергентными свойствами; они разрушают
мембраны клеток, вызывают гемолиз эритроцитов.
Тетратерпены. Всего известно около 500 соединений. Типичным
представителем их является фитоин, в молекулу которого включено
восемь остатков изопрена. К тетратерпенам принадлежат пигменты каро-
тиноиды> которые локализованы в хлоропластах зеленых листьев. Они
участвуют в процессах светопоглощения при фотосинтезе; важна их
защитная роль от разрушения хлоропластов на свету. Желтая и оранжевая
окраска тетратерпенов связала с наличием длинного ряда сопряженных
двойных связей. К группе каротиноидов относятся каротины а, (}, у —
провитамины витаминов группы А (см. гл. "Витамины"). Цвет моркови
обусловлен этими каротинами. К каротинам следует отнести ликопин —
алифатический тетратерпен, содержащийся в плодах томатов.
Циклизация у одного конца молекулы ликопина может привести к образованию
одного р-ионового кольца, как, например, в у-каротине; р-каротин имеет
на обоих концах молекулы два кольца р-ионона; а-каротин содержит на
одном из концов кольцо (3-ионона, на другом — сс-ионона; самый
активный из этих каротинов — р-каротйн, молекула которого в
пищеварительном тракте животных и человека способна дать две молекулы
витамина Ах; молекулы а- и у-каротинов способны дать по одной молекуле
витамина Ах (см. гл. "Витамины").
Тетратерпены, в молекулах которых имеются
кислородсодержащие группы, называются ксантофиллами. От них, в частности,
зависит окраска семян различных сортов кукурузы.
Следует отметить, что в терпенах, за редким исключением,
двойные связи находятся в траяс-конфигурации.
Политерпены. Типичными представителями политерпенов
являются каучук и гутта. Эти соединения образуются в латексе многих
14*
211
Часть I. Биомолекулы: структура, свойства, функции
Пинен — важный компонент многих эфирных масел, скипидара,
легко окисляется на воздухе, превращаясь в смолообразные
продукты. Камфен содержится в пихтовом, лавандовом, кипарисовом
маслах.
Ворнеол — вторичный монотерпеновый спирт — содержится в
камфорном, лавандовом, пихтовом эфирных маслах и представляет собой
твердое тело. При окислении борнеола образуется камфора — твердое
вещество, содержится в древесине и листьях камфорного лавра.
Камфора широко используется в медицине как возбудитель сердечной
деятельности. Ее получают из листьев лавра, из полыни, растущей в
степях южной России, а также синтетическим путем из скипидара.
Сескеитерпены — довольно обширная группа соединений. Они
часто встречаются в эфирных маслах с монотерпенами. Одним из
наиболее интересных соединений является сесквитерпеновый димер —
госсипол, обнаруженный в корнях и семенах хлопчатника.
Госсипол является токсическим соединением. При производстве
из семян хлопчатника хлопкового масла госсипол переходит в жмых
и частично попадает в масло. Это препятствует использованию жмыха
для кормления животных; масло же для пищевых целей
используется только в рафинированном виде.
/У
НО
но^А^—снз н8сЛА/0Н
Госсипол
Дитерпены* В природе обнаружено свыше 500 дитерпенов. Они
почти не летучи с водяным паром. К ним принадлежат геранилгерани-
0Лу спирт фитол (С20Н39ОН). Последний можно рассматривать как
дитерпеновый спирт. Моноциклическим дитерпеновым спиртом
является ретинол (витамин Ах), содержащий р-ионон и две изопреновые
группы (см. гл. "Витамины"). Дитерпены содержатся в выделениях
растений — бальзамах и смолах. Производные дитерпенов —
циклические кислоты, составляющие примерно три четверти смолистых
выделений хвойных растений (живицы). При переработке живицы
отгоняют с водяным паром скипидар. Оставшаяся твердая часть состоит в
основном из циклических (смоляных) кислот и называется канифолью,
широко используемой в химической промышленности.
Тритерпены. К тритерпенам относятся стеролы> широко
распространенные у растений и животных. Стеролы — это одноатомные
циклические спирты, в основе химической структуры молекул которых
лежит циклопентанопергидрофенантрен, построенный из четырех
колец; причем три конденсированных кольца шестичленные и
представляют собой остаток пергидрофенантрена, а четвертый — пятичлен-
ный является остатком молекулы циклопентана (см. структурную фор-
210
Глава 6. Вещества вторичного метаболизма растений
/V
V
ОН
ОН
он
он
чл
он
он
Фенол
Пирокатехин Гидрохинон Резорцин
К трехатомным фенолам относятся пирогаллол (1,2,3-тригидро
ксибензол) и флороглюцин (1,3,5-тригидроксибензол):
ОН
ОН
НО
ОН
НО
лл
ОН
Пирогаллол
Флороглюцин
В природе фенолы редко встречаются в свободном виде. Особенно
много производных фенолов в плодах цитрусовых. В небольшом
количестве фенолы присутствуют во всех плодах и фруктах. С пищей
человек получает в сутки около 50 мг фенолов.
Фенолы в большинстве случаев являются бесцветными
кристаллическими веществами; легко окисляются, причем их способность к
окислению возрастает по мере увеличения числа гидроксильных групп.
Большое разнообразие фенольных соединений (~ 1000) может быть
разделено по химической структуре на две основные группы:
монофенолъные — содержащие от Сг- до С4-углеродных боковых
фрагментов;
полифенолъные — содержащие от 2 до п фенольных остатков,
связанные С2- и С3-фрагментами или непосредственно конденсированные.
Группа монофенольных соединений подразделяется на следующие
подгруппы: 1) Се—Щ 2) С6—С3; 3) Сб—С4.
Св—С ^соединения. К ним принадлежат фенольные кислоты. В
растениях широко распространены п-гидроксибензойная, протокатехо-
вая, ванилиновая, галловая и сиреневая кислоты:
СООН
соон
а
^
У
А
соон
\
у\ш
но
он
он
тг-Гидроксибензойная
кислота
ОН
Протокатеховая
кислота
ОН
Галловая
кислота
213
Часть I. Биомолекулы: структура, свойства, функции
покрытосеменных растений. Из них только гевея используется в
промышленном масштабе. У гевеи каучук синтезируется и
накапливается в специализированных клетках или сосудах, называемых
млечниками, находящимися в коре дерева. Благодаря наличию перемычек
между соседними сосудами при подрезе коры гевеи (подсечке) латекс,
содержащий каучук, вытекает из надрезов и собирается.
Молекула каучука содержит 1500—60 000 изопреновых остатков
(молекулярная масса 1*105—1ф106). Двойные связи изопреновых
остатков находятся в цис-форме > что обусловливает растяжимость и
эластичность этого соединения.
цис~Форм& молекулы каучука
Гутта, как и каучук, образуется в млечном соке различных
деревьев, произрастающих в Малайзии. Для сбора гутты деревья срубают. В
настоящее время в небольших масштабах сбор гутты осуществляется
из гваюлы — серебристо-пушистого кустарника, произрастающего в
Мексике и Техасе (США).
Молекула гутты имеет более низкую молекулярную массу, чем
молекула каучука; сопряженные связи находятся в транс-форме:
транс -Форма молекулы гутты
При температурах ниже 65 °С гутта является твердым, плотным,
неэластичным веществом, но при более высоких температурах она
становится мягкой и эластичной. В качестве изолирующего материала
гутта используется в производстве кабеля.
6.2. Фенолы
К группе растительных фенолов относятся органические
соединения ароматического ряда, которые содержат одну или несколько гид-
роксильных групп, связанных с атомом углерода ароматического ядра.
Фенолы и продукты их превращений в большинстве случаев
являются антиоксидантами в растительных и животных клетках. Они
обладают бактерицидными свойствами, используются в пищевой
промышленности в качестве консервантов.
По числу гидроксильных групп, присоединенных к бензольному
кольцу, фенолы делят на одно-, двух- и трехатомные. Первым
представителем, по которому и названа группа фенольных соединений, является
фенол {гидроксибензол), часто называемый карболовой кислотой. К
двухатомным фенолам относятся пирокатехин (0~дигидроксибензол)>
гидрохинон (п-дигидроксибензол), резорцин (м-дигидроксибензол);
212
Часть I. Биомолекулы: структура, свойства, функции
СООН
СООН
ОСН3 Н3СО
ОСН
3
ОН
Ванилиновая
кислота
ОН
Сиреневая
кислота
Они обнаружены у всех исследованных к настоящему времени
покрытосеменных растений. Известны также альдегиды и спирты,
соответствующие этим кислотам; важнейшие их представители — вани
лин из стручков ваниллы и салициловый спирт из ивы.
СНО
А
осн.
А
%
у^ сн2он
он
Ванилин
ОН
Салициловый
спирт
гс-Гидроксибензойная кислота обычно присутствует в растениях в
связанной форме и высвобождается при гидролизе.
С6—С3-соединения. К этой подгруппе соединений относятся п-ку-
маровая, кофейная, о-гидроксикоричная кислоты и их
метилированные производные — феруловая и синаповая кислоты:
сн=сн-соон
сн=сн-соон
%
сн=сн-соон
У
л
ЧЛ
он
ОСН
ОН
п-Кумаровая
кислота
ОН
Кофейная
кислота
ОН
Феруловая
кислота
СН=СН~СООН
ОН
СН=СН-СООН
V
н3со
осн;
о-Гидроксикоричная
кислота
ОН
Синаповая
кислота
214
Глава 6. Вещества вторичного метаболизма растений
Эти соединения присутствуют как в свободной форме, так и в виде
эфиров, имеют #гра#с~конфигурацию. Характерной особенностью
о-гидроксикоричной кислоты является образование лактона. Оно
осуществляется после транс-цис-пзомеризации этой кислоты в
результате замыкания кольца между о/шо-гидроксильной и карбоксильной
группами:
н
соон
соон
игра^с-о-Гидроксикоричная цис-Ку маринован
кислота кислота
АА,
\А0А
Кумарин
(лактон)
Кумарин — бесцветное кристаллическое вещество с приятным
запахом, напоминающим запах сена* Он содержится в цветах донника;
как сам кумарин, так и эти цветы используются в качестве аромати-
заторов в некоторых сортах курительного табака.
Св—С^-соединения. Эта группа включает нафтохиноны> которые
в растениях находятся в восстановленной и гликозилированной
формах. В процессе выделения они, переходя в окрашенные соединения,
часто окисляются. Пример производных нафтохинона — глюкозид
восстановленного юелона, выделенный из ореха. Свободные нафтохино-
ны содержатся в древесине.
ОН
ОН О-Глюкоза
Глюкозид юглона
Из группы полифенольных соединений следует отметить
следующие подгруппы:
1) С6-С3-С6; 2) (С6-С3-Сб)2; 3) (С^-СД,; 4) (С6)п.
С6—С3—Отсоединения. К этой подгруппе относятся флавоно-
иды — чрезвычайно широко распространенные водорастворимые
фенол ьные производные. Многие из них ярко окрашены в красный,
темно-красный, пурпурный или желтый цвет. Флавоноиды
представляют собой гликозиды. Основу структуры их неуглеводной
группы {агликона) составляет флаван, состоящий из двух
ароматических колец А и В, соединенных в хромановую структуру через
кислородсодержащую С3~единицу:
215
Часть I. Биомолекулы: структура, свойства, функции
8
6
А/?
^в ^
А
5 4
Флаван
Таким образом, флавоноиды — это фенилпропановые производные.
Свободные флаваны находятся в омертвевших деревянистых тканях
растений, где они, вероятно, образуются в результате
ферментативного гидролиза флавоноидов. Флаваны имеют различную степень окис-
ленности:
А н
н н
Катехин
О. Н
/У%
Флаванон
ОН
Антоци&н
О
Флавонол
Широкое разнообразие в растениях флавоноидов связано с тем,
что к кольцам А и В способны присоединяться различные группы
(-ОН, -ОСН3, —СН3) и находиться в них в разнообразных
положениях, а также с тем, что гидроксильные группы флаванов могут
образовывать гликозидные связи (обычно в (^-конфигурации) с
широким набором Сахаров.
Из всех флаванов катехины — менее окисленные соединения. Это
бесцветные кристаллические вещества, легко окисляющиеся и
склонные к полимеризации. Они содержатся во многих плодах (яблоки,
216
Глава 6. Вещества вторичного метаболизма растений
груши, вишня, айва, персики, абрикосы) и в ягодах (брусника,
виноград, ежевика, земляника, малина, смородина). Особенно богаты кате-
хинами молодые листья чая (до 30 % их сухой массы).
Окисление катехинов важно в виноделии, в производстве чая и
какао. В результате этой реакции продукты окисления приобретают
приятный, слабо вяжущий аромат и привлекательную окраску. Кате-
хины обладают наиболее высокой Р-витаминной активностью по
сравнению с другими флаваноидами.
Флавононы — бесцветные кристаллические вещества, особенно
часто встречающиеся в плодах цитрусовых. В кожуре грейпфрута
содержится флавонон нарингин, имеющий горький вкус.
Наринеин: нирингин (К = ОН; К1==Н); эриедиктион (К=К1 = ОН);
гесперетин (К-ОСН.; К1вОН):
НО
\Агу
ОН О
Вкус горечи зависит от строения сахарного остатка, входящего в
флавонон* Так, у наргинина таким остатком является 2-о-рамно-
зид-Д-глюкопиранозид.
Антоцианы (от греч. апНюз — цвет + куапоз — лазоревый) —
гликозиды, сахарными компонентами в которых могут служить
остатки глюкозы, галактозы, рамнозы, в некоторых случаях — остатки
дисахаридов. Аглюконом (неуглеводной частью) в них являются
производные флавона, в котором кольца А и В связаны гетероциклом,
содержащим четырехвалентный кислород (оксоний).
Аглюконы антоцианов называются антоцианидинами, в которых
кольца А и В флавана связаны гетероциклом, содержащим
четырехвалентный кислород:
НО
он
он
Антоцианидины:
цианидин (К = ОН; К1=-Н); пеларгонидин (К^К^Н); пеонидин
(К = ОСН3, К1а=Н); дельфинидин (К«К1 «ОН); петунидин (К = ОСН3,
К1 - ОН); мальвидин (К = К1 = ОСН3).
Всего известно около двух десятков антоцианидинов. Название
многих из них произошло от названия растения. Антоцианы являют-
217
Часть /. Биомолекулы: структура, свойства, функции
ся гликозидами — комплексами антоцианидинов с сахарамн. Антоци-
анидины и близкие к ним по структуре флаванолы оказывают
большое влияние на окраску растений. Цветочный ковер лугов, газонов,
осенняя палитра лесов связаны с наличием в растениях этих
соединений.
Окраска антоцианидинов зависит от многих факторов. У
лепестков цветков многих растений большую роль играет так называемая
копигментация и способность антоцианидинов образовывать
комплексы с металлами. Синяя окраска васильков и многих других
цветков обусловлена копигментацией между антоцианами и другими
флавоноидными гликозидами или гидролизуемым таннином.
Главным пигментом пурпурной розы является цианидин-3,5-диглюко-
зид* В лепестках темно-красной розы он копигментирован с галло-
таннином.
Окраска многих плодов и ягод обусловлена в основном
антоцианами. С присутствием цианидина связана окраска некоторых сортов
вишни, малины, красной смородины, кожуры некоторых сортов яблок;
дельфинидина — окраска баклажанов, гранатов; пеларгонидина —
земляники; от комплекса цианидина и дельфинидина — окраска черной
смородины. С антоцианами связана и окраска продуктов переработки
плодов и овощей (варенья, компотов, фруктовых соков). В сочетании
с дубильными веществами они играют важную роль в технологии
столовых и красных вин.
Флавоноиды играют важную биологическую роль в организме
человека. Вместе с антоцианами, катехинами их относят к витаминопо-
добным соединениям — так называемому витамину Р. Многие
стороны действия флавоноидов на метаболизм еще неясны. Они способны
снижать проницаемость и ломкость капилляров крови.
Взаимодействуя с аскорбиновой кислотой, они участвуют в регуляции синтеза
коллагена у животных и человека, активируют дыхание тканей.
Недостаток флавоноидов ведет к точечным кровоизлияниям под кожу и
кровоточивости десен.
Несмотря на широкое распространение в растениях антоцианов их
биологическая роль изучена недостаточно.
Соединения (Св—С3—Св)2. К ним относятся бифлавоноиды>
образующиеся путем димеризации флавоноидов за счет образования
углерод-углеродных связей. Известно около 60 таких соединений, причем
их распространение в значительной степени ограничено
голосеменными растениями.
Соединения (С6—С,—С$)а* В эту группу входят так называемые
гидролизуемые таннины. Основной структурной единицей гидроли-
зуемых таннинов (ядром) является полиоксиалкоголь. Простейшим
примером гидролизуемого таннина является галлотаннин,
выделенный из растения сумаха, чаще называемый китайским таннином:
218
Глава 6. Вещества вторичного метаболизма растений
НСГ ^Г ^ОН
НО ОН ОН
Таннин из сумаха
При осторожном гидролизе кислотой или ферментом танназой из гал-
лотаннина можно получить галловую кислоту и глюкозу. Таннины
используются для дубления шкур животных при выделке кож. Особенно
богаты таннинами кора и листья дуба. Поэтому таннины часто называют
дубильными веществами. Высоким дубящим эффектом обладают
таннины с молекулярной массой от 500 до 3000 и содержащие в достаточном
количестве фенольные ОН-грулпы. Таннин, взаимодействуя с белком,
вызывает его денатурацию* За счет фенольных ОН-групп между танни-
ном и белком возникают поперечные связи, ведущие к образованию
прочного таннино-белкового комплекса, практически нерастворимого в воде,
кислотных и щелочных растворах. Для образования комплекса
низкомолекулярные таннины малоэффективны из-за возникновения
недостаточного количества перекрестных связей. Таннины с большой
молекулярной массой также малоэффективны, так как они не способны
проникать между волокнами коллагена шкуры животного.
Вяжущее действие таининов основано на физико-химических
процессах: вступая в реакцию с тканевыми белками, они отнимают воду
(дегидратация) и тем самым повышают вязкость и плотность белков в
жидких средах и тканях или вступают в химические реакции,
образуя белково-таннинные комплексы, выпадающие в осадок. Это
свойство таннинов широко используется в медицине. При нанесении тан-
нинов на рану образующиеся комплексы выпадают в осадок и
покрывают тонким слоем ее поверхность. Уплотняя поверхностный слой
тканей, таннины нарушают условия существования бактерий и,
таким образом, снижают их жизнедеятельность, затрудняют
размножение, уменьшают образование токсинов.
219
Часть I. Биомолекулы: структура, свойства, функции
Соединения (С6—С3—С€). Сюда относится вторая группа танни-
нов, известная под названием конденсированные таннины. Они
образуются только из фенолов флавонового типа, флаван-3-ол, поэтому их
часто называют флаволанами. К ним, например, относится димер прос-
дианидин, к которому присоединены другие молекулы флавана:
он
но
он
он
но
он
чАЛ
ОН
он
Просдианидин
В отличие от гидролизуемых таннинов, конденсированные
таннины при нагревании с разбавленными кислотами подвергаются
дальнейшей конденсации с образованием аморфных, часто окрашенных в
красный цвет соединений, называемых флобафенами* Их источником
является кора ивы, сосны, ели, каштана, дуба.
Строение конденсированных таннинов изучено недостаточно.
Соединения (С6—С3)п. К ним относится лигнин — полимер очень
сложного состава, построенный из гидроксифенилпропионовых
звеньев; его молекулярная масса — более 10 000.
Лигнин широко распространен в растительном мире, особенно в
растениях древесных пород. Он весьма устойчив и не растворяется
даже в горячей 70 %-ной серной кислоте. Вместе с целлюлозой, геми-
целлюлозами лигнин образует весьма прочный к механическим
воздействиям комплекс, выполняющий функцию "скелета" растений.
Окислительное разрушение лигнина в природе ведет к
образованию гуминовых кислот^ являющихся органическим компонентом почв.
6*3. Алкалоиды
К алкалоидам относят азотсодержащие гетероциклические
соединения основного характера, обладающие ярко выраженной
физиологической активностью. В растительном мире известно около 10 000 видов
алкалоидов* В большинстве случаев алкалоиды, как вещества основного
характера, в растениях содержатся в виде солей яблочной, винной,
лимонной и других кислот. В свободном виде они нерастворимы в воде, но
растворяются в органических растворителях. По химической структуре
азотистого гетероцикла они разделяются на следующие группы:
220
Глава 6. Вещества вторичного метаболизма растений
1) производные пиридина
А
%/
N
3) производные хинолина
2) производные пироллидина
и
АА
V/
4) производные индола
\ /
N
I
Н
изохинолина
аа
А
^ N
I
Н
5) производные пурина, к которым относятся кофеин и теобромин,
содержащиеся в кофе и чае
о
и
А
н3с-ы е
I и
о=е. .с
V"4/
И-СНз
СН
Л
н.
Кофеин
О
II
г<
1ВД С —
СН3
Теобромин
]ч-сн3
Как правило, в растениях содержится смесь нескольких
алкалоидов, иногда до 15—20, близких по своему строению.
Некоторые алкалоиды представляют собой комбинацию разных
гетероциклов. Так, молекулы алкалоидов листьев табака (никотина
и анабазина) являются комбинацией пиридина и пирролидина,
пиримидина и гидрированного пиримидина соответственно:
А
N
N
I
СН3
А
А
/
N
I
Н
Никотин
N
Анабазин
Никотин представляет собой бесцветную или светло-желтую про
зрачную жидкость жгучего вкуса, с неприятным запахом. Хорошо
растворим в воде, спирте, хлороформе и эфире. На воздухе легко окис-
221
Часть I. Биомолекулы: структура, свойства, функции
ляется, окрашиваясь в коричневый цвет* Его действие направлено на
центральную и периферическую нервную систему курящих и носит
двухфазный характер: вызывает непродолжительное возбуждение,
которое сменяется длительным торможением. Особенно сильно
никотин возбуждает центр дыхания. Частота сердечных сокращений
сначала замедляется, затем ускоряется.
При повторных введениях никотина в организм человека к нему
развивается привыкание. Он хорошо всасывается через слизистые оболочки
и кожу. Большая часть поступает в кровь. Никотин обезвреживается
главным образом в печени, а также частично в почках и легких. При
лактации никотин выделяется молочными железами в материнское
молоко. Хроническое отравление никотином ведет к воспалительным
процессам слизистых оболочек, снижению кислотности желудочного сока.
Он способствует развитию приступов стенокардии, нарушению зрения,
развитию раковых заболеваний дыхательных путей. В больших дозах
(для человека от 0,01—0,08 г) смерть наступает от паралича дыхания.
К производным хинолина относится хинин, содержащийся в коре
хинного дерева; применяется при лечении малярии.
Из изохинолиновых производных следует отметить морфин,
входящий в состав опия — сгущенного млечного сока опийного мака.
Применяется в медицине как успокаивающее и болеутоляющее
средство, а в больших дозах — как наркотик.
К производным индола относится группа алкалоидов, синтезируемая
грибом С1аи1серз ригригеа. Этот гриб развивается на зерне ржи в виде
выростов, называемых рожками спорыньи. Алкалоиды спорыньи весьма
ядовиты, особенно в большом количестве они накапливаются в перезимо-*
вавшем зерне ржи. Их наличие в ржаной муке может привести к
тяжелым отравлениям. Поэтому одной из важнейших технологических
операций в мукомолье является удаление рожков спорыньи из зерна. Гриб
С1ах>1сер$ ригригеа способен синтезировать более десятка родственных
алкалоидов, в основе строения которых лежит лизергиновая кислота:
соон
^ Ы-СНз
N
Н
Лизергиновая кислота
6.4. Гликозиды
Гликозиды — широко распространенные в природе вещества, в
молекулах которых остатки сахара связаны с молекулой вещества неуг-
222
Глава 6, Вещества вторичного метаболизма растений
л вводной природы — агликоном. Гликозиды часто имеют горький вкус
и специфический аромат, благодаря чему играют большую роль в
пищевой промышленности.
В зависимости от химической природы связи сахара с агликоном
различают О-, 8- и И-гликозиды: гликозил-О-К; гликозил-8-К, гли-
козил-№~К, где К — остаток агликона.
0-Гликозиды можно рассматривать как производные Сахаров, в
полу ацетальном гидроксиле которых радикалом является алифатическое,
карбоциклическое или гетероциклическое соединение. Хотя многие 0-
гликозиды гликозидной частью имеют монозы, однако ею могут быть и
остатки олигосахаридов (ди-, три- и т.д. сахаридов). Напомним, что по
типу гликозидов построены олигосахариды и полисахариды.
Встречающиеся в природе О-гликозиды являются (3-гликозидами.
Так, в гликозиде глюкованилине р-В-глюкоза связана с
производным фенола — ванилином. Глюкованилин содержится в плодах
ванили. При действии Р-глюкозидазы глюкованилин гидролизуется на
глюкозу и ванилин:
сно сно
н*о
оса
Р-Глюкозидаза
+ (З-В-Глюкоза
\
ОСН;
О-р-В-Глюкоза
Глюдованилин
ОН
Ванилин
Ванилин — альдегид ванилиновой кислоты, имеет характерный
приятный запах, в связи с чем широко используется в кондитерской и
мыловаренной промышленности. Специфический букет выдержанного
коньяка связан с наличием в нем ванилина. При многолетней выдержке
коньяков ванилин образуется в результате окисления кониферилового
спирта, источником которого является клепка дубовых бочек.
К гликозидам, агликоном которых является азотсодержащая
группа, относится амигдалин. Он содержится в семенах горького миндаля
(2,5—3,5 %), косточках персика (2—3 %), абрикоса, сливы (1—2 %),
вишни (0,8 %), яблок, груш, в листьях черемухи. Углеводной частью
амигдалина является дисахарид гентибиоза, агликоном — остатки
синильной кислоты и бензальдегида:
СН2ОН
сн.
) А п"0-^-0 №
^У°^У~
I
о-сн
I
он
он
* *■ 1
Остаток гентибиозы Агликон
Амигдалин
223
Часть I. Биомолекулы: структура, свойства, функции
Амигдалин представляет собой кристаллический порошок (белые
блестящие пластинки) без запаха, хорошо растворим в воде и спирте
при нагревании, гидролизуется под влиянием кислот, а также
фермента эмульсина, содержащегося в горьком и сладком миндале с
образованием р-О-глюкозы, бензальдегида и синильной кислоты*
Кроме амигдалина в растениях обнаружены еще 22 цианогенных
гликозида, дающих при гидролизе синильную кислоту.
К 0-гликозидам относятся весьма токсические соединения — сола-
кипы и чаконины, находящиеся в растениях семейства пасленовых.
Они содержатся в ботве, клубнях и особенно в ростках картофеля,
баклажанах (0,01—0,02 %). Во внутренней части клубня картофеля
их количество невелико — сконцентрированы они главным образом в
кожуре и удаляются при очистке картофеля. При длительном
лежании клубней картофеля на солнце они приобретают зеленый оттенок.
При этом количество соланинов резко возрастает во внутренней части
клубней, они приобретают резкий, горький вкус и становятся
непригодными в пищу.
Соланины и чаконины относятся к группе гликозидов, агликоном в
которых является соланоидин — соединение стероидной природы. В
клубнях картофеля синтезируется шесть видов соланинов, агликон в
которых один и тот же, а различаются они углеводной частью. Ниже
представлено химическое строение соланина, углеводной частью которого
является трисахарид, содержащий глюкозу, галактозу, рамнозу:
СИ
Н3С
/ЧА/
Глюкоза
Галактоза
Рам попа
Н3С
СН:
Углеводная часть
соланина
Соланидин - агликон
стероидной природы
Соланин
В отличие от а-соланина, у р-соланина углеводной частью
является дисахарид, содержащий галактозу и глюкозу; у-соланин содержит
только галактозу. Другие два гликозида, известные под названием а-,
$-чаконины, вместо галактозы содержат дополнительную молекулу
рамнозы; углеводной частью а-чаконина является глюкоза.
В диком картофеле 8о1апит йегпьввит содержится гликозид де-
миссин, отличающийся от ос-соланина лишь тем, что вместо рамнозы
224
Глава 6. Вещества вторичного метаболизма растений
в него входит ксилоза* а остаток соланидин не содержит двойной
связи. Любопытно в связи с этим отметить: в то время как листья
культурного картофеля поедаются личинками колорадского жука, листья
дикого остаются не тронутыми.
К 0-гликозидам относятся сапонины. Они близки по структуре и
свойствам к соланинам. Агликоны у них также стероидной природы.
Сапонины (от греч. заро — мыло) — это аморфные, хорошо
растворимые в воде ядовитые вещества, способные давать сильно пенящиеся
растворы. При введении в кровь они вызывают растворение
эритроцитов. При гидролизе кислотами сапонины образуют агликон сапогенин,
а из углеводной части — глюкозу, галактозу, арбинозу, метилпентозы
и галактуроновую кислоту.
Сапонины содержатся в семенах куколя — сорняка хлебных
злаков, в корнеплодах сахарной свеклы. На мельничных комбинатах
зерновые массы очищают от семян куколя на специальных машинах —
куколеотборниках.
В корнеплодах сахарной свеклы основная масса сапонинов
сосредоточена в хвостике и поверхностных слоях, В свеклосахарном
производстве значительное количество сапонинов переходит в
диффузионный сок. Однако в процессе его очистки известью они образуют с ионами
Са2+ нерастворимый осадок, удаляемый при фильтрации сока.
К 8-гликозидам (тиогликозидам) относится синигрин:
^ 8-В-]> Глюкоза
сн2=с!ьсн2-а
*К-0-80зК
Синигрин
Синигрин содержится в семенах горчицы и корнях хрена. Под
действием фермента тиогликозидазы, содержащейся в семенах горчицы
и корнях хрена, синигрин расщепляется с образованием
эфирно-горчичного масла, придающего горчице и хрену жгучий вкус.
Находящийся в хрене витамин С сильно стимулирует это расщепление.
Известно свыше 40 природных 8-гликозидов, близких к синигрину.
М-гликозиды (первичные или вторичные гликозиламины)
рассматриваются как производные гликозимина (первичные гликозиламины). Они
образуются при замене одного или двух атомов водорода в аминогруппе
остатков соединений алифатического или гетероциклического ряда.
Напомним, что к Ы-гликозидам принадлежат нуклеозиды и нуклеотиды.
6.5. Фитогормоны
Гормоны (от греч, НогтаЬпо — приводить в движение, побуждать) —
биологически активные соединения, играющие роль регуляторов
обмена веществ и функций человека, животных, растений.
Термин "гормоны" впервые введен английскими физиологами
М. Бейлиссом и Э. Старлингом в 1902 г. для известного в то время
биологически активного соединения секретина, стимулирующего вы-
15, Заказ 3486
225
Часть I. Биомолекулы: структура, свойства, функции
деление сока поджелудочной железы. Однако, как сейчас известно, не
все гормоны обладают стимулирующим действием. Некоторые из них
играют роль ингибиторов того или иного процесса.
Вопросы стимуляции синтеза растительных гормонов, их
секреции и перемещения от места синтеза к клеткам-мишеням, механизма
их действия на молекулярном уровне мало изучены.
Растительные гормоны можно подразделить на два типа:
стимуляторы роста растений; ингибиторы их роста.
К первому типу относятся три группы гормонов — ауксины, гиббе-
реллинЫу цитокинины; ко второму — абсцизовая кислота и этилен.
Ауксины (от греч. аихегп — расти). Из тканей растений и
микроорганизмов выделен гепьероауксин^ являющийся $-индолилуксусной
кислотой (ИУК), близкой по структуре к триптофану:
СНо-СООН
Гетероауксин (ИУК)
Близкими к ИУК стимуляторами роста растений являются (5-индо
лилацетонитрил, (З-нафтилуксусная и р-фенилуксусная кислоты:
СНо-СН=Ы-ОН
СН2-СООН
сн2-соон
)3-Инд о л и л ацетонит рил
р - Нафтилуксусная
кислота
А-
V
Р-Фенилуксусная
кислота
Обработка растений ИУК приводит к нарушению нормальных
соотношений в росте отдельных органов и тканей, вызывает
образование придаточных корней, увеличивает относительную массу
наземных частей растения, задерживает цветение; причем для этого
необходимы ничтожно малые концентрации (1:10000, 1:100000), а
в более высоких концентрациях она действует угнетающе. В сельс-
кохозяйственной практике ИУК широко применяется для ускоре-
■
ния образования корней у черенков и целых растений,
уничтожения сорняков, а также для задержки прорастания картофеля при
хранении.
Ауксины, по-видимому, образуются в основном в верхушках
стеблей и корней, в развивающихся листьях, цветках и плодах. От места
синтеза они перемещаются к месту воздействия на растущий участок.
Однако механизм процесса стимулирующего действия ауксинов
изучен недостаточно. Предполагают, что их действие аналогично действию
стероидных гормонов.
226
Г лава 6, Вещества вторичного метаболизма растений
Гиббереллины. Это большая группа тетрациклических терпенов,
Гиббереллины впервые были обнаружены как продукты
жизнедеятельности гриба аЬЪегеИа /идкигог, который поражает молодые растения
риса, вызывая удлинение стеблей и листьев, а затем их гибель*
Выделенное из культуральной среды в кристаллическом виде соединение
было названо гибберелином А9 при нанесении которого на растения
риса наблюдалась картина, типичная для поражения их грибом
ОгЬЬегеИа /щ1кигои Затем гиббереллины были выделены из многих
растений, включая побеги, корни, листья, пестики и тычинки цветков.
При нанесении гиббереллинов в процессе солодоращения на
ячмень они ускоряют его прорастание и стимулируют синтез а-амилазы
и других гидролаз.
В настоящее время известно более 50 видов гиббереллинов. Все
они получили номера в порядке их выделения: ГАр ГА2 ...ГАП, По
химической природе гиббереллины являются карбоковыми
кислотами, поэтому их называют также еибберелловыми кислотами.
Основной структурной единицей их является гиббан, который, в свою
очередь, образуется из тетрациклического дитерпена:
О .О
но
Гиббан
СНз СООН
Гиббереллин А1
-СНз
СНз СООН он
Гиббереллин Аз
Цитокинины — группа веществ, оказывающих стимулирующее
действие на деление клеток. Впервые они были обнаружены в солоде.
Первым выявленным цитокинином был фуриладенин (другие
названия: 6-фурфуриламинапурин, кинетин):
НС
сн
о
сш
ин
**
НС
^н
Фуриладенин
•И
%
СН
н
/
В незрелых семенах кукурузы найден цитокинитин, названный цеа
типом. Этот гормон является производным пурина и имеет
следующую структурную формулу:
15*
227
Часть I. Биомолекулы: структура, свойства, функции
Ш-СНо-СН=С-СЩОН
СН3
Цеатин
В растениях найден ряд производных цеатина, оказывающих
аналогичное действие.
Наличие пуринового кольца, по всей видимости, не имеет суще
ственного значения для проявления цитокинетической активности
Найдено значительное количество соединений, не имеющих
пуринового кольца, но обладающих цитокинетической активностью.
Особенно высокой активностью среди них обладает производное фенилмоче-
вины — М-З-нитрофенил-Ы'-фенилмочевина:
N0.
О
МН-С-ИН
И-З-Нитрофени л-Ы'-фенил мочевина
Достаточно много цитокининов обнаружено в активно
развивающихся органах и тканях, в прорастающих семенах, в почках клубней
картофеля в начальный период его прорастания.
Абсцизовая кислота (АБК) — гормон-ингибитор,
стимулирующий опадение листьев, вводит древесные растения в фазу покоя. Это
сесквитерпен, содержащий по одной карбоксильной, гидроксильнои и
карбонильной группе:
Н3С СН3 СООН
но X он
сн с
(Д/^Нз (!нз
Абсцизовая кислота
Эта кислота содержится в корнях, стеблях, листьях растений. АБК
ингибирует прорастание семян, стимулирует старение листьев,
созревание молодых плодов, повышает устойчивость растений к морозам.
Она синтезируется в хлороплаетах.
Этилен. Этот гормон подавляет рост растений, ускоряет процесс
созревания плодов, цветение. Он образуется многими видами
растений, зелеными водорослями, некоторыми грибами. Синтез этилена
возрастает при ранении растения, дефиците воды. Он успешно приме-
228
Глава 6. Вещества вторичного метаболизма растений
няется при хранении плодов и овощей, для ускорения их созревания.
Наиболее общая картина, связанная с процессом созревания, —
следующая: 1) размягчение тканей; 2) изменения в пигментации и
аромате; 3) гидролиз высокомолекулярных соединений; 4)
интенсификация дыхания.
Краткое содержание главы
В растениях осуществляется биосинтез различных веществ, не
входящих в состав компонентов важнейших метаболических путей. Эти
соединения получили название веществ вторичного происхождения.
Исследования последних лет показали, что многие вещества
вторичного происхождения имеют большое значение в метаболизме
растений. Однако механизм их действия на молекулярном уровне во
многом остается не выясненным*
По химической природе вещества вторичного происхождения можно
подразделить на следующие группы: 1) терпены; 2) растительные
фенолы; 3) гликозиды; 4) алкалоиды и 5) фитогормоны.
Терпены — углеводороды алифатического или циклического ряда.
В основе их химического строения находится молекула Отсоединения
изопрена. Эти вещества являются важнейшими компонентами
эфирных масел и смол растений, обусловливают определенный аромат
многих растений* Терпены классифицируются по числу содержащихся в
их молекуле атомов углерода* Это число кратно числу изопреновых
единиц. Терпены делятся на монотерпены С10, сесквитерпены С15> ди-
терпены С20, тритерпены С30, тетратерпены С40 и политерпены с
числом углеродных атомов от 7,5-103 до Зф105.
Монотерпены широко распространены в растениях, обладают
сильным ароматом. К производным монотерпенов относятся линалоол,
гераниол, содержащиеся в цветках ландыша и герани соответственно. К
монотерпенам относится также ментол — вторичный спирт,
содержащийся в эфирном масле перечной мяты, а также карвон, выделенный
из эфирных масел укропа и тмина.
Сесквитерпены часто встречаются вместе с монотерпенами. Сес-
квитерпеновый димер госсипол содержится в корнях и семенах
хлопчатника, является токсическим соединением.
В природе обнаружено до 500 дитерпенов. Дитерпеновым спиртом
является ретинол (витамин Ах). Производные дитерпенов —
циклические кислоты — являются основными компонентами живицы —
смолистых выделений хвойных растений.
К тритерпенам относятся стеролы — циклические одноатомные
спирты, производные циклопентанопергидрофенантрена. В дрожжах, грибах
находится эргостерол — провитамин 02. К производным стеролов
относятся гликозиды, сапонины, обладающие детергентным действием.
К тетратерпенам принадлежат каротиноиды. Представителями ка-
ротиноидов являются а-, р-, у-каротины — провитамины А.
229
Часть I. Биомолекулы: структура, свойства, функции
Типичными представителями политерпенов являются каучук,
накапливающийся в коре гевеи, и гутта.
Растительные фенолы содержат одну или несколько гидроксиль-
ных групп, связанных с бензольным кольцом. В природе фенолы
редко встречаются в свободном виде. Большое разнообразие фенольных
соединений (около 1000) по химической структуре может быть
разбито на две группы: монофенольные и полифенольные — содержащие от
2 до я фенольных остатков. К полифенольным соединениям относятся
флавоноиды, чрезвычайно широко распространенные в растениях.
Многие из них ярко окрашены от желтого до темно-красного цвета.
Флавоноиды представляют собой гликозиды. Основу структуры их
неуглеводной части (агликона) составляет флаван, состоящий из двух
ароматических колец А и В. Флаваны имеют различную степень окис-
ленности от наименее окисленного катехина до сильно окисленного
флавонола. К группе флавоноидов относятся гликозиды — комплексы
антоцианидинов с сахарами. Антоцианидины и близкие к ним по
структуре флаванолы оказывают большое влияние на окраску растений.
Окраска плодов и ягод обусловлена в основном антоцианами.
Флавоноиды играют важную биологическую роль в организме человека; их
относят к витаминоподобным соединениям — так называемому
витамину Р.
К группе полифенолов относятся гидролизуемые таннины, или
дубильные вещества. Взаимодействуя с белками, они образуют таннино»
белковый комплекс, практически нерастворимый ни в воде, ни в
кислотах, ни в щелочах; обладают вяжущим вкусом. Ко второй группе
таннинов относятся конденсированные таннины, не поддающиеся
кислотному гидролизу. В отличие от гидролизуемых таннинов,
конденсированные таннины при нагревании с разбавленными кислотами
подвергаются дальнейшей конденсации с образованием аморфных, часто
окрашенных в красный цвет соединений, называемых флобафенами.
Вяжущее действие таннинов основано на том, что, вступая в реакцию
с тканевыми белками, они отнимают воду (дегидратация) и тем
самым повышают вязкость и плотность белков в жидких средах и
тканях. Таннины способны также взаимодействовать с белками, образуя
белково-танниновые комплексы, выпадающие в осадок.
К полифенолам принадлежит лигнин — соединение сложного
состава, построенное из оксифенилпропионовых звеньев. Лигнин —
важнейший компонент растений древесных пород; устойчив и
нерастворим даже в горячей 70 %-ной серной кислоте. Вместе с целлюлозой,
гемицеллюлозами лигнин образует весьма прочный к механическим
воздействиям комплекс, играющий роль "скелета" растений.
Окислительное разрушение лигнина в природе ведет к образованию
гуминовых кислот, являющихся органическим компонентом почв.
К алкалоидам относят азотсодержащие гетероциклические
соединения основного характера, обладающие ярко выраженной физиоло-
230
Глава 6. Вещества вторичного метаболизма растений
гической активностью. В растениях они находятся в виде солей
органических кислот. По химической структуре азотсодержащего гетеро-
цикла они подразделяются на группы пиридина, пирролидина, хино-
лина, индола. Как правило, в растениях содержится смесь
нескольких алкалоидов, близких по своему строению. Некоторые алкалоиды
представляют собой комбинацию разных гетероциклов. Так,
например, молекула алкалоида листьев табака — никотина является
комбинацией пиридина и пирролидина*
Гликозиды — вещества, в молекулах которых остатки сахара
связаны с молекулами вещества неуглеводной природы, называемого аг-
ликоном. В зависимости от химической природы связи сахара с агли-
коном различают О-, Ы- и 8-гликозиды. Гликозиды часто имеют
горький вкус и специфический аромат; многие из них используются в
пищевой промышленности. Так, гликозид ванилин широко
применяется в кондитерской промышленности. В образовании
специфического аромата семян горького миндаля, косточек персиков, абрикосов,
слив, вишни участвует гликозид амигдалин. о-Гликозид соланин
содержится в кожуре клубней картофеля; в полежавшем на солнце
картофеле он накапливается в большом количестве во внутренних слоях
клубня, и из-за резкого горького вкуса картофель становится
непригодным в пищу. Близки по структурным свойствам к соланинам —
сапонины, хорошо растворимые в воде ядовитые вещества. Они
содержатся в семенах куколя — сорняка хлебных злаков, в хвостиках и
поверхностных слоях корней сахарной свеклы. В семенах горчицы и
корнях хрена содержится 8-гликозид синигрин, придающий горчице
и хрену жгучий вкус.
Растительные гормоны можно подразделить на стимуляторы роста
растений (гетероауксин, р-индолилацетонитрил, р-нафтилуксусная
кислота, фенилуксусная кислота, гиббереллины, цитокинины) и
ингибиторы роста (абсцизовая кислота, этилен) растений.
Хронология важнейших дат в исследовании
веществ вторичного происхождения
1806 г. — Ф. Сертюрнер выделил морфий и установил его свойства.
1819 г. — Ф. Рунге открыл кофеин,
1842 г. — А. А. Воскресенский открыл теобромин.
1886 г. — А. Ланденбург синтезировал алкалоид конеин.
1902 г. — Э. Фишеру присвоено почетное звание лауреата Нобелевской
премии. Им были осуществлены исследования многих природных гликозидов,
изучено строение и осуществлен синтез таннинов.
1925 г, — А. П. Орехов разработал классификацию алкалоидов.
ЛИТЕРАТУРА
Гудвин Т., Мецлер Э. Введение в биохимию растений. — М.: Мир> 1986,—
Т. 2 — 312 с.
231
Часть I. Биомолекулы: структура, свойства, функции
Запрометнов М. Н. Основы биохимии фенольных соединений. — М.: Наука,
1974. — 173 с.
Кретович В. Л. Биохимия растений. — М,: Высшая школа, 1986. — 497 с.
Мецлер Д. Биохимия. — Мл Мир, 1980, — Т. 3, — 488 с.
ВОПРОСЫ
1. Что лежит в основе химической структуры терпенов? Какова структура
монотерпенов, тритерпенов, каучука и гутты?
2, Предшественниками какого витамина являются тритерпены, тетратерпе-
ны?
3- Расскажите о растительных фенолах на примерах флавоноидов.
4. Что такое алкалоиды, какова их физиологическая роль, на какие группы
они подразделяются?
5. На какие группы подразделяются гликозиды в зависимости от связи
сахара с агликоном?
Часть II
МЕТАБОЛИЗМ
ВЕЩЕСТВ И ЭНЕРГИИ
Глава 7. ПРИНЦИПЫ
БИОЭНЕРГЕТИКИ КЛЕТКИ
Метаболизм веществ и энергии представляет собой совокупность
процессов превращения веществ и энергии в.живой клетке, а также
между клеткой и окружающей средой. С точки зрения биоэнергетики
метаболизм подразделяется на катаболизм — совокупность
биохимических реакций, связанных с извлечением химической энергии,
аккумулированной в расщепляемом соединении, и анаболизм .— ее
использование.
У всех организмов метаболизм включает в себя следующие четыре
основные функции:
1. Извлечение энергии из окружающей среды путем распада
поступивших в клетку органических соединений или поглощения
квантов света с образованием высокоэнергетических соединений в
количестве, достаточном для обеспечения всех энергетических
потребностей клетки.
2. Использование энергии для синтеза эндогенных соединений (или
транспорта их уже в готовом виде в клетку), являющихся
предшественниками макромолекулярных компонентов клетки; для
совершения работы (осмотической, электрической, механической).
3. Синтез из предшественников структурных компонентов клетки
(белков, нуклеиновых кислот, углеводов, липидов и других
соединений), а также их распад, обновление.
4. Синтез и распад биомолекул со специальным назначением
(гормоны, ферменты).
Метаболизм веществ и энергии в живой клетке характеризуется
определенным своеобразием. Все части клетки имеют примерно
одинаковую температуру, т.е. клетка по существу изотермична. Разные
части клетки мало отличаются и по величине давления. Это значит,
что клетка не способна использовать тепло в качестве источника
энергии. Иными словами, клетка не похожа на обычную тепловую или
электрическую машину. Ее можно представить как изотермическую
химическую машину.
Биоэнергетика представляет собой совокупность процессов
превращения энергии в биологических системах; извлечение энергии из
окружающей среды, ее аккумулирование и использование для
жизнедеятельности клетки. В основе биоэнергетики лежат законы физики и
химии, к ней приложимы первый и второй законы термодинамики.
235
Часть II. Метаболизм веществ и энергии в клетке
■ ■ ■!■■■■! М-Е^^^Д! ! ,ц ! Ц | —^^^^ 1-» II ■■ 1^И ■
Но поскольку живая клетка представляет собой открытую,
неравновесную, стационарную систему, проявление этих законов имеет ряд
характерных особенностей, изучение которых тесно связано с
выяснением наиболее общих и основных характеристик жизни.
В данной главе мы рассмотрим биохимическую термодинамику с
приложением классической термодинамики к биохимическим
явлениям, т.е. биоэнергетику клетки. Затем при анализе метаболизма
углеводов, жиров, белков будут обсуждены некоторые аспекты частной
термодинамики»
7*1. Биохимическая термодинамика
Задачи биохимической термодинамики состоят в изучении
взаимоотношений между химической и другими формами энергии:
тепловой, механической, электрической. Энергетическая характеристика
химических реакций, происходящих в клетке, позволяет дать ценную
научную информацию о механизме процесса и его направленности,
В термодинамику вводится понятие системы, которая
представляет собой совокупность веществ, подлежащих изучению. Все лежащее
за пределами системы называется окружающей средой. Энергия
может переходить из системы в окружающую среду или из окружающей
среды в систему. Характеристику перехода системы из одного
состояния в другое можно дать такими термодинамическими функциями,
как внутренняя энергия Е, энтальпия Н, энтропия 8 и свободная
энергия С Поскольку для живых систем трудно определить
абсолютные значения Е, Н, 8 и О, здесь имеют дело с изменением функций
состояния системы; ДЕ, АН, А8 и АО, где А представляет собой
разность между значениями функции состояния продуктов и
реагирующих веществ (например, АН = Н продуктов - Н реагентов).
Термодинамический анализ энергетических изменений
основывается на изучении указанных характеристик. Знания молекулярного
состава системы и окружающей среды или молекулярных
механизмов, лежащих в основе такого процесса, для термодинамического
анализа не требуются. Более того, в подавляющем большинстве случаев
термодинамический анализ энергетических изменений не требует
знания скорости процесса; достаточно знать лишь начальное и конечное
состояния системы.
Каждая термодинамическая функция имеет свои смысл и область
применения, поэтому представляет интерес остановиться на них более
подробно.
Внутренняя энергия представляет собой полную энергию
системы. Согласно первому закону термодинамики в замкнутой системе
она всегда должна быть величиной постоянной либо увеличиваться,
либо уменьшаться за счет окружающей среды, В этой системе при
взаимных превращениях различных форм энергии имеет место их
строгое количественное соотношение. Внутренняя энергия определяется
236
Глава 7. Принципы биоэнергетики клетки
только состоянием системы в данный момент и не зависит от
предыстории системы, ее нельзя ни создать, ни уничтожить.
Энтальпия — теплосодержание системы. Термин "энтальпия"
введен для того, чтобы отличить ее от внутренней энергии системы, В
классической термодинамике
Н « Е + АРУ,
где АРУ — работа системы, связанная с изменением давления АР и
(или) объема ДУ(РДУ).
Поскольку биохимические реакции протекают, как правило, при
постоянном давлении и мало изменяющемся объеме системы, разница
между значениями Е и Н пренебрежительно мала. Многое из того, что
в биохимической литературе говорится об энергии, фактически
относится к энтальпии, т.е. Е равна Н.
Энтропия — термодинамическая функция, характеризующая
степень неупорядоченности системы. Она отражает физический смысл
второго закона термодинамики, согласно которому в замкнутой
системе могут происходить лишь те превращения, при которых энтропия
либо остается постоянной (в случае обратимых процессов), либо
возрастающей (в случае необратимых процессов); самопроизвольное
уменьшение энтропии не имеет места. Увеличение энтропии является
мерой необратимости реальных процессов в замкнутой системе. Каждый
раз, когда в такой системе происходит какой-либо самопроизвольный
процесс, имеет место не только перераспределение различных форм
энергии, но и определенное увеличение энтропии; оно является как
бы платой за использование энергии.
Величина энтропии какого-либо соединения при данной
температуре зависит от того, какие колебательные и поступательные
движения могут совершать атомы, входящие в состав молекулы этого
соединения. Энергия колебаний квантована, т.е. молекула обладает
определенным набором энергетических уровней. Чем больше вероятность
существования данной молекулярной структуры, тем больше
энтропия системы. Эту зависимость можно выразить уравнением
8 = к-1п^,
где к — газовая постоянная на одну молекулу (т.е. газовая
постоянная К, деленная на число Авогадро; к называют постоянной Больцма-
на: она выражает соотношение между эрг и град)\ "\У
(термодинамическая вероятность) — число энергетических уровней, которым
обладает молекула при определенной общей энергии. С повышением
температуры это число увеличивается и энтропия возрастает, С
понижением температуры Д^Г уменьшается, при абсолютном нуле становится
равным единице и энтропия убывает до нуля.
В структурировании систем, состоящих из высокомолекулярных
соединений, энтропия вносит ценную информацию в характеристику
этих систем.
237
Часть II. Метаболизм веществ и энергии в клетке
Следует отметить, что энтропия открытой системы, которую
представляет собой живая клетка, изменяется не только за счет ее
нарастания в результате необратимых процессов, происходящих в клетке, Д§,,
но и за счет обмена энтропией между клеткой и окружающей средой А8 .
Таким образом, для живой клетки полное изменение энтропии
Д8 = Л8, + АЗ .
1 е
При Д81 > 0 член Д8е может иметь различный знак. Если А8е > 0 или
отрицателен, но по абсолютной величине меньше члена Д8., то общее
изменение энтропии клетки А8 > 0, т.е. общая энтропия такой клетки
возрастает. В случае А8е < 0 клетка как бы поглощает из среды
отрицательную энтропию, и если этот член по абсолютной величине больше А8.,
то общее изменение энтропии клетки отрицательное, несмотря на
продуцирование энтропии внутри клетки (Л8. > 0). Отрицательное значение
энтропии свидетельствует о том, что в клетке возможен синтез более
упорядоченных структур (например, белков, полисахаридов) из менее
упорядоченных (например, аминокислот, моносахаридов). Такое
положение может иметь место в том случае, когда во внешней среде
происходят сопряженные изменения с увеличением энтропии; это означает лишь,
что соответствующая часть внешней среды также должна быть включена
в границы открытой системы.
Свободная энергия биохимических реакций представляет собой часть
внутренней энергии клетки, которая может быть превращена в
полезную работу химического синтеза, механического движения,
транспорта веществ, преобразована в электрический потенциал. Собственно
говоря, поглощение пищевых продуктов организмом человека с
термодинамической точки зрения представляет собой извлечение из
среды свободной энергии.
Хотя изменения энтропии сопровождают все физические и
химические процессы, однако они не определяют ни направления процессов в
клетке, ни природы упорядоченности структур и слаженности процессов
метаболизма веществ. Определяющая роль здесь принадлежит
свободной энергии. Свободная энергия представляет собой термодинамическую
функцию, прямо связанную с биохимической стабильностью клетки.
Высокий уровень свободной энергии присущ потенциально неустойчивым
системам, которые при определенных условиях самопроизвольно будут
переходить на более низкий уровень этой энергии. Иначе говоря, в
биохимических реакциях, в которых наблюдается переход от неустойчивого
состояния с высокой химической энергией веществ к более устойчивому
состоянию с меньшей химической энергией продуктов реакции, будет
иметь место выделение энергии, ее убыль; изменение свободной энергии
будет отрицательным (-АС-), Такие биохимические реакции называются
экзергоническими, они являются термодинамически выгодными и могут
быть использованы для совершения полезной работы. Напротив,
биохимические реакции (например, реакции синтеза), требующие затраты энер-
238
Глава 7. Принципы биоэнергетики клетки
гни, характеризуются переходом веществ из устойчивого состояния с
низкой химической энергией к менее устойчивому состоянию с большей
химической энергией; изменение свободной энергии будет
положительным (+ДО). Такие реакции называются эндергоническими, они не могут
протекать в клетке без притока энергии извне. В соответствии с первым
и вторым законами термодинамики взаимосвязь между АО, АН и А8
может быть описана уравнением
АО - АН - ТАЗ,
где Т — абсолютная температура, член ТД8 представляет собой
связанную энергию. Она не может быть использована для совершения
работы. Следует отметить, что ТА8 для большинства биохимических
реакций составляет небольшую величину. Поэтому АН может служить
приближенно мерой совершаемой работы» Если АН велика (например,
при сгорании питательных веществ), то она не слишком отличается от
ЛС того же процесса. Этим определяется использование калорийности
пищи как приблизительной меры работы, совершаемой в результате
усвоения пищи организмом.
В живых организмах свободная энергия играет решающую роль.
Высокомолекулярные органические вещества обладают значительным
запасом свободной энергии и именно ее деградация лежит в основе
жизнедеятельности, а не изменение упорядоченности, т.е. энтропии
поглощенных продуктов.
Стандартная свободная энергия. Величина АО для той или иной
реакции является функцией концентрации реагирующих веществ: чем
она выше, тем больше изменение свободной энергии. Чтобы легче было
вычислить изменение свободной энергии, нужно знать изменение
свободной энергии в стандартных условиях (АО0). За такие условия
принимают температуру 25 °С, концентрацию растворенных веществ,
соответствующих одномолярной активности.
Рассмотрим следующее обобщенное химическое уравнение,
описывающее реакцию, в ходе которой а молей вещества А реагируют с Ъ
молями вещества В, давая продукты С, О и т.д.:
аА + ЬВ + ... = сС + <Ш + ... .
В ходе химической реакции изменение АО для каких-либо
определенных условий можно рассчитать, исходя из следующего уравнения:
СС а "г ССд ...
Здесь ас — активность компонента С и т.д.
Используя данное уравнение, можно рассчитать АО при низких
концентрациях веществ, что обычно имеет место в биохимических системах
(чаще всего эти концентрации близки к миллимолярным и существенно
ниже концентрации гипотетического стандартного раствора).
239
Часть II. Метаболизм веществ и энергии в клетке
В биохимической термодинамике обычно используют концентра
ции реагирующих компонентов, принимая а — 1:
ДО~ДО° + КТ1п!-СЗС+[Е,]С1-
[А]а + [В]ь
В этом уравнении квадратные скобки означают молярную
концентрацию; К — газовая постоянная, Т — абсолютная температура.
Таким образом, при определенных температурах и давлении
величина АО Судет изменяться при изменении концентрации
компонентов, тогда как АО0 постоянна для данной реагирующей системы.
Логарифмический член уравнения ответствен за наблюдаемую аномалию,
В самый начальный момент реакции концентрация С, & будет весьма
мала, АО будет отрицательным, а система — крайне неустойчива. В
состоянии равновесия ДО ~ 0; при этом условии
ДО«=-ЕТ1п^+™а
» т ш
[ А]а + [В]ь
* V
Поскольку система находится в равновесии, то правый член
уравнения фактически представляет собой константу равновесия К
системы. Отсюда следует, что
АО0 = -ЕТ 1п К.
При значении К — 1 АО0 = 0; если К > 1, то стандартная свободная
энергия равна -АО0 (реакция экзергоническая); если К < 1, то АО
будет +АО° (реакция эндергоническая). Данное уравнение — одно из
важных в термодинамике. Область его применения очевидна: при любой
температуре АО0 можно рассчитать из константы равновесия К,
которую в большинстве случаев удается определить экспериментально.
В биохимической термодинамике в качестве стандартного
состояния принимается состояние при рН 7,0. Эта величина подобрана с
учетом того, что большинство реакций в живой клетке протекает при
рН 7,0. Изменение свободной энергии при рН 7,0 обозначается
символом АО0'.
Использование АО0' означает, что за стандартное состояние
каждого исходного реагента и каждого образовавшегося продукта
принимают то соотношение его ионизированных и неионизированных форм,
какое существует при рН 7,0. С изменением величины рН может
изменяться степень ионизации компонентов реакции, поэтому значение
ДО0 (рассчитанное для рН 7,0) не всегда может использоваться при
других значениях рН, так как для некоторых биохимических реак*
ций значение АО0 довольно сильно изменяется с изменением
величины рН.
240
Глава 7; Принципы биоэнергетики клетки
7.2. Роль адениловой системы в энергетике клетки
После ознакомления с общими принципами биохимической
термодинамики и важнейшей термодинамической характеристикой —
свободной энергией системы необходимо рассмотреть второй вопрос
биоэнергетики — термодинамические характеристики специальной
группы соединений, играющих важную роль в передаче химической
энергии внутри клетки и известных под названием
"высокоэнергетические соединения". Химические превращения этих веществ
сопровождаются освобождением большого количества энергии, которая затем
расходуется на различные процессы жизнедеятельности; биосинтез,
транспорт веществ через мембраны, движение.
Высокоэнергетические соединения занимают центральное положение
в превращениях веществ и энергии клетки. Химическую связь в
молекуле высокоэнергетического соединения принято называть
высокоэнергетической (макроэргической) связью и обозначать символом —. Вокруг
нее и локализована свободная энергия. Количественная характеристика
этой связи дает возможность оценить термодинамическую вероятность
протекания процесса» в котором принимает участие
высокоэнергетическое соединение. Разрыв высокоэнергетической связи всегда
сопровождается отщеплением от высокоэнергетического соединения химической
группы и переносом ее на молекулу какого-либо акцептора (например, при
гидролитическом распаде таким акцептором является молекула воды).
Энергия, освобождающаяся при расщеплении высокоэнергетического
соединения, не эквивалентна самой энергии высокоэнергетической
связи; она измеряется изменением свободной энергии в реакции переноса
химической группы с высокоэнергетической связью на молекулу
акцептора. Изменение свободной энергии в реакциях переноса групп зависит
от химической природы донора и акцептора энергии.
К важнейшим веществам с высокой энергией переноса групп
относится обширный класс фосфорорганических соединений, включающий
в себя ангидриды фосфорной кислоты,,ацилфосфаты, гуанидинфосфа-
ты. В этих соединениях высокоэнергетические фосфатные группы
часто обозначают символом -Р. К высокоэнергетическим соединениям
относится также целый ряд тиоловых эфиров и эфиров аминокислот.
Центральное место среди них занимает АТР — аденозинтрифосфат,
значительные количества которого содержатся в животных,
растениях и микроорганизмах.
АТР был впервые получен К. Ломаном в 1929 г. из мышц
лягушки. Это соединение — сильная четырехосновная кислота, легко
растворимая в воде, содержит два высокоэргических остатка фосфорной
кислоты* АТР образуется из АВР путем фосфорилирования за счет
энергии, освобождающейся при окислении органических веществ. При
гидролизе АТР образуются АВР и АМР с выделением большого
количества химической энергии:
16. Заказ 3486 241
Часть II. Метаболизм веществ и энергии в клетке
о" о" о" о" о" о"
I I I _ . I I _ . I .
А~0~Р^О~Р-0~Р-0 +нон ^ А-О-Р-О-Р-0 +нон г» А-0~Р-0
II II II II II II
ООО 00 О
АТР АБР; АМР;
ДО0' = -30,5 кДжмоль"1 ДО0 = -30,5 кДж-моль"1;
А — остаток молекулы аденозина.
АТР вместе с АВР и АМР образуют адениловую систему. Энергия,
накопленная в виде АТР, используется в огромном числе различных
эндергонических процессов клетки, к числу которых относятся
биосинтез белков, нуклеиновых кислот, углеводов, жиров; транспорт
веществ и ионов через мембраны, различные формы движения.
АТР образуется в ходе эндергонической реакции, называемой
реакцией фосфорилирования:
АВР + Н3Р04 -> АТР.
В этой реакции используется либо энергия, извлекаемая при
окислении фрагментов питательных веществ, связанном с субстратным
фосфорилированивМу либо энергия, выделяющаяся при переносе
электронов по дыхательной цепи, сопряженном с окислительным фосфори-
лированием* либо энергия, получаемая за счет фотосинтетического
фосфорилирования.
ДО0' гидролиза АТР, идущего с отщеплением концевого остатка Н3Р04,
достаточно велика и составляет -30,5 кДж-моль"1; близкая величина
найдена для гидролиза АТР с образованием АМР и пирофосфата Н4Р2Ог
В то же время величина ДО0' для гидролиза АМР на аденозин и Н3Р04
составляет -12,6 кДжфмоль-1, т.е. почти в 2,5 раза ниже энергии макро-
эргической связи. В живой клетке с учетом реальных концентраций
реагентов АО0' может достигать -50 кДж-моль"1 и выше.
Почему АТР является термодинамически неустойчивым
соединением, имеет большую энергию фосфатной связи? Или: почему
равновесие гидролиза АТР {переноса фосфатной связи на воду) сильно
смещено в сторону завершения реакции?
Исчерпывающий ответ на этот вопрос можно получить при
рассмотрении структуры АТР (рис. 7Л, а, б). В структуре этого
соединения заложены два дестабилизирующих фактора:
электростатическое отталкивание и конкурентный резонанс.
При физиологических значениях рН, близких к 7,0, все четыре
-Р-ОН-связи АТР ионизированы, несут отрицательные заряды,
близко расположенные друг к другу. В результате во внешней сфере
структуры АТР возникает высокая плотность одноименных отрицательных
зарядов. Это приводит к стерическому напряжению всей молекулы, и
прежде всего связей -Р-0-Р-. При отщеплении концевой фосфатной
группы силы электростатического отталкивания ослабевают, стери-
242
Глава 7, Принципы биоэнергетики клетки
О О
О
0--Р-0-Р-0-Р-0-СШ
I [ I
О" О" О" НА Н
ОН ОН
>щ.
А-
АТР (при рН 7 тетразарядный анион)
а
б
Рис. 7Л. Химическая структура (а) и пространственная модель (б) молекулы
АТР
ческое напряжение снижается. Образовавшиеся продукты гидролиза,
а именно анионы АБР3" и НРО|", не могут вступить в обратную
реакцию, ведущую к синтезу АТР, потому что их сближению
препятствуют кулоновские силы отталкивания одноименных зарядов.
Из структуры АТР также видно: большее число электронов,
окружающих атом фосфора и кислорода концевой фосфатной группы АТР,
вступают в резонансную конкуренцию друг с другом за энергетически
более выгодные орбитали. Наличие такой конкуренции не позволяет
всем электронам концевой пирофосфатной группы АТР занять столь
низкие энергетические уровни» какие они могут иметь в ионах АВР3
и НРО|". Иными словами, при переносе с молекулы АТР фосфатной
группы на воду существенно снижается конкурентный резонанс
электронов в продуктах реакции.
7.3. Термодинамическая шкала фосфорорганических соединений
В табл. 7.1 приведены значения ДО0' для гидролиза некоторых
биологически важных фосфорилированных соединений, расположенных
в порядке снижения их абсолютной величины и образующих
термодинамическую шкалу стандартных свободных энергий. Как видно из этой
шкалы, АТР — вещество, уникальное еще и потому, что в
энергетическом ряду оно занимает промежуточное положение.
Все соединения, расположенные выше АТР, относят к классу
высокоэнергетических; они неустойчивы, легко отдают свои фосфатные
16
*
243
Часть II, Метаболизм веществ и энергии в клетке
Таблица 7.1
Стандартная свободная энергия (ДО0)
биологически важных фосфорорганических соединений
Соединения
Высокоэ нергет ические
Фосфоэн о л п иру ват
1,3-Бисфосфоглицерат
Креатинфосфат
Аргин инфосфат
Низкоэнергетические
АТР
АОР
1ШР-глюкоза
Глюкозо-1-фосфат
Г л юк озо-6-фосфат
АМР
-АО0', кДж-моль"1
61,7
49,2
42,5
32,2
28,3
24,2
21,8
15,8
14,1
13,8
группы. Соединения, расположенные ниже АТР, относят к классу
низкоэнергетических, они стремятся удержать фосфатные группы.
Функция АТР заключается именно в том, что в адениловой системе
она выполняет роль посредника, являясь переносчиком фосфатных
групп от высокоэнергетических соединений к низкоэнергетическим.
В системе АТР—АОР химическая энергия передается сначала от
высокоэнергетического донора фосфата к АБР и запасается там в форме
АТР, затем концевая фосфатная группа АТР передается на
низкоэнергетическую акцепторную молекулу, которая становится реакционно-
способной. В живой клетке нет пути переноса фосфатной группы от
высокоэнергетического соединения к низкоэнергетическому, равно как
не обнаружен также путь переноса этой группы от
высокоэнергетического донора к высокоэнергетическому, расположенному ниже по
термодинамической шкале. Таким образом, во всех реакциях, связанных
с переносом энергии в форме ~Р, роль общего промежуточного
продукта играет АТР; это соединение является связывающим звеном между
экзергоническими и эндергоническими реакциями, выполняет роль
универсального донора химической энергии, создает общий
клеточный фонд энергии.
АТР играет важную роль в синтезе 5'-трифосфатов рибонуклеози-
дов и 2'-дезоксирибонуклеозидов, (ЭТР, где N — нуклеозид), которые
не только служат предшественниками в синтезе РНК и ДНК, но и
играют важную роль в синтезе белков и полисахаридов (рис. 7.2).
Видно, что каждый нуклеозид-5'-трифосфат выполняет определенную роль
в биосинтезе биополимеров, получив от АТР предварительно
концевую фосфатную группу.
244
Глава 7, Принципы биоэнергетики клетки
~р
АТР ^ТР
:
Белки
Ы^-Гуанозин
"
АТР
♦
N2
1
-Р
ЫзТ]
1
РНК
-Гуанози]
~Р
1
АТР
1
Р А
н, М-Дк
-Р
ТР ЫТР
+ *
ДНК
юксигу анозин ,
~р
♦
АТР Ы1ТР
Полисахариды
Ш-Уридин
цитидин, дезокситимидин,
уридин дезоксицитидин
Рис. 7.2. Участие нуклеозидтрифосфатов (1ЧТР) в биосинтезе биополимеров
7.4. Характеристика важнейших высокоэнергетических соединений
В числе высокоэнергетических соединений прежде всего следует
назвать два: 1,3-6исфосфоелицерат и фосфоэнолпируват. Оба
являются промежуточными продуктами гликолиза (см. гл. 9) и с их
превращениями сопряжен процесс субстратного фосфорилирования.
1,3-Бисфосфоглицерат передает одну фосфатную группу на АВР, в
результате чего образуется АТР и 3-фосфоглицерат:
О
о
С-0
1
НСОН
I
л^
о
I
Н2С-0~Р=0
|_
о"
С-0
I
+ АБР - НСОН о" +АТР
I I
Н2С-0~Р=0
|_
о
1,3-Биефосфоглицерат З-Фосфоглицерат
Поскольку величина ДО0' гидролиза АТР известна, значение АО0
гидролиза 1-фосфатной группы 1,3-бисфосфоглицерата легко
рассчитать, определив предварительно константу равновесия реакции
переноса фосфатной группы. Такие данные были получены; в стандартных
условиях константа К оказалась равной 2,07; отсюда расчетным путем
было определено изменение АО0', которое оказалось равным -18,9 кДж.
Используя принцип аддитивности применительно к
последовательности реакций
1,3-бисфосфоглицерат + АБР %± 3-фосфоглицерат + АТР,
ДО0' = -18,7 кДж;
АТР + Н20 ^ АБР + Р., ДО0' - -30,5 кДж,
получаем величину ДО0' переноса 1-фосфатной группы
1,3-бисфосфоглицерата:
ДО0' = -30,5 + (-18,7) - -49,2 кДж.
245
Часть II. Метаболизм веществ и энергии в клетке
1,3-Бисфосфоглицерат представляет собой смешанный ангидрид
фосфорной и карбоновой кислот. Большая величина стандартной
свободной энергии гидролиза ацилфосфатной связи 1,3-бисфосфоглице-
рата обусловлена отрицательными зарядами продуктов гидролиза и
сильно выраженным эффектом стабилизации этих продуктов за счет
дел окал изации электронов.
Фосфоенолпируват также легко передает свою фосфатную группу
молекуле АБР:
СОО О" СОО
I I I
С-0~Р=0 + АОР = С-О + АТР
II I I
СН2 О" СН2
Фосфоенолпируват Пируват
Стандартная свободная энергия гидролиза фосфоенолпирувата
составляет -61,7 кДж. Столь большая отрицательная величина АО0,
фосфоенолпирувата обусловлена тем, что это соединение представляет собой
ангидрид двух кислот — фосфорной и енолпировиноградной. В ходе
гидролиза енольная форма пирувата переходит в более устойчивую кетофор-
му. Это и приводит к сильному сдвигу реакции в правую сторону.
Фосфоенолпируват и 1,3-бисфосфоглицерат не только играют роль
доноров химической энергии, но и являются важными интермедиата-
ми гликолиза.
Два других высокоэнергетических фосфорилированных
соединения — креатинфосфат и аргининфосфат — важны прежде всего как
"аккумуляторы" химической энергии:
О' Н СНа О" Н Н Н
III III I
-0-р~:к-с-к-сн2-с-сг о-р~к-с-к-(сн,)3-с— с-о"
I! 11+ Н II 11+ I II
о *ш2 о о гш3 мн2о
Креатинфосфат Аргининфосфат
Эти соединения представляют собой производные гуанидина, в
молекуле которых атомы фосфора непосредственно связаны с
атомами азота. Высокое значение АС-0' гидролиза этих соединений
обусловлено трудностями стабилизации гуанидиновой и фосфатной групп в
составе их молекул. При отщеплении фосфатной группы эти
трудности устраняются за счет сильной стабилизации продуктов
гидролиза — креатина и аргинина.
К группе высокоэнергетических соединений относятся также тио~
эфиры. В то время как высокоэнергетические фосфорные соединения
являются в основном промежуточными продуктами углеводного
обмена, то тиоэфиры — это промежуточные продукты обмена жирных кис-
о
лот, метаболическая форма которых — ацильная группа (К-С-). В
природе основным тиосодержащим соединением является кофермент А
246
Глава 7. Принципы биоэнергетики клетки
(СоА, восстановленная форма: СоА-8Н, кофермент ацилирования) —
соединение аденозин-3\5'-бисфосфорной кислоты с фосфопантотенил-
аминоэтантиолом:
3\5' АБР
Пантотенат-4-фосфат
КН.
НС^ /С-
*ч
сн
N
/
Н3С ОН
3| I
ад
г ?н ?н
сн„о- р-о-р-о-сн,-с-сн-со-ын
2 I! 1
о
I
о
сн
3
I
н
н
I ГЗ'
но <р
нсы^он
I
о
I
сн,
I '
сн,
со
сн,
сн,
8Н
/
Аминоэтантиол
В реакциях образования тиоэфиров кофермента А последний
предварительно синтезируется за счет переноса энергии с молекулы АТР:
СоА-8Н + АТР <с> СоА-8~Р~Р + АМР.
Данное высокоэнергетическое соединение легко вступает в
реакцию ацилирования с жирной кислотой:
О
а а
К~СН2-СООН + Р~Р~8-СоА <* К-СН2-С-8-СоА + РР1
Жирная кислота Ацилкофермент А Пиро-
фосфат
В результате этой реакции существенно возрастает реакционная
способность как атома углерода тиоэфирной связи, так и атома
углерода, находящегося в ос-положении. Существенный вклад в
уменьшение стабильности эфирной группы вносит атом серы в связи с его
большими размерами.
При участии кофермента А протекает ряд промежуточных
реакций клеточного дыхания, биосинтеза и окисления жирных кислот.
7.5. Биохимические реакции сопряжения
Две последовательные реакции являются сопряженными, если
продукт первой реакции служит субстратом второй:
А + В -> С + В,
В + Е -> М + N.
Сопряжение осуществляется благодаря наличию общего
промежуточного продукта В.
247
Часть II. Метаболизм веществ и энергии в клетке ^ ^
Реакции сопряжения занимают центральное место в
биоэнергетике клетки• Они тесно связаны с процессом энергетического
сопряжения, благодаря которому молекулярные превращения, приводящие к
возрастанию свободной энергии системы, — эндергонические реакции,
сопрягаются во времени и пространстве с экзергоническими
реакциями — донорами энергии* Благодаря энергетическому сопряжению в
клетке реализуются такие реакции, изолированное протекание кото-
рых невозможно. Энергия лабильной химической связи в молекуле
АТР переходит в химическую энергию стабильных биологических
соединений. Реакция сопряжения составляет основу синтезов
различных соединений, в том числе биополимеров — белков, нуклеиновых
кислот, полисахаридов.
В суммарном виде перенос энергии с АТР может осуществляться
двумя путями;
1. А + В + АТР -> А - В + АВР + Р.;
2. А + В + АТР -> А - В + АМР + РР..
Здесь энергетически невыгодный процесс А + В —> А - В, имеющий
положительную АО0', сопряжен с энергетически выгодным процессом
расщепления АТР (до АВР и Р1 или до АМР и РР^, имеющим
отрицательное значение ДО^, в ходе одной реакции, протекающей в две стадии:
1. А + АТР -> [А~Р] + АВР;
2. [А~Р] + В -» А - В + Р.,
Пример: глюкоза + фруктоза + АТР -> сахароза + АВР + Р., или:
1. А + АТР -> [А-АМР] + РР.;
2. [А-АМР] + В -> А - В + АМР.
Пример: ацетат + СоА-8Н + АТР -> ацетил-8СоА + АМР + РР..
Реакции сопряжения могут протекать и до третьему (АТР-незави-
симому) пути. В таких реакциях не происходит действительного
переноса энергии, АТР в них не участвует, а сопряжение объясняется только
с помощью понятия о равновесии. Неблагоприятное равновесие одной
реакции может быть смещено, если продукт этой реакции
используется в качестве реагента в последующей реакции.
Краткое содержание главы
Биохимическая термодинамика ставит перед собой цель изучить
взаимодействие между химической и другими формами энергии,
позволяет дать ценную информацию об интенсивности и направлении
обмена веществ. Важную роль в достижении этой цели играют такие
термодинамические характеристики, как внутренняя энергия,
энтальпия, энтропия и свободная энергия. Каждая из них имеет свой смысл
и область применения.
Внутренняя энергия представляет собой полную энергию системы
{условно замкнутого пространства с совокупностью веществ, подлежа-
248
Глава 7. Принципы биоэнергетики клетки
щих изучению). Согласно первому закону термодинамики внутренняя
энергия должна быть величиной постоянной либо увеличиваться, либо
уменьшаться за счет окружающей среды. При взаимных превращениях
различных форм энергии имеет место их строгое количественное
соотношение. В химической термодинамике энтальпия представляет собой
теплосодержание системы. Она включает в себя энергию и работу системы.
Поскольку биохимические реакции протекают при постоянном
давлении и мало изменяющемся объеме системы, энтальпия в биологических
системах фактически является внутренней энергией системы.
Энтропия — термодинамическая функция, характеризующая
степень неупорядоченности системы* В замкнутой системе могут
происходить лишь те превращения, при которых энтропия либо остается
постоянной (в случае обратимых процессов), либо возрастает (в случае
необратимых процессов). Увеличение энтропии является мерой
необратимости реальных процессов в. замкнутой системе. В
структурировании биологических систем, состоящих из высокомолекулярных со-
единений, энтропия вносит ценную информацию в характеристику этих
систем. Отрицательные значения энтропии свидетельствуют о том, что
в системе возможен синтез более упорядоченных структур из менее
упорядоченных. Это компенсируется за счет увеличения энтропии
внешней среды.
Свободная энергия является частью внутренней энергии
(энтальпии), которая может быть превращена в полезную работу. С
термодинамической точки зрения поглощение пищевых продуктов
организмом человека представляет собой извлечение из среды свободной
энергии. Свободная энергия играет определяющую роль в направлении
процессов в организме, в упорядочении структур. Высокий уровень
свободной энергии присущ потенциально неустойчивым системам,
которые будут переходить на более низкий уровень этой энергии;
изменение свободной энергии (ее убыль) будет отрицательным, а
реакция называется экзергонической. Напротив, биохимические реакции,
требующие затраты энергии, характеризуются переходом веществ из
устойчивого состояния с низкой свободной энергией к менее
устойчивому состоянию с большой свободной энергией. Изменение свободной
энергии (ее приход) будет положительным. Такие реакции
называются эндергоническими.
Для большинства биохимических реакций энтальпия близка к
свободной энергии. Этим определяется калорийность пищи как
приближенной меры работы, совершаемой в результате усвоения пищи
организмом.
Чтобы вычислять изменение свободной энергии, в биохимическую
термодинамику введено понятие изменения свободной энергии в
стандартных условиях. За такие условия принимают температуру 25 °С,
рН 7,0, концентрацию растворенных веществ, соответствующую одно-
молярной активности.
249
Глава 7. Принципы биоэнергетики клетки
Паттон А. Энергетика и кинетика биохимических процессов. — М.: Мир,
1968. — 157 с.
Скулачев В. П. Аккумуляция энергии в клетке. — М.: Наука, 1969. — 440 с.
Скулачев В. П. Энергетика биологических мембран. — М.: Наука, 1989, —
341 с.
Страйер Л. Биохимия. — М.: Мир, 1985. — Т. 2. — 308 с,
Строев Е* А. Биологическая химия. — М,: Высшая школа, 1986. — 479 с.
ВОПРОСЫ И ЗАДАЧИ
1. Изменение СР реакции гидролиза глюкозо-1 -фосфата равно -21 кДж-моль"1;
изменение О0^ гидролиза глюкозо~6-фосфата равно -13,9 кДясмоль"1. Вычислите С0,
следующей реакции:
Глюкозо-1-фосфат —> глкжозо-6-фосфат.
2. Какая из реакций, приведенных ниже, может быть сопряжена с
образованием АТР из АВР и Р.:
а) 1,3-бисфосфоглицерат + Н20 -» 3-фосфоглицерат + Р.;
О0' = -47,5 кДжмоль"1;
б) фруктозо-6-фосфат + Н20 —> фруктоза + Р.;
О0' = -13,9 кДж-моль'1.
3. Почему при гидролизе глюкозо-1-фосфата О0' - -21 кДж-моль"1, а для
гидролиза глюкозо-6-фосфата — лишь -13,9 кДж'Моль"1? Дайте объяснение,
исходя из положения фосфатной группы в молекуле глюкозы.
4. Что такое энтропия и какую характеристику она дает системе (например,
процессу денатурации белка)?
Часть II. Метаболизм веществ и энергии в клетке
Важную роль в передаче химической энергии внутри клетки
играют вещества, известные под названием высокоэнергетических
соединений, при превращении которых освобождается большое количество
энергии, расходуемое на различные процессы жизнедеятельности. К
числу таких соединений относится обширный класс фосфороргани-
ческих соединений. Центральное место среди них занимает АТР -—
аденозинтрифосфат. Энергия, накопленная в АТР, используется в
огромном числе различных эндергонических процессов клетки. В
термодинамической шкале стандартных свободных энергий АТР
занимает промежуточное положение и выполняет роль посредника в
передаче энергии от высокоэнергетического соединения к
низкоэнергетическому, что играет важную роль в синтезе белков, нуклеиновых кислот,
углеводов.
К группе высокоэнергетических соединений относятся также тио-
эфиры — промежуточные продукты обмена жирных кислот. В
природе основным тиоэфирным соединением является кофермент А.
В биоэнергетике живой клетки большую роль играют реакции
сопряжения, тесно связанные с процессом энергетического сопряжения.
Благодаря этому процессу эндергонические реакции сопрягаются во
времени и пространстве с экзергоническими реакциями. Реакции
сопряжения составляют основу синтеза различных соединений, в том
числе биополимеров.
Хронология важнейших дат в исследовании биоэнергетики клетки
1936 г. — 1$. А. Энгельгард впервые показал возможность преобразования
химической энергии, аккумулированной в АТР, в механическую работу
(сокращение мышечного волокна).
1941 г- — Ф. Липман выдвинул обобщающую концепцию, согласно которой
АТР в клетках играет роль главного универсального переносчика химической
энергии.
1953 г. — Ф. Липману присуждена Нобелевская премия за открытие
функции АТР, выступающего в качестве аккумулятора и переносчика энергии.
1957 г. — А. Сент-Дьердьи ввел в науку понятие "биоэнергетика".
1964 г. — П. Митчелл предложил хемиосмотическую гипотезу
преобразования биомембранного потенциала в химическую энергию фосфатной связи.
1969 г. — В. П. Скулачев экспериментально обосновал биохимические
механизмы аккумулирования энергии в клетке.
ЛИТЕРАТУРА
Бохински Р. Современные воззрения в биохимии. — М.: Мир, 1987. — 543 с.
Гудвин 7\, Мерсер Э. Введение в биохимию растений. — М.: Мир, 1986. —
Т. 1. — 392 с.
Ленинджер А. Основы биохимии. — М.: Мир, 1985. — Т- 2. — 352 с.
Марри Р„ Греннер Д., Мейес П., Родуэлл В. Биохимия человека. — М.: Мир,
1993. — Т. 1. — 381 с.
Мецлер Д. Биохимия. — Мл Мир, 1980. — Т. 1. — 407 с,
Николе Д. Биоэнергетика. — М.: Мир, 1985. — 527 с.
250
Глава 8. ФЕРМЕНТЫ
8*1» Общая характеристика
Ферменты (от лат. /егтепШт — бродило, закваска; синоним —
энзимы) — биологические катализаторы белковой природы,
способные во много раз ускорять химические реакции, протекающие в
живом организме» но сами не входящие в состав конечных продуктов
реакций. Термин "фермент" был предложен в начале XVII в,
голландским ученым Ван-Гельмонтом. Вещества, на которые действуют,
ферменты, называются субстратами.
Ферменты — инструменты обмена веществ в живой клетке. Все
многообразие биохимических реакций, протекающих в
Микроорганизмах, растениях и животных» катализируется соответствующими
ферментами. Переваривание, всасывание и усвоение пищевых веществ,
синтез и распад белков, нуклеиновых кислот, углеводов, жиров и
других соединений в тканях и клетках любого организма, дыхание,
механическая работа обеспечиваются действием соответствующих
ферментных систем. Одной из характерных особенностей ферментов является
их способность ускорять реакции в обычных физиологических
условиях, характерных для жизнедеятельности клеток.
Действие ферментов подчиняется общим законам термодинамики.
Они катализируют только энергетически выгодные реакции и
никогда не изменяют направления реакции; они не изменяют равновесие
обратимой реакции и лишь ускоряют его наступление.
В отличие от неорганических катализаторов ферменты имеют свои
особенности. Во-первых, скорость ферментативного катализа на
несколько порядков выше (от 103 до 109), чем скорость
небиологического катализа. Во-вторых, действие каждого фермента высокоспецифично,
т.е. каждый фермент действует только на свой субстрат или группу
родственных субстратов, тогда как неорганические катализаторы
вызывают ускорение различных реакций. Высокая специфичность дает
возможность ферментам направлять обмен веществ в определенное
русло; никаких побочных продуктов в ходе ферментативного процесса
не образуется, поэтому отпадает необходимость их удалять в течение
всего процесса с целью повышения его эффективности. В-третьих,
ферменты катализируют химические реакции в мягких условиях, т.е.
при обычном давлении, невысокой температуре (20—50 °С) и при
значениях рН среды, в большинстве случаев, близких к нейтральному.
252
Глава 8. Ферменты
В настоящее время учение о ферментах — этимология является
важнейшей и стремительно развивающейся областью биохимии. Ее
достижения широко используются в новейшей биотехнологии, в
пищевой промышленности, в медицине и сельском хозяйстве. Ферменты
являются важным инструментом в изучении механизмов обмена
веществ, в расшифровке структуры химических соединений.
С точки зрения локализации ферменты подразделяют на
внеклеточные и внутриклеточные. Внеклеточные ферменты выделяются
клеткой во внешнюю среду. Многие из них синтезируются
микроорганизмами и вызывают расщепление биополимеров, продукты которых
становятся доступными для транспорта через биологические
мембраны и их дальнейших превращений внутри микробной клетки. Сюда
же относятся ферменты растений, способные мигрировать к местам
локализации субстратов и там осуществлять их деструкцию. К
внеклеточным ферментам относятся ферменты пищеварительного
тракта, а также некоторые ферменты плазмы крови.
Внутриклеточные ферменты локализованы либо в клеточных орга-
неллах, либо в комплексе с надмолекулярными структурами.
Установлено, что в ядре клетки локализованы ферменты, принимающие
участие в синтезе РНК, ДНК; с митохондриями связаны ферменты
окисления жирных кислот, продуктов распада углеводов, т.е.
ферменты, играющие важную роль в биоэнергетике клетки; в лизосомах в
основном содержатся ферменты гидролиза; с рибосомами связаны
ферменты белкового синтеза.
Особую группу ферментов составляют мультиферментные
комплексы , в состав которых входит целый ряд ферментов,
катализирующих последовательные реакции превращения какого-либо субстрата.
Эти комплексы локализованы во внутримолекулярных структурах
таким образом, что каждый фермент располагается в
непосредственной близости от фермента, катализирущего реакцию в цепи данной
последовательности реакций. Благодаря такому расположению
ферментов процесс диффузии субстрата и продуктов реакции сводится к
минимуму. Локализация мультиферментных комплексов в
определенных внутриклеточных структурах, или компартментализация,
обеспечивает протекание в клетке в одно и то же время химически
несовместимых реакций. Так, окисление жирных кислот под действием
мультиферментной системы протекает в митохондриях, а их синтез —
при участии другой системы ферментов — в цитоплазме.
Сборка мультиферментных ансамблей, объединяющих ферменты
общего метаболического пути, на клеточных структурах обеспечивает
формирование метаболонов. Метаболой представляет собой
надмолекулярный комплекс ферментов, катализирующих последовательные
стадии метаболического пути, и структурных элементов клетки.
Ферменты играют важную роль в регуляции клеточного обмена
веществ. Простейший путь регуляции — изменение скорости фермен-
253
Часть II. Метаболизм веществ и энергии в клетке
тативной реакции, осуществляющееся при вариациях рН среды,
температуры, концентрации субстрата, присутствия специфических
активаторов или ингибиторов ферментов.
Другой путь регуляции метаболизма осуществляется под
действием так называемых аллостерических ферментов. Снижение или
повышение активности таких ферментов т ул.уо обычно является след-,
ствием конформационных переходов фермента в результате
присоединения к нему активаторов или ингибиторов, роль которых выполняют
различные метаболиты. Активность ферментов может также
регулироваться путем ковалентной модификации* заключающейся в
химическом изменении структуры молекулы (например, за счет фосфори-
лирования и дефосфорилирования, метилирования, аденилирования
определенных аминокислотных остатков).
Третий путь регуляции ферментативной активности — изменение
концентрации фермента в результате стимулирования или
подавления его биосинтеза либо изменение скорости его катаболизма*
В совокупности эти механизмы регуляции представляют сложную
систему, посредством которой организм контролирует все свои
функции. Индивидуальная особенность живой клетки определяется в
основном уникальным набором ферментов, которые она
запрограммирована производить.
8.2, Структура ферментов
Химическая природа ферментов. Ферменты представляют собой
высокомолекулярные соединения. Такое определение еще не говорит о
белковой природе ферментов- На первых этапах развития энзимологии,
напротив, исследователи их относили к какому-то еще неизвестному
классу соединений биологического происхождения, а присутствие белков в
препаратах ферментов объясняли загрязнением последних. Р. Вилып-
теттер с сотруд. пытались очистить ферментные препараты от белков.
Однако Дж. Самнеру, а затем Дж. Нортропу удалось показать,
что.кристаллические ферменты уреаза и пепсин представляют собой белки и что
ферментативная активность связана со специфической структурой
макромолекулы белка. Это было поистине выдающееся открытие,
подчеркнувшее огромную биологическую роль белков в живой материи.
В 80-х гг. XX выбыли обнаружены катализаторы, лишенные белка
и состоящие только из РНК. В ходе процесса модификации структуры
молекул РНК, а именно в ходе сплайсинга, сами молекулы РНК
выполняют функции катализаторов расщепления рибонуклеотидных
цепей, т.е. по сути дела проявляют ферментативную активность.
Построенные из РНК ферменты называют рибозимами. Однако около
трех тысяч открытых и в той или иной степени изученных к
Настоящему времени ферментов имеют белковую природу.
Как и другие белки, ферменты имеют первичный, вторичный,
третичный и часто четвертичный уровни структурной организации; им при-
254
Глава 8. Ферменты
сущи все физико-химические свойства белков и лишь одна
отличительная особенность — это способность ускорять химические реакции.
Ферменты могут быть как простыми — однокомпонентными, так
и сложными — двухкомпоиентными. Первые построены из
полипептидных цепей и при гидролизе распадаются только на аминокислоты;
вторые состоят из белковой части — апофермента и небелковой
части — кофактора. Оба компонента в отдельности лишены
ферментативной активности. При соединении этих компонентов образуется хо-
лофермент, обладающий каталитической активностью. Роль
кофактора выполняет какой-либо ион или органическое соединение. В
молекулу апофермента чаще всего входят двухвалентные ионы,
например 2п2+, М§2+, Ге2+, Си2+, реже — одновалентные К* и ^+« К
органическим кофакторам принадлежит примерно десяток соединений
различной структуры. Кофакторы органической природы
называются коферментами. Предшественниками большей части кофермен-
тов являются витамины. Тип связи между кофактором и апофер-
ментом может быть различным. В некоторых случаях они
существуют отдельно и связываются друг с другом только во время
протекания реакции; в других случаях кофактор и апофермент
связаны постоянно, иногда прочно, ковалентными связями. В последнем
случае говорят о наличии простетической группы в составе
фермента. Роль кофактора сводится либо к изменению трехмерной
структуры белка, способствующей лучшему связыванию фермента с
субстратом, либо к непосредственному участию в реакции в качестве
еще одного субстрата. Именно коферменты, как правило,
выступают в качестве дополнительных субстратов. Они обычно играют роль
промежуточных переносчиков электронов, а также некоторых
атомов или химических групп, которые в результате ферментативной
реакции переносятся с одного соединения — донора на другое —
акцептор.
Как правило, апофермент обладает гораздо меньшей
стабильностью, чем холофермент. Очевидно, что кофермент оказывает защитное
действие по отношению к апоферменту, пока они соединены друг с
другом, выполняя функцию "скрепки", которая не позволяет
пептидным цепям апофермента развертываться и тем самым препятствует
его денатурации.
Ионы металлов, входящие в состав ферментов, выступают в роли
координационных центров комплекса, в котором с металлом связаны
и апофермент, и субстрат. В результате образования такого комплекса
субстрат тесно соединяется с апоферментом, что облегчает
каталитическое действие. В некоторых ферментах ионы металлов выполняют и
другую роль. Так, в амилазах бактерий, растений, животных
содержится от 1 до 4 атомов кальция на молекулу фермента. Кальций
стабилизирует амилазы от кислотной, термической денатурации
(инактивации), от действия протеаз — ферментов, гидролизирующих бел-
255
Часть II. Метаболизм веществ и энергии в клетке
ки; этот металл выполняет важную функцию в создании третичной
структуры белка.
Активный центр ферментов. Ферментативный катализ
^происходит на расстоянии длины химической связи, поэтому вполне понятно,
что акт катализа должен совершаться на определенном участке белковой
глобулы. Этот локальный участок и носит название активного центра
фермента. В однокомпонентных ферментах активный центр образуется в
результате определенной ориентации аминокислотных остаткор
полипептидной цепи. Обычно в формировании этого центра принимает участие
небольшое количество аминокислот, в пределах 12—16.
Функциональные группы этих аминокислот могут принадлежать звеньям
полипептидной цепи, весьма удаленным друг от друга. Их сближение связано с
формированием третичной структуры фермента. В двухкомпонентных
ферментах активный центр представляет собой комплекс кофактора и
некоторых примыкающих к нему аминокислотных остатков. Ион
металла может либо находиться в активном центре апофермента и быть
непосредственно связанным с какой-либо функциональной группой
аминокислотного остатка, либо входить в состав простетической группы
(например, гема в цитохромах). В первом случае он способствует
формированию каталитически активной конформации апофермента; стабилизация
белковой молекулы возможна путем образования солевых мостиков между
ионами металла и карбоксильными группами кислых аминокислот при
формировании третичной структуры молекулы фермента. Во втором
случае он служит мостиком между алоферментом и органической
молекулой кофактора.
Функциональные группы активного центра ферментов. В
формировании активного центра ферментов важную роль играют:
СООН-группы дикарбоновых аминокислот или концевые группы
полипептидной цепи; имидазольная группа гистидина, 8Н-группа цис-
теина, ОН-группы серина, тирозина, треонина, е->Ш2-группа лизина и
концевые группы полипептидной цепи, гуанидиновая группа
аргинина, фенольная группа тирозина, а также гидрофобные остатки
алифатических аминокислот и ароматическое кольцо фенилаланина.
8.3. О механизме ферментативного катализа
Энергия активации. Как уже отмечалось в гл. 7, протекание
химической реакции возможно в том случае, если свободная энергия
системы АО отрицательна. Однако этот факт еще не свидетельствует о
том, какова будет скорость этой реакции. Скорость любой химической
реакции определяется энергетическим барьером, который
необходимо преодолеть реагирующим молекулам, причем высота этого барьера
неодинакова для разных реакций. По Аррениусу, химическая
реакция с точки зрения энергетики процесса описывается уравнением
Е,
'акт
256
Глава 8. Ферменты
где N — число активированных молекул; ЗМ^ — общее число
реагирующих молекул; е — основание натурального логарифма; К — газовая
постоянная; Т — абсолютная температура; Еакт (энергия активации) —
количество энергии, необходимое для того, чтобы все молекулы 1 моля
вещества при определенной температуре достигли переходного
состояния, соответствующего вершине энергетического барьера реакции.
Эта энергия представляет собой разность общей энергии реагирующих
молекул и энергии возбужденного переходного состояния. Чем
больше энергия активации в реагирующей системе, тем выше
энергетический барьер, тем ниже скорость (эффективность) реакции* Важнейшая
функция фермента — это снижение энергии активации
катализируемого процесса. На рис* 8.1 представлен график изменения уровня
энергии неферментативной и ферментативной реакций. Фермент снижает
высоту энергетического барьера (Е < Е' ).
а
Активированное
состояние
(А=В>
+
Исходное
состояние
Конечное
состояние
Е'
акт
* *^акт у
Продукты
реакции
®® (т)
да
::
Ход реакции
Рис, 8Л. Энергетическая схема нефермеятативной (1) и ферментативной (2)
реакций; ▼ — фермент
Современные представления о молекулярном механизме
ферментативного катализа. Несмотря на большое число исследований
по изучению механизма ферментативного катализа, данная научная
проблема во многом остается пока не решенной * Большую роль в этом
отношении сыграли работы М. Михаэлиса и М. Ментен, в которых
было развито представление о фермент-субстратном комплексе.
Образование этого комплекса и ведет к снижению энергии активации
реакции.
Процесс ферментативного катализа можно условно подразделить
на три стадии:
1. Стерическое связывание субстрата 8 с активным центром
фермента Е (образование фермент-субстратного комплекса Е8).
17. Заказ 3486
257
Часть II. Метаболизм веществ и энергии в клетке
2. Преобразование первичного комплекса Е8 в активированный
переходный комплекс Е8**
3. Отделение конечного продукта Р реакции от фермента.
Первая стадия непродолжительна по времени и зависит от
концентрации субстрата и фермента в среде, от скорости диффузии субстрата к
активному центру фермента. В образовании комплекса Е8 могут
участвовать в различных сочетаниях как ковалентные, координационные,
ионные связи, так и менее прочные формы связей — электростатическое
притяжение полярных групп, ван-дер-ваальсовы силы сцепления между
неполярными участками молекул, водородные связи. Характер этих
связей обусловлен химическими особенностями и субстрата, и
функциональных групп, входящих в активный центр фермента.
Вторая стадия является, собственно, актом катализа, т.е. актом
разрыва или образования в субстрате новых связей; она наиболее медленная
и лимитирует скорость протекания химической реакции. На этой стадии
и происходит снижение энергии активации. В расшифровке этой стадии
наиболее широкое признание получили следующие теории: теория
сближения и ориентации, теория деформации субстрата (теория "дыбы")»
теория индуцированного соответствия, теория кислотно-основного
катализа и теория ковалентного катализа. Однако все теории в
конечном счете сводятся к одной концепции — к образованию активного
переходного комплекса Е8*, который и обусловливает снижение энергии
активации ферментативной реакции.
Считают, что важнейшими факторами, определяющими способность
ферментов ускорять биохимические реакции, являются сближение и
ориентация фермента и субстрата. Фермент связывает молекулу
субстрата таким образом, что определенная связь в молекуле субстрата,
на которую нацелено действие, оказывается не только расположенной
в непосредственной близости от каталитической группы активного
центра, но и правильно ориентированной по отношению к ней.
В соответствии с теорией деформации после связывания субстрата
с активным центром молекула субстрата переходит в "напряженную",
"деформированную" форму, в которой изменяются длины и
валентные углы межатомных связей. Чем больше длина межатомной связи
и изменение ее валентного угла, тем меньше требуется энергии для ее
разрыва. Место деформации легко атакуется другим субстратом,
например, молекулой воды.
По теории индуцированного соответствия, при взаимодействии
фермента с субстратом происходят конформационные изменения
молекулы фермента, приводящие к напряжению структуры активного
центра, а также в определенной степени деформируется связанный
субстрат. Это облегчает достижение фермент-субстратным
комплексом переходного состояния.
На молекулярном уровне более четкое представление о механизме
действия ферментов дает теория кислотно-основного катализа. Лю-
258
Глава 8. Ферменты
бая реакция, идущая с разрывом ковалентных связей, предполагает
участие двух противоположных по характеру электронных
компонентов. Электроны разрываемой связи должны оттягиваться к электро-
фильному компоненту и уходить от нуклеофильного. Реагенты,
которые могли бы обусловить такую электронную перестройку — это
кислота и основание. Однако в одном и том же растворе создать
одновременно высокие концентрации обоих компонентов невозможно,
поскольку они нейтрализуют друг друга. В белковой молекуле фермента
благодаря закреплению на каталитической площадке электрофильных и
нуклеофильных групп не происходит прямой реакции нейтрализации.
Это, собственно, и определяет акт катализа. Находясь на
определенном расстоянии друг от друга, электрофильные и нуклеофильные
группы каталитического участка фермента не только связываются с
реагирующими группами субстрата, но и оказывают сильное
поляризующее действие на группы субстрата. К этому следует добавить
возможность флуктуации зарядов в комплексе Е8, которая создает высокую
степень эффективности данной поляризации. Это и является причиной
снижения величины энергии активации при ферментативном катализе.
На рис. 8.2 представлен предполагаемый механизм расщепления
а-1,4-глюкозидных связей в молекуле крахмала под действием амилаз.
- В активном центре этих ферментов находится отрицательно
заряженная карбоксильная группа и положительно заряженная имида-
зольная группа, которые образуют мощную нуклеофильно-электрофиль-
ную систему — карбоксилат-имидазолий.
Атом кислорода обладает большим отрицательным индукционным
эффектом, чем атом углерода. В связи с этим атом кислорода
а-1,4-глюкозидной связи будет иметь большую плотность
электронного облака, чем атом С-1. Снижение плотности электронного облака у
атома С-1 связано также с индукционным воздействием атома
кислорода глкжозидного кольца. Видимо, это и является причиной элект-
рофильно-нуклеофильной атаки связи С2-0 в молекуле крахмала
системой карбоксилат-имидазолий. Штриховые линии изображают
соединение фермента с субстратом, ведущее к перераспределению
электронов в фермент-субстратном комплексе и к исчезновению перекрытия
электронных орбит между Сх и О. Снижение энергии активации в дан-
махамм/гг//'
Г
5"
Н'ьИм
N
16-
СОО"
УУ^УУУУУУУУУ^УА/
НтИм
—о
У
С1
•о х
1
1
СОСГ
ууууууууууууууууу
Им
-о
с
но
\л
У
СОСГ
4
7777777777777777?
х
77777777777777777
а
77?//7/////////77
Н"Им
г*
но18н * |
о18н
но
А
у
СОСГ
4
77777777777777777
Рис. 8.2. Предполагаемый механизм расщепления а-1,4-гликозидных связей
в молекуле крахмала под действием амилаз (Им — имидазол)
17
А
259
Часть II. Метаболизм веществ и энергии в клетке ___
ном случае есть следствие атаки системой карбоксилат-имидазолий
связи Сх-0. Вопрос о том, что расщеплению подвергается связь С^-О,
а не 0-С4, был решен при гидролизе крахмала в Н2018.
В активных центрах ферментов, как правило» находятся
функциональные группы специфических аминокислотных остатков,
являющиеся хорошими донорами или акцепторами протонов (например, ими-
дазольная группа гистидина, карбоксильные группы аспарагиновой и
глутаминовой кислот и др.)»
В соответствии с теорией ковалентного катализа некоторые
ферменты взаимодействуют со своими субстратами, образуя нестабильные,
ковалентно связанные фермент-субстратные комплексы. Из этих
комплексов в ходе последующей реакции образуются продукты реакции,
причем значительно быстрее, чем в случае некатализируемых реакций.
Таким образом, третья стадия, завершающаяся образованием
продуктов реакции, обеспечивается процессами, протекающими на
предыдущих стадиях.
Субстратная специфичность действия ферментов. Важным
свойством ферментов является способность не только ускорять
реакцию, но и избирательно действовать на субстрат и определять путь
его превращения. Это свойство ферментов получило название
субстратной специфичности ферментов. Каждый фермент действует только на
свой определенный субстрат или группу субстратов.
По признаку специфичности действия все ферменты можно
разделить на две группы: ферменты, обладающие абсолютной
специфичностью, и ферменты, обладающие относительной специфичностью.
Абсолютная специфичность проявляется тогда, когда фермент действует
лишь на одно-единственное вещество и катализирует только
определенное превращение этого вещества. Например, уреаза катализирует
только одну реакцию — гидролиз мочевины; сукцинатдегидрогеназа
действует только на янтарную кислоту, окисляя ее в фумаровую кислоту. К
абсолютной специфичности относится также стереохимическая
специфичность; фермент действует только на один из оптических изомеров
(О- или Ь-изомер, цис- или транс-форму молекулы субстрата). Так, лак-
татдегидрогеназа специфична к Ь-лактату, глутаматдегидрогеназа — к
Ь-глутамату; фумаратгидратаза катализирует превращение фумаровой
кислоты (г^с-формы), присоединяя к ней молекулу воды, а на ее стерео-
изомер — малеиновую кислоту (тракс-форму) — она не действует.
Ферменты, имеющие относительную или групповую специфичность,
действуют сразу на многие субстраты, обладающие рядом общих
структурных свойств. Для этих ферментов важны тип связи и химическая
структура лишь одного из компонентов, образующих эту связь, тогда
как природа другого компонента данной связи может изменяться. К
таким ферментам относятся фосфатазы, гидролизующие различные
эфиры фосфорной кислоты, протеиназы, такие как трипсин, химо-
трипсин, расщепляющие пептидные связи в белках.
260
Глава 8, Ферменты
Существуют две точки зрения, объясняющие специфичность
действия ферментов. Одна из них развита Э. Фишером. По его образному
выражению, "фермент подходит к субстрату так, как ключ к замку".
В соответствии с этой гипотезой топография активного центра не только
высокоупорядочена, но и жестко закреплена. Активный центр
фермента соответствует топографии одного-единственного субстрата,
поэтому он и может подвергаться действию активного центра.
Гипотеза "ключа и замка" вполне удовлетворительно объясняет
абсолютную специфичность. Однако с позиций жесткой структуры
трудно объяснить относительную специфичность, поскольку в этом
случае группа субстратов имеет определенные отличительные черты в
своей структуре.
Вторая точка зрения, предложенная Д. Кошландом (теория
индуцированного соответствия фермента и субстрата), устраняет это
противоречие. В данном случае используется представление о гибкой
структуре молекулы фермента. Конформация фермента, в особенности
ориентация его активного центра, должна быть способной к определенным
модификациям. В зависимости от конформационной подвижности активного
центра фермент способен взаимодействовать либо с немногими, либо с
самыми разными субстратами. Иными словами, неодинаковая
специфичность разных ферментов является следствием малых или больших кон-
формационных перестроек активного центра: если она ограничена —
фермент высокоспецифичен; если конформационные переходы
активного центра широки, то фермент обладает и более широким спектром
действия на структурные аналоги субстратов.
Естественно, для ферментов весьма важно наличие у них
упорядоченной структуры, комплементарной конфигурации их субстратов. Однако
структура глобулярных белков не жесткая; молекулярно-динамические
исследования указывают, что атомы в белках совершают небольшие и
быстрые колебания. Более того, сравнение трехмерной структуры
ферментов со структурой комплексов "фермент - лиганд" показывает, что
при связывании лиганда происходят определенные конформационные
изменения. Некоторые ферменты в действительности испытывают
значительные изменения структуры при связывании молекулы субстрата.
При этом они переходят из неактивной конформации в активную.
Д. Кошланд предположил, что гибкость конформации ферментов
взаимосвязана с их каталитической активностью и субстратной
специфичностью. Его предположение о существовании явления
индуцированного соответствия было основано на кинетических
экспериментах с гексокиназой.
Гексокиназа катализирует фосфорилирование глюкозы с помощью
АТР:
Глюкоза + АТР %± Глюкозо-6-фосфат + АВР.
Вода (НОН), которая имеет сходство с группой при С-6 атоме
глюкозы (КОН), в некоторой степени соответствует активному центру и,
261
Часть П. Метаболизм веществ и энергии в клетке __^_
следовательно, должна бы быть подходящим субстратом для
гексокиназы. Однако гидролиз АТР протекает медленнее, чем фосфорилиро-
вание глюкозы, в 40 000 раз. Добавление пятиуглеродного сахара лик-
созы, который не имеет гидроксиметильной группы, аналогичной
группе при С-6 атоме глюкозы, и в этой связи не может подвергаться фос-
форилированию, увеличивает скорость гидролиза АТР в 20 раз. Этот
эксперимент подтверждает, что присутствие специфического
субстрата в активном центре гексокиназы воздействует на каталитические
группы, делая фермент активным. Рентгеноструктурные
кристаллографические исследования показывают, что гексокиназа существует в
двух конформациях: в открытой форме, когда глюкоза отсутствует, и
в закрытой форме, когда глюкоза связана в активном центре (рис. 8.3).
Связывание такого лиганда, как глюкоза, изменяет угол между двумя
доменами гексокиназы до 12° и закрывает щель при образовании
комплекса "фермент — глюкоза". Такой тип движения двух доменов
относительно друг друга может быть назван "затягиванием петли".
Таким образом, особенности структуры гексокиназного активного
центра предотвращают ненужный, расточительный гидролиз АТР; в этом
случае индуцированное соответствие имеет значение для сохранения
клеточной энергии* Ряд других киназ также функционирует в
соответствии с механизмом индуцированного соответствия. Так, рентгено-
структурный кристаллографический анализ показал, что конформа-
ционное изменение аденилаткиназы при переходе ее из открытой в
закрытую конформацию осуществляется в два этапа, сопряженные со
связыванием каждого из двух субстратов.
Рис. 8.3. Образование фермент-субстратного комплекса при
функционировании гексокиназы: а — "открытая" форма фермента; б — "закрытая" форма
фермента
Более стабильная форма гексокиназы —» это неактивная форма Ег
Связывание "хорошего" субстрата, такого как глюкоза, но не
"плохого" субстрата, такого как вода, способствует переходу фермента в
активный фермент-субстратный комплекс Б 8, как показано на
следующей схеме:
Е1 + 8 ^ ЕуЗ ?=* В 8 ^ Е8*
262
Глава 8. Ферменты
Поскольку Еа имеет менее стабильную конформацию, чем Ер необ^
ходима энергия для поддержания Еа-состояния. Энергия,
используемая для связывания 8 при образовании Еа8, — это энергия, которая
не может быть использована для катализа. Следовательно, фермент,
функционирующий по механизму индуцированного соответствия,
будет менее эффективен, чем гипотетический фермент, находящийся
всегда в активной конформации. Разницу эффективности отражает
отношение [Еа] к [Е^, которое для гексокиназы составляет 1:40 000.
В представлениях о механизме катализа и строении
фермент-субстратных комплексов придается большое значение впадинам, расщелинам,
полостям на поверхности белковой глобулы. Представление об активной
полости занимает центральное место в теории Кошланда. В эту полость,
по Кошланду, втягивается реагирующее вещество- Возникает строгая
адаптация субстрата и фермента и, следовательно, синхронное
взаимодействие каталитических групп с молекулой субстрата.
Важной особенностью теории индуцированного соответствия
является разделение способности фермента связывать молекулу субстрата, быть
специфичным к ней, и способности катализировать превращение субстрата.
Следствием различных механизмов связывания субстратов с
ферментами могут являться и различные продукты химической реакции.
Типичным примером этому могут быть амилазы, гидролизующие
крахмал; действуя на один и тот же субстрат, они образуют различные
продукты гидролиза: глюкоамилаза — глюкозу, (5-амилаза —
мальтозу; а-амилаза — декстрины. Теория индуцированного соответствия в
сочетании с гипотезой активной полости дает вполне
удовлетворительное объяснение такой особенности специфичности катализа.
Образование глюкозы при гидролизе крахмала глюкоамилазой можно
объяснить возникновением в глобуле фермента активной полости,
способной вместить в себя лишь один глюкозидный остаток (рис. 8.4, а).
Аналогичную картину можно представить для р-амилазы, с той лишь
разницей, что в белковой глобуле возникает полость в размерах,
необходимых для вмещения двух глюкозидных остатков (рис. 8.4, б).
Важной контактной группой для обеих форм амилаз является Е-группа,
способная связываться с нередуцирующим концом молекулы крахмала.
Однако глюкоамилаза осуществляет полный гидролиз крахмала,
расщепляя как а-1,4-, так и а-1,6-глюкозидные связи, в то время как
(3-амилаза действует лишь на а-1,4- глюкозидные связи. Как видно из
рис* 8.4, а, для активной полости глюкоамилазы фактически
безразлично, какой глюкозидный остаток может быть втянут в нее: остаток,
связанный с С-4 или С-б-атомом соседнего звена. Важно лишь, чтобы
связь С^-0 была комплементарна системе карбоксилат-имидазолий.
Иное для р-амилазы. Поскольку ее активная полость способна
вмещать два глюкозидных остатка, то ответвление в виде связи 0-Сб
является, вероятно, серьезным стерическим препятствием для сближения
системы карбоксилат-имидазолий со связью 0,-4) в молекуле крахмала.
263
Часть II. Метаболизм веществ и энергии в клетке
////////А
6сн2 -9
ъ
он \
7.
Ск^Х^™^
ш//Ш1Ш/тш/штщхс
ф
ш/ш//1//и
ОоОЫЗс
7777777777777777777777/ ^777777777777
А
НО 4к
в
Рис. 8.4. Специфичность действия глкжоамилазы (а), р-амилазы (б) и сс-ами-
лазы (в) на молекулу крахмала; I — отрицательно заряженая карбоксильная
группа; II — положительно заряженная имидазольная группа; III — контактные
группы К
Аналогичным образом можно объяснить образование декстринов
при гидролизе крахмала а-амилазой, если представить активный центр
этого фермента в виде "активной щели", не содержащей контактной
группы К (рис. 8.4, в).
Протеолитические ферменты трипсин, химотрипсин, эластаза
расщепляют в белковых молекулах пептидные связи. Однако действие каждого
из них строго специфично: трипсин гидролизует пептидные связи,
образованные лизином или аргинином; химотрипсин расщепляет
полипептидную цепь по фенилаланину, триптофану, тирозину; специфичность
эластазы проявляется в ее действии на такие небольшие гидрофобные
молекулы, как аланин. Установлено, что разница в специфичности
действия этих ферментов обусловлена небольшими изменениями в строении
"кармана", связывающего боковую цепь аминокислоты.
Субстратная специфичность — уникальная особенность ферментов.
Она является основой направленности биохимических процессов в
264
Глава 8. Ферменты
клетке в строго определенном русле; без нее немыслима стройная
гармония обмена веществ в организме.
Специфичность действия ферментов имеет и большое прикладное
значение. Она позволяет получать определенные продукты высокой
степени чистоты и с высоким количественным выходом.
*
8.4. Кинетика ферментативных реакций
Химическая кинетика (общие понятия). Химическая
кинетика — это раздел химии, изучающий закономерности изменения
скоростей химических реакций. Кинетические исследования лежат в
основе математического моделирования механизма реакции. Они дают
возможность получить информацию о превращении веществ на
каждой стадии, о лимитирующей (самой медленной) стадии,
определяющей скорость реакции, о влиянии факторов среды на эту скорость.
Химические реакции зависят прежде всего от числа молекул,
которые реагируют между собой с образованием продуктов реакции. С
этой точки зрения они подразделяются на мономолекулярные,
бимолекулярные и тримолекулярные реакции. Молекулярность реакций
имеет прямое отношение к механизму реакции и характеризуется
числом молекул, участвующих в элементарном акте реакции. Если
реакция А^В означает, что одна молекула преобразуется по
единственному пути, то это будет мономолекулярная реакция. Если для
образования продуктов реакции реагируют две молекулы (А+В -ч»С+Д),
то такую реакцию называют бимолекулярной.
В химической кинетике введено также понятие о порядке реакций,
которым характеризуют изменение скорости реакции. Порядок
реакции — это число, равное сумме показателей степени концентрации
реагирующих веществ в уравнении скорости реакции. Различают
реакции первого, второго, третьего, нулевого и дробного порядков. Так,
если скорость реакции описывается уравнением
V = к[А],
где V — скорость реакции; [А] — концентрация вещества А; к —
константа пропорциональности, показатель степени [А] равен единице,
следовательно, реакция будет первого порядка. Если
V = к[А]2,
то показатель степени [А] равен 2, следовательно, это реакция второго
порядка. Если
V « к[А] [В],
то сумма показателей степени [А] и [В] равна 2, это будет реакция
второго порядка. Наконец, если
V - к[А][В]2[С]3,
то данная реакция (в действительности невероятная) имеет шестой
порядок. Последнее уравнение можно также интерпретировать как
265
Часть П> Метаболизм веществ и энергии в клетке ____
уравнение первого порядка по веществу А, второго порядка — по
веществу В и третьего — по С.
Реакция, протекающая с постоянной скоростью и не зависящая от
концентрации реагирующих веществ, получила название реакции
нулевого порядка.
На первый взгляд, порядок реакции должен соответствовать ее мо-
лекулярности, т.е. мономолекулярная реакция должна бы быть
реакцией первого порядка, а бимолекулярная — реакцией второго
порядка. Однако во многих случаях кинетический порядок реакции,
определяемый экспериментально, не соответствует ее молекулярности.
Общие принципы химической кинетики применимы и к
ферментативным реакциям. Однако у последних имеется одна отличительная
особенность — образование комплекса Е8, насыщение фермента
субстратом. Это явление оказывает существенное влияние на
кинетические закономерности ферментативного катализа.
Использование эффекта насыщения привело Л. Михаэлиса и
М. Ментен в 1913 г. к созданию общей теории действия ферментов и
ферментативной кинетики, расширенной затем в работах Дж. Бриггса
и Дж. Холдейна. Было установлено, что скорость ферментативной
реакции зависит от химической природы и условий взаимодействия
фермента и субстрата и физико-химических факторов среды (температу-
Л
ры, величины рН, природы веществ, находящихся вместе с
ферментом в растворе). Скорость ферментативной реакции является мерой
каталитической активности фермента и обозначается просто как
"активность фермента". Измерить активность фермента можно либо по
количеству превращаемого субстрата, либо по нарастанию
концентрации продукта реакции в единицу времени.
Влияние концентрации фермента на скорость реакции.
Зависимость скорости ферментативной реакции V от концентрации
фермента [Е] описывается простым уравнением
V = к[Е] ,
где [Е] — концентрация фермента; к — константа
пропорциональности. Графически это уравнение представляет собой прямую линию
(рис. 8.5). Однако такая прямо пропорциональная зависимость имеет
место в том случае, когда концентрация субстрата [8] находится в
избытке по отношению к концентрации фермента [Е]. Из рис. 8.5
видно, что Ъ% а = к; при прочих равных условиях величина к зависит от
химической природы фермента и субстрата.
Влияние концентрации субстрата на скорость ферментатив-
ной реакции. Исходя из представлений Л. Михаэлиса и М* Ментен,
любую ферментативную реакцию можно описать уравнением
к,
Е + 8 ^ Е8 + Р,
266
Глава 8. Ферменты
где кх и к2 — константы скорости у
образования и распада комплекса Е8
соответственно; Р — продукты
реакции,
С позиций кинетики
ферментативную реакцию можно разбить на три
стадии:
1. Развитие реакции (йу/йх > 0),
где V — скорость реакции; х — ее
продолжительность. Предполагается, что
эта стадия протекает быстро и
обратимо и не сопровождается какими-
[Е]
Рис. 8.5. Зависимость скорости
ферментативной реакции от
концентрации фермента
либо химическими изменениями.
2. Стационарная стадия, в которой скорость исчезновения
субстрата равна скорости образования продуктов реакции и концентрация
комплекса Е8 постоянна (с1у/ск = 0). Стационарное состояние можно
определить как период, во время которого образуется продукт
реакции и концентрация всех промежуточных продуктов остается
постоянной. Стационарная стадия является основной, в ней протекают
химические процессы, ее продолжительность зависит от концентрации
субстрата.
3. Спад реакции (йу/оЧ < 0).
Выведем уравнение зависимости скорости реакции V от
концентрации субстрата [8] для основной стационарной стадии ферментативной
реакции. Обозначим: [Е0] — исходная концентрация фермента, тогда
([Е0] - [Е8]) - концентрация свободного фермента. Согласно закону
действия масс скорость образования комплекса Е8 описывается уравнением
а[Е8]
их
= к1([Е0]-[Е8])[8].
Аналогично скорость распада комплекса Е8 описывается
уравнением
а[Е8]
о!т
= к2[Е8].
При йу/оЧ = 0 ([Е8] = сопз!;) будем иметь равенство
кДЕ^ - [Е8])[8] = к2[Е8],
ИЛИ
([Е0]~[Е8])[8]==^-[Е8].
Обозначим к2/к1 - Кд; К^ — константа диссоциации комплекса Е8;
она представляет собой обратную величину химического сродства
фермента к субстрату: чем меньше К^, тем больше [Е8]; [Е8] — величина,
трудно определяемая экспериментально, от нее необходимо избавиться:
267
Часть П. Метаболизм веществ и энергии в клетке
[Е8]= [Е°][8]
К8 + [8]
Скорость ферментативной реакции V в общем случае определя
ется величиной [Е8]; если же в частном случае все молекулы фер
мента находятся в комплексе фермент—субстрат, то скорость реак
ции будет максимальной ("\гтах) и определяется величиной [Е0]; еле
довательно:
V [Е8] __ [Е8]
^ [Во] — [Во]
Подставив значение [Е8] в это уравнение, будем иметь:
у= Vтаx[8] .
К8 + [8]
Это уравнение получило название уравнения Михаэлиса—Ментен
в честь авторов, предложивших его и впервые разработавших
теоретические основы кинетики ферментативного катализа.
Л. Михаэлис и М. Ментен .показали, что их теоретические
предпосылки находятся в полном соответствии с экспериментальными
данными. Так, они установили величину К3 для реакции гидролиза
сахарозы инвертазои дрожжей в опытах, где количество субстрата
было одинаковым и менялось только количество взятого фермента:
Количество инвертазы (относительные единицы) 2,0 1,5 1,0 0,5
К8 ' 0,0167 0,0167 0,0167 0,0167
Как видно, величина К5 во всех случаях была одной и той же, что и
подтверждает правильность полученного уравнения.
Как меняется скорость реакции в зависимости от [8]? Для анализа
уравнения Михаэлиса—Ментен возьмем три случая:
1. [8] » К8; тогда величиной К8 можно пренебречь и V = V .
Иными словами, пока концентрация субстрата велика, скорость реакции
близка к максимальной и практически мало изменяется до тех пор,
пока уменьшение концентрации субстрата не будет значительным.
Реакция будет иметь нулевой порядок (п = 0).
2. [8] « К8; тогда практически скорость реакции будет
описываться уравнением
V
где Утах и Кд — кинетические константы и, следовательно, данное
уравнение — уравнение прямой линии; скорость ферментативной
реакции пропорциональна концентрации субстрата. Это типичная
реакция первого порядка (п = 1);
268
Глава 8. Ферменты
шах
3. Величина [8] — промежуточная; пусть [8] = К8, тогда V = 1/2У
В данном случае порядок реакции будет промежуточным между
нулевым и первым, т.е. будет иметь дробный характер (0 < п < 1).
Графически уравнение Михаэлиса—Ментен будет выражаться
гиперболой — кривой, асимптотически приближающейся к
горизонтальной оси (см. рис. 8.6).
В начальный момент
ферментативной реакции при высокой
[8] п = 0, а V « Утах (верхний
участок кривой). При уменьшении
[8] реакция имеет сначала
дробный порядок (0 < п < 1) (на
графике изгиб линий), а затем при
низких [8] она становится
реакцией первого порядка (п=1);
наклон кривой при п = 1 равен
*еа =
У
_ *тах
к,
. Для разных фермен-
[8]
Рис. 8.6. Зависимость скорости
ферментативной реакции от концентрации
субстрата
тов кривые будут различными в
зависимости от значения V _ и К«.
тах о
Таким образом, в
ферментативных реакциях порядок
реакции меняется в ходе самой реакции, В неферментативных
химических реакциях порядок реакции от ее начала до конца практически не
меняется. В этом состоит принципиальное отличие кинетики
ферментативных реакций от обычных химических реакций.
Уравнение Михаэлиса—Ментен справедливо для начала
ферментативной реакции, когда в реагирующей системе весьма мало продуктов
реакции. Оно не учитывает того факта, что распад комплекса Е8
происходит не только в сторону образования Е и 8, но и в сторону
образования ЕиР. Дж. Бриггс и Дж. Холдейн модифицировали уравнение
Михаэлиса—Ментен, они исходили из следующего уравнения
ферментативной реакции;
В нем авторы учли распад комплекса Е8 на Е и Р, введя в
уравнение константу скорости распада кг Проведя аналогичные
преобразования уравнения Михаэлиса—Ментен, они получили
модифицированное уравнение
V =
Ушах [5]
КМ + [3]
Величину К^ они назвали константой Михаэлиса; она не является
истинной константой равновесия, а равняется отношению (к2+к3)/к1.
Так как К8=к2/к1, то Км=Кд+к3/к1.
269
Часть II. Метаболизм веществ и энергии в клетке
Характеристика кинетических констант Км и Утах. Для
характеристики любого фермента важное значение, имеет Км. Она легко
воспроизводится и не зависит от концентрации фермента.
Физический смысл Км можно выяснить из уравнения Михаэлиса—Ментен. В
частном случае, когда у = 1/2 Vтаx, после соответствующего
преобразования этого уравнения мы получим Км = [8], Таким образом, Км
равна концентрации субстрата, при которой скорость реакции
составляет половину максимальной; Км имеет размерность моль*л"1.
Для ферментов, имеющих относительную специфичность,
величина Км может изменяться в зависимости от структуры субстрата; она
также изменяется от значения рН и температуры. Поэтому эта
кинетическая константа может дать ценную информацию в исследовании
механизма ферментативного катализа, дает возможность оценить
действие определенного фермента по отношению к данному субстрату.
Вторая кинетическая константа V" ^ довольно четко выражает
ШиЛ
эффективность действия фермента. Для сопоставления активность
различных ферментов чаще всего выражают через молярную
активность или число оборотов фермента. Она представляет собой число
молей субстрата или в случае субстрата-биополимера — число
разрываемых (синтезируемых) связей, реагирующих с одним молем
фермента в единицу времени (обычно за 1 мин или 1 с). В табл. 8*2
приведены величины V, для некоторых ферментов.
111 Й_Х
Таблица 8.2
Значения V ферментов
тах
Фермент
Алкогольдегидрогеназа
а-Амилаза солода
Фосфорилаза картофеля
Изомераза фосфотриоз
Карбоангидраза
Катализируемая реакция
Уксусный альдегид —» этанол
Гидролиз крахмала
Фосфоролиз крахмала
Фосфоглицеринальдегид о
дигидроксиацетонфосфат
н2со3 <* со2+н2о
Число оборотов в мин
4700
16000
40 000
500 000
36 000 000
Молярная активность ферментов весьма велика; ферменты в сотни и
тысячи раз активнее неорганических катализаторов. У разных
ферментов У1ШХ варьирует в широких пределах. Кроме того, как и в случае с Км,
~У"тах зависит от структуры субстрата, величины рН и температуры.
Графоаналитический метод определения Км и Утах. На кривой
(рис. 8.6), построенной в координатах V и [8] , мы видим, что V ,
является асимптотической величиной и определить ее, а также Км
точно нельзя. Для этой цели чаще всего используют метод Лайнуиве-
ра—Берка. Он позволяет получить ценную информацию о деталях
механизма ферментативного катализа при изменении условий
реакции, наличия другого субстрата или благодаря обработке фермента,
приводящей к изменению его структуры. Как мы увидим ниже, из
270
Глава 8. Ферменты
этого графика можно получить данные, касающиеся действия
ингибитора на фермент. Значения Км и Утах находят путем анализа
кинетических данных. По Лайнуиверу—Берку, для этих целей широко
используется зависимость 1/у от 1/[8] — метод двойных обратных
величин; уравнение Михаэлиса—Ментен принимает следующий вид:
1
V
К
м
1 1
+
Утах [3] V
тах
тангенс
Это уравнение прямой: у = ах+Ь (рис. 8.7), где а = Км/ \^
угла наклона прямой; Ь = 1/Утах, у = 1/у, х = 1/[8].
Другой часто используемый график — график Эди—Хофсти.
Преобразование уравнения Михаэлиса—Ментен приводит к выражению:
^^тах-Км-у/[В].
V
тах
Наклон - Км/Утах
ш&х
Наклон = -К
V/КМ
-1/К
1/[8]
м
у/[8]
Рис, 8.7, График Лайнуивера—Берка
Рис. 8.8. График Эди—Хофсти
:_* ~т
График, построенный в координатах {у; у/[8]) (рис* 8.8), представ-
яет собой прямую, отсекающую на оси ординат (при у/[8] *= 0) отре-
эк. равный Ут0Х> Тангенс угла наклона прямой равен -Км, а отрезок,
геекаемый на оси абцисс, у/Км.
График Лайнуивера—Берка имеет тот недостаток, что при высо-
х концентрациях субстрата экспериментальные точки попадают на
небольшой отрезок, в результате чего точкам, соответствующим
низким концентрациям субстрата, придается большее значение. Достоин-
:тзо же этого графика состоит в том, что он позволяет просто оценить
значение у при данной концентрации [8].
График Эди—Хофсти лишен указанного выше недостатка, однако
:"ределить с его помощью значение скорости протекания реакции
труднее. Считается, что этот график дает более точные результаты,
поэтому для определения Км и Утах исследователи часто
предпочитают пользоваться именно им.
Влияние температуры на активность ферментов. Важнейшим
фактором, от которого зависит активность ферментов, является тем-
271
Часть II. Метаболизм веществ и энергии в клетке
пература. При О °С, а тем более при температурах ниже О °С действие
большинства ферментов прекращается. Повышение температуры выше
О °С способствует повышению активности ферментов.
Изменение скорости химической реакции при повышении
температуры исследуемой системы на 10 °С обычно принято выражать
температурным коэффициентом <^10, который представляет собой
отношение скорости реакции при данной температуре у1+10 к скорости
реакции при температуре на 10 °С ниже данной:
<3ю =
_ Ут+ю
Температурный оптимум
Как правило, величина <510 для химических реакций лежит в
пределах 2—4, для ферментативных реакций — между 1 и 2; <310
ферментативных реакций заметно снижается при повышении температуры.
Характер влияния температуры на активность ферментов показан
на рис. 8.9.
В пределах физиологических условий и близких к ним скорость
реакции растет с увеличением температуры (кривая 2), но выше
определенной температуры начинается тёп-
ловая денатурация белковой молекулы
— инактивация фермента (кривая 2).
Эти два явления объясняют форму
кривой, близкую к кол околообразной, и
появление температурного оптимума
реакции.
Не все ферменты одинаково меняют
свою активность с изменением
температуры. Максимальная активность
ферментов колеблется в зоне температур
30—50 °С. Вполне отчетливо
наблюдается снижение активности при 60—70 °С;
Рис. 8.9. Влияние температуры ПРИ 10° °С каталитическое действие
больна активность фермента шинства ферментов прекращается
практически мгновенно.
Влияние температуры на скорость ферментативной реакции
чрезвычайно многообразно. Оно проявляется и в действии на белковую
структуру фермента, и в изменении скорости образования комплекса
Е8, и в изменении скорости образования комплекса ЕР, и в
изменении скорости расщепления комплекса ЕР; температура влияет на
число столкновений молекул субстрата с молекулами фермента.
Влияние рН среды на активность ферментов. Вторым важным
фактором, влияющим на активность ферментов, является значение
рН, Каждый фермент проявляет свое действие в пределах довольно
узкой зоны рН. В прямоугольной системе координат кривая
зависимости V = Г(рН) для большинства ферментов имеет колоколообразную
30 40 50 60 70 80 С
272
Глава 8. Ферменты
форму (рис. 8 Л О). Оптимальной
активности соответствует
определенная область рН> причем
вершина кривой с точкой утах
соответствует оптимальному
значению рН в действии фермента; эта
величина рН весьма различна для
разных ферментов. Большое
число ферментов в клетках имеют
оптимум рН, близкий к
нейтральному, т.е. совпадающий с
физиологическими значениями рН
живой клетки.
Характер кривой у=^(рН)
объясняется амфотерной природой
ферментов; восходящая и
нисходящая ветви этой кривой являются
типичными кривыми титрования и
определяются значениями рК
ионных групп, которые находятся в
активном центре фермента;
Пепсин
Сахараза
Трипсин
6
8
10 рН
Рис. 8,10. Влияние величины рН
среды на активность ферментов. Для
различных ферментов значения
оптимума рН колеблются в широких
пределах — от сильно кислой среды
(например , для пепсина) до щелочной
(например, трипсина). Многие ферменты, как,
например, сахараза, гидролизующая
сахарозу, имеют оптимум рН в слабо
кислой среде
где ЕН2+
ЕН*
к* 1* К8 к3
ЕН + 8 <* ЕН8 -* ЕН+Р,
кът;
Е"
— неактивная (катионная) форма фермента; Е" —
неактивная (анионная) форма фермента; ЕН — активная форма фермента. К
и Кь — константы кислотной диссоциации кислотных и основных групп
соответственно.
Представим в форме математического моделирования это
уравнение.
Общая концентрация фермента [Е0] будет равна сумме трех
состояний фермента:
[Е0] = ЕН2+[ЕН] + [Е-].
а
Найдем долю активного фермента:
а =
[ЕН]
[ЕН] =
[Е0] [ЕН^] + [ЕН] + [Е]
или
а =
•+
+
1 + рГ]/Кв+Кь/[Нт]
18. Заказ 3486
273
Часть П. Метаболизм веществ и энергии в клетке
При оптимальном рН (рНоц) [Е0]=[ЕН], а активность фермента
V —Утау. При значениях рН, отличающихся от рНоп, ^~аМтах или:
тах
V
.у _ тах
1 + [Н+]/Ка+Кь/[Н+]
Это уравнение справедливо при условии, что Н+-ионы оказывают
влияние лишь на ионизацию каталитически активных групп
фермента и не затрагивают его структуры в целом. При значениях
рН < рНоп[Н+]/Ка» КЬ/[Н+] и для восходящей ветви кривой V = ДрН)
уравнение принимает более простой вид:
V
,. = *тах
1 + [Н+]/Ка]
При значениях рН > рНоп [Н+]/Ка« КЬ/[Н+] и нисходящая ветвь
кривой может быть описана уравнением:
V
д, утах
1 + Кь/[Н+]
При построении кривой V = 1(рН) это уравнение удобно выразить
через рНа, рКь и рН:
V
,. _ *тах
1+10РКа-РН'
т* — . утах
1 + юрН'рКь '
Из уравнений вытекает важное следствие: при рН = рКа или при
рН = р!^ V » 1/2^^, т.е. при скорости расщепления субстрата
ферментом, равной половине максимальной, константа ионизации
каталитически активной группы фермента численно равна* концентрации
Н+-ионов среды. Исходя из этого, на кривой V = ?(рН) можно
определить величины рКа и рКь, а по ним — природу ионных групп,
участвующих в акте катализа. В табл. 8.3 представлены значения рК
ионных групп в ферментах, участвующих в катализе.
Таблица 8.3
Значения рК для ионных групп ферментов,
принимающих участие в акте катализа
Группа
а-Карбоксильная
Удаленная карбоксильная
Имидазольная
а-Аминная
Гидроксильная тирозина
Удаленная аминная
Гуанидиновая
Диапазон значений рК
1,8-2,2
3,9—4,3
5,0—7,8
8,3—9,8
9,7—10,1
9,5—10,5
9,0—12,0
274
Глава 8. Ферменты
Активаторы и ингибиторы ферментов. На активность
ферментов большое влияние оказывают активаторы —- химические
соединения, повышающие действие ферментов, и ингибиторы — соединения,
подавляющие их активность.
Действие многих протеаз активируется в присутствии
незначительных количеств веществ, содержащих 8Н-группы (цистеин, глутатион);
их активирующий эффект заключается в том, что они восстанавливают
3-8-связи неактивных ферментов с образованием свободных 8Н-групп,
которые играют важную роль в механизме ферментативного катализа.
Другим примером активации является превращение проферментов
или зимогенов — неактивных предшественников ферментов — в
активные ферменты. В этом случае в основе активации лежит гидролиз
одной или нескольких пептидных связей в молекуле фермента под
действием протеолитических ферментов. Так, неактивные проферменты
(пепсиноген, трипсиноген, химотрипсиноген) в процессе пищеварения
превращаются в активные пепсин, трипсин, химотрипсин.
Эффективными активаторами являются ионы С1~, в присутствии
которых резко повышается действие се-амилазы слюны человека —
фермента, гидролизуюгцего крахмал пищи.
Ингибиторы принадлежат к специфическим химическим
соединениям; каждое из них подавляет действие одного или группы
ферментов со специфическими функциями*
Процесс ингибирования может быть обратимым и необратимым.
В свою очередь, обратимые ингибиторы бывают конкурентного,
неконкурентного , бесконкурентного и смешанного действия.
Конкурентное торможение возникает тогда, когда ингибитор
взаимодействует с функциональными группами активного центра
фермента. Этот тип ингибирования связан с отсутствием абсолютной
специфичности фермента. Ингибирование в данном случае зависит от
концентрации субстрата; если [8] велика, то влияние ингибитора I может
не проявляться совсем; если же [8] мала, то ингибитор может
вытеснить субстрат из соединения с ферментом, действие которого при этом
затормаживается. Тройной комплекс Е81 при конкурентном ингиби-
ровании никогда не образуется.
В большинстве случаев ингибиторы конкурентного типа
представляют собой структурные аналоги субстратов. Например, структурные
аналоги янтарной кислоты — щавелевая, малоновая, щавелево-уксусная
кислоты — являются конкурентными ингибиторами сукцинатдегидро-
геназы, осуществляющей окисление янтарной кислоты в фумаровую:
СООН
СН2
сн*
СООН
Янтарная
кислота
СООН
СООН
Щавелевая
кислота
СООН
сн2
СООН
Малоновая
кислота
СООН
с=о
сн2
соон
Щавелево-уксусная
кислота
18*
275
Часть П. Метаболизм веществ и энергии в клетке
Конкурентным ингибитором р-амилазы, гидролизующей амилозу
до мальтозы,., является циклоамилоза. Индол, фенол, бензол
блокируют центр связывания ("гидрофобный карман") химотрипсина и инги-
бируют гидролиз производных триптофана, тирозина, фенилаланина.
В качестве конкурентных ингибиторов выступают часто и
продукты ферментативной реакции.
Процесс конкурентного ингибирования может быть представлен в
виде следующей схемы:
К8 к3
Е + 8 <> Е8 -> Е+Р
I ^ Е1,
где I — ингибитор конкурентного типа; ^ =
[Е][1]
[Е1]
константа
равновесия системы фермент—ингибитор.
Конкурентное ингибирование в стационарных условиях
описывается уравнением
1 _ Км
У
шах
1 +
V
Ш
+
/
[3] V,
тах
где [I] —• концентрация ингибитора; [8] — концентрация субстрата.
При конкурентном ингибировании происходит кажущееся увели-
/
чение Кк, в
м
1 +
ш
к;
раз, У„„ остается неизменной.
На рис. о. 11 представлен конкурентный тип ингибирования в виде
схемы (а) и графика Лайнуивера—Берка (б).
Неконкурентное ингибирование отличается от конкурентного тем,
что оно не может быть ослаблено или устранено увеличением [8]. Если
С ингибитором
1Д8]
а б
Рис. 8.11. Конкурентный тип ингибирования ферментативной реакции в виде
схемы (а) и графика Лайнуивера—Берка (б)
276
Глава 8. Ферменты
конкурентный ингибитор оказывает воздействие на процесс
связывания фермента с субстратом, то неконкурентный ингибитор подавляет
каталитическое превращение субстрата в продукты реакции. Видимо,
при неконкурентном торможении ингибитор связывается с
ферментом на участке, отличающемся от центра связывания субстрата;
однако при этом деформирует этот центр, подавляя его каталитическую
функцию. Иными словами, когда в реакционной среде находятся и
субстрат, и ингибитор, они присоединяются к ферменту
одновременно, а не конкурируют за один и тот же центр связывания. Это можно
изобразить в виде схемы:
Е + 8
+ 1ь
I
к5 П
Е1 + 8 ^ Е81,
Кй к3
Е8 -> Е+Р
+
Е1 I
где Е1 и Е81 — неактивные комплексы.
На рис* 8.12 представлены схема (а) и график Лайнуивера—Берка
(б) для неконкурентного ингибирования.
Кинетика неконкурентного ингибирования описывается
уравнением:
/
К
м
V
^ утах
1 1
+
\
[8] V,
тах
/
1 +
/
V
ш
к5
\
У
В присутствии ингибитора константа К„ не изменяется, а V.
м
тах
/
уменьшается в
1 +
V
Ш
к;
\
раз. К неконкурентным ингибиторам относят-
>
С ингибитором
Е
а>
а
1/[5]
Рис. 8.12. Неконкурентный тип ингибирования ферментативной реакции в
виде схемы (а) и графика Лайнуивера—Берка (б)
277
Часть II. Метаболизм веществ и энергии в клетке
ся ионы металлов (Си2*, Н§2+, А§+) или их производные, подавляющие
действие тиоловых ферментов в результате блокирования 8Н-группы:
Е-8Н + Н§2+ ^ Е-8Н§+Н+.
Сульфгидрильные группы могут оказывать существенное влияние
на активность фермента не только в тех случаях, когда они
локализованы в активном центре, но и когда они расположены вне его; в этом
случае роль их сводится к поддерживанию стабильной конформации
молекулы фермента.
Бесконкурентное ингибирование наблюдается в том случае, когда
ингибитор не способен присоединяться к ферменту, но он может
связываться с комплексом Е8, переводя его в неактивную форму. Иными
словами, бесконкурентный ингибитор в одинаковой мере уменьшает
Км и V,,».
При смешанном ингибировании, как и при бесконкурентном инги-
бировании, ингибитор действует как на участок связывания, так и на
каталитический центр фермента, причем независимо от того,
перекрываются они или нет. Ингибитор смешанного типа действия тоже
изменяет и Км, и ~^пшх, но, в отличие от бесконкурентного ингибирова-
ния, кажущаяся Км может или увеличиваться, или уменьшаться, а
величина V" всегда снижается.
П1&Х
Действие необратимых ингибиторов существенно отличается от
действия обратимых ингибиторов. Для кинетического анализа действия
необратимых ингибиторов теория Михаэлиса—Ментен неприменима. На
скорость этого ингибирования существенное влияние оказывают процесс
диффузии компонентов, частота столкновения молекул фермента и.
ингибитора. В связи с этим необратимый ингибитор, взятый в
концентрации, превышающей эквимолярную (по отношению к ферменту), может
сначала не давать полного ингибирования, но со временем степень
ингибирования будет возрастать; количество активных молекул фермента с
течением времени будет все меньше и меньше.
Одним из широко известных типов необратимого ингибирования
является действие алкилирующих агентов, образующих прочные ко-
валентные связи с 8Н-группами фермента, в результате чего
равновесие сильно смещено в сторону образования алкильного производного
фермента. К таким ингибиторам относится, например, йодацетамид;
Е-8Н + <ТСН2-С(ЖН2 -» Е-8-СН2-СОКН2+Н<1.
Необратимыми ингибиторами также являются цианиды,
подавляющие активность ферментов цитохромов, содержащих железо; этилендиа-
минтетраацетат (ЭДТА), подавляющий действие а-амилазы, в составе
молекулы которой находится ион Са2+. Мощные необратимые
ингибиторы сериновых ферментов (Е-СН2-ОН) — производные фосфорной
кислоты, хлорметилкетоны, сульфонилфториды. Так, производные фосфорной
кислоты блокируют активную ОН-группу остатка серина, ковалентно
связываясь с ней, и выводят фермент из активного состояния:
278
Глава 8. Ферменты
1 х I 1
Е-СН2ОН + Х-Р=0 -> Е~СН2~0~Р-0 + НХ
ок2 ок2
Из соевых бобов и других бобовых культур выделен
низкомолекулярный белковый ингибитор (молекулярная масса — около 20 000),
подавляющий действие трипсина. После автоклавирования при
высоких температурах сырой муки белки сои становятся
перевариваемыми для человека и домашних животных* Наличие такого ингибитора
следует учитывать при скармливании сырой муки из соевых бобов
цыплятам, свиньям и крупному рогатому скоту. Казеин, сухое
обезжиренное молоко, рыбная мука и автоклавированная мука соевых бобов
легко перевариваются трипсином. Внесение сырой соевой муки к ка-
кому-либо из названных белковых кормов подавляет переваривание
белков этих кормов. На гидролиз белков пепсином ингибитор
влияния не оказывает.
Существуют соединения, которые непосредственно не влияют на
активность фермента, т.е. они не являются ни ингибиторами, ни
активаторами ферментативных реакций. Однако эти вещества могут
оказывать опосредованное действие на ферментативную активность
путем подавления влияния активатора или ингибитора. Такие
соединения называются либераторами.
8.5. Номенклатура и классификация ферментов
В настоящее время известно около 3000 различных ферментов. Из
них около 250 выделены в кристаллическом виде. Систематизация
всех этих ферментов представляет собой довольно сложную задачу.
Если учесть, что почти для каждой реакции требуется свой фермент, а
ферменты, катализирующие одну и ту же реакцию у различных
организмов (животных, растений, микроорганизмов), отличаются по
своим свойствам, то становится понятным, что общее количество
существующих ферментов может достигать сотен тысяч и более.
Номенклатура ферментов. В настоящее время приняты два типа
названия ферментов: рабочее, или тривиальное, и систематическое.
Рабочее название впервые было предложено в 1898 г. Ж. Дюкло.
Оно складывается из названия субстрата, к корню которого
добавляется окончание аза {например, амилоза — амилаза, целлюлоза — цел-
люлаза, протеин — протеиназа). В названии многих ферментов
указывается также тип катализируемой реакции (например, лактат + де-
гидрогеназа —> лактатдегидрогеназа; аланин + рацемаза -> аланинра-
цемаза). За некоторыми давно известными ферментами оставлены
тривиальные названия, предложенные авторами, впервые открывшими
их; пепсин, трипсин, ренин и т. д.
Систематическое название фермента складывается из названия
субстрата, названия типа катализируемого превращения и окончания
279
Часть II. Метаболизм веществ и энергии в клетке
аза. Например, систематическое название лактатдегидрогеназы
пишется так:
Ь-лактат: КАВ+ — оксидоредуктаза
субстрат I субстрат II тип химического
превращения
Систематические названия даются только изученным ферментам. Они
более четки в представлениях о механизме катализа. Однако в силу того,
что многие субстраты представляют собой сложные соединения,
некоторые из названий оказываются слишком громоздкими. Поэтому наряду с
систематическими названиями ферментов сохраняются и тривиальные
названия, удобные для повседневного употребления.
Классификация ферментов. Согласно классификации,
разработанной Комиссией по ферментам Международного биохимического
союза в 1961 г., все ферменты делятся на шесть основных классов. В
каждый класс включены ферменты, катализирующие общий тип ре^
акций (табл. 8.4).
Таблица 8.4
Классы ферментов согласно рекомендациям
Комиссии Международного биохимического союза
по номенклатуре и классификации ферментов
№ п/п
1
2
3
4
5
6
Класс
Оксидоредуктазы
Транеферазы
Гидролазы
Лиазы
Изомеразы
Ли газы
Тип катализируемой реакции
Окислительно-восстановительные реакции всех типов
Перенос групп атомов от донорной молекулы к
акцепторной
Гидролитическое (с участием молекулы воды)
расщепление связей
Отщепление от субстратов групп (не путем гидролиза)
с образованием двойной связи или, наоборот,
присоединение групп по двойным связям
Взаимопревращения различных изомеров
Образование связей в реакциях конденсации
двух.различных соединений с участием АТР как источника
химической энергии
Согласно этой классификации каждый фермент имеет свое
кодовое число (шифр), перед которым стоят две буквы — КФ. Шифр
четырехзначный, цифры шифра разделены точками и характеризуют
следующие признаки фермента;
1. Первая цифра указывает класс, к которому принадлежит
фермент.
2. Вторая — подкласс, У оксидоредуктаз она указывает природу той
группы в молекуле донора, которая подвергается окислению; у трансфе-
раз -— природу переносимой группы; у гидролаз — тип гидролизуемой
связи; у лиаз — тип связи, подвергающейся разрыву (между
отщепляемой группой и остатком молекулы); у изомераз — тип катализируемой
реакции изомеризации; у лигаз — тип вновь образуемой связи.
280
Глава 8. Ферменты
3. Третья — подподкласс. У оксидоредуктаз эта цифра указывает
тип участвующего в реакции акцептора: кофермент ИАВ+ или МАВР+;
цитохром; молекулярный кислород. Таким образом, первые три
цифры шифра точно определяют тип фермента. Например, КФ1.2.3
обозначает оксидоредуктазу, для которой донором водорода служит
альдегид, а акцептором — кислород. У трансфераз третья цифра
указывает на тип транспортируемой группы; у гидролаз это число уточняет
тип гидролизуемой связи, у лиаз — тип отщепляемой группы; у изо-
мераз — уточняет характер превращения субстрата, а у лигаз —
природу образующегося соединения.
4. Четвертая цифра означает порядковый номер фермента в
данном подподклассе.
Приведем пример:
а-Амилаз а
Класс гидролазы
Подкласс карбогидразы
Подподкласс гликоназы
Порядковый номер в подподклассе
КФЗ. 2. 1.
I
8.6. Характеристика отдельных классов ферментов
Оксидоредуктазы (КФ1). Систематическое название
оксидоредуктаз составляется по схеме: донор водорода — акцептор электронов —
оксидоредуктаза.
Класс оксидоредуктаз включает 21 подкласс. Наиболее важными,
хорошо изученными являются следующие:
КФ
Группа, на которую действует оксидоредуктаза
-сн~
I
н
>с-он
-сн2-сн2-
-сн-мн2
-сн-ин
I I
1.1
1.2
1.3
1.4
1.5
1.6 ХАБН; ИАВРН
Однако среди оксидоредуктаз более широкое распространение
получили тривиальные названия. Оксидоредуктазы, катализирующие
перенос атомов водорода от донора к любому акцептору, получили
название дегидроееназ. В тех случаях, когда акцептором водорода
является кислород, оксидоредуктазы называются оксидазами. К классу
оксидоредуктаз относятся цитохромы, — группа ферментов,
переносящих электроны от окисляемого субстрата к кислороду.
Дегидрогеназы — это самая распространенная группа ферментов,
играющая важную роль в процессах фотосинтеза, дыхания, броже-
281
Часть II. Метаболизм веществ и энергии в клетке
ния. Дегидрогеназы обнаружены в органах и тканях животных и
растений, а также у микроорганизмов* Реакция ферментативного
дегидрирования обратима, отличается высокой специфичностью.
Дегидрогеназы представляют собой двухкомпонентные ферменты.
В зависимости от химической природы кофактора они
подразделяются на пиридинзависимые дегидрогеназы, коферментом которых
является никотинамидадениндинуклеотид (ИАВ) и никотинамиддинукле-
отидфосфат (ЫАОР), и на флавинзависимые дегидрогеназы (флаво-
протеины)* роль простетической группы в которых выполняют фла-
винадениндинуклеотид (ГАВ) и флавинаденинмононуклеотид (ГМЫ).
Реакции, катализируемые пиридинзависимыми дегидрогеназами,
можно представить в общем виде следующей схемой:
вн,
в
пд
пда
X
КАВН + Н+(КАОРН+Н+)
МАП+(КАВР+)
Здесь ВН2 — восстановленный субстрат (донор атомов водорода); В —
окисленный субстрат; ПД — пиридинзависимая дегидрогеназа; КАВ4"
(ЫАВР+) и ИАВ-Н + Н+ (ЫАВРН + Н+) — кофермент, соответственно
окисленный и восстановленный.
На рис. 8.13 представлена структура МАО в окисленной и
восстановленной формах. Что касается ЫАБР, то она идентична по структуре ЫАВ
с той лишь разницей, что в ЫАВР у С-3-атома риббзы ОН-группа
замещена остатком молекулы фосфорной кислоты, Никотинамидное кольцо
данных коферментов выполняет функцию промежуточного переносчика
гидрид-иона, который отщепляется от субстрата под действием дегидро-
геназ* В результате образуется ИАОН или ЫАВРН. Второй атом
водорода отщепляется от субстрата в виде иона Н+.
н
л
НС С-СОИНг
II I
нс^сн
НС
<
с
I
N
N
С
I
N
С-ИНг
нс-
носн
носн
I
о о
НС
I
сн2
сн
-сн
неон
неон
I
-СН
СН2
н н
л
НС С-СОЛНа
II II
нсч сн
N
НС
<
С
I
N
N
+2Н
не-
I
-2Н
•-_ НОСН
НОСН
I
_ •
-с
I
N
//
СН
СН
I
О-ИНг
Гк
О О
НС
о
о
II
О —Р
I
о
СН2
неон
неон
I
-сн
сн2
+ н
+
О—Р—О—Р—О
I
о
о
II
р-
1
о
-0-
0
II
-р
1
О"
О—Р—О—Р—О
Рис. 8.13. Химическая структура молекул МАО и МА1)Н
282
Глава 8. Ферменты
МАО и ЫАВР впервые были выделены и идентифицированы с 1933
по 1936 г. О. Варбургом и Л. Эйлером. Они обнаружены почти во всех
типах клеток, причем ЫАГ) содержится обычно в больших
количествах, чем ЫАОР. N АО-зависимые дегидрогеназы принимают участие
главным образом в процессах дыхания и брожения;
ИАВР-дегидрогеназы принимают участие в окислительно-восстановительных
процессах, сопряженных с биосинтетическими процессами. Поскольку связь
КАО и ИАОР с белком дегидрогеназы обратимо разрушается и вое-
станавливается в процессе каталитического цикла, эти коферменты
можно рассматривать как специализированные субстраты. Они
играют роль промежуточных акцепторов (переносчиков) атомов водорода
при сопряженных действиях двух дегидрогеназ. Кофермент
поочередно присоединяется к молекулам этих белков в двух формах —
акцепторной и донорной. В приведенной ниже схеме показан механизм
этого процесса между молекулой окисляемого субстрата (ВН2) и
молекулой акцептора (А) при действии двух дегидрогеназ (ПД1 и ПД2) и КАВ:
1. Катализ дегидрогеназой ПД1:
ПДХ + ИАВ+ ^ ПД1^АВ+ (1)
ПД1КАВ+ + ВН2 ^ ПД^АОН + Н* + В (2)
1Щ- КАВН + Н+ ^ ПДХ + ИАРН + Н+ (3)
2. Катализ дегидрогеназой ПД2:
ПД2 + ЫАРН + Н+ ?=* ПД2КАВН + Н+ (4)
ПД2^АВН + Н+ + А ^ ПД2-КАВ+ + АН2 (5)
ПД2 ЫАО+ ^ ПД2 + ИАВ+ (6)
Как видно из схемы, в последовательных фазах цикла,
протекающих каждая с участием одного фермента (фазы 1—3 и 4—6), ИАВ+ и
NА^Н + Н+ участвуют наравне с молекулами ВН2, В, А, АН2 в
качестве второго субстрата.
Многие пиридинзависимые дегидрогеназы прочно связаны с
ионами двухвалентных металлов; примером может служить алкогольде-
гидрогеназа, содержащая ионы 2п2+. Предполагают, что металл
служит для связывания молекул ЫАР или ЫАОР с ферментным белком.
Некоторые из них, в частности глицеральдегид-3-фоефатдегидрогена-
за, лактатдегидрогеназа, малатдегидрогеназа, были получены в
кристаллическом виде.
В качестве примеров в табл. 8.5 приведены систематические и
тривиальные названия некоторых пиридиновых дегидрогеназ и
катализируемые ими реакции.
Действие флавинзависимых дегидрогеназ, содержащих простети-
ческие группы ГАО и БЧ\Ш, можно представить в виде следующей
схемы:
283
Часть II Метаболизм веществ и энергии в клетке
ВН2 ФП ^ р ГАВ * Н2(ГЬШ ♦ Н2)
В *^ ФП-Нз ^ ГАВ(ГМЫ)
Структура и механизм присоединения-отщепления атомов
водорода у РАВ и РЬШ представлены на рис. 8.14. Оба соединения были
изучены (Х.Варбургом и Р. Куном в середине 30-х гг., именно в то
время, когда был идентифицирован рибофлавин. К АХ) и Р1УПЧ весьма
прочно присоединены к белковой молекуле фермента, поэтому в прин-
Таблица 8.5
N АО (Р )• зависимые дегидрогеназы
Шифр
(КФ)
1.1.1.1
1.1.1.25
1.1.1.37
1.4.1.6
Название
систематическое
Алкоголь:КА1)*-оксидо-
редуктаза
Ъ~лак;гат:КА1)+-оксидо-
редуктаза
Ь-малат:КАО+-оксидо-
редуктаза
Ь-глутамат: N АВР-окси-
доредуктаза
тривиальное
Алкогольдегидрогеназа
Лактатдегидрогеыаза
Малатдегидрогеназа
NА^Р-глутаматдегид-
рогеназа
Катализируемая реакция
Алкоголь + МАВ+ &
Ацетальдегид + ЫА0Н+Н+
Ь-лактат + ЫАО+ <=^
Пируват + ИАОН + Н+
Ъ-малат + ЫАВ*#
Оксалоадетат +ЫАЮН + Н+
Ь-глутамат Н- Н20+МАВР ?±
2-гидроксиглутарат + МН3 +
+ЫАБРН + Н+
ципе их нельзя называть коферментами, хотя некоторые авторы и
относят эти простетические группы к коферментам.
Активной частью ГАВ и ГЬШ является изоаллоксазиновое кольцо.
Реакцию представляют обычно как прямой перенос пары атомов
водорода от субстрата к БАХ) и РМ1М. В окисленной'форме флавиновые
дегидрогеназы интенсивно окрашены в красный, коричневый или
зеленый цвета. Для них характерны широкие полосы поглощения при
370 и 450 нм. При ферментативном и химическом восстановлении
полоса поглощения при 450 нм исчезает. Восстановленные формы этих
дегидрогеназ с трудом окисляются молекулярным кислородом. В
клетках конечным акцептором электронов, отщепляемых от флавиновых
дегидрогеназ, является система цитохромов. Эти ферменты легко
окисляются химическими окислителями (например, феррицианидом) или
восстанавливающимися красителями (например, метиленовым синим;
2,6-дихлорфенолиндофенолом). Указанные соединения широко
используются для количественного определения ферментов.
Многие пиридинзависимые и флавинзависимые дегидрогеназы
имеют четвертичную структуру.
Оксидазы катализируют реакции, окислителем которых является
молекулярный кислород воздуха. Наиболее изученными из них
являются глюкозооксидаза, пируватоксидаза, полифенолоксидаза, аскор-
бинагпоксидаза, оксидаза В- и Ь-аминокислот. В качестве
кофакторов оксидазы чаще всего содержат флавиновые (БТ\Ш, БАХ)) и гемсо-
284
Глава 8. Ферменты
Н О
I II
С N С
Н3С-С/ С ^С NН
НзСЛ\ЛЛ/с=0
С N N
I I
н сн2
н-с-он
н-с-он
н-с-он
сн2
о
~0-Р=0
I
о
"0-Р=0
I
о
I
н н о
I I II
С N С
н8с-с/Чс/^с/ нн
С N N
I I I
. н сн2 н
н-с-он
+2Н
-2Н
N.
НС
//
гш2
I
9Н2^о
\
к
-с
ОН ОН
н о
н8сч/чс-"\^мн
СН N N
СН2
Н-С-ОН
Н-С-ОН
I
Н-С-ОН
1
СН2
I
О
о=р-о~
I
о~
\ //
N
СН
Н-С-ОН
Н-С-ОН
сн2
I
о
I
о-р=о
I
о
_о-р=о
I
о
I
N.
НС
//
*ш2
с
\
Ц ^н
он он
-сч ^сн
N
а
+2
^
-2Н
Н Н О
I I II
С N С
ШС-С^С^С^КН
СН N N
1 I
СН2 Н
Н-С-ОН
Н-С-ОН
I
Н-С-ОН
I
СН2
I
О
"0-Р=0
I
О"
V
Рис. 8.14, Химическая структура молекул ГАБ (а) и КМЫ (б)
держащие группировки, металлы переменной валентности (медь,
молибден, железо) или сочетание переходных металлов с органическими
кофакторами {флавинами, темами). Отнимая водород от окисляемого
субстрата и передавая его затем кислороду, оксидаза может
образовывать при этом воду или пероксид водорода:
ВН,
В
Ох
Ох-Н,
X
н*о
ВН,
1/20,
В
Ох
Ох-Н.
Н202
О.
285
^ Глава 8. Ферменты
вых групп цитохромоксидазы (гем а3) может связывать ингибиторы —
монооксид углерода или цианиды; гем а эти ингибиторы не связывает.
Локализация цитохромов в клетках варьирует в зависимости от
специализации клеток и интенсивности их функционирования*
Большая часть клеточных цитохромов сосредоточена во внутренней мито-
хондриальной мембране» а также частично в ядерной мембране.
Цепь переноса электронов в митохондриях выглядит в виде
следующей схемы:
Субстрат (ЫАОН, сукцинат и др.) —» ФП —> Со<3 —> цитохром Ь —»
цитохром с$ —> цитохром с —> цитохром оа3-» кислород, где ФП —
специфичный к субстрату флавинзависимый фермент, Со(} — убихинон.
Трансферазы (КФ2) ■— класс ферментов, катализирующих
перенос химических групп от одного соединения (донора) к другому
(акцептору). Трансферазы занимают ключевые позиции в
энергетическом и биосинтетическом (пластическом) обмене веществ. В
зависимости от химической природы переносимых групп трансферазы
подразделяются на 8 подклассов:
КФ
2.1
2.2
2.3
2.4
2.5
2.6
2.7
2.8
Переносимые группы
Одноуглеродные
Альдегидные, кетонные
Ацильные
Гликозильные
Алкильные
Азотистые
Фосфорные
Содержащие серу
В принципе к трансферазам можно отнести и оксидоредуктазы,
если считать главным не процесс окисления-восстановления, а
перенос водорода или электронов от донора к акцептору,
сопровождающийся окислением-восстановлением.
Трансферазы примерно столь же распространены, как и
оксидоредуктазы. К важнейшим из них можно отнести метилтрансферазы
(КФ 2.1.1), трансферазы гидроксиметильных и формильных групп
(КФ 2.1.2), карбоксилтраясферазы (КФ 2.1*3). В обмене углеводов из
гликозилтрансфераз важную роль играют гексозотрансферазы (фос-
форилазы, КФ 2.4Л), акцептором которых служит неорганический
фосфат, К подклассу, катализирующему перенос азотистых групп,
относится обширный и очень важный в обмене аминокислот и белков
подкласс аминотрансфераз (КФ 2.6.1), переносящих аминогруппы,
донором которых являются аминокислоты, а акцептором — кетокисло-
ты. Аминотрансферазы относят также к числу пиридоксалевых
ферментов в связи с тем, что в их активный центр входит пиридоксаль-
фосфат — активная форма витамина В6.
Обычно перенос химических групп, катализируемый трансферазам и,
осуществляется с помощью специфических переносчиков — коферментов
287
Часть II. Метаболизм веществ и энергии в клетке
Цитохромы (от греч. ку108 -— клетка; сНгдта — окраска). Это группа
железосодержащих белков, участвующих в переносе электронов в
аэробных клетках от флавиновых ферментов к кислороду воздуха.
Цитохромы — двухкомпонентные ферменты, содержащие железопорфириновые
простетические группы — гемы. Группировка гема представляет собой
сложную компланарную циклическую систему, состоящую из
центрального атома железа, образующего координационные связи с четырьмя
остатками пиррола А, В, С, В, соединенными мостиками =СН~.
Присоединение и передача электронов осуществляются за счет обратимого
изменения валентности атома железа в теме (Ре3+ & Ре2+). Группа цитохромов
образует цитохромную систему > функционирующую путем
последовательного окисления и восстановления компонентов системы. Движущей
силой передачи электронов от флавопротеина к кислороду через
цитохромы является увеличение окислительно-восстановительного потенциала.
Цитохромы открыл в 1886 г. Мак-Мунн, а в 1925 г. Д. Кейлин
установил их биологическую роль и показал, что цитохромная
система играет важную роль в процессе биологического окисления.
Цитохромы содержатся во всех организмах, использующих для своей
жизнедеятельности энергию, освобождающуюся за счет окисления
субстратов кислородом воздуха. Большинство цитохромов (за
исключением цитохрома с) прочно связаны с мембранами. Только цитохром с
можно выделить путем экстракции из митохондрий растворами
солей; для него установлена аминокислотная последовательность.
Классификация цитохромов основана на различиях в природе их
гема. В соответствии с четырьмя типами гема выделяют четыре
группы цитохромов: а, Ъ, с, й, которые различаются спектрами
поглощения восстановленной формы в видимой области спектра, боковыми
цепями при порфириновых кольцах. У
цитохрома а гем содержит формильную
боковую группу; у цитохрома с гем
связан с белком двумя остатками цистеи-
на (рис. 8.15). Связь образуется
присоединением 8Н-группы цистеина к ви-
нильной группе порфина. Пятая и
шестая координационные связи железа
соединены с остатками аминокислот: ги-
стидина и метионина.
Комплекс цитохромов аав (цигпо-
хромоксидаза) передает электроны
молекулярному кислороду и является
конечным компонентом цитохромной
системы. Цитохромоксидаза прочно
связана с мембраной, обладает гидрофоб-
Рис. 8.15. Химическая струк- ными свойствами; молекулярная мае-
тура гема цитохрома с са комплекса — 240000, одна из гемо-
соон
I
сн2
сн2
СН3
н8с
сн3
286
Часть II. Метаболизм веществ и энергии в клетке
или кофакторов, образующих ковалентные промежуточные соединения
с субстратом. В этих случаях донором является кофактор, присоединя ю-
щий группу, подлежащую переносу- Трансферазы весьма разнообразны
по механизму катализа, специфичности к субстратам, рН-зависимости,
Молекулы многих трансфераз имеют четвертичную структуру;
некоторые из них выполняют регуляторную функцию в клеточном метаболизме.
Гидролазы (КФЗ) — класс ферментов, катализирующих реакции
разрыва в субстрате химических связей с участием воды. Многие
гидролазы локализованы в клеточных органеллах — лизосомах, где под
их действием осуществляется расщепление высокомолекулярных
соединений (белков, углеводов, липидов).
Молекулярная масса гидролаз колеблется в широких пределах — от
10000 до 300000. Они проявляют каталитическую активность в отсут^
ствие каких-либо кофакторов. Лишь немногие из них осуществляют
катализ в присутствии ионов металлов — главным образом 2п+2> Со2+, Са2+,
М#2+.Для небольшого числа гидролаз с молекулярной массой 10000—
30 000 известна первичная структура, а для некоторых — и конформа-
ция молекулы (например, для рибонуклеазы, пепсина, трипсина, химо-
трипсина, лйзоцима). Отмечено значительное сходство структуры
ферментов одного подкласса, особенно в области активного центра.
Все гидролазы делятся на 9 подклассов; ниже представлены
наиболее важные из них:
КФ Разрываемые связи
3.1 Сложноэфирные
3.2 Гликозильные
3.4 Пептидные
В принципе гидролазы также можно отнести к трансферазам, если
гидролиз рассматривать как перенос специфической группы
субстрата, являющегося донором, на молекулу воды, служащей акцептором:
перенос
I 1
Щ-Ъ2 + НОН -» Ех-Н + К2-ОН,
где Кх — субстрат-донор группы К2.
Гидролазы, катализирующие гидролиз сложноэфирных связей, —
эстеразы — действуют на сложные эфиры карбоновых и тиокарбоно-
вых кислот, моноэфиры фосфорных кислот. К этому подклассу
относятся, в частности, ферменты, играющие важную роль в метаболизме
липидов, нуклеиновых кислот, нуклеозидов (например, липазы,
рибонуклеазы, фосфотазы, фосфолипазы).
Эстеразы существенно различаются между собой по степени гидро-
фобности гидролизуемых субстратов: так, субстратами холинэстераз
являются водорастворимые соединения, а липазы эффективны только
на поверхности раздела фаз вода — жир. Эстеразы обладают широкой
субстратной специфичностью. Их максимальная активность лежит в
диапазоне значений рН от 5 до 8.
288
Глава 8. Ферменты
Карбогидразы {0-гликозилгидролазы, КФЗ>2) катализируют
гидролитическое расщепление О-гликозидных связей в самых различных гликози-
дах, о л иго- и полисахаридах. Специфичность действия карбогидраз
определяется главным образом конфигурацией расщепляемой связи (а- или
3-связь), а также природой гликозидного остатка. Карбогидразы,
содержащиеся в лизосомах, имеют ряд общих свойств, и прежде всего свойства
слабых кислот; для большинства из них оптимум рН лежит в пределах
4,5—5,5, что, по-видимому, совпадает с рН среды внутри лизосом.
По механизму действия различают следующие основные типы
карбогидраз: гликозидазы, эндогликаназьц экзогликаназы. Первые
отщепляют от олигосахаридов концевые невосстанавливающие остатки
моносахаридов. Для некоторых из них доказан механизм, включающий
образование промежуточного соединения, в котором фермент и
углевод ковалентно связаны между собой* а-Гликозидазы катализируют
гидролиз мальтозы, сахарозы, трегалозы, в которьдх нередуцирующая
глюкоза находится в а-форме> Благодаря способности а-гликозидаз
гидролизовать мальтозу их часто называют мальтазами. Из
некоторых биологических источников а-гликозидазы выделены в высоко-
очищенном состоянии; они представляют собой однокомпонентные
белки, хорошо растворимые в воде. Связывание молекулы субстрата с
лх активным центром осуществляется с помощью гидроксильных групп
у 2-го, 3-го, 4-го и 6-го атомов углерода концевого остатка глюкозы.
Это определяет абсолютную специфичность к глюконовой части
субстрата. Оптимальные значения рН находятся либо в области от 4 до 5
(кислые а-гликозидазы), либо от 6 до 7 (нейтральные а-гликозидазы).
(^-Гликозидазы катализируют гидролиз (3-гликозидной связи в
различных олигосахаридах (целлобиозе, гентибиозе). В отличие от
а-гликозидаз связывание молекулы субстрата осуществляется с помощью
гидроксильных групп у 2-го, 3-го, 4-го атомов углерода остатков глюкозы.
Многие свойства р-гликозидаз аналогичны свойствам а-гликозидаз.
Эндогликоназы (например, а-амилаза) расщепляют в
полисахаридах гликозидную связь, удаленную от концов молекулы. Как
гликозидазы, так и эндогликоназы расщепляют гликозидную связь с
сохранением конфигурации аномерного углерода (С-1 в циклической форме
моносахарида). Экзогликаназы (например, глюкоамилаза, р-амилаза)
отщепляют от невосстанавливающего конца полисахарида моно- или
олигосахаридные остатки с образованием аномерной р-конфигурации.
Протеазы (протеолитические ферменты, пептидгидролазы,
КФ 3.4) — подкласс ферментов, расщепляющих пептидные связи в
белках, пептидах. Их разделяют на два больших подподкласса: пептидазы
(экзопептидазыу КФ 3.4.11—15), действующие на КН2- или СООН-кон-
цевые участки пептидной цепи, и протеиназы (эндопептидазы,
КФ 3.4.21—25), расщепляющие в белках внутренние пептидные связи.
Пептидазы в соответствии с их субстратной специфичностью
разделяются на амипопептидазы и карбоксипептидазы, соответственно
19. Заказ 3486
289
Часть II. Метаболизм веществ и энергии в клетке
отщепляющие И- или С-концевые аминокислоты; дипептидазы, гид-
рол изующие дипептиды. Многие пептидазы в качестве кофактора
содержат металлы. Амино- и карбоксипептидазы широко используются
при установлении первичной структуры белков.
Протеиназы классифицируют не по специфичности к субстрату,
как остальные гидролазы, а по типу каталитических групп в
активном центре; в соответствии с этим различают сериновые» тиоловые,
карбоксильные и металлсодержащие протеиназы.
К многочисленным сериновым протеиназам, в активные центры
которых входит ОН-группа серина, относятся трипсин, химотрипсин,
эластаза — ферменты желудочно-кишечного тракта, а также
бактериальные протеиназы. В большинстве случаев эти ферменты активны
в нейтральной среде. К тиоловым ферментам, в активных центрах
которых находится 8Н-группа остатка цистеина, относятся
растительные и бактериальные протеиназы; наиболее хорошо изученными из
них являются папайи и фицин. К карбоксильным протеиназам, в
активном центре которых находятся карбоксильные группы аспараги-
новой кислоты, относятся пепсин, химозин, ряд протеиназ бактерий и
микромицетов; многие из них проявляют максимальную активность в
кислой среде. К металлопротеиназам относится коллагеназа.
Многие протеиназы синтезируются в виде неактивных
предшественников — проферментов или зимогенов. В результате ограниченного
избирательного протеолиза, протекающего либо под действием
специфических протеиназ, либо автокаталитически, происходит
отщепление пептидов, экранирующих активный центр протеиназы, и она из
зимогена переходит в активную форму.
Протеиназы имеют различную субстратную специфичность, гидро
лизуя в белках строго определенные пептидные связи, что
обусловлено химической структурой активного центра (табл. 8.6).
Таблица 8.6
Специфичность действия протеиназ
Протеиназы
Трипсин
Химотрипсин
Пепсин
Химозин
Папаин
Источник
Тонкий
кишечник
Тонкий
кишечник
Желудок
Желудок телят
Плоды папайи
Активный центр
ОН-группа серина;
имидазольная группа
гистидина
СООН; имидазольная
группа гистидина;
ОН-группа серина
СООН
СООН
8Н
Оптимум рН
7,5—8,5
8,5—9,0
1,3—3,0
5,0
Специфичность
аргинин-лизин
тирозин-фенил-
аланин
тирозин -фен и л-
аланин
фенилаланин-
метионин
(Х-казеин)
аргинин—лизин
Протеолитические ферменты разнообразны по своим
физико-химическим свойствам. Среди них есть простые белки с молекулярной
массой около 20000 (трипсин, химотрипсин, папаин) и ферменты,
290
Глава 8, Ферменты
имеющие четвертичную структуру с молекулярной массой,
превышающей 300000 (например, лейцинаминопептидаза); некоторые из них
относятся к гликопротеинам.
Многие протеиназы наряду с пептидными связями способны гид-
ролизовать и сложноэфирные связи.
Лиазы (КФ4) — ферменты, отщепляющие от субстратов ту или
иную группу (не путем гидролиза) с образованием двойной связи или,
наоборот, присоединяющие группы по двойным связям. В
зависимости от химической природы расщепляемой связи они подразделяются
на 4 подкласса:
КФ Расщепляемая связь
4.1 Углерод—углерод
4.2 Углерод—кислород
4.3 Углерод—азот
4 * 4 Углерод—сера
Лиазы — менее распространенный класс ферментов; они
участвуют в реакциях промежуточного распада продуктов обмена.
К лиазам, расщепляющим С~С-связи, относится подподкласс алъ~
долаз {альдегид-лиаз, КФ 4.1.2). Альдолазы участвуют в расщеплении
Сахаров. Хорошо изучена фруктозо-1,6-бисфосфатальдолаза из
дрожжей, катализирующая обратимую реакцию:
В-фруктозо-1,6-бисфосфат ^ дигидрооксиацетонфосфат +
+ О-глицеральдегид-3-фосфат.
Особенно активна альдолаза в тканях с интенсивным обменом
углеводов (например, в печени, сердце, поперечно-полосатых мышцах).
Из мышц она получена в кристаллическом виде. Альдолаза,
выделенная из бактерий и микромицетов, имеет молекулярную массу 70000,
состоит из двух субъединиц; в активном центре находится прочно
связанный ион 2п2+. Альдолаза растений и животных имеет
молекулярную массу 150000—160000; построена из четырех субъединиц.
Декарбоксилазы (КФ 4.1/1) — подподкласс С-С-лиаз;
катализируют реакции отщепления карбоксильных групп от аминокислот и ке-
токислот. Декарбоксилазы аминокислот являются двухкомпонентны-
ми ферментами. Роль кофермента выполняет фосфорилированная
форма витамина В6 — пиридоксаль-5-фосфат, который легко отделяется
простым диализом от апофермента. Декарбоксилазы строго
специфичны и катализируют отщепление карбоксильных групп только от
Ь-а-аминокислот, превращая их в физиологически активные амины:
Е-СН-СООН -> К-СН2-ЫН2 + С02
ин2
Аминокислота Амин
Декарбоксилазы широко распространены у микроорганизмов,
вызывающих "гниение" белков. Их высокая специфичность
используется при определении микроколичеств аминокислот.
19*
291
Часть II. Метаболизм веществ и энергии в клетке
Декарбоксилазы кетокислот были впервые обнаружены К. Кейли-
ным в 1911 г. В экстрактах пивных дрожжей им была найдена декар-
боксилаза пировиноградной кислоты (а-карбоксилаза, КФ 4.1.1.1),
осуществляющая реакцию:
О О
СН3-С-СООН -► СН3-С + С02
н
Пировиноградная Уксусный
кислота альдегид
Позднее были найдены декарбоксилазы ое-кетоглутаровой и щаве-
лево-уксусной кислот.
Все декарбоксилазы а-кетокислот — двухкомпонентные фермен-
ты, коферментом в которых является фосфорилированная форма
витамина Вх — тиаминпирофосфат.
К ферментам, расщепляющим С-О-связь, относятся гидролиазы
(КФ 4.2.1). В этот подподкласс входят такие важные ферменты, как
фумарат- и аконитатгидратазы.
Подподкласс полисахарид-лиазы (КФ 4.22) — ферменты,
катализирующие распад полисахаридов путем реакции элиминирования; в
результате на одном из концов продукта образуется ненасыщенное кольцо
сахара. Эти ферменты следует противопоставить ферментам подподклас-
са 3.2.1, которые расщепляют полисахариды путем гидролиза.
К подклассу С-К-лиаз относятся аммиак-лиазы (КФ 4.3.1); напри-
л ш
мер, Ь-аспартат-аммиак-лиаза (КФ 4,3.1.1) катализирует обратимую
реакцию синтеза аспарагиновой кислоты из фумаровой кислоты и аммиака.
Ферменты подкласса (КФ 4.4) катализируют реакции отщепления
Н28 или его производных.
Изомеразы (КФ 5) вызывают превращения в пределах одной
молекулы, катализируют внутримолекулярные перестройки. В
зависимости от специфических особенностей этой перестройки они
подразделяются на 5 подклассов:
КФ Название подкласса
5.1 Рацемазы и эшшеразы
5.2 Цис-т/>аяс-изомеразы
5.3 Внутримолекулярные оксидоредуктазы
5.4 Внутримолекулярные трансферазы
5-5 Внутримолекулярные лиазы
Рацемазы и эпимеразы осуществляют рацемацию аминокислот
(например, аланинрацемаза катализирует реакцию Ь-аланин ^ В-ала-
нин) или эпимеризацию Сахаров (например, ХГОР-глюкозоэпимераза
катализирует реакцию 1ЮР-глюкоза ?^ 1ШР-галактоза).
Цис-транс-изомеразы вызывают изменение геометрической
конфигурации у двойной связи субстрата. Например, малеинат-цис-
292
Глава 8. Ферменты
транс-изомераза катализирует превращения фумаровой и малеино-
вой кислот.
Внутримолекулярные оксидоредуктазы катализируют
превращения альдоз в кетозы, осуществляют окисление СНОН-группы с
одновременным восстановлением соседней С=0-группы. Типичным
представителем третьего подкласса изомераз является триозофосфатизо-
мераза (КФ 5.3 Л Л), катализирующая реакцию:
О
с-н сн2-он
I I *
НЧ>ОН з* С=0
ЗСН20~Р СН20-Р
О-Глицеринальдегид-3-фосфат Дигидроксиацетонфосфат
Внутримолекулярные трансферазы известны как мутазы; они
осуществляют перенос группы с одной части молекулы субстрата на
другую часть той же молекулы.
Внутримолекулярные лиазы осуществляют реакцию либо децикли-
зации молекулы субстрата, либо превращения одного типа кольца этой
молекулы в другой.
Лигазы (синтетазы) (КФ 6) катализируют присоединение друг
к другу двух молекул. Этот процесс сопряжен с разрывом пирофос-
фатной связи в молекуле АТР. Энергия, освобождаемая при
расщеплении АТР, используется для синтеза.
В зависимости от химической природы образуемой связи данный
класс подразделяется на 5 подклассов:
КФ Связь
6Л С-0
6.2 С-8
6.3 С-К
6.4 ОС
6.5 Фосфоэфирные связи
К подклассу КФ 6.1 относятся весьма важные лигазы,
катализирующие присоединение остатков аминокислот к тРНК при синтезе белка.
Так, аланил-тРНК-синтетаза (КФ 6.1.1.7) катализирует реакцию:
Алании + тРНК + АТР ^ Аланил-тРНК + АМР + РРГ
К подклассу КФ 6.2 принадлежат лигазы, катализирующие
присоединение остатков различных органических кислот к коферменту А
(СоА-8Н). Например, под действием фермента ацетилкофермент А-син-
тетазы (КФ 6.2.1.1) образуется ацетилкофермент А:
О
II
СН3-СООН + Н8-СоА + АТР ?± СН3-С-8СоА + АМР + РР;
Уксусная Кофермент А Ацетилкофермент А
кислота
293
Часть II. Метаболизм веществ и энергии в клетке
К третьему подклассу относится, например, глутамин-синтетаза
(КФ 6.3.1*2), катализирующая реакцию синтеза глютамина из глюта-
миновой кислоты и аммиака.
Подкласс КФ 6.4 включает содержащие биотин карбоксилазы. Эти
ферменты катализируют присоединение диоксида углерода к
органическим кислотам. К этой группе ферментов относится пируваткарбо-
ксилаза (КФ 6*4.1.1), катализирующая реакцию:
С=0 + С02 + АТР ^> СН2 + АОР + Р:
соон с=о
соон
Пируват Оксалоацетат
Этот фермент ингибируется реактивами, связывающими ЗН-груп-
пу, и для его действия необходимы ионы М§2+ и К+.
8.7. Регуляция активности ферментов
Регуляция ферментативной активности может достигаться следу-
ющими путями: 1) путем регуляции количества фермента за счет
регуляции скорости его синтеза и распада; 2) с помощью аллостеричес-
кой регуляции; 3) путем ковалентной модификации ферментов.
Регуляция содержания фермента за счет регуляции скорости
его синтеза и распада. Вполне очевидно, что количество фермента
увеличивается при повышении скорости его синтеза. Скорость
синтеза может возрастать в ответ на действие специфических
низкомолекулярных соединений — индукторов. Процесс ускорения синтеза
фермента под действием индуктора называется индукцией. Снижение
скорости синтеза фермента может происходить в результате репрессии.
Например, при наличии определенного соединения в питательной
среде в бактериальных клетках снижается или полностью подавляется
синтез данного соединения. Это происходит за счет блокирования
синтеза фермента под действием корепрессора (например, какой-либо
аминокислоты). Концентрация ферментов в клетке зависит также от
скорости их деградации, происходящей под действием специфических
протеаз.
Аллостерический механизм регуляции активности
ферментов. Важную роль в регуляции обмена веществ в клетке играют алло-
стерические ферменты (от гр. айо — другой, 81егеоз — место), т.е.
ферменты, имеющие кроме каталитического центра дополнительный
центр связывания — аллостерический центр. Активность аллостери-
ческих ферментов регулируется путем изменения конформации
молекул ферментов, вызванного присоединением к ним специального
метаболита-регулятора к аллостерическому центру. Метаболит-регул я-
294
Глава 8. Ферменты
тор, называемый аллостерическим эффектором или модулятором,
выполняет функцию либо активатора, либо ингибитора. Если в роли
активирующего модулятора выступает сама молекула субстрата, то
такие аллостерические ферменты относят к гомотропным. Если роль
эффектора играет какая-либо молекула, отличающаяся по строению
от молекулы субстрата, то такие аллостерические ферменты
называют гетеротройными. Аллостерический центр физически не совпадает
с каталитическим центром, он пространственно локализован в другом
месте; однако связывание эффектора с ним приводит к таким конфор-
мационным изменениям в молекуле фермента, которые затрагивают и
каталитический центр» изменяя его активность.
Необходимые для организма метаболиты образуются в
большинстве случаев в результате цепочки биохимических превращений. Если
в этом метаболите организм испытывает недостаток, то усиливается
его биосинтез за счет активации ферментов этой цепочки. В случае
накопления метаболита в избытке выключается вся цепочка реакции,
ведущая к его синтезу. Поэтому в такие цепи, как правило, включены
одна или несколько реакций, скорость которых регулируется аллосте-
рическими ферментами.
Ингибирование аллостерического фермента, катализирующего одну
из реакций цепи, конечным продуктом этой цепи называют ингибиро-
ванием по принципу обратной связи. В цепи реакций биосинтеза О из
А, катализируемой ферментами Ег„Еп, при высокой концентрации В
обычно наблюдается ингибирование превращения А в В. Накопившийся
конечный продукт В способен связываться с ферментом Ех как
аллостерический ингибитор по принципу обратной связи. Таким образом,
ингибированием Ех под действием ТУ регулируется синтез О.
Аллостерический ингибитор
4Г 1
А-*||-> В->С-*... ->0
Е1 Е2 Еп
Существует несколько вариантов ингибирования по принципу
обратной связи. Так, кумулятивное ингибирование осуществляется
действием двух или более конечных продуктов и носит аддитивный
характер. При кооперативном ингибировании подавляющее действие
сразу двух и более конечных продуктов носит синергетический
характер - суммарное действие конечных продуктов намного превышает
аддитивный эффект, характерный для кумулятивного ингибирования.
Кинетическая зависимость активности аллостерического фермента от
концентрации субстрата, V = ^([8]), отличается от таковой для обычных
ферментов. На рис. 8.16 представлена зависимость V — ОД8]) в отсутствие
и в присутствии аллостерического ингибитора. Если для обычных
ферментов эта зависимость носит гиперболический характер, то для алло-
стерических ферментов она имеет сигмоидальную форму (кривая 1). Эта
295
Часть II. Метаболизм веществ и энергии в клетке
ЕЗ]
Рис* 8.16. Зависимость
активности аллостерического фермента от
концентрации субстрата в отсутствие
(кривая 1)ив присутствии
(кривая 2) аллостерического ингибитора
форма еще отчетливее выражена при
действии на фермент аллостерического
ингибитора (кривая 2).
Сигмоидная форма кривых
свидетельствует о том, что кинетика
действия аллостерических ферментов
имеет достаточно сложный характер и не
описывается уравнением Михаэлиса—
Ментен* В объяснении сигмойдного
характера кинетической кривой получил
широкое признание эффект
кооперативное™. Как правило, молекула
аллостерического фермента олигомерна,
она может состоять из двух и более
субъединиц, содержать несколько
каталитических и аллостерических центров* Основываясь на эффекте кооператив-
ности и олигомерности аллостерических ферментов, в 1965 г, Жан Моно,
Джефри Уайман и Жан-Пьер-Шанже создали модель, достаточно четко
объясняющую сигмоидную форму кинетической кривой действия
аллостерического фермента. Эту модель авторы назвали моделью
согласованного механизма.
Используя эту модель, рассмотрим действие аллостерического
фермента, состоящего из двух идентичных субъединиц. Предположим, что
обе субъединицы могут находиться в двух конформациях — N и А. Кон-
формация N обладает низким сродством к субстрату, тогда как конфор-
мация А — высоким (рис. 8Д7 ).
Формы N и А могут переходить
одна в другую, но при допущении:
либо в №, либо в А-состояния; не
разрешено состояние КА. На рис. 8.18
показан процесс активации
аллостерического фермента субстратом. В
отсутствие субстрата молекула
фермента находится в К-форме. Внесение
субстрата сдвигает конформационное
равновесие в сторону образования
И-форма
А~форма
Рис. 8.17. Схематическое
изображение конформации
аллостерического фермента в неактивной (И)
и активной (А) формах
+3
Ы-форма
<2>
А8-форма
+8
А88~форма
Рис. 8.18. Модель согласованного механизма кооперативного связывания
субстрата аллостерическим ферментом. Присоединение молекулы субстрата 8 ведет
к переходу неактивной Ы-формы в активную А-форму с высоким сродством к
субстрату
296
Глава 8. Ферменты
А8-формы. Молекула субстрата» присоединяясь к каталитическому
центру одной субъединицы фермента, индуцирует активность
каталитического центра другой субъединицы и ведет к образованию А88-формы*
Не вызывает затруднений представить такую же модель аллосте-
рической активности и для ферментов с более высоким уровнем
четвертичной структуры.
На основании предложенной авторами модели можно понять
действие аллостерических ингибиторов и активаторов, Аллостерический
ингибитор переводит А-форму в Ы-форму, тогда как аллостерический
активатор переводит Ы-форму в А-форму (рис. 8Л9).
А-форма N-форма Ы- форма А-форма
Рис. 8.19. В соответствии с моделью согласованного механизма
аллостерический ингибитор • переводит А-форму в М-форму; аллостерический активатор ▼
переводит N-форму в А-форму
Ковалентиая модификация ферментов. Регуляция
каталитической активности ферментов может осуществляться за счет ковалентного
присоединения фосфатной группы или нуклеотида. Для млекопитающих
преобладающим является механизм фосфорилирования-дефосфорилиро-
вания, и одна из форм фермента (дефосфофермент или фосфофермент)
характеризуется более высокой каталитической активностью. Например,
более высокой каталитической активностью обладает фосфорилирован-
ная форма гликогенфосфорилазы. Для гликогенсинтазы характерна
более высокая активность в дефосфорилированнои форме. Присоединение
фосфатной группы обычно осуществляется к специфическим остаткам
серина или тирозина. Фосфорилирование катализируется протеинкина-
зами, а дефосфорилирование — протеинфосфатазами; активность
конвертирующих ферментов часто подвержена гормональному контролю-
Адсорбционный механизм регуляции активности ферментов.
Структурно-функциональное значение образования метаболонов.
Известно, что ферменты способны обратимо связываться со
структурными компонентами клетки (структурными белками мышц,
мембранами, цитоскелетом), что может приводить к изменению их
каталитических и регуляторных свойств. Предположение, что адсорбция
ферментов на субклеточных структурах может иметь регуляторное
значение, впервые было высказано А. И. Опариным. В настоящее время
известны многочисленные факты, подтверждающие эту точку зрения.
Например, при адсорбции б-фосфофруктокиназы на мембранах
эритроцитов снижается чувствительность фермента к ингибированию под
действием АТР, а также исчезают кооперативные свойства по
отношению к субстрату — фруктозо~6-фосфату.
297
Часть II. Метаболизм веществ и энергии в клетке
Изменения свойств ферментов при связывании с субклеточными
структурами могут быть сопряжены со стерическим экранированием
аллостерических или активных центров или же с модификациями
конформации белковой молекулы, происходящими при адсорбции
фермента. Важное значение для реализации адсорбционного
механизма регуляции ферментативной активности имеет существование
обратимого равновесия между связанной и свободной формами фермента.
Причем это подвижное равновесие чувствительно к концентрациям
клеточных метаболитов.
С субклеточными структурами могут связываться не только
индивидуальные ферменты, но и комплексы ферментов. Предположение о
существовании на мембране эритроцитов комплекса ферментов,
катализирующих процесс гликолиза {см. гл. 9), было высказано в 1965 г.
Д. Е. Грином с сотрудниками; позднее это предположение было
подтверждено рядом экспериментальных данных, свидетельствующих о
существовании полиферментных комплексов гликолитических
ферментов при физиологических условиях рН и ионной силы. В настоя-
щее время Б. И. Кургановым с сотрудниками созданы гипотетические
модели структур комплекса гликолитических ферментов в скелетных
мышцах, а также на внутренней поверхности мембраны эритроцитов.
Считают, что возможность объединения гликолитических ферментов
в комплекс обеспечивается существованием у них центров узнавания
своих "соседей". Специфичность структурной и пространственной
организации метаболона обеспечивается за счет наличия у формирующих
его ферментов не только активного центра, но и двух центров, с
помощью которых осуществляется взаимодействие с соседними
макромолекулами. При этом однозначность сборки достигается тем, что в ней
принимает участие так называемый якорный белок подложки, на
которой формируется комплекс. Комплекс гликолитических ферментов
формируется на тонких нитях миофибрилл мышечных волокон,
образованных Г-актином и регуляторными белками — тропомиозином и
тропонином, или на димерах белка, образующих так называемую
полосу 3 в мембране эритроцитов. Полагают, что физиологическое
значение формирования комплекса ферментов, участвующих в одном
метаболическом пути, на подложке биологической природы состоит в
том, что клетка получает возможность регулировать метаболическую
систему как единое целое под действием определенных воздействий
на "якорный" белок подложки. Метаболоны являются мобильными
образованиями, находящимися в подвижном равновесии со
свободными ферментами; и это равновесие контролируется клеточными
метаболитами. Ферменты, связанные общими метаболитами или кофер-
ментами, находятся в метаболоне рядом друг с другом, и это
обеспечивает эффективное продвижение интермедиатов по конвейеру
активных центров в микрокомпартменте, образующемся при сборке
метаболона*
298
Глава 8. Ферменты
А. Е. Любаревым и Б, И* Кургановым предложена также
гипотетическая структура метаболона, объединяющего ферменты цикла три-
карбоновых кислот (см. гл. 9). Сборка метаболона осуществляется на
внутренней митохондриальной мембране, и комплекс ферментов как
бы "зажат" между противоположными поверхностями внутренней
мембраны митохондрий. Ключевую роль при сборке метаболона
играет адсорбция ос-кетоглутаратдегидрогеназного комплекса на
мембране. В состав якорной площадки входит один из ферментов цикла три-
карбоновых кислот — сукцинатдегидрогеназа, являющийся
интегральным белком внутренней митохондриальной мембраны. Взаимодействия
ферментов, входящих в состав метаболона цикла трикарбоновых
кислот, обеспечивают функционирование этого надмолекулярного
комплекса ферментов как целостной, кооперативно действующей системы.
8.8. Множественные молекулярные формы ферментов
Многие ферменты существуют в нескольких молекулярных
формах, присутствующих в разных тканях одного организма, в разных
типах клеток одной и той же ткани или даже в одной и той же клетке.
Эти формы фермента катализируют одну и ту же реакцию, но они
могут существенно различаться по свойствам: молекулярной массе,
значению Км, оптимуму рН, чувствительности к активаторам и
ингибиторам, субстратной специфичности, аминокислотному составу,
иммунологическим характеристикам, электрофоретической
подвижности. Такие формы ферментов называют изоформами (изозимами) или
изоферментами. Однако, строго говоря, термин "изофермент" может
употребляться только в отношении различно генетически
детерминированных форм фермента. Поскольку эти формы являются продуктами
разных генов, то, естественно, они отличаются по первичной структуре.
Изоформы, кодируемые одним геном и имеющие одинаковую
аминокислотную последовательность, приобретают некоторые отличия в
структуре в ходе посттрансляционной модификации белковых молекул.
Известно несколько механизмов образования изоферментов,
основным из которых считают различия в четвертичной структуре
молекул. При этом образуются гетерополимеры (гибриды) двух или
большего числа нековалентно связанных полипептидных цепей.
Примером может служить лактатдегидрогеназа (ЛДГ), которая представляет
собой тетрамер, состоящий из двух типов субъединиц: Н и М. В
тканях животных ЛДГ представлена в виде пяти изоферментов ЛДГ: Н4 —
ЛД1\; НЗМ1 — ЛДГ2; Н2М2 — ЛДГ3; Н1МЗ — ЛДГ4; М4 — ЛДГ5.
Синтез субъединиц находится под контролем соответствующих генов.
Изоферменты ЛДГ значительно различаются по максимальной
скорости (Ушах ) и константам Михаэлиса (Км) для субстратов. Другой
причиной полиморфизма ферментов может быть генетическая
вариабельность, определяемая синтезом на разных аллелях одной гетерозяготы.
299
Часть II, Метаболизм веществ и энергии в клетке ^ _____
Генетические варианты (аллелозимы) характерны, например, для глю-
козо-6-фосфатдегидрогеназы человека. В результате химической
модификации ферментов (фосфорилирование или аденилирование)
возникают такие формы, как метазимы. Формы ферментов,
различающиеся по конформации, образуют группу так называемых конформе-
ров. Такие ферменты способны существовать в разных конформацион-
ных состояниях, характеризующихся различными модификациями
третичной структуры при одной и той же аминокислотной
последовательности полипептидной цепи. К таким формам принято относить
все аллостерические модификации ферментов. К изоформам могут быть
также отнесены разные полимеры, образующиеся из одной
субъединицы. Так, например, глутаматдегидрогеназа может существовать в
виде таких полимерных форм с различной молекулярной массой.
Распространение множественных молекулярных форм ферментов
в разных тканях и органах может быть сопряжено с различными
факторами. Так, часто существование изоформ связано с различной
субклеточной локализацией и физиолого-биохимической ролью
конкретного фермента в клетке. Формы ферментов, различающиеся
чувствительностью к аллостерическим эффекторам, необходимы для тонкой
и плавной регулировки обменных процессов в клетке. Другим
фактором, определяющим распространение изоформ, является дифферен-
цировка тканей, происходящая при развитии взрослого организма из
эмбрионов. Существование разных форм ферментов может быть
также связано с особенностями обмена веществ в разных органах. Так,
изофермент М4 — ЛДГ5 в скелетной мышце предпочтительно
катализирует реакцию восстановления пирувата в лактат при низких
концентрациях пирувата. Изофермент Н4 предпочтительно катализирует
окисление лактата в пируват в сердечной мышце.
Характерно, что изоферментные спектры могут значительно
изменяться при адаптации организмов к условиям окружающей среды или
при развитии различных форм патологий. Последнее обстоятельство
представляет интерес для клинической диагностики некоторых
заболеваний. Так, при остром инфаркте миокарда в сыворотке крови обычно
нарастают фракции ЛД1\ и ЛДГ2. Увеличение фракций ЛДГ4 и ЛДГ5
свидетельствует о патологических процессах в печени (гепатиты,
циррозы и др.). Диагностическое значение имеют изоферментные
спектры и других ферментов: креатинкиназы, щелочной фосфатазы, алко-
гольдегидрогеназы и т.д.
8.9. Методы исследования ферментативного катализа
Прежде всего необходимо отметить: поскольку ферменты —
вещества белковой природы, подходы к исследованию конформации их
молекул остаются в принципе теми же, что и для белков (гл. 1). Здесь
речь будет идти о специфических особенностях этой конформации,
связанной с биологической функцией ферментов.
зоо
Г лив а 8. Ферменты
Исследование механизма ферментативного катализа — одна из
важнейших проблем энзимологии. К сожалению, пока ни для одного
фермента не дано исчерпывающего описания механизма его действия; во
многом еще не разгадан столь огромный каталитический эффект
действия ферментов.
Активность ферментов. Определение количественного
содержания ферментов в биологических объектах представляет известные
трудности, поскольку, за редким исключением, ферменты в клетках
находятся в ничтожно малых концентрациях. Поэтому непосредственно
определяют не концентрацию фермента, а его активность. Об
активности фермента судят по скорости ферментативной реакции, т.е. по
скорости убыли субстрата или скорости образования продуктов
реакции (последнее предпочтительнее). Единица активности фермента
определена как количество фермента, катализирующего превращение
одного микромоля субстрата за одну минуту при оптимальных
значениях рН и температуры. Удельная активность представляет собой
число единиц фермента в 1 мг белка. Она широко используется при
очистке ферментов, поскольку служит критерием чистоты и
возрастает по мере его очистки. Молекулярная активность (число оборотов)
фермента представляет собой число молекул субстрата, которое
превращается молекулой фермента за 1 мин. В системе СИ за единицу
активности фермента принят катал (кат) — это такое количество
фермента, которое катализирует превращение 1 моля субстрата за 1 с.
При определении активности фермента в растворе, в культураль-
ной жидкости или экстракте, выделенном из органов животных,
растений, а также бактерий, необходимо знать химическое уравнение,
описывающее катализируемую реакцию; возможную потребность в
кофакторе; область температур, при которых фермент устойчив и со-
храняет высокую активность. Определение активности
предпочтительнее проводить в стандартных условиях, при которых поддерживаются
оптимальное значение рН и концентрации субстрата, превышающие
концентрации насыщения фермента. В этом случае начальная
скорость соответствует реакции нулевого порядка и пропорциональна
только концентрации фермента.
Выделение и очистка ферментов. В клетках животных,
растений, микроорганизмов, в биологических и культуральных жидкостях
ферменты находятся вместе с большим количеством других
соединений. В ходе исследований ферментов они могут вносить существенные
погрешности в полученные данные, в связи с чем часто оказывается
необходимым получение ферментов в очищенном состоянии.
Очищенный от балластных примесей фермент называется ферментным
препаратом.
Используют различные методы очистки. Какой-либо стандартной,
универсальной схемы очистки ферментов нет, так как структурные и
физико-химические различия ферментов чрезвычайно велики.
301
Часть II. Метаболизм веществ и энергии в клетке
Если мы имеем дело с внутриклеточными ферментами, прежде всего
необходимо разрушить клетки и перевести фермент в раствор.
Клеточную оболочку можно разрушить замораживанием и оттаиванием
клеточной массы, гомогенизацией с измельченным стеклом, действием
ультразвука и другими специальными методами. В некоторых случаях
целесообразна обработка клеток ацетоном, при атом клеточные стенки
разрушаются, биомасса обезвоживается, а фермент остается в составе сухого
порошка, из которого его затем экстрагируют буферным раствором.
Независимо от способа разрушения клеток первичный экстракт,
содержащий фермент, обычно представляет собой суспензию
разрушенных клеток в буферном растворе, часто содержащем добавки
различных солей, антиоксидантов, стабилизаторов выделяемого фермента.
На следующем этапе очистки ферментов эту суспензию
центрифугируют, чтобы отделить неразрушенные клетки, обрывки клеточных
оболочек, а в ряде случаев ядра, митохондрии и другие клеточные
органеллы. Надосадочная жидкость, полученная после
центрифугирования, содержит слойсную смесь ферментов, структурных белков,
нуклеиновых кислот, полисахаридов и других соединений. Для
удаления балластных веществ часто применяют методы их осаждения.
Например, нуклеиновые кислоты осаждают стрептомицинсульфатом.
Для удаления белковых примесей применяют их осаждение
сернокислым аммонием, органическими растворителями (ацетоном, этанолом,
изопропанолом). Удаление сопутствующих веществ может быть
достигнуто с помощью термической обработки или же путем обработки
кислотой. Для удаления низкомолекулярных примесей применяют диа-
лиз или фильтрацию через интактные, химически инертные,
диспергированные в стеклянной колонке гелеобразные наполнители декстрано-
вой природы, например, сефадексы различных марок (О-10, 0-15, 0-25).
Для глубокой очистки используют более тонкие методы очистки —
электрофорез, изоэлектрофокусировку, тонкослойную хроматографию,
аффинную хроматографию и др.
К наиболее эффективным методам очистки относится колоночная
хроматография.
Ионообменная хроматография является одним из способов
колоночной хроматографии, используемых для разделения белков с
близкими физико-химическими свойствами. В основе ионообменной
хроматографии лежат реакции ионного обмена между белками и
различными ионообменниками. Ионообменники представляют собой
нерастворимые в воде матрицы, имеющие положительно и отрицательно
заряженные функциональные группы. Принято различать анионооб-
менные и катионообменные адсорбенты. К катионообменникам
относится, например, карбоксиметилцеллюлоза (КМ) — целлюлоза,
содержащая отрицательно заряженную группировку: -0-СН2-СОО-. К
довольно широко используемым анионообменникам относится диэтилами-
ноэтил (ДЭАЭ) — целлюлоза, содержащая группу: -0~СН2-СН2-К(С2Н5)2.
302
г
Глава 8. Ферменты
При взамодействии белков с ионообменниками в ходе адсорбции
образуются множественные электростатические связи между
функциональными группами на поверхности матрицы и группами белковых
молекул, несущими противоположный заряд. Прочность связывания
каждого белка на матрице зависит от ряда факторов и, в первую очередь,
от величины заряда белка, зависящей от рН и ионной силы раствора.
При пропускании через колонку определенного элюирующего раствора
происходит хроматографическое разделение белков в зависимости от
степени их взаимодействия с ионообменником. Разрыв связей достигается
путем изменения рН или ионной силы протекающего раствора.
Весьма высокоэффективным методом очистки ферментов является
аффинная хроматография (хроматография по сродству). Белковые
молекулы, как известно, способны к специфическому связыванию с
определенными соединениями (лигандами), с которыми белок
взаимодействует 1п ишо, либо со структурными их аналогами,
синтезированными в лабораторных условиях. Лиганд, роль которого часто играют
субстрат, ингибитор или эффектор, иммобилизуют в полимерной
стационарной фазе, заполняющей колонку. Матрикс колонки
избирательно связывает фермент из раствора. По мере продвижения белковой
смеси по колонке фермент оказывается единственным отстающим
белком. Все остальные белки проходят через колонку без задержки. Для
эффективной избирательной элюции фермента с матрикса часто бывает
достаточно изменить рН элюирующего раствора или включить в его
состав соединение, конкурентно вытесняющее фермент из комплекса с ли-
гандом, связанным с носителем. Аффинная хроматография часто
используется для очистки ДНК-связывающих белков; при этом на носителе
иммобилизуют олигонуклеотиды ДНК определенной структуры.
Широкое применение получил метод хроматографии
гель-фильтрацией. При гель-хроматографии наиболее широко используются се-
фадексы, биогели. Заполняющий колонку материал выполняет роль
своеобразного "молекулярного сита". Выпускаются сефадексы с
разными размерами частиц и различной частотой поперечных связей. На
колонках с сефадексами 0-75, О-100, 0-150, О-200 можно
осуществлять фракционное разделение белков по размерам. Поэтому
гель-фильтрация может использоваться не только для очистки, но и для
определения молекулярной массы ферментов. Для проведения
гель-хроматографии может быть использован также биогель, представляющий
собой сшитый полиакрил амид. Подобно сефадексам разные марки
биогелей отличаются по пределам фракционирования белков в
соответствии с их молекулярными массами.
Существует также гидрофобная хроматография, применяемая для
разделения белков в зависимости от степени их гидрофобности.
Колонки для гидрофобной хроматографии заполнены специальными
носителями с выступающими гидрофобными цепями. На таком матрик-
се задерживаются белки с обнаженными гидрофобными участками. В
зоз
Часть II. Метаболизм веществ и энергии в клетке
качестве носителей при гидрофобной хроматографии могут
использоваться октил- или фенилсефароза*
Электрофорез является ценным физико-химическим методом,
применяемым для разделения белковых смесей, а также для
характеристики чистоты ферментных препаратов. Этот метод обладает большой
разрешающей способностью и применяется обычно для
препаративного выделения ферментов на последних стадиях очистки. Если белки
не находятся в изоэлектрической точке, где суммарный заряд равен
нулю, то под действием электрического поля они будут перемещаться
с определенной скоростью, которая зависит от суммарного заряда, а
также формы и размера белковой молекулы. Это явление и лежит в
основе многих вариантов электрофоретического разделения белковых
смесей в свободных водных растворах и твердом пористом матриксе,
применяемых как с целью разделения и очистки определенных форм
ферментов, так и с целью подтверждения их гомогенности.
Исследование механизма ферментативного катализа.
Механизм ферментативной реакции можно считать установленным, если
охарактеризованы все промежуточные соединения, комплексы и кон-
формационные состояния фермента и определены константы
скоростей их взаимопревращений. В развитии представлений о механизме
действия ферментов важную роль играет исследование активных
центров ферментов и, в первую очередь, идентификация функциональных
групп, участвующих в акте катализа и связывания субстрата. По
кривым зависимости "активность — рН" можно приближенно найти
значения рК и определить химическую природу функциональных групп,
принимающих участие в акте катализа. Однако данные по величинам
рК пригодны лишь для предварительного заключения. Вследствие
образования ковалентных, ионных или координационных связей и
индуктивного воздействия соседних групп в молекуле фермента
величина рК одной и той же группы может колебаться в широких
пределах и перекрывать константу диссоциации другой группы.
Для однозначной идентификации групп, входящих в активный
центр фермента, необходимо сочетание нескольких независимых
экспериментальных критериев. Лишь их совокупность может привести к
достаточно надежному заключению. Одним из критериев может быть
расчет теплоты ионизации этих групп.
Важным методом является подавление активности фермента
специфическими ингибиторами. При помощи блокирования
функциональных групп химическими реагентами получено немало данных о
природе, расположении и роли групп, существенных для действия
отдельных ферментов. Однако и в данном случае трудно найти такой
реагент, который вступил бы во взаимодействие лишь с какой-либо
одной группой. Поэтому данные по идентификации групп
химическими методами не всегда надежны, иногда противоречивы. Правда,
некоторые из специфических реагентов зарекомендовали себя практи-
304
Глава 8. Ферменты
чески безошибочными метчиками функциональных групп. Например,
п-хлормеркурибензоат и диизопропилфторфосфат ковалентно и
весьма специфично присоединяются к сульфгидрильным группам цистеи-
на и гидроксильным группам серина соответственно. Таким образом,
если один из упомянутых реагентов — ингибитор, то это хорошее
экспериментальное свидетельство в пользу участия 8Н-групп цистеина
пли ОН-групп серина в механизме ферментативного катализа..
Типичной реакцией на имидазольную группу гистидина является
ее фотоокисление в присутствии метиленового синего. В имидазоль-
ком кольце световая энергия вызывает большой эффект возбуждения,
в результате чего это кольцо окисляется с разрывом. Таким образом,
если в активный центр фермента входит группа имидазола, то
фермент в растворе метиленового синего на свету подвергается
необратимой инактивации. Достаточно специфическим реагентом на
имидазольную группу гистидина является диэтилпирокарбонат.
Величины рК и теплоты ионизации функциональных групп,
инактивация фотоокислением в присутствии метиленового синего
позволили установить, что активными группами амилаз являются имида-
зольная и карбоксильная- На основании этих данных, а также гидро-
лиза крахмала в тяжелой воде (Н2018) был предложен механизм
разрыва ос-1,4-глюкозидной связи в молекуле крахмала под действием
амилаз, описанный в разд. 8.3.
Одним из наиболее эффективных подходов к идентификации и
изучению роли функциональных групп активного центра ферментов
является метод направленного мутагенеза, заключающийся в
замене определенных аминокислотных остатков в белковой молекуле с
целью выяснения их значения в акте катализа.
Краткое содержание главы
Ферменты — биологические катализаторы белковой природы,
способные во много раз ускорять химические реакции, но не входящие в
остав конечных продуктов реакции. Они катализируют только
энергетически выгодные реакции и не изменяют направление реакции. В
отличие от неорганических катализаторов, ферменты имеют
следующие особенности: 1) эффективность их действия на несколько
порядков выше, чем небиологических катализаторов; 2) действие их
высокоспецифично; 3) они катализируют реакции в мягких условиях.
Ферменты играют важную роль в регуляции клеточного обмена
веществ. Регуляция ферментативной активности осуществляется под
действием температуры, рН среды, концентрации субстрата,
специфических активаторов или ингибиторов. Особую роль в регуляции обменных
процессов играют регуляторные ферменты, к которым относятся: а) ал-
лостерические ферменты, активность которых изменяется с помощью
нековалентно связываемых аллостерических модуляторов; б) ферменты,
изменяющие свою активность путем ковалентной химической модифи-
20. Заказ 3486 305
Часть II. Метаболизм веществ и энергии е клетке
кации. Существенное значение для регуляции клеточных процессов
имеют изменения концентрации ферментов в результате стимулирования
или подавления их биосинтеза или же изменения скорости деградации.
Установление белковой природы ферментов было выдающимся
открытием, подчеркнувшим огромную биологическую роль белков в
живой материи. Ферменты могут быть как однокомпонентными
(протеинами), так и двухкомпонентными. Двухкомпонентные ферменты
состоят из белковой части и небелковой части — кофактора. Оба
компонента в отдельности лишены ферментативной активности. В
качестве кофактора могут быть либо ионы металлов, либо витамины, либо
нуклеотиды. В большинстве случаев ферменты являются
глобулярными белками и акт катализа совершается на определенном участке
белковой глобулы, называемым активным центром фермента.
Важнейшая функция ферментов в акте катализа — это снижение
энергии активации, необходимой для протекания химической
реакции, что связано с образованием фермент-субстратного комплекса и
его переходом в активное состояние. В молекуле фермента электро-
фильные и нуклеофильные группы активного центра оказывают
сильное поляризующее действие на группы субстрата, что и является
причиной снижения энергии активации в акте катализа.
Важное свойство ферментов — не только ускорять реакцию, но и
избирательно действовать на субстрат и определять путь его
превращения, что получило название субстратной специфичности. Для
ферментов может быть характерна абсолютная, относительная
(групповая) и стереохимическая специфичность.
К ферментативным реакциям приложимы общие принципы
химической кинетики. Однако у этих реакций имеется отличительная
особенность, оказывающая существенное влияние на кинетические
закономерности действия ферментов — образование комплекса
фермент-субстрат. Скорость ферментативной реакции зависит от
химической природы субстрата и условий взаимодействия с ним фермента
(температуры, рН, сопутствующих веществ), от концентрации
фермента и субстрата. В замкнутой системе при полном насыщении
фермента субстратом в начальный период химическая реакция проходит
через стадию нулевого порядка, а затем, по мере снижения
концентрации субстрата, — через стадии дробного и первого порядков.
Важнейшими кинетическими характеристиками ферментов
являются константы Михаэлиса Км и максимальная скорость реакции У^ .
Константа Км представляет собой концентрацию субстрата, при
которой скорость реакции составляет половину максимальной, ее
размерность — моль'л-1. Она является мерой сродства фермента к субстрату:
чем меньше Км, тем выше это сродство. Константа Утах выражает
эффективность действия фермента.
На активность ферментов большое влияние оказывают
активаторы — химические соединения, повышающие действие ферментов, и
306
Глава 8. Ферменты
ингибиторы — специфические химические соединения, подавляющие
их активность. Процесс ингибирования может быть обратимым и
необратимым. Обратимые ингибиторы бывают конкурентного,
неконкурентного, бесконкурентного и смешанного действия.
Классификация ферментов осуществлена по типам реакций,
которые они катализируют. Согласно этому принципу все ферменты
делятся на шесть классов: оксидоредуктазы, осуществляющие
окислительно-восстановительные реакции; трансферазы, переносящие
различные химические группы от одного соединения к другому; гидролазы,
расщепляющие субстраты с участием воды; лиазы — ферменты,
осуществляющие реакции присоединения групп по двойным связям и
обратные реакции; изомеразы — переводящие соединение в его
изомер; лигазы — ферменты синтеза.
Ферменты могут существовать в виде физически различимых
множественных молекулярных форм, обладающих одним и тем же видом
каталитической активности, но различающихся по своим свойствам
(например, Км и V . рН-оптимум, чувствительность к ингибиторам и актива-
IV! ХИН л.
торам и т.д.). Эти формы могут присутствовать в разных тканях
организма, в разных клетках ткани или даже в одной и той же клетке.
Хронология важнейших дат в исследований фермейтов
1814 г, — К. Кирхгоф показал, что в прорастающем зерне пшеницы
содержится вещество, способное превращать крахмал в сахар и декстрины.
1833 г. — А. Пайен и Ж. Ф. Персо выделили из солода вещество, названное
ими диастазом (ныне известное как амилаза),
1836 г. — Т. Шванн описал фермент пепсин (рерЪо — варю),
1839 г. — Ю. Либих развил представление о метастабильном (неустойчивом)
состоянии веществ, заложил основы представлений о механизме
ферментативного катализа.
1877 г. — Ф. Кюне предложил термин "энзим" (фермент).
1894 г. — Э. Фишер продемонстрировал специфичность ферментов и
показал, что взаимодействие между ферментом и субстратом осуществляется по
принципу ключ—замок.
1897 г, — Г. Э. Бертран предложил термин "коэнзим" (кофермент),
1913 г. — Л. Михаэлис и М. Ментен разработали кинетическую теорию
действия ферментов.
1925 г. — Д. Кейлин показал, что цихромы — это переносящие электроны
белки, молекулы которых содержат простетические группы — темы.
1925 г. — Дж. Бриггс и Дж. Холдейн внесли существенные изменения в
теорию кинетики ферментативных реакций.
1926 г. — Д. Самнер получил в кристаллической форме фермент уреазу и
доказал, что он является белком.
1930^1933 гг. — Дж. Нортроп получил пепсин и трипсин в
кристаллическом виде и доказал их белковую природу.
1933 г. — О. Варбург открыл "желтый дыхательный фермент" (флавопротеин),
1933 г. — Д. Кейлин выделил цитохром с и воспроизвел процесс переноса
электронов в препаратах сердечной мышцы,
1935 г. — Р. Кун показал, что рибофлавин (витамин В2) входит в состав
"желтого фермента".
20*
307
Часть II, Метаболизм веществ и энергии в клетке
1935—1936 гг. — О. Варбург и Л. Б. Эйлер выделили пиримидиновые нуклео-
тиды, изучили их структуру и функции в дегидрогеназах.
1937 г. — К. Кори и Г. Кори начали свои исследования гликоген-фосфори-
лазы.
1943 г. — В. Чане впервые использовал Чувствительные фотометрические
методы для изучений фермент-субстратных взаимодействий.
1946 г. — Дж. Самнеру, Дж. Нортропу, У. Стенли присуждена Нобелевская
премия за открытие белковой природы ферментов.
1967 г. — Д. Филлипс, используя рентгеноструктурный анализ, установил
трехмерную структуру лизоцима и выяснил характер присоединения лизоцима
к субстрату (триацетилглюкозамину).
ЛИТЕРАТУРА
Албертс А, Брей Д„ Льюис Дж., Рэфф М., Роберте К., Уогпсон Дж.
Молекулярная биология клетки: В 3 т. — 2-е изд., перераб. и доп. — М.: Мир, 1994. —
Т. 1, — 515 с.
Аргпюхов В. Г., Ковалева Т. А., Шмелев В. Л. Биофизика. — Воронеж: Изд-во
Воронежского ун-та, 1994. — С, 88—138.
Березов Т. Т., Коровкин Б. Ф- Биологическая химия: Учебник.— 3-е изд.,
перераб. и доп. — М.: Медицина, 1998. — 704 с.
Брауншпгейн А. Е. На стыке химии и биологии. — М.: Наука, 1987. — 239 с.
Варфоломеев С. Д., Гуревич К. Г. Биокинетика: Практический курс. — М.;
ФАИР -Пресс, 1999. — 720 с.
Волькеншшейн М. В. Биофизика: Учеб- руководство, — 2-е изд., перераб. и
доп. — М.: Наука, 1988. — 592 с.
Диксон М., Уэбб Э. Ферменты: В 3 т. — М.: Мир, 1982. — 1117 с.
Дюга Э.> Пенни К. Биографическая химия. — М.: Мир, 1983. - 346 с.
Кнорре Д. Г., Мызина С. Д- Биологическая химия: Учебник. — 2-е изд., пе-
раб. и доп. — М.: Высш. шк., 1998. — 479 с.
Курганов Б. И. Аллостерические ферменты. — М.: Наука, 1978. — 234 с.
Ленинджер А. Основы биохимии: В 3 т. — М.: Мир, 1985. — Т.1. — С. 226—
269.
Марри Р., Греннер Д., Мейес П., Родэм В. Биохимия человека: В 2-х томах, —
М.: Мир, 1993. Т. 1. — С. 63—110.
Мецлер Д. Биохимия: В 3 т. — М.: Мир, 1980. ~~ Т.2. — С. 5—39.
Номенклатура ферментов / Под ред. А. Е. Браунштейна. — М,: ВИНИТИ,
1979. — 387 с.
Рубин А. Б. Биофизика: Учебник: В 2 т. — 2-е изд., испр. и доп. — М.:
Книжный дом "Университет", 1999. — Т. 1.: — 448 с.
Фридрих Л". Ферменты: четвертичная структура и надмолекулярные
комплексы. — М.: Мир, 1986. — 366 с.
ВОПРОСЫ И ЗАДАЧИ
1. Какой из коферментов принимает участие в превращении аминокислот:
а) тиаминпирофосфат; б) пиридоксальфосфат; в) ЫАВ; г) ГАВ; д) биотин?
2. В состав какого кофактора входит витамин В2: а) тиаминпирофосфата;
б) КАО; в) РАО?
3. Что такое абсолютная и относительная специфичность ферментов? Как их
объяснить с точки зрения теории индуцированного соответствия фермента и
субстрата?
308
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
4. Как зависит скорость действия ферментов от концентрации фермента,
субстрата?
5. В чем сущность ферментативного катализа? Как изменяется энергия
активации катализируемой реакции?
6. В реакции первого порядка сахароза —> инвертный сахар концентрация
сахарозы в начальный момент — 10 мМ, спустя 1 мин стала равной 5 мМ. Какой
она станет спустя 5 мин?
7. Определите тип ингибирования (конкурентный или неконкурентный)
ферментативной реакции по следующим данным:
Концентрация субстрата, мМ 2 4 8 16
Скорость реакции в отсутствие ингибитора, мМ>с-1 0,5 0,7 1,0 1,5
Скорость реакции в присутствии ингибитора, мМ*с-1 0,1 0,4 0,8 1,4
Глава 9 . КАТАБОЛИЗМ УГЛЕВОДОВ.
ГЕНЕРИРОВАНИЕ ЭНЕРГИИ ФОСФАТНОЙ СВЯЗИ
Отдельные биохимические реакции в живой клетке тесно связаны
между собой, образуют метаболические пути, или циклы, каждый из
которых выполняет определенную функцию. Принято различать
центральные и специальные метаболические пути. Первые являются
общими для распада и синтеза основных макромолекул, они
удивительно сходны у любых представителей живого мира. Вторые характерны
для синтеза и распада индивидуальных мономеров, макромолекул
(например, алкалоидов, терпенов, и т.д.), выполняющих какие-либо ре-
гуляторные или иные функции или просто являющиеся конечными
продуктами обмена.
В этой главе описаны основные пути катаболизма углеводов —
анаэробный и аэробный. Анаэробный путь катаболизма осуществляется
клеткой без доступа кислорода воздуха и называется брожением;
аэробный — идет с участием кислорода воздуха, представляет собой
окислительный процесс и называется дыханием. Конечные цели обоих
путей: а) синтез АТР за счет утилизации энергии, аккумулированной в
углеводах; б) получение из этих углеводов пластических соединений,
являющихся исходным материалом для синтеза компонентов клетки.
9Д. Гликолиз
Гликолиз (от греч. §1усуз — сладкий, 1у$1$ — разрушение) —
универсальный и основной процесс катаболизма углеводов для
большинства организмов; это последовательность реакций, приводящих к
превращению гексозы в.пируват с одновременным образованием АТР.
Благодаря исследованиям, проведенным К. Нейбергом, О. Варбургом,
О. Мейергофрм, Г. Эмбденом, Я. Парнасом, С. Костычевым, А.
Лебедевым, была выяснена последовательность реакций гликолиза,
представленная на рис, 9.1. Важнейшими моносахаридами, катаболизм
которых осуществляется по гликолитическому пути, являются В-глю-
коза и В-фруктоза. Однако и другие моносахариды способны.катабо-
лизироваться по пути гликолиза, поскольку они легко превращаются
в эти сахара. Гликолиз — процесс анаэробный, однако он может про-
текать как в отсутствие, так и в присутствии кислорода. Он является
ключевым метаболическим путем, генерирующим энергию в форме
АТР в клетках, где отсутствует фотосинтез.
310
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
сн2-он
Гликоген
{^^У у \| Глюкоао-1- фосфат
ноЧр^о-®
Н>ОН
НО
©
он
он
/\
АТР
Глюкоаа
АОР
Глюкоэо-6-фосфат
(р)-о-сна сн3-он
Фруктозоб-фосфат
©
(Р>Ч)-СН2 СНгО-<Р)
Фруктоз©-1 т6-бясфосфат
Н2С-0-@
С=0
Н/ХШ *щ
Д и га дрокеиацет онфосфат
Ы О
нс-он
I
Глицералъдегид-3-фосфат
Н3Р04
КАО+
АОР
ИАВН + Н*
С-О-®
I
НС-ОН 1,8-Бисфосфоглацерат
I
н^с-о-®
©
соо~
I
НС-ОН З-Фосфоглицерат
НгС-О^р)
соо"
I
нс-о-(р)
| 2-Фосфоглвцерат
Н2С-0Н
г©
АСР
СОО"
\
нс-он
СН3 Лактат
соо
I
с-см®
I Фосфоеволпнруват
сн.
Пируват
Рис. 9.1. Гликолитическии путь. Ферменты, функционирование которых
сопряжено с данным путем: / — гликогенфосфорилаза; 2 — гексокиназа; 3 —
фосфоглюкомутаза; 4 — глюкозофосфатизомераза; 5 — фосфофруктокиназа; 6 —
альдолаза; 7 — триозофосфатизомераза; 8 — глицеральдегидфосфатдегидрогена-
за; 9 — фосфоглицераткиназа; 10 — фосфоглицеромутаза; 11 — енолаза; 12 —
пируваткиназа; /3 — лактатдегидрогеназа
311
Г' -
Часть II. Метаболизм веществ и энергии в клетке
Исследования химизма гликолиза показали, что начальные этапы
процессов брожения и дыхания имеют общий путь. Это открытие было
уникальным, потому что оно вскрывало существование внутреннего
единства в живой материи. При дыхании у аэробных организмов
гликолиз предшествует циклу трикарбоновых кислот и цепи переноса
электронов. Пируват проникает в митохондрии, где он полностью
окисляется до С02, в результате чего с высокой эффективностью из гексо-
зы извлекается свободная энергия. При брожении, в анаэробных
условиях (дрожжи, молочнокислые бактерии), пируват превращается в
продукты брожения (этанол, лактат).
Спиртовое брожение
Гликолиз
О
с6н12о6
Глюкоза
СНз-С-СОО
Пируват
** СН3-СН2-ОН
Этанол
Молочнокислое брожение
СНз-СН-СОО
Дыхание
ОН
Лактат
*- СО» + НоО
Ниже представлены стадии процесса гликолиза. Все реакции
протекают в цитоплазме и катализируются десятью различными
ферментами > большинство из которых получены в кристаллическом виде и
хорошо изучены. Эти ферменты растворимы в воде, легко
экстрагируются из клеток*
Первая стадия гликолиза начинается с фосфорилирования гек-
соз и является "пусковой". Она требует затраты химической энергии в
виде АТР и обусловливает высокую реакционную способность
молекулы гексозы. Пусковая стадия включает: а) фосфорилирование
глюкозы (и других гексоз); б) их изомеризацию; в) второе
фосфорилирование; г) расщепление на триозофосфаты.
На этапе "а" осуществляется реакция между В-глюкозой и АТР,
приводящая к образованию глюкозо-6-фосфата:
ОН
СН2-0-Р=0
он
Гексокиназа
+ АТР
+ АВР
ОН
но4^—^ он
он
Глюкозо 6-фосфат
Реакция катализируется гексокиназой. В клетках имеется
несколько форм гексокиназ, некоторые из них относительно неспецифичны и
312
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
осуществляют также фосфорилирование других гексоз. Глюкозо-6-
фосфат может образовываться и без затраты АТР путем отщепления
глюкозильных остатков от крахмала или гликогена с помощью
специфических фосфорилаз.
На этапе "6м осуществляется изомеризация гексозо-б-фосфатов во
фруктозо-б-фосфат. На этом этапе все гексозы превращаются в один
фосфорилированный продукт. При действии на глюкозо-6-фосфат фос-
фоглюкоизомеразы образуется смесь, состоящая на 20 % из фруктозо-
6-фосфата и на 80 % — из глюкозо-б-фосфата:
о
н
СН2ОН
н-с-он с=о
НО-С-Н Фосфоглюкоизомераза ^.^н
н-с-он н-с-он
н-с-он н-с-он
СН2ОР СН2ОР
Глюкозо-6-фосфат Фруктозо-6-фосфат
За изомеризацией следует второе фосфорилирование (этап "в"). Под
действием фосфофруктокиназы и при участии АТР
фруктозо-б-фосфат превращается в фруктозо-1,6-бисфосфат:
СН2ОН СН2ОР
С=Ю С-0
НО-С-Н Фосфофруктокиназа >_н
I + АТР ► I + АВР
Н-С-ОН Н-С-ОН
Н-С-ОН Н-С-ОН
СН2ОР СН2ОР
Фруктозо-б-фосфат Фруктозо-1,0-бисфосфат
Почему метаболизм гексозы начинается с термодинамически
невыгодного этапа — присоединения двух макроэргических связей?
Ответ на этот вопрос прост: с кинетической точки зрения выгодно
запускать длинную последовательность реакций с практически
необратимой реакции, каковой и является фосфорилирование гексозы.
Потребление АТР на стадии фосфорилирования компенсируется его
образованием на поздних стадиях гликолиза.
Образование фруктозо-1,6-бисфосфата является наиболее
медленно текущей реакцией гликолиза. Она в целом и определяет скорость
всего процесса гликолиза, которая зависит от активности
фосфофруктокиназы. Таким образом, этот фермент играет важную роль в
регуляции скорости гликолиза, о чем будет сказано в дальнейшем.
313
Часть П. Метаболизм веществ и энергии в клетке
Далее следует превращение фруктозо-1,6-бисфосфата в триозофос-
фаты. Основная цель пусковой реакции сводится к образованию
соединения, легко расщепляемого на фосфорилированные трехуглерод-
ные фрагменты. Реакция изомеризации, перемещающая
карбонильную группу из положения С-1 в положение С-2, делает возможным
расщепление гексозы на два фрагмента. Из этих фрагментов на
последующих этапах извлекается энергия. Фруктозо-1,б-бисфосфат
расщепляется с образованием глицеральдегид-3-фосфата и дигидроксиацетон-
фосфата; эту реакцию катализирует альдолаза:
СН2ОР
СН2ОР
с=о
о-о
СН2ОН
гтп-г-тт Альдолаза ..
пи «_• л. Дигидроксиацетонфосфат 1 Триозофосфат
Н-С-ОН
Н-С-ОН ?"Н
СН2ОР
Фруктозо-1, б-бисфосфат
Н-С-ОН
+ / / изомераза
О
СН2ОР
Глицеральдегид-3-фосфат
После расщепления фруктозобисфосфата альдолазой в каждой из
образовавшихся половин оказывается фосфатная группа. Под действием
триозофосфатизомеразы происходит взаимопревращение фоефотриоз.
Равновесие этой реакции сдвинуто в сторону дигидроксиацетонфосфата:
на 96 % дигидроксиацетонфосфата приходится всего 4 % глицераль-
дегид-3-фосфата, но именно последний участвует в дальнейших
превращениях в процессе гликолиза. Благодаря высокой активности
триозофосфатизомеразы образование дигидроксиацетонфосфата не
лимитирует скорость гликолиза в целом. В то же время для
дигидроксиацетонфосфата существует и другой путь, связанный с
восстановлением в глицерофосфат — предшественник липидов.
Вторая стадия гликолиза начинается с окисления
глицеральдегид-3-фосфата, сопряженного с генерированием энергии в форме АТР.
Глицеральдегид-3-фосфат в присутствии неорганического фосфата
окисляется в 1,3-бисфосфоглицерат:
* Глицеральдегид-3- *
ЧН фосфат дегидрогеназа V \)~Р
Н-С-ОН ^Г ^\ Н-С-ОН
ЗСН2ОР р4 ИАВ+ ^БН + Н+ зСН^ОР
Глицеральдегид-3-фосфат 1,3-Бисфосфоглицерат
В этой окислительно-восстановительной реакции происходит
генерирование высокоэнергетического фосфатного радикала. Альдегидная
группа при С-1-атоме превращается в смешанный ангидрид фосфор-
314
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
ной и 3-фосфоглицериновой кислот. Энергия, высвобождающаяся при
окислении альдегидной группы, аккумулируется в этом ангидриде в
виде высокого потенциала переноса фосфатной группы. Реакция,
ведущая к образованию макроэргического фосфатного радикала,
получила название субстратного фосфорилирования. ДО0' гидролиза ацил-
фосфатной связи 1,3-бисфосфоглицерата равна -49,3 кДж-молеГ1*
Фосфорилирование на субстратном уровне катализирует глицераль-
дегид-3-фосфатдегидрогеназа* Эта дегидрогеназа была выделена из
дрожжей в кристаллическом виде, состоит из 4 идентичных
субъединиц, каждая из которых имеет молекулярную массу 35 000 и
содержит одну прочно связанную молекулу ЫАВ+. Каждая субъединица
имеет также 8Н-группу, играющую важную роль в акте катализа.
В следующей реакции 1,3-бисфосфоглицерат используется для
генерирования АТР; фосфатная группа высокоэнергетического ацилфос-
фата с участием фосфоглицераткиназы переносится на АВР с
образованием АТР и 3-фосфоглицерата:
1С 1С
I Х>~Р Фосфоглицераткиназа IХГ
н-с-он ^^ ^*\ н-с-он
ЗСН2ОР ДПР АТР ЗСН2ОР
1,3-Бисфосфоглицерат З-Фосфоглицерат
ДО0' реакции переноса — -49,3 + (+30,5) - -18,8 кДж-моль"1;
иными словами, эта реакция сильно сдвинута в сторону образования АТР.
В ходе последующих реакций гликолиза происходит образование
пирувата и второй молекулы АТР. Превращение 3-фосфоглицерата в
пируват начинается с переноса фосфатной группы от кислорода при С-3
на кислород при С-2, в результате чего 3-фосфоглицерат превращается в
2-фосфоглицерат. Эта реакция катализируется фосфоглицератмутазой:
,° *°
С С
I X)" Фоефоглицератмутаза _ | X)
Н-С-ОН « * Н-С-ОР
ЗСН9ОР СНоОН
З-Фосфоглицерат 2-Фосфоглицерат
Далее осуществляется реакция дегидратации 2-фосфоглицерата до
фосфоенолпирувата. Реакция катализируется енолазой (фосфопиру-
ватгидратазой) и представляет собой обычное а,р-злиминирование:
С
О ,0
, X)- Енолаза \ X)
Н-С-ОР \ " Н-С-О-Р
сноон ,; „ сн
Н*0
2
2-Фосфоглицерат Фосфоенолпируват
315
Часть II. Метаболизм веществ и энергии в клетке
Фосфоенолпируват обладает высоким потенциалом переноса
фосфатной группы; АО0' гидролиза высокоэнергетического енолфосфата
составляет -61,9 кДж-моль"1,
Образовавшийся фосфоенолпируват отдает фосфатную группу АОР
под действием пируваткиназы. Оставшийся при этом енол
самопроизвольно превращается в пируват:
I
СН3
Пируват
АО0 этой реакции = -61,9 + (+30,5) = -31,4 кДж-моль"1; столь
высокое отрицательное значение АО0' делает ее практически необратимой.
Регуляция гликолиза. Гликолиз выполняет две функции: 1)
генерирует АТР за счет расщепления гексозы и 2) поставляет строительные
блоки для реакций синтеза. Его регуляция и направлена на
удовлетворение этих двух потребностей клетки. Реакции, катализируемые гексоки-
назой, фосфофруктокиназой и пируваткиназой, практически
необратимы; они выполняют не только каталитическую, но и регуляторную
функцию. Особая роль в выполнении регуляторной функции отводится фос-
фофруктокиназе. Она действует на пусковой стадии гликолиза.
Превращение фруктозо-б-фосфата в фруктозо-1,6-бисфосфат, катализируемое
фосфофруктокиназой, является наиболее медленно текущей реакцией
гликолиза, определяющей скорость всего процесса. Активность фосфо-
фруктокиназы аллостерически контролируется рядом важных
метаболитов, а именно: 1) фосфорилированными промежуточными
продуктами, такими как глицеральдегид-3-фосфат, 2-фосфоглицерат и
фосфоенолпируват; 2) адениннуклеотидом и ортофосфатом. Фосфофруктоки-
наза ингибируется высокими концентрациями АТР, снижающими ее
сродство к фруктозо-6-фосфату. Активность фермента возрастает при
снижении отношения АТР/АМР. Иначе говоря, гликолиз стимулируется в
условиях низкого уровня энергии в клетке. На активность фосфофрукто-
киназы влияют также избыток или недостаток строительных блоков.
Так, она ингибируется цитратом — промежуточным продуктом на
начальных стадиях цикла трикарбоновых кислот (см. разд. 9.4). Избыток
цитрата означает, что соединения, играющие при биосинтезе роль
предшественников, присутствуют в больших количествах и, следовательно,
необходимо снижение интенсивности гликолиза. Таким образом, когда
клетка нуждается в энергии и строительных блоках, о чем
свидетельствуют низкое значение отношения АТР/АМР и низкое содержание
цитрата, фосфофруктокиназа наиболее активна. Если в клетке и энергия, и
строительные углеродные фрагменты присутствуют в избытке,
активность фермента резко снижается.
с с
I X)" Пируваткиназа | X)"
Н-С-О-Р /-" %^ С-ОН
СН2 , АОР АТР СН2
Фосфоенолпируват Енолпируват
316
•
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
Катализирующая неравновесную реакцию пируваткиназа ингиби-
руется АТР и цитратом и активируется своим субстратом и АВР.
Повышение уровня АТР, глюкозо-б-фосфата, как и цитрата,
приводит к ингибированию гексокиназы.
Особое значение для регуляции гликолиза имеет поступление
глюкозы, играющей роль "метаболического топлива". Регуляция
вхождения глюкозы осуществляется под действием гексокиназы,
катализирующей первую из гликолитических реакций, и гликоген-
фосфорилазы, обеспечивающей образование глюкозо-1-фосфата,
представляющего собой предшественник глюкозо-6-фосфата. В
некоторых тканях (например, скелетные мышцы) гексокиназа
является аллостерическим ферментом, и роль отрицательного
модулятора играет глюкозо-6-фосфат. Благодаря такому характеру
регуляции скорость образования глюкозо-б-фосфата постоянно
поддерживается в соответствии со скоростью его утилизации. Гликоген-
фосфорилаза регулируется как путем ковалентной модификации,
так и аллостерически. За счет ковалентной модификации
осуществляется переход активной фосфорилазы а в неактивную фосфоры-
лазу Ь. Взаимопревращения этих двух форм происходят под
действием киназы фосфорилазы 6, катализирующей фосфорилирова-
ние с помощью АТР специфических остатков серина в активном
центре фосфорилазы 6, и фосфатазы фосфорилазы а9
обеспечивающей удаление от фосфорилазы а фосфатных групп, необходимых
для каталитической активности. Кроме того, малоактивная фосфо-
рилаза Ъ может активироваться под действием аллостерического
модулятора АМР; АТР является отрицательным модулятором.
Однако следует иметь в виду, что у разных организмов регулятор-
ные этапы гликолиза могут значительно различаться характером
регуляции.
9.2. Брожение
У многих микроорганизмов, растущих в анаэробных условиях,
гликолиз является основным катаболическим путем,
предназначенным для извлечения энергии из углеводных субстратов; дальнейшие
превращения пирувата приводят к образованию определенных
конечных продуктов метаболизма — продуктов брожения. Химическая
природа этих продуктов зависит от вида микроорганизма и условий
протекания процесса, в которых один и тот же микроорганизм
осуществляет брожение*
Основными типами брожений, широко распространенными в
природе, являются спиртовое, молочнокислое, маслянокислое.
Спиртовое брожение осуществляется дрожжами рода Засека-
готусез и дрожжеподобными организмами (МопШта, 014шт и др.),
а также некоторыми микромицетами. Этанол способны продуцировать
317
Часть II. Метаболизм веществ и энергии в клетке
и клетки высших растении, если они находятся в среде, лишенной
кислорода.
Превращение пирувата в этанол идет в двух последовательных
реакциях. В первой происходит его декарбоксилирование:
?
-о
Пируватдекарбоксилаза
(ТРР)
сн3
Пируват
Н
+
Реакция полностью
необратима; она катализируется
пиру ватдекарбоксилазой, которая
содержит в качестве кофермеи-
та тиаминпирофосфат (ТРР).
Декарбоксилирование
пирувата осуществляется через
ряд промежуточных стадий
(рис. 9.2). Сначала а-С-атом
пирувата связывается с С-2-ато-
мом тиазолового кольца ТРР,
обладающим резко выраженной
нуклеофильностью, в
результате чего образуется 2-а-лак-
тильное производное ТРР.
Затем от этого производного
отщепляется С02 с
образованием 2-оксиэтильного
производного. На последнем этапе ок-
сиэтильная группа отделяется
от ТРР в виде свободного аце-
тальдегида.
Вторая реакция состоит в
восстановлении ацетальдегида в
этанол за счет КАБН:
сн3-сч
о
н
+ со.,
Ацетальдегид
О
II
СНз-С-СОО"
+
К—К—С—СНз
II II
2 СН С—СН2—СН2-ОР-ОР
V
I
ТТАПГ* " N С'~СНз
Ч*ми7 Я II
сн8—с -— с с—сн2—сн2-ор-ор
ОН Х87
к-
со.
н
СНя—С
К—Г*—С—СН,-
и
— с с—сн2—сн2-ор-ор
I \ /
он V
ь-
о
I
н
К—К—С—СНз
II II
НС С—СН2—СН2-0Р-0Р
V
Рис. 9.2. Декарбоксилирование
пирувата под действием пируватдекарбокси-
лазы
сн3-с
//
о
Алкоголь-
дегидрогеназа
сн3-сн2-он
•н . / >
Ацетальдегид ^АОН + Н ИАБ Этанол
Эта окислительно-восстановительная реакция катализируется ал
когольдегидрогеназой.
Суммарная реакция спиртового брожения имеет следующий вид:
Глюкоза + 2Р. 4- 2М)Р -> 2 этанол + 2С02 + 2АТР + 2Н20.
318
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
В это уравнение не входят КАВН и ЫАГ)+, так как образованный в
процессе гликолиза ЫАВН вновь превращается в ЫА1)* в результате
восстановления уксусного альдегида в этанол. Такая регенерация КАВ+
поддерживает в анаэробных условиях непрерывное течение гликоли-
тического процесса. Если бы этого не происходило, то гликолиз не
мог бы идти дальше образования глицеральдегид-3-фосфата и,
следовательно, невозможно было бы образование АТР.
При спиртовом брожении, вызываемом дрожжами,
используются гексозы; пентозы не сбраживаются. Легче всего дрожжи
сбраживают глюкозу и фруктозу, значительно труднее — маннозу и
особенно галактозу. Сахароза и мальтоза сбраживаются только после
предварительного гидролиза до гексоз, осуществляемого (3-фрукто-
фуранозидазой и а-глюкозидазой соответственно. Лактозу
сбраживают только особые дрожжи ЗассНаготусез /гацШз, содержащие
р-галактозидазу, которая гидролизует этот дисахарид, с
образованием глюкозы и галактозы. Дрожжи сбраживают весьма высокие
концентрации сахара, достигающие 60 %, и накапливают в среде
10—15 % этанола. Высокомолекулярные полисахариды (крахмал,
инулин, гемицеллюлозы, целлюлоза) и продукты их неполного
гидролиза не сбраживаются дрожжами, поскольку они не способны
проникать через клеточные мембраны, а сами дрожжи не синтезируют
ферменты, которые могли бы выделиться в окружающую среду- и
осуществить гидролиз этих полисахаридов до сбраживаемых
Сахаров.
Молочнокислое брожение отличается от спиртового тем, что пи-
руват не декарбоксилируется как при спиртовом брожении, а
непосредственно восстанавливается лактатдегидрогеназой с участием ЫАВН.
с _ с _
чО Лактатдегидрогеназа I чО
с=о ^—^-—^ нсьон
СН3 ИАОН + Н+ КАВ+ снз
Пируват Ь-Лактат
Суммарная реакция превращения глюкозы в лактат такова:
Глюкоза + 2 АВР + 2Р. ~> 2Лактат + 2АТР + 2Н20.
Возбудителями молочнокислого брожения являются
молочнокислые бактерии родов ЬасЬоЬасШиз и 81гер1ососси8* Молочнокислые
бактерии строго ограничены в своем обмене процессом брожения,
представляют собой факультативные анаэробы, т.е. способны проявлять
свою жизнедеятельность в присутствии кислорода воздуха; в их
клетках отсутствуют цитрхромы, каталаза. Известны две группы этих
бактерий. В одну из них входят гомоферментативные бактерии, которые
образуют только лактат. Некоторые из них образуют его не менее 90 %
319
Часть II, Метаболизм веществ и энергии в клетке
от количества продуктов брожения. Особенно большое количество лак-
тата накапливают некоторые термофильные бактерии, например
ТегтоЬаЫегшт сегеа1е9 широко применяемые в пищевой
промышленности в производстве молочной кислоты.
Молочнокислые бактерии второй группы являются гетерофермен-
тативными* Кроме молочной кислоты, они образуют уксусную
кислоту, а также этанол и диоксид углерода. При гетероферментативном
брожении катаболизм глюкозы до глицеральдегид-3-фосфата
осуществляется по фосфоглюконатному пути:
АТР
Глюкоза
АВР
Глюкозо-6-фосфат
КАО+—
ЫАБН
6-Фосфоглюконат
С02
Пентозо-5-фосфат
Ра
> Ацетилфосфат
Глицеральдегид- 3-фосфат
АЭР
КАВ+-
Этанол Ацетат
АТР
КАОН
Пируват
ЫАОН
ИАВ"
Лактат
Образовавшийся ангидрид уксусной и фосфорной кислот
превращается в уксусную кислоту или восстанавливается в этанол:
КАБН КАВ
+
2СНЯ-С
ч
О
ОР
Р1
Ацетилфосфат
СНз-СОО'
Ацетат
СН3-СН2ОН
Этанол
Маслянокислое брожение. Возбудителями маслянокислого
брожения являются бактерии рода С1о8Ш<Иит. Оно осуществляется коми
лексом ферментов по схеме:
320
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
О
II
2СН3-С-СОО
Пируват
2СоА-8Н-
О
2СН3-С-8-СоА + 2С02 + 2Н2
Ацетил-СоА
СоА-ЗН
Тиолаза
О
КАВН + Н
ИАВ+
НоО
ЫАОН +- Н
ИАП
+
СН3-С-СН2-СО-8СоА
Ацетоацетил-СоА
(3-гидроксибутирил-СоА-
дегидрогеназа
СН3"СНОН-СН2-СО"8СоА
Р-Гидроксибутирил-СоА
Кротоназа
СН8-СН=СН-СО-8СоА
Кротонил-СоА
Бутирил-СоА-
дегидрогеназа
СН3-СН2-СН2-СО-8СоА
СоА-8Н
Тиолаза
сн8-сн2-сн2-соон
Масляная кислота
Суммарное уравнение маслянокислого брожения:
сбн12°б + 2АЕ)Р + 2Р1 + 2КАШ + 2Н+ ~> СН3-СН2-СН2-СООН +
+ 2МАВ + 2Н2 + 2С02 + 2АТР + 2Н20.
Маслянокислые бактерии являются облигатными анаэробами,
отличаются резко выраженным бродильным типом обмена веществ.
Токсическое действие кислорода связано с отсутствием у данных бактерий
цитохромов и каталазы и с высоким содержанием флавиновых
ферментов. Последние переносят водород от субстрата на кислород с
образованием пероксида водорода, который накапливается в токсических
концентрациях. В природе маслянокислое брожение происходит в местах, куда
ограничен доступ кислорода. Особенно интенсивно оно развивается в
илистых днищах болот, прудов, озер, где в результате деятельности мас-
лянокислых бактерий разлагаются останки водных растений.
Субстратами служат полисахариды, моносахариды, органические кислоты,
спирты. Наряду с масляной кислотой, диоксидом углерода, водородом
маслянокислые бактерии могут образовывать в достаточно больших количе-
21. Заказ 3486
321
Часть II. Метаболизм веществ и энергии в клетке
ствах и другие продукты брожения. Так, С1о$Мс1шт асе16Ьи1у11сит при
сбраживании глюкозы вначале образует масляную кислоту. Однако с
подкислением среды начинают синтезироваться ферменты, действие
которых приводит к накоплению ацетона и бутанол а. Так,
синтезированная в бактериальной клетке ацетоацетатдекарбоксилаза совместно с тио-
лазой из ацетоацетил-СоА образуют ацетон:
о
и
СН3-С~СН2-ССК8СоА
СоА-8Н <—-Н Тио лаза
О
II
сн3-с-сн2-соо
Ацетоацетат
Ацетоацетатдекарбоксилаза
О
II
СНз- С~ СНд
Ацетон
В результате декарбоксилирования части ацетоацетата
утрачивается потенциальный акцептор водорода от ЫАВН ацетоацетат-СоА,
который при восстановлении в масляную кислоту мог бы дважды
использовать МАВН. Водород ЫАОН неизбежно должен быть передан на
другие акцепторы, в том числе на масляную кислоту, которая,
восстанавливаясь, образует бутанол. Другим промежуточным продуктом у
С1. асе1оЪи1уИсит является ацетальдегид, образующийся из ацетил-Со А,
который при участии ЫАОН и алкогольдегидрогеназы
восстанавливается в этанол. Эта разновидность брожения получила название ацето-
но-бутилового брожения и нашла использование в производстве
растворителей — ацетона и бутанола. Спиртовое, гомоферментативное
молочнокислое и маслянокислое брожения являются основными
типами брожений. Все другие многочисленные виды брожений
представляют собой комбинацию этих основных типов♦ Так, гетерофермента-
тивное молочнокислое, пропионовокислое брожения можно
представить как комбинированное гомоферментативное молочнокислое и
спиртовое брожения; ацетоно-бутиловое — смешанное маслянокислое и
спиртовое брожения; брожение пектиновых веществ, клетчатки —
разновидность маслянокислого брожения.
Различные типы и разновидности брожений играют большую роль
в круговороте веществ в природе, в создании экологически чистой
среды. Они весьма важны в практической деятельности человека. Так,
большое промышленное значение имеет брожение пектиновых веществ,
происходящее при замочке стеблей льна, конопли и получении
волокна. Метановое брожение, вызываемое специфическими
микроорганизмами, широко используется в промышленных и бытовых очистных
322
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
сооружениях для обеззараживания органических веществ•
Образующийся при этом метан используется как топливо. Важна роль
брожения в технологии ряда пищевых продуктов (см. гл. 17).
9.3. Биоэнергетика анаэробного разложения углеводов
В процесс гликолиза входят две АТР-зависимые реакции: первая —
образование глюкозо-6-фосфата и вторая — фруктозо-1,6-бисфосфата,
и две реакции синтеза АТР, сопряженные с образованием глице-
рол-3-фосфата и пирувата. Поскольку в ходе гликолиза каждая
молекула гексозы расщепляется на 2 трех углеродных фрагмента, то,
следовательно, в этом процессе синтезируется 4 молекулы АТР. В сумме,
таким образом, затрачиваются 2 молекулы АТР и образуются 4
молекулы АТР; итого генерируются 2 молекулы АТР.
В процессе гликолиза прослеживаются две фазы — эндергоничес-
кая и экзергоническая. Эндергоническая фаза, представляющая
собой начальную (пусковую) стадию гликолитического пути, содержит
три энергетических барьера: первый — образование глюкозо-6-фосфа-
та; второй — образование фруктозо-1,6-бисфосфата; третий — лежит
между фруктозо-1,6-бисфосфатом и 1,3-бисфосфоглицератом. Хотя
величина третьего барьера довольно значительна, тем не менее в
процессе гликолиза этот барьер не приводит к блокированию
метаболизма и накоплению фруктозо-1,6-бисфосфата. Это объясняется тем, что
за указанным барьером следует реакция превращения 1,3-бисфосфо-
глицерата в 3-фосфоглицерат; как уже отмечалось, бисфосфоглице-
рат — это ацилфосфат, который представляет собой нестабильное
высокоэнергетическое соединение. Невыгодное эндергоническое
превращение фруктозо-1,6-бисфосфата в 1,3-бйсфосфоглицерат проходит до
конца благодаря термодинамически выгодной реакции превращения
бисфосфоглицерата в 3-фосфоглицерат.
Термодинамически выгодной также является реакция
превращения фосфоенолпирувата в пируват. Здесь вновь высокоэнергетическое
соединение — фосфоенолпируват используется в качестве донора
фосфатной группы при образовании АТР.
Перечень этапов, на которых происходит потребление и
образование АТР, приведен в табл. 9.1.
Образующиеся две молекулы АТР и представляют собой тот
чистый выигрыш в энергии, который клетка приобретает в сложной цепи
процесса гликолиза.
Рассмотрим энергетические соотношения процесса брожения. Он
сопряжен с синтезом двух молекул АТР и для этого синтеза ДО0'
является положительной величиной:
ДО0' = 30,5-2 = +61 кДж-моль"1.
При спиртовом брожении глюкоза —> 2С02 +2 • этанол (ДО0' ™
= -235 кДж-моль"1). При молочнокислом брожении глюкоза —» 2 • лак-
21*
323
Часть II. Метаболизм веществ и энергии в клетке
Таблица 9.1
Потребление и образование АТР при гликолизе
Реакция
Глюкоза —» Глюкозо-6-фосфат
Фруктозо-6-фосфат -^ Фруктозо-1,б-бисфосфат
2-Висфосфоглицерат —» 2-3-Фосфоглицерат
2'Фосфоенолпируват —> 2Пируват
Итого
Количество молей АТР
в расчете на 1 молекулу глюкозы
-1
»1
+2
+2
+2
тат (АО0, = -196 кДж-моль"1). Суммарное изменение свободной энергии
при спиртовом брожении будет АО0' = АО^ + АО° = +61 + (-235) =
= -174 кДж'моль-1; при молочнокислом брожении — соответственно
ДО0' = АО°; + МЩ - +61 + (-196) - -135 кДж-моль"1.
Из данных по изменению свободной энергии видно, что
расщепление глюкозы при спиртовом или молочнокислом брожении
сопровождается освобождением такого количества энергии, которого гораздо
больше, чем требуется для синтеза двух молекул АТР. Лишь около
одной трети энергии, освобождающейся при сбраживании гексозы,
генерируется в АТР. Брожение представляет собой, таким образом,
практически необратимый процесс, идущий почти полностью в
сторону образования продуктов брожения. Две трети извлекаемой
свободной энергии теряется в форме тепла. Интересно отметить, что
последние реакции брожения (ацетальдегид — этанол; пируват — лактат) в
принципе могли бы быть сопряжены с образованием АТР. Однако
этого не происходит и выделяющаяся энергия не сохраняется. Видимо,
эти реакции, замыкающие многостадийный процесс брожения,
дополнительно подстраховывают интенсивное превращение промежуточных
интермедиатов в конечный продукт* Ради этого клетка
"расплачивается" потерей части свободной энергии — рассеиванием ее в
сбраживаемой среде.
Чем объяснить столь большое снижение свободной энергии в
процессе брожения? Это на первый взгляд кажется загадкой,
поскольку общего окисления при этом не происходит. Однако оба процесса
брожения включают окислительно-восстановительные реакции. Об
этом отчетливо свидетельствуют конечные продукты спиртового
брожения: этанол можно рассматривать как относительно
восстановленное соединение, поскольку его молекула содержит
определенное количество водорода, а С02 — как конечный продукт
окисления, поскольку в его молекуле водород отсутствует. При
молочнокислом брожении окислительно-восстановительный процесс хотя
и не проявляется в столь отчетливой форме, однако в химической
структуре лактата можно видеть, что один конец его молекулы —
СН3-группа — находится в восстановленном состоянии, другой —
СООН-группа — в окисленном. В исходном соединении — гексозе,
324
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
вовлекаемой в процесс брожения, атомы водорода распределены
более равномерно. Другими словами, в процессе брожения
наблюдается только перераспределение существовавших связей между
атомами субстрата. Имеет место поток атомов водорода и
кислорода в противоположных направлениях, что и дает столь большое по
величине отрицательное значение АО0'.
9.4. Цикл трикарбоновых кислот
В предыдущем разделе мы рассмотрели гликолитический путь,
конечным продуктом которого является пируват. У аэробных
организмов (животные, растения, многие микроорганизмы) пируват под-
вергается дальнейшему превращению — окислительному декарбок-
силированию с образованием ацетил-СоА. Этот активированный
ацетильный комплекс далее полностью окисляется до С02 и Н20,
вовлекаясь в так называемый цикл трикарбоновых кислот (ЦТК). Этот
цикл известен также под названием цикла лимонной кислоты или
цикла Кребса в честь Ганса Кребса (лауреата Нобелевской премии
1953 г.), определившего последовательность реакций цикла.
Сущность цикла и заключается в окислительном разложении
ацетильного остатка, в результате чего освобождаемая энергия запасается
в виде АТР.
Хотя цикл трикарбоновых кислот мы рассматриваем во
взаимосвязи с катаболизмом углеводов, однако его роль в метаболизме
веществ гораздо шире. Он выполняет следующие функции:
1. Интегративную — объединяет пути катаболизма углеводов,
жиров и белков; во всех аэробных организмах он выступает в роли
центрального метаболического пути углерода.
2. Амфиболическую — выполняет не только катаболическую
функцию распада ацетильных остатков, но и анаболическую, поскольку
субстраты цикла используются для синтеза других веществ.
3. Энергетическую — совместно с цепью переноса электронов
(см. разд. 9.8) является основным поставщиком химической энергии
в форме АТР.
Окислительное декарбоксилирование пирувата с образованием
ацетил-СоА и реакции цикла трикарбоновых кислот
осуществляются в клеточных органеллах — митохондриях. Биохимические
функции цикла указывают на то, что ацетат и любой компонент
цикла должны быть хорошими источниками энергии и их можно
употреблять с пищей как ценные энергетические вещества, лишь
бы они, поступив в клетку, могли достигнуть ферментной системы,
находящейся в митохондриях. Однако основным источником
энергии аэробных организмов является углеводный материал, из
которого она извлекается в ходе совместных процессов гликолиза и цикла
трикарбоновых кислот. Сопряжение этих путей происходит на уровне
превращения пируват —> ацетил-СоА, катализируемого пируватде-
325
Часть II. Метаболизм веществ и энергии в клетке
гидрогеназным комплексом. В ходе реакции происходят
окислительное декарбоксилирование а-оксокислоты (пирувата) и образо
вание тиоэфира:
Цитоплазма
Гексоза
О
сн3-с-соо"
Пируват
Пируватдегид роге-
Митохондрии
О
назныи комплекс
СН3-08СоА
Ацетил-Со А
—*/цтк)
Окислительное декарбоксилирование пирувата. Пируватдегид-
рогеназный комплекс представляет собой мультиферментную
систему, включающую три фермента, каждый из которых катализирует
определенную стадию окислительного декарбоксилирования. Этот
комплекс хорошо изучен (табл. 9.2).
Таблица 9.2
Пируватдегидрогеназный комплекс
Фермент
Пируватдегидрогеназный компонент
Дигидролипоил-
трансацетилаза
Дегидролипоил-
дегидрогеназа
Число цепей
в молекуле
24
12
12
Кофактор
ТРР
Липоамид
РАВ
Катализируемая реакция
Декарбоксилирование
пирувата
Окисление С2-фрагмента
и перенос на СоА-8Н
Регенерирование
окисленной формы липоамида
В процессе окислительного декарбоксилирования принимают
участие пять кофакторов: тиаминпирофосфат (ТРР), липоамид, СоА-8Н,
РАО, ЫА1) и три фермента: пируватдегидрогеназа, трансацетилаза и
дигидролипоилдеги дрогеназа.
На первой стадии пируват соединяется с ТРР и подвергается де-
карбоксилированию (см. разд. 9.2).
На второй стадии гидроксиэтильная группа, связанная с ТРР,
окисляется с образованием ацетильной группы и одновременно
переносится на липоамид. Окисление катализируется дигидролипоилтрансаце-
тилазным компонентом. Липоамид представляет собой остаток липое-
вой кислоты, присоединенный к специфической лизиновои боковой
цепи трансацетилазного компонента:
/СНЧ « ф*
СН2 СН-(СН2)4-СОтМН-(СН2)4-СН
чм/
I
со
ч
Остаток липоевой
кислоты
у
Остаток лизина
;
Окислителем в реакции служит 8-8-группа липоамида, восстанав
ливающаяся в 8Н-группы;
326
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
ОН
СНз-СН
I
3
! I
с=с
I I
К СНд
8-8
+ Н2С СН-К"
х /
Н8
Н2С
сна
I
8
СН-К" +
0
С
\
сн2
8
I I
! I
К СН-з
Гидроксиатил-ТРР
(ионизированная
форма)
Липоамид
Ацетиллипоамид Карбанион
Затем ацетильная группа переносится с ацетиллипоамида на СоА,
образуя ацетил-СоА:
СН3
с=о
Н8 8
Н2С СН-Е" + Н8-СоА
% /
сн2
Ацетиллипоамид
Н8 8Н
Н2С СН-Е" + СН3-СО~8-СоА
СН2
Дигидролипоамид Ацетил-СоА
При переходе ацетильной группы на СоА сохраняется богатая
энергией тиоэфирная связь*
На третьей, завершающей, стадии происходит восстановление
окислительной формы липоамида. Реакция катализируется дигидролипо-
илдегидрогеназным компонентом:
Н8 8Н
1 1 ■
сн2
ЫАО+
8—8
^^ Н2Сч СН-К"
N АРН + Н' СН2
Все промежуточные продукты окислительного декарбоксилирова-
ния пирувата прочно связываются с комплексом. Скорость их
превращения весьма тонко регулируется: она подавляется АТР и КАВН и
возрастает в присутствии АВР и КАБ+.
Реакции цикла трикарбоновых кислот. Последовательность всех
реакций цикла Кребса показана на рис. 9.3* Данная схема
представлена перед рассмотрением отдельных реакций цикла с целью создания
целостного представления о данном пути. Упрощенно описывая этот
достаточно сложный метаболический процесс, можно отметить, что в
ходе его функционирования цитрат, представляющий собой
Отсоединение, изомеризуется до изоцитрата. Последний последовательно
превращается в Отсоединение — 2-оксоглутарат и затем в Отсоединения:
сукцинат, фумарат, малат и оксалоацетат, являющийся исходным
интермедиатом цикла. Таким образом, цикл завершается, а
ацетильный остаток, вступающий в данный путь в виде ацетил-СоА,
окисляется, что обеспечивает генерирование высокоэнергетических восста-
327
Часть II. Метаболизм веществ и энергии в клетке
о-с-сосг
Н2С-СОО_
Оксало&цегат
СНз 8-СоА
Ацетил СоА
Н2С~СОО'
но-с-соо-
I
Н2С-С00"
Н-С-СОО'
I
с-соо-
няс-соо~
Н'°-$Е)
н
I
НО-С-СОО"
I
н-с-соо-
Н2ОСОО"
О-С-СОО'
I
н-с соо-
Н2С-СОО*
4— о-с-соо
КАР+
ЫАЮН + Н*
СВР
СТР
Н8-СаА
КАВ
ГАОНа
нр
Н2С-С0О"
н2с-соа
ис-соо
I
"ООС-СН
~1@
ЫАО+
н
НО-С-СОО"
I
Н2С-ОХГ
Г^А1>Н + Н*
^=с©
0=С~С00"
Н^С-СОО"
Цитрат
цис~ Аконит ат
Изоцитрат
Оксалосук ци ыат
2-0ксоглутарат
Сукциынл СоЛ
Сукцинат
Фумарат
Малат
Оксалоацетат
Рис. 9.3. Цикл трикарбоновых кислот. Ферменты, функционирующие в
данном метаболическом пути: 1 — цитратсинтаза; 2, 2' — аконитаза; 3, 3' — изоцит-
ратдегидрогеназа; 4 — 2-оксоглутаратдегидрогеназный комплекс; 5— сукцинил-
СоА-синтетаза; 6 — сукцинатдегидрогеназа; 7 — фумараза; 8 — малатдегидрогеназа
328
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
новленных коферментов — NА^Н и РАБН2, а также молекулы СТР в
тканях животных или АТР в растительных тканях.
Цикл трикарбоновых кислот состоит из 8 стадий, катализируемых
8 специфическими ферментами. Он начинается с конденсации
ацетильного остатка (ацетил-СоА) с оксалоацетатом, в результате чего
образуется цитрат.
Реакция конденсации ацетил-СоА с оксалоацетатом —
синтетическая, ведущая к образованию С-С-связи и к появлению трикарбоновой
кислоты — цитрата:
СОО"
СН2
СОО
1
с=о
СН.р
] -
СОО"
Оксалоацетат
+
сн3
1
сю
8-СоА
Ацетил-СоА
>
НО-С-СОО" + Н+
Н20 СоА-8Н ^Н2
СОО~
Цитрат
Необходимая энергия для синтеза С-С-связи обеспечивается
гидролизом тиоэфирной связи ацетил-СоА. Реакция катализируется цит-
ратсинтазой, которая высокоспецифична к ацетил-СоА. Поскольку
это первая реакция цикла, именно она играет важную роль в
регуляции цикла в целом, о чем будет сказано ниже.
Вторая реакция цикла трикарбоновых кислот — обратимое
превращение цитрата в изоцитрат. Реакция протекает через стадию
образования цис-аконитата и катализируется ферментом аконитатгидра-
тазой. Под действием этого фермента цитрат теряет молекулу воды и
превращается в ненасыщенный ц&с-аконитат, который, не отделяясь
от активного центра аконитазы, вновь присоединяет воду, но уже иным
путем и образует изоцитрат:
СОО" СОО" СОО"
I I I
ОН? _ СНо ^Н.9
I _ -Н20 I +Н20 I
НО-С-СОО *~ С-СОО" *- Н-С-СОО"
СН2 СН НО-С-Н
СОО СОО" СОО'
Цитрат ц ис- Аконитат Изоцитрат
Равновесие этой реакции в физиологических условиях сильно
сдвинуто влево. На долю цитрата приходится 90 %, а изоцитра-
та — лишь 6 %. Течение процесса в нужном направлении
обеспечивается постоянным уходом изоцитрата в результате его
последующих превращений.
Третья реакция цикла трикарбоновых кислот — окислительное
декарбоксилирование изоцитрата под действием фермента изоцит-
ратдегидрогеназы, в результате чего образуется 2-оксоглутарат:
329
Часть II. Метаболизм веществ и энергии в клетке
СОСГ гл. СОО
СН, 4 СН
■2
I
1
2
н-осоо" ^7* ^* V1*2
НО-С-Н КАВ+ МАШ + Н* ?=°
соо соо"
Изоцитрат 2-Оксоглутарат
Это первая окислительно-восстановительная реакция в цикле три-
карбоновых кислот; в цикле она играет основную регуляторнуто роль.
КАВ-зависимая изоцитратдегидрогеназа, функционирующая в ЦТК,
является олигомерным ферментом, состоит из 8 субъединиц, имеет
молекулярную массу 330000, локализована только в митохондриях.
Ее активатором является АВР, ингибитором — МАХ)Н.
Четвертая реакция цикла — окислительное декарбоксилирова-
ние 2-оксоглутарата с образованием сукцинил-СоА:
900 КАВР+ КАБРН + Н+ ?00
СН2 V А СН2
сн2 —^>"<Г ■* сн2
?=0 СоА-8Н С02 ?=0
СОО 8-СоА
2-Оксоглутарат Сукцинил-СоА
Реакция катализируется мультиферментным комплексом,
аналогичным пируватдегидрогеназному комплексу, называемым 2-оксоглу-
таратдегидрогеназным комплексом.
Пятая реакция — превращение сукцинил-СоА в сукцинат —
осуществляется при участии фермента сукцинил-СоА-синтетазы:
?°° аор + р, отр
СН2 ^ л ?00
сн2 > <Г—* Т
I
оо
8-СоА
сн2
Н*0 СоА-8Н
СН2
I
СОО
Сукцинил-СоА Сукцинат
Выделяющаяся при этом энергия сохраняется путем образования
ОТР. Эта реакция является реакцией субстратного фосфорилирова-
ния? протекает в три стадии, в которых участвует один и тот же
фермент Е:
1) Сукцинил-СоА + В + Р. ^ Е-сукцинил~Р + СоА-8Н;
2) Е-сукцинил~Р ?^ Е~Р + сукцинат;
3) Е~Р + ОВР «=* Е + ОТР.
Образовавшийся ОТР при участии митохондриальной нуклеозид-
дифосфаткиназы вступает в реакцию перефосфорилирования с АОР, в
результате чего образуется АТР:
330
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
ОТР + АОР ^ ОВР + АТР.
Шестая реакция — дегидрирование сукцината до фумарата под
действием сукцинатдегидрогеназы. Акцептором водорода в этой
реакции является ГАВ; поскольку РАВ ковалентно связан с боковой
цепью сукцинатдегидрогеназы, ее часто называют флавопротеином.
Фермент обладает абсолютной специфичностью; атомы водорода
отщепляются только в транс-положении. Если бы такая специфичность
отсутствовала, образовывалась бы смесь (50:50) двух изомеров: малеиновой
(цис-) и фумаровой (транс-) кислот. Однако под действием
сукцинатдегидрогеназы образуется только фумарат:
нн ГАО *АОН* н соо-
оос-с-с-соо" Чч^ -^ » с-с
I I ^ / \
н н "оос н
Сукцинат Фумарат
Сукцинатдегидрогеназа локализована на митохондриальной
мембране. Все остальные ферменты цикла трикарбоновых кислот
локализованы в матриксе митохондрий.
Седьмая реакция — гидратация фумарата с образованием малата,
катализируемая фумаразой (фумаратгидратазой). Подобно сукцинат-
дегидрогеназе, этот фермент также обладает абсолютной стереоспеци-
фичностью: ионы Н+ и ОН" присоединяются к фумарату только по
транс-туту, в результате чего образуется Ь-малат:
Н СОО"
\ /
с=с
/ \
н
I
оос-с-н
I
оос н г но-с-соо
н2о н
Фумарат Ь-Малат
Восьмая реакция — регенерация исходного соединения цикла —
оксалоацетата в результате окисления Ь-малата под действием малат-
дегидрогеназы. Акцептором водорода здесь является ЫАО4-:
Н
1 Н
ООС-С-Н . I
, . . _ ^ ООС-С-Н
НО-С-СОО /^ ^Ч '
I * О-С-СОО
Н КА1)+ КАШ + Н+
Ь-Малат Оксалоацетат
Образовавшийся оксалоацетат вступает в реакцию конденсации с
новой молекулой ацетил-СоА и начинается следующий виток цикла,
показанный в упрощенном виде на рис. 9.4,
Итак, основными реакциями цикла трикарбоновых кислот являются
реакции декарбоксилирования и дегидрирования, в результате которых
происходит освобождение энергии, аккумулированной в ацетил-СоА. В
ходе одного полного оборота цикла в процессе декарбоксилирования (реак-
331
Часть //, Метаболизм веществ и энергии & клетке
Ацетил-СоА
Оксалоацетат
Н20
N^11 + Н+
Малат
Н20
Фумарат
Электрон-
транспортная
цепь
•РАШг
Н5-СоА
Цитрат
ЫАВН + Н
С02
РАО
МАЛИ + Н
Сукцинат
Н8-СоА
Н20
АТРг±ОТР
ОБР + Р1
Сукцинил-СоА
Н8-СоА
С02
Рис. 9.4, Схема цикла трикарбоновых кислот
ции 3, 4) два атома углерода ацетила превращаются в С02;
осуществляются четыре реакции дегидрирования (реакции 3, 4, б и 8). В трех
реакциях дегидрирования участвует КАО+, в одной реакции (шестой) — РАО.
И, наконец, в пятой реакции в результате субстратного фосфорилирова-
ния образуется одна молекула ОТР, что эквивалентно одной молекуле
АТР. МАО+ и РАО регенерируются в цепи переноса электронов (разд. 9.8),
в которой терминальным акцептором электронов является кислород.
Именно в этом смысле цикл трикарбоновых кислот представляет собой
аэробный (зависимый от кислорода) путь.
Промежуточные соединения цикла трикарбоновых кислот играют
большую роль в процессах анаболизма; они принимают участие в
синтезе аминокислот, липидов (см* гл. 11 и 12). Оксалоацетат является
исходным соединением в синтезе углеводов, известным под названием
глюконеогенеза. Образовавшийся из оксалоацетата фосфоенолпиру-
ват— предшественник синтеза глюкозы. Конечный продукт цикла,
С02, частично используется в реакциях карбоксилирования.
Таким образом, цикл трикарбоновых кислот является одной из
важнейших "узловых станций" обмена веществ и энергии, на которой
пересекаются пути превращения различных соединений, что
обеспечивает единство и неразрывную связь различных типов обмена
веществ в организме.
332
Глава 9, Катаболизм углеводов. Генерирование энергии фосфатной связи
9,5. Биоэнергетика цикла трикарбоновых кислот
Величины изменений ДО0' отдельных реакций цикла
представлены в табл. 9.3.
Таблица 9.3
Свободная энергия АС0' реакций цикла трикарбоновых кислот
1.
2.
3.
4.
5.
6.
7.
Реакции
Ацетил-СоА
Цитрат
Изоцитрат + ЫАО+
2-Оксоглутарат + ЫАО+
Сукцинил-СоА + (ГОР + Р;
Сукцинат + РАБ
Фумарат + Н20
—>
Е2
Е3
—>
—>
Ее
Е7
—>
Еа
Цитрат + СоА
Изоцитрат
2-Оксоглутарат + С02 + ИАОН
Сукцинил-СоА+С02 + ИАВН
Сукцинат + ОТР + СоА
Фумарат + РАБН3
Малат
ДО0', кДж
-38,1
+6,7
-7,1
-39,2
-8,9
0,0
-3,7
8.
Малат + ЫАБ+ -» Оксалоацетат + ИАВН
+28,1
Суммарно:
О
СН3-С~8-СоА
+
ЗЫАО+
+
РАО
+
ОБР + Р1 + 2Н20
8 ферментов
2С02 + ЗЫАБН + Н+
+
РАБН2
+
ОТР
+
СоА-8Н
ДО0' - -62,2 кДж
Суммарное изменение АО0' при окислении ацетил-СоА составляет
-62,2 кДж на 1 моль, т.е. цикл трикарбоновых кислот
термодинамически выгоден. Реакции 2 и 8 должны были бы препятствовать
осуществлению остальных реакций. Однако реакция 2 сопряжена с
реакциями 3,4,5, имеющими отрицательное значение ДО0', а реакция 8 — с
реакцией 1, имеющей ДО0' = -38,1 кДж-моль"1, что и предопределяет
ход цикла трикарбоновых кислот в целом.
9.6. Регуляция превращения пирувата в ацетил-СоА
и цикла трикарбоновых кислот
Поскольку цикл трикарбоновых кислот занимает центральное
положение в обмене веществ и энергии, его регуляция играет
кардинальную роль у аэробных организмов. Интенсивность
функционирования цикла в первую очередь определяется скоростью поступления
333
Часть II. Метаболизм веществ и энергии в клетке
"топлива", т.е. ацетил-СоА, предшественниками которого являются
пируват и жирные кислоты. Важную роль в регуляции поставки аце-
тил-СоА играет ковалентная модификация лируватдегидрогеназного
комплекса. В качестве вспомогательных ферментов в регуляции пиру-
ватдегидрогеназного комплекса служат киназа пируватдегидрогена-
зы и фосфатаза фосфопируватдегидрогеназы. Киназа пируватдегид-
рогеназы при достаточно высокой концентрации АТР в клетке
катализирует реакцию фосфорилирования серинового остатка в активном
центре пируватдегидрогеназы, что приводит к образованию
неактивной формы фермента — фосфопируватдегидрогеназы.
Активация неактивной пируватдегидрогеназы осуществляется при
снижении уровня АТР в клетке, что происходит в результате
отщепления фосфатной группы от серинового остатка под действием фосфа-
тазы фосфопируватдегидроееназы. Важно отметить, что
активаторами фосфатазы фосфопируватдегидрогеназы являются ионы Са2+,
концентрация которых увеличивается при дефиците АТР. Кроме того,
роль аллостерических ингибиторов лируватдегидрогеназного комплекса
играют КАОН и ацетил-СоА. Вполне логично можно было бы считать,
что и "входная" реакция в цикл — конденсация ацетил-СоА с оксало-
ацетатом — важная регуляторная реакция цикла. Действительно, ее
скорость находится в тесной взаимосвязи с наличием в матриксе
митохондрий АТР, который является специфическим ингибитором цит-
ратсинтазы в ряде типов клеток.
Установлено, что АТР повышает константу Км для ацетил-СоА;
иными словами, с увеличением содержания АТР в реакционной
системе снижается насыщение фермента ацетил-СоА и в результате
уменьшается скорость синтеза цитрата. Уровень АТР в клетке
определяется скоростью окисления ЫАВН в цепи переноса
электронов, которая, в свою очередь, зависит от концентрации АВР и Р.«
Если в процессе катаболизма образуется больше АТР, чем это
необходимо для энергетических потребностей клетки, концентрация АВР
падает до низкого уровня, выключая, таким образом, процесс
синтеза АТР.
Важную роль в регуляции активности цитратсинтазы играет
также концентрация второго субстрата — оксалоацетата,
концентрация которого в митохондриях очень низка и зависит от условий
метаболизма* Существенное влияние на цитратсинтазу оказывает
также концентрация сукцинил-СоА, ингибирующего
ферментативную активность за счет снижения сродства к ацетил-СоА. Ингиби-
рующий эффект на фермент могут оказывать также жирные
кислоты, являющиеся предшественником ацетил-СоА. Кроме того, роль
ингибиторов цитратсинтазы у некоторых организмов могут играть
ЫАВН и цитрат.
Второй регуляторной реакцией является окисление изоцитрата,
катализируемое КАО-специфичной изоцитратдегидрогеназой. Действие
334
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
фермента в клетках животных, некоторых микроорганизмов
стимулируется АОР или АМР, которые повышают его сродство к субстрату.
В растениях роль положительного модулятора изоцитратдегидрогеназ-
ной активности играет цитрат. В клетках различных типов ЫАЮН и
NА^РН ингибируют фермент путем конкуренции с МАВ+.
Третьей регуляторной реакцией цикла является окислительное
декарбоксилирование 2-оксоглутарата, катализируемое 2-оксоглута-
ратдегидрогеназным комплексом; ингибиторами этого комплекса
являются сукцинил-СоА и ЫАОН.
Еще одним способом регуляции цикла трикарбоновых кислот,
связанным с фосфорилированием адениловои системы, является реакция
субстратного фосфорилирования, требующая ОВР. В этом случае
определяющую роль играет соотношение ОТР/ОВР.
В заключение можно сказать, что когда величины отношений
\АВН/МАВ+, АТР/АВР, ацетил-СоА/СоА, сукцинил-СоА/СоА высо^
ки, клетка в достаточной мере обеспечена энергией, и поток
превращаемых соединений в цикле замедлен. Если эти соотношения низки,
клетка испытывает потребность в энергии, и этот поток веществ через
цикл ускоряется. Короче говоря, поступление двухуглеродных
фрагментов в цикл трикарбоновых кислот и скорость цикла снижаются
при высоком уровне энергии в клетке; эти процессы усиливаются при
интенсивном расходе энергии.
9.7* Гексозомонофосфатный путь
Помимо катаболизма углеводов по пути гликолиза существует
другой альтернативный путь, получивший название гексозомонофосфат-
ного пути, или шунта; поскольку в нем принимают участие в
основном пентозы, он известен также под названием пентозофосфатного
пути. Слово "шунт" указывает на ответвление или на обходной путь
от другого, более прямого пути, каким является гликолиз.
Гексозомонофосфатный путь имеет многоплановое значение; достаточно,
например, напомнить о химической стадии процесса фотосинтеза (гл. 10),
где он играет важную роль в реакциях, приводящих к образованию
различных форм Сахаров.
Процесс носит окислительный характер, в ходе которого от
углеродной цепи происходит отщепление по одному атому углерода,
который освобождается в форме С02. В катаболизме гексоз по гексозомо-
нофосфатному пути принимают участие три системы ферментов: 1) де-
гидрогеназно-карбоксилазная; 2) изомеризующая и 3) система
структурной перестройки сахара.
Дегидрогеназно-карбоксилазная система расщепляет глюкозо-6-фос-
фат на рибулозо-5-фосфат и С02; для этого требуются три фермента:
глюкозо-6-фосфатдегидрогеназа, б-фосфоглюконатдегидрогеназа и глю-
конолактоназа; коферментом дегидрогеназ служит ЫАОР+:
335
Часть II. Метаболизм веществ и энергии в клетке
СН2ОР
Глюкозо-6-фосфат-
дегидрогеназа
^БР+ < КАВРН + Н+
ОН
Глюкозо-б-фосфат 6-Фосфоглюконолактон
Образовавшийся промежуточный продукт окисления 6-фосфоглю-
конолактон быстро превращается в 6-фосфоглюконат под действием
лактоназы:
сн2ор соон
I
6-Фосфоглюконолактоназа Н—С-ОН
0 ^ НО-С-Н
но >1^^/Г н-с-он
ОН Н-С-ОН
6-Фосфоглюконолактон ^Н ^р
6-Фосфоглюконат
Далее 6-фосфоглюконат подвергается окислительному декарбокси-
лированию под действием 6-фосфоглкжонатдегидрогеназы, в
результате чего образуется кетопентоза — Б-рибулозо-5-фосфат и еще одна
молекула КАВРН:
СООН 6-Фосфоглюкокат- СЩОН
Н-С-ОН дегидрогеназа С=0
I .. ... ^ I
НО-С-Н /^ "\ ^^ Н-С-ОН
н-с-он / \ со2 н-с-он
Н-С-ОН НАБР+ МАОРН + Н+ СН2ОР
СН2ОР
6-Фосфоглюконат Рибу лозо- 5-фосфат
Значение АО0' в результате окисления глюкозо-6-фосфата в рибу-
лозо-5-фосфат за счет ЫАХ)Р+ составляет -30,8 кДж'моль"1. Дальше
идут неокислительные реакции.
Изомеризующая система, состоящая из двух ферментов: рибуло-
зо-5-фосфатизомеразы и рибулозо-5-фосфатэпимеразы, обеспечивает
превращение рибулозо-5-фосфата в рибозо-5-фосфат и ксилулозо-5-фосфат:
Изомераза _
•*- Рибозо-о-фосфат
— ■ *- Ксилулозо-5-фосфат
Зпимераза
Дальше систему структурной перестройки катализирует цепь
сложных превращений моносахаридов, которые весьма сходны с
превращениями в процессе фотосинтеза (см, гл. 10); важную роль здесь
играют два фермента: трансальдолаза и транскетолаза:
336
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
Рибулозо-5-
фосфат
Рибозо-5-
фосфат
Транскетолаза
Седогепту-
лозо-7-фосфат
+
Глицеральде-,
гид-3-фосфат*
Седогепту-
+
Глицераль-
Трансал ь до л аза
лозо- 7-фосфат де гид- 3-фосфат
Фруктозо-6-
* фосфат
+
Эритрозо-4- в
фосфат '
Ксилулозо-5-
фосфат
+
Эритрозо-4-
фосфат
Тр анскето л аз а
Глицераль- Глюкозо-6
> дегид-3-фосфат фосфат
В целом гексозомонофосфатныи путь окисления углеводов
представлен на рис. 9.5.
Гексозомонофосфатныи путь и гликолиз имеют много общих
ферментов. Таким образом, оба пути могут реализоваться в одной клетке
Глюкоза
Глюкозо-6-фосфат (Се)
С02
Рибулозо-5-фосфат Рибозо-5-фоефат
Глгокозо-6-фосфат (Се)
Седогептулозо-7-
фосфат (С?)
\
Фруктозо-6-фосфат (Сб)
Фруктозо- 6
-фосфат (Сб)*
Транскетолаза
Глицеральдегид-3-
фосфат (Са)
Трансальдолаза
Рибулозо-5-
фосфат (Сз)
Эритрозо-4-фосфат
(С4
Транскетолаза
С02
Ацетил-СоА(С2>
Глицеральдегид-3 -
фосфат (Сз)
С02
цтк
СО-
Рас. 9.5. Гексозомонофосфатныи путь окисления углеводов
22. Заказ 3486
337
Часть П. Метаболизм веществ и энергии в клетке
одновременно. При превращении 6 молекул глюкозо-6-фосфата по гек-
созомонофосфатному пути суммарный эффект будет тот же, что и при
полном окислении 1 молекулы гексозы в С02 по пути гликолиза и
цикла трикарбоновых кислот. Суммарное уравнение окисления
глюкозы по гексозомонофосфатному пути выглядит следующим образом:
6 глюкозо-6-фосфат + 12КАВР+ -> 6С02 + 6Н20 + 12ИАВРН +
+ 5 глюкозо-6-фосфат.
Одна молекула глюкозо-6-фосфата полностью окисляется до С02 и
Н20, а пять молекул регенерируют и могут вновь вступать в гексозо-
монофосфатный путь.
Этот путь выполняет три основные функции. Во-первых, он
поставляет ЫАВРН, который используется в процессах
восстановительного биосинтеза. В частности, в связи с этим он имеет важное
значение в нефотосинтезирующих тканях, например в тканях
прорастающих семян, в часы темнового периода. Во-вторых, гексозомонофос-
фатный путь является источником рибозо-5-фосфата, необходимого для
синтеза нуклеиновых кислот. В третьих, он генерирует эритрозо-4-фос~
фат, который используется для синтеза ароматических аминокислот:
фенилаланина, тирозина, триптофана.
1
9,8. Транспорт электронов и окислительное фосфорилирование
Окислительное фосфорилирование представляет собой
генерирование химической энергии в АТР за счет окисления
высокоэнергетических (восстановленных) соединений, таких, как ЫАВН и РАВН2,
образованных в ходе гликолиза, в цикле трикарбоновых кислот и ряде
других внутриклеточных процессах. Только небольшая часть
потенциально доступной энергии, содержащейся в молекуле глюкозы,
извлекается в ходе гликолиза. В цикле трикарбоновых кислот
происходит генерирование восстановленных коферментов: ЫАВН и РАОН2.
Окисление жирных кислот и аминокислот, рассмотренные в
соответствующих разделах (см. гл. 11 и 12), ведут к образованию тех же
самых богатых энергией молекул. Теперь мы рассмотрим процесс
окислительного фосфорилирования, в котором происходит окисление
восстановленных коферментов и образование АТР. Аналогичный
процесс — фотосинтез, протекающий в хлоропластах растений, также
сопряжен с переносом электронов и превращением энергии.
Окислительное фосфорилирование является завершающим этапом
процесса дыхания. ЫАВН и РАВН2 окисляются молекулярным
кислородом не сразу и не прямо. Процесс идет постепенно с помощью
последовательно функционирующих окислительно-восстановительных
систем, переносящих электроны и протоны к молекулярному
кислороду. Совокупность этих систем получила название дыхательной цепи
переноса электронов. Главная функция дыхательной цепи —
преобразование энергии переноса электронов, аккумулированной в ИАВН
338
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
и КА1)Н2, в энергию переноса фосфатной связи в результате синтеза
АТР из АВР и Р.,
Окислительное фосфорилирование требует наличия нескольких
ферментных комлексов, локализованных в мембране. Некоторые из
них создают градиент концентрации протонов, а один из комплексов
использует протонный градиент для синтеза АТР из АВР и Р.. В
клетках эукариот окислительное фосфорилирование осуществляется в
митохондриях с помощью ферментов, локализованных во внутренней
митохондриальной мембране. В бактериях эти ферменты находятся в
плазматической мембране. В хлоропластах мембрана тилакоидов
содержит компоненты, участвующие в фотосинтезе. Рассмотрим
детально окислительное фосфорилирование, происходящее в митохондриях
животных, которое наиболее хорошо изучено.
Прежде всего следует отметить, что окислительное
фосфорилирование включает два тесно связанных процесса:
1) Митохондриальные восстановительные эквиваленты
окисляются в дыхательной электронтранспортной цепи> и серии мембранно-
связанных ферментных комплексов служат в качестве переносчиков
электронов. Электроны передаются от восстановительных кофермен-
тов к конечному акцептору электронов при аэробном метаболизме,
т.е. к молекулярному кислороду (02). При движении электронов через
комплексы энергия, извлекаемая за счет окисления
восстановительных эквивалентов, используется для транспорта протонов через
внутреннюю мембрану из матрикса (т.е. области, окруженной внутренней
мембраной) в наружное пространство. При этом происходит
генерирование протонного концентрационного градиента, в результате чего
матрйкс оказывается более щелочным и отрицательно заряженным,
чем пространство снаружи мембраны.
2) Протонному концентрационному градиенту присуща свободная
энергия. Эта энергия выделяется, когда протоны перемещаются назад
сквозь митохондриальную мембрану через другой внутренний
мембранный ферментный комплекс, а именно АТР-синтетазу. Этот
комплекс катализирует фосфорилирование АОР, когда протоны
перемещаются по концентрационному градиенту: АВР + Р. —» АТР + Н20.
Оба процесса энергетически сопряжены. Молекулы, участвующие
в каждом из процессов, находятся в одном компартменте и
представляют собой плотноупакованный и высокоупорядоченный ансамбль.
Движущей силой потока электронов от одного
высокоэнергетического соединения к другому и в конце дыхательной цепи к
терминальному кислороду является постепенное возрастание окислительной
способности компонентов цепи, известной под названием окислительно-
восстановительного потенциала. В данном разделе мы рассмотрим
структуру и функцию дыхательной цепи митохондрий, механизм и
энергетику окислительного фосфорилирования. Прежде чем
приступить к рассмотрению этих вопросов, следует остановиться на некото-
22*
339
Часть II. Метаболизм веществ и энергии в клетке
рых представлениях об окислительно-восстановительном потенциале.
Здесь этот вопрос будет описан применительно к биологическим
системам.
Окислительно-восстановительный потенциал биологических систем
(или редокс-потенциал) представляет собой показатель окислительной
или восстановительной способности любой
окислительно-восстановительной реакции, находящейся в состоянии равновесия.
Окислительно-восстановительный потенциал определяется по уравнению
Р Сгей
где Е — окислительно-восстановительный потенциал, В; К — газовая
постоянная; Т — температура, К; Р — число Фарадея; Е° — величина,
характерная для каждой окислительно-восстановительной системы и
соответствующая ее окислительно-восстановительному потенциалу при
отношении окисленной формы вещества (Сох) к восстановленной (Сгеа),
равном 1; Е° принято считать стандартной величиной.
Биологическая система является открытой. В ней биохимические
процессы протекают необратимо, а постоянство свойств системы (ее
стационарное состояние) во времени поддерживается непрерывным
притоком химических веществ и энергии извне. Поэтому в
биологической системе величина окислительно-восстановительного
потенциала является стационарной. Однако она зависит от многих причин:
активности оксидоредуктаз, концентрации молекулярного кислорода,
величины рН среды и т.д.
Значение потенциала в паре Сох и Сге(1 измеряется относительно
стандартной водородной пары (2Н+/Н2), которая является
общепринятой точкой отсчета. При строго стандартных условиях (давлении Н2
0,1 Па; концентрации [Н+] в 1 моль, температуре 25 °С) окислительно-
восстановительный потенциал водородной пары условно принят за
0,00 В. В дыхательной цепи окислительно-восстановительные
реакции идут в среде, близкой к нейтральной, и соответствующий
потенциал водородной пары (Е°) равен -0,42 В:
2Н+ (Ш; рН 0) / Н2(0,1 Па); Е° = 0,0 В;
2Н+ (Ю-7; рН 7) / Н2 (ОД Па); Е° = -0,42 В.
Измерение редокс-потенциала для других пар производится по паре
2Н+/Н2. Возникшее напряжение будет представлять собой разность
между редокс-потенциалами обеих пар.
В стандартных условиях сильный восстановитель (например, ЫАОН)
обладает отрицательным редокс-потенциалом, тогда как сильный
окислитель (02) имеет положительный редокс-потенциал.
Ред оке-потенциалы биологически важных
окислительно-восстановительных пар, входящих в дыхательную цепь, представлены в
табл. 9.4.
340
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
Таблица 9.4
Стандартные окислительно-восстановительные потенциалы
компонентов электронтранспортной цепи (рН 7, 25 °С)
Субстрат или комплекс
ИАВН
Комплекс I
ГШГ
Ге-8 кластер
Сукцинат
Комплекс II
РАО
Ге-8 кластер
ОП2/Я
&№
<ЭН2/Ср
Комплекс III
Ре-8 кластер
Цитохром &5б0
Цитохром &566
Цитохром сх
Цитохром с ф
Комплекс ГУ
Цитохром а
<Ч
Цитохром а3
Спв
о2
Е0', В
-0,32
-0,30
-0,25 до-0,05
+0,03
0,0
-0,26 до 0,00
+0,04
-0,16
+0,28
+0,28
-0,10
+0,05
+0,22
+0,23
+0,21
+0,24
+0,39
+0,34
+0,82
Любая пара, представленная в данной таблице с более положи-
тельным редоке-потенциалом, будет самопроизвольно
восстанавливаться, принимая электроны*
Митохондрии. Конечные стадии аэробного окисления
биомолекул у эукариот осуществляются в митохондриях (от греч. тИоз —
нить + Нопйпоп — зернышко) . Это относительно автономные
структуры, играющие роль "силовых станций" клетки. Данная органелла
является местом, где протекают цикл трикарбоновых кислот и
окисление жирных кислот, генерирующие восстановленные коферменты,
которые затем окисляются с помощью дыхательной
электрон-транспортной цепи. Структура митохондрии показана на рис. 9.6.
Количество митохондрий в клетках может значительно варьировать.
Некоторые водоросли содержат только одну митохондрию, тогда как
простейшее СНаоз скаоз — полмиллиона. Клетка печени животных
содержит около 5000 митохондрий. Количество митохондрий соотносится
в целом с энергетическими потребностями клетки. Ткань белой мышцы
содержит относительно мало митохондрий и обеспечивает свои
энергетические потребности в основном путем анаэробного гликолиза. Одним из
наиболее ярких примеров в этом отношении являются быстро реагирую-
341
Часть II. Метаболизм веществ и энергии в клетке
щие, но медленно
восстанавливающие свой энергетический
потенциал мышцы аллигаторов.
Аллигатор может наброситься
на свою жертву с огромной
скоростью и силой, но не может
повторить это несколько раз.
Ткань красных мышц содержит
много митохондрий, что
необходимо, в частности, для
летательных мышц перелетных птиц,
которые должны преодолевать
большие расстояния.
Митохондрии значительно
различаются по размерам и
форме в различных тканях и даже
в одной клетке. Типичная
митохондрия животных имеет
диаметр от 0,2 до 0,8 мкм и длину
от 0,5 до 1,5 мкм, подобно
размерам клетки ЕвсНепсЫа соИ.
Митохондрия окружена двумя мембранами с сильно
различающимися свойствами. Содержащийся во внешней мембране белок порин
образует каналы, что обеспечивает свободную диффузию ионов и большинства
водорастворимых метаболитов. Внутренняя митохондриальная
мембрана является барьером для протонов. Она способна пропускать такие
молекулы, как вода, молекулярный кислород, диоксид углерода, но
фактически непроницаема для больших полярных и ионизированных веществ
(К+, №\ СГ, Вг", «Г", КАВ\ ЫАПН, ИАБРН, АМР, СоА-8Н). Чтобы
пересечь внутреннюю мембрану, такие вещества, как Р., АВР, АТР, пи-
руват, сукцинат, мадат, цитрат, должны быть транспортированы
специальными мембранно-связанными транспортерами. Вход анионных
метаболитов в отрицательно заряженное внутреннее митохондриальное
пространство нежелателен с энергетической точки зрения. Такие
метаболиты обычно обмениваются на другие анионы из внутреннего митохондри-
ального пространства или же сопутствуют движению протонов по
концентрационному градиенту, генерируемому за счет окисления.
Внутренняя митохондриальная мембрана очень богата белками
(соотношение белков к липидам — 4:1 по массе). Внутренняя мембрана
образует частые складки, что обеспечивает значительное увеличение
площади занимаемого пространства. Эти складки называются криста-
ми. Число крист для митохондрий различных клеток варьирует в
широких пределах; так, в митохондриях клеток печени крист очень
мало и они короткие, в митохондриях мышечных клеток кристы
многочисленны, а матрикса мало. Видимо, число крист коррелирует с
Рис. 9.6. Структура митохондрии.
Внешняя митохондриальная мембрана
свободно проницаема для небольших соединений.
Внутренняя мембрана, которая фактически
непроницаема для полярных и
ионизированных веществ, образует кристы. Белковые
комплексы, катализирующие реакции,
вовлеченные в окислительное фосфорилирова-
ние? локализованы во внутренней мембране
342
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
энергетической нагрузкой митохондрии. Компоненты,
обеспечивающие реакции окисления, присущие окислительному фосфорилирова-
нию, локализованы во внутренней митохондриальнои мембране, но
некоторые из их субъединиц находятся в матриксе. На долю
ферментов дыхательной цепи приходится 20—25 % белков мембраны.
Остальные белки принимают участие в структурной организации
дыхательной цепи. В каждой дыхательной цепи ферменты расположены в
виде отдельных самостоятельно функционирующих групп,
называемых дыхательными ансамблями. Чем интенсивнее потребление
энергии в ткани или органе, тем большее число ансамблей в митохондрии:
так, в митохондриях печени их число составляет около 5000, а в
митохондриях сердца — достигает 20 000- Дыхательные ансамбли
распределены по плоскости внутренней мембраны равномерно,
расстояние между их центрами примерно 20 нм. Они находятся в липидном
окружении {"плавают" в липидном слое), прочно связаны с
мембраной. Исключение составляет цитохром с, который слабо связан с
внешней поверхностью мембраны и относительно легко извлекается
водными растворами; очевидно, он может выходить в межмембранное
пространство. В организации структуры дыхательных ансамблей
участвуют липиды бислоя мембраны. Хотя они и не являются
переносчиками электронов непосредственно, тем не менее они существенны для
функционирования ансамблей, создавая необходимое окружение для
активных центров переносчиков электронов. Удаление липидов из
митохондрий подавляет активность дыхательной цепи; добавление к
системе экстрагированных липидов восстанавливает активность цепи.
Область между внешней и внутренней митохондриальными
мембранами называется внутримитохондриальным пространством. Наружная
мембрана свободно проницаема для соединений с молекулярной массой
около 10 000. Так как внешняя мембрана проницаема для небольших
молекул, межмембранное пространство имеет тот же состав ионов и
метаболитов, что и цитозоль. Содержимое матрикса включает пируватде-
гидрогеназный комплекс, ферменты цикла трикарбоновых кислот (за
исключением сукцинатдегидрогеназного комплекса, который
локализован во внутренней мембране) и большинство ферментов,
катализирующих окисление жирных кислот. Концентрация белков в матриксе
настолько высока (достигает 500 мг*мл"х), что он представляет собой
субстанцию наподобие геля. Матрикс также содержит метаболиты,
неорганические ионы и собственный пул КА1)+ и ЫА1)Р+, что обеспечивает
отделение цитозольных пиридиннуклеотидных коферментов.
Электронное микроскопирование позволило обнаружить еще одну
характерную особенность внутренней мембраны — наличие в ней
сферических частиц диаметром 8—10 нм, сидящих на коротких ножках,
обращенных в сторону матрикса и названных Гх-частицами. Частицы
располагаются на расстоянии 10 нм друг от друга, и в каждой
митохондрии их насчитывается около 100. Они играют важную роль в со-
343
Часть II. Метаболизм веществ и энергии в клетке
пряжении электронного транспорта и фосфорилирования; удаление
частиц ведет к прекращению синтеза АТР.
Хемиосмотическая теория. Концепция о том, что градиент
концентрации протонов служит своеобразным хранилищем энергии и
используется для образования АТР, носит название хемиосмотической
теории, которая была сформулирована П. Митчеллом в начале 60-х гг.,
хотя с середины 20-х гг. механизм, с помощью которого клетки
осуществляют окислительное фосфорилирование, уже был предметом
исследований и дискуссий. Эксперименты В. А. Энгельгарда, показавшие, что
эритроциты голубя (они имеют митохондрии, в отличие от безъядерных
эритроцитов человека) одновременно с поглощением кислорода
используют неорганический фосфат, знаменовали открытие окислительного
фосфорилирования, т.е. сопряжения дыхания с фосфорилированием
(1931 г). В 1939—1940 гг. В. А. Белицером и Е. Т. Цыбаковой в
биоэнергетику было введено соотношение Р/О как показателя сопряжения
дыхания и фосфорилирования. Выло показано, что при поглощении одного
атома кислорода поглощается примерно три атома неорганического
фосфата, т.е. при переносе пары электронов от окисляемого субстрата на
кислород коэффициент Р/О равен примерно 3. Другими словами, в
дыхательной цепи имеется как минимум три пункта фосфорилирования,
где неорганический фосфат используется для образования АТР,
Существовало множество гипотез, пытавшихся объяснить механизм
сопряжения окисления и фосфорилирования. Так, например, химическая
гипотеза объясняла образование АТР подобно тому, как это имеет место при
субстратном фосфорилировании, т.е. через образование промежуточного
высокоэнергетического продукта. Но такой интермедиат не был
идентифицирован, П. Митчелл впервые предположил, что превращение
энергии связано с мембранным электронным транспортом в митохондрии.
Революционная идея Митчелла, что градиент концентрации протонов
обладает потенциальной энергией, в настоящее время стала
центральным положением в биоэнергетике. Митчелл был удостоен Нобелевской
премии в 1978 г. за исследования в данной области.
Согласно хемиосмотической теории энергия переноса электронов по
электрон-транспортной цепи сначала сосредоточивается в виде
протонного потенциала, называемого также электрохимическим градиентом
ионов Н+ (рис, 9,7). Этот потенциал создается движением заряженных
протонов через мембрану, которые выбрасываются из матрикса в
наружное пространство, что достигается за счет функционирования электрон-
транспортной цепи, действующей наподобие помпы. Диффузия протонов
обратно через мембрану, т.е. в область с их меньшей концентрацией,
происходит по концентрационному градиенту и сопряжена с
фосфорилированием, осуществляемым АТР-синтетазой. Таким образом, электрон-
транспортная цепь совершает два вида работы: осмотическую
(концентрирует протоны снаружи митохондриальной мембраны) и
электрическую (создает разность электрических потенциалов). Энергия, сосредото-
344
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
чиваемая в протонном потенциале,
используется АТР-синтетазой для
выполнения химической работы,
т.е. синтеза АТР, Протонный
потенциал принято обозначать как
А|д^. Он состоит, в соответствии с
отмеченным выше, из двух
компонентов: электрического (разность
электрических потенциалов, Д\}/) и
осмотического (разность
концентраций Н+ по обе стороны
внутренней митохондриальной мембраны,
ДрН). Протонный градиент иногда
называют рН~градиентом. Но это
слишком большое упрощение; в
данном случае упускается
градиент заряда, являющийся также
источником свободной энергии,
заключенной в протонном
концентрационном градиенте. Например, в
митохондриях печени Д\|/ = 0,17 V,
Электронный транспорт обеспечивает
перенос наружу Н+ и создает
электрохимический градиент
Выше рН,
ниже [Н+] в матриксе
Ниже рН,
выше [Н+] в окружающем пространстве
Рис. 9.7. Протонный и
электрохимический градиенты, существующие по обе
стороны внутренней митохондриальной
мембраны. Электрохимический градиент
генерируется за счет транспорта
протонов через мембрану
а АрН — 0,5; и количество
свободной энергии, доступной из электрического градиента, значительно
больше, чем из химического, а именно: 85 % против 15 %.
В свое время П. Митчеллом были предложены несколько
постулатов, подтверждение которых могло бы экспериментально поддержать
его теорию:
1. Целостность внутренней митохондриальной мембраны является
необходимым требованием для сопряжения дыхания и
фосфорилирования. Мембрана должна быть непроницаема для заряженных
соединений; в противном случае протонный градиент будет разрушен.
Ионизированные метаболиты могут проникать через мембрану только с
помощью специфических переносчиков.
2. Перенос электронов по электрон-транспортной цепи приводит к
генерированию протонного потенциала с более высокой
концентрацией Н+ с наружной стороны внутренней митохондриальной мембраны.
3. Мембранно-связанный фермент, АТР-синтетаза, катализирует
фосфорилирование АВР в реакции, сопряженной с переносом
протонов через внутреннюю митохондриальную мембрану.
Следует отметить, что в присутствии так называемых разобщающих
агентов происходит разобщение дыхания и фосфорилирования, а
окисление субстратов осуществляется без фосфорилирования АВР. Одним из
наиболее хорошо известных разобщителей является 2,4-динитрофенол.
Разобщающие агенты, являясь жирорастворимыми соединениями,
легко .проникают через внутреннюю митохондриальную мембрану; при этом
345
Часть II. Метаболизм веществ и энергии в клетке
они связывают протоны в цитозоле, переносят их через мембрану и
высвобождают в матрикс. Это приводит к разобщению (рассеиванию)
протонного потенциала. Такой перенос протонов разобщает окисление и фосфо-
рилирование; хотя протоны и поступают в матрикс, однако при этом они
не проходят через АТР-синтетазу. Таким образом, действие
разобщающих агентов подтверждает первый постулат.
В 1965 г. П. Митчелл и Дж. Мойле подтвердили второй постулат,
продемонстрировав, что, если митохондрии в анаэробных условиях
получают небольшое количество кислорода, то происходит закисле-
ние среды.
АТР-синтетазная активность была впервые обнаружена в 1948 г.
Несколько позже многочисленными работами была показана обратимость ее
действия. В настоящее время АТР-синтетаза достаточно хорошо изучена.
Комплексы электрон-транспортной цепи. Во внутренней
мембране митохондрии обнаружено пять комплексов белков,
обеспечивающих окислительное фосфорилирование. Они были изолированы в
активной форме путем осторожной солюбилизации с использованием
детергентов. Каждый ферментный комплекс катализирует отдельную
стадию энергопередающего процесса. Эти комплексы обозначаются
номерами от I до V. Четыре из них, с I по IV, содержат множество
кофакторов и вовлечены в перенос электронов. Комплекс V — это
АТР-синтетаза. Комплексы физически не связаны друг с другом.
Четыре ферментных комплекса содержат разнообразные окислительно-
восстановительные кофакторы. Транспорт электронов осуществляется
благодаря окислению и восстановлению этих кофакторов, и перенос
происходит от восстановленного переносчика к окисленному.
Некоторые параметры комплексов I—IV представлены в табл. 9.5.
Таблица 9.5
Характеристика комплексов электрон-транспортной цепи из сердца быка
Комплекс
X*. \-#4тХ ДХ^ли А1^*
I. КАВН-убихинон
оксидоредуктаза
П. Сукцинат-убихинон
оксидоредуктаза
III. Убихинол-цитохром с
оксидоредуктаза
IV. Цитохром с оксидаза
Субъединицы
>41
4
9—10
13
Молекулярная масса
~800 000
125000
«250 000
400 000
(димер)
Окислите льно-восстанови -
тельные компоненты
1ПШ
22—24 Ре-8 центров
в 5—7 кластерах
1РАБ
7—8 Ге-8 центров
в 3 кластерах
Цитохром Ьш
1 Ре-8 кластер
Цитохром Ь
Цитохром сг
Цитохром а
Цитохром а3
2 иона меди
Комплексы физически не связаны друг с другом. Четыре
ферментных комплекса содержат разнообразные окислительно-восстановитель-
346
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
ные кофакторы. Транспорт электронов осуществляется благодаря
окислению и восстановлению этих кофакторов, и перенос происходит от
восстановленного переносчика к окисленному.
Направление движения электронов определяют
окислительно-восстановительные потенциалы. Перенос электронов осуществляется от
МАВН (Е° = -0,32 В) к кислороду (Е° = +0,82 В). Редокс-потенциалы
переносчиков электрон-транспортной цепи указаны в табл. 9.4.
Убихинон (^ и цитохром с служат как связывающие звенья между
комплексами электрон-транспортной цепи. Кофермент (} переносит
электроны от комплексов I и II на комплекс III. Цитохром с связывает
комплексы III и IV* Комплекс IV использует электроны, катализируя
восстановление кислорода до воды. Перенос электронов по электрон-
транспортной цепи изображен на рис. 9.8 напротив шкалы
стандартных редокс-потенциалов (слева) и шкалы изменения стандартной
свободной энергии (справа). Стандартный редокс-потенциал (в вольтах)
непосредственно взаимосвязан с изменением стандартной свободной
энергии (в кДж/моль) в соответствии со следующей формулой:
АО0' - -пГАЕ0'.
Как видно из рис. 9.8, существенное количество энергии
выделяется в ходе переноса электронов. Эта энергия, выделяющаяся
порциями во время транспорта электронов, и запасается в форме энергии
протонного потенциала.
Указанные величины действительно справедливы только при
стандартных условиях. Взаимосвязь между настоящим редокс-потенциа-
лом (Е) и стандартным потенциалом (Е°) подобна взаимосвязи между
реальной и стандартной свободными энергиями:
Е = Е0, + КТ1п(ге<1/ох).
Здесь сокращения гей и ох обозначают две стадии электронного
переноса.
Стандартная свободная энергия, которая становится доступной
благодаря реакциям, катализируемым комплексами
электрон-транспортной цепи, показана в табл. 9.6.
Таблица 9.6
Стандартная свободная энергия, выделяемая в окислительных реакциях,
катализируемых комплексами электрон-транспортной цепи
Комплекс
I. (МАЭН/<Э)
П. (сукцинат/С})
III. (<ЗН2/Цитохром с)
IV. (Цитохром с/02)
Е* восстановитель,
В
-0,32
+0,03
+0,04
+0,23
Е0" окислитель,
В
+0,04
+0,04
+0,23
+0,82
ДЕ°\ В
+0,36
+0,01
+0,19
+0,59
ДО0',
кДж/моль
-70
-2
-37
-ПО
Обратите внимание, что только реакции, катализируемые
комплексами I, III и IV, дают энергию, достаточную для превращения АВР+Р. в
347
Часть II. Метаболизм веществ и энергии в клетке
МАШ -* ГОШ -> Ге-8 ><3 -» [^ ь] -> СуЪ сх -> Су* с -> Су* с3 -> Су1 а -» 02
Сукцинат ~> РАО -► Ге-8
Е°', В
-0,4 п
С0', кДж/моль
-0,2 -
0 ■
0,2 -
0,4 -
0,6 -
0,8-
МАВН-м
ЫАР
+
Комплекс I
МАОН-убихинон
оксидоредуктаза
- 200
Перенос
электронов
©
(
Сукцинат
Фу марат
Комплекс Ш
Убихинон-
цитохром с
оксидаза
Пит. с
Комплекс II
Сукцинат-
убихинон
оксидоредуктаза
Комплекс IV
Цитохром с
оксидаза
1/202 + 2Н®
1,0 ^
• 100
■ 0
Рас. 9.8. Митохондриальный электронный транспорт. Каждый из четырех
комплексов электрон-транспортной цепи, состоящий из нескольких белковых
субъединиц и кофакторов, подвергается попеременному восстановлению и
окислению. Комплексы взаимосвязаны с помощью мобильного переносчика убихино-
на (<^) и цитохрома с. Высота каждого комплекса соответствует Е0' между его
восстановленным агентом (субстратом) и его окисленным агентом (продуктом).
Перенос электронов от NА^Н и сукцината к 02 сопровождается транслокацией
протонов из матрикса в пространство, окружающее мембрану, что создает
градиент концентрации протонов через мембрану
АТР. Эти три комплекса обепечивают передачу протонов через
внутреннюю митохондриальную мембрану, когда через них проходят
электроны. Комплекс И, т.е. сукцинатдегидрогеназныи комплекс, являющийся
также компонентом цикла трикарбоновых кислот, не вносит вклада в
образование градиента концентрации протонов. Комплекс II переносит
электроны от сукцината к коферменту (^ и, таким образом, служит
своеобразным "поставщиком" для электрон-транспортной цепи.
Функционирование отдельных комплексов может быть
избирательно подавлено с помощью специфических ингибиторов. Ингибиторами
348
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
комплекса I являются ротенон, пиерицидин А. Интересно, что
ротенон как высокотоксичное вещество издавна использовался
американскими индейцами для ловли рыб. В настоящее время он применяется
как инсектицид. Ротенон легко проходит через дыхательные трубки
насекомых и жабры рыб. Полагают, что он блокирует перенос
электронов от железосерных центров на убихинон. Для млекопитающих
ротенон относительно нетоксичен. Ингибиторами комплекса II
являются малонат (конкурентный ингибитор сукцинатдегидрогеназы), те-
ноилтрифторацетон. Функционирование комплекса III подавляется в
присутствии антимицина А, амитала. Комплекс IV ингибируется под
действием цианида, азида, оксида углерода, сероводорода.
Переносчики электрон-транспортной цепи. Основная часть де-
гидрогеназ переносит водородные атомы от дыхательных субстратов
на КАВ+. Данный кофермент выполняет как бы коллекторную
функцию; в форме ЫАВН собираются восстановительные эквиваленты от
различных субстратов. Причем КАВ* собирает восстановительные
эквиваленты не только от субстратов, окисленных под действием
КАО-зависимых дегидрогеназ, но и под действием Т^АОР-зависимых
дегидрогеназ, что оказывается возможным благодаря действию
фермента трансгидрогеназы, катализирующего реакцию:
ЫАВРН + NА^+ & ЫА1)Р+ + ИАВН.
Движение электронов по комплексам электрон-транспортной цепи
обеспечивается за счет переноса между сопряженными кофакторами.
Электроны вступают в электрон-транспортную цепь двумя путями: от
МАОН и сукцината. Флавиновые коферменты комплексов I и II, РМЫ
и РАО соответственно, восстанавливаются. Восстановленные
коферменты Г]\ШН2 и ЕАОН2 в определенный момент времени отдают один
электрон, и на всех последующих этапах в электрон-транспортной цепи
осуществляется одноэлектронный перенос. Железосерные кластеры
присутствуют в комплексах I, II и III. Каждый железосерный кластер
может принимать или отдавать электрон, и при этом осуществляется
изменение валентности железа (Б*е3+ ^ Ге2+).
Цитохромы представляют собой гемопротеины, которые при
переносе электронов подвергаются обратимому окислению-восстановлению. При
этом меняется степень окисления железа от Ге2+ до Ге 3+. Все
цитохромы, за исключением цитохрома с, представляют собой интегральные
мембранные белки. Цитохромы были впервые открыты С. А. Максмун-
ном в 1886 г. Затем они были как бы вновь открыты Д. Кейлином в
1925 г. Кейлин выяснил, что эти гемопротеины вовлечены в процесс
дыхания. Он классифицировал их на основе абсорбционных спектров по
категориям а, Ъ и с. Гемовые простетические группы этих трех типов
цитохромов имеют небольшие структурные различия. Цитохромы
различаются не только по спектрам поглощения, но и, что особенно важно,
по их редокс-потенциалам. Цитохром Ь имеет различные формы. В элек-
349
Часть II. Метаболизм веществ и энергии в клетке
трон-транспортной цепи, очевидно, функционируют цитохромы 6560 и Ъш,
называемые так по максимуму поглощения света. Они образуют
комплекс, передекающий липидный бислой мембраны от внутренней к
наружной поверхности. Цитохром сг находится в липидном слое ближе к
наружной поверхности мембраны, Цитохромы а и а3 образуют
комплекс IV, называемый также цитохромоксидазой, которая является
терминальной оксидазой и переносит электроны непосредственно на
кислород* В отличие от других цитохромов, цитохромокслдаза содержит медь.
Перенос электронов сопровождается не только изменением валентности
железа в железопорфириновых группах, но и обратимым изменением
валентности меди (Си+ $± Си2+). Комплекс IV пересекает мембрану
поперек от внешней стороны (здесь в липидной части мембраны находится
цитохром а) до внутренней стороны (здесь находится цитохром ав).
Электрон-транспортные комплексы внутренней митохондриальной
мембраны функционально связаны с мобильными переносчиками
электронов — убихиноном <3 и цитохромом с*
Убихинон (или кофермент <^) представляет собой
жирорастворимый хинон с изопреноидной боковой цепью (рис. 9,9). Часто
встречаются обозначения С}10 или Со(^10, что связано с наличием в
большинстве тканей млекопитающих убихинона с 10 изопреновыми звеньями
в боковой цепи. Он способен одновременно принимать и отдавать два
электрона, Убихинон <5 диффундирует внутри липидного бислоя,
принимая электроны от комплексов I и II и передавая их на комплекс III,
При переходе восстановительных эквивалентов на убихинон он
восстанавливается в убихинол (Со(^Н2). В ходе этого процесса происходит
окисление РОДШ2 в составе ЫАХЩ-дегидрогеназы до ГМЫ. В
дыхательной цепи убихинон выпол-
НзОО-С
н8с-о-с
с
II
о
н3с-о-с
II
НаС-О-С
с.
с~сн3
С-ЧСНг-СН^С-СНг^Н
Убихинон
(окисленная форма,
^ или Со<3)
2Н+ + 2е~
•*■
ОН
^С-ЧСНг-СН-С-СНа^Н
С
I Убихинол
ОН (восстановленная форма,
<ЗН2 или Со<ЭН2)
Рис. 9.9. Убихинон (кофермент (^)
няет коллекторную функцию. Он
принимает восстановительные
эквиваленты как от МАВН-де-
гидрогеназы, так и от ряда
других флавиндегидрогеназ,
локализованных в митохондриях, в
частности от фермента цикла
Кребса — сукцинатдегидрогена-
зы, локализованного во
внутренней митохондриальной
мембране, от ацил-СоА-дегидрогеназы,
катализирующей одну из стадий
окисления жирных кислот.
Другой мобильный
электронный переносчик — это
цитохром су который является
периферическим мембранным
белком, ассоциированным с наруж-
350
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
ной поверхностью мембраны. Цитохром с переносит электроны с
комплекса III на комплекс IV.
Функционирование комплекса I. Комплекс I, МАВН-убихинонок-
сидоредуктаза, называемый также часто ЫАОН-дегидрогеназой,
осуществляет перенос двух электронов от МАОН к убихинону. Это весьма
сложный ферментный комплекс. Например, в состав комплекса I из
митохондрий быка входит по меньшей мере 41 субъединица. Порядок
реакций, катализируемых комплексом I, показан на рис. 9.10. ЫАСН
отдает одновременно два электрона на комплекс I в качестве гидрид-
иона (Не, т.е. иона, состоящего из двух электронов и протона). Убихи-.
нон принимает электроны от КАОН-дегидрогеназы, но сначала он
принимает один электрон, превращаясь в промежуточный анион — семи-
хинон, прежде чем превратиться в полностью восстановленную
форму— убихинол (СЩ2). Это осуществляется по следующей схеме:
+е^ в
+е~, +2Н+
— ><№
На первом этапе переноса электронов через комплекс I КАВН
передает гидрид-ион на РМ1М", в результате чего образуется ЕЧ\ШН2. РЪШН2
окисляется в два этапа через промежуточную форму — семихинон,
отдавая два электрона по одному следующему переносчику электрон-
транспортной цепи — [Ге-8]-кластеру:
-Н",~е
клш —> гмшь —-
>ГЬЩН
•жаш
Таким образом, на уровне РМК происходит конвертирование двух-
электронного переноса в одноэлектронный, который характерен для
всего последующего транспорта по электрон-транспортной цепи.
Затем электроны передаются на убихинон.
Во время движения электронов через комплекс I протоны трансло-
цируются из матрикса в наружное пространство. Считают, что при
Комплекс I
Окружающая среда
1
РММН2
I
2 одноэлектронных
переноса
И>Ш
ЫАОН
,©
1
2 одноэлектронных
переноса
Ре~8
2Н
Ф
,©
ЫАБ
4Н
©
Матрикс
Рис. 9.10. Перенос электронов и протонов через комплекс I. Электроны
передаются от ЫАБН к С} через РМК и серию Ге-8 кластеров. Два протона
необходимы для восстановления <3 до С}НГ Стехиометрически на пару переносимых
электронов осуществляется транслокация около четырех протонов. Химический
механизм переноса протонов через мембрану неизвестен
351
Часть II. Метаболизм веществ и энергии в клетке
передаче пары электронов от ЫАОН к коферменту (} через мембрану
переносятся четыре протона. Однако еще не известно, как протоны
транспортируются через мембрану.
Функционирование комплекса II. Комплекс II, сукцинат-убихи-
нон оксидоредуктаза, называемый также сукцинатдегидрогеназным
комплексом, принимает электроны от сукцината и, подобно комплек-
су I, катализирует восстановление убихинона (<^) до убихинола (<ЗН2).
Комплекс II млекопитающих состоит из четырех субъединиц. Две
наибольшие субъединицы образуют сукцинатдегидрогеназу, которая
содержит РАО в качестве простетическои группы и три железосерных
кластера. Две другие субъединицы, очевидно, необходимы для
связывания сукцинатдегидрогёназы с внутренней митохондриальной
мембраной и передачи электронов к их акцептору — убихинону. Ранее мы
уже рассматривали катализируемое сукцинатдегидрогеназой
окисление сукцината до фумарата в качестве одной из реакций цикла три-
карбоновых кислот (см. раздел 9.4).
При переносе двух электронов от сукцината к убихинону
происходит восстановление ГАВ. Затем два электрона путем одноэлектронно-
го переноса передаются от восстановленного флавина на серию трех
железосерных кластеров (рис. 9.11). Цитохром &560,
скооперированный с данным комплексом, связан с одной из меньших субъединиц.
Однако его роль в транспорте электронов остается не вполне ясной.
Поскольку в ходе реакций, катализируемых комплексом II,
выделяется небольшое количество свободной энергии, то этот комплекс не
вносит вклада в формирование градиента концентрации протонов
через внутреннюю митохондриальную мембрану. Его функция
заключается в том, что он вводит электроны от окисляемого сукцината в
электрон-транспортную цепь на уровне убихинона. Убихинон принимает
электроны от комплексов I и II и передает их комплексу III.
Функционирование комплекса III. Комплекс III, убихинол-ци-
тохром с оксидоредуктаза, состоит из 9 или 10 различных субъеди-
Комплекс II
Окружающая среда
2 од ноэлек тронных
переноса
Г
?АБН2
О
2е
О
2 одноэлектронных\:;,;:~;
перено.са
Ре-8 **
„О
'560
Сукцинат
Фумарат + 2Н®
ш
2Н
©
Матрикс
Рис. 9.11. Перенос электронов через комплекс II. Комплекс II не вносит
вклада в протонный потенциал, но служит поставщиком^ который вводит электроны
на уровне убихинона
352
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
Ь5бо и Ъ$№ и Дитохром с
ниц, включая белок с железосерным кластером, цитохром Ъ,
содержащий на полипептидной цепи два гема —
Окисление одной молекулы <}Н2 сопряжено с переносом четырех
протонов на наружную сторону внутренней митохондриальной мембраны
(рис. 9.12), но только два из них — из матрикса; два другие — от <ЗН2.
Электроны принимаются из данного комплекса одноэлектронным
переносчиком, цитохромом су который способен перемещаться вдоль
стороны внутренней мембраны, граничащей с цитозолем, и переносить
электроны на комплекс IV.
2Н
е
\
Окружающая среда
2 одноэлектронных
переноса
тт^
Комплекс III
^
2Н
©
Матрикс
Рис, 9.12. Перенос электронов и протонов через комплекс III. Электроны
передаются от С^Н2 к цитохрому с. Четыре протона транслоцируются через
мембрану: два — из матрикса и два — от <^Н0
Процесс переноса электронов через комплекс III весьма сложен*
Осуществляющиеся здесь реакции окисления — восстановления и их
сопряжение с движением протонов обеспечивают функционирование Я~цикла>
существование которого было предположено в 1975 г. П. Митчеллом.
Сложившееся в настоящее время понимание функционирования
С^-цикла, объясняющее, как электроны движутся между убихинолом,
<ЗН2 и цитохромом с, представлено на рис. 9.13. В процесс вовлечены
три формы убихинона: <^Н2, $ и семихинон <5е. Первые две формы
растворимы в липидном бислое и движутся между цитозольной и мат-
риксной сторонами мембраны, а <3'е может находиться в двух
различных центрах связывания около каждой из сторон мембраны. С^-цикл
осуществляется в два этапа. Сначала <Щ2 диффундирует к цитозоль-
ной стороне мембраны, где окисляется, отдавая электрон Ре-8 белку
и затем цитохрому сг (рис. 9.13, а). Когда (}Н2 окисляется до убисеми-
хинона (С^*0), два протона высвобождаются в цитозоль. Анион семихи-
нона, (^*0, восстанавливает гемовую группу цитохрома &566, которая
переносит электрон к гемовой группе Ь,ЙП. Восстановленный Ь.ЙЛ затем
560'
560
восстанавливает убихинон (<3) на матриксной стороне до <310.
На втором этапе <3-цикла (рис. 9.13, б) вторая молекула 0|Н2
повторяет процесс, описанный выше, второй электрон отдается цитохрому с. и
два дополнительных протона высвобождаются в цитозоль. Другой элект-
23. Заказ 3486
353
Часть II. Метаболизм веществ и энергии в клетке
а) Первая стадия $-цикла 2Н
Ф
Окружающая среда
$Н2
Матрикс
б) Вторал стадия <5~цикла
О
Ге-8
*1
е) Полный Ч-цикл
2^
2х1е©
2хЬ
О
2х1е®
Ре-В
2х1е©
с\
Рис. 9.13. Предполагаемое функционирование (^-цикла:
<ЗН2 на цитозольной стороне мембраны отдает электрон Ге-8-белку комплекса III
и 2Н+ в цитозоль, образуя ^'0. Ге-8-белок затем восстанавливает цитохром с1, и
электроны продолжают дальше двигаться по дыхательной цепи* (3*° отдает электрон
гемовой группе &566, в результате чего образуется ($, который диффундирует к
матриксной стороне мембраны. Восстановленный Ьш отдает электрон через Ь560 к (^,
находящемуся на матриксной стороне мембраны, в результате чего образуется Фе(а).
Для завершения цикла необходимо, чтобы вторая молекула ^Н2 повторила
первую часть цикла, образовав еще один восстановленный цитохром сг> другие 2Н"\
другой <3 и восстановленный Ь5б0, который тут же восстанавливает ф9,
образовавшийся на первом этапе цикла, до (^Н2, поглощая 2Н+ из матрикса (б), В результате
происходит окисление двух молекул (}Н2 и образуется одна молекула <ЗН2, как
показано на рисунке, отражающем весь путь в общем (в). Электроны с комплекса Ш
передаются мобильному переносчику — цитохрому с (Моран Л. А. и др,, 1994)
354
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
рон, поступающий на Ььт, может быть использован вместе с двумя
протонами из матрикса для превращения ранее образовавшегося на матрик-
сной стороне мембраны <5*е в <ЗН2. Суммарный результат этих двух
циклических этапов (рис, 9.13, в) — это окисление двух молекул <ЗН2,
образование одной молекулы <ЗН2, восстановление двух молекул цитохро-
ма су перенос четырех протонов через внутреннюю мембрану
митохондрии и поглощение двух протонов из матрикса. Поскольку разница в
заряде, создаваемая между матриксом и цитозолем, в итоге составляет
только 2, то протондвижухцая сила, генерируемая комплексом III, меньше,
чем это характерно для комплексов I и IV.
Функционирование комплекса IV. Комплекс IV, цитохром е-ок-
сидаза, — это последний компонент электрон-транспортной цепи. Этот
комплекс катализирует четырехэлектронное восстановление
молекулярного кислорода (02) до воды (2Н20) и осуществляет перенос
протонов в наружное пространство, окружающее митохондриальную
мембрану. В тканях млекопитающих мембраносвязанная форма фермента
представляет собой димер, который содержит 13 типов
полипептидных цепей с общей молекулярной массой около 400 000. Среди
полипептидных цепей комплекса IV есть цитохромы а и #3, чьи гемовые
простетические группы имеют идентичные структуры, но различные
стандартные восстановительные потенциалы, обеспечиваемые
различным микроокружением внутри олигомерного комплекса. Другие
окислительно-восстановительные кофакторы комплекса IV — это два иона
меди, называемые Сид и Сив, участие которых в транспорте
электронов сопряжено с изменением валентности меди: Си2+ ^ СиЛ
Цитохром с-оксидаза вносит свой вклад в создание протонного
потенциала двумя путями (рис. 9.14). Один представляет собой меха-
♦
Внешнее пространство
Комплекс IV
2 одноэлектронных
V переноса
а-Си
е
аз-С\1в
2 одноэлектронных
переноса
2Н
©
1/202
Матрикс
н2о
Рис. 9.14. Перенос электронов и протонов в комплексе IV. При переносе
электронов происходит окисление и восстановление атомов железа в гемовых группах
цитохромов и атомов меди. Комплекс IV осуществляет свой вклад в градиент
концентрации протонов двумя путями: за счет транслокации протонов из
матрикса в окружающее мембрану пространство в ходе переноса электронов между
цитохромами и забирая протоны из матрикса при образовании воды
23*
*
355
Часть II. Метаболизм веществ и энергии в клетке
низм, обеспечивающий транслокацию двух протонов при передаче пары
электронов (т.е. на каждый атом восстанавливаемого 02). Другой
механизм, с помощью которого цитохром с-оксидаза обеспечивает вклад
в градиент концентрации протонов, — это поглощение двух Н+ из
матрикса во время восстановления кислорода до воды. Хотя этот
механизм не вовлечен в реальную транслокацию Н+ через мембрану, тем
не менее, данный путь вносит вклад в формирование Др. Эффект
равносилен переносу четырех Н+ на каждую пару электронов.
Для восстановления молекулы 02 необходимы четыре электрона
(что эквивалентно двум молекулам ЫАВН) и четыре протона:
02 + 4е + 4Н+ -> 2Н20.
Восстановление 02 происходит в каталитическом центре, который
включает атом гемового железа цитохрома с и соседний атом меди,
Сив. Это так называемый двухлокусный центр, в котором
связываются токсичные соединения, такие как цианид и оксид углерода. Другой
гем (цитохром а) и атом меди (СиА) осуществляют одноэлектронный
перенос от молекул цитохрома с, находящихся на цитоплазматичес-
кой стороне комплекса, к кислородвосстанавливающему центру.
Функционирование комплекса V. В отличие от других
комплексов, комплекс V не вносит вклада в создание протонного потенциала,
а использует его энергию для синтеза АТР из АСР и Рг Комплекс V,
называемый также Г0Р1—АТР-синтетазой, имеет характерную
грибовидную структуру. АТР-синтетаза обеспечивает сопряжение
окисления субстратов и фосфорилирования АВР в митохондриях, и Г
означает "сопрягающий фактор". Компонент Рх, изолированный из
мембраны путем солюбилизации, катализирует гидролиз АТР, По этой
причине его традиционно называют В^АТРаза. Е0-компонент — это
протонный канал, пронизывающий мембрану. Перенос протонов
через канал в матрикс сопряжен с образованием АТР. Стехиометрия
переноса протонов в расчете на синтезируемый АТР оценивается как
ЗН+ на одну молекулу АТР. Р0-компонент чувствителен к олигомици-
ну, антибиотику, который предотвращает перенос протонов и,
следовательно, ингибирует синтез АТР.
На рис. 9.15 показано расположение Г1 и Р0 в плазматической
мембране, а также субъединичный состав наиболее хорошо изученного
комплекса V Е. соЫ. Предполагается, что в состав бактериального
Г1-компонента входят субъединицы а3^3у§е, а в состав
^-компонента — а1^2сю—12 ■ Субъединицы с компонента Р0 расположены так, что
образуют канал для переноса ионов Н+ через мембрану.
Гх-компонент митохондриальной АТР-синтетазы подобен
бактериальному. Рентгеноструктурные кристаллографические исследования
К1АТРазы как из печени крысы, так и из сердца быка показали, что
симметричный а3р3 олигомер связан с трансмембранным Р0-каналом
посредством мультисубъединичного "стебля". Ориентация Г1-компо-
356
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
нента в митохондрии такова, что
"шляпка" всей "грибовидной"
структуры Р^АТР-синтетазы находится
с внутренней стороны мембраны.
В 1979 г. П. Бойер предложил
так называемый механизм
чередующегося связывания* объясняющий
синтез АТР из АВР и Р.,
катализируемый комплексом V.
Существование данного механизма основано на
предположении, что используемая
энергия требуется не на образование
фосфоангидридной связи, а на
освобождение АТР из каталитического
центра. В активном центре АО
реакции: АВР + Р. ^ АТР + Н20,
около!; это значит, что образование
АТР в активном центре может быть
вполне осуществлено. Однако
образовавшийся АТР оказывается в
термодинамической яме и, таким
образом, имеет такую высокую энергию
связывания, что его высвобождение
невозможно. Конформационные
изменения, вызываемые движением
протонов, значительно ослабляют
связывание АТР и позволяют продукту высвободиться из активного
центра фермента. Константа диссоциации для АТР увеличивается
примерно от 10"*12 до 10~5, когда происходит изменение конформации
одной из трех ар субъединиц АТР-синтетазы.
Предполагается, что а3(33 олигомер АТР-синтетазы содержит три
каталитических сайта, т.е. три области, сопряженные с каталитическим
образованием АТР. В любой момент времени каждый из сайтов
находится в разной конформации: открытой, свободной и напряженной. Все три
каталитических сайта последовательно проходят через эти три
конформации. Предполагается, что образование и высвобождение АТР
включает следующие этапы, представленные на рис. 9Л6: 1) одна молекула
АВР и Р. связываются в открытом сайте; 2) происходящий через
внутреннюю митохондриальную мембрану перенос протонов приводит к тому,
что каждый из трех каталитических сайтов меняет свою конформацию.
Сайт с открытой конформацией (содержащий только что связанный АВР
и Р. ) становится свободным. Свободный сайт, уже заполненный АВР и
Р., становится напряженным сайтом. АТР-несущий сайт становится
открытым сайтом; 3) АТР высвобождается из открытого сайта, а АВР и Р.
конденсируются, образуя АТР в напряженном сайте.
Рис. 9.15. Схематическое
изображение "грибовидной" структуры
Г^АТР-синтетазы Е. соИ. Р0
компонент пронизывает мембрану,
образуя канал для протонов.
Предполагается, что Р4 состоит из трех а и
трех (3 субъединиц, организованных
так, что они образуют гексамерную
структуру наподобие "шляпки
гриба", и одной у, одной 8 и одной е
субъединиц, которые формируют
"стержень", соединяющий Р1 с К0
каналом. Считают, что Р0 Е. соИ
состоит из ^1&2сю—12 субъединиц, Мито-
хондриальный Р0 имеет более
сложную структуру и субъединичный
состав, но он недостаточно хорошо
охарактеризован
357
Часть II. Метаболизм веществ и энергии в клетке
Открытая Свободная Свободная Напряженная Напряженная Открытая
Напряженная Открытая I \ Свободная
АТР АВР+Р5
Рис. 9.16. Предполагаемые этапы образования и высвобождения АТР
согласно механизму "чередующегося связывания" (Моран Л. А. и др., 1994), Три
различных конформации связывающего центра АТР-синтетазы показаны в разных
формах, АОР и Р. связываются в открытом сайте, Конформационное изменение,
вызываемое движением протонов, приводит к их еще более сильному
связыванию в сайте со свободной конформацией. Тем временем сайт, который прочно
связывал АТР, становится открытым, а свободный сайт, содержащий другую
молекулу АОР и Р., становится напряженным. АТР высвобождается из
открытого сайта и синтезируется в напряженном сайте
Вклад комплексов, осуществляющих конверсию энергии, в
синтез АТР. Ранее предполагалось, что комплексы I, III и IV вносят
вклад в образование АТР в соответствии со стехиометрией "один к
одному", т.е. каждый комплекс обеспечивает синтез одной молекулы
АТР. Согласно современным представлениям выход АТР не должен
быть эквивалентным для каждого протонтранслоцирующего электрон-
транспортного комплекса, а также выход АТР в расчете на молекулу
окисляемого субстрата не должен выражаться целым числом.
Естественно, что половина молекулы АТР не может синтезироваться.
Многие электрон-транспортные цепи в митохондрии одновременно вносят
вклад в протонный концентрационный градиент, который является
общим хранилищем энергии, используемым многими АТР-синтетаз-
ными комплексами. С учетом этого образование одной молекулы АТР
из А1)Р и Р. , катализируемое АТР-синтетазой, требует переноса
приблизительно трех протонов; один дополнительный протон необходим
для транспорта Р., АВР и АТР. Соотношение протонов, транслоциру-
емых в пространство, окружающее мембрану, на пару электронов,
переносимых сопряженными электрон-транспортными комплексами,
составляет 4/1 для комплекса I, 2/1 — для комплекса III и 4/1 — для
комплекса IV* Эти значения могут быть использованы для расчета
соотношения молекул фосфорилируемого АХ)Р к атомам
восстанавливаемого кислорода, называемым Р/О отношением. Когда в качестве
субстрата для дыхательной электрон-транспортной цепи используется
митохондриальный ИА1Ш, 10 протонов экспортируются в цитозоль, и
отношение Р/О будет 10/4, или 2,5. Отношение Р/О для сукцината
будет 6/4, т.е. 1,5. Эти показатели Р/О должны рассматриваться как
максимальные значения — эффективность превращения энергии не
358
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
составляет 100 % по нескольким причинам, включая медленную утечку
протонов обратно внутрь митохондрий и энергию, используемую аде-
ниннуклеотидтранслоказой, а также другими транспортными
процессами, зависящими от протонного градиента.
Отношение Р: О, приведенное выше, соответствует современным
оценкам и отличается от традиционно используемых величин,
которые составляют 3 в расчете на молекулу ЫАЮН и 2 в расчете на
молекулу сукцината. Традиционные величины были получены, исходя из
данных ранних экспериментов. Однако даже после признания хемио-
осмотической теории величины 3 и 2 продолжают использоваться в
некоторых случаях, возможно, в связи с привлекательностью целых
чисел по сравнению с дробными ♦
9.9. Транспортные системы
внутренней митохондриальной мембраны
Транспорт АТР, АВР, Р. и других метаболитов через
внутреннюю митохондриальную мембрану. Во внутренней
митохондриальной мембране имеются специфические транспортные системы,
обеспечивающие перенос определенных соединений между цитозолем
и матриксом митохондрий.
Синтез АТР осуществляется в митохондриальном матриксе, но так
как его основная часть используется в цитозоле, он должен сюда
экспортироваться. Необходима специальная
транспортная система, позволяющая АОР
войти, а АТР выйти из митохондрии,
поскольку внутренняя митохондриальная
мембрана непроницаема для АТР и АОР.
АОР/АТР-переноечик, называемый также
адениннуклеотидтранслоказой,
осуществляет обмен митохондриального АТР на
цитозоАный АХ>Р (рис* 9.17). Адениннук-
леотидтранслоказа представляет собой
белок, локализованный во внутренней
митохондриальной мембране; его специфичным
ингибитором является растительный гли-
козид атрактилозид. Обмен АВР3' на АТР4*
носит электрогенный характер, так как
приводит к потере заряда в цитозоле (-1).
Такой тип обмена влияет на
электрическую составляющую ДрН, а именно на Ау.
Таким образом, некоторая доля свободной
энергии, сохраняемой в градиенте
концентрации протонов, расходуется на данный
транспортный процесс. Для образования
Окружающая
среда
е
II
Матрикс
АБР
©
О
АТР
е
н
Р;
0
О
Рис. 9.17. Транспорт АТР,
АВР и Р. через внутреннюю
митохондриальную мембрану.
Обмен АТР на АБР,
осуществляемый
адениннуклеотидтранслоказой, имеет
электрогенный характер. Симпорт Р.
и Н+ — электронейтральный
359
Часть II. Метаболизм веществ и энергии в клетке
АТР из АОР и Р. в митохондриальной матриксе также необходим
фосфатный переносчик, транспортирующий Р. из цитозоля. Этот
переносчик называется фосфаттранслоказой. Фосфат (Н^О^)
транспортируется в митохондрию путем электронейтрального симпорта вместе
с Н+ (см. рис. 9.17). Данный перенос не влечет за собой изменения
электрического компонента протондвижущей силы, Ду, но влияет на
химическую составляющую АрН. Таким образом, обе транспортные
системы, необходимые для образования АТР, используют АрН. В
целом энергетическая цена транспорта АТР из матрикса, а АОР иР. — в
матрикс приблизительно эквивалентна переносу одного протона.
Следовательно, синтез одной молекулы АТР в ходе окислительного фос-
форилирования требует притока четырех протонов из пространства,
окружающего мембрану (один — для транспорта и три, которые
передаются через Р0, — компонент АТР-синтетазы).
Во внутренней митохондриальной мембране функционируют
специфичные системы для переноса дикарбоновых кислот, а именно сук-
цината и малата; трикарбоновых кислот — цитрата и изоцитрата, а
также пирувата, аспартата и глутамата.
Челночные механизмы, обеспечивающие аэробное окисление
цитозольного МАОН. Восстановительные эквиваленты,
образовавшиеся в ходе гликолиза в цитозоле, должны войти в митохондрию для
того, чтобы участвовать в процессе окислительного фосфорилирова-
ния. Поскольку ни КАВН, ни NА^ не могут диффундировать через
внутреннюю митохондриальную мембрану, восстановительные
эквиваленты поступают в митохондрию с помощью челночных
механизмов. Эти челночные механизмы представляют собой циклические пути,
с помощью которых восстановленные коферменты в цитозоле
передают свою восстановительную способность метаболиту,
транспортируемому в митохондрию; восстановительные эквиваленты вновь
передаются другой молекуле кофермента в матриксе; и реокисленный
метаболит возвращается в цитозоль, в результате чего цикл может
повторяться.
В одном из таких механизмов переносчиком является глице-
рол-3-фосфат, который легко проходит через внутреннюю мембрану
митохондрии. Глицеролфосфатный челночный механизм весьма
активен в летательных мышцах насекомых, в которых очень высока
скорость окислительного фосфорилирования. Он также функционирует,
хотя и менее активно, в большинстве клеток млекопитающих, причем
наиболее интенсивно в скелетных мышцах и мозге.
В данный челночный механизм вовлечены два фермента: цитозоль-
ная МАХ)-зависимая глицерол-3-фосфатдегидрогеназа и мембраносвя-
занный глицерол-3-фосфатдегидрогеназный комплекс, содержащий
РАО в качестве простетической группы и имеющий субстратсвязыва-
ющий центр на внешней стороне внутренней митохондриальной
мембраны. В цитозоле ИАВН восстанавливает дигидроксиацетонфосфат в
360
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
реакции, катализируемой цитозольнои глицерол-3-фосфатдегидроге-
С_Р
назои:
сн2он
ИАБН + Н+ + 0=С
~>
2-
СН2ОРОз
Дигидроксиацетон-
фосфат
СН2ОН
но-с-н
СН2ОРОз
Глицерол-3 -
фосфат
ИАБ
2-
Глицерол-3-фосфат затем превращается обратно в дигидроксиаце-
тонфосфат с помощью мембраносвязанного глицерол-3-фосфатдегид-
рогеназного комплекса. В ходе этого процесса два электрона
переносятся на простетическую группу ГАВ фермента:
Е-РАВ + Глицерол-3-фосфат ?^ Е~ЕАВН2 + Дигидроксиацетонфосфат.
РАХ)Н2 передает два электрона мобильному электронному
переносчику, коферменту С}, который затем транспортирует электроны к уби-
хинол-цитохром г-оксидоредуктазе (комплекс Ш). В конечном итоге
глицеролфосфатный челнок окисляет цитозольный КАВН и
генерирует <5Н2 во внутренней митохондриальной мембране (рис. 9.18).
Окисление цитозольного КАОН этим путем обеспечивает меньшее
производство энергии (1,5 АТР на молекулу цитозольного ЫАОН) по
сравнению с окислением внутримитохондриального КАХ)Н, потому что в
последнем случае восстановительные эквиваленты вводятся без
помощи челнока непосредственно к митохондриальной МАСН-убихинон-
оксидоредуктазе (комплекс I).
маш + н
©
Д игидроацетон -
Цитозольная
глицерол-3-фосфат-
дегидрогеназа
у
фосфат
МАП
©
*К
Глицерол-3-
фосфат
Окружающая
среда
ГАСН^ч
Глицерол-3-фосфат
дегидрогеназный
комплекс
РАО
1111»
Матрикс
Рис. 9.18. Глицеролфосфатный челночный механизм. Цитозольный МАВН
восстанавливает дигидроксиацетонфосфат до глицерол-3-фосфата в реакции,
катализируемой цитозольнои глицерол-3-фосфатдегидрогеяазой. Обратная реакция
катализируется митохондриальным глицерол-3-фосфатдегидрогеназным
комплексом, содержащим флавопротеин, который переносит электроны на убихинон
Другой челночный механизм — малат-аспартатный —
функционирует в клетках сердца, почек и печени. В данном случае помимо
специфичных транспортных систем в процесс вовлечены и в цитозоле, и
в митохондриальном матриксе малатдегидрогеназа и аспартаттранс-
361
Часть II. Метаболизм веществ и энергии в клетке
аминаза. Действие данного челнока изображено на рис. 9Л9 ЫАОН в
цитозоле восстанавливает оксалоацетат до малата в реакции,
катализируемой цитозольной малатдегидрогеназой. Малат поступает в мито-
хондриальный матрикс путем дикарбоксилаттранспортирующего
электронейтрального обмена с 2-оксоглутаратом. В матриксе митохондри-
альная малатдегидрогеназа катализирует реокисление малата до ок-
салоацетата, в ходе чего происходит восстановление митохондриаль-
ного ИАВ до КАОН. ЫАЮН окисляется с помощью комплекса I
дыхательной электрон-транспортной цепи.
Дикарбоксилат-транс-
портирующая система
Малат
МАО
ЫАОН-Н
2е
0
Митохондри -
альная
малатдегидрогеназа
Электрон
-транспортная цепь
(во внутренней
мембране)
Оксал-
ацетат
Малат
^*- ЫАР
©
2-оксо-
глутарат
А
Митохондри- <
альная аспартат-
трансаминаза
Цитоплазма-
тическая
малатдегидрогеназа
2-оксо-
глутарат
А
^-кг.
ЫАБН-Н
©
Глутамат I
А
Глутамат
Оксалоацетат
А
Аспартат
Цитоллаз-
матическая
аспартат-
трансам иназа
Аспартат
Глутаматаспартат-
транслоказа
Рис. 9.19. Малат-аспартатный челночный механизм. ЫАВН в цитозоле
восстанавливает оксалоацетат до малата, который транспортируется в митохондри-
альный матрикс. Реокисление малата сопровождается генерированием ЫАОН,
который может отдавать электроны в электрон-транспортную цепь. Для
завершения цикла челночного механизма необходимо также функционирование ми*
тохондриальной и цитозольной аспартаттрансаминазы
Для возобновления действия челнока необходимо возвращение ок-
салоацетата в цитозоль. Это не происходит прямым путем, так как
оксалоацетат сам по себе не транспортируется через внутреннюю ми-
тохондриальную мембрану. Поэтому оксалоацетат взаимодействует с
глутаматом в реакции, катализируемой митохондриальной аспартат-
трансаминазой, в результате чего образуются 2-оксоглутарат и
аспартат. 2-Оксоглутарат выходит из митохондрии с помощью дикарбокси-
латтранспортирующей системы в обмен на входящий малат. Аспартат
362
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
»■!■!■ I ■ ■ т I ■ I I ' ■ * I ***»■ ■ I 1 III I 1Щ1 И^-и*—+■ II ■ I ■ I | ■ ■ ■ ■ I
выходит через глутамат/аспартат транспортирующую систему в
обмен на входящий глутамат. Оказавшись в цитозоле, аспартат и 2-ок-
соглутарат становятся субстратами цитозольной аспартаттрансамина-
зы, которая катализирует образование глутамата и оксалоацетата.
Глутамат возвращается в митохондрию в антипорте с аспартатом, и
оксалоацетат реагирует с другой молекулой цитозольного ЫАОН,
повторяя цикл. Поскольку при восстановлении оксалоацетата в цитозо-
ле забирается один протон, который высвобождается в матриксе при
окислении малата, то непрямое окисление цитозольного КАОН через
электрон-транспортную цепь обеспечивает меньший вклад протонов в
протонный концентрационный градиент, чем прямое окисление
митохондриального КАОН (9 вместо 10). Выход АТР при окислении двух
молекул цитоплазматического ЫАВН с помощью данного челнока,
следовательно, будет скорее не 5, а около 4,5.
9.10, Эффективность окисления глюкозы
и контроль окислительного фосфорилирования
Выход АТР при окислении молекулы глюкозы. Отношение Р/О
представляет собой количество молей АТР, синтезируемых при
окислительном фосфорилировании в расчете на пару электронов,
переносимых через определенный участок электрон-транспортной цепи.
Несмотря на интенсивное изучение этого отношения, его действительное
значение остается вопросом, требующим внимания. Если мы
принимаем, что при переносе двух электронов по электрон-транспортной
цепи от ЫАВН к 02 из матрикса транспортируются 10Н+, а также
соглашаемся, что 4Н+ транспортируются в матрикс в расчете на
молекулу синтезируемого и, подчеркнем еще раз, также транслоцируемого
через мембрану АТР, то отношение Р/О будет 10/4, или 2,5, в том
случае, когда электроны поступают в электрон-транспортную цепь на
уровне КАОН. Это несколько ниже по сравнению с ранними
оценками, в соответствии с которыми отношение Р/О при окислении
митохондриального ЫАОН равно 3. Для участка цепи от сукцината до 02
число протонов, переносимых в расчете на пару электронов, будет 6, и
Р/О отношение в этом случае будет 6/4, или 1,5, тогда как согласно
ранним оценкам — 2. Результаты экспериментальных измерений
отношения Р/О для двух рассматриваемых случаев ближе к
современным значениям, т.е. 2,5 и 1,5,
Принимая соответствующие Р/О отношения, можно оценить
количество молекул АТР, синтезируемых в результате полного окисления
молекулы глюкозы. Учитывая, что Р/О отношения должны
рассматриваться как приблизительные по причинам, указанным выше,
можно принять значения 2,5 и 1,5 для митохондриального окисления ЫАВН
и сукцината соответственно. В эукариотических клетках за счет
совместного функционирования гликолиза, цикла трикарбоновых кис-
363
Часть II, Метаболизм веществ и энергии в клетке
лот, электрон-транспортной цепи и окислительного фосфорилирова-
ния выход АТР будет 30—32 молекулы АТР в расчете на молекулу
окисленной глюкозы в зависимости от используемого челночного
механизма (табл. 9.7).
Общее уравнение для окисления глюкозы с использованием глице-
ролфосфатного челночного механизма будет иметь вид:
Глюкоза + 602 + -30АВР + ~Р. ~> 6С02 + -30АТР + ~36Н20.
Так как 2 КАОН, образованные в гликолизе, транспортируются
глицеролфосфатным челночным механизмом, то в этом случае в
расчете на каждую из них выход АТР будет 1,5, как уже указывалось
выше. В то же время если эти 2 ЫАПН переносятся в митохондрию с
Таблица 9.7
Выход АТР при окислении глюкозы
Метаболический путь
Гликолиз: превращение глюкозы до пирувата (цитозолъ)
Фосфорилирование глюкозы
Фосфорилирование глюкозо-6-фосфата
Дефосфорилирование 2 молекул 1,3-бисфосфоглицерата
Дефосфорилирование 2 молекул фосфоенолпирувата
Окисление 2 молекул глицеральдегад-3-фосфата дает 2
молекулы Ш0Н
Превращение пирувата до ацетил-СоА (митохондрии)
2ЫАПН
Цикл трикарбоновых кислот (митохондрии)
2 молекулы ОТР из двух молекул сукцинил-СоА
Окисление 2 молекул изоцитрата, 2 молекул 2-оксоглутарата
и 2 молекул малата дает 6 МАОН
Окисление 2 молекул сукцината дает 2 РАОН2
Окислительное фосфорилирование (митохондрии)
В расчете на каждую молекулу из 2 ИАОН, образовавшихся
в гликолизе, синтезируется 1,5 АТР, если для их транспорта
в митохондрию используется глицеролфосфатный челночный
механизм, и 2,5 — в случае использования малат-аспартатно-
го челнока
В расчете на каждую молекулу из 2 КАВН, образовавшихся
при окислительном декарбоксилировании 2 молекул
пирувата до 2 молекул адетил-СоА, синтезируется 2,5 АТР
В расчете на каждую молекулу из 2 ГА0Н2, образовавшихся
в цикле трикарбоновых кислот, синтезируется 1,5 АТР
В расчете на каждую из 6 ИАВН, образовавшихся в цикле
трикарбоновых кислот, синтезируется 2,5 АТР
Общий выход
Выход АТР в расчете
на молекулу глюкозы
глицеролфосфатный
челнок
-1
-1
+2
+2
+2
+3
+5
+3
+15
30
малат-
аспартатный
челнок
-1
-1
+2
+2
+2
+5
+5
+3
+15
32
Примечание, Отношения Р/О 2,5 и 1,5 для митохондриального окисления
ЫАОН и ГА1)Н2 представляют собой "конценсусные значения". Хотя они могут и не
отражать действительных значений, и хотя эти отношения могут меняться в
зависимости от метаболических условий, данная оценка выхода АТР при окислении
молекулы глюкозы может рассматриваться как приблизительная.
364
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
участием малат-аспартатного челнока, то выход АТР в расчете на
каждую молекулу составит 2,5, обеспечивая в этом случае в целом 32 АТР
при окислении молекулы глюкозы. Большая часть молекул АТР — 26
из 30 или 28 из 32 — образуются в ходе окислительного фосфорилиро-
вания; только 4 молекулы АТР образуются путем прямого синтеза в
гликолизе и цикле трикарбоновых кислот.
Ситуация в бактериях несколько другая. Клетки прокариот не
нуждаются в системах, обеспечивающих транспорт и обмен АТР/АВР*
Поэтому, бактерии потенциально могут продуцировать около 38 АТР
в расчете на молекулу глюкозы.
Подходя гипотетически к данному вопросу, рассмотрим, какую долю
энергии клетки эукариот извлекают из молекулы глюкозы.
Принимая во внимание значение 30,5 кДж/моль для гидролиза АТР в
стандартных условиях, мы можем считать, что образование 30 АТР при
окислении одной молекулы глюкозы дает 915 кДж/моль. Для
клеточного окисления ("сгорания") глюкозы АО = -2937 кДж/моль. Мы
можем рассчитать эффективность гликолиза, цикла трикарбоновых
кислот, электронного транспорта и окислительного фосфорилирования:
915
2937
-100-31%.
Контроль окислительного фосфорилирования в зависимости
от энергетических потребностей в клетке. Контроль
окислительного фосфорилирования не осуществляется путем простого ингибиро-
вания по принципу обратной связи или же с помощью какого-либо
аллостерического механизма. Общий контроль скорости
окислительного фосфорилирования сопряжен с доступностью субстратов и
энергетическими потребностями клетки. Скорость дыхания зависит от
концентраций внутримитохондриального КАЮН, 02, Р. и АВР. Ин-
тактные митохондрии суспендированные в фосфатном буфере, дышат
только в присутствии АВР. В ходе потребления АТР в реакциях
биосинтеза в цитозоле появляется АОР, транспортируемый с помощью
адениннуклеотидтранслоказы, и электронный транспорт, который
непосредственно связан с доступностью АРР, ускоряется* Окисление
субстратов усиливается тогда, когда возрастание концентрации АОР
сигнализирует, что фонд АТР нуждается в восполнении.
В некоторых ситуациях субстратное окисление не связано с
генерированием АТР. Такой тип окисления субстратов иногда называют
нефосфорилирующим или свободным окислением. При нефосфорили-
рующем окислении дыхание не сопряжено с фосфорилированием, и
энергия окисляемых субстратов превращается в теплоту. Это имеет
значение для поддержания температуры тела при охлаждении
теплокровных организмов. Превращение митохондрий, в которых
происходит нефосфорилирующее окисление, в своеобразную клеточную
"печку" было впервые продемонстрировано В. П. Скулачевым в экспери-
365
Часть II, Метаболизм веществ и энергии в клетке
ментах, в которых голубей подвергали многократному охлаждению
при минусовой температуре. При этом в митохондриях мышечной ткани
происходило разобщение дыхания и фосфорилирования, и
образование теплоты препятствовало замерзанию организма. Терморегулиру-
ющая функция мышечных митохондрий позже была обнаружена и у
морских котиков при естественных изменениях температуры.
Имеющее физиологическое значение нефосфорилирующее окисление
осуществляется в бурой жировой ткани, необычный цвет которой
является следствием наличия большого количества митохондрий. Бурая
жировая ткань имеется в обилии у новорожденных млекопитающих.
Много бурой жировой ткани у зимоспящих животных. Разобщение
дыхания и фосфорилирования в митохондриях этой ткани у
животных приводит к образованию большого количества теплоты,
согревающей протекающую кровь. Разобщителями являются свободные
жирные кислоты, повышающие проницаемость мембран для протоновI
Выделение эпинефрина играет роль триггерного механизма,
приводящего к активации липазы, что вызывает образование жирных кислот,
которые затем окисляются. Образующиеся в ходе окисления жирных
кислот восстановленные коферменты используются в качестве
субстратов электрон-транспортной цепи, но свободная энергия
восстановленных соединений не запасается в форме АТР, а выделяется в виде
тепла. Несопряжение окисления и фосфорилирования является следствием
активации термогенина, белка, образующего канал для перехода
протонов внутрь митохондрий. Термогенин не активен в качестве канала
до тех пор, пока не свяжется с жирными кислотами, индуцирующими
конформационные изменения в данной протон-транспортирующей
структуре. Когда термогенин активен, жирные кислоты окисляются в
митохондриях, и выделяющаяся свободная энергия рассеивается в виде
тепла, повышая температуру тела животного.
9.11. Токсичные формы кислорода и их роль в апоптозе
Образование токсичных форм кислорода и их роль в
окислительном повреждении клеточных структур. В предыдущем
разделе мы рассмотрели, каким образом восстановление молекулярного
кислорода связано с генерированием энергии в клетке. Некоторые
клетки крови, а именно фагоциты, также способны восстанавливать
молекулярный кислород, но с целью производства агентов,
уничтожающих микроорганизмы. Фагоциты удаляют чужеродные клетки,
например, бактерии, поглощая и затем разрушая их, используя при этом
как протеолитические ферменты, так и активные (токсичные) формы
кислорода. Во время стимуляции функционирования фагоцитов
происходит вспышка их дыхатальной активности, при этом потребление
кислорода увеличивается примерно в 50 раз. В то же время глюкоза
быстро метаболизируется через пентозофосфатный путь (см. раздел
366
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
9.7), что приводит к генерированию МАОРН. Мембраносвязанная
КАОРН-оксидаза плазмы катализирует одноэлектронное
восстановление кислорода путем переноса электронов от МАВРН к РАЛ) и затем,
по-видимому, к цитохрому Ъ, участвующему в восстановлении
кислорода. Продукт этой реакции — свободный радикал, называемый еуперок-
сиданионрадикал, -02".
ЫАВРН + 202 -> МАВР+ + 2-02" + Н+.
■02" может самопроизвольно диемутировать с образованием перокси-
да водорода, Н202; эти две формы кислорода представляют собой
исходный материал для образования еще более токсичных соединений, таких
как гидроксильный радикал ОН4. На рис. 9.20 представлена схема
взаимопревращений активных форм кислорода внутри клетки.
Небольшие количества
токсичных форм кислорода, Ю2, Н202
и ОН' образуются как продукты
окислительного метаболизма и в
других типах клеток. На
заключительном этапе передачи
электронов на кислород,
осуществляемом цитохромоксидазой, в норме
должно происходить
восстановление молекулы кислорода до двух
молекул воды за счет
присоединения четырех электронов. В
случае неполного восстановления
кислорода при присоединении
двух электронов образуется Н202;
при присоединении одного
электрона — супероксидный радикал (0~). Концентрация супероксида в
печени — около 10~и М, а концентрация Н202 — примерно в 1000 раз выше.
Активные формы кислорода весьма токсичны для клеток, так как они
повреждают многие клеточные структуры. Кислородные радикалы и пер-
оксиды способны разрушать липиды, белки и нуклеиновые кислоты.
Основными мишенями свободных радикалов являются ненасыщенные
связи в липидах мембран* Полиненасыщенные жирные кислоты,
входящие в состав мембран, — это основные субстраты процессов пероксидно-
го окисления липидов (ПОЛ). Первичными продуктами ПОЛ являются
диеновые конъюгаты: "СН2-СН2-СН=СН-СН=СН-СН2"* Образование
этих продуктов ведет к увеличению полярности хвостов жирных
кислот, что в свою очередь ведет к их вытеснению из толщи мембраны на
поверхность. В определенной степени эти процессы способствуют
самообновлению клеточных мембранных структур, а также оказывают
существенное воздействие на проницаемость мембран. Однако
чрезмерное образование активных форм кислорода ведет к значительным
НОо Пероксидаза
Катал аз)
(а)4;©'
о2 ~» Оз -Г1-*=' н2о2 —** он"^^-»^ н«>о
I 1—~*^1 \
Реакция
Фентона
Рис. 9.20. Схема, отражающая
взаимосвязь между активными формами
кислорода, существующими в клетке. 80В —
супероксиддисмутаза; 02~ — супероксид-
анионрадикал; НО^ — гидроксипероксид-
радикал; ОН* — гидроксильный радикал
367
Часть II, Метаболизм веществ и энергии в клетке
|_Ц_1 I * I I МП I * ~~^"«И1 II I * '~Т1—I ...
нарушениям структуры и функций липидов, белков, нуклеиновых
кислот, что в конечном итоге приводит к гибели клетки. Пероксида-
ция остатков жирных кислот нарушает структуру мембран и может
вести к лизису клеток. Тиоловые группы цистеиновых остатков в
полипептидных цепях белков могут также окисляться, приводя к
инактивации белков. Окислительное повреждение ДНК может
индуцировать мутации. В эритроцитах, где концентрация кислорода
относительно высока, потенциальная возможность окислительного повреждения
достаточно велика. Супероксид может скорее, чем 02, высвобождаться
непосредственно из оксигемоглобина и затем превращаться в Н202.
У млекопитающих повреждение сердца при сердечном приступе
связано с усилением образования свободных радикалов в месте
повреждения ткани. Предполагается, что разрушение молекул под действием
токсичных форм кислорода может быть также важнейшим
неспецифическим фактором в патогенезе широкого круга заболеваний, включая
артриты, эмфизему, некоторые виды рака и даже процесс старения.
Антиоксидаптные защитные системы клетки. В клетке
функционируют специальные защитные механизмы от повреждающего
воздействия свободнорадикальных форм кислорода. Существуют различные
ферменты, способствующие защите клеток от окислительного
повреждения. Один из них — супероксиддисмутаза, которая обнаружена как у
прокариот, так и эукариот, Супероксиддисмутаза катализирует реакцию
дисмутации супероксиданиона до пероксида водорода:
20^+2Н+^Н202 + 02.
Пероксид водорода может затем превращаться в Н20 и 02 под
действием каталазы:
2Н202 -> 2Н20 + Ог
В эукариотических клетках фермент глутатионпероксидаза более
эффективно осуществляет детоксикацию Н202, чем каталаза.
Глутатионпероксидаза разлагает Н202 за счет окисления восстановленного
глутатиона, участвующего в данной реакции. Действие глутатионпероксидазы
сопряжено с функционированием глутатионредуктазъц
обеспечивающей восстановление глутатиона за счет КАВРН* Глутатион (С8Н),
представляющий собой трипептид — у-глутамилцистеинилглицин, играет роль
восстановленного кофактора в глутатионпероксидазной реакции:
О О
11 {|
Н3Н+"СН-СН2"СН2-С-МН"СН-С-КН-СН2-СОО-
сосг сн2
8Н
Глутатион (08Н)
В присутствии глутатионпероксидазы и Н202 сульфгидрильные
группы двух молекул глутатиона взаимодействуют с образованием
дисульфида глутатиона (0880):
368
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
О О
II II
Н8Н+"СН-СН2-СН2-С-МН-СН-С»КН-СН2'СОО-
сосг сн2
8
I
8
I
о сн2о
II ! II
НзН+-сн-сн2-сн2-с-ын-сн-с-ын-сн2-соо-
СОО"
Дисульфид глутатиона (0880)
Для реставрации восстановленного глутатиона с помощью
реакции, катализируемой глутатионредуктазой, необходим КАВРН:
С88С + МАВРН + Н+ -» 208Н + КАОР+.
Для обеспечения потребности в эритроцитах в восстановленном
КА1)Р до 10 % от общего потребления глюкозы может быть метаболи-
зировано через пентозофосфатный путь. Помимо детоксикации
кислорода глутатион играет существенную роль в различных клеточных
процессах. Так, глутатион вовлечен в биосинтез лейкотриенов и в
транспорт некоторых аминокислот в клетку. Восстановленный глутатион
может также способствовать поддержанию конформации белков, в
которых свободные сульфгидрильные группы способны
самопроизвольно окисляться.
У людей мутация гена, кодирующего глюкозо-б-фосфатдегидроге-
назу (первый фермент пентозофосфатного пути), может приводить к
гемолитической анемии, сопровождающейся усиленным гемолизом
эритроцитов. Клинические проблемы возникают в том случае, когда
эти люди принимают некоторые лекарственные препараты (например,
антималярийный препарат примахин), что приводит к высокому
уровню пероксидов. В данной ситуации потребность в КА1)РН начинает
превосходить возможности мутантных клеток метаболизировать
глюкозу через пентозофосфатный путь.
Существенную роль в защите от токсичных форм кислорода играет
также неферментативная антиоксидантная система. Антиоксидантной
активностью обладает ряд природных соединений: аскорбиновая
кислота, серотонин, токоферолы, а также вещества, связывающие
железо. Особое значение имеют антиоксиданты — витамин Е и
аскорбиновая кислота. Витамин Е перехватывает свободные радикалы, отдавая
атом водорода радикалу и превращаясь в менее реакционноспособныи
Е-О* радикал. Аскорбиновая кислота восстанавливает как
радикальную форму витамина Е, так и КОО* пероксильные радикалы. Роль в
защите от токсичных форм кислорода помогает объяснить
необходимость наличия в питании ряда микроэлементов. Например, селен
присутствует в аминокислоте селеноцистеине; важный для активности
селеноцистеиновый остаток находится в активном центре глутатион-
24, Заказ 3486
369
Часть II. Метаболизм веществ и энергии в клетке
пероксидазы. Цинк, медь и марганец представляют собой
существенные компоненты изоферментов супероксиддисмутазы.
Роль токсичных форм кислорода в апоптозе. Образование
токсичных форм кислорода играет важную роль в биологическом
процессе, известным под названием апоптоз. Апоптоз представляет собой
запрограммированное в организме разрушение клеток и их гибель. В
настоящее время биохимические механизмы этого явления
интенсивно изучаются В. П. Скулачевым и его сотрудниками.
Первый существенный вклад в изучение запрограммированной
гибели клеток у эукариот внесли исследования Дж* Керра, А. Вилли и
их коллег, начатые в 70-х гг. На основании исследования
морфологических изменений гибнущих клеток была предложена концепция о
существовании двух основных типов клеточной смерти у животных:
апоптоз, тонко регулируемый, энергозависимый процесс, и некроз,
возникающий в результате травматического повреждения.
При повреждении клеток животных вследствие некроза
происходит разрушение плазматических мембран, вызываемое
гидролитическими ферментами и другими факторами (рис. 9.21).
Некротическая смерть клетки на ранних стадиях характеризуется
увеличением объема клетки и субклеточных органелл. В конечном итоге
имеет место автолиз и происходит дезинтеграция клетки. Апоптоз
представляет собой более распространненое биологическое явление, чем
некроз. Для клеток животных апоптоз — общее универсальное
явление с четко выраженными морфологическими признаками {см.
рис. 9.21), включая конденсацию хроматина, уменьшение объема
клетки, пузырчатость, расщепление ДНК на фрагменты (что происходит
часто, но не всегда под действием Са2+-зависимых эндонуклеаз) и
формирование апоптотических телец, захватываемых и деградируемых
соседними клетками и макрофагами.
Рис. 9.21. Различные типы клеточной смерти, выявленные в клетках
животных. Апоптоз — форма запрограммированной клеточной смерти, при которой
происходят конденсация хроматина, образование апоптозных телец,
поглощаемых в конечном итоге фагоцитами. Некроз имеет место при физическом повреж-
дении клеток; это приводит к автолизу мембран и воспалению тканей
370
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
Апоптоз — форма запрограммированной клеточной смерти,
характерная для животных, начиная от нематод до млекопитающих. Он
может иметь место как в ходе нормального развития, так и в качестве
ответной реакции на патогены. Апоптоз в организме выполняет
двоякую роль. С одной стороны, он полезен, так как лежит в основе
формирования многоклеточных организмов и обнаруживается на самых
ранних стадиях эмбриогенеза при формировании органов, замене
одной ткани на другие. Апоптоз может играть жизненно важную роль в
процессе эмбрионального и онтогенетического развития, при
различных морфогенетических процессах. Так, изучена
запрограммированная клеточная гибель при метаморфозе насекомых, в процессе
эмбриогенеза высших позвоночных — при развитии глаз млекопитающих,
сердца, нервной системы. Апоптоз устраняет клетки организма,
способные к активному образованию токсичных форм кислорода.
Положительная роль апоптоза заключается в удалении клеток, выживание
которых нежелательно для организма, например, мутантных клеток или
клеток, зараженных вирусом. С другой стороны, во многих случаях
апоптоз играет отрицательную роль. Его интенсивное функционирование
ведет к гибели такого большого количества клеток, что нормальная
деятельность жизненно важного органа становится невозможной.
Ключевую роль в процессе апоптоза играет регуляция синтеза
белков в организме. При этом одни специфические гены важны для
программируемой гибели клеток организма, другие необходимы для
окончательного удаления мертвых клеток. Регуляция экспрессии
специфических генов обеспечивает синтез определенных активаторов и
ингибиторов апоптоза, к которым относится довольно обширная группа
белков — ферментов, гормонов. В запуске и развитии процессов
апоптоза важную роль играют каспазы, относящиеся к типу цистеиновых
протеаз. Они участвуют как в инициации программы апоптоза, так и
в демонтаже клеточных компонентов. Каспазы синтезируются в виде
неактивного предшественника и активируются путем протеолитичес-
кого расщепления за счет автокатализа. Существуют каскадные
процессы активации каспаз, включающие ряд участников и
инициирующиеся под действие апоптозного сигнала. В настоящее время
очевидно, что апоптоз также сопряжен с продуцированием активных форм
кислорода, и эти формы прямо или опосредованно воздействуют на
процесс запрограммированной клеточной смерти.
Особо реакционноспособной и в этой связи губительной формой
является гидроксильный радикал ОН". Его высокая реакционная
способность обусловлена большим положительным редокс-потенциалом
(+1,5 В), что гораздо выше, чем у супероксидрадикала и пероксида
водорода. Основной источник ОН" в клетке — реакция пероксида
водорода с двухвалентным ионом железа:
Н.0о + Ге2+ -> 0Нв + ОН^ + Б*е8+.
24*
371
Часть II. Метаболизм веществ и энергии в клетке ^ ^^
Гидроксильный радикал оказывает разрушительное действие на
ДНК и такие клеточные структуры, как ядра и мембраны. Всплеск
его образования может инсценировать программу апоптоза не только
в клетке-продуценте, но и в ее ближайшем окружении.
Оксид азота и его участие в свободнорадикальных процессах. По
своей химической природе оксид азота относится к двухатомным
нейтральным молекулам (свободнорадикальный газ). Малые размеры и
отсутствие заряда обеспечивают этой неполярной молекуле высокую
проницаемость через мембраны клеток и субклеточных структур. Молекулы
N0 имеют неспаренный электрон на внешней тс-орбитали. Они легко
диффундируют в биологических средах и являются достаточно долгоживу-
щими: среднее время жизни в биологических тканях — 5,6 с (время
полужизни Т1/2 в почечной ткани крыс -~ 6,4 с, в миокарде — ОД с, в
физиологическом растворе — от 6 до 30 с, а в воде, из которой удален
кислород, N0 сохраняется в течение нескольких суток). Наличие одного
электрона с неспаренным спином придает молекуле N0 высокую
реакционную способность. Взаимодействуя с другими свободными радикалами,
молекула N0 может образовывать ковалентные связи.
Известно, что N0 образует стабильные комплексы с гемоглобином,
сывороточным альбумином, а также с негемовыми железосерными
белками. Свободнорадикальная природа позволяет оксиду азота как
активировать цепные свободнорадикальные реакции, так и ингибиро-
вать их. Кроме того, оксид азота способен вступать в окислительно-
восстановительные превращения, образуя многочисленные
азотсодержащие соединения, в которых валентность атома азота может
изменяться от -3 до +6.
Оксид азота обладает широким спектром биологического действия.
Его рассматривают в качестве одного из мессенджеров (посредников)
внутри- и межклеточной сигнализации в центральной и периферической
нервных системах. N0 вызывает расслабление гладкой мускулатуры стенок
сосудов, регулирует работу органов дыхания, желудочно-кишечного тракта
и мочеполовой системы. Он проявляет и цитотоксическую (цитостати-
ческую) функцию как один из основных компонентов клеточного
иммунитета. Образование этого соединения обеспечивает защиту организма от
бактериальных и злокачественных клеток.
Оксид азота участвует в процессе апоптоза, что связано с
токсическим воздействием избытка N0 на клетки. Кроме того, N0
рассматривают как регулятор пролиферации лимфоцитов. Эндогенный оксид
азота — важный компонент кальциевого гомеостаза в клетках и
соответственно активности Са2+-зависимых протеинкиназ. Однако
основными физиологическими мишенями для N0 считают гуанилатцикла-
зу и АЛР-рибозил-трансферазу.
Решающую роль обнаружения N0 в клетках и тканях различных
животных и микроорганизмов сыграл метод ЭПР. Это связано с тем,
что оксид азота содержит неспаренный электрон и обладает парамаг-
372
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
нитными свойствами. N0 — короткоживущий радикал, который
обладает высокой реакционной способностью, и поэтому концентрация
оксида азота в клетке очень мала. Сигнал ЭПР, обусловленный
радикалом N0, был открыт в нашей стране А. Ф. Ваниным и Р. М. Нал-
бандяном (1963) в лаборатории Л. А. Влюменфельда.
Оксид азота непрерывно образуется в организме человека и
животных. Синтез N0 в них происходит при ферментативном окислении
Ь-аргинина (рис. 9.22).
^"^Ш! ь"аргинин
X (ге)
ИАВРН + Н+'^ я-Н2-биоптерин>|КН20
-р_р^ Ы-гидрокси-Ь-аргинин
1Ш2
ГАБН
КАБР ^ ^
2Т°2 I ,N=0
+ ^ ГАВН'фнО, +Е-С-Н
КАОРН + Я+У ^ У ^2 "1Ш
РАБ фт2 ш.
к-с-н
—I *
о-он
н2о2
|^Н20
*°
Ь-цитруллин К-С
Рис. 9.22, Образование N0 из Ь-аргинина
Основной фермент биосинтеза оксида азота — 1М"0-синтаза. Этот
фермент достаточно селективен по отношению к субстратам: синтез
N0 может происходить лишь из Ь-аргинина, Ь-гомоаргинина. Ы-ме-
тил-Ь-аргинина. Предполагаемая суммарная реакция записывается
следующим образом:
2Ь~аргинин + ЗЫАЛРН + 402 + ЗЕТ -> Ы-ОН-Ь-аргинин ->
-> 2Ь-цитруллин + 2Ж> + ЗЫАОР+ + 4Н20.
гТО-синтазы — это семейство цитохром-Р-450-подобных гемопро-
теинов. Для функционирования этих ферментов необходим целый ряд
кофакторов: тиолсвязывающий гем, РАО, ГМЫ, (6К)-5,6,7,8-тетрагид-
ро-Ь~биоптерин (см. схему). Молекулы гГО-синтаз содержат домены с
редуктазнои и оксигеназнои активностью. По характеру индукции и
действия данные ферменты разделяются на два вида: конститутивные
и индуцибельные. Первые ферменты постоянно присутствуют в
клетках, вторые могут быть индуцированы под действием ряда факторов.
Увеличение внутриклеточной концентрации Са2+ приводит к
активации фермента, так как кальций является одним из его кофакторов.
373
Часть П. Метаболизм веществ и энергии в клетке
Основное разрушение N0 происходит путем его окисления
кислородом до N0^ и N0^. При повышении концентрации N0 до наномо-
лярных величин происходит его активное взаимодействие с перокси-
дами и гидропероксидами с образованием неактивных соединений.
Степень селективности оксида азота по отношению к пероксидам
примерно в 3 раза ниже, чем у супероксиддисмутазы, поэтому при
значительном накоплении N0 в тканях он может конкурировать с этим
ферментом за субстрат (0|). Оксид азота может связываться с теми же
внутриклеточными мишенями, что и 0|. Малые концентрации N0
могут оказывать цитопротекторное действие, а недостаток N0
приводит к свободнорадикальному повреждению мембран клеток и, в
частности, к развитию атеросклероза. Лишь концентрация оксида азота
порядка нескольких наномолей оказывается оптимальной для
процесса жизнедеятельности клеток.
9.12. Взаимосвязь процессов гликолиза и дыхания
Источником электронов в дыхательной цепи является ЫАВН,
генерируемый в матриксе, в основном, в результате функционирования
цикла трикарбоновых кислот. Окисление МАОН осуществляется фла-
вопротеиновым комплексом, локализованным на внутренней стороне
внутримитохондриальной мембраны; этот комплекс назван
внутренней МАЛШ-дегидрогеназой,
Каким же образом осуществляется окисление ЫАВН,
генерируемого в цитоплазме в процессе гликолиза?
В растениях и грибах находится второй флавопротеиновый
комплекс, локализованный на внешней поверхности
внутримитохондриальной мембраны; этот комплекс назван наружной ЫАОН-дегидроге-
назой. Он может катализировать окисление ИАОН, генерированного
при гликолизе и локализованного в межмембранном пространстве. Хотя
ИАОН и ЫА1)+ не могут диффундировать через внутреннюю митохонд-
риальную мембрану, они легко диффундируют через наружную
мембрану, проницаемую для молекул с молекулярной массой менее 104.
Таким образом, в растительной клетке внутренняя ЫАОН-дегидроге-
наза окисляет эндогенный МАОН, внешняя — экзогенный КАВН,
образованный в цитоплазме. Оба фермента передают электроны (и Н+) в-
дыхательную цепь через убихинон.
Клетки животных и насекомых лишены наружной ИАОН-дегид-
рогеназы. Взаимосвязь гликолиза и дыхания осуществляется с
помощью челночных механизмов, рассмотренных выше. Суть этих
механизмов заключается в том, что через внутреннюю мембрану
митохондрий переносится не сам МАОН, а отдаваемые им электроны.
Эффект Пастера. В анаэробных условиях дрожжи ЗассНаготусез
сегеоьзгае сбраживают глюкозу в этанол и диоксид углерода. Если же
дрожжи находятся в аэробных условиях, то скорость распада глюко-
374
. Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
зы резко уменьшается и она превращается в глубоко окисленные
продукты — С02 и Н20. Этот переход жизнедеятельности дрожжей с
брожения на дыхание получил название эффекта Пастера* Л, Пастер
открыл этот эффект при исследовании роли брожения в виноделии. В
чем же сущность эффекта Пастера? Вспомним: при брожении этанол
накапливается в среде в результате восстановления уксусного
альдегида с участием МАПН, образующегося на стадии окисления глице-
ральдегид-3-фосфата при гликолизе. Если не происходит регенерации
МАО* за счет этой реакции, то на стадии триоз гликолиз был бы
приостановлен. В аэробных условиях ЫАВН, образующийся в результате
окисления глицеральдегид-3-фосфата, вовлекается в систему
окисления с помощью митохондрии. Насыщенная кислородом митохондрия
весьма активна и в конкуренции за цитоплазматический ЫАОН
перехватывает последний от менее активной алкогольдегидрогеназы.
Каков механизм окисления КАОН здесь — действует ли наружная
МАОН-дегидрогеназа, подключается ли челночный механизм, — пока
неизвестно. Однако в любом из этих случаев вовлечение митохондрий
в окисление экзогенного ЫАВН резко повышает эффективность
извлечения свободной химической энергии из глюкозы и превращения
ее в форму АТР. Если при спиртовом брожении синтезируются только
две молекулы АТР, то при подключении в процесс окисления
глюкозы, например, глицерофосфатного челночного механизма будет
синтезировано в 30/2 = 15 раз больше молекул АТР.
Эффект Пастера позволяет дать объяснение принципиальной
разнице технологии продуктов брожения (спирт, вино, пиво) и
технологии прессованных хлебопекарных дрожжей. Интенсивная аэрация
питательной среды и высокая скорость генерирования свободной
энергии в АТР за счет глубокого окисления глюкозы способствуют
эффективному биосинтезу компонентов дрожжевой клетки, в результате чего
резко возрастает скорость накопления биомассы дрожжей.
Выделенные из жидких сред прессованные дрожжи представляют собой
готовый продукт в виде массы живых микроорганизмов, которые
используются в хлебопечении в качестве разрыхлителей теста.
Эффект Пастера характерен не только для дрожжей. Он имеет
место для любых факультативно анаэробных микроорганизмов, для
клеток высших растений, животных. При хранении влажных зерновых
масс интенсивное дыхание семян приводит к быстрому исчезновению
кислорода в межзерновом пространстве и к переключению дыхания
на спиртовое брожение, что пагубно влияет на качество семян. В
интенсивно работающей мышце человека или животного накапливается
повышенное количество молочной кислоты. Эффект Пастера
представляет собой один из важных механизмов взаимосвязи и взаимного
контроля гликолиза и дыхания.
375
Часть II. Метаболизм веществ и энергии в клетке
9.13. Исследование процессов катаболизма углеводов
В исследовании биокаталитических систем, участвующих в
катаболизме углеводов, использовались весьма разнообразные пути и
подходы. Развитие этих путей, логическое осмысливание результатов
экспериментов особенно поучительно, если рассматривать эти вопросы в
историческом аспекте. Уже с начала XX в. особое значение приобрели
попытки выделить интермедиаты процесса гликолиза и
преобразующие их ферменты. В 1918 г. О. М. Мейергоф выяснил важный факт,
что ферменты, участвующие в спиртовом брожении, содержатся в
тканях мышц. Бесклеточные экстракты мышц способны превращать
углеводы в лактат. После открытия А. Гарденом и У. Ионгом
фосфорных эфиров гексоз Г. Эмбден в 1921—1927 гг. обнаружил, что они
образуются при распаде гликогена в дрожжевых клетках и мышцах*
Тем самым было экспериментально подтверждено принципиальное
сходство процессов брожения и дыхания. Это способствовало
возникновению идеи единства химических механизмов катаболизма
углеводов и осознания единой схемы распада углеводов.
В этот же период установлено, что при катаболизме углеводов как
в мышцах, так и при брожении образуются фосфорилированные трех-
углеродные фрагменты. Эмбден (1922 г.) обнаружил в продуктах
распада гексоз фосфоглицерат. Это позволило ему и Мейергофу
(Нобелевская премия, 1922 г.) построить первую обоснованную схему
гликолиза, которая впоследствии была расширена и дополнена Парнасом. В
установлении последовательности отдельных стадий гликолиза
авторы широко использовали метод специфического ингибирования.
Выдающимся достижением биохимии первой половины XX в. было
выявление Г. Эйлером (20-е гг.) важной роли в гликолизе аденилатов,
что дало повод для систематических поисков новых ферментов и
кофакторов. В начале 30-х гг. О. Варбург, Г. Эйлер и В. Христиан при
исследовании ферментативного окисления гексозофосфорных эфиров
открыли ЫАО и ЫАВР;
Первые шаги в исследовании аэробного окисления углеводов
сделал в 30-х гг. Э. Сент-Дьердьи (Нобелевская премия, 1937 г.).
Исследуя дыхание измельченной сердечной мышцы голубя, он установил,
что в этот процесс вовлекаются сукцинат, фумарат, малат, оксалоаце-
тат, а Т. Тунберг обнаружил в мышцах особые ферменты — дегидроге-
назы, окисляющие эти кислоты. В дальнейшем Г. А. Кребс устано-
вил, что в процесс окисления гомогенатами мышц вовлекаются также
2-оксоглутарат и пируват. Это открытие навело на мысль> что
перечисленные кислоты последовательно превращаются друг в друга с
постепенным их окислением. Следующим шагом явилась расшифровка
пути окисления цитрата К. Мартиусом и Ф* Кноопом. В 1937 г. они
показали, что цитрат изомеризуется в изоцитрат через цис-аконитат и
что изоцитрат подвергается декарбоксилированию в 2-оксоглутарат.
376
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
В расшифровке цикла трикарбоновых кислот большую роль
сыграл конкурентный ингибитор сукцинатдегидрогеназы малонат. Было
известно, что малонат является дыхательным ядом. Кребс
предполагал, что сукцинатдегидрогеназа может иметь ключевое значение в
дыхании. Это было подтверждено: при добавлении сукцината к
мышце, отравленной малонатом, в ней накапливался сукщгаат* Более того,
накопление сукцината в такой мышце происходило также при
добавлении фумарата. Первый факт свидетельствовал о существовании
реакции цитрат —» сукцинат; второй — о существовании другого пути:
фумарат -н> сукцинат, однако, отличающегося от реакции,
катализируемой сукцинатдегидрогеназой.
В развитие представлений о биоэнергетике катаболизма углеводов
большой вклад внесли супруги К. и Г. Кори. Они выяснили роль АТР
как донора фосфатных групп, а также открыли процесс
восстановления АВР и АТР (Нобелевская премия, 1947 г.). В дальнейшем было
установлено, что фосфорные эфиры являются интермедиатами
катаболизма углеводов.
Накопившиеся факты нуждались в обобщении, в создании стройной
системы катаболизма углеводов, что и осуществил Кребс,
предложивший в 1937 г. схему цикла превращений органических кислот, их
окисления и образования АТР на узловых этапах этого процесса
(Нобелевская премия, 1953 г.). Цикл Кребса связал процессы поэтапного окисле-
ния органических веществ и постепенного выделения из них энергии.
Открытие реакций декарбоксилирования пирувата и переноса
ацетильной группы на оксалоацетат — важнейший этап исследований,
позволивший связать процессы анаэробного и аэробного распада углеводов.
В 30—40-е гг. происходит формирование основных представлений
о дыхательной цепи. Открытие Д. Кейлиным роли цитохромов в этой
цепи (1925 г.) позволило построить единую схему катаболизма
углеводов с включением в него кислорода. Схема объединяла процесс
активирования водорода (дегидрирование), идею о котором впервые
высказал еще в 1908—1912 гг. В. И. Палладии, а экспериментально
доказал ее в 1925 г. Г. Виланд, и идею активирования кислорода,
развиваемую А. Н. Бахом и О. Варбургом.
Окончательное формирование представлений о дыхательной цепи
открыло путь к развитию исследований в области биоэнергетики-
Термин "биоэнергетика" ввел в науку Э. Сент-Дьердьи (1957 г.). В
исследованиях стояли два вопроса: 1) какие молекулы среди природных
соединений ответственны за превращение энергии в клетке и 2) каким
образом они выполняют свою функцию.
В середине 40-х гг. В. А. Энгельгардт и М. Н. Любимова показали,
что миозин мышц обладает АТРазной активностью. Авторы сделали
вывод: миозин ответствен за превращение химической энергии АТР в
механическую работу мышц. В дальнейших исследованиях
установлено, что процесс трансформации энергии осуществляется в нераство-
377
Часть Л. Метаболизм веществ и энергии в клетке
римых белковых комплексах, встроенных в мембраны. Встал вопрос о
тесной взаимосвязи биоэнергетических процессов со структурой
мембран, В 1930 г. Энгельгардт открыл, что аккумулирование энергии в
АТР сопряжено с окислительным фосфорилированием, а в 1949 г,
А, Ленинджер показал, что этот сопряженный процесс локализован в
митохондриях, имеющих две мембраны — внешнюю и внутреннюю* В
начале 60-х гг. выяснилось, что внутренняя мембрана митохондрий
служит носителем ферментов окислительного фосфорилирования. В
1949—1950 гг. Ленинджер и сотрудники выявили, что в дыхательной
цепи при переносе электронов от КАОН к кислороду существуют три
точки, в которых энергия, освобождающаяся при
окислении-восстановлении, трансформируется в энергию фосфатной связи. На
основании термодинамических расчетов и экспериментальных данных Ленинд-
жера и Чанса была установлена примерная локализация трех участков
дыхательной цепи, в которых происходит запасание энергии.
Однако роль мембран в объяснении механизма окислительного
фосфорилирования в рамках появившихся химической и конфор-
мационной гипотез была игнорирована. В дальнейшем П. Митчелл
показал несостоятельность этих гипотез и разработал в 1961—
*
1966 гг. хемиосмотическую гипотезу, в которой постулировал
взаимосвязь между окислительным фосфорилированием и структурой
внутренней митохондриальной мембраны, развив представления о
мембранном потенциале. Большая серия обстоятельных
исследований механизмов аккумуляции энергии в клетке была осуществлена
В. П, Скулачевым.
Выяснение механизмов действия ферментных систем —
трансформаторов энергии — является важнейшей задачей биоэнергетики и на
сегодняшний день.
В исследованиях путей катаболизма углеводов были использованы
остроумные и изящные методы и приемы, среди которых особое
внимание заслуживали: 1) метод изотопных меток; 2) каталитическая
стимуляция дыхания; 3) метод спектрофотометрии и специфического
ингибирования промежуточных стадий; 4) использование детергентов;
5) выделение митохондриальных фрагментов и их реконструкция.
В методах изотопных меток был использован изотоп 14С. С
помощью этого метода удалось выяснить полную стехиометрию некоторых
реакций гликолиза и цикла трикарбоновых кислот,
В стимуляции дыхания широко использовались гомогенаты
грудной мышцы голубя, кролика, куда вводились ди- и трикарбоновые
кислоты. Резкое увеличение интенсивности дыхания позволило
установить их непосредственное участие в реакциях окисления и последо-
■
вательность их взаимопревращений.
Спектрофотометрический метод в сочетании со специфическим
ингибированием был впервые введен Б. Чансом. Метод позволил
проследить последовательность восстановления компонентов дыхательной
378
Глава 9, Катаболизм углеводов. Генерирование энергии фосфатной связи
цепи. Он дал возможность отличить окисленную и восстановленную
формы большинства компонентов цепи, поскольку переход пары
электронов цепи занимает определенное (конечное) время и при этом имеет
место последовательное восстановление переносчиков. При
добавлении специфических ингибиторов к препарату митохондрий в
стационарном состоянии дыхания электроны накапливаются на
компонентах дыхательной цепи, расположенных до точки ингибирования
{компоненты полностью будут восстановлены). В то же время будет
продолжаться перенос электронов от компонентов дыхательной цепи к
кислороду после точки ингибирования, в результате чего эти
компоненты будут полностью окисленными. Таким образом, в точке
действия ингибитора наблюдается резкий переход от восстановленных
компонентов цепи к окисленным.
Использование спектрофотометрического метода в сочетании со
специфичностью действия ингибиторов позволило установить
локализацию в дыхательной цепи пунктов фосфорилирования.
Выделение митохондриальных фрагментов дало возможность
расположить входящие в их состав дыхательные комплексы в
определенной последовательности и установить все компоненты системы
окислительного фосфорилирования в митохондриях. Хотя точное
положение всех частей еще неизвестно, в этом направлении достигнуты
успехи. Электронная микроскопия высокого разрешения и разработка
мягких методов разрушения митохондрий способствовали познанию
уникальной структуры этих органелл. Предпринимались попытки
реконструировать отдельные фрагменты. Фрагментация обычно
осуществляется обработкой детергентами типа дезоксихолата натрия, диги-
тонина, тритона Х-100 или ультразвуком. Полная реконструкция
дыхательной цепи была произведена смешиванием эквимолекулярных
количеств фрагментов. В этом подходе исследований была получена
ценная информация о роли фактора сопряжения Кг Его удаление
приводит к потере субмитохондриальными частицами способности
синтезировать АТР, но перенос электронов остается незатронутым. Фосфо-
рилирующая способность вновь восстанавливается при добавлении
фактора Б,1 к мембранным препаратам; иными словами, было
доказано, что этот фактор теснейшим образом связан с синтезом АТР.
Попытки расчленить и вновь реконструировать систему
окислительного фосфорилирования имеют важное значение в плане
постановки будущих экспериментов. Однако при интерпретации
результатов возникают значительные трудности, поскольку немногие из
компонентов были выделены в совершенно гомогенном состоянии.
Необходимо разработать щадящие методы очистки этих компонентов,
научиться так работать с каждым белком, чтобы избежать его
денатурации. Только в этом случае станет возможным, смешивая высокоочи-
щенные компоненты митохондриальных мембран, реконструировать
активную систему переноса электронов и фосфорилирования. Экспе-
379
Часть II, Метаболизм веществ и энергии в клетке
рименты такого рода помогут дать исчерпывающую информацию о
взаимосвязи структуры мембран и окислительного фосфорилирования.
В расшифровке механизма сопряжения переноса электронов в
дыхательной цепи и окислительного фосфорилирования широко
используются специфические соединения, способные нарушать это сопряжение. Их
называют разобщающими агентами. Агенты подразделяются на два класса:
первый — это липидорастворимые соединения, содержащие кислотную
группу и ароматическое кольцо; к ним относятся 2,4-динитрофенол, ди-
кумарал и др. Второй класс — вещества, вызывающие разобщающий
эффект в присутствии специфических катионов (обычно К+ или Ка+); к
ним относятся валиномицин, нонактин А, грамицидин А.
Разобщающие агенты типа 2,4-динитрофенола способствуют
переносу протонов через внутреннюю митохондриальную мембрану в мат-
рикс, снимая таким образом их электрохимический градиент,
генерируемый переносом электронов, В основе их разобщающего действия
лежит способность растворяться в липидном слое мембраны и,
следовательно, диффундировать через нее. Так как они имеют кислотную
группу, которая может ионизироваться с образованием ионов Н+, то
поэтому способны выравнивать концентрацию ионов Н+ по обе
стороны внутренней мембраны. Действие разобщающих агентов
убедительно согласуется с хемиосмотической гипотезой.
Таким образом, различные пути и подходы к исследованию,
использование остроумных методов экспериментирования позволили
получить представления о стройной системе катаболизма углеводов и
порционном, поэтапном извлечении в этом процессе химической
энергии, ее аккумулировании в форме АТР.
Краткое содержание главы
1
Катаболизм углеводов и связанное с ним генерирование энергии
фосфатной связи идут двумя путями — анаэробным (брожение) и
аэробным (дыхание). Начальная стадия катаболизма углеводов,
называемая гликолизом, складывается для этих путей из идентичных
последовательно идущих реакций. Конечной целью обоих путей являются:
а) синтез АТР за счет утилизации энергии, аккумулированной в
углеводах; б) получение из этих углеводов пластических соединений,
являющихся исходным материалом для синтеза компонентов клетки.
Гликолиз — основной процесс катаболизма углеводов для
большинства организмов. Последовательность его реакций приводит к
превращению гексозы в пируват с одновременным образованием двух
молекул АТР (субстратное фосфорилирование).
В анаэробных условиях гликолиз завершается брожением,
основными формами которого являются спиртовое, молочнокислое, масля-
нокислое,
У аэробных организмов в процессе дыхания пируват подвергается
окислительному декарбоксилированию с образованием ацетил-СоА,
380
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
который вовлекается в цикл трикарбоновых кислот. Образование аце-
тил-СоА и реакции цикла трикарбоновых кислот осуществляются в
митохондриях. Основными реакциями этого цикла являются декар-
боксилирование и дегидрирование, в ходе которых происходит
освобождение энергии, аккумулированной в ацетил-СоА. В ходе одного
оборота цикла два атома углерода ацетила превращаются в С02,
осуществляются четыре реакции дегидрирования с образованием трех
КАВН, одного ГАВН2 и в результате субстратного фосфорилирова-
ния — одной молекулы ОТР, эквивалентной одной молекуле АТР.
Помимо катаболизма углеводов по пути гликолиза существует
другой альтернативный путь, получивший название "гексозомонофосфат-
ный путь", или "шунт". Процесс носит окислительный характер, в
ходе которого от углеродной цепи идет отщепление по одному атому
углерода в форме С02.
Превращение энергии, запасенной в восстановленных кофермен-
тах, осуществляется в митоходриях в процессе окислительного фос-
форилирования, являющегося заключительным этапом генерирования
химической энергии в АТР за счет окисления МАЗОН и РАВН2.
Данный процесс включает два тесно сопряженных явления: 1) окисление
субстратов с помощью дыхательной электрон-транспортной цепи,
сопровождающееся транслокацией протонов через внутреннюю митохонд-
риальную мембрану, что обеспечивает генерирование градиента
концентрации протонов; и 2) образование АТР, осуществляемое за счет
энергии протонов, поступающих в матрикс через канал АТР-синтета-
зы. Оба процесса энергетически сопряжены. Движущей силой этих
процессов является возрастание окислительно-восстановительного
потенциала по пути потока электронов к кислороду.
Хемиосмотическая теория объясняет, каким образом фосфорили-
рование АХ)Р сопряжено с электронным транспортом. Она также
объясняет действие разобщающих агентов, в присутствии которых
субстратное окисление не сопровождается фосфорилированием АВР.
Постулаты этой теории включают также необходимость целостности митохонд-
риальной мембраны, градиента концентрации протонов и мембрано-
связанной АТРазы, действие которой обратимо*
Протондвижущая сила (Ар) зависит от разницы концентраций
протонов (АрН) и разницы заряда (Ду) по обе стороны мембраны,
В окислительное фосфорилирование вовлечены пять ансамблей
белков и кофакторов, включая электронпереносящие комплексы 1—1V и
АТР-синтетазу. Перенос электронов через комплексы в основном
происходит в соответствии с относительными восстановительными
потенциалами различных компонентов. Электроны от КА1ЭН проходят
через комплексы I, III и IV. Электроны от сукцината вводятся через
комплекс II. В электронном переносе участвует несколько
кофакторов, включая Р1\Ш, РАО, железосерные кластеры, атомы меди,
цитохромы и убихинон, Цитохромы дифференцируются в зависимости
381
Часть II. Метаболизм веществ и энергии в клетке
от их абсорбционных спектров и восстановительных потенциалов.
Мобильный переносчик убихинон связывает комплексы I и II с
комплексом III, а цитохром с связывает комплекс III с комплексом IV.
На каждые два электрона, переносимые от КАВН к убихинону (^
комплексом I, четыре протона транслоцируются в окружающее
мембрану пространство. Электронный перенос, осуществляемый
комплексом II, не вносит вклада в протонный потенциал. Перенос злектронов
от <Щ2 к цитохрому с комплексом III приводит в конечном итоге к
транслокации двух протонов из матрикса. Последовательность
переноса электронов в комплексе III описывается двухэталным (^-циклом.
Комплекс IV переносит электроны от цитохрома с к 02, конечному
окисляющему агенту электрон-транспортной цепи. Электронный
перенос и восстановление 02 до Н20 вносят еще два протона в градиент.
Протоны возвращаются в митохондриальный матрикс, проходя
через Р0 компонент Р0РХ АТР-синтетазы. Механизм, с помощью
которого течение протонов обепечивает синтез АТР из АРР и Р. не ясен.
Предполагается существование механизма чередующегося связывания,
с помощью которого объясняется, как перенос трех протонов
взаимосвязан с конформационными изменениями в синтетазе.
Транспорт АПР и Р. внутрь и АТР из митохондриального матрикса
эквивалентен потреблению одного протона. Выход АТР в расчете на
пару электронов, переносимых комплексами I—IV, может быть
рассчитан исходя из числа транслоцируемых протонов. Окисление
митохондриального ИАВН дает 2,5 АТР; окисление сукцината — 1,5 АТР.
Эти значения ниже тех, которые традиционно использовались. Цито-
зольный КАВН может вносить вклад в окислительное фосфорилиро-
вание, если его восстановительные эквиваленты переносятся в
митохондрии с помощью глицеролфосфатного или малат-аспартатного
челночного механизма.
В некоторых типах клеток окисление восстановленных субстратов
не сопряжено с фосфорилированием, что приводит к образованию
тепла. Фагоциты используют восстановительный потенциал ЫАВРН для
образования супероксида, который необходим для борьбы с
бактериальными инфекциями. Небольшие количества токсичных форм
кислорода, О^, Н202 и ОН*, образуются как продукты окислительного
метаболизма и в других типах клеток. Активные формы кислорода
весьма токсичны для клеток, так как они повреждают многие
клеточные структуры. Кислородные радикалы и лероксиды способны
разрушать липиды, белки и нуклеиновые кислоты. Существуют ферменты,
обеспечивающие защиту организма от активных форм кислорода:
супероксиддисмутаза, каталаза, глутатионпероксидаза и др. Клетки
используют восстановленные молекулы, такие как глутатион,
витамин Е и аскорбиновую кислоту для защиты от окислительного
разрушения активными формами кислорода. Образование токсичных форм
кислорода играет важную роль в биологическом процессе, известным
382
Глава 9. Катаболизм углеводов. Генерирование энергии фосфатной связи
под названием апоптоз, представляющим собой
запрограммированное в организме разрушение клеток и их гибель.
Хронология важнейших дат в исследовании катаболизма углеводов
1776 г. — А. Лавуазье установил, что процесс дыхания связан с окислением
веществ. Он впервые коснулся проблем энергетики обмена веществ.
1810 г. — Гей-Люссак вывел уравнение спиртового брожения.
1837 г. — Берцелиус постулировал каталитическую природу брожения.
1845 г. — X. Ф. Шонбейн развил обобщающую теорию биологического
окисления (теорию "активации кислорода").
1854—1864 гг. — Л. Пастер доказал, что брожение вызывается
микроорганизмами, и привел решающие аргументы против гипотезы о самозарождении жизни.
1897 г. — Э. Бухнер открыл, что спиртовое брожение может происходить в
бесклеточных дрожжевых экстрактах,
1905 г. ™ А. Гарден и У. Ионг показали, что для спиртового брожения
необходимо присутствие фосфата и выделили первый кофермент ™ козимазу,
который, как было в дальнейшем установлено, представляет собой МАО4".
1907—1913 гг. — В. И. Палладии показал важную роль в дыхании процесса
дегидрирования окисляемых субстратов.
1910 г. — С. П. Костычев впервые высказал представление о единстве
процессов брожения и дыхания.
1912 г. — К. Нейберг предложил схему химических реакций процесса
брожения.
1912 г. — О. Варбург высказал предположение, что в процессе дыхания
участвует особый фермент, активирующий кислород; установил, что этот фермент
ингибируется цианидом, и доказал необходимость железа для дыхания.
1912—1922 гг. ~ Т. Виланд показал, что в реакциях дегидрирования
происходит активация водорода.
1918 г. — О. Мейергоф установил важный факт, что козимаза (кофермент
ИАО), необходимый для спиртового брожения, содержится в мышцах.
1921—1927 гг. — Г. Эмбден обнаружил, что как и при брожении, при
распаде гликогена в мышцах образуются гексозофосфорные .эфиры. Этим было
экспериментально подтверждено единство процессов брожения и дыхания*
1922 г. — Г. Эмбдену и О. Мейергофу была присуждена Нобелевская премия
за первую экспериментально обоснованную схему анаэробного распада углеводов
(гликолиза).
1930 г. — С. П. Костычев представил важнейшие труды по биохимии брожения.
1931 г. — В. А. Энгельгардт показал, что фосфорилирование сопряжено с
дыханием.
1933 г. — Г. Эмбден и О. Мейергоф выделили наиболее важные
промежуточные продукты процесса гликолиза и брожения.
1935 г. — Э. Сент-Дьердьи показал, что дикарбоновые кислоты оказывают
каталитическое действие на процесс дыхания.
1937 г. — К. Кори и Г, Кори начали свои исследования по изучению
функций гликогенфосфорилазы.
1937 г. — Э. Сент-Дьердьи присуждена Нобелевская премия за расшифровку
механизма дыхания.
1937—1938 гг. — О. Варбург показал, что образование АТР сопряжено с
процессом дегидрирования глицеральдегид-3-фосфата.
1937—1941 гг. — В. Белицер, С. Калыгар независимо друг от друга провели
количественное изучение окислительного фосфорилирования.
383
Часть II. Метаболизм веществ и энергии в клетке
1939—1942 гг. — В. А. Энгельгардт и М. Н. Любимова открыли АТРазную
активность миозина.
1940—1943 гг. — Б. Клод выделил митохондриальную фракцию клеток
печени.
1947—1950 гг. — Ф. Липман выделил и охарактеризовал кофермент А.
1961—1966 гг. — П. Митчелл предложил гипотезу хемиосмотического фос-
форилирования.
1978 г. — П. Митчелл удостоен Нобелевской премии за исследования в
области биоэнергетики и создание хемиосмотической теории,
ЛИТЕРАТУРА
Берегов Т. 7\, Коровкин Б, Ф, Биологическая химия: Учебник. — 3-е издм
перераб. и доп. — М.: Медицина, 1998. — 704 с.
Бохински Р. Современные воззрения в биохимии. — М.: Мир, 1987. — 544 с.
Гудвин 7\, Мерсер Э. Введение в биохимию растений: В 2 т. —М.: Мир, 1986.
—Т. 1. — 392 с.
Кретович В. Л. Биохимия растений: Учебник. — 2-е изд., перераб. и доп. —
Мл Высш. шк., 1986. — 502 с.
Кнорре Д. Г., Мызина С. Д. Биологическая химия: Учебник. — 2-е изд.,
перераб. и доп. — М.: Высш. шк., 1998. — 479 с.
Марри Р., Греннер Д., Мейес 27., Родуэлл В. Биохимия человека: В 2 т. — М.:
Мир, 1993. — Т. 1. — 378 с.
Мецлер Д. Биохимия: В 3 т. — М.: Мир, 1980. — Т. 2. — 606 с.
Страйер Л. Биохимия: В 3 т. — М.: Мир, 1985. — Т. 2. — 307 с.
Скулачев В, П. Аккумулирование энергии в клетке. — М.: Наука, 1969. —
440 с.
Эллиот В., Эллиот Д. Биохимия и молекулярная биология / Под ред. А. И. Ар-
чакова, М. П. Кипичникова, А. И. Медведева, В. П. Скулачева — М.: Изд-во
НИИ Биомедицинской химии РАН, 1999. — 372 с.
ВисНапап В. В., Огиьзвеп ТТ., Зопев В. ^. ВюсЬет181гу ап<1 то!еси1аг Вю1о^у о^
Р1ап1з. — РиЬ. КоскуШе, 2000. — 1368 р.
Могап Ь, А, 8сг1гп§еоигК. С, НоНоп Н. Д., ОсНз А $., Вашп ^. В. ВюсЪеппз^гу.
— 2 ЕЙ. — N611 Райемоп РиЬ., Епе1е*гоос1 СШ*в, 1994. — 1200 р.
ВОПРОСЫ И ЗАДАЧИ
1. Реакция: глюкоза + АТР —> глюкозо-6-фосфат + АОР осуществляется при
участии какого фермента: 1) альдолазы; 2) фосфоглюкомутазы; 3) оксоизомера-
зы; 4) фруктокиназы; 5) гексокиназы?
2. Какие ферменты участвуют в гликолизе: 1) альдолаза; 2) енолаза; 3) пируват-
декарбоксилазный комплекс; 4) сукцинатдегидрогеназа; 5) фосфофруктокиназа?
3. Какую реакцию блокирует малонат: 1) сукциыат —> фумарат; 2) малат -»
оксалоацетат; 3) реакцию конденсации ацетил-СоА с оксалоацетатом?
4. Рассчитайте выход этанола при спиртовом брожении, выход лактата при
гомоферментативном молочнокислом брожении, если в среде находилось 100 г
глюкозы.
5. Если глюкоза, содержащая радиоактивную 14С-метку в положении 3 и 4
вовлекается в гомоферментативное молочнокислое брожение, где она будет в лак-
тате? Где она будет при спиртовом брожении?
6. Рассчитайте ДО0', высвобождаемую при окислении молекулы сукцината
до фумарата в интактной митохондрии бактериальной клетки.
I
р
Глава 10. БИОСИНТЕЗ УГЛЕВОДОВ
Биосинтез В-глюкозы — необходимый жизненно важный процесс
для всех высших животных. Постоянное поступление глюкозы в
качестве источника энергии необходимо для нервной системы, а также
эритроцитов, почек, всех тканей эмбриона. Мозг человека потребляет
более 120 г глюкозы в сутки. При понижении концентрации глюкозы
в крови ниже критического уровня происходит нарушение
функционирования мозга; тяжелая гипогликемия приводит к коматозному
состоянию. Очевидно, глюкоза играет существенную роль в
поддержании эффективных концентраций интермедиатов цикла Кребса. Она
является необходимым метаболитом липидного обмена, так как
служит источником глицерола — структурного элемента глицеридов.
Биосинтез глюкозы в организме животных постоянно
осуществляется благодаря совокупности строго регулируемых реакций.
Предшественниками В-глюкозы являются лактат, пируват, глицерол,
промежуточные продукты цикла трикарбоновых кислот и большинство
аминокислот. У животных синтез глюкозы из неуглеводных
предшественников называют глюконеогенезом* что означает образование "нового"
сахара. Глюкоза служит предшественником других физиологически
важных углеводов. В этой связи в первую очередь следует отметить
биосинтез гликогена, осуществляющийся главным образом в печени и
мышцах. Гликоген печени выполняет резервную функцию: глюкоза,
отщепляемая от цепей гликогена, поступает в кровь» Распад
мышечного гликогена обеспечивает потребности в АТР в ходе мышечного
сокращения.
В растениях глюкоза, а также другие углеводы образуются в
процессе фотосинтеза за счет восстановления диоксида углерода.
ЮЛ. Глюконеогенез
У позвоночных глюконеогенез протекает главным образом в
печени, значительно менее интенсивно — в почках (в корковом веществе).
Если центральным путем катаболизма углеводов является
превращение глюкозы в пируват, то центральным путем глюконеогенеза
принято считать превращение пирувата в глюкозу. В этой связи гликолиз
иногда называют "путем, ведущим вниз", а глюконеогенез — "путем,
ведущим вверх". Большинство реакций глюконеогенеза представляют
собой обращение соответствующих реакций гликолиза. Однако три
25. Заказ 3486
385
Часть П. Метаболизм веществ и энергии в клетке
реакции гликолиза (гексокиназная, фосфофруктокиназная и пиру-
ваткиназная) необратимы, поэтому в обход этих этапов в глюконеоге-
незе используются другие реакции, катализируемые другими
ферментами. Эти обходные этапы тоже необратимы* Благодаря этому и
гликолиз, и глюконеогенез представляют собой необратимые процессы.
Первый обходной путь: превращение пиру вата в
фосфоенолпируват. Образование фосфоенол пиру вата осуществляется в
несколько этапов. Первый этап протекает в митохондриях, где под действием
биотинсодержащего фермента пируваткарбоксилазы при участии АТР
и С02 происходит образование оксалоацетата из пирувата:
соон
СН
з
СН-
СмЭ +С02 + АТР ^ I + АОР + Р;
С—О
соон
Пируват
СООН
Оксалоацетат
Пируваткарбоксилаза является аллостерическим ферментом. Ее
аллостерическим активатором служит ацетил-СоА, в отсутствие кото-
рого фермент практически не проявляет активность.
Оксалоацетат восстанавливается в митохондриях до малата под
действием митохондриальной малатдегидрогеназы:
соон соон
СНг СН$
С-0 + НАШ + Н* =5=2: Н(1он + КА1>*
СООН СООН
Оксалоацетат Малат
Малат легко выходит из митохондрии с помощью дикарбоксилат-
транспортирующей системы и поступает в цитоплазму. Здесь он вновь
окисляется при помощи цитоплазматической малатдегидрогеназы:
СООН СООН
СН2 СН2
неон +шш+ з=ь с=о +*авн + н-
соон соон
Малат Оксалоацетат
Далее на оксалоацетат действует фосфоенолпируваткарбоксикина-
за. Путем декарбоксилирования и фосфорилирования (донором
фосфата является ОТР) оксалоацетат превращается в фосфоенолпируват
(ФЕП):
386
Глава 10. Биосинтез углеводов
СООН
СН.
С=0
+ ОТР
сн2 о
II • I
ОО-Р-0
I I
СООН О"
+ ели» + со2
СООН
Оксалоацетат Фосфоенолпируват
Второй обходной путь: превращение фруктозо-1,6-бисфосфа
та во фруктозо-6-фосфат. Данный этап глюконеогенеза
обеспечивается за счет действия фермента фруктозобисфосфатазы:
СН«ОР
с=о
СНоОН
С=0
но-с-н
но-с-н
н-с-он
н*о
н-с-он
СН2ОР
Фруктозо-1,6-бифосфат
Рх
н-с-он
н-с-он
СНоОР
Фруктозо-6-фосфат
Фруктозобисфосфатаза является регуляторным ферментом.
Ингибитором активности фермента является АМР, а его активатором - АТР*
Третий обходной путь: превращение глюкозо-6-фосфата в
глюкозу. Третья обходная реакция глюконеогенеза — это дефосфорилиро-
вание глюкозо-6-фосфата с образованием свободной глюкозы, катали-
зируемое глюкозо-6-фосфатазой.
с-°
н-с-он
Ч
о
н
н-с-он
но-с-н
н-с-он
н-с-он
СН?ОР
Глюкозо-6-фосфат
НоО
Р1
НО-С-Н
I
Н-С-ОН
Н-С-ОН
сн2он
Глюкоза
На рис. 10.1 показаны противоположно направленные пути
гликолиза и глюконеогенеза.
10.2. Регуляция глюконеогенеза и гликолиза
Первым регуляторным пунктом глюконеогенеза является пируват-
карбоксилазная реакция. В отсутствие ацетил-СоА, выполняющего
функцию аллостерического активатора пируваткарбоксилазы, фермент
практически лишен активности. В то же время ацетил-СоА является
25*
387
Часть II. Метаболизм веществ и энергии в клетке
Глюкоза
АТР
ЧУ"
Глюкозо-6-фосфат
Фруктозо-6-фосфат
Глюкозо-1 -
фосфат
Гликоген
Икгибируется
АМР
Путь
гликолиза
Фруктозо-1,в-бисфосфат
и
Глицеральдегид-3- ч^ ^ Дигидрокси-
фосфат V—^ ацетонфосфат
1, З-Бнсфосфоглицерат
и
З-Фосфоглицерат
и
2-Фосфоглицерат
и
Фосфоенолпируват
Оксалоадетат
Путь
глюконеогенеза
АОР
АТР
ИАВ
+
Малат
Пиру ват
Малат
V
ЫАПН
Оксалрацетат
АТР
Пируват Г\
Стимулируется
ацетил-СоА
Рис. ЮЛ. Гликолиз ("путь, ведущий вниз") и глкжонеогенез ("путь, ведущий
вверх")
ингибитором пируватдегидрогеназного комплекса. Таким образом, при
нараетании концентрации ацетил-СоА в митохондриях происходит
интенсификация биосинтеза глюкозы из пирувата при одновременном
замедлении окислительного декарбоксилирования пирувата.
388
Глава 10. Биосинтез углеводов
Второй регуляторный пункт — реакция, катализируемая фруктозо-
бисфосфатазой. Как отмечалось выше, АМР оказывает резкое ингиби-
рующее действие на данный фермент, а АТР — стимулирующее.
Поскольку соответствующий фермент гликолиза, фосфофруктокиназа>
активируется АМР и АОР, а торможение его активности происходит под
действием АТР и цитрата, то эти два противоположных этапа
глюконеогенеза и гликолиза регулируются координированным образом, как
говорят, реципрокно. Реципрокной регуляции гликолиза и глюконеогенеза
способствует также действие фруктозо~2у6-6исфосфатау играющего роль
мощного регулятора активностей фосфофруктокиназы и фруктозо-1,6-
бисфосфатазы* Увеличение концентрации фруктозо-2,6-биефосфата
приводит к усилению гликолиза за счет активации фосфофруктокиназы и
торможению глюконеогенеза путем ингибирования фруктозо-1>6-бисфос-
фатазы. Падение концентрации фруктозо-2,6-бисфоефата, наоборот,
вызывает подавление гликолиза и интенсификацию глюконеогенеза*
Важным моментом этого регуляторного пункта является зависимость
активности бифункционального фермента, обеспечивающего синтез и распад
фруктозо-2,6-бисфосфата, от г^и/сло-АМР-зависимого фосфорилирования.
С этим сопряжено эффективное воздействие гормонов, в частности глюка-
гона, на уровень фруктозо-2,б-бисфосфата в клетке. Условия,
способствующие фосфорилированию бифункционального фермента, обеспечивают
возрастание его фосфатазной активности, в результате чего происходит
распад фруктозо-2,6-бисфосфата на фруктозо-6-фосфат и неорганический
фосфат. Фосфокиназная активность бифункционального фермента,
возрастающая при снижении темпа фосфорилирования, обеспечивает
синтез фруктозо-2,6-бисфосфата из фруктозо-6-фосфата при участии АТР.
В определенной степени регуляция глюконеогенеза может
осуществляться за счет изменений активности гликолитического фермента
пируваткиназы, хотя этот фермент и не участвует в глюконеогенезе. Одна из
форм пируваткиназы, а именно Ъ-форма, преобладающая в тканях, где
протекает глюконеогенез, ингибируется АТР и рядом аминокислот.
Причем из аминокислот наибольший ингибирующий эффект оказывает ала-
нин, являющийся глюкогенной аминокислотой и играющий роль
предшественника глюкозы в глюконеогенезе. Благодаря регуляции на уровне
Ь-формы пируваткиназы при торможении гликолиза при достаточном
обеспечении клетки АТР и при наличии предшественников глюкозы
создаются условия, благоприятствующие интенсификации глюконеогенеза.
10.3. Фотосинтез
Фотосинтез — это синтез углеводов и других органических
соединений из неорганических — С02 и Н20 — под действием энергии
солнечного света. В данном процессе используется видимый, или белый,
свет, нижний предел длины волны которого равен примерно 400 нм,
верхний — примерно 700 нм. Этот свет и несет энергию всему живому
389
Часть II. Метаболизм веществ и энергии в клетке
на Земле- По М. Планку, энергия кванта света (Е) определенной
световой волны зависит от частоты излучения V:
Е = Ьу,
где И. — постоянная Планка (6,65-Ю"34 Дж'с-1).
По А. Эйнштейну, энергия световой волны испускается и
поглощается как величина, кратная Ьу (Игу, 2Ьу, ..., пЬу), Зависимость
между частотой излучения волны и ее длиной А, определяется
соотношением
с
где с — скорость света, равная 3'1010см-с_1.
Тогда энергия одного кванта (фотона)
ПС
ЕФ=ЬУ=Х
Энергия п фотонов
Из этого уравнения вытекает, что с увеличением длины волны
энергия света уменьшается и что энергия фотона поглощается веществом
целиком, а не по частям. Это необходимо учитывать при анализе
механизма фотосинтеза.
Принципиальное уравнение фотосинтеза следующее:
6С02 +6Н20—^ет(пьу) )Сбн1206 +б02.
В данном уравнении С02 представляет собой низкоэнергетический
окисленный углерод; Н20 -— слабый восстановитель; С6Н12Об — гексо-
за — высокоэнергетический восстановленный углерод; 02 — сильный
окислитель. Таким образом, фотосинтез — это процесс
преобразования энергии света в энергию химических связей органических
соединений. Он является эндергоническим процессом. Образованные при
фотосинтезе углеводы — важнейшая форма запасания и хранения
энергии. По образному выражению К. А. Тимирязева, они представляют
собой "консерв" лучистой энергии.
Фотосинтез осуществляется различными группами автотрофных
организмов: высшими зелеными растениями, красными и бурыми
водорослями, цианобактериями, зелеными и пурпурными
бактериями. При синтезе каждой молекулы гексозы АО°/ = 2865,5 кДж; ДН
этой реакции равна 2808,9 кДж-моль"1, отсюда ДС0'=ДН- ТД8 дает
ТД8 = -56,6 кДж. Так как Т > 0, значит Д8 < 0, т.е. синтез гексозы из
С02 и Н20 идет и с аккумулированием химической энергии, и с
упорядочиванием системы. Фотосинтез идет в огромных масштабах. Его
продукция на Земле составляет в год 4-Ю10 т связанного углерода! Биоло-
390
Глава 10, Биосинтез углеводов
гическая роль фотосинтеза велика; он является единственным
источником органических веществ и молекулярного кислорода* Появление
кислорода и озонового экрана в толще атмосферы нашей планеты
создало условия для биологической эволюции •
Хотя вопрос фотосинтеза и внесен в настоящую главу учебника,
однако полный метаболический процесс растительной клетки
представляет собой нечто большее, чем фиксация и превращение С02 в углеводы.
Тогда как растительная часть ассимилированного углерода будет хра-
ниться и использоваться в форме углеводов, остальная же часть его
путем включения в центральные метаболические пути будет служить
химическим источником энергии и углерода для синтеза других
соединений — аминокислот и белкбв, нуклеотидов и нуклеиновых кислот,
жирных кислот и липидов. Иными словами, углерод в С02 в конечном счете
включается в метаболические процессы всего организма*
Продукты фотосинтезирующих организмов затем используются
нефотосинтезирующими организмами, которые извлекают
химическую энергию восстановленного углерода, входящего в состав
образовавшихся при фотосинтезе органических соединений (т.е.
превращенную солнечную энергию), окисляя ее опять до С02. Эти
противоположные потоки химических превращений в биосфере называются
углеродно-кислородным циклом. При фотосинтезе преобразование
световой энергии в химическую включает окислительно-восстановительный
перенос электронов и сопряженное с ним образование АТР,
называемое фотосинтетическим. фосфорилированием, и восстановление
КАВР* в ИАОРН. Донором электронов служит вода, тогда как
восстановленный конечный акцептор их — КАВРН включается в процесс
восстановления диоксида углерода до углеводов. При фотосинтезе
молекулярный кислород происходит из молекулы воды.
У всех фотосинтезирующих организмов поток электронов от
доноров к акцепторам, вызванный действием света, направлен против
градиента восстановительного потенциала, котором характеризуется до-
норно-восстановителъная система. Энергия света поворачивает поток
электронов вспять и направляет его в сторону более отрицательного и
более богатого энергией состояния.
Процесс фотосинтеза осуществляется в клеточных органеллах —
хлоропластах. На рис. 10.2 представлена электронная
микрофотография хлоропласта из листа шпината (а) и дан© схематическое
изображение его структуры (б).
Хлоропласты, называемые также хлорофилловыми зернами,
имеют обычно пластинчатую форму длиной 5—10 мк и диаметром 0,5—
2 мк. В одной клетке зеленого листа содержится от 20 до 100 хлоро-
пластов. Это высокоупорядоченные тонкие структуры. Каждый
хлоропласт окружен внешней и внутренней мембранами. Внутри него
находится набор электронно-плотных тел, имеющих форму
приплюснутых дисков, получивших название тилакоидов. Последние собраны в сто-
391
Часть II. Метаболизм веществ и энергии в клетке
I х м«м ; мембрана
а б
Рис. 10.2. Хлоропласт из листа шпината (а) и схематическое изображение его
структуры (б)
почные структуры — граны; в каждом гране содержится от 10 до 100 ти-
лакоидов* Граны связаны между собой мембранными перемычками,
которые называются ламеллами. Пространство между гранами заполнено
жидким содержимым — стромой. В расчете на сухие вещества в
хлоропласте содержится 50 % белка, 35 % липидов, 7 % пигментов.
10.4* Пигменты хлоропластов высших растений
Пигменты хлоропластов высших растений относятся к двум
основным группам — хлорофиллам (зеленые пигменты; от греч. сЫогоз —
зеленый + рНуйоп — лист) и каротиноидам (желтые пигменты); к
последним принадлежат каротины и оксигенированные каротиноиды —
ксантофиллы. Все они входят в состав пигментных систем в виде хро-
мопротеинов. Фотосинтетические пигменты погдощают энергию света
разных длин волн: хлорофиллы — в синей и красной части спектра
(что соответствует длинам волн около 430—450 и 650—700 нм),
каротиноиды — в области 400—500 нм. В нормальном зеленом листе хло-
рофиллов гораздо больше, чем каротиноидов; их соотношение иногда
достигает 5:1. Количество хлорофиллов в растениях колеблется от 1,5
до 5 % на сухую массу.
Именно хлорофиллы играют роль фотосенсибилизатора —
преобразователя энергии фотонов света в химическую энергию
органических соединений; в этом процессе каротиноиды выполняют
вспомогательную роль. Структура молекулы хлорофилла изображена на
392
Глава 1 0. Биосинтез углеводов
рис. 10.3. Жирной линией выделена возможная система электронного
возбуждения в порфириновом ядре молекулы. Порфириновое ядро (1)
состоит из четырех соединенных СН-мостиками остатков пиррола (I,
II, III, IV), которые соединены двумя основными и двумя
координационными связями с атомом магния, занимающего центральное
положение в ядре. В молекулу хлорофилла входит остаток
высокомолекулярного непредельного спирта фитола (2). Существует не менее пяти
типов хлорофилла. Сине-зеленый хлорофилл а в остатке пиррола II
содержит СН3-группу, а желто-зеленый хлорофилл в вместо нее в этом
остатке содержит формильную группу — СНО. В высших растениях
хлорофиллы представлены в основном этими двумя формами.
Предполагают, что только хлорофилл а способен быть донором энергии
непосредственно для фотосинтетической реакции, а все остальные
пигменты передают поглощенную ими энергию хлорофиллу а*
Порфириновое ядро имеет вид плоскости размером примерно
1,5x1,5 нм и толщиной 0,4 нм; фитольный остаток длиной 1,5—2,0 нм
имеет ярко выраженные гидрофобные свойства; он связывает
молекулу хлорофилла с гидрофобным участком тилакоида.
Характерной особенностью химической структуры молекулы
хлорофилла является наличие в порфириновом ядре замкнутой системы
двойных сопряженных связей. Это обусловливает важнейшую физико-хими-
сн2
II
СН „ У
I Н I
V V %л-
п
СНз-С
I
С~~СН2—СНз
II
НС
N
\
М-
ч
щ
\
N
IV II
И'
.•
СН
III
СШ-
3_<?\ /°Ч <^°\ #
н х-н чг сг
С-СНз
сн2
сн2
I
с=о
НС—
I
оо
осн.
с-о
СН1
СН;
СНз СНз
0-СН2СН=С(СН2)зСН(СН2)зСН(СН2)зСНСНз
^ V '
2
Рис. 10.3. Химическая структура молекулы хлорофилла: У= -СН3 в
хлорофилле а: У= -СНО в хлорофилле в; 1 — порфириновое ядро, состоящее из
четырех соединенных СН-мостиками остатков пиррола (I, II, III, IV); 2 —
гидрофобная боковая цепь высокомолекулярного спирта фитола, соединяющая молекулу
хлорофилла с гидрофобным участком мембраны тилакоида
393
Часть П. Метаболизм веществ и энергии в клетке .
ческую особенность хлорофилла, имеющую биологическое значение —
способность поглощать энергию света в видимом диапазоне
электромагнитного спектра, преобразовывать эту энергию в энергию электронного
возбуждения и равномерно распределять ее по тетрапиррольному ядру.
Системы двойных сопряженных связей представляют собой гибридизи-
рованные о-связи и негибридизированные л-связи. Особенностью тс-связи
является способность к делокализации электронов, т.е. способность
легко отдавать и присоединять электроны. Энергия фотона переводит
молекулу пигмента в возбужденное состояние, сопровождающееся делокали-
зацией электрона.
Обнаружено, что длина волны света, вызывающего переход связи
из нормального в возбужденное состояние (л-»л*-переход), растет
(фотон требуется меньшей энергии) по мере увеличения числа двойных
связей в сопряженной системе молекулы. Объясняется это тем, что
при увеличении системы сопряженных связей в молекуле возрастает
число я-электронов, способных к делокализации. Поэтому длины волн
фотонов, вызывающих эти переходы, увеличиваются. При наличии
семи или более сопряженных двойных связей длины волн фотонов,
вызывающих переход 71->тс*, соответствуют видимой области спектра.
Поскольку молекулы всех фотосинтезирующих пигментов содержат
семь или более сопряженных двойных связей, ясно, что переходы и
объясняют способность этих молекул поглощать видимый свет.
10«5.« Стадии фотосинтеза
Суммарный процесс фотосинтеза складывается из двух стадий,
которые могут быть описаны следующими реакциями:
Н20 + АВР + Р. + КАВР+ ^5 02 + АТР + ИАБРН + Н+
С02 + АТР 4- ЫАРРН + Н+ ~> (СН20) + АРР + Р( + ЫАРР+
Суммарно: С02 + Н20 —> (СН20) + 02,
где (СН20) — углевод.
В первом процессе, часто называемом "световые реакции",
протоны, извлекаемые из воды, используются для хемиосмотического
синтеза АТР из АОР и Р., тогда как гидридион из водной среды
используется для восстановления КАОР+ до ЫАОРН. В ходе реакций данного
процесса происходит светозависимое образование газообразного
кислорода, возникающего за счет расщепления молекул воды. Эти
реакции возможны благодаря тому, что фотосинтетические организмы могут
улавливать энергию света и использовать ее для осуществления
метаболических реакций.
Второй процесс фотосинтеза включает использование МАОРН и АТР
для ассимиляции углерода; этот процесс обеспечивает восстановление
394
Глава 10. Биосинтез углеводов
газообразного диоксида углерода до углевода. Поскольку реакции
данного процесса непосредственно не зависят от света, а только от
поставки АТР и ЫАВРН, они часто называются "темновые реакции*'.
Термины "световые реакции" и "темновые реакции" довольно
широко используются, но следует иметь в виду, что оба процесса в норме
осуществляются одновременно. Более того, свет необходим для
максимальной скорости ассимиляции углерода в темновых реакциях.
В хлоропластах эти стадии пространственно разобщены: световая
осуществляется в мембранах тилакоидов, а темновая — в водной
среде стромы.
Взаимосвязь между световой и темной стадиями можно выразить
в виде следующей схемы;
12НоО
6СО» 12КАВРН
т
6НоО
всо.
пЬу
18АТР
Световая стадия
1
СбН130е
Темновая стадия
В световой стадии фотосинтеза происходят стабилизация и
запасание световой энергии и ее трансформация в химическую энергию. Она
включает четыре основных события: 1) фотохимическое возбуждение
хлорофилла; 2) фотоокисление воды до кислорода; 3)
фотовосстановление ИАОР* до ЫАБРН; 4) фотофосфорилирование (синтез АТР).
Именно в реакциях образования трех веществ (АТР, ИАОРН,
молекулярного кислорода) происходит, по существу, превращение световой
энергии в химическую.
Далеко не каждая мйле&ула хлорофилла или другого пигмента,
поглотившая свет, способна к преобразованию световой энергии в
химическую. Установлено, что это преобразование осуществляется
комплексом, состоящим примерно из 200—250 молекул хлорофилла; этот
комплекс работает как единая светоулавливающая система,
представляющая собой один реакционный центр. Такое обстоятельство
является необходимым следствием квантовой природы света. Фотоны
света, подобно каплям падающего дождя, поглощаются разными
молекулами хлорофилла. Такой ансамбль обеспечивает непрерывную работу,
подобно тому, как присоединение одного водостока к достаточно
значительной поверхности крыши позволяет получить из отдельных
капель дождя непрерывный поток воды.
Важное значение в функционировании реакционного центра имеет
процесс миграции энергии между различными формами пигментов.
Их различные спектральные формы образуют лестницу
энергетических уровней, по которой перенос энергии происходит в направлении
квантов с малой энергией (длинноволновых квантов). Так как часть
энергии по пути растрачивается в виде тепла, обратный перенос ста-
395
Часть П. Метаболизм веществ и энергии в клетке
новится невозможным. В конечном счете поглощенная энергия
сообщается пигменту с наиболее длинноволновой полосой поглощения —
хлорофиллу а, пигменту Р700.
10*6. Два типа фотосинтезирующих систем
В тилакоидной мембране присутствуют два типа фотосистем —
фотосистема I (ФС1) и фотосистема II (ФСП). ФС1 локализована
преимущественно в ламеллах стромы, тогда как ФСП локализована
преимущественно в ламеллах гран. Пара особых молекул хлорофилла а в
реакционном центре ФС1 обладает абсорбцией в длинноволновой
области спектра при 700 нм; этот первичный донор иногда называют Р700.
Пара особых молекул хлорофилла а в реакционном центре ФСП имеет
максимум поглощения при 680 нм, и поэтому этот донор называют
Р680.
Хотя эти две фотосистемы локализованы в различных областях
тилакоидной мембраны, они связаны с помощью специфических
электронных переносчиков и работают как единое целое. Такое разделение
ФС1 и ФСП предотвращает спонтанный перенос энергии возбуждения
между двумя фотосистемами, обеспечивая в то же время их
взаимосвязь только за счет переноса электронов. Энергия света, необходимая
для фотосинтеза, улавливается антенными пигментами,
сопряженными с реакционными центрами и пигментным комплексом,
называемым хлорофилл а/Ь светособирающий комплекс (ССК). Антенные
хлорофиллы ФСП являются частью пигмент-белкового комплекса,
расположенного вне реакционного центра данной фотосистемы. На-
против, антенные хлорофиллы ФС1 связаны с белковыми субъедини-
цами реакционного центра. ССК содержит около половины всего
хлорофилла, находящегося в хлоропласте. ССК присутствует в
тилакоидной мембране в виде трех фракций: одна из них взаимосвязана с ФС1,
другая — с ФСП, а третья представляет собой мобильную фракцию,
которая может собирать свет как для ФС1, так и для ФСП. Помимо
фотосистем и светособирающих комплексов существуют и другие
компоненты, участвующие в фотосинтезе, локализованные в тилакоидной
мембране или же ассоциированные с ней определенным образом
(рис. 10.4). К ним относятся кислородобразующий комплекс,
комплекс цитохромов &/ и хлоропластная АТР~синтетаза.
Кислородобразующий комплекс, состоящий из нескольких периферических белков и
четырех атомов марганца, взаимосвязан с ФСП. Периферические
белки выполняют роль помощников в кислород образующих процессах,
они нековалентно связаны с реакционным центром. Комплекс
цитохромов Ы пронизывает тилакоидную мембрану, он локализован как в
ламеллах стромы, так и ламеллах гран. АТР-синтетаза также
пронизывает тилакоидную мембрану и локализована исключительно в
ламеллах стромы.
396
АТР
Ламелла
ФСП
антенный
комплекс
ФСп' 2Н20
реакционный
центр
ФС1 реакционный центр
и антенные пигменты
АТ Р - синтетаза
Внутритилакоидное пространство
со
Рис. 10,4. Фотосинтетические компоненты тилакоидной мембраны. Процессы поглощения света указаны волнистыми стрел-
С)
^ нами, электронного транспорта — сплошными стрелками, транслокации протонов — пунктирными стрелками
с
о
3
то
Си
то
о
Од
О
Часть II, Метаболизм веществ и энергии в клетке _____
10-7, Циклический и нециклический транспорт электронов
Восстановление NА^Р* до МАОРН при нециклическом
транспорте электронов. Фотосинтетический перенос электронов может
осуществляться по одному из двух путей. Электроны могут следовать по
линейному пути через серию мембраносвязаняых переносчиков,
достигая в конечном итоге ЫАВР+, или же они могут транспортироваться
через переносчики циклическим образом, так что в данном случае
терминальный перенос на ЫАОР+ не имеет места- Во время как
нециклического, так и циклического электронного транспорта протоны транслоциру-
ются через тилакоидную мембрану из стромы во внутреннее тилакоид-
ное пространство (т.е. внутрь тилакоидных пузырьков), генерируя про-
тондвижущую силу, используемую для образования АТР. Однако только
нециклический электронный поток обеспечивает образование КАОРН.
Первоначальным явлением, предшествующим
фотосинтетическому электронному транспорту, является поглощение фотона света
пигментной молекулой. Фотоны могут поглощаться пигментами ССК или
антенными пигментами. Для иллюстрации мы проследим перенос
энергии возбуждения (экситона) от антенного комплекса на ФСП. Когда
пигмент поглощает фотон, то низкоэнергетический, расположенный
на определенном уровне, электрон переходит на более высокую
молекулярную орбиталь, обеспечивая поглощение энергии возбуждения
пигментом. Молекулярные орбитали имеют дискретные
энергетические уровни, и разница в энергии между основным состоянием
пигмента и состоянием, сопряженным с переходом электрона на более
высокоэнергетическую орбиталь, должна соответствовать энергии фотона
для того, чтобы этот фотон был поглощен. Энергия фотона обратно
пропорциональна длине световой волны, и поэтому пигменты
поглощают только определенные волны света.
Поглощение фотона ССК или антенным комплексом ФСП и
последующий быстрый перенос (КГ15 с) энергии возбуждения через
примыкающие пигменты может приводить к возбуждению реакционного
центра ФСП {рис. 10.5). Перенос энергии к одной из пар особых
молекул хлорофилла ФСП (т.е. к Р680) переводит этот пигмент в
возбужденное состояние. Возбужденный реакционный центр обозначается
Р680*. Перейдя в возбужденное состояние, Р680* становится
относительно сильным восстановителем, и реакционный центр хлорофилла
легко отдает электрон феофитину, являющемуся первым
электронным акцептором. Разделение заряда, которое имеет место вследствие
окисления реакционного центра до Р680+ и восстановления феофити-
на до РЬ% представляет собой этап превращения поглощенной энергии
света в химическую энергию. Возбуждение и электронный перенос в
ФС1 приводит к подобному разделению заряда. Фотоокисленный
реакционный центр Р680+ становится очень мощным окисляющим
агентом. Окисленный реакционный центр хлорофилла Р680 восстанавли-
398
Глава 10. Биосинтез углеводов
СЫ
СНГ
СО
а>
КС
3
1
<:
4
Р680 Электронный
\
\
\
\
*
перенос
Р680©
РЬ©
СЫ
Светособи-
рающий
комплекс
СЫ Р680
Антенные *
пигменты
Рис, 10.5. Перенос энергии возбуждения от пигментных молекул светособи-
рающего комплекса и антенного комплекса к реакционному центру Р680 и
последующий перенос электронов от Р680 к феофитину (РЬ). Такой же процесс
характерен для Р700
вается с помощью электрона, поступающего из кислородобразующего
комплекса. Источником электронов для кислородобразующего
комплекса служит вода. Четыре электрона из двух окисленных молекул
воды используются в фотосинтетическом электронном транспорте, тогда
как оставшиеся четыре протона высвобождаются во внутритилакоид-
ное пространство и участвуют в создании трансмембранного
протонного градиента. Молекула кислорода (02) из воды высвобождается в
атмосферу. Таким образом, во время фотосинтеза все фототрофы
(исключая анаэробные бактерии) выделяют кислород.
Способ, с помощью которого кислородобразующий комплекс
катализирует окисление воды — изъятие из нее электронов и
высвобождение кислорода, весьма интересен. Кислородобразующий комплекс
содержит кластер из четырех ионов марганца, характеризующийся
особым расположением, способствующим аккумуляции и последующему
поочередному переносу четырех электронов на окисленный Р680+. Как
только Мп-кластер сбрасывает четыре электрона, происходят
следующие процессы, сопряженные с расщеплением воды: 1) две молекулы
воды окисляются и их четыре электрона восстанавливают
Мп-кластер; 2) четыре протона высвобождаются во внутритилакоидное
пространство; 3) образуется одна молекула 02. Однако восстановление
Р680* происходит не только с помощью Мп-кластера, а сопряжено
также с процессами окисления-восстановления тирозинового остатка
белковой субъединицы реакционного центра, который принято
обозначать 2. Электронный перенос от 2 возвращает Р680* в его основное
состояние, благодаря чему он может быть легко возбужден снова.
Транспорт электронов через мембраносвязанные
фотосинтетические переносчики осуществляется в соответствии с разницей их восста-
399
Часть II. Метаболизм веществ и энергии в клетке
новительных потенциалов. Электронный перенос происходит спонтанно
от переносчиков, являющихся более сильными восстановителями, к
переносчикам, представляющим собой более сильные окислители. Тот
же самый принцип лежит в основе митохондриального электронного
транспорта (гл. 9). При фотосинтезе, однако, поглощение энергии
света временно снижает восстановительный потенциал первичного
донора; таким образом, Р680* и Р700* являются более сильными
восстановителями или донорами электронов, чем их исходные эквиваленты —
Р680 и Р700.
При графическом изображении серии фотосинтетических
компонентов электронного транспорта напротив шкалы восстановительных
потенциалов получается зигзагообразная фигура, называемая
^-схемой (рис. 10*6). На указанном рисунке также показаны электронные
переносчики между Р680 и Р700. Следует обратить внимание на
порядок электронного переноса, осуществляющегося от ФСП к ФС1, а не
от ФС1 к ФСП. (Терминология ФС1 и ФСП отражает
последовательность открытия фотосистем, а не порядок электронного переноса.)
Возбудившись, Р680* отдает электрон феофитину а (РЬа). Затем
происходит электронный перенос от восстановленного феофитина а к
пластохинону Р<3А, который прочно связан с ФСП. Подобно
митохондриальному убихинону (см. рис. 9.6), пластохинон может быть
восстановлен путем двух одноэлектронных переносов (рис. 10.7). В течение
двух последовательных фотохимических процессов Р(^А передает два
электрона путем одноэлектронного переноса второй молекуле пласто-
хинона Р<3В, обратимо связанной с ФСП. РС}В сначала принимает один
электрон от Р<5А и в результате протонирования превращается в семи-
хинол (т.е. пластосемихинол -Р(5ВН). Вследствие второго
фотохимического процесса (переноса второго электрона) пластосемихинол
восстанавливается и протонируется снова, образуя пластохинол (Р<3ВН2).
Полностью восстановленный, протонированный Р(3ВН2 затем
высвобождается в хиноновый пул. Длинные гидрофобные хвосты этих молекул
обеспечивают растворимость пластохинонового пула в тилакоидной
мембране.
Фотосинтетический (}-цикл, подобно митохондриальному (^-циклу
(гл. 9), осуществляет окисление восстановленного хинона. Окисление
Р(^Н2 происходит с помощью цитохромного комплекса &/. Этот
комплекс состоит из гемсодержащих цитохромов и железосерных белков и
аналогичен митохондриальному комплексу III (убихинол-цитохром с
оксидоредуктаза). Двухэтапный (5-цикл (рис* 10.8) приводит к
окислению двух молекул Р($Н2, восстановлению одной молекулы Р<5 до
РС}Н2, передаче двух электронов на пластоцианин путем
одноэлектронного переноса и транспортировке четырех протонов во внутрити-
лакоидное пространство. На первом этапе <Э-цикла Р<^Н2 окисляется в
центре, примыкающем к цитохрому Д и два протона из окисленного
Р<5 высвобождаются во внутритилакоидное пространство. Один из элек-
400
КЗ
л
СО
рэ
Р>
со
СО
00
"1,5
ФС I
"1>0
Я
-0,5 ~
я
о
н
о
с
3
я
А
Ф
Н
3
ш
о
2 +0,6
о
о
од
о-
+1,0-
+1,5-
Р700*
♦ \
ФС II
Р680
РЬа
Н20
Кислород-
образующий
комплекс
г-
Р9а
р^в
\
Комплекс
цитохромов Ь/
Протонный
градиент
Пластоцианин
А0
V
м
?х
ГдМАБР ®
оксидоредуктаза
ЫАБР
©
Р700
^Щ,
Свет
Р680
о2
Протонный
градиент
~^
Свет
л.
о
Рис. 10,6. 2-схема, называемая так в связи с ее характерным видом. Представляет собой модель, иллюстрирующую
восстановительные потенциалы компонентов цепи в сопряжении с потоком электронов через фотосинтетические электронные
переносчики- Сокращения: 2 — донор, отдающий электроны Р680; РЬа — феофитин а, акцептор электронов ФСП; РС^А — пластохинон,
прочно связанный с ФСП; Р(^в - обратимо связанный РС}, восстанавливаемый ФСП; Р<3 — пластохиноновый фонд, образуемый
Р(*> и Р^Н2; А0 — хлорофилл а, первичный электронный акцептор ФС1; Аг —
железосерные кластеры; Р<1 — ферредоксин
филлохинон (или витамин К); Г , Гй и Г
Часть II. Метаболизм веществ и энергии в клетке
НЯС
Н3С
НаС
НяС
+Н©
+е
НЯС
НЯС
Пластохинон (РЯ)
СНд
(СН2-СН=С-СН2)б-1оН
+н©
+еО Т
-н©
-еО
он
Пластосемихинол (*Р(ЗН)
сн3
о "
-н©
он
Плаетохинол (РС^ЭД)
СНз
2~СН^С—СН2)б-1 ()Н
он
Рис. 10.7. Восстановление плас-
тохинона до пластохинола путем
двух последовательных одноэлект-
ронных переносов
тронов, образующийся в результате
окисления Р(Щ2, проходит через
цитохром / и восстанавливает пластоци-
анин. Второй электрон плюс один
протон из стромы превращают молекулу
Р<3 в РС^Нв центре, удаленном от
места окисления Р<ЗН2. На втором
этапе окисляется вторая молекула
Р<ЗН2. Как только вновь один
электрон восстанавливает пластоцианин,
другой электрон плюс один протон
из стромы превращают Р<ЗН в Р(^Н2.
Полностью восстановленный Р(^Н2
затем высвобождается в пластохино-
новый пул. Поскольку цитохромный
комплекс 6/ может акцептировать
только электроны, но не протоны от
Р<^Н2, то данный циклический
процесс, осуществляемый с помощью
пластохинона и цитохромного
комплекса 6/ служит в качестве
протонной помпы, создавая протондви-
жущую силу, которая необходима
для синтеза АТР,
Первый
этап
Строма
2Н®
Внутритилакоидное
пространство
Второй
утап
Строма
Пластоцианин
Внутритилакоидное
пространство
Пластоцианин
Рис, 10.8. Фотосинтетический С^-цикл. Это двухэтапный процесс. На первом
этапе Р(^Н2 окисляется до Р(^. В этом процессе происходит высвобождение двух
протонов во внутренний компартмент тилакоида: один электрон передается на
пластоцианин через цитохром Д а другой вместе с протоном из стромы превращает молекулу
Р(} в 'РС^Н. На втором этапе окисляется вторая молекула Р(}Н2. Снова два протона
поступают во внутритилакоидное пространство; один электрон передается на
пластоцианин, а другой восстанавливает -Р(ЗН до Р<ЗН2, который затем высвобождается
в пластохиноновый пул. Два этапа вместе обеспечивают окисление двух молекул
РС^Н2, восстановление одной молекулы Р(} до РСЩ2, перенос двух электронов на
пластоцианин и перенос четырех протонов во внутритилакоидное пространство
402
Глава 10. Биосинтез углеводов
Пластоцианин представляет собой небольшой медьсодержащий
белок, который восстанавливается цитохромом / цитохромного
комплекса Ъ? и окисляется Р700* ФС1. Перенос электрона через
пластоцианин сопряжен с изменениями степени окисления его атома меди. Фонд
пластоцианина очень маленький (менее пяти молекул в расчете на
Р700). Эти молекулы пластоцианина перемещаются латерально в ти-
лакоидной мембране, перенося электроны между цитохромным
комплексом Ь? и Р700.
Как только Р700+ восстанавливается до Р700 с помощью
пластоцианина, он может энергезироваться, превращаясь в Р700*, за счет
возбуждения, передаваемого от антенных пигментов, ССК, связанного
с ФС1, или, при определенных условиях, подвижной фракции ССК,
Следует обратить внимание на то, что Р700*, имеющий
восстановительный потенциал около -1,3 В, является самым сильным
восстановителем в цепи переносчиков электронов. Электрон от Р700* легко
передается на электронный акцептор хлорофилла а, известный как
А0. Восстановленная форма, А~, отдает свой электрон молекуле филло-
хинона (витамин Кх), известному как Аг От Ах электрон передается на
серию железосерных кластеров — сначала на Гх и затем или на РА, или
на Гв. Гх представляет собой часть реакционного центра ФС1, тогда как
Рл и Рв локализованы на периферическом полипептиде, расположенном
на стороне тилакоидной мембраны, обращенной к строме* Из железосер-
ного кластера (РА или Рв) электроны переносятся на железосерный
белок — ферредоксин (Гй). Ферредоксин — это низкомолекулярный
небольшой водорастворимый белок, локализованный в строме
хлоропласта. Восстановительный потенциал ферредоксина (Е° = -0,43 В)
позволяет ему легко восстанавливать КАВР* до ЫАОРН (Е° — -0,32 В).
Восстановление КАВР+ с помощью восстановленного ферредоксина
катализируется ферредоксин-ЫАОР+~оксидоредуктазой, Данный фермент слабо
связан со стороной тилакоидной мембраны, обращенной к строме. Окси-
доредуктаза содержит простетическую группу РАВ, которая
восстанавливается до РАОН2 за счет восстановленного ферредоксина путем двух
одноэлектронных переносов. ЕАЮН2, в свою очередь, восстанавливает
МАХ)Р+, отдавая два электрона и протон в форме гидридиона. На этом
этапе возрастает разница значений рН по обе стороны тилакоидной мем-
браны, так как протоны стромы используются для превращения КАВР+
в ЫАОРН. Таким образом, путь нециклического электронного
транспорта завершается образованием ЫАОРН.
Распределение протонов между стромой и внутритилакоидным
пространством изменяется в ходе трех различных этапов
фотосинтетического электронного транспорта. Во-первых, протоны из воды
высвобождаются во внутритилакоидное пространство с помощью кислород-
образующего центра. Во-вторых, протоны стромы транслоцируются
во внутренний компартмент тилакоидов во время окисления РС^Н2
цитохромным комплексом &/. В-третьих, поглощение протонов в ходе
26*
403
Часть //. Метаболизм веществ и энергии в клетке ___^^_
восстановления КАОР+ снижает их концентрацию в строме. В то
время как протоны перемещаются из стромы во внутритилакоидное
пространство, ионы магния движутся через тилакоидную мембрану из
внутритилакоидного пространства в строму, в результате чего
сохраняется баланс заряда между двумя компартментами. Хлорид-ионы
транслоцируются во внутренний компартмент тилакоидов для
компенсации более высокой концентрации электронов в строме,
возникающей в результате электронного транспорта. При наличии
трансмембранного градиента концентрации протонов локализованная в
мембране АТР-синтетаза катализирует образование АТР из АОР и Р..
Поскольку этот процесс в растениях имеет светозависимый характер, то
фотосинтетическое образование АТР называется фотофосфорилирова-
нием. АТР-синтетаза хлоропластов представляет собой мультибелко-
вый комплекс, который подобно митохондриальной синтетазе состоит
из двух основных частей — СР0 и СРГ (Обозначения СР0 и СР%
отличают компоненты хлоропластной АТР-синтетазы от соответствующих ми-
тохондриальных компонентов, Р0 и Рх.) СГ0-частица пронизывает
тилакоидную мембрану и образует канал для протонов. СР1 выступает в
строму и катализирует образование АТР из АВР и Р.. Хлоропластная
АТР-синтетаза локализована исключительно в ламеллах стромы.
Циклический поток электронов. Установлено, что в расчете на
каждую пару электронов, транспортируемых для восстановления
одной молекулы КА1)Р+ до ЫАВРН, создается сила, движущая протоны
через тилакоидную мембрану, достаточная для синтеза одной
молекулы АТР. Стехиометрия "один к одному" (АТР: КАВРН), характерная
для световых реакций, могла бы создавать дисбаланс со
стехиометрией образующих углеводы темновых реакций, поскольку две молекулы
КАВРН и три молекулы АТР используются при восстановлении
одной молекулы С02 до углевода. В условиях, когда сопряжение между
световыми и темновыми реакциями становится несбалансированным,
функционирует модифицированный циклический путь электронного
транспорта, выполняющий компенсаторную роль и обеспечивающий
синтез АТР без образования КАОРН. Эта модифицированная
последовательность реакций называется циклический электронный
транспорт. Как и при нециклическом электронном транспорте, электроны
в данном случае передаются через С$-цикл от пластохинона на цито-
хромный комплекс Ь/ и затем на ФС1 и на ферредоксин. Однако при
циклическом электронном транспорте растворимый ферредоксин
отдает свой электрон обратно в фонд РС^ в ходе процесса, протекающего
с участием цитохромного ^/-комплекса через цитохром, который
вовлечен в функционирование только циклического электронного
переноса. Через С$-цикл циклический электронный транспорт дополняет про-
тондвижущую силу, генерируемую в течение нециклического
электронного переноса и, таким образом, увеличивает образование АТР без
сопутствующего продуцирования МАВРН.
404
Глава 10. Биосинтез углеводов
В целом, относительные скорости циклического и нециклического
электронных потоков определяются относительными количествами
ХАПРН и КАВР+. Когда в строме отношение МАПРН к ЫАОР*
высокое, скорость нециклического электронного транспорта
лимитирована вследствие низкой доступности КАОР+. При таких обстоятельствах
циклический электронный перенос преобладает.
10,8. Темновая стадия фотосинтеза.
Взаимопревращения моносахаридов
Ассимиляция С02 в углеводы в реакциях восстановительного
пентозофосфатного цикла. Второй основной фазой фотосинтеза
является превращение диоксида углерода в углеводы — процесс, кото*
рый осуществляется за счет АТР и ЫАВРН, образующихся в течение
световых реакций фотосинтеза. Образование углеводов происходит в
строме хлоропластов в результате цикла каталитических реакций,
которые приводят к трем важнейшим последствиям: 1) фиксация
атмосферного С02; 2) восстановление С02 до углевода; 3) регенерация
молекулы, акцептирующей С02. Метаболический путь, ведущий к
ассимиляции углерода, имеет несколько названий, включая:
восстановительный пентозофосфатный цикл; С3-дуть (указывающее, что.
первый интермедиат этого пути представляет собой трех углеродное
соединение); цикл Кальвина, или Кальвина—Бенсона*
Основу представлений о том, как из одноуглеродного глубокоокис-
ленного соединения — С02 образуется шестиуглеродное
восстановленное соединение — глюкоза, аккумулирующее в себе химическую
энергию из продуктов световой стадии фотосинтеза ~ ЫАОРН и АТР, дали
исследования М. Кальвина и его сотрудников. В своей работе они
использовали одноклеточную зеленую водоросль СМогеИа, биомасса
которой воспроизводится с высокой скоростью. Эксперименты
проводили с помощью меченого углерода (14С); радиоактивный 14С02 вводили
в освещенную суспензию водоросли. По прошествии определенного
времени водоросль убивали, вливая суспензию в кипящий этанол, что
быстро подавляло все ферментативные реакции.
Меченые по углероду соединения водоросли разделяли и
идентифицировали методом двухмерной хроматографии на бумаге. После
кратковременного освещения (5 с) был обнаружен только 3-фосфогли-
церат, на основании чего был сделан вывод о том, что 3-фосфоглице-
рат — первый стабильный продукт фиксации С02« Чем
продолжительнее была световая экспозиция, тем разнообразнее был набор
^-соединений* Кальвин и сотрудники пришли к мнению о существовании
сложного циклического механизма фиксации С02.
Рубиско — фермент, катализирующий первоначальный этап
восстановительного пентозофосфатного цикла. Фиксация
газообразного С02 в органический продукт совершается на первом этапе вос-
405
Часть II. Метаболизм веществ и энергии в клетке
становительного пентозофосфатного цикла. Эта реакция катализируется
рибулозо-1,5-6'шгфосфаткарбоксилазой-оксигеназой, сокращенно
называемой рубиско. На долю рубиско приходится около 50 % растворимого
белка в листьях растений. Хотя концентрация рубиско в строме может
быть вьппе 4 мМ, этот фермент столь строго регулируем, что активность
рубиско может лимитировать скорость ассимиляции углерода.
Рубиско растений, водорослей и цианобактерий состоит из восьми
больших субъединиц (молекулярная масса каждой 56 000) и восьми
малых субъединиц (молекулярная масса каждой 14 000) с общей
молекулярной массой около 560 000. Эта композиция обозначается Ь888, Ассо-
циат из восьми больших субъединиц формирует сердцевину молекулы.
Для катализа фиксации С02 рубиско должна быть в активном
состоянии. Для активации рубиско необходимы С02, М§2+ и
определенное значение рН стромы. На свету активность рубиско возрастает в
ответ на повышение величины рН и концентрации ионов М§2+,
обусловленных транслокацией протонов. Молекула С02, необходимая для
активирования рубиско, реагирует с лизиновым остатком (Ьуз-202)
участка полипептидной цепи, удаленного от активного центра
фермента. Помимо активации С02, М^2+ и рН, рубиско также
активируется непосредственно светом.
Субстратами реакции, катализируемой рубиско, являются свободная
молекула С02 (но не С02, связанная с лизиновым остатком) и пятиугле-
родный фосфорилированный сахар рибулозо-1,5-бысфосфат. Реакция
завершается образованием двух молекул 3-фосфоглицерата.
СН2ОР
с=о
I
н-с-он
н-с-он
СН2ОР
В-Рибулозо-1,5-
бисфосф&т:
(Ру-1,5-БФ)
СШОР
с-он
II
с-он
н-с-он
СН2ОР
(Ру-1,5-БФ)
(енольная
форма)
СНаОР
НООС-С-ОН
1
С=0
Н-С-ОН
СН2ОР
СН2ОР
Н-С-ОН
СОО"
+
СОО"
Н-С-ОН
СН2ОР
Э- З-Фосфогл ицерат,
две молекулы
Это сложная реакция; рибулозо-1,5-6'исфосфат (РуБФ) сначала
переходит в енольную форму, которая затем карбоксилируется по
второму атому углерода с образованием промежуточного продукта — р-кето-
кислоты, которая затем распадается на две молекулы 3-фосфоглицерата.
Процессы восстановления углерода и регенерации молекулы,
акцептирующей СОг. Рис. 10.9. иллюстрирует все реакции
восстановительного пентозофосфатного цикла. Следует обратить внимание, что
представлены этапы ассимиляции не одной, а трех молекул диоксида
углерода. Это связано с тем, что наименьшим углеродным интермедиатом вос-
406
зсо2
ЗНаО 611®
Рубиско
СОО
(6) Н-С-ОН
6АОЗ
СНаОР03® Фосфоглицерат-
6АТР
V
киназа
(6) Н-С-ОН
сн2оро3®
бИАОРН + 6Н
6Р
Глицеральдегид-3-фосфат-
дегидрогсиаза
3-Фосфоглииерат
1,3-Яысфосфог ли дерат
Т р но з офосфат
(глицеральдегид-3-фосфат
или дигидроксиацетонфосфат)
(о) ^с^
Н-С-ОН
СН2ОН
СН2ОРОз"
Рибулозо-1,5-
бисфосфат
ЗАОР
ЗАТР.
03
г
X
о
I
СН2ОН
Рибулоэо-5- С=0
<?Н«°Н 1иврШа НО-С-Н
НС ОН
1
с=о
но-с-н
I
СНгОРО^
н2о с=*о
но- с н
&
СН^ОРОй
©
Альдолаза
с=о
н-с-он
I
Н-С-ОН
I
СН2ОРОз
Рибулозо-5-
фосфат
©
скорой
Ксилулозо-5-
фосфат
©
Н-С-ОН Фруктозой ,6. Н-С-ОН
1 дггсфосфа-
таза
Н-С-ОН
СН2ОР03®
Фруктозо-6-
фосфат
Н-С-ОН
СН20Р03
Фруктозо-1 ,6-
бисфосфат
©
Глицеральдегид-3-фосфат
Тркозск
фосфат-
изомвраза
СН2ОН
С=0
1
СНгОН
с«о
©
СН2ОИ
I
с=о
I
Н-С-ОН
Н-С-ОН
Рибулозо-5-
фосфат-3-
эпимараза
Т ране кето лаза
СНзОН
I
С-О
о н
V
I
н-с-он
I
н-с-он
СН2ОРО^ СН2ОРОэч
Дигидрокси- Дигидрокси*
ацетокфосфат ацвтонфосфат
<э
СН2ОРО*
Эритро зо - 4-фосфа
е
т
Ал ьд о л аз а
СНаОРО^
©
СН2ОРО;>4
Рибулозо-5-
фосфат
О
но с н
н-с-ои
н9о
СИ2ОН
СН2ОРО^
Ксилулоэо-5-
фосфат
©
С=0 Рибулозо-5-фосфат-
Н-С-ОН л изомараза
н-с-он
<э
о н
у
н-с-он
I
н-с-он
н-с-он
с=о
I
но-с-н
I
н-с-он
I С ед огеххту л озо-I» 7 -
Н-С—ОН бифосфатаза Н—С-ОН
X
Н-С-ОН
СН2ОРОа®
Седоголтулозо-1,7-
Йысфосфат
Транскетолаза
Н-С-ОН
I
СН2ОРО.}
Седогептулоэо-7-
фосфат
.©
СН2ОРОз
Рибулозо-5-фосфат
СНаОРОз®
Рибулозо-5-фосфат
4^
О
Рис. 10.9. Восстановительный пентозофосфатный цикл. Включает три стадии: фиксацию С02, восстановление углерода до
(СН20) и регенерацию С02-акцептирующей молекулы. Концентрация интермедиатов восстановительного пентозофосфатного цикла
сохраняется за счет того, что одна молекула триозофосфата (глицеральдегид-3-фосфата или дигидроксиацетонфосфата) выходит
из цикла после того, как фиксируются три молекулы С02
о
о
с
со
Си
о
о
0)
Часть II. Метаболизм веществ и энергии в клетке
становительного пентозофосфатного пути является С3-молекула. Таким
образом, должны быть фиксированы три молекулы С02 до того, как одна
С3-единица может быть удалена из цикла без уменьшения объема фондов
интермедиатов восстановительного пентозофосфатного цикла*
Восстановительная фаза С3-пути осуществляется в течение двух
реакций, следующих за реакцией, катализируемой рубиско. Сначала,
3-фосфоглицерат превращается в 1,3-<шсфосфоглицерат в АТР-зави-
симой реакции, катализируемой фосфоглицераткиназой. Затем
1,3-#исфосфоглицерат восстанавливается с помощью ЫАВРН в
реакции, катализируемой глицеральдегид-3-фосфатдегидрогеназой.
Реакцию изомеризации между глицеральдегид-3-фосфатом и дигидрокси-
ацетонфосфатом осуществляет триозофосфатизомераза:
соон <х
Н-С-ОН + АТР - » Н-С-ОН + АВР
СН2ОР СН2ОР
З-Фосфоглицерат 1,3 -Бисфосфоглицерат
(1,3-БФГК)
*° ^°
*рОР *рН
н-с-он _, н-с-он
| + ЫАВРН +Н+ « »' | + ИАВР+ + ?1
СН2ОР СН2ОР
1,3-Бисфосфоглицерат 3-Фосфоглицеральдегид
(3-ФГА)
Последовательность реакций, протекающих после
восстановительной фазы цикла, завершается регенерацией молекулы,
акцептирующей молекулу С02 — рибулозо-1,5-б^сфосфата. Большинство реакций
восстановительного пентозофосфатного цикла сопряжено с
регенерирующей фазой. Глицеральдегид-3-фосфат направляется по трем
разным ветвям данного пути и испытывает взаимопревращения между
4-, 5-, 6- и 7-углеродными фосфорилированными сахарами. В
регенерирующей фазе функционируют различные изомеразы, эпимеразы, аль-
долазы, фосфатазы и транскетолаза. Последний этап — фосфорилиро-
вание рибулозо-5-фосфата с помощью АТР с образованием рибулозо-
1,5-#исфосфата в реакции, катализируемой фосфорибулокиназой.
По первому пути 3-ФГА изомеризуется в дигидроксиацетонфосфат
(ДАФ); эту реакцию катализирует фермент триозофосфатизомераза:
с__ сн2он
[
н-с-он ^ оо
СН2ОР СН2ОР
З-Фосфоглицеральдегид Дигидроксиацетон.
(3-ФГА) фосфат (ДАФ)
408
Глава 10, Биосинтез углеводов
По второму пути 3-ФГА и ДАФ образуют фруктозо-1,6~о*исфосфат
(Фру-1,6-БФ):
СН2ОР
с=о
сн2он
+
сно
н-с-он
СН2ОР
СН2ОР
!
с=о
т но-с-н
н-с-он
I
н-с-он
I
СН2ОР
В-фруктозо~1,6-#исфосфа,г
(Фру-1,6-БФ)
Реакцию катализирует фруктозобисфосфатальдолаза. Она
осуществляет альдольную конденсацию между 3-ФГА и ДАФ.
Фру-БФ в цикле Кальвина дефосфорилируется с образованием фрук-
тозо-б-фосфата (Фру-Ф); эта реакция катализируется фруктозобисфос-
фатазой:
Фру-1,6-БФ + Н20 # Фру-6-Ф + Р..
Таким образом, второй путь — это путь образования гексоз.
Поскольку последние являются структурными блоками (мономерами) в
биосинтезе большинства олиго- и полисахаридов в растительном мире,
этот путь — основной (генеральный) путь биосинтеза органической
массы на Земле.
По третьему пути 3-ФГА реагирует с Фру-1-Ф, в результате
получаются Б-ксилулозо-5-фосфат (Ксу-5-Ф) и эритрозо-4 -фосфат (Э-4-Ф);
реакцию катализирует транскетолаза, активная только в присутствии
тиаминпирофосфата (ТПФ) и ионов М§2+; она переносит двухуглерод-
ный фрагмент (СН2ОН-СО-) с кетозы на альдозу:
СН2ОН
но-с-н
I .1
н-с-он
н-с-он
СН2ОР
Фру-6-Ф
+
СНО
Н-С-ОН
СН2ОР
3-ФГА
СНоОН
с=о
НО-С-Н
н-с~он
СШОР
+
СНО
н-с-он
н-с-он
СНоОР
О-ксилулозо-5- О-эритрозо-4-
фосфат (Ксу-5-Ф) фосфат (Э-4-Ф)
Э-4-Ф реагирует затем с ДАФ с образованием семиуглеродного
соединения — В-седогептулозо-1,7-бисфосфата (Су-1,7-БФ):
409
Часть П. Метаболизм веществ и энергии в клетке
СН>ОР
сно <^0
Н-С-ОН + СНоОН
СНоОР
с=о
но-с-н
н-с-он
н-с-он н-с-он
СН2ОР Н-С-ОН
г I
СН2ОР
О - се д огепту л озо-1,7-
бисфосфат (Су-1,7-БФ)
После этого Су-1,7-БФ дефосфорилируется под действием
специфической фосфорилазы до седогептулозо-7-фосфата (Су-7-Ф):
ме2*
Су-1,7-БФ + Н20 ?± Су-7-Ф + Р..
При участии специфической транскетолазы Су-7-Ф реагирует с
3-ФГА с образованием Ксу-5-Ф и Б-рибозо-б-фосфата (Р-5-Ф):
Су-7-Ф + ЗФГА ^ Р-5-Ф + Ксу-5-Ф.
Затем Ксу-5-Ф, а также Р-5-Ф при участии специфической эпиме-
разы превращаются в Б-рибулозо-5-фосфат (Ру-5-Ф), который затем
фосфорилируется за счет фотосинтезированного АТР с образованием
Ру-1,5-БФ:
СН2ОН СН2ОР
с=о с=о
н-с-он —;т*—с::т^ н-с-он
Н-С-ОН АТР АОР Н-С-ОН
СН2ОР СН2ОР
Ру-5-Ф Ру-1,5-БФ
Реакцию осуществляет фермент фосфорибулозокиназа. С
получением Ру-1,5-БФ цикл Кальвина замыкается.
Таким образом, третий путь — это, собственно, путь регенерации
акцептора С02 — рибулозо-1,5-0ш:фосфата.
После трех оборотов данного пути (т.е. после того, как три
молекулы С02 будут фиксированы) один триозофосфат может быть удален из
цикла. Оставшиеся пять триозофосфатов используются для
регенерации трех молекул рибулозо-1,5-бысфосфата. Такая стехиометрия
способствует поддержанию необходимого уровня интермедиатов данного
метаболического пути.
Суммируя все реакции третьего пути, получим:
410
Глава 10. Биосинтез углеводов
Фру-6-Ф + 2(3~ФГА) + 2ДАФ + ЗАТР -» ЗРу-1,5-БФ + ЗАВР
Из всех реакций цикла Кальвина только две специфичны для
зеленых растений: 1) акцептирование С02 рибулозо~1,5-#ш:фосфатом и
быстрое преобразование шестиуглеродистого соединения в 3-фоефо-
глицериновую кислоту; 2) фосфорилирование рибулозо-5-фосфата в
рибулозо-1,5-<жсфосфат. Остальные реакции идут и в тех клетках, где
фотосинтеза нет. Многие из этих реакций являются обратимыми и
имеют место при брожении и дыхании.
Как видно из цикла Кальвина, на каждую ассимилированную
молекулу С02 затрачивается 2 молекулы ЫАОРН и три молекулы АТР.
Поскольку на фотосинтез одной молекулы фруктозо-6-фосфата
затрачивается 6 молекул С02, то необходимо, следовательно, использовать
12 молекул МАВРН и 18 молекул АТР. Суммарную реакцию
фотосинтеза можно записать так:
6С02 + 12КАСРН + 18АТР + 11Н20 -> Фруктозо-6-фосфат +
+ 12NА^Р+ + 18АБР + 17Р..
Чтобы разобраться, каким образом в цикле Кальвина
осуществляется основной путь — синтез гексоз, рассмотрим стехиометрию
реакций, протекающих в нем.
Введем в цикл Кальвина 6 молекул С02, для чего потребуется 6
молекул рибулозо-1,5-б#сфосфата. Это дает возможность получить 12
молекул 3-фосфоглицерата. Последние, в свою очередь фосфорилируясь,
образуют 12 молекул 1,3-#&сфосфоглицерата, а затем они
восстанавливаются в 12 молекул 3-фосфоглицеринового альдегида.
Напомним: в цикле Кальвина 3-фосфоглицериновый альдегид
используется тремя различными путями. По первому — 5 из 12 молекул
3-фосфоглицеринового альдегида изомеризуются в 5 молекул дигидр-
оксиацетонфосфата; по второму — 3 молекулы 3-фосфоглицеринового
альдегида конденсируются с 3 из 5 молекул дигидроксиацетонфосфа-
та, в результате чего получаются 3 молекулы фруктозо- 1,6-б#сфосфа-
та, которые, дефосфорилируясь, превращаются в 3 молекулы фрукто-
зо-6-фосфата. После реализации второго пути остаются четыре
молекулы 3-фосфоглицеринового альдегида и две молекулы дигидрокси-
ацетонфосфата.
Если теперь 6 молекул С02 в виде фруктозо-6-фосфата основного пути
направить на синтез и превращение углеводов, то для третьего пути —■
пути регенерации рибулозо-1,5-#исфосфата — останутся 2 молекулы
фруктозо-б-фосфата, 4 молекулы 3-фосфороглицеринового альдегида и
2 молекулы дигидроксиацетонфосфата, в совокупности содержащие 30
атомов углерода. Для замыкания цикла должно быть регенерировано 6
молекул рибулозо-1,5-#ш:фосфата, в сумме содержащих 30 атомов
углерода. Ясно, что из оставшихся соединений это осуществить вполне
возможно. Третий путь цикла Кальвина направлен именно на распределение
этих атомов углерода в молекулах рибулозо-5-фосфата.
411
Часть II. Метаболизм веществ и энергии в клетке
Приведенный выше цикл Кальвина свидетельствует о
чрезвычайной легкости, с которой происходит взаимное превращение моноз,
крайними членами которого являются триозы и гептозы; важной
формой моносахаридов в этих превращениях служат фосфорные эфиры
моноз — соединения, характерной чертой которых является высокий
уровень свободной химической энергии.
Помимо фосфорных эфиров моносахаридов важную роль во взаи-
мопревращениях играет второй тип производных моносахаридов —
нуклеозиддифосфатсахара (МБР-сахара).
В ЫБР-сахарах сахар соединен гликозидной связью с концевым
фосфатным остатком нуклеозиддифосфата. В качестве примера
таких производных Сахаров можно назвать уридиндифосфатглюкозу
(ИВР-Б-глюкозу):
о о
II I!
СН2-0-Р-0-Р-0
О" О"
он н
он он
Уридиндифосфатглюкоза (ШЭР-глюкоза)
Известно множество таких соединений, в которых различные са-
хариды присоединяются чаще всего к уридин-, цитидин-, аденозин- и
гуанозиндифосфатам (1ГОР, СОР, АВР, ОБР). Синтез МВР-сахаров
осуществляется через их 1-фосфаты с участием нуклеозидтрифосфа-
тов (ЫТР):
Моносахарид-1-фосфат + ЫТР <^ ИБР-сахар + РРГ
Выделившийся РР1 (пирофосфат) происходит из двух конечных
остатков ЫТР; реакция осуществляется под действием ферментов пи-
рофосфори лаз.
Константа равновесия этой реакции равна примерно 1;
следовательно, эта реакция легко обратима. Однако в клетке она сдвинута в
сторону синтеза МБР-сахара, поскольку происходит непрерывное
удаление РР, под действием фермента пирофосфатазы. ДО0' гидролиза
МБР-сахара на ЫВР и сахар составляет около -30 кДж • моль"1 в
отличие от -20 кДж-моль"1 для гидролиза 1-фосфорного эфира
моносахарида. Таким образом, ЫБР-сахара более активны в качестве доноров
гликозильных остатков, чем фосфорные эфиры моносахаридов; они
широко используются для синтеза олиго- и полисахаридов.
Фосфорилирование моносахаридов, синтез МБР-сахаров, их эпи~
меризация тесно взаимосвязаны между собой, что можно видеть на
примере взаимопревращения галактозы и глюкозы:
412
Глава 10. Биосинтез углеводов
ИВР-глюкозо-эпимераза
Щ)Р-0~галактоза -« : ^ Щ)Р-0~ глюкоза
I
ООР-галактозопирофосфорилаза
РР! 11ТР
_. Гоксокиназа ...
и-галактоза —^—=^——*- и-галактозо-1-фосфат
' А
АТР АБР
Аналогичные превращения происходят с фруктозой, глюкозой,
маннозой, а также с пентозами.
Следовательно, образовавшийся в цикле Кальвина фруктозо-б-фое-
фат может легко превращаться в глюкозу, галактозу, маннозу.
Важную роль во взаимопревращениях моносахаридов играет
окисление альдогексоз с образованием уроновых кислот, в которых первичная
спиртовая группа у шестого углеродного атома превращается в
карбоксильную группу* Из глюкозы в этом случае образуется глюкуроновая
кислота, из галактозы — галактуроновая, из маннозы — маннуроновая.
В эту реакцию вовлекаются ЫОР-гексозы. Так, 1ГОР-глюкоза при
окислении по шестому углеродному атому под действием "ОВР-глюкозодегид-
рогеназы превращается в ЦТ)Р-глюкуроновую кислоту, а последняя, де-
карбоксилируясь, дает производную пентозы — Ш)Р-ксилозу:
СН2ОН СООН
2ИАВ+ 2МАБН
"СШР-глюкоза 1ШР- глюкуроновая
кислота
Н
Декарбоксилаза I / Л
НО
со*
он н
ИВР-ксилоза
Подобным же путем 1ГОР-галактоза превращается в Щ)Р~галакту-
роновую кислоту, а из последней образуется производная другой
пентозы — 1ЮР-арабиноза.
В растениях найдены эпимеразы, катализирующие
взаимопревращения 1ГОР-уроновых кислот:
1ГОР-глюкуроновая кислота ^ 1ШР-галактуроновая кислота.
413
Часть II. Метаболизм веществ и энергии в клетке |
Аналогичным образом ЦТ)Р-манноза окисляется в 11ВР-маннуро-
новую кислоту, которая претерпевает эпимеризацию у четвертого
углеродного атома и превращается в Х1ВР-гулуроновую кислоту.
Легко образующиеся в растениях КВР-уроновые кислоты имеют
важное значение не только в процессах образования пентоз. Они
входят в состав пектиновых веществ и других полисахаридов,
получивших название полиуронидов.
10.9- Оксигеназная активность рубиско
и ее снижение при функционировании С4-пути
Оксигенация рибулозо-1,5-бисфосфата под действием рубиско.
Рибулозо-1,5-#исфосфат карбоксилаза-оксигеназа катализирует как
карбоксилирование, так и оксигенирование рибулозо-1,5-^цсфосфата.
Эти две реакции носят конкурентный характер — С02 и 02
конкурируют за активные центры рубиско. Кажущееся сродство рубиско по
отношению к С02 значительно больше, чем для кислорода, и С02
более растворим в строме, чем 02, но атмосферная концентрация
кислорода (21 %) значительно выше, чем диоксида углерода (0,03 %). В
результате обе реакции вносят значительный вклад в потребление
рибулозо-5-фосфата гп рй>о. При нормальных условиях отношение
карбоксилирования к оксигенированию — от 3:1 до 4:1.
Продуктами реакции оксигенирования, катализируемой рубиско,
являются одна молекула З-фосфоглицерата и одна молекула
фосфогликолата. На рис* 10.10 показан результат двух процессов
оксигенирования. Две молекулы З-фосфоглицерата вступают непосредственно
в восстановительный пентозофосфатный цикл, а две молекулы 2-фос-
фогликолата метаболизируются до одной молекулы С02 и одной
молекулы З-фосфоглицерата, которая вступает опять же в С3-путь.
Процессы потребления кислорода рубиско и метаболизма фосфогликолата
принято называть фотодыханием. Таким образом, при фотодыхании
происходит светозависимое .поглощение кислорода и выделение С02.
Метаболизм фосфогликолата осуществляется в трех различных
клеточных компартментах — хлоропласте, пероксисоме и митохондрии.
Фосфогликолат дефосфорилируется до гликолата в хлоропласте; глико-
лат затем входит в пероксисому, где он реагирует с 02, образуя глиокси-
лат и пероксид водорода. Пероксид водорода легко превращается в 02 и
Н20 с помощью фермента катал азы, и глиоксилат в ходе трансаминиро-
вания с глутаматом дает глицин. Глицин выходит из пероксисомы и
входит в митохондрию, где он может подвергаться окислительному де-
карбоксилированию и расщепляться до 5,10-метилентетрагидрофолата,
С02 и МН3. С02 или фотосинтетически рефиксируется, или
высвобождается из листа, а ЫН3 реассимилируется, образуя глутамат. 5,10-метилен-
тетрагидрофолат отдает свою метиленовую группу второй молекуле
глицина (также происходящую из глиоксилата), в результате чего образует-
ся серии. Серии выходит из митохондрии и входит в пероксисому, где он
414
Глава 10. Биосинтез углеводов
Н2С-ОРО^
I
с=о
н-с-он
н-с-он
Н2С-ОР03®
(2) Рибулоэо-1,5-
бифосфат
_С
Хлоропласт
2С02
ххР
.©
Рубиско
СОО
I
Н-С-ОН
I
н2с-он
Глицерат
(2) З-Фосфоглицерат
Восстановительный
пентозофосфатны й
цикл
АВР СОО°
\-Уг Н-С-ОН
Глицерат- Н?С-ОРО^
Г
Н2С-ОРОач
(2) Фосфогликолат
НоО
\
У
Фосфогли*
колатфосфа-
таза
АТР
киназа
З-Фосфоглицерат
СОО©
Н2С-ОН
(2) Гликолат
СОО
О
Пероксисома
Н-С-ОН
н2с-он
Глицерат
СО(Р
Н2С-ОН
(2) Гликолат
Гидрокси-
пируват-
редуктаза
ч
ыаеР
МАЛИ + Н®
02
Т Каталаза
г
202
Н202
Гликолат-
оксидаза
соо4-'
с=о
н2с-он
Гидроксипируват
н2о
СОО°
о=с-н
(2) Глиоксилат
^
Глутамат
Транс- Транс-
аминаза аминааа.
(^— а-Кетоглутарат
СО
<Р
н
,^-с-н
н2с-он
Серии
сосР
н3#-с-н
н
Глицин
/<*~~ Глутамат ■<-
' Транс-
^аминаза
I —►а-Кетоглутарат
сод3
н8ьР-с-н
и
Глицин
Нз^С-Н
н2с-он
Серия
РО^Н) Серингидроксил-
метилтрансфераза
ххР
т
Н20
НзГ^-С-Н
I
н
Глицин
Тетрагидро-
сосР
Нз^-С-Н
н
Глицин
Митохондрия
Н*0
фолат ^
т Глицин-
декарбоксилаэа
5,10-Метилентетрагидрофолат <
СО.
а-Кетоглутарат
Глутамат
Рис. 10.10. Реакции фотодыхания
415
Часть П. Метаболизм веществ и энергии в клетке ^___^^^
подвергается деаминированию до гидроксипирувата; гидроксипируват
затем восстанавливается до глицерата. Глицерат входит в хлоропласт,
где он фосфорилируется с помощью АТР до 3-фосфоглицерата, который
может включаться в восстановительный пентозофосфатный цикл.
Снижение оксигеназной активности рубиско при
функционировании С4-пути* В некоторых видах растений совместно с
восстановительным пентозофосфатным циклом функционирует другой путь
фиксации углерода. В данном метаболическом пути первоначальным
продуктом фиксации углерода является четырехуглеродное
соединение, и в этот процесс вовлечены два различных типа клеток.
Поскольку первоначальным продуктом фиксации С02 является С4-кислота, то
метаболический путь называется С4путем> тогда как, напомним,
восстановительный пентозофосфатный цикл также называется С3-путем.
К С4-растениям относятся такие экономически важные виды, как
кукуруза, сорго, сахарный тростник, а также многие из сорных
растений. Растения, в которых функционирует С4~дуть, по существу, не
обладают фотодыхательной активностью.
С4-путь, который может рассматриваться как прелюдия С3-пути,
имеет две фазы: карбоксилирования и декарбоксилирования. На
первоначальной фазе С02 фиксируется в виде Отсоединений в клетках
мезофилла (обычное место протекания восстановительного пентозофос-
фатного цикла в С3-растениях). Во второй фазе (^-соединения декар-
боксилируются в клетках обкладки, где С02 высвобождается, рефик-
сируется рубиско и включается в восстановительный
пентозофосфатный цикл. Рис. 10.11 иллюстрирует особенности клеточной
организации листьев С3- и С4-растений. Две важнейшие структурные
особенности, взаимосвязанные с ассимиляцией углерода, отличают листья С4
от С3-типа растений. Во-первых, в листьях С4~растений клетки
мезофилла полностью окружают клетки обкладки, которые, в свою
очередь, окружают васкулярную (сосудистую) ткань. Во-вторых, в
(^-растениях обкладочные клетки большие и содержат хлоропласты.
Напротив, обкладочные клетки С3-растений нефотосинтезирующие.
Последовательность реакций С4-пути в мезофилле и в клетках
обкладки схематично представлена на рис. 10.12. С02 гидратируется до
бикарбоната (НСО^") в цитозоле мезофилла. Бикарбонат и фоефоенол-
пируват являются субстратами для реакции карбоксилирования,
катализируемой фосфоенолпируваткарбоксилазой (ФЕП- карбоксилазой),
цитозольным ферментом, не имеющим оксигеназной активности.
Следует обратить внимание на то, что клетки мезофилла С4-растений
содержат ФЕП-карбоксилазу, но не рубиско, тогда как клетки обкладки
содержат рубиско, но не ФЕП-карбоксилазу.
В зависимости от вида С4~растения оксалоацетат, образующийся под
действием ФЕП-карбоксилазы, либо восстанавливается до малата, либо
трансаминируется до аспартата. С4-кислоты транспортируются через плаз-
модесму (каналы, связывающие клетки двух типов) в примыкающие
416
Глава 10. Биосинтез углеводов
— Верхний эпидермис
Внутриклеточное
пространство
Клетки обкладки
(содержат хдороплисты)
Васкулярная область
Клетки мезофилла
(окружают клетки
обкладки)
Нижний эпидермис
';**_
Срез листа С4-растения
обкл ад очные клетки.
Поскольку стенки клеток
обкладки совершенно
непроницаемы для. газов, то де-
карбоксилирование
Отсоединения значительно
увеличивает концентрацию
С02 внутри клеток и
создает высокое отношение С02
к 02. Поскольку в клетках
мезофилла рубиско
отсутствует и отношение С02 к
02 в крайней степени
высокое в клетках обкладки,
то оксигеназная
активность рубиско
минимальна. Таким образом,
фотодыхательная активность в
С4-растениях практически
отсутствует.
С4-растения делятся на
три основные подгруппы
на основании
преобладания у них одного из трех
декарбоксилирующих
ферментов в клетках
обкладки (хотя следует отметить,
что не все виды
(^-растений обязательно имеют
один преобладающий де-
карбоксилирующий фермент). К этим трем декарбоксилазам, каждая
из которых сопряжена с определенным метаболическим путем,
относятся: КАПР+-малик~энзим, МАВ+-малик-энзим и ФЕП-карбоксики-
наза (рис. 10.13).
КАОР^-малик-энзим локализован в цитозоле, тогда как КАВ+-
малик-энзим в митохондриях. Обе декарбоксилазы катализируют
образование С02 и пирувата из малата. Пируват затем декарбоксилиру-
ется до фосфоенолпирувата. В этой связи напомним, что в глюконео-
генезе пируват превращается в фосфоенолпируват в ходе двух
реакций: сначала пируват карбоксилируется до оксалоацетата, затем
происходит декарбоксилирование оксалоацетата с образованием
фосфоенолпирувата (см. разд. 10.1). В С4-цикле карбоксилирования и де-
карбоксилирования не происходит; пируват превращается в
фосфоенолпируват в ходе единственной реакции, катализируемой пируват-
фосфатдикиназой. В этой реакции пируват, АТР и Р. реагируют, обра-
Верхкий эпидермис
Клетки мезофилла
Внутри к л еточ н ое
п ростре нет зо
Васкулярная область
Клетки обкладки
(хлоропласты отсутствуют)
Клетки мезофилла
Нижний эпидермис
Срез листа Сз-растения
Рис, 10.11. Структурные особенности
листьев С3- и С4~растений. Следует обратить
внимание на то, что большие клетки обкладки
^-растений содержат хлоропласты и полностью
окружают клетки мезофилла
27, Заказ 3486
417
Глава 10. Биосинтез углеводов
а
нсор
Р1
Фосфоенол
иируват
*■ Оксалоацетат
Пируватфос-*^ АМР, Р^ \^ *АВР^ + ^
фат,цикиназа Г Клетка т^ИАВР^
|^ АТР, Р1 мезофилла Малат
%Ш??ЙШШШШ^
ЫАВРН 4 *Г ИАВ!
Пир у ват
Клетка обкладки
ЫАОР-малик-энзим
СОг * Восстановительный
пентозофосфатн ый
цикл
б
нсо
©
р|
Фосфоенол-
пируват
Оксалоацетат
Пируватфос-1^ АМР, РР1
фатдикинаэа Г
^ АТР, Р!
Пируват
Глутамат "^
а-Кетоглутарат 4—*Ч
Аспартат
Клетка
мезофилла
Алании
Са-Кетоглутарат
Глутамат
2
Клетка обкладки
Пируват
АОН
ИАО
.©
Оксалоацетат
Малат
ЫАО -малик-энзим
СО-
Восстановительный
пентозофосфатн ый
цикл
в
НСОз
О
Р,
Фосфоенол -
пируват
Клетка
мезофилла
Е
ШШШШ///ШШШ/Л
Клетка
обкладки
АОР АТР
Фосфоенол \.
пируват
* Оксалоацетат
Глутамат ,^ЛП иРУват
Кетоглутарат
Аспартат
Е
]>*а-:
I
У
ФЕП -карбоксикиназа
Алании
а-Кетоглутарат ^ч4
Глутамат -И
Оксалоацетат ^ Пируват*'
Е
Ш//Ш/////Ш-
со<
Восстановительный
пентоэофосфаткый
цикл
Рис, 10.13. Особенности метаболических процессов в трех основных
подгруппах С4-растений, основанные на преимущественном функционировании опреде-
ленного декарбоксилирующего фермента в клетках обкладки: а — С4-путь в
видах, в которых функционирует МАВР+-малик-энзим; б — С4-путь в видах с
КАБ^-малик-энзимом; в — С4-путь в видах с ФЕП-карбоксикиназой
27'
419
Часть II. Метаболизм веществ и энергии в клетке
Атмосферный СОг
Н20 + С02 ^ НСОз
Фосфоенол
пируват
(С8)
о
Р1
Оксалоацетат
(С4)
ФЕП-карбоксилаза
Восстановление или трансаминирование
Пируват-
фосфат-
киназа
ч
АМР, РР;
АТР, Р!
ЫАМН, 1Р ^
малат- *
дегидрогеназа
МАОР®
Пируват
Глутамат
Аспартат-
трансаминаза
а-Кетогл утарат
Малат
(С4)
Аспартат
(С4)
Сз-соединекие
шт
\
Клетка мезофилла
Клетка обкладки
Сз-соединение
г
Отсоединение
СО-
Рибулозо-1,5-фосфат
Тр иозофосфат
Углеводородные
Восстановительный ^^ продукты
пентозофосфатный
цикл
Рис. 10.12, Общая схема С4-пути. Четырехуглеродные соединения образуются в
клетках мезофилла, транспортируются в клетки обкладки и декарбоксилируются в
этих клетках. Высвободившийся С02 рефиксируется с помощью рубиско и вступает
в восстановительный пентозофосфатный цикл. Оставшееся трехуглеродное
соединение вновь превращается в С02-акцептирующую молекулу — фосфоенолпируват
зуя фосфоенолпируват, АМР и РРГ Вторая молекула АТР необходима
для образования АБР из АМР:
Аденнлаткичаза
АМР + АТР —> 2АОР
Энергетическая цена превращения одной молекулы пирувата в
фосфоенолпируват — две молекулы АТР.
Третья декарбоксилаза, ФЕП-карбоксикиназа, локализована в ци-
тозоле. Этот фермент отличается~6т МА0+-малик-энзима и ЫАВР+-ма-
лик-энзима тем, что вместо малата оксалоацетат декарбоксилируется
418
Часть II. Метаболизм веществ и энергии в клетке ^^^
и фосфорилируется с образованием фосфоеноллирувата* В отличие от
малик-энзима, ФЕП-карбоксикиназа регенерирует фосфоенолпируват
в течение одного этапа, на что затрачивается только одна молекула
АТР, Фосфоенолпируват транспортируется в клетку мезофилла.
Поскольку С4-аминокислота (аспартат) импортируется в клетку
обкладки, а С3-кислота (фосфоенолпируват) впоследствии экспортируется, то
для сохранения фонда глутамата* необходимого для трансаминирова-
ния оксалоацетата в клетке мезофилла, нужен дополнительный
челночный механизм для транспортировки пирувата и аланина.
Хотя для фиксации молекулы С02 с помощью С4-пути требуются
дополнительные затраты АТР по сравнению с С3-путем, тем не менее
(^-растения образуют больше биомассы на единицу листовой поверхности.
10.10, Биосинтез олигосахаридов
Биосинтез олигосахаридов осуществляется за счет реакции транс-
гликозилирования. Работами аргентинского биохимика Луиса Лелуа-
ра и его сотрудников было показано, что в этом процессе донорами
гликозильных групп являются ИВР-сахара.
Наиболее широко распространенные и встречающиеся в свободном
виде в живых клетках — дисахариды: сахароза, трегалоза, лактоза,
мальтоза, изомальтоза, целлобиоза. Сахароза, трегалоза, лактоза
синтезируются в живой клетке йе поио; что касается остальных дисаха-
ридов, то они являются продуктами гидролиза полисахаридов —
крахмала и целлюлозы.
Сахароза — важнейший пищевой сахар, наиболее
распространенный в растительном мире. Это главное соединение, в форме которого
связанный углерод и энергия транспортируются по растению. Синтез
сахарозы осуществляется с участием двух ферментов — сахарозосин-
тазы (КФ 2.4.1Л9) и сахарозофосфатсинтазы (КФ 2.4-1.14),
катализирующих соответственно реакции:
1ШР-глюкоза (или АВР-В-глюкоза) +1)-фруктоза ?± сахароза +1ГОР (или АЮР)
АО°'= -4 кДж-моль"1;
!ШР-глюкоза +0-фруктозо-6-фосфат ?^ сахарозо-6-фосфат +11ВР
ДО0'- -20 кДж-моль"1;
сахарозо-6-фосфат +Н20 ?± сахароза +РГ
Это типичные реакции трансгликозилирования.
Многочисленные данные свидетельствуют о том, что биосинтез
сахарозы в растениях идет в основном под действием
сахарозофосфатсинтазы: об этом говорит большая АО0', чем в случае реакции,
осуществляемой сахарозосинтазой. Предполагают, что сахарозосинтаза иг-
г
рает роль не в синтезе сахарозы, а в ее превращении в крахмал,
катализируя обратную реакцию, образуя при этом ЮТР-глюкозу или
АВР-глюкозу из сахарозы.
Достаточно большая величина АС-0' при биосинтезе сахарозы из
11ВР-глюкозы и фруктозо-6-фосфата обусловливает быстрое вовлече-
420
Глава 10. Биосинтез углеводов
ние моносахаридов в этот синтез* Поэтому во многих фруктах, овощах
она накапливается в достаточно большом количестве при
относительно минимальном количестве моносахаридов. В этих растениях она
играет роль аккумулятора и углерода, и энергии.
Трегалоза синтезируется в результате последовательных реакций,
которые катализируются трегалозофосфатсинтазой и трегалозофосфа-
тазой:
1ГОР-В-глюкоза +В-глюкозо~6-фосфат ^ трегалозо-5-фосфат + Ш)Р;
Трегалозо-б-фосфат +Н20 ^ трегалоза +Р..
Ферменты, катализирующие эти реакции, были выделены из
дрожжей, гороха.
Лактоза синтезируется в животных организмах; она также
образуется путем переноса гликозильного остатка "0Т)Р-галактозы
непосредственно на глюкозу:
1ШР-галактоза + глюкоза ^ лактоза +1ЮР.
Три-, тетра- и пентасахариды синтезируются многими
растениями. Обычно эти сахариды накапливаются в растительных тканях в
небольшом количестве, за исключением трисахарида рафинозы,
которая в значительных количествах содержится в сахарной свекле и
семенах хлопчатника. При хранении корней сахарной свеклы
количество рафинозы может достигать более 2 %.
Рафинозу можно считать представителем семейства олигосахаридов,
в состав которых входит остаток молекулы сахарозы и один или несколько
остатков В-галактозы. Биосинтез этих олигосахаридов осуществляется в
результате последовательного переноса остатков В-галактозы от галак-
тинола [0~аФ*галакпгопиранозил~( 1-^1) мио-инозитола] к С-6-гидро-
ксильной группе конечного остатка В~глюкозы или В-галактозы олиго-
сахарида. Галактинол синтезируется из 1ЮР-В-глюкозы и инозита;
НО
сн»он
о, н
-О-ГОР +
н он
1ШР~В-галактоза
ОН
Мп
2+
он он
Мио-иноаит
НО
сшон
он
н он он он
Галактинол
+ 1ЮР
421
Часть II. Метаболизм веществ и энергии в клетке
Реакция осуществляется под действием фермента 1ГОР-В-галакто-
за: лшо-инозитол-галактозилтрансферазы, для активности которой
необходимы ионы Мп2+. Биосинтез олигосахаридов семейства рафино-
зы представлен на схеме:
Сахароза + галактинол
Рафиноза (трисахарид) + галактинол лшо-инозит
Стахиоза (тетрасахарид) + галактинол лшо-инозит
Вербаксоза (пентасахарид) лшо-инозит
Что касается олигосахаридов более высокого порядка, то в
свободном виде они в клетке растений не встречаются* Эти олигосахариды
после синтеза присоединяются к остаткам аминокислот боковых
цепей белков с образованием гликопротеинов, которые в дальнейшем
включаются в состав плазматической мембраны.
10.11. Биосинтез полисахаридов
В клетках животных, растений, микроорганизмов биосинтез
полисахаридов осуществляется путем последовательных реакций трансгли-
козилирования. В реакциях участвует большое число
молекул-доноров и молекула акцептора, называемая "затравкой". Донорами био-
_ синтеза служат ЫОР-сахара, затравкой — низкомолекулярный
фрагмент синтезируемого полисахарида. Цепь молекулы затравки
удлиняется путем последовательного переноса к одному из концов молекулы-
затравки гликозильных остатков молекул-доноров, как это показано
на схеме, в которой О — остаток гликозила:
ЫВР-О + затравка ^ О-затравка + ИВР
ИВР-О + О-затравка ^ 0-0-затравка + ОТР
ЫБР-О + О-О-затравка ~> О-О-О-затравка + N0?
и т.д.
Суммарная реакция биосинтеза полисахарида может быть
представлена уравнением
пКОР-6 + затравка ?± 01-02-03-...-Сп-затравка + пКВР.
Число переносов п здесь может быть от сотен до тысячи и более,
причем все они катализируются одним и тем же ферментом. Этот
фермент специфичен к переносимому остатку и определяет природу гли-
козидной связи. Как правило, гликозидная связь возникает между
422
Глава 10. Биосинтез углеводов
аномерным атомом О-остатка и нуклеофильным концевым остатком
молекулы-затравки; наращивание цепи молекулы-затравки идет с ее
нередуцирующего конца и завершается им.
Включение сахарного мономера в полисахарид требует
расщепления двух макроэргических фосфатных связей * Первой стадией этой
реакции, как и в случае биосинтеза олигосахаров, является фосфори-
лирование мономера за счет молекулы АТР; во второй стадии
фосфорный эфир мономера реагирует с Ш*Р, в результате чего образуется
донор гликозильных радикалов ЫВР-О и РР.; затем РР. под действием
фермента пирофосфатазы превращается в 2Р., т.е. осуществляется
разрыв еще одной макроэргической связи, в результате чего равновесие
реакций биосинтеза полисахарида резко сдвигается в сторону
образования полисахарида*
Биосинтез крахмала. Крахмал представляет собой запасной
полисахарид. В процессе фотосинтеза он накапливается в хлоропластах,
быстро превращается в них в сахарозу и оттекает из листьев растений
в семена, клубни, корневища, где крахмал длительное время
хранится до момента, когда эти органы прорастают. Для нового
развивающегося растения он является поставщиком углерода и энергии. Крахмал
запасается в виде крахмальных зерен в нефотосинтезирующих
клеточных органе л лах — амилопластах. Зерна образуются путем
последовательного отложения молекул крахмала вокруг ядра.
Синтез компонентов крахмала — амилозы и амилопектина —
начинается с амилозы, затем часть ее претерпевает перестройку,
формируя основу для синтеза более сложного компонента крахмала —
амилопектина. Образование амилозы осуществляется под действием
фермента крахмалсинтазы, катализирующей перенос а-В-глюкозы от
АВР-В-глюкозы к невосстанавливающему концу а-1,4-глюкана —
молекулы-затравки, представляющей собой олиго- или полисахарид,
состоящий из остатков а-В-глюкозы, соединенных а-1,4-гликозидными
связями (рис. 10.14).
В этой реакции образуется новая а-1,4-гликозидная связь между
1-м углеродным атомом добавляемого остатка глюкозы и 4-м
углеродным атомом концевого глюкозного остатка цепи амилозы.
Установлено, что в синтезе амилозы принимают участие две
крахмалсинтазы. Одна из них, тесно связанная с крахмальными зернами,
может использовать в качестве донора глюкозы как ЦВР-В-глюкозу,
так и АВР-В-глюкозу, хотя последнее соединение для фермента
предпочтительнее. Вторая крахмалсинтаза является растворимым
ферментом, находится в хлоропластах фотосинтезирующей системы, в ами-
лопластах созревающих семян, клубней, корневищ; она специфична
только к АВР-В-глюкозе. Затравкой для обеих синтаз могут быть
а-1,4-глкжаны, начиная от мальтозы и кончая амилозой (п>2), а
также а-1,4-цепи разветвленных декстринов (с а-1,6-гликозидными
связями). Таким образом, крахмалсинтазы участвуют в биосинтезе как
423
Часть II. Метаболизм веществ и энергии в клетке
сн2он
сн2он
0-АОР +
ОН
АОР-Б-глюкоза
СН2ОН
сн2он
Крахмалсинтаза
4 И
АБР
Нередуцирующий
конец а-1,4-глюкана К,
содержащего п остатков
глюкозы
О—К
ОН
ОН
Удлиненная цепь а-1,4-глюкана,
содержащая п+1 остатков
а-Б-глюкозы
Рис. 10.14. Синтез молекулы амилозы крахмалсинтазой. П-глюкозильная хруп-
па АВР-О-глюкозы переносится на нередуцирующий конец а~1,4-глюкана с
образованием ос-1,4-глюкозидной связи
амилозы, так и амилопектина, но катализируют образование только
а-1,4-гликозидных связей.
Крахмалсинтаза не способна катализировать образование а~1,6-гли-
козидных связей, находящихся в точках ветвления цепей
амилопектина. Существует специальный "ветвящии" фермент, называемый также
С^-ферментом, — гликозил-(1—»6)-трансфераза; он был получен в
кристаллическом виде из клубней картофеля* (^-фермент превращает
амилозу в амилопектин, катализируя перенос ос-1,4~фрагмента, состоящего из
15—20 глюкозных остатков, с нередуцирующего конца цепи амилозы.
Восстанавливающая группа пе-
(вохоюхххохохохххх^хохсст
I
+
«сххооссооссссахос*
Рис. 10.15. Биосинтез амилопектина под
действием ое-1,6-глюканветвящего фермента:
© — восстанавливающий конец переносимого
фрагмента ое-1,4-глюкозильной цепи; • —
восстанавливающие концы амилозы и
амилопектина; ® — невосстанавливающий конец; ОО —
вновь образованная сс-1,6-глюкозидная связь
реносимого фрагмента
образует а-1,6-гликозидную связь с
той же или другой цепью
амилозы {рис. 10.15).
Акцептором а-1,4-глюко-
зильного фрагмента может
быть не только молекула ами^
лозы, но и наружная ветвь
ранее синтезированной
молекулы амилопектина. Невос-
станавливающие концы
молекулы амилопектина могут
дальше удлиняться под
действием крахмалсинтазы и
давать новые ветви под действи-
ем (^-фермента.
Хотя крахмал и является
резервным полисахаридом, он
легко может подвергаться
различным превращениям,
основную роль в которых играет
взаимопревращение
сахароза <г* крахмал. В фотосинтези-
424
Глава 10. Биосинтез углеводов
рующих клетках листьев это превращение происходит в направлении
крахмал —> сахароза; крахмал образуется в ходе активного
фотосинтеза, а его превращение в сахарозу — необходимую транспортную
форму углеводов — происходит в последующей темновой стадии. Ниже
представлена последовательность реакций этого превращения:
Крахмал —> глюкозо-1-фосфат -» глюкозо-6-фосфат
I
АТР фруктозо-6 -фосфат
АВР-В-глюкоза -*
АВР
Сахарозо-6-фосфат
Сахароза + Р1
Гидролиз РРХ под действием пирофосфатазы сдвигает весь процесс
в сторону синтеза сахарозы.
Такой же процесс имеет место в созревающих фруктах (яблоках,
грушах), чем и объясняется их сладкий вкус.
В созревающих семенах, клубнях, корневищах это превращение
направлено от сахарозы к крахмалу. Важную роль здесь играет саха-
розосинтетаза. Она действует в направлении превращения сахарозы и
МТР в КВР-В-глюкозу и В-фруктозу, где в роли N0? могут быть 1ТВР
или АВР:
Сахароза +АВР ^ АВР-В-глюкоза +В-фруктоза*
Хотя для этой реакции ДО0' равна +4 кДж • моль"1, однако при
присоединении одного глюкозного остатка к молекуле крахмала АО0'
составляет -12 кДж*моль-1; суммарное значение АО0' равно
-8 кДж-моль"1, что и сдвигает реакцию в сторону синтеза крахмала.
Процесс превращения крахмал о сахароза в результате реакции
трансгликозилирования является для растений термодинамически
выгодным. Если бы сахароза гидролизовалась на глюкозу и фруктозу,
глюкоза затем должна была бы превратиться в глюкозо-6-фосфат с
участием АТР и гексокиназы, изомеризоваться в глюкозо-1-фосфат; и
только после этого глюкозо-1-фосфат должен превратиться в Ш)Р- (или
АВР-) глюкозу с использованием 17ТР или АТР. Замена гидролиза
сахарозы трансгликозилированием позволяет сохранить в этом
взаимопревращении две молекулы ЫТР.
425
Часть П. Метаболизм веществ и энергии в клетке
Глубокие превращения крахмала происходят при прорастании семян
и клубней; резко возрастает активность а- и р-амилаз, которые гидроли-
зуют крахмал с образованием декстринов и мальтозы, происходит
накопление сахарозы в результате реакций трансгликозилирования.
Биосинтез гликогена. Биосинтез гликогена осуществляется у
животных практически во всех тканях, но особенно активны в этом
отношении печень и скелетные мышцы. Источником глюкозных единиц
при образовании гликогена является уридиндифосфатглюкоза —
1ШР-глюкоза (см. раздел 10.8), которая образуется в ходе трех
реакций. Сначала свободная глюкоза фосфорилируется под действием гек-
сокиназы до глюкозо-6-фосфата. Затем в результате реакции
изомеризации, катализируемой фосфоглюкомутазой, глюкозо-6-фосфат
превращается в глюкозо-1-фосфат. В третьей реакции под действием глю-
козо-1-фосфатуридилтрансферазы из глюкозо-1-фосфата и ОТР
образуются 1ШР-глюкоза и пирофосфат. Последний гидролизуется пиро-
фосфатазой до ортофосфата, в связи с чем процесс сдвигается в
сторону образования 1ШР-глюкозы;
В-глюкоза
1^-АТР
Гексокиназа
АОР
-фосфат
г
й
Фосфоглюкомутаза
-фосфат
Глюкозо-1 -фосфат-
уридилтрансфераза
Ш)Р-глюкоза
С 1ШР-глюкозы гликозильные группы переносятся на затравку
гликогена с помощью фермента гликогенсинтазы (рис. 10.16).
Данный фермент катализирует образование а(1^4)-связи между первым
С-атомом присоединяемого глюкозного остатка и четвертым С-атомом
концевого остатка глюкозы боковой цепи гликогена, используемой в
качестве затравки:
(Глюкоза)п + 1ШР-глюкоза —» 1ГОР + (Глюкоза)п+1.
Ветвь молекулы гликогена, использующаяся в качестве затравки,
должна содержать не менее четырех глюкозных остатков. Затравка
может содержать до 8 остатков глюкозы, присоединенных к особому
белку - гликогенину (молекулярная масса 37 000). Образование
затравки осуществляется в два этапа. На первом этапе происходит
присоединение первого глюкозного остатка. Для данного процесса необходимы
426
Глава 10. Биосинтез углеводов
Уридин
СН2ОН
СН»ОН
?
о
-О-Р-О-Р-0 +
' I I
О" О"
о-к
он
Т1БР-глюкоза
ПБР
н он н
Нередуцирующий конец боковой
цепи гликогена, содержащий п
остатков глюкозы
сшон
Гликогенсинтаза
т
СН2ОН
СНоОН
о. н н
,хО-К
Удлиненная боковая цепь
гликогена, содержащая
п + 1 остатков глюкозы
Рис. 10Л6. Удлинение цепи гликогена под действием гликогенсинтазы
1ШР-глюкоза в качестве донора гликозильной группы и
специфический фермент гл икози л трансфераза. Наращивание длины затравки на
втором этапе путем присоединения последующих ИВР-глюкозных
единиц катализируется самим гликогенином. Дальнейшее удлинение
затравки, используемой в синтезе гликогена, происходит под
действием гликогенсинтазы. Синтезированная молекула гликогена,
состоящая из нескольких сотен тысяч глюкозных остатков, содержит также
одну молекулу гликогенина.
Гликогенсинтаза катализирует образование только а(1—>4)-связей.
Образование <х(1—»6)-связей, находящихся в точках ветвления цепей
гликогена, происходит под действием "ветвящего фермента" — ами-
ло-(1,4^1,6)-трансгликозилазы. Данный фермент обеспечивает
перенос концевых фрагментов, включающих б—7 глюкозных остатков, с
нередуцирующего конца какой-либо боковой цепи, содержащей не
менее 11 остатков, на 6-гидроксильную группу этой же или другой
цепи гликогена, которая находится ближе к внутренней части
молекулы- Так образуется новая боковая ветвь. Далее к этой ветви могут
добавляться новые остатки глюкозы под действием гликогенсинтазы.
Увеличение числа нередуцирующих концов в молекуле гликогена
обеспечивает большую его доступность как для гликогенсинтазы, так и
для гликогенфосфорилазы.
Биосинтез инулина. По аналогии с биосинтезом амилозы можно
было бы предполагать, что в синтезе инулина донором фруктозы должна
быть ИВР-В-фруктоза. Однако до сих пор экспериментально это не под-
427
Часть II. Метаболизм веществ и энергии в клетке
тверждено. Имеющиеся данные свидетельствуют о том, что инулин, по-
видимому, синтезируется из сахарозы, которая играет одновременно роль
и акцептора, и донора остатков фруктозы. Доводом для этого
утверждения является тот факт, что на одном конце полифруктозидной цепи
всегда находится остаток глюкозы. Это вполне вероятно и с
термодинамической точки зрения, поскольку ДО0' для гидролиза сахарозы составляет
-29,3 кДж-моль"1, а присоединение одного гликозильного остатка к по-
лигликозидной цепи требует +20кДж-моль-1. Для переноса фруктозно-
го остатка от молекулы сахарозы-донора к молекуле полифруктозида
ДО0' будет равна (-29,3) + (+20) = -9,3 кДж*моль~*. Эта свободная
энергия и обусловливает синтез инулина и других фруктозаяов.
Синтез инулина можно представить уравнением
Сахароза + п сахароза ^- еахароза-[р(2~-»1)-В-фруктоза]п+ п глюкоза.
Реакцию катализирует фермент инулосинтаза, которая переносит
остаток фруктозы от сахарозы к концевой фруктозной молекуле
акцептора с образованием Р-2,1-гликозидной связи. Акцепторной
молекулой могут быть сахароза или растущая цепь молекулы инулина.
Инулосинтаза обнаружена в молодых побегах топинамбура, в клубнях
артишока.
Глубокие превращения инулина происходят при прорастании
клубней, корневищ, луковиц; синтезируемая в этом процессе инулиназа
интенсивно гидролизует инулин с образованием фруктозы и
продуктов неполного гидролиза инулина — инулидов. Благодаря действию
трансгликозилирующих ферментов в процессе прорастания этих
органов растений происходит накопление сахарозы, которая является
источником углерода и энергии для вновь развивающихся молодых
растений.
Биосинтез целлюлозы. Несмотря на то, что целлюлоза — один из
наиболее распространенных полисахаридов, о ее биосинтезе известно
мало. Установлено, что в этом синтезе принимает участие ферментная
система трансгликозилирования.
Ферментные препараты, полученные из бактерий Асе1оЬас1ег
хуИпит, катализируют синтез целлюлозы из ЦВР-В-глюкозы в
присутствии целлодекстринов, растворимых высокомолекулярных
продуктов ферментативного расщепления целлюлозы. Фермент,
катализирующий синтез целлюлозы, был выделен из высших растений
(проростков маиса, корней гороха, из кукурузы, волокнистой фасоли, из
незрелых семян хлопчатника). Он, по-видимому, катализирует реакцию
пЦВР-В-глюкоза + затравка ?=*• [(|31-»4)-В-глюкоза] -затравка + пЦВР.
Природа затравки неизвестна. К тому же продуктом реакции
является не целлюлоза, а более короткий {3-1,4-глюкан. Ни один
продукт, синтезированный до последнего времени т 1>Иго9 не был
идентифицирован как целлюлоза. Неудачи в биосинтезе целлюлозы в
бесклеточных системах заключаются, видимо, в том, что в этом процессе
428
Глава 10. Биосинтез углеводов
принимает участие сложная ферментная система, разрушающаяся при
получении бесклеточных ферментных препаратов.
Биосинтез гемицеллюлоз и пектиновых веществ* О биосинтезе
этих полисахаридов также известно мало; к тому же детальная
структура многих полисахаридов клеточной стенки до сих пор не
установлена. Достоверно известно, что донорами гликозильных остатков
гемицеллюлоз и пектиновых веществ являются КВР-сахара и ЫВР-уро-
новые кислоты соответственно. Однако слабо изучены ферменты
трансгликозилирования, участвующие в биосинтезе этих соединений,
природа акцепторных молекул. Ферментативные превращения, лежащие
в основе биосинтеза гемицеллюлоз и пектиновых веществ, могут быть
представлены в виде следующей схемы:
Галактоза —> В-галактуроновая кислота -» ОВР-В-галактуроновая кислота
В-глюкуроновая
кислота
ШЭР- В-глюкуроновая
кислота
ИВР-В-ксилоза
Ш)Р-Ь-арабиноза
Гемицеллюлозы
Однако эта схема синтеза гемицеллюлоз и пектиновых веществ пока
еще не реализована т иИго, но ее можно рассматривать в качестве
наиболее вероятной.
Ферментная система, содержащаяся в суспендированных частицах
маиса, катализирует включение остатка В-галактуроновой кислоты
из Ш)Р-В-галактуроновой кислоты в ое-1,4-В-полигалактуроновую
кислоту. Реакция трансгликозилирования, видимо, осуществляется по
уравнению
п"ВВР-В-галактуроновая кислота + затравка $$ [ос(1—>4)-В-галактуроно-
вая кислота]п-затравка + пЦПР.
Получена полигалактуроновая кислота, содержащая около 30
соединенных а-1,4-связями остатков В-галактуроновой кислоты.
Природа затравки неизвестна. Донором метальных групп при синтезе
пектина является 8-аденозинметионин, от которого они переносятся
особой трансферазой. Препарат метилтрансферазы был получен из
субклеточных структур проростков маиса.
429
Часть II. Метаболизм веществ и энергии в клетке
Было показано также, что ксилан образуется в растениях под
действием фермента, катализирующего перенос остатков ксилозы с
Ш)Р-В-ксилозы к олигоксиланам, содержащим 2—5 остатков
ксилозы, соединенных р-1,4-связями. Однако фермент переноса мало
изучен. В опытах, проведенных на растениях пшеницы, показано, что
ксилан особенно легко образуется из глюкуроновой кислоты,
Сахара, связанные с липидами, участвуют в биосинтезе
полисахаридов у бактерий, дрожжей и животных. Липидным компонентом этих
соединений является полипренол:
СН3
СНз"~С=СН—СН 2
сн3
С-Г12- 0=СН— С/Н^
Полипренол
<рн3
Ксн2-с=сн-сн2он
п
У бактерий п = 9, млекопитающих в большинстве случаев п = 18,
высших растений п = 4—11, В связывании Сахаров принимает
участие как полипренолфосфат, так и полипренолпирофосфат. Ниже
приведена реакция с полипренол фосфатом:
О
II
Полипренол-О-Р-0 + ИОР-сахар
О
О
Полипренол-О-Р-О-сахар + ИОР
О'
Полипренолфосфаты обладают такой же способностью к переносу
Сахаров, как и ЫБР-сахара. Однако, обладая длинной углеводородной
цепью, в отличие от ИВР-сахаров, они могут действовать в
гидрофобной среде мембраны. При биосинтезе полисахаридов на мембранах эти
гликозиды являются эффективными донорами Сахаров.
Краткое содержание главы
Глюконеогенез — синтез глюкозы из пирувата представляет собой
путь, обратный процессу гликолиза.
Фотосинтез — это синтез углеводов и других органических
соединений из неорганических — С02 и Н20 — под действием энергии
солнечного света. Он представляет собой процесс преобразования энергии
света в энергию химических связей органических соединений.
Преобразование световой энергии в химическую происходит с помощью
хлорофилла. Фотосинтез является единственным в природе процессом,
который идет с запасанием энергии, единственным источником
органических веществ и молекулярного кислорода. Продукты фотосинте-
зирующих организмов затем используются нефотосинтезирующими
организмами, превращая их опять до С02,
Пигментным аппаратом фотосинтеза является хлоропласт.
Помимо хлорофилла в хлоропластах находятся каротиноиды (желтые
пигменты) — каротины и ксантофиллы. Функция последних —
собирательная; они "улавливают" фотоны света и направляют их к хлоро-
430
Глава 1 0. Биосинтез углеводов
филлу. Хлорофиллу же отводится роль фотосенсибилизатора —
преобразователя фотонов света в химическую энергию органических
соединений. Характерной особенностью химической структуры
молекулы хлорофилла является наличие в порфириновом ядре замкнутой
системы двойных сопряженных связей, что обусловливает способность
хлорофилла поглощать энергию света в видимом диапазоне
электромагнитного спектра. Эта же система сопряженных связей находится у
вспомогательных пигментов.
Фотосинтетические процессы включают реакции, сопряженные с
светозависимым электронным транспортом, ведущие к образованию
КАОРН и АТР, и последующее использование КАВРН и АТР в ходе
превращения атмосферного С02 в углеводы.
Суммарный процесс фотосинтеза складывается из двух стадий:
световой (или энергетической) и темновой (или метаболической).
Световая стадия осуществляется на мембранах хлоропластов; темновая — в
водной среде хлоропластов. В световой стадии идет запасание
световой энергии и ее трансформация в химическую в виде АТР и КАОРН*
Эти соединения используются в дальнейшем в синтезе углеводов из
С02.
В фотосинтезе принимают участие две фотосинтезирующие
системы. Они выполняют разные, но взаимодополняющие функции. Тила-
коидная мембрана хлоропласта содержит компоненты
электрон-транспортной цепи, включая фотосистему I (ФС1), фотосистему II (ФСП) и
АТР-синтетазу. Тилакоидная мембрана находится в строме. Строма
содержит все ферменты и метаболиты, которые участвуют в
превращении С02 в углеводы в ходе пентозофосфатного восстановительного
цикла (цикла Кальвина).
Хлорофилл и другие пигменты, улавливающие световую энергию
для фотосинтеза, являются компонентами фотосистем —
функциональных единиц, обеспечивающих светозависимые реакции фотосинтеза.
Каждая фотосистема состоит из светособирающих антенных
пигментов и реакционного центра. Реакционный центр содержит особую пару
молекул хлорофилла.
Поглощение световой энергии хлорофиллом и определенными
пигментами приводит к возбуждению особой пары молекул хлорофилла
реакционного центра, что снижает их
окислительно-восстановительный потенциал. Возбуждение хлорофиллов реакционного центра
происходит в каждой из двух фотосистем растений и обеспечивает
нециклический перенос электронов от воды через фотосистемы к МА1)Р+.
Также осуществляется циклический электронный транспорт, при
котором электроны не переносятся на ЫАОР*, а возвращаются обратно к
ФС1. Как при нециклическом, так и циклическом электронном
транспорте протоны транслоцируются через фотосинтетическую мембрану,
генерируя протонный концентрационный градиент, который
обеспечивает хемиосмотическое превращение АВР и Р. в АТР.
431
Часть II. Метаболизм веществ и энергии в клетке __^_
Продукты светозависимых реакций, ИАБРН и АТР используются
в ходе восстановительного пентозофосфатного цикла, который
включает три стадии: фиксацию С02 с помощью рубиско, восстановление
С02 до углевода и регенерацию молекулы, акцептирующей С02 — ри-
булозо-1,5-бцсфосфата.
Фундаментальные исследования темновой стадии фотосинтеза были
проведены Кальвином и его сотрудниками. Для этой цели
использовали радиоактивный 14С02. Было установлено, что первым стабильным
продуктом фотосинтеза оказался 3-фосфоглицерат. Фиксация С02
осуществляется по циклическому механизму. Акцептором С02 в цикле
является рибулозо-1,5-бш:фосфат. Первый продукт цикла Кальвина —
3-фосфоглицерат восстанавливается в моносахарид — 3-фосфоглице-
ральдегид, который вовлекается в дальнейший процесс синтеза
углеводов.
В клетках растений функционируют метаболические пути,
сопряженные с восстановительным пентозофосфатным циклом. Один из них
непосредственно связан с оксигеназной активностью рубиско. Продукты
реакции, которые возникают в результате реакции оксигенации рибу-
лозо-1,5-бцефосфата под действием рубиско, метаболизируются через
фотодыхательный путь. Реакции фотодыхательного пути протекают в
трех различных клеточных компартментах: хлоропласте, пероксисо-
ме и митохондрии. Другой путь — С4-путь, функционирующий в
таких растениях, как кукуруза, сорго, сахарный тростник, сводит до
минимума оксигенадную активность рубиско. С4-путь функционирует
совместно с восстановительным пентозофосфатным путем. В данном
пути первоначальным продуктом является четырехуглеродное
соединение. Последующее декарбоксилирование данного четырехуглерод-
ного соединения приводит к высвобождению С02, который рефикси-
руется рубиско. В ходе С4-пути процессы фиксации С02 с помощью
ФЕП-карбоксилазы и под действием рубиско осуществляются в двух
разных типах клеток.
Биосинтез олиго- и полисахаридов осуществляется за счет
реакции трансгликозилирования. Исходными соединениями в этом
синтезе являются производные нуклеотидов и моносахаридов (Ш)Р-,
АВР-сахара). Основной транспортной формой углеводов в растениях
является сахароза. Она поступает в семена хлебных знаков или
клубни картофеля, где превращается с помощью систем трансгликозили-
рования в крахмал.
Хронология важнейших дат в исследовании анаболизма углеводов
1842 г. — Ю- Р« Майер показал, что источником энергии для образования
фотосинтетических продуктов служит солнечный свет; это дало возможность
сформулировать первый закон термодинамики (закон сохранения энергии). Майер:
"Растения потребляют один вид энергии — свет и преобразуют ее в другой вид
энергии — химическую".
432
€
Глава 10. Биосинтез углеводов
1862 г. — Ю. Сакс показал, что крахмал — продукт фотосинтеза.
1913 г. — Р. Вилыытеттер и А. Штоль выделили и изучили хлорофилл.
1915—1920 гг. — К. А. Тимирязев опубликовал классические исследования
з области фотосинтеза; работы по физике и химий хлорофилла,
1945 г. — М. Кальвин и его сотрудники начали серию исследований темно-
зых реакций фотосинтеза.
1948 г, — Л. Лелуар и его сотрудники выяснили роль уридиновых нуклеоти*
дов в биосинтезе углеводов,
1954 г. — Д. Арнон открыл процесс фотосинтетического фосфорилирования.
1957 г. — Л. Лелуар и его сотрудники осуществили синтез гликогена.
1961 г. — М. Кальвин разработал схему превращения углерода в процессе
фотосинтеза,
1980 г. — Г. Хере обнаружил в ткани печени фруктозо-2>6-бисфосфат,
являющийся мощным регулятором интенсивности гликолиза и глнжонеогенеза.
ЛИТЕРАТУРА
Гудвин Т., Мерсер Э. Введение в биохимию растений: В2т. — М.: Мир, 1986.
— Т. 1. — 392 с.
Кретович В. А. Биохимия растений: Учебник. — 2-е изд., перераб. и доп. —
М.: Высш. шк., 1986. — 503 с.
Ленинджер А. Основы биохимии: В 3 т. — М.: Мир, 1985. — ТЛ. — 365 с.
Рощупкин Д. И., Артюхов В. Г. Основы фотобиофизики: Учеб. пособие. —
Воронеж: ВГУ, 1997. — 116 с.
Рубин А, Б, Лекции по биофизике: Учеб. пособие. — М.: Изд-во МГУ, 1998.
— 168 с.
Степаненко Б. Н. Химия и биохимия углеводов. Моносахариды. — М.: Высш,
шк., 1977. — 224 с.
Степаненко В. Н. Химия и биохимия углеводов. Полисахариды. — М.: Высш..
шк., 1978. — 256 с.
Эллиот В., Эллиот Д. Биохимия и молекулярная биология / Под ред. А. И. Ар-
чакова, М, П. Кирпичникова, А. Й. Медведева, В. П. Скулачева — М.: Изд-во
НИИ Биомедицинской химии РАН, 1999. — 372 с.
ВисНапап В, В., Огшззеп Ж., Лопев К. ^^ ВюсЪегшзЪгу апй то1еси!аг ВЫоду о?
Р1ап18, — РиЬ. КоскуШе» 2000. — 1368 р.
Могап Ь. А, Зсптёеоиг К. С, НоНоп Я. Д., ОсНв К. 8.9 Каюп ^. 2). ВюсЪеппз^гу.
— 2 Ей. — N611 Ра«егвоп РиЬ., Епе1ешоос1 СИМз, 1994. — 1200 р.
ВОПРОСЫ И ЗАДАЧИ
1. Зеленая водоросль СМотеИа освещалась в отсутствие диоксида углерода,
затем в темноте она инкубировалась с 14С02. Почему 14С02 в течение короткого
промежутка включался в 14С-глкжозу, а затем это включение быстро
прекращалось?
2. Напишите уравнение синтеза амилозы из глюкозы. Какое соединение
играет роль затравки в этом синтезе?
3. Рассчитайте расход энергии при включении в молекулу амилозы
молекулы глюкозы.
4. Объясните, почему в молекуле инулина всегда содержится один остаток
глюкозы,
5. Какую роль играет в обмене углеводов система крахмал <р* сахароза?
28, Заказ 3486
Глава 11. МЕТАБОЛИЗМ ЛИПИДОВ
В гл. 4 было охарактеризовано большое число липидов различной
структуры. Каждое из этих соединений имеет свой специфический
путь метаболизма. В наши цели не входит подробное описание этой
специфичности. Рассмотрим здесь основные вопросы, касающиеся
прежде всего метаболизма жирных кислот, поскольку они
представляют собой важнейшие структурные компоненты простых и сложных
липидов. Будет обращено внимание на различие путей катаболизма и
анаболизма липидов, описаны мультиферментные комплексы,
участвующие в осуществлении этих путей, стереоспецифичность ферментов.
Наиболее интенсивно процессы метаболизма липидов протекают в
семенах масличных растений и в жировой ткани человека и животных.
Основную массу липидов в этих органах составляют нейтральные
жиры, или ацилглицеролы. Ацилглицеролы — важный источник
химической энергии, поэтому метаболизм этих соединений мы увяжем с
их мобилизацией и депонированием в них энергии.
Ацилглицеролы в организме человека и животных являются
запасными веществами и выполняют такую же функцию, как и
гликоген, служат источником энергии. Их запасы пополняются после
потребления пищи, и они являются своеобразным буфером .в процессе
накопления и использования энергии в организме. Однако роль
гликогена и ацилглицеролов различна; если гликоген вовлекается в
процессы катаболизма при кратковременных и внезапно возникающих
нагрузках, то ацилглицеролы необходимы для обеспечения организма
животных, человека на длительное время. Сложные липиды (фосфо-
липиды, сфинголипиды, гликолипиды), входящие в состав
биологических мембран, подвергаются процессам обмена менее интенсивно,
чем триацилглицеролы.
11.1. Катаболизм ацилглицеролов
Катаболизм триацилглицеролов можно разделить на три фазы:
1) гидролитическое расщепление трех эфирных связей; 2) катаболизм
глицерина и 3) катаболизм жирных кислот.
Гидролиз триацилглицеролов, В первой фазе распада
ацилглицеролы подвергаются внутриклеточному гидролизу под действием липаз
на свободные жирные кислоты и глицерин. Действие липаз можно
представить в виде следующей схемы:
434
Глава 11. Метаболизм липидов
О
II
-о-
СНз-0
сн
!
СНгО
О
II
С-Е1
О
II
С~Кз
Триацилглицерол
О
к2-с-о
К1-СООН
СН2ОЙ
сн
I
СНа-О
О
II
С~Ез
Диацилглицерол
Кг-СООН
НО-
СН2ОН
СН
I
СНгО
О
II
-С-Ез
Моноацилглицерол
н2о
Л
СН2ОН
** но-
Кз-СООН
сн
\
СН2ОН
Глицерин
К1? К2 и К3 — углеводородные радикалы высокомолекулярных
насыщенных и ненасыщенных жирных кислот*
Имеется несколько разновидностей липаз, содержащихся в
клетках животных и растений* Кислая липаза находится в лизосомах,
щелочная — в микросомах, нейтральная — в цитоплазме. Липазы
широко распространены в семенах и вегетативных органах растений.
Особенно богаты липазой бобы клещевины.
Известно, что гидролиз внутриклеточных триацилглицеролов не
приводит к накоплению жирных кислот и глицерина. Это
свидетельствует о том, что скорость гидролиза сбалансирована со скоростью
окисления продуктов гидролиза внутри клетки.
Скорость, с которой гидролизуются нейтральные жиры, как
правило, повышается с увеличением числа остатков жирной кислоты в
молекуле жира, с длиной цепи и со степенью непредельности жирной
кислоты.
Значение липазы в катаболизме триацилглицеролов
подтверждается тем, что в семенах существенно возрастает ее активность по мере
их прорастания.
В значительных количествах липазы присутствуют в
поджелудочной железе и в слизистой кишечника. Они наиболее активны в
присутствии желчных кислот или их солей, а также белкового
кофактора — колипазы. Колипаза связывает липазу в присутствии желчных
кислот, в результате чего рН-оптимум действия липазы смещается с 9
до 6.
Липазы поджелудочной железы отщепляют жирные кислоты только
у а- и а'-атомов ацилглицеролов, а липазы кишечника — также и у
р-атома углерода.
Окисление глицерина. Образующийся в результате гидролиза
ацилглицеролов глицерин вовлекается в цитоплазме в процесс гликолиза.
Сначала глицерин при участии глицерофосфаткиназы превращается в
28*
435
Часть II. Метаболизм веществ и энергии в клетке
СН2ОН
Глицерин
АТР
АБР
а-глицеролфосфат. Последний под действием ИАВ-зависимой а-гли~
церофосфатдегидрогеназы превращается в дигидроксиацетонфосфат:
СНгОН Глицерофосфат- СН2ОН а-Глицерофосфат- СН2ОН
I киназа | дегидрогеназа I
НО-СН » НО-СН о ^ ^ С=0 0
I и ^ \ I II
СН2-0-Р-ОН кав+ ^ВН+Н+ СН2-0-Р-ОН
он он
а-Глицеролфосфат
Дигидроксиацетонфосфат
Дигидроксиацетонфосфат, являясь обычным метаболитом
гликолиза, включается в данный метаболический путь.
Катаболизм жирных кислот. Катаболизм жирных кислот
может осуществляться несколькими путями. Из них /3-окисление
является наиболее важным источником аккумулирования химической
энергии в форме АТР. Тем не менее процессы а- и ю-окисления также
играют определенную роль в распаде и других превращениях жирных
кислот.
Активация жирных кислот. Подобно первой стадии гликолиза
Сахаров перед р-окислением жирные кислоты подвергаются
активации, в которой участвуют АТР и СоА; активация катализируется ацил-
СоА-синтетазой:
Ра М§2+
К-(СН2)ц-СН2-СН2-СООН + АТР + СоА-8Н
Ацил-СоА-синтеааза
Р а О
*- К-(СН2)П-СН2-СН2-С +АМР + РР!
Ч8~СоА
Активация жирной кислоты происходит в два этапа. Сначала
жирная кислота реагирует с АТР с образованием ациладенилата. В этом
смешанном ангидриде карбоксильная группа жирной кислоты
присоединяется к фосфатной группе АМР- Другие две фосфатные группы
АТР освобождаются в виде пирофосфата:
К-С' + АТР <± Н-С/ + РР!
ЧОН ЧАМР
Жирная кислота Ациладенилат
Далее сульфгидрильная группа СоА действует на прочно
связанный с ферментом ациладенилат с образованием ацил-СоА и АМР:
О О
II II
К-С-АМР + Н8-СоА ?± К-С-8-СоА + АМР
В реакции активации жирной кислоты важную роль играет тот факт,
что образовавшийся пирофосфат быстро гидролизуется под действием
пирофосфатазы. В результате реакция оказывается необратимой.
436
Глава И, Метаболизм липидов
Транспорт жирных кислот. Гидролиз ацилглицеролов
осуществляется в цитоплазме, а Р-окисление жирных кислот — в матриксе
митохондрий. Однако в матрикс могут проникнуть лишь жирные
кислоты с короткой углеродной цепью (4—10 атомов углерода).
Внутренняя митохондриальная мембрана непроницаема для жирных кислот с
длинной углеродной цепью (12 и более углеродных атомов). Для этих
кислот существует специальный механизм транспорта,
осуществляемый по типу облегченной диффузии. В качестве переносчика ациль-
ных остатков служит карнитин (у-триметиламино-р-оксибутират):
Н*С\ 7 Р <*
н3счч+-сн2-сн-сн2-соон
н3с/ он
Карнитин
Карнитин функционирует по типу челночного механизма (рис. 11.1).
Ацильная группа переносится с атома серы СоА на гидроксильную
группу карнитина с образованием ацилкарнитина, который проходит
через внутреннюю митохондриальную мембрану. На внутренней
стороне последней ацильная группа вновь вступает в реакцию с СоА; эта
реакция трансацилирования катализируется ацил-СоА; карнитин-
аци лтрансферазой.
Т*1*<* /А 4 < 4 *« +
шш-
2
3
^ЧГ
\*ч 4 4ъ: га
111111
; л ^г*л
>№х^?.1г.^;у-';^
.. ^_ ^—>«> ^.
о
К~С~8-СоА
СоА-8Н
^л*
^ 4ЧЧЧГ " ^^/} с^Г^Л Л ь
тШтШ^тШШт^
* П*К ЧЛ <*Г* Л
■йхЖ;Ь
* ЧЛУ^У<« "* ' < «Ч* -ь
'—Г ' ^ '~Г -|
>^ф.фМ'
Ъ^ # ^ > # Л # 4 # А /АП| # ■ ■■ ■ */> Л> Л^ ЧГЛТ А ^Л ^ » ^ * Л4 ^ УЛ> - ^ > > ' ^Л - %Т
.а:<м.
4
О
I!
К-С-8-СоА
СоА-8Н
Рис. 11 Л. Схема транспорта высокомолекулярных жирных кислот через
внутреннюю митохондриальную мембрану с участием карнитина: 1 — внешняя
митохондриальная мембрана; 2 — межмембранная полость; 3 — внутренняя
митохондриальная мембрана; 4 — матрикс
^-Окисление жирных кислот. Этот путь окисления связан с
присоединением атома кислорода к углеродному атому жирной кислоты,
находящемуся в (3-положении (-С^*-Са-СООН).
При р-окислении происходит последовательное отщепление от
карбоксильного конца углеродной цепи жирной кислоты двухуглеродных
фрагментов в форме ацетила и соответствующее укорачивание цепи
жирной кислоты (...-С-С-|С-С-|С"С-|С»СООН).
В матриксе ацил-СоА распадается в результате повторяющейся
последовательности четырех реакций: 1) окисления с участием ацил-СоА-де-
437
Часть II. Метаболизм веществ и энергии в клетке
р
а
К-(СН2)а-СН2-СН2-С
#
о
\
Адил-СоА
РАО
8-Со А
ГАВН
Окисление (1)
К-(СН2)а-С=С-С
I
н
ч
8-СоА
Еноил-СоА (транс-форма.)
Н„0
Гидратация (2)
НО Н 0
К-(СН2)П-С-С-С
Н Н 8~СоА
Ь-гидроксиацил-СоА
ЫАВ
ИАВН+Н+
Второе окисление (3)
О
О
В-(СН2)П-С-СН2-С^
8-СоА
Оксоацил-СоА
СоА-ЗН
Е-(СН2)„-С
Тйолиз(4)
О
\
8-СоА
СН8-С-8~СоА
Ацетил-СоА
О
СН3-С-8-СоА
О
СН3-С-8-СоА
О
СН3-С-8-СоА
В-С
Ч8-СоА
Рис. 11*2. /^-Окисление жирной
кислоты- Четыре последовательных
реакции: окисление (1); гидратация (2);
второе окисление (3); тиолиз (4)
438
гидрогеназы (РАВ-зависимой дегид-
рогеназы); 2) гидратации,
катализируемой еноил-СоА-гидратазой;
3) второго окисления под
действием 3-гидроксиацетил-СоА-дегидро-
геназы (МАС-зависимой дегидро-
геназы) и 4) тиолиза с участием
ацетил-СоА-ацилтрансферазьь
Совокупность этих четырех
последовательностей реакций составляет
один оборот р-окисления жирной
кислоты (рис. 11.2).
В результате одного оборота
цепь жирной кислоты
укорачивается на два атома углерода с
образованием ацетил-СоА и
происходит генерирование ГАВН2 и
МАВН. Отщепившийся ацетил-
СоА включается дальше в цикл
трикарбоновых кислот.
Характерной особенностью
{5-окисления жирных кислот
является тот факт, что все его
промежуточные продукты являются
производными кофермента А. В
данном случае роль кофермента А
можно сравнить с ролью фосфата
в метаболизме углеводов. Как
молекула сахара, перейдя в фосфо-
рилированное производное,
вовлекается в цепь реакций гликолиза
с образованием пирувата, так и
жирная кислота, превратившись в
производное кофермента А, в
процессе р-окисления преобразуется в
конечный продукт — ацетил-СоА.
Затем оба пути катаболизма —
гликолиз и (3-окисление —
сливаются в общий поток, которым
является цикл трикарбоновых кислот.
^-Окисление жирных кислот с
нечетным числом атомов
углерода. Если жирная кислота
содержит четное число углеродных
атомов, то конечным результатом
V
Глава 11. Метаболизм липидов
р-окисления является полное превращение ацил-СоА в ацетил-СоА. Однако
в состав ацилглицеролов, хотя и весьма редко, включаются жирные
кислоты с нечетным числом углеродных атомов. Конечным продуктом р-
окисления этих кислот, наряду с ацетил-СоА, является пропионил-СоА*
Пропионил-СоА не может подвергаться Р-окислению. Его катаболизм идет
по другому пути, по которому он превращается в сукцинил-СоА:
о со2 соон
СН8-СН2-С-8-СоА V- ^-- »• СН3-СН-С-8~СоА
АТР АБР+Р1
О
Пропионил-СоА Метилмалонил-СоА
О
и
»► НООС-СН2-СН2-С-8-СоА
Сукцинил-СоА
Диоксид углерода вступает в реакцию карбоксилирования пропио-
нил-СоА, осуществляемую биотинзависимой пропионил-СоА-карбокси-
лазой. Энергетические затраты на карбоксилирование покрываются
за счет АТР. Образовавшийся мети л мал они л-Со А под действием
фермента метилмалонил-СоА-мутазы (кофактором которой является
производное витамина В12 — дезоксиаденозилкобаламин) превращается в
сукцинил-СоА, который вступает в цикл трикарбоновых кислот.
Окисление ненасыщенных жирных кислот. Олеиновая,
линолевая и линоленовая кислоты, являющиеся важными компонентами
ацилглицеролов, также подвергаются р-окислению. Однако
указанные кислоты содержат двойную связь в положении 9 и 10, в цис~форме:
10 9
Е-С-.. .-С=С-С-С-С-С-С-С-С-СООН
I I
н н
Их окисление идет через дополнительные стадии, включающие
перемещение двойной связи в углеводородной цепи жирной кислоты
и перевод этой связи в транс-форму.
р-Окисление этих кислот может быть осуществлено до следующего
ацил-СоА-производного:
8
1 ю 9 I II
К-С-. ..-С=С-С-С-8-СоА
I III
н н нн
Оно имеет двойную связь в р, у-положении и цис-форжу вместо
а, р-положения и торакс-формы, которые требуются для нормального
(3-окисления. Это препятствие преодолевается с помощью
дополнительного фермента (цис-транс )-шошр&зы, которая сдвигает двойную связь
в ос, р-положении и формирует необходимую для р-окисления
трансформу:
439
Часть II. Метаболизм веществ и энергии в клетке
ТТ ТТ О Т-Г ТТ Т-Т О
I | || цис-транс-Изомераза | Т Т
К-С-...-С=Ю-ОС-8-СоА щ » Е-С-...-С-С=С-С-8-СоА
I ||| I I |
н н н н н н н
Чис-Еноил-СоА тра не -Еноил-СоА
Вторая двойная связь в линолевой кислоте» расположенная между
12-м и 13-м углеродными атомами, переходит в нормальное а, (5-поло-
жение, требуемое для р-окисления. Третья двойная связь также
находится в несоответствующем для р-окисления положении и
подвергается рассмотренной выше изомеризации.
Скорость окисления ненасыщенных жирных кислот выше, чем
насыщенных. Например, если взять за эталон скорость окисления
стеариновой кислоты, то скорость окисления олеиновой — в 11 раз,
линолевой — в 114, линоленовой — в 170 раз выше, чем стеариновой.
а- и о-Окисление. а-Окисление является окислением а-углерод-
ного атома жирной кислоты. Оно приводит к удалению
карбоксильной группы в виде диоксида углерода; окисленный углеродный атом
превращается в карбоксильную группу жирной кислоты, длина цепи
которой становится меньше на один атом углерода* Этот путь
окисления встречается в растительных клетках.
Детали се-окисления пока не установлены, но известно, что первой
стадией является гидроксилирование а-углеродного атома. Во второй
стадии образовавшаяся гидроксикислота подвергается
окислительному декарбоксилированию:
лг~ Окислительное
а I декарбоксилирование
Е-(СН2)п-СН2-СООН -** К-(СН2)п-СН-СООН
со2
К^СН^-СНа-СООН
За первым витком а-окисления осуществляется следующий.
При ю-окислении происходит окисление противоположного конца
цепи жирной кислоты с образованием дикарбоновой кислоты.
Следует отметить, что а- и со-окисление не имеют такого значения в
энергообеспечении организма, как (3-окисление. По всей вероятности, они
играют какую-то иную роль в специфических функциях клетки.
11.2. Глиоксилатный цикл
Растения и некоторые бактерии могут использовать ацетил-СоА не
только в цикле трикарбоновых кислот, но и в цикле, получившем
название глиоксилатного. Этот цикл играет важную роль в качестве
связующего звена в метаболизме жиров и углеводов. Особенно интенсивно
глиоксилатный цикл функционирует в особых клеточных органеллах —
глиоксисомах — при прорастании семян масличных растений. При этом
440
Глава П. Метаболизм липидов
происходит превращение жира в углеводы, необходимые для развития
проростка семени. Этот процесс функционирует до тех пор, пока у
проростка не разовьется способность к фотосинтезу. Когда в конце
прорастания запасной жир истощается, глиоксисомы в клетке исчезают.
Возможность функционирования глиоксилатного цикла связана с
тем, что растения и бактерии способны синтезировать такие
ферменты, как изоцитратлиаза и малатсинтаза, которые вместе с
некоторыми ферментами цикла трикарбоновых кислот участвуют в глиокси-
латном цикле (рис. 11.3).
Две начальные реакции глиоксилатного цикла идентичны
таковым цикла трикарбоновых кислот. В первой реакции ацетил-СоА
конденсируется с оксалоацетатом под действием цитратсинтазы. Во
второй реакции цитрат изомеризуется в изоцитрат при участии акони-
татгидратазы. Следующие реакции, специфичные для
глиоксилатного цикла, катализируются дополнительными ферментами. В ходе
одной из них под действием изоцитратлиазы расщепляется С2-С3-связь
в изоцитрате с образованием глиоксилата и сукцината:
Жирная кислота
т
Ацетил-8-СоА
СоА-8Н
Оксалоацетат
Цитрат
Жирная кислота
Ь-Малат
Ацетил-8-СоА
Биосинтез
углеводов
Изоцитрат
СоА-8Н
Фумарат
Глиоксилат
Реакция Сукцинат
сопряжения с циклом
трикарбоновых кислот
С -С
-расщепление
Рис. 11.3. Глиоксилатный цикл в растениях и бактериях
441
Часть II. Метаболизм веществ и энергии в клетке
соо"
Н-С-ОН йзоцитратлиаза СОО у^°
оос-с-н ** н~оо + 9Н:
I
1
сн, сн
2
| * I
соо" соо
Изоцитрат Глиоксилат Сукцинат
В ходе другой реакции, катализируемой малатсинтазой, глиокси
лат конденсируется с ацетил-СоА (второй молекулой ацетил-СоА,
вступающей в глиоксилатный цикл) с образованием Ь-малата:
8-СоА
СН3 - СОО
Ацетил-СоА Дтт
+ ч:\-^ ? 2
н-оо > но-с-н
соо" СоА"8Н соо-
Глиоксилат Ь-Малат
Затем Ь-малат окисляется до оксалоацетата. Эта реакция
идентична конечной реакции цикла трикарбоновых кислот; она же является
конечной реакцией глиоксилатного цикла*
Поскольку в глиоксисоме отсутствуют ферменты, участвующие в
превращении сукцината (сукцинатдегидрогеназа, фумаратгидратаза),
то эта кислота не может подвергаться дальнейшим превращениям в
цикле. Из глиоксисомы она поступает в митохондрию и включается в
цикл трикарбоновых кислот, превращаясь сначала в фумарат, а затем
в оксалоацетат. Сопряжение глиоксилатного цикла с циклом
трикарбоновых кислот приводит к интенсивному накоплению в растительной
клетке оксалоацетата. Под действием фосфоенолпируваткарбоксилазы он
превращается в фосфоенолпируват, а последний — в В-глюкозу,
включаясь в процесс гликолиза, действующий в обратном направлении.
В сущности, глиоксилатный цикл направлен на синтез сукцината из
двух ацетильных остатков. Пять его последовательных реакций,
катализируемых цитратсинтазой, аконитатгидратазой, изоцитрат л иазой, малат-
синтазой и малатдегидрогеназой, идут с изменениями ДО0', равным!! -
31,4; +6,7; +8,7; -44,5; и +27,9 кДж-моль"1 соответственно. Суммарный
энергетический эффект глиоксилатного цикла равен -32,6 кДж-молъ~:
Иными словами, реакция конденсации двух ацетильных остатков с
образованием сукцината является термодинамически выгодной.
11 *3. Энергетика окисления жирных кислот
В случае ^-чжжедещш жжрных наслот с четных чксзом гт*:*;- уг-
Глава 11. Метаболизм липидов
КАПН, Б*А1Ш2 и ацетил-СоА. В дыхательной цепи ИАОН и РАПН2
при окислении дают 4 молекулы АТР (2,5 молекулы — ИАКН; 1,5 —
ГАОН2, см. гл. 9); ацетил-СоА, окисляясь в цикле трикарбоновых
кислот и дыхательной цепи, дает около 10 молекул АТР. Превращение
молекулы жирной кислоты в ацетил-СоА в стадии активации требует
затраты двух макроэргических связей, эквивалентных двум АТР, При
четном числе атомов углерода п образуется п/2 молекул ацетил-СоА,
а число витков (^окисления будет (п/2)~1. В результате окисления
КАОН и ГАВН2 в дыхательной цепи будет генерировано 4-(п/2 - 1)
молекул АТР, а окисление п/2 молекул ацетил-СоА ведет к
образованию Ю-п/2 (или 5п) молекул АТР* Таким образом, суммарное
количество молекул АТР при ^окислении жирной кислоты с п числом
атомов углерода будет 4*(п/2 - 1) + 5п- 2. Так, р-окисление бутирата
С3Н7СООН способно дать 22 молекулы АТР, пальмитата С15Н31СООН —
106, стеарата — С17Н33СООН — 120.
Эффективность запасания энергии при ^-окислении жирных
кислот можно рассчитать, исходя из количества образованных молекул
АТР и свободной энергии ее окисления, определяемой
калориметрически. Так, ДО0' при гидролизе 106 молекул АТР, синтезированных
при р-окислении пальмитата, равна -3233 кДж-моль"1* Суммарное
уравнение полного окисления пальмитата может быть представлено
следующим образом:
С15Н31-СООН + 2302 -> 16С02 + 16Н20;
ДО0, - -9800 кДж-моль"1.
Эффективность выхода энергии в результате р-окисления
пальмитата при стандартных условиях составляет около 34 %; она близка к
эффективности аккумулирования энергии при гликолизе и
окислительном фосфорилировании.
Однако жирные кислоты в силу своего ярко выраженного
восстановленного состояния представляют собой соединения с большим
запасом метаболической энергии. Так, окисление жирной кислоты,
содержащей шесть атомов углерода, дает около 36 молекул АТР, а
молекулы глюкозы — 30. Но для окисления в цикле трикарбоновых
кислот молекул ацетил-СоА, отщепляемых от жирной кислоты,
необходимо достаточное количество оксалоацетата, поставщиком'которого
являются сахара. Не случайно среди биохимиков существует летучая
фраза: "Жиры сгорают в пламени углеводов".
Ненасыщенные жирные кислоты имеют менее восстановительный
характер; на каждую двойную связь синтезируется на 2 молекулы
АТР меньше по сравнению с окислением соответствующей ей
насыщенной жирной кислоты. Это связано с тем, что при р-окислении по
двойной связи выпадает стадия образования РА1)Н2 (реакция первого
окисления, рис. 11.2), окисление которого в дыхательной цепи ведет
к синтезу примерно 1,5 молекул АТР.
443
Часть II. Метаболизм веществ и энергии в клетке
11.4. Катаболизм фосфолипидов
Гидролиз фосфолипидов осуществляют несколько фосфолипаз,
различающихся по расщеплению определенных связей и получивших
название фосфолипаз А, С и Д:
Фосфолипаза А,
Фосфолипаза Ах
О
0 СН2""От - С— Кг
к-с^о-сн
Фосфолипаза С
сн3-о-
о
II
Фосфолипаза О
11
К,
о
Здесь Кх и К2 — углеводородные радикалы; К3 — остатки
азотсодержащих одноатомных спиртов (холина, этаноламина, серина).
Фосфолипаза А1 отщепляет жирные кислоты по а-положению;
фосфолипаза А2 осуществляет ту же самую реакцию по р -положению;
фосфолипаза С отщепляет фосфорилированные азотсодержащие
спирты от фосфолипидов; фосфолипаза ТУ гидролизует фосфолипиды с
образованием азотсодержащего спирта и фосфатидной кислоты,
В растениях обнаружены все типы расщепления. Фосфолипазы А
обнаружены также у животных и микроорганизмов. Фосфолипазы
локализованы преимущественно в лизосомах. Конечными
продуктами гидролиза фосфолипидов являются глицерин, жирные кислоты,
азотистые спирты и фосфорная кислота.
11.5. Биосинтез жирных кислот
В принципе биосинтез насыщенных жирных кислот происходит в
направлении, противоположном их Р-окислению; наращивание
углеводородных цепей жирных кислот осуществляется за счет
последовательного присоединения к их концам двухуглеродного фрагмента —
ацетил-СоА. Однако имеются и специфические особенности,
различающие эти два процесса. О них будет сказано более подробно после
того, как мы рассмотрим в деталях процесс биосинтеза жирных
кислот. Здесь лишь отметим, что если (5-окисление осуществляется в мат-
риксе митохондрий, то биосинтез жирных кислот — в основном в
микросомах, представляющих собой фрагменты внутриклеточных
мембран цитоплазмы* В цитоплазме идет синтез насыщенных кислот с
длинной углеводородной цепью до пальмитата (С16); в митохондриях
происходит дальнейшее наращивание цепи; на эндоплазматических
мембранах насыщенные кислоты превращаются в ненасыщенные.
При синтезе жирных кислот в цитоплазме источником ацетил-СоА
являются два пути: ^-окисление и гликолиз. Напомним, что
превращение образовавшегося при гликолизе пирувата в ацетил-СоА и его образо-
444
Глава 11. Метаболизм липидов
вание при р-окислении жирных кислот происходят в митохондриях;
внутренняя мембрана митохондрий относительно непроницаема для ацетил-
СоА. Его поступление в цитоплазму осуществляется по типу облегченной
диффузии в виде цитрата и ацетилкарнитина, которые в цитоплазме
превращаются в ацетил-СоА, оксалоацетат и карнитин.
Образование малонил-СоА. Эта реакция является реакцией
присоединения к ацетил-СоА диоксида углерода. Она необратима и
представляет собой решающий этап в синтезе жирных кислот. Группа СН3
ацетил-СоА превращается в более реакционноспособную СН2-группу
малонил-СоА, принимающую участие в дальнейшей реакции
конденсации. В процессе сборки углеводородной цепи молекулы жирной
кислоты принимают участие молекулы малонил-СоА, за исключением
лишь одной молекулы ацетил-Со А, необходимой в начале синтеза.
Синтез малонил-СоА катализируется биотинзависимой ацетил-СоА-
карбоксилазой. Реакция карбоксилирования идет в две стадии. В
первой стадии при участии АТР образуется промежуточный продукт —
карбоксибиотин:
Е-биотин + НС03" + АТР & Е-биотин~С02 + АБР + Р..
Затем активированная С02-группа переносится на ацетил-СоА с
образованием малонил-СоА:
Е-биотин~С02 + Ацетил-СоА ^ Малонил-СоА + Е-биотин.
Биотин ковалентно связан с ферментом. Его функция сводится к
переносу активированного с помощью АТР одноуглеродного фрагмента.
Суммарная реакция, катализируемая ацетил-СоА-карбоксилазой:
О Ацетил-СоА-кар- о
" боксилаза " .
СНз-ОЗ-СоА + С02 + АТР + Н20 +- ~ООС-СН2-С-8-СоА + АБР + Рь + Н+
Ацетил-СоА Малонил-СоА
Цикл элонгации в синтезе жирных кислот. Цикл элонгации
(удлинение цепи жирной кислоты) включает четыре реакции: 1)
конденсации ацетил-СоА с малонил-СоА; 2) восстановления; 3) дегидратации и
4) второго восстановления. Формально они идут в обратном направлении
реакциям р-окисления жирных кислот. Реакции цикла элонгации
катализируются мультиферментным комплексом, называемым синтетазои
жирных кислот. Важной особенностью синтеза жирных кислот является
участие в реакциях элонгации промежуточных продуктов синтеза,
ковалентно связанных с ацилпереносящим белком (АПБ-8Н). АПБ —
низкомолекулярный белок (молекулярная масса около 10 000), который
термостабилен, содержит активную 8Н-группу. В процессе элонгации
функция АПБ аналогична функции кофермента А при р-окислении жирных
кислот; интермедиаты цикла элонгации участвуют в реакциях синтеза в
виде тиоэфиров жирных кислот с АПБ. Под действием ацетилтрансаци-
лазы ацетильная группа ацетил-СоА переносится на 8Н-группу цистеи-
нового остатка синтетазы Е, находящейся в комплексе с АПБ:
445
Часть II. Метаболизм веществ и энергии в клетке
V
О Н8-Суз д СН3-С-8-Су8
у \ Ацетилтрансацилаза . \
СН3~С-8-СоА + /Е ^— *- Е
Н8-АПВ ^ Н8-АПБ
СоА-8Н
Малонильная группа малонил-СоА реагирует с 8Н~группой АПБ.
Эта реакция катализируется малонилтрансацилазой:
О
?
О СН8-С-8-Су8 Малонил- СНа-С-6-Су8
II \ трансацилаза \
ОООСН0~С-8-СоА + Е V. »» О Б
Н8-АПБ ^ ООС-СН2-С-8-АПБ
СоА-ВН
На рис. 11.4 представлена схема синтеза жирных кислот. Один
цикл элонгации включает четыре последовательных реакции.
В первой реакции — реакции конденсации — ацетильная и
малонильная группы, ковалентно связанные с 8Н-группами синтетазы,
взаимодействуют между собой с образованием ацетоацетил-АПБ с
одновременным выделением С02. Эту реакцию катализирует
конденсирующий фермент р-кетоацил-АПБ-синтетаза. Отщепляемый от мало-
нил-АПБ С02 — это тот же самый С02, который принимал участие в
реакции кар^оксилирования ацетил-АПБ. Таким образом, в
результате реакции конденсации происходит образование из двух- и трехугле-
родных компонентов четырехуглеродного соединения. Декарбоксили-
рование малонил-АЛБ приводит к существенному снижению
свободной энергии и делает необратимой реакцию конденсации. Реакция
образования малонил-СоА из ацетил-СоА и С02 идет с участием АТР;
на самом деле реакция конденсации запускается с помощью АТР.
Во второй реакции — реакции восстановления, катализируемой
Р-кетоацил-АПБ-редуктазой, ацетоацетил-АПБ превращается в р~гид-
роксибутирил-АПБ. Восстанавливающим агентом служит МАОРН.
В третьей реакции цикла элонгации — дегидратации — от Р-гидро-
ксибутирил-АПБ отщепляется молекула воды с образованием ос, р-бутеи-
нол-АПБ. Реакция катализируется р-гидроксиацил-АПБ-дегидратазой.
Четвертой (конечной) реакцией цикла является восстановление
а, р-бутеноил-АПБ в бутирил-АПБ; ненасыщенный интермедиат
жирной кислоты превращается в насыщенный. Реакция идет под
действием а, р-еноил-АПБ-редуктазы. Роль восстановителя здесь выполняет
вторая молекула NА^РН.
В следующем втором цикле элонгации группа бутирила переносится
на 8Н-группу цистеина синтетазы, т.е. занимает положение при той же
8Н-группе, с которой начиналась элонгация:
Н8-Суз х СН8-СН2-СН2-С -8-Суз ч
СН8-СН2-СН2-С -8-АПБ ' Н8-АПБ /
О
446
к
Глава 11. Метаболизм липидов
Последовательность
реакций элонгации во втором и
следующих циклах
повторяется.
В синтезе жирной кислоты
только одна молекула ацетил-
СоА непосредственно вступает
в реакцию конденсации, играя
роль "затравки", остальные п
молекул сначала
превращаются в п молекул малонил-СоА,
на что используются п молекул
АТР, В каждом цикле
элонгации требуются 2 молекулы
ХАСРН и, следовательно, на п
циклов необходимо затратить
2п молекул ЫАВРН-
Источником последних являются
реакции окисления малата в окса-
лоацетат и гексозофосфатныи
шунт.
Как правило, процесс
элонгации идет вплоть до
образования С16-ацил-АПБ. Этот
конечный продукт не может
служить субстратом для
конденсирующего фермента и
гидролизу ется с образованием паль-
митата и АПБ.
Различие циклов
^окисления и элонгации жирных
кислот. Механизмы реакций
элонгации и (3-окисления
осуществляются в принципе в
противоположных направлениях:
в первом случае идет процесс
наращивания ацильной цепи за
счет двухуглеродных
фрагментов от метильного конца к
карбоксильной группе, во
втором — ацилъная цепь
укорачивается в каждом обороте
окисления на два атома углерода от
карбоксильного конца к
метальной группе:
о
СНа-С-8-Суэ
V
\
ООС-СНо-С-8~АПБ
/
Е
1 — конденсация
(Р-кетоацил-АПБ-синтетаза
Н8-Суз
О
\
Е
О
а и /
СН8-С-СН2-С-8-АПБ
Ацетоацетил-АПБ
2 - восстановление V НАВРН+Н*
(р-кетоацил-АПБ-редуктаза) |^- ИАВР'
1+
Н8-Суз
Н ,Е
СН3-С-СН2-С-8-АПБ
ОН О
Р-Гидроксибутирил-АПБ
3 — дегидратация
(Р-гидроксиацил-АПБ-дегид
ратаза)
Н
>
НЗ-Суз
\
/
Е
СН8-С=С-С-8~АПБ
Н О
а, р- Бутеноил- АПБ
4 — восстановление I/ КАВРН + Н+
(а,Р-еноил-АГШ-редуктаза)
СН3-СН2-СН2-С-8-АПБ /
О
Бутирил-АПБ
СН8-СН2-СН2-С-8-Су8 ^
А
Н8-АПБ
/
Е
Циклы
элонгации
Н8-Суз
СН8-СН2-.. .-СН2-С-8-АПБ
О
\
/
Б
Рис. 11.4. Последовательность реакций
в синтезе жирных кислот (первый цикл
элонгации): конденсация, восстановление,
дегидратация, восстановление, насыщение
447
Часть П. Метаболизм веществ и энергии е клетке
Элонгация
—>
сн3-сн2-.. .-сн2-сн2-соон
<—
р-Окисление
Сближает эти процессы и тот факт, что в них принимают участие
интермедиаты одного типа, за исключением малонил-СоА. Однако
ферментативные превращения в них совершенно иные. Пути синтеза
и окисления жирных кислот не пересекаются даже частично: если
синтез жирных кислот происходит в основном в цитоплазме, то
расщепление — в митохондриях. Хотя интермедиаты в обоих процессах
сходны, однако промежуточные продукты синтеза жирных кислот
ковалентно связаны с АПБ, а продукты расщепления жирных
кислот — с коферментом А, Роль восстановителя в цикле элонгации
выполняет ИАОРН, роль окислителя в цикле (3-окисления — КАО+ и
ГАВ. В цикле элонгации промежуточное р-гидроксиацильное
соединение образуется в О-форме, при Р-окислении — в Ь-форме.
11.6. Регуляция биосинтеза жирных кислот
Скорость биосинтеза жирных кислот определяется содержанием
Сахаров в клетке. Она достигает максимального уровня при избытке
Сахаров и низком содержании жирных кислот. Увеличение
концентрации глюкозы в жировой ткани человека, животных и повышение
скорости гликолиза стимулируют процесс синтеза жирных кислот. Это
свидетельствует о том, что жировой и углеводный обмен тесно
взаимосвязаны друг с другом. Важную роль здесь играет реакция карбокси-
лирования ацетил-СоА с его превращением в малонил-СоА,
катализируемая ацетил-СоА-карбоксилазой, Активность последней зависит от
двух факторов: наличия в цитоплазме высокомолекулярных жирных
кислот и цитрата. Ф. Линен показал, что длинноцепочечные ацил-СоА
в концентрации 10"7 моль/л подавляют активность карбоксилазы.
Иными словами, само накопление жирных кислот оказывает
тормозящее влияние на их биосинтез.
Особая роль в регуляции биосинтеза жирных кислот отводится
цитрату, который является эффективным активатором ацетил-СоА-кар-
боксилазы. Цитрат в то же время играет роль связывающего звена
углеводного и жирового обмена. В его отсутствие ацетил-СоА-карбоксилаза
проявляет весьма низкую активность. Интенсивный катаболизм
углеводов в клетке способствует повышению концентрации цитрата в митохонд- •
риях и переходу его в цитоплазму. В цитоплазме цитрат вызывает
двойной эффект в стимулировании синтеза жирных кислот: во-первых, как
активатор ацетил-СоА-карбоксилазы и, во-вторых, как источник
ацетильных групп. Установлено, что в отсутствие цитрата ацетил-СоА-карбокси-
лаза находится в виде малоактивного мономера с молекулярной массой
448
Глава 11, Метаболизм липидов
540 000. В присутствии цитрата фермент превращается в активный три-
мер с молекулярной массой около 1 800 000, обеспечивающий
15-кратное увеличение скорости синтеза жирных кислот.
11*7. Биосинтез липидов
Биосинтез ацилглицеролов. В биосинтезе ацилглицеролов
принимают участие два предшественника: ацил-СоА и
глицерол-3-фосфат. Исходными соединениями" для синтеза данных
предшественников в организме человека и животных являются углеводы,
поступающие с пищей, в растениях — сахароза, поступающая из фотосинтези-
рующих тканей. При действии соответствующих гликозидаз
углеводы подвергаются гидролизу с образованием моносахаридов, которые в
цитоплазме клеток включаются в процесс гликолиза.
Глицерол-3-фосфат образуется в результате восстановления дигидр-
оксиацетонфосфата — промежуточного продукта гликолиза; реакцию
катализирует глицеролфосфатдегидрогеназа:
СН2ОН Глицеролфосфат- СН2ОН
С-0 дегидрогеназа но_<1__н
! /""" > I
СН2ОР ЫАОН+Н+ ЫАО+ СН2ОР
Дигидрокси- Ь-глицерол-3-
ацетонфосфат фосфат
В фотосинтезирующих клетках дигидроксиацетонфосфат
образуется также в хлоропластах и диффундирует из них в цитоплазму.
Другой предшественник ацилглицеролов — ацил-СоА —
синтезируется из ацетил-СоА, который образуется двумя путями: либо в
результате окислительного декарбоксилирования пирувата —
конечного продукта гликолиза, либо в результате расщепления цитрата,
поступающего из митохондрий в цитоплазму.
На первом этапе синтеза ацилглицерола образуется 3-фосфатидная
кислота (диацилглицерол-3-фосфат), при этом ацильные остатки
СоА-производных жирных кислот переносятся специфическими ацил-
трансферазами на спиртовые группы глицерол-3-фосфата:
р р
СН2ОН Ацнлтранс- СН2-0 С^ Ацилтранс- О СГН2"°~Сч«к1
но-с-н фераза - но-с-н Фераза - е,-с-о~с-н
зСН2ОР /> СоА-8Н зСН2ОР /> СоА-8Н зСН2ОР
ь.Г1ииеТ)ОЛ.3К1~Сч8-СоА Моноацил- К2_Сч8_СоА Фосфатидная
ь глицерол о глицерол-3- кислота
Ф°сФат фосфат
На втором этапе фосфатидная кислота гидролизуется фосфатидат-
фосфатазой с образованием 1,2-диацилглицерола:
>
9. Заказ 3486
449
Часть //. Метаболизм веществ и энергии в клетке
Р Р
о ?Н2~0-Ч о <ГНй-0-с-Е
'А ! : Фосфат и датфосфатаза И 21 1
к2-с-о-с-н " .7 » к2~с-о-с-н
! Т' ^\ |
зСН2ОР Н20 Р» СН2ОН
Фосфатидная 1,2-Диацилглицерол
кислота
Затем 1,2-диацилглицерол превращается в триацилглицерол
путем переноса ацильного остатка от СоА-производного третьей жирной
кислоты. Этот перенос катализируется ферментом диацилглицерол-
аци лтрансферазой:
# • //
О СГН2"°~С^К Диацилглицерол- О '?Н2~°~Сч>Е
о -Й-О-С-Н ' ацилтрансфераза к "_0Д>Н
Р
СН*0Н Р СоА-8Н СН2-0-СЧк
1,2-Диацилглицерол Кз С 3
Ь-СоА Триацилглицерол
Синтез ацилглицеролов требует затраты большого количества
свободной энергии. Для образования одной еложноэфирной связи жир-
ная кислота должна перейти в активную форму ацил-СоА, на что
необходима энергия двух макроэргических фосфатных связей, так как
реакция образования ацил-СоА протекает благодаря пирофосфатному
расщеплению АТР с последующим гидролизом пирофоефата.
Триацилглицеролы составляют энергетическое депо организма. Они
обладают высокой теплотой окисления, равной 35—40 кДж-моль"1,
локализуются в жировых клетках (адипоцитах) и характеризуются
высокой скоростью метаболизма.
Биосинтез фосфолипидов. Подобно биосинтезу ацилглицеролов,
в биосинтезе фосфолипидов ключевым промежуточным соединением
является глицерол~3-фосфат. В настоящее время предполагают, что
биосинтез фосфолипидов протекает в мембранах эндоплазматической
сети. В этом биосинтезе принимает участие цитидинтрифосфат (СТР),
выполняющий роль активатора.
Основными компонентами мембран являются фосфатидилэтанол-
амин и фосфатидилхолин.
В процессе синтеза фосфатидилэтаноламина вначале происходит
активация этаноламина под действием этаноламинкиназы:
. Этаноламинкиназа , I
Н3К+-СН2-СН2-ОН —* ■■ *! т Нз1Ч+-СН2-СН2-0-Р=0
Этаноламин АТР АВР ^^
Этаноламинфосфат
Этаноламинфосфат реагирует затем с цитидинтрифосфатом (СТР),
в результате чего образуется цитидиндифосфатэтаноламин и пиро-
450
Глава 11. Метаболизм липидов
фосфат. Эту реакцию катализирует фосфоэтаноламинцитидинтранс-
фераза:
Фосфоэтаноламин-
I цитидинтрансфераза
Нз^-СНз-СНз-О-Р-О ^—^- ** Н3М+-СН2-СН2-СВР
_ ОН гугр рт>. СВР-этаноламин
Этаяолашгафосфат
Активированный СВР-этанол амин при участии фермента этанол-
аминфосфаттрансферазы вступает в реакцию с Ь-диацилглицеролом с
образованием фосфатидилэтаноламина:
р
Н31Ч+-СН2-СН2--СБР + К2-С-0-С-Н -*~
СВР-этаноламин зСН ОН
Ь-диацилглицерол
Р
Этаноламинфосфо* 0 СНя-О-С^
трансферааа II 1 ^1
к2~с-о-с-н 0
СМР СН2~0-РНЭНЗН2~СН2-:М+Нз
он
Фосфатидилэтаноламин
Ь-диацилглицерол образуется при гидролизе фосфатидной
кислоты. Аналогичные реакции ведут к образованию СВР-холйна;
последний вступает в реакцию с Ь-диацилглицеролом, в результате чего
образуется фосфатидилхолин:
р
Н3С-К+-СН2-СН2-СВР + К2-С-0-С-Н *■
НзС/ СБР-холин *СН2ОН
Ь-диацилглицерол
/>
Холннфосфо- 0 СНд-О-С^
трансфераза II I ^
К2—С-~0—С~Н г. /-.гг
СМР Сн2-0-Р-0-СН2-СН2'Ы+-СНз
ОН ХСН3
Фосфатидилхолин
В синтезе фосфатидилэтаноламина и фосфатидилхолина важную
роль играет СТР. Подобно синтезу олиго- и полисахаридов, мы и здесь
видим, каким образом нуклеотиды могут выполнять функцию
переносчиков определенных химических групп в обмене веществ клеток.
29*
451
Часть II. Метаболизм веществ и энергии в клетке
Аналогичная активация серина и инозита с участием АТР и СТР
ведет к синтезу фосфатидилсерина, фосфатидилинозитола,
используемых также в сборке биомембран.
Помимо синтеза фосфатидов йе поро в клетке они легко
подвергаются взаимопревращениям. Так, в печени происходит образование
фосфатидилхолина при участии 8-аденозинметионина:
Метилтрансфераза
Фосфатидилэтаноламин + 38-адено.зинметионин —-* —•*-
— » фосфат и дилхолин + 38-аденозингомоцистеин.
В переносе метильных групп важную роль играют тетрагидрофо-
лиевая кислота и метилкобаламин. При декарбоксилировании фосфа-
тидилсерина при участии фосфатидилсериндекарбоксилазы
образуется фосфатидилэтаноламин;
Фосфатидилсерин-
декарбоксилаза
Фосфатидилсерин — —^^ •*• Фосфатидилэтаноламин
С02
Синтезированные фосфолипиды поступают с помощью липидперено-
сящих белков из цитоплазмы к мембранам и участвуют в их построении,
11.8. Исследования процессов метаболизма липидов
Началом исследований метаболизма липидов можно считать 1823 г.,
когда М. Шеврель установил химическую природу триацилглицеро-
лов. Это открытие послужило отправной точкой исследований по
установлению химического состава различных жиров. Впервые синтез
липидов осуществили П. Бертло в 1854 г. и Ш. Вюрц в 1859 г.
Дальнейшую историю изучения липидов можно разделить на три
периода, различающиеся по методическому уровню исследований.
На первом этапе (1880—1950 и\) липиды исследовали
традиционными методами органической химии, второй этап (1950—1970 гг.)
характеризуется широким применением методов хроматографии, а
последний —- использованием таких физических и физико-химических
методов, как масс-спектрометрия, оптическая спектроскопия,
флуоресцентный анализ*
Исследования по метаболизму глицерина. Метаболизм глицерина
раскрыт на основании результатов опытов, проведенных с семядолями
клещевины и с бесклеточными экстрактами из семядолей арахиса.
В 1956 г. Н. Биверс показал, что срезы ряда растительных тканей
превращают 1,3-14С-глицерин в 14С-сахарозу и иС02. Путь обмена
глицерина был детально исследован в этиолированных семядолях клещевины,
отделенных от молодых проростков- На протяжении 6-часовой
инкубации иС-еахароза содержала в 3—4 раза больше меток, чем 14С02.
Анаэробные условия, малонат, соединения фтора сильно подавляли процесс
452
Глава 11, Метаболизм липидов
утилизации глицерина. Установлено, что 14С-еахароза мечена в
одинаковой степени в глюкозной и фруктоз ной частях своей молекулы. Распад
глюкозной части показал, что 97 % радиоактивности равномерно
распределено между 1-м, 3-м, 4-м и 6-м углеродными атомами. Такое
распределение позволило предполагать, что молекула глицерина
включается в глюкозу целиком, без предварительного расщепления.
14
?•
3 2
Штумпф исследовал распад глицерина до С02 в бесклеточных
экстрактах из семядолей проростков гороха. В присутствии
митохондрий, растворимой цитоплазматической фракции и ряда кофакторов
1,3-14С-глицерин окислялся до ИС02. Необходимыми кофакторами
служили АТР, а-оксоглутарат, тиаминпирофосфат, М§2+ и ЫАО+.
Участие процессов гликолиза и ЦТК в этом распаде согласуется с
наблюдаемым подавлением реакций с соответствующими ингибиторами
(соединениями фтора и малонатом).
Исследования по ^-окислению жирных кислот. В 1904 г. Ф. Кнооп
выдвинул гипотезу, объясняющую механизм окисления жирных
кислот в животном организме. Гипотеза Ф. Кноопа получила в
дальнейшем убедительное экспериментальное подтверждение. В настоящее
время она является основой современных представлений о механизме
окисления жирных кислот.
Теория р-окисления была построена на основании определения
химической природы конечных продуктов катаболизма жирных кислот. В
опытах Ф. Кноопа животным вместе с пищей вводились фенильные
замещенные жирных кислот. Эти соединения, содержащие четное число
углеродных атомов, в качестве конечного продукта в моче давали фени-
луксусную кислоту. Введение жирных кислот с нечетным числом атомов
углерода приводило к образованию конечного продукта — бензойной
кислоты. Ф. Кнооп интерпретировал полученные данные как
доказательство последовательного отщепления двухутлеродных фрагментов после
окисления р-метиленовой группы в р-кетоформу:
н н н н н н
^Ьс|с-с]-С-С-СООН-*-<^>-С-СООН + 2"С2"
н н н н н н
Фени луксу сная
кислота
н н н н
<^-с4с~С-|-С- СООН -»* ^3~ СООН + 2"С;
н н н н
Бензойная
кислота
453
Часть II. Метаболизм веществ и энергии в клетке
■ I» I1 I II II1 ■ ■! I" ■ |М111 |^1 III ,,—^^»^ ,, ■*■■ -^^д^_ ,-^^^^Ш | ,■ Ш 1ЧИИ>* ■ »■" »Ч^
Кнооп получил данные о том, что а, р-ненасыщенные, р-окси- и
Р-кетопроизводяые являются промежуточными продуктами р-окисле-
ния жирных кислот.
В развитии теории р-окисления важную роль сыграло введение
радиоактивной метки (14С) в молекулу жирной кислоты. Многими
исследователями было показано, что срезы животных и растительных
тканей превращают меченые 14С-жирные кислоты, содержащие от 2
до 18 атомов углерода, в 14С02. В исследованиях установлены
следующие факты: 1) для окисления жирных кислот клеточными гомогена-
тами требуются те же самые кофакторы, что и для окисления пирува-
та (неорганический фосфат, ионы М#2+, цитохром с, АТР и какой-
либо субстрат цикла трикарбоновых кислот); 2) субстрат цикла
выполняет две функции: является источником образования АТР,
необходимого для активации жирной кислоты, и служит партнером для
конденсации остатков уксусной кислоты, которые в цикле
превращаются в С02.
Тесную взаимосвязь между Р-окислением и циклом трикарбоновых
кислот показало исследование процесса окисления 1-14С-маеляной
кислоты. Открытие Ф. Липманом и Ф. Линеном кофермента А и
установление того факта, что промежуточные продукты окисления жирных
кислот являются производными этого кофермента, существенно облегчило
изучение этого процесса с помощью ферментных препаратов.
В 1954 г. М. Барнер и его сотрудники, применив растворимые
экстракты С1о81гШит Ыиуиеп, в основном выяснили путь р-окисления.
Позднее Дисдейн и Ларди, используя растворимую систему
митохондрий печени, осуществили полное окисление жирных кислот,
содержащих от 4 до 18 атомов углерода. Ферменты, осуществляющие р-окис-
ление, были выделены и очищены Д. Грином и Ф. Линеном и их
сотрудниками из тканей животных. Однако эти факты были
установлены в основном на животных тканях- Данные о р-окислении жирных
кислот в растениях впервые получил в 1939 г. Грейс. Методы и
принципы, использованные им, были в основном такими же, которые
применил Кнооп в своих опытах по изучению катаболизма жирных
кислот у животных, Грейс установил, что при укоренении черенков
растений только соединения, содержащие в боковой цепи четное число
углеродных атомов, обладали активностью, сходной с активностью
стимулятора прорастания — 1-нафтилуксусной кислоты. Гомологи,
содержащие в боковых цепях нечетное число углеродных атомов, не
обладали такой функцией.
Аналогичные результаты, подтверждающие р-окисление,
получили и другие исследователи, применявшие целый ряд химических
производных жирных кислот и различные растительные ткани.
Исследование процессов биосинтеза яипидов. Большим успехом
в исследовании биосинтеза жирных кислот явилась обнаруженная
С. Уокилом потребность в диоксиде углерода> Превращение ацетил-СоА
454
•
Глава 11, Метаболизм липидов
в малонил-СоА с участием диоксида углерода представляет собой
решающий этап в синтезе жирных кислот* В экстрактах печени голубя
их синтез из ацетата протекал только в присутствии АТР и
бикарбоната. Бикарбонат оказался абсолютно необходимым компонентом, хотя
сам он в молекулу жирной кислоты не включался.
Благодаря исследованиям С. Уокила, Ф. Липмана и П. Вагелоса
было установлено, что фактической единицей биосинтеза жирных
кислот является не ацетил-СоА, а малонил-СоА. В присутствии АТР,
Мп2+, ЫАОРН и С02 растворимая ферментная система печени голубя
катализировала йе поуо синтез высокомолекулярных жирных кислот
из ацетил~СоА. Основным продуктом синтеза была пальмитиновая
кислота. Тот факт, что при использовании в качестве субстрата ацетил-
СоА требовались АТР и С02, позволил установить важную роль в
синтезе жирных кислот реакции карбоксилирования, идущей с затратой
энергии. Исходным соединением для синтеза кислот является не аце-
тил-СоА, а малонил-СоА. Включение малонильных остатков в
жирные кислоты убедительно показано на созревающих плодах и
семядолях арахиса, развивающихся коробочках льна.
Исследования метаболизма фосфолипидов, сфинголипидов, восков
в растениях немногочисленны. Имеющиеся экспериментальные
данные свидетельствуют, что исходным веществом в синтезе восков
является пальмитиновая кислота. Под действием соответствующих еинте-
таз она удлиняется с образованием жирных кислот, обычно эти
кислоты содержат 18—32 углеродных атома. Однако о синтетазах
известно немного. Возникающие при их действии удлиненные цепи
изменяются самыми различными путями с образованием всех компонентов
воска. Свободные жирные кислоты образуются в результате
гидролиза тиоэстеразами СоА-производных жирных кислот.
Высокомолекулярные альдегиды и спирты образуются из жирных кислот в
результате действия ЫАО-зависимых редуктаз. Это подтверждают опыты,
проведенные изотопным методом с листьями капусты.
Главными компонентами воска являются эфиры. Установлено, что
они могут образовываться двумя путями: переносом ацильного
остатка от фосфолипидов и переносом ацильного остатка от
СоА-производного жирной кислоты на спирт жирного ряда.
Краткое содержание главы
Ацилглицеролы в организме человека и животных являются
запасными веществами и служат источником энергии. Сложные липи-
ды (фосфолипиды, сфинголипиды, гликолипиды), входящие в состав
биологических мембран, подвергаются процессам обмена менее
интенсивно, чем триацилглицеролы.
Катаболизм триацилглицеролов включает три стадии: 1) гидролиз
под действием липаз на свободные жирные кислоты и глицерин; 2) ка-
455
Часть II, Метаболизм веществ и энергии в клетке ^^
таболизм глицерина и 3) катаболизм жирных кислот. Скорость
гидролиза четко сбалансирована со скоростью катаболизма.
Образовавшийся при гидролизе ацилглицеролов глицерин в цитоплазме вовлекается
в процесс гликолиза.
Основным путем извлечения химической энергии из жирных
кислот является их р-окисление. Этот путь связан с присоединением
кислорода к углеродному атому жирной кислоты, находящемуся в (5-по-
ложении. Конечный продукт р-окиеления — ацетил-СоА; последний
включается в цикл трикарбоновых кислот. Молекула жирной
кислоты с четным числом углеродных атомов полностью превращается в
ацетил-СоА. Продуктом окисления жирных кислот с нечетным
числом углеродных атомов наряду с ацетил-СоА служит пропионил-СоА,
который превращается в сукцинил-СоА, последний вовлекается в цикл
трикарбоновых кислот.
Эффективность выхода энергии в результате р-окисления жирных
кислот близка к эффективности аккумулирования энергии при
гликолизе и окислительном фосфорилировании. Однако жирные кислоты в
силу своего ярко выраженного восстановительного состояния
представляют собой соединения с большим запасом метаболической
энергии. Ненасыщенные жирные кислоты имеют меньший
восстановительный характер, на каждую двойную связь синтезируется на две
молекулы АТР меньше по сравнению с окислением соответствующей ей
насыщенной жирной кислоты.
Гидролиз фосфолипидов осуществляют фосфолипазы. Конечными
продуктами гидролиза фосфолипидов являются глицерин, жирные
кислоты, азотистые спирты и фосфорная кислота.
В принципе биосинтез жирных кислот происходит в
направлении, противоположном р-окислению. Однако имеются и
специфические особенности, различающие эти два процесса; и, по сути, это
различные ферментативные процессы. Синтез жирных кислот
происходит в основном в цитоплазме, расщепление их — в
митохондриях. Хотя интермедиаты в обоих процессах идентичны, однако
промежуточные продукты синтеза жирных кислот ковалентно связаны с
ацилпереносящим белком (АПБ), а продукты расщепления жирных
кислот — с коферментом А. Роль восстановителя в синтезе жирных
кислот выполняет МАВРН, роль окислителя при р-окислении — ЫАО+
и РАБ.
Скорость биосинтеза жирных кислот определяется содержанием
Сахаров в клетке. Она достигает максимального уровня при избытке
Сахаров и низком содержании жирных кислот. Важную роль здесь
играет реакция карбоксилирования ацетил-СоА в малонил-СоА,
катализируемая ацетил-СоА-карбоксилазой. Активность данного
фермента подавляется длинноцепочечными ацил-СоА. Эффективным
активатором этого фермента является цитрат. Последний вызывает
двойной эффект стимулирования синтеза жирных кислот, во-первых,
456
_ Глава 11. Метаболизм липидов
как активатор карбоксилазы и, во-вторых, как источник
ацетильных групп.
В биосинтезе ацилглицеролов принимают участие два
предшественника: ацил-СоА и глицерол-3-фосфат. Ацильные остатки ацил-СоА
переносятся ацилтрансферазами на спиртовые группы глицерол-3-фос-
фата, в результате чего образуется фосфатидная кислота. От
последней отщепляется фосфорная кислота с образованием 1,2-диацил-
глицерола, к которому присоединяется третий остаток ацила от
ацил-СоА.
Подобно биосинтезу ацилглицеролов в биосинтезе фосфолипидов
ключевым промежуточным соединением является глицерол-3-фосфат.
Он играет важную роль в сборке мембран, в синтезе основных
компонентов мембран — фосфатидилэтаноламина и фосфатидилхолина. В
этом биосинтезе принимает участие цитидинтрифосфат, выполняющий
роль активатора интермедиатов.
Хронология важнейших дат в исследовании метаболизма липидов
1904 г. — Ф. Кнооп установил, что процесс расщепления жирных кислот
г.роисходит путем последовательного отщепления двухуглеродных фрагментов,
;г назвал его (5-окислением.
1949 г. — Ю, Кеннеди и А. Ленинджер установили, что окисление жирных
кислот происходит только в митохондриях.
1954—1958 гг. — Ф, Линен с сотрудниками описали основные
ферментативные процессы окисления жирных кислот,
ЛИТЕРАТУРА
Березов Т. Т., Коровкин Б. Ф. Биологическая химия; Учебник. ~ 3-е изд.,
перераб, и доп. — М.: Медицина, 1998. — 704 с.
Бохински Р. Современные воззрения в биохимии. — М.: Мир, 1987, — 544 с.
Верещагин А. Г. Биохимия триацилглицеридов. — М.: Наука, 1972. — 214 с.
Гудвин Т., Мерсер Э. Введение в биохимию растений: В 2 т. — Мл Мир, 1986.
— Т. 1, — 393 с.
Кнорре Д. Г., Мызина С. Д. Биологическая химия: Учебник. — 3-е изд., испр.
— М.: Высш. шк., 2000. — 479 с,
МецлерД. Биохимия: В 3 т. — М.: Мир, 1980. ~ Т. 2. — 606 с.
Плешков Б. П, Биохимия сельскохозяйственных растений. — М.: Колос,
1965, — 414 с.
Страйер Л. Биохимия: В 3 т. — М.: Мир, 1985. — Т. 2. — 239 с.
ВОПРОСЫ И ЗАДАЧИ
1. В чем аналогия и разница анаболизма и катаболизма жирных кислот с
четным числом углеродных атомов в углеродной цепи?
2. Какова роль образования малонил-СоА из ацетил-СоА и С02 в синтезе
жирных кислот?
3. В каком триацилглицероле больше биологически доступной энергии при
одном и том же количестве атомов углерода: а) триацилглицерол с высоким
йодным числом; б) триацилглицерол с низким йодным числом?
457
*
Часть II. Метаболизм веществ и энергии в клетке
4. Где больше аккумулировано свободной химической энергии: в масляной
или пальмитиновой кислоте?
5. Жир может служить источником воды для животных пустыни.
Рассчитайте, сколько воды образуется при полном окислении 100 г триацилстеарилгли-
церола? о
о
6. В реакционной среде, содержащей пальмитил-СоА (С15Н31-С-8-СоА), ме-
О
14 "
ченый малонил-СоА (НОО С-СН2-С-8-СоА) и синтетазный комплекс, образует-
О
II
ся стеарил-СоА (С17Нзз~С-8-СоА). Обладает ли продукт этой реакции
радиоактивностью?
7. Почему при полном окислении лауряновой кислоты СН3-(СН2)10-СООН
выход АТР выше, чем при полном окислении сахарозы (С12Н22Оп), содержащей
то же количество атомов углерода?
Глава 12. Метаболизм аминокислот и нуклеотидов
Глава 12, МЕТАБОЛИЗМ АМИНОКИСЛОТ
И НУКЛЕОТИДОВ
Объединение процессов метаболизма казалось бы весьма
различных классов органических соединений аминокислот и нуклеотидов в
одну главу имеет определенный биологический смысл. Во-первых,
важнейший элемент в их структуре — азот — получен из одних и тех же
биологических источников. Аминокислоты и нуклеотиды —
ключевые звенья круговорота азота в природе/В-анаболических процессах
аминокислоты являются предшественниками пуриновых и пирими-
диновых оснований. Во-вторых, оба класса органических соединений
служат исходными элементами в синтезе белков и нуклеиновых
кислот и, следовательно, участвуют в формировании важной
биологической функции любого живого организма — хранении и передаче
генетической информации.
Биосинтез аминокислот, пуриновых и пиримидиновых оснований —
в большинстве случаев довольно сложный процесс, включающий от 5
до 15 стадий. Мы не будем описывать подробно эти стадии. Основное
внимание будет обращено на главные метаболические принципы,
касающиеся путей движения азота и углерода. Сначала мы познакомимся с
путями преобразования атмосферного азота в аммиак — химическую
форму, в которой он легко вступает в реакции биосинтеза аминокислот.
Затем опишем пути синтеза заменимых аминокислот и ознакомимся
кратко с более сложным синтезом незаменимых аминокислот,
ферментативные системы которого содержатся в растениях и микроорганизмах,
но отсутствуют у человека и животных. Хотя организм животных и
человека способен к синтезу заменимых аминокислот, основным
экзогенным источником аминокислот у них является поступающий с пищей
белок. Аминокислоты переводятся в доступную для организма форму
при переваривании белка под действием протеаз, входящих в состав
желудочно-кишечных секретов — пепсина, трипсина, химотрипсина, кар-
боксипептидазы, аминопептидазы. Свободные аминокислоты затем
всасываются через стенку тонкого кишечника и разносятся по всему
организму с потоком крови. Но недостаток в организме заменимых
аминокислот компенсируется их синтезом йе пох>о.
12.1. Круговорот азота в природе
Растения и микроорганизмы способны усваивать азот почвы,
переводя его из неорганической формы в органическую. Источником азота
459
Часть II. Метаболизм веществ и энергии в клетке
в почве являются два пути: 1) разложение останков вымерших
организмов (микроорганизмов, растений, животных) и 2) фиксация
атмосферного молекулярного азота (1Ч2).
Разложение останков организмов осуществляется
микроорганизмами, переводящими органическую форму азота в неорганическую, в
результате чего часть азота переходит в молекулярную форму N3 и
возвращается обратно в атмосферу. Восполнение этих потерь осуществляется
за счет фиксации азота атмосферы. На его долю приходится около 80 %
всех газов атмосферы или около 4 млрд т; иными словами,
молекулярный азот атмосферы является неисчерпаемым источником для
биосинтеза органических форм азота. Однако переводить молекулярный азот в
форму, доступную для живых организмов, могут специфические группы
бактерий. Ежегодно на Земле они связывают более 100 млн т
атмосферного азота. Именно эти бактерии обеспечивают реутилизацию доступной
формы азота, охраняя живую природу от экологической катастрофы,
поскольку растения и животные лишены этой способности.
Фиксация молекулярного азота из воздуха с образованием
аммиака осуществляется азотфиксирующими бактериями. Аммиак легко
переводится в органическую форму в виде аминокислот растениями и
микроорганизмами. Затем останки животных и растений
подвергаются микробиологическому разложению, органические формы азота вновь
переводятся в МН3. Часть азота под действием специфических
бактерий превращается в молекулярный азот и возвращается в атмосферу;
потерянный азот компенсируется его фиксацией из атмосферы.
Процесс разложения в почве белков, аминокислот, мочевины и других
азотистых соединений в результате жизнедеятельности
микроорганизмов называется аммонификацией. Поступивший в почву аммиак
окисляется бактериями — хемосинтетиками. Процесс окисления,
называемый нитрификацией, идет в две ступени: сначала аммиак
окисляется до азотистой кислоты бактериями ИИгозотопаЗу затем до азотной
кислоты — бактериями ЫИгоЪа&ег. Зеленые растения и многие виды
почвенных бактерий вновь восстанавливают нитраты до аммиака и
используют его в синтезе аминокислот.
Круговорот азота в природе представлен на рис, 12.1.
12.2. Азотфиксация
Превращение молекулярного азота атмосферы в аммиак
осуществляется азотфиксирующими бактериями. Некоторые из этих
микроорганизмов, а именно бактерии ЕЫгоЫит, проникают из почвы в корневую
систему растений, главным образом зернобобовых культур (соя, горох,
фасоль, люцерна), образуют на корнях клубеньки, в которых и происходит
связывание атмосферного азота (рис. 12.2). Эти бактерии находятся в
симбиозе с растением. Они питаются органическими веществами,
доставляемыми растением, а сами снабжают растение азотистыми соедине-
460
Глава 12. Метаболизм аминокислот и нуклеотидов
Азот атмосферы
Фиксация азота
бактериями
N11
Восстановление
растениями
Синтез
в растениях
Аминокислоты
н2
Нитраты
Разложение
останков
растений,
животных
Окисление
бактериями
(род ЙИгоЪас1ег)
ИНо
Нитриты
Окисление
бактериями
(род МИгонотопав)
Рис, 12.1. Схема круговорота азота в природе
ниями. Атмосферный азот могут
усваивать и некоторые свободно
живущие в почве микроорганизмы,
например анаэробные бактерии С1озШ(Иит^
сине-зеленые водоросли, аэробные
бактерии рода АгоЪоЪасЫг,
Превращение молекулярного
азота в аммиак представляет собой
сложный ферментативный процесс.
Энтальпия ковалентных связей в ]Ч2 (М^И)
весьма велика (^900 кДж * моль"1),
поэтому молекулярный азот устойчив к
химическим воздействиям.
Неслучайно А. Л. Лавуазье назвал этот элемент
из-за его инертности "азот", что
означает "безжизненный". Превращение
молекулярного азота в аммиак
описывается уравнением
Ы2 + ЗН2 ^ 2КН3.
Разработанный в 1910 г. Ф. Габе-
ром промышленный синтез
азотистых удобрений основан на этом
уравнении. Он идет при высокой
температуре (около 500 °С) и большом дав-
Рис. 12.2. Клубеньки на корневой
системе сои — место, где происходит
азотфиксация бактериями ЯЫгоЫит
461
Часть II. Метаболизм веществ и энергии в клетке
лении (около 30 Па). Поэтому не удивительно> что биологическая
фиксация атмосферного азота нуждается в затрате большого количества
энергии в виде АТР- Механизм участия АТР в превращении
молекулярного азота в аммиак неизвестен. Пока не удалось обнаружить
никаких фосфорилированных промежуточных продуктов в этом
процессе. Предполагается, что для преодоления высокого энергетического
барьера в молекуле 1^2 и восстановления ее до двух молекул ЬШа
требуется гидролиз 12 молекул АТР до АОР и Р..
Ферментная система, участвующая в фиксации атмосферного азота,
называется нитрогеназой. Этот сложный комплекс состоит из двух
компонентов, которые известны как белок 1 и белок 2. Белок 1 состоит из
двух различных полипептидных цепей с молекулярной массой 50 000 и
60 000, ассоциированных в смешанный полимер с субъединичной
структурой тетрамера а2Р2. Белок 2 является димером, но состоит из двух
идентичных пептидов с молекулярной массой 30 000. Димер
чувствителен к кислороду, при доступе которого он инактивируется. Связано это,
видимо, с тем, что он представляет собой железосерный белок,
содержащий 4 атома железа, 4 атома серы и 12 титруемых тиоловых групп.
Тетрамер (белок 1) содержит 2 атома молибдена, 24 атома железа, столько
же атомов серы и около 30 титруемых тиоловых групп.
Для восстановления 1Ч2 в ЫН3 требуется 6 электронов, источником
которых должен быть мощный восстановитель. У большинства азот-
фиксирующих микроорганизмов такую роль выполняет
восстановленный ферредоксин, который переводит МАВР+ в МАОРН.
Восстановление молекулярного азота идет в три этапа:
2е~ 2е~ 2е~
2Н+ 2Н+ 2Н+
Молекулярный Дйимид Гидразин Аммиак
азот
Последовательность реакций образования N13^:
АОР + Р1
КАВРН+Н+ ч , Белок 2окисл %Ь Белок 1^ х , Щ
(М
°2+) ~ У
док !„.._ ^ V
КАБР+ ^ ^ Белок 2^ ' > Белок 10висл + ^ 2NНд
(Мо6+)
АТР
Сначала КА1)РН отдает свои электроны белку 2. На втором этапе
АТР связывается с белком 2 и сдвигает его
окислительно-восстановительный потенциал с -0,29 до -0,40 В путем изменения его конформа-
ции. На третьем этапе происходит перенос электронов на белок 1,
гидролизуется АТР, и белок 2 отделяется от белка 1. Наконец, М2
связывается с белком 1 и восстанавливается до МН3.
462
Глава 12. Метаболизм аминокислот и нуклеотидов
Изучению нитрогеназной системы посвящено большое число работ,
что объясняется исключительно важным практическим значением
фиксации атмосферного азота. Процесс фиксации азота клубеньками бобо-
зых растений стимулируется молибденом и кобальтом. Нитрогеназа,
полученная из бесклеточных экстрактов клубен&ковых бактерий, быстро
лигибируется кислородом воздуха. Для предотвращения этого процесса
з клубеньках находится особый белок легоглобин> который подобно
гемоглобину крови легко присоединяет молекулярный кислород,
превращаясь в оксилегоглобин* В результате процесс азотфиксации идет в
строго анаэробных условиях, а оксилегоглобин является донором кислорода
при окислительном фосфорилировании. Однако механизм процесса
превращения Ы2 в КН3 до конца не выяснен. Исследование природы и
механизма действия нитрогеназы позволяет понять не только сущность этого
процесса, но и выявить новые пути совершенствования энергоемкой
технологии химического синтеза аммиака из молекулярного азота. В
настоящее время предпринимаются различные подходы в решении этой
проблемы. Были, например, предприняты попытки переноса клубеньковых
бактерий или их мутантов в небобовые культуры, в частности пшеницу,
кукурузу; предприняты попытки создания полезных симбиотических
ассоциаций. В связи с успехами генной инженерии проводятся
исследования по выделению бактериальной ДНК, ответственной за синтез
нитрогеназы, и ее вводу в геном микроорганизмов, не способных к такому
синтезу, или в геном растений.
12.3. Нитрификация
Соли азотной кислоты, образуемые в почве в результате окисления
аммиака почвенными бактериями — хемосинтетиками, являются
наиболее предпочтительной формой усвоения экзогенного азота для
большинства растений. Под действием бактерий ИНготбпа.8 N11 +
превращается в N0^":
N11/ + 1,5 02 -> N0" + 2Н+ + Н20;
АО/ = -272 кДж-моль"1.
Другой вид бактерий ИНгоЬааег окисляет N0" до N0":
N0 - + 0,5 02 -> К03-;
ДО/ 76 кДж • моль"1.
Выделенная при окислении энергия используется бактериями для
своей жизнедеятельности, а нитрат в виде солей азотной кислоты
поступает в корневую систему растений. Эти бактерии были открыты В. Н. Ви-
ноградским; они вездесущи, чрезвычайно активны и быстро окисляют
попадающий в почву аммиак. Транспорт нитрата в растительную клетку
осуществляется с помощью специфической пермеазы. Восстановление
нитрата происходит в два этапа: на первом — под действием нитратре-
дуктазы нитрат восстанавливается до нитрита; на втором — под
действием нитритредуктазы нитрит восстанавливается до аммиака.
463
Часть II. Метаболизм веществ и энергии в клетке
Препараты редуктаз были выделены из Е. соИ, из гриба Ыеигозрога
сгазза, из листьев сои, пшеницы, из проростков семян бобовых
растений. Оба фермента представляют собой металлопротеины. Для
действия нитратредуктазы необходим молибден. В качестве донора
электронов используется ИАОРН и ферредоксин.
12.4. Синтез аминокислот
В синтезе аминокислот источником азота служит аммиак в форме
N1^, источником углерода являются промежуточные продукты
цикла трикарбоновых кислот, гликолиза, гексозомонофосфатного цикла,
цикла Кальвина.
Синтез заменимых аминокислот. Существуют три основные пути
синтеза заменимых аминокислот: 1) восстановительное аминирование
(синтез глутамата и глутамина; по этому пути неорганический азот
превращается в органический); 2) трансаминирование — перенос
аминогруппы от аминокислоты — донора этих групп к ос-кетокислоте —
акцептору групп; 3) превращение одной аминокислоты в другую.
Восстановительное аминирование. В ассимиляции аммиака
особая роль отводится промежуточному продукту ЦТК — 2-оксоглутара-
ту. Под действием фермента глутаматдегидрогеназы из N11 + и 2-оксо-
глутарата синтезируется глутамат, в качестве восстановителя
используется ЫАБРН:
9°° КАВРН + Н+ ИАОР* 9°°
С=0 ^ у НзИ^-С-Н
сн2 —^>—<Г »' сн2
?н* гш4+ н2о * ?Нй
соо соо
2-Оксоглутарат Глутамат
Эта реакция имеет фундаментальное значение в азотистом обмене
растений, микроорганизмов. Глутамат является донором аминогрупп
при биосинтезе всех белковых аминокислот. Он же —
предшественник синтеза пролина и оксипролина. Глутаматдегидрогеназа
локализована в матриксе митохондрий, где функционирует ЦТК.
Глутамат является также предшественником в синтезе
глутамина — второго важного соединения, участвующего в ассимиляции
аммиака (перевода его в органическую форму):
?°°" АТР АБР + Н+ ?°°" мн+ ?00~
НзИ'-С-Н ч а Н^Г-С-Н *^4 Н3Ы+-С-Н
I У^ ^у* \ Ч^ I
С-Нд СН2 ~~^\. СНг
СН2 СН2 р.^ н+ СН2
соо с=о с=о
Глутамат О-Р *Ша
Глутамил-5- Глутамин
фосфат
464
Глава 12. Метаболизм аминокислот и нуклеотидов
Реакцию катализирует фермент глутаминсинтетаза. Связанный
с ферментом глутамил-5-фосфат соединяется с аммиаком в активном
центре фермента; при этом образуется глутамин и высвобождается
фосфат. Глутаминсинтетаза была выделена и очищена из клубеньков
сои. Ее молекулярная масса — 330 000; она состоит из 8 мономеров;
проявляет активность в присутствии Мп2+.
Глутамин служит затем источником азота в биосинтезе
биомолекул, наиболее важными из которых являются пиримидины и пурины.
По аналогичному пути в растениях идет синтез аспаратна.
Глутаматдегидрогеназа и глутаминсинтетаза являются весьма
активными ферментами и препятствуют накоплению токсического
аммиака в живой клетке в повышенных концентрациях, переводя его в
связанную органическую форму — глутамат и глутамин.
Синтез аминокислот трансаминированием. В живых
организмах наиболее распространенным путем синтеза заменимых
аминокислот является реакция трансаминирования. В этой реакции донором
аминогрупп служит в основном глутамат, их акцептором 2-оксокис-
лоты — промежуточные продукты катаболизма моносахаров; при этом
образуются 2-оксоглутарат и соответствующая аминокислота. Эта
реакция катализируется пиридоксальзависимым ферментом — транс-
аминазой:
ХТтт+ л Трансаминаза
I II (шфидоксальфосфат)
ООС-СН^СНо-СН-СОО" + К-С-СОО
Глутамат 2-Оксокислота
о нна*
^ »■ оос-сн2-сн2-с-соо- + к-сн-ооо-
2-Оксоглутарат Аминокислота
Реакция широко используется в метаболическом потоке азота. В
качестве оксокислот могут быть следующие соединения: пируват —
конечный продукт гликолиза, из которого при трансаминировании образуется
аланин; оксалоацетат — конечный продукт цикла трикарбоновых
кислот, из которого синтезируется аспартат. Эти аминокислоты
синтезируются с помощью весьма простых одностадийных реакций:
Пируват + глутамат ^ Аланин + 2-оксоглутарат;
Оксалоацетат + глутамат & Аспартат + 2-оксоглутарат.
У многих бактерий.аспартат служит предшественником амида ас-
парагина в реакции, катализируемой аспарагинсинтетазой. Эта
реакция аналогична глутаминсинтетазной:
Аспартат + N11 + + АТР & Аспарагин + АХ)Р + Р. + Н+.
Глутамат является донором аминогрупп при синтезе серина,
углеродным скелетом которого является промежуточный продукт
гликолиза — 3-фосфоглицерат. В первой стадии 3-фосфоглицерат окисляет-
30, Заказ 3486
465
Часть П. Метаболизм веществ и энергии в клетке
ся до 3-фосфопирувата, который подвергается трансаминированию с
образованием 3-фосфосерина; последний под действием фосфатазы
гидролизуется с образованием серина:
СОО"
!
н-с-он
зСН2ОР
З-Фосфоглицерат
ИАОР+ ИАВРН + Н
+
СОО"
I
с=о
I
зСН20Р
Глутамат
З-Фосфопи ру ват
2-Оксоглутарат
СОО"
чад-с-н
зСН2ОР
З-Фосфосерин
НоО
+
СОО
I
НзИ-С-Н
СН2ОН
Серин
Весьма сложен синтез гистидина — аминокислоты, входящей в
состав активных центров многих ферментов. Исходным веществом в
формировании имидазольной группы гистидина является АТР, а
донором углеродного скелета боковой цепи гистидина служит фосфори-
бозилпирофосфат, донором аминогрупп — глутамин. Реакция
включает 11 стадий; лишь на последних стадиях синтеза осуществляется
процесс трансаминирования.
Превращение одной аминокислоты в другую. Пролин, тирозин,
цистеин образуются в результате биохимических превращений
других аминокислот, в которых принимают участие специфические
ферментные системы.
Предшественником пролина является глутамат. В первой стадии
реакции глутамат восстанавливается в у-полуальдегид, затем
происходит спонтанное образование пятичленного гетероцикла с его
дальнейшим восстановлением:
СОО
+Н3КГ-СН
I
СН2
СОО"
Глутамат
+
ИАБРН + Нт ЫАБР
,+
+
АТР
АБР + Р,
СОО"
вд-сн
снг -
сн2
с=о
I
н
у-Полу альдегид
СОО
в СИ
/ \
N СН2
II !
НС — СН,
Пирролин-5-
карбоксилат
КАБРН + ВТ ЫАВР
> ,
>
Н20 + Н"
СОО
I
СН
/ \
ны сн2
I I
Н2С СН2
Пролин
466
Глава 12, Метаболизм аминокислот и нуклеотидов
Предшественником тирозина является незаменимая
аминокислота фенилаланин. Он образуется путем гидроксилирования фенильной
группы в положении 4. Эта реакция катализируется фенилала-
нин-4-монооксигеназой в присутствии КАВРН и кислорода:
Фенилаланин + МАВРН + Н+ + 02 -» Тирозин + ЫАВР+ + Н20.
Таким образом, отсутствие в пище фенилаланина ведет к
отсутствию в организме и тирозина.
Предшественником глицина является серии. Основной путь
превращения заключается в переходе трехуглеродной молекулы серина в
двухуглеродную молекулу глицина путем отщепления (3-углеродного
атома:
СОО" Серингидроксиметил- СОО~
+Н3К-С»Н ' - тр^сфераоа^^^^ +Н3К-С-Н
СН2ОН ' > ^^ Н
Серии Тетрагидро- К5-, К10-Метилен- Н20 Глицин
фолат тетрагидрофолат
Эта реакция катализируется ферментом серингидроксиметилтранс-
феразой, коферментом которой является тетрагидрофолат,
представляющий собой активную форму фолиевой кислоты (см. гл. 5). Он
является переносчиком группы -СН2ОН от молекулы серина на какой-
либо другой акцептор. Отщепляемый от серина {3-углеродный атом
образует метиленовыи мостик между атомами азота тетрагидрофолата
в положениях 5 и 10, в результате чего синтезируется 1Ч5-,К10-мети-
лентетрагидрофолат:
ОН Н ^Ш^^СО-К№- СН-С% СНгСОО"
N
/г*г6н° **
н
Но0
сн2~он
он
СН^ч^ЬСО-Ш-СН-СН^СН^СОО"
сн2 со°
Н2Гч N ^ N -^ -Метилентетрагидрофолат
н
Важнейшим элементом в составе многих биологически активных
соединений является сера. Она входит в состав двух аминокислот —
30
■к
467
Часть II. Метаболизм веществ и энергии в клетке
цисгпеина и метионина. Предшественником цистеина является серии,
а в качестве источника серы для синтеза цистеина используется
неорганический сульфат 8042~. При поступлении в клетку сульфат
превращается в метаболически утилизируемую форму аденозин-5-фосфосулъфагп:
Аденин
Остаток
рибозы
~1 1—
ОН ОН
АТР
О О О
II I! II
О-Р-О-Р-О-Р
I I I
О" О" О
АТР-сульфорилаза
80
2-
р^
Аденин
О О
Т I
ОН ОН
О-Р-0-8-0"
! II
О" О
Аденозин- 5'-фосфосу льфат
АТР
АЭР
Аденин
У
О
о о
II II
О-Р-0-8-0
I и
(Г
о
ОН 0-"Р=0
х0"
3'-Фосфоаденозин-5'-фосфосульфат
В этой реакции сульфат из свободного нереакционного состояния
переходит в более реакционноспособную форму, для чего на каждой
стадии необходим источник энергии в виде АТР.
В растениях и бактериях сульфатная группа восстанавливается в
сульфит (80|~), а затем в сульфид (82~). Реакция катализируется
двумя редуктазами, каждая из которых использует в качестве донора
электронов КАОРН; всего необходимо 4 молекулы КАОРН (8
электронов). Синтез цистеина из серина и сульфида идет через
промежуточное производное о-ацетилсерин:
ИН3+
I
но-сн*-сн-соо
Серии
СоА-8Н
СНз-С-СоА
О
О
+
вд
сн3-с-о-сн2-сн-соо
0-Ацетилсерин
I
Н8-СН-С00
СНя-С-О" Цистеин
И
О
Синтез незаменимых аминокислот. В исследованиях биосинтеза
незаменимых аминокислот использовались в основном микроорганизмы.
468
Глава 12. Метаболизм аминокислот и нуклеотидов
Установлено, что биосинтез каждой из них имеет специфические
особенности; он гораздо сложнее, чем биосинтез заменимых аминокислот.
Поскольку синтез каждой из незаменимых аминокислот имеет свою
особенность, мы не будем подробно его рассматривать и ограничимся
лишь некоторыми общими положениями. Так, три незаменимые
аминокислоты лизин, треонин и метионин в растениях и
микроорганизмах синтезируются из аспартата.
Изолейцин образуется у бактерий из незаменимой аминокислоты
треонина.
Многостадийный синтез у растений фенилаланина осуществляется
из эритрозо-4-фосфата и фосфопирувата; на предпоследних стадиях
осуществляется перенос аминогруппы от глутамата. Биосинтез гисти-
дина — незаменимой аминокислоты для детей — полностью изучен у
бактерий и грибов. Завершающей стадией является реакция транс-
аминирования, роль донора аминогруппы выполняет также глутамат.
Путь биосинтеза гистидина в высших растениях не изучен.
Регуляция синтеза аминокислот. Скорость синтеза
аминокислот определяется двумя основными факторами: 1) количеством
ферментов биосинтеза и 2) их ферментативной активностью. Регуляция
синтеза ферментов описывается в гл. 13. Здесь мы рассмотрим
регуляцию ферментативной активности. В основу этого типа регуляции
положен аллостерический механизм ингибирования первой реакции
конечным продуктом данной последовательности реакции:
г
Первая реакция (А—»В) обычно является важным участком
регуляции. Конечный продукт (Ь) биосинтетического пути ингибирует
фермент, катализирующий эту решающую реакцию. Она обычно
необратима и катализируется аллостерическим ферментом. Примером этого
принципа регуляции является регуляция биосинтеза изолейцина из
треонина, который был обнаружен в клетках Е. соЫ. Решающей
реакцией в этом синтезе является превращение треонина в а-кетобутират
под действием треониндезаминазы. Конечный продукт — изолейцин —
аллостерически ингибирует треониндезаминазу в случае его
накопления в клетке в большой концентрации, т.е. имеет место ингибирова-
ние по типу обратной связи.
12.5. Катаболизм аминокислот
В отличие от Сахаров и жирных кислот, аминокислоты в избытке
не запасаются в организме. Их роль в организме определяется тем,
что они служат строительными блоками для биосинтеза белков.
Избыточные аминокислоты подвергаются расщеплению. Существуют два
пути их диссимиляции: 1) окислительное дезаминирование и 2) де-
карбоксилирование.
469
Часть II. Метаболизм веществ и энергии в клетке
Окислительное дезаминироваиие аминокислот. В этом типе
реакций аминогруппа отделяется от аминокислоты, а остающийся
углеродный скелет превращается в промежуточные продукты обмена
веществ. Избыточные аминокислоты используются как источник
метаболической энергии. Хотя большую часть метаболической энергии,
вырабатываемой в организме, поставляет окисление углеводов и
жиров, 10—15 % ее общего количества приходится на окисление
аминокислот. Избыток аминокислот может возникнуть в организме либо
при катаболизме белков, причем образующиеся аминокислоты не
используются для синтеза новых белков, либо при поступлении
аминокислот с пищей в большем количестве, чем требуется организму, либо
при поступлении с пищей недостаточного количества углеводов —*
источника углерода и энергии.
Окислительное дезаминироваиие аминокислот приводит к
образованию аммиака и углеродного скелета в форме а-кетокислоты. Эта
реакция идет в две ступени: в первой — аминокислота окисляется до
иминокислоты:
Дегидрогеназа
К-СН-СОО" -^—^- *~ К-С-ССХГ
1 + /* % Хтт+
ИЦ? ' Л 1ОДГ
КА1)+ ЫАВН + Н+
во второй — иминокислота превращается в кетокислоту;
?
к-с-соо- + н2о ш* к-с-соо- + ]*н4+
ын2+
Аммиак для всех живых клеток является токсичным соединением.
В растениях и микроорганизмах прямого выделения аммиака не
происходит; ос-КН2-группа расщепляемой аминокислоты переносится
на 2-оксоглутарат, в результате чего образуются глутамат и
соответствующая 2-оксокислота:
СОО" СОО"
К С=0 Трансамшшрование К Н3М+-С-Н
I 1 II
+
2
1ЫГ-С-Н + СЩ — ** С=0 + СН
СОО" СН2 СОО" <^н
СОО" СОО"
Аминокислота 2-Оксоглутарат 2-Оксокислота Глутамат
Дезаминироваиие различных аминокислот приводит к
образованию единственного продукта — глутамата, выполняющего роль
"коллектора" аминогрупп, предотвращающего их превращение в аммиак.
Реакция трансаминирования 2-оксоглутарата легко обратима;
величина ДО0 для нее близка к нулю, поэтому она играет важную роль как
в расщеплении, так и синтезе аминокислот: при синтезе она выполня-
470
Глава 12. Метаболизм аминокислот и нуклеотидов
ет роль донора аминогрупп, при расщеплении аминокислот — роль
акцептора этих групп.
Окислительное дезаминирование самого глутамата катализирует-
ся глутаматдегидрогеназой, в результате чего образуется ион
аммония, Глутаматдегидрогеназа обладает необычной способностью: в ка-
таболических процессах она передает водород на ЫАП+, в
анаболических — на ИАВР+:
СОО" СОО"
+ I I
НзЫ -С-Н Глутаматдегидрогеназа С=0
СН2 —рг—7 "X т СН2 + ИН^
I
СНй — + тт л *т * т\тт тт+ СН.2
ИАО+ Н20 ЫАВН + Н
СОО (или МАВР+) (или КАОРН + Н+) со°
Глугамат 2-Оксоглутарат
Активность глутаматдегидрогеназы подавляется АТР и ОТР,
тогда как АОР и ОВР служат для нее активаторами. Иными словами,
снижение энергетического заряда в живой клетке ускоряет
окислительное дезаминирование глутамата.
В какой форме образованный в этой уникальной реакции КГН4*
обезвреживается и выводится из организма?
У высших растений и многих микроорганизмов такой формой
являются амиды аминокислот; у человека и большинства наземных
позвоночных животных — мочевина, безвредное, хорошо растворимое в
воде соединение, которое выводится с мочой; у рыб N11^ выводится
через обильно орошаемые водой жабры; у птиц и пресмыкающихся
МН4+ превращается в мочевую кислоту, а выводится из организма
вместе с калом;
0 н
II Г
ИН4+ ВДГ-С-КН2 | и Чс==ю
Ион аммония Мочевина о^ Чч№"''''' -у
н н
Мочевая кислота
Синтез амидов. Характерной особенностью высших растений и
многих микроорганизмов является строжайшая экономия азота.
Появившийся в клетке аммиак не выводится из нее, а быстро
вовлекается в синтез амидов — глутамина и аспарагина, рассмотренный в
разд. 12,2. Глутамин и аспарагин образуются в различных органах и
тканях высших растений: в корнях, стеблях, листьях и плодах. При
избыточном питании азотистыми соединениями, в частности
аммонийными солями, высшие растения могут накапливать достаточно
большое количество глутамина и аспарагина. Особенно много этих
471
Часть II. Метаболизм веществ и энергии в клетке
амидов образуется при прорастании семян бобовых растений,
содержащих сравнительно небольшой резерв углеводов и большое
количество белка. Так, прорастающий в темноте люпин может накапливать
до 11 % аспарагина от сухой массы проростков. Интенсивный синтез
глутамина идет в сахарной свекле. При внесении в почву больших
количеств аммонийных солей в качестве удобрения в корнях свеклы
может накапливаться более 5 % глутамина от сухой массы. В
свеклосахарном производстве глутамин относят к числу "вредного азота",
поскольку он снижает чистоту диффузионного сока, является
источником образования меланоидинов и тем самым препятствует
кристаллизации сахара.
Синтез мочевины. Образующийся в организме позвоночных
животных аммиак обезвреживается в цикле мочевины (рис. 12.3).
Мочевина синтезируется специфической группой ферментов,
координированное действие которых осуществляется в ряде последователь-
2АТР
ОН
Карбамоил-
фосфат
ин2
(СН2)3
нсш2
соон
Орнитин
* ^ .-. -л ■.■■■.■■■.-. л - /*^^ *г* ■>
ш * Н ■■■ I ' * _■ { 4 ф ■ ■ 1 Л ш *
'" .!•,* 'у.*/^< . < ,
. { . { *^ . ^ * л , «. * V* ' X"
.-.■..■ ^^-■■■■■■■ ■ ^^ .-■■,■
ш
(СН2)3
нс>га2
соон
Цитру ллин
АТР
СООН Аспартат
Ж&&ШЖФ.
Ж * ._.■.■.-. • у* Ш* / *, * \ *ц/ '4
....4.. >* 1Щ.. .^^ >'-V>• ■ ■
ын
(СН2)8
НСИНг
СООН
СООН
I
- СН
СН2
СООН
Аргинино-
сукцинат
Оксалоацетат
Малат
тт
(СН2)8
Фумарат
Мочевина
НШН2 Аргинин
I
СООН
Рис. 12.3, Цикл мочевины
472
Глава 12. Метаболизм аминокислот и нуклеотидов
ных реакции: орнитин —» цитруллин —> аргининосукцинат —>
аргинин. Эта последовательность была постулирована Г, Кребсом и К. Гей-
деляйтом в 1932 г. — за пять лет до открытия цикла трикарбоновых
кислот.
Источником углерода мочевины является С02, который можно
рассматривать как акцептор атомов азота. Из N11+, С02, Н20 и АТР в
сложной реакции, катализируемой карбамоилфосфатсинтетазой,
образуется карбамоилфосфагп (реакция 1 на схеме):
Карбамоилфосфатсинтетаза О
N11/ + С02 —т>~ -р~ ^— *- Н2Ы-С-0~Р
2АТР Н20 2АВР + Р, Карбамоилфосфат
Потребление двух молекул АТР ведет к необратимости синтеза
карбамоилфосфата и обусловливает в нем достаточно большой запас
свободной энергии благодаря наличию ангидридной связи. Затем
карбамоилфосфат конденсируется с орнитином, образуя цитруллин;
реакция катализируется орнитинкарбамоилтрансферазой (реакция 2):
О
II
ИНз л НК-ОЫН2
I Орнитин- | *
9^2 карбамоил- 9^2
■2
СН2 ? трансфераза ^
! _!_ Ы_ХТ_ГП_/Ч.ЛЭ №~ 1
+ н2*г-с-<ьр ** ■ + р5
СНо ' ***** ~ СН
2
I I
Н8Ы+-С-Н ВД^-С-Н
СООН СОО"
* Орнитин Цитруллин
В ходе второй АТР-зависимой реакции цитруллин конденсируется
с аспартатом (вторым источником азота в цикле мочевины) с
образованием аргининсукцината. Реакция катализируется аргинипсукци-
нагпсингпетазой (см. на схеме этап 3):
вд+ СОО"
о соо" с-кн-сн
„„ " . I Аргининсукцинат- I I
1Ш-С-га2 Щ^-С-Н еинтетаза *Н Ш2
сн2 сн2 ~7^ ^ *"" сн2 соо^
+
СН2 СОО АТР АМР + РР^ СН2
СН2 » СНо
I Аспартат , 2
Н81Г-С-Н НзМ'-С-Н
соо~ соо"
Цитруллин Аргининсукцинат
Аргининсукцинат затем расщепляется на аргинин и фумарат под
действием аргининосукцинатлиазы (реакция 4):
473
Часть II. Метаболизм веществ и энергии в клетке
н2ы+ соо" вд
С-1Ш-СН Аргининосук- С~НН2
N11 СН2 цинатлиаза N11
сн2 соо" "■"* сн2 н соо
1 | ^
СН2 СН2 • |
1 ■ С
СН2 СН2 _ / \
"оос н
I
Н8Ы+-С-Н Н8КР-С-Н
СОО" СОО" Фумарат
Аргининсукцинат Аргинин
Завершающей стадией синтеза мочевины является гидролиз
аргинина ферментом аргиназой (реакция 5):
Н2И+ ЫН
с-ын2 н2ы сн2
^н Аргиназа с=0 ^Н
+
3
I , ш I +
2
СН2 X Н2Ы ' СН2
I ' I
СН2 Н20 Мочевина Н3К+-С-Н
СН2 СОО"
■ Н3Ы+-С-Н Орнитин
СОО"
Аргинин
Хотя ЗЧН^* и аспартат являются непосредственными источниками
мочевины, они оба могут быть образованы из глутамата- Глутаматде-
гидрогеназа катализирует образование аммиака из глутамата.
Аспартат же может получать азот в виде аминогрупп в процессе трансами-
нирования от глутамата, где оксалоацетат выполняет роль акцептора
аминогрупп.
Поскольку 2-оксоглутарат, оксалоацетат, фумарат являются
важными метаболитами цикла трикарбоновых кислот, цикл мочевины
тесно связан с последним. У рыб в процессе окислительного дезамини-
рования различных аминокислот аммиак быстро включается в глута-
мат с образованием глутамина. Последний кровью переносится в
жабры рыб, где гидролизуется ферментом глутаминазой:
Глутамин + Н20 -> Глутамат + ЗЧН^.
Аммиак из жабр удаляется водой.
У птиц и пресмыкающихся аммиак выводится в форме мочевой
кислоты. Синтез ее сложной молекулы представляет собой
многоэтапный процесс и тесно связан с синтезом пуринов, который мы
рассмотрели в разд. 12.7.
Расщепление углеродных скелетов аминокислот. После дез-
аминирования для каждой из 20 белковых аминокислот характерен
свой путь расщепления. В конечном счете углеродные скелеты всех
аминокислот поступают в цикл трикарбоновых кислот, где они окис-
474
Глава 12. Метаболизм аминокислот и нуклеотидов
ляются до С02 и Н20. Полученные восстановительные эквиваленты в
форме ЫЛОН в дыхательной цепи расходуются на синтез АТР.
Декарбоксилирование аминокислот. Второй важный тип
диссимиляции аминокислот — их декарбоксилирование. В результате этой
реакции удаляется а-карбоксильная группа аминокислот и
образуются амины — весьма физиологически активные соединения. Реакция
катализируется пиридоксальфосфатзависимыми декарбоксилазами:
Е-СН-СОО" Декарбоксилаза^ к_СНг_№ + ^
2ущ + Пиридоксальфосфат
Декарбоксилирование имеет место во всех видах организмов. Хотя
эта реакция не является количественно преобладающей в расщепле-
нии аминокислот, однако продукты декарбоксилирования имеют
большое физиологическое значение. Амины активны даже в очень низких
концентрациях.
У животных в результате декарбоксилирования глутамата
образуется у-аминомасляная кислота:
С02
>.
оос-сн-сн2-сн2~соо~ —-^—** н3н+-сн2-сн2-сн2-соо
хттт + Декарбоксилирование А
^«з * у-Аминомасляная
Глутамат кислота
у-Аминомасляная кислота относится к нейромедиаторам —
химическим соединениям, влияющим на передачу нервных импульсов.
Весьма интенсивно образуют у-аминомасляную кислоту некоторые
молочнокислые бактерии и растения.
В высших растениях и грибах аспарагиновая кислота, декарбокси-
лируясь, превращается в р-аланин:
со2
"ООС-СН-СН2-СОО" —^ *- Н3К+-СН2-СН2-СОО~
^Нз+ Декарбоксилирование р.Аланин
Аспартат
В ничтожных количествах р-аланин является эффективным
стимулятором роста дрожжей; он входит в состав лантотеновой кислоты.
Декарбоксилирование является важной реакцией при разложении
белков в результате жизнедеятельности гнилостных бактерий. Так,
например, при декарбоксилировании лизина образуется кадаверин
(пентаметилендиамин):
С02
Н8Н+-(СН2)4-СН-СОО" ~—-^ *- НЙН+-(СН2)4-СН2-КН3+
2^^ + Лизиндекарбоксилаза ™
Лизин
При декарбоксилировании орнитина образуется путресцин (тетра-
метилендиамин), из тирозина — гпирамин, из гистидина — гнета-
мин, из триптофана — триптамин.
475
Часть Л. Метаболизм веществ и энергии в клетке
Образующиеся при декарбоксилировании кадаверин, путресцин,
триптамин являются основной причиной порчи мясных и рыбных
продуктов. При дальнейших превращениях триптамин дает ряд
промежуточных продуктов, а затем — скатол и индол — ядовитые
соединения, от которых в основном зависит запах гниющего мяса. Реакции
превращения триптофана:
со2
N
Н
-сн2-сн~соо"
^ „
Декарбоксилирование
-СН2~СН2-ЫН
2
N
Н
Триптофан
Триптамин
2Н
СН3 + СН3-Ш2
Метиламин
Скатол
Индол
Декарбоксилирование аминокислот у растений имеет прямое
отношение к биосинтезу алкалоидов — азотсодержащих органических
соединений, обладающих более выраженной физиологической
активностью, чем амины.
-В высших растениях и микроорганизмах расщепление
аминокислот может включать одновременно процессы и дезаминирования, и
декарбоксилирования. Именно такой путь отмечен для дрожжей
8асскаготусе$ сегеьшаеу вызывающих спиртовое брожение. В
результате дезаминирования и декарбоксилирования некоторых
аминокислот образуются побочные продукты спиртового брожения, сивушные
масла — смесь различных одноатомных спиртов, влияющих на
качество вина, пива, этилового спирта. Так, лейцин в ходе
последовательных реакций дезаминирования, декарбоксилирования и
восстановления превращается в изоамиловый спирт:
ИХ
ин4+
НяС
сн-сн2-сн-соо
ЗЯНз
Лейцин
+
НЯС
н»с
\
/
о
со.
сн-сн2-с-соо
ЫАР ИАОН + Н
+
НзС^
н*с
/
СН-СНо-СНО
КАЭН + Н+ ИАБ
н,с
\
/
СН-СН.>-СНоОН
н8с
Изоамиловый спирт
Аналогичный путь превращения валина приводит к образованию
изоб у тылового спирта.
476
Глава 12. Метаболизм аминокислот и нуклеотидов
12,6. Биосинтез нуклеотидов
Предшественниками биосинтеза нуклеотидов являются
аминокислоты: аспарагин, глицин, глутамин. Помимо аминокислот в синтезе
используются С02, МН3 и рибоза.
В расшифровке путей биосинтеза нуклеотидов большую роль
сыграл метод меченых изотопов. Животным скармливали различные
меченые метаболиты, после чего определялось место включения метки в
пуриновое или пиримидиновое кольцо. Было установлено, что пути
биосинтеза пуриновых и пиримидиновых нуклеотидов различны. В
каждом из них образуется свое гетероциклическое азотное основание —-
гипоксантин (6-оксипурин) и урацил (2,6-диоксипиримидин):
О
II
С-
0
\' II >сн
1 ■> в II
2
6.
н
Гипоксантин
н
Урацил
Биосинтез пуриновых нуклеотидов. В синтезе пуринового
кольца принимают участие три аминокислоты: глицин, который является
донором двух атомов углерода (С-4 и С-5) и одного атома азота (N-7);
донором двух атомов азота (N-3 и N-9) является глутамин, а
последний атом азота (N-1) поступает из аспарагина. Активированные
производные тетрагидрофолата поставляют атомы С-2 и С-8, тогда как
С02 служит источником атома С-6 (рис. 12.4),
Глицин
Аспартат
5 10
N -, N -формил-
тетрагидрофолат
Глутамин
5 10
N -, N -метилен-
тетрагидрофолат
Рис. 12.4. Происхождение атомов пуринового кольца
Общая схема синтеза пуриновых нуклеотидов показана на рис. 12.5.
Особенностью синтеза пуриновых нуклеотидов является тот факт,
что он начинается с рибозо-5'-фосфата, на котором идет сборка
пуринового кольца. Источником энергии в сборке кольца являются АТР и
СТР.
477
он
НО-Р-О-СНг
О
он он
а-Е-Рибозо-5-фосфат
Г
АТР
Фосфорилаза
АМР
ОН
НО-Р-О-СНз
о I/ \ он он
, , о-р-о-^-он
он он А А
а-5-Фосфорибозил-1~пирофосфат (ФРПФ)
СТР.
АЛ*
Ж
*
ФРПФ-ажидо-
трансфераза
ОН
НО-Р-О-СНа
О
сн2-сн2-сн-соо
N1*3
Ь-Глутамин
ООС-СН2-СН2-СН-СОО
Ь-Глутамат
р-5 -Фосфорибонилами н
Фосфорибозил-глицинамий*
синтетаза
АТР +ШШ6?ШЩ&<
у 2' ГЛИЦИН
АОР + (Р
нчжвь
ГЛИЦИНАМИД- (р)_|цЬ
(ГАР) I
5~Аминоимидазол-
рибонуклеотид-
карбоксилаза
С02
еоо"
5-Амвноимидазол- Н0^^»:Щ^
4-карбокси лат- л '
р иб о н ук леоти д
(р)-тъ
А
$-Аминоимидазол~4-№
сукциткарбоксиамид-
рибонуклеотидсинтетаза
/*
-«ЖгСН-СНа-СООН + АТР
М^
соон
Ь-Аспартат
АВР + Ш + Н
<л
5- Ами ной ми даэол -4-Ы -
сук ця но к арбокс иами Д'
рибонуклеотид (А ИКАР)
О
;■©■
р»-С-СН2-СООН
|;.. Й СООН
©-ШЪ
Аденилсукциназа
I Н С
у ноос' н
соон
Фумарат
5-Ам и кои м идазол-
4-карбоксиамнд- *-^ :*С
рибонуклеотид (АИКАР) (Е>-КаЬ
5-Ам и поим ида зол- 4-карбокси амид-
рибонуклеотид транформилаза
Г
-10
N -Формил-гаи
к+
рн,
жм
5-Формамидимидазол- Н ^Ц^ч
4-карбоксиамидрибонуклеотид ^-^ "Г
(ФАИКАР) Оу-ШЪ
ЩН3
■О
н
|Л V,10
Глицинамид- > г
рибонухлеотид' у
трансформилаза
Ишкшпикимм
лазаАк
V
о .*;!■■
а-К'Формилглициыамид-
рибояуклеотид (ФГАмР)
Е*АВ+
Ипозинатдегидрогеназа
еназа ]
ШК-С-СН2-СН2-СН-СОО'
а- №Форм ил ел ицинамид-
рибонукмотид -
амидолцгаза
ШЯПя
1щтшр- о
Ксантилат (р)_#1Ъ
Глутамат
1)
РЫЦЬ
АТР ^^ч ?Ш^С-СНг-СН2'СН-С00"
Гуанилатсинтетаза ^ц к Глутамин
/Ч ^^ Глутамат
^ШШ^*н
ам;
а-К-Формилгллцпвадшд-
рибонуклеотид (ФГАР)
АТР + Н20
5-Амцноимидазол- (
рибонуклеотид- Г М^2*, К+
синтетаза К,
I ^ АЮР + (?) + 2НГ*
Гуанилат (ГМФ)
;1й;;^-">ЧмД1
5-Амнкоимидазол- Н %гШ^';$Йй-
рябонуклеотид (р)-К1Ь "''
+
авр
1ЩН-СН-СНг-СО0Н
соон
Аспартат
Аденилосущиштсишпетаза
<рОН
НС-СНг-СООН
•Я
щ-н
щ&щвгъ
Аденнлосукцияат
АдениА
к Н
АдениЛ осу кцчн&зи
СООН
/
р"С^ Фумарат
ноос н
11 1 < Л/^\
Адевилат (АМФ)
со
Рис. 12.5. Общая схема биосинтеза пуриновых нуклеотидов
Часть II. Метаболизм веществ и энергии в клетке
Образованная в результате субстратного или окислительного фос-
форилирования АТР участвует в синтезе ОТР:
ОМР + АТР <± ОВР + АВР
ОВР + АТР ?* ОТР + АВР.
Обе реакции катализируются специфическими нуклеозидкиназами.
Источником пуриновых оснований при синтезе нуклеотидов в
клетке является также гидролитическое расщепление нуклеиновых
кислот и нуклеотидов, поступивших с пищей, а также повторное
использование эндогенных нуклеиновых кислот, подвергшихся частичному
расщеплению. Этот путь синтеза гораздо проще, чем синтез йе поро9
где сборка пуринового кольца осуществляется на рибозо-5-фосфате.
При синтезе нуклеотидов на освободившихся пуриновых
основаниях рибозофосфатная группа б'-фосфорибозил-а-В-пирофосфата
{ФРПФ) переносится на пурин и образуется соответствующий пурин-
рибону к леотид:
Р-О-СН
Пурин
О-Р-Р
ОН
ОН
5'~Фосфорибозил-а-В-пирофосфат
(ФРПФ)
Р-О-СН^^Х. Пурин
ОН
ОН
Пуринрибонуклеотид
Реакция протекает под действием специфических фосфорибозил-
трансфераз. Она сопряжена с расщеплением пирофосфата, которое резко
сдвигает эту реакцию в сторону синтеза нуклеотида.
Биосинтез пиримидиновых нуклеотидов. Синтез пиримидино-
вых нуклеотидов отличается от синтеза пуриновых нуклеотидов:
сначала собирается пиримидиновое кольцо, а затем это кольцо
присоединяется к рибозофосфату. Донором рибозофосфата служит ФРПФ. Само
пиримидиновое кольцо синтезируется из карбамоилфосфата и аспар-
тата (рис, 12.6).
Общая схема биосинтеза
пиримидиновых нуклеотидов представлена
на рис. 12.7.
Синтез карбамоилфосфата
аналогичен его синтезу в цикле мочевины
(см. разд. 12.3). Однако в обоих
случаях синтез компартментализован:
к ^ пиримидиновый карбамоилфосфат
фосфат синтезируется в цитоплазме; карба-
Рис. 12.6. Происхождение ато- моилфосфат, идущий на синтез мо-
мов пиримидинового кольца чевины, — в митохондриях.
Аспартат
480
Глава 12. Метаболизм аминокислот и нуклеотидов
сн2
сн2
I
Н-С-ЫНг
I
соон
Глутамин
ЦТР
4
2АТР _,_Ч>„_
?
А0Р
Н
Карбамоилфосфат-
синтетаза
О ОН
* ^ * АУ V* ,г Ъ 4 ЧГ I
ЯййгС-0-Р-ОН
СООН
сн2
сн2
I
Н-С-КНй
I
соон
Глутамат
г у
^а;и^Ш
?2
®
н
у| Аспартаттранс-
>*» * карбомоилаза
нощш
* * у * <♦^^
г6Н2
ШШ Щ, соон
. ?«*<гъ^>4> •■-*• '* ** т^
У1. <■ ->*&/<> *V** ■■■■■■■■ ***<г^
» *»Г<^*1ГЛ *' '*ЧР ' * ' * ■"■
■* * г Ль*^* ***И**л> 11
** '*'А'А** * * * *
н
Дшидрооратаза
НгО
й
К-Карбамоил-
аепартат
О
Ч^шшчнг ,,-■ ■* -1к- ;'";'иг* 1
Ой! а;:: ^\%г\Х1
гфл ^ЩН^СООН
. ^ъ^ ■.,■■.■■.. ■;.■■.;■.; _вГ' "
Ч*^' АЪ< ■* < "* ■* '_1г^' ' ''' '*' Т Т
■ У * * \* V ■ '
X Ь-Днгидро-
КАР* *ч. | оротат
Диеидроорогпат- ]
дегидрогтаза ^т (*"АО, Ре )
КАШ + Н+^*
О
* ■
Н. ..+* -_А--. ^/а- |Т
II. Л
ОН ОН
0-Р-0-1>-0Б
о^^ШР-соон
Орогат
■ * н * ■■ ■■ ■'
И
5-Фосфорнбоэилпирофосфат
Ьат (ФРПФ) А
Оротатфосфорибозил-
трансфераза
ОН
но-р-о-дн2
6
Оротидилат
СООН
Р)ЧРМР)-Рибоза
С02
А
АОР -^» ♦ ^г Глутамат
О
-сикшетпаза
АТР
Н
б Я
А ?
^ | ^ ^Нр-С-(СН2)2-СН-СООН
0Л.МАН
Оротидин-5'-фосфатп-
декарбоксилаза
Н
Уридин-5'-фосфат
^^
О Глутамнн
Н
N1^
(Р)-Рибоза (ЦМР)
X 1
Р)ЧР)ЧР)-Ри6оза
АТР*х1
МяН ПМР-киназа
Нуклеозид- ддр-^и»
дифосфаткиназа
У^' V. Уридин-5'-днфосфат
^М^\ ОШР)
АОР АТР
Рис. /2.7. Общая схема биосинтеза пиримидиновых нуклеотидов
31. Заказ 3486
481
Часть II. Метаболизм веществ и энергии в клешне
Цитоплазматический карбамоилфосфат вступает в реакцию с ас-
партатом, в результате чего образуется N-карбамоиласпартат:
О
II
СН2
соо
Н^-ОСКР +
Карбамоилфосфат Н-С-МЩ
СОО"
Аспартат
Аспартаттранс
карбамоилаза
+
Р1
~ООС
Н2К СН2
I I
о=с нс-соо
вш
>Т-Карбамоиласпартат
Эта реакция катализируется аспартаттранскарбамоилазой.
Затем карбамоиласпартат циклизуется с отщеплением молекулы воды и
образованием дигидрооротата, из которого через ряд промежуточных
реакций образуется уридин-5'-фосфат (1ШР):
О" О
X
+Н Л х СН2 Дигидро-
оротаза
Н
Ж^
о
II
с
о
о
НС-СОО"
I
н
о"
н„о
I
н
сн2
Рибоза
-СОО".
иГ"
сн
О^
сн
I
Рибоза-Р
N - карбамои л аспарт ат
Дигидрооротат
Уридин-5'-фосфат (Х1МР)
Образовавшийся Х1МР с участием АТР фосфорилируется и
превращается в ОТР; последний присоединяет аминогруппу от глутамина и
превращается в цитидинтрифосфат (СТР):
О О
II
Киназа -. С-
н
^
сн
О
сн
I
Рибоза-Р
2АТР
2АБР ^С.
О'
СН
сн
N
Рибоза-Р-Р-Р
Уридин-5'-фосфат (1ШР)
Уридин-5'-трифосфат (1ГГР)
КН.
СТР-синтетаза
2АТР 2АБР
+ +
Глутамин Глутамат
НК
о^\ ^
сн
II
сн
Рибоза-Р-Р-Р
Цитидин-5'-трифосфат (СТР)
482
Глава 12. Метаболизм аминокислот и нуклеотидов
Биосинтез дезоксирибонуклеотидов. Эти предшественники ДНК
образуются в реакциях восстановления рибонуклеотидов. Гидроксиль-
мая группа при 2'-атоме углерода остатка рибозы замещается атомом
зодорода в соответствии с уравнением
Рибонуклеозиддифосфат + ЫА1)РН + Н* ~»
-» Дезоксирибонуклеозиддифосфат + КАВР+ + Н20.
Таким путем АВР восстанавливается до дАВР, а ОВР — в сЮВР*
Механизм протекания этой реакции достаточно сложен. Так, у Е. сой
з передаче водородных атомов участвует белок тиоредоксин. В его
молекуле находятся две 8Н-группы, при участии которых и осуществляется
эта передача. Окисленная (дисульфидная) форма тиоредоксина
восстанавливается за счет ЫАОРН в реакции, катализируемой тиоредукта-
зой. Затем восстановленный тиоредоксин передает атомы водорода рибо-
нуклеозидфосфату, переводя его в дезоксирибонуклеозиддифосфат:
ИАВРН + Н+ + Тиоредоксин | *- ЫАОР+ + Тиоредоксин
Ч8' Ч8Н
Тиоредоксин / + Рибонуклеозид- ^ Тиоредоксин^ | + Н20 + Дезоксирибо-
\о-и дифосфат \о нуклеотид
Процесс восстановления рибонуклеозиддифосфатов в дезоксирибо-
нуклеозиддифосфаты регулируется аллостерически. Предполагается,
что рибонуклеотидредуктаза имеет множество конформационных
состояний, характеризующихся различными каталитическими
свойствами. Это обеспечивает синтез всех четырех дезоксирибонуклеотидов в
необходимых количественных соотношениях для синтеза ДНК.
Напомним, что в состав ДНК не входит урацил. Вместо него ДНК
содержит метилированный аналог урацила — тимин. В реакции
метилирования принимает участие дезоксирибоуридилат (сШМР).
Последний превращается в дезокситимидиннуклеотид (с1ТМР). Под
действием фермента тимидилатсинтетазы метильная группа
переносится от К5-,1Ч10-метилентетрагидрофолата на остаток урацила сШМР:
<11ШР + К5-,Ы10-метилентетрагидрофолат -> ЙТМР + Тетрагидрофолат
Биосинтез дезоксирибонуклеозидтрифосфатов —
предшественников ДНК осуществляется в результате реакций фосфорилирования дез-
оксирибонуклеозиддифосфатов в дезоксирибонуклеозидтрифосфаты.
Фосфатная группа переносится от АТР соответствующими киназами.
12.7. Распад нуклеотидов
В клетке РНК и ДНК подвергаются непрерывному обновлению. В
результате совместного действия эндо- и экзонуклеаз они
распадаются на нуклеозидмонофосфаты:
31*
483
Часть II, Метаболизм веществ и энергии в клетке
РНК 1 ^йдо" и экзонуклеазы
ДНК
}
Нуклеозидмонофосфаты
Хотя основная масса нуклеозидмонофосфатов вновь вовлекается в
процесс биосинтеза РНК и ДНК, определенное их количество
подвергается дальнейшему расщеплению.
Распад пуринов. В большинстве организмов первичные процессы
расщепления ОМР и АМР различны, но приводят к образованию
одного продукта — ксантина.
Гуанин из ОМР непосредственно превращается в ксантин, тогда как
образование ксантина из АМР происходит через промежуточную стадию:
после дефосфорилирования АМР адениновый фрагмент аденозина в
результате дезаминирования превращается в инозин. Последний гидроли-
зуется с образованием свободного основания гипоксантина, который
затем окисляется до ксантина под действием ксантиноксидазы. Ксантин-
оксидаза —■ флавопротеин, содержащий молибден и железо.
Особенностью функционирования этого фермента является то, что в процессе
окисления образуется высокотоксичный радикал О;;. Его детоксикацию
осуществляет супероксиддисмутаза:
Супероксиддисмутаза
202" + 2Н+ ** Н202 + 02.
Пероксид водорода разлагается под действием каталазы или пер-
оксидазы:
Н
Рибоза
о
к/кк^
Рибоза-Р
Рибоза
ОМР
Ш,
Н.0
Н
1Чк/\г/
%
Гуанозин
О
О
А н
Ксантин
,о,
о.
Гипокеантин
Р,
ОС
о
ш.
I
Рибоза-Р
АМР
Рибоза
Аденозин
Гуанин
Г^""^>---Нч Ксантиноксидаза н*Г^*-""
кЛ
Рибоза
Рибоза
Инозин
484
Глава 12, Метаболизм аминокислот и нуклеотидов
У человека и других приматов, некоторых рептилий ксантин под
действием ксантиноксидазы превращается в мочевую кислоту,
которая в виде конечного продукта распада пуринов выводится из
организма. У других млекопитающих мочевая кислота окисляется в
аллантоин. В организме рыб и амфибий аллантоин превращается в ал-
лантоиновую кислоту, которая образуется путем гидрирования ал-
лантоина. Затем аллантоиновая кислота гидролизуется на мочевину и
оксалат:
Ксантиноксидаза
О.
н+, о; о
Ксантин
Мочевая кислота
НоИ
О
нл
соо
N11,
СОО"
I
СОО"
Оксалат
+
О
Аллантоин
Аллантоиновая кислота
Н2М-С-*Ш2
Мочевина
Распад пиримидинов. Распад пиримидиновых оснований на
первой стадии включает реакцию гидрирования колец урацила и тимина.
Цитозин после дезаминирования восстанавливается с образованием
урацила. Затем осуществляется раскрытие пиримидинового кольца и
образование карбамоил-р-аланина; последний далее гидролизуется до
С02, N1^, Р-аланина:
НИ'
о
I!
С
о
"ООС
сн
2Н
НИ'
С\
сн
2
Н,
О
\„/СН
О
\м/сн*
Т
\
сн,
о
\„/СН,
кн
+
со
+
К
н
Урацил
Н
Н
МН2-СН2-СН}
СООН
Дигидроурацил Карбамоил-^-аланин (З-Аланин
Конечные продукты распада либо выводятся из организма, либо
повторно утилизируются в других метаболических процессах.
Краткое содержание главы
Для синтеза азотсодержащих соединений — аминокислот и
нуклеотидов необходим азот. Азот накапливается в почве в виде аммиака
485
Часть II. Метаболизм веществ и энергии в клетке
двумя путями: азотфиксацией из воздуха и за счет разложения
останков вымерших организмов. Процесс превращения атмосферного Ы2 в
МН3 идет с затратой АТР и с участием сложной нитрогеназной
системы бактерий. Затем соли аммония используются высшими
растениями для синтеза аминокислот, нуклеотидов и других азотсодержащих
соединений. Основными соединениями, в состав которых включается
аммиак, являются глутамат, глутамин, карбамоилфосфат. Пути
биосинтеза заменимых аминокислот хотя и разнообразны, но достаточно
просты. Важную роль в этом синтезе играют глутамат и глутамин —
доноры ЫН2-групп. Глутамат образуется в реакции восстановительно-
го аминирования 2-оксоглутарата; он является предшественником глу-
тамина, пролина и карбамоилфосфата — важного звена цикла
мочевины. В реакциях трансаминирования образуются аланин из пирува-
та и аспартат из оксалоацетата. Предшественником серина является
3-фосфоглицерат, а сам серии — предшественник глицина и цистеи-
на. Пути биосинтеза незаменимых аминокислот у растений и
бактерий гораздо длиннее и более сложнее, чем заменимых. Они
образуются из некоторых заменимых аминокислот, а также из других
метаболитов. Синтез как заменимых, так и незаменимых аминокислот
регулируется по механизму обратной связи, в котором решающая реакция
ингибируется конечными продуктами — аминокислотами.
Распад избыточных аминокислот осуществляется двумя путями:
окислительным дезаминированием и декарбоксилированием. В
результате окислительного дезаминирования у большинства аминокислот
МН2-группа не превращается в токсическое соединение — аммиак, а
переносится на 2-оксоглутарат, в результате чего образуется глутамат
и 2-оксокислота. У высших растений и микроорганизмов формой
обезвреживания аммиака является образование амидов — глутамина и
аспарагина. У человека, большинства наземных позвоночных
животных конечный продукт распада аминокислот — нетоксическая,
хорошо растворимая в воде мочевина, образующаяся в цикле мочевины и
выводимая из организма. У рыб аммиак быстро выводится через
обильно орошаемые водой жабры. У птиц и пресмыкающихся аммиак
превращается в мочевую кислоту и вывод^те^^меете с калом.
Образованные в результате окислительного лейаминирования 2-оксокислоты
вовлекаются в реакции гликолиз^и ЦТК. Декарбоксилирование
аминокислот ведет к образованию физиологически активных аминов —
предшественников еще более физиологически активных соединений — ал-
калоидов.
Аминокислоты являются предшественниками пуриновых и пири-
мидиновых оснований. Биосинтез гетероциклов пурина и
пиримидина различен. Гетероцикл пурина, входящий в состав пуриннуклеоти-
да, строится в несколько стадий на 5'-фосфорибозе. Гетероцикл
пиримидина синтезируется самостоятельно из аспартата, С02 и аммиака.
Присоединение к нему рибозо-5'-фосфата приводит к синтезу пирими-
486
Глава 12. МетабЬли^м аминокислот и нуклеотидов
~нннуклеотидов. Предшественники ДНК — дезоксирибонуклеотиды
образуются путем восстановления рибонуклеотиддифосфатов при
участии фермента рибонуклеотидредуктазы. Образующиеся при распаде
зуклеотидов свободные пурины и пиримидины вновь вовлекаются в
биосинтез нуклеотидов. Избыточное количество пурина превращается
з конечные продукты: у человека — в мочевую кислоту, у рыб и
амфибий — в аллантоиновую кислоту, распадающуюся затем на
мочевину и оксалат; пиримидин распадается на аммиак, С02 и (3-аланин.
Хронология важнейших дат
в исследовании азотсодержащих соединений
1806 г. — Л. В. Ваклен и П. И. Робике получили из сока спаржи
кристаллический аспарагин.
1810 г. — В. Г. Валластоном был открыт цистеин.
1819г. — Ж» Л. Пруст выделил кристаллы лейцина.
1820 г. — Г. Браконно получил из гидролизата желатины глицин.
В дальнейшем из гидролизатов белков были выделены и
идентифицированы остальные аминокислоты.
1933 г. — А. Е. Браунштейн и М. Г. Крицман открыли реакции
переаминирования аминокислот, катализируемые аминотрансфераза-
ми. Это открытие сыграло ключевую роль в познании процессов
биосинтеза аминокислот, их превращений во вторичные и конечные
азотистые соединения.
1945 г. — Г. М. Калькар установил фосфоролитический характер
расщепления нуклеозидов.
1951 г. — С. Е. Картер выделил нуклеозидазы, способные гидро-
лизовать нуклеозиды. Было определено наличие двух систем распада
нуклеозидов — гидролитической и фосфоролитической.
1952—1954 гг. — Ю. А. Овчинников и А. Е. Браунштейн
установили первичную структуру аспартатаминотрансферазы.
ЛИТЕРАТУРА
Березов Т. Т., Коровкин Б. Ф. Биологическая химия: Учебник. — 3-е изд.,
перераб. и доп. — М.: Медицина, 1998. — 704 с,
Бохински Р. Современные воззрения в биохимии. — М.: Мир, 1987. — 544 с,
Гудвин Г., Мерсер Э. Введение в биохимию растений: В 2 т. — М.: Мир, 1986.
— Т. 1. — 393 с.
Кнорре Д. Г., Мызина С. Д. Биологическая химия: Учебник. — 3-е изд., испр.
— М.: Высш. шк., 2000. ~ 479 с.
Ленинджер А. Основы биохимии: В 3 т. — М.: Мир» 1985. — Т. 2, — С. 337—
731.
Страйер Л. Биохимия: В 3 т. ~ М,: Мир, 1985. — Т. 2. — 239 с.
Эллиот В., Эллиот Д. Биохимия и молекулярная биология / Под ред. А. И. Ар-
чакова, М. П. Кирпичникова, А. Е. Медведева, В, П. Скулачева. — М.: Изд-во
НИИ Биомедицинской химии РАМН, 1999. — 372 с.
487
Часть II. Метаболизм веществ и энергии в клетке
ВОПРОСЫ И УПРАЖНЕНИЯ
1. Какие существуют пути накопления азота в почве как источника
биосинтеза органических форм азота?
Перечислите основные стадии круговорота азота в природе.
2. Расскажите о действии ферментного комплекса нитрогеназы в процессе
восстановления атмосферного азота в аммиак.
3. Напишите реакцию:
Трансаминаза
Алании + 2-оксоглутарат —*~
4. Напишите путь синтеза глутамата из глюкозы и аммиака.
5. Какие основные пути расщепления аминокислот Вам известны?
6. Напишите реакцию:
Глутаминаза
Глутамин + Н20 *»
7. Напишите реакцию образования аспартата, содержащего 15М? из фумара-
та. В каком положении окажется атом 15Ы в кольце пурина?
Глава 13, СИНТЕЗ ДНК, РНК И БЕЛКОВ,
ПЕРЕНОС ГЕНЕТИЧЕСКОЙ ИНФОРМАЦИИ
Одним из наиболее важных достижений 70—80-х гг. XX в.
является выяснение путей синтеза нуклеиновых кислот и белков и
расшифровка на молекулярном уровне передачи наследственных признаков
от организма к его потомкам. Перенос генетической информации —
важнейшая составляющая принципов молекулярной логики живого;
в его основе лежит особая биологическая память, заложенная в
нуклеиновые кислоты.
Хранение генетической информации осуществляется в ДНК
хромосом. В ДНК каждого вида организма закодирована структура всех типов
РНК, всех белков. ДНК регулирует во времени и пространстве биосинтез
всех компонентов клеток и тканей, определяет развитие и
функционирование организма в течение его жизненного цикла и обеспечивает его
индивидуальность. Велика ее роль в процессах апоптоза.
Во всех организмах перенос генетической информации
осуществляется по типу матричного механизма, важная роль в котором
отводится матрице. Матричный механизм включает три этапа:
1. Перенос информации от ДНК к ДНК. Механизм этого этапа
сводится к самокопированию, к удвоению молекулы ДНК и называется
репликацией (от лат. герИсаНо ■— повторение); роль матрицы здесь
выполняет сама молекула ДНК. Репликация осуществляется при делении клеток
высших организмов и размножении бактерий и вирусов.
2. Перенос информации от ДНК к РНК, называемый
транскрипцией; роль матрицы выполняет определенный участок ДНК, который
называется геном (или же может транскрибироваться группа генов).
Гены несут информацию о всех типах РНК: мРНК, тРНК, рРНК и др.
3. Перенос генетической информации от РНК к белку,
называемый трансляцией*, матрицей здесь является мРНК. В сущности,
механизм трансляции представляет собой перевод генетической
информации от одного типа веществ к другому, а именно: перевод
информации с "языка" полирибонуклеотидной цепи мРНК на "язык"
полипептидной цепи белка.
Итак, перенос генетической информации в биологической системе
можно представить в виде следующей схемы:
[—. днк »~ РНК *- Белок
Репликация Транскрипция Трансляция
489
Часть /./. Метаболизм веществ и энергии в клетке
Перенос информации в клетке в направлении ДНК -ч> РНК —>
Белок именуется основным постулатом молекулярной биологии.
В основе представлений о матричном механизме переноса
генетической информации лежит принцип комплементарности нуклеиновых
кислот как результат спаривания аденина с тимином (аденина с ура-
цилом в РНК) и гуанина с цитозином (см. гл. 2). Именно эти
основания несут генетическую информацию* Что касается сахарных и
фосфатных групп, то в нуклеиновых кислотах они выполняют
структурную роль.
Матричный механизм синтеза нуклеиновых кислот и его
подчинение условиям комплементарности имеет огромные преимущества
перед обычным биохимическим синтезом веществ. Во-первых, он
позволяет точно копировать генетическую информацию, обеспечивает вы-
сокую стабильность наследуемых признаков при их переносе от
организма к потомкам. Во-вторых, он создает строгую молекулярную
экономию на всех трех эталах биосинтеза нуклеиновых кислот и белков.
В-третьих, он исключает большой набор ферментов и кофакторов,
который был бы необходим в случае обычного биосинтеза.
13.1. Репликация ДНК
Репликация ДНК — сложный процесс, для осуществления
которого необходимо участие многих специальных ферментов и особых
белковых факторов, выполняющих некаталитические функции; наличие
в реакционной среде дезоксирибонуклеотидтрифосфатов и источника
энергии АТР. Матричный механизм репликации теоретически
возможен по трем путям: 1) консервативный — когда дочерняя двухцепо-
чечная молекула ДНК образуется на родительской нативной ДНК без
разделения цепей последней; 2) полуконсервативный — цепи
родительской ДНК расходятся и на каждой из них синтезируются
комплементарные цепи, в результате чего образуются две дочерние
молекулы ДНК, каждая из которых содержит одну родительскую и одну вновь
синтезированную цепь; 3) дисперсивный — расщепление
родительской ДНК в нескольких местах и образование на ней новых цепей
ДНК; по дисперсионному механизму каждая дочерняя цепь ДНК
должна состоять из коротких участков как родительской, так и
новообразованной ДНК, соединенных между собой случайным образом.
Исследования М. Мезелсона и Ф. Сталя (1957 г.) показали, что
репликация происходит полуконсервативным путем (рис. 13.1).
Их опыты заключались в следующем. Клетки Е. сой выращивали
на среде с 15ЬШ4С1 в течение 14 поколений и анализировали
распределение радиоактивной метки. Оказалось, что ДНК этих клеток почти
целиком была помечена 15К. После этого клетки переносил^ в среду,
I
содержащую обычный ЗЧН4С1, и синтезированную ДНК исследовали в
течение нескольких поколений в градиенте плотности хлористого це-
490 \
\
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
На:;равленис
ч.: и мс нтации
"Тяжелая
ДНК
Исходная
("тяжелая") ,51*-ДНК
"Легкая"
ДНК
14
"Легкая" 14Ы-ДНК
Результаты эксперимента
',
Гибридная
ДНК
"Легкая" Гибридная
ДНК ДНК
ДНК после
одного
удвоения на
После двух
удвоений на
[14МЭгШ4С1
Первое удвоение: обе
молекулы ДНК
содержат по одной "легкой"
и по одной "тяжелой"
цепи
Второе удвоение:
образовались две
молекулы
гибридной ДНК и две —
"легкой"
Рис. 13.1. Результаты эксперимента Мезелсона и Сталя. При
центрифугировании в градиенте плотности СвС1 "тяжелая" [15Ы] ДНК достигает равновесия в
полосе, которая расположена ближе ко дну пробирки, чем полоса равновесия
"легкой" [14Г4] ДНК. Равновесное положение гибридной ДНК оказывается
промежуточным. Определение плотности дочерних ДНК после первого и второго
удвоения показало, что репликация ДНК осуществляется по полуконсервативному
механизму
зия. Наблюдаемая картина показала, что после одной генерации в
среде присутствовал гибрид 15К-,14]Ч-ДНК, который при нагревании
разделяли на 14Ы- и 15Ы-цепи; в последующих поколениях
преобладают 14М"-цепи. Этот факт нашел подтверждение в опытах с клетками из
корней проростков фасоли.
ДНК-полимераза. Этот фермент, осуществляющий синтез цепей
ДНК, был открыт и изучен А. Корнбергом и его сотрудниками.
Фермент катализирует реакцию
(<ШМР)П + <ШТР
(сШМР)
п+1
+ РР.
ДНК удлиненная ДНК
Для действия фермента необходимо присутствие ионов М^2+. В его
активном центре содержится ион 2п2+. Для начала
функционирования ДНК-полимеразы необходима предсуществующая ДНК, одна из
цепей которой играет роль затравкщ а другая — роль матрицы. К
491
Часть II. Метаболизм веществ и энергии в клетке __
З'-концу цепи-затравки присоединяются новые нуклеотиды под
действием ДНК-полимеразы; таким образом, синтез новой цепи идет в
направлении 5' -> 3'. Причем присоединение нуклеотидов к
цепи-затравке осуществляется в соответствии с принципом комплементарно-
сти по отношению к цепи-матрице. Для работы ДНК-полимеразы
необходимо наличие 5'-трифосфатов всех четырех дезоксирибонуклеози-
дов, которые не могут быть заменены соответствующими 5'-дифосфа-
тами и б'-монофосфатами* Энергия, необходимая для образования фос-
фодиэфирной связи при присоединении каждого нового нуклеотида,
обеспечивается за счет расщепления пирофосфатной связи между а- и
р-фосфатными группами дезоксирибонуклеозид-5'-трифосфатов,
используемых в качестве предшественников строительных блоков ДНК.
В клетках Е. соИ обнаружены три различных ДНК-полимеразы,
обозначаемые I, II и III соответственно. За удлинение цепей ДНК в
ходе репликации отвечает в основном ДНК-полимераза III,
представляющая собой большой комплекс с молекулярной массой 550 000.
Характерно, что ДНК-полимераза I и ДНК-полимераза III обладают
помимо полимеразной активности, обеспечивающей ковалентное
связывание новых дезоксирибонуклеозидов, 3'—»6'- и 5'—»3'-экзонуклеаз-
ными активностями. Свойства и функции ДНК-полимеразы II пока
изучены недостаточно.
Механизм полуконсервативной репликации ДНК. Репликация
по полуконсервативному пути включает следующие этапы: 1)
образование репликативной вилки или, иными словами, локальное
расплетание цепей ДНК; 2) синтез РНК-затравки на матричных цепях ДНК
и синтез фрагментов ДНК, называемых фрагментами Оказаки; 3)
удаление РНК-затравок и 4) "сшивание" фрагментов ДНК (завершение
синтеза ДНК) — рис. 13.2, Все эти ступени протекают с высокой
скоростью и исключительной точностью.
Образование репликативной вилки. Вспомним, что молекула ДНК
представляет собой скрученную двойную спираль и что основания,
несущие генетическую информацию, находятся внутри спирали.
Чтобы ферменты репликации имели доступ к несущим генетическую
информацию основаниям, цепи матричной ДНК должны быть
разделены хотя бы на коротком участке. Локальное разделение цепей ДНК
осуществляется под действием специального фермента хеликазы. Этот
фермент узнает точку начала репликации и разрывает водородные связи
между комплементарными основаниями двойной спирали. В
результате двойная спираль расплетается и расходится на отдельные цепи.
Расплетенный участок ДНК называется репликативной вилкой. Ее
образование идет с затратой энергии. На разделение одной
комплементарной пары оснований требуется энергия примерно двух молекул
АТР, гидролизующихся до АОР и Р.. После образования
расплетенного участка к каждой из его цепей присоединяется несколько молекул
ДНК-связывающего белка. Роль этого белка — стабилизировать одно-
492
/
/
Г лава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
3'
5'
3'
I * •
Щ--Щ 5'
I I
I I
Сьй 3'
**^г^г** з' ^^Т^лТТТТ'*'?^
5
8--"ч«я Цепь о 0 аза
цепь 3
5'
3'
М 5'
I I
I I
... йчй 3'
М5'
1 I
I I
йчй 3'
йчй з'
цепочечные фрагменты Л„/?Т?ТГТГТТТГ?Г?ТТТТ?-",ГТ 5'
*• тттттл Дг1К | | | 1 | | I | | * I | I 1 I ■ | | | I II
родительской ДНК, пред- б' йч]^м^ы>^ - г^й з-
отвратить обратное
воссоединение
комплементарных пар оснований.
Благодаря действию хе~
ликазы и связывающего
белка ^хукЯеотвдЗБ».
последовательности
становятся доступными для их
считывания. Все ступени
репликации совершаются
вслед за движением реп-
ликативной вилки вдоль
молекулы ДНК.
Существует еще одна
проблема, связанная с
образованием реплика-
тивной вилки. Как
известно, спирализация ДНК
имеет плектонимичес-
кий характер, т.е. цепи
могут быть разделены,
если одну из них
раскручивать относительно
другой. Быстрое
раскручивание цепей ДНК должно
приводить к вращению
всей хромосомы,
расположенной впереди реплика-
тивной вилки. Однако
полагают, что существуют
специальные шарнирные
устройства, роль которых
выполняют ферменты —
топоизомеразы. Под
действием этих ферментов происходит кратковременный разрыв одной из
цепей ДНК, который затем точно восстанавливается после нескольких
оборотов. Благодаря этому с большой скоростью вращается только
короткий фрагмент ДНК.
Синтез затравки и фрагментов ДНК (фрагментов Оказаки) на
матричных цепях ДНК. Синтез новых цепей ДНК катализируется
ДНК-полимеразой. Однако она не способна инициировать синтез
новых цепей на одноцепочечной ДНК-матрице. Было установлено, что
инициацию репликации осуществляет фермент РНК-полимераза (прай-
Рис. 13.2. Предполагаемый механизм
репликаций ДНК: 1 — образование репликатив-
ной вилки (расплетание двойной спирали); оо —
расплетающие белки; | — фермент хеликаза;
2— синтез ведущей цепи ДНК (|>П\0\.„);
синтез фрагментов Оказаки отстающей цепи ДНК
с участием фермента праймазы (мчш\шч8г«\.,.);
вча\... — РНК-затравка; 3 — удаление рибонук-
леотидов с участием ДНК-полимеразы I (экзо-
нуклеазяая реакция, ведущая к образованию
бреши х); 4 — связывание фрагментов ДНК
отстающей цепи, катализируемое ДНК-лигазой
(заделывание бреши х)
493
Часть II. Метаболизм веществ и энергии в клетке
маза), способная осуществлять синтез цепей йе ггоио. При ее участии в
репликативной вилке на освобожденных цепях синтезируется
затравка-инициатор, представляющая собой короткий участок РНК (10—
60 пар оснований), называемый РНК-праймером; он комплементарен
свободному участку ДНК. Синтез РНК-затравки идет от б'-ОН-конца
к З'-ОН-концу (рис. 13.3). Первоначально обнаруженный у Е4 соИ
такой принцип образования затравки является общим для прокариот и,
возможно, для эукариот.
Синтез фрагментов ДНК. Для синтеза цепи ДНК необходимы все
четыре дезоксирибонуклеозидтрифосфата (й-АТР, й-ОТР, ё-ТТР и
с!~СТР) и ионы М§2+. Механизм действия ДНК-полимеразы включает
два акта: 1) сначала она присоединяет соответствующий нуклеозид-
трифосфат к матричной цепи с образованием комплементарной пары
между основаниями нуклеозидтрифосфата и матричной цепи; 2)
после этого ДНК-полимераза катализирует отщепление пирофосфата,
присоединяя одновременно дезоксирибонуклеотид к З'-ОН-концу
РНК-праймера (рис, 13.4), Гидролиз пирофосфата до фосфата ведет к
полному завершению реакции присоединения нуклеотида к растущей
цепи ДНК. Присоединяя, таким образом, один дезоксирибонуклеотид за
другим, ДНК-полимераза образует смешанные фрагменты РНК—ДНК.
Напомним, что цепи родительской ДНК антипараллельны. Общее
направление синтеза новых цепей ДНК осуществляется на обеих
матричных цепях вслед за движением репликативной вилки. Логично было
бы ожидать, что синтез одной
дочерней цепи должен идти в
направлении 5/~>3/, другой —
в направлении $'—>Ь\ Однако
все известные
ДНК-полимеразы синтезируют ДНК только
в направлении 5'->3'.
Проблема несоответствия
специфичности действия
ДНК-полимеразы и направления движения
репликативной вилки была
решена Р. Оказали, который
обнаружил, что значительная
часть новосинтезированной
ДНК появляется в виде
коротких отрезков (длиной около
1000—2000 нуклеотидов),
получивших название
фрагментов Оказаки, содержащих
РНК-праймеры длиной от 10
до 200 нуклеотидов. Этз^фраг-
менты существу^дз^ороткое
494
ОТР
РНК-праймер
к/ (затравка) «»
—ии
*т
3' 5' 3' 5'
Фрагмент одной
матричной цепи
ДНК в точке начала
репликации
Рис. 13.3. Синтез РНК-праймера
(затравки): МТР — рибонуклеозидтрифосфат; ■,
К!— основания фрагмента матричной цепи
ДНК и РНК-праймера соответственно
б' РНК-праймер 3' I ЦУ з'
И |^ № ЕЗ И ? (Р)-0-Вг-И-ОН
11111 V Л
11111 \^>^
3' Матричная цепь 5'
Рис. 13.4. Механизм действия
ДНК-полимеразы: Й — основания РНК-праймера;
■ — основания остатков дезоксирибонук-
леотидов; Вг — остаток дезоксирибозы
Глава 13. Синтез_^ЩК+..РНК и белков. Перенос генетической информации
время и локализованы около репликативной вилки. Они
синтезируются на матрич^й цепи ДНК, имеющей направление 3'—»5';
наращивание цепи иде^ в направлении 5'—»3', т.е. против движения
репликативной вилки. Другая новая цепь синтезируется непрерывно. Та цепь,
которая синтезируется из фрагментов Оказаки, называется отстающей
цепью, а та, что синтезируется без разрывов или почти без разрывов,
называется ведущей цепью (см. рис. 13,2). Таким образом, синтез
фрагментов Оказаки позволяет осуществлять репликацию в направлении б'^З'.
Удаление фрагментов РНК. Наращивание фрагментов Оказаки идет
до длины их цепей 1000—2000 нуклеотидов, после чего РНК-прайме-
ры удаляются. Удаление осуществляется путем последовательного
отщепления рибонуклеотидов, катализируемого одной из форм ДНК по-
лимеразы, а именно ДНК-полимеразой I, обладающей б'-^З'-экзонук-
леазной активностью. По существу в данном случае ДНК-полимераза
катализирует реакцию замещения: на место удаленного рибонуклео-
тида она присоединяет к З'-ОН-концу укороченного фрагмента
Оказаки соответствующий дезоксирибонуклеотид. Последовательное
присоединение дезоксирибонуклеотидов к месту удаленных
рибонуклеотидов приводит к тому, что новый фрагмент Оказаки содержит лишь
дезоксирибонуклеотидные остатки.
Сшивание фрагментов Оказаки завершает синтез отстающей цепи
ДНК. Эту реакцию катализирует фермент ДНК-лигаза, под действием
которой образуется фосфоэфирная связь между З'-ОН-концом ДНК,
подлежащим удлинению, и 5'-фосфатной группой новосинтезирован-
ного фрагмента Оказаки. Этот фермент заделывает одноцепочечные
разрывы (бреши) в двуцепочечной ДНК.
Так, в результате последовательного действия ДНК-полимераз и
ДНК-лигазы идет наращивание отстающей цепи ДНК в направлении,
противоположном движению репликативной вилки. На образование
одной фосфодиэфирной связи требуется энергия двух молекул АТР.
Точность репликации ДНК и механизм исправления допущенных
ошибок. Специфическая особенность ДНК-по лимеразы — проявлять
как полимеразную, так и экзонуклеазную активность. Это характерно
как для ДНК-полимеразы I, так и для ДНК-полимеразы III.
Ошибки, возникшие в процессе репликации, передаются
потомству, что неизбежно приводит к негативным последствиям для его
жизнеспособности. Было установлено, что частота ошибок при
репликации ДНК Е. сой не превышает 1 на 109—1010 нуклеотидов. Обладая
З'-ОН-нуклеазной активностью, ДНК-полимераза III способна
осуществлять коррекцию (находить и исправлять ошибки). Механизм
коррекции заключается в отщеплении З^концевых нуклеотидов (при этом
фермент как бы возвращается назад) и замене ошибочно встроенного
нуклеотида нормальным нуклеотидом. Таким образом, в ходе
репликации ДНК-полимераза проверяет каждый проведенный акт
полимеризации, прежде чем перейти к следующему акту, В основе механиз-
495
Часть II. Метаболизм веществ и энергии в клетке
ма ее действия лежит уотсон-криковское спаривание оснований нук-
леотидов матричной и дочерней цепей. В процессе коррекции всегда
удаляются с конца цепи-затравки некомплементарные остатки нукле-
отидов. Присоединив очередной нуклеотид, ДНК-полимераза
"сверяет" его с партнером на цепи матрицы и в случае несоответствия паре
(А—Т или О—С) та же полимераза проявляет 3'—>5/-экзонуклеазную
активность, удаляя ошибочно присоединенный нуклеотид.
С высокой точностью копирования ДНК связан и
предварительный синтез РНК-праймера. На первый взгляд кажется странным,
зачем нужно было синтезировать эту затравку, а затем удалять ее.
Однако если бы ДНК-полимераза могла осуществлять репликацию без
затравки, то корреляция была бы невозможна. Ведь прежде чем
образовать фосфодиэфирную связь, ДНК-полимераза должна проверить
комплементарность предшествующей пары оснований. Поскольку РНК-
полимераза начинает синтез цепей без затравки и, следовательно, не
проверяет предыдущую пару оснований, частота ошибок в данном
случае гораздо выше (на несколько порядков), чем для ДНК-полимеразы.
Но синтезированная РНК-затравка хотя и с ошибками дает
возможность осуществлять функцию корреляции ДНК-полимеразе, а затем и
удалять эту затравку, заменяя ее последовательностью нуклеотидов
1
ДНК. Синтез, а затем расщепление ДНК-затравки усложняют
механизм репликации, но эти процессы являются своего рода "платой" за
точность и надежность репликации ДНК.
13.2. Репарация генетических повреждений в ДНК
В процессе жизнедеятельности клетки ДНК может подвергаться
существенным повреждениям как спонтанно, так и под действием
физических и химических факторов среды. Они связаны с потерей
макромолекулой ДНК азотистых оснований, их дезаминированием, димеризаци-
ей пиримидиновых оснований, разрывом фосфодиэфирных связей остова
молекулы. Эти повреждения особенно часто имеют место при
ультрафиолетовом облучении, под действием ионизирующей радиации и
различных химических веществ, получивших название мутагенов. Наиболее
часты потери пуриновых оснований (апуринизация), связанные с их
отщеплением от остатков дезоксирибозы. При дезаминировании цитозина,
аденина, гуанина образуются основания, не характерные для ДНК.
Разрыв фосфодиэфирных связей может привести к образованию двух цепей,
которые могут попеременно смешиваться друг с другом.
Высокая частота появления повреждений неизбежно привела бы
клетку к гибели. Поэтому клетка имеет ферментные системы, которые
исправляют возникшие повреждения. Процесс исправления повреждений
в ДНК называется репарацией. Репарация является вторым способом
после полуконсервативной репликации, обеспечивающим сохранность
генетической информации в ДНК в неизменном состоянии.
496
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
Среди механизмов репараций наибольшее значение имеет так
называемая эксцизиционная репарация, осуществляемая с
использованием неповрежденной комплементарной цепи как матрицы.
Эксцизиционная репарация хордшо изучена при устранении повреждений,
возникающих в цепях/ДНК при ультрафиолетовом облучении (260—
300 нм)> Под действием УФ-лучей развивается фотохимическая
реакция, ведущая к образованию ковалентных сшивок между двумя со-
I
седними остатками тимина и переходом их в тиминовый димер:
о
о
Л..Л
"И"
соон ноос
УФ-лучи
N^4)
сн3 нх
сАн
3'
Соседние димеры цепи ДНК
Тиминовый димер не
укладывается в двойную
спираль молекулы ДНК и
не может выполнять
функцию матрицы при
репликации.
Процесс репарации ти-
минового участка
включает четыре стадии (рис. 13,5):
1) инцизии: эндонуклеаза
находит димер и
осуществляет разрыв полинуклео-
тидной цепи; 2) эксцизии:
экзонуклеаза удаляет
поврежденный участок цепи,
катализируя
последовательный гидролиз сахарофое-
фатных связей; 3)
ограниченной репликации: ДНК-
пол и мераза заполняет
образовавшуюся брешь,
используя нормальные дезоксири-
бонуклеозидтрифосфаты, а
в качестве матрицы —
комплементарную интактную
цепь ДНК; 4)вшивки вновь
синтезированного участка с
основной цепью,
катализируемой ДНК-лигазой.
и и и и и
Тиминовый димер
УФ-лучи
и и и и и
\
Фотохимическая
реакция
Специфическая
эндонуклеаза
I
Ц~иТТ
Инцизия
(разрыв одной цени
вблизи пир им
плисового димера)
и и и
Экзонуклеаза
Эксцизня
(удаление поврежденного
участка)
ДНК-полимераза
ш
Реиаративный синтез
(заполнение бреши)
ггп и и ы ы и и и и и
111111111
тт
ДНК-ли газа
и1 а и и
I
ИГЕГ
Соединение цени
и и и и и
П
3'
5'
•тт
и
и п
• г •
и
-тт
Рис. 13.5. Репарация ДНК, поврежденной
ультрафиолетовыми лучами
32. Заказ 3486
497
Часть II. Метаболизм веществ и энергии в клетке _
Неспаренные основания в нитях ДНК точно также удаляются по
механизму эксцизиционнои репарации. Важную роль здесь играют
специфические эндонуклеазы, распознающие соответствующие
повреждения. Разрывы в цепях ДНК, возникающие при воздействии
радиации, также заделываются по механизму эксцизиционнои репарации,
с той лишь разницей, что первая стадия репарации здесь отсутствует.
Второй тип механизма репарации — репарация с использованием
генетической информации гомологичной молекулы ДНК — имеет
место в случае повреждения в каком-либо месте обеих цепей ДНК. Чаще
всего она возникает при действии ионизирующего излучения. В этом
случае клетка должна иметь не менее двух копий генетического
материала. Механизм этой репарации на молекулярном уровне не изучен.
К третьему типу репарации относится фотохимическое
расщепление пиримидинового димера под действием фотореактивирующего
фермента — фотолиазы. Так, фотолиаза Е. еоН находит на ДНК
образовавшийся под действием ультрафиолетового облучения димер и
прочно связывается с ним* При облучении такого комплекса
преимущественно видимым светом (350—600 нм) происходит восстановление
остатков тимина в нормальное состояние. Установлено, что клетки
большинства организмов, подвергающиеся воздействию
ультрафиолетовых лучей, содержат фотореактивирующую лиазу. Фермент играет
важную роль в устранении повреждений, возникающих при действии
ионизирующих излучений. К этому же типу механизмов репарации
следует отнести сшивку одиночных разрывов сахарофосфатного
остова молекулы, катализируемую ДНК-лигазами,
Все механизмы репарации генетических повреждений в клетке
многократно продублированы и могут идти разными путями. Эти пути
находятся под сложным, пока еще слабо изученным генетическим
контролем. Благодаря этому выживание мутагенных клеток,
дефектных по какому-либо ферменту репарации, обеспечивается
включением резервного пути защиты организма (клетки).
13,3. Рекомбинация ДНК
Помимо репарации ДНК подвергаются также изменениям и
перестройкам другого рода, получившим название рекомбинации.
Рекомбинация — это образование новых молекул ДНК путем разрывов и
пере воссоединений фрагментов предсуществующих молекул ДНК. С
биологической точки зрения она представляет собой процесс
перегруппировки генетического материала, в результате которого возникает
новое сочетание генетических структур (генов, хромосом, участков
хромосом).
Способы рекомбинации. Принципиально процесс рекомбинации
можно представить как обмен отдельными участками между двумя
I
гомологичными молекулами ДНК: /
/
/
/
/
498 /
/
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
Рекомбинация
-X
Гомологичные ДНК
Рекомбинантныо ДНК
Однако процесс рекомбинации является сложным и до конца не
выясненным.
Наиболее вероятны три способа рекомбинации.
По первому способу (рис. 13.6) две гомологичные молекулы ДНК
образуют тетраду. В одной из цепей каждой молекулы ДНК
происходит разрыв под действием фермента эндонуклеазы. В полинуклеотид-
ных цепях появляются свободные концы, к которым ДНК-полимераза
может присоединить новые нуклеотиды, вытесняя при этом предсу-
ществующие участки цепей. В результате образуются два свободно
выступающих одноцепочечных хвоста. Если эти хвосты комплектен-
3'
5'
ЕЕ
и
5'3''
3'5'с
П
±
щ
Спаривание двух
гомологических молекул
(образование тетрады) ДНК
{
Разрыв цепей под действием
эндонуклеазы (►)
{
Наращивание целей под дей
ствием ДНК-полимеразы (•)
I
Образование на гомологичных
участках двухцепочечного
мостика
♦
Разрыв цепей под действием
эндонуклеазы {►)
+
5' Действие ДНК-полимеразы
3' и спаривание оснований
I
б' ДНК-полимераза (•)
3' заделывает бретаъ
^"Т^Т^ ДНК-лигаза сшивает
разрывы
Рис. 13.6. Образование рекомбинантных ДНК в результате наращивания по
линуклеотидных комплементарных хвостов
32
499
Часть II. Метаболизм веществ и энергии в клетке
тарны, они спариваются, что приводит к образованию двух
цепочечного мостика (перекреста). Затем под действием эндонуклеазы вторично
осуществляются одиночные разрывы в противоположных цепях и
образуются два фрагмента родительских молекул с перекрывающимися
концевыми последовательностями* Процесс рекомбинации завершают
ДНК-полимераза, заделывающая бреши в перекрывающихся концах,
и ДНК-лигаза, сшивающая эти концы.
Второй способ рекомбинации заключается в прямом обмене
генетическим материалом между параллельно расположенными
спиралями гомологичных молекул ДНК (рис. 13.7). Обмену цепей
предшествует их разрыв под действием эндонуклеазы- Разорванные концы
обмениваются между собой и образуют перекрестный мостик с
удалением разрывов ДНК-лигазой. Благодаря перемещению перекрестного
мостика и свободному вращению остатков нуклеотидов вокруг связей,
образующих углеводно-фосфатный остов молекулы ДНК,
обменивающиеся цепи переходят из одной молекулы ДНК в другую с
последующей спирализацией. Затем следуют удаление перекрестного мостика с
помощью дополнительных разрывов под действием эндонуклеазы и
сшивка разрывов ДНК-лигазой. В результате образуются две
молекулы рекомбинантных ДНК.
I
♦
ПП
Гомологичные молекулы ДНК
+
Разрыв цепей иод действием
эндонуклеазы
I
Обмен цепями, приводящий
к образованию перекрестного
мостика
I
Перемещение перекрестного
мостика
Удаление перекрестного
мостика с помощью дополнительного
разрыва нуклеазой, последующее
ааполненйе^брешей ДНК-полиме-
разой и сЬздгака разрывов
ДНК~лЪгазой
Рис. 13.7. Рекомбинация в результате обмена между д^умя двойными
спиралями ДНК \
500
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
А6АСТА6ТАССТ
ТСТСАТСАТеСА
♦
Ц.1 II 1,1
1
пхг
и.1 I 1
А6АСТА6ТАССТ
ТСТСАТСАТ66А
ГОЛ
]
♦
Третий способ известен под названием сайтепецифической
рекомбинации (рис. 13.8). Его сущность заключается в следующем. На
определенном участке ДНК (сайте) происходят два смещенных
относительно друг друга разрыва двойной епщшш ДНК под действием
эндонуклеазы, обладающей специфичностью только к нуклеотидной
последовательности сайта. В результате такого действия эндонуклеазы в
каждой цепи молекул ДНК возникает один з^асток узнавания (комп-
лементарности), называемый липким концом\ Связывание по липким
концам фрагментов молекул родительских ДНК и ведет к
образованию рекомбинантных ДНК.
Эволюционная роль рекомбинации. Рекомбинация, связанная с
обменом между генами или объединение генов из разных источников с
образованием измененной
хромосомы, — явление,
широко распространенное
в природе.
Одним из видов такой
рекомбинации является
трансформация бактерий
под действием экзогенной
ДНК. Она была открыта в
1942 г. О. Эвери, К. Мак-
Леодом и М. Мак-Карти. Их
исследования показали, что
ДНК, перешедшая из
вирулентного штамма гнойного
пневмококка в
невирулентный, переводила последний
в вирулентный. По всей
вероятности, содержавшийся
в ДНК вирулентной клетки
ген вирулентности
включался в ДНК невирулентной
клетки.
Другим видом
рекомбинации, протекающей в
природе, является лизоге-
ния, или бактериофагия.
Многие бактерии
заражаются специфичными к ним
фагами. ДНК этих фагов
встраивается в
бактериальную хромосому. Такая ре-
комбинантная хромосома
Действие специфической
эндонуклеазы и
образование фрагментов ДНК с
липкими концами
А6АСТАСТА ССТ
ТСТ 6АТСАТ6СА
' I "И I
] Г
I I .11 I. I. I
II. I I II
А6АСТА6ТА ССТ
ТСТ САТСАТбОА
I м;:ц
Обмен фрагментами и
ИХ сшивка
II I И
и 1111
АСАСТАеТАССТ
ТСТОАТСАТбвА
Ш I 1
А0АСТА6ТАССТ
тстедтсАтесА
1 I I I I Г >
Рис. 13.8. Схема сайтспецифической реком-
может реплицироваться и бинации молекул ДНК
501
Часть II. Метаболизм веществ и энергии в клетке
при размножении бактерий переносится из поколения в поколение.
При воздействии какого-либо фактора гены фага могут возобновить
свою функцию, в результате чего образуются фаговые частицы и
наступает лизис бактериальной клетки. Таков, возможно, механизм
действия онкогенных вирусов.
Рекомбинация ДНК может осуществляться также при конъюгации
(временном воссоединении) бактерий. Обычно бактерии
размножаются простым делением клеток. Однако некоторые виды бактерий
осуществляют иногда половую конъюгацию. В этом случае между
клеткой-донором и клеткой-реципиентом возникает длинный
соединительный канал — пиль, через который в реципиентную клетку от донор-
ной передается хромосома или часть ее. В результате
клетка-реципиент получает новые гены, которые соединяются с ее хромосомой.
У эукариотов генетическая рекомбинация происходит при половом
слиянии яйцеклетки и сперматозоида. В появляющемся потомстве
клеток дочерние хромосомы будут представлять собой комбинацию генов
яйцеклетки и сперматозоида. В результате новое потомство получает
фенотипические признаки, принадлежащие обоим родителям.
Генетическая рекомбинация является неотъемлемой
характеристикой всех живых организмов. Она представляет собой материальную
основу изменения наследственных признаков и играет большую роль
в эволюционном развитии живой материи. Особо следует
остановиться на роли рекомбинации при половом размножении организмов. Этот
процесс обеспечивает комбинативную изменчивость геномов, т.е.
создает возможность осуществлять разнообразные сочетания
родственных вариантов генов. Видимо, он сыграл большую роль в организации
рациональной структуры генома, участвующего в биосинтезе
полиферментных комплексов, принимающих участие в сопряженных
биохимических реакциях, метаболических циклах. Рекомбинация
половым путем дает возможность осуществлять отбор высокоактивных
геномов. Именно она оказалась более эффективной относительно других
видов рекомбинации*
13.4. Генная инженерия ,
I
/
Технология создания и использования рекЬмбинантных ДНК.
Детальное изучение механизма рекомбинации позволило осуществить в
лабораторных условиях синтез новых молекул ДНК, в которых гены
одного организма оказываются объединенными''с генами другого. Такой
синтез явился величайшим достижением молекулярной биологии,
приведшим к возникновению нового научного направления — генной
инженерии. Генная инженерия дала возможность получать новые
искусственные комбинации генов, не встречающиеся в природе.
Большой прогресс был сделан после открытия сайтспецифических
эндонуклеаз, названных в дальнейшем эндонуклеазами рестрикции.
502
Глава 23. Синтез ДНК, РНК и белков. Перенос генетической информации
Эндонуклеазы рестрикции имеют две характерные особенности: они
способны узнавать лишь участки достаточно протяженной нуклеотид-
ной последовательности; они катализируют процесс образования
смещенных относительно друг друга/разрывов в обеих цепях молекулы
ДНК (разрывы наискосок). Эта Уникальная особенность эндонуклеаз
рестрикции позволила получит^ фрагменты ДНК с одноцепочечными
липкими концами и переносить)их из одного организма в другой. Так,
Е. еоИ продуцирует эндонуклеа^у рестрикции (ЕсоШ), действующую
на последовательность ...ОААТТК^.. и расщепляющую связь между О
и А. В составе дуплекса ДНК эта\последовательность комплементарна
последовательности .... СТТААСг ....,ч другой цепи и в то же время при
прочтении ее справа налево она идентична последовательности нуклео-
тидов первой цепи. Такой участок молекулы ДНК называется
палиндромом (так называют выражения, читающиеся одинаково в обоих
направлениях, например: "А роза упала на лапу Азора").
В результате действия фермента ЕсоШ на вновь образованных
фрагментах ДНК возникают липкие концы. Один из фрагментов имеет на
конце одноцепочечный участок, комплементарный одноцепочечному
участку на другом фрагменте. Возникает возможность взаимного
узнавания и реконструирования таких фрагментов благодаря
взаимодействию комплементарных оснований.
Для переноса в клетку-хозяина фрагмента чужеродной ДНК
необходима другая ДНК, способная объединиться с этим фрагментом и
осуществить его перенос. Эта ДНК получила название вектора.
Функцию вектора способны выполнить ДНК плазмиды и ДНК-вирусы.
Плазмиды встречаются у бактерий и представляют собой интактные
кольцевые ДНК, отделенные от основной хромосомы бактерии. Плазмида
гораздо мельче хромосомы, в клетке может находиться до десятка ее
копий. Каждая плазмида содержит несколько, а иногда много генов,
которые реплицируются, транскрибируются и транслируются
независимо от хромосомных генов, но одновременно с ними. Бактерии могут
существовать и без плазмид, однако, их присутствие сообщает клетке
специфические свойства. Так, многие плазмиды содержат гены,
кодирующие синтез ферментов, способных разлагать антибиотики и
обеспечить устойчивость бактериальной клетки к ним* Такие плазмиды
важны как маркеры при скрининге (отборе) трансформированных
клеток, образовавшихся в процессе эксперимента по получению ре-
комбинаитных ДНК.
В качестве вирусного вектора заслуживает внимания ДНК фага X.
Она легко переносит чужеродный ген в Й. соИ. Если ДНК фага X,
несущую чужеродный ген, смешать с белком оболочки этого фага, то
образуются фаговые частицы, весьма эффективно инфицирующие клетку-
хозяина.
Технология создания и использования рекомбинантных ДНК
включает следующие основные этапы;
503
Часть II. Метаболизм веществ и энергии в клетке
1) получение целевого гена;
2) соединение этого гена с вектором (получение рекомбинантной
ДНК);
3) введение рекомбинантной ДНК в клетку-хозяина;
4) отбор клеток, содержащих целевой ген; клонирование гена.
Получение целевого гена основано на использовании фермента РНК-
зависимой ДНК-полимеразы (обратной транскриптазы),
осуществляющей синтез ДНК с использованием в качестве матрицы РНК.
Клетки, интенсивно синтезирующие целевой ген и его мРНК, лизируют;
освободившиеся полирибосомы (полисомы) собирают
центрифугированием и их обрабатывают антигенами против того белка, ген
которого необходимо выделить. Полирибосомы вместе с синтезированным
ими белком дают нерастворимый комплекс с антигеном, откуда
выделяют мРНК. Бе используют в качестве матрицы для синтеза гена с
помощью обратной транскриптазы.
Соединение гена с вектором (получение рекомбинантной ДНК).
Существуют два варианта встраивания гена в вектор: 1) встраивание,
когда оба компонента имеют липкие концы; 2) встраивание, когда
компоненты несут тупые (некомплементарные) концы.
По первому варианту, получившему название "шотган" (от англ.
8Но1§ип — дробовик), всю клеточную ДНК обрабатывают эндонуклеа-
зой рестрикции, образующей в месте разрыва одноцепочечные липкие
концы. Полученные фрагменты ДНК встраивают затем в плазмиды,
переведенные в линейную форму той же самой эндонуклеазой
рестрикции. В результате образуется сложная смесь гибридов фрагмент —
ДНК-плазмида. Для поиска гибрида, несущего целевой ген,
используют процедуры скрининга. Как видим, этот метод объединяет два
этапа — выделение целевого гена и его встраивание в вектор.
По второму варианту ДНК необходимо снабдить липкими концами. С
этой целью к З'-концам ее цепей присоединяют дезоксирибонуклеотид-
ные остатки одного типа, например, остатки аденозина. Синтез липкого
конца идет в присутствии АТР с помощью специфической трансферазы.
Эта трансфераза обладает групповой специфичностью к дезоксирибонук-
леотидам, не нуждается в матрице; на обоих концах ДНК она
катализирует синтез ро1у (А)-хвоста, состоящего из 50—100 остатков.
В свою очередь, плазмиду переводят в линейную форму,
расщепляя ее в одной точке рестрикционной эндонуклеазой с
образованием в месте расщепления тупых концов (некомплементарных
хвостам ДНК). К тупым концам линейной плазмиды присоединяют
липкие ро!у (Т)-хвосты (комплементарные ро1у (А)-хвостам ДНК). После
смешивания обоих компонентов встроенные липкие концы
соединяются по комплементарным парам А-Т ичих сшивают с помощью ДНК-
лигазы, в результате чего образуется гибридная ДНК.
Введение гибридной ДНК в клетку-хо&^ина* Этот этап основан на
том, что ДНК способна проникать в большинство эукариотических
I
504 /
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
клеток. Однако эффективность этого проникновения весьма невелика
(примерно 1—2 клетки из 106). Ее увеличивают, подбирая
специальные условия. Так, например, существенно возрастает проникновение
ДНК в клетки Е. сой, если в среду ввести ионы Са2+.
С целью эффективного включения целевого гена, имеющего
небольшую молекулярную массу, часто используют ДНК фага X. Ген и
ДНК фага X, предварительно обработанные реетриктирующей эндо-
нуклеазой, легко образуют рекомбинантную ДНК. Последнюю
одевают в белковую оболочку фага X, в результате чего образуется вирусная
частица, эффективно инфицирующая клетки Е. сой.
Отбор клеток с целевым геном и клонирование гена.
Завершающий этап технологии рекомбинантной ДНК состоит в том, чтобы
определить, какие клетки несут целевой ген; их выделить, создать
оптимальные условия для их размножения, т.е. осуществить
клонирование (воспроизводство) гена в необходимых масштабах.
Отбор можно осуществить по специфическим признакам либо
вектора, либо встроенного гена. Напомним: некоторые плазмидные
векторы сообщают клетке устойчивость к какому-либо антибиотику. Если
в культуральную среду внести антибиотик, то можно осуществить
размножение только тех клеток, которые устойчивы к антибиотику и
подавить^эазмножение и развитие клеток, в которые плазмида не
проникла. ДругЫ^ подход, основанный на специфическом признаке гена,
состоит в том,\1тобы определить, какие клетки синтезируют белок,
кодируемый целевым геном. Клетки можно отобрать из их колонии,
развившейся на твердой питательной среде, однако, с той оговоркой,
что эта колония явилась продуктом одной клетки.
Принципиальная технологическая схема синтеза и клонирования
рекомбинантной молекулы ДНК представлена на рис. 13.9.
Значение генной инженерии для науки и практики. В
результате интенсивного развития методов генной инженерии получены
клоны множества генов рибосомной, транспортной и 58 РНК, гистонов,
глобина мыши, кролика, человека, коллагена, овальбумина,
инсулина, интерферона человека и прочее. Это позволило создавать штаммы
бактерий, производящих многие биологически активные вещества,
используемые в медицине, сельском хозяйстве и микробиологической
промышленности.
Так, в настоящее время разработана и внедрена в промышленность
технология синтеза таких гормонов, как инсулин, соматотропин, и
некоторых противовирусных агентов (например, интерферона). Гены
этих биологически активных соединений достаточно эффективно экс-
прессируются в клетке Е. со1и В Японии и США на основе генно-
инженерных методов производится эффективный противораковый
препарат — интерлейкин. К началу XXI в. около 200 новых
диагностических препаратов введено в медицинскую практику и более 100
лекарственных средств находилось на стадии клинических испытаний.
505
Чаешь II, Метаболизм веществ и энергии в клетке
+
Фрагмент ДНК, содержащий
целевой ген. Получен с
помощью эндоиуклеазы ЕсоК I
Плазмидный вектор,
обработанный зндо-
нуклеазой ЕсоК I
I Встраивание
А гена в вектор
Рекомбинантная
молекула ДНК
Хромосома
клетки-хозяина
1 Внедрение в клетку-хозяина с помощью
" трансформации или заражения вирусом
"
Отбор клеток, содержащих ре комби нантную
молекулу ДНК (среда с антибиотиком)
Клонирование
+ ...
Рис. 13.9. Технологическая схема синтеза и клонирования-^комбинантной
молекулы ДНК ч^____
К ним относятся лекарства, излечивающие артрозы,
сердечно-сосудистые заболевания, некоторые опухолевые процессы. Ведутся
исследования по предотвращению и подавлению СПИДа.
Большая роль отводится генной инженерии по выведению новых
сортов культурных растений, способных к интенсивному синтезу
целевого продукта, устойчивых к болезням и различным
экстремальным воздействиям. С этой целью разрабатываются системы переноса в
растения специфических чужих генов и клонирование таких
растений. Успешно осуществлен перенос генов с помощью плазмид вируса
А§гоЬас1егшт Ште^асьепз. Растения, в хромосому которых
встраивается целевой ген и способные к дальнейшему клонированию,
называются трансгенными. Впервые трансгенные растения были получены
в 1982 г. в Институте растениеводства в Кельне и компании Мопзап{;о.
Эти растения приобрели устойчивость к антибиотику канамицину,
тормозящему рост, В настоящее время получены трансгенные
растения, способные противостоять воздействию различных вирусных
инфекций. В генно-инженерных лабораториях Бельгии и США осуще-
506
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
ствлены работы по внедрению в растительные клетки генов,
отвечающих за синтез инсектицидов, защищающих растения от
насекомых-вредителей. Получены и клонированы трансгенные растения картофеля
и томатов, устойчивых к колорадскому жуку. С помощью методов
генной инженерии делаются попытки создать растения, способные
усваивать атмосферный азот; улучшать белковый состав растительной
пищи, осуществлять синтез незаменимых аминокислот, ферментов,
витаминов, антибиотиков.
Методы клонирования успешно применяются при выращивании
млекопитающих. В настоящее время с помощью различных способов
клонируют овец, мышей, коров. Часто животные, полученные с
применением генно-инженерных методов, имеют уникальные свойства и
представляет'значительный научный интерес. Так, например, могут
быть клс^ированы животные, клетки которых моложе или старше их
физио^гического возраста.
За/короткий срок генная инженерия оказала огромное влияние на
развитие молекулярно-генетических методов и позволила
существенно продвинуться по пути познания строения и функционирования
генетического аппарата. Достижения молекулярной биологии и генной
инженерии дали человеку возможность читать генетические тексты —
сначала вирусов, бактерий, дрожжевых грибков. После восьми лет
работы многих исследовательских групп удалось точно определить
97 млн пар нуклеотидов и их местонахождение в спирали ДНК,
хранящей полную наследственную информацию микроскопического
червячка СаепогНаЬйШз е1е§ап8. Расшифровка (секвенирование) генома
С. е1е§ат была осуществлена по совместному проекту двумя
исследовательскими группами: из Центра геномного секвенирования
Вашингтонского университета (США) и Сенгеровского центра (Кембридж,
Англия). Однако если у дрожжей функция половины генов в геноме
неизвестна (так называемые молчащие гены), то у червя эта доля еще
больше: из 19 тыс. генов 12 тыс. остаются пока загадочными. Тем не
менее впервые удалось полностью прочитать геном многоклеточного
животного — обитающего в почве микроскопического червячка
длиной около миллиметра. Геном этой нематоды состоит из 97 млн пар
нуклеотидов ДНК, округленно ОД млрд пар. Геном человека — 3 млрд
нуклеотидных пар. Разница в 30 раз. Однако работа по прочтению
генома С. е1е$ап8 убедила даже самых закоренелых скептиков, что
расшифровка генома человека вполне возможна.
Международная программа по расшифровке генома человека была
начата в 1989 г., тогда же благодаря инициативе и энергии А. А. Бае-
ва к программе подключилась и Россия. Сейчас геномная программа
уже доказала свое выдающееся значение для развития наших знаний
о жизни в целом.
Считают, что программа по расшифровке генома человека в
значительной степени способствовала и развитию микробиологии. В насто-
507
Часть II, Метаболизм веществ и энергии в клетке
ящее время расшифрованы более 20 полных геномов, в том числе
возбудителей многих опасных болезней (туберкулеза, сыпного тифа, язвы
желудка и др.)* Знание геномной структуры патогенных бактерий очень
важно для создания вакцин, для диагностики и других медицинских
целей. Значительное влияние генная инженерия оказывает на
медицинскую генетику, которая занимается генодиагностикой
наследственных болезней, генетическими основами предрасположенности ко
многим распространенным болезням.
Геномные методы идентификации личности имеют далеко идущие
последствия для общества. Действительно, криминалистика
получила в свое распоряжение абсолютно надежный метод доказательства
виновности или невиновности человека. Для анализа, называемого
геномной дактилоскопией, достаточно одной капли крови, одного
волоса, кусочка ногтя, следов пота, слюны и т.д.
6 апреля 2000 г. американская компания Се1ега Сепоппсз объявила о
том, что закончила работу над декодированием трех миллиардов пар
генов, составляющих геном человека. Таким образом, Се1ега Оепоипсв
опередила серьезный международный проект, финансируемый
правительствами нескольких стран. Однако необходимо определить функции
каждого конкретного гена человека, что может оказаться более сложным,
нежели просто нарисовать схему расположения генов.
Совершенно очевидно, что расшифровка генома человека сыграет
важную, если не самую решающую роль в медицине настоящего века,
поскольку его открытие облегчит исследования и лечение многих
наследственных заболеваний.
13,5. Транскрипция
В процессе транскрипции с помощью ферментной системы
происходит синтез цепи РНК на одной из нитей матричной ДНК, в результате
чего образуется РНК — точная копия нити ДНК по последовательности
азотистых оснований в полирибонуклеотидной цепи. Транскрипция
обеспечивает синтез всех трех типов РНК: мРНК, тРНК, рРНК.
Механизм синтеза РНК в основных чертах сходен с механизмом
синтеза ДНК. Непосредственными предшественниками синтеза РНК
являются рибонуклеозидтрифосфаты. Однако в отличие от
репликации ДНК в синтезе РНК отсутствуют какие-либо механизмы
корреляции, и транскрипция РНК не обладает такой точностью копирования,
как репликация ДНК. Случайно возникшие при транскрипции
ошибки не могут иметь решающего значения, поскольку эти ошибки не
воспроизводятся в последующих поколениях. В отличие от
репликации, где копируется вся ДНК с образованием дочерних ДНК, транс-,
крипция осуществляется избирательно лишь на отдельном участке
ДНК, соответствующем гену или группе генов. Она направляется
особыми регуляторными последовательностями нуклеотидов ДНК, ука-
508
Глава 13, Синтез ДНК, РНК и белков. Перенос генетической информации
зывающих начало и конец участков гена, подлежащего
транскрипции.
Транскрипция катализируется ДНК-зависимой РНКполимеразой.
Для синтеза необходимы все четыре рибонуклеозид-5'-трифосфата (АТР,
ОТР, 1ТТР и СТР), а также ионы М#2+. РНК-полимераза удлиняет цепь
РНК, присоединяя молекулы рибонуклеотидов к З'-ОН-концу
молекулы РНК:
(ИМР)П + ЫТР 3=Ь ^МР)п+1 + РР1
РНК Рибонуклеотид- Удлиненная Пирофосфат
5'-трифосфат Цепь ДНК
В растущих цепях РНК рибонуклеотидмонофосфаты
присоединяются к З'-концу, а исходный (первый) нуклеотид остается с Р~Р~Р-хвостом.
Поскольку аденин и урацил являются комплементарной парой, в
участки мРНК, соответствующие местам ДНК-матрицы, где находятся
остатки аденина, включаются остатки урацила; в места же,
соответствующие остаткам тимина ДНК-матрицы, в мРНК включаются остатки аде-
нина. Остатки гуанина и цитозина ДНК-матрицы определяют
включение в цепь РНК соответственно остатков цитозина и гуанина. Таким
образом, нуклеотидная последовательность цепи РНК комплементарна
последовательности нуклеотидов матричной цепи ДНК. В отличие от
каталитического действия ДНК-полимеразы, РНК-полимераза в затравке не
нуждается. Однако этот фермент начинает функционировать после того,
как свяжется со специфическим участком матричной цепи ДНК.
Процесс транскрипции включает четыре стадии: связывание РНК-
пол имеразы с ДНК, инициацию — начало синтеза цепи РНК,
элонгацию (удлинение цепи РНК) — собственно процесс синтеза полинуклео-
тидной цепи и терминацию — завершение этого синтеза (рис. 13 Л О).
Связывание РНК-полимеразы с ДНК происходит на определенном
участке последней, обладающим наибольшим химическим сродством
к ферменту. Этот участок, имеющий специфическую
последовательность нуклеотидов, называется промотором, В узнавании промотора
важную роль играет специфический белок — 0-фактор. Сам по себе он
не обладает каталитической функцией. В присутствии а-фактора
происходит прочное связывание РНК-полимеразы с промотором.
Связывание сопровождается частичным локальным плавлением цепей ДНК
и их расхождением. Природа сигнала начала транскрипции пока
неясна. Установлено, что промоторные участки многих ДНК имеют
длину 20—50 нуклеотидов, в состав которых входят в основном АТ-пары.
Большое количество этих пар в промоторе объясняется, вероятно, тем,
что такие последовательности плавятся значительно легче, чем
участки с большим количеством ОС-пар. Поэтому они являются хорошими
мишенями для РНК-полимеразы, в функционировании которой
важную роль играет локальная денатурация ДНК. Она переводит
закрытый комплекс полимераза-промотор в открытый, в котором происхо*
509
Часть II. Метаболизм веществ и энергии в клетке
Сигнал Днк
инициации Связывание
РН К- полимеразы
0ШШШ с ДНК
[ ■ ■ **' ■■ *' ■ ^^Ч ■Л' * э ■ ■. ■■ ,■■■■■■■■■ С ' ■
МТР
Инициация
синтеза РНК
РРР
РНК
ЫТР
Элонгация
РРР<г
Терминаци
* 1 *
РРР а
Сигнал
термииации
* * *
• - I
Сход с матрицы
Рис. 13.10. Схематическое изображение стадий транскрипции РНК: (ЛЭ —
РНК-полимераза; Ш — с-фактор; ^ — р-фактор; ШР — рибонуклеозид-5'-три-
фосфаты
дят значительные конформационные изменения, с разрушением
водородных связей между комплементарными основаниями
ДНК-матрицы. Последние становятся доступными для спаривания с
поступающими рибонуклеозидтрифосфатами.
Инициация синтеза РНК начинается не на участке, более прочно
связанном с РНК-полимеразой, а на расстоянии приблизительно 6—
7 оснований от него. На этой стадии первыми в синтез РНК
вовлекаются нуклеотиды рррА либо рррО. Эти нуклеотиды вступают в
реакцию со второй молекулой рибонуклеозидтрифосфата, приводящей к
образованию динуклеотида, с б'-концом которого все еще связан три-
фосфат.
На стадии элонгации а-фактор освобождается из комплекса, в
состав которого входят РНК-полимераза, ДНК-матрица и
синтезирующаяся цепь РНК. Освободившийся о-фактор может снова
присоединяться к другим молекулам РНК-полимеразы. В результате
отщепления а-фактора создаются условия для элонгации цепи РНК. Нуклео-
тидные субъединицы присоединяются к З'-концу РНК* Для
образования правильной пары оснований соответствующий нуклеозидтрифос-
фат должен быть пристроен к цепи ДНК-матрицы до того, как про-
510
Глава 13. Синтез ДНК, РНК и белков, Перенос генетической информации
■ | | ~ Т ■ ' Г" —I ' I ' " 1—Г I * ' I ' ***«■■■ ■ <■ |
изойдет его связывание с растущей цепью РНК. В процессе элонгации
РНК-полимераза локально расщепляет двойную спираль ДНК. Ново-
синтезируемый транскрипт РНК и матричная ДНК временно
образуют короткие участки гибридной двойной спирали ДНК—РНК, что
способствует правильности считывания цепи ДНК. Длина гибрида ДНК—
РНК составляет примерно 10 нуклеотидных пар. Образуется он за счет
спаривания оснований новосинтезируемой РНК с основаниями ДНК,
По мере продвижения РНК-полимеразы вдоль цепи ДНК растущая
цепь РНК отходит от матрицы, двухспиральная структура ДНК после
прохождения фермента вновь восстанавливается.
Терминация транскрипции осуществляется под действием
специфического р-фактора, который узнает закодированный в ДНК сигнал
терминации. Однако в некоторых случаях присутствие р-фактора
необязательно. Значение этих различий пока неясно. По достижении
РНК-полимеразой сигнала терминации синтезированная цепь РНК
окончательно отделяется от ДНК-матрицы.
Процессинг РНК. Сошедшие с ДНК-матрицы новосинтезирован-
ные РНК претерпевают дальнейшие модификации, называемые
посттранскрипционным процессингом или созреванием. Такие модифика-
ции связаны либо с удалением определенных нуклеотидов путем
гидролиза, либо с присоединением нуклеотидов к концевым группам, либо
с химической модификацией (например, метилированием).
При транскрипции мРНК эукариот на З'-конце появляются
ро1у (А)-хвосты, содержащие 100—200 остатков адениловой
кислоты. Предполагается, что эти хвосты необходимы для транспорта мРНК
из ядра. При поступлении мРНК в цитоплазму они отщепляются.
Важной операцией процессинга мРНК эукариот является образование в
них б'-концевого "кэпа" (от англ. сар — шапка). Он представляет
собой остаток 7-метилгуанозина. Предполагается, что он принимает
участие в связывании мРНК с рибосомой.
При транскрипции тРНК образуются крупные молекулы пре-тРНК.
Иногда молекулы пре-тРНК содержат не одну, а несколько молекул
различных видов тРНК. В процессинге тРНК участвуют рибонуклеа-
зы, "обетригивающие" пре-тРНК. Помимо обработки тРНК этими
ферментами в процессинг включается также модификация азотистых
оснований. Одной из основных модификаций является
метилирование, которое может происходить как по основанию, так и по З'-ОН-груп-
пе остатка рибозы. К тем тРНК, у которых в транскрипте не
содержится З'-концевая тринуклеотидная последовательность -С-С-А,
данный тринуклеотид присоединяется в ходе посттранскрипционного
процессинга.
рРНК образуются также из более длинных предшественников. Перед
процессингом пре-рРНК метилируются по специфическим
основаниям, затем расщепляются нуклеазами. У прокариот из 308-пре-рРНК
образуются 168- и 238-РНК. Пре-рРНК эукариот имеет большую мо-
511
Часть II. Метаболизм веществ и энергии в клетке ^^__^__
лекулярную массу (около 440е)- После метилирования она
распадается на ряд фрагментов, одни из которых включаются в рибосому, а
другие разрушаются.
Весьма сложен посттранскрипционный процессинг мРНК у эука-
риот. Это сопряжено с особенностями организации эукариотических
генов. В их нуклеотидной последовательности вставлены участки, не
кодирующие аминокислотную последовательность полипептидного
продукта. Эти вставки, прерывающие строгое соответствие между
нуклеотидной последовательностью ДНК и аминокислотной последователь-
ностью кодируемого полипептида, называются интронами. Участки
ДНК, несущие информацию об аминокислотной последовательности
лолилептида, называются экзонами* Характерно, что интроны, как
правило, значительно длиннее экзонов (их общая длина составляет
около 80 % длины гена). Предполагают, что с помощью интронов гены
как бы делятся на мини-гены, которые могут рекомбинировать с
образованием новых генов. Другое предположение заключается в том> что
интроны играют роль своеобразных регуляторных сигналов. Эукарио-
тическая РНК-полимераза переписывает подряд всю нуклеотидную
последовательность транскрибируемого гена, включая и экзоны, и
интроны. В результате в ядре образуются длинные предшественники мРНК,
йазываемые гетерогенными ядерными РНК (гяРНК). Однако в
зрелой мРНК блоки интронов отсутствуют. Это достигается за счет
удаления интронов в ходе процесса, называемого сплайсингом.
Сплайсинг — довольно сложный процесс, осуществляемый макро-
мол екулярным комплексом, который называется сплайсосомои. Суть
данного процесса заключается в том, что под действием особого типа
РНК, малых ядерных РНК (мяРНК) интроны сворачиваются в петлю
(образуется структура типа "лассо"), в результате этого экзоны
сближаются и происходит "вырезание" интрона; мяРНК способны
удерживать интрон в петле за счет комплементарного спаривания нуклео-
тидных последовательностей на концах интрона и нуклеотидных
последовательностей самой мяРНК. Благодаря тому, что жяРНК играют
роль как бы временных матриц, удерживающих рядом экзоны,
становится возможным их ферментативное соединение.
Зрелая мРНК переходит из ядра в цитоплазму, чему способствуют
специфические белки, проводящие зрелые мРНК сквозь поры в
ядерной мембране*
При изучении сплайсинга в ряде систем была показана его аутока-
талитическая природа. Так, при исследовании процессинга про-рРНК
у ТеиаНутепа ШегторНЫа обнаружено, что процесс вырезания
интрона происходит независимо от наличия какого-либо белкового фактора
и требует присутствия только М§2+ и гуанозина в качестве кофактора,
Было выяснено, что в данном случае сама РНК обладает
каталитической активностью. РНК-молекулы, обладающие каталитической
активностью, подобные обнаруженной у ТеЪгакутепа 1НеггпорЫ1ау часто
512
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
называют рибозимами для того, чтобы отличать их от ферментов
белковой природы.
Процесс сплайсинга интрона у Те1гаНутепа сходен с
ферментативными реакциями с участием белковых катализаторов по ряду
признаков: скорость реакции, лежащей в основе сплайсинга, на несколько
порядков выше по сравнению с некатализируемой реакцией; рибозим
высокоспецифичен по отношению к своему субстрату. Однако процесс
сплайсинга нельзя считать истинно энзиматическим, поскольку
первоначальный катализатор не возобновляется в ходе реакции- После
его высвобождения от исходной рРНК рибозим под действием
автокаталитической активности укорачивается с двух концов, превращаясь
в форму, известную как Ь-21. Эта форма обладает рядом свойств
первоначального рибозима, в частности, сохраняются гуанозинсвязываю-
щий центр и большинство так называемых внутренних
направляющих последовательностей, способствующих образованию петли
интрона. Ь-21 может функционировать как специфическая эндонуклеаза,
расщепляя другие РНК-молекулы в участках, содержащих
последовательность ССС11С1Т. Эта реакция аналогична начальному этапу
сплайсинга, она требует присутствия гуанозинового кофактора, и субстрат —
РНК, связывается с внутренней направляющей последовательностью.
Реакция, протекающая под действием Ь-21, истинно каталитическая,
так как рибозим остается интактным. Константа скорости для реак-
ции отщепления — 2,1*103 с"1, что в 1011 раз выше, чем для некатал и-
зируемого гидролиза. Следует отметить, однако, что число оборотов для
этого рибозима очень низкое, поскольку продукт остается тесно
связанным с внутренней направляющей последовательностью. Маловероятно,
что эта активность значительна 1п V^VО. 1п V^^^о намного более
эффективны полученные с помощью метода мутагенеза производные Ь-21.
Таким образом, была открыта возможность биокатализа при
участии РНК. Следует отметить, что на самом деле т ишо в сплайсинге
про-рРНК участвует также и белок. Но считают, что он выполняет не
каталитическую функцию, а его роль заключается в поддержании
оптимальной для сплайсинга конформации про-рРНК*
13,6- Обратная транскрипция
Обратная транскрипция характерна для так называемых ретрови*
русое — обширной группы вирусов, различающихся как по
биологическим свойствам (в частности, по способности вызывать
злокачественные опухоли), так и по морфологии. К данной группе относятся
хорошо известные вирус иммунодефицита человека, вирус саркомы Рауса.
Их вирионы содержат фермент обратную транскриптазу (РНК-за-
висимую ДНК-полимеразу). Когда вирус попадает в клетку-хозяина, с
помощью обратной транскриптазы осуществляется синтез ДНК с
использованием в качестве матрицы вирусной РНК. Новосинтезирован-
33. Заказ 3486
513
Часть II. Метаболизм веществ и энергии в клетке _
ная (одноцепочечная) ДНК при участии ферментов клетки-хозяина
превращается в двунитевую структуру в результате достраивания
комплементарной ДНК (кДНК). Эта двунитевая ДНК встраивается в
геном клетки-хозяина с помощью специфических рекомбинационных
механизмов, где она может в течение длительного времени (в ряду
поколений) оставаться в скрытом неэкспрессируемом состоянии*
Однако под действием определенных факторов (ионизирующие
излучения, канцерогенные агенты и т.д.) эти бездействующие гены могут
активироваться и способствовать превращению клетки в раковую»
13.7. Синтез белка (трансляция)
Синтез белка кодируется структурными генами; информация,
содержащаяся в этих генах, определяет первичную структуру белка.
Первоначально информация передается на мРНК; последние,
соединяясь с рибосомами, образуют комплексы — полисомы, на которых и
протекает синтез полипептидных цепей. Трансляция осуществляется
в соответствии с генетическим кодом, представляющим собой
своеобразный текст, записанный в нуклеиновой кислоте с помощью четырех
нуклеотидов и предоставляющий информацию о белковом тексте,
включающем 20 аминокислот. Большая часть информации о механизме
биосинтеза белка была получена в опытах с бактериями.
Прежде чем перейти к рассмотрению деталей процесса
трансляции, необходимо отметить его следующие особенности. Во-первых,
важную роль в биосинтезе белка играют активация аминокислот и
образование их комплексов с тРНК. Во-вторых, в основе трансляции,
осуществляемой рибосомами, лежит спаривание оснований трех
нуклеотидов (триплетов) мРНК, получивших название кодонов, и
комплементарных им триплетов тРНК — антикодонов. В-третьих,
исключительно важным фактором в синтезе белка является феномен двои
ной специфичности ферментов, участвующих в трансляции. Это
четко видно на примере действия аминоацил-тРНК-синтетаз; каждая из
них, с одной стороны, высокоспецифична к определенной
аминокислоте, а с другой — к определенной тРНК. Именно двойная
специфичность синтетаз обеспечивает реализацию генетического кода.
Основные свойства и функции генетического кода.
Исследования, проведенные Ф. Криком и С. Бреннером, позволили установить
следующие основные свойства генетического кода:
1. Код триплетен: каждой аминокислоте соответствует набор,
содержащий три нуклеотида. Это легко заключить из следующего
рассуждения. Поскольку каждая молекула ДНК и соответственно мРНК
содержит четыре вида оснований, то в случае кодирования одним
основанием одной аминокислоты могли бы быть детерминированы
только четыре аминокислоты. При кодировании одной аминокислоты
набором из двух оснований можно детерминировать лишь 16 аминокис-
514
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
лот (42=16). Поскольку в состав белка входят 20 аминокислот, то ясно,
что одна аминокислота должна кодироваться группой, содержащей не
менее чем три основания (43 = 64). Действительно, экспериментально
было установлено, что одну аминокислоту кодирует группа из трех
оснований — триплет. Эта группа нуклеотидов получила название
кодон. Кодоны называют смысловыми, если они соответствуют какой-
нибудь аминокислоте, и бессмысленными (незначащими), если они
никакой аминокислоты не кодируют.
2. Были открыты все 64 кодона: 3 осуществляют стадию терминации
при синтезе белка: ИАА, 11А6, 1ЮА; они не кодируют в норме ни одну
аминокислоту; 61 — кодируют аминокислоты, т.е. на каждую из 20
аминокислот приходится около 3 кодонов. Иными словами, генетический
код вырожден (избыточен): одной аминокислоте соответствует более чем
один кодон. Бывает 2, 3, 4 и 6-кратная вырожденность.
3. Код специфичен: каждую аминокислоту детерминируют только
определенные кодоны, которые не могут быть использованы для
другой аминокислоты.
4. Генетический код может быть перекрывающимся и неперекры-
вающимся. В случае перекрывающегося кода триплет АБВ кодирует
аминокислоту ак1, триплет БВГ — аминокислоту ак2, триплет ВГД —
аминокислоту ак3. В случае неперекрывающегося кода основания
каждого триплета специфичны только к одной аминокислоте (схема 1):
АБВ—ГДЕ—ЖЗИ—КЛМ—НОП ... Последовательность триплетов
акх— ак2— ак3— ак4— ак5 ... Последовательность аминокислот
Исследования показали, что генетический код, за исключением
редких случаев, является неперекрывающимся.
5. Коллинеарность кода: последовательность кодонов мРНК
однозначно определяет последовательность аминокислот в полипептидной
цепи. Основания мРНК считываются с определенной точки
(например, с триплета АБВ) и строго последовательно. Другими словами:
код называют коллинеарным, если кодоны располагаются в том же
порядке, что и остатки кодируемых ими аминокислот. Допустим, что
в какой-либо точке мРНК произошла мутация: удаление (делеция)
или вставка какого-либо основания (схема 2):
АБВ ГДЕ ' ЗИК ЛМН ОПР — Делеция Ж
ак, — ак2 — ак3* — ак4* — ак&*
гс
АБВ ГДЕ т СЖЗ ИКЛ МОР Р — Вставка С
ан, — ак, — ак3* — ак4* —• ак5*
33*
515
Часть II. Метаболизм веществ и энергии в клетке
Первые две аминокислоты полипептидной цепи остаются те же,
что и в цепи схемы 1. При дальнейшем наращивании полипептидной
цепи в связи с изменением состава оснований триплетов будут
принимать участие иные аминокислоты ак3*, ак4*, ак5*. В результате
возникает так называемый сдвиг рамки считывания оснований.
6. Генетический код универсален: один и тот же код используется
всеми видами живых организмов. Некоторые отклонения были
обнаружены только для ДНК митохондрий, что представляет интерес с
точки зрения эволюции данных органелл.
Важной проблемой трансляции явилась расшифровка
генетического кода. В результате работ, проведенных в начале 60-х гг. М, Ни-
ренбергом, эта проблема была решена. Автор обнаружил, что
добавление полиуридилата [ро1у (И)] в бесклеточную систему синтеза белка
приводит к синтезу полифенилаланина.
Это явилось основанием сделать заключение: ро1у (II) выполняет
функцию мРНК; иными словами, триплет ХЛШ является кодоном,
детерминирующим фенилаланин. Это открытие указало путь к расшифровке
нуклеотидного состава всех существующих кодонов. В дальнейших
исследованиях в качестве основных компонентов были использованы
бесклеточные экстракты Е. соИ, способные к активному синтезу белка, и
синтетические полирибонуклеотиды, сыгравшие роль мРНК. При
добавлении АТР, ОТР и аминокислот к этой системе синтезировался
полипептид, аминокислотный состав которого однозначно определялся нуклео-
тидным составом синтетического полинуклеотида. Так, ро1у (А)
выполнял роль матрицы при синтезе полилизина и, следовательно, для лизина
кодоном оказался ААА; ро1у (С) был матрицей для синтеза полипролина
и кодоном для пролина оказался триплет ССС.
Использование смешанных сополимеров, содержащих два разных
нуклеотида в случайной последовательности, позволило установить
нуклеотидный состав других кодонов. Однако последовательность
оснований в этих кодонах оставалась не выясненной. Ее удалось
установить с помощью двух различных экспериментальных подходов: 1) с
помощью зависимого от кодона специфического связывания молекул
тРНК с рибосомами; 2) с использованием синтетических полирибо-
нуклеотидов с упорядоченной последовательностью.
В 1964 г, М. Ниренберг установил, что тринуклеотид, подобно
триплету мРНК, специфически связывается с определенной молекулой
тРНК, для которой он является кодоном. Были синтезированы 64 три-
нуклеотида и проверены на связывание с молекулами тРНК,
специфичными для каждой из 20 аминокислот. Этот простой подход
позволил расшифровать около 50 кодонов.
Второй подход был использован Г. Кораной. Им были
синтезированы полирибонуклеотиды с определенной повторяющейся
последовательностью из двух, трех и четырех оснований. Эти регулярные
сополимеры были использованы в качестве матриц в системе синтеза бел-
516
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
ка. Итак, внесенный в эту систему сополимер с повторяющимся тет-
рануклеотидом (11А11С) выполнял функцию матрицы для синтеза
полипептида с повторяющейся последовательностью Туг-11е-8ег-Ьеш
ПАП | АПС | ПСП | С1ТА | ИАТ! | АИС | ИСИ | СИА
ф- ф ф ф ф ф ф ф—
Туг Не 8ег Ьеи Туг Не 8ег Ьеи
Как видно, в синтезе полипептида из аминокислот тирозина, изо-
лейцина, серина, лейцина принимают участие четыре специфичных
для них ко дона.
Функция кодонов IIАА, IIАО, ИОА ( не кодирующих ни одну
аминокислоту) связана с терминацией синтеза белка.
В результате проведения фундаментальных работ, выполненных в
лабораториях Ниренберга и Кораны, к 1966 г. был полностью
расшифрован генетический код (табл. 13.1).
Таблица 13.1
Генетический код (последовательность оснований в триплетах мРНК,
кодирующих аминокислоты)
Первое
положение
(5'-конец)
V
С
А
С
Второе положение
и
РЬе
РЬе
Ьеи
Ьеи
Ьеи
Ьеи
Ьеи
Ьеи
Не
Не
Не
Ме1
Уа1
Уа1
Уа1
Уа1
С
8ег
8ег
Бег
8ег
Рго
Рго
Рго
Рго
Т1гг
ТЬг
ТЬг
ТЬг
А1а
А1а
А1а
А1а
А
Туг
Туг
Стоп
Стоп
Н1з
Н1з
вы
01п
Азп
Азп
Ьуз
Ьуе
Анр
Азр
С-1и
01и
О
Суз
Суз
Стоп
Тгр
Аг^
Агё
Агё
8ег
8ег
Агё
С1у
01у
01у
С1у
Третье
положение
(З'-конец)
1
V
С
А
О
V
С
А
О
С
А
С
С
А
О
Образование аминоацил-тРНК осуществляется в цитоплазме в
две стадии под действием аминоацил-тРНК-синтетазы. На первой
стадии образуется аминоацил-АМР и пирофосфат; в результате этой
реакции аминокислота активируется по карбоксильной группе:
К-СН-СООН + АТР 2 К-СН-СО-АМР + РР1ф
Пирофосфатаза гидролизует РР1 на 2 Р.. Затем следует перенос ами-
ноацильной группы с АМР на тРНК:
К-СН-СО-АМР + тРНК г К-СН-СО~тРНК + АМР
517
Часть II. Метаболизм веществ и энергии в клетке
Антикодон тРНК
\ ХУ2 ~/~
Кодон
мРНК
У * Молекула тРНК переносит акти-
с-а-о-с-сн-к Остаток вированную аминокислоту к рибо-
Г МНЛ аМИНОКИСЛОТЫ . " ттгтт*
14112 соме, Антикодон тРНК узнает
кодон мРНК. Таким образом, компле-
ментарность в системе
кодон—антикодон является основой механизма
формирования первичной
структуры белка (рис. 13.11), и тРНК
играет роль адаптера (промежуточного
звена) при переводе
четырехбуквенного кода нуклеиновых кислот на
двадцатибуквенный код белков.
Механизм работы рибосом.
Работа рибосом включает три
последовательных этапа: инициацию
(начало синтеза белка), элонгацию
(удлинение полипептидной цепи),
терминацию (окончание синтеза
белка). Следует подчеркнуть, что
полной ясности в работе рибосом
пока нет. Это связано с тем, что до сих пор не установлена роль как
некоторых рРНК, так и белковых компонентов, входящих в состав
субчастиц рибосомы.
Процессы инициации, элонгации, терминации более обстоятельно
исследованы у прокариот, в частности у Е. соИ, поэтому они взяты за
основу при дальнейшем описании синтеза белка.
Инициация — сложный процесс (рис. 13.12). У прокариот роль
инициирующей аминокислоты (]Ч-концевой аминокислоты) играет
производное метионина — Ы-формилметионин; у эукариот — метио-
нин:
СН3-8-СН2-СН2-СН-СООН СН3~8-СН2-СН2-СН-СООН
Рис. 13Л1. Схематическое
изображение комплекса аминоацил—тРНК—
мРНК. Показан участок прикрепления
аминокислоты и кодона мРНК (ХУ2) с
антикодоном тРНК (Х'У'2')
ИН2
Метионин
N13
ноо
К-Формилметионин
В процессе инициации принято выделять три стадии. Для
протекания этих стадий необходимы ОТР как источник энергии и три бел-
+
ковых фактора инициации: ГР-1, 1Г-2, 1Г-3 (1Г — 1П1С1айоп ?аск>гз).
Предварительно 308-субчастица рибосомы связывается с
фактором инициации 1Р-3, что препятствует ассоциации 308- и 508-субчае-
тиц. Затем малая 308-субчастица рибосомы связывается с мРНК.
Причем кодон инициации А1ТО (реже 0110), находящийся на б'-конце
мРНК, оказывается в строго определенном месте. Это достигается за
счет наличия с б'-стороны от инициирующего кодона специального
инициирующего сигнала, представляющего собой последовательность
518
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
508
303
1Г-3
+
мРНК
4**
508 Т
3'
Ин и ци ирующи й
к од он
3'
ОТР
1Р-2
Ше*
Аитикодон
3'
из 6—8 остатков адени-
на или гуанина. Эта
сигнальная
последовательность узнается
комплементарной
последовательностью 168—рРНК
малой субчастицы
рибосомы.
Затем на второй
стадии к образовавшемуся
комплексу присоединяв
ется Ы-формилметио-
нин-тРНК,
предварительно связанная с
фактором инициации 1Г-2,
а также с ОТР,
играющим роль источника
энергии. Полагают, что
связыванию 1Р~2-фак-
тора помогает фактор
ГР-1, роль которого,
однако, до конца не
выяснена.
Рибосома имеет два
участка связывания
тРНК: Р-участок (пеп-
тидильный, или донор-
ный) и А-участок
(акцепторный, или амино-
ацилъный). Эти
участки расположены
частично на малой (308),
а частично на большой
(508)-субчастицах.
Инициирующая Г-МеЪ-тРНЕ связывается в Р-участке. Однако все
остальные аминоацил-тРНК присоединяются к А-участку,
Процесс инициации завершается связыванием образованного
комплекса с 508-субчастицей рибосомы. На этой стадии ОТР гидролизу-
ется на ОБР и Р.; продукты гидролиза и белковые факторы выводятся
в окружающую среду. В результате образуется инициирующий 708-
рибосомный комплекс.
Элонгация пептидной цепи включает три стадии: 1) связывание
второй аминоацил-тРНК (А2-тРНК) со следующим кодоном мРНК,
находящимся на А-участке рибосомы; 2) образование пептидной связи и
3) транслокация (рис. 13.13).
1Р-2 1Г-1 1Р-3
Ини ци и рующий
кодон
3'
Следующий
кодон
Рис, 13.12. Схематическое изображение
инициации синтеза белка. 1Р-1, 1Р-2, 1Р-3 — факторы
инициации; Р и А — соответственно пептидиль-
ный и аминоацильный участки рибосомы
519
г
Часть II. Метаболизм веществ и энергии в клетке
ССР
Пептидил-
трансфераза
Я1е*
Процесс связывания
идет с участием трех
белковых факторов Ти, Тз,
О и источника энергии
ОТР. На первой стадии в
свободный А-участок
поступает А2-тРНК в виде
комплекса с Ти—ОТР.
Связывание антикодона
А2-тРНК с кодоном мРНК
сопровождается
освобождением фактора Ти и
гидролизом ОТР на ОВР и
Р.- Высвободившийся
комплекс Ти-ОВР
соединяется со вторым
фактором Тз, который
вытесняет из комплекса ОВР.
Затем к бинарному
комплексу Ти—Те присоединяется
ОТР, в результате чего
фактор Тв
высвобождается* Образованный
комплекс Ти—ОТР вновь
участвует в присоединении
аминоацил-тРНК к
комплексу мРНК-рибосома.
На второй стадии
процесса элонгации
осуществляется образование
пептидной связи.
Реакция катализируется
ферментом пептидилтранс-
феразой, входящей в состав 508-субчастицы рибосомы. Из Р-участка
М-формилметионшговый остаток перебрасывается от тРНК в А-учас-
ток на А2-тРНК. Карбоксильная группа Ы-формилметионина
реагирует с аминогруппой А2-тРНК с образованием дипептидил-тРНК (см.
рис. 13.13).
На третьей стадии — транслокации — происходит перемещение
рибосомы на один кодон в направлении к З'-концу мРНК. На этом
этапе деацилированная тРНК вытесняется с Р-участка в окружающую
среду; осуществляется перенос с А-участка на Р-участок дипепти-
дил-тРНК. Важную роль здесь играет третий фактор элонгации О.
Продвижение на один кодон сопровождается освобождением фактора
О. Источником энергии на транслокацию является реакция гидроли-
Свободная у^ I <$ .Л
тРНК Г
+ ОБР + Р1
4!№1
Рис. 13.13. Элонгация синтеза белка:
связывание аминоацил-тРНК, образование пептидной
связи, транслокация
520
Глава 13. Синтез ДНК, РНК и белков, Перенос генетической информации
за ОТР на ОВР и Р.; при каждом акте транслокации расщепляется
одна молекула ОТР.
Транслокацией завершается первый цикл элонгации. Затем
следует второй цикл. В освободившийся А-участок поступает новая амино-
ацил-тРНК, антикодон которой комплементарен вновь поступившему
в А-участок кодону мРНК. В результате образуется трипептид.
Циклы элонгации повторяются до тех пор, пока не завершится синтез
полипептидной цепи, аминокислотная последовательность которой
четко контролируется поступающими на рибосому кодонами мРНК.
Терминация — окончание процесса трансляции — происходит после
того, как А-участок рибосомы связывается с терминирующим кодоном
мРНК (11АА, Х1АО, 1ГОА). С этим кодоном не может связаться ни одна
тРНК, так как таких тРНК с соответствующими антикодонами нет.
Действие терминирующих кодонов осуществляется совместно с
белковыми рилизинг-ф&ктор&ми: Кг К2 и 8. Эти факторы
способствуют отщеплению образовавшегося полипептида от конечной тРНК, его
высвобождению и отделению ненагруженной тРНК от Р-участка,
после чего рибосома диссоциирует на составляющие ее субчастицы*
В процессе трансляции мРНК функционирует в форме
полирибосомы (рис. 13.14), На молекуле мРНК рибосомы движутся одна за дру-
Рис. 13.14. Схематическое изображение работающей полирибосомы
(комплекс из мРНК и четырех рибосом): МС — малая субъединица рибосомы; БС —
большая субъединица рибосомы; Д — аминоацил-тРНК; КИ — кодон
инициации; ПЦЭ — полипелтидная цепь на этапе элонгации; СБ — синтезированный
белок; КТ — кодон терминации
521
Часть II. Метаболизм веществ и энергии в клетке
гой, "считывая" кодоны матрицы, в результате чего полипептидная
цепь на каждой рибосоме постепенно растет.
Благодаря образованию полирибосом отсутствует необходимость в
большом числе копий мРНК. В то же время синтез белка протекает
быстрее, что особенно важно в фазе интенсивного развития клетки. В
полирибосоме возрастает продолжительность жизни мРНК, так как
после ее отделения от рибосомы она тут же гидролизуется цитоплаз-
матическими рибонуклеазами и, таким образом, для синтеза того же
белка потребовалась бы транскрипция новой молекулы мРНК. В
результате функционирования полирибосомы молекула мРНК может быть
использована много раз для воспроизведения одной и той же
последовательности аминокислот. Эта способность молекулы мРНК
объясняет нам, почему клетка нуждается в сравнительно небольшом
количестве мРНК (обычно 1—2 % от общего количества РНК).
В процессе элонгации и по окончании трансляции белки
подвергаются модификации. После того как цепь достаточно удлинилась,
концевая 14-формильная группа удаляется под действием фермента пеп-
тидилформилазы, а метионин от полипептидной цепи отщепляется
специфичной аминопептидазой, В ходе элонгации белки начинают
укладываться в третичную структуру, которую они окончательно
принимают после схода с рибосомы. Некоторые белки, выделяемые
клеткой в окружающую среду, как, например, внеклеточные ферменты,
имеют на И-конце пептидные участки, облегчающие транспорт
ферментов через мембраны; эти участки удаляются в результате
ограниченного протеолиза.
К посттрансляционным модификациям относятся окисление
8Н-групп в 8-8-группы, присоединение к остатку пролина ОН-груп-
пы, фосфорилирование ОН-группы серина, присоединение к белку
остатков Сахаров и образование гликопротеинов; иными словами,
происходит специфическое "дозревание" белков.
Термодинамический анализ процесса трансляции мало изучен.
Однако уже сейчас можно ответить на вопрос: каков источник
энергии, за счет которого осуществляется синтез пептидной связи? По-
видимому, этим источником является связь аминоацил-тРНК. Она
образуется при расщеплении АТР в синтетазной реакции. В процессе
трансляции используется некоторое, пока точно не установленное,
количество молекул ОТР. Вероятно, ОТР участвует в образовании
комплексов мРНК с рибосомами, в перемещении рибосомы к З'-концу
молекул мРНК.
13.8- Адресный транспорт белков
Новосинтезированные белки должны достичь своего места
предназначения в клетке. Это становится возможным благодаря тому, что
многие белки содержат на 14-конце так называемые "лидерные
последовательности", включающие 15—30 аминокислотных остатков. С по-
522
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
мощью таких сигнальных последовательностей белки доставляются к
определенному месту в клетке, где узнаются специфическими рецеп-
торными участками. После "доставки" белка "лидерные
последовательности" отщепляются под действием пептидаз.
Белки, функционирующие вне клетки (секретируемые белки),
переносятся через клеточную мембрану также с помощью сигнальных
последовательностей. Особенностью этих участков полипептидной цепи
является высокое содержание остатков гидрофобных аминокислот,
благодаря чему они легко проникают через бислойную липидную
мембрану или же встраиваются в мембрану. Транспорт осуществляется за
счет того, что формируется комплекс, включающий мембранный ре-
цепторный белок, мРНК и рибосому, благодаря чему образуется
канал в мембране, через который и проникает сигнальная
полипептидная последовательность внутрь цистерны эндоплазматического рети-
кулума, увлекая за собой экспортируемый белок. После отщепления
лидерной последовательности зрелый белок через аппарат Гольджи
уходит из клетки в виде секреторного пузырька. В транспорте белков
через мембраны могут участвовать также особые белки — порины.
13.9. Регуляция синтеза белка
Индукция и репрессия синтеза белков. Регуляция синтеза
белков находится под контролем генетической системы клетки и
осуществляется с помощью механизмов, известных под названием
индукции и репрессии. Эти механизмы реализуются двумя путями. Один из
них представляет собой регуляцию на уровне транскрипции (т.е. она
направлена на стадию образования мРНК). Другой путь регуляции —
это регуляция на уровне трансляции (на стадии синтеза белка).
Впервые модель индукции-репрессии была предложена Ф. Жакобом и
Ж. Моно в 1961 г. Основная идея этой модели была сформулирована
учеными при объяснении способности Е. сой использовать лактозу
((3-галактозид). Авторы установили, что при наличии в питательной
среде в качестве единственного источника углерода лактозы или
какого-либо другого (3-галактозида Е. соИ синтезирует $~галактозидазу,
гидролизующую лактозу на глюкозу и галактозу. При выращивании
на лактозе клетка Е. соИ содержит несколько тысяч молекул Р-галак-
тозидазы. Если же выращивать Е. сой на глюкозе, то синтез р-галак-
тозидазы практически прекращается. Одновременно и согласованно с
синтезом р-галактозидазы синтезируются еще два белка — галакто-
зид-пермеаза и галактозид-трансацетилаза, Пермеаза необходима для
переноса лактозы через клеточную мембрану, роль трансацетилазы
пока не установлена.
Такая стимуляция синтеза либо одного белка, либо группы
белков, вызываемая прибавлением к среде специфического вещества,
получила название индукции; соответственно синтезируемые белки, в
523
Часть II. Метаболизм веществ и энергии в клетке
данном случае р-галактозидаза, пермеаза и трансацетилаза, —
индуцируемыми белками, а вещество, стимулирующее синтез белков
(лактоза), — индуктором.
В отличие от индуцируемых белков, в клетке синтезируются и
другие белки без внесения в питательную среду индуктора; они получили
название конститутивных. Конститутивные белки присутствуют в
бактериальных клетках в постоянных количествах независимо от того,
есть ли в них потребность или нет. К конститутивным белкам
.относятся все ферменты, участвующие в главных путях катаболизма,
например, таких, как гликолиз.
Репрессия синтеза белка осуществляется с помощью
специфического вещества — репрессора. Каждый репрессор блокирует синтез
какого-либо одного или нескольких определенных белков, Репрессоры
имеют белковую природу. Их структуру определяет ген-регулятор,
который может существовать автономно от регулирующей системы. В
молекуле репрессора находятся два центра связывания; один — с
индуктором, другой — со специфическим участком ДНК. Связываясь с
определенными участками молекулы ДНК, репрессор блокирует
инициацию транскрипции мРНК. Специфический участок ДНК, с
которым связывается репрессор, называется оператором. Однако
репрессоры не всегда способны связываться с оператором и блокировать
синтез мРНК. Они могут существовать как в активной, так и в
неактивной формах. Если репрессор взаимодействует с индуктором, он инак-
тивируется и не способен взаимодействовать с оператором. В
отсутствие индуктора репрессор прочно связывается с оператором.
Ген, кодирующий синтез белка, называется структурным геном.
В некоторых случаях репрессор может контролировать действие
только одного гена, однако один и тот же репрессор может воздействовать
и на несколько генов. Так, в уже отмеченном нами выше синтезе
р-галактозидазы, пермеазы и трансацетилазы репрессор
контролирует транскрипцию трех генов.
Поскольку все три белка необходимы для утилизации лактозы и
накапливаются они в клетках Е. соИ в строго определенных
соотношениях, их синтез осуществляется координироеанно благодаря тому, что
три гена расположены рядом, вследствие чего структурная
информация для трех ферментов транскрибируется в виде одной молекулы
мРНК. Участок молекулы ДНК, кодирующий одну молекулу мРНК и
содержащий один контролирующий оператор, представляет собой
структурную единицу, называемую опероном. В опероны могут
входить один, два или несколько структурных генов. В этой структурной
единице оператор очень близко расположен к месту, где начинается
транскрипция мРНК; он тесно сцеплен со структурными генами.
В состав оперона входит также определенная последовательность
нуклеотидов, примыкающая непосредственно к оператору; она
называется промотором. В совокупности с репрессором оператор препят-
524
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
ствует процессу синтеза мРНК. Промотор выполняет функцию
инициации этого синтеза, является местом связывания РНК-полимеразы.
Блокирование оператора репрессором препятствует связыванию РНК-
полимеразы с промотором, В опытах по утилизации лактозы
клетками Е. соЫ установлено, что лактозныи промотор слегка перекрывается
оператором и поэтому репрессор при связывании с оператором
блокирует промотор и препятствует его связыванию с РНК-полимеразой.
Механизм регуляции экспрессии генов. На основании своих
исследований по утилизации лактозы клетками Е. сой Жакоб и Моно
предложили модель индуцируемого синтеза ферментов, объясняющую
механизм экспрессии генов (рис. 13.15). Индуцируемый синтез р-га-
лактозидазы, пермеазы и трансацетилазы осуществляет лактозныи
оперон (/ас-оперон). Автономно от /ас-оперона находится регулятор-
ный ген и В самом /ас-опероне находятся структурные гены г, у, а,
последовательность нуклеотидов которых кодирует эти три белка.
Непосредственно со структурными генами сцеплен оператор о вместе
с промоторным участком р. Промотор обеспечивает "вход"
РНК-полимеразы в &2С-оперон. Если в питательной среде вместо лактозы
находится глюкоза, регуляторный ген г обеспечивает с помощью
специфической мРНК (1-мРНК) синтез белка-репрессора. Этот белок имеет два
центра связывания: один — для оператора, другой — для индуктора.
Активная форма репрессора присоединяется к оператору, препятствуя
тем самым связыванию РНК-полимеразы с промоторным участком и
Направление тралскрипции -■—■■■■ |»
Регуляторный Участки
ген контроля
Структурные гены
а
1ША^^^
ДНК
I
РНК-
полимераза
\
{
{
мРНКдля
репрессора \у \/ \у
[ААЛ|АЛЛ!ЛАА^
Полигенная
мРНК
Рибосомы
Велок-
репрессор
(активный)
Индуктор
I
Рибосомы
{
\
\
Кодируемые
белки
+
р-Галактозидаза Пермеаза Трансацетилаза
Индуктор-репрессорный
комплекс (неактивный)
Рис. 13.15. Модель индуцированного синтеза ферментов, объясняющая меха
низм экспрессии генов
525
Часть II. Метаболизм веществ и энергии в клетке
транскрипции структурных генов г, у, а. В этих условиях три белка
&гс-оперон не транскрибирует*
Если глюкоза будет заменена индуктором лактозой, индуктор
соединяется с репрессором и инактивирует его. Комплекс индуктор—
репрессор не способен связываться с оператором, остается свободным
и промоторный участок. РНК-полимераза связывается с ним, прохо-
дит через оператор, транскрибирует гены г, у, а с образованием
полигенной мРНК, которая кодирует синтез Р-галактозидазы, пермеазы и
трансацетилазы.
В регуляции экспрессии генов может также принимать участие
особый белок — так называемый катаболитный генактивирующий
белок. Данный белок часто называют сокращенно САР (в соответствии
с аббревиатурой от англ, саЫЪоШе §епе асИиаНоп рШегп). Он
способен специфически взаимодействовать с цикло-АМР (сАМР), который
может образовываться из АТР под действием фермента
аденилатциклазы (рис. 13.16). Комплекс САР—сАМР связывается со
специфическим САР-участком промотора и тем самым обеспечивает доступ РНК-по-
лимеразе в участок первоначального связывания в области промотора.
Благодаря этому при наличии комплекса САР—сАМР может
начаться процесс транскрипции структурных генов. Так, если клетки Е. сой
находятся в среде, содержащей в достаточном количестве глюкозу, а
также лактозу, то /ас-белки не синтезируются и клетки используют
только глюкозу. При истощении запасов глюкозы в клетках бактерий
резко увеличивается количество сАМР. Последний часто называют
"сигналом голода". В данных условиях легко образуется комплекс САР—
сАМР, и становится возможной транскрипция /ас-генов и синтез /ас-
белков, необходимых для утилизации лактозы.
к<?
ЫН,
I •
с
н
I О-Р
! &
I
о
I
о
I
I?
о-р-о-сн*
I I ^О.
о
I/
•и
%
^м/С^ь/
сн
н
I I
он он
АТР
Аден и латциклаза
РР;
• *Н2
НС
Чк/'
5'
о—сн,
*м
%
>/
сн
17°
&#
13' I
О-Р О ОН
I
о
3', 5 '-Ци кл ическ ий
адеиозинмонофосфат (сАМР)
Рис. 13.16. Образование 3',5'-циклического аденозинмонофосфата (сАМР) из
АТР под действием аденилатциклазы. При отщеплении пирофосфата от АТР
образуются сложноэфирные связи между а-фосфатной группой и 3'- и 5'-гид-
роксильными группами рибозы и происходит замыкание кольца
526
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
Таким образом, транскрипция ^ас-генов возможна только в
присутствии лактозы; &с-оперон находится как под позитивным
контролем, осуществляемым через промотор, так и под негативным
контролем с участием оператора.
Рассмотренный выше механизм действия /ас-оперона в
дальнейшем удалось подтвердить и уточнить в целом ряде других
исследований на других оперонах. Если действительно этот механизм
приложим для всех случаев, то можно прийти к заключению, что в любой
живой клетке находятся сотни оперонов, но они не активны до тех
пйр, пока находятся под воздействием своих репрессоров. Появившись в
клетке, индуктор инактивирует репрессор и становится возможным
синтез белков. Однако достаточно обоснованные экспериментальные
доказательства пока немногочисленны и касаются в основном прокариот.
У некоторых прокариот имеются опероны, механизм регуляции в
которых отличается от механизма регуляции 2ас-оперона. Белок-реп-
рессор у них первоначально синтезируется в неактивной форме; опе-
рон экспрессируется в отсутствие индуктора. Активация репрессора
осуществляется специфическим веществом — корепрессором.
Комплекс репрессор—корепрессор ингибирует экспрессию оперона.
Примером такого механизма регуляции является репрессия гистидинового
оперона (/и$-оперона), который кодирует синтез 10 разных
ферментов, участвующих в синтезе аминокислоты гистидина, причем сам
гистидин выполняет роль корепрессора.
Регуляция синтеза белка у эукариот недостаточно изучена. Их
хромосомы гораздо сложнее, чем у бактерий; к тому же хромосомы
локализованы в ядре, окруженном мембраной. Это существенно
осложняет процесс передачи генетической информации с хромосом в
цитоплазму, где происходит синтез белка. У эукариот не найдены регуля-
торные белки, обладающие функцией репрессоров.
Синтез конститутивных белков. Почти во всех живых клетках
присутствуют наборы основных белков, необходимых для реализации
главных путей метаболизма. К ним следует отнести и конститутивные
ферменты, количество которых не зависит от окружающей среды.
Например, в клетке Е. соЦ количество ферментов, участвующих в
гликолизе, остается неизменным, если в среду вносят вместо глюкозы
лактозу или какой-либо другой источник углерода и энергии; иными
словами, эти ферменты являются конститутивными, скорость синтеза
которых не регулируется ни индукторами, ни корепрессорами. Пока
для этих ферментов не известны специфические регуляторные
механизмы их синтеза.
В общих чертах интенсивность конститутивного синтеза зависит
от скорости транскрибирования структурных генов, скорости синтеза
белка в стадиях инициации и элонгации, от продолжительности
жизни мРНК, т.е. контроль осуществляется на уровне трансляции.
Механизм регуляции на этом уровне имеет, вероятно, для бактерий второ-
527
Часть II. Метаболизм веществ и энергии в клетке
степенное значение, но он очень важен для эукариот. Однако мало
известно о факторах, влияющих на кинетику процесса трансляции.
Регуляция синтеза белка представляет собой отражение принципа
клеточной экономии, поэтому она имеет большое биологическое
значение. В результате действия регуляторных систем в любой клетке
присутствует необходимое количество каждого белка, позволяющее
ей осуществить метаболические процессы плавно и с максимальной
эффективностью. Благодаря регуляции синтеза ферментов в клетке
создается их рациональное соотношение, обеспечивающее нормальное
течение основных биохимических процессов. Индуцированный синтез
ферментов микроорганизмами не только является экономически
целесообразным процессом, но и носит приспособительный характер для
клетки с окружающей ее средой. Индуцированные ферменты
синтезируются лишь в том случае, когда они необходимы для утилизации
питательных веществ среды.
Регуляция экспрессии генома и процесс старения. В настоящее
время существует точка зрения, что старение организма представляет
собой не результат поломки живой системы, а особую биологическую
функцию. Как это не парадоксально, но смерть определенной части
клеток является существенной частью роста и развития многих эука-
риотных организмов, включая и растения, и животных. Клеточная
смерть может быть компонентом ответа на биотический и
абиотический стрессы. Поскольку организм контролирует инициацию и
осуществление процесса клеточной смерти, то этот процесс получил
название запрограммированная смерть клетки.
Доказательством запрограммированной смерти на клеточном
уровне является так называемый "лимит Хейфлика". Л. Хейфлик
показал, что естественная продолжительность жизни человека
обусловлена числом митозов (делений) клеток, иными словами, числом
репликаций ДНК. Было выявлено, что клоны клеток, выделенных из
эмбрионов человека, совершают около 50 удвоений популяции. У
диплоидных клеток человека число удвоений популяций обратно
пропорционально возрасту донора. Эти данные навели на мысль, что
ограниченная способность к удвоениям нормальных клеток в культуре
является выражением старения на клеточном уровне.
Другим доказательством запрограммированной клеточной смерти
является апоптоз (см. гл. 9). В клетках, зараженных вирусом,
включается каскад событий, сопровождающихся деградацией
биополимеров и приводящих в конечном итоге к гибели таких клеток.
Интересно, что с помощью подобных механизмов организм может
освобождаться от вредных или ненужных клеток и даже органов. В качестве
примера можно привести исчезновение хвоста у головастика.
Существует предположение, что с помощью апоптоза могут выбраковываться
клетки, которые представляют опасность для жизни или нормального
функционирования организма. Согласно точке зрения В. П. Скулаче-
528
Г лава 13, Синтез ДНК, РНК и белков. Перенос генетической информации
ва, старение есть не что иное, как запрограммированная смерть на
надклеточном уровне, т.е. на уровне организма; это явление получило
название "феноптоз".
Существенным шагом в понимании причин лимитированной
способности большинства нормальных соматических клеток делиться 1п
1)и)0 и 1п г>Иго явились наблюдения укорочения теломер при делении и
открытие фермента теломеразы. Теломеры — это структуры,
расположенные на концах линейных хромосом и состоящие из
повторяющихся последовательностей ДНК. В связи с особенностями
функционирования ДНК-полимераза не может реплицировать полностью линейные
концы ДНК; несколько нуклеотидов на З'-конце матрицы ДНК
остаются недореплицированными. Как рассматривалось в разделе 13.1,
ДНК-полимеразы ведут синтез в направлении 5'—>3'; при этом они
нуждаются в одноцепочечной ДНК-матрице и З'-конце затравки,
образующейся под действием праймазы. Удаление РНК-затравок после
завершения синтеза фрагментов Оказаки и заделывание брешей дезоксири-
бонуклеотидами приводит к тому, что дочерние цепи ДНК
оказываются короче материнских на размер первого РНК-праймера,
включающего 10—20 нуклеотидов. Образуются так называемые З'-оверхенги, т.е.
выступающие З'-концы материнских цепей. "Проблема концевой недо-
репликации" была сформулирована в 1971 г. А. М. Оловниковым,
который предположил, что старение клетки может быть связано с потерей
повторяющихся последовательностей ДНК на концах хромосом из-за их
концевой недорешгакации* Им была также высказана идея о
существовании особого механизма, предотвращающего этот эффект.
Действительно, впоследствии в ряде типов клеток (половых,
раковых, а также клетках организмов, размножающихся вегетативным
путем) был открыт фермент, обеспечивающий компенсацию
укорочения ДНК. Этот фермент был назван тпеломеразой. Данный фермент
наращивает на концах ядерной ДНК многократно повторяемый гекса-
нуклеотид (у человека это ТТАООО), что и приводит к образованию
теломер. Помимо белковой части теломераза содержит в своем составе
РНК, которая используется в качестве матрицы при наращивании
повторяющихся последовательностей. Это обстоятельство позволяет
отнести теломеразу к своеобразной обратной транскриптазе.
Сформировавшиеся при "концевой недорепликации" З'-оверхенги узнаются
теломеразой, использующей их З'-концы в качестве затравок.
Образующиеся длинные (у человека включающие сотни повторов) одноцепо-
чечные концы в свою очередь служат матрицами для синтеза
дочерних цепей с помощью обычного репликативного механизма.
Благодаря этому при последующем укорочении ДНК сокращается только этот
нетранскрибируемый текст теломерного участка хромосомы;
наследственная информация не утрачивается и механизм экспрессии не
нарушается. В период раннего эмбриогенеза происходит выключение гена,
кодирующего теломеразу в большинстве соматических клеток челове-
34. Заказ 3486
529
Часть II. Метаболизм веществ и энергии в клетке
ка. Вместе с этим упраздняется защитный механизм,
предотвращающий "ук°РОчение" теломер. Это приводит к ухудшению
функционирования хромосом, а с исчезновением всей теломеры, обеспечивающей
защиту от потери генетического материала при репликации,
начинается деградация смысловых участков ДНК. Показано, что существует
корреляция между укорочением теломер и "лимитом Хейфлика".
Измененная ДНК в случае неспособности к нормальному
функционированию ведет к гибели клетки. Такая клетка подвергается апопто-
зу и разрушается. Много исследований проведено по
пострадиационному апоптозу. В ответ на воздействие радиации в клетках
развивается определенная последовательность биохимических и
морфологических изменений, завершающихся ферментативной деградацией ДНК и
распадом ядра. Совершенно очевидно, что высокие дозы радиации
вызывают быструю гибедь клеток путем некроза; под влиянием
малых доз радиации, когда изменения в цитоплазме менее
существенны, на первый план выступает апоптическая гибель. Ключевую роль в
процессах апоптоза играет регуляция синтеза определенных белков,
обеспечиваемого специфическими генами. Одни из них важны для
программируемой гибели клеток организма, другие - для окончательного
удаления мертвых клеток. Специфические гены участвуют в синтезе
активаторов и ингибиторов апоптоза, к которым относится обширная
группа белков - ферментов, гормонов. Так, в деградации ядра клеток
важную роль играют протеазы, называемые каспазами. Они
участвуют в запуске и развитии процессов апоптоза. Каспазы расщепляют
находящиеся в комплексе с ДНК гистоны. Последующая деградация
ДНК происходит под действием эндонуклеаз. Для того чтобы
запустить процесс апоптоза, клетка должна осуществить синтез этих
ферментов, в ней должны быть активированы специфические гены,
ответственные за их транскрипцию и трансляцию.
Вероятность повреждения ДНК и апоптической гибели клетки
увеличивается с интенсификацией образования активных форм
кислорода (см. гл. 9). Характерно, что с возрастом происходит
интенсификация продуцирования токсичных форм кислорода. Это отражается на
состоянии белков, являющихся мишенью их воздействия. Таким
образом, наряду с синтезом дефектных белков, обусловленным
накоплением ошибок в процессах транскрипции и трансляции, увеличивается
и степень их окислительной денатурации. При этом механизмы
репарации не могут справиться со все возрастающим количеством
дефектов, процесс апоптоза становится более интенсивным.
Следует отметить, что в отличие от нормальных смертных клеток
линии аномальных бессмертных клеток не стареют и синтезируют те-
ломеразу. Теломеры этих так называемых иммортализованных
клеток не укорачиваются при последовательных пассажах гп г)Иго.
Экспрессия теломераз была обнаружена в некоторых типах нормальных
клеток, например, в стволовых клетках костного мозга, тканях пло-
530
Глава 13. Синтез ДНК, РНК и белков. Перекос генетической информации
да, лимфоцитах периферической крови. Характерно, что все эти
клетки имеют высокие темпы обновления или же находятся в постоянно
размножающемся фонде дифференцирующихся клеток. Однако тело-
меразная активность в этих нормальных клетках значительно ниже,
чем в популяциях раковых клеток. Поскольку уровень экспрессии
теломеразы на сегодняшний день представляется наиболее
специфической характеристикой, отличающей раковые клетки от нормальных,
то данный факт инициировал поиск ингибиторов теломеразы с целью
разработки методов и подходов в лечении раковых заболеваний.
13.10. Ингибиторы транскрипции и трансляции
и их биомедицинское значение
Ингибиторы белкового синтеза служат полезными инструментами для
изучения особенностей пластического обмена в живых организмах.
Процесс транскрипции ингибируется токсичным антибиотиком ак-
тиномицином В как у эукариот, так и у прокариот. Этот антибиотик
оказывает тормозящее действие на ДНК-зависимую РНК-полимеразу, так
как плоская часть его молекулы способна внедряться в двойную спираль
ДНК между соседними парами О и С, что приводит к локальной
деформации матрицы и невозможности движения полимеразы вдоль нее. Ак-
тиномицин О обладает противоопухолевым эффектом, однако он
применяется крайне редко ввиду высокой токсичности. ,
Токсическое действие ос-аманитина, синтезируемого ядовитым
грибом АтапИа рНаНоШез, состоит в ингибирующем эффекте на процесс
транскрипции в клетках животных. Тем не менее это вещество не
влияет на синтез РНК у прокариот.
Антибиотик рифампицин также является ингибитором синтеза РНК.
Его эффект достигается за счет связывания с РНК-полимеразой и
торможения ее функционирования. Рифампицин оказывает сильное ингиби-
рующее действие на РНК-полимеразу прокариот, однако у эукариот этот
антибиотик практически не тормозит синтез РНК* В этой связи
рифампицин применяется для лечения туберкулеза. Известно также
противовирусное действие рифампицина. В частности, он используется для
лечения трахомы, относящейся к ДНК-содержащим вирусам.
К ингибиторам белкового синтеза на уровне трансляции относится
пуромицин, который синтезируется грибом 81гер1отусез а1Ьоп1§ег. Суть
механизма действия пуромицина состоит в том, что он, являясь
структурным аналогом З'-концевого участка аминоацил-тРНК, замещает ее
собой в А-участке рибосомы. Происходит образование пептидилпуро-
мицина, к которому не способна присоединиться ни одна
аминокислота. Синтез полипептида прекращается.
Другая группа антибиотиков ™ тетрациклины также тормозит
белковый синтез путем блокирования А-участка рибосомы.
Тетрациклины широко применяются в клинике, так как, ингибируя синтез белка
в 708-рибосоме, они значительно меньше влияют на 808-рибосомы.
34* 531
Часть II. Метаболизм веществ и энергии в клетке
).ц 1 ■ I ■ »т^1— ■■ I I МИН ■■■ М**1 Ч и ■■ ■ и I ■ й\ Ы*Ы "' — ■ Л' '
Существуют антибиотики, ингибирующие синтез белка в 808-рибо-
сомах, но не влияющие на 708-прокариотичеекие и митохондриальные
рибосомы* Это, например, относится к циклогексимиду, специфически
ингибирующему транслоказу. Путем подавления активности транс лока-
зы также ингибируют синтез белка в 808-рибосомах (но не в 708-рибо-
сомах) эритромицин и олеандомицин. Хлорамфеникол, наоборот,
нарушает процесс элонгации при синтезе белка в 708-рибосоме за счет инги-
бирования пептидилтрансферазы, однако на 808-рибосомы он не действует.
Этот антибиотик используется при лечении тифозных инфекций.
Такие антибактериальные и противотуберкулезные антибиотики,
как неомицин и стрептомицин, действуют на белоксинтезирующую
систему бактерий, вызывая ошибки при считывании генетической
информации. Это приводит к синтезу аномальных белков в
бактериальной клетке и снижению ее жизнеспособности.
Краткое содержание главы
Во всех организмах перенос генетической информации
осуществляется в цепи ДНК <н> РНК -> белок по типу матричного механизма.
Перенос информации с РНК на ДНК возможен благодаря обратной
транскрипции с помощью РНК-зависимой ДНК-полимеразы.
Матричный синтез нуклеиновых кислот и белка обеспечивает высокую
стабильность наследуемых признаков при их переносе от организма к
потомкам, создает строгую молекулярную экономию на всех этапах
синтеза нуклеиновых кислот и белков.
Репликация (воспроизведение) ДНК является сложным процессом,
идет по полуконсервативному пути с участием специфических
ферментов, белковых факторов, источника энергии (АТР) и включает три
ступени: образование репликативной вилки, синтез затравки из РНК
и фрагментов ДНК, удаление фрагментов РНК и "сшивание"
фрагментов ДНК с участием ДНК-полимераз и лигаз. Все ДНК-полимеразы
синтезируют ДНК в направлении 5'*-»3'. В процессе
жизнедеятельности клетки ДНК может подвергаться повреждениям (мутациям) как
спонтанно, так и под действием физических и химических факторов.
Репарация (исправление) повреждений является вторым способом после
полуконсервативной репликации, обеспечивающим сохранность
генетической информации в ДНК в неизменном состоянии. Существуют
три типа молекулярно-генетического механизма репарации: 1) с
использованием информации неповрежденной цепи ДНК; 2) с
использованием информации гомологичной полинуклеотидной цепи и 3)
репарации ДНК без использования матричной цепи ДНК.
ДНК подвергается изменениям и перестройкам, получившим
название "рекомбинация". Рекомбинация представляет собой
образование новых молекул ДНК путем разрывов и перевоссоединений
исходных молекул ДНК; в результате рекомбинации возникает новое соче-
532
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
тание генетической информации. Генетическая рекомбинация
является неотъемлемой характеристикой всех живых организмов. Она
составляет материальную основу изменения наследственных признаков.
Подробное изучение генетической рекомбинации позволило
осуществить в лабораторных условиях синтез новых молекул ДНК, в
которых гены одного организма оказываются объединенными с генами
другого. Такой синтез привел к возникновению нового научного
направления —■ генной инженерии, которая дала возможность получать
новые искусственные комбинации генов, не встречающиеся в
природе. Технология рекомбинантных ДНК включает следующие основные
этапы: выделение или получение целевого гена, получение рекомби-
нантной ДНК, введение этой ДНК в клетку-хозяина и клонирование
(размножение) гена.
Следующий этап в передаче генетической информации —
транскрипция. В этом процессе с помощью ферментной системы
происходит синтез цепи РНК на одной из нитей матричной ДНК. В результате
транскрипции образуется РНК — точная копия нити ДНК по
последовательности азотистых оснований в полинуклеотидной цепи.
Механизм синтеза РНК в основных чертах сходен с механизмом синтеза
ДНК. Однако в отличие от репликации, где копируется вся молекула
ДНК с образованием дочерних молекул, транскрипция
осуществляется избирательно лишь на отдельном участке молекулы ДНК — гене
(или группе генов). Она направляется особыми регуляторными
последовательностями нуклеотидов, указывающих начало и конец
участков гена, подлежащего транскрипции. Транскрипция катализируется
ДНК-зависимыми РНК-полимеразами; они наращивают цепь РНК в
направлении 5'—»3'. Процесс транскрипции включает четыре стадии:
связывание РНК-полимеразы с ДНК, инициацию — начало синтеза
цепи РНК, элонгацию (удлинение цепи РНК) — собственно процесс
синтеза полинуклеотидной цепи и терминацию — завершение этого
синтеза. Сошедшая с ДНК-матрицы новосинтезированная РНК
подвергается дальнейшей модификации — посттранскрипционному про-
цессингу, который заключается либо в удалении, либо в
присоединении нуклеотидов к цепи РНК, либо в метилировании оснований, а
также в удалении нитронов путем сплайсинга.
Завершающим этапом передачи генетической информации
является трансляция — синтез белка. Важную роль в этом процессе играет
генетический код, в котором в виде нуклеотидов содержится
информация об аминокислотной последовательности белка. Код определяет
первичную структуру белка. Первоначально он передается с генов на
мРНК. Местом синтеза белка являются рибосомы. Соединяясь с мРНК,
рибосомы образуют полирибосомы, на которых и протекает синтез
полипептидных цепей с помощью предварительно активированных
аминокислот. Одну аминокислоту кодирует группа нуклеотидов,
включающая три основания — триплет. Триплет получил название "ко-
533
Часть II. Метаболизм веществ и энергии в клетке
дон". Открыты 64 кодона, из которых 61 кодируют аминокислоты.
Три кодона, неспецифичных к аминокислотам, осуществляют стадию
терминации. Стадия инициации у прокариот всегда начинается с
М-формилметионина, у эукариот — с метионина.
В процессе элонгации и по ее окончании белки подвергаются
модификации; идет удаление формильной группы и метионина,
формирование третичной структуры белка, окисление 8Н-групп в 8-8-группы,
фосфорилирование ОН-групп серина, присоединение к белку остатков
Сахаров.
Транспорт белков к месту их назначения осуществляется
благодаря наличию на их концах сигнальных последовательностей,
специфически узнаваемых рецепторными зонами клетки.
В клетке существуют тонкие механизмы регуляции синтеза белка,
известные под названием индукции и репрессии синтеза. Эти механизмы
осуществляются как на уровне транскрипции, так и на уровне
трансляции. Классические работы Жакоба и Моно с Е. соИ позволили
разработать механизм регуляции на уровне транскрипции, в основу которого
положено функционирование структурной единицы — оперона. В состав
оперона входят промоторный участок, являющийся местом связывания
ДНК с РНК-полимеразой, оператор и структурные гены. Функция
оператора заключается в выключении структурных генов из процесса
синтеза мРНК. Это выключение осуществляется репрессором, блокирующим
оператор. Белок-репрессор кодируется геном-регулятором, существующим
автономно от оперона. Оперон включается в работу в том случае, когда
репрессор блокирован специфическим веществом-индуктором.
Существенную роль в регуляции экспрессии генома может играть
особый белок — катаболитный генактивирующий белок (САР),
способный к образованию активного комплекса САР—сАМР. В клетках
бактерий, например Е. со119 сАМР играет роль "сигнала голода",
образуясь при недостаточном содержании глюкозы в питательной среде.
Способность комплекса САР—сАМР связываться с определенным
участком промотора обеспечивает доступ РНК-полимеразы к
структурным /ас-генам и синтез /ас-белков, благодаря чему становится
возможной утилизация лактозы питательной среды. Белки,
синтезируемые под действием специфического индуктора, называются
индуцируемыми. В живых клетках существуют наборы белков-ферментов,
необходимых для реализации основных путей метаболизма. Эти
ферменты называются конститутивными; их количество в клетке не
зависит от окружающей среды, скорость их синтеза не регулируется на
уровне транскрипции. К конститутивным ферментам относятся, в
частности, ферменты гликолиза. Интенсивность конститутивного
синтеза зависит от скорости транскрибирования генов, скорости синтеза
белка, продолжительности жизни мРНК, т.е. контроль
осуществляется в основном на уровне трансляции. Апоптоз тесно взаимосвязан с
регуляцией синтеза специфических белков.
534
Глава 13. Синтез ДНК, РНК и белков. Перенос генетической информации
Хронология важнейших дат в исследовании синтеза
нуклеиновых кислот и белков
1941—1942 гг. — Г. Касперсон и Ж. Браше показали, что в
синтезе белка принимает участие РНК.
1944 г, — О. Эвери, К. Мак-Леод и М. Мак-Карти
экспериментально показали, что в ДНК закодирована генетическая информация.
1950 г. — М. Лясковский подразделил нуклеазы на эндо- и экзо-
нуклеазы.
1953 г. — Е. Гейл и Д. Факс получили прямое доказательство
непосредственного участия ДНК в синтезе белка,
1954 г. — Г. Гамов предположил, что для кодирования одной
аминокислоты используется сочетание трех нуклеотидов ДНК, Эта
элементарная единица наследственности получила название кодон.
1954 г. — Ф, Крик выдвинул адапторную гипотезу, согласно
которой функцию перевода языка нуклеиновой кислоты на язык белков
выполняют адапторные РНК (т-РНК).
1955 г. — А. Корнберг и его сотрудники открыли ДНК-полимеразу
(удостоен Нобелевской премии в 1959 г.).
1957 г. — Г. Шрамм и X, Френкель-Конрат выделили из вируса
табачной мозаики РНК и показали ее инфекционность.
1957 г. — М. Мезелсон и Ф. Сталь экспериментально доказали
полуконсервативный механизм репликации ДНК.
1958 г. — М. Хогланд и П. Замечник обнаружили, что для
активации каждой аминокислоты, участвующей в синтезе белка,
необходимы специфический фермент, АТР и тРНК.
1961 г. — М* Ниренберг и Г. Маттеи расшифровали структуру ко-
донов.
1961 г. ■— Ф. Жакоб и Ж. Моно предложили схему регуляции
активности генов; в 1965 г. им была присуждена Нобелевская премия*
1962 г. — Ф. Шапвиль показал, что положение аминокислоты в
синтезируемой белковой молекуле определяется специфической тРНК,
к которой аминокислота присоединяется.
1963 г. — А. Рич и Дж. Уотсон выдвинули предположение, что
синтез полипептидной цепи происходит на рибосоме, которая
считывает информацию с мРНК. Открыли полисомы.
1964 г. — М. Ниренберг установил, что тринуклеиды (кодоны)
способствуют связыванию определенных молекул т-РНК,
1964 г. — Г. Корана синтезировал полирибонуклеотиды с
определенной последовательностью нуклеотидов.
1966 г. — В. Л. Карриер и Р. Бсетлоу показали, что повреждения
в молекуле ДНК под действием УФ-лучей могут быть исправлены в
результате каталитического действия нуклеаз.
1967 г. — С. Бреннер установил, что окончание синтеза белка (тер-
минация) зашифровано в кодонах м-РНК (ПАА, ИАСг и 1ТОА).
1968 г. — Г. Корана и М. Ниренберг удостоены Нобелевской
премии за исследование генетического кода.
535
Часть II. Метаболизм веществ и энергии в клетке
1969 г. — Р. Мерифилд впервые осуществил синтез белка
(панкреатической рибонуклеазы) в лабораторных условиях.
ЛИТЕРАТУРА
Албертс В., Брей Д., Рэфф М„ Роберте К., Уопгсон Дж. Молекулярная
биология клетки: В 3 т. — М.: Мир, 1994 — Т.1. — 515 с,
Ашмарин И. П. Молекулярная биология. — Л,; Изд-во ЛГУ, 1977. — 373 с.
Березов Т. Т., Коровкин Б. Ф. Биологическая химия: Учебник, — 3-е изд.,
перераб. и доп. — М.: Медицина, 1998. — 704 с.
Бохински Р. Современные воззрения в биохимии. — М.: Мир, 1987. — 544 с,
Дэвидсон Дж. Биохимия нуклеиновых кислот. — М.: Мир, 1976. — 341 с.
Кнорре Д. Г., Мызина С. Д. Биологическая химия: Учебник для хим., биол. и
мед. спец. вузов. — 3-е изд., испр. — М.: Высш. шк., 2000. — 479 с.
Ленинджер А Основы биохимии: В 3 т. — М.: Мир, 1985. — Т. 3. — С.745—
1056.
Молекулярная биология: Структура и биосинтез нуклеиновых кислот:
Учебник для биол. спец. вузов / В. Н. Агол, А. А. Богданов, В. А. Гвоздев и др.; Под
ред. А. С* Спирина. — М.: Высш. шк., 1990. — 352 с.
Спирин А С. Молекулярная биология: Структура рибосомы и биосинтез
белка, _ м.: Высш. шк., 1986. — 300 с.
Спирин А С. Принципы функционирования рибосом // Соросовский
образовательный журнал. — 1999. — № 4. — С. 65—70.
Спирин А С. Биосинтез белка: инициация трансляции // Соросовский
образовательный журнал. — 1999. — № 5. — С. 2—7.
Спирин А С. Биосинтез белка: элонгация полипептида и терминация
трансляции // Соросовский образовательный журнал. — 1999. — № 6. — С. 2—7.
Спирин А С. Принципы структуры рибосом // Соросовский образовательный
журнал. — 1999. — № 11. — С. 65—70. '
Страйер Л. Биохимия: В 3 т. — М.: Мир, 1985. — Т. 3. — 396 с.
Эллиот В., Эллиот Д. Биохимия и молекулярная биология / Под ред. А. И. Ар-
чакова, М. П. Кипичникова, А, И, Медведева, В. П. Скулачева. — М.: Изд-во
НИИ Биомедицинской химии РАН, 1999. — 372 с.
ВОПРОСЫ И ЗАДАЧИ
1. Репликация. Напишите нуклеотидную последовательность участка ДНК,
синтезируемого ДНК-полимеразой, если матрицей для него была
последовательность (5')-АТС|ААА)ССТ|ССА|ТТТ-(3/).
2. Транскрипция. Напишите нуклеотидную последовательность РНК,
синтезируемой ДНК-зависимой РНК-полимеразой, если матрицей для нее является
синтезированный участок, полученный в п. 1.
3. Трансляция. Напишите аминокислотную последовательность пептида, если
в качестве матрицы для его синтеза будет РНК, полученная вп.2.
4. Какие последствия вызывает точечная мутация ДНК: а) делеция; внесение
нуклеотида в молекулу ДНК; б) делеция и внесение нуклеотида на одном из
локальных участков ДНК.
5. Какова роль рекомбинации ДНК в эволюции живой материи?
Перечислите стадии технологии получения рекомбинантных ДНК.
6. Какой результат был бы получен при полуконсервативной репликации
ДНК, если исходная ДНК была тяжелой (15М-ДНК):
а) Е. сой в 1-й генерации развивалась на легкой среде (14Ы-среде);
б) во 2-й — на тяжелой среде (15Ы-среде).
Глава 14. МОЛЕКУЛЯРНЫЕ ОСНОВЫ ДЕЙСТВИЯ
КЛЕТОЧНЫХ МЕДИАТОРОВ
14Л- Передача сигналов через клеточные мембраны с помощью
специфических рецепторов и эффекторных систем
Клетки всех организмов должны быть способны реагировать на
сигналы, поступающие из окружающей среды. Ответные реакции
клеток различаются в зависимости от поступающих сигналов и могут
оказывать воздействие на метаболизм, секрецию веществ,
дифференциацию, клеточный рост и деление. Многие молекулы, передающие
сигналы, такие, например, как гормоны, не проходят через
плазматическую мембрану, а взаимодействуют со специфическими белковыми
рецепторами плазматической мембраны клетки - мишени. Это
взаимодействие представляет собой первый этап в механизме сигнальной
трансдукции, которая обеспечивает превращение инициирующего
внешнего сигнала во внутриклеточный сигнал, в конечном счете
приводящий к соответствующему ответу клетки.
Принципиальная схема сигнальной трансдукции представлена на
рис. 14.1. После связывания первичного медиатора, называемого
также иногда мессенджером, например, гормона или ростового фактора,
сигнал передается через мембранный белок — трансдуктор
(внутриклеточный передатчик)> мембраносвязанному эффекторному
ферменту. Действие эффекторного фермента приводит к образованию вто-
Внешний сигнал
Первичный медиатор
А
Мембранный
рецептор
;;-*,
Переносчик
Эффек-
торный
фермент
■ .*■'■>■:■*■■*■ ■ ^ /
ШъШ Плазматическая
< * * * * * * ■* ■ *•'
ШШ& мембрана
4
Вторичный медиатор
Цитозольяхае или
ядерные эффекторы
Клеточный ответ
Рис. 14.1. Сигнальная трансдукция через плазматическую мембрану клетки
537
Часть П. Метаболизм веществ и энергии в клетке
ричного медиатора, который обычно представляет собой небольшую
молекулу или ион. Вторичный медиатор ответствен за перенос
сигнала к конечному пункту назначения, которым могут быть ядро,
внутриклеточный компартмент или цитозоль. Существует огромное
разнообразие медиаторов, мембранных рецепторов и трансдукторов, но
известно всего несколько эффекторных ферментов и вторичных
медиаторов. Таким образом, клетки обладают эксклюзивными
специфичными системами для восприятия внешних сигналов на их
поверхности, но используют общие пути для передачи этих сигналов внутри
клетки. Основными сигнальными путями у животных являются
системы, включающие аденилатциклазу, инозитолфосфолипиды и рецеп-
торные тирозинкиназы.
Стероидные гормоны, которые не связываются с рецепторами
плазматической мембраны, представляют собой исключение из общей схемы
сигнальной трансдукции, показанной на рис. 14.1. Поскольку стероиды
гидрофобны, то они способны диффундировать через плазматическую
мембрану в клетку, где они связываются с рецепторными белками в
цитоплазме. Стероидрецепторные комплексы затем переносятся в ядро,
Тироидные гормоны (тироксин и трийодтиронин), а также витамин О
тоже действуют непосредственно на уровне ядра. Гормонрецепторные
комплексы связываются со специфическими последовательностями ДНК,
называемыми элементами гормонального ответа, и посредством этого
усиливают или подавляют экспрессию регулируемых генов.
Следует отметить, что общей нитью, пронизывающей все процессы
при передаче сигнала, является образование вторичных медиаторов,
приводящее с почти неизменным результатом к прямой или
опосредованной активации протеинкиназ. Это достаточно большое и разнообразное
семейство ферментов, катализирующее процессы фосфорилирования
различных белковых субстратов в мембране, цитозоле или ядре. Хотя
известны свойства только нескольких таких белков, многие из них, как
оказалось, играют критическую роль в регуляции метаболизма и контроле
клеточного роста и деления. В некоторых случаях белки путем
фосфорилирования активируются; в других случаях они инактивируются.
Протеинкиназы делятся на две группы. Первая группа — серин-трео-
ниновые киназы — катализирует фосфорилирование гидроксильной
группы специфического серинового или треонинового остатка в белках.
Вторая группа — тирозиновые протеинкиназы — специфична по
отношению к гидроксильной группе тирозиновых остатков. Но обе эти группы
используют АТР в качестве донора фосфатной группы.
14.2. Активация аденилатциклазного сигнального пути
с помощью гормонов
Многие гормоны, регулирующие внутриклеточный метаболизм,
оказывают свое воздействие на клетки-мишени, активируя аденилат-
циклазную сигнальную систему. Например, адреналин играет цент-
538
Глава 14. Молекулярные основы действия клеточных медиаторов
ральную регуляторную роль во многих метаболических путях,
включая метаболизм гликогена. Адреналин выделяется из мозгового слоя
надпочечников и действует на все клетки, плазматическая мембрана
которых содержит р-адренорецептор. Когда адреналин связывается,
конформация рецептора изменяется, способствуя взаимодействию
между рецептором и специфическим белком - трансдуктором, а именно,
белком С, локализованным на цитозольной стороне плазматической
мембраны. Комплекс "рецептор — адреналин" активирует О-белок,
который способен обратимо связываться и активировать эффекторный
фермент аденилатциклазу.
Аденилатциклаза представляет собой интегральный мембранный
фермент, активный центр которого обращен к цитозолю. Он
катализирует образование вторичного медиатора — сАМР (см. гл. 13) из АТР.
Затем сАМР диффундирует с поверхности мембраны в цитозоль и
активирует сАМР-зависимый фермент протеинкиназу А, Эта киназа
состоит из двух регуляторных субъединиц и
двух каталитических субъединиц и
неактивна, когда все субъединицы объединены.
Когда концентрация цитозольного сАМР
возрастает в результате сигнальной трансдукции
через аденилатциклазу, четыре молекулы
сАМР связываются с регуляторными
субъединицами протеинкиназы А, что приводит
к освобождению каталитических субъединиц,
которые обладают ферментативной
активностью (рис. 14.2). Протеинкиназа А,
относящаяся к серин-протеиновой группе протеин-
киназ, может вносить глубокие изменения в
клеточный метаболизм, катализируя фосфо-
рилирование ряда ферментов-мишеней.
Фосфорилирование аминокислотных
остатков в полипептидных цепях
ферментов-мишеней имеет обратимый характер благодаря
действию протеинфосфатазы,
катализирующей гидролитическое отщепление фосфатной
группы. Гормончувствительная липаза
представляет собой хорошо изученный пример
фермента, активность которого регулируется
путем фосфорилирования-дефосфорилирова-
ния (рис. 14.3). Липаза активируется в ответ
на гормональные сигналы; активированный
фермент превращает триацилглицеролы в
жирные кислоты, которые могут быть
окислены с образованием энергии. Многие другие
ферменты, играющие ключевую роль в мета-
539
"I
-I
,1
Неактивный комплекс
Активные каталитические
субъединицы
Рис. 14.2. Активация
протеинкиназы А сАМР.
Комплекс из 4 субъединиц
(2 каталитических (С) и
2 регуляторных (К)
субъединиц) неактивен. Когда
4 молекулы сАМР
связываются с димером,
состоящим из регуляторных
субъединиц,
каталитические субъединицы
высвобождаются
Часть II. Метаболизм веществ и энергии в клетке
Ра Н20
Рис, 14.3. Регуляция активности липазы путем фосфорилирования й дефос-
фори л ирован ия
болизме, также регулируются путем фосфорилирования с помощью
протеинкиназы А.
Важнейшая особенность аденилатциклазного пути (и многих
других сигнальных путей) — это усиление сигнала. Единственный гор-
монрецепторный комплекс способен взаимодействовать и
активировать многие С-белки. Единственная активированная молекула
фермента аденилатциклазы обеспечивает синтез многих молекул
вторичного медиатора — с АМР. В дальнейшем потоке сигнального пути
единственная молекула протеинкиназы А может фосфорилировать многие
белки-мишени. Эта серия процессов усиления действует как
каскадный механизм.
Возможность выключения пути сигнальной трансдукции
представляет собой существенный элемент регуляции сигнальных процессов.
Например, концентрация сАМР в цитозоле возрастает только в
определенный момент регуляции. Затем растворимая фосфодиэстераза
катализирует гидролиз сАМР до АМР, таким образом лимитируя
эффект вторичного медиатора. Кофеин и теофиллин, относящиеся ко
вторичным метаболитам растительного происхождения (см, разд. 6), при
высоких концентрациях ингибируют сАМР-фосфодиэстеразу,
уменьшая скорость превращения сАМР до АМР. Эффекты сАМР и
активирующее действие стимулирующего гормона пролонгируются и
интенсифицируются под действием этих ингибиторов.
14.3. Роль О-белков как сигнальных трансдукторов
Семейство внутриклеточных гуаниннуклеотидсвязывающих белков,
или О-белков, служит в качестве передатчиков сигнала между
гормональным рецептором плазматической мембраны и эффекторными
ферментами. О-белки связывают гуаниновые нуклеотиды (ОТР или ОБР)
и обладают ОТРазной активностью; поэтому они способны медленно
катализировать гидролиз связанного ОТР до ОВР. О-белки являются
периферическими мембранными белками, расположенными на внут-
540
Глава 14. Молекулярные основы действия клеточных медиаторов
ренней поверхности плазматической мембраны. Существует два
класса О-белков: гетеротримерные и мономерные. Только класс гетеротри-
мерных белков вовлечен в сигнальные процессы через гормональные
рецепторы. О-белки функционируют как молекулы, способные как
бы переключаться и существовать в двух состояниях: ОВР-связанная
форма (неактивная) и ОТР-связанная форма (активная). Циклический
процесс активации и
дезактивации О-белков
иллюстрируется рис. 14.4. Гетеротримерные
О-белки состоят из а-, (3-й у-
субъединиц. Оа, обычно
содержит ОВР, связанный с а-
субъединицей; этот комплекс
неактивен. Когда гормонрецеп-
торный комплекс
взаимодействует с ОаРу, О-белок
претерпевает конформационные
изменения, превращаясь в
активную форму. Связанный
СВР заменяется на ОТР,
вызывая диссоциацию Оа-ОТР из
Оа.. Оа-ОТР затем
взаимодействует в мембране с аденилат-
циклазой, значительно
увеличивая образование сАМР.
ОТРазная активность
О-белков обеспечивает их
функционирование по принципу
"сделал — отключайся", что связано с медленным гидролизом ОТР до
ООР, Гидролиз дезактивирует О-белок и позволяет О-ОВР объединить-
ся с О^, прекращая действие С-белка на аденилатциклазу.
Многие гормоны действуют, используя сАМР-сигнальный путь.
Гормоны, связываясь со стимулирующими рецепторами, активируют
аденилатциклазу и увеличивают внутриклеточный уровень сАМР; тогда
как, связываясь с ингибиторными рецепторами, тормозят аденилат-
циклазную активность. Различают два типа рецепторов по их
способности связываться с С-белками. Стимулирующие О-белки (Оа)
активируют аденилатциклазу, а ингибирующие О-белки (О.) подавляют
фермент. Соматостатин -— гипоталамический полипептидный гормон,
который связывается с рецепторами, сопряженными с Ог Действие со-
матостатина снижает уровень сАМР в клетке и подавляет фосфорили-
рование белков протеинкиназой А. Когда адреналин связывается с
В-адренорецептором, который взаимодействует с О . аденилатциклаз-
ная активность стимулируется. Однако адреналин может также свя-
Гормонрецепторный
комплекс
\ ОВР СТР
Неактивный
Активный
\
ОТРазная
активность
Неактивный
Рис. 14А. О-белковый цикл. О-белки
активируются после связывания с лиганд-ре-
цепторным комплексом и инактивируются
под действием их собственной ОТРазной
активности
541
Часть И. Метаболизм веществ и энергии в клетке
зыватьсл с другим типом рецепторов, с а-адренорецепторами,
которые взаимодействуют с 0[9 приводя к ингибированию аденилатцикла-
зы (два класса адренорецепторов обнаружены в различных типах
клеток). Таким образом, конечный ответ клетки на действие гормона
зависит от типа присутствующих рецепторов и от типа О-белка, с
которым они сопряжены. Основные особенности аденилатциклазного
сигнального пути, включающего О-белки, представлены на рис. 14.5.
Гетеротримерные О-белки играют центральную роль как
передатчики сигнала и в других сигнальных путях. Так, О-белки (С )
вовлечены также в инозитолфосфолипидный сигнальный путь (который
будет рассмотрен ниже). Другой О-белок, Ок, контролирует активность
калиевых каналов в сердечной мышце. Для О-белков характерно
структурное сходство. Они содержат различные а-субъединицы, но их р- и
у-субъединицы подобны и часто взаимозаменяемы.
О-белки представляют собой биологические мишени для
некоторых бактериальных токсинов, в частности холерного токсина,
продуцируемого вызываемой заболевание бактерией УгЬпо сНо1егае*
Холерный токсин состоит из а- и |3-субъединиц и существует как ос(35-комп-
лекс. Вследствие связывания |3-субъединиц с олигосахаридными це-
Стимулирующий
гормон
Ад еп и л атцик лаза
Ингибирующий
гормон
♦
Протеинкиназа А
(неактивная)
и
б'-АМР
)
Протеипкиназа А
(активная)
Фосфодиэстераза
I
Белок—ОН * Белок-(?) •*****. Клеточный ответ
Рис. 14.5. Схематическое изображение аденилатциклазного сигнального пути.
Связывание гормона со стимулирующим трансмембранным рецептором (Е8)
ведет к активации стимулирующего С-белка, О . Другие гормоны могут связывать-
ся с ингибиторными рецепторами (К.), которые сопряжены с аденилатциклазои
через ингибиторный О-белок, О.. 0$ активирует интегральный мембранный
фермент аденилатциклазу, тогда как О. ингибирует ее, сАМР активирует протеин-
киназу А, что приводит к фосфорилированию клеточных белков
542
Глава 14. Молекулярные основы действия клеточных медиаторов
пями молекул ганглиозида Сш на поверхности клеток а-субъединицы
пересекают плазматическую мембрану и попадают в цитозоль. Эта
а-субъединица представляет собой АВР-рибозилазу, которая
катализирует перенос АВР-рибозильной группировки от кофактора МАО на
аргининовый остаток а-субъединицы белка 6 . Данная ковалентная
модификация приводит к инактивации СТРазной активности С*з
Таким образом, когда холерный токсин оказывает свое воздействие на
клетки, аденилатциклаза постоянно активна, и уровень сАМР
остается высоким. В организме, зараженном V. сНо1егаеу сАМР стимулирует
некоторые транспортеры в плазматической мембране клеток, приводя
к массивному выделению ионов и воды. В конечном итоге быстрая
дегидратация может привести к летальному исходу.
14.4. Медиаторы инозитолфосфолипидного сигнального пути
Другой важнейший сигналпередающий путь, используемый
некоторыми гормонами, ростовыми факторами и другими лигандами,
сопряжен с функционированием двух различных вторичных
медиаторов, образующихся из мембранного фосфолипида — фосфатидилино-
зитол-4,5-бисфосфата (ФИФ2). Этот компонент плазматической
мембраны синтезируется из фосфатидилинозитола в ходе двух
последовательных этапов фосфорилирования, катализируемых киназами,
требующих для своей активности АТР.
Вследствие связывания лиганда со специфическим рецептором
сигнал передается через О-белок, 6_, который испытывает превращение в
ОТР-связанную форму. Ор затем активирует эффекторный фермент
фосфоинозитидитспецифичную фосфолипазу С, которая локализована
на цитоплазматической стороне мембраны. Фосфолипаза С
катализирует гидролиз ФИФ2 на два продукта: инозитол-1,4,5-трифосфат (ЙФ3),
представляющий собой водорастворимую молекулу, и диацилглице-
рол (рис. 14.6). Как ИФ3, так и диацилглицерол, подобно сАМР,
являются вторичными медиаторами, преобразующими первоначальный
сигнал во внутриклеточный. Действие ИФ3 приводит к возрастанию
концентрации ионов Са2+ в цитозоле, а диацилглицерол активирует
протеинкиназу.
ИФ3 диффундирует через цитозоль и связывается со
специфическим рецептором в мембране эндоплазматического ретикулума. Для
внутреннего содержимого эндоплазматического ретикулума
характерна высокая концентрация ионов Са2+, обусловливаемая действием
Са2+-АТРазы. Рецепторный белок ИФ3 выполняет как бы функцию
ворот в кальциевом канале, который открывается на короткое время
и пропускает Са2+ в цитозоль. Концентрация Са2+ в цитозоле при этом
быстро возрастает менее чем с 500 нМ до примерно 1 мкМ. Этот поток
Са2+ не постоянен, так как ионы Са2+ закачиваются обратно
Са2+-АТРазой вскоре после закрытия канала. Возрастание концентра-
543
Часть II. Метаболизм веществ и энергии в клетке
Н Н
Диацилг лицерол Инозитол-1,4,5-трифосфат
(ИФ3)
Рис. 14.6. Реакция, катализируемая фосфолипазой С.
Фосфатидилинозитол- 4,5-бисфосфат (ФИФ2) гидролизуется до инозитол-1,4,5-трифосфата (ИФ3) и
диацилгдицерола под действием фосфоинозитидспецифичной фосфолипазы С
ции Са2+ обеспечивает стимуляцию Са2+-зависимых протеинкиназ, фос-
форилирующих различные белки-мишени. Многие Са2+-кальмодулин-
зависимые ферменты также активируются в данных условиях, Каль-
модулин представляет собой Са2+-связывающий белок с
молекулярной массой около 17 000. Он обнаруживается как в свободной форме в
цитозоле, так и в качестве регуляторной субъединицы некоторых
ферментов. Связывание Са2+ с кальмодулином индуцирует конформаци-
онные изменения в белке, в результате которых он ассоциируется с
различными ферментами и модулирует их активность.
Уровень Са2+ в цитозоле может быть определен с применением
специальных кальцийчувствительных флуоресцентных веществ. Эти
вещества могут быть введены внутрь живых клеток, где они с высоким
сродством связываются с Са2+. Флуоресценция веществ в свободном и
кальцийсвязывающем состояниях не одинакова и чувствительна к
изменениям концентраций Са2+ от наномолярных до микромолярных
концентраций- Изменения флуоресценции внутри живой клетки могут,
таким образом, быть использованы для измерения возрастания
концентрации Са2+, происходящего во время реализации сигнального процесса.
Кальцийспецифичные ионофоры, такие, например, как иономи-
цин, представляют собой также полезные инструменты для
исследования значения ионов Са2+в сигнальных процессах. Если к клеткам,
находящимся в среде с высокой концентрацией Са2+, добавить эти
вещества, то происходит быстрый приток ионов Са2+ в клетку в
отсутствие каких-либо внешних стимуляторов. Такие ионофоры
используются для калибровки уровня кальция, измеренного с использованием
флуоресцентных веществ.
544
Глава 14. Молекулярные основы действия клеточных медиаторов
Другой продукт гидролиза ФИФ2 — диацилглицерол остается в
плазматической мембране и активирует кальций- и фосфолипидзави-
симую протеинкиназу, известную как протеинкиназа С* Эта киназа
активна только тогда, когда и фосфотид ил серии, и Са2+ связаны с
ней. Протеинкиназа С в норме неактивна в присутствии низких
внутриклеточных концентраций кальция. Однако, когда диацилглицерол
связывается с киназой, ее сродство к кальцию значительно
возрастает, так что киназа оказывается способной связывать Са2+ при низких
концентрациях и становится активной. Растворимая цитозольная форма
протеинкиназы С существует в равновесии с ее
мембранно-периферической формой» Активация с помощью диацилглицерол а приводит к
транслокации киназы к внутренней стороне плазматической
мембраны, где она остается связанной в течение некоторого времени.
Протеинкиназа С, принадлежащая к семейству серин-треониновых киназ,
фосфорилирует многие как цитозольные, так и мембраносвязанные
белки-мишени, изменяя их каталитическую активность. Существует
несколько изоферментов протеинкиназы С, различающихся по
каталитическим свойствам и распределению в тканях. Сигнальная транс-
дукция через инозитол-фосфолипидный путь может быть выключена
с помощью ряда механизмов. Во-первых, когда С-ТР гидролизуется,
О возвращается в свою неактивную форму и уже не стимулирует фос-
фолипазу С. Во-вторых, диацилглицерол — это короткоживущий
вторичный медиатор; он быстро превращается в фосфатидат под
действием ферментов, ответственных за биосинтез фосфолипидов и, в
конечном итоге, используется для синтеза фосфатидилинозитола. ИФ3 —
тоже недолгоживущий вторичный медиатор; под действием цитозоль-
ных ферментов — фосфатаз, он быстро превращается в инозитол,
который также идет на биосинтез фосфатидилинозитола.
Инозитол-фосфолипидный сигнальный путь представлен на рис. 14.7. Следует
также подчеркнуть, что действие ИФ3 и диацилглицерол а имеет синер-
гичный характер, т.е. один интермедиат усиливает действие другого.
14.5. Активация тирозинкиназных рецепторов
под действием ростовых факторов
Ростовые факторы — это белки, регулирующие клеточную
пролиферацию путем стимулирования покоящихся клеток к делению.
Ростовые факторы специфичны по отношению к определенным типам
клеток и действуют только на те клетки, которые имеют
соответствующие поверхностные рецепторы. К ростовым факторам, действующим
только на определенные типы клеток, относятся: эпидермальный,
нервный, фибробластный, лимфоцитный (последний известен также под
названием интерлейкины). Другие ростовые факторы, такие,
например, как инсулин, действуют на многие различные типы клеток, так
как их рецепторы присутствуют во всех этих клетках.
35. Заказ 3486
545
Часть II. Метаболизм веществ и энергии в клетке
Рис. 14.7. Инозитол-фосфолипидный сигнальный путь. Связывание гормона
с его трансмембранным рецептором (К) активирует С-белок, а именно С , Это
обратимо стимулирует специфичную мембраносвязанную фосфолипазу С (ФЛС),
которая катализирует гидролиз фосфолипида ФЙФ2 на внутренней стороне
плазматической мембраны. Вторичные медиаторы, ИФ3 и диацилглицерол (ДАГ)
ответственны за перенос сигнала внутрь клетки. ИФ3 диффундирует к эндоплазма-
тическому ретикулуму, где связывается и открывает ИФ3-зависимые ворота
кальциевого канала в мембране, высвобождая находящийся там Са2+.
Диацилглицерол остается в плазматической мембране, где он активирует кальций- и фосфо-
липидзависимый фермент протеинкиназу С (ПКС). ИФ3 иод действием фосфатаз
в конечном итоге превращается в инозитол (И)
Для многих ростовых факторов характерен общий сигнальный путь.
Связывание ростового фактора с его рецептором на поверхности
клетки приводит к активации тирозинкиназной каталитической
активности внутриклеточного домена рецептора. Выделение и секвинирова-
ние генов многих тирозинкиназных рецепторов свидетельствует, что
эти белки характеризуются сходной молекулярной топологией.
Общими чертами являются: наличие большого гликозилированного
внеклеточного домена, в котором связывается ростовой фактор;
гидрофобного, пронизывающего мембрану домена; внутриклеточного тиро-
зинкиназного каталитического домена. Для многих ростовых
факторов активация каталитической активности киназного домена, —
очевидно, результат димеризации. В случае инсулинового рецептора,
изначально являющегося димером (рис. 14.8), связывание инсулина с
а-субъединицей индуцирует конформационное изменение,
передающееся тирозинкиназным доменам (3-субъединицы, которые вследствие
этого активируются. Многие активированные киназные домены рецепто-
546
Глава 14. Молекулярные основы действия клеточных медиаторов
ров ростовых факторов
используют АТР для фосфорилирования
гидроксильных групп тирозино-
вых остатков в самом рецепторе;
данный процесс известен под
названием автофосфорилирование.
Вероятно* каждый рецептор ди-
мерной структуры катализирует
фосфорилирование своего
партнера. Другие белки также фосфори-
лируются с помощью
активированного тирозинкиназного
рецептора, что ведет к стимуляции
вторичных сигнальных путей. Так,
часто происходит гидролиз ФИФ2
с последующей активацией про-
теинкиназы С и возрастанием
концентрации кальция в цито-
золе. Клеточная стимуляция
ростовыми факторами — это
комплексный процесс, который
вовлекает триггерные системы
множества сигнальных путей (рис.
14.9). Отщепление фосфатных
групп как от рецепторов
ростовых факторов, так и от их
белков-мишеней происходит под
действием ферментов,
называемых тирозинфосфатазами. Хотя только несколько ферментов этой
группы достаточно хорошо изучены, но очевидно, что они играют
важнейшую роль в сигнальных процессах, воздействуя на тирозин-
киназу.
14.6. Некоторые онкогенные продукты,
участвующие в сигнальных процессах
Онкогены представляют собой гены, трансформирующие
нормальные клетки в раковые. Эти гены часто многократно и
несоответствующим образом экспрессируются по сравнению с нормальными клеточны-
ми генами, которые кодируют белки, вовлеченные в сигнальные пути,
ведущие к пролиферации. Онкогены были впервые открыты в онкоген-
ных РНК-содержащих вирусах. Раковые гены во многих из этих
вирусов были, очевидно, приобретены ими из организма хозяина и в ходе
эволюции интегрированы в вирусный геном. Первоначальный ген
хозяина, протоонкогену мог кодировать ростовой фактор, рецептор ростово-
35*' 547
Инсулин
Тирозинкиыааные домены
Рис, 14.8. Молекулярная структура
инсулийового рецептора. Рецептор
состоит из двух внеклеточных ос-цепей,
каждая из которых имеет инсулинсвязыва-
ющий центр, и двух трансмембранных
р-цепей с цитозольными тирозинкиназ-
ными доменами. Вследствие связывания
инсулина с сс-цепями р-цепи
катализируют автофосфорилирование тирозяно-
вых остатков в примыкающих
внутриклеточных доменах. Другие клеточные
белки также фосфорилируются под
действием тирозинкиназы, что вызывает
каскад событий в клетке
со
Ростовой фактор
л
Рецептор
Димеризадия
Фосфолипаза С
Тирозинкиназныи
домен
ИФа А
I
Активирован пая
протеинкиназа С
Белок-ОН
Белок—®
Клеточный
ответ
Са®-высвобожденный
Клеточный
ответ
Клеточный
ответ
г?
3
§,
О
!а
СИ
го
то
о
3
50
Рис. 14.9. Общая схема сигнальной транедукции под действием мембранного тирозинкиназного рецептора- Фактор роста
связывается с рецептором, активируя внутриклеточный тирозинкиназныи домен. Рецептор автофосфорилируется, и тирозино-
вые остатки в других клеточных белках также фосфорилируются. Другие сигнальные пути активируются с помощью не вполне
изученных механизмов- Часто имеет место гидролиз ФИФ2, катализируемый фосфолипазой С, что приводит к высвобождению
ИФ3 и диацилглицерола (ДАГ)
и-егЬВ
Онкогенный
продукт
с-егЬВ
Рецептор эпидермального
ростового фактора
Постоянно
активе и
•Тироаин-
киназные
домены
Неактивен в отсутствие
эпидермального ростового
комплекса
Глава 14, Молекулярные основы действия клеточных медиаторов
го фактора или какой-нибудь промежуточный компонент сигнального
пути, например, такой, как ядерный фактор транскрипции,
вовлеченный в генную экспрессию. Заражение клетки хозяина онкогенным
вирусом приводит к экспрессии дефектных белков и неконтролируемой
клеточной пролиферации. Например, птичий эритробластный вирус,
вызывающий рак у цыплят, содержит онкоген и-егЬВ (и указывает на
вирусный ген). Белковый продукт этого онкогена гомологичен
рецептору эпидермального ростового фактора, геном которого является прото-
онкоген с-егЬВ (с указывает, что
это клеточный ген). Как
показано на рис. 14.10, продукт и-
егЬВ представляет собой
видоизмененный рецептор
эпидермального ростового фактора.
Аминокислотные остатки (с 551 по
1154) рецептора ростового
фактора на 90 % идентичны
таковым для белка и-егЬВ, который,
однако, утрачивает внешний ли-
гандовязывающий домен.
Онкогенный белок постоянно активен
в отсутствие ростового фактора
и передает сигналы для клеточ- Рис. 14:10. Белковый продукт вирусно-
ного деления. Более половины го онкогена и-егЬВ и протоонкогена с-егЪВ.
известных онкогенных белков Обратите внимание, что внеклеточный ли-
как бы подменяют тирозинки- гандсвязывающий домен отсутствует в ви-
русном белке, аминокислотная последова-
назныи рецептор. телыюсть которого более чем на 90 % иден-
Изучение онкогенов и сиг- тична соответствующей области клеточного
нальных процессов способству- продукта — рецептора эпидермального фак-
ет пониманию причин и разра- тора роста. Предполагается, что онкогенный
ботке возможных средств пред- белок передает сигнал при отсутствии акти-
отвращения рака у человека. пирующего воздействия ростового фактора
14.7. Гормоны — медиаторы на уровне целостного организма
Термин "гормон" происходит от греч. "Ногтато", что означает
"возбуждать", "приводить в действие". Гормоны представляют собой
химические агенты с очень высокой биологической активностью. Как
правило, достаточно их очень малых количеств (иногда десятка
микрограмм), чтобы поддержать и сохранить жизнедеятельность
организма. Гормоны образуются в специализированных железистых клетках
тканей одного типа, выделяются в кровь или лимфу и доставляются к
другим тканям-мишеням, где они проявляют свою физиологическую
активность. Действие гормонов многосторонне. Они не только
регулируют клеточный метаболизм, но и влияют на ростовые процессы, ра~
549
Часть II. Метаболизм веществ и энергии в клетке
боту сердца, почек, кишечника, репродуктивной системы, а также на
кровяное давление. В следующих разделах остановимся на тех
гормонах, которые регулируют основные пути обмена веществ.
Когда гормоны, высвободившись из эндокринных желез, в
которых осуществляется их синтез, поступают в кровяное русло, то, в
принципе, потенциально они могли бы контактировать со всеми клетками
организма. Избирательность гормональных ответов достигается
благодаря присутствию в определенных клетках специфических
гормональных рецепторов. Многие клетки, например, обладают инсулино-
выми рецепторами; такие клетки называются инсулинчувствительны-
ми. Под действием инсулина увеличивается поглощение глюкозы
этими клетками, в результате чего концентрация глюкозы в крови
понижается. В связи с недостатком инсулиновых рецепторов эритроциты и
клетки мозга называются инсулиннечувствительными.
Клетки печени — это клетки, богатые рецепторами глюкагона, что
делает глюкагон исключительно селективным по отношению к
данным мишеням. Напротив, многие ткани чувствительны к адреналину
и норадреналину. Адреналин и норадреналин связываются с
различными типами адренорецепторов. Распространение различных адрено-
рецепторов варьирует между разными видами клеток. Наиболее
хорошо изучено действие а1- и ^-адренорецепторов, на котором мы
остановимся еще раз ниже. Но если в клетке присутствуют оба типа
рецепторов, то один из них, как правило, доминирует и клеточный
ответ может оказаться не однозначным.
Глюкагон и адреналин относятся к гормонам, имеющим
мембранные рецепторы, сопряженные с О-белком. Глюкагон связывается со
своим рецептором, который, активируясь, стимулирует О-белок, а
именно С3. Связывание адреналина или норадреналина с р-адреноре-
цептором также стимулирует 08. 03 обратимо активирует аденилат-
циклазу, и этот мембраносвязанныи фермент катализирует
образование вторичного медиатора, сАМР. Циклический АМР активирует про-
теинкиназу А (называемую также сАМР-зависимой протеинкиназой).
Таким образом, данные этапы выполняют функции триггерных
механизмов (реле), обеспечивающих возрастание внутриклеточной
концентрации сАМР.
Связывание адреналина или норадреналина с о^-адренорецепто-
ром активирует О-белок, а именно О , который вызывает
совершенно другой регуляторныи каскад. В этом случае мембраносвязанныи
фермент фосфолипаза С активируется, приводя к образованию инози-
тол-1,4,5-трифосфата и диацилглицерола (этот процесс описан в
разд. 14.4). Образование инозитол-1,4,5-трифоефата обеспечивает
высвобождение кальция из содержимого эндоплазм атич ее кого ретикулу-
ма в цитозоль, где он определяет разнобразные физиологические
эффекты. Другой продукт, диацилглицерол, представляет собой
молекулу липидной природы, которая активирует мембраносвязанныи регу-
550
Глава 14. Молекулярные основы действия клеточных медиаторов
ляторный фермент — протеинкиназу С. Одной из известных мишеней
протеинкиназы С является инсулиновыи рецептор. Находясь в фосфо-
рилированном состоянии, инсулиновыи рецептор плохо связывает
инсулин. Таким образом, действие протеинкиназы С, обусловленное
адреналином, как бы снимает эффект инсулина.
Как инозитол-1,4,5-трифосфат, так и диацилглицерол являются
вторичными медиаторами, переносящими информацию от первичного
медиатора — гормона. Ионы Са2+, также участвующие в передаче
сигнала, могли бы рассматриваться как третичные медиаторы, хотя
данная терминология не используется. Все эти молекулы представляют
собой сигнальные молекулы, контролирующие активности регулятор-
ных белков.
14.8. Классификация и общие биологические признаки гормонов
Принято выделять три класса гормонов: пептидные, амины и
стероиды,
К пептидным гормонам относятся гормоны гипоталамуса и
гипофиза, гормоны поджелудочной железы и щитовидной железы.
К классу аминов принадлежат гормоны, представляющие собой
производные аминокислот, — это гормоны, секретируемые мозговым
слоем надпочечников, тиреоидные гормоны.
К стероидным гормонам относятся половые гормоны, гормоны коры
надпочечников.
Все гормоны функционируют в рамках тонко регулируемой
эндокринной системы, координирующим центром которой является
гипоталамус. На уровне этой специализированной области мозга
происходит объединение потоков информации от нервной и гормональной
систем. В результате восприятия и интегрирования нервных импульсов
гипоталамус осуществляет секрецию ряда гипоталамических
гормонов, которые, в свою очередь, регулируют выделение тройных
гормонов гипофиза. Последние выделяются в кровь и, достигая
периферических эндокринных желез (поджелудочной железы, щитовидной
железы, коры надпочечников и др.), влияют на секрецию гормонов в
этих тканях. Гормоны периферических эндокринных желез с током
крови доставляются к рецепторам клеток-мишеней. Таким образом,
для гормонов характерно такое свойство, как дистантность действия.
Регуляция процессов, связанных с эндокринной системой,
осуществляется по принципу обратной связи. В то время, когда тропные гормоны
активируют процессы выделения гормонов периферическими
эндокринными железами, последние, циркулируя в крови, угнетают образование
тропных гормонов, воздействуя на гипофиз или гипоталамус. Регуляция
может происходить также на основе метаболитно-гормональных
взаимосвязей. Изменения концентраций каких-либо метаболитов в крови могут
непосредственно или через гипоталамус и гипофиз влиять на выделение
551
Часть 77. Метаболизм веществ и энергии а клетке
гормонов периферическими эндокринными железами. Очевидно, роль
таких метаболитов могут выполнять глюкоза, некоторые аминокислоты,
нуклеотиды, жирные кислоты, ионы Са2+, Ыа+, К* и др.
При стимуляции выделения гормонов их концентрация в крови
может возрастать на несколько порядков, тогда как нестимул ирован-
ный так называемый базалъныи уровень гормонов в крови низок (от
микромолярных до пикомолярных концентраций).
Для гормонов характерно также такое общее свойство, как строгая
специфичность действия. Действие одного гормона не может быть
полностью заменено действием другого.
Одни гормоны способны вызывать немедленный физиологический
ответ. Так, под действием адреналина клетки печени через несколько
секунд начинают выбрасывать глюкозу в кровь. Другие гормоны
действуют более медленно. Например, эффект половых и тиреоидных
гормонов достигает максимума в течение более длительного промежутка
времени (часы или даже дни).
Характерно, что некоторые пептидные гормоны первоначально
синтезируются в виде неактивных предшественников — прогормонов.
Например, инсулин первоначально синтезируется в виде проинсули-
на, представляющего собой одну полипептидную цепь, содержащую
80 аминокислотных остатков. В секреторных гранулах под действием
селективного протеолиза он превращается в двухцепочечную
структуру, стабилизируемую дисульфидными мостиками и включающую
51 аминокислотный остаток. В активной форме инсулин секретирует-
ся в ответ на повышение уровня глюкозы в крови.
14.9. Характеристика гормонов, продуцируемых различными
эндокринными системами
Гормоны гипоталамуса и гипофиза. Гормоны гипоталамуса по
специальным кровеносным сосудам попадают непосредственно в
гипофиз, не поступая в общий кровоток. По своей структуре они являются
короткими пептидами, в состав которых входит 3—15
аминокислотных остатков. На рис. 14.11 в качестве примера приведена
структурная формула одного из гипоталамических гормонов — соматостатина.
Среди других гипоталамических гормонов можно назвать тиролибе-
рин, способствующий выделению тиротропина передней долей
гипофиза; соматолиберин, высвобождающий соматотропин; пролактолибе-
рин, вызывающий секрецию пролактина. Наряду с указанными
существуют и другие гормоны, вырабатываемые гипоталамусом.
Достигнув расположенного рядом гипофиза, каждый гипоталамический
гормон регулирует секрецию какого-то одного гипофизарного гормона.
Некоторые гормоны гипоталамуса оказывают стимулирующий эффект
на секрецию определенных гормонов гипофиза; другие, наоборот,
тормозят процесс выделения. В передней и средней доле гипофиза» так
552
Глава 14. Молекулярные основы действия клеточных медиаторов
называемом аденогипофизе,
происходит образование
тройных гормонов; задняя
доля — нейрогипофиз секре-
тирует нейрогормоны (вазо-
прессин и окситоцин),
синтезирующиеся в гипоталамусе.
Тройные гормоны
представляют собой достаточно
длинные полипептиды с
молекулярной массой от 1070
до 34 000. Такие гипофизар-
ные гормоны, как тиротро-
пин, фолликулостимулиру-
ющий и лютеинизирующий
гормоны, являются глико-
протеинами. В их состав
входят два типа субъединиц:
а-субъединицы, одинаковые у разных гормонов, и (3-субъединицы,
имеющие некоторые различия и определяющие специфичность действия
данных гормонов* Окситоцин и вазопрессин представляют собой
циклические октапептиды. Тройные гормоны реализуют свое действие на
функционирование периферических эндокринных желез или же
каких-либо тканей организма путем связывания с мембранными
клеточными рецепторами и активации аденилатциклазы. Образование под
действием эффекторного фермента вторичного медиатора, сАМР,
оказывает, в свою очередь, воздействие на гормонообразование или
метаболизм в клетках-мишенях. Тропины не только обеспечивают
регуляцию синтеза и секреции гормонов периферическими эндокринными
системами, но и воздействуют на функционирование нервной
системы, образование половых клеток, а также на метаболизм ряда тканей
и органов.
Непосредственное действие на периферические ткани оказывает ряд
гипофизарных гормонов. Так, вазопрессин умеренно повышает
кровяное давление и увеличивает обратное всасывание воды в почках.
Недостаточность вазопрессина приводит к такому заболеванию, как
несахарный диабет, сопровождающемуся выделением большого
количества мочи (4—10 л в сутки) низкой плотности. Как следствие,
развивается жажда. Препараты вазопрессина оказывают положительный
эффект при данном заболевании. В результате действия вазопрессина
происходит стимуляция протеинкиназ под действием вторичного
медиатора, сАМР, и активированные протеинкиназы обеспечивают фос-
форилирование ряда белков клеточных мембран. Это приводит к уве-
личению проницаемости мембран для воды. Более интенсивное
обратное всасывание воды в почках снижает диурез. Окситоцин оказывает
Пироглутаминовая
кислота
Гистидин
Пролинамид
N-конец
I
А1а
01у
СУ8-
)
Ьуз
I
Аап
РЬе
8
8
Тиролиберин (ТЛ)
РЬе
|гр
Ьуз
I
ТЬг
I
РЬе
I
ТЬг
I
8ег
Суз-
Соматостатин
Рис. 14.11. Структурные формулы тиро
либерина и соматостатина
553
Часть II. Метаболизм веществ и энергии в клетке
воздействие на некоторые гладкие мышцы, и особенно на мыщцу
матки. Его действие сопряжено с повышением внутриклеточной
концентрации ионов Са2+. Этот гормон стимулирует также и лактацию.
Окситоцин находит применение в акушерстве.
Соматотропин (гормон роста) вызывает увеличение массы
внутренних органов и тканей лица, стимуляцию клеточного деления в
хрящах и рост костей в длину. Этот гормон таже реализует свое
действие через активацию аденилатциклазы. Под его влиянием
образуются также полипептиды, регулирующие ростовые процессы, — это
так называемые соматомедины трех типов: А, В и С. Соматомедины
типов А и С стимулируют деление клеток хрящей, синтез ДНК, РНК
и белков, способствуют образованию протеогликанов. Соматомедины
типа В активируют синтез ДНК и белка в клетках нервной системы.
Недостаточность соматотропина приводит к карликовости.
Избыточная его секреция в молодом возрасте вызывает гигантизм, а в зрелом
возрасте — акромегалию, характерными признаками которой
являются усиленный рост лицевых костей (массивные нижняя челюсть,
носа, тяжелые надбровья), а также увеличение размеров ступней и
кистей рук. Соматотропин оказывает также сильное влияние на
углеводный обмен. Избыток соматотропина вызывает гипофизарный
диабет, что, по-видимому, связано с подавлением секреции инсулина по
сравнению с выделением глюкагона поджелудочной железой.
Кортикотропин непосредственно воздействует на жировую ткань,
обеспечивая освобождение жирных кислот и глицерина и вызывая
поглощение глюкозы. Глицерин и жирные кислоты образуются под
действием триацилглицероллипазы, которая активируется под
действием сАМР.
Жиромобилизующие эффекты оказывают также гонадопгропины и
а- и $-липопгропины.
В гипофизе синтезируются также эндорфины. Эти пептиды
образуются путем селективного гидролиза из длинного предшественника —
проопиокортина, содержащего 260 аминокислотных остатков и
служащего источником образования и других гипофизарных гормонов:
литотропинов и меланотропинов. Эндорфины — это гормоны,
обладающие обезболивающим действием. По характеру своего воздействия
они подобны морфину, но их эффект может быть выражен сильнее. К
эндорфиновой группе соединений относятся также энкефалины,
способные связываться с теми же рецепторами мозга, что и опиаты.
Предполагается, что эти нейропептиды могут воздействовать на
функционирование нейронов. Существуют предположения, что они участвуют
в биохимических механизмах, вызывающих отклонения психической
деятельности при шизофрении и наркомании.
Гормоны щитовидной железы. Щитовидная железа секретирует
йодтиронины — тироксин и трийодтиронин а также кальциотонин
(рис. 14.12). Поскольку последний образуется также в паращитовид-
554
Глава 14. Молекулярные основы действия клеточных медиаторов
ных железах, то его действию
внимание будет уделено ниже при
рассмотрении гормонов этих
эндокринных желез. Что касается тироксина
(сокращенно обозначаемого Т4) и
трийодтиронина (ТЗ), то эти
соединения являются производными
тирозина. Под действием гипоталамичес-
кого гормона тиролиберина
происходит стимуляция выделения тиротро-
пина в аденогипофизе. Тиротропин
доставляется кровью к клеткам
щитовидной железы и связывается с их
специфическими рецепторами, что
приводит к активации синтеза и
секреции тиреоидных гормонов. Синтез
тироксина и трийодтиронина
происходит в ходе ряда ферментативных
процессов, начинающихся с йодиро-
н
СНо-С-СОО
+
ин3
I I
Тироксин (Ь-3,5,3'?5'-тетрайодтиронйн)
НО
Тряйодтиронин (Ь-3,5,3'-трийодтиропин)
НО-/ %-СЩ-С-СОО"
Ь-3-моиойодтирозин
Рис. 14.12. Структурные
формулы тироксина, трийодтиронина и
Ь-3-монойодтирозина
вания тирозиновых остатков,
входящих в тироглобулин, с образованием монойодтирозиновых или дий-
одтирозиновых остатков в составе этого гликопротеина* Необходимый
для данных реакций йод поступает в щитовидную железу путем ак-
тивного транспорта с кровью. Иод связывается тироглобулином,
называемым также коллоидным белком, накапливается здесь и
используется для йодирования тирозиновых остатков. Далее в результате
конденсации моно- и дийодтирозинов происходит образование три-
йодтиронина и тироксина в молекуле тироглобулина. Практически
весь новообразованный тироксин и трийодтиронин остаются
связанными с тироглобулином в тиреоидных пузырьках до тех пор, пока не
произойдет перемещение тироглобулина к внешней поверхности
мембраны, омываемой внеклеточной жидкостью, и не осуществится
секреция иодтиронинов благодаря их высвобождению из состава белка
под действием протеолитических ферментов. Поступив в кровь, йод-
тиронины участвуют в формировании комплексов с альбуминами
плазмы, а также с тироксинсвязывающим глобулином и в виде таких
комплексов транспортируются к различным тканям организма.
Полагают, что более значительный физиологический эффект
трийодтиронина, который в 3—5 раз сильнее, чем эффект тироксина, обусловлен
меньшим связыванием ТЗ с белками плазмы. Существует также
предположение, что тироксин является прогормоном трийодтиронина.
и
Иодтиронины оказывают влияние на различные ткани организма.
Особенно сильно тиреоидные гормоны воздействуют на метаболизм
печени, сердца, почек, скелетных мышц. Их действие менее выражено
по отношению к нервной и жировой тканям. В большинстве случаев
555
Часть II. Метаболизм веществ и энергии в клетке . .. . ■
влияние йодтиронинов имеет стимулирующий характер; исключение
составляют только мозг взрослого человека и некоторые
репродуктивные ткани. Процессы, запускаемые йодтиронинами, сопровождаются
клеточной пролиферацией, ростом и дифференцировкой клеток,
интенсификацией энергетического обмена. Для их действия характерен
также калоригенный эффект, связанный с тем, что в митохондриях
возрастает не только производство энергии, но и теплообразование.
Йодтиронины связываются со специфическими клеточными
рецепторами, и взаимодействие образовавшихся комплексов с
определенными генами обеспечивает интенсификацию синтеза ряда ферментов.
Причем функционирование большинства из этих ферментов и
ферментных систем сопряжено с энергетическим обменом. Так, йодтиронины
стимулируют ферменты окислительного метаболизма митохондрий,
ферменты "челночных" систем, обеспечивающих транспортировку
восстановленных кофакторов из цитоплазмы в митохондриальный мат-
рикс. Имеются данные об увеличении числа митохондрий в клетках,
а также размеров крист внутри них в результате действия тиреоид-
ных гормонов. В процессы мобилизации энергетических ресурсов
вовлечена аденилатциклаза, благодаря активированию которой
возрастает содержание сАМР в клетках, что стимулирует гликогенолиз в
мышцах и печени, а также липолиз в жировой ткани. Таким образом,
результатом действия йодтиронинов является увеличение скорости
основного обмена. Возрастание скорости основного обмена, или базалъ-
ной скорости метаболизма, сопровождается повышенным
потреблением кислорода.
Определение базальной скорости метаболизма осуществляется при
диагностике заболеваний, обусловленных нарушением функций
щитовидной железы. При гиперфункции щитовидной железы
развивается гипертиреоз. Выраженные формы такого состояния получили
название базедовой болезни. Заболевание может быть связано с
недостаточностью йода, являющегося незаменимым микроэлементом. Это
сопровождается увеличением щитовидной железы вследствие
накопления коллоидного белка, необходимого для захватывания
минимальных количеств йода, присутствующего в кровяном русле. Скорость
основного обмена при этом заболевании выше, чем в норме.
Температура тела также может увеличиваться, что связано с калоригенными
свойствами тиреоидных гормонов. Для больных характерны
повышенная возбудимость, пучеглазие. Несмотря на то, что тиреоидные
гормоны способствуют синтезу необходимых ферментных систем, постепен-
но процессы распада белков-приобретают преобладающий характер. В
результате может иметь место отрицательный баланс азота в
организме. Недостаточность секреции йодтиронинов приводит к
гипотиреозу, сопровождающемуся снижением скорости основного обмена. Для
больных характерны, наоборот, более медленное сгорание пищи в
организме по сравнению с нормой, выделение меньшего количества тепла,
556
Глава 14. Молекулярные основы действия клеточных медиаторов
некоторое енижейве памяти, нарушение обновления кожного
эпителия, общая медлительность. Гипотиреоз в раннем возрасте или у
новорожденных приводит к кретинизму, сопровождающемуся физической
и умственной отсталостью.
Гормоны паращитовидпых желез. В паращитовидных железах
образуются пептидные гормоны: калъциотонин> о котором уже
упоминалось выше, так как он образуется и в щитовидной железе, и па-
рагпирин. Первоначально эти гормоны синтезируются в виде пропро-
гормонов, представляющих собой длинные полипептидные цепи. В
результате протеолиза образуются прогормоны, а затем путем
последующего ферментативного гидролитического отщепления фрагмента по-
липептидной цепи — активные гормоны. Тройные гормоны не
принимают участия в регуляции образования и секреции этих гормонов из
секреторных гранул аппарата Гольджи. Секреция этих двух гормонов
регулируется под действием ионов Са2+. Выделение кальциотонина
усиливается при увеличении концентрации Са2+ в крови. Секреция
паратирина стимулируется, наоборот, при снижении уровня Са24~ . В
то же время кальциотонин и паратирин сами регулируют баланс ионов
Са2+ и неорганического фосфата. Действие кальциотонина приводит к
снижению концентрации и кальция, и фосфата в крови, тогда как
паратирин повышает содержание кальция и снижает уровень
неорганических фосфатов.
Гормоны надпочечников. Адреналин (или эпинефрин) и норадре*
налип (или норэпинефрин) образуются в мозговом веществе
надпочечников. Это сходные по структуре водорастворимые амины,
образующиеся из тирозина. Промежуточными интермедиатами являются
3,4-дигидроксифенилаланин (называемый сокращенно дофа) и 3,4-ди-
гидроксифенилэтиламин (известный как дофамин). Дофамин,
адреналин и норадреналин относятся к катехоламинам. Известно, что кате-
холамины могут функционировать в качестве нейромедиаторов в
нервной системе и мозге. Нарушение синтеза дофамина в мозге имеет
место при болезни Паркинсона, и введение его предшественника — 3,4-ди-
гидроксифенилаланина приводит к некоторому улучшению состояния
больных.
Адреналин и норадреналин накапливаются в секреторных
гранулах; их повышенная секреция происходит при стрессе, а также при
понижении уровня глюкозы в крови. Мозговой слой надпочечников
воспринимает нервные импульсы, и из хромаффиновых секреторных
гранул за счет экзоцитоза выделяется адреналин, поступающий затем
в кровь. Адреналин вызывает сАМР-зависимое действие (механизм
которого рассмотрен выше) на печень и скелетные мышцы, а также
на жировую ткань, которые являются гормональными мишенями. В
конечном итоге изменения затрагивают углеводный и липидный
обмен. Адреналин вызывает распад гликогена в печени, что приводит к
возрастанию уровня глюкозы крови. Происходит также интенсифика-
557
Часть II. Метаболизм веществ и энергии в клетке
ция расщепления гликогена в мышцах за счет анаэробного
гликолиза. Помимо этого адреналин действует на сердечно-сосудистую
систему. Под его влиянием повышается кровяное давление, увеличиваются
сила и частота сердечных сокращений, В этой связи адреналин
применяется как средство, вызывающее стимуляцию сокращений сердца;
поэтому его применяют в критических ситуациях, например, при
сердечном коллапсе.
Адреналин не только обеспечивает стимуляцию распада гликогена
за счет сАМР- и Са2+-зависимого активирования соответствующих
киназ, ведущего к возрастанию активности гликогенфосфорилазы
(рис, 14.13), но и подавляет синтез гликогена из глюкозы. Благодаря
этому весь доступный гликоген, а также сахарофосфаты, являющиеся
предшественниками глюкозы, могут быть использованы для
образования глюкозы. С помощью сАМР в качестве вторичного медиатора
адреналин вызывает процесс фосфорилирования гликогенсинтазы, в
результате чего активная дефосфорилированная форма данного
фермента превращается в неактивную фосфорилированную форму. Таким
образом, одна и та же цепь событий, вызываемая связыванием
адреналина с клеточными рецепторами, приводит, с одной стороны, к
возрастанию гликогенфосфорилазной активности, и соответственно, к
Адреналин
Рецептор адреналина
Клеточная мембрана
АТР сАМР + РР1
Неактивная у Активная
лротеинкиназа протеинкиназа + Ко - сАМ?4
(С2К2) (2С)
Неактивная у Активная
АТР + киназа г*" киназа 4- АОР
фосфорилааы С а фосфорилазы
Фосфорилаза Ь
1^
Фосфорилаза а
АТР + (неактивная) Са2+ (активная) + АБР
Гликоген + Р[
+
Глюк 030-1-фосфат
Глюкоза + Р1
Глюкоза крови
Рис, 14.13. Каскадный процесс, включающийся в клетках печени под дей
ствием адреналина
558
Глава 14. Молекулярные основы действия клеточных медиаторов
стимуляции образования глюкозы, а с другой стороны — к
торможению гликогенсинтазной активности и подавлению синтеза гликогена.
Гормоны коры надпочечников. Гормоны коры надпочечников
представляют собой жирорастворимые стероидные гормоны,
образующиеся из холестерола. Их называют корлгикоспгероидами. Регуляция
секреции гормонов коры надпочечников осуществляется
гипоталамусом. Выделение кортиколиберина гипоталамусом является ответной
реакцией на стресс. Кортиколиберин, в свою очередь, вызывает
стимуляцию выделения кортикотропина аденогипофизом. Кровотоком кор~
тикотропин доставляется к поверхностным рецепторам клеток коры
надпочечников и активирует процесс использования эфиров
холестерола для синтеза стероидных гормонов.
Среди кортикостероидов принято выделять
три группы. К первой группе относятся
глюкокортикоиды, действие которых направлено в
основном на углеводный обмен. Представителем
данной группы является кортизол (рис. 14.4).
Глюкокортикоиды транспортируются к
своим тканям-мишеням (печени, почкам,
скелетным мышцам, соединительной и лимфойдной
тканям) в виде комплекса с а-глобулином
плазмы крови. Характерно, что образующиеся в этих
тканях комплексы гормон—рецептор могут
оказывать противоположно направленное действие
на процессы транскрипции и трансляции в различных типах тканей.
Так, в лимфойдной ткани происходит торможение этих процессов, что
приводит к подавлению новообразования белков и, более того, идет
активный протеолиз. Все это в совокупности приводит к так называемому
лимфоцитозу, т.е. распаду лимфойдной ткани. Образующиеся в
результате расщепления белков аминокислоты поступают в кровь и
доставляются в печень и почки. В этих органах под действием глюкокортикои-
дов, наоборот, происходит стимуляция транскрипции и трансляции
определенных генов, причем преимущественно ферментов глюконеогенеза.
Таким образом, доставленные аминокислоты активно используются в глю-
конеогенезе, а синтезирующаяся глюкоза идет на образование гликогена
в печени и мышцах.
Глюкокортикоиды оказывают также жиромобилизующий эффект
и стимулируют образование кетоновых тел, что взаимосвязано с
усилением секреции адреналина под их действием.
Поскольку глюкокортикоиды оказывают выраженное иммунодеп-
рессивное действие за счет подавления синтеза антител лимфоидными
клетками, а также антиаллергическое и противовоспалительное
действие, то они находят достаточно широкое применение в медицине.
Гормональные препараты на основе глюкокортикостероидов и их
аналогов применяют для предупреждения отторжения органов после пе-
Рис, 14.14,
Структурная формула кортизола
(гидрокортизона)
559
Часть II. Метаболизм веществ и энергии в клетке
ресадки, используют их также при бронхиальной астме, дерматозах,
ревматизме и т.д.
Нарушения секреции глюкокортикоидов приводят к различным
заболеваниям. Так, при избыточном выделении этих гормонов
развивается болезнь Иценко—Кушинга, симптомами которой являются
потеря мышечной массы, утомляемость, связанные с повышенными
темпами превращений аминокислот в глюкозу, а также с мобилизацией
жировых депо. При недостаточности глюкокортикоидов развивается
аддисонова болезнь, признаками которой являются гипогликемия,
нарушения водно-солевого баланса, снижение устойчивости к
инфекциям, эмоциональным стрессам, мышечная слабость и т.д.
Ко второй группе глюкокортикоидов относятся минералокоргпикои-
ды. Их важнейшим представителем является альдостерон (рис. 14.15).
Альдестерон регулирует водно-солевой баланс. С помощью
альбуминов плазмы он транспортируется к своим тканям-мишеням, а именно
к почкам, где способствует выведению ионов К+ и задержке ионов
Ыа+. Эта регуляция сопряжена со стимуляцией транскрипции генов,
кодирующих белки, вовлеченные в транспорт ионов Ма+ через
мембраны клеток эпителия дистальных канальцев почек» В результате
наряду с усилением реабсорбции Ыа* происходит также задержка ионов
С1" и воды» Ионы К+ выделяются в мочу в обмен на ионы Ка+.
Следует отметить, что альдостерон имеет также слабо
выраженную глюкокортикоидную активность. В то же время кортизол
обладает слабой минералокортикоидной активностью, чем и объясняются, в
частности, нарушения баланса ионов Ыа+, К+ и воды при
недостаточности гормональной функции надпочечников, приводящей к аддисо-
новой болезни.
К третьей группе принадлежат стероиды,
характеризующиеся'промежуточными свойствами между минералокортикоидами и глюкокор-
тикоидами. Кортикостерон является представителем данной группы
(рис. 14.16).
Рис. 14.15. Структурная форму- Рис. 14.16. Структурная
формула альдостерона ла кортикостерона
Половые гормоны. Половые гормоны так же, как и кортикостеро-
иды> относятся к стероидным гормонам и образуются из холестерола.
Синтез этих гормонов осуществляется в половых железах: эстрогенов
•
560
Глава 14. Молекулярные основы действия клетояных медиаторов
(женские половые гормоны) — в основном в яичниках; андроеенов
(мужские половые гормоны) — в основном в семенниках. Яичники и
семенники являются железами смешанной секреции,
продуцирующими и половые клетки, и половые гормоны. Однако эстрогены
образуются не только в яичниках, но и, хотя и в значительно меньшем
количестве, в коре надпочечников и в семенниках. Андрогены тоже
образуются и в коре надпочечников, и в яичниках, хотя опять же в
меньших количествах, чем в семенниках. Структурные формулы
важнейших половых гормонов представлены на рис. 14.17.
Тестостерон (5-Эстрадиол Прогестерон
Рис. 14.17. Структурные формулы тестостерона, {З-эстрадиола и прогестерона
Прогестерон — половой гормон, являющийся предшественником
в синтезе половых гормонов, а также других стероидов коры
надпочечников. Этот гормон способствует имплантации яйца.
Соотношение еекретируемых половых гормонов определяет
половые признаки организма. Прогестерон и эстрогены дополняют
функции друг друга, воздействуя на клеточный метаболизм, рост и
развитие тканей. Эстрогены осуществляют регуляцию женской
репродуктивной системы. Они обеспечивают формирование половых
признаков, основными из которых являются формирование скелета, тембра
голоса и рост волос по женскому типу, развитие молочных желез.
Физиологические эффекты эстрогенов основаны на их воздействии на
определенные гены. Так, основной эстроген — {3-эстрадиол,
связываясь со своим внутриклеточным рецептором в тканях-мишенях
(молочные железы и матка) так называемым эстрофиллином /, вызывает
его молекулярную перестройку, в результате чего рецептор
превращается в эстрофиллин II. Последний выполняет функцию вторичного
медиатора в действии эстрогена. Он способен входить в клеточное ядро
и вызывать активацию определенных генов, что обеспечивает
индукцию синтеза специфических белков, определенным образом влияющих
на клеточный метаболизм, рост и дифференцировку клеток. При
развитии рака молочной железы у женщин снижается число рецепторов
эстрогенов, что иногда используется в качестве диагностического
теста с целью определения стадии заболевания.
Андрогены поддерживают функционирование репродуктивной
системы мужчин, стимулируют рост и способствуют формированию вто-
36. Заказ 3486
561
Часть Л, Метаболизм веществ и энергии в клетке
ричных половых признаков. Они приводят к развитию мощной
скелетной мускулатуры, роста волос на лице и теле, формированию
мужского тембра голоса. Андрогены, связываясь с андрогенными
рецепторами в тканях-мишенях (простата, семенные пузырьки, мышцы),
воздействуют на хроматин, что приводит к стимуляции репликации ДНК,
транскрипции определенных генов, и в первую очередь к резкому
возрастанию белкового синтеза.
Следует отметить, что как андрогены, так и эстрогены могут
оказывать различное по характеру влияние на многие ткани организма, не
связанное с функционированием репродуктивной системы. В этом
отношении прежде всего надо сказать об анаболическом действии гормонов,
т.е. способности обеспечивать положительный азотистый баланс путем
стимуляции белкового синтеза. Но этот эффект выражен в значительно
большей степени при действии андрогенов, чем эстрогенов. Так
называемые анаболические стероиды, являющиеся химическими производными
андрогенов, принимают иногда спортсмены (борцы, штангисты) для
увеличения силы и мышечной массы. Препараты на основе анаболических
стероидов применяют также при заболеваниях, сопровождающихся
отрицательным азотистым балансом. За счет индукции образования
белков под действием андрогенов происходит интенсификация аэробного
расщепления углеводов, липидов и синтеза энергии. Андрогены
снижают общее содержание липидов и выводят холестерин из организма. Но
последний эффект андрогенов выражен в значительно меньшей степени,
чем эстрогенов. Эстрогены весьма активно выводят холестерин и
снижают его уровень в крови, что, возможно, взаимосвязано с более редким
развитием атеросклероза коронарных и других сосудов у женщин по
сравнению с мужчинами. Эстрогены стимулируют также функционирование
пентозофосфатного пути и гликолиза, что способствует интенсификации
биосинтетических процессов с участием ИАВРН и рибозо-5-фосфата, а
также синтеза АТР.
Гормоны поджелудочной железы. В поджелудочной железе
помимо синтеза пищеварительных ферментов осуществляются также
образование и секреция ряда полипептидных гормонов, играющих
важнейшую роль в регуляции углеводного и липидного обмена. Синтез
гормонов происходит в специализированных клетках эндокринной
ткани поджелудочной железы, называемых островками Соболева—Лан-
герганса. В островках имеются клетки нескольких типов: А-типа,
образующие глюкагон; В-типа (или р-клетки), вырабатывающие
инсулин; В-типа, выделяющие соматостатин; РР-типа (или Р-клетки),
секретирующие панкреатический полипептид.
Глюкагон представляет собой одноцепочечный полипептид с
молекулярной массой около 3500. Активный глюкагон синтезируется из
неактивного предшественника —- проглюкагона, содержащего 37
аминокислотных остатков, путем протеолитического отщепления 8
аминокислотных остатков от С-конца проглюкагона. Проглюкагон, в свою очередь,
562
Глава 14. Молекулярные основы действия клеточных медиаторов
синтезируется из предшественника — препроглюкагона за счет
отщепления сигнальной полипептидной последовательности с Г^-конца (см. гл. 1).
Высвобождение глюкагона происходит в ответ на понижение уровня
глюкозы в крови. Под действием глюкагона повышается концентрация
глюкозы в крови за счет активации деградации гликогена. Эффект
глюкагона прямо противоположен действию инсулина. Тканями-мишенями
глюкагона являются, в первую очередь, печень, жировая ткань и, в
значительно меньшей степени, мышцы, что обусловлено наличием и
количеством определенных рецепторов (см. разд. 14.7). Мобилизация гликогена
печени и скелетных мышц, а также триацилглицеролов жировой ткани
осуществляется путем стимуляции аденилатциклазы и образования сАМР,
Наряду с повышением концентрации глюкозы в крови происходит
также возрастание содержания жирных кислот и кетоновых тел. Последние
образуются из ацетил-СоА, высвобождающегося при расщеплении
жирных кислот. Гипергликемический эффект глюкагона сопряжен также с
двумя взаимосвязанными процессами, а именно с подавлением гликоли-
тического распада глюкозы за счет ингибирования Ь-изофермента пиру-
ваткиназы (см. гл. 9, 10), с одной стороны, и со стимуляцией глюконео-
генеза — с другой. Вызываемая глюкагоном интенсификация распада
белков в печени способствует стимуляции глюконеогенеза, так как
образующиеся из белков аминокислоты могут быть использованы этим путем
для синтеза глюкозы.
Инсулин — это белок с молекулярной массой 5700,
синтезирующийся в виде одноцепочечного предшественника — проинсулина, из
препроинсулина путем отщепления И-концевой (лидерной)
аминокислотной последовательности. Проинсулин разных видов животных
содержит от 78 до 86 аминокислотных остатков. Он превращается в
активный инсулин за счет селективного протеолиза, в ходе которого
образуется полипептид, состоящий из двух цепей (А-цепь, содержащая
21 аминокислотный остаток, и В-цепь, содержащая 30 остатков),
удерживаемых вместе с помощью двух дисульфидных мостиков (рис. 14*18).
Инсулин
(показан серым)
К-конец
23 остатка
Дису л ьфид н ые
мостики
В-цепь
(30 остатков)
Сигнальная последовательность:
отщепляется при образовании
проинсулина
Выстригаемый
дипептид
С-пептид
(30 остатков)
Выстригаемый
дипептид
Рас. 14.18. Образование инсулина из предшественника — препроинсулина
36
*
563
Часть II. Метаболизм веществ и энергии в клетке
Секреция инсулина из р-клеток, происходит в результате
сложного процесса, при участии ионов Са2+, и зависит от концентрации
глюкозы в крови. Чем выше уровень глюкозы в крови, тем больше
выделение инсулина* Инсулин в крови может находиться в двух формах:
свободной и связанной с белками плазмы. Тканями-мишенями
инсулина являются мышечная и жировая ткани, печень. Рецепторы
инсулина представляют собой гликопротеины.
Под действием инсулина усиливается активный транспорт
глюкозы в печень и мышцы, где она превращается в гликоген. В результате
происходит снижение содержания глюкозы в крови и, как следствие,
уменьшается выделение инсулина. Но под действием инсулинрецеп-
торного комплекса происходит изменение проницаемости клеточных
мембран не только для глюкозы, но и для аминокислот и ряда ионов:
Ка+, К+, Са2+. Активация Ка+, К+-насоса, ведущая к возрастанию
мембранного N3+/ К+-градиента, способствует транспорту аминокислот в
клетку* При недостаточности инсулина развивается комплекс
метаболических сдвигов, обусловливающий сахарный диабет. При данном
заболевании уровень глюкозы в крови повышен; имеет место гипер-
гликемия. Наряду с этим наблюдается глюкозурия> т.е. большое
содержание глюкозы в моче. Кроме того, в организме больных
утрачивается способность к синтезу жирных кислот из липидов. Мобилизация
липидов резко возрастает, жирные кислоты быстро сгорают. При этом
развивается кетоз> обусловленный накоплением кетоновых тел в
организме, что сопровождается снижением рН крови и может привести к
летальному исходу. При дефиците инсулина может происходить
также торможение белкового синтеза, так как углеродные скелеты
аминокислот используются для образования глюкозы в процессе глюко-
неогенеза. Таким образом, в организме все доступные питательные
вещества используются для биосинтеза глюкозы крови, так как все
ткани нуждаются в глюкозе, но она выводится с мочой.
Для лечения сахарного диабета применяются препараты
инсулина. Однако не все сдвиги обменных процессов могут быть нормализо-
ваны с помощью инсулина. Так, поскольку при сахарном диабете
происходит нарушение синтеза базальной мембраны кровеносных
капилляров, то это приводит к повреждению коронарных сосудов, а также
сосудов сетчатки глаза, почек и т.д. В этой связи на поздних стадиях
диабета могут развиваться слепота, почечная недостаточность и
другие нарушения, которые уже не могут быть устранены путем введения
инсулина. При избытке инсулина в организме также возникают
патологические состояния, сопровождающиеся выраженной
гипогликемией* При наиболее тяжелых формах могут иметь место судороги со
смертельным исходом. В таких ситуациях своевременное введение
глюкозы или гормонов, способствующих повышению уровня глюкозы в
крови, устраняет симптомы заболевания. Избыточная секреция инсулина
может быть, в частности, при развитии опухолей островковой ткани.
564
Глава 14. Молекулярные основы действия клеточных медиаторов
,'■!■■ ,1 II ,1 I !■ ■ ,1 ■ ■■ 1| ,1 М ■ I1 ИМИ ,1 ■ | ■' I —**» I !-■
Соматостатин образуется не только в поджелудочной железе, но
и, как упоминалось выше, в гипоталамусе, откуда он впервые и был
выделен. Соматостатин, вырабатываемый в поджелудочной железе,
оказывает воздействие на образование глюкагона и инсулина. Его
положительный эффект при сахарном диабете, очевидно, взаимосвязан
с подавлением выделения глюкагона.
Панкреатический полипептид включает 36 аминокислотных
остатков. Его действие способствует выделению ферментов слизистой
желудка и экзокринными клетками поджелудочной железы.
Краткое содержание главы
Гормоны и ростовые факторы связываются со специфическими
рецепторами плазматической мембраны, которые конвертируют
внешний сигнал во внутриклеточный. После связывания лиганда с
рецептором О-белок, выполняющий роль передатчика сигнала,
активируется путем обмена связанного ОРР на СТР. Активированный С-
белок передает сигнал мембранному эффекторному ферменту,
который продуцирует один или более вторичных медиаторов. Эти
небольшие молекулы или ионы затем переносят сигнал внутрь клетки.
Вторичные медиаторы часто активируют протеинкиназы. Фосфорилиро-
вание аминокислотных остатков в полипептидных цепях этих киназ
приводит к изменениям функционирования белков-мишеней, что
вызывает клеточный ответ на первоначальный внешний сигнал.
В сигнальных процессах используется несколько общих путей
передачи сигнала. Один сигнальный путь приводит к опосредованной О-
белком активации мембраносвязанной аденилатциклазы,
продуцирующей вторичный медиатор сАМР. Это обратимо активирует протеин-
киназу А, относящуюся к серин-треониновым протеинкиназам. В
другом важнейшем пути О-белок активирует фосфолипазу С, которая
катализирует гидролиз липида фосфатидилинозитол-4,5-бисфосфата.
Один продукт (инозитол-1,4,5-трифосфат) увеличивает концентрацию
Са2+ в цитоплазме, тогда как другой продукт (диацилглицерол)
активирует протеинкиназу С. Многие факторы роста активируют тирозин-
киназный домен мембранных рецепторов, расположенный на участке
белковой молекулы, обращенном к цитоплазме клетки. Эта
активация приводит к фосфорилированию тирозиновых остатков рецептора
так же, как и других белков-мишеней.
Онкогены — гены, трансформирующие нормальные клетки в
раковые, часто несут информацию о видоизмененных белках,
обслуживающих сигнальные процессы, что ведет к неконтролируемой
клеточной пролиферации.
Гормоны — химические агенты с весьма высокой биологической
активностью, присутствующие в организме в малых количествах. Их
действие многосторонне. Они регулируют не только клеточный мета-
565
Часть П. Метаболизм веществ и энергии в клетке
болизм, но и работу внутренних органов, кровяное давление. Их
действие на тот или другой орган достигается благодаря присутствию в
определенных клетках специфических рецепторов.
Принято выделять три класса гормонов: пептидные, стероидные и
амины. К пептидным относятся гормоны гипоталамуса и гипофиза,
гормоны щитовидной и поджелудочной желез. К аминам
принадлежат гормоны, секретируемые мозговым слоем надпочечников, тиро-
идные гормоны- К стероидам относятся половые гормоны, гормоны
коры надпочечников.
Все гормоны функционируют в системе эндокринных связей,
координирующим центром которой является гипоталамус. Регуляция
гормональных процессов, связанных с эндокринной системой,
осуществляется по принципу обратной связи. Для гормонов характерна
строгая специфичность их действия.
Гормоны щитовидной железы секретируют йодтиронины, активно
воздействующие на метаболизм в тканях печени, сердца, почек,
скелетных мышц. В паращитовидной железе образуются пептидные
гормоны — кальциотонин и паратирин, участвующие в регуляции
уровня ионов Са2+ и фосфатов крови. Гормоны надпочечников — адрена-
лин и норадреналин участвуют в метаболизме углеводов и липидов.
Кора надпочечников синтезирует кортикостероиды. Их принято
подразделять на три группы. К первой группе относятся глюкокортикои-
ды, действие которых направлено на углеводный обмен. Ко второй
группе относятся минералокортикоиды, регулирующие в организме
водно-солевой баланс. К третьей группе принадлежат стероиды,
характеризующиеся промежуточными свойствами между первой и
второй группами. К стероидам относятся половые гормоны, соотношение
которых определяет половые признаки организма. В поджелудочной
железе помимо синтеза пищеварительных ферментов осуществляются
также образование и секреция ряда полипептидных гормонов, играю-
щих важную роль в регуляции обмена углеводов и липидов. К этим
гормонам относятся инсулин и глюкагон.
Хронология важнейших дат в исследовании молекулярных основ
действия клеточных медиаторов
1901 г. — Дж. Такамине с сотр. выделили из мозгового слоя
надпочечников адреналин.
1902 г. — В. Бейлисс, Э. Старлинг впервые предложили термин
"гормон'* (от греч. Ногтато — привожу в движение, побуждаю).
1902 г. — Л. В. Соболев установил, что островки Лангерганса
выполняют внутрисекреторную функцию.
1910 г, — М. Н. Чебоксаров впервые показал влияние нервного
раздражения на секрецию адреналина.
1910 г. — Г. Эппингер, Л. Гесс впервые предложили
адреналиновую пробу как метод диагностики функциональных нарушений нер-
566
Глава 14. Молекулярные основы действия клеточных медиаторов
вной и эндокринной систем организма по характеру реакций этих
систем на парентеральное введение адреналина,
1921 г. — Ф. Бантинг, С. Бест получили экстракт из островковой
ткани поджелудочной железы, содержащий инсулин.
1929 г. — А. Бутвнандт, Э. Дойзи выделили женский половой
гормон эстрон и получили его в кристаллическом состоянии.
1931 г. — А. Бутенандт получил в кристаллическом виде мужской
половой гормон андростерон.
1953 г. — Ф. Штрауб получил глюкагон в гомогенном состоянии.
1955 г. — Ф. Сенгер изучил аминокислотную последовательность
инсулина и установил его структуру. В 1958 г, ему присуждена
Нобелевская премия.
1955 г. — Э. Сазерленд выяснил механизм действия адреналина и
глюкагона на распад гликогена печени.
1962 г. — Д. Кооп впервые указал на способность кальцитонина
поддерживать определенный уровень кальция в крови.
1966 г. — Д. Стайнер впервые выделил проинсулин.
1967 г. — Л. Крулих открыл гормон соматостатин, подавляющий
секрецию соматотропина.
1992 г. — Э. Кребс и Э. Фишер удостоены Нобелевской премии за
открытие класса протеинкиназ и фосфатаз, играющих важные роли в
сигнальных процессах.
ЛИТЕРАТУРА
Албертс Б., Брей Д., Льюис Дж.% Рэфф М., Роберте К., Уотсон Дж.
Молекулярная биология клетки. — М.: Мир, 1994, — Т. 2. — 540 с,
Березое Т. Т., Коровник Б. Ф. Биологическая химия: Учебник. — 3-е изд.,
перераб. и доп. — М.: Медицина, 1998. — 704 с,
БышевскийА. ЯГ., Терсенов О. А. Биохимия для врача. — Екатеринбург: Изд-
во "Уральский рабочий", 1994. — 383 с.
Ленинджер А. Основы биохимии: В 3 т. — М.: Мир, 1985, — Т. 3. — С- 743—
1056.
Марри Р., Гренер Д., Мейес П., Родуэлл В, Биохимия человека: В 2 т. — М.:
Мир, 1993. — Т. 2. — 414 с,
Маршалл В. Дж. Клиническая биохимия. — М.: Изд-во "БИНОМ", СПб.:
Изд-во "Невский Диалект", 1999. — 357 с.
Сгпрайер Л. Биохимия: В 3 т. — М.: Мир, 1985. — Т. 3, — 397 с.
Строев Е. А. Биологическая химия. — М.: Высшая школа, 1986. — 478 с.
Могап X,. А, 8спт§еоиг К. С, НоПоп Н* Л. ОсНв В. 8„ Каюп ^^ В. ВюсЬетийгу.
— 2 Ей. — ЫеИ РаПегзоп РиЬ., Епв1еигоос1 СИЯв, 1994. — 1200 р.
ВОПРОСЫ И УПРАЖНЕНИЯ
1. Охарактеризуйте основные ступени принципиальной схемы сигнальной
транедукции. Какую роль выполняют мессенджеры, транедукторы, эффектор-
ные ферменты, вторичные медиаторы?
2. Какие факты свидетельствуют о специфичности систем, воспринимающих
клеточные сигналы на поверхности клетки? Что указывает на общность путей
передачи этих сигналов внутри клетки?
567
Часть II. Метаболизм веществ и энергии в клетке
3, Механизм действия каких веществ представляет собой исключение из об*
щей схемы сигнальной трансдукции?
4* Перечислите основные сигнальные пути в клетках животных и дайте им
краткую характеристику.
5. Роль протеинкиназы А в аденилатциклазном сигнальном пути.
Объясните, почему полный комплекс субъединиц (две регуляторные
субъединицы плюс две каталитические субъединицы) не способен обеспечивать фос-
форилирование ферментов-мишеней. Что необходимо для активации
протеинкиназы А:
а) повышение концентрации серина;
б) повышение концентрации сАМР;
в) снижение концентрации сАМР;
г) снижение концентрации серина?
6. Какая форма О-белков активна:
а) ОТР-связанная;
б) ОВР-связанная;
в) ОТР-связанная;
г) мономерная?
7. Какой из продуктов гидролиза фосфатидилинозитол-4,5-бисфосфата (ФИФ3)
обеспечивает:
а) возрастание концентрации ионов Са2+;
б) активирует протеинкиназу?
8. За счет чего достигается избирательность гормональных ответов?
9. Классификация гормонов.
1) Тиреоидные гормоны принадлежат к:
а) стероидным гормонам;
б) аминам;
в) пептидам.
2) Гормоны гипофиза принадлежат к:
а) аминам;
б) стероидам;
в) пептидам.
10. Значение прогормонов.
Опишите особенности структуры активного и неактивного инсулина.
Секреция инсулина в активной форме усиливается при повышении концентрации:
а) протеинкиназы А;
б) глюкозы крови;
в) Са2+ в цитоплазме.
Часть III
ЭЛЕМЕНТЫ
ПРИКЛАДНОЙ
БИОХИМИИ
Выделение
ДНК
Амплификация
мкмг-
"*!:;;
А. *
■/ #«;
^У^
—*а!Ш
А*4 >^ . 1»ч.! ■ _„| \*.\:Л ■ — *^
*«
ОМАТес1то1оду Цс1
Глава 15. НЕКОТОРЫЕ АСПЕКТЫ
ЭКОЛОГИЧЕСКОЙ БИОХИМИИ
Экология (от греч. оЫгоз — дом, место обитания) — наука,
изучающая совокупности живых организмов, взаимодействующих между
собой и со средой обитания и образующих с нею единую систему, в
которой протекают процессы трансформации вещества и энергии.
В недалеком прошлом экология изучала только высшие уровни
организации, начиная с организма и кончая экосистемой. Вопросы
взаимодействия экосистем на молекулярном уровне не затрагивались.
Однако единство всех уровней организации биологической материи
обусловливает необходимость комплексного рассмотрения экосистем,
что приводит к выводу о целесообразности специального изучения роли
химических и физико-химических процессов в механизмах
функционирования экосистем.
В предмет экологической биохимии входит изучение конкретных
биохимических механизмов, с помощью которых осуществляется
приспособление (адаптация) организмов к окружающей среде.
Экологическая биохимия включает биохимические аспекты метаболизма
экологически важных веществ и биохимические механизмы
обезвреживания токсинов. Иными словами, в экологической биохимии акцент
делается на химические взаимосвязи между живыми системами. В
настоящее время наряду с названием "экологическая биохимия"
существуют близкие к ней термины "биохимическая экология" или даже
"химическая экология". Следует отметить, что цели и задачи
экологической биохимии и биохимической экологии различны.
Экологическая биохимия на первый план выдвигает биохимическое описание
предмета. В задачу биохимической экологии входит описание
вопросов общей экологии с включением определенных аспектов биохимии.
Проблемы экологической биохимии весьма важны сегодня. Их
решение позволит достаточно глубоко представить ту роль, которую иг-
рают в биосфере метаболические циклы. Особенно важны
биохимические исследования загрязнений, связанных с деятельностью
человека. Они дают возможность выработать ответственные решения,
способные повлиять на будущее всего живого на Земле. Биохимические
подходы имеют большое значение в решении проблемы мутаций и
устранения тех последствий, которые вносят в биосферу
синтетические химические материалы, число которых растет из года в год.
Особенно опасны загрязнения биосферы радионуклидами. Никто иной,
571
Часть III. Элементы прикладной биохимии
как биохимики, хорошо представляют себе, к каким йоследствиям
могут привести мутации, порождаемые радиоактивным облучением.
Именно они должны указать человечеству на грядущую опасность для
его существования.
В одной главе представить исчерпывающий материал по
проблемам экологии — весьма трудная задача. Однако ознакомление с
предыдущими главами книги дает возможность решить некоторые из них.
В этой главе рассматриваются следующие вопросы:
1. Адаптация организмов к изменениям условий внешней среды,
типы биохимической адаптации.
2. Роль веществ вторичного происхождения в экологических
процессах.
3. Токсины растений.
4. Загрязнение окружающей среды и механизмы процессов деток-
сикации.
15,1. Адаптация организмов к изменениям условий внешней среды
Адаптация — способность живых организмов приспосабливаться
к изменяющимся условиям окружающей среды, ведущая к
сохранению (или даже повышению) их выживаемости и
самовоспроизведению. Адаптация — одна из центральных проблем современной
биологии. В литературе, однако, по вопросам адаптации чаще всего
обсуждаются высокоспециализйрованные приспособления на
физиологическом, морфологическом, поведенческом уровнях. В своем подходе к
проблеме адаптации мы сосредоточим внимание на биохимических
механизмах, которые определяют ход метаболических процессов,
постоянство состава внутриклеточной среды. С позиции биохимии
адаптация сохраняет в клетке функции макромолекул (ферментов, белков,
нуклеиновых кислот), обеспечивает целостность надмолекулярных
структур, в том числе и биоэнергетических, необходимых для
нормального протекания метаболических процессов.
На адаптацию растений оказывают влияние два основных типа
факторов — климатические и почвенные. К ним относятся
температура, интенсивность освещения, длина солнечного дня, влажность и
засоленность почвы.
Многие растения способны выживать в суровых условиях
северных широт и арктических районов Земли. Адаптация к холоду у них,
по-видимому, тесно связана с повышенным содержанием Сахаров в
клеточном соке. Состав Сахаров у холодоустойчивых растений
варьирует от растения к растению. Чаще всего это три обычных сахара:
глюкоза, фруктоза, сахароза с примесью таких олигосахаридов, как
р^финоза. По всей вероятности, они обеспечивают растениям защиту
от мороза по крайней мере двумя способами: 1) повышенным
осмотическим давлением, которое препятствует образованию в вакуолях боль-
572
Глава 15. Некоторые аспекты экологической биохимии
ших количеств кристаллов льда; 2) метаболическим эффектом; в этом
случае сахара превращаются в цитоплазме в различные строительные
блоки и тем самым выполняют дополнительную защитную функцию*
Многие растения (ольха, осина, лоза и др.) растут в долинах рек,
их корневая система хорошо адаптировалась к затоплению.
Установлено, что корни таких растений обладают способностью перестраивать
свою дыхательную систему при переходе из аэробных условий в
анаэробные. При затоплении корни растения лишаются кислорода, и здесь
срабатывает эффект Пастера: образовавшийся при гликолизе пируват
в конечном счете превращается в этанол, который как токсический
продукт из корней выбрасывается в воду. Предполагается также, что
у некоторых растений в процессе гликолиза промежуточные
продукты, например фосфоэнолпируват, превращаются не в токсичный
этанол, а в иные продукты, такие, как малат, лактат, аланин,
безвредные для затопленных корней.
Другая форма адаптации — у пустынных и высокогорных
растений, произрастающих в условиях острого недостатка воды: эти
растения преодолевают засуху благодаря морфологическим и
анатомическим приспособлениям. Поверхность таких растений покрыта Толстым
воскообразным слоем; листья у них заменены колючками, которые
сводят до минимума потерю воды на испарение.
Многие растення-галофиты произрастают на засоленных почвах
(соленые болота, морские побережья, пустыни-солончаки). Галофиты
устойчивы к высоким концентрациям МаС1, некоторые из них
способны выдержать до 20 % ИаС1 в почве, хотя уровень хлорида натрия в
наиболее засоленной почве много ниже (2—6 %)♦ Эти растения имеют
специфические механизмы, ограничивающие поступление в них соли.
Многие из них способны накапливать соль в вакуолях.
Как у галофитов, так и у засухоустойчивых растений, способных
противостоять большим осмотическим давлениям в почве,
обнаружено большое количество пролина: его концентрация может в 10 раз
превышать нормальный уровень. Однако каким образом высокие
концентрации пролина в цитоплазме растений обеспечивают соле- и
засухоустойчивость, точно не установлено. Существенный вклад в
устойчивость к солевому стрессу вносят четвертичные аммонийные
соединения — холин и бетаин.
Продолжительность биохимической адаптации варьирует в
широких пределах — от длительных периодов, необходимых для
эволюционного изменения организма, до долей секунды. С точки зрения
скорости биохимической адаптации её можно подразделить на
генетическую адаптацию, акклиматизацию, немедленную адаптацию.
Генетическая адаптация является длительным процессом. Она
идет на протяжении многих поколений. Популяция может
использовать различные пути приспособления. Мутации генов могут привести
к изменению активности ферментов; аминокислотные замены в бел-
573
Часть III. Элементы прикладной биохимии
ках обусловят появление новых изоферментов, а в некоторых
случаях — ферментов совсем нового типа. Генетическая адаптация может
обеспечить выработку новых способов регуляции метаболических про-
цессов, привести к синтезу белков нового типа, придающих
организму устойчивость к экстремальным условиям внешней среды.
Акклиматизация — такой процесс адаптации, при котором
организм приспосабливается к нескольким изменяющимся параметрам
внешней среды. Главное отличие этой адаптации от генетической
состоит в том, что в этом случае используются те характеристики
организма, которые уже закодированы в его геноме. Здесь имеют место
существенная активизация отдельных сторон метаболического
процесса, значительная перестройка клеточных структур.
Немедленная адаптация происходит очень быстро и, вполне
понятно, не связана с какими-либо изменениями в геноме; не
происходят и существенные перестройки клеточных структур. Немедленная
адаптация нередко осуществляется путем изменения активности
ферментов или существенного возрастания биосинтеза ферментов, в
которых организм нуждается в данный момент. Особенно примечательны
примеры по немедленной адаптации при культивировании
микроорганизмов на различных питательных средах. Об индуцированном
биосинтезе Р-галактозидазы бактериями Е. сой мы уже говорили в гл.13,
рассматривая общую теорию индукции и репрессии биосинтеза
ферментов у микроорганизмов. При внесении в питательную среду
лактозы синтез р-галактозидазы возрастает в тысячу раз в весьма короткий
промежуток времени. Как правило, индуцированный биосинтез
ферментов микроорганизмами развивается на средах, в которые вносятся
специфические для синтеза этих ферментов субстраты.
15.2. Роль веществ вторичного происхождения
в процессах экологических взаимодействий
Вещества вторичного происхождения в экологических процессах
выполняют самые разнообразные функции. Установлено, что вещества
вторичного происхождения принимают участие в опылении растений, в
размножении насекомых. Многие из них являются пищевыми аттрактанта-
ми, участвуют в защите растений от насекомых и животных.
Опыление растений. Взаимоотношения насекомых с цветущим
растением устанавливаются благодаря трем основным биохимическим
факторам: окраске цветков, запаху и питательной ценности нектара и
пыльцы. Процесс опыления приносит пользу каждой из сторон:
нектар и пыльца растений используются насекомым как источник пита*
ния, а цветки при переносе пыльцы оплодотворяются.
Окраска цветков обусловлена в основном флавоноидами и кароти-
ноидами, находящимися в хлоропластах и вакуолях клеток
лепестков. Окраску от оранжевой и красной до белой, желтой, голубой при-
574
Глава 15. Некоторые аспекты экологической биохимии
дают лепестку цветка флавоноиды. Желтая окраска с оттенками
оранжевой и красной зависит от наличия в лепестках каротиноидов. Циа-
нидины дают малиновую, ярко-красную окраску; они же на фоне
каротиноидов дают коричневую окраску.
Важную роль в опылении играет аромат цветка, К нему очень
чувствительны пчелы. У многих растений максимум образования
пахучих веществ приходится на момент созревания пыльцы и готовности
цветка к оплодотворению. Особую роль в создании аромата цветков
играют моно- и сесквитерпены, простые алифатические спирты, кето-
ны, сложные эфиры.
Иногда специфический аромат цветка может быть обусловлен лишь
одним соединением. Однако, как правило, аромат представляет собой
комплекс нескольких компонентов; один из них может существенно
усиливать эффективность остальных, создавая при этом
неповторимую специфичность аромата.
С помощью окраски и аромата цветков растение привлекает
насекомых к себе и использует их для опыления* Однако основной
причиной, побуждающей насекомых посещать цветущие растения,
является использование в качестве пищи нектара и пыльцы. Нектар
представляет собой простой раствор углеводов, имеющий сладковатый вкус.
Концентрация Сахаров обычно варьирует от 15 до 75 % на сухую
массу, основными из которых являются глюкоза, фруктоза и сахароза. В
нектаре некоторых растений обнаружены в небольших количествах
рафиноза, трегалоза, мелибиоза. Содержание белка, аминокислот и
липидов в нектаре незначительно. Однако химический состав нектара
вполне обеспечивает потребности насекомого-опылителя в пищевых
компонентах и энергии.
Ценной пищей для пчел, жуков является пыльца. Она содержит
белки (15—30 %), крахмал (1—7 %), сахара (2—15 %) и жиры (5—
10%). В следовых количествах присутствуют витамины и
минеральные вещества. Пыльца большинства растений окрашена, что связано
с наличием в ней каротиноидов и флавоноидов.
Аттрактанты и репелленты. Все вещества, определяющие
пищевое поведение животных, подразделяются на два типа: 1) аттрак-
тантыу или пищевые стимуляторы и 2) репелленты и токсины.
В настоящее время хорошо изучены пищевые аттрактанты
насекомых. Они включают широкий круг веществ вторичного
происхождения: гликозиды, алкалоиды, флавоноиды, терпены, эфирные масла.
Эти соединения выполняют в организме насекомых и ряд других
полезных функций, например, служат имитаторами гормонов или
являются эффективными антиоксид антами, препятствующими развитию
нежелательных процессов свободнорадикального окисления.
Действие аттрактантов можно рассмотреть на примере тутового
шелкопряда, источниками пищи у которого являются исключительно
листья шелковицы. Монотерпены листьев шелковицы играют роль
575
Часть III. Элементы прикладной биохимии
обонятельных аттрактантов. Инозитол, систостерол и сахароза
служат для гусениц вкусовыми факторами.
Интересным веществом, содержащимся в листьях капусты,
является тиогликозид синигрин. В результате его гидролиза и химической
перегруппировки выделяется горчичное масло — аллилизоцианат,
обладающее едким, острым вкусом. Синигрин играет роль аттрактанта
у бабочки-капустницы и, более того, служит стимулятором для
откладки яиц. Синигрин также привлекает к капусте капустную тлю.
Пищевое поведение тлей осуществляется преимущественно с помощью
двух факторов — концентрации синигрина и соотношения свободных
аминокислот в листьях. Для многих же насекомых синигрин
является репеллентом.
Практически любому вторичному соединению отведена роль
специфического стимулятора питания. Так, ядовитый алкалоид спарте-
ин действует как стимулятор при кормлении тлей. Тли умеют
выискивать на растении места для кормления с наиболее высокой
концентрацией спартеина.
Флавоноиды также играют роль пищевых аттрактантов для
насекомых. Так, ильмового жука-короеда привлекает к коре вяза кате-
хин-7-ксилозид. Флавоны, по-видимому, служат пищевыми аттрак-
тантами для жуков, кормящихся на коре плодовых деревьев.
Важным средством защиты растений от поедания являются танни-
ны, содержащиеся в относительно высоких концентрациях в листьях
древесных растений. Особенность таннинов заключается в том, что
они имеют вяжущий вкус, вызывают раздражение языка. Поэтому
таннины можно отнести к репеллентам для высших животных, пре-
смыкающих и, видимо, насекомых. Вяжущий вкус является для
животных своеобразным сигналом о низкой питательной ценности
данных веществ. Действительно, питательная ценность белков листа
резко снижается в связи с образованием трудноперевариваемого белково-
таннинового комплекса.
Активным репеллентом для колорадского жука является
алкалоид демиесин, выделенный из диких видов картофеля,
произрастающего в Южной Америке, предохраняющий картофель от поедания этим
насекомым. Алкалоид соланин культурных сортов картофеля хотя и
близок по структуре к демиссину, однако от нападения колорадского
жука не предотвращает. Было установлено, что между структурой
молекулы демиссина и ее способностью быть репеллентом существует
корреляция. Для проявления репеллентности необходимы три
особенности структуры демиссина: 1) присутствие тетрасахарида; 2)
присутствие в качестве одного из компонентов тетрасахарида ксилозы; 3)
отсутствие двойной связи в стерольном остатке молекулы демиссина.
Если, как в соланине, находится трисахарид и отсутствует ксилоза
или имеется двойная связь в остатке стерола, то вещество теряет
свойства репеллента.
576
Глава 15. Некоторые аспекты экологической биохимии
Главный алкалоид томатов и некоторых сортов картофеля тома-
тин, как и демиссин, содержит тетрасахарид с ксилозой и не имеет в
молекуле двойной связи. Это соединение является таким же
активным репеллентом, как и демиссин.
Репеллентные свойства демиссина важны с практической точки
зрения. Скрещивание диких видов картофеля с культурными сортами
может дать гибриды, устойчивые против колорадского жука, который в
настоящее время наносит немалый вред при выращивании картофеля.
К группе веществ, защищающих растения от чрезмерного
истребления животными, относятся цианогенные гликозиды. В
естественных условиях травоядные животные предпочитают нецианогенные
формы растений. К довольно сильным репеллентам относятся
алкалоиды, обладающие горьким вкусом.
Особой группой аттрактантов являются феромоны — вещества,
используемые для общения насекомых в пределах вида. Феромоны
имеют небольшую молекулярную массу (в пределах 80—300) с числом
атомов углерода от 5 до 20, обладают высокой летучестью, стойким
запахом, переносятся по воздуху.
Наиболее хорошо изучены половые феромоны — соединения,
выделяемые самками насекомых для привлечения самцов. Большинство
феромонов являются производными углеводородов (ненасыщенные
спирты, ацетаты, кетокислоты). Среди них лучше всего изучена 9-
кетодецековая кислота, выделяемая пчелиной маткой и
привлекающая трутней.
Половые феромоны заслуживают внимания в сельском хозяйстве
для борьбы с насекомыми-вредителями. Установка на месте обитания
насекомых-вредителей специальных ловушек с феромоном дает
возможность отловить и уничтожить самцов и тем самым
воспрепятствовать размножению насекомых-вредителей.
Интересная разновидность феромонов — следовые феромоны. Это
вещества-метчики, испускаемые общественными насекомыми
(пчелами, муравьями, термитами); служат ориентирами на пути насекомого
от гнезда к пище и обратно. Химическая структура следовых
феромонов разнообразна. Муравьи-листорезы вырабатывают очень активное
вещество — метиловый эфир метилпирролкарбоксилата. Монотерпен
гераниол является следовым феромоном для медоносной пчелы.
Некоторые сесквитерпены являются еще одной разновидностью
феромонов — феромонов тревоги. Эти вещества выделяются
насекомыми, заметившими опасность. У некоторых муравьев они
представлены простыми углеводородами, такими, как ундекан, тридекан, пен-
тадекан, и производными этих соединений — альдегидами и кетона-
ми. У тлей феромоном тревоги служит моноциклический сесквитер-
пен гермапрен А.
Многие терпены относятся к токсинам и используются
насекомыми как средство для защиты. Они летучи и сильно пахнут, их запаха
37. Заказ 3486
577
Часть III. Элементы прикладной биохимии
может быть достаточно, чтобы отпугнуть нападающее животное. Пары
этих терпенов могут оказывать раздражающее действие, а их капли,
попавшие на нападающего, могут вызвать жжение и зуд. Одним из
ярких примеров использования млекопитающими феромонов
является реакция скунса на испуг. Животное выбрасывает пахучий секрет
из анальных желез. Запах секрета для человека весьма неприятен и
заставляет его удаляться от животного. Этот запах предупреждает
других скунсов и об их опасности; феромон представляет собой
серосодержащее соединение изопентилмеркаптан: СН3-СН-СН2-СН2-8Н.
СНз
В смоле сосны и в защитном секрете некоторых насекомых
содержится смесь моно- и дитерпенов. Среди этих соединений найдены ос- и
Р-пинен. Для сосны смола — эффективное средство защиты от
насекомых.
Известны стероидные токсины, действующие на сердечную
мышцу позвоночных. Они используются лягушками и жабами как
защитные вещества. Стероидной природы голотурии синтезирует морской
огурец, чтобы отпугивать хищников, особенно рыб. Защитные
стероиды обнаружены в крови некоторых светлячков, предохраняющих их
от поедания птицами, ящерицами и некоторыми млекопитающими.
Очень эффективные стероидные соединения имеют жуки-плавунцы.
Хищные рыбы, съевшие такого жука, впадают в наркотическое
состояние, и у них вырабатывается рефлекс в дальнейшем избегать
нападения на плавунца.
Некоторые алкалоиды, фенолы также служат средством защиты.
Божьи коровки синтезируют алкалоид Ы-оксидкокцинеллин, который
защищает их от хищников (муравьев, птиц). Защитные фенолы
вырабатывают жуки-бомбардиры. При нападении хищника они
выбрасывают на него горячее облако токсина. Температура секрета
поднимается до 100 °С. При выбрасывании токсина развивается реакция, в
которой участвуют гидрохинон, Н202 и фермент каталаза:
ОН о
Каталаза .11
+ Н202 *- Г || +2 Н20
ОН О
Гидрохинон Бензхинон
Гидрохинон окисляется до бензхинона — главного защитного веще
ства. Реакция экзотермическая, происходит со взрывом и
сопровождается звуком, подобным выстрелу из пистолета. Пары бензхинона
вызывают сильное раздражение, прежде всего повреждая ткани глаза.
Благодаря стойкому и неприятному запаху хиноны оказывают ре-
пеллентное действие на нападающих врагов.
578
Глава 15. Некоторые аспекты экологической биохимии
15.3. Аллелопатические хемоэффекторы и токсины растении.
Механизмы их обезвреживания
В борьбе за существование растения конкурируют друг с другом за
свет, влагу и питательные вещества почвы. В этой конкуренции они
вырабатывают разнообразные средства защиты друг от друга. Защита
растения с помощью синтезированного им химического вещества
называется аллелопагпией.
Химические вещества, участвующие во взаимодействиях высших
растений, называют соответственно аллелопатическими веществами.
Большинство из них являются терпенами или фенолами — летучими
соединениями относительно простого строения. Чаще всего
аллелопатические вещества используются растением одного типа для борьбы с
растениями другого типа: например, кустарник или дерево
конкурируют с злаком или травянистым растением.
Приведем примеры аллелопатического действия химических
соединений. Черный орех оказывает токсическое действие на растения,
находящееся рядом с ним (сосна, картофель, злаки, яблони, томаты).
Растения погибают под действием токсина, попавшего в почву вместе
с листьями и ветвями ореха. Этот токсин представляет собой 4-глюко-
зид-1,4,5-тригидроксинафталин, который в почве гидролизуется и
окисляется, превращаясь в нафтохинон — юглон:
ОН
Гидролиз
Окисление
ОН О-Глюкоза
4-Глюкозид-1,4,5-три-
гидроксинафталин
ОН О
Юглон
(5-гидроксинафтохинон)
Юглон представляет собой водорастворимый желтый пигмент. Его
токсичность связана с ингибированием прорастания семян.
Пустынные растения конкурируют за воду, подавляя вблизи себя рост
однолетних растений. Так, кустарник энселия (пустыня Могаба,
Центральная Калифорния) выделяет токсин З-ацетил-6-метоксибензальдегид,
который образуется в листьях, а затем, когда листья опадают на землю,
он высвобождается и сохраняется в неизменном виде, предотвращая рост
других растений рядом. Другой токсин, выделенный из растений
пустынь, — коричная кислота — обладает таким же эффектом. Аллелопати-
ческое действие имеют гидроксибензойная и гидроксикоричная
кислоты, эффективно подавляющие прорастание семян.
Важным видом защиты растений является биосинтез
специфических химических соединений, защищающих растения от
болезнетворных организмов и получивших название фитоалексины.
37*
579
Чкгстъ III. Элементы прикладной биохимии _„
? —
Из сердцевины древесных растений выделены терпены и
фенолы, предохраняющие деревья от грибковых заболеваний. Из яблок
выделен цианидин, предохраняющий их от гниения. У многих
растений вблизи места бактериального заражения накапливаются
различные ароматические соединения. Так, у картофеля в местах
заражения концентрация этих соединений увеличивается в 2—3 раза.
Многие фитоалексины образуются, когда растение-хозяин входит в
контакт с грибом-паразитом, подавляя его развитие. К таким
соединениям относятся пизатин, бензойная кислота. Их биосинтез
резко возрастает в условиях стресса (УФ-радиация, температурный
шок).
Токсины играют ключевую роль в защите растений от
травоядных. Стероидный алкалоид соланин, например, присутствует во всех
видах картофеля. В небольшом количестве он редко представляет
опасность при употреблении картофеля. Смерть от отравления соланином
реальна только при чрезмерно высоких концентрациях его в тех
клубнях, которые либо проросли, либо находились на поверхности почвы
и в результате чего "позеленели". В таких случаях наступает
летальный исход от расстройства дыхания.
Животные, как правило, "осведомлены" о присутствии токсинов в
растении, даже не прикасаясь к нему. Так, предупредительным
сигналом о наличии синильной кислоты является запах "горького
миндаля", алкалоидов и сапонинов — горький вкус. Сигналом
токсичности ягод белладонны служит их зловещий темно-пурпурный цвет.
Все токсины растений можно подразделить на два класса: 1)
азотсодержащие и 2) без азотистые токсины. -
Наиболее простой группой азотсодержащих токсинов являются
небелковые аминокислоты. Они широко распространены в
растительном мире, их обнаружено около 300. Являясь антиметаболитами той
или иной белковой аминокислоты, они участвуют в образовании
дефектного биологически неактивного белка либо подавляют синтез
какой-либо белковой аминокислоты. В частности, в семенах бобовых
обнаружена азитидин-2-карбоновая кислота, ингибирующая синтез и
утилизацию пролина.
К группе азотсодержащих токсинов относятся цианогенные гли-
козиды, их более 600. Сами по себе цианогенные токсины
безвредны. При ферментативном разрушении они выделяют синильную
кислоту, которая блокирует цитохромную систему, подавляя
дыхание. Это приводит к гибели организма. Одним из характерных
источников НСИ являются семена горького миндаля, содержащие
гликозид — амигдалин. Токсичность 1КДО такова, что ежегодно
регистрируются случаи отравления людей и домашнего скота. Путь
образования НСИ из цианогенных гликозидов можно представить в
виде следующей реакции:
580
Глава 15. Некоторые аспекты экологической биохимии
Кл О-Глюкоза Глюкозидаза К, ОН Спонтанно ^1
/Сч —-=^- ^ /Сч — *- С=0 + НСИ
2
К* ™ Глюкоза К* С* К
Гликозид Цианогидрин Кетон
Все растения, в которых образуются цианогенные гликозиды,
содержат специфические гликозидазы, осуществляющие гидролиз гли-
козидов. В результате гидролиза образуются сахар (обычно глюкоза)
и промежуточный продукт цианогидрин, который затем спонтанно
разлагается с образованием кетона или альдегида и НСЫ",
У некоторых растений накапливаются гликозиды, которые при
расщеплении выделяют токсичные нитраты, Азотсодержащие
гликозиды горчичного масла, присутствующие в растениях в достаточно
большом количестве, относятся также к токсинам. Выделяемые при
расщеплении этих гликозидов изоцианаты вызывают слюнотечение,
раздражение полости рта.
Самой известной группой азотсодержащих токсинов являются
алкалоиды. Об их физиологическом действии мы уже говорили в гл. 6.
Поглощаемого с пищей количества алкалоидов может быть
недостаточно для летального исхода животного, но оно может быть вполне
достаточным, чтобы вызвать врожденные уродства потомства. К
таким соединениям относятся алкалоиды пиролизиновой и
никотиновой групп, а также производные пиперидина — кониин болиголова.
Безазотистые токсины имеют структуру терпенов или
достаточно простых углеводородов. Из терпенов следует отметить две особо
токсичные группы — сердечные гликозиды и сапонины, а также
токсины микробного происхождения — афлотоксины. Сердечные
гликозиды имеют стероидную структуру и обладают кардиотонической
активностью. Их примерами служат строфантин, олеалдрин, дигиток-
син. Они ингибируют мембранные АТРазы. Гриб Азрег§Ши8 /1аии8
синтезирует четыре афлотоксина, вызывающие разрушение печени.
Выделяемые микромицетами афлотоксины представляют собой
значительную угрозу для животных, поскольку они заражают корма.
Для обезвреживания токсинов в организме животных
функционируют специфические системы детоксикации. Детоксикация сводится
к тому, что токсин переводится в безвредную форму и вместе с мочой
или фекалиями выводится из организма. Важную роль в
детоксикации играют оксигеназы; как правило, они локализованы в
микросомах печени.
Оксигеназы делятся на монооксигеназы и диоксигеназы.
Содержание монооксигеназ в тканях относительно велико. Монооксигеназы
катализуют реакции, в которых происходит включение одного из
атомов кислорода молекулы 02 в молекулу органического субстрата, а
581
Часть III. Элементы прикладной биохимии
второй атом кислорода восстанавливается до Н20. Большинство моно-
оксигеназ использует в качестве донора водорода, необходимого для
образования воды, КАВРН. В этом случае суммарное уравнение
реакции имеет вид:
КН + КАВРН + Н+ + 02 -> КОН + МАОР+ + Н20.
В связи с тем, что эти ферменты окисляют одновременно два
субстрата, их часто называют оксигеназами со смешанной функцией.
Существуют монооксигеназы, использующие в качестве косубстратов
ЫАВН, восстановленные флавиновые нуклеотиды — КАОН2 и ГЬШН2
а-кетоглутарат. К монооксигеназным реакциям относятся
многочисленные реакции, протекающие с участием цитохромов Р-450. Цито-
хромы Р-450 относятся к группе гемопротеинов. Название этих
ферментов обусловлено тем, что комплекс, образуемый восстановленной
формой тема в составе цитохрома с оксидом углерода, имеет
максимум поглощения при 450 нм. Для гемоглобина и миоглобина
максимум поглощения аналогичного комплекса — при 420 нм, Цитохромы
Р-450 в печени катализируют реакции гидроксилирования чуждых
для организма соединений. Такие вещества называют
ксенобиотиками (от греч. хепоз — чужой). Это, как правило, липофильные
соединения, попадающие в организм извне или же образующиеся как
побочные продукты в ходе различных реакций. С помощью цитохромов Р-450
может осуществляться, например, гидроксилирование лекарственных
препаратов. Образующиеся продукты имеют большую растворимость
в воде и значительно легче выводятся из организма. Еще одним
примером реакций, протекающих с участием цитохромов Р-450, является
гидроксилирование стероидов в процессе образования гормонов коры
надпочечников. Обычно функционирование цитохромов Р-450
сопряжено с работой цитохром Р-450 редуктаз, обеспечивающих
восстановление за счет водорода NА^РН флавинового нуклеотида в составе
белка. Цитохром Р-450 существует в различных формах, специфично
взаимодействующих с теми или иными субстратами. Диоксигеназы
катализируют реакции, в ходе которых оба атома молекулы 02
включаются в состав органического субстрата. В качестве примера можно
рассмотреть реакцию, катализируемую пирокатехазой:
О
Пирокатехаза ^—- С—ОН
+ 0=0 *-
^'ХШ ^— С— ОН
Катехол Молекулярный О
кислород
В ходе данной реакции происходит окисление катехола молеку
лярным кислородом, что приводит к раскрытию его кольцевой струк
туры.
582
Глава 15. Некоторые аспекты экологической биохимии
Многочисленными исследованиями показано, что домашние
животные приспосабливаются к НСМ. Так, способность к детоксикации
этого соединения выявлена у крупного рогатого скота и овец. Этот
процесс осуществляется с участием фермента роданезы, который
катализирует превращение цианида в тиоцианат:
Роданеза
СЫ" + 8
СЖ
О
Источником серы является р-меркаптопируват Н8-СН2-С-СОО ,
который, в свою очередь, превращается в пируват.
У большинства растений механизм детоксикации сводится к
образованию безвредного гликозида. Он протекает с участием ХШР-глюко-
зы и фермента гликозилтрансферазы. Особенно токсичны для
растений и микроорганизмов фенолы. Они детоксицируются прямым
путем с образованием гликозидов. В детоксикации могут принимать
участие реакции окисления, декарбоксилирования, метилирования, аце-
тилирования, образования сложных эфиров. Реже в процессе
детоксикации вместо Сахаров участвуют аминокислоты.
По аналогичному механизму идет детоксикация фунгицидов и
гербицидов.
Фунгициды — вещества, используемые в борьбе с грибковыми
заболеваниями растений- Из этих заболеваний особый вред наносит
мучнистая роса у злаковых и тыквенных. Фунгициды используются для
протравливания семян.
Одним из широко используемых для ячменя фунгицидов является
этиримол, производное пиримидина. Его детоксикация в листьях
ячменя на свету идет по следующей схеме:
Глюкоза
Окисление , Глюкозили-
боковой цепи | 9^ рование , |
С4Н9 ™т/\ /Ан8 «,/Х /С4Н
ОН
он о
3
'2"5
Этиримол
Боковая цепь этиримола сначала окисляется до соответствующего
спирта, а затем образуется глюкозид этого спирта — естественный
продукт метаболизма.
Особый интерес заслуживает гибкость растений к гербицидам —
веществам, применяемым для уничтожения сорняков. В
эффективности уничтожения сорняков решающую роль играют скорость и способ
детоксикации. Так, использование в качестве гербицида 2,4-дихлор-
583
Часть III. Элементы прикладной биохимии
феноксиуксусной кислоты обусловлено тем, что
сельскохозяйственные культуры способны быстро ее обезвреживать, а сорняки погибают
из-за отсутствия такой способности. Ферментный аппарат сорняков
при обработке их гербицидом активируется не сразу, и именно
разница в скорости детоксикации определяет избирательное действие
гербицидов.
Растения обладают хорошей способностью приспосабливаться к
высокому содержанию в почвах токсических металлов. Известно,
например, что на отработанных после добычи тяжелых металлов
отходах быстро заселяются такие злаки, как полевица и овсяница;
полевица хорошо растет на почвах, содержащих до 1 % свинца.
Точный механизм адаптации растений к тяжелым металлам
неизвестен. В первую очередь они действуют на поверхность корней. Уста-
новлено, что ферменты корней приспосабливаются к высоким
концентрациям металлов, у толерантных к металлу растений шире набор изо-
ферментов, чем у нетолерантных. Возможно, что толерантные растения
обладают способностью осуществлять биосинтез специфических белков,
связывающих тяжелые металлы в безвредные металлопротеины.
Имеются данные о том, что некоторые толерантные растения ограничивают
проницаемость ионов цинка и меди через клеточные мембраны-
Способность растений расти на почвах, содержащих тот или иной
тяжелый металл, успешно используется человеком в поисках новых
месторождений ценных минеральных отложений. Есть
специфические растения-индикаторы, с помощью которых можно обнаружить
залежи никеля, серебра, золота, урана в той или иной зоне.
15.4. Загрязнение окружающей среды. Мутации
В результате человеческой деятельности многие химически
активные соединения попадают в окружающую среду, оказывая
губительное действие на живые организмы, в том числе и на человека.
Необходимо отметить две важные особенности загрязняющих веществ в
экосистемах и организмах. Во-первых, высокая устойчивость многих
соединений в биосфере в реальных условиях. Так, ДДТ (дихлордифенил-
трихлорэтилен) исчезает из почвы за время, доходящее до 30 лет, хлор-
дан — за 14 лет, гептахлор — до 17 лет. Во-вторых, некоторые
чужеродные соединения могут подвергаться распаду или трансформации
таким образом, что получающиеся продукты оказываются
значительно более устойчивыми или токсичными.
По механизму действия химические соединения можно
подразделить на две группы: а) токсические, действие которых связано с
нарушением отдельных звеньев метаболизма; оно происходит в
большинстве случаев за короткий период времени; б) мутагенные,
действующие на генетическом уровне; они ярко проявляются в потомстве и
носят особо опасный характер для человека.
584
(
Глава 15. Некоторые аспекты экологической биохимии
В основе действия химических соединений на генетическом
уровне лежит процесс мутации. Причиной мутации является изменение
последовательности азотистых оснований на каком-либо участке ДНК,
причем для этого достаточно, чтобы произошло изменение одной-един-
ственной пары оснований.
Вещества-мутагены впервые были открыты в 1942 г. И* А.
Раппопортом в нашей стране и Ш. Ауэрбахом в Великобритании. Они
получили доказательства высокой мутагенной активности формалина, эти-
ленимина* Затем мутагенная активность была обнаружена у нитросо-
единений: нитратов, нитритов, нитрозоаминов. Мутагенная активность
азотистой кислоты обусловлена, главным образом, вызываемым ею
дезаминированием аденина, гуанина, цитозина. Помимо этого
азотистая кислота способна вызывать аномальные сшивки в молекуле ДНК.
Нитриты, вступая во взаимодействие с вторичными аминами,
образуют высокотоксичные нитрозоамины:
*МН + НЖ)2 -^ /N-N=0 + Н20.
К2 К2
Эта реакция легко может протекать в желудке, причем
образующиеся нитрозоамины легко всасываются и способствуют развитию
онкологических заболеваний. Во многих растениях содержится
определенное количество нитратов, причем в некоторых из них, например
в свекле и шпинате, их количество достаточно велико. Хорошо
отлаженных механизмов их обезвреживания у человека и животных нет.
Многие соединения в обычных условиях не проявляют
активности* Однако в организме они часто подвергаются гидроксилированию,
превращаясь в активные мутагены. Активность химических
мутагенов существенно возрастает под влиянием физических воздействий
(УФ-лучи, ионизирующая радиация), а также при случайном
включении вирусной ДНК в хромосомы.
Источниками азотсодержащих мутагенов могут быть минеральные
и органические удобрения, бытовые сточные воды, промышленные
отходы. К сильным мутагенам относятся дезинфицирующие
вещества— дихлорэтан, оксид этилена, фенол, алкилирующие соедине-
ния. Их мутагенное действие связано с многочисленными
повреждениями цепей ДНК. Мутагенами являются также триэтиленмеланин,
используемый в производстве резины, целлофана, эпоксидной смолы
и акролеина, ацетальдегид — в производстве консервантов,
фотохимикатов. С выхлопными газами автомашин в окружающую среду
поступают такие мутагены, как свинец, оксиды азота, ароматические
углеводороды, триметилфосфат. Промышленные отходы могут
вступать во взаимодействие с другими мутагенами, такими, как соли
тяжелых металлов, образуя мутагенные комплексы высокой
активности. В районах с интенсивным применением пестицидов у детей чаще
585
Часть III. Элементы прикладной биохимии
диагностируются железодефицитная анемия, активный туберкулез,
вирусный гепатит, заболевания верхний дыхательных путей, нервной
системы.
15.5, Основные реакции биотрансформации ксенобиотиков
Все реакции, участвующие в биотрансформации ксенобиотиков,
можно, в основном, разделить на 4 типа: 1) окисление; 2)
восстановление; 3) расщепление; 4) конъюгация.
Реакции окисления. Среди ферментативных систем, участвующих
в окислении, особое место занимают оксигеназы. Они представляют
собой комплексы мембраносвязанных ферментов, осуществляющих
окисление субстратов за счет активации молекулярного кислорода.
Оксигеназы катализируют окисление спиртов, альдегидов, аминов,
ароматических соединений. У многих организмов имеются активные
оксигеназные системы, катализирующие реакции окисления с
участием цитохрома Р-450. Эти системы способны окислять широкий круг
экзогенных соединений, включая пестициды, фенолы.
Реакции восстановления. Под действием специфических дегидроге-
наз могут восстанавливаться альдегиды и кетоны в соответствующие
спирты, ароматические нитрозо- и азосоединения. Они обычно
восстанавливаются с участием микросомальных и бактериальных ферментов.
Реакции расщепления. Под действием таких ферментов, как хо-
линэстераза, арилэстераза, осуществляется гидролиз эфиров.
Гидролиз эфирной связи является начальным этапом микробиологической
деградации многих пестицидов. С участием эстераз происходит
деградация фосфорорганических инсектицидов. Алкильные группы,
связанные с кислородом, азотом или серой, могут удаляться микросо-
мальными ферментами. В результате образуются соответствующие
фенолы, амиды, тиолы. Эти реакции важны при деградации многих
пестицидов.
Чрезвычайно большой интерес представляют реакции
расщепления лигнина, природного полимера, имеющего циклическую
структуру. Его содержание в растительных тканях составляет 20—30 %
сухой массы. В деградации лигнина основную роль играют микромице-
ты. На первых стадиях происходит отщепление метоксильных групп
от молекулы лигнина. На второй стадии о-дифенольные остатки
окисляются оксигеназами грибов, в результате чего происходит разрыв
ароматических колец полимера с образованием алифатических карбо-
новых кислот.
Глубокое исследование процесса деградации лигнина имеет
большое значение для отраслей, связанных с переработкой целлюлозосо-
держащего сырья, поскольку лигнинсодержащие отходы медленно
разлагаются и накапливаются в больших количествах на местах
переработки этого сырья.
586
Глава 15. Некоторые аспекты экологической биохимии
1
Реакции конъюгации. Многие из этих реакций весьма
эффективны при детоксикации загрязняющих соединений.
Важную роль в конъюгировании играет ацетил-СоА. При участии
специфических ацетилтрансфераз остаток ацетата переносится на
алифатические амины или сульфонамиды. Сульфгидрильные соединения
(глутатион, цистеин) весьма эффективно связываются с
конденсированными кольцевыми системами — нафталином, антраценом. В
конъюгации с глутатионом принимают участие более 40 химических
соединений. С участием метионина и соответствующих ферментов
осуществляются реакции метилирования алкилфенолов, аминов,
пиридина.
В реакциях конъюгации принимают участие широко
распространенные в растениях рибоза, глюкоза, глюкуроновая кислота, причем
последняя имеет преимущество перед глюкозой как детоксицирую-
щий агент, поскольку содержит ионизирующуюся СООН-группу.
Конъюгации с этими соединениями подвергаются спирты, кетоны,
фенолы, амины, тиолы, арилмеркаптаны. Активным конъюгантом для
пестицидов является лигнин.
В организме животных биологический смысл конъюгации
заключается в том, чтобы придать чужеродным веществам повышенную во-
дорастворимость и вывести их в виде водорастворимых конъюгатов из
организма. Однако с экологической точки зрения проблема все равно
остается, поскольку загрязняющее вещество в форме конъюгата
попадает либо в почву, либо в воду и продолжает циркулировать в
окружающей среде.
В биосфере особую опасность представляют неразлагающиеся
соединения. К ним относятся тяжелые металлы, входящие в состав
различных соединений. Эти соединения остаются в окружающей среде до
тех пор, пока не будут связаны с донными осадками водоемов или не
окажутся захороненными в виде нерастворимых соединений в почве.
Тяжелые металлы могут накапливаться в организме живых существ,
в том числе человека.
Еще один вид загрязнений, весьма медленно исчезающий в
биосфере, — радиоактивные соединения — радионуклиды. Период полураспада
радионуклидов длится десятки, сотни, тысячи лет. Они могут оказывать
вредные мутагенные воздействия на многие поколения людей.
Экологическая опасность неразлагающихся соединений связана с
тем, что они нарушают структуру, стабильность и продуктивность
экосистем. Отравленная река или морская акватория с мертвой
водой — это не только утраченные пищевые, рыбохозяйственные
ресурсы, но и утраченные возможности использовать природные
биоочистные системы. Поступление в экосистему больших количеств трудно-
удаляемых загрязняющих соединений ведет к изменению
физико-химических параметров среды обитания. Нарушение структуры
экологической системы вследствие возникновения новых субстратов ведет
587
Часть III. Элементы прикладной биохимии
к увеличению численности окисляющих и утилизирующих их групп
бактерий, что особенно важно для водной среды, где могут резко
снижаться содержание кислорода и величина рН.
Ограниченность способности экосистем и организмов к
обезвреживанию загрязняющих соединений ведет к тому, что для проявления
негативных эффектов достаточно накопления в среде небольших
количеств этих веществ, на первый взгляд не опасных из-за их низкой
концентрации. Однако сублетальные концентрации загрязняющих
веществ также опасны, поскольку вызывают медленное, хроническое
отравление, ведут к потере репродуктивной способности организма,
нарушают естественный экологический баланс в экосистемах,
приводят к ненормальному соотношению численности видов, повышают
действие других токсинов.
15.6. Некоторые прикладные аспекты экологической биохимии
В решении проблемы уменьшения загрязнения биосферы
биологическое значение могут иметь некоторые виды и штаммы
микроорганизмов. Большую роль в решении этой проблемы может сыграть
генная инженерия. Промышленность способна производить
специфические ферментные препараты, разрушающие токсичные загрязняющие
соединения. Одной из перспективных задач генной инженерии
является введение в геном сельскохозяйственных растений генов,
увеличивающих устойчивость растений к грибным и бактериальным
заболеваниям; создание сортов растений, резистентных к определенным
гербицидам. На биологическом уровне такую резистентность можно
обеспечить, например, путем включения в геном растения генов,
синтезирующих ферменты, разрушающие данный гербицид.
В борьбе с насекомыми-вредителями сельского хозяйства
целесообразно использовать природные экологически активные вещества,
получать их в промышленном масштабе. К таким соединениям, в
частности, относятся феромоны. Этот вопрос заслуживает особого
внимания в связи с тем, что здесь используются природные соединения,
легко вовлекаемые в метаболические процессы.
Однако следует отметить, что наиболее экологически
обоснованным является такой подход к охране окружающей среды, который
базируется на концепции интегральной защиты растений. В любой
экосистеме есть свои специфические механизмы регуляции. Задачей
человека является познание этих механизмов, дающее возможность
эффективно использовать комплекс средств воздействия на данную
экосистему с тем, чтобы сохранить ее структуру и функционирование,
сделать стабильной экосистему. Поэтому экологическую биохимию
можно охарактеризовать как науку о биохимической стабилизации и
дестабилизации экологического равновесия в биосфере.
588
Глава 15. Некоторые аспекты экологической биохимии
Краткое содержание главы
Важным разделом общей экологии является экологическая
биохимия. В предмет этой науки входит изучение конкретных
биохимических механизмов, с помощью которых осуществляется адаптация
организмов к окружающей среде. Экологическая биохимия включает
также биохимические аспекты метаболизма экологически важных веществ
и биохимических механизмов обезвреживания токсинов.
С позиций биохимии адаптация сохраняет в клетке функцию мак-
ромолекул (ферментов, белков, нуклеиновых кислот), обеспечивает
целостность надмолекулярных структур, необходимых для
протекания метаболических процессов. Биохимическую адаптацию можно
подразделить на генетическую адаптацию, акклиматизацию,
немедленную адаптацию.
Генетическая адаптация является длительным процессом, идущим
на протяжении многих поколений. В ее основе лежит мутация генов,
которая может обеспечить выработку новых способов регуляции
метаболических процессов, привести к синтезу белков нового типа,
придающих организму устойчивость к экстремальным условиям внешней
среды.
Акклиматизация — это такой процесс адаптации, при котором
организм приспосабливается к нескольким изменяющимся параметрам
внешней среды. В этом случае используются те характеристики
организма, которые уже закодированы в геноме организма. Здесь имеют
место существенная активация отдельных сторон метаболизма и
перестройка клеточных структур.
Немедленная адаптация происходит очень быстро. Она не связана
с какими-либо изменениями в геноме, с перестройкой клеточных
структур, а осуществляется путем изменения активности или
существенной интенсификации биосинтеза ферментов, в которых организм
нуждается в данный момент.
В экологических процессах большую роль играют вещества
вторичного происхождения. При опылении растений насекомых
привлекает прежде всего окраска цветков. Она обусловлена в основном фла-
воноидами и каротиноидами, находящимися в хлоропластах и
вакуолях клеток лепестков. Важную роль в опылении играет аромат
цветка, обусловленный моно- и сесквитерпенами, простыми
алифатическими спиртами, кетонами, сложными эфирами. Однако основной
причиной, побуждающей насекомых посещать цветущие растения, —
нектар и пыльца. Их химический состав вполне обеспечивает
потребности насекомого-опылителя в необходимых пищевых компонентах и
энергии.
Вторичные вещества оказывают прямое воздействие на пищевое
поведение животных. Эти вещества можно подразделить на два
класса: 1) аттрактанты, или пищевые стимуляторы и 2) репелленты и ток-
589
Часть III. Элементы прикладной биохимии
сины. В настоящее время хорошо изучены пищевые аттрактанты
насекомых. Они включают широкий круг различных веществ:
гликозиды, алкалоиды, флавоноиды, терпены, эфирные масла. К
репеллентам можно отнести широко распространенные в растениях таннины.
Вяжущий вкус для животных является сигналом того, что
питательная ценность белков листа резко снижается в связи с образованием
трудно перевариваемого белково-таннинового комплекса. Особой
группой аттрактантов являются феромоны — вещества, используемые для
общения насекомых в пределах вида. Феромоны имеют небольшую
молекулярную массу (в пределах 80—300). Поскольку они переносятся
по воздуху, они обладают высокой летучестью. Наиболее хорошо
изучены половые феромоны — соединения, выделяемые самками
насекомых для привлечения самцов и побуждения их к спариванию.
Большинство феромонов являются производными углеводородов
(ненасыщенные спирты, ацетаты, оксокислоты). Интересная разновидность
феромонов — следовые феромоны. Это вещества-метки, выделяемые
общественными насекомыми (пчелами, муравьями, термитами),
служат ориентирами на пути насекомого от гнезда к пище и обратно; к
ним относятся сложные эфиры, монотерпены. Разновидностью
феромонов являются феромоны тревоги. Эти вещества выделяются
насекомыми, заметившими опасность. К ним относятся простые
углеводороды и их производные — альдегиды, кетоны. Некоторые алкалоиды,
фенолы служат средством защиты насекомых от хищников.
В борьбе за существование растения вырабатывают разнообразные
средства защиты друг от друга, к которым относятся вещества
вторичного происхождения. Защита растений с помощью
синтезированного ими химического вещества называется аллелопатией. В
природных условиях аллелопатия представляет собой биохимические
взаимодействия между растениями. Большинство из хемоэффекторов с
аллелопатической активностью являются терпенами или фенольными
соединениями относительно простого строения, имеющими высокую
летучесть.
В защите растений от травоядных важную роль играют токсины.
Они дают о себе знать с помощью предупредительных сигналов
зрительной или обонятельной природы. Все токсины растений можно
подразделить на два класса: 1) азотистые (небелковые аминокислоты,
цианогенные гликозиды, алкалоиды и др.); 2) безазотистые токсины
(сердечные гликозиды, сапонины, афлотоксины и др.).
В организме животных имеются специфические системы детокси-
кации. Механизм детоксикации сводится к тому, что токсин
переводится в безвредную форму и выводится из организма. Важную роль
здесь играют оксигеназы со смешанной функцией.
В результате человеческой деятельности многие химически
активные соединения попадают в окружающую среду, оказывая
губительное действие на организмы, в том числе на самого человека. По меха-
590
Глава 15. Некоторые аспекты экологической биохимии
низму действия химические соединения делятся на две группы: а)
токсические, действие которых связано с нарушением отдельных звеньев
метаболизма; б) мутагенные — соединения, действующие на
генетическом уровне, вызывая мутации ДНК* В присутствии химических
мутагенов скорость мутирования возрастает под влиянием
физических воздействий (УФ-лучей, рентгеновских лучей и др.).
Все реакции биотрансформации чужеродных веществ, т.е.
ксенобиотиков, в том числе загрязняющих соединений, можно разделить
на 4 типа: 1) окисление; 2) восстановление; 3) расщепление; 4)
конъюгация. Вещества, не подвергающиеся этим типам превращений,
представляют особую опасность для биосферы. К ним относятся тяжелые
металлы, входящие в состав различных соединений. Медленно
исчезают в биосфере радиоактивные загрязнения, период полураспада
которых длится десятки, сотни, тысячи лет. Они могут оказывать
вредные мутагенные действия на многие поколения людей.
В решении проблемы уменьшения загрязнения биосферы
биологическое значение могут иметь специфические микроорганизмы,
полученные методами генной инженерии* Промышленность способна
производить специфические ферментные препараты, разрушающие
токсины. В борьбе с насекомыми-вредителями сельского хозяйства
целесообразно использовать природные экологически активные вещества,
например, феромоны.
Хронология важнейших дат в развитии экологической биохимии
1859 г. — Е. Пфлюгер впервые отметил, что адаптация организма
развивается при действии механических и термических раздражений.
1866 г. — Е, Геккель ввел понятие "экология" как науки об
отношениях организмов к окружающей среде.
1908 г. — В. Нернст установил, что при длительном раздражении
электрическим током в организме происходит увеличение порога
раздражения. Ввел термин "аккомодация" (приспособление).
1930—1945 гг. — В. И. Вернадский показал роль живых
организмов биосферы в круговороте химических элементов земной коры.
1940 г. — В. И, Сукачев ввел понятие биогеноценоза как
наименьшей целостной морфологической части географического ландшафта.
1974 г. — В. В. Ковальский развил научные основы
биогеохимической экологии.
ЛИТЕРАТУРА
Арчаков А. И, Микросомальное окисление. — М.: Наука, 1975. — 214 с.
Билъ К. Я. Экология фотосинтеза. — М.: Наука, 1993. — 220 с.
Васнецова А. Л.. Гладышев Г. П. Экологическая биофизическая химия. —
М.: Наука, 1989. — 134 с.
Ганасси Е. Э. Радиационные повреждения и репарация хромосом. — М.:
Наука, 1976. — 103 с.
591
Часть III. Элементы прикладной биохимии
Остроумов С. А. Введение в биохимическую экологию. — М.: Изд-во
Московского ун-та, 1986. — 176 с.
Телитченко М. М.. Остроумов С, А Введение в проблемы биохимической
экологии. — М.: Наука, 1990. — 285 с.
Харборн Дж. Введение в экологическую биохимию. — М.: Мир, 1985. —
311с.
Хочачка П., Сомеро Дж. Биохимическая адаптация. — М.: Мир, 1988 —*
568 с,
ВОПРОСЫ
1. Дайте краткую характеристику типам биохимической адаптации.
2. Какую роль играют вещества вторичного происхождения в экологических
процессах?
3. Охарактеризуйте сущность действия аттрактантов, репеллентов.
4. В чем сущность аллелопатии? Приведите примеры этого явления.
5. Токсины, их классификация и биохимический механизм действия.
6. Каковы причины мутации генетического фонда живого организма?
Назовите вещества-мутагены,
7. Перечислите типы реакций, обеспечивающих биотрансформацию
ксенобиотиков.
Глава 16. НЕКОТОРЫЕ АСПЕКТЫ МЕДИЦИНСКОЙ
И ФАРМАЦЕВТИЧЕСКОЙ БИОХИМИИ
16*1* Некоторые аспекты медицинской биохимии
Медицинская биохимия представляет собой раздел биохимической
науки, который рассматривает биохимические механизмы, лежащие
в основе процессов, протекающих в организме человека в норме и при
их нарушениях в связи с возникновением болезней. В ее задачи
входят расшифровка биохимических основ развития заболеваний как на
молекулярном уровне, так и на более сложных уровнях организации
живой материи, разработка приемов активного воздействия с
помощью лекарственных препаратов на ход определенных биохимических
реакций в организме с целью подавления или предупреждения тех
или иных патологических состояний, расшифровка особенностей
механизма нарушений функционирования ферментов.
16.1.1. Биохимические основы наследственных болезней
В настоящее время у человека известно более 2000
наследственных нарушений обмена веществ. Врожденные пороки обмена веществ,
или так называемые молекулярные болезни, обусловливаются
молекулярными дефектами структурных генов. Эти дефекты вызываются
различными факторами или же возникают спонтанно. У человека
наиболее частая форма мутаций, приводящая к дефектам ДНК, — это
точечная мутация. Обычно такие мутации сопряжены с трансверсией,
приводящей к замене пиримидинового основания на пуриновое или
наоборот. Как следствие, происходит синтез дефектных белков с
полной или частичной утратой способности выполнять свои функции, В
этой связи молекулярные болезни часто называют протеинопатия-
ми. Протеинопатии принято делить на две группы: энзимопатии и
неферментные протеинопатии. В результате нарушения
нормального функционирования белка происходит дезорганизация клеточного
метаболизма, что приводит к возникновению определенного
заболевания с характерными симптомами.
Энзимопатии аминокислотного обмена. Фенилкетонурия — это
типичный пример наследственных болезней, связанных с
нарушением обмена аминокислот. Причиной болезни является нарушение
метаболизма фенилаланина. При нормальном метаболизме фенилаланин
превращается в тирозин. Реакцию катализирует фермент фенилала-
38. Заказ 3486
593
Часть III. Элементы прикладной биохимии ^_^_^__^
нинмонооксигеназа, под действием которого один из атомов
молекулы кислорода 02 присоединяется к фенилаланину, в результате чего
образуется гидроксильная группа тирозина. Второй атом кислорода
восстанавливается до Н20 за счет окисления ЯАОН:
Фенилаланин + 02 + ИАВН + Н+ -> Тирозин + ЫАВ+ + Н20.
Фенилкетонурия возникает при наследственном дефекте,
связанным с синтезом фенилаланинмонооксигеназы. Ее отсутствие или
недостаток приводит к развитию второстепенного пути метаболизма
фенилаланина. На этом пути фенилаланин передает 3>Ш2-группу
а-кетоглуторату и превращается в фенилпируват:
Фенилаланин + а-Кетоглуторат о Фенилпируват + Глутамат.
Однако это тупиковая реакция: фенилпируват, а также такие
кислоты, как фениллактат и фенилацетат, образующиеся на
второстепенных путях превращений, накапливаются в крови и выводятся с
мочой. Заболевание выявляется по повышению уровня фенилаланина в
крови и фенилпирувата в моче.
Избыток фенилпирувата у новорожденных детей нарушает
нормальное развитие мозга и служит причиной умственной отсталости.
Симптомы болезни — повышенная возбудимость, приступы судорог, экземо-
подобные сыпи. Содержание фенилаладина в крови выше 40 мг/л
служит основанием для подозрения на данное заболевание, 200 мг/л —
подтверждает диагноз. При достаточно раннем выявлении болезни можно
создать условия для нормального развития ребенка. Из пищи должны
быть исключены белки с высоким содержанием фенилаланина. В
рационе можно использовать гидролизаты белков после удаления из них
определенного количества фенилаланина, поскольку полное удаление ведет к
дефициту этой незаменимой аминокислоты. При отсутствии лечения
многие больные фенилкетонурией не доживают до 25 лет.
Тирозинемия — это энзимопатия, обусловленная дефектом фермента
/г-гидроксифенилпируватоксидазы* Вследствие этого нарушается
образование гомогентизиновой кислоты. Происходит накопление ее
предшественников — тирозина и гс-гидроксифенилпировиноградной
кислоты — в крови и повышается их выделение с мочой. При данном
заболевании у детей наблюдается сниженная активность, отставание в
развитии.
Гиспгидинемия — энзимопатия, связанная с дефектом гистидазы,
обеспечивающей окислительное дезаминирование гистидина.
Заболевание начинает выявляться к концу первого года жизни ребенка.
Могут иметь место судороги, дефекты речи, шатающаяся походка,
снижение интеллекта. Ребенок отличается малым ростом. Повышается
содержание гистидина в крови и моче. У больных содержание
гистидина в крови — 20—270 мг/л (норма — 4—10 мг/л).
Имеют место врожденные нарушения метаболизма аминокислот:
глицина, лизина, диоксифенилаланина, гомоцистеина, валина, лей-
594
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
цина, изолейцина. Они связаны с недостаточностью активности и (или)
концентрации соответствующих ферментов.
Эизимопатии углеводного обмела. Галактоземия — энзимопа-
тия, связанная с нарушением функционирования ферментов,
обеспечивающих вхождение галактозы в углеводный метаболизм. В
метаболизме галактозы важную роль играет галактокиназа. Ее дефицит
ведет к тому, что галактоза, не подвергаясь фосфорилированию, не
вступает в дальнейшие превращения и появляется в моче. Характерным
симптомом заболевания является ухудшение зрения в результате
образования катаракты. Ее возникновение связано с усиленным
превращением галактозы в галактинол- Накапливаясь в стекловидном теле
глаза, галактинол связывает большое количество воды, что приводит
к разрыву зонулярных волокон.
Дефицит другого важного фермента углеводного метаболизма —
галактозо-1-фосфат-уридилтрансферазы ведет к накоплению галакто-
зо-1-фосфата и галактозы. Их непереносимость вызывает рвоту, жел-
тушность, дегидратацию, диарею. Если галактоза не исключена из
рациона, у детей наблюдается задержка умственного и физического
развития и возможен летальный исход.
Непереносимость лактозы. Дисахариды усваиваются организмом
человека после их предварительного ферментативного гидролиза. Так,
лактоза должна быть прогидролизована под действием фермента лак-
тазы на а-глюкозу и р-галактозу. Максимальная активность лактазы
наблюдается в момент рождения ребенка. Затем у многих людей ее
активность быстро снижается. Исследования показали, что 70 %
населения планеты страдают недостаточностью данного фермента.
Предварительный гидролиз лактозы молока ферментом решает эту
проблему. От приобретенной недостаточности лактазы наследственная
форма отличается появлением признаков болезни в первые дни после
рождения. Основные симптомы: диарея, боли после приема пищи,
вздутие живота.
Непереносимость сахарозы, мальтозы обусловлена дефицитом
сахаразы и мальтазы. Обладая высокой гидрофильностью, эти
дисахариды усиленно связывают воду, повышают осмотическое давление в
желудочно-кишечном тракте, вызывают диарею.
Непереносимость фруктозы может быть обусловлена дефектом
фруктозо-1-фосфатальдолазы, катализирующей превращение фрукто-
зо-1-фосфата в триозы. Накопление фруктозо-1-фосфата приводит к
-> подавлению гликогенолиза и нарушению функционирования
гликолиза. В результате наряду с гипофосфатемией имеет место дефицит
АТР. Происходит интенсивное расщепление адениловых нуклеотидов,
повышается содержание инозинмонофосфата и мочевой кислоты.
Нередко после приема фруктозы с пищей непереносимость развивается
быстро и остро: уже через 30 мин возникают рвота, боли в животе;
возможны кома и судороги. Непереносимость фруктозы может быть
38*
595
Часть III. Элементы прикладной биохимии
обусловлена также недостаточностью фруктозо-1,6-бисфосфатазы. При
этом в клеточном метаболизме происходят примерно те же сдвиги,
что и при дефиците фруктозо-Ьфосфатальдолазы.
Гликогенозы. Причиной этих патологических процессов являются
нарушения распада и синтеза гликогена. Различают три основные
формы гликогенозов: печеночную, мышечную и генерализованную.
Пример печеночного гликогеноза — болезнь Гирке, Ее причиной
является отсутствие или крайне низкая активность в печени, почках
и тканях кишечника глюкозо-6-фосфатазы. Дефицит глюкозо-6-фос-
фатазы ограничивает выход глюкозы в кровь при расщеплении
гликогена в печени. Первыми проявлениями заболевания являются
гипогликемия между приемами пищи, судороги, связанные с низким
содержанием глюкозы в крови, кетонемия и кетонурия. У ребенка
наблюдается отставание в росте, развивается диспропорция тела
(большая голова, короткие шея и ноги, гипотония мышц).
Примером мышечного гликогеноза является болезнь Мак-Ардля.
Нарушение обмена мышечного гликогена сопряжено с отсутствием
фосфорилазной активности. В этом случае имеет место быстрая общая
утомляемость, прогрессирующая миопатия и судороги после
физической нагрузки.
Для болезни Помпе характерны формы генерализованного и
мышечного гликогеноза. Гликогеноз развивается в связи с дефицитом
кислой а-1,4-глюкозидазы — лизосомного фермента,
обеспечивающего расщепление гликогена до глюкозы. Имеет место также
недостаточность а-(1—»6)-гликозидазы (кислой мальтазы). Обнаруживается
отложение гликогена во всех органах и тканях. Размеры сердца,
печени, селезенки и почек увеличены. Первые симптомы выявляются
через несколько дней или недель после рождения ребенка. Характерны-
ми для болезни признаками являются поверхностное, ускоренное
дыхание, отсутствие аппетита, беспокойство, бронхиты. Больные
погибают обычно на третьем году жизни.
Эизимопатии липидного обмена. Эти нарушения касаются прежде
всего липидов мембран. Причиной их возникновения являются
дефекты ферментов, участвующих в метаболизме липидов. При этом
биосинтез липидов протекает нормально, а их расщепление
нарушено, что приводит к накоплению и липидов, и продуктов их частичного
распада в тканях и органах. Заболевания, обусловленные данной
причиной, получили название лизосомных болезней. Объясняется это тем,
что многие стадии катаболизма липидов протекают в клеточных орга-
неллах — лизосомах. При генетических нарушениях функций
лизосомных ферментов не полностью расщепленные липиды
накапливаются в лизосомах, объем их резко возрастает, что ведет к нарушению
функционирования клеток организма. При лизосомной болезни
наблюдается задержка умственного развития ребенка, так как мозг
особенно богат липидами.
596
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
Примером лизосомных болезней является синдром Херлера, или
так называемый гаргоилизм. Заболевание обусловлено дефектом
фермента, ответственного за расщепление мукополисахаридного фрагмента
некоторых протеогликанов. При этом заболевании у детей на коже
образуются грубые складки, что сильно изменяет черты лица.
Развивается слепота, характерна умственная отсталость. Летальный исход —
в раннем возрасте.
Болезнь Тея-Сакса характеризуется накоплением в мозге и
селезенке ганглиозидов. Это обусловлено генетическим дефектом
фермента К-ацетилгексозаминидазы, катализирующего гидролиз
связи между остатками В-галактозы и 14-ацетил-В-галактозамина в
молекуле ганглиозида. Данное заболевание также характеризуется
поражением мозга, слепотой. Больные обычно не доживают до
пятилетнего возраста.
Другим примером является болезнь Нимана-Пика,
обусловливаемая дефектом фермента сфингомиелиназы и накоплением сфингомие-
лина в тканях мозга, селезенки и печени. Болезнь проявляется вскоре
после рождения, сопровождается задержкой умственного развития,
летальный исход наступает на втором-третьем году жизни.
Неферментные протеинопатии. Серповидноклеточная анемия.
Это наглядный пример генетического заболевания, для которого
характерно наличие в крови дефектных, серповидных по форме
эритроцитов (рис. 16Л). Такая форма эритроцитов приводит к
периодической закупорке кровеносных капилляров в различных органах и
тканях организма. Перенос кислорода к ним резко уменьшается, что
ведет к анемии. Обращает на себя внимание не только изменение
формы эритроцитов, но и снижение их количества в крови. В
результате анемии у больного
возникают приступы слабости,
тошноты и одышки* Анемия
является причиной смерти в
раннем возрасте.
Исследования показали,
что серповидная форма
эритроцитов обусловлена
наличием в них аномального
гемоглобина, получившего
название гемоглобина 8 (НЪ8). В
эритроцитах здоровых людей
присутствует нормальный
гемоглобин А,
обусловливающий форму эритроцитов в
виде дисков.
Аминокислотный анализ гемоглобина 8
Рис. 16 Л. Эритроцит больного серповид-
ноклеточной анемией; микрофотография,
полученная с помощью сканирующего электрон-
показал, что в его молекуле ного микроскопа
597
Часть III. Элементы прикладной биохимии
остаток глутаминовой кислоты, содержащейся в гемоглобине А,
заменен на остаток валина. Эта замена приводит к возрастанию гидрофоб-
ности гемоглобина 8, к агрегации его молекул и образованию
длинных нитей, обусловливающих серповидную форму эритроцитов.
Серповидноклеточная анемия широко распространена у людей,
живущих в так называемом малярийном поясе Земли. Она
обеспечивает защиту организма от тропической малярии. В эпидемических
очагах малярии (например, в некоторых районах Центральной
Африки) серповидноклеточной анемией поражено 40 % населения. На
основе полученных сведений об аномалии НЬ8 предпринимаются попытки
разработки способов рационального лечения этой болезни. Ведется
поиск специфических лекарственных средств, которые были бы
способны взаимодействовать с функциональными группами молекулы
гемоглобина 8 и предотвращать этой модификацией вредные
последствия замены глутаминовой кислоты на валин. Так, обработка
эритроцитов цианатом (N^0-0") приводит к нормализации формы клеток
и кривой насыщения гемоглобина кислородом, а также устранению
симптомов заболевания.
К неферментным протеинопатиям относятся и так называемые
транспортные протеинопатии, сопряженные с дефектами белков,
ответственных за транспорт определенных соединений через мембраны.
В качестве примера можно привести цистинурию — заболевание,
обусловленное дефектом белка, транспортирующего цистин. Это приводит
к повышенному выделению цистина с мочой и образованию цистино-
вых камней в мочевыводящих путях. Повышение уровня цистина
сопровождается избыточным содержанием в моче лизина, цистеин-го-
моцистеин-дисульфида, орнитина и аргинина.
К неферментным протеинопатиям относятся также агаммаглобу-
линемии, сопровождающиеся нарушением защитных реакций
организма, что связано с дефектами образования антител.
Существуют заболевания, связанные с недостаточностью синтеза
гормонов. Так, наиболее широко распространенной формой
нарушения метаболизма углеводов в организме человека является сахарный
диабет, связанный с недостатком гормона инсулина. Сахарный
диабет бывает двух основных форм - врожденный и неврожденный.
Сейчас мы рассмотрим генетическую форму этой болезни* В следующем
разделе, в котором будут рассмотрены вопросы патологии питания,
опишем ее неврожденную форму.
Основным биохимическим показателем углеводного обмена
является содержание глюкозы в крови. В норме плазма крови содержит
65—100 мг% глюкозы* Некоторое превышение этого уровня в
здоровом организме после приема значительных количеств глюкозы или
сахарозы может составить около 50 %. При диабете и в состояниях,
угрожающих его развитием (преддиабет), это повышение содержания
глюкозы достигает 80 % и более. При выраженном диабете уровень
598
Глава 16, Некоторые аспекты медицинской и фармацевтической биохимии
глюкозы может достигать очень высоких значений (до 2000 мг%).
Начиная с уровня около 200 мг%, преодолевается почечный барьер, и
глюкоза появляется в моче. Вместе с глюкозой почки выбрасывают из
организма повышенные количества воды. Отсюда жажда диабетиков,
способных иногда выпивать 10—20 л воды в сутки.
Причиной врожденной патологии сахарного диабета является на-
рушение биосинтеза гормона инсулина. Структура инсулина
обстоятельно исследована лауреатом Нобелевской премии Ф. Сенджером
(1945—1956 гг.) и рассмотрена нами в гл. 14. Инсулин —
универсальный анаболический гормон* Он оказывает влияние на все виды
обмена веществ. Конечный эффект его действия складывается из
непосредственной стимуляции биосинтетических процессов и одновременного
снабжения клеток строительным материалом (например,
аминокислотами) и энергией (глюкозой).
Важнейшими молекулярными дефектами синтеза инсулина
являются мутация на участке соединения А- и В-цепей при переходе про-
инсулина в инсулин, замена фенилаланина на лейцин вблизи С-конца
Вщепи, а также дефекты действия рецепторов клеток.
Главная функция инсулина — участие в транспорте глюкозы в
клетки организма: этот гормон повышает проницаемость клеточных мембран
для глюкозы, способствуя ее переходу в ткани. Несмотря на высокий
уровень глюкозы во внеклеточных жидкостях, клетки испытывают ее
острый дефицит (в буквальном смысле "голод среди изобилия")-
Дефицит глюкозы в клетках вызывает ускоренный распад гликогена.
Поэтому больной испытывает чувство жажды и голода. Дефицит глюкозы в
клетках ведет к более интенсивному использованию липидов в качестве
источника энергии. Образующийся при усиленном (^-расщеплении
жирных кислот ацетил-КоА не окисляется полностью в цикле Кребса: его
избыток в виде кетоновых тел выделяется вместе с мочой. Дефицит
глюкозы в клетках обусловливает отрицательный азотистый баланс в
организме в связи с усиленным распадом глюкогенных аминокислот (алани-
на, глутаминовой и аспарагиновой кислот). Это ведет, с одной стороны, к
снижению уровня аминокислот в клетках и нарушению синтеза белка, с
другой — к усиленному образованию мочевины.
Во врачебной практике для снятия патологии сахарного диабета
широко используется инъекция препаратов инсулина. Инсулин легко
соединяется с ионами двухвалентных металлов, особенно с цинком,
кобальтом, кадмием; может образовывать комплексы с
полипептидами, в частности с протаминами. Это свойство используется для
создания препаратов инсулина пролонгированного действия.
16.1.2. Элементы патологической биохимии питания
Патология белкового питания. Важным критерием в оценке
нормального режима питания является определение соотношения
вводимого и выводимого из организма азота. Равенство между ними сви-
599
Часть III. Элементы прикладной биохимии
детельствует о нормальном режиме использования белков пищи.
Преобладание выводимого из организма белка имеет место при дефиците
белков в пище. Это обусловлено тем, что аминокислоты организма
используются не для ресинтеза белков, а как источник энергии.
Положительный белковый баланс наблюдается при развитии организма
ребенка, при выздоровлении человека от истощающих заболеваний.
Патологические процессы, возникающие в условиях
неполноценного белкового питания, особенно тяжелы у детей. Прекращение пи-
тания грудным молоком и переход на растительную пищу ведут к
задержке роста, физического и умственного развития, гиподинамии,
малокровию, дисфункции печени и почек, отечности. Подавление
образования пищеварительных ферментов еще более усиливает
белковую недостаточность. Болезнь в целом, называемая квашиоркору
прогрессирует и приводит нередко к гибели ребенка.
У взрослых белковое голодание ведет к подавлению физической и
психической активности, к снижению в организме уровня белков,
особенно альбуминов плазмы крови; возникают отеки, параличи
мышечной ткани; ослабляется деятельность сердца, снижается активность
иммунной системы; возникает малокровие.
Патологии переваривания белков пищи возникают в связи с
агрессивным действием переваривающих ферментов. Они оказывают
повреждающее действие на слизистую оболочку желудочно-кишечного
тракта в случае нарушения ее структуры под действием каких-либо
факторов. К этому следует еще добавить высокую концентрацию ионов
водорода желудочного сока за счет соляной кислоты (рН 1,5—2,5).
Устойчивость слизистой оболочки желудка и двенадцатиперстной
кишки к агрессивному действию пепсина и соляной кислоты
обеспечивается рядом защитных факторов: адекватной продукцией слизи,
активной секрецией бикарбонатов поджелудочной железой,
слизистой оболочкой желудка и двенадцатиперстной кишки, регенерацией
специфических клеток в слизистой оболочке. Повреждение любого из
факторов защиты снижает устойчивость слизистой оболочки к
агрессивному воздействию желудочного сока. Основными формами
патологии желудка и кишечника являются воспалительные процессы.
Патология углеводного питания. Широко распространенной
формой патологии углеводного питания является неврожденный сахарный
диабет. Причины его возникновения многообразны. Выдвигаются,
например, представления о вирусных инфекциях, вызывающих побочные
деструктивные процессы в поджелудочной железе, об аутоиммунной
природе заболеваний, когда антитела атакуют ее р-клетки. У взрослых
важнейшей причиной возникновения неврожденного сахарного диабета
являются избыточная масса тела, первичная гипертония.
Основным показателем этой формы сахарного диабета, также как и
врожденного, является глюкозурия. Она возникает при употреблении
чрезмерных количеств Сахаров с пищей (например, сахарозы), особенно
600
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
быстро всасываемых после расщепления в кишечнике. Это приводит к
увеличению сахара в крови, превышающего порог реадсорбции глюкозы
в почках. У здоровых людей глюкозурия наблюдается в течение 2—4 ч
после приема пищи. Она может возникать и при нормальной
концентрации глюкозы в крови в связи с нарушением метаболических процессов в
почках, обусловливающих реадсорбцию глюкозы.
Наиболее доступной формой лечения неврожденного сахарного
диабета является регулирование и ограничение потребления Сахаров в
питании. Чрезмерное потребление Сахаров связано, как правило, с их
вкусовыми качествами как сладких веществ. В настоящее время в
пищевой промышленности с целью сокращения уровня Сахаров и
уменьшения калорийности продукта используются заменители Сахаров,
обладающие высокой степенью сладости. К ним относятся, например,
сахарин, аспартам, монелин.
Сахарин в 400 раз слаще сахарозы. Вредных для людей
последствий, вызванных употреблением в пищу сахарина, не выявлено.
Сахарин используют для приготовления диетических безалкогольных
напитков. Хорошо исследован на токсичность аспартам — метиловый
эфир дипептида аспарагилфенилаланина. Поскольку в его состав
входят остатки белковых аминокислот, нет оснований опасаться наличия
токсичных свойств у данного вещества. Аспартам в 180 раз слаще
сахарозы, поэтому он используется для приготовления диетических
пищевых продуктов.
Особо высокой сладостью обладает монелин. Он в 2000 раз слаще
сахарозы. Монелин — низкомолекулярный белок (молекулярная
масса — 11000), добываемый из плодов растения АМсап зегепсНрИу Ьеггу.
Сладкий вкус монелина обусловлен специфической конформацией его
белковой молекулы. При нагревании или других видах денатурации
монелин утрачивает сладость.
Патологии, связанные с питанием жировыми продуктами.
Около 40 % поступающих с пищей триацилглицеролов подвергаются
гидролизу в тонком кишечнике на глицерин и жирные кислоты; 3—
10% триацилглицеролов всасываются не расщепляясь, остальные —
поступают в кровь в виде моноацилглицеролов.
Причиной возникновения патологии могут быть дефицит липазы,
выделяемой поджелудочной железой; закупорка желчных протоков
печени и фистула желчного пузыря, приводящие к дефициту желчи в
кишечнике, снижению функций слизистой оболочки, где
локализованы ферменты гидролиза триацилглицеролов.
Как гидрофобные соединения липиды транспортируются кровью в
виде надмолекулярных комплексов — .тйгаопротеинов, в состав
которых входят не только триацилглицеролы, но и холестерол, фосфоли-
пиды. Нормальное содержание липидов в плазме крови составляет
500—1500 мг/л. Повышение их концентрации наблюдается при
обильном использовании в пищу жировых продуктов, а также при нефроти-
601
Часть III. Элементы прикладной биохимии
ческом синдроме, гипотериозе, переломе костей, алкоголизме. Это
основные причины широко распространенного заболевания —
атеросклероза, характерной чертой которого являются изменения,
происходящие в стенках кровеносных сосудов. В стенках сосудов формируются
кашеобразные липидные отложения и происходит их дальнейшее
уплотнение. Важнейшим компонентом этих отложений является холес-
терол. Характерной чертой атеросклероза являются ишемическая
болезнь сердца, инфаркт миокарда, инсульт и тромбозы.
Ишемический синдром возникает также и при поражении клеток
крови. Увеличение содержания холестерола в мембранах эритроцитов
приводит к нарушению функций мембраносвязанных ферментов;
увеличиваются скорость агрегаций эритроцитов и их размеры,
снижается деформируемость клеток, ухудшаются их реологические свойства.
Однако холестерол играет большую роль в метаболизме веществ в
организме человека. Он является важным компонентом клеточных
мембран, источником витамина В, гормонов, желчных кислот.
Поэтому холестерол в определенных количествах необходим для
нормальной жизнедеятельности организма. Лишь его избыток способствует
развитию атеросклероза.
Потребление в пищу животных жиров ведет к существенному
увеличению уровня холестерола в крови, насыщенных жирных кислот.
Наиболее богаты холестеролом печень, почки, мозг, яйца, икра. В
100 г почек его содержится 1120 мг, в печени трески — 740 мг,
говяжьей печени — 440 мг, рыбной икре — 300 мг, в яичном желтке —
200 мг. Норма потребления холестерола для взрослого человека —
около 300 мг в сутки. Съеденные два яичных желтка уже полностью
обеспечат суточную потребность в холестероле. Однако установлено,
что человек постоянно получает с пищей 500—1000 мг холестерола.
Стойкое превышение нормы холестерола в крови ведет к росту
вероятности летального исхода от инсульта или инфаркта миокарда.
В лечении больных атеросклерозом и его профилактике
существенное значение имеет правильно составленная, индивидуально
подобранная диета. У людей значительное распространение получили вредные
привычки — переедание, повышение нормы животных жиров,
гиподинамия. Избыток веществ-энергоносителей трансформируется и
откладывается в жировой ткани. Важно включение в пищу
растительных масел, содержащих полиненасыщенные жирные кислоты. При
отношении в пище поли ненасыщенных жирных кислот к
насыщенным, равном 0,4, вариация холестерола 300—1000 мг/сутки не
влияет у здоровых людей на его уровень в крови. Ненасыщенные жирные
кислоты ограничивают всасывание холестерола пищи в тонком
кишечнике, стимулируют в печени синтез желчных кислот. Включение
в пищу целлюлозы, гемицеллюлоз, пектиновых веществ, содержащихся
в овощах, фруктах, черном хлебе, способствует образованию их комп-
лексов с холестеролом и удалению его из организма.
602
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
Патологические состояния, связанные с нарушением липидного
обмена, являются причиной самой высокой смертности и
инвалидности населения планеты, особенно жителей больших городов
экономически развитых стран. Проблема атеросклероза является в настоящее
время не только медицинской, но и социальной проблемой.
Атеросклероз, если не считать наследственного фактора, во многом обуслов-
лен вредными привычками, такими как переедание, пристрастие к
очень жирной и высококалорийной пище.
Алкоголизм. Элементы патобиохпмии. Алкоголизм — это
совокупность патологических изменений в организме под влиянием
длительного и неумеренного потребления алкоголя, что вызывает
нарушение психики, межличностных отношений, социального и
экономического статуса больного. По данным исследователей США, люди,
потребляющие алкоголь, составляют 90 % населения, но лишь 5—
7 % мужчин и 0,5 % женщин из них можно отнести к алкоголикам.
Тем не менее эта цифра достаточно высока и дает основание признать
алкоголизм как одну из распространенных в мире болезней.
Прекращение приема алкоголя больными вызывает у них сильные
мышечные боли, галлюцинации, судороги. Пути метаболизма, а так-
же действие алкоголя на функционирование различных органов
человека хорошо изучены. Однако окончательно не установлено, какие
изменения физиологических функций приводят к привыканию к
алкоголю и формированию физической зависимости от него.
Этиловый спирт постоянно образуется в организме человека. В норме
уровень эндогенного этанола в крови человека колеблется в пределах
0,001—0,01 г/л. В небольших количествах (4—5 г/сутки) этанол
поступает с некоторыми пищевыми продуктами. 10—100-кратное
увеличение этой концентрации приводит к опьянению человека. При
высоких концентрациях в крови (0,4—0,7 %) могут наступать
коматозное состояние и остановка дыхательного центра.
Попадая в организм, этанол через желудочно-кишечный тракт
всасывается в кровь. Небольшое количество его удаляется через почки и
легкие, а остальное (около 99 %) окисляется в печени. Поэтому
вполне понятно, что печень наиболее подвержена токсическому действию
этанола и его метаболитов. Под действием алкогольдегидрогеназы
печени этанол окисляется в высокотоксичный ацетальдегид:
О
СН^-СНо-ОН ^—^—*» СНо-С^
н
2 V/!! уГ -^ ^-*3 Ч
КАО+ ЫАЮН + Н+
Второй путь окисления осуществляется специфической оксигеназой,
О
СН,-СНо-ОН + Оо —^ ^ » СН3-С^ + Н20
ИАБРН + Н+ КАСР+
603
Часть III. Элементы прикладной биохимии
Вклад этого пути в метаболизм этанола невелик, однако он
возрастает при поступлении в организм больших доз алкоголя и становится
существенным (50—70 %) при хроническом алкоголизме.
Существует и третий, хотя менее значительный, путь окисления
этанола в печени с участием пероксида водорода:
/О
сн3~сн2~он + н2о2 ^ сн3-с' + 2н2о
В митохондриях печени ацетальдегид превращается в уксусную
кислоту под действием N АО-зависимой алкогольдегидрогеназы:
Алкоголь-
О дегидрогеназа
СН„-С' +Н20 ^—^ » СНя-СООН
ЧН 2 / \ 3
з
КАЮ + ИАВН + Н+
Небольшая часть ацетата поступает в кровоток и в
периферических тканях окисляется до С02 и Н20, а остальная часть при участии
АТР превращается в ацетил-СоА, который вовлекается в цикл Кребса
или используется в синтезе жирных кислот, триацилглицеролов, хо-
лестерола.
Если доза этанола в организме была небольшой (0,2—0,5 г/кг
массы тела), то повышение концентрации ацетальдегида в клетке не
приводит к токсикозу. Этанол и ацетальдегид быстро и согласованно
окисляются в печени с образованием ацетил-СоА. При значительных
дозах этанола (1,5—2 г/кг и более) концентрация ацетальдегида в
печени резко возрастает. Это вызывает неприятные ощущения
отравления, тахикардию, потливость. Высокие концентрации ацетальдегида
ингибируют действие алкогольдегидрогеназы; это и вызывает
"синдром похмелья" и отвращение к алкоголю. Аналогичные действия на
алкогольдегидрогеназу оказывает антиалкогольный препарат дисуль-
фирам (тетурам). В то же время ацетальдегид ингибирует ИАВ-дегид-
рогеназу дыхательной цепи, что, в свою очередь, вызывает
подавление функционирования цикла Кребса.
Этанол — высококалорийный продукт. Длительное потребление его
в количествах, составляющих половину от общей поступающей
энергии, ведет к жировому перерождению печени. Это связано с тем, что в
результате избыточного образования ацетил-СоА не вовлекается
полностью в цикл Кребса и часть его используется в печени для синтеза
жирных кислот, триацилглицеролов, холестерола, липопротеинов. При
хроническом алкоголизме развивается фиброз печени, усиливается
синтез коллагена, что приводит в конечном счете к циррозу печени и к
смертельному исходу.
Основным принципом терапии этого заболевания является
внушение отвращения к этанолу, что достигается его приемом совместно с
некоторыми препаратами (например, апоморфином и эмитином), об-
604
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
ладающими рвотным действием, а также использование тетурама.
Однако тетурам в виде имплантированной лекарственной формы (эс-
пераль) оказывает скорее психологический эффект, так как его
уровень в крови весьма мал, чтобы понизить активность алкогольдегид-
рогеназы; повышение дозы тетурама нежелательно вследствие его
токсического действия на организм. Перспективной задачей является
получение лекарственных средств длительной коррекции алкогольного
влечения, не требующих постоянного введения и соответствующего
контроля их приема.
16.1.3. Биохимические механизмы,
лежащие в основе нервно-психических заболеваний человека
Наркомании. Наркомании (от греч. пагке — онемение,
оцепенение; тата — безумие, сумасшествие) представляют собой группу
токсикомании, возникающих вследствие злоупотреблений наркотически-
ми средствами. При наркотической эйфории помимо подъема
настроения, благодушия, чувства радости возникают различные приятные
телесные ощущения. Нарушается активное внимание. Мышление
становится непоследовательным, образным. В течение наркотической
эйфории обычно имеют место две фазы: 1) собственно эйфория — фаза
острых ощущений психического и физического возбуждения (1—
5 мин); 2) фаза успокоения и расслабленности (1—3 ч),
завершающаяся дремотой и сном.
Как правило, злоупотреблять наркотиками начинают в молодом
возрасте. Подверженность злоупотреблениям наркотиками
характерна для лиц морально неустойчивых, внушаемых, психически
незрелых, с малой волей, не способных к оценке своих действий. Когда
подобные личности попадают в среду наркоманов, они не способны
противостоять ей*
В основе действия наркотиков лежит подмена экзогенным
химическим агентом естественного внутреннего биохимического сигнала,
вызывающего приятные ощущения. В норме этот сигнал срабатывает
тогда, когда обстановка действительно благоприятна, т.е. когда
вокруг и внутри организма "все хорошо". Наркотические средства
вызывают состояния умиротворенности, удовлетворения, благодушия,
неоправданные ситуацией.
С биохимической точки зрения действие наркотиков на организм
многообразно и трудно выделить ведущее звено патогенеза
наркомании. Считают, что в формировании наркотической зависимости
существенную роль играет активация детоксицирующих ферментов
печени» Наркотики оказывают сильное воздействие на обмен нейропепти-
дов (эндорфинов, энкефалинов), а также на содержание циклических
нуклеотидов. Так, морфин из опиумного мака и родственные ему
вещества взаимодействуют с теми же рецепторами головного мозга, что
и опиоидные нейропептиды. Однако влияние наркотика значительно
605
Часть III. Элементы прикладной биохимии
ОН
Морфин
сильнее по сравнению с эндогенными аналогами, так
как он воздействует в существенно большей дозе и
более длительное время благодаря своей
стабильности. В этой связи морфин используется в
клинической практике как болеутоляющее средство* Канна-
биоиды, представляющие собой психомиметически
активные компоненты марихуаны (рис. 16.2),
подавляют образование сАМР. Кроме того, эти соединения значительно
изменяют проницаемость клеточных мембран. Экспериментально
доказано, что какнабиоиды (тетрагидроканнабинол, каннабинол и канна-
бидиол) существенно повышают частоту хромосомных нарушений у
животных. Причем это характерно не только для подопытных
животных, но и для их потомства.
СД1(п)
СбНп(п)
$-9-Тетрагидроканнабинол Каннабинол
ОН
Каннабидиол
СвНп(п)
Рис, 16.2. Каннабиоиды из конопли
Наркомании не сопровождаются локальным поражением какой-
либо одной системы или органа. Считают, что в организме нет
системы, на которую бы они не оказывали воздействия. Вмешательство
наркотиков в нейромедиаторный обмен приводит к функциональным
нарушениям центральной и периферической нервной системы.
Физическое истощение, низкая функциональная активность всех
физиологических систем — характерные общие признаки всех форм наркоманий.
Поражение различных систем организма обусловливается также
торможением окислительных процессов в тканях. Полагают, что в основе
механизма развития наркотической зависимости лежит взаимодействие
определенных рецепторов с не свойственными для них лигандами, что
приводит к структурной перестройке рецепторов. После этого они уже не
могут нормально взаимодействовать с эндогенными агентами.
Наркотическая зависимость является практически необратимой.
Эпилепсия. Эпилепсия (от греч. ерйерзга — схватывание)
представляет собой хроническое заболевание, характеризующееся судорожными
и психопатологическими приступами; иногда сопровождается
изменением личности. Результаты рентгенографических, томографических и кли-
нико-электрофизиологических исследований свидетельствуют, что
развитие эпилепсии сопряжено с наличием стабильного очага
эпилептической активности, обусловленного органическими поражениями головного
мозга. Причиной судорожных и психопатологических состояний могут
быть энергетические нарушения в отдельных участках мозга, которые и
служат очагами эпилептической активности.
606
Глава 26. Некоторые аспекты медицинской и фармацевтической биохимии
Энергия в форме АТР необходима нервным клеткам (нейронам)
для поддержания электрического потенциала на мембране и, в том
числе, на мембране аксонов и дендритов, представляющих собой
длинные отростки нейронов и образующих "линии передач" в нервной
системе. Благодаря волнообразным изменениям электрических свойств
мембраны, т.е. потенциала действия, осуществляется передача
нервных импульсов по нейронам. Большое количество АТР в мозге
расходуется для синтеза нейромедиаторов — веществ, обеспечивающих
передачу нервных импульсов от одной нервной клетки к другой через
участки контакта между ними — синапсы. В настоящее время
известно значительное число веществ, выполняющих функции
нейромедиаторов и ингибиторов передачи нервных импульсов и действующих
специфично в отношении различных типов нейронов. Это
аминокислоты: глутамат, аспартат, глутамин, у^аминобутират, глицин, таурин.
Медиаторные функции различного характера могут выполнять также
ацетилхолин, некоторые пептиды, моноамины (адреналин, норадре-
налин, серотонин). Механизм действия нейромедиаторов состоит в том,
что находящийся в везикулах (пузырьках) пресинаптических нервных
окончаний медиатор в ответ на потенциал действия изливается в си-
наптическую щель и взаимодействует с рецепторами постсинаптичес-
кого нейрона; в результате индуцируется дальнейшая передача
импульса. Глутамат, ацетилхолин выполняют возбуждающую функцию;
у-аминобутират, глицин, таурин — тормозную функцию.
При эпилепсии нарушается деятельность тормозных структур
центральной нервной системы. В эпилептическом очаге имеет место
дисбаланс между тормозными и возбуждающими медиаторами. Так,
изменения в глутаматергической системе играют важную роль в
патогенезе эпилептических приступов. При введении глутамата в
определенные отделы мозга возникают судорожные приступы. Известно, что
действие барбитуратов, обладающих антисудорожной активностью,
основано на их способности связываться с глутаматными рецепторами
и подавлять их функционирование.
Возможной причиной возникновения судорожных состояний
могут быть также изменения в у-аминобутиратергической системе. Как
отмечалось выше, у-аминобутират обладает противоположным
нейрофизиологическим действием по сравнению с глутаматом. Введение
у-аминобутирата в мозг подавляет эпилептические приступы.
Характерно, что на у-аминобутиратных рецепторах также имеются участки,
связывающиеся с барбитуратами, однако в данном случае это
взаимодействие приводит к усилению эффекта медиатора.
Блокада рецепторов тормозных медиаторов специфическими
соединениями вызывает судорожные приступы. Так действует пикроток-
син, подавляющий функционирование рецепторов у-аминобутирата;
аналогичным образом действует стрихнин, блокирующий рецепторы
глицина.
607
Часть III. Элементы прикладной биохимии _^_____
В патогенезе эпилепсии существенную роль могут играть и опиоид-
ные пептиды (энкефалин, р-эндорфин).
Отдельные факты свидетельствуют о том, что в генезе
психопатологических приступов определенное значение может иметь и
генетический фактор.
Шизофрения. Шизофрения (от греч. всЫго — раскалывать,
разделять; рНгеп — разум) — это психическая болезнь с приступообразным
или непрерывным течением, проявляющаяся изменениями личности —
нарушением мышления, снижением психической активности; могут
возникать бред, галлюцинации. Однако сохраняются формальные
способности интеллекта: приобретенные знания, память и т.д.
Для данного заболевания характерны изменения ряда
биохимических процессов в организме. Существенную роль в патогенезе
шизофрении могут играть перестройки метаболизма. В частности, при
шизофрении происходят изменения ферментных систем, участвующих
в обмене биогенных аминов. Метаболические нарушения приводят к
накоплению метилированных продуктов обмена аминов. Кроме того,
в головном мозге образуется избыточное количество дофамина, что
может быть результатом нарушения механизмов его распада или
усиления синтеза* Интересно, что галоперидол, применяющийся в
качестве антипсихотического средства, является специфическим
ингибитором рецепторов дофамина. Однако "дофаминовую гипотезу" нельзя
считать полностью доказанной, так как ни в головном мозге, ни в
цереброспинальной жидкости не обнаружено изменения уровня гомо-
ванильной кислоты, являющейся основным продуктом обмена
дофамина, а также активности дофамин-р-гидроксилазы, метаболизирую-
щей дофамин.
Существует гипотеза, согласно которой гиперактивность дофами-
нергических нейронов может быть вторичным процессом,
индуцированным первичными изменениями в метаболизме эндогенных опио-
идов. Предполагается, что эндорфины играют роль модуляторов в
процессе передачи информации через дофаминовые синапсы, В пользу
этого свидетельствует существование тесной взаимосвязи между эн-
дорфиновой и дофаминовой системами в мозге.
Превращения биогенных аминов по метаболическим путям,
практически не функционирующим в норме, при шизофрении могут
приводить к образованию соединений, обладающих психомиметической
активностью. Так, окисление катехоламинов по хиноидному пути
приводит к возникновению адренолютина и адренохома, способных
вызывать психомиметические эффекты.
По некоторым данным, изменения в глутаматергической системе
могут также иметь отношение к развитию заболевания*
В целом можно констатировать, что несмотря на недостаточность
сведений, позволяющих оценить механизмы патогенеза шизофрении,
это заболевание сопряжено с нарушениями нейрохимических процес-
608
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
сов. В основе этих нарушений могут быть как генетические причины,
так и результаты воздействия каких-либо внешних факторов.
16.1.4. Некоторые аспекты патобиохимии
онкологических заболеваний
Некоторые особенности метаболизма при опухолевом росте.
Характерной особенностью опухолевых клеток является интенсивное
клеточное деление и рост. Это сопряжено с ускорением синтеза
клеточных макромолекул, причем с одновременной утратой способности
к выполнению своих специфических функций.
В опухолевых клетках значительно интенсифицируется
образование белков, что предположительно связано с повышением
проницаемости клеточных мембран для субстратов, необходимых для
белкового синтеза. Интенсивно растущие новообразования извлекают из
крови необходимые аминокислоты, лишая тем самым здоровые клетки
жизненно важных субстратов. Нарушения азотного метаболизма яв-
ляются, очевидно, одной из причин развития раковой кахексии —
синдрома, характеризующегося общим истощением организма,
снижением массы тела, мышечной слабостью» Наряду с изменениями в
белковом спектре крови (рост уровня гликопротеинов во фракциях
глобулинов, снижение уровня сывороточного альбумина) развитие
новообразований сопровождается появлением аномальных белков (а~> (3-,
у-фетопротеинов, канцероэмбрионального антигена), синтез которых
в норме прекращается в процессе онтогенеза. Например, (3-фетопроте-
ин не обнаруживается у взрослых здоровых людей, однако
выявляется при карциноме легких; а-фетопротеин выявляется при
карциномах печени; для у-фетопротеина, обнаруживаемого с достаточной
частотой при развитии новообразований, не выявлена специфичность по
отношению к определенным видам опухолей. Увеличение содержания
канцероэмбрионального антигена типично при опухолевых процессах
желудочно-кишечного тракта (толстый кишечник, поджелудочная
железа, печень).
При развитии злокачественных новообразований происходит
накопление нуклеиновых кислот, активируется ряд ферментов,
ответственных за синтез пуриновых и пиримидиновых оснований: ДНК-
полимераза, аденилсукцинатсинтетаза, аденилсукциназа, дигидрооро-
таза, уридинкиназа и др.
Для большинства опухолевых клеток характерен анаэробный
гликолиз, сопровождающийся продуцированием лактата. При этом чем
выше скорость роста опухоли и чем менее она дифференцирована, тем
интенсивнее протекает гликолиз и слабее окислительное фосфорили-
рование. По-видимому, такие сдвиги метаболизма сопряжены с
недостаточностью оксигенации опухолевых клеток, что связано с их
быстрым размножением при слабой васкуляризации, т.е. обеспеченности
кровеносными сосудами.
39. Заказ 3486
609
Часть III. Элементы прикладной биохимии
Интенсифицируется также утилизация глюкозы через пентозофос-
фатяый путь, что приводит к повышенной продукции рибозо-5-фосфа-
та, служащего ключевым интермедиатом в синтезе нуклеотидов и
нуклеиновых кислот.
Отмеченные особенности углеводного обмена сопряжены с
изменениями изоэнзимного спектра ряда важнейших ферментов
энергетического обмена: лактатдегидрогеназы, гексокиназы, пируваткиназы и
др. Так, например, индуцируется изофермент III гексокиназы,
характеризующийся очень высоким сродством к глюкозе. Это позволяет
ассимилировать глюкозу даже в очень низких концентрациях и
успешно конкурировать с нормальными клетками за этот жизненно
важный метаболит. Таким образом, опухоль действует как фактор,
вызывающий гипогликемию, что сопровождается сдвигами гормонального
баланса, в частности сверхпродукцией глюкокортикоидов.
Повышение уровня глюкокортикоидов приводит к стимуляции
глюконеогенеза. Основными субстратами глюконеогенеза являются продукты
превращения аминокислот. Такие метаболические перестройки
сопряжены с потерей энергии, так как в результате гликолиза образуются
2 молекулы АТР в расчете на одну молекулу глюкозы, в то время как
для глюконеогенеза необходимо 6 молекул АТР. Дефицит АТР
компенсируется ускорением распада глюкозы. Содержание гликогена в
опухолевых тканях снижается. Опухоль действует как ловушка не
только для глюкозы и азотистых соединений, но и для витаминов,
особенно витамина Е, обладающего антиоксидантными свойствами
(см. гл. 5).
Для организма с прогрессирующей опухолью характерны
существенные метаболические сдвиги, индуцируемые, с одной стороны,
дефицитом ряда важнейших жизненно важных соединений, с другой —
продуцированием опухолевыми клетками биологически активных
соединений, дезорганизующих обмен веществ. Так, некоторые виды опу-
холей секретируют эктопические гормоны. Например, в отличие от
нормальных тканей, злокачественные опухоли легкого и
дыхательных путей способны к эктопической секреции адренокортикотропного
гормона или, реже, глюкагона и инсулина. Кроме того, в организм
поступают продукты распада опухолевой ткани, образующиеся в
некротических очагах внутри опухоли. Формирование зон некроза в
опухолях связано с недостаточной обеспеченностью кислородом и
питательными веществами.
Нарушения обмена веществ у онкологических больных являются
ключевым звеном развития иммунодепрессивного состояния. Один из
путей снижения жизнеспособности иммунокомпетентных клеток (ти-
моцитов, лимфоцитов) — это гиперпродукция глюкокортикоидов,
приводящая к лизису этих клеток и распаду ДНК, ответственной за их
формирование. Подавление функциональных способностей
иммунокомпетентных клеток коррелирует со снижением потребления глюкозы
610
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
тканями организма, возрастанием в сыворотке крови уровня холесте-
рола, неэтерифицированных ненасыщенных жирных кислот,
возникновением резистентности к инсулину.
Молекулярные аспекты развития опухолей. В процессе
развития опухоли, или так называемого канцерогенеза* принято выделять
две фазы: трансформация клеток и прогрессирующий опухолевый рост.
Злокачественное перерождение клетки может происходить под
действием вирусов, химических и физических канцерогенов. Основой
трансформации клетки является повреждение ДНК, что может быть
результатом воздействия различных факторов.
Химический канцерогенез может быть вызван действием
различных канцерогенов, находящихся в окружающей среде. К
канцерогенам относятся, например, полициклические углеводы, способные
внедряться между основаниями ДНК; алкилирующие соединения,
ароматические ариламины и ариламиды, образующие с пиримидиновы-
ми и пуриновыми основаниями ковалентные связи. Образование
комплекса "канцероген—ДНК" отрицательно сказывается на
функционировании клетки. Если этот дефект не будет устранен, клетка может
либо погибнуть, либо трансформироваться в опухолевую клетку с
модифицированным геномом. Деление такой клетки приводит к
переносу мутации на последующие генерации. Это и может стать ключевым
моментом злокачественного перерождения. Однако трансформация не
может возникнуть в результате одноразового воздействия на
структуру ДНК. Процесс трансформации происходит достаточно
продолжительное время. Латентный период, т.е. время от начала
трансформации до проявления клинических признаков прогрессии опухоли,
может составлять от 10 до 50 дней.
Физический канцерогенез может быть обусловлен
ультрафиолетовым и радиационным облучением. Причем доказано
преимущественное появление опухолей в тканях, непосредственно подвергшихся
облучению. Трансформация клетки в опухолевую зависит от дозы и вида
облучения.
Вирусный канцерогенез. Онкогенные РНК-содержащие вирусы (см.
гл. 13) способны вызывать опухолевую трансформацию клеток, что
определяется наличием в их геноме онкогенов. Эти онкогены исходно
не присущи вирусам. Они приобретают их из генома клеток, в
которых они существуют. Онкогены могут значительно
модифицироваться в результате множественных мутаций и вновь попадать в клетки
животных, в результате чего последние приобретают способность к
нерегулируемому делению и индукции опухолей. Следует отметить,
что в геноме эукариотных организмов присутствуют протоонкогены,
являющиеся предшественниками онкогенов. Именно эти гены и
приобретаются вирусами и затем подвергаются мутациям. Однако прото-
онкогены обычно не вызывают опухолевую трансформацию клеток.
Убедительно доказано, что достаточно замены одного основания за
39*
611
Часть III. Элементы- прикладной биохимии „_^
счет точечной мутации, чтобы протоонкоген превратился в онкоген и
приобрел способность вызывать опухолевую трансформацию.
Индуцирующим фактором злокачественных новообразований может быть
транслокация протоонкогена из одной хромосомы в другую;
оказавшийся в другом геномном окружении протоонкоген попадает под
действие регулятора генной активности (промотора), активируется сверх
нормы, в результате появляется способность к нерегулируемому
росту. Согласно теории "аутокринной регуляции", нерегулируемое
размножение клетки является следствием аутостимуляции этого
процесса за счет дефектов в системе переноса митогенного сигнала.
Следует отметить, что нормальная функция протоонкогенов — это
участие в регуляции клеточного деления, что, естественно, жизненно
важно для многоклеточного организма. В норме свою функцию они
выполняют адекватно времени и месту. Однако под действием каких-
либо неблагоприятных воздействий протоонкогены способны выходить
из-под контроля и приводить к нерегулируемому клеточному
делению.
16.1.5. Патобиохимия нарушений функций сердца при ишемии
В нормальных условиях для сердца характерен аэробный обмен.
Источником кислорода является коронарная кровь, из которой при
прохождении миокарда извлекается до 70 % кислорода. Высокая
степень извлечения кислорода сопряжена с высокой интенсивностью
окислительных процессов, в первую очередь цикла трикарбоновых кислот
и р-окисления жирных кислот. Почти всю энергию сердце получает за
счет окислительного фосфорилирования. АТР — источник энергии для
его сокращения. Недостаточность кислорода в каком-либо участке
сердечной мышцы, где кровеносные сосуды оказались
заблокированными, например, отложениями липидов (см. разд. 16.3), приводит к
ишемии сердца. Прекращение кровоснабжения может привести к
некрозу определенного участка сердечной мышцы, в результате чего
развивается процесс, известный как инфаркт миокарда.
Для метаболизма ишемизированного миокарда характерны
снижение интенсивности окислительного фосфорилирования и стимуляция
анаэробного расщепления углеводов. Активация анаэробного
гликолиза является дополнительной возможностью восполнения
энергетических потребностей в условиях ишемии. Интенсификация гликолиза
и гликогенолиза на ранних этапах ишемии за счет использования
имеющегося в миокарде гликогена и усиленно поглощаемой глюкозы
индуцируется в результате повышения внутриклеточной
концентрации сАМР и катехоламинов. Повышение уровня сАМР
обеспечивается активацией аденилатциклазы (см. гл. 13), очевидно, за счет
выброса эндогенных катехол аминов. Это, в свою очередь, приводит к
образованию активной формы гликогенфосфорилазы, т*е. фосфорилазы а
и активации фосфофруктокиназы — регуляторного фермента глико-
612
Глава 16, Некоторые аспекты медицинской и фармацевтической биохимии
лиза. Однако усиленный анаэробный метаболизм не способен
длительное время поддерживать близкий к норме уровень АТР. Запасы
гликогена быстро истощаются. Внутриклеточный ацидоз, связанный с
накоплением лактата, ведет к торможению гликолиза вследствие ин-
гибирования фосфофруктокиназы. Снижение уровня АТР
сопровождается накоплением продуктов ее распада: АВР и АМР. При
снижении уровня АТР более чем на 80 % происходит деструкция клеточных
мембран кардиомиоцитов, нарушается система транспорта АТР,
наблюдается усиленный выход АТР из цитоплазмы. Снижение
концентрации АТР в кардиомиоцитах до критического уровня обычно
происходит через 40—60 мин после полной окклюзии коронарной артерии.
При длительной ишемии начинают интенсифицироваться процессы
распада аденозина с образованием гипоксантина и ксантина.
Появление этих соединений в крови может служить показателем
выраженности ишемии сердечной мышцы.
Наряду со снижением содержания АТР при ишемии в
кардиомиоцитах резко уменьшается концентрация креатинфосфата,
выполняющего функцию транспорта макроэргов в миокарде. Это
сопровождается ингибированием активности креатинфосфокиназы и
существенными изменениями ее изоферментного спектра. В результате повышения
проницаемости клеточных мембран происходит выход этого фермента
в кровь, и по активности этого фермента в крови можно судить о раз-
мерах некротического очага миокарда. Весьма чувствительным
тестом повреждения миокарда при инфаркте является определение изо-
фермента МВ креатинфосфокиназы. Помимо креатинкиназного теста
в диагностике инфаркта миокарда часто используется определение в
сыворотке крови активности лактатдегидрогеназы, аспартатамино-
трансферазы, а также миокардиально специфичных белков — тропо-
нина Т и миоглобина.
В норме в миокарде процессы пероксидного окисления липидов
(ПОЛ), обеспечивающие регуляцию липидного состава клеточных
мембран, координируются под действием прооксидактов и антиоксидан-
тов, стимулирующих и подавляющих эти процессы соответственно.
Ключевым звеном в цепи патологических изменений в ишемизиро-
ванном миокарде является чрезмерная активация ПОЛ.
Значительная активация ПОЛ, вызываемая дисбалансом прооксидантной и ан-
тиоксидантной систем, имеет место уже на ранних этапах ишемии
(через 10—20 мин). В дальнейшем ПОЛ приобретает лавинообразый
характер, распространяясь из зоны ишемии на отдаленные от нее
участки миокарда. Это сопровождается существенными модификациями
структуры и функций клеточных мембран, включая связанные с
мембраной ферментные системы. Повышение мембранной проницаемости
приводит к выходу из клетки ионов К+ и накоплению Са2+ и Иа4",
чему способствует подавление функционирования энергозависимых
ионных насосов. Нарушение ионного баланса является причиной
613
Часть III. Элементы прикладной биохимии
развития аритмии, депрессии сократительной функции сердечной
мышцы.
Кислородная недостаточность приводит к разбуханию кардиомио-
цитов и клеточных органоидов, в том числе митохондрий; происходит
расширение митохондриальных крист и разрыв митохондриальной
мембраны.
На поздней стадии ишемии миокарда (в течение первых двух
суток от начала инфаркта) синтез нуклеиновых кислот и белков
подавлен. Затем содержание ДНК и белков начинает постепенно нарастать.
Увеличивается интенсивность пентозофосфатного пути и
генерирование ЫАВРН. Это, в свою очередь, способствует ускорению синтеза
липидов, в ходе которого потребляются лактат и пиру ват.
Однако и в поздние сроки (более 10 дней после инфаркта)
содержание АТР и креатинфосфата остается сниженным, и показатели
функционального состояния миокарда не достигают нормальных значений.
16.2. Некоторые аспекты фармацевтической биохимии
16.2.1. Лекарственные и диагностические средства
Лекарственные средства — это лекарственные соединения,
используемые для диагностики, ликвидации или профилактики
заболевания. Изучением методов и приемов биосинтеза лекарственных средств,
эффективности их действия занимается фармацевтическая биохимия.
Лекарственные средства используются в качестве лекарственных
препаратов в виде форм, обеспечивающих оптимальное лечебное действие
и удобных для применения и хранения. Одно и то же лекарственное
средство может существовать в различных формах: в твердом
(таблетки, драже, порошки, капсулы), в жидком (суспензии, эмульсии,
настои, отвары, сиропы и др.)> в газообразном (аэрозоли) состояниях. В
лекарственные препараты наряду с основным действующим веществом
вносят вещества-добавки, обеспечивающие более рациональный
характер действия, регулирующие скорость поступления в организм,
повышающие устойчивость препарата при хранении*
В зависимости от специфики действия лекарственные средства
подразделяют на ряд групп: а) на основании локализации действия в
отдельных органах (например, сердечные гликозиды); б) по механизму
действия (например, ингибиторы аминооксидазы); в) по
принадлежности к классу веществ биогенной природы (например, гормоны,
ферменты, витамины); г) по содержанию в них активных элементов
(например, железо, йод, натрий и др.).
В подгруппах лекарственные средства классифицируют по
химическому строению (производные барбитуровой кислоты), по принципу
действия (антикоагулянты), по источникам получения (препараты
ландыша, наперстянки). Противомикробные, противопаразитарные
лекарственные средства подразделяют преимущественно по спектру
614
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
действия (например, противотуберкулезные), по химическому
строению (например, сульфамидные препараты), по особенностям
применения (например, антисептические, дезинфицирующие средства).
Современная медицина все шире использует ферменты в качестве
перспективных средств медикаментозного лечения вследствие их
исключительно высокой активности и специфичности. Имеют место
следующие направления применения ферментов в медицине: устранение
дефицита ферментов с целью компенсации врожденной или
приобретенной функциональной недостаточности; удаление нежизнеспособных
денатурированных структур, клеточных и тканевых фрагментов;
лизис тромбов; комплексная терапия злокачественных новообразований;
детоксикация организма. Например, протеазы (в основном, трипсин,
а-химотрипсин, террилитин, коллагеназа, субтилизин) находят
применение при лечении ожогов и удалении патологических продуктов,
скапливающихся в очагах гнойно-некротических процессов; хорошие
результаты получают при лечении лучевых язв кожи, трофических
язв. Тромболитические ферменты используют при лечении тромбозов
конечностей, коронарных сосудов, легких. Так, с большим успехом
используется препарат "стрептодеказа", представляющий собой
фермент стрептокиназу, иммобилизованный на полисахариде. Этот
фермент способствует активации плазминогена, являющегося
предшественником протеиназы плазмина, который предотвращает тромбообразо-
вание. Ферментный препарат Ь-аспарагиназы применяют при
лечении некоторых онкологических заболеваний. Раковые клетки
некоторых типов не могут осуществлять синтез аспарагина, и расщепление
аспарагина, поступающего с током крови, под действием Ь-аспараги-
назы лишает злокачественные новообразования ресурсов необходимой
для них аминокислоты. При лечении различных патологий часто
используются также ингибиторы ферментов, в том числе белковой
природы.
Широкое применение в медицине получили гормональные
препараты* Так, пептидный гормон инсулин представляет собой основное
средство лечения при сахарном диабете. Другой пептидный гормон -
соматотропин используется для лечения карликовости; этот гормон
способствует также заживлению ран и в комплексе с гормоном
щитовидной железы кальцитонином регулирует обмен Са2+ в костной
ткани. Кортизон, являющийся стероидным гормоном надпочечников,
используется при лечении ревматоидного артрита. Другой стероидный
гормон — преднизолон применяется при лечении различных кожных,
глазных и других заболеваний.
Лечение ряда заболеваний оказывается возможным только
благодаря применению антибиотиков — веществ, представляющих собой
продукты жизнедеятельности и характеризующихся высокой
биологической активностью к определенным группам микроорганизмов,
задерживающих или полностью подавляющих их развитие. К антибио-
615
Часть III. Элементы прикладной биохимии
тикам терапевтического назначения принадлежат, например, тетра-
циклинЫу механизм действия которых рассмотрен в гл. 13. Их
продуцентами являются актиномицеты рода 8№ер№тусез. Антибиотики
данной группы эффективны против грамположительных и грамотри-
цательных бактерий, хламидий, риккетсий. Пенициллины, цефало-
спорины относятся к классу ф-лакгпамных антибиотиков.
Продуцентами являются грибы родов РепьсШшт, СерНа1озрвгит* Эти
антибиотики подавляют грамположительные и грамотрицательные бактерии
путем нарушения синтеза клеточной стенки. Широко применяемые
антибиотики стрептомицин, канамицин, тобрамицин, гентамицин
действуют в основном на грамотрицательные бактерии за счет
необратимого подавления белкового синтеза. Данные антибиотики
относятся к классу аминогликозидных; продуцентами являются
актиномицеты рода 8Шр1отусез, бактерии родов Мгсготопо8рогау ВасШиз.
Приведенные примеры далеко не исчерпывают многообразия
антибиотиков.
Для борьбы с инфекционными заболеваниями используются еак
цины. Препараты вакцин производят на основе инактивированных или
ослабленных возбудителей болезни. С помощью вакцинации
ликвидировано такое распространенное в прошлом заболевание, как оспа,
значительно ограничена возможность распространения полиомиелита,
бешенства и ряда других заболеваний.
В медицине находят применение также интерфероны^
обладающие антивирусной активностью. Выделяют следующие группы интер-
феронов: а-, р-, у-, е-интерфероны, образуемые лейкопластами, фиб-
робластами соединительной ткани, Т-лимфоцитами и
эпителиальными клетками соответственно. Интерфероны построены из 146—166
аминокислотных остатков. В состав (3- и у-интерферонов входят остатки
сахароз• Интерфероны могут быть полезны при лечении
онкологических заболеваний, болезней, вызываемых вирусами гепатитов,
бешенства, герпеса.
Интерлейкины — лечебные средства, применяемые при
иммунных расстройствах. По структуре они представляют собой
полипептиды, содержащие около 150 аминокислотных остатков. Макрофаги,
являющиеся специфической группой лейкоцитов крови, продуцируют
интерлейкин-1. При попадании в организм антигена интерлейкин-1
обеспечивает стимуляцию пролиферации Т-хелперов, представляющих
собой субпопуляцию Т-лимфоцитов. Т-хелперы образуют интерлей-
кин-2, стимулирующий размножение В-лимфоцитов (продуценты
антител) и разных субпопуляций Т-лимфоцитов: Т-киллеров,
Т-хелперов, Т-супрессоров (рис» 16.3.)- Под действием интерлейкина-2 из Т-лим-
фоцитов высвобождаются так называемые лимфошны,
представляющие собой регуляторные белки, активирующие определенные звенья
иммунной системы. Интерлейкин-1 рекомендуют для лечения
некоторых злокачественных заболеваний.
616
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
Макрофаг
Т-Хелпер
Интерлейкин-1
Антигсп
Интерлейкин-2
и
Пролиферация
Т-лимфоцитов
Пролиферация
В-лимфоцитов
Синтез факторов
иммунной защиты
Т-Киллеры Т-Хелперы Т-Супрессоры
Рис. 16.3. Роль интерлейкинов в иммунном ответе на антиген (Кетлинская С. А.
и др., 1985)
Моноклональные антитела могут быть использованы для
диагностики ряда патологий. Так как они обладают высокой
специфичностью и избирательностью, то с их помощью можно идентифицировать
различные белковые соединения, гормоны и интермедиаты
клеточного метаболизма. В этой связи моноклональные антитела могут быть
успешно использованы для дифференциальной диагностики
различных форм рака. Они позволяют также диагностировать
наследственные заболевания, связанные с отсутствием или недостаточностью
определенных ферментов. Моноклональные антитела могут быть
использованы и для лечебных целей. Так, антитела, конъюгированные с
токсичными соединениями, могут обеспечить адресную доставку яда,
например, к раковой опухоли, и не затронуть при этом здоровые клетки.
Диагностическое значение имеют так называемые
ДНК/РНК-зонды. Они представляют собой короткие фрагменты ДНК и РНК,
содержащие радиоактивную метку. С помощью ДНК/РНК-зондов могут быть
выявлены различные аномалии в геноме пациента.
16.2,2. Биотехнология лекарственных средств
Это раздел фармацевтической биохимии, изучающий вопросы
биосинтеза, выделения, хранения и анализа лекарственных средств. Он
включает важный подраздел — биофармацию, который выявляет
влияние физико-химических свойств лекарственных форм, технологии
617
Часть III. Элементы прикладной биохимии ^ ^
лекарственных препаратов на биологическое действие лекарств.
Биофармация рассматривает лекарственный препарат как сложную
систему, взаимодействующую с организмом.
В приготовлении лекарственных препаратов большое значение
имеют природа и количество вспомогательных веществ. Необходимо
учитывать тот факт, что эти вещества могут вступать в реакцию с
лекарственным средством и существенно снижать его активность.
Поэтому использование вспомогательного вещества требует проведения
специальных исследований для выяснения его влияния не только на
технологию лекарственного препарата, но и на процессы всасывания
лекарственных средств и характер их распределения в организме.
На эффективность действия лекарств существенное влияние
оказывают производственные факторы: технологические операции,
температура, вид используемой энергии. Так, величина давления при
прессовании таблеток влияет на их распад и скорость растворения
компонентов. Грануляция (особенно влажная) может обусловить
замедление всасывания. Количество смачивающей жидкости,
применяемой при грануляции, величина и форма гранул существенно влияют
на физиологическую доступность таблетированных препаратов.
Одно и то же лекарственное вещество в кристаллическом или
аморфном состоянии обладает различным эффектом действия. Хорошо
растворимые вещества быстро высвобождаются из лекарственных форм,
легко диффундируют, быстро проявляют лечебное действие. В то же
время для пролонгирования действия более пригодны менее раствори*
мые вещества.
В фармацевтической промышленности для синтеза лекарственных
соединений нашли применение иммобилизованные ферменты.
Обладая высокой специфичностью, ферменты позволяют получить
лекарственные соединения без побочных продуктов. Так,
иммобилизованная пенициллинамидаза используется при получении аминопеницил-
линовой кислоты, которая является предшественником в синтезе пе-
нициллинов широкого спектра действия.
С помощью иммобилизации ферментов на полимерных носителях
решаются многие проблемы ферментной терапии. Естественно, что
ферменты, введенные в организм пациента, находятся в иных
условиях и в ином окружении, чем ферменты, синтезируемые в клетках
живого организма. Внутриклеточные ферменты существуют в
микроокружении, способствующем поддержанию их стабильности и
эффективному функционированию. Они могут находиться в связи с
мембранами и другими внутриклеточными белками. Введенный в организм
фермент, являясь высокомолекулярным соединением, будет медленно
диффундировать в нужное место, он может быть атакован протеиназа-
ми, ингибиторами, утилизирован фагоцитами и лейкоцитами.
Проблемы адресной доставки ферментов к пораженному органу,
снижения иммуногенности, повышения стабильности решаются путем про-
618
Глава 16, Некоторые аспекты медицинской и фармацевтической биохимии
изводства иммобилизованных ферментных препаратов. Антигенность
иммобилизованных ферментных препаратов выражена в меньшей
степени, чем обычных ферментов. Это объясняется меньшей
доступностью поверхности белка для рецепторов иммунной системы в связи с
экранированием матрицей носителя, Так, например, антигенность
иммобилизованной на полиэтиленгликоле аспарагиназы примерно в
50 раз ниже, чем нативного белка. Однако следует подчеркнуть, что
способ иммобилизации значительно влияет на иммуногенность
ферментных препаратов. Во многих случаях к снижению иммуногеннос-
ти препаратов приводит ковалентное связывание с полиэтиленглико-
лем, полисахаридами. Связывание фермента с
носителями-полиэлектролитами часто приводит к возрастанию степени антигенности. При
приготовлении лекарственных препаратов на основе ферментов выбор
носителя для иммобилизации определяется не только оценкой его
воздействия на иммуногенность, но и отсутствием токсичности,
возможностью вывода почками. В связи с последним обстоятельством
молекулярная масса полимера-матрицы не должна превышать 60 кДа или
матрица должна представлять собой достаточно легко биодеградируе-
мый полимер. В противном случае возможны осложнения, связанные
с закупоркой почечных канальцев.
Для иммобилизации протеиназ, используемых для очищения ран,
чаще всего применяют полиамидное и коллагеновое волокно,
волокнистые материалы на основе целлюлозы, поливинилового
спирта. Протеолитические ферменты включают в некоторые шовные
материалы.
Весьма перспективным направлением развития фармацевтической
промышленности является производство ферментных препаратов типа
"контейнер". Проблема введения лекарственного препарата в организм
решается путем создания для ферментов специальных
"контейнеров-переносчиков" типа липосом или так называемой "искусственной
клетки". Такие препараты представляют собой микросферы с
проницаемой оболочкой. "Искусственные клетки" получают путем микрокап-
сулирования. Для получения микрокапсул используются оболочки
эритроцитов, так называемые "тени" эритроцитов. Внутреннее
содержимое эритроцитов удаляется, а оставшаяся "тень" заполняется
белковым содержимым. Если "тени" приготовить на основе клеток,
выделенных из крови пациента, то можно добиться полной совместимости
при введении их с необходимым содержимым этому больному. В
настоящее время препараты аспарагиназы в "тенях" эритроцитов
используются для лечения опухолей. Другим способом получения
ферментных препаратов типа "контейнер" является введение лекарственного
вещества в липосомы. Гидрофильные вещества вводятся внутрь липо-
сомы, гидрофобные — в стенку. Сами липосомы образуются при
обработке ультразвуком водной дисперсии липидов выше температуры их
застывания. Липосома, несущая ферментный препарат, может быть
619
Часть III. Элементы прикладной биохимии
Эмульсия вода/ масло
8&Ш. В
а
в
поглощена клеткой путем эн-
доцитоза. Иммобилизация
ферментов с использованием
полупроницаемых оболочек
(мембран) схематично
изображена на рис, 16.4.
Ферментные препараты
типа "контейнер" имеют ряд
преимуществ. Во-первых, они
могут быть стабилизированы в
инкапсулированном состоянии
за счет введения в капсулу
стабилизирующих соединений. Во-
вторых, фермент, защищенный
"контейнером", не подвержен
разрушающему воздействию
протеиназ, ингибиторов. Он не
вызывает иммунной ответной
реакции. Наконец,
инкапсулированные ферментные
препараты могут быть достаточно
легко имплантированы в нужное
место. Поскольку оболочка
"контейнеров" проницаема для
низкомолекулярных субстратов
и продуктов их превращений,
то, находясь, например,
поблизости от опухолевой ткани,
фермент будет осуществлять
каталитическое превращение
определенных метаболитов,
необходимых для развития опухоли,
и злокачественное образование
будет лишено необходимых для
ее развития ресурсов.
Биотехнология, основанная на достижениях биохимии,
предоставляет фармацевтической промышленности новые методы получения
гормональных препаратов. Особое значение имеют методы генной
инженерии. Так, в результате включения в геном бактериальной клетки
ЕзсНепсЫа сой специфических генов осуществлен в промышленных
условиях биосинтез инсулина, соматотропина и других гормонов.
Раньше гормональные препараты получали из донорской крови,
удаленных при операциях органов и т.д. Причем для получения
гормональных препаратов требовалось очень много исходного материала.
Например, в гипофизе человека содержится не более 4 мг соматотропи-
д
Рис, 16А. Иммобилизация ферментов с
использованием полупроницаемых оболочек
(мембран): а — микрокапсулирование; б —
двойное эмульгирование; в — включение в
волокна; г — включение в полые волокна
с полупроницаемыми стенками; д ~
включение в липосомы; точками обозначены
молекулы фермента, символами 8 и Р —
субстрат и продукт ферментативной реакции
соответственно (Березин И. В. и др., 1987)
620
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
на, тогда как для лечения ребенка от карликовости требуется не
менее 7 мг в неделю, и курс лечения длится несколько лет. Инсулин
ранее получали из поджелудочной железы свиньи или быка.
Терапевтическое применение препарата инсулина было ограничено, что было
связано с его дефицитом и высокой стоимостью. К тому же
существовала опасность аллергических реакций при применении данного
препарата, обусловленная заменой некоторых аминокислотных остатков
в полипептидных цепях инсулина из тканей животных по сравнению
с человеческим гормоном. В настоящее время путем клонирования у
Е. сой гена человеческого инсулина получают гормон, идентичный
человеческому. К тому же стоимость препарата резко снижена.
С использованием генно-инженерных штаммов Е. соИ, дрожжей,
культивируемых клеток насекомых, млекопитающих получают а-, (3-,
у-интерфероны. С помощью методов генной инженерии получают
также модифицированные интерфероны, в которых производят замены
определенных аминокислотных остатков. Аминокислотные замены
изменяют антивирусную активность интерферонов. Так, замена цистеи-
на в положении 17 на серии в р-интерфероне приводит к повышению
терапевтической эффективности действия препарата за счет
предотвращения возможности образования неактивного димера р-интерферона
при возникновении дисульфидной связи между соответствующими ци-
стеиновыми остатками.
Путем клонирования определенных генов в Е. соИ или
культивирования лимфоцитов производят интерлейкины.
Методы генной инженерии оказываются полезными при создании
новых антибиотиков. Так, с целью получения более эффективных
антибиотиков в геном микроорганизмов вводится информация о
ферменте, который будет способствовать модификации продуцируемого
антибиотика. Постоянно ведется работа по поиску новых
антибиотиков. Причинами необходимости создания новых антибиотиков
являются: возрастание резистентности патогенных микроорганизмов к
существующим антибиотикам, а также токсичность многих препаратов
и их способность вызывать аллергические реакции. Для получения
модифицированных антибиотиков применяют также методы
клеточной инженерии, благодаря чему могут быть созданы гибридные
антибиотики, например, с новыми комбинациями агликона и Сахаров.
Другим подходом к повышению эффективности действия антибиотиков
является их инкапсулирование. В таком виде, например в виде липо-
сом, антибиотики могут быть доставлены к определенному органу или
ткани, и при этом наряду с повышением эффективности будет
снижено побочное действие.
На генно-инженерных подходах основаны также методы
получения рекомбинантных вакцин и вакцин-антигенов. При получении
рекомбинантных вакцин гены, кодирующие иммуногенные белки
возбудителей болезней (например, гликопротеин Х> вируса герпеса, анти-
621
Часть III. Элементы прикладной биохимии
—-^Т»^^^^»— ■** '' НИМ Щ ^^*^^- I1 ИИ! 1|' III > ,' II Щ I I1 I1 —ВДЛш**., I ,1
гены малярийного плазмодия, гепатита В), встраивают в ДНК какого-
либо хорошо известного вируса (чаще всего используют вирус
коровьей оспы). Благодаря этому получают вакцины против
соответствующих инфекций. Клонируя гены возбудителей заболеваний в Е. соИ9
клетках млекопитающих» дрожжах, получают вакцины-антигены.
Например, клонирован ген поверхностного антигена НВЗ-вируса
гепатита В. Вакцины-антигены, как правило, не вызывают аллергических
реакций. Их преимуществом является также высокая стабильность
при хранении.
Большой интерес представляет разработка биотехнологии иммуно-
препаратов, в основе которой лежит использование антител.
Некоторые аспекты биосинтеза и особенности структуры антител будут
рассмотрены в следующем разделе при ознакомлении с иммунохимичес-
кими методами анализа.
16.2.3. Биохимические методы, применяемые для мониторинга
и контроля качества лекарственных средств
Мониторинг, оценка качества и стандартизация лекарственных
средств являются важнейшими аспектами деятельности
фармацевтической службы, поскольку создают необходимую основу для
назначения лекарственных препаратов пациентам, контроля эффективности
лечения и выявления возможных побочных эффектов. Для решения
этих задач наряду с химическими методами часто применяются
также методы анализа биологической активности. Так, биохимические
методы применяются для анализа гормональной активности
препаратов. Например, показателем активности кортикотропина является
снижение содержания холестерола и аскорбиновой кислоты в
надпочечниках; инсулина — снижение содержания глюкозы в крови; сома-
тотропина — включение сульфата в хрящи. Биохимические тесты дают
возможность определения индекса терапевтической реакции,
например, путем тестирования функционального состояния щитовидной
железы у больных тиреотоксикозом, принимающих карбимазол.
Наиболее часто используют специальные методики, позволяющие
определить концентрацию лекарственного вещества в плазме крови. При этом
содержание вещества в плазме сравнивают со "стандартными
значениями", которые часто также называют "терапевтическими
значениями". Эти значения представляют собой диапазон концентраций между
минимальной эффективной концентрацией лекарственного препарата
и его максимальной безопасной концентрацией. Границы данного
диапазона не абсолютны и зависят от состояния пациента и эффекта
других препаратов.
Для иллюстрации значения мониторинга лекарственных средств
рассмотрим некоторые примеры. Применение препарата метотрексата
с целью подавления опухолевых процессов становится более
безопасным при его мониторинге в плазме. Метотрексат значительно снижает
622
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
запасы восстановленного фолата в организме за счет ингибирования
фермента дигидрофолатредуктазы, В высоких концентрациях мето-
трексат действует губительно не только на опухоль, но и на самого
человека, нарушая функцию костного мозга. Поскольку
максимальное поражение костного мозга происходит позднее, чем опухолевых
клеток, то можно предотвратить нежелательный эффект данного
препарата, назначив "препарат-спасатель" (лейковорин) в случае, когда
содержание метотрексата в плазме указывает на риск повреждения
костного мозга. Другим примером может служить мониторинг
лекарственного препарата фенитоина с целью повышения эффективности
его действия. Данное противосудорожное средство имеет низкий
терапевтический индекс и обладает специфическими фармакокинети-
ческими свойствами, так как фермент, обеспечивающий его
элиминацию, насыщается при концентрациях, обычно используемых в
терапии. Это приводит к тому, что зависимость между принимаемой дозой
и содержанием препарата в плазме крови имеет нелинейный
характер. В результате незначительное увеличение дозы препарата или же
снижение активности метаболизирующего его фермента за счет
подавления активности (например, какими-либо другими препаратами) может
привести к весьма существенному повышению концентрации
лекарственного вещества в плазме. Это сопровождается появлением признаков
токсичности, напоминающих неврологические заболевания (эпилепсия).
Для проведения высокоспецифичного анализа используются
ферментные электроды, которые представляют собой электрохимические
датчики, работающие на основе иммобилизованных ферментов. С
помощью ферментных электродов возможно не только многократное
определение содержания тех или иных веществ в отдельных пробах,
но и непрерывный анализ, что особенно важно в промышленных
условиях.
Биохимические автоматические анализаторы разрабатываются на
основе проточных анализаторов с иммобилизованными ферментами. Так,
с использованием иммобилизованной пенициллиназы разработаны
методы определения пенициллина в фармацевтических препаратах.
Широкое распространение получают иммунохимические методы,
основанные на образовании комплексов антител с антигеном.
Антитела — белки сыворотки крови, синтезирующиеся в
организме позвоночных в ответ на появление чужеродного вещества
антигена. По своей структуре иммуноглобулины (понятия антитела и
иммуноглобулины являются практически синонимами) представляют
собой четырехцепочечные молекулы, состоящие из двух пар легких (Ь)
цепей (молекулярная масса — 22 000) и из двух пар тяжелых (Н)
цепей (50 000—70 000), связанных с олигосахаридными
фрагментами. Соединение цепей происходит за счет дисульфидных связей
(рис, 16.5). Выделяют 5 классов иммуноглобулинов человека: 1^0,1§М,
1#А, 1&Е, 1$Т>. Классификация основана на различиях в последова-
623
Часть III. ЭЛементы прикладной биохимии
Шарнирная
область
Рис. 16.5. Схематическая структура молекул иммуноглобулина: Ь
Н — тяжелая цепи; А — активный центр антитела
легкая,
I С
1 1
тельности и количестве аминокислотных остатков Н-цепей,
содержании олигосахаридных фрагментов, положении и количестве дисуль-
фидных мостиков, а также способности к полимеризации. Понятие
антиген обозначает химический или биологический объект, против
которого могут быть получены антитела. Роль антигенов могут играть
низкомолекулярные вещества (например, антибиотики), а также
белки, нуклеиновые кислоты, вирусы, микроорганизмы. Антитела
образуются не против всей молекулы антигена, а к их небольшим
участкам, называемым антигенными детерминантами.
Для получения антител используются различные виды животных:
кролики, мыши, морские свинки, овцы, лошади. Инъекции антигена,
осуществляемые в присутствии стимуляторов иммунного ответа,
приводят к накоплению в сыворотке крови специфических антител. Основные
этапы получения поликлональных и моноклональных антител показаны
на рис. 16.6. При иммунном ответе на введение одного антигена у
разных особей синтезируются различные антитела, что является
отражением генетических особенностей организмов. Антитела могут быть
выделены в виде у-глобулиновой фракции путем осаждения спиртом, полиэти-
ленгликолем или сульфатом аммония. Очистка полученных таким
образом антител может быть осуществлена с помощью ионообменной
хроматографии, а также аффинной хроматографии на иммуносорбентах.
624
\
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
Полиспецифический
антиген
Иммунные
В-лимфодиты
Поликлоналъные
антитела
(А, В, С, О)
Миеломные
клетки
Слияние
Клоны
лимфоцитов
Моноклональные
антитела
Клонирован ие
и размножение
>Анти>
Анти
С
V
/
Рис. 16.6. Получение поликлональных и моноклональных антител (Бере-
зинИ. В. и др., 1987)
Следует отметить, что ограниченность использования антител для
количественного анализа антигенов сопряжена с определенными
трудностями, возникающими при получении стандартных препаратов
антисывороток. Это сопряжено с зависимостью состава этих препаратов
от ряда трудно контролируемых факторов, включая индивидуальные
особенности животного, цикла иммунизации и т.д* Даже после
очистки с применением достаточно высокоэффективных методов сыворотка
остается гетерогенной, что ограничивает ее использование. Проблема
гетерогенности сыворотки может быть решена с помощью получения
40, Заказ 3486
625
Часть III. Элементы прикладной биохимии
моноклональных антител, основанного на методе, включающем
клонирование В-лимфоцитов, образующих только один тип антител.
Время жизни В-лимфоцитов при переводе их из организма-хозяина в куль-
туральную среду очень ограничено. Поэтому осуществляют слияние
В-лимфоцитов с "бессмертными" опухолевыми клетками В-лимфоци-
тарного происхождения (миеломные лимфоциты), способными
неограниченно долго жить в культуре. В результате образуется смесь
гибридных клеток. Из них отбирают гибриды, синтезирующие антитела
определенного вида и способные размножаться в культуре. Такие
гибридные клетки, или гибридомы, клонируют. Полученные клоны
являются источниками моноклональных антител, однородных по
структуре и специфичности.
Определение антигенов и антител осуществляется с помощью
иммунологического анализа, для которого разработаны многочисленные
варианты, предусматривающие применение меченых реагентов. С этой
целью могут быть использованы разнообразные метки. Применяют
радиоактивное мечение изотопами: 1311, 1251, 3Н. Однако данные
реагенты нестабильны из-за радиоактивного распада и представляют
определенную опасность для здоровья и окружающей среды. В этой
связи в последнее время все более широкое использование в качестве
маркеров находят ферменты. Например, применяют пероксидазу и
фосфатазу, которые при добавлении к соответствующим субстратам
образуют окрашенные продукты. На использовании ферментов
основан твердофазный иммуноферментный анализ (ИФА, или ЕЫ8А, от
англ. епгуте-Ипкей гттиповогЪеШ аввау), позволяющий определять
антитела и антигены. Схема проведения основных этапов иммунофер-
ментного анализа показана на рис. 16.7.
Методы иммуноферментного анализа применимы для контроля
лекарственной терапии, особенно препаратов, влияющих на
сердечнососудистую систему, антибиотиков. Эти методы позволяют выявлять
наличие психотропных препаратов в организме, наркотиков. Методы
иммуноферментного анализа используются в контроле
биотехнологических процессов и качества лекарственных препаратов. Так, в
микробиологических производствах иммуноферментный анализ может
применяться для выявления высокоэффективных микроорганизмов —
продуцентов биологически активных веществ (антибиотиков,
ферментов и т.д.). Иммуноферментный анализ может также использоваться
для контроля появления посторонних микроорганизмов,
бактериофагов в ферментерах. Классический радиоиммунологический анализ
(РИА) является примером жидкофазного иммунологического
анализа. Разработка методов мечения антигенов до высокой удельной
активности открывает возможности выявления очень низких
концентраций антигена (до 10"12г/мл). РИА успешно используется для
выявления вируса гепатита, большинства белковых гормонов и
низкомолекулярных веществ (стероиды, простагландины, соединения группы
626
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
Отмывка
Культуральные жидкости,
содержащие антитела
Антиген
Твин-20,
альбумин
Отмывка
Добавление конъюгирован-
ных с ферментом антител
против 1& мыши
=с=с=с
Инкубация и отмывка
Фермент
Антитела
против 1& мыши
о
о
о
о
Субстрат
Добавление субстрата
4
Инкубация и отмывка
от о# о#
^ Ч^ Ч_/
■Продукт
=€=!=€
о
о
о
о
Инкубация
Изменение окраски
означает положительную
реакцию
Рис. 16.7. Схема проведения основных этапов иммуноферментного анализа
(Бутенко Р. Г. и др., 1987)
морфина). Иммунорадиометрическое определение антигена основано
на твердофазном иммунологическом анализе. Твердую поверхность,
например пластик, нагружают антителами и добавляют исследуемый
раствор. После отмывания количество антигена определяют, добавляя
избыток радиоактивно меченых антител.
40*
627
Часть III. Элементы прикладной биохимии ^^
16.2.4. Некоторые аспекты метаболизма лекарств
С биохимической точки зрения лекарственные соединения
относятся к ксенобиотикам. Метаболизм лекарственных соединений
зависит от наличия в организме ферментов, которые способны
катализировать их превращение. Эти ферменты должны обладать ярко
выраженной групповой специфичностью к различным субстратам. Если
такие ферменты отсутствуют, то лекарственные соединения
выводятся из организма практически в неизменном виде.
Существуют два основных пути превращения лекарственных
соединений в организме — модификация и конъюгация.
Модификация включает реакции окисления, восстановления,
гидролиза, а также ацетилирования, алкшгарования. Под действием
специфических ферментов исходная структура соединения переходит в
модифицированную форму. В молекулу лекарственного соединения
вводятся дополнительные функциональные группы (например, гидро-
ксильная) или выводятся собственные функциональные группы
(например, путем гидролиза гликозидных, эфирных, пептидных связей).
В результате биотрансформации лекарственное соединение переходит
в гидрофильное состояние, что способствует его дальнейшим
превращениям и выделению из организма* Во многих случаях
биотрансформация является предшествующей стадией для конъюгации
лекарственного соединения.
Конъюгация представляет собой процесс образования ковалентных
связей между молекулой лекарственного соединения с
биомолекулой-метаболитом (например, с глюкуроновой кислотой, сульфгидриль-
ным соединением). При конъюгации образуется новое вещество,
частями которого являются, с одной стороны, лекарственное соединение,
с другой — молекула конъюгирующего вещества.
В ряде случаев лекарственные средства используют в различных
сочетаниях, что приводит к усилению их действия, простому
сложению действия каждого из компонентов или наоборот, подавлению
действия одного компонента другим. Последний вид взаимодействия
применяют для лечения отравлений или передозировок, а также для
коррекции побочных эффектов лекарственных средств.
Наряду с лечебным эффектом лекарственные средства могут
вызвать аллергию или другие нежелательные реакции, связанные с
неизбирательностью их действия, индивидуальными особенностями
организма, а также токсическими эффектами при дозировках
лекарственных средств, которые приводят к нарушению функций органов и
систем организма. Эти побочные эффекты возникают в основном при
нарушениях врачебных указаний и самолечении.
Подвергшись различным химическим превращениям, лекарственные
средства выводятся из организма. В неизменном виде выделяются
главным образом гидрофильные соединения; липофильные — средства инга-
628
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
ляции, наркоза, не вступая в химические реакции с веществами систем
и органов, выводятся через легкие с выдыхаемым воздухом.
Краткое содержание главы
Медицинская биохимия рассматривает молекулярные механизмы,
лежащие в основе метаболических процессов, протекающих в
организме в норме, а также при их нарушениях в связи с возникновением
заболеваний.
К группе так называемых молекулярных болезней относятся
заболевания, обусловленные дефектами определенных структурных генов.
Как следствие, это приводит к синтезу дефектных белков,
утрачивающих способность выполнять свои функции. Молекулярные болезни
подразделяются на энзимопатии и неферментные протеинопатии.
Примером энзимопатии является фенилкетонурия. В ее основе лежит на-
рушение метаболизма фенилаланина в связи с недостаточностью фе-
нилаланингидроксилазы. Отсутствие специфических ферментов
является причиной возникновения тирозинемии, непереносимости
отдельных Сахаров (лактозы, сахарозы и др.)- К энзимопатиям
относятся также гликогенозы (печеночный, мышечный, генерализованный),
лизосомные болезни, связанные с нарушениями липидного обмена.
Причиной неферментативных патологий являются нарушения
транспорта веществ через мембраны, дефекты образования антител и
синтеза гормонов; так, недостаток гормона инсулина ведет к
тяжелому заболеванию — сахарному диабету.
Примером патологии питания может служить дефицит белка в пище. !|
Это приводит к болезни, называемой квашиоркор, встречающейся у де- !|
тей, не имеющих полноценной диеты. Причиной патологии питания мо- [
жет быть также подавление образования пищеварительных ферментов [
или дефицит желчи. Нарушение липидного обмена лежит в основе ши- |
роко распространенного заболевания — атеросклероза. Оно связано с от- !
ложением на стенках кровеносных сосудов холестерола и проявляется в \
виде ишемической болезни сердца, инсульта, тромбозов. I
Ряд нервно-психических заболеваний человека также сопряжен с I
биохимическими изменениями организации и регуляции клеточного )
обмена веществ. \
Характерной чертой возникновения и развития злокачественных 1
4
опухолей являются интенсивное клеточное деление и рост, причем с
одновременной утратой способности клеток к выполнению своих спе~ !
цифических функций. В опухолевых клетках идет интенсивное про- 5
дуцирование биологически активных соединений, дезорганизующих |
нормальный обмен веществ. Злокачественное перерождение клеток 1
может происходить под действием вирусов, химических канцероге- |
нов, физических факторов (например, ультрафиолетового и
радиационного облучения). |
г
I
1
1
629
Часть III. Элементы прикладной биохимии
Нарушения функционирования сердца часто сопряжены с
недостаточностью снабжения кислородом какого-либо участка сердечной
мышцы, например, вследствие блокирования сосудов отложениями липи-
дов. Прекращение кровоснабжения может привести к некрозу
участка сердечной мышцы и развитию процесса, известного как инфаркт
миокарда. Для метаболизма ишемизированного миокарда характерны
снижение интенсивности окислительного фосфорилирования и
интенсификация анаэробного гликолиза* Однако усиленный анаэробный
метаболизм не способен длительное время поддерживать близкий к
норме уровень АТР. Уменьшается также концентрация креатинфос-
фата, выполняющего функцию транспорта макроэргов в миокарде.
Ключевым звеном в цепи патологических изменений в ишемизиро-
ванном миокарде является чрезмерная активация пероксидного
окисления липидов, что сопровождается изменениями
структурно-функциональной организации клеточных мембран, включая связанные с
ними ферментные системы.
Фармацевтическая биохимия изучает методы и приемы
биосинтеза лекарственных средств, эффективности их действия при
профилактике или подавлении заболеваний. Лекарственные средства
используются в виде лекарственных препаратов в формах, обеспечивающих
оптимальное лечебное действие и удобных для применения и
хранения. В зависимости от специфики действия лекарственные средства
подразделяются на ряд групп: в зависимости от влияния на какой-
либо орган, по механизму действия, по специфической биологической
активности, по содержанию активных элементов.
Важным разделом фармацевтической биохимии является
биотехнология лекарственных средств, изучающая вопросы биосинтеза,
выделения и анализа лекарственных средств, природу и количество
вспомогательных веществ, вносимых в лекарственный препарат. На
эффективность действия лекарств существенное влияние оказывают
производственные факторы: технологические операции, температура и т.д.
В биотехнологии лекарственных препаратов весьма перспективно
применение иммобилизованных ферментов. Особое значение имеют
методы генной инженерии. На их основе осуществлен в промышленных
условиях биосинтез инсулина, соматотропина, интерферонов, интер-
лейкинов. Методы генной инженерии широко используются при
создании новых антибиотиков, вакцин-антигенов, рекомбинантных
вакцин.
В деятельности фармацевтической службы важное место занимает
мониторинг, задача которого — оценка качества и стандартизация
лекарственных средств. Он дает возможность определить, в
частности, индекс терапевтической реакции. Причем биохимические
методы и подходы часто незаменимы для получения необходимой
информации. Так, широкое распространение получают иммунологические
методы, основанные на образовании комплексов антител с антигеном.
630
Глава 16. Некоторые аспекты медицинской и фармацевтической биохимии
На молекулярном уровне исследуется механизм действия антигенных
детерминантов —• специфических участков антител.
Лекарственные соединения являются ксенобиотиками. Они
выводятся из организма либо в неизменном виде, либо подвергаются
модификации или конъюгации* Модификация в организме человека
включает самые разнообразные типы реакций — окисление,
восстановление, гидролиз, ацелирование, алкилирование* В результате
биотрансформации лекарственное соединение переходит в гидрофильное
состояние и выводится из организма. В основе конъюгации лежат реакции
образования ковалентных связей между молекулой лекарственного
соединения и молекулой-метаболитом, что способствует
обезвреживанию чужеродного вещества.
Хронология важнейших дат в развитии
медицинской и фармацевтической биохимии
1581 г. — царь Иван IV издал указ об открытии в Москве первой
аптеки. При Петре I зарождается фармацевтическая промышленность,
основную базу которой составили лекарственные огороды.
1783—1788 гг. — Н. М. Амбодик-Максимович составил описание
целительных растений.
1883 г. — И. И. Мечников (лауреат Нобелевской премии)
открыл явление фагоцитоза, показал, что функцию фагоцитоза
выполняют лейкоциты; он является создателем фагоцитарной теории
иммунитета.
1908 г. — А. Е. Гаррод сформулировал концепцию
наследственных нарушений обмена веществ.
1920 г. — основан Всесоюзный научно-исследовательский химико-
фармацевтический институт.
1928 г. — С. Ван-Кревельд описал клиническую картину гликоге-
ноза.
1929 г. — А. Флеминг открыл пенициллин, а в 1940—1942 гг.
Г* Флори, Е* Чейн, 3. В. Ермольева и Т. И. Балезина получили его в
чистом виде.
1931 г. — организован Всесоюзный научно-исследовательский
институт лекарственных и ароматических растений.
1934 г. — С, Н. Давиденков установил существование
генетической гетерогенности наследственных болезней и причин их
клинического полиморфизма*
1952 г. — И. Помпе провел энзимологическое исследование глико-
геноза.
1952 г. — учрежден Институт фармакологии АМН СССР.
1953 г. — П. Л. Сенов исследовал лекарственные растения
Дальнего Востока, Средней Азии; разработал методы физико-химического
анализа сложных лекарственных средств.
631
Часть III. Элементы прикладной биохимии
1964 г. — Л. Полинг (лауреат Нобелевской премии) выдвинул
концепцию "молекулярных болезней", вызываемых нарушением синтеза
того или иного белка или образованием функционально
неполноценных белков. Показал, что серповидноклеточная анемия связана с
наличием в крови аномального гемоглобина.
1970 г. — А. Н. Климов открыл фермент гидроксиметилглутарил —
СоА-редуктазу, играющий ключевую роль в регуляции скорости
биосинтеза холестерола; он является автором автоиммунной теории
патогенеза атеросклероза.
ЛИТЕРАТУРА
Барашнев Ю. И.» Вельпгищев Ю. Е. Наследственные болезни обмена у детей.
— Л.: Медицина, 1978. — 319 с.
Березов Т. Т., Коровкин Б. Ф. Биологическая химия: Учебник. — 3-е изд.,
перераб. и доп. — М.: Медицина, 1998. — 704 с.
Бышевский А Ш.ш Терсенов О, А. Биохимия для врача. — Екатеринбург:
Изд-во "Уральский рабочий", 1994. — 383 с.
Вилкинсон Д. Принципы и методы диагностической энзимологии. — М.:
Медицина, 1981. — 624 с.
Виноградов А, 5. Дифференциальный диагноз внутренних болезней. — М.:
Медицина, 1987. — 592 с.
Зилва Дж., Пэннел 77, Р. Клиническая химия в диагностике и лечении, —
М.: Медицина, 1988. — 528 с.
Кнорре Д. Г., Мызина С.Д* Биологическая химия: Учебник. — 3-е изд., испр.
— М.: Высш. шк., 2000. — 479 с.
Колов В- 17., Фирсова В. И, Фармацевтическая биохимия: Учеб, пособие. —
СПб.: Изд-во Химико-фармацевтического института, 1992. — 77 с.
Ленинджер А. Основы биохимии; В 3 т. — М.: Мир, 1985. — Т. 3. — С. 743—
1056.
Марри Р., Тренер Д., Мейес П., Родуэлл В. Биохимия человека: В 2 т. — М.:
Мир, 1993. — 753 с.
Маршалл В. Дж. Клиническая биохимия. — М.: Изд-во "БИНОМ"; СПб.:
Изд-во "Невский Диалект", 1999. — 357 с.
Мусил Я. Основы биохимии патологических процессов. — М*: Медицина,
1985. — 314 с.
Ройпг А. Основы иммунологии. — М.: Мир, 1991. — 327 с.
Сливкин А. И., Селеменев 5. Ф., Суховеркова Е. А. Физико-химические и
биологические методы оценки качества лекарственных средств: Учеб. пособие /
Под ред. В, Г. Артюхова, А. И. Сливкина. — Воронеж: Изд-во ВГУ, 1999. —
368 с.
Стпрайер Л". Биохимия: В 3 т. — М.: Мир, 1985. — Т. 2 — 307 с.
Строев Е* А Биологическая химия. — М.: Высш, шк., 1986. — 478 с.
Элементы патологической физиологии и биохимии / Под ред. И. П. Алшари-
на. — 2-е изд. — М.: Изд-во МГУ, 1997. — 238 с.
Эллиот А, Эллиот Д. Биохимия и молекулярная биология / Под ред. А. И. Ар-
чакова, М. П. Кирпичникова, А. Е. Медведева, В. П. Скулачева — М.: Изд-во
НИИ Биомедицинской химии РАН, 1999. — 327 с.
632
Глава 16, Некоторые аспекты медицинской и фармацевтической биохимии
ВОПРОСЫ
1. Назовите и охарактеризуйте молекулярные болезни человека и причины
их возникновения.
2. Назовите и охарактеризуйте болезни, возникающие при нарушениях
употребления белков, жиров, углеводов в ходе питания.
3. Какие биохимические механизмы лежат в основе развития
нервно-психических заболеваний человека? Назовите соединения, обладающие
психомиметической активностью.
4. Какие нарушения азотного метаболизма взаимосвязаны с развитием
новообразований? С чем связано развитие раковой кахексии?
5. К каким сдвигам метаболизма приводит слабая васкуляризация
опухолевых клеток?
6. Назовите основные фазы развития опухоли (канцерогенеза)? Какие типы
канцерогенеза Вам известны?
7. Каковы основные нарушения метаболических процессов при ишемии
миокарда?
8. Каковы основные направления использования в медицине ферментных и
гормональных препаратов?
9. Приведите примеры антибиотиков терапевтического назначения.
Охарактеризуйте их продуцентов и механизм действия.
10. Для каких целей в медицине применяются интерфероны, интерлейкины,
вакцины, моноклональные антитела?
11. Расскажите о современных направлениях биотехнологии лекарственных
средств. Каким образом иммобилизация ферментов позволяет решать проблемы
энзимотерапии? Каковы основные подходы биотехнологий, основанных на
достижениях генетической инженерии?
12. Какие биохимические методы применяются для мониторинга и контроля
качества лекарственных препаратов?
Глава 17. БИОХИМИЧЕСКИЕ ПРЕВРАЩЕНИЯ
ВАЖНЕЙШИХ ПИЩЕВЫХ КОМПОНЕНТОВ
ПРИ СОЗРЕВАНИИ, ХРАНЕНИИ И ПЕРЕРАБОТКЕ
СЕЛЬСКОХОЗЯЙСТВЕННОГО СЫРЬЯ
Важнейшими пищевыми компонентами продукции
растениеводства являются белки, углеводы и жиры. Они же служат запасными
питательными веществами семян, клубней, корнеплодов, при
прорастании которых эти вещества — источник пластического
материала и энергии. Существенные преобразования происходят с ними
при биосинтезе в растениях, хранении и переработке растительного
сырья.
17Л. Биохимические превращения
при отложении запасных веществ в растениях
Белки в зерновых злаках. Зерновые злаки являются основным
поставщиком белков в питании населения Земли. Среди них следует
особо выделить пшеницу, мука из которой широко используется в
производстве хлебобулочных и мучных кондитерских изделий. Эта
культура обладает высокой эффективностью синтеза белков,
количество которых в зрелом зерне может доходить до 25 % от массы зерна,
в то время как в других зерновых культурах (рожь, ячмень, овес,
просо) — до 15 %, а в рисе — около 10 %. Биосинтез белка в
зерновых культурах зависит от многих факторов, основными из которых
являются сортовые (генетические) особенности растения,
климатические условия, технология выращивания.
При выращивании пшеницы особое внимание уделяется
биосинтезу белков клейковины — глиадина и глютенина.
В период созревания зерна в нем идет интенсивный биосинтез
аминокислот и формируется клейковина. В конце фазы молочной
спелости клейковина имеет плохую связность и низкую гидратационную
способность, а к полной спелости зерна она приобретает нормальные
реологические (физико-механические) свойства. Количество небелковых
азотистых веществ и неклейковинных белков увеличивается только
до конца молочной спелости, а далее остается практически
постоянным. Наоборот, абсолютное содержание клейковины в зерне
значительно увеличивается. Установлено, что с самого начала налива зерна
пшеницы глиадин и глютенин существуют в виде комплекса.
634
Глава 17. Биохимические превращения важнейших пищевых компонентов,..
Большое влияние на биосинтез белков в зерне злаковых оказывают
климатические условия. Содержание белка в зерне одного и того же
сорта в разных районах может отличаться на 10 % и более. Для территории
России характерно, что количество белка в зерне при продвижении с
северо-запада на юго-восток, как правило, повышается; чем жарче лето
и меньше осадков, тем больше белков в зерне злаковых. Повышается
синтез белка при внесении азотных и фосфорных удобрений — первые
являются источником пластического материала в метаболизме растений,
вторые важны для интенсификации процессов биоэнергетики.
Важен для урожая и качества зерна его сорт (генетический
фактор). При оптимальных условиях азотного питания увеличивается
количество клейковины, улучшается качество. Однако превратить
генетически слабый сорт пшеницы в сорт сильный за счет усиленного
азотного питания невозможно, поскольку признак качества, во
многом определяющий хлебопекарные свойства зерна, находится под
жестким генетическим контролем. Сортовые особенности четко
проявляются при формировании клейковины в зерне различных сортов
пшеницы в одних и тех же условиях. Установлено, что различия между
сортами пшеницы достаточно высоки — до 40 % по содержанию
сырой клейковины.
Белки в зернобобовых культурах. Зернобобовые культуры —
ценнейший источник белка. Важным для них признаком является
способность фиксации азота воздуха при помощи клубеньковых
бактерий, находящихся в симбиозе с корневой системой растения. В связи
с этим зернобобовые культуры богаты белками, количество которых
может достигать 30 % общей массы зерна. Они являются в то же
время прекрасным источником азота в почве, повышения ее
плодородия. Синтезируются в основном белки группы глобулинов (60—80 %
от общей массы белков). В начальный период роста и развития в
бобовых растениях сравнительно мало азота, затем при усилении
интенсивности фиксации бактериями азота из воздуха его количество
значительно повышается и достигает максимума в период цветения растения.
Биосинтез белка в зерне бобовых культур зависит от почвенно-
климатических условий. Для гороха, фасоли, чечевицы пределы
изменчивости составляют 10—15 %, а для сои — превышают 20 %.
Интенсивный биосинтез белка идет на черноземах в жаркое и сухое
лето (Кубань, Волгоградская область). Северо-западные районы
России с их бедными почвами, более холодным и влажным климатом
дают урожай зернобобовых с относительно меньшим содержанием
белков. Таким образом, и здесь выявляется такая же закономерность,
что и для зерновых. Однако в семенах зернобобовых культур эта
относительная изменчивость значительно ниже, чем в зерне злаковых. Она
связана с симбиозом корневой системы растения с
жизнедеятельностью клубеньковых бактерий. Повышают интенсивность биосинтеза
белка фосфорные и калийные удобрения.
635
1
Часть III. Элементы прикладной биохимии
Углеводы в важнейших сельскохозяйственных культурах.
Источником углеводов в питании человека являются зерновые
культуры, картофель, сахарная свекла, плоды и овощи. Основным запасным
углеводом злаковых является крахмал. Он используется в качестве
источника углерода и энергии при прорастании семян, является
источником энергии и углерода в пище человека. Особенно много
крахмала в семенах кукурузы, пшеницы и риса.
Рреднее содержание крахмала зерна злаков (в % от сухой массы)
Пшеница — 60 Ячмень — 55
Рожь — 55 Просо — 48
Кукуруза — 70 Овес — 45
Рис — 68
В зависимости от сорта и условий выращивания культур содержание
крахмала в зерне может изменяться на 10—15 %* Обычно содержание
крахмала уменьшается при увеличении в зерне количества белков.
В начальный период налива в зерне идет интенсивное накопление
полифруктозидов (около 1/3 сухой массы), сахарозы и
моносахаридов. Сахароза и моносахариды играют роль транспортных углеводов и
оттекают из листьев в колос. В период молочно-восковой спелости
увеличивается приток этих углеводов в семена, резко повышается
синтез крахмала. Важную роль в превращении указанных углеводов
играет система трансгликозилирования. В период полной зрелости
количество растворимых углеводов в зерне снижается. Однако в нем
остаются, иногда в значительном количестве, сахароза, глюкоза,
фруктоза. Эти сахара используются в первый период прорастания
развивающегося зародыша.
На рис. 17.1 представлено строение ячменного зерна, состоящего
из эндосперма, алейронового слоя, плодовой оболочки, цветочной
пленки, щитка и зародыша. Максимальное количество Сахаров
содержится в зародышах семян, их много также в алейроновом слое, тогда как
в центральной части эндосперма находится в основном крахмал.
Общее содержание Сахаров в зрелом зерне отдельных видов злаков
составляет 3—5 %; лишь в семенах ржи оно может достигать 10 %. На
долю сахарозы приходится не менее половины Сахаров, причем почти
вся она находится в зародыше семян, что свидетельствует о ее важной
биохимической роли в первый период развития зародыша при
прорастании зерна.
В зерне ржи, пшеницы, кукурузы количество целлюлозы
достигает 2—3 %♦ Она синтезируется главным образом в семенных оболочках
и клеточных стенках. Целлюлозе в семенах злаков сопутствуют геми-
целлюлозы.
Поскольку целлюлоза и гемицеллюлозы входят в состав
клеточных стенок растений, казалось бы, они должны относиться к числу
соединений, принимающих ограниченное участие в обмене веществ
636
Глава 17. Биохимические превращения важнейших пищееых компонентов...
клеток. Однако это не так: установлено,
например, что при созревании ржаного
зерна происходит уменьшение
абсолютной массы этих полисахаридов в стеблях
и листьях растения, отток и
преобразование их системой трансгликозилирования
в крахмал и другие полисахариды зерна.
На биосинтез углеводов в зерне
оказывают существенное влияние
климатические факторы. Установлено, что ;зри
продвижении по нашей стране на юг и
восток количество крахмала в зерне
уменьшается. При повышенной влажности,
низких температурах интенсифицируется
синтез углеводов.
Вторым источником углеводов в
питании человека является картофель.
Высокое содержание крахмала — основной
показатель качества картофеля. В
пищевой промышленности картофель идет на
производство крахмала, патоки,
глюкозы, спирта. В процессе биосинтеза
крахмал в клубнях накапливается от 10 до
25 %; среднее содержание его в клуб-
2
3
Рис. 17Л. Строение
ячменного зерна: 2 — эндосперм;
2 — алейроновый слой; 3 —
плодовая оболочка; 4 —
цветочная пленка; 5 — щиток;
6 — зародыш
нях — 18 %. Интенсивность биосинтеза
крахмала и его накопление в клубнях зависит от сортовых
особенностей картофеля и условий выращивания. В ранних сортах картофеля
крахмала меньше, чем в средне- и позднеспелых сортах. На пищевые
цели выращивают среднекрахмалистые сорта, для промышленной
переработки — высококрахмалистые.
Крахмал синтезируется в хлоропластах, затем превращается в
сахарозу, которая перемещается в запасные органы -— клубни и вновь
преобразуется в крахмал- Образование клубней и, следовательно,
синтез крахмала начинается примерно через месяц после появления
всходов, В начальный период клубнеобразования превращение сахарозы в
крахмал идет с невысокой скоростью и значительная часть сахарозы
остается в свободном состоянии. Поэтому молодые клубни
характеризуются невысоким отношением крахмал/сахароза. В период
интенсивного клубнеобразования, совпадающий с отмиранием листьев,
скорость биосинтеза крахмала в клубнях резко повышается и
соотношение крахмал/сахароза увеличивается до 60 и более.
Интенсивность биосинтеза крахмала в различных частях клубня
картофеля неравномерна. Наибольшее его количество накапливается
в камбии и во внешних слоях сердцевины (рис. 17.2). Различаются по
содержанию крахмала клубни одного и того же растения. Как прави-
637
Часть III. Элементы прикладной биохимии
_^,_ ло, клубни среднего размера более высо-
/^^^^^^^Шг>к 1 кокрахмалисты, чем мелкие и крупные.
1-^ШШШЩг^^^Ш^^ Накопление крахмала в клубнях может
т-'^-;^ 2 существенно изменяться в заБисимости от
\^^Ш^^:^^ШРг^* з района выращивания, типа почвы, в носи-
^^^Ш^Щ^^/ мых в нее удобрений, влажности. Измен-
^--*. ;^зк^^ чивость содержания крахмала в клубнях
< т л „ в зависимости от условий выращивания од-
Рис. /7.2. Распределение ' 0 п/
крахмала в клубне картофеля: ного й того же С0Рта может Достигать 8 %.
1 _ сердцевина; 2 - камбий; в южных районах России содержание
3 — кожура крахмала в клубнях выше, чем в
северных; выше его содержание в засушливые
годы, хотя урожайность картофеля снижается. При недостатке в почве
фосфора наблюдается уменьшение содержания крахмала в клубнях.
Калийные удобрения, умеренные дозы азотных удобрений повышают крах-
малистость клубней и сбор крахмала с единицы посевной площади.
Третьим важным источником углеводов в питании является
сахарная свекла. Она служит сырьем в производстве сахарозы — пищевого
сахара. В зависимости от сорта свеклы и условий выращивания
содержание сахарозы может изменяться от 12 до 24 % и в среднем
составляет 16—20 % на сырую массу.
Накопление сахарозы в корнеплодах и их рост начинается после
развития достаточно мощного листового фотосинтетического
аппарата. В молодых корнеплодах отношение сахароза/моносахариды
находится на низком уровне. Появление в листьях сахарозофосфатсинте-
тазы приводит к увеличению и абсолютного, и относительного
содержания сахарозы.
Синтез сахарозы в корнеплоде идет в различных частях с
различной интенсивностью. В хвостовой части и особенно в головке корня
накопление сахарозы наиболее низкое, чем в центральной части.
Максимальное количество сахарозы накапливается несколько ниже
наиболее широкой части корйя (рис. 17.3).
Биосинтез сахарозы в сахарной свекле зависит от условий ее
выращивания. На этот процесс оказывают влияние интенсивность и
длительность освещения. При выращивании свеклы в более северных
районах накапливается меньше сахарозы, чем на юге. Качество сахарной
свеклы тем выше, чем больше в корнях сахарозы и меньше других
растворимых в воде компонентов. При переработке такой свеклы на
сахарных заводах чистота свекловичного сока будет выше, выше и
выход кристаллического сахара. На биосинтез сахарозы и качество
свеклы влияют агротехника возделывания, состав и дозы удобрений,
вносимых в почву. Снижение влажности почвы вызывает повышение
содержания моносахаридов, пектиновых веществ, снижается
биосинтез сахарозы. Внесение одних азотных удобрений хотя и ведет к
повышению урожайности свеклы, но ухудшает ее качество. Фосфорные и
638
Глава 17. Биохимические превращения важнейших пищевых компонентов...
97-98
99-99,8
96-96
97-98
99-99,8
97-98
93-94
95-96
91-92
калийные удобрения повыша- 50-60
ют биосинтез сахарозы, улуч- 88-90
шают качество свеклы. Фосфор
усиливает синтез ферментов,
участвует непосредственно сам
в синтезе сахарозы,
уменьшается содержание "вредного
азота" в корне.
Четвертым источником
углеводов в пище являются
плоды и овощи. На долю
углеводов приходится подавляющая
часть органических веществ.
Количество Сахаров в плодах
колеблется от 8 до 20 %.
В период формирования и
роста плодов на материнском
растении из моноз идет синтез
крахмала, затем при
созревании и послеуборочном
дозревании крахмал почти полностью
превращается в сахарозу,
которая при последующем хранении
плодов под действием Р-фрук-
тофуранозидазы превращается
в глюкозу и фруктозу.
Имеется обширная группа плодов, в которых отсутствует синтез
крахмала. Однако в их тканях содержание Сахаров во время
созревания увеличивается. В этом процессе важную роль играет реакция транс-
гликозилирования, в которой гемицеллюлозы, целлюлоза и
пектиновые вещества частично превращаются в сахара.
В большинстве овощных культур (капуста, огурцы, томаты,
перцы) сахара накапливаются до 3—5 %, в некоторых сортах лука — до
15 % на сырую массу. Синтезируются в основном моносахариды,
сахароза накапливается в малых количествах, за исключением лука,
содержащего в основном сахарозу. Крахмала в овощах очень мало (от
ОД до 0,5 %).
Повышенное содержание Сахаров весьма важно при засолке
огурцов, квашении капусты, мочке яблок. В результате этих
технологических операций происходит молочнокислое брожение, сахара
превращаются в молочную кислоту, и чем больше в этих продуктах
образуется молочной кислоты, тем дольше они хранятся и имеют лучшие
вкусовые качества.
Кроме Сахаров, в овощах содержатся целлюлоза, гемицеллюлоза,
пектиновые вещества. Целлюлоза синтезируется в количестве от 0,2
Рис. 17.3. Распределение сахара в
корне сахарной свеклы (% от максимального
количества)
639
Часть III. Элементы прикладной биохимии
до 2,5 %♦ При более высоком содержании целлюлозы, имеющем место
при старении растений, качество овощей ухудшается.
Пектиновые вещества синтезируются в большинстве овощей: так,
в плодах томатов их количество находится в пределах ОД—0,2 %, в
капусте — от 0,5 до 2,5 %. При переработке плодов томатов они
играют определенную роль, обусловливая плотность плодов и
консистенцию продуктов, получаемых при консервировании и переработке. У
овощей, выращиваемых в более южных районах, содержание сахара
повышается; такая же картина имеет место в сухие, жаркие годы.
Существенное влияние на синтез Сахаров в овощах оказывают
удобрения. Фосфорные удобрения, наряду с увеличением урожая,
повышают содержание Сахаров. Как правило, при избыточном,
одностороннем азотном удобрении хотя урожайность овощей и возрастает,
содержание Сахаров уменьшается; это обусловливает накопление
азотсодержащих соединений, особенно небелковых форм, что делает
небезопасным использование этих овощей в пищу.
Липиды в масличных культурах. Главным компонентом липи-
дов масличных культур являются ацилглицеролы и свободные
жирные кислоты — пищевые растительные масла* Важнейшим
источником масел являются подсолнечник, лен, конопля, горчица,
хлопчатник, в семенах которых накапливается 20—45 % масла на сухую
массу. Основная масличная культура в России — подсолнечник, в зрелых
семенах которого содержится 25—45 % масла. Низкомолекулярные
насыщенные жирные кислоты (капроновая, каприловая, лауриновая)
образуются преимущественно в масличных культурах тропических
стран (оливковое, пальмовое масла). В масличных растениях
умеренных и северных широт они не найдены, здесь синтезируются главным
образом высокомолекулярные насыщенные и ненасыщенные жирные
кислоты. Для большинства масличных культур, выращиваемых в
России, характерно наличие в масле двух насыщенных кислот —
пальмитиновой и стеариновой и трех ненасыщенных — олеиновой, лино-
левой и линоленовой, на долю которых приходится около 80 % всех
жирных кислот.
Во время созревания семян масличных культур интенсивный
синтез масел идет из углеводов. Вскоре после цветения в семенах
отмечается высокое содержание поли- и олигосахаридов, и особенно
сахарозы, а количество липидов незначительно. После окончания стадии
вегетации начинается интенсивное превращение углеводов через
процесс гликолиза в липиды. У льна, подсолнечника интенсивный
биосинтез масел и расход на них углеводов наступают через 10—15 дней
после цветения.
В период созревания семян масличных культур существенно
меняется и качественный состав масел: уменьшается количество
свободных жирных кислот в результате биосинтеза ацилглицеролов,
возрастает степень ненасыщенности жирных кислот. В зависимости от почв,
640
Глава 1 7. Биохимические превращения важнейших пищевых компонентов...
влажности, внесения удобрений, агротехники интенсивность
биосинтеза и накопления масел может изменяться в 1,5—2,0 раза. При
низких температурах и повышенной влажности возрастает биосинтез
масел с высоким йодным числом. В северных и западных районах
России больше синтезируется ненасыщенных жирных кислот. При
внесении в почву фосфорных и калийных удобрений биосинтез жиров
возрастает. Азотные удобрения способствуют увеличению синтеза
ненасыщенных кислот.
Как правило, более интенсивный синтез жиров в семенах
масличных культур ведет к большому накоплению ненасыщенных кислот и,
следовательно, повышению пищевой ценности жиров.
17.2. Биохимические превращения
при хранении сельскохозяйственного сырья
Основным биохимическим процессом при хранении растительного
сырья является дыхание. Различают два типа дыхания: с доступом
кислорода воздуха — аэробное дыхание и без доступа кислорода
воздуха — анаэробное дыхание. Следовательно, этим организмам присущ
эффект Пастера (см. гл. 9).
Анаэробное дыхание описывается уравнением спиртового брожения.
Оно возникает в экстремальных условиях, когда в газовой среде,
окружающей дышащий объект, нет кислорода* По всей вероятности,
возможен и смешанный тип дыхания (в зависимости от концентрации
кислорода в газовой среде) — аэробный и анаэробный. Судить о типе дыхания
можно по так называемому дыхательному коэффициенту,
представляющему собой отношение количества выделившегося диоксида углерода к
количеству поглощенного кислорода К = [С02]/[02]* В случае аэробного
типа дыхания К = 1. При развитии анаэробного дыхания К > 1.
При хранении картофеля, сахарной свеклы, влажного зерна потери
их сухой массы могут достигать больших величии. Интенсивное
дыхание этих растительных объектов может привести к существенному
изменению газового состава среды: исчезновению кислорода, накоплению
диоксида углерода, увлажнению дышащего объекта. Выделяемое тепло
интенсифицирует процесс дыхания. Все это в совокупности способствует
переходу от аэробного типа дыхания к анаэробному.
Различные продукты растительного происхождения резко
отличаются по интенсивности дыхания. Сухие семена слабо дышат, более
интенсивно дышат сочные плоды и овощи, картофель и сахарная
свекла. Наибольшую интенсивность дыхания обнаруживают
микроорганизмы, особенно микромицеты. Поэтому эпифитная микрофлора
зерна, картофеля, сахарной свеклы может оказывать существенное
влияние на качество хранящегося объекта.
Биохимические процессы, происходящие в зерне при хранении.
В зерне после уборки продолжаются процессы метаболизма, связан-
41. Заказ 3486 641
I
1
Часть III. Элементы прикладной биохимии
ные с его дозреванием. Полная физиологическая зрелость зерна
злаковых культур наступает через б—8 недель хранения. При дозревании
снижается влажность зерна» продолжается синтез белков из аминокислот,
крахмала из Сахаров, происходит снижение общего количества
водорастворимых соединений, уменьшение гидрофильности биополимеров.
Влажность зерна является основным фактором, обусловливающим
интенсивность его дыхания. Вода в зерне находится в двух
состояниях: 1) свободная вода и 2) связанная (адсорбционная) вода.
Связанная вода прочно удерживается белками, крахмалом и
другими биополимерами. Она не может перемещаться из клетки в клетку
и почти не участвует в метаболизме. С повышением влажности в
клетках зерна появляется свободная вода, слабо или совсем не
удерживаемая биополимерами, она может перемещаться из клетки в клетку. С
появлением свободной воды резко возрастает активность гидролаз,
оксидоредуктаз, а вместе с этим и процесс дыхания.
На рис. 17.4 представлена зависимость интенсивности дыхания зер-
у
на проса от его влажности. По данным В. Л. Кретовича, зерно с влаж-
ностью 14—15 % дышцт в 3—4 раза интенсивнее, чем сухое зерно с
влажностью меньше 14 %. Сырое зерно с влажностью, превышающей
17 %, дышит в 20:—30 раз интенсивнее сухого. Именно поэтому
влажное зерно может очень быстро согреться и испортиться.
Влажность зерна, при превышении которой в нем появляется
свободная вода и резко возрастает интенсивность дыхания, называется
критической, или базисной. Для злаковых культур она равна 14,5—
15,5 %♦ Критическая влажность масличных культур равна 8—10 %.
Это связано с тем, что в масличных семенах содержится в больших
количествах жир — гидрофобное соединение. Если пересчитать
содержание влаги на массу зерна без жира, то влажность его будет той же
величиной, что и критическая влажность зерна злаковых культур.
Вторым важным фактором, определяющим интенсивность дыхания
зерна, является температура.
При повышении температуры
интенсивность дыхания
возрастает и достигает максимума
при 50—55 °С. Дальнейшее
повышение температуры
приводит к ослаблению
интенсивности дыхания и
отмиранию зерна в связи с
разрушением протоплазменных
структур клеток (рис. 17.5).
Однако доминирующую роль
в определении
эффективности дыхания зерна играет
влажность.
о
о
>>
о
о
1-4
к
■=;
О
ад
о
11 12 13 14
15 16 17 18
Влажность, %
Рис. 17.4. Зависимость интенсивности
дыхания семян проса от влажности
642
Глава 17. Биохимические превращения важнейших пищевых компонентов...
Е-
О р-
%
2 »
► ь
ё%
° я
н
Я о
«1 С
ч о
Ф X
3 >>
ъ °
И
^да
180
160
140
120
100
80
60
40
20
1 «-^
0 10 20 30 40 50 60 70
*, °С
Рис. ./7.5. Влияние температуры на
интенсивность дыхания пшеничного зерна с
влажностью 14 % (1), 16 % Г2/, 18 % ^З; и
22 % (4)
При влажности зерна,
превышающей критическую,
процесс дыхания
приобретает автокаталитический
характер. В результате
выделения воды и дальнейшего
повышения влажности зерна,
выделения тепла и плохой
теплопроводности зерна
температура зерновой массы
резко возрастает. Этот процесс
получил название
самосогревания зерновых масс. Уже на
первой стадии
самосогревания, при повышении
температуры до 25—30 °С»
наблюдается снижение прорастае-
мости зерна, что особенно •
важно, если зерно идет на посев или солодоращение. В следующей,
второй, стадии самосогревания, при повышении температуры до 35—
38 °С, в зерне появляется солодовый залах; оно отпотевает, сыпучесть
его понижается. Наблюдается нарастание растворимых углеводов,
повышение кислотности зерна, появление спирта; начинается процесс
денатурации белка. На этой стадии интенсивно развиваются
микроорганизмы, особенно грибы из родов А&рег§Шиз и РетсШшт. И,
наконец, третья стадия самосогревания характеризуется повышением
температуры до 50 °С и выше, резким снижением сыпучести зерна,
появлением затхлого и плесневого запаха. В зерне активно
функционируют гидролазы, которые вместе с термофильными микроорганизмами
вызывают расщепление биополимеров, гниение белка.
Зерно, находящееся во второй и третьей стадиях самосогревания,
для пищевых целей непригодно.
Биохимические процессы, происходящие в картофеле при
хранении. Клубень картофеля покрыт кожурой, пропитанной суберином;
снаружи кожура имеет пробковый слой. Такая кожура не пропускает
влагу и недоступна для большинства микроорганизмов.
Дышит картофель через чечевички-почки, которые образуются в
глубоких слоях тканей, но врастают в кожуру. В отличие от зерна,
форма дыхания картофеля — аэробная. Он не приспособлен к
анаэробной форме. Даже при кратковременном анаэробиозе клубни
картофеля подвергаются быстрой порче. Как и при хранении зерна,
жизненное состояние клубня связано с неизбежными потерями наиболее
важного пищевого компонента — крахмала. Очевидно, что задача
сводится к отысканию таких условий, при которых сохранялась бы
жизнеспособность клубня, но интенсивность дыхания была бы наимень-
41*
643
Часть III. Элементы-прикладной биохимии
шей. Важнейшим фактором в регулировании дыхания картофеля
является температура.
Клубни картофеля после уборки проходят три стадии: дозревание,
покой и пробуждение (прорастание).
В стадии дозревания картофель особенно интенсивно дышит и
выделяет влагу. Примерно через месяц кожура покрывается прочным
пробковым слоем; раны, нанесенные клубням при копке и перевозке,
покрываются субериновым защитным слоем, который, однако, более
проницаем для микроорганизмов, чем пробковый. Этот процесс
хорошо идет при температуре 15—20 °С и достаточном доступе кислорода.
Стадия покоя характеризуется снижением интенсивности дыхания
и влагоотдачи* При хранении картофеля стремятся не только осла-
бить процесс дыхания, чтобы уменьшить траты крахмала, но и про-
длить срок покоя. Продолжительность покоя — 3—5 месяцев.
Наиболее простой способ продления периода покоя — хранение картофеля
при низких температурах.
При хранении идет интенсивный обмен углеводов в клубнях, и
особенно взаимопревращение крахмал ^ сахароза. При температуре,
близкой к нулевой, крахмал превращается в сахарозу; увеличивается
количество глюкозы и фруктозы. Клубни приобретают
несвойственный им сладкий вкус. Это связано с тем, что при низкой температуре
процессы распада крахмала преобладают над его синтезом. При
повышении температуры синтез крахмала начинает преобладать над его
распадом и содержание Сахаров снижается.
Температурный режим хранения картофеля играет важную роль
при дальнейшей его переработке. Качество вареного, сушеного,
хрустящего картофеля, картофельной крупки, как правило, тем выше, чем
ниже содержание Сахаров в клубнях. Весьма заметное ухудшение
качества готового продукта наступает, когда в клубнях накапливается
5 % и более Сахаров. При тепловой обработке такого картофеля
развивается процесс образования меланоидинов, ведущий к потемнению
продукта и ухудшению его вкуса; в крахмало-паточной
промышленности будут повышенные потери в производстве, поскольку сахара из
мезги уходят вместе с соковой водой.
Для уменьшения интенсивности дыхания понижают температуру
хранения картофеля. Оптимальная температура хранения клубней
находится в интервале 3—5 °С, в котором процессы превращения
крахмала в сахар и расходование последнего на дыхание находятся
практически в равновесии и потери крахмала наименьшие. Относительная
влажность воздуха в картофелехранилище должна быть 75—80 %,
т.е. должна быть равна содержанию воды в самом картофеле. При
более высокой влажности усиливается дыхание картофеля.
Биохимические процессы, происходящие в свекле при
храпении. В свекловичном корне при хранении идут многочисленные
процессы метаболизма, связанные с деструкцией низко- и высокомолеку-
644
Глава 17. Биохимические превращения важнейших пищевых компонентов...
лярных соединений. Это неизбежно приводит к снижению
технологических качеств свеклы: тем больше, чем дольше хранится свекла.
На интенсивность дыхания свеклы оказывают влияние
температура и влажность окружающей среды, физиологическое состояние
корней, удельная поверхность корней, наличие повреждений,
агротехника возделывания свеклы и сроки ее уборки. Особое значение имеет
температура- Оптимальной является 0—2 °С. При этом свекла
практически находится в состоянии анабиоза, технологические качества ее мало
изменяются, до минимума снижены траты сахарозы на дыхание.
При повышения температуры возрастают потери сахарозы.
Сложные углеводы под действием гидролаз превращаются в более простые
растворимые сахара и другие продукты неполного гидролиза,
накапливается трисахарид рафиноза; в результате гидролиза белков в
свекле появляются пептиды и аминокислоты- Поэтому длительное
хранение сахарной свеклы нежелательно.
На дыхание расходуется сахароза. Траты сахарозы неизбежны, но
их можно уменьшить, понижая температуру хранения. На рис. 17.6
приведены данные о суточных потерях сахарозы на дыхание корней
свеклы в зависимости от температуры.
Следует также учесть, что при дыхании свеклы выделяется тепло,
что очень часто влечет за собой дальнейший рост температуры с
последующим ускорением дыхания. Температурный режим хранения
свеклы нужно соблюдать особенно тщательно, так как замороженная
свекла гибнет, а при оттаивании из нее вытекает сок и теряется
большое количество сахарозы. Сахароза является прекрасной питательной
средой для множества микроорганизмов, вызывающих порчу свеклы.
Необходим достаточный приток к
корням свеклы кислорода воздуха.
Траты сахарозы усиливаются, когда
свекла переходит на анаэробный тип
дыхания. Из микроорганизмов
особенно распространены грибы Во1гуИ& селе-
геа> РНота ЬеЫеу вызывающие
гниение свеклы. После того как грибы
разрушили ткань свеклы, на ней
появляются бактерии. Из бактериальных
возбудителей болезней свеклы известны
Вас. ЪеЫе огзсозит и Вас. ЬеШе ?1аиигп9
синтезирующие из сахарозы декстра-
ны, обусловливающие слизистый
бактериоз свеклы. Декстраны сильно
повышают вязкость диффузионного сока,
затрудняют его фильтрацию.
При переработке свеклы особо
важную роль играют (3-фруктофуранози-
1,2 г
3 0,8 ■
а
о
и
я
о
О 0,2
0,6 ■
0,4 •
о
10 15 20 25 30 35
Температура, °С
Рис. 17.6. Суточные потери
сахара при хранении корней
сахарной свеклы в зависимости от
температуры
645
Часть III. Элементы прикладной биохимии
даза (инвертаза, КФ 3.2.1.26) и пектинрасщепляющие ферменты. Они
содержатся в корнях свеклы, но особенно активными продуцентами
этих ферментов являются микроорганизмы. Попадая в
диффузионный сок, р-фруктофуранозидаза вызывает гидролиз (инверсию)
сахарозы, что оказывает отрицательное влияние на дальнейшую очистку
сока и кристаллизацию сахарозы.
Инвертный сахар является не только прямой потерей сахарозы, но
и исходным соединением для образования меланоидинов,
повышающих вязкость растворов. Меланоидины и инвертный сахар резко
снижают процесс кристаллизации сахарозы.
Из пектинрасщепляющих ферментов особенно отрицательно
действие пектиназы. Сам свекловичный корень ее не содержит, однако
при заражении грибами, в частности Во1гуИ$ сепегеа, корень
размягчается, происходит гидролиз пектина. Значительная часть
пектиновых веществ переходит в диффузионный сок* При действии извести
на дефекат происходит щелочной гидролиз пектина с образованием
пектиновой (полигалактуроновой) кислоты. Последняя дает с ионами
Са2+ кальциевую соль. Эта соль забивает поры ткани, через которые
сок фильтруется. Скорость фильтрации резко снижается. Пектиновые
вещества замедляют кристаллизацию сахарозы. Оптимальная
температура развития грибов и бактерий — 25—30 °С; понижение
температуры до 0—-2 °С подавляет их развитие.
17.3. Ферменты в технологии пищевых продуктов
В технологии пищевых продуктов особенно велико значение кар-
богидраз и протеаз. Технологические режимы получения продукта в
основном определяются действием этих ферментов; они
обусловливают выход и качество продукта.
Амилазы в промышленной переработке крахмала. В основе
промышленной переработки крахмала, а также крахмалсодержащего сырья
(картофеля, семян хлебных злаков) лежит гидролиз этого полисахарида
до сахара и декстринов, которые используются в изготовлении большого
числа пищевых продуктов и напитков, а также являются источником
углерода при ферментациях. Существуют два метода гидролиза:
кислотный и ферментативный. Однако последний имеет определенные
преимущества, среди которых необходимо отметить следующие: 1) высокое
качество изготовляемого продукта, поскольку при ферментативном
гидролизе образуется меньше побочных соединений, придающих неприятный
привкус продукту; 2) специфичность действия ферментов позволяет
получить продукт с заданными физическими свойствами, например
сладостью, устойчивостью к кристаллизации, с определенной окраской, пено-
стойкостью; 3) достигается более высокий выход продукта.
Ферментативный гидролиз крахмала осуществляется с помощью
амилаз: а-амилазы, (5-амилазы и глюкоамилазы. Каждый из этих ти-
646
Глава 1 7, Биохимические превращения важнейших пищевых компонентов...
пов ферментов имеет свои специфические особенности, которые
обусловливают определенные качественные характеристики пищевого
продукта. Эти особенности широко используются в технологических
операциях»
а-Амилаза (а-1,4-глюкан-глюкангидролаза, КФ 3.2.1.1) — эндо-
фермент, гидролизующии а~1,4~связи внутри молекулы амилозы или
амилопектина, в результате образуются продукты неполного
гидролиза крахмала — а-декстрины (рис. 17.7). Поэтому а-амилазу называют
декстринирующим ферментом; она интенсивно разжижает
крахмальный клейстер, резко снижая его вязкость; не расщепляет ос-1,6-связи.
Она действует не только на клейстеризованный, но и на нативный
крахмал, разрушая крахмальные зерна. При действии а-амилазы на
амилозу можно получить при полном гидролизе около 85 % мальтозы
и 15 % глюкозы, при действии на амилопектин — около 70 %
мальтозы, 10 % изомальтозы и 20 % глюкозы.
а-Амилаза найдена у животных (слюна, поджелудочная железа), у
высших растений (солод — проросшие семена ячменя, ржи,
пшеницы, проса), у микромицетов (родов Азрег§И1и$9 КЫгориз) и у бактерий
{Вас. зиЫШз, Вас, сИаз1аисиз).
^-Амилаза (ос-1,4-глюкан-мальтогидролаза, КФ 3.2.1.2) — экзофер-
мент, гидролизует ос-1,4-связи с нередуцирующих концов молекул
амилозы, амилопектина с
образованием мальтозы;
является сахарофицирующим
ферментом; она также не
расщепляет а-1,6-связи.
При действии р-амила-
зы на амилозу можно
получить 100 % мальтозы;
при действии на
амилопектин — 60 % мальтозы;
40 % продуктов неполного
гидролиза получили
название р-амилодекстрин, на
оставшихся ответвлениях
которого находятся а-1,6-глю-
козидные связи.
Предельный декстрин гидроли-
зуется (3-амилазой только
в том случае, если в
реакционную смесь добавить
а-амилазу. При
совместном действии а- и {5-ами-
лаз на крахмал около 95 %
его превращается в маль-
Гидролиз амилозы
*
* О-О-О-О-О-А А-0-СН>0<><>-0-А (!н>0-ОЧ)ЮЧ>ЮЮ Сн>ОЮЮЮ
* А-А А-см>А А-о-о-о-А Ао-о-А А-<хьА А-о-о-о-А А-о-А А-о-А
*<йд<йй>|оД>д»й>«и»а0|аао<йд,о
1 2
Гидролиз амилопектина
СЧ>°ОООООооо<><)*0л ^
О-О-О-О-О-О-о
'о~->
*
Рис. 17.7. Действие а-амилазы на амилозу: /
— мальтоза; 2 — глюкоза; 3 — нормальный
а-декстрин; 4 — конечный а-декстрин; * —
редуцирующий конец амилозы или
амилопектина; —» — действие а-амилазы
647
Часть III. Элементы прикладной биохимии
тозу и 5 % — в низкомолекулярные предельные декстрины,
содержащие а-1,б-гликозидные связи*
Схематически гидролиз амилозы и амилопектина р-амилазой по-
казан на рис. 17.8.
Название "р-амилаза" указывает на то, что при гидролизе
крахмала первоначально образуется Р-мальтоза в результате того, что в ходе
ферментативной реакции происходит вальденовская реверсия.
р-Амилаза распространена в тканях высших растений. Хорошо
известными их источниками являются ячменный солод, пшеница, батат
и соевые бобы.
Глюкоамилаза (а-1,4-глюкан-глюкогидролаза, КФ 3.2.1.3) является
экзоферментом. Действуя с нередуцирующих концов молекулы амилозы
или амилопектина, она отщепляет молекулу глюкозы. Фермент
расщепляет не только а-1,4-, но и а-1,6-глюкозидные связи. Поэтому для глю-
коамилазы предложено систематическое название а-1,4; 1,6-глюкан-глю-
когидролаза. Амилоза и амилопектин полностью превращаются в
глюкозу* Теоретически из 100 г крахмала можно получить 111 г глюкозы*
Однако, как правило, гидролиз крахмала, особенно в высококонцеитри-
рованных растворах субстрата, не идет до конца. Препараты глюкоами-
лазы часто содержат активную трансгликозилазу, которая катализирует
синтез олигосахаридов с а-1,6-глюкозидными связями. Действие транс-
гликозилазы особенно сильно возрастает при повышении концентрации
крахмала и продуктов его гидролиза*
На рис. 17.9 показана схема действия глюкоамилазы на амилозу и
амилопектин.
Гидролиз амилозы
* с5ооооооооооооооооооооЗ)
* <5оооооооооо2) <5с 5о 5о 6<> 5о
<0 Гидролиз амилопектина
1
Рис. 17.8. Действие (5-амилазы на
амилозу и амилопектин: 1 — мальтоза; 2 —
конечный р-декстрин; * — редуцирующий конец
амилозы или амилопектина; —> — действие
В-амилазы
Гидролиз амилозы
* <5оооооооо^оооооооооооЗ)
*00ООООО0000ОО000ОООО0О 1
Гидролиз амилопектина
а
а
&
дс
1\Х
ч
Рис. 17.9. Действие
глюкоамилазы на амилозу и амилопектин:
1 — глюкоза; * — редуцирующий
конец амилозы или
амилопектина; ~> — действие глюкоамилазы
648
Глава 1 7. Биохимические превращения важнейших пищевых компонентов...
Глюкоамилаза редко встречается у бактерий; она найдена у
микромицетов родов Азрег&Шиз, НЫхориз. Из микромицетов производятся
промышленные препараты глюкоамилазы.
Способность глюкоамилазы расщеплять в крахмале а-1,4- и
а-1,6-глюкозидные связи ставит ее в разряд весьма перспективных
ферментов при промышленном гидролизе крахмала.
Амилазы в хлебопечении. Основной операцией в технологии
хлебобулочных изделий является брожение теста, вызываемое
дрожжами; его цель — разрыхление теста за счет диоксида углерода,
выделяемого при сбраживании сахара. В этом процессе амилазы играют
исключительно важную роль. Мука содержит небольшое количество
сбраживаемых Сахаров, которых хватает на весьма короткий период
брожения теста. Поэтому амилазы, гидролизуя крахмал до
сбраживаемых Сахаров, крайне необходимы для продолжения брожения теста и
его насыщения диоксидом углерода. Активность амилаз в муке
обусловливает ее сахарообразующую способность. От нее зависит
интенсивность брожения теста, количество остаточных Сахаров в нем и в
конечном счете качество хлебобулочных изделий. Однако часто саха-
рообразующая способность муки недостаточна, особенно это касается
высших сортов муки, что вызывает серьезные осложнения в
технологии хлеба, приводит к недостатку сахара в тесте, к плотному
малопористому мякишу. Поэтому для улучшения хлебопекарных свойств муки
широко применяют препараты амилаз из микромицетов. Их
добавление в количестве 0,002—0,004 % к массе муки не только повышает
скорость брожения теста, но и приводит к образованию достаточного
количества Сахаров. Повышенное содержание Сахаров в
выпекаемом хлебе улучшает его вкус и аромат, уменьшает скорость черст-
вения.
Однако высокая активность се-амилазы, полезная в процессе
брожения, может сыграть отрицательную роль при выпечке хлеба
вследствие ее дезагрегирующего действия на клейстеризованный крахмал.
Такое явление, например, имеет место в случае переработки муки из
проросшего зерна, содержащей высокоактивную, термостабильную
а-амилазу. В результате хлеб получается с липким, заминающимся
мякишем.
Следует отметить, что конечным продуктом действия а- и р-амилаз
муки на крахмал является мальтоза. При действии на крахмал а-амила-
зы и глюкоамилазы микромицетов конечным продуктом является
глюкоза. Это имеет определенное положительное значение при
использовании дрожжей с низкой активностью а-глюкозидазы (мальтазы).
Амилазы в крахмалопаточной промышленности. Крахмал и
крахмалсодержащее сырье (кукуруза, картофель) — прекрасные
источники Сахаров, получаемых в виде патоки или глюкозы.
Технология этих продуктов включает две основные стадии: клейстеризацию
крахмала и разжижение крахмального клейстера; осахаривание крах-
649
Часть III. Элементы прикладной биохимии
мала. Применяют три метода гидролиза крахмала: кислотный,
кислотно-ферментативный, ферментативный. При кислотном гидролизе
как разжижение крахмального клейстера, так и осахаривание
крахмала осуществляют одним катализатором — соляной кислотой; при
кислотно-ферментативном гидролизе разжижение осуществляют
соляной кислотой, осахаривание — амилазами; при ферментативном —
обе стадии ведут амилазами. В России получили распространение
кислотный и кислотно-ферментативный способы гидролиза; в Японии,
США широко используется ферментативный гидролиз.
Хотя соляная кислота — недорогостоящий катализатор, однако ее
применение имеет ряд существенных недостатков: 1) жесткие
факторы воздействия на крахмал (низкие значения рН, высокая
температура), что приводит к частичной деградации продуктов гидролиза; 2)
реверсия глюкозы с образованием продуктов, придающих неприятный
вкус гидролизату; 3) относительно низкая чистота гидролизатов,
обусловленная введением соляной кислоты и ее последующей
нейтрализацией. Поэтому предпочтение следует отдать ферментативному методу
гидролиза, позволяющему получить продукт более высокого качества.
При ферментативном гидролизе крахмала применяют а- и р-амилазы
ячменного солода, глюкоамилазу микромицетов, бактериальную а-
амилазу.
Первая стадия в технологии патоки — клейстеризация крахмала и
разжижение крахмального клейстера — является подготовительной
для успешного проведения второй стадии — осахаривания крахмала.
При ферментативном способе осахаривания крахмала для
разжижения крахмального клейстера применяют термостабильные
бактериальные а-амилазы. Их высокая разжижающая активность позволяет
использовать 35—40 %-ные концентрации крахмала. Поскольку
а-амилазы быстро инактивируются при высоких концентрациях Н+-ионов,
разжижение ведут при рН 5,5—7,0 — в стабильной для ферментов
зоне рН. Нагревание осуществляется ступенчато, особенно важна
пауза при температуре клейстеризации крахмала, в которой
одновременно с образованием крахмального клейстера под действием
термостабильной а-амилазы идет его разрушение; конечная
температура варьирует от 80 до 100 °С. Разжижение занимает от 2 до 4 ч
и обычно завершается, когда количество редуцирующих веществ
составит 20—25 % ♦
Вторая стадия — ферментативное осахаривание — осуществляется
препаратами микромицетов, содержащими активную глюкоамилазу.
Осахаривание ведется при температуре 55—58 °С и рН 5,0—5,5.
Дозировка препарата — ОД5—0,20 % к сухому крахмалу.
Продолжительность осахаривания колеблется от 3 до 6 ч в зависимости от
назначения вырабатываемой патоки. После осахаривания сироп нагревают до
90 °С с целью инактивации амилазы, обесцвечивают с помощью
активного угля и уваривают.
650
Глава 17. Биохимические превращения важнейших пищевых компонентов,..
Патоку используют в производстве карамельных конфет,
мармелада, пастилы, халвы, варенья, джемов, столовых сиропов, а также
алкогольных и безалкогольных напитков.
Основная задача в технологии глюкозы — осуществить глубокий
гидролиз крахмала, получить максимальный выход глюкозы.
В технологии пищевой глюкозы осахаривание крахмала
ферментативным способом проводится глюкоамилазой в течение 48—60 ч при
58—60 °С. В результате получают сироп, содержащий 96—98 %
редуцирующих веществ. Затем сироп нагревают для инактивации
фермента, уваривают до 70 % сухих веществ, охлаждают до 50 °С,
смешивают с утфелем от предыдущей кристаллизации (20—30 % от всей
массы утфеля). При постепенном снижении температуры до 15 °С утфель
затвердевает в течение 3 суток; его измельчают в порошок,
подсушивают нагретым воздухом до влажности 9 %.
Пищевая глюкоза находит широкое применение в пищевой и
медицинской промышленности.
Кристаллическую глюкозу получают аналогичным путем. Однако
главная цель здесь — получить гидролизат с высокой степенью
чистоты; от этого показателя зависит успех кристаллизации и выход
кристаллической глюкозы. Для этого используют хорошо очищенный
кукурузный крахмал. При ферментативном гидролизе в
кристаллической форме может быть выделено более 90 % глюкозы.
Кристаллическая глюкоза нашла широкое применение в медицине,
Амилазы в технологии продуктов брожения. Гидролиз
крахмала в сбраживаемые сахара является важнейшим биохимическим
процессом в технологии продуктов брожения — пива и спирта. Он
осуществляется либо под действием а- и р-амилаз солода, либо под
действием а-амилазы и глюкоамилазы, содержащихся в препаратах
микрошщетов.
Задача пивоварения — получить высокий выход экстрактивных
веществ из перерабатываемого сырья. Основным сырьем является
ячменный солод. Выход экстракта находится в тесной взаимосвязи с
активностью амилаз, образованных при солодоращении. Роль амилаз
заключается в максимальном извлечении крахмала из солода и его
переводе в сбраживаемые сахара сусла. Однако в пивоварении не
стремятся осахарить конечные декстрины; напротив, они необходимы в
сусле, поскольку в дальнейшем создают полноту вкуса пива, в
определенной степени обусловливают его пеностойкость.
Гидролиз крахмала в сбраживаемые сахара (мальтозу, глюкозу)
и конечные декстрины осуществляется под действием амилаз в
процессе затирания. Основной фактор, с помощью которого
регулируются специфичность и интенсивность действия амилаз, —
температура. Интервал температур 60—65 °С является оптимальным для
образования сбраживаемых Сахаров. В интервале температур 70—75 °С
происходит быстрый перевод крахмала в экстракт с образованием
651
Часть III. Элементы прикладной биохимии
большого количества декстринов. Регулируя продолжительность этих
двух температурных фаз, можно добиваться оптимального
соотношения между количеством сбраживаемых Сахаров и декстринов,
определенной степени сбраживания пива, его коллоидной стойкости и
пеностойкости.
В технологии спирта при переработке крахмалсодержащего сырья
(зерно хлебных злаков, картофель) основная цель — перевести
полностью крахмал в сбраживаемые сахара. Для осахаривания крахмала
используют солод, полученный после 8—10-суточного проращивания
зерна ячменя. Его расход составляет около 15 % от массы осахарива-
емого крахмала. Вместе с ячменным солодом широко используется
смесь солодов — ячменного и просяного в соотношении примерно 3:1
соответственно. В просяном солоде находится а-1,6-амилоглюкозида-
за, способная гидролизовать конечные декстрины с а-1,6-глюкозид-
ными связями. В технологии спирта широко используемым в
настоящее время источником амилаз являются микромицеты родов АврегёШиз
и НЫгориВу а также бактерии Вас. зиЫШз. Их культивирование
осуществляют либо поверхностным методом на увлажненных
пшеничных отрубях, либо глубинным методом с применением жидких
питательных сред. Поверхностную или глубинную культуру используют
либо непосредственно, либо из нее готовят препараты амилаз.
Так как действие амилаз становится более эффективным после
перевода крахмала в клейстеризованное состояние, сырье в спиртовом
производстве подвергают развариванию. Для разжижения крахмала
разваренной массы на многих заводах используют термостабильную
а-амилазу Вас. зиЫШ$ (в виде препарата амилосубтилина).
Осахаривание разваренной массы проводят при температуре 56—
58 °С в аппарате-осахаривателе. Однако осахаривание здесь не идет до
конца; оставшиеся недоосахаренными декстрины (30—40 %) доосаха-
риваются в процессе брожения. Поэтому важно сохранить амилазы в
активной форме. Повышение температуры в осахаривателе выше 60 °С
приводит к инактивации амилаз солода, к недоосахариванию
декстринов в процессе брожения, к увеличению продолжительности
брожения и снижению выхода спирта. С другой стороны, слишком низкая
температура в осахаривателе (ниже 55 °С) ведет к развитию в процессе
спиртового брожения кислотообразующей микрофлоры; концентрация
Н+-ионов в сбраживаемой среде резко возрастает, что вызывает
инактивацию кислотонеустойчивой сс~амилазы. Это ведет к тем же
последствиям, что и в случае термической инактивации амилаз в осахаривателе.
И все же даже в случае активных а- и р-амилаз в сбраживаемой
среде остаются неиспользованными около 5 % конечных декстринов,
содержащих главным образом а-1,6-гликозидные связи; они
являются прямыми потерями сбраживаемого материала. Весьма эффективно
применение препаратов микромицетов, в состав которых входит
активная глюкоамилаза. Этот фермент имеет два важных преимущества
652
Глава 17. Биохимические превращения важнейших пищевых компонентов...
перед амилазами солода: 1) относительно высокая стабильность к
Н+-ионам и температуре; 2) способность гидролизовать а~1,6-глико-
зидные связи. Поскольку гидролиз декстринов в сбраживаемый сахар
является лимитирующей реакцией процесса брожения, то высокая
активность глюкоамилазы обусловливает существенное сокращение
длительности брожения.
Ферменты в производстве фруктозноглюкозных сиропов. В
настоящее время широко распространено производство фруктозно-глю-
козных сиропов, основная цель которого — получить в продукте
повышенное количество фруктозы. Фруктоза в 1,5 раза слаще сахарозы,
почти в 2 раза слаще глюкозы. Поэтому для достижения
определенного уровня сладости в пищевом продукте можно использовать меньшее
ее количество и тем самым снизить количество потребляемого сахара,
что весьма ценно при разработке рационов ограниченной
калорийности* Ее использование особенно важно в лечебном питании больных
сахарным диабетом.
Фруктозно-глюкозные сиропы получают двумя способами: 1)
гидролизом сахарозы и инулина; 2) изомеризацией глюкозы.
Гидролиз сахарозы на фруктозу и глюкозу (инверсия), осуществ-
ляемый под действием (3-фруктофуранозидазы, обеспечивает
образование инвертного сахара, в котором фруктоза и глюкоза находятся в
эквимолекулярном соотношении.
Для промышленного производства инвертазы используют дрожжи
ЗассНаготусез сегеиьзьа и ЗассНаготусез саг1зЬег§епз189 грибы рода
Азрег§Шиз<
Оптимум рН дрожжевой инвертазы находится в зоне 4,0—5,5.
Оптимальная температура действия фермента в разбавленных растворах
сахарозы — 55 °С, в концентрированных — 65—75 °С,
Влияние концентрации сахарозы на активность инвертазы имеет
практический интерес, так как сахароза обладает высокой
растворимостью и возможно получение высококонцентрированного
инвертного сиропа. Максимальная скорость инверсии — при гидролизе 5—
8 %-ных растворов сахарозы; при гидролизе 70 %-ного раствора
скорость этой реакции составляет только четверть максимальной.
Инвертный сахар применяется в кондитерской промышленности
при производстве отливных помадных корпусов и жидких фруктоз-
ных начинок для конфет.
Вторым важным источником получения фруктозы является
инулин и другие низкомолекулярные полифруктозиды, содержащиеся до
15 % и более в топинамбуре, цикории.
Гидролиз инулина до фруктозы осуществляется с помощью
специфического фермента инулазы ф-1,2-фруктан-фруктогидролазы, КФ 3.2.1.7).
Расщепление а-1,2-фруктозидной связи начинается с р-О-фруктозного
конца полимера и продолжается до последней связи. Пррг полном
гидролизе инулина образуется 95 % фруктозы и 5 % глюкозы.
653
Часть III. Элементы прикладной биохимии
Активная инулаза содержится в грибе АзрегдШиз аюатоп.
Ферментный препарат получают поверхностным или глубинным
культивированием продуцента. Препарат инулазы обладает высокой
стабильностью к Н+-ионам и температуре, способен осуществлять глубокий
гидролиз инулина (97—98 %)♦
Полученные из инулина фруктозные сиропы могут быть широко
использованы в кондитерской промышленности, а также в лечебном
питании больных сахарным диабетом.
В технологии фруктозо-глюкозных сиропов из крахмала его гид-
ролизуют до глюкозы. Глюкозу подвергают изомеризации с
превращением во фруктозу под действием глюкозоизомеразы:
ос-Амилаза
Крахмал *- Разжиженный крахмальный клейстер ——*-
Глюкоамилаза Глюкозоизомераза
*•* Глюкоза *- Фруктоза + Глюкоза
55 % 45 %
Глюкозоизомераза обладает высокой термостабильностью. Ее
препараты используются в реакции изомеризации, осуществляемой при
температуре 60 °С. Сродство к глюкозе довольно низкое, поэтому
используются ее 50 %-ные растворы. Реакция изомеризации обратима.
При равновесии концентрация фруктозы варьирует от 50 % при 60 °С
до 55 % при 85 °С.
Промышленные препараты глюкозоизомеразы получают из штаммов
ВЪгерЫтусев. Оптимум активности фермента — около рН 7,0; он
стабилен в интервале рН 4—12. Температурный оптимум — около 80 °С.
Полученные из крахмала фруктозо-глюкозные сиропы с высоким
содержанием фруктозы являются прекрасными подсластителями и
могут быть использованы вместо сахарозы в напитках и пищевых
продуктах. Целесообразно вносить глюкоизомеразу в глюкозную патоку,
где содержание фруктозы может достигать 50 % от исходного
количества глюкозы.
Пектолитические ферменты и их роль в плодоовощной
промышленности. При переработке фруктов и овощей используются
пектолитические ферменты, специфически расщепляющие
пектиновые вещества.
Основной целью в производстве фруктовых и овощных пюре,
соков, в виноделии является расщепление протопектина, пектина,
приводящее к разрушению межклеточных структур, улучшению
качества продукта и увеличению сокоотдачи перерабатываемых фруктов и
овощей.
Для получения промышленных препаратов пектолитических
ферментов широко используют микроскопические грибы рода АзрегдШиз^
особенно часто — А т§ег.
654
Глава 17. Биохимические превращения важнейших пищевых компонентов...
Пектиновые вещества обладают высокой гидрофильностью и
играют роль защитных коллоидов для суспендированных частиц. Поэтому
при производстве фруктовых соковых концентратов фильтрация и
осветление их затрудняются; частички, вызывающие мутность, после
обработки пектолитическими ферментами могут быть легко отделены
при седиментации или фильтрации. Обработка пектолитическими
ферментами фруктовой пульпы повышает эффективность отделения сока
от растительной ткани, увеличивает его выход при прессовании
пульпы. В зависимости от активности пектолитических ферментов расход
их препаратов при обработке фруктов колеблется от 0,05 до 0, 2 % от
массы сырья.
Пектолитические ферменты играют и отрицательную роль в
пищевой промышленности- При хранении фруктов и овощей находящаяся
на их поверхности эпифитная микрофлора выделяет данные
ферменты, которые вызывают деструкцию и гниение этих продуктов. При
хранении соленых консервированных овощей пектолитические
ферменты вызывают их размягчение (мацерацию) и делают не
пригодными в пищу. Весьма отрицательна роль пектолитических ферментов в
технологии свеклосахарного производства, о чем говорилось в
разделе, посвященном вопросам хранения свеклы (разд. 17.2.),
Протеолитические ферменты в пищевой промышленности. В
технологии пищевых продуктов широко используются препараты
микробных протеаз. Их получают из культуральных сред. Однако не
утратили своего значения протеазы животных и растений, поскольку
здесь они находятся уже в готовом виде, и в случае комплексной
переработки сырья экономически целесообразно, а порой и необходимо
получить из них препараты протеаз. Из протеаз, содержащихся в
различных тканях животных, широко применяются реннин и пепсин.
Эти ферменты получены в кристаллическом состоянии (рис. 17.10),
их свойства хорошо изучены.
Реннин (химоэзин, сычужный фермент, КФ 3.4.23.4) содержится
в пищеварительном соке четвертого отдела желудка (сычуга) молодых
а б
Рис, 17.10. Кристаллы реннина (а) и пепсина (б)
655
Часть III. Элементы прикладной биохимии
жвачных животных. Обладает чрезвычайно высокой активностью в
свертывании (створаживании) казеина молока. Препараты реннина
нашли широкое применение в сыроделии. Свертывание казеина
молока является важной технологической операцией в производстве
сыров. Из всех азотистых веществ, содержащихся в коровьем молоке, на
долю казеина приходится около 80 %. Принято считать, что реннин
превращает казеин в параказеин, который осаждается в присутствии
ионов Са2+.
Действие реннина высокоспецифично• Имеются данные,
свидетельствующие о том, что этот фермент расщепляет лишь пептидную
связь между метионином и фенилаланином. Другие протеазы также
свертывают казеин молока, но они обладают более широким
спектром действия на пептидные связи, осуществляют глубокий гидролиз
казеина.
Узкая специфичность реннина к протеолизу и его высокая
коагулирующая способность являются основным критерием,
обусловливающим его широкое применение в сыроделии. Поскольку источник
фермента (желудок телят) ограничен, важное значение имеет замена
реннина микробными протеазами. Однако эта проблема пока
остается нерешенной. Пепсин и многие протеазы растений и
микроорганизмов хотя и обладают высокой свертывающей активностью
казеина, однако глубокий гидролиз казеина ведет к снижению выхода и
качества сыра, к появлению в процессе созревания сыра горьких
пептидов.
Пепсин (КФ 3.4.23.1) — протеаза желудочного сока, обладает
широкой субстратной специфичностью, расщепляет почти все белки
растительного и животного происхождения. Оптимум действия
пепсина— при рН 2,0. При рН около 5,0 пепсин интенсивно свертывает
казеин молока. В связи с тем, что он менее дефицитен, чем реннин,
пепсин находит применение в производстве творога, сыра как в
отдельности, так и совместно с реннином. Фермент используется также
для растворения белковой мути в пиве, которая часто выпадает в виде
осадка при хранении пива на холоде. В промышленности препараты
неочищенного пепсина получают из кислотного экстракта (автолиза-
та) слизистой оболочки желудка свиней путем высаливания 15 %-ным
раствором ЫаС1 и последующего высушивания. Очищенный пепсин
выделяют этанолом.
Из растительных протеаз широкое применение получили папаин и
бромелин. Эти протеазы обладают широкой специфичностью.
Папаин (КФ 3.4.22.2) содержится в млечном соке дынного дерева
Сапса рарща, является хорошо изученным ферментом. Препараты
неочищенного папаина получают путем высушивания млечного сока
дынного дерева. Высокая рН- и термостабильность папаина
обусловливают его эффективное использование в технологии пищевых
продуктов, В мясной промышленности он применяется .для умягчения
656
Глава 17. Биохимические превращения важнейших пищевых компонентов...
мяса. Кусок мяса либо погружают в раствор ферментного препарата,
либо наносят порошок препарата на поверхность куска, либо вводят
раствор в ткань с помощью шприца. Папаин гидролизует не только
мышечные белки, но и значительное количество коллагена и эластина;
их гидролиз резко возрастает при температуре вьппе 40 °С, что особенно
важно при тепловой обработке мяса. В пищевой промышленности
папаин используется также для предотвращения белковой мути пива.
Бромелин используется для тех же целей, что и папаин. Бромелин
получают в качестве побочного продукта при сборе ананасов из их
стеблей. Отпрессованный из стеблей сок фильтруют и подвергают
фракционному осаждению ацетоном. Наиболее богатую фракцию отделяют
и высушивают.
Близки по своим свойствам к папаину протеазы семян злаковых.
Они играют важную роль в технологии хлеба и пива.
В технологии хлеба при замесе и брожении теста активные
протеазы дезагрегируют клейковину* Если мука содержит достаточное
количество и высокого качества клейковину ("сильная" мука), то
действие протеаз будет положительно. Частичная дезагрегация
клейковины способствует равномерному распределению в тесте диоксида
углерода, лучшему формированию мякиша хлеба. Если же клейковина
муки обладает низкой упругостью ("слабая" мука), то действие
протеаз будет носить отрицательный характер, снижается газоудерживаю-
щая способность теста, увеличивается расплываемость подового
хлеба, снижается пористость мякиша.
Ярким примером отрицательного действия протеаз является
переработка муки из пшеницы, пораженной клопом-черепашкой. В
семенах такой пшеницы содержится весьма активная протеаза,
введенная туда этим насекомым. При замесе и брожении теста она
интенсивно дезагрегирует клейковину. Хлеб из такой муки будет
дефектным.
Особая роль отводится зерновым протеазам в производстве пива.
Во время солодоращения активность протеаз резко возрастает. В
результате их действия в солоде накапливаются растворимые белки,
полипептиды и аминокислоты. Во время получения сусла идет
дальнейший гидролиз белков. Продукты гидролиза оказывают
существенное влияние на цвет, пенистость, стойкость и вкус пива. В процессе
приготовления пивного сусла существенное значение имеет
регулирование действия протеаз, что достигается изменением температуры и
продолжительности действия протеаз ("белковая пауза").
Оксидоредуктазы* В формировании вкуса, цвета и аромата
пищевых продуктов среди оксидоредуктаз заслуживают особого внимания
глюкозооксидаза, о-дифенолоксидаза, липоксигеназа.
Глюкозооксидаза (р-В-глюкозо: 02-оксидоредуктаза, КФ 1.1.3.4)
окисляет глюкозу до глюконовой кислоты в присутствии
молекулярного кислорода в соответствии со следующей схемой:
42. Заказ 3486 657
/
Часть III. Элементы прикладной биохимии
НО
он
он
{3- Б- Глюкоза
+ Е-КАБ (глюкозооксидаза)
НО
ОН
Глюконо-8-лактон
+ Е-КМШ
О
СНоОН
НО
СООН
он
Н202 + Е-РАБ
Каталаза
Глюконовая кислота Н20 + 1/202
Глюкозооксидаза является флавопротеином (Е-ЕАВ). [З-В-Глюкоза
окисляется до глюконо-8-лактона, который спонтанно превращается с
участием воды в глюконовую кислоту. Восстановленная
глюкозооксидаза (Е-РАБН2) восстанавливает молекулярный кислород до перокси-
да водорода, который под действием каталазы расщепляется на воду и
кислород.
Глюкозооксидаза микроскопических грибов получена в
кристаллическом виде, ее молекулярная масса — 186 000, она
высокоспецифична: окисляет лишь (З-В-глюкозу и практически не затрагивает
другие сахара. Оптимальная зона рН для действия фермента лежит
между 5 и 6.
Поскольку глюкозооксидаза позволяет удалять как кислород, так
и глюкозу, она широко используется при обработке тех пищевых
продуктов, вкус, окраска и аромат которых ухудшаются в результате
окислительных процессов. Часто при высушивании продуктов
питания глюкоза вступает в реакцию меланоидинообразования с белками
или их продуктами расщепления, что вызывает снижение качества
продукта, изменение его окраски. Обработка такого продукта глюко-
зооксидазой предотвращает этот процесс. Наличие, например,
глюкозы в яйцах приводит к тому, что при сушке и хранении яичного по-
658
Глава 17. Биохимические превращения важнейших пищевых компонентов.
рошка продукт темнеет и приобретает неприятные вкус и запах;
яичный белок теряет способность к растворению. Поэтому до сушки в
яичную массу вносят препарат глюкозооксидазы, в результате чего
этот дефект устраняется.
Применение глюкозооксидазы было предложено для удаления
следов кислорода из вина, пива, фруктовых соков, майонеза. Это
позволяет повысить качество данных продуктов в течение более
длительного периода хранения.
Промышленные препараты глюкозооксидазы получают из
штаммов грибов АзрегдШиз.
о-Дифенолоксидаза (о-дифенол: 02-оксидоредуктаза) (тривиальные
названия: полифенолоксидаза, тирозиназа, КФ 1.10.3.1)
катализирует реакцию окисления дифенолов, полифенолов. В результате действия
этого фермента о-дифенол окисляется в хиной:
Пирокатехин о-Хинон
(о-дифенол)
о-Дифенолоксидаза катализирует также окисление монофенолов:
тирозина; фенола; тг-крезола; 3,4-диметилфенола, а также трифенола
пирогаллола. Дальнейшее окисление хинонов и полимеризация
продуктов окисления приводят к образованию темноокрашенных
веществ — меланинов.
о-Дифенолоксидаза имеет большое значение в пищевой
промышленности. Особенно важна ее роль в технологии черного чая, где
чайный лист разрушают, дубильные вещества под действием о-дифенол-
оксидазы окисляются кислородом воздуха до темноокрашенных
соединений, которые определяют вкус, цвет и аромат черного чая.
Активная о-дифенолоксидаза содержится в картофеле, яблоках,
корнеплодах. Здесь она играет отрицательную роль. При сушке
нарезанных плодов и овощей последние приобретают бурую окраску. Для
предотвращения потемнения перед сушкой нарезанный продукт
подвергают бланшировке, т.е. ошпаривают кипятком или острым паром,
в результате чего о-дифенолоксидаза инактивируется. Цвет ржаного
хлеба является результатом действия о-дифенолоксидазы на тирозин.
Действие данного фермента отрицательно в макаронной
промышленности. Макаронные изделия, приготовленные из муки с высокой
активностью о-дифенолоксидазы, при сушке темнеют и приобретают
непривлекательный вид.
Липоксигеназа {линолеат: 02-оксидоредуктаза, КФ 1.13.1.13)
катализирует реакцию окисления кислородом воздуха ненасыщенных
жирных кислот, Липоксигеназа широко распространена в раститель-
42*
659
Часть III. Элементы прикладной биохимии
ном мире. Она найдена в семенах хлебных и масличных культур,
бобовых. Особенно велико ее содержание в соевой муке. При хранении
муки, крупы, растительных масел и других жировых продуктов ли-
поксигеназа вызывает их порчу. Первой стадией этого процесса
является гидролиз жира на глицерин и жирные кислоты под действием
липазы. Последние под действием липоксигеназы превращаются в
гидропероксиды, которые окисляют различные вещества, в первую
очередь жир, с образованием альдегидов и кетонов, обладающих
резкими и неприятными вкусом и запахом.
Процесс ферментативного окисления ненасыщенных жирных кислот
часто сопряжен с разрушением каротина (провитамина А), что также
ведет к снижению биологической ценности пищевого продукта. В
продуктах с высоким содержанием жира разрушительное действие
липоксигеназы предотвращается токоферолами, которые взаимодействуют с
промежуточными продуктами процессов окисления жирных кислот.
17.4. Процессы брожения и дыхания
в технологии пищевых продуктов
Биохимические процессы, протекающие при брожении и дыхании,
играют большую роль в технологии многих пищевых продуктов. От
направленности и интенсивности этих процессов в большинстве слу-
чаев зависят выход продукта и его качество.
Роль спиртового брожения в технологии пищевых продуктов.
Спиртовое брожение является важнейшей стадией в технологии
хлеба, спирта, пива, вина. Цель спиртового брожения в хлебопечении —
разрыхление теста диоксидом углерода, придание тесту физических
свойств, необходимых при его разделке и выпечке изделий.
Вследствие большого содержания белков и витаминов группы В дрожжи
существенно повышают биологическую ценность хлеба. Наряду с
диоксидом углерода и этанолом при брожении теста образуются
побочные продукты, обусловливающие вкус и аромат хлеба.
Брожение ведется^в интервале температур 25—35 °С, величина рН
при брожении изменяется от 6 до 5 и ниже, что связано с
растворением в тесте диоксида углерода и накоплением органических кислот. В
начальный период брожения сбраживаются собственные сахара муки,
после чего в процесс брожения вовлекается мальтоза, образующаяся в
результате гидролиза крахмала а- и р-амилазами, поступившими в
тесто с мукой.
В технологии этанола основная цель брожения — получить
высокий выход спирта. Сырьем являются зерно, картофель и меласса —
отход свеклосахарного производства. Крахмалсодержащее сырье
подвергают тепловой обработке, что связано с получением гомогенной
сваренной массы, с клейстеризацией и превращением крахмала в
растворимое легкоподвижное состояние. В таком виде крахмал легко гидро-
660
Глава 1 7. Биохимические превращения важнейших пищевых компонентов...
лизуется амилазами в сбраживаемые сахара. Осахаренная среда
(сусло) сбраживается специальными расами дрожжей ЗассНаготусез
сегеигзте при 28—30 °С. Важно, чтобы сбраживаемые сахара
полностью превратились в этанол, для чего необходимы три условия: 1)
высокая активность дрожжей; 2) высокая активность амилаз,
превращающих несбраживаемые декстрины и остаточный крахмал в
сбраживаемые сахара; 3) предотвращение развития в процессе спиртового
брожения посторонней микрофлоры, особенно кислотообразующих
бактерий.
Сбраживание мелассы осуществляют после ее разбавления водой
до необходимой концентрации сахара (около 18—20 % сухих веществ)
и тепловой обработки (стерилизации) полученного сусла.
С биохимической точки зрения более сложной является
технология пива. Специфический вкус и аромат его зависят не только от
жизнедеятельности дрожжей, но и от правильно подготовленного к
сбраживанию пивного сусла. Основным сырьем в пивоварении является
ячменный солод. Подготовка (варка) пивного сусла представляет
собой комплекс биохимических реакций, протекающих в измельченном
солоде под действием гидролаз (в основном, амилаз и протеаз). От
правильного ведения гидролитических реакций зависят эффективность
процесса брожения, выход, вкус и аромат пива.
Процесс сбраживания пивного сусла идет в две стадии: 1) главное
брожение, продолжительность которого 6—8 дней, протекает при
температуре 5—8 °С; 2) дображивание — от 20 до 100 дней при
температуре около 1 °С и абсолютном давлении 0,14—0,15 МПа. На последней
стадии идет формирование специфических вкусовых свойств пива и
насыщение его диоксидом углерода. Важную роль в достижении этой
цели играет раса дрожжей.
В технологии вин возбудителями спиртового брожения являются
специальные винные дрожжи. Широкий ассортимент вин
определяется прежде всего физиолого-биохимическими особенностями винных
дрожжей данной местности; от них в значительной степени зависят
букет и вкусовые достоинства вин. Сырьем в виноделии служат виног-
рад, фрукты и ягоды.
Роль молочнокислого брожения в технологии пищевых
продуктов. Молочнокислое брожение является основной стадией в
производстве ржаного хлеба, в консервировании овощей и плодов, в
изготовлении кисломолочных продуктов.
Молочнокислое брожение в технологии ржаного хлеба
необходимо, во-первых, для накопления достаточно большого количества
молочной кислоты, осуществляющей инактивацию а-амилазы (этот
фермент весьма активен в ржаной муке и проявляет интенсивное декст-
ринирующее действие на крахмал при выпечке хлеба); во-вторых, для
придания хлебу кисловатого вкуса и приятного аромата; в-третьих,
для разрыхления теста диоксидом углерода.
661
Часть III. Элементы прикладной биохимии
Для брожения используется часть старого выбродившего теста,
получившего название закваски и содержащего как гомофермента-
тивные, так и гетероферментативные молочнокислые бактерии. В
зависимости от количественного соотношения этих видов бактерий,
условий их жизнедеятельности в тесте будут накапливаться в разных
количествах и продукты брожения. Более высокая интенсивность ге-
тероферментативного брожения обусловливает и лучшую разрыхляв-
мость теста. Однако она ведет к накоплению большего количества
уксусной кислоты, в результате чего ржаной хлеб приобретает
отчетливо выраженный кислый вкус. Напротив, более интенсивное гомо-
ферментативное брожение способствует накоплению в тесте в
больших количествах молочной кислоты, придающей хлебу мягкий
кисловатый вкус.
Молочнокислое брожение играет большую роль в засолке огурцов
и помидоров, квашении капусты, мочке яблок. Молочная кислота
является консервирующим фактором, предотвращающим развитие
посторонней, особенно гнилостной, микрофлоры, придает продукту
специфические вкус и аромат.
В технологии молочной кислоты используются термофильные
молочнокислые бактерии (ЬЬт. сегеа1е9 по старой номенклатуре —
ВеШгиШ). Основным сырьем является свекловичная меласса.
Оптимальная температура действия этих бактерий лежит в пределах 48—-
55 °С. Мелассу разбавляют водой до концентрации 3—4 % (по
сахару), пастеризуют при 70 °С, затем раствор охлаждают до 48—50 °С,
вносят культуру молочнокислых бактерий. Для поддержания
оптимального значения рН (5,5—6,0) используют СаС03. В ходе брожения
вводят такое количество сахара, чтобы содержание лактата в культу-
ральной жидкости было около 15 %. Брожение продолжается 6—8
суток. Выход молочной кислоты составляет около 90 % от массы
сахара* Выделяют молочную кислоту из культуральной жидкости в виде
лактата кальция, из которого разложением серной кислотой
получают чистую молочную кислоту. Молочная кислота — слабая кислота с
приятным кислым вкусом и без запаха. Она широко используется в
пищевой промышленности как подкислитель в джемах, желе,
кондитерских изделиях, наливках, при консервировании плодов и овощей
для придания аромата и остроты готовому продукту.
Неполные окисления в технологии органических кислот.
Многие аэробные микроорганизмы осуществляют неполное окисление
глюкозы, при котором в качестве продуктов обмена выделяются лишь
частично окисленные органические соединения. Важными
продуктами неполного окисления являются органические кислоты, широко
используемые в пищевой промышленности и наряду с молочной
кислотой получившие название пищевых кислот. К ним относятся
лимонная, глюконовая, уксусная кислоты. Поскольку при неполном
окислении из окисляемого субстрата извлекается относительно неболь-
662
Глава 17. Биохимические превращения важнейших пищевых компонентов...
шое количество энергии и для энергообеспечения продуцента
необходимо окислить большое количество субстрата, неполное окисление
называется также окислительным, или аэробным, брожением.
При аэробном брожении одним из важнейших условий является
обеспечение возбудителя брожения в период синтеза кислоты
кислородом воздуха. Кратковременное прекращение его доступа
приостанавливает процесс накопления кислоты. Другое важное условие —
необходимость поддержания рН среды на оптимальном уровне, как
правило, близком к нейтральному, за исключением уксуснокислого
брожения, протекающего при низких значениях рН. При соблюдении
оптимальных условий жизнедеятельности продуцента и удачном его
подборе субстрат почти полностью переходит в целевой продукт. Так,
в процессе производства глюконовой кислоты ее выход достигает 95 %,
лимонной — 80 %, уксусной — 90—95 %.
Лимоннокислое брожение представляет собой ферментацию на
основе цикла трикарбоновых кислот. Общими предпосылками для
накопления лимонной кислоты являются: 1) введение "экстра-углерода"
в цикл в результате фиксации диоксида углерода на пирувате с обра-
зованием оксалоацетата; 2) активация цитратсинтазы; 3) ингибирова-
ние дегидрогеназ цикла трикарбоновых кислот.
Определенные штаммы Азр. п1§ег способны осуществлять
сверхсинтез лимонной кислоты; эти штаммы вполне соответствуют
указанным предпосылкам: они обладают высокой активностью
цитратсинтазы и в культуральной среде могут накапливать до 15 % кислоты.
Сырьем служит меласса, из которой готовят сусло, содержащее 10—
20 % сахарозы. Оптимальная температура лимоннокислого брожения —
31—32 °С, которую необходимо поддерживать на этом уровне,
поскольку при брожении выделяется тепло. При отклонениях температуры от
оптимального уровня снижается активность продуцента, образуется
большее количество побочных кислот.
Химизм лимоннокислого брожения показан на рис. 17.11.
Лимоннокислое брожение осуществляется двумя способами:
поверхностным и глубинным, длительность которых — 3—5 и 5—10 дней
соответственно. Из культуральной среды лимонную кислоту удаляют
в виде кальциевой соли. Свободную кислоту получают путем
разложения цитрата кальция разбавленной серной кислотой.
Лимонная кислота нашла широкое использование при
производстве напитков.
Глюконовокислое брожение представляет собой простейшую
реакцию превращения глюкозы в глюконовую кислоту. Возбудителями
этого брожения являются микромицеты родов РепШШит иАзрег§Ши$9
некоторые штаммы которых способны к активному синтезу глюкозо-
оксидазы. У Азр. т§ег этот процесс происходит с высоким выходом
даже в 30—35 %-ном растворе глюкозы, если кислоту нейтрализовать
СаС03.
663
Часть III. Элементы прикладной биохимии
^6^12^6
с
Гликолиз
>
сн3-со~соон
Пиру ват
сн3-со-соон
Пируват
СО,
НХ-СО
2| I
ноос соон
Оксалоацетат
СоА-8Н
СН3-СО~8СоА
^
СоА-8Н
СН2
СООН СООН СООН
Лимонная кислота
Рис. 17.11. Схема процесса лимоннокислого брожения
Сбраживание ведется культурой грцба при температуре 30—32 °С
и интенсивном аэрировании среды стерильным воздухом. Среда
содержит 10—15 % глюкозы и небольшое количество солей,
содержащих бор, магний, фосфор, азот и СаС03; последний задается с целью
нейтрализации глюконовой кислоты. Готовый продукт получают в виде
глюконата кальция. После фильтрации сбраженную массу охлаждают
до 20 °С и выдерживают в течение 24—48 ч для кристаллизации.
Кристаллы глюконата кальция отделяют центрифугированием.
Кальциевая соль глюконовой4 кислоты используется как пищевая добавка при
аллергических заболеваниях и в детском питании.
Уксуснокислое брожение вызывается уксуснокислыми
бактериями. Они обладают высокой толерантностью (устойчивостью) к
кислотам. Исходным субстратом является этанол. Химизм уксуснокислого
брожения может быть представлен в виде схемы (рис. 17.12). Важную
роль в окислении этилового спирта до уксусной кислоты играет
ЫАОР-зависимое дегидрирование. Выигрыш энергии в результате этого
окисления составляет б молекул АТР.
Уксуснокислое брожение используется в производстве уксуса,
являющегося в принципе водным раствором уксусной кислоты. Уксус
широко применяется в пищевой промышленности в качестве
вкусовой добавки к пище.
В производстве уксуса используют два вида бактерий, обладающих
высокой кислотообразующей способностью — Вас1. ЗсНигепЬасЫ и
Вас1. сигиит. Они хорошо развиваются при рН 3. Но уксусная кислота,
накапливаясь в сбраживаемой среде, угнетающе действует на них. Бро-
664
Глава 17. Биохимические превращения важнейших пищевых компонентов.
СН3-СН2-ОН
Этанол
МАОР
+
*- NА^РН + Н+
СН8-С*
о
н
Ацетальдегид
КАБР
+
сн3-с-он
3 чон
Ацетальдегидгидрат
КАСРН + Н+
СН.-СООН
Уксусная кислота
2Н20
О.
г 2МАВРН + Н
1 +
^ 6А0Р + 6Р,
+
Рис. 17.12. Схема процесса уксуснокислого брожения
жение полностью приостанавливается при содержании 14 % кислоты. В
связи с этим концентрация спирта в сбраживаемой среде должна
находиться в пределах 10—12 %. Окисление спирта в уксусную кислоту
осуществляют генераторным методом или методом глубинного
культивирования бактерий. В первом случае раствор спирта протекает через слой
стружки (чаще буковой), в котором в закрепленном состоянии находятся
уксуснокислые бактерии; во втором, более совершенном методе,
бактерии культивируются в ферментаторах в интенсивно перемешиваемой и
аэрируемой среде. Температура брожения поддерживается на уровне 32—
34 °С. Для нормальной жизнедеятельности бактерий используют не
чистый водно-спиртовой раствор, а сусло, в состав которого входят, кроме
воды, спирта, уксусной кислоты, минеральные соли, легкоусвояемые
углеводы и азотсодержащие вещества.
17,5, Проблемы прикладной энзимологии
в технологии пищевых продуктов
Одной из основных проблем применения ферментов в технологии
пищевых продуктов является снижение стоимости ферментных пре-
665
Часть III, Элементы прикладной биохимии __^^_
л аратов. В связи с большими объемами производства пищевых
продуктов расход ферментных препаратов может составить
внушительную величину, что приводит к существенному повышению стоимости
продукта. Эта проблема может быть решена совершенствованием
технологии ферментных препаратов, подбором дешевых сред в случае
микробного биосинтеза ферментов, выбором активных продуцентов
целевых ферментов, разработкой рациональных режимов биосинтеза,
использованием иммобилизованных ферментных препаратов.
Иммобилизованные ферменты (от лат. ШтоЬШз —
неподвижный) — это препараты ферментов, молекулы которых связаны с
нерастворимым носителем (матрицей) и сохраняют при этом полностью
или частично свои каталитические функции.
Иммобилизованные ферменты обладают многими преимуществами по
сравнению с обычными препаратами ферментов. К ним относятся: 1)
более низкая стоимость, связанная с возможностью повторного
применения фермента; 2) повышенная стабильность при хранении и
использовании препаратов иммобилизованных ферментов; 3) отсутствие примесей
фермента в готовом продукте; 4) возможность организации непрерывно-
поточного процесса катализа и более строгого контроля за ним; 5)
возможность применения иммобилизованных мультиферментных систем.
Все методы иммобилизации ферментов можно подразделить на
физические и химические.
К методам физической иммобилизации можно отнести адсорбцию
фермента на нерастворимом носителе, включение фермента в поры
геля, пространственное отделение фермента от остального объема
реакционной системы с помощью полупроницаемой мембраны. В
качестве носителей используются кремнезем, оксиды алюминия, глины,
пористое стекло, керамика, активированный уголь, полисахариды,
коллаген. К основным преимуществам следует отнести доступность и
дешевизну адсорбентов-носителей, простоту иммобилизации. Однако
применение метода ограничивается недостаточной прочностью
связывания фермента с носителем.
Суть метода иммобилизации ферментов путем включения в гель
состоит в том, что молекулы фермента находятся в трехмерной сетке
тесно переплетенных полимерных нитей, образующих гель. Широкое
распространение получили гели из крахмала, агар-агара, полимеры
акриловой и кремниевой кислот.
Иммобилизация ферментов с использованием мембран состоит в
том, что водный раствор фермента отделяется от водного раствора
субстрата полупроницаемой мембраной, через которую легко проходят
молекулы субстрата с низкой молекулярной массой, но молекулы
фермента мембрана не пропускает* К этому методу относятся микрокап-
сулирование и включение ферментов в волокна.
Основным недостатком мембранных систем и систем полимерных
гелей является невозможность действия ферментов на высокомолеку-
666
Глава 1 7. Биохимические превращения важнейших пищевых компонентов,.,
лярные субстраты, молекулы которых не могут пройти через поры
мембраны.
Химические методы иммобилизации заключаются в ковалентной
сшивке между белком фермента и носителем. В качестве носителя
обычно используют целлюлозу, декстрановые гели (сефарозу, агаро-
зу), микропористое стекло, кремнезем, а также синтетические
полимеры. При ковалентном связывании фермента носитель предварительно
активируют, обрабатывая его, например, бромцианом, азотистой
кислотой. Благодаря этому в носителе появляются активные
группировки, которые способны вступать в реакцию с функциональными
группами молекулы фермента. Однако при химической иммобилизации
необходимо, чтобы активные группы модифицированного носителя не
блокировали активный центр фермента.
Препараты, полученные химическим методом иммобилизации,
обладают важным достоинством. Ковалентная связь с носителем
обеспечивает высокую прочность образующегося комплекса. При
достаточно широком варьировании физико-химических факторов, таких, как
рН, температура, фермент не десорбируется с носителя и не
загрязняет целевые продукты катализируемой реакции. Однако ковалентное
связывание ферментов обычно включает довольно сложные
процедуры, поэтому око не используется для промышленных препаратов. В
большинстве случаев оно приводит к существенной деформации
молекулы фермента, к снижению или потере каталитической активности.
Препараты иммобилизованных ферментов пока не нашли
широкого применения в пищевой промышленности, так как во многих
случаях высокомолекулярные субстраты подвергаются ферментативным
превращениям.
При ферментативной обработке низкомолекулярных компонентов
пищевых продуктов проблема диффузии субстрата к ферменту
становится вполне разрешимой. Так, нашла промышленное применение
глюкозоизомераза, иммобилизованная адсорбцией на оксиде
алюминия. В таком виде ее используют в непрерывно-поточном колоночном
реакторе. Иммобилизованная р-галактозидаза применяется для
гидролиза лактозы. Препараты иммобилизованной р-галактозидазы
получают в виде сферических частиц силикагеля, покрытых поливинил-
ацетатом и содержащих фермент.
Важной проблемой использования ферментов в пищевой
промышленности является повышение их стабильности в технологических
средах, и особенно в отношении таких важных физико-химических
факторов, как температура и рН. В случае длительного пребывания в
реакционной среде фермента стабильность имеет доминирующее
значение относительно активности ферментов. Термическая устойчивость
многих гидролаз лежит в интервале 40—50 °С. Однако известно
довольно большое число ферментов, продуцируемых термофильными
микроорганизмами, температура инактивации которых довольно высока
667
Часть III. Элементы прикладной биохимии
(70—80 °С). Поэтому подбор микроорганизмов, которые обладали бы
способностью к эффективному биосинтезу таких ферментов, является
актуальным вопросом.
В технологии глюкозо-фруктозных сиропов исследования
направлены главным образом на способы повышения концентрации
фруктозы в изомеризованном продукте при помощи комплексирования
фруктозы или сдвига глюкозо-фруктозного равновесия путем изменения
условий реакции. Методы повышения эффективности биосинтеза глю-
козоизомеразы требуют подробных исследований. Значительного
увеличения продуктивности штаммов можно достичь с помощью генной
инженерии.
Краткое содержание главы
Важнейшими пищевыми компонентами продукции
растениеводства являются белки, углеводы и жиры. Они же играют роль
запасных питательных веществ семян, клубней, корнеплодов, при
прорастании которых эти вещества служат .источником пластического
материала и энергии.
Зерновые злаки — основной поставщик белков в питании
человека. Среди них следует особо выделить пшеницу, мука из которой
широко используется в хлебопечении. При выращивании пшеницы
особое внимание уделяется биосинтезу белков клейковины — глиадина и
глютенина. Установлено, что содержание белка в зерне при
продвижении с северо-запада на юго-восток России повышается. Чем жарче
лето и меньше осадков, тем больше белков в зерне злаковых. Качество
формируемой клейковины в созревающем зерне пшеницы находится
под жестким генетическим контролем.
Ценнейший источник белка — зернобобовые культуры. Их
способность фиксировать азот воздуха при помощи клубеньковых
бактерий дает возможность накапливать большое количество белка.
Источником углеводов в питании человека являются зерновые
культуры, картофель, сахарная свекла, плоды и овощи,
В начальный период налива в зерне мало крахмала, идет
интенсивный биосинтез полифруктозидов, сахарозы и моносахаридов. В
период полной зрелости количество растворимых углеводов в зерне
снижается, увеличивается содержание крахмала. В процессе
биосинтеза в зерновых злаках накапливается от 45 до 70 % крахмала от
массы зерна.
На биосинтез углеводов в зерне оказывают существенное влияние
климатические факторы. Установлено, что при продвижении в нашей
стране на юг и восток количество крахмала в зерне уменьшается.
Важнейшей овощной культурой является картофель. Содержание
крахмала — основной показатель качества картофеля. В пищевой
промышленности картофель идет на производство пищевого крахмала,
668
Глава 17. Биохимические превращения важнейших пищевых компонентов...
патоки, глюкозы, спирта. В процессе биосинтеза в клубнях
накапливается от 10 до 20 % крахмала.
Сахарная свекла является сырьем в производстве сахарозы —
пищевого сахара. В зависимости от сорта свеклы и условий
выращивания содержание сахарозы может изменяться от 12 до 24 % на сырую
массу. Накопление сахарозы в корнеплодах и их рост начинается
после развития достаточно мощного листового фотосинтетического
аппарата. Интенсивность и длительность освещения этого аппарата
увеличивают содержание сахарозы в корнеплоде. Качество сахарной
свеклы тем выше, чем больше в корнях сахарозы и меньше растворимых в
воде компонентов. Повышают качество свеклы фосфорные и
калийные удобрения.
Важнейшим источником пищевых растительных масел в нашей
стране является подсолнечник, в зрелых семенах которого
содержится 25—45 % липидов. Конечный продукт синтеза масел —■ смесь
ацилглицеролов и свободных жирных кислот. Пищевая ценность
растительных масел определяется наличием ненасыщенных жирных
кислот, на долю которых в семенах подсолнечника приходится 80 % всех
жирных кислот. Синтез ацилглицеролов идет через углеводы. После
окончания стадии вегетации масличных культур начинается
интенсивное превращение углеводов через процесс гликолиза в жиры. При
низких температурах, повышенной влажности больше синтезируется
ненасыщенных жирных кислот.
Основным биохимическим процессом при хранении
сельскохозяйственного сырья является дыхание; оно может быть аэробным (с
доступом кислорода воздуха) и анаэробным, которое возникает в
экстремальных условиях, когда в газовой среде, окружающей дышащий
объект, нет кислорода. Дыхание ведет к трате ценных питательных
веществ, к изменению газового состава среды, выделению тепла и воды.
При хранении зерна важнейшими факторами, от которых зависит
интенсивность дыхания, являются влажность и температура; при
хранении картофеля, сахарной свеклы, плодов и овощей — температура
и достаточный доступ к ним кислорода воздуха.
В зерне с влажностью выше критической (14,5—15,5 %)
появляется свободная вода, что обусловливает резкое повышение
интенсивности дыхания. Такое же явление имеет место при хранении масличных
культур, критическая влажность которых гораздо ниже (8—10 %),
При влажности зерна выше критической процесс дыхания
приобретает автокаталитический характер, что ведет к самосогреванию зерна и
его порче.
Клубни картофеля при хранении проходят три стадии —
дозревание? покой и пробуждение (прорастание). Стадия покоя
характеризуется снижением интенсивности дыхания, уменьшаются траты
крахмала. Поэтому при хранении картофеля стремятся продлить срок
покоя, что достигается за счет снижения температуры до 3—5 °С.
669
Часть III. Элементы прикладной биохимии
В свекловичном корне при хранении идут многочисленные
процессы метаболизма, связанные с деструкцией низко- и
высокомолекулярных соединений. Это неизбежно приводит к снижению
технологических качеств свеклы. Особое влияние оказывает температура.
Оптимальной является температура 0—2 °С, при которой свекла находится
в состоянии анабиоза, технологические качества ее мало изменяются,
до минимума снижаются траты сахара на дыхание.
В основе переработки крахмалсодержащего сырья лежат
превращения крахмала в сахара. Амилазы, содержащиеся в муке, гидроли-
зуют крахмал при брожении теста в сбраживаемый сахар, что
необходимо для насыщения теста диоксидом углерода. Обогащение муки
амилазами не только повышает скорость брожения теста, но и
приводит к образованию достаточного количества сахара, к улучшению
качества хлеба.
В производстве патоки, пищевой и кристаллической глюкозы
крахмал гидролизуют с помощью ос-амилазы, (3-амилазы, глюкоамилазы.
Соотношение различных фракций углеводов в патоке можно изменять в
широких пределах, используя специфичность действия этих ферментов.
Гидролиз крахмала в сбраживаемые сахара является важнейшим
биохимическим процессом в технологии продуктов брожения — пива,
спирта. Он осуществляется под действием амилаз. Выход экстракта
из перерабатываемого солода в производстве пива зависит от
активности амилаз. Важным показателем вкусовых достоинств пива, его пе-
ностойкости является соотношение сахара/декстрины. Это
соотношение также регулируется изменением температурного режима
получения пивного сусла. В технологии спирта при переработке крахмалсо-
держащего сырья основная цель — перевести крахмал полностью в
сбраживаемые сахара.
Основная цель в производстве фруктозно-глюкозных сиропов —
получить в продукте повышенное количество фруктозы. При
гидролизе сахарозы р-фруктозидазой образуются эквимолекулярные
количества фруктозы и глюкозы; при гидролизе инулина инулазой — 95 %
фруктозы и 5 % глюкозы. Фруктоза усваивается быстрее, чем
сахароза, отличается большей сладостью, чем сахароза, является
диетическим продуктом для больных сахарным диабетом, Фруктозо-глюкоз-
ные сиропы получают из крахмала с использованием глюкоамилазы и
глюкозоизомеразы.
В получении плодово-овощных соков большую роль играют пекта-
зы и пектиназы. Разрушая пектиновые вещества, эти ферменты
увеличивают сокоотдачу пульпы плодов и овощей, увеличивают выход
сока, осветляют его.
Пектолитические ферменты играют отрицательную роль в
технологии свеклосахарного производства. Под их действием идет
превращение протопектина в растворимый пектин. Это ведет к повышению
вязкости диффузионного сока, затрудняет его фильтрацию, кристал-
670
Глава 17. Биохимические превращения важнейших пищевых компонентов...
лизацию сахарозы. Особенно интенсивно деградация пектинов идет
при гниении корней сахарной свеклы.
Ферментные препараты протеаз нашли широкое применение в
технологии пищевых продуктов. Растительные протеазы (папаин, фицин,
бромилин) широко используются для умягчения мяса.
Высокая активность протеаз в хлебопечении ведет к ухудшению
качества хлеба, поскольку в процессе брожения теста протеазы
дезагрегируют его клейковину.
В формировании вкуса, аромата пищевых продуктов принимают
участие оксидоредуктазы. Глюкозооксидаза окисляет глюкозу до глю-
коновой кислоты, предохраняя тем самым при термической обработке
пищевые продукты от неприятных вкуса и запаха.
В технологии пищевых продуктов широко используются
различные типы брожений: спиртовое — в получении пива, спирта, вина;
молочнокислое — в хлебопечении, консервировании плодов и овощей;
лимоннокислое — в производстве лимонной кислоты; уксуснокислое —
уксуса; глюконовокислое — глюконовой кислоты.
Хронология важнейших дат в исследовании биохимических
процессов при созревании, хранении и переработке
сельскохозяйственного сырья
1935 г. — А. И. Опарин совместно с А. Н. Бахом организовал
Институт Биохимии АН СССР, где впоследствии были осуществлены
многочисленные работы, посвященные биохимическим основам
переработки сельскохозяйственного сырья, энзимологии технологий
пищевых продуктов.
1935—1946 гг. — А. Н. Бах и его ученики заложили фундамент
технической биохимии, изучающей биохимические процессы в
различных отраслях пищевой промышленности.
1960—1965 гг. — Р. В. Фениксова провела фундаментальные
исследования по технической энзимологии.
1961—1976 гг. - А. А. Покровский (директор Института питания
АМН СССР) разработал биохимические основы концепции
сбалансированного и лечебного питания, ряд специализированных продуктов
для питания детей, лиц пожилого возраста.
ЛИТЕРАТУРА
Березин И. В. Исследование в области ферментативного катализа и
инженерной энзимологии, — М.: Наука, 1990. — 382 с.
Жеребцов Н. А. Амилолитические ферменты в пищевой промышленности. —
М.: Легкая и пищевая пром-сть, 1984. — 160 с.
Иммобилизованные клетки и ферменты / Под ред. Дж, Вудворда, — М.:
Мир, 1988. — 235 с.
Кретович В. Л. Биохимия зерна и хлеба // Техническая биохимия / Под ред.
В. Л. Кретовича. — М.: Высш. шк., 1973. — С, 73—77.
671
ПРИЛОЖЕНИЕ
*М
ОТВЕТЫ НА ВОПРОСЫ И ЗАДАЧИ
Глава 1. Белки
1. а) По биуретовой реакции и содержанию азота в белке;
б) 12,5 % при содержании азота в белке 16 %.
2. а) Ковалентные связи (пептидные, дисульфидные), водородные связи
(а-спирали и р-складки), гидрофобные взаимодействия, солевые мостики, ван-
дер-ваальсовы силы;
б) Трипептиды:
1) А1а-Ме^Азр; 2) А1а-Авр-Ме1;; 3) Ме*-А1а-Азр;
4) Ме1-Аар-А1а; 5) Азр-А1а-Ме1; 6) Авр-Ме*-А1а;
в) структура пептидов:
1) СН3-8-СН2-СН2-СН-СО-КН-СН-СН2-СН2»СО-КН- СН2-СООН
КН3+ СООН
2)
1^1
V
Ме1;-01и-С1у
НО-СН-СНз
гсн2-сн-со-кн-сн-сн2-со-ын-сн-соон
№* соон
РЬе-Азр-ТЬг
г) Первый гексапептид не содержит ни одной незаменимой аминокислоты;
второй содержит валин; третий — валин, треонин; четвертый — валин, лизин,
треонин, фенилаланин; пятый — все незаменимые аминокислоты и является
наиболее биологически активным.
3. Азр-1л8-01и.
4. а) Лейцин остается на стартовой точке или вблизи нее, аспарагиновая
кислота будет перемещаться к аноду, аргинин к катоду;
б) Полипептид (Ьуз)10-(Аг&)5 при рН 3 заряжен положительно, поэтому он
сорбируется на катионообменнике. Буфер со значением рН 10 снимает заряд с
полепептида, что ведет к его десорбции.
Глава 2. Нуклеиновые кислоты
1. В состав ДНК входят основания аденин, гуанин, тимин, цитозин; в состав
РНК — аденин урацил, гуанин, цитозин.
2. Нуклеозид — это основание (пуриновое или пиримидиновое), связанное с
молекулой сахара ф-В-рибозы или [З-В-дезоксирибозы); нуклеотид — это
нуклеозид, связанный с молекулой фосфорной кислоты. С появлением в молекуле нук-
леозида сахара, имеющего полярные ОН-группы, растворимость нуклеозидов
выше, чем у оснований; она существенно возрастает в нуклеотиде, содержащем
фосфорную кислоту с ярко выраженными гидрофильными свойствами.
3. Нуклеотиды в клетке выполняют следующие основные функции:
1) являются строительными блоками ДНК и РНК;
2) выполняют функцию аккумуляторов химической энергии;
43. Заказ 3486
673
3) выполняют функции коферментов;
4) участвуют в синтезе белков, углеводов, жиров.
4. Комплементарность отражает зависимость последовательности азотистых
оснований одной нити молекулы ДНК от последовательности оснований другой
нити данной молекулы. Ее обусловливают водородные связи, возникающие
между основаниями аденин—тимин, гуанин—цитозин. Эти связи возникают между
Н2- и ОН-группами оснований.
5. Образование двух копий ДНК из одной молекулы ДНК. Репликация
осуществляется в соответствии с полуконсервативным механизмом.
6. Расстояние между парами оснований — 0,34 нм; следовательно, длина
молекулы будет 51000 нм (51 мкм).
7. 3 530 пар оснований.
Глава 3. Углеводы
1. а) Альдогексоза; б) кетогексоза; в) инулин, леван.
2. 20 мМ.
3. 68,4 %.
4. а) Гидрофильные соединения; б) крахмал, гликоген.
5. Крахмалистость картофеля — 16, 2 %.
6. Сахаристость 17,1 %.
Глава 4. Липиды
1. Триацилглицерол, содержащий олеиновую, пальмитиновую и
стеариновую кислоты.
3. а) „ б)
О
СН2-0-С-(СН2)14-СН3
о
II
СН2-0-С-(СН2)1б-СНа
о
II
СН-0-С-(СН2)7-СН=СН-(СН2)7-СНз СН-0-С-(СН2)гб-СН3
о
II
ОН
I
СН2-0-Р=0
ОН
4. Соотношение белки : липиды 1:100.
5. Стеародиолеинглицерол:
О
II
СН2-0-С-(СН2)16-СН3
О СНд
СН2-0-Р-0-СН2-СН2- Ы"СН3
ОН
СН:
о
СН-0-С-(СН2)7-СН=СН-(СН2)7-СН3
о
СН2-0-С-(СН2)7-СН=СН-(СН2)7~СН3
Глава 5. Витамины
1. Авитаминоз — полное отсутствие витамина в организме; гиповитаминоз —
его недостаток; гипервитаминоз — его избыток.
2, Водорастворимые витамины — гидрофильные вещества, входящие в со
став ферментов и функционирующие в качестве коферментов. Они играют клю
674
чевую роль в метаболизме веществ организма- Жирорастворимые витамины —
гидрофобные вещества, их биохимические функции изучены недостаточно.
Провитамины — предшественники витаминов. Витамеры — группа витаминов с
общей биологической функцией. Витаминоподобные вещества также обязательно
необходимы организму, но, в отличие от витаминов, они несут пластическую
функцию и требуются организму в гораздо больших количествах, чем витамины.
Антивитамины — вещества, подавляющие функции витаминов.
3* Н3С СН3 СНз СНз
сн=сн-с=сн-сн-сн-с=сн-сн2-он
ЧЛи,
Нерастворимость в воде (гидрофобность) объясняется тем, что р-иононовое
кольцо и два остатка изопрена ациклической части витамина А1 являются
углеводородами •
4. Витамин В — антирахитический; витамин Е защищает липиды мембран
от разрушающего действия активных форм кислорода; витамин К участвует в
синтезе тромбина, играющего важную роль в свертывании крови.
Витамин Е — антиоксидант — вещество, подавляющее окисление липидов в
жировых продуктах.
5. Витамин Вх играет важную роль в углеводном обмене; витамины В2 и РР
принимают участие в окислительно-восстановительных процессах; витамин В6
участвует в обмене аминокислот.
6. Витамин С принимает участие в окислении тирозина, фенилаланина, в
синтезе белка коллагена, являющегося важным компонентом кровеносных
сосудов и костной ткани. Авитаминоз С ведет к их хрупкости, разрушению
кровеносных сосудов,
7. Витамин Е является производным углеводородного циклического
соединения токола и длинной углеводородной цепи, иными словами, это типичное
гидрофобное соединение. Витамин С представляет собой лактон гексоновой
кислоты, содержащий полярные группы (четыре ОН-группы и одну СО-группу), что
обусловливает его высокую гидрофильность.
8. Неустойчивость витамина С связана с наличием в его молекуле двойной
связи и со способностью легко отдавать атомы водорода при окислении.
9. Суточная потребность витамина С для взрослого человека — 75 мг. При
использовании только одного источника витамина С потребуется следующее
количество свежих овощей в г: картофеля — 300, капусты — 200, яблок — 300,
смородины — 33.
Глава 6. Растительные вещества вторичного происхождения
1. В основе химической структуры терпенов лежит группа изопрена
сн2-с-сн=сн2.
Монотерпены содержат 10 атомов углерода, тритерпены — 30, политерпены
(каучук и гутта) — от 7,5-103 до 3405.
2. Тритерпены — предшественники витамина Т)у тетратерпены —
предшественники витамина А,
3. Флавоноиды — водорастворимые фенольные соединения, в основе
структуры которых лежит флаван, состоящий из двух ароматических колец А и В,
соединенных через кислородсодержащую С3-единицу:
43
А
675
4, Алкалоиды — азотсодержащие гетероциклические соединения основного
характера, обладающие ярко выраженной физиологической активностью.
Подразделяются на производные пиримидина, пирролидина, хинолина, индола,
пурина и др.
5. В зависимости от химической природы связи сахара с агликоном
различают О-, 8-, И-гликозиды.
Глава 7. Принципы биоэнергетики клетки
1. ДС°'= -7,1 кДж-моль"1 -
2. Сопряжена реакция а.
3. В глюкозо-1-фосфате фосфатная группа примыкает к углеродному атому
карбонильной группы, благодаря чему плотность электронного облака у этой
группы выше, чем в глюкозо-6-фосфате.
4* Энтропия является характеристикой упорядоченности системы. При
денатурации белковая глобула переходит в хаотический (неупорядоченный) клубок,
что обусловливает резкое возрастание энтропии.
Глава 8. Ферменты
1. Пиридоксальфосфат.
2. В состав РАБ.
3. Фермент, обладающий абсолютной специфичностью, катализирует
реакцию превращения только одного субстрата. Фермент с относительной
специфичностью катализирует превращение группы сходных по химической природе
субстратов, он имеет более лабильный активный центр, способный образовывать
комплексы с этой группой субстратов,
. 4. V - к[Е] при [8] » [Е] и V - У[8]/(КШ + [8]).
5. Энергия активации при ферментативном катализе уменьшается, что ведет
к резкому увеличению скорости реакции.
6. 0,5 мМ.
7. Конкурентный тип ингибирования.
Глава 9. Катаболизм углеводов.
Генерирование энергии фосфатной связи
1. Гексокиназа.
2. Альдолаза, енолаза, фоефофруктокиназа*
3. Малонат блокирует реакцию сукцинат —» фумарат, являясь конкурентным
ингибитором сукцинатдегидрогеназы.
4. Этанола — 51,1 г; лактата — 100 г.
5. При молочнокислом брожении Х4С-метка будет в карбоксильной группе
лактата, при спиртовом — в С02.
6. ДС°' = -61 кДж.
Глава 10. Биосинтез углеводов
1. При освещении водоросли СМогеЫа шел синтез NА^РН и АТР, которые в
темновой реакции использовались на синтез Х4С-глюкозы из НС02. Поскольку в
676
темноте не образуются ЫАВРН и АТР, их исчезновение в водоросли прекращает
синтез глюкозы.
2. Синтез амилозы идет под действием амилосинтазы из АВР-глкжозы.
Необходима затравка (С6Н10О5)п, где п > 2:
АВР-глюкоза + (СбН10О5)п -> (СбН10Об)п+1 + АВР.
3. Присоединение молекулы глюкозы к затравке идет с затратой двух макро-
энергетических связей АТР:
1) Глюкоза + АТР —> глюкозо-1-фосфат +АВР:
ДО°' = -30,5 кДж-моль"1;
2) Глюкозо-1-фосфат + АТР -> АВР-глюкоза + РР^
3) РР^Н/) -> 2Р5:
ДО(Г= -30,5 кДжмоль'1.
(С6Н10О5)п + АВР-глюкоза -» (С6Н10О5)п+1 + АВР.
4. Затравкой в синтезе инулина является сахароза. Наращивание цепи
инулина идет со стороны остатка молекулы фруктозы, с которым реагирует
АВР-фруктоза.
5. Крахмал в зеленом листе после фотосинтеза превращается в сахарозу,
которая поступает в клубни и корневища растений и вновь превращается в
крахмал.
Глава 11. Метаболизм липидов
1. Аналогия: исходным продуктом анаболизма и конечным продуктом
катаболизма является ацетил-СоА.
Различие: анаболизм идет через малонил-СоА и осуществляется в
цитоплазме. Катаболизм идет в митохондриях; малонил-СоА не является интермедиатом
данного процесса.
2. В результате образования малонил-СоА, идущего с использованием АТР, и
последующей его конденсацией с ацил-СоА, сопровождаемой отщеплением С02,
процесс синтеза жирной кислоты приобретает необратимый характер.
3. В триацилглицероле с низким йодным числом.
4. Энергии больше аккумулировано в пальмитиновой кислоте.
5.
О
II
СН2_0-С~С х 7Н35
?
СН-СМ>С17Н35 + 81,502 -► 57С02 + 55Н20
О
II
СН2-0-С-С17Нз5
Выделится 111 г воды.
6. Не обладает, иС-атом выделяется в виде 14С02.
7. Молекула лауриновой кислоты содержит 2 атома кислорода, молекула
сахарозы — 11 атомов.
Глава 12* Метаболизм аминокислот и нуклеотидов
1. Два пути: азотфиксадия из воздуха с помощью специфических
микроорганизмов и разложение растительных и животных останков.
2. Ферментный комплекс нитрогеназа состоит из двух компонентов —
белка 1 и белка 2. Их действие можно представить в виде следующей схемы:
677
КАБРН + Н+ . У Белок 2окисл ^ ^ Белок 1аоая х , N0
(Мо2+)
ЫАБР+ Белок 2восст Белок 1окисл 21Ш3
АТР (Мо6+)
Трансаминаза
3. Алании + 2-Оксоглуторат — *- Пируват + Глутамат,
СоА-8Н
4. Глюкоза—•►-Пируват —^—*-Ацетил-СоА—МЗксалоацетат —*~Цитра'
Изоцитрат —*~2-Оксоглутарат ^^- » Глутамат,
5. Окислительное дезаминирование и декарбоксилирование.
Глутаминаза
6. Глутамин + Н20 *- Глутамат + КН3.
7,
СОО" СОО"
сн « нс-1^н2
II + 15МН8 +~ I
сн сн2
СОО" СОО"
Фу марат Аспартат
Глава 13. Синтез ДНК, РНК и белков.
Перенос генетической информации
1. ТАС ТТТ ОСА СОТ ААА.
2. АШ ААА ССУ ОСА ШШ.
3. Ме*-Ьу8-Аг2-А1а-РЬе.
4. а) До делеции или внесения нуклеотида в цепь ДНК синтез полипептидной
цепи остается неизменным. При дальнейшем наращивании полипептидной цепи
в связи с изменением состава оснований триплетов будут принимать участие
иные аминокислоты.
б) Делеция и внесение нуклеотидов на локальном участке ДНК ведет к
изменению аминокислотной последовательности только на участке полипептидной
цепи, комплементарной локальному участку РНК.
5. Рекомбинация ДНК составляет материальную основу изменения
наследуемых признаков и играет большую роль в эволюционном развитии живой
материи, в организации рациональной высокоактивной структуры генома
живого организма.
Получение целевого гена, получение рекомбинантной ДНК, введение ее в
клетку хозяина, клонирование гена.
6. а) Первая генерация Е. сой даст полутяжелую ДНК (|5Ы,14М-ДНК)-
б) Вторая генерация даст тяжелую и полутяжелую ДНК.
678
Глава 14, Молекулярные основы действия
клеточных медиаторов
1. Мессенджеры — это первичные медиаторы, связывающиеся в рецептор-
ной зоне на клеточной поверхности и передающие сигнал на мембранные транс-
д у кторы.
Трансдукторы — внутриклеточные передатчики, обеспечивающие передачу
сигнала мембраносвязанному эффекторному ферменту.
Вторичные медиаторы — это вещества, образующиеся под действием эффек-
торного фермента и обеспечивающие перенос сигнала к конечному пункту
назначения в клетке.
3. Стероидные гормоны - гидрофобные соединения, способные
диффундировать через плазматическую мембрану в клетку, где они связываются с рецептор-
ными белками в цитоплазме.
5. Повышение концентрации сАМР в цитозоле благодаря активации аденилат-
циклазы приводит к связыванию двух регуляторных субъединиц протеинкиназы А
с четырьмя молекулами сАМР и высвобождению двух каталитических субъединиц
фермента, в результате чего происходит активация протеинкиназы А.
6. Активна ОТР-связанная форма О-белков.
7. а) инозитол-1,4,5-трифосфат (ИФ3); б) диацилглицерол.
9. 1) б; 2) в.
10. б).
Глава 15. Некоторые аспекты экологической биохимии
1. Биохимическая адаптация подразделяется на генетическую адаптацию,
акклиматизацию, немедленную адаптацию*
2. Вещества вторичного происхождения в экологических процессах
выполняют разнообразные функции: принимают участие в опылении растений
насекомыми, являются пищевыми аттрактантами, репеллентами, феромонами и т.д.
3. Аттрактанты являются пищевыми стимуляторами. Они включают
широкий круг веществ вторичного происхождения: гликозиды, терпены, флавонои-
ды, алкалоиды.
Репелленты — вещества, защищающие растения от поедания животными и
насекомыми.
4. Химические вещества, участвующие во взаимодействиях высших растений,
используются растением одного типа для борьбы с растениями другого типа (юглон,
гидроксибензойная, гидроксикоричная кислоты, фитоаллексины, соланин).
5. Токсины подразделяются на два класса: I) азотсодержащие и 2) безазотистые
токсины» К азотсодержащим токсинам относятся небелковые аминокислоты,
участвующие в синтезе биологически неактивных белков; цианогенные гликозиды, при
ферментативном расщеплении выделяющие синильную кислоту, и др.
К безазотистым токсинам относятся сердечные гликозиды, ингибирующие
мембранные АТР-азы; микробные афлотоксины, вызывающие разрушение
печени; сапонины, вызывающие гемолиз эритроцитов.
6. Причиной мутаций, возникающих под действием химических
соединений, является изменение последовательности азотистых оснований на каком-
либо участке ДНК. К мутагенам относятся формалин, этиленимин, нитраты,
нитриты, нитрозамины, дихлорэтан, оксид этилена, фенол, свинец, триметил-
фосфат, соли тяжелых металлов, радионуклиды,
7. Обезвреживание токсинов осуществляется в реакциях окисления,
восстановления, расщепления, конъюгации.
679
Глава 16. Некоторые аспекты медицинской
и фармацевтической биохимии
1. К молекулярным болезням относятся: фенилкетонуряя, тирозинемия, энзи-
мопатии углеводного и азотного обмена, серповидноклеточная анемия. В основе
молекулярных болезней лежат дефекты структурных генов и, как следствие, синтез
белков с полной или частичной утратой способности выполнять свои функции.
6. Б процессе канцерогенеза принято выделять две фазы: трансформация
клеток и прогрессирующий опухолевый рост. Различают химический,
физический и вирусный канцерогенез,
Глава 17. Биохимические превращения важнейших пищевых
компонентов при созревании, хранении и переработке
сельскохозяйственного сырья
1. Основной биохимический процесс при хранении зерна, картофеля,
сахарной свеклы — дыхание. Интенсивность дыхания зерна зависит от его влажности.
При влажности выше критической (14—15 %) интенсивность дыхания резко
возрастает, что ведет к самосогреванию зерновых масс, ухудшению семенных и
продовольственных характеристик зерна.
Интенсивность дыхания картофеля и сахарной свеклы зависит от
температуры. Чем выше температура хранения, тем выше интенсивность дыхания и
потери углеводов на этот процесс. Наилучшей температурой при хранении картофеля
и сахарной свеклы является 0—+5 °С.
2. Амилазы, р-фруктофуранозидаза, протеазы.
3. а-Амилаза превращает крахмал в декстрины; неустойчива к Н+-ионам
среды, но термостабильна; {3-амилаза превращает крахмал в мальтозу;
термолабильна, но устойчива к Н+-ионам; глюкоамилаза превращает крахмал в глюкозу;
имеет высокую кислотную и термическую устойчивость.
4. Пектинрасщепляющие ферменты используются при получении
фруктовых соков. Разрушая пектиновые вещества, они резко повышают сокоотдачу. В
технологии получения сахара из сахарной свеклы они играют отрицательную
роль, способствуют повышению вязкости получаемых продуктов.
5. Гидролиз сахарозы на глюкозу и фруктозу осуществляется (3-фруктофурано-
зидазой. Производство глюкозо-фруктозных сиропов из крахмала идет по схеме:
а-Амилаза Глюкоамилаза Глюкозоизомераза
Крахмал *- Декстрины ► Глюкоза ** Фруктоза + Глюкоза
6. По высокой активности и устойчивости к физико-химическим факторам
среды, по специфичности их действия.
7. Иммобилизованные ферменты — это препараты ферментов, молекулы ко-
торых связаны с нерастворимым носителем и сохраняют при этом полностью или
частично свои каталитические функции.
Все методы иммобилизации подразделяются на физические (включение
фермента в поры геля, пространственное отделение фермента с помощью
полупроницаемой мембраны) и на химические (ковалентное связывание фермента с носителем).
8. Разработка установок с использованием иммобилизованных ферментов,
отыскание и получение штаммов микроорганизмов — эффективных
продуцентов ферментов с высокой рН- и термостабильностъю.
9. 11,1 г глюкозы.
10. 22,2 г мальтозы.
11. 5,4 г глюконовой кислоты.
ПРЕДМЕТНЫЙ
А
Абсцизовая кислота 226, 228
Авитаминоз 181, 196
Авенин 70
Авидин 197
Агароза 129
Адаптация генетическая 573—574
Аденилаткиназа 418
Аденин 78,79
Аденипатциклаза 526, 538—540, 556, 559
Аденилатциклазная сигнальная
система 538—543
Аденилосукцинатсинтетаза 477
Аденилсукциназа 479—480
Адениннуклеотидтранслоказа 359—360
Адениловая система 242, 244
8-Аденозингомоцистеин 452
8-Аденозинметионин 429, 452
Аденозинтрифосфатсульфорилаза
467—468
Аденозин-5-фосфосульфат 468
Адреналин 538—539, 541—542, 551—
552, 610
Аденозинтрифосфат 244—247, 315—
316, 323—324, 334—335, 338—
339, 356-360, 363—366, 377-
380, 404, 447, 450, 453, 456
Азитидин-2-карбоновая кислота 580
Азотфиксация 460—463
Акклиматизация 574
Аконитатгидратаза (см. также акони-
таза) 294, 328—329, 441—442
Активные формы кислорода 204,
366—371
Актиномицин О 531
Акцепторная ветвь тРНК 94
Алании 30—31,389, 465
р-Аланин 190, 475, 485
Аланил-тРНК-синтетаза 293
Аллелозимы 300
Альбумины 69
Альдаровые кислоты 112
Альдоновые кислоты 112
Альдостерон 159, 560
Ал ьд о лаза 407, 409
Алкалоиды 220—222
Аллантоин 485
Аллантоиновая кислота 485
Алкогольдегидрогеназа 270, 283—
284, 318, 604
УКАЗАТЕЛЬ
Аллостерические ферменты 254,
294—297
ос-Аманитин 531
Аммйак-лиаза 292
Амигдалин 223, 580—581
Амилаза 255, 270, 281, 646—653
Амилоза 121, 423—426, 647—648
Амилопектин 121, 423—426, 647—
648
Аминирование восстановительное
464—465
Аминоацил-тРНК 95, 517
Аминоацил-тРНК-синтетаза 517
п-Аминобензойная кислота 191, 197
Ами ноими д азол -4 -к арбокс иами д ри бо -
нуклеотидформилаза 478
Амин оими д аз ол рибону к л еоти д к арб ок -
сил аза 478
Ами ноими д аз ол - 4 Г^-сук цин ок арб оке и-
амидрибонуклеотидсинтетаза
478—479
Аминолимонная кислота 31
у-Аминомасляная кислота 479, 607
Аминопептидазы 289, 522
Аминосахара 114
Амитал 349
Аммонификация 460—461
Амфотерная природа белков 53
Анабазин 221
Аномерный углеродный атом 110,
116—118
Андрогены 159, 560—561
Антибиотики 615, 621
Антивитамины 182, 189, 196—197
Антикодон 95, 96
Антимицин А 349
Антиоксиданты 177, 185, 186, 194,
613
Антипорт 167—168
Антоцианы 217, 218—219
Антроновый метод 135
Апоптоз 370—372
Апофермент 255
Арабан 129, 130
Арабаногалактан 129
Арабиноза 121, 129
Арахидоновая кислота 175
Аскорбиновая кислота 193—196, 200
Аскорбиноген 194
Аскорбиноксидаза 194, 200
681
Аспарагин 465, 471—472, 477
Аспарагиназа 619, 624
Аспарагинсинтетаза 465
Аспартат 362, 418—419, 465, 474,
477, 480
Аспартаттрансаминаза 362
Аспартаттранскарбомоилаза 481
Аргиназа 474
Аргинин 30—31, 373—374
Аргининсукцинат 473—474
Аргининсукщшатлиаза 473
Аргининсукцинатсинтетаза 473
Аргининфосфат 244
Арилэстераза 586
Атеросклероз 601—602
Атрактилозид 359
Аттрактанты 575—577
АТР-синтетаза 344, 345
Ауксин 226—227
Аффинная хроматография 61
Афлотоксины 581
Ацетальдегид 318, 603—604
Г^-Ацетилгексозаминидаза 597
Ацетил-СоА 321,325—327,333, 387—
388, 443, 444, 445, 448, 587
Ацетил-СоА-ацилтрансфераза 438
Ацетил-СоА-карбоксилаза 445, 448
Ацетил-СоА-синтетаза 293
З-Ацетил-6-метоксибензальдегид 579
Ацетилсерин 468
Ацетилтрансацилаза 446
Ацетилфосфат 320
Ацетилхолин 155, 168, 607
Ацетоацетат 322
Ацетоацетил-СоА 321—322
Ацетоацетатдекарбоксилаза 322
Ацетон 322
Ацилглицеролы (см. также жиры)
146—150, 434—435, 449—451
Ацил-СоА 438—439
Ацил-СоА-дегидрогеназа 350, 437—
438
Ацил-СоА: карнитинацилтрансфераза
437
Ацилпереносящий (АПБ) белок 445—
447
Ацилтрансфераза 449
Ацильная группа 146, 152, 162,
246—247
Б
Базальная скорость метаболизма 556
Базальный уровень гормонов 552
Бактериофаги 99, 100
Барьер энергетический 257
Бацитрацин А 22
Белки индуцируемые 523—527
Белки конститутивные 527
Белки, структура 44—49
Бензойная кислота 453
Биосинтез ацилглицеролов 449—450
Биосинтез гемицеллюлоз 429—430
Биосинтез гликогена 426—427
Биосинтез инсулина 427—428
Биосинтез крахмала 423—425
Биосинтез олигосахаридов 420—422
Биосинтез фосфолипидов 450—451
Биосинтез целлюлозы 428—429
Биотин 196—197, 445
1,3-Бисфосфоглицерат 244—245, 311,
314
Биоэнергетика анаэробного
разложения углеводов 323—325
Биоэнергетика цикла трикарбоновых
кислот 333
Биуретовая реакция 20
Борнеол 209—210
Брожение глюконовокислое 663—664
— лимоннокислое 663
— маслянокислое 320—323
— молочнокислое 319—320, 661—662
— спиртовое 317—319, 660—661, 667
— уксуснокислое 664—665
В
Вазопрессин 552—553
Валиномицин 167, 380
Вакцины 621—622
Ван-Слайка реакция 27
Ванилин 214
Вербиоза 118
Ветвящий фермент 424
Вещества витаминоподобные 197—
199
Вещества вторичного происхождения
207
Вирионы 98
Вирусы 98—100
Витамеры 182—185, 189
Витамин А 183—184
Витамин В12 192—193
Витамины группы В 187—193 (см.
также по названиям)
Витамин В 184—185
Витамин Е 185—186
Витамины жирорастворимые 183—187
682
Витамин К 187
Витаминизация 201—203
Витаминоподобные вещества 182,
197—199
Влажность критическая 642—643
Вода связанная 642
Воски 146, 149, 455
Г
Галактаны 128—129
Галактаровая кислота 112
Галактинол 421—422, 595
Галактоза 106, 118, 119, 129, 132,
319, 412—413, 596
Галактокиназа 596
(З-Галактозидаза 319, 523—526, 667
Галактозид-пермиаза 523—526
Галактозид-трансацетилаза 523—526
Галактозо-1-фосфат 412—413
Галактозо-1-фосфатуридилтрансфера-
за 595
Галактозилдиацилглицеролы 157
Галактоманнаны 129
Галактоцереброзид 156
Галактуроновая кислота 112, 130,
131, 413, 429
Галловая кислота 219
Галлотаннин 218—219
Ганглиозиды 155, 157
Гексозомонофосфатный путь (см.
также пентозофосфатный путь)
335—338, 369
Гексокиназа 261—262, 311—312,
316, 388, 413, 426, 610
Гель-фильтрация 41—43
Гемицеллюлозы 119, 128—129, 429—
430
Ген-регулятор 524
Ген структурный 524
Гентибиоза 117
Гераниол 208—209
Геранилгераниол 210
Геспертин 217
Гетерогенная ядерная РНК 512
Гетероолигосахариды 114
Гетерополисахариды 119
Гиберелловые кислоты 227
Гиббереллины 227—228
Гибридомы 626—627
Гидроксиацил-СоА 438
Гидроксиацил-СоА-дегидрогеназа 438
Гидроксибутирил-СоА 321
Гидроксилизин 23
Гидроксильный радикал 367, 371—
372
Гидрид-ион 282
о-Гидроксикоричная кислота 214
Гидроксипролин 23, 194
Гидроксипируват 415—416
Гидроксипируватредуктаза 415
Гидроксифенилпируватоксидаза 594
Гидролазы 280, 288—291
Гидрофильность белков 55
Гидрофобные взаимодействия 51
Гидрохинон 212—213
Гипервитаминоз 184
Гиповитаминоз 181, 183, 188—189
Гипоксантин 477, 484, 613
Гипотиреоз 556
Гипохромный эффект 67, 68, 88, 92
Гистамин 475
Гистидаза 594
Гистидин 30—31, 475
Гистоны 71
Глиадин 634—635
Гликоген 119, 123—126, 426—427,
596
Гликогенин 426
Гликогенфосфорилаза 317, 612
Гликогенозы 596
Гликозидазы 289, 449
Гликозидная связь 114, 428
Гликозиды 111, 222—225
Гликозилтрансфераза 427, 583
Гликолат 415, 442
Гликолатоксидаза 415
Глиоксилат 415—417, 440—442
Глиоксилатный цикл 440—442
Гликолиз 245, 298, 310—317, 387—
389, 449, 595, 609
Гликолипиды 156
Гликопротеины 132, 553, 564
Гликосфиноголипиды 156
Гликопротеины 115, 132
Глицин 30—31, 71, 415, 467, 477,
607
Глицинамидрибонуклеотидтрансфор-
милаза 479
Глициндекарбоксилаза 415
Глицерат 415
Глицерин 113, 435—436, 444, 452—
453
Глицеролфосфат 436
Глицерофосфаткиназа 435—436
Глицеральдегид 105
683
Глицеральдегид-3-фофат 311, 314,
320, 324, 337, 407-410, 455
Глицеральдегид-3-фосфатдегидрогена-
за 311, 315, 407—409
Глицерофосфатный челночный
механизм 360—361
Глицерол-3-фосфатдегидрогеназный
комплекс 360
Глицерол-3-фосфатдегидрогеназа 360,
436
Глобулины 169
Глутамат 30—31, 191, 464—466, 470,
474, 481, 607
Глутаматдегидрогеназа 284, 466, 474
Глутамин 30—31, 464, 474—475, 477,
481—482
Глутаминаза 474
Глутаминсинтетаза 464—465
Глутатион 368—369
Глутатиопероксидаза 368—369
Глутатиоредуктаза 368
Глюкагон 389, 562—563
Глюканы 119
Глюкаровая кислота 112
Глюкоамилаза 648—649, 654
Глюкоза 107—108, 118, 121, 132,
168, 218—219, 318—321, 387—
388, 413, 426, 442, 523—527,
587, 600^601, 651, 657—659,
662—664
сс-Глюкозидаза 319, 596
Глюкокортикоиды 559—560
Глюкозойзомераза 654
Глюкозооксидаза 657—659
Глюкозо-1 -фосфат 244, 311, 317, 426
Гл юкозо-1 -фосфат-уридилтрансфераза
426
Глюкозо-6-фосфат 244, 312—313,
314, 318, 321, 324, 336, 387, 425
Глюкозо-6-фосфатаза 387, 596
Глюкозо-6-фосфатдегидрогеназа 300,
335—336, 369
Глюконеогенез 332, 385—389, 559,
563
Глюконовая кислота 657—658, 663
Глюконолактоназа 336
Глюкуроновая кислота 112, 413, 429
Глютелины 70
Глютенин 634—635
Гомоолигосахариды 114
Гомополисахариды 119
Гордеин 70
Госсипол 175
Грамицидин 166, 380
Гуанин 77, 80
Гуминовые кислоты 220
д
Дансйльный метод 62
Дегидроаскорбиновая кислота 194,
200
7-Дегидрохолестерол 184—185
Дегидрогеназы 281—285
Дегидрооротаза 481
Дезаминирование окислительное
470—471
Дезоксиаденозилкобаламин (см.
также цианокобаламин) 192, 439
Дезоксирибонуклеиновая кислота 77
Дезоксирибонуклеозиды 77
Декарбоксшщрование аминокислот
469, 475—476
Декарбоксилазы 291—292, 417—418
Декстраны 128
Декстрины 647
Дельфинидин 217—218
Демиссин 224—225, 577
Детергенты 146, 173
Диабет сахарный 589—599, 600—601
Диализ 61
Диацилглицерол 450—452
Диацилглицеролтрансфераза 450
Дигидроксиацетон 105
Дигидроксиацетонфосфат 311, 314,
361, 407—408, 436, 449
3,4-Дигидроксифенилаланин (дофа)
557
3,4-Дигидроксифенилэтиламин
(дофамин) 557
Дигидрооротат 482—483
Дигидрооротатдегидрогеназа 482
Дигидроурацил 79, 485
Дигидроуридиловая ветвь 94
Дигидроуридин 94
Диеновые коньюгаты 367
Дикарбоксилаттранспортирующие
системы 362—363, 386
Диметилгидроксимасляная кислота
190
Динитробензольные производные
аминокислот 28
Дипальмитоилфосфатидилхолин
152—153
Дисахариды 115
Дисперсия оптического вращения 66
684
Дитерпены 208, 210
о-Дифенолоксидаза 659
Дифосфатидилглицерол 151
Дисульфидные связи 51
ДНК-лигазы 487, 493, 495—496, 497,
499—500
ДНК-плазмиды 504
ДНК-полимеразы 491—496, 499, 501
ДНК рекомбинантная 498, ёЙО
ДНК-связывающие белки 303, 492—
493
Дыхание 310
Дыхательный коэффициент 344, 358,
363
Дыхательная цепь 338—339
Домен 44, 48
Дульцит 113
Е
Единица активности фермента 301
Еноил-СоА 438—440
Еноил-СоА-гидратаза 438
Еноил-АПБ-редуктаза 446
Енолаза 311, 315
Ж
Жидкостно-мозаичная модель
мембраны 162—164
Жирные кислоты 142, 367, 436—440,
442—449
3
Зёренсена реакция 27—28
Значение рК функциональных групп
в белках 53
И
Излучение ионизирующие 406
Изоамиловый спирт 476
Изолейцин 469
Изомальтоза 117
Изомеразы 280, 292—293*439—440
Изомеры лолепептидов 33
Изомальтоза 117
Изопрен 159, 187
Изоферменты 299—300
Изоцитрат 329—330, 442
Изоцитратдегидрогеназа 330
Изоцитратлиаза 441
Изоэлектрическая точка 25, 30
Иммобилизованные ферменты 666—
667
Ингибирование по типу обратной
связи 469
Индол 476
Индуктор 294, 524—527
Индукция 294, 523—527
Инвертаза 268
Йнвертный сахар 115
Ингибиторы ферментов 275—279
Инозин 484
Инозинах 479
Инозинатдегидрогеназа 479
Инозитол-1,4,5-трифосфат 153, 443,
550
Инозитолфосфолипидныи сигнальный
путь 543—546
Инозит 198, 452
Ионофоры 165—166
Инсулин 550, 562-564, 598-599,
620
Интерлейкины 616
Инулин 119, 124—125, 137, 427—
428, 653—654
Инулиназа 428, 653—654
Йнтерфероны 616, 621
Интроны 512
Инфаркт миокарда 612—614
Ишемия 612—614
й
Йодное число 173-
Йодтиронины 554—557
К
Кадаверин 475—476
Казеин 655—656
Кальциотонин 557
Кальциферолы 184—185
Камфен 209—210
Камфора 209—210
Канцерогенез 611—612
Карбамоил-|3-аланин 485
Карбамоиласпартат 482—483
Карбамоилфосфат 473—474, 482
Кат^Замоилфосфатсинтетаза 473
Карбоангидраза 270
Карбогидразы 289
у-Кйрбоксиглутаминовая кислота 22
Карбоксилазы 294
Карбоксипептидаза 62, 289—290
Карнитин 437
Каротиноиды 211, 392—393
Каротины 183, 211, 392
Карвон 209
Кардиолипин 151, 154
Каспазы 371
Катаболитный генактивирующий
белок 526
685
Катал 301
Каталаза 368, 415, 658
Катехоламины 557, 612
Катехины 216—217
Каучук 211—212
Квашиоркор 600
Кератины 72
а-Кетоглутарат (см. также 2-оксоглу-
тарат) 415
а-Кетоглутаратдегидрогеназный
комплекс (см, также 2-оксоглута-
ратдегидрогеназный комплекс)
299
3-Кетоацил-АПБ-редуктаза 447
3-Кетоацил-АПБ-синтетаза 447
Киназа пируватдегидрогеназы 334
Кинетика ферментативных реакций
265—269
Кислотное число 172
Кислоты жирные незаменимые 175—
176
Кислоты уроновые 112
Классификация углеводов 104—105
Клейковина 70
Клонирование гена 503—505
Клупеин 71
Ковалентная модификация ферментов
254, 297, 317
Код генетический 514—517
Кодон 515—517
Коллаген 71—73
Коллагеназа 290, 615
Компартаментализация 253
Комплекс фермент-субстрат 257
Комплексы электронтранспортной
цепи 346—349
Конкурентный резонанс 243
Константа Михаэлиса 269—271
Конститутивные белки 524, 527—528
Конформацил белков 44
Коричная кислота 579
Кортизол 559
Кортиколиберин 559
Кортикостерон 560
Кортикостероиды 559
Кортикотропин 554, 559
Кофактор 255
Кофеин 221, 540
Кофейная кислота 214
Кофермент А 191, 246—247, 436—
437
Коэффициент седиментации 40
Крахмал 122—124, 423—425, 636—
639, 643—644
Крахмалсинтаза 423—424
Креатинфосфат 244, 246, 613
Креатинфосфокиназа 613
Кротоназа 321
Кротонил СоА 321
Ксантилат 479
Ксантин 484, 613
Ксантиноксидаза 484
Ксантопротеиновая реакция 20
Ксантофиллы 211, 392
Ксенобиотики 586
Ксиланы 129—130, 430
Ксилоза 119, 129
Ксилит 137
Ксилулозо-5-фосфат 407—409
Кумарин 215
Кумариновая кислота 215
Кумаровая кислота 214
Кумулятивное ингибирование 295
Л
Лактальбумин 69
Лактат 319—320, 613
Лактатдегидрогеназа 284, 299, 319,
613
Лактоза 116—117, 319, 421, 595
Леван 125
Легоглобин 463
Легумелин 69
Легумин 69
Лейцин 476
Лиазы 280, 291—293
Либераторы 279
Лигазы 280, 293—294
Лигнин 220, 587
Лизергиновая кислота 222
Лизин 30—31, 475
Лизиндекарбоксилаза 475
Лизосомы 288
Лизофосфоглицериды 153
Ликопин 211
Лимит Хейфлика 528—529
Лимонен 209
Линалоол 208—209
Линолевая кислота 145
Линоленовая кислота 440
Липоеомы 162
Липазы 288, 366, 434—435
Липиды 142—180, 596—597
Липидные бислои 161
Липоевая кислота 198—199
686
Липоксигеназа 659—660
Липопротеины 72
Липотропины 554
Лютеинезирующий гормон 853
М
Малат 327, 331, 418—420, 441—442
Малат-аспартатный челночный
механизм 361—363
Малатдегидрогеназа 284, 328, 331,
362—363, 386, 442—443
Малатсинтаза 442
Малая ядерная РНК 93
Малик-знзим 417—420
Малонат 349, 377
Малонил-СоА 445, 447, 456
Малонилтранеацилаза 446
Мальвидин 217
Мальтоза 117, 420, 595, 647—648
Мальтотриоза 119
Маннаны 119, 129
Манноза 106, 119, 129, 413
Маннуроновая кислота 112, 413
Масляная кислота 321, 454
Матричная РНК 95—96
Медиаторы 154, 537—538
Мезосома 77
Меланоидинообразование 29, 644
Мембранные транспортные системы
166—176
Мембраны биологические 150, 160—
171
Мембраны митохондрий 342
Ментол 209
Метаболоны 298—299
Метазимы 300
Металлопротеины 72
5 Д 0-Метилентетрагидрофолат 414
Метилирование ДНК 90—93
Метилкобаламин 452
Метилмалонил-СоА 439
Метилмалонил-СоА-мутаза 439
Метилметионин 199
Метилтрансфераза 452
5-Метилцитозин 79
Метионин 30—31, 469, 587
Метод Лайнуивера—Берка 270—271
Метод Эдмана 29
Миллона реакция 20
Минералокортикоиды 560
Минорные азотистые основания 79
Миоинозит 198
Мирицилпальмитат 149
Митохондрии 341—344
Мицеллы 160—161
Модифицированные аминокислоты
22—23
Монелин 601
Моноацилглицерол-3-фосфат 449
Моносахариды 105—110
Монотерпены 208—210
Морфин 222
Мочевая кислота 471, 485
Мочевина 471—472, 474, 485, 599
Мутагенез 91
Мутагены 496, 584—586
Мутазы 293
Мут-аротация 108—109
Н
Нарингин 217
(5-Нафтилуксусная кислота 226, 454
Нафтохинон 187, 215
Нейтронография 65
Некроз 370
Нектар 574—575
Неомицин 532
Нефосфорилирующее окисление
365—366
Никотин 221—222
Никотиновая кислота 190
Никотинамидадениндинуклеотид
282—283
Никотинамидадениндинуклеотидфос-
фат 282—283
Никотинамидадениндинуклеотид
восстановленный ^АВН)-убихи-
нон-оксидоредуктаза (комплекс I
электронтранспортной цепи)
351—352
Нингидриновая реакция 28
Нитратредуктаза 463—464
Нитритредуктаза 463—464
Нитрификация 463—464
Нитрогеназа 463—464
Нонактин 380
Норадреналин 550, 557—558
Нуклеозиддикиназа 481—482
Нуклеазы 76
Нуклеозиды 244
Нуклеозиддифосфатсахара 412—414
Нуклеопротеины 72
Нуклеотиды 76, 80—81, 477—483
О
Облегченная диффузия 166—167
З'-Оверхенги 529
687
Порядок химических реакций 265—
266
Посттрансляционная модификация 522
Правила Чаргаффа 83—84
Праймаза 493—494
Препараты гормональные 615
Препараты ферментов 615
Провитамины 182—184, 211
Прогестерон 561
Прогормоны 552
Пролактин 552
Пролактолиберин 552
П рол амины 70
Пролин 30—31, 466
Промотор 509, 524
Пропионил-СоА 439
Пропионил-СоА-карбоксилаза 439
Простетическая группа 68, 72
Протамины 71
Протеазы 289—291
Протеиназы 289—291
Протеин киназы 538—540, 545
Протеинопатия 593—599
Протеинфосфатаза 539
Протеноиды 71
Протонный потенциал (см. также .
электрохимический градиент
ионов Н+) 344—346
Протопектины 131, 132
Протромбин 187
Проферменты 275, 290
Процессинг РНК 511—512
Псевдоуридин 94
Пури новые азотистые основания 78—
79
Пуромицин 531
Путресцин 475—476
Р
Радикал супероксидный 367
Радионуклиды 571, 587
Распад нуклеотидов 483—485
Рафиноза 118, 136, 422
Рацемазы 292
Реакции сопряжения 247—248
Редокс-потенциал 347
Резорцин 212—213
Резорциновый метод 135
Рекомбинация 498—504
Реннин 655—656
Рентгенографический метод 64, 133
Репарация генетических повреждений
496—502
Репарация эксцизионная 497
Репелленты 575—577
Репликативная вилка 493—494
Репликация 492—493
Репрессия синтеза белка 294, 523—
525
Ретиеновая кислота 183
Ретиналь 183—184
Ретинол 184
Рибозимы 512—513
Рибозо-5-фосфат 336, 480
Рибонуклеиновая кислота 77
Рибонуклеазы 288, 522
Рибонуклеозиды 79
Рибосомная РНК 93
Рибосомы 93
Риботимидин 94
Рибофлавин 189
Рибулозо-1,5-бисфосфат 406—407,
414—415
Рибулозо-5-фосфат 336, 407, 410
Рибулозо~1,5-бисфосфаткарбоксилаза-
оксигеназа (см. также рубиско)
405—406, 408, 415—421
Рилизинг-факторы 521
Рифампицин 531
РНК-зависимая ДНК-полимераза
(обратная транскриптаза) 504,
513—514, 528
РНК-полимераза 509—510, 525
РНК-праймер 494—495
Роданеза 583
Ротенон 348—349
С
Салициловый спирт 214
Сальмин 71
Самосогревание зерновых масс 643
Сапогенин 225
Сапонины 211, 225, 581
Сахароза 115, 420, 428
Сахарозо-6-фосфат 420
Сахарозо-фоефат-синтаза 420
Связь макроэргическая 241
Седогептулозо-1,7-бисфосфат 407, 410
Седогептулозо-7-фосфат 337, 407, 410
Секвенирование 63
Селеноцистеин 369
Семихинон 351—353
Сердечные гликозиды 581
Серии 30—31, 415, 452, 466—467
Серингидроксиметилтрансфераза 415,
467
44. Заказ 3486
689
Овальбумин 69
(З-Окисление жирных кислот 437—440
Оксалат 485
Оксалоацетат 328, 362, 386, 418—
420, 441—442, 465
Оксалосукцинат 328
Оксигеназы 586—587
Оксид азота 372—374
Оксидазы 284
Оксидоредуктазы 280—292, 657—660
Оксилегоглобин 463
Оксиметилфурфурол 107, 135
Окситоцин 553
Оксоацил-СоА 438
2-Оксоглутарат (см. также ос-кетоглута-
рат) 328, 330, 362—363, 464—466,
470
2-Оксоглутаратдегидрогеназный
комплекс 328—330
Олеандрин 581
Олеандомицин 531
Олеиновая кислота 145—146, 440
Олигосахариды 114—119
Оператор 525
Оперон 525
Оптическая активность 21, 107
Орезенин 70
Орнитин 472—474
Орнитинкарбамоилтрансфераза 473
Оротатфосфорибозилтрансфераза 481
Оротидинфосфатдекарбоксилаза 481
Оротовая кислота 198, 481—482
П
Палиндром 96, 503
Панкреатический полипептид 565
Паноза 119
Папаин 290, 656—657
Паратирин 557
Патология белкового питания 559—600
Патология жирового питания 600—603
Патология углеводного питания 600—
601
Пальмитат 144, 447
Пантотеновая кислота 190—191
Пиерицидин А 348—349
Пектин 131—132, 429—430, 654—655
Пектолитические ферменты 654—655
Пектиновая кислота 130
Пеларгонидин 217
Пенициллинамидаза 618
Пентозофосфатный путь 405—408,
416—417, 610
Пеонидин 217
Пепсин 290, 600, 655—656
Пептидазы 289, 522
Пептидилтрансфераза 520, 531
Пептидилформилаза 522
Пептиды 38
Пептидная связь 20, 32, 45
Пероксид водорода 367—368, 604,
658
Пероксидаза 626
Пероксидное окисление липидов 367,
613—614
Пероксиды 176—177, 186
Петунидин 217
Пинен 209—210
Пиранозы 109
Пиридинзависимые дегидрогеназы
282—283
Пиридоксаль 189—190, 475
Пиридоксамин 189
Пиридоксин 189
Пиримидиновые азотистые основания
77—78
Пирогаллол 213
Пирокатехаза 582
Пирокатехин 212—213
Пирофосфатаза 423, 425, 436, 517
Пирофосфорилазы 412
Пируват 311—312, 315—316, 318—
321, 323—324, 386—389, 465
Пируватдегидрогеназньш комплекс
326—328, 387—388
Пиру ватдекарбокси л аза 318
Пируваткарбоксилаза 386—388
Пируваткиназа 311—312, 316—317,
610
Пируватоксидаза 284
Пируватфосфатдикиназа 417—418
Плазмиды 77
Пластохинон 400—402
Пластоцианин 401
Полипептидная теория 32—34
Полипренол 430
Полирибосомы (полисомы) 518, 521—
522
Полисахариды 119—132
Полисахарид—лиазы 292
Полифенолоксидаза 194, 284—285
Политерпены 208, 211—212
Полиурониды 130
Поляриметния 107—108, 134
Порины 523
688
Порядок химических реакций 265—
266
Посттрансляционная модификация 522
Правила Чаргаффа 83—84
Праймаза 493—494
Препараты гормональные 615
Препараты ферментов 615
Провитамины 182—184, 211
Прогестерон 561
Прогормоны 552
Пролактин 552
Пролактолиберин 552
Проламины 70
Пролин 30—31, 466
Промотор 509, 524
ПропионшьСоА 439
ПропионшьСоА-карбоксилаза 439
Простетическая группа 68, 72
Протамины 71
Протеазы 289—291
Протеиназы 289—291
Протеинкиназы 538—540, 545
Протеинопатия 593—599
Протеинфосфатаза 539
Протеноиды 71
Протонный потенциал (см, также .
электрохимический градиент
ионов Н+) 344—346
Протопектины 131, 132
Протромбин 187
Проферменты 275, 290
Процессинг РНК 511—512
Псевдоуридин 94
Пуриновые азотистые основания 78—
79
Пуромицин 531
Путресцин 475—476
Р
Радикал супероксидный 367
Радионуклиды 571, 587
Распад нуклеотидов 483—485
Рафиноза 118, 136, 422
Рацемазы 292
Реакции сопряжения 247—248
Редокс-потенциал 347
Резорцин 212—213
Резорциновый метод 135
Рекомбинация 498—504
Реннин 655—656
Рентгенографический метод 64, 133
Репарация генетических повреждений
496—502
Репарация эксцизионная 497
Репелленты 575—577
Репликативная вилка 493—494
Репликация 492—493
Репрессия синтеза белка 294, 523—
525
Ретиеновая кислота 183
Ретиналь 183—184
Ретинол 184
Рибозимы 512—513
Рибозо-5-фосфат 336, 480
Рибонуклеиновая кислота 77
Рибонуклеазы 288, 522
Рибонуклеозиды 79
Рибосомная РНК 93
Рибосомы 93
Риботимидин 94
Рибофлавин 189
Рибулозо-1,5~бисфосфат 406—407,
414—415
Рибулозо-5-фоефат 336, 407, 410
Рибулозо-1,5-бисфосфаткарбоксилаза-
оксигеназа (см. также рубиско)
405—406, 408, 415—421
Рилизинг-факторы 521
Рифампицин 531
РНК-зависимая ДНК-полимераза
(обратная транскриптаза) 504,
513—514, 528
РНК-полимераза 509—510, 525
РНК-праймер 494—495
Роданеза 583
Ротенон 348—349
С
Салициловый спирт 214
Сальмин 71
Самосогревание зерновых масс 643
Сапогенин 225
Сапонины 211, 225, 581
Сахароза 115, 420, 428
Сахарозо-6-фосфат 420
Сахарозо-фосфат-синтаза 420
Связь макроэргическая 241
Седогептулозо-1,7-бисфосфат 407, 410
Седогептулозо-7-фосфат 337, 407, 410
Секвенирование 63
Селеноцистеин 369
Семихинон 351—353
Сердечные гликозиды 581
Серии 30—31, 415, 452, 466—467
Серингидроксиметилтрансфераза 415,
467
44. Заказ 3486
689
Серотонин 607
Сесквитерпены 208—2-10, 229
Сиаловая (ЗМ-ацетилнейраминовая)
кислота 157
Сигнальная трансдукция 537
Симпорт 167—168, 360
Синаповая кислота 214
Синапсы 607
Синигрин 225, 576
Синтаза жирных кислот 445—449
Синтез амидов 472
Синтез аминокислот 464—469
Синтез белка 514—521
Синтез сахарозы 638—639
Система адениловая. 242
Системы транспортные 166—170
Скатол 476
Слоисто-складчатая структура
(р-структура) белков 47—48
Соединения высокоэнергетические
243—244
Соединения фосфоорганические 241 —
243
Соланины 224, 580
Сояаноидин 224
Соматолиберин 552
Соматомедины 554—555
Соматостатин 552, 565
Соматотропин 554
Сопряжение энергетическое 248
Сорбит 113, 137, 195
а-Спираль Полинга-Кори 46—47
Сплайсинг 512
Сплайсосома 512
Стахиоза 118, 422
Стеарат 145, 443
Стрептокиназа 615
Стероиды 157—160
Стеролы 210—211
Стигмастерол 159
Структура моносахаридов 109—110
Субстратная специфичность 260—265
Субтилизин 615
Субъединицы белка 49
Сукцинат 327—328, 330, 440—442
Сукцинатдегидрогеназа 299, 328, 331,
350—352
Сукцинат-убихинон-оксидоредуктаза
(комплекс II электронтранспорт-
ной цепи) 352
Сукцинил-СоА 328, 330, 439
Сукцинил-СоА-синтетаза 328, 330
Супероксиддисмутаза 367
Супероксиданионрадикал 367—368
Сфингозин 154
Сфинголипиды 154—157
Сфингорлиелиназа 597 :
Сфингомиелины 155—156
Т
Танназа 219, 221
Таннины 218—220, 576
Теломеры 529—530
Теломераза 520—530
Теноилтрифторацетон 349
Температурный коэффициент 272
Термодинамика биохимическая 286—
288
Теобромин 221
Теофиллин 540
Терминация синтеза полипептида 518
Термогенин 366
Терпены 207—210
Террилития 615
Тетрагидрофолат 191 — 192, 414, 452,
467, 483
Тетратерпены 209—211
Тетрациклины 616
Тиамин 188—189
Тиаминпирофосфат 292, 318, 409
Тимидилатсинтетаза 483
Тимин 77—78, 483
Тиоурацил 79
Тиоэфиры 246—247
Тирамин 475
Тироксин (тетрайодтиронин, Т4) 23,
538, 554—555
Тиолаза 321—322
Тиоредоксин 483
Тиоредуктаза 483
Тиреоидные гормоны 554—556
Тироглобулин 555
Тирозин 30—31, 467, 475, 555, 557
Тирозинкиназа 538, 547
Тирозинкиназные рецепторы 545, 547
Тирозинфосфатазы 547
Тиролиберин 552, 554
Тиротропин 553
Токоферолы 185—186, 369
Томатин 577
Топоизомеразы 493
Травсальдолаза 336—337
Трансаминирование 416, 465—466, 470
Трансгидрогеназа 349
Трансгликозирование 420—422, 426
690
2-Фосфоглицерат 315
З-Фосфоглицерат 245, 315, 323, 407,
415, 466
Фосфоглицераткиназа 311, 313, 407
Фосфоглицератмутаза 311, 315
Фосфоглюкоизомераза 311, 313
Фосфоглюкомутаза 426
Фосфоглюконатный путь 320
6-Фосфоглкжонат 320, 336
6-Фосфоглюконатдегидрогеназа 335
6-Фосфоглгоконолактон 336
Фосфодиэстераза 540
Фосфод и эфирная связь 76
Фоефоенолпируват 315—316, 324,
332, 387, 417, 442
Фосфоенолпируваткарбоксикиназа
386, 417—421
Фосфоенолпируваткарбоксилаза
(ФЕП)-карбоксилаза 416, 442
Фосфолипаза 153, 288, 444, 539, 543
Фосфолипиды 450—452
Фосфопротеины 72
Фосфорибозил-глицинамидсинтетаза
478—479
Фосфорибозилпирофосфат 480
Фосфорибозилтрансфераза 480
Фосфорибулокиназа 407, 410
Фосфорилирование окислительное
242, 338^360
Фосфорилирование субстратное 242,
245
Фосфорилирование фотосинтетачес-
кое 242, 391, 398—405
Фосфатаза 288
Фосфофруктокиназа 297, 311, 313,
316, 389, 612
Фосфоэтаноламинцитидинтранефераза
451
Фотодыхание 414—415
Фотолиаза 498
Фотосинтез 335, 389—411
Фотосистемы I, II 396
Фрагменты Оказакй 493—496
Фракционирование белков 58
Фруктаны 124т-125
Фруктоза 106, 118, 137, 319, 413,
420, 595—596
Фруктозо-1 -фосфат 595—596
Фруктозо-1-фосфатальдолаза 595—
596
Фруктозо-6-фосфат 297, 313—314,
316, 387, 407—409, 411, 420
692
Фруктозо-1,6-бисфосфат 311, 313,
, 316, 323—324, 387, 409
Фруктозо-2,6-бисфосфат 389
Фрукрозобисфосфатаза 387, 389, 409,
411, 596
Фру ктозо-1,6-бисфосфатальдо лаза
291, 409
)3-Фруктофуранозидаза 319, 645—646
Фунгициды 583—584
Функциональные группы 21
Функции белков 35—36
Фумарат 328, 441—442
Фумаратгидратаза (см. также фумара-
за) 292, 328, 331
Фуранозы 109
Фуриладенин 227
Фурфурол 107, 130, 135
X
Хеликаза 492—493
Хемиосмотическая теория 344—346
Хеуорса проекционные формулы
109—110
Химозин 290
Химотрипсин 290, 615
Хинин 222
Хлорамфеникол 531
Хлоропласт 391—392, 615
Хлорофилл 392—394
Хлорофилл а/Ь светособирающий
комплекс (ССК) 396
Холат натрия (желчная соль) 159
Хо лекал ьциферол 185
Холестерол 158—160, 160, 211, 601 —
602, 611
Холин 197—199
Холинэстераза 288, 586
Холофермент 255
Хроматография газо-жидкостная 136,
172, 201
Хроматография гидрофобная 303—
304
Хроматография ионообменная 61, 302
Хроматография тонкослойная 171,
302
Хромопротеины 72
ц
Цветные реакции на белки 20
Цеатин 227—228
Целлюлоза 119, 125—127, 136, 428—
429, 428—429
Церамид 155
Цереброзиды 155—156
Цианидин 217
Цианиды 349
Цианокобаламин 192—193
Цикл глиоксилатный 440—442
Цикл Кальвина 405—410
Цикл мочевины 472—474
Цикл трикарбоновых кислот (см.
также цикл Кребса) 325—335,
438, 441—442, 604
Циклопентан оперги дрофе н антрен
211—212
Цистеин 30—31, 468—470, 587
Цистин 23, 30—31, 598
Цитидиндифосфат (СВР)-фосфат 450
Цитидиндифосфат (СВР)-холин 451
Цитидиндифосфат (СВР)-этаноламин
450—451
Цитидинтрифосфат (СТР)-синтетаза
481—482
Цитозин 77—78, 485
Цитокинины 227—228
Цитохромоксидаза 286—287, 350,
355—356
Цитохромы 285, 349—350
Цитохром ад3 355
Цитохром Ь 352
Цитохром Ь560 352
Цитохром с 353
Цитохром / 400—402
Цитохромный комплекс &/ 396, 400—
401
Цитохром Р-450 582
Цитраль 209
Цитрат 316, 329—330, 332—333, 441,
448
Цитратсинтаза 329, 441
Цитронеллол 208
Цитруллин 373—374, 472—473
Ч
Чаконины 224
Челночные механизмы переноса
ИАВН 360—363
Число оборотов фермента 270
Число омыления 173
Ш
Шкала термодинамическая 343—344
Шпильки РНК 93—96
Э
Эдестин 70
Эдмана метод 63
Экзонуклеазы 483, 497
Экзоны 512
Эластаза 70, 200
Эластин 71
Электронная микроскопия 68
Электростатическое отталкивание 242
Электрофорез белков 55, 304
Элементы гормонального ответа 538
Эмульгаторы 149
Эндогликоназы 289
Эндонуклеазы 83, 370, 483, 497—500,
502—503
Эндорфины 554, 605, 608
Энергетический барьер реакции 256—257
Энергия активации 257
Энергия свободная 236—239
Энзимопатия 593—597
Энкефалины 554, 605, 608
Энтальпия 236—237
Энтропия 236—237
Эпимеразы 292, 412—414
Эпимеры 107
Эпинефрин (см. также адреналин) 366,
557
Эргокальциферол 185
Эргостерол 185, 211
Эриедиктион 217
Эритрозо-4-фосфат 337, 409—410
Эритромицин 532
Эстеразы 288, 586
Эстрадиол 159, 561
Эстрогены 159, 561
Эстрофиллин 561
Этанол 318, 320, 603—605
Этаноламин 450—451
Этаноламинкиназа 450
Этаноламинфосфат 450
Этаноламинфосфотрансфераза 451
Этилен 228—229
Этиленимин 585
Этиримол 583
Эффект Пастера 374—375
Ю
Юглон 215, 579
693