/




























































































































































































































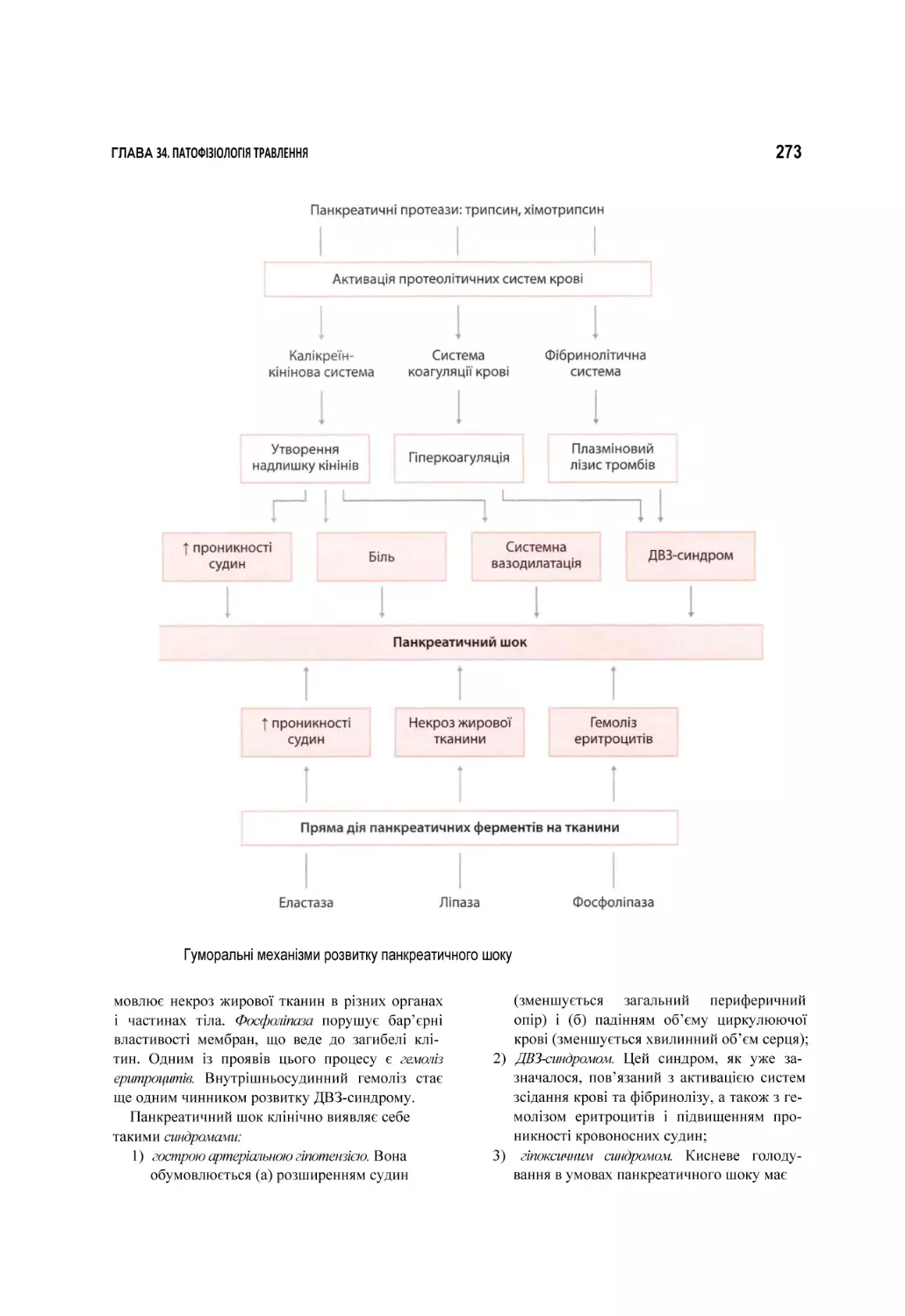


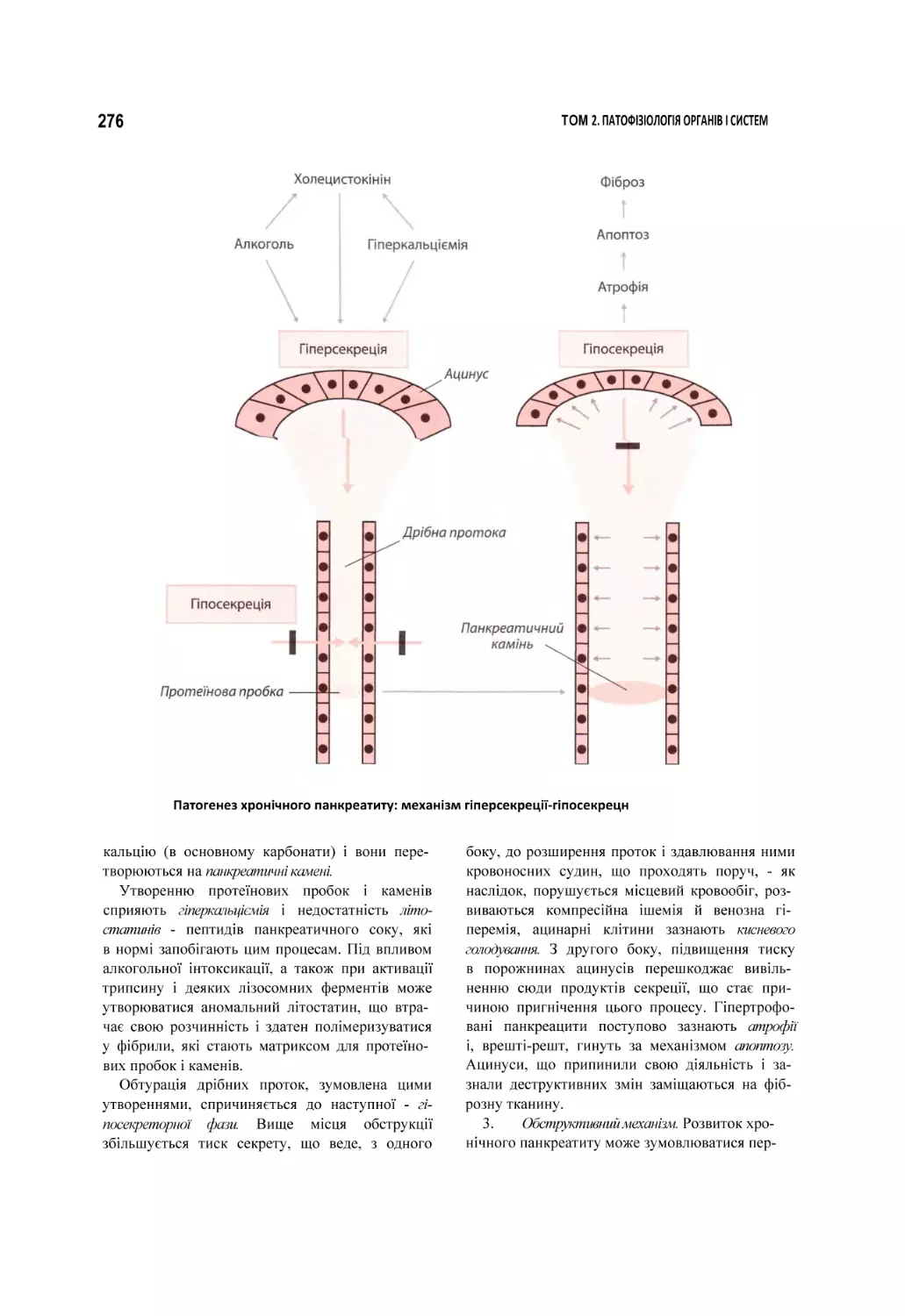



















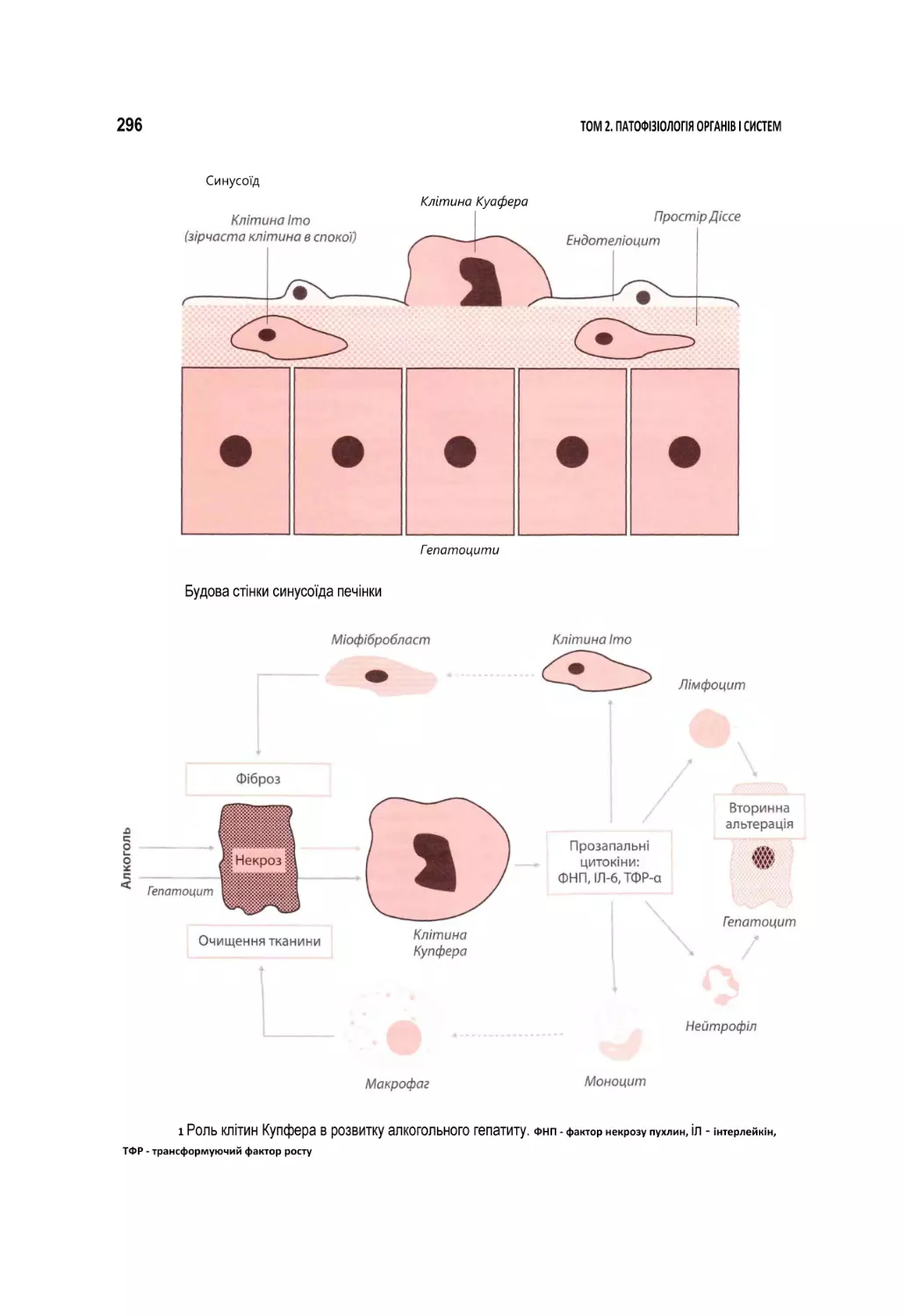







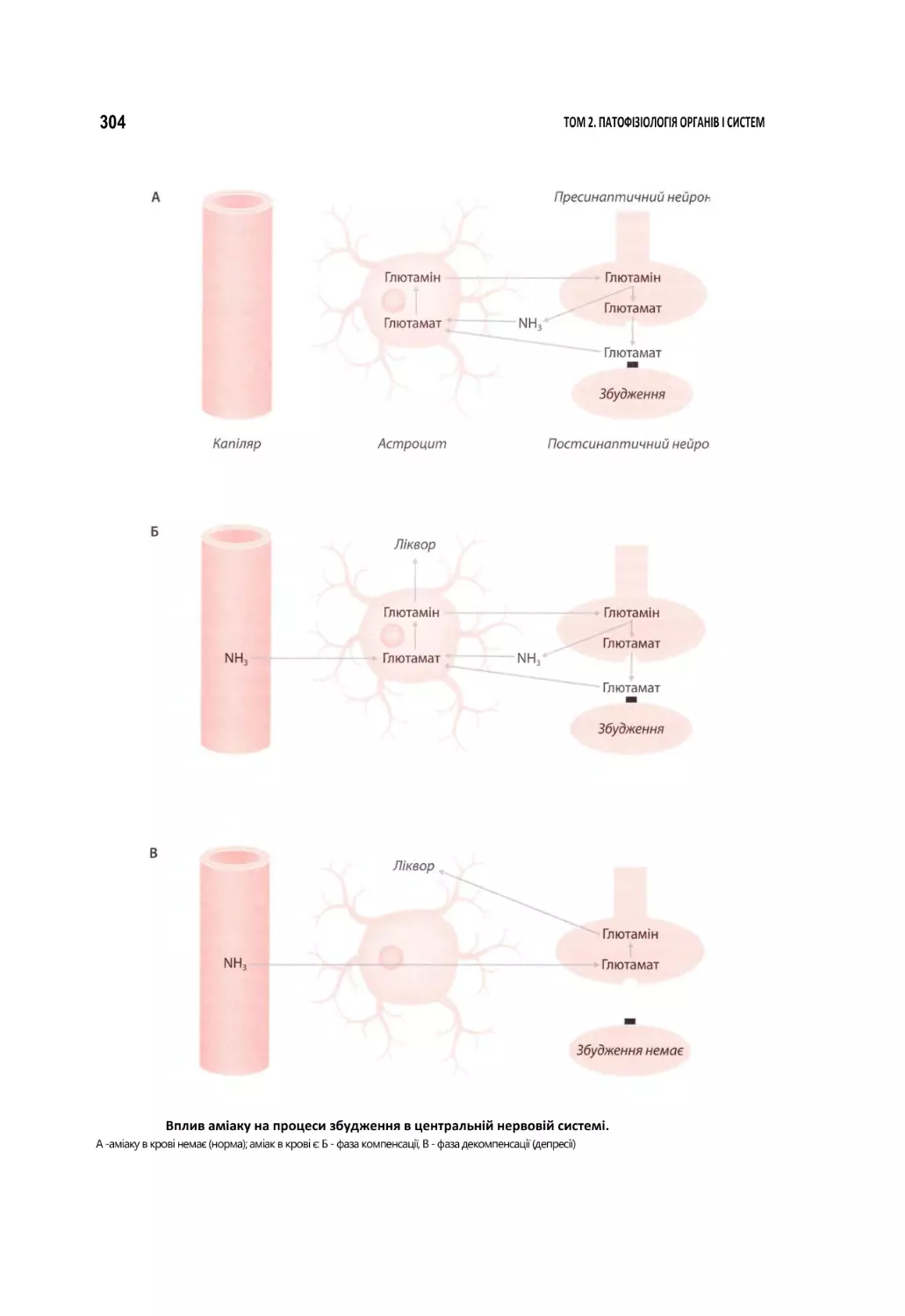
































































































































Автор: Атаман О.В.
Теги: медицина фізіологія фізіологія людини практична медицина патофізіологія
Год: 2016




Текст
МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ'Я УКРАЇНИ
О. В. АТАМАН
ПАТОФІЗІОЛОГІЯ
У ДВОХ ТОМАХ
ТОМ 2
ПАТОФІЗІОЛОГІЯ
ОРГАНІВ І СИСТЕМ
Підручник для студентів вищих медичних
навчальних закладів IVрівня акредитації
Вінниця
Нова Книга
2016
ерепік умовних скорочень та позначень....................................................................................................................................................5
Патофізіологія системи крові................................................................................................................................8
30.1. Зміни загального об'єму крові. Крововтрата................................................................................................................................... 9
30.2. Порушення системи еритроцитів....................................................................................................................................................... 15
30.2.1. Еритроцитоз................................................................................................................................................................................... 19
30.2.2. Анемії................................................................................................................................................................................................. 21
303. Порушення системи лейкоцитів. Лейкоцитози і лейкопенії................................................................................................... 53
30.4. Лейкози.............................................................................................................................................................................................................64
303. Порушення гемостазу. Геморагічний діатез....................................................................................................................................74
51. Патофізіологія серця...................................................................................................................................................................... 92
31.1. Недостатність серця...................................................................................................................................................................................92
31.1.1. Недостатність серця від перевантаження........................................................................................................................ 93
31.1.2. Міокардіальна недостатність серця.................................................................................................................................. 104
31.1.3. Позаміокардіальна недостатність серця.........................................................................................................................112
31.1.4. Основні прояви недостатності серця...............................................................................................................................113
31.2. Аритмії............................................................................................................................................................................................................114
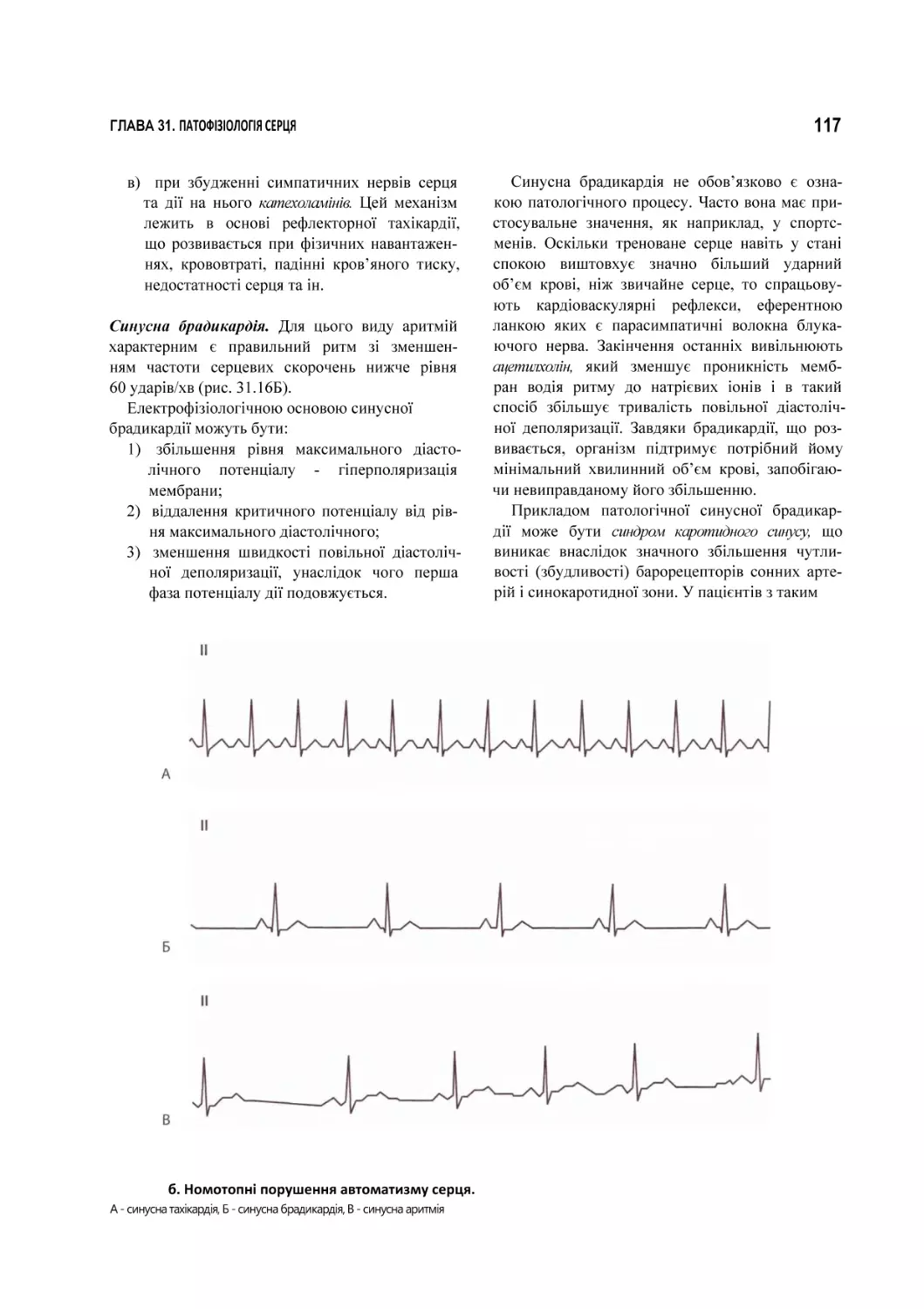
31.2.1. Порушення автоматизму........................................................................................................................................................115
31.2.2. Порушення збудливості..........................................................................................................................................................118
31.2.3. Порушення провідності.......................................................................................................................................................... 122
31.2.4. Порушення збудливості та провідності...........................................................................................................................126
31.3. Недостатність вінцевого кровообігу. Ішемічна хвороба серця........................................................................................ 131
2. Патофізіологія кровоносних судин.......................................................................................................................................155
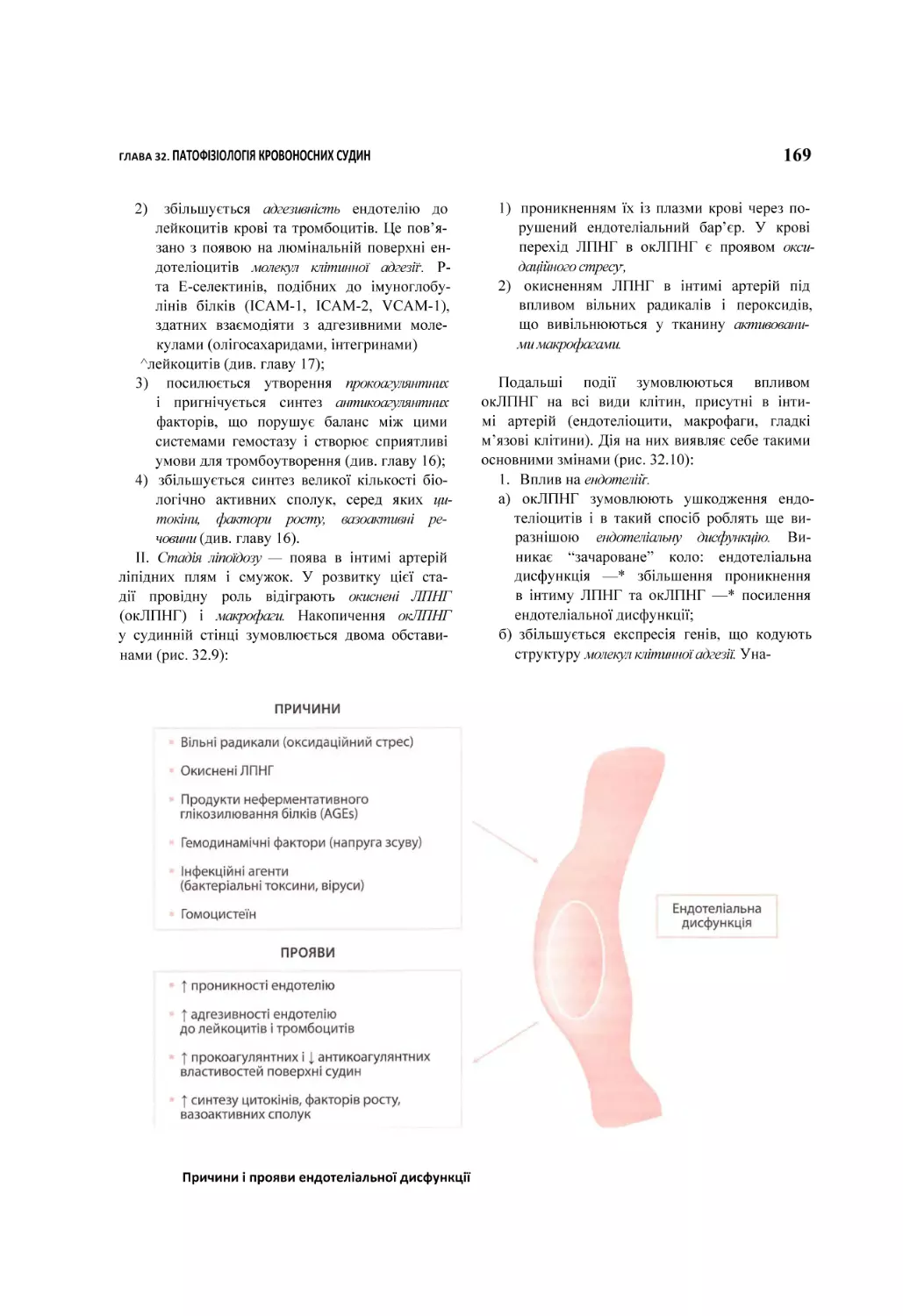
32.1. Артеріосклероз.......................................................................................................................................................................................... 155
32.1.1. Атеросклероз................................................................................................................................................................................156
32.1.2. Артеріосклероз Менкеберга.................................................................................................................................................178
322. Артеріальна гіпертензія...........................................................................................................................................................................186
3223. Патофізіологія венозних судин......................................................................................................................................................... 200
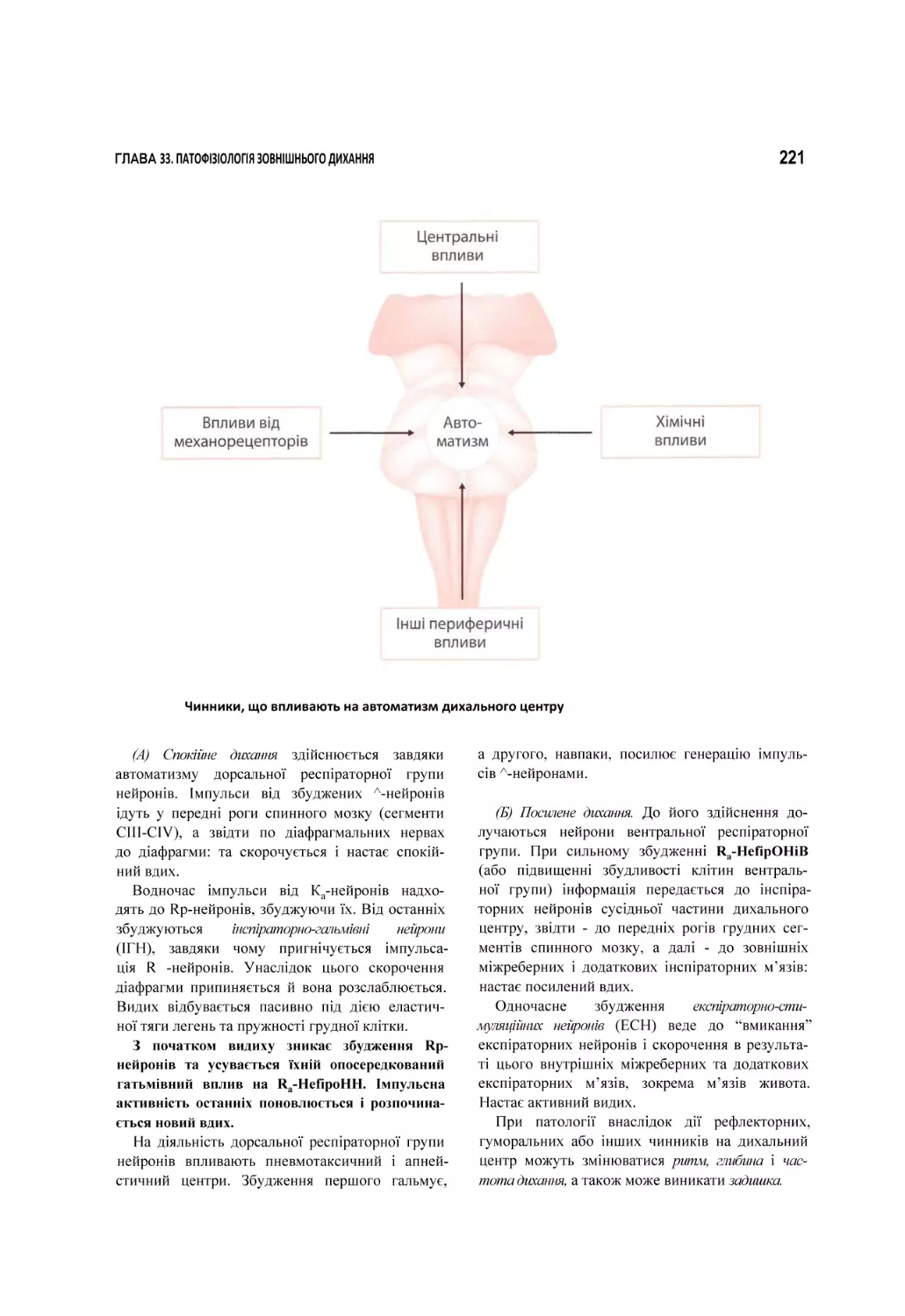
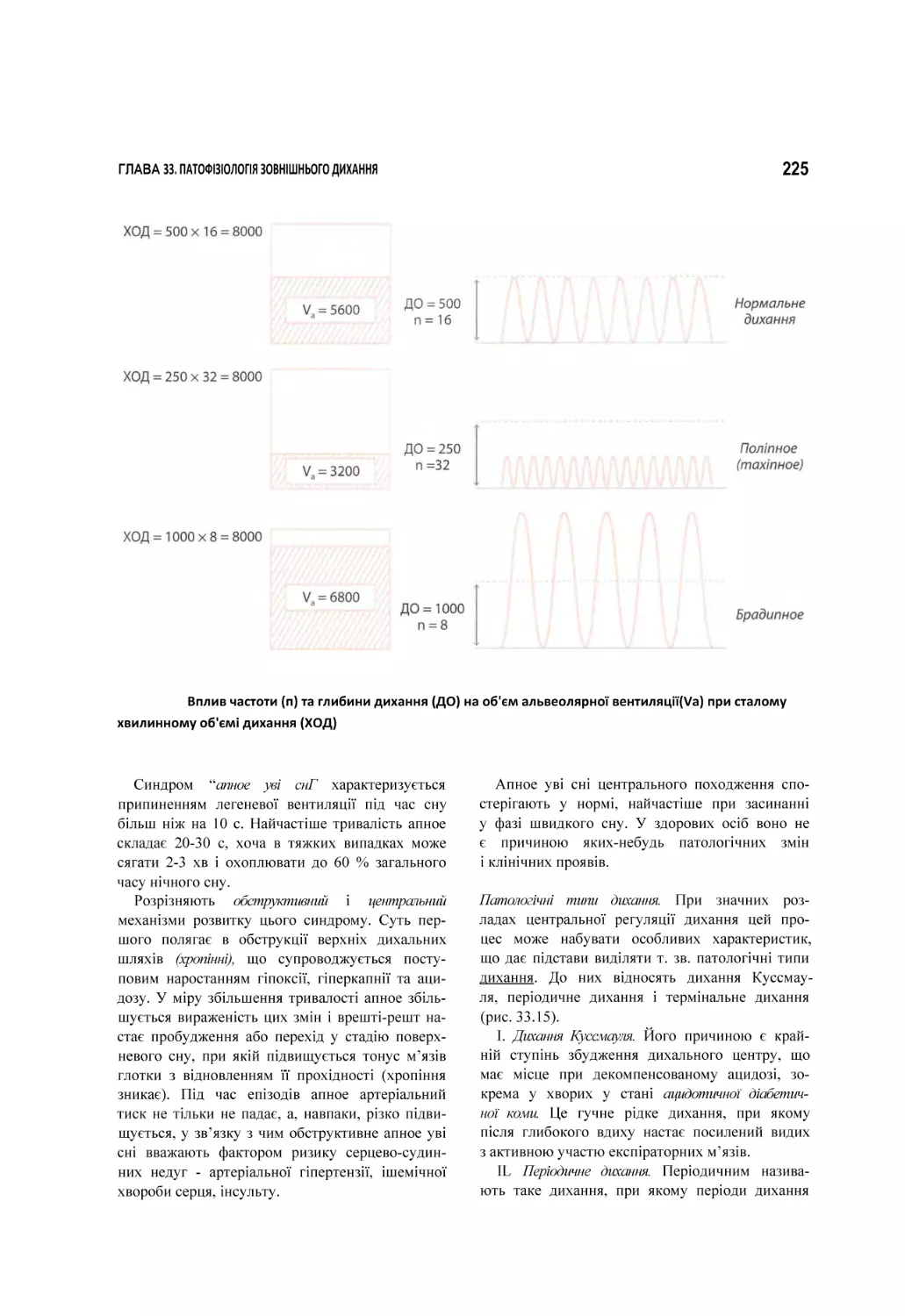
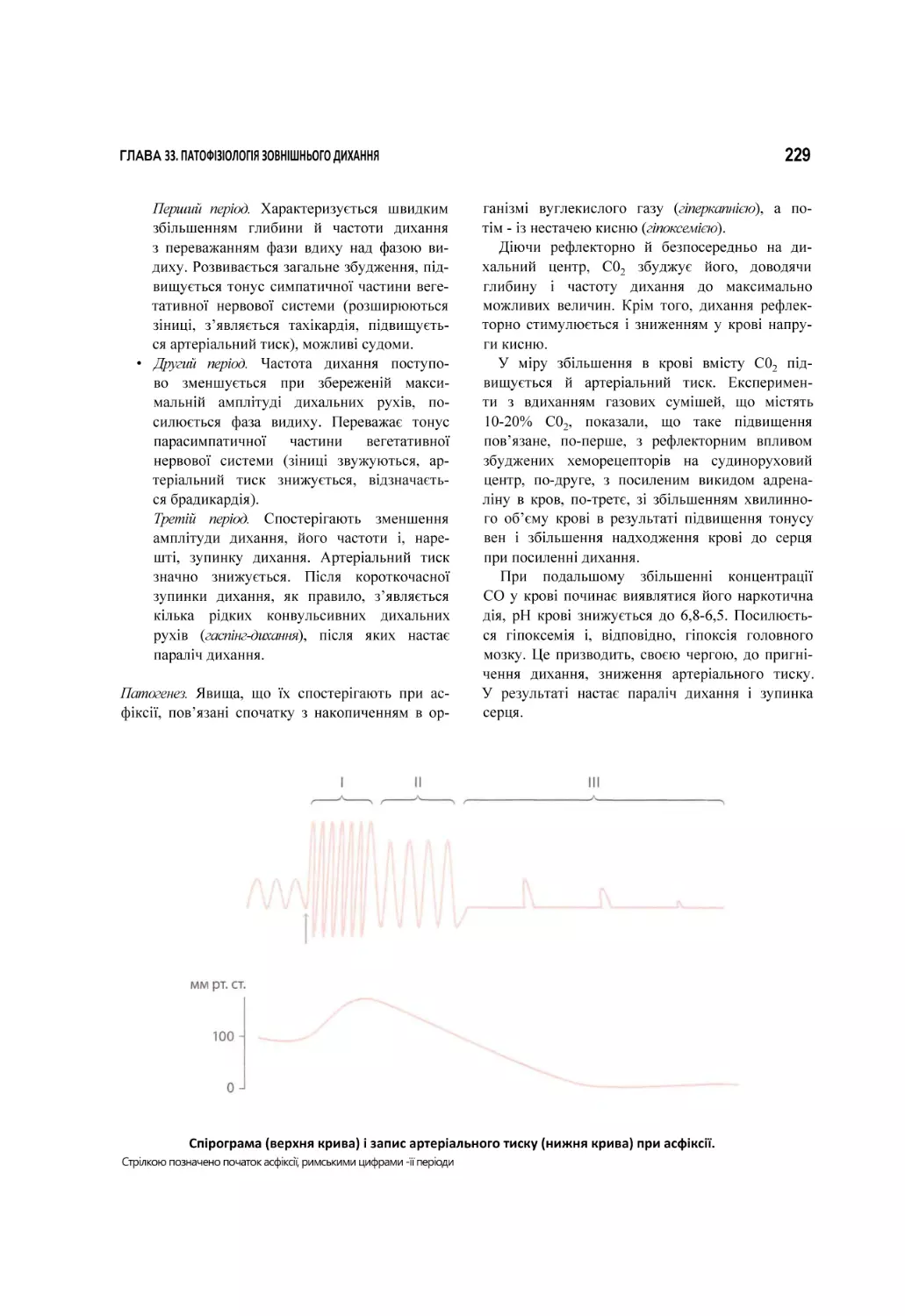
33. Патофізіологія зовнішнього дихання.................................................................................................................................. 209
33.1. Вентиляційна недостатність дихання..............................................................................................................................................211
33.2. Паренхіматозна недостатність дихання......................................................................................................................................... 230
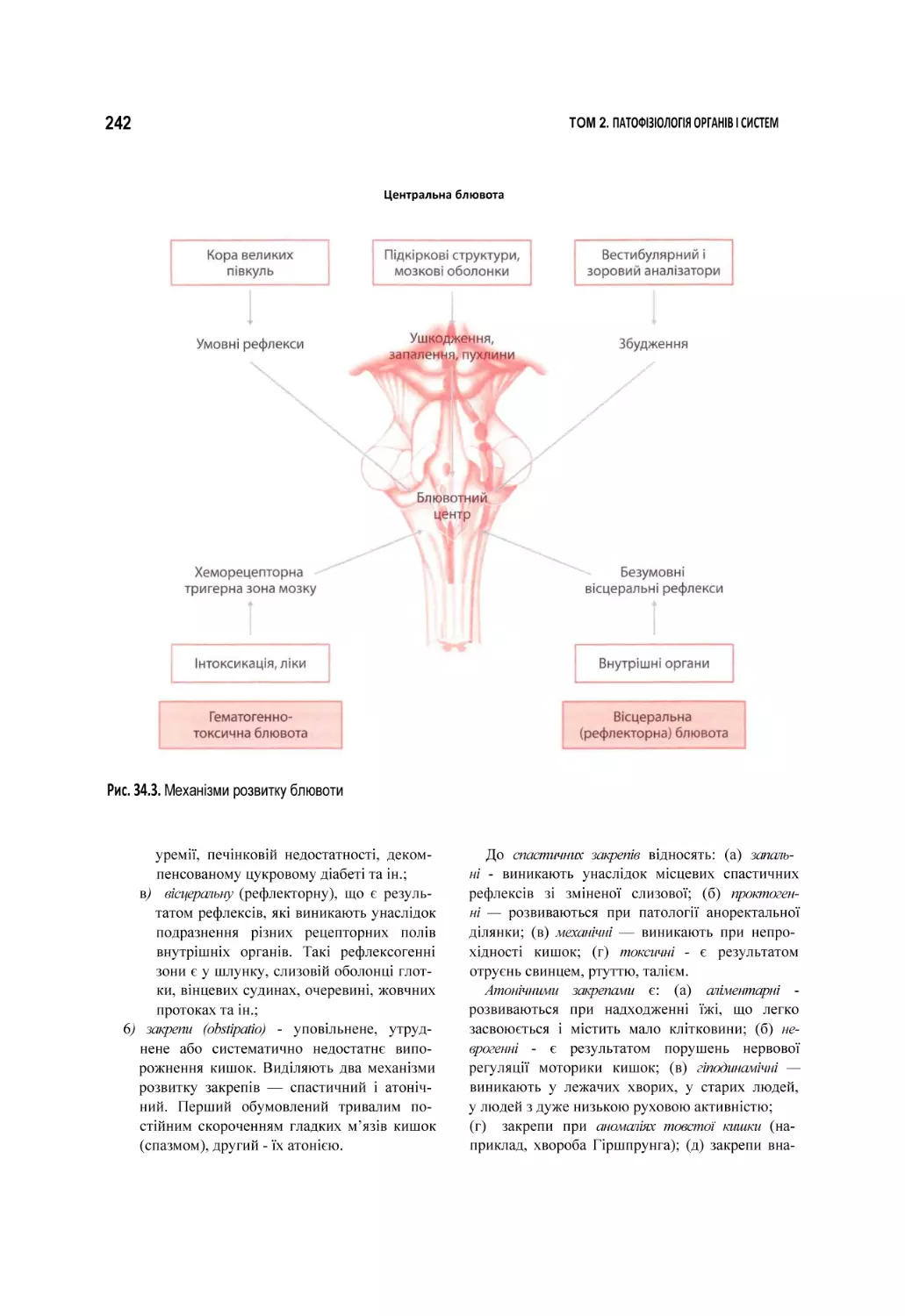
34. Патофізіологія травлення.......................................................................................................................................................... 236
34.1. Порушення травлення в порожнині рота................................................................................................................................. 244
3-2. Порушення травлення у шлунку..........................................................................................................................................................250
З-3. Патофізіологія підшлункової залози..................................................................................................................................................264
ЗА4. Порушення травлення в кишках.........................................................................................................................................................278
4
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Глава 35. Патофізіологія печінки.........................................................................................................................................................284
35.1. Недостатність печінки.............................................................................................................................................................................284
35.2. Алкогольні ураження печінки. Вірусні гепатити.........................................................................................................................289
35.3. Порушення окремих печінкових функцій.................................................................................................................................... 299
35.3.1. Порушення метаболічних функцій.................................................................................................................................... 300
35.3.2. Порушення антитоксичної функції. Печінкова кома................................................................................................ 302
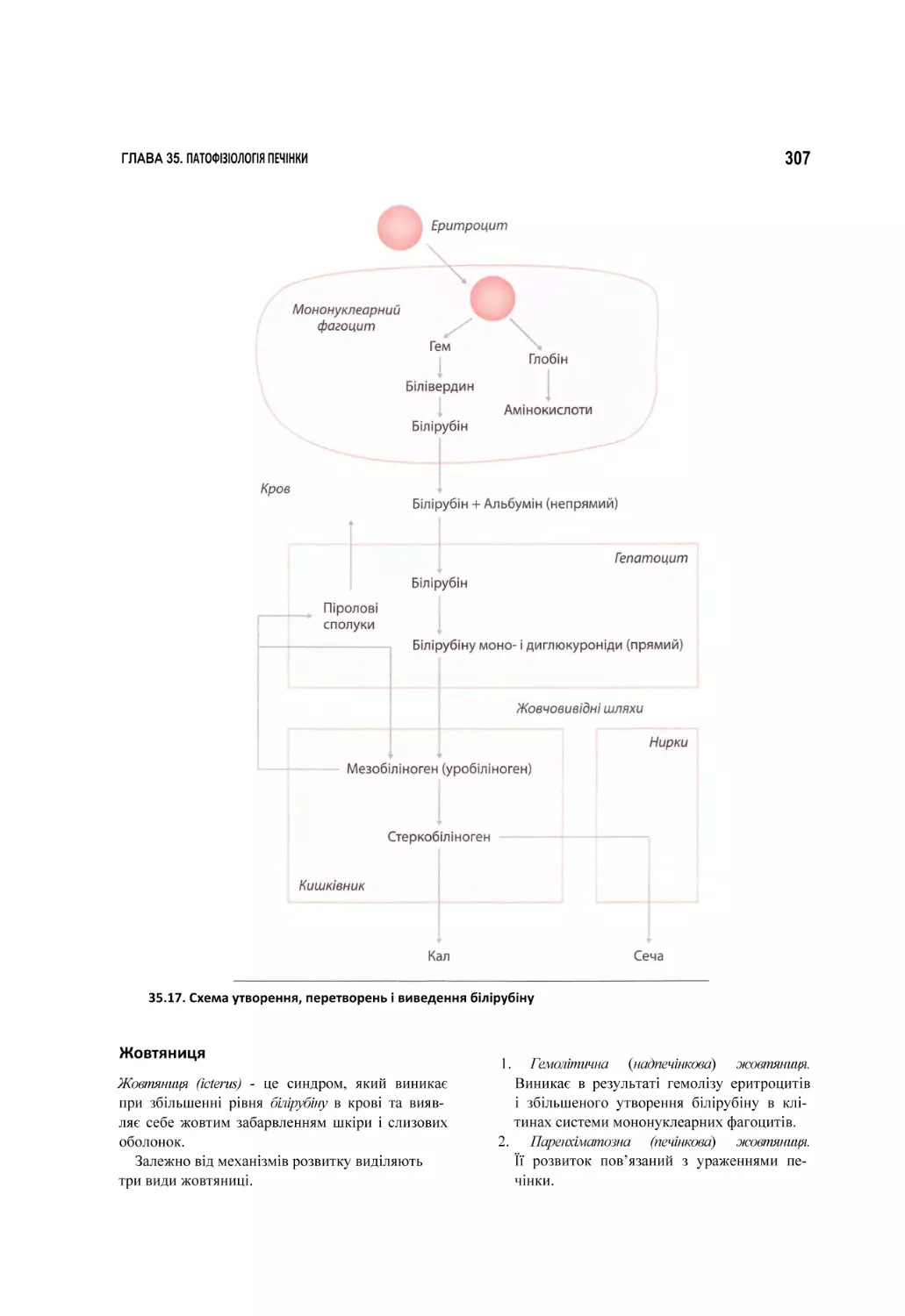
35.3.3. Порушення екскреторної функції. Жовтяниці............................................................................................................. 305
35.3.4. Порушення гемодинамічних функції................................................................................................................................313
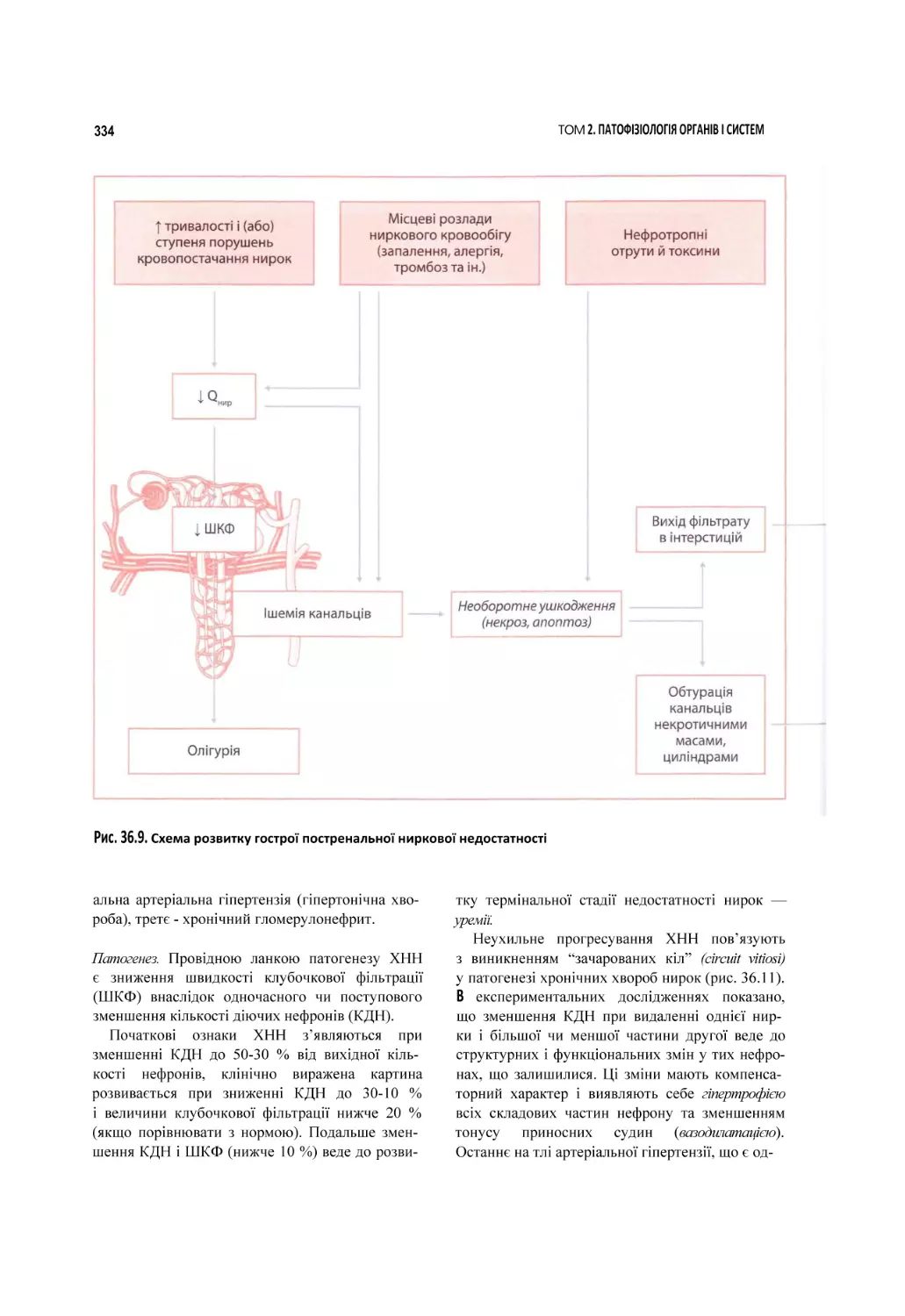
Глава 36. Патофізіологія нирок........................................................................................................................................................... 316
36.1. Механізми порушень функцій нирок..............................................................................................................................................316
36.2. Основні клінічні прояви уражень нирок.......................................................................................................................................320
36.3. Ниркова недостатність. Великі ниркові синдроми і захворювання нирок................................................................... 330
57. Патофізіологія ендокринної системи...................................................................................................................347
37.1. Загальні закономірності порушень ендокринної функції......................................................................................................347
37.2. Патофізіологія гіпоталамо-гіпофізарної системи......................................................................................................................353
37.3. Патофізіологія надниркових залоз................................................................................................................................................... 360
37.3.1. Патофізіологія кори надниркових залоз.........................................................................................................................360
37.3.2. Патофізіологія мозкової речовини надниркових залоз..........................................................................................370
37.4. Патофізіологія щитоподібної залози...............................................................................................................................................372
37.5. Патофізіологія прищитоподібних залоз.........................................................................................................................................377
37.6. Патофізіологія статевих залоз.............................................................................................................................................................379
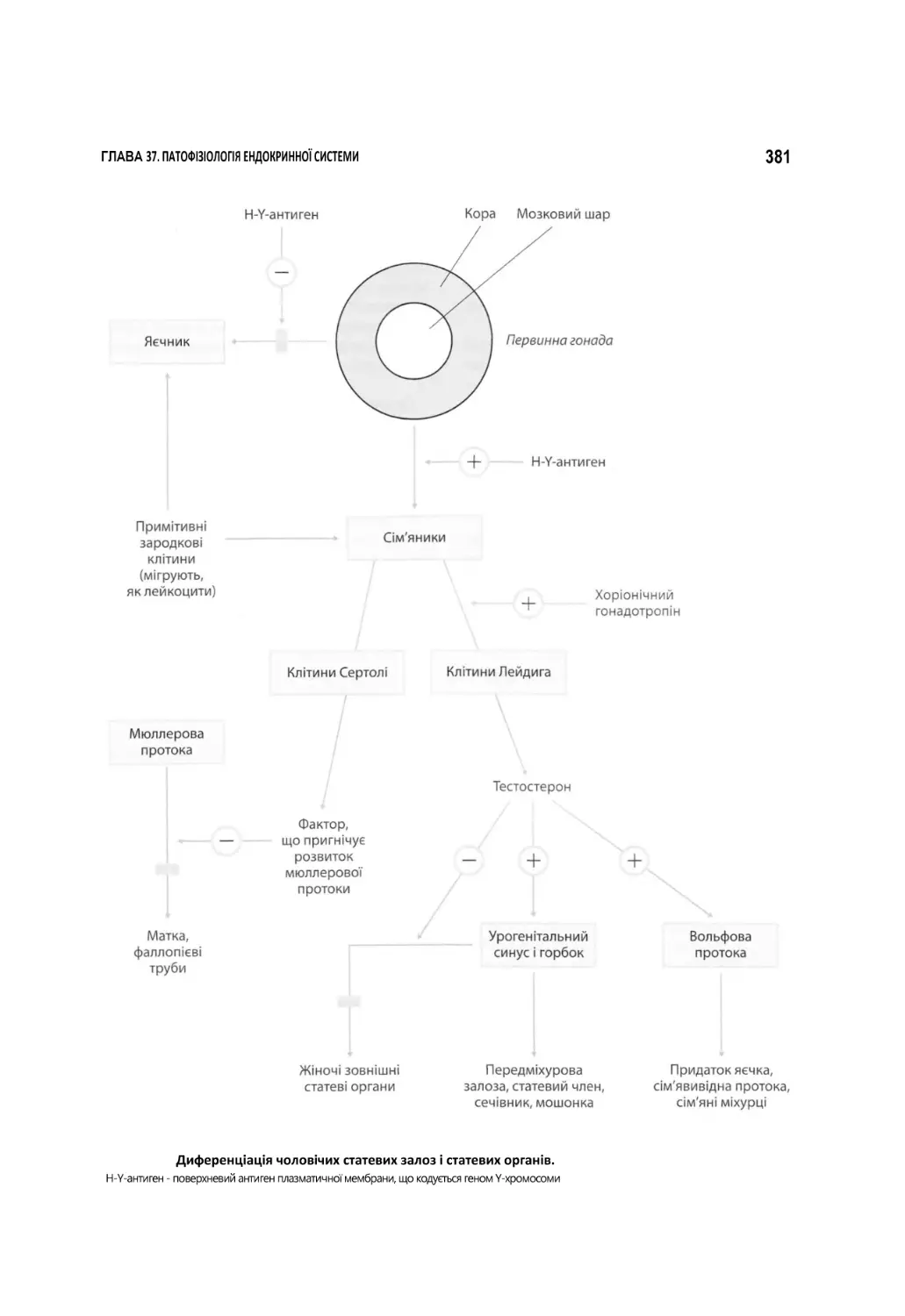
37.6.1. Патофізіологія чоловічих статевих залоз........................................................................................................................379
37.6.2. Патофізіологія жіночих статевих залоз............................................................................................................................ 382
37.7. Патофізіологія інших ендокринних залоз..................................................................................................................................... 384
J8. Патофізіологія нервової системи..............................................................................................................................386
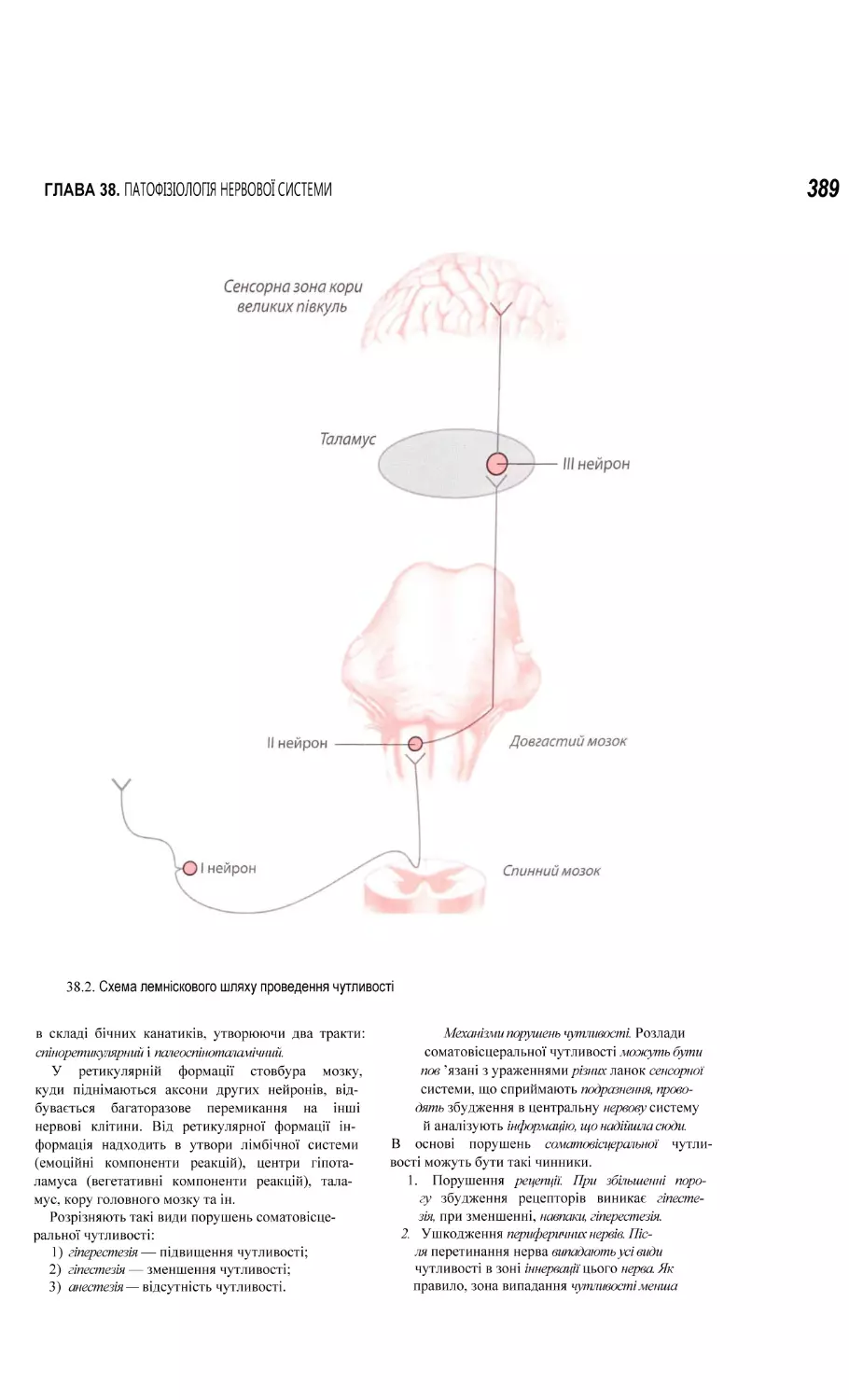
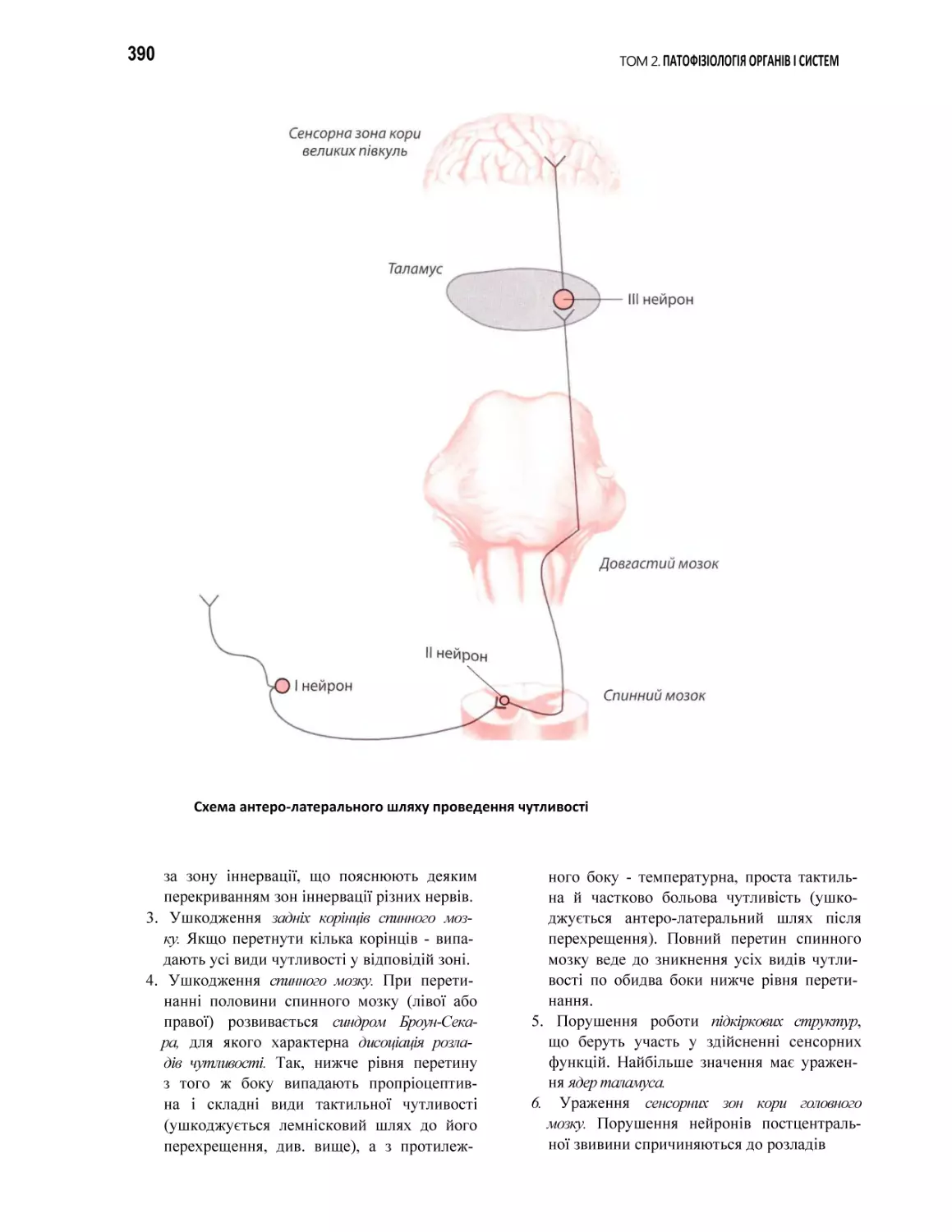
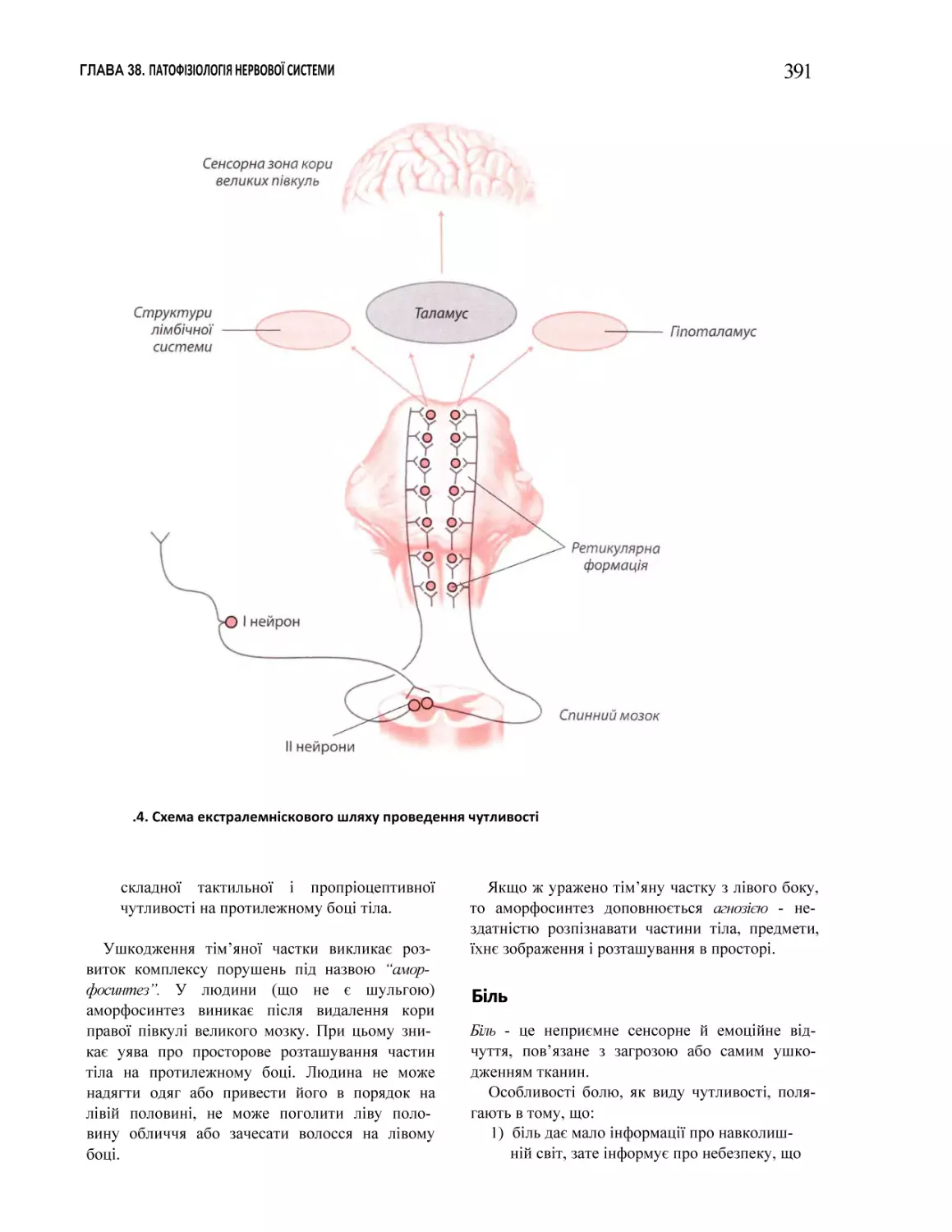
38.1. Порушення соматовісцеральної чутливості. Біль..................................................................................................................... 387
38.2. Порушення ефекторних функцій нервової системи................................................................................................................404
38.2.1. Порушення рухових функцій................................................................................................................................................ 404
38.2.2. Порушення вегетативних функцій..................................................................................................................................... 409
38.2.3. Нервова трофіка та її порушення........................................................................................................................................409
38.3. Порушення інтегративних функцій нервової системи............................................................................................................ 412
Покажчики...............................................................................................................................................................................................................423
Об'єднаний предметний покажчик (для т. І та т.ІІ).........................................................................................................................424
Об'єднаний іменний покажчик (для т. І та т.ІІ)...................................................................................................................................444
ПЕРЕЛІК
умовних
скорочень
та позначень
KSHV/
HHV-8
autoimmune polyendocrinene syndrome
type І (автоімунний поліендокринний
синдром I типу)
Kaposi sarcoma herpesvirus, human
herpesvirus-8 (герпесвірус саркоми
Капоті)
LATS
autoimmune polyendocrine syndrome type
II (автоімунний поліендокринний синдром
II типу)
long acting thyroid stimulator (тривало
діючий стимулятор щитоподібної
залози)
MEN
multiple endocrine neoplasia (множинна
ендокринна неоплазія)
MEN-1
multiple endocrine neoplasia-1
(синдром множинних ендокринних
пухлин І типу)
MEN-2
multiple endocrine neoplasia-2
(синдром множинних ендокринних
пухлин І типу)
MGP
matrix Gla protein (матриксний білок)
NK-клітини
natural killer (природні кілери)
або
груповий фактор крові
APS1
APS2
APUD«стема
CNPs, NPs
APUD-System - Amine Precursor Uptake
and Decarboxylation (дифузна ендокринна
система)
calcifying nanopartfcles, nanoparticles /
nanobacteria (кальцифікуючі наночастки,
нанобактерії)
CPPs
calciprotein padiides (кальцій-протеїнові
частки)
EBV
Epstein - Barr virus (вірус Епштейна - Барр)
Hb
haemoglobin (гемоглобін)
PG
prostaglandins (простагландини)
HbA, HbA2
haemoglobin A, haemoglobin A2
(гемоглобін дорослих)
РРі
PyroPhosphate inorganic (неорганічний
пірофосфат)
haemoglobin F (фетальний гемоглобін
плода і новонароджених)
Rag
Recombination activating genes
(гени, що активують рекомбінацію (ДНК))
haemoglobin S, haemoglobin H
(патологічні форми гемоглобіну)
Rh
резус-фактор крові
HBV
hepatitis В virus (Вірус гепатиту В)
SCF
HCV
hepatitis С virus (Вірус гепатиту С)
serum calcifi cation factor (кальцифікуючий
фактор сироватки)
HJV
hemojuvelin (гемоювелін)
ТВІІ
HMGB1
high-mobility group protein B1
(ядерні негістонові протеїни)
TSH-binding inhibitor immunoglobulins
(імуноглобуліни, що перешкоджають
зв'язуванню ТТГ з рецепторами до
цього гормону)
HTLV-1
humanТ-сеІІ leukemia virus-1
(вірус Т-клітинної лейкемії людини)
TGI
thyroid growth stimulating lmmunonoglobulins (імуноглобуліни, що стимулюють ріст
щитоподібної залози)
ід, іг
immunoglobulin (імуноглобулін)
TLR
Toll-like receptor (Toll-подібні рецептори)
IRIDA
Iron-refractory iron deficiency anemia
(залізорефрактерна анемія
з дефіцитом заліза)
HbF
HbS, HbH
б
TSI
ПЕРЕЛІК УМОВНИХ СКОРОЧЕНЬ ТА ПОЗНАЧЕНЬ
thyroid stimulating immunoglobulin
(імуноглобуліни, що стимулюють
функцію щитоподібної залози)
ксо
кінцевосистолічний об'єм
КСФ
колонієстимулюючий фактор
КТРГ
кортикотропіновий рилізинг-гормон
лг
лютеїнізуючий гормон
лпвг
ліпопротеїни високої густини
лпднг
ліпопротеїни дуже низької густини
лпнг
ліпопротеїни низької густини
мдп
максимальний діастолічний потенціал
М-КСФ
моноцитарний колонієстимулюючий
фактор
синдром Вольфа - Паркінсона - Уайта
мРНК,
ІРНК
матрична рибонуклеїнова кислота,
інформаційна РНК
внутрішній фактор Кастла
НАДФ,
NADP
н и коти нам ідаден і н д и ну кл еотидфосфат
ВФК
Г-6-ФДГ
глюкозо-б-фосфатдегідрогеназа
окЛПНГ
Г-КСФ
гранулоцитарний колонієстимулюючий
фактор
окиснені ЛПНГ, окиснені ліпопротеїни
низької густини
ОФВ
об'єм форсованого видиху
гмк
гладка м'язова клітина
оцк
об'єм циркулюючої крові
ГМ-КСФ
гранулоцит-моноцитарний
колонієстимулюючий фактор
ПАТ
пульсовий артеріальний тиск
гнн
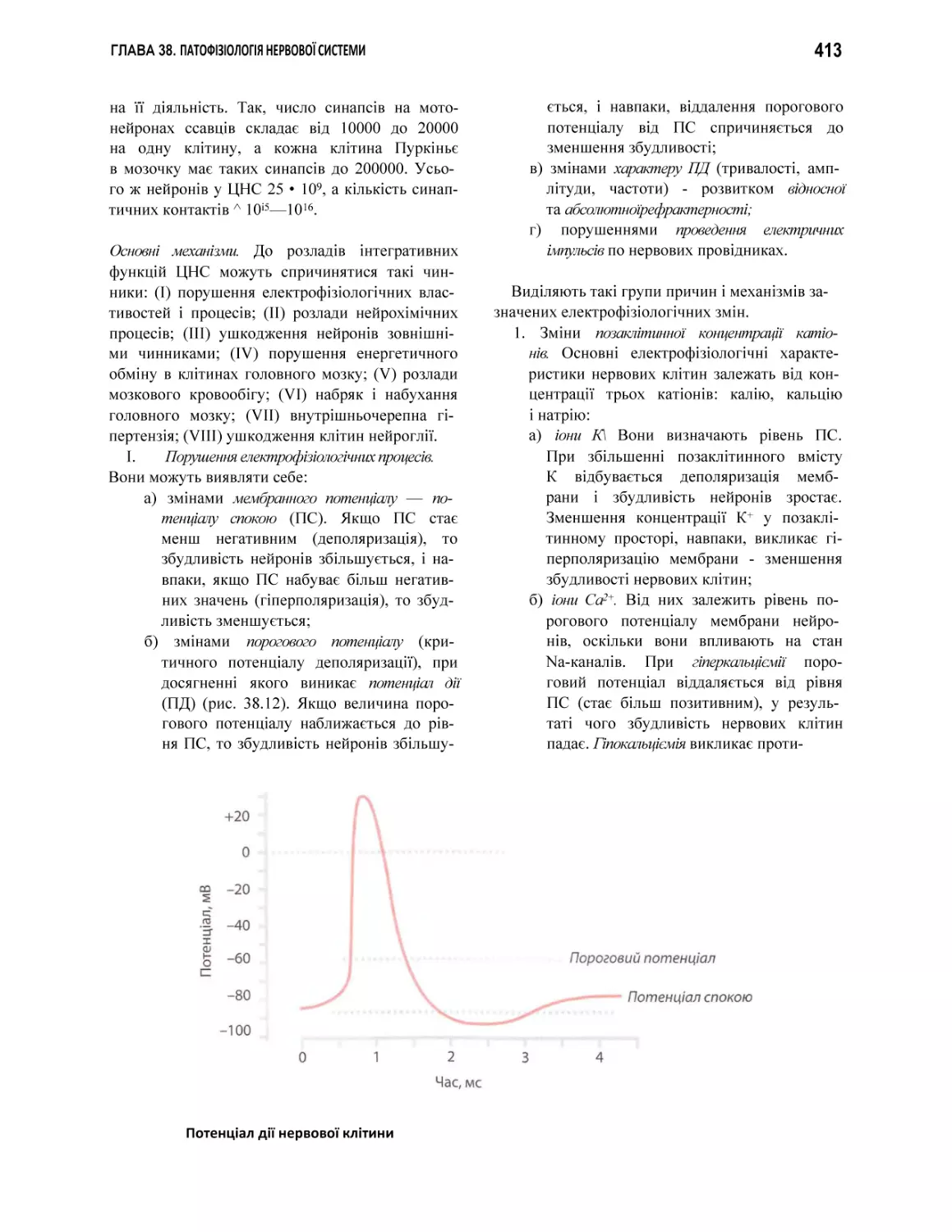
пд
потенціал дії нервової клітини
гостра ниркова недостатність
ПНУГ
передсердний натрійуретичний гормон
Гп
глікопротеїни
пол
пероксидне окиснення ліпідів
ДАТ
діастолічний артеріальний тиск
двз-
пп
пороговий потенціал
синдром
синдром дисемінованого
внутрішньосудинного зсідання крові
ПРЛ
пролактин
ЕЛС
екстралемніскова система
пс
потенціал спокою
ЕСН
експіраторно-стимуляторні нейрони
ПФ
потенціал фосфорилювання
ЕХП-клітини
ентерохромафіноподібні клітини
РНК
рибонуклеїнова кислота
ІГН
інспіраторно-гальмівні нейрони
CAT
систолічний артеріальний тиск
ІСАГ
ізольована систолічна артеріальна
гіпертензія
свд
синдром вегетосудинної дистонії
ІФС
інтенсивність функціонування структур
свпт
суправентрикулярна
пароксизмальна тахікардія
ІХС
ішемічна хвороба серця
кдн
стг
кількість діючих нефронів
соматотропний гормон,
гормон росту
кдо
кінцеводіастолічний об'єм
ТАГ
триацилгліцероли
КоА, СоА
кофермент А, коензим А
ТДМ1
транспортер двовалентних металів 1
КП
колірний показник
ттг
тиреотропний гормон
АВблокада
атріовентрикулярна блокада
АВВ
атріовентрикулярний вузол
АДГ
алкогольдегідрогеназа
АКТГ
адренокортикотропний гормон
АлДГ
альдегіддегідрогеназа
AT
артеріальний тиск
ВЖК
вільні жирові кислоти
ВП
венозне повернення
впвсиндром
7
ПЕРЕЛІК УМОВНИХ СКОРОЧЕНЬ ТА ПОЗНАЧЕНЬ
ТФР
тромбоцитарний фактор росту
ТФР-а
трансформуючий фактор росту-а
УДФГТ
уридиндифосфоглюкуронілтрансфераза
УО
ударний об'єм
ФАТ
хнн
хронічна недостатність нирок, хронічна
ниркова недостатність
хвилинний об'єм дихання
фактор активації тромбоцитів
ход
хос
цтк
ФВ
фактор Віллебранда
ШКФ
швидкість клубочкової фільтрації
ФНП
фактор некрозу пухлин
ШОЕ
швидкість осідання еритроцитів
ФРФ
фактор росту фібробластів
шпт
шлуночкова пароксизмальна тахікардія
ФСГ
фолікулостимуляційний гормон
ЮГА
юкстагломерулярний апарат
хвилинний об'єм серця
циклтрикарбонових кислот
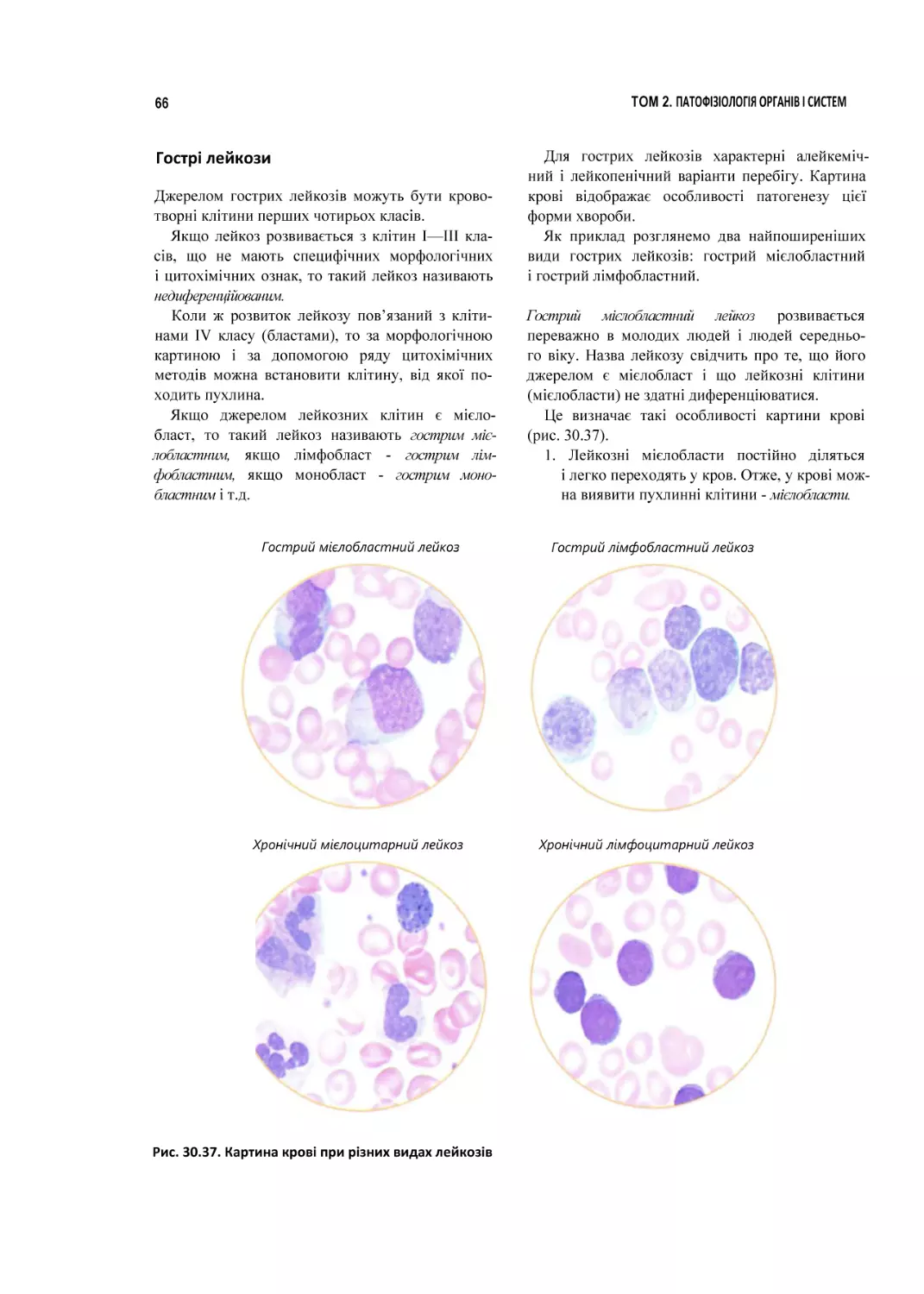
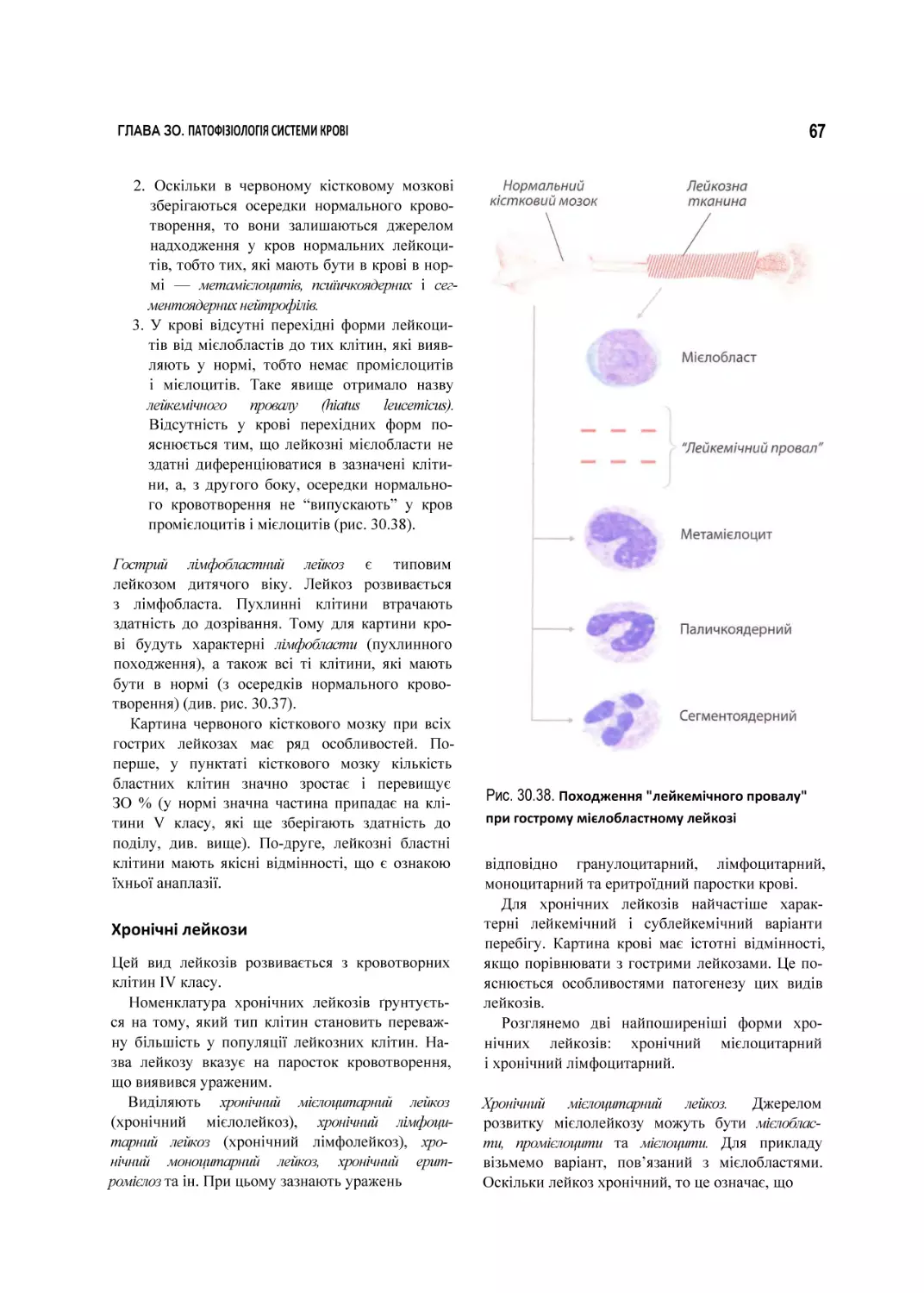
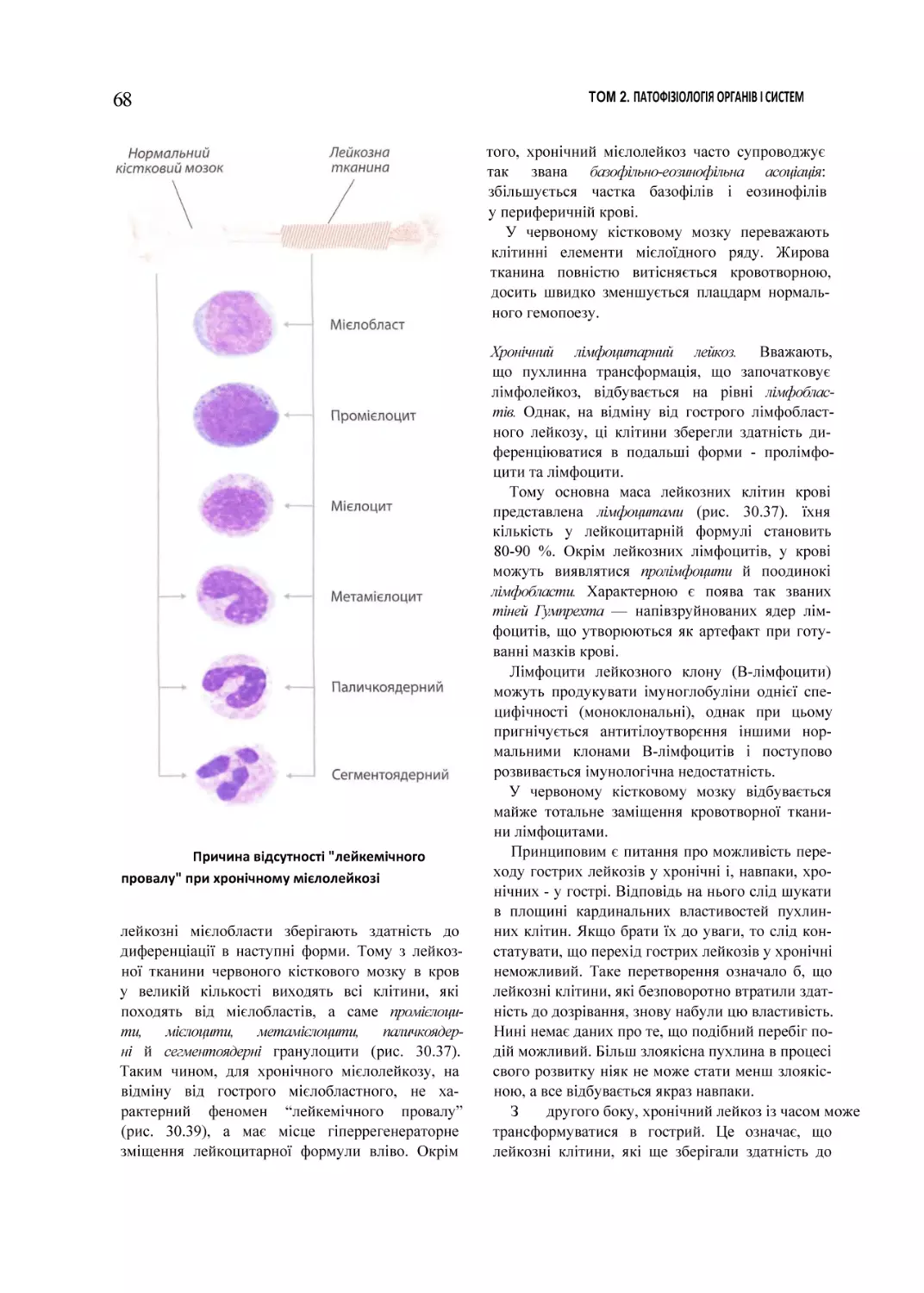
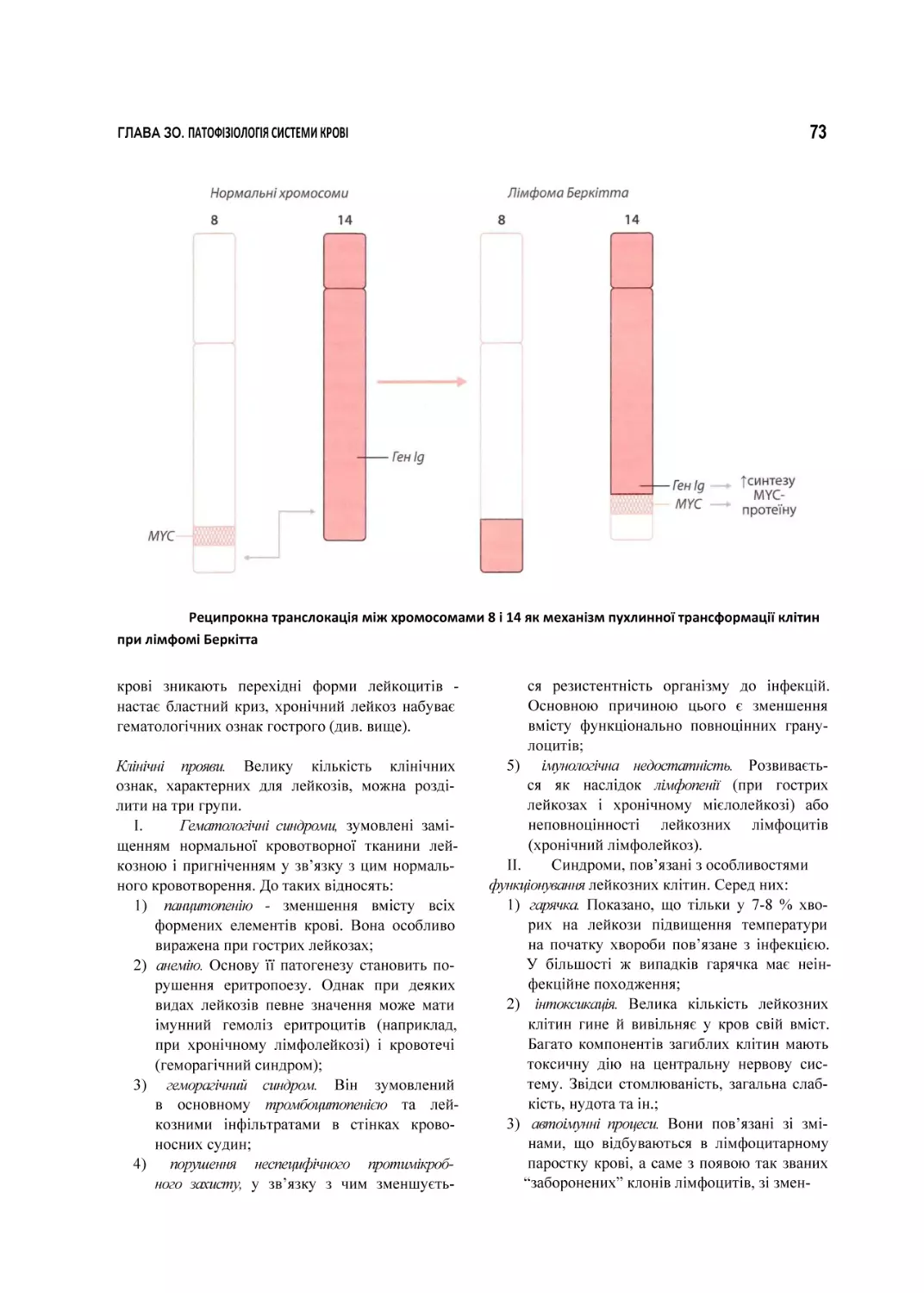
ГЛАВА ЗО
Патофізіологія
системи
коові
Система крові в широкому розумінні охоплює:
1) периферичну кров;
2) органи кровотворення та руйнування крові;
3) механізми регуляції.
клеарних фагоцитів. Такими є селезінка, черво
ний кістковий мозок, печінка (клітини Купфера).
Периферична кров
Провідну роль у регуляції системи крові відігра
ють гуморальні механізми. Вони спрямовані на
регуляцію (а) кровотворення, (б) взаємодії між
клітинами крові й (в) розподілу формених еле
ментів в організмі.
Кров виконує дві основі групи функцій:
(1) транспортну і (2) гомеостатичну.
* Кров є головним засобом транспорту газів
(дихальна функція), поживних речовин, ві
тамінів, мікроелементів (нутритивна або
трофічна функція); проміжних і кінцевих
продуктів обміну речовин {реутилізаційна
та екскреторна функції); гормонів, біоло
гічно активних речовин, ферментів, інгі
біторів {регуляторна функція); імунокомпетентних клітин, фагоцитів, імуноглобулінів {захисна функція); теплоти {термо
регуляторна функція). В умовах патології
кров може виступати засобом транспорту
патогенних чинників: (а) мікроорганізмів
(найпростіших, бактерій, рикетсій) і віру
сів (гематогенні шляхи передавання й по
ширення інфекції) (див. главу 11); (б) екзо
генних та ендогенних токсичних речовин
(участь у розвитку інтоксикацій) (див. гла
ву 10); (в) невластивих крові часток — ем
боліє (див. главу 16); (г) пухлинних клітин
(гематогенне метастазування) (див. главу
19); (д) лікарських препаратів.
У це поняття входить (а) кров, що циркулює
в кровоносних судинах організму, і (б) депоно
вана кров, тобто така, що тимчасово виведена
з циркуляції й перебуває в певній частині судин
печінки, селезінки та деяких інших органів.
Периферична кров, об’єм якої в дорослої лю
дини становить 6-8 % від маси тіла, або 4-6 л,
складається з плазми (55-60 %) і клітин (фор
мених елементів), на які припадає 40-45 % від
загального об’єму.
Основними компонентами плазми є вода
(90-91 %), білки (6,5-8 %) і низькомолекуляр
ні речовини (2 %). Формені елементи скла
даються з еритроцитів (4-5-1012/л у чоловіків
і 3,5-4,5-1012/л у жінок), лейкоцитів (4-9-109л)
і тромбоцитів (150-300- 109/л).
Органи кровотворення
та руйнування крові
Процеси утворення еритроцитів і всіх видів
лейкоцитів, крім лімфоцитів, проходять у черво
ному кістковому мозку (мієлопоез). Генерування
лімфоцитів (лімфопоез) здійснюється в черво
ному кістковому мозку, вилочковій залозі, лім
фатичних вузлах, селезінці, мигдаликах, пеєрових бляшках.
Руйнування формених елементів крові відбу
вається в органах і тканинах, багатих на макро
фаги - клітини, що входять до системи монону-
Механізми регуляції
Транспорт речовин кров’ю здійснюється:
1) у вільному, розчиненому у воді вигляді;
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
9
2) у хімічно зв’язаній формі (з білками
плазми, з іншими органічними сполуками);
З і за участю клітин (адсорбція речовин на
поверхні формених елементів, перебу
вання всередині клітин).
■ Кров разом із лімфою і міжклітинною рілнною утворює внутрішнє середовище
організму, склад і властивості якого мають
бути сталими і не залежати від впливу зов
нішніх умов. Таку властивість позначають
як гомеостаз. До гомеостатичних функцій
крові відносять:
і підтримання сталості хімічного складу
й фізичних властивостей крові. Пара
метрами гомеостазу, що їх контролює
організм, є (а) об’єм циркулюючої кро
ві, (б) осмотичний і онкотичний її тиск,
(в) концентрація іонів натрію, калію
і кальцію; (г) концентрація глюкози;
(д) водневий показник (pH); (е) темпе
ратура;
21 гемостатичну функцію - підтримання
крові в рідкому стані в неушкоджених
судинах і тромбоутворення та коагуля
ція крові при кровотечах;
3) захисну функцію, спрямовану на збере
ження сталості хімічного і антигенного
сктаду організму. Захист від чужорідних
хімічних та біологічних об’єктів досяга
ється за участю лейкоцитів і білків плаз
ми крові.
30.1._ ЗМІНИ ЗАГАЛЬНОГО
ОБ’ЄМУ КРОВІ. КРОВОВТРАТА
Порушення об’єму крові виявляють себе гіповолемією або гіперволемією — зменшенням або
збільшенням об’єму крові, якщо порівнювати
з нормою (нормоволемією) (рис. 30.1).
Гіпо- і гіперволемію поділяють на просту
(зберігається нормальне співвідношення плазми
і клітин крові), поліцитемічну (переважають клі
тини крові) і олігоцитемічну (переважає плазма).
Крім того, до порушень об’єму крові від
носять зміни об’ємного співвідношення між
клітинними елементами та плазмою при нор
мальному загальному об’ємі крові - оліго- і по
ліцитемічну нормоволемію (гемодилюція і гемоконцентрація). Показником об’ємного спів
відношення є гематокрит, що визначає частку
клітинних елементів (переважно еритроцитів)
у загальному об’ємі крові (у нормі 0,36-0,48,
або 36-48 %).
Гіповолемія проста — зменшення об’єму кро
ві без зміни гематокриту. Виникає відразу після
гострої крововтрати і зберігається доти, доки рі
дина не перейде із тканини в кров.
Гіповолемія олігоцитемічна — зменшення
об’єму крові з переважним зменшенням у ній
клітин — еритроцитів. Спостерігається при
гострій крововтраті в тих випадках, коли над
ходження крові й тканинної рідини в кровонос
не русло не компенсує об’єм і особливо склад
крові.
V------------------------- --- ------------------------- '
V------------------- V-------------------------- '
Гіповолемія
Гіперволемія
Нормоволемія
1. Зміни загального об'єму крові.
і -проста, б - олігоцитемічна, в - поліцитемічна
10
Гіповолемія поліцитемічна - зменшення об’є
му крові внаслідок зменшення об’єму плазми
при відносному збільшенні вмісту еритроцитів.
Розвивається при зневодненні організму (про
нос, блювота, посилене потовиділення, гіпервентиляція), шоку (вихід рідини в тканини вна
слідок підвищення проникності стінок судин).
Гіперволемія проста - збільшення об’єму
крові при збереженні нормального співвідно
шення між еритроцитами та плазмою. Виникає
відразу ж після переливання великої кількості
крові. Однак незабаром рідина виходить з кро
воносного русла, а еритроцити залишаються,
що веде до згущення крові. Проста гіперволемія
при посиленій фізичній роботі обумовлена над
ходженням у загальний кровообіг крові з депо.
Гіперволемія олігоцитемічна - збільшення
об’єму крові за рахунок плазми. Розвивається
при затримці води в організмі у зв’язку із за
хворюваннями нирок, при введенні кровоза
мінників. Її можна моделювати в експерименті
шляхом внутрішньовенного введення тваринам
ізотонічного розчину натрію хлориду.
Гіперволемія поліцитемічна збільшення
об’єму крові через наростання кількості еритро
цитів. Спостерігається при зниженні атмосфер
ного тиску, а також при різних захворюваннях,
пов’язаних з кисневим голодуванням (вади сер
ця, емфізема). Її розглядають як компенсатор
не явище. Однак при еритремії поліцитемічна
гіперволемія є наслідком пухлинного розрос
тання клітин еритроцитарного ряду кісткового
мозку.
Олігоцитемічна нормоволемія виникає при
анемії внаслідок крововтрати (об’єм крові нор
малізувався за рахунок тканинної рідини, а кіль
кість еритроцитів ще не відновилася), при гемо
лізі еритроцитів, порушеннях гемопоезу.
Поліцитемічна нормоволемія спостерігається
при переливанні невеликих кількостей еритроцитарної маси.
Значення змін загального об’єму крові по
лягає в тому, що гіповолемія веде до порушень
дихальної й транспортної функцій еритроцитів;
трофічної, екскреторної, захисної, регуляторної
функцій крові. Це так чи інакше відображається
на сталості внутрішнього середовища організму
(гомеостазі).
Гіперволемія обумовлює збільшення наванта
ження на серце. У випадку одночасного збіль
шення гематокриту (поліцитемічна гіперволе
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
мія) збільшується в’язкість крові, що може бути
причиною порушень мікроциркуляції й прово
кувати утворення тромбів.
Крововтрата - це патологічний процес, що ви
никає внаслідок кровотечі і характеризується
складним комплексом патологічних порушень
та компенсаторних реакцій, спрямованих проти
зменшення об’єму циркулюючої крові й гіпоксії,
обумовленої зниженням дихальної функції крові.
До етіологічних факторів, що викликають
кровотечу, відносять:
1) порушення цілісності судин при пораненні
або ураженні патологічним процесом (ате
росклероз, пухлина, туберкульоз);
2) підвищення проникності судинної стінки
(гостра променева хвороба);
3) зниження зсідання крові (геморагічний
діатез).
Перебіг і результат крововтрати залежать від
особливостей самої кровотечі (швидкості, ве
личини, виду ушкодженої судини, механізму
ушкодження); швидкості включення й вираженості компенсаторних реакцій організму; ста
ті, віку, станів, що передують і супроводжують
крововтрату (охолодження, травма, захворю
вання серця, глибокий наркоз). Серйозну небез
пеку для життя людини становить втрата 50 %
об’єму циркулюючої крові, смертельною є втра
та крові понад 60 %.
У патогенезі гострої крововтрати виділяють
три стадії.
1. Початкова стадія. Характеризується змен
шенням об’єму циркулюючої крові - про
стою гіповолемією, зниженням артеріаль
ного тиску, гіпоксією переважно циркуляторного типу.
2. Компенсаторна стадія. Обумовлена здійс
ненням комплексу захисно-компенсатор
них реакцій, спрямованих на ліквідацію
наслідків втрати крові.
3. Термінальна стадія. Характеризується на
ростанням патологічних змін в організмі
аж до настання смерті. Розвивається при
недостатності компенсаторних реакцій,
а також при інтенсивній і швидкій крово
втраті, на тлі дії несприятливих факторів
(охолодження, велика травма, серцево-су
динні захворювання) і при відсутності лі
кувальних заходів.
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
У патогенезі гострої крововтрати центральне
місце посідають порушення загальної гемодияаміки — зменшення об’єму циркулюючої крові
гіповолемія) і зумовлене цим падіння артері
ального тиску (гіпотензія) (рис. 30.2).
Зазначені порушення, з одного боку, є при
чиною розвитку власне патологічних змін в ор
ганізмі (анемії, гіпоксії, ацидозу, інтоксикації),
з другого - вмикають складний комплекс реф
лекторних і гуморальних захисно-компенсаторніїх реакцій, спрямованих на відновлення й збе
реження гомеостазу.
Захисно-компенсаторні реакції. Залежно від
строків виникнення їх поділяють на термінові
і відстрочені.
11
За призначенням розрізняють такі групи ме
ханізмів компенсації.
• Спрямовані на зменшення об’єму судинно
го русла. Мета цієї групи реакцій - привес
ти у відповідність об’єм судинного русла
зменшеному об’єму циркулюючої крові
(;централізація кровообігу). Це дає можли
вість на деякий час підтримати необхідний
тиск крові й забезпечити кровопостачання
життєво важливих органів. Зазначені реак
ції — термінові, у їхньому розвитку провід
не значення мають рефлекси.
До зменшення об’єму судинного русла при
крововтраті ведуть такі зміни (рис. 30.3):
Рис. 30.2. Етіологічні чинники та головна ланка патогенезу гострої крововтрати
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
12
Рис. 30.3 Активація симпатоадреналової системи як провідний механізм компенсаторних реакцій, спрямо
ваних на зменшення об'єму судинного русла при гострій крововтраті.
ОЦК - об'єм циркулюючої крові, AT - артеріальний тиск
1) спазм артеріол шкіри, м’язів, органів трав
ної системи;
2) відкриття артеріовенозних анастомозів за
значених органів і тканин у результаті спаз
му прекапілярних сфінктерів;
3) веноконстрикція (скорочення гладких
м’язів вен), що збільшує надходження кро
ві до серця і зменшує ємність венозного
відділу кровообігу.
В основі цих змін лежать такі механізми:
а) зниження артеріального тиску —»збудже
ння барорецепторів —* активація симпа
тоадреналової системи —> дія катехола-
мінів на а-адренорецептори гладких
м’язів артерій, артеріол, прекапілярних
сфінктерів і вен;
б) зменшення об’єму циркулюючої кро
ві й артеріального тиску —» збудження
волюмо- і барорецепторів —» активація
нейросекреторних клітин гіпоталамуса,
що продукують вазопресин —* дія цього
гормону на Vj-рецептори гладких м’язів
судин з наступною вазоконстрикцією;
в) зменшення об’єму циркулюючої крові
й активація симпатоадреналової системи
—> вивільнення клітинами юкстагломерулярного апарату нирок реніну —> ак-
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
тивація ренін-ангіотензинової системи
з утворенням ангіотензину II —> спазм
гладких м’язів кровоносних судин.
• Спрямовані на збільшення об’єму циркулю
ючої крові.
Реакції цієї групи здійснюються в період
часу від кількох годин до кількох діб після
кровотечі.
Збільшення об’єму циркулюючої крові в за
значений проміжок часу досягається за допомо
гою таких механізмів.
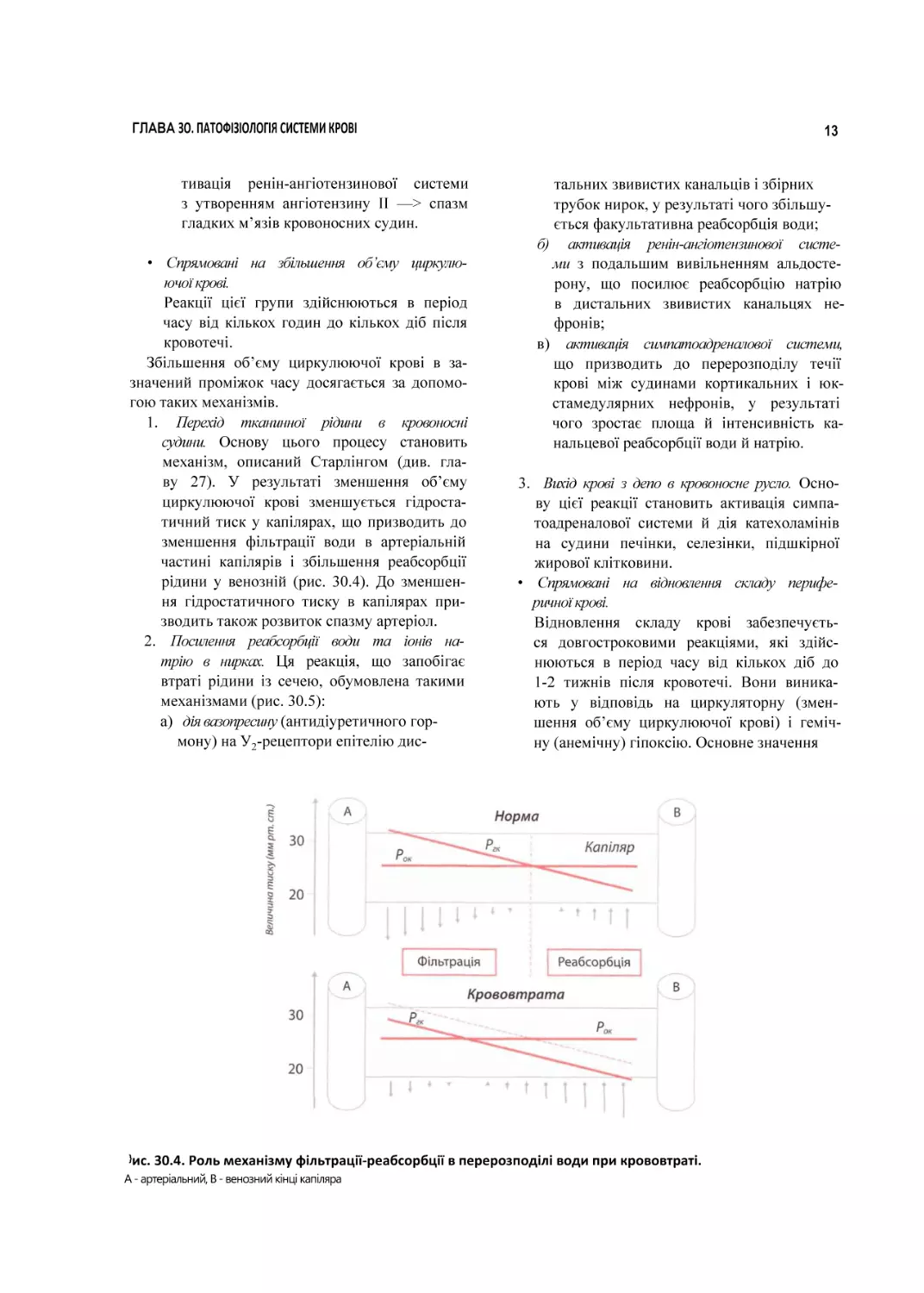
1. Перехід тканинної рідини в кровоносні
судини. Основу цього процесу становить
механізм, описаний Старлінгом (див. гла
ву 27). У результаті зменшення об’єму
циркулюючої крові зменшується гідроста
тичний тиск у капілярах, що призводить до
зменшення фільтрації води в артеріальній
частині капілярів і збільшення реабсорбції
рідини у венозній (рис. 30.4). До зменшен
ня гідростатичного тиску в капілярах при
зводить також розвиток спазму артеріол.
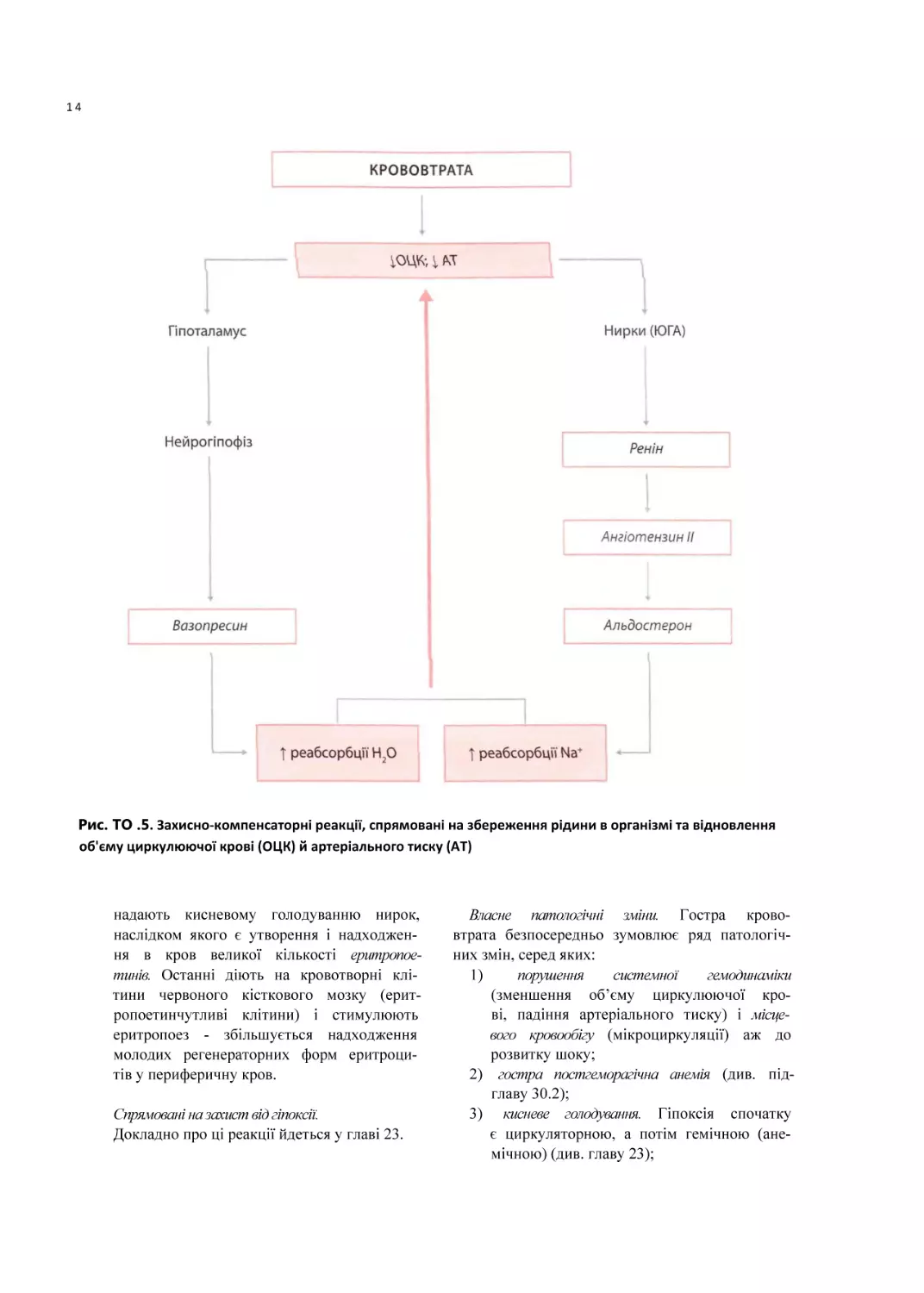
2. Посилення реабсорбції води та іонів на
трію в нирках. Ця реакція, що запобігає
втраті рідини із сечею, обумовлена такими
механізмами (рис. 30.5):
а) дія вазопресину (антидіуретичного гор
мону) на У2-рецептори епітелію дис
}ис.
13
тальних звивистих канальців і збірних
трубок нирок, у результаті чого збільшу
ється факультативна реабсорбція води;
б) активація ренін-ангіотензинової систе
ми з подальшим вивільненням альдосте
рону, що посилює реабсорбцію натрію
в дистальних звивистих канальцях нефронів;
в) активація симпатоадреналової системи,
що призводить до перерозподілу течії
крові між судинами кортикальних і юкстамедулярних нефронів, у результаті
чого зростає площа й інтенсивність канальцевої реабсорбції води й натрію.
3. Вихід крові з депо в кровоносне русло. Осно
ву цієї реакції становить активація симпа
тоадреналової системи й дія катехоламінів
на судини печінки, селезінки, підшкірної
жирової клітковини.
• Спрямовані на відновлення складу перифе
ричної крові.
Відновлення складу крові забезпечуєть
ся довгостроковими реакціями, які здійс
нюються в період часу від кількох діб до
1-2 тижнів після кровотечі. Вони виника
ють у відповідь на циркуляторну (змен
шення об’єму циркулюючої крові) і гемічну (анемічну) гіпоксію. Основне значення
30.4. Роль механізму фільтрації-реабсорбції в перерозподілі води при крововтраті.
А - артеріальний, В - венозний кінці капіляра
14
Рис. ТО .5. Захисно-компенсаторні реакції, спрямовані на збереження рідини в організмі та відновлення
об'єму циркулюючої крові (ОЦК) й артеріального тиску (AT)
надають кисневому голодуванню нирок,
наслідком якого є утворення і надходжен
ня в кров великої кількості еритропоетинів. Останні діють на кровотворні клі
тини червоного кісткового мозку (еритропоетинчутливі клітини) і стимулюють
еритропоез - збільшується надходження
молодих регенераторних форм еритроци
тів у периферичну кров.
Спрямовані на захист від гіпоксії.
Докладно про ці реакції йдеться у главі 23.
Власне патологічні зміни. Гостра крово
втрата безпосередньо зумовлює ряд патологіч
них змін, серед яких:
1)
порушення
системної
гемодинаміки
(зменшення об’єму циркулюючої кро
ві, падіння артеріального тиску) і місце
вого кровообігу (мікроциркуляції) аж до
розвитку шоку;
2) гостра постгеморагічна анемія (див. підглаву 30.2);
3) кисневе голодування. Гіпоксія спочатку
є циркуляторною, а потім гемічною (ане
мічною) (див. главу 23);
15
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
4) негазовий ацидоз. Він обумовлений гіпо
ксією й надходженням у кров молочної
кислоти (див. главу 29);
5) порушення екскреторної функції нирок.
При падінні артеріального тиску зменшу
ється інтенсивність клубочкової фільтра
ції й розвиваються явища гострої ниркової
недостатності: оліго- й анурія, інтоксика
ція (азотемія) (див. главу 36).
Крайнім проявом власне патологічних змін
при крововтраті є геморагічний шок. Провід
ний механізм його розвитку полягає у змен
шенні об’єму циркулюючої крові, що веде до
падіння артеріального тиску, порушення мікроциркуляції, розладів кровопостачання жит
тєво важливих органів (головного мозку, серця,
нирок). Наслідком цього є розвиток гіпоксії,
ацидозу й інтоксикації, що погіршує перебіг
шоку, створює “зачаровані кола” у його пато
генезі та в кінцевому підсумку веде до смерті
(рис. 30.6).
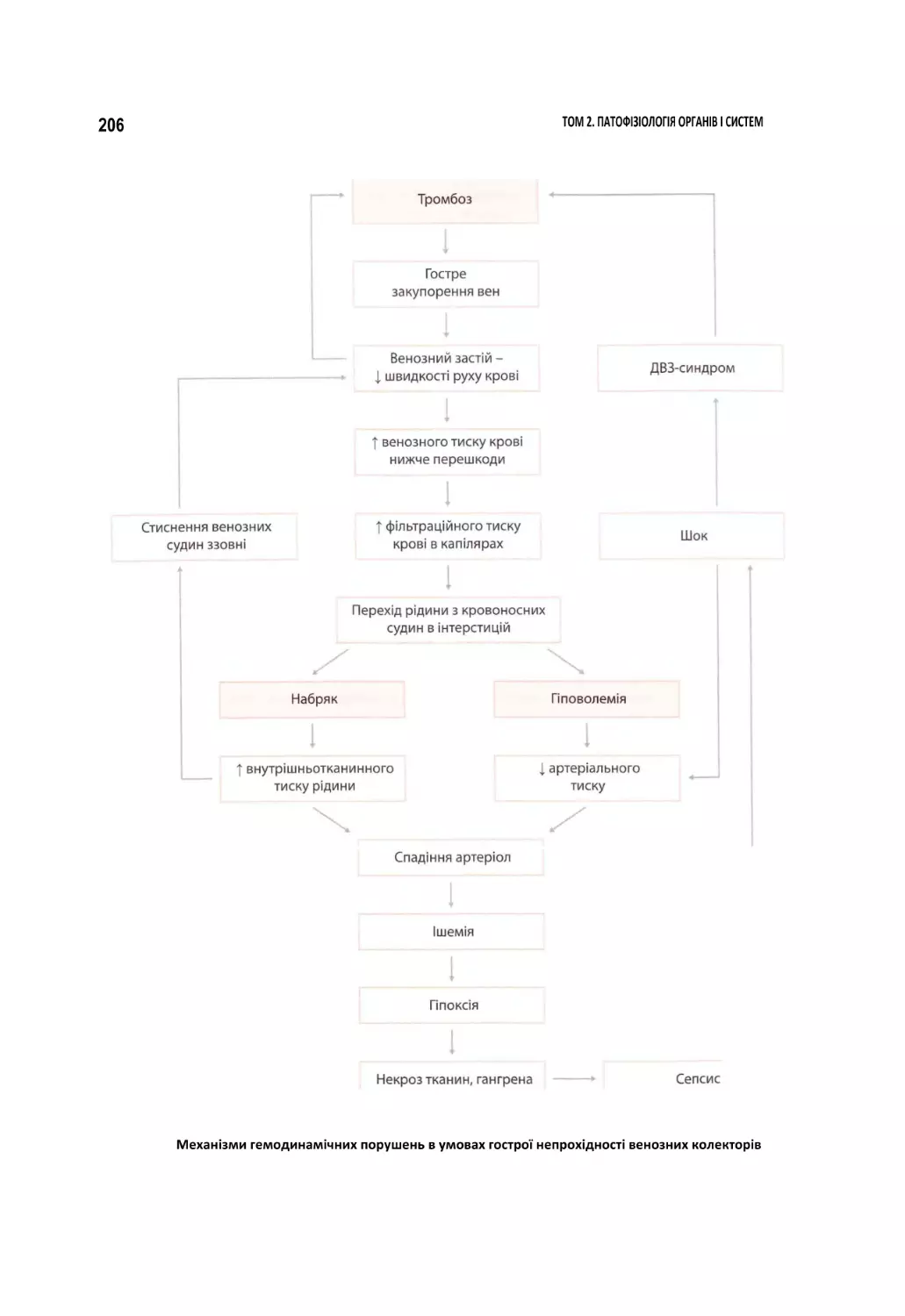
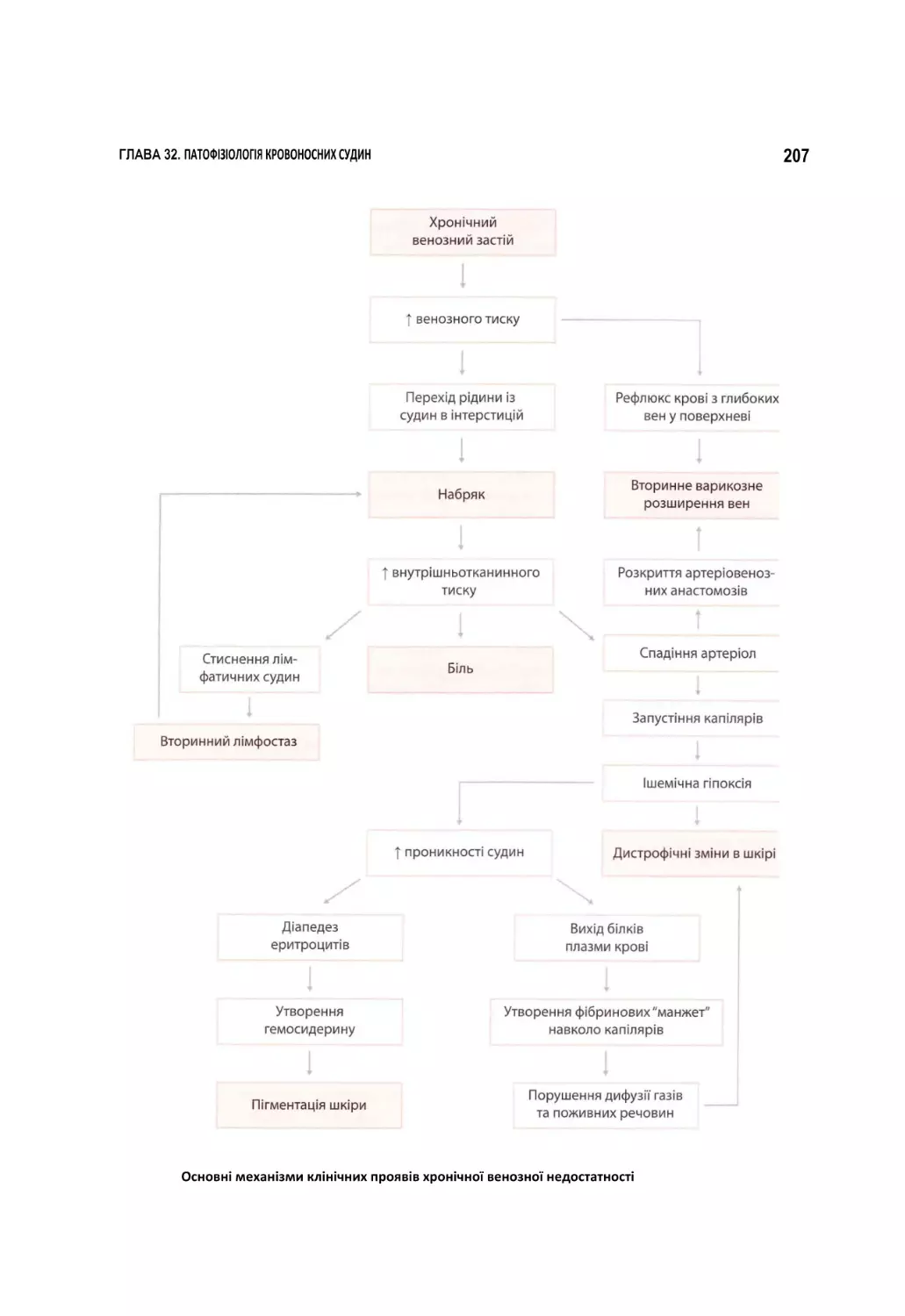
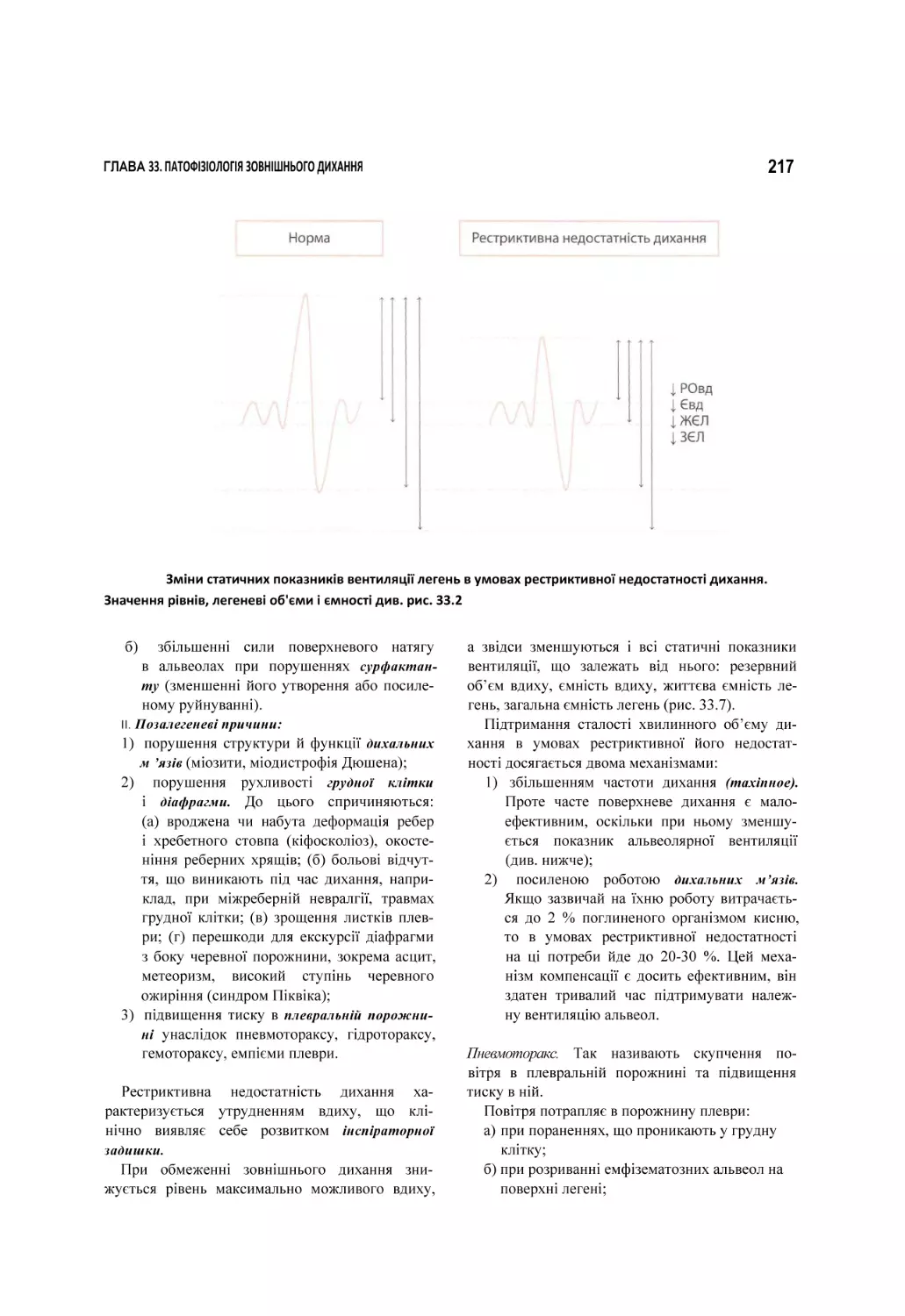
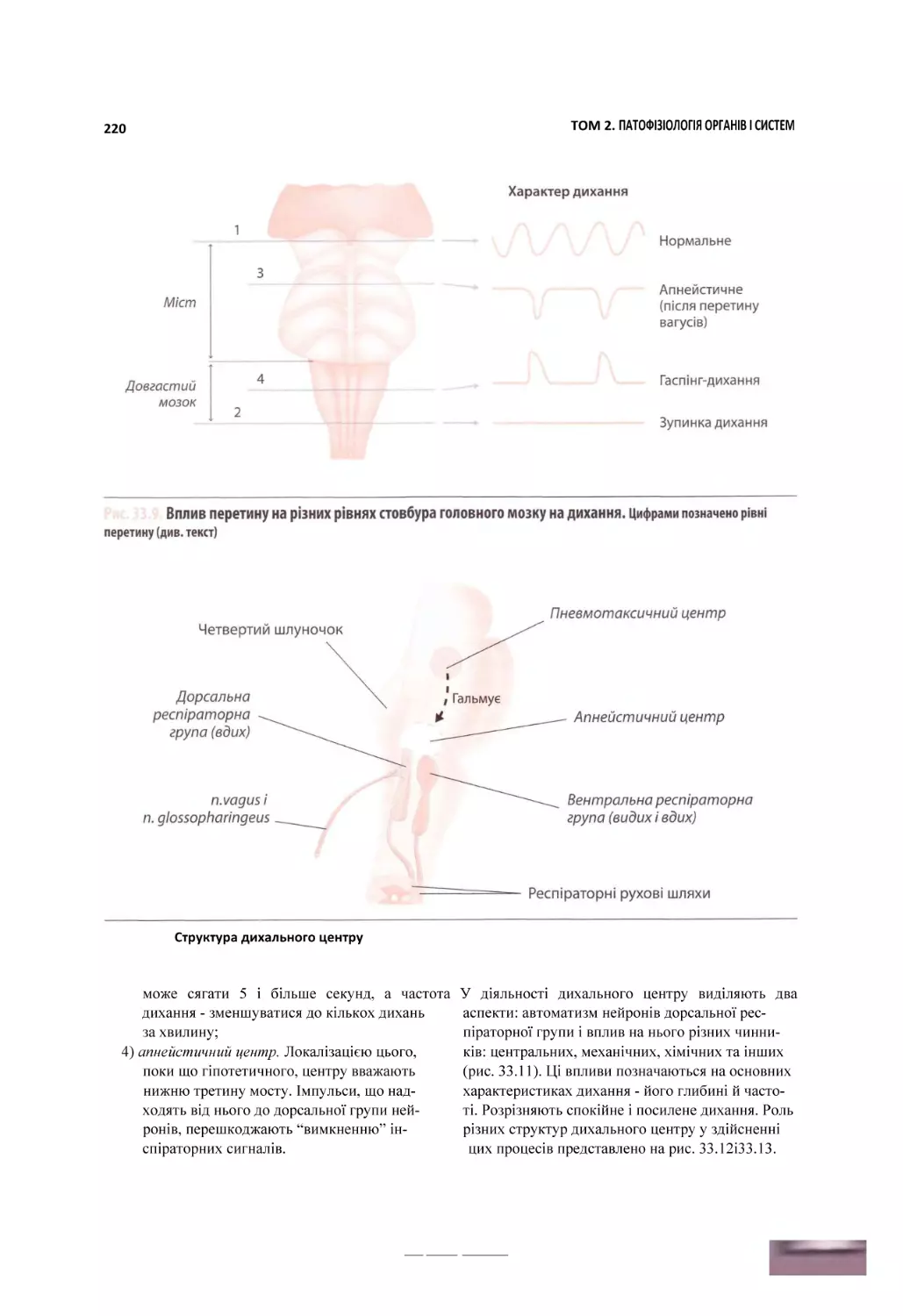
б Схема патогенезу геморагічного шоку
K1IIW ПОРУШЕННЯ СИСТЕМИ
ЕРИТРОЦИТІВ
У нормі кількість еритроцитів у чоловіків дорів
нює 4-1012—5 • 1012, у жінок - 3,5 • 1012^4,5 • 1012
віл крові. Концентрація гемоглобіну в чолові
ків становить 130-160, у жінок - 120-140 г/л.
В умовах патології можливі два види змін
кількості еритроцитів і гемоглобіну в перифе
ричній крові:
1) еритроцитоз - збільшення вмісту еритро
цитів і гемоглобіну;
2) анемія - зменшення їх кількості.
Кількісні зміни еритроцитів можуть бути
обумовлені:
а) порушенням співвідношення між їхнім
утворенням (еритропоезом) і руйнуванням
(еритродіерезом);
б) втратою еритроцитів при порушенні ціліс
ності судин (крововтрата);
в) перерозподілом еритроцитів.
16
До якісних змін еритроцитів відносять:
1) їхні регенераторні форми',
2) дегенеративні зміни',
3) клітини патологічної регенерації.
Причинами якісних змін еритроцитів можуть
бути:
а) порушення дозрівання еритроцитів у чер
воному кістковому мозку або збільшення
проникності кістковомозкового бар’єра,
у результаті чого збільшується надходжен
ня в кров незрілих клітин з низьким вмістом
гемоглобіну (регенераторні форми еритро
цитів);
б) зміна типу кровотворення з еритробластичного на мегалобластичний, коли в кіст
ковому мозку й крові з’являються мегалобласти і мегалоцити (клітини патологічної
регенерації);
в) набуті й спадкові порушення обміну ре
човин, складу й структури еритроцитів,
у тому числі синтезу гемоглобіну (змен
шення утворення або синтез аномальних
гемоглобінів), що веде до появи в крові де
генеративних форм еритроцитів.
Регенераторні форми еритроцитів
Клітини фізіологічної регенерації (регенератор
ні форми) - це молоді незрілі клітини червоного
паростка крові, надходження яких у перифе
ричну кров свідчить про посилення регенерації
клітин еритроїдного ряду в червоному кістко
вому мозку або збільшення проникності кістко
вомозкового бар’єра.
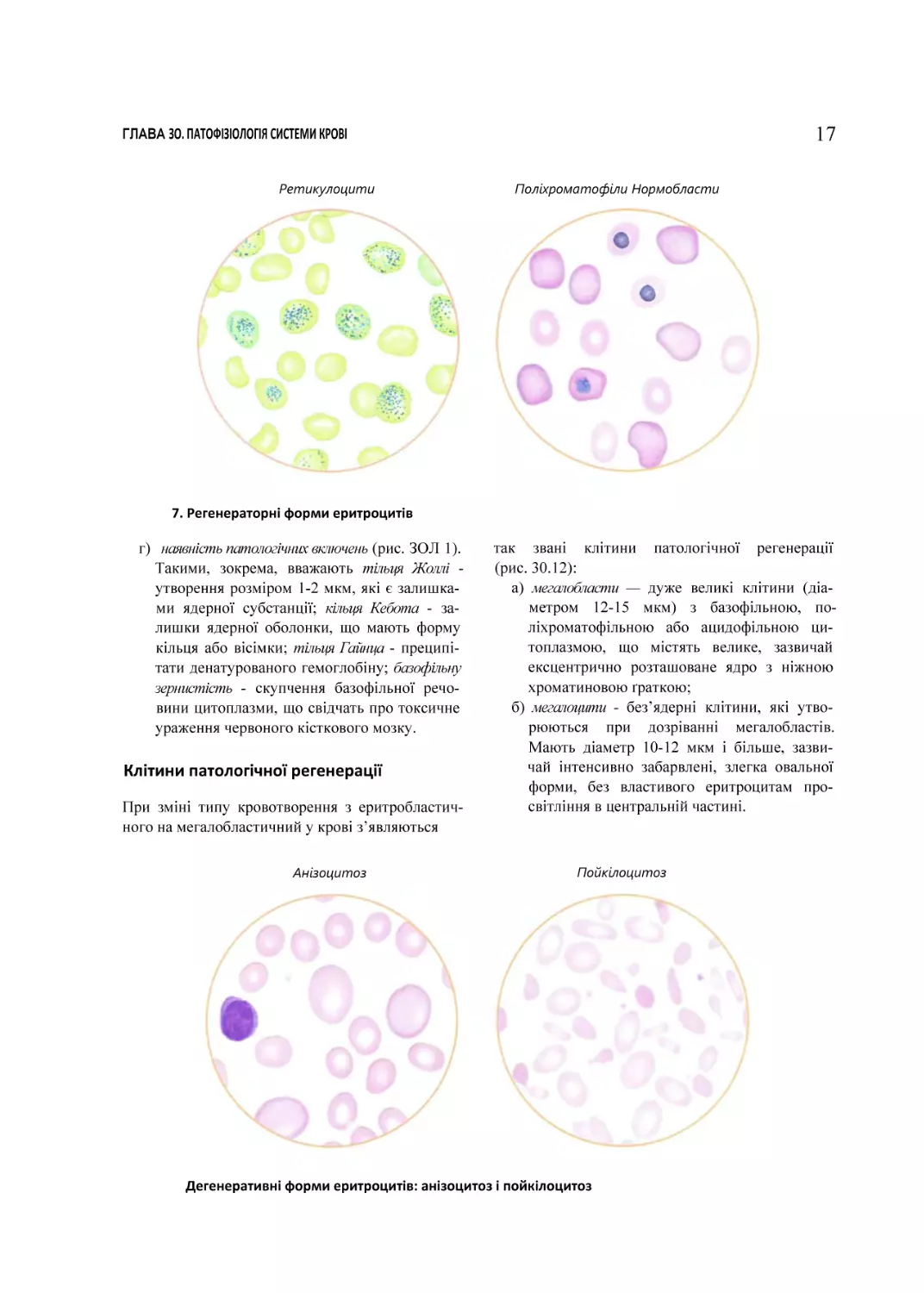
До регенераторних форм відносять (рис. 30.7):
а) ретикулоцити. їх виявляють у мазку кро
ві при суправітальному фарбуванні (барв
ник — бриліанткрезилблау). Це без’ядерні
клітини
брудно-зеленого
забарвлення
(кольори “болотної зелені”) із чорними
включеннями у вигляді гранул (substantia
granulofilamentosa). У нормі їхній вміст
у крові становить 0,2-2 %. При посиленій
регенерації клітин червоного паростка кро
ві їхня кількість може зростати до 50 %;
б) поліхроматофіли. їх виявляють у мазку
крові при фарбуванні за Романовським Гімза. Є без’ядерними клітинами, цито
плазма яких виявляє властивість поліхроматофілії, тобто сприймає як кислотні, так
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
і основні барвники. Тому поліхроматофіли
відрізняються від зрілих еритроцитів ціанотичним відтінком свого забарвлення.
Власне кажучи, ретикулоцити і поліхро
матофіли є клітинами однакового ступеня
зрілості - безпосередніми попередниками
еритроцитів. Різні назви пов’язані з різни
ми їхніми властивостями, які виявляються
при різних способах фарбування;
в) нормобласти (ацидофільні, поліхроматофільні, базофільні). Це ядерні попередники
еритроцитів. У нормі в периферичній крові
їх немає, вони містяться тільки в червоно
му кістковому мозку. При посиленій реге
нерації клітин еритроїдного ряду можуть
з’являтися в крові ацидофільні та поліхроматофільні, рідше базофільні нормоблас
ти. Іноді при гіперрегенераторних анеміях
у крові можна виявити еритробласти (по
передники нормобластів).
Дегенеративні форми еритроцитів
Дегенеративними називають якісні зміни ери
троцитів, що свідчать про неповноцінність цих
клітин. До таких відносять:
а) анізоцитоз - зміну величини еритроци
тів (рис. 30.8). Можлива поява макроцитів - еритроцитів з діаметром понад 8 мкм
і мікроцитів - клітин, діаметр яких мен
ший 6,5 мкм (середній діаметр нормально
го еритроцита близько 7,2 мкм);
б) пойкілоцитоз - зміну форми еритроцитів
(рис. 30.8). У нормі еритроцити мають фор
му дисків, увігнутих всередину з обох бо
ків. В умовах патології можуть з’являтися
грушоподібні,
витягнуті,
серпоподібні,
овальні еритроцити, клітини сферичної
форми (сфероцити) та інші (рис. 30.9);
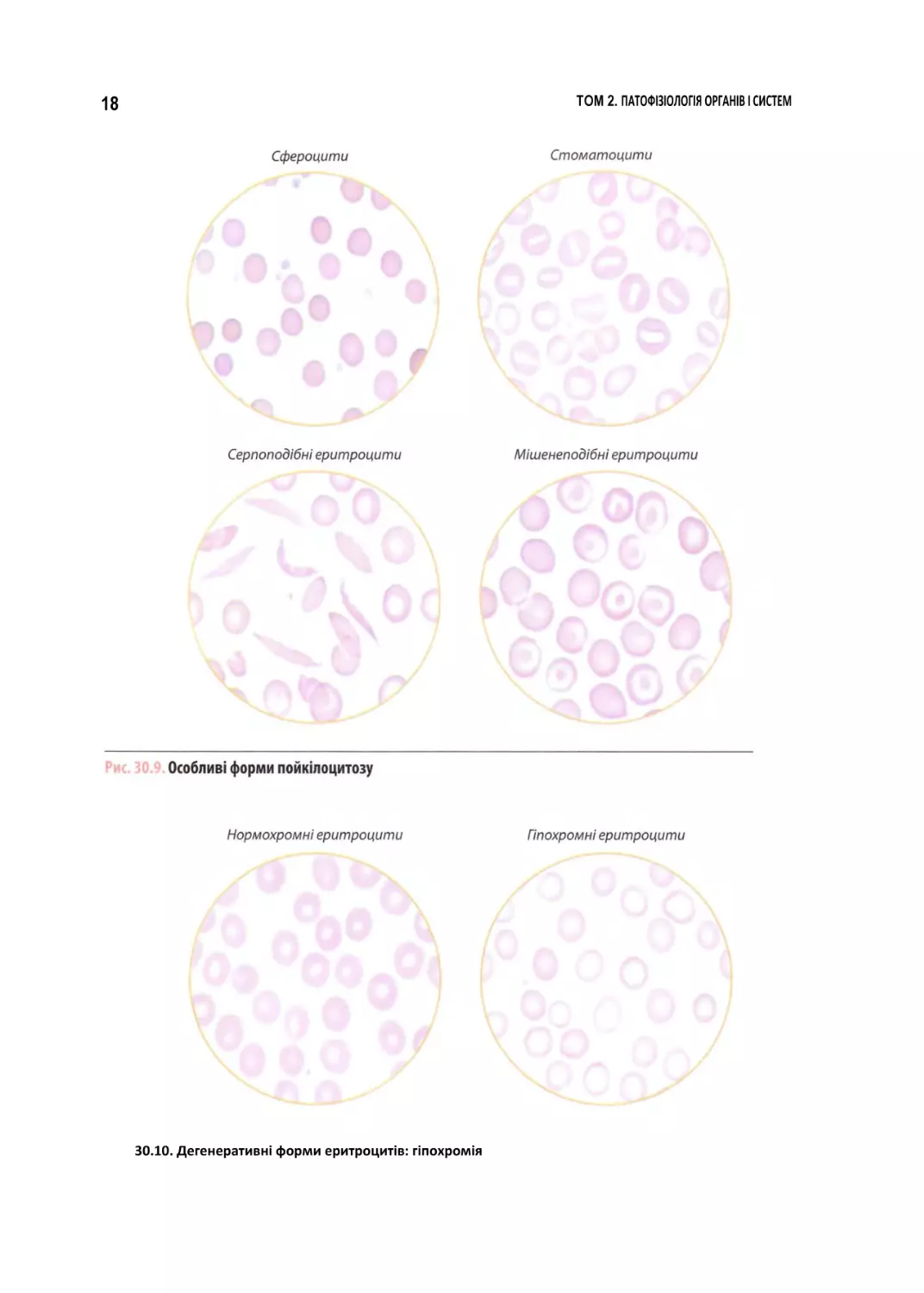
в)
зміну
забарвлення
еритроцитів,
що
залежить від вмісту в них гемоглобіну
(рис. 30.10). Еритроцити, інтенсивно за
барвлені, називають гіперхромними, із
блідим
забарвленням
гіпохромними.
Еритроцити, у яких забарвлена у вигляді
кільця тільки периферична частина, де
міститься гемоглобін, а в центрі — про
світління, мають назву анулоцитів. У ви
падку, коли еритроцити виявляють різну
інтенсивність забарвлення, ведуть мову
про анізохромію;
17
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Ретикулоцити
Поліхроматофіли Нормобласти
7. Регенераторні форми еритроцитів
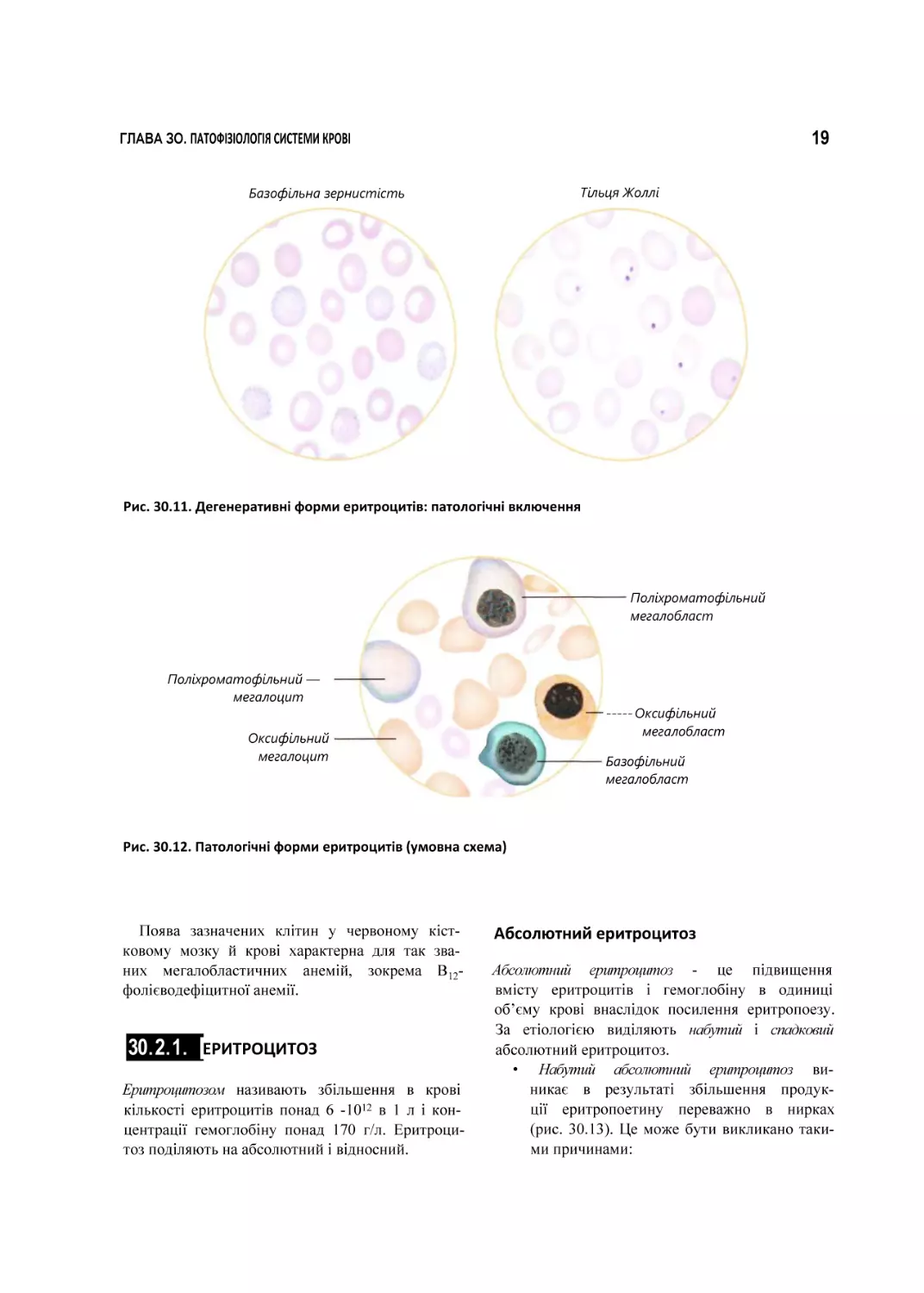
г) наявність патологічних включень (рис. ЗОЛ 1).
Такими, зокрема, вважають тільця Жоллі утворення розміром 1-2 мкм, які є залишка
ми ядерної субстанції; кільця Кебота - за
лишки ядерної оболонки, що мають форму
кільця або вісімки; тільця Гайнца - преципі
тати денатурованого гемоглобіну; базофільну
зернистість - скупчення базофільної речо
вини цитоплазми, що свідчать про токсичне
ураження червоного кісткового мозку.
Клітини патологічної регенерації
При зміні типу кровотворення з еритробластичного на мегалобластичний у крові з’являються
Анізоцитоз
так звані клітини патологічної регенерації
(рис. 30.12):
а) мегалобласти — дуже великі клітини (діа
метром 12-15 мкм) з базофільною, поліхроматофільною або ацидофільною ци
топлазмою, що містять велике, зазвичай
ексцентрично розташоване ядро з ніжною
хроматиновою ґраткою;
б) мегалоцити - без’ядерні клітини, які утво
рюються при дозріванні мегалобластів.
Мають діаметр 10-12 мкм і більше, зазви
чай інтенсивно забарвлені, злегка овальної
форми, без властивого еритроцитам про
світління в центральній частині.
Пойкілоцитоз
Дегенеративні форми еритроцитів: анізоцитоз і пойкілоцитоз
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
18
30.10. Дегенеративні форми еритроцитів: гіпохромія
19
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Тільця Жоллі
Базофільна зернистість
Рис. 30.11. Дегенеративні форми еритроцитів: патологічні включення
Поліхроматофільний
мегалобласт
Поліхроматофільний —
мегалоцит
----- Оксифільний
мегалобласт
Оксифільний
мегалоцит
Базофільний
мегалобласт
Рис. 30.12. Патологічні форми еритроцитів (умовна схема)
Поява зазначених клітин у червоному кіст
ковому мозку й крові характерна для так зва
них мегалобластичних анемій, зокрема В12фолієводефіцитної анемії.
30.2.1.
ЕРИТРОЦИТОЗ
Еритроцитозом називають збільшення в крові
кількості еритроцитів понад 6 -1012 в 1 л і кон
центрації гемоглобіну понад 170 г/л. Еритроцитоз поділяють на абсолютний і відносний.
Абсолютний еритроцитоз
Абсолютний еритроцитоз - це підвищення
вмісту еритроцитів і гемоглобіну в одиниці
об’єму крові внаслідок посилення еритропоезу.
За етіологією виділяють набутий і спадковий
абсолютний еритроцитоз.
• Набутий абсолютний еритроцитоз ви
никає в результаті збільшення продук
ції еритропоетину переважно в нирках
(рис. 30.13). Це може бути викликано таки
ми причинами:
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
20
Набутий абсолютний еритроцитоз: компенсаторне значення
і власне патологічні зміни в організмі
а) порушеннями нейрогуморальної регуля
ції - збудженням симпатичної частини нер
вової системи, гіперфункцією деяких ен
докринних залоз. Так, катехоламіни, АКТГ,
тиреоїдні гормони, глюкокортикоїди поси
люють утилізацію кисню, що сприяє роз
витку кисневого голодування й утворенню
еритропоетинів у нирках;
б) гіпоксичною, дихальною, циркуляторною
гіпоксією - у випадку гірської хвороби, не
достатності зовнішнього дихання й крово
обігу (див. главу 23);
в) місцевою гіпоксією нирок унаслідок їхньої
ішемії (гідронефроз, стеноз ниркових артерій);
г) гіперпродукцією еритропоетинів деякими
пухлинами (гіпернефрома, рак печінки та ін.).
Окрім того, абсолютний еритроцитоз розвива
ється при істинній поліцитемії (еритремія або хво
роба Вакеза), що є різновидом хронічного лейкозу.
»
Спадковий
абсолютний
еритроцитоз.
Причиною його виникнення може бути
генетично обумовлений дефект глобіну
в молекулі гемоглобіну або дефіцит в ери
троцитах 2,3-дифосфогліцерату, що є ре
гулятором оксигенації й дезоксигенацїї
гемоглобіну. При цьому підвищується спо
рідненість гемоглобіну до кисню і зменшу
___________________________________________
21
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
ється віддача останнього тканинам (крива
дисоціації оксигемоглобіну зміщена вліво).
Розвивається гіпоксія, стимулюється про
дукція еритропоетинів, під впливом яких
посилюється еритропоез.
І гіпоксичне, і дисрегуляторне посилення
утворення еритропоетинів супроводжується
підвищенням функціональної активності еритроцитарного паростка червоного кісткового
мозку, що виявляється збільшенням у крові
вмісту еритроцитів і гемоглобіну, зростанням
гематокриту. При цьому може збільшуватися
об’єм циркулюючої крові (поліцитемічна гіперволемія), її в’язкість; сповільнюється швидкість
течії крові, порушується серцева діяльність. Ар
теріальний тиск підвищується, відзначається
повнокрів’я внутрішніх органів, гіперемія шкі
ри й слизових оболонок, спочатку посилюється
тромбоутворення, а потім виникають кровотечі
(розвивається ДВЗ-синдром, див. підглаву 30.4).
Зміни в крові при набутих абсолютних еритроцитозах часто носять компенсаторний харак
тер, сприяють поліпшенню кисневого постачан
ня тканин в умовах гіпоксії. Із припиненням дії
етіологічного фактора відбувається нормалізація
кількості еритроцитів і гемоглобіну. Саме в цьо
му полягає корінна відмінність абсолютного еритроцитозу, що характеризується підвищенням
фізіологічної регенерації еритроцитарного паро
стка кісткового мозку, від еритремії (хвороба Вакеза), при якій первинне й необоротне зростання
кількості еритроцитів обумовлене гіперплазією
еритроцитарного паростка пухлинної природи.
Відносний еритроцитоз
Відносний еритроцитоз - це збільшення вміс
ту еритроцитів і гемоглобіну в одиниці об’єму
крові внаслідок зменшення об’єму плазми.
Ного розвиток пов’язаний з дією факторів, які
обумовлюють зневоднення організму (див. гла
ву 27) або перерозподіл крові, що викликає поліцитемічну гіповолемію (наприклад, шок, опіки).
АНЕМІЇ
Анемія - це клінічно-гематологічний синдром
або самостійна хвороба, що характеризується
зменшенням кількості еритроцитів і (або) вміс
ту гемоглобіну в одиниці об’єму крові, а також
якісними змінами еритроцитів.
Ознаки анемій
Анемія може виявляти себе гематологічними
і загальними клінічними ознаками.
Гематологічні ознаки анемій поділяють на
кількісні і якісні.
До кількісних відносять:
• зменшення вмісту еритроцитів в одини
ці об’єму крові (у чоловіків нижче 4- 10і2,
у жінок нижче 3,5 • 1012 в 1 л крові);
» зменшення концентрації гемоглобіну (у чо
ловіків нижче 130 г/л, у жінок нижче 120 г/л);
• зменшення гематокриту (у чоловіків ниж
че 43 %, у жінок нижче 40 %);
• зміни колірного показника (норма 0,85-1).
Якісними ознаками анемій є поява:
« регенераторних форм еритроцитів (ретикулоцитів, поліхроматофілів, нормобластів);
• дегенеративних змін у клітинах еритроци
тарного ряду (анізоцитоз, пойкілоцитоз, гі
похромія, патологічні включення);
» клітин патологічної регенерації (мегалобластів, мегалоцитів).
Загальні клінічні прояви анемій зумовлюються:
• гіпоксією, що виникає при будь-якому виді
анемії (див. главу 23);
• синдромами, що пов’язані з особливостя
ми патогенезу кожного окремого виду ане
мії (наприклад, при В12-фолієводефіцитній
анемії - неврологічні розлади й ураження
травної системи, при гемолітичній анемії жовтяниця тощо).
Класифікація
Анемії можна класифікувати, беручи за основу
різні їхні характеристики.
І. Патогенетична класифікація. Залежно
від механізмів розвитку розрізняють:
►
постгеморагічні анемії (напр., анемія
після гострої крововтрати);
►
гемолітичні анемії (напр., серпоподібноклітинна);
» анемії, обумовлені порушеннями еритропоезу (напр., залізодефіцитна, В12-фолієводефіцитна).
22
За етіологією анемії можуть бути:
• спадковими (напр., таласемія);
• набутими (напр., хронічна постгеморагічна анемія).
III. За інтенсивністю регенерації червоного
кісткового мозку виділяють анемії:
• регенераторні (напр., гостра постгеморагічна анемія);
» гіперрегенераторні (напр., набута гемолі
тична анемія);
• гіпорегенераторні (напр., залізодефіцитна анемія);
• арегенераторні (напр., апластична анемія).
II.
Ознаками посиленої регенерації клітин еритроїдного ряду з боку (а) периферичної крові
є збільшення вмісту ретикулоцитів і поліхроматофілів та поява нормобластів (регенератор
ні форми еритроцитів), а з боку (б) червоного
кісткового мозку - зміщення лейкоеритроїдного співвідношення від 3:1 до 1:1 і навіть до 1:2
і 1:3.
IV. За колірним показником (КП) анемії можуть
бути:
• нормохромними (КП = 0,85-1; напр.,
гостра постгеморагічна анемія в перші
кілька діб після крововтрати);
• гіпохромними (КП < 0,85; напр., залізодефіцитна анемія);
» гіперхромними (КП > 1; напр., В12-фолієводефіцитна анемія).
V. За типом кровотворення виділяють:
• анемії з еритробластичним типом еритропоезу (напр., залізодефіцитна ане
мія);
• анемії з мегалобластичним типом крово
творення (напр., В12-фолієводефіцитна
анемія).
VI. За розмірами еритроцитів анемії бувають:
• мікроцитарними (напр., мікросфероцитарна анемія Мінковського - Шофара);
» нормоцитарними (напр., гостра постге
морагічна анемія);
• макроцитарними (напр., В]2-фолієводефіцитна анемія).
VII. За клінічним перебігом розрізняють анемії:
■ гострі (напр., анемія після гемотрансфузійного шоку);
■ хронічні (напр., гіпопластична анемія).
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
30.2.2.1. Постгеморагічні анемії
Постгеморагічними називають анемії, що роз
виваються внаслідок крововтрати.
Залежно від характеру крововтрати виділя
ють два види анемій цієї групи:
1) гостру постгеморагічну;
2) хронічну постгеморагічну анемію.
Гостра постгеморагічна анемія
Вона виникає після швидкої масивної крововтра
ти при пораненні судин або їхньому ушкоджен
ні патологічним процесом.
Картина крові при гострій постгеморагічній
анемії зазнає змін залежно від часу, що пройшов
після крововтрати. З урахуванням цього можна
виділити три періоди, кожний з яких характери
зується певного картиною периферичної крові
(рис. 30.14).
1. Перші кілька годин після гострої крово
втрати. У цей період зменшується загаль
ний об’єм крові, а також загальна кількість
еритроцитів в організмі. Однак в одиниці
об’єму крові вміст еритроцитів і концентра
ція гемоглобіну не змінюються. Це поясню
ється тим, що відразу ж після крововтрати
спрацьовують термінові компенсаторні ре
акції, спрямовані на зменшення об’єму су
динного русла, і ще недостатньо виражені
реакції, спрямовані на поповнення об’єму
циркулюючої крові (перехід рідини із тка
нин у кров).
2. Період часу від кількох годин до кількох діб
після гострої крововтрати. У результаті пе
реходу рідини з інтерстиціального простору
в кровоносні судини відбувається розведен
ня крові (гемодилюція). Як результат, змен
шується кількість еритроцитів і гемоглобі
ну в одиниці об’єму крові, падає гематокрит.
Колірний показник залишається без змін
(нормохромна анемія). Якісні зміни еритро
цитів у мазку крові ще не виявляються.
3. Період часу від кількох діб до 1—2 тижнів
після гострої крововтрати. Найбільш ха
рактерною рисою картини крові в цей пері
од є поява великої кількості регенераторних
форм еритроцитів, що пов’язане з посилен
ням еритропоезу в червоному кістковому
мозку. Оскільки молоді незрілі еритроцити
містять гемоглобіну менше в порівнянні
23
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Компенсаторні реакції
Норма
Крововтрата
І. Зменшення
об'єму
судинного
русла
II. Поновлення
об'єму
циркулюючої
крові
III. Відновлення
клітинного
складу
крові
ис. 30.14. Картина периферичної крові в різні періоди після гострої крововтрати
зі зрілими клітинами, колірний показник
зменшується й анемія стає гіпохромною.
Місце анемії після гострої крововтрати в різ
них класифікаціях анемій показано на рис. 30.15.
(мікроцитоз і пойкілоцитоз, гіпохромія). Кіль
кість еритроцитів і гематокрит можуть залиша
тися без змін. Дефіцит заліза, як провідна ланка
патогенезу, визначає місце анемії після частих
повторних крововтрат у різних класифікаціях
анемій (рис. 30.16).
Хронічна постгеморагічна анемія
Хронічна постгеморагічна анемія розвивається
внаслідок повторних, часто невеликих крово
втрат, викликаних ураженням кровоносних су
дин при деяких хворобах (дисменорея, виразко
ва хвороба шлунка, геморой та ін.) і порушення
ми судинно-тромбоцитарного та коагуляційного
гемостазу (геморагічний діатез). Втрата заліза
при частих кровотечах надає цій анемії зстізодефіцитного характеру.
У зв’язку із втратою заліза при частих крово
течах розвиваються гематологічні ознаки залізодефіцитної анемії: зменшується концентрація
гемоглобіну й колірний показник, у мазку крові
з’являються дегенеративні форми еритроцитів
Гемолітичні анемії
Гемолітичними називають анемії, що виникають
унаслідок руйнування (гемолізу) еритроцитів.
Класифікація
Особливості розвитку цих анемій додають до
даткові критерії до їхніх класифікацій, що наво
дяться нижче.
І. За походженням гемолітичні анемії, як
і анемії загалом, можуть бути:
• набутими;
• спадково обумовленими.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
24
Анемія після гострої крововтрати. Місце в різних класифікаціях анемій
За патогенезом
Постгеморагічна
За етіологією
Набута
За інтенсивністю регенерації
червоного кісткового мозку
Гіпорегенераторна
За колірним показником
Гіпохромна
За типом кровотворення
3 еритробластичним типом
еритропоезу
За розмірами еритроцитів
Мікроцитарна
За клінічним перебігом
Хронічна
Анемія після повторних крововтрат. Місце у різних класифікаціях анемій
П. За причинами гемолізу розрізняють:
анемії, обумовлені екзоеритроцитарними
факторами (екстракорпускулярні);
анемії, обумовлені ендоеритроцитарними
факторами (корпускулярні). До таких від
носять (а) дефекти мембрани (мембранопатії); (б) порушення ферментів (ферменте-,
або ензимопатії); (в) зміни структури гемо
глобіну (гемоглобінопатії).
III. За механізмами гемолізу виділяють:
* анемії з внутрішньосудинним гемолізом;
* анемії з внутрішньоклітинним гемолізом.
IV. За клінічним перебігом гемолітичні анемії
бувають:
- гострими;
* хронічними.
Механізми гемолізу
Розрізняють два основних механізми гемолізу
еритроцитів:
* внутрішньосудинний;
• внутрішньоклітинний.
Внутрішньосудинний гемоліз виникає в кро
воносних судинах унаслідок дії факторів, що
ушкоджують еритроцити. Ці фактори отримали
назву гемолітичних. До них відносять:
а) фактори фізичної природи (механічна травма,
іонізуюча радіація, ультразвук, температура);
б) хімічні агенти (гемолітичні отрути)',
в) біологічні фактори (збудники інфекційних
хвороб, токсини, ферменти);
г) імунні фактори (антитіла).
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Внутрішньосудинний гемоліз може здійсню
ватися такими механізмами.
І. Механічний гемоліз. Розвивається внаслідок
механічного руйнування мембран еритроци
тів, наприклад, при роздавлюванні еритро
цитів у судинах стопи (маршовий гемоліз).
II. Осмотичний гемоліз. Виникає тоді, коли
осмотичний тиск усередині еритроцита
більший, ніж осмотичний тиск плазми кро
ві. У цьому випадку вода за законами осмосу
надходить в еритроцит, об’єм його зростає,
і в підсумку відбувається розрив мембра
ни. Причиною осмотичного гемолізу може
бути або зменшення осмотичного тиску
середовища, у якому перебувають еритро
цити (гіпотонічні розчини), або збільшення
осмотичного тиску в самих еритроцитах.
Останнє, як правило, пов’язане зі збільшен
ням концентрації іонів натрію всередині
еритроцитів у результаті підвищення про
никності їхньої мембрани або внаслідок
порушення роботи Na-K-насосів.
III. Окисний гемоліз. Розвивається внаслідок
вільнорадикального
окиснення
ліпідів
і білків плазматичної мембрани еритроци
тів. Результатом цього є збільшення проI никності еритроцитарної мембрани, що
потім веде до реалізації осмотичного меха
нізму гемолізу.
Основу окисного гемолізу становлять реакції
вільнорадикального окиснення і, зокрема, про
цеси пероксидного окиснення ліпідів еритроци
тарної мембрани (див. главу 7). Існує два шляхи
активації цього механізму:
1) посилене утворення вільних радикалів, що
буває при дії екзогенних речовин-окислювачів (деякі лікарські препарати, гемолітич
ні отрути, токсичні дози вітаміну D; про
дукти, що містяться в бобах Vicia fava), під
впливом іонізуючої радіації та гіпероксії;
2) порушення діяльності антиоксидантних
систем еритроцитів, що може бути зу
мовлено спадковими або набутими пору
шеннями активності ферментів глютатіонової антиоксидантної системи (глютатіонпероксидази і глютатіонредуктази),
дефіцитом селену — мікроелемента, необ
хідного для функціонування глютатіонпероксидази, пригніченням реакцій пентозного циклу (наприклад, дефіцит глюкозо-6фосфатдегідрогенази).
25
IV. Детергентний гемоліз. Пов’язаний з розчи
ненням ліпідних компонентів мембрани ери
троцитів речовинами-детергентами. Цей вид
гемолізу викликають жовчні кислоти (холемічний синдром), жиророзчинні хімічні
агенти, деякі токсини бактерій (лецитинази).
V. Комплементзалежний гемоліз. Обумовле
ний руйнуванням (перфорацією) мембрани
еритроцитів активним комплементом. Цей
механізм лежить в основі імунного гемолізу.
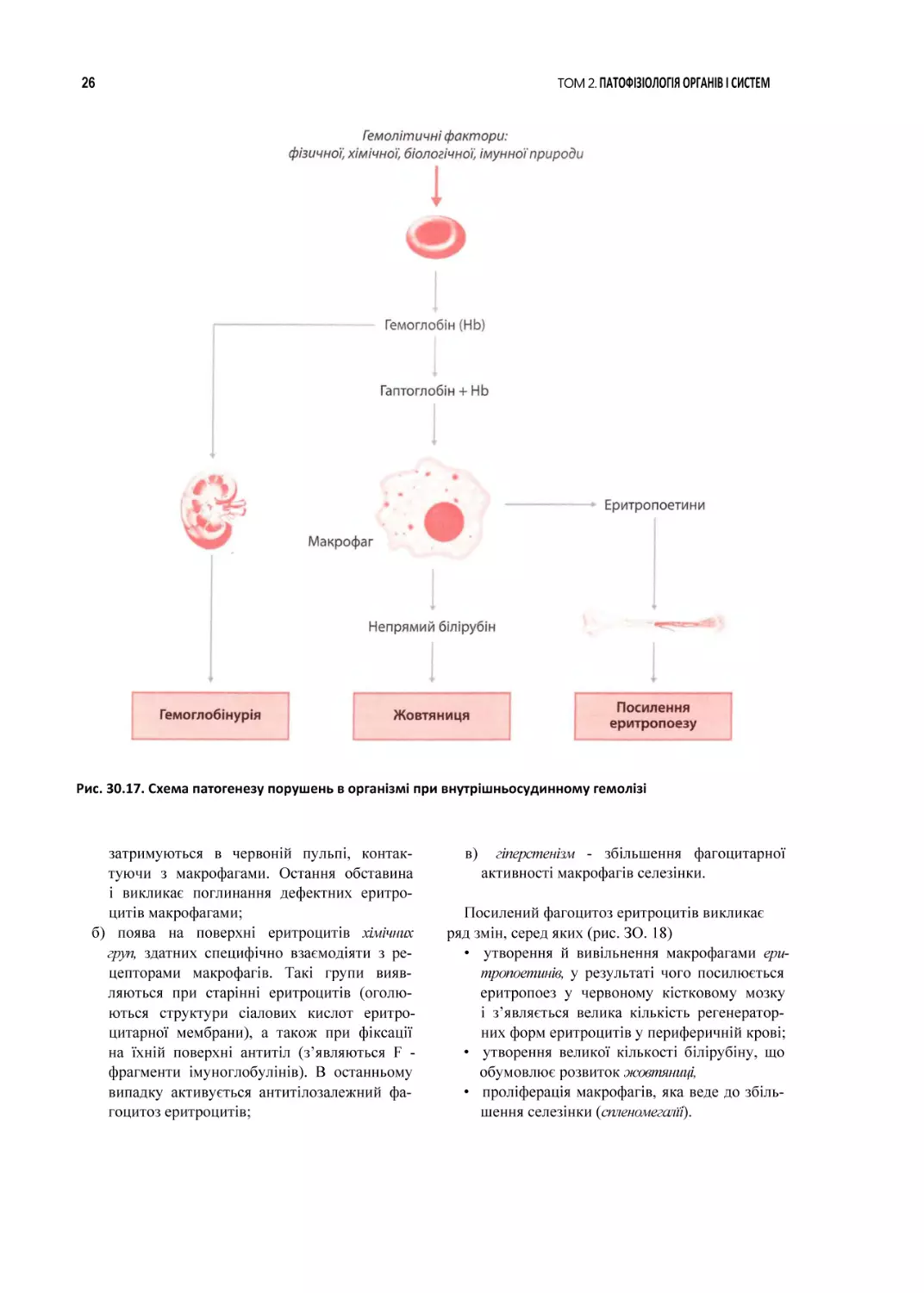
Внутрішньосудинний гемоліз супроводжу
ється виходом гемоглобіну з клітин у плазму
крові, де він частково з’єднується з білком гаптоглобіном. При цьому відбуваються такі про
цеси (рис. 30.17).
L Комплекс гемоглобін — гаптоглобін погли
нається макрофагами і викликає утворення
й вивільнення останніми макрофагальних
еритропоетинів. Еритропоетини, вплива
ючи на червоний кістковий мозок, стиму
люють еритропоез. У результаті в червоно
му кістковому мозку й периферичній крові
з’являються ознаки посиленої регенерації
клітин еритроїдного ряду.
2. Поглинений макрофагами гемоглобін зазнає
біохімічних перетворень, у результаті яких
білкова частина молекули розщеплюється до
амінокислот, а з гема утворюється білірубін.
Останній зв’язується з білками й надходить
у кров (непрямий білірубін). У результаті
розвивається синдром, відомий під назвою
гемолітична жовтяниця (див. главу 35).
3. Частина не зв’язаного з гаптоглобіном ге
моглобіну фільтрується в нирках. Це при
зводить, з одного боку, до появи гемогло
біну в сечі {гемоглобінурія), з другого - до
“забивання” пор ниркового фільтра, що
може бути причиною появи ознак гострої
ниркової недостатності.
Внутрішньоклітинний
гемоліз
еритроцитів
розвивається внаслідок поглинання і перетрав
лювання еритроцитів макрофагами.
У його основі можуть лежати такі причини:
а) поява дефектних еритроцитів. Зменшення
пластичності еритроцитів, їхньої здатності
до деформації, набряк призводять до того,
що вони не можуть вільно проходити через
міжендотеліальні щілини венозних синусів
селезінки (“селезінковий фільтр”) і надовго
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
26
Рис. 30.17. Схема патогенезу порушень в організмі при внутрішньосудинному гемолізі
затримуються в червоній пульпі, контак
туючи з макрофагами. Остання обставина
і викликає поглинання дефектних еритро
цитів макрофагами;
б) поява на поверхні еритроцитів хімічних
груп, здатних специфічно взаємодіяти з ре
цепторами макрофагів. Такі групи вияв
ляються при старінні еритроцитів (оголю
ються структури сіалових кислот еритроцитарної мембрани), а також при фіксації
на їхній поверхні антитіл (з’являються F фрагменти імуноглобулінів). В останньому
випадку активується антитілозалежний фа
гоцитоз еритроцитів;
в)
гіперстенізм - збільшення фагоцитарної
активності макрофагів селезінки.
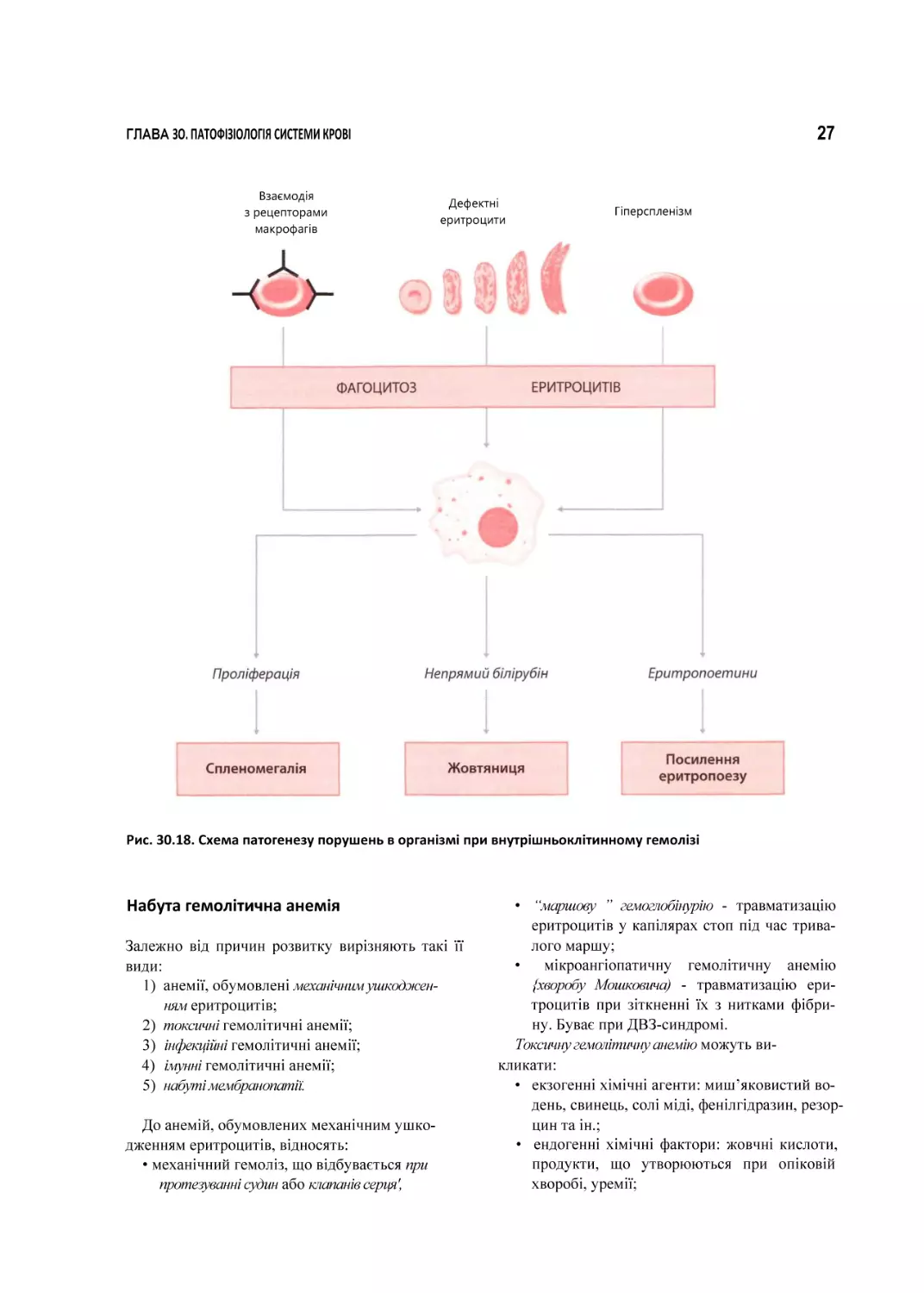
Посилений фагоцитоз еритроцитів викликає
ряд змін, серед яких (рис. ЗО. 18)
• утворення й вивільнення макрофагами еритропоетинів, у результаті чого посилюється
еритропоез у червоному кістковому мозку
і з’являється велика кількість регенератор
них форм еритроцитів у периферичній крові;
• утворення великої кількості білірубіну, що
обумовлює розвиток жовтяниці,
• проліферація макрофагів, яка веде до збіль
шення селезінки (спленомегалїі).
27
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Взаємодія
з рецепторами
макрофагів
Дефектні
еритроцити
Гіперспленізм
Рис. 30.18. Схема патогенезу порушень в організмі при внутрішньоклітинному гемолізі
Набута гемолітична анемія
Залежно від причин розвитку вирізняють такі її
види:
1) анемії, обумовлені механічним ушкоджен
ням еритроцитів;
2) токсичні гемолітичні анемії;
3) інфекційні гемолітичні анемії;
4) імунні гемолітичні анемії;
5) набуті мембранопатії.
До анемій, обумовлених механічним ушко
дженням еритроцитів, відносять:
• механічний гемоліз, що відбувається при
протезуванні судин або клапанів серця',
• “маршову ” гемоглобінурію - травматизацію
еритроцитів у капілярах стоп під час трива
лого маршу;
• мікроангіопатичну гемолітичну анемію
{хворобу Мошковича) - травматизацію ери
троцитів при зіткненні їх з нитками фібри
ну. Буває при ДВЗ-синдромі.
Токсичну гемолітичну анемію можуть ви
кликати:
• екзогенні хімічні агенти: миш’яковистий во
день, свинець, солі міді, фенілгідразин, резор
цин та ін.;
• ендогенні хімічні фактори: жовчні кислоти,
продукти, що утворюються при опіковій
хворобі, уремії;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
28
» отрути біологічного походження: зміїна,
бджолина, отрута деяких видів павуків.
Причинами інфекційної гемолітичної ане
мії можуть бути гемолітичний стрептокок, ма
лярійний плазмодій, токсоплазма, лейшманії.
Гемоліз еритроцитів при інфекційних хворобах
виникає або (а) внаслідок розмноження збудни
ків в еритроцитах (малярійний плазмодій), або
(б) в результаті дії токсинів-гемолізинів (гемолі
тичний стрептокок).
Імунні гемолітичні анемії - це анемії, що ви
никають за участі специфічних імунних механіз
мів. Вони обумовлені взаємодією гуморальних
антитіл з антигенами, фіксованими на поверхні
еритроцитів, і тому є проявом II типу алергічних
реакцій за класифікацією Кумбса і Джелла (див.
главу 15).
Залежно від причин розвитку виділяють такі
види імунних гемолітичних анемій:
•
алоімунні (ізоімунні) гемолітичні анемії.
їх причиною можуть бути: (а) надходжен
ня ззовні антитіл проти власних еритро
цитів (гемолітична хвороба новонародже
них) або (б) поява в організмі еритроцитів,
проти яких у плазмі є антитіла (перели
вання крові, не сумісної за групами АВО
або Rh);
• аутоімунні гемолітичні анемії. Вони обу
мовлені утворенням в організмі антитіл
проти власних еритроцитів. Це може бути
пов’язано або (а) з первинними змінами са
мих еритроцитів (поява аутоантигенів), або
(б) зі змінами в імунній системі (скасуван
ня імунологічної толерантності, поява “за
боронених” клонів лімфоцитів);
• гетероімунні (гаптенові) гемолітичні ане
мії. Виникають при фіксації на поверхні
еритроцитів чужорідних антигенів (гаптенів), зокрема, лікарських препаратів (пені
цилін, сульфаніламіди), вірусів.
Одним з найпоширеніших різновидів імун
них гемолітичних анемій є гемолітична хво
роба новонароджених — недуга, що виникає
в результаті гемолізу еритроцитів плода й ново
народженого, викликаного антитілами матері.
Найчастіше бувають два варіанти цієї хвороби:
1) резус-конфлікт;
2) АВО-конфлікт.
Резус-конфлікт розвивається у випадку вагіт
ності Rh -матері ИГ-плодом (найчастіше при
повторній вагітності). Спочатку відбувається
імунізація матері Rh-еритроцитами плода, які
можуть потрапляти в організм матері під час
пологів або при дефектах плаценти. Найбільш
імовірною є імунізація під час пологів, тому ре
зус-конфлікт виникає найчастіше в умовах по
вторної вагітності Rh-плодом.
У відповідь на надходження RlT-еритроцитів
в організмі матері синтезуються антитіла проти
D-антигену. Ці антитіла (Ig G) здатні проникати
через плаценту в організм плода й викликати ге
моліз його еритроцитів.
АВО-конфлікт найчастіше виникає в ситуаці
ях, коли мати має групу крові 0(1), а плід - А(ІІ)
або В(ІІІ). Нормальні ізоаглютиніни в системі
АВО належать до класу IgM. Ці антитіла не про
никають через плаценту й тому не можуть бути
причиною АВО-конфлікту. Однак у 10 % здоро
вих людей, що мають групу крові 0(1), є анти
тіла проти аглютиногенів А і В, представлені
IgG. Наявність цих антитіл не залежить від по
передньої імунізації. Аглютиніни IgG проника
ють через плаценту і можуть викликати гемоліз
еритроцитів плода з групами крові А(ІІ), В(ІІІ).
Серед дітей-первістків гемолітична анемія як
результат АВО-конфлікту буває з такою ж час
тотою, як і у дітей, народжених після других,
третіх і наступних пологів, на відміну від резус-конфлікту, при якому частота гемолітичної
анемії збільшується зі збільшенням кількості
пологів.
Імунний гемоліз еритроцитів може зумовлю
ватися такими видами антитіл:
• гемаглютинінами. Вони належать до імуноглобулінів класів IgM і IgG. Викликають
аглютинацію (склеювання) еритроцитів.
Відомі теплові й холодові гемаглютиніни
з оптимумом реакції зв’язування антигенів
при звичайній температурі тіла або при зни
женні температури до 32 °С (напр., у кін
цівках за холодної погоди). У цих умовах
механізм гемолізу еритроцитів - внутріш
ньоклітинний: агрегати еритроцитів при
проходженні через селезінку зазнають фа
гоцитозу, що його здійснюють макрофаги;
• гемолізинами. Вони є антитілами переваж
но класу IgG. Здатні фіксувати комплемент.
Відомі гемолізини з оптимумом реакції
зв’язування антигенів при звичайній тем
29
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
пературі тіла (теплові) і при зниженні тем
ператури крові (холодові).
Гемоліз еритроцитів під дією гемолізинів має
два механізми:
1)
внутрішньосудинний
комплементзалежний гемоліз. У результаті фіксації, а потім
і активації комплементу відбувається руй
нування мембрани еритроцитів;
2) внутрішньоклітинний — антитілоопосередкований фагоцитоз. Макрофаги своїми F рецепторами взаємодіють із Гс-фрагментами гемолізинів, фіксованих на еритро
цитах. При цьому або відбувається повне
поглинання еритроцитів макрофагами, або
макрофаг “відкушує” частину еритроцита,
перетворюючи його на мікросфероцит.
Набуті мембранопатії - це гемолітичні ане
мії, які виникають унаслідок набутих у проце
сі індивідуального розвитку дефектів мембран
еритроцитів.
Прикладом може бути пароксизмальна нічна
гемоглобінурія (хвороба Маркіафави - Мікелі).
Це захворювання виникає в результаті соматич
ної мутації кровотворних клітин, унаслідок якої
з’являються аномальні популяції еритроцитів,
лейкоцитів, тромбоцитів з дефектами мембра
ни. Вважається, що порушення мембран зазна
чених клітин пов’язані зі зміною співвідношен
ня жирових кислот, що входять до складу їхніх
фосфоліпідів (зменшується вміст ненасичених
і збільшується — насичених жирових кислот).
Еритроцити аномальної популяції набувають
здатності фіксувати комплемент, що є передумо
вою комплементзалежного гемолізу. Зменшення
pH середовища є чинником, який провокує вну
трішньосудинний гемоліз. Цим пояснюється
той факт, що руйнування еритроцитів розвива
ється найчастіше вночі (у нічний час pH крові
трохи зменшується).
Картина крові при набутій гемолітичній
анемії характеризується зменшенням кілько
сті еритроцитів і концентрації гемоглобіну, хо
ча при внутрішньосудинному гемолізі вміст
останнього може не зменшуватися за рахунок
гемоглобіну, що перебуває в плазмі крові. Ко
лірний показник, як правило, у нормі, однак
може бути й більший за одиницю, що пов’язано
із позаеритроцитарним гемоглобіном. У мазку
крові виявляється значна кількість регенера
торних форм еритроцитів: ретикулоцитів, поліхроматофілів, нормобластів, що свідчить про
регенераторний, а іноді й гіперрегенераторний
характер анемії.
У червоному кістковому мозку лейкоеритроцитарний індекс становить не 3:1, як у нормі,
а2:1, 1:1, 1:2. Такі зміни є ознакою посиленої
регенерації клітин еритроїдного паростка крові.
Показано, що здоровий червоний кістковий мо
зок може компенсувати 6-8-кратне збільшення
темпів руйнування еритроцитів без розвитку по
мітної анемії.
Спадкові гемолітичні анемії
Залежно від механізмів розвитку всі спадково
обумовлені гемолітичні анемії поділяють на три
групи:
1) мембранопатії. Основу цієї групи анемій
становлять дефекти мембран еритроцитів;
2) ферментопатії (ензимопатії). Ці анемії
обумовлені
спадковими
порушеннями
ферментів еритроцитів;
3) гемоглобінопатії*— анемії, що виникають
унаслідок якісних змін гемоглобіну.
Мембранопатії
Розвиток цієї групи анемій пов’язаний зі спад
ково зумовленими порушеннями цитоскелета
і мембрани еритроцитів, унаслідок чого зміню
ються форма клітин, втрачаються такі важливі
властивості, як пластичність (здатність зміню
вати свою форму під дією зовнішніх сил), осмо
тична резистентність, тривалість життя.
Найпоширенішою формою мембранопатій
є сфероцитоз і, зокрема, мікросфероцитарна
анемія Мінковського — Шоффара. У % випад
ків сфероцитоз є аутосомно-домінантною хво
робою, решта тяжких варіантів перебігу цієї
анемії припадає на аутосомно-рецесивний тип
спадкування.
Генетичні дефекти, що зумовлюють розвиток
сфероцитозу, пов’язані з білками цитоскелета
еритроцитів (рис. 30.19). Форма, еластичність
і довговічність клітин червоної крові багато
в чому залежать від фізично-хімічних власти
востей цитоскелета, що має численні контакти
з білками еритроцитарної мембрани.
Основним білком цитоскелета є спектрин,
молекули якого складаються з переплетених
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
зо
Рис. 30.19. Білки мембрани і цитоскелета еритроцита
а- і Р-ланцюгів. Велика кількість таких молекул
зв’язується в комплекси з олігомерами актину
і утворює власне спектрин-актиновий скелет клі
тин. “Прив’язування” цього скелета до мембрани
відбувається через два види контактів. Перший,
що здійснюється через білки анкирин і band 4.2,
з’єднує спектрин із трансмембранним білкомтранспортером band 3. Другий вид контактів за
безпечує зв’язок актину з іншим трансмембран
ним білком глікофорином А. Проміжною ланкою
в цьому контакті виступає протеїн 4.1.
Сьогодні з’ясовано, що спадковий сфероцитоз
може виникати через мутації, що відбуваються
в генах (1) анкирину, (2) а- і р-ланцюгів спектрину, (3) band 3 і (4) band 4.2, тобто в генах білків,
що беруть участь в утворенні першого виду кон
тактів між цитоскелетом і клітинною мембраною.
Найчастіша аутосомно-домінантна форма сфероцитозу пов’язана з мутаціями гена анкирину.
Незалежно від того, у яких молекулах цито
скелета виникає дефект, порушується стабіль
ність мембрани, що веде до відриву її фрагмен
тів при проходженні еритроцитів через вузькі
капіляри тканин та їхньому контакті з елемен
тами судинної стінки під час циркуляції в ар
теріальних судинах (дія механічного чинника).
При цьому змінюється співвідношення площі
мембрани до об’єму цитоплазми таким чином,
що єдино можливою формою клітин, у якій міг
би ще вміститися весь об’єм цитоплазми, стає
сферична. Іншими словами, унаслідок втрати
частини мембрани еритроцити, що мають нор
мальну дископодібну форму, перетворюються
на сфероцити.
Такі зміни мають щонайменше два патогене
тичні наслідки.
1. Тривала затримка в селезінці. Еритроци
ти, ставши сфероцитами, втрачають свою
пластичність, а отже, і здатність до дефор
мації. Через це вони не можуть проходити
через вузькі міжендотеліальні щілини ве
нозних синусів селезінки й на тривалий час
затримуються в ній. Макрофаги селезінки,
довго контактуючи з такими клітинами,
“відкушують” частину мембрани еритроци
тів і перетворюють останніх на мікросфероцити.
При подальших проходженнях мікросфероцитів через селезінку макрофаги повністю фагоцитують змінені еритроцити — відбувається
внутрішньоклітинний гемоліз. Таким чином,
тривалість життя еритроцитів зменшується до
8-12 діб замість 120.
З урахуванням цієї ланки патогенезу непога
ний лікувальний ефект має видалення селезін
ки — спленектомія.
2.
Порушення
енергозабезпечення.
Через
тривале перебування сфероцитів у ткани
нах селезінки та виведення їх з циркуляції
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
порушується надходження глюкози всере
дину клітин і виведення молочної кислоти
(кінцевого продукту гліколізу) з еритро
цитів. Це веде до (а) внутрішньоклітин
ного ацидозу і (б) зменшення ресинтезу
АТФ. Ацидоз є чинником ушкодження
мембрани (див. главу 7), а дефіцит АТФ
спричиняється до пригнічення діяльності
Na-K-насоса еритроцитів. Порушення ви
ведення іонів Na+ з клітин назовні веде до
зростання осмотичного тиску всередині
еритроцитів і, як наслідок, до переміщення
води в цитоплазму. Це зумовлює ще один
механізм руйнування сфероцитів — осмо
тичний внутрішньосудинний гемоліз.
Ферментопатії
Метаболізм, що відбувається в еритроцитах, не
є таким багатим, як у багатьох інших видах клі
тин організму. Проте він представлений кілько
ма важливими шляхами для існування й вико
нання транспортної функції.
Причиною розвитку спадкових ферментопатій можуть бути порушення таких ферментних
систем еритроцитів:
1) пентозного циклу. Найпоширенішою ферментопатією є глюкозо-6-фосфатдегідрогеназодефіцитна анемія, обумовлена пов
ного відсутністю або значним зменшенням
активності глюкозо-6-фосфатдегідрогенази. Відомо близько 250 мутантних форм
цього ферменту (див. далі);
2) гліколізу. Описано спадкові дефекти 11 з 13
ферментів гліколізу. Найбільш поширеним є
дефіцит піруваткінази. Основою патогене
зу анемій, що виникають у результаті пору
шень реакцій гліколізу, є дефіцит АТФ, що
закономірно призводить до порушення енер
гозабезпечення Na-K-насосів плазматичних
мембран. Наслідком цього є збільшення
концентрації іонів натрію і, як результат,
осмотичного тиску всередині еритроцитів,
що, своєю чергою, викликає надходження
води в клітини. При цьому еритроцити на
бухають і перетворюються на сфероцити,
які зазнають фагоцитозу з боку макрофагів;
3) циклу глютатіону (глютатіонсинтетази,
глютатіонредуктази, глютатіонпероксидази). Порушення цього циклу призводять
до пригнічення роботи глютатіонперок-
31
сидазної антиоксидантної системи, на
слідком чого є активація процесів пероксидного окиснення ліпідів в еритроцитах.
Реакції вільнорадикального окиснення
ліпідів мембран порушують їхні бар’єрні
властивості й значно збільшують проник
ність еритроцитарної мембрани до іонів,
зокрема, іонів натрію. Останні за граді
єнтом концентрації надходять у клітини,
збільшуючи осмотичний тиск усередині
еритроцитів. Подальші процеси аналогічні
тим, які розвиваються при інших описаних
тут ферментопатіях;
4) утилізації АТФ. Прикладом цієї групи
ферментопатій є дефіцит білкових ком
понентів Na-K-АТФ-ази еритроцитарної
мембрани. Первинні порушення роботи
Na-K-насосів
еритроцитів
призводять
до збільшення концентрації іонів натрію
в клітинах з усіма наслідками, що звідси
випливають.
Серед спадкових ферментопатій найчасті
ше, і то в певних регіонах земної кулі (країни
середземноморського
басейну,
представни
ки негроїдної раси в Північній Америці), зу
стрічаються різні клінічні форми глюкозо6-фосфатдегідрогеназодефіцитної
анемії
(Г бФДД-анемїї).
Ця анемія успадковується за рецесивним,
зчепленим з Х-хромосомою типом. Генетичні
дефекти виявляють себе тим, що порушується
фолдинг (формування остаточної просторової
структури) молекул синтезованого ферменту,
унаслідок чого відбувається руйнування (проте
оліз) більшої чи меншої частини ензиму в протеосомах клітин, що є попередниками еритро
цитів. Як результат, зрілі клітини червоної крові
мають значно меншу, ніж потрібно, кількість
цього ферменту, і його не вистачає на весь пері
од життя еритроцитів. Цим власне пояснюється
те, що в процесі фізіологічного старіння еритро
цитів істотно зростає їхня чутливість до факто
рів, що провокують гемоліз (див. нижче).
Еритроцити, будучи переносниками кис
ню, постійно перебувають в умовах, сприятли
вих для активації процесів вільнорадикального
окиснення. Саме запобіганню цьому слугує сис
тема антиоксидантного захисту, представлена
в еритроцитах глютатіонпероксидазною систе
мою (див. главу 7). Ефективне її функціонування
32
є можливим лише за умов достатньої активності
пентозного циклу (рис. 30.20).
Дефіцит глюкозо-6-фосфатдегідрогенази стає
причиною порушень прямого окиснення глю
кози, унаслідок чого зменшується утворення
НАДФН і відновлення (регенерація) глютатіону
в еритроцитах. Брак відновленого глютатіону
унеможливлює інактивацію пероксидних спо
лук, зокрема Н О що утворюються в реакціях
вільнорадикального окиснення за участю моле
кулярного кисню (02) і заліза (Fe2+) - компонен
та молекул гемоглобіну.
Безпосередньою причиною гемолізу еритро
цитів при ГбФДД-анемії є оксидаційний стрес
(див. главу 7). Його можуть започатковувати:
а) лікарські препарати (напр., протималярій
ні засоби, сульфонаміди, антибіотики ряду
нітрофурантоїну та ін.);
б) деякі харчові продукти. Одним з таких
є боби Vicia fava, які містять речовини з ви
раженими окисними властивостями. У ре
гіонах, де споживання бобових є дуже по
ширеним, ендемічна форма ГбФДД-анемії
отримала назву фавізм;
в) інфекції, що супроводжуються вираженою
лейкоцитарною реакцією - продукуванням
і вивільненням у тканини вільних радика
лів і пероксидів. Окисний гемоліз у хворих
часто провокується при вірусному гепатиті,
пневмоніях, тифозній гарячці.
У патогенезі гемолізу еритроцитів при
ГбФДД-анемії важливе значення мають два ме
ханізми:
1. Пероксидне окиснення ліпідів мембран.
Цей процес, що є компонентом оксидаційного стресу, призводить до збільшення про
никності еритроцитарних мембран і надхо
дження в клітини іонів натрію й води. Роз
вивається набухання й набряк еритроцитів,
а потім — їх осмотичний внутрішньосудинний гемоліз.
2. Денатурація молекул гемоглобіну. При
збільшенні вище певного критичного рів-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ня кількості вільних радикалів і пероксидів
відбувається окиснення SH-груп ланцю
гів гемоглобіну, що веде до його денату
рації й утворення зв’язаних з мембраною
преципітатів, відомих як тільця Гайнца
(рис. 30.21). Поява останніх може мати такі
наслідки для еритроцитів:
а) пряме ушкодження мембрани, що зумов
лює внутрішньосудинний гемоліз',
б) у менш тяжких випадках зменшується плас
тичність еритроцитів і їхня здатність до де
формації. Це веде до затримки еритроцитів
у тканинах селезінки, до тривалого контак
ту їх з макрофагами. Як наслідок, остан
ні “відкушують” частини еритроцитів, що
містять тільця Гайнца, - з’являються “по
кусані” червонокрівці неправильної форми.
Частина з них поновлює свою цілісність,
перетворюючись у сфероцити. І “покусані”
еритроцити, і сфероцити у підсумку зазна
ють внутрішньоклітинного гемолізу в се
лезінці.
Гемоглобінопатп
Гемолітичні анемії, пов’язані з порушеннями
структури молекул гемоглобіну, отримали назву
гемоглобінопатій.
Гемоглобін, як відомо, являє собою тетрамер,
утворений двома парами однакових поліпептидних ланцюгів (а і (З), кожен з яких зв’язаний
з гем-групою. До складу останньої входить за
лізо (Fe2+) (рис. 30.22). У дорослих 96 % гемо
глобіну припадає на НЬА (а2Р2), 3 % - на НЬА2
(а282) і 1 % - на фетальний гемоглобін HbF (а2у2).
В основі спадкових гемоглобінопатій можуть
бути дві групи порушень: якісні та кількісні.
Сутність якісних гемоглобінопатій полягає
в порушенні первинної структури ланцюгів ге
моглобіну, наприклад, заміна амінокислот, по
довження або вкорочення ланцюгів молекул
гемоглобіну. Найпоширенішою клінічною фор
мою цієї групи є серпоподібноклітинна анемія.
На сьогодні ідентифіковано кілька сотень ано
мальних гемоглобінів, що виникають унаслідок
точкових мутацій або делеції в генах гемоглобінових ланцюгів.
Кількісні
гемоглобінопатії
характеризують
ся порушеннями утворення окремих видів лан
цюгів гемоглобіну. Прикладом можуть бути аі /3-таласемїї.
33
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
0. Функціонування антиоксидантноїглютатіонпероксидазноїсистеми в еритроцитах
Серпоподібноклітинна анемія. Причиною цієї
анемії є генетичний дефект, сутність якого по
лягає в тому, що в результаті точкової мутації
відповідного гена в р-ланцюгу молекули гемо
глобіну в 6-му положенні від N-кінця глютамі
нову кислоту заміщено на валін. Це призводить
до появи патологічної форми гемоглобіну, яку
позначають як HbS. Оскільки валін має непо
лярний радикал, що міститься на поверхні моле
кули, то в результаті заміни амінокислот розчин
ність гемоглобіну різко падає. У цьому полягає
основна функціональна відмінність HbS від зви
чайних форм гемоглобіну. У відновленому стані
розчинність HbS зменшується майже в 100 разів.
Це призводить до того, що HbS випадає в осад утворюються кристали, які деформують еритро
цити. Як наслідок, клітини червоної крові набу
вають серпоподібної форми (див. рис. 30.9).
Серпоподібноклітинна анемія успадковуєть
ся за типом неповного домінування. У гомо
зигот за патологічним геном увесь гемоглобін
представлено HbS. У гетерозигот на HbS припа
дає до 40 %, решта - це нормальний гемоглобін
НЬА. Заповнені HbS (чи сумішшю нормального
НЬА і HbS) еритроцити мають коротший строк
життя і швидше руйнуються в селезінці. Це дає
перевагу гетерозиготам у регіонах з високою за
хворюваністю на малярію, оскільки мерозоїти
малярійного плазмодія не встигають завершити
свій розвиток у таких еритроцитах.
Кристалізація HbS і зміна форми еритроцитів
з нормальної на серпоподібну є процесом обо
ротним і залежить від оксигенації і деоксигенацїї гемоглобіну. За великої кількості таких цик
лів серпоподібність клітин стає необоротною.
Необоротним змінам еритроцитів сприяють
(рис. 30.23):
а) гостра гіпоксія різного походження;
б) зменшення водневого показника (pH) у кро
ві й тканинах;
в) проходження еритроцитів через капіляри
тканин з низькими фізіологічними значен-
34
Рис. 30.21. Тільця Гайнца в еритроцитах
при ГбФДД-анеми
нями показника напруги кисню (р02) (жи
рова тканина, селезінка);
г) збільшення часу проходження еритроцитів
через судини мікроциркуляторного русла,
що є характерним для порушень місцевого
кровообігу, зокрема венозної гіперемії (див.
главу 16);
д) дегідратація еритроцитів, унаслідок якої
вони ущільнюються і в них зростає кон
центрація гемоглобіну. Дегідратація клітин
виникає через порушення бар’єрних влас
тивостей деформованих кристалами HbS
мембран, у результаті чого іони К+, а за
ними і вода виходять з еритроцитів.
У патогенезі змін, що виникають при серпоподібноклітинній анемії, виділяють дві основні
ланки.
1. Гемоліз еритроцитів (хронічна гемолітич
на анемія). Еритроцити зі зміненою, серпо
подібною формою втрачають здатність до
деформації, а тому важко проходять через
вузькі міжендотеліальні проміжки веноз
них синусів селезінки і надовго затриму
ються там. Цим пояснюється інтенсивний
фагоцитоз серпоподібних еритроцитів ма
крофагами (внутрішньоклітинний гемоліз),
що власне й зумовлює зменшення кількос
ті червонокрівців та гемоглобіну в одини
ці об’єму крові (анемію) і появу клінічних
ознак, характерних для гемолітичних ане
мій (див. нижче).
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Рис. 30.22. Будова молекули гемоглобіну
2. Трофічні порушення в тканинах (мікротромбози, некрози). їхній розвиток пов’язаний
з еритроцитами, що мають як необоротну,
так і оборотну серпоподібність.
Ущільнення, втрата пластичності й здатності
до деформації унеможливлюють проходження
серпоподібних еритроцитів через вузькі капіля
ри. Останні забиваються такими клітинами, ко
трі й зупиняють рух крові по мікросудинах.
Свій внесок до розвитку трофічних пору
шень додають і еритроцити, які ще зберігають
нормальну форму (оборотна серпоподібність).
Встановлено, що такі клітини, на відміну від
нормальних з НЬА, мають на зовнішній по
верхні своєї мембрани значно більшу кількість
адгезивних молекул, здатних взаємодіяти з від
повідними молекулами ендотеліальних клітин.
Завдяки цьому значна кількість еритроцитів
з HbS, що мають звичайну форму, затримуєть
ся на поверхні ендотелію капілярів, зменшуючи
при цьому просвіт мікросудин і створюючи пе
решкоду для руху крові.
Крім того, унаслідок збільшення часу перебу
вання HbS в капілярах відбувається його повна
деоксигенація і він кристалізується. Це, своєю
чергою, стає причиною необоротної зміни фор
ми еритроцитів на серпоподібну з усіма подаль
шими наслідками.
Таласемія. Анемії під назвою “таласемії”
є кількісними гемоглобінопатіями, оскільки
35
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Рис. 30.23. Патогенез порушень, що виникають при серпоподібноклітинній анемії
їхній розвиток пов’язаний з порушеннями син
тезу певних поліпептидних ланцюгів гемогло
біну.
У дорослої людини основною формою ге
моглобіну є НЬА, білкова частина якого скла
дається з двох а- і двох р-ланцюгів (ааРР). Поліпептидні Р-ланцюги кодуються двома алельними генами, що містяться в 11-й хромосомі,
а а-ланцюги - чотирма (двома парами) генами,
локалізованими в 16-й хромосомі.
Залежно від того, синтез яких ланцюгів пору
шено, розрізняють Р- і а-таласемію.
/З-Таласемія. Розрізняють два варіанти цієї ане
мії: (1) Р°-таласемію, коли у гомозигот повністю
відсутній синтез Р-ланцюгів, і (2) [Ґ-таласемію,
за якої нормальні Р-ланцюги утворюються, але
в значно меншій кількості.
Причиною цих форм Р-таласемії є різні точ
кові мутації, кількість яких ще донедавна сяга
ла 100. Залежно від локалізації та своєї сутності,
вони можуть мати один із трьох наслідків:
1) порушення транскрипції. Виникають у разі
мутацій, локалізованих у промоторній час
тині гена. Найчастіше зменшується спорід
неність промотора з РНК-полімеразою, що
веде до значного (на 75-80 %) зменшення
швидкості транскрипції, а отже, і синтезу
нормальних Р-ланцюгів, - розвивається Р+таласемія;
2) порушення сплайсингу РНК. Більшість му
тацій з такими наслідками локалізована
в інтронах гена. Якщо порушується вирі
зання якогось інтрона, то така мРНК за
знає деградації ще в ядрі, і Р-ланцюг з неї
не синтезується (Р°-таласемія). Якщо ж
всередині інтрона виникає ще один сайт
для зв’язування відповідного фермен
ту при збереженні нормальних сайтів, то
якась частина мРНК після сплайсингу буде
зміненою (міститиме ділянку невирізано-
36
го інтрона), а якась залишиться нормаль
ною (якщо сплайсинг буде започатковано
із звичайних сайтів). У цьому разі синтез
Р-ланцюгів відбувається, хоча кількість їх
стає набагато меншою (Р+-таласемія);
3) порушення трансляції. До цього веде по
ява стоп-кодона в молекулі мРНК. Причи
ною можуть бути (а) заміна однієї азотис
тої основи іншою в якомусь з екзонів або
(б) втрата (делеція) чи вставляння (інсерція) азотистої основи, що веде до зміщен
ня рамки зчитування (див. главу 12).
При всіх варіантах Р-таласемії синтез а-ланцюгів не зазнає змін, а тому ще в попередниках
еритроцитів (еритробластах) створюється зна
чний їх надлишок. Вони об’єднуються в не
розчинні агрегати, що набувають у клітинах
вигляду включень, з якими пов’язані основні
патогенетичні механізми цієї анемії (див. далі).
Оскільки синтез у- і 8-ланцюгів також не по
рушується, то в клітинах червоної крові плода
і новонароджених фетального гемоглобіну HbF
(аауу) буде в достатній кількості, а у дорос
лих можна спостерігати значне компенсаторне
збільшення його концентрації. Це ж саме стосу
ється і гемоглобіну А2 (аа88).
Патогенез р-таласемії складний і пов’язаний
з двома основними механізмами анемій: (1) по
рушеннями еритропоезу і (2) гемолізом еритро
цитів (рис. 30.24).
1. Ще на стадії еритробласта через відсутність
або значний дефіцит Р-ланцюгів не синтезу
ється або зменшується утворення НЬА, вна
слідок чого для клітин усього ряду червоної
крові характерною ознакою стає гіпохромія.
Водночас великий надлишок а-ланцюгів
стає причиною їхньої агрегації. Такі агрега
ти через не зовсім поки що зрозумілу сис
тему (можливо, ушкоджуються клітинні
мембрани) запускає механізми апоптозу
(див. главу 7), і більшість еритробластів (від
70 до 85 %) гине, припиняючи кровотворен
ня на цьому етапі. Отже, можна вести мову
про неефективний еритропоез, як про один
із патогенетичних механізмів анемії.
2. З тих еритробластів, що не загинули, утво
рюється якась кількість зрілих еритроцитів.
Однак вони не повноцінні, оскільки також
містять агрегати а-ланцюгів. Ці агрега
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ти концентруються в центральній частині
клітин, що надає останнім мішенеподібної
форми. Пластичність, а отже, здатність та
ких еритроцитів до оборотної деформації
порушуються, що стає причиною їхньої за
тримки в тканинах селезінки та поглинан
ня макрофагами. Внутрішньоклітинний
гемоліз, що відбувається при цьому, робить
свій внесок у патогенез анемії.
Анемія через зумовлене нею кисневе голо
дування органів і тканин спричиняється до двох
відносно специфічних для Р-таласемії синдромів:
а) при кров’яній формі гіпоксії значно змен
шується утворення в печінці гепцидину
білка, що регулює вміст заліза в крові через
гальмування його всмоктування в кишковику й вивільнення із клітин, де залізо де
понується (такими зокрема є макрофаги).
(Докладно про гепцидин мова піде далі.)
Зменшення гепцидину закономірно веде до
зростання всмоктування аліментарного за
ліза та посиленого вивільнення у кров того
заліза, що було депоноване у макрофагах
після перетравлювання гемолізованих ери
троцитів. Як наслідок, розвивається пере
вантаження організму залізом, що виявляє
себе розвитком вторинного гемохроматозу
(див. далі);
б) клітини нирок у відповідь на гіпоксію
утворюють і вивільнюють у кров великі
кількості еритропоетину. Останній, діючи
на еритропоетинчутливі клітини, значно
посилює еритропоез. Ступінь посилення
цього процесу такий, що відбувається роз
ростання (гіперплазія) і збільшення об’єму
червоного кісткового мозку через зменшен
ня товщини і маси кортикальних пластин
кісток. Крім того, з’являються осередки
екстрамедулярного кровотворення, зокрема
в печінці і селезінці. Порушення кісткової
тканини, зумовлені експансією червоного
кісткового мозку, стають причиною пригні
чення росту кісток та різних їхніх аномалій,
що ведуть до деформації скелета.
Варто зазначити, що розвиток розглянутих
двох синдромів зумовлюється по суті звичайни
ми фізіологічними механізмами, що вмикають
ся при анеміях різного походження. Як прави
ло, ці механізми мають захисне, компенсаторне
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
37
Схема патогенезу р-таласемії
значення. Проте в умовах Р-таласемії через не
ефективність еритропоезу вони стають не тіль
ки зайвими, а й шкідливими, оскільки ведуть до
вторинних, власне патологічних змін.
Вираженість порушень в організмі при Р-тала
семії залежить від суті генетичного дефекту (Р°
чи р+) та від генотипу за відповідним геном. По
вна картина хвороби (велика таласемія) розви
вається в гомозигот за патологічним геном (р°/
Р° і Р+/р+). У гетерозигот (Р°/р і Р+/р) наявність
нормального гена може забезпечувати синтез
такої кількості р-ланцюгів, що клінічно прояви
хвороби мало виражені (мала таласемія) або їх
зовсім немає.
а-Таласемія. Ця
хвороба
характеризується
+
зменшенням синтезу (а -таласемія) або повним
припиненням утворення (а°-таласемія) глобінових а-ланцюгів.
На відміну від Р-таласемії, причиною недуги
є не точкові мутації, а втрата (делеція) генів, що
кодують структуру а-ланцюга. Кожен з таких чо
38
тирьох генів (див. вище) в нормі є відповідаль
ним за синтез приблизно 25 % продукту. Звідси
випливає, що вираженість хвороби напряму за
лежить від кількості втрачених генів (див. далі).
При а-таласемії, як і при Р-варіанті, (1) змен
шується утворення нормального гемоглобіну НЬА та (2) утворюється надлишок інших, ніж
а, глобінових ланцюгів. У новонароджених
це виявляє себе появою тетрамерів (уууу), що
отримали назву гемоглобіну Барта, а у дорос
лих - інших тетрамерів (РРРР), відомих як НЬН.
Оскільки Р-, у- і 5-глобінові ланцюги більш роз
чинні, ніж а-ланцюги, то менша кількість їхніх
агрегатів зумовлює меншу ушкоджувальну дію,
а отже, за однакового ступеня генетичних по
рушень вираженість неефективного еритропоезу та інтенсивність гемолізу при а-таласемії
є меншими, якщо порівнювати з Р-таласемією.
Клінічні прояви хвороби визначаються обся
гом генетичних дефектів. Розрізняють:
» безсимптомну форму (а-таласемія-2). Вона
виникає при делеції одного з чотирьох генів.
Три інших забезпечують синтез достатньої
кількості нормальних а-ланцюгів;
» малу форму (а-таласемія-1). Є наслідком
втрати двох із чотирьох генів, прояви ане
мії незначні;
• гемоглобінопатію Н. Делеція трьох із чо
тирьох генів має істотні наслідки. Один
нормальний ген, що залишився, не в змо
зі забезпечити синтез достатньої кількос
ті а-ланцюгів, а отже, і утворення НЬА.
В умовах надлишку Р-ланцюгів з’являється
НЬН (РРРР), який є функціонально непо
вноцінним, оскільки має дуже високу спо
рідненість з киснем і не може його віддава
ти тканинам. Накопичення великої кількос
ті окисненого НЬН веде до його агрегації,
зміни форми і пластичності еритроцитів,
унаслідок чого такі клітини вилучаються із
крові макрофагами при проходженні через
селезінку - настає гемоліз;
• водянку плода. Причиною є втрата всіх
чотирьох генів. Такий гомозиготний стан
несумісний із життям і найчастіше закін
чується внутрішньоутробною загибеллю
плода. В еритроцитах пуповинної крові
знаходять гемоглобін Барта (уууу), окиснена форма якого не здатна віддавати кисень
тканинам. За повної відсутності нормаль
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
них HbF і НЬА настає аноксія з відповідни
ми наслідками.
На завершення огляду гемолітичних анемій,
наведемо їхні основні клінічні прояви.
Крім гематологічних ознак - змін пери
феричної крові та червоного кісткового мозку,
що можуть мати свої особливості залежно від
форми хвороби (див. вище), - для гемолітичних
анемій характерні такі клінічні ознаки й син
дроми:
а) гіпоксія. Вона зумовлена анемією й виявляє
себе різкою слабкістю, неприємними відчут
тями в ділянці серця, серцебиттям, задиш
кою;
б) гемолітична (надпечінкова) жовтяниця
(див. главу 35);
в) посилене утворення жовчних каменів, особ
ливо білірубінових, що пояснюється зна
чним збільшенням вмісту білірубіну в жов
чі та збільшенням її в’язкості;
г) гемоглобінурія. Розвивається при внутрішньосудинному гемолізі. Гемоглобін, що
вивільняється зі зруйнованих еритроци
тів, зв’язується з білком плазми крові гаптоглобіном. 100 мл плазми крові містить
стільки гаптоглобіну, що він може зв’язати
125 мг гемоглобіну. Якщо ж концентрація
гемоглобіну в плазмі перевищує 125 мг %,
то незв’язаний гемоглобін проходить через
нирковий фільтр і з’являється в сечі;
д) спленомегалія. Збільшення селезінки є ха
рактерним для внутрішньоклітинного меха
нізму гемолізу еритроцитів. В основі цього
явища лежить підвищення функціональної
активності макрофагів, що викликає їхню
інтенсивну проліферацію. Спленомегалія
часто супроводжується збільшенням печін
ки (проліферацією печінкових макрофагів);
е) гемосидероз - відкладення гемосидерину
в макрофагах. Гемосидерин - це частково
денатурований і депротеїнізований феритин, тобто білок, що містить багато заліза
в негемовій формі (вміст Fe у гемосидерині - 25-30 %);
є) порушення мікроциркуляції. Часто виника
ють при інтенсивному внутрішньосудинному гемолізі й обумовлені розвитком ДВЗсиндрому (див. главу 30.4);
ж) гарячка. Розвивається в результаті різкої
активації фагоцитарної функції макрофагів,
39
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
унаслідок чого вони вивільняють інтерлейкін-1 (див. главу 18).
30.2.2.3. І Анемії, пов’язані
з порушеннями кровотворення
Класифікація
I. За походженням анемії, зумовлені розлада
ми еритропоезу, можуть бути:
• набутими;
• спадково обумовленими.
II. За причинами виникнення виділяють такі
групи анемій:
•
мієлотоксичні. Розвиваються внаслідок
ушкодження кровотворних клітин під дією
екзогенних (радіація, хімічні агенти, віру
си) та ендогенних чинників (імунні факто
ри, токсичні продукти обміну речовин);
• дефіцитні. Причиною їх виникнення є де
фіцит факторів, необхідних для кровотво
рення, - недостатність заліза, фолієвої кис
лоти, білків, вітамінів Вб і В12;
• дисрегуляторні. Виникають як наслідок
розладів регуляції еритропоезу (порушен
ня співвідношення між еритропоетинами
й інгібіторами еритропоезу при недостат
ності нирок, ушкодження елементів стро
ми, що є мікрооточенням для кровотворних
клітин);
• пов’язані зі зменшенням плацдарму ерит
ропоезу. Є наслідком заміщення кровотвор
ної тканини лейкозними клітинами, спо
лучною тканиною (фіброз), метастазами
пухлин.
III. Залежно від сутності процесів, що лежать
в основі розвитку анемій, виділяють:
• порушення утворення еритроцитів', дефі
цит кровотворних клітин унаслідок їхньо
го ушкодження або заміщення, порушення
розмноження клітин кровотворення (роз
лади ресинтезу ДНК), дефекти дозрівання
еритроцитів і виходу їх у кровоносні суди
ни (неефективний еритропоезу,
•
порушення синтезу гемоглобіну: дефі
цит заліза, розлади синтезу порфіринів
(спадкові порушення ферментів, отруєн
ня свинцем, дефіцит вітаміну В6, розлади
синтезу білкових ланцюгів молекул гемо
глобіну).
Серед анемій, зумовлених порушеннями еритропоезу, найбільше клінічне значення мають
(а) залізодефіцитна, (б) Вп-фолієводефіцитна
і (в) гіпопластична анемії.
Залізодефіцитна анемія
В організмі дорослої людини міститься від 2 г
(у жінок) до 6 г (у чоловіків) заліза. Воно в гемовій і негемовій формах входить до складу ці
лого ряду білків.
У формі гему залізо міститься в таких проте
їнах, як (а) гемоглобін, (б) міоглобін, (в) цитохроми; (г) каталаза, (д) лактоферин. У негемовій
формі залізо є компонентом (а) феритину, (б) гемосидерину; (в) трансферину, (г) ферментів: аконітази, ксантиноксидази і НАДН-дегідрогенази.
За участю у фізіологічних і біохімічних про
цесах розрізняють (а) функціонально активне та
(б) депоноване залізо. 80 % першого припадає
на гемоглобін, решта 20 % - на міоглобін та фер
менти.
Депонування заліза відбувається у двох біл
ках: феритині і гемосидерині. У складі цих комп
лексів міститься до 20 % від загальної кількості
заліза в організмі. Феритин знаходять у клітинах
майже всіх тканин, але основна його кількість
припадає на печінку, селезінку і червоний кіст
ковий мозок. У печінці феритин міститься пере
важно у гепатоцитах (якась кількість перебуває
і в клітинах Купфера), а в селезінці та кровотвор
ній тканині - усередині макрофагів. В останніх
незначна кількість феритину в умовах норми
зазнає денатурації й часткової деградації з утво
ренням гемосидерину.
Баланс заліза в організмі залежить від його над
ходження ззовні та від його втрат. Спеціальних
фізіологічних механізмів виведення заліза (екс
креції) не існує. Воно втрачається в нормі тіль
ки в результаті злущування епітеліальних клітин
(зокрема ентероцитів), що містять певну кіль
кість феритину (щодоби організм у такий спосіб
втрачає 1-2 мг заліза), а у жінок - ще й унаслідок
фізіологічних крововтрат (менструацій).
Збалансований добовий раціон сучасної лю
дини містить від 10 до 20 мг заліза, що у гемовій
формі входить до продуктів тваринного похо
дження, і різну кількість негемового заліза, що
є в рослинній їжі. Гемове залізо всмоктується на
20 %, а тому нормального його вмісту в їжі ціл
ком достатньо, щоб компенсувати добові втра
40
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ти. Значення негемового заліза рослин невелике,
оскільки тільки 1-2 його відсотки всмоктується
в кишках.
Надійшовши в організм, залізо раціонально
використовується, чому слугує особливий фізіо
логічний цикл (рис. 30.25). Після всмоктування
залізо зв’язується зі спеціальним транспортним
білком плазми крові — траснферином. У його
складі воно переноситься в червоний кістковий
мозок, де кровотворні клітини - попередники
еритроцитів - захоплюють трансферин, маючи
особливі мембранні рецептори до нього. Невилучена частина трансферину надходить у печін
ку, де відбувається депонування аліментарного
заліза у складі феритину.
У ході еритропоезу залізо йде на синтез гему
та гемоглобіну. Утворені зрілі еритроцити цир
кулюють у периферичній крові протягом 120
днів, після чого вони захоплюються макрофага
ми і зазнають гемолізу. Унаслідок деградації ге
моглобіну та розщеплення гему вивільнене залі
зо залишається в макрофагах у складі феритину.
При зменшенні забезпеченості організму за
лізом або при збільшенні потреб у ньому (на
приклад, після крововтрати) депоноване в гепатоцитах і макрофагах залізо може переходити
у кров і за допомогою трансферину переносити
ся до клітин, що його потребують.
Цикл заліза в організмі
Регуляція вмісту заліза в організмі орієнтова
на на підтримання такої його кількості, яка здатна,
перш за все, забезпечувати належне кровотворен
ня. Об’єктами такої регуляції є (1) всмоктування
заліза в кишках і (2) мобілізація заліза з місць
його депонування (з гепатоцитів і макрофагів).
Всмоктування заліза (рис. 30.26). Залізо, як
уже зазначалося, надходить в організм у складі
продуктів тваринного (гемова форма) і рослин
ного походження (негемова форма). У мембра
нах ворсинок на апікальній поверхні ентероцитів (здебільшого у дванадцятипалій кишці)
вмонтовані особливі транспортні білки, через
які переносяться всередину клітин гем і вільне
негемове залізо. Відповідні білки мають назву
транспортер гему і транспортер двовалент
них металів 1 (ТДМ 1). Останній може перено
сити тільки Fe2+, а тому всмоктуванню вільного
заліза має передувати його відновлення до Fe2+,
яке здійснюється за участю мембранного ентероцитарного цитохрому В. Далі залізо (Fe2+), ви
вільнене із гему й те, що надійшло через ТДМ
1, транспортується з ентероцитів назовні за до
помогою вмонтованого у мембрану базальної
частини клітин транспортного білка - феропортину 1. Після окиснення ще одним мембранним
ферментом - гефестином - залізо у формі Fe3+
зв’язується з трансферином і переноситься до
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
місць використання (червоний кістковий мозок)
і депонування (печінка). Те залізо, що не змогло
бути перенесеним у кров через феропортин 1,
депонується в ентероцитах у складі феритину.
На початку 2000-х років було відкрито меха
нізм, за допомогою якого відбувається регуляція
процесів всмоктування заліза. Виявилося, що
в печінці утворюється протеїн гепцидин, який,
зв’язуючись з феропортином 1, зменшує перене
сення заліза через базальну частину мембрани
ентероцитів (рис. 30.27). Показано, що приєд
нання гепцидину до феропортину 1 веде до фосфорилювання останнього, після чого той вилу
чається з мембрани і зазнає протеолізу в протеасомах. При зменшенні утворення гепцидину, що
настає при гострій вираженій гіпоксії (наприклад,
після великої крововтрати), кількість умонтова
них у мембрану молекул феропортину 1 зростає
і всмоктування заліза в кишковику може сягати
максимально можливого рівня. Навпаки, посиле
на секреція гепцидину пригнічує всмоктування
заліза й може спричинятися до залізодефіцитного стану навіть при достатньому вмісті цього еле
мента в продуктах харчування (див. далі).
Депонування заліза. Вихід Fe2+ з гепатоцитів
та макрофагів у кров також відбувається через
феропонтин 1 і регулюється гепцидином згідно
з тим самим механізмом, що й в ентероцитах.
Причини. Дефіцит заліза в організмі, що веде
до розвитку анемії, може бути зумовлений пев
ними причинами.
Аліментарна недостатність. Вважається,
що для підтримання нормального балан
су заліза в організм, його щоденне над
ходження у складі їжі має становити не
менше 1 мг. З урахуванням того, що тільки
15-20 % заліза харчових продуктів може
всмоктуватися, добова потреба в цьому
елементі складає від 7 до 10 мг у чоловіків
і від 7 до 20 мг — у жінок. Якщо продукти
містять заліза менше, ніж потребує орга
нізм, то розвивається залізодефіцит. Такий
стан може виникати у грудних дітей, що ви
годовуються коров’ячим чи козячим моло
ком, оскільки залізо, що міститься в ньому,
погано всмоктується. У багатьох країнах
світу з низьким рівнем економіки та вели
кою кількістю бідних людей дефіцит за
ліза пов’язаний з незбалансованим харчу
ванням - відсутністю в раціоні продуктів
41
тваринного походження, що містять залізо
у формі гему.
2. Порушення всмоктування. Основною при
чиною є ураження органів травної систе
ми, за яких зазнають розладів секреторна,
рухова і всмоктувальна функції. Це зокре
ма є характерним для станів після резекції
шлунка і великої частини кишок, гастритів,
ентеритів.
3. Крововтрати. У високорозвинених країнах
вони є найпоширенішою причиною дефіци
ту заліза. До нього ведуть невеликі, але по
вторні зовнішні кровотечі, що бувають при
багатьох хворобах органів травлення (напр.,
виразкова хвороба шлунка і дванадцятипа
лої кишки, геморагічний гастрит, злоякісні
пухлини шлунка і кишок, геморой), сечо
виділення (напр., пухлини нирок, сечового
міхура) і жіночого статевого тракту (напр.,
менорагії, рак матки). Анемія, що при цьо
му розвивається, є хронічною постгеморагічною, проте в її патогенезі провідну роль
відіграє не зменшення об’єму крові як такої,
а втрата заліза, що порушує утворення ге
моглобіну.
4. Посилене використання заліза. Ризик дефі
циту заліза в організмі значно зростає в жі
нок під час вагітності й лактації, оскільки
материнське залізо йде на розвиток плода
і секретується в складі молока.
Патогенез. Недостатність заліза в організмі
веде до порушення синтезу гему і білків, що
містять залізо в гемовій формі (див. вище). Як
наслідок, виникають розлади тих функцій, у ви
конанні яких беруть участь ці білки. Звідси мож
на виділити кілька напрямів у патогенезі пору
шень, зумовлених дефіцитом заліза.
1. Порушення синтезу гемоглобіну стають пря
мою причиною зменшення концентрації цьо
го білка в крові, а отже, і анемії (див. далі).
2. Зменшення утворення цитохромів у мітохондріях веде до розладів клітинного
дихання (порушується утилізація кисню),
розвитку тканинної гіпоксії (див. главу 23).
3. Падіння активності каталази супроводжу
ється порушенням функції антиоксидант
них систем. Це стає причиною активації
реакцій вільнорадикального окиснення
і веде ро ушкодження клітин (див. главу 7).
За таким механізмом можуть виникати
42
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Ентероцити дванадцятипалої кишки
Втрата зі злущеним епітелієм
Рис. 30.26. Регуляція всмоктування заліза у дванадцятипалій кишці
гемоліз еритроцитів і дистрофічні зміни
у тканинах.
4. Зменшення синтезу міоглобіну знижує
пристосувальні можливості м’язових клі
тин щодо гіпоксії та може ставати причи
ною слабкості й швидкої втомлюваності
м’язів.
Картина крові та червоного кісткового мозку.
Для залізодефіцитної анемії характерне зни
ження концентрації гемоглобіну в периферич
ній крові та зменшення колірного показника, що
свідчить про меншу насиченість кожного окре
мого еритроцита гемоглобіном. Кількість ери
троцитів в одиниці об’єму крові при цьому або
трохи зменшується, або залишається без змін.
При біохімічних дослідженнях виявляється
зниження вмісту заліза в сироватці крові, а та
кож ступеня насичення залізом трансферину
(сидеропенія).
У мазках крові зменшується кількість реге
нераторних форм еритроцитів (ретикулоцитів,
поліхроматофілів) і з’являються дегенеративні
форми. Характерні гіпохромія (з’являються на
віть анулоцити), анізоцитоз (мікроцитоз), пойкілоцитоз (див. рис. 30.10).
43
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Червоний
кістковий мозок
Пояснення ДИВ. у тексті. ТДМ1 - транспортер двовалентних металів 1
У червоному кістковому мозку зменшується
вміст сидеробластів і сидероцитів (еритроблас
тів і нормоцитів, що містять гранули заліза),
частка яких у нормі становить 20—40 %. У той
же час збільшується вміст базофільних і поліхроматофільних форм клітин еритроїдного
ряду при одночасному зменшенні оксифільних.
Зазначений феномен отримав назву “синього
кісткового мозку
Клінічні прояви. Основні прояви залізодефіцитної анемії зумовлені її патогенезом. їх можна
об’єднати в кілька синдромів.
Гематологічний синдром. Він охоплює
описані вище порушення з боку перифе
ричної крові та червоного кісткового мозку.
2. Гіпоксія. Виявляється загальною слабкістю,
запамороченнями, серцебиттям, задишкою,
непритомностями. Кисневе голодування
в умовах дефіциту заліза має два механіз
ми: кров яний (зменшення кисневої ємнос
ті крові) і тканинний (порушення клітин
ного дихання й утилізації кисню).
3. Синдром трофічних порушень. Виявляє
себе такими ознаками, як сухість і тріщини
шкіри, тріщини в кутиках рота, ангулярний
1.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
44
Участь гепцидину в регуляції вмісту заліза в крові
стоматит, ураження нігтів (зміни форми,
стоншення), атрофія сосочків язика (атро
фічний глосит), атрофічний гастрит. Вва
жають, що розвиток зазначених порушень,
з одного боку, пов’язаний з гіпоксичним
і вільнорадикальним ушкодженням клітин,
з другого — із розладами вторинних мета
болічних шляхів, у реалізації яких беруть
участь ферменти, що містять залізо.
4. Сидеропенічний синдром. Це специфічний
симптомокомплекс, що виникає при дефі
циті заліза. Його ознаками є спотворення
смаку й нюху. Хворі часто їдять крейду, зуб
ний порошок, вугілля, глину, пісок, лід, сирі
крупи, тісто, сирий м’ясний фарш. Мають
пристрасть до запахів гасу, бензину, ацето
ну, вихлопних газів автомобілів. Патогенез
зазначених порушень досі невідомий.
5. Синдром м’язової слабкості. М’язова слаб
кість, що виникає при дефіциті заліза, за
вжди більш значна, ніж та, якої варто було
б очікувати, зважаючи на ступінь анемії.
Для цього синдрому характерні слабкість
і стомлюваність скелетних м’язів, слабкість
міокарда (міокардіопатія), порушення ков
тання (дисфагія), розлади сечовипускання.
Очевидно, що важливе значення в розвит
ку зазначених симптомів мають гіпоксія
(кров’яна і тканинна), а також зменшення
вмісту міоглобіну в м’язовій тканині.
Залізорефрактерні анемії
До цієї групи належать анемії, що мають ознаки
залізодефіцитних, проте на відміну від остан
ніх погано піддаються або зовсім не піддаються
лікуванню введенням препаратів заліза ззовні
(звідси назва - рефрактерні). Більшість з них за
походженням є спадково зумовленими і виника
ють унаслідок мутацій генів, що мають стосу
нок до обміну заліза в організмі.
Сьогодні можна виділити два різновиди та
ких анемій: (1) залізорефрактерна анемія з де
фіцитом заліза, відома як IRIDA (iron-refractory
iron deficiency anemia) і (2) залізорефрактерна
анемія з надлишком заліза.
• Залізорефрактерна анемія з дефіцитом
заліза (IRIDA). Ця анемія характеризуєть
ся всіма ознаками дефіциту заліза, який
не може бути ліквідований пероральним
введенням препаратів, що містять даний
елемент. Тільки внутрішньовенне застосу
вання лікарських форм заліза дає певний
лікувальний ефект.
45
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Сьогодні встановлено, що в основі IRIDA мо
жуть лежати численні мутації генів, продукти
яких причетні до всмоктування заліза в тонкому
кишечнику. Особлива увага вчених прикута до
мембранного ферменту гепатоцитів - матриптази-2 (рис. 30.28). Цей ензим, який є подібною до
трипсину протеїназою, має прямий стосунок до
регуляції експресії вже відомого нам гепцидину.
У нормі матриптаза-2 перешкоджає збіль
шенню утворення гепцидину в печінкових клі
тинах завдяки розщепленню (гідролізу) мемб
ранного білка гемоювеліну (HJV - hemojuveliri),
що виступає додатковим рецептором (корецептором) до деяких гуморальних регуляторних
сигналів. Нині описано кілька десятків мутацій
у різних частинах гена матриптази-2, що викли
кають дефіцит цього ферменту. За таких умов
гемоювелін не розщеплюється, а як корецептор
зв’язується з клітинними рецепторами і разом
з ними взаємодіє із сигнальними молекулами.
Наслідком цього стає значне посилення експре
сії гена гепцидину. Як результат, порушується
всмоктування заліза у дванадцятипалій кишці
та вивільнення його з гепатоцитів і макрофагів
(див. вище).
• Залізорефрактерна анемія з надлишком за
ліза. Вона виникає в результаті порушеного
введення заліза в гем при зниженні актив
ності ферментів, що каталізують синтез
порфіринів і гему.
Дефіцит матриптази-2
З0.2 о Регуляція експреси гепцидину і механізм розвитку залізорефрактерної анемії з дефіцитом
заліза (IRIDA). HJV - гемоювелін
46
Причинами розвитку цієї анемії можуть бути:
а) генетично обумовлене зниження активнос
ті
декарбоксилази копропорфіриногену ферменту, що забезпечує один з кінцевих
етапів синтезу гему (успадковується реце
сивно, зчеплено з Х-хромосомою);
б) зменшення вмісту піридоксальфосфату активної форми вітаміну В6, унаслідок чого
залізо, що міститься в мітохондріях ери
тробластів, не може бути введене в гем;
в) блокада свинцем сульфгідрильних груп фер
ментів, що беруть участь у синтезі гему
(буває при побутовому й виробничому
отруєнні свинцем).
Зменшення активності ферментів, що беруть
участь в утворенні порфіринів і гему, призво
дить до зниження утилізації заліза й порушен
ня синтезу гемоглобіну, що викликає недостат
ність еритропоезу і розвиток гіпохромної анемії
з низьким вмістом гемоглобіну.
Порушення використання заліза веде до вто
ринного гемохроматозу: підвищується вміст
сироваткового заліза, воно відкладається у вну
трішніх органах у складі гемосидерину (гемосидероз), вторинно розростається сполучна тка
нина, розвивається цироз печінки, порушуються
функції багатьох органів (серця, підшлункової
залози, суглобів та ін.).
Мегалобластичні анемії
Мегалобластичними називають анемії, в основі
яких лежать порушення синтезу ДНК, унаслідок
чого пригнічується кровотворення і з’являються
патологічні форми клітин червоної крові - мегалобласти і мегалоцити (див. вище).
До мегалобластичних відносять:
1) В12-дефіцитну анемію, однією з форм якої
є перніціозна анемія (анемія Аддісона Бірмера);
2) фолієводефіцитну анемію',
3) В]2-рефрактерну анемію - стійку до ліку
вання препаратами вітаміну В]2.
За походженням мегалобластичні анемії мо
жуть бути (а) набутими (виявляються звичайно
в дорослих та осіб літнього віку) і (б) спадково
зумовленими. В останньому випадку порушення
можуть мати стосунок до системи транспорту
речовин (усмоктування в травному каналі) і ре
акцій метаболізму (ферментопатії).
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Фізіологічна роль вітаміну В]2 і фолієвої кис
лоти. Вітамін В,2 (ціанокобаламін) надходить
в організм у складі харчових продуктів тварин
ного походження (м’ясо, яйця, сир, печінка)
у зв’язаному з білками вигляді (див. рис. 30.29).
У результаті гідролізу білків пепсином вітамін
В12 вивільняється в шлунку, де утворює комп
лекси з продуктами секреції слинних залоз — кобалофілінами (R-зв’язувачами). Білкова частина
цих комплексів зазнає гідролізу у дванадцятипа
лій кишці під впливом трипсину панкреатично
го соку. Водночас парієтальні клітини слизової
оболонки фундальної частини шлунка секретують гастромукопротеїн, відомий як внутрішній
фактор Кастла. Цей фактор після надходження
у дванадцятипалу кишку зв’язується з вільним
вітаміном В12 (1 мг гастромукопротеїну зв’язує
25 мг ціанокобаламіну), і утворені комплекси
досягають клубової кишки, епітеліальні кліти
ни якої містять на своїх мембранах рецептори,
специфічні до гастромукопротеїну. Після по
глинання комплексів вітамін В[2 переноситься
від внутрішнього фактора Кастла на молекули
транскобаламіну II і в такому вигляді надходить
у кров, а звідти до місць використання (черво
ного кісткового мозку, клітин слизової оболонки
органів травного каналу) і депонування. Печінка
є важливим депо вітаміну В]2. Запаси його тут
настільки великі, що потрібно 3-6 років, аби
при порушеннях надходження вітаміну В]2 в ор
ганізмі виникли ознаки його дефіциту.
У клітинах з вітаміну В)2 утворюються дві
його біологічно активні форми: (1) метилкобаламін і (2) 5-дезоксіаденозилкобаламін.
• Метилкобаламін. Ця форма вітаміну В|2
є конче необхідною для синтезу ДНК у клі
тинах, а тому вона істотно впливає на про
цеси проліферації клітин загалом і крово
творення зокрема.
Вплив метилкобаламіну на синтез ДНК опо
середковується рядом біохімічних реакцій
(рис. 30.30). Перша з них - це утворення меті
оніну з гомоцистеїну. У цій реакції метилкоба
ламін служить донатором метальної групи, від
даючи яку, перетворюється в кобаламін. Регене
рація метилкобаламіну відбувається завдяки ме
тальній групі, що входить до складу молекули
№-метюттетрагідрофолієвої кислоти (№-метилFH4) - основної форми фолієвої кислоти, що
міститься в плазмі крові. При цьому №-метил-
47
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Слизова оболонка
травного каналу
Регенерація
П
Печінка
Депонування
Рис. 30.29. Можливі причини недостатності вітаміну В12.
ВФК - внутрішній фактор Кастла. Пояснення див. у тексті
Червоний
кістковий мозок
Кровотворення
48
FH4 перетворюється в тетрагідрофолієву кис
лоту (FH4). Остання у формі ще однієї похід
ної - N5’1 °-метилен- FH4 - бере участь у другій
важливій реакції ** перетворенні дезоксіуридинмонофосфату (дУМФ) у дезокситимідинмонофосфат (дТМФ). Оскільки дТМФ є безпосеред
нім попередником однієї з чотирьох азотистих
основ — тиміну, то стає зрозумілим, чому від на
ведених вище реакцій залежить синтез ДНК.
• 5-Дезоксіаденозилкобаламін. Ця форма ві
таміну В12 виступає простетичною групою
ферменту метилмалоніл-КоА-мутази, що
здійснює ізомеризацію метилмалоніл-КоА,
унаслідок якої останній перетворюєть
ся в сукциніл-КоА. При цьому токсичний
продукт - метилмалонова кислота - пере
ходить у сукцинат, який, “згораючи” в цик
лі Кребса, дає необхідну клітинам енергію.
Причини дефіциту вітаміну В]Т 3 поданої на
рис. 30.29 схеми випливають такі можливі при
чини В 2-дефіцитної анемії:
1) екзогенна (аліментарна) недостатність —
ненадходження вітаміну В12 в організм
з продуктами харчування. Може розвива
тися в маленьких дітей при вигодовуванні
їх козячим молоком або сухими молочни
ми сумішами, у вегетаріанців;
2) порушення утворення і вивільнення в сли
ну кобалофілінів (R-зв’язувачів);
3) недостатність пепсину при шлунковій гіпосекреції та ахілії (див. главу 34);
4) дефіцит внутрішнього фактора Кастла.
Буває при спадково обумовлених пору
шеннях, атрофії слизової оболонки шлун
ка, автоімунних ушкодженнях парієтальних клітин слизової шлунка, після гастректомії або видалення понад 2/3 шлунка;
5) порушення панкреатичної секреції зі змен
шенням вмісту й активності протеолітич
них ферментів;
6) розлади функції тонкої кишки: хронічні
проноси (целіакія, спру), резекція великих
ділянок кишки;
7) порушення утворення транскобаламінів
у печінці та транспорту їх в ентероцити;
8) недостатнє депонування вітаміну В12 у пе
чінці, наприклад, при цирозі.
Крім наведених вище причин, до дефіциту ві
таміну В12 можуть спричинятися:
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
► конкурентне використання вітаміну В12
гельмінтами й мікрофлорою кишок (дифілоботріоз, синдром “сліпої петлі”);
* посилене використання вітаміну В12
у жінок при вагітності.
Патогенез. Можна виокремити дві гілки патоге
незу порушень, що настають при дефіциті віта
міну ВІ2. Вони пов’язані з недостатністю осно
вних біологічно значимих форм вітаміну - метилкобаламіну та 5-дезоксіаденозилкобаламіну.
1. Дефіцит метилкобаламіну веде до накопи
чення гомоцистеїну та недостатнього утво
рення тетрагідрофолієвої кислоти (FH4)
(див. рис. 30.30). Брак останньої спричиня
ється до порушення утворення тимінових
нуклеотидів, а отже, і синтезу ДНК. Най
більше від цього страждають клітини з ви
сокою проліферативною активністю. До та
ких належать кровотворні клітини та епіте
лій слизових оболонок органів травлення:
а) порушення кровотворення. У червоному
кістковому мозку має місце парадоксальна
ситуація. З одного боку, під впливом еритропоетину, утворення і секреція якого
зростають при зумовленій анемією гіпоксії,
стимулюється проліферація клітин-попередників усіх трьох паростків крові (еритро
цитів, лейкоцитів і тромбоцитів). Це веде до
розростання кровотворної тканини, аж до
повного витіснення нею жовтого кістково
го мозку. З другого боку, через обмеження
синтезу ДНК подальший поділ клітин-попередників стає неможливим. Як результат,
вони гинуть через запуск механізмів апоптозу, а кількість зрілих клітин крові стрім
ко зменшується - розвивається панцитопенія. Стосовно червоного паростка крові
мову ведуть про неефективний еритропоез.
Невелика кількість еритроцитів, яка все
ж таки утворюється, має змінені розміри й фор
му (див. далі). Через це змінюється їхня плас
тичність, і еритроцити стають “жертвами” ма
крофагів селезінки - так до основного механіз
му анемії додається ще й гемолітичний.
Виражена анемія та гіпоксія пригнічують
утворення в печінці гепцидину (див. вище), уна
слідок чого значно зростає всмоктування заліза
в кишках та вивільнення його з депо. У резуль
таті цього неефективний еритропоез супрово-
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
49
Біохімічна роль метилкобаламіну
джується перевантаженням організму залізом —
виникає вторинний гемохроматоз з відкладан
ням великої кількості гемосидерину в тканинах:
б) порушення в органах травлення. Обмежена
можливість синтезу ДНК зумовлює пригні
чення проліферативної активності клітин
епітелію травного каналу. Як результат, на
явні епітеліоцити набувають гігантських
розмірів, а після злущування залишають
після себе дефекти, які не можуть бути за
повненні новоутвореними клітинами. Та
ким чином, з одного боку, розвивається
атрофія слизових оболонок, а з другого через порушення бар’єрних властивостей
останніх мікробні та інші фактори стають
причиною запалення. Це виявляє себе від
повідними клінічними ознаками (див. далі).
Слід зазначити, що запально-атрофічні про
цеси в шлунку можуть порушувати секрецію
внутрішнього фактора Кастла, а в тонких
кишках - всмоктування його комплексів з віта
міном В12. Це, безумовно, посилює дефіцит ві
таміну В12 і запускає “зачароване коло” патоге
незу анемії.
Описані порушення в системах крові й трав
лення можуть бути ліквідовані парентеральним
введенням препаратів вітаміну В12 чи тетрагідрофолієвої кислоти.
Ще одним наслідком недостатнього утво
рення метилкобаламіну, як зазначалося вище,
є накопичення гомоцистеїну, котрий не пере
творюється в метіонін. Сьогодні відомо, що ви
сокі концентрації гомоцистеїну мають здатність
ушкоджувати ендотелій кровоносних судин
і в такий спосіб сприяти розвитку атеросклеро
зу та пов’язаних з ним тромбозів (див. главу 32).
2. Дефіцит 5-дезоксіаденозилкобаламіну. Біо
хімічну роль цієї сполуки ми вже обгово
рювали. Тут зазначимо, що при недостат
ності вітаміну В12, через брак коферментної його форми в організмі накопичуються
метилмалонова та її попередник — пропіонова кислоти, які є токсичними для нерво
вих клітин. Крім того, у нервових волокнах
синтезуються жирові кислоти зі зміненою
структурою, що призводить до порушення
утворення мієліну, посиленого його розпа
ду (демієлінізація) та ушкодження аксонів.
Розвивається дегенерація задніх і бічних
стовпів спинного мозку (фунікулярний мі
єлоз), уражуються черепні й периферичні
нерви.
Проте не все так просто. Досить рідко у хворих
зі спадковою недостатністю метилмалоніл-КоАмутази розвиваються неврологічні ознаки, харак
терні для В12-дефіцитної анемії. Це ставить під
50
сумнів наведене вище пояснення неврологічних
розладів при недостатності вітаміну В .
Про самостійність обговорюваної гілки пато
генезу В12-дефіцитної анемії свідчить той факт,
що притаманні їй порушення нервової системи
усуваються тільки введенням препаратів вітамі
ну В)2, а препарати фолієвої кислоти є абсолют
но неефективними.
Картина крові та червоного кісткового мозку.
Найхарактернішою рисою В12-дефіцитної анемії є
поява в крові й червоному кістковому мозку клітин
патологічної регенерації — мегалобластів та їхніх
без’ядерних форм-мегалоцитів (див. рис. 30.12).
На тлі істотного зменшення вмісту еритро
цитів і концентрації гемоглобіну колірний по
казник збільшений, що пояснюється більшим,
ніж у нормі, діаметром еритроцитів. Характер
ними є явища дегенерації еритроцитів: анізоцитоз (макроцитоз), пойкілоцитоз (поява клітин
овальної форми), патологічні включення (тільця
Жоллі, тільця Кебота). Ретикулоцитів і поліхроматофілів майже немає.
Крім того, у крові зменшений вміст грануло
цитів (особливо нейтрофілів) і тромбоцитів.
У мазках можна виявити гігантські нейтрофіли
з гіперсегментованими ядрами.
У червоному кістковому мозку еритробластичний тип кровотворення змінюється на мегалобластичний. Про це свідчить поява вели
кої кількості гігантських клітин - мегалоблас
тів, характерною рисою яких, окрім розмірів,
є структура ядра. Воно надто велике і має чітко
виражені ядерця та ніжні ниткоподібні утво
рення хроматину (незріле ядро). У міру появи
гемоглобіну в таких клітинах ядро не зазнає
характерних для еритробластів і нормобластів
змін - конденсації хроматину та пікнозу, а зали
шається незрілим.
Клінічні прояви. Підсумовуючи наведений
вище матеріал, можемо виділити три основні
групи проявів В12-дефіцитної анемії.
1. Гематологічний синдром. Він зумовлений
панцитопенією - зменшенням кількості всіх
формених елементів крові. Його самостій
ними складовими є:
а) анемія та пов’язана з нею гіпоксія;
б) лейкопенія (особливо нейтропенія), що
стає причиною зниження резистентнос
ті організму до інфекцій;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
в) тромбоцитопенія, яка може зумовлюва
ти розвиток геморагічного синдрому.
2. Ураження травного каналу, що виявляють
себе розвитком запально-атрофічних змін
в епітелії язика (глосит) та у слизових обо
лонках порожнини рота {стоматит), стра
воходу (езофагіт), шлунка {ахілічний га
стрит), тонких кишок {дуоденіт, ентерит).
3. Неврологічний синдром. Характерними
є ураження задніх і бічних стовпів спинно
го мозку, зумовлені дегенерацією мієліну
{фунікулярний мієлоз). Вони зумовлюють
появу рухових (спастичні парези) і сен
сорних (тяжкі парестезії, сенсорна атаксія)
розладів, найчастіше в нижніх кінцівках.
Рідше розвиваються дегенеративні зміни
в спинномозкових гангліях і периферичних
нервах.
Перніціозна анемія (анемія Аддісона - Бірмера). Ця хвороба є одним із найпоширеніших
варіантів В(2-дефіцитної анемії. Її розвиток зу
мовлений недостатністю внутрішнього факто
ра Кастла, внаслідок чого стає неможливим
всмоктування вітаміну В12 у тонких кишках.
Центральне питання патогенезу перніціозної
анемії стосується причин і механізмів такої не
достатності. Сьогодні вважають, що в її основі
лежить хронічний атрофічний гастрит, що
має, найімовірніше, автоімунне походження.
Про це зокрема свідчать такі факти:
а) зменшення кількості парієтальних клітин,
що утворюють внутрішній фактор Кастла,
відбувається на тлі інфільтрації слизової
оболонки шлунка лімфоцитами і плазма
тичними клітинами',
б) у плазмі та шлунковому сокові часто вияв
ляють кілька видів імуноглобулінів: анти
тіла, що блокують зв’язування внутріш
нього фактора Кастла з вітаміном В,2, і такі,
що перешкоджають взаємодії комплексу
цих сполук з рецепторами ентероцитів;
в) при моделюванні у тварин автоімунно
го гастриту за участю CD4+ Т-лімфоцитів
у крові виявляють автоантитіла, аналогічні
тим, що є у хворих з перніціозною анемією;
г) така анемія нерідко асоційована з іншими
автоімунними хворобами, зокрема авто
імунними ураженнями щитоподібної зало
зи і мозкової речовини надниркових залоз.
51
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
При зменшенні кількості парієтальних клі
тин, що утворюють внутрішній фактор Кастла,
нижче від критичного рівня, і повному висна
женні депо вітаміну В12 у печінці настають клі
нічні прояви, механізми розвитку яких ми вже
обговорювали.
Єдина відмінність перніціозної анемії від не
достатності вітаміну В]2, зумовленої іншими
чинниками, полягає в тому, що введення пре
паратів ціанокобаламіну усуває всі клінічні про
яви хвороби, окрім хронічного атрофічного гас
триту. Це вказує на те, що зміни слизової обо
лонки шлунка мають не вторинний характер, як,
наприклад, при екзогенній недостатності ціано
кобаламіну, а є власне причиною недостатності
вітаміну В]2 в організмі.
Фолієводефіцитна анемія. Причиною дефіциту
фолієвої кислоти в організмі можуть бути:
а) екзогенна (аліментарна) недостатність.
При добовій потребі в 50-200 мг фоліє
вої кислоти цілком достатньо в рослинній
(особливо зеленій) їжі. Дуже незбалансований раціон у немовлят, старих людей, бід
них верств населення та алкоголіків є час
тою причиною дефіциту цього вітаміну;
б) порушення всмоктування як один із компо
нентів синдрому мальабсорбції (див. главу 34);
в) посилене використання, що не компенсу
ється збільшенням надходження фолієвої
кислоти ззовні. Такі обставини можуть ви
никати у вагітних, немовлят, при дисемінованих формах злоякісних пухлин, при знач
ному посиленні еритропоезу, характерному
для гемолітичних анемій;
г) втрата вітаміну, наприклад, при гемодіалі
зі, при порушеннях процесів реабсорбції
в нирках.
Патогенез анемії, картина крові та основні
клінічні прояви хвороби нічим не відрізняють
ся від тих, що характерні для дефіциту вітамі
ну В.,. Є один лише виняток: при недостатнос
ті фолієвої кислоти не розвиваються невроло
гічні ураження. І це зрозуміло, адже фолієва
кислота, на відміну від вітаміну В12, не має
відношення до порушень обміну метилмалонової кислоти та її накопичення. Оскільки змі
ни кровотворення і зміни в системі травлення
є наслідком первинного дефіциту фолієвої кис
лоти, то введення препаратів ціанокобаламіну
не може їх усунути. Лікувальний ефект матиме
тільки застосування препаратів самої фолієвої
кислоти.
Вп-рефрактерна
анемія.
Мегалобластичні
анемії, стійкі до лікування препаратами ціа
нокобаламіну, відносять до В,2-рефрактерних
анемій. Основними їхніми причинами є хіміч
ні сполуки, що порушують утворення пурино
вих і піримідинових азотистих основ, а отже,
і синтез ДНК. До таких, зокрема, належать ін
гібітори обміну фолієвої кислоти (метотрексат).
Оскільки інгібування синтезу ДНК вважається
однією із засад протипухлинної терапії, то стій
кість до лікування анемії вітаміном В|2 часто
є побічним ефектом застосування хіміотерапев
тичних засобів.
Гіпопластична анемія
Гіпопластична (апластична) анемія - це хво
роба системи крові чи окремий синдром, що
характеризується пригніченням кровотворної
функції червоного кісткового мозку та виявляє
себе недостатнім утворенням еритроцитів, Гра
нулоцитів і тромбоцитів (панцитопенією) або
одних тільки еритроцитів (.парціальна гіпопластична анемія).
Причини. Розрізняють (1) набуті й (2) спадково
зумовлені форми гіпопластичної анемії.
Набуті форми можуть мати такі причини:
а) фізичні фактори (іонізуюча радіація);
б) хімічні агенти (бензол, свинець, пари ртуті,
лікарські препарати: цитостатичні засоби,
левоміцетин, сульфаніламіди);
в) біологічні фактори (віруси гепатиту, герпе
су, цитомегаловірус).
Окрім того, до набутих відносять так звану ідіопатичну форму, причину якої встановити не
вдається. На цю форму припадає 50-75 % усіх
випадків гіпопластичної анемії.
Усі наведені етіологічні чинники поділяють
на дві групи:
1) фактори з облігатним (обов’язковим) мієлотоксичним ефектом. До них відносять
іонізуючу радіацію, бензол та його похідні,
протипухлинні препарати (цитостатики),
неорганічні сполуки миш’яку та ін. Ушко-
52
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
вих антигенів, до яких немає імунологіч
джувальна дія цих чинників на червоний
ної толерантності. Причиною цього стають
кістковий мозок залежить від дози та трива
спонтанні (можливо, при ідіопатичній фор
лості. У більшості випадків кровотворення
мі анемії) або індуковані мієлотоксичними
відновлюється, якщо їхній вплив припиня
факторами мутації. Експресія додаткових
ється;
антигенів стає причиною імунних реак
2) фактори з факультативним мієлотоксичцій, спрямованих на знищення змінених
ним впливом. Останній може виявляти
стовбурових клітин. У цих реакціях беруть
себе лише в окремих поодиноких випад
участь антигенспецифічні Т-лімфоцити
ках (наприклад, апластична постмедикай цитокіни, що мають цитотоксичну дію
ментозна анемія). У розвитку анемії після
(зокрема, фактор некрозу пухлин - ФНП).
прийому лікарських препаратів велике,
Настає імунне ушкодження, механізми яко
якщо не вирішальне значення мають ін
го описано в главі 15.
дивідуальні особливості організму. Вста
новлено, що не існує прямого зв’язку між
Результатом загибелі стовбурових клітин
розвитком гіпопластичної анемії, з одного
стає зменшення їхньої кількості, що веде до іс
боку, і дозою, а також тривалістю засто
сування лікарських препаратів, з другого. тотного зменшення пулів проліферуючих і до
До гіпопластичної анемії може спричи
зріваючих клітин - червоний кістковий мозок
спустошується та заміщається жировою тка
нятися приймання деяких антибіотиків
(левоміцетин), протисудомних, антитире- ниною. Такий його стан позначають терміном
оїдних, антигістамінних засобів, транкві
аплазія.
Сьогодні відомо, що на втягування стовбуро
лізаторів.
вих клітин у процес кровотворення сильно впли
вають клітини мікрооточення, що належать
Прикладом спадково зумовленої форми гіпо
до строми червоного кісткового мозку. Цілком
пластичної анемії є анемія Фанконі - захворю
вання з аутосомно-рецесивним типом успадку можливо, що деякі форми гіпопластичної анемії
вання. Сутність дефекту полягає в порушеннях в основі свого патогенезу мають ураження саме
цих клітин. Про існування такого варіанта свід
систем репарації ДНК. Унаслідок цього легко
чить той факт, що для всіх представників чи
виникають невідновлювані ушкодження ДНК
під впливом ультрафіолетового випромінюван стої лінії мишей (“сталеві” миші) з первинними
спадково зумовленими дефектами стромальних
ня й хімічних агентів. Порушується кровотво
рення, часто розвиваються гострі лейкози, ано клітин характерним є розвиток гіпопластичної
анемії.
малії нирок, селезінки, кісток.
Патогенез. Об’єктом уражень при гіпопластичній анемії є стовбурові клітини червоного кіст
кового мозку. Доказом цього є панцитопенія,
тобто порушення утворення всіх формених еле
ментів крові, що мають спільного попередника
(еритроцитів, гранулоцитів, тромбоцитів).
Розрізняють два можливі механізми ушко
дження стовбурових клітин (рис. 30.31).
1. Необоротне ушкодження ДНК, яке за
пускає механізми програмованої загибелі
клітин - апоптоз (див. главу 7). Вважають,
що в такий спосіб діють фактори з облігатним, дозозалежним мієлотоксичним
ефектом.
2. Імунне ушкодження клітин. Виникає в разі
появи на поверхні стовбурових клітин но
Картина крові та червоного кісткового моз
ку. Для картини периферичної крові характер
не зменшення вмісту еритроцитів і концентра
ції гемоглобіну, при цьому колірний показник
у межах норми. У мазку крові, як правило, не
виявляють регенераторних форм еритроцитів
(ретикулоцитів, поліхроматофілів). Зменшуєть
ся вміст гранулоцитів (особливо нейтрофілів)
і тромбоцитів. Кількість лімфоцитів може зали
шатися без змін. У червоному кістковому мозку
зменшується кількість кровотворних клітин зі
збільшенням вмісту жирової тканини (карти
на спустошення червоного кісткового мозку).
У зв’язку з тим, що залізо не використовуєть
ся для кровотворення, збільшується його вміст
в еритробластах і позаклітинно.
53
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Т-лімфоцит
Механізми розвитку аплазії червоного кісткового мозку
Клінічні прояви. Гіпопластична анемія виявляє
себе ознаками, поява яких зумовлена зменшен
ням утворення трьох видів формених елемен
тів крові: еритроцитів, Гранулоцитів і тром
боцитів. Відповідно можна виокремити три
основні клінічні синдроми.
1. Анемія і пов’язаний з нею гіпоксичний син
дром.
2. Запальні процеси, зумовлені інфекційними
агентами (пневмонія, отит, пієліт тощо).
У їхній основі лежить зменшення резис
тентності організму до інфекцій, що настає
внаслідок лейкопенії (див. підглаву 30.3).
3. Геморагічний синдром (див. підглаву 30.4).
ПОРУШЕННЯ СИСТЕМИ
ЛЕЙКОЦИТІВ. ЛЕЙКОЦИТОЗИ
І ЛЕЙКОПЕНІЇ
У широкому розумінні система лейкоцитів охоп
лює (а) клітини-попередники зрілих клітин бі
лої крові, (б) власне лейкоцити і (в) клітини, що
є похідними лейкоцитів (макрофаги, плазматич
ні клітини).
Клітини білої крові містяться в організмі
в трьох “відсіках”: у червоному кістковому моз
ку, периферичній крові й тканинах.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
54
Етапи лейкопоезу в червоному кістковому мозку
1. У червоному кістковому мозку вони утво
рюють 4 пули (рис. 30.32):
а) пул стовбурових клітин, що перебу
вають у стані спокою. Він дуже малий
і є резервом кровотворення;
б) мітотичний пул. Це клітини, що всту
пили на шлях мітотичного поділу. Від
стовбурової клітини до утворення клі
тин, що дозрівають, звичайно проходить
від 4 до 11 поділів;
в) дозріваючий пул. Він містить клітини,
диференціація яких триває в середньо
му 3-5 діб;
г) резервний пул складається зі зрілих лей
коцитів, що можуть переходити в кров.
• Лейкоцити периферичної крові утворюють
два приблизно однакових за кількістю клі
тин пули. Це - (а) циркулюючі клітини і (б)
ті, що перебувають у стані крайового сто
яння. Пул останніх позначають як пристін
ковий або маргінальний.
• У периферичних тканинах можна знайти
(а) мігруючі лейкоцити і (б) лейкоцити
у стані спокою, що перетворюються чи
вже перетворилися у функціонально актив
ні клітини сполучної тканини (макрофаги,
мастоцити, плазматичні клітини).
Підраховано, що загальна маса лейкоцитів
периферичної крові становить близько 10 г, тимчасом як у червоному кістковому мозку — 500 г,
у периферичних тканинах - 600 г. Це ще раз до
водить, що кров слугує лише засобом транспор
ту лейкоцитів від місць їхнього утворення до
тканин, де власне вони й виконують свої функції.
Основна функція лейкоцитів - захист ор
ганізму від чужорідних біологічних об’єктів,
якими є бактерії, віруси, найпростіші, гриби,
гельмінти тощо. Цей захист досягається ме
ханізмами неспецифічної (фагоцитоз) і спе
цифічної (імунологічної) резистентності (див.
главу 14).
Функціонування системи лейкоцитів, узго
джене з потребами організму, забезпечується
засобами гуморальної регуляції. Об’єктами та
кої регуляції є:
1) лейкопоез. Група речовин-цитокінів, що
стимулює цей процес, отримала назву лейкопоетинів (див. далі);
2) міграція лейкоцитів у тканини. Вона здій
снюється під впливом речовин-хемотаксинів (хемоатрактантів);
3) міжклітинні взаємодії. їхня регуляція
здійснюється цитокінами лімфоцитарного
(.лімфокіни) і моноцитарного (монокіни)
походження.
Лейкоцити периферичної крові поділяють на
гранулоцити (базофіли, еозинофіли, нейтро
філи) і агранулоцити (лімфоцити, моноцити)
(рис. 30.33).
* Нейтрофіли. Вони становлять найчисленнішу популяцію лейкоцитів. Циркулюють
у крові протягом 4-8 год., після чого пе
реходять у тканини, де живуть до 4-5 діб.
Основні їхні функції:
а) фагоцитоз мікроорганізмів з подальшим
знищенням їх (звідси ще одна назва - мікрофагоцити)',
55
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
ис. 30.33. Лейкоцити периферичної крові. NK-клітина - природний кілер
б) вивільнення у тканини бактерицидних
речовин, що вбивають мікробів на від
стані (дистантна бактерицидна дія);
в) участь у розвитку гострого запалення
(див. главу 17).
» Еозинофіли. У своїх утвореннях (великі
і малі гранули, ліпідні тільця) містять чи
малу кількість речовин, що мають цитоток
сичну дію (великий основний білок, каті
онний білок еозинофілів, еозинофільний
нейротоксин) і ферментативну активність
(пероксидаза, арилсульфатаза Б, циклоксигеназа, 5-ліпоксигеназа та ін.). Серед функ
цій цих клітин: (а) знешкодження паразитів
(у тому числі гельмінтів), (б) інактивація
біологічно активних речовин, (в) участь
у розвитку алергічних реакцій І типу (ана
філактичних) (див. главу 15).
• Базофіли. Являють собою циркулюючу
форму тканинних базофілів (мастоцитів),
що є головними дійовими особами в алер
гічних реакціях І типу. Характеристику цих
клітин та їхню участь у патогенезі алергії
подано в главі 15.
• Моноцити. Ці клітини започатковують роз
виток у тканинах цілої групи клітин, що
об’єднуються в систему мононуклеарних
фагоцитів (стара назва: ретикулоендотеліальна система). Вийшовши із червоно
го кісткового мозку, моноцити циркулю
ють у крові протягом 10-20 год., а потім
мігрують у тканини, де перетворюються
в макрофаги, строк життя яких сягає міся
ців і навіть років. Із моноцитів у тканинах
утворюються макрофаги сполучної ткани
ни, клітини Купфера в печінці, макрофаги
селезінки, макрофаги червоного кістково
го мозку, альвеолярні макрофаги легень,
макрофаги серозних порожнин (черевної,
плевральної), остеокласти, клітини мікроглії в головному мозкові. Основна функ
ція утворених макрофагів - (а) фагоцитоз
56
(див. главу 14). За цією ознакою моноцитів
та їх похідні називають облігатними фаго
цитами. Окрім того, ці клітини є (б) учас
никами початкових етапів специфічної
імунної відповіді, оскільки вони поглина
ють, переробляють і презентують антигени
імунокомпетентним клітинам (див. главу
14). І, нарешті, похідні моноцитів (в) віді
грають важливу роль у патогенезі запален
ня (див. главу 17) і гарячки (див. главу 18).
• Лімфоцити. Ці клітини постійно рециркулюють в організмі. Тривалість їхнього
життя різна - від 1 доби до багатьох ро
ків і навіть усього життя (клітини імунної
пам’яті). Лімфоцити забезпечують реак
ції специфічного імунного захисту орга
нізму. За функціональною спеціалізацією
розрізняють
В-лімфоцити,
Т-лімфоцити
і NK-клітини (природні кілери). Докладну
характеристику функцій усіх цих клітин
подано в главі 14.
Утворення лейкоцитів - лейкопоез - це склад
ний регульований процес, що поєднує в собі (а)
розмноження кровотворних клітин та (б) їх ди
ференціацію (рис. 30.34).
За сучасними уявленнями, всі формені еле
менти крові походять з одного джерела - стов
бурових клітин крові. На шляхах диференціації
розрізняють 6 класів клітин:
• І клас - стовбурові клітини крові. Ці клі
тини діляться рідко, в основному вони пе
ребувають у О0-періоді клітинного циклу,
а тому їхня частка від загальної кількості
гемопоетичних клітин у кровотворних ор
ганах є дуже низькою (КИ-10"5).
• II клас — напівстовбурові клітини. Від по
передніх вони відрізняються тим, що є комітованими, тобто вже запрограмовани
ми на певний напрям диференціації. Від
наступних же клітин їх відрізняє те, що
вони зберігають можливість диференцію
ватися не в одному, а в кількох напрямах.
Крім того, ці клітини набувають чутливості
до регуляторів гемопоезу (еритропоетину,
лейкопоетинів, тромбопоетину), від яких
і залежить напрям диференціації.
• III клас —уніпотентні клітини. На відміну
від попередніх клітин, кожна клітина цьо
го класу може розвиватися тільки в одному
напрямі, а тому природно, що за числом
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
різних видів формених елементів крові іс
нує 8 видів уніпотентних клітин-попередників (окремо для В- і Т-лімфоцитів).
Варто зазначити, що клітини перших трьох
класів мають багато спільних рис:
а) вони знаходяться переважно в червоно
му кістковому мозку, але при цьому здат
ні потрапляти в кров і після циркуляції
знову поселятися в кровотворних орга
нах;
б) усі клітини цих класів схожі на малі лім
фоцити, а отже, їх не можна відрізнити
одну від одної за морфологічними озна
ками. Причина полягає в тому, що на да
них стадіях диференціація йде лише на
рівні геному;
в) клітини I—III класів мають здатність до
самопідтримання: при їхніх поділах час
тина дочірніх клітин ідентична материн
ським (тобто поповнюється пул клітин
того класу, до якого належали батьківські
клітини), і лише друга частина зазнає ди
ференціації — перетворюється у клітини
наступних класів;
г) завдяки попереднім властивостям (самопідтриманню і диференціації) клітини
цих класів здатні утворювати колонії при
введенні їх у селезінку мишей-реципієнтів, що попередньо отримали дозу раді
ації, яка вбиває всі власні гемопоетичні
клітини. Цим власне і пояснюється те, що
для позначення клітин перших трьох кла
сів використовують термін колонієутворюючі одиниці (КУО).
• IV клас - бласти. Через початок процесів
специфічного синтезу речовин змінюється
морфологія клітин: вони відрізняються від
клітин I—III класів (схожих на малі лімфо
цити) більшим розміром, світлішим ядром
і світлою цитоплазмою, появою в цито
плазмі перших продуктів специфічних син
тезів. Незважаючи на останню обставину,
розрізнити між собою (тобто “по горизон
талі”) бластні клітини за морфологічними
ознаками практично неможливо. На від
міну від клітин попередніх класів, бласти
не здатні до самопідтримання. Це означає,
що при поділі їх утворюються тільки більш
диференційовані клітини, а клітини, подіб
ні батьківським, не відтворюються.
_____________ ____________________________________
57
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
» V клас - дозріваючі клітини. Цей клас
у кожному напрямі гемопоезу представ
лений цілим рядом клітин, що переходять
одна в одну. При цьому з’являються чіткі
морфологічні відмінності не тільки “по
вертикалі” — між суміжними клітинами
кожного ряду, а й “по горизонталі” - між
клітинами різних напрямів диференціації.
На певному етапі дозрівання здатність клі
тин до поділу повністю втрачається і вони
лише змінюють свою будову, наближаю
чись за морфологічними й функціональ
ними ознаками до зрілих форм. Останніми
представниками клітин цього класу, що
здатні ділитися, є поліхроматофільні нормоцити для еритропоезу та мієлоцити для мієлопоезу. Самі ці клітини станов
лять більшість у нормальному кістковому
мозку.
» VI клас - зрілі клітини. Це формені еле
менти крові (еритроцити, лейкоцити, тром
боцити), здатні виконувати притаманні їм
функції.
Для характеристики стану системи лейкоци
тів використовують ряд показників, серед яких:
1) вміст лейкоцитів в одиниці об’єму кро
ві. У нормі цей показник становить від
4 • 109/л до 9 • 109/л. Збільшення вмісту лей
коцитів у крові отримало назву лейкоцито
зу, зменшення - лейкопенії;
2) лейкоцитарна формула — кількісне співвід
ношення різних форм лейкоцитів у пери
феричній крові. Для норми характерні такі
значення: базофіли - 0-1 %, еозинофіли 2-4 %, мієлоцити - 0 %, метамієлоцити
(юні нейтрофіли) - 0-1 %, паличкоядерні
нейтрофіли - 3-5 %, сегментоядерні ней
трофіли 50-70 %, лімфоцити - 20-35 %,
моноцити - 2-8 %. За умов патології мож
ливими є зміни лейкоцитарної формули,
зокрема зміщення її вліво чи вправо (ядер
ний зсув) (див. далі);
3) абсолютний вміст кожної форми лейкоци
тів в одиниці об’єму крові. Цей показник
розраховують на підставі загального вміс
ту лейкоцитів і лейкоцитарної формули.
На відміну від останньої, він дає уявлення
про абсолютне, а не відносне збільшення
чи зменшення вмісту певного виду лейко
цитів у периферичній крові;
4) якісні характеристики лейкоцитів. їх ви
значають, вивчаючи мазки крові. В умовах
патології можлива поява різних дегенера
тивних форм лейкоцитів, що можуть ви
являти себе анізоцитозом, наявністю в ци
топлазмі вакуоль, токсигенної зернистості,
включень типу тілець Князькова — Деле
(базофільно забарвлені грудочки цитоплаз
ми); великою азурофільною зернистістю;
зникненням звичайної зернистості, пікнозом або набряканням ядра, його гіпер- і гіпосегментацією, а також невідповідністю
ступеня дозрівання ядра й цитоплазми, каріорексисом, цитолізом (рис. 30.35);
5) мієлограма. Цей показник характеризує
кількість і якісний склад клітин червоного
кісткового мозку.
Лейкоцитоз
Цим терміном позначають збільшення кількості
лейкоцитів в одиниці об’єму крові понад 9 • 109/л.
Лейкоцитоз не має самостійного значення, він
є лише симптомом, що супроводжує розвиток
багатьох хвороб.
Класифікація:
1. Залежно від причин розвитку виділяють (а)
фізіологічний і (б) патологічний лейкоцитози.
Фізіологічний лейкоцитоз є звичайною ре
акцією організму на ті чи інші впливи. До його
різновидів відносять:
а) емоціогенний лейкоцитоз — виникає під час
сильних емоцій;
6) міогенний - розвивається під час інтенсив
ної фізичної роботи;
в) статичний - характерний для переходу
людини з горизонтального у вертикальне
положення;
г) аліментарний — розвивається після при
ймання їжі;
д) лейкоцитозу вагітних',
е) лейкоцитоз у новонароджених.
Патологічний лейкоцитоз пов’язаний з пе
ребігом в організмі патологічних процесів. Він,
як правило, розвивається при: (а) інфекційних
хворобах; (б) запальних та алергічних процесах;
(в) інтоксикації екзо- й ендогенного походження.
2. Лейкоцитоз може бути (а) абсолютним і (б)
відносним. Для абсолютного лейкоцитозу
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
59
характерне збільшення абсолютної кількос
ті лейкоцитів в одиниці об’єму крові. Про
відносний лейкоцитоз ідеться в тому випад
ку, коли зростає відносний вміст окремих
форм лейкоцитів у периферичній крові.
3. За механізмом розвитку лейкоцитоз бу
ває: (а) реактивним; (б) перерозподіленим
і (в) пухлинного походження (див. далі).
4. Залежно від видів лейкоцитів, вміст яких
у крові збільшений, виділяють:
» Нейтрофільний лейкоцитоз (нейтрофільоз). Він характерний для: (а) гнійнозапальних процесів, спричинених гноє
творними бактеріями (абсцеси, флегмони,
сепсис); (б) асептичного запалення, зу
мовленого некрозом тканин (наприклад,
інфаркт міокарда, опіки); (в) важкого кис
невого голодування (велика гостра крово
втрата, гострий гемоліз); (г) ендогенної
інтоксикації (уремія).
•
Еозинофільний лейкоцитоз (еозинофілія). Розвивається при (а) алергічних
реакціях І типу (анафілактичних); (б) па
разитарних інфекціях, у тому числі гель
мінтозах; (в) деяких злоякісних пухли
нах, зокрема хронічному мієлолейкозі.
• Базофільний лейкоцитоз. Буває дуже
рідко, може супроводжувати розвиток
(а) хронічного мієлолейкозу; (б) гемофі
лії; (в) хвороби Вакеза (поліцитемії).
•
Лімфоцитарний лейкоцитоз (лімфоцитоз). Характерний для (а) деяких гострих
вірусних інфекцій (вірусний гепатит,
цитомегаловірусна та герпесвірусна ін
фекції), (б) ряду хронічних інфекційних
хвороб, що супроводжуються тривалою
стимуляцією імунної системи (туберку
льоз, сифіліс, бруцельоз), (в) хронічного
лімфолейкозу.
• Моноцитарний лейкоцитоз (моноцитоз).
Його виявляють при (а) деяких хроніч
них інфекціях (туберкульоз, бруцельоз),
(б) інфекційному мононуклеозі, (в) ін
фекціях, збудниками яких є рикетсії та
найпростіші (висипний тиф, малярія).
Механізми. Розрізняють три механізми розвитку
лейкоцитозів.
І. Перехід лейкоцитів із червоного кісткового
мозку у кров. Цей механізм лежить в осно
ві реактивного лейкоцитозу, що виникає як
60
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Гіпосегментація ядер
Токсигенна зернистість
Гіперсегментація ядер
Вакуолізація цитоплазми
Дегенеративні форми гранулоцитів
реакція червоного кісткового мозку на пато
генні впливи. Він закономірно розвивається
при інфекційних захворюваннях, запаленні,
дії низьких доз токсичних речовин.
Реактивний лейкоцитоз зумовлюється (а) по
силенням лейкопоезу (проліферації та дозріван
ня лейкоцитів) і (б) збільшенням переходу лей
коцитів із резервного пулу червоного кісткового
мозку в кров.
• Посилення лейкопоезу має в своїй основі дію
на кровотворні клітини II класу (див. вище)
речовин, що мають узагальнену назву лей
ко поетини. Сьогодні до цієї групи сполук
відносять (а) цитокіни та (б) фактори росту.
До цитокінів, з якими пов’язують стимуля
цію лейкопоезу, відносять інтерлейкін-1 (ІЛ-1)
та фактор некрозу пухлин (ФНП), що є важли
вими для посилення утворення нейтрофілів,
а також інтерлейкін-5 (ІЛ-5), що зумовлює еозинофілію, та інтерлейкін-7, який стимулює лімфопоез, та інші.
Фактори росту, що посилюють розмноження
й диференціацію кровотворних клітин, отрима
ли назву колонієстимулюючих факторів. Най
більшим їхнім джерелом є стромальні клітини
червоного кісткового мозку і Т-лімфоцити, хоча
здатність продукувати ці речовини мають також
макрофаги, ендотеліальні клітини судин, фібро
бласти.
Роль лейкопоетинів у розвитку лейкоцитозу
продемонстровано на рис. 30.36. У вогнищі за
палення, як відомо, відбувається активація ма
крофагів (див. главу 17), одним із проявів якої
є вивільнення в тканини і кров цитокінів, зокре
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
61
ма ІЛ-1 та ФНП. Останні, діючи на клітини стро
ми червоного кісткового мозку, Т-лімфоцити
і клітини ендотелію судин, зумовлюють виді
лення кількох видів факторів росту: (а) гранулоцит-моноцитарного
колонієстимулюючого
фактора (ГМ-КСФ), (б) гранулоцитарного ко
лонієстимулюючого фактора (Г-КСФ) і (в) моноцитарного
колонієстимулюючого
фактора
(М-КСФ). Вплив цих факторів на кровотворні
клітини, запрограмовані на шлях мієлопоетичного розвитку, спричиняється до посилення по
ділу їх і диференціацію в клітини гранулоцитар
ного ряду й моноцити.
Продукти активованих макрофагів (цитокіни
та колонієстимулюючі фактори) можуть і безпо
середньо впливати на червоний кістковий мозок,
посилюючи описаним вище способом мієлопоез.
Дискусійним до сьогодні залишається питан
ня про існування фізіологічних інгібіторів лейкопоезу. На роль останніх свого часу претенду-
вали високомолекулярний інгібітор сироватки
крові (ліпопротеїн), кейлони, лактоферин. Якщо
ж все-таки існують антагоністи лейкопоетинів,
то зменшення утворення їх в організмі могло би
бути ще одним чинником посилення лейкопоезу.
• Збільшення переходу лейкоцитів із ре
зервного пулу червоного кісткового мозку
в кров є другим механізмом реактивного
лейкоцитозу. Цілий ряд прозапальних цитокінів, і насамперед уже згадувані ІЛ-1
та ФНП, що посилено продукуються при
гострому запаленні, збільшують проник
ність ендотелію судин червоного кістко
вого мозку й у такий спосіб значно полег
шують вихід зрілих лейкоцитів та безпосе
редніх їхніх попередників (метамієлоцитів
та паличкоядерних клітин) у периферичну
кров. Подібну дію мають і бактеріальні
ендотоксини, здатні викликати реактивний
лейкоцитоз.
Активований макрофаг
ІЛ-1
ФНП
Механізми реактивного лейкоцитозу. Пояснення і умовні позначення - див. у тексті
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
62
II. Перерозподіл лейкоцитів крові. Суть цього
механізму полягає в тому, що лейкоцити
переходять із пристінкового пулу в цирку
люючий, збільшуючи в такий спосіб кіль
кість лейкоцитів в одиниці об’єму крові.
Більшість форм фізіологічного лейкоцитозу
має в своїй основі перерозподільний механізм.
Його особливостями є:
а) короткочасний характер зі швидким повер
ненням кількості лейкоцитів до норми піс
ля завершення дії причини;
б) збереження нормального кількісного спів
відношення різних видів лейкоцитів (лей
коцитарна формула не міняється);
в) відсутність дегенеративних змін лейкоцитів.
III. Вихід у кров пухлинних клітин. Розвиток лей
козів - злоякісних пухлин із кровотворних
клітин — може зумовлювати перехід великої
кількості лейкозних клітин різного ступеня
зрілості з пухлинних осередків у кров. Осо
бливо характерним є лейкоцитоз такого по
ходження для хронічних лейкозів (див. далі).
Зміщення лейкоцитарної формули.
Розвиток
лейкоцитозу нерідко супроводжується зміщен
ням лейкоцитарної формули (ядерним зсувом).
Таке поняття використовують для позначення
порушень співвідношення між незрілими й зрі
лими формами нейтрофілів.
При дослідженні лейкограми встановлюють
наявність ядерного зсуву нейтрофільних гра
нулоцитів уліво або вправо. Ця термінологія
пов’язана з розташуванням незрілих нейтро
фільних гранулоцитів (мієлоцитів, метамієлоцитів, паличкоядерних нейтрофілів) у лівій час
тині лейкоцитарної формули (формули Арнета
й Шилінга), тимчасом як зрілі, сегментоядерні
нейтрофіли умовно поставлені в крайнє праве
положення. Збільшення вмісту в крові молодих
форм нейтрофільних гранулоцитів свідчить про
ядерний зсув уліво, переважання зрілих нейтро
філів з великою кількістю сегментів (5-6) на тлі
зникнення більш молодих клітин - про ядерний
зсув управо.
Зміщення лейкоцитарної формули вліво бу
ває набагато частіше, і воно може виявляти себе
різними варіантами. Розрізняють такі різновиди
ядерного зсуву вліво:
1. Регенераторний зсув є показником реак
тивної активації гранулоцитопоезу (на тлі
помірного загального лейкоцитозу підви
щений вміст паличкоядерних нейтрофілів
та метамієлоцитів, можуть зустрічатися
поодинокі мієлоцити).
2. Гіперрегенераторний зсув відображає над
мірну гіперплазію лейкопоетичної тка
нини з порушенням дозрівання клітин
і вираженим омолодженням складу крові.
При цьому різко зростає число паличко
ядерних гранулоцитів та метамієлоцитів,
з’являються мієлоцити, промієлоцити; за
гальна кількість лейкоцитів може бути
збільшена, незмінена і навіть знижена вна
слідок того, що розвивається виснаження
мієлоїдного паростка після попередньої
активізації. Картина крові часто нагадує
таку при хронічних лейкозах, а тому гіперрегенераторне зміщення лейкоцитарної
формули інколи позначають як лейкемоїдну
реакцію. На відміну від лейкозів, лейкемоїдна реакція зникає, якщо припиняється дія
чинників, що її викликали.
3. Дегенеративний зсув свідчить про при
гнічення й глибокі порушення лейкопоезу,
коли на тлі загальної лейкопенії в лейкограмі збільшується число паличкоядерних
нейтрофільних гранулоцитів з дегенера
тивними змінами в їхній цитоплазмі та ядрі
при зменшенні кількості сегментоядерних
форм і відсутності метамієлоцитів.
4. Регенераторно-дегенеративний зсув спо
стерігають при гіперпродукції в червоному
кістковому мозкові патологічно змінених
лейкоцитів і порушенні їхнього дозрівання.
У цьому випадку виявляють лейкоцитоз,
а в мазку крові зростає число паличкоядер
них гранулоцитів, метамієлоцитів та мієло
цитів з ознаками дегенерації.
Лейкопенія
Зменшення кількості лейкоцитів у периферич
ній крові нижче 4-109/л називають лейкопенією.
Вона часто виступає симптомом якої-небудь
хвороби, проте є нозологічні форми, при яких
саме лейкопенія складає суть патологічного
процесу і визначає картину недуги та всі інші
симптоми.
■■■■■наМ
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Класифікація
I. За походженням лейкопенії бувають (а) на
бутими і (б) спадково зумовленими.
Набуті лейкопенії можуть виникати під
впливом фізичних (іонізуюча радіація), хіміч
них (бензол, цитостатики, лікарські препарати),
біологічних (віруси гепатиту, інфекційного мо
нонуклеозу) та імунних факторів.
Прикладами спадкових лейкопеній є нейтропенія Костмана, спадкова нейтропенія аутосомно-домінантного типу, синдром “лінивих лейко
цитів”, циклічна нейтропенія.
II. За видом лейкоцитів, кількість яких змен
шена, виділяють:
а) нейтропенію. Причин зменшення кількості
нейтрофільних лейкоцитів у периферичній
крові багато, і ми на них зупинимося при
обговоренні механізмів розвитку лейкопе
ній. При падінні цього показника нижче
0,75-109/л ведуть мову про агранулоцитоз
(див. далі);
б) лімфопенію. Цей варіант лейкопенії буває
значно рідше, ніж попередній. Основні його
причини: спадково зумовлені й набуті імуно
дефіцити, зокрема ВІЛ-інфекція (див. главу
14), стан після лікування глюкокортикоїдами
та препаратами цитотоксичної дії, автоімун
ні хвороби, деякі гострі вірусні інфекції;
в) еозинопенію. Зменшення кількості еозино
філів є характерним для стресових реакцій
(див. главу 8) і дії високих доз глюкокортикоїдів.
Механізми
Загалом можна виділити дві групи механізмів
лейкопенії (мова йтиме про зменшення кількос
ті клітин мієлоїдного ряду, оскільки механізми
лімфопенії та пов’язаної з нею імунологічної
недостатності розглядалися в главі 14).
І.
Порушення надходження лейкоцитів із
червоного кісткового мозку в кров. В основі цього
можуть лежати такі патологічні зміни:
1. Ушкодження кровотворних клітин. За цих
обставин розвивається так звана мієлотоксична лейкопенія, яка є одним із компонентів
синдрому гіпопластичної анемії, що ми вже
обговорювали (див. підглаву 30.2). Виділя
ють три основні механізми ушкодження й
загибелі клітин червоного кісткового мозку:
63
а) цитолітичний. Він пов’язаний з дією на
клітини іонізуючої радіації, цитостатичних препаратів, імунних факторів (анти
тіл, Т-лімфоцитів) та ін. Ступінь ураження
червоного кісткового мозку при цьому за
лежить від дози й тривалості дії зазначених
факторів;
б) антиметаболічний. У його основі лежить
дія агентів, що втручаються в обмін пури
нових і піримідинових основ, порушуючи
при цьому процеси поділу клітин мієлоїд
ного ряду. За таким принципом діють деякі
протипухлинні препарати й антибіотики
(левоміцетин);
в) ідіосинкразичний. Реалізується при повтор
ному введенні лікарських препаратів, чут
ливість організму до яких підвищена (ідіо
синкразія). Найчастіше це препарати, що
містять у своїй структурі бензольні кільця.
У випадку ідіосинкразії немає зв’язку між
імовірністю розвитку лейкопенії та дозою,
а також тривалістю дії лікарських засобів.
2. Порушення мітозу — неефективний лейкопоез. Основна причина цього - дефіцит
необхідних для клітинного поділу речо
вин, зокрема вітаміну В]2 і фолієвої кисло
ти. Брак зазначених сполук веде водночас
до розвитку В]2- і фолієводефіцитної ане
мії (неефективний еритропоез). Патоге
нез останньої був предметом обговорення
в підглаві 30.2. Крім того, порушення мі
тозу можуть виникати внаслідок недостат
ності лейкопоетинів. Така ситуація цілком
імовірна при втраті здатності стромальних
та інших клітин червоного кісткового моз
ку утворювати й вивільняти колонієстимулюючі фактори.
3. Порушення дозрівання лейкоцитів. їхню
основу можуть складати генетично зумов
лені дефекти як самих кровотворних клі
тин (наприклад, нейтропенія Костмана),
так і стромальних клітин “мікрооточення”
(наприклад, лейкопенія у “сталевих” ми
шей лінії SL/SLa). При цьому дозрівання
клітин крові досягає певної стадії (напри
клад, промієлоцитів) та зупиняється.
4. Порушення виходу лейкоцитів із червоно
го кісткового мозку в кров. Подібні зміни
часто пов’язані з генетичними дефектами
лейкоцитів, що порушують основні їхні
функції й властивості, зокрема здатність до
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
64
міграції. Прикладами можуть бути синдром
“лінивих лейкоцитів”, нейтропенія “ємен
ських євреїв ”.
5. Зменшення плацдарму лейкопоезу. Цей ме
ханізм має місце при заміщенні кровотвор
ної тканини лейкозними клітинами, мета
стазами пухлин та ін.
II. Скорочення часу перебування лейкоцитів
у периферичній крові. Розвиток лейкопенії за
цим механізмом може бути пов’язаний з такими
чинниками:
1. Деструкція (руйнування) лейкоцитів. Лейко
пенія подібного походження зумовлюється:
а) автоімунними механізмами (наприклад,
ревматоїдний артрит, системний червоний
вовчак). Руйнування лейкоцитів здійсню
ється за участю антилейкоцитарних антитіл,
специфічних до антигенів клітин білої крові
(II тип алергічних реакцій, див. главу 15);
б) імунними гаптеновими механізмами (на
приклад, амідопіринова гостра нейтропе
нія). У цьому випадку деструкцію лейкоци
тів викликають антитіла, утворені на лікар
ські препарати (гаптени), що фіксуються на
поверхні клітин. Механізми ушкодження
лейкоцитів такі ж, як і в інших алергічних
реакціях II типу;
в) гіперспленізмом - підвищенням фагоцитар
ної активності макрофагів селезінки.
2. Посилене використання лейкоцитів. Цьо
му передує прискорений перехід їх із
циркулюючого пулу в пристінковий з по
дальшою еміграцією в тканини. Висока
інтенсивність утилізації лейкоцитів, як
правило, пов’язана з вогнищами трива
лого хронічного запалення, зумовленого
бактеріями, патогенними грибами, рикет
сіями.
3. Виведення великої кількості лейкоцитів
з організму. Виражену хронічну втрату
нейтрофілів спостерігають у курців. У них
під час ранкового кашлю з мокротинням
втрачається від 0,5 до 2-Ю8 гранулоцитів
і 0,8-1,6-108 макрофагів.
Агранулоцитоз
Так називають клінічно-гематологічний синдром,
що характеризується різким зменшенням вмісту
нейтрофілів нижче 0,75Т09/л при зменшенні за
гальної кількості лейкоцитів нижче 1 • 109/л.
В основі розвитку агранулоцитозу можуть ле
жати два механізми: (а) мієлотоксичний - ура
ження червоного кісткового мозку та (б) імун
ний - руйнування клітин гранулоцитарного ряду
антилейкоцитарними антитілами (див. вище).
Агранулоцитоз супроводжується значним
ослабленням реактивності організму у зв’язку
з порушенням захисної функції лейкоцитів. Це
виявляє себе бурхливим розвитком бактеріаль
ної і грибкової інфекцій, особливо в тканинах
порожнини рота (виразково-некротичний гінгі
віт, стоматит, ангіна).
ЛЕЙКОЗИ
Велику категорію, що має важливе клінічне зна
чення, складають ураження системи крові пух
линного походження, іншими словами, злоякіс
ні пухлини, які ростуть із кровотворних клітин,
їх позначають терміном “гемобластози”.
Залежно від того, де міститься первинне дже
рело пухлини, гемобластози поділяють на дві
великі групи:
1) з первинним ураженням червоного кістко
вого мозку - лейкози (лейкемії);
2) з первинною локалізацією поза червоним
кістковим мозком (найчастіше в лімфатич
них вузлах) - лімф оми, лімфогранулема
тоз, лімфосаркома та ін. При цих хворо
бах червоний кістковий мозок також утя
гується в процес, але вторинно.
Предметом нашого обговорення будуть лей
кози т злоякісні пухлини, що виникають із кро
вотворних клітин і первинно уражують черво
ний кістковий мозок.
Які існують докази того, що лейкози - дійсно
хвороби пухлинної природи? Наведемо їх.
• При лейкозах, як і при злоякісних пухли
нах, відбувається безмежний і нерегульований поділ клітин.
• Лейкоз, як і будь-яка злоякісна пухлина,
виникає з одної-єдиної первинно зміненої,
трансформованої клітини. Усі лейкозні клі
тини, якими б різними вони не були, похо
дять із однієї клітини.
• Для лейкозів, як і для інших злоякісних
пухлин, характерне явище анаплазії - по
ява морфологічних, біохімічних та інших
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
властивостей, що наближають лейкозні
клітини до ембріональних.
• Лейкозам, як і іншим злоякісним пухлинам,
притаманна пухлинна прогресія, тобто на
буття із часом лейкозними клітинами все
більш і більш злоякісних властивостей.
• Однаковість причин розвитку лейкозів
і злоякісних пухлин. Одні й ті ж етіологічні
фактори (фізичні, хімічні, біологічні) здат
ні викликати виникнення як лейкозів, так
і інших злоякісних пухлин.
Проте лейкози мають цілий ряд істотних від
мінностей, що надає їм особливої специфічності.
• У випадку лейкозу неможливо точно вста
новити первинну локалізацію пухлини,
а отже, радикально видалити її вже на ран
ніх етапах розвитку.
• Лейкози - це пухлини, які від самого по
чатку метастазують. Лейкозні клітини лег
ко проникають у кров і розносяться нею по
всьому організму. Вони заселяють все нові
й нові ділянки, на яких виникають вторин
ні лейкозні інфільтрати.
• Для лейкозів характерна системність ура
ження. У зв’язку з раннім метастазуванням
зазнає змін уся система крові: червоний
кістковий мозок, лімфатичні вузли, селе
зінка, печінка.
» При лейкозах пригнічується нормальне
кровотворення. Це пов’язано з витісненням
нормальної кровотворної тканини лейкоз
ними клітинами. З другого боку, має значен
ня токсична дія продуктів лейкозних клітин
на клітини гемопоезу.
• Лейкози - це різновид пухлин, що уражу
ють людину переважно в дитячому ї моло
дому віці.
Важливим є питання, з яких кровотворних
клітин можуть виникати лейкози. Відповідаючи
на нього, слід мати на увазі, що будь-яка пухли
на може розвиватися тільки з тих клітин, які ма
ють первісну здатність до поділу. Не є винятком
і лейкози.
На рис. 30.34 було представлено сучасну
схему кровотворення, відповідно до якої кліти
ни гемопоезу поділяють на шість класів. Як за
значалося, здатність до поділу притаманна всім
представникам перших чотирьох класів і пер
шим “по вертикалі” клітинам V класу. Останні
65
ми клітинами мієлоїдного ряду, які ще можуть
ділитися, а отже, ставати джерелом лейкозів,
є мієлоцити (нейтрофільні, еозинофільні, базофільні), промоноцити, промегалоцити й поліхроматофільні нормоцити. Клітини V і VI класів
(дозріваючі й зрілі), що втратили здатність до
проліферації, трансформуватися в лейкозні не
можуть, оскільки в них уже немає структур,
необхідних для здійснення мітотичного поділу.
Що стосується клітин лімфоїдного ряду, то всі
вони можуть ставати джерелом лейкозів та інших
гемобластозів. Це пояснюється тим, що після ан
тигенної стимуляції настає бласттрансформація
зрілих В- і Т-лімфоцитів, тобто вони перетворю
ються на певного виду лімфобласти (імунобласти) - клітини IV класу, що проліферують.
Класифікація:
I. Залежно від особливостей патогенезу
і пов’язаної з ними гематологічної картини
лейкози поділяють на (а) гострі й (б) хро
нічні. Основна відмінність між ними по
лягає в тому, що при гострих лейкозах
пухлинні клітини, набувши здатності до
безмежного неконтрольованого росту, пов
ністю втрачають здатність дозрівати, тобто
диференціюватися в інші форми. У той же
час при хронічних лейкозах лейкозні кліти
ни поряд зі здатністю до безмежного рос
ту зберігають властивість дозрівати й пе
реходити у зрілі форми. Іншими словами,
при гострих лейкозах пухлинні клітини
тільки діляться і не дозрівають, при хро
нічних - діляться і дозрівають. З ураху
ванням цієї обставини гострі лейкози варто
вважати більш злоякісним видом хвороби.
II. Залежно від того, які кровотворні клі
тини втягуються в пухлинний процес, лей
кози поділяють на дві великі категорії: (а)
ураження мієлоїдної гілки гемопоезу (міє
лоз ейкози, еритромієлоз та ін.) і (б) пухли
ни клітин лімфоїдного паростка кровотво
рення (лімфолейкози).
III. Залежно від вмісту лейкоцитів у перифе
ричній крові лейкози бувають (а) лейкемічними (виражений лейкоцитоз - від 20 • 109/л
до 100 • 109/д), (б) сублейкемічними (помір
ний лейкоцитоз до 20 • 109/л), (в) алейкемічними (вміст лейкоцитів не міняється),
(г) лейкопенічними (кількість лейкоцитів
зменшується).
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
66
Гострі лейкози
Джерелом гострих лейкозів можуть бути крово
творні клітини перших чотирьох класів.
Якщо лейкоз розвивається з клітин I—III кла
сів, що не мають специфічних морфологічних
і цитохімічних ознак, то такий лейкоз називають
недиференційованим.
Коли ж розвиток лейкозу пов’язаний з кліти
нами IV класу (бластами), то за морфологічною
картиною і за допомогою ряду цитохімічних
методів можна встановити клітину, від якої по
ходить пухлина.
Якщо джерелом лейкозних клітин є мієлобласт, то такий лейкоз називають гострим мієлобластним, якщо лімфобласт - гострим лімфобластним, якщо монобласт - гострим монобластним і т.д.
Для гострих лейкозів характерні алейкемічний і лейкопенічний варіанти перебігу. Картина
крові відображає особливості патогенезу цієї
форми хвороби.
Як приклад розглянемо два найпоширеніших
види гострих лейкозів: гострий мієлобластний
і гострий лімфобластний.
Гострий мієлобластний лейкоз розвивається
переважно в молодих людей і людей середньо
го віку. Назва лейкозу свідчить про те, що його
джерелом є мієлобласт і що лейкозні клітини
(мієлобласти) не здатні диференціюватися.
Це визначає такі особливості картини крові
(рис. 30.37).
1. Лейкозні мієлобласти постійно діляться
і легко переходять у кров. Отже, у крові мож
на виявити пухлинні клітини - мієлобласти.
Гострий мієлобластний лейкоз
Гострий лімфобластний лейкоз
Хронічний мієлоцитарний лейкоз
Хронічний лімфоцитарний лейкоз
Рис. 30.37. Картина крові при різних видах лейкозів
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
67
2. Оскільки в червоному кістковому мозкові
зберігаються осередки нормального крово
творення, то вони залишаються джерелом
надходження у кров нормальних лейкоци
тів, тобто тих, які мають бути в крові в нор
мі — метамієлоцитів, псиїичкоядерних і сегментоядерних нейтрофілів.
3. У крові відсутні перехідні форми лейкоци
тів від мієлобластів до тих клітин, які вияв
ляють у нормі, тобто немає промієлоцитів
і мієлоцитів. Таке явище отримало назву
лейкемічного провалу (hiatus leucemicus).
Відсутність у крові перехідних форм по
яснюється тим, що лейкозні мієлобласти не
здатні диференціюватися в зазначені кліти
ни, а, з другого боку, осередки нормально
го кровотворення не “випускають” у кров
промієлоцитів і мієлоцитів (рис. 30.38).
Гострий лімфобластний лейкоз є типовим
лейкозом дитячого віку. Лейкоз розвивається
з лімфобласта. Пухлинні клітини втрачають
здатність до дозрівання. Тому для картини кро
ві будуть характерні лімфобласти (пухлинного
походження), а також всі ті клітини, які мають
бути в нормі (з осередків нормального крово
творення) (див. рис. 30.37).
Картина червоного кісткового мозку при всіх
гострих лейкозах має ряд особливостей. Поперше, у пунктаті кісткового мозку кількість
бластних клітин значно зростає і перевищує
ЗО % (у нормі значна частина припадає на клі
тини V класу, які ще зберігають здатність до
поділу, див. вище). По-друге, лейкозні бластні
клітини мають якісні відмінності, що є ознакою
їхньої анаплазії.
Хронічні лейкози
Цей вид лейкозів розвивається з кровотворних
клітин IV класу.
Номенклатура хронічних лейкозів ґрунтуєть
ся на тому, який тип клітин становить переваж
ну більшість у популяції лейкозних клітин. На
зва лейкозу вказує на паросток кровотворення,
що виявився ураженим.
Виділяють хронічний мієлоцитарний лейкоз
(хронічний мієлолейкоз), хронічний лімфоцитарний лейкоз (хронічний лімфолейкоз), хро
нічний моноцитарний лейкоз, хронічний еритромієлоз та ін. При цьому зазнають уражень
Рис. 30.38. Походження "лейкемічного провалу"
при гострому мієлобластному лейкозі
відповідно гранулоцитарний, лімфоцитарний,
моноцитарний та еритроїдний паростки крові.
Для хронічних лейкозів найчастіше харак
терні лейкемічний і сублейкемічний варіанти
перебігу. Картина крові має істотні відмінності,
якщо порівнювати з гострими лейкозами. Це по
яснюється особливостями патогенезу цих видів
лейкозів.
Розглянемо дві найпоширеніші форми хро
нічних лейкозів: хронічний мієлоцитарний
і хронічний лімфоцитарний.
Хронічний мієлоцитарний лейкоз.
Джерелом
розвитку мієлолейкозу можуть бути мієлоблас
ти, промієлоцити та мієлоцити. Для прикладу
візьмемо варіант, пов’язаний з мієлобластями.
Оскільки лейкоз хронічний, то це означає, що
68
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
того, хронічний мієлолейкоз часто супроводжує
так звана базофільно-еозинофільна асоціація:
збільшується частка базофілів і еозинофілів
у периферичній крові.
У червоному кістковому мозку переважають
клітинні елементи мієлоїдного ряду. Жирова
тканина повністю витісняється кровотворною,
досить швидко зменшується плацдарм нормаль
ного гемопоезу.
Причина відсутності "лейкемічного
провалу" при хронічному мієлолейкозі
лейкозні мієлобласти зберігають здатність до
диференціації в наступні форми. Тому з лейкозної тканини червоного кісткового мозку в кров
у великій кількості виходять всі клітини, які
походять від мієлобластів, а саме промієлоцити, мієлоцити, метамієлоцити, паличкоядерні й сегментоядерні гранулоцити (рис. 30.37).
Таким чином, для хронічного мієлолейкозу, на
відміну від гострого мієлобластного, не ха
рактерний феномен “лейкемічного провалу”
(рис. 30.39), а має місце гіперрегенераторне
зміщення лейкоцитарної формули вліво. Окрім
Хронічний лімфоцитарний лейкоз. Вважають,
що пухлинна трансформація, що започатковує
лімфолейкоз, відбувається на рівні лімфобластів. Однак, на відміну від гострого лімфобластного лейкозу, ці клітини зберегли здатність ди
ференціюватися в подальші форми - пролімфоцити та лімфоцити.
Тому основна маса лейкозних клітин крові
представлена лімфоцитами (рис. 30.37). їхня
кількість у лейкоцитарній формулі становить
80-90 %. Окрім лейкозних лімфоцитів, у крові
можуть виявлятися пролімфоцити й поодинокі
лімфобласти. Характерною є поява так званих
тіней Гумпрехта — напівзруйнованих ядер лім
фоцитів, що утворюються як артефакт при готу
ванні мазків крові.
Лімфоцити лейкозного клону (В-лімфоцити)
можуть продукувати імуноглобуліни однієї спе
цифічності (моноклональні), однак при цьому
пригнічується антитілоутворєння іншими нор
мальними клонами В-лімфоцитів і поступово
розвивається імунологічна недостатність.
У червоному кістковому мозку відбувається
майже тотальне заміщення кровотворної ткани
ни лімфоцитами.
Принциповим є питання про можливість пере
ходу гострих лейкозів у хронічні і, навпаки, хро
нічних - у гострі. Відповідь на нього слід шукати
в площині кардинальних властивостей пухлин
них клітин. Якщо брати їх до уваги, то слід кон
статувати, що перехід гострих лейкозів у хронічні
неможливий. Таке перетворення означало б, що
лейкозні клітини, які безповоротно втратили здат
ність до дозрівання, знову набули цю властивість.
Нині немає даних про те, що подібний перебіг по
дій можливий. Більш злоякісна пухлина в процесі
свого розвитку ніяк не може стати менш злоякіс
ною, а все відбувається якраз навпаки.
З
другого боку, хронічний лейкоз із часом може
трансформуватися в гострий. Це означає, що
лейкозні клітини, які ще зберігали здатність до
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
диференціації, таку властивість втрачають. При
цьому лейкоз із менш злоякісної перетворюється
в більш злоякісну форму. Подібне явище по суті
відображає пухлинну прогресію - загальну влас
тивість усіх злоякісних пухлин (див. главу 19).
Гематологічним проявом переходу хронічно
го лейкозу в гострий є так званий “бластний
криз”, коли в крові й червоному кістковому
мозкові різко зростає кількість бластних клітин,
а в крові поступово зникають перехідні форми.
Причини лейкозів
Етіологічні фактори, що мають стосунок до лей
козів, поділяють на дві групи: (а) екзогенні й (б)
ендогенні.
Екзогенними причинами можуть бути всі чин
ники, які здатні викликати розвиток злоякісних
пухлин узагалі (див. главу 19). Такими є (1) фі
зичні фактори (іонізуюча радіація); (2) хімічні
агенти (хімічні канцерогени); (3) фактори біоло
гічного походження (онкогенні віруси).
До ендогенних факторів відносять: (1) гене
тичні чинники (спадкову схильність, хромосом
ні хвороби); (2) тривалу імунологічну стимуля
цію, зумовлену хронічними запальними проце
сами; (3) імунологічну недостатність.
Докази того, що іонізуюча радіація може бути
причиною лейкозів, було наведено у відповідній
частині глави 19.
Про роль хімічних канцерогенів у виникненні
лейкозів свідчить можливість відтворення остан
ніх у тварин (миші, щури, кури) за допомогою
поліциклічних ароматичних вуглеводнів, нітрозосполук та ін., а також розвиток лейкозів у лю
дей після тривалої інтоксикації бензолом та його
похідними, після приймання цитостатичних засо
бів, що використовувалися з лікувальною метою.
Значення вірусів було доведено ще в 1908
році Елерманом і Бангом, які вперше одержали
лейкоз у курей після введення їм безклітинних
фільтратів лейкозних клітин. У 1950 році Гросс
аналогічним способом індукував лейкоз у ссав
ців (дитинчат мишей).
Нині відомо багато вірусів, здатних викликати
трансформацію кровотворних клітин у пухлинні.
У тварин (птахів, мишей, щурів, кішок, мавп)
лейкози зумовлюються вірусами, більшість
з яких належить до РНК-вмісних, а саме - до
ретровірусів. Залежно від характеру онкогенної
дії їх поділяють на
69
а) гостротрансформуючі, тобто такі, що ви
кликають розвиток пухлин після короткого
латентного періоду (віруси гострих лейко
зів). Ці віруси містять у своїй структурі онкоген (див. главу 19);
б) повільнотрансформуючі. Вони зумовлюють
розвиток пухлин після тривалого латентно
го періоду (віруси хронічних лімфолейкозів). Геном цих вірусів, на відміну від по
передніх, не містить онкогена.
Стосовно лейкозів людини сьогодні з упевненіс
тю можна сказати, що виявлено три віруси, з яки
ми пов’язують виникнення хвороби. Такими є:
1) вірус Епштейна — Барр (Epstein — Barr
virus - EBV). Він належить до ДНКвмісних вірусів сімейства герпесвірусів.
Викликає розвиток лімфоми Беркітта
у дітей віком від 2 до 14 років у деяких
країнах Центральної Африки. Лімфома
Беркітта не є в буквальному значенні сло
ва лейкозом, оскільки розвивається без
первинного ураження червоного кістко
вого мозку. Джерело цієї пухлини - у під
щелепних лімфатичних вузлах. Пухлина
росте дуже швидко, збільшуючись у масі
в 2 рази через кожні 2 дні. Через 6-12
тижнів дитина гине. Крім того, геном EBV
виявляють у 30-40 % випадків лімфоми
Ходжкіна, а також у багатьох В-клітинних
лімфомах, що виникають при Т-клітинних
імунодефіцитах, і дуже рідкому різновиді
лімфом, зумовлених природними кілерами (NK-клітинами);
2) вірус Т-клітинної лейкемії людини (human
T-cell leukemia virus-1 - HTLV-1). Будучи
ретровірусом, що належить до групи так
званих Т-лімфотропних вірусів, він ви
кликає розвиток у дорослих Т-клітинної
лімфоми-лейкемії — лейкозу, поширено
го в Японії, країнах басейну Карибсько
го моря, у Південній Америці, на Алясці.
За своїми властивостями HTLV-1 подібний
до вірусу імунодефіциту людини (ВІЛ), що
викликає СНІД;
3) герпесвірус саркоми Капоті / герпесвірус людини-8 (Kaposi sarcoma herpesvirus/
human herpesvirus-8 - KSHV/HHV-8). Цей
вірус є причиною розвитку однієї з рідкіс
них форм В-клітинної лімфоми, пов’язаної
з тканинами плеври.
70
Про роль спадкових чинників у виникненні
лейкозів свідчить цілий ряд фактів.
• У людей із хромосомними хворобами істот
но збільшується частота виникнення лей
козів. Так, в осіб з хворобою Дауна (трисомія по 21-й хромосомі) частота розвит
ку гострих лейкозів зростає в 18-20 разів.
Таку ж закономірність виявлено й серед
хворих на синдроми Клайнфельтера, Шерешевського - Тернера, Фанконі.
• Майже при всіх видах лейкозів у пухлин
них клітинах виявляють ознаки хромосом
них аберацій: делеція, транслокація, інвер
сія. Дуже характерною ознакою хронічного
мієлолейкозу є поява так званої “філадель
фійської” хромосоми (вона виявляється
в 92 % хворих з даним видом лейкозу).
Ця хромосома являє собою одну з 22-ї пари
хромосом, у якої відбулося вкорочення
приблизно половини довгого плеча внаслі
док обміну ділянками (транслокації) з 9-ю
хромосомою (див. далі).
• Неодноразово в літературі описано випад
ки так званої сімейної форми хронічних
лейкозів.
• Виведено чисті лінії тварин (мишей), у яких
висока вразливість спонтанними лейкозами.
Сьогодні є докази того, що тривала імуно
логічна стимуляція, яка супроводжує хронічні
запальні процеси, має стосунок до виникнен
ня пухлин лімфоїдного походження. Ось деякі
з них:
• Встановлено тісний зв’язок між хронічним
запаленням слизової оболонки шлунка, зу
мовленим Helicobacter pylori, і В-клітинною
лімфомою, що має локалізацію у стінках
цього органа.
• Таку ж асоціацію виявлено між особливим
різновидом хронічного запалення тонкої
кишки - залежною від глютену ентеропатією (див. главу 34) та інтестинальною
Т-клітинною лімфомою.
До виникнення гемобластозів може бути при
четна не тільки стимуляція імунної системи,
а й розвиток імунологічної недостатності. Так,
при спадково зумовлених імунодефіцитних ста
нах, пов’язаних з ураженням В- і Т-лімфоцитів
(синдроми Брутона, Луї - Барр, Віскотта - Олдрича), значно зростає частота розвитку гос
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
трого лімфобластного лейкозу й лімфосарком,
а у ВІЛ-інфікованих хворих — ризик появи
В-клітинних лімфом.
Застосування цитостатиків, що викликають
розвиток імунодепресивного стану (вторинних
імунодефіцитів), супроводжується збільшенням
частоти виникнення ракових пухлин у 2,5 рази,
а пухлин лімфоїдної тканини - у 35 разів.
Механізми лейкозів
Механізми розвитку лейкозів підпорядковують
ся загальним закономірностям злоякісного пух
линного росту, що ми їх у деталях розглядали
в главі 19. Проте є ряд особливостей, на яких
і сконцентруємо свою увагу.
Для зручності аналізу поділимо патогенез
лейкозного процесу на три послідовні етапи чи
стадії.
1, Пухлинна трансформація, тобто пере
творення нормальної клітини в лейкозну.
Для розвитку цієї події в клітинах лімфо
їдної системи особливо велике значення
має явище, що отримало назву регульо
ваної нестабільності геному. Суть його
полягає в тому, що під впливом анти
генної стимуляції в геномі попередників
В- і Т-лімфоцитів відбувається перемі
щення генетичного матеріалу — рекомбі
нація (реаранжування) генів, що кодують
імуноглобуліни у В-клітинах і специфічні
рецептори Т-клітин. Цей механізм дуже
складний і схематично може бути позна
чений як процес розрізування і зшивання
у нових місцях специфічних фрагментів
ДНК, об’єднаних у три групи з умовним
позначенням V, D, J. Формування специ
фічних послідовностей важких ланцюгів
відбувається завдяки рекомбінації генів
з усіх трьох зазначених вище ділянок ДНК
(VDJ-механізм), а легких - лише двох: V та
J. Завдяки реаранжуванню генів досягаєть
ся надзвичайно велика кількість можливих
комбінацій (наприклад, для Т-клітинного
рецептора - 3 • 1022), чим і пояснюється ви
сока різноманітність і специфічність імуноглобулінів та рецепторів Т-лімфоцитів.
VDJ-механізм рекомбінації здійснюється
за допомогою Rag-білків, що є продуктами
відповідних генів (Recombination activating
genes) (рис. 30.40). Вважають, що помилки
71
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
в діяльності цих протеїнів можуть вести до
зшивання вирізаних фрагментів ДНК у не
правильних місцях. Якщо це відбувається
поблизу протоонкогенів, то може змінитися
експресія останніх, і вони набудуть власти
востей клітинних онкогенів (див. главу 19).
Для зрілих, стимульованих антигеном В-лімфоцитів нестабільність геному характерна
й для стадії диференціації їх в зародкових цен
трах лімфатичних вузлів. Тут гени імуноглобулінів зазнають подальшої модифікації, яка
зрештою визначає клас антитіл, що буде синте
зуватися. Поряд з цим для клітин, що перебува
ють в зародкових центрах, характерна т. зв. гіпермутантність - висока частота спонтанних
соматичних мутацій. Цілком можливо, що і по
рушення регульованої модифікації імуноглобулінових генів, і спонтанні мутації в В-клітинах
змінюють експресію розташованих поблизу
протоонкогенів таким способом започаткову
ють пухлинну трансформацію. Хоча точний
механізм цього ще не відомий, притаманна
В-лімфоцитам нестабільність геному на етапі їх
диференціації в лімфатичних вузлах може пояс
нювати, чому частота лімфом, що розвиваються
саме з цих клітин, набагато вища, ніж пухлин,
джерелом яких є зрілі Т-лімфоцити.
Переміщення генетичного матеріалу між
хромосомами є важливим механізмом розвитку
пухлин як мієлоїдного, так і лімфоїдного по
ходження. Як приклад розглянемо дві хвороби:
хронічний мієлолейкоз і лімфому Беркітта.
• Хронічний мієлолейкоз. Генетичним мар
кером цієї недуги є так звана філадельфій
ська хромосома. Причина її появи в транслокації, що відбувається між 9-ю і 22-ю
хромосомами (рис. 30.41). Річ у тім, що
у кровотворних клітинах у передаванні ін
формації від клітинних рецепторів до ядра
беруть участь цитоплазматичні тирозинові протеїнкінази (див. главу 8), одна з яких
кодується геном ABL, що міститься у 9-й
хромосомі. При транслокації цього гена
у 22-гу хромосому відбувається злиття
генів ABL і BCR, унаслідок чого утворю
ється новий гібридний ген, продукт якого
Abl-Bcr є онкопротеіном. На відміну від
нормального білка АЬІ, новий протеїн збе
рігає високу ферментну активність постій
но, незалежно від того, діють на клітину чи
ні стимулятори клітинного поділу.
Рис. 30.40. Участь Rag-білків у рекомбінації V, D та J фрагментів ДНК імуноглобулінових генів
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
72
Реципрокна транслокація між хромосомами 9 і 22 як механізм пухлинної трансформації клітин
при хронічному мієлолейкозі
Лімфома Беркітта. У виникненні цієї
пухлини має значення переміщення протоонкогена МУС з 8-ї хромосоми у 14-ту
(рис. 30.42). Унаслідок такої транслокації
він потрапляє в ділянку розташування гена,
що кодує важкий ланцюг молекули імуноглобуліну. Дія на МУС потужних підсилю
вачів зазначеного гена спричиняється до гіперекспресії протоонкогена: значно зростає
кількість його білкового продукту, що має
відношення до регуляції клітинного поділу.
2. Моноклонова стадія. У результаті пухлин
ної трансформації утворюється клон одна
кових пухлинних клітин, для яких харак
терні безмежний ріст і порушена здатність
до диференціації. Швидкий ріст лейкозних
клітин призводить до поширення (мета
стазування) їх по всій системі крові, вклю
чаючи кровотворні органи й кров. У лей
козних клітинах, що циркулюють у крові,
виявляють однакові хромосомні маркери,
такі як уже згадувана філадельфійська хро
мосома для хронічного мієлолейкозу.
•
Нестабільність генотипу лейкозних клітин
є важливою умовою, що полегшує виникнення
нових мутацій # як спонтанних, так і зумовле
них тривалим впливом канцерогенних факторів.
У результаті цього утворюються нові пухлинні
клони, та пухлинний процес переходить у нову,
злоякіснішу стадію.
3. Поліклонова стадія. Перехід моноклонової
стадії в поліьслонову є проявом загальної
властивості злоякісних пухлин — пухлинної
прогресії (див. главу 19). Лейкозні клітини
набувають більшої злоякісності. Вони ста
ють такими, що їх не можна диференціюва
ти ані морфологічними, ані цитохімічними
методами. У кровотворних органах і кро
ві збільшується кількість бластних клітин
з дегенеративними змінами ядра і цитоплаз
ми. Лейкозні клітини поширюються за межі
кровотворних органів, утворюючи лейкозні
інфільтрати в різних тканинах. Унаслідок
добору знищуються клітини тих клонів, на
які діяли імунна система й гормони організ
му, цитостатичні засоби (хімічні, гормональ
ні, променеві). Домінують клони пухлинних
клітин, найбільш стійких до цих впливів.
При хронічних лейкозах клітини поступово
втрачають здатність до дозрівання, а отже, із
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
73
Реципрокна транслокація між хромосомами 8 і 14 як механізм пухлинної трансформації клітин
при лімфомі Беркітта
крові зникають перехідні форми лейкоцитів настає бластний криз, хронічний лейкоз набуває
гематологічних ознак гострого (див. вище).
Клінічні прояви. Велику кількість клінічних
ознак, характерних для лейкозів, можна розді
лити на три групи.
І.
Гематологічні синдроми, зумовлені замі
щенням нормальної кровотворної тканини лейкозною і пригніченням у зв’язку з цим нормаль
ного кровотворення. До таких відносять:
1) панцитопенію - зменшення вмісту всіх
формених елементів крові. Вона особливо
виражена при гострих лейкозах;
2) анемію. Основу її патогенезу становить по
рушення еритропоезу. Однак при деяких
видах лейкозів певне значення може мати
імунний гемоліз еритроцитів (наприклад,
при хронічному лімфолейкозі) і кровотечі
(геморагічний синдром);
3) геморагічний синдром. Він зумовлений
в основному тромбоцитопенією та лейкозними інфільтратами в стінках крово
носних судин;
4)
порушення неспецифічного протимікроб
ного захисту, у зв’язку з чим зменшуєть
ся резистентність організму до інфекцій.
Основною причиною цього є зменшення
вмісту функціонально повноцінних грану
лоцитів;
5) імунологічна недостатність. Розвиваєть
ся як наслідок лімфопенії (при гострих
лейкозах і хронічному мієлолейкозі) або
неповноцінності лейкозних лімфоцитів
(хронічний лімфолейкоз).
II.
Синдроми, пов’язані з особливостями
функціонування лейкозних клітин. Серед них:
1) гарячка. Показано, що тільки у 7-8 % хво
рих на лейкози підвищення температури
на початку хвороби пов’язане з інфекцією.
У більшості ж випадків гарячка має неінфекційне походження;
2) інтоксикація. Велика кількість лейкозних
клітин гине й вивільняє у кров свій вміст.
Багато компонентів загиблих клітин мають
токсичну дію на центральну нервову сис
тему. Звідси стомлюваність, загальна слаб
кість, нудота та ін.;
3) автоімунні процеси. Вони пов’язані зі змі
нами, що відбуваються в лімфоцитарному
паростку крові, а саме з появою так званих
“заборонених” клонів лімфоцитів, зі змен-
74
шенням кількості й функціональної актив
ності Т-супресорів.
III.
Синдроми, зумовлені метастазуванням
лейкозних клітин і розвитком лейкозних проліфератів у різних органах і тканинах. Такими є:
1) збільшення розмірів лімфатичних вузлів,
печінки й селезінки;
2) шкірний синдром. Його зумовлює поява
в шкірі проліфератів лейкозних клітин лейкемідів;
3) виразково-некротичні ураження слизових
оболонок (виразково-некротичні стоматит,
ангіна, ентеропатії);
4) кістково-суглобовий синдром, що виявляє
себе болем у кістках і суглобах;
5) синдром нейролейкозу. Він може мати фор
му менінгеального синдрому, синдрому
підвищення внутрішньочерепного тиску,
різноманітних неврологічних порушень:
парезів, паралічів, парестезій. В основі
його розвитку - поява лейкозних проліфе
ратів в оболонках головного й спинного
мозку, речовині мозку, нервових стовбурах,
вегетативних гангліях;
6) лейкозний пневмоніт. Лейкозні проліферати порушують дихальну функцію легень розвивається недостатність зовнішнього
дихання;
7) серцева недостатність. Може бути на
слідком розмноження лейкозних клітин
у м’язі серця.
30.5. ПОРУШЕННЯ ГЕМОСТАЗУ.
ГЕМОРАГІЧНИЙ ДІАТЕЗ
В організмі існують механізми, що забезпечу
ють, з одного боку, збереження рідкого стану
крові, а з другого - зупинку кровотечі при ушко
дженні кровоносних судин. їх об’єднують по
няттям система гемостазу.
Виділяють два механізми гемостазу: (1) судинно-тромбоцитарний і (2) коагуляційний.
Судинно-тромбоцитарний
(первинний,
мікроциркуляторний) гемостаз забезпечує зу
пинку кровотечі з судин мікроциркуляторного русла, що мають діаметр до 100 мкм. Він
здійснюється завдяки взаємодії судинної стінки
з тромбоцитами. Унаслідок активації судиннотромбоцитарного гемостазу утворюється білий
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
тромбоцитарний тромб. Порушення цього меха
нізму гемостазу є причиною майже 80 % крово
теч і 95 % випадків тромбоутворення.
Коагуляційний (вторинний, макроциркуляторний) гемостаз є продовженням судинно-тромбоцитарного і здійснюється на його основі. Коагу
ляційний гемостаз бере участь у зупинці крово
теч із судин, діаметр яких перевищує 100 мкм.
У результаті його активації утворюється черво
ний тромб, що складається з фібрину і форме
них елементів крові.
Для розуміння сутності різноманітних пору
шень гемостазу важливим є розуміння механіз
мів, що лежать в основі функціонування систе
ми гемостазу в нормі. Коротенько розглянемо їх.
Судинно-тромбоцитарний гемостаз. Як
ви
пливає з назви, до його здійснення причетні су
динна стінка і тромбоцити.
Про роль стінки кровоносних судин в ак
тивації механізмів гемостазу і в забезпеченні
тромборезистентності докладно йшлося в главі
16. Тут лише окреслимо деякі основні аспекти
участі тромбоцитів у процесах гемостазу.
Тромбоцитам притаманний ряд властивос
тей, що мають прямий зв’язок з гемостазом.
1. Ангіотрофічна функція. Так позначають
здатність тромбоцитів підтримувати нор
мальну структуру й функцію стінок мікросудин. Вважають, що тромбоцити є
фізіологічними “годувальниками” ендо
телію. Ендотеліальні клітини поглинають
тромбоцити, з яких вивільняються речови
ни, необхідні для підтримки структурної
цілісності й функціональної активності
мікросудин.
2. Вазоконстрикторна функція. Це здатність
викликати спазм ушкоджених судин завдя
ки вивільненню із тромбоцитів вазоактивних речовин: адреналіну, норадреналіну,
серотоніну.
3. Адгезивно-агрегаційна функція. Її суть по
лягає в тому, що тромбоцити можуть при
липати до ушкоджених ділянок судинної
стінки і склеюватися один з одним, утво
рюючи тромбоцитарну пробку.
4. Участь у зсіданні крові. Вона пов’язана
з вивільненням так званих тромбоцитарних факторів зсідання крові, зокрема, фак
тора 3, фактора 4, фактора 6 (тромбостеніну) та ін.
75
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Рис. 30.43. Схема процесів, що лежать в основі судинно-тромбоцитарного гемостазу
Судинно-тромбоцитарний гемостаз склада
ється з ряду послідовних процесів (рис. 30.43).
L Спазм артеріол. Розрізняють (а) первин
ний (початковий) і (б) вторинний (відстро
чений) спазм. Перший виникає відразу ж
після ушкодження судинної стінки, триває
кілька секунд, основний механізм його роз
витку - рефлекторний. Причиною вторин
ного відстроченого спазму є біогенні аміни,
які вивільняються тромбоцитами, — катехоламіни, серотонін.
2. Адгезія тромбоцитів - прилипання тром
боцитів до ушкоджених ділянок судинної
стінки. Основною причиною цього є ого
лення (демаскування) колагену внаслідок
ушкодження ендотелію судин. Розрізняють
(1) доконтактну і (2) контактну фази адгезії
тромбоцитів.
Під час доконтактної фази в крові ще до
взаємодії з ушкодженою стінкою судини прохо
дить первинна активація тромбоцитів. Спочатку
змінюється форма тромбоцитів від дископодіб
ної до сферичної (набряк). Потім викидаються
довгі ниткоподібні відростки (від 3 до 10 у кож
ному тромбоциті).
Під час контактної фази відбувається взає
модія відростків активованих тромбоцитів з еле
ментами базальної мембрани судинної стінки.
При цьому мають значення:
а) безпосередній контакт відростків тромбо
цитів з колагеном',
б) опосередкований контакт тромбоцитів
з колагеном через фактор Віллебранда;
в) реверсія електричного заряду інтими при її
ушкодженні (заряд міняється з на “+”),
у результаті чого можлива електростатична
взаємодія тромбоцитів, що мають негатив
ний поверхневий заряд, зі стінкою судини;
г) уповільнення течії крові в ушкодженій су
дині.
3.
Агрегація тромбоцитів - набрякання
і склеювання кров’яних пластинок. Причи
ною агрегації є поява речовин-агрегантів.
Вони можуть бути тромбоцитарного (вихо
дять з активованих тромбоцитів) і нетромбоцитарного (вивільняються ушкоджени
ми клітинами, лейкоцитами, утворюються
в плазмі крові) походження.
Найбільше значення мають такі агреганти:
а) АДФ. Вивільняється з ушкоджених клітин
судинної стінки, гемолізованих еритроци
тів, тромбоцитів у процесі їхньої активації;
б) тромбоксан А2 і арахідонова кислота.
Тромбоксан А, є продуктом циклоксиге-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
76
назного шляху перетворення арахідонової
кислоти в тромбоцитах (див. главу 17);
в) біогенні аміни - адреналін, серотонін. їхні
джерела - плазма крові і тромбоцити;
г) фактор активації тромбоцитів (ФАТ).
Цей фактор є фосфоліпідом, вивільняється
тканинними базофілами, базофілами й ней
трофілами крові (див. главу 17);
д) тромбін. Він є потужним агрегантом у до
зах значно менших, якщо порівнювати
з тими, що викликають зсідання крові. Такі
малі кількості тромбіну завжди утворю
ються в місцях ушкодження судинної стін
ки завдяки зовнішньому механізму зсідан
ня крові, пов’язаному з вивільненням клі
тинного тромбопластину;
е) тромбоспондин. Після вивільнення з акти
вованих тромбоцитів адсорбується на їхній
мембрані, взаємодіє з білками крові.
Окрім речовин-агрегантів, існують фактори,
що мають протилежні, антиагрегантні власти
вості. До них, зокрема, належать простациклін,
продукти фібринолізу, простациклінозалежний
макромолекулярний білок, білковий фактор
Барнес - Ліана.
У самому процесі агрегації тромбоцитів роз
різняють ряд послідовних етапів:
початкова агрегація. Вона відбувається
одночасно з адгезією тромбоцитів. Її зу
мовлює АДФ нетромбоцитарного похо
дження (вивільняється з ушкоджених клі
тин судинної стінки);
2) оборотна агрегація. На цьому етапі агре
гація може бути припинена, тромбоцити
ще не ушкоджені. Оборотну агрегацію ви
кликають тромбоцитарний АДФ, а також
тромбоксан А2 та арахідонова кислота;
3) необоротна агрегація. У цей період тром
боцити ушкоджуються й гинуть. Вважають,
що основною причиною необоротної агре
гації є тромбін, що утворюється локально
в місці ушкодження судинної стінки.
1)
Механізми агрегації тромбоцитів охоплюють
дві групи процесів: (І) активацію тромбоцитів
і (II) власне склеювання.
Активація тромбоцитів відбувається під
впливом речовин-агрегантів, які збільшують
проникність тромбоцитарної мембрани до іонів
кальцію (рис. 30.44). У результаті цього концен
трація останніх у цитоплазмі кров’яних пласти
нок зростає. Це викликає щонайменше чотири
функціонально важливих ефекти:
1) скорочення мікрофібрил, у результаті чого
утворюються довгі ниткоподібні відростки;
Механізм активації тромбоцитів, що передує їх склеюванню
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
посилення гідролізу АТФ, наслідком чого
є утворення потужного агреганта - АДФ;
3) викид гранул тромбоцитів (див. далі);
4) активація фосфоліпази А2, що викликає
утворення арахідонової кислоти, а потім
і тромбоксану Аг
2)
У процесі власне склеювання тромбоцитів
мають значення:
а) утворення між тромбоцитами “містків ”,
що складаються з АДФ та іонів кальцію',
б) білкове склеювання — поява “містків” із біл
ків плазми крові. Останні отримали назву
плазмових кофакторів агрегації. До них від
носять фібриноген, альбуміни, агрексони А
і В. Усі ці протеїни склеюють тромбоцити
завдяки взаємодії з глікопротеїновими рецеп
торами тромбоцитів (існує 5 типів таких ре
цепторів) і тромбоспондином — агрегантом,
адсорбованим на тромбоцитарній мембрані.
4. Реакція вивільнення. Так називають виді
лення із тромбоцитів гранул чотирьох ти
пів. Розрізняють реакції вивільнення І і ви
вільнення II. Перша - це реакція раннього
вивільнення, вона здійснюється на етапі
початкової агрегації тромбоцитів. Наслід
ком її є вихід гранул І типу (містять біоген
ні аміни) і II типу (містять білки, зокрема,
фактор 4 пластинок, фактор Віллебранда).
Реакція вивільнення II - це реакція пізнього
вивільнення, відбувається на етапі необоротної
агрегації тромбоцитів. При ушкодженні тромбоцитарних мембран із пластинок виходять
гранули III і IV типів, що містять лізосомні фер
менти, фактор 3.
5. Консолідація тромбу. Суть цього явища
полягає в ущільненні тромбу, у результаті
чого формується остаточний його варіант.
В основі процесу лежить в’язкий мета
морфоз тромбоцитів та їхня ретракція під
впливом тромбостеніну.
Коагуляційний гемостаз. Коагуляція (зсідання
крові) є складним багатоетапним ферментатив
ним процесом, що в кінцевому підсумку закін
чується утворенням фібринового згустку.
Основу процесу становлять реакції протео
лізу, у яких прямо або опосередковано беруть
участь 12 факторів зсідання (усі вони, за винят
77
ком ф. III, мають плазмове походження) і фактор
З тромбоцитів.
До факторів зсідання крові відносять: ф. І —
фібриноген, ф. II — протромбін, ф. III — тканин
ний тромбопластин\ ф. IV - іони кальцію; ф. V —
проакцелерин, ф. VII» проконвертин, ф. VIII антигемофільний глобулін, ф. IX - фактор
Крістмаса, ф. X - фактор Стюарта - Прауера,
ф. XI - плазмовий попередник тромбопластину,
ф. XII - фактор Хагемана, ф. XIII — фібринстабілізуючий фактор.
За функціональними властивостями всі фак
тори, що беруть участь у зсіданні крові, можна
поділити на такі групи:
1) білки-ферменти. Це в основному протеолі
тичні ферменти: ф. II, III, VII, IX, X, XI, XII;
один фактор (ф. XIII) є трансферазою. Усі
зазначені ферменти містяться в крові і тка
нинах у неактивній формі. їхня активація
здійснюється шляхом протеолітичного від
щеплення пептидів, що закривають активні
центри ферментів. Таке відщеплення відбу
вається за участю активованого попередньо
го фактора зсідання (активної протеази). Та
ким чином, реакції активації зсідання крові
мають каскадний, ланцюговий характер;
2) неферментні білки — акселератори (білки-прискорювачі). Такими є ф-V і ф.УШ.
Ці фактори в сотні разів прискорюють фер
ментативні реакції зсідання крові. На від
міну від ферментів, вони споживаються
в процесі коагуляції крові;
3) фосфоліпідна матриця (мікромембрана),
на якій відбуваються впорядковані реакції
зсідання крові. Роль такої матриці можуть
виконувати: (а) фактор 3 тромбоцитів; (б)
фосфоліпідні фрагменти мембран ушко
джених клітин; (в) фосфоліпідні фрагмен
ти мембран гемолізованих еритроцитів;
4) іони кальцію. їхня участь у зсіданні крові
полягає у прив’язуванні білкових факторів
до фосфоліпідних матриць;
5) субстрат зсідання крові - фібриноген (ф. І).
Більшість факторів зсідання синтезується
в печінці. їх поділяють на дві групи: (а) вітамінК-залежні: ф. II, VII, IX, X (вітамін К у коферментній формі входить до складу печінкових
карбоксилаз, які беруть участь в утворенні за
значених факторів) і (б) вітамін-К-незалежні:
78
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Зовнішній механізм
................................... Внутрішній механізм
Рис. 30.45. Механізми активації системи зсідання крові
ф. I, V, XI (їхнє утворення не вимагає вітамі
ну К).
Процес зсідання крові проходить три фази.
І
фаза - утворення протромбінази. Існує
три різних механізми активації цього процесу
(рис. 30.45):
а) зовнішній (тканинний) механізм. Він ак
тивується при ушкодженні клітин. При
цьому вивільняються ферменти - тканин
ний тромбопластин (ф. III) і фосфоліпідні
фрагменти мембран, що стають матрицею,
на якій фіксуються фактори зсідання крові.
Це дуже швидкий механізм активації зсі
дання. Він забезпечує зсідання крові поза
кровоносними судинами (при крововили
вах у тканини) і локальне утворення малих
доз тромбіну, необхідного для необоротної
агрегації тромбоцитів (рис. 30.46);
б) внутрішній (кров’яний) механізм. Почат
ком його є контактна або ферментативна
активація ф. XII крові. Внутрішній меха
нізм зсідання досить тривалий, тому пер
ша його фаза по суті визначає час усього
процесу коагуляції крові. Внутрішній
і зовнішній механізми пов’язані між со
бою через калікреїн-кінінову систему, ак
тивація першого з них веде до активації
другого, і навпаки;
в)
макрофагально-моноцитарний
механізм.
На відміну від двох попередніх є механіз
мом патологічної активації зсідання крові.
Його викликає дія на макрофаги ендотокси
нів бактерій, імунних комплексів, компле
менту, продуктів розпаду тканин. При цьо
му з макрофагів вивільняється уже активна
протромбіназа (ф. Ха). Цей механізм має
певне пристосувальне значення, оскільки
завдяки зсіданню крові обмежується поши
рення патогенних факторів в організмі.
II фаза — утворення тромбіну. Вона відбува
ється за участю активної протромбінази і ф.У
(рис. 30.47).
III фаза - утворення фібрину — складається
з кількох послідовних процесів, під час яких
утворюються (рис. 30.48): (а) фібрин-мономер
із фібриногену під дією тромбіну; (б) розчинний
фібрин-полімер (фібрин S); (в) нерозчинний фіб
рин (фібрин І) під дією активного ф. XIII.
Порушення гемостазу
Розлади функціонування системи гемостазу, або
гемостазіопатії, поділяють на три групи: (І) гемо
рагічні діатези', (II) тромбогеморагічні гемостазі
опатії — синдром дисемінованого внутрішньосу-
79
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Фосфоліпідні фрагменти
клітинних мембран
Рис. 30.46. Зовнішній і внутрішній механізми активаціїзсідання крові
динного зсідання крові (ДВЗ-синдром)', (III) тромбофільні діатези - тромбози і тромбоемболії.
Геморагічний діатез
Схильність організму до повторних кровотеч
і крововиливів, що виникають спонтанно або
після незначних травм, називають геморагічним
діатезом, а його клінічні прояви — геморагічним
синдромом.
Геморагічні діатези поділяють на дві великі
групи (рис. 30.49): (І) порушення судинно-тромбоцитарного гемостазу і (II) розлади коагуляційнош гемостазу.
Залежно від того, що є об’єктом первинних
уражень, порушення першої групи поділяють на
(а) вазопатії, (б) тромбоцитопенії, (в) тромбоцитопатії. Розлади коагуляційного гемостазу
позначають як коагулопатії.
Для оцінки стану гемостазу та його порушень
використовують багато показників, найважливі
шими з яких є такі:
а) час кровотечі. Дає загальне уявлення про
те, нормальною чи порушеною є функція ге
мостазу. Визначають in vivo як час, який три
ває капілярна кровотеча після проколу шкіри
голкою, наприклад, у м’якуш пальця. Залеж
но від обраної методики визначення цього
показника його значення в нормі становить
від 2-х до 9-ти хвилин. Збільшення часу кро
вотечі вказує на порушення судинно-тромбоцитарного гемостазу і є характерним для
тромбоцитопеній і тромбоцитопатій;
б) кількість тромбоцитів у периферичній
крові. У нормі цей показник складає від
150 до 300- 107л;
в) протромбіновий час. Характеризує зовніш
ній механізм ініціювання коагуляції крові.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
80
II фаза процесу зсідання крові
Фібрин нерозчинний (фібрин І)
1 8 III фаза процесу зсідання крові
Його визначають як час утворення згустка
плазми після додавання до неї зовнішніх
джерел тканинного тромбопластину (на
приклад, екстракту мозку тварин) та іонів
Са2+. Подовження цього показника свід
чить про розлади коагуляційного гемостазу
і може бути зумовлене дефіцитом чи пору
шеннями функцій факторів V, VII, X, про
тромбіну чи фібриногену;
г) парціальний протромбіновий час. Засто
совують для оцінювання внутрішнього
(кров’яного) механізму зсідання крові. Ви
значають як час утворення згустку плазми
після додавання до неї каоліну (слугує кон
тактним активатором фактора XII) і цефа
ліну (заміщує фосфоліпідний компонент
тромбоцитів - фактор 3). Цей показник
подовжується при коагулопатіях, зумовле
них дефіцитом чи дисфункцією факторів V,
VIII, IX, X, XI, XII, протромбіну чи фібри
ногену.
Вазопатії. Спадково зумовлені чи набуті гемо
рагічні діатези, що виникають як наслідок пер-
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
81
Рис. 30.49. Класифікація геморагічних діатезів
винних порушень судинної стінки, називають
вазопатіями.
Цей вид порушень гемостазу буває відносно
часто, проте не є причиною тяжких, смертельно
небезпечних кровотеч. Звичайно він характеризу
ється точковими геморагіями (петехії і пурпура)
у шкірі і в слизових оболонках, зокрема у яснах;
інколи, однак, набуває форми менорагій, кровотеч
з носа, шлунково-кишкових кровотеч чи гематурії.
При вазопатіях усі згадувані вище показники
гемостазу — час кровотечі, кількість тромбоци
тів, протромбіновий час і парціальний протромбіновий час — не відрізняються від норми.
Залежно від механізму розвитку вазопатії по
діляють на дві групи: (І) запальні вазопатії - вас
куліти і (II) диспластичні вазопатії-ураження
судин, пов’язані з порушеннями їхньої сполуч
ної тканини (неповноцінність судинної стінки).
І. Запальні вазопатії. До цієї категорії від
носять:
1) інфекційні васкуліти. Вони є проявом ці
лого ряду інфекційних хвороб (вірусних
геморагічних гарячок, висипного тифу, ме
нінгококової інфекції, сепсису);
2) імунні васкуліти. Розвиваються як наслі
док імунокомплексних недуг (алергічні
реакції III типу), наприклад, при систем
ному червоному вовчаку, вузликовому періартеріїті, геморагічному васкуліті (хво
роба Шенляйна - Геноха). До цієї категорії
можна віднести і шкірні геморагії, що ін
коли виникають як реакція на ліки;
3) інфекційно-імунні васкуліти. Виникають
при інфекційних хворобах, що супроводжу
ються імунним ураженням судинної стінки.
У патогенезі запальних вазопатій провід
на роль належить ушкодженню ендотелію, що
може бути зумовлено: (а) цитопатичною дією
ендотеліотропних вірусів; (б) токсинами мік
робів, наприклад, веротоксином, що його виді
ляють бактерії кишкової групи; (в) комплексами
“антиген — антитіло ” і комплементом.
Унаслідок ушкодження ендотелію судин роз
вивається ряд таких порушень:
а) діапедез еритроцитів, що клінічно вияв
ляє себе точковими крововиливами (петехії
і пурпура);
б) інтенсивне мікротромбоутворення, що стає
причиною порушень мікроциркуляцїї та
живлення тканин;
4) тромбоцитопенія споживання — як ре
зультат утворення великої кількості мікротромбів.
82
II. Диспластичні вазопатії. В основі цього
виду вазопатій лежать набуті і спадково зумовле
ні порушення сполучної тканини стінки крово
носних судин. їхніми прикладами можуть бути:
Гіповітаміноз С. Аскорбінова кислота
є необхідним компонентом реакції гідроксилювання проліну, унаслідок якої той
перетворюється в оксипролін. Ця реакція
вважається однією з ключових в утворен
ні колагену. При гіповітамінозі С з ураху
ванням сказаного порушується утворення
повноцінного колагену (ламкість судин, ви
падання зубів і т.д.).
Телеангіектазії. Це спадково зумовлені ло
кальні дефекти сполучної тканини судин,
що спричиняються до стоншення їхніх сті
нок і розширення просвіту. Телеангіектазії,
на відміну від інших вазопатій, є джерелом
небезпечних для життя кровотеч, особливо
при локалізації у внутрішніх органах.
Гемангіоми. Судинні пухлини мають тен
денцію до частих кровотеч.
Синдром Елерса - Дато. Його основу скла
дають генетично зумовлені дефекти кола
гену.
У патогенезі диспластичних вазопатій мають
значення:
1) стоншення стінок мікросудин і розши
рення їхнього просвіту;
2) неповноцінний локальний гемостаз. Через
недостатню кількість або неповноцінність
колагену в субендотелії судин порушуєть
ся адгезія тромбоцитів;
3) легка ранимість судин.
Тромбоцитопенія. Зменшення вмісту тромбо
цитів в одиниці об’єму периферичної крові ниж
че 150- 109/л позначають як тромбоцитопенію.
Геморагічні прояви тромбоцитопенії у вигля
ді спонтанних кровотеч з’являються при змен
шенні кількості кров’яних пластинок нижче
20- 109/л. Тромбоцитопенія в діапазоні від 20 до
50- 109/л значно посилює кровотечі, зумовлені
травмою судин.
Клінічно геморагії тромбоцитопенічного по
ходження виявляють себе петехіями та пурпурою на шкірі, кровотечами зі слизових оболонок
шлунково-кишкового й сечостатевого трактів,
крововиливами в головний мозок і сітківку ока.
При цьому збільшується час кровотечі при не-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
змінених показниках протромбінового і парці
ального протромбінового часу.
За походженням тромбоцитопенії можуть
бути (а) спадково зумовленими і (б) набутими.
За механізмом розвитку виділяють такі види
тромбоцитопенії.
I. Тромбоцитопенії, пов ’язані з порушеннями
утворення тромбоцитів. Джерелом кров’яних
пластинок у червоному кістковому мозку, як ві
домо, є гігантські клітини - мегакаріоцити, що
завершують тромбоцитарну лінію клітин-попередників, до якої ще входять промегакаріоцити
і мегакаріобласти. Власне, ураження цих кро
вотворних клітин і зумовлюють групу тромбо
цитопеній, у яку об’єднують:
а)
мієлотоксичні тромбоцитопенії.
Вони
виникають унаслідок ушкодження крово
творних клітин. Як правило, поєднуються
з анемією й лейкопенією. Причинами їхньо
го розвитку є ті ж само фактори, що викли
кають розвиток гіпопластичної анемії (див.
підглаву 30.2);
б) дефіцитні тромбоцитопенії. їхній розвиток
обумовлюється недостатністю вітаміну ВІ2
або фолієвої кислоти. Є одним з гематоло
гічних проявів В,2-фолієводефіцитної анемії
(див. підглаву 30.2). В умовах порушено
го синтезу ДНК страждає розвиток клітин
тромбоцитарної лінії, мегакаріоцити зазна
ють деструктивних змін і гинуть шляхом
апоптозу (неефективний мегакаріопоез);
в) дисрегуляторні тромбоцитопенії. Основ
ною причиною порушень регуляції тромбоцитопоезу є недостатність тромбопоетинів - речовин, що стимулюють утворення
тромбоцитів у червоному кістковому мозку;
г) тромбоцитопенії, пов’язані зі зменшенням
тацдарму
кровотворення.
Розвиваються
при лейкозах і кісткових метастазах злоякіс
них пухлин разом із анемією та лейкопенією.
II.
Тромбоцитопенії, зумовлені посиленим
руйнуванням тромбоцитів. Причиною такого
руйнування можуть бути:
а) імунне ушкодження, обумовлене антитромбоцитарними антитілами на власні компо
ненти кров’яних пластинок або на лікар
ські препарати, адсорбовані на тромбоци
тах. Автоімунне ушкодження вважають
найбільш імовірним механізмом розвитку
так званої ідіопатичної тромбоцитопенічної пурпури (хвороби Верльгофа). У 80 %
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
хворих на цю недугу у плазмі крові й на
поверхні кров’яних пластинок виявляють
антитіла (переважно IgG) до мембранних
глікопротеїнів тромбоцитів, зокрема комп
лексів ІІЬ-ІІІа і ІЬ-ІХ;
б) гіперспленізм «* гіперфункція селезінки,
що часто супроводжується спленомегалією. У результаті підвищення фагоцитарної
активності макрофагів відбувається інтен
сивне руйнування всіх формених елементів
крові, у тому числі й тромбоцитів;
в) механічне ушкодження тромбоцитів. Час
то виникає при гемангіомах і накладенні
штучних клапанів серця;
г) набуті мембранопатії (гемолітична ане
мія Маркіафави — Мікелі). Соматичні му
тації кровотворних клітин спричиняються
до утворення пулів клітин (еритроцитів,
гранулоцитів, тромбоцитів) з дефектами
мембрани. У результаті збільшується чут
ливість таких клітин до дії комплементу
й відбувається їхнє руйнування.
III.
Тромбоцитопенії споживання. Виника
ють у результаті посиленого використання
тромбоцитів на утворення тромбів (хвороба
Шенляйна - Геноха, хвороба Мошковича, ДВЗсиндром).
У патогенезі геморагічного синдрому при
тромбоцитопеніях мають значення:
1) порушення ангіотрофічної функції тром
боцитів, у результаті чого виникають дис
трофічні зміни в ендотелії та збільшується
ламкість мікросудин. Це веде до збільшен
ня ранимості судин, діапедезу еритроцитів,
крововиливів;
2) послаблення адгезії й агрегації тромбоци
тів, унаслідок чого порушуються основні
етапи формування тромбу;
3) порушення вторинного спазму ушкодже
них артеріол. При тромбоцитопеніях ви
вільняється мало біогенних амінів (катехоламінів, серотоніну), з дією яких пов’язане
скорочення гладких м’язів судин;
4) уповільнене зсідання крові. Його обумов
лює недостатнє вивільнення фактора З
пластинок і тромбостеніну. У результаті
порушується внутрішній механізм І фази
зсідання крові та ретракція згустку.
Тромбоцитопатії. В основі цієї категорії по
рушень судинно-тромбоцитарного гемостазу
83
лежать якісні зміни тромбоцитів, їхня функці
ональна неповноцінність. При цьому кількість
кров’яних пластинок у крові може залишатися
в нормі.
За походженням тромбоцитопатії бувають
(а) спадково зумовленими і (б) набутими.
За характером якісних дефектів кров’яних
пластинок тромбоцитопатії поділяють на (а) ендотромбоцитарні й (б) екзотромбоцитарні.
Ендотромбоцитарні
тромбоцитопатії
обумовлюються порушеннями основних скла
дових частин тромбоцитів: мембрани, гранул
і ферментів. Звідси поділ їх на три категорії:
1) мембранопатії,
2) гранулопатії,
3) ферментопатії.
Мембранопатії виникають при спадкових
аномаліях мембранних глікопротеїнів, що
виконують функції клітинних рецепторів;
при блокаді цих рецепторів аномальними
білками плазми крові (парапротеїнами),
при ушкодженні мембрани кров’яних плас
тинок патогенними факторами.
Гранулопатії виявляють себе дефіцитом
гранул І і II типів.
В основі ферментопатії може лежати змен
шення активності ферментів циклу Кребса,
гліколізу, порушення функцій АТФ-аз, циклоксигенази і тромбоксансинтетази.
При
екзотромбоцитарних
тромбоцитопатіях причини порушення функцій тромбоцитів
лежать поза кров’яними пластинками. У зв’язку
з цим екзотромбоцитарні тромбоцитопатії мо
жуть бути:
а) пов’язаними зі змінами плазми крові (де
фіцит плазмових білків, що є плазмовими
кофакторами агрегації тромбоцитів);
б) зумовленими змінами в судинній стінці
(порушення утворення фактора Віллебранда ендотелієм судин, розлади зовнішнього
механізму зсідання крові).
Залежно від сутності порушень гемостазу ви
діляють:
1) тромбоцитопатії з первинними розладами
адгезії тромбоцитів',
2) тромбоцитопатії з первинними порушен
нями агрегації тромбоцитів',
3) тромбоцитопатії з первинними дефектами
реакцій вивільнення вмісту тромбоцитів.
84
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Перш ніж дати характеристику наведеним
вище тромбоцитопатіям, розглянемо молеку
лярні механізми адгезії й агрегації кров’яних
пластинок (рис. 30.50).
У цих процесах важливу роль відіграють фак
тор Віллебранда і глікопротеїнові рецептори
тромбоцитів.
Фактор Віллебранда (ФВ) - це дуже великий
білковий комплекс, які складається зі 100 субодинитть і має молекулярну масу, яка перевищує
20-106 дальтонів. Він здатен взаємодіяти з ба
гатьма протеїнами, що беруть участь у гемостазі.
Такими, зокрема, є колаген, гепарин і глікопротеїни тромбоцитарної мембрани Ib-ІХ (Г п ІЬ-ІХ).
Останні виступають рецепторами для ФВ. Осно
вним місцем утворення ФВ є ендотеліальні клі
тини, у яких він запасається у так званих тіль
цях Вейбеля—Паладе. Секреція ФВ відбувається
у двох напрямках: (1) з базальної поверхні ендотеліоцитів у субендотеліальний простір і (2)
з люмінального боку - у кров. При ушкодженні
судин оголюється їхня поверхня (денудація ен
дотелію), і субендотеліальний, зв’язаний з кола
геном базальної мембрани, ФВ стає доступним
тромбоцитам. Водночас відбувається адсорб
ція на оголеній судинній стінці циркулюючо
го в крові ФВ. Взаємодія кров’яних пластинок
з ФВ започатковує процес адгезії тромбоцитів,
який переростає згодом в їх агрегацію.
Проте участь ФВ у гемостазі цим не вичерпу
ється. Циркулюючий пул ФВ, який підтримується
не тільки ендотеліальними клітинами, а й тром
боцитами, утворює комплекси з фактором VIII
(антигемофільним глобуліном) плазми. Лише
в такій формі останній є доступним для здійснен
ня реакцій І фази зсідання крові, що відбуваються
0. Участь фактора Віллебранда у здійсненні судинно-тромбоцитарного і коагуляційного гемостазу
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
за внутрішнім (кров’яним) механізмом на фосфоліпідних фрагментах тромбоцитів (ф. 3).
Таким чином, ФВ є важливим не лише для
здійснення судинно-тромбоцитарного гемоста
зу, а й для реакцій коагуляції крові. Саме цим
і пояснюється розмаїта картина порушень ге
мостазу, що виникають при дефіциті ФВ - хво
робі Віллебранда (див. далі).
Глікопротеїни (Гп) тромбоцитарних мемб
ран відіграють роль рецепторів до факторів, що
беруть участь в адгезії та агрегації тромбоцитів.
Серед них важливе місце посідають два комп
лекси: Гп ІЬ-ІХ і Гп ІІЬ-ІІІа. Перший з них за
безпечує взаємодію кров’яних пластинок з ФВ,
а другий *8* з білками плазми крові (зокрема фі
бриногеном), за допомогою яких здійснюється
склеювання тромбоцитів (агрегація).
Деякі причини розвитку тромбоцитопатій по
казано нарис. 30.51.
Тромбоцитопатії з первинними розладами
адгезії тромбоцитів. До таких належать:
• Хвороба Віллебранда. Вона зумовлюється
генетичними розладами синтезу й секре
ції ФВ ендотеліальними клітинами і ме
гакаріоцитами (переважний тип спадку
вання аутосомно-домінантний). Є одним
із найпоширеніших спадково зумовлених
порушень гемостазу в людини. Описано
понад 20 варіантів цієї недуги, які можна
розділити на дві групи: (1) зменшення кіль
кості утворюваного ФВ (75 % від усіх ви
падків хвороби) і (2) якісні зміни ФВ, що
роблять його неповноцінним (решта 25 %).
З урахуванням участі ФВ у здійсненні ге
мостазу (див. вище) можна виділити дві
патогенетичні лінії у розвитку його по
рушень. Перша з них - це розлади адгезії
тромбоцитів, що ведуть до порушень су
динно-тромбоцитарного
гемостазу.
Дру
га - порушення коагуляційного гемоста
зу через дефіцит комплексів ВФ — ф-VIII,
унаслідок чого антигемофільний глобулін
не має доступу до реакцій зсідання крові.
З останнім пов’язана схожість клінічної
картини хвороби Віллебранда з гемофілією
А (див. далі). Звідси ще одна назва хворо
би - ангіогемофілія. Наведені вище напря
ми патогенезу пояснюють, чому при хворо
бі Віллебранда подовжується час кровотечі
на тлі незміненої кількості тромбоцитів
85
і збільшеного показника парціального протромбінового часу.
ш Хвороба Бернара — Сульє, або макротромбоцитодистрофія. Причиною є спадково
обумовлений дефект глікопротеїнів тромбоцитарної мембрани Гп ІЬ-ІХ, що вза
ємодіють з фактором Віллебранда. При
цьому тромбоцити набувають гігантських
розмірів. Тип спадкування аутосомно-рецесивний.
Тромбоцитопатії з первинними порушен
нями агрегації тромбоцитів або дизагрегаційні. їх прикладом може бути:
Тромбастенія Гланцмана. Вона виникає як
наслідок зменшення утворення або якісних
дефектів мембранних глікопротеїнів Гп ІІЬІІІа, що беруть участь в агрегації. При цьо
му адгезія тромбоцитів і вивільнення їхніх
гранул відбуваються, а агрегація - ні, не
зважаючи на дію таких потужних агрегантів, як АДФ, адреналін, тромбін. Тип спад
кування: аутосомно-рецесивний.
Тромбоцитопатії з первинним порушен
ням реакцій вивільнення вмісту тромбоцитів.
Розлади цього процесу викликають порушення
другої хвилі агрегації тромбоцитів. При цьому
початкова агрегація кров’яних пластинок відбу
вається нормально, але не переходить у наступ
ну стадію і завершується дезагрегацією тромбо
цитів. Прикладом тромбоцитопатій цієї групи є:
Парез реакції вивільнення - стан, що вини
кає під впливом ацетилсаліцилової кислоти
та інших нестероїдних протизапальних за
собів, що інгібують утворення тромбоксану
А2. Показано, що одноразове приймання ас
пірину необоротно “паралізує” вивільнен
ня гранул на час від 3,5 до 6 діб, аж поки не
з’являться нові тромбоцити. Саме цим по
яснюється широке застосування цього пре
парату для попередження тромбозів.
Коагулопатії. В основі порушень коагуляційного
гемостазу можуть лежати:
1) зменшення активності системи зсідання
крові,
2) підвищення активності антикоагулянтної
системи,
3) збільшення активності фібринолітичної
системи.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
86
Субендотеліальний шар
ис. 30.51. Деякі причини розвитку тромбоцитопатій
І.
Зменшення активності системи зсідання
крові зумовлюється недостатністю або якісни
ми змінами факторів, що беруть участь у цьому
процесі. За походженням ці зміни можуть бути:
а) набутими,
б) спадково зумовленими.
Набуті порушення, як правило, є комплекс
ними і пов’язані з недостатністю багатьох фак
торів зсідання крові. Найчастіше їхньою причи
ною є:
а) гіповітаміноз К, при якому порушуються
реакції карбоксилювання, необхідні для
утворення функціонально активних форм
факторів II, VII, IX, X і протеїну С;
б) недостатність печінки - органа, у якому
синтезується більшість факторів зсідання;
в)
синдром дисемінованого внутрішньосудинного зсідання крові (ДВЗ-синдром), при
якому йде значне споживання факторів зсі
дання, унаслідок чого створюється їхній
дефіцит (див. далі).
Спадково
зумовлені
коагулопатії
мають
у своїй основі дефіцит одного якогось фактора
зсідання крові. Сьогодні описано генетичні де
фекти для кожного з них.
Клінічно порушення зсідання крові істотно
відрізняються від розладів судинно-тромбоцитарного гемостазу. Якщо для останніх характер
ними є спонтанні капілярні (точкові) кровотечі
з крововиливами у формі петехій і пурпури, то
для коагулопатій - тривалі зовнішні та внутріш
ні кровотечі із судин значно більшого діаметра
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
(артерій і вен), які виникають у результаті травм
або хірургічних втручань (наприклад, після
видалення зуба). Такі кровотечі часто виявля
ють себе гематомами - великими крововили
вами в м’язи, під шкіру, у порожнину суглобів
(гемартрозами).
З огляду на те, що процес зсідання крові від
бувається у три фази, розглянемо можливі пору
шення, які виникають на кожному з етапів.
І фаза. Залежно від характеру порушень цієї
фази виділяють три групи розладів:
1) ізольовані порушення зовнішнього меха
нізму активації зсідання. Виникають при
дефіциті ф.УІІ - гіпопроконвертинемії.
Цей дефіцит може бути спадково зумов
леним (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К,
ураження печінки). Характерним є подов
ження протромбінового часу на тлі незміненого показника парціального протромбі
нового часу й часу кровотечі;
2) ізольовані порушення внутрішнього меха
нізму активації зсідання. У зв’язку з тим,
що в цей механізм втягується велика кіль
кість факторів, то і ряд можливих розладів
теж великий. Він охоплює:
дефіцит ф.УШ - гемофілію А. Фактор VIII,
що входить до комплексу з фактором Віллебранда (див. вище), синтезується, на від
міну від інших факторів, не в паренхіма
тозних клітинах печінки, а в епітеліальних
клітинах синусоїдів (клітинах Купфера)
і в клітинах гломерулярного та тубулярного
епітелію нирок. Антигемофільний глобулін
виступає кофактором фактора IX у реакції
перетворення ф. X у ф. Ха. При цьому, на
відміну від ферментів, відбувається його
споживання, тобто зменшується його кіль
кість, що може в подальшому лімітувати
процес зсідання крові. Причиною гемофі
лії А є генетичний дефект, який успадкову
ється зчеплено з Х-хромосомою, - звідси
особливості передавання хвороби від жі
нок, що є носіями патологічної хромосо
ми, особам чоловічої статі (див. главу 12).
У 15 % хворих утворюються автоантитіла
проти ф. VIII, що свідчить про значні зміни
структури білка, унаслідок чого той сприй
мається імунною системою як чужорідний;
дефіцит ф. IX — гемофілію В. Причиною
розвитку є генетичні дефекти, що успадко
87
вуються зчеплено з Х-хромосомою. Клініч
но нічим не відрізняється від гемофілії А:
оскільки ф. IX і ф. VIII виконують свої
функції в єдиному комплексі, то дефіцит
будь-якого з них має одні й ті ж наслідки;
» дефіцит ф. XI —гемофілію С. Виникає при
генетичних порушеннях, що передаються
за аутосомно-рецесивним типом;
» дефіцит ф. XII буває дуже рідко. Завдя
ки калікреїн-кініновій системі цей дефект
добре компенсується, оскільки запуск вну
трішнього механізму зсідання відбувається
через зовнішній. А тому брак цього факто
ра, на відміну від усіх інших, не супрово
джується геморагічним діатезом.
Для ізольованих порушень внутрішнього ме
ханізму коагуляції характерним є подовження
парціального протромбінового часу при незмінених показниках тромбінового часу і часу кро
вотечі.
3) поєднані порушення зовнішнього і внут
рішнього механізмів зсідання. Вони розви
ваються при дефіциті ф. X, що може бути
спадково зумовленим (тип спадкування
аутосомно-рецесивний) або набутим (гіпо
вітаміноз К, ураження печінки).
II фаза. Основними причинами порушень
цієї фази коагуляції є:
1) дефіцит ф.ІІ — гіпопротромбінемія. Най
частіше має набутий характер і розвива
ється внаслідок гіповітамінозу К або ура
жень печінки;
2) дефіцит ф-V — парагемофілія. Порушення
утворення проакцелерину може бути спри
чинено ураженнями печінки або автоантитілами проти ф-V
Для порушень II фази зсідання крові харак
терним є подовження як протромбінового, так
і парціального протромбінового часу без змін
часу кровотечі.
III фаза. Завершальний етап зсідання крові
може порушуватися внаслідок таких причин:
1) дефіциту фібриногену. Розрізняють афібриногенемію — повну відсутність фібри
ногену (спадкова хвороба з аутосомно-ре
цесивним типом спадкування) і гіпофібриногенемію — зменшення синтезу фібрино
гену в печінці при її ураженнях;
2) дисфібриногенемії “ якісних змін фібри
ногену. їх причиною є генетичні дефекти
88
(тип спадкування аутосомно-домінантний), що виявляють себе утворенням ано
мального фібрину;
3) порушень полімеризації фібрину. Розви
ваються в результаті утворення комплексів
фібриногену з фібрином-мономером і про
міжними продуктами, від яких ще пов
ністю не відщепилися пептиди А і В. При
цьому утворюється так званий заблокова
ний фібриноген (тромбінрезистентний фі
бриноген), що не піддається дії тромбіну;
4) дефіцит ф. XIII. Виникає як наслідок спад
кових порушень (тип спадкування аутосомно-рецесивний). Виявляє себе пору
шеннями перетворення розчинного фібри
ну (фібрину S) у нерозчинний (фібрин І).
II.
Підвищення активності антикоагулянтної системи. Поняттям “антикоагулянтна сис
тема” об’єднують цілий ряд природних сполук,
що перешкоджають здійсненню зсідання крові,
їх поділяють на дві категорії:
1) первинні;
2) вторинні антикоагулянти.
Первинні антикоагулянти постійно синте
зуються в організмі й тому завжди містяться
в плазмі крові. До них відносять:
а) антитромбін III - основний універсальний
антикоагулянт, що є інгібітором протеаз.
Синтезується ендотелієм судин. Пригнічує
активність усіх протеолітичних ферментів
крові, у тому числі тромбіну, калікреїну, плаз
міну, ф. ХІІа, ф. ХІа, ф. Ха, ф. ІХа, ф.УПа;
б) гепарин (антитромбін II) - глікозаміноглікан, що вивільняється тканинними базофі
лами і базофілами крові при їхній дегрануляцїї. Антикоагулянті властивості має не
сам гепарин, а комплекс гепарину з анти
тромбіном III. Завдяки гепарину антитром
бін III фіксується на поверхні ендотелію
судин, де його антикоагулянті властивості
в багато разів зростають;
в) а ^антитрипсин, а2-макроглобулін, інгі
бітор СІ-компонента комплементу — усі
вони є неспецифічними інгібіторами про
теаз, у тому числі й факторів зсідання крові.
Вторинні антикоагулянти у плазмі крові
в нормі не містяться. Вони утворюються в про
цесі зсідання крові й фібринолізу. Такими вва
жають:
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
а) антитромбін І. Так власне позначають фі
брин за його здатність адсорбувати і в та
кий спосіб інактивувати велику кількість
тромбіну;
б) продукти фібринолізу. Вони перешко
джають процесам полімеризації фібрину
і утворенню фібринових структур;
в) протеїн С. Цей білок в активованому стані
руйнує фактори V і VIII. Активація ж його
відбувається під впливом мембранного
протеїну ендотеліоцитів — тромбомодуліну
в разі приєднання до останнього молекул
тромбіну (див. главу 16).
До підвищення активності антикоагулянтної
системи крові найчастіше спричиняються такі
обставини:
1) збільшення вмісту гепарину в крові - гіпергепаринемія. Причинами останньої мо
жуть бути посилена дегрануляція тканин
них базофілів і базофілів крові, зокрема,
при алергічних реакціях І типу; руйнуван
ня базофільних лейкоцитів при лейкозах;
введення гепарину ззовні;
2)
поява “патологічних” антикоагулянтів,
до яких відносять антитромбін V, що по
рушує полімеризацію фібрину-мономеру;
“вовчаковий” антикоагулянт, який пере
шкоджає утворенню протромбіназного
комплексу; парапротеїни, що унеможлив
люють полімеризацію фібрину.
III.
Збільшення активності фібринолітичної системи. Система фібринолізу при її ак
тивації здійснює лізис (протеоліз) фібрину
в кровоносному руслі й у такий спосіб бере
участь у підтриманні рідкого стану крові
й у відновленні кровообігу в тромбованих су
динах. До складу системи фібринолізу входять
(рис. 30.52):
1) плазміноген (профібринолізин) — неактив
ний протеолітичний фермент, що завжди
міститься в плазмі крові;
2) плазмін (фібринолізин) - активна форма
плазміногену. Утворюється в результаті
дії активних протеаз на плазміноген і від
щеплення від його молекули пептиду, який
“прикриває” активний центр;
3) активатори фібринолізу — велика група
речовин, які або самі є протеазами і здатні
перетворювати плазміноген у плазмін, або
викликають появу таких протеаз;
89
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
Механізми активації фібринолізу
4)
інгібітори фібринолізу. До них відносять
інгібітори протеаз, серед яких найбільше
значення має а2-антиплазмін.
Розрізняють внутрішній і зовнішній механіз
ми активації фібринолізу.
Внутрішній механізм обумовлений актива
цією фактора XII зсідання крові й утворенням
калікреїну, унаслідок чого в крові з’являється
велика кількість активаторів фібринолізу.
Зовнішній механізм пов’язаний з надходжен
ням у кров готових активаторів фібринолізу: ендотеліального, тканинного (урокіназа), бактері
ального (стрептокіназа).
Причинами підвищення активності фібринолітичної системи є:
1. Посилене утворення й надходження в кров
активаторів фібринолізу. Таке відбува
ється при значних ушкодженнях тканин:
великі травми, ушкодження клітин токси
нами, операційні втручання, лейкози та ін.
2. Зменшення вмісту в крові інгібіторів про
теолізу. Воно має місце при недостатньо
му їх утворенні або посиленому викорис
танні.
ДВЗ-синдром
Синдром
дисемінованого
внутрішньосудинного
зсідання крові (ДВЗ-синдром) - це генералізоване зсідання крові всередині судин, що викли
кає утворення великої кількості мікрозгустків
і агрегатів клітин, які порушують мікроциркуляцію в органах і тканинах. Цей синдром ха
рактеризують як катастрофу для організму.
Причини. Залежно від причин розвитку виді
ляють такі різновиди ДВЗ-синдрому:
а) інфекційно-септичний (розвивається при
сепсисі);
б) посттравматичний (при краш-синдромі,
опіковій хворобі, множинних переломах
кісток);
в) шокогенний (при всіх видах шоку);
г) хірургічний (після операцій з великою
травматизацією тканин);
д) акушерський (при передчасному відшару
ванні плаценти, надходженні в кров навко
лоплідних вод);
е) токсигенний (після укусу змії);
є) пухлинний (при злоякісному пухлинному
рості);
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
90
Рис. 30.53. Гуморальний протеазний вибух як механізм ініціювання ДВЗ-синдрому
ж) алергічний (при імунному ушкодженні тка
нин) та ін.
Механізми. В основі патогенезу ДВЗ-синдрому
лежить так званий “гуморальний протеазний
вибух”, тобто одночасна активація всіх протео
літичних ферментів плазми крові, що входять
до складу чотирьох позаклітинних біохімічних
систем (рис. 30.53):
1) системи зсідання крові',
2) фібринолітичної системи',
з) калікреїн-кінінової системи;
4) системи комплементу.
Основний принцип активації позаклітинних
протеаз - відщеплення пептидів, що прикривають
їхні активні центри. Утворення активних протео
літичних ферментів крові має свої особливості:
а) можлива самоактивація ферментів - ак
тивний фермент, впливаючи на неактивну
форму, перетворює її в активну;
б) одні активні протеази здатні активувати
інші (перехресна активація)',
в)
ланцюговий характер активації. Теоретич
но поява навіть однієї молекули активної
протеази може викликати активацію всіх
наявних протеаз крові.
Однак у нормі реакції активації протеолітич
них ферментів мають обмежений характер, що
пояснюється існуванням великої групи інгібі
торів протеаз. При патології, коли у кров над
ходять великі кількості активних протеаз, по
тужність існуючих інгібіторів може стати недо
статньою. Отоді й виявляє себе ланцюговий ха
рактер активації протеолітичних систем плазми
крові. Така активація набуває генералізованого
характеру, втягує всі протеази крові — відбува
ється “гуморальний протеазний вибух”.
Існує три основних джерела надходження
протеаз у кров:
1) ушкоджені клітини. Має значення го
стре ушкодження великої кількості клітин,
з яких у позаклітинний простір і кров над
ходять лізосомні протеази, тканинний тромбопластин. Запалення, як місцевий процес,
ГЛАВА ЗО. ПАТОФІЗІОЛОГІЯ СИСТЕМИ КРОВІ
що виникає при альтерації клітин, обмежує
надходження продуктів розпаду в кров, ло
калізуючи ушкодження і в такий спосіб по
переджаючи розвиток ДВЗ-синдрому;
2) надходження в кров великої кількості по
заклітинних протеаз, наприклад, трипси
ну при гострому панкреатиті, ферментів,
що містяться в навколоплідних водах;
3) екзогенні протеази. їхніми джерелами мо
жуть бути бактеріальні клітини при сепси
сі, зміїна отрута та ін.
У патогенезі ДВЗ-синдрому розрізняють дві
фази.
I
фаза - фаза гіперкоагуляції і агрегації
тромбоцитів. її основу становить генералізована активація системи зсідання крові. Поява
всередині судин тромбіну (тромбінемія) спри
чиняється до утворення фібрину й агрегатів
тромбоцитів.
Існує три механізми запуску цієї фази:
1) ферментативний механізм - надходження
в кров великої кількості активних протеаз
і тканинного тромбопластину;
2) контактний механізм - активація ф. XII
при контакті його з чужорідними поверх
нями (екстракорпоральний кровообіг, ге
модіаліз, штучні клапани серця);
3) тромбоцитарний механізм — первинна ак
тивація агрегації тромбоцитів при генералізованому ушкодженні ендотелію судин,
порушеннях реологічних властивостей
крові, гострому внутрішньосудинному ге
молізі еритроцитів.
У результаті реалізації зазначених механіз
мів утворюється велика кількість мікрозгустків і агрегатів клітин, що призводить до розла
дів мікроциркуляції, розвитку сладж-синдрому
(див. главу 16). Це все веде до появи таких клі
нічних проявів, як гіпоксія, ацидоз, інтоксика
ція продуктами розпаду, гостра недостатність
зовнішнього дихання (мікрозгустками закупо
рюються капіляри легень), гостра ниркова не
достатність (забиваються капіляри клубочків),
порушення мозкового кровообігу.
II фаза - фаза гіпокоагуляції (геморагічний
синдром). Ця фаза розвивається як наслідок ви
снаження механізмів судинно-тромбоцитарного
і коагуляційного гемостазу.
91
У її виникненні мають значення:
а) зменшення активності системи зсідання
крові (споживання факторів I, V, VIII);
б) активація фібринолітичної системи (над
ходження в кров великої кількості актива
торів фібринолізу);
в) підвищення антикоагулянтної активності
крові за рахунок утворення продуктів фіб
ринолізу;
г) розвиток тромбоцитопенії споживання;
д) підвищення проникності стінки судин (має
значення утворення великих кількостей кінінів).
Фаза гіпокоагуляції клінічно виявляє себе ма
сивними кровотечами, що їх важко зупинити.
Тромбофільні діатези
Ще одну категорію первинних порушень гемо
стазу складають хвороби і синдроми, при яких
зростає схильність до утворення тромбів. їх
об’єднують поняттям тромбофільні діатези або
тромбофілії.
В основі патогенезу тромбофілій можуть ле
жати:
1) ендотеліальні механізми, що виявляють
себе зменшенням тромборезистентності
стінки судин;
2) тромбоцитарні механізми, пов’язані
зі збільшенням агрегаційної
здатності тромбоцитів;
3) зниження антикоагулянтної активності
крові (зменшення утворення
антитромбіну III);
4) падіння активності фібринолітичної
системи (зменшення утворення
ендотеліального активатора плазміногену,
поява потужних інгібіторів плазміну,
дефіцит плазміногену);
5) порушення системи зсідання крові
(дисфібриногенемії).
Клінічно тромбофільні діатези виявляють
себе тромбозами і тромбоемболіями венозних
і артеріальних судин у різних органах та ткани
нах (див. главу 16).
ГЛАВА 31
Патофізіологія серця
Система кровообігу, призначення якої - транс
порт крові до органів і тканин в об’ємі, що від
повідає потребам організму, складається із сер
ця, кровоносних судин та апарату центральної
й місцевої регуляції їхньої діяльності.
Серце як орган виконує функцію двох насо
сів - правого і лівого - і забезпечує циркуляцію
крові відповідно у малому і великому колах кро
вообігу. Насосна функція серця грунтується на
чергуванні скорочення серцевого м’яза (систо
ли) і його розслаблення (діастоли).
Велика кількість чинників, про які мова піде
далі, можуть порушувати діяльність серця та
кровоносних судин і спричинятися до недостат
ності системного кровообігу.
Недостатність кровообігу - це стан, при
якому серцево-судинна система не здатна забез
печити органи й тканини організму необхідною
кількістю крові. Вона може бути обумовлена:
1) недостатністю серця;
2) порушеннями функції кровоносних судин;
3) одночасними ураженнями серця і судин
(ісерцево-судинна недостатність).
Наслідки недостатності кровообігу для орга
нів і тканин можна розділити на дві групи:
1. Порушення трофічного забезпечення орга
нів і тканин. Вони виникають у результаті
(а) зменшення доставки кисню (циркуляшорна гіпоксія, див. главу 23) і (б) змен
шення доставки поживних речовин;
2. Порушення видалення з органів і тканин
кінцевих продуктів обміну речовин. Наслід
ком цього є розвиток: (а) негазового ацидо
зу та (б) інтоксикації.
31.1. НЕДОСТАТНІСТЬ СЕРЦЯ
Недостатність серця — це патологічний стан,
суть якого полягає в нездатності серця забез
печувати хвилинний об’єм крові, що відповідає
потребам органів і тканин. Іншими словами,
мова йде про недостатність насосної функції
серця, унаслідок чого страждає кровопостачан
ня структур організму.
Класифікація:
L Залежно від клінічного перебігу розрізня
ють (а) гостру і (б) хронічну недостатність
серця. Останню ще називають застійною.
2. За вираженістю клінічних проявів недо
статність серця може бути (а) прихованою
(компенсованою) і (б) явною (декомпенсо
ваною).
3. Залежно від порушення функції переваж
но того чи того відділу серця розрізняють:
(а) лівошлуночкову, (б) правошлуночкову
і (в) тотальну недостатність серця.
4. За патогенезом виділяють: (а) недостатність
серця від перевантаження; (б) міокардіальну недостатність серця; (в) позаміошрдіальну недостатність.
Недостатність серця може мати у своїй осно
ві первинні порушення одного з двох компо
нентів насосної функції — систоли чи діастоли
(рис. 31.1).
Систолічна дисфункція виникає як наслідок
зменшення скорочувальних можливостей сер
цевого м’яза, що характерно для різних варі-
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
93
Об'єм крові у венах
Серцевий виштовх
Нормальна функція
Систолічна дисфункція
Діастолічна дисфункція
Недостатність серця
Рис. 31.1. Порушення компонентів насосної функції серця, як основа його недостатності, д - діастола,
С - систола. Товщина й довжина стрілок відображають зміни показників порівняно з нормою
антів перевантаження серця (див. нижче) і для
уражень міокарда при його ішемічній хворобі та
дилатаційних кардіоміопатіях. Систолічна дис
функція від самого початку виявляє себе змен
шенням серцевого виштовху, що веде до падін
ня хвилинного об’єму серця.
Діастолічна дисфункція пов’язана з чинника
ми, що зменшують наповнення порожнин серця
кров’ю під час діастоли. Такими є порушення
процесів розслаблення міокарда, зменшення
здатності стінок серця до розтягнення, напри
клад, при кардіосклерозі); перешкоджання на
повненню шлуночків кров’ю, наприклад, при
ексудативних перикардитах. За цих обставин
збільшується об’єм крові у венозній частині
кровоносного русла і, відповідно, зростає веноз
ний тиск крові.
Незалежно від того, що є первинним, а що вторинним, недостатність серця характеризу
ється двома кардинальними функціональними
ознаками: (1) зменшенням серцевого виштов
ху і (2) накопиченням крові у венозних суди
нах.
31.1.1.
НЕДОСТАТНІСТЬ СЕРЦЯ
ВІД ПЕРЕВАНТАЖЕННЯ
Цей патогенетичний варіант недостатності роз
вивається в результаті дії на здорове серце ве
ликих навантажень опором або об’ємом, тобто
коли збільшується опір серцевому виштовху
або приплив крові до певного відділу серця. Це
буває при вадах серця, гіпертензії великого або
малого кола кровообігу, артеріовенозних фісту
лах, при виконанні дуже важкої фізичної роботи.
При цьому до серця з нормальною скорочуваль
ною здатністю висуваються надмірні вимоги.
Виділяють два типи перевантажень серця
(рис. 31.2).
1. Перевантаження об’ємом, або переднавантаження
(англ.,
preload),
виникає
тоді, коли до серця або до окремих його
порожнин притікає збільшений об’єм кро
ві. У цих умовах серце або його відділ, що
зазнає перевантаження, мають переміщати
збільшений об’єм крові в артеріальну сис-
/
94
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Рис. 31.2. ВИДИ перевантаження серця. ВП - венозне повернення, ХОС - хвилинний об'єм серця
тему. Це досягається збільшенням хвилин
ного об’єму серця відповідно до збільше
ного венозного повернення.
Перевантаження об’ємом виникає при:
а) збільшенні венозного повернення крові до
серця, зокрема при збільшенні об’єму цир
кулюючої крові (гіперволемія) або збіль
шенні тонусу венозних судин (зменшення
ємності венозної системи);
б) вадах серця - недостатності його клапа
нів. Так, при недостатності аортального
і двостулкового клапанів розвивається пе
ревантаження лівого шлуночка, при недо
статності клапана легеневої артерії і три
стулкового клапана - перевантаження пра
вого шлуночка.
2.
Перевантаження опором, або післяна
вантаження (англ., afterload), має місце,
коли серце або окремі його відділи змуше
ні виконувати роботу проти збільшеного
опору, що перешкоджає переміщенню всієї
крові в артеріальну систему. При переван
таженні опором серце намагається зберег
ти свій хвилинний об’єм, незважаючи на
збільшений опір вигнанню крові.
Перевантаження опором розвивається при:
а) підвищенні артеріального тиску (збільшен
ні периферичного судинного опору). В умо
вах гіпертензії великого кола кровообігу пе
ревантаження опором діє на лівий шлуночок,
а при гіпертензії малого кола - на правий;
б) вадах серця — стенозах клапанних отворів.
Так, при стенозі отвору аорти розвивається
перевантаження лівого шлуночка, при сте
нозі отвору двостулкового клапана - лівого
передсердя, при стенозі отвору легеневої ар
терії— правого шлуночка, при стенозі отвору
тристулкового клапана—правого передсердя.
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
95
Механізми компенсації
При дії на серце навантажень об’ємом і опором
посилення роботи серця забезпечується двома
типами компенсаторних механізмів:
1) негайними механізмами компенсації серця;
2) механізмами довготривалої адаптації сер
ця - гіпертрофією міокарда.
Негайні механізми компенсації серця вми
каються одразу після початку дії перевантажень.
Вони є генетично детермінованими і здійсню
ються наявними в серці структурами, інтенсив
ність функціонування яких може зростати до
максимально можливого рівня. До таких меха
нізмів відносять:
1) гетерометричний;
2) гомеометричний;
3) хроноінотропний механізми;
4) інотропну дію катехоламінів.
1. Гетерометричний механізм забезпечує не
гайну компенсацію серця до дії переванта
жень об’ємом. Його суть полягає в збільшен
ні сили серцевих скорочень в умовах надхо
дження до серця збільшеного об’єму крові.
Фізіологічну основу цього механізму складає
закон Франка - Старлінга. Він має два форму
лювання: для окремих м’язових волокон і для
серця в цілому. У першому варіанті його сут
ність відображається положенням: що більша
вихідна довжина м’язового волокна (у певних
межах), то більша сила його скорочень. Для
серця в цілому вживаним є формулювання: що
більший кінцеводіастолічний об’єм шлуночків
серця, то більший їхній ударний об ’єм.
На молекулярному рівні закон Франка - Стар
лінга пояснюється тим, що сила скорочення
м’язового волокна визначається кількістю міст
ків, що утворюються між актиновими і міозиновими філаментами. Ця кількість, у свою чергу,
залежить від взаємного розташування тонких
актинових і товстих міозинових волокон. Опти
мальним таке розташування є при досягненні
саркомером довжини 2,0-2,2 мкм: при цьому
кількість актоміозинових містків, а отже, і сила
скорочення м’язового волокна, стає максималь
ною. Подальше розтягнення м’яза веде до змен
шення кількості містків і до зменшення сили
скорочень.
3. Гетерометричний механізм негайної
компенсації серця: показники кардіодинаміки.
ВП - венозне повернення, КСО - кінцевосистолічний об'єм,
КДО - кінцеводіастолічний об'єм, У0 - ударний об'єм
Якщо ж мова йде про серце в цілому, то збіль
шення надходження крові у шлуночки під час
діастоли стає причиною розтягування стінок
міокарда і збільшення т. зв. кінцеводіастолічного тиску (рис. 31.3). Завдяки наведеному вище
механізму сила скорочення серцевого м’яза
зростає, що виявляє себе збільшенням серцево
го виштовху, тобто ударного об ’єму серця.
/
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
96
КДО =
130 мл
Рис. 31.4. Становлення гомеометричного механізму негайної компенсації серця.
Пояснення див. у тексті, умовні
позначення - на рис. 31.3
Отже, основними функціональними ознака
ми гетерометричного механізму компенсації є:
а) підвищення кінцеводіастолічного тиску
через збільшення надходження крові в по
рожнини шлуночків;
б) зростання ударного, а отже, і хвилинного
об’ємів серця за рахунок збільшення сили
серцевих скорочень.
Напруга м’язових волокон міокарда при цьо
му не міняється. Змінюється тільки їхня довжи
на, звідси назва механізму - гетерометричний.
2. Гомеометричний механізм є негайним ме
ханізмом компенсації серця до дії наванта
жень опором. Його сутність полягає в зрос
танні сили серцевих скорочень в умовах
збільшення опору вигнанню крові.
Нині показано, що в основі цього механізму,
як і гетерометричного, лежить закон Франка Старлінга, тобто збільшення вихідної довжини
волокон міокарда і пов’язане з цим підвищення
кінцеводіастолічного тиску.
Становлення гомеометричного механізму від
бувається в такій послідовності (рис. 31.4). При
збільшенні опору вигнанню крові різко падає
ударний об’єм, унаслідок чого зростає кінцевосистолічний об’єм шлуночків.
Оскільки надходження крові в шлуночки про
довжує залишатися попереднім, то в наступному
циклі скорочень серця збільшується кінцеводіастолічний об’єм. А це за законом Франка - Стар
лінга веде до збільшення сили скорочень серця.
Для гомеометричного механізму характерні
такі зміни показників кардіодинаміки:
97
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
Частота скорочень серця
ис. 31.5. Хроноінотропний механізм негайної компенсації: показники кардіодинаміки при збільшенні
навантаження об'ємом. Умовні позначення див. на рис. 31.3
а) збільшення кінцевосистолічного об’єму;
б) збільшення кінцеводіастолічного об’єму
за рахунок первинного підвищення попе
реднього показника, а не через збільшення
припливу крові, як при гетерометричному
механізмі;
в) ударний, а отже, і хвилинний об’єми завдя
ки збільшенню сили серцевих скорочень
залишаються на попередньому рівні, не
зважаючи на збільшення опору вигнанню
крові.
При гомеометричному механізмі збільшуєть
ся напруга м’язових волокон міокарда, тимчасом як їхня довжина не змінюється. Звідси на
зва механізму компенсації - гомеометричний.
З. Хроноінотропний механізм, відомий ще
як феномен “сходинок ” або феномен Боудича, є ще .одним негайним механізмом
компенсації серця до дії підвищених на
вантажень. Його сутність полягає в тому,
що при зростанні частоти скорочень сер
ця збільшується сила його скорочень. При
цьому одночасно зменшується час роз
слаблення міокарда, що сприяє швидкому
наповненню шлуночків серця кров’ю.
Нині вважають, що основу хроноінотропного
механізму становить збільшення надходження
іонів кальцію в саркоплазму кардіоміоцитів під
час потенціалів дії, сумарна тривалість яких при
тахікардії зростає. Підвищення концентрації іо
нів кальцію в саркоплазмі веде до збільшення
кількості утворюваних кальцій-тропонінових
комплексів і, як наслідок, до збільшення сили
скорочень м’язових волокон.
Для хроноінотропного механізму характерні
такі зміни показників кардіодинаміки (рис. 31.5):
а) ударний об’єм збільшується (при наванта
женні об’ємом) або залишається постійним
(при навантаженні опором). У результаті
збільшення частоти серцевих скорочень
зростає хвилинний об’єм серця;
б) кінцеводіастолічний об’єм зменшується,
якщо приплив крові до серця постійний,
або залишається без змін, якщо приплив
крові зростає;
в) кінцевосистолічний об’єм зменшується.
4. Інотропна дія катехоламінів. їхня участь
у здійсненні негайної адаптації серця до
підвищених навантажень пов’язана зі
здатністю адреналіну й норадреналіну
безпосередньо збільшувати силу серцевих
скорочень - з позитивним інотропним
ефектом.
Установлено, що під впливом катехоламінів
збільшується кількість Са-каналів сарколеми,
здатних відкриватися під час потенціалу дії
(катехоламіни через цАМФ-опосередкований
механізм викликають фосфорилювання білків
Са-каналів, завдяки чому останні й набувають
такої властивості).
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
98
Дія катехоламінів
Позитивний інотропний ефект катехоламінів: ЗМІНИ об'ємів серця. Умовні позначення див. на рис. 31.3
У результаті цих процесів збільшується над
ходження іонів кальцію в саркоплазму, де підви
щується їхня концентрація, і, як наслідок, збіль
шується сила скорочень кардіоміоцитів, оскіль
ки зростає кількість утворюваних кальцій-тропонінових комплексів, а отже, й актоміозинових
містків.
Інотропна дія катехоламінів (а не закон Фран
ка - Старлінга) є провідним механізмом адапта
ції серця до фізичних навантажень. При цьому
показники кардіодинаміки змінюються таким
чином (рис. 31.6):
а) збільшується ударний об’єм;
б) зростає хвилинний об’єм серця через
збільшення ударного об’єму та завдяки
збільшенню частоти серцевих скорочень
{позитивний хронотропний ефект катехо
ламінів);
в) зменшується кінцеводіастолічний об’єм
(при рентгенівському дослідженні можна
спостерігати зменшення об’єму серця);
г) знижується кінцевосистолічний об’єм.
Механізми довготривалої адаптації серця.
Тривала в часі адаптація серця до наванта
жень, залежно від їхнього рівня й періодич
ності, може відбуватися за трьома сценаріями
(Ф. Меєрсон).
1. Гіпертрофія серця у спортсменів (“адап
товане” серце). Розвивається при пері
одичних
навантаженнях,
інтенсивність
яких поступово зростає, тобто в умовах
тренувань. Цей варіант гіпертрофії є зба
лансованим і характеризується рівномір
ним збільшенням усіх складових компо
нентів серця. Завдяки такій гіпертрофії
істотно зростають функціональні резер
ви серця - у стані спокою збільшується
кінцевосистолічний об’єм, що дає змогу
значно підвищити ударний об’єм при фі
зичних навантаженнях, які завжди супро
воджуються активацією симпатоадреналової системи (рис. 31.7).
2. Компенсаторна гіпертрофія серця (“переадаптоване” серце). Є наслідком патологіч-
99
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
"Звичайне серце"
"Серце спортсмена"
Стан спокою
Катехоламіни
Фізичне
навантаження
ис. 31.7. Гіпертрофія серця у спортсменів: об'єми серця в стані спокою і при фізичному навантаженні.
Умовні позначення див. на рис. 31.3
них процесів, що пов’язані з серцем. Роз
різняють два види компенсаторної гіпер
трофії:
а) гіпертрофію від перевантажень (розви
вається при вадах серця, артеріальній гі
пертензії);
б) гіпертрофію від ушкодження (характерна
для атеросклеротичних уражень, міокардіопатії).
Розвиток компенсаторної гіпертрофії серця
характеризується такими особливостями:
» патогенний фактор, що викликає гіпертро
фію, діє постійно;
* компенсаторна гіпертрофія, на відміну від
гіпертрофії серця у спортсменів, є незбалансованою (див. далі);
* при компенсаторній гіпертрофії згодом
розвивається недостатність серця.
3. Атрофія міокарда (“деадаптоване” серце).
Характеризується зменшенням маси серця
в результаті тривалої гіпокінезії та змен
шення навантажень на серце.
Компенсаторна гіпертрофія серця. При трива
лому підвищенні навантаження на серце розви
вається його гіперфункція, що згодом викликає
структурні зміни в серці -гіпертрофію міокарда.
За динамікою змін обміну, структури й функ
ції міокарда в розвитку компенсаторної гі
пертрофії серця виділяють три основні стадії
(Ф. Меєрсон).
\.Аварійна стадія. Розвивається безпосе
редньо після підвищення навантаження,
100
характеризується
поєднанням
патоло
гічних змін у міокарді (зникнення глі
когену, зниження рівня креатинфосфату,
зменшення вмісту внутрішньоклітинного
калію й підвищення вмісту натрію, по
силення гліколізу, накопичення лактату)
з мобілізацією резервів міокарда та орга
нізму в цілому. У цій стадії підвищується
навантаження на одиницю м’язової маси,
а
отже,
інтенсивність
функціонування
структур (ІФС), відбувається швидке,
протягом тижнів, збільшення маси серця
через посилений синтез білків і потовщен
ня м’язових волокон.
2. Стадія завершеної гіпертрофії й віднос
но стійкої гіперфункції. У цій стадії про
цес гіпертрофії завершений, маса міокарда
збільшена на 100-120 % і далі не зростає,
ІФС нормалізується. Патологічні зміни
в обміні речовин і структурі міокарда не
виявляються, споживання кисню, утворен
ня енергії, вміст макроергічних сполук не
відрізняються від норми. Нормалізуються
гемодинамічні порушення. Гіпертрофоване серце пристосувалося до нових умов
навантаження і протягом тривалого часу
компенсує їх.
3. Стадія поступового виснаження і прогре
суючого кардіосклерозу. Характеризується
глибокими метаболічними і структурними
змінами, які поволі накопичуються в енергоутворюючих і скорочувальних елементах
клітин міокарда. Частина м’язових воло
кон гине й заміщається сполучною тка
ниною, ІФС знову зростає. Порушується
регуляторний апарат серця. Прогресуюче
виснаження компенсаторних резервів при
зводить до виникнення хронічної недостат
ності серця, а надалі — до недостатності
кровообігу.
Що стосується механізмів гіпертрофії міокар
да, то центральним було і залишається питання
про те, у який спосіб гіперфункція м’язових во
локон спричиняється до збільшення їхньої маси.
Незважаючи на велику кількість “білих плям”
у цій проблемі, неспростовним є твердження,
що гіпертрофія міокарда має в основі посилен
ня синтезу білків, з яких складаються м’язові
волокна. Необхідною умовою такого посилення
є не що інше, як активація генетичного апара
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ту клітин, що виявляє себе збільшенням тран
скрипції відповідних генів. Останнє є феноме
ном регуляторним і пов’язане з дією на геном
особливих білків, що їх називають транскрип
ційними факторами (див. главу 6).
Пошук конкретних молекулярних посеред
ників між гіперфункцією клітин та їхньою гі
пертрофією триває досі. Не втратила своєї при
вабливості концепція Ф. Меєрсона, відповідно
до якої провідною ланкою в активації геному
м’язових клітин є збільшення т. зв. потенціалу
фосфорилювання (ПФ):
де [АДФ], [Р.], [АТФ] - концентрації в цитоплаз
мі клітин відповідно АДФ, неорганічного фос
фату і АТФ.
ПФ закономірно збільшується у двох випад
ках (рис. 31.8):
а) при посиленому використанні АТФ, що зав
жди спостерігається при збільшенні функ
ціонального навантаження на клітини (при
їхній гіперфункції);
б) при порушеннях утворення АТФ, що харак
терно, зокрема, для кисневого голодування.
Збільшення показника ПФ через невідомі
поки що механізми викликає появу в клітинах
транскрипційних факторів, які, впливаючи на
геном клітини, посилюють синтез інформацій
ної РНК на матриці генів, що кодують структуру
функціонально важливих білків клітини, у тому
числі скорочувальних протеїнів і ферментів.
Отже, за Ф. Меєрсоном, розвиток гіпертро
фії серця можна описати такою послідовністю
процесів: збільшення навантаження на серце
(гіперфункція) —> посилене використання АТФ,
що перевищує інтенсивність його ресинтезу
—* збільшення потенціалу фосфорилювання —*
поява або збільшення вмісту в клітинах тран
скрипційних факторів ~* зростання інтенсив
ності синтезу інформаційної РНК і процесів
трансляції в рибосомах —»• посилення біосинте
зу структурних, функціональних білків і білківферментів —» збільшення маси міокарда, його
гіпертрофія.
Останнім часом за допомогою новітніх моле
кулярно-генетичних методів показано, що зміна
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
101
31.8. Роль порушень енергозабезпечення клітин у розвитку гіпертрофії міокарда (за Ф. Меєрсоном)
102
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
31.9. Молекулярно-генетичні механізми гіпертрофії міокарда, пнуг - передсердний натрійуретичний гормон
діяльності геному м’язових клітин під час гіпер
функції пов’язана щонайменше з двома групами
чинників: (1) механічним розтягненням волокон
і (2) дією на клітини гуморальних сигнальних
молекул (рис. 31.9).
Так, під впливом механічного розтягнення
в кардіоміоцитах змінюється експресія 185 генів,
при цьому діяльність 164-х посилюється, а 21-го
пригнічується. Дія адреналіну зумовлює зміну
експресії 450 генів, з них більше половини ста
ють активнішими, а менше - зазнають репресії.
Окрім збільшення синтезу властивих^’ яловим
клітинам білків, у кардіоміоцитах з’являються
нові протеїни, що є наслідком дерепресії від
повідних ембріональних генів. У такий спосіб
важкі ланцюги а-міозину замінюються важкими
ланцюгами ембріонального (І-міозину. Останній
виявляє меншу АТФ-азну активність, а отже, зу
мовлює повільніше, зате енергетично більш еко
номне скорочення серцевого м’яза
У серці ембріона ген передсердного натрійуретичного гормону (ПНУГ) є активним як
у клітинах передсердь, так і шлуночків. Піс
ля народження такий ген у шлуночкових кар
діоміоцитах “вимикається” і знову починає
працювати лише під впливом чинників, що
супроводжують довготривалу гіперфункцію
міокарда. Поновлення експресії гена ПНУГ
у шлуночках сприяє виведенню солі та води
із організму, а отже, зменшує навантаження на
гіпертрофоване серце, що можна вважати од
ним з механізмів його захисту від надмірної
роботи.
Важлива роль генетичного апарату в роз
витку гіпертрофії міокарда доводиться існуван
ням гіпертрофічної кардіоміопатії — спадкової
аутосомно-домінантної хвороби, що характери
зується значним потовщенням стінки лівого і/
або значно рідше правого шлуночка. Причиною
недуги є мутації одного з дев’яти генів, що ко
дують білки саркомеру, зокрема важкі ланцюги
(3-міозину, тропонін Т, протеїн С, що зв’язує мі
озин. У результаті порушення балансу між окре
мими скорочувальними білками м’язові волокна
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
в міокарді набувають неправильного, хаотично
го розташування, гіпертрофія стає асиметрич
ною з переважним потовщенням міжшлуночкової перегородки. Гіпертрофічна кардіоміопатія, тривалий час не маючи жодних клінічних
проявів, є основною причиною раптової смерті
у спортсменів молодого віку.
Ще одне принципове питання, на яке немає
остаточної відповіді, стосується причин деком
пенсації гіпертрофованого серця: які чинники
зумовлюють серцеву недостатність в умовах
тривалої компенсаторної гіпертрофії міокарда.
Учені сходяться в тому, що такими чинника
ми є особливості гіпертрофованого серця, що
в кінцевому підсумку виявляють себе значними
порушеннями балансу між багатьма сторонами
життєдіяльності як окремих кардіоміоцитів, так
і міокарда в цілому.
Відомо, що гіпертрофоване серце відрізня
ється від нормального низкою метаболічних,
функціональних і структурних ознак, які, з од
ного боку, дають йому можливість тривалий час
долати підвищене навантаження, а з другого створюють передумови для виникнення патоло
гічних змін.
Передумовою декомпенсації серця може бути
сукупність таких особливостей гіпертрофова
ного міокарда:
1. Потовщення кожного м’язового волокна
супроводжується зміною співвідношен
ня внутрішньоклітинних структур. Об’єм
клітини при цьому збільшується пропо
рційно кубу лінійних розмірів, а поверхня —
пропорційно їх квадрату, що призводить до
зменшення клітинної поверхні на одиницю
маси клітини. Відомо, що саме через по
верхню кардіоміоцита відбувається його
обмін з позаклітинною рідиною — погли
нання кисню, поживних речовин, виведен
ня продуктів метаболізму, обмін води та
електролітів. Зазначені вище зміни створю
ють умови для погіршення забезпечення
м’язового волокна необхідними йому ком
понентами і видалення непотрібних йому
продуктів життєдіяльності.
2. Клітинна мембрана відіграє важливу роль
у проведенні збудження і спряженні про
цесів збудження й скорочення, що здійс
нюється через тубулярну систему та саркоплазматичний ретикулум. Оскільки ріст
цих утворень при гіпертрофії м’язового
103
волокна також відстає, то створюються
передумови для порушення процесів ско
рочення та розслаблення кардіоміоцитів:
унаслідок уповільнення виходу іонів каль
цію в саркоплазму погіршується скорочен
ня, а в результаті утруднення зворотного
транспорту іонів кальцію в саркоплазматичний ретикулум - розслаблення, іноді
можуть виникати локальні контрактури
окремих кардіоміоцитів.
3. У процесі розвитку гіпертрофії маса мітохондрій спочатку збільшується швидше,
ніж маса скорочувальних білків, створю
ючи умови для достатнього енергетично
го забезпечення та належної компенсації
функції серця. Однак надалі, у ході поси
лення процесу, збільшення маси мітохондрій починає відставати від росту маси
цитоплазми. Унаслідок граничного наван
таження в мітохондріях розвиваються де
структивні зміни, знижується ефективність
їхньої роботи, порушується окисне фосфорилювання. Це веде до погіршення енер
гетичного
забезпечення
гіпертрофованої
клітини.
4. Збільшення маси м’язових волокон не су
проводжується адекватним збільшенням
мережі кровоносних судин, що живлять
серце. Це веде до погіршення оксигенації та трофічного забезпечення міокарда,
що може бути особливо критичним під
час збільшення навантаження на нього.
А тому слід вести мову про погіршення
судинного
забезпечення
гіпертрофованого
міокарда.
5. При розвитку гіпертрофії міокарда до про
цесу обов’язково долучається нервовий
апарат серця. Особливо на початкових ста
діях спостерігається посилення внутрішньосерцевих і екстракардіальних рефлек
сів. Проте кількість нервових закінчень
у гіпертрофованому міокарді змінюється
мало, по суті розвивається часткова денервація серця, воно стає більш чутливим до
гуморальних регуляторних чинників, зо
крема катехоламінів.
6. Гіпертрофоване серце через збільшення
маси його скорочувального апарату і сис
тем енергозабезпечення здатне тривалий
час виконувати значно більшу роботу, ніж
серце нормальне, зберігаючи при цьому
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
104
метаболізм у нормі. Однак здатність при
стосовуватися до навантажень, що зміню
ються, діапазон адаптаційних можливостей
у гіпертрофованого серця обмежені, змен
шений функціональний резерв. Це робить гіпертрофоване серце через зазначену незбалансованість внутрішньоклітинних і тка
нинних структур більш уразливим у різних
несприятливих обставинах.
Наведені
особливості
гіпертрофованого
серця стають передумовою функціональних
і структурних змін, характерних для його де
компенсації. Значна частина м’язових воло
кон гине механізмами апоптозу чи некрозу.
У зв’язку з цим навантаження на ті клітини, що
залишилися, зростає; робота, здатних до скоро
чення кардіоміоцитів та окремих їхніх струк
тур, збільшується до максимально можливого
рівня. Врешті-решт настає виснаження меха
нізмів функціонування клітин і запускається
програма апоптозу. Живих, діяльних кардіомі
оцитів стає все менше, а елементів сполучної
тканини, що вона їх заміщує, - все більше: роз
вивається кардіосклероз з усіма ознаками недо
статності серця.
МІОКАРДІАЛЬНА
НЕДОСТАТНІСТЬ СЕРЦЯ
Міокардіальною називають недостатність сер
ця, що розвивається в результаті первинного
ураження серцевого м’яза. Оскільки міокард
складається з двох типів м’язових волокон атипових, що становлять провідникову систему
серця, і скорочувальних клітин робочого міо
карда, — то розвиток недостатності серця може
бути пов’язаний з ушкодженням як перших, так
і других. Звідси можна виділити два варіанти
міокардіальної недостатності серця: (а) зумов
лену ураженням провідникової системи сер
ця - аритмічну і (б) викликану ушкодженням
робочого міокарда. Останню іноді називають
міокардіопатичною.
Причиною розвитку міокардіальною варіан
та серцевої недостатності часто є інфекції, ін
токсикація, гіпоксія, авітамінози, порушення
вінцевого кровообігу, деякі спадкові дефекти
обміну речовин. При цьому недостатність роз
вивається навіть за нормального або зниженого
навантаження на серце.
Порушення скорочувальної функції робочого
міокарда можуть бути зумовлені як (а) кількісни
ми — зменшення числа кардіоміоцитів унаслідок
їхньої загибелі, так і (б) якісними - зменшення
здатності до скорочення - змінами (про ураження
провідникової системи серця див. підглаву 31.2).
Ушкодження та загибель м’язових волокон
найчастіше настають внаслідок порушень кро
вопостачання серця, а тому їхні механізми бу
дуть обговорені в одній із наступних підглав
(див. 31.3). Тут ми зупинимося лише на змінах,
що зумовлюють функціональні розлади кардіо
міоцитів, не викликаючи напряму їх ушкоджен
ня і смерті.
У клітинах робочого міокарда відбувається
п’ять процесів, що визначають скорочувальну
функцію серця. Усі вони можуть порушуватися
і становити основу недостатності серця.
Об’єктами можливих первинних функціо
нальних розладів у серцевому м’язі є процеси:
(1) збудження кардіоміоцитів; (2) електромеха
нічного спряження; (3) власне скорочення; (4)
розслаблення; (5) енергозабезпечення міокарда.
1. Порушення збудливості і збудження кар
діоміоцитів. Електрофізіологічні
властивості
клітин серця характеризують поняттями збудли
вість і збудження. Перший з цих термінів озна
чає здатність клітин збуджуватися, тобто гене
рувати потенціали дії, а другий - власне процес
генерування таких потенціалів та їхнього поши
рення.
Причиною збудження клітин робочого міо
карда є імпульси, які надходять до нього через
провідникову систему серця. Потенціали дії, що
виникають в кардіоміоцитах, мають характерну
форму і складаються з трьох послідовних фаз
(рис. 31.10):
І фаза - фаза швидкої деполяризації (“на
трієва”). Триває 1-2 мс, під час яких мемб
ранний потенціал стрімко змінюється від
-90 мВ до +(30-40) мВ. Основу цієї фази
складає швидке входження іонів Na+ в клі
тину через мембранні потенціалзалежні Naканали, які відкриваються при надходженні
імпульсів по волокнах Пуркіньє. Перемі
щення іонів Na+ в клітину швидко припи
няється внаслідок того, що Na-канали за
знають інактивації, тобто закриваються.
105
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
Рис. 31.10. Потенціал дії ізольованого шлуночкового кардіоміоцита
II фаза - фаза повільної реполяризації або
фаза “тато” (“кальцієва”). Вона є дуже
тривалою (у клітинах передсердь - до 0,2 с,
шлуночків — до 0,3 с) і власне визначає за
гальну тривалість потенціалу дії. Її розви
ток зумовлений відкриттям повільних потенціалзалежних Са-каналів, через які іони
Са2+ (а також і Na+) входять іззовні в клітину
завдяки високому концентраційному граді
єнту. У цю фазу проникність мембрани до
іонів К+ зменшується в 5 разів, проте на
явний реполяризуючий калієвий струм усе
ж таки залишається більшим за деполяризуючий кальцієвий, а тому на загал настає
не деполяризація, а повільна реполяризація
мембрани.
• III фаза - фаза швидкої реполяризації (“ка
лієва”). Після закриття Са-каналів входжен
ня в клітини іонів Са2+ і Na+ припиняється,
натомість відкриваються К-канали, через
які іони К+ за градієнтом концентрації ви
ходять з кардіоміоцитів назовні, у такий
спосіб реполяризуючи мембрану.
•
Основні характеристики потенціалу дії клі
тин робочого міокарда (тривалість, амплітуда,
крутість наростання деполяризації) і збудли
вість кардіоміоцитів залежать від таких чинни
ків:
а) характеру електричної імпульсації, що
надходить до кардіоміоцитів через про
відникову систему серця. При аритміях
можливе порушення основних параметрів
потенціалу дії волокон робочого міокарда
і його збудливості;
б) стану (властивостей) сарколеми кардіоміо
цитів, і насамперед, іонних каналів. Нині
відомі хімічні агенти, здатні вибірково по
рушувати провідність цих каналів. Це блокатори Na-каналів, Са-каналів, К-каналів;
в) концентрації позаклітинних іонів, що бе
руть участь у формуванні електричних
потенціалів на мембрані кардіоміоцитів.
В умовах in vivo найбільше значення має
позаклітинна концентрація іонів калію.
Зміни збудливості волокон міокарда можуть
виявляти себе:
1) змінами тривалості потенціалів дії, а отже,
і сили серцевих скорочень. Зменшення три
валості потенціалів дії виникає при збіль
шенні частоти стимуляції, збільшенні поза
106
клітинної концентрації іонів калію, при дії
ацетилхоліну. Збільшення тривалості по
тенціалу дії характерне для охолодження;
2) змінами тривалості періодів абсолютної
і відносної рефрактерності, а отже, здат
ності сприймати імпульсацію (засвоювати
ритм). Тривалість цих періодів залежить
від тривалості потенціалів дії.
Важливе значення для підтримання збудли
вості кардіоміоцитів має позаклітинна концен
трація іонів калію. Вона впливає на збудли
вість міокарда через рівень потенціалу спокою
м’язових клітин.
Збільшення концентрації іонів калію - гіперкаліємія — викликає деполяризацію мембрани
м’язових волокон. При цьому характер змін
збудливості міокарда залежить від рівня гіперкаліємїї. Виділяють такі діапазони концентрацій
К+, для яких характерні певні картини порушень:
1) від 4 до 8 ммоль/л. Відбувається незначна
деполяризація, мембранний потенціал па
дає від -90 мВ до -80 мВ. При цьому стан
Na-каналів сарколеми істотно не зміню
ється. Унаслідок того, що величина мем
бранного потенціалу наближається до кри
тичного рівня деполяризації, збудливість
м’язових волокон і швидкість проведення
імпульсів зростають;
2) від 8 до 35 ммоль/л. Мембранний потенці
ал падає від -80 мВ до -40 мВ. При такому
рівні деполяризації значно зменшується
провідність швидких потенціалзалежних
Na-каналів (період відносної рефрактер
ності). Наслідком є зменшення збудливос
ті та провідності, а також зміни характеру
потенціалів дії (зменшення тривалості,
амплітуди, крутості);
3) понад 35 ммоль/л. Мембранний потенціал
стає менше -40 мВ. При цьому всі швидкі
потенціалзалежні Na-канали перебувають
у стані інактивації (абсолютна рефрактерність) - відбувається зупинка серця.
Зменшення позаклітинної концентрації іонів
калію — гіпокаліємія — викликає гіперполяризацію мембрани. Якщо гіпокаліємія досягає рівня
2-3 ммоль/л, дещо збільшується поріг деполя
ризації, зменшується збудливість, збільшують
ся тривалість потенціалів дії і сила скорочень
серця.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
У хірургічній практиці нерідко доводиться
зупиняти серце, щоб виконати операції на ньо
му. Штучну зупинку серця для цієї мети назива
ють кардіоплегією. Один із методів кардіоплегії
ґрунтується на тому, що у вінцеві артерії вводять
розчини, до складу яких входять високі концен
трації іонів калію, ацетилхолін і деякі інші пре
парати. Цей метод дозволяє проводити операції
на “сухому” серці протягом 40-60 хв.
2.
Порушення електромеханічного спряжен
ня. Поєднання процесів збудження зі скорочен
ням клітин одержало назву електромеханічного
спряження.
Основною дійовою особою цього проце
су є іони кальцію, що надходять у цитоплазму
м’язових клітин під час збудження. У кардіоміоцитах існує два важливих джерела цих іонів:
а) внутрішньоклітинне — структури саркоплазматичного ретикулуму: цистерни і по
здовжні трубочки (як і в скелетних м’язах).
У них всередині накопичуються великі кон
центрації кальцієвих іонів завдяки роботі
спеціальних Са-насосів;
б) позаклітинне — поперечні Т-трубочки і се
редовище, що оточує м’язові волокна.
Оскільки Т-трубочки є інвагінаціями плаз
матичної мембрани, то вміст іонів Са2+ у них
такий самий, як і в інтерстиції, тобто на кіль
ка порядків вищий, ніж у саркоплазмі. Ве
лика кількість кальцієвих іонів депонується
в Т-трубочках завдяки зв’язуванню з ані
онними залишками глікопротеїнів мембра
ни (глікокаліксом). Позаклітинне джерело
кальцію є дуже важливим: у середовищі,
позбавленому Са2+, сила серцевих скорочень
поступово зменшується і вони, врешті-решт,
припиняються. Це означає, що внутрішньо
клітинних запасників Са2+ недостатньо, щоб
підтримувати скорочувальну активність
кардіоміоцитів.
Сьогодні виділяють два механізми електро
механічного спряження в серці.
1. Потенціалзалежне спряження (рис. 31.11).
Суть його полягає в тому, що поширення
збудження (потенціалу дії) уздовж плазма
тичної мембрани зумовлює деполяризацію
мембран Т-трубочок, а через них— мембран
цистерн і поздовжніх трубочок саркоплазматичного ретикулуму. Така деполяризація
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
107
Поперечні
трубочки
Рис. 31.11. Спряження збудження кардіоміоцитів зїх скороченням: потенціалзалежний механізм, пд - потенціал дії
веде до відкриття потенціалзалежних каль
цієвих каналів, унаслідок чого іони Са2+
завдяки високому градієнту концентрацій
швидко виходять із названих вище струк
тур у цитоплазму.
2. Механізм кальцієвих залпів (рис. 31.12). Під
час фази плато потенціалів дії позаклітинні
іони Са2+ входять через потенціалзалежні
Са-канали в клітину. Цей первинний каль
цієвий залп або безпосередньо, або через
утворення вторинних посередників (месенджерів) - 1,4,5-інозитолтрифосфату - діє
на мембрани структур саркоплазматичного
ретикулуму, викликаючи відкриття наяв
них там Са-каналів і вихід іонів Са2+ у ци
топлазму як наслідок.
На характер електромеханічного спряження
в міокарді впливає ряд факторів.
• Тривалість і частота потенціалів дії.
Зміни тривалості потенціалів дії в кардіоміоцитах відбуваються за рахунок укоро
чення або подовження фази плато, під час
якої іони Са2+ через потенціалзалежні Саканали сарколеми надходять у саркоплазму.
З урахуванням того, що концентрація іонів
Са2+ у цитоплазмі м’язових волокон визначає
кількість актоміозинових комплексів, що утво
рюються, а отже, і силу скорочень, можна зро
бити такий висновок. При зменшенні тривалості
потенціалу дії зменшується надходження іонів
Са2+ у саркоплазму і, як наслідок, зменшується
сила скорочення м’язового волокна. При збіль
шенні тривалості потенціалу дії - все навпаки.
Тривалість потенціалу дії кардіоміоци
тів зменшується при гіперкаліємїї, при дії на
м’язові волокна ацетилхоліну. Збільшення три
валості потенціалу дії характерне для гіпокаліємії, охолодження серця.
При збільшенні частоти потенціалів дії, не
зважаючи на те, що тривалість кожного окре
мого потенціалу, а отже, і фази плато зменшу
ється, сумарна тривалість зазначеної фази за
одиницю часу збільшується. Це означає, що
в кардіоміоцитах створюються більш високі
концентрації Са2+ і, як наслідок, збільшується
сила скорочення м’язових волокон. Цим, зо
крема, пояснюється хроноінотропний механізм
негайної компенсації серця.
При зменшенні частоти потенціалів дії (на
приклад, при брадикардії) сумарна тривалість
фази плато за одиницю часу зменшується, що
веде до зменшення концентрації Са2+ у сарко
плазмі та зменшення сили скорочень серця.
• Концентрація іонів кальцію в позаклітин
ній рідині. На початку XX ст. Рінгером було
показано, що в розчині, позбавленому іонів
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
108
Рис. 31.12. Спряження збудження кардіоміоцитів з їх скороченням: механізм кальцієвих залпів.
ФІ-Ф2 - фосфатидилінозитолдифосфат, ІФЗ - інозитолтрифосфат, ДАГ - діацилгліцерол, СПР - саркоплазматичний ретикулум
кальцію, ізольоване серце швидко зупи
няється. Нині відомо, що причиною цього
є повне роз’єднання збудження і скорочен
ня. В умовах норми іони кальцію, що надхо
дять під час фази плато потенціалу дії з по
заклітинного середовища в саркоплазму
кардіоміоцитів, “запускають” вивільнення
Са2+ із саркоплазматичного ретикулуму
й поповнюють його запаси в цих структу
рах м’язових волокон. При зменшенні по
заклітинної концентрації Са2+ його запаси
в саркоплазматичному ретикулумі швидко
виснажуються, концентрація Са2+ у сар
коплазмі падає й, отже, зменшується сила
серцевих скорочень.
При вивченні ролі іонів Са2+ у діяльності
серця було відкрито явище, яке отримало назву
“кальцієвого парадоксу”. Суть цього експери
ментального феномену полягає в тому, що після
того як у безкальцієвий розчин, яким здійсню
вали перфузію серця протягом кількох хвилин,
вносять іони кальцію, розвивається необоротне
ушкодження міокарда: зменшується вміст АТФ
і креатинфосфату в тканинах серця, з кардіоміо
цитів виходять білки, у тому числі ферменти
(міоглобін, креатинкіназа), руйнується сарко
плазматичний ретикулум.
“Кальцієвий парадокс” пояснюють тим, що
в безкальцієвому розчині відбувається розша
рування глікокаліксу кардіоміоцитів (зовнішній
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
шар глікокаліксу відокремлюється від внутріш
нього), у результаті чого значно зростає проник
ність сарколеми до іонів кальцію. При наступно
му внесенні в середовище іонів Са2+ спостеріга
ється їхнє масивне надходження у клітини, різко
зростає концентрація цих іонів у саркоплазмі,
що “запускає” кальцієві механізми ушкодження
(див. главу 7).
- Стан Са-каналів сарколеми кардіоміоцитів. У плазматичній мембрані м’язових клі
тин серця потенціалзалежні повільні Са
нтали - це білкові молекули, здатні пропус
кати через себе іони кальцію в тому випадку,
коли вони відкриті. Відкриття Са-каналів
відбувається при деполяризації мембрани,
але відкриватися здатні не всі існуючі кана
ли, а тільки фосфорильовані (активовані).
Суть активації Са-каналів полягає в їхньому
фосфорилюванні, у результаті чого збільшується
кількість Са-каналів, здатних відкриватися при
виникненні потенціалу дії. В основі цього біо
хімічного процесу лежить збільшення концен
трації цАМФ у саркоплазмі м’язових волокон.
Усі фактори, що викликають утворення цАМФ,
ведуть до активації Са-каналів. До таких, зо
крема, належать: (а) катехоламіни і фармаколо
гічні агенти, що активують Р-адренорецепторщ
(б)
інгібітори фосфодіестерази (метилксантини: теофілін, кофеїн та ін.). Зазначені агенти
збільшують вміст цАМФ або через активацію
аденілатциклази
(через
Р-адренорецептори),
або через пригнічення руйнування цАМФ (інгі
бітори фосфодіестерази).
З активацією Са-каналів пов’язаний позитив
ний інотропний ефект катехоламінів. Основу
його складає така послідовність подій: катехо
ламіни —> активація Р-адренорецепторів —► ак
тивація аденілатциклази —* утворення цАМФ —>
активація протеїнкіназ —» фосфорилювання Саканалів сарколеми —> збільшення надходження
Са2+ у саркоплазму під час потенціалів дії —>
збільшення концентрації Са2+ у саркоплазмі —*
збільшення сили скорочень серця.
В умовах блокади Са-каналів, навпаки, змен
шується надходження іонів Са в саркоплазму
кардіоміоцитів і, отже, зменшується сила серце
вих скорочень.
Блокаду Са-каналів викликають:
а) ендогенні фактори - брак АТФ, дефіцит
цАМФ, іони водню (ацидоз);
109
б) екзогенні фактори - двовалентні іони (Ni2+,
Со2+, Мп2+), деякі тривалентні іони (La3+),
органічні сполуки, що їх застосовують
у практичній медицині (верапаміл, ніфедипін тощо).
* Стан систем видалення іонів кальцію із
цитоплазми м’язових волокон. На нього,
зокрема, впливають (а) серцеві глікозиди —
фармакологічні препарати рослинного по
ходження, що підвищують силу скорочень
серця (препарати дигіталісу, строфантин
та ін.) та (б) ендогенні дигіталісоподібні
і строфантиноподібні фактори. Вважа
ють, що серцеві глікозиди, які здавна вико
ристовуються в медицині, імітують дію цих
ендогенних природних факторів. Фізіо
логічне значення останніх сьогодні ще не
встановлено.
Механізм дії серцевих глікозидів і подібних
до них ендогенних факторів пов’язаний з при
гніченням активності Na-K-АТФ-ази кардіоміо
цитів. Наслідком цього є порушення роботи
Na-K-насосів сарколеми, зменшення градієнта
концентрацій іонів Na+ по обидва боки плаз
матичної мембрани м’язових волокон. Це при
зводить до порушення Na-Ca-обмінного механіз
му в кардіоміоцитах (див. нижче), у результаті
чого зменшується видалення Са2+ із саркоплаз
ми в позаклітинне середовище та збільшується
внутрішньоклітинна концентрація Са2+. Остан
нє й обумовлює збільшення сили серцевих ско
рочень.
3.
Порушення процесу власне скорочення.
Суть скорочення кардіоміоцитів полягає в пе
реміщенні актинових і міозинових філаментів
одних відносно інших (рис. 31.13). Цьому пе
редує утворення містків між молекулами ак
тину й головками міозину. Такі актоміозинові
комплекси з’являються тільки тоді, коли зни
кає блокування білком тропоміозином діля
нок актину, здатних зв’язуватися з головками
міозину. Вивільнення таких ділянок від тропоміозину настає після утворення комплексів
ще одного білка - тропоніну С - з іонами Са2+
при збільшенні концентрації останніх в цито
плазмі. Отже, поява комплексів “Са-тропонін”
спричиняється до зміщення молекул тропоміозину відносно актинових філаментів, уна
слідок чого центри актину стають доступними
3. Механізм скорочення м'язових волокон серця: участь різних білків та іонів кальцію в цьому
процесі. ТрС - тропонін с
для міозинових головок - процес скорочення
починається.
З урахуванням цього на силу скорочень
м’язових волокон серця впливають такі чинни
ки:
1) концентрація іонів Са2+ у саркоплазмі. За
лежність тут така: що вищий вміст Са2+, то
більше утворюється комплексів Са2+ з тропоніном С, то більше вивільняється центрів
зв’язування (активних центрів) на актинових міофіламентах, більше утворюється
“містків” між актином і головками міозину,
більшою буде сила скорочення м’язового
волокна. При зменшенні концентрації Са2+
у саркоплазмі - усе навпаки;
2) ступінь спорідненості тропоніну С до
іонів кальцію. Іони водню й неорганічно
го фосфату, зв’язуючись з тропоніном С,
унеможливлюють взаємодію цього білка
з Са2+, у результаті чого сила скорочень
кардіоміоцитів зменшується;
3) стан скорочувальних білків - актину і мі
озину. Велике значення має взаємне розта
шування актинових і міозинових міофіламентів. Воно лежить в основі залежності,
що її описує закон Франка — Старлінга.
При дуже сильному розтягненні м’язових
волокон кількість утворюваних актоміозинових “містків” зменшується — зазначений
закон не “спрацьовує”, сила скорочень сер
ця падає;
4) концентрація АТФ, енергія гідролізу якого
забезпечує ковзання м’язових філаментів
один відносно одного.
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
4.
Порушення процесу розслаблення. Основ
на подія, що визначає розслаблення кардіоміоцитів, — це видалення іонів кальцію із сарко
плазми, у результаті чого концентрація Са2+
у ній зменшується і стає нижче 10-7 моль/л. При
цьому комплекси Са2+ з тропоніном С розпада
ються, тропоміозин зміщується по відношенню
до актинових філаментів і закриває їхні активні
центри - скорочення припиняється.
Описано три механізми видалення іонів Са2+
із саркоплазми кардіоміоцитів:
1)
Са-насоси плазматичної мембрани та
саркоплазматичного
ретикулуму.
Вони
2+
видаляють Са , відповідно, у позаклітин
не середовище і цистерни саркоплазма
тичного ретикулуму. Складовою їхньою
частиною є Са-АТФ-аза, яка для здійс
нення активного транспорту іонів Са2+
використовує енергію АТФ. Посилення
роботи Са-насосів пов’язано з фосфорилюванням білків, зокрема фосфоламбану,
що входять до їхнього складу. Таке фосфорилювання, у свою чергу, відбуваєть
ся під впливом протеїнкіназ, що можуть
активуватися щонайменше двома меха
нізмами. Перший з них - кальмодулінзалежний - зумовлений власне збільшенням
концентрації іонів Са2+ у цитоплазмі. При
цьому відбувається зв’язування кальцію
з кальмодуліном з подальшою активаці
єю певної протеїнкінази. Другий меха
нізм - цАМФ-залежний — *‘запускається”
катехоламінами. Під їхнім впливом, який
опосередковується
Р-адренорецепторами
й аденілатциклазою, зростає утворення
цАМФ і активуються цАМФ-залежні про
теїнкінази, які ведуть до фосфорилювання
вже згадуваних білків Са-насосів. Таким
чином, катехоламіни збільшують не тіль
ки силу серцевих скорочень, а й швидкість
розслаблення кардіоміоцитів, сприяючи
кращому наповненню порожнин серця
кров’ю під час діастоли. При надходженні
значної кількості іонів кальцію в структу
ри саркоплазматичного ретикулуму вони
зв’язуються з білком кальсеквестрином
і в такий спосіб депонуються в клітинах;
2) Na-Ca-обмінний механізм. Видаляє іони
Са2+ у позаклітинне середовище. Є різно
видом вторинного активного транспорту
111
(антипорту). Використовує енергію граді
єнта концентрацій іонів натрію по обидва
боки плазматичної мембрани, тому зале
жить від роботи Na-K-насоса, що створює
цей градієнт;
3)
Са-акумулятивна функція мітохондрій.
Активується тільки при значному під
вищенні вмісту іонів Са2+ у саркоплазмі,
що найчастіше буває в умовах патоло
гії. Видалення Са2+ із саркоплазми в матрикс мітохондрій відбувається за допо
могою енергії, що вивільняється в про
цесі транспорту електронів дихальним
ланцюгом. Використання цієї енергії на
активний транспорт іонів Са2+ у мітохондрії є альтернативою окисному фосфорилюванню.
Основними причинами порушень розслаб
лення кардіоміоцитів є:
а) дефіцит АТФ. При цьому порушується
енергозабезпечення Са- і Na-K-насосів,
а також не відбувається розщеплення актоміозинових “містків”, що утворилися в про
цесі скорочення. Останнє пояснюється тим,
що для розриву зв’язків між молекулами
актину й міозину необхідне приєднання но
вих молекул АТФ. За відсутності достатньої
кількості АТФ такі зв’язки не розриваються
і настає феномен трупного задубіння',
б) порушення роботи Са-транспортних сис
тем. Відомі спадково зумовлені дефекти
білків Са-насосів, що призводять до розвит
ку кардіопатії.
Порушення розслаблення кардіоміоцитів ви
являють себе розвитком м ’язових контрактур.
5.
Порушення енергозабезпечення. Енергія
гідролізу АТФ у функціонально активних кардіоміоцитах забезпечує:
1) механічну роботу скорочень міофібрил
(ковзання міофіламентів один відносно
одного);
2) осмотичну роботу - активний транспорт
іонів Са2+, Na +, К+ проти градієнтів кон
центрацій (робота іонних насосів);
3) фосфорилювання білків Са-каналів плазма
тичної мембрани, фосфоламбану (білка Санасосів саркоплазматичного ретикулуму).
Унаслідок дефіциту АТФ порушується вико
нання всіх названих вище робіт.
112
Розлади енергозабезпечення кардіоміоцитів
можуть бути пов’язані з порушеннями:
а) ресинтезу АТФ (гіпоксія, голодування, де
фіцит вітамінів, зменшення активності
ферментів енергетичного обміну);
б) транспорту АТФ із мітохондрій до місць
його використання (порушення креатинкіназної транспортної системи);
в) утилізації АТФ (зменшення АТФ-азної ак
тивності структур кардіоміоцитів).
З наведеної вище інформації випливає, що на
всіх етапах процесу скорочення м’язових клітин
серця (збудження, електромеханічне спряження,
власне скорочення, розслаблення) важливу роль
відіграють іони кальцію.
Сучасний рівень знань про молекулярні ме
ханізми скорочувальної функції кардіоміоци
тів дозволяє виділити два принципово різних
патогенетичних варіанти недостатності серця:
гіпокальцієвий і гіперкальцієвий, для яких ха
рактерне відповідно зменшення й збільшення
концентрації іонів Са2+ у саркоплазмі кардіоміо
цитів.
і Гіпокальцієвий варіант розвивається в ре
зультаті порушень збудження та електроме
ханічного спряження у волокнах міокарда.
Це буває при аритміях (брадикардії різного
походження, блокади), короткочасній іше
мії міокарда (порушується фосфорилювання Са-каналів у результаті дефіциту АТФ),
ацидозі (блокада Са-каналів іонами водню),
гіпокальціємії. Виявляє себе зменшенням
сили серцевих скорочень. Основний прин
цип лікування - підвищення вмісту іонів
Са2+ у саркоплазмі кардіоміоцитів під час
систоли серця. Для цього використовують:
(а) серцеві глікозиди; (б) катехоламіни
й Р-адреноміметики; (в) парні електричні
стимули.
* Гіперкальцієвий варіант розвивається в ре
зультаті посиленого надходження іонів
Са2+ у саркоплазму кардіоміоцитів з по
заклітинного середовища (усі види ушко
дження сарколеми, при яких підвищується
її проникність) або внаслідок порушень
видалення Са2+ із саркоплазми (дефіцит
АТФ, порушення функції Са-транспортних
систем). Виявляється розвитком контракту
ри (перескорочення) міофібрил, у резуль
таті чого стає неможливим розслаблення
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
м’язових волокон, а отже, і подальше їх
скорочення. Основний принцип лікування т
зменшення вмісту іонів Са2+ у саркоплазмі
кардіоміоцитів. Для цього використовують:
(а) (3-адреноблокатори і (б) блокатори Саканалів.
ПОЗАМІОКАРДІАЛЬНА
НЕДОСТАТНІСТЬ СЕРЦЯ
Позаміокардіальною називають недостатність
серця, що розвивається в результаті дії причин,
не пов’язаних безпосередньо з міокардом. Вона
характеризується
первинною
діастолічною
дисфункцією (див. вище) і може виникати вна
слідок дії одного з двох чинників:
1) зменшення притоку крові до серця по ве
нах. Основними причинами цього є гіповолемія (крововтрата) або різке розширення
судин (колапс);
2) неможливості прийняти всю кров, що над
ходить до серця по венах. Така ситуація
виникає при накопиченні рідини в порож
нині перикарда (запальний ексудат при пе
рикардитах, кров - при пораненнях), що
утруднює розширення камер серця під час
діастоли.
Накопичення рідини в порожнині перикарда
може відбуватися швидко і повільно.
Швидке накопичення відбувається внаслідок
крововиливу при пораненні або розриві серця,
при перикардиті, що швидко розвивається. Че
рез погану розтяжність перикарда в його порож
нині підвищується тиск, що перешкоджає діастолічному розширенню серця, виникає гостра
тампонада серця.
В експерименті цей процес моделюють
уведенням рідини в порожнину перикарда
(О. Фохт). При цьому спостерігають:
а) зменшення кровонаповнення порожнин
серця;
б) зниження ударного об’єму;
в) падіння артеріального тиску. Між внутрішньоперикардіальним і артеріальним тиском
існує чітка обернена залежність: що біль
шим є перший, то нижчим буде другий;
г) підвищення венозного тиску.
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
Умикання компенсаторних механізмів при
перикардиті відбувається рефлекторно за учас
ті сигналів, що надходять з трьох рецепторних
полів: (1) отворів порожнистих і легеневих вен —
підвищення тиску на шляхах припливу; (2) аор
ти і сонних синусів (синокаротидні зони) - зни
ження тиску на шляхах відтоку; (3) перикарда —
підвищення внутрішньоперикардіального тиску.
При перетинанні блукаючих і депресорних нер
вів, а також при вимиканні рецепторних полів
за допомогою новокаїну пристосувальні реакції
не здійснюються - порушення кровообігу ма
ють набагато тяжчий перебіг.
При тампонаді серця мобілізація потужних
механізмів компенсації, які ведуть до посилення
скорочень серця (гомео- і гетерометричні меха
нізми, інотропний ефект катехоламінів), мало
ефективна або неможлива. Тому діють тільки
відносно малопотужні й енергетично невигідні
механізми компенсації та підтримки артеріаль
ного тиску ш (а) збільшення частоти серцевих
скорочень і (б) звуження периферичних судин.
Цим і пояснюється тяжкий клінічний перебіг
гострої тампонади серця.
Повільне накопичення рідини в перикарді
спостерігається при хронічному ексудативному
перикардиті та гідроперикарді, воно супрово
джується поступовим розтягуванням перикарда
і збільшенням об’єму навколосерцевої сумки.
Унаслідок цього внутрішньоперикардіальний
тиск змінюється порівняно мало, а порушення
системного кровообігу не виникають тривалий
час завдяки більш ефективній у цих умовах ро
боті компенсаторних механізмів.
ОСНОВНІ ПРОЯВИ
НЕДОСТАТНОСТІ СЕРЦЯ
У разі неспроможності негайних і довготрива
лих механізмів компенсації підтримувати належ
ну роботу серця й виконання ним насосної функ
ції розвивається декомпенсація, яка виявляє себе
порушеннями показників кардіо- і гемодинаміки,
а також розвитком цілого ряду клінічних проявів.
Для декомпенсованого серця характерними є
такі зміни показників його діяльності:
1) зменшується серцевий виштовх (ударний
об’єм)',
2) збільшується кінцевосистолічний об’єм',
113
3) зростає кінцеводіастолічний об’єм',
4)
підвищується кінцеводіастолічний тиск,
у результаті чого розвивається міогенна
дилатація серця (розширення його порож
нин);
5) зменшуються серцеві індекси — показни
ки, що характеризують скорочувальну ак
тивність серця. Серед них велике значен
ня має зниження dP/dt —max
максимальної
швидкості приросту тиску під час періоду
ізоволемічного скорочення;
6) зростає частота серцевих скорочень. Та
хікардія розвивається рефлекторно в ре
зультаті збудження рецепторів устя пере
повнених кров’ю порожнистих вен (ре
флекс Бейнбріджа). Має також значення
безпосереднє збудження клітин синуснопередсердного вузла в результаті підви
щення тиску крові в порожнині правого
передсердя.
Унаслідок порушень роботи серця страждає
вся система кровообігу, що виявляє себе зміна
ми таких показників гемодинаміки:
1) зменшується хвилинний об’єм крові;
2) якщо недостатність серця розвивається за
лівошлуночковим типом, то
і падає артеріальний тиск у великому колі
кровообігу,
» збільшується загальний периферичний опір
(реакція спрямована на зменшення падін
ня артеріального тиску);
* підвищується тиск крові в малому колі
кровообігу. Гіпертензія малого кола при
зводить до застою крові в легенях;
3) якщо недостатність серця розвивається за
правошлуночковим типом, то
» знижується артеріальний тиск у малому
колі кровообігу,
* збільшується опір судин малого кола',
* зростає центральний венозний тиск — роз
вивається застій крові у великому колі кро
вообігу;
4) збільшується об’єм циркулюючої крові —
розвивається гіперволемія. Вона є резуль
татом затримки води в організмі й поліцитемії (див. нижче).
Клінічні прояви. Для декомпенсованої недо
статності серця характерні певні клінічні озна
ки та синдроми.
114
Циркуляторна гіпоксія. Кисневе голоду
вання, що розвивається при недостатнос
ті серця, має характер застійної гіпоксії.
Основні її характеристики й наслідки опи
сано в главі 23.
2. Задишка. Для розвитку цього симптому ма
ють значення:
а) вплив надлишку іонів водню на хеморецеп
тори судин і безпосередньо на дихальний
центр;
б) набряк інтерстиціальної тканини легень
і пов’язане з цим збудження J-рецепторів.
3. Ціаноз. Ціанотичний відтінок шкіри і ви
димих слизових оболонок у хворих з де
компенсованим серцем обумовлений збіль
шенням концентрації в крові відновленого
гемоглобіну в результаті більш повного ви
лучення кисню тканинами.
4. Набряки. При правошлуночковій недостат
ності розвиваються набряки нижньої поло
вини тіла; при лівошлуночковій - інтерстиціальний набряк легень (синдром серцевої
астми) або альвеолярний набряк (синдром
набряку легень). Механізм розвитку серце
вих набряків є досить складним, і він до
кладно аналізується у главі 27.
5. Кардіальний цироз печінки. Характерний
для правошлуночкової недостатності сер
ця. Виявляє себе порушеннями функції пе
чінки й синдромом портальної гіпертензії
(див. главу 35).
6. Порушення кислотно-основної рівноваги.
В умовах декомпенсованої серцевої недо
статності можливими є всі чотири варіанти
змін цього параметру гомеостазу:
а) негазовий ацидоз. Унаслідок циркуляторної
гіпоксії у крові накопичуються кислі про
дукти обміну речовин, зокрема, молочна
кислота;
б) газовий ацидоз. У разі розвитку альвеоляр
ного набряку легень настає недостатність
зовнішнього дихання, що спричиняється
до гіперкапнії, а отже, і газового ацидозу;
в) негазовий алкалоз. Тривала хронічна недо
статність серця супроводжується вторин
ним гіперальдостеронізмом і обумовленою
ним гіпокаліємією. Остання і стає причи
ною алкалозу;
г) газовий алкалоз. Це порушення може бути
наслідком рефлекторної задишки (гіпервентиляції), що призводить до гіпокапнії.
1.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
7. Вторинний гіперальдостеронізм (див. гла
ву 37). Посилена секреція альдостерону
клітинами кори надниркових залоз є при
чиною електролітних порушень в організ
мі - гіпернатріємії, гіпергідрії та гіпокаліємії — і пов’язаних з ними клінічних проявів
(див. глави 27 і 29).
8. Поліцитемічна гіперволемія. Кисневе голо
дування, що супроводжує серцеву недо
статність, веде до посиленого утворення
в клітинах нирок еритропоетинів, які че
рез стимулювання кровотворення в черво
ному кістковому мозку збільшують кіль
кість еритроцитів у периферичній крові.
Еритроцитоз, що розвивається, збільшує
в’язкість крові й через це утруднює мікроциркуляцію (див. главу ЗО). Як наслідок,
у тканинах виникають трофічні розлади,
посилюється кисневе голодування.
АРИТМІЇ
Аритміями серця називають порушення частоти,
ритму, узгодженості й послідовності його ско
рочень.
Узгоджена й ритмічна робота серця з певною
частотою скорочень забезпечується провіднико
вою системою, яку утворюють атипові м’язові
волокна. Ця система складається з таких струк
тур (рис. 31.14):
1) синусно-передсердний або синоатріальний
вузол розташований у стінці правого пе
редсердя поблизу місця впадіння верхньої
порожнистої вени;
2)
передсердно-шлуночковий, або атріовентрикулярний, вузол. Міститься в стінці
правого передсердя, якраз вище фіброзної
тканини, що розділяє праве передсердя
і правий шлуночок;
3) міжвузлові пучки — тонкі волокна, що
з’єднують синоатріальний та атріовентрикулярний вузли. Таких є три: передній, се
редній і задній;
4) пучок Гіса - товстий пучок, що йде від
атріовентрикулярного вузла через фіброз
ну тканину, яка розділяє праве передсердя
і правий шлуночок, до міжшлуночкової
перегородки. Там він ділиться на дві ніж
ки - праву та ліву;
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
115
2) пов’язані з порушеннями збудливості;
3) спричинені розладами провідності;
4) викликані поєднаними порушеннями збуд
ливості й провідності.
31.2.1.
Рис. 31.14. Будова провідникової системи серця
5)
волокна Пуркіньє - тонкі волокна, на
які розгалужуються гілки ніжок пучка
Гіса. Вони контактують безпосередньо
з м’язовими клітинами робочого міокарда
і через них збудження досягає кардіоміоцитів.
Для атипових м’язових волокон провіднико
вої системи властиві три функції: (1) автома
тизм, (2) збудливість і (3) провідність. Ці волок
на, на відміну від клітин робочого міокарда, не
здатні скорочуватися, а тому й отримали назву
атипових.
Залежно від того, порушення яких функцій
провідникової системи лежать в основі аритмій,
порушення ритму серця поділяють на 4 групи.
Так, розрізняють аритмії:
1) обумовлені змінами автоматизму;
ПОРУШЕННЯ АВТОМАТИЗМУ
Автоматизм, тобто здатність клітин спонтан
но генерувати потенціали дії, складає основу
функціонування провідникової системи серця.
Така властивість притаманна всім волокнам
цієї системи, однак за умов норми спонтан
на генерація імпульсів здійснюється тільки
в клітинах синоатріального вузла, у зв’язку
з чим він отримав назву водія ритму, або пейсмекера.
Електрофізіологічні властивості клітин цього
вузла характеризуються низькими значеннями
(від -55 до -60 мВ) потенціалу спокою, останній
отримав тут назву максимального діастолічного
потенціалу (МДП). Завдяки існуванню великої
кількості мембранних неселективних каналів
відбувається постійне входження в клітини іонів
Na+, чим і пояснюється низький рівень потенці
алу спокою у структурах синоатріального вузла
і розвиток його спонтанної електричної актив
ності.
Потенціали дії, що їх генерують клітини про
відникової системи, складаються з трьох фаз
(рис. 31.15):
• І фаза — повільна діастолічна деполяриза
ція. Вона зумовлюється входженням іонів
Na+ в клітини, внаслідок чого мембранний
потенціал поступово зменшується до кри
тичного, порогового рівня, який становить
-40 мВ;
• II фаза & швидка деполяризація мембрани.
При досягненні критичного рівня депо
ляризації відкриваються кальцієві канали,
через які іони Са2+, а разом з ними і Na+
входять в клітини, зумовлюючи швидке па
діння потенціалу;
• III фаза — реполяризація мембрани. Вона
настає в результаті закриття кальцієвих
і відкриття калієвих каналів. Через остан
ні іони К+ виходять із клітин назовні,
відновлюючи
початковий
мембранний
потенціал.
116
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Спонтанне генерування потенціалів дії в клітинах синоатріального вузла. Римськими цифрами
позначено фази потенціалу дії (див. у тексті), МДП - максимальний діастолічний потенціал, ПП - пороговий потенціал
Таких циклів у клітинах синоатріального вуз
ла відбувається 70/хв, що й зумовлює за відсут
ності будь-яких регуляторних впливів таку саму
частоту серцевих скорочень.
Усі аритмії, зумовлені розладами автоматизму,
поділяють на дві групи.
І. Номотопні аритмії. За цих порушень ритму
серця генерування імпульсів до скорочення від
бувається так само, як і в нормі - у синоатріальному вузлі. До номотопних аритмій відносять:
(а) синусну тахікардію, (б) синусну брадикар
дію та (в) синусну (дихальну) аритмію.
Синусна тахікардія характеризується збіль
шенням частоти серцевих скорочень від 90 до
150 ударів/хв унаслідок посиленої імпульсації
з синоатріального вузла (рис. 31.16А).
Частота генерації імпульсів клітинами водія
ритму залежить від трьох чинників: (а) макси
мального діастолічного потенціалу; (б) рівня
критичного потенціалу, після досягнення якого
виникає потенціал дії; (в) швидкості діастолічної деполяризації (рис. 31.15).
Звідси випливає, що збільшення частоти сер
цевих скорочень — синусна тахікардія — може
мати у своїй основі один з таких чинників:
1) зменшення рівня максимального діасто
лічного потенціалу, тобто наближення
його до порогового;
2) зміну порогового потенціалу деполяриза
ції у бік наближення його до потенціалу
спокою;
3) збільшення швидкості повільної діастолічної деполяризації, унаслідок чого перша
фаза потенціалу дії вкорочується.
Описані електрофізіологічні зміни розвива
ються:
а) під впливом підвищеної температури тіла.
При збільшенні температури тіла на 1 °С
частота серцевих скорочень зростає на 18
ударів/хв. Це пояснюють посиленням об
мінних процесів і руху іонів у клітинах водія
ритму, завдяки чому змінюються наведені
вище електрофізіологічні характеристики;
б) при розтягненні ділянки синоатріального
вузла. Переповнення правого передсердя
кров’ю веде до механічного розтягнення
його стінки і структур водія ритму. При
цьому зростає проникність клітинних
мембран до іонів Na+ і, як наслідок, при
скорюється фаза повільної діастолічної
деполяризації;
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
в) при збудженні симпатичних нервів серця
та дії на нього катехоламінів. Цей механізм
лежить в основі рефлекторної тахікардії,
що розвивається при фізичних навантажен
нях, крововтраті, падінні кров’яного тиску,
недостатності серця та ін.
Синусна брадикардія. Для цього виду аритмій
характерним є правильний ритм зі зменшен
ням частоти серцевих скорочень нижче рівня
60 ударів/хв (рис. 31.16Б).
Електрофізіологічною основою синусної
брадикардії можуть бути:
1) збільшення рівня максимального діастолічного потенціалу - гіперполяризація
мембрани;
2) віддалення критичного потенціалу від рів
ня максимального діастолічного;
3) зменшення швидкості повільної діастолічної деполяризації, унаслідок чого перша
фаза потенціалу дії подовжується.
б. Номотопні порушення автоматизму серця.
А - синусна тахікардія, Б - синусна брадикардія, В - синусна аритмія
117
Синусна брадикардія не обов’язково є озна
кою патологічного процесу. Часто вона має при
стосувальне значення, як наприклад, у спортс
менів. Оскільки треноване серце навіть у стані
спокою виштовхує значно більший ударний
об’єм крові, ніж звичайне серце, то спрацьову
ють кардіоваскулярні рефлекси, еферентною
ланкою яких є парасимпатичні волокна блука
ючого нерва. Закінчення останніх вивільнюють
ацетилхолін, який зменшує проникність мемб
ран водія ритму до натрієвих іонів і в такий
спосіб збільшує тривалість повільної діастолічної деполяризації. Завдяки брадикардії, що роз
вивається, організм підтримує потрібний йому
мінімальний хвилинний об’єм крові, запобігаю
чи невиправданому його збільшенню.
Прикладом патологічної синусної брадикар
дії може бути синдром каротидного синусу, що
виникає внаслідок значного збільшення чутли
вості (збудливості) барорецепторів сонних арте
рій і синокаротидної зони. У пацієнтів з таким
118
синдромом навіть незначний зовнішній тиск на
ділянку шиї може спричинитися до вираженого
барорецепторного рефлексу, що супроводжуєть
ся збудженням блукаючого нерва і виділення
його закінченнями ацетилхоліну. Іноді цей реф
лекс буває настільки сильним, що серце може
зупинятися на 5—10 сек.
Синусна аритмія характеризується непра
вильним синусним ритмом, при якому частота
серцевих скорочень змінюється в межах від 60
до 90 ударів/хв (рис. 31.16В). Причиною її ви
никнення є періодичні зміни тонусу блукаючо
го нерва під час глибокого дихання, а тому таку
аритмію ще позначають як дихальну. Під час
вдиху число серцевих скорочень зростає, під час
видиху - серцебиття уповільнюється. Дихальна
аритмія в нормі буває у дітей, але зрідка може
спостерігатися і в дорослих.
II. Гетеротопні аритмії. До цієї категорії від
носять аритмії, при яких генерування імпульсів
до скорочення відбувається не в синоатріальному вузлі, а в інших структурах провідникової
системи, що є водіями ритму II (атріовентрикулярний вузол) і III порядку (клітини провіднико
вих шляхів шлуночків).
Здатність до спонтанної генерації потенці
алів дії (автоматизму) притаманна всім кліти
нам провідникової системи серця. Проте лише
синоатріальний вузол в умовах норми є водієм
ритму. Це пояснюється законом "градієнта ав
томатизму ”, відповідно до якого частота спон
танних розрядів клітин провідникової системи
є тим менша, чим далі від синоатріального вуз
ла вони розташовані. Так, клітини передсердношлуночкового вузла за відсутності будь-яких
впливів на нього генерують 70 імп/хв, атріовентрикулярного вузла - 40-60 імп/хв, волокна гі
лок пучка Гіса - 20-40 імп/хв.
Зменшення активності або припинення діяль
ності водія ритму І порядку зумовлює розвиток
аритмій, що отримали назву синдрому слабкос
ті синоатріального вузла. Цей синдром може
виявляти себе такими видами патологічних рит
мів серця:
а) передсердний повільний ритм - водій ритму
міститься в структурах лівого передсердя,
частота серцевих скорочень менше 70/хв.;
б) атріовентрикулярний ритм — джерелом
імпульсів є водії ритму II порядку (верхня,
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
середня або нижня частина атріовентрикулярного вузла), частота серцевих скорочень
залежно від місця генерації імпульсів змен
шується від 70 до 40/хв;
в) ідіовентрикулярний гмлуночковий ритм —
генерація імпульсів відбувається у водіях
ритму III порядку (пучок Гіса або його ніж
ки), частота скорочень серця менше 40/хв.
ПОРУШЕННЯ ЗБУДЛИВОСТІ
В основі аритмій, пов’язаних з порушеннями
функції збудливості, лежить поява розташова
них поза синоатріальним вузлом ектопічних
осередків збудження, що генерують позачергові
імпульси до скорочення. При цьому клітини во
дія ритму функціонують нормально, на відміну
від синдрому слабкості синоатріального вузла,
що ми його вже розглядали.
Підвищення здатності до генерації імпульсів
в клітинах провідникової системи, розташова
них нижче синоатріального вузла, може бути на
стільки значним, що нормальний водій ритму не
в змозі підпорядкувати собі їхній автоматизм. Це
веде до того, що виникають або передчасні ско
рочення (екстрасистоли), або функцію водія рит
му повністю перебирає на себе ектопічний осе
редок збудження (пароксизмальна тахікардія).
Серед основних причин підвищення збудли
вості окремих структур провідникової системи:
а) локальна ішемія міокарда;
б) невеликі кальцифікати в різних частинах
серця, які тиснуть на прилеглі кардіоміоцити, викликаючи їхнє збудження;
в) дія на клітини провідникової системи ток
сичних доз деяких ліків, нікотину, кофеїну;
г) механічне подразнення стінок серця при
його катетеризації (особливо при входжен
ні катетера в правий шлуночок).
Одним з механізмів формування ектопічних
осередків збудження є виникнення різниці по
тенціалів між розташованими поруч міоцитами
внаслідок, наприклад, неодночасного закінчен
ня реполяризацїї в них, що може викликати збу
дження у волокнах, які вже вийшли з фази рефрактерності. Це явище спостерігається, зокре
ма, при локальній ішемії міокарда і при отруєн
ні серцевими глікозидами.
119
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
Екстрасистолія. Вид аритмій, для якого ха
рактерним є виникнення позачергових, перед
часних скорочень серця або тільки шлуночків,
отримав назву екстрасистолії, а позачергові
скорочення - екстрасистол.
Залежно від локалізації осередку, з якого ви
ходить позачерговий імпульс, розрізняють кіль
ка видів екстрасистол: (а) передсердну, (б) атріовентрикулярну і (в) шлуночкову (рис. 31.17).
Оскільки хвиля збудження, що виникла в не
звичному місці, поширюється в зміненому на
прямку, це впливає на структуру електричного
поля серця й знаходить відображення на елек
трокардіограмі. Кожний вид екстрасистоли має
свою електрокардіографічну картину, що дає
можливість визначити місце ектопічного осе
редку збудження.
Передсердна екстрасистола виникає за наяв
ності осередку ектопічного збудження в різних
ділянках передсердь (рис. 31.17А). Характери
зується: (а) зміною форми зубця Р (знижений,
двофазний, негативний - залежно від локаліза
ції ектопічного осередку) при (б) збереженому
комплексі QRS і (в) деякому подовженні діастолічного інтервалу після екстрасистоли. Останнє
обумовлено тим, що, поширюючись ретроград
ним шляхом, потенціал дії передчасно збуджує
клітини синоатріального вузла, і нормальний
синусний імпульс, що виникає при цьому, збі
гається в часі з передчасним збудженням шлу
ночків. Наступний синусний імпульс генеруєть
ся через нормальний інтервал, але виявляється
трохи віддаленим від моменту закінчення попе
реднього збудження шлуночків - неповна ком
пенсаторна пауза.
Атріовентрикулярна екстрасистола має сво
їм джерелом структури передсердно-шлуночкового вузла (рис. 31.17Б). Хвиля збудження,
що виходить з верхньої і середньої частин вуз
ла, поширюється у двох напрямках: у шлуноч
ках - у нормальному, у передсердях - у ретро
градному. При цьому негативний зубець Р може
накладатися на комплекс QRS, який за формою
подібний до передсердних екстрасистол. Діа-
Рис. 31.17. Екстрасистолія. А - передсердна, Б - атріовентрикулярна, В - шлуночкова
120
столічний інтервал після екстрасистоли трохи
подовжений - неповна компенсаторна пауза.
Екстрасистола може супроводжуватися одно
часним скороченням передсердь і шлуночків.
Якщо джерелом атріовентрикулярної екстра
систоли є нижня частина вузла, то збудження
не переходить на передсердя (через однонаправленість проведення імпульсів через атріовентрикулярний вузол), а поширюється тільки
в шлуночках. З цієї причини виникає повна ком
пенсаторна пауза, така ж, як і при шлуночковій екстрасистолі (див. нижче). На відміну від
останньої, тривалість комплексу QRS істотно не
змінюється.
Шлуночкова екстрасистола виникає в ре
зультаті появи ектопічних осередків збудження
в шлуночках. Її ознаками є такі зміни електро
кардіограми:
а) відсутність зубця Р, що пояснюється не
можливістю поширення хвилі збудження
на передсердя;
б) комплекс QRS екстрасистоли деформова
ний і має більшу, ніж у нормі, тривалість.
Для деформованого QRS характерне збіль
шення вольтажу, що пояснюється про
веденням передчасного збудження через
лівий і правий шлуночок в одному і тому
ж напрямку (у нормі направленість поши
рення деполяризації в різних шлуночках не
збігається, що зумовлює ефект часткової
нейтралізації електричних імпульсів). По
довження комплексу QRS екстрасистоли
пов’язують з тим, що передчасне збуджен
ня проходить переважно не волокнами
Пуркіньє, а через м’язові клітини шлуноч
ків, швидкість проведення потенціалів дії
в яких значно менша;
в) зубець Т має направленість протилеж
ну комплексу QRS. Причина цього також
у повільному проведенні імпульсів через
м’язові волокна міокарда. Через цю обста
вину реполяризація, так само як і деполя
ризація, у різних відділах шлуночків від
бувається асинхронно: першими зазнають
реполяризації ті клітини, які першими деполяризуються;
г) після екстрасистоли настає повна компен
саторна пауза. Вона виникає внаслідок
того, що збудження, яке охопило шлуночки,
не передається через атріовентрикулярний
вузол назад на передсердя, водночас чер
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
говий нормальний імпульс збудження, що
йде від синоатріального вузла, не поширю
ється на шлуночки, бо вони перебувають
у фазі рефрактерності. Наступне скоро
чення шлуночків виникає тільки після при
ходу до них чергового нормального імпуль
су. Тому тривалість компенсаторної паузи,
разом з інтервалом, що передує екстрасис
толі, дорівнює тривалості двох нормальних
діастолічних пауз. Однак якщо скорочення
серця настільки рідкі, що до моменту при
ходу чергового нормального імпульсу шлу
ночки встигають вийти зі стану рефрактер
ності, то компенсаторної паузи не буває.
Позачергове скорочення потрапляє в інтер
вал між двома нормальними, і в цьому ви
падку воно має назву вставної екстрасис
толи.
Шлуночкові екстрасистоли можуть правиль
но чергуватися з нормальними скорочення
ми - у такому разі ведуть мову про алоритмію.
Окремими її варіантами є бігемінія, тригемінія
і квадригемінія.
При бігемінії після кожного нормального
скорочення йде екстрасистола (рис. 31.18А), це
найчастіше буває при передозуванні препаратів
дигіталісу. Якщо ж екстрасистола виникає після
кожних двох нормальних скорочень, то таку ало
ритмію називають тригемінією (рис. 31.18 Б).
Ще один варіант тригемінії: після кожного нор
мального скорочення підряд ідуть дві екстра
систоли. Відповідно, квадригемінія характери
зується тим, що після кожних трьох нормальних
скорочень настає позачергове, або після кожного
нормального - три екстрасистоли.
Пароксизмальна тахікардія. Значні порушен
ня збудливості клітин міокарда можуть спричи
нятися до виникнення групи екстрасистол, що
швидко повторюються і повністю пригнічують
фізіологічний ритм. Таку аритмію називають
пароксизмальною тахікардією.
Місцем виникнення осередків патологічного
збудження можуть бути передсердя, атріовентри
кулярний вузол і шлуночки. Звідси розрізняють
пароксизмальну тахікардію: (а) передсердну, (б)
атріовентрикулярну і (в) шлуночкову. Перші дві
об’єднують у групу суправентрикулярних.
Суправентрикулярна
пароксизмальна
тахі
кардія (СВПТ). При цій аритмії нормальний
Рис. 31.18. Варіанти алоритмії: бігемінія (А), тригемінія (Б)
ритм серця раптово переривається нападом ско
рочень частотою від 140 до 250 ударів/хв. Три
валість нападу може бути різною - від кількох
секунд до кількох хвилин, після чого він так
само раптово припиняється й установлюється
нормальний ритм.
Найчастіше буває передсердна форма СВПТ
(рис. 31.19, А). Для неї є характерними правиль
ний ритм, зміна форми зубця Р (двофазний або
інвертований), тривалість і форма комплексів
QRS без змін, зубець Т часто спотворений че
рез накладання на нього зубця Р. Оскільки три
валість потенціалів дії та рефрактерних періодів
збільшується за ходом провідникової системи, то
дистально розташовані її ділянки не завжди здат
ні відтворити частоту імпульсації, що надходить
із проксимальних відділів. Тому більша частина
імпульсів при передсердній СВПТ не може про
водитися атріовентрикулярним вузлом.
Для атріовентрикулярної форми СВПТ, на
відміну від передсердної, характерна відсут
ність зубців Р, усі інші електрокардіографічні
ознаки дуже схожі. У розвитку даного варіанта
тахікардії великого значення надають форму
ванню стійкого осередку збудження механізмом
“повторного входження імпульсів”, про який
мова піде далі.
Шлуночкова
пароксизмальна
тахікардія
(ШПТ) характеризується появою серії екстрасистол (рис. 31.19, Б), джерелом яких можуть
бути різні частини одного зі шлуночків. При
чиною такої аритмії є, як правило, значне іше
мічне ушкодження серцевого м’яза. При ШПТ
імпульси до скорочення можуть виходити
з одного і того ж осередку збудження (мономорфна ШПТ) або з кількох різних (поліморф
на ШПТ).
Найчастішою причиною мономорфної ШПТ
є ушкодження й загибель кардіоміоцитів (ін
фаркт міокарда), що веде до утворення рубців.
Останні не можуть проводити через себе елек
тричні потенціали, що створює умови для фор
мування патологічних кіл циркуляції збудження
та реалізації механізму “повторного входження
імпульсів” (див. далі).
Поліморфна ШПТ має у своїй основі частіше
не морфологічні, а функціональні зміни в міо
карді. Вони характеризуються уповільненням
проведення певними ділянками шлуночків збу
дження через подовження періодів відносної
та абсолютної рефрактерності (збільшується
тривалість комплексів QRS). Причинами цього
можуть бути вроджений синдром довгого QT,
токсична дія деяких лікарських препаратів, змі
ни концентрації електролітів (калію, кальцію,
магнію), ішемія міокарда.
На електрокардіограмі ШПТ характеризується
регулярним ритмом, збільшенням частоти скоро
чень шлуночків від 180 до 250/хв, зубець Р від
сутній, комплекс QRS деформований, його три-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
122
II
III
Рис. 31.19, Пароксизмальна тахікардія: А - суправентрикулярна (передсердний варіант), Б - шлуночкова
валість звичайно є більшою за 0,12 с, зубець Т
має направленість, протилежну комплексу QRS.
ШПТ, на відміну від СВПТ, є смертельно не
безпечним порушенням ритму серця, оскільки
робота передсердь і шлуночків втрачає свою
узгодженість. Значно скорочується час напов
нення шлуночків кров’ю, а отже, і серцевий
виштовх, ШПТ може з часом переходити у фі
бриляцію шлуночків і завершуватися раптовою
зупинкою серця і смертю.
31.2.3.
ПОРУШЕННЯ ПРОВІДНОСТІ
У нормі імпульси, що виникають у синоатріальному вузлі, проводяться до волокон робочого
міокарда через структури провідникової систе
ми серця з різною швидкістю (V). Можна ви
ділити чотири послідовні етапи фізіологічного
поширення збудження в серці:
1) проведення від синоатріального вузла. Від
клітин водія ритму імпульси поширюють
ся у трьох напрямках: (1) до кардіоміоцитів правого передсердя (V = 0,3 м/с); (2) до
м’язових волокон лівого передсердя через
міжпередсердні пучки (V = 1 м/с); (3) до
атріовентрикулярного вузла через три між
вузлові пучки (передній, середній і задній)
(V = 1 м/с);
проведення через атріовентрикулярний
вузол (АВВ). У нормі збудження від перед
сердь до шлуночків не може пройти інак
ше, як через цей вузол, бо передсердя від
ділені від шлуночків фіброзною тканиною,
яка не проводить імпульсів. Проведення
потенціалів дії через АВВ має дві важливі
особливості:
• дуже низька швидкість проведення, яка обу
мовлює т. зв. атріовентрикулярну затрим
ку (0,13 с). Фізіологічне значення останньої
полягає в тому, що передсердям надається
час для переміщення крові, що в них міс
титься, у шлуночки до скорочення останніх.
Атріовентрикулярна затримка складається
із затримки на вході ABB (V = 0,02-0,05
м/с), на рівні вузла (V = 0,05 м/с) і на виході
з нього (V = 0,02-0,05 м/с). Причини такої
затримки вбачають у дуже низькому потен
ціалі клітин АВВ, що є значно меншим, ніж
в інших клітинах міокарда; а також у тому,
що волокна АВВ є дуже тонкими і мають
малу кількість нексусів, унаслідок чого
збільшується опір проведенню імпульсів;
• однонаправленість проведення збудження.
Імпульси не можуть проходити у зворот
ному напрямку - від шлуночків до перед
сердь;
3) проведення через пучок Гіса та волокна
Пуркіньє. Оскільки ці структури склада-
2)
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
ються з товстих волокон, то швидкість
проведення імпульсів по них є дуже висо
кою і становить від 1,5 до 4 м/с. Це дозво
ляє майже миттєво й одночасно проводити
збудження до всіх, навіть до найбільш від
далених ділянок міокарда;
4) проведення через волокна робочого міо
карда. Термінальні волокна Пуркіньє за
кінчуються на поверхні скорочувальних
волокон ендокардіальної частини серцево
го м’яза. Далі потенціали дії поширюють
ся вже через волокна робочого міокарда
від ендокардіальної до епікардіальної по
верхні міокарда зі швидкістю 0,3-0,5 м/с.
Порушення проведення збудження через на
ведені вище структури є причиною аритмій, які
можна розділити на дві групи: (1) блокади і (2)
прискорене проведення імпульсів.
Блокади серця
Це аритмії, обумовлені уповільненням (неповна
блокада) або повним припиненням (повна блока
да) проведення імпульсів по провідниковій сис
темі. їхньою причиною є, як правило, оборотне
й необоротне ушкодження клітин, що входять до
складу різних структур провідникових шляхів.
При оборотному ушкодженні настає стійка
деполяризація клітин, унаслідок чого подовжу
ються періоди абсолютної та відносної рефрактерності, що веде до уповільнення або повного
припинення проведення потенціалів дії. Заги
бель клітин (необоротне ушкодження) обумов
лює формування рубця, через який проходжен
ня імпульсів стає неможливим.
Порушення провідності можуть виникати на
різних рівнях провідникової системи: між синоатріальним вузлом і передсердями, між перед
сердями і шлуночками, а також в одній із ніжок
пучка Гіса. Тому виділяють такі види блокад:
(а) синоатріальну; (б) атріовентрикулярну; (в)
внутрішньошлуночкову - блокаду ніжок пучка
Гіса.
Синоатріальна блокада. У досить рідких ви
падках імпульс, що виник у клітинах синоатріального вузла, не може передаватися на м’язові
волокна передсердь, унаслідок чого останні не
скорочуються. Найхарактернішою електрокар
діографічною ознакою повної такої блокади є
123
випадіння зубця Р. На час блокади функцію
водія ритму переймає на себе атріовентрикулярний вузол, частота імпульсів від якого є
меншою, що відображається розвитком бради
кардії.
Атріовентрикулярна
блокада
(АВ-блокада).
Проведення імпульсів через АВВ порушується
за таких умов:
а) при ішемії АВВ в цілому чи окремих пуч
ків його волокон;
б) при запаленні міокарда (міокардитах), яке
поширюється на структури АВВ;
в) при здавленні пучків АВВ рубцевою ткани
ною чи кальцифікатами;
г) при надмірно сильній стимуляцій серця
блукаючим нервом, наприклад, через барорецепторні рефлекси при синдромі каротидного синуса (див. вище).
Залежно від вираженості порушень провід
ності та особливостей електрокардіографічної
картини розрізняють три ступені АВ-блокади
(рис. 31.20).
I ступінь АВ-блокади (рис. 31.20А). Для ньо
го характерним є збільшення часу проведення
імпульсів від передсердь до шлуночків, що су
проводжується сталим подовженням інтервалу
P-Q. У нормі цей показник дорівнює 0,16 с при
нормальній частоті серцевих скорочень. Збіль
шення його понад 0,2 с свідчить про затримку
проведення збудження через АВВ, проте всі ім
пульси, які генеруються в синоатріальному вуз
лі, проводяться до шлуночків.
II ступінь АВ-блокади. Якщо внаслідок по
рушень провідності не всі імпульси проходять
у шлуночки, то мову ведуть про II ступінь АВблокади. Розрізняють два типи електрокардіо
графічних змін, характерних для нього.
Тип 1 (МобітцІ) (рис. 31.20Б) характери
зується прогресуючим збільшенням ін
тервалу P-Q доти, доки одне із збуджень,
найчастіше восьме-десяте, не прово
диться у шлуночки і випадає. Після тако
го випадіння інтервал P-Q відновлюєть
ся, поступово подовжуючись із кожним
подальшим скороченням серця (періоди
Венкебаха, або Мобітца). Вважають, що
цей феномен пов’язаний з прогресую
чим утрудненням проведення імпульсів
через АВВ.
724
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Г
31.20. Атріовентрикулярна блокада: А - перший ступінь, Б - другий ступінь (Мобітц І), В - другий
ступінь (Мобітц її), Г - третій ступінь (повна блокада)
Tun II (Мобітц II) (рис. 31.20В). Випадін
ня комплексів QRS і скорочень шлуночків
відбувається без прогресуючого подов
ження інтервалу P-Q. Випадати можуть
кожне друге-третє скорочення або, навпа
ки, проводяться тільки кожне друге, третє
або четверте збудження передсердь. Цей
тип є прогностично несприятливим, він
часто передує повній АВ-блокаді.
III ступінь АВ-блокади, або повна попереч
на блокада. Розвивається тоді, коли жоден ім
ки. У такому разі активуються центри автома
тизму, розташовані нижче АВВ (водії ритму III
порядку). Частота генерованої ними імпульсації є низькою (від 20 до 40/хв), що й зумовлює
таку ж низьку частоту скорочень шлуночків
(ідіовентрикулярний ритм). Отже, при повній
поперечній блокаді передсердя та шлуночки
скорочуються незалежно одне від одного, ко
жен відділ у своєму ритмі: передсердя з часто
тою близько 70, шлуночки - 20-40 скорочень/
хв. На електрокардіограмі реєструються два
пульс із передсердь не може пройти у шлуноч
незалежні ритми: передсердний (зубці Р) і шлу-
І
125
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
ночковий (комплекси QRS), ніяк не пов’язані
між собою.
Особливе значення має момент переходу не
повної АВ-блокади в повну, коли до шлуночків
припиняється надходження імпульсів від пе
редсердь. Повільна діастолічна деполяризація
в потенційних водіях ритму виникає не одразу,
а тільки через певний час після такого припи
нення (5-30 с). Цей період, що має назву преавтоматичної паузи, характеризується асистолі
єю шлуночків. У результаті того, що тимчасово
припиняється надходження крові до головного
мозку, людина втрачає свідомість, розвивають
ся судоми (синдром Морганьї — Едемса — Стокса). Можлива смерть, але найчастіше при по
новленні скорочень шлуночків зазначені явища
проходять. Синдром може повторюватися бага
торазово.
Внутрішньошлуночкова блокада. Майже
всі
ті фактори, що зумовлюють АВ-блокаду, особ
ливо загибель ділянок міокарда внаслідок його
ішемічного ушкодження, можуть викликати по
рушення провідності в пучку Гіса: його ніжках
і гілках. У такому разі мову ведуть про внутрішньошлуночкову блокаду, яка може бути непов
ною і повного.
Розрізняють блокаду правої та лівої ніжок
пучка Гіса. При уповільненні або повному при
пиненні проведення імпульсів через праву ніж
ку кардіоміоцити правого шлуночка, на відміну
від лівого, не отримують своєчасно стимулів до
скорочення. їхнє збудження настає пізніше від
звичайного, і причиною цього є імпульси, що
прийшли сюди від уже збуджених м’язових во
локон лівого шлуночка.
При блокаді лівої ніжки пучка Гіса чи
окремих її гілок усе відбувається навпаки.
Кардіоміоцити лівого шлуночка, не отримавши
своєчасно імпульсів по волокнах Пуркіньє, збу
джуються дещо пізніше, отримавши потенціа
ли дії від збуджених м’язових клітин правого
шлуночка.
Описані зміни пояснюють основну елек
трокардіографічну
характеристику
блокади
ніжок пучка Гіса - подовження (понад 0,11 с)
комплексів QRS та їхня деформація. Додаткові
ознаки, що виявляються при вивченні грудних
відведень ЕКГ, дають можливість встановити
локалізацію внутрішньошлуночкової блокади
(рис. 31.21).
Прискорене проведення імпульсів
(синдром Вольфа - Паркінсона Уайта)
Якщо збудження проводиться від передсердь
до шлуночків швидше, ніж у нормі, то розви
вається синдром Вольфа —Ларкінсона - Уайта
(ВПВ-синдром).
Причиною ВПВ-синдрому вважають існу
вання додаткових провідникових шляхів, що
сполучають передсердя і шлуночки. До таких
відносять (рис. 31.22):
а) пучок Паладіно — Кента. Він є видозміне
ною міокардіальною тканиною, локалізо
ваною в зоні атріовентрикулярного кільця.
Може існувати два пучки (лівий і правий),
що містяться відповідно у кільцях двостул
кового і тристулкового клапанів. Зазначені
пучки проводять імпульси від передсердь
до шлуночків, минаючи атріовентрикулярний вузол;
б) пучок Махайма. З’єднує верхню частину
пучка Гіса зі шлуночками;
в) пучок Джеймса. Зв’язує передсердя з ниж
ньою частиною атріовентрикулярного вуз
ла або з пучком Гіса.
По додаткових провідникових шляхах імпульси
проводяться швидше і досягають шлуночків ра
ніше імпульсів, які йдуть звичайним чином че
рез атріовентрикулярний вузол. Це призводить
до передчасної активації частини шлуночків,
інша їхня частина збуджується пізніше імпуль
сами, що проходять через передсердно-шлуночковий вузол.
На ЕКГ ВПВ-синдром характеризується змен
шенням інтервалу P-Q (менше 0,12 с), появою
на висхідній частині зубця R хвилі Д (передчас
не збудження частини шлуночків через додат
ковий провідниковий шлях), за рахунок А-хвилі
розширюється комплекс QRS (стає більшим за
0,1 с) (рис. 31.23).
Для осіб з ВПВ-синдромом характерні на
пади передсердної пароксизмальної тахікардії,
спровоковані передсердними екстрасистолами.
їхній механізм вбачають у виникненні в перед
сердях стійких осередків електричної актив
ності внаслідок того, що імпульси із шлуночків
повертаються назад у передсердя через пучки
Паладіно - Кента, формуючи в такий спосіб
замкнені кола циркуляції потенціалів дії. Річ
126
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
FC
R
Блокада
правої ніжки
пучка Гіса
Блокада
лівої ніжки
пучка Гіса
Рис. 31.21. Блокада ніжок пучка Гіса: грудні відведення ЕКГ — V1 і V6
у тім, що волокна додаткових провідникових
шляхів мають коротший, якщо порівнювати
з волокнами пучка Гіса, період рефрактерності.
Саме тому імпульси, які пройшли атріовентрикулярний вузол, ідуть ретроградно через пуч
ки Паладіно — Кента в передсердя, бо не ма
ють можливості поширюватися до шлуночків,
оскільки потрапляють на період рефрактернос
ті волокон пучка Гіса.
31.2.4.
ПОРУШЕННЯ ЗБУДЛИВОСТІ
ТА ПРОВІДНОСТІ
В основі цієї групи аритмій лежить формування
замкнутих кіл, якими циркулює збудження, на
даючи серцю стійкого, автономного, не залеж
ного від імпульсів водія ритму, характеру. Такий
механізм розвитку аритмій отримав назву “по
X JL XJ
вторного входження імпульсів” (англ., механізм
re-entry).
Для розуміння суті цього механізму розгляне
мо поширення імпульсів у невеликій ділянці мі
окарда, що має форму кільця (рис. 31.24). Якщо
нанести імпульс у якійсь точці такого кільця
так, щоб він поширювався тільки в один бік, то,
пройшовши по колу, збудження в точці нанесен
ня імпульсу затухне, бо потрапить до неї тоді,
коли ця ділянка міокарда перебуватиме у фазі
абсолютної рефрактерності. Так відбувається
в умовах норми.
Існує три причини, з яких повернення ім
пульсів у точку первинного нанесення збуджен
ня не затухатиме, а викликатиме нові імпульси,
які циркулюватимуть по колу без затухання як
завгодно довго, підтримуючи самих себе.
1. Збільшення шляху, по якому циркулюють
імпульси (рис. 31.24). У цьому випадку
імпульси повертатимуться в точку на-
727
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
несення первинного збудження вже тоді,
коли кардіоміоцити даної ділянки вийдуть
з періоду рефрактерності й будуть у змо
зі проводити імпульси далі. Цей механізм
формування аритмій є характерним для
всіх уражень серця, що супроводжуються
розширенням його камер.
2. Зменшення швидкості проведення імпуль
сів, навіть при збереженні нормальної
довжини поширення збудження по колу.
Оскільки час, протягом якого імпульс буде
повертатися до вихідної точки, збільшува
тиметься, то потенціали дії, що надійшли
сюди, вже не застануть м’язові волокна
в стані рефрактерності, бо вона встигне
завершитися. Уповільнення проведення
імпульсів може стати передумовою даного
типу аритмій (а) при блокаді волокон Пуркіньє, (б) ішемії кардіоміоцитів, (в) гіперкаліємії.
3. Укорочення періоду рефрактерності м ’язових волокон. Якщо час, протягом якого три
ває рефрактерність, зменшується, то навіть
за нормальної довжини кола та швидкості
проведення імпульсів збудження може ді
йти до вихідної точки в момент, коли кар
діоміоцити цієї ділянки відновлять свою
збудливість і зможуть далі проводити по
тенціали дії. Така ситуація виникає при
повторній електричній стимуляції серця,
при дії деяких лікарських засобів, зокрема
адреналіну.
не. 31.22 Додаткові шляхи провідникової
системи серця
Формування патологічних кіл циркуляції збу
дження може відбуватися як у передсердях, так
і в шлуночках. Порушення ритму серця, що ви
никають при цьому, отримали назви тріпотін
ня і миготіння (фібриляції).
Тріпотіння і миготіння передсердь
Якщо
циркуляція
збудження
відбувається
в стінках передсердь, то розвивається миготли
ва аритмія, що може виявляти себе тріпотінням
або миготінням передсердь.
Про тріпотіння передсердь ведуть мову
тоді, коли частота їхніх скорочень досягає 200400/хв. Основний механізм тріпотіння полягає
в утворенні великих патологічних кіл циркуля
ції збудження в передсердях (рис. 31.25). При
цьому порушується їхня нагнітальна функція,
оскільки в той час, коли одна частина стінки
передсердь скорочується, друга, навпаки, роз
слабляється. Унаслідок цього у шлуночки над
ходить об’єм крові, значно менший за той, що
мав би надійти в умовах нормального функціо
нування передсердь.
Водночас унаслідок нездатності шлуночків
відтворювати високий ритм передсердь (геть
усі імпульси здатні проходити у шлуночки при
їхній частоті не більше 180/хв) розвивається
відносна серцева блокада: шлуночки відповіда
ють скороченням на кожне друге (2:1) або третє
( 3 : 1 ) скорочення передсердь, тому що інші хви
лі збудження потрапляють у фазу рефрактер
ності волокон атріовентрикулярного вузла. Ско
рочення шлуночків можуть виникати раніше,
128
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ис. 31.23. Електрокардіографічні характеристики ВПВ-синдрому
Механізм формування патологічних кіл циркуляції збудження в серці
129
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
Передсердя
Шлуночки
Осередок
збудження
А
Б
Механізми формування тріпотіння (А) та миготіння (Б) передсердь і шлуночків
ніж настане достатнє наповнення їх кров’ю - це
викликає значні порушення кровообігу.
На ЕКГ реєструється правильний ритм, зуб
ці Р заміщаються на хвилю F, частота коливань
у якій удвічі, а то і втричі більша за частоту
комплексів QRS; зубець Т деформується через
накладання хвилі F (рис. 31.26, А).
Миготіння (фібриляція) передсердь виникає
при утворенні малих патологічних кіл цирку
ляції імпульсів. Про такий варіант миготливої
аритмії ведуть мову, якщо кількість імпульсів
у передсердях досягає 400-600/хв. При цьому
діяльність серця порушується в цілому через
кілька обставин.
1. М’язові волокна передсердь скорочують
ся асинхронно, через що все передсердя
перебуває в стані неповного скорочення,
його участь у перекачуванні крові (насосна
функція) припиняється. Оскільки майже
1/3 крові в нормі надходить у шлуночки під
час їхньої діастоли в результаті скорочення
передсердь, то миготіння останніх змен
шує на таку ж частку діастолічне наповне
ння, а отже, і ударний об’єм шлуночків.
2. Імпульси, що безладно приходять до атріовентрикулярного вузла окремими м’язо
вими волокнами передсердя, здебільшого
нездатні викликати його збудження, тому
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
130
Рис. 31.26. Тріпотіння (А) та миготіння (Б) передсердь
що застають вузол у стані рефрактерності
або не досягають критичного рівня депо
ляризації. Тому атріовентрикулярний ву
зол збуджується нерегулярно, і скорочення
шлуночків мають випадковий характер. Як
правило, число скорочень шлуночків за
хвилину перевищує нормальне. Нерідко
скорочення шлуночків відбуваються до їх
нього наповнення кров’ю й не супроводжу
ються пульсовою хвилею. Тому частота
пульсу стає меншою за частоту скорочень
серця (дефіцит пульсу).
Миготіння передсердь є одним із найчасті
ших видів аритмій, особливо в осіб похилого
віку. Воно може мати постійний хронічний ха
рактер або виявляти себе окремими епізодами,
що тривають від кількох хвилин до годин.
Найхарактернішою ознакою миготіння пе
редсердь є неправильний ритм серця, що лег
ко можна визначити при пальпації пульсу чи
за допомогою ЕКГ (інтервали R-R не однако
ві, мають різну тривалість). Зубець Р відсутній,
він заміщається низькоамплітудною хвилею f
(рис. 31.26, Б). Кількість скорочень шлуночків
може змінюватися в широкому діапазоні, сягаю
чи 180/хв.
Миготіння і тріпотіння передсердь можуть
переходити одне в одне. Вони розвиваються
при багатьох патологічних процесах і хворобах,
але найчастішими їхніми причинами є: (а) вади
серця, зокрема стеноз лівого атріовентрикулярного отвору; (б) тиреотоксикоз; (в) атероскле
ротичний кардіосклероз. Розвитку миготливої
аритмії сприяють високий артеріальний тиск,
алкогольна інтоксикація, запальні процеси
(міокардити) і дистрофічні зміни (кардіоміопатія) у серцевому м’язі.
Миготлива аритмія, порушуючи повноцінне
діастолічне наповнення шлуночків кров’ю, знач
но погіршує системний кровообіг, може бути
одним із чинників його недостатності. Ще один
негативний наслідок пов’язаний з тим, що при
миготінні створюються сприятливі умови для
утворення на поверхні ендокарда передсердь
тромбів (не відбувається періодичного скоро
чення і розслаблення стінки!). Відрив тромбів
від стінки лівого передсердя може стати при
чиною тромбоемболії артерій головного мозку,
унаслідок чого розвивається кардіоемболічний
інсульт (див. главу 38).
Тріпотіння та миготіння
(фібриляція) шлуночків
Цей вид аритмій є найнебезпечнішим серед усіх
порушень ритму: якщо серце не вивести з тако
го стану протягом 1-3 хв, то майже завжди на
стає смерть.
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
131
її
27. Тріпотіння (А) та фібриляція (Б) шлуночків
Про тріпотіння ведуть мову, коли імпульси
виходять з якогось одного ектопічного осеред
ку збудження, локалізованого у стінці шлуноч
ка. Цей осередок генерує регулярні, повторні
імпульси з частотою 150-300/хв. За таких об
ставин створюються великі патологічні кола
циркуляції збудження, яке за механізмом “re
entry” підтримує саме себе (рис. 31.25). На ЕКГ
фіксуються гладкі хвилі з відносно регулярни
ми й однаковими за амплітудою коливаннями
(рис. 31.27, А).
Фібриляція шлуночків має в своїй основі ім
пульси, що виникають дуже швидко й нерегу
лярно у великій кількості ектопічних осередків
збудження у шлуночках. Це викликає хаотичні,
некоординовані скорочення окремих м’язових
волокон і унеможливлює синхронне скорочення
серцевого м’яза в цілому. На ЕКГ реєструється
низьковольтна хвиляста лінія з різною ампліту
дою і проміжками між окремим коливаннями такий стан позначають як дезорганізація елек
тричної активності серця (рис. 31.27, Б).
При фібриляції шлуночків одні ділянки міо
карда здійснюють процес скорочення, а інші розслаблюються. За таких умов шлуночки пе
ребувають у якомусь проміжному стані - і не
розслабляються, і не скорочуються. Певна річ,
унаслідок цього практично зникає пропульсивна сила серцевих скорочень, насосна функція
серця припиняється, кровообіг зупиняється,
швидко настає втрата свідомості (через 4-5 с)
і зрештою смерть.
Причини фібриляції шлуночків різні: (а) про
ходження електричного струму через серце, (б)
гостра недостатність вінцевого кровообігу, що
викликає ішемію міокарда та його інфаркт, (в)
наркоз хлороформом або циклопропаном та ін.
Фібриляції сприяє зменшення концентрації вну
трішньоклітинного калію, що веде до зниження
мембранного потенціалу кардіоміоцитів і лег
шого виникнення в них деполяризації.
При настанні фібриляції шлуночків необхідно
негайно проводити дефібриляцію - пропускати
через серце короткий сильний одиничний елек
тричний розряд. При цьому відбувається одно
часна деполяризація всіх волокон міокарда та
припиняються асинхронні збудження м’язових
волокон, настає тимчасова асистолія (3-5 с) з на
ступним відновленням скорочень серця в ритмі,
що його задає синоатріальний вузол.
КІІК1 НЕДОСТАТНІСТЬ
ВІНЦЕВОГО КРОВООБІГУ.
ІШЕМІЧНА ХВОРОБА СЕРЦЯ
Важливим чинником діяльності серця є належ
не кровозабезпечення його тканин, яке здійсню
ється вінцевими судинами. Аби зрозуміти мож
132
ливі причини й механізми порушень вінцевого
кровообігу, слід знати основні його особливості,
що мають місце в умовах норми. А такими є:
а) високий рівень екстракції кисню в капіля
рах серця. У серці екстрагується 70-75 %
02, що надходить з артеріальною кров’ю,
тимчасом як у тканинах головного мозку 25 %, у скелетних м’язах і печінці - близь
ко 20 %, у нирках - 10 %. Високий рівень
вилучення 02 у серці пояснюється знач
ною довжиною його капілярів і у зв’язку
з цим — більшим часом контакту крові зі
стінкою капілярів. При збільшенні потреби
серця в кисні вона не може бути задоволе
на посиленням екстракції О, (як у скелет
них м’язах), оскільки остання і так є мак
симально можливою в стані спокою. Тому
для забезпечення збільшених енергетич
них потреб серця залишається тільки один
шлях - посилення вінцевого кровообігу;
б) високий базальний тонус вінцевих судин.
Він дає можливість у стані спокою забезпе
чувати вінцевий кровообіг на рівні 250-300
мл/хв, що становить близько 5 % хвилинно
го об’єму крові. Високий базальний тонус
судин обумовлює високий резерв вінцевого
кровообігу. Так, при зменшенні базального
тонусу судин серця інтенсивність кровообі
гу у них може зростати в 7-10 разів;
в) фазний характер вінцевого кровообігу,
пов’язаний з періодами серцевого циклу.
Під час систоли відбувається здавлювання
інтрамуральних судин - кровообіг міні
мальний і становить близько 15 % загаль
ного вінцевого кровообігу. Під час діасто
ли здавлювання судин припиняється і кро
вообіг стає максимальним (близько 85 %
загальної величини). Фазність вінцевого
кровообігу найбільш виражена в субендокардіальній зоні міокарда (найбільше здав
лювання судин) і найменш виражена — у субепікардіальній;
г)
підпорядкованість вінцевого кровообігу
метаболічним потребам серця й відносна
незалежність його від нервових регуля
торних впливів. В умовах патології ця під
порядкованість часто порушується і збіль
шується чутливість вінцевих судин до нер
вових імпульсів;
О) винятково висока чутливість вінцевих су
дин до зменшення напруги кисню в крові.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Зменшення р02 артеріальної крові всього
лише на 5 % істотно збільшує інтенсив
ність вінцевого кровообігу;
е) недостатній розвиток колатеральних су
дин. При несприятливих умовах колатералі
в серці не можуть компенсувати порушен
ня течії крові у вінцевих судинах, тому ко
латеральний кровообіг тут функціонально
неповноцінний.
Регуляція вінцевого кровообігу здійснюється
трьома механізмами.
1. Міогенна авторегуляція. Її основу стано
вить закон Бейліса, відповідно до якого при
розтягненні гладких м’язів кровоносних су
дин збільшується сила їхнього скорочення.
Міогенна авторегуляція забезпечує сталість
вінцевого кровообігу та відносну незалеж
ність його від змін артеріального тиску. Так,
при збільшенні тиску крові в аорті збіль
шується розтягування гладких м’язових
клітин (ГМК) вінцевих артерій, що веде
до їхнього скорочення, підвищення тонусу
артерій і збереження сталості кровообігу.
При зменшенні артеріального тиску вінце
вий кровообіг підтримується на постійному
рівні завдяки розслабленню ГМК і розши
ренню артерій. Показано, що при розтягу
ванні судинних ГМК збільшується проник
ність їхньої плазматичної мембрани до іо
нів кальцію. Останні проникають у клітини
і викликають їхнє скорочення.
2. Метаболічна регуляція. Вона підпорядковує
вінцевий кровообіг метаболічним потребам
серця. Здійснюється за допомогою цілого
ряду іонів і метаболітів, серед яких іони
водню, калію, молочна кислота, проста
гландини. Однак найбільше значення мають
два фактори: зменшення напруги О, в арте
ріальній крові і аденозин. Останній утворю
ється в результаті гідролізу аденінових нуклеотидів при гіпоксії та при посиленій ро
боті серця. Будучи природним блокатором
Са-каналів, аденозин зменшує надходження
іонів Са2+ у цитоплазму ГМК вінцевих су
дин, унаслідок чого зменшується ступінь
їхнього скорочення й падає базальний то
нус - вінцеві судини розширюються, коро
нарний кровообіг зростає (рис. 31.28).
3. Нервова регуляція. Нейрогенний тонус він
цевих судин незначний, про що свідчить
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
133
Метаболічна регуляція вінцевого кровообігу за допомогою аденозину
майже повна відсутність змін вінцевого
кровообігу після повної денервації судин
серця. Набагато більше значення має опо
середкований вплив нервової системи на
коронарний кровообіг. Він здійснюється
через зміни роботи серця та інтенсивності
обміну речовин у ньому.
В експерименті все ж таки вдається виявити
безпосередній вплив нервів на тонус вінцевих
судин. Так, при подразненні парасимпатичних
нервів і введенні ацетилхоліну відбувається не
значне розширення вінцевих артерій. Медіатори
симпатичної нервової системи (катехоламіни) при
дії на а-адренорецептори викликають звуження
судин, а впливаючи на Р-адренорецептори — роз
ширення. Оскільки в нормі у вінцевих судинах
переважають Р-адренорецептори, то загальний
ефект симпатичних впливів - незначне розши
рення судин серця.
Недостатність вінцевого кровообігу — це па
тологічний стан, що характеризується нездатніс
тю вінцевих судин забезпечувати кровопостачан
ня серця відповідно до його енергетичних потреб.
Інакше кажучи, виникає невідповідність між
енергетичними потребами серця й доставкою
кисню та поживних речовин вінцевими судинами.
Недостатність вінцевого кровообігу може
бути (а) відносною й (б) абсолютною.
» Відносна коронарна недостатність вини
кає у випадку первинного збільшення енер
гетичних потреб серця (збільшення наван
таження на серце при фізичній роботі, ар
теріальній гіпертензії). За таких обставин
інтенсивність вінцевого кровообігу може
зростати, але цього буває замало, щоб задо
вольнити потреби серця (рис. 31.29).
• Абсолютна коронарна недостатність ви
никає у випадку первинного порушення
вінцевого кровообігу, у результаті чого
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
134
Стан спокою
(норма)
Фізична робота
(норма)
Фізична робота
(відносна недостатність
вінцевого кровообігу)
Рис. 31.29. Модель відносної недостатності вінцевого кровообігу (за А. і. Хомазюком)
зменшується доставка кисню та поживних
речовин міокарду як у стані спокою, так
і при збільшенні енергетичних потреб сер
ця (рис. 31.30).
Інтенсивність вінцевого кровообігу визнача
ється формулою
р -р
^ _____ арт вен
R’
де Q ~ об’ємна швидкість течії крові; Рарт і Рвен тиск крові відповідно на початку та в кінці сис
теми вінцевих судин; (Р - Реен) - перфузійний
тиск; R - опір вінцевих судин.
Оскільки абсолютна коронарна недостатність
характеризується зменшенням інтенсивності
вінцевого кровообігу (зменшенням Q), можли
вими є два патогенетичних варіанти її розвитку.
І.
Зменшення перфузійного тиску. При цьому
розвивається коронарна недостатність централь
ного походження. Її причинами можуть бути:
артеріальна гіпотензія (зменшення Р \
наприклад, при всіх видах шоку, колапсі,
недостатності аортальних клапанів. При
зменшенні артеріального тиску спрацьову
ють механізми міогенної авторегуляції він
цевого кровообігу, що підтримують його на
постійному рівні. Однак, коли артеріаль
ний тиск падає нижче 70 мм рт. ст., ці меха
нізми виявляються неспроможними;
б) порушення венозного відтоку (збільшен
ня Р_). Ця причина практично не має
значення, бо навіть при значному підви
щенні тиску у правому передсерді, куди
впадає коронарний венозний синус, кров
витискується із вен під час систоли: при
скороченні серцевого м’яза вени здавлю
ються і кров, що міститься в них, перемі
щується далі. В експерименті моделюють
перев’язуванням вен серця. Однак при
цьому венозний відтік порушується лише
частково, бо 1/3 венозної крові виходить
а)
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
135
Потреба
Стан спокою
(норма)
Стан спокою
(абсолютна недостатність
вінцевого кровообігу)
Фізична робота
(абсолютна недостатність
вінцевого кровообігу)
Рис. 31.30. Модель абсолютної недостатності вінцевого кровообігу (за А. і. Хомазюком)
з тканини міокарда через вени Тебезія
прямо в порожнини серця.
II.
Збільшення опору вінцевих судин. При цьо
му розвивається коронарна недостатність міс
цевого походження. Оскільки
де ц -- в’язкість крові; І - довжина судин; г - ра
діус судин, то збільшення опору може бути обу
мовлене;
а) збільшенням в ’язкості крові при порушен
ні її реологічних властивостей (зневоднен
ня, поліцитемія, ДВЗ-синдром). При цьому
розвиваються порушення мікроциркуляції,
аж до сладж-синдрому та істинного капі
лярного стазу (див. главу 16);
б) зменшенням радіуса судин. Радіус судин
є основним чинником, що визначає інтен
сивність вінцевого кровообігу. При змен
шенні цього параметра у 2 рази опір коро
нарних судин зростає в 16 разів. Зменшення
радіусу судин є основною причиною ішемії
серця і розвитку абсолютної коронарної не
достатності.
Ішемія міокарда може зумовлюватися одним
з трьох механізмів.
1. Обтураційний механізм — зменшення про
світу вінцевих артерій через їхнє перекриття
зсередини. Його причинами можуть бути:
а) стенозуючий атеросклероз (є причиною
ішемії міокарда в 90 % випадків);
б) тромбоз вінцевих артерій (найчастіше
є наслідком атеросклерозу);
в) емболія вінцевих артерій;
г) запальні процеси в стінці судин серця - коронарити (бувають при ревматизмі, сифілісі).
Для розвитку клінічних ознак ішемії міокар
да має значення величина “критичного стено
136
зу”. Це мінімальне зменшення просвіту судин,
при якому виникає ішемія. У людини цей по
казник становить 75 % (у свиней — менше, у со
бак - більше).
2. Ангіоспастичний механізм - спазм вінце
вих судин. Його причинами можуть бути:
а) збудження а-адренорецепторів на тлі бло
кади (3-адренорецепторів;
б) вазопресин;
в) ангіотензин II;
г) тромбоксан А2;
д) гіпокапнія;
е) ендотелій — біологічно активна речовина
ендотеліального походження.
Існує клінічна форма коронарного ангіоспаз
му - стенокардія Принцметала.
3. Компресійний механізм — здавлювання
вінцевих судин ззовні. Може мати місце при
тахікардії (збільшується загальна тривалість
періоду здавлювання вінцевих судин під час
систоли серця). Негативна дія тахікардії на він
цевий кровообіг виявляє себе при збільшенні
частоти серцевих скорочень понад 200/хв. Іноді
причиною компресії є рубці та пухлини. В екс
перименті її використовують для моделювання
ішемії й інфаркту міокарда шляхом накладення
лігатури на вінцеві артерії.
Вплив недостатності вінцевого
кровообігу на міокард
При порушенні кровообігу у вінцевих судинах
серця на міокард діють три основні фактори, що
зумовлюють розвиток патологічних змін у ньому:
1) гіпоксія. Є прямим наслідком недостатньо
го надходження крові по артеріальних су
динах - ішемії;
2) ацидоз. Розвивається в результаті накопи
чення кислих продуктів обміну речовин
унаслідок порушення відтоку крові та ак
тивації гліколізу;
3) збільшення позаклітинної концентрації іо
нів калію в зоні ішемії. Його виявляють від
самого початку порушень вінцевого крово
обігу (через кілька хвилин), коли ще немає
ушкодження кардіоміоцитів. У цей період
відбувається пасивний вихід іонів калію
з клітин, і його позаклітинна концентрація
зростає до 8 ммоль/л і більше.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Сьогодні встановлено причину ранньо
го “витікання” іонів К+ з кардіоміоцитів. Вона
пов’язана з існуванням т.зв. АТФ-залежних калі
євих каналів (КДТф-каналів) у плазматичній мемб
рані м’язових волокон (рис. 31.31). Стан цих ка
налів (закриті чи відкриті) залежить від вмісту
АТФ у клітинах. При концентрації АТФ в кардіоміоцитах > 1 ммоль/л (у фізіологічних умовах
вона становить 3-5 ммоль/л) К ф-канали закри
ті і не пропускають через себе іони К+. В умовах
кисневого голодування, що настає при ішемії
міокарда, рівень АТФ у клітинах стає меншим
за 1 ммоль/л і канали відкриваються. Іони калію
виходять назовні, збільшуючи свою концентра
цію в інтерстиції та гіперполяризуючи мембрану.
Останнє має важливе захисне значення, про що
мова піде далі.
Через кілька годин від початку ішемії “виті
кання” іонів К+ із клітин може бути обумовле
не порушенням роботи Na-K-насосів (дефіцит
АТФ) та збільшенням неселективної проник
ності ушкоджених мембран.
Наведені вище фактори, діючи на міокард,
викликають цілий ряд негативних для нього на
слідків.
1. Порушення скорочувальної здатності міо
карда з розвитком недостатності серця.
При ішемії міокарда розрізняють ранні й піз
ні розлади його скорочувальної функції.
Ранні порушення виникають дуже рано, вже
при незначному дефіциті АТФ. Вони є відо
браженням гіпокальцієвого варіанта порушень
скорочувальної функції серцевого м’яза (див.
вище) і розвиваються в результаті блокади Саканалів сарколеми кардіоміоцитів. Зменшен
ня провідності Са-каналів в умовах ішемії має
кілька механізмів:
а) дефіцит АТФ —» відкриття КДТф-каналів —>
“витікання” іонів К+ з клітин —* гіперполяризація сарколеми —» зменшення тривалості
фази “плато” потенціалів дії —» зменшення
надходження іонів Са2+ у саркоплазму через
потенціалзалежні Са-канали (рис. 31.31);
б) дефіцит АТФ —> порушення фосфорилювання білків Са-каналів. Останні в нефосфорильованому стані пропускають через
себе значно менше іонів Са2+, ніж після
процесу фосфорилювання;
в) активація гліколізу —*• накопичення в кліти
нах іонів Н+ -+ безпосередня блокада Саканалів.
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
137
Збільшення позаклітинної концентрації іонів калію як ранній прояв ішемії міокарда
Наслідком зазначених вище змін є зменшення
надходження іонів Са2+ у саркоплазму м’язових
волокон, що веде до розладів електромеханіч
ного спряження і зменшення сили скорочень
кардіоміоцитів.
Порушення здатності кардіоміоцитів до ско
рочень у початковий період ішемії має до певної
міри захисне пристосувальне значення, оскіль
ки при цьому зменшуються витрати АТФ на
виконання механічної роботи і заощаджений
АТФ може використовуватися для підтримання
структурної цілісності клітин. Це якийсь час за
побігає розвиткові необоротного ушкодження
м’язових волокон та їх некрозу.
Пізні порушення скорочувальної функції
виникають при тривалих розладах вінцевого
кровообігу - понад ЗО хв. їх відносять до гіперкальцієвого (контрактурного) типу порушень.
Концентрація іонів кальцію в саркоплазмі кар
діоміоцитів збільшується через дві основні
причини: (а) порушення видалення іонів Са2+
із саркоплазми внаслідок дефіциту АТФ і (б)
збільшення надходження іонів Са2+ у м’язові во
локна через ушкоджену плазматичну мембрану
клітин. У результаті збільшення вмісту Са2+ по
рушуються процеси розслаблення кардіоміоци
тів, настає контрактура міофібрил, зменшується
скорочувальна здатність серця.
2. Поява аномальної електричної актив
ності - електрична нестабільність сер
ця, розвиток аритмій. Розлади ритму сер
ця виникають уже через кілька хвилин від
початку ішемії міокарда. Основна причина
електричної нестабільності — вихід іонів
калію з кардіоміоцитів, у результаті чого
збільшується їхня позаклітинна концентра
ція. Це викликає часткову деполяризацію
сарколеми клітин, що перебувають поруч
в неішемізованій ділянці.
Залежно від рівня підвищення вмісту поза
клітинного калію можливі такі наслідки:
а) у ділянках міокарда, де концентрація
позаклітинного калію збільшується до
8 ммоль/л, мембранний потенціал зменшу
ється від -90 мв до —80 мв, наближаючись
до рівня критичного потенціалу деполя
ризації. Тому в цих ділянках збільшується
138
збудливість клітин і швидкість проведення
імпульсів;
б) у зонах міокарда, де позаклітинна концен
трація калію перевищує 8 ммоль/л, мемб
ранний потенціал м’язових волокон стає
менше -80 мв, зменшується провідність
Na-каналів (відбувається їхня інактива
ція), у результаті зменшується збудливість
кардіоміоцитів та швидкість проведення
імпульсів (відносна і абсолютна рефрактерність).
Одночасне існування різних ділянок міокар
да з різними типами електрофізіологічних по
рушень (з одного боку, збільшення збудливості
й провідності, з другого боку - зменшення цих
характеристик) створює умови для реалізації
механізмів
повторного входження імпульсів
(див. вище), у результаті чого можуть розви
ватися тріпотіння й фібриляція шлуночків, що
призводять до смерті.
Фібриляція шлуночків, що виникає при іше
мії міокарда, є основною причиною раптової
смерті. При цьому на розтині, як правило, жод
них ознак ішемічного ушкодження міокарда не
виявляють.
3.
Ушкодження кардіоміоцитів, обумовлене
ішемією. Провідною патогенетичною лан
кою ушкодження м’язових волокон серця
при ішемії безумовно є гіпоксія, яка може
мати гострий (перекриття вінцевої артерії
тромбом) або хронічний (зменшення про
світу вінцевої артерії неускладненою ате
росклеротичною бляшкою) характер.
(А) Гостра ішемічна гіпоксія зумовлює го
стре необоротне ушкодження клітин та їхню за
гибель переважно механізмом некрозу (див. гла
ву 7). Некроз кардіоміоцитів позначають тер
міном інфаркт міокарда. На місці загиблих
клітин формується сполучнотканинний рубець,
який стає структурною основою постінфарк
тного кардіосклерозу. При великому обсягові
ішемічних уражень постінфарктний кардіо
склероз може бути причиною хронічної недо
статності серця (рис. 31.32).
Провідною патогенетичною ланкою некро
тичної загибелі клітин при гострій ішемічній
гіпоксії є порушення бар’єрних властивостей
клітинних мембран, що настає внаслідок реалі
зації цілого ряду механізмів (рис. 31.33):
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
1) зменшення напруги кисню (рО.) в кардіоміоцитах має кілька наслідків. По-перше, воно
веде до пригнічення окисного фосфорилювання в мітохондріях і розвитку різкого де
фіциту АТФ. Показано, що при зменшенні
вмісту АТФ до рівня нижче 10 % від вихід
ного значення, ушкоджуються мембрани
і настає некроз. По-друге, компенсаторно
зростає інтенсивність гліколізу, унаслідок
чого збільшується в клітинах концентрація
лактату і розвивається внутрішньоклітин
ний ацидоз (|рН). І по-третє, зменшення
напруги кисню в мітохондріях нижче рівня
1 мм рт. ст. припиняє роботу дихального
ланцюга через втрату цитохромоксидазою
спорідненості з киснем. Унаслідок цього
відбувається “скидання” електронів без
посередньо на молекули кисню, які, хоч
і в незначній кількості, а все ж таки ще є
в мітохондріях. Неповне відновлення кис
ню в обхід цитохромоксидази і дихального
ланцюга стає причиною появи вільних ра
дикалів - активних форм кисню (супероксидного аніон-радикалу, синглетного кис
ню, пероксиду водню). Оскільки потуж
ності наявних антиоксидантних систем
недостатньо, то започатковуються реакції
вільнорадикального окиснення, серед яких
важливе значення має пероксидне окиснен
ня ліпідів (ПОЛ). Його активація є універ
сальним механізмом ушкодження мембран
(докладно див. главу 7);
2) зменшення вмісту АТФ, що настає в ре
зультаті пригнічення клітинного дихання
і окисного фосфорилювання, не тільки
“відкриває” АТФ-залежні калієві канали
(див. вище), а й спричиняється до по
рушення роботи енергозалежних іонних
насосів: натрій-калієвого та кальцієвих.
Наслідком цих порушень є збільшення
внутрішньоклітинної концентрації іонів
натрію та кальцію;
3) збільшення внутрішньоклітинної концен
трації іонів Na+ веде до підвищення осмо
тичного тиску в кардіоміоцитах і їх набря
ку. У таких клітинах відбувається механіч
не розтягування сарколеми, що порушує
бар’єрні властивості останньої, аж до
можливого розриву;
4)
зростання
внутрішньоцитоплазматичної
концентрації іонів кальцію, крім того, що
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
Підвищення проникності мембран мітохондрій веде до входження у їхній матрикс іонів Н+
і К+. Це, з одного боку, робить неефективною
роботу “водневої помпи” і припиняє пов’язані
з нею реакції окисного фосфорилювання,
а з другого - зумовлює набухання і набряк мітохондрій, аж до повного їхнього руйнування.
Ушкодження мембран лізосом на тлі внутріш
ньоклітинного ацидозу спричиняється до вихо
ду в цитоплазму активних лізосомних фермен
тів, які започатковують перетравлення (реакції
гідролізу) спочатку внутрішньоклітинних, а по
тім, після виходу за межі зруйнованих кардіоміоцитів, і позаклітинних структур. Так настає
некроз ділянки ішемізованого міокарда.
Загибель кардіоміоцитів шляхом некрозу зу
мовлює розвиток запалення, спрямованого на
“розчищення” осередку інфаркту і формуван
ня сполучнотканинного рубця. Проте цей про
цес у міокарді має серйозні негативні наслідки.
Оскільки в запалення обов’язково втягуються
прилеглі до інфаркту ділянки серцевого м’яза,
то в них виникає вторинне, пов’язане вже не
з ішемією, а із запаленням, ушкодження (вто
ринна альтерація, див. главу 17), що збільшує
зону ураженого міокарда.
Вторинна альтерація в сусідніх з інфарктом
ділянках зумовлюється лейкоцитами (ней
трофілами, моноцитами), що емігрують сюди
з крові. Донедавна було незрозумілим, що саме
викликає хемотаксис і активацію лейкоцитів
у вогнищі інфаркту, адже запалення, що розви
вається, є асептичним, тобто жодним чином не
пов’язане з відомими хемоатрактантами бакте
ріального походження.
Нині показано, що вплив на лейкоцити чинять
мітохондрії загиблих клітин. З них у навколиш
нє середовище вивільнюються мітохондріальна
ДНК і формілпептиди. Маючи еволюційне похо
дження від бактерій, ці органели зберегли мож
ливість впливати на лейкоцити так само, як і бак
теріальні клітини (рис. 31.34). Мітохондріальна
ДНК, як і бактеріальна, діючи на Toll-подібні
рецептори лейкоцитів (див. главу 14), зумовлює
активацію останніх, а мітохондріальні форміл
пептиди, що є подібними до бактеріальних, че
рез взаємодію із серпентиновими формілпептидними рецепторами започатковують хемотаксис.
Надходження великої кількості нейтрофілів
у вогнище інфаркту має своїм негативним на
слідком вивільнення у тканину великої кількос
141
ті (а) вільних радикалів і пероксидних сполук
(секреторна дегрануляція), а також появу (б) ме
діаторів запалення, зокрема таких, як лейкотрієни. Розвиток запалення у прилеглих до інфарк
ту ділянках міокарда веде до розширення зони
некрозу та збільшення маси м’язових волокон,
не здатних скорочуватися. Безперечно, це дуже
погано для серця в цілому, а тому один із напря
мів лікування інфаркту міокарда полягає у змен
шенні запальної реакції в ураженому серці, на
що спрямовується дія лікарських препаратів
з антиоксидантною і протизапальною дією. По
казано, що високоефективними в цьому сенсі
є біофлавоноїди, які, маючи виражені антиок
сидантні властивості (інактивують вільні ради
кали і пероксиди лейкоцитарного походження),
водночас пригнічують активність ліпоксигенази
(зменшується утворення лейкотрієнів), а також
цілого ряду катаболічних ферментів, у тому чис
лі й лізосомних (О. О. Мойбенко).
(Б) Хронічна ішемічна гіпоксія. Тривале по
рушення надходження крові до серця, як прави
ло через атеросклеротичні ураження вінцевих
артерій, зумовлює хронічне кисневе голодуван
ня кардіоміоцитів, унаслідок чого зменшується
окисний ресинтез АТФ. У цій ситуації клітини
мають дві стратегії: або вижити шляхом при
стосування, або загинути, не завдаючи шкоди
сусіднім клітинам.
Звісно, перевагу має перша стратегія, і вона
реалізує себе через відомий уже нам механізм.
При зменшенні вмісту АТФ у клітинах, зростає
концентрація продуктів його гідролізу - АДФ
і неорганічного фосфату, а отже, і потенціал
фосфорилювання (див. вище). Це, у свою чергу,
зумовлює активацію цілого ряду транскрипцій
них факторів, які, пройшовши у ядро, активують
генетичний апарат кардіоміоцитів у напрямку
збільшення синтезу основних білків, від яких
залежить функціонування та виживання клітин
(рис. 31.35). Крім того, сам собою дефіцит кис
ню спричиняється до появи в саркоплазмі актив
ного транскрипційного фактора HIF-1 (hipoxiainducible factor-1), що посилює транскрипцію
генів, важливих для адаптації до кисневого го
лодування (міоглобіну, ферментів біологічного
окиснення і гліколізу тощо). Іншими словами,
спрацьовують усі можливі механізми адаптації
клітин до гіпоксії, докладно описані у главі 23.
Реалізація цих механізмів виявляє себе струк
турними змінами в міокарді - розвивається його
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
142
Хемотаксис
Хемотаксис
Нейтрофіл
Секреторна дегрануляція
Рис. 33.34. Вплив мітохондрій загиблих клітин на лейкоцити.
FRP - рецептор до формілпептидів, TLR - Тої Гподібний рецептор
гіпертрофія. Збільшення маси м’язових воло
кон при тривалій ішемії має ще й другу причину.
В умовах загибелі одних кардіоміоцитів більше
навантаження лягає на інші, у яких зміни вну
трішньоклітинного гомеостазу ще не набули ка
тастрофічного характеру. Отже, додається чин
ник компенсаторної гіперфункції — негативний
для окремих м’язових клітин, але конче необхід
ний для збереження насосної функції серця як
одного цілого.
Якщо в результаті хронічної гіпоксії та ком
пенсаторної гіперфункції дефіцит АТФ стає
значним і кардіоміоцити вже не в змозі викону
вати свою скорочувальну функцію, вмикають
ся механізми запрограмованої загибелі клітин,
розвивається апоптоз. Він ініціюється мітохондріальним і ядерним шляхами (див. главу 7).
1) Мітохондріальний шлях пов’язаний зі
збільшенням проникності мембран міто
хондрій і відкриттям т. зв. мітохондріальної
пори (рис. 31.35). Одним з важливих меха
нізмів цих змін є відкриття АТФ-залежних
калієвих каналів мітохондрій. Встановлено,
що згадувані вже нами КДТф-канали є не
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
143
ис. 31.35. Активація мітохондріального і ядерного шляхів апоптозу при хронічній ішемічній гіпоксії.
ПФ - потенціал фосфорилювання, МП - мітохондріальна пора
тільки в сарколемі кардіоміоцитів, а й у мітохондріальних мембранах. При значному
зменшенні концентрації АТФ ці канали
відкриваються, й іони К+ за градієнтом кон
центрації й електричного заряду перехо
дять із саркоплазми в матрикс мітохондрій.
При цьому відбувається деполяризація
внутрішньої мітохондріальної мембрани
(це впливає на роботу дихального ланцюга
та систем окисного фосфорилювання) і пе
реміщення води всередину органел - роз
вивається набухання й набряк мітохондрій.
Механічне розтягнення мітохондріальних
мембран, як результат набряку, стає при
чиною відкриття мітохондріальної пори,
через яку в цитоплазму починають перехо
дити цитохром С та AIF (apoptosis inducing
factor) — чинники, що започатковують на
ступні фази апоптозу (див. главу 7).
2) Ядерний шлях ініціювання апоптозу
“вмикається” через нездатність генетич
ного апарату кардіоміоцитів реалізувати
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
144
ті численні стимули, що надходять у ядро
через активовані транскрипційні факто
ри. Така нездатність пов’язана насампе
ред зі значним дефіцитом АТФ. У ядрі
ця сполука є не тільки джерелом енергії
для процесів біосинтезу й переміщення
нуклеїнових кислот, а й пластичним ма
теріалом, без якого, через брак азотистих
основ і неорганічного фосфату, не можуть
будуватися молекули всіх видів РНК і від
новлюватися дефекти ДНК, що виника
ють. За таких умов “безладу” в ядрі свою
роль “кризового менеджера” починає ви
конувати білок р53 (див. главу 7). Будучи
транскрипційним фактором, він посилює
експресію проапоптичних білків і змен
шує “ антиапоптичних. Крім того, цей
протеїн переходить із ядра в цитоплазму,
а потім і в мітохондрії, де після взаємодії
з антиапоптичним білком Вс1-2 відкриває
мітохондріальну пору і в такий спосіб за
пускає мітохондріальний шлях ініціюван
ня апоптозу.
Порівнюючи два основних механізми заги
белі кардіоміоцитів в умовах ішемії міокарда некроз і апоптоз, слід позначити такі моменти.
(А) Загибель кардіоміоцитів механізмом апоп
тозу є, безумовно, кращим варіантом, ніж не
кроз. Це пояснюється тим, що апоптоз, на від
міну від некрозу, ніколи не викликає запалення,
а отже, і не ускладнює цим процесом уражен
ня міокарда (див. вище). (Б) Крім того, апоптоз
відбувається асинхронно в різних клітинах, за
вдяки чому серцю легше перенести таку втра
ту, ніж одночасну втрату такої ж кількості кар
діоміоцитів, що зазнали некрозу. (В) Оскільки
некрозу зазнають не поодинокі окремі клітини,
а цілі ділянки міокарда, то завершується цей
процес утворенням чітко окресленого сполуч
нотканинного рубця; — постінфарктним карді
осклерозом. Апоптична загибель кардіоміоци
тів також завершується формуванням сполуч
ної тканини, але цей процес носить дифузний
характер, бо гине не якась ділянка міокарда, а
окремі клітини і в різних частинах серцевого
м’яза. Такі зміни в міокарді позначають як ате
росклеротичний кардіосклероз.
Слід зазначити, що крім двох основних меха
нізмів смерті кардіоміоцитів при ішемії, певне
значення, особливо при хронічних порушен
нях коронарного кровообігу, що супроводжу
ються недостатністю поживних речовин, має
ще один — автофагічний (див. главу 7). Цей
механізм, будучи за багатьма ознаками схожим
на апоптоз, є сьогодні предметом вивчення ба
гатьох учених.
4. Реперфузійний синдром. Так називають
синдром, що виникає внаслідок поновлен
ня кровообігу в ішемізованій ділянці міо
карда, тобто в результаті реперфузії.
Поновлення вінцевого кровообігу може бути
обумовлене припиненням коронарного ангіо
спазму, лізисом тромбу, руйнуванням агрегатів
клітин крові, хірургічним видаленням тромбу,
зняттям лігатури.
Клінічно реперфузійний синдром виявляє
себе значним збільшенням обсягу та інтен
сивності ушкодження міокарда відразу ж піс
ля усунення перешкод вінцевому кровообігу
(рис. 31.36). Внаслідок цього стан хворого різ
ко погіршується. Мінімальна тривалість ішемії,
після якої виникає виражений реперфузійний
синдром, становить 40 хв. Якщо тривалість іше
мії менше 20 хв., зазначений синдром не розви
вається (така тривалість характерна для нападів
стенокардії). При тривалості ішемії 20—40 хв.
іноді можуть розвиватися реперфузійні ушко
дження серця.
Патогенетичною основою реперфузійного
синдрому є так званий “кисневий парадокс”.
Якщо здійснювати перфузію серця розчином,
що не містить кисню (або містить його мало),
а через 40 хв. і більше перейти на перфузію
розчином з нормальною напругою 02, то в ре
зультаті такої перфузії порушення, обумовлені
попередньою гіпоксією, не тільки не зменшу
ються, як цього варто було б очікувати, а ста
ють більш вираженими (парадокс!). В основі
зазначеного парадокса — різка активація про
цесів вільнорадикального окиснення, обумов
лена надходженням кисню в клітини, у яких
міститься велика кількість відновлених компо
нентів дихального ланцюга. Відбувається ски
дання електронів в обхід дихального ланцюга
безпосередньо на молекули кисню, внаслідок
чого утворюється велика кількість вільних ра
дикалів. Вони ініціюють реакції пероксидного
окиснення ліпідів, що є важливим молекуляр
ним механізмом ушкодження клітинних мемб
ран (див. главу 7).
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
Кількість загиблих клітин
145
Реперфузія
Кількість загиблих клітин
Ішемія
Кількість загиблих клітин
В
ис. 31.36. Вплив тривалості ішемії та реперфузії на кількість загиблих клітин у міокарді.
А - тривалість ішемії < 20 хв; Б - тривалість ішемії > 40 хв; В - посткондиціонування. І - ішемія, Р - реперфузія
146
Природні механізми захисту
серця від ішемічного та
реперфузійного ушкодження пре- і посткондиціонування
Кардіологи давно помітили, що повторний ін
фаркт міокарда переноситься легше, ніж пер
ший, а в людей похилого віку з тривалою істо
рією ішемічної хвороби серця він проходить не
так гостро, має меншу летальність, ніж у молод
ших осіб.
В експериментах на собаках було показано, що
кілька короткочасних періодів місцевої ішемії
міокарда не викликають, як очікувалося, ефек
ту додавання (кумуляції) ушкоджень, а, навпаки,
підвищують стійкість серцевого м’яза до наступ
ної тривалої ішемії. Це виявляло себе зменшен
ням ділянки інфаркту та навколоінфарктної зони.
Отже, клінічні спостереження й результати
дослідів свідчили про здатність серця до сво
єрідного тренування під впливом ішемії, про
наявність певних природних механізмів, що за
пускаються ішемією й ведуть до підвищення
стійкості міокарда до цього патологічного про
цесу в подальшому. Різні варіанти такої стійкос
ті отримали назву прекондиціонування (передпідготовка) і посткондиціонування (післяпідготовка).
Прекондиціонування. Це феномен підвищеної
стійкості міокарда до ішемії, який виникає вна
слідок попередньої дії короткочасної ішемії або
інших факторів і здійснюється за участю ряду
внутрішньоклітинних механізмів.
Прекондиціонування розділяють на (а) раннє
і (б) пізнє (рис. 31.37).
Раннє прекондиціонування у класичному ва
ріанті відтворюють в експерименті кількома ко
роткочасними періодами ішемії (від 1 до 5 хв),
розділеними такими ж за тривалістю періодами
реперфузії з наступною тривалою ішемією (ЗО хв
і більше). При цьому захисні ефекти наведеної
схеми виявляють себе одразу ж після проведення
циклів “ішемія - реперфузія”, а тому таке пре
кондиціонування отримало назву раннього іше
мічного.
Пізнє
прекондиціонування
проводять
так
само, але проміжок часу між епізодами “іше
мія - реперфузія” і наступною тривалою іше
мією складає від 18 до 96 годин. Підвищення
стійкості серця до ушкодження, що виникає за
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
цих умов, наводить на думку про участь гене
тичного апарату кардіоміоцитів у реалізації та
кого явища.
Сьогодні встановлено, що феномен прекон
диціонування можуть викликати не тільки ко
роткі цикли “ішемія - реперфузія” (ішемічне
прекондиціонування), а й велика кількість екзо
генних факторів фізичної (гіпоксія, гіпероксія,
гіпертермія), хімічної (засоби для інгаляційно
го наркозу, активатори К -каналів, інгібітори
протеасоми та ін.) і біологічної природи (ліпополісахариди бактерій). Серед ендогенних чин
ників, здатних індукувати прекондиціонування,
називають аденозин, оксид азоту, брадикінін,
ацетилхолін та ін. Нині для вивчення механізмів
феномену широко застосовують фармакологічні
препарати з різними й добре відомими точками
докладання їхньої дії (фармакологічне прекон
диціонування).
Посткондиціонування. Якщо після тривалої
ішемії (понад 40 хв) реперфузію починати не
відразу, а провести спочатку кілька дуже ко
ротких циклів “реперфузія - ішемія” (по ЗО с
кожна), то реперфузійне ушкодження, що розви
вається після повного відновлення коронарного
кровообігу, буде значно меншим (рис. 31.36, В).
Такий феномен отримав назву посткондиціону
вання. Як і прекондиціонування, його можуть
викликати не тільки короткі цикли “ішемія реперфузія”
(ішемічне
посткондиціонування),
а й інші, наведені вище чинники.
Вивчення механізмів пре- і посткондиціону
вання на клітинному рівні показало, що вони
є дуже складними і в загальних рисах можуть
бути описані схемою, до якої входять такі еле
менти (рис. 31.38):
1) індуктори, або тригери. Це всі ті екзоген
ні й ендогенні чинники, що можуть запо
чатковувати пре- і посткондиціонування.
Ми вже про них вели мову;
2) рецептори. Велика кількість індукторів
(біологічно активні речовини, фармаколо
гічні агенти тощо) чинять свою дію через
мембранні циторецептори, хоча є й такі,
що обходяться без цього;
3) посередники. Серед них особливо важливу
роль відіграють протеїнкінази (протеїнкіназа С, мітогенактивована протеїнкіназа
та інші), у зв’язку з чим їх ще назвали кі
назами виживання (servival kinases)',
147
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
А
Кількість загиблих клітин
Кількість загиблих клітин
Кількість загиблих клітин
Ішемія
----------------4*
Час
18-96 год
Рис. 31.37. Вплив тривалості ішемії (А) та раннього (Б) і пізнього (В) прекондиціонування на кількість
загиблих КЛІТИН У міокарді. Умовні позначення див. рис. 33.36
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
148
Б
А
Індуктори
Рецептори
Посередники
Ефектори
Протеїнкінази та інші ферменти
...
...
М КАТФ-канали КАХФ-канали
n
Сарколема Мітохондри ! Ядро j * с™колеми мЦохондрій
▼ їі
Ефекти
Уповільнення
метаболічних реакцій
Підтримання
іонного гомеостазу
і конц. іонів Са2+ і К+ в цито
плазмі, уповільнення реакцій
окиснення в мітохондріях
Механізми пре- і посткондиціонування: А - загальна схема, Б - кардіопротекторна дія аденозину
4) ефектори. Основні ефекти пре- і посткон
диціонування пов’язані з такими струк
турами кардіоміоцитів, як сарколема, мітохондрії та ядро. Фосфорилювання біл
ків цих структур під впливом численних
протеїнкіназ спричиняється до змін їхніх
властивостей і функціональної активності;
5) ефекти. Важливе значення для захисту
серця від ішемічного та реперфузійного
ушкодження мають уповільнення метабо
лічних реакцій і підтримання іонного го
меостазу, на що, власне, і спрямовані роз
глянуті вище зміни у клітинах серцевого
м’яза.
У підготовці кардіоміоцитів до можливої
ішемії особлива роль належить аденозину, що є
продуктом повного гідролітичного розщеплен
ня АТФ. Його посилене утворення має місце
як при інтенсивній роботі серця, так і при по
рушенні окисного ресинтезу АТФ. Діючи че
рез аденозинові рецептори, ця сполука активує
протеїнкіназу С, а та у свою чергу фосфорилює велику кількість білків, серед яких компо
ненти КАТф-каналів сарколеми й мітохондрій
(рис. 31.38Б). Будучи фосфорильованими, ці ка
нали відкриваються і пропускають через себе
іони калію із цитоплазми відповідно назовні та
в мітохондрії. Про роль АТФ-чутливих калієвих
каналів ми вже вели мову (див. вище), тут лише
зазначимо, що завдяки їхньому відкриванню
в кардіоміоцитах зменшується цитоплазматич
на концентрація іонів Са2+ і К+, уповільнюються
реакції окиснення в мітохондріях, тобто вини
кають ефекти, що мають захисне значення для
серця.
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
Некоронарогенні некрози серця
Масова некротична загибель клітин міокарда
може бути зумовлена не тільки його ішемією,
а й деякими іншими чинниками, що безпосеред
ньо не мають стосунку до вінцевого кровообігу.
Такі некрози отримали назву некоронарогенних.
Існує ряд експериментальних моделей не
крозу серцевого м’яза, причина виникнення
якого не пов’язана з патологією вінцевих судин.
Ці моделі певною мірою відображають ситуа
цію, що спостерігається в природних умовах.
1. Гіпоксичний некроз міокарда. Може бути
відтворений за допомогою різних видів
гіпоксії: гіпоксичної, гемічної. При цьо
му на тлі загальної недостатності кисню
в організмі, що сама по собі веде до під
вищення навантаження на систему кро
вообігу, розвивається необоротне ушко
дження м’язових волокон серця. Розвитку
некрозу сприяє фіксація тварини в не
зручній позі, наприклад, розтягування
у верстаті, або додаткове навантаження біг у тредбані.
2.
Електролітно-стероїдна
кардіопатія
з некрозом. За спостереженнями Сельє,
при введенні щурам значної кількості
солей натрію разом з деякими аніона
ми (сульфатними, фосфатними) в серці
з’являються осередки ушкодження деге
неративно-некротичного типу, що часто
супроводжуються гіалінозом судин інших
органів. Ці ушкодження стають більшими
або виникають при введенні меншої кіль
кості солей, якщо одночасно вводити де
які стероїдні гормони надниркових залоз.
На цьому тлі легше розвиваються і мають
тяжчий перебіг ушкодження серця, викли
кані іншими причинами. Так, введення
навіть невеликих доз норадреналіну, по
хідних кальциферолу, гіпоксія, м’язове на
пруження або, навпаки, значне обмеження
рухливості ведуть до розвитку великого
некрозу міокарда. Солі калію і магнію при
цьому мають захисну дію.
3. Імунні ушкодження серця. Можливі при
введенні в організм експериментальної
тварини гетерогенної сироватки, що міс
тить антитіла проти білків серця тварини
цого виду (кардіоцитотоксини). Доведено
також, що в організмі за певних умов мо
149
жуть виникати антитіла і сенсибілізовані
лімфоцити, які діють на тканини власно
го серця й викликають його ушкодження.
Цьому сприяє проникнення в кров дена
турованих компонентів некротизованих
кардіоміоцитів. В експерименті аналогіч
ний процес можна викликати введенням
тварині суспензії міокарда зі стимулято
ром імунологічної реакції (ад’ювантом
Фрейнда). Серце може бути ушкоджене
і циркулюючими імунними комплексами
“антиген — антитіло”, а також при фіксації
на його структурах цитофільних антитіл
типу IgE з подальшою їхньою реакцією
з антигеном.
4. Нейрогенні ураження серця. Дистрофічні
зміни та некроз міокарда можна відтво
рити гострим або хронічним подразнен
ням шийно-грудного вузла симпатичного
стовбура, блукаючого нерва, гіпоталаму
са, мозкового стовбура або інших відді
лів головного мозку. Введення в кров ве
ликих доз адреналіну або норадреналіну
також веде до ураження серця. В основі
механізму нейрогенних ушкоджень ле
жить невідповідність між рівнями функції,
метаболізму та кровопостачання тканин.
Подразнення серцевих симпатичних нер
вів супроводжується значним збільшен
ням споживання кисню міокардом. При
цьому збільшення вінцевого кровообігу
є недостатнім (відносна коронарна недо
статність), а тому розвивається гіпоксія
міокарда. При склерозуванні вінцевих ар
терій невідповідність інтенсивності кро
вообігу рівневі обміну речовин виявляєть
ся ще більшою мірою, що може виявитися
катастрофічним як для серця, так і для ор
ганізму в цілому.
Ішемічна хвороба серця
Найпоширенішою хворобою в економічно роз
винених країнах світу є ішемічна хвороба серця
(ІХС), що являє собою абсолютну або відносну
недостатність кровопостачання міокарда вна
слідок уражень вінцевих артерій серця. При
цьому порушується життєво важлива рівновага
між коронарним кровообігом і метаболічними
потребами серця, що й спричиняється до ушко
дження кардіоміоцитів.
150
ІХС може мати гострий (інфаркт міокарда)
і хронічний (періодичні напади стенокардії) пе
ребіг та виявляти себе в різних клінічних фор
мах, серед яких основними є:
1) раптова коронарна смерть (первинна
зупинка серця). Її причиною, як правило,
є електрична нестабільність серця, що
супроводжує розвиток недостатності він
цевого кровообігу (див. вище). Із стану
клінічної смерті, зумовленого зупинкою
серця, можна вивести засобами реаніма
ції. Якщо цього не відбувається, то орга
нізм помирає;
2) стенокардія — напади короткочасної (до
20 хв.) гострої коронарної недостатнос
ті, які супроводжуються больовим син
дромом, відчуттям страху і пов’язаними
з цим вегетативними реакціями. Розріз
няють різні варіанти стенокардії, серед
них (а) стабільна стенокардія напруги,
(б) коронарний синдром X (стенокардія
напруги при ангіографічно інтактних
судинах), (в) стенокардія Принцметала (вазоспастична стенокардія), (г) різні
форми нестабільної стенокардії',
3) інфаркт міокарда - некроз серцевого
м’яза, обумовлений порушеннями він
цевого кровообігу. Виникає при обо
ротній (транзиторній) ішемії, що триває
понад 40-60 хв., або при необоротних
порушеннях
коронарного
кровообігу.
Порушення в міокарді, що розвивають
ся при тривалості його ішемії від 20 до
40 хв, часто позначають як передінфар
ктний стан (проміжний коронарний
синдром, гостра вогнищева дистрофія
міокарда);
4) кардіосклероз - склеротичні зміни серце
вого м’яза, що ведуть до розвитку недо
статності серця. Можуть бути дифузни
ми
(атеросклеротичний
кардіосклероз)
і вогнищевими (постінфарктний кардіо
склероз).
У клінічній практиці для швидкого прийняття
рішень щодо тактики лікування послуговують
ся терміном гострий коронарний синдром, яким
об’єднують такі форми ІХС, як нестабільна сте
нокардія, гострий інфаркт міокарда й раптова
коронарна смерть.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Причини. Основною причиною ІХС (у більш
ніж 90 % пацієнтів) є атеросклероз вінцевих
артерій серця. Докладна мова про цей процес
піде у главі 32. Інші причини: запалення вінце
вих артерій (коронарити), порушення нервової
та гуморальної регуляції їхнього тонусу - ма
ють набагато менше клінічне значення.
Безпосередніми причинами гострого коро
нарного синдрому є:
1) оклюзія (закупорювання) вінцевих арте
рій, що виникає внаслідок розриву ате
росклеротичної бляшки й утворення на її
поверхні тромбу. Фрагменти тромбу, утво
реного на початкових ділянках головних
артерій серця, можуть відриватися і, пе
ретворившись на емболи, закупорювати
дрібніші гілки коронарних судин. При
неповній оклюзії артерій тромбом можуть
розвиватися нестабільна стенокардія, го
стрий субендокардіальний інфаркт міо
карда (ця зона серцевого м’яза найгірше
кровопостачається, особливо в систолу)
або ж раптова коронарна смерть. Повна
оклюзія веде до гострого трансмурального
інфаркту міокарда чи до раптової смерті;
2) збільшення навантаження на серце (фі
зичне напруження, артеріальна гіпертен
зія). Як уже зазначалося (див. вище), іс
нує “критичний стеноз” вінцевих артерій,
при якому з’являються клінічні ознаки
ішемії міокарда. У людей такі симптоми
виникають при дозованому фізичному
навантаженні, якщо просвіт артерій стає
менший за 25 % (обтурація становить
75 %), а в стані спокою — при обтурації,
що перевищує 90 %. Звісна річ, що різке
збільшення навантаження на серце значно
збільшує його потребу в кровопостачанні.
Якщо вінцеві артерії мають просвіт мен
ший за критичний, то в цій ситуації вини
кає різкий дисбаланс між потребами міо
карда в кисні та можливостями його до
ставки. Як наслідок, можуть розвиватися
ознаки гострого коронарного синдрому;
3) стрес (див. главу 8). Стрес, особливо пси
хоемоційний, супроводжується значною
активацією
симпатоадреналової
систе
ми та дією на серце катехоламінів (адре
наліну й норадреналіну). Для розвитку
гострого коронарного синдрому мають
значення такі їхні ефекти:
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
а) порушення вінцевого кровообігу. Катехоламіни, сприяючи розвиткові атеросклерозу
(стрес і катехоламіни є фактором ризику
цієї хвороби) здатні викликати контрактурний спазм гладких м’язів вінцевих ар
терій (катехоламінове ушкодження клітин
судинної стінки) і активувати тромбоутво
рення та зсідання крові;
б) збільшення потреби серця в кисні. Енерге
тичні потреби серця зростають у результаті
реалізації позитивного іно- і хронотропного ефектів катехоламінів та збільшення за
гального периферичного опору, внаслідок
чого збільшується навантаження на серце;
в)
некоронарогенні
катехоламінові
ушко
дження кардіоміоцитів (див. вище). Вони
не пов’язані з порушенням вінцевого кро
вообігу. Виникають у результаті того, що
великі дози катехоламінів активують лі
підні та кальцієві механізми ушкодження
клітин (див. главу 7).
Основні клінічні прояви інфаркту міокарда.
Некроз серцевого м’яза клінічно виявляє себе
розвитком таких основних синдромів:
1. Больовий синдром. У його розвитку мають
значення (а) хімічні фактори, що з’явля
ються в тканинах при ушкодженні клітин,
серед них іони Н+, К+, простагландини, лізосомні ферменти, а також (б) зміни ско
рочувальних властивостей ішемізованої
ділянки міокарда, у результаті чого відбу
вається патологічне розтягування (пролабування) стінки серця при його скорочен
ні. Це веде до подразнення механорецепторів серця та розвитку болю.
Патогенетичне значення больового синдро
му в розвитку інфаркту міокарда полягає в тому,
що:
а) біль є потужним чинником ініціації стресу
та активації симпатоадреналової системи.
Великі дози катехоламінів, що вивільня
ються при цьому, сприяють ушкодженню
міокарда (див. вище);
б) сильний біль викликає спочатку збуджен
ня, а потім і перезбудження життєво важ
ливих центрів головного мозку (дихаль
ного, серцево-судинного). Ця обставина
є важливим чинником розвитку кардіогенного шоку (див. далі).
151
2. Гостра серцева недостатність. Розвива
ється при ураженні великих ділянок міо
карда. Може виявляти себе синдромом
серцевої астми і набряку легень (див. гла
ву 33) або кардіогенним шоком.
3. Аритмічний синдром. Можливий розвиток
усіх видів аритмій, про які йшлося у цій
главі. Найнебезпечнішою є поява фібриля
ції шлуночків, що веде до зупинки серця.
4.
Резорбційно-некротичний синдром. Він
є наслідком надходження в кров продуктів
розпаду змертвілої тканини серця. Вияв
ляє себе такими ознаками:
а) гарячкою (див. главу 18);
б) нейтрофільним лейкоцитозом',
в) збільшенням швидкості осідання еритро
цитів (ШОЕ). Ці перші три ознаки є від
дзеркаленням загальної реакції організму,
що настає у відповідь на запалення у при
леглій до інфаркту зоні міокарда;
г) ферментемією - появою в крові ферментів,
що надходять із ушкоджених кардіоміоци
тів (креатинкіназа, аспартатамінотрансфераза, лактатдегідрогеназа І типу та ін.);
д) автоімунним синдромом (синдромом Дреслера). Вважають, що в його основі лежать
конформаційні зміни білків ураженого міо
карда, внаслідок чого вони набувають анти
генних для даного організму властивостей.
Проявом цього синдрому є запаленням се
розних оболонок організму - полісерозит
(перикардит, плеврит, перитоніт).
Безпосередніми причинами смерті хворих
з інфарктом міокарда можуть бути:
1) різке зменшення серцевого виштовху
з розвитком гострої недостатності серця
і системного кровообігу - кардіогенний
шок (див. далі);
2) застій крові в судинах малого кола крово
обігу з розвитком набряку легенів і гострої
дихальної недостатності (див. главу 33);
3) фібриляція шлуночків, що припиняє насос
ну функцію серця і може стати причиною
раптової коронарної смерті;
4) розрив серця та його тампонада. Через
кілька днів після інфаркту в результаті
розсмоктування змертвілих клітин стінка
серця в ділянці некрозу починає все біль
ше і більше розтягуватися під дією тиску
крові в період систоли. Це веде до по-
152
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ґ
9, Перша (первинне падіння артеріального тиску) і друга (компенсаторний спазм артеріол)
фази патогенезу кардіогенного шоку
ступового стоншення і вибухання (анев
ризми) стінки аж до настання її розриву.
Кров через дефект, що утворився, швидко
наповнює порожнину перикарда, і розви
вається тампонада серця. Різке збільшен
ня
внутрішньоперикардіального
тиску
унеможливлює наповнення камер серця
кров’ю під час діастоли. Це, у свою чергу,
веде до падіння серцевого виштовху, гос
трої серцевої недостатності та смерті.
Кардіогенний шок
Шок, що виникає в результаті різкого падіння
нагнітальної (насосної) функції серця, назива
ють кардіогенним.
Розрізняють 4 форми кардіогенного шоку.
1) рефлекторну (больовий шок). Основним
механізмом її розвитку є тривалий біль,
що викликає активацію симпатоадреналової системи, яка переходить у гальму
вання. Це призводить до депресії ско
рочувальної функції серця, брадикардії,
зменшення тонусу периферичних судин
і падіння артеріального тиску;
2)
гіпокінетичну (справжній кардіогенний
шок). Основним фактором її розвитку
є різке зменшення скорочувальної функ
ції серця в результаті ішемічного ушко
дження кардіоміоцитів. Справжній кар
діогенний шок розвивається, коли площа
ураженого міокарда перевищує 40 %;
3) дискінетичну. Виникає в результаті аси
нергії (неузгодженості) скорочень міокар
да. Причиною такої асинергії є грубі ушко
дження серця - аневризми, розрив міжшлуночкової перегородки, відрив хорд клапанів;
4) аритмічну. Є наслідком тяжких аритмій,
зокрема блокад серця.
Патогенез. У патогенезі кардіогенного шоку
розрізняють кілька фаз.
• І фаза - первинне падіння артеріального
тиску (рис. 31.39). Усі патогенетичні фак
тори кардіогенного шоку (рефлекторна
депресія, збільшення площі ушкодженого
ГЛАВА 31. ПАТОФІЗІОЛОГІЯ СЕРЦЯ
опір. Зазначена реакція є компенсаторною
та спрямована на попередження подаль
шого падіння артеріального тиску.
• III фаза — вторинне падіння артеріаль
ного тиску (рис. 31.40). Тривалий спазм
артеріол у периферичних тканинах викли
кає порушення мікроциркуляції та гіпоксію.
Наслідком кисневого голодування є:
а) ацидоз, що викликає депресію скорочу
вальної функції міокарда (блокада каль
цієвих каналів, конкурентне зв’язування
іонів Н+ з тропоніном);
б) розширення артеріол, що виникає в ре
зультаті накопичення в тканинах мета-
IV фаза
III фаза
міокарда, асинергія серцевих скорочень,
аритмії) викликають зменшення серцево
го виштовху. Це, за законами гемодинаміки, призводить до зменшення хвилинного
об’єму серця й падіння артеріального тиску.
• II фаза - компенсаторний спазм артеріол
(рис. 31.39). Характеризується активацією
симпатоадреналової системи, надходжен
ням у кров катехоламінів, вазопресину,
глюкокортикоїдів,
утворенням
ангіотензину II. Вивільнення потужних судино
звужувальних факторів викликає генералізований спазм артеріол, у результаті чого
збільшується
загальний
периферичний
153
40. Третя (вторинне падіння артеріального тиску) і четверта (термінальна) фази патогенезу
кардіогенного шоку
154
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Рис. 31.41. "Зачароване коло" (circulus vitiosus) у патогенезі кардіогенного шоку
болітів-вазодилататорів
(“метаболічний
симпатоліз ”)\
в) надходження у кров із тканин так званих
ішемічних токсинів. Серед них велике па
тогенетичне значення має фактор депре
сії міокарда, що вивільняється з підшлун
кової залози.
Усі зазначені зміни, погіршуючи скорочу
вальну функцію серця й “знімаючи” компенса
торний спазм артеріол, зумовлюють подальше
падіння артеріального тиску.
• IV фаза - термінальні зміни (рис. 31.40).
У результаті істотного падіння артеріаль
ного тиску (нижче 40 мм рт. ст.):
а) ще більше порушується коронарний кро
вообіг і збільшується ішемія міокарда —
зменшення скорочувальної функції міокар
да прогресує, оскільки замикається “зача
роване коло” (рис. 31.41);
б) розвивається гостра ниркова недостат
ність (повністю припиняється клубочкова
фільтрація, виникають анурія, інтоксика
ція);
в) порушується мозковий кровообіг, розвива
ється гіпоксія головного мозку, виникають
розлади функції життєво важливих центрів.
Сукупність зазначених змін стає причиною
смерті.
ГЛАВА 32
Патофізіологія
кровоносних
судин
Відповідно
до
функціональної
класифікації,
кровоносні судини поділяють на кілька груп.
1. Компенсаційні судини, або судини-амортизатори, — аорта й артерії еластичного типу,
їхня функція полягає насамперед у тому,
щоб перетворювати поштовхоподібні ви
киди крові з серця у рівномірну течію кро
ві. Еластичні та колагенові структури цих
судин визначають напругу їхніх стінок,
необхідну для протидії значній розтягувальній дії крові. При цьому важливо те,
що підтримання постійної функціональної
напруги за рахунок зазначених структур не
вимагає витрат енергії.
2. Резистивні судини, або судини опору, — ар
теріоли і венули, розташовані в посткапілярних ділянках судинного русла. Опір
течії крові в зазначених судинах здійсню
ється завдяки їхнім структурним особли
востям (відносно товста стінка, якщо по
рівнювати з величиною просвіту), а також
здатності м’язових структур стінки пере
бувати в стані постійного тонусу й актив
но змінювати величину просвіту під дією
додаткових нейрогуморальних впливів.
Цим забезпечується відповідність просвіту
резистивних судин об’єму крові, що пере
буває в них, а також сталість і адекватність
кровопостачання органів і тканин.
3. Судини обміну - капіляри й венули. На рів
ні цих судин здійснюється двосторонній
обмін між кров’ю і тканинами водою, газа
ми, електролітами, необхідними поживни
ми речовинами та метаболітами.
4. Ємнісні судини, якими є вени, депонують
кров з метою її розподілу і повернення до
серця. Основна маса крові (75-80 %) зосе
реджена саме в цих судинах. Викид крові
з ємнісних судин здійснюється як активним
скороченням м’язових волокон, так і пасив
но-еластичною віддачею.
5. Судини перерозподілу — судини-сфінктери
та артеріовенозні шунти. Регулюють кровонаповнення органів і тканин.
З патологічними змінами в різних типах су
дин пов’язаний розвиток тих чи інших патоло
гічних процесів і хвороб. Так, артеріосклероз
характеризується ураженням артеріальних су
дин еластичного й еластично-м’язового типу.
Тому він є хворобою переважно компенсаційних
судин. Відповідно артеріальну гіпертензію та
гіпотензію відносять до порушень функції ре
зистивних судин, а генералізовані зміни проник
ності судинних стінок - до характерних проявів
патології судин обміну. Порушення ємнісних су
дин виявляють себе розладами центральної гемодинаміки широкого діапазону - від розвитку
артеріальної гіпертензії до виникнення колапсу,
а також специфічними для вен недугами (вари
козним розширенням, тромбофлебітами та ін.).
Термін “артеріосклероз”, уведений в обіг ще на
початку XIX століття, буквально означає затвер
діння артерій. Воно настає в результаті розвитку
сполучної тканини в судинній стінці та характе
ризується втратою артеріями своїх еластичних
властивостей.
Відповідно до рекомендацій експертів Все
світньої організації охорони здоров’я (ВООЗ)
усі склеротичні ураження артерій поділяють на
дві групи:
156
1) власне артеріосклероз, який охоплює такі
форми, як атеросклероз, артеріосклероз
Менкеберга, артеріолосклероз, вікові скле
ротичні зміни артерій;
2) хвороби артерій запальної і запально-алер
гічної природи. До них відносять сифілітич
ний аортит, облітеруючий ендартеріїт, алер
гічні васкуліти, ревматоїдний артеріїт та ін.
Предметом нашого обговорення буде власне
артеріосклероз, оскільки через свою пошире
ність і тяжкі наслідки (ішемічна хвороба серця,
ішемічні інсульти) він є однією з центральних
наукових і клінічних проблем сьогодення. Адже
у майже всіх економічно розвинених країнах
ураження серця та головного мозку, зумовлені
склеротичними змінами артерій, посідають пер
ше місце серед причин смертності населення.
Уражені склерозом артерії вирізняються під
вищеною щільністю й крихкістю. Унаслідок
зниження еластичних властивостей вони не
в змозі адекватно змінювати свій просвіт залеж
но від потреби органа або тканини у кровопоста
чанні. Спочатку функціональна неповноцінність
склеротично змінених судин, а отже, органів
і тканин виявляється тільки при підвищенні до
них вимог, тобто при збільшенні навантаження.
Подальше прогресування артеріосклеротичного
процесу може призвести до зниження праце
здатності й у стані спокою.
Патогенетичну сутність артеріосклерозу скла
дають чотири процеси: інфільтрація, проліфера
ція, дегенерація і склерозування. Різні поєднання
цих процесів у різних судинах визначають “мо
заїчний” характер артеріосклеротичних уражень.
1. Інфільтрація - проникнення із плазми кро
ві в судинну стінку й відкладення в ній лі
підів, складних вуглеводів і білків.
2. Проліферація - розмноження гладких
м’язових клітин артеріальної стінки, у ре
зультаті чого формуються так звані фіброз
ні “бляшки”, що виступають у просвіт ар
терій і порушують течію крові в них.
З .Дегенерація. Цим терміном позначають
ушкодження й загибель клітин судинної
стінки, а також розвиток дистрофічних
змін, у тому числі кальцинозу.
4. Склерозування - посилене утворення спо
лучної тканини, що виявляє себе синтезом
її основної інтерстиціальної речовини й во
локнистих структур.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Найпоширенішими формами артеріосклеро
зу є атеросклероз і артеріосклероз Менкеберга.
Основні відмінності між ними полягають у на
ступному:
1) при атеросклерозі місцем ураження є вну
трішній шар судинної стінки - інтима,
тимчасом як при артеріосклерозі Менке
берга - середня оболонка артерій -медіа',
2) у патогенезі атеросклерозу переважають
процеси інфільтрації та проліферації, на
томість при менкебергівському склерозі дегенеративні зміни і склерозування',
3) атеросклеротичні ураження характеризу
ються відкладанням у судинну стінку пе
реважно ліпідів (холестеролу), тимчасом
як при артеріосклерозі менкебергівського
типу відкладаються солі кальцію - розви
вається кальцифікація артерій (медіакальциноз);
4) атеросклеротичний процес через утворен
ня “бляшок” веде до стенозування - змен
шення просвіту артерій; основним про
явом артеріосклерозу Менкеберга є змен
шення еластичності судин;
5) унаслідок атеросклерозу розвивається
ішемія органів і тканин, зокрема такі не
безпечні для життя процеси, як інфаркт
міокарда, ішемічний інсульт', тяжким на
слідком менкебергівського склерозу є фор
мування аневризм артерій, розрив яких
майже невідворотно стає причиною смерті.
АТЕРОСКЛЕРОЗ
Спочатку поняття “атеросклероз", запропонова
не Маршаном у 1904 р., використовували для по
значення лише двох типів змін: накопичення лі
підів у вигляді кашкоподібних мас у внутрішній
оболонці артерій (від грецьк. athere - каша) і влас
не склерозу - сполучнотканинного ущільнення
стінки артерій (від грецьк. scleros - твердий).
Сучасне тлумачення атеросклерозу набагато
ширше. За визначенням експертів ВООЗ, ате
росклероз - це різні поєднання змін інтими арте
рій, що виявляються у вигляді осередкового від
кладення ліпідів, складних сполук вуглеводів,
елементів крові та циркулюючих у ній продук
тів, утворення сполучної тканини і відкладення
кальцію.
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Морфогенез. У розвитку атеросклеротичного
процесу виділяють кілька послідовних стадій
(рис. 32.1).
• І стадія ™ доліпідні зміни в стінці артері
альних судин. Суть їх полягає у змінах
функціонування ендотелію - у розвитку
так званої ендотеліальної дисфункції (див.
157
нижче). Морфологічно судини ще не мають
жодних особливостей, ліпіди в їхніх стін
ках не виявляються, звідси й походить на
зва цієї стадії процесу.
II стадія — поява в інтимі артерій ліпідних
плям і смужок. Причиною цього є інфіль
трація судинної стінки плазмовими ліпо-
Морфогенез атеросклеротичних уражень і участь різних клітин у їхньому формуванні
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
158
А.Нормальна судина
Б. Початкові зміни
В. Середній ступінь
Г. Критичний стеноз
Рис. 32.2. Вплив атеросклеротичної бляшки на просвіт артеріальної судини
протеїнами - процес, що його називають
ліпоїдозом. На цій стадії важливу патогене
тичну роль відіграють макрофаги.
• III стадія - формування фібро-ліпідної
бляшки, для позначення якої використову
ють ще терміни атеросклеротична бляшка,
або атерома. Розвиток атероми відбуваєть
ся за участю гладких м’язових клітин арте
ріальної стінки.
• IV стадія - ускладнена бляшка. Дегене
ративні зміни, які відбуваються в атеро
склеротичній бляшці, ведуть до розриву
її фіброзно-м’язової покришки, що за
пускає процеси тромбоутворення з част
ковим або повним перекриттям просвіту
артерій.
Слід зазначити, що просвіт артерій при ате
росклерозі довгий час залишається незмінним,
незважаючи на прогресування цього процесу
(рис. 32.2). Річ у тім, що спочатку ріст бляш
ки відбувається не всередину просвіту судини,
а назовні, у бік адвентиції. Лише після того,
як можливості такого росту будуть вичерпані,
атеросклеротична бляшка, далі збільшуючись
у розмірі, починає зменшувати просвіт суди
ни — розвивається поступове її стенозування.
Кількість крові, що проходить через такі артерії,
зменшується, а органи і тканини, що отримують
кровопостачання від них, зазнають ішемії.
При повільному стенозуванні артерій в ор
ганах з порушеним живленням розвивають
ся атрофічні зміни з поступовим заміщенням
функціонально активної паренхіми сполучною
тканиною. Швидке звуження або повне пере
криття просвіту артерій тромбом часто веде до
змертвіння ділянки органа з порушеним крово
обігом, тобто до інфаркту. Інфаркт міокарда та
ішемічний інсульт є дуже частими й небезпеч
ними ускладненнями атеросклерозу вінцевих
і мозкових артерій.
Фактори ризику. Факторами ризику ате
росклерозу називають сукупність внутрішніх
і зовнішніх умов, які набагато підвищують імо
вірність розвитку цієї хвороби в людини.
Із середини минулого століття і донині три
ває без перебільшення видатне епідеміологічне
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
159
Рис. 32.3. Фактори ризику атеросклерозу
дослідження, відоме як фремінгемське. У ньому
бере участь вже третє покоління усіх мешкан
ців міста Фремінгем (США, Масачусетс). Більш
як півстолітнє спостереження за учасниками
дослідження, станом їхнього здоров’я, трибом
життя та ін. дало підстави вченим виділити ці
лий ряд як основних, так і менш важливих фак
торів ризику атеросклерозу та його ускладнень
(рис. 32.3).
Основні фактори ризику поділяють на дві
категорії. Перша ®Нце ті, на які не можна впли
нути: вік, стать, спадкова схильність. Друга ка
тегорія охоплює чинники, на які можна впли
вати: гіперліпідемія, артеріальна гіпертензія,
цукровий діабет, куріння. Докладніше розгля
немо їх.
1. Вік. Різке збільшення частоти і тяжко
сті атеросклеротичних уражень судин
у зв’язку з віком, особливо помітне після ЗО
років, стало підставою для того, щоб деякі
дослідники вважали атеросклероз функці
єю віку і винятково біологічною пробле
мою (І. Давидовський). Більшість учених,
однак, дотримуються думки, що вікові
й атеросклеротичні зміни судин — це різні
форми артеріосклерозу, особливо на пізніх
стадіях їхнього розвитку. При цьому вікові
зміни судин сприяють розвитку атероскле
ротичних уражень.
2. Стать. У віці від 40 до 70 років на ате
росклероз та інфаркт міокарда атероскле
ротичної природи чоловіки хворіють час
тіше, ніж жінки (у середньому в 3—4 рази).
Після 70 років захворюваність на цю не
дугу серед чоловіків і жінок приблизно
однакова. Зазначені відмінності, мабуть,
пов’язані, з одного боку, з нижчим вихід
ним рівнем холестеролу і тим, що у жінок
він міститься в плазмі крові в основному
у фракції неатерогенних ліпопротеїнів ви
сокої густини (ЛПВГ), а з другого «з анти
склеротичною дією жіночих статевих гор
монів - естрогенів.
3. Спадковість. Роль спадкового фактора
у виникненні атеросклерозу підтверджують
статистичні дані про високу частоту іше
мічної хвороби серця в окремих родинах,
а також у однояйцевих близнюків. Мова йде
про спадкові форми гіперліпопротеїнемії
і спадково обумовлені дефекти метаболіз
му артеріальної стінки. З відкриттям яви
ща однонуклеотидного поліморфізму генів
(див. главу 6) широко вивчається вплив
цього генетичного чинника на різні пато
генетичні механізми атерогенезу й тяжкі
ускладнення атеросклерозу (інфаркт міо
карда, ішемічний інсульт). Вважають, що
певні види і поєднання поліморфізму ге
160
нів-кандидатів складають основу спадкової
схильності до атеросклеротичного процесу.
4. Гіперліпідемія. Доведено, що збільшення
вмісту в плазмі крові т. зв. атерогенних лі
попротеїнів, що містять великі кількості
холестеролу, особливо, коли це відбуваєть
ся на тлі зменшення концентрації антиатерогенних ліпопротеїнів, є важливим факто
ром ризику атеросклерозу. Докладна мова
про роль ліпідів в атерогенезі піде далі. Тут
тільки зазначимо, що основною причиною
гіперліпідемії в економічно розвинених
країнах світу є надмірне споживання жирів,
що входять до складу харчових продук
тів. Так, досвід країн з високим життєвим
рівнем (США, Швеція, Чехія та ін.) пере
конливо доводить таку закономірність: що
більше потреба в енергії задовольняється
за рахунок тваринних жирів і продуктів,
які містять холестерол, то вищий вміст хо
лестеролу в крові й відсоток захворюванос
ті на атеросклероз. Навпаки, у країнах, де
на частку жирів тваринного походження
припадає незначна частина енергетичної
цінності добового раціону (близько 10 %),
захворюваність на атеросклероз низька
(Японія, Китай).
5. Артеріальна гіпертензія. Підвищений ар
теріальний тиск, особливо коли він пере
вищує 160/90 мм рт. ст., набуває значення
фактора ризику атеросклерозу, якщо діє
в комбінації з іншими. Так, при однако
вому рівні холестеролу захворюваність на
інфаркт міокарда при гіпертензії в п’ять
разів вища, ніж при нормальному артері
альному тиску. В експерименті на кролях,
у їжу яких додають холестерол, атероскле
ротичні зміни розвиваються швидше і до
сягають більшого ступеня на тлі артеріаль
ної гіпертензії.
6. Цукровий діабет. Атеросклероз є найпо
ширенішим варіантом діабетичної макроангіопатії (див. главу 24). 75-85 % хворих
на діабет мають атеросклероз і вмирають
від його ускладнень. З другого боку, у 4/5
хворих на атеросклероз виявляють знижен
ня толерантності до глюкози, а 1/3 з них пе
ребуває в предіабетичному стані.
7. Куріння. Куріння цигарок, особливо коли
воно поєднується з іншими факторами ри
зику (гіперліпідемією, артеріальною гіпер
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
тензією, діабетом), значно збільшує час
тоту й вираженість атеросклерозу, сприяє
розвиткові його ускладнень.
Існує ще цілий ряд чинників, стосовно яких
є дані щодо їхнього зв’язку з атеросклерозом.
Проте остаточної відповіді на питання про їхнє
значення в атерогенезі поки що не отримано.
До факторів, що можуть мати стосунок до роз
витку атеросклерозу, відносять також:
ожиріння. Очевидно, що не саме по собі
збільшення маси тіла має значення для ате
росклеротичного процесу, а тісно пов’язані
З ожирінням порушення ліпідного і ліпопротеїнового складу плазми крові, цукро
вий діабет, артеріальна гіпертензія;
гіподинамію. Малорухливий спосіб життя,
різке зменшення фізичного навантаження
(гіподинамія) є ще одним претендентом
на фактори ризику атеросклерозу. Про
це, зокрема, свідчать менша захворюва
ність на атеросклероз серед працівників
фізичної праці і більша — в осіб, робота
яких пов’язана з розумовою діяльністю;
швидша нормалізація рівня холестеролу
в плазмі крові - після надмірного його над
ходження ззовні — під дією фізичних на
вантажень;
» стрес. Є спостереження, які свідчать про
те, що захворюваність на атеросклероз
вища серед людей *стресових професій”,
тобто професій, що вимагають тривалої
і сильної нервової напруги (лікарі, учителі,
викладачі, працівники управлінського апа
рату, льотчики та ін.). У цілому захворюва
ність на атеросклероз вища серед міського
населення, якщо порівнювати з сільським.
Це може пояснюватися тим, що в умовах
великого міста людина частіше зазнає нейрогенних стресових впливів;
* гормональні порушення, хвороби обміну ре
човин, інтоксикації, інфекції. Якщо не бра
ти до уваги вже згадуваний цукровий діабет,
то розвиткові атеросклерозу сприяють гіпо
функція щитоподібної залози (мікседема)
і дефіцит естрогенів. Серед розладів обмі
ну речовин, окрім порушень обміну ліпідів
і ліпопротеїнів, певне значення для атерогенезу можуть мати подагра і гомоцистеїнемія - накопичення в організмі амінокислоти
гомоцистеїну, що є проміжним продуктом
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
обміну метіоніну й цистеїну. Встановлено,
що гомоцистеїн, діючи на ендотелій крово
носних судин, викликає його ушкодження,
унаслідок чого різко зростає надходження
в інтиму артерій холестеролу, ліпопротеїнів
та інших компонентів крові. Тривала дія ал
коголю, нікотину, інтоксикація бактеріаль
ного походження та інтоксикація, виклика
на різними хімічними сполуками (фториди,
CO, H2S, свинець, бензол, сполуки ртуті),
також сприяють атеросклеротичному про
цесу. Більшість наведених тут інтоксикацій
супроводжується не тільки загальними по
рушеннями жирового обміну, властивими
атеросклерозу, але й типовими дистрофіч
ними та інфільтративно-проліферативними
змінами в артеріальній стінці.
Патогенез. Відомі нині теорії патогенезу ате
росклерозу можна звести до двох, принципово
різних концепцій, що відрізняються між собою
відповіддю на питання: що первинне, а що вто
ринне при атеросклерозі, інакше кажучи, що
є причиною, а що наслідком - загальні порушен
ня ліпідного обміну в організмі, що зумовлюють
ліпоїдоз внутрішньої оболонки артерій {ліпідна
концепція), чи місцеві зміни судинної стінки, без
яких неможливий подальший перебіг подій (су
динна концепція).
І. Судинна концепція. Відповідно до уявлень
Р. Вірхова та його послідовників, при ате
росклерозі спочатку розвиваються дистро
фічні зміни внутрішньої оболонки стінки
артерій, а відкладення ліпідів і солей каль
цію - явище вторинного порядку. Перева
гою даної концепції є те, що вона спромож
на пояснити розвиток спонтанного й експе
риментального атеросклерозу як у тих ви
падках, коли є порушення ліпідного обміну,
так і в тих (що особливо важливо), коли їх
немає. Першорядну роль автори цієї кон
цепції відводять артеріальній стінці, тобто
субстрату, який безпосередньо втягується
в патологічний процес.
її.Ліпідна концепція. Від того часу як
М. Анічков і. С. Халатов провели свої пер
ші експерименти (див. нижче), успішно
розвиваються погляди на атеросклероз як
процес, який виникає внаслідок загальних
метаболічних порушень в організмі, що
супроводжуються гіперхолестеролемією
161
та гіперліпопротеїнемією. З цих позицій
атеросклероз є результатом первинної ди
фузної інфільтрації ліпідів, зокрема холес
теролу, у незмінену внутрішню оболонку
артерій. Подальші зміни в судинній стін
ці, що ведуть до формування атероскле
ротичної бляшки, розвиваються у зв’язку
з відкладенням у ній ліпідів, тобто є вто
ринними.
Роль ліпідів в спорогенезі
Вчення про роль загальних порушень ліпідно
го обміну в патогенезі атеросклерозу пройшло
кілька історичних етапів.
Перший етап - холестероловий — бере свій
початок від класичних експериментів М. Анічкова і С. Халатова, які у 1912 році вперше
відтворили атеросклероз у кролів уведенням
всередину холестеролу (через зонд або домі
шуючи його до звичайного корму). Виражена
ліпідна інфільтрація артерій розвивалася через
4-6 місяців при щоденному використанні 0,250,5 г холестеролу на 1 кг маси тіла. При цьому
істотно підвищувався рівень холестеролу в си
роватці крові (у 3-5 разів, якщо порівнювати
з вихідними значеннями), що стало підставою
для припущення про провідну патогенетичну
роль у розвитку атеросклерозу гіперхолестеролемії.
У людини принципово можливими механіз
мами підвищення рівня холестеролу в крові є
такі:
1) надмірне надходження холестеролу в ор
ганізм у складі їжі (аліментарна гіперхолестеролемія);
2) надто посилений синтез цієї сполуки в са
мому організмі;
3) порушення виведення холестеролу з орга
нізму в складі жовчі;
4) зменшення використання холестеролу пе
риферичними клітинами.
Отже, на першому етапі розвитку ліпідної
(плазмової) теорії атеросклерозу вона зводила
ся до положення “без холестеролу немає атеро
склерозу'’.
З’ясування ролі холестеролу в патогенезі ате
росклерозу спричинилося до того, що цю речо
вину в популярній і навіть науковій літературі
стали називати “головним убивцею XX століт
162
тя”. У багатьох лікарів холестерол і досі асоцію
ється з його патогенною дією в організмі й зов
сім поза увагою залишається його вкрай важли
ва фізіологічна й біохімічна роль.
Про важливе значення холестеролу для орга
нізму свідчать такі факти:
щодня з їжею в організм людини надходить
300-500 мг холестеролу;
в організмі синтезується (в основному в пе
чінці) ще 700-1000 мг холестеролу щодо
би;
• кожна клітина організму, за невеликим
винятком, має власні системи синтезу хо
лестеролу, здатні забезпечувати потреби
клітин у цій речовині без надходження її
ззовні.
Така висока надійність забезпечення організ
му холестеролом пояснюється його важливими
функціями. Серед них:
1) мембранна функція. Холестерол є важли
вим компонентом плазматичних мембран
усіх клітин. Його наявність у мембранах
забезпечує такі їхні властивості: (а) меха
нічну міцність; (б) рідинний стан ліпідів
мембрани; (в) проникність мембран для
іонів і метаболітів; (г) електроізоляційні
властивості; (д) активність деяких мемб
ранних ферментів; (е) здатність мембран
до злиття;
2) метаболічні функції. Холестерол є поперед
ником цілого ряду сполук, що виконують
важливі функції в організмі. Серед цих ре
човин:
• жовчні кислоти. На утворення їх викорис
товується 600 мг холестеролу щодоби;
• стероїдні гормони (глюко- і мінералокортикоїди, чоловічі й жіночі статеві гормо
ни). їхній синтез вимагає близько 40 мг
холестеролу щодоби;
вітамін D3. Синтезується в шкірі під дією
ультрафіолетового випромінювання з 7-дегідрохолестеролу.
Другий етап розвитку ліпідної (плазмової)
концепції атеросклерозу отримав назву ліпопротеїнового. Після того, як стало відомо, що
транспорт ліпідів, у тому числі й холестеролу,
здійснюється у складі ліпопротеїнів, виявилося,
що для розвитку атеросклерозу має значення не
стільки гіперхолестеролемія, скільки кількісні
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
зміни ліпопротеїнів плазми крові. Основою кон
цепції стало положення: “без атерогенних ліпо
протеїнів немає атеросклерозу”.
У главі 25 ми вже розглядали характеристи
ку окремих класів ліпопротеїнів плазми крові,
спадково зумовлені та набуті порушення їх
нього обміну. Тут тільки зазначимо, що розви
ток атеросклерозу пов’язаний зі збільшенням
у крові рівня багатих на холестерол ліпопротеї
нів, а саме ліпопротеїнів низької (ЛПНГ) і дуже
низької (ЛПДНГ) густини.
Зростання плазмової концентрації ліпопротеїнових міцел цих типів може бути зумовлено
як (а) збільшенням їх утворення, як правило,
при великому надходженні в організм холес
теролу і тригліцеридів (аліментарна гіперліпопротеїнемія), так і (б) порушенням їх вилучен
ня з крові й утилізації клітинами периферич
них тканин.
Перший механізм пов’язаний з переїданням,
надмірним вмістом жирів у раціоні людини,
а в експерименті відтворюється введенням тва
ринам холестеролу (класична модель М. Анічкова і С. Халатова) або тригліцеридів.
Другий механізм є провідним у розви
тку
спадкового
атеросклерозу,
зокрема
пов’язаного із сімейною гіперхолестеролемією
(типом ІІа гіперліпопротеїнемій за класифіка
цією ВООЗ, див. главу 25). В основі цієї не
дуги лежить збільшення в крові концентрації
ЛПНГ, зумовлене спадковими порушеннями
рецепторного апарату клітин, що забезпечує
надходження й утилізацію ними зовнішнього
холестеролу.
Розглянемо докладніше ці механізми.
І.
Зменшення використання ЛПНГ кліти
нами. У другій половині XX ст. лауреа
ти Нобелівської премії Дж. Гольдштейн
і М. Браун відкрили рецепторний механізм
надходження ліпопротеїнів плазми крові
в периферичні клітини. Було показано, що
в організмі існує два механізми транспорту
ЛПНГ в клітини: рецептор-опосередкований (специфічний) і неспецифічний.
Основні відмінності цих механізмів пред
ставлено в таблиці.
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
163
4)
Пов'язаний зі специ
Не пов'язаний зі специфічними рецепторами | фічними рецепторами
доЛПНГ
до ЛПНГ, є різновидом
і піноцитозу
Є у всіх клітинах
Інтенсивність його
регулюється кількістю
рецепторів
Є тільки в клітинах
макрофагального ряду
Інтенсивність його
і не регулюється
Мета: задоволення
потреби клітин
ухолестеролі
Мета: очищення крові
від надлишку ліпопротеїнів (гомеостатична
функція)
Не є причиною нако
пичення холестеролу
в клітинах
Є причиною накопи
чення холестеролу
в клітинах
Суть специфічного транспорту ЛПНГ в клі
тини представлено на рис. 32.4. У ньому можна
виділити кілька послідовних подій:
1) взаємодія міцел ЛПНГ зі специфічними
рецепторами плазматичної мембрани за
допомогою апопротеїну АроВ-100. Утво
рені комплекси завдяки процесам лате
ральної дифузії зосереджуються в певних
ділянках плазматичної мембрани, покри
тих ізсередини білком клатрином, - цей
процес отримав назву кластеризації;
2) після утворення кластерів починається
процес ендоцитозу комплексів “ЛПНГ рецептор”: спочатку в плазматичній мемб
рані виникають покриті клатрином по
глиблення, з яких формуються покриті
везикули. Ці везикули при злитті з лізосомами віддають останнім свій вміст, після
чого поновлюють свою структуру, повер
таються назад до плазматичної мембрани
і вмонтовуються в неї (т. зв. рециркуляція
везикул). Завдяки цьому процесу специ
фічні рецептори знову стають доступними
і можуть зв’язуватися з наступними міце
лами ЛПНГ;
3) гідроліз компонентів ЛПНГ в лізосомах із
вивільненням амінокислот (розщеплення
апопротеїнів) і холестеролу з його ефірів;
використання холестеролу на утворення
клітинних мембран, синтез стероїдних
гормонів (клітини надниркових і статевих
залоз) і жовчних кислот (гепатоцити).
Якщо ж холестеролу виявляється забагато
для цих потреб, то клітина починає захищати
себе від нього. У таких умовах одночасно спра
цьовують три механізми:
• пригнічується синтез рецепторів до ЛПНГ,
унаслідок чого зменшується надходження
ЛПНГ, а отже і холестеролу, ззовні;
• інгібується синтез власного холестеролу
з ацетил-КоА через вплив на ключовий
фермент цього процесу - З-гідрокси-Зметилглютарил-КоА-редуктазу;
• посилюється зв’язування вільного холесте
ролу через утворення його ефірів із жиро
вими кислотами.
Дж. Гольдштейн і М. Браун показали, що по
рушення рецепторного апарату клітин можуть
бути причиною розвитку спадкового атероскле
розу {рецепторна теорія). Було встановлено,
що у хворих зі спадковою гіперліпопротеїнемією Па типу має місце генетично обумовлений
дефіцит специфічних рецепторів до ЛПНГ —
у гетерозигот їх менше, ніж у нормі, а у гомо
зигот вони взагалі відсутні. Внаслідок цього
ЛПНГ не може надходити в периферичні кліти
ни (останні забезпечують ним самі себе завдяки
власним системам синтезу), їхній вміст у крові
збільшується.
З поданої на рис. 32.5 схеми випливає, що де
фіцит рецепторів до ЛПНГ може мати у своїй
основі генетично зумовлені порушення таких
процесів:
1) синтезу рецепторів в рибосомах
ендоплазматичного ретикулуму;
2) їх транспорту до структур апарату Ґольджі;
3) вмонтовування рецепторів у плазматичну
мембрану;
4) утворення кластерів на поверхні
плазматичної мембрани;
5) рециркуляції везикул, тобто повторного
використання рецепторів, що містяться
в їхніх мембранах.
Недостатність специфічних рецепторів до
ЛПНГ і зростання внаслідок цього рівня остан
ніх у плазмі крові стає причиною активації
164
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Протеїн
Ефіри холестеролу
АроВ-100
Рецептор до ЛПНГ
Рецептор
Ендоплазматичний
ретикулум
Рецепторний
білок
4. Специфічний транспорт ЛПНГ у клітини та його регуляція (за Дж. Гольдштейном і М. Брауном).
АроВ-100 - апопротеїн, ГМГ-КоА-редуктаза - З-гідрокси-З-метилглютарил-КоА-редуктаза
неспецифічного механізму транспорту ЛПНГ
у макрофагах. Цей механізм є центральним
у розвитку атеросклеротичних змін в судинах,
і про нього мова піде далі.
II.Посилене утворення ЛПНГ. Безпосеред
ньою причиною посиленого утворен
ня ЛПНГ є збільшення вмісту в плазмі
крові ліпопротеїнів проміжної густини
(ЛППГ) - перехідної форми між ЛПДНГ
і ЛПНГ (рис. 32.6).
ЛППГ утворюються в капілярах жирової тка
нини і м’язів в результаті вилучення більшої час
тини тригліцеридів із міцел ЛПДНГ (тут відбу
вається гідролітичне розщеплення тригліцеридів
ліпопротеїновою ліпазою ендотеліальних клітин).
Далі одна частина ЛППГ захоплюється гепатоцитами завдяки взаємодії рецепторів до ЛПНГ
і апопротеїну В-100. Слід зазначити, що для та
кої взаємодії конче необхідна присутність ще од
ного апопротеїну - АроЕ: без нього ЛППГ не мо
жуть бути захоплені печінковими клітинами.
165
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Основні причини спадково зумовленого дефіциту рецепторів ДО ЛПНГ. Пояснення див. у тексті
Друга частина ЛППГ перетворюється в крові
в ЛПНГ. Таке перетворення передбачає подаль
ше зменшення вмісту в міцелах тригліцеридів
і втрату апопротеїну АроЕ. Беручи це до уваги,
можна виділити дві причини збільшення вмісту
в крові ЛППГ, а отже, і ЛПНГ, що з них утворю
ються.
1. Посилене утворення ЛППГ внаслідок
збільшення вмісту в крові ЛПДНГ. Най
частішою причиною цього є аліментарний
фактор - надмірне споживання жирів. При
великому надходженні тригліцеридів в ор
ганізм печінка через утворення ЛПДНГ
“переганяє” їх у жирову тканину, де вони
відкладаються. Так, з одного боку, попе
реджається жирова інфільтрація печінки,
а з другого - жири знаходять природне міс
це для свого депонування (див. главу 25).
2. Зменшення вилучення ЛППГ із крові гепатоцитами. Цей механізм найчастіше має
у своїй основі генетичні порушення, що
доводиться цілим рядом експерименталь
них моделей. Сьогодні встановлено такі
можливі спадково зумовлені дефекти:
а) дефіцит рецепторів до ЛПНГ. При сімей
ній гіперхолестеролемії, яку ми вже розгля
дали, гетерозиготи мають тільки половину,
а гомозиготи взагалі не мають таких рецеп
торів не тільки в периферичних клітинах, а
й у печінці. Це означає, що гепатоцити не
можуть вилучати з крові ні ЛПНГ, ні ЛППГ,
а отже, вміст як одних, так і других у крові
зростає.
Японські вчені вивели породу кролів
Watanabe, у яких розвивається атеросклероз без
введення ззовні холестеролу. Причина хворо
би, як з’ясувалося, у відсутності рецепторів до
ЛПНГ.
Застосування методики “генетичного нокау
ту” (див. главу 12) дозволило вивести лінію ми
шей (LDL-receptor -/- mice), у яких немає гену,
що кодує рецептор до ЛПНГ. У таких “нокауто
ваних” мишей розвиваються атеросклеротичні
зміни в судинах.
Сучасна клінічна практика свідчить про те,
що трансплантація печінки пацієнтам-гомозиготам із сімейною гіперхолестеролемією веде до
швидкої нормалізації ліпопротеїнового складу
крові, оскільки гепатоцити трансплантата, маю
чи рецептори до ЛПНГ, успішно вилучають із
крові ЛПНГ і ЛППГ;
166
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
клітинами
Рис. 32.6 Роль печінки в очищенні крові від ліпопротеїнів проміжної густини (Л ППГ) і низької (Л ПНГ)
б)
відсутність чи якісні зміни апопротеїну
В-100. Генетичні дефекти цього білка уне
можливлюють захоплення ЛПНГ і ЛППГ
печінковими клітинами. Водночас з цієї
ж причини ЛПНГ не можуть взаємодіяти
з відповідними рецепторами периферич
них клітин.
У мишей, позбавлених гена В-100 (В-100 -/mice), в артеріях розвиваються зміни, характер
ні для атеросклерозу;
в) відсутність чи якісні зміни апопротеїну
АроЕ. Як зазначалося вище, дефіцит цьо
го білка унеможливлює взаємодію ЛППГ
з рецепторами гепатоцитів. Як наслідок,
зменшується чи не відбувається зовсім
захоплення ЛППГ печінкою, а отже, зрос
тає частка цих міцел, що перетворюють
ся в ЛПНГ. Виведено чисту лінію мишей,
у яких відсутній ген АроЕ (АроЕ -/- mice).
У “нокаутованих” тварин з таким дефек
том розвивається атеросклероз без будьяких зовнішніх додаткових впливів, а вве
дення холестеролу значно посилює цей
процес.
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Нині вчення про роль загальних порушень лі
підного обміну в патогенезі атеросклерозу про
ходить третій етап свого розвитку. Суть його
полягає в тому, що головним фактором атерогенезу вважають не стільки гіперхолестеролемію
та гіперліпопротеїнемію, скільки порушення
співвідношення між окремими класами ліпопротеїнів плазми крові й появу якісно змінених
їхніх міцел. У зв’язку з цим виникло поняття
“дисліпопротеїнемія атерогенного характеру”.
За сучасними уявленнями, розвиткові атеро
склерозу сприяє поєднання таких змін ліпопротеїнового складу плазми крові:
1) збільшення вмісту багатих на холестерол
і тригліцериди ЛПНГ і ЛПДНГ. Холесте
рол, що входить до їхнього складу, стали
називати “поганим” холестеролом, а самі
ліпопротеїни - атерогенними\
2) зменшення вмісту ліпопротеїнів високої
густини (ЛПВГ). Ці ліпопротеїни теж міс
тять холестерол, але, на відміну від ЛПНГ,
вони транспортують його не в клітини,
а навпаки, від клітин до печінки. Холесте
рол ЛПВГ позначають як “гарний” холес
терол, а самі ЛПВГ — антиатерогенними
ліпопротеїнами;
3) поява в крові не властивих для норми лі
попротеїнів, що одержали назву модифіко
ваних. До них відносять глікозильовані та
ацетоацетильовані ліпопротеїни (див. гла
ву 24), ліпопротеїни, зв’язані з продуктами
ПОЛ; комплекси “ліпопротеїн - антитіло”,
ліпопротеїн Lp(a) та ін. Усі модифіковані
ліпопротеїни мають високу атерогенну
здатність.
Серед якісно змінених ліпопротеїнів особли
ву увагу привертають до себе окиснені ЛПНГ
(окЛПНГ). Вважають, що саме вони, потрапив
ши чи утворившись в інтимі артерій, запуска
ють усі наступні процеси, характерні для атерогенезу.
Причиною окисної модифікації ЛПНГ як у су
динах, так і в крові, є вільні радикали і пероксиди, що з’являються у великій кількості в умовах
оксидаційного стресу (див. главу 7). У судинній
стінці такими є активні форми кисню ("02", ОН',
О,,, Н202) і азоту (N0‘, 00N0 ). їх утворення
пов’язують з діяльністю цілого ряду ферментів
судин і макрофагів, що емігрували сюди із крові.
Серед таких ензимів - НАД(Ф)Н-оксидаза, ні-
167
тритоксидсинтази (eNOS, iNOS), мієлопероксидаза, ксантиноксидаза, ліпоксигеназа та інші.
Поява в інтимі артерій окЛПНГ спричиняєть
ся до змін, які ще стануть предметом нашого об
говорення.
Завершуючи розгляд ліпідної концепції атеро
склерозу, наведемо, як підсумок, основні докази
ролі холестеролу і атерогенних ліпопротеїнів
у розвитку цієї хвороби. Такі докази можна роз
ділити на чотири категорії:
1) морфологічні: в атеросклеротичних бляш
ках завжди міститься холестерол;
2) експериментальні: введення кролям холес
теролу веде до розвитку атеросклеротич
них уражень (модель М. Анічкова і С. Халатова). Існують також моделі генетично
зумовленого атеросклерозу (лінія кролів
Watanabe, генетично “нокаутовані” миші);
3) клінічні: розвиток спадкової (сімейна гіперхолестеролемія) і набутої (цукровий
діабет, гіпотиреоз) гіперхолестеролемії
і гіперліпопротеїнемії супроводжується
атеросклеротичними ураженнями судин та
їхніми ускладненнями (інфарктом міокар
да, ішемічним інсультом);
4) епідеміологічні: існує кореляція між рівнем
холестеролу і ЛПНГ, з одного боку, і час
тотою атеросклерозу та його ускладнень,
з другого. Зменшення рівня цих показни
ків уповільнює прогресування атероскле
розу і може навіть вести до його регресії.
Місцеві механізми атерогенезу
Атеросклеротичний процес у своєму розвитку
проходить чотири основні стадії (див. вище).
Кожна з них має свої механізми і пов’язана з пе
реважною участю тих чи інших клітин судинної
стінки.
І.
Стадія доліпідних змін. Центральна роль
у розвитку первинних порушень, що започатко
вують атерогенез, належить ендотелію крово
носних судин. Численні фактори, що діють на
нього з боку крові, можуть ушкоджувати ендотеліальні клітини і в такий спосіб збільшувати
проникність ендотеліального бар’єру до ком
понентів крові. Ушкодження ендотелію судин
може бути обумовлене:
а) механічними і фізичними факторами (тиск
крові, її турбулентний рух, іонізуюча радіа
ція);
168
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
б) хімічними сполуками (нікотин, гомоцистеїн, великі дози вітаміну D);
в) ендотеліотропними вірусами, токсинами
бактерій (наприклад, веротоксин);
г) імунними факторами (комплекси “антиген антитіло”).
Про роль ушкодження ендотелію в атерогенезі свідчать такі факти:
» в експерименті в місцях руйнування ендо
телію спеціальним катетером розвивається
ліпоїдоз артеріальної стінки;
* у людини ліпоїдоз найчастіше виникає
в тих ділянках артерій, які зазнають дії гемодинамічних факторів, таких як кров’яний
тиск, турбулентна течія крові, удар пульсо
вої хвилі. Це дуга і біфуркація аорти, місця
відходження і розгалуження артерій;
» у хворих на артеріальну гіпертензію значно
зростає ймовірність розвитку атеросклерозу.
Вплив на ендотелій патогенних чинників, за
лежно від їхньої сили й тривалості дії, може
мати один із двох наслідків: (а) загибель ендотеліоцитів з повним оголенням внутрішньої
поверхні судинної стінки (денудація) і (б) по
рушення властивостей та діяльності ендотеліоцитів, що позначають терміном “ендотеліальна
дисфункція”.
Що саме - денудація чи ендотеліальна дис
функція - має більше значення для атерогенезу?
Відповідь на це запитання дали експерименти,
у яких відтворювали атеросклероз введенням
кролям холестеролу, після того як балонним
катетером було знято ендотелій з поверхні од
нієї із загальних сонних артерій. Несподівано
виявилося, що відкладання ліпідів було біль
шим у тих місцях, де вже розпочалася регенера
ція ендотелію, а там, де артеріальна стінка все
ще залишалася денудованою, інтенсивність ліпоїдозу була менш високою, проте вищою, ніж
у другій сонній артерії з інтактним ендотелієм
(рис. 32.7).
Отже, сьогодні вважається, що саме ендоте
ліальна дисфункція є провідним патогенетичним
механізмом, що започатковує подальші події
в інтимі артерій.
Причини ендотеліальної дисфункції можуть
бути різними (рис. 32.8). Серед них:
а) вільні радикали і пероксиди, що утворю
ються у великій кількості в умовах оксидаційного стресу',
б) окиснені ЛПНГ, про які у нас вже була мова;
в) продукти неферментативного глікозилювання білків (AGEs), що утворюються при
цукровому діабеті внаслідок тривалої гі
перглікемії (див. главу 24);
г) характерні для гемодинаміки фізичні фак
тори, зокрема напруга зсуву;
д) інфекційні агенти: бактеріальні токсини,
віруси;
е) шмоцистеїн при його накопиченні в крові.
Ендотеліальна дисфункція виявляє себе змі
нами властивостей і діяльності ендотеліоцитів:
1) підвищується проникність ендотелію до
високомолекулярних компонентів плазми
крові, зокрема ліпопротеїнів;
Вираженість атеросклерозу при денудації та дисфункції ендотелію
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
збільшується адгезивність ендотелію до
лейкоцитів крові та тромбоцитів. Це пов’я
зано з появою на люмінальній поверхні ендотеліоцитів молекул клітинної адгезії-. Рта Е-селектинів, подібних до імуноглобулінів білків (ІСАМ-1, ІСАМ-2, VCAM-1),
здатних взаємодіяти з адгезивними моле
кулами (олігосахаридами, інтегринами)
^лейкоцитів (див. главу 17);
3) посилюється утворення прокоагулянтних
і пригнічується синтез антикоагулянтних
факторів, що порушує баланс між цими
системами гемостазу і створює сприятливі
умови для тромбоутворення (див. главу 16);
4) збільшується синтез великої кількості біо
логічно активних сполук, серед яких цитокіни, фактори росту, вазоактивні ре
човини (див. главу 16).
II. Стадія ліпоїдозу — поява в інтимі артерій
ліпідних плям і смужок. У розвитку цієї ста
дії провідну роль відіграють окиснені ЛПНГ
(окЛПНГ) і макрофаги. Накопичення окЛПНГ
у судинній стінці зумовлюється двома обстави
нами (рис. 32.9):
2)
Причини і прояви ендотеліальної дисфункції
169
1) проникненням їх із плазми крові через по
рушений ендотеліальний бар’єр. У крові
перехід ЛПНГ в окЛПНГ є проявом оксидаційного стресу-,
2) окисненням ЛПНГ в інтимі артерій під
впливом вільних радикалів і пероксидів,
що вивільнюються у тканину активовани
ми макрофагами.
Подальші події зумовлюються впливом
окЛПНГ на всі види клітин, присутні в інти
мі артерій (ендотеліоцити, макрофаги, гладкі
м’язові клітини). Дія на них виявляє себе такими
основними змінами (рис. 32.10):
1. Вплив на ендотелій-.
а) окЛПНГ зумовлюють ушкодження ендотеліоцитів і в такий спосіб роблять ще ви
разнішою ендотеліальну дисфункцію. Ви
никає “зачароване” коло: ендотеліальна
дисфункція —* збільшення проникнення
в інтиму ЛПНГ та окЛПНГ —* посилення
ендотеліальної дисфункції;
б) збільшується експресія генів, що кодують
структуру молекул клітинної адгезії. Уна-
170
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Оксидаційний
Макрофаги
Рис. 32.9. Шляхи накопичення окиснених ЛПНГ (окЛПНГ) в інтимі артерій та їхня дія на клітини судинної
стінки
слідок цього на поверхні ендотеліоцитів
з’являються адгезивні білки (див. вище), що
створює умови для адгезії моноцитів крові;
в) посилюється експресія генів макрофагального
колонієстимулюючого
фактора
(МКСФ) і моноцитарного хемоатрактантного протеїна-1 (МХП-1). Завдяки цьо
му стимулюється утворення й дозрівання
моноцитів у червоному кістковому мозку,
вихід їх у кров, прилипання до поверхні су
дин та направлене переміщення в тканинах
судинної стінки.
2. Вплив на макрофаги. Окиснені ЛПНГ зу
мовлюють активацію таких процесів:
а) хемотаксису — направленого руху клітин
до місця скупчення ліпопротеїнових міцел;
б) екзоцитозу, що забезпечує секреторну дегрануляцію — викид у тканини вільних ра
дикалів, пероксидів, ферментів та інших
сполук, що мають бактерицидну та цито
токсичну дію;
в) ендоцитозу — процесу поглинання і на
ступного перетравлювання окЛПНГ.
3. Вплив на гладкі м’язові клітини (ГМК) ви
являє себе активацією:
а) міграції ГМК із середнього шару судинної
стінки (медії) у внутрішній;
б) проліферації - розмноження ГМК, що вже
перейшли із медії в інтиму;
в) ендоцитозу, подібного до того, що відбува
ється в макрофагах;
г) синтезу позаклітинних компонентів спо
лучної тканини.
Роль макрофагів у розвитку пов’язаних з ліпоїдозом подальших подій є визначальною.
171
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
окЛПНГ
Рис. 32.10. Дія окисненихЛПНГ (окЛПНГ) на клітини судинної стінки, гмк-гладкі м'язові клітини, МКСФмакрофагальний колонієстимулюючий фактор, МХП-1 - моноцитарний хемоатрактантний протеїн-1
Накопичення цих клітин в інтимі артерій від
бувається внаслідок надходження моноцитів із
крові в судинну стінку. При цьому події, у яких
беруть участь макрофаги, мають таку послідов
ність (рис. 32.11):
1) адгезія моноцитів до поверхні ендотелію.
Вона зумовлена зв’язуванням адгезивних
молекул ендотеліоцитів (див. вище) з олігосахаридними хімічними групами та інтегринами лейкоцитів;
2) проходження моноцитів через ендотеліальний бар’єр і переміщення їх в інтимі
(міграція);
3) проліферація моноцитів, що веде до збіль
шення їхньої кількості в тканині;
4) диференціація моноцитів у макрофаги;
5) здійснення активованими макрофагами
екзоцитозу (секреторна дегрануляція) і ендоцитозу (поглинання окЛПНГ);
6) перетворення макрофагів у пінисті кліти
ни внаслідок накопичення великої кількос
ті захоплених окЛПНГ;
7) загибель пінистих клітин з виходом у тка
нинний детрит неперетравлених ліпідів,
серед яких кристали холестеролу - основ
ний компонент некротичного ядра атеро
склеротичної бляшки.
Діяльність макрофагів в осередку атерогенезу пов’язана з дією окЛПНГ на два види клітин
них рецепторів (рис. 32.12):
1. Toll-подібні рецептори. Взаємодія окЛПНГ
з ними веде до активації макрофагів, що
виявляє себе значним посиленням процесів
екзоцитозу. Унаслідок секреторної дегрануляції в інтиму вивільнюються: (а) вільні
радикали й пероксиди; (б) прозапальні цитокіни; (в) хемокіни; (г) протеолітичні фер
менти. Іншими словами, відбуваються події,
характерні для процесу запалення (див. гла
ву 17). Ще одним важливим наслідком акти
вації макрофагів є посилення експресії гена,
що кодує інший різновид макрофагальних
рецепторів - scavenger-рецептори.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
172
Участь моноцитів і макрофагів в атерогенезі
2.
Scavenger-рецептори, або рецептори “при
бирання сміття”. Взаємодія цих рецепторів
з окЛПНГ стимулює процеси ендоцитозу,
завдяки яким відбувається поглинання ліпопротеїнів, накопичення в клітинах холестеролу, перетворення макрофагів у пінис
ті клітини.
III.
Стадія атероми - формування фіброліпідних бляшок. В атеросклеротичній фіброліпідній бляшці виділяють некротичний центр
і фіброзну покришку (рис. 32.13).
Некротичний центр складається із загиблих
клітин, певної кількості ще живих пінистих клі
тин, кристалів холестеролу та відкладень солей
кальцію.
До складу фіброзної покришки входять гладкі
м’язові клітини (ГМК), макрофаги, пінисті клі
тини, позаклітинні компоненти сполучної тка
нини - колаген, еластин, протеоглікани.
Формування фіброзної покришки відбуваєть
ся за участю ГМК, які на цьому етапі стають го
ловними учасниками атерогенезу.
Залучення ГМК артерій до атеросклеро
тичного процесу відбувається під впливом
окЛПНГ і факторів росту, що вивільнюються
ендотеліоцитами, макрофагами і тромбоцитами
(рис. 32.14). Дія зазначених чинників на ГМК
медії веде - через зміни експресії відповідних
генів - до зміни фенотипу цих клітин: вони втра
чають скорочувальні білки (актин і міозин) і на
бувають здатності до (а) міграції, (б) проліфе
рації, (в) ендоцитозу і (г) синтезу компонентів
сполучної тканини. Іншими словами, відбува
ється перетворення типових ГМК скорочуваль
ного типу у модифіковані, здатні переходити із
середнього шару артеріальної стінки в інтиму.
Там вони і долучаються до атеросклеротичного
процесу.
Основна подія, що відбувається в інтимі ар
терій за участю модифікованих ГМК, - це про
ліферація.
Для пояснення механізмів розмноження ГМК
у судинній стінці в процесі формування атеро
склеротичних бляшок було запропоновано ряд
теорій.
1. Теорія факторів росту (Росс). У результа
ті ушкодження ендотелію судинної стінки
відбувається адгезія й агрегація тромбо
цитів, унаслідок чого останні вивільняють
тромбоцитарний фактор росту (ТФР).
Він діє на специфічні рецептори ГМК,
пов’язані з тирозиновою протеїнкіназою.
Наслідком активації останньої є розмно
ження зазначених клітин.
Як доказ цієї теорії наводять той факт, що
у свиней зі спадковою хворобою Віллебранда
(ендотеліальні клітини не утворюють фактора
Віллебранда) не відбувається адгезія і агрега-
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
173
окЛПНГ
Вплив окиснених ЛПНГ на діяльність макрофагів судинної стінки
Схематичне зображення структури атеросклеротичної бляшки (атероми).
ГМК - гладка м'язова клітина
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
174
ис. 32.14. Причини перетворення скорочувального типу гладких м'язових клітин (ГМК) у модифікований.
ТФР - тромбоцитарний фактор росту, ФРФ-2 - фактор росту фібробластів-2, ТФР-а - трансформуючий фактор росту-а
ція тромбоцитів, не вивільняється ТФР, ніколи
не розвиваються спонтанні й індуковані атеро
склеротичні бляшки.
Крім ТФР, стимулом до розмноження ГМК
у бляшці є фактор росту фібробластів (ФРФ)
і трансформуючий фактор росту-a (ТФР-а), які
виділяються ендотеліоцитами та макрофагами.
2. Мембранна теорія (Р. Джексон, А. Готто). Як тільки в клітинах зростає кількість
вільного холестеролу, він перетворюється
в естерифіковану форму. Якщо для цього
виявляється недостатньо жирових кислот
чи відповідних ферментів, то надлишковий
холестерол спрямовується в плазматич
ні мембрани, до складу яких він входить.
У результаті змінюється рідинний стан
мембрани, її основні властивості. Інформа
ція про це — у поки що нез’ясований спо
сіб - надходить у ядро, і клітина починає
ділитися, щоб утилізувати надлишковий
холестерол, “пустивши” його на утворення
мембран нових клітин.
Доказом теорії є те, що in vitro при інкубації
ГМК у середовищі з підвищеним вмістом ЛПНГ
активуються процеси клітинного поділу.
3.
Моноклональна теорія (Бендітт). Відповід
но до цієї теорії, атеросклеротичні бляшки
є по суті доброякісними пухлинами. Під
дією мутагенних факторів (тютюновий
дим, віруси та ін.) відбуваються зміни гено
му у якійсь одній ГМК, а згодом під впли
вом промоторних факторів (артеріальна гі
пертензія, гіперхолестеролемія) ця клітина
починає проліферувати.
Доказом теорії служить той факт, що всі клі
тини атеросклеротичної бляшки походять з од
нієї клітини. Це було встановлено при вивченні
ізоферментного складу ряду ферментів, зокре
ма глюкозо-6-фосфатдегідрогенази (Г-6-ФДГ),
у нормальних і атеросклеротично змінених су
динах деяких популяцій людей.
Існує дві ізоферментні форми Г-6-ФДГ: А і Б.
У жінок негроїдної раси, що є гетерозиготами,
має місце клітинна мозаїка за цим фермен
том: одні клітини містять тільки ізофермент
А, а інші - тільки ізофермент Б (рис. 32.15).
У нормальній стінці аорти така мозаїка є зви
чайним явищем, проте в атеросклеротичних
бляшках ситуація зовсім інша: частина з них
містить ГМК з ізоферментом А і не має ГМК
175
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Рис. 32.15. Моноклональне походження атеросклеротичних бляшок. Г-б-ФДГ - глюкозо-б-фосфатдегідрогеназа
з ізоферментом Б, у решти бляшок все навпа
ки: можна виявити лише ГМК з ізоферментом
Б і немає клітин з ізоферментом А. Усе це має
свідчити про те, що всі ГМК конкретної ате
росклеротичної бляшки мають моноклональне
походження.
IV.
Стадія ускладненої бляшки. Основу де
генеративних змін, характерних для цієї стадії
атерогенезу, складають такі процеси.
1. Загибель клітин, що утворюють фіброз
ну покришку атеросклеротичної бляшки.
Одна з причин цього — ушкоджувальна дія
продуктів, що вивільнюються в тканину ак
тивованими макрофагами. Такими є вільні
радикали і пероксиди, а також цілий ряд
гідролітичних ферментів лізосомного по
ходження. Ушкодження клітин особливо
прогресує в умовах антиоксидантної не
достатності. Про це свідчить бурхливий
розвиток холестеролового атеросклеро
зу у кролів при тривалому перебуванні
їх (протягом 4-6 місяців) на штучній ді
єті, повністю позбавленій антиоксидантів,
а також сезонний характер загострень ате
росклерозу в людини. Навесні, коли у ба
гатьох розвивається стан антиоксидантної
недостатності, такі загострення виникають
частіше.
Загибель клітин механізмами некрозу і апоптозу веде до розширення некротичного центру
бляшки і, коли втрата клітинних елементів стає
вищою за їхнє поповнення шляхом проліфера
ції, фіброзна покришка бляшки починає посту
пово стоншуватися.
Ще один наслідок некрозу клітин полягає
в тому, що в атеросклеротичну бляшку починають
проростати vasa vasorum із медії та адвентиції ар
теріальної стінки (реакція на альтерацію). Через
дію на них тих самих ушкоджувальних факторів
вони можуть розриватися, у результаті чого вини
кають геморагії, які дестабілізують бляшку, тобто
роблять її більш схильною до розриву.
2. Гідроліз позаклітинних компонентів спо
лучної тканини. Причиною цього явища
є вивільнення активованими макрофагами
ферментів, здатних розщеплювати колаген,
еластин, протеоглікани та інші білки поза
клітинного матриксу. Такими ферментами
є (рис. 32.16):
1)
матриксні металопротеїнази - цинкзалежні протеолітичні ферменти;
2) цистеїнові (тіолові) протеїнази — ензи
ми, каталітична активність яких потребує
сульфгідрильної групи цистеїну;
3) еластази — ферменти, здатні гідролізувати
еластин.
Після надходження цих ферментів у поза
клітинний матрикс відбувається їхня актива
ція, яку здійснює плазмін. Слід уважити, що
в кінцевому підсумку активність зазначених
ензимів залежить від балансу між плазміном
та тканинними інгібіторами протеїназ. В ате-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
176
Роль макрофагальних протеолітичних ферментів в атерогенезі
росклеротичній бляшці цей баланс зміщується
в бік плазміну, а отже, відбувається гідролітич
не розщеплення волокнистих структур (колаге
ну, еластину) і компонентів основної аморфної
речовини (протеогліканів) сполучної тканини.
Сам по собі гідроліз цих структур має своїм
наслідком руйнування фіброзної покришки ате
росклеротичної бляшки й веде врешті-решт до
її розриву з усіма наслідками, що з цього випли
вають.
З другого боку, продукти такого гідролізу,
особливо еластинові пептиди, впливають на
всі типи клітин, присутні в бляшці: ендотеліо
цити (викликають ушкодження, ендотеліальну дисфункцію), ГМК (стимулюють міграцію),
моноцити (активують хемотаксис), лімфоцити
(стимулюють поділ), тромбоцити (активують
агрегацію). Цей вплив здійснюється через спеці
альні еластинові рецептори, вмонтовані в плаз
матичну мембрану клітин.
3. Кальцифікація. У некротичному ядрі атеро
склеротичної бляшки з часом починаються
процеси кальцифікації — утворення й ріст
кристалів гідроксіапатиту. На причинах
і механізмах цього явища в судинній стін
ці ми зупинимося згодом, розглядаючи па
тогенез артеріосклерозу Менкеберга (див.
підглаву 32.1.2). Тут лише зазначимо, що іс
нують два протилежні погляди на значення
обвапнення в атерогенезі. На думку одних,
кальцифікація бляшок відіграє захисну роль,
оскільки стабілізує їх, робить стійкіши
ми до ушкодження. Інші ж учені, навпаки,
вважають, що обвапнення є процесом не
сприятливим, воно передує деструктивним
змінам, які лежать в основі розриву бляшки
та наступного тромбоутворення. У кожному
разі дослідження показали, що наявність
осередків обвапнення в атеросклеротично
змінених вінцевих судинах серця є поганою
_____________________
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
прогностичною ознакою і свідчить про ви
соку ймовірність інфаркту міокарда.
4. Тромбоутворення. Описані вище події (за
гибель клітин, геморагії, гідроліз компо
нентів сполучної тканини) ведуть до по
ступового стоншення фіброзної покришки
атеросклеротичної бляшки і врешті до її
розриву. Унаслідок цього порушується ці
лість ендотелію, атероматозні маси вихо
дять у кров, а на місці утвореного дефекту
починається адгезія і агрегація тромбоци
тів (див. главу 16). Як результат, утворю
ється тромб, який частково чи повністю
перекриває просвіт артерії.
На думку деяких дослідників тромбоутворення
є не тільки завершальним акордом атерогенезу, а
й механізмом якщо не формування, то прогресу
вання атеросклеротичної бляшки. Так, прихиль
ники тромбогенної теорії (Рокитанський, Дьюгід) вважали, що утворення тромбів на внутріш
ній поверхні артерій з наступною їхньою органі
зацією є структурною основою розвитку бляшки.
Постійне виявлення в атеросклеротичних бляш
ках людини продуктів крові, зокрема фібрину, ро
бить цю концепцію життєздатною й нині.
Атеросклероз як хронічне запалення. У другій
половині XIX століття видатний німецький па
толог Рудольф Вірхов започаткував теорію ло
кального запалення, яку якщо й згадували про
тягом більш як ста років, то тільки в історично
му аспекті.
Нині стало зрозуміло, наскільки правий був
геніальний Вірхов. Адже всі ті зміни в артері
альній стінці, що їх було описано вище, є по суті
проявами хронічного запалення (див. главу 17).
В інтимі артерій можна простежити всі три ком
поненти, характерні для запального процесу:
(1) альтерацію, (2) ексудацію з еміграцією лей
коцитів і (3) проліферацію (рис. 32.17).
Альтерація, як і при будь-якому запаленні,
передує всім наступним подіям. Вона виникає
вже на першому етапі атерогенезу - стадії доліпідних змін і виявляє себе ушкодженням
ендотелію та розвитком його дисфункції. Далі
процес альтерації — уже вторинної — триває про
тягом усіх стадій атеросклерозу. Він пов’язаний
в основному з діяльністю макрофагів, які стають
джерелом вільних радикалів і пероксидів, цілої
низки ферментів, що завдають ушкодження
177
клітинним і позаклітинним елементам інтими
артерій. У результаті вторинної альтерації фор
мується некротичний центр атеросклеротичної
бляшки, руйнується її фіброзна покришка, на
стають ускладнення (тромбоз).
Ексудація й еміграція лейкоцитів — це проце
си, що складають основу II стадії атерогенезу —
формування ліпідних плям і смужок. Ліпідна ін
фільтрація, тобто перехід ліпопротеїнів із плаз
ми крові в інтиму, є не що інше, як ексудативний
процес, що відбувається завдяки збільшенню
проникності ендотеліального бар’єра. Він су
проводжується еміграцією моноцитів, які, по
трапивши в інтиму, розмножуються і диферен
ціюються в макрофаги. Тут ці клітини після ак
тивації починають свою фагоцитарну діяльність,
спрямовану на очищення тканини від патоген
ного агента, яким виступають окиснені ЛПНГ, а
потім і вільний кристалічний холестерол. Проте
фагоцитоз в осередках атерогенезу не є завер
шеним, оскільки клітини організму не мають
ферментів, здатних розщеплювати холестерол.
Як наслідок, через нездатність до утилізації ця
сполука у вільній і естерифікованій формах на
копичується всередині макрофагів, зумовлюючи
утворення заповнених ліпідами вакуоль - посту
пово макрофаги перетворюються в пінисті клі
тини. У цих клітинах розвиваються необоротні
зміни і вони гинуть, вивільнюючи в тканину неперетравлений холестерол, який кристалізуєть
ся та стає об’єктом фагоцитозу для наступних
макрофагів. Таким чином процес повторюється
багато разів, у нього втягуються все нові й нові
макрофаги, відбувається формування некротич
ного центру атеросклеротичної бляшки.
Оскільки утилізація чи видалення кристаліч
ного холестеролу з інтими артерій неможливі,
то причина активації макрофагів не усувається,
а отже, запалення носить хронічний характер.
Воно стає подібним до хронічного запального
процесу, що відбувається в легенях при накопи
ченні в їхніх тканинах таких не здатних до ути
лізації речовин, як окисли кремнію (силікоз), ву
гільний пил (антракоз) та інші неорганічні спо
луки, що викликають розвиток пневмоконіозу.
Проліферація є основою росту атеросклеро
тичних бляшок. Міграція в інтиму й поділ там
ГМК, очевидно, спрямовані на локалізацію па
тологічного процесу шляхом створення фіброз
ної капсули (щось подібне до пневмоконіозу,
тільки там у цьому процесі беруть участь фібро-
178
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Рис. 32.17. Атеросклеротичний процес як хронічне запалення інтими артерій
бласти). Якщо надходження в інтиму ліпідів та
інших компонентів крові припиняється (це мож
ливе при нормалізації ліпопротеїнового складу
крові), то ріст бляшки припиняється, вона ста
білізується і може навіть зменшуватися в об’ємі
за рахунок розсмоктування різних компонентів
некротичних мас, звісно, крім кристалів холестеролу. У цьому випадку ведуть мову про част
кову регресію атеросклерозу.
Якщо ж дія на судини атерогенних факторів
продовжується, то атеросклеротична бляшка
росте, аж поки не настане розрив її фіброзної
покришки. Тільки після цього накопичений
в некротичному ядрі холестерол має можливість
залишити інтиму артерій, потрапляючи в кров.
На жаль, таке “очищення” судинної стінки від
бувається на тлі тромбоутворення, яке може ста
ти фатальним, якщо проходить в артеріях жит
тєво важливих органів (серця, головного мозку).
32.1.2.
МЕНКЕБЕРГА
У 1903 році (на рік раніше, ніж Маршан увів
в обіг термін “атеросклероз”) німецький пато
лог Менкеберг докладно описав на прикладі
артерій нижніх кінцівок людини, відому ще
з часів Вірхова, форму склерозу артерій, що
отримала згодом назву менкебергівського арте
ріосклерозу.
Цей тип артеріосклерозу характеризується
ураженням середньої оболонки (медії) артерій
еластичного та еластично-м’язового типу й ви
являє себе тріадою морфологічних ознак: медіанекрозом, медіакальцинозом і медіасклерозом.
Причини. Сьогодні виділяють три основні
причини артеріосклерозу Менкеберга.
L Старіння. Біологічний вік людини є чин
ником, від якого залежить розвиток скле
ротичних уражень судин. Дифузне відкла
дання солей кальцію в середній оболонці
артерій можна виявити ще в грудному і ди
тячому віці, а після ЗО років медіакальциноз є постійною морфологічною знахідкою
в аортальній стінці людини. Приблизно
у третини людей віком менше 50 років ви
являють кальцифікацію артерій. У жінок
цей процес починається дещо пізніше, ніж
у чоловіків. При досягненні 70-річного
віку відмінності між жінками та чоловіка
ми зникають. У приблизно 90 % пацієнтів
із серцево-судинними хворобами є ознаки
кальцифікації артерій.
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
2. Цукровий діабет II типу. Практично у всіх
пацієнтів, що мають синдром “діабетичної
стопи”, виявляють кальцифікацію артерій
при вивченні рентгенограм нижніх кінці
вок. Вважають, що менкебергівський ар
теріосклероз є однією з причин розвитку
цього синдрому (див. главу 24).
З .Хронічна ниркова недостатність, особли
во на термінальних стадіях свого розвитку,
коли розвивається уремія (див. главу 36).
Інтенсивність кальцифікації артерій у та
ких хворих сильно зростає після переве
дення їх на сеанси гемодіалізу.
Крім названих вище трьох основних причин,
менкебергівський склероз розвивається при гі
перфункції прищитоподібних залоз, гіперфосфатемії, гіпервітамінозі D, гіповітамінозі К,
синдромі Марфана (спадкова хвороба, зумовле
на дефіцитом глікопротеїну фібриліну-1).
Патогенез. Провідними патогенетичними лан
ками менкебергівського склерозу є (1) ушко
дження гладких м’язових клітин (ГМК) артері
альної стінки і (2) кальцифікація медії.
Механізми
ушкодження
судинної
стінки.
Ушкодження ГМК та їхня загибель можуть мати
у своїй основі такі головні механізми.
1.
Порушення енергетичного обміну. Ви
сновок про роль цього механізму в пато
генезі менкебергівського артеріосклерозу
випливає з експериментальних дослі
джень українських патофізіологів на чолі
з Ю. В. Бицем. Так, було встановлено, що
пригнічення енергетичного обміну артері
альної стінки отрутами, що є інгібіторами
метаболізму (монойодацетат, фторид на
трію, етилмеркурхлорид), викликає розви
ток виражених дистрофічно-склеротичних
уражень артеріальних судин. Водночас
у венозних судинах, стійких до уражень
менкебергівського типу, істотно зменшу
ється, а інколи і повністю зникає висока
резистентність до дії ушкоджувальних
факторів, зокрема таких, як великі дози ві
таміну D та адреналіну.
На підставі одержаних експериментальних
даних було сформульовано основні положення
енергодефіцитної теорії артеріосклерозу Мен-
179
кеберга (Ю. В. Биць, О. В. Атаман). Суть її по
лягає в тому, що багато патогенних факторів (гі
поксія, голодування, дефіцит вітамінів, отрути,
токсини та ін.), діючи на судинну стінку, первин
но порушують процеси енергетичного обміну
в ній. Це викликає обумовлене дефіцитом АТФ
ушкодження клітин і зменшує активну резис
тентність судин до дії інших ушкоджувальних
чинників. Кінцевим результатом є загибель ГМК
з наступною кальцифікацією судинної стінки.
Доказами цієї теорії є не тільки результати
наведених вище експериментів, а й аналіз чис
ленних наукових даних, який дозволив виявити
певну закономірність. Сутність її можна визна
чити таким положенням: що нижча інтенсив
ність енергетичного обміну стінки кровоносних
судин, то вища їхня чутливість до розвитку де
генеративних і склеротичних уражень.
Про це свідчить цілий ряд фактів:
» види тварин з високою інтенсивністю енер
гетичного обміну в стінці артерій (щури,
собаки) характеризуються високою резис
тентністю до артеріосклерозу, і навпаки,
кролі й людина, у яких відзначають низь
кий рівень енергозабезпечення судин, є високосприйнятливими до розвитку склеро
тичних уражень;
* молоді тварини, у яких інтенсивність енер
гетичного обміну судинної стінки значно
вища, ніж у старих, стійкіші до розвитку
артеріосклерозу;
* артерії з нижчим рівнем енергетичного об
міну (грудна й черевна аорти) уражують
ся частіше, якщо порівнювати з артеріями,
в яких енергетичний обмін інтенсивніший
(легенева артерія);
* вени, інтенсивність енергозабезпечення
яких у 4-5 разів вища, ніж великих артерій,
ніколи не уражаються процесом, подібним
до артеріосклерозу.
2. Оксидаційний стрес. Активація вільнорадикального окиснення в судинній стінці
закономірно веде до ушкодження клітин
(див. главу 7). В експериментах установ
лено, що моделювання артеріосклерозу за
допомогою таких агентів, як монойода
цетат, високі дози вітаміну D, адреналіни
супроводжується
вираженою
активаці
єю пероксидного окиснення ліпідів на тлі
зменшення активності систем антиокси
дантного захисту.
180
Розвиток дегенеративних змін, схожих на
ураження менкебергівського типу, відтворюєть
ся також переведенням тварин на дієту, повніс
тю позбавлену антиоксидантів. Перебування
кролів протягом 4-6 місяців на такій дієті веде
до ушкодження ГМК судин та їхньої загибелі
переважно механізмами апоптозу.
3. Перевантаження клітин кальцієм. Каль
цієві механізми ушкодження ГМК відігра
ють важливу роль у розвитку дистрофічних
змін в артеріальній стінці. Про це свідчать
експерименти, в яких доведено, що блокатори кальцієвих каналів (верапаміл, ніфедипін та ін.) попереджають появу прита
манних артеріосклерозу Менкеберга змін
в артеріях тварин, яким вводили великі
дози вітаміну D й адреналіну.
Перевантаження клітин кальцієм може бути
зумовлено (1) збільшеним надходженням його
в цитоплазму з інтерстицію і (2) порушенням
видалення кальцієвих іонів із цитоплазми на
зовні та в місця їх депонування (причини цьо
го - див. главу 7). Накопичення кальцію в цито
плазмі запускає “кальцієву тріаду” ушкодження
клітин (див. главу 7), зумовлює перескорочення
(контрактуру ГМК) та відповідні функціональ
ні ефекти. Крім того, “закачування” іонів каль
цію в мітохондрії та цитоплазматичні везику
ли може створювати там, за наявності великої
кількості фосфатних аніонів, умови для випа
діння в осад солей кальцію та подальшої їхньої
кристалізації - іншими словами, започатковува
ти процеси внутрішньоклітинної кальцифікації.
Механізми кальцифікації судин. Розвиток медіакальцинозу, як основної ознаки артеріоскле
розу Менкеберга, підпорядковується загальним
закономірностям ектопічної кальцифікації, що
були предметом нашого обговорення в главі 28.
Тут ми лише зупинимося на особливостях цього
процесу в артеріях, і то тільки тих, що стосу
ються механізмів його ініціювання. А таких іс
нує декілька.
1. Активна генетично запрограмована кальцифікація. Якщо раніше вважалося, що
кальцифікація судин — це пасивний, деге
неративний, необоротний процес, який ха
рактеризує кінцеві стадії розвитку уражень
артерій, то нині ця точка зору зазнала кри
тичного переосмислення, оскільки здобу
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
то чимало доказів того, що кальцифікація
артеріальної стінки в багатьох випадках
є процесом активним, генетично запрогра
мованим, подібним до мінералізації хрящо
вої та кісткової тканини.
Виділяють три послідовні стадії активної
мінералізації судинної стінки: (1) остеогенна
стимуляція мезенхімних клітин адвентиції (адвентиціальна стадія)', (2) поява кальцифікуючих судинних клітин у медії і (3) власне каль
цифікація через утворення спеціальних везикул
(рис. 32.18).
Причинами остеогенної стимуляції мезен
хімних клітин адвентиції можуть бути (а) запа
лення, (б) оксидаційний стрес і (в) висока гіпер
глікемія.
Запалення, яке розвивається внаслідок ушко
дження клітин та їхнього некрозу, впливає на
процес кальцифікації через: (а) цитокіни (TNF-a,
інтерлейкіни, адипокіни), (б) вільні радикали
і пероксиди, що вивільнюються в тканину ак
тивованими лейкоцитами; (в) новоутворення та
проростання в медію vasa vasorum, до складу
яких входять ендотеліоцити та перицити.
Процес кальцифікації судинної стінки власне
розпочинається з того, що прозапальні цитокіни,
продукти вільнорадикального окиснення і висо
кі концентрації глюкози, діючи на ендотеліальні
клітини й перицити мікросудин адвентиції, сти
мулюють утворення і секрецію ними сигналь
них молекул, серед яких кістковий морфогенетичний протеїн ВМР-2. Він діє на мезенхімні
клітини адвентиції, які у відповідь утворюють
і вивільнюють сигнальні молекули, що входять
до сімейства Wnt.
Wnt-молекули після секреції надходять у vasa
vasorum, а через них у медію, і чинять паракринний ефект на судинні стовбурові клітини - клітини-остеопрогенітори. Ті після такого впливу
диференціюються в кальцифікуючі клітини, по
дібні до остеобластів.
Кальцифікуючі клітини генерують високу ак
тивність лужної фосфатази і утворюють велику
кількість так званих мінералізуючих матриксних везикул, діаметр яких дорівнює приблиз
но 100 нм. Усередині цих везикул відбувається
нуклеація, тобто утворюються перші кристали
оксіапатиту. За умов високої концентрації іонів
Са2+ і Р043' відбувається ріст кристалів, форму
ються спочатку дрібні розпорошені кальцифіка-
181
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Рис. 32.18. Механізм активної генетично запрограмованої кальцифікації судинної стінки.
ГМК - гладка м'язова клітина
ти, а потім, шляхом злиття менших, великі, ма
сивні ділянки обвапнення судин.
2.
Залежна від апоптозу кальцифікація
(рис. 32.19). При хронічному ушкодженні
гладких м’язових клітин судин активують
ся процеси апоптозу (див. главу 7) і, на від
міну від некротичної загибелі клітин, запа
лення не розвивається, а якщо так, то й не
виникають основні стимулятори описаної
вище остеогенетичної програми — прозапальні цитокіни, вільні радикали і перок
сиди. Через це кальцифікація розвивається
дещо іншим шляхом.
Завершальною фазою апоптозу є утворен
ня апоптичних тілець, діаметр яких (250 нм)
у 2,5 рази перевищує діаметр мінералізую
чих матриксних везикул (100 нм). Через від
сутність у медії макрофагів (тут немає vasa
vasorum, якщо в адвентиції не розвивається
запалення) ці тільця не утилізуються шляхом
фагоцитозу, а стають місцем утворення пер
ших кристалів гідроксіапатиту та подальшого
їхнього росту.
3. Порушення організації та обміну еластину
(рис. 32.20). Про можливу роль еластину
в розвитку медіакальцинозу свідчить той
факт, що структурами артеріальної стінки,
з яких власне розпочинається менкебергівський склероз, є еластичні волокна і мем
брани. Особливо це характерно для зумов
леної віком кальцифікації артерій, що дало
182
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Рис. 32.19. Залежна від апоптозу кальцифікація судинної стінки
підстави для введення терміну “віковий
еластокальциноз меді'ї”.
Порушення структури еластину та його де
градація пов’язані головним чином з некро
тичною загибеллю ГМК. Сам собою некроз
спричиняється до вивільнення із загиблих клі
тин протеолітичних ферментів, серед яких
еластаза— фермент, що здійснює гідролітичне
розщеплення еластину. Окрім того, зменшення
кількості життєздатних ГМК веде до зменшен
ня утворення в медії природних антикальциногенних факторів, серед яких білок MGP (див.
далі). Дефіцит цього протеїну робить еластичні
волокна здатними осаджувати на собі фосфати
кальцію й перетворювати їх на кристали гідроксіапатиту.
Другий шлях, через який некроз ГМК впли
ває на стан еластичних волокон і мембран,
пов’язаний з розвитком запалення в адвентиції
артерій. Еміграція моноцитів у медію із vasa
vasorum, що проросли сюди, веде до вивіль
нення ферментів, зокрема матриксних металопротеіназ і еластази. Ці ензими чинять гідро
літичну дію на еластин, розщеплюють його на
еластинові пептиди, що мають широкий спектр
біологічних ефектів (див. підглаву 32.1.1). При
деградації еластичних волокон на їхній поверхні
з’являються хімічні структури, здатні зв’язувати
кальцій та фосфатні іони і в такий спосіб запо
чатковувати кальцифікацію.
4.
Недостатність
природних
інгібіторів
кальцифікації. Загалом обвапнення судин
за будь-яким із патогенетичних варіантів
відбувається тільки тоді, коли порушується
баланс між чинниками, що ініціюють каль
цифікацію, і факторами, що захищають
судинну стінку від відкладання солей каль
цію. До останніх належать неорганічний
пірофосфат (РР.), матриксний білок MGP
{matrix Gla protein), фетуїн, остеопонтин,
остеопротегерин та інші (див. главу 28).
Про роль недостатності природних інгібіто
рів у кальцифікації артерій свідчить розвиток
медіакальцинозу у генетично “нокаутованих”
мишей, зокрема в тих, що не мають гена MGP
(MGP -/- mice). Протягом двох місяців від на
родження такі тварини помирають через крово
течі, спричинені розривом грудної або черевної
183
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Адвентиція
Медіа
Власне кальцифікація - медіакальциноз
Рис. 32.20. Порушення організації та обміну еластину як механізм ініціації медіакальцинозу
аорти. При вивченні кровоносних судин виявля
ють сильно виражену кальцифікацію аорти та її
гілок, аналогічну артеріосклерозу Менкеберга.
Антикальциногенна активність MGP зникає,
якщо порушується залежний від вітаміну К про
цес у-карбоксилювання цього білка (див. главу
28). Застосування в експерименті варфарину антикоагулянту, що є антагоністом вітаміну К,
позбавляє MGP його інгібіторних властивостей
і веде до кальцифікації медії артерій.
5. Загальні порушення фосфорно-кальцієвого
обміну. Розвиткові кальцифікації артерій
сприяє збільшення концентрації в крові та
в інтерстиції іонів кальцію (гіперкальціємія) та фосфатів (гіперфосфатемія). Мож
ливі причини цих порушень обговорюва
лися в главі 28.
При гіперкальціємії та гіперфосфатемії ство
рюються фізично-хімічні умови, сприятливі для
осадження фосфатних солей кальцію і утворен
ня кристалів гідроксіапатиту у судинній стінці.
Крім того, гіперкальціємія збільшує кальцієве
перевантаження клітин - один з механізмів їх
ушкодження і загибелі (див. вище), а гіперфос
фатемія впливає на вже описані процеси актив
ної генетично запрограмованої, а також залеж
ної від апоптозу кальцифікації, посилюючи їх.
Загальні порушення фосфорно-кальцієвого об
міну є провідним механізмом кальцифікації судин
у хворих з хронічною нирковою недостатністю.
6.
Циркуляція сироваткових прокальциногенних факторів. Численні спостереження
свідчать про тісний зв’язок між змінами
кісток, зокрема остеопорозом, і артеріо
184
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
склерозом Менкеберга. Так, резорбція
кісткової тканини, що має місце при ста
рінні, при порушенні секреції жіночих
статевих гормонів, за умов цукрового діа
бету, хронічної ниркової недостатності,
при гіпервітамінозі D та ін., закономірно
супроводжується кальцифікацією артерій.
В експериментальних дослідженнях було
встановлено, що застосування препаратів,
які пригнічують діяльність остеокластів
і запобігають резорбції кісток (бісфосфонати та ін.), попереджає розвиток медіакальцинозу, безпосередньо не впливаючи
на процеси, що відбуваються в судинній
стінці. Вважають, що при резорбції кіст
кової тканини у кров потрапляють білкові
комплекси, які відіграють роль прокальциногенного фактора завдяки здатності бути
нуклеатором формування кристалів гідроксіапатиту (SCF, serum calcification factor).
SCF після надходження в судинну стінку
спричиняється до утворення початкових,
малих за розмірами кальцифікатів, які по
тім, проникаючи всередину колагенових
і еластичних структур або осідаючи на їх
ній поверхні, стають місцем подальшого
обвапнення, що здійснюється фізично-хі
мічними механізмами. Останні забезпечу
ють ріст кристалів мінералу та формування
більших чи менших осередків кальцинозу.
Нині ідентифікація сироваткових прокальциногенних факторів триває, але вже одержані
результати дають підстави виділити ряд претен
дентів на роль нуклеаторів кальцифікації судин:
а) великий сироватковий нуклеатор кістко
вого походження, яким, мабуть, є SCF. До
кази його наявності було наведено вище;
б) кальцій-протеїнові частки (CPPs, calciprotein particles). Відповідно до однієї з гі
потез, у крові постійно циркулюють CPPs,
що складаються з кальцію, фосфатів і білка
фетуїну-А. Ці частки є засобом транспорту
нерозчинних у воді солей кальцію. CPPs за
своїм функціональним призначенням є по
дібними до ліпопротеїнів, а фетуїн-А - до
апопротеїну. Вміст CPPs у крові визнача
ється балансом процесів їх утворення, з од
ного боку, і видалення чи деградації, з дру
гого. Основним джерелом CPPs є кісткова
тканина, у якій під впливом остеокластів
відбувається резорбція мінеральних і ор
ганічних компонентів тканини. Після пере
ходу CPPs у м’які тканини вони захоплю
ються макрофагами та, можливо, й іншими
клітинами (ендотеліоцитами, ГМК), після
чого відбувається розчинення їх у кислому
середовищі лізосом.
Згідно з наведеною гіпотезою ектопічна кальцифікація може розвиватися за двох обставин:
(1) при надмірному утворенні в кістках і над
ходженні в тканини CPPs (аналогічно розвитку
атеросклерозу, зумовленого гіперліпопротеїнемією) і (2) при порушеннях фагоцитарної функ
ції макрофагів та подібних до них клітин. Абсо
лютна чи відносна неспроможність здійснювати
деградацію CPPs веде до апоптозу макрофагів,
ендотеліоцитів і ГМК. Апоптичні тільця стають
місцем відкладання гідроксіапатиту і в такий
спосіб започатковують процеси патологічної мі
нералізації структур судинної стінки;
в)
кальцифікуючі нанобактерії. Наприкін
ці минулого століття у крові людини було
вперше виявлено наночастки (25—250 нм),
що мають у своїй оболонці чи клітин
ній стінці фосфат кальцію та істотно від
різняються за своїми характеристиками
від інших відомих сьогодні біологічних
об’єктів. Цей вид істот (не віруси, не бакте
рії і не пріони) було названо Nanobacterium
sanguineum, нині ж у літературі можна
зустріти й інші терміни: наночастки (NPs,
nanoparticles), кальцифікуючі наночастки
(CNPs, calcifying nanoparticles) або просто
нанобактерії (nanobacteria).
Спочатку було встановлено, що CNPs екскретуються з сечею і можуть бути причетні до
хронічних запальних процесів у нирках і до
утворення в них каменів. Потім з’ясувалося, що
CNPs можна виявити в різних ділянках серце
во-судинної системи, де відбуваються процеси
патологічного обвапнення, і, зокрема, в кальцифікованих атеросклеротичних бляшках. В екс
перименті було показано, що введення кролям
нанобактерій, виділених з крові людини, зу
мовлює розвиток атеросклеротичних змін у по
збавлених ендотелію сонних артеріях тварин.
Наведені факти дали підстави авторам від
криття припустити, що описані раніше сиро
ваткові фактори нуклеації є ніщо інше, як CNPs.
185
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Висловлено думку, що нанобактерії можуть не
тільки слугувати місцем зародження кристалів
гідроксіапатиту в судинній стінці, а й посилю
вати її кальцифікацію через інші механізми, зо
крема через індукування апоптозу.
Значення артеріосклерозу Менкеберга. Менкебергівський склероз є не менш поширеною фор
мою уражень артерій, ніж атеросклероз. Проте
йому до останнього часу приділялося значно
менше уваги, ніж атеросклерозу, оскільки він не
має таких яскравих клінічних проявів і досить
частих небезпечних наслідків (інфаркт міокарда,
ішемічний інсульт).
Однак кальцифікація, що відбувається в ар
теріальних судинах, не є безневинним явищем.
Вона може виявляти себе рядом функціональ
них і структурних змін, що зумовлюють пору
шення діяльності організму в цілому.
Передусім,
унаслідок
менкебергівського
склерозу змінюються біомеханічні властивості
артерій: зменшується їхня еластичність, роз
тяжність, збільшується жорсткість, ригідність.
Це зумовлює такі гемодинамічні зміни, як збіль
шення швидкості поширення пульсової хвилі,
зростання артеріального тиску.
Сам по собі артеріосклероз Менкеберга, зо
крема у людей похилого і старечого віку, зумов
лює розвиток ізольованої систолічної артері
альної гіпертензії (ІСАГ), що характеризується
збільшенням систолічного і пульсового арте
ріального тиску на тлі незміненого або зниже
ного діастолічного (рис. 32.21). Такий варіант
гіпертензії слід розглядати як компенсаторний,
оскільки завдяки ІСАГ периферичні тканини
можуть і далі одержувати необхідний їм об’єм
крові, незважаючи на втрату артеріями елас
тичності і здатності адекватно збільшувати свій
просвіт. Лікування ІСАГ у цьому разі усуватиме
компенсаторне її призначення та вестиме до роз
витку ішемічних порушень в органах і тканинах
на тлі вже “нормального” артеріального тиску.
Якщо ж до кальцифікації артерій додаються
інші патогенетичні чинники, що впливають на
артеріальну гіпертензію, збільшуючи діастолічний і зменшуючи пульсовий тиск (хвороби
нирок, цукровий діабет тощо), то в таких ви
падках периферичні тканини зазнаватимуть
ішемічної гіпоксії через нездатність кальцифікованих судин пропускати через себе по
трібний тканинам об’єм крові. Такий стан пе
риферичного кровообігу можна розглядати як
Рис. 32.21. Вплив артеріосклерозу Менкеберга на центральну гемодинаміку і місцевий кровообіг.
CAT, ДАТ, ПАТ - відповідно систолічний, діастолічний і пульсовий артеріальний тиск
186
декомпенсований. Він зумовлює розвиток ди
строфічних змін в органах і тканинах, зокрема
таких, як синдром діабетичної стопи у хворих
на цукровий діабет.
Артеріосклероз Менкеберга може стати при
чиною аневризм артерій. Розрив стінки кальцифікованої судини, наприклад, аорти, веде до
того, що кров затікає між шарами артеріальної
стінки й розшаровує їх далі. Якщо розшару
вання прориває стінку аорти повністю (усі три
шари), то відбувається швидка масивна крово
втрата, яка більш ніж у 90 % випадків веде до
смерті, навіть якщо вчасно було розпочато пра
вильне лікування.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
правило, має есенціальна гіпертензія з трива
лим безсимптомним перебігом. При належному
контролі артеріального тиску вона не несе смер
тельної загрози організму.
Про злоякісну артеріальну гіпертензію ве
дуть мову тоді, коли різко підвищується тиск
до 200/120 мм рт. ст. За відсутності належного
лікування вона призводить до смерті протягом
1-2 років. Злоякісна гіпертензія може виникати
в осіб, що мали нормальний тиск, але частіше
їй передують доброякісні варіанти есенціальної
і вторинної гіпертензії.
Гем один им ічпі варіанти. Оскільки величина
артеріального тиску визначається загальним за
коном гемодинаміки, відповідно до якого
32.2. АРТЕРІАЛЬНА ГІПЕРТЕНЗІЯ
Артеріальною гіпертензією вважають стале під
вищення артеріального тиску понад 140/90 мм
рт. ст. Вона може бути первинною і вторинною.
При первинній артеріальній гіпертензії під
вищення артеріального тиску не може бути
пов’язане з конкретною хворобою чи патологіч
ним процесом у тих чи інших органах та систе
мах: причина підвищення артеріального тиску
залишається неясною. Для позначення цієї фор
ми гіпертензії використовують два рівнозначних
терміни: “есенціальна гіпертензія” і “гіперто
нічна (гіпертензивна) хвороба”. На первинну
гіпертензію припадає 90-95 % випадків сталого
підвищення кров’яного тиску.
Вторинна артеріальна гіпертензія, частота
якої набагато менша і становить 5-10 %, вини
кає як наслідок патологічних процесів у різних
органах і системах. Вона характерна для:
а) хвороб нирок (гломерулонефрит, пієлонеф
рит, полікістоз нирок та ін.) (див. главу 36);
б) пухлин надниркових залоз (феохромоцитома, альдостерома);
в) уражень серця й судин (деякі вади серця,
коарктація аорти);
г) хвороб нервової системи (бульбарний поліо
мієліт, енцефаліти; травми, струси мозку та ін.).
У всіх цих випадках причина гіпертензії ясна.
Її усунення, як правило, веде до нормалізації
артеріального тиску.
Артеріальна гіпертензія може бути доброякіс
ною та злоякісною. Доброякісний характер, як
де Р — артеріальний тиск; Q - хвилинний об’єм
серця; R — загальний периферичний опір, то
артеріальна гіпертензія може бути обумовлена
збільшенням хвилинного об’єму серця Q, збіль
шенням загального периферичного опору R або
тим та іншим одночасно.
Відповідно до цього виділяють три гемодинамічних варіанти артеріальної гіпертензії.
1. Гіперкінетичний тип. Обумовлений іс
тотним збільшенням роботи серця, у ре
зультаті чого зростає його хвилинний
об’єм Q. Прикладом може бути ізольована
систолічна артеріальна гіпертензія, що
розвивається в людей похилого та старе
чого віку в результаті ураження судин ар
теріосклерозом Менкеберга (див. підглаву
32.1.2).
2. Еукінетичний тип. Виникає при помірному
збільшенні хвилинного об’єму серця Q і за
гального периферичного опору R.
3. Гіпокінетичний тип. Його розвиток пов’я
заний з істотним збільшенням загального
периферичного опору R.
Регуляція артеріального тиску. Рівень арте
ріального тиску має бути таким, щоб забезпечу
вати системний кровообіг відповідно до потреб
організму. Для цього існують системи регуляції,
що здійснюють свій вплив на гемодинаміку че
рез (а) нервові, (б) гуморальні та (в) ниркові ме
ханізми (рис. 32.22).
187
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Рис. 32.22. Фактори регуляції артеріального тиску
Оскільки рівень артеріального тиску ви
значається двома параметрами: (1) хвилинним
об’ємом серця і (2) загальним периферичним
опором (див. вище), то кожен з них може бути
об’єктом такої регуляції.
Хвилинний об’єм серця залежить від (а)
сили серцевих скорочень і (б) венозного по
вернення (переднавантаження), на рівень яко
го прямо впливає об ’єм циркулюючої крові. З
огляду на це зміни хвилинного об’єму можуть
обумовлюватися регуляторними впливами як
на скорочувальну функцію міокарда (симпа
тичні нерви, катехоламіни), так і на об’єм кро
ві, що повертається до серця (ниркові механіз
ми регуляції).
Основним чинником, що визначає загальний
периферичний опір, є тонус резистивних су
дин - дрібних артерій, артеріол і посткапілярних венул. Його зміни (вазоконстрикція чи вазодилатація), що виникають під впливом нервових
і гуморальних факторів, позначаються на вели
чині артеріального тиску.
Залежно від спрямованості дії всі системи,
що беруть участь у регуляції кров’яного тиску,
поділяють на дві групи: (І) ті, що підвищують
артеріальний тиск (пресорні), і (II) ті, що пони
жують його (депресорні).
І. Пресорні системи
1.
Симпатоадреналова система, до якої від
носять симпатичні адренергічні нервивазоконстриктори і катехоламіни (адрена
лін та норадреналін).
Механізм підвищення артеріального тиску під
впливом катехоламінів складний і пов’язаний
з дією їх на всі структури, від яких залежить
характер системного кровообігу (рис. 32.23).
До таких структур належать:
а)
артерії та артеріоли. Діючи через
а-адренорецептори, адреналін і норадре
налін зумовлюють скорочення гладкої мус
кулатури і, як наслідок, звуження судин.
Саме по собі зменшення просвіту резис
тивних судин веде до істотного зростан
ня загального периферичного опору (R)
(якщо радіус судин зменшується в 2 рази,
то опір збільшується в 16 разів). Крім того,
звуження артеріол спричиняється до змен
шення тиску крові в капілярах і, за законом
Стерлінга (див. главу 27), до зменшення
фільтрації та збільшення реабсорбції води
через капілярну стінку. Такі зміни обміну
рідини між плазмою крові та інтерстицієм
ведуть до збільшення об’єму циркулюючої
крові, а в результаті цього — і до збільшення
хвилинного об’єму серця (Q);
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
188
Рис. 32.23. Вплив катехоламінів на артеріальний тиск. АР-адренорецептори; ОЦК-об'єм циркулюючої крові;
Q - хвилинний об'єм серця; R - загальний периферичний опір
б) вени і венули. Скорочення ГМК цих судин
має прямим своїм наслідком зменшення
ємності венозного русла. Це, у свою чергу,
веде до збільшення венозного повернен
ня до серця і відповідного зростання сили
серцевих скорочень. Останнє стає причи
ною збільшення хвилинного об’єму серця,
а отже, і артеріального тиску;
в) серце. Впливаючи на серце через Pj-адренорецептори, катехоламіни збільшують силу
серцевих скорочень та їхню частоту (див.
главу 31). Оскільки хвилинний об’єм серця є
добутком цих показників, то зростання кож
ного з них спричиняється до збільшення Q;
г)
нирки. Катехоламіни здатні впливати на
ниркові механізми регуляції кровообігу
прямо і опосередковано через ренін-ангіотензинову систему. Прямий вплив полягає
в тому, що адреналін і норадреналін, зумов
люючи спазмування кровоносних судин
кортикальних нефронів, перенаправляють
кров у судини юкстамедулярних нефронів.
Оскільки останні мають набагато довшу
петлю Генле, то площа реабсорбції іонів
натрію та води істотно зростає, що веде
до зменшення діурезу і збільшення об’єму
циркулюючої крові (див. главу 27). Крім
того, активація симпатоадреналової систе-
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
ми є однією з причин секреції реніну кліти
нами юкстагломерулярного апарату нирок
(див. главу 27). Запуск ренін-ангіотензинової системи, як це буде показано далі, є важ
ливим чинником підвищення артеріального
тиску.
Слід зазначити, що вираженість пресорних
ефектів катехоламінів значно зростає у присут
ності глюкокортикоідів, які чинять стосовно
адреналіну та норадреналіну так звану пермісивну дію (див. главу 37), Саме цим пояснюють
здатність препаратів глюкокортикоїдних гормо
нів підвищувати артеріальний тиск.
2. Ренін-ангіотензинова система. Причини
та суть її активації були докладно описані
в главі 27. Тут лише нагадаємо, що кін
цевий продукт цієї системи — ангіотензин II - чинить свою пресорну дію через
такі механізми:
а) викликає скорочення ГМК артеріол, у ре
зультаті чого судини звужуються й зростає R;
б) зумовлює затримку натрію та води через
пряму дію на нирковий кровообіг і процеси
реабсорбції;
в) стимулює секрецію альдостерону кліти
нами кори надниркових залоз і вже через
цей гормон опосередковано збільшує реаб
сорбцію натрію і води, а отже, і об’єм цир
кулюючої крові.
3. Альдостерон-вазопресинова система. Роль
альдостерону і вазопресину в регуляції вод
но-сольового обміну було описано в главі
27. При посиленій секреції цих гормонів
збільшується реабсорбція натрію й води,
що веде до збільшення об’єму крові в суди
нах і хвилинного об’єму серця, як наслідок.
Крім того, вазопресин, як це видно з назви
гормону, здатен безпосередньо впливати на
тонус артеріол, істотно збільшуючи його.
Щоправда, такий ефект виявляє себе тільки
при значному зниженні артеріального тиску
і, мабуть, має невелике значення, коли йдеть
ся про патогенез артеріальної гіпертензії.
4. Пресорні фактори ендотеліального, тромбоцитарного і лейкоцитарного походжен
ня. У цю групу об’єднано сполуки, що чи
нять переважно місцевий вплив на тонус
резистивних судин, проте, при значній
активації їх утворення і вивільнення мож
ливою є системна дія, яка виявляє себе
189
підвищенням артеріального тиску. Про су
динозвужувальні речовини тканинного по
ходження вже йшлося у відповідних главах
підручника. Тут ми тільки зробимо невели
ке узагальнення, ще раз назвавши ці сполу
ки та джерела їх утворення:
а) ендотеліальні клітини здатні вивільнювати
при ушкодженні судинної стінки, а також
під впливом катехоламінів, ангіотензину II
і вазопресину ендотелій - пептид, що має
потужну і стійку судинозвужувальну дію
(див. главу 16). Крім того, вазоконстрикторний ефект притаманний таким сполукам
ендотеліального походження, як фактор
активації тромбоцитів (ФАТ), проста
гландин F,, ендотеліальний аноксемічний
фактор констрикції, ендопероксиди;
б) тромбоцити є джерелом тромбоксанів —
похідних арахідонової кислоти, що утво
рюються в кров’яних пластинках під впли
вом ферменту циклооксигенази (див. главу
17). Тромбоксани, крім здатності активува
ти агрегацію тромбоцитів, мають виражену
судинозвужувальну дію;
в) у лейкоцитах перетворення арахідонової
кислоти ідуть через ліпоксигеназний шлях,
кінцевими продуктами якого є лейкотрієни (LT) (див. главу 17). Значна частина цих
сполук, зокрема LT С4, LT D4, LT Е4, викли
кають вазоконстрикторні ефекти.
II. Депресорні системи
1,
Барорецепторні рефлекси. Вони почи
наються
подразненням
барорецепторів
дуги аорти та синокаротидної зони при
підвищенні тиску в судинах. Збільшен
ня імпульсації від цих рецепторів веде до
(а) активації депресорного і (б) гальму
вання пресорного відділів судинорухового
центру, нейрони якого містяться в стовбу
ровій частині головного мозку. Активація
депресорного відділу спричиняється до по
силення імпульсації з кардіоінгібіторного
центру і збудження внаслідок цього пара
симпатичних нервів, що регулюють роботу
серця. Під впливом медіатора цих нервів
ацетилхоліну зменшується частота сер
цевих скорочень, падає хвилинний об’єм
серця, а отже, і артеріальний тиск. З дру
гого боку, гальмування пресорного відділу
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
190
судинорухового центру веде до зменшення
активності симпатоадреналової системи
і в такий спосіб усуває пресорні ефекти
останньої.
2. Калікреїн-кінінова система. Причини і ме
ханізми активації цієї системи були пред
метом обговорення в главі 17 підручника.
Основний представник кінінів — брадикінін - має виражену судинорозширюваль
ну дію, яка опосередковується активацією
брадикінінових рецепторів на поверхні ендотеліальних клітин та утворенням унаслі
док цього оксиду азоту. Очевидно, участь
калікреїн-кінінової системи в регуляції ар
теріального тиску тісно пов’язана з функці
онуванням ренін-ангіотензинової системи.
Про це зокрема свідчить той факт, що один
і той же ангіотензинперетворювальний
фермент (конвертаза), з одного боку, зу
мовлює появу біологічно активного ангіотензину II, а з другого - інактивує брадикінін, гідролізуючи його до менших пептидів
і амінокислот.
3. Ниркові депресорні фактори. Вважають,
що такими є похідні арахідонової кисло
ти - простагландини (PG D2, PG Е2) і простациклін (PGІ2). Судинорозширювальний
ефект простагландинів, як і більшості при
родних вазодилататорів, опосередковуєть
ся оксидом азоту, що його утворюють ендотеліальні клітини.
4.
Передсердний натрійуретичний гормон
(атріопептин). При збільшенні об’єму цир
кулюючої крові та концентрації іонів Na+
в ній міоендокринні клітини передсердь
вивільнюють атріопептин (див. главу 27),
депресорна дія якого пов’язана з такими
ефектами:
а) збільшення виведення з організму натрію
і води, внаслідок чого зменшується об’єм
циркулюючої крові, а отже, хвилинний
об’єм серця (Q). Водночас атріопептин
гальмує секрецію альдостерону, що поси
лює наведений вище ефект натрійуретичного гормону;
б) зменшує тонус гладких м’язів кровоносних
судин, завдяки чому знижується загальний
периферичний опір (R);
в) через негативну інотропну і хронотропну дію на серце зменшує хвилинний його
об’єм (Q).
5.
Депресорні фактори ендотеліального по
ходження. В ендотелії судин утворюється
оксид азоту (N0), через який опосеред
ковується судинорозширювальна дія пе
реважної більшості біологічно активних
речовин. Докладну характеристику оксиду
азоту та механізмів його дії подано в главі
16 підручника.
Крім того, ендотеліоцити є джерелом ще ці
лої низки сполук-вазодилататорів. Такими, зо
крема, є ендотеліальний фактор гіперполяризації, аденозин, простациклін (PG І2), натрій
уретичний пептид С. Існування в організмі
великої кількості чинників, що мають пресорну
і депресорну дію, вимагає підтримання певного
балансу між ними, аби величина артеріального
тиску підтримувалася на певному сталому рівні.
Звісна річ, що порушення цього балансу
в бік переважання пресорних систем над де
пресорними (збільшення активності перших
і зменшення активності других) буде вести до
підвищення артеріального тиску, тобто до ар
теріальної гіпертензії (рис. 32.24). Порушен
ня балансу в інший бік (зменшення активності
пресорних систем і збільшення активності де
пресорних) матиме своїм наслідком артеріаль
ну гіпотензію.
Експериментальні моделі артеріальної гіпер
тензії. Жодна хвороба людини не має такої ве
ликої кількості різних експериментальних моде
лей, як артеріальна гіпертензія.
За методами відтворення всі її моделі можна
поділити на кілька великих груп.
I. Порушення функції центральної нервової
системи. До таких відносять:
а) зіткнення процесів умовного збудження
та гальмування, що призводить до розвитку
у тварин (собак, мавп) неврозу;
б) моделювання психоемоційної напруги шля
хом створення зоосоціального конфлік
ту (у мавп), змін біоритмів, іммобілізації
тварин;
в) електричну й хімічну стимуляцію лімбічних структур головного мозку.
II. Порушення мозкового крово- і лімфообігу.
Підвищення артеріального тиску можна викли
кати:
а) одно- і двостороннім перев’язуванням сон
них і вертебральних артерій, що живлять
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
191
Рис. 32.24. Регуляція артеріального тиску: порушення балансу між пресорними й депресорними системами
як причина розвитку артеріальної гіпер- і гіпотензії
мозок
(центрально-ішемічна
артеріальна
гіпертензія)',
б) блокадою лімфовідведення по периневральних і периваскулярних лімфатичних
шляхах за допомогою каоліну, що його вво
дять у велику цистерну мозку.
ПІ. Порушення функції депресорних регуля
торних систем. Зменшення активності цих сис
тем можна відтворювати:
а) двостороннім перетинанням у кролів і со
бак депресорних і синусних нервів, у ре
зультаті чого знімаються гальмівні впливи
з барорецепторів рефлексогенних зон дуги
аорти і каротидного синуса (рефлексогенна
гіпертензія, або гіпертензія розгальмуван
ня);
б) центральною деаферентацією барорецеп
торів, що її викликають ушкодженням ядра
солітарного тракту;
в) пригніченням синтезу простагландинів за
допомогою, наприклад, індометацину.
IV.
Порушення функції нирок. Артеріальна
гіпертензія ниркового походження виникає при:
а) звужуванні обох ниркових артерій або
звужуванні однієї ниркової артерії з ви
даленням другої контрлатеральної нирки
(реноваскулярна
гіпертензія).
Виникнен
ня гіпертензії в цьому випадку пов’язане
з активацією ренін-ангіотензинової систе
ми;
б) видаленні обох нирок і переведенні тварин
на гемодіаліз для запобігання уремії (ренопривна гіпертензія). її розвиток пояснюють
повним припиненням депресорних функ
цій нирок (не утворюються ниркові про
стагландини та інші сполуки, що знижують
тиск);
в) обгортання нирок целофаном, шовком. При
цьому виникає перинефрит: здавлюється
ниркова паренхіма, розвивається венозний
застій і гіпоксія нирок, активується ренінангіотензинова система.
V. Порушення гормонального стану. До арте
ріальної гіпертензії спричиняються:
а) введення тваринам адреналіну,
б) введення вазопресину,
в) субтотальне видалення кори надниркових
залоз. При цьому відбувається посилення
регенерації залозистої тканини з підвище
ною продукцією кортикостероїдів, особли
во альдостерону (наднирково-регенерацій
на гіпертензія).
VI. Порушення водно-сольового обміну. Арте
ріальну гіпертензію можна моделювати:
192
а) введенням тваринам великої кількості ку
хонної солі (сольова гіпертензія);
б) застосуванням мінералокортикоїдів (дезоксикортикостерону, альдостерону) ш мінералокортикоїдна гіпертензія',
в) поєднаним введенням кухонної солі й міне
ралокортикоїдів.
VII.
Моделі генетично обумовленої арте
ріальної гіпертензії. У багатьох лабораторіях
світу виведено чисті лінії щурів, характерною
рисою яких є гіпертензія як ознака, що спадкується. Це, зокрема, щури зі спонтанною гіпер
тензією (лінія Окамото - Аокі); щури, схильні
до інсультів; новозеландські щури, міланські
щури; щури, чутливі до сольової дієти, та ін.
Нині ведуться пошуки конкретних генів, що
мають стосунок до стійкого підвищення артері
ального тиску. Для цього використовують новіт
ні технології “генетичного нокауту” (див. гла
ву 13). За їхньою допомогою виведено чисті лі
нії мишей, у яких артеріальна гіпертензія є ста
лою ознакою (наприклад, дефіцитні за геном
ендотеліальної NO-синтази миші).
Причини
первинної
артеріальної
гіпертензії.
Есенціальна артеріальна гіпертензія, як і біль
шість поширених недуг людини, належить до
полігенних (мультифакторіальних) хвороб, тоб
то таких, у виникненні яких мають значення як
чинники зовнішнього середовища, так і спад
кова схильність (див. главу 13). З огляду на це
можна виділити дві групи етіологічних факторів
гіпертензивної хвороби.
І. Екзогенні фактори. Численні клінічні спо
стереження та епідеміологічні дослідження да
ють підстави вважати, що в основі есенціальної
гіпертензії можуть лежати такі причини.
1. Тривала психоемоційна перенапруга, або
хронічний стрес (див. главу 8). Стрес у по
всякденному житті (“стрес-планктон ”),
який складається з маси негативних момен
тів: неблагополуччя на роботі, домашня ру
тина, проблеми в сім’ї тощо є чинниками,
що сприяють артеріальній гіпертензії. Уста
новлено більш значну поширеність гіпер
тензії серед людей, характер роботи яких
пов’язаний з постійною психоемоційною
напругою, наприклад серед телефоністок
і телеграфістів, студентів у період екзаме
наційної сесії, у дітей і підлітків, що навча
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ються у спеціалізованих математичних і де
яких інших школах з напруженим режимом
занять.
Російський учений Ланг уперше був висло
вив думку, що першопричиною гіпертонічної
хвороби є психоемоційні перенапруги, які ве
дуть до розвитку невротичних порушень вищої
нервової діяльності та підвищення артеріально
го тиску як вегетативного компонента цих пору
шень. Основну роль Ланг відводив так званим
невідреагованим негативним емоціям, тобто та
ким, при яких сильні вегетативні реакції, зокре
ма з боку серцево-судинної системи, не супро
воджуються адекватними руховими реакціями.
2. Збільшення індексу маси тіла, ожиріння.
В епідеміологічних дослідженнях показано,
що існує зв’язок між наведеними фактора
ми й есенціальною гіпертензією. На думку
вчених, один з механізмів такого зв’язку по
лягає в тому, що при збільшенні маси жиро
вої тканини посилюється утворення в адипоцитах лептину (див. главу 25). Останній,
пригнічуючи центри апетиту, істотно акти
вує гіпоталамічні центри симпатоадреналової системи, унаслідок чого збільшується
тонус симпатичних вегетативних нервів
і секреція катехоламінів клітинами мозко
вої речовини надниркових залоз.
При ожирінні часто має місце посилена про
дукція інсуліну (гіперінсулінемія). Надлишок
інсуліну завдяки ростовій активності цього гор
мону стимулює проліферацію гладких м’язових
клітин кровоносних судин. Цей процес, якщо
він відбувається у резистивних судинах, веде до
потовщення судинної стінки і, як наслідок, до
зменшення радіусу судин, збільшення загально
го периферичного опору, а отже, й артеріально
го тиску.
Сидячий триб життя (гіподинамія) також вва
жається фактором ризику гіпертензивної хво
роби. Проте навряд чи він відіграє самостійну
роль, оскільки люди з обмеженою руховою ак
тивністю, як правило, мають підвищений індекс
маси тіла, а часто й ожиріння.
3. Надмірне споживання кухонної солі. Низь
кий рівень захворюваності на есенціальну
гіпертензію реєструють у представників
деяких етнічних груп, що споживають малу
кількість кухонної солі. До таких належать
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
ґренландські ескімоси, аборигени гір Ки
таю та Австралії, деякі племена індіанців,
що живуть у Панамі. Індивідуальне спожи
вання солі в цих групах є набагато меншим
(1-2 г на добу), ніж у представників інших
націй та народностей (10-15 г на добу).
Водночас там, де солі споживається більше,
первинну артеріальну гіпертензію виявляють
частіше. Висока захворюваність на гіпертензивну хворобу характерна для негритянського на
селення Багамських островів, деяких регіонів
Японії, де добове споживання солі сягає 20-50 г.
Ми вже зазначали, що в експерименті арте
ріальну гіпертензію можна відтворити згодову
ванням тваринам кухонної солі.
II.Ендогенні фактори. Як доказ значення
спадковості в етіології гіпертензивної хворо
би наводять той факт, що в парах однояйцевих
близнюків артеріальну гіпертензію виявляють
набагато частіше, ніж у парах близнюків двояйцевих. Крім того, поширеність артеріальної
гіпертензії серед родичів хворих на гіпертензивну хворобу значно більша, ніж у популяції
в цілому. Нарешті, експериментальні моделі
генетично обумовленої гіпертензії у тварин
за основними своїми характеристиками є най
ближчими до первинної артеріальної гіпертен
зії людини.
Важливим фактором спадкової схильності
людей до гіпертензії є однонуклеотидний полі
морфізм генів (див. главу 7). Сьогодні зв’язок
цього явища з гіпертензивною хворобою вивча
ється на великій кількості генів-кандидатів, що
мають стосунок до різних систем і механізмів
регуляції кров’яного тиску. Серед таких - гени
ангіотензину і ангіотензинових рецепторів, ангіотензинперетворювального ферменту, різних
форм NO-синтази, простагландинсинтаз, ендотеліну, адренорецепторів та багатьох інших.
Патогенез. Нині уявлення про патогенез есенціальної гіпертензії розвиваються в основному
в рамках двох концепцій, що їх можна позначи
ти як дисрегуляторна і власне судинна.
Дисрегуляторна концепція пояснює виник
нення первинної артеріальної гіпертензії по
рушеннями механізмів регуляції артеріального
тиску.
В основі власне судинної концепції лежить по
ложення про те, що гіпертензивна хвороба вини
193
кає як наслідок первинних порушень у гладких
м’язових клітинах резистивних судин (артеріол).
Дисрегуляторна концепція. Патогенез первин
ної артеріальної гіпертензії, відповідно до основ
них положень цієї концепції, проходить дві фази:
гіперкінетичну і гіпокінетичну.
І. Гіперкінетична фаза характеризується пе
реважно збільшенням хвилинного об’єму сер
ця, наслідком чого і є підвищення артеріаль
ного тиску. На цьому етапі хвороби гіпертензія
є оборотною, оскільки зумовлюється функціо
нальними, а не структурними змінами в орга
нізмі. При усуненні зовнішніх факторів ризику
та нормалізації регуляторних процесів показни
ки артеріального тиску ще можуть повертатися
до нормальних величин. У розвитку гіперкінетичної фази можна виділити ряд важливих по
слідовних подій.
1)
активація симпатоадреналової системи
(рис. 32.25). Вона відбувається в резуль
таті хронічного стресу (“стресу-планктону”), психоемоційних перенапруг, що
зумовлюють появу осередків постійного
тривалого збудження в центральній нер
вовій системі (патологічна домінанта).
Катехоламіни, які виділяються при цьо
му, викликають щонайменше три важли
вих для подальшого розвитку гіпертензії
ефекти:
збільшують хвилинний об’єм серця (Q);
підвищують загальний периферичний
опір (R);
викликаючи спазм приносних артеріол
нирок і безпосередньо діючи на клітини
юкстагломерулярного апарату, сприяють
виділенню в кров реніну;
2) активація ренін-ангіотензинової системи
(рис. 32.26). Надходження реніну в кров
зумовлює ряд послідовних біохімічних ре
акцій, у результаті яких утворюється ангіотензин II. Поява цього пептиду зумовлює
такі зміни:
скорочення гладких м’язів артеріол (ангіо
спазм);
збудження структур центральної нервової
системи, що беруть участь у регуляції ар
теріального тиску;
вивільнення в кров альдостерону кліти
нами клубочкової зони кори надниркових
залоз;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
194
Гіперкінетична фаза патогенезу первинної артеріальної гіпертензії: активація
СИМпатоадреналової системи. ЦНС - центральна нервова система, ЮГА - юкстагломерулярний апарат
3)
активація
альдостерон-вазопресинової
(рис. 32.27). Надходження в кров
альдостерону, а також надмірне надхо
дження в організм хлориду натрію ви
кликають розвиток гіпернатріємії і під
вищення у зв’язку з цим осмотичного
тиску плазми крові. Збудження централь
них і периферичних осморецепторів, що
настає в цих умовах, активує секрецію
вазопресину
(антидіуретичного гормону)
у ядрах гіпоталамуса. Вазопресин, впли
ваючи на нирки, викликає збільшення фа
культативної реабсорбції води. Це, у свою
чергу, веде до збільшення об’єму цирку
люючої крові (гіперволемія), хвилинного
об’єму серця, а отже, і артеріального тиску.
Зазначений механізм доповнюється безпо
середньою судинозвужувальною дією ва
зопресину.
системи
II.
Гіпокінетична фаза характеризується не
оборотними структурними змінами резистивних судин, у результаті чого загальний перифе
ричний опір і артеріальний тиск постійно збіль
шені. У розвитку цієї фази можна виділити ряд
послідовних стадій (рис. 32.28):
1)
ауторегуляторний
спазм
артеріол.
Ви
никає як наслідок збільшення хвилинного
об’єму серця. Є реакцією, спрямованою на
підтримку сталості кровообігу в тканинах
(попереджає надходження надлишкової
кількості крові);
2)
гіпертрофія
гладких
м’язів
артеріол.
Є структурним проявом гіперфункції глад
ких м’язів, що виникає при часто повторю
ваних спазмах;
3) артеріолосклероз. Гіпертрофовані гладкі
м’язові клітини поступово зазнають дис
трофічних змін і гинуть, відбувається за-
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
195
Гіперкінетична фаза патогенезу первинної артеріальної гіпертензії:
активація ренін-ангіотензинової системи. AT - ангіотензинові, ЦНС - центральна нервова система
міщення їх сполучною тканиною - розви
вається артеріолосклероз. Артеріоли пе
ретворюються в ригідні сполучнотканин
ні трубки, не здатні ні до скорочення, ні до
розслаблення. Загальний периферичний
опір, а отже, і артеріальний тиск постійно
збільшені. Порушується живлення жит
тєво важливих органів: головного мозку,
серця, нирок. Можливий розрив змінених
артеріол — тоді розвивається крововилив.
Найнебезпечнішим ускладненням є кро
вовилив у мозок - геморагічний інсульт.
порядком подій, що відбуваються: спочатку йде
гіпокінетична, вже потім за нею гіперкінетична
фаза (рис. 32.29).
Суть первинних уражень резистивних судин
можуть складати такі зміни:
1) тривалий стійкий спазм артеріол. Він
часто розвивається внаслідок ендотеліальної дисфункції, при якій зменшується
утворення в ендотеліоцитах оксиду азоту
та інших вазодилататорів і зростає ви
вільнення вазоконстрикторів — ендотеліну та інших.
Власне судинна концепція. Згідно з її по
ложеннями в основі есенціальної гіпертензії
лежать первинні зміни в кровоносних судинах,
а розлади регуляції системної гемодинаміки ма
ють вторинний характер. По суті ця концепція
відрізняється від дисрегуляторної зворотним
Тривале скорочення гладких м’язів артеріол
може мати в своїй основі й генетичні дефекти.
Так, неповноцінність Са-насосів ГМК спричи
няється до порушень видалення іонів кальцію із
цитоплазми клітин і збільшення їхньої внутріш
ньоклітинної концентрації. Це викликає постій-
196
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Гіперкінетична фаза патогенезу первинної артеріальної гіпертензії:
активація альдостерон-вазопресинової системи
197
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Гіпокінетична фаза патогенезу первинноїартеріальної гіпертензії:
розвиток структурних змін артеріол
ну контрактуру (перескорочення) ГМК артеріол.
Крім того, надлишок іонів Са2+ у цитоплазмі
клітин викликає їх ушкодження (див. главу 7)
і є, отже, передумовою розвитку артеріолосклерозу;
2) гіпертрофія гладких м ’язів артеріол. Вона
може розвиватися не тільки як наслідок
їхньої гіперфункції, а й у результаті дії на
судини факторів, що мають ростову ак
тивність. До таких, як відомо, належить
інсулін (див. главу 24). З огляду на це по
товщення стінок резистивних судин, зу
мовлене гіпертрофією ГМК, закономірно
розвивається при гіперінсулінемії, що су
проводжує початкові етапи цукрового діа
бету II типу, синдром інсулінорезистентності, поширені варіанти ожиріння;
3) артеріолосклероз. Його розвиткові пере
дує ушкодження ГМК артеріол, а тому ве-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
198
Власне судинна концепція патогенезу первинної артеріальної гіпертензії.
ОЦК - об’єм циркулюючої крові
ликий ряд патогенних факторів, здатних
запускати різні молекулярні механізми
ушкодження клітин (кальцієві, пероксидні,
енергодефіцитні та інші) є потенційними
чинниками артеріальної гіпертензії.
Важливу роль у виникненні структурних
змін в артеріолах надають порушенням роботи
Na-K-насосів плазматичних мембран ГМК. На
таку думку наводить той факт, що у багатьох хво
рих на гіпертензивну хворобу виявляють так зва
ний ендогенний строфантиноподібний фактор,
що, як і відомі серцеві глікозиди (строфантин, оуабаїн), пригнічує роботу Na-K-насосів.
У результаті порушень діяльності цього насос
ного механізму в цитоплазмі поступово збільшу
ється концентрація іонів Na+ і виникає набряк
ГМК артеріол. Це має кілька наслідків:
а) потовщення стінки і зменшення просвіту
артеріол;
б) збільшення чутливості ГМК до дії ендоген
них катехоламінів;
в) ушкодження й загибель клітин з подаль
шим розвитком артеріосклерозу.
Усі наведені вище зміни резистивних судин
(тривалий спазм ГМК, їхня гіпертрофія та артеріолосклероз) стають причиною стійкого збіль
шення загального периферичного опору, а отже,
і підвищення артеріального тиску.
Проте не тільки цим виявляють себе первинні
порушення функції й структури периферичних
судин. Прямим наслідком зменшення їхнього
просвіту є розвиток ішемічної гіпоксії органів
і тканин. Така гіпоксія має стійкий характер,
оскільки ішемія, через особливий характер її
199
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
причин, не усувається метаболітами-вазодилататорами, що накопичуються в тканинах при кис
невому голодуванні.
Дальше прогресування артеріальної гіпертен
зії пов’язане з ішемією нирок і головного мозку.
У нирках гіпоксія зумовлює вивільнення ре
ніну клітинами юкстагломерулярного апарату:
відбувається запуск ренін-ангіотензинової, а по
тім і альдостерон-вазопресинової систем.
Ішемія головного мозку веде до активації
симпатоадреналової системи з притаманними
їй впливами на всі складові системи кровообі
гу (див. вище). Збільшення реабсорбції натрію
і води, що настає в цих умовах, стає причиною
зростання об’єму циркулюючої крові та, як на
слідок, збільшення хвилинного об’єму серця.
Останній зростає і в результаті прямого впливу
катехоламінів на функції серця.
Значення
артеріальної
гіпертензії.
Основ
на небезпека артеріальної гіпертензії полягає
в тому, що вона може стати причиною розри
ву структурно змінених судин і зумовлювати
крововиливи. Якщо це відбувається в тканинах
головного мозку, то розвивається геморагічний
інсульт, який виявляє себе порушенням тих чи
інших функцій центральної нервової системи,
що залежить від локалізації інсульту. Настан
ню таких тяжких наслідків часто передує різке
стрибкоподібне підвищення артеріального тис
ку - гіпертонічний криз.
Тривала ішемія нирок зумовлює розвиток
нефросклерозу та виявляє себе ознаками хро
нічної ниркової недостатності (див. главу 36).
Ішемія головного мозку веде до появи невро
логічних розладів, гіпертонічної енцефалопатїї,
порушень зору.
Артеріальна гіпертензія є чинником, що збіль
шує навантаження на серце (післянавантаження). У зв’язку з цим вона стає причиною гіпер
трофії міокарда лівого шлуночка, а потім і недо
статності серця (див. главу 31).
Підвищення артеріального тиску, як уже за
значалося, є одним з основних факторів ризику
атеросклерозу. Проте й атеросклероз сприяє
розвиткові артеріальної гіпертензії через утво
рення “зачарованих кіл” (рис. 32.30).
Слід, однак, зауважити, що стійке підвищен
ня артеріального тиску може мати й компенса
торне значення, наприклад, у людей похилого
і старечого віку. Завдяки підвищенню у них
систолічного і пульсового тиску (ізольована
систолічна артеріальна гіпертензія) вдається
підтримувати адекватний периферичний кро
вообіг через склеротично змінені судини (див.
підглаву 32.1.2).
Зв'язок артеріальної гіпертензії з атеросклерозом: "зачароване коло" (circulus vitiosus)
200
Артеріальна гіпотензія
Зниження артеріального тиску може бути (а) фі
зіологічним (не супроводжується хворобливими
симптомами) і (б) патологічним.
Фізіологічна артеріальна гіпотензія характер
на для тренованих спортсменів, вона може роз
виватися при довготривалій адаптації до гіпок
сії, до змін кліматичних умов тощо.
Патологічна артеріальна гіпотензія за своїм
перебігом може бути (а) гострою і (б) хронічною.
Гостра гіпотензія є провідною ознакою гос
трої судинної недостатності, що настає в умо
вах різних видів шоку і при колапсі (див. главу 20).
Про хронічну артеріальну гіпотензію ведуть
мову, коли має місце стале зменшення артері
ального тиску нижче 90/60 мм рт. ст. і спосте
рігаються симптоми, такі як загальна адинамія,
швидка стомлюваність, тахікардія, задишка,
запаморочення, головний біль, непритомності,
депресивний стан з періодичним підвищенням
нервової збудливості тощо.
За походженням хронічна гіпотензія може
бути (а) первинною (есенціальна гіпотензія)
і (б) вторинною (симптоматичною).
Причини есенціальної гіпотензії невідомі.
Є думка, що в її основі лежить перенапружен
ня основних процесів кори великого мозку (збу
дження і гальмування). Однак, на відміну від
первинної артеріальної гіпертензії, переважає
гальмування, яке поширюється на підкіркові ве
гетативні утворення, зокрема на судиноруховий
центр.
Симптоматична хронічна артеріальна гіпо
тензія є ознакою багатьох соматичних хвороб,
зокрема серця (вади клапанів, міокардит), го
ловного мозку (комоція), легень (крупозна пнев
монія), печінки (гепатит, механічна жовтяниця),
крові (анемія), ендокринних залоз, а також екзо
генних інтоксикацій.
З огляду на те, що рівень артеріального тиску
визначається величиною серцевого виштовху,
об’ємом циркулюючої крові та тонусом резистивних судин, можливі три гемодинамічні фор
ми артеріальної гіпотензії:
1) пов’язана з недостатністю скорочувальної
функції серця;
2) викликана зменшенням об’єму циркулюю
чої крові;
3) така, що виникає внаслідок зниження то
нусу резистивних судин.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ПАТОФІЗІОЛОГІЯ
ВЕНОЗНИХ СУДИН
Венозні судини, як уже зазначалося, належать
до ємнісних судин: вони депонують кров з ме
тою її розподілу і повернення до серця. Вста
новлено, що у венах міститься 70-80 % загаль
ного об’єму крові. Про важливе функціональне
значення венозного відділу свідчить хоча б той
факт, що одномоментне зменшення його ємнос
ті всього лише на 3 % подвоює венозне повер
нення до серця, а при однакових за величиною
змінах тиску в артеріальній і венозній системах
об’єм останньої змінюється приблизно в 30 ра
зів більше, ніж артеріальної.
З урахуванням сказаного було зроблено ви
сновок про те, що вже незначні порушення
функції з боку ємнісних судин можуть призво
дити до істотних порушень загальної гемодинаміки. Такі розлади можуть виявляти себе двома
типами змін:
а) розвитком артеріальної гіпертензії при
збільшенні тонусу гладких м’язів венозних
судин, унаслідок чого збільшується веноз
не повернення крові до серця, а в результа
ті цього і хвилинний його об’єм;
б) виникненням артеріальної гіпотензії - гос
трої (колапсу) або хронічної — при швидко
му або тривалому збільшенні ємності ве
нозної системи.
Патологічні процеси, що розвиваються у ве
нозних судинах, мають свої особливості, якщо
порівнювати з іншими типами судин.
Це в першу чергу стосується високої стійкос
ті вен до тих типів склеротичних уражень, до
яких особливо чутливі артерії, - атеросклерозу
і артеріосклерозу Менкеберга (див. вище).
У венах людини, на відміну від артерій, ні
коли не розвивається атеросклероз. Для судин
венозного відділу не характерні ліпідна інфіль
трація, формування фіброзних і атероматозних
бляшок.
Цікаво зазначити, що неуражені артерії та ве
нозні судини за вмістом ліпідів та ліпопротеїнів
у їхніх стінках не набагато відрізняються між
собою. Однак у процесі розвитку атеросклерозу
концентрація ліпопротеїнових комплексів в ар
теріальній стінці зростає в 3-5 разів, тимчасом
як у венозній залишається без змін.
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
201
По суті, жодну теорію атеросклерозу не може
бути визнано за доведену, якщо вона не пояснює
факт високої стійкості вен до атеросклеротично
го процесу. Сьогодні можна виділити такі най
поширеніші пояснення високої резистентності
венозних судин до атерогенних впливів.
Умови гемодинаміки у венозних судинах
істотно відрізняються від таких в артері
ях. Високий кров’яний тиск, а також особ
ливості течії крові в артеріальній системі
частково сприяють ушкодженню внутріш
нього ендотеліального шару і створюють
умови для проникнення в артеріальну стін
ку плазмових білків, у тому числі й ліпо
протеїнів. При цьому як основний доказ
наводять факт про те, що вени після транс
плантації в артеріальну систему нарівні
з артеріями стають об’єктом атеросклеро
тичних уражень.
ку артерій і вен у напрямку до адвентиції
здійснюється постійний, хоча й повільний,
рух плазми крові разом з великомодекулярними сполуками, серед яких ліпопротеїни.
Однак в артеріальній стінці, на відміну від
венозної, ці сполуки проходять через товсті
шари колагенових, еластичних і м’язових
волокон - через бар’єри, що затримують
надходження великих молекул в адвен
тицію. До того ж в артеріальних судинах
менш розвинена, ніж у венах, мережа vasa
vasorum і лімфатичних судин, що мають
стосунок до виведення ліпопротеїнів із су
динної стінки.
Підґрунтям різної стійкості артерій і вен до
атеросклеротичних уражень можуть бути
функціональні
особливості
основних клі
тинних елементів судин ендотеліальних
і гладких м’язових клітин.
Однак пояснення високої резистентності вен
до атеросклерозу більш “сприятливими” гемодинамічними умовами в судинах низького
тиску зазнає серйозної й обґрунтованої крити
ки. Річ у тім, що вени та венозний трансплан
тат - це не одне й те ж. Ціла низка обставин,
таких як позбавлення кровопостачання через
vasa vasorum, порушення іннервації, механіч
не ушкодження стінки під час приготування
трансплантата, а також у результаті дії на стін
ку незвичних для неї гемодинамічних умов,
і нарешті, попереднє зберігання - це все зага
лом спричиняється до того, що венозний тран
сплантат, пересаджений в артеріальну систему,
поводить себе не як активна венозна тканина, а
як ушкоджена судинна трубка, що зазнала тяж
ких дистрофічних змін, зумовлених самим про
цесом трансплантації.
Про те, що умови низького кров’яного тис
ку не є вирішальним чинником резистентності
вен до атеросклерозу, свідчить ще й такий факт.
У собак з гіперхолестеролемією трансплантація
найчутливішої до атеросклерозу ділянки черев
ної аорти в яремну вену жодним чином не запо
бігає розвиткові в пересадженій артерії тяжких
атеросклеротичних змін.
» Є спроби пояснити різну чутливість арте
рій і вен до атеросклерозу існуванням знач
них структурних відмінностей між арте
ріальною та венозною стінкою. Припуска
ють, що й за нормальних умов через стін
У запобіганні венозної стінки від ліпідної
інфільтрації, певно, важливе значення має по
рівняно низька транспортна активність ендоте
ліальних клітин вен. Про це, зокрема, свідчить
той факт, що в ендотелії вен кількість плазмалемальних везикул у 2 рази менша, ніж в ендотелі
альних клітинах артерій. Проте варто підкресли
ти, що інтенсивності поглинання радіоактивно
мічених нативних ліпопротеїнів низької густини
(ЛПНГ) артеріями і венами приблизно однако
вого діаметра майже не відрізняються. У той
же час швидкість розщеплення ЛПНГ, коли роз
рахунок здійснювати на одиницю площі поверх
ні судини, в артеріях значно вища, ніж у венах.
Швидкість проліферації венозних гладких міоцитів і ендотеліальних клітин у культурі тканин
завжди нижча за інтенсивність поділу клітин ар
терій.
Венозні судини характеризуються значно
вищою інтенсивністю енергетичного об
міну, ніж артерії. Це відповідно до “енергодефіцитної” теорії (див. підглаву 32.1.2)
робить більш ефективними механізми ак
тивної резистентності до ушкодження у ве
нозній стінці, якщо порівнювати з артері
альною.
Отже, є багато спроб пояснити, чому у венах,
на відміну від артерій, попри загальні порушен
ня обміну речовин в організмі, попри одні й ті ж
самі зміни ліпідного та ліпопротеїнового скла
ду крові, ніколи не розвивається атеросклероз.
202
Цілком імовірно, що успішне розв’язання цього
питання буде наближати нас до розуміння при
чин високої уразливості артерій атеросклеро
тичним процесом.
Патологічні процеси, що розвиваються у ве
нах, ведуть до розладів венозного кровообігу,
які залежно від басейну уражених судин можуть
мати загальний чи місцевий характер. Одна
з найчастіших частих причин цього - варикозне
розширення вен.
Варикозне розширення вен
Варикозним розширенням називають зміни ве
нозних судин, що виявляють себе нерівномір
ним збільшенням їхнього просвіту з утворенням
випинань венозної стінки в місцях її стоншення,
подовженням і розвитком вузлоподібної звивис
тості, функціональною недостатністю клапанів
та порушеннями руху крові у венах.
Варикозного розширення здебільшого зазна
ють вени, розташовані в тканинах, що легко
стискуються: у підшкірній жировій клітковині,
підслизовому шарі стінки стравоходу, шлун
ка, кишок. Найчастіше варикозне розширення
буває в поверхневих венах нижніх кінцівок, де
його поділяють на первинне та вторинне.
Первинне — розвивається без будь-якого
зв’язку з жодною хворобою, воно не пов’язане
з ураженнями магістральних вен. Вторинний
варикозний симптомокомплекс є наслідком пер
винних порушень глибоких і комунікативних
вен нижніх кінцівок: тромбозу, компресії, дисплазії й аплазії венозних судин.
Причини. Є підстави вважати, що для виник
нення варикозного розширення вен мають зна
чення такі чинники:
1) спадкова схильність. У багатьох клініч
них спостереженнях установлено безпе
речний зв’язок між розвитком варикоз
ного розширення вен у батьків і дітей.
У пацієнтів з варикозною хворобою знач
но частіше, ніж у здорових людей, вияв
ляють HLA-антиген В 7 і рідше - HLAантигени Aw 19, Cw5 і Cw6. В основі
спадкової схильності до варикозного роз
ширення може лежати однонуклеотидний поліморфізм генів (див. главу 6), що
мають стосунок до структурних, функ
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ціональних і метаболічних властивостей
венозної стінки;
2) вплив екзогенних токсичних, інфекційних
та інших патогенних факторів. Пока
зано, що в стінці варикозно розширених
вен відбувається перетворення “скоро
чувального” типу гладких м’язових клі
тин у “метаболічний” тип (див. підглаву
32.1.1). Тобто має місце неспецифічна ре
акція, характерна для дії багатьох ушко
джувальних агентів на стінку кровонос
них судин;
3) зміни гормональної регуляції. Саме цією
обставиною пояснюють розвиток вари
козного розширення вен у вагітних жі
нок. Естрогени, рівень яких зростає під
час вагітності, через невідомі поки що
механізми спричинюють гіпотонію глад
кої мускулатури матки та інших органів
і тканин. Цілком можливо, що під впли
вом цих гормонів зменшується тонус
гладких м’язів венозних судин, зміню
ється структура колагенових та еластич
них волокон їхньої стінки. Поновлення
гормонального балансу після пологів
сприяє зворотному розвиткові варикоз
них змін;
4) порушення нервової регуляції. їхня при
четність до варикозного розширення вен
може бути пов’язана як зі змінами функці
ональних властивостей гладких м’язів, так
із розвитком нейродистрофічного процесу,
що виявляє себе комплексом метаболічних
змін у венозній стінці (див. главу 38).
Патогенез. У розвитку варикозного розширен
ня вен центральне місце посідає “зачароване
коло” (circulus vitiosus), що поєднує між собою
три провідні ланки патогенезу: збільшення пе
риферичного венозного тиску, власне розши
рення вен і функціональну недостатність веноз
них клапанів (рис. 32.31).
1. Збільшення тиску крові у венозних судинах.
Венозна гіпертензія майже завжди пов’язана зі
збільшенням кровонаповнення венозних судин.
Основними причинами цього можуть бути:
а) утруднення відтоку крові (збільшення внут
рішньочеревного тиску, звуження магі
стральних вен);
б) перерозподіл крові між системами глибо
ких і поверхневих вен нижніх кінцівок, що
203
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Порушення енергозабезпечення
венозної стінки
"Зачаровані" кола (circuli vitiosi) у патогенезі варикозної хвороби вен.
ГМК - гладкі м'язові клітини
здійснюється через комунікативні (перфорантні) венозні судини;
в) скидання крові з артеріальної системи у ве
нозну, пов’язане з відкриттям артеріоловенулярних анастомозів.
2.
Слабкість венозної стінки. Розширення
венозних судин настає під впливом тис
ку крові в разі зменшення тонусу гладких
м’язів венозної стінки. Цьому сприяють та
кож зміни еластичних властивостей колаге
нових структур.
Одна з гіпотез патогенезу варикозного розши
рення вен ґрунтується на фактах про первинні
розлади метаболізму венозної стінки. Зокрема,
великого значення надають порушенням енер
гетичного обміну в гладких м’язах венозних су
дин. Розлади енергопостачання венозної стінки,
на думку прихильників гіпотези, спричиняють
204
ся до функціональних і структурних порушень
гладких міоцитів. Перші з них виявляють себе
зменшенням тонусу венозної стінки і неспро
можністю відповідати скороченням на нервові
й гуморальні регуляторні стимули, а другі - че
рез ушкодження клітин зумовлюють розвиток
змін позаклітинних компонентів венозної стін
ки, зокрема колагену.
3.
Функціональна недостатність венозних
клапанів. Вона виникає внаслідок розши
рення венозних судин, тобто найчастіше
є вторинною за походженням. Одначе існує
думка, що у декотрих хворих неповноцін
ність клапанів глибоких і перфорантних
вен зумовлена їхніми анатомічними вада
ми, що мають спадкову природу. Незалеж
но від конкретних причин недостатність
клапанного апарату вен веде до збільшення
кровонаповнення венозних судин, а отже,
і до підвищення тиску крові в них — “зача
роване коло” замикається.
Отже, незважаючи на те, який із трьох вищенаведених факторів започатковує розвиток хво
роби, виникає стійкий circulus vitiosus, завдяки
якому патогенез варикозного розширення вен
набуває рис самодостатності, а патологічний
процес як такий прогресує і стає необоротним.
Недостатність венозного кровообігу
Розлади, що виникають унаслідок порушень
руху крові по венозних судинах, називають ве
нозною гіперемією, або венозним застоєм. Про
основні причини й механізми їхнього розвитку
йшлося в главі 16.
Неспроможність вен відводити кров від ор
ганів і тканин можна також позначити як недо
статність венозного кровообігу. За клінічним
перебігом вона може бути гострою і хронічною.
Найчастішою причиною першої є гострий тром
бофлебіт, а друга — виникає, як правило, внаслі
док набутих чи вроджених уражень венозних
судин.
Гострий тромбофлебіт. У цій хворобі поєдна
ні два патологічних процеси - тромбоз веноз
них судин і запалення венозної стінки. При цьо
му спочатку може розвиватися тромбоз, а потім
запалення (флебіт), або навпаки - запалення
з наступним долученням тромбозу.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Патогенез тромбозу й запалення венозної
стінки в цілому підпорядкований загальним за
кономірностям розвитку цих патологічних про
цесів (див. глави 16 і 17).
Запалення венозної стінки (флебіт) залеж
но від своєї причини може спочатку зачепити
тільки внутрішню (ендофлебіт) або зовнішню
(перифлебіт) оболонку венозної судини. Завдя
ки явищам вторинної альтерації запалення час
то поширюється на всю товщу венозної стінки
(панфлебіт).
Різні патогенні чинники (інфекція, алергія,
механічна травма тощо), спричиняючи ушко
дження стінки вен, ведуть до появи в ній ве
ликої кількості різних медіаторів запального
процесу: лізосомних факторів, біогенних амі
нів (гістаміну, серотоніну), кінінів (брадикініну, калідину), похідних арахідонової кислоти
(простагландинів, лейкотрієнів), продуктів ак
тивації комплементу, цитокінів (лімфо- й монокінів); сполук, що утворюються під час ак
тивації зсідання крові та внаслідок деградації
фібрину.
Під впливом зазначених медіаторів у веноз
ній стінці закономірно порушуються процеси
мікроциркуляції в системі vasa vasorum, підви
щується проникність самої венозної стінки та
її власних судин, відбувається еміграція лей
коцитів. Саме цим і пояснюються характерні
для гострого тромбофлебіту набряк венозної
стінки та її лейкоцитарна інфільтрація.
Вивільнення лейкоцитами цитокінів спричи
няється до розвитку загальних порушень, що
виявляють себе гарячкою, збільшенням вмісту
в крові “білків гострої фази запалення”, зміна
ми показників периферичної крові (лейкоци
тоз, зсув лейкоцитарної формули вліво, підви
щення ШОЕ).
Розлади кровообігу в периферичних ткани
нах, що виникають при гострому тромбофлебіті
в результаті тромбозу венозних судин та набря
ку венозної стінки, часто позначають термінами
“гострий венозний застій ”, “гостра венозна не
прохідність На наслідки повної оклюзії вен по
суті впливають два фактори: (а) функціональна
важливість венозного колектора, що в ньому
утворився тромб, та (б) розвиток колатерально
го кровообігу. Залежно від співвідношення між
ними розрізняють три варіанти подій:
1) тромбоз не надто важливого венозного ко
лектора з добре розвиненими колатераля-
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
ми. За цих умов не виникає значного ве
нозного застою, а гемодинамічні наслідки
венозного тромбозу непомітні;
2) тромбоз важливої магістральної вени
з добре розвиненими колатералями. При
цьому на самому початку виникають ви
ражені порушення місцевого кровообігу,
що виявляють себе розвитком значного
набряку тканин, але згодом, завдяки по
ступовій адаптації бічних венозних гілок
і відведенню крові обхідними шляхами,
клінічні симптоми гострого венозного за
стою спадають;
3) тромбоз важливого венозного колекто
ра з погано розвиненими колатералями.
Ця ситуація найнесприятливіша, бо запо
чатковує розвиток значних місцевих і за
гальних гемодинамічних порушень.
На рис. 32.32 представлено послідовність по
дій та “зачаровані кола”, що характеризують па
тогенез розладів кровообігу за умов венозного
тромбозу. З певним припущенням можна виді
лити дві стадії розвитку гемодинамічних пору
шень, спричинених гострою непрохідністю ве
нозних судин, — венозну й артеріальну.
I. Венозна стадія. Провідною ланкою патоге
незу цієї стадії можна вважати перехід ве
ликого об’єму рідини з кровоносних судин
у тканини. До цього спричиняється збіль
шення фільтраційного тиску в капілярах
унаслідок венозного застою. Перерозподіл
рідини між судинним руслом та інтерстицієм має щонайменше два важливі наслідки:
набряк і гіповолемію.
Розвиток набряку істотно погіршує умови
місцевого кровообігу на рівні як венозних, так
і артеріальних судин. Збільшення внутрішньотканинного тиску рідини, з одного боку, веде
до здавлювання венозних судин ззовні, що по
силює венозний застій, а з другого - зумовлює
зменшення трансмурального тиску в артеріолах.
II. Артеріальна стадія. Зниження артеріаль
ного тиску, зумовлене гіповолемією, і збіль
шення внутрішньотканинного тиску, діючи
одночасно, створюють умови, що порушу
ють рух крові по артеріальних судинах. Як
тільки тиск крові в артеріолах стає рівним
або меншим за суму внутрішньотканин
ного тиску і так званого критичного тиску
205
закриття (мінімальний трансмуральний
тиск, що при ньому артеріоли ще відкриті),
то настає спадіння артеріол і кровопоста
чання тканин припиняється. Ішемія, через
зумовлену нею гіпоксію, веде до розвитку
некрозу тканин (ішемічний тромбофлебіт),
а за умов повної непрохідності всіх веноз
них колекторів кінцівки та приєднання
гнильної інфекції настає найнебезпечніше
ускладнення - венозна гангрена.
Хронічна венозна недостатність. Основними
причинами її розвитку є зміни венозних судин,
зумовлені тромбофлебітом (посттромбофлебітний синдром); уроджена відсутність чи недо
статність клапанів глибоких вен кінцівок, вари
козна хвороба вен.
Поява
посттромбофлебітного
синдрому
пов’язана з неповним поновленням руху крові
по магістральних венах та декомпенсацією ко
латерального кровообігу.
Патогенез порушень, що виникають у ткани
нах при хронічній венозній недостатності, до
сить складний (рис. 32.33). Він торкається всіх
ланок місцевого кровообігу й мікроциркуляції:
венозної, артеріальної, капілярної, лімфатичної.
Чинником, що започатковує розвиток основ
них клінічних проявів хронічного венозного
застою, є підвищення венозного тиску крові.
Саме воно зумовлює набряк тканин і зворотний
рух крові з глибоких вен кінцівок у поверхневі.
Останнє спричинює появу вторинного варикоз
ного розширення вен. Перехід рідини з кровонос
них судин в інтерстицій веде до підвищення вну
трішньотканинного тиску. Ця обставина долучає
до патогенезу хронічної венозної недостатності
розлади артеріального кровозабезпечення (іше
мія) та лімфообігу (вторинний лімфо стаз).
Спадіння артеріол, що настає через значне
зменшення трансмурального тиску, має два важ
ливі наслідки. З одного боку, розкриваються ар
теріовенозні анастомози, через які кров перехо
дить з артерій безпосередньо у венозну систему.
При цьому збільшується кровонаповнення вен,
що сприяє варикозному розширенню їх. З дру
гого боку, кров не надходить у капіляри - розви
вається ішемічна гіпоксія. Вона, своєю чергою,
через ушкодження ендотеліальних клітин та
накопичення продуктів метаболізму на перших
порах веде до збільшення проникності стінки
кровоносних судин (капілярів, венул). Відбува-
206
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Механізми гемодинамічних порушень в умовах гострої непрохідності венозних колекторів
ГЛАВА 32. ПАТОФІЗІОЛОГІЯ КРОВОНОСНИХ СУДИН
Основні механізми клінічних проявів хронічної венозної недостатності
207
208
ється діапедез еритроцитів та вихід у тканини
білків плазми крові. Згодом навколо судин мікроциркуляторного русла утворюються щільні
фібринові “манжети”, що перешкоджають ди
фузії газів та поживних речовин до клітин. Як
наслідок ішемічної гіпоксії та описаних розла
дів мікроциркуляції розвиваються дистрофічні
зміни в шкірі та гіподермальний склероз. Шкіра
стоншується, стає нерухливою (не збирається
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
в складку), зникає волосся; незначні травми,
розчісування викликають поранення; виника
ють трофічні виразки, послаблюються процеси
загоювання.
Вторинний лімфостаз, що супроводжує хро
нічну венозну недостатність, посилює розвиток
набряків і через них, як це випливає з рис. 32.33,
усі інші прояви венозного застою.
ГЛАВА 33
Сукупність процесів, що забезпечують спо
живання організмом кисню й видалення вуг
лекислого газу, називають диханням. Воно
здійснюється в кілька послідовних етапів (сто
совно кисню, а для вуглекислого газу - навпа
ки): (І) вентиляція легенів (легенева вентиля
ція) <■> обмін газів між зовнішнім середовищем
та легеневими альвеолами; (II) дифузія газів
через альвеоло-капілярну мембрану, завдяки
чому забезпечується обмін газів між легеневи
ми альвеолами та кров’ю легеневих капілярів;
(III) транспорт газів кров’ю до капілярів тка
нин; (IV) дифузія газів із капілярів у тканину
до її клітин; (V) внутрішнє (клітинне) дихання,
що відбувається в мітохондріях клітин.
Перші два етапи позначають терміном зов
нішнє дихання. Саме його порушення і будуть
предметом обговорення у цій главі підручника.
Зовнішнє дихання виконує дві групи функцій:
дихальні й недихальні.
До дихальних відносять:
а) підтримання газового гомеостазу, тобто
сталості напруги кисню та вуглекислого
газу в крові. Звісно, що ця функція є основ
ною;
б) підтримання кислотно-основної рівноваги
завдяки регуляції вмісту С02 в крові (див.
главу 29);
в) участь у процесах терморегуляції.
Недихальними вважаються такі функції:
а) метаболічні - участь у перетворенні та
інактивації біологічно активних речовин
крові;
б) захисні. До таких належить бар ’єрна функ
ція повітроносних шляхів;
в) генерація звуків.
Структурними елементами системи зовніш
нього дихання є:
1) грудна клітка - рухливий каркас, що зда
тен змінювати свій об’єм, і замкнена по
рожнина, що захищає легені від прямих
зовнішніх впливів;
2) дихальні м’язи (діафрагма, зовнішні та
внутрішні міжреберні м’язи), що забезпе
чують зміни об’єму грудної клітки;
3) плевральна порожнина, через зміну тиску
у якій відбувається передавання дихаль
них рухів від грудної клітки до легенів;
4) легені — центральні органи системи зов
нішнього дихання, у яких здійснюється
альвеолярна вентиляція і відбувається об
мін газів між альвеолами та кров’ю;
5) повітроносні шляхи (трахея, бронхи, брон
хіоли), через які здійснюється вентиляція
легенів. Крім того, вони очищають, зволо
жують і зігрівають повітря, що надходить
ЗЗОВНІ.
Регуляція зовнішнього дихання здійснюєть
ся нервовими механізмами через спеціалізовані
нейрони стовбурової частини головного мозку,
об’єднані поняттям дихальний центр.
Недостатність зовнішнього дихання
Так позначають нездатність дихальної системи
забезпечувати нормальний склад газів крові (га
зовий гомеостаз).
Класифікація:
І. За клінічним перебігом розрізняють гостру
та хронічну недостатність дихання.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
210
» Гостра недостатність виникає раптово,
триває протягом кількох днів, годин і навіть
хвилин. Її прикладом може бути асфіксія
(див. далі).
»
Хронічна недостатність
розвивається
протягом тривалого часу і є наслідком
хвороб бронхів та легень (хронічна пнев
монія, пневмосклероз, емфізема легень
та ін.).
II. За вираженістю клінічних ознак недостат
ність дихання може бути компенсованою і де
компенсованою.
При компенсованій недостатності газовий
склад крові ще не змінений (спрацьовують ком
пенсаторні захисні механізми); при декомпенсо
ваній — газовий гомеостаз порушений.
III. Декомпенсована дихальна недостатність
може бути різної тяжкості. За вираженістю змін
газового складу артеріальної крові виділяють
три ступені недостатності зовнішнього дихання
(див. табл.)
1 ступінь
>70
<50
II ступінь
70-50
50-70
III ступінь
<50
>70
IV. За патогенезом розрізняють два різнови
ди: (1) вентиляційну, вона ж гіперкапнічна, і (2)
паренхіматозну, вона ж гіпоксемічна, недостат
ність зовнішнього дихання (рис. 33.1).
V. Залежно від причин розвитку виділяють:
а) центральну,
б) периферичну, або нервово-м’язову;
в) торако-діафрагмальну, або парієтальну;
г) легеневу дихальну недостатність.
Основні клінічні прояви. Недостатність зовніш
нього дихання виявляє себе змінами, більшість
Патогенетична класифікація недостатності зовнішнього дихання.
КОР - кислотно-основна рівновага
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
з яких пов’язані з порушеннями газового складу
крові. До таких належать:
1) порушення регуляції дихання, серед яких
найчастіше мають місце різні варіанти за
дишки (див. далі);
2) гіпоксемія (зменшення рО, артеріальної
крові). Вона є причиною (а) гіпоксії (див.
главу 23) і (б) ціанозу. Останній виникає,
коли насичення гемоглобіну киснем стає
нижче 80 % (синюшний відтінок шкіри
та видимих слизових оболонок обумовлю
ється відновленим гемоглобіном);
3) гіперкапнія (збільшення рС02 артеріальної
крові). З її розвитком пов’язані: (а) збуджен
ня дихального центру і задишка (коли рС02
стає вище 90-100 мм рт. ст,, збудження ди
хального центру змінюється його гальму
ванням); (б) розширення мозкових судин
і звуження судин м’язів, нирок; (в) зміщен
ня кривої дисоціації оксигемоглобіну впра
во; (г) газовий ацидоз.
4) газовий ацидоз. Він обумовлює: (а) пору
шення функцій білкових молекул (конформаційні зміни), що є особливо небезпечним
для центральної нервової системи (може
розвиватися дихальна кома)', (б) збудження
дихального центру; (в) збільшення про
никності клітинних мембран (розвиваєть
ся набряк та ушкодження клітин); (г) змі
щення кривої дисоціації оксигемоглобіну
вправо - ефект Бора (див. главу 23).
211
ВЕНТИЛЯЦІЙНА
НЕДОСТАТНІСТЬ ДИХАННЯ
Вентиляційна недостатність виникає внаслі
док порушень обміну газів між атмосферним
повітрям і альвеолами легень, тобто в результаті
порушень легеневої вентиляції (гіповентиляції).
Для цього виду дихальної недостатності ха
рактерні такі порушення гомеостазу:
а) зменшення напруги кисню (р02) в артері
альній крові - гіпоксемія',
б) збільшення напруги вуглекислого газу (рС02)
в артеріальній крові - гіперкапнія',
в) газовий ацидоз.
Для характеристики вентиляційної недо
статності використовують статичні (легеневі
об’єми та ємності) і динамічні показники. Пер
ші - відображають потенціальні можливості
органів зовнішнього дихання здійснювати вен
тиляцію, а другі - інтенсивність цього процесу.
Статичні показники вентиляції представ
лено на рис. 33.2. До динамічних відносять:
(а) частоту дихання', (б) хвилинний об’єм ди
хання (ХОД), що дорівнює добуткові від мно
ження дихального об’єму на частоту дихання;
(в) показник альвеолярної вентиляції (V ), що
є тією частиною ХОД, яка досягає альвеол
і бере участь у газообміні (визначається як
різниця між ХОД і вентиляцією мертвого про-
Легеневі об'єми та ємності: ДО - дихальний об'єм; РОвд - резервний об'єм вдиху; РОвид - резервний об'єм
видиху; ЗОЛ - залишковий об'єм легень; ЖЄЛ - життєва ємність легень; Євд - ємність вдиху; ФЗЄ - функціональна залишкова
ємність; ЗЄЛ - загальна ємність легень
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
212
стору); (г) максимальну вентиляцію легень —
об’єм повітря, що проходить через легені за
певний проміжок часу при диханні з макси
мально можливою частотою і глибиною.
Розрізняють три патогенетичні варіанти вен
тиляційної недостатності зовнішнього дихан
ня: обструктивну, рестриктивну і дисрегуляторну.
Обструктивна недостатність
Обструктивною називають дихальну недостат
ність, яка виникає внаслідок збільшення аероди
намічного опору повітроносних шляхів.
Оскільки за законом Пуазейля аеродинаміч
ний опір (R) визначається формулою
то основним чинником, від якого він залежить,
є радіус повітроносних трубок (г). Так, змен
шення цього показника вдвічі веде до зростання
опору в 16 разів.
Причини. Зменшення радіуса трубок повітро
носних шляхів може бути зумовлене такими
змінами:
1) спастичним скороченням гладких м’язів
бронхів і бронхіол (бронхіальна астма);
2) набряком їхніх слизових оболонок (брон
хіти, бронхіоліти);
3) здавлюванням бронхів чи бронхіол, напри
клад, при емфіземі легень.
Порушення прохідності бронхів і бронхіол
лежить в основі недуг, що їх об’єднують по
няттям хронічні обструктивні хвороби легень.
До таких відносять бронхіальну астму, емфізе
му та хронічні бронхіти.
Бронхіальна астма. Це алергічне захворюван
ня, що характеризується повторними нападами
експіраторної задишки (утруднений видих), ви
кликаної дифузним порушенням прохідності
бронхів.
Бронхіальна астма належить до атонічних
хвороб і є однією з клінічних форм алергічних
реакцій І типу (анафілактичних) (див. главу 15).
У її виникненні важливе значення має спадкова
схильність.
шшшш
Медіатори алергії, що вивільнюються тканин
ними базофілами сенсибілізованого організму при
взаємодії фіксованих на них антитіл (IgE) з анти
генами, чинять вплив на клітини і тканину бронхів
та бронхіол. Так, під впливом гістаміну і лейкотрієнів у цих структурах (а) відбувається спастич
не скорочення гладких м’язів, (б) виникає набряк,
(в) збільшується утворення слизу. Це все зменшує
прохідність бронхів і бронхіол та зумовлює об
структивний характер порушень вентиляції легень.
Під час тривалих нападів бронхіальної астми
розвивається недостатність дихання, для якої
характерним є утруднення видиху (експіратор
на задишка).
Це пояснюється тим, що при зменшенні діамет
ру бронхів і бронхіол видих, який у нормі здійс
нюється пасивно за рахунок пружності грудної
клітки й еластичної тяги легень, неможливий без
участі експіраторних м’язів, при скороченні яких
активно зменшується об’єм грудної клітки та
збільшується внутрішньоплевральний тиск. Коли
тиск іззовні на бронхіоли стає вищим за тиск зсе
редини, їхній просвіт повністю перекривається,
і значна кількість повітря залишається в альвео
лах, будучи неспроможною вийти назовні.
Описані події позначаються на статичних по
казниках вентиляції легень (рис. 33.3). Так, рі
вень максимально можливого видиху підніма
ється вгору й наближається до рівня спокійно
го видиху. У зв’язку з цим змінюються об’єми
та ємності легень, що залежать від величини
максимального видиху, а саме: (1) зменшується
резервний об’єм видиху; (2) зменшується жит
тєва ємність легень; (3) збільшується залишко
вий об’єм легень.
Обструктивний характер порушень вентиля
ції легень у хворих на бронхіальну астму ви
являють за допомогою функціональної проби
Тіффно шляхом визначення об’єму форсованого
видиху (ОФВ) (рис. 33.4). ОФВ - це об’єм повіт
ря, що видихається із легенів при форсовано
му видихові за 1 секунду. У нормі відносно до
життєвої ємності легень він становить 70-75 %.
При обструктивній недостатності дихання цей
показник істотно зменшується.
Емфізема легень. Це захворювання легеневої
паренхіми, що супроводжується руйнуванням
альвеолярних перегородок і тонкої мережі ле
геневих капілярних судин, а також звуженням
просвіту термінальних бронхіол.
213
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
Зміни статичних показників вентиляції легень при бронхіальній астмі та емфіземі.
Значення рівнів, легеневі об'єми та ємності див. рис. 33.2
Об'єм форсованого видиху (ОФВ, тест Тіффно) при обструктивній вентиляційній
недостатності дихання
У виникненні емфіземи легень мають значен
ня такі фактори:
а) інфекції, що зумовлюють хронічний обструк
тивний бронхіт;
б) тривала дія токсичних речовин, велика кіль
кість яких міститься в цигарковому димі;
в) старіння організму;
г) спадковість.
Центральною ланкою патогенезу емфіземи
вважають деструкцію еластинових структур
легеневої тканини, яка настає в результаті по
рушення рівноваги між активністю протеаз, що
розщеплюють еластин, та їхніми інгібіторами
(еластолітична концепція).
Ця концепція ґрунтується як на клінічних, так
і на експериментальних доказах. Відомо, що де-
214
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Еластолітична концепція розвитку емфіземи легень
фіцит активної форми а ^інгібітору протеїназ
((Xj-ІП), зумовлений мутацією відповідного гена,
значно збільшує вірогідність розвитку емфізе
ми легень - особливо в осіб, що палять. 0,-111
є основним інгібітором еластолітичних фермен
тів, що вивільнюються нейтрофілами і макро
фагами під час запалення.
В експериментах показано, що введення
в трахею протеолітичного ферменту папаїну
веде до деградації еластину легеневої ткани
ни та розвитку у тварин емфіземи. З позицій
еластолітичної концепції патогенез емфіземи
легень вбачається так (рис. 33.5). Під впли
вом зовнішніх чинників (інфекцій, токсичних
речовин цигаркового диму та ін.) виникає
первинне ушкодження бронхів, бронхіол і ле
геневої паренхіми, що започатковує процес
запалення (див. главу 17). Його важливим ком
понентом є еміграція нейтрофілів і активація
альвеолярних макрофагів. Ці клітини стають
джерелом ферментів (еластази, матриксних
металопротеїназ), що розщеплюють (гідролізують) еластин, викликаючи в такий спосіб
деградацію еластинових структур легеневої
тканини, тобто вторинне ушкодження легень
(вторинну альтерацію).
Припинити цей процес можуть тільки інгі
бітори протеолітичних ферментів - а,-ІП, а,макроглобулін та деякі інші. Проте в умовах
спадково зумовленого їх дефіциту такий захист
не спрацьовує. Слід зазначити, що деякі компо
ненти цигаркового диму, а також вільні радикали
як лейкоцитарного, так і зовнішнього походжен
ня мають здатність до інактивації інгібіторів
протеаз, а отже, усувають перешкоду на шля
ху деструктивних процесів, що відбуваються
215
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
Механізм розвитку обструктивної недостатності дихання при емфіземі легень
в легенях. Старіння організму сприяє розвитко
ві емфіземи легень, оскільки в людей похило
го і старечого віку значно зменшується синтез
еластину з одночасним посиленням активності
еластолітичних ферментів.
Формування емфіземи легень стає причиною
обструктивної недостатності дихання. Механіз
ми розвитку останньої є доволі складними, що
випливає зі схеми, представленої на рис. 33.6.
Насамперед слід мати на увазі, що стінка
бронхіол через відсутність хрящового каркаса
є м’якою і податливою. Бронхіоли збільшують
свій просвіт під час вдиху під дією транспуль-
монального тиску, тобто тиску зсередини (у цей
час альвеоли розправляються), і зменшують
його під час видиху за рахунок тиску ззовні
(у цей час альвеоли спадаються).
Обструкція бронхіол при емфіземі має два
механізми:
1) обструкція зсередини. Її причини - посиле
на секреція слизу й набряк, що є компонен
тами хронічного бронхіту, як результату дії
інфекцій та токсичних компонентів цигар
кового диму. Крім того, під час вдиху вини
кає і і,функціональна,:' обструкція. Річ у тім,
216
що просвіт бронхіол опосередковано зале
жить від еластичної тяги легень: що більша
їхня еластичність, то більше розрідження
необхідно створити в плевральній порож
нині під час вдиху (і тим самим підвищити
транспульмональний тиск), щоб перебороти
еластичну тягу легень і розтягнути їх. Якщо
ж легені втрачають свою еластичність, то
вони розтягуються набагато легше, тобто
при набагато меншому транспульмональному тиску. У результаті зменшується сила, що
діє на стінки бронхіол зсередини і розправ
ляє їх, - просвіт бронхіол під час вдиху стає
меншим, ніж у нормальних легенях;
2) обструкція ззовні. Зменшення еластичної
тяги легень, зумовлене розпадом еласти
нових структур, і обструкція бронхіол зсе
редини унеможливлюють пасивний видих.
Для його здійснення залучаються експіра
торні м’язи, при скороченні яких відбува
ється активне зменшення об’єму грудної
клітки й підвищення внутрішньоплеврального тиску. У міру видихання збільшуєть
ся сила, що діє на бронхіоли ззовні. Настає
такий момент, коли стінки бронхіол пов
ністю спадаються ще до того, як відбу
деться повне спадіння альвеол: унаслідок
цього продовження видиху стає неможли
вим і він припиняється. Таким чином, при
емфіземі стінки бронхіол відіграють роль
клапана, який під час вдиху відкривається
(хоча й менше, ніж у нормі), а під час види
ху закривається, і повітря опиняється не
наче в пастці. Як результат, альвеоли зали
шаються постійно роздутими, у них збіль
шується кількість залишкового повітря.
Описані вище зміни в легенях позначаються на
статичних показниках вентиляції (див. рис. 33.3).
Так, зміщуються стосовно норми практично всі
рівні, що характеризують цей процес. Через
зменшення еластичної тяги легень, яка в нормі
заважає вдихові, дещо піднімається вгору рівень
максимального вдиху. Зменшення еластичнос
ті легеневої тканини обумовлює підняття рівня
спокійного видиху. Здавлювання бронхіол під
час видиху наближає рівень максимально до рів
ня спокійного видиху.
У результаті при емфіземі змінюються всі,
крім дихального об’єму, легеневі об’єми та єм
ності:
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
а) зменшуються резервний об’єм вдиху й ре
зервний об’єм видиху, життєва ємність ле
гень і ємність вдиху;
б) збільшуються залишковий об’єм легень,
функціональна залишкова ємність і загаль
на ємність легень.
При емфіземі легень порушується не тільки
їхня вентиляція, а й перфузія (кровообіг). Це
обумовлено тим, що протеолітичні ферменти та
вільні радикали руйнують не лише альвеолярні
перегородки, їх об’єктами стають також стінки
капілярних судин. Унаслідок цього зменшується
кількість капілярів у легеневій тканині, що, з од
ного боку, порушує газообмін між альвеолами
і кров’ю, а з другого - веде до збільшення тис
ку (гіпертензії) в малому колі кровообігу з усіма
наслідками, що з цього випливають (див. далі).
Зменшення альвеолярної вентиляції та перфузії в різних ділянках легень відбувається не
рівномірно, що виявляє себе різнонаправленими
змінами вентиляційно-перфузійного відношення
(див. далі). Це стає ще одним чинником, що по
силює недостатність зовнішнього дихання при
емфіземі легень.
Рестриктивна недостатність
Рестриктивною називають дихальну недостат
ність, пов’язану з обмеженням зовнішнього ди
хання.
Причини. Обмеження дихання може зумов
люватися легеневими й позалегеневими при
чинами.
І.
Легеневі причини :
1) зменшення дихальної поверхні легень, що
буває після видалення сегмента, частки
або цілої легені; при руйнуванні великих
ділянок легень (туберкульоз); у результаті
розсмоктування повітря в альвеолах і спа
діння легеневої тканини (ателектаз);
2) збільшення пружного опору легень — пору
шення їхньої здатності розправлятися під
час вдиху. Така ситуація закономірно ви
никає при:
а) зменшенні розтяжності легень у зв’язку із
заміщенням еластичних структур легене
вої тканини колагеновими {пневмосклероз,
пневмоконіоз), при інтерстиційному запа
ленні легень;
217
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
Зміни статичних показників вентиляції легень в умовах рестриктивної недостатності дихання.
Значення рівнів, легеневі об'єми і ємності див. рис. 33.2
б)
збільшенні сили поверхневого натягу
в альвеолах при порушеннях сурфактанту (зменшенні його утворення або посиле
ному руйнуванні).
II. Позалегеневі причини:
1) порушення структури й функції дихальних
м ’язів (міозити, міодистрофія Дюшена);
2) порушення рухливості грудної клітки
і діафрагми. До цього спричиняються:
(а) вроджена чи набута деформація ребер
і хребетного стовпа (кіфосколіоз), окосте
ніння реберних хрящів; (б) больові відчут
тя, що виникають під час дихання, напри
клад, при міжреберній невралгії, травмах
грудної клітки; (в) зрощення листків плев
ри; (г) перешкоди для екскурсії діафрагми
з боку черевної порожнини, зокрема асцит,
метеоризм, високий ступінь черевного
ожиріння (синдром Піквіка);
3) підвищення тиску в плевральній порожни
ні унаслідок пневмотораксу, гідротораксу,
гемотораксу, емпієми плеври.
Рестриктивна недостатність дихання ха
рактеризується утрудненням вдиху, що клі
нічно виявляє себе розвитком інспіраторної
задишки.
При обмеженні зовнішнього дихання зни
жується рівень максимально можливого вдиху,
а звідси зменшуються і всі статичні показники
вентиляції, що залежать від нього: резервний
об’єм вдиху, ємність вдиху, життєва ємність ле
гень, загальна ємність легень (рис. 33.7).
Підтримання сталості хвилинного об’єму ди
хання в умовах рестриктивної його недостат
ності досягається двома механізмами:
1) збільшенням частоти дихання (тахіпное).
Проте часте поверхневе дихання є мало
ефективним, оскільки при ньому зменшу
ється показник альвеолярної вентиляції
(див. нижче);
2) посиленою роботою дихальних м’язів.
Якщо зазвичай на їхню роботу витрачаєть
ся до 2 % поглиненого організмом кисню,
то в умовах рестриктивної недостатності
на ці потреби йде до 20-30 %. Цей меха
нізм компенсації є досить ефективним, він
здатен тривалий час підтримувати належ
ну вентиляцію альвеол.
Пневмоторакс. Так називають скупчення по
вітря в плевральній порожнині та підвищення
тиску в ній.
Повітря потрапляє в порожнину плеври:
а) при пораненнях, що проникають у грудну
клітку;
б) при розриванні емфізематозних альвеол на
поверхні легені;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
218
Зміни дихання при пневмотораксі: механізм частого поверхневого дихання (тахіпное)
в) у результаті розпаду легеневої тканини (ту
беркульоз, пухлина, абсцес);
г) при введенні повітря в плевральну порож
нину з лікувальною метою (методика ліку
вання кавернозного туберкульозу легень).
Якщо повітря, яке потрапило в плевральну по
рожнину, не сполучається з атмосферним пові
трям, пневмоторакс називають закритим, якщо
сполучається - відкритім. Нарешті, якщо осо
бливості вхідного отвору в порожнині плеври до
пускають потрапляння повітря під час вдиху, але
перешкоджають його виходу при видиху, пнев
моторакс називають клапанним, або вентильним.
Характерною рисою пневмотораксу є часте
поверхневе дихання (тахіпное). Його рефлек
торний механізм показано на рис. 33.8. Спадін
ня однієї легені при пневмотораксі веде до збу
дження іритантних рецепторів, що містяться
в епітелії та субепітеліальному шарі бронхів
і бронхіол. Імпульси від цих рецепторів збу
джують нейрони дихального центру, що від
повідають за вдих. Унаслідок вдиху альвеоли
неушкодженої легені розтягуються більше, ніж
звичайно. На це реагують рецептори розтяг
нення, імпульси від яких гальмують вдих, і той
передчасно припиняється.
Ателектаз. Припинення вентиляції альвеол
веде до розсмоктування в них повітря, унаслі
док чого частина легеневої тканини спадається.
Такий стан називають ателектазом.
В основі ателектазу може бути або (а) змен
шення транспульмонального тиску, або (б)
збільшення сили поверхневого натягу в альвео
лах. До цього спричиняються:
1) фактори, що зменшують тиск всередині
альвеол через повну обтурацію повітроносних шляхів: закупорка чи перекриття
бронхів більшого або меншого діаметра
при пневмонії, злоякісних пухлинах тощо;
2) фактори, що підвищують тиск у плевраль
ній порожнині (скупчення ексудату, пнев
моторакс, гемоторакс);
3) дефіцит сурфактанту або внаслідок пору
шень його утворення, або через посилене
його руйнування.
Сурфактант є сумішшю поверхнево-активних
речовин, що містяться на межі “повітря - рі
дина” в легеневих альвеолах, тобто вистилає
альвеоли зсередини. Основними компонентами
сурфактанту є фосфоліпіди і полісахариди. За
вдяки своїм властивостям сурфактант перешко
джає спадінню та злипанню альвеол.
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
Продуцентами сурфактанту є клітини аль
веолярного епітелію II типу. Порушення їхньої
секреторної діяльності у плода стає причиною
респіраторного дистрес-синдрому
(гіалінового
мембранозу) у новонароджених. При народжен
ні дитини легені через брак сурфактанту не мо
жуть розправитися (не вистачає зусиль, щоб по
долати сили поверхневого натягу в альвеолах)
і настає асфіксія, а згодом смерть.
Зменшення утворення сурфактанту у плода
пов’язують з дефіцитом глюкокортикоїдних гор
монів, від рівня яких залежить синтетична і се
креторна активність альвеолоцитів II типу (див.
главу 37).
У дорослих недостатність сурфактанту може
бути зумовлена посиленим його руйнуванням,
що трапляється при диханні чистим киснем та
отруєннях, коли активуються процеси вільнорадикального окиснення ліпідів.
Дисрегуляторна недостатність
Розвиток вентиляційної недостатності дихання
може бути пов’язаний з порушеннями регуляції
цього процесу. У такому разі можна вести мову
про дисрегуляторний характер респіраторної
недостатності.
Регуляція зовнішнього дихання є складним
процесом, вона здійснюється за обов’язковою
участю нейронів центральної нервової системи,
об’єднаних поняттям дихальний центр. Ці ней
рони завдяки функції автоматизму (здатності ге
нерувати спонтанні потенціали дії) є джерелом
імпульсів, що запускають вдих і видих.
Практично нервова регуляція зовнішнього ди
хання здійснюється через вплив на ці дві складі
процесу вентиляції, які визначають два основні
його параметри - глибину й частоту дихання.
Застосовуючи в експерименті метод перетину
стовбура головного мозку на різних його рівнях,
вдалося з’ясувати місце розташування нейронів
дихального центру (рис. 33.9).
1. Перетин вище мосту. Він не впливає на ха
рактер дихання, що свідчить про локаліза
цію дихального центру нижче цього рівня.
2. Перетин, що відділяє довгастий мозок від
спинного. За таких умов дихання зупиня
ється. Отже, нейрони, що мають стосунок
до діяльності дихального центру, містять
ся в ділянці, яка охоплює довгастий мозок
і міст.
219
3. Перетин на межі верхньої та середньої тре
тин мосту. Якщо при цьому перетнути ще й
обидва блукаючі нерви, то виникає апнейстичне дихання: дихання зупиняється на
фазі вдиху, лише рідко переривається екс
піраторними рухами (видихами). Вважа
ють, що при такому перетині усуваються
гальмівні впливи верхніх відділів мосту на
нейрони, що відповідають за вдих.
4. Перетин довгастого мозку між верхньою
та нижньою його половинами веде до роз
витку гаспінг-дихання: тривалий видих
переривається рідкими і короткими вдиха
ми. Звідси випливає висновок, що основна
маса нейронів дихального центру, що за
безпечують автоматичну регуляцію вдиху
й видиху, локалізується у верхній половині
довгастого мозку.
Методом реєстрації електричної активності
нейронів стовбура головного мозку їх вдало
ся розділити на інспіраторні та експіраторні.
Перші виявляють свою електричну активність
під час вдиху, а другі - під час видиху.
За локалізацією і функціями нейрони ди
хального центру поділяють на чотири групи
(рис. 33.10):
1) дорсальна респіраторна група. Вона скла
дається виключно з інспіраторних нейронів
(збуджуються під час вдиху). Ці клітини
представлені R-нейронами, що посилають
імпульси до діафрагми, і R^-нейронами, що
чинять гальмівний вплив на Ка-нейрони.
Дорсальну групу вважають центром авто
матизму, де відбувається спонтанна, неза
лежна ритмічна генерація імпульсів, що за
безпечують спокійний вдих і видих;
2) вентральна респіраторна група. До неї
входять як інспіраторні, так і експіраторні
нейрони. Нейрони цієї групи починають
“працювати”, коли виникає потреба у по
силенні дихання (див. далі);
3) пневмотаксичний центр. Розташований
у верхній третині мосту. Він регулює мо
мент вимкнення імпульсації інспіраторних
нейронів, завдяки чому обмежує трива
лість вдиху, а отже, чинить вплив на часто
ту дихання. Якщо сигнали з цього центру
сильні, то вдих може тривати 0,5 с, а часто
та дихання зростати до ЗО—40/хв. І навпа
ки, тривалість вдиху при слабких сигналах
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
220
Структура дихального центру
може сягати 5 і більше секунд, а частота
дихання - зменшуватися до кількох дихань
за хвилину;
4) апнейстичний центр. Локалізацією цього,
поки що гіпотетичного, центру вважають
нижню третину мосту. Імпульси, що надходять від нього до дорсальної групи нейронів, перешкоджають “вимкненню” інспіраторних сигналів.
У діяльності дихального центру виділяють два
аспекти: автоматизм нейронів дорсальної респіраторної групи і вплив на нього різних чинників: центральних, механічних, хімічних та інших
(рис. 33.11). Ці впливи позначаються на основних
характеристиках дихання - його глибині й частоті. Розрізняють спокійне і посилене дихання. Роль
різних структур дихального центру у здійсненні
цих процесів представлено на рис. 33.12i33.13.
__ ____ ______
221
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
Чинники, що впливають на автоматизм дихального центру
(А) Спокійне дихання здійснюється завдяки
автоматизму дорсальної респіраторної групи
нейронів. Імпульси від збуджених ^-нейронів
ідуть у передні роги спинного мозку (сегменти
CIII-CIV), а звідти по діафрагмальних нервах
до діафрагми: та скорочується і настає спокій
ний вдих.
Водночас імпульси від Ка-нейронів надхо
дять до Rp-нейронів, збуджуючи їх. Від останніх
збуджуються
інспіраторно-гальмівні
нейрони
(ІГН), завдяки чому пригнічується імпульсація R -нейронів. Унаслідок цього скорочення
діафрагми припиняється й вона розслаблюється.
Видих відбувається пасивно під дією еластич
ної тяги легень та пружності грудної клітки.
З початком видиху зникає збудження Rpнейронів та усувається їхній опосередкований
гатьмівний вплив на Ra-HefipoHH. Імпульсна
активність останніх поновлюється і розпочина
ється новий вдих.
На діяльність дорсальної респіраторної групи
нейронів впливають пневмотаксичний і апнейстичний центри. Збудження першого гальмує,
а другого, навпаки, посилює генерацію імпуль
сів ^-нейронами.
(Б) Посилене дихання. До його здійснення до
лучаються нейрони вентральної респіраторної
групи. При сильному збудженні Ra-HefipOHiB
(або підвищенні збудливості клітин вентраль
ної групи) інформація передається до інспіраторних нейронів сусідньої частини дихального
центру, звідти - до передніх рогів грудних сег
ментів спинного мозку, а далі - до зовнішніх
міжреберних і додаткових інспіраторних м’язів:
настає посилений вдих.
Одночасне
збудження
експіраторно-сти
муляційних нейронів (ЕСН) веде до “вмикання”
експіраторних нейронів і скорочення в результа
ті цього внутрішніх міжреберних та додаткових
експіраторних м’язів, зокрема м’язів живота.
Настає активний видих.
При патології внаслідок дії рефлекторних,
гуморальних або інших чинників на дихальний
центр можуть змінюватися ритм, глибина і час
тота дихання, а також може виникати задишка.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
222
Механізми СПОКІЙНОГО дихання. ПТЦ - пневмотаксичний центр, АПЦ-апнейстичний центр,
ІНГ - інспіраторно-гальмівні нейрони
Ці зміни є або (а) проявом компенсаторних
реакцій організму, спрямованих на підтримку
сталості газового складу крові, або (б) відобра
женням порушень нормальної регуляції дихан
ня, що ведуть до розладів альвеолярної вентиля
ції, до недостатності дихання.
Причинами дисрегуляторних змін дихання
можуть бути:
а) порушення функції дихального центру. Во
ни часто є наслідком прямих ушкоджуваль
них впливів ішемії мозку, черепно-мозкової
травми, пухлин, різних інтоксикацій. Проте
можуть мати і рефлекторний характер, як,
приміром, при синдромі "апное уві снГ.
Описано рідкісний синдром “прокляття Ун
дини”, при якому неможлива автоматична діяль
ність дихального центру. Хвора людина дихає
лите тоді, коли свідомо контролює акт дихан
ня (на час сну потрібне штучне дихання). Цей
синдром асоціюється з хворобою Гіршпрунга
(мегаколон), дисфагією і нейробластомою. Його
причиною є мутація одного з генів, що визначає
диференціацію певних структур нервової систе
ми в ембріогенезі.
б) порушення функції мотонейронів спин
ного мозку. Іннервація дихальних м’язів
може зазнавати змін при таких хворобах, як
поліомієліт, сирингомієлія, бічний аміотрофічний склероз, а також при травмах і пух
линах спинного мозку. Характер і ступінь
порушення зовнішнього дихання при цьому
залежать від місця ушкодження спинного
мозку (наприклад, при ураженні патоло
гічним процесом верхньої шийної частини
спинного мозку порушується робота діа-
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
223
Механізми посиленого дихання. ІНГ—інспіраторно-гальмівні нейрони, ЕСН - експіраторно-стимуляційні
нейрони, ІН - інспіраторні нейрони, ЕН - експіраторні нейрони
фрагми) і від кількості уражених мотонейронів;
в)ураження нервово-м’язового апарату. По
рушення вентиляції можуть виникати при
ушкодженні нервів, що іннервують дихаль
ні м’язи (запалення, авітаміноз, травма),
при утрудненні передачі м’язам нервового
імпульсу (міастенія, ботулізм, правець).
Із м’язів, що беруть участь в акті дихання,
велике значення має діафрагма. При ураженні
п. frenicus порушується її робота, можуть ви
никати парадоксальні рухи: догори - при вди
ху, вниз - при видиху. При клонічних судомах
м’язів діафрагми з’являється гикавка, під час
якої повітря втягується в легені.
Зміни частоти і глибини дихання. Різні фізіо
логічні і патологічні впливи на центральну ре
гуляцію дихання найчастіше виявляють себе та
кими варіантами змін його частоти та глибини.
1. Брадипное - рідке дихання. Механізм його
розвитку полягає в зміні характеру нерво
вої імпульсації, що йде від різних рецепто
рів до дихального центру, або в первинно
му порушенні діяльності самих дихальних
224
нейронів. Розрізняють кілька варіантів брадипное:
а) рефлекторне зменшення частоти дихання.
Може спостерігатися при підвищенні арте
ріального тиску (рефлекс із барорецепторів
дуги аорти й каротидного синуса), при гіпероксії;
б) глибоке рідке дихання. Виникає при під
вищенні опору руху повітря у верхніх ди
хальних шляхах ■» стенотичне дихання.
У цьому випадку вдих і видих відбуваються
повільніше, ніж звичайно. У встановленні
такого типу дихання певну роль відіграють
імпульси, що надходять у дихальний центр
від міжреберних м’язів, що працюють
з підвищеним навантаженням. Крім того,
має значення запізнювання в цьому випад
ку гальмівного рефлексу Герінга - Брейєра
(див. нижче);
в) поверхневе рідке дихання. Часто є резуль
татом зниження збудливості респіраторних
нейронів у результаті прямої дії патоген
них факторів на дихальний центр (тривала
й тяжка гіпоксія різного походження, вплив
речовин з наркотичною дією, органічні
ураження головного мозку при запаленні,
порушеннях мозкового кровообігу, набря
ку; функціональні розлади центральної
нервової системи, такі як невроз, істерич
ні реакції тощо). У всіх цих випадках рід
ке дихання супроводжується зменшенням
його глибини, що призводить до зниження
альвеолярної вентиляції та розвитку недо
статності зовнішнього дихання.
2. Гіперпное — глибоке часте дихання, що
веде до гіпервентиляції легень. В умовах
патології гіперпное розвивається при ін
тенсивній рефлекторній або гуморальній
стимуляції дихального центру, наприклад,
при зниженні парціального тиску кисню
у вдихуваному повітрі, при підвищенні
напруги С02 у крові, при анемії, ацидозі
тощо.
Гіпервентиляція легень супроводжується по
силеним виведенням з організму вуглекислого
газу і розвитком унаслідок цього газового алка
лозу (див. главу 29).
3. Поліпнеє (тахіпное) «* часте поверхне
ве дихання. В основі розвитку поліпное
лежить рефлекторна перебудова роботи
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
дихального центру. У деяких тварин (на
приклад, у собак) часте поверхневе дихан
ня виникає при дії високої температури.
У людини поліпное може спостерігатися
при гарячці, функціональних порушеннях
центральної нервової системи (істерія),
ураженні легень (ателектаз, пневмоторакс,
пневмонія, застійні явища тощо).
Крім того, до розвитку поліпное може спри
чинятися біль, що локалізується в ділянках тіла,
які беруть участь у дихальному акті (грудна
клітка, черевна стінка, плевра). Біль призводить
до обмеження глибини дихання і збільшення
його частоти (щадне дихання).
Поліпное, на відміну від брадипное, знижує
ефективність дихання, тому що при ньому знач
но зменшується альвеолярна вентиляція, газо
обмін в основному відбувається у “мертвому”
просторі, тобто в повітроносних шляхах (трахеї,
бронхах, бронхіолах), а не в альвеолах.
Ілюстрацію цього наведено на рис. 33.14.
Оскільки об’єм альвеолярної вентиляції (V)
є різницею між хвилинним об’ємом дихання
(ХОД) і вентиляцією мертвого простору (VM):
V = ХОД - Уи.«= ДО • п - ОМП • п,
де ДО - дихальний об’єм (у нормі - близько
500 мл), ОМП - об’єм мертвого простору (при
близно дорівнює 150 мл), п — частота дихання,
то можна визначити ефективність дихання при
різній його частоті та глибині.
Із рис. 33.14 випливає, що за однакового хви
линного об’єму дихання його ефективність різ
на: при поліпное альвеолярна вентиляція змен
шується, а при брадипное, навпаки, зростає.
4. Апное - тимчасова зупинка дихання. Може
бути пов’язане зі зменшенням рефлектор
ної або безпосередньої хімічної стимуляції
дихального центру. Наприклад, апное ви
никає у тварини або людини після пасив
ної гіпервентиляції під наркозом унаслідок
зменшення в артеріальній крові напруги
С02 та припиняється одразу ж, як тільки
вміст С02 нормалізується. При швидкому
підвищенні артеріального тиску, напри
клад, при введенні в кров адреналіну, також
спостерігається тимчасова зупинка дихан
ня (рефлекс із барорецепторів).
П
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
225
Вплив частоти (п) та глибини дихання (ДО) на об'єм альвеолярної вентиляції(Va) при сталому
хвилинному об'ємі дихання (ХОД)
Синдром “апное уві снГ характеризується
припиненням легеневої вентиляції під час сну
більш ніж на 10 с. Найчастіше тривалість апное
складає 20-30 с, хоча в тяжких випадках може
сягати 2-3 хв і охоплювати до 60 % загального
часу нічного сну.
Розрізняють обструктивний і центральний
механізми розвитку цього синдрому. Суть пер
шого полягає в обструкції верхніх дихальних
шляхів (хропінні), що супроводжується посту
повим наростанням гіпоксії, гіперкапнії та аци
дозу. У міру збільшення тривалості апное збіль
шується вираженість цих змін і врешті-решт на
стає пробудження або перехід у стадію поверх
невого сну, при якій підвищується тонус м’язів
глотки з відновленням її прохідності (хропіння
зникає). Під час епізодів апное артеріальний
тиск не тільки не падає, а, навпаки, різко підви
щується, у зв’язку з чим обструктивне апное уві
сні вважають фактором ризику серцево-судин
них недуг - артеріальної гіпертензії, ішемічної
хвороби серця, інсульту.
Апное уві сні центрального походження спо
стерігають у нормі, найчастіше при засинанні
у фазі швидкого сну. У здорових осіб воно не
є причиною яких-небудь патологічних змін
і клінічних проявів.
Патологічні типи дихання. При значних роз
ладах центральної регуляції дихання цей про
цес може набувати особливих характеристик,
що дає підстави виділяти т. зв. патологічні типи
дихання. До них відносять дихання Куссмауля, періодичне дихання і термінальне дихання
(рис. 33.15).
І. Дихання Куссмауля. Його причиною є край
ній ступінь збудження дихального центру, що
має місце при декомпенсованому ацидозі, зо
крема у хворих у стані ацидотичної діабетич
ної коми. Це гучне рідке дихання, при якому
після глибокого вдиху настає посилений видих
з активною участю експіраторних м’язів.
IL Періодичне дихання. Періодичним назива
ють таке дихання, при якому періоди дихання
226
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
чергуються з періодами його пригнічення і ап
ное. Найчастіше бувають такі види періодично
го дихання:
1) дихання Чейна — Стокса. Воно характери
зується наростанням амплітуди дихання до
вираженого гіперпное, а потім зменшен
ням її до апное, після чого знову настає
цикл дихальних рухів, що закінчуються
новою зупинкою вентиляції легень.
Циклічні зміни дихання у людини можуть су
проводжуватися потьмаренням і втратою свідо
мості в період апное і його нормалізацією в пе
ріод збільшення вентиляції. Артеріальний тиск
при цьому також коливається, як правило, під
вищуючись у фазі посилення дихання і знижу
ючись у фазі його ослаблення.
У більшості випадків дихання Чейна - Сток
са є ознакою гіпоксії головного мозку. Його мо
жуть викликати деякі лікарські препарати (на
приклад, морфін), перебування на великій висо
Види патологічного дихання
ті (особливо під час сну). У недоношених дітей
його виникнення пов’язують з недосконалістю
нервових центрів.
Патогенез дихання Чейна - Стокса поясню
ють сильним пригніченням діяльності дихаль
ного центру, що виникає при гіпоксії. Тяжке кис
неве голодування веде до зниження збудливості
нейронів дихального центру, унаслідок чого фі
зіологічних рефлекторних і гуморальних подраз
ників стає недостатньо, щоб підтримувати дихан
ня - і воно зупиняється. Під час апное С02 не ви
водиться з організму, напруга його в крові зростає
до такого рівня, при якому вже може наставати
збудження клітин дихального центру - дихання
поступово поновлюється, його амплітуда зростає.
Однак при цьому збільшується виведення С02
з видихуваним повітрям, його напруга в крові
зменшується, а отже, і зменшується його стиму
ляційний вплив на дихальний центр - амплітуда
дихання починає зменшуватися і зрештою настає
нове апное. І так повторюється раз за разом.
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
Під час апное гіпоксія посилюється і ще біль
ше пригнічуються функції клітин кори велико
го мозку та підкіркових структур. Як результат,
людина втрачає свідомість, падає артеріальний
тиск. Поновлення дихання веде до оксигенації
крові, що зменшує гіпоксію головного мозку
і поліпшує функцію нейронів судинорухового
центру. Свідомість прояснюється, підвищується
артеріальний тиск;
2) дихання Біота. Цей вид періодичного ди
хання відрізняється від дихання Чейна —
Стокса тим, що дихальні рухи мають по
стійну амплітуду. Вони раптово припиня
ються, так само як і раптово починаються.
Найчастіше дихання Біота спостерігається
при менінгіті, енцефаліті та інших захво
рюваннях, що супроводжуються ушко
дженням центральної нервової системи,
особливо довгастого мозку;
3) хвилеподібне дихання, або дихання Грокка. Воно нагадує дихання Чейна — Стокса
з тією лише різницею, що замість періо
дів апное реєструють слабке поверхневе
дихання з наступним наростанням його
глибини, а потім поступове зменшення ди
хальних рухів. Вважають, що таке дихання
виникає при тих само патологічних проце
сах, що викликають дихання Чейна - Сток
са, і, можливо, є його початковою стадією.
III.
Термінальне дихання. Так називають ди
хання, характерне для термінальних станів, тоб
то умов, коли організм перебуває на межі життя
і смерті. Найчастіше реєструють два типи тер
мінального дихання:
а) апнейстичне дихання. Такий тип характе
ризується конвульсивним зусиллям вдих
нути, що не припиняється, але зрідка пере
ривається видихом. В експерименті його
спостерігають після перетинання у твари
ни обох блукаючих нервів і мозкового стов
бура на межі між верхньою і середньою
третинами мосту (див. рис. 33.9);
б) гаспінг-дихання. Це поодинокі, рідкі вдихи,
частота й амплітуда яких поступово змен
шуються аж до повної зупинки дихання.
Такий вид дихання спостерігають при аго
нії, наприклад у заключній стадії асфіксії.
Звичайно, характерні для гаспінг-дихання
рідкі та низькоамплітудні вдихи виника
ють після тимчасової зупинки дихання
227
(претермінальної паузи). Поява їх, мож
ливо, пов’язана зі збудженням клітин, що
містяться в каудальній частині довгастого
мозку, після вимикання функції вищерозташованих відділів мозку.
Задишка, або диспное, - це відчуття нестачі по
вітря, що викликає потребу посилити дихання.
У механізмах задишки розрізняють суб’єк
тивний і об’єктивний компоненти.
Суб ’єктивний компонент полягає в тому, що,
відчуваючи нестачу повітря, людина не тільки
мимовільно, але й свідомо збільшує активність
дихальних рухів, прагнучи позбутися цього
тяжкого відчуття, наявність якого і є найбільш
істотною відмінністю диспное від інших видів
порушення регуляції дихання (гіперпное, поліпное та ін.). Тому в людини, що знепритомніла,
задишки не буває.
Іноді суб’єктивний чинник стає провідним
механізмом задишки. Таке відбувається при нев
розах, істерії, психосоматичних розладах.
Об’єктивним компонентом диспное є реф
лекторні й гуморальні впливи на дихальний
центр. Задишка виникає у тих випадках, коли
впливи, що збуджують вдих, є сильнішими за
впливи, що його пригнічують, а також у разі
підвищення чутливості дихального центру до
чинників, які стимулюють дихання. Найважли
вішими з цих впливів є такі.
1. Збудження іритантних рецепторів леге
невої тканини. Цей вид механорецепторів,
що містяться в епітелії та субепітеліальному шарі бронхів і бронхіол, активуєть
ся при сильних або дуже швидких змінах
об’єму легень (спадінні альвеол чи їхньо
му значному розтягненні). В умовах пато
логії може виникати постійна імпульсація
від іритантних рецепторів. Таке буває, на
приклад, при застійних явищах у легенях
(недостатність серця, пневмонія), коли
переповнені кров’ю судини, що оточують
альвеоли, здавлюють їх, унаслідок чого
об’єм альвеол зменшується, що й веде до
збудження іритантних рецепторів.
2. Рефлекси з повітроносних шляхів. Можуть
викликатися твердими частинками, тютю
новим димом, парами хімічних сполук, що
потрапили в повітроносні шляхи і подраз
нюють уже згадувані іритантнірецептори.
Останні в цьому випадку виявляють влас-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
228
тивості хеморецепторів. Значне збудження
іритантних рецепторів спостерігають при
бронхітах, бронхопневмонії, бронхіаль
ній астмі та хворобах, при яких у бронхах
і альвеолах з’являються слиз, ексудат чи
транссудат.
3. Збудження рецепторів інтерстиціальног
тканини легень (J-рецепторів). Усі патоло
гічні процеси, що ведуть до застійних явищ
у легенях (пневмонія, недостатність сер
ця), можуть викликати тривале збудження
J-рецепторів і підвищену стимуляцію ди
хальних нейронів.
4. Рефлекси з барорецепторів аорти і сонної
пазухи. Ці рефлекси долучаються до пато
генезу задишки при крововтраті, шоку, ко
лапсі. В умовах зменшення артеріального
тиску до рівня 70 мм рт. ст. і нижче різко
зменшується імпульсація від барорецепто
рів, яка в нормі чинить гальмівний вплив
на дихальний центр, пригнічуючи вдих
і активуючи видих.
5 . Рефлекси з хеморецепторів аорти і сонної
пазухи. При зниженні в крові напруги 02,
підвищенні напруги С02, при збільшенні
концентрації іонів водню посилюється ім
пульсація від хеморецепторів кровоносних
судин, унаслідок чого збуджується дихаль
ний центр і посилюється вдих. Цей меха
нізм відіграє важливу роль у розвитку за
дишки при ацидозі, недостатності дихання,
при анемії.
6. Пряма стимуляція нейронів дихального цен
тру. У довгастому мозку є хеморецептори,
вибірково чутливі до вуглекислого газу.
Сильне збудження цих рецепторів при гіперкапнії також сприяє розвитку задишки.
7. Рефлекси з дихальних м ’язів. Відчуття
нестачі кисню може виникнути при над
мірному розтягненні міжреберних м’язів
і сильному збудженні рецепторів розтягу,
імпульсація від яких надходить у вищі
відділи головного мозку. Цей механізм
діє під час виконання тяжкої фізичної ро
боти, що потребує значної роботи інспіраторних м’язів, при зменшенні еластич
ності легень, звуженні верхніх дихальних
шляхів.
8. Стимуляція дихального центру продукта
ми власного метаболізму. Ідеться про нако
пичення вуглекислого газу, кислих продук
тів обміну і зниження напруги кисню без
посередньо в нервових центрах унаслідок
порушення мозкового кровообігу (спазм
або тромбоз судин головного мозку, набряк
мозку, колапс).
Дихання при задишці, як правило, часте і гли
боке. Посилюється як вдих, так і видих, який за
умов задишки носить активний характер і від
бувається за участю експіраторних м’язів (див.
рис. 33.13). Однак у деяких випадках може пе
реважати або вдих, або видих; тоді ведуть мову
про інспіраторну (посилений вдих) або експіра
торну (посилений видих) задишку.
Інспіраторну задишку спостерігають, напри
клад, у першій стадії асфіксії, при загальному
збудженні центральної нервової системи, при
фізичному навантаженні у хворих з недостат
ністю кровообігу, при пневмотораксі.
Експіраторна задишка буває рідше і виникає
головним чином при бронхіальній астмі, емфізе
мі, коли при видиху збільшується опір потокові
повітря в нижніх дихальних шляхах (див. вище).
Асфіксія
Крайній, загрозливий для життя, ступінь гострої
недостатності дихання, який виявляє себе тим,
що у кров зовсім не надходить кисень, а з крові
не виводиться вуглекислий газ, позначають тер
міном асфіксія, або ядуха.
Причини. В основі асфіксії може лежати будьякий з обговорених нами механізмів порушення
вентиляції легень:
а) обструктивний: здавлювання дихальних
шляхів (задушення), закупорка їхнього
просвіту (сторонні предмети, запальний
набряк), поява рідини в дихальних шляхах
і альвеолах (утоплення, набряк легень, по
трапляння блювотних мас);
б) рестриктивний: двосторонній пневмоторакс,
різке обмеження рухомості грудної клітки;
в) дисрегуляторний: сильне пригнічення ди
хального центру (центральний параліч ди
хання), порушення проведення нервових
імпульсів до дихальних м’язів (периферич
ний параліч дихання).
Перебіг. У розвитку асфіксії виділяють три пе
ріоди (рис. 33.16).
229
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
Перший період. Характеризується швидким
збільшенням глибини й частоти дихання
з переважанням фази вдиху над фазою ви
диху. Розвивається загальне збудження, під
вищується тонус симпатичної частини веге
тативної нервової системи (розширюються
зіниці, з’являється тахікардія, підвищуєть
ся артеріальний тиск), можливі судоми.
• Другий період. Частота дихання поступо
во зменшується при збереженій макси
мальній амплітуді дихальних рухів, по
силюється фаза видиху. Переважає тонус
парасимпатичної
частини
вегетативної
нервової системи (зіниці звужуються, ар
теріальний тиск знижується, відзначаєть
ся брадикардія).
Третій період. Спостерігають зменшення
амплітуди дихання, його частоти і, наре
шті, зупинку дихання. Артеріальний тиск
значно знижується. Після короткочасної
зупинки дихання, як правило, з’являється
кілька рідких конвульсивних дихальних
рухів (гаспінг-дихання), після яких настає
параліч дихання.
Патогенез. Явища, що їх спостерігають при ас
фіксії, пов’язані спочатку з накопиченням в ор
ганізмі вуглекислого газу (гіперкапнією), а по
тім - із нестачею кисню (гіпоксемією).
Діючи рефлекторно й безпосередньо на ди
хальний центр, С02 збуджує його, доводячи
глибину і частоту дихання до максимально
можливих величин. Крім того, дихання рефлек
торно стимулюється і зниженням у крові напру
ги кисню.
У міру збільшення в крові вмісту С02 під
вищується й артеріальний тиск. Експеримен
ти з вдиханням газових сумішей, що містять
10-20% С02, показали, що таке підвищення
пов’язане, по-перше, з рефлекторним впливом
збуджених хеморецепторів на судиноруховий
центр, по-друге, з посиленим викидом адрена
ліну в кров, по-третє, зі збільшенням хвилинно
го об’єму крові в результаті підвищення тонусу
вен і збільшення надходження крові до серця
при посиленні дихання.
При подальшому збільшенні концентрації
СО у крові починає виявлятися його наркотична
дія, pH крові знижується до 6,8-6,5. Посилюєть
ся гіпоксемія і, відповідно, гіпоксія головного
мозку. Це призводить, своєю чергою, до пригні
чення дихання, зниження артеріального тиску.
У результаті настає параліч дихання і зупинка
серця.
Спірограма (верхня крива) і запис артеріального тиску (нижня крива) при асфіксії.
Стрілкою позначено початок асфіксії, римськими цифрами -її періоди
230
ПАРЕНХІМАТОЗНА
НЕДОСТАТНІСТЬ ДИХАННЯ
Паренхіматозною називають недостатність ди
хання, що виникає як наслідок порушень газо
обміну між альвеолами легень і кров’ю.
Її причинами є:
а) вогнищеві ураження легеневої паренхіми
(ексудативне чи продуктивне запалення)*
б) порушення легеневого кровообігу.
Виділяють три основних механізми паренхі
матозної недостатності дихання:
1) порушення дифузії газів',
2) розлади легеневої перфузії (кровообігу);
3) зміни загальних і регіонарних вентиляційно-перфузійних відношень.
У зв’язку з високим коефіцієнтом дифузії
С02, що в 20-25 разів перевищує аналогічний
показник для 02, перехід С02 із крові в альвео
ли практично не страждає. Тому основною ха
рактеристикою паренхіматозної недостатності
дихання є порушення надходження 02 з альвеол
у кров, у результаті чого зменшується р02 крові,
тобто розвивається гіпоксемія. Вона рефлектор
но через хеморецептори синокаротидної зони
та дуги аорти викликає гіпервентиляцію легень,
унаслідок чого може настати гіпокапнія і газо
вий алкалоз. Цим, власне, паренхіматозна недо
статність дихання відрізняється від вентиляцій
ної, для якої характерними є гіперкапнія й газо
вий ацидоз (див. рис. 33.1).
Отже, паренхіматозна недостатність дихання
виявляє себе такими змінами газового складу
крові:
а) зменшується р02 артеріальної крові (гіпок
семія);
б) рС02 артеріальної крові не міняється або
зменшується (гіпокапнія);
в) кислотно-основна рівновага не порушена
або розвивається газовий алкалоз.
Порушення дифузії газів у легенях
Дифузія газів через альвеоло-капілярну мембра
ну здійснюється відповідно до першого закону
Фіка:
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
r = kj(P,-P 2 ),
де V - кількість газу, що дифундує за одиницю
часу; k — коефіцієнт дифузії; S - загальна пло
ща, через яку відбувається дифузія; І - товщина
мембрани; Р} і Р2т парціальний тиск газів з різ
них боків мембрани.
З урахуванням цього можна виділити такі
причини порушень дифузії газів у легенях:
1) зменшення коефіцієнта дифузії (А). Ве
личина його залежить як від природи газу,
так і від середовища, у якому відбувається
дифузія. Практично має значення змен
шення коефіцієнта дифузії кисню у зв’язку
зі зміною властивостей легеневої тканини.
При цьому перехід С02 із крові в альвеоли,
як уже зазначалося, не міняється, оскіль
ки коефіцієнт його дифузії дуже високий
(у 20-25 разів вищий, ніж кисню);
2) зменшення площі дифузії (S). Має місце
при зменшенні дихальної поверхні легень.
У нормі загальна поверхня легеневих аль
веол складає 50-80 м2;
3) збільшення товщини альвеоло-капілярної
мембрани (/), що складається з таких еле
ментів: альвеолярний епітелій, інтерстиціальний простір, стінка капіляра, плазма
крові, мембрана та внутрішнє середовище
еритроцита. З урахуванням цього можна
виділити ряд основних причин збільшен
ня товщини альвеоло-капілярної мембра
ни. Такими є:
а) потовщення стінки альвеол;
б) потовщення капілярної стінки;
в) внутрішньоальвеолярний набряк;
г) інтерстиціальний набряк;
д) розширення капілярних судин;
4) зменшення різниці між парціальним тис
ком газів в альвеолярному повітрі та їх на
пругою в крові легеневих капілярів (Р—Р 2 ).
Така ситуація виникає при всіх порушен
нях вентиляції легень;
5) зменшення часу контакту крові з альвео
лярним повітрям. У нормі час проходжен
ня крові по легеневих капілярах становить
0,5-0,6 с. Для здійснення повної дифузії
необхідний контакт еритроцита з газами
протягом щонайменше 0,3 с. В умовах нор
ми часу проходження крові по капілярах
231
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
з надлишком вистачає для повної дифузії.
Інша річ в умовах патології: дифузія кис
ню порушується, коли час контакту стає
менше 0,3 с. Це буває при потовщенні альвеоло-капілярної мембрани (збільшується
час проходження кисню через неї) і при
збільшенні швидкості легеневого крово
обігу (наприклад, при фізичній роботі).
Порушення легеневої перфузії
За основним законом гемодинаміки об’ємна
швидкість легеневого кровообігу (Q M J визнача
ється різницею тисків на початку і в кінці систе
ми легеневих судин, тобто у правому шлуночку
і лівому передсерді (Р п ш — Рлп), а також величи
ною опору судин (R).
У нормі величина Qiee дорівнює хвилинному
об’єму крові й становить 4,5-5 л/хв.
З наведеної формули випливають такі причи
ни зменшення інтенсивності легеневої перфузії:
1) зменшення тиску крові в правому шлуноч
ку (недостатність правого серця, зменшен
ня венозного повернення при крововтраті,
шоку, колапсі);
2) збільшення тиску в лівому передсерді (сте
ноз отвору двостулкового клапана, лівошлуночкова недостатність серця);
3) збільшення опору судин малого кола кро
вообігу. Воно може бути обумовлене:
а) рефлекторним збільшенням тонусу ар
теріол легень. Відомо багато рефлексів,
що завершуються спазмуванням гладких
м’язових клітин легеневих артеріол. Серед
них ті, що забезпечують відповідність пер
фузії рівню альвеолярної вентиляції. Так,
зменшення парціального тиску 02 в альве
олах веде до звуження артеріол і зменшен
ня кровообігу у відповідній ділянці легень,
при збільшенні вмісту О — усе навпаки;
б) збільшенням в’язкості крові, що харак
терно для зневоднення, агрегації форме
них елементів крові, наприклад, при ДВЗсиндромі (див. главу ЗО);
в) наявністю перешкод для руху крові як із
середини (тромбоз, емболія), так і ззовні
(здавлювання судин при запаленні легень,
їх набряку).
У клініці порушення легеневої перфузії най
частіше виявляють себе розвитком таких основ
них синдромів: гіпертензії малого кола крово
обігу, набряку легень, тромбоемболії легеневої
артерії.
Синдром гіпертензії малого кола кровообігу.
У нормі середній тиск в легеневій артерії скла
дає 13-15 мм рт. ст. (систолічний - 20 мм рт. ст.,
діастолічний - 8 мм рт. ст.). Такий досить низь
кий тиск пов’язаний з низьким загальним пери
феричним опором легеневих судин.
Про гіпертензію малого кола кровообігу
йдеться тоді, коли тиск крові в легеневій артерії
стає більшим за 25 мм рт. ст. Він може зростати
у 5-15 разів і ставати навіть вищим за тиск крові
у великому колі кровообігу.
Відповідно до основного закону гемодинамі
ки тиск у системі легеневої артерії (Р) залежить
від двох чинників: (а) хвилинного об’єму серця
(в) І (б) периферичного опору кровоносних су
дин (R).
P = QR
Звідси випливає, що гіпертензія малого кола
кровообігу може мати у своїй основі або (1) збіль
шення Q, або (2) зростання R, або (3) одночасне
підвищення обох показників-QiR. Найчастіше
причиною легеневої гіпертензії є збільшення пе
риферичного опору судин, зумовлене зменшен
ням їхнього просвіту.
Залежно від того, на якій ділянці судинного
русла зростає цей опір, розрізняють дві форми
гіпертензії:
а) прекапілярну — збільшення опору відбува
ється на рівні артеріол і капілярів;
б) посткапілярну - опір зростає на рівні ве
нозних судин.
Слід зазначити, що одна форма гіпертензії
нерідко ускладнюється другою, і навпаки (на
приклад, при ураженнях лівого серця).
Розвиток легеневої гіпертензії може бути
обумовлений такими механізмами:
1) тривалий нерефлекторний спазм артері
ол легень. Найчастіше виникає в результа
ті зменшення парціального тиску кисню
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
232
2)
3)
4)
5)
6)
в альвеолярному повітрі, що буває при
гіпоксичній гіпоксії (див. главу 23) і пору
шеннях вентиляції легень. Показано, що та
кий спазм не є рефлекторним, оскільки збе
рігається після перетину блукаючих нервів,
видалення симпатичних вузлів, уведення
речовин, що блокують вивільнення медіато
рів вегетативних нервів. Тривале спастичне
скорочення гладких м’язів артеріол веде до
легеневої гіпертензії через збільшення пе
риферичного опору (R) і зростання хвилин
ного об’єму серця НВ (рис. 33.17);
гострий рефлекторний спазм легене
вих артеріол. Розвивається при емболії
судин легень (див. далі), стенозі отвору
двостулкового клапана. В останньому
випадку вмикається рефлекс Китаєва:
збільшення тиску в лівому передсерді
та легеневих венах викликає збудження
барорецепторів і спазм легеневих артері
ол, що попереджає збільшення гідроста
тичного тиску в капілярах легень і розви
ток набряку;
збільшення тиску повітря в бронхах і аль
веолах. Викликає здавлювання легеневих
капілярів і, як наслідок, збільшення судин
ного опору в малому колі кровообігу. Бу
ває у людей під час важких нападів кашлю.
При цьому тиск у легеневій артерії може
зростати до 250 мм рт. ст.;
облітерація легеневих судин (артеріол, ка
пілярів, венул) внаслідок ураження їхніх
стінок (наприклад, при емфіземі легень).
В експерименті показано, що гіпертензія
малого кола кровообігу виникає при вими
канні не менше 2/3 судинного русла. Отже,
видалення однієї легені ще не призводить
до розвитку цього синдрому;
порушення відтоку крові по легеневих ве
нозних судинах (вади двостулкового кла
пана серця, особливо стеноз мітрального
отвору; недостатність лівого шлуночка,
здавлювання легеневих вен). При цьому не
тільки зростає опір кровообігу у венозно
му відділі, а й вмикається рефлекс Китаєва,
що веде до спазмування легеневих артері
ол (див. вище);
збільшення в’язкості крові (наприклад,
при поліцитемії) закономірно веде до під
вищення периферичного опору всіх судин,
у тому числі й малого кола кровообігу;
7) збільшення хвилинного об ’ему серця біль
ше ніж у 3 рази стає причиною легене
вої гіпертензії, оскільки саме при такому
збільшенні повністю вичерпуються ре
зервні можливості судин легень розши
рюватися та підтримувати завдяки цьому
нормальний тиск у малому колі крово
обігу через адекватне зменшення перифе
ричного опору;
8) уроджені вади, пов’язані зі скиданням
крові зліва направо (незарощення боталлової протоки, дефекти міжшлуночкової пе
регородки). У цих умовах через вилучення
з кровообігу значної частини судинного
русла сильно зростає периферичний опір,
а отже, і тиск у легеневій артерії.
Тривала легенева гіпертензія веде до підви
щення навантаження на правий шлуночок, що
виявляє себе спочатку гіпертрофією його міо
карда, а потім і розвитком правошлуночкової
недостатності серця (див. главу 31).
Синдром набряку легень. Вихід рідини з крово
носних судин в інтерстиціальну тканину легень
і альвеоли позначають як набряк легень. Він є од
нією з причин гострої дихальної недостатності.
Розрізняють кілька основних механізмів його
розвитку (докладно про набряки див. главу 27).
1. Гідростатичний механізм - різке збіль
шення гідростатичного тиску в капілярах
легень. Набряк розвивається, коли гідро
статичний тиск стає вищим за ЗО мм рт. ст.
(у нормі 6-9 мм рт. ст.), тобто його величи
на стає більшою за онкотичний тиск крові.
Така ситуація може виникати при гострій
лівошлуночковій недостатності серця, обу
мовленій великим інфарктом міокарда, при
стенозі отвору двостулкового клапана (кардіогенний механізм), при введенні великих
кількостей (кілька літрів) крово- і плазмозамінних розчинів хворим з порушеним ді
урезом (гіперволемічний механізм).
2. Мембраногенний механізм - збільшення
проникності легеневих капілярів. Буває при:
а) екзогенній інтоксикації (отруєння фос
форорганічними сполуками, наприклад,
фосгеном);
б) ендогенній інтоксикації (уремія, печін
кова недостатність);
в) алергічних реакціях І типу.
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
Механізм розвитку легеневої гіпертензії в умовах тривалого спазму артеріол легень
233
234
3. Онкотичний механізм — зменшення онко
тичного тиску плазми крові. Відносно час
то виникає у хворих з нефротичним син
дромом (див. главу 36).
Розвиток набряку легень проходить дві фази.
1. Інтерстиціальний набряк — накопичен
ня набрякової рідини в інтерстиціальній
тканині легень. Клінічно виявляє себе на
падами серцевої астми. При цьому розви
вається
паренхіматозна
недостатність
дихання з притаманними їй гіпоксемією та
гіпервентиляцією легень, що може супро
воджуватися гіпокапнією й газовим алка
лозом.
2. Альвеолярний набряк — перехід набрякової
рідини в альвеоли. У цих умовах порушу
ється їхня вентиляція — розвивається вен
тиляційна недостатність дихання з яви
щами гіпоксемії, гіперкапнії та газового
ацидозу.
Синдром тромбоемболіїлегеневої артерії. Цей
синдром виникає внаслідок закупорювання ле
геневої артерії чи її гілок відірваними тромбами
(тромбоемболами), що утворилися у венах ве
ликого кола кровообігу (найчастіше у великих
венах нижніх кінцівок і таза).
Поява емболів у судинах легень викликає
розвиток таких змін:
1) генералізований спазм артеріол усього ма
лого кола кровообігу (а не тільки тих, що
пов’язані із судинами, де містяться емболи). Це зумовлює різку гіпертензію малого
кола і розвиток гострої правошлуночкової
недостатності серця (синдром гострого
легеневого серця);
2) зменшення артеріального тиску у вели
кому колі кровообігу. Пов’язане зі змен
шенням хвилинного об’єму серця (мен
ше крові надходить до лівого шлуночка)
і зниженням тонусу артеріол у перифе
ричних відділах (рефлекс Швачка - Паріна);
3) порушення зовнішнього дихання. Через
різке зменшення перфузії легень розви
вається
паренхіматозна
недостатність
дихання.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Порушення загальних і регіонарних
вентиляційно-перфузійних відношень
Вентиляційно-перфузійне відношення для ле
гень у цілому визначається як відношення хви
линного об’єму альвеолярної вентиляції (V) до
хвилинного об’єму крові (Q). У нормі
Можливі три варіанти порушень цього показ
ника.
1. Якщо зазначене відношення більше за
одиницю, то це свідчить про збільшен
ня функціонального мертвого простору
й зменшення внаслідок цього ефективнос
ті вентиляції легень. Така ситуація вини
кає при гіпервентиляції, не підкріпленій
збільшенням перфузії, або при нормальній
вентиляції, але порушеному легеневому
кровообігу.
2. Вентиляційно-перфузійне відношення мен
ше за 0,8 свідчить про так званий ефект
шунтування, коли кров, не збагачена кис
нем, потрапляє в легеневі вени і потім у ве
лике коло кровообігу. Це буває тоді, коли
інтенсивність легеневої перфузії значно пе
ревищує показник альвеолярної вентиляції.
3. В умовах патології у зв’язку з нерівномір
ністю вентиляції та перфузії різних альве
ол вентиляційно-перфузійні відношення
на різних ділянках легень часто бувають
різними. При цьому вентиляційно-перфу
зійне відношення для легень у цілому може
бути в нормі, незважаючи на розвиток ознак
дихальної недостатності. Остання зумов
люється існуванням в уражених легенях
трьох типів альвеол з різними регіонарними
вентиляційно-перфузійними відношеннями
(рис. 33.18):
1) альвеоли, у яких вентиляція і перфузія
є оптимальними (V/Q = 0,8-1). Вони утво
рюють ефективний альвеолярний об’єм
і становлять більшість альвеол здорових
легень;
2) альвеоли, які вентилюються, але не отри
мують кровопостачання (F/Q>1). їхня
ГЛАВА 33. ПАТОФІЗІОЛОГІЯ ЗОВНІШНЬОГО ДИХАННЯ
сукупність становить альвеолярний мерт
вий простір',
3) альвеоли, у яких відбувається перфузія, але
вони не вентилюються (VJQ<0,8). З ними
пов’язане артеріовенозне шунтування.
235
Збільшення кількості альвеол другого і тре
тього типів може бути причиною гіпоксемії. При
цьому виділення СО, не порушується завдяки
високому коефіцієнту його дифузії - розвиваєть
ся паренхіматозна недостатність дихання.
Варіанти регіонарних вентиляційно-перфузійних відношень
ГЛАВА 34
Травлення — це складний фізіологічний процес,
під час якого їжа, що надійшла в травний канал,
зазнає механічних та хімічних перетворень,
а поживні речовини, що містяться в ній, після
розщеплення всмоктуються у кров і лімфу.
Травлення здійснюється органами травної
системи, призначення якої полягає в забезпе
ченні організму речовинами, що задовольняють
його енергетичні, пластичні та гомеостатичні
потреби.
В основі травлення лежать три основні функ
ції органів травної системи:
1) рухова {моторна) функція. Її здійсню
ють м’язи - поперечносмугасті (жу
вальні, м’язи глотки, сфінктери прямої
кишки) і гладкі (м’язи стравоходу, шлун
ка, тонкої і товстої кишки). Завдяки цій
функції відбуваються процеси жування,
ковтання; тонічні, перистальтичні і ма
ятникоподібні рухи шлунка і кишок. Як
результат, відбувається подрібнення, пе
ремішування і переміщення компонентів
їжі по травному каналу;
2) секреторна функція. У результаті секре
торної діяльності клітин травних залоз та
епітелію слизових оболонок утворюють
ся і потрапляють у просвіт органів трав
лення (а) ферменти, що здійснюють гід
ролітичне розщеплення речовин, (б) елек
троліти, які створюють оптимальне для
функціонування ферментів значення pH,
(в) слиз, що захищає слизові оболонки від
ушкоджувальної дії власних травних со
ків та екзогенних чинників, що містяться
в їжі;
3) всмоктування. Надходження речовин із
травного каналу у внутрішнє середови
ще організму здійснюється через епітелій
слизових оболонок. В основі цього про
цесу лежать два механізми: (а) пасивний
транспорт (дифузія) і (б) активний тран
спорт, який забезпечується спеціалізова
ними системами клітин, що використову
ють для цього енергію.
Крім основної - травної - функції система
травлення виконує й ряд важливих додаткових
(нетравних) функцій. До таких зокрема відно
сять:
а) видільну {екскреторну) функцію. Через
травний канал з організму виводяться деякі
кінцеві продукти обміну речовин (жовчні
пігменти, сечовина та ін.), речовини екзо
генного походження;
б) захисну {бар’єрну) функцію. Вона пов’я
зана з тим, що стінки порожнистих орга
нів травної системи утворюють структур
ний бар’єр між зовнішнім і внутрішнім
середовищем організму. Крім того, бакте
рицидну дію мають деякі ферменти (на
приклад, лізоцим) і високі концентрації
іонів водню (кисле середовище у шлун
ку). В окремих відділах травної системи
(глотка, апендикс) зосереджені утворення
лімфоїдної тканини, які відіграють важли
ву роль у специфічній імунологічній реак
тивності організму;
в) ендокринну функцію. Розкиданні у різних
органах травлення ендокринні клітини
входять до складу дифузної ендокринної
системи (APUD-системи), яка продукує
цілу низку пептидних гастроінтестинальних гормонів. Вони впливають не тільки
на процеси травлення, але й на різні інші
функції організму.
У регуляції діяльності травної системи мають
важливе значення як нервові, так і гуморальні
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
механізми. Існує закономірність, яка полягає
в тому, що в міру віддалення від ротової по
рожнини роль нервової регуляції зменшується,
а значення гуморальної, навпаки, зростає.
Основу нервової регуляції травлення станов
лять рефлекси: умовні та безумовні, централь
ні й периферичні, стимуляційні й гальмівні.
Еферентною ланкою переважної більшості
рефлексів є парасимпатичні вегетативні нерви
(медіатор — ацетилхолін). Симпатична нерво
ва система (медіатор — норадреналін) регулює
в основному інтенсивність кровообігу в органах
травлення.
Серед найважливіших гормонів, що регулю
ють процеси травлення, слід назвати:
а) гастрин. Утворюється G-клітинами слизо
вої оболонки антрального відділу шлунка.
Стимулює секрецію шлункового соку, зо
крема НС1, та має цілу низку інших ефектів;
б) секретин. Є продуктом секреції епітеліаль
них клітин (S-клітин) слизової оболонки
верхніх відділів тонкої кишки, зокрема два
надцятипалої кишки. Посилює секрецію
бікарбонатів і води як складових частин
панкреатичного соку;
в) холецистокінін. Продукується І-клітинами
слизової оболонки дванадцятипалої киш
ки. Має два важливих ефекти: посилює
скорочення жовчного міхура і жовчовиді
лення (звідси назва - холецистокінін) і сти
мулює секрецію ферментів підшлункового
соку (звідси ще одна назва цього гормону панкреозимін).
Крім названих гормонів, у регуляції травлення
беруть участь і інші. Серед них шлунково-галь
мівний пептид, мотилін, вазоінтестинальний
пептид, нейротензин, панкреатичний поліпептид,
вілікінін, соматостатин, ентероглюкагон та ін.
Недостатність травлення
Так позначають патологічний стан, при якому
травна система не забезпечує засвоєння пожив
них речовин, що надходять в організм. Наслід
ком цього є розвиток різного ступеня голодуван
ня (див. главу 22).
Класифікація
1. За клінічним перебігом виділяють гостру
і хронічну недостатність травлення.
237
2. Відповідно до анатомічного принципу не
достатність травлення може бути обумов
лена порушеннями цього процесу: (а) у по
рожнині рота; (б) у шлунку; (в) у кишках.
3. Недостатність травлення може бути загаль
ною (тотальною) і селективною {парці
альною). При загальній недостатності по
рушено засвоєння всіх поживних речовин,
при селективній - тільки окремих їхніх
класів (наприклад, ліпідів, лактози, вітамі
ну В]2 та ін.).
4. За етіологією розрізняють спадково обу
мовлену (деякі види мальабсорбції) і набу
ту недостатність травлення. Остання може
бути:
а) інфекційного походження;
б) обумовленою впливами фізичних фак
торів (наприклад, при гострій промене
вій хворобі);
в) пов’язаною із впливами хімічних аген
тів;
г) диерегуляторною;
д) аліментарною.
5. Патофізіологічний принцип передбачає по
діл недостатності травлення на три варіан
ти. Це недостатність, обумовлена порушен
нями:
1) рухової функції травної системи. До цієї
категорії відносять: (а) порушення жу
вання; (б) розлади ковтання - дисфагію,
(в) шлункові дискінезії, (г) кишкові дискінезії, (д) дискінезії жовчного міхура
та жовчних проток, (е) порушення дефе
кації;
2) секреторної функції. Виділяють гіперсекреторні та гіпосекреторні стани. До
перших відносять: (а) гіперсалівацію,
(б) шлункову гіперсекрецію, (в) панкре
атичну гіперсекрецію, (г) гіперхолію.
До других — (а) гіпосалівацію, (б) шлун
кову гіпосекрецію, (в) панкреатичну гіпосекрецію, (г) ахолію;
3) процесів усмоктування. Розлади цього
компоненту травлення виявляють себе
синдромом мальабсорбції.
Причини порушень травлення. Залежно від
походження можна виділити такі групи етіоло
гічних чинників.
1. Аліментарні (харчові) фактори. До таких
відносять: (а) приймання недоброякісної
238
і грубої їжі; (б) сухоїдіння; (в) нерегулярне
приймання їжі; (г) незбалансоване харчу
вання (наприклад, зменшення вмісту віта
мінів у раціоні); (д) зловживання алкоголем.
2. Фізичні фактори. Серед чинників цієї групи
найбільше значення має іонізуюча радіація,
яка уражує епітеліальні клітини травної
трубки, що мають високу мітотичну актив
ність. При одноразовому загальному опро
міненні організму дозою, що перевищує
10 Гр, розвивається так звана кишкова фор
ма гострої променевої хвороби, що швидко
завершується смертю (див. главу 9).
3. Хімічні агенти. Вони є причиною розладів
травлення при отруєннях неорганічними
і органічними сполуками на виробництві
та в побуті.
4. Біологічні фактори. Такими є чинники, що
зумовлюють розвиток інфекційних шлун
ково-кишкових хвороб: (а) бактерії (на
приклад, холерний вібріон, збудники ди
зентерії, черевного тифу, паратифу та ін.);
(б) бактеріальні токсини (наприклад, при
сальмонельозах, стафілококовій інфек
ції); (в) віруси (наприклад, аденовіруси);
(г) гельмінти.
5. Органічні ураження. До розладів трав
лення можуть спричинятися (а) уродже
ні аномалії органів травлення; (б) після
операційні стани; (в) пухлини травної
системи.
6. Порушення нервової й гуморальної регу
ляції. Розлади травлення можуть розви
ватися при: (а) психоемоційних порушен
нях (невротичні і неврозоподібні стани),
(б) психічних захворюваннях (шизофре
нія, маніакально-депресивний синдром),
(в) органічних ураженнях центральної нер
вової системи (енцефаліти, діенцефаліт);
(г) ушкодженні периферичних структур
вегетативної нервової системи; (д) рефлек
торних порушеннях (різні вісцеро-вісцеральні рефлекси).
Порушення гуморальної регуляції травлення
можуть бути пов’язані з розладами синтезу і се
креції гастроінтестинальних гормонів (гастри
ну, секретину, холецистокініну та ін.).
Експериментальне
моделювання
порушень
травлення. Для відтворення і вивчення в екс
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
перименті порушень діяльності різних відділів
травної системи використовують ряд принци
пів.
1. Принцип видалення. Полягає в хірургічно
му видаленні тих чи інших органів травної
системи (шлунка, тонкої або товстої киш
ки). Найбільш фундаментальні досліджен
ня в цьому напрямі було виконано Є. Лон
доном. Він та його учні показали, що ви
далення різних відділів травного каналу
призводить до розвитку різного ступеня
недостатності травлення. Однак травна
система має досить високі компенсаторні
можливості. Про це свідчить той факт, що
життя тварин (собак) можливе навіть після
поетапного видалення у них шлунка, всієї
клубової і більшої частини порожньої киш
ки, а також майже всієї товстої кишки, за
винятком її початкового відділу та прямої
кишки.
2. Принцип ізолювання. Набув широкого ви
користання у фізіологічних і патофізіо
логічних дослідженнях завдяки роботам
І. Павлова. Цей учений, зокрема, запро
понував методику ізольованого шлуночка
(рис. 34.1). Крім того, часто застосовують
ся метод ізольованої петлі кишок, накла
дення анастомозів між різними відділами
системи травлення. На відміну від мето
дів видалення, ці методики моделювання
дають можливість одержувати додаткову
інформацію про характер і механізми змін
в ізольованих ділянках травного каналу,
про зміни нервової та гуморальної регуля
ції в цих умовах.
3. Принцип відведення. Полягає у відведен
ні на різних рівнях травної системи їжі,
що надходить в організм, та хімусу, а та
кож у відведенні за межі травного каналу
або в інші його відділи травних секретів.
Ілюстрацією цього принципу можуть бути
такі методики: (а) “удаване годування” (на
кладання езофагостоми за І. Павловим)
(рис. 34.2); (б) створення фістул кишок;
(в) виведення назовні проток слинних залоз,
панкреатичної протоки, загальної жовчної
протоки.
4. Принцип екзогенних ушкоджень. При мо
делюванні патологічних процесів у трав
ній системі застосовують різні ушкоджу
вальні впливи: фізичні (висока і низька
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
Методика створення ізольованого малого шлуночка за І. Павловим
Езофаготомована собака з фістулою шлунка ("удаване годування ")
239
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
240
температури, іонізуюча радіація), хімічні
(кислоти, луги, солі важких металів, орга
нічні сполуки), біологічні (бактерії та їхні
токсини).
5. Принцип порушення нервової регуляції. Для
експериментального вивчення розладів
травлення використовують такі методи:
а) відтворення експериментальних невро
зів — метод “зіткнення” умовних реф
лексів за І. Павловим. При одночасному
“вмиканні” процесів умовного збуджен
ня і умовного гальмування виникає поза
межне гальмування в корі головного моз
ку. Наслідком цього є розгальмовування
підкіркових структур, що виявляється
в собак-ваготоніків підвищенням актив
ності центрів парасимпатичної нервової
системи. Останнє викликає збільшення
секреторної й рухової активності органів
травлення;
б) вплив на підкіркові структури централь
ної нервової системи (наприклад, ядра
блукаючого нерва). Для їхнього збу
дження проводять вживляння електродів
з наступною електричною стимуляцією,
для одержання гальмування руйнують
ці структури;
в) вплив на нервові провідники. Для припи
нення рефлекторної регуляції перетина
ють блукаючий нерв та його гілки, для
посилення нервової імпульсації прово
дять їх електричну стимуляцію;
г) порушення передавання збудження в па
расимпатичних гангліях. Воно виникає
при застосуванні фармакологічних пре
паратів, що є гангліоблокаторами;
д) втручання в медіаторні механізми пе
редавання збудження на ефекторні
клітини. Цей метод передбачає вико
ристання речовин, що мають власти
вості т-холіноміметиків (для поси
лення функції органів травлення) або
т-холінолітиків (для послаблення їхньої
діяльності).
6.
Принцип порушення гуморальної регуляції.
Для відтворення в експерименті розладів
гуморальної регуляції травлення послуго
вуються такими підходами:
а) введення в організм ззовні високих доз
гастроінтестинальних гормонів (гастри
мтзш
ну, секретину, холецистокініну та ін.) або
їхніх аналогів;
б) впливи, що активують утворення влас
них таких гормонів (наприклад, застосу
вання так званих сокогінних речовин для
гуморального стимулювання секреції
шлункового соку);
в) пригнічення утворення й секреції гормо
нів травної системи;
г) фармакологічна блокада чутливих до га
строінтестинальних гормонів рецепторів
на поверхні ефекторних клітин.
Клінічні синдроми уражень травної системи.
Порушення діяльності органів травлення мо
жуть виявляти себе великою кількістю клініч
них ознак, що їх зручно об’єднати в ряд синдро
мів.
I. Голодування. Порушення засвоєння пожив
них речовин, що надходять з їжею, є одним
із проявів хронічної недостатності травлен
ня і може стати причиною типового патоло
гічного процесу, що має назву голодування
(див. главу 22). Голодування при уражен
нях травної системи найчастіше має риси
неповного (кількісного) або часткового
(якісного).
II. Диспепсичний синдром. Так позначають
різні поєднання симптомів, що виникають
внаслідок порушень діяльності різних від
ділів травної системи. До таких симптомів
зокрема відносять:
\)анорексію — повну відсутність апетиту
при об’єктивній потребі в харчуванні.
Виділяють такі види анорексії: (а) інток
сикаційну — розвивається при гострих
і хронічних отруєннях (наприклад, соля
ми ртуті, лікарськими препаратами, бак
теріальними токсинами); (б) диспепсич
ну - виникає при захворюваннях органів
травної системи, має найчастіше умов
норефлекторну природу; (в) нейродинамічну - розвивається в результаті реципрокного гальмування центру апетиту
при перезбудженні окремих структур
лімбічної системи (наприклад, больовий
синдром при інфаркті міокарда, кольках,
перитоніті); (г) невротичну — пов’язана
з надмірним збудженням кори головно
го мозку і сильними емоціями (особливо
негативними); (д) психогенну — є резуль-
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
татом свідомого обмеження споживання
їжі (наприклад, з метою схуднення або
як результат нав’язливої ідеї при пору
шеннях психіки); (е) нейроендокринопатичну — обумовлену органічними ура
женнями центральної нервової системи
(гіпоталамуса) і ендокринними захворю
ваннями (гіпофізарна кахексія, адисонова хвороба).
В основі розвитку анорексії можуть лежати
два механізми: (а) зменшення збудливості хар
чового центру (інтоксикаційна, диспепсична,
нейроендокринопатична анорексія) і (б) галь
мування нейронів харчового центру (нейродинамічна, невротична, психогенна анорексія);
2) печію (pyrosis) - відчуття жару по ходу
стравоходу. Розвиток цього симптому
пов’язаний з подразненням рецепторів
стравоходу при закиданні вмісту шлун
ка у стравохід (рефлюкс). Це може бути
обумовлено: (а) великою кількістю утво
рюваного шлункового соку; (б) функці
ональною недостатністю кардіального
сфінктера;
3) відрижку (eructatio) — раптове мимовіль
не виділення в порожнину рота газу зі
шлунка або стравоходу, іноді з невелики
ми порціями вмісту шлунка. Збільшення
вмісту газів у шлунку може бути викли
кано двома причинами: (а) надходжен
ням великої кількості газів з їжею і пит
вом (наприклад, газовані напої), заков
туванням повітря (аерофагія); (б) утво
ренням газів у самому шлунку, особливо
при тривалій затримці там їжі (буває при
виразковій хворобі, раку шлунка). У ре
зультаті збільшення вмісту газів у шлун
ку збільшується внутрішньошлунковий
тиск. Це рефлекторно викликає: (а) ско
рочення м’язів стінки шлунка; (б) спазм
воротаря; (в) розслаблення м’язів стра
вохідно-шлункового сфінктера. Наслід
ком зазначених змін є витискання газів
з порожнини шлунка у стравохід, глотку,
а потім у порожнину рота;
4) нудоту (nausea) - своєрідне тяжке від
чуття в ділянці під грудьми, у грудях
і порожнині рота, що нерідко передує
блювоті й часто супроводжується за
гальною слабкістю, пітливістю, підви
241
щенням слиновиділення, похолоданням
кінцівок, блідістю шкіри, зниженням
артеріального тиску, тобто ознаками
активації
парасимпатичної
нервової
системи. В основі нудоти лежить збу
дження блювотного центру, яке, од
нак, ще не достатнє для виникнення
блювоти;
5) блювоту (vomitus) — складнорефлекторний акт, що призводить до вивер
ження вмісту шлунка назовні через рот.
Виникає в результаті збудження блю
вотного центру, що міститься в довгас
тому мозку.
У механізмі блювоти виділяють ряд послі
довних стадій. Спочатку відбувається глибокий
вдих з опущенням діафрагми, одночасно опус
кається надгортанник і піднімається гортань
(перекривається вхід у гортань), піднімається
м’яке піднебіння (перекривається вхід у носо
глотку).
Потім скорочується воротар, тіло шлунка
і стравохідно-шлунковий сфінктер розслаблю
ються, кардія підтягується догори, стравохід
розширюється і коротшає.
І нарешті, відбувається сильне скорочення
діафрагми та м’язів черевного преса, у резуль
таті чого підвищується внутрішньочеревний
і внутрішньошлунковий тиск. Це, у свою чер
гу викликає виверження вмісту шлунка через
стравохід і порожнину рота назовні - виникає
блювота.
Розрізняють такі патогенетичні варіанти блю
воти (рис 34.3):
а) центральну - пов’язану з підвищенням
збудливості блювотного центру. Це бу
ває при захворюваннях центральної нер
вової системи (менінгіти, енцефаліти,
пухлини мозку), у разі надходження збу
дження з кори великих півкуль (умовно
рефлекторна блювота) або рецепторів
лабіринту (вестибулярна блювота)-,
6) гематогенно-токсичну - обумовлену
прямою дією токсичних речовин, що
містяться у крові, на хеморецептори
структур блювотного центру. Це можуть
бути екзогенні речовини (чадний газ,
алкоголь, лікарські препарати, токсини
бактерій) або токсичні продукти власно
го метаболізму, що накопичуються при
242
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Центральна блювота
Рис. 34.3. Механізми розвитку блювоти
уремії, печінковій недостатності, деком
пенсованому цукровому діабеті та ін.;
в) вісцеральну (рефлекторну), що є резуль
татом рефлексів, які виникають унаслідок
подразнення різних рецепторних полів
внутрішніх органів. Такі рефлексогенні
зони є у шлунку, слизовій оболонці глот
ки, вінцевих судинах, очеревині, жовчних
протоках та ін.;
6) закрепи (obstipatio) - уповільнене, утруд
нене або систематично недостатнє випо
рожнення кишок. Виділяють два механізми
розвитку закрепів — спастичний і атоніч
ний. Перший обумовлений тривалим по
стійним скороченням гладких м’язів кишок
(спазмом), другий - їх атонією.
До спастичних закрепів відносять: (а) запаль
ні - виникають унаслідок місцевих спастичних
рефлексів зі зміненої слизової; (б) проктогенні — розвиваються при патології аноректальної
ділянки; (в) механічні — виникають при непро
хідності кишок; (г) токсичні - є результатом
отруєнь свинцем, ртуттю, талієм.
Атонічними закрепами є: (а) аліментарні розвиваються при надходженні їжі, що легко
засвоюється і містить мало клітковини; (б) неврогенні - є результатом порушень нервової
регуляції моторики кишок; (в) гіподинамічні —
виникають у лежачих хворих, у старих людей,
у людей з дуже низькою руховою активністю;
(г) закрепи при аномаліях товстої кишки (на
приклад, хвороба Гіршпрунга); (д) закрепи вна
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
слідок порушень водно-сольового обміну (напри
клад, при зневодненні);
7)
метеоризм - надмірне скупчення газів
у травному каналі через їх підвищене
утворення або недостатнє виведення з ки
шок. Джерелами газів у кишках є повітря,
що його заковтують; процеси нейтралі
зації шлункового соку гідрокарбонатами
(утворення вуглекислого газу), діяльність
мікрофлори.
Надмірне утворення газів лежить в основі
розвитку таких видів метеоризму: (а) алімен
тарного — розвивається при прийманні їжі, що
містить багато клітковини, крохмалю (бобові,
капуста, картопля); (б) при розладах травлення
(ферментопатіях, порушеннях всмоктування,
кишкових дисбактеріозах).
Порушення виведення газів становить сут
ність метеоризму: (а) механічного - розвиваєть
ся в результаті порушення прохідності кишок
(спазми, спайки, пухлини); (б) динамічного —
виникає при розладах рухової функції кишок
(при закрепах); (в) циркуляторного - є наслід
ком загальних і місцевих розладів кровообігу;
8) проноси (діарея) - прискорене випорож
нення кишок з виділенням розріджених,
а в частині випадків і великих за об’ємом
випорожнень. Проноси виникають при по
рушенні нормальних співвідношень між
секрецією та всмоктуванням рідини в киш
ках, при розладах моторики кишок.
Виділяють такі патогенетичні варіанти про
носів:
а) осмотична діарея. Розвивається при
збільшенні осмотичного тиску кишково
го вмісту після приймання всередину ре
човин, які погано або зовсім не всмокту
ються (наприклад, проносних), а також
при порушеннях травлення і всмокту
вання (синдроми мальдигестії й мальабсорбції, див. далі);
б) секреторна діарея. Пов’язана з активаці
єю секреції іонів електролітів (Na+, СІ ),
що викликає посилену секрецію води
в просвіт кишок (наприклад, при холері,
див. далі);
в) діарея, зумовлена гальмуванням актив
ного транспорту іонів через клітинні
243
мембрани в кишках (наприклад, уро
джена хлордіарея - генетичний дефект
усмоктування аніонів хлору в клубовій
кишці);
г) діарея при підвищенні проникності киш
кової стінки (запальна, або ексудативна);
д) діарея, викликана порушеннями кишко
вої моторики. Може розвиватися як при
посиленні, так і при ослабленні рухової
активності кишок.
III. Зневоднення організму. Основними його
причинами при розладах травлення мо
жуть бути:
1) гіперсалівація — збільшення утворення
й секреції слини (див. далі);
2) нестримна блювота',
3) проноси.
При гіперсалівації розвивається гіперосмолярне зневоднення (слина гіпотонічна стосовно
крові), а при нестримній блювоті й проносах гіпоосмолярне (електролітів втрачається біль
ше, якщо порівнювати з водою; докладно див.
главу 27).
Втрата рідини через травний канал може бути
обумовлена специфічною дією деяких патоген
них факторів. Так, установлено, що при холері
зневоднення є результатом дії холерного екзо
токсину на складну систему аденілатциклази
ентероцитів. При цьому настає стійке необорот
не підвищення активності ферменту, унаслідок
чого різко, у багато разів, збільшується секреція
кишкового соку. Велика кількість кишкового се
крету шляхом впливу на рецептори тонкої киш
ки викликає діарею.
IV.
Порушення кислотно-основної рівноваги.
При розладах функцій травної системи мо
жуть розвиватися:
1) негазовий алкалоз — як наслідок нестрим
ної блювоти;
2) негазовий ацидоз. Виникає в результаті
втрати великих кількостей гідрокарбонатів
підшлункового соку та жовчі при діареї.
Докладну характеристику зазначених пору
шень подано в главі 29.
V. Кишкова автоінтоксикація. Її розвиток,
як правило, пов’язаний з дисбактеріозами
й утворенням великих кількостей токсич
них продуктів бродіння і гниття.
244
Дисбактеріоз - це порушення співвідношен
ня між окремими видами кишкової мікрофлори.
При цьому часто збільшується кількість бакте
рій, які викликають процеси гниття і бродіння.
У результаті зростає утворення в кишках ток
сичних продуктів - сірководню, скатолу, індолу,
фенолів, путресцину, кадаверину та ін. Якщо
утворення цих продуктів перевищує функціо
нальні можливості печінки щодо їхньої деток
сикації, розвиваються ознаки печінкової недо
статності (див. главу 35).
Розвитку кишкової автоінтоксикації сприя
ють ослаблення перистальтики кишок (закре
пи), зменшення секреції кишкового соку, киш
кова непрохідність.
VI. Больовий синдром. Біль часто супрово
джує розвиток хвороб травного каналу.
Він може мати різний характер залежно від
причин і патогенезу.
Розрізняють такі механізми виникнення болю
при ураженнях органів травлення:
а) спастичний механізм. Біль спричиняється
спазмом гладкої мускулатури різних відді
лів травного каналу. Вважають, що в цьому
випадку причиною болю є перетискання
судин, що проходять у товщі стінки порож
нистих органів, унаслідок чого розвиваєть
ся ішемія. Остання викликає появу продук
тів, що діють на больові нервові закінчен
ня (див. главу 38). У разі різкого сильного
спазму, що виникає раптово, розвиваються
болі за типом кольки;
б) гіпотонічний механізм. При зменшенні то
нусу гладкої мускулатури (гіпотонії) болі
виникають у результаті розтягнення стін
ки порожнистих органів (шлунка, кишок,
жовчного міхура) їхнім вмістом. При цьому
механічне розтягнення тканин обумовлює
подразнення закладених у них нервових за
кінчень;
в) вплив біологічно активних речовин (гіста
міну, серотоніну, кінінів, простагландинів
та ін.) на нервові закінчення. Ці речовини
утворюються і вивільнюються при ушко
дженні клітин і запаленні (гастрити, дуоде
ніти, ентерити, коліти, холецистити). Особ
ливо багато їх з’являється при гострому
панкреатиті (див. далі).
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ПОРУШЕННЯ ТРАВЛЕННЯ
В ПОРОЖНИНІ РОТА
У процесах травлення, що відбуваються в рото
вій порожнині, беруть участь три групи струк
тур:
1) руховий апарат, до якого входять зуби,
щелепи, жувальні м’язи, м’язи стінок по
рожнини рота (у тому числі язика), глотки.
Функціями цього апарату є:
а) механічна обробка їжі, її подрібнення.
Ці процеси здійснюються під час акту
жування',
б) переміщення їжі в стравохід - ковтан
ня',
2) секреторний апарат, представлений слин
ними залозами і залозами слизової оболон
ки рота. Завдяки секреції слини відбува
ються:
а) формування харчової грудки. У цьому
процесі беруть участь основні компо
ненти слини — вода і муцин;
б) хімічна обробка їжі - первинний гідро
ліз полісахаридів за участю а-амілази
слини;
в) захист від мікробів, що здійснюється
завдяки бактерицидній дії лізоциму.
Крім того, слина до певної міри відіграє тро
фічну функцію, зволожуючи слизову оболонку
порожнини рота, впливаючи ззовні на ткани
ни зубів. Через слину можуть виводитися дея
кі кінцеві продукти обміну речовин в організмі
(сечовина, аміак, креатинін);
3) слизова оболонка та її рецепторний апа
рат. Ці структури здійснюють:
а) хімічний аналіз складу їжі (смаковий
аналізатор);
б) рефлекторну сигналізацію. Рецептори
слизової оболонки є рецепторним по
лем, що започатковує мозкову фазу ре
гуляції травлення;
в) початковий етап всмоктування деяких
речовин.
Порушення рухових функцій
Ураження структур, що беруть участь у здій
сненні механічних перетворень їжі, можуть ви
являти себе розладами актів жування і ковтання.
245
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
(I) Порушення жування. Причинами розла
дів цього акту можуть бути:
а) ушкодження зубів та їхня відсутність (карі
єс, пародонтит) (див. далі);
б) ураження жувальних м’язів (міозит);
в) порушення іннервації жувальних м’язів
(бульбарні паралічі, неврити);
г) ураження скронево-нижньощелепних су
глобів;
д) травматичне ушкодження кісток (нижньої,
верхньої щелепи);
е) патологічні процеси в слизовій оболонці
порожнини рота і ясен (стоматит, гінгівіт);
є) ураження м’язів і слизової язика;
ж) гіпосалівація.
Порушення жування мають ряд негативних
наслідків:
зменшується рефлекторна секреція шлун
кового й підшлункового соків;
• уповільнюється травлення в шлунку;
• травматизується слизова оболонка ротової
порожнини, стравоходу, шлунка;
людина відмовляється від споживання де
яких продуктів харчування, що потрібні
організму, але потребують пережовування.
(II)
Порушення ковтання — дисфагія.
Складний рефлекторний акт ковтання відбува
ється в три фази: ротову (довільну), глоткову
(швидку мимовільну) і стравохідну (повільну
мимовільну). Кожна з них може зазнавати змін,
що порушують акт ковтання в цілому. Причина
ми дисфагії можуть бути:
а) зміни рецепторів слизової порожнини рота
(наприклад, при стоматитах) і глотки (при
ангінах);
б) ураження чутливих аферентних і рухових
еферентних нервових провідників, що бе
руть участь у здійсненні ковтальних реф
лексів (волокна V, VII, IX, X, XII пар череп
но-мозкових нервів);
в) ушкодження нервових центрів «в у корі го
ловного мозку (порушується довільна фаза
ковтання) і центру ковтання, розташова
ного в ділянці дна IV шлуночка;
г) ураження м’язів язика, глотки і стравохо
ду. Порушення функції гладкої мускулату
ри стравоходу можуть виявлятися як спас
тичними станами (ідіопатичний дифузний
спазм, ахалазія - спазм кардії), так і гіпото
нічними (атонічними), для яких характер
не порушення перистальтики стравоходу;
д) уроджені та набуті дефекти м’якого й твер
дого піднебіння;
е) механічні перешкоди (пухлини, рубці, здав
лювання стравоходу ззовні).
При дисфагії вкрай утруднене приймання
їжі, у результаті чого можуть розвиватися го
лодування і виснаження. Крім того, можливе
потрапляння слини і часток їжі в дихальні шля
хи, що викликає аспіраційну пневмонію, а іноді
й гангрену легень.
Карієс зубів
Патологічний процес, що виявляє себе демінералізацією і прогресуючим руйнуванням твер
дих тканин зуба з утворенням дефектів у ви
гляді порожнин, називають карієсом (рис. 34.4).
Поширеність карієсу зубів становить 80-90 %,
а в деяких районах земної кулі - 100 %.
Етіологія. Вважають, що у виникненні карі
єсу головне значення мають три чинники: мі
кроорганізми, характер їжі та сприятливі для
цього процесу умови на поверхні зубної емалі
(рис. 34.5). Розглянемо їх.
1. Мікроорганізми. У ротовій порожнині лю
дини проживає величезна кількість різно
видів бактерій, проте до розвитку карієсу
мають стосунок тільки ті, що здатні утво
рювати кислоти в процесі своєї життєді
яльності. їх називають карієсогенними бак
теріями. До таких відносять деякі штами
стрептококів (Str. mutans, Str. viridans, Str.
sanguis, Str mitis, Str. salivarius), лактобактерїї (Lactobacillus) та ін.
В експерименті доведено, що у тварин-гнотобіонтів (безмікробних тварин) карієс не ви
никає.
2. Надмірне споживання харчових продуктів,
що можуть проникати в зубний наліт і лег
ко ферментуються бактеріями. Такими,
зокрема, є вуглеводи (сахароза, у меншій
мірі - глюкоза, фруктоза). Крохмаль у чис
тому вигляді, який є полісахаридом, не вва
жають карієсогенним, оскільки його моле
кули не проникають у зубний наліт. Проте
при харчовій обробці відбувається гідроліз
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
246
Каріозний процес
руйнує дентин
Періодонтит
(запалення на верхівці
кореня зуба)
ис. 34.4. Карієс та його ускладнення
Рис. 34.5. Основні причини і умови розвитку карієсу зубів
крохмалю й утворюються продукти, здатні
індукувати та посилювати каріозний процес.
В експерименті використовують карієсоген
ні дієти (містять від 25 до 65 % сахарози), що
викликають розвиток карієсу у щурів, хом’яків,
мишей.
3.
Карієсочутливість зубної поверхні. Вона
по суті визначається двома чинниками:
(а) наявністю зубного нальоту і (б) власти
востями емалі зубів.
Зубний наліт {зубна бляшка) - це плівка, що
утворюється на поверхні зубів і складається
247
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
із скупчень бактерій і позаклітинної матриці.
Остання містить глікопротеїди, полісахариди
та інші речовини, які потрапляють на поверхню
зуба із слини.
Наявність небезпечного для зубів нальоту зу
мовлюється такими чинниками:
а) зубний наліт легко утворюється. Це буває,
коли людина споживає в основному м’яку,
багату на вуглеводи їжу. Мала кількість
в’язкої слини сприяє утворенню зубного
нальоту;
б) зубний наліт легко фіксується. Поверхня
зуба має складний рельєф і складається
з бугрів і поглиблень між ними (фісур). Фісури є слабкими місцями в зубі, оскільки
вони досить глибокі й вузькі. Залишки їжі,
а разом з ними і мікроорганізми, легко про
никають сюди. Ще одним слабким місцем є
бічні поверхні зубів у проміжках між ними;
в) зубний наліт погано видаляється. Недо
тримання гігієни порожнини рота, непра
вильне чищення зубів є причиною того,
що зубний наліт своєчасно не видаляється,
а стає місцем, звідки починає розгортатися
каріозний процес.
Властивості зубної емалі багато в чому ви
значають стійкість до карієсу. Підвищення чут
ливості зубів до цього процесу може бути зу
мовлене:
а) недостатнім насиченням емалі зубів фто
ром. Відомо, що надходження цього хіміч
ного елементу у тверді тканини зуба веде
до утворення певної кількості фторапатитів - мінералів, що є стійкішими, ніж гідроксіапатити, до дії кислот;
б) генетичними факторами, що впливають
на закладку зубних зачатків, формування
органічних компонентів твердих тканин
зуба та інші процеси, від яких залежить
фізіологічна мінералізація. Про роль спад
кового чинника свідчить факт виведення
кількох чистих ліній щурів, з яких одні чут
ливі, а інші - стійкі до розвитку карієсу;
в) загальними порушеннями фосфорно-каль
цієвого обміну (гіпокальціємічні і гіпофосфатемічні стани; див. главу 28).
Патогенез. У виникненні й розвитку карієсу про
відного значення надають продуктам життєдіяль
ності бактерій, що входять до складу зубного на
льоту. До карієсогенних факторів, що тут утво
рюються і діють на поверхню емалі, відносять:
а) органічні кислоти (молочна, піровино
градна та ін.), що є продуктами бродіння
й анаеробного окиснення вуглеводів. Істот
но зменшуючи pH середовища, ці кислоти
зумовлюють розчинення гідроксіапатиту
емалі, а потім і дентину, унаслідок чого роз
вивається їхня демінералізація. Зменшення
буферних властивостей слини (не відбува
ється нейтралізація кислот) сприяє цьому
процесу;
б) речовини-хелатори (пептиди, амінокис
лоти). Вони здатні утворювати комплекси
з кальцієм, забираючи його від гідроксі
апатиту і викликаючи цим демінералізацію
твердих тканин зуба;
в) протеолітичні ферменти. Діючи на ор
ганічні компоненти зуба, вони руйнують
матрикс емалі та дентину. Відбувається де
струкція твердих тканин, порушується їхня
ремінералізація.
Утворення дефектів у твердих тканинах зуба
спричиняється до того, що інфекція легко про
никає через них у пульпу і в глибокі тканини,
що оточують зуб. Як наслідок, розвивається
запалення - пульпіт, періодонтит. Повне руй
нування зуба стає причиною його хірургічного
видалення і зумовлює необхідність подальшого
протезування.
Пародонтит
Пародонтит — це деструктивно-запальне ураже
ння пародонта, тобто тканин, що оточують ко
рінь зуба (періодонта, кісткової тканини зубної
комірки, ясен, окістя). Хвороба виявляє себе ре
зорбцією зубних комірок, гноєтечею з ясен, роз
хитуванням і випадінням зубів.
Етіологія. Причини пародонтозу можна розді
лити на місцеві й загальні (рис. 34.6).
До місцевих відносять:
а) мікроорганізми порожнини рота. Серед
них Porphyromonas gingivalis, Actinobacillus actinomycetemcomitans, Prevotella inter
media, Treponema denticola. Вони є джере
лом ферментів і біологічно активних речо
вин зубного нальоту і сприяють утворенню
зубного каменю;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
248
Причини й механізми пародонтиту
б) аліментарні фактори — постійне харчу
вання продуктами, що чинять недостат
нє навантаження на зубощелепний апарат
(м’яка, механічно подрібнена і термічно
оброблена їжа), дефіцит вітамінів, особли
во С, Р і Е;
в) аномалії прикусу, при яких відбувається
травматизація ясен, зменшується або, на
впаки, надмірно збільшується навантажен
ня на жувальний апарат.
Розвиткові пародонтозу сприяють загальні
порушення в організмі:
а) нейрогенні фактори - емоційна перенапру
га, що викликає стрес; порушення нервової
трофіки;
б) ендокринні розлади — гіпогонадизм, гіпо
тиреоз, гіпоінсулінізм (цукровий діабет),
гіперпаратиреоз;
в) порушення імунної системи, що виявляють
себе розвитком автоалергічних реакцій.
Патогенез. У розвитку пародонтиту важливого
значення надають зубному каменю, до утворен
ня якого причетні мікроорганізми ротової по
рожнини і їжа. Зубний камінь являє собою за
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
тверділий зубний наліт і складається із залишків
їжі, бактерій, змертвілих клітин, фосфатних та
інших солей кальцію і заліза.
Ріст над’ясенних і під’ясенних зубних каме
нів зумовлює механічний тиск на м’які тканини
і порушує внаслідок цього кровообіг у них. Це
стає важливою умовою розвитку двох складо
вих пародонтиту - атрофії тканин і власне за
палення.
Атрофія тканин пародонту настає в резуль
таті недостатнього навантаження на зубощелепний апарат при вживанні м’яких, механічно
подрібнених і термічно оброблених продуктів,
їй сприяють порушення кровопостачання па
родонту внаслідок росту зубних каменів та дії
деяких загальних чинників (наприклад, катехоламінів в умовах емоційного та інших стре
сів). Атрофічні зміни в пародонті у свою чер
гу є фактором, що зменшує кровообіг у його
тканинах.
Запалення розвивається внаслідок ушко
дження (альтерації) клітин пародонта про
дуктами діяльності мікроорганізмів і травма
тизації ясен при аномаліях прикусу. У запа
лених тканинах порушується кровообіг (див.
главу 17), еміграція лейкоцитів стає причи
ною вторинної альтерації, веде до формуван
ня гною. Лізосомні ферменти нейтрофілів
і макрофагів (металопротеїнази, колагенази)
зумовлюють гідролітичне розщеплення ор
ганічних компонентів пародонта, унаслідок
чого руйнуються зубні зв’язки, розвивають
ся деструктивні зміни у твердих тканинах,
що оточують зуб. Врешті-решт процес веде
до розхитування зубів, яке завершується їх
втратою.
Порушення слиновиділення
Утворення слини відбувається в привушних,
підщелепних і під’язикових слинних залозах,
а також у великій кількості маленьких залоз, що
містяться на поверхні язика, у слизовій оболон
ці піднебіння і щік.
За добу в нормі утворюється 0,5—2 л слини,
одна третина з яких припадає на привушні зало
зи. Регуляція слиновиділення здійснюється нер
вовими (рефлекторними) механізмами за учас
тю парасимпатичної і симпатичної вегетативної
нервової системи.
249
Гіперсалівація. Збільшення утворення і секре
ції слини називають гіперсалівацією. Вона може
бути обумовлена такими причинами:
а) збудженням рецепторів порожнини рота,
стравоходу і шлунка (рефлекторний меха
нізм). Гіперсалівація супроводжує розви
ток запальних процесів у порожнині рота
(гінгівіти, стоматити, пульпіти, періодонтити), є реакцією на дію бормашини;
б) збудженням центру слиновиділення, що
міститься в довгастому мозку (бульбарні
паралічі);
в) подразненням вегетативних нервів, що іннервують слинні залози. При стимуляції па
расимпатичних нервів виділяється багато
рідкої слини, при активації симпатичних мала кількість, але густої, в’язкої слини;
г) впливом т-холіноміметиків — фармаколо
гічних препаратів, що активують /и-холінорецептори;
д) дією гуморальних регуляторних факторів.
Цей механізм лежить в основі так званої
парадоксальної, або паралітичної, гіперсалівації. Вона починається через добу після
повної денервації слинних залоз, досягає
максимуму через 6-7 діб, з 15-ї доби по
чинає зменшуватися і через 35^4-0 діб пов
ністю зникає. У патогенезі парадоксальної
гіперсалівацїї провідне значення має під
вищення чутливості денервованих залоз до
циркулюючих у крові гуморальних факто
рів (ацетилхоліну, гістаміну).
При гіперсалівацїї виділяється від 5 до 20 л
слини за добу. Це призводить до:
1) зневоднення організму — розвивається гіперосмолярна гіпогідрія, оскільки слина
гіпотонічна стосовно крові (див. главу 27);
2) нейтралізації шлункового соку, що пов’я
зано зі слабколужним середовищем слини.
Ця обставина викликає порушення трав
лення у шлунку.
Гіпосалівація. Зменшення утворення і виділен
ня слини — гіпосалівація - може мати такі при
чини:
а) центральне гальмування секреції слин
них залоз (страх, переляк, біль);
б) дія т-холінолітиків — фармакологічних
агентів, що блокують т-холінорецептори
периферичних тканин;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
250
в) ушкодження секреторних клітин слин
них залоз (запалення, пухлини);
г) порушення виведення секрету (закупор
ка проток слинних залоз каменями);
д) зневоднення організму.
Серед наслідків гіпосалівації:
1) порушення жування, формування харчо
вої грудки, ковтання, що спричиняється до
розладів наступних етапів травлення;
2) травматизація слизової оболонки рота
з розвитком її запалення (стоматиту);
3) посилене розмноження мікроорганізмів.
Воно обумовлене зменшенням бактерицид
ної дії слини (мало лізоциму) і утворенням
нальоту зі злущеного епітелію на поверхні
язика (прекрасне поживне середовище для
мікробів);
4) порушення трофічних впливів слини на
зуби, недостатня нейтралізація органічних
кислот, що є продуктами діяльності бакте
рій зубного нальоту. Це все сприяє розвит
кові карієсу зубів.
ПОРУШЕННЯ
ТРАВЛЕННЯ V ШЛУНКУ
Шлункові, як одному з центральних органів
травної системи, притаманні всі функції, що
у своїй сукупності забезпечують процес трав
лення: (1) рухова, (2) секреторна і (3) всмокту
вальна.
Тут, у порожнині цього органа, відбувається
подальша фізична обробка їжі: набухання, роз
рідження і розчинення окремих компонентів
харчових продуктів. Хімічна обробка їжі полягає
в тому, що продовжується дія ферментів слини
на вуглеводи, які перебувають у центрі харчової
маси, а під впливом ферментів шлункового соку
починається гідролітичне розщеплення білків
поверхневих шарів подрібненої їжі.
Крім основних, власне травних, шлунок ви
конує й ряд вторинних (додаткових) функцій.
Такими можна вважати (а) захисну (бар ’єрну)
функцію, пов’язану з бактерицидними влас
тивостями компонентів шлункового соку;
(б) участь у здійсненні кровотворення через
пряму причетність до всмоктування вітаміну
В12, конче необхідного для цього процесу (див.
главу ЗО); (в) ендокринну функцію — синтез
і секрецію гормону гастрину, який впливає не
тільки на процеси травлення, а й на багато ін
ших; утворення греліну - гормону, що регулює
харчову поведінку і впливає на перебіг жиро
вого обміну (див. главу 25).
Шлункові дискінезії
Значення рухової функції шлунка полягає в то
му, що вона забезпечує:
1) депонування їжі (резервуарна функція);
2) перемішування її зі шлунковим соком у зоні,
що прилягає до слизової оболонки шлунка.
Завдяки цьому утворюється хімус - харчові
маси, змішані зі шлунковим соком;
3) переміщення вмісту шлунка до пілоричного його відділу;
4) порційна евакуація хімусу у дванадцяти
палу кишку.
Гладкі м’язи стінки шлунка завдяки проце
сам скорочення і розслаблення зумовлюють
такі види рухової активності:
а) розслаблення (релаксація) наповнення. Піс
ля надходження їжі в шлунок його гладка
мускулатура розслаблюється і він може
вміщати до 1,5 л без зміни тиску в ньому;
б) тонічні скорочення. Вони мають у своїй
основі підвищення тонусу гладких м’язових
клітин;
в) перистальтичні скорочення - скорочен
ня у вигляді хвиль, що поширюються від
кардіальної до пілоричної ділянки шлунка.
Вважають, що “водіями ритму” для таких
скорочень є клітини зі спонтанною елек
тричною активністю (здатні самостійно
генерувати потенціали дії), що містяться
у верхній частині тіла шлунка в ділянці ве
ликої його кривини.
Рухова активність шлунка має певну пері
одичність. Розрізняють (а) голодні його рухи
і (б) рухи, пов’язані з травленням.
Регуляція моторики шлунка здійснюється за
допомогою як нервових, так і гуморальних ме
ханізмів. До неї причетні:
а) периферичні рефлекси. їхньою централь
ною ланкою є нервові клітини гангліїв
шлункової стінки. Завдяки периферичним
рефлексам відбувається холінергічне ско
251
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
рочення і пуринергічне розслаблення глад
кої мускулатури шлунка;
б) рефлекси за участю центральної нервової
системи. Еферентною їх ланкою є вегета
тивні гілки блукаючого нерва, закінчення
яких вивільнюють ацетилхолін. Той, діючи
на m-холінорецептори гладких м’язових
клітин, викликає спочатку розслаблення
наповнення, а потім запускає процес ско
рочення;
в) гуморальні фактори. Серед них посилю
ють рухову активність шлунка гастрин
і мотилін, а гальмують - секретин, холецистокінін і шлунково-гальмівний пептид.
Порушення рухової функції шлунка назива
ють шлунковими дискінезіями. Виділяють два їх
варіанти: гіпертонічний і гіпотонічний.
Гіпертонічні дискінезій Для гіпертонічного ва
ріанта шлункових дискінезій характерне збіль
шення тонусу м’язів шлунка (гіпертонія) і по
силення його перистальтики (гіперкінезія).
Причинами рухових розладів шлунка гіпер
тонічного типу можуть бути:
а) деякі харчові фактори (груба їжа, алкоголь);
б) підвищення шлункової секреції;
в) збільшення тонусу блукаючого нерва;
г) деякі гастроінтестинальні гормони
(гастрин, мотилін).
Гіпертонія і гіперкінезія шлунка спричиня
ються до:
1) тривалої затримки харчових мас у шлун
ку, що сприяє підвищенню шлункової се
креції та розвитку виразок на слизовій
оболонці (див. далі);
2) розвитку антиперистальтики (зворотної
перистальтики) шлунка, що обумовлює
появу таких ознак диспепсії, як відрижка,
нудота, блювота.
Однією з форм гіпертонічних дискінезій є пілороспазм - спастичне скорочення пілоричної
частини шлунка. Його спостерігають переваж
но у грудних дітей, частіше в перші тижні та мі
сяці життя.
Вважають, що пілороспазм у дітей обумов
лений функціональними розладами нервовом’язового апарату пілоричної частини шлунка.
Він буває головним чином у збудливих дітей,
що зазнали внутрішньоутробної гіпоксії, що на
родилися в асфіксії, з ознаками пологової трав
ми центральної нервової системи.
При пілороспазмі відзначають слабкий роз
виток мускулатури кардіальної частини шлунка
і більш виражений її розвиток у ділянці воро
таря. Це сприяє легкому виникненню блювоти
і зригувань.
Гіпотонічні
дискінезій
Розлади
моторики
шлунка цього типу характеризується гіпотоні
єю і гіпокінезією.
Зменшення рухової активності шлунка може
бути обумовлене:
а) аліментарними факторами (жирна їжа);
б) зменшенням шлункової секреції (гіпоацидні гастрити);
в) зменшенням тонусу блукаючого нерва;
г) дією гастроінтестинальних гормонів, що
пригнічують моторику шлунка, — секрети
ну, холецистокініну, шлунково-гальмівного
пептиду;
д) видаленням пілоричної частини шлунка;
е) загальним ослабленням організму, висна
женням, гастроптозом (опущенням шлунка).
При гіпотонічних дискінезіях зменшується
час перебування їжі в шлунку, що веде до по
рушення її перетравлювання. Дія неперетравлених компонентів їжі на рецептори слизової
оболонки кишок викликає підвищення їхньої
перистальтики і проноси.
Порушення шлункової секреції
Секреторна активність слизової оболонки
шлунка, що виявляє себе утворенням шлунко
вого соку, пов’язана з діяльністю трьох типів
структур:
1) власне шлункових залоз — трубчастих залоз,
розташованих у слизовій тіла і дна шлун
ка. Вони складаються з трьох видів секре
торних клітин: (а) головних — продукують
пепсиногени, (б) парієтальних (обкладинних) — здійснюють секрецію хлористовод
невої (соляної) кислоти і внутрішнього
фактора Кастла та (в) додаткових (мукоцитів) - секретують слиз (мукоїдний секрет)
і деякі пепсиногени;
2) пілоричних залоз, розташованих у слизо
вій оболонці антрального відділу шлунка.
252
Ці залози не мають парієтальних клітин,
а тому не можуть секретувати НС1. Вони
продукують велику кількість слизу, деякі
пепсиногени. G-клітини цих залоз утворю
ють гормон гастрин;
3) клітин поверхневого епітелію. Вони є ще
одним джерелом слизу шлункового соку
і його електролітів, зокрема бікарбонатів.
У нормі утворюється 2—2,5 л шлункового
соку за добу, він має сильно кислу реакцію pH = 0,8-1,5.
Основними, функціонально важливими ком
понентами шлункового соку є (1) хлористовод
нева кислота, (2) ферменти та (3) слиз.
1. Хлористоводнева (соляна) кислота. Саме
вона створює кисле середовище у порож
нині шлунка. Низькі величини pH необхід
ні для:
а) денатурації й набухання білків, що значно
полегшує їх дальший гідроліз;
б) початкової активації пепсиногенів;
в) створення оптимального середовища для
ферментної дії пепсинів;
г) бактерицидної дії шлункового соку;
д) регуляції діяльності травного каналу в ці
лому (вплив на нервові й гуморальні її ме
ханізми).
Сучасні уявлення про механізми утворення
і секреції НС1 парієтальними клітинами зобра
жено на рис. 34.7.
Джерелом іонів водню є вугільна кислота
(Н2С03), що утворюється із вуглекислого газу
та води під впливом карбоангідрази. Про це,
зокрема, свідчить той факт, що пригнічення ак
тивності цього ферменту деякими специфічни
ми інгібіторами значно зменшує секрецію НС1.
Крім того, секреція одного іона Н+ супроводжу
ється появою в плазмі крові одного іона НС03',
іншими словами, під час секреції НС1 відбува
ється залужнення крові.
Сам процес секреції хлористоводневої кисло
ти є активним і потребує значних витрат енергії,
а тому парієтальні клітини містять дуже багато
мітохондрій. Механізмами активного транспор
ту, що переміщують іони водню і хлору через
плазматичну мембрану, є насоси, здатні гідролізувати АТФ для цих потреб.
На першому етапі завдяки роботі хлорних
насосів іони СІ' переміщуються із цитоплазми
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
клітин назовні. При цьому на зовнішню по
верхню мембрани переноситься й негативний
електричний заряд - вона стає негативно заря
дженою з потенціалом від -40 до -70 мВ. Далі
відбувається пасивний вихід із клітин іонів
калію, оскільки, по-перше, їхня концентрація
в цитоплазмі набагато вища, ніж ззовні, а подруге, до цього спричиняється негативний
електричний заряд зовнішньої поверхні мемб
рани, створений, як було показано вище, робо
тою хлорних насосів.
На другому етапі починає працювати “водне
ва помпа” - Н+,К+-насос, який, використовуючи
енергію АТФ, виводить із клітин іони водню, од
ночасно повертаючи назад іони калію.
Переміщення води із інтерстицію через па
рієтальні клітини в протоки залоз відбувається
пасивно за законами осмосу.
2. Ферменти. Основними ферментами шлун
кового соку є пепсиногени - неактивні про
теолітичні ензими, що мають властивості
ендопептидаз (руйнують внутрішні пептидні зв’язки в молекулах білків).
За властивостями розрізняють дві групи пеп
синогенів. Пепсиногени І групи (п’ять видів)
утворюються головними клітинами залоз фун
дальної частини шлунка. Оптимум ферментної
дії їхніх активних форм припадає на діапазон
pH від 1,5 до 2. Пепсиногени II групи (таких два
види) секретуються залозами антрального відді
лу шлунка і мають оптимум дії при pH 3,2-3,5.
Одразу після секреції пепсиногенів відбу
вається їхня початкова активація хлористо
водневою кислотою, а вже потім перетворен
ня неактивних ферментів у активні проходить
автокаталітичним шляхом — під впливом
пепсинів відщеплюється пептид-інгібітор від
молекул пепсиногенів і останні стають пепси
нами.
3. Слиз. У шлунковому сокові він є сумішшю
муцинів (мукопротеїнів), до складу яких
входять високомолекулярні глікопротеїни
й кислі полісахариди. Основне призначен
ня слизу - захист слизової оболонки від
самоперетравлювання власним шлунковим
соком, а також від механічної та хімічної дії
компонентів їжі.
У регуляції шлункової секреції має важливе
значення поєднання нервових і гуморальних
механізмів (рис. 34.8).
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
253
Механізм утворення НСІ
Механізми утворення й секреції хлористоводневої кислоти парієтальними клітинами шлункових
залоз.
Штриховими лініями позначено пасивний транспорт, літерою Р - мембранні насоси, що використовують для транспорту
енергію АТФ
Перші представлено периферичними реф
лексами, а також умовними й безумовними вагусними рефлексами. Важливо зазначити, що
в кінцевому підсумку всі ці рефлекси завер
шуються виділенням нервовими закінченнями
ацетилхоліну, який через т-холінорецептори
стимулює діяльність усіх без винятку секре
торних клітин слизової оболонки, що спричи
няється до значного посилення шлункової се
креції.
Серед гуморальних регуляторних чинни
ків найбільше значення мають гастрин і гіс
тамін. Будучи продуктом внутрішньої секре
ції G-клітин пілоричних залоз, гастрин прямо
й опосередковано (через виділення гістаміну)
стимулює діяльність головних, парієтальних
і додаткових клітин шлункових залоз, а отже,
і утворення шлункового соку.
Гістамін у стінці шлунка утворюється особ
ливими ентерохромафіноподібними клітина-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
254
Гастрин
Рис. 34.8.
Нервова і гуморальна регуляція секреції хлористоводневої кислоти.
АХ - ацетилхолін, ПК - парієтальна
клітина, ЕХП-клітина - ентерохромафіноподібна клітина, Psy - парасимпатичні нерви
ми (ЕХП-клітинами), що містяться в глибоких
виїмках шлункових залоз, перебуваючи майже
в прямому контакті з парієтальними клітина
ми. А тому саме ці клітини і секреція ними НСІ
стають об’єктами стимуляційної дії гістаміну,
виділення якого, своєю чергою, стимулюється
ацетилхоліном і гастрином.
Секрецію шлункового соку пригнічують про
стагландини (особливо продукування НСІ),
а також ряд гастроінтестинальних гормонів —
секретин, холецистокінін, шлунково-інгібіторний пептид, нейротензин.
У людини залежно від кількості шлункового
соку і його якісних характеристик виділяють
шлункову гіпер- і гіпосекрецію.
Шлункова гіперсекреція. Для такого стану ха
рактерне:
1) збільшення кількості шлункового соку як
після приймання їжі, так і натще;
гіперацидітас і гіперхлоргідрія — відпо
відно збільшення загальної кислотності
та вмісту вільної хлористоводневої кисло
ти в шлунковому сокові;
3) підвищення перетравлювальної здатнос
ті шлункового соку.
2)
Порушення травлення, пов’язані зі шлунко
вою гіперсекрецією, обумовлені тривалою за
тримкою їжі у шлунку (воротар закритий, тому
що нейтралізація дуже кислого хімусу, що над
ходить у дванадцятипалу кишку, вимагає багато
часу).
Ця обставина має ряд наслідків:
• у кишки надходить мало хімусу, що стає
причиною зменшення перистальтики ки
шок і розвитку закрепів;
• у шлунку посилюються процеси бродіння
і утворення газів. Це викликає появу від
рижки й печіїІ
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
• збільшується рухова активність шлунка розвивається гіпертонус і гіперкінезія його
гладкої мускулатури.
Утворення великої кількості активного шлун
кового соку є важливим чинником, що сприяє
розвиткові виразок у шлунку і дванадцятипалій
кишці (див. далі).
В експерименті збільшення утворення і се
креції шлункового соку моделюють, застосову
ючи такі підходи.
I. Вплив на нервові механізми регуляції шлун
кової секреції. Його здійснюють шляхом:
а) збудження рецепторів слизової оболон
ки шлунка (уведення в шлунок сокогінних екстрактів, алкоголю);
б) зменшення гальмівного впливу кори го
ловного мозку на центри блукаючого не
рва - метод “зіткнення ” умовних реф
лексів за І. Павловим (див. вище);
в) електричного подразнення центрів блу
каючого нерва або його еферентних во
локон, що іннервують шлунок;
г) введення т-холіноміметиків.
II. Вплив на гуморальні механізми регуляції
шлункової секреції. Посилене утворення шлун
кового соку викликають введенням:
а) гістаміну або фармакологічних агентів,
що стимулюють його утворення в слизо
вій шлунка;
б) гастрину чи його аналогів;
в) фармакологічних препаратів — стимуля
торів гістамінових Н2-рецепторів (аце
тилсаліцилової кислоти, індометацину);
г) речовин (глюкокортикоїдів, ацетилса
ліцилової кислоти), що пригнічують
утворення простагландинів, які в нормі
гальмують секрецію хлористоводневої
кислоти.
Шлункова гіпосекреція. Характерними її риса
ми є:
1) зменшення кількості шлункового соку на
тще і після приймання їжі;
2) зменшена або нульова кислотність шлун
кового соку (гіпо- або анацидітас), змен
шення вмісту в ньому або повна відсут
ність вільної хлористоводневої кислоти
(гіпо- або ахлоргідрія);
3) зменшення перетравлювальної здатнос
ті шлункового соку аж до ахілії (повного
255
припинення утворення хлористоводневої
кислоти й ферментів).
Зменшення шлункової секреції спричиня
ється до розладів травлення у всьому травному
каналі. Це пояснюється тим, що в умовах недо
статнього утворення шлункового соку воротар
постійно відкритий і вміст шлунка швидко пере
ходить у дванадцятипалу кишку, де середовище
внаслідок відсутності надходження зі шлунка
НС1 постійно лужне. Така обставина гальмує
утворення секретину, внаслідок чого зменшу
ється секреція підшлункового соку й порушу
ються процеси порожнинного травлення в киш
ках. Недостатньо перетравлені компоненти їжі
подразнюють рецептори слизової оболонки ки
шок, що призводить до посилення їхньої перис
тальтики, - розвиваються проноси.
Крім того, внаслідок відсутності хлористоводне
вої кислоти в шлунку посилено розмножується мі
крофлора. З цим пов’язана активація процесів гнит
тя й бродіння в шлунку і поява таких диспепсичних
симптомів, як відрижка, обкладений язик та ін.
Експериментальне моделювання шлункової
гіпосекреції ґрунтується на відомих уже нам
підходах.
I. Вплив на нервові механізми регуляції шлун
кової секреції. Він зокрема передбачає:
а) блокаду рецепторів слизової оболонки
шлунка (застосування речовин, що викли
кають місцеву анестезію);
б) перетинання блукаючого нерва або його гі
лок (селективна ваготомія);
в) використання гангліоблокаторів (п-холінолітиків);
г) застосування т-холінолітиків.
II. Вплив на гуморальні механізми регуляції
утворення та секреції шлункового соку. Його
можна здійснювати за допомогою:
а) блокади гістамінових Н2-рецепторів, на
приклад, циметидином;
б) антагоністів гастрину “-секретину, шлунково-інгібіторного пептиду та ін.
Виразкоутворення
Виразкою (ulcer) шлунка називають локальний
дефект його слизової оболонки, часто із захоп
ленням підслизового шару (рис. 34.9). Поверх
неві дефекти слизової, які не поширюються на
глибші шари, позначають як ерозії.
256
Утворення і розвиток виразок у шлунку
пов’язують з порушенням балансу між двома
групами чинників, які позначають як фактори
агресії та фактори захисту (рис. 34.10).
До факторів агресії відносять чинники, що
самі по собі здатні викликати або сприяти роз
виткові ушкодження слизової оболонки. Таки
ми є (рис. 34.11):
а) власне шлунковий сік, а саме: агресивні
його компоненти - хлористоводнева кисло
та й активні пепсини;
б) екзогенні фактори інфекційної природи.
У людини найбільше значення має Helico
bacter pylori (див. далі);
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
в) екзогенні фактори неінфекційного походже
ння: фізичні чинники (механічне ушкоджен
ня, температура, радіація), хімічні агенти
(кислоти, луги, різні органічні і неорганічні
сполуки, ліки, алкоголь тощо).
Захист слизової оболонки шлунка від дії
агресивних чинників здійснюється завдяки існу
ванню слизового бар’єра між просвітом шлунка
та клітинами його епітелію.
Слиз є продуктом діяльності всіх залоз слизо
вої оболонки: власне шлункових (тут його про
дукують мукоцити) і пілоричних, а також клі
тин поверхневого епітелію. Останні, крім того,
0.
Порушення балансу між факторами агресії та факторами захисту як причина
утворення виразок у шлунку
257
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
Фактори агресії та фактори захисту слизової оболонки шлунка
секретують ще й бікарбонати, які утворюють
тонкий прошарок між гелем слизу та поверхнею
епітеліальних клітин.
Слиз завдяки своїм фізично-хімічним влас
тивостям перешкоджає дифузії в глибину іонів
водню і сам є стійким до протеолітичної дії
пепсинів. Бікарбонатні аніони нейтралізують
ті водневі іони, яким все ж таки вдається про
никнути у глибокі шари слизу.
Стійкість слизової оболонки шлунка до утво
рення виразок залежить не тільки від здатності
утворювати необхідну кількість повноцінного
слизу, а й від чинників, що забезпечують нор
мальну структуру і життєдіяльність клітин епі
телію. Такими зокрема є:
а) належне кровопостачання, необхідне для
підтримання як структурної цілісності
клітин, так і їхньої функціональної актив
ності;
б) повноцінна регенерація клітин, яка потре
бує відповідного пластичного, енергетич
ного і регуляторного забезпечення;
в) нервова трофіка — вплив периферичних
нервів на перебіг обміну речовин у кліти
нах (докладно див. главу 38).
Важливе місце в забезпеченні стійкості шлун
ка до виразкоутворення посідають простаглан
дини (рис. 34.12). їхня захисна дія пов’язана
з такими ефектами:
а) посилюють продукування слизу клітинами
залоз шлунка;
б) збільшують утворення бікарбонатів клітина
ми поверхневого епітелію слизової оболонки;
в) пригнічують секрецію хлористоводневої
кислоти парієтальними клітинами шлунко
вих залоз;
г) посилюють кровообіг у стінці шлунка, ви
кликаючи розширення судин.
Патогенез виразок шлунка досить складний,
у ньому, як правило, поєднуються різні факто
ри - як ті, що збільшують агресивні впливи на
слизову оболонку, так і ті, що зменшують захис
ну її здатність.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
258
2. Роль простагландинів у захисті слизової оболонки шлунка від виразкоутворення.
НПЗЗ - нестероїдні протизапальні засоби
Проте все ж можна виділити ряд провідних
механізмів, які за певних умов відіграють голов
ну роль у виразкоутворенні. Це дає підстави ви
окремити кілька патогенетичних варіантів вира
зок шлунка.
Пептичні виразки. Пептичною називають ви
разку, що виникає в результаті перетравлювальної дії шлункового соку на слизову оболонку
шлунка і дванадцятипалої кишки.
Розрізняють два різновиди пептичної виразки:
1) виразка, що утворюється в результаті під
вищення агресивності шлункового соку
(збільшення кількості соку, його кислот
ності та перетравлювальної сили). В екс
перименті може виникати при моделюван
ні шлункової гіперсекреції методами, що
вже були описані в цій главі;
2) виразка, обумовлена зменшенням захис
них властивостей слизової оболонки. За
цих умов шлунковий сік, що має нормаль
ну агресивність, чинить ушкоджувальну
і перетравлювальну дію на змінену сли
зову. Існує два підходи до моделювання
в експерименті пептичної виразки цього
виду:
а) порушення утворення слизу (наприклад,
уведення глюкокортикоїдів, ацетилсалі
цилової кислоти чи інших нестероїдних
протизапальних засобів);
б) руйнування слизового бар’єра речовинами-детергентами (наприклад, уведення
в шлунок жовчних кислот, відтворення
дуодено-гастрального рефлюксу).
Екзогенні виразки. До екзогенних відносять
виразки, що виникають у результаті прямої дії
на слизову оболонку шлунка ушкоджувальних
факторів зовнішнього середовища.
В експерименті множинні виразки шлунка ек
зогенного походження моделюють за допомогою:
а) фізичних впливів - уведення в шлунок роз
дробленого скла (механічне ушкодження),
окропу, рідкого азоту і т.п.;
б) хімічних агентів — уведення сильних кис
лот і лугів.
259
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
Екзогенною можна вважати виразку в люди
ни, зумовлену мікробними факторами, зокрема
Helicobacter pylory. Про роль цього збудника
у розвитку виразкової хвороби мова піде далі.
Трофічні виразки. Так називають виразки, які
виникають у результаті порушення живлення
слизової оболонки шлунка. До трофічних від
носять:
а) судинні виразки. Вони утворюються вна
слідок порушень кровопостачання стін
ки шлунка. В експерименті їх моделюють
перев’язуванням артерій, що живлять шлу
нок, введенням у шлункові артерії атофану — речовини, яка викликає склерозування стінки й облітерацію просвіту судин;
введенням великих доз катехоламінів, що
спричиняють спазм артерій та ішемію сли
зової шлунка;
б) нейрогенні виразки, які є результатом по
рушень нервової трофіки і розвитку нейродистрофічного процесу (див. главу 38).
В експерименті моделюють перетинанням
нервів, що іннервують стінку шлунка.
Ще не так давно для хірургічного лікуван
ня виразкової хвороби пропонували опера
цію селективної ваготомії - перетин гілок
блукаючого нерва, що здійснюють іннерва
цію шлунка. При цьому досягалося зменшен
ня секреторної активності шлункових залоз
і спостерігали загоювання пептичних виразок.
Проте з часом розвивалися рецидиви старих
і з’являлися нові виразки, цілком імовірно через втрату нервових трофічних впливів на
слизову оболонку.
Гіпорегенераторні виразки. Вони виникають
у результаті порушення регенерації епітелію
слизової оболонки. У нормі покривний епітелій
і залозистий апарат слизової шлунка повністю
оновлюється через кожні 5 діб. Якщо процеси
регенерації з якихось причин порушуються, то
спочатку утворюються поверхневі дефекти ерозії, а потім і глибші - виразки.
Розвиток гіпорегенераторних виразок може
бути зумовлений:
а) дією високих доз глюкокортикоїдів, що
пригнічують біосинтез білка і клітинний
поділ (про механізми ульцерогенної дії глюкокортикоїдних гормонів див. главу 37);
б) білковим голодуванням і гіповітамінозами;
в) дією отрут - інгібіторів біосинтезу білків.
Виразкова хвороба
Виразковою хворобою називають хронічне рецидивуюче захворювання, що характеризується
утворенням виразки в шлунку або дванадцяти
палій кишці.
Для виразкової хвороби характерне утворен
ня пептичної виразки в “улюблених” місцях
шлунка і в початковому відділі дванадцятипалої
кишки (див. рис. 34.9), на відміну від гострих
виразок, що можуть виникати в результаті дії ці
лого ряду ульцерогенних факторів.
Етіологія. Сучасний рівень знань дає підстави
виділяти основну причину виразкової хвороби,
причини вторинних симптоматичних (найчас
тіше гострих) виразок і фактори, що сприяють
утворенню виразок у людини. Розглянемо їх.
І. Основна причина виразкової хвороби в лю
дини — бактерія Helicobacter pylori. Такий ви
сновок було зроблено на підставі відкриття ав
стралійських учених Маршсиїла і Уоррена, за
яке вони удостоїлися Нобелівської премії (2005).
Було встановлено, що приблизно 50 % на
селення Земної кулі інфіковано цим мікробом.
У більшості людей гелікобактерна інфекція ні
чим себе не виявляє, проте у 10-15 % інфіко
ваних розвивається виразкова хвороба (виразка
виникає частіше у дванадцятипалій кишці, ніж
у шлунку) і в кількох відсотків — рак шлунка
(уражується тіло цього органа) чи лімфома, яка
росте з клітин асоційованої із слизовою оболон
кою лімфоїдної тканини - MALT-лімфома (від
mucosa associated lymphoid tissue).
За різними даними вважається, що понад
90 % виразок дванадцятипалої кишки і до 80 %
виразок у шлунку зумовлено цим збудником.
Helicobacter pylori (Н. pylori) являє собою
спіралеподібну грамнегативну бактерію, що має
4-8 джгутиків і здатна швидко рухатися навіть
у густому слизу. Н. pylori може утворювати на
вколо себе плівку, що захищає бактерії від імун
ної відповіді хазяїна і робить їх нечутливими до
дії антибіотиків.
Геном Н. pylori складається з понад 1,5 тис.
генів, з яких більш ніж 40 об’єднано у т. зв.
‘‘’’острів патогенності”, який ще позначають
як Cag (cytotoxin-associated antigen), - ділянку
260
геному, що відповідає за патогенні властивості
бактерій. “Острів патогенності” звичайно від
сутній у штамів, виділених від людей, що є безсимптомними носіями Н. pylori.
Здатність Н. pylori викликати розвиток гас
тритів і виразок пов’язана з цілим рядом фак
торів вірулентності бактерій, на яких ми зупи
нимося далі.
II. Вторинні (симптоматичні) виразки шлун
ка і дванадцятипалої кишки мають, як правило,
чітко визначену причину, усунення якої веде до
загоювання виразок. До виразкоутворення мо
жуть спричинятися:
1) тривале вживання нестероїдних протиза
пальних лікарських препаратів, зокрема
аспірину (“аспіринові” виразки). Основ
ний механізм їхньої ульцерогенної дії —
зменшення утворення важливого захис
ного чинника слизової шлунка — проста
гландинів (див. вище) через пригнічення
ключового ферменту їхнього синтезу - циклоксигеназщ
2) високі дози глюкокортикоїдних гормонів
(“глюкокортикоїдні виразки ”). Ці гормони
та їхні препарати зменшують продукуван
ня вже згадуваних простагландинів (че
рез ліпокортиновий механізм, див. главу
37), а також пригнічують біосинтез білків
(білків слизу, ферментів шлункового соку)
і поділ клітин (регенерацію епітелію сли
зової оболонки шлунка);
3) стрес (“стресорні виразки”) (див. главу 8).
У розвитку стресорних виразок мають зна
чення тривалий спазм кровоносних судин
стінок шлунка, що виникає під впливом
катехоламінів, а також дія на слизові обо
лонки шлунка і кишок глюкокортикоїдних
гормонів, про ульцерогенні ефекти яких
уже йшла мова;
4) шокові стани з вираженою еректильною
фазою і больовим синдромом - травма
тичний шок, опіковий шок ("шокові вираз
ки”). Основним механізмом таких виразок
є порушення трофіки стінок шлунка через
тривалу ішемію, зумовлену надмірним збу
дженням
симпатоадреналової
системи
і виділенням великих кількостей катехола
мінів, що зменшують просвіт артеріальних
судин;
5) гіперпродукція гастрину, що веде до знач
ної шлункової гіперсекреції й посилення
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
агресивності шлункового соку (“гастринові пептичні виразки”). Надмірна секре
ція гастрину характерна для:
а) синдрому Золлінгера — Еллісона (гастриноми) — пухлини (найчастіше острівцевого апарату підшлункової залози), клі
тини якої набули здатність утворювати
і секретувати гастрин;
б) гіперпаратиреозу (див. главу 37). При цьо
му зростання продукції гастрину G-клі
тинами слизової оболонки шлунка зумов
лене гіперкальціємією (див. главу 28).
III.
Фактори, що сприяють розвитку вираз
кової хвороби. До таких можна віднести:
а) спадкову схильність. Той факт, що да
леко не всі інфіковані, навіть найбільш
вірулентними штамами Н. pyloris люди
мають виразкову хворобу, свідчить про
важливе значення генетичного чинни
ка і зокрема однонуклеотидного полі
морфізму генів для виразкоутворення.
На цій підставі виразкову хворобу слід
вважати полігенною (мультифакторіальною) недугою (див. главу 12).
Роль спадкового фактора підтверджується
відносно високою (40-60 %) частотою виразко
вої хвороби у батьків і родичів хворих, особли
во молодого віку. Установлено, що в пацієнтів
з обтяженою спадковістю в слизовій оболонці
шлунка у 1,5-2 рази більше парієтальних клі
тин, ніж у здорових людей. Ознаками генетич
ної схильності є також 0 (І) група крові, яку
часто виявляють у хворих на виразку, дефіцит
а,-антитрипсину і деяких глікопротеїнових ком
понентів слизу;
б) хронічну психоемоційну перенапругу, як
правило, негативного характеру (негатив
ні емоції, конфліктні ситуації, почуття по
стійної тривоги, перевтома і т.п.), яка часто
повторюється. У людей, що є симпатотоніками, така перенапруга нерідко веде до ар
теріальної гіпертензії, а у ваготоніків - зу
мовлює шлункову гіперсекрецію і сприяє
виразкоутворенню.
Про значення, якого ще донедавна надавали
нервовим чинникам у розвитку виразкової хво
роби, свідчить цілий ряд теорій, серед яких нер
вово-вегетативна (її прихильники вважали, що
причиною виразки є гіперсекреція шлункового
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
соку, гіпермоторика шлунка і судинні порушення
в ньому в осіб з конституціонально обумовленим
переважанням тонусу парасимпатичної нервової
системи), нервово-рефлекторна (пояснювала ви
никнення виразки рефлекторними впливами на
шлунок, наприклад, при хронічному апендициті,
коліті, жовчнокам’яній хворобі), кортиковісцеральна (підкреслювала провідну роль порушень
умовнорефлекторної діяльності головного моз
ку —неврозів - в утворенні виразок);
в) неправильне харчування - їжа всухом’ятку,
нерегулярне приймання їжі, вживання гру
бої або гострої їжі, погане її пережовуван
ня, їда поспіхом, відсутність зубів, недо
статній вміст у харчових продуктах білків
і вітамінів;
г) шкідливі звички - куріння, уживання алко
голю (особливо міцних спиртних напоїв).
Патогенез. Розвиток виразкової хвороби прямо
пов’язаний з факторами патогенності та віру
лентності, притаманними збудникові цієї неду
ги — Helicobacter pylori. А тому доцільно корот
ко охарактеризувати їх (рис. 34.13).
Джгутики. Наявність 4-8 джгутиків до
зволяє бактеріям швидко переміщуватися
у слизові й колонізувати клітини хазяїна.
Білки поверхні та ліпополісахариди мемб
ран. Завдяки їхнім молекулам відбувається
прикріплення бактерій до клітин слизової
оболонки. Вони є антигенами, здатними ін
дукувати імунну відповідь на себе;
Уреаза - фермент, що здійснює розщеплен
ня сечовини до вуглекислого газу й аміаку.
Останній, взаємодіючи з водневими іона
ми, нейтралізує хлористоводневу кислоту
шлункового соку.
Секреторні ферменти: муцинази, протеа
зи, ліпази. Виділення їх бактеріями веде до
розщеплення основних компонентів слизу,
руйнування захисного шару слизової обо
лонки шлунка.
Екзотоксини. Серед них особливо важливе
значення має вакуолізуючий токсин (VacA),
який індукує утворення вакуолей в епітелі
альних клітинах і в такий спосіб спричиняє
їхнє ушкодження.
Білки-ефектори. Так називають бактері
альні протеїни, які особливим способом
секреції “вприскуються” всередину клі
тин хазяїна і викликають у них цілий ряд
261
структурних і функціональних змін. До та
ких білків, зокрема, належить cagA. Потра
пивши в клітини слизової оболонки шлун
ка, він взаємодіє з актином, що входить
до складу цитоскелета (змінюється форма
клітин), посилює ріст клітин і пригнічує
апоптоз (можлива малігнізація), індукує
утворення і секрецію деяких цитокінів, зо
крема інтерлейкіну-8 (ІЛ-8). Останній ви
ступає медіатором запалення, що втягує
в цей процес моноцити (макрофаги) і лім
фоцити.
У патогенезі виразкової хвороби можна виді
лити ряд послідовних етапів (рис. 34.14).
I. Інфікування Helicobacter pylori. Потрапив
ши із зовнішнього середовища у шлунок, бак
терії завдяки своїм джгутикам легко проходять
через в’язкий слизовий бар’єр і за допомогою
білків поверхні та ліпополісахаридів прикріп
люються до епітеліальних клітин слизової
оболонки.
II. Запалення (хронічний гастрит) з гіперсекрецією. Після прикріплення Н. pylori до клітин
розвиваються такі події:
1) сечовина, що надходить у бактерії ззовні,
під впливом уреази розпадається на вуг
лекислий газ і аміак (рис. 34.15). Остан
ній дифундує в слизовий шар і, нейтра
лізуючи тут хлористоводневу кислоту,
зумовлює підвищення pH до 6-7. Це стає
сигналом для G-клітин, які починають
у відповідь продукувати гастрин. Над
ходження цього гормону у кров, а по
тім — до секреторних клітин шлункових
залоз спричиняється до посилення їхньої
активності та в підсумку - до шлункової
гіперсекрецїї;
2) ферменти, що виділяються бактеріями
в слиз, - муцинази, протеази, ліпази —
зумовлюють гідролітичне розщеплення
компонентів слизу, ведуть до руйнування
захисного слизового шару стінки шлунка
(рис. 34.16);
3) екзотоксини, зокрема VacA, діючи на епі
теліальні клітини, викликають їх ушко
дження, яке можна позначити як первинну
альтерацію',
4) білки-ефектори, такі як cagA, індукують
утворення і секрецію клітинами слизової
оболонки інтерлейкіну-8 - цитокіну, що
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
262
Фактори патогенності та вірулентності Helicobacter pylori
активує еміграцію моноцитів і лімфоцитів
в уражені бактеріями тканини;
5) макрофаги, що утворилися з моноцитів,
вивільнюють у слизову оболонку вільні
радикали, пероксиди й лізосомні фермен
ти - фактори, що зумовлюють вторинну
альтерацію (див. главу 17);
6) В-лімфоцити стають джерелом антитіл
проти антигенів Н. pylori і автоантитіл
проти власних змінених білків слизової
оболонки (автоантигенів), зумовлюючи
долучення до процесу імунних механізмів
ушкодження клітин.
Легко бачити, що всі описані вище події
(первинна альтерація, еміграція моноцитів
і лімфоцитів, вторинна альтерація) є суттю
процесу, що має назву запалення (див. главу
17), а тому може бути позначений як хронічний
гастрит.
III.
Утворення виразки. В основі виразок, що
виникають у слизовій оболонці шлунка і два
надцятипалої кишки при гелікобактерній інфек
ції, лежать два механізми:
1) запальний, тобто пов’язаний з ушкоджен
ням клітин епітелію факторами Н. pylori
(первинна альтерація) і продуктами макрофагального (вільні радикали й перок
сиди, лізосомні ферменти) та лімфоцитарного (автоантитіла) походження (вторинна
альтерація);
2) пептичний - зумовлений дією на слизо
ву оболонку власного шлункового соку
(хлористоводневої кислоти, пепсинів),
агресивність якого при гелікобактерній
інфекції, як було показано вище, зростає
(шлункова гіперсекреція). Крім того, агре
сивний шлунковий сік діє на позбавле
ний захисного слизового шару епітелій,
оскільки під впливом ферментів Н. pylori
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
відбувається руйнування основних ком
понентів слизу.
Наведені вище механізми виразкоутворення
визначають дві основні стратегічні лінії в ліку
ванні виразкової хвороби:
а) антибактеріальна терапія, спрямована на
знищення збудника недуги - Н. pylori;
б) зменшення агресивності шлункового соку
і захист клітин слизової оболонки від його
дії (антацидна терапія).
IV.
Ускладнення виразки. Поглиблення дефекту
слизової оболонки і перехід його на підслизовий
шар і м’язову оболонку стінки шлунка, а також
Рис. 34.14. Етапи патогенезу гелікобактерної інфекції
263
процеси рубцювання зумовлюють ряд усклад
нень виразкової хвороби. Такими зокрема є:
1) кровотечі. Оголення стінок кровоносних
судин і “роз’їдання” їх кислотою та фер
ментами стає причиною кровотеч, що мо
жуть мати летальний наслідок;
2) перфорація виразки - утворення наскріз
ного отвору у стінці шлунка чи дванад
цятипалої кишки. При цьому їхній вміст
потрапляє в черевну порожнину, і розви
вається запалення очеревини - перитоніт;
3) пенетрація. Це також утворення отвору,
одначе він відкривається не в черевну по
рожнину, а в розташовані поруч органи:
264
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Механізм шлункової гіперсекреції при гелікобактерній інфекції
підшлункову залозу, товсту кишку, печін
ку, малий сальник тощо;
4) пілоростеноз - непрохідність пілоричного
відділу шлунка, тобто утруднення прохо
дження їжі зі шлунку в кишки в результа
ті деформації та звуження місця переходу
шлунка у дванадцятипалу кишку. Виникає
як наслідок рубцювання виразки, розташо
ваної в кінцевому відділі шлунка чи в по
чатковому відрізку дванадцятипалої кишки.
ПАТОФІЗІОЛОГІЯ
ПІДШЛУНКОВОЇ ЗАЛОЗИ
Підшлункова залоза є органом, що виконує ек
зокринну і ендокринну функції. Щодо порушень
останньої, то мова про них ішла в главі 24. Тут
ми зупинимося тільки на розладах підшлунко
вої секреції, що мають стосунок до процесів
травлення.
Продуктом зовнішньосекреторної діяльнос
ті підшлункової залози є панкреатичний сік —
безбарвна прозора рідина з pH 8,0-8,5. За добу
в людини утворюється приблизно 2,0—2,5 л
соку.
Підшлунковий сік містить два функціонально
важливі компоненти: (І) ферменти й (II) бікар
бонати. З огляду на їхнє значення не тільки для
процесів травлення, а й для розвитку патологіч
них процесів, дамо коротку характеристику цих
компонентів.
І.
Ферменти. Місцем утворення ферментів
є залозисті ацинарні клітини підшлункової
265
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
Механізми розвитку хронічного гастриту і пептичної виразки при гелікобактерній інфекції
залози. Секреторний процес, що відбувається
в них, складається з кількох послідовних ета
пів:
1) біосинтезу молекул ферментів (більшість
з них утворюється у формі неактивних
проферментів);
2) формування секреторних гранул, заповне
них цими ферментами;
3) власне секреції - виділення гранул у про
світ ацинусів механізмом екзоцитозу.
Панкреатичні ферменти належать до класу
гідролаз і можуть розщеплювати різні компо
ненти їжі. Залежно від субстрату, на який вони
діють, розрізняють:
1.
Протеолітичні ферменти. їх поділяють
на ендопептидази (трипсин, хімотрипсин,
еластаза) і екзопептидази (карбоксиполіпептидаза А, карбоксиполіпептидаза В).
Перші - розщеплюють внутрішні пептидні зв’язки білкових молекул, зумовлюючи
розпад протеїнів до дрібніших поліпеп
тидів. Другі - діють на зовнішні пептидні
зв’язки з різних кінців поліпептидних лан
цюгів, вивільнюючи окремі амінокислоти.
Панкреатичні протеази секретуються у фор
мі неактивних проферментів — трипсиногену,
хімотрипсиногену, проеластази, прокарбоксиполіпептидаз. їхня молекулярна маса більша за
266
масу відповідних активних ензимів. Це свідчить
про те, що в процесі активації відбувається від
щеплення від молекул проферментів поліпептидних фрагментів, унаслідок чого активні цен
три ензимів стають доступними для субстратів.
Після надходження неактивних панкреатич
них протеаз у просвіт дванадцятипалої кишки
там відбуваються такі послідовні події:
1)
початкова
активація
трипсиногену.
Епітеліальні клітини слизової оболон
ки дванадцятипалої кишки вивільнюють
у її просвіт невелику кількість ферменту
ентерокінази. Маючи гідролітичні власти
вості, цей ензим перетворює перші порції
трипсиногену в трипсин;
2)
автокаталітична активація ферментів.
Вона відбувається під впливом уже утво
рених молекул активного трипсину. Уна
слідок цього стрімкого лавиноподібного
процесу всі протеази підшлункового соку
за короткий проміжок часу стають актив
ними;
3) дія активних протеаз на білки та гідролі
тичне розщеплення останніх;
4) самоінактивація трипсину. При великій
кількості активного трипсину й відсут
ності необхідних для нього субстратів
трипсин розпізнає певні місця в сусідніх
трипсинових молекулах (англ. critical self
recognition cleavage site) і розщеплює їх.
2. Ліполітичні ферменти. Підшлунковий сік
містить (а) панкреатичну ліпазу, що гідролізує нейтральні жири; (б) холестеролестеразу, що розщеплює ефіри холестеролу;
(в) фосфоліпази, які відщеплюють вільні
жирові кислоти від молекул фосфоліпідів.
Тільки останні - фосфоліпази - виділяють
ся в підшлунковий сік у неактивній формі
й активуються у дванадцятипалій кишці
трипсином. Решта від самого початку є ак
тивними.
3. Амілолітичні ферменти. До таких нале
жить панкреатична а-амілаза — ензим, що
гідролізує полісахариди (крохмаль, гліко
ген) до декстринів. а-Амілаза секретується
в підшлунковий сік у формі, що не потре
бує активації.
Важливим компонентом панкреатичного соку,
що запобігає передчасній активації ферментів
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
у протоках залози, є інгібітори трипсину, серед
яких SPINK1 (serine protease inhibitor Kazal type 1).
II.
Бікарбонати.
Секрецію
бікарбонатів
і води здійснюють епітеліальні клітини найдрібніших і дрібних проток підшлункової зало
зи. Завдяки бікарбонатам:
а) відбувається нейтралізація кислого шлун
кового соку, що надходить у дванадцятипа
лу кишку;
б) створюється лужне середовище, опти
мальне для дії ферментів у тонкій кишці;
в) підтримується кислотно-основна рівно
вага в організмі, яка могла би поруши
тися в результаті секреції НСІ у шлунку
(якби не було секреції бікарбонатів, то
мав би розвиватися негазовий алкалоз,
див. главу 29). Посиленому утворенню
хлористоводневої кислоти в шлунку має
відповідати збільшена секреція бікарбо
натів у панкреатичній залозі.
Джерелом бікарбонатів є вуглекислий газ, що
надходить у клітини з крові (рис. 34.17). Під
впливом карбоангідрази утворюється вугільна
кислота (Н2С03), яка дисоціює на водневі та бі
карбонаті іони. Шляхом активного транспорту
перші - повертаються у кров, а другі - разом
з іонам натрію секретуються у просвіт проток.
У регуляції панкреатичної секреції провідне
місце посідають три чинники:
1) ацетилхолін, що є медіатором вегетатив
них парасимпатичних нервів. Усі умовні
та безумовні рефлекси, що стимулюють
секрецію шлункового соку, одночасно по
силюють панкреатичну секрецію. При сти
муляції панкреатичних гілок блукаючого
нерва активується діяльність ацинарних
клітин і виділяється невелика кількість
соку, багатого на ферменти;
2) холецистокінін. Утворення цього гастроінтестинального гормону епітеліальними
клітинами дванадцятипалої і верхнього від
ділу порожньої кишки стимулюється жиро
вими кислотами і продуктами розщеплен
ня білків — пептидами та амінокислотами.
Холецистокінін діє на ацинарні клітини
підшлункової залози і стимулює секрецію
панкреатичних ферментів. У відповідь на
цей гормон виділяється невелика кількість
густого, багатого на ферменти соку;
267
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
Механізми утворення й секреції бікарбонату клітинами дрібних проток підшлунковоїзалози.
Штриховими лініями позначено пасивний транспорт, літерами AT - механізми активного трансмембранного транспорту
3)
секретин. Основним стимулятором се
креції цього гормону клітинами слизової
оболонки дванадцятипалої кишки є хло
ристоводнева кислота шлункового соку.
При зменшенні pH дуоденального хімусу нижче 4,5-5,0 починається виділення
у кров секретину, максимальною його се
креція стає, коли pH падає нижче 3,0. Під
впливом секретину посилюється секреція
бікарбонатів і води клітинами дрібних
проток підшлункової залози. Дія секрети
ну супроводжується виділенням у дванад
цятипалу кишку великої кількості бідно
го на ферменти і багатого на бікарбонати
панкреатичного соку.
Порушення зовнішньосекреторної діяльності
підшлункової залози можуть виявляти себе пан
креатичною гіперсекрецією і гіпосекрецією.
Панкреатична гіперсекреція. Збільшення утво
рення і секреції підшлункового соку може бути
обумовлено:
1) підвищенням тонусу парасимпатичної
нервової системи (блукаючого нерва) в ре
зультаті рефлекторних чи центральних
впливів на її центри. Ацетилхолін, як уже
зазначалося, є стимулятором діяльності
ацинарних клітин, що продукують фер
менти підшлункового соку;
2) збільшенням утворення й секреції гастроінтестинальних гормонів, що стимулю
ють секрецію води і бікарбонатів у складі
підшлункового соку (секретин) та підви
щують вміст у ньому травних ферментів
(холецистокінін). Факторами, що посилю
ють утворення зазначених гормонів, є кис
лий вміст хімусу, що надходить із шлунка
в дванадцятипалу кишку, вільні жирові
кислоти і продукти початкового гідролізу
білків (пептиди, деякі амінокислоти).
Збільшення панкреатичної секреції поліпшує
процеси порожнинного травлення, однак за де
яких умов може сприяти розвитку гострого пан
креатиту, про що мова піде далі.
Панкреатична гіпосекреція. Причинами змен
шення утворення і секреції підшлункового соку
можуть бути:
1) нейрогенне гальмування зовнішньосе
креторної функції підшлункової залози,
(зменшення тонусу блукаючого нерва,
отруєння атропіном та ін.);
2) дуоденіт - запалення слизової дванадця
типалої кишки, унаслідок чого зменшуєть
268
ся утворення стимуляторів панкреатичної
секреції - секретину і холецистокініну;
3) порушення виведення підшлункового соку
(закупорка проток, їх здавлювання);
4) зменшення кількості ацинарних клітин
(руйнування, хронічні панкреатити);
5) спадково зумовлена недостатність ентерокінази, внаслідок чого порушується почат
кова активація протеолітичних ферментів
підшлункового соку (трипсину, хімотрипсину).
Головним наслідком панкреатичної гіпосекреції є порушення порожнинного травлення
в кишках - розвиток синдрому мальдигестії
(див. далі).
Гострий панкреатит
Гостре запалення підшлункової залози має ряд
важливих особливостей, з якими пов’язані тяж
кий перебіг цієї хвороби та часто смертельно
небезпечні ускладнення.
Такими особливостями є:
1) генералізований характер запалення: у за
пальний процес втягується, як правило,
вся залоза;
2) виражена судинна реакція, яка спричиня
ється до набряку органа і крововиливів
у нього (геморагій);
3) значна альтерація, яка виявляє себе не
крозом жирової тканини і паренхіми за
лози. Такий тотальний некроз, який по
ширюється і на інкреторний апарат під
шлункової залози (острівці Лангерганса),
веде до повної втрати органа, не сумісної
з життям.
Етіологія. До розвитку гострого панкреатиту
можуть вести різні чинники, серед них:
а) вживання алкоголю і переїдання, що його
супроводжує; приймання великої кількості
жирної їжі;
6) жовчні камені та інші фактори, що ство
рюють перешкоду для виділення панкре
атичного соку в дванадцятипалу кишку
(пухлини, аномалії розвитку головної про
токи підшлункової залози, деякі паразити,
наприклад, аскариди);
в) механічне ушкодження підшлункової зало
зи при травмах і хірургічних втручаннях',
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
г) інфекційні агенти (віруси епідемічного па
ротиту, коксакі, деякі бактерії);
д) інтоксикації, а також дія деяких лікарських
засобів (імунодепресанти, ііазиди, сульфонаміди та ін.). Загалом сьогодні налічують
понад 80 різних фармакологічних препа
ратів, приймання яких може спровокувати
гострий панкреатит;
е) гостра ішемія підшлункової залози, зумов
лена тромбозом її судин, емболією, васкулі
тами чи шоком;
є) метаболічні порушення в організмі, що
супроводжуються гіперксиїьціємією. Такі,
зокрема, виникають при гіперпаратиреозі
(див. главу 36);
ж) генетичні фактори. Оскільки гострий пан
креатит виникає не у всіх, хто зазнає впли
ву наведених чинників, то можна ствер
джувати, що для розвитку цієї недуги має
значення спадкова схильність, пов’язана
з явищем однонуклеотидного поліморфіз
му генів. Крім того, описано мутації ряду
генів, що зумовлюють розвиток особливих,
ідіопатичних форм панкреатиту, про які
мова піде далі.
Патогенез. Провідним механізмом розвитку
гострого панкреатиту будь-якої етіології є пе
редчасна активація ферментів підшлунково
го соку в протоках підшлункової залози. Най
більше значення має активація трипсину, тобто
утворення його із трипсиногену під дією ак
тивних протеолітичних ферментів клітинного
(лізосомного) або кишкового походження. Осо
бливістю активації ферментів у протоках під
шлункової залози є те, що цей процес має ла
виноподібний характер і, виникнувши в одному
місці залози, швидко охоплює всі її протоки. Це
обумовлено автокаталітичними реакціями, під
час яких молекули активних ензимів (трипсину,
хімотрипсину) перетворюють неактивні фер
менти (трипсиноген, хімотрипсиноген) в ак
тивні.
Таким чином, для ініціювання гострого пан
креатиту потрібна “іскра”, якою можуть стати
навіть поодинокі молекули активних протеаз,
що потрапили в протоки залози з ушкоджених
клітин або кишок. Залежно від механізмів пе
редчасної активації ферментів підшлункового
соку можна виділити кілька патогенетичних ва
ріантів гострого панкреатиту (рис. 34.18).
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
I. Первинно-альтеративний. Цей варіант роз
вивається в результаті ушкодження (альтерації)
тканини підшлункової залози різними патоген
ними факторами. Такими можуть бути механіч
не ушкодження (травма, операційні втручання),
вплив хімічних агентів (інтоксикації, отруєння,
у тому числі лікарськими препаратами), біоло
гічні чинники (віруси, бактерії); ішемічне ушко
дження.
Ініціаторами передчасної активації ферментів
підшлункового соку у всіх цих випадках є лізосомні ферменти, що вивільняються з ушкодже
них клітин залозистої тканини.
II. Гіпертензивний. В основі розвитку тако
го варіанту хвороби лежить підвищення тиску
панкреатичного соку в протоках підшлункової
залози. Залежно від причин виникнення гіпер
тензії розрізняють два різновиди цього варіанта:
1) гіперсекреторний панкреатит. Причиною
підвищення тиску в протоках залози є різке
збільшення секреції підшлункового соку
(жирна їжа й алкоголь, що стимулюють
утворення секретину та холецистокініну;
гіперкальціємія), коли панкреатичні про
токи не в змозі швидко пропустити весь
сік, що утворився (відносна недостатність
проток). У цьому випадку мають значення
анатомо-фізіологічні особливості вивід
них проток залози, тому панкреатит у та
кій формі може виникати не в усіх людей;
2)
обтураційний панкреатит. Гіпертензія
в протоках підшлункової залози виникає
при нормальному рівні секреції, але при
наявності перешкод (обтурації) відтоку
соку (жовчні камені, здавлювання проток
ззовні тощо).
Підвищений тиск соку є причиною ушко
дження клітин, що вистилають протоки залози.
Це призводить до виходу лізосомних ферментів
і активації трипсиногену з наступною реалізаці
єю автокаталітичного механізму (див. вище).
Крім того, гіпертензія у протоках залози
спричиняється до здавлювання судин, що про
ходять у паренхімі органа. Компресійна ішемія,
що виникає при цьому, може стати причиною
ушкодження ацинарних клітин і виходу з них
ще однієї порції лізосомних ферментів.
III. Рефлюксний. Він розвивається в резуль
таті закидання в протоки підшлункової залози
вмісту дванадцятипалої кишки або жовчі.
269
Дуодено-панкреатичний рефлюкс виникає при
збільшенні
внутрішньодуоденального
тис
ку (ікишкова непрохідність) або розслабленні
сфінктера Одді. Ініціатором передчасної акти
вації ферментів підшлункової залози в цьому
випадку є ентерокіназа - протеолітичний фер
мент, що утворюється клітинами слизової обо
лонки дванадцятипалої кишки і в нормі запо
чатковує активацію панкреатичних ферментів
у порожнині цієї кишки (див. вище).
Умовою виникнення жовчно-панкреатично
го рефлюксу є наявність спільної кінцевої ді
лянки панкреатичної і загальної жовчної про
ток (спільна ампула). При цьому жовчні кисло
ти, що мають детергентну дію, при потраплян
ні в протоки підшлункової залози ушкоджують
їхні клітини. З останніх виходять лізосомні
ферменти, які й запускають процес актива
ції панкреатичних ензимів. Крім того, жовчні
кислоти можуть підвищувати активність пан
креатичної ліпази, хоча остання секретується
ацинарними клітинами у формі вже активного
ферменту.
IV.
Ідіопатичний. У 10-20 % випадків не ви
являють жодного чинника, що міг би бути при
чиною гострого панкреатиту. У частини таких
хворих розвиток недуги зумовлюється гене
тичними факторами — мутаціями деяких генів,
а тому можна вести мову про спадковий панкре
атит. Сьогодні описано кілька варіантів такого
панкреатиту:
а) мутації гена PRSS1, що кодує одну з трьох
форм трипсиногену - катіонний трипсиноген. Хвороба передається за аутосомнодомінантним типом і характеризується на
падами гострого панкреатиту, починаючи
з дитячого віку. При одній з таких мута
цій у молекулах катіонного трипсиногену
змінюється структура згадуваного вище
критичного місця саморозпізнавання (criti
cal self-recognition cleavage site), з яким
пов’язана інактивація трипсину самим
трипсином. У цьому випадку трипсиноген
і трипсин стають стійкими до самоінактивації. Ненормально висока активність пан
креатичних протеаз і стає, на думку деяких
учених, причиною панкреатиту. Проте все
ж таки не зрозуміло, як у такому разі запус
кається основний механізм хвороби - пе
редчасна активація ферментів у протоках
залози;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
270
Основні механізми розвитку гострого панкреатиту
б) мутації гена інгібітору трипсину SPINK1
(serine protease inhibitor Kazal type 1).
У нормі продукт цього гена запобігає пе
редчасній активації ферментів у протоках
залози. У гомозигот за патологічним геном
інгібітор трипсину відсутній, а тому ви
падкова (спонтанна) активація навіть кіль
кох молекул трипсину в протоках може
бути цілком достатньою для ініціювання
лавиноподібного процесу автокаталітичної активації ферментів підшлункового
соку.
Механізми місцевих змін. Запалення, що роз
вивається у підшлунковій залозі, пов’язане з пе
редчасною активацією ферментів панкреатич
ного соку в протоках підшлункової залози і са
моперетравлюванням у зв’язку з цим залозистої
тканини (рис. 34.19).
Так, під дією трипсину й хімотрипсину від
бувається гідролітичне розщеплення тканинних
білків і білкових компонентів клітин. Еластаза
спричиняє гідроліз еластину — складової час
тини базальних мембран судин. Фосфоліпаза,
розщеплюючи фосфоліпіди клітинних мембран,
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
271
вої залози, підвищення проникності судин з роз
витком набряку, геморагій', виникнення болю.
Розвивається гостре запалення, що має ряд ха
рактерних ознак, про які вже йшла мова.
Загальні зміни - панкреатичний шок. Пан
креатичний шок є тяжким загальним проявом
гострого панкреатиту і, як і будь-який інший
шок, характеризується порушеннями загальної
гемодинаміки (падінням артеріального тиску)
та генералізованими розладами мікроциркуляції
(див. главу 20).
У патогенезі панкреатичного шоку мають
значення два механізми.
1. Больовий механізм. Причиною різкого гос
трого оперізуючого болю, що виникає при
панкреатиті, з одного боку, є набряк під
шлункової залози (тиск на сонячне спле
тення), а з другого — дія активних травних
ферментів (трипсин, фосфоліпаза та ін.) та
біологічно активних речовин - медіаторів
запалення (кініни, простагландини) на нер
вові закінчення залози. Інтенсивний біль
викликає спочатку збудження, а потім по
замежне гальмування в життєво важливих
центрах головного мозку. Це веде до при
гнічення зовнішнього дихання і порушення
системного кровообігу - розвивається бо
льовий шок (див. главу 20).
2. Гуморальний механізм. Він обумовлений
ферментемією — надходженням у кров ак
тивних панкреатичних ензимів.
обумовлює порушення їхньої бар’єрної функції,
а панкреатична ліпаза, гідролізуючи тригліцериди, викликає некроз жирової тканини. Із фосфоліпідів мембран вивільняється арахідонова
кислота, з якої утворюються простагландини.
З крові в тканину залози надходять калікреїноген і а2-глобуліни. Під дією трипсину і хімотрипсину відбувається активація калікреїну і в кінце
вому підсумку утворюються кініни.
Активні ферменти підшлункового соку, про
стагландини, кініни “ усі ці фактори виклика
ють вторинну альтерацію тканини підшлунко
Ферменти, що потрапили в кров, інактивуються
природними
інгібіторами
про
теаз (а.-інгібітор протеаз, антитромбін III,
а2-макроглобулін,
а2-антиплазмін,
інтер-аінгібітор трипсину та ін.). Однак, якщо потуж
ність цих інгібіторів недостатня і всі ферменти,
що надійшли, не можуть бути інактивовані, то
наступною подією є активація протеолітичних
систем крові (рис. 34.20). Її зумовлюють трип
син і хімотрипсин.
При цьому активуються:
1) калікреїн-кінінова система. Кініни (калідин, брадикінін), що є продуктами актива
ції цієї системи, викликають генералізоване розширення судин, яке веде до зменшен
ня загального периферичного опору. З дру
гого ж боку, вони підвищують проникність
судин, унаслідок чого рідка частина крові
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
272
Механізми розвитку місцевих змін у підшлунковій залозі при гострому панкреатиті, тг - тригліцериди
переходить у тканини (плазморагії) і, отже,
зменшується об’єм циркулюючої крові.
Зазначені зміни гемодинаміки обумовлю
ють падіння артеріального тиску — одного
з основних проявів шокового стану;
2) система зсідання крові;
3) фібринолітична система.
Активація двох останніх систем є причиною
відповідно гіперкоагуляцїї і плазмінового лізису
тромбів - процесів, що складають суть синдро
му дисемінованого внутрішньосудинного зсі
дання крові (ДВЗ-синдрому) і пов’язаних з ним
розладів мікроциркуляції (див. главу ЗО).
Крім активації протеолітичних систем крові,
панкреатичні ферменти чинять пряму ушко
джувальну дію на клітини і тканини організму.
Так, під впливом еластази підвищується про
никність кровоносних судин з наслідками, які
ми вже розглядали. Панкреатична ліпаза зу-
п
273
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
Гуморальні механізми розвитку панкреатичного шоку
мовлює некроз жирової тканин в різних органах
і частинах тіла. Фосфоліпаза порушує бар’єрні
властивості мембран, що веде до загибелі клі
тин. Одним із проявів цього процесу є гемоліз
еритроцитів. Внутрішньосудинний гемоліз стає
ще одним чинником розвитку ДВЗ-синдрому.
Панкреатичний шок клінічно виявляє себе
такими синдромами:
1) гострою артеріальною гіпотензією. Вона
обумовлюється (а) розширенням судин
(зменшується загальний периферичний
опір) і (б) падінням об’єму циркулюючої
крові (зменшується хвилинний об’єм серця);
2) ДВЗ-синдромом. Цей синдром, як уже за
значалося, пов’язаний з активацією систем
зсідання крові та фібринолізу, а також з ге
молізом еритроцитів і підвищенням про
никності кровоносних судин;
3) гіпоксичним синдромом. Кисневе голоду
вання в умовах панкреатичного шоку має
274
у своїй основі ряд механізмів (див. гла
ву 23):
а) циркулят орну гіпоксію — порушення за
гальної гемодинаміки (падіння артері
ального тиску) і мікроциркуляції (ДВЗсиндром);
б) дихальну гіпоксію - пригнічення діяльності
дихального центру (позамежне гальмуван
ня життєво важливих центрів при сильному
больовому синдромі), а також ушкодження
легеневої тканини, що настає під впливом
еластази, ліпази і фосфоліпази (гострий
респіраторний дистрес-синдром);
в) гемічну гіпоксію, що виникає в результаті
гемолізу еритроцитів, обумовленого пан
креатичними ферментами;
4) інтоксикаційним синдромом. Його роз
виток пов’язаний з надходженням у кров
продуктів автолітичного розпаду тканини
підшлункової залози.
Хронічний панкреатит
Тривалий запальний процес у підшлунковій за
лозі може бути пов’язаний з повторними напа
дами гострого панкреатиту, проте найчастіше
він має самостійне значення і притаманні тіль
ки йому особливості етіології та патогенезу.
Основними морфологічними ознаками хро
нічного панкреатиту є:
1) деструктивні зміни зовнішньосекреторної
паренхіми, що виявляють себе атрофією
ацинусів, некрозом і апоптозом Цанкреацитів, стенозом одних та розширенням ін
ших дрібних проток залози;
2) заміщення паренхіми фіброзною ткани
ною (фіброз) з явищем ектопічної кальцифікаціі’;
3) на пізніх стадіях у деструктивний процес
втягуються панкреатичні острівці, що ви
конують ендокринну функцію.
Морфологічні зміни при хронічному панкре
атиті, на відміну від гострого, є необоротними,
вони не зникають після припинення дії пато
генних факторів, мають тенденцію до прогре
сування.
Етіологія. Розвиток хронічного панкреатиту
може зумовлюватися цілим рядом чинників. Се
ред них найбільше значення мають такі:
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
а) хронічна алкогольна інтоксикація, особливо
в людей молодого й середнього віку. Вона є
причиною 70-80 % випадків хвороби;
б) довготривала обструкція панкреатичних
проток, що може бути зумовлена пухлина
ми, кістами, каменями, аномаліями розвит
ку підшлункової залози тощо;
в) токсична дія хімічних сполук. Так, вжи
вання продуктів з маніоку, що є джерелом
синильної кислоти та її солей (ціанідів),
вважається однією з причин т. зв. тропіч
ного панкреатиту, поширеного серед бід
них верств населення країн Азії та Африки.
Сприяють розвитку цієї форми панкреати
ту аліментарні фактори - недостатність
білків і мікроелементів у продуктах харчу
вання;
г) хронічна гіперкальціємія. У 10-15% хво
рих з гіперпаратиреозом розвиваються
ураження підшлункової залози з ознаками
хронічного її запалення;
Я) спадковість. Хронічний панкреатит є од
ним з основних проявів системної спад
кової хвороби. — муковісцидозу. Крім того,
у нього може переходити рецидивуючий
гострий панкреатит, зумовлений мутаціями
гена PRSS1 (див. вище);
е) у багатьох випадках встановити причи
ну хвороби не вдається. У такому разі
мову ведуть про ідіопатичний хронічний
панкреатит.
Патогенез. В основі розвитку хронічного пан
креатиту можуть лежати кілька механізмів.
1.
Первинно-альтеративний механізм. Суть
його полягає в необоротному ушкодженні клі
тин паренхіми, що розвивається в результа
ті прямої дії на залозу патогенних чинників
(рис. 34.21). Причиною некрозу ацинарних клі
тин можуть бути:
а) токсичний вплив хімічних сполук, на
приклад, ціанідів (тропічний панкреа
тит);
б) оксидаційний стрес і пов’язане з ним пероксидне окиснення ліпідів (див. главу 7).
Вони можуть бути зумовлені як поси
леним генеруванням вільних радикалів
(хронічна алкогольна інтоксикація), так
і недостатністю антиоксидантних сис
тем (білкове голодування, недостатність
мікроелементів);
275
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
Ішемічна гіпоксія Оксидаційний стрес
Первинно-альтеративний механізм розвитку хронічного панкреатиту
в) хронічна ішемія та інші розлади місце
вого кровообігу, що ведуть до кисневого
голодування;
г) автоімунне ушкодження клітин під
шлункової залози.
Первинна альтерація, що виникає під впливом
перелічених вище чинників, започатковує за
пальний процес з притаманними йому порушен
нями місцевого кровообігу, процесами еміграції
лейкоцитів і проліферації клітин сполучної тка
нини. Діяльність останніх зумовлює заміщення
змертвілих ацинусів фіброзною тканиною.
2.
Механізм гіперсекреції-гіпосекреції. В ос
нові цього механізму лежать різноспрямовані
зміни секреторної активності клітин підшлун
кової залози: гіперсекреція - в ацинарних кліти
нах і гіпосекреція - у клітинах дрібних панкреа
тичних проток (рис. 34.22).
У початкову фазу процесу тривала дія чинни
ків, що стимулюють діяльність ацинарних клі
тин прямо (холецистокінін, гіперкальціємія) чи
опосередковано (алкоголь), веде до гіпертрофії
панкреацитів і виділення ними в порожнину
ацинусів великої кількості густого білкового
секрету.
Якщо на цьому тлі зменшується секреція
води і бікарбонатів епітеліальними клітинами
дрібних панкреатичних проток (наприклад, не
достатність секретину чи парасимпатичної сти
муляції), то із в’язкого, дуже густого секрету
утворюються протеїнові пробки, які згодом за
знають мінералізації: у них відкладаються солі
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
276
Патогенез хронічного панкреатиту: механізм гіперсекреції-гіпосекрецн
кальцію (в основному карбонати) і вони пере
творюються на панкреатичні камені.
Утворенню протеїнових пробок і каменів
сприяють гіперкальціємія і недостатність літостатинів - пептидів панкреатичного соку, які
в нормі запобігають цим процесам. Під впливом
алкогольної інтоксикації, а також при активації
трипсину і деяких лізосомних ферментів може
утворюватися аномальний літостатин, що втра
чає свою розчинність і здатен полімеризуватися
у фібрили, які стають матриксом для протеїно
вих пробок і каменів.
Обтурація дрібних проток, зумовлена цими
утвореннями, спричиняється до наступної - гіпосекреторної фази. Вище місця обструкції
збільшується тиск секрету, що веде, з одного
боку, до розширення проток і здавлювання ними
кровоносних судин, що проходять поруч, - як
наслідок, порушується місцевий кровообіг, роз
виваються компресійна ішемія й венозна гі
перемія, ацинарні клітини зазнають кисневого
голодування. З другого боку, підвищення тиску
в порожнинах ацинусів перешкоджає вивіль
ненню сюди продуктів секреції, що стає при
чиною пригнічення цього процесу. Гіпертрофовані панкреацити поступово зазнають атрофії
і, врешті-решт, гинуть за механізмом апоптозу.
Ацинуси, що припинили свою діяльність і за
знали деструктивних змін заміщаються на фіб
розну тканину.
3.
Обструктивний механізм. Розвиток хро
нічного панкреатиту може зумовлюватися пер
277
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
винною обструкцією як дрібних, так і більших
проток.
Порушення прохідності перших найчастіше
є наслідком нападів гострого панкреатиту. Фіб
розна тканина і псевдокісти, що утворюються
на місці осередків некрозу інтерстиціальної
жирової тканини, здавлюють ззовні дрібні пан
креатичні протоки і порушують проходження
секрету через них. Панкреатичні протоки біль
шого калібру можуть зазнавати як компресії
(наприклад, пухлинами чи рубцями), так і обтурацїї (наприклад, жовчними каменями).
Незалежно від того, на якому рівні відбува
ються обструктивні зміни, у протоках залози
вище місця перешкоди і в порожнинах ацинусів підвищується тиск секрету з усіма тими на
слідками, що вже були обговорені. Досить час
то патогенез хронічного панкреатиту поєднує
в собі кілька, а то і всі наведені вище механізми.
Прикладом може бути алкогольна інтоксикація
(рис. 34.23).
Хронічний вплив алкоголю на підшлункову
залозу “вмикає”:
1)
первинно-альтеративний механізм. Ал
коголь, з одного боку, зумовлює поси
лене генерування вільних радикалів (ак
тивних форм кисню) в ацинарних клі
тинах залози, а з другого - є причиною
антиоксидантної недостатності в ор
ганізмі. Остання пов’язана в основному
з аліментарним чинником — недостатнім
споживанням
білків,
гіповітамінозами
Е, К, С; дефіцитом мікроелементів (се
лену, цинку), що є характерним для хро
нічних алкоголіків. Наведені причини
зумовлюють оксидаційний стрес і акти
вацію пероксидного окиснення ліпідів —
універсальних механізмів ушкодження
клітин;
2)
механізм гіперсекреції-гіпосекреції. Під
впливом алкоголю на початкових етапах
зростає підшлункова секреція, як унаслі
док прямої дії на панкреацити, так і опо
середковано, через утворення і секрецію
холецистокініну. При цьому зменшуєть
ся надходження в секрет літостатинів,
з’являються аномальні їх форми, які не
те що не запобігають, а навіть сприяють
утворенню протеїнових пробок і панкреа
тичних каменів у протоках залози;
3) обструктивний механізм. Як результат
зумовлених алкоголем повторних нападів
гострого панкреатиту, в інтерстицїї фор
муються осередки фіброзу та псевдокісти,
Механізми розвитку алкогольного хронічного панкреатиту
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
278
які здавлюють дрібні протоки залози. При
цьому порушується виведення секрету із
ацинусів і збільшується його тиск на стін
ки проток і на панкреацити.
Основні клінічні прояви
1. Болі в животі. Постійні чи періодичні
болі в животі є характерною ознакою хронічно
го панкреатиту. В основі їх виникнення лежать
різні механізми залежно від патогенетичного
варіанта хвороби. До больового синдрому спри
чиняються:
а) механічне розтягнення стінок панкреа
тичних проток при підвищенні тиску се
крету в останніх;
б) медіатори запалення (простагландини,
кініни та інші), що утворюються в осе
редках цього процесу та діють на нерво
ві закінчення;
в) ішемія, яка спричиняється до місцевого
ацидозу. Підвищення концентрації іонів
водню при накопиченні недоокиснених
продуктів зумовлює подразнення рецеп
торів і викликає біль;
г) активні травні ферменти (трипсин, хімотрипсин) і ферменти лізосом, що ста
ють причиною деструкції периневральних структур і нервових волокон;
д) здавлювання нервових волокон фіброз
ною тканиною і кальцифікатами, що
утворилися і ростуть поблизу.
2. Недостатність зовнішньосекреторної
функції підшлункової залози. Вона виявляє
себе порушеннями порожнинного травлен
ня - синдромом мальдигестії.
Цей синдром виявляє себе:
а) порушенням перетравлювання жирів
(відсутність панкреатичної ліпази, фосфоліпаз). Не засвоюється 60-80 % жиру,
він виводиться з калом - з’являється
симптом стеатореї (жир у калі);
б) розладами всмоктування жиророзчин
них вітамінів - розвиваються ознаки гі
повітамінозів А, Е, К;
в) недостатнім перетравлюванням біл
ків (відсутність травних протеаз).
Не засвоюється до 30—40 % харчового
білка. У калі з’являється велика кіль
кість м ’язових волокон',
г) порушенням перетравлювання вуглево
дів (відсутність амілази).
Недостатність ендокринної функції під
шлункової залози — вторинний цукровий
діабет.
Перехід хронічного запального процесу на
панкреатичні острівці веде до деструкції остан
ніх, загибелі p-клітин і розвитку діабету, який
характеризується абсолютною інсуліновою не
достатністю і за основними своїми ознаками
є подібним до цукрового діабету І типу (див.
главу 24).
3.
ПОРУШЕННЯ
ТРАВЛЕННЯ В КИШКАХ
Тонким кишкам повного мірою притаманні всі
три функції травної системи: секреторна, рухова
і всмоктувальна.
У порожнину дванадцятипалої кишки над
ходять панкреатичний сік і жовч. Крім того,
завдяки секреторній діяльності бруннерових
залоз слизової оболонки цієї кишки, а також
ліберкюнових залоз крипт тонкого кишечника
формується кишковий сік.
Значення цих секретів полягає в тому, що
вони:
а) нейтралізують кислоту шлункового соку;
б) забезпечують порожнинне травлення - де
полімеризацію компонентів їжі до олігомерів і мономерів, гідроліз ефірних сполук;
в) беруть участь у здійсненні пристінкового
травлення.
Рухова функція кишок направлена на (а) пе
ремішування хімусу з травними соками, (б) пе
реміщення вмісту кишок у дистальному на
прямку, (в) підвищення внутрішньокишкового
тиску, що сприяє фільтрації води й електролітів
із порожнини кишок у кров і лімфу.
У тонких кишках відбувається всмоктування
мономерів, тобто проходження їх через кишкову
стінку у кров і лімфу. Цей процес тісно пов’язаний
з пристінковим (мембранним) травленням.
У товстих кишках відбуваються такі про
цеси:
1) завершується перетравлювання тих речо
вин, що ще залишилися після проходжен
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
279
ня хімусу через тонку кишку. Цей процес
дуже слабкий, він здійснюється (а) фер
ментами, що потрапили в товсту кишку
із тонкої разом із вмістом останньої, і (б)
ферментами соку товстої кишки;
2) інтенсивно всмоктується вода;
3) поступово формуються калові маси.
г) тонічні скорочення, що пов’язані зі збіль
шенням тонусу гладких м’язових клітин
стінки кишок;
д) скорочення ворсинок, завдяки яким їхня по
верхня має можливість контактувати усе
з новими порціями вмісту кишок;
е) складний рефлекторний акт дефекації.
Важливе значення для організму має нор
мальна мікрофлора товстих кишок, яка на 90 %
представлена біфідобактеріями, решта припа
дає на лактобактерії, кишкову паличку, ентеро
коки та ін.
Завдяки нормальній мікрофлорі товстих ки
шок:
а) відбувається кінцевий розпад залишків неперетравленої їжі і компонентів травних
соків. Ці процеси проходять під впливом
бактеріальних ферментів. Останні забезпе
чують також розпад до 40 % целюлози хар
чових продуктів;
б) пригнічується розвиток і розмноження па
тогенних мікроорганізмів',
в) синтезуються деякі корисні для макроорга
нізму речовини, зокрема вітамін К, віта
міни групи В\
г) утворюються токсичні продукти перетво
рення {гниття) білків: аміак, індол, скатол,
путресцин, кадаверин та ін. Усі ці речовини
надходять у печінку, де й знешкоджуються.
Регуляція рухової активності кишок здійсню
ється за допомогою:
1) нервових механізмів. Такими є (а) пери
феричні рефлекси, що замикаються на
рівні внутрішньоорганних гангліїв, і (б)
центральні рефлекси, еферентною лан
кою яких можуть бути парасимпатичні
чи симпатичні нерви. Збудження пер
ших - викликає скорочення гладкої мус
кулатури кишок, а других - навпаки, її
розслаблення;
2) гуморальних механізмів. Посилення мото
рики кишок чи її послаблення можуть зу
мовлюватися великою кількістю гормонів
(у тому числі гастроінтестинальних) та
біологічно активних речовин (медіаторів
запалення і алергії). Такими зокрема є ва
зопресин, окситоцин, гастрин, мотилін, вілікінін, гістамін, серотонін, брадикінін та
багато інших;
3) міогенних механізмів авторегуляції, що
пов’язані з функцією автоматизму деяких
гладких м’язових клітин (пейсмекерів).
Кишкові дискінезії
Моторна функція кишок, що здійснюється цир
кулярною і поздовжньою їхньою мускулатурою,
виявляє себе різними видами рухової активнос
ті. До таких відносять:
а) ритмічну сегментацію, що має важливе
значення для перемішування вмісту кишок
і збільшення тиску в них;
б) великі і малі маятникоподібні рухи, завдя
ки яким хімус переміщується то вперед, то
назад, що сприяє перемішуванню вмісту
кишок і травних соків;
в) перистальтичні рухи, що поширюються
хвилеподібно в дистальному напрямку.
Ці рухи забезпечують переміщення хі
мусу вздовж кишок. В умовах патології
можуть
виникати
антиперистальтичні
рухи, що поширюються в протилежному
напрямку;
Порушення рухової функції кишок назива
ють кишковими дискінезіями. Виділяють два їх
варіанти: гіперкінетичний і гіпокінетичний.
1. Гіперкінетичний варіант. Для нього харак
терним є посилення перистальтики кишок,
ритмічної сегментації і маятникоподібних
рухів, що найчастіше виявляє себе проно
сами {діареєю).
Причинами кишкових дискінезій цього типу
можуть бути:
а) підвищення збудливості рецепторів кишок
до адекватних подразників під час розвит
ку запалення слизової оболонки кишок (ен
терити, коліти);
б) дія на рецептори кишок незвичайних, па
тологічних подразників - неперетравленої
їжі (наприклад, при ахілії), продуктів гнит
тя й бродіння, токсичних речовин тощо;
280
в) підвищення збудливості центрів блукаючо
го нерва;
г) збільшення утворення деяких гастроінтестинальних гормонів, що посилюють пери
стальтику кишок, наприклад, мотиліну.
Серед наслідків кишкових дискінезій гіперкінетичного типу:
а) розлади травлення (перетравлювання, всмок
тування);
б) зневоднення',
в) видільний негазовий ацидоз (втрата бікар
бонатів).
2. Гіпокінетичний варіант. Він характери
зується послабленням рухової активності
кишок, зокрема перистальтики, унаслідок
чого виникають закрепи.
Залежно від механізмів розвитку виділяють
спастичні Й атонічні закрепи. Спастичні за
крепи виникають у результаті стійкого трива
лого тонічного скорочення гладкої мускулатури
кишок (спазму) і можуть бути обумовлені вісцеро-вісцеральними рефлексами або дією ток
сичних факторів (наприклад, отруєння свин
цем).
Причиною
розвитку
атонічних закрепів,
пов’язаних зі зменшенням скорочувальної функ
ції гладких м’язів кишок, можуть бути:
а) бідне харчування, низький вміст кліткови
ни в харчових продуктах;
б) надмірне перетравлювання їжі в шлунку
(наприклад, при шлунковій гіперсекреції);
в) вікові зміни рецепторного апарату кишок
в осіб старечого віку, а також структурні
зміни кишкової стінки при ожирінні',
г) зменшення тонусу блукаючого нерва',
д) порушення внутрішньокишкової іннерва
ції, наприклад, при хворобі Гіршпрунга, для
якої характерна відсутність гангліонарних
клітин ауербахового сплетення в сигмопо
дібній і прямій кишках.
Кишкові дискінезії гіпокінетичного типу
призводять до:
а) розвитку кишкової автоінтоксикації',
б) виникнення метеоризму,
в) утворення калових каменів',
г) у крайніх випадках може розвиватися киш
кова непрохідність.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Кишкова непрохідність
Непрохідність кишок {ileus) — це захворювання,
що характеризується порушенням проходжен
ня хімусу по кишках. Причиною цього можуть
бути або обтурація, або здавлювання кишок, або
порушення їхніх функцій.
Класифікація. Виділяють механічну та дина
мічну кишкову непрохідність.
I. Механічна непрохідність може бути:
а) обтураційною. Розвивається внаслідок за
купорки просвіту кишки пухлиною, кало
вими каменями, клубком гельмінтів;
б) странгуляційною. Є результатом здавлюван
ня кишки ззовні (заворот, защемлення в гри
жових воротах, утворення вузлів). Особли
вістю цього виду непрохідності є здавлю
вання судин брижі, що порушує живлення
стінки кишки аж до некрозу.
II. Динамічна непрохідність буває:
а) спастичною. Обумовлена спастичним ско
роченням гладких м’язів кишок;
б) паралітичною. Розвивається внаслідок гли
бокого пригнічення рухової функції кишок.
Основні клінічні прояви
1. Больовий синдром. Він розвивається вна
слідок спастичного скорочення гладких
м’язів, некрозу стінки кишки, розтягнення
її рідиною. У результаті тривалого інтен
сивного болю можуть з’являтися ознаки бо
льового шоку (падіння артеріального тиску,
розлади зовнішнього дихання), обумовлені
позамежним гальмуванням судинорухово
го і дихального центрів після їхнього над
мірного збудження.
2. Зневоднення. В умовах кишкової непро
хідності відбувається накопичення великої
кількості рідини в кишках вище місця пе
решкоди. Причиною цього є:
а) збільшення секреції травних залоз у відпо
відь на розтягування стінки кишки рідиною
і газами;
б) перехід рідини з кровоносних судин
у кишки (транссудація) внаслідок застою
крові та збільшення проникності стінок
судин;
в) порушення процесів всмоктування води.
281
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
Накопичення рідини в кишках спричиняється
до збільшення внутрішньокишкового тиску, що
зумовлює подразнення великої кількості рецеп
торів і блювання. Що вище перешкода в киш
ках, то інтенсивніша блювота та більш вираже
не зневоднення.
3. Порушення обороту травних ферментів.
Збільшення внутрішньокишкового тиску
призводить до закидання вмісту кишок
(у тому числі й травних соків, ентерокінази) у протоки підшлункової залози.
Відбувається передчасна активація фер
ментів підшлункового соку, унаслідок
чого розвивається гострий панкреатит
з явищами панкреатичного шоку (див.
підрозділ 34.3).
4. Порушення кислотно-основної рівноваги.
Якщо при нестримному блюванні втрача
ються значною мірою хлориди — розвива
ється негазовий алкалоз, якщо бікарбона
ти - негазовий ацидоз (див. главу 29).
5. Кишкова автоінтоксикація. Вона особли
во виражена при низькій кишковій непро
хідності та виявляє себе ознаками недо
статності печінки (див. главу 35).
6. Гострий перитоніт. Запалення очереви
ни зумовлюється мікробами, які проника
ють усередину через некротизовану стінку
кишки. Перитоніт є чинником, що викли
кає і посилює біль та больовий шок, а та
кож, зумовлюючи рефлекторне блювання,
сприяє розвитку зневоднення, порушень
кислотно-основного стану і розладів сис
темної гемодинаміки.
7. Розлади загального кровообігу і мікроциркуляції. Порушення системної гемодинаміки
(падіння артеріального тиску, зменшення
хвилинного об’єму серця, зменшення за
гального периферичного опору) є проявом
больового й панкреатичного шоку, а також
зневоднення. Згущення крові (гемоконцентрація) стає причиною розладів мікроциркуляції.
8. Гіпоксія. Її розвиток обумовлений порушен
нями кровообігу і зовнішнього дихання.
9. Пригнічення функції життєво важливих
органів (серця, головного мозку, нирок).
Воно настає внаслідок кисневого голоду
вання та інтоксикації.
Синдром мальабсорбції
Мальабсорбцією називають комплекс змін, що
виникають у результаті порушень усмоктування
речовин у кишках.
Основним місцем, де відбувається всмокту
вання, є тонкий кишечник. Тут цьому процесу
сприяють:
а) велика площа поверхні всмоктування (до
200 м2), що створюється завдяки круговим
складкам слизової оболонки кишок, вор
синкам і мікроворсинкам;
б) добре розвинена система кровоносних
і лімфатичних судин у кишковій стінці;
в) здатність ворсинок до скорочення, завдяки
чому лімфа видавлюється в судини.
Всмоктування різних сполук відбувається
різними механізмами і шляхами. Транспорт
речовин здійснюється як активно (з викорис
танням енергії АТФ), так і пасивно (дифузія,
фільтрація, осмос). Одні сполуки мають про
ходити через ентероцити (трансцелюлярний
транспорт), інші (вода, електроліти) - можуть
всмоктуватися і через щілини між ними.
Порушення всмоктування в кишках можуть
бути обумовлені розладами, що виникають на
трьох рівнях. Тому виділяють:
1) передентероцитарні порушення. Розви
ваються внаслідок розладів процесів трав
лення, що передують всмоктуванню;
2) ентероцитарні. Виникають у результаті
порушення діяльності епітеліальних клі
тин слизової кишок;
3) постентероцитарні. Вони є наслідком по
рушення процесів, які забезпечують над
ходження речовин, що всмокталися, у вну
трішнє середовище організму (кров, лімфу).
Передентероцитарні порушення. Всмоктуван
ня речовин стає менш інтенсивним або зовсім
не відбувається, якщо порушується перетрав
лювання компонентів їжі та утворення сполук,
придатних до всмоктування. Причинами цього
можуть бути:
а) розлади рухової функції травного каналу,
що вже були предметом нашого обгово
рення;
б) порушення порожнинного травлення. За
походженням вони можуть бути гастрогенними, панкреатогенними, гепатогенними,
282
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ентерогенними, дисрегуляторними, ятро
генними (пов’язаними з тривалим застосу
ванням антибіотиків та інших лікарських
препаратів). Клінічно розлади порожнин
ного травлення виявляють себе синдромом
мальдигестії (див. підрозділ 34.3);
в) порушення пристінкового травлення. їх
ній розвиток найчастіше зумовлений розла
дами утворення і вмонтовування ферментів
у плазматичну мембрану мікроворсинок
ентероцитів.
Пристінкове (мембранне, контактне) трав
лення - це гідроліз речовин, що здійснюється
ферментами щіткової облямівки ентероцитів.
Остання являє собою сукупність мікроворси
нок, що є утвореннями плазматичної мембрани
клітин (рис. 34.24).
Остаточний гідроліз речовин здійснюють дві
групи ферментів щіткової облямівки: (1) ад
сорбовані глікокаліксом ензими підшлункового
і кишкового соків та (2) вмонтовані в мембрану
мікроворсинок власні ферменти ентероцитів.
Найчастіше порушення пристінкового трав
лення пов’язані зі спадково зумовленими роз
ладами синтезу саме мембранних ферментів
мікроворсинок. Такі порушення називають інтестинальними ферментопатіями.
Серед інтестинальних ферментопатій досить
поширеними є непереносимість дисахаридів
(лактози, сахарози, трегалози) і недостатність
пептидаз (глютенова хвороба).
Непереносимість
дисахаридів
обумовлена
недостатністю дисахаридаз. У деяких регіонах
Африки, Азії й Південної Америки 90 % на
селення не переносить молока і його не спо
живає. У представників зазначених етнічних
груп немає ферменту лактази, що розщеплює
лактозу молока. Через це нерозщеплена лакто
за, накопичуючись на поверхні слизової, по
дразнює рецептори кишок і викликає профузні
проноси.
Глютенову хворобу зумовлює недостатність
пептидаз. Ця недуга, що є поширеною в Євро
пі, зокрема в Голландії, характеризується непереносимістю продуктів, виготовлених зі злаків.
Глютен — білкова частина клейковини пшениці,
рису, ячменю, вівса - складається з нешкідливо
го глютеїну і гліадину, який токсично діє на сли
зову тонкої кишки (викликає ентерит, проноси).
При недостатності пептидаз гліадин не розщеп-
24. Мікроворсинки ентероцитів
люється і чинить свою токсичну дію, зумовлю
ючи клінічну картину хвороби.
Ентероцитарні порушення. Причинами
мальабсорбції може бути цілий ряд структурних,
функціональних і метаболічних змін епітелію
слизової оболонки тонких кишок. Серед них:
а) зменшення площі всмоктування (стан після
резекції кишки, атрофія ворсинок і мікро
ворсинок);
б) спадково обумовлені та набуті порушен
ня утворення білків, що є переносниками
моносахаридів (непереносимість глюкози,
галактози, фруктози), амінокислот (триптофанмальабсорбція), іонів кальцію (гіпо
вітаміноз D);
в) порушення функціонування іонних насо
сів ентероцитів (транспорт моносахаридів
і амінокислот пов’язаний з роботою Na-Kнасосів);
г) дефіцит енергії (всмоктування більшості
речовин - процес енергозалежний);
д) порушення формування в ентероцитах
комплексів, що транспортуються (хіломікронів, ліпопротеїдів).
Постентероцитарні порушення. До мальабсорбції можуть спричинятися чинники, що по
рушують перехід речовин у внутрішнє середо
вище організму вже після того, як вони потрапи
ли в ентероцити. Такими є:
ГЛАВА 34. ПАТОФІЗІОЛОГІЯ ТРАВЛЕННЯ
а) порушення кровообігу в стінках кишок.
Вони можуть бути зумовлені як розладами
загальної гемодинаміки в системі ворітної
вени, так і місцевими порушеннями (іше
мія, венозна гіперемія, тромбоз, емболія,
реакції судин при запаленні);
б) порушення лімфовідтоку. Крім загальних
розладів лімфообігу, вони можуть виника
283
ти при порушеннях скорочення ворсинок
кишкової стінки. Таке скорочення в нормі
здійснюється завдяки місцевим рефлексам
за участю підслизового нервового сплетен
ня (сплетення Месснера) і речовини віллікініну, що її утворює слизова оболонка
тонкої кишки.
ГЛАВА 35
Печінка виконує багато важливих фізіологічних
функцій, які для зручності можна розділити на
чотири групи.
1. Метаболічні функції передбачають участь
печінки у всіх видах обміну речовин в ор
ганізмі: (а) вуглеводному, (б) жировому,
(в) білковому, (г) обміні вітамінів, (д) гор
монів і біологічно активних речовин.
2. Захисні функції охоплюють (а) фагоци
тарну і (б) антитоксичну функції печінки.
Перша - здійснюється особливими ендотеліальними клітинами печінкових синусів —
клітинами Купфера, що входять до системи
мононуклеарних фагоцитів. Другу - вико
нують клітини паренхіми — гепатоцити.
3. Екскреторна функція полягає в утворенні
і виділенні жовчі. Завдяки жовчі печінка
бере участь у процесах травлення (травна
функція) і виділенні продуктів обміну з ор
ганізму (видільна функція).
4. Гемодинамічні функції забезпечують участь
печінки в підтриманні системного крово
обігу.
1 НЕДОСТАТНІСТЬ ПЕЧІНКИ
Недостатністю печінки називають нездатність
цього органа забезпечувати сталість внутрішнього
середовища організму відповідно до його вимог.
Класифікація.
І. Недостатність печінки може бути:
а) відносною;
б) абсолютною.
Відносна виникає при первинному підвищен
ні навантаження на печінку, коли вимоги орга
нізму щодо підтримання гомеостазу перевищу
ють нормальні функціональні можливості цього
органа.
Абсолютна недостатність розвивається при
первинному ураженні печінки, внаслідок чого
зменшуються її функціональні можливості
і вона не здатна забезпечувати сталість внутріш
нього середовища не тільки при навантаженні,
а й у звичайних умовах. Відносна недостатність
печінки згодом може переходити в абсолютну.
Послідовність подій у цьому випадку така: під
вищення навантаження на печінку —> відносна
недостатність печінки —* порушення сталості
внутрішнього середовища —*■ вторинне ураження
печінки —> абсолютна печінкова недостатність.
II. Залежно від причин ушкодження гепатоцитів абсолютна недостатність печінки може
бути:
а) печінково-клітинною (гепатоцелюлярною);
б) холестатичною;
в) печінково-судинною (гепатоваскулярною).
III. Залежно від кількості функцій, що пору
шуються при ураженнях печінки, недостатність
цього органа може бути:
а) тотальною (зазнають розладів усі види
функцій печінки);
б) парціальною (страждає одна або кілька
функцій).
IV. За клінічним перебігом недостатність пе
чінки може бути:
• гострою;
- хронічною.
Причини. Печінка є органом, чутливим до дії
багатьох зовнішніх і внутрішніх патогенних
факторів. До уражень печінки можуть спричи
нятися:
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
а) фізичні фактори - іонізуюче випроміню
вання, механічна травма;
б) хімічні агенти, що мають токсичну (гепатотропну) дію. Вони можуть бути як екзо
генного походження (алкоголь, промис
лові отрути - чотирихлористий вуглець,
фосфорорганічні
сполуки,
хлороформ,
миш’як; лікарські препарати - ПАСКнатрій, сульфаніламіди, цитостатики, де
які антибіотики; рослинні отрути - афлатоксин, мускарин, алкалоїди геліотропа),
так і ендогенного (продукти розпаду тка
нин при опіках, некрозах; токсикоз вагіт
них);
в) інфекційні агенти — віруси (вірусного ге
патиту, інфекційного мононуклеозу), збуд
ники туберкульозу, сифілісу, найпростіші
(лямблії, амеби), гриби (актиноміцети),
гельмінти (ехінокок, аскариди);
г) аліментарні фактори — білкове, вітамінне
голодування, дуже жирна їжа;
д) алергічні реакції на введення вакцин, сиро
ваток, харчових продуктів і лікарських пре
паратів;
е) порушення кровообігу в печінці місцевого
(ішемія, венозна гіперемія, тромбоз, ембо
285
лія) і загального (при недостатності крово
обігу) характеру;
є) ендокринні та обмінні порушення в організ
мі (цукровий діабет, гіпертиреоз, ожиріння);
ж) пухлини (гепатоцелюлярний рак) та їхні ме
тастази в печінку (рак шлунка, легень, мо
лочної залози, лейкозні проліферати);
з) генетично зумовлені дефекти обміну ре
човин (спадкові ферментопатії), уроджені
вади розвитку печінки.
Патогенез. Розрізняють три основні механізми
ушкодження гепатоцитів і відповідно три пато
генетичні варіанти абсолютної печінкової недо
статності (рис. 35.1).
1. Пряме ушкодження печінкових клітин па
тогенними агентами - печінково-клітинна
(гепатоцелюлярна) недостатність.
Його можуть викликати (а) фізичні фактори
(іонізуюча радіація), (б) гепатотропні отрути
(чотирихлористий вуглець, токсини блідої по
ганки, алкалоїди геліотропа), (в) віруси гепа
титу А, В, С. Конкретні механізми ушкодження
гепатоцитів при дії зазначених вище чинників
ми розглянемо нижче.
Патогенетичні варіанти абсолютної печінкової недостатності
286
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
В експерименті на тваринах гепатоцелюлярну
недостатність моделюють повним або частковим
видаленням печінки, а також за допомогою отрут,
що їх вводять в організм (чотирихлористого вуг
лецю, хлороформу, тринітротолуолу та ін.).
2. Ушкодження гепатоцитів, зумовлене три
валим холестазом, — холестатична недо
статність печінки.
Холестазом називають порушення утворен
ня, екскреції та виведення жовчі у дванадцяти
палу кишку. Ушкодження гепатоцитів в умовах
тривалого холестазу обумовлене принаймні
двома причинами: (а) збільшенням тиску жовчі
у жовчних капілярах, унаслідок чого печінкові
клітини здавлюються, відбувається механічний
розрив їхніх мембран; (б) роз’єднанням окиснення і фосфорилювання в мітохондріях під
впливом білірубіну та жовчних кислот, що не
можуть бути виведені з клітин. Як результат та
кого роз’єднання виникає дефіцит АТФ, а потім
і дегенеративні зміни в гепатоцитах.
В експерименті холестатичну недостатність
відтворюють перев’язуванням жовчовивідних
проток.
3. Первинні порушення кровообігу в печін
ці - печінково-судинна (гепатоваскулярна)
недостатність. Основним механізмом уш
кодження гепатоцитів у цих випадках є гі
поксія.
Найчастіше причинами гепатоваскулярної
недостатності є портальна гіпертензія та іше
мія печінки. В експерименті розлади кровообігу
в печінці моделюють за допомогою (а) накладан
ня портокавальних анастомозів (фістула Екка,
фістула Екка - Павлова); (б) перев’язування во
рітної вени; (в) перев’язування печінкових вен;
(г) накладання лігатури на печінкову артерію.
Експериментальне
моделювання.
Класични
ми методами дослідження печінкових функцій
і відтворення печінкової недостатності стали
хірургічні втручання, метою яких є моделюван
ня порушень кровообігу в печінці та повне ви
далення цього органа. До таких методів можна
віднести:
1) накладання фістули Екка. Метод полягає
в тому, що між нижньою порожнистою
і ворітною венами в собак створюють
анастомоз. Ворітну вену вище анастомозу
перев’язують, і вся кров, що відтікає від
органів черевної порожнини, надходить
безпосередньо в нижню порожнисту вену,
обминаючи печінку (рис. 35.2Б). Цей екс
перимент дозволяє вивчати знешкоджу
вальну, а також сечовиноутворювальну
функції печінки. Після операції у тварин
уже через 3-4 доби при згодовуванні м’яса
(м’ясне отруєння) або через 10-12 діб при
використанні молочно-рослинної дієти
виникають розлади рухових функцій цен
тральної нервової системи, аж до появи
періодичних клонічних і тонічних судом.
У крові зростає рівень аміаку, який у нор
мі знешкоджується в печінці. Накладанням
35.2. Фістули Екка та Екка - Павлова (за К. М. Виковим). А - розташування судин до операції, Б - фістула Екка,
В - фістула Екка - Павлова
287
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
фістули Екка по суті відтворюють екзоген
ну (портокавальну) печінкову кому, про
яку мова піде далі;
2) зворотна фістула Екка - Павлова. Після
створення анастомозу між ворітною і ниж
ньою порожнистою венами перев’язують
вище анастомозу не ворітну, а нижню по
рожнисту вену (рис. 35.2, В). При цьому
в печінку спрямовується кров не тільки від
органів травлення по ворітній вені, а й від
задньої половини тулуба. Тварини з такою
фістулою можуть жити довго;
3) повне видалення печінки. Його проводять
у два етапи. Спочатку накладають зворот
ну фістулу Екка — Павлова, завдяки якій
розвивається колатеральний кровообіг:
частина венозної крові від задньої частини
тіла через портокавальні анастомози від
водиться у верхню порожнисту вену, об
минаючи печінку. Через 3-4 тижні після
першої операції проводять другу: ворітну
вену перев’язують і печінку видаляють.
У найближчий час після видалення печінки
у собак з’являється м’язова слабкість, адинамія,
різко падає рівень глюкози в крові і розвиваєть
ся гіпоглікемічна кома, від якої тварина й гине.
Введенням глюкози можна дещо подовжити
її життя. Одночасно в крові зростає кількість
аміачних сполук і зменшується вміст сечовини.
Собаки після такої операції живуть не більше
12-15 годин і вмирають від печінкової коми.
Остання є моделлю ендогенної (печінково-клі
тинної) печінкової коми в людини (див. далі).
Для експериментального відтворення різних
патогенетичних варіантів печінкової недостат
ності використовують прийоми, які ми вже згаду
вали у відповідній частині цієї глави (див. вище).
Основні форми уражень печінки. Структурною
основою недостатності печінки є розвиток у ній
морфологічних змін, які описуються поняттями
стеатоз, гепатит і цироз. Ці форми пов’язані
між собою так, як це показано на рис. 35.3.
Залежно від сили і тривалості дії патогенного
фактора на печінку у ній може розвиватися сте
атоз (тривалий вплив помірних чинників) або
гепатит (дія сильних чинників протягом корот
шого часу). Якщо дія факторів ушкодження при
пиняється, то і стеатоз, і гепатит можуть мати
зворотний розвиток - у такому разі структура
печінки відновлюється і настає повне одужання.
Якщо ж патогенний фактор продовжує свою
дію, то розвивається незворотний процес - ци
роз. У нього може переходити як стеатоз печін
ки, так і гепатит у разі повторних його атак.
З. Форми уражень печінки та зв'язок між НИМИ. Штриховими стрілками показано зміни в бік відновлення
структури печінки після припинення дГі патогенних факторів
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
288
Стеатоз печінки. Стеатозам (син.: жирова ін
фільтрація, жирова дистрофія печінки, жировий
гепатоз) називають збільшення в кілька разів про
ти норми вмісту тригліцеридів у печінкових клі
тинах, унаслідок чого розвивається дифузне або
осередкове ожиріння печінки. Найчастіше причи
нами стеатозу є алкогольна інтоксикація, цукро
вий діабет, ожиріння, хронічні вірусні інфекції
та інтоксикації, гепатотропні отрути, аліментарні
фактори (білкове голодування, дефіцит у їжі так
званих ліпотропних речовин - холіну, метіоніну).
У патогенезі жирової інфільтрації печінки
можна виділити кілька механізмів (рис. 35.4):
1) збільшення надходження вільних жиро
вих кислот (ВЖК) у печінку. До цього
спричиняється гіперліпацидемія - підви
щення рівня ВЖК у крові, що характерно
для цукрового діабету І типу (див. главу
24). Крім того, вміст жирових кислот у гепатоцитах може зростати внаслідок поси
леного синтезу їх з ацетил-КоА;
2) порушенням катаболізму жирових кислот
у гепатоцитах, зокрема процесів їхнього
окиснення. Причиною цього можуть бути
гіпоксія, дефіцит коферментів, переважне
використання для енергетичних потреб ін
ших, ніж жирові кислоти, сполук, зокрема
етанолу.
Реалізація перших двох механізмів веде до
надмірного утворення триацилгліцеролів (ТАГ)
із жирових кислот і, якщо виведення ТАГ із пе
чінки у складі ліпопротеїнів відстає від інтен
сивності їхнього утворення, то надлишок ТАГ
відкладається в гепатоцитах у вигляді жирових
крапель;
3) порушення виведення ТАГ з печінки
в кров у складі ЛПДНГ (ліпопротеїнів
дуже низької густини). Причинами цього
можуть бути:
а) розлади синтезу основних компонентів
ліпопротеїнових часток (білкової
частини - апопротеїну, фосфоліпідів);
б) порушення формування міцел
ліпопротеїнів;
в) недостатність процесу секреції
ліпопротеїнів.
Гепатит
Гепатит - дифузне запалення печінкової ткани
ни - розвивається у відповідь на ушкодження
і загибель гепатоцитів.
Залежно від причин розрізняють первин
ні (самостійні нозологічні форми) і вторинні
(розвиваються при інших захворюваннях) ге
патити.
Три можливі механізми жирової інфільтрації печінки. Пояснення див. у тексті
289
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
Первинні гепатити за етіологією найчастіше
бувають (а) вірусними, (б) алкогольними, (в) ме
дикаментозними, (г) автоімунними.
За характером перебігу гепатити поділяють
на гострі та хронічні.
Гострий гепатит характеризується дистро
фічними змінами й некрозом гепатоцитів (аль
терація), характерними для запального процесу
змінами місцевого кровообігу, утворенням лей
коцитарних інфільтратів у сполучнотканинній
стромі органа.
Хронічний гепатит може бути самостійною
хворобою або результатом переходу гостро
го гепатиту в хронічну форму. Характерними
ознаками дифузного хронічного запалення
в печінці є дистрофічні зміни (зокрема стеатоз) у гепатоцитах на тлі інфільтрації строми
переважно лімфоцитами, проліферації зірчас
тих ендотеліальних клітин і фіброзу порталь
них трактів. При цьому звичайна структура
печінки (поділ паренхіми на часточки) збері
гається.
Беручи до уваги поширеність і важливе зна
чення алкогольного і вірусних гепатитів, ці фор
ми уражень печінки розглянемо окремо (див.
далі).
Цироз печінки. Поняттям цироз позначають
хронічне прогресуюче ураження печінки, що
характеризується, з одного боку, розростанням
сполучної тканини, а з другого - патологічною
регенерацією печінкової паренхіми (утворен
ням вузлів регенерації), внаслідок чого зміню
ється звичайна архітектоніка органа, поділ його
тканини на правильні структурні одиниці - пе
чінкові часточки. Розвиток наведених вище змін
супроводжується перебудовою судинного русла
печінки, порушенням кровопостачання осеред
ків ще життєздатної паренхіми.
Цироз є наслідком необоротного ушкодження
й загибелі великої кількості печінкових клітин,
його розвитку часто передують стеатоз печін
ки і гепатити (див. рис. 35.3).
Залежно від причин ушкодження печінкової
паренхіми розрізняють три патогенетичні варі
анти цирозу:
1) постнекротичний. Розвивається як наслі
док некрозу великої кількості гепатоцитів.
Клінічним його еквівалентом є гепатоцелюлярна недостатність печінки (див.
вище);
2) біліарний. Формується внаслідок тривало
го холестазу — порушень виведення жовчі
по внутрішньопечінкових і позапечінкових жовчовивідних шляхах (наприклад,
при обтурації останніх). Недостатність пе
чінки, яка розвивається при цьому, позна
чають як холестатичну;
3) портальний. Є наслідком хронічних пору
шень кровообігу в печінці (наприклад, при
застійній недостатності серця). Клінічно
виявляє себе ознаками печінкової недо
статності, що її відносять до гепатоваскулярного варіанта.
АЛКОГОЛЬНІ УРАЖЕННЯ
ПЕЧІНКИ. ВІРУСНІ ГЕПАТИТИ
Найпоширенішою причиною уражень печінки
у нашій країні та в країнах Заходу є надмірне
вживання алкоголю і крайній його вияв - алко
голізм. Це небезпечне медичне та соціальне яви
ще сьогодні посідає п’яте місце серед причин
смертності населення.
Алкоголь уражає всі без винятку органи і тка
нини організму, але особливо небезпечним він
є для центральної нервової системи і печінки.
Причинами патологічних змін у печінкових
клітинах при хронічній алкогольній інтоксика
ції є (рис. 35.5):
1) етанол, який впливає на клітини організ
му прямо й опосередковано через утворен
ня токсичних метаболітів (ацетальдегіду)
(див. нижче);
2) цілий ряд гепатотоксичних сполук, що
містяться в неякісних алкогольних напоях
(сивушні масла тощо) та в сурогатах алко
голю, до вживання яких часто долучають
ся хворі на алкоголізм;
3) аліментарний фактор. При алкоголізмі
через неякісне незбалансоване харчування
порушується надходження в організм ві
тамінів, антиоксидантів, мікроелементів,
незамінних амінокислот, поліненасичених
жирових кислот - сполук, конче необхід
них для забезпечення структурної цілості,
функціональної активності, обміну речо
вин у клітинах. Справу значно погіршують
порушення процесів травлення, що, як
правило, супроводжують хронічну алко-
290
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Основні чинники впливу алкоголізму на печінку
гольну інтоксикацію, - хронічний гастрит,
хронічний панкреатит, ураження слизової
оболонки тонких кишок.
Токсична дія етанолу. Розрізняють пряму
й опосередковану метаболітами дію етанолу на
клітини.
1. Пряма дія. Етанол (етиловий спирт,
С Н ОН) являє собою одноатомний спирт,
що має властивості змінювати фізичнохімічні властивості клітинних мембран.
У зв’язку з цим етанол відносять до мембранотропних токсичних речовин. Проника
ючи в плазматичні мембрани клітин, ця ор
ганічна сполука змінює трансмембранний
транспорт речовин, порушує міжклітинні
контакти і взаємодії, зв’язується з багатьма
клітинними рецепторами. Особливо вира
женими ці ефекти є в центральній нервовій
системі, вплив на яку виявляє себе галь
муванням (депресією) багатьох нервових
функцій, розвитком ефекту сп’яніння.
Депресивну дію алкоголю на центральну нерво
ву систему пояснюють, зокрема, тим, що етанол,
зв’язуючись з мембранними рецепторами нейро
нів, активує одні (переважно гальмівні) і блокує
інші (переважно збуджувальні). Так, етанол ви
ступає агоністам центральних нікотинових ацетилхолінових рецепторів (п-холінорецепторів),
рецепторів до у-аміномасляної кислоти (ГАМКрецепторів), серотонінових (5-НТЗ) та гаіцинових рецепторів. Водночас він є антагоністом
відомих сьогодні різновидів глютаматних рецеп
торів, зменшує провідність Na+, К+ і Са2+ іонних
каналів.
2. Опосередкована метаболітами дія. Ка
таболізм етанолу здійснюється головним
чином у печінці, де окиснюється від 75 %
до 98 % введеного в організм етилового
спирту.
Окиснення алкоголю - це біохімічний про
цес, що складається з трьох послідовних фер
ментативних перетворень.
1. Окиснення етанолу до ацетальдегіду
(рис. 35.6). У гепатоцитах є три компартменти, де може відбуватися цей процес:
а) у цитозолі під впливом НАД-залежного
ферменту, що містить цинк, - алкогольдегідрогенази (АДГ). Ця реакція відіграє
провідну роль у перетвореннях етилово
го спирту в організмі. У нормі наявність
у клітинах АДГ забезпечує окиснення
291
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
ШЛЯХИ утворення ацетальдегіду 3 етанолу. 1 - окиснення етанолу НАД-залежною алкогольдегідрогеназою
(АДГ) в цитозолі; 2 - мікросомальна етанолокислювальна система (МЕОС); 3 - окиснення етанолу каталазою в пероксисомах
невеликих кількостей етанолу, що утво
рюється мікрофлорою кишок у реакціях
бродіння. При надходженні алкоголю
ззовні потужність ферменту є недостат
ньою, щоб швидко метаболізувати ета
нол, а тому його концентрація в крові
зростає й виявляються його мембранотропні впливи на центральну нервову
систему - виникає ефект сп’яніння.
При хронічному алкоголізмі кількість АДГ
у печінці не збільшується, оскільки цей фермент
не є таким, що індукується. Активність АДГ за
лежить від його ізоферментної форми (у люди
ни таких є три), а також від характеристик від
повідного гена. Показано, що деякі варіанти однонуклеотидного поліморфізму гена АДГ збіль
шують стійкість організму до алкоголю.
Слід зазначити, що крім гепатоцитів АДГ міс
тять ще й епітеліальні клітини слизової оболон
ки шлунка. Встановлено, що в жінок активність
цього шлункового ферменту значно нижча, ніж
у чоловіків, а тому сп’яніння у них настає рані
ше і від менших доз алкоголю;
б) у структурах гладкого ендоплазматично
го ретикулуму завдяки діяльності мікросомальної етанолокислювальної систе
ми, у якій провідну роль відіграє один із
представників цитохрому Р450 (CYP2E1),
що здійснює окиснення не тільки ета
нолу, а й багатьох інших ксенобіотиків,
у тому числі й лікарських препаратів
(барбітуратів та ін.). При хронічному ал
коголізмі окиснення етанолу цим шляхом
прискорюється на 50-70 % завдяки гіпер
трофії ендоплазматичного ретикулуму та
індукції CYP2E1.
Крім основної реакції, цитохром Р450 каталі
зує утворення активних форм кисню (див. главу
7). З цієї причини при індукції етанолом мікросомального його окиснення паралельно зрос
тає утворення вільних радикалів і пероксиду
водню - чинників, що започатковують оксидаційний стрес і пероксидне окиснення ліпідів. За
значені процеси, як буде показано далі, є важли
вими механізмами ушкодження і загибелі гепа
тоцитів;
в) у пероксисомах під впливом катет ази.
Ця реакція має другорядне значення,
оскільки каталаза здатна розщеплювати
лише 2 % етанолу.
2. Окиснення ацетальдегіду до оцтової кис
лоти (ацетату). Відбувається в мітохондріях. Основна реакція каталізується НАДзалежним ферментом альдегіддегідрогеназою (АлДГ).
Описано цілий ряд поліморфізмів гена
АлДГ. Зокрема показано, що приблизно 50 %
населення Китаю, Японії та В’єтнаму мають непереносимість алкоголю, зумовлену точковими
мутаціями цього гена, при яких зменшується
активність АлДГ. Водночас у представників на
ведених вище етнічних груп переважає ізофер
мент алкогольдегідрогенази (АДГ2) з більшою,
292
ніж в інших ізоферментів (АДГ, та АДГ3), ак
тивністю. Це веде до того, що етанол швидше
перетворюється на ацетальдегід. З другого ж
боку, через низьку активність АлДГ затриму
ються дальші перетворення ацетальдегіду. Як
результат, рівень цієї сполуки значно зростає,
що й зумовлює її токсичні ефекти при вживан
ні навіть невеликих доз алкоголю. Серед них почервоніння обличчя й тіла людини, покрит
тя їх плямами, підвищення температури шкіри
(англ., alcoholflush reaction, Asian flush).
Крім АлДГ, окиснення ацетальдегіду в мітохондріях може здійснюватися ще одним фер
ментом - ФАД-залежною альдегідоксидазою.
Активність його значно зростає при збільшен
ні вмісту ацетальдегіду. У ході реакції утворю
ються оцтова кислота, пероксид водню та інші
активні форми кисню. Останні, як уже зазна
чалося, започатковують оксидаційний стрес
і пероксидне окиснення ліпідів і стають завдяки
цьому важливими чинниками ушкодження гепатоцитів.
3. Утворення ацетил-КоА з оцтової кислоти.
Реакцію каталізує фермент ацетил-КоАсинтаза. Для її здійснення потрібні кофер
мент А й АТФ. Власне, утворенням ацетил-КоА і завершується специфічна части
на катаболізму етанолу. Далі, залежно від
співвідношення АТФ/АДФ і концентрації
оксалоацетату в мітохондріях, ацетил-КоА
може “згорати” в циклі трикарбонових кис
лот, стаючи джерелом енергії для ресинтезу АТФ, або йти на синтез жирових кислот
чи утворення кетонових тіл (див. далі).
Важливим для розвитку зумовлених алкого
лем метаболічних змін у клітинах є збільшення
співвідношення в цитозолі та в мітохондріях
відновленого й окисненого НАД (НАДН/НАД)
(рис. 35.7).
Так, підвищення цього співвідношення в ци
тозолі спричиняється до того, що:
а) пригнічуються реакції глюконеогенезу, що
може стати однією з причин гіпоглікемії
(див. главу 24);
б) зростає вміст у клітинах молочної
кислоти, вихід якої у кров стає причиною
лактацидемічного ацидозу,
в) збільшується утворення гліцерол-3фосфату - необхідного для синтезу
триацилгліцеролів субстрату.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Підвищення
співвідношення
НАДН/НАД
у мітохондріях веде до пригнічення реакцій
циклу трикарбонових кислот. Унаслідок цього
ацетил-КоА не окиснюється до кінцевих про
дуктів СО, й Н20, а спрямовується на синтез
жирових кислот і утворення кетонових тіл.
Слід зазначити, що токсична дія алкоголю
на центральну нервову систему в першу чергу
пов’язана з прямим мембранотропним впливом
етанолу на нейрони. Натомість в ураженнях пе
чінки провідну патогенетичну роль відіграє ме
таболіт етанолу — ацетальдегід.
При надходженні великих доз алкоголю
(більш ніж 2 г/кг) через різні швидкості окис
нення етанолу й ацетальдегіду в цитозолі різко
підвищується концентрація останнього. Аце
тальдегід є сполукою з високою реакційною
здатністю, він неферментативно може ацетилювати SH- і NH -групи білків та інших сполук
у клітинах і порушувати їхні функції. У моди
фікованих (ацетильованих) протеїнах часто ви
никають “зшивки”, нехарактерні для нативної
структури (наприклад, у молекулах білків хро
матину, ліпопротеїнів). Ацетилювання струк
турних і функціональних білків, а також фер
ментів веде до зменшення синтезу “білків на
експорт”, порушує процеси їхньої секреції.
Накопичення в гепатоцитах ацетальдегіду
і розлади метаболізму, що супроводжують хро
нічну алкогольну інтоксикацію, стають при
чиною патологічних змін у печінці - стеатозу
та алкогольного гепатиту (рис. 35.8).
Алкогольний стеатоз печінки. У його розвит
ку мають значення всі механізми, що ми їх об
говорювали вище.
Так, з одного боку, посилюється утворення
триацилгліцеролів (ТАГ) через те, що:
а) збільшується надходження в печінку віль
них жирових кислот із крові, оскільки хро
нічна алкогольна інтоксикація супроводжу
ється посиленим гідролізом ТАГ (ліполізом) у жировій тканині (людина худне);
б) зменшується окиснення жирових кислот
в мітохондріях через пригнічення реакцій
циклу трикарбонових кислот в умовах під
вищення співвідношення НАДН/НАД у мі
тохондріях (див. вище);
в) посилюється біосинтез жирових кислот
з ацетил-КоА. Причиною цього є збіль
шення концентрації ацетил-КоА через
293
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
Метаболічні зміни в гепатоцитах, що виникають при збільшенні співвідношення НАДН/НАД
у ЦИТОЗОЛІ та мітохондріях КЛІТИН. АДГ - алкогольдегідрогеназа, АлДГ - альдегіддегідрогеназа,
ТАГ - триацилгліцероли, ЦТК - цикл трикарбонових кислот
посилене його утворення з етанолу і не
достатнє “спалювання” в циклі трикарбо
нових кислот. Біосинтезу ліпідів сприяє
збільшення співвідношення НАДН/НАД
у клітинах.
Підвищення концентрації жирових кислот
у гепатоцитах, а також збільшене утворення
гліцерол-3-фосфату, що його супроводжує, веде
до зростання синтезу ТАГ. На молекулярногенетичному рівні це забезпечується посилен
ням експресії генів, що відповідають за синтез
ліпідів.
З
другого боку, при хронічній алкогольній ін
токсикації порушується виведення ТАГ з печінки
у складі ліпопротеїнів. Причиною цього стає ток
сична дія ацетальдегіду: ацетилювання апопротеїнів порушує процес формування міцел ліпопро
теїнів дуже низької густини (ЛПДНГ), а модифі
кація тубуліну та інших білків цитоскелета робить
неповноцінними процеси їх секреції в кров.
Алкогольний гепатит. В основі гепатиту, як
і будь-якого запалення, лежить ушкодження
(альтерація) клітин (див. главу 17). Вважають,
що основним механізмом ушкодження гепатоцитів та їхнього некрозу при хронічній алко
гольній інтоксикації є пероксидне окиснення
ліпідів (ПОЛ), суть якого була предметом обго
ворення в главі 7.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
294
.8. Механізми патологічних змін, що виникають у гепатоцитах під впливом ацетальдегіду.
ЛПДНГ - ліпопротеїни дуже низької густини, ТАГ - триацилгліцероли, ЦТК - цикл трикарбонових кислот
В основі активації ПОЛ алкоголем лежать два
чинники (рис. 35.9).
1. Посилене утворення вільних радикалів і пероксидів (активних форм кисню). Як уже
зазначалося, це відбувається внаслідок:
а) індукції цитохрому Рт (CYP2E1), який
окиснює етанол в ацетальдегід у мікросомах гепатоцитів;
б) підвищення активності альдегідоксидази
в мітохондріях, де відбувається окиснення ацетальдегіду в ацетат.
2. Зменшення можливостей антиоксидант
ного захисту. До цього спричиняються:
а) інактивація глютатіону через зв’язува
ння його SH-груп з ацетальдегідом. Че
рез брак відновленого глютатіону при
пиняє свою діяльність глютатіонпероксидазна система — провідний фермен
тативний механізм захисту клітин від
вільних радикалів і пероксидних сполук;
б) недостатнє надходження в організм
хворих на алкоголізм вітамінів-антиоксидантів (токоферолу, каротинів, ас
корбінової кислоти) і мікроелементів
(селену, міді, цинку), необхідних для
функціонування антиоксидантних фер
ментів.
Очевидно, що активація ПОЛ є дуже важли
вим, але не єдиним механізмом ушкодження
печінки. Альтерації гепатоцитів при хронічній
алкогольній інтоксикації може сприяти й ряд ін
ших чинників.
» Під впливом етанолу значно полегшуєть
ся проникнення бактеріальних токсинів із
кишковика в печінку. В умовах порушеної
295
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
Пероксидне окиснення ліпідів як механізм альтерації гепатоцитів при хронічній алкогольній
інтоксикації
функції гепатоцитів накопичення цих ток
синів стає фактором альтерації клітин.
» Показано, що етанол, діючи на ендотеліальні клітини синусоїдів печінки, стиму
лює виділення ними ендотелінів — вазоактивних поліпептидів, що викликають ско
рочення гладких м’язових клітин, а отже,
і звуження судин (див. главу 16). Унаслідок
цього зменшується кровопостачання печін
кової паренхіми та додається ще один фак
тор ушкодження - гіпоксія.
» Неферментне ацетилювання білків, зумов
лене накопиченням в клітинах ацетальдегіду, може спричинятися до такої модифі
кації протеїнів, унаслідок якої вони стають
автоантигенами. У такому разі до меха
нізмів альтерації гепатоцитів долучається
й механізм автоімунного їхнього ушко
дження.
Некроз гепатоцитів через вивільнення назовні
продуктів розпаду клітин “запускає” подальші
процеси, характерні для запалення. Централь
не місце в цьому посідають клітини Купфера,
розташовані в стінках синусоїдних капілярів
печінки (рис. 35.10).
Під впливом продуктів первинної альтерації
гепатоцитів, а також унаслідок прямої дії ета
нолу та його метаболітів відбувається активація
клітин Купфера (рис. 35.11). Вона виявляє себе
вивільненням цілої низки прозапальних цитокінів - фактора некрозу пухлин (ФНП), інтерлейкіну-6 (ІЛ-6), трансформуючого фактора росту
а (ТФР-а) та інших.
Зазначені медіатори запалення стають причи
ною еміграції нейтрофілів, моноцитів і лімфо
цитів крові. Нейтрофіли і лімфоцити, перші через вивільнення вільних радикалів, пероксидних сполук і гідролітичних ферментів, а другі завдяки імунному механізму цитотоксичності,
викликають вторинну альтерацію гепатоцитів.
Моноцити перетворюються в макрофаги, які
шляхом фагоцитозу розчищають паренхіму пе
чінки від осередків некрозу.
Водночас прозапальні цитокіни діють на зір
часті клітини перисинусоїдального простору
(клітини Іто), активують їх, унаслідок чого
ті набувають властивостей міофібробластів.
296
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Синусоїд
Клітина Куафера
Гепатоцити
Будова стінки синусоїда печінки
1 Роль
клітин Купфера в розвитку алкогольного гепатиту. ФНП - фактор некрозу пухлин, іл - інтерлейкін,
ТФР - трансформуючий фактор росту
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
Останні продукують позаклітинні компоненти
сполучної тканини, що заповнюють очищені
місця тканини. Завдяки цьому гепатит завер
шується фіброзом, але, якщо дія токсичних доз
алкоголю продовжується, то поступово розвива
ється цироз печінки.
Алкогольний цироз. Фінальна стадія алкогольно
го ураження печінки, якою є цироз, розвивається
повільно, але невблаганно. їй найчастіше пере
дують стеатоз печінки та алкогольний гепатит.
Розвиток цирозу має в основі такі головні па
тологічні зміни.
1. Необоротне ушкодження і зумовлені ним
некроз та апоптоз великої кількості пе
чінкових клітин. Механізми цих змін при
хронічній алкогольній інтоксикації ми вже
обговорювали.
2. Розростання сполучної тканини фі
броз. Цей процес, як було показано вище,
пов’язаний з діяльністю міофібробластів —
активованих форм перисинусоїдальних
клітин Іто.
3. Осередкова регенерація паренхіми з утво
ренням регенераторних вузлів різної вели
чини. Вважають, що проліферація ще не
уражених гепатоцитів пов’язана з дією на
них факторів росту, утворення і вивіль
нення яких зростає при некрозі печінкових
клітин. Залишається невідомим, які саме
клітини є джерелом цих факторів: ушко
джені гепатоцити, клітини Купфера, кліти
ни Іто чи лейкоцити, що емігрували в печін
кову тканину в процесі її запалення. Таку
регенерацію паренхіми називають патоло
гічною, оскільки не відбувається відтворен
ня нормальної структури печінкових часто
чок з характерними для них особливостями
кровопостачання. Через це новоутворені
гепатоцити не можуть повного мірою вико
нувати притаманні їм функції, а через не
достатнє надходження крові зазнають дис
трофічних змін і, зрештою, гинуть.
4. Порушення кровообігу в печінці. їх розви
ток пов’язаний з такими двома основними
чинниками:
а) структурною перебудовою системи кро
вопостачання органа. Через загибель ве
ликої кількості клітин паренхіми і розви
ток фіброзу судини портального тракту
стають ближчими до центральної вени,
297
чим створюються умови для переходу
крові з печінкової артерії і ворітної вени
в центральну, обминаючи синусоїди.
Іншими словами, формуються судинні
анастомози, завдяки яким кров не дохо
дить до гепатоцитів неушкоджених діля
нок: розвивається ішемія останніх з на
ступним некрозом;
б) здавлюванням судин регенераторними вуз
лами й фіброзною тканиною.
Порушення кровообігу в печінці спричиня
ються до істотних змін гемодинамічних функ
цій цього органа та розвитку синдрому пор
тальної гіпертензії, мова про який піде далі.
Вірусні гепатити
Останнім часом, з огляду на високу пошире
ність і тяжкі наслідки, важливого значення на
була проблема вірусних гепатитів - інфекцій
них хвороб вірусного походження, при яких
уражується печінка. Хронічне ушкодження цьо
го органа, зумовлене вірусами гепатитів В і С,
є однією з основних причин смертності в світі.
Етіологія. Причиною більшості інфекційних
гепатитів є гепатотропні віруси, що належать до
різних сімейств. Вони позначаються літерами
латинської абетки: А, В, С, D, Е. Відповідно на
зивають і зумовлені ними гепатити.
З огляду на особливо тяжкі наслідки хво
роби ми зупинимося лише на двох її фор
мах — гепатитові В і С. Збудником гепатиту В
є ДНК-вмісний вірус HBV (hepatitis В virus) із
сімейства гепаднавірусів (рис. 35.12). Для цьо
го вірусу характерна надзвичайно висока стій
кість до різних фізичних і хімічних факторів:
низької та високої температури (у тому числі до
кип’ятіння), тривалого впливу кислого середо
вища тощо.
За різними оцінками у світі цим вірусом інфі
ковано від 3 до 6 % людей (близько 2 мільярдів).
Механізм передавання інфекції - парентераль
ний. Зараження відбувається природним (стате
вим, вертикальним, побутовим) та ін’єкційним
шляхами. Вірус можна виявити в крові та різних
біологічних рідинах - слині, сечі, спермі, піхво
вому секреті та ін. Контагіозність вірусу HBV
у 100 разів вища, ніж вірусу імунодефіциту лю
дини (ВІЛ).
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
298
12. Будова вірусів гепатиту В І гепатиту С. HBsAg, HBeAg і НВсАд - вірусні антигени
Вірус гепатиту С (HCV, hepatitis С virus) - це
РНК-вмісний вірус, що належить до сімейства
флавівірусів (рис. 35.12). Вірусні частки (віріони) мають оболонку й виявляються в крові
в дуже малих кількостях в асоційованих з ліпопротеїнами низької густини (ЛПНГ) чи з анти
тілами проти вірусних білків формах.
Джерелом інфекції є хворі з активним гепати
том С і латентні хворі - носії вірусу. Зараження
відбувається парентеральним шляхом через ін
фіковану кров та її компоненти. Інші (природні)
парентеральні шляхи інфікування є менш імо
вірними, ніж у випадку з гепатитом В. Гепатит
С називають “лагідним убивцею” через здат
ність маскувати справжню причину під вигля
дом багатьох інших хвороб. “Підступність” ві
русу HCV полягає ще й в тому, що він має дуже
нестабільний геном і пов’язану з цим велику
варіабельність структури антигенів. Це дозво
ляє вірусу легко уникати імунних атак на себе.
Виживанню вірусу сприяє й те, що деякі його
компоненти є інгібіторами клітинних білків учасників індукованої у-інтерфероном системи
противірусного захисту.
Патогенез. У переважній більшості випадків
інфікування вірусом гепатиту В або нічим себе
не виявляє (5-10 %, “здорове” носійство вірусу),
або розвивається недуга з невиразними симпто
мами і легким перебігом (60-65 %) чи картина
гострого гепатиту (20-25 %). Як правило, таке
інфікування завершується одужанням. Проте
у 4-5 % випадків інфекція затримується в орга
нізмі надовго і в третини таких людей може зу
мовлювати хронічний гепатит, прогресування
якого завершується цирозом печінки чи гепатоцелюлярним раком.
Вірус гепатиту С у 85 % випадків інфіку
вання спричиняється до хронічного гепатиту,
у решти інфікованих хвороба проходить безсимптомно, настає одужання.
Найбільша небезпека, яку несе в собі зара
ження вірусами гепатитів В і С, полягає в мож
ливості розвитку хронічного гепатиту з перехо
дом його в цироз печінки чи в гепатоцелюлярний рак.
Провідним патогенетичним механізмом ві
русного хронічного гепатиту є ушкодження гепатоцитів за участю імунної системи організму
(рис. 35.13). Так, при інфікуванні вірусом гепа
титу В синтез його компонентів у клітинах (т. зв.
проліферативна фаза інфекції) веде до експресії
вірусних антигенів на поверхні гепатоцитів. Це
зумовлює імунну відповідь, у якій беруть участь
механізми як клітинного, так і гуморального
імунітету (див. главу 14).
Взаємодія уражених печінкових клітин із ци
тотоксичними CD8+ Т-лімфоцитами запускає
процеси апоптозу (див. главу 7) і вивільнення із
загиблих гепатоцитів особливого білка з групи
299
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
ис. 35.13 Механізм імунного ушкодження інфікованих вірусом гепатиту В (HBV) гепатоцитів.
CXCL9, CXCL10 - хемокіни, HMGB1 - ядерний негістоновий протеїн, IFN-y - у-інтерферон; ММР8, ММР9 - металопротеїнази
ядерних негістонових протеїнів - HMGB1 (highmobility group protein В1), відомого ще під на
звою амфотерин. Цей білок виступає лігандом
по відношенню до т. зв. Toll-подібних рецепто
рів (TLR) (див. главу 14), активація яких у лей
коцитах і макрофагах запускає процеси фагоци
тозу та вивільнення ними великої групи прозапальних цитокінів.
Показано, що під впливом HMGB1 нейтрофільні лейкоцити вивільнюють матриксні ме
талопротеїнази (ММР8, ММР9), які активують
хемотаксис мононуклеарних фагоцитів. Останні
спрямовуються в осередки загибелі печінкових
клітин і фагоцитують їхні залишки (апоптичні
тільця).
Водночас цитотоксичні CD8+ Т-лімфоцити,
що взаємодіють з ураженими гепатоцитами,
стають джерелом у-інтерферону, який захищає
ще не інфіковані клітини від вірусних атак та
індукує вивільнення ними хемокінів (CXCL9,
CXCL10) - цитокінів, що стимулюють хемотак
сис уже згадуваних мононуклеарних фагоцитів.
До описаного вище механізму імунного ушко
дження клітин додається ще й механізм антиті-
лозалежної клітинної цитотоксичності (алер
гічна реакція II типу, див. главу 15), який поси
лює процеси альтерації й пов’язаного з нею за
палення.
Тривале вірусне та імунне ушкодження пе
чінкових клітин, особливо на тлі алкогольної чи
інших видів інтоксикації, стає причиною необо
ротних змін у печінці, веде до розвитку цирозу.
Основні його механізми мало чим відрізняються
від тих, що ми їх описали при обговоренні алко
гольних уражень печінки.
35.3. ПОРУШЕННЯ ОКРЕМИХ
ПЕЧІНКОВИХ ФУНКЦІЙ
Печінка, як уже зазначалося, виконує багато
різноманітних функцій, які ми об’єднали в чо
тири групи (див. вище). З урахуванням цього
і розглянемо основні їх порушення, що можуть
розвиватися як прояв загальної недостатності
печінки або бути відносно ізольованими.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
300
ПОРУШЕННЯ
МЕТАБОЛІЧНИХ ФУНКЦІЙ
Печінка є органом, у якому відбувається пере
важна більшість притаманних клітинним струк
турам біохімічних реакцій і перетворень. Вони
не тільки забезпечують власні потреби клітин
цього органа, а й працюють на потреби організ
му в цілому. З порушенням метаболічних функ
цій печінки пов’язаний цілий ряд клінічних про
явів (рис. 35.14).
Вуглеводний обмін. Участь печінки у вуглевод
ному обміні полягає переважно в підтримуванні
сталості концентрації глюкози в плазмі крові.
Це досягається завдяки тому, що в печінці від
бувається депонування глюкози у вигляді гліко
гену (на глікоген припадає близько 20 % маси
печінки).
Можливими є дві ситуації, при яких порушу
ється підтримування печінкою сталості концен
трації глюкози крові.
1. Зменшення вмісту глікогену в печінці.
До цього можуть спричинятися:
а) ненадходження глюкози з кишок у печін
ку - голодування',
б) порушення перетворення харчових моно
сахаридів (фруктози, галактози) у глюко
зу, що характерно для спадково зумовле
них хвороб - фруктоземії й галактоземії;
в) порушення глюконеогенезу (наприклад,
при гіпофункції кори надниркових залоз);
г) порушення синтезу глікогену з глюко
зи, обумовлене зменшенням активності
ферментів глікогенезу (наприклад, спад
кова хвороба - аглікогеноз)',
д) дефіцит АТФ, необхідного для транспор
ту глюкози в гепатоцити і реакцій біо
синтезу глікогену.
2. Порушення процесів вивільнення глюкози
з глікогену й надходження її в кров. Такі
розлади лежать в основі спадкових хвороб,
що одержали назву глікогенозів.
Оскільки утворення глюкози з глікогену в гепатоцитах може відбуватися двома шляхами
(фосфоролітичним і гідролітичним), то можливі
дві групи глікогенозів:
а) глікогенози, при яких порушується
фосфоролітичне розщеплення глікогену.
До них, зокрема, відносять дефіцит
кінази фосфорилази, фосфорилази
{хвороба Герша), глюкозо-6-фосфатази
{хвороба Гірке)',
б) глікогенози з розладами гідролітичного
розщеплення глікогену в лізосомах
гепатоцитів. У цю групу, зокрема, входять
хвороба Помпе, хвороба Форбса — Корі,
хвороба Андерсена.
Морфологічно глікогенози виявляють себе
значним збільшенням вмісту глікогену в гепатоцитах. Однак цей глікоген не може бути джере
лом глюкози крові.
Основні клінічні прояви порушень метаболічних функцій печінки
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
Основним проявом порушень вуглеводної
функції печінки є розвиток печінкової гіпогліке
мії, що у важких випадках може спричинятися
до гіпоглікемічної коми (див. главу 24).
Жировий обмін. Печінка посідає одне з цен
тральних місць у здійсненні обміну ліпідів (див.
главу 25). При ураженнях цього органа можуть
виникати такі розлади жирового обміну:
1) порушення перетравлювання і всмокту
вання жирів у тонкій кишці, що має місце
при розладах жовчоутворення й жовчови
ділення (див. далі);
2) порушення синтезу тригліцеридів, фосфоліпідів, холестеролу, а також утворен
ня ліпопротеїнів плазми крові (ЛПДНГ
ІЛПВГ);
3) надмірне утворення кетонових тіл',
4) жирова інфільтрація (стеатоз, жировий
гепатоз) печінки, механізми якої ми вже
обговорювали.
Білковий обмін. Печінка бере активну участь
у здійсненні обміну білків. У ній зокрема від
буваються:
1) розщеплення (дезамінування, декарбоксилювання) і перебудова (трансамінування)
амінокислот;
2) синтез сечовини з аміаку (див. главу 26);
3) утворення білків “на експорт” - білоксинтезуюча функція.
В основі порушень останньої можуть лежати
такі причини:
а) зміни структури генів, що несуть інформа
цію про будову відповідних білків (напри
клад, афібриногенемія)',
б) порушення процесів транскрипції— синте
зу інформаційної РНК на матриці ДНК (на
приклад, дія токсинів блідої поганки, анти
біотика актиноміцину D);
в) розлади процесів транскрипції-зчитуван
ня інформації з інформаційної РНК у рибо
сомах (наприклад, дія антибіотика пуроміцину);
г) зменшення кількості амінокислот, необ
хідних для біосинтезу білків (наприклад,
білково-енергетична недостатність, дефі
цит у їжі незамінних амінокислот);
д) дефіцит АТФ, енергія якого використову
ється в біосинтетичних процесах (гіпоксія,
ЗОЇ
голодування, гіповітамінози Bj В2, РР, дія
інгібіторів ферментів, роз’єднання окиснення й фосфорилювання).
У печінці синтезується переважна більшість
білків плазми крові: всі альбуміни, 75-90%
а-глобулінів, близько 50 % (3-глобулінів. Тому
найяскравіші прояви порушень білоксинтезуючої функції печінки пов’язані з розладами
утворення саме білків плазми крові. Такими
проявами є гіпопротеїнемічний і геморагічний
синдроми.
Гіпопротеїнемічний синдром виникає в ре
зультаті зменшення концентрації альбумінів
у плазмі крові. Це призводить до зниження он
котичного тиску плазми, наслідком чого може
бути розвиток набряків (див. главу 27).
Геморагічний синдром (підвищена крово
точивість) є наслідком порушень синтезу біл
ків - факторів зсідання крові (фібриногену, про
тромбіну, проконвертину, проакцелерину) (див.
главу ЗО).
Обмін вітамінів. При ураженнях печінки мо
жуть розвиватися такі розлади обміну вітамінів:
1) порушення утворення біологічно актив
них форм вітамінів з вітамінів-попередників (наприклад, тіамінпірофосфату з віта
міну Bj);
2) порушення депонування вітамінів у печін
ці (наприклад, вітаміну В12);
3) розлади всмоктування жиророзчинних ві
тамінів у результаті порушень функції
утворення жовчі в печінці.
Клінічно всі зазначені порушення виявляють
себе розвитком ознак відповідних гіповітаміно
зів.
Обмін мікроелементів. При хворобах печінки
порушуються:
а) депонування заліза (у формі феритину),
міді, цинку, кобальту, молібдену, марганцю
та ін.;
б) синтез транспортних білків, що забезпечу
ють перенесення мікроелементів в організ
мі (трансферину, церулоплазміну);
в) екскреція мікроелементів з жовчю.
Обмін гормонів і біологічно активних речовин.
У печінці відбувається інактивація багатьох гор
монів і біологічно активних речовин.
302
Інактивації зазнають усі стероїдні гормони
(гаюкокортикоіди, мінералокортикоїди, жіночі
й чоловічі статеві гормони), тиреоїдні гормони,
інсулін. При порушенні функції гепатоцитів змен
шується інтенсивність перетворення зазначених
гормонів, тому їхня концентрація в крові зростає
і з’являються ознаки гіперфункції відповідних
ендокринних залоз (вторинного гіперальдостеронізму, гіпертиреозу та ін.; див. главу 37). Поді
бні ознаки можуть бути пов’язані і з порушенням
синтезу в печінці транспортних білків (наприклад,
транскортину), що зв’язують вільні гормони.
Печінка є органом, де відбувається руйнуван
ня багатьох біологічно активних речовин, і зо
крема—біогенних амінів: катехоламінів, гістамі
ну, серотоніну. У зв’язку з цим у разі порушення
функції гепатоцитів концентрація зазначених
біологічно активних речовин у крові може зрос
тати. У результаті розвиваються ознаки актива
ції симпатоадреналової системи (підвищення
артеріального тиску, тахікардія та ін.), підвище
ного утворення гістаміну (свербіж шкіри, розви
ток виразок у травному каналі).
ПОРУШЕННЯ
АНТИТОКСИЧНОЇ ФУНКЦІЇ.
ПЕЧІНКОВА КОМА
Печінка виконує захисні (бар’єрні) функції, до
яких відносять фагоцитарну й антитоксичну.
Фагоцитарну функцію здійснюють клітини
Купфера, що належать до системи мононуклеарних фагоцитів. Можливі розлади цієї функції
описано в главі 14.
Антитоксична функція властива гепатоцитам. Вона полягає в інактивації кінцевих про
дуктів обміну речовин (сечовиноутворення)
і екзогенних сполук, що надходять з кишок
(продукти діяльності мікрофлори - фенол, кре
зол, індол, скатол, аміни), а також з навколиш
нього середовища. Детоксикація різних речовин
здійснюється за допомогою різних хімічних ре
акцій (синтезу, окиснення, відновлення, гідролі
зу, метилювання, кон’югації), що відбуваються
в структурах ендоплазматичного ретикулуму
(мікросомах), мітохондріях, пероксисомах і ци
топлазмі печінкових клітин.
До порушень антитоксичної функції печінки
можуть спричинятися:
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
1) зменшення кількості функціонуючих гепа
тоцитів (наприклад, при масивних некро
зах печінки). Так, установлено, що синтез
сечовини з аміаку порушується при ура
женні 80 % і більше паренхіми печінки;
2) зменшення активності ферментів, що бе
руть участь у реакціях детоксикації. Воно
може бути зумовлене генетичними дефек
тами відповідних ферментів (наприклад,
спадкове порушення ферментів синтезу
сечовини) або набутими порушеннями
їх утворення (розлади білоксинтезуючої
функції печінки);
3) зменшення концентрації речовин, що бе
руть участь в інактивації токсичних про
дуктів (кисню, глюкуронової та сірчаної
кислот, гліцину, таурину, цистеїну тощо);
4) дефіцит АТФ. При цьому можуть порушу
ватися процеси надходження й виведення
з гепатоцитів токсичних речовин, а також
реакції синтезу й кон’югації, які вимага
ють витрат енергії.
Синдром гепатоцеребральної недостатності.
Порушення антитоксичної функції печінки ви
являють себе ознаками інтоксикації, що уражує
насамперед центральну нервову систему. Тому
комплекс змін, що виникають, одержав назву
синдрому
гепатоцеребральної
недостатності,
відомого ще як печінкова енцефалопатія. Для
нього характерними є різні поєднання психіч
них і невротичних розладів аж до втрати свідо
мості й розвитку коматозного стану.
Залежно від ступеня порушень антитоксич
ної функції печінки зазначений синдром може
виявляти себе:
а) емоційно-психічними розладами: емоцій
ною нестійкістю (почергові зміни ейфорії
й депресії), безсонням уночі й сонливістю
вдень, головним болем, запамороченнями;
б) вираженими порушеннями свідомості - роз
витком ступору (сонливість, сплутаність
свідомості);
в) печінковою комою (див. нижче).
В основі синдрому гепатоцеребральної недо
статності лежить накопичення в крові так званих
церебротоксичних речовин. Серед них особливо
важливого значення надають таким сполукам:
1) аміаку. В організмі існує два джерела над
ходження аміаку в кров: (а) усі периферичні
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
303
тканини, де відбуваються процеси дезамі
нування амінокислот, і (б) кишки, де утво
рення аміаку обумовлене процесами гнит
тя, тобто діяльністю кишкової мікрофлори.
Аміак є токсичною сполукою, а тому в нор
мі його в крові практично немає, бо він пе
ретворюється в гепатоцитах у сечовину. Із
кишок аміак надходить одразу в печінку по
судинах портальної системи, а з перифе
ричних клітин — у зв’язаній формі у складі
глютаміну (т. зв. глютаміновий човниковий
механізм транспорту NH3). При недостат
ності печінки аміак, що надходить сюди із
двох зазначених джерел, не може повністю
перетворюватися в сечовину, а тому його
концентрація в крові зростає;
2) продуктам окиснення амінокислот у киш
ках. Такими є фенол, індол, скатол, аміни
(путресцин, кадаверин); сполуки сірки,
що утворюються при окисненні метіоні
ну (диметил сульфід, метил меркаптан).
З останніми, зокрема, пов’язують появу
специфічного симптому печінкової коми печінкового запаху,
3) низькомолекулярним жировим кислотам:
масляній, валеріановій, капроновій. Вони
з’являються внаслідок порушень окиснен
ня жирових кислот в ушкоджених гепато
цитах.
Центральне місце в патогенезі печінкової
коми належить надходженню в кров аміаку та
інших церебротоксичних речовин. Про це, зо
крема, свідчить той факт, що у хворих у стані
печінкової коми в багато разів збільшується
вміст зазначених сполук у крові. Крім того,
в експерименті показано, що введення тваринам
церебротоксичних речовин супроводжується
розвитком ознак печінкової коми.
Механізми порушень діяльності централь
ної нервової системи, що ведуть до печінкової
коми, досить складні. Проте можна виділити
ряд провідних патогенетичних чинників.
L Порушення діяльності синапсів і синоп
тичної передачі. Вони виявляють себе
послабленням процесів центрального збу
дження і посиленням центрального галь
мування. Поєднання цих змін стає причи
ною депресії, пригнічення всіх функцій
центральної нервової системи аж до втрати
свідомості, порушень регуляції життєво
важливих функцій - дихання і кровообігу.
Печінкова кома. Печінковою комою називають
патологічний стан, що виникає в результаті тяж
ких порушень антитоксичної функції печінки
і виявляє себе втратою свідомості, випадінням
рефлексів на внутрішні й зовнішні подразники,
розладами життєво важливих функцій організму
(кровообігу, дихання). Печінкова кома є крайнім
проявом синдрому гепатоцеребрсиїьної недо
статності, а тому має ті ж самі причини, що
і цей синдром.
Залежно від джерела й механізмів надхо
дження в кров церебротоксичних речовин виді
ляють два патогенетичних варіанти печінкової
коми:
I. Ендогенну {печінково-клітинну, або розпадну). У цьому випадку поява церебро
токсичних речовин у крові пов’язана з по
рушеннями антитоксичної функції печінки
при ушкодженні й загибелі гепатоцитів;
II. Екзогенну {портокавальну, або шунто
ву). Її розвиток обумовлений тим, що
Серед причин наведених змін такі:
а) дія аміаку. Тканини головного мозку ма
ють здатність оберігати себе від токсичної
дії аміаку завдяки тому, що гліальні кліти
ни — астроцити— захоплюють його і пере
творюють у форму глютаміну, який через
ліквор виводиться в кров, а потім у печінку
(рис. 35.15Б). Така властивість астроцитів
пов’язана з тим, що вони беруть участь
у нормальному обміні одного з основних
збуджувальних медіаторів центральної нер
вової системи - глютамату (рис. 35.15А).
В умовах, коли рівень аміаку в крові висо
кий, а запаси глютамату в астроцитах через
це зменшуються і, врешті-решт, зникають,
NH3 починає проникати в нервові клітини
і зв’язуватися там з глютаматом. Як ре
зультат, рівень цього медіатору в пресинаптичних везикулах падає і збуджувальні
сигнали не передаються на постсинаптичні
нейрони (рис. 35.15В);
церебротоксичні речовини потрапляють
у системний кровообіг з кишок через во
рітну вену та портокавальні анастомози,
минаючи печінку. При цьому антиток
сична функція печінки може істотно не
страждати.
304
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Вплив аміаку на процеси збудження в центральній нервовій системі.
А -аміаку в крові немає (норма); аміак в крові є: Б - фаза компенсації, В - фаза декомпенсації (депресії)
305
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
б)
несправжні нейромедіатори (псевдонейромедіатори). Так називають сполуки, що,
накопичуючись у тканинах головного моз
ку, заміщають нормальні медіатори нерво
вої системи або порушують утворення їх
з попередників. Такі зміни спричиняються
до припинення синаптичного передавання
нервових імпульсів і, як наслідок, до пору
шень міжнейронних взаємодій та розладів
інтегративних функцій центральної нерво
вої системи.
Цілий ряд церебротоксичних речовин (низь
комолекулярні жирові кислоти, тирамін, октопамін та ін.) мають властивості несправжніх
нейромедіаторів. Крім того, показано, що утво
ренню псевдонейромедіаторів сприяє зменшен
ня концентрації в нейронах амінокислот з роз
галуженим ланцюгом (валіну, лейцину, ізолей
цину) і збільшення рівня ароматичних аміно
кислот (фенілаланіну, тирозину, триптофану).
Останні метаболізуються переважно в печінці,
і їхня концентрація в крові зростає при недо
статності цього органа. Збільшене надходжен
ня в нейрони ароматичних амінокислот при
зводить до зниження синтезу збуджувальних
медіаторів - дофаміну і норадреналіну й утво
рення псевдонейромедіаторів — октопаміну,
(3-фенілетиламіну та ін.;
в) дія гальмівних медіаторів. Важливу роль
у розвитку нервових розладів при пе
чінковій комі відводять накопиченню
у-аміномасляної кислоти (ГАМК) — основ
ного гальмівного медіатора центральної
нервової системи. Одна з причин збіль
шення концентрації цієї речовини в крові
і тканинах головного мозку - порушення
в печінці процесів інактивації ГАМК, що
утворюється кишковою мікрофлорою.
Окрім того, вважають, що при недостатності
печінки в організмі зростає рівень ендогенних,
подібних до бензодіазепіну, сполук. Вони, як
і відповідні фармакологічні препарати, чинять
депресивну дію на функції центральної нерво
вої системи.
2.
Порушення енергетичного обміну та
пов’язаний з цим дефіцит АТФ. Провідна
роль у розвитку таких порушень належить
аміаку (рис. 35.16). Накопичуючись у вели
ких кількостях, аміак зв’язується з глюта-
матом і а-кетоглютаратом, перетворюю
чи їх в глютамін. Оскільки а-кетоглютарат
є одним із центральних метаболітів циклу
трикарбонових кислот, зв’язування його
з аміаком і виведення з цього циклу веде
до зменшення інтенсивності біологічного
окиснення субстратів, клітинного дихан
ня і в підсумку - до порушення ресинтезу
АТФ. Зменшення вмісту АТФ у нервових
клітинах спричиняється до розладів актив
ного транспорту катіонів, порушення ге
нерації нервових імпульсів, змін величини
мембранного потенціалу.
3. Зміни основних параметрів гомеостазу.
Порушення антитоксичної функції печінки
супроводжуються розвитком метаболіч
ного ацидозу і пов’язаних з ним порушень
обміну електролітів. Причинами ацидо
зу при печінковій комі є церебротоксичні
речовини (низькомолекулярні жирові кис
лоти), збільшення вмісту в крові вільних
амінокислот і лактату внаслідок пору
шень їхнього перетворення в гепатоцитах.
Метаболічний ацидоз закономірно веде до
розвитку гіперкаліємії та пов’язаних з нею
функціональних змін як у центральній нер
вовій системі, так і в організмі в цілому
(див. глави 27 і 29).
35.3.3. ПОРУШЕННЯ
ФУНКЦІЇ. ЖОВТЯНИЦІ
ЕКСКРЕТОРНОЇ
Сутність екскреторної функції печінки полягає
в утворенні й виділенні жовчі. Завдяки цьому
здійснюється:
1) виведення з організму продуктів метабо
лізму (білірубіну) і надлишку деяких речо
вин (холестеролу);
2) участь печінки в процесах травлення
(емульгування, перетравлювання і всмок
тування жирів).
Порушення екскреторної функції печінки
можуть виявляти себе такими синдромами, як
жовтяниця, холемічний синдром, ахолічний син
дром, а також жовчнокам ’яною хворобою.
Обмін жовчних пігментів. Основний жовчний
пігмент - білірубін — кінцевий продукт обміну
306
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
16. Аміак як причина порушень енергетичного обміну в нервових клітинах
гему. Головним джерелом білірубіну крові є ге
моглобін еритроцитів (рис. 35.17). Утворення бі
лірубіну з гему відбувається в клітинах системи
моноиуклеарних фагоцитів (макрофагах селезін
ки, червоного кісткового мозку, клітинах Купфера
в печінці), де фагоцитовані еритроцити зазнають
гемолізу. Білірубін, що утворився, нерозчинний
у воді, тому його транспорт у печінку здійсню
ється у зв’язаному з білками (альбумінами) ви
гляді. Такий білірубін одержав назву непрямого,
оскільки дає реакцію з діазореактивом Ерліха
тільки після попереднього осадження білків.
Непрямий білірубін з крові надходить у пе
чінку, де відбуваються три процеси, важливі
з погляду обміну жовчних пігментів:
1) захоплення гепатоцитами зв’язаного з біл
ками (непрямого) білірубіну. Його забезпе
чують специфічні білкові рецептори плаз
матичної мембрани печінкових клітин;
2) кон’югація білірубіну з глюкуроновою
кислотою, внаслідок чого утворюються
моно- і диглюкуроніди білірубіну {пря
мий білірубін)',
3) екскреція прямого білірубіну у складі жовчі.
Усі
зазначені
процеси
(захоплення,
кон’югація і екскреція) потребують витрат
енергії АТФ. Екскретований з жовчю білірубін
у жовчному міхурі і тонкій кишці під дією фер
ментів мікрофлори зазнає декон’югації та пере
ходить в уробіліноген. Основна кількість уробіліногену в товстій кишці далі перетворюється
в стеркобіліноген, що виводиться з калом. Не
значна кількість стеркобіліногену всмоктується
в кров у ділянці нижнього і середнього геморо
їдальних сплетень прямої кишки і, минаючи пе
чінку, потрапляє в нирки, де фільтрується і ви
водиться із сечею.
Друга, набагато менша частина уробіліногену
бере участь у так званому печінково-кишковому
кругообігу — всмоктується в тонкій кишці, по
трапляє в печінку, частково зазнає окиснення до
піролових сполук, а частково знову надходить
у жовчовивідні шляхи і в кишки.
Для розуміння порушень обміну жовчних піг
ментів та їх проявів важливими є відмінності
у властивостях прямого й непрямого білірубіну
(табл. 35.1)
307
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
35.17. Схема утворення, перетворень і виведення білірубіну
Жовтяниця
Жовтяниця (icterus) - це синдром, який виникає
при збільшенні рівня білірубіну в крові та вияв
ляє себе жовтим забарвленням шкіри і слизових
оболонок.
Залежно від механізмів розвитку виділяють
три види жовтяниці.
Гемолітична (надпечінкова) жовтяниця.
Виникає в результаті гемолізу еритроцитів
і збільшеного утворення білірубіну в клі
тинах системи мононуклеарних фагоцитів.
2. Паренхіматозна (печінкова) жовтяниця.
Її розвиток пов’язаний з ураженнями пе
чінки.
1.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
308
Таблиця 35.1
Властивості непрямого і прямого білірубіну
3.
Механічна (підпечінкова) жовтяниця. Ви
никає в результаті порушення відтоку жов
чі жовчовивідними шляхами.
Гемолітична
(надпечінкова)
жовтяниця.
Посилений фагоцитоз еритроцитів чи самого
гемоглобіну, що вивільнився зі зруйнованих
червонокрівців, спричиняється до утворення
у клітинах системи мононуклеарних фагоцитів
великої кількості білірубіну, який після утво
рення комплексів з альбуміном крові (непря
мий білірубін) надходить у печінку. Гепатоцити
в разі повноцінної їхньої діяльності перетво
рюють увесь цей непрямий білірубін у прямий
і екскретують останній у складі жовчі. Цим
власне пояснюється високий вміст стеркобіліногену в калі та підвищення його вмісту в сечі.
У зв’язку з тим, що непрямий білірубін не філь
трується в нирках (бо зв’язаний з білками), він
не з’являється в сечі (симптому білірубінурії
немає).
З урахуванням вищезазначеного, основними
ознаками порушення пігментного обміну при
гемолітичній жовтяниці є:
а) збільшення концентрації непрямого біліру
біну в крові;
б)
підвищення
вмісту
стеркобіліногену
в калі — гіперхолічний кал;
в) збільшення вмісту стеркобіліногену в сечі;
г) поява в сечі уробіліногену. У зв’язку з ве
ликим навантаженням на печінку вона не
в змозі екскретувати всю його кількість
у жовч (порушується печінково-кишко
вий кругообіг) і окиснювати до піролових
сполук.
Слід вказати, що при значному гемолізі ери
троцитів, наприклад, при гемолітичній хворобі
новонароджених, у крові починають з’являтися
аніони не зв’язаного з альбуміном білірубіну
(цього білка просто не вистачає). Вони дифун
дують в різні тканини, у тому числі проника
ють через гематоенцефалічний бар’єр у мозок.
Білірубін, який відкладається в ядрах великих
півкуль і стовбурі головного мозку (“ядерна”
жовтяниця), чинить токсичну дію, що виявляє
себе цілою низкою неврологічних симптомів.
У новонароджених на 3-6 день життя зникають
спінальні рефлекси, виникає гіпертонус м’язів
тулуба, з’являються сонливість, різкий плач, су
доми, порушення дихання, може настати його
зупинка і смерть. Якщо дитина виживає, то мо
жуть розвиватися глухота, паралічі, відставання
розумового розвитку.
Паренхіматозна
(печінкова)
жовтяниця.
В основі розвитку цього виду жовтяниць лежать
ізольовані або комбіновані порушення захоп
лення, кон’югації та екскреції білірубіну кліти
нами печінки.
Розрізняють (1) печінково-клітинну (гепатоцелюлярну) і (2) жовтяниці з ізольованими пору
шеннями процесів, що забезпечують виведення
білірубіну з організму.
І.
Печінково-клітинна жовтяниця. Харак
теризується порушеннями всіх трьох процесів,
309
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
що відбуваються в гепатоцитах: захоплення,
кон’югації та екскреції білірубіну. Виникає при
ушкодженні печінкових клітин (усі різновиди
гострих і хронічних гепатитів, цирози). Розви
ток жовтяниці за цих обставин пов’язаний з та
кими механізмами:
1) унаслідок загибелі печінкових клітин
утворюються сполучення між жовчними
і кровоносними капілярами. У результаті
жовч потрапляє в кров (холемія), а разом
з нею і прямий білірубін;
2) у клітинах, які не загинули, але ушкодже
ні, порушується захоплювання непря
мого білірубіну й екскреція жовчі. Вона
починає виділятися не тільки в жовчні
капіляри, але й у кров. При цьому змен
шується надходження жовчі в кишки (гіпохолія).
Зазначені порушення зумовлюють появу та
ких змін показників пігментного обміну:
а) збільшується вміст у крові непрямого бі
лірубіну (порушується його захоплювання
гепатоцитами);
б) у крові з’являється значна кількість прямо
го білірубіну (результат надходження жов
чі у кров через анастомози між жовчними
і кров’яними капілярами);
в)
зменшується
вміст
стеркобіліногену
в калі - гіпохолічний кал',
г) у сечі з’являється прямий білірубін, який
фільтрується в нирках, — білірубінурія. Че
рез порушення кишково-печінкового кру
гообігу уробіліногену та реакцій його окиснення в гепатоцитах ця сполука може по
трапляти через нирки в сечу;
д) зменшення або повна відсутність стеркобі
ліногену в сечі.
Крім того, у крові й сечі виявляють жовчні
кислоти (холалемія іхолалурія).
II. Печінкові жовтяниці з ізольованими пору
шеннями процесів, що забезпечують виведення
білірубіну з організму. Вони можуть бути обу
мовлені:
1) порушеннями захоплення непрямого білі
рубіну гепатоцитами. Надходження непря
мого білірубіну з крові в печінкові клітини
пов’язано з транспортною функцією мемб
ранних Y- і Z-білків (лігандинів), яким при
таманна і функція ферменту глютатіон-S-
2)
»
»
»
трансферази. Якщо в генах, що кодують
ці білки, виникають мутації, які порушу
ють транспортування білірубіну, то розви
вається жовтяниця, при якій збільшується
вміст непрямого білірубіну в крові й від
повідно зменшується утворення стеркобі
ліногену в калі;
розладами кон’югації білірубіну. Зв’язу
вання білірубіну з глюкуроновою кисло
тою каталізується ферментом уридиндифосфоглюкуронілтрансферазою
(УДФГТ),
зокрема 1А1 ізоформою цього ензиму.
Описано цілий ряд порушень УДФГТ, які
лежать в основі т. зв. ензимопатичних
жовтяниць. До таких відносять:
фізіологічну жовтяницю новонародже
них. Її появу в перші дні після народжен
ня пов’язують з незрілістю ферментних
систем печінки і, зокрема, УДФГТ. Крім
того, додається ще кілька механізмів: по
силений гемоліз еритроцитів, що містять
фетальний гемоглобін (HbF), незрілість
механізмів транспорту білірубіну в гепатоцити і екскреторної функції печінкових
клітин. Утворення з часом достатньої кіль
кості УДФГТ веде до нормалізації обміну
жовчних пігментів, і жовтяниця через 1-2
тижні поступово зникає;
синдром Жільбера. Він виявляє себе не
значним підвищенням рівня непрямого
білірубіну в сироватці крові, що нерідко
стає випадковою знахідкою при біохі
мічному аналізі крові у дорослих людей.
Появу клінічних ознак жовтяниці можуть
провокувати тривале голодування, низь
кокалорійна дієта, інфекційні хвороби,
хірургічні втручання, вживання алкого
лю. Причиною такого спадково зумовле
ного синдрому є мутації в промоторній
ділянці гена УДФГТ1А1, унаслідок яких
зменшується експресія цього гена і утво
рюється менше, ніж потрібно, активного
ферменту. Брак УДФГТ веде до зменшен
ня кон’югації білірубіну й, відповідно,
до зростання вмісту непрямого білірубі
ну в сироватці крові. Синдром Жільбера
дуже поширений. За різними даними, від
5 до 10 % людей мають його ознаки;
синдром Кріглера — Найяра. Цей спад
ково зумовлений синдром існує у двох
варіантах.
310
Тип І синдрому має у своїй основі повну від
сутність УДФГТ через мутації в змістовній час
тині відповідного гена. Жовтяниця має тяжкий
перебіг, від самого народження характеризуєть
ся значним збільшенням вмісту непрямого білі
рубіну в сироватці крові. Хворі діти найчастіше
помирають на першому році життя через ура
ження головного мозку (білірубінова енцефалопатія). Трансплантація печінки є чи не єдиним
способом ефективного лікування.
Тип II менш злоякісний. Він зумовлений му
таціями, при яких істотно зменшується актив
ність УДФГТ (менше 20 % від нормальної). При
цьому гепатоцити здатні утворювати тільки моноглюкуронід білірубіну;
3) порушеннями екскреції білірубіну. Ви
ведення кон’югованого білірубіну з гепатоцитів у складі жовчі зазнає змін при
кількох спадково зумовлених синдромах.
Серед них:
синдром Дабіна — Джонсона. Його при
чиною є мутації гена MRP2 (multidrug
resistance 2), що кодує структуру білка переносника глюкуронідів білірубіну через
мембрану гепатоцитів у жовчні капіляри.
Через дефекти, що виникли, прямий білі
рубін не може повністю виводитися з жов
чю, і певна його кількість потрапляє в кров,
зумовлюючи жовтяницю;
синдром Ротора. У своїй основі має склад
ні спадково зумовлені зміни механізмів
екскреції білірубіну. За клінічними озна
ками схожий на попередній, відмінність
полягає лише в морфологічних змінах пе
чінкової тканини (відсутнє характерне для
синдрому Дабіна - Джонсона накопичен
ня патологічного пігменту в гепатоцитах).
Синдром має доброякісний перебіг, тип
спадкування автосомно-рецесивний.
Механічна (підпечінкова) жовтяниця. Причи
ною цього виду жовтяниці є механічна перешко
да відтоку жовчі. Це може бути:
1) здавлювання (компресія) жовчовивідних
шляхів ззовні (пухлина головки підшлун
кової залози, дія рубця);
2) їх закупорка (обтурація) каменем, гель
мінтами, густою жовчю.
Механічне перешкоджання відтоку жовчі
призводить до застою й підвищення тиску жов
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
чі, розширення й розриву жовчних капілярів
і надходження жовчі в кров як прямо, так і че
рез лімфатичні шляхи.
У зв’язку з цим виникають такі зміни показ
ників обміну жовчних пігментів:
а) збільшується вміст у крові прямого біліру
біну (гіпербілірубінемія)',
б) у крові з’являються жовчні кислоти (холалемія);
в) збільшується вміст у крові холестеролу
(гіперхолестеролемія).
Характерна
поява
модифікованих ліпопротеїнів (ліпопротеїн X), що мають атерогенні властивості
(див. главу 32);
г) у сечі з’являється білірубін (білірубінурія),
унаслідок чого вона набуває темного за
барвлення (“колір пива”); крім того, у ній
виявляють жовчні кислоти (холалурія).
Із сечі зникає стеркобіліноген;
д) у калі немає стеркобіліногену (безбарвний
кал).
Зазначені зміни пігментного обміну зумовлю
ють розвиток двох важливих клінічних синдро
мів, характерних для механічної жовтяниці: холемічного й ахолічного (див. нижче).
Таким чином, наведені вище характеристи
ки різних видів жовтяниць вказують на те, що
особливості порушень обміну жовчних пігмен
тів залежать від механізмів розвитку жовтяниці.
У табл. 35.2 подано основні біохімічні кри
терії, що дають можливість встановити вид
жовтяниці. При цьому для наочності й кращого
розуміння процесів вважаємо, що в ідеальній
нормі прямого білірубіну в сироватці крові не
має (хоча насправді певна його кількість у кров
потрапляє - процес виділення білірубіну в жовч
не є абсолютно досконалим).
Аналіз даних таблиці дає підстави розділити
всі жовтяниці залежно від характеру гіпербілірубінемії на три групи:
1) зі збільшенням у сироватці крові непря
мого (некон ’югованого) білірубіну. До цієї
категорії віднесемо гемолітичну жовтяни
цю і печінкові жовтяниці з ізольованими
порушеннями захоплення і кон’югації бі
лірубіну (синдроми Жільбера, Кріглера Найяра);
2) з появою у крові й сечі прямого (кон’ю
гованого) білірубіну. Такими є механічна
жовтяниця та печінкові жовтяниці з ізольо-
311
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
Таблиця 35.2
Біохімічні характеристики різних видів жовтяниці
ваними порушеннями екскреції білірубіну
(синдроми Дабіна - Джонсона, Ротора);
3) з одночасним збільшенням у сироватці
крові непрямого та появою в крові й сечі
прямого білірубіну. Цим критеріям відпо
відає печінково-клітинна жовтяниця.
Холемічний синдром
Холемічним, або синдромом хол ест азу, нази
вають сукупність клінічних ознак, зумовлену
надходженням компонентів жовчі (жовчних кис
лот, прямого білірубіну, холестеролу) у кров,
у зв’язку з порушенням формування і відтоку
жовчі.
Цей синдром супроводжує розвиток меха
нічної та печінково-клітинної жовтяниць (див.
вище). Його прояви пов’язані з дією на органи
і тканини основних компонентів жовчі.
1. Поява в крові жовчних кислот (холалемія) зумовлює:
а) розлади діяльності центральної нервової
системи. Вони виникають як наслідок
загальнотоксичної дії жовчних кислот
(загальна астенія, дратівливість, що змі
нюється депресією; сонливість удень
і безсоння вночі; головні болі, стомлюва
ність);
б) артеріальну гіпотензію, брадикардію.
їх розвиток пов’язаний з підвищенням
тонусу блукаючого нерва і прямою дією
жовчних кислот на синусно-передсердний вузол та кровоносні судини;
в) свербіж шкіри, що виникає в результаті
подразнення нервових закінчень жовч
ними кислотами;
г) множинні ушкодження і загибель клі
тин, обумовлені детергентною дією жов
чних кислот. Цим, зокрема, пояснюють
гемоліз еритроцитів, запалення і некрози
в різних органах і тканинах (печінковий
некроз, перитоніт, гострий панкреатит
та ін.);
д) появу жовчних кислот у сечі (холалурію).
2. Надходження у кров прямого білірубіну ви
кликає появу жовтого забарвлення шкіри
і слизових оболонок, тобто власне жовтя
ницю.
3. Збільшення вмісту в крові холестеролу —
гіперхолестеролемія - стає причиною аку
мулювання цієї сполуки клітинами шкіри,
312
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
унаслідок чого з’являються підшкірні від
кладання ліпідів — ксантоми. З невідомих
поки що причин починає утворюватися
аномальний ліпопротеїн. X, що має вираже
ну атерогенну дію.
Ахолічний синдром
Сукупність клінічних ознак, що виникають че
рез ненадходження жовчі в кишки у зв’язку
з порушеннями її формування та відтоку, нази
вають ахолічним синдромом.
Для цього синдрому характерні:
1) розлади перетравлювання і всмоктування
жирів. Вони обумовлені порушенням про
цесів емульгування жирів, зменшенням ак
тивності панкреатичної ліпази (жовч під
вищує активність цього ферменту); пору
шенням утворення міцел, що всмоктуються
в тонкій кишці. Наслідком зазначених змін є:
а) поява жиру в калі - стеаторея;
б) розлади всмоктування жиророзчинних
вітамінів, у результаті чого розвивають
ся гіповітамінози А, Е, К;
в) зменшення надходження в організм ненасичених жирових кислот, необхідних
для побудови фосфоліпідів клітинних
мембран;
2) порушення рухової функції кишок - ослаб
лення перистальтики і зменшення тонусу
кишок (закрепи). Жовч, як відомо, є фізіо
логічним стимулятором скорочень гладкої
мускулатури органів травної системи;
3) посилення процесів гниття і реакцій бро
діння в кишках у результаті зменшення
бактерицидної дії жовчі. Це призводить до
збільшення навантаження на антитоксичні
системи печінки;
4) зміни характеристик калу - знебарвлення,
стеаторея.
Дисхолія. Жовчнокам’яна хвороба
Дисхолією називають порушення фізично-хіміч
них властивостей жовчі, унаслідок чого вона на
буває літогенних властивостей, тобто здатності
утворювати камені (конкременти) у жовчному
міхурі й жовчних протоках. Результатом цього
є розвиток жовчнокам ’яної хвороби. За різними
даними, від 10 до 20 % населення розвинених
країн мають жовчні камені. У понад 80 % ви
падків вони нічим себе клінічно не проявляють
протягом десятків років.
За хімічною структурою розрізняють (а) холестеролові та (б) пігментні (містять білірубін)
жовчні камені. У країнах Європи і СІЛА понад
80 % припадає на холестеролові.
Етіологія. До факторів ризику жовчнокам’яної
хвороби відносять:
а) вік і стать. Якщо в осіб віком до 40 років
камені виявляють у 5-6 %, то цей показних
у людей старечого віку (понад 80 років)
становить 25-30 %. У жінок європейських
країн частота жовчнокам’яної хвороби
у 3-4 рази вища, ніж у чоловіків;
б) спадкову схильність. Встановлено зв’язок
між поліморфізмом цілого ряду генів, що
мають стосунок до обміну холестеролу
і екскреторної функції печінки, з утворен
ням холестеролових жовчних каменів. Опи
сано випадки сімейної жовчнокам’яної хво
роби;
в) нераціональне харчування (їжа з великим
вмістом холестеролу і рафінованих вугле
водів), ожиріння;
г) інфекційно-запальні процеси в жовчному
міхурі та жовчних протоках, застій жовчі
(холестаз).
Патогенез. У розвитку холестеролових каменів
мають значення дві групи чинників: (І) з боку
жовчі - зміни її складу і фізично-хімічних влас
тивостей (дисхолія) і (II) з боку жовчного міхура
та жовчовивідних шляхів - порушення їхньої ру
хової та секреторної активності.
І.
Фактори жовчі. Жовч набуває літогенності
при її перенасиченні холестеролом, а точніше,
при зниженні холато-холестеролового і леци
тин-хол естеролового індексів (відношення від
повідно жовчних кислот і лецитину до холесте
ролу жовчі). Це може бути зумовлено:
1) зменшенням вмісту жовчних кислот
у жовчі через: (а) порушення печінковокишкового їх кругообігу при патології ки
шок і зміні їхньої мікрофлори; (б) пригні
чення синтезу жовчних кислот у печінці;
(в) прискорення їх всмоктування слизовою
оболонкою запаленого жовчного міхура;
2) зменшенням вмісту лецитину в жовчі. Се
ред причин цього недостатнє надходження
313
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
в організм ненасичених жирових кислот
і деяких незамінних амінокислот (метіо
ніну), потрібних для синтезу фосфоліпідів;
3) збільшенням синтезу холестеролу в гепатоцитах і посиленим виведенням його
з жовчю при гіперхолестеролемії.
Жовчні кислоти і лецитин забезпечують за
вислий (розчинний) стан холестеролу в жовчі.
При зменшенні їх кількості, а також при зрос
танні концентрації холестеролу останній випа
дає в осад.
Для наступного процесу - формування власне
каменів - потрібно, щоб осадження холестеролу
відбувалося на певних структурах, що відігра
ють роль матриці (нуклеатора). Такими структу
рами виступають мікропреципітати неорганіч
них і органічних кальцієвих солей.
Інфекція, застій жовчі сприяють процесу
утворення каменів, тому що змінюють фізич
но-хімічні властивості жовчі: (a) pH зміщується
в кислий бік; (б) знижується розчинність солей
і вони випадають в осад; (в) відбувається коа
гуляція білків, що вивільнюються зі злущених
клітин епітелію жовчовивідних шляхів.
II. Фактори жовчного міхура і жовчовивід
них шляхів. До таких можна віднести:
1) порушення рухової активності жовчного
міхура
гіпотонічного
(гіпокінетичного)
типу. Вони виникають в результаті ура
жень мускулатури цього органа (напри
клад, при запаленні) або є наслідком роз
ладів нервової та гуморальної регуляції:
зменшення тонусу блукаючого нерва, при
гнічення утворення холецистокініну. При
зменшенні тонусу і моторики жовчного
міхура створюються сприятливі для фор
мування каменів умови: жовч надовго за
тримується в міхурі, її компоненти погано
перемішуються;
2) посилення секреції слизу. Слиз, який секретується епітеліальними клітинами жовч
ного міхура та жовчовивідних шляхів, має
властивість захоплювати кристали холес
теролу, завдяки чому сприяє їхній агрегації
та утворенню каменів.
Ще одним різновидом жовчних каменів є піг
ментні камені. Так називають конкременти, що
містять білірубін і менше ЗО % холестеролу.
Розрізняють чорні й коричневі пігментні камені.
Чорні, як і холестеролові, формуються в жовч
ному міхурі, їх найчастіше виявляють у людей
похилого віку. Вони складаються в основному
з кальцієвих солей білірубіну, фосфорної (фос
фатів) і вугільної кислоти (карбонатів). Утворен
ня таких каменів характерне для хронічного ге
молізу еритроцитів при спадкових гемолітичних
анеміях, імплантації штучних клапанів серця.
Коричневі камені локалізуються головним чи
ном у жовчних протоках. Вони містять кальцієві
солі білірубіну, пальмітинової і стеаринової жи
рових кислот, холестерол. Утворення коричневих
каменів пов’язують з інфекціями (кишкова палич
ка, опісторхоз, лямбліоз та ін.). Під впливом мі
кробних глюкуронідаз відбувається декон’югація
прямого білірубіну жовчі, що веде до осадження
нерозчинного некон’югованош білірубіну з на
ступним формуванням жовчних каменів.
Основні клінічні прояви. Жовчні камені неза
лежно від їхнього хімічного складу стають при
чиною:
а) порушень жовчовиділення і розвитку меха
нічної жовтяниці (див. вище);
б) запалення жовчного міхура і жовчовивід
них шляхів—холециститу, холангіту,
в) больового синдрому, зумовленого спастич
ними скороченнями гладкої мускулатури
жовчного міхура, - печінкової коліки',
г) нападів гострого панкреатиту при об
струкції каменями спільної частини за
гальної жовчної та панкреатичної вивідних
проток (див. главу 34).
ПОРУШЕННЯ
ГЕМОДИНАМІЧНИХ ФУНКЦІЙ
Гемодинамічними називають функції печінки,
які забезпечують її участь у здійсненні систем
ного кровообігу. До них відносять:
а) колекторну функцію. Печінка збирає через
систему ворітної вени кров з великого ба
сейну —від органів черевної порожнини. Че
рез печінку проходить 30-35 % хвилинного
об’єму крові, що становить 1,5-1,8 л/хв;
б) депонування крові. У печінці може місти
тися до 700 мл крові, тимчасово виведеної
з кровообігу. При необхідності (наприклад,
після крововтрати) ця кров мобілізується;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
314
в)
участь у підтриманні тонусу кровоносних
судин через синтез білків, що є попередни
ками біологічно активних речовин - регу
ляторів артеріального тиску. Ідеться, зокре
ма, про синтез ангіотензиногену, з якого
утворюється ангіотензин II. Порушення
гемодинамічних функцій печінки виявля
ють себе розвитком синдрому портальної
гіпертензії.
Синдром портальної гіпертензії. Підвищення
тиску крові у системі ворітних судин - порталь
на гіпертензія - розвивається в результаті пору
шення відтоку крові з органів черевної порож
нини по цих судинах.
Залежно від того, де знаходиться перешкода
відтоку крові, виділяють такі форми портальної
гіпертензії:
1) підпечінкову - перешкода в стовбурі або
великих гілках ворітної вени (емболи,
здавлювання пухлиною);
2) внутрішньопечінкову - причина порушень
відтоку крові в самій печінці (тривалий
спазм гладком’язових сфінктерів сину
соїдних капілярів; здавлювання дрібних
печінкових вен вузлами регенеруючих гепатоцитів при ушкодженні печінки, її ци
розі);
3) надпечінкову — перешкода локалізована
у позаорганних відділах печінкових вен
або в нижній порожнистій вені проксимальніше місця впадіння в неї печінкових
вен. Сюди ж відносять портальну гіпер
тензію, що виникає при збільшенні тис
ку в системі нижньої порожнистої вени
в умовах недостатності правого шлуночка
серця.
До основних проявів синдрому портальної гі
пертензії відносять такі.
1. Розвиток колатерального кровообігу в ре
зультаті розкриття портокавальних анастомозів. Це зумовлює появу таких ознак:
а) варикозного розширення вен стравоходу
і кардіальної частини шлунка;
б) шлунково-кишкових кровотеч, причиною
яких є ушкодження варикозно розшире
них вен;
в) розширення підшкірних вен передньої
грудної та черевної стінки (“голова ме
дузи”);
г) скидання крові з ворітної вени в порож
нисті в обхід печінки, що викликає ін
токсикацію, а у важких випадках — роз
виток екзогенної (портокавальної, або
шунтової) печінкової коми (див. вище).
2. Гепатолієнальний синдром. Його важли
вими складовими є спленомегалія і гіперспленізм.
Спленомегалія - це збільшення розмірів селе
зінки. При портальній гіпертензії вона виникає
в результаті застою крові.
Гіперспленізмом називають збільшення функ
ціональної активності селезінки. Він виявляє
себе посиленим руйнуванням формених еле
ментів крові. Характеризується анемією, лей
копенією і тромбоцитопенією. В основі цього
явища лежить збільшення фагоцитарної актив
ності макрофагів селезінки в умовах уповіль
нення циркуляції крові.
3. Асцит. Про суть цього явища мова піде
далі.
4. Гепаторенальний синдром. Виявляє себе
порушеннями фільтраційної здатності нир
кових клубочків при збереженні функцій
канальцевого епітелію (див. главу 36). При
чиною цього, цілком імовірно, є зменшення
ниркового кровообігу у зв’язку зі зменшен
ням об’єму циркулюючої крові та зміною
тонусу кровоносних судин, що спостеріга
ється при розладах гемодинамічних функ
цій печінки.
Асцит. Асцитом називають накопичення віль
ної рідини (як правило, транссудату) у черевній
порожнині.
Причинами асциту можуть бути:
а) портальна гіпертензія різного походження
(див. вище);
б) набряки при хронічній недостатності сер
ця, захворюваннях нирок, аліментарній
дистрофії;
в) порушення відтоку лімфи грудною прото
кою (її поранення, здавлювання);
г) ураження очеревини пухлинним або тубер
кульозним процесом (асцит - перитоніт).
Асцитична рідина за своїм характером буває
зазвичай серозною, значно рідше - геморагічною.
У патогенезі асциту, зумовленого хворобами
печінки, мають значення такі механізми:
ГЛАВА 35. ПАТОФІЗІОЛОГІЯ ПЕЧІНКИ
1) гідростатичний. Пов’язаний з підвищен
ням тиску крові в капілярах судин ворітної
системи;
2) онкотичний. Обумовлений зменшенням
білоксинтетичної функції печінки, вна
слідок чого розвивається гіпопротеїнемія
й падає онкотичний тиск крові;
3) затримка натрію в організмі. Пов’язана зі
збільшенням вмісту альдостерону в крові.
Причиною цього є активація ренін-ангіотензинової системи (застій крові в судинах
ворітної системи —*> зменшення венозного
повернення —У падіння хвилинного об’єму
315
серця ~+ гіпоксія нирок —» вивільнення
реніну). Крім того, у зв’язку з розладами
метаболічних функцій печінки може по
рушуватися інактивація альдостерону, що
рівнозначно його гіперпродукції;
4) лімфогенний механізм. У зв’язку з пору
шенням лімфовідтоку відбувається пе
рехід багатої білками лімфи в черевну
порожнину. Це викликає підвищення он
котичного тиску рідини черевної порож
нини з наступним виходом сюди води із
кровоносних судин та інтерстиціального
простору.
ГЛАВА 36
Патофізіологія
нирок
Основною функцією нирок, як центральних ор
ганів видільної системи, є забезпечення сталос
ті внутрішнього середовища організму, тобто
гомеостатична функція.
Нирки причетні до підтримання таких показ
ників гомеостазу:
1) об’єму позаклітинної рідини (ізоволемії).
В умовах патології нирок цей показник
може змінюватися як в один (гіпергідрія),
так і в другий (гіпогідрія) бік;
2) осмотичного тиску цієї рідини (ізотонії).
При ураженнях нирок можуть розвиватися
як гіперосмія, так і гіпоосмія;
3) іонного складу плазми крові та інтерстиціального середовища (ізоіонії). Хвороби
нирок часто супроводжуються порушен
ням балансу електролітів (натрію, калію,
хлору та ін.) у позаклітинній рідині - дисіонією;
4)
кислотно-основної рівноваги (ізогідрії).
Найчастіше патологія нирок супроводжу
ється розвитком негазового ацидозу,
5) хімічного складу плазми крові. Нирки,
з одного боку, виводять кінцеві продукти
метаболізму і сполуки екзогенного похо
дження, а з другого - зберігають необхідні
організму речовини - білки, амінокислоти,
глюкозу. З огляду на це при ураженнях ни
рок відбувається накопичення продуктів
обміну азотовмісних речовин (азотемія)
і втрачаються важливі хімічні сполуки виникають
гіпопротеїнемія, гіпоаміноцидемія, гіпоглікемія',
6) артеріального тиску. Одним із важливих
проявів ниркових недуг є розвиток артері
альної гіпертензії',
7) кисневої ємності крові. Контроль цього
показника здійснюється через регуляцію
еритропоезу нирковими еритропоетинами
(див. нижче). Патологічні процеси в нирках
часто супроводжуються недостатнім утво
ренням останніх і розвитком анемії.
Підтримання зазначених вище показників
гомеостазу здійснюється завдяки двом групам
ниркових функцій: (1) сечоутворенню (екскре
торна функція) і (2) вивільненню в кров гор
монів, ферментів, біологічно активних сполук
(інкреторна функція).
Екскреторна функція
нирок
складається
з трьох пов’язаних між собою процесів:
1) клубочкової фільтрації',
2) кансиїьцевоїреабсорбції;
3) канальцевої секреції.
До інкреторних функцій відносять: (1) секре
цію реніну юкстагломерулярним апаратом ни
рок та ниркових депресорних факторів', (2) ви
вільнення еритропоетинів та інгібіторів ери
тропоезу', (3) утворення гормонально активної
форми вітаміну D.
36.1. МЕХАНІЗМИ ПОРУШЕНЬ
ФУНКЦІЙ НИРОК
Залежно від рівня, на якому розвиваються пору
шення, що ведуть до розладів ниркових функ
цій, можна виділити три групи патологічних
змін: (1) преренальні; (2) ренальні та (3) постренальні (рис. 36.1).
Преренальні порушення. До цього варіанта від
носять порушення ниркових функцій, що вини
кають унаслідок розладів ниркового кровообігу.
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
317
ис. 36.1. Три рівні порушень ниркових функцій
Кровообіг у нирках виконує дві важливі
функції:
1) специфічну, яка забезпечує фільтрацію
плазми крові в першій мережі капіляр
них судин (у клубочках) і повернення на
зад у кров реабсорбованих компонентів.
Останній процес здійснюється у другій
мережі капілярів, що обплутують канальці нефронів;
2) неспецифічну (нутритивну), суть якої по
лягає в забезпеченні клітин нирок киснем
і поживними речовинами.
Інтенсивність ниркового кровообігу в нормі
дуже висока (близько 1300 мл/хв, або 25 % хви
линного об’єму крові в стані спокою) і визна
чається його специфічною функцією (участю
у фільтрації та реабсорбції). Оскільки
Он =
а R=
87/
де QH, - об’ємна швидкість ниркового кровообі
гу:
Р - тиск на початку, Jа7 Р - наприкінці
J 7 apm
г сисвен
теми перфузії ниркових судин; R - опір ниркових
судин; у - в’язкість крові; І - довжина судин; г радіус судин, то зменшення інтенсивності нир
кового кровообігу (Q) може бути обумовлене:
1) зменшенням артеріального тиску (Ра т)
нижче 80 мм рт. ст. У нирках у діапазоні
величин артеріального тиску (AT) від 80
до 200 мм рт. ст. спрацьовують механізми
міогенної авторегуляції кровообігу (ефект
Бейліса): якщо AT підвищується, приносні артеріоли спазмуються, при зниженні
AT, навпаки, ці судини розширюються,
і це забезпечує сталість ниркового крово
обігу незалежно від коливань AT. Якщо ж
AT падає нижче рівня 80 мм рт. ст., то за
значений вище механізм стає неефектив
ним, і кровообіг в нирках зменшується.
Така ситуація є характерною для всіх видів
шоку (див. главу 20);
2) збільшенням венозного тиску ( Р ) . При
чиною цього можуть бути загальні пору
шення (наприклад, правошлуночкова не
достатність серця, що веде до збільшення
центрального й периферичного венозного
тиску) і місцеві розлади - венозна гіпере
318
мія (наприклад, при інтерстиціальному за
паленні ниркової тканини);
3) ішемією нирок (зменшенням радіуса су
дин), що буває при атеросклерозі й артері
альній гіпертензії;
4) збільшенням в’язкості крові (наприклад,
при ДВЗ-синдромі).
Усі зазначені порушення спричиняються до
зменшення фільтраційного тиску в ниркових
клубочках - це виявляє себе зменшенням швид
кості клубочкової фільтрації, а отже, ознаками
недостатності нирок.
Ренальні порушення. До таких відносять (І) по
рушення клубочкової фільтрації та (II) розлади
канальцевої реабсорбції і секреції.
І. Порушення клубочкової фільтрації. Інтен
сивність (швидкість) цього процесу (ШКФ) ви
значається рівнянням:
де ЕФТ - ефективний фільтраційний тиск; Кф коефіцієнт фільтрації. З огляду на це можна ви
ділити дві групи механізмів зменшення показ
ника ШКФ:
1) зменшення ЕФТ. Оскільки
де Рк - гідростатичний тиск у капілярах клубоч
ків; Ро - онкотичний тиск крові; Р — гідроста
тичний тиск у капсулі клубочків - так званий
тканинний тиск, то зменшення ШКФ може бути
обумовлено:
а) зниженням гідростатичного тиску в капі
лярах клубочків (PJ унаслідок загальних
і місцевих розладів кровообігу (див. вище);
б) збільшенням онкотичного тиску крові (Р),
що буває, наприклад, при зневодненні;
в) підвищенням тканинного тиску в нирках
(jР ). Причиною цього можуть бути пе
решкоди відтоку фільтрату або сечі при
ушкодженні канальців (закупорка канальців некротичними масами і циліндрами),
при інтерстиціальному запаленні (здав
лювання канальців набряковою рідиною),
при порушеннях прохідності сечоводів
і сечовивідних шляхів (камені, стриктури,
здавлювання пухлиною);
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
2) зменшення коефіцієнта фільтрації (К,).
Величина цього параметра залежить від
площі поверхні клубочків, що беруть
участь у фільтрації (кількості функціону
ючих нефронів), і від проникності стінок
клубочкового фільтра, на яку впливають
кількість пор на одиницю площі поверхні,
їхня геометрія і діаметр; електричний за
ряд базальної мембрани клубочків.
З урахуванням зазначеного можна виділити
дві основні причини зниження фільтраційної
здатності клубочків:
а) зменшення загальної площі фільтрації уна
слідок зменшення кількості діючих нефро
нів. Людина від народження має в кожній
нирці по 1 млн нефронів. Істотні порушен
ня фільтраційної здатності нирок виника
ють при зменшенні кількості нефронів до
ЗО %, тобто коли їхня загальна кількість
в обох нирках стає меншою за 600 тисяч;
б) зменшення проникності стінки клубочко
вого фільтра, що спостерігається при по
товщенні мембрани (наприклад, при діабе
тичній нефропатії), склерозуванні клубоч
ків (наслідок гломерулонефриту), забиванні
пор фільтра білками (гемоглобіном, міогло
біном відповідно при гемолізі еритроцитів
і роздавлюванні м’язової тканини).
Про здатність нирок здійснювати фільтрацію
роблять висновок на підставі величини швид
кості клубочкової фільтрації (ШКФ) - так по
значають об’єм плазми крові, що фільтрується
в ниркові канальці за одиницю часу.
ШКФ визначають за кліренсом інуліну об’ємом плазми, що повністю очищається від
цієї речовини нирками за 1 хв.
де С.п - кліренс інуліну; U.n - концентрація іну
ліну в сечі; Р.п - концентрація інуліну в плазмі;
V - діурез за 1 хв.
У нормі С.п, а отже, і ШКФ дорівнюють 100—
140 мл/хв. Зменшення ШКФ є основним показ
ником розвитку недостатності нирок. Так, при
гострій нирковій недостатності ШКФ швидко
зменшується від 100-140 до 10-1 мл/хв, а при
хронічній — поступово від 100-140 до 30 мл/
319
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
36.2. Особливості процесів реабсорбції та секреції в різних відділах нефрону
хв (початкова стадія), від ЗО до 10 мл/хв (ран
ня поліурична стадія), від 10 до 5 мл/хв (пізня
олігурична стадія), нижче 5 мл/хв (термінальна
стадія).
II. Порушення канальцевої реабсорбції та се
креції. Процеси реабсорбції і секреції тісно
пов’язані між собою як своїми механізмами, так
і місцем, де вони відбуваються.
Канальці різних відділів нефрону мають різні
властивості щодо здійснення реабсорбції й се
креції (рис. 36.2). Так, у проксимальних звивис
тих канальцях відбуваються процеси облігатної
(нерегульованої) реабсорбції. Тут реабсорбуються вода (80-85 % від об’єму фільтрату), ка
тіони натрію, калію, кальцію, магнію; аніони
хлору, фосфатів; усі білки, що в нормі фільтру
ються, амінокислоти, глюкоза. Клітини про
ксимальних канальців секретують іони водню,
а також цілий ряд екзогенних речовин (парааміногіпурову кислоту, діодраст та ін.), у тому
числі й лікарські препарати (наприклад, пеніци
лін). У петлі Генле за участю зворотно-протитокового механізму, що спочатку концентрує
первинну сечу, а потім її розбавляє, реабсорбується вода (у низхідному коліні) та іони натрію
і хлору (у висхідній частині). У дистальних зви
вистих канальцях відбувається факультатив
на (регульована) реабсорбція води та іонів на
трію й секреція іонів калію та водню. І нарешті,
у збиральних трубочках здійснюється остаточ
на реабсорбція води.
В основі порушень функцій ниркових каналь
ців можуть лежати такі механізми:
1) ушкодження клітин канальцевош епіте
лію. Воно може бути зумовлене ішемією,
нефротропними отрутами, дією фізичних
(радіація) і біологічних (інфекція) факто
рів. При ушкодженні канальців для пору
шення ниркових функцій мають значення:
а) вихід фільтрату через ушкоджені ка
нальці в інтерстицій, що веде до збіль
шення тканинного тиску та зменшення
клубочкової фільтрації;
б) обтурація канальців некротичними ма
сами і циліндрами, що також зумовлює
зменшення ефективного фільтраційного
тиску;
2) зменшення активності ферментів і транс
портних білків, що беруть участь у про
цесах реабсорбції й секреції. Це можуть
бути спадково обумовлені дефекти систем
транспорту глюкози (ренальна глюкозурія),
амінокислот
(аміноацидурія),
фосфатів
(фосфатний нирковий діабет)', складні
поєднані порушення реабсорбції глюкози,
амінокислот, бікарбонату і фосфатів (син
дром Фанконі). Можливі й набуті розлади
транспортних систем, що забезпечують
320
реабсорбцію і секрецію. Наприклад, при
отруєнні флоридзином, що пригнічує гек
сокіназу та глюкозо-б-фосфатазу, розвива
ється ренальна глюкозурія;
3) порушення енергозабезпечення. їх причи
ною можуть бути гіпоксія, голодування, гі
повітамінози, зменшення активності фер
ментів енергетичного обміну. Усі ці чин
ники ведуть до дефіциту АТФ у клітинах
ниркових канальців. При цьому стражда
ють енергозалежні механізми реабсорбції
і секреції (первинний і вторинний актив
ний трансмембранний транспорт, ендоі екзоцитоз);
4) надлишок речовин, що реабсорбуються.
Він зумовлює функціональне переванта
ження систем зворотного транспорту. Цей
механізм стосується так званих порогових
речовин, до яких належать глюкоза та бі
карбонати. Якщо концентрація цих спо
лук у крові перевищує пороговий рівень
(для глюкози - 10 ммоль/л, для бікарбона
тів - 27 ммоль/л), то їхній надлишок виво
диться із сечею - розвивається глюкозурія
(при цукровому діабеті), сеча стає лужною
(при алкалозах);
5) порушення гуморальної регуляції процесів
факультативної реабсорбції. Вони можуть
бути пов’язані зі змінами вмісту в крові
альдостерону, передсердного натрійуретичного гормону, вазопресину, паратгормону.
Постренальні порушення. Цей варіант змін
ниркових функцій пов’язаний з порушеннями
процесу виведення сечі по сечовивідних шля
хах. Основна причина цього — один із двох ме
ханічних чинників:
(1) обтурація сечовивідних шляхів каменями,
що є характерним для сечокам’яної хворо
би. Сутність цієї недуги ще стане предме
том нашого обговорення (див. далі);
(2) здавлювання (компресія) сечовивідних
шляхів ззовні. Воно може виникати вна
слідок стриктур сечоводів, при аденомі
передміхурової залози й інших пухлинах
органів малого таза.
Зазначені причини перешкоджають відтоку
сечі та викликають унаслідок цього підвищен
ня тиску рідини в капсулі клубочків. Це своєю
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
чергою спричиняється до зниження ефективно
го фільтраційного тиску і зменшення швидкості
клубочкової фільтрації.
ОСНОВНІ КЛІНІЧНІ
ПРОЯВИ УРАЖЕНЬ НИРОК
Порушення екскреторної та інкреторної функцій
нирок виявляють себе великою кількістю клініч
них ознак, різні поєднання яких між собою зу
мовлюють розвиток так званих великих нирко
вих синдромів, що ще стануть предметом нашого
обговорення. Тут ми зупинимося на походженні
та механізмах основних клінічних проявів, з яких
власне й складається картина хвороб нирок.
Кількісні та якісні зміни сечі
До кількісних змін сечі відносять:
1) олігурію та анурію;
2) поліурію;
3) ніктурію;
4) гіпо- та ізостенурію,
а до якісних - появу патологічних компонен
тів:
протеїнурію;
* гематурію;
циліндрурію;
лейкоцитурію (піурію).
Олігурія та анурія. В умовах нормальної жит
тєдіяльності з організму щодоби має виводитися
із сечею 700 мосм осмотично активних речовин
(сечовина, електроліти тощо). Щоб вивести таку
їхню кількість при максимально можливій осмоляльності сечі (1000 мосм/кг), необхідно як мі
німум 700 мл сечі на добу. Такий добовий об’єм
сечі одержав назву облігатного діурезу, або облігатного об’єму. Той об’єм сечі, що перевищує
700 мл/добу, називають факультативним.
Олігурія — це зменшення добового діурезу
нижче облігатного об’єму, тобто менше 700 мл/
добу. Анурія — це повна відсутність діурезу.
Олігурію не слід плутати з антидіурезом зменшенням діурезу до облігатного об’єму.
Антидіурез виникає не в результаті зменшення
швидкості клубочкової фільтрації, як олігурія,
а внаслідок посилення до максимально можли
вого рівня факультативної реабсорбції води (на
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
приклад, під впливом вазопресину). При цьому
сеча стає гіпертонічною.
Причиною олігурії є порушення клубочкової
фільтрації. Олігурія виникає тоді, коли під дією
преренальних, ренальних і постренальних фак
торів (див. вище) швидкість клубочкової філь
трації стає нижче 10 мл/хв.
Олігурія спричиняється до:
1) збільшення об’єму позаклітинної рідини гіпергідрії (див. главу 27);
2) підвищення в організмі рівня осмотично
активних речовин. Зокрема, розвиваються
гіпернатріємія, гіперкаліємія\
3) накопичення в крові кінцевих продуктів
обміну речовин - азотемії та пов’язаної
з нею інтоксикації.
Поліурія. Збільшення добового діурезу понад
1,8 л називають поліурією. У людини макси
мально можливий діурез за умови, що він не
осмотичний, дорівнює 25 л/добу, що становить
15 % від об’єму профільтрованої води.
Причинами поліурії можуть бути позаниркові
(психогенна полідипсія, порушення водно-со
льового обміну та його регуляції, наприклад,
нецукровий діабет) і ниркові (поліуричні стадії
гострої та хронічної недостатності нирок) фак
тори.
Залежно від механізмів розвитку виділяють
такі види поліурії.
1. Водний діурез. Він обумовлений зменшен
ням факультативної реабсорбції води. Ви
никає при водному навантаженні, нецукровому діабеті (див. главу 37). Сеча при такій
поліурії гіпотонічна, тобто містить мало
осмотично активних речовин.
2. Осмотичний діурез (салурез). Його причи
ною є збільшення вмісту в сечі осмотично
активних речовин, що не реабсорбувалися.
Це спричиняється до затримки води в нир
кових канальцях і вторинного порушення
її реабсорбції. Поліурія цього типу розви
вається при:
а)
порушенні реабсорбції електролітів.
Якщо в проксимальних звивистих ка
нальцях і висхідній частині петлі Генле
пригнічується активний транспорт іонів
натрію та хлору (порушується концен
траційна здатність нирок), то це веде до
зменшення осмотичного тиску в інтерстиції і його підвищення в рідині каналь-
321
ців. Як результат, зменшується пасивний
перехід води з канальців в інтерстицій
на рівні проксимальних звивистих ка
нальців і низхідного коліна петлі Генле —
кількість вторинної сечі зростає, при
цьому вона є майже ізотонічною;
б) збільшенні вмісту в первинній сечі так
званих трогових речовин (наприклад,
глюкози при цукровому діабеті, аміно
кислот і сечовини при дієті з високим
вмістом білків);
в) дії екзогенних речовин, що погано реабсорбуються (манітол) або порушують ре
абсорбцію електролітів (салуретики).
В умовах максимального осмотичного діуре
зу виділення сечі може досягати 40 % величини
клубочкової фільтрації. Великий осмотичний
діурез спричиняється до зневоднення (гіпогідрії) (див. главу 27).
3. Гіпертензивний діурез. Розвиток цього
виду поліурії пов’язаний з артеріальною
гіпертензією.
Підвищення
кров’яного
тиску веде до збільшення швидкості руху
крові в прямих судинах мозкового шару ни
рок (вони йдуть паралельно колінам петлі
Генле), що у свою чергу спричиняється до
посилення конвекційного транспорту ре
човин. Саме цей транспорт, а не дифузія,
стає провідним у переміщенні електролітів
та інших речовин із інтерстицію ниркової
тканини в кров. Наслідком посилення кон
векційного транспорту стає “вимивання”
натрію, хлору, сечовини з інтерстицію, що
веде до зниження осмотичного тиску поза
клітинної рідини й зменшення в результаті
цього реабсорбції води в низхідній ділянці
петлі Генле - розвивається поліурія.
Ніктурія. Суть цієї патологічної ознаки поля
гає в тому, що нічна частина діурезу перевищує
денну.
У нормі 60-80 % добової кількості сечі виді
ляється в період з 8 до 20 год., тобто відношен
ня нічного діурезу до денного становить 1:2.
Це пояснюють тим, що саме вдень в організм
надходять вода та їжа, а вночі у здорових людей
знижуються до нижньої межі норми артеріаль
ний тиск і швидкість клубочкової фільтрації,
а реабсорбція води в канальцях досягає макси
мально можливих рівнів (до 99,7 %).
r
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
322
При ніктурії нічна порція сечі може більше
ніж удвічі перевищувати денну. Залежно від
причин виділяють:
1) серцеву ніктурію, що розвивається при
серцевій недостатності. Удень у хворих
збільшуються навантаження на серце
й приймання води, що веде до застою кро
ві й затримки води в тканинах (набряки).
Уночі в горизонтальному положенні по
ліпшується венозний відтік і зменшується
навантаження на серце. Це викликає ви
ділення
передсердного натрійуретичного
гормону (атріопептину), збільшення діуре
зу та зменшення набряків;
2) ниркову ніктурію — характерну для уражен
ня нирок. Її пояснюють поліпшенням уночі
порушеного тканинного кровообігу. У ре
зультаті прискорюється рух крові по суцинах
нирок, розвивається гіпертензивний діурез.
Гіпостенурія та ізостенурія. При порушенні
здатності нирок концентрувати сечу розвиваєть
ся гіпостенурія - зменшення відносної густини
сечі до 1012-1006 з незначними змінами цієї
густини протягом доби (рис. 36.3).
Поєднання гіпостенурії з поліурією свід
чить про ушкодження канальців при відносно
достатній функції клубочків. Якщо ж гіпо
стенурія виникає на тлі олігурїї, то це ознака
ушкодження всіх структур нефронів (канальців
і клубочків).
При повній втраті нирками здатності концен
трувати і розводити сечу розвивається ізосте
нурія, при якій відносна густина сечі дорівнює
густині фільтрату, тобто 1010, і не міняється
протягом доби (монотонний діурез).
Ізостенурія є ознакою дуже тяжких порушень,
при яких ниркові канальці, по суті, перетворю
ються у звичайні трубки, що проводять фільтрат
без будь-яких його змін у ниркові миски.
Протеїнурія. У нормі в нирках фільтрується
30-50 г білка за добу. Увесь він реабсорбується
в проксимальних звивистих канальцях, а тому
в кінцевій сечі його не повинно бути.
Виділення білка із сечею — протеїнурія є патологічною ознакою. В основі її розвитку
можуть лежати такі механізми:
1) збільшення проникності клубочкового
фільтра у зв’язку з ураженням базальної
мембрани (клубочкова протеїнурія)',
2) зменшення канальцевої реабсорбції білка,
що профільтрувався (канальцева протеїнурія);
3) патологічне надходження білка у просвіт
канальців з ушкоджених клітин тубулярного епітелію або з перитубулярної лімфа
тичної рідини (секреторна протеїнурія).
Густина сечі
Рис. Зб.З. Добові коливання густини сечі в нормі, при гіпо- та ізостенурії. І - норма, II - слабке обмеження,
III - гіпостенурія, IV - ізостенурія
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
Протеїнурія може бути селективною, коли
в сечі виявляють тільки низькомолекулярні білки,
і неселективною, для якої характерна поява в сечі
як низько-, так і високомолекулярних білків.
За ступенем селективності розрізняють нефротичний тип протеїнурії (у сечі тільки альбу
міни або альбуміни+а-глобуліни) і нефритичний тип (у сечі визначаються всі класи білків
плазми крові - альбуміни, а-, (3- і у-глобуліни).
Гематурія. Поява в сечі крові - гематурія може бути обумовлена:
1) ушкодженням клубочкового фільтра і над
ходженням еритроцитів у первинну сечу.
При цьому в кінцевій сечі виявляють “ви
лужені”, тобто гемолізовані еритроцити;
2) ушкодженням сечовивідних шляхів. Ерит
роцити в сечі в такому разі будуть “свіжи
ми”, незміненими.
Лейкоцитурія. Якщо при мікроскопічному ви
вченні сечі кількість лейкоцитів у полі зору пе
ревищує 5, то таку ознаку називають лейкоцитурією. Якщо кількість лейкоцитів у сечі дуже
велика і серед них є зруйновані (гній), то в тако
му разі мову ведуть про піурію.
Основна причина лейкоцитурії — запальні
процеси в нирковій тканині та сечовивідних
шляхах.
Циліндрурія. Циліндри є зліпками ниркових
канальців. Вони утворюються при ушкодженні
епітелію канальців і складаються з осаджено
го білка і загиблих клітин. Залежно від будови
розрізняють гіалінові, зернисті, соскоподібні та
інші циліндри.
Циліндрурія є ознакою патологічних процесів
у нирках, що супроводжуються ураженням тубулярного епітелію.
Інтоксикація
При ураженнях нирок у крові можуть накопичу
ватися токсичні речовини - кінцеві продукти об
міну речовин, що в нормі виводяться з організму
із сечею. Причиною цього є гостра та хронічна
ниркова недостатність, мова про яку піде далі.
В основі інтоксикації лежать порушення про
цесів клубочкової фільтрації. Перші її ознаки
з’являються, коли швидкість клубочкової філь
трації (ШКФ) стає нижче 50 мл/хв, а виражені
323
клінічні симптоми - при падінні ШКФ нижче
10 мл/хв.
Нині відомо понад 200 речовин, здатних ви
кликати інтоксикацію при недостатності нирок.
Найбільше значення мають: (а) сечовина; (б) се
чова кислота; (в) похідні гуанідину - креатин,
креатинін,
метилгуанідин,
диметилгуанідин
та ін.; (г) ароматичні сполуки - феноли, індол,
ароматичні аміни; (д) аліфатичні аміни; (е) низь
комолекулярні пептиди.
Ознакою накопичення зазначених токсичних
речовин є азотемія - збільшення вмісту в крові
залишкового азоту.
Клінічно інтоксикація при ураженнях нирок
виявляє себе уремічним синдромом. Його прояви
пов’язані з ураженням практично всіх органів
і систем організму. Однак на перший план висту
пають ознаки токсичного ураження центральної
нервової системи: анорексія, нудота, блювота,
психічні порушення, ураження периферичних
нервів.
Украй важким проявом інтоксикації є розви
ток уремічної коми - повної втрати свідомості зі
зникненням рефлексів на зовнішні і внутрішні
подразники, пригніченням життєво важливих
функцій. Крім інтоксикації, у патогенезі уреміч
ного синдрому мають значення порушення обмі
ну води та електролітів, зміни кислотно-основ
ної рівноваги.
Набряки
Набряки, що розвиваються при хворобах нирок,
мають різні механізми. Патогенетично розрізня
ють три типи ниркових набряків.
1. Набряки при гострій і хронічній недостат
ності нирок. Основний механізм їх розвит
ку — гідростатичний (гіперволемічний)
(див. главу 27). Зменшення швидкості клу
бочкової фільтрації, характерне для нирко
вої недостатності, призводить до затримки
натрію й води в організмі (позитивний вод
ний баланс) і, як наслідок, до гіперволемії.
Остання, будучи причиною збільшення гід
ростатичного тиску в капілярах, викликає
розвиток набряків за механізмом Старлінга.
До посилення набряків при недостатності
нирок ведуть і деякі інші - вторинні механізми.
Так, в умовах гіперволемії виникає ефект розве
дення білків плазми крові, унаслідок чого падає
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
324
онкотичний її тиск (онкотичний механізм на
бряків/ Крім того, при уремії серед токсичних
продуктів, що накопичуються в крові, є такі, які
збільшують проникність судин, зокрема в леге
невій тканині (мембраногенний механізм). На
слідок цього - розвиток набряку легень.
2. Нефротичні набряки. Основний механізм
їх розвитку - онкотичний (гіпопротеїнемічний) (див. главу 27). Порушення клубочкового фільтра при нефрозі зумовлю
ють масивну протеїнурію, у результаті
якої розвивається гіпопротеїнемія і падає
онкотичний тиск крові. Це, своєю чергою,
за механізмом Старлінга викликає перехід
води із судин у тканини - розвиваються на
бряки (рис. 36.4).
Набряки посилюються через утворення “за
чарованого кола”: перехід води із судин у тка
нини веде до гіповолемії (зменшення об’єму
циркулюючої крові). Та, у свою чергу, спричи
няється до активації ренін-ангіотензинової сис
теми (див. главу 27) і посилення секреції аль
достерону. Останній викликає затримку натрію
в організмі, що через збудження центральних
і периферичних осморецепторів зумовлює се
крецію вазопресину (антидіуретичного гормо
ну). Вазопресин, діючи на дистальні звивисті
канальці нефронів і збиральні трубочки, збіль
шує факультативну реабсорбцію води, унаслі
док чого об’єм циркулюючої крові відновлю
ється, проте настає ефект розведення білків: їх
концентрація у плазмі крові падає, і гіпопротеї
немія стає більш вираженою.
3. Нефритичні набряки. їх розвиток пов’я
заний із запальними процесами в ниркових
клубочках - гострим і хронічним гломеру
лонефритом (див. далі). Патогенез цих на
бряків складний і в основі має кілька меха
нізмів (рис. 36.5):
а) активація ренін-ангіотензинової системи.
Запалення клубочків супроводжується за
стоєм крові в судинах нирок і зумовленою
цим гіпоксією юкстагломерулярного апа
рату. Як результат, настає активація ренінангіотензинової системи з наступним по
силенням секреції альдостерону, який
викликає затримку натрію в організмі та
підвищення осмотичного тиску крові.
У відповідь на це посилюється секреція ва-
4. Схема механізмів розвитку нефротичних набряків.
ОЦК - об'єм циркулюючої крові
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
зопресину, збільшується факультативна ре
абсорбція води, розвивається гіперволемія.
Остання стає провідним патогенетичним
механізмом набряків (див. главу 27);
б) порушення процесів клубочкової філь
трації. Уповільнення кровообігу в за
палених клубочках стає причиною змен
шення швидкості клубочкової фільтрації.
Це спричиняється до затримки виведення
325
електролітів і води з організму, унаслідок
чого настає гіперволемія - уже згадуваний
вище патогенетичний чинник набряків;
в) розвиток гіпопротеїнемії. Для запалення
клубочків характерним є підвищення про
никності ниркового фільтра. Унаслідок
цього розвивається протеїнурія, яка веде
до гіпопротеїнемії, зниження онкотичного
тиску крові та розвитку набряків.
ис. 36.5. Схема механізмів розвитку нефритичних набряків.
ШКФ - швидкість клубочкової фільтрації, ЮГА - юкстагломерулярний апарат
326
Порушення кислотноосновної рівноваги
Нирки відіграють важливу роль у підтриманні
кислотно-основної рівноваги в організмі. Тут
відбуваються три процеси, що мають прямий
стосунок до цього важливого параметра гомео
стазу:
1) ацидогенез;
2) амоніогенез;
3) реабсорбція бікарбонатів. Докладно суть
цих процесів ми розглядали в главі 29.
При ураженнях нирок можливими є різні
патогенетичні варіанти порушень кислотноосновної рівноваги. Найчастіше розвивається
негазовий ацидоз. Залежно від сутності патоло
гічних процесів у нирках розрізняють такі його
форми.
1. Клубочковий ацидоз (нирковий азотемічний
ацидоз). Виникає внаслідок розвитку нир
кової недостатності при зменшенні швид
кості клубочкової фільтрації нижче 25 мл/
хв. Він є метаболічним ацидозом зі збіль
шеною аніонною різницею (див. главу 29).
Його розвиток обумовлений накопиченням
іонів водню, що утворюються ендогенно,
в основному у формі сірчаної, фосфорної,
сечової та інших кислот.
2.
Проксимальний
нирковий
канальцевий
ацидоз. Є наслідком первинних порушень
реабсорбції бікарбонатів у проксималь
них звивистих канальцях нефронів. Його
відносять до видільних ацидозів з нор
мальною аніонною різницею (гіперхлоремічний).
3. Дистальний нирковий канальцевий ацидоз.
Обумовлений первинними порушеннями
процесів ацидогенезу в дистальних звивис
тих канальцях, де відбувається відтитровування буферів (збереження бікарбонатів)
і підкислення (ацидифікація) сечі. Є мета
болічним ацидозом з нормальною аніонною
різницею (гіперхлоремічний). Виявляє себе
нездатністю витримувати ендогенне наван
таження іонами водню. При цьому pH сечі
не опускається нижче 6.
Зміни функції нирок іноді можуть спричиня
тися до розвитку негазового гіпокаліємічного
алкалозу. Це, зокрема, буває при гіперальдо-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
стеронізмі. Альдостерон, посилюючи секрецію
іонів калію в дистальних звивистих канальцях,
викликає гіпокаліємію, яка, своєю чергою, обу
мовлює виникнення алкалозу (див. главу 29).
Порушення фосфорнокальцієвого обміну
Участь нирок у здійсненні фосфорно-кальцієво
го обміну пов’язана як з їхньою екскреторною
функцією (фільтрація, реабсорбція й виведення
кальцію та фосфатів), так і інкреторною (пере
творення 25-оксивітаміну D у його гормонально
активну форму - 1,25-діоксивітамін D).
При ураженнях нирок порушення фосфорнокальцієвого обміну в організмі виявляють себе
двома групами змін:
1) нирковою остеодистрофією',
2) ектопічною кальцифікацією.
Ниркова остеодистрофія. Нирковою остео
дистрофією, або остеопатією, позначають
комплекс дистрофічних змін у кістках, що є на
слідком розладів ниркових функцій.
Клінічно загальні порушення фосфорнокальцієвого обміну ниркового походження мо
жуть виявляти себе різними змінами в кістках:
а) резорбцією кісткової тканини (фібрознокістозним остеїтом). У кістках утворюють
ся порожнини, які заповнюються фіброз
ною тканиною. Такі зміни є наслідком гі
перпаратиреозу,
б) остеомаляцією — розм’якшенням кісток,
їхньою деформацією, болями в кістках.
У дітей порушується окостеніння хрящів.
Причиною цих змін є гіпокальціємія;
в) остеопорозом - зменшенням щільності
кісткової тканини без деформації кісток;
г) остеосклерозом - збільшенням щільності
тканини кісток.
Найчастіше причиною розвитку ниркової
остеодистрофії стає хронічна недостатність
нирок (ХНН), про яку мова піде далі. Вона за
пускає цілий ряд механізмів, які власне і зумов
люють розвиток дистрофічних змін у кістках
(рис. 36.6):
1) зменшення екскреції фосфатів. При па
дінні ШКФ нижче ЗО мл/хв порушується
виведення фосфатів з організму, унаслі
док чого зростає їх концентрація в крові.
327
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
Гіперфосфатемія веде до гіпокальціємії,
оскільки добуток концентрацій фосфатів
і кальцію (кальцій-фосфатний добуток)
має залишатися сталим (див. главу 28). Гіпокальціємія стимулює продукування паратгормону прищитоподібними залозами,
а останній зумовлює резорбцію кісткової
тканини та її демінералізацію, намагається
зменшити гіперфосфатемію, пригнічуючи
реабсорбцію фосфатів у ниркових канальцях (див. главу 28);
2) порушення перетворення в нирках 25-оксивітаміну D у його гормонально активну
форму— 1,25-діоксивітамін D. Недостатнє
утворення цієї сполуки спричиняється до
порушення процесів усмоктування каль
цію в кишках, унаслідок чого розвивається
гіпокальціємія. Остання стає прямою при
чиною остеомаляції, стимулює секрецію
паратгормону з усіма наслідками, що ви
никають з цього (див. вище);
3) клубочковий ацидоз. В умовах ацидозу від
бувається посилений обмін іонів Са2+ і Na+
кісток на іони Н+ крові (буферна функція
кісткової тканини). Це веде до демінералізації кісток і розвитку ознак остеопорозу.
Ектопічна кальцифікація. Звапніння м’яких
тканин — ектопічна кальцифікація — при ура
женнях нирок має в основному метастатичний
характер і зумовлена загальними порушеннями
фосфорно-кальцієвого обміну — гіперфосфатемією. Цей механізм у деталях ми розглядали
в главі 28.
Тут лише зазначимо, що при недостатності
нирок через накопичення в організмі токсичних
продуктів відбувається ушкодження клітин у різ
них органах і тканинах, що робить цілком імо
вірним і дистрофічний варіант кальцифікацїї.
Серед основних проявів цього патологічно
го процесу розвиток артеріосклерозу Менкеберга, що є хворобою артерій, при якій ура-
Рис. 36.6. Схема механізмів розвитку нирковоїостеодистрофії.
ШКФ - швидкість клубочкової фільтрації
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
328
жується середній шар судинної стінки (медіа)
із відкладанням туди солей кальцію (докладно
див. главу 32).
Артеріальна гіпертензія
Стійке підвищення артеріального тиску є одним
з проявів багатьох уражень нирок різної етіоло
гії й патогенезу. Залежно від механізмів розви
тку розрізняють такі варіанти артеріальної гі
пертензії ниркового походження (рис. 36.7).
1. Гіпертензія, залежна від об’єму. Цей вид
виявляють у 80-90 % хворих з хронічною
нирковою недостатністю. Її патогенез пояс
нюють таким чином: при порушеннях клубочкової фільтрації зменшується екскреція
води й електролітів, що веде до гіперволемії
та збільшення внаслідок цього хвилинного
об’єму серця (Q). Як результат - підвищен
ня артеріального тиску (див. главу 32).
Крім цього основного механізму, має значен
ня набряк стінок артеріол і збільшення їхньої
реактивності щодо пресорних агентів в умовах
накопичення іонів Na+ в інтерстиції судин. Зазна
чена обставина зумовлює збільшення загального
периферичного опору і зростання артеріального
тиску.
Гіперволемія, яка обумовлює цей варіант гі
пертензії, є чинником, що пригнічує активність
юкстагломерулярного апарату нирок, а отже, рі
вень реніну при цьому не збільшується.
2. Гіпертензія з високим рівнем реніну (високоренінова). Основний її механізм поля
гає в тому, що при порушеннях ниркового
кровообігу (нефросклероз, атеросклероз
ниркових артерій, гломерулонефрит) роз
вивається гіпоксія юкстагломерулярного
апарату, яка супроводжується вивільнен
ням реніну і активацією ренін-ангіотензинової системи. Наслідки цього ми вже об
говорювали в цій главі та в главах 27 та 32.
Тут тільки зазначимо, що підвищення арте
ріального тиску при цьому варіанті гіпертензії
відбувається за рахунок як збільшення хвилин
ного об’єму серця (гіперволемія), так і зростан
ня загального периферичного опору (вазоконстрикторна дія ангіотензину II і вазопресину).
З .Гіпертензія, не залежна від об’єму і ре
ніну. Вона характеризується нормальним
об’ємом циркулюючої крові й нормальним
рівнем реніну. Вважають, що розвиток цьо
го виду гіпертензії пов’язаний з порушен
ням депресорних функцій нирок, а саме — зі
зменшенням утворення ниркових проста
гландинів, зокрема ПГ Е2, зменшенням
активності
ниркової
калікреїн-кінінової
системи, пригніченням утворення інших
(гіпотетичних) сполук, що мають гіпотен
зивну дію.
Анемія
Одна з важливих інкреторних функцій нирок
полягає в утворенні сполук, що регулюють
кровотворення в червоному кістковому мозку еритропоетинів та інгібіторів еритропоезу.
Ознаки анемії у хворих з ураженнями нирок
з’являються при зменшенні швидкості клубочкової фільтрації нижче 40 мл/хв.
У патогенезі анемії ниркового походження
мають значення певні механізми.
1. Пригнічення еритропоезу. Утворення ери
троцитів у червоному кістковому мозку
зменшується внаслідок:
а) порушення регуляції еритропоезу, обу
мовленого пригніченням утворення нир
кових еритропоетинів і посиленим про
дукуванням інгібіторів кровотворення;
б) ушкодження кровотворних клітин уре
мічними токсинами',
в) дефіциту заліза, що розвивається як ре
зультат хронічних крововтрат при хворо
бах нирок і втрат трансферину в резуль
таті протеїнурії.
2. Крововтрати. Вони можуть бути пов’язані
з гематурією, з частим утворенням виразок
у шлунку й кишках хворих з ураженнями
нирок, з розвитком геморагічного діатезу.
3. Гемоліз еритроцитів. Його обумовлюють
уремічні токсини.
Порушення гемостазу
При ураженнях нирок можуть розвиватися як
тромбофілічні порушення-гіперкоагуляція кро
ві, так і геморагічний діатез (див. главу ЗО).
І.
Синдром гіперкоагуляції крові. В основі
його розвитку лежать:
а) зменшення активності фібринолітичної
системи внаслідок дефіциту ниркового
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
7. Схема механізмів розвитку артеріальної гіпертензії ниркового походження.
ЮГА - кжстагломерулярний апарат
329
330
ферменту — урокінази, що є потужним
активатором плазміногену;
б) зниження активності антикоагулянтної
системи через порушення синтезу і де
понування гепарину в нирках;
в) втрата із сечею внаслідок протеїнурії ан
титромбіну III - основного природного
антикоагулянти.
II. Геморагічний діатез. У його розвитку ма
ють значення розлади як судинно-тромбоцитарного гемостазу (тромбоцитопенія внаслідок мієлотоксичної дії уремічних токсинів), так і коагуляційного (втрата із сечею факторів зсідання
крові при протеїнурії).
НИРКОВА НЕДОСТАТНІСТЬ.
ВЕЛИКІ НИРКОВІ СИНДРОМИ
І ЗАХВОРЮВАННЯ НИРОК
Недостатність нирок - це патологічний стан,
для якого характерне порушення сталості вну
трішнього середовища організму внаслідок не
здатності нирок здійснювати свої гомеостатичні
функції.
Класифікація. Ниркову недостатність класифі
кують у такий спосіб.
I. За клінічним перебігом розрізняють гостру
і хронічну недостатність нирок.
II. Залежно від причин розвитку ниркова недо
статність може бути преренальною, ренальною, постренальною й аренальною.
III. Залежно від обсягу порушених функцій не
достатність нирок буває тотальною (пору
шено всі функції) і парціальною (порушено
лише окремі функції).
IV. За механізмами розвитку розрізняють не
достатність нирок:
1) пов’язану з первинним ураженням клу
бочків - гломерулярну;
2) зумовлену первинним ураженням канальців - тубулярну.
Гостра недостатність нирок
Цей вид ниркової недостатності характеризу
ється швидким виникненням і значними пору
шеннями екскреторної функції нирок.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Причини. Залежно від причин розвитку розріз
няють такі варіанти гострої ниркової недостат
ності (ГНН).
1. Преренальна ГНН. В основі її виникнення
лежать гострі порушення кровопостачан
ня нирок. При цьому нирки не ушкоджені,
але не функціонують через розлади сис
темної гемодинаміки. Причинами цього
можуть бути:
а) падіння артеріального тиску при різ
них видах шоку нижче 80 мм рт. ст. (див.
вище);
б) гіповолемія, що настає при зневодненні,
великій гострій крововтраті тощо (див.
глави 27 і ЗО);
в) масивний гемоліз еритроцитів (перели
вання несумісної крові) чи міоліз (трива
ле роздавлювання м’язів);
г) ДВЗ-синдром, що розвивається як усклад
нення багатьох патологічних процесів
і хвороб (див. главу ЗО).
2. Ренальна ГНН. Її причиною є пряма чи
опосередкована дія патогенних чинників на
нирки, зокрема на такі їхні структури:
а) клубочки. Ушкодження цих структур на
стає при гострому гломерулонефриті,
вузликовому періартеріїті, злоякісній ар
теріальній гіпертензії;
б) канальці. Некроз тубулярного епітелію
може розвиватися як наслідок гострої
ішемії нирок (тромбоз, емболія) або дії
на них нефротропних отрут чи токсинів
при екзогенних та ендогенних інтоксикаціях (важкі метали та їхні солі, оцтова кис
лота, хлороформ, грибна й зміїна отрути,
токсикоз вагітних, діабетична кома);
в) інтерстицій. Патологічні зміни в інтерстиціальній тканині нирок виникають
при запальних і алергічних процесах
у ній (гострий пієлонефрит, гострий
алергічний інтерстиціальний нефрит
тощо).
3. Постренальна ГНН. Цей варіант недостат
ності нирок виникає при гострих порушен
нях виділення сечі, зумовлених непрохід
ністю сечовивідних шляхів через їхню обтурацію (камені) або компресію (пухлини).
4. Аренальна ГНН. Так називають недостат
ність нирок, що виникає після видалення
однієї-єдиної нирки.
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
Патогенез. У розвитку ГНН провідну роль мо
жуть відігравати три механізми.
1. Порушення кровообігу в нирках (преренальна ГНН). При швидкому і значному
зменшенні кровопостачання нирок тиск
крові в капілярах клубочків падає, що веде
до зниження швидкості клубочкової філь
трації (ШКФ) і, як наслідок, до зменшен
ня діурезу (початкова стадія) (рис. 36.8).
Якщо кровообіг не відновлюється, а про
довжує погіршуватися, то, з одного боку,
ШКФ падає ще більше - до рівня, при
якому діурез стає меншим за облігатний
(700 мл за добу) або ж зовсім припиня
ється (стадія оліго-, анурії), а з другого
канальні нефронів починають зазнавати
ішемії.
При зменшенні кровообігу в нирках нижче
рівня 20-25 % від нормальної величини настає
необоротне ішемічне ушкодження тубулярного епітелію і, по суті, преренальна ГНН пере
ходить у ренальну форму (рис. 36.9). Якщо ж
відновлення кровопостачання нирок відбудеть
ся раніше, ніж настане некроз канальців, то ді
урез починає поступово зростати і стає навіть
вищим за вихідний рівень (поліурична стадія).
Це пояснюється тим, що на відновлення функ
ції тубулярного епітелію потрібен певний час
і доти, доки не настане повна регенерація його
клітин, процеси канальцевої реабсорбції будуть
недостатніми, щоб повністю нормалізувати ді
урез.
Необоротне ушкодження клубочків, ка
нальців і судин нирок (ренальна ГНН). За
гибель клітин ниркової паренхіми механіз
мами некрозу й апоптозу найчастіше вини
кає внаслідок таких причин (рис. 36.9):
а) порушень центральної гемодинаміки, що
спричиняються до значної і тривалої ішемії
канальців. У цьому випадку мова йде влас
не про перехід преренальної форми ГНН
у ренальну (див. вище);
б) місцевих розладів ниркового кровообігу, що
ведуть до кисневого голодування (веноз
ної гіперемії, ішемії, стазу) (див. главу 16).
Такі виникають при запаленні нирок (го
стрий гломерулонефрит, гострий пієлонеф
рит та ін.), алергічних реакціях, тромбозі
ниркових судин;
2.
331
в) прямої ушкоджувальної дії на клітини тубу
лярного епітелію нефротоксичних отрут
і токсинів.
Загибель клітин канальців зумовлює пору
шення процесів клубочкової фільтрації через
підвищення внутрішньотканинного тиску, який
перешкоджає цьому процесу (див. вище). При
чинами такого наслідку є (а) вихід фільтрату
через ушкоджені канальці в інтерстицій і (б) обтурація канальців некротичними масами та ци
ліндрами.
У результаті втягування в процес клубочків
падає ШКФ і розвивається олігурія.
3. Порушення відтоку сечі (постренальна
ГНН). Непрохідність сечовивідних шляхів
незалежно від її причин зумовлює накопи
чення сечі вище місця перешкоди і підви
щення внаслідок цього внутрішньотканин
ного тиску (тиску первинної сечі в капсулі
ниркових клубочків) (рис. 36.10). Якщо
при двох попередніх формах ГНН (преренальній і ренальній) оліго- і анурія були на
слідком падіння ШКФ, то при постренальній—усе навпаки: спочатку виникає анурія,
а вже потім, як її наслідок, починає змен
шуватися ШКФ.
Основні клінічні прояви. У клінічному пере
бігові ГНН виділяють чотири стадії: (1) почат
кову; (2) оліго-, анурії; (3) поліурії; (4) видужан
ня.
Найбільш характерні й значні порушен
ня в організмі спостерігають на стадії оліго-,
анурії. Вони зумовлені змінами всіх парамет
рів гомеостазу, до підтримання яких причетні
нирки. Проявами таких змін є (а) гіперволемія,
(б) відносна гіпонатріємія (вода затримується
більшою мірою, порівняно з іонами натрію, ре
абсорбція яких порушується), (в) гіперкаліємія,
(г) ацидоз, (д) азотемія (уремія).
Зазначені вище порушення гомеостазу найвідчутніше позначаються на життєво важливих
функціях центральної нервової, серцево-судин
ної та дихальної систем.
Гіпоосмолярна гіперволемія спричиняється
до водного отруєння організму (див. главу 27),
що виявляє себе набряком головного мозку, ін~
терстиціальним набряком легень.
Про порушення діяльності центральної нер
вової системи свідчить великий ряд ознак: від
332
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
1.8 Стадії гострої преренальної ниркової недостатності: А - початкова, Б - олігурична, В - поліурична,
Г - одужування. QHII|) - об'ємна швидкість ниркового кровообігу, ШКФ - швидкість клубочкової фільтрації.
головного болю, блювоти до арефлексії, розла
дів свідомості, судом, коми.
Ацидоз у декомпенсованій формі стає при
чиною порушень функцій дихального центру
та розвитку термінального дихання за типом
Куссмауля (див. главу 33).
Значні зміни виникають і в системі крово
обігу: пригнічується скорочувальна функція
серця, порушується серцевий ритм у вигля
ді екстрасистолії, брадикардії, блокади (див.
главу 31); розвивається артеріальна гіпотензія
з наступним переходом у гіпертензію. Більша
частина хворих з ГНН гине на висоті стадії олі»
го-, анурії. При сприятливому перебігові хво
роби, а головне - при проведенні ефективних
терапевтичних заходів, через 5-10 діб може
настати перехід у стадію відновлення діурезу
й поліурії (див. вище), що свідчить про процес
одужування.
Хронічна недостатність нирок
Повільне прогресуюче зниження видільної
функції нирок, зумовлене необоротним змен
шенням кількості діючих нефронів, називають
хронічною нирковою недостатністю (ХНН).
Основною ознакою ХНН є поступове, протягом
місяців і років, зниження швидкості клубочко-
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
вої фільтрації, ступінь якого й визначає тяжкість
перебігу хвороби та появу її основних клінічних
проявів.
Причини. В основі розвитку ХНН лежать хро
нічні прогресуючі хвороби, при яких ураження
нирок складають суть патологічного процесу
або вони є ускладненням системних порушень
в організмі. До розвитку ХНН можуть спричи
нятися:
1) ураження судин нирок, що порушують кро
вообіг у них: атеросклероз великих нирко
вих артерій, склеротичні зміни у дрібних
артеріях і артеріолах, що виникають при
тривалій артеріальній гіпертензії різного
походження;
333
2) хвороби нирок запально-алергічної (хроніч
ний гломерулонефрит, системний червоний
вовчак, вузликовий періартерїїт та ін.) і за
пальної (хронічний пієлонефрит) природи;
3) порушення обміну речовин в організмі:
цукровий діабет (діабетична нефропатія),
ожиріння, подагра тощо;
4) обструктивні зміни в сечовидільних шля
хах: сечокам’яна хвороба, аденома про
стати, звуження уретри;
5) спадково зумовлені порушення: полікістоз
нирок, їх гіпоплазія.
Сьогодні серед основних причин ХНН, що
ведуть до розвитку термінальної її стадії, перше
місце посідає цукровий діабет, друге - есенці-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
334
Рис. 36.9. Схема розвитку гострої постренальної ниркової недостатності
альна артеріальна гіпертензія (гіпертонічна хво
роба), третє - хронічний гломерулонефрит.
Патогенез. Провідною ланкою патогенезу ХНН
є зниження швидкості клубочкової фільтрації
(ШКФ) внаслідок одночасного чи поступового
зменшення кількості діючих нефронів (КДН).
Початкові ознаки ХНН з’являються при
зменшенні КДН до 50-30 % від вихідної кіль
кості нефронів, клінічно виражена картина
розвивається при зниженні КДН до 30-10 %
і величини клубочкової фільтрації нижче 20 %
(якщо порівнювати з нормою). Подальше змен
шення КДН і ШКФ (нижче 10 %) веде до розви
тку термінальної стадії недостатності нирок —
уремії.
Неухильне прогресування ХНН пов’язують
з виникненням “зачарованих кіл” (circuit vitiosi)
у патогенезі хронічних хвороб нирок (рис. 36.11).
В експериментальних дослідженнях показано,
що зменшення КДН при видаленні однієї нир
ки і більшої чи меншої частини другої веде до
структурних і функціональних змін у тих нефронах, що залишилися. Ці зміни мають компенса
торний характер і виявляють себе гіпертрофією
всіх складових частин нефрону та зменшенням
тонусу приносних судин (вазодилатацією).
Останнє на тлі артеріальної гіпертензії, що є од-
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
335
36.10. Схема розвитку гостроїренальної
ниркової недостатності
до критичної маси й не настане термінальна ста
дія, що завершується смертю.
ним з проявів ХНН, веде до значного зростання
тиску в капілярах клубочків і збільшення внаслі
док цього клубочкової фільтрації. У такий спо
сіб нефрони, що залишилися, намагаються запо
бігти падінню ШКФ. Проте, тривале збільшення
тиску крові в судинах клубочків через нез’ясовані
досі механізми спричиняється до склеротичних
змін у цих структурах - розвивається гломерулосклероз. Заповнення клубочків клітинними і по
заклітинними компонентами сполучної тканини
припиняє їхнє функціонування й веде зрештою
до загибелі нефронів і подальшого зменшення
КДН. Завдяки описаному “зачарованому колу”
ХНН прогресує доти, доки КДН не зменшиться
Основні клінічні прояви. Перебіг ХНН має чо
тири стадії:
1) початкову,
2) ранню поліуричну;
3) пізню олігуричну;
4) термінальну.
Основним критерієм ХНН є зменшення ШКФ
(у клініці визначають непрямим методом - за
збільшенням концентрації креатиніну в сиро
ватці крові) на тлі ознак хвороби, що спричини
лася до ниркової недостатності.
І.
Початкова стадія. Характеризується
зменшенням ШКФ до від 100 до ЗО мл/хв
і клінічними проявами основної хвороби.
Недостатність фільтраційної здатності ни
рок на цій стадії можна встановити тільки
при збільшенні функціонального наванта
ження на нирки.
336
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Механізми прогресування хронічної ниркової недостатності, що ведуть до її термінальної стадії
("зачароване коло"), кдн - кількість діючих нефронів
2. Рання поліурична стадія. Для неї характер
ним є зменшення ШКФ від ЗО до 10 мл/хв
і збільшення добового діурезу. Поліурія,
що виникає, має компенсаторне значення:
в умовах зменшення ШКФ вивести з орга
нізму всю необхідну кількість осмотично
активних речовин можна або надмірно кон
центруючи сечу, або екскретуючи їх з вели
кою кількістю води. Уражені нирки, часто
з порушеною концентраційною здатністю
нефронів, обирають саме другий, ощадли
вий з огляду на енергетичні витрати спосіб.
Основним механізмом поліурії на цій стадії
є збільшення артеріального тиску - гіпертензивний діурез (див. вище) і перерозподіл крово
постачання коротких (кортикальних) і довгих
(юкстамедулярних) нефронів у бік зменшен
ня надходження крові в останні (при цьому
зменшується загальна площа реабсорбції води
й електролітів).
3. Пізня олігурична стадія. Показник ШКФ
зменшується від 10 до 5 мл/хв, поліурія
змінюється на олігурію. Виразними стають
усі клінічні ознаки ХНН (див. нижче).
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
4. Термінальна стадія. Настає при зменшен
ні ШКФ нижче 5 мл/хв і характеризується
анурією, ознаками вираженої уремії, при
гніченням життєво важливих функцій (кро
вообігу та дихання). Завершується смертю
єдиний вихід з цієї ситуації — трансплан
тація нирки. Розвиток термінальної стадії
можна відстрочити пожиттєвим гемодіалі
зом (штучна нирка).
Основні клінічні прояви ХНН з’являються
тоді, коли ШКФ стає меншою за ЗО мл/хв. Крім
кількісних (див. вище) і якісних (характерних
для основної хвороби) змін сечі, ХНН виявляє
себе такими ознаками: (а) інтоксикацією; (б) на
бряками; (в) нирковим азотемічним ацидозом;
(г) порушеннями фосфорно-кальцієвого обміну
(ниркова остеодистрофія, артеріосклероз Менкеберга); (д) артеріальною гіпертензією; (е) ане
мією; (є) порушеннями гемостазу; (ж) недостат
ністю серця; (з) інфекційними ускладненнями.
Механізми переважної більшості наведених
вище змін при ХНН були предметом нашого об
говорення в одній з попередніх частин цієї глави
(див. 36.2).
Нефротичний синдром
Нефротичним називають синдром, що виникає
при ураженнях нирок різного походження і ви
являє себе (1) масивною протеїнурією, (2) гіпопротеїнемією, (3) розвитком генералізованих
набряків і (4) гіперліпідемією. Втрата білків із
сечею, головним чином альбумінів, сягає 3,5 г
і більше на добу, а концентрація альбумінів
у плазмі крові стає меншою за ЗО г/л.
Причини. За походженням нефротичний син
дром поділяють на первинний і вторинний.
Первинний нефротичний синдром виникає
внаслідок самостійних, не пов’язаних із сис
темними хворобами, патологічних процесів
у нирках, що спричиняються до змін структури
клубочків і підвищення внаслідок цього про
никності ниркового фільтра. Такими є:
а) набуті форми уражень нирок: мембранозна гломерулопатія, нефротичний синдром
з мінімальними проявами (ліпоїдний не
фроз), фокальний сегментарний гломерулосклероз, проліферативні форми хроніч
ного гломерулонефриту та ін.;
337
б) спадково зумовлені ураження клубочкового
апарату нирок - уроджений (сімейний) не
фротичний синдром.
Вторинний нефротичний синдром розвива
ється як один з проявів системних хвороб ор
ганізму. Його причиною можуть бути цукровий
діабет, пізні токсикози вагітних (нефропатія),
амілоїдоз, системний червоний вовчак, деякі
лікарські препарати (нестероїдні протизапальні
засоби, пеніцилін) та ін.
Патогенез. Провідною ланкою патогенезу нефротичного синдрому є збільшення проникнос
ті ниркового фільтра. Останній є стінкою капі
лярів клубочків, що складається з трьох шарів
(рис. 36.12):
1) ендотелію. Його особливістю є те, що ко
жен ендотеліоцит перфорований великою
кількістю (кілька тисяч) малих за діаме
тром (70-100 нм) наскрізних пор (фенестр). Поверхня ендотелію несе на собі
негативний електричний заряд;
2) базальної мембрани. Складається з колаге
нових волокон (колаген IV типу), ламініну,
протеогліканів, фібронектину та деяких
інших глікопротеїнів. Поліаніонний харак
тер протеогліканів зумовлює негативний
електричний заряд на поверхні мембрани;
3) вісцеральні епітеліальні клітини (подоцити). Вони покривають капіляри клубочків
ззовні, утворюючи, по суті, зовнішній шар
капілярної стінки. Особливість цих клі
тин - наявність відростків у вигляді “ні
жок”. “Ніжки” подоцитів прикріплюються
до зовнішньої поверхні базальної мембра
ни таким чином, що між ними утворюють
ся т. зв. фільтраційні щілини. їхня ширина
становить 20-30 нм, і через них, у вигляді
містків, проходять молекули білка нефрину, завдяки яким утворюється своєрідна
щілинна діафрагма. Взаємодія білків цитоскелета подоцитів (актину, CD2AP, подоцину) і нефрину дозволяє змінювати
ширину щілин, а отже, і проникність нир
кового фільтра.
Увесь простір між капілярами клубочків за
повнено матриксом, схожим за своїм складом
до базальної мембрани, і мезангіальними кліти
нами. Останні здатні до скорочення, фагоците-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
338
Рис. 36.12. Будова ниркового фільтра (стінки капілярів клубочка)
зу, проліферації, синтезу позаклітинних компо
нентів сполучної тканини, виділення біологічно
активних речовин, що є медіаторами запалення
й алергічних реакцій.
Наведена будова ниркового фільтра пояс
нює, чому через нього легко проходять вода,
електроліти, низькомолекулярні органічні спо
луки і не проходять білки. Непроникність капі
лярної стінки клубочків до білків плазми крові
зумовлюється двома чинниками: (1) структур
ним: розміром фенестр в ендотеліоцитах, пор
у базальній мембрані та щілин між “ніжками”
подоцитів; (2) фізично-хімічним: негативним
зарядом на поверхні усіх трьох компонентів
капілярної стінки (ендотелію, базальної мемб
рани, подоцитів). Молекули, що мають діа
метр, більший за розмір пор і щілин ниркового
фільтра, а також негативний електричний за
ряд на своїй поверхні, не можуть пройти через
клубочковий бар’єр у фільтрат. До таких зокре
ма належать альбуміни та інші білки плазми
крові.
Значне підвищення проникності ниркового
фільтра до білків, що складає основу нефротичного синдрому будь-якої етіології, може бути
пов’язане з такими механізмами.
1. Імунний механізм. У його основі лежать
імунокомплексні реакції (алергічні реакції
III типу за Кумбсом і Джеллом, див. гла
ву 15), які спричиняються до ушкодження
усіх структур стінки капілярів клубочків
(ендотелію, базальної мембрани, подоци
тів). Антигени в імунних комплексах мо
жуть мати як екзогенне (бактерії, віруси,
паразити, лікарські препарати, компоненти
їжі тощо, так і ендогенне (ДНК, денатуро
вані нуклеопротеїни, білки пухлин, тиреоглобулін) походження. Антитіла до зазна
чених антигенів являють собою переважно
імуноглобуліни класу Ig М. Циркулюючі
в крові імунні комплекси не можуть про
йти через нирковий фільтр і затримуються
в його структурах - на поверхні ендотелію,
у субендотеліальному і субепітеліальному просторах по обидва боки базальної
мембрани. Фіксація цих комплексів до за
значених структур спричиняється до акти
вації комплементу, утворення комплексу
мембранної атаки (див. главу 14) і цитолі
зу клітин, на поверхні яких відбуваються
ці події. З ушкоджених ендотеліоцитів і по
доцитів вивільнюються лізосомні фермен-
339
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
ти, які гідролізують компоненти базальної
мембрани (колаген, протеоглікани та ін.)
та роблять її проникною для високомолекулярних сполук.
Протеїнурія, що виникає внаслідок цього, не
є селективною — через ушкоджений нирковий
фільтр у сечу виходять білки різної молекуляр
ної маси.
Метаболічний механізм і пов’язані з ним
фізично-хімічні зміни. Проникність стінки
капілярів клубочків може підвищуватися
внаслідок зменшення негативного елек
тричного заряду всіх її структур (ендо
телію, базальної мембрани, вісцеральних
епітеліальних клітин). Такі зміни можуть
бути зумовлені зникненням сіалопротеїну, що в нормі тонким шаром покриває ен
дотелій капілярів та відростки подоцитів
і завдяки своїм аніонним групам створює
на їхній поверхні негативний електричний
заряд. Крім того, до подібного наслідку
ведуть порушення обміну протеогліканів
базальної мембрани, при яких зменшуєть
ся кількість аніонних груп у їхніх моле
кулах.
У місцях, де втрата аніонів, а отже, і негатив
ного електричного заряду, максимальна, нако
пичуються поліморфноядерні лейкоцити, про
дукти секреторної дегрануляції яких - металопротеїнази та інші гідролітичні ферменти ушкоджують базальну мембрану капілярних су
дин, а вільні радикали та пероксиди зумовлюють
вторинну альтерацію клітин ендотелію та вісце
рального епітелію.
2.
Молекулярно-генетичні механізми. Успіхи
сучасної науки дали можливість встанови
ти причини деяких спадково зумовлених
форм нефротичного синдрому, що харак
теризуються мінімальними структурними
змінами елементів ниркового фільтра (їх
можна виявити тільки при електронній мі
кроскопії).
Так, до змін функціонування фільтраційних
щілин між “ніжками” подоцитів (див. рис. 36.12),
а отже, і до порушень самого ниркового фільтра,
можуть спричинятися мутації таких генів:
а) гена нефрину (NPHS1). При цьому розви
вається вроджений нефротичний синдром
фінського типу, при якому в новонародже
них виявляють підвищений вміст білка
в сечі;
б) гена подоцину (NPHS2). Дефект цього гена
стає причиною стійкого до лікування пре
паратами стероїдних гормонів нефротич
ного синдрому в дітей.
3.
В експериментальних дослідженнях на “ге
нетично нокаутованих” мишах було показано,
що відсутність гена ще одного білка подоци
тів — CD2AP (CD2-associated protein) - веде до
розвитку протеїнурії у тварин.
Нефротичний синдром при великій його три
валості може вести до зменшення кількості ді
ючих нефронів і розвитку хронічної ниркової
недостатності. Це зокрема пояснюється тим,
що в умовах протеїнурії значно посилюється
реабсорбція білка в проксимальних звивистих
канальцях. Клітини цих структур нефронів не
в змозі перетравити увесь поглинений з про
світу канальців білок, а тому він накопичуєть-
Білки, що втрачаються з сечею
Наслідки зменшення вмісту білка в крові
Альбуміни
Гіпоонкія, набряки
Антитромбін III
Схильність до тромбозів і тромбоемболій
Фактори зсідання крові
Геморагічний діатез
Компоненти комплементу
Зменшення резистентності до інфекцій
Імуноглобуліни
(Ід)
Те ж саме
Ліпопротеїни високої густини (ЛПВГ)
Прискорений розвиток атеросклерозу
Білки, що транспортують мікроелементи
Дефіцит Fe, Cu, Zn
Білки, що транспортують гормони
Ендокринні порушення
340
ся в їхній цитоплазмі, згодом перетворюється
в гіаліноподібну субстанцію - розвивається гіаліново-крапельна і вакуольна дистрофія тубулярного епітелію, він гине, а разом з ним гинуть
і нефрони.
Основні клінічні прояви. Масивна протеїнурія,
що її спостерігають при нефротичному синдро
мі, спричиняється до зменшення вмісту в кро
ві майже всіх функціонально важливих білків
(гіпопротеїнемії). Цим і пояснюють широкий
спектр порушень, що виникають в організмі
(див. табл.)
Серед основних клінічних проявів нефротичного синдрому, зумовлених протеїнурією, - ге
нерал ізован і онкотичні набряки, що виявляють
себе накопиченням води не тільки в м’яких тка
нинах, а й у порожнинах організму - черевній
(асцит), плевральній. Механізми їх розвитку
ми вже розглядали (див. вище).
Що стосується ще одного компонента нефротичного синдрому - гіперліпідемії, то до
сьогодні ще немає задовільного пояснення цієї
ознаки. Порушення ліпідного обміну в більшос
ті хворих виявляють себе підвищенням концен
трації холестеролу, триацилгліцеролів, ліпопротеїнів дуже низької (ЛПДНГ) і низької (ЛПНГ)
густини, модифікованих ліпопротеїнів на тлі
зменшення вмісту ліпопротеїнів високої густи
ни (ЛПВГ). Іншими словами, розвивається дисліпопротеїнемія атерогенного характеру (див.
32], Рівень наших знань ще недостатній,
щоб встановити прямі причинно-наслідкові
зв’язки між цими розладами і первинними по
рушеннями фільтраційної здатності нирок, ха
рактерними для нефротичного синдрому.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
системних хвороб організму (наприклад,
системний червоний вовчак, вузликовий
періартерїїт та ін.).
II. За клінічним перебігом виділяють (а) го
стрий і (б) хронічний гломерулонефрит.
Для гострого варіанту хвороби характер
ний бурхливий початок, його тривалість до кількох тижнів.
Хронічний гломерулонефрит є прогресуючим
протягом тривалого часу (від кількох місяців до
року й більше) дифузним ураженням нирок, що
часто завершується хронічною нирковою недо
статністю та вимагає застосування гемодіалізу
і проведення трансплантації нирок.
Терміном “гломерулонефрит” позначають ве
лику групу різних за походженням хвороб, що
характеризуються запаленням клубочків обох
нирок, переважно імунної (алергічної) приро
ди.
Причини. До основних етіологічних чинників
гострого гломерулонефриту відносять такі:
1)
стрептококову інфекцію. Найчастіше
ураження нирок настають через 1-4 тиж
ні після хвороб, зумовлених специфічни
ми “нефритогенними” штамами (типи 1,
4, 12) гемолітичного стрептокока гру
пи А (ангіна, гнійничкова хвороба шкі
ри — імпетиго, особливо в дітей у віці від
6 до 10 років);
2) нестрептококові інфекції. Гострий гло
мерулонефрит може розвиватися під час
і після багатьох бактеріальних (стафілоко
ковий ендокардит, пневмококова пневмо
нія, менінгококова інфекція), вірусних (ге
патит В, гепатит С, епідемічний паротит,
ВІЛ-інфекція, інфекційний мононуклеоз)
та паразитарних (малярія, токсоплазмоз)
хвороб;
3) переохолодження організму. Під час війн
гломерулонефрит часто розвивався у воя
ків, що тривалий час перебували в окопах
у холодну сиру погоду. Таку форму хворо
би назвали окопним нефритом',
4) системні хвороби різного походження:
системний червоний вовчак, вузликовий
періартеріїт, синдром Гудпасчера, гемора
гічні васкуліти тощо.
Класифікація.
І. Розрізняють (а) первинний і (б) вторин
ний гломерулонефрит. Про перший ведуть
мову тоді, коли патологічний процес об
межується одними лише нирками. Вторин
ний гломерулонефрит є наслідком певних
Хронічний гломерулонефрит може бути на
слідком гострого, але частіше розвивається пер
винно. Залежно від причин виділяють такі його
форми:
1) інфекційного походження (постстрептококовий, при затяжному септичному ендо
Гломерулонефрит
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
кардиті, малярії, сифілісі, туберкульозі та
інших інфекціях);
2) неінфекційні (сироватковий, вакцинний,
медикаментозний,
травматичний,
при
отруєннях, охолодженні, тромбозі нирко
вих вен);
3) зумовлений системними хворобами (рев
матоїдний артрит, системний червоний
вовчак, геморагічний васкуліт та ін.);
4) особливі форми (після еклампсії, радіацій
ний, спадковий та ін.).
Експериментальні моделі Відтворення у тва
рин різних форм гломерулонефриту дає мож
ливість встановлювати причини й механізми
розвитку цієї хвороби. Велику кількість запро
понованих експериментальних моделей гломе
рулонефриту зручно розділити на кілька груп
залежно від основних патогенетичних механіз
мів, що лежать в їхній основі.
І.
Нефротоксичний нефрит. Відтворюється
введення тваринам нефроцитотоксичної сиро
ватки, тобто сироватки, що містить антитіла
проти антигенів ниркової тканини. Серед моде
лей цієї групи найчастіше використовують:
а) модель Ліндемана. Кролям внутрішньовен
но вводять нефроцитотоксичну сироватку,
отриману від морської свинки, попередньо
імунізованої суспензією нирки кроля. При
цьому відбувається фіксація протиниркових антитіл на базальних мембранах клу
бочків з наступним зв’язуванням компле
менту й розвитком ушкодження;
б) модель Масугі. Щурам внутрішньовенно
вводять нефроцитотоксичну сироватку,
одержану від кролів, імунізованих ткани
ною нирок щурів (перший варіант: щур кріль - щур), або кролям вводять сироват
ку, що її одержали від качок, імунізованих
нирковою тканиною кролів (другий варі
ант: кріль - качка - кріль). Особливість
цієї моделі полягає в тому, що антитіла
фіксуються на базальній мембрані клубоч
ків, але не можуть зв’язувати комплемент.
Це спричиняється до утворення власних
щурячих (кролячих) антитіл проти фіксо
ваних кролячих (качиних) імуноглобулінів (для цього потрібно 6-8 діб). Останні
в такому випадку виступають антигенами.
Щурячі (кролячі) антитіла, з одного боку,
взаємодіють з фіксованими на базальній
341
мембрані кролячими (качиними) імуноглобулінами, а з другого - зв’язують комп
лемент, активація якого й веде до ушко
дження клітин клубочків.
II. Імунізація компонентами ниркової ткани
ни. На цьому ґрунтуються такі експерименталь
ні моделі:
а) модель Хеймена. Щурів імунізують анти
генами, що містяться в препаратах, одер
жаних із щіткової облямівки клітин про
ксимальних звивистих канальців нефронів. В організмі утворюються антитіла на
ці антигени, які фіксуються не тільки на
структурах щіткової облямівки клітин тубулярного епітелію, а й на мембрані вісце
ральних епітеліальних клітин (подоцитів)
з боку прилягання її до базальної мембра
ни капілярів клубочків. Встановлено, що
у щурів антигенний комплекс Хеймена - це
великий білок (330 кДа), названий мегаліном. Він за своєю структурою дуже схожий
на рецептор до ліпопротеїнів низької гус
тини. У нирках людини виявити такий бі
лок при різних формах гломерулонефриту
не вдалося;
б) модель Стеблея. Овець імунізують антиге
нами базальних мембран капілярів клубоч
ків, одержаних із ниркової тканини люди
ни. Антитіла, що при цьому утворюються,
взаємодіють не тільки із введеними ззовні
антигенами, а й зі структурами ниркових
клубочків тварин.
III.
Індукований стрептококами нефрит.
Про роль стрептококів у розвитку гломеруло
нефриту свідчить модель подружжя Ковелті.
Її відтворюють введенням у черевну порожнину
кролів клітинної суспензії ниркової тканини ра
зом з культурою стрептококів.
IV. Автоалергічний нефрит. На можливість
розвитку автоалергічних уражень нирок вказу
ють такі моделі:
а) модель Герцена - заморожування однієї
нирки хлороформом. При цьому в кро
ві дослідних тварин (кролів) з’являються
специфічні протиниркові антитіла, і зазнає
уражень друга інтактна нирка. Зазначена
модель доводить роль автоімунних меха
нізмів у розвитку окопного нефриту (не
фриту воєнного часу);
б) виведення чистих ліній тварин (наприклад,
новозеландські миші лінії NZB/BL), у яких
342
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
спонтанно виникають автоімунні хвороби,
у тому числі й гломерулонефрит.
Патогенез. В основі розвитку гломерулонефри
ту лежать алергічні реакції II типу (цитотоксич
ні) і III типу (імунокомплексні) за класифікаці
єю Кумбса і Джелла (див. главу 15). Відповідно
розрізняють два основні патогенетичні механіз
ми хвороби.
І. Ураження структур клубочків вільними ан
титілами (реакції II типу). До розвитку гломе
рулонефриту можуть спричинятися:
а) антитіла проти компонентів базальної
мембрани капілярів клубочків. Саме такі
беруть участь у розвитку хвороби при екс
периментальному її відтворенні за допо
нефроцитотоксичних
сироваток
могою
(моделі Ліндемана і Масугі). Сьогодні
з’ясовано, що імуноглобуліни специфіч
но зв’язуються з неволокнистою ділян
кою (доменом NC1) а3-ланцюгів колагену
IV типу. Цей домен відіграє важливу роль
у формуванні комплексів між окремими
ланцюгами колагену та утворенні остаточ
ної кінцевої форми колагенових волокон.
Антитіла до компонентів базальних мемб
ран капілярів клубочків можуть перехрес
но взаємодіяти з подібними базальними
мембранами в інших органах і тканинах,
зокрема в альвеолах легень. Таке поєднане
ураження капілярів клубочків і легеневої
тканини характерне для синдрому Гудпасчера.
Гломерулонефрит, який індукується антитіла
ми проти базальних мембран, має, як правило,
стрімкий перебіг, швидко прогресує. На нього
припадає до 5 % випадків гострого гломеруло
нефриту в людини;
б) антитіла до компонентів клітин (вісце
ральних епітеліальних, ендотеліоцитів,
мезангіальних клітин), що входять до
складу клубочків. Імуноглобуліни до по
верхневих антигенів вісцеральних епітелі
альних клітин (подоцитів) започатковують
розвиток гломерулонефриту, який в експе
рименті відтворюється методом Хеймена
(див. вище). У людини, як і в експеримен
тальних тварин, такий варіант уражень ни
рок виявляє себе мембранозною гломерулопатією.
Утворення в організмі антитіл проти компо
нентів базальної мембрани та клітин клубочків
становить основу автоалергічних уражень ни
рок у людини. Причини появи таких автоантитіл можуть бути різними, про що йшла мова
у відповідній частині глави 15;
в) антитіла до нениркових антигенів, фіксова
них на структурах клубочків. Гломеруло
нефрит може розвиватися внаслідок взає
модії антитіл з антигенами нениркового по
ходження при затримці (осіданні) останніх
в клубочках. Такими антигенами можуть
виступати катіонні молекули, які завдяки
електростатичній взаємодії фіксуються на
структурах клубочків, що містять велику
кількість аніонних хімічних груп (поверх
ня ендотелію, базальна мембрана, мембра
на ніжок подоцитів). Імунні реакції цього
типу індукуються молекулами ДНК і нуклеопротеїдів, великими агрегатами біл
ків (зокрема імуноглобулінів), продуктами
бактеріального, вірусного й паразитарного
походження тощо.
II. Ураження клубочків циркулюючими імун
ними комплексами (реакції III типу). Фіксація
в клубочках (в основному в субендотеліальному просторі капілярів) комплексів “антиген —
антитіло” запускає механізми, що ведуть до
ушкодження клітин і базальної мембрани, пору
шуючи їхні бар’єрні властивості, - розвиваєть
ся імунокомплексний гломерулонефрит.
У патогенезі такого типу хвороби можуть
брати участь імунні комплекси, що містять ан
тигени як екзогенного (інфекційної чи неінфекційної природи), так і ендогенного (білок тка
нин, ДНК) походження. Антитіла (Ig G, Ig М)
прямо в крові вступають у взаємодію із зазна
ченими антигенами, потім у вигляді комплексів
“антиген - антитіло” й “антиген - антитіло комплемент” надходять у клубочки, відкладаю
чись на їхній базальній мембрані.
Імунокомплексний механізм мають уражен
ня нирок, що розвиваються після стрептококо
вої інфекції, при гепатитах В і С, сифілісі, ма
лярії, а також системному червоному вовчаку,
сироватковій хворобі та ін. Більшість випадків
гломерулонефриту в людини (не менше 80 %)
є імунокомплексними.
В ушкодженні структур клубочків як вільни
ми антитілами (реакції II типу), так і імунними
343
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
комплексами (реакції III типу) провідну роль ві
діграють дві події:
1) активація комплементу, яку започат
ковує фіксація його компонентів на F фрагментах антитіл;
2) прихід, фіксація та активація в клубочках
лейкоцитів крові (моноцитів і нейтрофі
лів). Взаємодія рецепторів цих клітин з Fcфрагментами імуноглобулінів та СЗЬ про
міжним продуктом активації комплементу
веде до вивільнення лейкоцитами факторів
альтерації (вільних радикалів, пероксидів,
протеаз), медіаторів запалення, що вплива
ють на місцевий кровообіг та проникність
судин (простагландини, оксид азоту), сти
мулюють хемотаксис (лейкотрієни, цитокіни) і проліферацію клітин (фактори рос
ту). Іншими словами, розвивається імунне
запалення, механізми якого описано в гла
ві 15.
Основні клінічні прояви. Гострий гломеруло
нефрит, що розвивається після перенесеної
стрептококової інфекції (на нього припадає по
над 90 % усіх випадків хвороби), характеризу
ється поєднанням ознак, що їх об’єднують по
няттям нефритичний синдром. Основними його
компонентами є гематурія, протеїнурія, артері
альна гіпертензія та азотемія на тлі олігурії. Час
то розвиваються т. зв. нефритичні набряки. По
ходження наведених вище клінічних ознак було
предметом обговорення в частині 36.2 цієї глави.
Особливу клінічну форму складає т. зв. зло
якісний гострий гломерулонефрит нез’ясованої
етіології. Він характеризується бурхливим по
чатком, швидким прогресуванням і завершу
ється настанням протягом кількох тижнів нир
кової недостатності. Його розвиток може бути
пов’язаний з (а) утворенням антитіл до базаль
них мембран клубочків (перший тип), (б) від
кладанням циркулюючих імунних комплексів
в нирковій тканині (другий тип), (в) антитілами
проти власних антигенів нейтрофільних лейко
цитів (третій тип).
Хронічний гломерулонефрит залежно від похо
дження та особливостей патогенезу може вияв
ляти себе в різних клінічних формах. Такими є:
а) латентна форма. Її основна ознака - по
мірна протеїнурія, гематурія. У частини
хворих бувають набряки і нестійка артері
альна гіпертензія;
б) гіпертензивна форма. Для неї характерні
стійке підвищення артеріального тиску, на
бряки, гематурія, протеїнурія, циліндрурія
і лейкоцитурія;
в) нефротична форма. Виявляє себе озна
ками нефротичного синдрому: масивною
протеїнурією, гіпопротеїнемією, генералізованими набряками, гіперліпідемією (див.
вище);
г) змішана, або нефротично-гіпертензивна
форма. Характеризується набряками й гі
пертензією, але, на відміну від нефротичної форми, немає характерних змін крові.
Хронічний гломерулонефрит є однією з основ
них причин хронічної ниркової недостатності,
що завершується термінальною стадією —уремі
єю.
Пієлонефрит
Пієлонефритом називають хворобу, що харак
теризується запаленням паренхіми нирок (пе
реважно інтерстиціальної тканини) і ниркових
мисок, як правило, інфекційної природи.
Залежно від клінічного перебігу розрізняють
гострий і хронічний пієлонефрит.
Причини. У переважній більшості випадків
(понад 85 %) причиною недуги є грамнегативні
бактерії, що складають нормальну мікрофлору
кишковика, зокрема Escherichia coli. Крім того,
будь-яка патогенна інфекція нижніх сечовивід
них шляхів (бактерії, віруси, гриби) здатна при
потраплянні в ниркову тканину викликати в ній
запалення.
Занесення інфекції в нирки може відбуватися
двома шляхами: (1) гематогенним (наприклад,
при сепсисі, інфекційному ендокардиті) або (2)
поширенням її у висхідному напрямку по сечо
вивідних шляхах. Більшість випадків пієлонеф
риту пов’язана саме з другим шляхом.
Виникненню хвороби, особливо її хронічної
форми, сприяють умови, що викликають застій
сечі (звуження, закупорка сечоводів, аденома
простати) та її закидання із сечового міхура
в сечівник (везикоуретральний рефлюкс), пору
шують трофіку сечовивідних шляхів, знижують
реактивність організму (цукровий діабет, хро
нічна інтоксикація тощо).
344
Патогенез. Потрапляння бактеріальної інфекції
в паренхіму однієї чи обох нирок зумовлює роз
виток в інтерстиції (переважно мозкового шару)
гострого запалення, основним компонентом
якого стає інфільтрація тканини нейтрофілами.
Некротичні зміни, що розвиваються, ведуть до
утворення гною і в частині випадків до форму
вання абсцесів. З часом у процес втягуються
канальці нефронів: ушкоджуються та гинуть
клітини тубулярного епітелію, гній потрапляє
в просвіт канальців.
На початкових стадіях хвороби, порушення
функцій канальців є провідними, якщо порів
нювати з клубочками (своєрідна функціональна
дисоціація). Про це свідчать:
а) зменшення здатності нирок до концентру
вання сечі внаслідок порушення процесів
реабсорбції в петлі Генле нефронів;
б) синдром втрати солей, в основі якого - різ
ке зниження реабсорбції іонів натрію, ка
лію, кальцію в проксимальних звивистих
канальцях;
в) ранній і важкий канальцевий ацидоз,
пов’язаний з порушенням ацидо- й амоніогенезу.
Як наслідок зазначених розладів можуть роз
виватися небезпечні для життя порушення вод
но-електролітного обміну і кислотно-основної
рівноваги.
При гострому пієлонефриті нейтрофільну ін
фільтрацію згодом змінює надходження в осе
редки запалення моноцитів, які, перетворив
шись на макрофаги, розчищають тканину від
некротичних мас і започатковують процес заго
ювання, що виявляє себе формуванням рубців.
Постійне інфікування ниркової паренхіми
веде до розвитку хронічного пієлонефриту.
Найчастіше це буває за двох обставин:
1) при закиданні сечі із сечового міхура в се
чівник (везикоуретральний рефлюкс) та
із ниркових мисок у збиральні трубочки
(інтраренальний рефлюкс). У такому разі
хронічний пієлонефрит, що розвивається,
позначають як рефлюксну нефропатію',
2) при звуженні (обструкції) сечовивідних
шляхів. Цей механізм лежить в основі хро
нічного обструктивного пієлонефриту.
Хронічна форма пієлонефриту веде до за
міщення нефронів рубцевою тканиною, нирки
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
зморщуються, зменшується кількість діючих
нефронів, розвивається хронічна ниркова недо
статність
Основні клінічні прояви. Залежно від похо
дження їх можна розділити на три групи.
1. Загальні ознаки важкого інфекційного і за
пального процесу: інтоксикація, гарячка,
зміни крові - лейкоцитоз (часто зі зміщен
ням лейкоцитарної формули вліво), підви
щення ШОЕ та ін.
2. Сечовий синдром. Залежно від стадії хворо
би він може виявляти себе поліурією (змен
шення концентраційної здатності каналь
ців на початкових стадіях) або олігурією
(порушення клубочкової фільтрації на за
вершальних етапах). Характерними є лейкоцитурія, гж до появи гною в сечі (піурія);
помірна протеїнурія (канальцевого й се
креторного походження, див. частину 36.2
цієї глави), циліндрурія, гіпо- та ізостенурія (порушення і повна втрата здатності
канальців концентрувати сечу), полакіурія
(часте сечовипускання, зумовлене супутні
ми запальними процесами в сечовому міху
рі та сечовивідних шляхах). Із сечі можна
висіяти бактерії, що спричинилися до запа
лення (бактеріурія).
3. Прояви, пов’язані з розладами екскретор
ної та інкреторної функцій нирок: набряки,
артеріальна гіпертензія, анемія, порушен
ня фосфорно-кальцієвого обміну (ниркова
остеодистрофія,
артеріосклероз
Менкеберга), негазовий ацидоз та ін.
Прогресування хвороби супроводжується
наростанням описаних порушень і веде до тер
мінальної стадії хронічної ниркової недостат
ності.
Сечокам’яна хвороба
Сечокам’яною називають хворобу, обумовлену
утворенням каменів у паренхімі нирок і в миско
во-сечовідному сегменті сечовивідних шляхів.
За хімічним складом камені поділяють на:
а)
кальцій-оксалатні. Вони складаються
з кальцієвої солі щавлевої кислоти (оксала
ту), часто з домішками фосфату кальцію. їх
утворення стає причиною близько 70 % ви
падків сечокам’яної хвороби;
ГЛАВА 36. ПАТОФІЗІОЛОГІЯ НИРОК
б) фосфатно-амонієво-магнієві
(15—20 %).
До їхнього складу входять магнієві та амо
нійні солі фосфорної кислоти;
в) уратні (5-10 %). Основний їхній компо
нент — натрієві та амонійні солі сечової
кислоти;
г) цистинові (1-2 %). Мають у своєму складі
амінокислоту цистин.
Причини. В етіології сечокам’яної хвороби ма
ють значення такі фактори.
1. Якісний склад води та їжі, режим пиття
і харчування. Від зазначених чинників за
лежать: (a) pH сечі; (б) величина діурезу,
(в) концентрація в сечі солей, з яких утво
рюються камені, а також (г) речовин, що за
безпечують їхню розчинність і стабільний
стан. Вважають, що споживання сильно
мінералізованої, жорсткої питної води з ве
ликим вмістом солей кальцію, вживання
їжі з надмірним вмістом солей, з яких утво
рюються камені, нестачею білків, вітамінів
групи В, особливо піридоксину, є фактором
ризику сечокам’яної хвороби.
2. Інфекції сечовивідних шляхів. Вони сприя
ють:
а) зміщенню реакції сечі в лужний бік уна
слідок уролітичної дії деяких бактерій
і осадження кристалів фосфатів;
б) зниженню поверхневого натягу між се
чею та ушкодженою слизовою оболон
кою органів сечовиведення;
в) утворенню в ушкоджених місцях фібри
ну, клітинного детриту, згустків крові, які
можуть служити кристалізаційними цен
трами;
г) зниженню колоїдної стабільності сечі
внаслідок обумовленого пієлонефри
том порушення ниркової екскреції ряду
речовин - сечовини, лимонної кислоти,
кальцію, фосфатів та ін.
3. Жаркий і сухий клімат, робота в гарячих це
хах. Такі умови викликають значну втрату
організмом рідини при потінні та підвищен
ня внаслідок цього густини сечі. Це сприяє
частішому виникненню сечокам’яної хво
роби в певних географічних районах і в осіб
відповідних професій.
4. Ендокринні хвороби (наприклад, первин
ний гіперпаратиреоз) і хвороби обміну ре
345
човин (цистиноз, оксалоз, гіперкальціурія,
гіпергліцинемія та ін.).
5. Спадково обумовлені канальцеві синдро
ми, що характеризуються порушенням ре
абсорбції амінокислот (цистину, гліцину
та ін.).
Патогенез. Наявність двох складових частин
у структурі каменів — неорганічної й органіч
ної - було покладено в основу кристалізаційної
і колоїдної теорій їх утворення.
Відповідно до першої, утворення каменів
цілком підпорядковане принципам кристаліза
ції, а утворення органічної матриці (на її частку
припадає 2-3 % сухої маси) є додатковим вто
ринним процесом.
На противагу цьому, прихильники колоїдної
теорії первинним при утворенні каменів вважа
ють формування органічної матриці, а кристалі
зацію на ній сечових солей розглядають як про
цес вторинний.
Так чи інакше, для настання кристалізації
потрібне зниження стабільності в сечі солей,
з яких утворюються камені. Підвищення їхньої
концентрації необхідне тільки на початкових
етапах кристалізації. Подальше здійснення цьо
го процесу може відбуватися і при нормальній
концентрації солей.
У цьому випадку провідну роль у патогенезі
сечокам’яної хвороби відіграють фактори, що
знижують розчинність солей. До їхнього числа
відносять:
а) зниження в сечі вмісту солюбілізаторів —
сечовини, креатиніну, гіпурової кислоти,
ксантину, хлориду натрію, цитратів, маг
нію, що в нормі підтримують розчинений
стан солей, з яких утворюються камені,
та перешкоджають їхньому осадженню;
б) зменшення в сечі вмісту речовин, що при
гнічують кристалізацію, — інгібіторів крис
талізації (неорганічний пірофосфат, іони
цинку, марганцю, кобальту та ін.);
в) зниження в сечі рівня компл ексоутворювачів, зокрема іонів магнію та цитратів, які
в нормі зв’язують відповідно близько 3040 % оксалатів та понад 50 % іонізованого
кальцію з утворенням розчинних комплек
сних сполук;
г) поява в сечі мукопротеїдів, піровиноград
ної кислоти, сульфаніламідів, продуктів ко
лагену, еластину, що створюють умови для
346
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
швидкої кристалізації солей у насиченому
розчині.
Утворення каменів різного хімічного складу
пов’язано з особливістю умов, що викликають
цей процес і йому сприяють.
Кальцій-оксалатні камені. У їхньому фор
муванні провідну роль відіграють порушення
кальцієвого обміну - гіперкальціємічні стани
(див. главу 28). Вони супроводжуються гіперкальціурією та підвищенням вмісту в сечі основ
ного компоненту каменів - кальцію. У більш
ніж половині випадків гіперкальціурія розви
вається без гіперкальціємії, вона пов’язана або
з підвищеним всмоктуванням кальцію в кишках
((абсорбційна гіперкальціурія), або з первинни
ми порушеннями його реабсорбції в канальцях
(ниркова гіперкальціурія).
Крім того, утворенню кальцій-оксалатних ка
менів сприяють:
а) посилена секреція сечової кислоти - гіперурикозурія, що веде до утворення криста
лів уратів. Останні виступають центрами
кристалізації (нуклеаторами) для оксалату
кальцію в збиральних трубочках;
б) збільшення в сечі концентрації щавлевої
кислоти - гіпероксалурія. Такий стан може
бути зумовлений спадковими порушення
ми обміну речовин {первинна гіпероксалу
рія) або посиленим всмоктуванням оксала
тів у кишках (ентеральна гіпероксалурія).
Остання форма може розвиватися у вегета
ріанців, дієта яких містить продукти, багаті
на оксалати;
в) зменшення в сечі концентрації цитратів —
гіпоцитратурія - через поки що невідомі
механізми може спричинятися до утво
рення кальцій-оксалатних каменів навіть
при нормальному вмісті в сечі кальцію
й оксалатів. Гіпоцитратурія часто асоці
йована з негазовим ацидозом і хронічною
діареєю.
Камені, що містять кальцій, інколи утво
рюються без будь-яких змін хімічного складу
сечі. У такому разі мову ведуть про ідіопатичну
сечокам’яну хворобу.
Фосфатно-амонієво-магнієві камені. Цей вид
каменів найчастіше утворюється після інфекцій
сечовивідних шляхів, викликаних бактеріями,
що розщеплюють сечовину до аміаку (протей
на інфекція, деякі види стафілококів). Зміщення
pH сечі в лужний бік при цьому зумовлює ви
падіння в осад магнієвих і амонійних солей фос
форної кислоти та подальшу їхню кристаліза
цію. Камені, що утворюються, мають особливо
великі розміри, оскільки концентрація сечовини
в сечі дуже висока і субстрату для діяльності
бактерій не бракує.
Уратні камені. їхнє утворення пов’язане
зі збільшенням концентрації сечової кислоти
в крові (гіперурикемією) і в сечі {гіперурикозурією). Це характерно для подагри та інших пору
шень пуринового обміну в організмі (див. главу
26). Проте у більш ніж половині випадків уратні
камені виникають без зазначених вище змін. Вва
жають, що причиною цього може бути тенденція
до утворення надто кислої сечі з pH нижче 5,5.
При таких значеннях водневого показника урати
втрачають свою розчинність і випадають в осад.
Цистинові камені. Виникають при спадково
зумовлених порушеннях реабсорбції амінокис
лот, зокрема цистину. Цистинурія стає причи
ною утворення каменів при зменшенні pH сечі.
Основні клінічні прояви. У вираженій формі
сечокам’яна хвороба характеризується:
а) нападами ниркової кольки, причиною яких
є спастичне скорочення гладкої мускулату
ри ниркової миски та сечоводу, зумовлене
їх закупоркою каменем;
б) гематурією, що виникає внаслідок ушко
дження сечовивідних шляхів каменями;
в) загальними ознаками запалення цих шля
хів - гарячкою, лейкоцитозом;
г) приєднанням інфекції та розвитком уна
слідок цього калькульозного пієлонефриту,
який при хронічному перебігові може при
звести до хронічної ниркової недостатності
(див. вище);
д) гостра затримка сечі, викликана механіч
ною закупоркою сечовивідних шляхів,
може стати причиною постренальної гос
трої ниркової недостатності (див. вище).
ГЛАВА 37
Потофізіологія
ендокринної
системи
Ендокринна система - це фізіологічна система,
основне призначення якої - регуляція структури,
функції та обміну речовин у периферичних ор
ганах і тканинах за допомогою спеціальних сиг
нальних речовин, що їх називають гормонами.
До складу цієї системи входять ендокринні
залози - спеціалізовані популяції секреторних
клітин, які утворюють і виділяють у кров (або
інші циркулюючі рідини) гормони або їхні най
ближчі біосинтетичні попередники.
Гормонами називають продукти діяльності
ендокринних залоз. Вони можуть синтезува
тися: (а) епітеліальними клітинами (власне за
лозистим епітелієм); (б) нейроендокринними
клітинами (клітини гіпоталамуса); (в) міоендокринними клітинами (м’язовими волокнами пе
редсердь).
Секрецію гормонів здійснюють:
а) ендокринні органи, що складаються із за
лозистих клітин одного типу (щитоподібна
залоза);
б) ендокринні органи, до складу яких входять
залозисті клітини різних типів (аденогіпофіз, кора надниркових залоз);
в) групи ендокринних клітин у неендокринних органах (підшлункова залоза).
Класифікація гормонів
I. За анатомічним принципом розрізняють
гормони гіпоталамуса, аденогіпофіза, нейрогіпофіза, кори і мозкової речовини над
ниркових залоз, щитоподібної та пршцитоподібних залоз і т.п.
II. За хімічною структурою виділяють:
а) стероїдні гормони (мінерало- і глюкокортикоїди, жіночі й чоловічі статеві гор
мони);
б) похідні амінокислот (тиреоїдні гормони,
катехоламіни, мелатонін);
в) білково-пептидні гормони (рилізинг-гормони, вазопресин, окситоцин, гормони
аденогіпофіза, інсулін, глюкагон, паратирин, кальцитонін).
III. За функціональними ефектами гормони
можуть бути:
а) ефекторними (діють безпосередньо на
органи-мішені);
б) тропними (регулюють синтез ефекторних гормонів);
в) рилізинг-гормонами (регулюють синтез
і секрецію тропних гормонів).
IV. За значенням для організму виділяють:
а) гормони, що забезпечують фізичний,
статевий і розумовий розвиток організ
му (соматотропний, гонадотропні, стате
ві гормони; нейропептиди);
б) адаптивні гормони, що забезпечують
довгострокову адаптацію організму до
змін зовнішнього середовища (тиреоїдні
гормони, АКТГ, глюкокортикоїди);
в) гомеостатичні гормони, які беруть
участь у підтриманні сталості внутріш
нього середовища організму (альдосте
рон, вазопресин, паратгормон, інсулін).
37.1.
ЗАКОНОМІРНОСТІ ПОРУШЕНЬ
ЕНДОКРИННОЇ ФУНКЦІЇ
Ендокринною функцією називають участь ен
докринних залоз у здійсненні гуморальної регу-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
348
.1. Ендокринна функція: її складові та варіанти порушень
ляції структурних змін, фізіологічних і біохіміч
них процесів в організмі.
Ендокринна функція має такі складові части
ни (рис. 37.1):
1) регуляція діяльності ендокринних залоз;
2) власне функція ендокринних залоз -утво
рення і секреція гормонів',
3) транспорт гормонів до клітин-мішеней;
4) взаємодія гормонів з периферичними клі
тинами - циторецепція;
5) руйнування гормонів та екскреція продук
тів їх інактивації.
Загалом існує три типи порушень ендокрин
ної функції (ендокринопатій):
а) гіперфункція ендокринних залоз (ендо
кринна гіперфункція);
6) гіпофункція (ендокринна гіпофункція);
в) дисфункція — різноспрямовані одночасні
зміни функції ендокринних залоз.
Залежно від того, порушення якої складової
ендокринної функції лежать в основі ендокри
нопатій, розрізняють три їх варіанти : дисрегу
ляторні, залозисті і периферичні.
Дисрегуляторні ендокринопатії. В основі їх
розвитку лежать розлади регуляції ендокринних
залоз.
Діяльність ендокринних залоз здійснюється
за допомогою чотирьох механізмів (рис. 37.2).
Такими є:
1) нервовий (імпульсно-медіаторний). За до
помогою прямих нервових впливів регу
люється діяльність: (а) мозкового шару
надниркових залоз; (б) нейроендокринних
структур гіпоталамуса; (в) епіфіза;
2) нейроендокринний (гіпоталамічний). Здійс
нюється нейроендокринними клітинами
гіпоталамуса, що перетворюють нервові
імпульси у специфічний ендокринний про
цес. При цьому утворюються і секретуються в систему портальних судин гіпофіза рилізинг-гормони, що регулюють діяльність
аденогіпофіза;
3) ендокринний. Його сутність полягає в без
посередньому впливі одних гормонів на
синтез і секрецію інших. Прикладом цього
механізму регуляції є дія тропних гормонів
аденогіпофіза на кору надниркових залоз,
щитоподібну залозу, статеві залози;
349
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
МЕХАНІЗМИ РЕГУЛЯЦІЇ
Рис. 37.2 Механізми регуляції ендокринних залоз
4)
неендокринний гуморальний. Здійснюється
неспецифічними гуморальними фактора
ми, зокрема метаболітами та іонами. Так,
концентрація глюкози в крові прямо впли
ває на синтез і секрецію інсуліну та глю
кагону; склад і рівень амінокислот - на
утворення соматотропного гормону; вміст
іонів калію - на виділення в кров альдо
стерону; концентрація кальцію — на секре
цію паратгормону і кальцитоніну.
Розвиток дисрегуляторних ендокринопатій
може бути пов’язаний з розладами всіх чоти
рьох механізмів регуляції. В одних випадках
при активації зазначених механізмів розвива
ється ендокринна гіперфункція, в інших, при
пригніченні регуляторних впливів, - ендокрин
на гіпофункція.
В основі регуляції ендокринної функції ле
жать два основних принципи:
а) прямих зв ’язків;
б) зворотних зв ’язків.
Принцип прямих зв ’язків покладено в основу
діяльності таких ендокринних функціональних
систем, як (а) гіпоталамо-гіпофізарно-наднирникова; (б) гіпоталамо-гіпофізарно-тиреоїдна;
і в) гіпоталамо-гіпофізарно-гонадальна.
Цей принцип реалізується через здійснен
ня трьох послідовних етапів: (1) сприйняття
гіпоталамусом вищих нервових регуляторних
сигналів і секреція відповідних рилізинг-гормонів; (2) секреція аденогіпофізом відповідних
тропних гормонів як відповідь на дію рилізинггормонів; (3) утворення й виділення перифе
ричними залозами (корою надниркових залоз,
щитоподібною залозою, статевими залозами)
ефекторних гормонів як відповідь на дію троп
них гормонів (рис. 37.3).
Порушення прямих зв’язків у регуляції ендо
кринних функцій можуть бути обумовлені роз
ладами синтезу й секреції:
а) рилізинг-гормонів;
б) тропних гормонів;
в) ефекторних гормонів.
Принцип зворотних зв 'язків. Розрізняють зво
ротні негативні зв’язки в системах “гіпотала
мус - аденогіпофіз - периферичні залози” й ко
роткі парагіпофізарні зворотні зв’язки.
Сутність перших полягає в тому, що утворюва
ні гормони пригнічують діяльність структур, які
здійснюють попередні етапи регуляції. Унаслідок
цього посилення секреції ефекторного гормону
через певні ланки призводить до зменшення його
утворення й надходження в кров, і навпаки, змен
шення вмісту гормону в крові викликає підви
щення інтенсивності його утворення та секреції.
Порушення негативних зворотних зв’язків
у системах “гіпоталамус - аденогіпофіз — пери-
350
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
феричні залози” мають велике значення в розви
тку ендокринної патології. Це положення можна
проілюструвати схематично такими прикладами.
• Система “гіпоталамус — аденогіпофіз —
щитоподібна залоза Дефіцит йоду в про
дуктах харчування і питній воді —» змен
шення утворення тиреоїдних гормонів —»
стимуляція гіпоталамуса й аденогіпофіза
—♦ збільшення утворення тиреоліберину
і тиреотропного гормону —* гіпертрофія
щитоподібної залози —> ендемічний зоб.
Система “гіпоталамус — аденогіпофіз — кора
надниркових залоз Тривале застосування з лі
кувальною метою глюкокортикоїдних препара
тів —» пригнічення діяльності відповідних струк
тур гіпоталамуса й аденогіпофіза -* зменшення
утворення кортиколіберину та АКТГ —*■ атрофія
пучкової зони кори надниркових залоз —» змен
шення утворення власних глюкокортикоїдів —>
при різкій відміні глюкокортикоїдних препаратів
розвивається синдром гострої недостатності
кори надниркових залоз (синдром відміни).
• Система “гіпоталамус - аденогіпофіз —
статеві залози”. Тривале застосування
(наприклад, спортсменами-важкоатлетами)
анаболічних стероїдів - похідних чолові
чих статевих гормонів —* пригнічення ді
яльності відповідних структур гіпоталамуса
й аденогіпофіза —* зменшення утворення
гонадотропних гормонів —* атрофія клітин
сім’яників, що продукують чоловічі статеві
гормони —► розвиток імпотенції та безплід
дя.
Короткі
парагіпофізарні
зворотні
зв’язки
(рис. 37.4) існують поза системою “гіпотала
мус - аденогіпофіз — периферичні залози”.
Вони відіграють провідну роль у регуляції ді
яльності прищитоподібних залоз, (3-клітин ост
рівців підшлункової залози. Ці зв’язки можна
описати схемами.
• Прищитоподібні залози: зменшення кон
центрації іонів кальцію в плазмі крові —>
збільшення продукції паратгормону —*■
збільшення вмісту іонів кальцію —¥ пригні
чення секреції паратгормону.
•
клітини острівців підшлункової залози:
збільшення концентрації глюкози в кро
ві —> активація утворення й секреції інсулі
ну —* зменшення вмісту глюкози в крові —►
пригнічення секреції інсуліну.
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
351
Рис. 37.4. Короткі парагіпофізарні зворотні зв'язки як механізми регуляції ендокринних залоз
Власне залозисті ендокринопатії. Порушення
діяльності залоз внутрішньої секреції можуть
бути зумовлені як кількісними, так і якісними
змінами ендокринних клітин.
1. Зміна кількості функціонально активних
клітин ендокринних залоз може спричи
нятися або до (а) ендокринної гіпофункції
(зменшення кількості клітин при видаленні
залози чи її частин, ушкодженні, некрозах
тощо), або до (б) ендокринної гіперфункції
(збільшення кількості клітин при добро
якісних і злоякісних пухлинах залозистого
епітелію).
2. Якісні зміни в ендокринних клітинах мо
жуть виявляти себе: (а) розладами біосин
тезу гормонів і (б) порушеннями процесів
їх секреції.
Причини порушень біосинтезу є різними для
гормонів з різною хімічною структурою.
Для білково-пептидних гормонів такими є:
а) розлади транскрипції;
б) порушення трансляції;
в) дефіцит необхідних амінокислот;
г) дефіцит АТФ;
д) порушення посттрансляційної модифікації
та активації продуктів синтезу.
Серед причин, що змінюють синтез стероїд
них гормонів:
а) порушення надходження в клітини, син
тезу і депонування холестеролу - вихідної
речовини для утворення цих гормонів;
б) набуті або спадково обумовлені дефекти
ферментів, що беруть участь у реакціях
біосинтезу стероїдів;
в) дефіцит кисню (гіпоксія), необхідного для
реакцій гідроксилювання;
г) дефіцит відновленого НАДФ (НАДФН) основного джерела електронів і протонів
у реакціях гідроксилювання стероїдів.
До порушень синтезу гормонів, що є похідни
ми амінокислот, ведуть:
а) дефіцит вихідних амінокислот (тирозину,
триптофану);
б) брак мікроелементів (йоду для утворення
тиреоїдних гормонів);
в) набуті або спадково обумовлені дефекти
ферментів синтезу цих гормонів;
г) дефіцит АТФ.
Секреція гормонів ендокринними клітинами
може здійснюватися одним із трьох механізмів:
1) вивільненням гормону із клітинних секре
торних гранул (секреція білково-пептид
них гормонів і катехоламінів);
2) вивільненням гормону з білковозв’язаної
форми (секреція тиреоїдних гормонів);
352
3) відносно вільною дифузією гормонів через
клітинні мембрани (секреція стероїдних
гормонів).
В основі розладів секреції гормонів можуть
лежати такі механізми:
а) порушення депонування гормонів. Це відбу
вається при неутворенні комплексів гормо
нів з речовинами, що їх називають факто
рами депонування (білками-нейрофізинами
для вазопресину й окситоцину, АТФ — для
катехоламінів, цинком — для інсуліну). Як
наслідок, гормони не здатні накопичувати
ся в необхідних кількостях у секреторних
гранулах;
б) порушення передачі сигналів, що стиму
люють секрецію. Вони часто пов’язані зі
зменшенням утворення в клітинах або над
ходження вторинних посередників (цАМФ,
іонів Са2+);
в) ураження контрактильних елементів (мікрофіламентів, мікротрубочок), що бе
руть участь у процесах екзо- й ендоцитозу.
Ці процеси становлять основу секреції білково-пептидних гормонів (екзоцитоз) і тиреоїдних гормонів (ендоцитоз);
г) дефіцит АТФ, унаслідок чого порушується
забезпечення енершзалежних процесів транс
порту гормонів у секреторних клітинах.
Периферичні ендокринопатії. В основі розвит
ку цього варіанта ендокринопатій можуть лежа
ти:
1) порушення транспорту гормонів в орга
нізмі;
2) розлади метаболічної інактивації гормо
нів;
3) порушення взаємодії гормонів з перифе
ричними клітинами-мішенями - патологія
циторецепції гормонів.
І.
Порушення транспорту гормонів в орга
нізмі. Існує чотири форми транспорту гормонів:
1) транспорт вільного гормону (розчиненого
у воді). Саме в такій формі гормон виявляє
свою біологічну активність, отже, від кон
центрації вільної форми гормону залежать
його функціональні, структурні й біохіміч
ні ефекти. У нормі вміст вільних гормонів
у крові не перевищує 10 % від загальної їх
кількості;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
2) комплекси гормонів зі специфічними транс
портними білками плазми крові. Вміст цієї
транспортної форми в крові становить 80 %
і більше від сумарної концентрації такого
гормону;
3) неспецифічні комплекси гормонів з білками
плазми крові (альбумінами, а-глобулінами);
4) адсорбція гормонів на поверхні формених
елементів крові (еритроцитів, лімфоцитів,
моноцитів).
Порушення транспорту гормонів в організмі
можуть виявляти себе двома типами розладів
ендокринної функції.
З одного боку, при збільшенні зв’язування
гормону зменшується вміст його вільної форми,
а отже, з’являються ознаки відповідної ендо
кринної гіпофункції.
З другого боку, зменшення зв’язування гор
мону викликає збільшення в крові концентрації
його вільної форми, що веде до появи ознак від
повідної ендокринної гіперфункції.
II.
Розлади метаболічної інактивації гормо
нів. Реакції біохімічних перетворень гормонів
залежать від їхньої структури. Так, руйнування
білково-пептидних гормонів відбувається в пе
чінці під дією ферментів пептидаз.
Метаболічна інактивація стероїдних гормо
нів здійснюється в печінці, кишках, нирках практично у всіх органах і тканинах, за винят
ком тиміко-лімфоцитарної системи. У реакціях
перетворення стероїдів беруть участь НАДФНзалежні ферменти. Інактивовані форми сте
роїдних гормонів, що утворилися в різних ор
ганах, надходять у печінку, де відбувається їх
кон’югація із сірчаною і глюкуроновою кис
лотами з подальшим виведенням з організму
у складі сечі та калу.
Інактивація катехоламінів може здійснюва
тися трьома шляхами:
1) окисненням, що його обумовлює
моноаміноксидаза (МАО-шлях);
2) дією катехолоксиметилтрансферази
(КОМТ-шлях);
3) хіноїдним окисненням з утворенням
адренохрому.
Метаболічні перетворення тиреоїдних гор
монів, що відбуваються переважно в печінці,
охоплюють:
гг
353
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
а) реакції дейодування;
б) окисне дезамінування
і декарбоксилювання залишків аланіну;
в) кон’югацію із сірчаною та глюкуроновою
кислотами.
У людини 65-95 % інактивованих метаболі
тів усіх гормонів виводиться з організму із се
чею.
Порушення метаболічних перетворень гор
монів можуть обумовлювати розвиток перифе
ричних розладів ендокринної функції. Так, при
уповільненні інактивації гормонів збільшується
вміст їх у крові, що виявляється ознаками від
повідної ендокринної гіперфункції. І навпаки,
прискорене перетворення гормонів у неактивні
форми супроводжується розвитком ендокринної
гіпофункції.
III.
Порушення взаємодії гормонів з перифе
ричними клітинами. Гормони є сигнальними
молекулами, дія яких на клітини-мішені реалі
зується через взаємодію з клітинними рецеп
торами (циторецепторами). Розрізняють два
принципово різних типи циторецепції гормонів:
а) внутрішньоклітинний тип, який лежить
в основі механізму дії стероїдних і тиреоїдних гормонів. Він пов’язаний з вільним
проходженням гормону через плазматичну
мембрану в клітину, у цитоплазмі якої від
бувається його взаємодія із внутрішньоклі
тинними білками-рецепторами. Ефекторною структурою клітини, на яку впливає
утворений комплекс, є ядро, а основним
біологічним ефектом - зміни інтенсивнос
ті процесів транскрипції, а отже, і синтезу
клітинних білків;
б) мембранний тип, що є основним механіз
мом дії білково-пептидних гормонів і катехоламінів. При цьому гормони не проника
ють усередину клітини, а зв’язуються з біл
ками-рецепторами на поверхні плазматич
ної мембрани. Далі передача регуляторного
сигналу з поверхні клітини до її ефекторних структур обумовлена появою в цито
плазмі так званих вторинних посередників,
або месенджерів. У результаті виникають
швидкі біохімічні ефекти, пов’язані з акти
вацією вже синтезованих ферментів чи ін
ших білків. Серед відомих сьогодні вторин
них посередників такі сполуки, як цикліч
ні нуклеотиди - цАМФ, цГМФ; іони Са2+,
фосфоліпідні месенджери—діацилгліцерол
(ДАГ) та інозитолтрифосфат (ІФ3). Доклад
но про механізми передавання сигналів від
циторецепторів до ефекторних структур
клітин ішлося в главі 8.
Порушення взаємодії гормонів з периферич
ними клітинами-мішенями може бути обумов
лене патологією клітинних рецепторів. Існує
два варіанти таких порушень:
1) зменшення кількості рецепторів або їхньої
спорідненості до гормону (десенситизація). При цьому, незважаючи на те, що
концентрація гормону в крові нормальна
або навіть підвищена, розвиваються озна
ки ендокринної гіпофункції',
2) збільшення кількості рецепторів до гормо
ну (сенситизація). Як правило, супрово
джується розвитком елементів відповідної
ендокринної гіперфункції.
ПАТОФІЗІОЛОГІЯ
ГІПОТАЛАМО-ГІПОФІЗАРНОЇ
СИСТЕМИ
Гіпоталамус є відділом центральної нерво
вої системи, де відбувається інтегрування нер
вових та ендокринних механізмів регуляції.
Це пов’язано з тим, що нейрони гіпоталамуса,
об’єднані в окремі ядра, є особливими нейрона
ми - нейроендокринними клітинами, здатними
синтезувати та вивільняти гормони.
Гіпоталамус анатомічно й функціонально
пов’язаний з передньою (аденогіпофізом) і зад
ньою (нейрогіпофізом) частками гіпофіза. Тому
виділяють дві функціональні системи: гіпоталамо-аденогіпофізарну
і
гіпоталамо-нейрогіпофізарну.
Гіпоталамо-аденогіпофізарна
система
Діяльність цієї системи пов’язана з утворенням
у гіпоталамусі гіпофізотропних гормонів. За
лежно від функціональних ефектів (активація
або пригнічення функції аденогіпофіза) їх поді
ляють на дві групи: -рилізинг-гормони (ліберини) і статини.
354
До активаторів секреторної функції клітин
аденогіпофіза відносять (рис. 37.5):
1) тиреотропіновий рилізинг-гормон
(тиреоліберин), який стимулює секрецію
тиреотропного гормону (ТТГ);
2) кортикотропіновий рилізинг-гормон
(кортиколіберин), що є активатором
секреції адренокортикотропного
гормону (АКТГ);
3) соматотропіновий рилізинг-гормон
(соматоліберин), дія якого спрямована
на посилення секреції соматотропного
гормону (СТГ);
4) гонадотропіновий рилізинг-гормон (гонадоліберин), що обумовлює секрецію обох
гонадотропних гормонів: фолікулостимуляційного (ФСГ) та лютеїнізуючого (ЛГ).
Інгібіторами секреції гормонів аденогіпофіза
є дві сигнальні сполуки:
1)пролактин-інгібіторний фактор, яким є
дофамін
(представник
катехоламінів).
Ця сполука пригнічує утворення і виділен
ня пролактину;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
2) соматотропін-інгібіторний фактор (соматостатин) - гормон, що інгібує секрецію
СТГ. Гормони гіпоталамуса вивільнюють
ся в гіпоталамо-гіпофізарну ворітну систе
му судин і з течією крові досягають клітин
аденогіпофіза.
Залежно від здатності сприймати барвники
ці клітини поділяють на (а) ацидофіли (мають
еозинофільну цитоплазму), (б) базофіли (їхня
цитоплазма сприймає основні компоненти фар
би) і (в) хромофобні клітини, які погано фарбу
ються як кислотними, так і основними барвни
ками.
Ендокринні клітини аденогіпофіза спеціалі
зуються на утворенні й секреції певного виду
тропних гормонів. З огляду на це виділяють:
а) соматотрофи, що продукують СТГ (гор
мон росту). Вони є ацидофілами, і на них
припадає майже половина клітин передньої
частки гіпофіза;
б) лактотрофи, що утворюють пролактин.
Як і попередні клітини, вони мають ацидо
фільну цитоплазму;
Рис. 37.5. Вплив рилізинг-гормонів гіпоталамуса на секрецію тропних гормонів аденогіпофізом.
КТРГ - кортикотропіновий рилізинг-гормон, АКТГ - адренокортикотропний гормон, ТТРГ - тиреотропіновий рилізинг-гормон,
ТТГ - тиреотропний гормон, ПІФ - пролактин-інгібіторний фактор, ПРЛ - пролактин, СТРГ - соматотропіновий рилізинг-гормон,
СІФ - соматотропін-інгібіторний фактор, СТГ - соматотропний гормон, ГТРГ - гонадотропіновий рилізинг-гормон,
ФСГ - фолікулостимуляційний гормон, ЛГ - лютеїнізуючий гормон
355
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇСИСТЕМИ
в) кортикотрофи, продуктом діяльності яких
є ціла низка пептидних сполук: АКТГ, пропіомеланокортин,
меланоцитстимуляційний гормон (МСГ), ендорфіни і ліпотропін;
г) тиреотрофи, що продукують ТТГ;
д) гонадотрофи, які утворюють ФСГ та ЛГ.
Кортикотрофи, тиреотрофи і гонадотрофи
є базофільними клітинами аденогіпофіза.
Для розуміння тих змін, що виникають в ор
ганізмі при порушеннях функціонування гіпоталамо-аденогіпофізарної системи, важливо знати
основні біологічні ефекти гормонів передньої
частки гіпофіза. Коротко розглянемо їх.
L Соматотропний гормон (СТГ, соматотропін, гормон росту). Має ростову і метабо
лічну активність.
Ростова активність пов’язана з дією СТГ на
рецептори гепатоцитів, клітин ниркового епі
телію та ін., унаслідок чого вивільняються тка
нинні фактори росту - соматомедини (інсуліноподібні фактори росту). Останні зумовлюють
такі ефекти:
а) посилюють поглинання сульфатів кліти
нами сполучної тканини та їх включення
в хондроїтинсульфат. Це викликає посиле
ний ріст хрящів;
б) збільшують кількість мітозів і стимулюють
клітинний поділ;
в) є гуморальними факторами, що беруть
участь у розвитку гіпертрофії різних органів.
Метаболічна активність. СТГ має цілий
ряд відносно швидких метаболічних ефектів,
не пов’язаних з утворенням соматомединів.
До них відносять:
а) вплив на вуглеводний обмін. СТГ, стимулю
ючи a-клітини острівців підшлункової за
лози, викликає збільшення продукції глю
кагону. При тривалій дії великих доз СТГ
розвивається інсулінорезистентність (див.
главу 24). Усе це в підсумку призводить до
гіперглікемії',
б) вплив на жировий обмін. СТГ, активуючи
ліполіз у жировій тканині, збільшує вміст
вільних жирових кислот у крові, сприяє
розвитку жирової інфільтрації печінки, ви
кликає гіперпродукцію кетонових тіл;
в) вплив на білковий обмін. СТГ має анаболічну дію. Він збільшує транспорт амінокис
лот у клітини та активує біосинтез білків.
Адренокортикотропний гормон (АКТГ,
кортикотропін). Має дію, пов’язану (гландотропну) і не пов’язану (негландотропну)
з впливом на кору надниркових залоз.
Гландотропна дія. АКТГ чинить вплив на
кору надниркових залоз (в основному на пучкову
зону), активує синтез і секрецію кортикостероїд
них гормонів, головним чином глюкокортикоїдів.
До негландотропних ефектів відносять:
а) посилення пігментації шкіри (відтворює
дію
меланоцитстимуляційного
гормо
ну);
б) мобілізацію жиру з жирових депо (ефект
подібний до впливу ліпотропіну).
3. Тиреотропний гормон (ТТГ). Діє на щи
топодібну залозу, стимулюючи утворення
і вивільнення тиреоїдних гормонів.
4. Фолікулостимуляційний гормон (ФСГ). Ви
кликає ріст фолікулів у яєчниках і утворен
ня естрогенів. У чоловіків регулює сперма
тогенез.
5. Лютеїнізуючий гормон (ЛГ). Зумовлює
утворення жовтого тіла в яєчниках і синтез
прогестинів. У чоловіків стимулює секре
цію тестостерону.
6. Пролактин. Стимулює ріст молочних за
лоз, утворення й секрецію молока.
2.
Узагальнені дані про гормони гіпоталамуса
та аденогіпофіза подано в табл. 37.1.
Порушення діяльності гіпоталамо-аденогіпофізарної системи можуть бути зумовлені таки
ми причинами (рис. 37.6).
1. Патогенна дія факторів зовнішнього і вну
трішнього середовища, які в нормі регу
люють стан цієї системи (негативні емоції,
біль, психічні порушення та ін.).
2. Ураження відділів центральної нервової
системи, які чинять регуляторний вплив
на гіпоталамус, — вищих центрів кори ве
ликих півкуль головного мозку, структур
лімбічної системи, ретикулярної формації.
Порушення центральної регуляції нейро
ендокринних зон гіпоталамуса настає при
збільшенні або зменшенні впливів на ньо
го. Так, зростання активуючих і послаблен
ня гальмівних сигналів веде до гіперфунк
ції гіпоталамо-аденогіпофізарної системи.
Відповідно, зменшення активуючої дії
і збільшення гальмівних впливів зумовлю
ють гіпофункцію цієї системи.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
356
Таблиця 37.1
Тиреотропіновий рилізинггормон (ТТРГ)
Пептид
(3 амінокислоти)
Стимулює секрецію ТТГ
Кортикотропіновий рилізинггормон (КТРГ)
1 пептидний ланцюг
(41 амінокислота)
Стимулює секрецію АКТГ
Соматотропіновий рилізинггормон (СТРГ)
1 пептидний ланцюг
(44 амінокислоти)
Стимулює секрецію СТГ
Гонадотропіновий рилізинггормон (ГТРГ)
1 пептидний ланцюг
(10 амінокислот)
Стимулює секрецію ФСГ та ЛГ
Пролактин-інгібіторний
фактор (ПІФ)
Дофамін
Інгібує секрецію пролактину
Соматотропін-інгібіторний
фактор (СІФ)
1 пептидний ланцюг
(14 амінокислот)
Інгібує секрецію СТГ
Аденогіпофіз
Тиреотропний гормон (ТТГ)
Глікопротеїн, складається з 2-х
ланцюгів: а (92 амінокислот),
Р (138 амінокислот)
Стимулює секрецію тиреоїдних
гормонів, див. 37.3
Адренокортикотропний
гормон (АКТГ)
1 пептидний ланцюг
(39 амінокислот)
Стимулює секрецію глюкокортикоїдів і андрогенів, див. 37.2
1 поліпептидний ланцюг
(191 амінокислота)
Стимулює ріст, впливає на вуглевод
ний, жировий і білковий обмін
Фолікулостимуляційний
гормон (ФСГ)
Глікопротеїн, складається з 2-х
ланцюгів: а (92 амінокислот),
Р (129 амінокислот)
Стимулює ріст фолікулів у яєчниках,
утворення естрогенів. У чоловіків
регулює сперматогенез
Лютеїнізуючий гормон (ЛГ)
Глікопротеїн, складається з 2-х
ланцюгів: а (92 амінокислот),
Р (141 амінокислот)
Стимулює утворення жовтого тіла
в яєчниках, синтез прогестинів; у чо
ловіків - секрецію тестостерону
Пролактин (ПРЛ)
1 поліпептидний ланцюг
(227 амінокислот)
Стимулює ріст молочних залоз,
утворення і секрецію молока
і Соматотропний гормон (СТГ)
3. Ураження гіпоталамуса, при яких порушу
ється утворення і виділення гормонів ней
роендокринними клітинами. При цьому
гіперфункція
гіпоталамо-аденогіпофізарної
системи розвивається при збільшенні се
креції рилізинг-гормонів і зменшенні утво
рення статинів, а гіпофункція - навпаки,
при пригніченні виділення ліберинів і по
силенні секреції статинів.
4. Ураження аденогіпофіза, що ведуть до по
рушення утворення і секреції тропних гор
монів. Залежно від спрямованості цих по
рушень можуть розвиватися ендокринна
гіпер- або гіпофункція.
Гіпопітуїтаризм
Гіпофункцію аденогіпофіза позначають термі
ном “гіпопітуїтаризм”. Розрізняють пангіпопітуїтаризм і парціальний гіпопітуїтаризм.
Пангіпопітуїтаризмом називають змен
шення утворення всіх гормонів аденогіпофіза.
357
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
Основні причини порушень діяльності гіпоталамо-аденогіпофізарної системи (див. текст)
В експерименті його моделюють видаленням
залози — гіпофізектомією.
Причини:
1) ушкодження гіпофіза. Зменшення кількос
ті функціонально активних ендокринних
клітин може бути зумовлено операціями
на гіпофізі з приводу видалення його пух
лин, а також застосуванням іонізуючої ра
діації при їх променевій терапії;
2) ішемія аденогіпофіза. Вона є причиною
післяпологового некрозу гіпофіза - синдро
му Шихана (Sheehan’s syndrome). Останній
зумовлюється зменшенням кровопоста
чання залози, що настає після ускладнень
пологів, які порушують системний крово
обіг (значна крововтрата, шок). Річ у тім,
що процес вагітності супроводжується
значним збільшенням (майже вдвічі) маси
й об’єму гіпофіза. При цьому васкуляризація залози залишається такою ж, як і була.
Іншими словами, при вагітності має місце
відносна недостатність кровопостачання
залози, яка стає абсолютною і значною при
падінні системного артеріального тиску,
що спостерігається при багатьох усклад
неннях пологів.
Ішемічний некроз розвивається саме
в передній частці гіпофіза, оскільки її кро
вопостачання здійснюється через мережу
венозних судин, що йдуть сюди з гіпотала
муса (ворітна система!), а отже, кров, що
в них міститься, несе кисню менше, ніж
кров артеріальна. Задня ж частка залози
є значно стійкішою і, як правило, не ура
жається, бо кровопостачається, як і біль
шість периферичних тканин, через арте
ріальні судини: з цієї причини зменшення
артеріального тиску не має для неї таких
фатальних наслідків;
3) хромофобні аденоми гіпофіза, тобто пух
лини, що ростуть із неендокринних хромофобних клітин. Пухлина, що збільшу
ється в об’ємі, здавлює та ушкоджує зало
зисті клітини аденогіпофіза, заповнюючи
собою все більший і більший простір, що
його займає гіпофіз, і витісняючи в такий
358
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
спосіб функціонально активну тканину
залози.
Клінічні прояви пангіпопітуїтаризму пов’язані
з дефіцитом гормонів аденогіпофіза і порушен
ням діяльності периферичних ендокринних за
лоз (щитоподібної, статевих, кори надниркових
залоз). Перші ознаки ураження аденогіпофіза на
стають при ушкодженні 70-75 % тканини залози,
а для розвитку повної картини пангіпопітуїтариз
му необхідне руйнування 90-95 % аденогіпофі
за. Синдроми, що розвиваються при тотальному
порушенні функцій аденогіпофіза, випливають
з наслідків дефіциту кожного з тропних гормо
нів, що тут продукується (див. табл. 37.2).
Парціальний гіпопітуїтаризм. Так назива
ють порушення утворення не всіх, а окремих
гормонів аденогіпофіза.
Причинами парціального гіпопітуїтаризму є, як
правило, спадкові порушення процесів, що лежать
в основі синтезу й секреції відповідних гормонів.
А тому прояви хвороби з’являються або в дитячо
му віці, або в період статевого дозрівання.
Описано такі варіанти парціального гіпопіту
їтаризму:
а) гіпофізарний нанізм (карликовість) - від
ставання і зупинка росту, зумовлені не
достатністю СТГ;
б) вторинний гіпогонадизм — порушення
статевого дозрівання внаслідок дефіциту
ФСГ і ЛГ (див. 37.6);
в) вторинний гіпотиреоз — гіпофункція
щитоподібної залози, зумовлена дефіци
том ТТГ (див. 37.4);
г)
вторинний гіпокортицизм — недостат
ність кори надниркових залоз через брак
АКТГ.
Гіперпітуїтаризм
Гіперфункцію аденогіпофіза позначають як гі
перпітуїтаризм. Основною причиною його роз
витку є доброякісні пухлини - аденоми ендо
кринних клітин.
В етіології аденом гіпофіза важливого значен
ня надають генетичним порушенням механізмів
регуляції проліферативної активності клітин.
Серед інших описано синдром множинної ендо
кринної неоплазії (multiple endocrine neoplasia,
MEN), що виявляє себе спадково обумовленою
гіперплазією багатьох залоз внутрішньої секре
ції, в т.ч. аденогіпофіза. Залежно від походжен
ня розрізняють дві групи аденом.
І.
Еозинофільні
аденоми.
Розвиваються
з ацидофільних клітин аденогіпофіза. До таких
аденом відносять:
а) пролактиному - пухлину, що росте з лактотрофів. На цей вид аденом припадає близь
ко ЗО % усіх клінічних випадків доброякіс
них пухлин аденогіпофіза. Виявляє себе
синдромом
гіперпродукцїі
пролактину:
припиненням менструацій (аменореєю),
витіканням молока, молозива чи молокоподібної рідини з молочних залоз, не зумов
леним вагітністю (галактореєю); відсутніс
тю лібідо, безпліддям;
б) аденому соматотрофів, що продукують
гормон росту - СТГ. Клінічно надмірне
Таблиця 37.2
Основні клінічні прояви пангіпопітуїтаризму
359
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
Таблиця 37.3
Функціональні ефекти гормонів нейрогіпофіза
утворення гормону виявляє себе гігантиз
мом (якщо аденома розвивається в дітей
і молодих людей до закриття епіфізарних
хрящів) або акромегалією (у дорослих).
Для гігантизму характерне більш-менш про
порційне збільшення всіх складових частин
тіла, хоча надмірна довжина верхніх та нижніх
кінцівок все ж таки порушує цю пропорцій
ність. Акромегалія виявляє себе посиленим
ростом акральних (кінцевих) ділянок рук, ніг,
підборіддя, носа, язика, а також збільшенням
об’єму внутрішніх органів: печінки, серця, щи
топодібної залози та ін.
Крім того, розвиваються ознаки підвищеної
метаболічної активності СТГ - гіперглікемія,
інсулінорезистентність, аж до розвитку метагіпофізарного іскрового діабету, жирова інфіль
трація печінки.
II. Базофільні аденоми. Ростуть із базофільних клітин аденогіпофіза - кортикотрофів, тиреотрофів і гонадотрофів. Із трьох можливих
варіантів пухлин найчастіше буває і виявляє
себе вираженими клінічними проявами адено
ма, що продукує АКТГ. При цьому розвиваєть
ся хвороба Іценка - Кушинга. Вона характери
зується:
а) вторинним гіперкортицизмом — збільшен
ням утворення кортизолу (див. 37.3);
б) посиленою пігментацією шкіри (негландотропна дія АКТГ, див. вище).
Досить рідко бувають пухлини, що продуку
ють інші гормони: ТТГ, гонадотропні гормони
(ФСГ та ЛГ), МСГ. Якщо вони і розвиваються,
то не мають виражених специфічних ознак, їх
часто виявляють випадково при інструменталь
них дослідженнях головного мозку.
Гіпоталамо-нейрогіпофізарна
система
Основним структурним елементом гіпоталамонейрогіпофізарної системи є супраоптичне і паравентрикулярні ядра гіпоталамуса, які у відпо
відь на надходження інформації від рецепторів,
що контролюють показники гомеостазу (осмо
тичний тиск, об’єм циркулюючої крові, артері
альний тиск), і від вищих нервових центрів про
дукують вазопресин та окситоцин. ЦІ гормони
по аксонах нейроендокринних клітин спуска
ються в задню частку гіпофіза, звідки надходять
у кров. Основні функціональні ефекти вазопре
сину й окситоцину наведено в табл. 37.3.
При порушенні функціонування гіпоталамонейрогіпофізарної системи може виникати кіль
ка варіантів розладів в організмі.
І. Синдром неадекватної секреції вазопреси
ну (синдром Пархона). Виникає при пух
линах різних тканин, що утворюють вазо
пресин (ектопічна продукція), а також при
розладах регуляції ендокринної функції
гіпоталамуса. Характеризується підвище
360
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ною секрецією вазопресину навіть тоді,
коли вона має гальмуватися (при зменшен
ні осмотичного тиску плазми крові).
Основним механізмом появи клінічних ознак
є вторинна гіпонатріємія, яка настає в резуль
таті посилення факультативної реабсорбції води
та ефекту розведення. Зменшення позаклітин
ної концентрації іонів натрію веде до перероз
поділу води: вона переміщується у внутрішньо
клітинний сектор (див. главу 27). Як наслідок,
розвивається набухання і набряк головного моз
ку з появою відповідних неврологічних симпто
мів. При цьому, хоча загальний об’єм води в ор
ганізмі збільшується, об’єм циркулюючої крові
істотно не міняється й набряки периферичних
тканин не виникають.
Прояви синдрому значно зменшуються при
обмеженні надходження води в організм.
II. Нецукровий діабет. Розрізняють два його
патогенетичних варіанти:
центральний
(нейрогенний), при якому утворюється
мало вазопресину, і нефрогений, в основі
якого - нечутливість епітеліальних клітин
дистальних відділів нефронів і збірних
трубок до дії вазопресину (відсутність або
мало У2-рецепторів).
У патогенезі основних проявів нецукрового
діабету провідну роль відіграє зменшення фа
культативної реабсорбції води в нирках. Це при
зводить до поліурії (добовий діурез зростає до
25 л — сечовиснаження) і зневоднення. Останнє
зумовлює спрагу (іполідипсію). У декомпенсо
ваному стані зменшується об’єм циркулюючої
крові (гіповолемія) і падає артеріальний тиск,
розвивається гіпоксія.
III. Зменшення продукції окситоцину. Виявляє
себе порушеннями лактації, слабкістю по
логової діяльності.
Синдромів, обумовлених збільшенням секре
ції окситоцину, не описано.
ПАТОФІЗІОЛОГІЯ
НАДНИРКОВИХ ЗАЛОЗ
Надниркові залози складаються з двох різних
структур:
1) кіркової речовини;
2) мозкової речовини.
На кору припадає приблизно 70-80 % від усієї
маси залоз, решта - це мозкова речовина, роз
ташована в центральній зоні кожного з двох ен
докринних органів.
Кора і мозок істотно відрізняються між собою
не тільки структурою, а й походженням, харак
тером гормонів, що тут продукуються; механіз
мами регуляції ендокринної функції.
Кора надниркових залоз секретує кортико
стероїди, що синтезуються з холестеролу,
а мозкова речовина є джерелом катехоламінів (адреналіну й норадреналіну), що тісно
пов’язані з функціонуванням вегетативних
симпатичних нервів і утворюють разом з ними
симпатоадреналову систему. З огляду на сут
тєві відмінності ендокринної функції кори над
ниркових залоз та їхньої мозкової речовини по
рушення діяльності цих двох структур будемо
розглядати окремо.
ПАТОФІЗІОЛОГІЯ КОРИ
НАДНИРКОВИХ ЗАЛОЗ
Кора надниркових залоз гістологічно складаєть
ся з трьох зон, кожна з яких синтезує свої стеро
їдні гормони.
У клубочковій (зовнішній) зоні утворюються
мінералокортикоїди - гормони, що регулюють
в основному водно-сольовий обмін. Основним
представником цієї групи є альдостерон, інтен
сивність секреції якого становить 0,125 мг/добу.
Пучкова (середня) зона є джерелом глюкокортикоїдів — гормонів, що впливають на обмін
речовин в організмі. У людини основний глюкокортикоїд - кортизол. Менше значення мають
кортизон і кортикостерон. Інтенсивність секре
ції глюкокортикоїдів становить 20-25 мг/добу.
Клітини сітчастої (внутрішньої) зони син
тезують чоловічі статеві гормони - андрогени
і в меншій кількості - жіночі статеві гормони естрогени (про них мова піде у частині 37.6).
Оскільки порушення, що виникають в орга
нізмі при ураженнях кори надниркових залоз,
прямо пов’язані зі зменшенням чи посиленням
дії її гормонів, то є сенс спочатку зробити неве
ликий огляд основних біологічних ефектів цих
361
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
гормонів, механізмів регуляції їхньої секреції та
дії на клітини-мішені.
Мінералокортикоїди
Альдостерон - основний представник мінералокортикоїдів — є життєво необхідним гормоном.
При видаленні обох надниркових залоз (адреналектомії) смерть тварин настає через відсутність
саме цього гормону.
В організмі функціонують три механізми ре
гуляції, що посилюють утворення і секрецію
альдостерону:
1) ангіотензиновий механізм. Він пов’язаний
з активацією ренін-ангіотензинової систе
ми та утворенням ангіотензину II. Цей пеп
тид по суті відіграє роль тропного гормону
для клубочкової зони кори надниркових
залоз, а отже, стимулює надходження аль
достерону в кров (докладно див. главу 27);
2) АКТГ-опосередкований механізм. Суть його
полягає в тому, що тропний ефект ангіотен
зину II виявляє себе в повній мірі лише при
наявності АКТГ. Такий вплив одного гор
мону на ефекти іншого позначають термі
ном “пермісивна дія”. За відсутності АКТГ
чи при значному пригніченні його секреції
істотно зменшується, аж до повного зник
нення, стимуляційна дія ангіотензину II на
клубочкову зону;
3) пряма стимуляція секреції альдостерону
високими концентраціями іонів калію плаз
ми крові. При гіперкаліємії відбувається пе
реміщення калієвих іонів всередину клітин,
що викликає деполяризацію плазматичної
мембрани з відкриттям потенціалзалежних
Са-каналів, входженням іонів Са2+ в цито
плазму. Останні запускають секреторний
процес, що завершується надходженням
альдостерону в кров.
Крім стимуляторів секреції альдостерону,
в організмі діють і механізми, що пригнічують
цей процес. До таких, зокрема, відносять (а) передсердний натрійуретичний гормон (атріопептин) і (б) дофамін.
Біологічні ефекти альдостерону можна розді
лити на дві групи.
І. Ниркові ефекти. Альдостерон, діючи на
дистальні звивисті канальці нефронів, збіль
шує: (а) реабсорбцію іонів натрію; (б) сек
рецію іонів калію; (в) секрецію іонів водню
(ацидогенез) (докладно див. глави 27 і 29).
II. Позаниркові ефекти. Вони пов’язані із
впливом альдостерону на слинні й потові
залози, дистальні відділи товстої кишки
і спрямовані на затримку натрію в організ
мі та виведення калію.
Глюкокортикоїди
Утворення й секреція кортизолу та інших глюкокортикоїдів, на відміну від альдостерону, пов
ністю залежать від АКТГ. Вплив цього гормону
на пучкову зону надниркових залоз виявляє себе
цілим рядом змін, що виникають у різний час
від початку дії АКТГ. Так, розрізняють:
1) гострий ефект (протягом кількох хвилин).
У його основі лежить залежне від вторин
них внутрішньоклітинних посередників
(цАМФ, фосфоліпідних месенджерів) фосфорилювання білків, що мають стосунок до
біосинтезу стероїдних гормонів. Це зокре
ма виявляє себе полегшеним зв’язуванням
холестеролу з цитохромом Р450 і посилен
ням трансляції наявної інформаційної РНК.
Як наслідок - істотно збільшується інтен
сивність утворення стероїдів;
2) підгострий ефект (через десятки годин).
Посилюються процеси транскрипції генів,
що кодують структуру ферментів стероїдогенезу. Оскільки кількість молекул та
ких ферментів зростає, то й збільшується
синтез кортикостероїдів;
3) хронічний ефект (через кілька діб). Акти
вація транскрипції багатьох генів веде до
структурних змін - розвивається гіпертро
фія й гіперплазія пучкової зони кори над
ниркових залоз.
Біологічна дія глюкокортикоїдів залежить від
їхнього рівня в крові, а тому велику кількість
різних ефектів цих гормонів поділяють на дві
групи: низькодозові й високодозові.
І. Низькодозові ефекти. Так називають дію
фізіологічних концентрацій глюкокортикоїдів.
Низькодозовими вважають ефекти замісних
доз глюкокортикоїдних гормонів, що усувають
зміни в організмі тварин після видалення у них
надниркових залоз. Експериментальні дослі
дження дали змогу встановити, що фізіологічні
концентрації глюкокортикоїдів:
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
362
1) чинять вплив на вуглеводний обмін. Вони,
з одного боку, посилюють глюконеогенез,
а з другого **# зменшують чутливість тка
нин до інсуліну (див. главу 24);
2) впливають на білковий обмін. Глюкокортикоїдам притаманна катаболічна дія:
вони активують протеоліз у периферичних
тканинах, унаслідок чого збільшується
надходження амінокислот у кров і розви
вається аміноацидемія. Водночас ці гор
мони пригнічують біосинтез білків у тих
самих тканинах;
3) мають пермісивну дію. Глюкокортикоїди
є конче необхідними для того, аби про
являлися біологічні ефекти деяких інших
гормонів. Так, за відсутності кортизолу
значно послаблюється, а то й зовсім зни
кає ліполітична, гіперглікемічна, пресорна, бронхорозширювальна і калоригенна
дія катехоламінів (див. частину 37.3.2),
а також гіперглікемічна дія глюкагону,
4) нормалізують діяльність центральної нер
вової системи і тонус скелетних м’язів
у тварин після адреналектомії. Механізми
такого тонізуючого впливу допоки невідомі;
5) впливають на процеси росту й диференці
ацію органів та тканин в ембріогенезі. Ме
ханізми цього ефекту ще не з’ясовано, але
відомо, що глюкокортикоїди усувають такі
прояви адреналектомії, як затримка росту
у тварин.
II. Високодозові ефекти. Високі концентрації
глюкокортикоїдів у крові можуть утворюватися
при:
а) дії на організм надмірних за силою або
патогенних агентів, що спричиняють роз
виток неспецифічної реакції під назвою
стрес (див. главу 8);
6) патологічній гіперфункції кори наднирко
вих залоз (див. нижче);
в) введенні глюкокортикоїдів та їхніх фарма
кологічних аналогів з лікувальною метою.
До високодозових ефектів глюкокортикоїдних гормонів відносять:
1) антианаболічну дію — пригнічення біо
синтезу білків у різних органах і ткани
нах. Клінічно вона виявляє себе слабкістю
м’язів (особливо рук і плечового пояса),
затримкою росту дітей, уповільненням
2)
3)
4)
5)
загоєння ран, переломів кісток через при
гнічення синтезу сполучнотканинних про
теїнів;
гіперглікемічну дію. Підвищення концен
трації глюкози в крові зумовлене цілим
рядом механізмів: (а) збільшенням інтен
сивності глюконеогенезу в печінці, (б) під
вищенням секреції глюкагону, (в) змен
шенням використання глюкози перифе
ричними клітинами, зокрема м’язовими,
лімфоцитами. Як реакція у відповідь, роз
вивається вторинний гіперінсулінізм з по
дальшим виснаженням P-клітин острівців
підшлункової залози і появою вторинної
інсулінової недостатності (див. главу 24).
Результат цього - розвиток метастероїдного цукрового діабету,
ліполітично-ліпотропну дію. Глюкокор
тикоїди стимулюють утворення й дію на
жирову тканину гормонів, що мають ліполітичні властивості (адреналін, глюкагон).
У результаті збільшується вміст вільних
жирових кислот у крові (гіперліпацидемія),
відбувається відкладення жиру на тулубі
й обличчі. Останнє набуває місяцеподібної форми, що й надає такому ожирінню
специфічних рис;
гіпертензивну дію. Підвищення артеріаль
ного тиску почасти пов’язане з тим, що
глюкокортикоїди у великих дозах мають
деякі властивості мінералокортикоїдів,
тобто, збільшуючи реабсорбцію іонів на
трію, сприяють підвищенню об’єму цир
кулюючої крові. Крім того, кортизол по
силює пресорну дію катехоламінів і при
гнічує утворення депресорних факторів
у нирках, зокрема простагландинів (див.
нижче);
антимітотичну 1 цитолітичну дію. Висо
кі дози глюкокортикоїдних гормонів при
гнічують клітинний поділ, унаслідок чого
порушуються процеси фізіологічної та
репаративної регенерації. Завдяки цьому
ефекту пригнічується і ріст пухлин. Змен
шення утворення соматомединів у печінці
є ще одним механізмом уповільнення реге
нераторних процесів. Крім того, глюкокор
тикоїди зумовлюють руйнування (цитоліз)
деяких типів клітин, зокрема лімфоцитів.
Вважають, що основний механізм такого
ефекту - запуск апоптозу (див. главу 7).
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
Цитолітична дія глюкокортикоїдних гор
монів виявляє себе інволюцією вилочкової
залози, еозинопенією, протилейкозними
ефектами;
6) їмунодепресивну дію. Вона обумовлена
пригніченням проліферації лімфоцитів
(антимітотична дія) і зменшенням утво
рення антитіл (прояв антианаболічної дії).
Завдяки впливу на імунну систему глюкокортикоїди запобігають розвиткові алер
гічних реакцій сповільненого типу (див.
главу 15);
7) ульцерогенну дію. Високі дози глюкокортикоїдів можуть викликати розвиток виразок
у шлунку і дванадцятипалій кишці. Це обу
мовлено тим, що ці гормони (а) пригнічу
ють утворення простагландинів, у резуль
таті чого збільшується шлункова секреція
(див. главу 34); (б) зменшують утворення
слизу (антианаболічна дія); (в) пригнічу
ють фізіологічну регенерацію епітелію
травного каналу (антимітотична дія);
8) остеопатичну дію. Під впливом глюко
кортикоїдних гормонів розвивається остеопороз, що часто виявляє себе компре
сійними переломами хребців і переломами
довгих кісток. Серед складних і ще не до
кінця з’ясованих механізмів цього ефек
ту — пригнічення синтезу білкових компо
нентів органічної матриці кісткової ткани
ни як результат загальної антианаболічної
дії гормонів;
9) психотропну дію. Механізми впливу глюкокортикоїдів на психічні процеси люди
ни ще не з’ясовано. У деяких схильних до
цього людей високі дози гормонів можуть
спричинятися до психозів-,
10) ліпокортинові ефекти. Так називають ці
лий ряд високодозових ефектів глюкокортикоїдів, пов’язаних з утворенням у кліти
нах білка ліпокортину.
Ліпокортин є ендогенним внутрішньоклітин
ним інгібітором фосфоліпази Аг Пригнічення ак
тивності цього ферменту припиняє вивільнення
арахідонової кислоти з фосфоліпідів мембран,
унаслідок чого не утворюються дві важливі групи
біологічно активних речовин: (а) продукти циклоксигеназного (простагландини, простациклін,
тромбоксани) і (б) ліпоксигеназного (лейкотрієни) шляхів перетворення арахідонової кислоти.
363
Сьогодні встановлено, що глюкокортикоїди є індукторами транскрипції гена, що кодує
структуру ліпокортину (рис. 37.7). Поява цього
білка в клітинах запобігає гідролітичному роз
щепленню мембранних фосфоліпідів і веде до
того, що не утворюються біологічно активні
речовини - похідні арахідонової кислоти. Саме
ця обставина й зумовлює ряд ефектів, що їх
об’єднують поняттям ліпокортинові. Нижче на
ведено основні з них.
Протизапальна дія. З одного боку, вона
пов’язана зі зменшенням утворення лізофосфоліпідів, які є сильними детергентами
(див. главу 7). Унаслідок цього зменшується
ушкодження мембран (стабілізація мемб
ран), а отже, і явища альтерації. З другого
боку, пригнічується утворення медіаторів
запалення (простагландинів, лейкотрієнів),
у результаті чого менш вираженими стають
судинні реакції у вогнищі запалення, під
вищення проникності судин, хемотаксис
лейкоцитів (див. главу 17).
Протиалергічна дія. Обумовлена змен
шенням утворення медіаторів алергічних
реакцій негайного типу (І типу за Кумбсом
і Джеллом), зокрема лейкотрієнів, що ви
кликають бронхоспазм і напади бронхіаль
ної астми (див. главу 15).
Жарознижувальна
дія.
Глюкокортикоїди
зменшують зумовлюване вторинними пірогенами
(інтерлейкіном-1)
утворення
простагландинів у центрі терморегуляції
в гіпоталамусі, унаслідок чого гарячка не
розвивається або, якщо вона вже виникла,
зменшується (див. главу 18).
Шлункова гіперсекреція. Пов’язана зі змен
шенням гальмівної дії простагландинів на
секрецію хлористоводневої кислоти парієтальними клітинами залоз шлунка (див.
главу 34).
• Антиагрегантна дія. Зменшення утворен
ня тромб океанів з арахідонової кислоти
веде до пригнічення агрегації тромбоцитів
і судинно-тромбоцитарного гемостазу.
Гіпертензивна дія. Пов’язана зі зменшен
ням утворення в нирках простагландинів,
що вважаються природними депресорними
факторами (див. главу 32).
Тератогенна дія - формування “вовчої
пащі”, “заячої губи”. У розвитку цих де
фектів має значення порушення виходу
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
364
Л іпокортиновий механізм дії високих доз глюкокортикоїдних гормонів
гідролітичних ферментів з клітин епітелію
слизової оболонки піднебіння у довкружну
тканину. Це пояснюють тим, що за відсут
ності активної фосфоліпази А2 мембрана
лізосом залишається непроникною для сво
їх ферментів, вони не можуть вийти за межі
клітин, зруйнувати епітелій країв двох по
ловин твердого піднебіння і створити тим
самим умови для їх зрощення.
Гіпофункція кори надниркових залоз
Зниження ендокринної функції кори наднирко
вих залоз, аж до повного її припинення, виявляє
себе розвитком синдрому адренокортикальної
недостатності.
Класифікація
I. За етіологією недостатність кори наднир
кових залоз може бути (а) спадково зумов
леною і (б) набутою.
II. За механізмами розвитку розрізняють пер
винну і вторинну адренокортикальну недо
статність. Первинна виникає як результат
власне ураження надниркових залоз, вто
ринна пов’язана або з ураженнями гіпота
ламуса (дефіцит кортикотропінового рилізинг-гормону), або з гіпофункцією аденогіпофіза (дефіцит АКТГ).
III. Залежно від характеру порушень виділя
ють (а) тотальну (порушено утворення
всіх гормонів) і (б) парціальну (як прави
ло, спадково обумовлені дефекти синтезу
окремих гормонів) недостатність кори над
ниркових залоз.
IV. За клінічним перебігом недостатність кори
надниркових залоз може бути гострою
і хронічною.
Гостра адренокортикальна недостатність.
Основними її причинами є:
а) стан після адреналектомії (видалення над
ниркових залоз);
б) крововиливи в надниркові залози, що ви
никають при сепсисі, особливо при менін
гококовій інфекції, - синдром Уотерхауса Фрідеріксена;
в) раптова відміна глюкокортикоїдних препа
ратів.
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
Перші дві причини зумовлюють первинну,
а остання - вторинну (пов’язану з гіпофункцією
аденогіпофіза) недостатність кори надниркових
залоз. Гостра адренокортикальна недостатність
може виникати у хворих з хронічною недостат
ністю під впливом різних стресорних агентів
(див. главу 8). У такому випадку мову ведуть
про адренаповий криз.
Хронічна
адренокортикальна
недостатність
відома під назвою хвороби Аддісона (бронзової
хвороби). Найчастіші її причини:
а) туберкульозне руйнування надниркових за
лоз;
б) автоімунне ушкодження їх (автоімунний
адреналіт);
в) синдром набутого імунодефіциту (СНІД);
г) метастази злоякісних пухлин.
В економічно розвинених країнах від 60 % до
70 % випадків хвороби Аддісона зумовлюєть
ся автоімунними механізмами: ушкодженням
секреторних клітин (алергічними реакціями
II і IV типу) та утворенням автоантитіл проти
основних ферментів стероїдогенезу (21-гідроксилази, 17-гідроксилази).
Автоімунний адреналіт може виявляти себе
у трьох клінічних формах:
автоімунний поліендокринний синдром
I типу (autoimmune polyendocrine syndrome
type 1; APS1). Цей синдром зумовлюється
мутаціями гена AIRE, продукт якого бере
участь у регуляції автоімунних процесів
в організмі. Характеризується множинни
ми ураженнями залоз внутрішньої секреції
(автоімунний адреналіт, автоімунний гіпопаратиреоз, ідіопатичний гіпогонадизм),
що поєднуються з кандидозом слизових
оболонок і шкіри, дистрофічними змінами
в органах і тканинах ектодермального по
ходження (шкіра, зуби, нігті);
автоімунний поліендокринний синдром
II типу (APS2). Для нього характерним
є поєднання хронічної адренокортикальної
недостатності з автоімунним ураженням
щитоподібної залози (автоімунний тиреоїдит) або з цукровим діабетом І типу. На від
міну від APS1, кандидоз та дистрофічні
ураження структур ектодермального похо
дження не розвиваються. APS2 не є моногенною спадковою хворобою, у виникненні
365
цього синдрому має значення поліморфізм
генів, зокрема локусу HLA. Ця обставина
дає підстави відносити APS2 до полігенних
хвороб (див. главу 12);
ізольований автоімунний адреналіт. Харак
теризується ураженнями кори надниркових
залоз без втягування у патологічний процес
інших залоз внутрішньої секреції. Так само,
як APS2, вважається полігенною хворобою.
Основні клінічні прояви адренокортикаль
ної недостатності. їх можна розділити на
дві великі групи, враховуючи ту обставину, що
при ураженні кори надниркових залоз зменшу
ється або ж зовсім припиняється секреція двох
основних видів гормонів — мінералокортикоїдів
і глюкокортикоїдів.
І. Прояви, пов’язані з випадінням мінералокортикоїдних функцій кори надниркових залоз
(рис. 37.8).
Зменшення секреції альдостерону веде до
змін тих процесів, у регуляції яких беруть
участь мінералокортикоїди (див. вище). Недо
статність цих гормонів зумовлює цілий ряд по
рушень, серед них:
1) зневоднення (дегідратація) (див. главу 27).
Розвивається внаслідок втрати іонів нат
рію (зменшується їхня реабсорбція) з на
ступною втратою води (поліурія). Збіль
шення діурезу зумовлюється зменшенням
факультативної реабсорбції води, оскільки
при гіпонатріємії знижується осмотичний
тиск плазми крові, а отже — і секреція вазо
пресину (антидіуретичного гормону);
2) артеріальна гіпотензія. Зниження арте
ріального тиску є наслідком зменшення
об’єму циркулюючої крові як результату
зневоднення;
3) гемоконцентрація. Згущення крові також
пов’язане із втратою рідини. Його наслід
ком є порушення реологічних властивос
тей крові, що призводить до розладів мікроциркуляції й гіпоксії (див. главу 16);
4) зменшення ниркового кровообігу. Падіння
системного артеріального тиску веде до
зниження фільтраційного тиску в капіля
рах клубочків, у результаті чого зменшу
ється швидкість клубочкової фільтрації
і розвивається інтоксикація (азотемія);
5) гіперкаліємія. При недостатності альдо
стерону зменшується канальцева секреція
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
366
Рис. 37.8. Походження основних клінічних проявів недостатності мінералокортикоїдів.
АДГ - антидіуретичний гормон
іонів К+, що веде до зростання їхньої кон
центрації в крові. Крім того, ушкоджені
внаслідок гіпоксії клітини стають ще од
ним джерелом калієвих іонів у позаклітин
ному середовищі. Гіперкаліємія порушує
діяльність усіх збудливих структур орга
нізму, зокрема серця й центральної нерво
вої системи, з розвитком відповідних клі
нічних ознак (див. главу 27);
6)
дистальний канальцевий ацидоз. Пригні
чення секреції іонів водню в умовах недо
статності альдостерону порушує процеси
ацидогенезу в дистальних звивистих канальцях нефронів, унаслідок чого сеча не
підкислюється і організм втрачає бікарбо
нат натрію з виснаженням відповідної бікарбонатної буферної системи (див. глави
29 і 36);
367
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
7) шлунково-кишкові порушення (нудота, блю
вота, проноси). У їхньому розвитку певне
значення мають втрата натрію (осмотична
діарея) та інтоксикація.
Зазначені порушення мінералокортикоїдної
функції кори надниркових залоз без відповідної
корекції стають причиною смерті.
II. Прояви, обумовлені порушенням глюкокортикоїдної функції кори надниркових залоз.
Значна частина ознак адренокортикальної не
достатності пов’язана з випадінням низькодозових ефектів глюкокортикоїдних гормонів (див.
вище), а отже, усувається введенням замісних
доз кортизолу чи його аналогів. До проявів глюкокортикоїдної недостатності відносять:
1) гіпоглікемію, що виникає при голодуванні
(непереносимість голодування);
2) артеріальну гіпотензію, у розвитку якої
має значення послаблення пермісивної дії
глюкокортикоїдів стосовно катехоламінів.
Пресорна дія останніх за відсутності кор
тизолу значно зменшується;
3) послаблення реакції жирової тканини на
звичайні ліполітичні стимули. За відсут
ності глюкокортикоїдів значно зменшу
ється чутливість жирової тканини до дії
гормонів, що активують реакції ліполізу
в адипоцитах (катехоламінів, глюкагону,
тироксину) (див. главу 25);
4) зменшення опірності організму до дії різ
них патогенних факторів. Причиною цього
є порушення механізмів неспецифічної ре
зистентності - стресу, у здійсненні якого
глюкокортикоїди відіграють провідну роль
(див. главу 8);
5) зменшення здатності виводити воду при
водному навантаженні — водне отруєння.
Можливо, одним із механізмів цього є при
гнічення секреції вазопресину (див. вище)
і послаблення його дії на клітини-мішені
за умов дефіциту глюкокортикоїдів (випа
дає їхня пермісивна дія);
6) м ’язова слабкість і швидка стомлюваність',
7) затримка росту й розвитку дітей',
8) емоційні розлади, що часто виявляють себе
депресією. Водночас збільшується схиль
ність до судом;
9) сенсорні порушення — втрата здатності роз
різняти окремі відтінки смакових, нюхо
вих, слухових відчуттів;
10)
дистрес-синдром у новонароджених (гіаліновий мембраноз). Недостатність глю
кокортикоїдів веде до порушень утворення
сурфактантів у легенях, унаслідок чого
легені не розправляються при народженні
дитини (див. главу 33).
Слід зазначити, що механізми розвитку бага
тьох клінічних ознак глюкокортикоїдної недо
статності (зокрема порушень з боку скелетних
м’язів, центральної нервової системи, сенсор
них органів тощо) ще не з’ясовано, проте відо
мо, що замісні дози кортизолу усувають ці про
яви, на підставі чого роблять висновок, що вони
пов’язані з недостатністю глюкокортикоїдної
функції кори надниркових залоз.
III.
Прояви, пов’язані з впливом інших гор
монів.
До таких, зокрема, відносять деякі ознаки,
обумовлені негландотропною дією АКТГ (див.
вище). Зменшення концентрації глюкокортико
їдів у крові спричиняється до активації певних
нейроендокринних структур гіпоталамуса і базофільних клітин аденогіпофіза. Це, у свою чер
гу, викликає гіперпродукцію АКТГ, який, діючи
на пігментні клітини сполучної тканини, стиму
лює утворення меланіну та обумовлює пігмен
тацію шкіри (звідси назва “бронзова хвороба”
для позначення хронічної адренокортикальної
недостатності).
Гіперфункція кори надниркових залоз
Розрізняють гіперфункцію клубочкової (гіперальдостеронізм) і пучкової зон кори наднирко
вих залоз.
Гіперальдостеронізм
Посилення секреторної діяльності клітин клу
бочкової зони кори надниркових залоз веде до
збільшення рівня мінералокортикоїдів у плазмі
крові та посиленого їхнього впливу на основні
клітини-мішені, якими є клітини дистальних
звивистих канальців нефронів (див. главу 27).
Розрізняють первинний і вторинний гіпераль
достеронізм.
Первинний
гіперальдостеронізм.
Первинним
називають гіперальдостеронізм, що розвиваєть
ся в результаті патологічних процесів, які від
368
буваються в клубочковій зоні кори надниркових
залоз.
Його причинами можуть бути:
1) доброякісні пухлини (аденоми), що скла
даються з клітин, які продукують альдо
стерон. Такий варіант, на який припадає по
над 80 % випадків первинного гіперальдостеронізму, відомий ще як синдром Конна;
2) вузликова двостороння гіперплазія клубочкового шару — ідіопатичний гіперальдостеронізм. Можливою причиною таких
змін є посилена експресія гена, що кодує
структуру ферменту альдостеронсинтази
(CYP11B2);
3) утворення т. зв. “химерного гена”, у ре
зультаті злиття гена альдостеронсинта
зи (CYP11B2) і гена одного з ферментів
синтезу кортизолу (CYP11B1). Унаслідок
цього клітини з такими генетичними по
рушеннями утворюють одночасно аль
достерон і кортизол, а також гібридні сте
роїди. При цьому секреція альдостерону
стає повністю залежною від стимуляцій
ної дії АКТГ, а тому пригнічення секреції
останнього введенням глюкокортикоїдів
нормалізує рівень альдостерону в орга
нізмі.
Основні прояви первинного гіперальдостеронізму зумовлюються нирковими ефектами аль
достерону. Великі кількості мінералокортикоїдів, що їх продукують клітини аденоми, зумов
люють розвиток таких ознак (рис. 37.9):
1) пригнічення активності ренін-ангіотензи
нової системи через гальмівний вплив аль
достерону на клітини юкстагломерулярного апарату нирок. Як результат — зменшу
ється активність реніну в плазмі крові;
2) артеріальної гіпертензії. За своїм патоге
незом вона є низькореніновою (див. вище),
її розвиток пов’язують зі збільшенням
вмісту натрію в крові (гіпернатріємією)
і в стінках кровоносних судин, унаслідок
чого підвищується чутливість гладких
м’язів артерій і артеріол до дії пресорних
факторів, зокрема катехоламінів;
3) гіпокаліємії, що є результатом посиленої
секреції іонів К+ у канальцях нирок. Зни
ження позаклітинної концентрації калі
євих іонів веде до порушень діяльності
збудливих органів і тканин (розлади авто
в
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
матизму та скорочувальної функції серця,
міастенія, парези) (див. главу 27);
4) негазового алкалозу, що розвивається вна
слідок посилення секреції іонів водню
(ацидогенезу) у дистальних звивистих ка
нальцях нефронів (див. главу 29);
5) поліурії. Вважають, що збільшення діуре
зу при первинному гіперальдостеронізмі
є результатом втрати чутливості епітелію
ниркових канальців до дії вазопресину
(антидіуретичного гормону). Цим, зокре
ма, пояснюють той факт, що при синдромі
Конна об’єм циркулюючої крові не зрос
тає і набряки не розвиваються.
Вторинний
гіперальдостеронізм.
Синдром
вторинного гіперальдостеронізму є результатом
тривалої активації ренін-ангіотензинової сис
теми факторами, що були предметом обгово
рення в главі 27. Виражена гіперфункція клітин,
що продукують альдостерон, у цьому випадку
зумовлюється стимуляційною дією на них ангіотензину II — біологічно активного продукту
ренін-ангіотензинової системи.
Для вторинного гіперальдостеронізму харак
терні такі прояви:
а) артеріальна гіпертензія. На відміну від
первинного гіперальдостеронізму, вона
є високореніновою, а отже, має зовсім ін
ший патогенез (див. главу 32);
6) набряки. Наявність цієї ознаки істотно
відрізняє клінічну картину вторинного гі
перальдостеронізму від синдрому Конна.
Активація ренін-ангіотензинової системи
зумовлює затримку води в організмі через
посилене продукування антидіуретичного
гормону (див. главу 27). Як результат, збіль
шується об’єм циркулюючої крові, що веде
до переходу рідини з кровоносних судин
у тканини;
в) гіпокаліємія;
г) негазовий алкалоз. Дві останні ознаки ма
ють таке ж саме походження, як і при пер
винному гіперальдостеронізмі.
Гіперфункція пучкової зони
Існує дві клінічні форми гіперфункції цієї зони
кори надниркових залоз:
1) хвороба Іценка - Кушинга - базофільна
аденома передньої частки гіпофіза;
369
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
9. Механізми основних проявів первинного гіперальдостеронізму - синдрому Конна.
АДГ - антидіуретичний гормон, ЮГА - юкстагломерулярний апарат
2)
синдром Іценка — Кушинга, причинами
якого можуть бути:
а) аденома пучкової зони кори наднирко
вих залоз;
б) ектопічна продукція АКТГ деякими зло
якісними пухлинами (наприклад, рак
легень);
в) ятрогенний чинник - введення глюкокортикоїдів в організм з лікувальною метою.
Гіперфункція пучкової зони кори наднирко
вих залоз виявляє себе утворенням і секрецією
великих кількостей глюкокортикоїдів, високодозові ефекти яких і визначають клінічну картину
(див. вище).
Для хвороби і синдрому Іценка - Кушинга ха
рактерні:
1) артеріальна гіпертензія,
2) гіперглікемія — метастерої'дний цукровий
діабет',
3) специфічний вид ожиріння з відкладан
ням жиру на тулубі й обличчі. Останнє на
буває місяцеподібної форми;
4) інфекції з мінімальними ознаками запа
лення або без них;
5) шлункова гіперсекреція та утворення ви
разок у шлунку і дванадцятипалій кишці;
6) остеопороз;
7) м ’язова слабкість',
8) уповільнене загоєння ран.
Дисфункція кори надниркових залоз
Прикладом різнонаправлених змін секреторної
діяльності клітин кори надниркових залоз може
бути адреногенітальний синдром. Він виникає
в результаті спадково обумовленої блокади син
тезу кортизолу та посиленого утворення андро
генів зі спільних проміжних продуктів.
Залежно від рівня блокади синтезу кортизолу
розрізняють три варіанти адреногенітального
синдрому (рис. 37.10).
1. Порушення ранніх етапів синтезу - дефі
цит глюкокортикоїдів, мінералокортикоїдів і гіперпродукція андрогенів. Прояви:
ознаки недостатності глюко- і мінералокортикоїдної функцій кори надниркових за
лоз, ознаки раннього статевого дозрівання
в осіб чоловічої статі, вірилізація у жінок
(поява чоловічих статевих ознак).
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
370
Рис. 37.10. Три варіанти адреногенітального синдрому (пояснення див. текст).
КТРГ - кортикотропіновий рилізинг-гормон, АКТГ - адренокортикотропний гормон
2. Порушення проміжних етапів — дефіцит
глюкокортикоїдів, надлишок андрогенів,
утворення мінералокортикоїдів не пору
шено (класичний адреногенітальний син
дром). Прояви ті ж самі, що і в першому
випадку, тільки без ознак недостатності мінералокортикоїдної функції.
3. Порушення на кінцевих етапах синтезу
кортизолу - дефіцит глюкокортикоїдів, гіперпродукція андрогенів і мінералокорти
коїдів. До проявів класичного адреногені
тального синдрому додаються ознаки гіперальдостеронізму.
37.3.2.
МОЗКОВОЇ РЕЧОВИНИ
НАДНИРКОВИХ ЗАЛОЗ
У мозковій речовині надниркових залоз утворю
ються два гормони, що належать до групи катехоламінів: адреналін і норадреналін. У людини
70-90 % припадає на адреналін і 10-30 % - на
норадреналін.
Джерелами катехоламінів є хромафінні кліти
ни, що є видозміненими клітинами симпатичних
гангліїв. Регуляція їхньої секреторної активнос
ті здійснюється нервовими механізмами - прегангліонарними симпатичними нейронами. Ви
вільнення катехоламінів у кров відбувається
при активації симпатичної нервової системи.
Функціональний синергізм цієї частини вегета
тивної нервової системи та мозкової речовини
надниркових залоз дає підстави розглядати їх як
одне ціле. Звідси термін — симпатоадреналова
система.
Катехоламіни є гормонами з мембранним ти
пом циторецепції. їхній вплив на периферичні
клітини здійснюється через:
1) а^адренорецептори. Вони є у кровонос
них судинах, матці, гладких м’язах кишок,
м’язах зіниці ока. Виникнення біологічних
ефектів при взаємодії катехоламінів з цим
видом рецепторів пов’язано з активацією
фосфоліпази С, утворенням інозитолтри-
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
фосфату і збільшенням надходження в ци
топлазму іонів кальцію (див. главу 8);
2) а2-адренорецептори. їх виявляють у тром
боцитах, пресинаптичних терміналах сим
патичних і парасимпатичних нервів.
Взаємодія катехоламінів з а2-адренорецепторами викликає зменшення активності
аденілатциклази і вмісту цАМФ у кліти
нах, з чим, власне, і пов’язані біологічні
ефекти, що виникають;
3) р -адренорецептори. Є в серці, гладких
м’язах травного каналу, жировій тканині,
печінці. Реалізація дії катехоламінів відбу
вається через активацію аденілатциклази
та збільшення вмісту цАМФ;
4) Р2-адренорецептори. Виявлені в крово
носних судинах, бронхах, матці. їхня вза
ємодія з катехоламінами супроводжується
активацією аденілатциклази й утворенням
цАМФ;
Дія катехоламінів на зазначені вище рецепто
ри зумовлює дві групи змін.
371
I. Функціональні ефекти. Вони пов’язані
із впливом адреналіну та норадреналіну на
м’язові органи й тканини. Головними з них є:
1) кардіотонічна дія (дія на серце). Катехоламіни, зумовлюючи позитивні іно-, хроно-,
батмо- і дромотропні ефекти, збільшують
силу й частоту серцевих скорочень, під
вищують збудливість і провідність у серці
(див. главу 31);
2) пресорна дія (дія на кровоносні судини).
Впливаючи на а -адренорецептори, катехоламіни викликають звуження судин,
а діючи на р2-адренорецептори, — їхнє
розширення. Оскільки загальна кількість
а,-адренорецепторів у судинах значно пе
ревищує число Р2-адренорецепторів, то
загальним ефектом є збільшення перифе
ричного судинного опору й підвищення
артеріального тиску (див. главу 32);
3) бронхорозширювальна дія. Вона обумовле
на розслабленням гладких м’язів бронхів
у зв ’язку з активацією |32-адренорецепторів.
II. Метаболічні ефекти. Виникають при вза
ємодії катехоламінів з |3, -адренорецепторами.
Серед них:
1) гіперглікемічна дія. В її основі - активація
фосфоролітичного розщеплення глікогену
в печінці (див. главу 35);
2) ліполітична дія. Обумовлена активацією
гормончутливої тригліцеридліпази. Вияв
ляє себе збільшенням вмісту в крові віль
них жирових кислот (гіперліпацидемією)
(див. главу 25);
3) теплоутворювальна дія. Пов’язана з ак
тивацією окиснення в мітохондріях бурої
жирової тканини, яке тут відбувається без
фосфорилювання, тобто без утворення
АТФ. У результаті цього істотно збільшу
ється теплоутворення (нескорочувальний
термогенез) (див. главу 18).
Порушення секреторної діяльності мозкової
речовини надниркових залоз можуть виявляти
себе двома типами змін: гіпофункцією і гіпер
функцією.
Гіпофункціональні
стани.
Такі
порушен
ня бувають рідко, мабуть, у зв’язку з тим, що
функції мозкової речовини надниркових залоз
можуть перебирати на себе хромафінні кліти
ни, розташовані за межами цих ендокринних
372
органів. Описано спадкову автосомно-рецесивну хворобу - сімейну дизавтономію (синдром
Райлі - Дея). Сутність генетичного дефекту по
лягає в порушенні структури або в повній від
сутності дофамін-Р-гідроксилази — ферменту,
що перетворює дофамін у норадреналін.
Гіперфункціональні стани. Значне збільшення
утворення й секреції катехоламінів мозковою
речовиною надниркових залоз має місце при
пухлині хромафінних клітин - феохромоцитомі. Стале підвищення рівня адреналіну й норадреналіну в крові викликає описані вище функ
ціональні й метаболічні ефекти, що клінічно ви
являють себе широким рядом симптомів, серед
яких артеріальна гіпертензія, тахікардія, гіпер
глікемія, гіперліпацидемія, гіпертермія та інші.
ПАТОФІЗІОЛОГІЯ
ЩИТОПОДІБНОЇ ЗАЛОЗИ
У щитоподібній залозі утворюються тиреоїдні
гормони - тироксин (Т4) і трийодтиронін (Т3).
Крім того, С-клітинами (парафолікулярними)
синтезується кальцитонін, який бере участь
у регуляції фосфорно-кальцієвого обміну (див.
частину 37.5).
У фолікулярних епітеліальних клітинах щито
подібної залози відбувається вивільнення Т4 (мен
шою мірою Т3) з тиреоглобуліну, після чого гор
мон надходить у кров, де основна його маса пере
буває у зв’язаній з білками - тироксинзв ’язуючим
глобуліном (ТЗГ) і транстиретином - формі.
Кількість гормону у вільній формі - незначна,
проте вона підтримується на сталому рівні завдя
ки вивільненню Т4 із ТЗГ.
У периферичних тканинах більшість молекул
Т зазнає дейодування і перетворюється в Т3.
З останнім і пов’язана дія гормону на клітинимішені та основні його біологічні ефекти.
Регуляція утворення й секреції тиреоїдних
гормонів здійснюється системою гіпоталамус-аденогіпофіз за схемою, що наведена на
рис. 37.3: гіпоталамус —* тиреотропіновий рилізинг-гормон —» аденогіпофіз —> тиреотропний
гормон (ТТГ) —»■ щитоподібна залоза. ТТГ, дію
чи на щитоподібну залозу, викликає такі ефекти:
а) збільшує захоплення і введення йоду в ор
ганічні сполуки;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
б) посилює протеоліз депонованого тиреогло
буліну;
в) стимулює секрецію Т4 і Т3;
г) у разі тривалої дії викликає гіпертрофію
й гіперплазію щитоподібної залози.
Тиреоїдні гормони є гормонами з внутріш
ньоклітинним типом циторецепції. Установлено
три внутрішньоклітинні мішені для їхньої дії:
плазматична мембрана, мітохондрії, ядро. На
плазматичній мембрані чутливих до тиреоїдних
гормонів клітин виявлено високоафінні ділянки
зв’язування Т3. Результатом взаємодії Т3 з таки
ми ділянками є стимуляція транспорту аміно
кислот. Відповідь виникає дуже швидко й не ви
магає синтезу інформаційної РНК і білка.
У мітохондріях Т3 зв’язується з ферментом
внутрішньої мембрани — транслоказою аденінових нуклеотидів — і активує його. Наслідком цьо
го є посилення транспорту АДФ із цитоплазми
в мітохондрії. У результаті концентрація АДФ
у мітохондріях зростає, що викликає збільшення
інтенсивності біологічного окиснення (принцип
акцепторного контролю).
Ядро є основною внутрішньоклітинною мі
шенню для Т3. Це визначає довгострокові ефекти
тиреоїдних гормонів. При зв’язуванні Т3 з ядер
ними рецепторами відбувається індукція тран
скрипції генів, що кодують цілий ряд функціо
нально важливих білків. Серед них:
а) Na-K-АТФ-аза плазматичних мембран;
б) ферменти ліпогенезу (зокрема, НАДФмалатдегідрогеназа);
в) ферменти мітохондрій (а-гліцерофосфатдегідрогеназа);
г) білкові компоненти p-адренорецепторів.
Усі біологічні ефекти, обумовлені дією тире
оїдних гормонів на клітини, можна поділити на
три групи.
I. Анаболічна дія — вплив на ріст і диферен
ціацію тканин. Цей ефект є низькодозовим.
Його відсутність або зменшення виявля
ються при гіпотиреозі (див. нижче).
II. Метаболічні ефекти — збільшення інтен
сивності катаболічних процесів (окиснен
ня, ліполізу). Будучи високодозовими, вони
виявляють себе в умовах гіпертиреозу
(див. нижче).
III. Сенсибілізуючі ефекти — збільшення чут
ливості клітин до дії інших гормонів, зо-
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
крема естрогенів і катехоламінів. Щодо
останніх, сенсибілізуючий вплив тиреоїдних гормонів пов’язаний зі збільшенням
кількості Р-адренорецепторів на поверхні
клітин.
Гіпотиреоз
В основі розвитку гіпофункції щитоподібної зало
зи - гіпотиреозу - можуть лежати такі причини.
I. Центральні порушення: зменшення утво
рення та секреції тиреотропінового рилізинггормону і ТТГ у зв’язку з розладами діяльності
гіпоталамуса й аденогіпофіза (вторинний гіпо
тиреоз).
II. Власне залозисті порушення, що зумовлю
ють розвиток первинного гіпотиреозу:
а) руйнування тканини залози, наприклад, ра
діоактивним йодом, видалення значної час
тини паренхіми при хірургічних операціях;
б) дефіцит йоду в питній воді та їжі - ендеміч
ний зоб;
в) автоімунне ушкодження клітин залози - ав
тоімунний тиреоїдит Хашимото;
г) уроджені порушення - гіпо- і аплазія щито
подібної залози, ензимопатії;
д) введення в організм тиреостатичних пре
паратів, що пригнічують синтез і секрецію
тиреоїдних гормонів.
III. Периферичні порушення:
а) нечутливість периферичних клітин до дії
тиреоїдних гормонів - синдром тиреоїдної
резистентності. Останній є автосомнодомінантною хворобою, при якій має місце
мутація гена, що кодує ядерний рецептор
до тиреоїдних гормонів;
б) підвищене зв’язування тиреоїдних гормо
нів білками плазми крові;
в) посилений метаболізм їх у печінці.
Патогенез основних проявів. У розвитку про
явів гіпотиреозу мають значення такі механізми.
І. Порушення росту і диференціації тканин
у дитячому віці. Такі зміни пов’язані з випадін
ням низькодозових (анаболічних) ефектів тире
оїдних гормонів і зменшенням секреції сомато
тропного гормону аденогіпофіза.
Оскільки тиреоїдні гормони потрібні для
нормального процесу енхондральної осифікації
на межі діафіза й епіфіза, то в умовах гіпоти
373
реозу порушується ріст кісток у довжину. При
цьому періостальний ріст кісток зберігається,
у зв’язку з чим вони стають товстими. Розви
вається комплекс змін скелета - гіпотиреоїдна
карликовість.
Поряд з цим затримується і розумовий розви
ток - поступово виникає кретинізм.
II. Зменшення теплоутворювальної дії тире
оїдних гормонів. Випадіння цього ефекту вияв
ляє себе:
а) зменшенням основного обміну (на 20—40 %)
внаслідок зниження інтенсивності біоло
гічного окиснення в мітохондріях і функці
ональної активності збудливих тканин;
б) зменшенням теплопродукції, у зв’язку
з чим падає температура тіла;
в) поганою адаптацією до холоду при збере
женні адаптації до високої температури;
г) гіпофагією — малим споживанням енерге
тичних ресурсів.
III. Зменшення функціональної активності
збудливих тканин. Воно пов’язане з падінням
активності Na-K-АТФ-аз та змінами процесів
активного транспорту іонів. З другого боку, має
значення зменшення чутливості тканин до ка
техоламінів, що обумовлено зменшенням кіль
кості p-адренорецепторів на клітинах. Функці
ональні зміни збудливих органів і тканин вияв
ляють себе:
а) порушеннями діяльності центральної нер
вової системи — уповільненням розумової
діяльності, млявістю, загальмованістю,
сонливістю і т.п.;
б) зменшенням функціональної активності
скелетних м ’язів - слабкістю, зменшенням
тонусу, швидкою стомлюваністю;
в) порушеннями діяльності серцево-судинної
системи - брадикардією, зменшенням хви
линного об’єму серця, падінням артеріаль
ного тиску;
г) зменшенням скорочувальної функції глад
ких м ’язів кишок — закрепами;
д) порушеннями процесів всмоктування
й екскреції. Зменшення всмоктування глю
кози в кишках призводить до гіпоглікемії,
а порушення екскреції холестеролу в скла
ді жовчі - до гіперхолестеролемії й атеро
склерозу.
IV. Порушення з нез ’псованими механізмами
розвитку. До них відносять слизовий набряк мікседему, яка характеризується збільшенням
374
у тканинах кількості глікозаміногліканів, що
зв’язують воду; потовщенням шкіри, одутлим
обличчям.
Існує гіпотеза, згідно з якою мікседема є на
слідком дії на сполучну тканину тиреотропного гормону, кількість якого при власне залозис
тій і периферичній формах гіпотиреозу істотно
зростає. Проте механізм такого впливу ТТГ за
лишається невідомим.
Гіпертиреоз
В основі гіперфункції щитоподібної залози - гі
пертиреозу — можуть лежати такі причини.
I. Центральні порушення — збільшення се
креції тиреотропінового рилізинг-гормону
і ТТГ при гіперфункції гіпоталамуса і аденогіпофіза (вторинний гіпертиреоз).
II. Власне залозисті порушення - первинний
гіпертиреоз. Найпоширенішими клінічни
ми формами первинного гіпертиреозу є ди
фузний токсичний зоб (хвороба Гревса) та
аденоми щитоподібної залози (див. нижче).
III. Периферичні порушення'.
а) збільшення чутливості клітин до дії Т3 і Т4;
б) зменшення зв’язування тиреоїдних гор
монів транспортними білками, унаслідок
чого збільшується їхній рівень у вільній
формі;
в) уповільнення метаболізму тиреоїдних
гормонів у печінці при її недостатності.
Патогенез основних проявів. У розвитку проя
вів гіпертиреозу мають значення такі механізми.
I. Антианаболічні ефекти. Вони є високодозовими і виявляють себе такими змінами:
а) затримкою росту в дітей;
б) атрофією м ’язів і слабкістю',
в) схудненням',
г) негативним азотистим балансом.
II. Посилення теплоутворювальної дії тирео
їдних гормонів. Його результатом є:
а) збільшення основного обміну,
б) посилене теплоутворення й підвищення
температури тіла',
в) погана адаптація до високої температури
при збереженій - до холоду;
г) гіперфагія — підвищене споживання їжі, як
енергетичного ресурсу.
III. Збільшення функціональної активності
збудливих тканин. Воно пов’язане з підвищен
TONI 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ням активності Na-K-насосів клітинних мемб
ран і збільшенням чутливості клітин до катехоламінів. Цим, зокрема, обумовлені такі прояви
гіпертиреозу:
а) порушення діяльності центральної нер
вової системи - прискорення психічних
процесів, занепокоєння, збудження, без
соння;
б) постійна спонтанна скорочувальна актив
ність волокон скелетних м ’язів — фібрилярні посмикування, тремор. З цим пов’язана
м’язова слабкість, стомлюваність;
в) зміни діяльності серцево-судинної систе
ми - тахікардія, збільшення хвилинного
об’єму серця, артеріального тиску;
г) підвищення скорочувальної активності
гладких м ’язів кишок - проноси;
д) збільшення інтенсивності процесів усмок
тування й екскреції. З цим, зокрема, пов’я
зана гіперглікемія і гіпохолестеролемія.
IV. Катехоламінові ефекти. У їхній осно
ві підвищення чутливості клітин до дії катехоламінів у зв’язку зі збільшенням на клітинній
поверхні кількості /3-адренорецепторів. У кліні
ці гіпертиреозу велике значення мають функціо
нальні ефекти катехоламінів, зокрема їх вплив на
серцево-судинну систему (див. частину 37.3.2),
і метаболічні зміни. З останніми пов’язані такі
порушення:
а) посилення глікогенолізу в печінці —> гіпер
глікемія ~+ гіперфункція Р-КЛІТИН острівців
підшлункової залози з подальшим їх висна
женням —> тиреоїдний цукровий діабет',
б) посилення ліполізу в жировій тканині -#
гіперліпацидемія —# збільшення кетогенезу
в печінці —> метаболічний ацидоз',
в) активація не пов’язаного з фосфорилюван-,
ням окиснення в бурій жировій тканині —і
збільшення теплопродукції —і підвищення
температури тіла та основного обміну.
V. Порушення з нез псованими механізмами
розвитку — орбітопатія і двосторонній екзо
фтальм (витрішкуватість). В основі їх розви
тку — набряк і лімфоїдна інфільтрація м’язів
очного яблука та ретробульбарної тканини.
Ці зміни не залежать від концентрації тиреоїд
них гормонів у плазмі крові та від рівня антитіл
до ТТГ-рецепторів (див. нижче). Є думка, що
в умовах гіпертиреозу виділяється особливий
екзофтальмічний фактор, однак його дотепер
не виявлено.
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
Зоб
Зобом називають видиме збільшення щито
подібної залози. Залежно від функціональних
змін, що супроводжують його розвиток, виділя
ють три види зобу:
1) гіпотиреоїднмй - збільшення маси залози
при одночасному зменшенні секреції тиреоїдних гормонів. До цього виду належать
ендемічний зоб та автоімунний тиреоїдит
Хашимото;
2) еутиреоїдний, при якому збільшення маси
щитоподібної залози не супроводжується
вираженими змінами її функціональної
активності. Прикладом такої форми є спо
радичний зоб;
3) гіпертиреоїдний, який супроводжується
ознаками гіперфункції щитоподібної зало
зи. Такий варіант характерний для дифуз
ного токсичного зобу (хвороба Ґревса, або
базедова хвороба).
Коротко розглянемо кілька найпоширеніших
хвороб, ознакою яких є розвиток зобу.
Ендемічний зоб. Причиною ендемічного зобу
є недостатній вміст йоду в питній воді та продук
тах харчування, що пов’язано з особливостями
грунту і підземних вод у певних регіонах та міс
цевостях земної кулі.
Йод є хімічним елементом, конче необхідним
для синтезу тиреоїдних гормонів. Він надходить
в організм у формі йодидів, мінімальна добова
потреба в яких становить 50-75 мкг. При змен
шенні цієї кількості нижче 50 мкг/добу ведуть
мову про йодидну недостатність.
Аніони йоду, що надійшли в організм, спочат
ку фіксуються клітинами щитоподібної залози,
а потім окиснюються до молекулярного йоду.
Останній йде на йодування тирозинових залиш
ків синтезованого білка — т иреогл обул і ну. Про
теоліз тиреоглобуліну в лізосомах епітеліальних
клітин фолікулів веде до вивільнення тироксину
і надходження його в кров.
Дефіцит йоду спричиняється до порушення
наведених вище процесів і зменшення рівня
тиреоїдних гормонів у крові (рис. 37.11). Це зу
мовлює посилення продукції тиреотропінового
рилізинг-гормону і ТТГ. Останній, впливаючи на
тканину щитоподібної залози, стимулює проце
си гіпертрофії та гіперплазії - розвивається зоб.
375
Оскільки збільшення маси залози не усуває
дефіцит тиреоїдних гормонів (причину - недо
статність йоду - не ліквідовано), то підвищена
продукція ТТГ зберігається, триває його дія на
тканину залози: зоб прогресує.
Клінічна картина ендемічного зобу може ви
никати при нормальному і навіть підвищеному
рівні йоду в організмі, якщо порушується діяль
ність білків, у тому числі ферментів, що беруть
участь у фіксації йодидів (“йодна помпа"), їх
окиснення, а також йодування тирозинових за
лишків тиреоглобуліну.
Тиреоїдит Хашимото. Цей різновид запален
ня щитоподібної залози має у своїй основі автоалергічні механізми ушкодження залозистих
клітин, пов’язані з первинними змінами в імун
ній системі (див. главу 15). Часто такі зміни
мають спадкову природу і асоційовані з полі
морфізмом генів, що кодують структуру HLAантигенів.
Ушкодження клітин щитоподібної залози при
тиреоїдиті Хашимото може спричинятися таки
ми механізмами:
а) алергічними реакціями II типу, зумовле
ними антитілами проти антигенів щитопо
дібної залози, зокрема білка-рецептора до
ТТГ. Імуноглобуліни, що зв’язуються з цим
рецептором, здатні фіксувати комплемент,
унаслідок чого започатковується комплементзалежний цитоліз — механізм ушко
дження і загибелі клітин (див. главу 15);
б) гіперчутливістю уповільненого типу, основ
ними дійовими особами якої є CD4(+)лімфоцити та прозапальні цитокіни, що ними
продукуються (алергічні реакції IV типу);
в) опосередкованою СБ8(+)-лімфоцитами цитотоксичністю. У цьому випадку йде без
посереднє ушкодження секреторних клітин
Т-лімфоцитами-кілерами, які започаткову
ють процеси апоптозу в клітинах-мішенях (див. главу 7). Цей механізм імунного
ушкодження також є різновидом алергійних реакцій IV типу.
Загибель клітин паренхіми щитоподібної за
лози веде до прогресуючого зменшення їхньої
кількості і появи ознак гіпотиреозу. Водночас
розвиток запалення супроводжується значною
інфільтрацією залози мононуклеарними кліти
нами і розростанням сполучної тканини - фіб-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
376
Схема патогенезу ендемічного зобу
розом. Такі зміни ведуть до збільшення маси
щитоподібної залози на тлі зменшення кількос
ті секреторних клітин — розвивається гіпотиреоїдний зоб.
Дифузний токсичний зоб. Ця недуга відома
ще як хвороба Ґревса, або базедова хвороба.
Вона характеризується проявами гіперфункції
щитоподібної залози, а тому зоб, що розвиваєть
ся, є гіпертиреоі'дним.
Вважають, що дифузний токсичний зоб є ав
тоімунною хворобою, у виникненні якої мають
значення так звані ТТГ-міметичні антитіла,
тобто антитіла, що імітують дію ТТГ при вза
ємодії з поверхневими антигенами клітин щито
подібної залози (стимуляторний тип антитілозалежної дисфункції клітин як варіант алергійних
реакцій II типу, див. главу 15).
До ТТГ-міметичних антитіл відносять:
а) імуноглобуліни, що стимулюють функ
цію щитоподібної залози: LATS (long
acting thyroid stimulator) і TSI (thyroid
stimulating immunoglobulin). Ці антитіла
здатні взаємодіяти із ТТГ-рецепторами
і через них впливати на секреторну ак
тивність клітин, підвищуючи утворен
ня й вивільнення тиреоїдних гормонів.
Оскільки ні LATS, ні TSI не мають місць
зв’язування комплементу, то клітини щи
топодібної залози не ушкоджуються (як
при тиреоїдиті Хашимото), а активно
функціонують;
б) імуноглобуліни, що стимулюють ріст щи
топодібної залози — TGI (thyroid growthstimulating immunoglobulins). Ці антитіла
також взаємодіють з рецепторами до ТТГ,
але, на відміну від попередніх, мають сто
сунок до активації проліферативних проце
сів у цьому органі;
в) імуноглобуліни, що перешкоджають зв’язу
ванню ТТГ з рецепторами до цього гор
мону - ТВІІ (TSH-binding inhibitor immu-
377
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
noglobulins). Одні варіанти таких антитіл,
блокуючи взаємодію ТТГ з рецепторами
до нього, зумовлюють розвиток гіпотире
озу, тимчасом як інші, навпаки, виявляють
ТТГ-міметичну дію і стають причиною гіпертиреоїдного зобу.
Ключову роль антитіл проти ТТГ-рецепторів
у розвитку дифузного токсичного зобу до
водять експерименти. Так, імунізація мишей
такими білками-рецепторами веде до утво
рення відповідних антитіл, що супроводжу
ється збільшенням щитоподібної залози та по
явою ознак її гіперфункції. У частини тварин
з’являється характерний для хвороби Ґревса
очний синдром - орбітопатія і двосторонній
екзофтальм.
ПАТОФІЗІОЛОГІЯ
ПРИЩИТОПОДІБНИХ ЗАЛОЗ
Більшість людей має по чотири невеликі (маса
кожної - 35-40 мг) прищитоподібні залози, роз
ташовані на задній поверхні щитоподібної зало
зи парами біля верхніх і нижніх полюсів остан
ньої. Проте приблизно в 10 % людей є лише три
чи дві прищитоподібні залози. Паренхіматозні
клітини прищитоподібних залоз спеціалізують
ся на утворенні паратиреоїдного гормону (ПТГ),
який ще називають паратгормоном або паратирином.
Секреція ПТГ регулюється вмістом іонів
кальцію в плазмі крові. Вона зростає при змен
шенні концентрації Са2+ у плазмі, і навпаки,
зменшується при збільшенні вмісту цих іонів.
Крім того, вивільненню паратгормону в кров
перешкоджає 24,25-(ОН)2 вітамін D, що утворю
ється в нирках.
Основна функція ПТГ полягає в забезпечен
ні сталості концентрації іонів кальцію в плазмі
крові, на що направлені основні його біологічні
ефекти. Суть останніх і механізми їхнього роз
витку були предметом докладного обговорення
в главі 28.
Тут лише нагадаємо, що, впливаючи на основ
ні свої мішені (кісткову тканину, структури ни
рок), ПТГ збільшує рівень іонізованого каль
цію в плазмі крові, а отже, і в позаклітинному
середовищі тканин.
Гіпопарсггиреоз
Гіпофункція прищитоподібних залоз, яку позна
чають як гіпопаратиреоз, може мати такі при
чини:
1) випадкове ушкодження або видалення
прищитоподібних залоз при операціях на
щитоподібній залозі;
2) ушкодження їх при лікуванні радіоактив
ним йодом хвороб щитоподібної залози;
3) автоімунні ушкодження залоз. У більшості
випадків так званого ідіопатичного гіпопаратиреозу у хворих виявляють антитіла
проти мембранних рецепторів, що віді
грають роль сенсора іонів кальцію у зало
зистих клітинах. Крім того, автоімунний
механізм лежить в основі гіпопаратиреозу,
що є складовою частиною автоімунного
поліендокринного синдрому І типу (APS1)
(див. частину 37.3.1);
4) уроджене недорозвинення прищитоподіб
них залоз, яке часто поєднується з аплазі
єю тимуса і дефектами серця;
5) нечутливість клітин-мішеней до дії ПТГ —
псевдогіпопаратиреоз. При цьому рівень
ПТГ у крові може бути нормальним і на
віть підвищеним. Вважають, що в пере
важній більшості випадків псевдогіпопаратиреозу його причиною є генетично
зумовлені дефекти передавання сигналу
від мембранних циторецепторів до ефекторних структур через систему внутріш
ньоклітинних посередників (месенджерів), пов’язаних з функціями G-білків (див.
главу 8).
Псевдогіпопаратиреоз є складовою частиною
так званої мультигормональної резистентності,
відомої як синдром уродженої остеодистрофії
Олбрайта. При ньому нечутливість до ПТГ по
єднується з нечутливістю й до інших гормонів,
що реалізують свою дію через однакові месенджерні механізми, - тиреотропного, фолікулостимуляційного і лютеїнізуючого гормонів.
Основні прояви. Кардинальною ознакою гіпо
паратиреозу, що визначає всі інші, є зменшення
рівня іонів кальцію в плазмі крові - гіпокальціємія.
Вона обумовлює розвиток паратиреопривної
тетанії, що виявляє себе різким підвищенням
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
378
нервово-м’язової
збудливості,
множинними
фібрилярними скороченнями м’язів усього тіла.
Потім виникають напади клонічних судом, що
переходять у тонічні. Судомні скорочення мо
жуть поширюватися і на внутрішні органи (пілороспазм, ларингоспазм). Під час одного з та
ких нападів настає смерть. Зменшення рівня
кальцію у крові веде до порушень центральної
нервової системи: виникає емоційна нестабіль
ність, з’являється відчуття страху, депресія;
можливі галюцинації, різні варіанти психозів
тощо.
При хронічному гіпопаратиреозі у тварин
розвивається клінічна картина паратиреопривної кахексії. Вона характеризується схудненням,
анорексією,
підвищеною
нервово-м’язовою
збудливістю, диспепсією й різноманітними тро
фічними порушеннями.
Гіперпаратиреоз
Гіперфункція прищитоподібних залоз - гіперпа
ратиреоз - може мати дві групи причин.
І.
Первинний гіперпаратиреоз. Він виникає
внаслідок патологічних процесів, що розви
ваються в прищитоподібних залозах. До таких
відносять:
а) доброякісні пухлини - аденоми (75-80 %
усіх випадків хвороби);
б) первинну гіперплазію залоз, яка може мати
дифузний чи вузловий характер;
в) злоякісні пухлини, що розвиваються із се
креторних епітеліальних клітин прищито
подібних залоз.
Крім того, первинний гіперпаратиреоз може
бути одним із багатьох проявів складних син
дромів, зумовлених цілим рядом генетичних де
фектів. Такими, зокрема, є:
а) синдром множинних ендокринних пух
лин І типу (multiple endocrine neoplasia-1,
MEN-1). Він виникає внаслідок мутації
одного з генів-супресорів пухлинного рос
ту та виявляє себе появою аденом і гі
перплазією прищитоподібних залоз. Може
мати як сімейний, так і спорадичний ха
рактер;
б) синдром множинних ендокринних пух
лин II типу (MEN-2). Його виникнення
пов’язане з мутаціями гена, що кодує тирозинкіназні рецептори клітин;
сімейна гіпокстьціурична гіперкальціємія —
автосомно-домінантна хвороба, що харак
теризується зменшенням чутливості клітин
прищитоподібних залоз до підвищення рів
ня іонів кальцію в крові. У її основі лежать
мутації гена, що кодує структуру чутливо
го до кальцію паратиреоїдного рецептора.
При цьому нормальний рівень іонів каль
цію сприймається як знижений, що веде до
посилення продукції ПТГ.
II.
Вторинний гіперпаратиреоз. Так позна
чають гіперфункцію прищитоподібних залоз,
зумовлену тривалим зменшенням рівня іонів
кальцію в плазмі крові — гіпоксиїьціємією (див.
главу 28). У більшості випадків причиною цьо
го є хронічна ниркова недостатність (див. гла
ву 36). Зниження концентрації кальцієвих іонів
у крові зумовлює компенсаторне посилення се
креції ПТГ, проте якщо не настає нормалізації
цього важливого гомеостатичного показника, то
клінічно такий варіант гіперпаратиреозу вияв
ляє себе ознаками, характерними для гіпокальціємічних станів (див. главу 28).
в)
Основні прояви. Для первинного гіперпарати
реозу характерними є дві групи пов’язаних між
собою змін.
I. Порушення кісткової тканини - генералізована фіброзна остеодистрофія.
Вона обумовлена підвищенням активності остеокластів і пригніченням функції остеоблас
тів. Виявляє себе болями в кістках і суглобах,
розм’якшенням кісток, різкою деформацією
скелета. Розвивається демінералізація кісткової
тканини (остеомаляція), що обумовлює підви
щення вмісту іонів кальцію в плазмі крові — гіперкальціємію.
II. Гіперкальціємія. З нею прямо пов’язані:
а) кальцифікація м’яких тканин (нирок, су
дин, легень). У важких випадках розвива
ється ниркова недостатність;
б) утворення кальцієвих каменів у нирках
і жовчному міхурі;
в) порушення збудливості нервової системи
та м’язів - м’язова слабкість, депресія, по
рушення пам’яті;
г) артеріальна гіпертензія;
д) посилення шлункової секреції з можливим
формуванням виразок у шлунку і дванад
цятипалій кишці.
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
ПАТОФІЗІОЛОГІЯ
СТАТЕВИХ ЗАЛОЗ
ПАТОФІЗІОЛОГІЯ
ЧОЛОВІЧИХ СТАТЕВИХ ЗАЛОЗ
Чоловічі статеві залози (сім’яники, яєчка) міс
тять п’ять типів клітин: клітини Лейдига (інтерстиціальні клітини), клітини Сертолі, зародкові
клітини (сперматогонії та їхні похідні), міоепітеліальні клітини, епітеліальні клітини проток.
Власне ендокринними клітинами сім’яників
є клітини Лейдига, що продукують гормони ан
дрогени, серед яких основним є тестостерон.
Біологічні ефекти тестостерону зумовлюють
ся як самим гормоном, так і метаболітами, що
утворюються з нього. До останніх відносять
дигідротестостерон (утворюється безпосеред
ньо в клітинах-мішенях, діє на передміхурову
залозу, сім’яні міхурці), естрадіол (утворюєть
ся в нейронах ЦНС і в клітинах Сертолі, діє на
нервові клітини), 5-р андростани (викликають
гемоліз еритроцитів у плода).
Регуляція діяльності сім’яників здійснюєть
ся системою “гіпоталамус - аденогіпофіз” і має
у своїй основі принцип негативних зворотних
зв’язків (рис. 37.12). У цій схемі важливу роль
відіграють клітини Сертолі, що активуються
тестостероном і продукують гормон інгібін,
який пригнічує секреторну діяльність відповід
них клітин гіпоталамуса та аденогіпофіза.
Біологічні ефекти тестостерону:
1. Ембріональна диференціація (рис. 37.13).
Андрогени забезпечують диференціацію
статевої системи в період ембріонального
розвитку. Утворюючись під впливом хоріонічного гонадотропіну (у період між 6 і 16
тижнями), тестостерон активує формуван
ня чоловічих статевих органів з вольфової
протоки, урогенітального синуса і горбка,
пригнічуючи розвиток зовнішніх жіночих
статевих органів.
Водночас клітини Сертолі вивільнюють фак
тор, що блокує диференціацію мюллерової про
токи в матку і фаллопієві труби. Якщо на цьому
етапі не відбувається синтез тестостерону, то
розвиваються статеві органи жіночого типу -
379
чоловічий
псевдогермафродитизм
(генетична
стать - чоловіча (XY), фенотип - жіночий).
Якщо в експерименті плоду-самці імплантува
ти кришталик тестостерону, то розвиваються ге
ніталії самця -жіночий псевдогермафродитизм.
Крім того, андрогени забезпечують диферен
ціацію структур центральної нервової системи
“за чоловічим типом”, визначаючи надалі чо
ловічий тип статевої поведінки. Така дія тес
тостерону пов’язана з його впливом на нервові,
зокрема гіпоталамічні, центри, які здійснюють
циклічну регуляцію секреції гонадотропних
гормонів і формують поведінкові реакції, прита
манні представникам чоловічої статі. Є докази
того, що вплив тестостерону на статеву дифе
ренціацію структур центральної нервової систе
ми пов’язаний із перетворенням його в естраді
ол у спеціалізованих нервових клітинах.
2. Статеве дозрівання та розвиток вторин
них статевих ознак в особин чоловічої ста
ті. Як зазначалося вище, перша хвиля по
силеної секреції тестостерону припадає на
2-4 місяць ембріонального розвитку. Після
завершення диференціації статевої систе
ми за чоловічим типом секреція андрогенів
припиняється.
Друга хвиля підвищеної секреції тестосте
рону супроводжує період статевого дозрівання,
під час якого формуються вторинні статеві озна
ки (зовнішні статеві органи, оволосіння шкіри,
ріст вусів і бороди, зміна тембру голосу тощо).
Оскільки андрогени стимулюють діяльність
сальних залоз, то в пубертатному періоді шкі
ра стає жирною, часто відбувається інфікування
сальних залоз - з’являються вугрі.
3. Регуляція сперматогенезу. Тестостерон
активує діяльність клітин Сертолі, міоепітеліальних клітин сім’яників та епіте
ліальних клітин проток, які у свою чергу
здійснюють вплив на інтенсивність поділу
статевих клітин та їх дозрівання. Така дія
тестостерону не пов’язана з надходженням
його в кров, а є місцевою.
4. Регуляція статевої поведінки. Андрогени
мають стосунок до формування статевого
потягу (лібідо) і здатності до копуляції. Це
пов’язано з тим, що тестостерон впливає
на преоптичну зону гіпоталамуса (центр
статевої поведінки) і полегшує спинномоз
кові рефлекси, важливі для копуляції. Тим-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
380
Рис. 37.12. Регуляція ендокринної функції чоловічих статевих залоз.
ГТРГ - гонадотропіновий рилізинг-гормон, ФСГ - фолікулостимуляційний гормон, ЛГ - лютеїнізуючий гормон
часова недостатність андрогенів у період
статевої диференціації та розвитку мозку
призводить до формування в подальшому
гомосексуальної орієнтації.
5. Анаболічна дія. Тестостерон активує проце
си білкового синтезу. Саме з цим пов’язаний
вплив андрогенів на ріст скелета та м’язів
(посилення росту кісток у довжину, збіль
шення м’язової маси).
Чоловічий гіпогонадизм
Гормональна недостатність чоловічих статевих
залоз - чоловічий гіпогонадизм - може мати такі
причини.
І.
Центральні (дисрегуляторні) порушення.
Такими можуть бути зменшення утворення гонадотропінового рилізинг-гормону в гіпотала
мусі та лютеїнізуючого гормону в аденогіпофізі.
До цього можуть спричинятися ураження зазна
чених структур, а також гіперфункція епіфіза,
у разі якої збільшення утворення мелатоніну
супроводжується пригніченням синтезу гонадо
тропних гормонів.
II. Власне залозисті порушення:
а) кастрація - хірургічне видалення сім’яників:
б) фіброз яєчок після деяких вірусних інфек
ційних хвороб, ускладнених орхітом (на
приклад, після епідемічного паротиту);
в) порушення розвитку яєчок.
Після втрати сім’яників розвивається комп
лекс порушень під назвою євнухізм. Гіпофунк
цію яєчок при збереженні їх позначають термі
ном “євнухоїдизм”.
III. Периферичні порушення. До таких відно
сять:
а) зменшення чутливості клітин-мішеней до
дії андрогенів;
б) збільшене зв’язування тестостерону з біл
ками плазми крові;
в) посилене руйнування андрогенів у печінці.
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
Диференціація чоловічих статевих залоз і статевих органів.
H-Y-антиген - поверхневий антиген плазматичної мембрани, що кодується геном Y-хромосоми
381
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
382
Основні прояви. Ознаки чоловічого гіпогонадизму залежать від того, в який період онтоге
незу він розвивається. А тому можна виділити
дві групи проявів недостатності статевих за
лоз.
I. Гіпогонадизм, що формується до періоду
статевого дозрівання, може виявляти себе:
а)
синдромом
тестикулярної
фемінізації
(спадково обумовленим чоловічим псевдогермафродитизмом, див. вище);
б) відсутністю або слабко вираженими вто
ринними статевими ознаками;
в) пригніченням сперматогенезу в разі наяв
ності сім’яників;
г) відсутністю статевого потягу;
д) порушенням окостеніння епіфізарних хря
щів при збереженому впливі соматотроп
ного гормону — розвивається євнухоїдний,
або гіпогонадний, гігантизм;
е) слабким розвитком скелетної мускулатури
(відсутність анаболічних ефектів андрогенів).
II. Гіпогонадизм, що виникає після завершен
ня статевого дозрівання, характерний, зокре
ма, для процесу старіння. Він виявляє себе:
а) деяким зменшенням вираженості вторин
них статевих ознак;
б) порушеннями сперматогенезу;
в) розвитком імпотенції;
г) анаболічними порушеннями - зменшенням
маси скелетних м’язів і працездатності.
Чоловічий гіпергонадизм
Гіперфункція чоловічих статевих залоз може
розвиватися (а) в дитячому віці до періоду ста
тевого дозрівання і (б) у дорослих.
У дітей гіпергонадизм найчастіше пов’язаний
з пухлинами і запальними процесами в гіпо
таламусі, а також гіпофункцією епіфіза. Усі
ці причини викликають збільшення продукції
гонадотропних гормонів і, як наслідок, андроге
нів. Клінічно такий гіпергонадизм виявляє себе
передчасним статевим дозріванням.
Причиною гіперфункції статевих залоз у до
рослих є добро- і злоякісні пухлини, що ростуть
з клітин Лейдига. При цьому виражених клініч
них ознак, пов’язаних з гіперандрогенемією, не
має. У випадку злоякісних новоутворень харак
терні відсутність кахексії, збереження м’язової
маси та сили м’язів.
37.6.2.
ЖІНОЧИХ СТАТЕВИХ ЗАЛОЗ
Місцем синтезу жіночих статевих гормонів
є яєчники. Тут утворюється три групи гормонів.
I. Естрогени: 17/3-естрадіол, естрон, естріол. Продукуються епітеліальними клітина
ми фолікулів.
Основним і найактивнішим естрогеном
є 17|3-естрадіол. У передовуляційну фазу клі
тини теки синтезують тестостерон, який потім
надходить у сусідні клітини гранульози, де від
бувається ароматизація тестостерону і він пере
творюється на17|3-естрадіол.
II. Прогестини, серед яких основним є про
гестерон. Утворюються клітинами жовто
го тіла.
III. Мінорні гормони: інгібін (пригнічує утво
рення ФСГ в аденогіпофізі) і релаксин (роз
м’якшує лобкове зрощення, тазові зв’язки,
шийку матки, розслаблює гладкі м’язи мат
ки). Секреція релаксину сильно зростає
перед початком пологів, тому що завдяки
своїм ефектам він сприяє кращому прохо
дженню плода через пологові шляхи.
У регуляції утворення й секреції жіночих
статевих гормонів вирішальне значення мають
система “гіпоталамус - аденогіпофіз — яєчникип і механізми позитивного та негативного зво
ротного зв’язку (рис. 37.14).
У передовуляційний період під впливом ФСГ
посилюється утворення і секреція естроге
нів, які за принципом позитивного зворотного
зв’язку стимулюють утворення гонадотропінового рилізинг-гормону (ГТРГ) в гіпоталамусі
та лютеїнізуючого гормону (ЛГ) в аденогіпофі
зі (забезпечують “пік ЛГ”). Останній викликає
підвищення секреторної активності клітин, що
продукують прогестини.
У післяовуляційний період естрогени та інгібін
за принципом негативного зворотного зв’язку при
гнічують утворення ГТРГ і гонадотропних гормо
нів аденогіпофіза, що веде в підсумку до зменшен
ня секреції і самих жіночих статевих гормонів.
Біологічні ефекти жіночих статевих гормонів:
1. Статеве дозрівання й розвиток вторинних
статевих ознак в особин жіночої статі.
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
Секреція естрогенів у дівчаток розпочина
ється і наростає в період статевого дозрівання,
тимчасом як у хлопчиків у цей час спостеріга
ють вже другу хвилю підвищення секреції тес
тостерону (див. вище).
Під впливом естрогенів збільшуються молоч
ні залози, змінюється будова тіла, відкладається
жир у підшкірну жирову тканину, ростуть і роз
виваються зовнішні статеві органи. Оволосіння
лобка є результатом дії андрогенів.
2. Забезпечення циклічних змін в організмі жін
ки — підготовка статевих органів до заплід
нення та імплантації яйцеклітини. Це здій
снюється завдяки участі жіночих статевих
гормонів разом з ФСГ і ЛГ у гормональному
контролі овуляції й менструального циклу.
3. Забезпечення вагітності, пологів і лакта
ції. Так, естрогени викликають швидкий
ріст м’язів матки, стимулюють ріст системи
проток молочних залоз. Прогестини блоку
383
ють скорочувальну активність вагітної мат
ки, стимулюють ріст залозистого епітелію
молочних залоз. Зі зменшенням продукції
прогестинів плацентою пов’язують настан
ня пологів.
4. Екстрагенітальні ефекти. Естрогени впли
вають на:
а) кісткову тканину, забезпечуючи умови
для своєчасного окостеніння епіфізарних хрящів;
б) печінку, активуючи синтез цілого ряду
білків (транспортних білків, факторів
зсідання, ангіотензиногену, ліпопротеїдів високої густини - ЛПВГ). Зі збіль
шенням у крові антиатерогенних ЛПВГ
пов’язують той факт, що жінки в передменопаузальний період менш схильні до
атеросклерозу та його ускладнень, ніж
чоловіки відповідного віку;
в) кровоносні судини, регулюючи процеси
ангіогенезу, судинний тонус.
Рис. 37.14. Регуляція ендокринної функції жіночих статевих залоз.
ГТРГ - гонадотропіновий рилізинг-гормон, ФСГ - фолікулостимуляційний гормон, ЛГ - лютеїнізуючий гормон
384
Прогестини зумовлюють збільшення базаль
ної температури тіла в післяовуляційний період.
Жіночий гіпогонадизм
Серед причин гіпофункції жіночих статевих за
лоз — жіночого гіпогонадизму — виділяють такі
групи чинників.
I. Центральні (дисрегуляторні) порушення.
Вони можуть бути обумовлені психогенними
факторами, ураженнями гіпоталамуса (дефіцит
гонадотропінового рилізинг-гормону), гіпо
функцією аденогіпофіза (дефіцит ФСГ і ЛГ),
гіперфункцією епіфіза (зменшення утворення
гонадотропних гормонів при збільшенні синте
зу мелатоніну).
II. Власне залозисті порушення:
а) спадково обумовлена аплазія яєчників;
б) дегенерація яєчників (запальна, кістоз
на);
в) автоімунне ушкодження жіночих стате
вих залоз;
г) хірургічне видалення яєчників.
III. Периферичні розлади. До таких відносять:
а) зменшення чутливості клітин-мішеней до
дії жіночих статевих гормонів;
б) підвищене зв’язування їх білками плазми
крові;
в) посилене руйнування естрогенів і прогестинів у печінці.
Основні прояви. Клінічні прояви гіпофункції
статевих залоз залежать від часу настання гіпо
гонадизму, а тому можна виділити три варіанти
недостатності жіночих статевих гормонів.
I. Якщо гіпогонадизм розвивається до на
стання статевого дозрівання, то форму
ється комплекс порушень під назвою “ова
ріальний євнухоїдизм>,\ слабкий розвиток
вторинних статевих ознак, первинна аме
норея, високий зріст через порушення осифікації епіфізів кісток.
II. Розвиток гіпогонадизму в дітородному віці
супроводжується порушеннями циклічних
процесів в організмі жінки (розлади мен
струального циклу), безплідністю, перед
часним клімаксом.
III. Після менопаузи виникають клімактеричні
зміни, пов’язані в основному з випадінням
екстрагенітальних ефектів естрогенів (див.
вище):
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
а) розвивається постменопаузальний остеопороз, що нерідко веде до патологічних
переломів кісток, навіть при незначних
їхніх ушкодженнях;
б) настає нестабільність судинного тону
су, що виявляє себе “припливами”, по
чуттям жару, киданням у піт тощо.
Жіночий гіпергонадизм
В основі жіночого гіпергонадизму, при якому
збільшується секреція жіночих статевих гормо
нів, можуть лежати дві групи причин.
I. Центральні (дисрегуляторні) порушення.
Вони обумовлюються збільшенням секреції
гонадотропінового рилізинг-гормону і гонадо
тропних гормонів. Проявами таких порушень
можуть бути:
а) синдром передчасного статевого дозрі
вання. Характерною є рання поява вто
ринних статевих ознак, ранній початок
менструацій та ймовірність завагітніти
(у віці 7-8 років);
б) синдром уявної вагітності. Він вияв
ляє себе аменореєю й усіма зовнішніми
ознаками вагітності. При цьому має міс
це підвищена продукція пролактину клі
тинами аденогіпофіза.
II. Власне залозисті порушення. До причин
таких порушень відносять кістозні процеси
і пухлини яєчників, що продукують гормони.
При цьому:
а) розвивається гіпертрофія ендометрію;
б) порушується менструальний цикл, понов
люються маткові кровотечі в менопаузі;
в) можливим є розвиток злоякісних пухлин
в естроген-залежних органах і тканинах
(матці, піхві, молочних залозах).
ПАТОФІЗІОЛОГІЯ
ІНШИХ ЕНДОКРИННИХ ЗАЛОЗ
Епіфіз
Ендокринні клітини епіфіза - пінестоцити продукують гормон мелатонін — похідну сполу
ку амінокислоти триптофану.
Рівень секреції гормону залежить від освіт
леності сітківки ока, інформація від якої через
ГЛАВА 37. ПАТОФІЗІОЛОГІЯ ЕНДОКРИННОЇ СИСТЕМИ
складні провідникові шляхи, у тому числі гі
поталамус і спинний мозок, надходить в епі
фіз. У світлий час доби секреція мелатоніну
гальмується, у темряві, навпаки, зростає. Так,
у людини в нічний час (з 23 до 7 год.) вивільня
ється 70 % загальної добової кількості мелато
ніну. З секрецією мелатоніну пов’язують функ
цію епіфіза, що її позначають як “біологічний
годинник
Серед відомих нині біологічних ефектів ме
латоніну - пригнічення утворення й секреції
гонадотропних гормонів аденогіпофіза, змен
шення продукції ТТГ, АКТГ і СТГ. Патологію
епіфіза мало вивчено. Відомо, що при збіль
шенні продукції мелатоніну відбувається за
тримка статевого розвитку, а при зменшенні
його секреції, навпаки - передчасне статеве
дозрівання.
Підшлункова залоза
У підшлунковій залозі ендокринну функцію ви
конують клітини острівців, що продукують ін
сулін (P-клітини) і глюкагон (а-клітини).
Зменшення синтезу й секреції інсуліну при
зводить до розвитку цукрового діабету, який
був предметом обговорення в главі 24.
Збільшення утворення інсуліну - гіперінсулінізм - буває:
а) у разі виникнення пухлин, що розвиваються
з P-клітин панкреатичних острівців і про
дукують інсулін;
385
при спонтанній ідіопатичній гіпоглікемії
у дітей;
в) на початкових стадіях цукрового діабету
II типу, при інсулінорезистентності (див.
главу 24);
г) при ожирінні, демпінг-синдромі (стан після
резекції шлунка), деяких ендокринних хво
робах (акромегалії, тиреотоксикозі, хворобі
Іценка - Кушинга).
б)
Основні прояви гіперінсулінізму зумовлені гі
поглікемічною, анаболічною і мітогенною дією
інсуліну (див. главу 24).
Під впливом надлишку інсуліну розвиваєть
ся синдром гіпоглікемії (аж до гіпоглікемічної
коми), ожиріння, склеротичні ураження крово
носних судин.
Другий гормон підшлункової залози - глю
кагон - має гіперглікемічну та ліполітичну дію,
стимулює секрецію інсуліну.
Збільшення секреції глюкагону буває у разі
розвитку пухлини з альфа-клітин острівців під
шлункової залози (глюкагонома) і виявляє себе
розвитком вторинного цукрового діабету (див.
главу 24).
Описано синдром недостатності глюкагону.
Для нього характерна спонтанна гіпоглікемія,
яку спостерігають у дітей.
ГЛАВА 38
Патофізіологія
нервової системи
Нервова система - це фізіологічна система ре
гуляції функцій організму, що забезпечує швид
ке його пристосування до дії численних факто
рів зовнішнього і внутрішнього середовища.
До основних рис нервової регуляції відно
сять:
а) її швидкість;
б) точність, або високу адресність регулятор
них впливів;
в) поєднання електричних і хімічних механіз
мів передавання інформації;
г) реалізацію принципів негативного і пози
тивного зворотних зв’язків;
д) вплив не тільки на функції органів і тка
нин, а й на трофічне їх забезпечення;
е) рефлекторний механізм здійснення регуля
ції.
Найпростішою структурою нервової систе
ми, що бере участь у її функціонуванні, є ней
рон, а найпростішим функціональним елемен
том нервової діяльності - рефлекс.
Основний принцип нервової регуляції і міжнейронних взаємодій полягає в поєднанні та
чергуванні процесів збудження і гальмування.
Рефлекторний характер нервової регуляції за
безпечують три групи процесів:
1) сенсорні процеси, або механізми чутливос
ті. Вони покликані сприймати інформа
цію про стан зовнішнього і внутрішнього
середовища, здійснювати контроль їхніх
параметрів і характеристик;
2) інтегративна діяльність, яка спрямову
ється на переробку отриманої інформації,
її аналіз і синтез регуляторних сигналів;
3) власне регулювання функцій органів і сис
тем організму, тобто передавання інфор
мації у вигляді синтезованих сигналів на
ефекторні структури. Звідси й поняття
“ефекторні функції” нервової системи.
До них відносять:
а) рухову,
б) вегетативну;
в) трофічну функції.
Класифікація порушень
діяльності нервової системи
I. За анатомічним принципом виділяють:
а) порушення периферичної нервової сис
теми',
б) порушення центральної нервової сис
теми, що охоплюють розлади функцій
спинного мозку, довгастого, середнього
і т.д., аж до кори великих півкуль.
II. За походженням розрізняють (а) спадково
зумовлені та (б) набуті порушення нерво
вої системи. Набуті можуть бути первин
ними і вторинними.
Первинні розлади виникають унаслідок пря
мої дії на нервову систему патогенних факто
рів: фізичних (травма, радіація, термічні впли
ви), хімічних (токсини, отрути), біологічних
(віруси, бактерії), соціальних (слово).
Вторинні розлади обумовлюються насампе
ред порушеннями гомеостазу (гіпоксія, гіпоглі
кемія, ацидоз і т.п.), імунними факторами (автоалергічні реакції), розладами мозкового крово
обігу.
III. Клітинний принцип передбачає такі види
порушень функції нейронів:
387
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
а) порушення електрофізіологічних
процесів',
б) розлади нейрохімічних (медіаторних)
процесів',
в) порушення аксоплазматичного
транспорту.
IV. Залежно від виду порушених функцій роз
різняють такі розлади діяльності нервової
системи:
а) порушення сенсорних функцій
(чутливості);
б) розлади ефекторних функцій: рухової,
вегетативної, трофічної;
в) порушення інтегративних функцій.
Останній принцип класифікації ми й покла
демо в основу розгляду головних закономірнос
тей розвитку патологічних процесів і хвороб
нервової системи.
Сенсорні системи організму
ккпт порушення
СОМАТОВІСЦЕРАЛЬНОЇ
ЧУТЛИВОСТІ. БІЛЬ
Сенсорні системи організму поділяють на дві
великі групи:
1) системи, пов’язані з органами чуття
(зорова, слухова, вестибулярна,
смакова, нюхова);
2) соматовісцеральну сенсорну систему
(рис. 38.1).
Патофізіологія органів чуття настільки спе
цифічна, що вона є предметом вивчення на спе
ціальних клінічних кафедрах. Тут ми зупини
мося тільки на соматовісцеральній системі, тоб
то чутливості тіла і внутрішніх органів. Цей тип
чутливості має кілька особливостей. По-перше,
рецепторні структури не зібрані в окремому
388
органі, а розкидані по всьому тілу, а по-друге,
аферентні волокна не утворюють спеціальних
нервів, а входять до складу багатьох нервових
провідників. Розрізняють такі види соматовісцеральної чутливості.
I. Чутливість шкіри. Рецептори, розташовані
в різних шарах шкіри, забезпечують три види її
чутливості:
1) тактильну, або механорецепцію. За
вдяки різним варіантам механорецепторів формуються чуття (а) тиску (диски
Меркеля в епідермісі й тільця Руффіні
в дермі), (б) дотику (тільця Мейсснера),
(в) вібрації (тільця Пачіні), (г) повзання
комах по шкірі, лоскоту (механочутливі
вільні нервові закінчення). Від рецепто
рів тиску, дотику й вібрації відходять
товсті мієлінізовані нервові волокна Ар з
високою швидкістю проведення імпуль
сів (30-70 м/с), а від вільних нервових
закінчень — немієлінізовані С-волокна
з повільним проведенням потенціалів дії
(0,5-1 м/с);
2) температурну, або терморецепцію. У шкі
рі містяться теплові та холодові терморецептори. Кількість других на одиницю
площі значно більша, ніж перших. Від
теплових і частково від холодових рецеп
торів відходять немієлінізовані С-волокна,
від другої частини холодових рецепторів мієлінізовані А -волокна зі швидкістю
проведення імпульсів 10-25 м/с;
3) больову, або ноцицепцію. Про цей вид чут
ливості ми поведемо мову нижче.
II. Глибока чутливість, або пропріорецещія.
Вона забезпечує надходження інформації від
м’язів (м’язові веретена), суглобів (суглобо
ві рецептори), сухожиль (рецептори Ґольджі).
Пропріорецепція забезпечує такі чуття:
а) положення - під яким кутом перебуває ко
жен суглоб, а отже, у якому положенні пе
ребувають кінцівки;
б) руху - у якому напрямку і з якою швидкіс
тю переміщуються кінцівки;
в) сили - оцінюється м’язова сила, необхідна
для руху або утримування суглобів у пев
ному положенні.
Нервові імпульси від різних видів пропріорецепторів проводяться по волокнах Аа-типу зі
швидкістю 70-120 м/с.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
III.
Больова чутливість усього тіла - ноцицепція (див. далі).
Інформація про різні види чутливості від пе
риферичних рецепторів у центральну нервову
систему передається трьома основними провід
никовими шляхами:
1) лемнісковим шляхом (рис. 38.2). Ним про
водяться всі види глибокої (пропріоцептивної) чутливості, а також тактильна чутли
вість від спеціалізованих механорецепторів шкіри (складна тактильна чутливість).
Цей шлях складається з трьох нейронів.
Перші нейрони являють собою псевдоуніполярні клітини спинномозкових гангліїв. Аксони
цих клітин входять через задні корінці в спин
ний мозок і піднімаються догори у складі задніх
канатиків, утворюючи пучки Голля і Бурдаха.
Тіла других нейронів містяться в довгастому
мозку (п. gracilis і п. cuneatus). їхні аксони пе
реходять на протилежний бік, перехрещуючись
з аксонами контрлатеральних нейронів, і утво
рюють медіальну петлю (lemniscus medial is).
Тіла третіх нейронів містяться в таламусі,
їхні аксони йдуть у кору головного мозку, у від
повідні сенсорні зони;
2) антеро-латеральний шлях (неоспіноталамічний тракт) (рис. 38.3). Проводить тем
пературну, найпростіші види тактильної
(від механочутливих вільних нервових
закінчень) і шкірну больову (ранній біль)
чутливість.
Перші нейрони є клітинами спинномозкових
гангліїв. Тіла других нейронів містяться в задніх
рогах спинного мозку. їхні аксони посегментно
переходять на протилежний бік спинного мозку,
перехрещуються і в складі бічних канатиків під
німаються в таламус. Там містяться тіла третіх
нейронів, що посилають свої відростки в сенсор
ні зони кори головного мозку;
3) екстралемнісковий шлях (рис. 38.4). Про
водить больову чутливість (пізній біль,
глибокий і вісцеральний біль). На відміну
від двох попередніх, є багатонейронним
і філогенетично більш давнім.
Тіла перших нейронів містяться в спинномозко
вих гангліях, а других - у задніх рогах спинного
мозку. Аксони останніх частково переходять на
інший бік, а частково з цього ж боку йдуть нагору
389
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
38.2. Схема лемніскового шляху проведення чутливості
в складі бічних канатиків, утворюючи два тракти:
спіноретикулярний і палеоспіноталамічний.
У ретикулярній формації стовбура мозку,
куди піднімаються аксони других нейронів, від
бувається багаторазове перемикання на інші
нервові клітини. Від ретикулярної формації ін
формація надходить в утвори лімбічної системи
(емоційні компоненти реакцій), центри гіпота
ламуса (вегетативні компоненти реакцій), тала
мус, кору головного мозку та ін.
Розрізняють такі види порушень соматовісцеральної чутливості:
1) гіперестезія — підвищення чутливості;
2) гіпестезія — зменшення чутливості;
3) анестезія — відсутність чутливості.
Механізми порушень чутливості. Розлади
соматовісцеральної чутливості можуть бути
пов ’язані з ураженнями різних ланок сенсорної
системи, що сприймають подразнення, прово
дять збудження в центральну нервову систему
й аналізують інформацію, що надійшла сюди.
В основі порушень соматовісцеральної чутли
вості можуть бути такі чинники.
1. Порушення рецепції. При збільшенні поро
гу збудження рецепторів виникає гіпесте
зія, при зменшенні, навпаки, гіперестезія.
2. Ушкодження периферичних нервів. Піс
ля перетинання нерва випадають усі види
чутливості в зоні іннервації цього нерва. Як
правило, зона випадання чутливості менша
390
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Схема антеро-латерального шляху проведення чутливості
за зону іннервації, що пояснюють деяким
перекриванням зон іннервації різних нервів.
3. Ушкодження задніх корінців спинного моз
ку. Якщо перетнути кілька корінців - випа
дають усі види чутливості у відповідій зоні.
4. Ушкодження спинного мозку. При перети
нанні половини спинного мозку (лівої або
правої) розвивається синдром Броун-Секара, для якого характерна дисоціація розла
дів чутливості. Так, нижче рівня перетину
з того ж боку випадають пропріоцептивна і складні види тактильної чутливості
(ушкоджується лемнісковий шлях до його
перехрещення, див. вище), а з протилеж
ного боку - температурна, проста тактиль
на й частково больова чутливість (ушко
джується антеро-латеральний шлях після
перехрещення). Повний перетин спинного
мозку веде до зникнення усіх видів чутли
вості по обидва боки нижче рівня перети
нання.
5. Порушення роботи підкіркових структур,
що беруть участь у здійсненні сенсорних
функцій. Найбільше значення має уражен
ня ядер таламуса.
6. Ураження сенсорних зон кори головного
мозку. Порушення нейронів постцентральної звивини спричиняються до розладів
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
391
.4. Схема екстралемніскового шляху проведення чутливості
складної тактильної і пропріоцептивної
чутливості на протилежному боці тіла.
Ушкодження тім’яної частки викликає роз
виток комплексу порушень під назвою “амор
фосинтез”. У людини (що не є шульгою)
аморфосинтез виникає після видалення кори
правої півкулі великого мозку. При цьому зни
кає уява про просторове розташування частин
тіла на протилежному боці. Людина не може
надягти одяг або привести його в порядок на
лівій половині, не може поголити ліву поло
вину обличчя або зачесати волосся на лівому
боці.
Якщо ж уражено тім’яну частку з лівого боку,
то аморфосинтез доповнюється агнозією - не
здатністю розпізнавати частини тіла, предмети,
їхнє зображення і розташування в просторі.
Біль
Біль - це неприємне сенсорне й емоційне від
чуття, пов’язане з загрозою або самим ушко
дженням тканин.
Особливості болю, як виду чутливості, поля
гають в тому, що:
1) біль дає мало інформації про навколиш
ній світ, зате інформує про небезпеку, що
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
392
може виникнути або вже виникла внаслі
док дії ушкоджувальних факторів, - за
хисна функція болю;
2) на відміну від інших видів чутливості, до
болю не розвивається адаптація. У зв’язку
з цим біль може бути причиною страждань
хворого;
3) біль супроводжується складними емоційни
ми, вегетативними й руховими реакціями;
4) біль може бути патогенетичним механіз
мом розвитку генералізованих патологіч
них процесів, зокрема шоку (див. главу 20).
Класифікація:
L За клінічною характеристикою (суб’єк
тивним відчуттям) біль може бути гострий
і тупий, локалізований і дифузний, мати ха
рактер пощипування, поколювання, жару
тощо.
II. Залежно від тривалості больових відчут
тів біль може бути гострім і хронічним.
Гострий біль швидко проходить після
припинення дії больових стимулів, хро
нічний є тривалим, він завдає страждань
хворому.
III. За значенням для організму біль може бути
фізіологічним і патологічним. Фізіологіч
ний біль має захисне значення. Він сигналі
зує про ушкодження або його можливість,
ініціює певні поведінкові реакції, спрямо
вані на усунення ушкодження, обмежує
функції ураженого органа. Патологічний
біль не несе сигнальної функції, він стає
механізмом порушення життєдіяльності,
у тому числі й мозку, призводить до роз
ладів функцій різних органів і систем.
IV. За локалізацією больових відчуттів виділя
ють:
а) місцевий біль, який локалізується в місці
дії подразника, тобто там, де розвиваєть
ся патологічний процес;
б) проекційний біль. Про такий варіант болю
ведуть мову, коли місце, на яке діє бо
льовий подразник, не збігається з тим, де
біль відчувається. Наприклад, при ушко
дженні міжхребцевих дисків здавлюють
ся спинномозкові нерви. При цьому бо
лить та ділянка, що отримує іннервацію
від защемленого нерва, тобто має місце
проекція болю на ділянки, іннервацію
яких здійснює ушкоджений нерв;
в)
відбитий (іррадійований, рефлектор
ний) біль. Больове відчуття, що виникає
внаслідок впливу на внутрішні органи,
часто локалізується не в даному органі
(або не тільки в ньому), а у віддалених
поверхневих ділянках шкіри. Біль за
вжди відбивається на ділянки перифе
рії, які отримують іннервацію від того
ж сегмента спинного мозку, що й ураже
ний внутрішній орган. Якщо йдеться про
поверхню шкіри, то біль відбивається на
певному дерматомі. Оскільки органи
одержують іннервацію більш ніж від од
ного спинномозкового сегмента, то біль
відбивається на кількох дерматомах. Ра
зом вони утворюють зону Геда для дано
го органа.
V. За швидкістю розвитку больових відчуттів
розрізняють швидкий (він же ранній) і по
вільний (він же пізній) біль. Швидкий біль
виникає майже відразу (приблизно через
0,1 с) після нанесення больового стимулу
і швидко проходить, а повільний - через
0,5-1 с і може тривати довго - від кількох
секунд до хвилин.
VI. За місцем дії больових стимулів виділяють
соматичний і вісцеральний біль. Соматич
ний біль поділяють на поверхневий і глибо
кий (рис. 38.5).
Соматичний поверхневий біль. Місцем його
виникнення є шкіра. При дії больових стимулів
на неї вдається виділити два компоненти болю:
" швидкий (ранній);
* повільний (пізній).
Якщо голкою вколоти шкіру, то відразу ж
(через 0,1 с) виникає гострий, різкий, добре
локалізований біль, що швидко минає після за
вершення дії больового стимулу - це так званий
швидкий, або ранній біль.
Через певний час (0,5-1 с) виникає повільний,
або пізній, біль. Це тупий, ниючий, дифузний
біль. Він триває ще якийсь час після припинен
ня дії механічного фактора.
Два види соматичного поверхневого болю —
швидкий і повільний - істотно відрізняються
між собою своїми ознаками, механізмами фор
мування больового відчуття і значенням для ор
ганізму (див. табл. 38.1).
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
393
Види болю за механізмами розвитку і місця його виникнення
Соматичний глибокий біль. До болю, що ви
никає в глибоких тканинах, відносять головний,
зубний біль, біль у м’язах і суглобах. Він за
основними своїми характеристиками є повіль
ним і має багато спільного з повільним (пізнім)
соматичним поверхневим болем (див. табл. 38.1).
Цей біль часто тупий, не має чіткої локалізації,
супроводжується афективними (загальне незду
жання, хворобливий стан) і вегетативними (ну
дота, потовиділення, зменшення артеріального
тиску) реакціями.
Виникнення соматичного глибокого болю
пов’язано зі збудженням вільних нервових за
кінчень, яких особливо багато в періості, на по
верхні суглобів, у листках твердої мозкової обо
лонки і в стінках артеріальних судин.
Вісцеральний біль. Біль, що виникає у внутріш
ніх органах, називають вісцеральним.
Розрізняють (а) власне вісцеральний і (б) “парієтальний” біль.
Власне вісцеральний біль виникає внаслідок
збудження вільних нервових закінчень у па
ренхіматозних чи порожнистих органах груд
ної та черевної порожнин. Він не виникає на
дію локальних больових стимулів (укол, розріз
тощо), а спричиняється впливом больових чин
ників на велику кількість рецепторів, дифузно
розкиданих у тканині.
Власне вісцеральний біль розвивається при:
а) ішемії органів, що супроводжується на
копиченням у тканинах молочної кислоти
й ушкодженням клітин, з яких виходять
речовини, що викликають біль (див. ниж
че);
б) запаленні. Для цього патологічного про
цесу характерна поява в тканинах великої
кількості біологічно активних сполук -ме
діаторів запального процесу, що, діючи на
нервові закінчення, зумовлюють біль (див.
главу 17);
в) спазмуванні порожнистих органів. При
сильному скороченні гладких м’язів, з од
ного боку, здавлюються нервові закінчен
ня, що зумовлює їхнє збудження, а з дру
гого - перетискуються артерії, внаслідок
чого порушується кровопостачання та роз
вивається ішемія;
г) швидкому й різкому розтягненні порож
нистих органів. І в цьому разі, як і при
спазмуванні, біль виникає через механіч
не збудження вільних нервових закінчень
та порушення кровопостачання стінки ор
гана.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
394
Таблиця 38.1
Порівняльна характеристика швидкого і повільного болю
Больові стимули
Механічний, термічний
Хімічні, тривала дія механічного
чи термічного стимулу
Час виникнення після нанесення
стимулу
Майже відразу - через 0,1 с
Через 0,5-1 с
Суб'єктивне сприйняття
Гострий, різкий
Тупий, ниючий
Локалізація
Чітко локалізований
Дифузний
Час зникнення
Швидко зникає після закінчення
дії больового стимулу
Зникає поступово
Поріг больового відчуття
Низький
Високий
Залежність від інтенсивності
больового стимулу
Ступінь болю зростає зі
збільшенням інтенсивності
стимулу (градуальність
больового відчуття)
Відсутність градуальності закон "усе або нічого"
Нервові волокна, що
проводять біль
Мієлінізовані А.-волокна зі
швидкістю проведення 10-25 м/с
Немієлінізовані С-волокна зі
швидкістю проведення 0,5-1 м/с
Шляхи проведення
в центральній нервовій
системі
Антеролатеральний шлях
(неоспіноталамічний тракт)
(див. рис. 38.3)
Основний медіатор
аферентних нейронів
Глутамат
Субстанція Р
Місце формування
больового відчуття
Сенсорні центри кори
великого мозку
Таламічні сенсорні центри
Вегетативний компонент
Збудження симпатичної
нервової системи
Збудження парасимпатичної
нервової системи
Призначення
Є засобом попередження про
ушкодження
Є засобом нагадування про
ушкодження
Значення
Захисне - вмикає захисні
рефлекси та реакції
Відіграє патогенну роль викликає афективні (емоційні)
реакції, загальне нездужання,
хворобливий стан
Філогенетичне походження
Є епікритичним (еволюційно
молодшим)
Є протопатичним (еволюційно
більш давнім)
Екстралемнісковий шлях
І (палеоспіноталамічний тракт)
(див. рис. 38.4)
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
Напади різкого болю, що виникає в порож
нистих органах унаслідок спазмування чи різ
кого перерозтягнення їхньої стінки, одержали
назву кольок.
“Парієтальний” біль пов’язаний зі збуджен
ням вільних нервових закінчень у парієтальних
листках плеври, очеревини чи перикарда.
Через те, що таких закінчень тут дуже багато,
то біль, як і в шкірі, виникає і на локальні стиму
ли (наприклад, розрізування парієтальних лист
ків) і має гострий різкий характер.
Паренхіма деяких органів (печінка, легені)
не містить вільних нервових закінчень і не є
джерелом болю, проте якщо патологічний про
цес переходить на їхню капсулу чи парієтальні
листки серозних оболонок або гладком’язові
структури (наприклад, жовчні протоки в пе
чінці, бронхіоли в легенях), то виникає біль.
За основними своїми характеристиками віс
церальний біль є повільним і схожим на по
вільний (пізній) соматичний поверхневий біль
та соматичний глибокий біль (див. вище).
Для вісцерального болю, крім больових від
чуттів, характерні:
1) афективні реакції— пригнічений
емоційний стан, загальне нездужання,
стан хвороби;
2) вегетативні реакції— нудота,
потовиділення, падіння артеріального
тиску;
3) рефлекторне скорочення скелетних
м ’язів - напруження м’язів черевної
стінки, вимушена поза та ін.
Причини болю. Біль викликають больові стиму
ли — фактори, що обумовлюють сильне подраз
нення або ушкодження тканини. До них відно
сять:
а) механічні больові стимули (> 40 г/мм2);
б) термічні стимули (температура < 15 °С
і > 45 °С);
в) хімічні сполуки (альгетики), до яких від
носять брадикінін, серотонін, гістамін,
кислоти (pH < 6), іони К+ (при концен
трації понад 20 ммоль/л), ацетилхолін,
протеолітичні ферменти. Ряд сполук
(простагландини, субстанція Р) прямо не
збуджують рецептори, а підвищують бо
льову чутливість нервових закінчень;
г) ушкодження нервових провідників (нев
ропатичний біль).
395
Механізми болю. Проблема механізмів виник
нення болю до кінця не розв’язана, про що свід
чить існування цілого ряду теорій. Серед них такі:
I. Теорія інтенсивності. Її прихильники вва
жали, що в організмі немає спеціальних
больових рецепторів. Біль виникає в тому
випадку, коли низькопорогові механоі терморецептори стимулюються з інтен
сивністю, що перевищує певний рівень.
Якщо фактор діє з низькою або середньою
інтенсивністю, то виникає тактильне або
температурне відчуття, якщо ж інтенсив
ність висока - то відчуття болю.
II. Теорія розподілу імпульсів. Її сутність по
лягає в тому, що больовий стимул викликає
особливий хід нервових імпульсів, що від
різняється від поширення розрядів, які вини
кають при дії неушкоджувальних факторів.
На цьому будується так звана “ворітна те
орія” болю (Р. Мелзак, П. Волл), що надає ве
ликого значення у формуванні больових від
чуттів желатинозній субстанції спинного мозку
{substantia gelatinosa, SG; рис. 38.6).
Нейрони SG здійснюють пресинаптичне
гальмування, блокуючи проходження імпульсів
у нейрони задніх рогів спинного мозку по тов
стих (А) і тонких (С) нервових волокнах. Якщо
нейрони SG збуджуються, відбувається преси
наптичне гальмування — “ворота” закриті. Якщо
ж нейрони SG загальмовані, то пресинаптичне
гальмування знімається —’’ворота” відкриті.
Інтенсивна стимуляція товстих мієлінізованих нервових волокон викликає збудження ней
ронів SG - “ворота” закриваються, і проведення
імпульсів у спинний мозок зменшується.
При інтенсивному збудженні тонких немієлінізованих волокон, що проводять біль, відбу
вається гальмування нейронів SG — знімається
пресинаптичне гальмування і полегшується над
ходження імпульсів у задні роги спинного мозку.
Як докази цієї теорії наводять кілька фактів.
Відомо, що при ушкодженні товстих мієлінізованих волокон збільшується больо
ва чутливість відповідної ділянки шкіри.
Із позицій ворітної теорії це пояснюють
таким чином: ушкодження А-волокон —■
нема стимулюючої дії на SG —*■ “ворота”
відчинені —» незначні підпорогові больові
стимули проводяться в ЦНС ««* виникає
відчуття болю.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
396
Схема, що ілюструє "ворітний" механізм болю.
А - товсті мієлінізовані; С - тонкі немієлінізовані волокна; SG - желатинозна субстанція; Т - нейрони задніх рогів спинного
м о з к у , з б у д ж е н н я , " - " - гальмування.
* У клініці для полегшення болю застосовують
деякі фізичні процедури, що діють на механо- і терморецептори. Лікувальний ефект, що
настає, пов’язують з такими подіями: збіль
шення імпульсації по товстих мієлінізованих
волокнах —» активація SG —» “ворота” зачи
нені —> зменшення імпульсації по тонких немієлінізованих волокнах, що проводять біль,
—»зменшення больового відчуття.
III. Теорія специфічності. Вона передбачає іс
нування специфічних больових рецепто
рів - ноцицепторів, які відповідають тільки
на інтенсивні стимули і таким чином беруть
участь у формуванні больових відчуттів.
Виділяють такі види ноцицепторів:
а) механочутливі ноцицептори (містяться
у шкірі, скелетних м’язах);
б) термочутливі ноцицептори: теплові й хо
лодові. Теплові збуджуються при темпе
ратурі вище 45 °С (рецептори гарячого),
а холодові реагують на зменшення темпе
ратури шкіри нижче 15 °С;
в) хемочутливі ноцицептори, що збуджують
ся хімічними больовими стимулами (альгетиками) (див. вище);
г) полімодальні ноцицептори, що здатні ре
агувати одночасно на подразники кількох
модальностей: механічні й температурні
(механотермочутливі ноцицептори), меха
нічні та хімічні (механохемочутливі ноци
цептори).
Усі ноцицептори являють собою вільні нер
вові закінчення. Соматичні ноцицептори здатні
сприймати механічні, температурні та хімічні
стимули, а вісцеральні - реагують на механічні
(розтягування порожнистих органів) і хімічні
подразнення, але не сприймають температур
них 90 % ноцицепторів розташовані в соматич
них тканинах (шкірі й глибоких тканинах тіла),
і лише 10 % належать вісцеральним органам.
Ноцицептори пов’язані або з Ад-волокнами,
або з волокнами групи С. Через Ад-волокна,
швидкість поширення імпульсів у яких стано
вить 10-25 м/с, проводиться швидкий (ранній)
біль, при цьому імпульсація проходить через
антеро-латеральний провідниковий шлях (неоспіноталамічний тракт) і має представництво
в сенсорній зоні кори великих півкуль головно
го мозку (встановлення локалізації болю).
Усі види повільного болю (пізній соматичний
поверхневий, соматичний глибокий, вісцераль
ний) зумовлюються подразненням ноцицепто
рів, пов’язаних з немієлінізованими волокнами
типу С, швидкість проведення імпульсів у яких
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
становить 0,5-1 м/с. По цих волокнах збуджен
ня надходить до ретикулярної формації стовбу
ра головного мозку через екстралемніскові про
відникові шляхи (палеоспіноталамічний тракт,
див. рис. 38.4), а звідти - до різних структур
центральної нервової системи, де формуються
загальні реакції організму на біль (див. нижче).
Патологічний біль. Патологічним, або хро
нічним, називають сильний, тривалий біль, що
виснажує та завдає страждань хворому. Виділя
ють такі основні форми хронічного болю.
Невралгія - больовий синдром, пов’язаний
з порушеннями функції периферичного
нерва при вірусних інфекціях (наприклад,
оперізуючий лишай), авітамінозах, пору
шеннях кровообігу, розладах обміну речо
вин (цукровий діабет). Особливо важкою
є невралгія трійчастого нерва, яка виявляє
себе нападами настільки сильного болю,
що хворі не в змозі приймати їжу та роз
мовляти. Виникнення такого болю прово
кує дія дуже слабких подразників, напри
клад, дотик до кута рота.
Каузалгія - сильний пекучий біль, що ви
никає при ушкодженні великих соматич
них нервів (неповне перетинання нерва).
У хворого виникає відчуття, начебто на
шкіру ллють окріп або прикладають руку
до розпечених предметів, або тримають її
у вогні. Навіть легкий дотик до шкіри, що
її іннервує ушкоджений нерв, викликає не
стерпний біль. Крім того, біль можуть про
вокувати несподівані зорові та слухові по
дразники.
Фантомний біль. Виникає після ампутації
кінцівок - “болить” кінцівка, якої вже не
має. При цьому біль дуже сильний і часто
нестерпний.
Таламічний біль - тяжкий спонтанний
біль у всій половині тіла з гіперпатією
(суб’єктивним враженням підвищеної чут
ливості). Розвивається при ураженнях ядер
таламуса.
В основі формування патологічного болю
може лежати ряд механізмів.
І.
Периферичні механізми. До таких відно
сять:
а) хімічне подразнення і збільшення чутли
вості больових рецепторів (сенситизація
397
ноцицепторів). Патологічні процеси з три
валим перебігом (травматизація, запален
ня, порушення кровообігу) спричиняються
до локального виділення хімічних стиму
ляторів болю - альгетиків (див. вище). При
цьому може збільшуватися чутливість но
цицепторів до дії звичайних хімічних, ме
ханічних та термічних подразників (змен
шується поріг чутливості вільних нервових
закінчень) - розвивається гіпералгезія: на
віть дотик стає причиною болю. Серед чин
ників, що збільшують больову чутливість,
важливе місце посідають простагландини
і субстанція Р;
б) здавлювання нервів. Больові нервові ім
пульси можуть виникати не тільки внаслі
док подразнення ноцицепторів, а й у будьякому місці нервового волокна при його
здавлюванні чи згинанні. Так, при зміщен
ні міжхребцевих дисків здавлюються задні
корінці спинномозкових нервів і виникає
больова імпульсація. Крім того, тривале
здавлювання нерва спричиняється до ло
кальної його ішемії, внаслідок чого змен
шується імпульсація по товстих мієлінізованих волокнах і - відповідно до “ворітної-”
теорії болю - зростає проведення больо
вих імпульсів по тонких немієлінізованих
С-волокнах;
в) регенерація нервів. Після перетину нерва
чи травматичного його ушкодження (на
приклад, після ампутації кінцівок) стиму
люються процеси регенерації нервових во
локон. З центральної частини ушкоджено
го нерва виростають тонкі волоконця, які
утворюють потовщення - невроми. Останні
стають джерелом больової імпульсації. Ак
тивація симпатичних нервів стимулює ріст
тонких волокон у невромі, а тому сприяє
виникненню больового відчуття, як, на
приклад, при каузальгії. Після того як реге
неруючий нерв досягне периферії, у шкірі
виникають явища гіпералгезії (збільшення
больової чутливості через зменшення по
рогу збудження рецепторів регенерованого
нерва) і гіперпатії. Остання - вкрай непри
ємне і дуже тривале відчуття, що виникає
у відповідь навіть на легке подразнення;
г) демієлінізація нервів. Мієлін у нормі за
безпечує ізольоване проведення імпульсів
по кожному нервовому волокну і високу
398
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
швидкість такого проведення. При демієлінізації - руйнуванні мієлінових оболо
нок нервів, зумовленому вірусами, дефек
тами обміну речовин тощо, - створюються
умови для переходу збудження від одного
нервового волокна на друге, що йде пара
лельно. Крім того, при різкому зменшенні
швидкості проведення імпульсів в окре
мих ділянках патологічно змінених нервів
вони можуть ставати джерелом стійкого
збудження і зумовлювати біль. Демієлінізація нервів є одним із провідних механіз
мів розвитку невралгії, зокрема при вірус
них інфекціях і цукровому діабеті.
II.
Периферично-центральні механізми. Та
кими вважають механізми, в яких беруть участь
як периферичні, так і центральні структури нер
вової системи. Серед них:
а) патологічні рефлекси. У нормі рухові й ве
гетативні рефлекси на больові стимули
спрямовані на протидію ефектам, що під
тримують біль (наприклад, рефлекторне
відсмикуваня кінцівки, гіперемія ушкодже
ного м’яза). В умовах патології ці рефлек
си можуть втрачати своє захисне значення
і функціонувати як механізми, що збільшу
ють ефекти больових стимулів. Так, зги
нальний рефлекс, зумовлений больовими
імпульсами від ноцицепторів шкіри, може
стати причиною тривалого скорочення
м’язів і привести до збудження у них влас
них вільних нервових закінчень, що викли
катиме посилення болю. Активація симпа
тичної нервової системи, що супроводжує
біль, з одного боку, стимулює утворення
ноцицепторів у тканинах, а з другого; ~
спричиняється до звуження кровоносних
судин та ішемії. Остання зумовлює появу
хімічних сполук (альгетиків), що подраз
нюють вільні нервові закінчення та спри
чиняють біль (див. вище);
б) порушення балансу аферентних входів.
Цей механізм власне пояснюється “воріт
ною” теорією болю, яку ми розглядали
вище. Так, при послабленні імпульсації
по товстих мієлінізованих волокнах (їхні
ушкодження, загибель) зменшується ак
тивація нейронів у substantia gelatinosa,
“ворота” відчиняються і посилюється над
ходження в центральну нервову систему
больових імпульсів. Цей механізм має зна
чення у виникненні фантомного болю, ка
узалгії, невралгії, діабетичної нейропатії,
больових синдромів, зумовлених механіч
ним здавлюванням нервів (випадіння міжхребцевих дисків, пухлини тощо);
в) зменшення гальмівного впливу ретику
лярної формації на проведення аферент
них імпульсів. Деякі відділи ретикулярної
формації стовбура головного мозку чинять
постійний гальмівний вплив на передаван
ня імпульсів у задніх рогах спинного моз
ку і завдяки цьому зменшують проведення
больової імпульсації. Вираженість такого
впливу залежить від загального обсягу сен
сорної інформації, що надходить сюди: що
більше такої інформації, то більше гальму
вання, і навпаки. При ушкодженні нервів
зменшується обсяг сенсорної інформації,
яку отримує ретикулярна формація, а отже,
зменшується гальмівний вплив останньої
на нейрони задніх рогів спинного мозку,
що веде до відкривання “воріт” і полегше
ного проходження в центральну нервову
систему больової імпульсації;
г) денерваційна гіперчутливість. При ушко
дженні периферичних нервів виникають
дегенеративні зміни в тілах первинних сен
сорних нейронів — хроматоліз. Для нерво
вих клітин у такому стані характерне різ
ке збільшення збудливості, через що вони
можуть ставати джерелом патологічної ім
пульсації, яка сприймається як біль.
III.
Центральні механізми. При хворобах, що
уражують певні структури центральної нерво
вої системи, нерідко виникає спонтанний пе
кучий біль з явищами гіпералгезії та гіперпатії
(див. вище). У формуванні такого “центрально
го” болю може мати значення ряд механізмів:
а) генерація патологічно посиленого збу
дження. При ушкодженні структур го
ловного мозку внаслідок ішемії, а також
при утворенні гліальних рубців після
інсультів може відбуватися хронічне по
дразнення ядер сірої речовини на шляхах
проведення і передавання ноцицептивної
інформації. Ці ядра стають “генератора
ми патологічно посиленого збудження”:
спонтанні розряди їхніх нейронів зумов
люють напади болю;
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
б) зняття гальмівного впливу кори головного
мозку на таламічні ядра. При порушенні
центрального гальмування клітин таламуса
останні переходять у стан гіперактивності,
а з другого боку, припиняється фільтрація
сенсорних сигналів. Як наслідок, форму
ються неадекватні стимулу, перебільше
ні сенсорні відчуття, у тому числі й біль.
В експерименті показано, що введення тва
ринам токсинів, що порушують процеси
гальмування в центральній нервовій сис
темі (правцевий токсин, стрихнін), веде до
різкого збільшення електричної активності
пулу так званих больових нейронів (нерво
вих клітин, що мають стосунок до форму
вання болю);
в) деаферентація нейронів. Руйнування шля
хів і трактів, що проводять аферентні ім
пульси в центральну нервову систему,
веде до часткової деаферентації тих нейро
нів, до яких ця інформація має надходити.
Таке явище характеризується перебудовою
функціональних зв’язків відповідних не
рвових клітин зі стійкими змінами синаптичної передачі. В експериментальних до
слідженнях встановлено, що деаферента
ція нейронів супроводжується введенням
в дію раніше неефективних (неробочих)
синапсів, збільшенням потужності тих,
що залишилися; стимуляцією росту нових
пресинаптичних терміналей від аксонів,
що збереглися; збільшенням чутливості
мембран деаферентованих нейронів до хі
мічних стимулів - нейротрансмітерів (ме
діаторів) і нейромодуляторів. Вважають,
що порушення нормальних синаптичних
зв’язків і утворення нових є одним із меха
нізмів болю центрального походження;
г) зміни якості больових відчуттів. Якість
больових відчуттів може змінюватися при
ізольованих порушеннях одного з двох
трактів проведення больової імпульсацїї:
неоспіноталамічного
(антеро-латеральна
система) чи палеоспіноталамічного (екстралемніскова система) (див. вище). Так,
при ізольованому ушкодженні першого ін
формація про больове подразнення надхо
дить у центральну нервову систему тільки
по палеоспіноталамічному тракту. Через
особливості проведення по ньому (мала
швидкість, немає представництва в сен
399
сорній зоні кори великих півкуль та ін.)
змінюється усвідомлення тяжкості больо
вого подразнення (перебільшене відчуття)
і з’являються особливі якості болю. Він
стає гіперпатичним: відчуття болю, що ви
никає через багато секунд після нанесення
больового подразника (великий латентний
період), прогресивно наростає; біль має
сильний, часто вибуховий, дифузний, по
гано окреслений характер; неприємні від
чуття, спричинені больовим стимулом,
тривають довго після завершення його дії;
д) дисоціація больового відчуття і зовніш
нього больового стимулу. У центральній
нервовій системі може зароджуватися “па
тологічна домінанта” без будь-яких орга
нічних порушень. Відчуття болю форму
ється без надходження сюди аферентних
сигналів з периферії. Так, фантомний біль
зберігається навіть тоді, коли повністю
припинити патологічну імпульсацію з кук
си ампутованої кінцівки. Цим механізмом
пояснюють і біль при деяких психічних
хворобах (істерії, депресії);
е)
умовнорефлекторний механізм. Він не
рідко є основою так званого психогенного
болю. При дії деяких больових стимулів
можуть утворюватися стійкі умовні реф
лекси. Згодом —уже при відсутності больо
вих сигналів,— біль може виникати при дії
умовних подразників, якими є слово, згад
ка, образи тощо.
Мігрень. Одним із найпоширеніших варіантів
соматичного глибокого болю є головний біль,
від якого у світі страждають мільйони людей.
За походженням він може бути (а) внутрішньо
черепним (інтракраніальним) і (б) позачерепним
(екстракраніальним).
Власне сама тканина головного мозку не є
джерелом больових імпульсів: навіть розрізу
вання чи електричне подразнення ділянок сен
сорних зон кори великих півкуль не викликає
болю — виникають лише парестезії у вигляді
поколювання в частинах тіла, що представлені
в ушкоджених ділянках сенсорної кори.
Джерелом внутрішньочерепного болю мо
жуть бути ноцицептори твердої мозкової обо
лонки, стінок венозних синусів і менінгеальних
артерій. Особливо чутливими до болю є менінгеальні артерії середнього калібру. Збудження
400
вільних нервових закінчень зазначених струк
тур настає при дії на них механічних (розтягу
вання, стягування) і хімічних (медіатори запа
лення, продукти мікробів тощо) чинників.
Причинами позачерепного головного болю
можуть бути: (а) тривале спастичне скорочення
м’язів обличчя і шиї, (б) імпульси, що форму
ються в тканинах носа та навколоносових сину
сів, (в) порушення функції очних м’язів.
Однією з частих причин головного болю є мі
грень - хвороба, причини й механізми розвитку
якої до сьогодні залишаються невідомими.
Мігрень являє собою неврологічну недугу,
основним проявом якої є епізодичні чи регуляр
ні сильні та нестерпні напади головного болю.
При цьому відсутні будь-які ознаки органічних
чи функціональних уражень голови й головного
мозку, які могли б бути причиною таких нападів.
Поширеність мігрені в людській популяції
досить висока - від 3 до 10 %. Вважають, що
у виникненні хвороби має значення спадкова
схильність. Наявність спадкового чинника ви
являють у 65-90 % випадків мігрені. Жінки хво
ріють частіше, ніж чоловіки.
Класичний перебіг нападів мігрені проходить
три фази:
I. Продромальна фаза. Характеризується по
явою ознак-провісників за кілька годин,
і навіть кілька днів, до розвитку больового
нападу. Такими ознаками є зміна емоційно
го стану, відчуття втоми, сонливість. У де
яких людей безпосередньо перед нападом
виникають порушення зору - зорова аура:
з’являється блимаюча точка у певному міс
ці поля зору (блимаюча скотома) або дефек
ти поля зору, деформується зорове сприй
няття (наприклад, видовження всіх предме
тів), можуть виникати зорові галюцинації.
Крім зорових порушень, аура може виявля
ти себе слуховими, тактильними, нюхови
ми, смаковими та іншими галюцинаціями;
II. Больова фаза. Виникаючий біль, як прави
ло, локалізується в одній половині голо
ви (гемікранія), інколи в обох половинах.
Точна локалізація болю може бути різною:
лоб, висок, навколоочні ямки, очне яблу
ко, тім’я, потилиця. Характер болю - та
кож: пульсуючий, тупий, розриваючий,
свердлячий тощо. Біль нерідко супрово
джується нудотою і блюванням, які інколи
покращують загальний стан хворих. Бо
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
льова фаза триває від кількох годин до 1-2
діб. Періоди між нападами мігрені можуть
бути різними »•* від поодиноких випадків
протягом життя до кількох на тиждень;
III. Фаза завершення нападу. Больовий напад,
як правило, припиняється раптово: люди
на засинає та прокидається цілком здоро
вого.
Фактори, що провокують напади мігрені,
дуже різні за своїм походженням і суттю: емо
ційний стрес, зміни погоди, період менструації
та овуляції, недостатній або надмірний сон, фі
зичне навантаження, деякі харчові продукти,
ліки, алкоголь, запори, сильне світло, шум, не
приємні запахи тощо.
Серед теорій, що намагаються пояснити па
тогенез мігрені, можна виділити такі.
Судинні теорії. Відповідно до вазомотор
ної теорії основу мігрені складають пору
шення тонусу мозкових судин. Розрізня
ють три патогенетичні стадії процесу:
1) спазм кровоносних судин у ділянці розга
луження сонної артерії, який веде до пору
шення живлення мозкової тканини. Клініч
но цей стан виявляє себе розвитком ознак
продромальної фази, зокрема зорової аури
(див. вище);
2) патологічне розширення великих судин
(особливо екстракраніальних) і атонія їхніх
стінок: удари хвиль крові в такі стінки зу
мовлюють характерний пульсуючий біль;
3) набряк судинної стінки внаслідок підви
щення її проникності (буває не завжди).
Є думка, що у больовій стадії нападів мігре
ні дрібні судини звужуються, а великі, навпаки,
розширюються. Спазм дрібних артерій збіль
шує периферичний опір і в такий спосіб поси
лює пульсацію великих артерій, а отже, й інтен
сивність болю.
' Біохімічні теорії. Згідно з цими теоріями
мігрень пов’язана з порушеннями обміну
біологічно активних речовин, що беруть
участь у регуляції тонусу мозкових су
дин. Найбільше доказів має причетність
до мігрені серотоніну, рівень якого у крові
змінюється під час нападів хвороби: збіль
шується в продромальну фазу і зменшуєть
ся - у больову. Дослідження показали, що
введення фармакологічних препаратів, що
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
знижують концентрацію серотоніну в крові
(наприклад, резерпіну), може зумовлювати
головний біль. Якщо ж ці препарати вводи
ти разом із серотоніном, то біль не виникає.
Участь серотоніну в розвитку головного
болю може бути пов’язана з кількома механіз
мами. З одного боку, при зниженні рівня серо
тоніну послаблюється тонус мозкових судин
(атонія), що, як ми вже зазначали, сприяє виник
ненню болю. З другого боку, серотонін є нейромедіатором сенсорних зон головного мозку.
Серотонінергічні нейрони входять до складу де
яких центрів нейрональної неопіатної антиноиицептивної системи (див. нижче), що гальмує
виникнення больових відчуттів. Якщо розвива
ються спадково зумовлені порушення обміну
серотоніну, то зменшується потужність цієї сис
теми, і фактори, які по суті є індиферентними
для цілком здорової людини, можуть провоку
вати головний біль у схильних до мігрені осіб.
Теорії центрального походження мігрені.
Деякі вчені вважають, що мігрень є різно
видом сенсорної епілепсії, при якому па
тологічні зміни локалізовані в сенсорному
центрі - таламусі. Напад мігрені почина
ється в корі великих півкуль (продромальна фаза), а потім переходить на таламус
(больова фаза).
Уроджені порушення функціонування гіпо
таламуса можуть бути опосередковано причет
ні до мігрені. При таких розладах, як правило,
змінюються вазомоторні реакції у головному
мозку і розвиваються різні вегетативні пору
шення (нудота, блювота й ін.), що провокують
напади болю.
Загальні реакції організму на біль. Трива
лий вісцеральний і глибокий соматичний біль,
у формуванні якого бере участь екстралемніскова провідникова система, супроводжується
загальними реакціями організму. У розвитку
останніх центральне місце посідає ретикуляр
на формація стовбура головного мозку, що є
зв’язаною з різними відділами центральної нер
вової системи (рис. 38.7).
До загальних реакцій організму на біль від
носять такі.
1. Емоційні реакції. Біль, як правило, супровод
жується негативними емоціями: депресією,
401
апатією, загальним нездужанням, хвороб
ливим станом. Виникнення цих ознак по
в’язують зі збудженням відповідних струк
тур лімбічної системи головного мозку.
2. Вегетативні реакції. Гострий швидкий
біль супроводжується активацією симпатоадреналової системи, що виявляє себе
тахікардією, підвищенням артеріального
тиску, гіперглікемією і гіперліпацидемією (аферентні больові імпульси йдуть
через
антеро-латеральну
провідникову
систему). Тривалий повільний біль (гли
бокий соматичний, вісцеральний) викли
кає активацію парасимпатичної нервової
системи, ознаками чого є брадикардія,
нудота, посилене потовиділення, падіння
артеріального тиску.
3. Рухові реакції. Біль часто супроводжуєть
ся рефлекторним скороченням скелетних
м’язів (напруження м’язів черевної стінки
при вісцеральному болі — симптом гостро
го живота; характерна поза при кольках).
4. Емоційно-больовий стрес. Біль і супут
ні йому механізми є причиною розвитку
стресу. Останній пов’язаний зі збуджен
ням відповідних гіпоталамічних центрів,
вивільненням тропних гормонів в аденогіпофізі, активацією секреторної функції
периферичних ендокринних залоз, зокрема
кори надниркових залоз (див. главу 8).
5. Больовий шок. Біль є важливим патогенетич
ним механізмом розвитку різних видів шоку.
Серед них травматичний, опіковий, кардіогенний, панкреатичний (див. главу 20).
Антиноцицептивні
механізми.
Антиноцицептивними, або аналгезивними, називають при
родні протибольові механізми, що обмежують
больові відчуття. Вони пригнічують проведення
больових сигналів на всіх рівнях нервової сис
теми, що беруть участь у формуванні відчуття
болю.
Виділяють нейрофізіологічні та нейрохімічні
антиноцицептивні механізми.
Нейрофізіологічні механізми пов’язані з гру
пами нейронів, електрична стимуляція яких
викликає пригнічення або повне вимикання ді
яльності різних рівнів аферентних систем, що
передають ноцицептивну інформацію у вищі
відділи мозку. Так, при стимуляції деяких груп
нейронів мигдалеподібного тіла, гіпокампа, се-
402
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
'’ Роль ретикулярної формації стовбура головного мозку в розвитку загальних реакцій організму на
біль. ЕЛС - екстралемніскова система
реднього мозку настає аналгезія. Ретикулярна
формація чинить гальмівний вплив на “воріт
ний” механізм передавання болю структурами
спинного мозку (див. вище).
Нейрохімічні механізми пов’язані з аналгезивною дією хімічних речовин - нейромодуляторів. До них відносять:
а) ендогенні опіоїдні пептиди (опіати) — ендорфіни (найбільше значення має Р-ендорфін), енкефаліни (мет- і лейенкефалін), динорфіни. Зазначені сполуки утворюються
з поліпептидів-попередників, якими є від
повідно проопіомеланокортин, проенкефалін, продинорфін. Вплив опіоїдних пеп
тидів реалізується через взаємодію з ош
атними рецепторами клітин (р-, к-, %- та
ін.). Найбільша щільність таких рецепторів
у деяких ядрах таламуса, мигдалеподібно
му тілі, центральній сірій речовині, гіпо
таламусі, ядрі солітарного тракту, задніх
рогах спинного мозку. Зв’язування опіатів
з опіоїдними рецепторами зумовлює змен
шення утворення в нейронах вторинних
посередників (цАМФ), пригнічення ви
вільнення медіаторів закінченнями аксо
нів, гальмування зумовленої цими медіа
торами спонтанної електричної активності
нервових клітин;
б) гіпоталамічні нейропептиди — вазопресин,
окситоцин, соматостатин, нейротензин;
в) ендогенні канабіноїди. їхніми представника
ми є похідні поліненасичених жирових кис
лот — анандаміди. При зв’язуванні анандамідів з канабіноїдними рецепторами в цен
тральних структурах — гіпокампі, корі вели
ких півкуль - виникає ефект знеболювання
на тлі заспокійливої дії та ознак ейфорії.
Завдяки взаємодії нейрофізіологічних і нейрохімічних механізмів в організмі функціонують
чотири антиноцицептивні системи (рис. 38.8).
1. Нейронна опіатна система. Її утворюють
енкефалінергічні нейрони трьох рівнів: се
реднього (центральна сіра речовина), до-
тша іг. ттітш wmm\
сшш
вгастого (велике ядро шва, ретикулярне
парагігантоклітинне ядро) і спинного (ней
рони задніх рогів) мозку.
2. Гормональна опіатна система. Складаєть
ся з п’яти рівнів: спинний мозок, довгастий
мозок, середній мозок, гіпоталамус, аденогіпофіз. В аденогіпофізі під впливом кортикотропінового рилізинг-гормону вивіль
няється /3-ліпотропін, з якого утворюється
Р-ендорфін. Останній надходить у кров,
досягає нервових структур і гальмує ноцицептивні нейрони спинного мозку і та
ламуса. Крім того, Р-ендорфін потрапляє
в спинномозкову рідину III шлуночка, за
вдяки чому чинить прямий гальмівний
вплив на нейрони таламуса і збуджує галь
мівні нейрони центральної сірої речовини
середнього мозку.
3. Нейронна неопіатна система. її пред
ставлено моноамінергічними структурами
стовбура мозку: серотонінергічними, норадренергічними, дофамінергічними. Ці
структури знаходяться у ядрах шва, бла
китній плямі, центральній сірій речовині.
Антиноцицептивні системи організму
403
їх електрична стимуляція у тварин зумов
лює ефект аналгезії. При цьому збільшу
ється вміст моноамінів у спинномозковій
рідині і гальмується ноцицептивне про
ведення у спинному мозку. Є докази того,
що нейрони зазначених моноамінергічних
структур у тварин, що не зазнають больо
вих впливів, мають низьку функціональну
активність, проте вона стає високою в умо
вах ноцицептивної стимуляції.
4. Гормональна неопіатна система. Відомо,
що люди, які перебувають у стані стре
су (бойові дії, сильні переживання тощо)
можуть не помічати болю. Аналгезивний
ефект стресу пояснюють вивільненням
гормонів, що зумовлюють загальний адап
таційний синдром (див. главу 8). Зокрема,
аналізується два механізми:
1) стресор —* гіпоталамус —» кортикотропіновий рилізинг-гормон —* аденогіпофіз
—► АКТГ -# кора надниркових залоз —>
глюкокортикоїди —* ліпокортинові ефек
ти (див. главу 37) —» зменшення утворен
ня простагландинів —* збільшення порогу
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
404
чутливості ноцицепторів. В експеримен
тах показано, що видалення гіпофіза різ
ко зменшує аналгезію, пов’язану зі стре
сом;
2) стресор —f гіпоталамус —* вазопресин. Ва
зопресин є дуже важливою ендогенною
аналгезивною речовиною. Про це свідчить
ряд доказів.
У щурів з генетично зумовленими пору
шеннями синтезу вазопресину різко збіль
шена чутливість до больових стимулів.
Введення вазопресину в кров або порож
нини шлуночків мозку викликає у тварин
глибоку і тривалу аналгезію.
Вазопресин чинить свою антиноцицептивну дію незалежно від ендогенної опіатної
системи.
Із нейронів гіпоталамуса, що утворюють
вазопресин, цей гормон не тільки потрапляє
в кров через нейрогіпофіз і зумовлює власне
гормональні ефекти (див. глави 17 і 37), а й
надходить у спинномозкову рідину і транспор
тується по аксонах вазопресинергічних нервів
до різних структур мозку - таламуса, гіпокампа, мозочка, мигдалеподібного тіла, чорної суб
станції, ретикулярної формації, до моноамінергічних антиноцицептивних структур. Завдяки
впливу на зазначені утвори мозку і формується
аналгезивний ефект вазопресину.
Розуміння механізмів формування болю і ен
догенних чинників, що його пригнічують, да
ють можливість окреслити основні принципи
боротьби з цим відчуттям. До таких можна від
нести:
1) зменшення больової аферентації. Це до
сягається:
а) зменшенням збудження рецепторів (іммо
білізація кінцівок, розслаблення м’язів);
б) збільшенням порогу больової чутливості
(пригнічення утворення простагландинів,
зменшення активності симпатоадреналової
системи);
в) порушенням проведення імпульсів від ре
цепторів по нервових провідниках (прин
цип анестезії)',
2) модуляція сенсорних входів. При збіль
шенні імпульсацїї по товстих нервових во
локнах зменшується больова аферентація
(див. вище). Цей принцип лежить в основі
фізичних методів знеболювання;
активація ендогенних антиноцицептивних
систем або імітування їхньої дії введенням
фармакологічних агоністів (введення нар
котичних аналгетиків);
4) пригнічення, руйнування або видалення
центрів патологічної больової імпульсацїї
в ЦНС;
5) усунення психогенної больової патологіч
ної домінанти.
3)
На цих принципах ґрунтуються основні ме
тоди знеболювання:
1) фармакологічні. Вони полягають у за
стосуванні наркотичних і ненаркотичних
аналгетиків, місцевих анестетиків, антиде
пресантів, транквілізаторів:
2) фізичні. Деякі з них: (а) черезшкірна елек
трична стимуляція нервів; (б) глибоке про
грівання тканин; (в) масаж; (г) акупункту
ра (голковколювання);
3) нейрохірургічні. Ці методи передбачають
видалення або руйнування структур ЦНС,
що беруть участь у формуванні болю, на
приклад, хордотомія - перетинання антеро-латерального тракту;
4) психогенні: навіювання, гіпноз, автогенне
тренування.
ПОРУШЕННЯ
ЕФЕКГОРНИХ ФУНКЦІЙ
НЕРВОВОЇ СИСТЕМИ
До ефекторних функцій нервової системи відно
сять (а) рухову, (б) вегетативну і (в) трофічну.
Розглянемо основні закономірності порушень
кожної з них.
ПОРУШЕННЯ
РУХОВИХ ФУНКЦІЙ
В експериментальних дослідженнях порушення
рухових функцій нервової системи вивчають,
використовуючи такі хірургічні методи:
а) перетинання периферичних нервів і перед
ніх корінців спинного мозку (відтворення
периферичних паралічів)',
б) ушкодження спинного мозку (відтворення
спінального шоку)',
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
в) перетинання стовбура мозку між середнім
і довгастим мозком (відтворення децеребраційної ригідності);
г) видалення мозочка;
д) перетинання пірамідних шляхів;
е) ушкодження структур екстрапірамідної
системи;
є) видалення або електрична стимуляція ру
хових зон кори головного мозку.
Порушення моторики, що розвиваються при
ураженнях нервової системи, виявляють себе
рядом синдромів, на короткій характеристиці
деяких з них ми і зупинимося.
Порушення нервово-м’язової передачі До бло
кування передачі імпульсів через нервовом’язові синапси можуть спричинятися такі чин
ники.
I. Механічне ушкодження нерва. Воно при
зводить до порушення проведення потенціалів
дії до нервових терміналей і розладів аксоплазматичного транспорту.
II. Токсини та отрути. Серед них:
а) ботулінічний токсин;
б) а-бунгаротоксин (зміїна отрута);
в) кураре-екстракт, який отримують з рослин
родів Strychnos і Chondodendron, що ростуть
у Південній Америці. Він здавна використо
вувався індіанцями як отрута для стріл;
г) інсектициди—засоби боротьби з комахами
(хлорофос, дихлофос, карбофос);
д) фосфорорганічні бойові отруйні речовини
(хімічна зброя).
III. Фармакологічні агенти — міорелаксанти,
інгібітори холінестерази.
IV. Спадкові фактори. Вони, зокрема, є при
чиною розвитку міастенії (myasthenia gravis).
Ця хвороба, що виникає з частотою 1/20000,
виявляє себе м’язовою слабкістю і швидкою
стомлюваністю у зв’язку з блокадою нервовом’язової передачі. Вважають, що в основі мі
астенії - зменшення кількості ацетилхолінових
рецепторів, можливо, обумовлене їхнім авто
імунним ушкодженням.
В основі порушень нервово-м’язової переда
чі може лежати ряд механізмів.
1. Припинення проведення збудження до пресинаптичних нервових закінчень. Воно на
стає при:
405
а) порушенні анатомічної цілісності нерва
(травма, перетин);
б) порушенні фізіологічної цілісності нер
ва — блокада проведення імпульсів у ре
зультаті зменшення амплітуди потенціа
лів дії або зменшення збудливості нерво
вих волокон;
в) пригніченні енергетичного забезпечення
нервових волокон;
г) демієлінізації нервів.
2. Розлади аксоплазматичного транспорту.
Вони виникають при механічному ушко
дженні нерва, порушеннях мікротрубочок,
дефіциті енергії. У результаті не тільки
настає блокада нервово-м’язової переда
чі, а й порушується нервова трофіка (див.
нижче).
3. Порушення синтезу і депонування ацетил
холіну в нервових терміналях. їх можуть
обумовлювати:
а) дефіцит вихідних продуктів синтезу аце
тилхоліну - ацетил-КоА, холіну;
б) зменшення активності холін-ацетилтрансферази;
в) порушення утворення синаптичних вези
кул;
г) пригнічення транспорту ацетилхоліну
з аксоплазми в синаптичні везикули.
4.
Пригнічення
вивільнення ацетилхоліну
в синаптичну щілину. Цей механізм лежить
в основі дії ботулінічного токсину.
5. Кількісні та якісні зміни ацетилхолінових
рецепторів
постсинаптичної
мембрани,
а саме:
а) зменшення їх кількості;
б) оборотна (дія кураре) або необоротна
(вплив а-бунгаротоксину) блокада аце
тилхолінових рецепторів;
в) їх інактивація (десенситизація) з розвит
ком нечутливості до ацетилхоліну.
6. Розлади енергетичного обміну. При цьому
страждають усі процеси, що потребують
витрат енергії:
а) проведення потенціалів дії до пресинаптичних терміналей;
б) синтез і утворення в тілі нейрона компо
нентів нервових терміналей;
в) здійснення аксоплазматичного транс
порту;
г) транспорт ацетилхоліну з аксоплазми
в синаптичні везикули;
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
406
д) вивільнення вмісту синаптичних вези
кул у синаптичну щілину - екзоцитоз;
е) новоутворення синаптичних везикул
із пресинаптичної мембрани - ендоцитоз;
є) захоплення нервовими терміналями про
дуктів гідролізу ацетилхоліну (холіну,
ацетату).
Периферичні паралічі та парези. Параліч — це
повна, а парез - часткова втрата довільних ру
хів. Периферичні паралічі виникають при ушко
дженні периферичних рухових нервів і при деге
нерації а-мотонейронів спинного мозку. Остан
нє характерне для розвитку поліомієліту.
Для периферичних паралічів і парезів харак
терні такі ознаки:
1) атонія (гіпотонія) м ’язів — зменшення їх
нього тонусу. Звідси ще одна назва - мля
вий параліч;
2) арефлексія (гіпорефлексія) — відсутність
або ослаблення спинномозкових рефлек
сів, у здійсненні яких бере участь ушко
джений нерв або мотонейрон (розрив реф
лекторної дуги);
3) атрофія м ’язів, обумовлена їхньою гіпо
функцією.
Центральні паралічі При ушкодженні цен
тральних рухових низхідних шляхів розвива
ються центральні паралічі. Найчастішими їхні
ми причинами є травми спинного мозку й роз
лади мозкового кровообігу - інсульти.
У розвитку основних ознак центральних па
ралічів провідну роль відіграє зменшення галь
мівних впливів з розташованих вище нервових
центрів на а-мотонейрони спинного мозку. Цим
пояснюють такі характеристики центрального
паралічу:
1) гіпертонію - збільшення тонусу м’язів
(спастичний параліч);
2) гіперрефлексію - посилення спинномоз
кових рефлексів (збільшення їхньої амплі
туди та розширення зони, з якої їх можна
викликати);
3) появу патологічних рефлексів (рефлекс
Бабінського та ін.).
Отже, існують суттєві відмінності в механіз
мах розвитку й клінічних ознаках периферич
них і центральних паралічів, подані на рис. 38.9.
Паркінсонізм. Синдром паркінсонізму є одним
із найпоширеніших гіпокінетико-гіпертонічних
синдромів. У його патогенезі провідна роль, як
вважають, належить недостатності дофамінергічних систем головного мозку. Відбуваєть
ся руйнування патологічним процесом чорної
субстанції, де утворюється дофамін.
У нормі дофамін із чорної субстанції по нігростріарних шляхах надходить у хвостате
ядро. Там він є гальмівним медіатором, що
пригнічує діяльність нейронів, які затримують
і обмежують рухові акти (гальмується гальму
вання).
При руйнуванні чорної субстанції утворення
дофаміну зменшується, у результаті чого збіль
шується активність нейронів хвостатого ядра
і, отже, посилюється гальмування рухових ак
тів - розвивається гіпокінезія (рис. 38.10).
До основних проявів паркінсонізму відно
сять:
1) гіпокінезію - малу рухову активність. При
цьому обличчя стає маскоподібним, жес
тикуляція бідною, рухи повільними. Ходь
ба характеризується дуже дрібними кро
ками та відсутністю природного хитання
рук;
2) м’язову ригідність (гіпертонус м’язів).
Вона виявляє себе своєрідним опором па
сивним рухам;
3) тремор - тремтіння пальців рук і кистей, а
також нижньої щелепи.
Гіперкінези. Гіперкінетичні синдроми, або гі
перкінези, - це надмірні мимовільні рухи в різ
них групах м’язів, що виникають, як правило,
раптово і пов’язані з органічними чи функціо
нальними ураженнями центральної нервової
системи.
Найчастіше причиною гіперкінезів стають
патологічні процеси у структурах екстрапірамідної системи.
До найпоширеніших гіперкінетичних син
дромів відносять:
а) хорею - безладні мимовільні рухи з вира
женим локомоторним ефектом, що супро
воджуються гіпотонією м’язів (хорея - та
нок, або хвороба, святого Вітта). Цей син
дром є прямою протилежністю паркінсо
нізму. У його основі морфологічні зміни
в смугастому тілі (шкарлупі і хвостатому
ядрі);
407
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
б) атетоз - повільні тонічні скорочення м’я
зів, зовні схожі на “червоподібні” рухи, що,
як правило, відбуваються в дистальних від
ділах рук;
в) гемібалізм - швидкі розгонисті рухи рук,
що нагадують кидання або штовхання
м’яча.
Рухові мозочкові порушення. При ураженнях
мозочка розвивається комплекс порушень, що
його позначають як мозочковий синдром. Його
основними проявами є такі неврологічні зміни:
1) атаксія - порушення координації рухів.
Розрізняють статико-локомоторну і ди
намічну атаксію. У разі першої з них по
рушуються в основному стояння і ходь
ба, у разі другої - виконання кінцівками
різних довільних рухів. Статико-локомоторна атаксія виникає при ураженнях
черв’яка, а динамічна - при патологічних
процесах у корі півкуль мозочка. До ознак
атаксії відносять:
а) асинергію - порушення синергізму ру
хів: рухи виконуються не одночасно,
а послідовно (розщеплення рухів);
б) дисметрію - рухи виконуються або
в надлишковому, або в недостатньому
обсязі;
2) інтенційний тремор. Його немає в стані
спокою, але він з’являється під час руху
руки, коли треба досягти якої-небудь мети,
наприклад, доторкнутися до кінчика носа;
3) гіпотонія м ’язів. У результаті зменшення
їхнього тонусу розвивається слабкість мус
кулатури, швидка стомлюваність;
Ураження рухових
периферичних нервів
А) ністагм - рухи очних яблук, що швидко
повторюються;
5) запаморочення (головокружіння);
6) дефекти мови — скандована мова.
Судоми. Мимовільні скорочення скелетних
м’язів, що мають характер нападів, називають
судомами. Вони є особливим різновидом гіпер
кінезів.
Розрізняють клонічні та тонічні судоми.
Клонічні
характеризуються
короткочасним
скороченням і розслабленням окремих груп
м’язів, що настають швидко одне за одним.
Для тонічних судом характерні тривалі скоро
чення м’язів, що створюють ефект “застиган
ня” тулуба й кінцівок у яких-небудь вимуше
них позах.
Можливе поєднання різних компонентів су
дом, тоді мова йде про клонічно-тонічні (пере
важає клонічний компонент) або тонічно-клонічні (більш виражені тонічні зміни) судоми.
За поширеністю судоми можуть бути місце
вими і генералізованими.
Причиною судом, як правило, є надмірне збу
дження підкіркових структур головного мозку.
Залежно від механізмів розвитку нападів су
дом розрізняють:
1) судомну реакцію. Вона виникає епізодич
но на дію надзвичайного для організму по
дразника. Може розвиватися у будь-якої
здорової людини, але частіше і легше ви
никає у людей із так званою судомною го
товністю. У дітей віком до 3 років судомні
реакції виникають у 4-5 разів частіше, ніж
у дорослих. Причиною судомних реакцій
Ураження рухових
низхідних шляхів у ЦНС
Порівняльна характеристика периферичних і центральних паралічів
408
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Механізм пригнічення рухів (гіпокінезії) при синдромі паркінсонізму.
" + " - з б у д ж е н н я , - гальмування
можуть бути: гіпертермія, гіпокальціємія
(тетанія), гіпоглікемія, гіпоксія, екзогенне
отруєння та ін.;
2) судомний синдром. Розвивається в умовах
активного патологічного процесу в цен
тральній нервовій системі. У його розвит
ку вирішальне значення має зменшення
порогу судомної готовності й формування
в рухових центрах головного мозку “па
тологічної домінанти” - осередку патоло
гічного збудження;
3) епілепсія - хронічна хвороба, що характе
ризується повторними судомними і (або)
психопатологічними нападами та нерідко
зміною рис особистості. Етіологію й пато
генез епілепсії ще не з’ясовано.
409
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
НІiWMI ПОРУШЕННЯ
ВЕГЕТАТИВНИХ ФУНКЦІЙ
Вегетативна нервова система (симпатична і пара
симпатична) відіграє важливу роль у підтриман
ні сталості внутрішнього середовища організму,
в адаптивних його реакціях, у регуляції діяльнос
ті внутрішніх органів та деяких інших структур.
В експериментальних дослідженнях для ви
вчення ролі вегетативних нервів у розвитку па
тологічних процесів і хвороб використовують
ряд методів:
1) хірургічні методи: подразнення або руй
нування центрів парасимпатичної та сим
патичної нервової системи, перетинання
вегетативних нервів, видалення вегетатив
них гангліїв;
2) фармакологічні методи: введення в орга
нізм симпатолітиків, адреноблокаторів, холінолітиків {фармакологічна денервація);
3) імунологічні методи: введення антитіл
проти білків, що входять до складу ганглі
їв вегетативної нервової системи (напри
клад, імунний симпатоліз).
Основні функціональні ефекти збудження
симпатичної та парасимпатичної нервової сис
теми подано в табл. 38.2.
Залежно від функціональної активності різ
них відділів вегетативної нервової системи ви
діляють два типи конституції людей.
Симпатикотонічна конституція: переважає
тонус симпатичної нервової системи. Характерна
блідість і сухість шкіри, холодні кінцівки, блиск
очей і м’який екзофтальм, нестійка температура
тіла, схильність до тахікардії та підвищення арте
ріального тиску, закрепи, висока працездатність
(особливо під вечір), ініціативність, тривожність,
фізична витривалість, неспокійний сон, погана
переносимість сонця, тепла, шуму, яскравого
світла, іноді неприємні відчуття в ділянці серця.
Ваготонічна конституція: переважає тонус
парасимпатичної нервової системи. Характерні
холодна, волога, бліда шкіра, гіпергідроз (пітли
вість), гіперсалівація, яскравий червоний дермо
графізм, брадикардія, схильність до падіння арте
ріального тиску, дихальна аритмія, схильність до
непритомностей, тенденція до збільшення маси
тіла, апатичність, астенія, схильність до депресії,
мала фізична витривалість, нерішучість, слабка
ініціативність, висока чутливість, боязкість, біль
ша працездатність у ранковий час.
Синдром вегетосудинної дистонії (СВД). Та
кий узагальнений термін використовують сьо
годні для позначення комплексних змін в орга
нізмі, зумовлених порушеннями вегетативної
нервової системи людини.
СВД може виявляти себе ознаками:
а) посиленої активності переважно симпатич
ної нервової системи (див. табл. 38.2);
б) збільшення активності переважно парасим
патичної нервової системи (див. табл. 38.2);
в) змішаними симптомокомплексами.
СВД може характеризуватися: (а) перманент
ними (постійними) порушеннями - так звана
вегетативна лабільність, або (б) пароксизмальними порушеннями - “вегетативні бурі
За поширеністю розладів в організмі виділя
ють такі види СВД:
а) генералізовані порушення;
б) системні розлади (нейроциркуляторна дис
тонія, нейрогастральна дистонія, порушен
ня терморегуляції та ін.);
в) місцеві порушення (у певній частині голо
ви, у дистальних відділах кінцівок).
Причинами СВД можуть бути:
• конституціональні особливості організму
(СВД починає виявляти себе в ранньому
дитячому віці);
» ендокринні перебудови організму (у періо
ди статевого дозрівання, клімаксу);
• первинні хвороби периферичних ендокрин
них залоз;
• первинні ураження вісцеральних органів;
• алергія;
» ураження вегетативної нервової системи;
• органічні ураження головного мозку;
• неврози.
НЕРВОВА ТРОФІКА
ТА ЇЇ ПОРУШЕННЯ
Серед ефекторних функцій нервової системи
окремо виділяють трофічну функцію, або нер
вову трофіку, — вплив нервової системи на об
мін речовин в органах і тканинах, що одержу-
410
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
Таблиця 38.2
Порівняльна характеристика функціональних ефектів парасимпатичної
та симпатичної нервових систем
Очі
Звужує зіниці
Сльозові залози
Стимулює секрецію
Слинні залози
Стимулює виділення великої кількості
рідкої слини
Стимулює виділення невеликої
кількості густої слини
Серце
Уповільнює ритм, зменшує інтенсивність
обміну речовин, звужує вінцеві судини
Збільшує частоту і силу серцевих
Система зовнішнього
дихання
Звужує бронхи, стимулює секрецію
слизових залоз бронхів
Розширює бронхи, пригнічує
секрецію слизових залоз бронхів
Печінка і жовчовивідні
шляхи
Стимулює скорочення жовчних
проток і жовчного міхура,
розслаблює сфінктер Одді
Стимулює глікогеноліз і збільшує
рівень глюкози в крові, розслаблює
жовчні протоки та жовчний міхур
Система травлення
Посилює перистальтику шлунка і кишок,
збільшує їхній тонус; стимулює секрецію
шлункового,
підшлунковогой
кишкового
соків; зменшує тонус сфінктерів
Послаблює перистальтику шлунка
й кишок, зменшує їхній тонус;
збільшує тонус сфінктерів, звужує
судини
Розширює зіниці
скорочень, посилює обмін речовин,
розширює вінцеві судини
Селезінка
Стимулює скорочення елементів
капсули й зумовлює перехід
у кровоносне русло багатої на
еритроцити депонованої крові
Мозковий шар
надниркових залоз
Стимулює секрецію адреналіну
Нирки
Викликає звуження судин, зменшує
діурез
Сечовий міхур
Збільшує тонус м'язів і зменшує тонус
сфінктерів (зумовлює випорожнення)
Статеві органи
Викликає ерекцію, посилює
перистальтику матки
Стимулює еякуляцію
Кровоносні судин
У більшості судин викликає
звуження, у деяких (судини м'язів,
вінцеві артерії серця) - розширення
Потові залози
Стимулює секрецію
Волосяні фолікули
Викликає реакцію пілоерекції
Метаболізм
Посилює
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
ють іннервацію. Великий внесок у розвиток
цього напряму патофізіології нервової системи
зроблено київською школою вчених на чолі
з М. Н. Зайком (1908-1991).
Виділяють три групи механізмів нервовотрофічних впливів (рис. 38.11):
1. Медіаторні механізми. Суть їх становить
вплив медіаторів периферичних нервів на
метаболізм органів і тканин. Вважають,
що в цьому бере участь не пов’язане з не
рвовими електричними імпульсами (неімпульсне) виділення медіаторів, що обумов
лює виникнення так званих мініатюрних
постсинаптичних потенціалів.
2. Немедіаторні механізми - власне трофічна
функція. У їхній основі лежить явище аксотазматичного транспорту. Роль останг
нього в здійсненні нервової трофіки дово
дять дослідами з перехресною реіннервацією, коли нерв, що йде до швидкого (білого)
411
м’яза, направляють до повільного (червоно
го), і навпаки. Після повної регенерації “чу
жий нерв” так міняє метаболізм, що білий
м’яз набуває ознак, характерних для чер
воного, червоний стає схожим на білий. Це
стосується не тільки швидкості скорочен
ня. Головне полягає в тому, що при цьому
змінюється активність ферментів і направ
леність обміну речовин. Перебудову його
виявляють раніше, ніж починається виді
лення ацетилхоліну, тобто до того, як м’яз
починає відповідати на подразнення нерва.
Крім того, пригнічення аксоплазматичного
транспорту колхіцином, вінбластином також
викликає трофічні зміни в тканинах, хоча ім
пульсна активність нерва істотно не міняється.
3. Судинні механізми. Нерви можуть вплива
ти на обмін речовин і опосередковано, че
рез зміни тонусу кровоносних судин.
Механізми мишву нервів на обмін речовин у клітинах
412
Трофічні впливи нервової системи реалізу
ють себе на різних рівнях організації клітин.
Об’єктами такого впливу можуть бути (а) мем
брани, (б) ферменти, (в) геном клітин.
Нейродистрофічний процес. Комплекс тро
фічних порушень в органах і тканинах, що ви
никає при ушкодженні периферичних нервів чи
інших структур нервової системи, позначають
як нейродистрофічний процес, або нейрогенну
дистрофію. Особливо тяжкі порушення розви
ваються при ушкодженні аферентних волокон
і нервів.
Нейродистрофічний процес характеризується
такими ознаками:
1) структурними порушеннями — розвитком
виразок на шкірі та слизових оболонках,
атрофією м’язів, дистрофічними змінами
тканин, явищами дегенерації й загибелі
клітин;
2) функціональними змінами — підвищенням
чутливості денервованих структур до дії
гуморальних факторів (закон Кеннона);
3) розладами обміну речовин — пригніченням
активності одних ферментів і підвищен
ням активності інших, активацією біохі
мічних процесів, характерних для ембріо
нального періоду розвитку.
У патогенезі нейрогенної дистрофії, що роз
вивається при травмуванні периферичного нер
ва, головну роль відіграють такі фактори.
1. Припинення надходження інформації від
денервованого органа в нервовий центр
(регіонарний вузол, спинний або головний
мозок) і відсутність у зв’язку з цим корек
ції трофіки з боку нервів, що збереглися.
2. Припинення утворення нервом нейромедіаторів, у тому числі й тих, доставка яких
до клітини здійснюється механізмом аксоплазматичного транспорту.
3. Патологічна імпульсація з центральної
кукси перерізаного нерва, що посилює
порушення функції нервових центрів,
а отже, і зміни обміну речовин, що вже ви
никли на периферії внаслідок денервації.
4. Проведення патологічної імпульсації пе
рерізаним чутливим нервом у зворотному
напрямку (антидромно).
5. Зміни генетичного апарату клітин у денер
вованому органі з порушенням синтезу біл
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
ків, що спричиняється до появи речовин автоантигенів. Імунна система при цьому
відповідає реакцією відторгнення.
6. Неадекватні реакції, найчастіше підвищені,
на біологічно активні речовини, лікарські
препарати та інші гуморальні впливи (закон
денервації Кеннона). Наприклад, після пе
ретинання блукаючого нерва м’язова обо
лонка шлунка стає чутливішою до впливу
нервових медіаторів. Крім того, у ній спо
стерігають незвичні зміни обміну речовин
у відповідь на дію деяких гормонів.
7. Травматичні впливи середовища (меха
нічна травма, інфекція), що сприяють
швидшому розвитку трофічних порушень
у денервованих тканинах.
ПОРУШЕННЯ
ІНТЕГРАТИВНИХ ФУНКЦІЙ
НЕРВОВОЇ СИСТЕМИ
Основу інтегративних функцій центральної
нервової системи складають міжклітинні вза
ємодії - обмін інформацією між нейронами.
Складовими такого обміну є:
1) сприймання інформації;
2) її переробка;
3) надсилання інформації іншим клітинам.
Існує два механізми передавання інформації
в ЦНС:
1) електрофізіологічні - електричні сигнали;
2) нейрохімічні - хімічні сполуки.
Інформаційні сигнали, що надходять від од
них нейронів до інших, можуть зумовлювати
чотири стандартні ефекти:
1) посилення збудження;
2) послаблення збудження;
3) посилення гальмування:
4) послаблення гальмування.
Різні поєднання цих чотирьох ефектів зу
мовлюють різноманітну картину можливих
результатів міжнейронних взаємодій. До цьо
го слід додати, що кожна нервова клітина має
численні зв’язки з нейронами свого та інших
відділів ЦНС, які по-різному можуть впливати
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
на її діяльність. Так, число синапсів на мотонейронах ссавців складає від 10000 до 20000
на одну клітину, а кожна клітина Пуркіньє
в мозочку має таких синапсів до 200000. Усьо
го ж нейронів у ЦНС 25 • 109, а кількість синаптичних контактів ^ 10і5—1016.
Основні механізми. До розладів інтегративних
функцій ЦНС можуть спричинятися такі чин
ники: (І) порушення електрофізіологічних влас
тивостей і процесів; (II) розлади нейрохімічних
процесів; (III) ушкодження нейронів зовнішні
ми чинниками; (IV) порушення енергетичного
обміну в клітинах головного мозку; (V) розлади
мозкового кровообігу; (VI) набряк і набухання
головного мозку; (VII) внутрішньочерепна гі
пертензія; (VIII) ушкодження клітин нейроглії.
І.
Порушення електрофізіологічних процесів.
Вони можуть виявляти себе:
а) змінами мембранного потенціалу — по
тенціалу спокою (ПС). Якщо ПС стає
менш негативним (деполяризація), то
збудливість нейронів збільшується, і на
впаки, якщо ПС набуває більш негатив
них значень (гіперполяризація), то збуд
ливість зменшується;
б) змінами порогового потенціалу (кри
тичного потенціалу деполяризації), при
досягненні якого виникає потенціал дії
(ПД) (рис. 38.12). Якщо величина поро
гового потенціалу наближається до рів
ня ПС, то збудливість нейронів збільшу
Потенціал дії нервової клітини
413
ється, і навпаки, віддалення порогового
потенціалу від ПС спричиняється до
зменшення збудливості;
в) змінами характеру ПД (тривалості, амп
літуди, частоти) - розвитком відносної
та абсолютноїрефрактерності;
г) порушеннями проведення електричних
імпульсів по нервових провідниках.
Виділяють такі групи причин і механізмів за
значених електрофізіологічних змін.
1. Зміни позаклітинної концентрації катіо
нів. Основні електрофізіологічні характе
ристики нервових клітин залежать від кон
центрації трьох катіонів: калію, кальцію
і натрію:
а) іони К\ Вони визначають рівень ПС.
При збільшенні позаклітинного вмісту
К відбувається деполяризація мемб
рани і збудливість нейронів зростає.
Зменшення концентрації К+ у позаклі
тинному просторі, навпаки, викликає гіперполяризацію мембрани - зменшення
збудливості нервових клітин;
б) іони Са2+. Від них залежить рівень по
рогового потенціалу мембрани нейро
нів, оскільки вони впливають на стан
Na-каналів. При гіперкальціємії пороговий потенціал віддаляється від рівня
ПС (стає більш позитивним), у резуль
таті чого збудливість нервових клітин
падає. Гіпокальціємія викликає проти-
414
лежний ефект - наближення порогового
потенціалу до рівня ПС і пов’язане з цим
збільшення збудливості нейронів (клі
нічно розвиваються судоми - тетанія).
в) іони Na+. Вони зумовлюють розвиток
ПД. В умовах in vitro при видаленні Na+
із позаклітинного середовища ПД не ви
никає. In vivo вміст позаклітинних іонів
Na+ може змінюватися в невеликому
діапазоні. У разі таких змін на перший
план виступають порушення електрофі
зіологічних процесів, зумовлені змінами
осмотичного тиску: збільшення позаклі
тинної концентрації Na+ —► збільшення
ОСМОТИЧНОГО тиску -9- зневоднення ней
ронів —► збільшення внутрішньоклітин
ної концентрації К+ (ефект концентру
вання) —* збільшення ПС —* зменшення
збудливості. І навпаки, зменшення по
заклітинної концентрації Na+ —* змен
шення осмотичного тиску —> набряк
нервових клітин —* зменшення внутріш
ньоклітинного вмісту К+ (ефект розве
дення) —*• зменшення ПС —і збільшення
збудливості.
2. Порушення провідності іонних каналів.
У розвитку розладів електрофізіологічних
процесів у нейронах мають значення змі
ни Na-каналів, К-каналів, Са-каналів і С1каналів клітинних мембран.
Найкраще вивчено порушення, пов’язані
з провідністю Na-каналів. Установлено такі
типи їхніх змін:
а) блокада (закупорка). В експерименті для
відтворення блокади Na-каналів викорис
товують отрути - тетродотоксин та ін.;
б) пригнічення активації (виникають при гіпокальціємії);
в) порушення інактивації та реактивації.
Блокаду К-каналів в експерименті виклика
ють за допомогою тетраетиламонію.
3. Пригнічення роботи іонних електрогенних
насосів (Na-K-насосів). Функціонування
цих механізмів первинного активного тран
спорту порушується при їх специфічній
інактивації та зміні хімічного складу ліпі
дів мембран, які створюють мікрооточення.
4. Порушення енергозабезпечення. Дефіцит
АТФ незалежно від його причини призво
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
дить до розладів функції згаданих вище
Na-K-насосів.
5. Зміни стану мембран, що проводять ім
пульси. Однією з причин таких змін є демієлінізація, при якій порушуються електрокабельні властивості нервових провідни
ків. При цьому спостерігають два ефекти:
а) зменшення швидкості проведення імпуль
сів;
б) ефаптичний ефект - перехід імпульсу з од
ного нервового волокна на інше, що йде
паралельно, у результаті утворених контак
тів між волокнами — ефапсів. Ефаптичний
ефект є однією з причин генералізації збу
дження.
II.
Порушення нейрохімічних процесів. Нейрохімічними називають процеси в центральній
нервовій системі, пов’язані з утворенням і ви
вільненням речовин-нейрорегуляторів, до яких
відносять нейротрансмітери, нейромодулятори
і нейрогормони.
1. Нейротрансмітери, або нейромедіатори, — це хімічні сполуки, що накопичу
ються у пресинаптичній частині синапсу
і вивільнюються у синаптичну щілину при
збудженні терміналей нервового волокна.
Нейротрансмітери змінюють іонну про
відність постсинаптичних мембран і, як
наслідок, мембранний потенціал. Якщо
мембранний постсинаптичний потенціал
зменшується (деполяризація), то настає
збудження, якщо ж, навпаки, збільшується
(гіперполяризація), то розвивається галь
мування.
Залежно від кінцевих ефектів усі нейротранс
мітери поділяють на дві групи: (1) збуджуваль
ні медіатори ЦНС (ацетилхолін, катехоламіни,
серотонін, субстанція Р, глутамат) і (2) гальмів
ні (гамма-аміномасляна кислота -ГАМК, глі
цин).
Нейротрансмітери здійснюють чітко локалі
зоване і швидке вмикання й вимикання окремих
нейронних систем.
Виділяють такі механізми порушень нейротрансмітерних функцій у центральній нервовій
системі:
а) порушення синтезу нейротрансмітерів
та (або) їхнього транспорту до закінчень
нервових волокон. Таке буває при блоку
ванні відповідних ферментних систем (на-
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
приклад, семікарбазид пригнічує синтез
ГАМК), при розладах механізмів аксоплазматичного транспорту (наприклад, руй
нування мікротрубочок і мікрофіламентів
колхіцином та вінбластином);
б) розлади депонування нейротрансмітерів
у нервових закінченнях, у тому числі ви
снаження їхніх депо (наприклад, резерпін
порушує депонування норадреналіну й се
ротоніну);
в) порушення секреції нейромедіаторів у синаптичні щілини. Причинами цього мо
жуть бути зменшення концентрації іонів
Са2+ і збільшення вмісту Mg2+, дефіцит
АТФ, ботулінічний токсин, який пригні
чує вивільнення ацетилхоліну; правцевий
токсин, що припиняє секрецію гальмівних
медіаторів, зокрема гліцину;
г) порушення взаємодії нейротрансмітерів
з постсинаптичними рецепторами. До цього
спричиняються сполуки, які або блокують
таку взаємодію (наприклад, стрихнін бло
кує гліцинові рецептори та зменшує галь
мування, кураре - n-холінорецептори і при-*
гнічує збудження), або імітують дію медіа
торів (агоністи відповідних рецепторів);
д) пригнічення зворотного захоплення ней
ромедіаторів нервовими закінченнями. Це
веде до поступового зменшення кількос
ті медіаторів у синаптичних везикулах,
а отже, і до зменшення їхнього вивільнення
у синаптичну щілину при надходженні по
тенціалів дії;
е) порушення руйнування нейротрансміте
рів відповідними ферментами. Так, бло
кування холінестерази фосфорорганічни
ми сполуками веде до підвищення рівня
ацетилхоліну в синаптичних щілинах, а
пригнічення активності таких фермен
тів, як моноаміноксидаза і катехол-Ометилтрансфераза, — до зростання вмісту
норадреналіну й серотоніну.
Порушення з боку нейротрансмітерів можуть
мати такі наслідки:
1) збільшення концентрації збуджувальних
медіаторів у синаптичній щілині (посиле
на секреція або послаблене видалення) чи
дія на рецептори постсинаптичної мемб
рани відповідних агоністів зумовлюють
ефект збудження',
415
2) зменшення вмісту збуджувальних медіа
торів у синаптичній щілині або порушен
ня їх рецепції стають причиною ефекту
гальмування',
3) збільшення концентрації гальмівних ме
діаторів у синаптичній щілині або дія на
рецептори агоністів викликають ефект
гальмування',
4) зменшення вмісту гальмівних нейротранс
мітерів у синаптичній щілині або пору
шення їхньої рецепції спричиняються до
ефекту збудження.
Можливі причини розвитку патологічного
збудження й патологічного гальмування нерво
вих центрів подано в табл. 38.3.
2. Нейромодулятори. До цієї категорії відно
сять нейропептиди, які, так само як і нейротрансмітери, вивільняються в синаптичну
щілину. Однак їх вплив на постсинаптичні
структури носить не прямий, а опосередко
ваний характер.
Дія нейромодуляторів на нейрони може
здійснюватися через:
а) аксон-аксонні синапси—ефект
пресинаптичної модуляції',
б) специфічні рецептори постсинаптичної
мембрани - ефект постсинаптичної
модуляції',
в) системи вторинних посередників
(месенджерів) — ефект
внутрішньоклітинної модуляції.
Найбільш вивченими нейромодуляторами є:
пептиди-аналгетики, що входять до гор
мональної
опіатної
антиноцицептивної
системи (енкефаліни, ендорфіни, динорфіни) (див. вище). Зменшення їх утворення
або блокада відповідних рецепторів ве
дуть до підвищення больової чутливості,
втрати почуття комфорту, пригніченого
настрою;
• гіпногенні пептиди (пептиди сну). Збіль
шене їх утворення зумовлює сонливість,
а зменшене, так само як і блокада відповід
них рецепторів, - безсоння;
модулятори харчової і питної поведінки
(нейротензин, мозкові аналоги холецистокініну, гастрину). Порушення секреції цих
речовин може бути механізмом розладів
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
416
Таблиця 38.3
Можливі причини патологічного збудження
й патологічного гальмування нервових центрів
Іонні порушення: збільшення позаклітинної
концентрації іонів К+ і зменшення - іонів Са2+
Іонні порушення: зменшення позаклітинної
концентрації іонів К+ і збільшення - іонів Са2+
Ефаптичні ефекти
Порушення роботи електрогенних насосів та їх
енергозабезпечення
Збільшення концентрації збуджувальних
нейромедіаторів у синаптичній щілині
Зменшення концентрації збуджувальних
нейромедіаторів у синаптичній щілині
Дія на збуджувальні постсинаптичні рецептори
речовин-агоністів
Блокування постсинаптичних збуджувальних
рецепторів
Зменшення концентрації гальмівних
нейромедіаторів у синаптичній щілині
Збільшення концентрації гальмівних
нейромедіаторів у синаптичній щілині
Блокування постсинаптичних рецепторів до
гальмівних медіаторів
Дія на гальмівні постсинаптичні рецептори
речовин-агоністів
апетиту (анорексія, поліфагія) і формуван
ня відчуття спраги (адипсія, полідипсія).
3. Нейрогормони. Таку назву отримали пептидні сполуки, що утворюються в не
йроендокринних клітинах гіпоталамуса
(див. главу 37). Дія нейрогормонів на різні
структури ЦНС може здійснюватися че
рез: (а) кров, (б) ліквор і (в) синапси. У гі
поталамусі є пептидергічні нейрони, які
посилають свої аксони не в нейрогіпофіз
і аденогіпофіз, а безпосередньо до певних
структур головного мозку (наприклад, до
структур лімбічної системи). Закінчення
цих нервових клітин вивільняють у синаптичні щілини пептиди, такі як вазопресин,
окситоцин, рилізинг-гормони.
З порушеннями утворення і вивільнення не
йрогормонів у ЦНС можуть бути пов’язані такі
розлади:
а) порушення нам ’яті та здатності до навчан
ня. Вазопресин посилює запам’ятовування,
позитивно впливає на консолідацію слідів
пам’яті й на мобілізацію інформації, що збе
рігається. Окситоцин має протилежну дію, а
тому його називають амнестичним гормоном',
б) розлади сексуальної поведінки. Посилення
статевого потягу й формування відповід
них поведінкових реакцій викликають гонадотропіновий рилізинг-гормон, оксито
цин, фрагменти АКТГ;
в) порушення больової чутливості. Вазопре
син, як уже зазначалося (див. вище), є важ
ливим елементом гормональної неопіатної
антиноцицептивної системи.
Порушення міжнейронних взаємодій внаслі
док розладів електрофізіологічних і нейрохімічних процесів зумовлюють формування осе
редків патологічного збудження і патологічного
гальмування, що можуть виявляти себе цілим
рядом функціональних змін. До таких зокрема
відносять:
1) сумацію збудження. Суть її полягає в тому,
що дія підпорогових стимулів спричиня
ється до збудження нервових центрів;
2) трансформацію ритму збудження. Роз
різняють вирівнювальну трансформацію,
коли слабкі та сильні стимули викликають
однаковий ступінь збудження нервового
центру, і парадоксальну трансформацію,
при якій слабкі стимули зумовлюють біль
ше збудження, ніж сильні;
3) збільшення стомлюваності нервових цен
трів;
4) тривалу післядію збудження. При цирку
ляції потенціалів дії збудження може під
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
тримувати само себе {ефект реверберації),
а отже, активний стан нервових центрів
може тривати довго, навіть за відсутності
аферентних сигналів;
5) патологічну домінанту. У центральній не
рвовій системі формуються осередки пато
логічного збудження, які підпорядковують
собі діяльність усіх інших нервових цен
трів. Таке явище отримало назву патоло
гічної домінанти. Осередки патологічної
домінанти пригнічують інші центри, а при
подразненні останніх збільшують свою
збудливість.
III. Ушкодження нейронів зовнішніми чинни
ками. Найчастіше такими чинниками є:
1) механічна черепно-мозкова травма. За
лежно від ступеня ушкодження виділя
ють: (а) струс мозку {commotio)', (б) удар
мозку (contusio); (в) здавлення мозку
{compressio). У переважній більшості ви
падків причинами здавлення мозку є гема
томи (епідуральні, субдуральні, внутрішньомозкові, внутрішньошлуночкові);
2) інфекційні агенти. Ушкодження нейронів
центральної нервової системи виникає при
енцефалітах (групі вірусних інфекцій),
поліомієліті (вірус уражує мотонейрони
спинного мозку і стовбура мозку), сказі,
сифілісі, токсоплазмозі, бруцельозі та ін
ших нейроінфекціях.
IV. Порушення енергетичного обміну в кліти
нах головного мозку. Енергетичний обмін у тка
нинах головного мозку має ряд особливостей.
1. Основним енергетичним субстратом є глю
коза. Саме цим пояснюють той факт, що в
умовах гіпоглікемії насамперед страждає
головний мозок.
2. Висока інтенсивність аеробного гліколізу.
Навіть в аеробних умовах 15 % поглине
ної глюкози перетворюється на молочну
кислоту (послаблений ефект Пастера). Це
свідчить про високу інтенсивність роботи
іонних насосів, що використовують АТФ,
який утворюється в реакціях гліколізу.
3. Високий рівень споживання кисню. У стані
спокою головний мозок використовує 20 %
кисню, що надходить в організм. Інтенсив
ність споживання кисню нейронами у 6-7
разів вища, ніж клітинами нейроглії.
4. Сталість високого рівня енергетичного об
міну. Великі енергетичні потреби головного
417
мозку практично не міняються в стані спо
кою і в умовах напруженої діяльності мозку.
5. Украй висока чутливість головного мозку
до зменшення енергозабезпечення. Цей ор
ган є найчутливішим до гіпоксії. Зменшен
ня споживання кисню тканинами головного
мозку на 20 % нижче норми спричиняється
до втрати свідомості. Порушення енер
гетичного обміну в клітинах ЦНС стають
причиною розвитку коматозних станів
(див. главу 20).
Пригнічення процесів енергетичного обміну
в нейронах може зумовлюватися такими причи
нами:
1) кисневим голодуванням - гіпоксією (див.
главу 23). Дефіцит кисню в нервових клі
тинах виникає при:
а) недостатньому вмісті кисню у вдихуваному повітрі (гіпоксична кома);
б) недостатності зовнішнього дихання (рес
піраторна кома);
в) порушеннях мозкового кровообігу (див.
нижче) (апоплексична кома);
г) розладах транспортної функції крові (ге
молітична кома, анемічна кома);
2) дефіцитом енергетичних субстратів. Цей
чинник лежить в основі гіпоглікемічної та
аліментарно-дистрофічної ком;
3) внутрішньоклітинними порушеннями. Та
кими зокрема є:
а) розлади циклу Кребса (печінкова кома, див.
главу 35);
б) пригнічення клітинного дихання при інак
тивації ферментів дихального ланцюга мітохондрій (коми при інтоксикаціях ціані
дами, CO, H2S).
Усі зазначені вище причини зумовлюють де
фіцит АТФ, який, у свою чергу, веде до пору
шень енергозалежних процесів у нервових клі
тинах: (а) транспорту іонів, (б) біосинтезу речо
вин, (в) скорочення внутрішньоклітинних струк
тур (рис. 38.13). Як наслідок, порушуються:
1) генерація та проведення нервових імпуль
сів (електрофізіологічні процеси);
2) синтез нейротрансмітерів, нейромодуляторів і нейрогормонів (нейрохімічні про
цеси);
3) транспорт медіаторів від місць їх синтезу
до нервових закінчень (аксоплазматич-
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
418
Порушення енергетичного обміну в нервових клітинах як механізм розвитку коматозних станів
ний транспорт) і вивільнення їх із синаптичних везикул у синаптичну щілину.
Комплекс виникаючих розладів стає провід
ним механізмом розвитку коми: людина втра
чає свідомість, зникають рефлекси на зовнішні
подразники, пригнічуються життєво важливі
функції організму (зовнішнє дихання, крово
обіг).
V.
Розлади мозкового кровообігу. Значним
енергетичним потребам головного мозку під
порядковується мозковий кровообіг, у зв’язку
з чим він має певні особливості:
а) у стані спокою об’ємна швидкість мозко
вого кровообігу дорівнює 15 % від хви
линного об’єму крові, хоча маса головно
го мозку становить менш ніж 2 % маси
тіла;
б) сталість мозкового кровообігу. Її визна
чає сталість енергетичних потреб голов
ного мозку (див. вище). Якщо вінцевий
кровообіг у серці може зростати в 7-10 ра
зів відповідно до збільшених його потреб
в енергії, то мозковий кровообіг має бути
постійним, незмінним;
в) з наведених вище причин у тканинах голов
ного мозку робоча артеріальна гіперемія
не виникає. Розвиток тут патологічної ар
теріальної гіперемії завжди має негативне
значення, оскільки є причиною збільшення
внутрішньочерепного тиску (див. нижче);
г) у тканинах головного мозку немає так зва
них резервних капілярів, проте досить роз
винений колатеральний кровообіг',
д) у регуляції мозкового кровообігу мають
значення міогенні — авторегуляція тонусу
судин (ефект Бейліса) - і метаболічні ме
ханізми. Останні пов’язані зі змінами рСО,
і р02 артеріальної крові. При збільшенні
рС02 і зменшенні р02 відбувається розши-
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇСИСТЕМИ
рення мозкових судин, а при зменшенні
рС02 і збільшенні р02, - навпаки, їхнє зву
ження. Нервові механізми в регуляції моз
кового кровообігу практичного значення не
мають.
Порушення регуляції мозкового кровообігу
можуть виявляти себе кількома синдромами.
Синдром надмірної перфузії - посилення моз
кового кровообігу після значного його обме
ження. У цьому випадку артеріальна гіперемія
мозку настає в результаті того, що порушуєть
ся міогенна авторегуляція місцевого крово
обігу. При тривалому кисневому голодуванні
та пов’язаному з ним ацидозі ушкоджуються
гладкі м’язові клітини судин, у зв’язку з чим
вони втрачають здатність скорочуватися у від
повідь на розтягнення. А тому при збільшенні
артеріального тиску не спрацьовує ефект Бейліса, порушується сталість мозкового крово
обігу, він стає надмірним.
* Синдром внутрішнього обкрадання. Суть
його полягає в тому, що при збільшенні
рС02 артеріальної крові чи введенні вазо
дилятаторів кровонаповнення збільшується
тільки в інтактних ділянках мозку (артерії
й артеріоли розширюються), а в ішемізованих, навпаки, воно зменшується через пере
розподіл крові (її значно більше надходить
в розширені судини, ніж у звужені).
» Спотворений синдром внутрішнього об
крадання - “синдром Робін Гуда”. При
зменшенні рС02 артеріальної крові або при
застосуванні судинозвужувальних засобів
кровонаповнення ішемізованих ділянок
мозку значно посилюється через те, що
відбувається звуження судин інтактних ді
лянок і туди менше надходить крові.
Порушення мозкового кровообігу поділяють
на гострі та хронічні.
1.
Гострі порушення мозкового кровообігу —
інсульти. До таких відносять розлади кровообі
гу, що призводять до стійких порушень функцій
головного мозку. Розрізняють:
1) геморагічні інсульти — крововиливи
в мозок. Найчастіше вони бувають ре
зультатом стійкої артеріальної гіпертен
зії (розрив зміненої стінки артеріальної
судини, мікроаневризм);
2) ішемічні інсульти (інфаркт мозку).
419
Ураховуючи те, що об’ємна швидкість моз
кового кровообігу (Qm) визначається форму
лою:
можна виділити ряд причин, що зумовлюють
недостатнє кровонаповнення мозку та розвиток
інсультів:
* зменшення артеріального тиску СРарт)- Усі
види шоків на термінальній стадії їх розви
тку ведуть до значного зменшення мозко
вого кровообігу і стають причиною коми
(див. главу 20). Якщо ж внаслідок падіння
артеріального тиску розвивається інсульт,
то такий його патогенетичний варіант по
значають як гемодинамічний;
* збільшення тиску у венозних судинах мозку (Рвен), що настає при здавлюванні вен
в умовах підвищеного внутрішньочерепно
го тиску (див. нижче);
* зменшення просвіту артерій, через які відбу
вається кровопостачання мозку (г). Цей чин
ник власне і зумовлює “класичну” ішемію
мозку, яка стає причиною його інфаркту.
Якщо в основі інсульту лежить атероскле
ротичний процес, ускладнений тромбоут
воренням, то такий варіант називають атеротромботичним. Якщо ж рух крові пору
шується через дрібні артерії кінцевого типу,
що не мають колатералей, то виникають не
великі, часто безсимптомні, інфаркти мозку
в деяких його структурах (смугасте тіло,
стовбур мозку, мозочок). У цьому разі мову
ведуть про лакунарні інсульти. І, нарешті,
причиною оклюзії мозкових артерій можуть
бути емболи серцевого походження — тром
би, що відірвалися від стінок серця, клапа
нів при різних формах їхніх уражень (вади
серця, інфекційний ендокардит, інфаркт мі
окарда, миготлива аритмія тощо). Такий ін
сульт позначають як кардіоемболічний;
розлади мікроциркуляції в тканинах мозку,
причиною яких є порушення реологічних
властивостей крові, зокрема збільшення її
в’язкості (rj). Така ситуація є характерною
для ДВЗ-синдрому (див. главу ЗО), зневод
нення організму (див. главу 27). Ішемічні
інсульти цього типу називають гемореологічними.
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
420
2.
Хронічні порушення мозкового кровообігу.
Тривалі розлади кровообігу в тканинах мозку,
найчастіше зумовлені атеросклеротичним про
цесом, ведуть до розвитку осередкових дис
трофічних змін у різних структурах головного
мозку. Ці зміни клінічно виявляють себе син
дромом судинної енцефалопатії — комплексом
різноманітних неврологічних і психічних по
рушень, що настають внаслідок розладів інте
гративних функцій центральної нервової систе
ми (див. нижче). Судинна енцефалопатія може
бути й наслідком значної кількості лакунарних
інсультів (мікроінсультів).
VI.
Набряк і набухання головного мозку. На
бряком мозку звичайно називають нако
пичення рідини в інтерстиціальній тканині
мозку, а набуханням - його внутрішньоклі
тинний набряк. Часто ці патологічні зміни
поєднуються.
За етіологією набряк мозку може бути
(а) травматичним, (б) пухлинним, (в) післяопе
раційним, (г) токсичним, (д) запальним та ін.
У патогенезі набряку мозку мають значення
дві групи факторів:
1) судинні фактори, тобто чинники, пов ’язані
з кров’ю та стінками кровоносних судин.
До таких відносять:
а) збільшення гідростатичного тиску в ка
пілярах (артеріальна гіперемія, венозна
гіперемія, гіперволемія);
б) зменшення онкотичного тиску крові;
в) збільшення проникності гематоенцефалічного бар ’єра;
2) тканинні фактори. Такими є:
а) збільшення онкотичного тиску в мозко
вій тканині (вихід білків з ушкоджених
клітин, розщеплення протеїнів);
б) зменшення
гідростатичного тиску
в мозковій тканині;
в) ушкодження гліальних елементів гематоенцефалічного бар’єра, унаслідок чого
підвищується проникність останнього.
Ушкодження нейронів при набряку та набу
ханні головного мозку настає в результаті того,
що підвищується внутрішньочерепний тиск
і порушується кровопостачання тканин мозку
через здавлювання венозних судин і розвиток
венозної гіперемії (рис. 38.14). До цього до
дається й механізм механічного ушкодження,
якщо відбувається вклинювання головного моз
ку в отвори черепа.
У патогенезі набряку мозку велике значення
мають “зачаровані кола” (circuli vitiosi). Одне
з них представлено нарис. 38.15.
Внутрішньочерепна гіпертензія. Внутріш
ньочерепний тиск - це тиск цереброспінальної рідини (ліквору) в порожнині че
репа і шлуночках мозку. В умовах норми
в горизонтальному положенні цей показ
ник дорівнює 150 мм вод. ст.
Внутрішньочерепна гіпертензія — це підви
щення внутрішньочерепного тиску, яке може
сягати 700-800 мм вод. ст.
Причинами внутрішньочерепної гіпертензії
можуть бути:
1) збільшення кровонаповнення головного
мозку (артеріальна гіперемія, венозна гі
перемія);
2)
збільшення кількості цереброспінальної
рідини (ліквору) - гідроцефалія. Воно мо
же бути зумовлене або (а) збільшенням
утворення ліквору, або (б) зменшенням йо
го резорбції та відтоку зі шлуночків мозку;
3) збільшення об ’єму мозкової тканини - на
бряк і набухання мозку (див. вище);
4)
поява додаткових об’ємних структур
у порожнині черепа. Це можуть бути за
пальний ексудат (при менінгітах), гемато
ми, пухлини, абсцеси.
VII.
Підвищення
внутрішньочерепного
тиску
призводить до здавлювання мозкових вен. Це,
у свою чергу, викликає порушення мозкового
кровообігу та гіпоксію, а з другого боку, є од
ним із чинників розвитку набряку мозку (див.
рис. 38.14 і 38.15).
VIII.
Ушкодження клітин нейроглії. Нейрогліальні клітини (олігодендроцити, астро
цити, епендимальні клітини, клітини мікроглії та ін.) є важливим компонентом
центральної нервової системи. На них при
падає 40-50 % об’єму тканин головного
мозку, а їх кількість тут майже в 10 разів
більша, ніж число нейронів.
На відміну від останніх, клітини нейроглії не
мають збудливості та не можуть генерувати по-
421
ГЛАВА 38. ПАТОФІЗІОЛОГІЯ НЕРВОВОЇ СИСТЕМИ
Рис. 38.14. Механізми ушкодження нервових клітин при набряку та набуханні головного мозку
тенціали дії, вони зберігають здатність до поді
лу та розмноження.
До основних функцій нейроглії відносять:
а) утворення мієліну. Ця функція здійснюєть
ся олігодендроцитами - аналогами шваннівських клітин у периферичній нервовій
системі;
б) створення гематоенцефалічного бар’єра.
До складу останнього входять астроцити
(виконують функцію селективного філь
тра) і мікрогліальні клітини (здійснюють
фагоцитоз);
в) забезпечення сталості позаклітинної концен
трації іонів ІС (функція калієвого буфера).
Ушкодження нейроглії може мати ряд наслід ків.
1. При порушенні утворення мієліну відбу
вається демієлінізація волокон провідни
кових шляхів. Це стає причиною хвороби
під назвою “розсіяний склероз”. При цьому
зменшується швидкість проведення нерво
вих імпульсів, виникають ефаптичні ефек
ти (див. вище).
2. Якщо порушується структура гематоенце
фалічного бар’єра, то можуть розвиватися
автоалергічні ушкодження нейронів і нер
вових провідників головного мозку - автоалергічний енцефаломієліт (див. главу 15);
3. Зменшення калієвої буферності нейроглії
може спричинятися до збільшення позаклі
тинної концентрації іонів К+, а це, у свою
чергу, - до підвищення збудливості не
йронів. Дехто вважає, що подібні зміни
можуть зумовлювати розвиток нападів епі
лептичних судом.
Наведені вище основні причини порушень
діяльності центральної нервової системи зумов
люють розлади багатьох інтегративних її функ
цій. Клінічно ці розлади можуть виявляти себе
великим спектром неврологічних і психічних
порушень. До таких, зокрема, відносять:
1) порушення відчуттів і сприйняттяь? ви
щих сенсорних функцій. Прикладом є аг
нозія — розлади розпізнавання. Розрізня
ють зорову, слухову та інші види агнозії;
422
розлади свідомості. Такими, зокрема, є
різного ступеня оглушення (форма потьма
реної свідомості): обнубіляція (потьмарен
ня), сомнолентність (сонливість), сопор
(непритомність, непробудний сон) і кома
(див. главу 20);
3) розлади мислення. Вони є ознакою бага
тьох психічних хвороб: шизофренії, епі
лепсії, маніакально-депресивного синдро
му. Однією з крайніх форм порушень мис
лення є деменція (слабоумство);
4) розлади мови - афазія. Розрізняють рухо
ву афазію (утруднене або неможливе від
творення мови, розуміння мови може бути
збережене) і сенсорну афазію (розлади
сприйняття мови);
5) порушення поведінкових реакцій. Прикла
дом може бути апраксія - порушення ці
леспрямованих дій (людина не може запа
лити сірника, змахнути рукою, розрізати
хліб, хоча руки не паралізовані);
2)
ТОМ 2. ПАТОФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
6) порушення емоцій. Серед таких гіпотимія
(депресивний синдром), гіпертимія (маніа
кальний синдром), неврози (неврастенія, не
вроз нав’язливих станів, істеричний невроз);
7) порушення мотивацій. Розрізняють не
нормально посилені мотивації (булімія,
полідипсія, гіперсексуалізм, статеві збо
чення), ненормально ослаблені мотивації
(анорексія, адипсія, імпотенція, фригідність), штучно створені мотивації (нарко
манія, алкоголізм, куріння);
8) порушення здатності до навчання. Не
здатність до навчання є однією з провід
них ознак олігофренії (уродженого слабо
умства). Залежно від ступеня інтелекту
ального дефекту розрізняють дебільність,
імбецильність, ідіотію;
9) розлади пам ’яті - амнезію',
10) розлади циклів сну - неспання. Вони мо
жуть виявляти себе безсонням і сонли
вістю.
Покажчики
Об’єднаний
предметний покажчик
Аберації хромосомні...............................................1225,1170
Автоімунний тиреоїдит Хашимото.................. 11373-375
Автоліз.............................................................................. І 75,86,87
Автофагія .....................................................184,95,420, II 744
Агаммаглобулінемія Брутона.................................1226,227
Аглікогеноз..............................................................................II300
Агнозія...................................................................................... II397
Агранулоцитоз....................... І 729, 759,254,275, II63-64
Агрегація тромбоцитів.................1295,312,317,472, II 75
• оборотна..........................................................................II76
• необоротна ................................................................... II76
• початкова........................................................................II76
Адаптація...............................142,55, 97, II 95-104,373-374
Адгезини................................................................ і 753, 754, 755
Адгезія.............................................................................. І 754, II 76,
• лейкоцитів...........................\ 312,322,331, \\ 170-172
• тромбоцитів....................................... 1267, II 75,85-87
Аденома (и)................................................................. II358-359
• базофільні..................................................................II 359
• еозинофільні.............................................................II 358
• соматотрофів............................................................II 358
• хромофобні гіпофіза............................................ 11358
Адреналін.............................. 1287,284,450,489,
II 74, 749,
188,189-191,237,370
Адреналіт................................................................................. II365
• автоімунний.............................................................. II365
• ізольований автоімунний...................................II365
Азотемія...........................................\ 469,470, \\ 321,323,337
Акромегалія............................................................. 1456, II359
Активатори фібринолізу...................................................1188
Активація тромбоцитів......................................................... II76
Алергени................................................................ 1242,243,253
Алергічні реакції . ...... ................1243, II285,342,375,386
• імунологічна стадія.........................................1243,246
• патофізіологічна стадія................................. 1243,245
• патохімічна стадія........................................ 1243,245
• типи .................................................................... \ 243,244
Алергія.............................................................. 1242,243, II204
Алімфоцитоз.......................................................................... 1228
Алкалоз .................................................. 1570,573,574,11 774
• газовий................................................................117 74,230
• негазовий.................................................\\ 114,243,368
Алкалоз негазовий гіпокаліємічний........................... II326
Алкаптанурія................................................................. 1504,505
Алоритмія.....................................................................II 120-121
Альбінізм .............................................................. 1504,505,506
Альдостерон..................... \ 516, \\ 189,192-199,361-368
Альтерація. . . 148,300,304,315, II 141,177,261,295
Аміак.............................................II261,282,302-306
Аміноацидемія.............................................. 1469,504, II362
Аміноацидурія....................................................................... II319
Амінокислоти ароматичні............................................... II323
Амінокислоти крові ......................... 1502,504, II 160,384
Амнезія......................................................................................II422
Амоніогенез................................................... 1565,568, II326
"Аморфосинтез"...................................................................II397
Ампліфікація................................................................. 1374,386
Анаплазія .................................................... 1349,352, II64-67
Анафілаксія.................................................................. 1245,246
Анацидітас............................................................................... II255
Анандаміди............................................................................. II402
Ангідремія............................................................................. 1523
Ангіогемофілія........................................................................ II 85
Ангіогенез.......................................................... І 93,278, II383
Ангіоневротичний набряк...................................... \215,246
Андрогени....................................................... І 180, II360,379
АнеміяСО............................................................... \ 32,510,425
• В12-дефіцитна.........................................................N27,46
• Аддісона - Бірмера..............................................N22,50
• алоімунні (ізоімунні) гемолітичні........................II22
• апластична.......................................................І 729, II22
• аутоімунні гемолітичні............................................ N22
• В12-рефрактерна...................................................... 1570
• В12-фолієводефіцитна.................................... 132,425
• гемолітична(і).................\254,257,275,\\ 22,24,51
• гетероімунні (гаптенові) гемолітичні . . N22,27
• гіпопластична(і)....................................................... І 729
• глюкозо-6-фосфатдегідрогеназодефіцитнаІ 779,
N31
• гостра постгеморагічна.........................................1122
• залізодефіцитна.............................................1426, II23
• імунні......................................................................... N22,28
425
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
•
•
•
•
інфекційні.....................................................................1122
мегалобластична.......................................... 1425,508
мікросфероцитарна........................................\ 81,177
мікросфероцитарна
Мінковського - Шоффара.......................................II29
• набута гемолітична.................................................... II22
• перніціозна Аддісона - Бірмера 1275,425, II50
• постгеморагічна гостра ...................................... 1708
• постгеморагічні........................................................... II23
• серпоподібноклітинна............................................. II33
• спадкові гемолітичні анемії................................... II29
• токсичні............................................................................II29
• фолієводефіцитна.......................................................II46
Анергія.................................................................... 1270,274,394
Анізоцитоз.......................................................................... I117,21
Анорексія............................................... \ 395,417,418, \\ 240
• інтоксикаційна....................................................... II2 40
• диспепсична............................................................. II247
• нейродинамічна..................................................... II247
• невротична................................................................ N247
• нейроендокринопатична................................... N247
Антигени.......................................... І 157,254,392, II202,298
Антидіурез............................................................................. II320
Антидіуретичний гормон..................1579,520, II359, Збб
Анти доти............................................................... \ 148,149, 151
Антикоагулянти...............................................................II88-90
• антитромбіні............................................................... 1189
• антитромбін III.............................................................II89
• "вовчаковий"............................................................... N89
• вторинні......................................................................... N 88
• гепарин (антитромбін II)........................................ II89
• інгібітор ^-компонента комплементу ... II89
• "патологічні"................................................................ II89
• первинні.........................................................................II 88
• продукти фібринолізу............................................. II 89
• протеїн С......................................................................... II89
• тромбомодулін.............................................................II89
• сц-антитрипсин...........................................................N89
• а2-макроглобулін...................................................... N 89
Антикоагулянтна система ............................. 1296, II88-89
Антионкогени ........................................................... \ 381,382
Антитіла...............................І 157,219,220, \\ 28,64,83,342
Анурія...................................................... \ 522,532, \\ 320,331
Апарат Ґольджі................................................ 184, 762, II766
Аплазія.............................................................................. II 52,378
• гіпопластична............................................................. N 52
• Фанконі.......................................................................... N 52
• залізорефрактерна.................................................... N45
• В12-рефрактерна........................................................ N57
Апное.................................................................І 7 79, II227-230
Апопротеїн АроЕ...................................................... II 167-169
Апопротеїн В-100...............................................................II 769
Апоптоз ..............................................\ 86,87, \\ 144-146,280
Арефлексія (гіпорефлексія)........................................... II47 7
АритміяСО.......................................................\ 400, \\ 114-120
• дихальна................................................... 148,299, II409
. серця.............................................................. 18,299,117 74
• гетеротопні................................................................. II 7 78
Артеріальна гіпертензія...............................N 160,186,194
• первинна....................................................................Ч 786
• вторинна.....................................................................II 786
• доброякісна...............................................................II 786
• злоякісна.....................................................................II 786
Артеріальна гіпотензія.................................................... И 200
• гостра...........................................................................Ч ^80
• хронічна......................................................................11200
Артеріолосклероз Менкеберга І 189,476,557, II 156,
178
Артеріосклероз............................................. 1205,289, II 755
Асинергія............................................................ Н 153-155,407
Асоціація базофільно-еозинофільна...........................N 68
Астенія.............................................................. \ 121, \\ 311,409
Астма................................................. \ 194,243,260,363 \\ 212
• бронхіальна..................................\ 26,250,251, \\ 212
• серцева........................................................................П 757
Асцит........................................................................... 1424,11374
Атаксія............................................................... \ 454,649, \\ 407
• статико-локомоторна...........................................N 407
• динамічна...................................................................N407
Ателектаз .................................................................. 1735, N278
Атеросклероз.......................................\ 17,37,28, \\ 156,177
Атетоз.......................................................................................II407
Атонія (гіпотонія) м'язів.........................................II406-407
Атріопептин.......................................... \ 517,518,519, \\ 190
Атрофія..........................................\ 97,138, 189,422, II49,99
Атрофія м'язів....................................................................... N406
Атрофія міокарда ("деадаптоване" серце) . . . . N99
Аутоалергічні реакції........................................................ І 783
Аура зорова..........................................................................N 400
Афазія...................................................................................... II422
Аферента ція бол ьова.................................................... II404
Афібриногенемія................................................................ N307
Афлатоксин В, .............................................. 1357,358, II289
Ахілія............................................................................II 48,50,279
Ахлоргідрія............................................................................. N255
Ацидоз..............................................\ 57, 150,290,570, II 7 74
• газовий..............................................................\\ 114,211
• видільний................................................................... II326
• канальцевий....................................................II348,366
• клубочковий.............................................................II326
• метаболічний........................................................... II374
• негазовий................................................\\ 114,283,348
Б
Бактеріемія.................................................................. І 160, II64
Бактеріурія............................................................................ 11344
Баланс ................................................................................................
• азотистий................................... \ 419,420,500, II316
• водний ................................................. \ 511,522, II324
Барорецептори...................................................II 12, 189,224
Безсоння....................................................................... II 302,311
ПОКАЖЧИКИ
426
Бігемінія........................................................................... II 120-121
Білірубін................................................. І 123,148,433, II306
• непрямий (некон'югований).............................. II306
• прямий (кон'югований).............................II 306,309
Білірубінурія................................................................II308-310
Білки......................................................................................................
• p53 ............................................ 188,94,377,396, II 744
• активатори....................................................I 89, II80-89
• гемостазу.......................................................... 1472, II76
• деградація........................... \ 295,318,503, \\ 40, 184
• захисні................................................................ 13/6,507
• лізосомні......................................................1307, II 77,90
• мембранні...................................................187,1140, 746
• регуляторні......................................................... \ 82,229
• скорочувальні.......................................I 79,216, II 772
• структурні.......................................................................187
• транспортні............................\ 77,156,179, \\40,319
• ферменти.............................................182,89, II 77,236
• фосфорилювання . . . . I 60,74,557, II 111, 148
• функціональні.................................... 1384, II 121,359
• циторецептори........................................... 1309, II 146
• ядерні................................................................................187
Біль...............................................................\ 98,107,332,, \\ 393
• відбитий (іррадійований,
рефлекторний)........................................................ II393
• власне вісцеральний............................................ II393
• місцевий...................................................................... N393
• патологічний (хронічний)................................... N393
• проекційний............................................................. II393
• психогенний....................................................II 399,404
• таламічний........................................................ N397,402
• фантомний.......................................................II 397-399
Біологічно активні
речовини ...........................144,281,289,302, II 746,363
Бластопатії................................................................................ І /82
Бласттрансформація............................................................II 65
Блокада(и)серця................................................................... 1289
• атріовентрикулярна (АВ-блокада)................. II 723
• внутрішньошлуночкова......................................II /24
• синоатріальна.......................................................... II /23
Блювання ............................................. І 135,469,576,4 281
Блювота (vomitus).................................................... 4 241,232
Брадикардія ..................................І 115,119,534, II 107,229
• синусна........................................................................II 117
Брадипное.............................................................................II223
В
Вади серця............................ І 78, 180, 183, 189, II 130, 186
Вазодилатація......................... . . . . \ 160,280,312,4 334
Вазоконстрикція.................... ............. І 340,405, II 72, 787
Вазопатії.................................... .......................................II 80,82
• диспластичні................ ............................................. II 82
• запальні (васкуліти) ............................................. II82
Вазопресин ............................. . . І 79,282,403, II 736, 789
Варфарин.................................. .......................................... N183
Васкуліт (и)............................................... 1254,26 7,294, II82
• імунні................................................................................ II82
• інфекційні....................................................................... II 82
• інфекційно-імунні.......................................................II 82
"Вегетативні бурі"............................................................... N409
Веротоксин...................................................................... N81,168
Видужання ........................................................................ 129,47
Випромінювання............................................................................
• інфрачервоне............................................. \36,120,121
• іонізуюче.......................................... 153,67, 123, II285
• ультрафіолетове . .1 720, 727, 170,363,4 52, 162
Виснаження теплове.............................................................17 75
Виразка (ulcer).............................................................. II256-259
Виразки............................................................................II256-259
• нейрогенні...................................................................II259
• судинні..........................................................................N259
• трофічні........................................................................ N259
• "аспіринові"................................................................N260
• "гастринові пептичні"...........................................II260
• "глюкокортикоїдні".................................................N260
• "стресорні"..................................................................N260
• "шокові"...................................................................... II260
Вівісекція ..................................................................................І 76
Відмороження........................................................... І 116,117
Відношення вентиляційно-перфузійне . . .4216,234
Відрижка (uctatio)........................................................N257,255
Вінбластин.....................................................................4 411,415
Вірогенія.................................................................................... І163
Вірулентність............................................................................ І753
Вірус (и)................................................................................................
• гепатиту С, HCV (hepatitis С virus)................... N298
• гепатиту В, HBV (hepatitis В virus).....................N298
• Епштейна -Барр...........................................................II69
• онкогенні..........................................\ 34,164,363,4 69
• Т-клітинної лейкемії людини................................N69
Віталізм....................................................................................... 137
Вітамін D................................................ \ 426,540,4 162,326
Водне отруєння..................................................................... II367
Водно-електролітний обмін ... .І 152,411,470,535
Волокна Пуркіньє....................................................... II 7 75, 722
"Ворітна теорія"болю (Мелзака, Волла)..................II395
В'язкість крові......................................................II 70, 7 74, 735
Г
Галактоземія ........................................................................... І777
Гаметопатії................................................................................ І769
Гаптоглобін........................................................................II 35,38
Гарячка............................ 1332,337,343,345, II38,73,344
Гастрин............................................................................ 4 237,251
Гастрит.................................................І 754,355,1147,50,244
Геліказа................................................................................... 1207
Гемангіоми................................................................................ II82
Гематоенцефалічний бар'єр........................................ II308
Гематокрит.....................................................................4 9,21,23
Гематома....................................................................................N87
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
Гематурія......................................................................II323,343
Гемібалізм............................................................................... II407
Гемобластози....................................................... І 347, II 64-65
Гемоглобінопатії............................................. II 24,29,34-38
Гемоглобінурія....................................................126,2/5,1125
• маршова...............................................................126, II27
• пароксизмальна нічна..............................................II29
Гемодилюція .............................................................1/5/,119,22
Гемодинаміка......................1278,290,400,519, II 113,168
Гемодіаліз (штучна нирка)............................ II 179, 191,340
Гемоконцентрація............................. 1293,571,522, II365
Гемоліз.......................................................... 142,155,179,254
• внутрішньоклітинний...........................142,179, \\ 25
• внутрішньосудинний............................1254,534II25
• детергентний............................................................... N26
• комплементзалежний............................................. 1126
• механічний................................................................... II26
• осмотичний..................................................................1126
• окисний.......................................................................... II26
Гемолітична хвороба новонароджених . 1254,1128
Гемопоез............................................... І 13О, 257,W10,56,65
Геморагія............................................................... І 131,254, II82
Гемосидероз................................................................І 198,1138
Гемостаз..............................................\ 261,297,472, \\ 74-87
• коагуляційний ..........................................1536, II86-87
• судинно-тромбоцитарний . 1297,501, II83,363
Гемостазіопатії........................................................................II 78
Гемофілія.........................................\ 177,179,187,192, \\ 87
• Гемофілія А................................................................... II 87
• Гемофілія В....................................................................II 87
• Гемофілія С.....................................................................II87
Ген нефрину (NPHS1).......................................................11339
Ген передсердного натрійуретичного
гормону (ПНУГ)...................................................................II102
Ген подоцину (NPHS2)....................................................... II339
Генералізація.......................................................................... І155
"Генетичний нокаут"........................ \ 275,556, \\ 192,339
Гени............................................................................................. II 72
• вимкнені...................................................... \ 189,373,397
• експресія.................... І 93,203,233,373,11102, 169
• конститутивні............................................ 1157,283,386
• міозину......................................................... 1285,286,287
• регульовані....................................................... 1487, II71
• структурні................................................................... 1373
Генотип......................................................... І 784, 190, \\ 37,72
Гепарин......................................................................................II 88
Гепатит (и).......................................\ 88,158,233,366, \\ 288
• алкогольний...................................................... N292,297
• вірусні.......................................................................... II289
• гострий........................................................................ II398
• хронічний.................................................................... N389
Геропротекція............................................................ 1207,208
Герпесвірус саркоми Капоші..........................................II 69
Гетерокатефтентність ..................................................... І 798
Гетерокінетичність............................................................ І /98
Гетерометрія.............................................................................132
427
Гетеротопність...................................................................... І /98
Гетерохронність................................................................... І /98
Гігантизм.......................................................................N359,382
Гідроцефалія........................................................................ N 420
Гіперазотемія............................................................. 1506,522
Гіпералгезії.................................................................. II397-398
Гїперальдостеронізм........................ 1522,532,575, II368
• первинний................................................................... II367
• вторинний................................................................... II368
Гіпераміноацидемія ........................................................ 1504
Гіперамоніємія.................................................................... 1507
Гіперацидітас....................................................................... II254
Гіпербілірубінемія............................................................. II310
Гіпервентиляція........................... 127,435,565, II224,230
Гіперволемія........................................... 15/2,527,529, II 10
• проста..............................................................................I110
• олігоцитемічна...................................................II 10,113
• поліцитемічна................................................ II 10, II 7/4
Гіпергепаринемія................................................................... II88
Гіпергідрія.......................................157 7,520,527,534, II376
Гіперглікемія..................................177,450,472,, II355,359
Гіпергонадизм жіночий.................................................. II384
Гіперемія................................................................144,288,317
• артеріальна........................................\44,286,317,\\418
• венозна....................................... І 107,289,317, \\ 383
Гіперергія................................................................................... 149
Гіперінсулінемія........................................... \ 460,473, \\ 192
Гіперінсулінізм..............................1454,462,497, II 362,387
Гіперкаліємія..................................1405,470,534, II33/, 365
Гіперкальціємічні стани....................................................II346
Гіперкальціємія .............................. І 70,537,550,553,11260
• сімейна гіпокальціурична.................................... II378
Гіперкапнія.....................................І 135,565,570, II 210,230
Гіперкінези..............................................................................II406
Гіперкоагулянтність ......................................................... 1296
Гіперліпацидемія . . . . \75,451,459,467, \\ 288,362
Гіперліпідемія .............................................. 1482, II160,337
Гіперліпопротеїнемія
. . . . І 467,480,482, II 759, 162
Гіпермагніємія...................................................................... 1535
Гіпермутантність...........................................................................N71
Гіпернатріємія...............................\ 74,524,532,579, \\ 194
Гіпероксія........................................... 167,135,462, II25, /46
Гіпероксалурія......................................................................N346
Гіперонкія...................................................................... 1306,514
Гіперосмія.................................................1305,332,521, N376
Гіперпаратиреоз..........................І 535,550,558,560, II 378
• первинний.................................................................N 378
• вторинний...................................................................N378
Гіперпатії...........................................................................................N397-398
Гіперпітуїтаризм................................................................. N 358
Гіперпное...............................................................................N 224
Гіперпротеїнемія ............................................................... 1501
Гіперсалівація .................................... \ 521,572, \\ 237,249
Гіперсекреція..................................................... І 77,11254,275
• шлункова............................................................ І 17,\\254
• панкреатична................................................... \17,\\267
428
Гіперспленізм................................................................ II 83,314
Гіпертензія .............................................. І 79,11113,231,332
• артеріальна.........................І 18, 187,473,475, II 328
• внутрішньочерепна................. І 123,289,412, II420
• малого кола кругообігу........................... 1438,11 113
• наднирково-регенераційна...............................11/9/
• портальна ...........................................І299,11114,314
• реноваскулярна...................................................... II /9/
• ренопривна...............................................................II 191
• рефлексогенна, або гіпертензія
розгальмування.......................................................11/9/
• сольова........................................................................II 792
• центрально-ішемічна артеріальна . . . . II 191
• внутрішньопечінкова.......................................... 11374
• надпечінкова........................................................... 11374
• підпечінкова.............................................................11374
Гіпертермія...........................146, 109, 113,345, II 146,372
Гіпертимія................................................................................II422
Гіпертиреоз....................................1205,255,275,345, II374
• первинний................................................................. II374
• вторинний.................................................................11374
Гіпертрофія........................................................................II 98-99
• від перевантажень............................................................ II98
• від ушкодження.................................................................. II99
• компенсаторна серця...................................................... II99
Гіпертрофія міокарда...................................................................II99
Гіпертрофія серця.......................................................І 782, II 99
Гіперурикемія .............................................. 1508,509, II 346
Гіперурикозурія..............................................................................II346
Гіперфосфатемія....................................І 553,560, II783,327
Гіперфункція кори надниркових залоз...................11367
Гіперхлоргідрія................................................................... 11254
Гіперхолестеролемія................................................\\ 311-313
• сімейна................................................................. II 162, 165
Гіперхлоремія .............................................. 1573,579, II326
Гіперхромія........................................................................ I117,22
Гіпоаміноацидемія............................................................. 1504
Гіпоацидітас..................................................................................... II254
Гіпобеталіпопротеїнемія................................................ 1487
Гіповентиляція.....................................................................II27 7
Гіповітаміноз А.......................................................... 1478, II372
Гіповітаміноз С...............................................................168, II82
Гіповітаміноз Е.............................. \ 67,68,478,, \\ 277,312
Гіповітаміноз К.................................................1478, II87,3 72
Гіповолемія.................................................................... І 7 7 7,522
• проста................................................................................ II 9
• олігоцитемічна.............................................................. II 9
• поліцитемічна......................................................................II10
Гіпогідрія......................................... І 511,521,524, II 249,316
Гіпоглікемія.................................................... 1395,454, II3 76
• спонтанна ідіопатична.........................................II385
Гіпогонадизм.................................І 784, 185,426, \\ 248,382
• жіночий.......................................................... І 784, II384
• чоловічий .................................................... І 785, II380
• вторинний................................................................. II358
Гіподинамія....................................\ 37,422,423, \\ 160,192
ПОКАЖЧИКИ
Гіпоергія...........................................................................149,303,
Гіпокаліємія............................................. 1533,579, II 106,368
Гіпокальціємія . .1537,540,546,550, II7 72,247,326,408
Гіпокапнія............................. 1435,438,573, II 7 74, 736,230
Гіпокінезія............................................. І 737, 138,11251,406
Гіпокоагуляція............................................................1267, N97
Гіпокортицизм вторинний.............................................. II358
Гіпоксемія . . І 732,408,432,442,11270-229,229-234
Гіпоксія................................................... 173,120,428,11 14,20
• гемічна............................................................................. II 73
• гостра ішемічна......................................................... II 738
• циркуляторна..............................................................II 7 74
Гіполіпопротеїнемія......................................................... 1487
Гіпомагніємія........................................................................ 1535
Гіпонатріємія........................1470,532,533,579, II33 7,360
Гіпоонкія................................................................... 1572, N339
Гіпоосмія................................................................................ II316
Гіпопаратиреоз......................1547,560, II382-383, II377
Гіпопітуїтаризм.......................................................................І 78
• пангіпопітуїтаризм.......................................... II356-358
• парціальний..............................................................II358
Гіпопроконвертинемії..........................................................II87
Гіпопротеїнемія.................................. 1507,527, II ЗОЇ, 315
Гіпопротромбінемія............................................................1187
Гіпосалівація...............................................................II 237,249
Гіпостенурія.......................................................................... II322
Гіпотензія...............................................................II 7 7, 134,200
• артеріальна....................................\ 45,403,422, \\200
• есенціальна...............................................................11200
Гіпотермія....................................... \ 109,110,117,118, \\422
Гіпотимія................................................................................11422
Гіпотиреоз...................................... \ 118,184,426, \\ 358,374
• первинний................................................................... II373
• вторинний....................................................................II358
Гіпотонія м'язів............................................................ II406-407
Гіпофібриногенемія ............................................... \ 177,178
Гіпофізектомія........................................................................II357
• гіпофізотропна...........................................................II357
Гіпофосфатемія ........................................................ 1557,558
Гіпохлоргідрія........................................................................ II254
Гіпохлоремія......................................................................... 1574
Гіпохолія................................................................................... II309
Гіпохромія.......................................................................... II 78,36
Гіпоцитратурія.......................................................................II346
Гістамін...................................І 722,247,370,527, II204,253
Гістосумісність........................................................................ І780
Глікогенози.............................................................. 1454, II300
Глікозиди серцеві...........................................II 709, 7 72, 798
Гліколіз..........................................\ 74,147,467, \\ 31,83,100
• аеробний................................................................... 11417
Гл і коп ротеїн и................. І 161,216,295,323,1185,252
Гломерулонефрит.......................................... 177, 758, II340
• гострий........................................................... 1258, II 340
• злоякісний гострий.................................................. II340
• імунокомплексний................................................... II342
• хронічний ..................................................... 1257,11340
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
Гломерулосклероз. . .
.............................................11337
Глюкагон............................ . . . \ 451,489, \\ 362,367,385
Глюкозурія ренальна . .......................1454, II319-320
Глюкокортикоїди. . . .
1253,454,489,504, II 361-367
Глюконеогенез................ ... 197,450,504, II292,359
Глютамат............................. ..............................................II303
Глютамін............................. .....................................11303,305
Голодування .................... . . . . 137,73,4/7,424,11240
• кисневе ................... .....................................11331,417
• білкове
II 259,274
Гомеостаз...........................
. . 160,409,472.1110-11, 142
• газовий.................... ...............................................N209
Гомоцистинурія . . . . ............................. І /77,294,506
Гомоцистеїнемія . . . .
.............................................. N160
• гонадотропна . .
............................................. I1160
Гонадотрофи.................... .............................................. N355
Гормони.............................. ....................................II 353,369
• 17(3-естрадіол . .
...............................................N382
• адаптивні ............... ...............................................N347
• адреналін ............... ................................................ II 76
• адренокортикотропний гормон (АКТГ) . II 356
• альдостерон............................................................ II 189
• амнестичний........................................................... II 359
• андрогени.........................................................II360,379
• атріопептин............................................................. II 190
• ацетилхолін......................................................II 106, 146
• білково-пептидні...................................................... II347
• брадикінін........................................................ II271,279
• вазопресин...........................................................II 12, 194
• гіпофізотропні........................................................II 353
• гомеостатичні.........................................................II 347
• гонадотропні...................................................II 347,382
• грелін............................................................................II250
• дофамін............................................................. II356,372
• ендотелій.......................................................... II 136,189
• естріол............................................................................II387
• естрогени......................................................................II387
• естрон.............................................................................II387
• кальцитонін.................................................................II347
• катехоламіни............................................. \\97,187
• кортизол........................................................... II359,367
• кортикостероїди........................................... II 191,360
• лютеїнізуючий (ЛГ)........................................ N354-355
• меланоцитстимуляційний.................................... II358
• мінералокортикоїди....................................II 361-367
• норадреналін...................................................... II 74,150
• окситоцин........................................................ II 279,359
• паратиреоїдний гормон (ПТГ),
паратгормон................................................... II327,377
• паратирин....................................................................II382
• передсердний натрійуретичний
гормон (атріопептин)............................................I1190
• пролактин.........................................................II 355-358
• соматотропний гормон (СТГ)................ II 354-358
• соматотропні............................................................. 1504
• справжні..........................................................................167
429
• стероїдні.................................................. N 149,258,337
• тестостерон......................................................II355,379
• тиреоїдні.............................................. 1504, II 347,372
• тиреотропний гормон (ТТГ)..................II354-359
• тироксин (Т4)............................................................... II372
• тканинні....................................................................... І 101
• трийодтиронін (Т3)................................................... II372
• фолікулостимуляційний (ФСГ) . . . . II354-355
Гостра недостатність нирок............................................ II330
• постренальна ГНН....................................................II330
• преренальна ГНН.................................................... N330
• ренальна ГНН.............................................................II330
• аренальна ГНН..........................................................N33/
Гранулопатії.............................................................................. II83
Гранулоцити......................................................................II 54,68
Грелін............................................................................... 1490,497
д
ДВЗ-синдром....................................................................1179,89
Дегенерація............................................................. 1395, II156
Дегідратація................................... 1408,464,470,515, II34
Деградація ............................................................ 184,203,390
Дегрануляція................................. 1215,221,309,326,11141
Декомпартменталізація....................................................... 160
Декомпенсація............................. 145,110,405,471, II 113
Декомпозиція ........................................................................ 185
Делеція................................................ І /75, /85,373, II32,36
Деменція.................................................................... 1425, II422
Демієлінізація................................................................................. 449,397
Демінералізація.........................................................II 247,397
Денервація фармакологічна...........................................II409
Денатурація.......................................І 58, 743, /44, /49, II32
Деполімеризація ............................................... 185,319,519
Депротеїнізація .................................................................... І161
Десенсибілізація......................................................... 1253,265
Десквамація........................................................................... 13/9
Детермінізм............................................................................... 134
Детоксикація..................................183, /42, /47,506, II302
Дефекти мови........................................................................ II407
Дефіцит енергетичних субстратів................................. II4/7
Дефіцит йоду........................................................ N350,373,375
Дизергія.......................................................................................149
Дилатація серця міогенна.............................................. II 113
Дисбактеріоз................................................................ N243-244
Дискінезії кишкові
перистальтичні............................................. \\ 237,279-280
Дисметрія...............................................................................II407
Дисемінація................................................................. І 155,391
Дисіонія ................................................................................. 1306
Дисліпопротеїнемія .................................. 1480,482, II 167
Диспротеїнемія .................................................................. 1501
Дистрес-синдром........................................ І 102, II219,367
• респіраторний.........................................................11229
Дистрофія............................................................................................
• клітинна...................................................... 185, II 289,326
430
• нейрогенна......................... ....................................114/2
Дисфагія........................................... ............................. II 44,237
Дисфібриногенемія.................... . ... І 777,178, II87,91
Дисфункція..................................... ....................................... II 93
• діастолічна.......................... .......................................II 93
• систолічна........................... .......................................II 93
• ендотеліальна.................... .......................... 1294,11 /69
Дисхолія........................................... ....................................113/2
Дихання ........................................... ............. І 74,80,431,511
• апнейстичне ...................... ....................................11227
• Біота....................................... ....................................11227
• гаспінг-дихання................ ............................112/9,227
• Грокка................................... .....................................N227
• Куссмауля............................ ............. 14/2,57/, II225
• термінальне........................ .......................... 1412,571
• Чейна - Стокса.................. ................ \ 412,432, \\ 226
Діабет................................................ І /77, /79,57/, II 159,292
• нецукровий ....................... ............. 1527,532,11327
• фосфатний нирковий . ............. \ 177,179, \\ 319
• цукровий ............................ . . І 187, 192,455, II 160
• цукровий вторинний . ....................................II /78
Діагноз.............................................. .......................\ 25,41, 199
Діапедез........................................... \ 291,325,326,328, \\81
.. \ 156,422,423, \\ 243
Діарея................................................
Діатез (и).......................................... І 792, /93, 794,426, II 74
• геморагічні (вазопатії). .......................................II 80
• тромбофільні..................... ..................................... 1197
.......................
1426,11330
• геморагічний.....................
...................................................
...... ■
* *-'■'Z
Діурез................................................
................................... 1132/
• водний..................................
.................................. 11327
• гіпертензивний.................
.................................. 11320
• облігатний...........................
.................................. 11327
• осмотичний........................
.................................. 11320
• факультативний................
................................... 1 7 5 7
• форсований .......................
....................................N355
Дія гландотропна........................
793,399
Домінанта патологічна. . ............................II
. .
Дофамін-(3-гідроксилаза.і...................................
..
11372
.............
\
329,384,385,
\\
342
Домени ............................................
.......................................
II
50,267
Дуоденіт...........................................
Дуплікація.......................................
................................... I 786
Е
Ейфорія................................................... \ 134,432, \\ 302,402
Екзонуклеази.............................................................. \ 172,173
Екзотоксини................................... 163,154,344, II261-262
Екзофтальм двосторонній (витрішкуватість) . II374
Екзоцитоз.............................. \ 69,161,304,552, \\ 170-171
Експериментальні..............................................І 13,270,462
• дослідження.......................... І 76, 19,270,462, \\ 184
• модель ........................................................І 16, II 162,341
• терапія ...............................................................................17
Екстрасистола.............................................................II 119-120
• атріовентрикулярна.............................................. II 720
• вставна.........................................................................11/2/
ПОКАЖЧИКИ
• передсердна................................................................її120
• шлуночкова................................................................. II727
Екстрасистолія CO................................................. 1534, II 120
Екстремальні стани........................................... I 107,156,399
Ексудат.................................... I 7 77, 794,327,337, II 772,228
Ексудація......................................... 17 7 7,300,316,382, II 777
Елементи контрактильні.................................................II352
Електротравма....................................................................... І730
Елонгація ................................................................................. І755
Емболи.................................... І 78,136,294,391, \\ 150,314
Емболія.............................................І 136,293,298,\\ 135,231
Еміграція................................................... \ 107,322,336, \\ 182
Емфізема.......................................................................І 733, II 70
• легень............................................................................N270
Ендартеріїт................................................................ 1298, II 756
Ендокардит............................................................... І 759, N340
Ендокринна ......................................................................................
• гіперфункція............................................................. II348
• гіпофункція................................................................II348
Ендокринопатії.................................................................................
• дисрегуляторні..........................................................N35/
Ендонуклеази..........................................................І 71,89,174
Ендотоксини.................................. \ 154, 156,218,405, \\ 61
Ендоцитоз.................................................................... I1170,352
Енергодефіцит.....................\ 73,279,412,462, \\ 179,198
Енхансери............................................................................... 1372
Енцефаломієліт автоалергічний....................................II42/
Енцефалопатія..................................................................................
• білірубінова................................................................ N3/0
• печінкова..................................................................... II302
Еозинопенія......................................................................II63,363
Епілепсія.................................1344,425,453,504, II40/, 408
Епітаксія................................................................................... 1553
Епітопне поширення........................................................ 1274
Еритема .................................................................І 131,122,123
Еритремії.............................................................................II /0,20
Еритромієлоз........................................................................... II 65
Еритропоетини ..................................... \ 13О, 395,436, \\ 25
Еритроцити.......................................................... II 17,29,32,57
Еритроцитоз......................................................... 1436, II 19-21
• абсолютний..................................................................11/9
• відносний.......................................................................N2/
Естрогени........................................ \ 296,361,398, \\ 205,387
Етіологічні фактори.................................................\38,\\65,69
• екзогенні.....................................................136,421, II69
• ендогенні............................................. 136,37,421,1169
Етіологія................................................................... 134, II /0-/ /
Еукаріоти ............................................................................... І /52
Ефапси.......................................................................................N4/4
Ефект (и).............................................................................N98,27/
• Бора.............................................................. 147,430,436
• внутрішньоклітинної модуляції.......................114/5
• гальмування................................................................N4/5
• ефаптичний.................................................................N4/4
• збудження...................................................................N475
• катехоламінові.........................................................II374
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
• ліпокортинові
II 363
• позитивний інотропний.......................................... || gs
• позитивний хронотропний
катехоламінів...............................................................II 98
• постсинаптичної модуляції.................................N475
• пресинаптичної модуляції.................................11475
• реверберації.............................................................. N477
• шунтування................................................................ N234
Є
Євгеніка .....................................................................................140
Євнухізм..................................................................................|| 380
Євнухоїдизм............................................................. І 185, II380
Ж
Жирова дистрофія печінки..................... 1486,487, II288
Жовтяниця (і) (icterus)...................... 1478,480,482, II307
• гемолітична (надпечінкова)..............................ц зо7
• ензимопатичні.........................................................N307
• механічна........................................................ 1478, || 203
• паренхіматозна (печінкова).............................. ц 308
• печінково-клітинна............................................... II308
• паренхіматозна (печінкова).............................. ц зов
• фізіологічна новонароджених.........................п зов
Жовч .......................................... 122, ЗО, 478,549, II243,268
Жовчні кислоти............................................................ || 272,317
З
Задишка, або диспное...................................................... N227
• експіраторна..............................................................N228
• інспіраторна................................................................II228
Залежність.......................................................................... 1 7 4 7
Залози.............................................................................. N257,355
• власне шлункові......................................................N257
• пілоричні.....................................................................N257
• прищитоподібні.......................................................N357
Заміщення............................................................. І 78,239,427
• медіатори.............................................І ЗО, 81,306,320
Закон ....................................................................................................
• "градієнта автоматизму".......................................117 78
• Кеннона........................................................................N472
• Франка - Стерлінга.............................................. II95-97
Закрепи (obstipatio)....................................1552, II242-243
• атонічні.........................................................................N280
• спастичні......................................................................N280
Запаморочення (головокружіння)...................II200,407
"Зачаровані кола".................146,76,114,406, II203,420
Звикання.......................................................................... І 141,338
Здавлювання нервів......................................................... N 397
Зоб............................................................................................. 1426
• дифузний токсичний.............................................. 1500
• ендемічний ............................................................... 1426
• спорадичний ........................................................... 1375
Зупинка серця...............................................................II 706, 750
431
І
Ізогідрія....................................................................................N376
Ізольована систолічна артеріальна
гіпертензія (ІСАГ)................................................................II 785
Ізостенурія...................................................................... II320-322
Імунітет................................................................... І 16,209,242
Імунодефіцити........................................1225,227,367, II 69
• В-клітинні.................................................................... 1225
• вторинні ............................................................ 1225,231
• гуморальні.................................................................. 1225
• клітинні......................................................................... 1227
• комбіновані..................................................... І 191,229
• Т-клітинні.......................................................... 1225,367
• Т- і В-клітинні поєднані........................................ 1225
Імуностимуляція................................................................. 1240
Імуносупресія ..................................................................... 1240
Інвазивність.......................................................................... 1354
Інверсія...................................................................................... І 785
Інгібітори кальцифікації................................................. 11182
Інгібітори протеаз природні............................................. II89
Інгібітори фібринолізу..........................................................II89
Індекс..........................................................................................II 7 75
• лецитин-холестероловий.................................... N372
• холато-холестероловий....................................... N372
Індекси серцеві......................................................................II 7 75
Ініціація.................................. 1285,360,387,553, II 151,183
Інсулін .............................................. 144,443,558, II792,302
Інсулінрезистентність...................................... 1454,456,498
Інсульт (и)........................................І 17,206,498, II419-420
• атеротромботичний...............................................N479
• гемодинамічний..................................................... 11479
• геморагічні................................................................. N479
• гемореологічний....................................................II420
• ішемічні.......................................................................II479
• кардіоемболічний...................................................N479
• лакунарний................................................................. N479
Інтерлейкін 1.....................................................1321,338, II67
Інтерферони ................................. \ 51,216,501, \\ 298-299
Інтима........................................................................................ II756
Інтоксикація................................... 17 7 7, 748,522, II274,288
Інфаркт.................................................................................................
• міокарда...............................\ 18,206,498,11150, 160
Інфекція ..............................................\ 27,161,322,\\ 60,313
Інфікування Helicobacter pylori..................................... N267
Інфільтрація............................................. 1257,379, II756, ЗОЇ
Іони Na+...................................................................................N474
Іони К+......................................................................................N473
ІониСа2+................................................................................... N473
Іонні електрогенні насоси (Na-K-насоси) . . . . II253
Іонофори......................................................................І 68,70,80
Ішемічна хвороба серця. . . .
125,37,481, II 131,149
Ішемічні токсини.................................................1400,405,406
Ішемія......................................................... 1291,516,11199,269
• міокарда .............................................. 1292,433, II 735
• печінки........................................................... 1408, II285
ПОКАЖЧИКИ
432
К
• ліпідні механізми.........................................................175
• нейроендокринні.............................................................. II347-353
Кал......................................................................................II309-310
• остеопрогенітори...................................................II 780
• безбарвний...............................................................11309
• пінисті...................................................................................... II171-177
• гіпохолічний............................................................. 11309
• старіння....................................................... \ 59, 197,207
Калікреїн................................ 1275,374,315, II 78,190,271
• стовбурові.................... І 130, 188,240,554, \\ 52,56
Кальсеквестрин..............................................................1 1 / 7 /
• ушкодження.......................... І 60,75,571, II 780,204
Кальцитонін................................... 1540,545,547, II347,372
• фактор втрати.......................................................... 1357
Кальцифікація..................... \ 86,553,560,179, \\ 180-182
Коагулопатії.................................................................І 179, II 85
• ектопічна....................................................................11327
Коагуляція....................................................................1501, II 77
Камені...............................................................................II316-317
Кодон............................................................................. І 775,1136
• кальцій-оксалатні...................................................II346
Коефіцієнт фільтрації (Кф)............................................. II378
• уратні............................................................................ II346
" Кола зачаровані"...................................II75,739,203,420
• фосфатно-амонієво-магнієві............................ II346
Колаген...................................................................II82, 155,204
• цистинові....................................................................II346
Колагенові волокна..........................1328, II 204-205,219
Камені жовчні............................................................... II269,313
Колапс ............................................. І 77, 7 75,470,11 7 72, 734
• пігментні......................................................................N313
Коліка печінкова................................................................11373
• холестеролові.................................................11372-373
Кольки........................................................................... II 244,346
Камені пігментні................................................................ 11373
Колхіцин....................................................................... 1147 7,475
• коричневі.................................................................. 11373
Кома.........................................І 115,411,471,W214,306,417
• чорні.............................................................................II373
• гіперсмолярна .............................................. 1412,471
Камені панкреатичні.......................................................... II276
• гіпоглікемічна..................................... \411,471,\\287
Канцерогени........................................... 1356,358,389, II69
• діабетична кетонемічна............................ \ 412,471
Канабіноїди.............................................................................II402
• дихальна............................................................\\211,422
Кардіоміопатія гіпертрофічна...................................... II 702
• лактацидемічна............................................. \ 412,471
Кардіоплегія.......................................................................... II 706
• печінкова.............................................. 147 7, II303-304
Кардіосклероз............................................... 1292, II 700, 730
• уремічна .................................................................... 147 7
• атеросклеротичний................................................II 750
Компенсація ...................................................................... 142,55
• постінфарктний........................................................II 750
• ліпідна.......................................................................... II 761
Карієс....................................................................1426, II245-247
• судинна........................................................................ II 755
Каріоліз..................................................................................185,87
Комплемент......................... 157,27 7 507, II25,82,90,341
Каріопікноз................................................................................185
Кондиціоналізм....................................................................... 139
Каріорексис......................................................................... 185,87
Конкордантність................................................................. І 790
Карликовість гіпотиреоїдна............................................II373
Конституціоналізм........................................................... 139,40
Каспази................................................................187,89,91,268
Конституція............................................................ 150,192,4409
Кастрація.................................................................................. II380
• симпатикотонічна.................................................... II409
Каузалгія................................................................................... II397
• ваготонічна............................................................... N 409
Кахексія .................................................................. \46,\\241,378
Кон'югації білірубіну........................................................11309
• опікова ....................................................... \ 110,111,112
Концепція.................................................... II 700, 161, 193,213
• ракова............................................................. 1395, II378
Коронарити.......................................................................... N 750
Квадригемінія..................................................................................II720 Кортикотрофи..................................................................... N 355
Квашіоркор................................................. 137,422,424,500
Кофактори плазмові агрегації..........................................II77
Кейлони........................................................................ 1327, II62
Крайовий стан лейкоцитів............................ 1275,322,325
Кетоз ..............................................................1456,456,469,471
Кріоглобуліни ........................................................... 1507,502
Кільця Кебота............................................................................II77
"Криз бластний"............................................................... 4 69,73
Кініни....................................... \ 44,260,314,320, \\ 271,278
Криз.......................................................................................................
Кластеризація..................................................................................II763
• адреналовий...............................................................II365
Клінічна патофізіологія..........................................................І75
• гіпертонічний............................................................II 799
Кліренс інуліну....................................................................11378
Кристали холестеролу......................................................II 777
Клітини....................................................... 159,74,95,207,554
Крові елементи................................................................ II 8,68
• автофагічна смерть......................................................І95
Крововтрата .............................. І 130,408,512,521,4 9, 15
• ацидотичні механізми ............................................... І74
• гостра................................................................................II59
• електролітно-осмотичні механізми..................І 74
Кровообіг колатеральний.............................................I I 4 7 8
• енергозабезпечення......................... 164,73,82,486
Кругообіг печінково-кишковий....................................II306
Ксантома(и).......................................................................... I I 3 7 2
• жирова дистрофія................................................... 1487
• ліпідна..........................................................................I I 3 7 2
• захисні компенсаторні реакції . . І 78, 7 72,435
,
433
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
Ксеродермія пігментна......................... \ 174,355,363,380
Кумуляція.................................................................. І 141, II 146
Л
Лабільність вегетативна..................................................II409
Лактотрофи...........................................................................11354
Ламінопатії............................................................................ 1207
Лейкеміди................................................................................. II 74
Лейкоз..................................................І 183,364,580, II64-65
Лейкози (лейкемії1)........................................................................
• гострі............................................................................... II 67
• хронічні.......................................................................... II 67
• гострий міелобластний...........................................II 66
• гострий лімфобластний..........................................1166
• гострий монобластний........................................... II66
• хронічний мієлоцитарний.....................................II66
• хронічний лімфоцитарний....................................1166
• хронічний моноцитарний......................................II66
• хронічний еритромієлоз.........................................II 66
Лейкопенія 0)..........................................І 128,224,396, II 63
• набуті...............................................................................II 63
• спадкові..........................................................................II63
Лейкотрієни.......................................... 1218,248,332, II 343
Лейкоцитоз.............................................. \ 128,156,332, \\ 57
• абсолютний.................................................................. II59
• аліментарний...............................................................II59
• базофільний................................................................. II60
• вагітних...........................................................................II59
• відносний...................................................................... II59
• емоціогенний.............................................................. II 59
• еозинофільний............................................................II59
• лімфоцитарний........................................................... II60
• міогенний......................................................................II59
• моноцитарний............................................................ 1160
• нейтрофільний............................................................II 59
• новонароджених....................................................... II59
• патологічний............................................................... 1159
• статичний.......................................................................II 57
• фізіологічний..........................................................II 59,62
Лейкоцитурія..............................................................II323,343
Лейциноз .............................................................................. 1506
Лихоманка.............................................................................. 1337
Ліберини.................................................................... 1/07,11356
Ліганди....................................................... І 79,228,487, II309
Лізосоми.......................................... 184,257,307, II 139, 163
Лізофосфоліпіди....................................... І 72,73,310, II 739
Ліміт Хейфліка......................................................1200,201,348
Лімфокіни................................................. 1308,312,488, II54
Лімфолейкози........................................................................1166
Лімфома(и)..........................................................................II69-72
• Беркітта............................................. 1366,374, II 70,72
• Т-клітинна лімфома-лейкемія..............................II 70
• В-клітинна.......................................................................II 70
Лімфобласти...............................................................II 65-66,69
Лімфогранулематоз............................................................... II 65
Лімфопенія .................................................\ 128,231, \\ 63,73
Лімфосаркома.........................................................................Н 65
Ліпіди.......................................................... 164, 745,428,11 757
• мембранні................................. \ 144,145,\\ 139,160
• пероксидне
окиснення........................... І 64,66,75,428, II32,297
"Ліпідна тріада"...................................................................... І 75
Ліпопротеїни..................................................... \ 141,480,\\ 162
• антиатерогенні............................................ 1473, II 762
• атерогенні..................................................... 1465, II760
• модифіковані............................................................II3 70
М
Макроангіопатії.................................................. 1477,473,476
Макрофаги .................................148,394,556, II ЗО, 54, 777
Маразм аліментарний........................................... 1422,423
Маргінація лейкоцитів..................................................... 1322
Мегакаріобласти.....................................................................II82
Мегакаріоцити.........................................................................II82
Мегалобласти ..................................................І 32, II 77,22,45
Мегалоцити................................................................. II 17,46,65
Медіа........................................................................................II 156
Медіатори гальмівні ЦНС.............................................. II 305
Медіатори збуджувальні ЦНС....................................... II305
Мембрана.......................................................... І 60,90,397,577
• мітохондріальна................................. І 77,83,90, II 742
• плазматична................................ \ 60,397,577, \\ 103
• ядерна................................................................... 185,207
Мембранопатії.........................................................................II29
• гострі.................................................................................II29
• набуті................................................................................П27
• хронічні.....................................................................II29-30
Месенджери..................................І 74,755,466, II353,367
Метаболізм........................\ 42,96,192,509, II3 7, 704,205
Метамієлоцити................................................................. II 57,68
Метаплазія .................................................................. І 355,425
Метастазування.................................. \ 21,354,390, \\ 65,74
Метеоризм .................................................... \ 133,534, \\ 243
Метилювання.......................................................... І 742, II302
Методи знеболювання......................................................II404
• нейрохірургічні......................................................... II404
• психогенні....................................................................II404
• фармакологічні..........................................................II404
• фізичні........................................................................... II404
Механізм Na-Ca-обмінний.................................. II 111,113
Механізм гемостазу.......................................................II 74-75
• вторинний.......................................................................II75
• коагуляційний............................................................... II77
• первинний...................................................................... II74
• судинно-тромбоцитарний...................................... II75
Механізми..........................................................................................
• антиноцицептивні, або аналгезивні. . . . N407
• нейромодулятори....................................................II474
• нейрофізіологічні.................................................... II407
Миготіння (фібриляція) передсердь . . . . I I 129-132
434
Міастенії (myasthenia gravis)............................... II223,368
Мігрень......................................................................... II399-400
Мієлобласти...................................................................... II 66-68
Мієлолейкози................................................................... II 67-68
• гострий.....................................................................II 67-68
• хронічний.................................................................II67-68
Мієлопоез....................................................................... II 8,57,62
Мієлоцити............................................................................II57-67
Мікроангіопатії................................................... 1466,473,476
Мікроби .......................................................... ... * і 752, 159
Мікроциркуляція.........................І 42,251,527, II 10, 14,81
Мікседема....................................................... І 527, II 160,373
Мімікрія молекулярна...................................................... 1274
Мінералокортикоїди........................................... І 101, \\ 365
Міокардит....................................................... 1257, II 123,200
Місенс-мутації..................................................................... І 175
"Мітохондріальні пори"........................ 190,93,11 142-144
Мітохондрії..................................................\ 70,82, НИЗ, 141
Модель
..............................N347
• Герцена....................................................
............................. II340
• Гудпасчера.............................................
• Ліндемана...............................................
..............................N347
............................. N347
• Масугі........................................................
• подружжя Ковелті..............................
..............................N347
• Стеблея ...................................................
............................. II347
............................. II347
• Хеймена ..................................................
Модифікація ..............................................
. . 182,170,203,329
18.
.......................182, 170
• ковалентна ............................................
• посттрансляційна.............................
... \ 203,329,\\
І
351
Мозаїцизм гонадальний.............................
............................. І 777
..........................175,507
Молекула ...........................................................
. \ 75, 172,557, \\ 342
• ДНК....................................................175,
І 75,775,557,
II35,69
• РИК................................................І 75,
/;
Монокаузалізм.................................................
..................................139
Мотивація...........................................................
.............................11422
Мотилін...............................................................
............. II237,257,280
Мутагени....................................................І ІУ69,
769, 770, 777, II 774
Мутації...............................................................
. .. \І 169,497,\\ 177
• генні........................................................
... \ І 169, 175,\\273
• хромосомні............................................
. . . . І 769, 787,1173
Муцини ..............................................................
.......................II 242,256
Н
Набряк(и) .............................................................................. 1525
• Квінке...........................................................І 194,215,246
• легень........................................... І 7 7 7, 735, 747, II 7 74
• мозку................................................................ 1524,11230
Набряк легень.....................................................................11232
• інтерстиціальний.................................................... II232
• альвеолярний...........................................................II232
Набухання мозку................................................................II420
Нанізм гіпофізарний (карликовість)......................... II358
Невагомість...........................................................136, 737, 138
Невралгія ....................................................... \ 192,288, \\ 397
• трійчастого нерва....................................................N397
ПОКАЖЧИКИ
Неврози..................................................І 17, 18,32,192,4 422
Невроми.................................................................................II397
Недокрів'я місцеве.................................................. \ 291,292
Недостатність ............................................І 63,112,463,548
• антиоксидантна............................. 163,68,426, II 775
• білково-енергетична............................... 1422, N307
• вінцевого кровообігу................................. 144, II 736
• зовнішнього дихання..................... І 112,225,4 224
• кори наднирників..................................... І 707, II364
• кровоносних судин.................................... 1433, II205
• кровообігу..............................................І 119,401,4 92
• печінки.................................................. \ 149,548,4 289
• нирок гостра ........................................І 78,408, II334
• нирок хронічна.......................................... 1550, II332
• серця......................................................... І 747,433, II 92
• травлення ........................................................ 150,4 236
Недостатність адренокортикальна............................II364
• гостра........................................................................... II364
• йодидна...................................................................... II375
• хронічна....................................................................... N365
Недостатність вінцевого кровообігу.....................................
• відносна коронарна..............................................II 733
• абсолютна коронарна......................................... II 733
Недостатність дихання.................................................... II27 7
• вентиляційна............................................................ N27 7
• дисрегуляторна.........................................................N279
• імунологічна................................................................. II73
• компенсована........................................................... N270
• кровообігу................................................................. II420
• некомпенсована...................................................... N270
• обструктивна............................................................. N272
• паренхіматозна........................................................ N230
• рестриктивна.............................................................N276
• серцево-судинна....................................................... II 92
Недостатність нирок.........................................................N330
• гостра............................................................................ N330
• хронічна.......................................................................N334
• гломерулярна.......................................................... II330
• парціальна.................................................................II330
• тотальна...................................................................... II330
• тубулярна.....................................................................N330
Недостатність серця............................................................ II 92
• гостра.............................................................................. II 92
• хронічна (застійна).......................................... II 92, 747
• прихована (компенсована)...................................II 92
• явна (декомпенсована)...........................................II 92
• лівошлуночкова................................................. 4 94,114
• правошлуночкова............................................ 4 94,114
• тотальна..........................................................................II 92
• від перевантаження...........................................N 92-93
• міокардіальна....................................................... N 92,704
• позаміокардіальна........................................... 4 92,112
Недостатність травлення................................................. N237
• гостра............................................................................ N237
• хронічна...................................................................... N237
• тотальна...................................................................... II237
435
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
• парціальна.................................................................II237
Нейрогіпофіз.............................. 1282,519,520, II347,353
Нейрогормони.......................................................... 11474,4/6
Нейродистрофічний процес.............................. 127, II472
Нейромедіатори..........................І 156,283,411, \\ 401,415
• несправжні................................................................. II 305
Нейрони....................................................... \ 79, 198,338,492
• інспіраторно-гальмівні (ІГН)............................. 11227
• експіраторно-стимуляційні (ЕСН)...................II223
• пептидергічні........................................................... N476
Нейромодулятори.............................................................II475
• модулятори харчової і питної поведінки 11475
• пептиди гіпногенні................................................ 11475
• пептиди-аналгетики..............................................N475
Нейропатії............................................. 1207,475,476, II 398
Нейтропенія ........................................................................ І 129
• амідопіринова гостра..................................................... N64
• Костмана....................................... \ 129,159,269, \\ 64
• "єменських євреїв"......................................................N64
Нейротрансмітери,
або нейромедіатори........................... І 156,283,411, \\ 414
Нейтрофіли........................................................................N54-57
• паличкоядерні.............................................................N57
• сегментоядерні............................................................ II57
Некроз.................................................................................. N52,59
Некрози серця.................................................................................
• некоронарогенні.................................................... II 749
• гіпоксичний некроз міокарда..........................II 749
• електролітно-стероїдна кардіопатія
з некрозом.................................................................II 749
Неоваскуляризація........................................... 1354,397,475
Нервізм................................................................................. 132,33
• розлади.......................................... 182,115, II387,422
Нервова трофіка.......................................... 1475, II257,405
Непрохідність кишок (ileus).............................................II280
Неспання..................................................................................II422
Нестабільність серця електрична..........................................II750
Нефрит........................................................................... N340-347
• автоалергічний........................................................ N347
• індукований стрептококами..............................N347
• нефротоксичний..................................................... N347
• окопний...................................................................... II 342
Нефритичні набряки.......................................................... II324
Нефропатія.....................................1469,471,474, II 333,337
Нефротичні набряки................................................................... II324
Нирки................................................І 737,142,517,538, \\ 326
Ніктурія....................................................................................N327
• ниркова....................................................................... II 322
• серцева.........................................................................N322
Ністагм.......................................................................................II407
Нозологічна одиниця ........................................... 123,25,34
Нозологія ...........................................................................122,26
«Нокаут генетичний».......................... II 165-167, 182, 192
Номотопні аритмії.....................................................................N776
Нонсенс-мутації............................................................................. І775
Норадреналін .............................. \ 101,281,489, \\ 74, 150
Нормоволемія.......................................................................... II70
• проста............................................................................... II70
• олігоцитемічна..............................................................II70
• поліцитемічна............................................................... II70
Ноцицептори.......................................................................II396
• механочутливі ноцицептори............................II396
• полімодальні ноцицептори.............................. II396
• термочутливі ноцицептори...............................N 396
• хемочутливі ноцицептори................................. II396
Ноцицепція...........................................................................N388
Нудота (ausea)...............................\ 115,432,524, \\ 73,367
О
Об'єм........................................................................................... II 98
• серця ударний..............................................................II 98
• шлуночків кінцевосистолічний.......................II 7 73
Обмін....................................................................................................
• азотистих основ ..................................
............. 1500,507
• амінокислот ..........................................
. . . . 1502, N307
• білковий .........................................144,
І 44,350,469, II302
• водно-сольовий ........................ 144,
144,570,574,11307
• електролітів...........................................
....................... N307
• енергетичний........................................
....................182,799
• жировий .................................................
............. 1350,11307
• основний ...............................................
................ 1344,419
• вуглеводний..........................................
............. 1350,11300
• фосфорно-кальцієвий....................
. . ІІ 536,553, II 300
Обнубіляція.......................................................
...........................N422
Оглушення.........................................................
...........................N422
Ожиріння .............................................. 1458,
458,495, II 160, 197
Оклюзія...............................................................
.............................II750
• неповна ..................................................
............................ II750
Олігодендроцити............................................
........................... N420
Олігонурія..........................................................
.......................... N331
Олігофренія.......................................................
............. 1784,11422
Олігурія................................................................
................ N320-327
Онкогени ...........................................................
. . . . 1 3 8 2 , II69-71
• вірусні.......................................................
................ 1369, II69
• клітинні....................................................
. . . . 1369,377,II 77
Опіки.....................................................................
. . . . І 709,,N27,59
• сонячні ....................................................
................ 1723,327
Опромінювання..............................................
................ І 724, 728
Опсонізація........................................................
. . . .\ 215,220,315
Опсоніни.............................................................
...................... 192,220
Орбітопатія........................................................
................ N374-377
Орозомукоїд.....................................................
................ 1332,333
Оротоацидурія.................................................
........................... 1508
Орхіт......................................................................
........................... N380
Осифікація..........................................................
............. 1554,11373
Остеодистрофія...............................................
. . . \\ 326-327,344
• ниркова ...............................................
. . \ 558,559, \\ 326
• генералізована фіброзна................
........................... N378
Остеомаляція................................................
. . ІІ 548, II 326-327
Остеопороз........................................... І 138,
138,205, II 326-327
Остеосклероз....................................................
...........................N326
436
Отруєння................................І 740,146,532, II279,338,280
Отрути гепатотропні............................................... II285,288
П
Панкреатит.....................................I 355,456,498,535, II272
• гострий.............................................................. 11268-27/
• хронічний......................................................... 11275-276
• первинно-альтеративний..................................11269
• гіпертензивний....................................................... 11269
• гіперсекреторний.................................................. 11269
• обтураційний.............................................................N269
• рефлюксний................................................................II269
• дуодено-панкреатичний рефлюкс...................II269
• жовчно-панкреатичний рефлюкс................. 11269
• ідіопатичний............................................................... II269
• спадковий..................................................................11269
Панкреозимін....................................................................... N237
Панцитопенія.......................................................І 729,1150-5/
Парабіоз................................................................................146,60
Парагемофілія........................................................................II 87
"Парадокс кальцієвий"....................................................I I / 0 8
"Парадокс кисневий"....................................................... II /44
Пародонтит............................................................................ N247
Параліч (і)................................................. \ 131,156,534, \\ 406
• периферичні..............................................................N406
• млявий..........................................................................N406
• центральні.................................................................. N406
Пара протеїни............................................150/, 502, II83,88
Паратгормон..............................1395,542,558, II 347-350
Парез (и).......................................................І 136,534, \\ 50,85
• периферичні...............................................................II406
Паркінсонізм...................................................... 195,205,11406
Патогенез..................................................................І 13,41,335
Патогенність......................................................................... І 153
"Патологічна домінанта"..........................................II /93,404
Патологічна реакція..............................................................129
Патологічний процес ..126,27,29, 118,10,248, N 365
Патологічний стан.......................126,28,27,471, II 92,237
Патологічні рефлекси............................................... \29,\\398
Патологія .....................................................................І 30,31,32
• гуморальна ................................................................... 130
• клітинна.....................................................................131,32
• молекулярна ...............................................................137
• целюлярна............................................................... 131,32
Пауза
• повна компенсаторна..........................................II 720
• неповна компенсаторна.....................................II 7 79
• преавтоматична......................................................II 720
Пентада Цельса - Галена...................................... 1302,337
Пепсиногени.........................................................................N257
Пептидази...............................................................................N265
Пептиди ендогенні опіоїдні (опіати).........................II402
Пептидилпролілізомераза................................................... І 9
Перевантаження.............................................................II 94-95
• об'ємом, або переднавантаження.................... II 94
ПОКАЖЧИКИ
• опором, або післянавантаження........................ II 95
Перикардит..............................................1327,95, II 7 72-7 73
Перинефрит......................................................................... II 797
Перитоніт гострий .....................І 729, 159,321,408,» 281
Періоди
• розвитку..................................... \ 350,441,545,» 412
• Венкебаха, або Мобітца....................................... II 723
Періодонтит...........................................................................N247
Пероксиди........................................... 165,257,555, I I 7 4 7 , 775
Печія (pyrosis)..................................................................... I I 2 4 7
Пієлонефрит................................................................ І 755, II343
Пілоростеноз......................................................................... II264
Пірогени.................................................................І 337,338,346
Піротерапія................................................................. 1345,346
Піурія....................................................................................... II 344
Плазмін................................................................................II 88,175
Плазміноген.............................................................II88,97
Плазморагії............................................................................ N272
Пневмоконіози............................................. 1224,11177,216
Пневмонітлейкозний...........................................................N74
Пневмосклероз........................................................... N270,276
Пневмоторакс..................................................\ 399,400,» 217
• відкритий...................................................................N 2 7 8
• закритий.................................................................... N 2 7 8
Подагра .......................................... І 780,508,509, II 760,333
Пойкілоцитоз....................................................................II 17,23
Покришка фіброзна................................................II 775-777
Поліетіологічність.................................................................. 134
Поліморфізм......................................1246,266,272, II 759,202
Поліома ................................................................................. 1364
Поліпное (тахіпное)............................................................ II224
Полісерозит........................................................................ N 1 5 1
Полісоми................................................................... 183,84
Поліурія .................................1464,469,521,533, II320,337
Поліфагія............................................................ 1469,497,II416
Поліцитемія.................................................... \ 436,438,» 135
Поріг больової чутливості............................................... II397
Порушення........................................................................................
• взаємодії.....................................................................II353
• депонування нейротрансмітерів.................... N 4 7 5
• електрофізіологічних процесів. » 387,413-414
• енергетичного обміну в клітинах
головного мозку.....................................................N 4 1 3
• зворотного захоплення.......................................N 4 7 5
• руйнування.......................................... II8, 109,261,352
• рухових функцій нервової системи . . . . N404
• рухові мозочкові.................................................... I I 4 0 7
• секреції........................................................................N 4 7 5
• синтезу........................................................................N 4 7 2
• транспорту................................................................. II352
Посткондиціонування............................................II 146, 148
• ішемічне..................................................................... II 746
Потенціал..........................................................II 104,136,413
• мембранний............................................................. II 106
• спокою (ПС).............................................................. II 7 75
• пороговий..................................................................II 7 76
437
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
•
•
•
•
Джеймса..................................................................... II 725
Махайма......................................................................II 725
міжвузлові................................................................. II 7 74
передсердно-шлуночковий,
або атріовентрикулярний.................................. N7 74
• синусно-передсердний,
або синоатріальний вузол................................. II 7 74
• Паладіно - Кента.....................................................II 727
• ДІЇ(ПД).....................................................................................II 7 76
Правило Бергоньє-Трибондо....................................... 1727
Прекондиціонування........................................................II 746
• раннє.............................................................................II 746
• пізнє...............................................................................II 746
Принцип анестезії............................................................... II404
Пріони ............................................................................. 136,752
Провідність іонних каналів .... II 105-106,
138,290
Прогерія.......................................................................... 1206,207
Прогестини.....................................................1398, II382-384
Прогресія пухлинна ...............................І 352,353,361,390
Прокаріоти............................................................................ І 752
Пролактинома.................................................................... 11358
Пролімфоцити........................................................................II 68
Проліферація..........................І 95,300,326,327, II48,156
Проліферати лейкозні.....................................................11285
Промегакаріоцити.............................................................. 1182
Промієлоцити...................................................................II 62,67
Промені .................................................................136,121,541
• інфрачервоні.................................... 136, 120,121,123
• ультрафіолетові.............................. 136, 120,121,170
Промотор....................................... І 162,360,387, \\ 35, 174
Проноси (діарея)......................................................... II243,282
Простагландини.................................... 145,217,249, II 757
• простагландини (PGD2,PGE2)..............................II790
Простацикліни................................................. 1284,310, II 76
• простациклін (PG12)................................................ II790
Протеази.............................................................................II 77-90
Протеасоми.............................................І 65,203,460, II 746
Протеїнурія.................................... 1500,507,502,11322-339
• клубочкова................................................................II322
• канальцева................................................................ II322
• секреторна.................................................................II322
• селективна................................................................. II322
Протеїндисульфідізомераза................................................ 19
Протеїнкіназа................................1236,347,446,465, II 146
Протеоліз........................................... \ 60,378,504, \\ 88,372
Протиотрути........................................................................... І748
Протоонкогени .............................. \ 370,371,388, \\ 71-72
Проферменти.............................................................II265-266
«Професії стресові»............................................................. II760
Профілактика..........................................................\ 13,19,195
Процесинг................................................................................ І799
Процес нейродистрофічний........................................ N472
Псевдогермафродитизм чоловічий......................... II379
Псевдогіпопаратиреоз ............................ І 547, II377-378
Псевдонейромедіатори..................................................II305
Психіка............................................. \ 115, 193,417,506, \\ 241
Пульпіт...........................................................................II 247,249
Пурпура ідіопатична тромбоцитопенічна
(хвороба Верльгофа).............................................................II82
Пухлини.................................................................. І 18,347,560
• доброякісні ........................................ \ 347,364, \\ 368
• злоякісні .............................. \ 18,347,350,560, \\ 378
Пучок (и)................................................................................ II 7 74
• Пса.........................................................................\\ 114, 118
Р
Радикали...................................І 64,65,204,309, II 768, 780
Радіація................................................................................................
• іонізуюча.................... \ 18,36,65,392, \\ 24,63,238
• сонячна............................................................... І 727, 723
Радіоліз води.................................................................... 164,65
Радіопротектори .................................................................. І727
Радіотоксини.......................................................................... І726
Радіочутливість......................................................... І 727, 728
Рахіт..........................................................................\ 122,548,560
Реабсорбція......................................................... N 73, 194,319
• канальцева................................................................. N378
• облігатна (нерегульована).................................. N379
Реактивність ........................................\ 37,47,48,225, II343
• етіологічний аспект....................................................150
• імунологічна................................................... 149,50,57
• клінічний аспект.......................................................... 150
• лікувальний аспект.................................................... 157
• патогенетичний аспект............................................ 150
Реа кції.................................................................... \ 74,215,246
• алергічні .............................................................. \243,245
• анафілактичні............................................ \215,246,251
• вегетативні.................................................................II395
• емоційні............................................................ \\ 394,401
• протеїнкіназні................................................ І 74, II 750
• протеолізу.......................................................................II77
• рухові............................................................................N407
• судомна....................................................................... II407
• Шульца - Дейла....................................................... 1246
Регенерація нервів..............................................................II397
Регуляція алостерична ......................................................... І78
Резистентність...............................................І 48,57, II 73, 779
Резорбція кісткової тканини...........................................II784
Резус-конфлікт......................................................................... II28
Ремісі...........................................................................129,36
Ренін-ангіотензинова система . 1518,519, II 184-186
Репарація..................................................\ 125,149,384, \\ 52
Реперфузія...................................................................II 146-148
Реплікація........................................................................ І 763, 772
Ретикулум...........................................................................................
• ендоплазматичний ..................І 69,329,464, II 763
• саркоплазматичний...................................... II 707-7 7 7
Ретинопатія................................................................. 1474,475
Рефлекс (и) ....................................\ 50,137,411,552, II286
• Бейнбріджа...............................................................II 7 73
• патологічні...................................................................II406
438
• Швачка - Паріна....................................................... II234
Рефлексії барорецепторні..............................................I1189
Рефрактерність..........................................................II /27,138
• абсолютна............................................................I1106,138
• відносна....................................................................... II 138
Рецептор (и)......................................................... 182,451,489
• scavenger-рецептори........................................... 11/77
• ТоІІ-подібні.................................................................II 170
• інсуліновий .............................................................. 1493
• іритантні.......................................................................N227
• клітинні.............................................................. 182, II374
• мембранні................................................1457, II40,146
• ядерні............................................................................ 1489
Рецидив............................................ 129,30,36,225,11259,274
Речовини............................................................................................
• біологічно активні........................................II 146,412
• церебротоксичні.......................................... II303-305
Речовини-нейрорегулятори........................................11474
Речовини-хелатори.......................................................... II247
Рилізинг-гормони.................................................... 11353,476
Ритми серця......................................................................................
• атріовентрикулярний........................................... II 718
• ідіовентрикулярний шлуночковий . . . . II 778
• патологічні.................................................................II 118
• передсердний повільний................................... 117 78
Рівновага кислотно-основна (ізогідрія). . 11270,376
Розлади мозкового кровообігу.........................\\ 413,418
Розрив серця....................................................................... II 757
"Розсіяний склероз".........................................................11427
Розширення вен варикозне......................................... II202
• вторинне..................................................................... N202
• первинне................................................................... 11202
Рухи антиперистальтичні............................................... II279
С
Салурез................................................................................... II32 7
Самоактивація ферментів................................................1190
Сатурація .............................................................................. І 734
Секретин.......................................................................II237,255
Секреція канальцева........................................................II368
Сенсибілізація......................................................І 141,245,263
Сепсис........................................ \ 108,129,158,408, II 59,81
Сечовина...................................................................... II26 7,320
Сечокам'яна хвороба.......................................................II344
Симптом......................................................................... 1115,531
• "сухої шкіри"............................................................. 17 75
• "ямки"........................................................................... 153 7
Симпатоліз імунний..........................................................II409
Синдром .............................................................................................
• "апное уві сні"...........................................................N222
• "голих"лімфоцитів.................................................. 1230
• "котячого крику"..................................................... І 787
• "краш"-синдром............................. \ 17,107,150,409
• "лінивих лейкоцитів"....................................1224, N64
• "прокляття Ундини".............................................„ II222
ПОКАЖЧИКИ
• "системної відповіді при запаленні" ... 1332
• "суперчоловіків"..................................................... І 785
• автоімунний поліендокринний
II типу (APS2)...............................................................II365
• автоімунний поліендокринний І типу
(autoimmune polyendocrine
syndrome type 1; APS1)............................................II365
• адреногенітальний..................................................N369
• адренокортикальноїнедостатності.
. . . N364
• Ангельмана .............................................................. І 777
• аритмічний................................................................ II 757
• ахолічний.................................................................... N372
• Броун-Секара............................................................N390
• вегетосудинної дистонії....................................... N409
• Вернера........................................................................ 1207
• відгодовування......................................................... 1427
• Віскотта - Олдрича ............................І 777,230, II 70
• Вольфа - Паркінсона-Уайта............................... N125
• втрати солей...............................................................II344
• гематологічний....................................... 1729,1144,50
• геморагічний..................................... І 737, 749, N307
• гепатолієнальний................................... ... ., . , ІІ314
• гепаторенальний.....................................................N314
• гепатоцеребральної недостатності.
. . . N302
• Гетчинсона - Гілфорда......................................... 1208
• Гіпер-ІдМ............................................................ 1226,228
• гіперкінетичні........................................................... N406
• гіперкоагуляції крові............................................. N328
• гіперпродукції пролактину.................................N358
• гострий коронарний............................................. II 750
• Гудпасчера...................................................................II340
• Дабіна - Джонсона................................................. N370
• Дауна ............................................................\182,183,\\70
. ДВЗ............................................ \ 160,261,334,410, II 89
• дефектної адгезії лейкоцитів......................... І 9,325
• диспепсичний........................................................... N240
• ДіДжорджі........................................................ \ 226,228
• діабетичної стопи. . \ 471,474,475,476, \\ 179,186
• Дреслера..................................................................... 1255
• Едвардса........................................................... І 182, 183
• Елерса-Данло................................................. \ 177,1182
• Жільбера.......................................................................II309
• загальний адаптаційний........................... 155,97
• Золлінгера - Еллісона.............................. 1350, II260
• Іценка - Кушинга........................................... 1395,496
• каротидного синусу.....................................II 117,191
• класичний адреногенітальний.......................... II370
• Кляйнфельтера
(Клайнфельтера)..................І 182, 184,185,456, II70
• Конна............................................................................ N368
• коронарний X.......................................................... II 750
• Кріглера - Найяра................................................... N370
• ламкої Х-хромосоми ........................................... І 776
• Леша-Ніхана.................................................... І 177,508
• Лі-Фраумені..................................................... 1356,379
• Лінча................................................................... 16,380
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
Луї - Бар..................................... 1230,381, II 70
мальабсорбції................. 1500,558, II237,281
мальдигестії................................ 150, II268,278
Марфана........................................ І 177, II 179
Меньера ................................................. І 136
множинної ендокринної неоплазії
(multiple endocrine neoplasia, MEN) . . II 358,378
мозочковий............................................11407
Морганьї - Едемса - Стокса..................... II 125
набряку легень........................................N232
набутого імунодефіциту . .137,232,239,365, II365
надмірної перфузії..................................II4 79
неадекватної секреції вазопресину
(синдром Пархона)................................. II359
недостатності глюкагону........................ II385
Незелофа.......................................... 1226,228
нейролейкозу.................................................. II74
нефротичний.......................... 1232,480, II337
Патау................................................ І 782, 783
передчасного старіння.................. 1426, II358
передчасного статевого дозрівання ... II369
портальної гіпертензії...............1299,117 74,374
посиленої секреції вазопресину .. 196,11367
посттромбофлебітний.............................II 205
Прадера - Віллі................................. І 177,497
Райлі-Дея.................................................N372
резорбційно-некротичний......................II 757
реперфузійний........................................ II 744
Ротора......................................................N370
сидеропенічний .............................. І 426, II44
слабкості синоатріального вузла............117 78
спотворений внутрішнього обкрадання "синдром Робін Гуда".............................II 479
судинної енцефалопатії.........................11420
судомний.................................................II408
тестикулярної фемінізації.......................N382
тиреоїдної резистентності...................... II373
тривалого стиснення...................І 107,409,410
тромбоемболії легеневої артерії............II234
трофічних порушень........................ 1426, II 44
Уотерхауса - Фрідеріксена...................... II364
уремічний................................................N323
уродженої остеодистрофії Олбрайта. . . N377
Фанконі.................................. 1558,559,11379
холемічний або синдром холестазу. ... N377
Чедіака - Хігасі......................................... 1224
Шегрена........................................... 1269,275
Шерешевського-Тернера *1 782, 184,207,456
Шихана
II357
шкірний........................................................... II74
Сироватка анти ретикулярна
цитотоксична....................................... \ 21,254, II 347
Сироватка Богомольця........................ 149,255,336
Система(и)
• NO-синтазна..................................... 1222,223
• альдостерон-вазопресинова.................. II 789
439
• антиоксидантні...................166,78,204, II25,31
• гемостазу.................................................I1169
• гіпоталамо-аденогіпофізарна................ N353
• гіпоталамо-нейрогіпофізарна................ N359
• гіпоталамус - аденогіпофіз статеві залози.......................................... || 350
• гіпоталамус - аденогіпофіз щитоподібна залоза..........................................................II
350
26,50,4
160,21
86,88
\ 50, 157,269,459, \\ 7
160,260,314,501, 190,2
158,211,260,314, 88
222,223,309
• депресорні.................................................................. I1189, 790
• ендокринна.............................................. 1
• зсідання................................І
•
•
•
•
•
•
•
7 7, II 347
7,375,507, II
імунна...................................
калікреїн-кінінова . 1
комплементу.............................І
кровообігу..................................................................................II 92
мієлопероксидазна......................................... 1
неопіатна гормональна...................................................11403
неопіатна нейронна..........................................................11403
11
II
100,317,496, \\ 38
• нервова....................................... 150,
• опіатна гормональна........................................................ II403
• опіатна нейронна............................................................... 11403
• пресорні.................................................................................. II 789
• ренін-ангіотензинова............................................II
• серця провідникова...............................................II
• симпатоадреналова . . . . 1
Сімейна дизавтономія (синдром Райлі - Дея). II
189,192
125-127
340,403,519,
372
II 787
Склерозування.................................................II 756
Сладж........................................................ 1292,406
Сладж-синдром.......................................... II 97, 735
Смерть.................................................. І ЗО, 253,506
• клітини................................................. 162,86
• раптова............................................ І 728, 733
• раптова коронарна................................. II 750
Солюбілізатори................................................II345
Соматотрофи................................................... II354
Соматотропін.......................1443,452,490, II354,356
Сомнолентність................................................II422
Сонливість.................................................II 302,422
Сонячний удар......................................................... І123,345
Сопор................................................................В 422
Спадковість.....................І 37,50,168,355, II 162,216
Спазмофілія..................................................... 1550
Спазмування ........................................... N237-232
Сплайсинг.............................................1207, II35-36
Спленомегалія................................... \ 482, \\ 38,314
Сполучна тканина..................149,292,297,300, II45
Спрага ........................................ \48,515,\\360,416
Стаз...................................... \ 61,159,402,407, \\ 331
Стани коматозні........................................II417-418
Старіння........................І 722, 795, 197,202, II 178,215
Стати ни............................................................N 353
Стеатоз печінки.........................................II 288,292
Стеаторея................................................ 1487, N372
Стеноз ........................................ \ 183,296,399, \\ 94
Стенозування............................................II 756, 758
Стенокардія...............................................................
440
• стабільна напруги...................................................II 750
• Принцметала............................................................II 736
Стеркобіліноген........................................................ 11306,57 7
Стрес..................................................................130,97, 101,466
• оксидаційний........................................................... II 179
• хронічний...................................................................II 192
• емоційно-больовий..............................................II401
"Структурний антиоксидант".....................................166,67
Ступор............................................................... \ 150,412, \\ 302
Судини ................................................................................................
• ємнісні..........................................................................II 755
• компенсаційні, або судини-амортизаториІІ 755
• обміну.......................................................................... II 755
• опору, або резистивні..........................................II 755
• перерозподілу......................................................... II 755
Судоми...................................................................... 157, 131,548
• клонічні.......................................................................ІІ407
• тонічні.......................................................................... II407
• місцеві......................................................................... II407
• генералізовані......................................................... 11407
Сумація збудження........................................................... N 4 7 6
Суперантигени.................................................................... І 757
Суправентрикулярна пароксизмальна
тахікардія (СВПТ).......................................................... II 120-121
• передсердна форма СВПТ................................. 1 1 7 2 7
• атріовентрикулярна форма СВПТ.................. II 727
Сфероцитоз........................................................................ II29-30
Т
Таласемія...........................................................................І 777, II35
• а-таласемія..................................................................... II38
• 3-таласемія.....................................................................II35
Тампонада серця ..................................1399,400, II 7 72, 752
Тахікардія...............................................\ 115,344,\\ 118-122
• пароксизмальна............................................. N120-122
• синусна........................................................................II 7 76
Таутомеризація ........................................................... І 770, 775
Тахіпное...........................................................................II217-224
Телеангіектазії.........................................1207,355,387, II82
Теломераза..................................................1207,349,371,386
Теорія....................................................................................................
• Вірхова ............................................................................159
• інтенсивності............................................................11395
• мембранна (Джексон, Готто)............................ II 774
• моноклональна (Бендітт).................................... II 774
• розподілу імпульсів.............................................. 11395
• специфічності...........................................................II396
• тромбогенна............................................................. II 777
• факторів росту (Росс)............................................II 772
• центрального походження мігрені . . . . I I 4 0 7
Тепловий удар..................................................................... 17 75
Тератогенні фактори........................................................ І 769
Термінальні стани............................................129,30,469,230
Тетанія ............................................. 1228,547,548, II408,414
Тип протеїнурії...................................................................... II323
ПОКАЖЧИКИ
• нефротичний..............................................................II323
• нефритичний..............................................................II323
Типові патологічні процеси ............................126,28,277
Тиреотрофи.......................................................................... II 355
Тиск атмосферний................................................................. 136
• підвищений...............................................\ 298,442,454
• понижений........................................................ 1298,529
Тиск
• барометричний..................................................155, 737
• кінцеводіастолічний.......................................II 97, 7 73
Тіла кетонові...........................................................................II292
Тільця....................................................................................................
• апоптичні..........................................................II 184, 299
• Вейбеля - Паладе......................................................I I 8 4
« Гайнца.............................................................................. II 77
• Жоллі................................................................................N77
Тіні Гумпрехта......................................................................... N69
Токсемія.......................................................................... \ 108,150
• опікова............................................................... І 7 70, 7 7 7
Токсикоз.......................... ... .1148,409,410, \\285,330
Токсини............................................ \ 154,157,321,\\ 168,261
Токсини і отрути................................................................... II405
• ботулінічний токсин...............................................N405
• а-бунгаротоксин (зміїна отрута)..................... II405
• кураре-екстракт...................................................... II405
• інсектициди...............................................................II405
• речовини отруйні фосфорорганічні
бойові (хімічна зброя)......................................... II405
Толерантність .....................................................І 141,270,273
• імунологічна..................................................... \ 210,270
• центральна........................................................ \ 270,273
• периферична................................................... \ 270,271
Травлення....................................... 150, 7 7 7, 728,574, II237
Травма........................................................... І 706, 709, 730,472
• механічна............................................. \ 106,107, \\ 204
• термічна ..................................................................... 1 7 0 9
• хімічна.......................................................................... І 740
Транскрипція.................................І 162,234,541, \\ 35,102
Транслокація.....................................\ 185,371,374, II 70-72
Трансляція................................................ І 164, 175, II36, 702
"Трансплантат проти хазяїна"......................1228,240,397
Транспозиція міжхромосомна............................. І 185,186
Транспозони.........................................................\ 169,372,385
Транспорт аксоплазматичний............................II 387,405
Трансформація............................................................. 185,764
• Вірхова ....................................................... 132,294,383
• ритму збудження...................................................N 4 7 6
Треморінтенційний...........................................................I I 4 0 7
Тригемінія.......................................................................II 720-727
Тріпотіння передсердь............................................. II 727, 730
Тромб..........................................................1293,298,334, II 74
Тромбастенія Гланцмана.................................................. I I 8 7
Тромбін...................................................................................... II 76
Тромбоемболія...................................... 1297,298,299, I I 9 7
Тромбоз...............................................1293,296,527, II50,87
Тромбоспондин...............................................................II 76-77
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
441
• ф. X I I I - фібринстабілізуючий фактор. . . II 78
Тромбоутворення.............................................................. II 9,2/
• ф. Ill-тканинний тромбопластин . . . 1 1 8 0 , 9 7
Тромбоксани................................. \ 73,310,318, \\ 189,363
Фактори...................................................................................
II 753
Тромбофільні діатези (тромбофілі'О..................... II 80,91
•
ризику...........................................................
136,37,11759
Тромбофілія.......................................................1296, II 97,328
• росту................................................1707,327,374, II 60
Тромбофлебіт.............................................................II204-205
Фармакологічні
агенти....................................................II 709
• гострий.......................................................................... II204
Фенілкетонурія
.........................................
\ 26,191,504,505
Тромбоцити.......................................................................II 75-76
Фенокопії...............................................................................
І 769
Тромбоцитопенія СО ............... І 7-29,230,254,269, II 83
•
Артюса
........................................................................
1258
• мієлотоксичні........................................................II 39,83
•
Овері...................................................................
1245,246
• споживання................................................................... II83
Феномен "сходинок" або феномен Боудича ... N97
• дефіцитні........................................................................II 83
Феохромоцитома............................................................... 1456
• дисрегуляторні . ........................................................ I I 8 3
Фермент протеолітичний........................................ N88,268
Тропоміозин.................................................................................... II109-111
Ферменти ...........................................................................................
Тропонін С.............................................................................II 110
• амілолітичні................................................................N266
• ліполітичні................................................................. N 266
У
• мембранні.............................................. \ 71,79,82,286
Уреаза..................................................................................... I I 2 6 7
Ферментемія............................................................... II 151,271
Уремія...................................................................1535, II59, 779
Ферментопатії (ензимопатії).............................................N29
Уробіліноген.....................................................................................II306-309 • гліколізу..........................................................................N37
Урокіназа.................................................................................. I I 8 9
• пентозного циклу......................................................N37
Ускладнення......................... І ЗО, 119,471,498, II 758, 795
• утилізації АТФ.............................................................. N37
Ушкодження ................................................. 142,60,402,571
• циклу глютатіону....................................................... N37
• нейроглії.....................................................................11427
Ферментопатії інтестинальні .......................................... І 779
Фетуїн-А.....................................................................................II 784
• нерва механічне..................................................... II405
Фібриляція .............................................................................. І 737
Ушкодження серця............................................................II 749
• шлуночків ....................................................... 1400, II 737
• імунні............................................................................II 749
• передсердь ..................................................\ 1 3 1 , \\ 129
• нейрогенні................................................................. W149
Фібрин нерозчинний (фібрин І)..................................... II 80
Фібрин-мономер.................................................................. N 80
Ф
Фібриноген......................................................................... N77,88
Фагоцити................................................... \ 216,217,223, \\ 54
Фібрин-полімер розчинний (фібрин S)......................N80
Фагоцитоз.............................. \ 27,53,223,556, \\ 26,34,54
Фібринолітична система................... І 160,297,315. \\274
Фавізм.........................................................................................II 32
Фіброз .................................... \ 129,177,327,328, II 7 74, 772
Фактор (и)...........................................................................................
Фібробласти ..................................1265,297,329,465, II 774
• активації тромбоцитів.............................................1176
Фільтрація клубочкова...................................................... і 754
• Віллебранда................................................................. II 76
Фістула Екка............................................................................Я 286
• депонування гормонів.........................................II352
Фолдинг.........................................................................1232, N37
• екзофтальмічний.................................................... II374
Фосфат-діабет............................................ І 777, 179,558,559
• ендогенний строфантиноподібний . . . . II298
Фосфоліпаза .............................................. І 77,87,346, I I 2 7 0
• жовчі.............................................................................II 372
Фосфор....................................................................І 740,426,538
• транскрипційні........................................................ II 744
Фосфорилювання потенціалу............................ I 1 1 0 0 , 743
• Фактори зсідання крові.......................II 79,343, II 78
Функції крові:..............................................................................N 8
• вітамін-К-залежні..................................................II 78
• гомеостатична....................................................N 8 , 3 7 6
• вітамін-К-незалежні.............................................II 78
• дихальна............................................................................N8
• ф. І - фібриноген.................................................... II 78
• захисна................................................................................... N8
• ф.У-проакцелерин............................................... 1178
• нутритивна, або трофічна.............................................N8
• ф. V I I I - антигемофільний глобулін . . . . I I 7 8
• регуляторна......................................................................... N8
• ф - V I I - проконвертин........................................1178
• реутилізаційна і екскреторна..................................... N8
• ф. I V - і о н и кальцію............................................. 1 1 7 8
• терморегуляторна............................................................ N8
• ф. I X - фактор Крістмаса............................. II 78,88
• транспортна.........................................................................N8
• ф. II - протромбін.................................................. II 78
Функція мітохондрій Са-акумулятивна................N 7 7 7
Функції тромбоцитів.............................................................N74
• ф. XI - плазмовий попередник
• адгезивно-агрегаційна.............................................N74
тромбопластину.....................................................II 78
• ангіотрофічна............................................................... N74
• ф. XII - фактор Хагемана.................................... II 78
• вазоконстрикторна....................................................N74
• ф. X - фактор Стюарта - Прауера...................II 78
ПОКАЖЧИКИ
442
X
Хашимото тиреоїдит................1269,275, II373-376
Хвороб моделювання.............................. І 75, II 255
• експериментальне ..175, 76, 18,498, її 238,255
• математичне..............................................І 79
Хвороба(и)
• автоімунні ......................... \ 258,269,501, II 73
• адаптації................................................198,99
• Аддісона (бронзова хвороба).................. II365
• Альцгеймера .............................. \ 95,183,205
• антиген-асоційовані................................ І 180
• Бернара - Сульє, або макротромбоцитодистрофія.......................................... N85
• Верльгофа..................................................II82
• виразкова........................................... її 41,259
• висотна................................................ І 133,136
• Віллебранда....................................... \ 177, її 85
• Гантінгтона........................... І 95,176,177, 158
• Герша.......................................................II300
• Гірке.........................................................II300
• гірська....................................... І 133,136, N20
• Гіршпрунга..........................................II242,280
• глютенова.............................................. 11282
• гостра променева..................... І 727, 728, II 10
• Ґревса,або
базедова хвороба . . 1254,255,275, II375-376
• Дауна .................................................. 126, II 70
• декомпресійні..................................... І135,136
• жовчнокам'яна....................... \ 192,498, її 312
• Іценка - Кушинга..................................... 1456
• кесонна.................................................... І 736
• класифікації......................... \ 25,26, її 155,387
• Крайньої Півночі........................................126
• Крейтцфельда-Якоба................................ І752
• Крона................................................. 1355,417
• Лебера........................................................ І7 77
• легень хронічні обструктивні................ II272
• Леша - Ніхана.......................................... І 789
• Маркіафави - Мікелі .................. \ 215, її 29,83
• мітохондріальні....................................... 1462
• моногенні ........................................... І177,496
• Мошковича........................................... її 27,83
• накопичення ............................................. І777
• новонароджених гемолітична................1128
• опікова............................................. І 7 70, 7 7 7
• Паркінсона............................................ 188,95
• полігенні (мультифакторіальні). . І 77, 779, 789
• Реклінгаузена..................................... І176,177
• сечокам'яна................................... І 509, II344
• сироваткова............................ \ 258,261, її 342
• сімейні........................................... І 769, II 762
• спадкові................................178, 768,1129, 759
• танжерська............................................ 1487
• уроджені................................. \ 169, її 232,373
• хромосомні................... 128, 782, 785,456, II70
• цивілізації................................................... 136
• Шенляйна - Геноха...............................N87-83
Хемокіни..................................... 1278,224,11 171,299
Хемотаксини............................................... \ 92,217
Хемота ксис......................... 12 77,264,331, її 141, 170
Холалемія........................................................ N309
Холалурія......................................................... N309
Холангіт.......................................................... N373
Холемія............................................................ N 309
Холестаз.................................................... N286,37 7
Холестерол.......................... \ 38,297,485, її 156,160
Холецистит................................................II 244,311
Холецистокінін.................................. її 237-240,266
Хордотомія.......................................................II404
Хорея...................................................... І 769, II406
Хроматоліз......................................................11398
Хромосома "філадельфійська".........................II72
Хронічна недостатність нирок...................II326,332
ц
Центр дихальний. . . .
Центр некротичний. .
Циліндри....................
• воскоподібні. . .
• гіалінові.............
• зернисті..............
Циліндрурія...............
Цикл Кребса ..............
Цироз печінки............
• біліарний...........
• портальний.. . .
• постнекротичний
Цитокіни.....................
Цитоліз.......................
Цитопенія...................
Цитоплазма ...............
Цитоскелет ................
Ціаноз.........................
........................II 7 74,220
........................I1172,775
...............................N323
...............................N323
...............................N323
...............................N323
........................ N323,343
............І 74,289,425,486
. . 1290,355,535, II7 74,289
............................... N289
...............................N289
............................... N289
.. \ 159,218,240, її 60,180
І 763,220,237,393, II57,362
............................. 12 67
.......................... 1 85,234
. А 72,85,217,329, її 29-30
.............. \ 290, її 114,211
ч
Час.............................................................................
• кровотечі............................................... II 79-82
• парціальний протромбіновий.................. II80
• протромбіновий.................................... її 79-81
Ш
Шаперони...........................................................І 84
Шлуночкова пароксизмальна тахікардія (ШПТ)
• (мономорфна ШПТ).................................II 727
• (поліморфна ШПТ)...................................II 727
Шок
1243,253,399
• анафілактичний........................... 124
443
ОБ'ЄДНАНИЙ ПРЕДМЕТНИЙ ПОКАЖЧИК
399,402,524
А
-конфлікт...................................................... 1128
399, 108, АВ
а,-адренорецептори................................................II371
• ангідремічний....................................................... I. 1
. .
• больовий ...................................................................
1708,11407
II 750
• геморагічний...................................................... І. 1
1399,400,11 757
• кардіогенний...................................................... ІЗ;
...
• опіковий.....................................................................
1399,400
а2-адренорецептори................................................II371
\ 399, \\ 273,278
160 В
108,400
В-ендорфін............................................... II402-403
• панкреатичний .................................................. ІЗ.
. . . . І 708,
• септичний ..................................................................
. . . .
• травматичний ...........................................................
І
Я
Ядерний зсув......................................................II62
• вліво...........................................................II62
• вправо........................................................II62
• гіперрегенераторний................................ II62
• дегенеративний........................................ N62
• регенераторний.........................................II63
• регенераторно-дегенеративний.............. N63
Ядерний фактор NF-kB......................... І 94,234,236
Ядерні
• ендонуклеази....................................... І 89, 97
Ядро............................................ \ 95,162,553, \\ 174
3,-адренорецептори................................................II371
32-адренорецептори............................................... II371
с
Са-насоси плазматичної мембрани
і саркоплазматичного ретикулуму............... II 7 / 7
Н
Helicobacter pylori.................................. II 70,256-264
R
Rag-білки............................................................................................II 70-71
ПОКАЖЧИКИ
444
Об’єднаний іменний покажчик
Меєрсон Ф............................................II 98, 700-707
Мелзак Р...........................................................II395
Мечников 1.1..................179,204, 208,302,326,331
Мойбенко О. О............................................. 11747
Асклепіад.....................................................
.....................................137
Морганьї Д.........................................................137
Банг О............................................................
...................................... І 364
Орбелі Л............................................................. 133
Бернар К...............................................
76
. . .1І 16,24-25,32-33,288,453
Павлов І. П................................120,24,27,33-34,193
Биць Ю. В..................................................
....................................II 779
Парацельс......................................................І ЗО, 740
Біттнер Дж...................................................
...................................... 1364
Пашутін В. В.................................................127,25
Біша М...........................................................
......................................137
Підвисоцький В. В......................................... І 19-20
Блюменбах Й.............................................
......................................137
Пірке К............................................................... 1258
Богомолець О. О. . . І
12,15-16,20-21,23-24,25,34,
Платон................................................................ 137
49-50,192
Портьє П............................................................1245
Браун М........................................................
.............................. II 162-164
Раус П.......................І 364-365,367,369-370,374,388
Бурхаве Г. ...................................................
........................................ І 37
Рейнке И............................................................ 137
Введенський М. Є...........................
........................................ 133
РепрьовО. В................................................... \ 19,21
Вірхов Р. ......................................17,24.І 7,24-25,32,59,294,302,355
Ріше Ш.............................................................. 1245
Волл П.................................................
....................................N395
Рокитанський К.....................................................І ЗО
Вороній В.....................................................
........................................ 127
Савченко І. Г....................................................... 127
Гален Клавдій............................................
................... І ЗО, 197,302,331
Санторіо............................................................. 137
Галлер А.......................................................
......................................137
СельєГ.............................................. І 97-98, 102,331
Гаральд цур Гаузен.................................
.................................... 1364
Сєченов І. М...................................................137-33
......................................127
Генес С..........................................................
СиротинінМ. М......................................... 127,33,49
Гіппократ ....................................................
..........17,27,30,148,192-193
Соран Ефеський................................................. 137
Гольдштейн Дж........................................
.............................. II162-164
Сперанський О. Д............................................... 133
.........................................127
Горєв М. М..................................................
Стерлінг Е...................................... \ 279,511,520,522
Готто А..........................................................
....................................II 774
Тальянцев
А. І............................... .. . .................. 127
Гофман Ф.....................................
. . і , „ п « . . , , ____.137
ТарасевичЛ.А................................................
120-27
.................................... 1364
Гросс Л..........................................................
Татаринов
Є.
0....................................................
127
Демокрит ....................................................
........................................ 17
Темісон
Лаодікейський
....................................
137
Джексон Р...................................................
....................................II 774
Трентін Дж........................................................ 1365
......................................137
ДрішГ. ...........................................................
Ухтомський 0.0................................................... 133
.................................... 1365
Едді Б.............................................................
Ушинський М..................................................... 127
.................................... 1364
Елерман В...................................................
Фессал................................................................ 137
Заболотний Д. К.......................................
........................................ 120
........................127,25 ,1147 7 Фохт О. Б............................................................ 127
Зайко М. Н...............................................
ФрейдЗ........................................................ 137,40
Ібн-Сіна (Авіценна).............................
..................................... 130
Фролькіс В. В.............................................. 1204-205
.................................... . .127
Кавецький Р Є.............................
ХалатовС. С................................................II 767-762
.....................................127
Комісаренко В. П.................................
Хржонщевський Н. А................................ І 73, 19-20
..................122,302,376,337
Конгейм Ю..............................
Целій Авреліан...................................................137
...................................... 302
Левіс Т. І....................................................
Цельс Авл Корнелій ................................... І 302,337
......................................127
Ліндеман В. К..........................................
ІІІаде Г...............................................................1302
......................................127
Лондон Ю. С.........................................
Шацило Б. А....................................................... 127
.................................... 1364
Люке Б.......................................................
ІІІоуп Р............................................................. 1364
..............................І 16,32-33
Мажанді Ф.................................
Адлер А.........................................................
......................................137
Альперн Д. Є..................................
......................................127
Анічков М. М.............................................
.................127II 164-165, 170
Аристотель..................................................
.....................................137
12,15-
Навчальне видання
Олександр Васильович Атаман
Патофізіологія
Том 2. Патофізіологія органів і систем
Підручник
Редактор О. В. Марчук
Технічний редактор Ж. С. Швець
Коректор Л. Я. Шутова
Дизайн та верстка: О. С. Парфенюк
Ілюстрації: Д. С. Волощук
Підписано до друку 18.08.15. Формат 60x84/8. Папір крейдований.
Гарнітура Міріад. Друк офсетний. Ум. друк. арк. 55,44.
Зам. № 761.
ПП “Нова Книга”
21029, м. Вінниця, вул. Квятека, 20
Свідоцтво про внесення суб’єкта видавничої справи
до Державного реєстру видавців, виготівників
і розповсюджувачів видавничої продукції
ДК № 2646 від 11.10.2006 р.
Тел. (0432) 52-34-80,52-34-82. Факс 52-34-81
E-mail: info@novaknyha.com.ua
www.novaknyha.com.ua
Від друковано згідно з наданим оригінал-макетом
у друкарні «Фактор-Друк».
61030, м. Харків, вул. Саратовська, 51.
Тел.; + 3 8 057 717 53 55.