/














































































































































































































































































Автор: Репинская И.Б. Шварцберг М.С.
Теги: органическая химия химия органический синтез
ISBN: 5-7615-0204-6
Год: 2000




Текст
И. Б. РЕПИНСКАЯ, М. С. ШВАРЦБЕРГ
ИЗБРАННЫЕ МЕТОДЫ СИНТЕЗА
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Под редакцией М. С. Шварцберга
Рекомендовано Министерством образования Российской Федерации
в качестве учебного пособия для студентов высших учебных
заведений, обучающихся по специальности „Химия"
НОВОСИБИРСК
ИЗДАТЕЛЬСТВО НОВОСИБИРСКОГО УНИВЕРСИТЕТА
2000
УДК 547
ББК Г22
Р41
Рецензенты:
|проф. Б. В. Иоффё~\
проф. Н. С. Простаков
проф. А. А. Мороз
Рекомендовано к печати ученым советом факультета естественных наук
Новосибирского государственного университета
и поддержано Минобразованием в рамках гранта „Интеграция"
Федеральная программа книгоиздания России
И. Б. Репинская, М. С. Шварцберг
Р41 Избранные методы синтеза органических соединений:
Учебное пособие. - Новосибирск: Изд-во Новосиб. ун-та,
2000,- 284 с.
ISBN 5-7615-0204-6
Книга представляет собой пособие для практического освоения ряда
фундаментальных методов органического синтеза. В ней рассмотрены
каталитическое гидрирование органических соединений, восстановление
комплексными гидридами металлов, реакции литийорганических соедине-
ний и применение жидкого аммиака в органическом синтезе. Каждая глава
включает обсуждение важнейших особенностей метода, а также описание
экспериментальной процедуры 10-15 синтезов с подробной химико-физи-
ческой характеристикой получаемых веществ (ИК, УФ, ПМР спектры).
Пособие предназначено для студентов старших курсов химических
факультетов университетов. Оно также может быть полезно аспирантам,
преподавателям и химикам-исследователям, занимающимся органичес-
ким синтезом.
УДК 547
ББК Г22
ISBN 5-7615-0204-6
© И. Б. Репинская, М. С. Шварцберг, 2000
ОГЛАВЛЕНИЕ
СПИСОК ПРИНЯТЫХ СОКРАЩЕНИЙ.......................................6
ПРЕДИСЛОВИЕ .................................................... 7
Глава 1. ГИДРИРОВАНИЕ............................................9
1.1. Восстановление органических соединений.................9
1.2. Гидрирование как метод восстановления.................13
1.3. Катализаторы гидрирования.............................18
1.4. О механизме каталитического гидрирования..............24
1.5. Влияние строения восстанавливаемого соединения, природы
катализатора и условий реакции на селективность гидрирования ... 33
1.6. Гидрирование важнейших классов соединений.............42
1.6.1. Ацетилены........................................42
1.6.2. Этилены..........................................46
1.6.3. Арены и гетарены.................................49
1.6.4. Карбонильные соединения..........................59
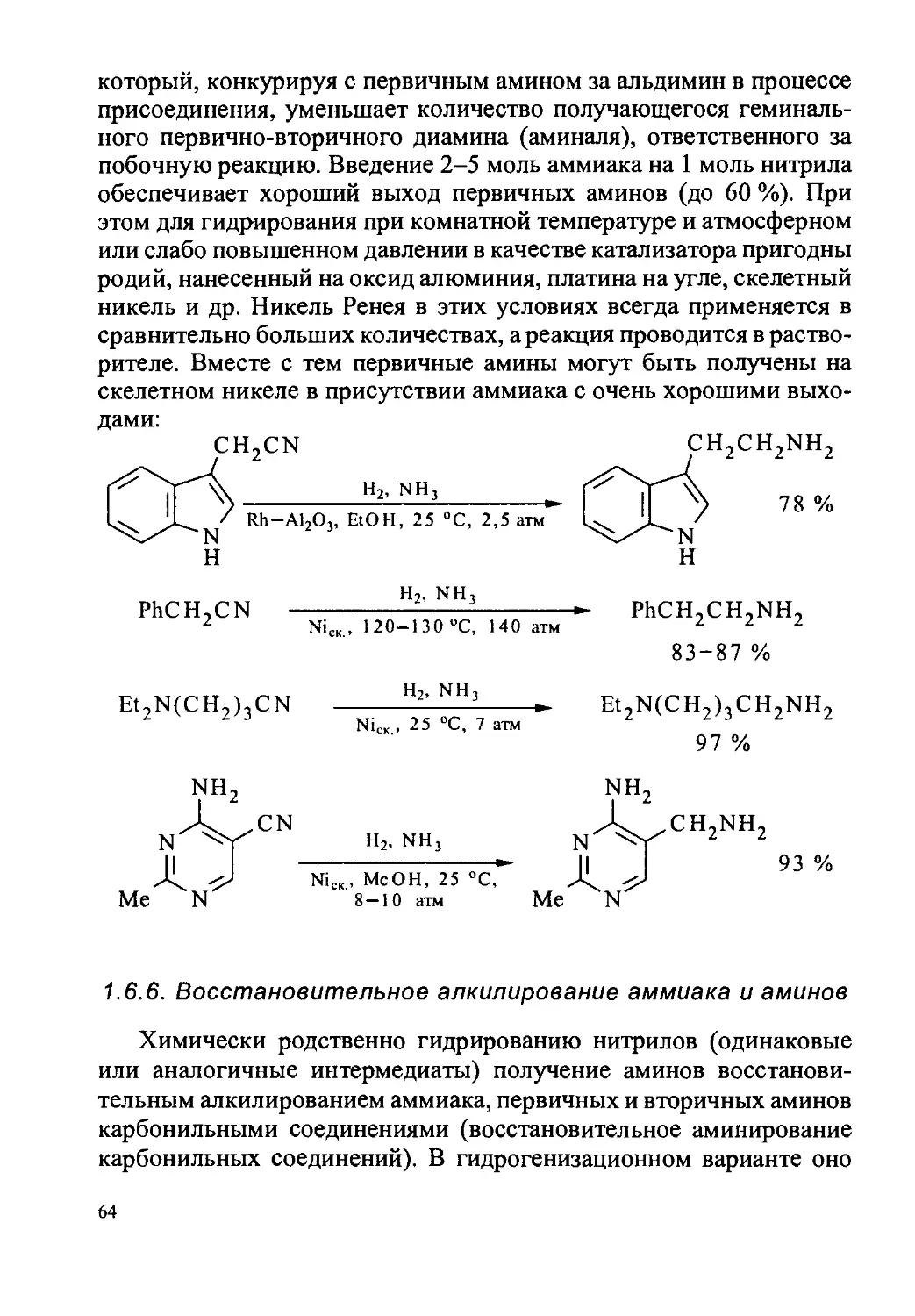
1.6.5. Нитрилы..........................................62
1.6.6. Восстановительное алкилирование аммиака и аминов . ... 64
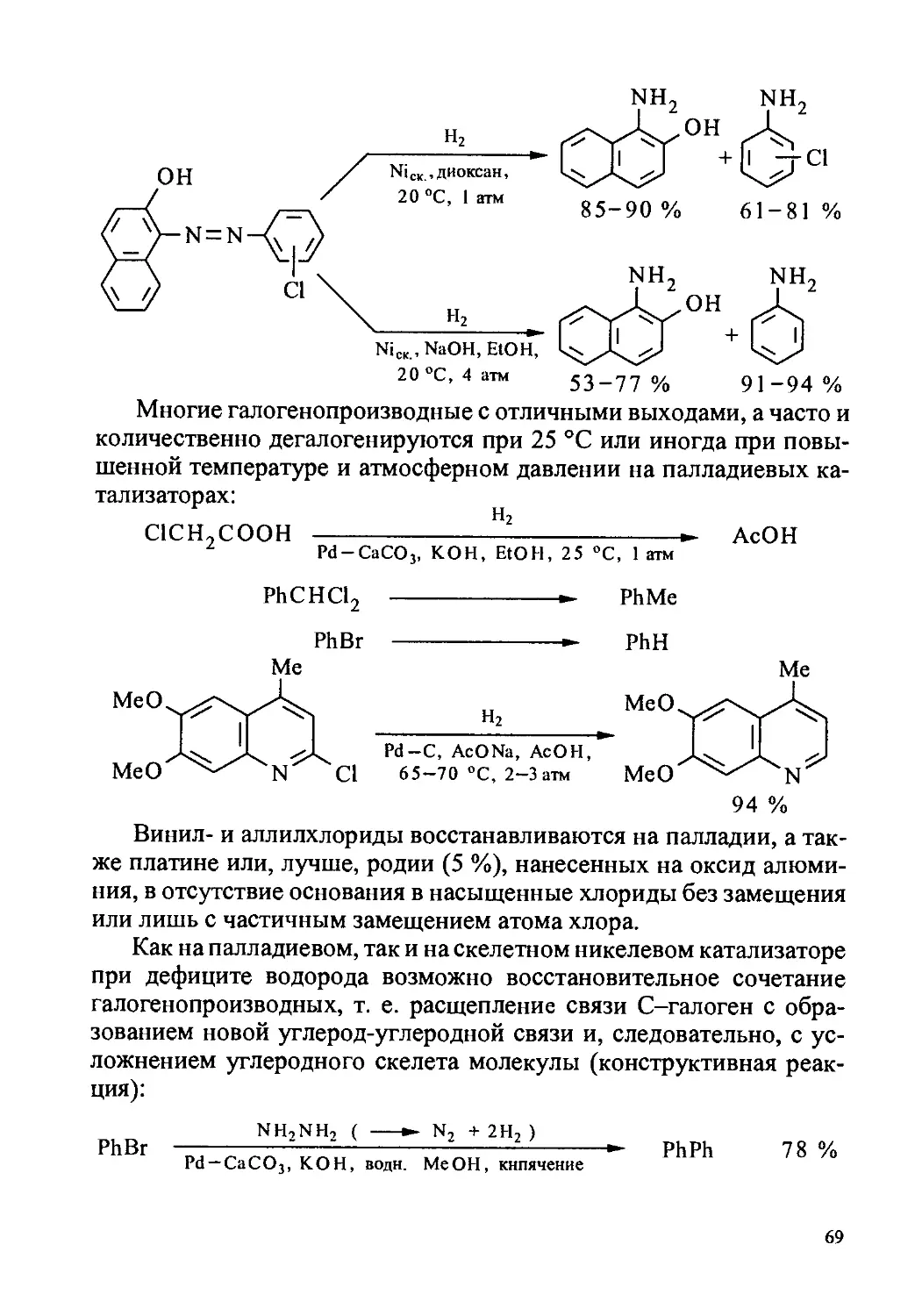
1.6.7. Гидрогенолиз галогенопроизводных.................67
1.6.8. Сложные эфиры....................................72
1.6.9. Амиды карбоновых кислот..........................75
1.7. Техника эксперимента..................................76
1.8. Экспериментальные работы..............................79
Получение катализаторов.................................80
Гидрокоричная кислота...................................83
5,6,7,8-Тетрагидро-1-нафтол.............................84
Бензилацетон............................................84
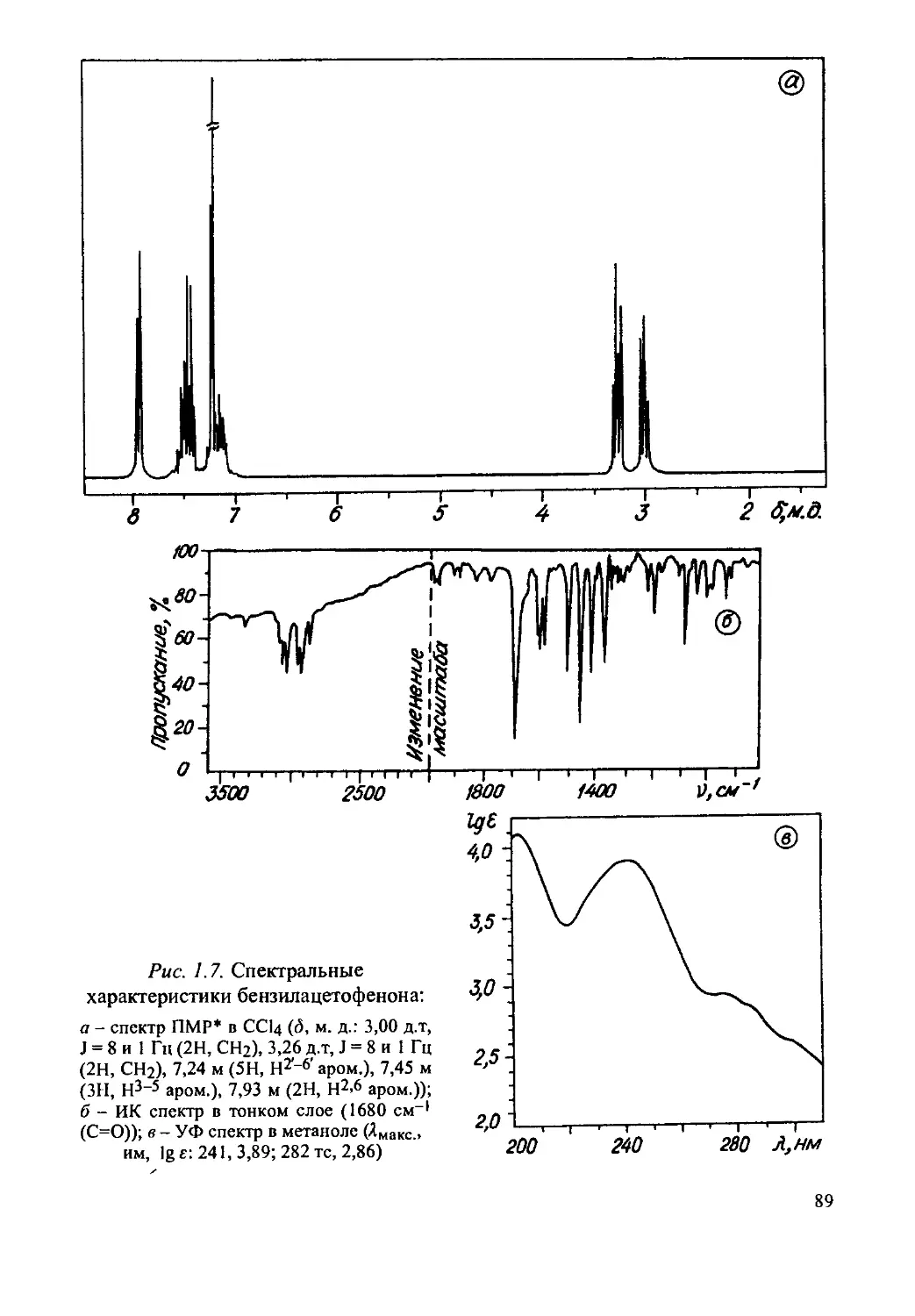
Бензилацетофенон........................................87
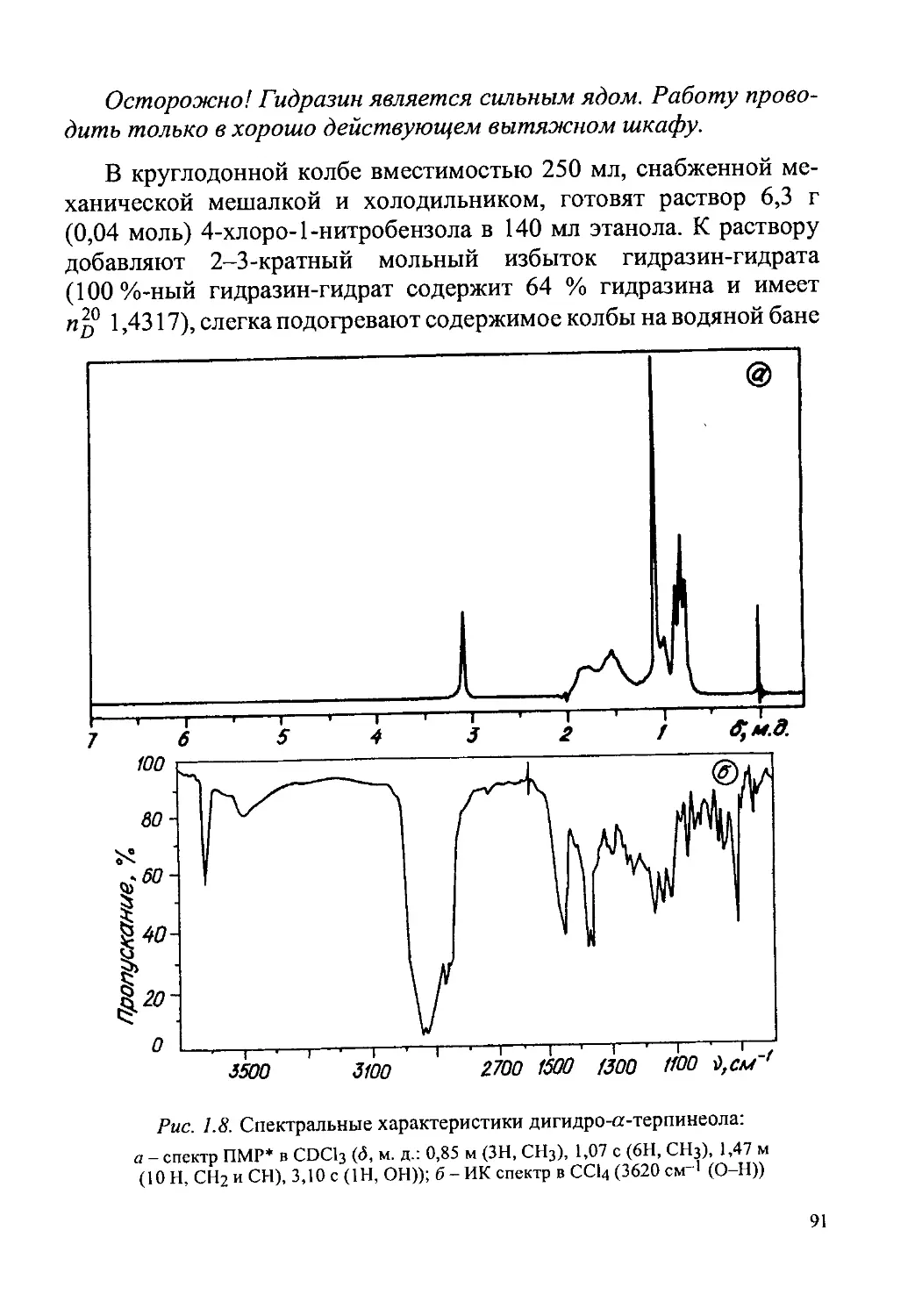
Дигидро-а-терпинеол.....................................90
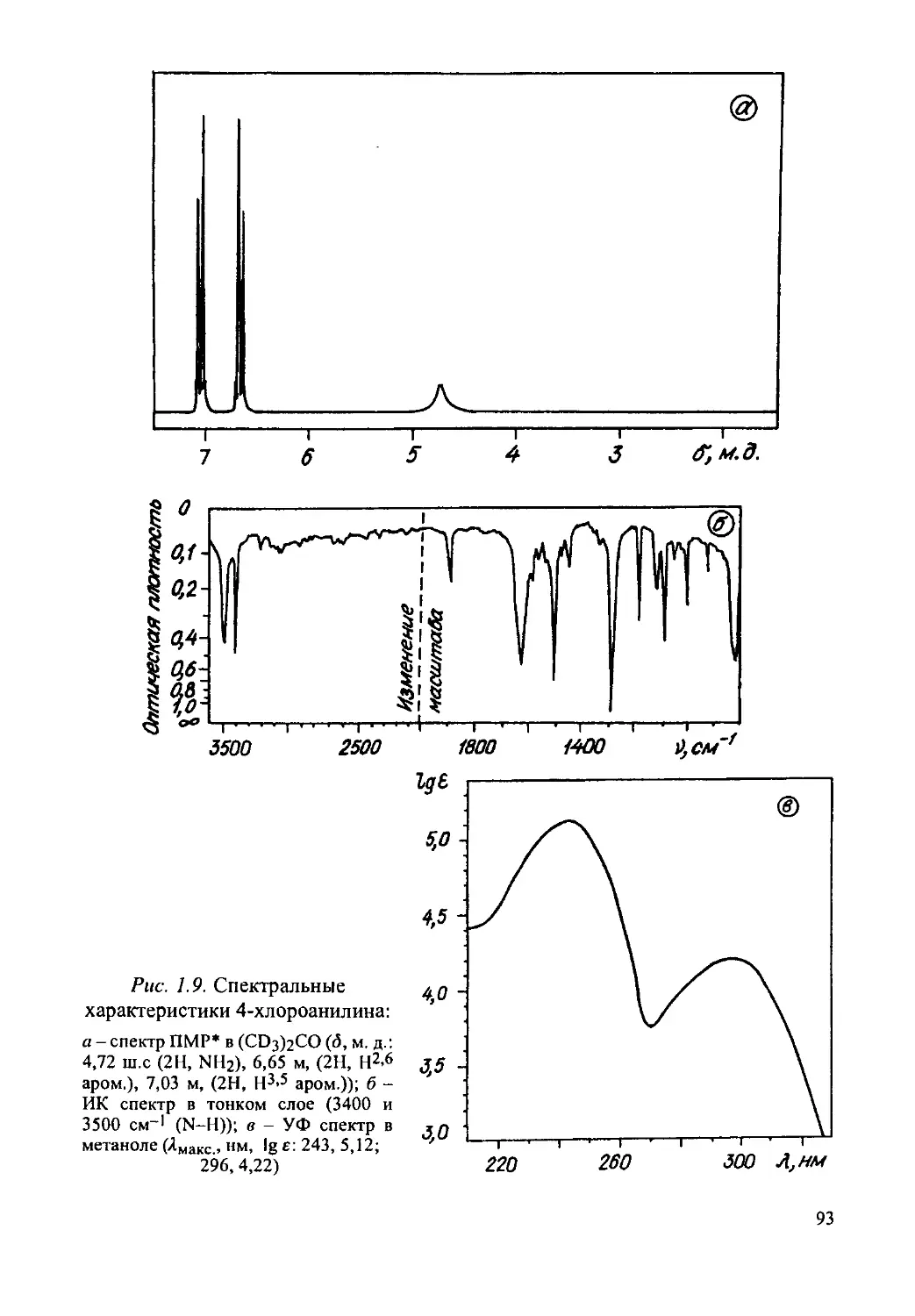
4-Хлороанилин...........................................90
Циклопентанол (способ 1)................................94
Циклогексиламин.........................................94
Метилциклогексан........................................96
Дигидрорезорцин.........................................96
Дициклогексано-18-краун-6..............................100
3
Диизопропиламин......................................100
Глава 2. ВОССТАНОВЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
КОМПЛЕКСНЫМИ ГИДРИДАМИ МЕТАЛЛОВ...............................103
2.1. Незамещенные алюмо- и борогидриды металлов..........104
2.2. Замещенные алюмо- и борогидриды металлов............107
2.3. Смешанные гидриды...................................112
2.4. Получение комплексных гидридов металлов. Выбор
растворителя ........................................... 115
2.5. Механизм восстановления.............................121
2.6. Восстановление важнейших классов органических соединений . 125
2.6.1. Альдегиды и кетоны............................125
2.6.2. Хлороангидриды и ангидриды кислот.............134
2.6.3. Сложные эфиры и лактоны.......................135
2.6.4. Амиды и лактамы...............................136
2.6.5. Нитрилы.......................................139
2.6.6. Карбоновые кислоты и их соли..................140
2.6.7. Нитро-, нитрозосоединения, оксимы и имины.....141
2.6.8. Эпоксиды и спирты.............................143
2.6.9. Галогенопроизводные...........................144
2.7. Промышленное применение комплексных гидридов металлов . . 145
2.8. Экспериментальные работы............................147
Получение комплексных гидридов металлов..............148
1-Фенилэтанол........................................151
Изоборнеол...........................................151
Фталиловый спирт.....................................153
N-Этиланилин.........................................155
4-Нитробензальдегид..................................157
Коричный спирт.......................................160
Циклопентанол (способ 2).............................163
4-Метилпентен-3-ол-2.................................163
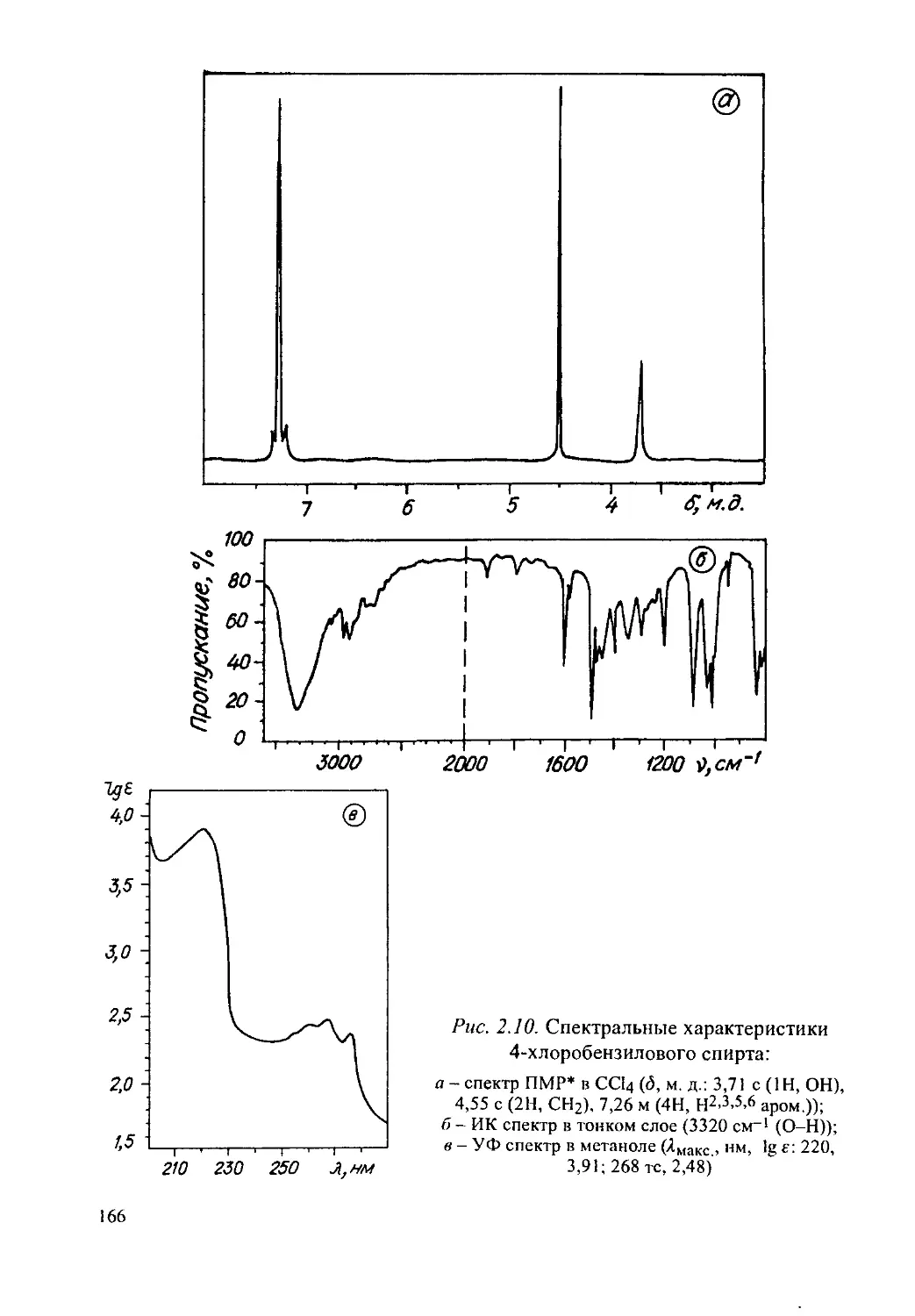
4-Хлоробензиловый спирт..............................164
Глава 3. МЕТОДЫ СИНТЕЗА С ПРИМЕНЕНИЕМ ЖИДКОГО АММИАКА
КАК РАСТВОРИТЕЛЯ..............................................167
3.1. Свойства жидкого аммиака............................167
3.2. Восстановление важнейших классов соединений.........170
3.2.1. Ароматические системы.........................171
3.2.2. Ацетилены и этилены...........................179
3.2.3. а,/З-Непредельныс карбонилсодержащие соединения . . . 181
3.2.4. Реакция восстановительного расщепления простых связей. 186
3.3. Реакции алкилирования...............................187
3.3.1. Алкилирование терминальных алкинов............188
3.3.2. Алкилирование активных метиленовых соединений .... 191
3.4. Реакции нуклеофильного замещения в ароматическом ряду . . . 195
3.5. Реакции элиминирования..............................199
4
3.6. Реакции присоединения..................................200
3.7. Промышленное применение жидкого аммиака................201
3.8. Экспериментальные работы...............................202
Получение амидов натрия и лития в жидком аммиаке........204
2,3-Дифенилпропионовая кислота..........................204
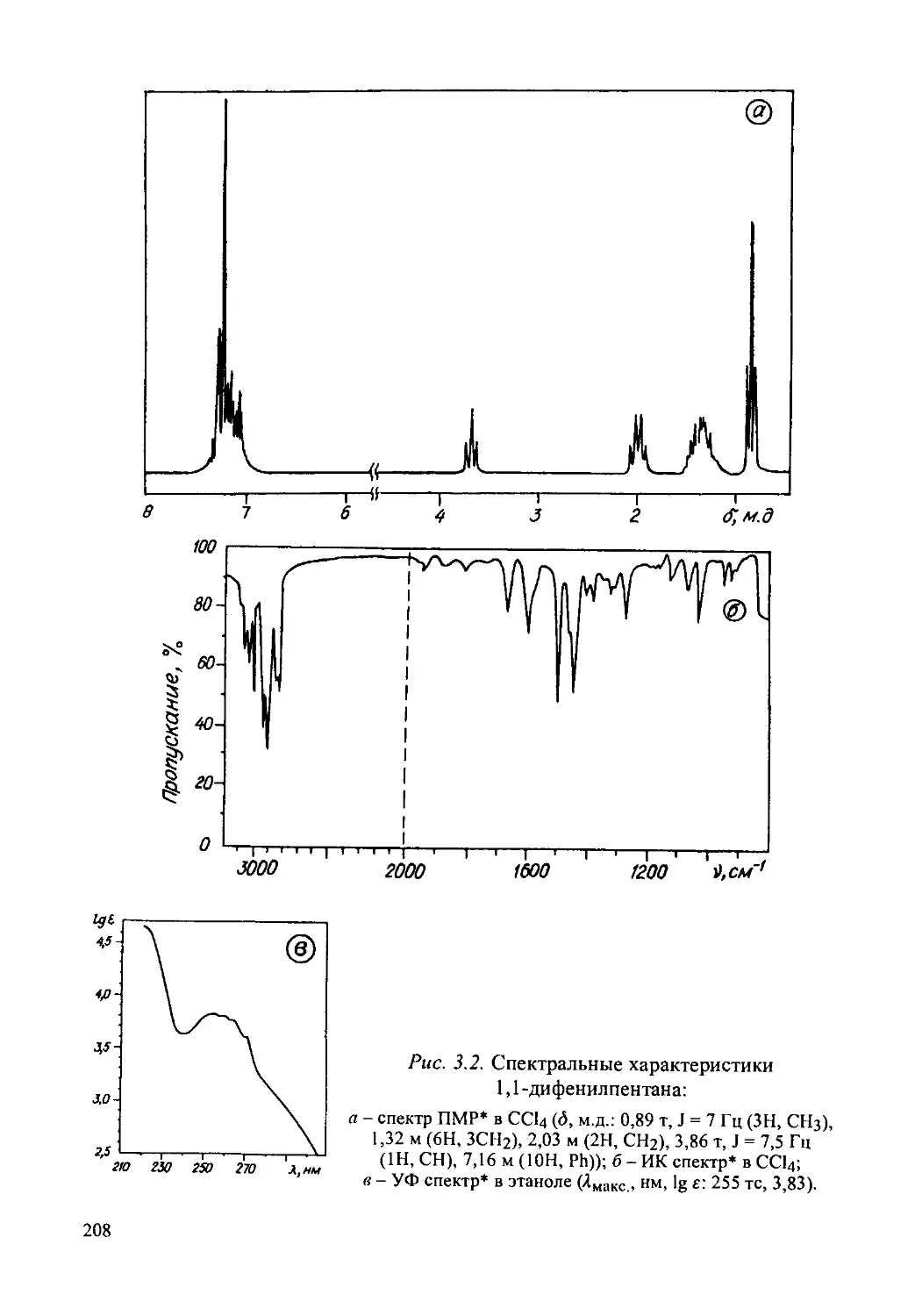
1,1-Дифенилпентан.......................................205
1,1,2-Трифенилэтан......................................207
Гептин-2-ол-1...........................................209
Гексин-1................................................210
1 -Этинилциклогексанол..................................212
2-Нитроанизол...........................................214
4-Нитрофенетол..........................................215
5,8-Дигидро-1-нафтол....................................217
Глава 4. ЛИТИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ В ОРГАНИЧЕСКОМ
СИНТЕЗЕ..........................................................220
4.1. Строение литийорганических соединений..................221
4.2. Методы синтеза литийорганических соединений............225
4.2.1. Замещение галогена литием („прямой" синтез)......225
4.2.2. Реакция металлирования...........................227
4.2.3. Замещение галогена литием с помощью
литийорганического соединения...........................233
4.2.4. Получение из других металлорганических соединений. . . 236
4.3. Литийорганические соединения в органическом синтезе....237
4.3.1. Присоединение к кратным связям углерод-углерод...238
4.3.2. Присоединение к кратным связям углерод-азот......240
4.3.3. Присоединение к кратным связям углерод-кислород . . . . 240
4.3.4. Реакции замещения при атоме углерода.............245
4.3.5. Протонирование литийорганических соединений......250
4.3.6. Получение реакционноспособных интермедиатов......251
4.3.7. Синтез других металлорганических соединений......252
4.3.8. Разрыв связи кислород-углерод простых эфиров.....254
4.4. Промышленное применение литийорганических соединений . . 256
4.5. Экспериментальные работы...............................257
Получение литийорганических реагентов...................258
1,1,3-Трифенилпропен-2-ол-1 (дифенилстирилметанол)......262
1,1,2-Трифенилэтанол-1..................................263
Дифенил(5-бромо-2-метоксифенил)метанол..................263
2,6-Диметоксибензальдегид...............................264
2,6-Диметокситолуол.....................................268
Дифенил(2,6-диметоксифенил)метанол......................270
4-Оксиацетофенон........................................272
Транс-4-нитро-4'-метоксистильбен........................275
М,М-Диметиламид-1-оксициклогексанкарбоиовой кислоты. . . . 276
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА.........................................280
5
СПИСОК ПРИНЯТЫХ СОКРАЩЕНИЙ
ГМФА Глим ДАБЦО Диглим ДИПАЛ ДМСО ДМФА ТГФ ТМЭДА - гексаметилфосфотриамид (гексаметапол) - диметиловый эфир этиленгликоля - 1,4-диазобицикло[2,2,2]октан - диметиловый эфир диэтиленгликоля - диизопропиламид лития - диметилсульфоксид - Ы,Ы-диметилформамид - тетрагидрофуран - N,N,N',N-тетраметил-1,2-этилендиамин (тетраметилэтандиамин)
Эфир Ас А1к Аг В: - диэтиловый эфир - ацетил - алкил - арил - основание
Ви втор-Ви - бутил - втор-бутл
трет-Ви - трет-бутнл
Et - этил
НА Hal М Me Ph Рг г/зо-Рг Ру Ts - протонная кислота - галоген - металл - метил - фенил - пропил - изопропил - пиридин - л-толуолсульфонил (тозил)
6
ПРЕДИСЛОВИЕ
Учебные планы химических факультетов университетов и от-
дельных специализаций химико-технологических вузов включают
курс современных методов органического синтеза. Предлагаемое
пособие призвано частично восполнить недостаток учебной лите-
ратуры по этому курсу, особенно остро ощущающийся в процессе
экспериментального освоения студентами методов синтеза. По-
следнее практическое руководство по современным методам орга-
нического синтеза на русском языке вышло в издательстве Ленин-
градского (ныне Санкт-Петербургского) университета более 20 лет
тому назад.
В четырех главах настоящего учебного пособия рассмотрены
методы каталитического гидрирования органических соединений и
их восстановления комплексными гидридами металлов, примене-
ние жидкого аммиака в органическом синтезе и реакции литийорга-
нических соединений. Каждая глава содержит обзор литературы, в
котором обсуждаются область применения метода, его важнейшие
особенности, механизмы реакций, экспериментальные условия их
реализации и зависимость реакционной способности реагентов от
строения. Обзоры тематически связаны с соответствующими
разделами лекционного курса и могут использоваться при их уг-
лубленном изучении, что существенно, так как по большинству из
рассмотренных методов в отечественной учебной литературе по-
добных обзоров нет. Перечень основных литературных источников,
использованных при написании книги, по-видимому, будет полезен
в большей степени преподавателям, чем студентам, поскольку
в него включены преимущественно труднодоступные издания и
специальные монографии, малопригодные в качестве учебного ма-
териала.
Помимо общего обсуждения синтетического метода, каждая
глава включает описание иллюстрирующих его возможности конк-
7
ретных методик получения 10-12 соединений. Они заимствованы
главным образом из оригинальных научных публикаций и сбор-
ников за разные годы, проверены нами и хорошо воспроизводимы.
В общеизвестных лабораторных практикумах они, как правило, не
встречаются. Предполагается, что при выполнении синтезов сту-
денты приобретают навыки работы с ацетиленом, жидким аммиа-
ком, водородом и реакционноспособными соединениями (гидриды
металлов, литийорганические соединения), применение которых
сопряжено с повышенной опасностью и требует определенного
профессионального мастерства и строгого следования правилам
безопасной работы. Вместе с тем из состава реагентов, используе-
мых в практических работах, по возможности исключены токсич-
ные вещества, в первую очередь токсичные растворители (метанол,
бензол, тетрахлорид углерода). В описаниях всех синтезов акцен-
тируется внимание на необходимых мерах предосторожности.
Осуществляя рекомендованные синтезы, студенты получают
возможность приобрести дополнительный опыт в части выделения,
разделения, очистки и идентификации органических веществ, а
именно, экстракции, перегонки, ректификации, кристаллизации,
возгонки, адсорбционной и газожидкостной хроматографии. В ме-
тодиках приведены данные тонкослойной хроматографии синтези-
руемых соединений на пластинках Silufol UV 254, что позволяет
максимально просто проводить первичную идентификацию про-
дуктов, устанавливать их индивидуальность и во многих случаях
следить за ходом реакций. Полная идентификация полученных ве-
ществ достигается после определения их физико-химических
констант и спектральных характеристик (ПМР, ИК и УФ спектров),
которые для сопоставления приводятся в пособии. Спектры, поме-
ченные звездочкой, записаны нами, остальные взяты из „Sadt-
ler Standart Spektra“ (Sadtler Research Laboratories. Philadelphia).
Спектры ПМР записаны M.M. Шакировым на приборе WP-200
фирмы Bruker (частота 200,13 МГц) с применением гексаметилди-
силоксана в качестве внутреннего стандарта (0,04 м. д.). ИК и УФ
спектры сняты на спектрометрах UR-20 и СФ-16.
Глава 1 написана профессором М. С. Шварцбергом, осталь-
ные главы - доцентом И. Б. Репинской.
Авторы будут благодарны за все замечания, советы и рекомен-
дации и с удовольствием учтут их в дальнейшей работе над посо-
бием.
8
Глава 1. ГИДРИРОВАНИЕ
1.1. ВОССТАНОВЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Реакции восстановления - это химические превращения, в ре-
зультате которых понижается степень окисления атома или сово-
купности атомов, представляющих реакционный центр исходного
соединения. Чтобы это определение было классификационно чет-
ким в приложении к органическим соединениям, при расчете степе-
ни окисления ковалентно связанных атомов каждую поделенную
пару электронов полностью относят к более электроотрицательно-
му из атомов-партнеров, а пару электронов связи между атомами
одного и того же элемента делят между ними. Остающийся на атоме
целочисленный заряд условно считают его степенью окисления (х),
пренебрегая количественными различиями в электронной плот-
ности на этом атоме (по существу, в его степени окисления) при свя-
зывании его с электроноакцепторными или электронодонорными
партнерами разной силы (рис. 1.1).
В рамках такой схемы образование связи атома углерода с лю-
бым более электроотрицательным атомом сопряжено с „потерей"
одного электрона, т. е. с
увеличением его степени
окисления на единицу и,
напротив, образование
связи с менее электроот-
рицательным атомом - с
„приобретением" элек-
трона, т. е. с уменьшени-
ем степени окисления на
Рис. 1.1. Степень окисления атомов в молекуле
трихлорэтилена
единицу, о- и тг-связи
между атомами углерода
9
не изменяют степень окисления этих атомов. Если z и h - число свя-
зей атома углерода соответственно с атомами относительно элект-
роотрицательных и электроположительных элементов, его степень
окисления легко определяется по формуле х = z - h.
В органической химии в соответствии с общим определением
реакциями восстановления принято называть реакции, протекаю-
щие с уменьшением суммарной степени окисления атомов углерода
или гетероатомов реакционного центра субстрата. Органические
соединения восстанавливаются в процессах присоединения по
кратным связям водорода, металлов, гидридов металлов и гидридов
электроположительных металлоидов (бора, кремния, фосфора), за-
мещения электроотрицательного гетероатома, гетероатомной или
углеродной группировки на атом водорода или металла, элимини-
рования электроотрицательных атомов или гетероатомных групп,
связанных с атомами реакционного центра через электроотрица-
тельные атомы, и сочетания с предшествующим (или одновремен-
ным) разрывом связей между атомами углерода или гетероатомами
и атомами более электроотрицательных элементов. Отдельные при-
меры таких реакций приведены ниже.
,0 Н р
И Li NH 1 11
СН3(СН2)7СнС(СН2)7С ’ СН3(СН2)7С = С(СН2)7С
ОН н ОН
С = ССМе2
ОН
_____ н2
Ru— А120р изо-РгОН,
50 °C, 3,5-4 атм
С = ССМе2
ОН
100 %
10
н н
\ /
Ме^сос-с^с^С00Ме
। I
н н
н н
н2 \ /
----------i► С=с
[Cr(PhCOOMe)(CO)3], / \
С5Н|2,160 °C, 30 атм Et СН2СООМе
> 90 %
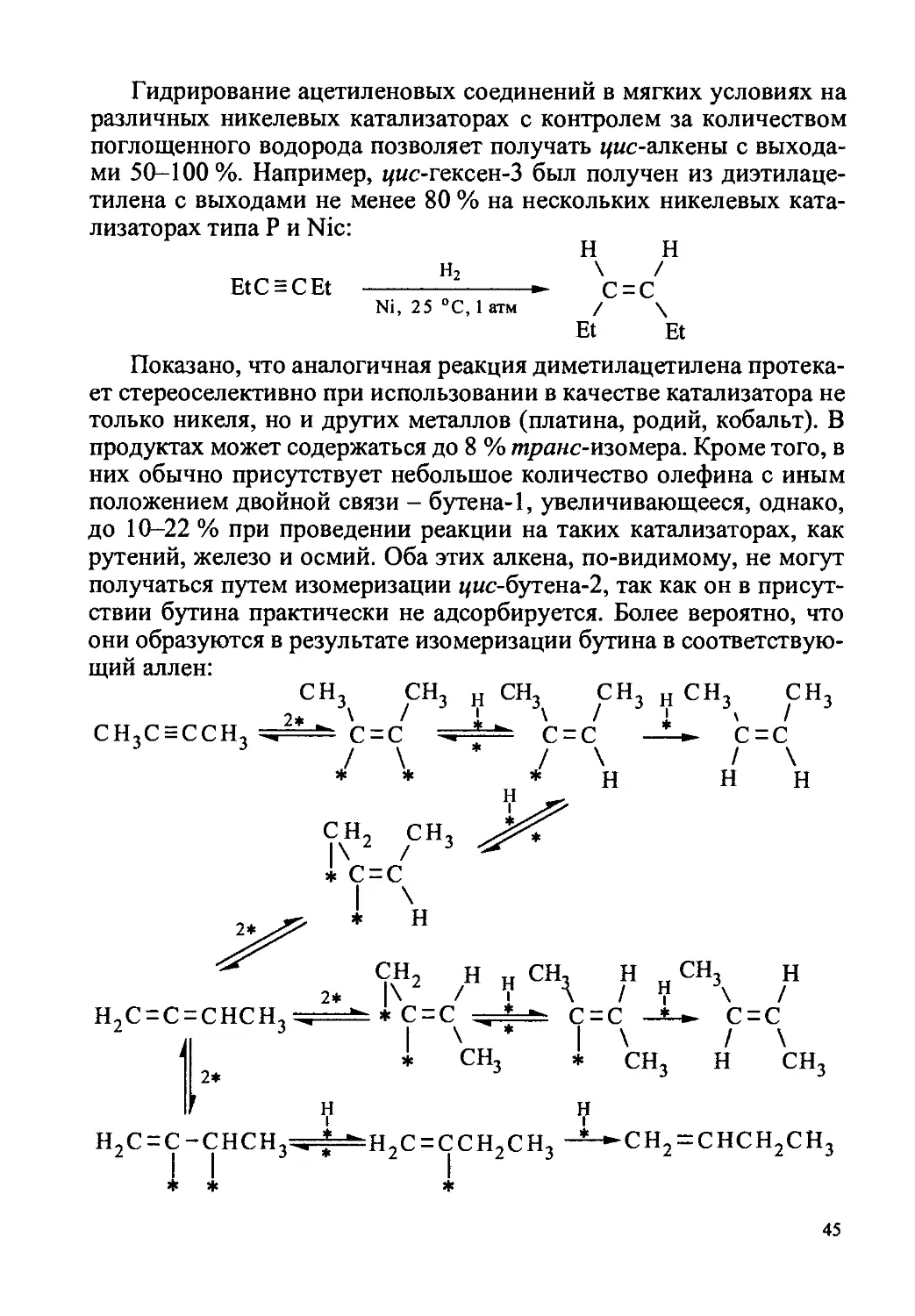
EtC = CEt -..2- 6»
Mg
ТГФ
Et^H АсОНг
EtC^JyB
84 %
MgBr
95 %
CONHCH,OH
2 Н2
Et Et
\ /
С = С
/ \
н н
80 %
-100 %
CONHMe
93 %
Pd-C, CFjCOOH, СНС13,20°C, 1 атм
Ph Ph
\ /
c-c
h2nz
N N
HgO
C,H,
О о
nh2
PhC = CPh
80 %
11
о
о
> 80 %
70 %
93 %
Реакции, в которых степень окисления одного из атомов реак-
ционного центра понижается, а другого на столько же повышается,
и которые протекают в отсутствие каких-либо восстанавливающих
или окисляющих реагентов, например реакции гидратации или де-
гидратации, относят к изогипсическим, т. е. не изменяющим сте-
пень окисления молекулы (от греческих изо - тот же самый и гип-
сос - уровень):
н,о
ВиС = СН -------------- ВиСОМе 80 %
HgSO4, H2so4
Ме,ССН,СОМе ------„ ► Ме2С = СНСОМе 65 %
2 I 2 12, 130 °C 2
ОН
CI7H„CH=NOH НС| вон; gre- Ci7HJ5C = N 95%
Изогипсическими принято считать также реакции, в которых обра-
зуются связи между углеродными атомами или гетероатомами ор-
ганических субстрата и реагента с сохранением суммарной степени
окисления этих атомов:
О
II
PhCMe + EtMgBr ---------►
SO °C
СН3СОСН3
Ba(OH)2
Et Et
' HO I
PhCMe ——PhCMe 70 %
I H+ I
OMgBr OH
(CH3)2CCH2COCH3 68-74 %
OH
12
-н2о,~ю °C
82 %
NH,CONHMe --------------- NH,CON-NO 80 %
H2so4,-ю°с I
Me
Методы восстановления обычно разделяют на две группы: вос-
становление молекулярным водородом в присутствии катализато-
ров - гидрирование и восстановление прочими неорганическими и
органическими реагентами - „химическое" восстановление. Мето-
ды второй группы весьма разнообразны и различаются между собой
природой восстановителя, экспериментальными условиями и меха-
низмом реакций, областью применения. Каталитическое гидриро-
вание, напротив, представляет, по существу, единый синтетический
метод, базирующийся на применении простейшего и универсаль-
ного восстановителя, ограниченного круга катализаторов и отли-
чающийся значительной общностью техники эксперимента при
широком диапазоне изменений отдельных параметров химического
процесса.
1.2. ГИДРИРОВАНИЕ КАК МЕТОД ВОССТАНОВЛЕНИЯ
Гидрирование используется для решения синтетических и,
реже, аналитических задач и реализуется в микро-, полумикро-,
стандартном препаративном и промышленном масштабах. При-
соединяя этим методом водород по кратным связям, можно исчер-
пывающе или частично восстанавливать различные углеродные и
гетероатомные ненасыщенные фрагменты молекулы, такие как аце-
тиленовая и этиленовая связи, карбонильная и нитрильная группы,
ароматическое и гетероароматическое ядра и др., и тем самым за-
менять одни функциональные группы другими или удалять нена-
сыщенные реакционные центры. Во многих случаях в условиях
13
гидрирования протекает гидрогенолиз, т. е. полный разрыв связи
между атомами реакционного центра, например С-О, C-S, C-N,
С-С1, С-С, N-N и т. д., с присоединением водорода по месту рас-
щепления.
Гидрогенолиз связи С-С1 лежит в основе одного из способов по-
лучения альдегидов - реакции Розенмунда:
СОС1
Pd— BaSO4, хинолин-S,
Ме,С.Н., 150 °C, 1 атм
2 6 4’ ’
сно
74-81 %
Применение палладиевого катализатора (Pd-BaSO4, Pd-C),
обычно частично дезактивированного небольшим количеством
сернистого соединения, и мягкие условия гидрирования позволяют
практически избежать дальнейшего восстановления образующего-
ся альдегида.
Способность связи С-галоген к гидрогенолизу используется
при удалении атомов галогена из ароматических циклов. Так,
4,5-дииодо-1-метил- и 2,4,5-трииодо-1-метилимидазол легко вос-
станавливаются в 4-иод-1-метилимидазол рассчитанным количест-
вом водорода при атмосферном давлении и температуре 20 °C на
скелетном никелевом катализаторе в присутствии основания:
I I
I
I
Me
Н2
NiCK, СаО, МеОН
Me Me
63-69 %
Направленно получить тот же моноиодид прямым иодированием
1 -метилимидазола не удается, так как скорости вступления первого,
второго и третьего атомов иода в гетероцикл близки между собой.
. Легко подвергаются гидрогенолизу связи С-гетероатом (O,N) в
бензильном положении. Этим определяется возможность приме-
нения бензильной и родственных групп как защитных при синтезе
полифункциональных соединений. Снятие такой защиты достига-
ется гидрированием соединения на палладиевом катализаторе при
температуре 20 °C и атмосферном давлении:
14
~ 100 %
Me
\
mpem-Bu-SiO
/
Me
Гидрирование как аналитический метод сыграло заметную роль
в истории химии при установлении строения непредельных орга-
нических соединений. В настоящее время оно иногда использует-
ся для количественного определения степени ненасыщенности
веществ, особенно смесей (например жиров), непредельных карбо-
новых кислот, ацетиленовых соединений и др. Степень ненасыщен-
ности при этом обычно характеризуется так называемым числом
гидрирования - массой водорода в граммах, необходимой для
гидрирования 10 кг вещества.
Особенность гидрирования как синтетического метода заклю-
чается в применении для восстановления веществ разных классов
одного и того же восстановителя и общих или химически родст-
венных катализаторов. Эта особенность ограничивает возможность
избирательного гидрирования соединений, если необходимо, на-
пример, обеспечить заданную глубину восстановления группы,
способной к более глубокому восстановлению, или прогидрировать
одну из нескольких восстанавливающихся групп, не затрагивая
остальных. Тем не менее возникающие затруднения во многих слу-
чаях успешно преодолеваются подбором катализатора и условий
реакции:
Me Me
\ н, \
Ме-СС = СН------- ркт'дТ;----Ме~ССН = СН, 94 %
у Pd— CaCOj, Ро(ОАс)2, у 2
цд хинолин, петролейный эфир, uq
10 °C, 1 атм
15
Гидрирование при атмосферном давлении термодинамически
разрешено при относительно невысоких температурах. Ниже
100 °C присоединение водорода по кратным связям необратимо и
является сильно экзотермической реакцией. С повышением темпе-
ратуры начинает играть роль процесс дегидрирования насыщенных
групп, и, таким образом, гидрирование становится обратимым. При
высоких температурах преобладает обратная реакция дегидрирова-
ния. Так, алкины и алкены могут гидрироваться в алканы, а бензол в
циклогексан при умеренных температурах и атмосферном давле-
нии, поскольку в этих условиях насыщенные углеводороды ста-
бильнее соответствующих ненасыщенных, хотя и те и другие (при
числе атомов углерода более двух) термодинамически неустойчивы
по отношению к распаду на углерод и водород. При достаточно вы-
соких температурах относительная стабильность этих соединений
изменяется на противоположную и гидрирование оказывается не-
возможным.
Положение равновесия процесса гидрирования - дегидрирова-
ния зависит от давления водорода. Верхняя температурная граница
„выгодных" для гидрирования условий с повышением давления
смещается в сторону высоких температур. Например, при гидриро-
вании октилового эфира каприловой кислоты при 260 °C:
С7Н|5СООС8Н,7 + 2Н2 2С8Н|7ОН
СиО • Сг2О3
содержание сложного эфира в реакционной смеси по достижении
равновесия с увеличением давления водорода от 10 до 85 и 280 атм
падает с 80 % соответственно до 10 и 1 %.
16
Свободная энергия гидрирования бензола
С6н6 + зн2 с6н12
при температуре 271 °C с повышением давления каждого из реа-
гентов от 1 до 200 атм изменяется от 0 до -71,2 Дж/моль
(-17000 ккал/моль).
Практически в зависимости от активности катализатора и строе-
ния субстрата гидрирование проводят при температурах от -100 до
500 °C и давлениях от 0,01 до 400 атмосфер и более, причем высо-
кие температуры применяются, как правило, в сочетании с высоким
давлением.
Единство метода гидрирования, о котором говорилось выше,
отнюдь не абсолютно и потому в известной мере условно. Сущест-
вуют две разновидности каталитического восстановления водоро-
дом - гетерогенное и гомогенное гидрирование. При гетерогенном
гидрировании субстрат находится в жидкой или газовой фазе, а
реакция протекает на поверхности твердого катализатора. Катали-
заторами являются переходные металлы в мелкодисперсном или
пористом состоянии, а также оксиды или сульфиды металлов.
При гомогенном гидрировании активация молекулярного водо-
рода и реакция происходят в растворе субстрата и катализатора -
соединения переходного металла. Важнейший из таких катализато-
ров - хлоротрис(трифенилфосфин)родий КЬС1(РРЬз)з (катализатор
Уилкинсона). Комплексы переходных металлов, по-видимому,
повторяют каталитические свойства этих металлов, но действуют в
гомогенной системе, что значительно облегчает исследование ме-
ханизма катализа.
Гетерогенное гидрирование традиционно и, являясь хорошей
основой экологически чистой технологии, широко применяется не
только в лабораторной практике, но и в промышленности (гидро-
генизация жиров, получение многоатомных спиртов из полисаха-
ридов, анилина из нитробензола, циклогексанона из фенола, про-
изводство бензола и нафталина гидродеалкилированием, гидро-
очистка и гидрообессеривание нефтяных фракций и т. д.). Далее
рассматривается только этот вариант метода*.
* О каталитическом гомогенном гидрировании см. Макквилпин Ф.Дж. Гомо-
генное гидрирование в органической химии / Пер. с англ. М.: Химия, 1980. 160 с.
17
1.3. КАТАЛИЗАТОРЫ ГИДРИРОВАНИЯ
Важнейшими катализаторами реакций гидрирования являются
элементы VIII группы периодической системы: платиновые метал-
лы Pt, Pd и менее употребительные Rh, Ru и Os, а также Ni, Fe и Со.
Используются, особенно в промышленности, оксидные, сульфид-
ные, в том числе и многокомпонентные, катализаторы (СиО • СггОз,
ZnO • СГ2О3; FeS, CoS, M0S2, Re2S? и др.). Все эти катализаторы не
отличаются большой специфичностью, и на них, за редким исклю-
чением, можно восстанавливать самые разные органические соеди-
нения.
К числу наиболее активных относятся платиновые катализа-
торы, позволяющие в мягких условиях гидрировать большинство
функциональных групп. Не восстанавливаются на этих катализато-
рах карбоновые кислоты, их амиды и эфиры.
Эффективным и удобным в работе является катализатор на
основе оксида платины (IV) - катализатор Адамса. Его получают
осторожным нагреванием до 400-500 °C хлороплатиновой кисло-
ты H2PtCI6 или хлорида платины(^) PtCU с избытком нитрата
натрия:
PtCl4 + 4NaNO3 —► PtO2 + 4NO2 + 4 NaCl +О2
После растворения плава и тщательной промывки водой катали-
затор, представляющий собой коричневый порошок РЮг, высуши-
вают над концентрированной серной кислотой или в вакууме.
В реакционной среде оксид платины (IV) восстанавливается
водородом до металла, который, образуясь в виде тонкой диспер-
сии, действует как катализатор. Для восстановления оксида, дис-
пергированного в растворителе, требуется несколько секунд, но в
присутствии субстрата продолжительность восстановления возрас-
тает и может достигать 10-15 мин.
Наряду с катализатором Адамса иногда используются (в настоя-
щее время крайне редко) металлические платиновые катализаторы,
приготавливаемые восстановлением солей платины. Так, из хлоро-
платиновой кислоты, применяя в качестве восстановителя форма-
лин в щелочной среде, получают мелкодисперсную платиновую
чернь:
H2PtCl6 + 2СН2О + 8КОН —► Pt + 6КС1 + 2НСООК + 6Н2О
18
Восстановлением той же кислоты слабощелочным раствором
гидразина в присутствии защитных коллоидов (белковые вещества,
их гидролизаты, поливиниловый спирт и т. п.) можно приготовить
коллоидную платину.
Свободные металлические платиновые катализаторы не очень
удобны в обращении, требуют специальных предосторожностей
при хранении, в процессе получения трудно воспроизводимы по
активности. Лучше в этих отношениях платиновые катализаторы на
носителях, или поверхностные катализаторы. Применяют разнооб-
разные носители: активированный уголь, оксид алюминия, сили-
кагель, сульфаты и карбонаты бария, кальция и других металлов,
асбест, пемзу, кизельгур и др. Обычно при приготовлении поверх-
ностных платиновых катализаторов металл осаждают на носитель
из раствора соли, в котором суспендирован или которым пропитан
носитель (например активированный уголь или асбест соответст-
венно). Как и при получении платиновой черни, соль часто восста-
навливают формалином. Весьма активен катализатор Pt-C, при-
готовленный непосредственно перед гидрированием путем вос-
становления хлороплатиновой кислоты борогидридом натрия в
этаноле в присутствии активированного угля.
Роль инертного носителя состоит в увеличении поверхности
контакта металла или другого активного компонента катализатора с
реагирующими веществами. Поэтому удельная поверхность самого
носителя и его структура влияют на активность катализатора. Кро-
ме того, его активность, селективность и стабильность нередко мо-
гут быть повышены добавлением небольшого количества других
металлов, солей, оксидов или минеральных кислот, называемых
промоторами (активаторами). Для платиновых катализаторов это
обычно соли платины, палладия, олова, железа, цинка или мине-
ральные кислоты. Так, промотирование катализатора Адамса хло-
ридами железа или олова (6,5-7 % по массе) увеличивает скорость
гидрирования валерианового альдегида в 8-10 раз:
Н2
Bu С НО pto2; FeC1^ (SnCl2); 25 °C, 1атм^ ВиСН2ОН
Считают, что действие промоторов в основном связано с изме-
нениями микрокристаллической структуры каталитически актив-
ного компонента, его химического состава и электронного строе-
ния, проявляющимися в изменении свойств его поверхности.
19
Платиновые и другие катализаторы на основе благородных ме-
таллов полностью или частично дезактивируются многими вещест-
вами - контактными, или каталитическими, ядами. Особенно они
чувствительны к соединениям двухвалентной серы (H2S, RSH, CS2,
тиофен и др.), мышьяка и фосфора. Отсюда высокие требования к
чистоте как реактивов, применяемых при получении этих катали-
заторов, так и всех компонентов реакционной системы гидриро-
вания (водород, восстанавливаемое соединение, растворитель).
Следует заметить, что в некоторых случаях одна и та же добавка к
катализатору может играть роль либо промотора, либо дезактивато-
ра в зависимости от ее количества и температуры реакции.
Методика приготовления существеннейшим образом отражает-
ся на свойствах не только платиновых, но и любых других катализа-
торов. Значение имеют чистота, концентрации, порядок и скорость
смешения реагентов, температура осаждения, промывки, сушки,
природа носителя и промоторов и т. д. Поэтому получение катали-
затора - тонкая и ответственная процедура, во многом определяю-
щая успех реализации запланированной каталитической реакции.
Разновидности и способы приготовления палладиевых катали-
заторов аналогичны описанным для платиновых. Широко употреб-
ляется в лабораториях палладий, нанесенный на карбонат кальция
(бария) или сульфат бария. Для получения этих катализаторов све-
жеосажденный карбонат кальция (сульфат бария) замешивают с
раствором хлорида палладия при температуре 50-60 °C и после
адсорбции соли палладия осадок отфильтровывают, тщательно
промывают водой и высушивают. Адсорбированная на поверхнос-
ти носителя соль восстанавливается до металлического палладия
водородом в процессе гидрирования. Палладиевая соль может быть
также восстановлена щелочным формалином или водородом сразу
после смешения ее раствора с горячей суспензией носителя в про-
цессе приготовления катализатора.
Поверхностный палладиевый катализатор отличается тем, что
достаточно легко поддается частичной дезактивации. Выше
(см. 1.2) был приведен пример использования такого частично от-
равленного сернистыми соединениями катализатора для повыше-
ния избирательности восстановления хлорангидридов карбоновых
кислот в альдегиды. Селективно гидрировать ацетилены в этилены
и кумулены в полиены обычно удается с помощью катализатора
Линдлара - палладия на карбонате кальция, дезактивированного
ацетатом свинца, например:
20
О Me
ОМе С1(СН2)6 СН2СН
I н2 \ / 2 \
С1(СН2)6С = ССН2СН pd_CaCo3> рь(ОАс)2Г /С-С\ ®Ме
I петролейный эфир, и и
ОМе 20°С, 2 атм П М
95%
Н2
Ph2C = C = C = CPh2 м_СаСОз.РЬ(ОАс)2;» Ph2C = CH-CH = CPh2
ТГФ , 20 °C, 1 атм 85%
В процессе приготовления этого катализатора палладий восста-
навливают водородом; дезактивацию проводят в водной суспензии
при температуре 20-100 °C.
Отдельную группу катализаторов составляют скелетные метал-
лические катализаторы, из которых наиболее известен скелетный
никелевый катализатор, или никель Ренея. Общий принцип получе-
ния таких катализаторов заключается в вымывании из двухкомпо-
нентного сплава подходящим реактивом неактивного компонента.
Так, никель Ренея чаще всего получают выщелачиванием алюми-
ния из измельченного никель-алюминиевого сплава (30-50 % Ni)
20-40 %-ным раствором гидроксида натрия. Вместо алюминиевого
сплава никеля можно использовать кремниевый, магниевый или
цинковый.
Во всех случаях никель получается в виде пирофорного крис-
таллического порошка, и поэтому его хранят под слоем спирта
или воды. Он обладает высокой пористостью и большой удельной
поверхностью. Свежеприготовленный катализатор содержит 25-
100 мл/г водорода, причем с потерей водорода активность ката-
лизатора снижается; известное влияние на каталитическую актив-
ность оказывает остающийся после выщелачивания алюминий. По-
этому, изменяя условия выщелачивания алюминия и промывки
катализатора, можно получать различающиеся по активности сорта
скелетного никелевого катализатора. Кроме того, катализатор про-
мотируется добавлением в сплав хрома, молибдена или кобальта в
количестве 3-10 % от массы никеля, введением солей благородных
металлов в ходе промывки катализатора или при гидрировании, а
также небольших количеств щелочи или органических оснований
при гидрировании. Например, продолжительность гидрирования
21
бензальдегида в бензиловый спирт при атмосферном давлении и
20 °C на одном из активных сортов скелетного никелевого катали-
затора с введением добавок триэтиламина и тетрахлорида платины
уменьшается более чем в 20 раз:
Промотор
Et3N
[Et3N]2[H2PtC16]
Et3N + PtC14
Время реакции, мин
44
30
22
2
В отдельных случаях гидрирование с успехом проводят на ката-
лизаторах, приготовленных выщелачиванием из никель-алюми-
ниевого сплава лишь небольшой части алюминия. Так, обработкой
сплава 3-10 %-ным едким натром, при которой вымывается около
8 % алюминия, получают катализатор Бага, отличающийся от обыч-
ного скелетного никелевого катализатора механической проч-
ностью (куски, зерна), способностью к реактивации при повторном
выщелачивании и потому более удобный для применения в уста-
новках гидрирования непрерывного действия.
Никель Ренея - активный и универсальный катализатор реакций
гидрирования. В зависимости от строения восстанавливаемого сое-
динения он используется при температурах от 20 до 200 °C и дав-
лениях до 250 атм и более. Повышенная по сравнению с другими
никелевыми катализаторами активность никеля Ренея проявляется
преимущественно при гидрировании в мягких условиях. Например,
ацетон легко гидрируется на скелетном никеле под давлением
1-3 атм уже при 20 °C, в то время как на поверхностном катализа-
торе - только при температуре около 100 °C:
н / Ni„_, го °с
сн3сосн3 ----------2----( сщснонсн,
3 ~ 1-3 атм \ 3 3
Ni—кизельгур, ~ 100 °C
При температуре выше 100 °C никель Ренея близок, а иногда и усту-
пает по активности поверхностным или высокодиспергированным
металлическим никелевым катализаторам (см. 1.6.3).
При высоких температурах и давлениях во избежание развития
бурной реакции скелетный катализатор рекомендуется применять в
22
количестве не более 5 % от массы субстрата. Если температура гид-
рирования близка к 200 °C, нельзя в качестве растворителя исполь-
зовать диоксан, так как в этих условиях его реакция с водородом и
никелем Ренея сопровождается взрывом.
Металлические никелевые, кобальтовые, медные и другие ката-
лизаторы приготавливают восстановленим солей (хлоридов, ацета-
тов и др.), оксидов, гидроксидов или основных карбонатов, терми-
ческим разложением солей (нитратов, карбонатов, формиатов,
оксалатов и т. п.) обычно с последующим восстановлением. Терми-
ческое разложение органических солей, например формиатов,
сопровождается восстановлением и приводит непосредственно к
получению металлов. Эффективный катализатор для гидрирования
под давлением (катализатор Сабатье) получают разложением
формиата никеля в токе диоксида углерода при температуре
200-250 °C:
(HCOO)2Ni*2H2O -------► Ni + ЗН2О + СО + СО2
СО + н2о Н2 + СО2
Катализаторы, приготовленные восстановлением хлорида ни-
келя алюминиевой или цинковой пылью и ацетата никеля гидридом
или борогидридом натрия (в последнем случае катализатор содер-
жит значительное количество NiB2), сравнимы по активности с
никелем Ренея и пригодны для гидрирования при комнатной темпе-
ратуре и атмосферном давлении.
Активными, относительно избирательными и малочувствитель-
ными к ядам высокотемпературными катализаторами гидрирова-
ния являются оксидные катализаторы состава СиО • Сг20з (СиСггОд),
2СиО • Сг20з (СиО • СиСггОд), ZnO • Сг20з (ZnCr2O4) и др. - так
называемые хромитные катализаторы Адкинса. В лабораторных ус-
ловиях их обычно получают термическим разложением хроматов
соответствующих металлов. Самый употребительный из этих ката-
лизаторов - хромит меди - можно приготовить осторожным нагре-
ванием основного хромата меди и аммония при 350-450 °C. Хромат
предварительно синтезируют в ходе обменной реакции между
нитратом меди и дихромовокислым аммонием в водно-аммиачном
растворе и высушивают при температуре 75-100 °C. Промотором
медно-хромитного катализатора является оксид бария, поэтому в
реакцию на этой стадии наряду с нитратом меди дополнительно
вводят некоторое количество нитрата бария.
23
2Cu(NO3)2 + (NH4)2Cr2O7 + 4NH4OH —
—* 2NH4Cu(OH)CrO4 + 4NH4NO3 + H20
2NH4Cu(OH)CrO4 -------► CuO’CuCr2O4 + N2 + 5H2O
Гидрирование на хромите меди протекает при температуре
100-300 °C и давлении 40-400 атм. В противоположность никелю
он значительно более активен при восстановлении углерод-кис-
лородных связей, чем при восстановлении ароматических циклов;
используется в промышленности как катализатор гидрирования
эфиров высших карбоновых кислот в спирты:
н,
EtOOC(CH2)4COOEt -------------2--------► НО(СН,)АОН
274 CuCr,O,, 250 С, 150атм V 2'6
2 90 %
95 % 97 %
Приведенные сведения об общих принципах и способах приго-
товления катализаторов на примерах наиболее часто употребляю-
щихся в лабораторной практике, позволяют составить представле-
ние о значении этой неотъемлемой составляющей каталитического
гидрирования как синтетического метода. Более полное описание
методов получения катализаторов и их рецептуры можно найти в
специальной литературе.
1.4. О МЕХАНИЗМЕ КАТАЛИТИЧЕСКОГО ГИДРИРОВАНИЯ
Любые химические превращения протекают лишь по термоди-
намически разрешенным направлениям, т. е. с уменьшением сво-
бодной энергии системы, что, в свою очередь, определяется только
начальным и конечным состояниями этой системы. Поэтому, если
катализатор вводится в реакционную систему и по окончании взаи-
модействия выводится из нее, он не изменяет свободной энергии
24
реакции, но может значительно влиять на ее скорость. Маршруты
одной и той же реакции, протекающей в присутствии и в отсутствие
катализатора, различны, и следовательно, различны эффективные
энергии активации того и другого процессов. Важно, что катализа-
тор обычно существенно ускоряет одно из нескольких термодина-
мически разрешенных превращений реагентов и, избирательно
направляя тем самым химический процесс, обеспечивает преиму-
щественное образование определенных продуктов из большого
числа принципиально возможных.
Гексен-1 при температуре 20-50 °C в атмосфере водорода впол-
не устойчив и не изменяется достаточно продолжительное время. В
то же время теоретически для него в подобных условиях возможен
ряд реакций: распад на углерод и водород, изомеризация, полиме-
ризация, присоединение водорода. В присутствии платинового или
скелетного никелевого катализатора гексен энергично и почти
количественно гидрируется в гексан:
н2
ВиСН=СН,----------*- СЛН.,
Pt(NiCK. )
Скорости же конкурентных реакций (распад, полимеризация) оста-
ются ничтожно малыми, и эти реакции практически не реализу-
ются.
В общих чертах представление о механизме гетерогенного гид-
рирования заключается в следующем. На начальных стадиях водо-
род и восстанавливаемый органический субстрат взаимодействуют
с катализатором, образуя на его поверхности неустойчивые соеди-
нения. При адсорбции водорода катализатором выделяется значи-
тельное количество теплоты (на никеле ЛЯ ~ -125 кДж/моль), и
вследствие взаимодействия водорода с металлом происходит либо
ослабление связи Н-Н, либо диссоциация молекулы на атомы (звез-
дочкой в уравнениях обозначены активные центры катализатора):
Ц---Ц
Н2 + 2* ;
* *
Ц
Н2 + 2* 2 ;
*
Образование такого типа „поверхностных14 гидридов приводит к
активации водорода, необходимой для дальнейшего развития про-
цесса восстановления.
25
Адсорбция органического субстрата также приводит к дефор-
мации его молекулы с разрыхлением и разрывом связей и к обра-
зованию связей с катализатором. Имеющие место химические взаи-
модействия сопровождаются выделением большого количества
теплоты (например, при адсорбции этилена на никеле - около
250 кДж/моль). Изучение нестойких соединений, образующихся
при взаимодействии органических веществ с катализатором на его
поверхности и неразрывно связанных с ним, является очень труд-
ной задачей. Тем не менее аналогии между гетерогенными и гомо-
генными, катализируемыми и некатализируемыми реакциями в
сочетании с данными физико-химических исследований позволили
представить вероятную структуру поверхностных соединений и,
используя эти представления, объяснить важнейшие эксперимен-
тально установленные химические особенности процесса гидриро-
вания.
Рассмотрим в качестве примера схему восстановления алкенов
и циклоалкенов. При хемосорбции этих соединений возможно об-
разование тг-адсорбированных (А) и (или) сг-адсорбированных
частиц (Б):
Не исключено, что эти формы находятся в равновесии.
Высказано предположение, что алкены, содержащие атомы
водорода в аллильном положении, могут адсорбироваться также с
разрывом СН-связи и образованием тг-аллильного поверхностного
комплекса (В):
В
При такой диссоциативной адсорбции вероятным предшест-
венником комплекса В является форма А или Б. Принципиальная
возможность разрыва СН-связи при адсорбции подтверждается
26
обнаружением этана в газовой фазе над никелевым катализатором,
взаимодействующим с этиленом в отсутствие водорода. Наиболее
просто и рационально этот факт объясняется диссоциативной хемо-
сорбцией этилена:
Н НС=СН Н
СН2 = СН2 + 4* - | || |
* * * *
Так или иначе, при адсорбции на катализаторе происходит актива-
ция водорода и восстанавливаемого органического субстрата, пре-
вращение их в поверхностные соединения, способные реагировать
друг с другом по одному или нескольким определенным маршру-
там, не требующим преодоления высокого активационного барье-
ра. Согласно предложенным механизмам, ненасыщенное соедине-
ние, связанное с поверхностью катализатора (А или Б), может
присоединять атом водорода и образовывать „полугидрированный“
интермедиат Г - моноадсорбированный радикал, напоминающий
металлорганическое соединение. Эта реакция, как и предшествую-
щие ей процессы, обратима, и интермедиат либо присоединяет еще
один атом водорода и превращается в насыщенный продукт, кото-
рый немедленно десорбируется, либо теряет атом водорода и вновь
переходит в адсорбированную форму исходного ненасыщенного
соединения или его изомера, отличающегося положением двойной
связи. В условиях низкотемпературного гидрирования стадия пре-
вращения полугидрированной формы Г в алкан практически не-
обратима:
Н
+ 2|
*
н
+ 2|
*
27
Очевидно, что эта схема допускает возможность ряда сопутст-
вующих гидрированию реакций: аллильного сдвига двойной связи,
цис-транс-изомерации, обмена атомов водорода или в присутствии
дейтерия каталитического дейтерирования олефина с образованием
продуктов, содержащих не только два, но и больше или меньше
двух атомов дейтерия в молекуле. Нужно заметить, что к тем же
выводам позволяет прийти и альтернативная схема механизма гид-
рирования, постулирующая образование тг-аллильного интерме-
диата В:
Перечисленные сопутствующие процессы действительно име-
ют место в полном соответствии с предложенными схемами. Так, во
время восстановления 1,2-диметилциклопентена на оксиде плати-
ны из реакционной смеси может быть выделен его 2,3-диметилизо-
мер, а при восстановлении пентена-1 на скелетном никеле - цис- и
/пранс-пентены-2. В зависимости от применяемого катализатора,
температуры и давления водорода изомеризация алкенов протекает
или быстрее, или медленнее, чем гидрирование. На никеле, являю-
щемся активным катализатором изомеризации, при температуре
60-130 °C миграция двойной связи в бутене-1 происходит в 2 раза
быстрее гидрирования, а г/ис-ш/?аис-изомеризация бутена-2 - гораз-
до быстрее миграции двойной связи. Наоборот, на платиновом
катализаторе при температуре 20 °C и атмосферном давлении гид-
рирование гексена происходит в 30 раз быстрее миграции двойной
связи. Обмен атома водорода алкена на атом водорода с поверх-
ности катализатора обнаруживается при гидрировании соединений,
меченных дейтерием, или при каталитическом восстановлении дей-
терием. Наиболее высока скорость такого обмена в аллильных по-
ложениях.
28
В приведенных схемах в неявном виде предполагается, что оба
атома водорода подходят к адсорбированной молекуле субстрата со
стороны катализатора. При этом молекула субстрата, по-видимому,
должна быть обращена к поверхности катализатора, как правило,
своей пространственно менее затрудненной стороной:
| катализатор
| катализатор |
сн-сн
| катализатор
Если интервал времени между присоединением первого и вто-
рого атомов водорода достаточно мал (кинетический контроль),
следует ожидать, что в результате гидрирования исключительно
или преимущественно образуются продукты i/wc-присоединения. В
противном случае между актами переноса первого и второго атомов
водорода к субстрату в полной мере разыгрываются процессы изо-
меризации и обмена водорода (термодинамический контроль) и
реакция теряет стереоселективность. Таким образом, когда гидри-
рование проводится в условиях, в которых кинетический контроль
является определяющим, пространственное строение продуктов
может быть предсказано путем конформационного анализа ад-
сорбции восстанавливаемого соединения. Это означает также, что
стереохимический результат гидрирования зависит от природы
катализатора и условий реакции.
При гидрировании 1,2-диметилциклогексена в адсорбирован-
ном на катализаторе соединении в форме Б (или А) метильные
группы обращены в сторону, противоположную поверхности ката-
лизатора, и в „полугидрированном“ интермедиате Г они находятся
в i/uc-положении. Очевидно, что, если скорость гидрогенолиза свя-
зи металл (катализатор)-углерод в Г много выше скорости переноса
к катализатору атома водорода связанной с этим углеродом ме-
тиленовой группы, продуктом реакции будет исключительно цис-
1,2-диметилциклогексан:
29
Стереохимический результат гидрирования не изменится и при
условии, что скорости этих процессов (гидрогенолиза и переноса
атома водорода) соизмеримы, но значительно превышают скорость
десорбции 2,3-диметилциклогексена, образующегося из адсорби-
рованной формы Б' (или А'). Если же его десорбция существенна,
т. е. изомеризация 1,2-диметилциклогексена протекает сравнитель-
но быстро, гидрирование должно приводить к получению смеси
цис- и тл/?анс-диметилциклогексанов. 2,3-Диметилциклогексен в
отличие от исходного 1,2-изомера способен адсорбироваться в двух
стерически различных формах Б' и Б", из которых вторая при
г/цс-присоединении (сии-присоединении) двух атомов водорода
дает транс-продукт.
Экспериментально найдено, что 1,2-диметилциклогексан, полу-
ченный гидрированием 1,2-диметилциклогексена на РЮд в уксус-
ной кислоте при температуре 25 °C и атмосферном давлении водо-
рода, состоит на 81,8 % из цис- и на 18,2 % из транс-изомера. Гид-
рирование в тех же условиях 2,3-диметилциклогексена приводит к
образованию продукта, содержащего 77,6 % цис- и 22,4 % транс-
изомера. С повышением давления элементарные реакции, в кото-
зо
рых участвует водород, должны относительно ускоряться, а обрат-
ные реакции, для которых нужны свободные активные центры на
поверхности катализатора, и следовательно, процессы изомериза-
ции исходного циклоалкена, наоборот, должны замедляться. В со-
ответствии с этим при гидрировании 1,2-диметилциклогексена под
давлением водорода 500 атм получается 1,2-диметилциклогексан с
95 %-ным содержанием г/мс-изомера, в то время как 2,3-диметил-
циклогексен дает продукт, еще более обогащенный транс-изоме-
ром (> 30 %), чем полученный при атмосферном давлении.
Стереохимический результат гидрирования во многих случаях
существенно зависит от природы катализатора. Так, при гидриро-
вании на Pd-C в уксусной кислоте при 25 °C и атмосферном
давлении 1,2- и 2,3-диметилциклогексен образуют одну и ту же
смесь геометрических изомеров 1,2-диметилциклогексана, в кото-
рой преобладает уже транс-изомер (73 %). Причины столь сильной
зависимости не вполне ясны. По-видимому, приходится принять,
что на палладиевом катализаторе значительно интенсифицируется
превращение 1,2-диметилциклогексена в 2,3-изомер, десорбция и
реадсорбция которого в процессе гидрирования и определяют, в
конечном счете, стереохимию реакции.
При прогнозировании или интерпретации стереохимического
результата гидрирования, не осложненного сопутствующими реак-
циями, решающее значение имеет корректность анализа прост-
ранственного строения, включая положение на поверхности ката-
лизатора, возможных адсорбированных форм субстрата.
Например, можно с достаточной уверенностью предсказать по-
ложение на катализаторе адсорбированной молекулы непредельной
карбоновой кислоты ряда пинена, имеющей жесткий бицикличес-
кий каркас, благодаря которому двойная связь пространственно эк-
ранирована с одной стороны значительно больше, чем с другой:
В согласии с этим в процессе гидрирования присоединение водо-
рода действительно происходит с более доступной стороны двой-
ной связи, и выход ожидаемого изомера насыщенной кислоты до-
стигает 90 %.
31
Аналогично бицикло[2,2,1]гепт-2-ен-2,3-дикарбоновая кислота
при гидрировании присоединяет водород с пространственно менее
затрудненной стороны, которой, очевидно, адсорбированная моле-
кула обращена к катализатору, и образует почти исключительно
насыщенную элдо-цис-кислоту:
На стереохимию гидрирования могут оказывать влияние функ-
циональные заместители в восстанавливаемом соединении, спо-
собные взаимодействовать непосредственно с катализатором или
носителем („якорный" эффект). Так, гидрирование двойной связи
в 1 -бензилоксикарбонил-4-пропилиденпирролидин-2-карбоновой
кислоте на платиновом катализаторе приводит в основном к обра-
зованию щю-изомера. Следовательно, эта непредельная кислота в
ходе реакции адсорбируется на катализаторе большей частью таким
образом, что ее карбоксильная группа обращена в сторону, проти-
воположную поверхности катализатора. Чтобы изменить положе-
ние молекулы кислоты на катализаторе при адсорбции и тем самым
стереонаправленпость гидрирования, используют в качестве носи-
теля катализатора не нейтральный пористый материал, как обычно,
а основную ионообменную смолу. Благодаря солеобразованию с
такой подложкой карбоксильная группа начинает играть роль свое-
го рода якоря, ориентирующего адсорбирующуюся молекулу кар-
боксильной группой вниз, к поверхности катализатора. Теперь уже
атом водорода, перемещаясь от катализатора к С4-атому гетероцик-
ла, образует с ним связь с той стороны, в которую обращена карбо-
ксильная группа, т. е. занимает по отношению к ней гщс-положение,
тогда как пропильный заместитель оказывается в тралс-поло-
жении:
CO2CH,Ph
CHEt
Н
Z
32
т/?а«с-4-Пропилпирролидин-2-карбоновая кислота является основ-
ным продуктом реакции в указанных условиях и получается с высо-
ким выходом.
1.5. ВЛИЯНИЕ СТРОЕНИЯ ВОССТАНАВЛИВАЕМОГО СОЕДИНЕНИЯ,
ПРИРОДЫ КАТАЛИЗАТОРА И УСЛОВИЙ РЕАКЦИИ
НА СЕЛЕКТИВНОСТЬ ГИДРИРОВАНИЯ
Восстанавливающиеся группы в органическом соединении гид-
рируются с различной легкостью. По этому признаку они могут
быть расположены в некоторой ориентировочной последователь-
ности (табл. 1.1). Чем ниже положение группы в этом ряду, тем
более жесткие условия требуются для ее гидрирования, так как
реакционная способность любой функциональной группы зависит
от структурного окружения, а при гидрировании еще и от природы
катализатора. Группы, находящиеся в начале ряда, как правило,
можно селективно гидрировать в присутствии находящихся в его
конце, но не наоборот. Например, ненасыщенные сложные эфиры
легко восстанавливаются на платиновом, палладиевом или скелет-
ном никелевом катализаторе в эфиры насыщенных кислот, но их
каталитическое гидрирование в ненасыщенные спирты удается
лишь в исключительных случаях. Чтобы осуществить это превра-
щение, чаще всего обращаются к „химическим" методам восста-
новления:
| CuCr,O,, 250 °C, 200 атм С 18 Н3 7 0Н
Н 1 Н 86 %
\ / Н,
С = С —-—Д C17H7SCOOBu
/ \ Nic,,(PtQ2) 17 3 5
CH3(CH2)7Z (CH2)7COOBu ~100 /о
Н Н
\ /
_________Нг_____С = С
ZnCr2O4, 300°С, СН3(СН2)/ \сн2)8он
200 атм j 1 1 4 278
63-65 %
о
Г, Н2 II
ft /Pd.'^. PhCH2CH2COM« -100%
PhCH=CHCOMe
\ LiAlH,
—- PhCH=CHCH2OH 73%
2 Заказ № 137
33
Избирательное гидрирование соседних групп ряда либо не уда-
ется вообще, либо достигается путем тщательного подбора условий
реакции. При этом иногда оказывается возможным найти условия
селективного восстановления любой из двух одновременно присут-
ствующих в соединении групп, занимающих разное положение в
сравнительном ряду активности.
Так, при гидрировании цитраля на катализаторе Адамса в пер-
вую очередь восстанавливается а,/3-С=С-связь, затем альдегидная
группа и, наконец, вторая С=С-связь. На том же катализаторе, но
модифицированном добавками сульфата железа (II) и ацетата цин-
ка, порядок восстановления изменяется, и сначала гидрируется аль-
дегидная группа, а уже после нее a,fl -С=С-связь:
Из приведенных примеров „обращения" активности восстанав-
ливающихся групп при гидрировании, очевидно, следует, что, хотя
их относительная реакционная способность в основном определя-
ется химическим строением, некоторую селективность действия
проявляет и катализатор, т. е. металл катализатора и модифицирую-
щие добавки (промоторы и дезактиваторы). Платиновые катализа-
торы, на которых при комнатной температуре и атмосферном или
слегка повышенном давлении гидрируются почти все типы органи-
ческих соединений, полностью неэффективны при восстановлении
карбоновых кислот и их эфиров в спирты. Хромит цинка, на ко-
тором при высокой температуре и давлении гидрируется алкокси-
карбонильная группа, неактивен при восстановлении легко гидри-
рующейся на других катализаторах С=С-связи. Поверхностные
осмиевые катализаторы, в отличие от скелетного никелевого ка-
тализатора или оксида платины, обеспечивают первоочередное
восстановление карбонильной группы в а,fl -ненасыщенных аль-
дегидах:
34
PhCH=CHCHO 6s_CJ00oC)30aTM" PhCH = CHCH2OH 95 %
Кислород- или азотсодержащие группы в бензильном поло-
жении особенно легко и, как правило, без затрагивания ароматичес-
кого ядра молекулы подвергаются гидрогенолизу на палладиевых
катализаторах. На родии, нанесенном на оксид алюминия или
уголь, проявляющем повышенную активность по отношению к аро-
матическим системам, с успехом удается восстанавливать бензоль-
ное кольцо с сохранением функциональных групп в а-положении
боковой цепи:
Селективность гидрирования полифункционального субстрата
или субстрата, в котором имеется группа, способная восстанавли-
ваться частично, как и результативность гидрирования любого
соединения вообще, зависит не только от его строения и природы
Таблица 1.1. Ориентировочная относительная реакционная способность
функциональных групп при каталитическом гидрировании
Функциональная группа (восстанавливаемое соединение) Продукт восстановления Функциональная группа (восстанавливаемое соединение) Продукт восстановления
RCOC1 RCHO, RCH2OH Полициклические Частично вое-
rno2 rnh2 ароматические • углеводороды становленные продукты
RC=CR RCH=CHR, RCH2CH2R RCOOR' RCH2OH(R'OH)
RCHO rch2oh RCONHR' RCH2NHR'
RCH=CHR rch2ch2r О
RCOR RCH(OH)R, RCH2R ROH RH
PhCH2OR PhCHj(ROH) RCOOH RCH2OH
RC=N RCH2NH2 RCOO-Na+ -
35
катализатора, но и от условий реакции: продолжительности, темпе-
ратуры, давления, растворителя, количества катализатора.
Температура обычно влияет на скорость гидрирования меньше,
чем на скорость других реакций. Так, повышение температуры с
50 °C до 100 °C вызывает лишь 4-кратное увеличение скорости гид-
рирования сложных эфиров на скелетном никелевом катализаторе.
Вместе с тем избирательность восстановления с повышением тем-
пературы падает, и максимальная региоселективность достигается
при возможно более низкой температуре. Например, в 1-фенил-
ундека-1,3-диен-5-оне на никеле Ренея под давлением водорода
100 атм гидрируются при температуре 40 °C практически только
сопряженная диеновая группировка, при 130 °C - эта группировка и
карбонильная функция, при 260 °C - все восстанавливающиеся
структуры, включая бензольный цикл; гидрогенолиз С-0-связи не
происходит:
Ph(CH = CH)2CC6H13
н2
NiCK , AcOEt,
100 атм
7^* Ph(CH2)4COC6H13
он
7^7^ Ph(CH2)4CHC6H13
— Q-(CH2)4CHC6H13
На том же катализаторе при температуре 50 °C этилбензоат
может быть селективно прогидрирован в гексагидробензоат. Одна-
ко при более высокой температуре избирательность реакции теря-
ется, и в результате’восстановления наряду с эфиром образуются
бензиловый спирт и толуол:
PhCOOEt
Н;
Nicl[ ,100 атм
50 °C
>50 °C
С6НИСООЕ1
C6HnCOOEt + PhMe +
+ PhCH2OH
Осуществление частичного гидрирования рассчитанным коли-
чеством водорода при температурах, близких к нижней темпера-
турной границе реакции, в целом ряде случаев обеспечивает доста-
точную селективность восстановления:
36
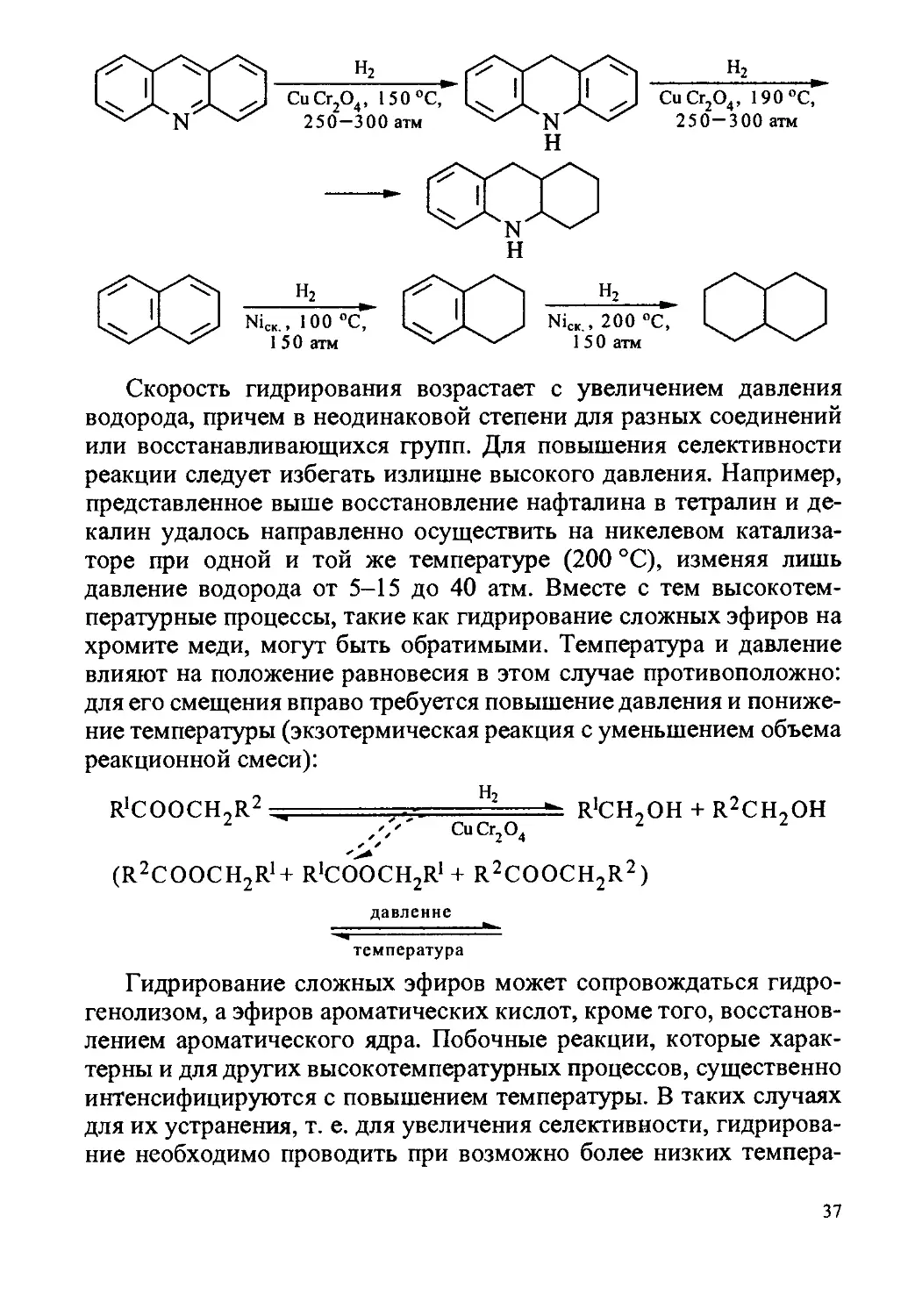
Скорость гидрирования возрастает с увеличением давления
водорода, причем в неодинаковой степени для разных соединений
или восстанавливающихся групп. Для повышения селективности
реакции следует избегать излишне высокого давления. Например,
представленное выше восстановление нафталина в тетралин и де-
калин удалось направленно осуществить на никелевом катализа-
торе при одной и той же температуре (200 °C), изменяя лишь
давление водорода от 5-15 до 40 атм. Вместе с тем высокотем-
пературные процессы, такие как гидрирование сложных эфиров на
хромите меди, могут быть обратимыми. Температура и давление
влияют на положение равновесия в этом случае противоположно:
для его смещения вправо требуется повышение давления и пониже-
ние температуры (экзотермическая реакция с уменьшением объема
реакционной смеси):
r'cooch,r2 , ... .Hz R’CH,OH + R2CH,OH
2 CuCr2O4 2 2
(R2COOCH2R1+ r^ooc^r1 + R2COOCH2R2)
давление
- -
------------
температура
Гидрирование сложных эфиров может сопровождаться гидро-
генолизом, а эфиров ароматических кислот, кроме того, восстанов-
лением ароматического ядра. Побочные реакции, которые харак-
терны и для других высокотемпературных процессов, существенно
интенсифицируются с повышением температуры. В таких случаях
для их устранения, т. е. для увеличения селективности, гидрирова-
ние необходимо проводить при возможно более низких темпера-
37
турах. Вынужденное снижение температуры приводит к падению
скорости основной реакции. Чтобы обеспечить приемлемую ско-
рость гидрирования, снижение температуры компенсируют в пер-
вую очередь увеличением количества катализатора, а часто также и
повышением давления:
250 °C, 175 атм
COOEt ВиСН — 1 Си Сг,О, COOEt (эфир : кат. =15:1) 150 °C, 350 атм
(эфир: кат. =1:1,5)
сн2он
ВиСН + Ви(СН2)2ОН
сн3
64%
ВиСН(СН2ОН)2
80%
Скорость гидрирования зависит от количества катализатора.
Сложные эфиры практически не гидрируются при температуре
100 °C на скелетном никеле, если его количество не превышает
10 % от массы эфира, но энергично восстанавливаются даже при
50 °C, когда масса катализатора достигает 70 %. За исключением
экстраординарных случаев, подобных вышеприведенному, когда
реакция вынужденно проводится при заведомо слишком низкой
температуре и ее необходимо ускорить, избыточное количество ка-
тализатора ухудшает избирательность гидрирования. При заданном
типе катализатора (скелетный никель, палладий на носителе и др.)
чем более он активен, тем менее селективно его действие. При этом,
по-видимому, нужно отличать общий уровень активности катализа-
тора (разные сорта скелетного никеля, частично дезактивирован-
ные сернистыми соединениями или хинолином палладиевые ката-
лизаторы и др.) от активации или дезактивации его по отношению к
восстановлению той или иной функции (оксид платины, активиро-
ванный сульфатом железа (II), селективно восстанавливает карбо-
нильную группу, дезактивированный ацетатом цинка, - этилено-
вую группу и т. п.).
Таким образом, для достижения максимальной селективности
гидрирования, как правило, целесообразно использовать наименее
активный катализатор, выбранный с учетом строения восстанавли-
ваемого соединения, и проводить реакцию в как можно более мяг-
ких условиях, т. е. с небольшим количеством катализатора при ми-
нимальных температуре и давлении, обеспечивающих приемлемую
скорость процесса, контролируя поглощение водорода (непо-
38
средственно по объему, по падению давления в аппарате или по вре-
мени реакции).
На скорость, направление и селективность гидрирования неко-
торое влияние оказывает и реакционная среда, т. е. природа и ко-
личество растворителя. Наиболее часто в качестве растворителя
используются этиловый и метиловый спирты, уксусная кислота,
реже - диоксан, бензол (очищенный от тиофена), циклогексан и др.
Лучшие растворители водорода - насыщенные углеводороды, в
которых его растворимость в 3 раза выше, чем в спиртах, однако
они не всегда достаточно хорошо растворяют восстанавливаемые
органические соединения. Слишком летучие растворители, в част-
ности эфир, при высоких температурах создают дополнительное
давление в реакторе (автоклаве), при низких - затрудняют точное
измерение количества поглощенного водорода. Вода иногда при-
меняется при гидрировании кислот, их солей и других растворимых
в ней веществ. Обнаружено, что она ухудшает избирательность вос-
становления винилгалогенидов, способствуя гидрогенолизу связи
С-галоген.
Разбавление реакционной смеси растворителем смягчает усло-
вия восстановления и, следовательно, благоприятно отражается на
его селективности. Растворители могут также содействовать, при-
чем в разной степени в зависимости от своей природы, десорбции
исходных, промежуточных и конечных соединений с поверхности
катализатора и тем самым влиять на результат реакции. Так, веро-
ятно, конкурентная адсорбция лежит в основе дезактивирующего
действия растворителя - пиридина на палладиевый катализатор при
селективном гидрировании тройной связи в этинилтестостероне
39
В аналогичных условиях при использовании в качестве раство-
рителя диоксана гидрирование тройной связи до двойной в том же
соединении протекает неизбирательно. Однако восстановление
двумя молями водорода затрагивает только ненасыщенный углево-
дородный заместитель, но не сопряженную еноновую систему и
приводит к образованию 17-этилтестостерона.
Кислотность реакционной среды может существенно сказы-
ваться на скорости, глубине и стереохимическом результате гидри-
рования. Так, нитростирол быстро и с 90 %-ным выходом гидриру-
ется на платиновом катализаторе в уксусной кислоте с добавкой
серной кислоты, но в отсутствие последней реакция протекает
медленно, а выход 2-фенилэтиламина резко падает:
PhCH = CHNO, ------------------PhCH2CH7NH,
1 PtO,, АсОН, H,SO. ill
2 ’ ’24
Спирты, этилацетат и уксусная кислота с небольшими примесями
серной или хлороводородной кислот, ускоряющих реакцию, яв-
ляются лучшими средами при гидрогенолизе карбонильной группы
в альдегидах и кетонах на поверхностных палладиевых катализа-
торах или на оксиде платины. Гидрогенолизом в этих условиях,
например, была получена (1-метилинданил-1)уксусная кислота:
Me СН2СООН Me СН2СООН
[Г^Т> -----------------—-
U Pd—С, EtOH, H2SO4, U
90 °C, 5 атм
О
Попытки восстановить исходный инданон другими методами -
амальгамой цинка в разбавленной хлороводородной кислоте (по
Клемменсену) или щелочным разложением гидразона (по Кижне-
ру - Вольфу) окончились безрезультатно.
Интересно, что спирты, по-видимому, совсем не обязательно яв-
ляются интермедиатами при гидрогенолизе карбонильных соедине-
ний в кислых средах. Так, в тех же условиях, в которых некоторые
азабициклические кетоны гидрируются в азауглеводороды, соот-
ветствующие спирты гидрогенолизу не подвергаются и, следова-
тельно, едва ли образуются промежуточно в процессе превращения
кетонов в углеводороды:
40
но
Кислотные растворители, протонируя исходное соединение,
промежуточные или конечный продукты, могут изменять природу
восстанавливаемой и образующихся молекул. Так, пиридин превра-
щается в кислой среде в пиридиниевую соль и гидрируется уже в
виде соли. При этом пиридиниевый катион адсорбируется на по-
верхности катализатора „плашмя” за счет л-электронов цикла, по-
добно бензолу. Нейтральная же молекула пиридина адсорбируется
посредством неподеленных электронов атома азота и располагается
под углом к поверхности катализатора, что затрудняет перенос к
ней хемосорбированного водорода. Поэтому пиридин наиболее лег-
ко гидрируется на платиновом или родиевом катализаторе в уксус-
ной кислоте или в смеси метанола и хлороводородной кислоты:
_____________Нг_____________
PtO2 (Rh-C), АсОН (МеОН-НС1),
25 °C, 1-4 (до 25) атм
Кроме того, когда продукт гидрирования представляет собой силь-
ное основание, он, прочно хемосорбируясь на катализаторе, вызы-
вает дезактивацию, или так называемое „отравление” последнего.
Кислота протонирует образующееся основание и препятствует его
адсорбции на катализаторе, т. е. действию в качестве „каталитичес-
кого яда”.
Взаимодействие промежуточных продуктов восстановления с
кислотным растворителем может быть причиной вывода их из сфе-
ры реакции и прекращения дальнейшего гидрирования. Например,
гидрирование пиримидина в разбавленных кислотах протекает с
поглощением лишь 2 моль водорода и приводит к образованию со-
лей 1,4,5,6-тетрагидропиримидина:
41
Кислотность среды может влиять на общий стереохимический
результат гидрирования, например:
В последнем примере, используя в качестве растворителя EtOH,
получали смесь из 53 % цис- и 47 % /иранс-изомера, а используя
EtOH с добавлением 10 % НС1, - из 93 % и 7 % соответственно.
Обычно кислые среды благоприятны для образования продук-
тов 1/пс-присоединения водорода. При гидрировании в нейтраль-
ных и основных растворителях увеличивается относительное коли-
чество продуктов транс-присоединения. Однако это положение
часто не выполняется и отражает скорее некую тенденцию, чем оп-
ределенную стереохимическую закономерность гидрирования.
1.6. ГИДРИРОВАНИЕ ВАЖНЕЙШИХ КЛАССОВ СОЕДИНЕНИЙ
1.6.1. Ацетилены
Ацетилены могут быть гидрированы исчерпывающе в насы-
щенные соединения или частично в этиленовые. Полное гидриро-
вание обычно не вызывает затруднений и осуществляется в мягких
условиях (25 °C, 1-5 атм) на палладиевых, платиновых и активных
никелевых катализаторах, но в синтетических целях используется
сравнительно редко:
i-(CH2)gCsC-C = C(CH2)2-i н2
СО
II
о
PtO2 (Pd—СаСО3), AcOEt
(C6H14), 25 °C, 1 атм
-(CH2)l4-
—со—
II
о
42
Гораздо большее значение имеет парциальное (частичное) гид-
рирование ацетиленов. Возможность селективного осуществления
этой реакции определяет, как правило, высокий выход олефинов.
Наиболее подходящими катализаторами являются поверхностные
палладиевые катализаторы, особенно частично дезактивированные
ацетатом свинца (катализатор Линдлара, см. 1.3), хинолином или
гидроксидом калия, и никелевые катализаторы (скелетный, а также
так называемые бориды никеля Р-1 и Р-2 и Nic-катализатор, полу-
чаемые восстановлением ацетата никеля соответственно борогид-
ридом натрия в водно-спиртовом растворе и гидридом натрия в
тетрагидрофуране в присутствии третичного амилового спирта).
Скорость гидрирования тройной связи на этих катализаторах выше,
чем двойной, в то время как на других катализаторах такого разли-
чия или нет, или, наоборот, двойная связь гидрируется с большей
скоростью (особенно если это концевые связи). Замедление реак-
ции гидрирования алкинов после поглощения 1 моль водорода зна-
чительно облегчает необходимое его дозирование.
Вместе с тем селективность восстановления алкинов в алкены
обеспечивается не разницей в скоростях первой и второй ступеней
гидрирования, а значительно большей прочностью адсорбции ис-
ходных соединений. Пока в реакционной смеси имеется непрореа-
гировавший алкин, он вытесняет образующийся алкен с поверхнос-
ти катализатора и занимает сам освободившиеся активные центры.
Таким образом, дальнейшее восстановление алкена становится воз-
можным практически только после полного превращения в него ис-
ходного ацетилена. Благодаря мягким условиям реакции и повы-
шенному сродству к катализатору (адсорбируемости) ацетиленовой
группы тройную связь удается избирательно гидрировать до двой-
ной в соединениях, содержащих наряду с ней изолированные или
сопряженные двойные связи и другие достаточно легко восстанав-
ливающиеся или лабильные группы, такие как нитрильная, сложно-
эфирная, ацетальная и др.; из диацетиленовых соединений получа-
ются диены:
н2
Nic- Me ОН,
25 °C, 1атм
-сн=сн2 84 %
(80 %- ной чистоты)
43
(СН2)4^С С^(СН2)4
^с=с^
н2
Pd-BaSO4, Me ОН,
хииолин
Нч /Н
с=с
(СН2)4^ "(СН2)4
хс=с^
Нх ЧН 87 %
РгС^С(СН2)4С1
___________Н2__________
Pd—CaCO3(NiCK ), АсОМе,
1 4 °C, 1 атм
нч zh
с=с
Ргх Ч(СН2)4С1
~80%
OEt
I
РгС = С(СН2)4С^ССН
OEt
н2
Pd—CaCOj, AcOEt,
25 °C, 1 атм
Pr z(CH2^4 HC(OEt)2
C=C C=C 91 %
/ \ / \
н H H H
При гидрировании к адсорбированной на катализаторе молеку-
ле ацетиленового соединения атомы водорода переносятся и при-
соединяются со стороны поверхности катализатора, на которой они
до того также были адсорбированы (рис. 1.2). Поэтому парциальное
гидрирование ацетиленов с внутренней тройной связью приводит к
образованию исключительно или, по меньшей мере, преиму-
щественно термодинамически менее стабильных геометрических
изомеров - цис-алкенов и представляет собой удобный и высоко-
стереоселективный метод их синтеза. Так, при восстановлении
стеароловой кислоты на катализаторе Линдлара получается про-
дукт, содержащий 95 % олеиновой кислоты (цис-изомер):
ОН
/
он
/
СН,(СН,)7С = С(СН7)7С -
J \\ Pd-СаСО3, Pb(OAc)2,
о AcOEt
н2
СН3(СН2)\7 (СН2)7Сх
о
С = С
/ \
н н
Эйкозатетраин-5,8,11,14-овая кислота в аналогичных условиях
дает соответствующую полностью цис-тетраеновую (арахидоно-
вую) кислоту, являющуюся биопредшественником простагланди-
нов - физиологически весьма активных природных соединений,
регуляторов обмена веществ в клетках:
СООН
V\A=A=/WwV\
СООН
44
Гидрирование ацетиленовых соединений в мягких условиях на
различных никелевых катализаторах с контролем за количеством
поглощенного водорода позволяет получать i/uc-алкены с выхода-
ми 50-100%. Например, цис-гексен-3 был получен из диэтилаце-
тилена с выходами не менее 80 % на нескольких никелевых ката-
лизаторах типа Р и Nic:
Н Н
н2 \ /
Et С = С Et --------------► С = С
Ni, 25 °C, 1 этм / \
Et Et
Показано, что аналогичная реакция диметилацетилена протека-
ет стереоселективно при использовании в качестве катализатора не
только никеля, но и других металлов (платина, родий, кобальт). В
продуктах может содержаться до 8 % транс-изомера. Кроме того, в
них обычно присутствует небольшое количество олефина с иным
положением двойной связи - бутена-1, увеличивающееся, однако,
до 10-22 % при проведении реакции на таких катализаторах, как
рутений, железо и осмий. Оба этих алкена, по-видимому, не могут
получаться путем изомеризации г/ис-бутена-2, так как он в присут-
ствии бутина практически не адсорбируется. Более вероятно, что
они образуются в результате изомеризации бутина в соответствую-
щий аллен:
3 V
сн3с=ссн3
сн, сн,
л / 3
► с=с
/ \
н н
СН3 н
\ /
— с=с
/ \
н сн3
н н
H,C=C-CHCH3^f=|=^H,C = CCH9CH. —*-*-СН2 = СНСН2СН3
Z । । j * 2 । Z 3 z
* * *
45
Рис. 1.2. Схема присоединения атомов
водорода к адсорбированной на
поверхности катализатора молекуле
ацетиленового соединения
В отдельных случаях при гидри-
ровании ацетиленовых производных
возникают осложнения. Так, затруднено или невозможно гидриро-
вание полиарилацетиленов, у которых ароматические заместители
расположены вблизи тройной связи (1,1,4,4-тетрафенилбутин-2-ди-
ол-1,4; 1,1,1,4,4,4-гексафенилбутин-2 и др.). Иногда, если структура
ацетиленового соединения специфична, например, включает гидро-
ксильные группы в пропаргильном положении или сближенные в
пространстве и напряженные тройные связи, гидрирование даже в
очень мягких условиях может сопровождаться гидрогенолизом,
трансаннулярными и другими побочными реакциями:
1.6.2. Этилены
Присоединение водорода по изолированной двойной углерод-
углеродной связи в присутствии благородных металлов, скелетного
никелевого и других активных никелевых катализаторов (см. 1.6.1)
в большинстве случаев происходит легко при температуре 20-25 °C
и давлении водорода 1-4 атм. Скорость реакции зависит от стро-
ения соединения и в ряду алкенов и циклоалкенов снижается при
увеличении степени замещения этиленовой группы (правило Ле-
бедева). Эту зависимость иллюстрируют приведенные для каждого
типа алкенов значения относительных скоростей, полученные
обобщением данных по жидкофазному конкурентному гидриро-
46
ванию многочисленных соединений на Pt-SiO2 при температуре
20 °C; скорость гидрирования циклогексена (стандарт) принята
за 104:
R1R2C=CH2
R1CH=CH2 > (1’3440>1°3 > r1r2C=CHR3 > R1R2C=CR3R4
(4,74-7,5)-105 R1=CHR2 754-3700 0,54-20
(3,34-130) - юз
Большие различия в относительных скоростях при конкурент-
ном гидрировании определяют возможность избирательного вос-
становления менее замещенной этиленовой группы, в первую оче-
редь однозамещенной, в полиенах, содержащих изолированные
двойные связи:
I Л „ н2 I
95%
zz xz Ni(P-2), EtOH, 25 °C, 1 атм Z/ v
Нг
Pt-C, 25-60 °C, 3,7 атм
97,6 %
> 95 %
Однако сравнение скоростей гидрирования отдельно взятых
олефинов показывает, что степень замещения в этом случае влияет
существенно меньше, чем при конкурентном гидрировании пар
соединений. Так, время полуреакции (гидрирования 50 % субстра-
та) для алкенов С5-С6 разного строения различается не более чем в
45 раз. Длина цепи, связанной с этиленовой группой, и ее развет-
вленность мало отражаются на скорости гидрирования (выражена
как относительное время полуреакции):
1,0 1,6 16 45
Очевидно, относительные скорости реакций в условиях конку-
рентного гидрирования отражают различия не только в константах
скорости непосредственно присоединения водорода, как при гидри-
47
ровании каждого вещества в отдельности, но и различия в адсорби-
руемости соединений. По-видимому, способность адсорбироваться
на катализаторе зависит от характера замещения ненасыщенного
фрагмента молекулы в большей мере, чем константы скорости при-
соединения водорода к адсорбированным алкенам. Гидрирование
некоторых полностью замещенных этиленов протекает с трудом и
требует достаточно жестких условий:
Ni„., 250 °C, 200 атм
Напротив, двойные связи, сопряженные с ароматическими цик-
лами, карбонильной, карбоксильной, нитрильной и другими груп-
пами или включенные в напряженные циклы, восстанавливаются
очень легко:
PhCH = CH2
Ni—кизельгур , 20 °C, 2—3 атм
PhEt
100 %
/CN
Pd—С, EtOH, 25 °C,
1 атм
CN
/
сн
COOEt
94%
COOEt
НССН=СНСООМе
НС(СН2)2СООМе
МеО
NiCK,EtOH, АсОН,
25 °C, 1 атм
Me О
96%
Сопряженные диены и полиены при парциальном гидрировании
обычно дают смеси всех возможных продуктов.
Боковая цепь в стироле гидрируется на N1-AI2O3 почти в 3 раза
быстрее, чем двойная связь в гексене-1, и в 900 раз быстрее, чем
ароматический цикл в бензоле (данные по конкурентному восста-
новлению). При повышенном давлении водорода даже трифенил-
этилен восстанавливается на поверхностном никелевом катализа-
торе при температуре 20 °C:
Ph2C = CHPh
Ni —кизельгур, чикло-С6НцМе,
20 °C, 95 атм
Ph2CHCH2Ph
-100%
48
Вопросы стереохимии гидрирования двойной связи были рас-
смотрены в 1.4. В дополнение здесь уместно заметить, что примени-
тельно к этиленовым соединениям металлы как катализаторы про-
цессов изомеризации, сопутствующих восстановлению и ответст-
венных за отклонения от правила i/ис-присоединения водорода, по
активности могут быть разделены на 3 группы:
Ni, Pd, Fe > Ru, Rh, Os > Pt, Ir.
Скорость реакции изомеризации на никелевых и палладиевых ката-
лизаторах обычно выше скорости присоединения водорода, в то
время как на платиновых чаще всего наоборот. Кроме того, соотно-
шение скоростей зависит от температуры и при ее увеличении
может возрастать в случае одних металлов и уменьшаться в случае
других.
1.6.3. Арены и гетарены
Ароматические циклы (арены) гидрируются труднее, чем двой-
ные и тройные связи, нитро-, карбонильные и нитрильные группы.
Присоединение первых двух атомов водорода к бензолу приводит к
разрушению ароматической системы и связано с большими энер-
гозатратами. Энергия сопряжения (резонанса) бензола составляет
150,73 кДж/моль (36 ккал/моль), циклогексадиена-1,3 - менее
12,5 кДж/моль (3 ккал/моль). Сопоставление разности этих величин
с теплотой гидрирования циклогексена (двойной связи в цикле) -
119,75 кДж/моль (28,6 ккал/моль) показывает, что при ступенчатом
восстановлении бензола начальная стадия должна быть эндотер-
мической реакцией. Теплота гидрирования бензола в циклогек-
садиен-1,3, рассчитанная на базе экспериментальных величин теп-
лоты гидрирования бензола и циклогексадиена в циклогексан,
ДЯ1 = + 23,45 кДж/моль (5,6 ккал/моль):
49
&Н\ — KHi — AH 4 = — 208,51 —(—231,96) = 23,45 кДж/моль
AH2 = AH4 - AH3 = - 231,96 - (-119,75) = - 112,21 кДж/моль
(—26,8 ккал/моль).
Необходимостью компенсации потери энергии резонанса, по-
видимому, объясняется сравнительно низкая реакционная способ-
ность ароматических соединений при гидрировании. В достаточно
жестких условиях гидрирования бензолов циклические диены и
алкены, если они и образуются в качестве свободных (т. е. десор-
бированных) интермедиатов, должны восстанавливаться далее с
высокой скоростью (возможность образования небольших коли-
честв цикленов при гидрировании ароматических углеводородов
установлена). Поэтому неудивительно, что практически единствен-
ными продуктами гидрирования бензолов, как правило, являются
циклогексаны.
При гидрировании полициклических ароматических соеди-
нений с нарушением ароматичности одного из колец потеря энер-
гии сопряжения значительно меньше, чем при частичном вос-
становлении бензола. Разность резонансных энергий нафталина и
1,2- или 1,4-дигидронафталина не превышает 104,7 кДж/моль
(25 ккал/моль), превращение фенантрена и антрацена в их 9,10-ди-
гидропроизводные сопровождается проигрышем в энергии сопря-
жения соответственно в 83,74 и 50,24 кДж/моль (20 и 12 ккал/моль).
Поэтому нафталин восстанавливается легче бензола, но труднее
антрацена и фенантрена; при этом конденсированные ароматичес-
кие углеводороды в отличие от бензола легко подвергаются частич-
ному гидрированию.
Наиболее часто применяются при гидрировании бензоидных
циклов никелевые катализаторы. Восстановление на них протекает
в жестких условиях: при температуре 100-200 °C и давлении
100-300 атм. Никель, нанесенный на носитель, как катализатор вы-
сокотемпературного гидрирования ароматических соединений по
эффективности сопоставим с никелем Ренея, а в отдельных случаях,
возможно, даже несколько превосходит его, как следует из при-
веденных ниже данных по восстановлению бензола в циклогексан:
Температура, °C Время, мин
Ni-кизельгур (5 %) 125 10
NiCK(18%) 150 60
50
Для низкотемпературного гидрирования бензола, его гомологов
и производных используются платиновые и родиевые катализа-
торы, в отдельных, исключительных случаях - высокоактивные
сорта скелетного никеля. Палладий при низких температурах не
катализирует восстановления бензольных колец и широко исполь-
зуется при гидрогенолизе связей С-гетероатом в бензильном по-
ложении, не затрагивающем ароматических циклов, а также при
дегалогенировании ароматических соединений:
Вг
При давлении водорода 1-3 атм и температуре 25-50 °C на
платиновых катализаторах ароматические углеводороды восста-
навливаются с отличными выходами, хотя и сравнительно медлен-
но. 0,2 Моль соединения в уксусной кислоте гидрируются с 0,2 г
оксида платины в этих условиях за 2-26 ч. Время реакции резко
сокращается с увеличением давления (до 12-30 мин при 215 атм).
Скорость восстановления на родиевых катализаторах выше, чем
на платиновых. Так, бензол на ЯЬ-А^Оз (5 % Rh) при прочих рав-
ных условиях реагирует в 4 раза быстрее, чем на аналогичном
платиновом катализаторе. Предложен смешанный катализатор
RhCh-PtCh, получаемый сплавлением хлорида родия и хлоропла-
тиновой кислоты (3 : 1) с нитратом натрия (подобно катализатору
Адамса) и позволяющий проводить гидрирование с приемлемой
скоростью при атмосферном давлении и температуре 25 °C. Про-
иллюстрировать преимущества этого катализатора можно на при-
мере гидрирования толуола в метилциклогексан (при 25 °C):
Давление Н2, атм Время, мин
РЮ2 2-3 195
PtO2 215 12
RhO2-PtO2 1 1Ю
51
Наилучшим образом реакция на платиновых и родиевых ката-
лизаторах протекает в кислой среде. Часто в качестве растворителя
применяется ледяная уксусная кислота. Гидрирование ароматичес-
ких углеводородов в этаноле облегчается добавкой малого коли-
чества хлороводородной кислоты.
Детальный механизм гидрирования ароматических соединений
не выяснен. Предполагается, что присоединение атомов водорода,
поступающих с поверхности катализатора, к адсорбированной мо-
лекуле происходит ступенчато. Так, при частичном насыщении
после образования л- или сг-адсорбированного циклогексена воз-
можна десорбция и затем реадсорбция циклоолефина:
С6Н1 о
Эти стадии должны играть важную роль в стереохимии гидрирова-
ния. Соответственно отмечен некоторый параллелизм в соотноше-
нии t/wc- и транс-изомеров, образующихся при гидрировании кси-
лолов и диметилциклогексенов (указано содержание t/wc-изомера в
продуктах гидрирования на РЮг в АсОН):
CH
Me Me
На катализаторах гидрирования имеет место цис-транс-изоме-
ризация продуктов.
52
Таким образом, стереохимический результат гидрирования оп-
ределяется природой и положением заместителей в ароматическом
кольце, катализатором и условиями реакции.
Скорость гидрирования ароматических соединений также зави-
сит от характера и числа заместителей в цикле, причем при замеще-
нии изменяются как адсорбируемость соединения, так и скорость
присоединения водорода к адсорбированному субстрату. Так, толу-
ол гидрируется на Ni-АЬОз (140 °C, 35 атм) в 2 раза, ксилолы -
в 3,2-4,3 раза, а пентаметилбензол в 200 раз медленнее бензола; на
РЮг в уксусной кислоте (30 °C, 2,4—4 атм) относительные скорости
„собственно** восстановления, т. е. восстановления адсорбиро-
ванных на катализаторе бензола, толуола, орто-, мета- и пара-
ксилола, составляют соответственно 1,00; 0,71; 0,44; 0,61 и 0,77;
адсорбируемость соединений в указанной последовательности так-
же падает.
Из функциональных производных бензола легче других гидри-
руются одно- и многоатомные фенолы. На никелевых катализа-
торах при повышенном давлении водорода реакция протекает
достаточно энергично и почти количественно уже при температуре
120-150 °C:
Ni—кизельгур, 150 °C,
100—300 атм
Ni—кизельгур, 125 °C,
100—300 атм
н2
Н2
Возможно частичное восстановление фенолов в циклогекса-
ноны. Направленно циклические кетоны удается получать, проводя
реакцию в присутствии скелетного никелевого катализатора с до-
бавлением щелочи:
ОН
н2
NiCK., NaOH, 50 °C,
70 — 110 атм
53
Анилины и анилиды гидрируются отосительно трудно - для их
успешного восстановления на никелевых катализаторах нужны
достаточно жесткие условия:
NHR NHR
R = Н, Ас
Вместе с тем на скелетном никеле гидрирование анилина про-
текает уже при температуре 120-130 °C и давлении 100 атм (ср. с
восстановлением бензола).
На платиновых катализаторах ароматические амины наилуч-
шим образом восстанавливаются в виде гидрохлоридов, очевидно,
благодаря изменению характера адсорбции при переходе от нейт-
ральной молекулы амина к аммониевому катиону и протонирова-
нию продукта:
NH, NH2
-----------------------► 90 %
PtOyHCl, АсОН, 25 °C, 1 атм I 1
Эфиры ароматических карбоновых кислот гидрируются с от-
личными выходами примерно в тех же условиях, что и анилины:
COOEt
COOEt
__________Нг_________
Ni, 200 °C, 100-170 атм
COOEt
COOEt
93%
Гидрирование полициклических ароматических углеводородов
протекает ступенчато. Как было сказано выше, соединения с кон-
денсированными циклами гидрируются быстрее, чем бензол. Бла-
годаря исключительно низкой активности при восстановлении
изолированных бензольных колец хромит меди и палладий являют-
ся удобными катализаторами для частичного гидрирования конден-
сированных ароматических систем:
Pd-C (CuCr2O4), 25 °C(1 00—1 50 °C),
1.5—2 атм (150—200 атм) inn о/
54
При использовании для этой цели других катализаторов (ни-
кель) проявляется тенденция к сохранению ароматичности цент-
ральных колец:
Гетероароматические соединения (гетарены) восстанавливают-
ся на тех же катализаторах и в целом в условиях, близких условиям
гидрирования ароматических карбоциклов.
Пиррол восстанавливается труднее бензола. На скелетном ни-
келе реакция протекает с низким выходом и останавливается до
полного превращения субстрата. Для достижения удовлетворитель-
ных результатов при применении в качестве катализатора оксида
платины необходимо повышенное давление водорода (не менее
5 атм): N N Н Н
Катализатор NicK PtO2 Гомологи Давление, атм Температура, °C Выход, % 200-300 180 47 ~6 20 60-80 и производные пиррола, особенно N-замещенные,
обычно гидрируются легче:
55
Niclt, 50 °C, 200-300 атм
98%
COOEt
I
COOEt
COOEt
Индол и его гомологи в зависимости от условий реакции гидри-
руются исчерпывающе или селективно в 2,3-дигидропроизводные с
сохранением бензольного цикла:
н2
Си Cr2O4(PtO2), 190 °C
(25 °C), 250-300 (1 атм )
Н2
NiCK., 230 °C, 250-300 атм
Для парциального восстановления применяют при высоких тем-
пературах медно-хромитный катализатор, при комнатной - оксид
платины.
Гидрирование тиофена трудноосуществимо, так как он является
„каталитическим ядом“ для благородных металлов и подвергается
десульфуризации скелетным никелем. В жестких условиях тиофен
восстанавливается на таких катализаторах, как гептасульфид и геп-
таселенид рения, дикобальт октакарбонил, полисульфид кобальта и
сульфид молибдена:
_____________Нг______________
Re2S7(Re2Se7), 230-260 °C,
14 0 атм (320 атм )
70-100 %
56
Восстановительная десульфуризация производных тиофена ни-
келем Ренея используется в синтетических целях, например:
°2N\____ NH,
Л А ^2 I
// \\ ----------► изо-Ви С Н, С Н(СНЭ), С ООН
Ni 60 °C 2 ' 2'2
изо-Ви S СООН 84 о/о
Реакцию можно осуществить, прибавляя никель-алюминиевый
сплав в водно-щелочной раствор исходного соединения, что осо-
бенно удобно, если это тиофенкарбоновая кислота.
Другой пятичленный цикл - фуран - гидрируется легче бензола.
На палладиевом катализаторе и даже на активном скелетном никеле
реакция протекает с выходом более 90 % при температуре 25 °C и
небольшом давлении:
--------------- / \ 90-93 %
NiCK.(PdO), 2 5 °C, 2—4 атм (7 атм )
При использовании менее активных сортов скелетного никеле-
вого катализатора или других никелевых катализаторов требуются
повышенные температура (50-190 °C) и давление (не менее 90 атм).
Фурфуриловый спирт на никелевом катализаторе при темпе-
ратуре 125 °C и давлении 100-200 атм и на RJ1-AI2O3 при 20 °C и
1 атм восстанавливается в тетрагидрофурфуриловый спирт, кото-
рый может быть получен также гидрированием фурфурола на пла-
тиновом катализаторе с добавкой соли Fe (II) при 20 °C и атмо-
сферном давлении или на никеле Ренея:
Фурановое кольцо легко подвергается гидрогенолизу, и во избе-
жание его раскрытия гидрирование следует проводить в возможно
более мягких условиях. Вместе с тем гидрогенолиз фурановых сое-
динений на палладиевых, платиновых и медно-хромитных катали-
заторах имеет самостоятельное синтетическое значение:
н2, н2о
Pd, 50-60 °C, I атм
СН3СОСН2СН2СН2ОН
57
Пиридин гидрируется при температуре 25 °C и небольшом дав-
лении водорода на платине, родии и палладии в кислых средах,
лучше всего в уксусной кислоте (см. 1.5). При восстановлении на
скелетном никеле необходима несколько более высокая темпера-
тура, чем для бензола. Эту реакцию, как и гидрирование на другом
пригодном для этой цели катализаторе - хромите меди, нельзя про-
водить в спиртовом растворе из-за возможного N-алкилирования
продукта. Более эффективно и не осложнено алкилированием гид-
рирование пиридина на оксиде рутения:
Катализитор Температура, °C Давление, атм Выход пиперидина, %
NiCK 200 130-300 83
Систем 220 100-150 50
RuO2 95 70-100 100
Производные пиридина гидрируются быстрее соответствую-
щих производных бензола. В соединениях, содержащих изолиро-
ванные или конденсированные бензольное и пиридиновое кольца, в
первую очередь обычно восстанавливается гетероцикл:
NiCK., 160 °C, 150 атм
58
1.6.4. Карбонильные соединения
Для восстановления карбонильных соединений в спирты при-
годны все металлические катализаторы. Наиболее эффективны пла-
тина и активные сорта скелетного никеля. На этих катализаторах, а
также на родии и рутении большинство альдегидов и кетонов
гидрируется при температуре 25 °C и давлении 1-4 атм. С менее
активными разновидностями никеля Ренея достаточная скорость
восстановления достигается при температурах до 100-125 °C и
давлениях до 100 атм:
СН3(СН2)5СНО - PtO2(NiCK ), 25 °C, 3 атм ► СН3(СН2)5СН2ОН 70-90 %
RCOCHj «2 RCH(OH)CH3
NiCK., 25-30 °C, 1-3 атм
R = Me, Ph 60-100 %
сн3сн=снсно Н2 NiCK., 125 °C, 100 атм сн3сн2сн2сн2он 94 %
Чтобы восстановление карбонильной группы на оксиде плати-
ны было полным, рекомендуется промотировать катализатор не-
большим количеством хлорида или сульфата железа(П). При гидри-
ровании алифатических альдегидов на недостаточно отмытом от
щелочи скелетном никеле интенсифицируются побочные реакции
конденсации; вместе с тем щелочь активирует этот катализатор.
Рутений применим для восстановления в водных растворах. Пал-
ладий весьма активен при гидрированиии ароматических, но не
очень эффективен при гидрировании алифатических и алицикли-
ческих карбонильных соединений:
({ СНО ------------—------► ( V-CHO 81 %
\__/ Pd-C, 75 °C, 14 атм \_/
Н2
СН, = СНСН,СМе,СНО -------------- СН,СН,СН,СМе,СНО
2 2 2 PJ-А 1,0,, 25 °C, 3 2 2 2
2 атм 88,5 %
Альдегиды восстанавливаются легче кетонов. Если карбониль-
ная группа непосредственно связана с ароматическим ядром, обра-
зующийся при гидрировании спирт может далее в тех же условиях
подвергаться гидрогенолизу:
59
ArCOR -------► ArCH(OH)R ----------ArCH2R
При низкотемпературном восстановлении эту побочную реак-
цию удается в основном предотвращать, останавливая гидриро-
вание по поглощении рассчитанного количества водорода. При
использовании в качестве катализатора благородных металлов, в
наибольшей степени способствующих гидрогенолизу, следует про-
водить гидрирование в нейтральных растворителях - этаноле и
этилацетате, но не в уксусной кислоте, так как в кислых средах ско-
рость гидрогенолиза возрастает.
Во многих случаях можно избежать гидрогенолиза, применяя
медно-хромитный катализатор, на котором в типичных для него
условиях гидрирования ароматические альдегиды и жирноаромати-
ческие кетоны восстанавливаются в бензиловые спирты обычно
значительно быстрее, чем последние подвергаются гидрогенолизу,
гидрогенолиз же интенсивно протекает лишь при температуре
выше 200 °C:
_____________ /---V _____^2____» ,---.
CuCr2O„, 150 °C, // \\ CuCr2O4, // \\
100-150 атм ЧПА 200—300 °C, 1 атм
СН2ОН и СНО U Me
97 % 90-95 %
В то же время ряд ароматических карбонильных соединений (и
соответствующих им спиртов) на хромите меди, а также на никеле
Ренея быстро гидрогенолизуется уже при температуре 130-175 °C и
давлении 70-350 атм:
___________Нг__________
CuCr2O4, EtOH, 160 °C,
200—300 атм
R = Me, Ph
92-93 %
PhCOPh ----------------------------------
CuCr2O4, EtOH, 175 °C, 100-1 50 атм
PhCH2Ph
97 %
OH
I
PhCOCHPh
_________________h2 .______________
CuCr2O4, EtOH, 175°C, 100-150 атм
PhCH2CH2Ph
~ 100 %
60
Me R
EtOOC N Me
H
R = COMe, CH(OH)Me
h2
NiCK., EtOH, 170 °C,
200—300 атм
Восстановление ароматических альдегидов и кетонов еще на
одном высокотемпературном катализаторе - скелетной меди прак-
тически не сопровождается гидрогенолизом; оптимальные условия
получения спиртов с применением этого катализатора - темпера-
тура 175-185 °C и давление 180-200 атм.
С другой стороны, направленное исчерпывающее гидрирование
карбонильной группы в метиленовую и метильную наилучшим
образом осуществляется на палладиевых катализаторах при ис-
пользовании в качестве растворителей уксусной кислоты, а также
этанола, этилацетата или уксусной кислоты с добавками мине-
ральной кислоты при температуре 20-90 °C и давлении до 5 атм
(см. 1.5). В тех же условиях применяют платиновые катализаторы.
Однако в этих случаях, и особенно при гидрировании на плати-
но-родиевых катализаторах, возрастает вероятность восстановле-
ния бензольных циклов:
НОСНМе
94 %
н2
RhO2-PtO2>
EtOH (АсОН)
НОСНМе
н2
Pto2 (PdO),
EtOH
Н2
PdO (PtO2),
АсОН, HC1
Для гидрогенолиза карбонильной группы при высокой темпе-
ратуре и давлении чаще всего применяется скелетный никель (при-
меры см. выше).
Необходимо заметить, что реакционная способность связи С-0
по отношению к водороду увеличивается в молекуле не только в
61
бензильном положении, но и под влиянием близко расположенных
карбонильной, алкоксикарбонильной или гидроксильной групп.
Поэтому некоторые полифункциональные карбонил- или гидро-
ксилсодержащие соединения аналогично ароматическим сравни-
тельно легко подвергаются гидрогенолизу (см. 1.5).
При значительном повышении температуры и давления в про-
цессе восстановления различных альдегидов и кетонов, например,
на скелетном никеле до 200-250 °C и более чем до 100 атм, начи-
нают заметную роль играть реакции гидрогенолиза С-С-связей
(деструктивное гидрирование):
। । ।
R‘COR2— R^CEPOHjR2—► R'H (R]CH3) + R2CH3 (R2H) + H2O
I I I
Так, октадеканаль и 3-циклогексилпропаналь (или соответст-
вующие спирты) могут быть таким путем превращены в углеводо-
роды, содержащие в молекуле на один атом углерода меньше, чем
субстрат, с выходом около 80 %:
C,7H3,R --------------► С.7Н3,
" NiCK , 25 0 °C, 100—200 атм и
<^\>-CH2CH2R
-сн4, -Н2О
R =СНО, СН2ОН
1.6.5. Нитрилы
Нитрилы гидрируются в амины на платиновых и родиевых ката-
лизаторах при температуре 25 °C и давлении 1-3 атм и на скелетном
никеле при 25-150 °C и 1-270 атм. Применяются также бориды
никеля, кобальтовые и поверхностные палладиевые катализаторы.
Восстановление протекает ступенчато. Первичный амин, образую-
щийся в результате присоединения к нитрилу 2 моль водорода,
способен присоединяться к промежуточному имину, что в конеч-
ном счете приводит к получению смеси первичного, вторичного и
даже третичного аминов; реакции конденсации с повышением тем-
пературы интенсифицируются:
62
RC=N ”2 > RCH=NH ”2 ► RCH2NH2 M~n4.
rchnh2
hnch2r
- rch=nch2r
_Яз____(RCH,),NH
-NH3 z
RCH—NH
rchnh2 h2
N(CH2R)2 "NH3
(RCH2)3N
Содержание побочных продуктов снижается при проведении
реакции в кислой среде, очевидно, благодаря протонированию пер-
вичного амина, затрудняющему его присоединение по связи C=N.
Этот подход пригоден только в отношении катализаторов, не
реагирующих с кислотами. Практически применяют оксид платины
и как растворитель уксусную кислоту; высокие выходы первичных
аминов (до 90 %) могут быть получены при использовании палла-
дия, нанесенного на уголь, и спирта в присутствии 1-3 эквива-
лентов минеральной кислоты (НС1, H2SO4, НСЮд). Аналогично, но
за счет ацилирования, подавляется присоединение первичного ами-
на к ненасыщенному интермедиату при гидрировании нитрилов в
уксусном ангидриде; продукт реакции в этом случае представляет
собой N-ацетилпроизводное первичного амина:
H2(R = H)
/Pd-C, EtOH, НС1,'
/ 25 °C, 1 атм
PhCH2NH2-HCl
-100 %
H2, Ас2О
PtO2> 25 °C, 1-3 атм
ги мц
R = Н, n-Ме, о-Ме
63-87 %
н2, Ас2О н3о+
PhCH,CN ---------—7-------► Ph(CH,),NH » Ph(CH,),NH,
2 PtO2, 25 "С, 1 атм v 272 | v 2>2 2
Ac 62 %
Образование вторичного и третичного аминов при гидриро-
вании нитрилов эффективно ингибируется в присутствии аммиака,
63
который, конкурируя с первичным амином за альдимин в процессе
присоединения, уменьшает количество получающегося геминаль-
ного первично-вторичного диамина (аминаля), ответственного за
побочную реакцию. Введение 2-5 моль аммиака на 1 моль нитрила
обеспечивает хороший выход первичных аминов (до 60 %). При
этом для гидрирования при комнатной температуре и атмосферном
или слабо повышенном давлении в качестве катализатора пригодны
родий, нанесенный на оксид алюминия, платина на угле, скелетный
никель и др. Никель Ренея в этих условиях всегда применяется в
сравнительно больших количествах, а реакция проводится в раство-
рителе. Вместе с тем первичные амины могут быть получены на
скелетном никеле в присутствии аммиака с очень хорошими выхо-
дами:
PhCH2CN
Н2, NH3
NiCK., 120-130 °C, 140 атм
PhCH2CH2NH2
83-87 %
Et,N(CH,),CN --------------’----► Et,N(CH,),CH,NH
z z J NiCK, 25 °C, 7 атм 2 z J z
97 %
NiCK., McOH, 25 °C,
8 — 10 атм
H2, NHj
1.6.6. Восстановительное алкилирование аммиака и аминов
Химически родственно гидрированию нитрилов (одинаковые
или аналогичные интермедиаты) получение аминов восстанови-
тельным алкилированием аммиака, первичных и вторичных аминов
карбонильными соединениями (восстановительное аминирование
карбонильных соединений). В гидрогенизационном варианте оно
64
заключается в каталитическом восстановлении водородом альдеги-
да или кетона в присутствии аммиака или амина*. Применяются
обычно скелетный никелевый (40-150 °C, 3-150 атм) или плати-
новый (25 °C, 1-3 атм) катализаторы. На начальных стадиях про-
цесса происходит конденсация карбонильного соединения с ами-
ном (аммиаком):
R1. Л 4 R’v н, r’
С = О HNR C-NR3R4 --------------► -?CHNR3R4
R2 R2/1 R2/
OH
R4 = H
'H2° (R3=R4=H)
< , H2
, C=NR3 ---------------------------
Реакция, как правило, сопровождается образованием аминов
более высокой, чем заданная, степени алкилирования, так как
первичный амин, получающийся при восстановительном алкилиро-
вании аммиака, частично реагирует с карбонильным соединением и
дает вторичный амин, который, в свою очередь, может тем же путем
превращаться в третичный амин. Кроме того, как и при гидриро-
вании нитрилов, возможны процессы конденсации с участием ин-
термедиатов (см. 1.6.5), увеличивающие, особенно при повышен-
ных температурах, выход тех же побочных продуктов. Преиму-
щественное получение амина заданной степени алкилирования
определяется соотношением реагентов. Его доля в продуктах алки-
лирования заметно зависит также от строения карбонильного сое-
динения и условий реакции. Например, гидрирование фурфурола в
присутствии избытка аммиака на скелетном никеле приводит к об-
разованию первичного амина с выходом 79 %, тогда как при моль-
ном отношении альдегида к аммиаку 2:1 в основном получается уже
вторичный амин:
* О восстановительном алкилировании аминов с применением других
восстановителей см.: Марч Д. Органическая химия: В 4 т. М.: Мир, 1987. Т. 3.
460 с.
3 Заказ № 137
65
4 V
NH3 (изб.)
/\ NiCK., 40-75 °C,
О СНО 20—100 атм
о ch2nh2
79 % (ди-6)%
NH3(0,5 моль )
о сн2
2nh
4 V
Ч V
66 % (моно —12)%
При восстановительном алкилировании аммиака простейшими
кетонами (ацетон, метилэтилкетон) из-за склонности последних к
сложным побочным конденсациям с аммиаком выходы первичных
аминов обычно невелики (30-50 %). При алкилировании низшими
альдегидами вследствие их высокой реакционной способности
часто наблюдается образование значительного количества аминов
более высокой, а при бис-алкилировании и более низкой степени
алкилирования, а также гетероциклических соединений. В подоб-
ных случаях необходимы разумный подход к выбору методики и
тщательное регулирование условий синтеза. Так, при восстанови-
тельном алкилировании аммиака двумя молями уксусного альде-
гида на скелетном никелевом катализаторе наряду со вторичным
амином получается значительное количество первичного и третич-
ного. Однако, если осуществить вместо одного серию таких син-
тезов, перенося образующийся этиламин из каждого предыдущего
цикла в последующий, оказывается возможным повысить суммар-
ный выход диэтиламина до 84 %.
В синтезе вторичных аминов из первичных рекомендуется вы-
делять промежуточный альдимин или кетимин (основание Шиф-
фа), а затем его гидрировать. Поступая таким образом при восстано-
вительном алкилировании первичных аминов ароматическими аль-
дегидами, получают вторичные амины с выходами, как правило,
превышающими 70 %. Если выделение шиффова основания затруд-
нительно, смесь реагентов перед гидрированием нагревают некото-
рое время для завершения стадии конденсации:
R
с=о + nh9ch,ch,oh
э/ Z X X
R -|
€=NCH9CH9OH
2/ 2 2
R
\
66
R1 = H, R2 = Pr, Bu, С H (-70 %)
R1= R2 = изо-Bu; R1= Me, R2 = C6H13 (-95%)
1.6.7. Гидрогенолиз галогенопроизводных
Гидрогенолиз связи С-галоген является важным методом дега-
логенирования органических соединений. Реакция протекает легко
на различных катализаторах. Поэтому прогидрировать ненасыщен-
ные функции в галогеносодержащем субстрате и при этом избежать
потери галогена нередко не менее сложно, чем удалить атом гало-
гена, сохранив неизменными другие восстанавливающиеся группы.
Решающее значение в каждом конкретном случае имеет характер
связи С-галоген (тип галогенопроизводного) и условия реакции, в
первую очередь температура.
В целом устойчивость к гидрогенолизу возрастает в следующем
ряду: иодо- < бромо- < хлоро- < фторопроизводные. Иодо- и бромо-
алканы легко восстанавливаются на скелетном никелевом катализа-
торе в присутствии гидроксида калия для связывания галогеноводо-
рода при температуре 25 °C и атмосферном давлении. Хлориды в
этих условиях гидрогенолизуются медленнее. Многие из них на
платиновых, родиевых и никелевых катализаторах при 25 °C не
восстанавливаются. Алкилфториды гидрировать с приемлемыми
выходами даже в жестких условиях практически не удается. В не-
насыщенных галогенопроизводных восстановительное дегалогени-
рование протекает особенно легко, если атом галогена находится в
аллильном или пропаргильном положении. При этом во всех слу-
чаях восстанавливается и непредельная группа. Атомы галогена,
связанные непосредственно с атомами углерода винильной и эти-
нильной групп, замещаются водородом несколько труднее, но лег-
че, чем расположенные изолированно от кратной связи или в гало-
геналканах. Эта закономерность, по-видимому, объясняется учас-
тием кратной связи в формировании переходного состояния при
гидрогенолизе связи С-галоген в галогеновинильных, -этиниль-
ных, -аллильных и -пропаргильных группах. В ароматических сое-
динениях атом галогена в бензильном положении замещается водо-
родом много быстрее, чем находящийся в ароматическом цикле.
Дегалогенирование может быть осуществлено без восстановления
бензольного кольца.
67
Прекрасным катализатором гидрогенолиза галогенопроизвод-
ных разных типов является никель Ренея. Дегалогенирование арил-
галогенидов проводят, как уже указано выше для иодо- и бромоал-
канов, в присутствии щелочи и обычно при температуре 25 °C и
давлении 1-3 атм:
н2
NiCK., КОН, МеОН, 25 °C, 1 атм
Н2
NiCK., КОН, МеОН, 25 °C, 1-3 атм
R= СН2ОН, CH2OEt, СООН, СН2СООН, CH2CONH2
X = Вг, С1
__________________Н2___________________
NiCK., КОН, МеОН, 25 °C, 1 атм
75-100 %
Однако отнюдь не все арилгалогениды достаточно энергично реа-
гируют при температуре 25 °C. Например, для гидрогенолиза неко-
торых наиболее устойчивых арилхлоридов необходима более высо-
кая температура (до 100 °C и более). Ароматические полигалогено-
производные частично дегалогенируются на скелетном никелевом
катализаторе в присутствии ограниченного, эквивалентного числу
удаляемых атомов галогена количества основания или с контролем
за количеством поглощенного водорода. В отсутствие основания
восстановительное дегалогенирование на никеле Ренея тормозится.
Так, галогеносодержащие азосоединения на этом катализаторе при
температуре 25 °C и давлении 1-3 атм в присутствии щелочи под-
вергаются дегалогенированию и одновременно расщеплению по
связи N=N, без добавления щелочи происходит только гидрогено-
лиз азогруппы:
68
н2
20 °C, 4 атм
85-90 % 61-81 %
53-77 % 91-94%
Многие галогенопроизводные с отличными выходами, а часто и
количественно дегалогенируются при 25 °C или иногда при повы-
шенной температуре и атмосферном давлении на палладиевых ка-
тализаторах:
н2
С1СН9СООН ---------------------------------- АсОН
2 Pd-CaCO,, КОН, EtOH, 25 °C, 1 этм
PhCHCl2 --------------- PhMe
_________Hl________
Pd-C, AcONa, АсОН,
65-70 °C, 2-3 атм
Винил- и аллилхлориды восстанавливаются на палладии, а так-
же платине или, лучше, родии (5 %), нанесенных на оксид алюми-
ния, в отсутствие основания в насыщенные хлориды без замещения
или лишь с частичным замещением атома хлора.
Как на палладиевом, так и на скелетном никелевом катализаторе
при дефиците водорода возможно восстановительное сочетание
галогенопроизводных, т. е. расщепление связи С-галоген с обра-
зованием новой углерод-углеродной связи и, следовательно, с ус-
ложнением углеродного скелета молекулы (конструктивная реак-
ция):
NH2NH2 ( --► N2 + 2Н2 )
PhBr
Pd —СаСО3, KOH, водн. MeOH, кипячение
PhPh 78 %
69
PhCH2-C^
Me
CH2I
CH2I
(H2), n2
NiCK., EtOH, 80 °C
Арилгалогениды на оксиде платины при температуре 50-70 °C
и давлении водорода 3 атм не только дегалогенируются, но и пол-
ностью гидрируются; выходы циклоалканов при этом составляют
70-95 %:
н2
PtO2, EtOH, 5 5 °C, 3 атм
X = Br, С1
Наиболее легко происходит гидрогенолиз связи С-С1 в хлоро-
ангидридах карбоновых кислот. Реакция осуществима в мягких
условиях, хотя и при повышенной температуре, и используется как
метод синтеза альдегидов (см. 1.2). В раствор ацилхлорида в ксило-
ле, толуоле или бензоле вводят сернистое соединение, являющееся
дезактиватором катализатора, поверхностный палладиевый катали-
затор, чаще всего Pd-BaSO4, и пропускают в реакционную смесь
водород, постепенно поднимая температуру до оптимальной. Де-
зактиватор, так называемый хинолин-S (строение неизвестно), го-
товят растворением серы в хинолине при кипячении; применяются
также промышленные серосодержащие реактивы (NH2CSNH2,
Me2NCSNMe2, PhNCS). Вместо палладиевых катализаторов с успе-
хом может применяться и оксид платины, дезактивированный тио-
мочевиной. Для этой реакции оптимальной является температура
80-180 °C, при которой начинается выделение хлороводорода.
Последний удаляется из реакционной смеси током водорода. По-
глощая его раствором щелочи и определяя количественно, контро-
лируют ход реакции. В указанных условиях не восстанавливаются
70
даже такие реакционноспособные функции, как нитрогруппа и
двойная углерод-углеродная связь. Выходы альдегидов составляют
50-95 %:
СОС1
_____________Нг____________
Pd—кизельгур, хинолин-S,
Ме2С6Н4, 150 °C
СНО
РЬСН = СНСОС1
Pd — BaSO4, хинолин-S,
Ме2С6Н4, 122 °C, 0,7 атм
no2
PhCH = CHCHO
50-60 %
PhC О Cl ----------------------------*-
PtO2, NHjCSNH,, PhMe, 110 °C
PhC HO
96 %
Побочными продуктами при получении альдегидов этим мето-
дом являются спирты, углеводороды, сложные и иногда простые
эфиры. Высокая температура благоприятна для образования спир-
тов. Значение полного гидрогенолиза хлорокарбонильной группы
(с разрывом связей С-С и С-С1), как правило, невелико. Лишь в
отдельных случаях, например при восстановлении трифенилаце-
тилхлорида, оно может возрастать настолько, что основная реакция
подавляется и углеводород, содержащий на один углеродный атом
меньше, чем исходное соединение, становится практически единст-
венным органическим продуктом гидрирования:
н2
Ph3CCOCl pd-BaSO4, PhH (Ме2С6Н4)
Ph3CH + СО + НС1
В настоящее время для селективного гидрирования хлороангид-
ридов в альдегиды наряду с классической используется модифици-
рованная и, по-видимому, более удобная методика. В соответствии
с ней реакцию проводят в ТГФ, ацетоне или бензоле на палладие-
вом катализаторе, иногда частично дезактивированном сернистым
соединением, при температуре 0-50 °C, а для связывания выделяю-
щегося хлороводорода добавляют амин (2,6-диметилпиридин,
wTO-PrjNEt):
С1СО(СН7)4СОС1 ------------------------- ОНС(СН2)4СНО
2 4 Pd —С, ТГФ , 2,6-Ме,Ру, 2 4
0 °C, 1 атм ’ 74 %
71
COCI
сно
Pd—С, хинолин-S, C6H6, 2,6-Ме2Ру,
40-50 °C, 1 атм
С1
Me СОС1
А ------------------±------------
I J Pd—С, Ме2СО, »3o-Pr2NEt, 25°С, 1 атм
77 %
С1
Me СНО
78 %
1.6.8. Сложные эфиры
Для гидрирования сложных эфиров в спирты применяют высо-
котемпературные оксидные катализаторы - хромит меди и, реже,
хромит цинка (катализаторы Адкинса) (см. 1.3). В промышленности
этим способом из жиров и жирных кислот получают высшие
спирты, перерабатываемые далее в моющие средства - алкилсуль-
фаты ROSOsNa, и глицерин. Благоприятное положение равновесия
процесса гидрирования - дегидрирования (см. 1.2, 1.5) при темпе-
ратурах, обеспечивающих на хромите меди необходимую скорость
восстановления эфиров (200-250 °C), достигается при давлении
водорода 200-400 атм. Однако скорость гидрирования зависит от
количества катализатора, и, увеличивая его сверх обычного (до
1,0-1,5 от массы эфира), можно понижать температуру реакции на
100 °C и даже более без существенного изменения длительности
восстановления. Это, в свою очередь, позволяет в зависимости от
конкретных условий либо уменьшать давление водорода, сохраняя
высокую степень превращения эфиров в спирты (положение равно-
весия), либо повышать его, чтобы дополнительно ускорить процесс.
Оптимальная продолжительность гидрирования эфиров составляет
1-8 ч.
В присутствии большого количества катализатора реакцию про-
водят в растворителе - спирте или диоксане, в противном случае -
обычно без него. Для удаления примесей, способных дезакти-
вировать катализатор (кислоты, основания, сернистые и галогено-
содержащие соединения), рекомендуется эфир предварительно пе-
регнать над скелетным никелем. Выходы спиртов, как правило, со-
ставляют не менее 80-90 %.
Эфиры ненасыщенных кислот на хромите меди гидрируются в
насыщенные спирты, но при восстановлении на цинк-хромитном
72
катализаторе при температуре 280-300 °C и давлении 200 атм
удается получать с выходами 35-65 % непредельные спирты
(см. 1.5).
Основными побочными процессами при гидрировании эфиров
на хромите меди являются гидрогенолиз образующихся спиртов и
частичное восстановление ароматических гетероциклов и конден-
сированных полициклических ядер. Легко гидрогенолизуются
гликоли, склонность которых к гидрогенолизу возрастает в ряду
1,2 < 1,4 < 1,3, и а- и /?- арилзамещенные спирты. Реакция иногда
сопровождается и расщеплением связи С-С. Для предотвращения
гидрогенолиза спиртов гидрируют эфиры в присутствии большого
количества катализатора при температуре 125-150 °C; выходы
двухатомных и ароматических спиртов составляют 60-80 %:
н2
PhCH(OH)COOEt ———./г)|ЕЮн/ PhCH(OH)CH2OH
125 °C, 350 атм (15 мии ) 80 %
По-видимому, труднее избежать частичного восстановления
конденсированного ароматического ядра, которое на катализаторе
Адкинса часто протекает уже при 100-200 °C (см. 1.6.3). Более или
менее удовлетворительный результат дает понижение температуры
реакции до 85-110 °C:
Ш COOEt „ /х. ^СНЭОН
п 2
CuCr2O4 (1,5 г/г )Л L JI J 0/
108 °C, 315 атм
Для восстановления сложных эфиров в спирты наряду с хро-
митными катализаторами применяется скелетный никель. В при-
сутствии избытка этого катализатора (до 1,5 г/г) эфиры гидриру-
ются при температуре 25-125 °C и давлении 350 атм с выходами не
менее 80 %; имеющиеся в молекуле исходного соединения арома-
тические кольца также восстанавливаются. Эфиры а-аминокислот
на активном никеле Ренея при 50 °C и 150-200 атм с удовлет-
ворительными или хорошими выходами дают аминоспирты; повы-
шение температуры более чем до 100 °C недопустимо, так как при
этом реакция может протекать со взрывом:
nh2 nh2
I н, I
RCHCOOEt —-—-г-* RCHCH2OH 52-93 %
N1CK, EtOH z
R = Bu, изо-Ви, PhCH2, Ph
73
Гидрирование эфиров аминокислот на хромите меди в спирте
сопровождается N-алкилированием:
NH, NMe,
I Н2 I 2
ГЬССООЙ CuC,A Ме0Н PhCCH2OH 88 %
Me Me
Специфично и имеет особое значение как метод удаления за-
щитной группы (см. 1.2) гидрирование бензиловых эфиров карбо-
новых кислот на палладиевых катализаторах при температуре 20 °C
и атмосферном давлении. Продуктами реакции являются соответст-
вующая карбоновая кислота и толуол. Бензиловые эфиры гидроге-
нолизуются легче, чем восстанавливаются двойная углерод-угле-
родная связь и нитрогруппа, причем реагируют даже соединения,
содержащие в молекуле атом двухвалентной серы. Вместо молеку-
лярного водорода при дебензилировании могут быть использованы
доноры водорода - циклогексен и циклогексадиен, легко арома-
тизирующиеся на палладии; реакцию проводят в инертной атмос-
фере с реагентами, взятыми в стехиометрическом отношении, в
уксусной кислоте или этаноле в присутствии палладия (10 %), нане-
сенного на уголь:
74
CH2OCH2Ph
сн2он
PhCH?OOCNHCHCOOH —------—-------- NH?CHCOOH
2 Pd—C, EtOH, 25 °C 2
99 %
При расщеплении этим способом бензилалкил(арил)карбонатов
или -карбаматов образующиеся моноэфир угольной кислоты или
N-замещенная карбаминовая кислота самопроизвольно декарбок-
силируются соответственно в окси- или аминосоединение. Бензил-
оксикарбонильная и и-нитробензилоксикарбонильная защитные
группы широко используются в химии аминокислот:
NHCOOCH2Ph
PhCH2CHCONHCH2COO-< />~NO2
nh2-hci
83,9 %
1.6.9. Амиды карбоновых кислот
Амиды гидрируются в амины на хромите меди при температуре
210-260 °C и давлении 100-350 атм:
r‘conr2r3
Си Сг2О4
r‘ch2nr2r3 + Н2О
Часть амида может гидролизоваться выделяющейся водой в карбо-
новую кислоту, которая, в свою очередь, вызывает дезактивацию
катализатора. Чтобы избежать этого, восстановление проводят в
смешивающемся с водой растворителе, таком как диоксан, мак-
симально интенсифицируя процесс применением значительного
количества катализатора (15% массы субстрата) и ужесточением
условий. Интенсификация восстановления амидов, не замещенных
по атому азота, одновременно способствует частичному подавле-
нию побочных реакций, приводящих к образованию соответствую-
щих вторичных аминов и могущих играть существенную роль:
75
60 %
ch2nh2
CuCr2O4, диоксан
2 50 °C, 200-300 атм
33 %
2nh
N-Моно- и М,М-дизамещенные амиды в аналогичных условиях
дают соответственно вторичные и третичные амины. Реакция со-
провождается процессами гидрогенолиза связей С-N, восстанови-
тельного алкилирования и др.:
c..h23conh-0 r г 1
11 \__/ CuCr2O4, диоксан
250 °C, 200-300 атм
Ci2H25NH4Q +
62 %
+ (C12H25>2NH +
24 %
15 %
C12H25NH2
16 %
О о
/—S II II /—\
< NC(CH2)8CN )
<2n(ch2)10n3 + НО(СН2)10ОН
94 % 4 %
67 % 7 %
1.7. ТЕХНИКА ЭКСПЕРИМЕНТА
Гетерогенное гидрирование проводится при нормальном или
повышенном давлении в „замкнутом" реакторе (периодический
процесс) или в проточной установке. Восстанавливаемое вещество,
контактирующее с твердым катализатором, может находиться в
жидкой (жидкость, раствор) или газовой фазе.
Лабораторное гидрирование в жидкой фазе при атмосферном
давлении обычно осуществляется в простейшей установке. В ка-
76
честве реактора используется круглодонная колба или так называе-
мая „утка“ - горизонтально расположенный цилиндрический сосуд
с трубкой для ввода газа и горлом с пробкой для загрузки реагентов
в верхней части (на боковой стенке цилиндрического корпуса).
Перемешивание реакционной смеси достигается энергичным
встряхиванием реактора, который для этого располагается на
качалке, или при помощи магнитной мешалки. Водород поступает в
реактор из газовой бюретки, связанной резиновым шлангом с урав-
нительной склянкой (напорным резервуаром). До подачи водорода
в бюретку ее заполняют водой, газовую линию от бюретки к реак-
тору перекрывают, а линию от газометра с водородом к бюретке
открывают. Воду вытесняют из бюретки водородом в уравнитель-
ную склянку, после чего водородный резервуар (газометр) отклю-
чают и открывают кран на газовой линии, соединяющей бюретку и
реактор; в который помещены реагенты и который предварительно
„промыт" водородом. Опуская уравнительную склянку, выравни-
вают уровень воды в ней и бюретке и тем самым устанавливают в
системе давление, равное атмосферному. Измерив начальный объ-
ем водорода в бюретке, начинают гидрирование. Если для гидриро-
вания необходимо небольшое нагревание (до 50-70 °C), исполь-
зуется утка с двойными стенками (с „рубашкой"), между которыми
циркулирует вода заданной температуры, подаваемая из термоста-
та. Иногда вместо этого обычная утка вставляется в металлический
блок с электрообогревом.
Гидрирование при нормальном давлении в газовой фазе прово-
дится в проточных установках. Реактор такой лабораторной уста-
новки представляет собой кварцевую или стеклянную трубку, час-
тично заполняемую зернами катализатора. Каталитическая трубка
помещается в специальную электропечь с терморегулятором, обес-
печивающую одинаковый обогрев (температурное плато) части
реактора, в которой находится катализатор. Гидрируемое вещество
подается в трубку дозирующим устройством, испаряется в ее на-
чальной части и вместе с вводимым сюда же водородом поступает в
зону катализатора. Продукты реакции принимаются в охлаждае-
мый приемник-сепаратор, в котором собираются жидкие вещества
и происходит их отделение от непрореагировавшего водорода и
образовавшихся газов. Давление в системе контролируется по пока-
заниям манометра, температура - при помощи термопары, находя-
77
щейся в защитной трубке - „кармане14 в слое катализатора. Легко
испаряющиеся жидкости могут подаваться в реактор в виде паров в
смеси с водородом. Для получения такой смеси водород до введе-
ния в реактор барботируют через промывную склянку с гидрируе-
мым веществом; соотношение реагентов регулируется темпера-
турой жидкости в испарителе. Для поддержания в реакторе нужной
температуры вместо электропечи применимы нагревательные бани.
В этом случае используется U-образная контактная трубка, у кото-
рой отверстия для ввода реагентов и выхода продуктов находятся
вверху.
Проточная установка для гидрирования должна быть тщательно
герметизирована. Охлаждение аппаратуры после опыта и хранение
катализатора осуществляются в атмосфере водорода или инертного
газа. Попадание воздуха приводит к окислению и потере активнос-
ти катализатора. Подтекание водорода и паров органических ве-
ществ из установки недопустимо, так как грозит образованием
взрывоопасных смесей с воздухом.
Реакционными аппаратами периодического действия для гид-
рирования под давлением являются автоклавы - сосуды цилиндри-
ческой формы, рассчитанные на определенное давление. Автокла-
вы многообразны по конструкции, различаются объемом, значени-
ями рабочего давления и температуры, способами перемешивания
реакционной смеси (качающиеся, вращающиеся, с мешалкой) и
обогрева и др. Конструкционный материал автоклава должен быть
инертен по отношению к реакционной среде. Автоклавы снабжены
контрольно-измерительными приборами, предохранительными
устройствами, вентилями для ввода газа и т. д.
Лабораторный автоклав для работы при высоких давлениях
чаще всего заполняется водородом из баллона, что позволяет созда-
вать давление в нем немногим больше 100 атм при температуре
20 °C. Это приблизительно соответствует 175-190 атм при 250 °C.
Для сжатия водорода до более высоких давлений используются
компрессоры. Максимальное начальное давление в автоклаве долж-
но быть таким, чтобы по достижении верхнего предела рабочего
интервала температуры оно не могло превысить допустимый
уровень. После загрузки гидрируемого вещества, растворителя и
катализатора автоклав закрывают, удаляют из него воздух, несколь-
ко раз создавая и сбрасывая давление азота. Оставляя на время авто-
клав под давлением, проверяют его на герметичность. После запол-
78
нения автоклава водородом под давлением начинают гидрирова-
ние. За процессом следят по снижению давления. Знание свободно-
го объема автоклава, начальных и конечных значений давления и
температуры позволяет определять количество поглощенного в
реакции водорода.
По окончании гидрирования автоклав охлаждают и сбрасывают
избыточное давление водорода. При разгрузке следует иметь в ви-
ду, что некоторые катализаторы пирофорны и во избежание возго-
рания и возможных при этом вспышек паров органических веществ
должны постоянно находиться под слоем жидкости. Практическое
осуществление гидрирования при высоких давлениях требует от
исполнителя знания применяемой для этого аппаратуры и специ-
альных правил безопасной работы с автоклавами. Сведения о про-
точных установках, работающих под давлением, и более подробное
описание аппаратуры для гидрирования можно найти в специаль-
ной литературе.
1.8. ЭКСПЕРИМЕНТАЛЬНЫЕ РАБОТЫ
Особенности работы с водородом. Водород относится к горю-
чим и взрывоопасным газам. Он поступает в лаборатории в балло-
нах, окрашенных в зеленый цвет. Баллон рекомендуется устанавли-
вать вне здания в специальных металлических шкафах. При такой
установке газ к рабочим местам подается по медным или стальным
трубам. Допустимое количество водорода в лабораторных помеще-
ниях не должно превышать 5 л (при давлении 150 атм). Основная
опасность, связанная с применением водорода, заключается в обра-
зовании взрывчатых водородно-воздушных смесей, пределы взры-
ваемости которых по объемной доле водорода очень широки:
3,3-81,5 %. Наибольшую опасность представляет смесь, образован-
ная двумя объемами водорода и 4,8 объемами воздуха. Учитывая
это, при работе с водородом следует проявлять особую осто-
рожность. Необходимо избегать утечек газа, тщательно проверяя
герметичность всех соединений рабочей установки. Должно быть
исключено также присутствие поблизости открытого огня и нагре-
тых предметов. Приступать к выполнению работ с использованием
водорода следует после ознакомления с инструкциями по технике
безопасности при работе с газовыми баллонами и водородом.
79
Получение катализаторов
а) Скелетный никелевый катализатор
NiAl2 + 6 NaOH---- Ni + 2 Na3A103 + 3 H2
Реактивы
Никель-алюминиевый сплав Ренея 10 г
Гидроксид натрия 16 г
Осторожно! При реакции выделяется водород, и работу следу-
ет проводить в хорошо действующем вытяжном шкафу. Катализа-
тор пирофорен и на воздухе самовоспламеняется. Поэтому все
остатки катализатора, в том числе и на фильтре, следует временно,
до полного уничтожения, хранить под слоем воды.
В стакане емкостью 1 л готовят взвесь 10 г никель-алюминие-
вого сплава Ренея, содержащего 30-50 % никеля, в 100 мл дистил-
лированной воды. Затем прибавляют твердый гидроксид натрия с
такой скоростью, чтобы вспенивание не происходило слишком бур-
но. Энергичное течение реакции начинается после некоторого ин-
дукционного периода, поэтому вначале следует проявить осторож-
ность. Когда дальнейшее прибавление гидроксида натрия не будет
больше приводить к заметному вскипанию (до этого момента
необходимо добавить примерно 16 г), смесь выдерживают 10 мин
при комнатной температуре и затем 30 мин на водяной бане при
70 °C. По истечении указанного времени жидкость, находящуюся
поверх осевшего на дно стакана никеля, тщательно декантируют и
катализатор промывают водой, перемешивая и сливая воду с отсто-
явшегося осадка. Промывку ведут до достижения нейтральной
реакции промывных вод по фенолфталеину. Таким же способом вы-
тесняют воду спиртом (20-30 мл). Катализатор хранят в стеклянной
посуде под слоем спирта. Хотя катализатор можно сохранять таким
образом некоторое время, более целесообразно готовить его
непосредственно перед употреблением (в количестве, необходимом
для каждого конкретного синтеза), избегая заметного падения его
активности при хранении. Активность катализатора можно прове-
рить, положив несколько крупинок его на лист бумаги: через
1-2 мин должно наблюдаться самовозгорание катализатора.
80
б) Палладий, сорбированный на угле
Pd+2 -*Pd°
Реактивы
Дихлорид палладия 0,5 г
Уголь активированный 4 г
В сосуде для гидрирования к 4 г угля марки „сибунит“ прибав-
ляют водный раствор 0,5 г дихлорида палладия. Восстановление
проводят в установке, показанной на рис. 1.3. После того как погло-
щение водорода закончится, катализатор отфильтровывают, про-
мывают водой, спиртом и, наконец, эфиром. Полученный катали-
затор содержит 4 % палладия, нанесенного на уголь.
Рис. 1.3. Установка для каталитического гидрирования
при атмосферном давлении:
1 - газометр; 2 - трехходовый кран; 3 - газовый цилиндр на 1 л; 4 - уравнительная
склянка; 5 - реакционная колба с насадкой Дрекселя
81
в) Медно-хромовый катализатор Адкинса
Cu(NO3)2
Прокаливание
Ba(NO3)2 -------► Комплексные ------------to-
СиО Сг2О3
ВаО • СггО3
г
г
хроматы
(NH4)2CrO4
Реактивы
Нитрат меди 21,8
Нитрат бария 2,6
Аммиак (25 %-ный водный раствор) 15 мл
Дихромовокислый аммоний 12,6 г
В колбе вместимостью 0,5 л при температуре 70 °C растворяют
2,6 г нитрата бария в 80 мл дистиллированной воды, затем до-
бавляют в раствор 21,8 г нитрата меди при перемешивании до пол-
ного растворения соли. В другой колбе готовят раствор из 12,6 г
дихромовокислого аммония и 60 мл воды с добавлением 15 мл
25 %-ного водного раствора аммиака. Этот раствор медленно, тон-
кой струей прибавляют к нагретому первому раствору, осторожно
встряхивая колбу. В ней выпадает красновато-коричневый осадок
смешанных хроматов меди, бария и аммония, который отфильт-
ровывают и тщательно отжимают на воронке Бюхнера, а затем вы-
держивают в сушильном шкафу при температуре 100-120 °C в
течение 10-12 ч. Сухой осадок переносят в фарфоровую чашку,
покрывают ее часовым стеклом и осторожно, чтобы содержимое не
было выброшено выделяющимися газами, нагревают в муфельной
печи в течение 1 ч при температуре 350-450 °C. После окончания
разложения солей и охлаждения около 16 г полученного катали-
затора, представляющего собой синевато-черную массу, переносят
в ступку и растирают в порошок. Далее порошок трехкратно обра-
батывают 10 %-ным раствором уксусной кислоты (по 120 мл),
растворяющей избыточную окись меди. Жидкость декантируют и
промывают осадок четырьмя порциями воды, не менее чем по
120 мл каждая. Указанные операции следует проводить непрерывно
друг за другом, так как катализатор при стоянии имеет тенденцию
образовывать коллоидные растворы. Сырой катализатор после про-
мывки отфильтровывают на воронке Бюхнера, высушивают в су-
шильном шкафу при температуре ПО °C в течение 10 ч и вновь
82
растирают в порошок. В итоге получается 130-140 г тонкого черно-
го порошка, устойчивого к воздействию воздуха и сырости.
Гидрокоричная кислота
(3-фенилпропионовая кислота)
н,
PhCH = CHCOOH ----------- PhCH2CH2COOH
N1CK.
Реактивы
Коричная кислота 3,7 г
Никель скелетный 0,5 г
Водород (из баллона)
Этанол (ректификат) 20 мл
Работа проводится в установке для каталитического гидриро-
вания (рис. 1.3). Газометр, наполненный водородом, соединяют при
помощи трехходового крана с газовым цилиндром, который запол-
няют водородом. Замечают объем газа, для чего уравнительную
склянку устанавливают таким образом, чтобы уровень воды в ней
был одинаков с уровнем воды в цилиндре. Затем переводят трех-
ходовой кран в положение, при котором газовый цилиндр соединя-
ется с реакционной колбой, и проверяют установку на герметич-
ность. После этого, перекрыв соединение установки с колбой, вно-
сят в нее 3,7 г (0,025 моль) коричной кислоты, 20 мл этанола и
немного скелетного никелевого катализатора (на конце шпателя).
Далее проводят вытеснение воздуха из реакционной колбы, для
чего ее соединяют с газовым цилиндром и медленно пропускают
200-300 мл водорода. Последний выходит под тягу через шланг,
одетый на отросток насадки Дрекселя. После промывки систему
закрывают заглушкой, а трехходовой кран переводят в положение,
при котором газовый цилиндр соединен с реакционной колбой.
Отмечают исходный объем водорода в цилиндре, пускают в ход
мешалку. Гидрирование можно считать законченным, когда погло-
тится рассчитанное количество водорода. Контроль полноты гид-
рирования проводят методом ТСХ. Элюент - смесь хлороформа и
этилацетата, 20:1; Rj коричной кислоты 0,5, Ry гидрокоричной
кислоты 0,35. Раствор фильтруют, этанол тщательно удаляют на
ротационном испарителе. Остаток на холоде затвердевает. Его очи-
щают перекристаллизацией из разбавленной хлороводородной кис-
лоты (концентрация примерно 10 %). Получают бесцветные крис-
таллы в форме длинных игл; т. пл. 47-49 °C. Выход 3,4 г (90 %).
83
Дополнительная очистка может быть произведена методом ваку-
умной сублимации при температуре 100 °C и давлении 2 мм рт. ст.
Спектральные характеристики приведены на рис. 1.4.
5,6,7,8-Тетрагидро-1-нафтол
5,8-Дигидро-1 -нафтол
Палладий на угле
Водород (из баллона)
Этилацетат
н2
Pd—С
ОН
4,4 г
0,3 г
20 мл
Гидрирование проводят в установке, показанной на рис. 1.3.
Подготовительные операции аналогичны описанным при получе-
нии гидрокоричной кислоты. В реакционную колбу помещают
раствор 4,4 г (0,03 моль) неочищенного 5,8-дигидро-1-нафтола (по-
лучение см. 3.8) в 20 мл этилацетата и 0,3 г катализатора. После
поглощения рассчитанного количества водорода катализатор от-
фильтровывают, растворитель удаляют в вакууме. Остаток быстро
затвердевает. Перекристаллизацией его из петролейного эфира
(т. кип. 40-60 °C) получают 3,7 г (84 %) почти бесцветных крис-
таллов; т. пл. 68-69 °C. При необходимости может быть проведена
их дополнительная очистка методом вакуумной сублимации при
температуре 100 °C и давлении 4 мм рт. ст. Хроматография: элю-
ент - хлороформ; Rf 0,5. Спектральные характеристики приведены
на рис. 1.5.
Бензилацетон
(4-фенилбутанон-2)
PhCH = CHCOMe ------=--►
Nlc.
Реактивы
Бензальацетон
Никель скелетный
Водород(из баллона)
Этанол (ректификат)
PhCH2CH2COMe
3,7 г
0,5 г
20 мл
84
Рис. 1.4. Спектральные
характеристики
гидрокоричной кислоты:
а - спектр ПМР* в ССЦ (<5, м. д.:
2,62 т, J = 8 Гц (2Н, СН2СО), 2,92 т,
J = 8 Гц (2Н, CH2Ph), 7,26 с (5Н, Ph),
10,92 с (1Н, СООН)); б - ИК спектр в
тонком слое (3050 см-1, ш. (О-Н));
в- УФ спектр в метаноле (2макс , нм,
1g с: 257,5,2,46)
85
100
Ь-
$ §
ill
о
1800
1400
tooo
Рис. 1.5. Спектральные характеристики
5,6,7,8-тетрагидро-1-нафтола:
а - спектр ПМР* в ССЦ (<5, м. д.: 1,81 м (4Н, 6- и
7-СН2), 2,62 т, J = 6 Гц (2Н, 5(8)-СН2), 2,74 т,
J = 6 Гц (2Н, 8(5)-СН2), 4,70 с (1Н, ОН), 6,59 д,
J = 8 Гц (1Н, Н2 аром.), 6,69 д, J = 8 Гц (1Н, Н4
аром.), 6,98 т, J = 8 Гц (1Н, Н3 аром.)); б - ИК
спектр в КВг (3350 см-1, ш.); в - УФ спектр* в
этаноле (Амакс., нм, 1g г: 260, 2,77)
86
Гидрирование бензальацетона осуществляют на установке, по-
казанной на рис. 1.3. Подготовительные операции аналогичны опи-
санным при получении гидрокоричной кислоты. В реакционную
колбу помещают 3,7 г (0,025 моль) бензальацетона, 20 мл этанола и
немного (на конце шпателя) скелетного никелевого катализатора.
Гидрирование проводят до поглощения рассчитанного количества
водорода. Контроль полноты реакции осуществляют методом ТСХ.
Элюент - смесь хлороформа и этилацетата, 10:1;/?/ бензальацетона
0,7, R/ бензилацетона 0,8. Полученный после гидрирования раст-
вор фильтруют, этанол удаляют в вакууме. Остаток перегоняют,
используя прибор для микроперегонки. Т. кип. 235 °C при 760 мм
рт. ст. и 130 °C при 15 мм рт. ст., п^0 1,5111. Выход 3,2 г (87 %).
Спектральные характеристики приведены на рис. 1.6.
Бензилацетофенон
(1,3-дифенилпропанон-1)
н,
PhCH = CHCOPh -----— ----- PhCH9CH7COPh
NiCK. 2 2
Реактивы
Бензальацетофенон 5,2 г
Никель скелетный 0,5 г
Водород(из баллона)
Этилацетат 20 мл
Процедура гидрирования бензальацетофенона аналогична опи-
санной для получения гидрокоричной кислоты. Реакционная смесь
состоит из 5,2 г (0,025 моль) бензальацетофенона, 20 мл этилацетата
и небольшого количества скелетного никеля; реакцию проводят
при комнатной температуре. После поглощения рассчитанного ко-
личества водорода рекомендуется провести контроль полноты
реакции методом ТСХ. Элюент - хлороформ; Rj бензальацетофе-
нона 0,7, R/ бензилацетофенона 0,8. После завершения процесса
гидрирования реакционную смесь фильтруют, этилацетат частично
удаляют. При охлаждении раствора происходит кристаллизация
бензилацетофенона. Т. пл. 69-71 °C , выход 5 г (95 %). Спектраль-
ные характеристики приведены на рис. 1.7.
87
3500 2500 1800 1400
Рис. 1.6. Спектральные характеристики
бензилацетона:
a - спектр ПМР* в ССЦ (д, м. д.: 2,07 с (ЗН,
СНз), 2,77 м (4Н, СН2), 7,21 м (5Н, Ph)); б-ИК
спектр в тонком слое (1720 см-1 (С=О)); в -
УФ спектр в метаноле (2Макс нм> 1g е: 209,
3,91; 258 тс, 2,37) '
88
3 7 6 5 4 3 2 fyf.d.
Рис. 1.7. Спектральные
характеристики бензилацетофенона:
а - спектр ПМР* в ССЦ (<5, м. д.: 3,00 д.т,
J = 8 и 1 Гц (2Н, СН2), 3,26 д.т, J = 8 и 1 Гц
(2Н, СН2), 7,24 м (5Н, Н2'-6' аром.), 7,45 м
(ЗН, Н3“5 аром.), 7,93 м (2Н, Н2-6 аром.));
б - ИК спектр в тонком слое (1680 см-1
(С=О)); в - УФ спектр в метаноле (2Макс ,
им, 1g с: 241,3,89; 282 тс, 2,86)
89
Дигидро-а-терпинеол
(2-(4-метилциклогексил)пропанол-2)
Me Me
Реактивы
а-Терпинеол 5,1 г
Никель скелетный 1 г
Водород (из баллона)
Этилацетат 20 мл
В реакционную колбу установки для гидрирования (см. рис. 1.3)
загружают 5,1 г (0,033 моль) а-терпинеола (свежеперегнанный, с
содержанием основного вещества 96 %), небольшое количество
скелетного никелевого катализатора и 20 мл этилацетата. Подго-
товительные операции аналогичны описанным при получении гид-
рокоричной кислоты. После поглощения рассчитанного количества
водорода катализатор отфильтровывают, раствор промывают не-
сколько раз 2 %-ным раствором КМпОд, затем водой и сушат суль-
фатом магния. Затем растворитель осторожно удаляют в вакууме
водоструйного насоса. Остаток перегоняют, собирая фракцию с
температурой кипения 93-100 °C при 20 мм рт. ст., 1,4631. Про-
дукт обладает сильным цветочным запахом и кристаллизуется при
охлаждении в бесцветные кристаллы; т. пл. 35 °C. Выход состав-
ляет 4,3 г (84 %). Спектральные характеристики даны на рис. 1.8.
4-Хлороаиилин
Способ 1. Восстановление гидразин-гидратом
Реактивы
4-Хлоро-1-нитробензол 6,3 г
Гидразин-гидрат 4-6 г
Никель скелетный 1 г
Этанол (ректификат) 140 мл
90
Осторожно! Гидразин является сильным ядом. Работу прово-
дить только в хорошо действующем вытяжном шкафу.
В круглодонной колбе вместимостью 250 мл, снабженной ме-
ханической мешалкой и холодильником, готовят раствор 6,3 г
(0,04 моль) 4-хлоро-1 -нитробензола в 140 мл этанола. К раствору
добавляют 2-3-кратный мольный избыток гидразин-гидрата
(100%-ный гидразин-гидрат содержит 64 % гидразина и имеет
1,4317), слегка подогревают содержимое колбы на водяной бане
Рис. 1.8. Спектральные характеристики дигидро-а-терпинеола:
<7-спектр ПМР* в CDC13 (i, м. д.: 0,85 м (ЗН, СН3), 1,07 с (6H, СН3), 1,47 м
(ЮН, СН2 и СН), 3,10 с (1Н, ОН)); б- ИК спектр в СС14 (3620 см~' (О-Н))
91
и вносят небольшое количество скелетного никелевого катализато-
ра. При этом наблюдается вспенивание раствора. По мере протека-
ния реакции (время может варьироваться от 5 до 60 мин) реакцион-
ная смесь становится из желтой почти бесцветной. Завершение
реакции можно проконтролировать методом ТСХ. Элюент - хлоро-
форм; Rf 4-хлоро-1-нитробензола 0,8, Rf 4-хлороанилина 0,3; про-
межуточно образующийся 4-хлорофенилгидроксиламин имеет Rf
0,2.
После окончания процесса в реакционную колбу вносят еще не-
много катализатора для разложения избытка гидразин-гидрата и
раствор нагревают до кипения для удаления растворенных газооб-
разных продуктов реакции. Горячий раствор фильтруют для осво-
бождения от никеля, кипятят с активированным углем и снова
фильтруют. Охлажденный фильтрат выливают в большой избыток
воды и выделяют амин в виде соли хлороводорода обычным спо-
собом. Выход 4-хлороанилина составляет 4,1 г (80 %). Т. пл.
71-73 °C. Спектральные характеристики приведены на рис. 1.9.
Способ 2. Гидрирование при повышенном давлении
Реактивы
4-Хлоро-1-нитробензол 47,3 г
Никель скелетный 3-5 г
Водород (из баллона)
Этанол (ректификат) 150 мл
Работа проводится в автоклаве вместимостью 0,5 л, снабженном
механической мешалкой. В автоклав загружают смесь 47,3 г
(0,3 моль) 4-хлоро-1-нитробензола, 150 мл этанола и 3-5 г катали-
затора - никеля скелетного. Давление водорода доводят до
70-100 атм, включают перемешивание и обогрев (50-70 °C). Когда
поглощение водорода прекратится, перемешивание и обогрев
прекращают, из охлажденного автоклава выгружают реакционную
смесь. Катализатор отфильтровывают, этанол при необходимости
частично удаляют на ротационном испарителе. Выделение продук-
та осуществляют аналогично описанному в способе 1. Выход 4-хло-
роанилина составляет 28,7 г (75 %). Спектральные характеристики
приведены на рис. 1.9.
92
Рис. 1.9. Спектральные
характеристики 4-хлороанилина:
а-спектр ПМР* в (СОз)2СО (<5, м. д.:
4,72 ш.с (2Н, NH2), 6,65 м, (2Н, Н2-6
аром.), 7,03 м, (2Н, Н2’-’ аром.)); б -
ИК спектр в тонком слое (3400 и
3500 см-1 (N-Н)); в - УФ спектр в
метаноле (2макс > им, 1g е: 243, 5,12;
296, 4,22)
93
Циклопентанол
(способ 1)*
О ОН
6^6
Реактивы
Циклопентанон 84 г
Никель скелетный 5-6 г
Водород (из баллона)
В автоклав вместимостью 0,5 л, снабженный механической
мешалкой, загружают 84 г (1 моль) циклопентанона и 5-6 г ске-
летного никелевого катализатора. Предварительно спирт, под сло-
ем которого хранился катализатор, сливают и катализатор про-
мывают небольшим количеством циклопентанона. Гидрирование
циклопентанона проводят при давлении 100 атм и температуре
120-130 °C. Первоначально давление в автоклаве быстро падает,
затем падение замедляется. Рассчитанное количество водорода
поглощается обычно в течение 2-4 ч. После охлаждения автоклав
разгружают. Реакционную смесь отфильтровывают от катализатора
и продукт реакции выделяют на ректификационной колонне эффек-
тивностью 10 теоретических тарелок. Состав отобранных фракций
анализируют методом ГЖХ. Т. кип. 98-100 °C, Лд° 1,3949. Выход
60,2 г (70 %).
При необходимости гидрирование циклопентанона можно про-
вести в растворе этанола. В этом случае реакционную смесь реко-
мендуется предварительно промыть водой, высушить сульфатом
магния и выделить конечный продукт аналогично вышеописанно-
му. Спектральные характеристики приведены на рис. 1.10.
Циклогексил амии
Н2
NicK.
* Способ 2 см. в разделе 2.9.
94
Реактивы
Анилин 93 г
Никель скелетный 5-6 г
Водород(из баллона)
В автоклав вместимостью 0,5 л, снабженный механической
мешалкой, загружают 93 г (1 моль) свежеперегнанного анилина и
5-6 г скелетного никелевого катализатора. Предварительно спирт,
Рис. 1.10. Спектральные характеристики циклопентанола:
а -спектр ПМР* в CD3C1 (б, м. д.: 1,47 м (8Н, СН2), 3,85 с (1Н, СИ), 4,07 уш. с
(1Н, ОН)); б - ИК спектр в тонком слое (3320 см-1 (О-Н))
95
под слоем которого хранился катализатор, сливают и катализатор
промывают небольшим количеством анилина.
Гидрирование проводят при давлении 100 атм и температуре
120-130 °C. Обычно рассчитанное количество водорода погло-
щается в течение 4-6 ч. После остывания автоклав разгружают.
Катализатор отфильтровывают и продукт реакции выделяют фрак-
ционной перегонкой. Т. кип. 132-134 °C, 1,4372. Индивиду-
альность соединения контролируют методом ГЖХ. Выход 74,3 г
(75 %). Спектральные характеристики приведены на рис. 1.11.
Метилциклогексан
Реактивы
Толуол
Никель скелетный
Водород (из баллона)
92 г
5-6 г
Гидрирование толуола в присутствии скелетного никеля прово-
дят аналогично описанному для анилина (см. получение циклогек-
силамина). Метилциклогексан выделяют ректификацией па колон-
не эффективностью 10-15 теоретических тарелок. Состав отобран-
ных фракций анализируют методом ГЖХ. Из 92 г (1 моль) толуола
получают 84,3 г (86 %) метилциклогексана. Т. кип. 141 °C,
1,4190. Спектральные характеристики приведены на рис. 1.12.
Дигидрорезорцин
(циклогександион-1,3)
н2
Nic)t.
Реактивы
Резорцин 55 г
Никель скелетный 10 г
Гидроксид натрия 24 г
Водород(из баллона)
96
В автоклав вместимостью 0,5 л, снабженный механической
мешалкой, помещают 24 г (0,6 моль) гидроксида натрия, 100 мл
дистиллированной воды, 55 г (0,5 моль) резорцина и 10 г скелетного
никеля. В автоклаве создают давление 70-100 атм и устанавливают
температуру не выше 50 °C (при более высокой температуре обра-
зуются сложные продукты конденсации). Через 10-12 ч поглоща-
ется рассчитанное количество водорода.
Реакционную смесь, выгруженную после охлаждения из ав-
токлава, фильтруют для удаления катализатора, добавляют кон-
центрированную хлороводородную кислоту до получения кислой
реакции по конго, охлаждают до 0 °C. Выкристаллизовавшийся ди-
Рис. 1.11. Спектральные характеристики циклогексиламина:
а-спектр ПМР* в СС14 (<5, м. д.: 1,4 м (ЮН, СН2), 1,51 м (2Н, NH2), 2,64 м (1Н, CH));
б - НК спектр в топком слое (3270 и 3360 см-' (N-H))
97
гидрорезорцин отфильтровывают и сушат. Получают 50-60 г про-
дукта с примесью хлорида натрия. Для удаления последнего полу-
ченный продукт растворяют в хлороформе, раствор фильтруют.
Выделившийся дигидрорезорцин выделяют и сушат. Получают
47,6 г (85 %) почти бесцветного вещества; т. пл. 104-106 °C. До-
полнительную очистку можно провести перекристаллизацией из
Рис. 1.12. Спектральные характеристики метилциклогексана:
а - спектр ПМР* в СС14 (<5, м. д.: 0,85 д, J = 7 Гц (ЗН, СН3), 1,2 и 1,6 м (НН, СН2 и СП));
б - ИК спектр в тонком слое
98
этилацетата либо возгонкой при температуре 80 °C и давлении
2 мм рт. ст.
Дигидрорезорцин - вещество нестойкое. Сохранять его в те-
чение длительного времени следует под инертным газом в темном
сосуде на холоде. Спектральные характеристики приведены на
рис. 1.13.
Рис. 1.13. Спектральные характеристики дегидрорезорцина:
а - спектр ПМР* в CDCI3 соответствует содержанию в растворе смеси кето-енольной
формы и дикето-формы в соотношении примерно 2 : 1; <5, м. д.: 1,96 м (ЗН, $СН2 и $СН2),
2,40 т, J = 6 Гц (4Н, 6СН2 и 4'.6'СН2), 2,58 т, J = 6 Гц (2Н, 4СН2), 3,39 с (1Н, 2СН2), 5,58 с
(2Н, СН=), 0,12 с (Ш, ОН)); б-ИК спектр в КВг (1605 см-1 (С=О))
99
Дициклогексано-18-краун-6
Реактивы
Дибензо-18-краун-6 18 г
Палладий (4 %) на угле 10 г
Водород(из баллона)
Декан 120 мл
В автоклав вместимостью 0,5 л, снабженный механической ме-
шалкой, помещают 18 г (0,05 моль) дибензо-18-крауна-6, 120 мл
декана или бензина и 10 г катализатора. Устанавливают давление
100 атм и автоклав нагревают до 170 °C, а затем выдерживают в
этих условиях до поглощения рассчитанного количества водорода.
Реакционную смесь выгружают, автоклав промывают 15 мл раство-
рителя. Промывную жидкость объединяют с реакционной смесью и
фильтруют. Остаток на фильтре промывают 50 мл используемого
растворителя. Шлам собирают для регенерации палладия.
Объединенный фильтрат упаривают на ротационном испарите-
ле при температуре 130-150 °C. Получают 18 г жидкого продукта.
Его растворяют в небольшом объеме гексана и хроматографируют
на оксиде алюминия (2-я активность) с использованием колонки
длиной 10-15 см. Элюент - гексан. После отгонки гексана остаток
затвердевает. Его можно перекристаллизовать из бензина. Из раст-
вора медленно выпадают бесцветные кристаллы дициклогекса-
но-18-крауна-6. Выход составляет 7,4 г (40 %). Спектральные ха-
рактеристики приведены на рис. 1.14.
Ме2СО
Диизопропиламин
ж-.NHj, Н2
СиО-СцО, + ВаО-Сг2О3
Реактивы
Ацетон
Медно-хромовый катализатор Адкинса
Аммиак жидкий
Водород (из баллона)
»3O-Pr2N Н
58 г
4 г
15 мл
100
В автоклав вместимостью 0,5 л помещают 58 г (1 моль) ацетона,
5 мл дистиллированной воды, 4 г меднохромового катализатора и
15 мл (0,59 моль) жидкого аммиака. Автоклав закрывают, нагне-
тают в него водород до давления 100-120 атм, нагревают до 160 °C
Рис. 1.14. Спектральные характеристики дициклогексано-18-крауна-6;
а - спектр ПМР* в (CD3)2CO 0, м. д.: 1,53 м (16H, СН2), 3,58 м (2ОН, СН2О и СНО));
б - ИК спектр* в КВг (1100 см1 (С-О))
4 Заказ № 137
101
и выдерживают при этой температуре 3 ч. После охлаждения авто-
клава выгружают реакционную смесь. Ее фильтруют для удаления
катализатора, катализатор, осевший на фильтре, промывают 50 мл
горячей воды (70 °C). Фильтрат насыщают хлоридом натрия и экст-
рагируют эфиром (100 мл). Органический слой отделяют и трижды
промывают водой (по 10 мл), сушат таблетированным гидроксидом
натрия и фракционируют, собирая фракцию с температурой кипе-
ния 80-85 °C. Ее сушат гидроксидом натрия и повторно перего-
няют. Получают 35 г (70 %) диизопропиламина с т. кип. 83-85 °C,
Пд° 1,3922. Спектральные характеристики диизопропиламина даны
на рис. 1.15.
Рис. 1.15. Спектральные характеристики диизопропиламина:
а - спектр ПМР* в СС14 (<5, м. д.; 0,82 д.д, J = 6,5 и 1 Гц (1Н, СН), 2,63 епт, J = 6,5 Гц (6Н,
СНз)); б - ИК спектр в тонком слое (2980 см(N-H))
102
Глава 2. ВОССТАНОВЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
КОМПЛЕКСНЫМИ ГИДРИДАМИ МЕТАЛЛОВ
Среди реакций восстановления, широко используемых в орга-
нической химии, важное место занимают реакции, связанные с
передачей восстанавливаемому соединению гидрид-иона. Такого
типа реакции крайне важны и для биологических систем, многие из
которых претерпевают восстановление за счет передачи субстрату
гидрид-иона от молекул, содержащих 1,4-дигидропиридиновый
фрагмент.
В органической химии в качестве синтетического эквивалента
нуклеофильного синтона Н“ наиболее часто используют комплекс-
ные гидриды металлов. Последние приобрели в настоящее время
исключительное значение прежде всего благодаря тому, что реак-
ции с ними протекают в мягких условиях, имеют высокую селек-
тивность и дают хороший выход продуктов. Введенные более 50
лет назад в практику органического синтеза, комплексные гидриды
резко расширили область применения реакций восстановления.
В настоящее время используется большое число разнообразных
по строению комплексных гидридов металлов. Все они благодаря
отчетливо выраженному нуклеофильному характеру взаимодейст-
вуют преимущественно с полярными кратными связями, такими
как С=О, C=N, C=N, N=O и т. п. Наибольшее значение имеет вос-
становление карбонилсодержащих соединений. Во всех случаях,
как и следовало ожидать, атака гидрид-иона происходит по элект-
рофильному атому углерода или гетероатому. Нуклеофильный ха-
рактер присущ всем комплексным гидридам металлов и отличает их
от восстановителей других типов. Однако в зависимости от строе-
ния они имеют разную реакционную способность и селективность.
103
2.1. НЕЗАМЕЩЕННЫЕ АЛЮМО- И БОРОГИДРИДЫ МЕТАЛЛОВ
Наиболее широкое практическое применение находят алюмо-
гидриды и борогидриды металлов. Из этой группы соединений ча-
ще других используют алюмогидрид лития UAIH4 и борогидрид
натрия ИаВНд.
Впервые полученный в 1947 г., алюмогидрид лития в настоящее
время является одним из самых универсальных реагентов. Он вос-
станавливает в мягких условиях различные по характеру ненасы-
щенные группировки и оставляет без изменения в большинстве
случаев лишь кратные С-С-связи и связи С-0 простых эфиров.
Обращает на себя внимание разнообразие способных к восстанов-
лению алюмогидридом лития функциональных групп (табл. 2.1). Из
карбонилсодержащих соединений альдегиды, кетоны, кислоты, их
производные могут быть легко восстановлены до спиртов. Подда-
ются восстановлению оксимы, нитросоединения и галогеноуглево-
дороды.
Мощные восстановительные свойства и связанный с этим ши-
рокий спектр действия алюмогидрида лития оказываются нежела-
тельными при восстановлении полифункциональных соединений,
так как могут препятствовать селективности реакции: при наличии
в молекуле нескольких групп зачастую не удается восстановить
одну из них, не затрагивая при этом другие, способные к восстанов-
лению группы. И здесь становятся очевидными преимущества
борогидрида натрия. Он является мягким восстановительным
реагентом, доступен, не реагирует в отличие от ЫАШд с водой и
спиртами, и поэтому условия работы с ним чрезвычайно просты.
Благодаря ослабленным восстановительным свойствам ЫаВНд пре-
вращает альдегиды и кетоны в соответствующие спирты в при-
сутствии различных функциональных групп, таких как нитро-,
карбалкокси-, галогене-, нитрильная группы. Например:
COOEt
CH3C(CH2)2NO2
NaBH4
МеОН
NaBH4
МеОН
О
COOEt
CH3CH(CH2)2NO2
он
104
PhCCH2Br
II
NaBH4
МеОН
PhCHCH2Br
О ОН
О высокой селективности борогидрида натрия свидетельствует
тот факт, что он может быть использован для восстановления кар-
бонильной группы насыщенного кетона в присутствии в той же мо-
лекуле сопряженной карбонильной группы. Делокализация элект-
ронной плотности в ненасыщенной системе уменьшает ее чувстви-
тельность к нуклеофильной атако, и борогидрид натрия атакует
лишь атом углерода с наименьшей электронной плотностью, како-
вым и является атом углерода карбонильной группы насыщенного
кетона:
В определенных экспериментальных условиях борогидридом
натрия удается даже восстановить альдегидную группу в присутст-
вии кетонной:
Me
Me---
Me С
II
О
О
II
СН2СН
NaBH4
Me
СН2СН2ОН
EtOH, СН2С12, -78 °C
Me С
II
О
97 %
В целом алюмогидрид лития и борогидрид натрия заметно отли-
чаются друг от друга по восстановительным свойствам - реакцион-
ной способности, хемоселективности и, как будет показано ниже,
также по стереоселективности. В то же время свойства одного хоро-
шо дополняют свойства другого. Именно поэтому в лабораторной
практике часто обходятся лишь этими двумя комплексными гидри-
дами металлов.
Находят, однако, применение и алюмогидриды других метал-
лов - натрия NaAlH4, магния Mg(AlH4)2, а также борогидриды ли-
нь
тия ЫВНд, калия КВН4, кальция Са(ВН4)2 и тетрабутиламмония
BU4NBH4. Из перечисленных реагентов наиболее доступен боро-
гидрид лития. Сравнением его восстанавливающих свойств со
свойствами борогидрида натрия удается проследить влияние катио-
на на реакционную способность комплексных гидридов металлов.
Так, борогидрид натрия практически не восстанавливает сложные
Таблица 2.1. Соединения, восстанавливаемые LiAlH4
Исходное соединение1) Продукт Расход (гидридный эквивалент)2)
Альдегид RCHO Спирт RCH2OH 1
Кетон R2C=O Спирт R2CHOH 1
Хлороангидрид RCOC1 Спирт RCH2OH 2
_ r2c-cr2 Эпоксид \ / О Спирт R2CH-C(OH)R2 1
Сложный эфир R'CC^R2 Спирты R’CH2OH + R2OH 2
Лактон RCOO 1 1 Диол HO-R-CH2OH 2
Амид RCONR2 Третичный амин RCH2-NR2 2
(дизамещенный) или альдегид RCHO 1
Амид RCONHR (монозамещенный) Вторичный амин RCH2NHR 3
Амид RCONH2 (незамещенный) Первичный амин RCH2NH2 4
Нитрил RCN Первичный амин RCH2NH2 2
или альдегид RCHO 1
Кислота RCOOH Спирт RCH2OH 3
Оксим RCH=NOH Амин RCH2-NH2 3
Нитросоедннение RNO2 (алифатическое) Амин RNH2 и др. 6
Галогениды RHal Углеводород RH 1
') Порядок перечисления соответствует примерной реакционной способ-
ности.
2) Li AIH4 использует для восстановления четыре атома водорода, и его гид-
ридный эквивалент равен 0,25 моль.
106
эфиры (реакция идет слишком медленно), а борогидрид лития вос-
станавливает их очень быстро и поэтому особенно удобен для пе-
ревода сложных эфиров в спирты:
LiBH,
CH3(CH2)16COOEt -------—-------- СН3(СН2)16СН2ОН
74 %
X X
PhCH = CHCOOEt --------------► PhCH = CHCH,OH 98 %
ДИГЛИМ z
Все синтезированные алюмогидриды оказались мощными вос-
становителями, и это обстоятельство затруднило получение данных
о влиянии металла на активность алюмогидридного аниона. Тем не
менее кинетические исследования процессов восстановления кето-
нов алюмогидридами лития и натрия в ТГФ позволили установить,
что LiAlEU в 10 раз более реакционноспособен, чем NaAlEU. Найде-
но также, что NR4AIH4 менее активен по отношению к камфоре,
чем NaAlH4. Таким образом, для алюмогидридной серии оказы-
вается верен следующий ряд реакционной способности: LiAlH4 >
NaAlH4 > NR4AIH4. Понять до конца роль катиона металла помо-
гает знание механизма реакции (см. 2.5). Здесь лишь отметим, что
изменение природы металла является одним из способов модифи-
кации свойств комплексного гидрида металла.
2.2. ЗАМЕЩЕННЫЕ АЛЮМО- И БОРОГИДРИДЫ МЕТАЛЛОВ
Альтернативным путем модификации реакционной способ-
ности комплексных гидридов является замена одного или несколь-
ких атомов водорода на алкокси-, алкил-, ацетокси- или цианогруп-
пы. По восстановительным свойствам такие соединения заметно
отличаются от незамещенных.
Наибольшее распространение получили алкоксизамещенные
гидриды. Три(яу2ея1-бутокси)алюмогидрид лития LiAlH(OBu-
трет)у, например, легко восстанавливает альдегиды, кетоны и хло-
107
роангидриды кислот до спиртов, но не восстанавливает сложные
эфиры и нитрилы:
Замечательной особенностью этого алюмогидрида является
также то, что в растворе диглима при температуре - 78 °C он восста-
навливает хлороангидриды и диметиламиды кислот до альдегидов.
Присутствие в молекуле таких групп, как CN, NO2, COOR и др. не
препятствует селективному восстановлению:
Li Al H (О Ви - трет )3
диглим , -78 °C
88 %
Повышенная хемоселективность три(/ире/и-бутокси)алюмогид-
рида лития оказывается особенно полезной в химии сложных орга-
нических соединений, например, стероидного ряда. Так, небольшая
скорость восстановления а,/3-ненасыщенных кетонов делает воз-
можным селективное восстановление кетогрупп стероида в при-
сутствии сопряженной кетогруппы. Таким образом 3,17-диоксоанд-
ростен-4 был превращен в 17/3-ацетокси-3-оксоандростен-4:
Однако ни алюмогидрид лития, ни три(яу2ея1-бутокси)алюмо-
гидрид лития не являются подходящими реагентами для превраще-
ния нитрилов и амидов в альдегиды. Очевидным преимуществом
обладает здесь триэтоксиалюмогидрид лития ЫА1Н(ОЕ0з - он
108
селективно восстанавливает ароматические и алифатические нит-
рилы, а также М,М-дизамещенные амиды в альдегиды с высокими
выходами:
LiAlH(OEt),
CH3(CH2)2C = N —СН3(СН2)2СНО 68%
CH3(CH2)2CONMe2 .СН3(СН2)2СНО 90 %
Г.12О, о с
Другой алкоксизамещенный алюмогидрид - триметоксиалюмо-
гидрид лития LiAlH(OMe)3 является сильным восстановителем,
близким по свойствам алюмогидриду лития. Ди(2-метоксиэток-
си)алюмогидрид лития LiAlH2(OCH2CH2OMe)2 выгодно отлича-
ется от алюмогидрида лития, с которым он обладает примерно
одинаковыми восстановительными свойствами, и от других алко-
ксиалюмогидридов прекрасной растворимостью во многих раство-
рителях, в том числе, что особенно ценно, в неполярных раствори-
телях типа бензола, толуола и т. п. Хорошая растворимость этого
комплексного гидрида даже при низких температурах позволяет
восстанавливать сложные эфиры до альдегидов:
трет -Bu С О О Me
LiAlH2 (ОСН2 СН2 ОМе)2
ТГФ , -78 °C
трет -ВиСНО
Таким образом, в целом алкоксизамещенные гидриды более
селективны, чем незамещенные. Однако четкой корреляции между
природой алкоксигруппы, входящей в состав гидрида, и его селек-
тивностью нет. Селективность часто зависит от концентрации раст-
воров, от способности комплексного гидрида ди- и тримеризовать-
ся в разных растворителях. Положение осложняется способностью
некоторых алкоксизамещенных алюмогидридов диспропорцио-
нировать с образованием более реакционноспособного тетрагидро-
алюминатного иона:
2Н3А1ОРг-изо .......А1Н4 + Н2А1(ОРг-изо)2
Необычными свойствами обладают также комплексные гидри-
ды металлов, в которых водород замещен алкильной группой. Хотя
эти соединения известны давно, их свойства изучены лишь в по-
следние десятилетия. Из этого типа реагентов все более широкое
применение находят триэтилборогидрид лития LiBHEt3, три(в/иор-
109
бутил)борогидрид лития Ь1ВН(Ви-в/иор)з и три(в/иор-бутил)бо-
рогидрид калия КВН(Ви-етиор)з, а также бутилборогидрид лития
ЫВНзВи:
Li+
Триэтилборогидрид лития (,,супергидрид“) восстанавливает те
же функциональные группы, что и борогидрид лития. Однако
благодаря индуктивному эффекту трех алкильных групп его нук-
леофильность оказалась в 20 раз больше, чем тиофеноксидного
иона, и в 1 000 раз больше, чем борогидрида лития. Оказалось также,
что триэтилборогидрид лития является прекрасным реагентом для
восстановительного расщепления первичных и вторичных алкил-
бромидов и тозилатов. Он, однако, не восстанавливает арилгалоге-
нидов, и это дает возможность селективного восстановления алкил-
и арилгалогенидов. Далее, триэтилборогидрид лития с высокой
степенью стерео- и региоселективное™ раскрывает эпоксиды, да-
вая спирты, соответствующие присоединению воды к алкену по
правилу Марковникова, практически без примеси изомерного
спирта:
99 %
99 %
Другие перечисленные выше триалкилборогидриды исполь-
зуют главным образом для стереоселективного восстановления
кетонов (см. 2.6.1). Здесь ограничимся лишь одним примером, из
которого можно видеть повышенную стереоселективность три-
ио
(вдаор-бутил)борогидрида лития, что связано, по-видимому, с раз-
ветвленностью его алкильного радикала:
NaBH4
КВН(ОВи-л1/2ел1)з
ЫВН(Ви-в/лор)з
31 %
92%
99,3 %
69%
8%
0,7 %
Замена атома водорода комплексных гидридов металлов на ци-
аногруппу приводит к получению цианогидридов, из которых наш-
ли применение цианоборогидриды натрия NaBt^CN и тетрабутил-
аммония BU4NBH3CN. Электроноакцепторные свойства циано-
группы делают эти реагенты мягче и селективнее, чем борогидрид
натрия. Уникальность цианоборогидрида натрия состоит в том, что
он, в отличие от других гидридов, стабилен в кислых растворах
вплоть до pH 3. Такая устойчивость к кислотам часто бывает очень
полезной в органическом синтезе. Так, селективное восстановление
этим реагентом может проводиться, если в соединении присутст-
вуют такие функциональные группы, которые чувствительны к
более основным условиям восстановления борогидридом натрия. В
нейтральном растворе при использовании в качестве растворителя
гексаметилфосфотриамида NaBH3CN удаляет иодо-, бромо- и този-
локсигруппы, при этом другие восстанавливаемые группы не затра-
гиваются:
PhCH-CHCH.Br NaBH3CN^ phCH-CHCH, 63 %
\ / z 70 °C \ / J
О О
Альдегиды и кетоны в нейтральных средах также оказываются
инертными по отношению к этому восстановителю. Лишь в кислых
средах они легко восстанавливаются через промежуточное образо-
вание протонированной формы:
+
ОН
PhCHO рН 3~4 - с NaBH3cN phCH2OH
НС1, МеОН у 2
Ph Н
111
Иминиевые соли, являясь аналогами протонированных карбо-
нильных соединений, также восстанавливаются NaBt^CN. Эти
соединения могут быть получены из альдегида и амина в нейтраль-
ной среде и далее восстановлены в тех же условиях. Это позволило
предложить удобный вариант проведения реакции восстановитель-
ного аминирования:
, Н
пН 6 +/ NaBH.CN
PhCHO + EtNH2 =5£=±± PhCH=N -------------2---- PhCH,NHEt
\ МеОН, 25 °C 2
Et 80 %
Реакция наилучшим образом протекает в присутствии молеку-
лярных сит с размером пор 0,3 нм, селективно адсорбирующих
воду; применима также к получению а-аминокислот:
nh2
PhCH2CHCOOH
49 %
PhCH2COCOONa
NH4Br, NaBH3CN
МеОН
2.3. СМЕШАННЫЕ ГИДРИДЫ
Другим приемом модификации восстановительных свойств
комплексных гидридов металлов является использование их в сме-
си с некоторыми солями (чаще - кислотами Льюиса). Такие систе-
мы получили название „смешанные гидриды14. Природа их не всег-
да может быть установлена и меняется в зависимости от соотно-
шения гидрид : соль. Так, часто применяется добавление к LiAlH4
хлорида алюминия, обычно в соотношении от 1 : 1 до 1 : 4. По-ви-
димому, в таких системах в качестве восстановителя выступает ряд
гидридов алюминия:
LiAlH4 + А1С13 -> LiCl + 2А1Н2С1
LiAlH4 + 3 AICI3 -* LiCl + 4 А1НС12
LiAlH4 + 4 AICI3 -> LiCl + 4 A1HC12 + AICI3
Эти уравнения лишь приблизительно описывают состав реакци-
онной системы LiAlH4 - AICI3. Хлороалюминийгидриды в растворе
представляют собой сольваты (эфираты). В процессе их обра-
зования возникают ионизированные промежуточные продукты,
которые диспропорционируют. Поэтому система может содержать
хлорид алюминия. Его присутствие ослабляет нуклеофильный
112
характер восстановительной системы и одновременно увеличивает
электрофильные, кислотные свойства. Поэтому система LiAlbU -
AICI3 обладает более слабой восстановительной активностью, чем
алюмогидрид лития. Кроме того, действие ее оказывается специ-
фическим в отношении ряда реакций. Например, связь углерод-
галоген не затрагивается смешанными гидридами.
Это преимущество может быть использовано при восстановле-
нии полифункциональных соединений. Например, в то время как в
результате восстановления метилового эфира 3-бромопропионовой
кислоты алюмогидридом лития образуется лишь пропанол-1, сме-
шанный гидрид дает 3-бромопропанол-1:
ВгСН2СН2СООМе .-L.lA1H4~А1С'3 (1:1)^ ВгСН2СН2СН2ОН
EtjO
Необычно ведут себя в присутствии смешанных гидридов также
диарил- и арилалкилкетоны. Их карбонильная группа восстанав-
ливается при этом до метиленовой, что является основой удобного
синтетического метода, альтернативного восстановлению по Клем-
менсену (амальгама цинка и хлороводородная кислота).
Восстановление смешанным гидридом 1лА1Н4-А1С1з (3:1) пред-
ставляет собой удобный путь синтеза ненасыщенных спиртов из
а,/3 -ненасыщенных карбонильных соединений, которые трудно по-
лучить, используя лишь LiAlH4, из-за конкурентного восстановле-
ния кратных углерод-углеродных связей. Действующим реагентом,
по-видимому, здесь является гидрид алюминия (алан), образован-
ный in situ:
3 LiAlH4 + AICI3 -> 3 LiCl + 4 AIH3
LiAlH,- AICI, (3:1)
PhCH^CHCOOEt --------------------— PhCH = CHCH,OH
Et,0 2
90 %
Примечательно также, что рассматриваемая восстановительная
система полностью изменяет направление раскрытия кольца эпок-
сида и в результате с высоким выходом образуется спирт, соот-
ветствующий присоединению воды к алкену против правила Мар-
ковникова:
Ph2C(OH)CH2Ph
Ph2C—CHPh
LiAlH4 — AICI3 (3:1)
Ph2CHCH(OH)Ph
О
113
Необычны и уникальны свойства систем, полученных совмест-
ным использованием IJAIH4 или NaBH4 и галогенидов бора. Они
могут восстанавливать сложные эфиры и лактоны до простых эфи-
ров, особенно если спиртовая компонента имеет структуру третич-
ного спирта. Реальным восстанавливающим агентом, возможно,
является в таких системах диборан:
3 NaBH4 + 4 BF3 -» 3 NaBF4 + 2 В2Н6
Особенно успешным оказалось совместное использование с
комплексными гидридами металлов солей лантаноидных металлов,
прежде всего потому, что они существенно модифицируют свойст-
ва борогидрида натрия. Например, хлорид церия (III) в составе
таких систем селективно восстанавливает менее реакционноспо-
собные карбонильные группы, не затрагивая более реакционноспо-
собные. Так, кетоны могут быть восстановлены в присутствии аль-
дегидов:
МеС
II
1. NaBH4 - СеС13-6Н2О
EtOH, Н2О
2. Н3О +
Me
Me----
МеСН
I
ОН
В этом случае, возможно, альдегидная группа защищена от восста-
новления образованием гидрата, который стабилизируется комп-
114
лексообразованием с ионом церия, а затем регенерируется в процес-
се выделения продукта.
Таким образом, комплексные гидриды металлов обладают ши-
роким диапазоном действия. Благодаря этому неодинаковые функ-
циональные группы можно восстанавливать с разной скоростью,
что позволяет выбрать реагент, а также растворитель и условия ре-
акции так, чтобы достичь желаемой хемоселективности, а часто и
стереоселективности восстановления. Выбор подходящего комп-
лексного гидрида металла можно сделать на основании данных
табл. 2.2.
2.4. ПОЛУЧЕНИЕ КОМПЛЕКСНЫХ ГИДРИДОВ МЕТАЛЛОВ.
ВЫБОР РАСТВОРИТЕЛЯ
Преимуществом комплексных гидридов как восстановителей
является их доступность и легкость получения в лаборатории.
Растворенный в эфире алюмогидрид лития получают в резуль-
тате взаимодействия гидрида лития с хлоридом или, лучше, броми-
дом алюминия в эфире:
4 LiH + AlHaL --------► LiAlH4 + 3 LiHal
J Et2O 4
Полагают, что реакция протекает через промежуточное образо-
вание гидрида алюминия (алана), который быстро реагирует с гид-
ридом лития:
3LiH + А1На13 ------► 3 LiHal + А1Н3
LiH + А1Н3 -------► LiAlH4
Раствор, который отфильтровывают от выпавшего в осадок га-
логенида лития и непрореагировавшего гидрида алюминия, готов к
употреблению. После его выпаривания и удаления в вакууме соль-
ватирующего эфира может быть получен LiAlH4 95 %-ной чистоты.
Этот способ применяется в промышленности.
При необходимости знать точную концентрацию алюмогидри-
да лития ее определяют либо газометрическим способом (измере-
нием объема водорода, выделившегося при осторожном разло-
жении точно отмеренного объема реагента раствором воды в ТГФ),
либо йодометрическим способом (добавлением избытка бензольно-
го раствора иода и обратным титрованием непрореагировавшего
иода тиосульфатом, см. 2.8).
115
116
Таблица 2.2. Реакционная способность различных функциональных групп в реакциях
с комплексными гидридами металлов при температуре 0 4-25 °C
Реакция Восстановитель
LiAlH4 в эфире NaBH4 в метаноле LiBH4* 1>B диглиме LiAlH(OBu- трет)з вТГФ LiAlH (ОМе)з вТГФ LiAlH2(OCH2 СН2ОМе)2 LiBHEtj в эфире NaBH3CN
RCHO -* RCH2OH 4- + + 4- 4- 4- 4- 4-
r2co -» R2CHOH 4- + + 4- 4- 4- 4- 4-
rcoci-*rch2oh 4- +2) 4- +3) 4- 4- 4-
Лактон -* диол 4- - 4- - 4- 4- 4- -
Эпоксид -* спирт 4- - 4- - 4- 4- 4- -
R1COOR2 *-»RCH2OH + r2oh 4- - 4- -4) 4- 4- 4- -
rcooh-*rch2oh 4- - - - 4- 4- - -
rcoo--»rch2oh 4- - - - 4- 4- - -
rconr2-»rch2nr2 4- - - - 4- 4- -
rcn-»rch2nh2 4- - - - 4- - 4- -
RNO2 -» RNH25) +3) - - - 4- 4-
RHal-»RH 4- - - - 4- 4- +6) 4-
rch=ch2 -» rch2ch3 - - - - - - - -
Примечание. Плюсом обозначено наличие реакции, минусом - очень медленная реакция или ее отсутствие.
1) Получен из NaBH4 и Li Cl. 2) Реакция с растворителем; восстановление легко протекает в гидроксилнесодержащих
средах. 3) Реакция проходит лишь до стадии образования альдегида при -78 °C. 4) Сложные эфиры фенолов дают
альдегиды. 5) Алифатический радикал. 6) В случае, если R- арильный, реакция отсутствует.
Если при получении алюмогидрида лития использовать LiD или
LiT, то удается получить LiAlD4 и LiAlT4, которые имеют большое
значение при установлении строения органических соединений и
при изучении механизмов реакций.
Алюмогидриды других металлов (натрия, магния и т. д.) можно
получить аналогично тому, как это было описано для LiAlH4, а так-
же другим, более эффективным (но и более дорогим) способом,
который основан на действии хлоридов металлов на алюмогидрид
лития в подходящем растворителе:
MCI + LiAlH4 ---------► LiCl + МА1Н4
Борогидрид натрия можно приготовить различными способами.
В ряде случаев исходным соединением служит триметоксибор. При
действии на него гидрида натрия, взятого в избытке, и диборана
образуется борогидрид натрия:
NaH + В(ОМе)3 ---------- NaBH(OMe)3
2NaBH(OMe)3 + В2Н6 ---------- 2 NaBH4 + 2 В(ОМе)3
Борогидриды других металлов можно получить аналогичным
образом, однако чаще используют доступный NaBH4, обменивая в
нем катион обработкой соединениями другого металла в подходя-
щем растворителе:
NaBH4 + LiCl ----------- LiBH4 + NaCl
диглим
3 NaBH4 + A1C13 ► A1(BH4)3 + 3 NaCl
Алкоксизамещенные алюмо- и борогидриды металлов также
оказываются достаточно доступными реагентами. Их удобно полу-
чать действием стехиометрического количества соответствующего
спирта на алюмогидрид лития или борогидрид натрия:
LiAlH4 + 3 ROH -------► LiAlH(OR)3
Триалкоксизамещенные гидриды обычно не склонны к диспро-
порционированию и устойчивы в растворе. При необходимости они
могут быть выделены в индивидуальном состоянии, перекристал-
лизованы или сублимированы.
Как правило, работа с комплексными гидридами металлов не
требует создания специальных условий эксперимента. Она прово-
дится с использованием обычной лабораторной посуды при ком-
117
натной, повышенной или пониженной температуре. Гидриды вно-
сятся в реакционную смесь растворенными, и только ИаВНд и
некоторые другие можно прибавлять порциями в твердом виде. В
случае алюмогидрида лития используют его небольшой избыток
(5-10 %), учитывая, что часть соединения реагирует со следами
воды и других гидроксилсодержащих примесей. После завершения
восстановления непрореагировавший гидрид и образовавшийся
комплекс гидролизуют, для чего осторожно прибавляют воду или
водные растворы хлорида натрия, хлорида аммония или гидроксида
натрия. Если реакционная смесь содержит большой избыток
LiAlH4, то для разложения реакционной смеси рекомендуется ис-
пользовать этилацетат, который медленно реагирует с гидридом,
давая этиловый спирт. Последний не мешает дальнейшему выделе-
нию продуктов реакции.
Условия эксперимента могут сильно влиять на ход реакций вос-
становления. Например, нитрилы образуют амины, если нитрил до-
бавлять в раствор L1A1H4, и альдегиды, если раствор L1A1H4 добав-
лять к нитрилу.
Особое внимание при работе с комплексными гидридами метал-
лов уделяется подбору растворителя. Здесь решающее значение
имеют устойчивость комплексных гидридов металлов, температура
кипения растворителя (в том случае, если реакция требует нагре-
вания), а также растворимость исходных соединений (табл. 2.3).
Алюмогидрид лития легко реагирует с водой и спиртами, поэто-
му они не могут быть использованы в качестве растворителей.
Более того, вещества, содержащие подвижный водород, следует
исключать из реакционной среды во избежание разложения LiAlH4.
Наиболее широко в качестве растворителя для проведения реак-
ций с алюмогидридом лития применяют диэтиловый эфир, в кото-
ром он достаточно хорошо растворим. В других простых эфирах с
более высокой температурой кипения алюмогидрид лития раство-
ряется хуже, в бензоле, толуоле, хлороформе он нерастворим вовсе.
Если возникает необходимость проведения реакции при относи-
тельно высокой температуре, то используют ТГФ, глим или диглим,
реже - диоксан. Использовать температуру выше 100 °C, однако,
нежелательно, так как становится заметным термическое разложе-
ние алюмогидрида лития:
LiAlH4 -> LiH + Al + 1,5 Н2
118
Таблица 2.3. Растворимость некоторых комплексных гидридов металлов
(г на 100 г растворителя) при 25 ± 5 °C
Гидрид Растворитель
H2O MeOH EtOH Et2O ТГФ Диоксан Глим Диглим
LiAlH4 35-40 13 0,1 7(12)') 5(8)'’
LiBH4 2,52) 2,52) 2,5 28 0,06
NaBH4 55(88,5)3) 16,42) 202) 0 0,1 0,8 5,5
NaBHjCN 212 СИЛЬНО раство- римый слабо раство- римый 0 37,2 17,6
NaBH(OMe)3 1,6(4,5)')
LiAlH(OBu- mpem)^ 2 36 4 41
О При 75 °C.
2) Сопровождается разложением.
3) При 60 °C.
Вещества, растворимость которых в эфире ограничена, можно
перевести в раствор при помощи экстрактора Сокслета. В некото-
рых случаях бывает удобным применять смесь растворителей или
суспензию твердого вещества в эфире. Восстановление суспензий
является, как правило, наиболее безопасным приемом работы.
В противоположность алюмогидриду лития борогидрид натрия
(а также калия, но не лития) очень медленно реагирует с водой и
спиртами, обладает высокой стабильностью, и восстановление с его
помощью можно проводить в этих средах. Это обстоятельство
делает борогидрид натрия наиболее подходящим реагентом для
восстановления альдегидов и кетонов. Стабильность водных раст-
воров борогидрида натрия зависит от содержания щелочи в раст-
воре. При комнатной температуре водные растворы при pH > 9 ус-
тойчивы в течение длительного времени, но при температуре около
100 °C быстро разлагаются. В этаноле борогидрид натрия более
устойчив, однако при нагревании также разлагается (при 60 °C на
33 % за 4 ч). Устойчивость в метаноле меньше: при комнатной
температуре борогидрид натрия медленно реагирует с метанолом,
выделяя водород. Поэтому использовать МеОН рекомендуется
лишь при пониженных температурах, что возможно в том случае,
когда восстановление идет быстро.
119
Следовательно, этанол как растворитель имеет преимущества:
реакция проходит в гомогенном растворе с относительно малым
расходом восстанавливающего агента на побочную реакцию с раст-
ворителем. Часто для восстановления борогидридом натрия ис-
пользуют изопропанол, в котором он умеренно растворим (0,1 М
при 25 °C) и совершенно устойчив. Этот растворитель целесообраз-
но применять, если для реакции требуется более высокая темпера-
тура или длительное время.
Приведем наиболее распространенные растворители для неко-
торых комплексных гидридов металлов:
Реагент Растворитель
L1AIH4 NaAlH4 NaAlH2(OCH2CH2OMe)2 LiAlH(OBu-m/?ezn)3 NaBH4 Et20, ТГФ, глим, диглим, CH2CI2 ТГФ, глим, диглим PhH, PhMe, СбН4Ме2, Et2O, ТГФ ТГФ, диглим Н2О, МеОН, EtOH (реже 2-метоксиэтиловый эфир)
NaBHjCN NaBH(OMe)3 ЫВН4 Me4NBH4 Н2О (рН>3), МеОН, ТГФ, ДМСО Et2O, ТГФ ТГФ, диглим EtOH, PhH
Небезынтересным является вопрос о влиянии растворителя на
восстановительную силу комплексных гидридов металлов. Для
алюмогидрида лития такую зависимость проследить не удается, так
как его высокая реакционная способность ограничивает выбор
растворителей, сводя его лишь к простым эфирам, в которых он яв-
ляется мощным реагентом; различия в восстановительной силе при
этом незначительны. Напротив, использование борогидрида нат-
рия, являющегося мягким восстановителем, позволяет заключить,
что роль растворителя может быть чрезвычайно большой. Так, вос-
становление ацетона заканчивается за несколько минут в водном
или спиртовом растворе и вовсе не наблюдается при проведении
реакции в растворителях эфирного типа - ТГФ, диглиме и триг-
лиме, хотя ЙаВЩ хорошо растворим в них. Следовательно, раство-
ритель важен не только для достижения гомогенности среды. Роль
его более сложна и может быть осмыслена лишь с учетом механиз-
ма реакции.
120
2.5. МЕХАНИЗМ ВОССТАНОВЛЕНИЯ
Представления о нуклеофильном характере комплексных гид-
ридов металлов вполне согласуются с экспериментальными данны-
ми по восстановлению органических соединений, различающихся
природой функциональных групп и строением углеродного скеле-
та. Так, способность к восстановлению карбоновых кислот и их
производных изменяется в ряду
RCOO - < RCOOH < RCONR2 < RCN < RCOOR < RCOHal.
Известно также, что константа скорости восстановления бензаль-
дегида борогидридом натрия в диглиме в 400 раз больше константы
скорости восстановления ацетофенона; это позволяет считать, что
легкость восстановления увеличивается при переходе от альдеги-
дов к кетонам.
Эти факты вполне согласуются с общими представлениями об
атаке гидрид-иона, входящего в состав комплексного аниона, на
электронодефицитный атом функциональной группы. Однако бо-
лее детальные исследования показали, что механизм восстановле-
ния зависит не только от природы субстрата, но в значительной
степени и от природы комплексного гидрида (реагента).
Восстановление альдегидов и кетонов борогидридом натрия в
изопропаноле (так же как и в других гидроксилсодержащих раство-
рителях) происходит путем постепенного замещения гидрид-ионов
борогидридного аниона молекулами растворителя с одновремен-
ной атакой гидрид-иона на электронодефицитный атом углерода
карбонильной группы:
R2C=O + изо-РгОН + ВН4"----------- ГвН3(О Рг- изоТ ^С° ’ “то'Рг0Н„
-r2choh L J -r2choh
г -|- R2CO, изо-PrOH r ")~R,CO,u3o-PrOH
----- ВН2(ОРг-мзо)2 " |ВН(ОРг-озо)з| 2 -
L J K^VtlUrl L -J Kj^HUrl
В результате используются все четыре гидрид-иона борогид-
рида натрия, но стадией, определяющей скорость, является первая,
т. е. та, на которой происходит перенос гидрид-иона от незамещен-
ного борогидрида. Увеличение скорости реакции на каждой после-
дующей стадии связано с тем, что алкоксиборогидриды, как было
121
показано выше, являются более сильными восстановителями, чем
борогидрид натрия. Учитывая, что в условиях восстановления
алкоксиборогидриды натрия не диспропорционируют (однозначно
это удалось показать лишь для моноалкоксиборогидрида натрия),
следует признать, что реакция последовательного восстановления
карбонильного соединения осуществляется четырьмя разными вос-
станавливающими агентами. В результате реакции образуется
свободный спирт и тетраалкоксибораты, имеющие алкоксигруппы
растворителя. Роль растворителя, однако, этим не ограничивается.
По-видимому, он выполняет одновременно функцию электрофиль-
ного катализатора, координируясь с кислородным атомом группы
С=О. В целом для борогидридного восстановления в спирте пред-
ложен синхронный механизм с ациклическим переходным состоя-
нием следующего вида:
_ R
4 Pi
изо-РгО ’ ’ • Н3В—Н ’ • • ’ • • Н—ОРг-изо
R
Кинетические исследования восстановления кетонов борогид-
ридом натрия показывают, что переходное состояние по энергии
ближе к продуктам реакции и состав реакционной смеси обуслов-
лен термодинамической стабильностью продуктов. Тонкие детали
механизма остаются, однако, не ясными.
Как видно из структуры переходного состояния, катион натрия
не играет особой роли при борогидридном восстановлении в гидро-
ксилсодержащих растворителях. Поэтому добавление к раствору
борогидрида натрия в этих растворителях дибензо-18-крауна-6,
захватывающего катион натрия, не меняет скорости реакции. На-
против, добавление литиевых солей катализирует восстановление
борогидридом натрия, обнаруживая таким образом, что катион
лития является лучшим катализатором, чем протон спирта. Этот
каталитический эффект исчезает в водных растворах, где катион
лития сильно сольватирован.
Хотя детальное изучение восстановления кетонов борогидри-
дом лития в апротонных растворителях не проводилось, все же
полагают, что реакции проходят аналогично восстановлению боро-
гидридом натрия, за исключением того, что роль электрофильного
катализатора выполняет катион лития.
122
Иным путем происходит восстановление альдегидов и кетонов
алюмогидридом лития. Атака свободным ионом или ионной парой
приводит к образованию моноалкоксиалюмогидрида. Этот проме-
жуточный продукт реагирует со второй молекулой карбонильного
соединения, и образуется диалкоксиалюмогидрид, который далее
реагирует с третьей молекулой карбонильного соединения, превра-
щаясь в триалкоксиалюмогидрид. Наконец, последний реагирует с
четвертой молекулой карбонильного соединения, в результате чего
образуется тетраалкоксисодержащий анион, который и освобож-
дает при взаимодействии с водой четыре молекулы спирта:
Rx
/С5=<3 . R2C = O
R ] •, + ------► Li+H3A1 -OCHR2 ---------------
’Li
А1Н3
. _ R,C = O , _ R,C = O
—►Li + H2A1 (OCHR2)2 —-------*-LiHAl (OCHR2)3 —------►
---- Li+AF(OCHR2)4 —H--° - 4R2CHOH + LiAl(OH)4
Изучение кинетики реакции показало, что в противоположность
борогидридному восстановлению вхождение в анион алкоксигруп-
пы уменьшает восстановительную способность алюмогидрида, и
поэтому каждая последующая стадия передачи гидрид-иона про-
ходит медленнее предыдущей, так что самой быстрой стадией ока-
зывается первая. При этом передача гидрид-иона на каждой из
стадий облегчается координацией карбонильного атома кислорода
с катионом металла, которая поляризует карбонильную группу,
вследствие чего глубина восстановления зависит от координирую-
щих свойств катиона металла. Можно полагать, что катион лития
из-за меньшего размера поляризует карбонильную группу больше,
чем катион натрия, и поэтому гидрид-ион более легко переносится
от алюминия к углеродному атому в случае LiAlH4, чем NaAlH4.
Это вполне согласуется с их относительной реакционной способ-
ностью, отмеченной выше.
Однако следует учитывать, что при восстановлении с помощью
LiAlH4 вопрос о природе истинного восстанавливающего агента
часто не может быть решен однозначно. Это связано с возмож-
123
ностыо диспропорционирования промежуточных алкоксиалюмо-
гидридов:
2 LiAlH3(OR) -> LiAlH2(OR)2 + LiAlH4
2 LiAlH2(OR)2 -> LiAl(OR)4 + LiAlH4
Получающийся в таком случае алюмогидрид лития восстанавлива-
ет новую порцию карбонильного соединения и является, возможно,
единственным восстановителем в процессе всей реакции. Однако
некоторые алкоксигидриды оказываются вполне стабильными и не
склонными к диспропорционированию. В таком случае на второй,
третьей и четвертой стадиях реакции восстановителем является не
тетрагидроалюминатный анион, а анионы типа A1(OR),!H4.,I.
Природа переходного состояния в случае восстановления
LiAlH4 остается во многом неясной. Полученные кинетические до-
казательства свидетельствуют лишь, что переходное состояние по
энергии ближе исходным реагентам, и в целом реализуется кинети-
ческий состав продуктов реакции.
Механизм восстановления кислот и их функциональных произ-
водных известен лишь в самых общих чертах. Общую схему восста-
новления этих соединений можно представить следующим обра-
зом. По-видимому, вначале происходит нуклеофильное присоеди-
нение гидридного эквивалента по карбонильной группе с образова-
нием алкоголята А:
о ом у—r’ch2om + mx
r'-c-x 1МН - r'-ch-x -н Б
А \—- r'ch2x + м2о
X = Cl, OR2, ОН, NH2, NHR2, NR2 В
MH = V4LiAlH4
Естественно, что при восстановлении соединений, имеющих сво-
бодный водород, происходит также его замещение металлом. При
гидролизе соответствующего алкоголята А образуются альдегиды.
На второй ступени восстановления происходит нуклеофильное
замещение с участием второго гидридного эквивалента и обра-
зуется алкоголят Б. Гидролиз его приводит к образованию спиртов.
И лишь в случае амидов возможен гидрогенолиз связи С-0 в алко-
124
голяте А, в результате чего получаются амины В. Аналогичный
процесс, в ходе которого сложные эфиры преобразуются в простые,
идет только в присутствии такой сильной кислоты Льюиса, как BF3.
Такой механизм более вероятен, чем предположение, согласно
которому из алкоголята А при гетеролизе связи С-Х in situ обра-
зуется альдегид, который восстанавливается далее до спирта.
Восстановление других классов органических соединений ком-
плексными гидридами металлов может происходить иным путем.
Так, восстановление алкилгалогенидов, эфиров сульфокислот и
эпоксидов протекает как нуклеофильное замещение 5у2 типа, в
процессе которого перенос гидрид-иона осуществляется атакой
аниона AIH4 _. Как и следовало ожидать, при восстановлении эпок-
сидов происходит обращение конфигурации атома углерода, ата-
куемого алюмогидридом лития, а в случае несимметричного эпок-
сида связь углерод-кислород разрывается со стороны наименее
замещенной связи в соответствии со значимостью стерических пре-
пятствий в 8^2 реакциях. Восстановление винил-, циклопропил- и
арилгалогенидов может проходить другим путем: по карбанионно-
му, четырехцентровому или радикальному механизму в зависимос-
ти от природы восстановителя и условий реакции.
2.6. ВОССТАНОВЛЕНИЕ ВАЖНЕЙШИХ КЛАССОВ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
2.6.1. Альдегиды и кетоны
Комплексные гидриды металлов чаще других реагентов исполь-
зуют для восстановления альдегидов и кетонов. Наиболее употреб-
ляемые алюмогидрид лития и борогидрид натрия восстанавливают
альдегиды и кетоны соответственно до первичных и вторичных
спиртов. Как следует из уравнения реакции, на восстановление
1 моль карбонильного соединения требуется всего 0,25 моль реа-
гента:
4 RCHO + LiAlH4------- (RCH2O)4LiAl Н2° -4RCH2OH
4 R2CO + LiAlH4 ------ (R2CHO)4LiAl Hz° 4 R2CHOH
Алюмогидрид лития и борогидрид натрия восстанавливают
алифатические, ароматические и гетероциклические альдегиды и
125
кетоны. Даже стерически затрудненные кетоны типа димезитилке-
тона без труда восстанавливаются алюмогидридом лития.
Изучение относительной скорости восстановления кетонов и
альдегидов позволило заключить, что в целом большинство кето-
нов восстанавливается медленее. На основании этого были разрабо-
таны методы хемоселективного восстановления альдегидной функ-
ции в присутствии кетонной. Применение для этой цели борогидри-
да натрия рассмотрено выше. Другой подход состоит в использо-
вании модифицированных гидридов, таких как Li А1Н(ОВи-«гре«г)з,
но особую селективность здесь проявили три(алкилтио)борогидри-
ды и цианоборогидриды тетрабутиламмония:
СНО
R1
ЧС = О
r2/
R1 - двухвалентный радикал
Na+BH( SBu-mpem)3
(Bu4N+BH3CN-)
СН2ОН
R1
чс=о
r2/
90 %
СН2ОН
Z
чснон
r2/
5 %
Восстанавливаемое карбонильное соединение может содержать
двойные и тройные связи, а также такие группы, как NR3, ОН, OR, F
и др., которые, как правило, остаются без изменения:
NaBH4
LiAlH4
Et2O
ОН
Однако если карбонильное соединение содержит группу, кото-
рая способна восстанавливаться, такую как COOR, CONR2, SH и
др., то при использовании L4AIH4 они восстанавливаются одновре-
менно с карбонильной группой. Лишь при использовании МаВЩ
эти группы остаются без изменения:
СНО
Вг
/
СН = С
СН2ОН
126
Галогенозамещенные кетоны и альдегиды можно восстановить
до соответствующих галогеноспиртов даже с помощью ГлАЦ-Ц,
причем связи С-галоген не претерпевают гидрогенолиза:
С13С-С-СС13 —-1А1Н4 - С13С-СН-СС13 90 %
О ОН
Лишь в жестких условиях галоген может быть замещен на водород.
Из приведенных примеров можно видеть, что при восстанов-
лении полифункциональных соединений не всегда просто вос-
становить другие функции, не затрагивая карбонильной группы
альдегидов и кетонов. Однако, как известно, ацетальные группы не
восстанавливаются даже алюмогидридом лития. Это обстоя-
тельство имеет большое значение в органическом синтезе, позволяя
использовать метод защиты карбонильной группы альдегидов и
кетонов при восстановлении полифункциональных соединений.
Например, ацетоуксусный эфир превращают в 4-оксибутанон-2
восстановлением его ацеталя алюмогидридом лития с последую-
щим мягким кислым гидролизом:
9 О О О
II НО ОН, Н+ \ / L1AIH4
Me- С-CH2COOEt ------------► Me-C-CH2COOEt --------►
О О ®
\ / н2о, н+ II
-----«► Me- С-СН2СН2ОН —- Ме-С-СН2СН2ОН
80%
Благодаря хорошей растворимости в воде и метаноле борогид-
рид натрия находит применение для восстановления сахаров. При
этом гликозидные связи не затрагиваются. Селективно протекает
восстановление альдегидных групп сердечных гликозидов и агли-
конов.
Лишь в редких случаях восстановление группы С=О альдегидов
и кетонов осложняется процессом гидрогенолиза (восстановления
до СНз- и СНг-групп соответственно). К такому типу превращений
склонны, в частности, ароматические альдегиды, содержащие в
ароматическом кольце электронодонорные группы, а также многие
диарил- и алкиларилкетоны. Для проведкения гидрогенолиза осо-
бенно эффективным оказывается применение системы LiAlH4 -
AICI3. Возможно, А1С1з как кислота Льюиса образует комплекс с
127
карбонильной группой и, увеличивая электрофильность атома угле-
рода, таким образом способствует гидрогенолизу:
МеО—~СНО ... L1A1»4 ~ А1С1з (1 :2) Ме0_^А~^_ Ме
Эта реакция, очевидно, идет через промежуточное образование
бензилового спирта, в котором электронодонорные заместители
благоприятствуют реакции замещения гидрид-ионом по S/yl или
SnI механизму.
Некоторые затруднения, которые, однако, удается преодолеть,
возникают при восстановлении а,/3-непредельных карбонильных
соединений. Как было отмечено выше, комплексные гидриды
металлов, как правило, не восстанавливают двойные связи. Такая
инертность в целом полезна, так как позволяет селективно про-
водить восстановление полярных групп. Например, каротиноидные
спирты, которые долгое время не удавалось получить, сейчас
успешно синтезируют из соответствующих карбонильных соеди-
нений:
LiAlH4
Et2O, О "С
Me Ме
ОН
I
СН = СНСНМе
90%
Ме
Однако результат восстановления а,/3-непредельных альдеги-
дов и кетонов не всегда бывает однозначным. В зависимости от
строения субстрата, природы реагента и условий реакции конечный
продукт может представлять собою непредельный или предельный
спирт. Так, если сравнить наиболее часто употребляемые алюмо-
гидрид лития и борогидрид натрия (табл. 2.4), видно, что последний
менее подходит для селективного восстановления «^-непредель-
ных соединений: он чаще, чем алюмогидрид лития, восстанавли-
вает одновременно карбонильную группу и двойную связь. При
использовании LiAIH4 осложнений иногда удается избежать благо-
даря тщательному подбору условий (см. данные для коричного
альдегида). В некоторых случаях хорошие результаты дает приме-
нение смеси LiAlH4 и AICI3, а также борогидрида натрия и хлоридов
церия:
128
Таблица 2.4. Выход (%) непредельных (А) и предельных (Б) спиртов
при восстановлении а,/?-непредельных карбонильных соединений
алюмогидридом лития и борогидридом натрия
Соединение LiAlH4 NaBH4
Общий А Б Общий А Б
СН2=СНСНО 70 98 2 70 85 15
МеСН=СНСНО 94 100 0 91 92 8
СН2=СНСОМе 79 92 8 86 57 43
МеСН=СНСОМе 85 99 1 90 65 35
PhCH=CHCHO 98 0 100 97 100 0
871) 100 0
PhCH=CHCOPh 65 юо2) 0 83 183) 82
PhCH= С НСОС6Н4ОМе 504) 0 100 87 37 63
{=}=о 97 98 2 90 59 41
4—7 О
Ме-^ Рг-шо 100 100 0 69 38 62
о=° 100 85 15 100 0 100
Примечание. Соотношение соединений А и Б в реакционной смеси определялось
методом ГЖХ. ') Обратный порядок прибавления реагентов, - 10 °C. 2) - 78 °C,
2ч- выход А 99 %. 3) - 20 °C, 1 ч - выход А 65 %. 4) 31 % исходного соединения не
прореагировало.
129
Важным аспектом реакций восстановления карбонильных сое-
динений комплексными гидридами металлов является их стерео-
селективность. В результате восстановления несимметричных
ациклических кетонов получают, как и следовало ожидать, рацеми-
ческие спирты. Однако восстановление кетонов, имеющих рядом с
группой С=О хиральный центр, приводит к предпочтительному
образованию одного из диастереомерных спиртов. Так, при восста-
новлении алюмогидридом лития кетонов, имеющих три различных
a-заместителя, «грео-форма спирта оказалась преимущественным
продуктом реакции (R = Ме, 74 %; R = Et, 76 %; R = нзо-Рг, 83 %;
R = трет-Ви, 98 %):
А1Н3
/Н
н С О HR HR
\ ^// ч / Ъ /
Ме^О-С --------- Ме*-'С—С ЮН + Ме*~С —С "Н
/ \ /V /V
Ph R Ph Н Ph ОН
трео эритро
Результаты хорошо объясняются с учетом пространственных
факторов. Реагент подходит со стороны, противоположной самой
объемной группе (Ph). При этом конформация молекулы кетона в
момент реакции такова, что относительно небольшой карбониль-
ный кислород располагается ближе к группе среднего размера (Ме),
а наиболее объемная группа (Ph) оказывается ближе к самой ма-
ленькой группе (Н):
130
Аналогично в случае стерически затрудненных циклических
кетонов реагент подходит преимущественно с более доступной сто-
роны. Так, восстановление камфоры алюмогидридом лития приво-
дит в основном к образованию экзо-спирта (изоборнеола), в то
время как норкамфора, в которой подход гидрид-аниона со стороны
метиленового моста легче, чем в камфоре, дает преимущественно
энЭо-спирт (борнеол):
Me Me' Me Me
Y Me Y Me
У У OH
LiAlH4 Л''|^'"У
Z—*'''^/ Et^O
LiAlH4
Et2O OH
Стереоселективность восстановления возрастает с увеличением
объема и нуклеофильности комплексного гидрида, а также зависит
от природы растворителя. Это можно видеть на примере реакции
восстановления камфоры (табл. 2.5). Отклонение от отмеченных в
таблице закономерностей, характерное для три(/ире/и-бутокси)алю-
могидрида лития, связано с тем, что его истинный объем меньше,
чем триметоксиалюмогидрида: в растворе ТГФ первый мономерен,
второй - полимерен, так что, по-видимому, для объема этих сое-
динений верен ряд ЫА1Н(ОМе)з > LiAlH4 > 1лА1Н(ОВи-лгрелг)з.
Таблица 2.5. Восстановление камфоры комплексными гидридами металлов
Гидрид, растворитель Выход, %
изоборнеол борнеол
LiAlH4, Et2O 90 10
LiAlH4, ТГФ 92 8
LiAlH(OMe)3, ТГФ 99 1
LiAlH(0Bu-mpem)3, ТГФ 93 7
LiAlH4-AlC13 (1:4), Et2O 73 27
LiBH(Bu-emop)3, ТГФ 99,6 0,4
LiBH(C6Hi 1)3, ТГФ 99,3 0,7
131
Стереохимический результат восстановления циклических ме-
нее затрудненных кетонов, например ряда циклогексанона, пред-
сказать несколько труднее. В зависимости от направления атаки
гидрид-иона на замещенные циклогексаноны можно ожидать обра-
зования двух стереоизомерных спиртов: аксиальная атака приводит
к образованию экваториального спирта, в то время как эквато-
риальная - аксиального спирта (циклогексановое кольцо должно
содержать заместители, чтобы избежать конформационных пере-
ходов):
Из двух возможных спиртов преимущественно образуется эква-
ториальный, что связано, скорее, не с его большей устойчивостью, а
с особенностями переходного состояния. Так, восстановление
борогидридом натрия холестан-3-она, имеющего фиксированную
структуру, дает выход экваториального спирта 94 %:
Преобладание экваториального спирта наблюдается также в
ряду пространственно незатрудненных циклогексанонов (эквато-
риальный заместитель в положении 4 не создает стерических за-
труднений):
О // X] LiAlH4^ mpem-Qu^Js^ 8 н ОН трет !9 % 1 1 %
132
Однако в случае пространственно затрудненных кетонов важ-
ным оказывается фактор, связанный со стерическим затруднением
подхода к карбонильной группе. В результате аксиальный спирт
может оказаться главным продуктом реакции.
Восстановление, например, 3,3,5-триметилциклогексанона (ак-
сиальные заместители в положениях 3 и 5 создают пространствен-
ные затруднения) дает два стереоизомерных спирта, из которых
преобладает аксиальный. Последний является практически единст-
венным продуктом, если используют более селективный восстано-
витель - три(трет-бутокси)алюмогидрид лития:
Me Ме
Для получения аксиального спирта подходящими оказываются
не алюмогидрид лития или борогидрид натрия, а более селективные
восстановители. Широкое распространение в последнее время
находит три(в/пор-бутил)борогидрид лития. Этот реагент восста-
навливает 4-т/?ет-бутилциклогексанон до аксиального спирта с
96 %-ным выходом. Получить экваториальный спирт из прост-
ранственно затрудненного циклогексанона сложнее, но здесь ока-
залось полезным наблюдение, что смесь аксиального и экватори-
ального спиртов, полученную восстановлением ЕлАШд или NaBbU,
можно превратить в экваториальный спирт, выдержав ее со скелет-
ным никелевым катализатором:
Me Ме
Удается решить вопрос стереоселективности восстановления в
химии стероидов и других сложных природных соединений с регу-
лярной структурой.
5 Заказ № 137
133
2.6.2. Хлороангидриды и ангидриды кислот
Из производных карбоновых кислот хлороангидриды относятся
к наиболее легко восстанавливаемым соединениям. При этом ис-
пользование L1AIH4 обычно приводит к превращению галогепоан-
гидридов и ангидридов кислот в первичные спирты:
2 RCOC1 + LiAlH4 ----► (RCH2O)2CI2AlLi Н2°— 2 RCH2OH
(RCO)2O + LiAlH4 ----► (RCHO)2OAlLi H2° - 2 RCH2OH
Выходы продуктов восстановления обычно высокие. Особенно лег-
ко восстанавливаются хлороангидриды, и это используют, когда не
удается восстановить соответствующую кислоту:
Li Al Н4
Me.CCOCl —- Ме4ССН,ОН 86 %
J Et2O 3 z
В синтетическом плане наибольший интерес представляет воз-
можность частичного восстановления хлороангидридов кислот до
альдегидов. Одним из наиболее подходящих реагентов для этого
является три(ти/>ети-бутокси)алюмогидрид лития, так как он при
низких температурах не реагирует с альдегидами, что исключает
дальнейшее восстановление их в спирты:
_ LiAlH(OBu-mpe„i), f) Al—/ т. Н,О
RCOC1 ------------। Li —RCHO
- 78 °C I <
RCHC1
Реакция применима к хлороангидридам кислот алифатического
и ароматического ряда, имеющим различные заместители, включая
группы NO2, CN, COOR, МеСО, а также двойные углерод-углерод-
ные связи. Например:
Ангидриды кислот реагируют труднее. Чаще используют вос-
становление циклических ангидридов дикарбоновых кислот, кото-
рые в зависимости от условий могут давать гликоли или лактоны.
Так, более сильные комплексные гидриды восстанавливают фтале-
вый ангидрид до фталилового спирта, в контролируемых условиях,
134
избегая избытка комплексных гидридов, удается восстановить фта-
левый ангидрид до лактона:
LiAlH4, Et2O, 35 °C 87 %
NaAlH2(OCH2CH2OMe)2, PhH, 80 °C 88,5 %
NaBH4, ДМФА, 0-25 °C 97 %
LiBHEt3, ТГФ, 0 °C 76 %
2.6.3. Сложные эфиры и лактоны
Наиболее часто используется восстановление сложных эфиров
до спиртов:
2 r'cOOR2 + LiAlH4 ----- [(R'CH2O)2(R2O)2]AlLi
----- 2 ^СН2ОН + 2 R2OH
При получении спиртов восстановлением сложных эфиров ком-
плексные гидриды металлов применяются сейчас чаще, чем другие
восстановители. В тех случаях, когда целью является получение
спирта из соответствующей кислоты, целесообразно восстановле-
нию подвергать не саму кислоту, а ее метиловый или этиловый эфи-
ры. Выходы спиртов обычно высокие:
LiAlH,
МеСН(ОМе)СН2СООМе ------- МеСН(ОМе)СН2СН2ОН
2 70 %
Реакция восстановления сложных эфиров может быть осущест-
влена также под действием борогидрида лития. Использование
этого реагента предпочтительно, если эфир содержит другие вос-
станавливаемые группы. Борогидрид натрия восстанавливает лишь
сложные эфиры фенолов, причем особенно хорошо в тех случаях,
когда в ароматическом кольце содержатся электроноакцепторные
группы. С другими сложными эфирами взаимодействие NaBH4
протекает слишком медленно. Поэтому обычно удается восстано-
135
вить карбонильную группу альдегида и кетона, не затрагивая слож-
ноэфирной функции в той же молекуле. Но если очень большой
избыток (до 16 моль) NaBH} кипятить со сложным эфиром в мета-
ноле 1-2 ч, то можно получить высокий выход спирта (72-93 %).
Сложные эфиры удается восстановить и до альдегидов:
С5НиСООМе
С5НИСНО
Реакцию, однако, удается осуществить лишь при низких температу-
рах и с помощью более мягких реагентов, чем алюмогидрид лития:
LiAlHi, ТГФ, -78 °C
NaAlEU, ТГФ, -60 ч- - 45 °C
49 % (22 % гексанола)
85%
NaAlH2(Bu-iwo)2, Et2O, - 70 °C 72 %
Эфиры фенолов восстанавливаются до альдегидов при более
низких температурах и более длительное время с выходом на
10-15 % ниже, чем эфиры алифатических спиртов. Например,
ЫА1Н(ОВи-и1/?ети)з восстанавливает селективно эфиры фенолов в
альдегиды с хорошими выходами:
1. Li AlH(OBu-mpem),
CH3(CH2)4COOPh ———СН3(СН2)4СНО + PhOH
62 %
Лактоны, подобно сложным эфирам, легко восстанавливаются
алюмогидридом лития, причем продуктами восстановления явля-
ются двухатомные спирты:
ОН
,0
LiAlH4
сн=снсн2он
Et2O
75 %
2.6.4. Амиды и лактамы
Амиды карбоновых кислот с помощью алюмогидрида лития
можно превратить в соединения трех типов: альдегиды, амины или
спирты. При этом все реакции проходят через стадию образования
аминосодержащего алкоголята (см. 2.5).
136
-----------------► r'cho + R?2NH + МОН
ом
R'CONR^ -^Ur'cHNR22-------► имин R1CH2Nl62 + M2O
MH - l/4LiAlH„ R‘CH20H + ^NH + 2M°H
Полное восстановление амидов до аминов употребляется наи-
более часто. При этом промежуточный алкоголят стабилизируется
элиминированием кислородсодержащей группы, так как она явля-
ется лучше уходящей, чем азотсодержащая группа. Образующийся
в результате имин восстанавливается далее.
Согласно суммарному уравнению реакции, учитывающему
взаимодействие гидридного реагента с водородом аминогруппы,
для восстановления дизамещенного амида требуется 0,5 моль, мо-
нозамещенного - 0,75 моль и незамещенного - 1 моль алюмогид-
рида лития:
2R'CONR22 + LiAlIH ->2R'CH2NR22 + LiA102
4R'CONHR2 + 3LiAlH4 -> LiAl(NR2CH2R')4 + 2LiA102 + 4H2
2RCONH2 + 2UAIH4 -> LiAl(NCH2R)2 + LiA102 + 4H2
Практически для восстановления амидов до аминов следует
применять избыток ГлАЩд. Дизамещенные амиды уже при
25-30 %-ном избытке LiAlHt восстанавливаются количественно и
быстро. При восстановлении монозамещенных амидов их следует
длительное время (12-20 ч) кипятить с 200-250 %-ным избытком
LiAlbU. Предполагается, что комплекс, образующийся при выделе-
нии активного водорода первичного и вторичного амида, реагирует
дальше с большим трудом.
Возможность восстановления амидов с образованием альдеги-
дов зависит от структуры амида и гидрида, а также от условий реак-
ции. Иногда бывает достаточно смешивать реагенты в обратном
порядке при низкой температуре или использовать стехиометри-
ческое количество алюмогидрида лития. Но решающую роль играет
электронное влияние заместителей при атоме азота. Селективное
образование альдегидов из амидов кислот облегчается, если ско-
рость нуклеофильного присоединения по карбонильной группе с
образованием аминоалкоголята превышает скорость нуклеофиль-
137
ного замещения с образованием спирта и амина. Виттит установил,
что такое условие выполняется , если нуклеофильность атома азота
амидной группы понижена, вследствие чего нормальная мезомерия
амидной группы нарушена сдвигом электронов в обратном направ-
лении к атому азота. Это обстоятельство облегчает нуклеофильную
атаку LiAlFL» на карбонильный углерод. Атом азота в результате
заполняет свою электронную оболочку за счет присоединения гид-
рид-иона, а не в результате сдвига электронной пары:
R’-C-NR2, ------R1-C=NR2 ----------► r’-c-nr2 ------►
II 2 I 2 Л I
о о ( _ о"
Н •”H3AlLi
----► r’-ch-nr2 Н2° ► ^СН=О + Й>ЫН
1^Э
О
Практически увеличение доли положительного заряда на карбо-
нильном атоме углерода достигается переводом кислот в соответст-
вующие N-метиланилиды и производные азотсодержащих гетеро-
циклов, главным образом ацильные производные азолов (свободная
пара электронов азота делокализована в ароматической л:-системе):
ЫА1Н4
н2о
RCHO
Удобным способом получения альдегидов является восстановление
N-ацилазиридинов, которые могут быть генерированы in situ:
LiAlH4
Et2O
60 %
Селективности при восстановлении амидов можно достигнуть
применением алкоксизамещенных алюмогидридов, о чем уже упо-
миналось выше. Так, три(/и/?ет-бутокси)- и триэтоксиалюмогидри-
ды лития легко восстанавливают М,М-диметиламиды до альдеги-
дов:
CH3cH,scH,coNMe, ..E1AIH(OEt2?..«. CH3CH2SCH2CHO
3 2 2 2 Et2O, 0 °C 270%2
Лактамы ведут себя аналогично амидам в том смысле, что
обычно восстанавливаются алюмогидридом лития до циклических
138
вторичных аминов, а при стехиометрическом количестве гидрида в
некоторых случаях образуют аминоспирты. Реакция представляет
собой один из важнейших методов синтеза азотсодержащих гете-
роциклов. Так, восстановление лактама 4-метил-4-аминопентано-
вой кислоты приводит к образованию 2,2-диметилпирролидина:
LiAlH4
ТГФ
Как и в других реакциях гидрогенолиза, часто оказывается
полезной комбинация алюмогидрида лития с хлоридом алюминия:
LiAlH4 — А1С13
Et2O
94 %
2.6.5. Нитрилы
В результате восстановления нитрилов алюмогидридом лития
можно получить либо амин, либо альдегид. Полагают, что проме-
жуточным продуктом восстановления является имин (координа-
ционно связанный с металлом), и, следовательно, если остановить
реакцию на этой стадии, то после гидролиза получается альдегид;
дальнейшее восстановление имина дает первичный амин:
I. LiAIH4
2.Н2О
4RC = N + LiAlH4----- LiAl(N-CHR)4
\ Н2°
4RCH2NH2
4RCHO
Таким образом, теоретически на исчерпывающее восстанов-
ление 1 моль нитрила в амин расходуется 0,5 моль LiAIRt. Однако
многие нитрилы требуют для восстановления большее количество
реагента. Так, для полного восстановления нитрилов до первичных
аминов рекомендуется применять не менее 200 %-ного избытка
LiAlH4 и, кроме того, проводить процесс при низких температурах,
139
так как при более высоких температурах могут образоваться вто-
ричные амины:
rch2nh2 rcho„ rch2n=chr -Ю.». rch2nhch2r
В целом восстановление нитрилов - удобный способ получения
первичных аминов (без примеси вторичных и третичных):
CH3(CH2)7CN L'A°4 - CH3(CH2)8NH2 90 %
<Q>-cn L1A'H4 » CH2NH2 88 %
Me Me
Для получения альдегидов помимо L1AIH4 используются также
NaAlH4 и LiAlH(OEt)3. Наиболее универсальным реагентом явля-
ется последний. Он легко превращает в альдегиды ароматические,
гетероциклические и алифатические нитрилы:
LiAIH(OEt)3
трет-BuC=N -----------------► трети-Bu С НО 74 %
Et2O, -30 °C
2.6.6. Карбоновые кислоты и их соли
Монокарбоновые кислоты реагируют с LiAlH4 согласно сле-
дующему уравнению:
4 RCodH + 3 LiA!H4 ------ LiAl(OCH2R)4 + 2 LiA102 + 4 H2
LiAl(OCH2R)4 + 4 H2O ----«► 4 RCH2OH + LiAl(OH)4
Таким образом, один гидридный эквивалент алюмогидрида лития
расходуется на реакцию с активным водородом кислоты и два гид-
ридных эквивалента на восстановление карбоксильной группы до
спиртовой.
Во многих случаях восстановление карбоновых кислот идет с
трудом. Поэтому их рекомендуется превращать в сложные эфиры
или хлороангидриды, которые восстанавливаются легче. Причина
инертности карбоновых кислот заключается, во-первых, в низкой
растворимости их литиевых солей в эфире, во-вторых, в понижен-
ной электрофильности углеродного атома карбоксильной группы.
140
В ненасыщенных кислотах двойные связи в общем не восста-
навливаются. Однако в том случае, когда двойная связь сопряжена с
ароматическим кольцом, она может восстановиться:
О
МеСН = СН-СН = СНС -bi-'--4-» МеСН=СН-СН=СНСН,ОН
\ Et2o 2
ОН 92 %
PhCH=CHCOOH L1A'H4 ► PhCH,CH,CH,OH 85 %
Et2O
2.6.7. Нитро-, нитрозосоединения, оксимы и имины
Нитросоединения алифатического ряда восстанавливаются до
первичных аминов с помощью LiAlHj, 1лА1Н(ОМе)з, LiAlH(OEt)3:
н,о
2 RNO2 + LiAlH4 ------«► LiAl(NR)2 -—- 2 RNH2
Двойная углерод-углеродная связь, сопряженная с нитрогруп-
пой, в обычных условиях восстанавливается до простой. Интерес-
ной особенностью этой реакции является то, что в отличие от
сопряженной карбонильной группы нитрогруппа способна присое-
динять водород после того, как восстановится двойная связь. Поэ-
тому при осторожном ведении реакции оказывается возможным
превращать ненасыщенные нитросоединения в насыщенные, не
затрагивая нитрогруппы:
МеО
МеО
ОМе
CH=CHNO2
LiAiH4
Et2O
МеО
МеО
ОМе
PhCH-CHNO2 L1A'-4-» PhCH2CH2NO2 40-60 %
По-другому проходит восстановление ароматических нитросое-
динений - реакция останавливается на стадии образования азо-
соединений:
»-BrC6H4NO2 LiA'H4 ► »-BrC6H4N=NC6H4Br-n 88 %
До азосоединений идет также восстановление ароматических
нитрозо- и азоксисоединений. С-Нитрозосоединения алифатичес-
кого ряда восстанавливаются в амины:
141
2 RNO + LiAlH4
H,o
LiAl(NR)2 -—*- 2 RNH2
Восстановление оксимов представляет собой наиболее удобный
путь перехода от альдегидов и кетонов к аминам:
н,о
2 R2C=NOH + LiAlH4------ LiAl(NCHR2)2——2 R2CHNH2
На восстановление 1 моль оксима требуется 0,75 моль LiAlH4, при
этом 0,25 моль гидрида расходуется на реакцию с активным водоро-
дом оксима. Рассматриваемая реакция является косвенным мето-
дом получения первичных аминов из соответствующих альдегидов
и кетонов. Однако выходы первичных аминов часто бывают невы-
сокими из-за возможности побочных процессов. В частности, арил-
кетоксимы образуют значительное количество вторичных аминов.
Вероятно, это происходит потому, что оксим вступает в перегруп-
пировку Бекмана:
перегруппировка
PhCHMe -----PhCMe ——-—!------------►PhNHCOMe *- PhNHEt
I II
NH2 NOH
В приведенном примере общий выход аминов составляет 75 %,
соотношение первичного и вторичного амина 4:1. Оксимы кетокис-
лот, имеющие заместитель в а- или /3-положении к оксиминогруп-
пе, и альдоксимы с ароматическими заместителями в /3-положении
к оксиминогруппе, реагируют с алюмогидридом лития аномально,
превращаясь в азиридины:
PhCH,CCH9Ph
II
NOH
Li Al H4
ТГФ
PhCH2 Ph
H NH н
Имины, основания Шиффа, гидразины и другие соединения, со-
держащие связь C=N, восстанавливаются под действием L1AIH4 и
NaBH4. Алюмогидрид лития и борогидрид натрия восстанавливают
также и иминиевые соли. Реакции проводят в нейтральной или, луч-
ше, в слабокислой среде. Поскольку циаиоборогидриды обладают в
этих условиях большей стабильностью, чем другие комплексные
гидриды, их применение предпочтительнее.
142
2.6.8. Эпоксиды и спирты
Восстановление эпоксидов протекает по Sn2 механизму и при-
водит к раскрытию цикла эпоксида с образованием спирта. В не-
симметричных эпоксидах гидрид-ион преимущественно атакует
менее замещенный углеродный атом, давая более замещенный
спирт. Селективность восстановления может быть увеличена при-
менением алкоксиалюмогидридов, как это можно видеть на при-
мере восстановления оксида стирола:
он
CH2CH2OH
LiAlH4, Et2O
LiAlH(OBu-m/?ewi)3
LiAlH(OMe)3, ТГФ
100 % о
99 % 1 %
В согласии с Sn2 механизмом восстановление эпоксидов проте-
кает с обращением конфигурации атакуемого гидрид-ионом угле-
родного атома. Поэтому эпоксид 1,2-диметилциклогексена дает
транс-1,2-диметилциклогексанол:
LiAlH4
Et2O
В присутствии кислот Льюиса направление раскрытия кольца
может быть изменено на противоположное. Так, восстановление
оксида 1-метилциклогексена цианоборогидридом натрия в при-
сутствии эфирата фторида бора дает не 1-метилциклогексанол, а
гщс-2-метилциклогексанол. Направление раскрытия кольца здесь
определяется образованием более стабильного карбониевого иона.
Восстановление первичных и вторичных спиртов проводится
обычно путем восстановления образованных из них тозилатов.
Последние легко восстанавливаются алюмогидридом лития, и это
часто используется в синтезе для замены гидроксильной группы на
водород (гидрогенолиз спиртов). Первичные спирты образуют то-
зилаты легче, чем вторичные, что дает возможность хемоселек-
143
тивного восстановления первичной гидроксильной группы. Это об-
стоятельство часто используется в химии природных соединений, в
частности сахаридов:
MeS SMe MeS SMe MeS SMe
но— TsCl но— LiAlH4 но—
но— ( Ру — ОН —он :н2он но — ( —он —он :h2ots “ но— ( —он — он :н3
23 %
2.6.9. Галогенопроизводные
При действии алюмогидрида лития на первичные и вторичные
алкилгалогениды происходит замещение галогена водородом (ре-
акция гидрогенолиза).
Легкость протекания этой реакции существенно зависит от при-
роды субстрата. В ряду неактивированных алкил- и циклоалкил-
галогенидов связь C-F обычно устойчива к действию комплексных
гидридов металлов, другие галогены по реакционной способности
располагаются в следующем порядке: I > Br > С1. Алюмогидрид
лития является вполне подходящим реагентом для восстановления
этого класса соединений. Борогидрид натрия (в апротонных раство-
рителях) также часто используется и имеет преимущества перед
LiAlH4 - он обладет большей селективностью по отношению к дру-
гим восстанавливаемым группам в той же молекуле. Наиболее
селективным, однако, является цианоборогидрид натрия, который
при pH 6 восстанавливает лишь немногие функциональные группы:
Br(CH2)4COOEt NaBH3CN » CH3(CH2)3COOEt 88 %
Полагают, что реакция идет как замещение Sn2 типа, однако
получены доказательства, что восстановление алкилгалогенидов,
особенно иодидов, может протекать по механизму, включающему
одноэлектронный перенос.
Третичные алкилгалогениды часто дают алкены, которые в ус-
ловиях восстановления подвергаются гидроборированию; при об-
ид
работке продукта гидроборирования кислотой образуются алканы,
так что в результате имеет место непрямой гидрогенолиз гало-
генида.
Бензил- и аллилгалогениды претерпевают восстановительное
расщепление легче, что иногда позволяет провести процесс селек-
тивно:
Комплексные гидриды металлов в определенных условиях
являются удобными и эффективными восстановителями и для
арилгалогенидов. Скорость реакции возрастает в ряду: фториды <
хлориды < бромиды < иодиды, а также в том случае, если галоген
активирован орто- и пара- электронодонорными группами или
присоединен к гетероциклической системе. Неактивированные
арилфториды и хлориды не восстанавливаются комплексными гид-
ридами металлов, а бромиды и, особенно, иодиды реагируют очень
легко. Это позволяет селективно восстанавливать полигалогено-
производные:
CI
LiAlH4
95 %
Обычным реагентом, используемым для гидрогенолиза, явля-
ется алюмогидрид лития в кипящем эфире, в ТГФ или диглиме.
Борогидрид натрия в неводных растворителях, таких как диметил-
сульфоксид или диглим, эффективен лишь при восстановлении
активированных бензилгалогенидов:
NaBH,
«-NO2C6H4CH2Br «-NO2C6H4CH3 98 %
2.7. ПРОМЫШЛЕННОЕ ПРИМЕНЕНИЕ
КОМПЛЕКСНЫХ ГИДРИДОВ МЕТАЛЛОВ
В настоящее время ЫАШд и другие сложные гидриды являются
соединениями, широко используемыми в органической химии и
145
применяются прежде всего там, где требуются хороший выход и
высокая степень чистоты продукта. К тому же некоторые процессы
восстановления трудно осуществляются с другими восстанавли-
вающими агентами. К таким процессам следует отнести восстанов-
ление кислот и сложных эфиров в спирты, восстановление амидов,
нитрилов и имидов в соответствующие амины. Применение LJAIH4
имеет здесь явные преимущества. Высокая реакционная способ-
ность комплексных гидридов порождает повышенные требования к
работе с ними. Каждый конкретный процесс предварительно тща-
тельно исследуется в лаборатории.
В настоящее время обращение с наиболее реакционноспособ-
ными гидридами типа LJAIH4 облегчается благодаря применению
различных видов упаковок для этих соединений. Гидрид может
поставляться в виде таблеток в защитных оболочках. Их бросают
целиком в реактор, оболочка растворяется в ТГФ или эфире, при
этом соприкосновение гидрида с воздухом исключается. Для
использования в промышленном масштабе L1AIH4 выпускается в
специальных контейнерах, которые можно присоединить прямо к
загрузочному отверстию реактора. Удобно также применять уже
готовые растворы в инертных растворителях. Так, 15 %-ный раст-
вор LJAIH4 в толуоле не является пирофорным в нормальных ус-
ловиях.
Примером процессов, осуществляемых в промышленности, яв-
ляется синтез витамина А из соответствующей кислоты или ее
эфира:
Используется ЫА1Н4 и при промышленном производстве стерои-
дов (прегнан), аминокислот (лизин), алкалоидов (мескалин, мор-
фин). Промышленный синтез морфина удалось осуществить благо-
даря получению /3-6-дигидродезоксикодеина из соответствующего
лактама:
146
МеО
Me О
ОМе
LiAlH4
ОМе
NH
Устойчивость пиримидинового кольца к действию IJAIH4
позволила синтезировать витамин Вь На промежуточной стадии
соответствующий этиловый эфир был селективно восстановлен до
спирта:
Особенно широко используются комплексные гидриды метал-
лов для промышленного синтеза фармацевтических и парфюмер-
ных препаратов.
2.8. ЭКСПЕРИМЕНТАЛЬНЫЕ РАБОТЫ
Особенности работы с комплексными гидридами металлов.
Несмотря на то, что с комплексными гидридами металлов обычно
несложно работать, следует помнить, что небрежное обращение с
этими богатыми энергией соединениями с большим содержанием
водорода может привести к пожару и взрывам. Поэтому при работе
со всеми комплексными гидридами металлов следует проявлять
особую осторожность.
На воздухе при комнатной температуре все комплексные гидри-
ды металлов достаточно стойки. Алюмогидрид лития, вероятно,
вследствие реакции его поверхностного слоя с влагой и углекис-
лотой воздуха, образует поверхностную защитную пленку гидро-
ксида алюминия, что предотвращает его разложение. Однако при
продолжительном хранении он превращается в серый порошок,
очень легко воспламеняющийся и способный взрываться. Таким
препаратом алюмогидрида лития пользоваться не рекомендуется, и
он подлежит уничтожению.
Комплексные гидриды металлов нужно тщательно защищать от
попадания на них влаги. Только борогидрид и цианоборогидрид
натрия медленно реагируют с водой. Напротив, алюмогидрид лития
147
и многие другие гидриды при контакте даже с небольшим коли-
чеством воды могут возгораться и взрываться. Для хранения алю-
могидрида лития лучше использовать стеклянные сосуды, которые
закрываются резиновыми пробками или плотными завинчивающи-
мися крышками. Сосуды предварительно должны быть заполнены
азотом. Алюмогидрид лития может воспламениться также при пе-
ресыпании с помощью стеклянных воронок и измельчении. Для из-
мельчения целесообразно использовать обернутые алюминиевой
фольгой ступки и пестики из твердой резины. Для пересыпания сле-
дует применять воронки из меди.
Разлитые реакционные смеси могут воспламеняться на воздухе
после того, как часть растворителя испарится, поэтому они должны
быть тщательно засыпаны песком.
Для работы с комплексными гидридами металлов следует при-
менять только свежеперегнанные, свободные от пероксидов раст-
ворители. Особенно опасны в этом смысле ТГФ и диметиловые
эфиры, из которых часто употребляется диглим. Реакция между
пероксидами и алюмогидридами плохо контролируется, поэтому
возможны взрывы.
При работе с большими количествами комплексных гидридов
металлов следует обеспечить отвод выделяющегося при их разло-
жении водорода от близко расположенного пламени, электромо-
торов, которые могут давать искру и т. п. Подлежащие уничто-
жению остатки комплексных гидридов металлов небольшими пор-
циями смешивают с сухим эфиром, не содержащим пероксидов,
переносят в колбу с мешалкой и обратным холодильником и по
каплям добавляют раствор сухого этилацетата в эфире, пока не
прекратится кипение эфира. Для тушения воспламенившегося гид-
рида очаги пожара рекомендуется засыпать песком, измельченным
мелом или поваренной солью. Применение воды, пенных и углекис-
лотных огнетушителей запрещается.
Приступать к выполнению работ следует после ознакомления с
инструкцией по технике безопасности при работе с гидридами ме-
таллов.
Получение комплексных гидридов металлов
а) Эфирный раствор алюмогидрида лития
4LiH + AlBr. —- LiAlH4+3LiBr
J Et-,O 4
148
Реактивы
Г идрид лития 1,8г
Бромид алюминия 10,1г
Эфир сухой 100 мл
80 мл сухого эфира помещают в плоскодонную колбу вмести-
мостью 0,3 л, закрывают колбу пробкой, в которую вставлена по-
глотительная трубка, наполненная хлоридом кальция, и охлаждают
в бане со льдом и солью. Небольшими порциями в колбу с эфиром
вносят через широкую воронку 10,1 г (0,038 моль) бромида алюми-
ния, стараясь, чтобы он не попадал на шлиф, и встряхивая колбу
после каждого добавления. Полученный раствор бромида алюми-
ния используют для приготовления эфирного раствора алюмогид-
рида лития.
В трехгорлую колбу, снабженную капельной воронкой, механи-
ческой мешалкой и эффективным холодильником, защищенным от
влаги воздуха поглотительной трубкой, наполненной хлоридом
кальция (рис. 2.1), помещают 20 мл сухого эфира и быстро вносят
через боковой тубус 1,8 г (0,22 моль) порошкообразного гидрида
лития. Пускают в ход мешалку и к перемешиваемой суспензии гид-
рида лития в эфире из капельной воронки прибавляют раствор бро-
мида алюминия с такой скоростью, чтобы эфир слабо кипел. Для
завершения реакции смесь нагревают дополни- -о-
телыю при слабом кипении в течение 1,5-2 ч, после
чего колбу охлаждают или оставляют до следую-
щего дня. J
Полученный эфирный раствор, не встряхивая U р>
колбу, осторожно сливают через воронку с там- j
поном из стеклянной ваты. Осадок с воронки и из хх ",
колбы (смесь бромида лития с избытком гидрида
лития при хранении на воздухе может воспламе- _| Г X
ниться) небольшими порциями переносят под -Z- н
тягой в сосуд с водным (1:1) этанолом. Содержание к 112/ 7
алюмогидрида лития в растворе определяют мето- Wjf г 4 х, |
дом йодометрического титрования. С этой целью в го*''
коническую колбу вместимостью 100 мл наливают / \
20 мл 0,4 н. бензольного раствора иода и быстро _!_J> I
добавляют к нему 1 мл эфирного раствора алюмо-
Рис. 2.1. Установка для получения алюмогидрида лития
149
гидрида лития, отмеренного с помощью приспособления для заса-
сывания жидкостей. После непродолжительного встряхивания кол-
бы прибавляют 5-10 мл воды, 4-5 капель уксусной кислоты и
оттитровывают избыток иода 0,2 н. раствором тиосульфата до
обесцвечивания бензольного слоя. В конце титрования смесь энер-
гично встряхивают. Расчет проводят по формуле
2(Ш[, - И№,8,О3 ‘ ^Na2S,O3
М =------:--777^---------’
4Иыа1н4
где М- молярность анализируемого эфирного раствора алюмогид-
рида лития; P'Na.s.o, - объем тиосульфата, израсходованного на
титрование избытка иода; Л^а,з2о, нАj, - нормальность растворов
тиосульфата и иода. Нормальность растворов иода предварительно
определяют титрованием, для чего 10 мл бензольного раствора иода
титруют тиосульфатом в тех же условиях, что и при анализе.
Определив молярность анализируемого раствора и измерив его
объем мерным цилиндром, рассчитывают количество и выход полу-
ченного алюмогидрида лития. Он обычно составляет 85-90 %.
б) Три(трет-бутокси) алюмогидрид лития
Реактивы
Алюмогидрид лития 0,1 моль
трет-Бутанол 22,2 г
Эфир сухой 110 мл
В круглодонную колбу вместимостью 250 мл, снабженную ме-
ханической мешалкой, холодильником и капельной воронкой, по-
мещают 30 мл эфирного раствора, содержащего 0,1 моль алюмогид-
рида лития. К перемешиваемому содержимому колбы из капельной
воронки медленно добавляют раствор 22,2 г (0,3 моль) безводного
отре/и-бутапола в 80 мл сухого эфира. В процессе добавления пос-
ледней трети раствора наблюдается образование белого осадка.
После окончания добавления растворитель декантируют, а его ос-
татки удаляют из реакционной колбы с помощью водоструйного
насоса при нагревании на водяной бане. Твердый остаток пред-
ставляет собой три(трет-бутокси)алюмогидрид лития. Выход поч-
ти количественный. Вещество сохраняют в колбе, защищая от
действия влаги воздуха. Раствор три(/и/?е/7/-бутокси)алюмогидрида
150
лития концентрации 2 М готовят растворением полученного твер-
дого гидрида в диглиме.
1-Фенилэтанол
PhCОМе —1Н4; PhCH(OH)Me
2. Н2О
Реактивы
Алюмогидрид лития
Ацетофенон
Эфир сухой
0,03 моль
12,1 г
120 мл
В трехгорлую колбу, снабженную капельной воронкой, механи-
ческой мешалкой и обратным холодильником (см. рис. 2.1), поме-
щают эфирный раствор, содержащий 0,03 моль алюмогидрида ли-
тия в 100 мл сухого эфира. Из капельной воронки вводят раствор
12,1 г (0,1 моль) ацетофенона в 20 мл сухого эфира с такой ско-
ростью, чтобы эфир умеренно кипел. После прибавления всего ко-
личества ацетофенона колбу нагревают 0,5 ч. Затем смесь охлаж-
дают в ледяной бане и, продолжая перемешивание, добавляют по
каплям воду до тех пор, пока не прекратится выделение водорода.
После этого прибавляют 10 %-ный раствор серной кислоты до пол-
ного или почти полного растворения осадка гидроксида алюминия.
Органический слой отделяют от водного, из водного делают экст-
ракцию эфиром. Объединенные эфирные растворы промывают
последовательно 10 %-ным раствором карбоната натрия, водой и
сушат сульфатом натрия. Эфир удаляют и остаток перегоняют в
вакууме. Получают 11,7 г (95 %) 1-фенилэтанола. Т. кип. 95 °C при
12 мм рт. ст., Пр 1,5244. Хроматография: элюент - хлороформ,
R/ 0,4. Спектральные характеристики приведены на рис. 2.2.
Изоборнеол
Me Me
1. LiAlH4, Et2O
2. H2O
151
. 0>f-
1^2-
*0,3-
0,5-
0,7-
1,0-
MOO
3000
2000
1600
~1——I—"
1200 1И,см
i
9?
о
Puc. 2.2. Спектральные характеристики
1-фенилэтанола:
a - спектр ПМР в ССЦ (<5, м. д.: 1,3 д, J = 8 Гц (ЗН,
СНз), 2,76 с (1Н, ОН), 4,65 кв, J = 8 Гц (1Н, СН),
7,18 с (5Н, Ph)); б - ИК спектр в тонком слое
(3350 см-1 (О-Н)); в - УФ спектр в циклогексане
(АМакс.> нм, 1g е: 257 тс, 2,24)
152
Реактивы
Алюмогидрид лития 0,012 моль
Камфора 1,5 г
Эфир сухой 60 мл
В колбу вместимостью 100 мл (см. рис. 2.1) помещают 50 мл
эфирного раствора, содержащего 0,012 моль алюмогидрида лития.
Раствор не должен содержать гидрида лития. В противном случае
процесс восстановления протекает нестереоселективно. К пере-
мешиваемому содержимому колбы прибавляют раствор 1,5 г
(0,01 моль) камфоры в 10 мл сухого эфира. Прибавление ведут с та-
кой скоростью, чтобы раствор не кипел. Реакционную смесь пере-
мешивают при комнатной температуре в течение 2 ч. Затем, про-
должая перемешивание, охлаждают колбу ледяной водой, добав-
ляют необходимое количество воды и 20 %-ного раствора хлорово-
дородной кислоты до растворения осадка (pH 5-6). Эфирный слой
отделяют, из водного слоя продукт извлекают эфиром. Объеди-
ненные эфирные растворы промывают водой и сушат сульфатом
магния. Эфир упаривают и получают 1,4 г (90 %) изоборнеола,
[а]р - 26,0° (в этаноле). Значение величины оптического вращения
соответствует содержанию в изоборнеоле 10 % борнеола. Состав
продукта может быть подтвержден методом ГЖХ. Изоборнеол
очищают перекристаллизацией из гексана. Получают вещество с
т. пл. 212-213 °C, [а]р - 30,6°. По литературным данным т. пл.
214 °C, [а]р - 32,3°. Спектральные характеристики изоборнеола
даны на рис. 2.3.
Фталиловый спирт
1. LiAIH4, Et2O
2. Н2О
/х;.СН2ОН
^^СН2ОН
Реактивы
Алюмогидрид лития
Фталевый ангидрид
Эфир сухой
0,12 моль
14,8 г
250 мл
153
Установка состоит из двугорлой колбы, снабженной экст-
рактором Сокслета с обратным холодильником (рис. 2.4). Верхний
конец холодильника защищают поглотительной трубкой, напол-
ненной хлоридом кальция. В колбу помещают 250 мл сухого эфира,
содержащего 0,12 моль алюмогидрида лития, в гильзу экстрактора
Сокслета помещают 14,8 г (0,1 моль) фталевого ангидрида. Раствор
Рис. 2.3. Спектральные характеристики изоборнеола:
а - спектр ПМР* в СС14 0, м. д.: 0,8 с, 0,90 с и 1,02 с (3 х ЗН, ЗСН3), 1,18 м, 1,5 м
(2 х 2 х 2Н, ЗСН2), 2,67 с (1Н, ОН), 3,59 м (1Н, СП)); б - ИК спектр в Kbr
(3400 см-1, ш. (О-Н))
154
Рис. 2.4. Установка для получения фталилового спирта
нагревают и кипятят до тех пор, пока весь ангидрид не
перейдет в реакционную колбу (на это потребуется
4-6 ч). После этого экстрактор убирают, в боковые
тубусы помещают обратный холодильник и капельную
воронку, включают мешалку и при охлаждении осто-
рожно прибавляют воду для разложения избытка алю-
могидрида лития, а затем 10 %-ный раствор серной
кислоты до полного растворения осадка гидроксида
алюминия. Эфирный слой отделяют, а из водного слоя
продукт тщательно экстрагируют эфиром. Полное из-
влечение продукта реакции может быть достигнуто в
экстракторе непрерывного действия. Объединенные
эфирные растворы сушат, эфир удаляют и в остатке
получают 6,9 г (50 %) фталилового спирта. После пере-
кристаллизации из петролейного эфира получают бес-
цветное вещество; т. пл. 63-65 °C. Возможна дополни-
нительная очистка методом сублимации при температуре 100 °C и
давлении 2 мм рт. ст.
Хроматография: элюент - петролейный эфир и хлороформ 1:5,
Rf 0,5. Спектральные характеристики даны на рис. 2.5.
N-Этиланилин
NHCOMe
1. LiAlH4, Et2O
2. Н2О
NHCH2Me
Реактивы
Алюмогидрид лития
Ацетанилид
Эфир сухой
ТГФ сухой
0,09 моль
9,5 г
50 мл
50 мл
В трехгорлую колбу (см. рис. 2.1) помещают 50 мл эфирного
раствора, содержащего 0,09 моль алюмогидрида лития и прибав-
ляют по каплям раствор 9,5 г (0,07 моль) ацетанилида в 50 мл сухого
ТГФ. Скорость прибавления регулируют таким образом, чтобы
эфир слабо кипел. По окончании прибавления смесь нагревают 6 ч,
затем охлаждают ледяной водой и добавляют в нее по каплям 3,6 мл
155
Рис. 2.5. Спектральные характеристики
фталилового спирта:
а - спектр ПМР* в ССЦ (<5, м. д.: 4,69 с (4Н,
СН2), 5,0 с (2Н, ОН), 7,27 м (2Н, Н3-6 аром.),
7,38 м (2Н, Н4-5 аром.)); 6 - ПК спектр*
в КВг (3380 см-' (О—Н)); в - УФ спектр*
в этаноле (Лмакс_, нм, 1g е: 275 тс, 2,98)
156
15 %-ного раствора гидроксида натрия и 10 мл воды. Реакционную
смесь фильтруют, осадок промывают несколько раз небольшими
порциями эфира и отбрасывают. Эфирный фильтрат сушат твердым
гидроксидом калия, эфир упаривают и остаток перегоняют в ваку-
уме. Собирают фракцию с т. кип. 98 °C при 18 мм рт. ст. Получают
5,5 г (60 %) N-этиланилина, п2^ 1,5519. Хроматография: элюент -
хлороформ, Rf 0,7. Спектральные характеристики приведены на
рис. 2.6.
4-Нитробензальдегид
// \ LiAlH(OBu-mpem)3 // \
(bN-C Х>—СОС1 ---------------—& O2N—\ х>—СНО
\ __ / диглим \ __ /
Реактивы
Три(трет-бутокси)алюмогидрид лития 0,1 моль
4-Нитробензоилхлорид 18,6 г
Диглим сухой 100 мл
В круглодонную колбу вместимостью 500 мл, снабженную низ-
котемпературным термометром, трубкой для подвода азота и
капельной воронкой с уравнительной трубкой, помещают раствор
18,6 г (0,1 моль) 4-нитрохлорбензилхлорида в 50 мл диглима. Реак-
ционная колба заполняется сухим азотом и охлаждается примерно
до -78 °C. К перемешиваемому раствору в течение 0,5-1 ч добавля-
ют 2 М раствор, содержащий 0,1 моль три(т/?ет-бутокси)алюмо-
гидрида лития в 50 мл диглима, следя, чтобы не происходило замет-
ных колебаний температуры. Затем охлаждающую баню убирают и
реакционной смеси в течение 1 ч дают нагреться до комнатной
температуры, после чего содержимое колбы выливают на толченый
лед. 4-Нитробензальдегид (с небольшим количеством непрореа-
гировавшего хлороангидрида) отсасывают в вакууме водоструйно-
го насоса и несколько раз экстрагируют этанолом. После удаления
этанола из объединенных экстрактов получают 12,3 г (81 %) 4-нит-
робензальдегида с температурой плавления 103-104 °C, который
затем перекристаллизовывают из воды или водного спирта. Выход
10,1 г (67 %), т. пл. 104-105 °C. Спектральные характеристики при-
ведены на рис. 2.7.
157
1-.-1-.-1-1-1-1-1->-г-' I
-/65432 в;м.д.
3500 2500 1600 1400
Puc. 2.6. Спектральные
характеристики N-этиланилина:
a - спектр ПМР* в ССЦ 0, м. д.:
1,22 т, J = 8 Гц (ЗН, СН3), 3,12 кв,
J = 8 Гц (2Н, СН3), 4,6 уш. с (1Н,
NH), 6,61 м (ЗН, Н2А6арОМ.) 7,13 м
(2Н, I-P-5 аром.)); б - ИК спектр в
тонком слое (3400 см-1 (N-H)); в -
УФ спектр в метаноле (2маиг, нм,
1g е: 246, 4,04; 295,3,32)'
158
Рис. 2.7. Спектральные
характеристики
4-нитробензальдегида:
а - спектр ПМР* в ССЦ 0, м. д.:
8,05 д. т, J = 8,5 и 2 Гц (2Н, Н3-5
аром.), 8,37 д. т, J = 8,5 и 1,5 Гц(2Н,
Н2.6 аром.), 10,14 с (1Н, СН=О)); б -
ИК спектр в КВг (1350 и 1540 см-1
(ЫО2), 1705 см-' (С=О)); в - УФ
спектр* в этаноле (2Макс , нм> lg т:
265. 3,03)
159
Коричный спирт
1. LiAlH4, Et2O
/ 2. Н2О V
PhCH = CHCHO ------< PhCH = CHCH9OH
\ NaBH4, EtOH / 2
4' (способы 1, 2)
LiBH4
PhCH = CHCOOEt ----------*-—► PhCH = CHCH2OH
ДИГЛИМ z
(способ 3)
Способ J. Восстановление коричного альдегида алюмогидридом
лития
Реактивы
Алюмогидрид лития 0,033 моль
Коричный альдегид 13,2 г
Эфир сухой 60 мл
Раствор 13,2 г (0,1 моль) коричного альдегида в 40 мл сухого
эфира помещают в трехгорлую колбу вместимостью 200 мл, снаб-
женную механической мешалкой, капельной воронкой и термомет-
ром, погруженным в жидкость. Колба сообщается с атмосферой че-
рез боковой отвод капельной воронки, защищенной поглотитель-
ной трубкой с хлоридом кальция. Раствор охлаждают до температу-
ры -10 °C, погружая колбу в смесь льда с солью, после чего
добавляют к нему из капельной воронки в течение 0,5 ч 20 мл
эфирного раствора, содержащего 0,033 моль алюмогидрида лития.
В процессе добавления поддерживают температуру реакционной
смеси не выше -10 °C. Когда эта операция будет закончена, смесь
выдерживают в течение 10 мин, после чего, не прекращая переме-
шивания, добавляют к ней (вначале осторожно) воду для разложе-
ния избытка алюмогидрида лития, а затем 80 мл 10 %-ного раствора
серной кислоты (до полного или почти полного растворения гидро-
ксида алюминия). Продукт реакции извлекают эфиром. Объединен-
ные эфирные экстракты промывают раствором карбоната натрия и
водой до нейтральной реакции, сушат сульфатом магния. После
отгонки растворителя остаток медленно перегоняют из колбы с
небольшим дефлегматором. Получают 12 г (90 %) коричного спир-
та. Т. кип. 140 °C при 14 мм рт. ст. Препарат медленно кристаллизу-
ется. Т. пл. 33-34 °C. Хроматография: элюент - хлороформ и
петролейный эфир, 20 : 1; Rf 0,5.
160
Способ 2. Восстановление коричного альдегида борогидридом
натрия
Реактивы
Борогидрид натрия
Коричный альдегид
Этанол
4,7 г
15,9г
40 мл
В колбу вместимостью 100 мл помещают 15,9 г (0,12 моль) ко-
ричного альдегида и прибавляют по каплям раствор 4,7 г (0,12 моль)
борогидрида натрия в 40 мл этанола при перемешивании с по-
мощью магнитной мешалки. Температуру реакционной смеси конт-
ролируют так, чтобы она не превышала 20-30 °C, в случае необ-
ходимости охлаждая колбу ледяной водой. Этанол удаляют, к ос-
татку прибавляют 30 мл 10 %-ного раствора гидроксида натрия.
Продукт реакции извлекают эфиром и выделяют, как описано выше
(см. способ 1). Получают 15,6 г (97 %) коричного спирта, индиви-
дуального по данным ТСХ и ГЖХ.
Способ 3. Восстановление этилового эфира коричной кислоты
борогидридом лития
Реактивы
Борогидрид натрия 2,37 г
Бромид лития 5,4 г
Этиловый эфир коричной кислоты 17,6 г
Диглим сухой 125 мл
В колбу, снабженную механической мешалкой, капельной во-
ронкой и холодильником, помещают 2,4 г (0,06 моль) борогидрида
натрия и 125 мл диглима. В полученный раствор прибавляют 5,4 г
(0,06 моль) хорошо растертого сухого бромида лития. Содержимое
колбы перемешивают 0,5 ч и затем к нему прибавляют в течение
30 мин 17,6 г (0,1 моль) этилового эфира коричной кислоты. Реак-
ционную смесь для завершения реакции нагревают на водяной бане
в течение 3 ч, затем охлаждают и выливают ее в смесь 100 г толче-
ного льда и 10 мл хлороводородной кислоты. Продукт реакции вы-
деляют, как описано выше. Получают 11 г (82 %) коричного спирта,
индивидуального по данным ТСХ и ГЖХ. Спектральные характе-
ристики коричного спирта даны на рис. 2.8.
161
п-------1-----1-------’-----1-------'-----1------>------1----
7 6 5 4 м.д.
0,3
I
g 0,5
£ 0,7
£ 1,0
oo
3000 2000 1600 1200 ^CM'1
Puc. 2.8. Спектральные
характеристики коричного спирта:
а - спектр ПМР* в ССЦ (<5, м. д.: 3,1 уш. с
(1Н, ОН), 4,22 д.д, J = 6 и 1,5 Гц (2Н, СН2),
6,32 д. т, J = 16 и 6 Гц (1Н, =СН), 6,53 д. т,
J = 16 и 1,5 Гц (1 И, PhCH=), 7,28 м (5Н, Ph));
б - ИК спектр* в тонком слое (3350 см-1
(О-Н)); в - УФ спектр в метаноле (2макс ,
нм, Ige: 251,4,08; 282, 3,22; 91,3,12)'
162
Циклопентанол
(способ 2)
Реактивы
Борогидрид натрия
Циклопентанон
1,5 г
8,4 г
В колбу, снабженную магнитной мешалкой, помещают 30 мл
воды и 5 капель 10 %-ного раствора гидроксида натрия. При пере-
мешивании присыпают по порциям 1,5 г (0,04 моль) борогидрида
натрия. Помещают колбу в баню с холодной водой и прибавляют по
каплям 8,4 г (0,1 моль) циклопентанона, поддерживая температуру
смеси не выше 40 °C. Затем перемешивают смесь в течение 30 мин
при комнатной температуре, после чего медленно добавляют
10 %-ный раствор хлороводородной кислоты до достижения кис-
лой реакции среды. Затем вносят 10 г хлорида натрия и переме-
шивают раствор до насыщения. Декантируют раствор в делитель-
ную воронку, экстрагируют продукт эфиром. Объединенный эфир-
ный экстракт сушат сульфатом магния. Отгоняют эфир из колбы с
дефлегматором и остаток перегоняют, отбирая фракцию с т. кип.
137-140 °C. Получают 6,5 г (75 %) циклопентанола, 1,4531.
Спектральные характеристики приведены выше (см. рис. 1.10).
4-Метилпентен-3-ол-2
NaBH,
Ме,С = СНСОМе ----------—► Ме,С = СНСН(ОН)Ме
2 EtOH, н2о 2
Реактивы
Борогидрид натрия 3,7 г
4-Метилпентен-3-он-2 (мезитилоксид) 17,7 г
Этанол 60 мл
Способ 1 см. в разделе 1.9.
163
В плоскодонную колбу помещают 90 мл смеси этанол - вода
(2 : 1), добавляют несколько капель 10 %-ного раствора гидроксида
натрия и вносят 3,7 г (0,1 моль) борогидрида натрия. Раствор
нагревают до 60-70 °C при перемешивании с помощью магнитной
мешалки, прибавляют по каплям 17,7 г (0,18 моль) 4-метилпентен-
З-она-2 (мезитилоксида). При той же температуре перемешивание
продолжают в течение 1 ч. Затем реакционную смесь разбавляют
водой, продукт реакции экстрагируют эфиром. Объединенные
эфирные растворы промывают водой, сушат сульфатом натрия.
Эфир удаляют и остаток перегоняют, собирая фракцию с т. кип.
138-140 °C. Получают 13,9 г (77 %) 4-метилпентен-3-ола-2, /°
1,4310. Спектральные характеристики даны на рис. 2.9.
4-Хлоробензиловый спирт
С1Л\ //-COOEt д^им - C1-Z >сн2он
Реактивы
Борогидрид натрия
4-Хлороэтилбензоат
Бромид лития
Диглим сухой
2,37 г
18,5г
5,4 г
125 мл
В колбу, снабженную механической мешалкой, капельной во-
ронкой и холодильником (см. рис. 2.1) помещают 2,4 г (0,06 моль)
борогидрида натрия и 125 мл диглима. После того как весь гидрид
растворится, прибавляют 5,4 г (0,06 моль) хорошо растертого
бромида лития. Содержимое колбы перемешивают 0,5 ч и затем к
нему прибавляют в течение 30 мин 18,5 г (0,1 моль) 4-хлороэтил-
бензоата. Реакционную смесь нагревают на кипящей водяной бане в
течение 3 ч, затем охлаждают и выливают ее в смесь 100 г толченого
льда и 10 мл хлороводородной кислоты. Твердый 4-хлоробензило-
вый спирт отфильтровывают, промывают водой от свободной кис-
лоты, сушат. Получают 14,2 г (97 %) 4-хлоробензилового спирта,
т. пл. 73-74 °C. После перекристаллизации из воды получают 13,9 г
(85 %) продукта, т. пл. 74-75 °C. Хроматография: элюент -
хлороформ, Rf 0,3. Спектральные характеристики приведены на
рис. 2.10.
164
Рис. 2.9. Спектральные характеристики 4-метилпентен-3-ола-2:
а - спектр ПМР* в ССЦ (д, м. д.: 1,12 д, J = 8 Гц (ЗН, СН3), 1,64 д. д., J = 6 и 1,5 Гц
(6Н, (СН3)2С=), 3,27 уш. с (1Н, ОН), 4,45 м (2Н, СН=), 5,14 м (1Н, СН));
б - ИК спектр в тонком слое (1680 см-1 (С=С), 3400 см-1, ш. (О-Н))
6 Заказ № 137
165
100
!
во-
60-
40-
£
20-
0
3000
2000 1600 1200 $,см
Рис. 2.10. Спектральные характеристики
4-хлоробензилового спирта:
а - спектр ПМР* в СС14 (<5, м. д.: 3,71 с (1Н, ОН),
4,55 с (2Н, СН2), 7,26 м (4Н, Н23,5,6 аром.));
б - ИК спектр в тонком слое (3320 см-1 (О-Н));
в - УФ спектр в метаноле (2макс , нм, 1g е: 220,
3,91; 268 тс, 2,48)
166
: i л yQ;«HqoaT.-.:.q
снммь глоев к и». '
'Ш> ЩТЗ нщ.
умет’•.
Глава 3. МЕТОДЫ СИНТЕЗА С ПРИМЕНЕНИЕМ
ЖИДКОГО АММИАКА КАК РАСТВОРИТЕЛЯ
3.1. СВОЙСТВА ЖИДКОГО АММИАКА
Жидкий аммиак относится к числу наиболее известных невод-
ных растворителей. Это объясняется тем, что он обладает почти
уникальной комбинацией физических свойств:
Температура плавления, °C -77,8
Температура кипения при 760 мм рт. ст., °C -33,5
Плотность при 0 °C, кг/дм3 0,771
Диэлектрическая проницаемость при -33 °C 23,0
Дипольный момент, Д 0,93
Вязкость в парах, мПа • с 0,2543
Теплота испарения, кДж • моль-1 23,42
Растворимость в воде, г/дм3 при 760 мм рт. ст. и 25 °C 320
Низкие вязкость (1/4 вязкости воды) и плотность жидкого амми-
ака обусловливают подвижность ионов в нем и легкость проведения
химических реакций, в том числе гетерогенных, в которых веду-
щую роль играют процессы диффузии растворенных соединений.
Высокое значение дипольного момента облегчает химическое
взаимодействие между полярными молекулами аммиака и ионами,
а также между самими молекулами аммиака. Диэлектрическая
проницаемость аммиака значительно меньше, чем диэлектрическая
проницаемость воды (е = 78,5), однако она гораздо больше, чем
диэлектрическая проницаемость уксусной кислоты (е = 6,4). Поэто-
му естественно ожидать, что значения растворимости ионных солей
167
в аммиаке находятся в общем между значениями растворимости тех
же солей в воде и уксусной кислоте.
Вследствие высоких донорных свойств атома азота аммиак лег-
ко образует водородные связи, о чем свидетельствует и аномально
высокая температура его кипения. Это приводит к тому, что в ам-
миаке хорошо растворяются не только ионные, но также многие
органические (неионизованные) соединения. Особенно легко раст-
воримы соединения, образующие водородные связи (амины, фено-
лы, сложные эфиры, углеводы). Для соединений, трудно раство-
римых в аммиаке, осложнений удается избежать использованием
сорастворителей, таких как эфир, тетрагидрофуран, диоксан или
глим.
Сильные водородные связи придают аммиаку диполярный ха-
рактер, вследствие чего в нем облегчается образование заряженных
частиц типа катионов, а также радикал-анионов, анионов и дианио-
нов. Известно, что катионы сольватированы как в протонных, так и
в диполярных апротонных растворителях, но по способности соль-
ватировать анион эти два типа растворителей заметно различаются.
И хотя формально по классификации Паркера аммиак относится к
протонным растворителям, значительно большая его способность,
чем, например, гидроксилсодержащих растворителей, сольватиро-
вать анионные реагенты позволяет считать его, по существу, апро-
тонным растворителем. Важно также то, что протонироваться ам-
миаком могут лишь относительно основные анионные частицы. В
случае необходимости кислотность среды может быть увеличена
добавлением соединений - доноров протонов, таких как кислые
углеводороды, спирты или вода. Все это создает основу для прове-
дения в аммиаке реакций, в которых важную роль играют интер-
медиаты анионного типа.
Реакции сольволиза (аммонолиза), в которых аммиак участвует
как реагент, редко осложняют течение реакций. Аммонолиз в
жидком аммиаке, так же как и гидролиз в водных системах, зависит
от степени самоионизации аммиака, поскольку инициируется ата-
кой аммоний- или амид-иона на субстрат:
2 NH3 NH4 + NH2~
Жидкий аммиак ионизуется лишь в очень малой степени. При
температуре -33 °C его $Ка 34. Согласно этому он и оказывается
168
при обычных экспериментальных условиях заметно более слабым
сольволизующим агентом, чем, например, вода (рАГа » 16).
Преимуществом аммиака является также то, что он обладает
специфической способностью растворять щелочные и щелочнозе-
мельные металлы. Растворение щелочноземельных металлов и ли-
тия протекает экзотермично, в то время как такие щелочные метал-
лы, как натрий и калий, имеют отрицательную теплоту растворе-
ния. Разбавленные растворы металлов в аммиаке имеют голубой
цвет, концентрированные растворы легко растворимых металлов -
бронзовый блеск. Полагают, что разбавленные растворы металлов в
жидком аммиаке (менее 0,005 М) содержат сольватированные ка-
тионы металла и специфические анионы - растворенные элект-
роны:
М --------► М+ + е
„Свободные** электроны, по-видимому, занимают полость, ок-
руженную молекулами аммиака, протоны которых ориентированы
внутрь полости. В более концентрированных растворах (от 0,005 до
1 М) появляются частицы М, М_, М2, которые, вероятно, возни-
кают за счет простого электростатического взаимодействия сольва-
тированных электронов и катионов. В еще более концентриро-
ванных растворах (свыше 1 М), по существу, присутствуют соль-
ватированные катионы, которые удерживаются вместе электрона-
ми, т. е. природа этого раствора становится близка природе металла.
С точки зрения практического использования растворов металлов в
аммиаке наиболее важно, что своеобразие их свойств обусловли-
вает их высокую восстановительную способность.
Не менее важной для системы аммиак - металл оказывается спо-
собность аммиака реагировать с растворенным металлом. И хотя в
отсутствие катализаторов процесс не имеет практического
значения, он легко ускоряется переходными металлами, такими как
железо, кобальт, никель, а также ультрафиолетовым светом. Эта ка-
талитическая реакция представляет собой удобный метод полу-
чения амидов различных металлов, которые сами по себе широко
используются в органическом синтезе как сильные основания:
м + nh3 --------► mnh2 + V2 н2
Аммиак является не только реагентом, но и средой для полу-
чения амидов, а также прекрасным растворителем для многих реак-
ций с их участием.
169
Далее будут продемонстрированы свойства аммиака как высо-
коэффективного растворителя на примере некоторых важнейших
химических реакций.
3.2. ВОССТАНОВЛЕНИЕ ВАЖНЕЙШИХ КЛАССОВ СОЕДИНЕНИЙ
Растворы металлов в жидком аммиаке являются прекрасной
средой для гомогенного химического восстановления. Сольватиро-
ванные электроны в растворе щелочного металла в жидком аммиаке
можно рассматривать как простейший восстанавливающий агент.
Сам раствор представляет собой сильную восстановительную сис-
тему.
Механизм восстановления растворенным металлом включает
передачу электрона от металла на вакантную орбиталь субстрата
(А) с образованием анион-радикала. Последний может далее прото-
нироваться, давая радикал, который в свою очередь, принимая дру-
гой электрон, превращается в анион. Протонирование последнего
приводит к образованию продукта реакции. Альтернативный путь
включает одновременное или постадийное присоединение к субст-
рату двух электронов с образованием дианиона, который, присое-
диняя последовательно два протона, дает тот же продукт реакции:
Очевидно, что последовательность и согласованность этих
стадий будет зависеть от таких факторов, как природа субстрата,
гомогенность и восстановительный потенциал среды, а также от
наличия и природы источника протона. Детальное обсуждение ме-
ханизма приводится ниже на примере субстратов, для восстанов-
ления которых наиболее часто используют систему металл - жид-
кий аммиак, а именно ароматических соединений, а, Д-непредель-
ных карбонильных соединений и алкинов. Во всех случаях реакция
восстановления становится возможной благодаря способности ам-
миака как диполярного и мощного ионизирующего растворителя
стабилизировать за счет сольватации возникающие анион-радика-
лы и анионы.
170
3.2.1. Ароматические системы
Одним из наиболее полезных применений системы металл -
аммиак для целей органического синтеза является восстановление
ароматических колец. Растворы металлов в жидком аммиаке в
присутствии спирта в качестве донора протона или без него
выступают как достаточно мощные агенты для того, чтобы
восстановить ароматическое кольцо, и одновременно достаточно
специфичные, чтобы восстановление провести лишь частично до
дигидробензолов (циклогексадиенов). Этот тип реакции известен
как восстановление по Берчу. Легкость восстановления в первом
приближении коррелирует с восстановительным потенциалом
соединения и уменьшается в порядке > антрацен > фенантрен >
> нафталин > дифенил > бензол. Сам бензол не удается восстано-
вить щелочным металлом в жидком аммиаке, и его восстановление
может быть успешно проведено до 1,4-дигидробензола лишь в при-
сутствии более эффективного донора протонов, такого как этанол:
Такой тип восстановления наблюдается и для замещенных про-
изводных бензола. Хемоселективность восстановления является
уникальной особенностью восстановления по Берчу: главными
продуктами оказываются несопряженные диены. В согласии с при-
веденной выше общей схемой восстановления растворенными ме-
таллами, механизм восстановления на примере бензола может быть
представлен следующим образом:
Н НН НН НН
Металл, окисляясь до катиона, передает электрон ароматичес-
кому кольцу, что приводит к образованию анион-радикала. Интер-
медиаты такого типа могут быть идентифицированы по спектрам
ЭПР, из которых в настоящее время получен большой объем ин-
формации относительно их строения. Первая стадия реакции - при-
соединение электрона - представляет собой равновесный процесс,
положение которого для бензола существенно сдвинуто влево. Для
171
протекания восстановления необходимо, чтобы образовавшийся
анион-радикал быстро и необратимо протонировался. Аммиак как
донор протонов оказывается для этого слишком слабой кислотой,
однако спирты, имеющие более высокую кислотность ($Ка 16-19),
адекватную, по-видимому, основности анион-радикала, способны
смещать равновесие практически необратимо вправо. Поэтому вос-
становление самого бензола и его производных требует присутст-
вия спирта, в то время как полициклические ароматические соеди-
нения легко переходят в соответствующие анион-радикалы или
даже в дианионы в отсутствие добавляемого источника протонов (в
качестве такового для этих соединений выступает аммиак).
Образовавшийся анион-радикал, отщепив протон от подходя-
щего донора, превращается в радикал. Восстановление радикала
из-за значительного сродства его к электронам, избыток которых
присутствует в реакционной среде, проходит быстро и необратимо.
Димеризация радикалов редко осложняет процесс восстановления.
Возникший анион основен и, присоединяя протон от внесенного в
реакционную среду донора протонов, дает конечный продукт -
несопряженный дигидробензол.
В ароматическом ряду лишь в некоторых случаях, например в
реакциях дифенила и полициклических ароматических соединений,
может реализоваться альтернативный путь с участием дианиона,
образующегося за счет присоединения электрона к анион-радикалу.
Это присоединение, очевидно, идет медленно, так как в кольце уже
присутствует отрицательный заряд, но скорость его может возрас-
тать, если структура субстрата способствует эффективной делока-
лизации заряда.
Реакция восстановления растворами металлов в жидком аммиа-
ке относится к числу реакций, для которых тщательный выбор
условий и строгий контроль за их соблюдением являются важными
факторами, позволяющими провести восстановление с достаточной
скоростью и в желаемом направлении. Прежде чем переходить к
рассмотрению конкретных примеров, остановимся на этом под-
робнее.
В качестве растворенного металла используют натрий и ли-
тий, реже - калий и кальций. Растворимость этих металлов в аммиа-
ке при температуре -33 °C составляет:
172
г металла/100 г
аммиака
моль металла/моль
аммиака
Li 10,9 0,25
Na 24,6 0,18
К 46,5 0,20
Са 33,6 0,14
При использовании перегнанного аммиака и при тщательном
подборе условий реакции ни один из щелочных металлов не имеет
преимущества. На практике литий иногда предпочитают другим
металлам. Связано это, по-видимому, с тем, что он отличается от
других металлов замечательно высокой молярной растворимостью.
Он также имеет более высокий потенциал восстановления в ам-
миаке (-2,99 В при -50 °C), чем натрий (-2,59 В) и калий (-2,73 В).
Но важнее всего оказывается крайне слабая тенденция лития всту-
пать в катализируемую коллоидным железом (всегда присутствую-
щим в неперегнанном жидком аммиаке) побочную реакцию с обра-
зованием амида, а также меньшая основность солей лития (включая
амид), что позволяет избежать вторичных процессов.
Важной проблемой оказывается также выбор соединения, ко-
торое выступает в роли донора протонов. Даже различие между эта-
нолом и /и/>е/и-бутанрлом (первый - более сильная кислота) часто
оказывается важным. Соединения, вносимые в реакционную систе-
му в качестве донора протона, могут ускорять реакцию, менять ее
ход или предотвращать образование сильно основного амид-иона.
Скорость реакции таких кислот с восстанавливающей системой в
общем много меньше, чем скорость восстановления, так как пере-
нос протона к аммиаку сильно замедляется по мере того, как увели-
чивается концентрация основания:
НА + NH3 .......—А" + NH4+
NH4+ + е ---------► NH3 + !/2 Н2
На ход процесса восстановления ароматических соединений
оказывает влияние и ряд других факторов, таких как наличие при-
месей (поэтому аммиак рекомендуется перегнать), порядок смеше-
ния реагентов, отсутствие пероксидов в растворителях и т. п.
Заместители в бензольном кольце влияют как на скорость вос-
становления, так и на ориентацию присоединения протонов. В при-
173
сутствии электроноакцепторных заместителей скорость восстанов-
ления ароматического кольца увеличивается и протонирование
проходит в положения 1 и 4, в результате чего заместитель оказы-
вается в положении, подвергшемся протонированию (а). Электро-
нодонорные заместители дезактивируют кольцо и направляют про-
тоны в положения 2 и 5, в результате чего заместитель оказывается
в положении, не подвергшемся протонированию (б)*:
R = СООН, CONH2, SiMe3, Ar
Na, EtOH
ж. NH,
R = NR2, OR1, OH, Aik
Когда оба типа групп (акцепторные и донорные) присутствуют
в молекуле, ход реакции контролирует электроноакцепторный
заместитель:
СООН
Рг-изо
1. Li, ж. NHj
2. EtOH
3. NH4C1
92 %
Исключение составляет группа OR, которая, по-видимому, стабилизирует
анион-радикал за счет отрицательного индуктивного эффекта. Этим можно
объяснить тот факт, что метоксибензол восстанавливается в 3 раза быстрее, чем
бензол.
174
Строение продуктов реакции может быть понято, если учесть,
что оно определяется местом присоединения протонов сначала к
анион-радикалу и затем к карбаниону, возникающим как интер-
медиаты в процессе восстановления. При этом следует учесть сле-
дующие обстоятельства. Во-первых, протонирование анион-ради-
калов и карбанионов в кинетически контролируемом процессе про-
исходит предпочтительно по положению, в котором больше плот-
ность отрицательного заряда. Данные по распределению плотности
заряда в анион-радикалах, полученные из спектров ЭПР, указыва-
ют, что для анион-радикалов, содержащих электроноакцепторный
заместитель, таким положением оказывается лора-положение по
отношению к заместителю, для анион-радикалов, содержащих
электронодонорный заместитель, - мета- и орто-положения. В
этих положениях и протекает первый акт протонирования. Во-вто-
рых, следует учесть, что образовавшийся карбанион является мезо-
мерным:
Распределение электронной плотности, как это показывают
данные спектроскопии ЯМР 13С, таково, что электронная плотность
в положении 4 оказывается несколько большей, чем в положении
2(6). Именно в этом положении и происходит второе протонирова-
ние. Направленность обоих процессов обусловливает природу про-
дуктов реакции, например:
СООН СООН СООН СООН СООН
НН НН
Предсказание места присоединения протона может быть сдела-
но также на основании принципа наименьшего движения. В соот-
ветствии с этим принципом легче протекают те элементарные реак-
ции, в которых происходит наименьшее изменение взаимного
расположения атомов и электронной конфигурации. К рассматри-
175
ваемому случаю этот принцип в упрощенном виде может быть при-
менен следующим образом. Согласно методу валентных схем
порядок шести углерод-углеродных связей (в предположении, что
каждая из трех мезомерных форм вносит равный вклад) следующий
(при движении по кольцу): 1,5/3,4/3,4/3, 5/3 и 1. При превращении
карбаниона в диен порядок этих связей изменяется следующим
образом:
Легко видеть, что две связи, имевшие порядок 1, остаются неиз-
менными в обоих продуктах, но в остальных четырех связях
происходят изменения. При образовании 1,4-диена это изменение
выражается следующей суммой: 1/3+1/3+1/3+1/3, тогда как при
образовании 1,3-дйена соответствующее изменение составляет 2/3+
+2/3+1/3+1/3. Получение 1,3-диена должно сопровождаться боль-
шим изменением порядка связей, поэтому согласно принципу наи-
меньшего движения продуктом будет 1,4-диен.
Восстановление по Берчу полициклических аренов иногда про-
ходит сложно, однако с помощью различных вариантов метода уда-
ется получить конечные продукты с высокими выходами. Так, если
восстановление проводят лишь раствором металла в жидком ам-
миаке без внесения донора протонов, то продуктами реакции ока-
зываются анион-радикалы и иногда - дианионы. Затем с целью
протонирования образовавшихся интермедиатов вносят донор
протона - спирт. Это дает возможность получить частично гидро-
генизированные продукты, даже если они сами по себе могут быть
восстановлены далее.
Нафталин, например, реагирует с натрием в жидком аммиаке,
образуя комплекс красного цвета, разложение которого метанолом
дает 1,4-дигидронафталин. Если при восстановлении вводить одно-
временно этанол и нафталин, то можно получить 1,4,5,8-тетрагид-
ронафталин. При таких условиях 1,4-дигидронафталин также пре-
вращается в это соединение:
176
1. Na, ж. NH3
2. МеОН
Na, EtOH
ж. Nil,
Селективное восстановление 1-нафтола до 5,8-дигидропроиз-
водного и нафтойной-1 кислоты до 1,4-дигидропроизводного явля-
ется примером действия соответственно электронодонорного и
электроноакцепторного заместителей:
Ряд полициклических ароматических соединений, которые
имеют потенциал восстановления меньше, чем бензол, восстанав-
ливаются щелочными металлами в жидком аммиаке в отсутствие
спирта. Так, антрацен легко дает дианион (в присутствии солей
железа, которые понижают восстановительную силу растворов ли-
тий - аммиак), обработка которого спиртом приводит к получению
9,10-дигидроантрацена:
Н
В отсутствие солей железа идет дальнейшее восстановление до
тетрагидро- и гексагидроантраценов.
Значимость восстановления ароматических систем по Берчу для
органического синтеза связана не только с непосредственным полу-
чением соединений с шестичленным кольцом, но также с возмож-
ностью дальнейших превращений продуктов восстановления. Так,
эфиры енолов, получаемые восстановлением алкоксибензолов, мяг-
ким гидролизом превращают в несопряженные кетоны, а более
жестким гидролизом (разбавленная хлороводородная кислота) - в
сопряженные кетоны:
177
Н20 , АсОН
О
PhOMe -
ж. nh3
Эта реакция широко используется при получении стероидных
Аналогично енамины, получаемые восстановлением анилинов,
удается превращать в сопряженные кетоны:
Синтетические возможности восстановления по Берчу были
недавно расширены введением реакции восстановительного алки-
лирования. Реакция основана на том, что промежуточный анион
может улавливаться алкилгалогенидом:
Me COONa Me СООН
1. NH4C1
2. НС1, Н2О
178
Восстановительное алкилирование антрацена литием в жидком
аммиаке и алкилбромидом протекает стереоселективно и дает в
качестве основного продукта гуис-9,10-диалкил-9,10-дигидроант-
рацен:
Li , EtBr
».NH3
3.2.2. Ацетилены и этилены
Сродство к электрону простых алкенов столь мало, что, как пра-
вило, не позволяет проводить их восстановление растворами метал-
лов, в том числе и в жидком аммиаке. Однако сопряжение двойной
связи заметно облегчает процесс восстановления. Так, сопряжен-
ные диены могут легко восстанавливаться натрием в жидком ам-
миаке, давая продукты 1,4-присоединения (Е- и Z-изомеры):
СНЭ=СН-СН=СН, ---------► СН,СН = СНСН,
2 2 ».NH3 2 3
Сопряжение двойной связи с ароматическим кольцом также
повышает ее сродство к электрону и делает возможным восстанов-
ление стиролов и их производных. Этот процесс был использован
для синтеза 19-норстероидов, некоторые из которых являются
медицинскими препаратами. Процесс протекает стереоселективно
с образованием транс-изомера:
Me О Me О
I IН I)
JL \ Й *.NHj * Il Й I Й
Растворы металлов в жидком аммиаке оказывают специфичес-
кое действие на соединения, содержащие тройную углерод-угле-
родную связь. Реакция идет высокоселективно с образованием
двойной углерод-углеродной связи, при этом никаких насыщенных
продуктов не образуется. Замечательно также, что процесс оказы-
вается полностью стереоселективным и продуктами восстановле-
179
ния дизамещенных алкинов являются алкены, имеющие А-конфи-
гурацию:
СН3(СН2)6 Н
\ /
СН3(СН2)6С = С(СН2)7СО2Н Li Г С = С
ТГФ, ».NH, / \
Н (СН2)7СО2Н
Этот метод, таким образом, удачно дополняет метод каталити-
ческого гидрирования, позволяющего получать алкены, имеющие
Z-конфигурацию.
Реакция восстановления, возможно, протекает через промежу-
точное образование дианиона, более стабильной конфигурацией
которого из-за отталкивания двух заряженных sp1 -орбиталей ока-
зывается ш/>днс-конфигурация. Протонирование дианиона может
проходить в две стадии:
Рг
Н Pr Н Рг
\ / \ /
-----► /С = С^ _______ 3 /С = С\ 80-90 %
Рг Рг Н
Восстановление терминальных тройных связей имеет некото-
рые особенности. Реакция с раствором металла приводит к образо-
ванию ацетиленид-иона, который не подвергается дальнейшему
восстановлению, так как атому, уже несущему заряд, трудно при-
нять электрон. Этот факт позволяет селективно восстанавливать
дизамещенную тройную связь в присутствии концевой тройной
связи:
Н (СН2)4С = СН
1. Na, ж-NH,
РгС^С(СН,),С = СН -------------С = С 750/0
24 2.NH4C1 / \
Рг Н
Однако, если реакцию проводить в присутствии сульфата аммо-
ния, который способен освобождать концевую СН-группу, то это
препятствует образованию ацетиленид-иона и восстановление лег-
ко дает соответствующий алкен:
1. Na, «.Nil,
СН = С(СН2)2С^СН -- СН2 = СН(СН2)2СН=СН2
Z. ( 1N Пд)2 &'-'4
180
3.2.3. a, ft-Непредельные карбонилсодержащие соединения
a, ft -Ненасыщенные кислоты, эфиры, кетоны и альдегиды также
могут быть восстановлены раствором металла в жидком аммиаке.
Рассмотрим особенности этого процесса на примере «^-непре-
дельных альдегидов и кетонов.
Восстановление a, ft -непредельных кетонов металлом в жидком
аммиаке (содержащем обычно эфир как сорастворитель) приводит
к образованию енолятов металла. Последние обычно стабильны в
жидком аммиаке, но под действием мягкого донора протона (напри-
мер хлорида аммония; рКа аммоний-катиона равен 9,3) могут быть
превращены в соответствующие насыщенные кетоны. Они и явля-
ются конечными продуктами восстановления. Если в реакционной
среде присутствует избыток металла и в качестве донора протона
используется кислота более сильная, чем насыщенный кетон (на-
пример этанол), то конечным продуктом восстановления оказыва-
ется насыщенный спирт:
Образование енолята как первичного продукта восстановления
удалось показать путем проведения реакции С-алкилирования. Ре-
акционную смесь до внесения в нее донора протонов обрабатывали
алкилирующим агентом. Продуктом реакции являлось а-алкилза-
мещенное соединение:
Приведенная реакция восстановительного алкилирования стала
использоваться для региоселективного введения заместителя в
а-положение по отношению к карбонильной группе, что не всегда
удается сделать при основно-катализируемом алкилировании насы-
щенного кетона:
181
PhCH=CHCOPh
1. Li, ж.Nil-,, Et2O
2. RHal
PhCH2CH(R)COPh
Преимущество восстановления а,/?-непредельного кетона раст-
вором металла в жидком аммиаке до насыщенного кетона состоит в
том, что его удается осуществить для соединений, имеющих разно-
образные функциональные и защитные группы. Например, благо-
даря тому, что бензольное кольцо восстанавливается лишь очень
медленно в отсутствие донора протона (спирта), удается провести
селективное восстановление кратных С-С-связей «^-непредель-
ного кетона, имеющего бензольное кольцо:
1. Na, ж. NH ,, Et2O
2. NH4C1
3. CrOj-AcOH
Достоинством рассматриваемого метода восстановления явля-
ется также то, что реакции в общем случае не сопровождаются пе-
регруппировками, что иногда затрудняет использование других
химических методов восстановления. Особенно важно это обсто-
ятельство для химии природных соединений - стероидов и терпе-
ноидов, для которых восстановление в жидком аммиаке применя-
ется особенно часто.
Восстановление «,Д-непредельных кетонов согласно общей
схеме химического восстановления начинается с передачи одного
электрона из раствора на антисвязывающую лг-орбиталь сопряжен-
ной системы с образованием анион-радикала (показан в виде ион-
ной пары с металлом):
\ / е м+ \ /
с - с . с = С
// \\ \ / \ ,
— с О ^.С О“М+
Далее в зависимости от структуры восстанавливаемого кетона и
других факторов может осуществляться один из приведенных или
конкретно оба пути превращения анион-радикалов в еноляты:
182
Путь А:
е’ м+
\ / \
^,С О’М+
Путь Б:
^,С О- М+
М+, основание
\ / \ \ / \
ОН .Ск О“М +
н н
Первый путь А включает образование дианиона (показана
вероятная координация с металлом по кислороду и углероду) и его
протонирование по более основному ^-положению. Для осуществ-
ления этого пути важна способность системы делокализовать
отрицательный заряд. При реализации второго пути Б образовав-
шийся радикал-анион медленно протонируется, давая окси-ради-
кал, который быстро присоединяет второй электрон с образованием
оксиаллильного аниона. Последний подвергается протонированию,
давая енол. Передача протона от этого енола основанию (до кето-
низации) будет приводить к образованию енолята.
Поскольку восстановление а,Д-непредельных кетонов включа-
ет образование интермедиатов карбанионного типа, структурные
особенности исходного соединения могут способствовать внутри-
Одной из интересных особенностей рассматриваемой реакции
является то, что ее стереохимия подчиняется другим закономернос-
тям, нежели стереохимия часто употребляемого альтернативного
183
метода восстановления - каталитического гидрирования. Результа-
том этих двух реакций оказываются продукты различного стро-
ения:
Стереохимия восстановления конденсированных циклогексе-
нонов, в которых ^-углеродный атом находится в месте сочленения
циклов (например стероидных ен-4-онов-З), почти всегда подчи-
няется стереоэлектронному контролю. Главным продуктом являет-
ся более устойчивый из двух изомеров (цис- или транс-'), в котором
введенный ^-водородный атом занимает аксиальное положение. В
условиях установления равновесия между енолятом и кетоном (тер-
модинамический контроль) получается продукт, имеющий ста-
бильную конфигурацию при а-углеродном атоме. Кинетическое
протонирование промежуточного енолята дает менее стабильный
эпимер, как это видно на примере замещенного декалона:
184
Стереоселективность реакции определяется прежде всего ста-
бильностью промежуточного карбаниона. При восстановлении за-
мещенного декалона, например, образуется дианион, для которого
можно предположить три конформации А, Б, и В. Из них конфор-
мация А наиболее стабильна, так как i/wc-декалиновая структура Б
имеет неблагоприятные внутренние (аксиаль-аксиальные) отталки-
вания, а структура В не имеет никакой возможности для делокали-
зации отрицательного заряда. В результате протонирование приво-
дит к образованию тщ?ш/с-декалоновой системы.
Как было отмечено выше, если в процессе восстановления в
реакционной среде присутствует донор протонов (или он вносится
в реакционную среду, когда в ней еще присутствует взятый в
избытке металл), то может иметь место частичное или полное вос-
становление a, fl -ненасыщенного кетона в насыщенный вторичный
спирт. Восстановление проходит через стадию образования насы-
щенного кетона и требует присутствия более сильного протони-
рующего агента, чем кетон, например такого, как хлорид аммония
или этанол.
Восстановление насыщенных кетонов до насыщенных спиртов
иногда представляет самостоятельный интерес. Механизм такого
восстановления хорошо изучен. В результате присоединения элект-
рона образуется анион-радикал (называемый кетил-радикалом),
который имеет большую часть неспаренной электронной плотности
на атоме углерода. После протонирования по кислороду возникает
окси-радикал, способный принять второй электрон с образованием
карбаниона. Протонирование последнего и приводит к образова-
нию спирта:
О О- ОН
R1 — C-R2 —R’-C-R2 R’-C-R2 е -
ОН ОН
----- R'-C-R2 R’-CH-R2
185
Важной особенностью реакции является то, что она приводит к
получению термодинамически более стабильного аксиального
спирта
/71/>е/и-Ви
Поэтому реакцию часто используют как альтернативную восста-
новлению кетонов комплексными гидридами металлов, которое
имеет тенденцию давать экваториальный спирт (см. 2.6.1).
3.2.4. Реакция восстановительного расщепления
простых связей
Растворы металлов в жидком аммиаке вызывают в некоторых
случаях расщепление простых связей, и это их свойство использу-
ется в органическом синтезе. Можно полагать, что реакция протека-
ет путем прямого присоединения электронов к простой связи,
которая в результате расщепляется. Промежуточные анионы стаби-
лизируются благодаря сопряжению или присутствию электроноак-
цепторных групп, поэтому разрыв характерен для простых связей
между углеродом и гетероатомом (С-Х, где Х=На1, О, S и др.):
ОМе Н ОМе
О Me
SBu
2. NH4C1
3. НС1, Н2О
186
Реакции такого рода используют в структурных исследованиях,
для снятия защитных групп, в пептидных синтезах и т. п.
3.3. РЕАКЦИИ АЛКИЛИРОВАНИЯ
Амиды щелочных металлов в жидком аммиаке являются силь-
нейшими основаниями, способными отрывать протон от некоторых
органических соединений. Это приводит к возникновению карб-
анионов - важных промежуточных соединений органического син-
теза. Последние способны образовывать новую углерод-углерод-
ную связь, взаимодействуя с разнообразными электрофилами, в том
числе с алкилгалогенидами:
RH -------- R1- R2Hal > R'-R2
Для того чтобы реакция введения алкильной группы (реакция
алкилирования) протекала с заметной скоростью, должна быть со-
здана достаточная концентрация карбанионов. Для реакции
RH + NH£ „ R" + NH3
кислотно-основное равновесие смещено вправо, если соединение
RH является достаточно сильной кислотой, во всяком случае,
заметно более сильной, чем аммиак. Так как кислотность аммиака в
1018 раз меньше кислотности воды, можно ожидать, что кислоты,
рКа которых в водных растворах меньше 16, являются сильными
кислотами в жидком аммиаке, а кислоты, рКа которых в водных
растворах больше 34, в жидком аммиаке практически не проявляют
кислотных свойств. Некоторые органические соединения имеют
следующие приближенные значения кислотности (рАГа):
СН4 48 Инден 20
РЬСНз 41 (СН3)2СО 19
Ph2CH2 34 CH3CONH2 17
Ph3CH 31 ch3no2 10
Флуорен 23 CH3COCH2COOEt И
CH3COOEt 24 СН3СОСН2СОСНз 9
СН2=СН2 25 (СН3СО)2СН2 9
PhOCH 21
187
Приведенные соединения депротонируются за счет С-Н-связи,
и их часто называют С-Н кислотами. Сила С-Н кислоты зависит от
валентного состояния атома углерода и природы связанных с ним
заместителей.
3.3.1. Алкилирование терминальных алкинов
Повышение 5-характера карбанионного углерода определяет
возрастающую устойчивость карбанионов:
R3C-CH£ < Ar" = R2C = CH~ < RC^C"
Поэтому в противоположность алканам и алкенам терминаль-
ные алкины в аммиаке проявляют кислотные свойства (рКа состав-
ляет 18-27) и могут быть использованы в реакции алкилирования.
Обменивая водород на щелочной металл, они образуют соли - аце-
тилениды:
NH,-
RCHCH --------RC = C~+NH3
Ацетилениды реагируют далее с алкилгалогенидами с образо-
ванием алкинов, имеющих внутреннюю тройную связь:
R2Hal । -
R*C = CM --------► R>CHCR2 + MHal
Особенно важное значение имеет алкилирование самого аце-
тилена, которое дает возможность через моносоль (однозарядный
анион) получать терминальные алкины:
НС=С-М+ RHa‘ » RC = CH + MHal
В простейшем случае незамещенный ацетиленид получают,
пропуская ацетилен в раствор натрия в жидком аммиаке. В про-
цессе реакции происходит восстановление части ацетилена в эти-
лен, тогда как другая его часть, выполняющая функцию донора
протонов, дает ацетиленид-анионы:
НС^СН ——- [НС-СН] нс=сн». СН2 = СН2 + 2NaC = CH
3 2 Na+
Реакция протекает медленно из-за чисто механических затруд-
нений при растворении газа в кипящем растворе аммиака. Труднос-
ти усугубляются экзотермичностью реакции, а также увлечением
ацетилена из реакционной смеси этиленом и парами аммиака. Тем
не менее этот способ благодаря его простоте достаточно часто ис-
188
пользуют при получении в лабораторных масштабах незамещен-
ных ацетиленидов натрия и лития. Синтез же таким путем замеще-
нных ацетиленидов нерационален из-за потери третьей части ис-
ходного терминального алкина вследствие его восстановления и в
связи с загрязнением образующимся алкеном продуктов после-
дующего алкилирования ацетиленида. Более удобным методом в
этом случае представляется реакция обмена между терминальным
алкином и амидом щелочного металла. Так как аммиак является
существенно более слабой кислотой, чем алкины-1, образование
ацетиленидов проходит до конца и, как правило, с количественным
выходом.
Для получения из ацетилена алкинов с внутренней тройной
связью целесообразно сначала алкилировать моноацетиленид нат-
рия или лития, а затем промежуточный алкин-1, не выделяя, пере-
вести в металлическое производное действием амида металла и да-
лее алкилировать повторно:
HC = CLi BuBr - BuC^CH L‘NH2*> BuCECLi
ж NH
------ BuCaCBu 67 %
Реакционная способность алкилгалогенидов падает от иод- к
хлоропроизводным, но практически лучшие результаты дают
бромиды, так как они активнее хлоридов и в отличие от иодидов при
температуре -33 °C образуют лишь несущественное количество
аминов - продуктов побочной реакции аммонолиза. Нужно, однако,
иметь в виду, что непрореагировавшие бромиды, как и хлориды,
трудно отделяются от продуктов реакции, и для очистки получен-
ных алкинов нередко необходимо применять фракционирование на
эффективных колонках. Скорость реакции также быстро падает с
ростом молекулярной массы бромида, возможно, из-за понижения
его растворимости в жидком аммиаке. Иногда эту трудность уда-
ется преодолеть, повышая температуру реакции до комнатной
(проведение реакции в автоклаве) или используя сорастворитель
(часто ТГФ, ГМФА или ДМСО):
Со Н 1 7 ВГ
HC=CNa -------—------*- НС = СС»Н|7 87 %
)K.NHj ДМСО 8 17
Рассматривая реакцию алкилирования терминальных алкинов,
следует отметить, что область ее применения ограничена. Практи-
189
чески этим способом в молекулу исходного ацетиленового соеди-
нения удается вводить только первичные алкильные радикалы,
причем не имеющие в ^-положении разветвления или активирую-
щей группы типа Ph, Hal, RS, RC=C и т. п. Например:
HC=CNa —-» HC = CBu 85-90 %
x.NH3
C7H.,Br
CH, = CH-C = CNa —---------- CH, = CH-CsCC7H,,
2 ж N H 2 / 1Э
' 3 80 %
Третичные и вторичные алкилгалогениды, как и первичные
галогенопроизводные с активирующей группой в ^-положении, в
условиях алкилирования главным образом подвергаются дегидро-
галогенированию. Непригодны для препаративного использования
и первичные бензил-, аллил- и пропаргилгалогениды, так как про-
дукты алкилирования, первоначально образующиеся при их взаи-
модействии с ацетиленидами, благодаря тлрш/с-металлированию по
активной метиленовой группе претерпевают сложные вторичные
превращения.
Хотя во всех вышеприведенных примерах в качестве алкили-
рующих средств указаны алкилгалогениды, нередко для этих целей
используют диалкилсульфаты и алкилтозилаты. Их применение
иногда позволяет избежать осложнений, например при введении в
молекулу вторичных алкильных радикалов.
Из функциональных производных алкинов, которые с успехом
могут быть алкилированы в аммиаке, следует упомянуть пропарги-
ловый и некоторые родственные ему спирты. Эти соединения при
взаимодействии с двумя молями LiNH2 образуют дилитиевые соли,
в анионах которых имеются существенно различающиеся нуклео-
фильные центры: более „мягкий11 углеродный и „жесткий11 кисло-
родный. По отношению к галоидным алкилам „мягкий11 центр более
реакционен, и поэтому такие соли при взаимодействии с экви-
мольным количеством галогенида дают исключительно продукты
С-алкилирования (с динатриевым производным реакция проходит
хуже):
НОСН,С=СН .......-.У”2-». LiOCH.C^CLi CH?1 -
2 x.NH3 2 2. H2O
------► HOCH2CSCCH3 80 %
190
3.3.2. Алкилирование активных метиленовых соединений
Относительно устойчивые карбанионы возникают также в слу-
чае, если карбанионный атом связан с двумя или тремя электроно-
акцепторными группами. Стабилизация аниона происходит за счет
сопряжения неподеленной пары электронов карбанионного центра
с кратной связью, как правило, входящей в состав электроноак-
цепторной группы
/С.-/- ,
Xх С -------------- X ''С
Электроноакцепторные группировки облегчают гетеролиз С-Н
связи под действием основания и далее протекание реакций по
СНз-, СН2- и СН-группам, поэтому содержащие их соединения
часто называют активными метиленовыми соединениями. По спо-
собности активировать соседние С-Н связи группы могут быть
расположены в ряд
NO2 > COR > SO2R > CO2R > CN > Ph.
В соответствии с этим такой заместитель, как фенил, не очень
сильно стабилизирует карбанион, вследствие чего толуол оказыва-
ется более слабой кислотой, чем аммиак (см. 3.3.1). Однако полифе-
нилзамещенные алканы, в которых, по крайней мере, две фениль-
ные группы присоединены к одному и тому же атому углерода,
становятся заметно более кислыми. Дифенил- и трифенилметаны
образуют с амидами натрия и калия соли красного цвета, которые
сохраняются в растворах аммиака неопределенно долгое время. С
подходящим алкилгалогенидом они вступают в реакцию алкилиро-
вания по механизму бимолекулярного нуклеофильного замещения:
Ph-CH, ---NH2»> Ph, CH Na BuBr > Ph9CHBu 92 %
2 2 x.NH3 2 2
Приведенная реакция успешно протекает с первичными и вто-
ричными, но не с третичными алкилгалогенидами, которые в ус-
ловиях реакции имеют склонность претерпевать элиминирование.
Другой возможной побочной реакцией является диалкилирование,
так как получающееся соединение обычно не сильно отличается по
кислотности от исходного. Тем не менее удается получать высокие
191
выходы моноалкилированных продуктов, контролируя количество
амида натрия.
Алкилирование успешно осуществляется, если помимо фениль-
ной к тому же самому атому углерода присоединена другая элект-
роноакцепторная группа. Так, фенил- и дифенилацето нитрилы ус-
пешно алкилируются алкилгалогенидами по «-положению:
NaNH,
PhCH,CN ---------U- PhCHCN
2 >k.NH,
Na+
Устойчивость карбанионов возрастает, когда карбанионный
центр оказывается сопряжен с кратной связью С=С или C=N,
поскольку электроотрицательные атомы кислорода и азота способ-
ны лучше делокализовать отрицательный заряд, чем атом углерода:
Н Н
I I
С. R ------------► .Сх R
R " С R ''С
II I
О О
Наиболее важными с точки зрения синтеза активирующими
группами являются карбонильная и сложноэфирная группы. Уда-
ление протона от «-углеродного атома карбонилсодержащего сое-
динения под действием основания приводит к образованию соот-
ветствующего енолят-аниона. Последний наиболее часто вовлека-
ется в реакции бимолекулярного нуклеофильного замещения, в том
числе и в реакцию алкилирования.
Алкилирование соединений, содержащих одну функциональ-
ную группу, - кетонов, альдегидов, сложных эфиров и нитрилов
(рКа > 18), требует присутствия сильного основания, и в качестве
такового часто используют амид металла в жидком аммиаке. Сила
этого основания достаточна, чтобы полностью превратить назван-
ные соединения в сопряженные основания - енолят-ионы; это по-
зволяет избежать реакций типа альдольной конденсации:
0 0 О
192
PhCH2COOEt
NaNH2
».NH3, Et2O
Na+
PhCHCOOEt
PhCH2CH2Br
PhCH2CH2CHCOOEt 77-81 %
Ph
Me Me
Me
Аналогично этому алкилирование фенил- и дифенилуксусных
кислот по a-положению можно провести, превращая их действием
амидов металлов в дисоли, которые в действительности имеют
структуру енолятов:
О" Ph
NaNH, 7 PhCH2Cl I
PhCH2COOH ——PhCH = C -----------------—► PhCH,CHCOOH
z «.NHj \ 2
O" 84 %
Важнейшими в органическом синтезе субстратами, подвергаю-
щимися алкилированию, являются ^-бифункциональные соедине-
ния: /3-кетоэфиры, /3-дикетоны и т. д. Для этих соединений доста-
точную концентрацию енолят-аниона могут создавать относитель-
но слабые основания. Алкилирование при обычных условиях с
использованием иного, чем аммиак, растворителя или без него
происходит по наиболее кислому атому водорода. Например, аце-
тилацетон при обработке щелочным металлом или алкоксидом
щелочного металла образует моноанион, который алкилируется по
метиленовой, а не по метильной группе, так как кислотность первой
больше и именно она отдает протон основанию.
Принципиально иной результат дает использование системы
амид натрия - жидкий аммиак. Основные свойства этой системы
оказываются достаточно сильными, чтобы отщепить протон не
только от наиболее кислой метиленовой группы, но и от следующей
по кислотности группы. Образовавшиеся дикарбанионы представ-
ляют собой амбидентные нуклеофилы, так как в них имеются два
способных атаковаться атома углерода (помимо атома кислорода,
атака по которому возможна для любого енолят-аниона). Важно,
однако, что реакция с одним молем галоидного алкила происходит
193
по наиболее нуклеофильному центру. Так, алкилирование ацетил-
ацетона в присутствии амида натрия в жидком аммиаке проходит
по метильной, а не по метиленовой группе:
МеСОСН2СОМе
NaNH2
О О
К /I
/с
С
BuBr
Me
— н о н+
---► С5Н[ [СОСНСОМе —----- С5Н[ [СОСН2СОМе
82 %
Алкилирование несимметричных дикетонов через дианионы
приводит в большинстве случаев к образованию одного продукта
или, реже, смеси, в которой существенно преобладает продукт
алкилирования по метильной, а не по метиленовой группе:
СОМе i.knh2, >k.nh3
COCH2CH2Ph
2. PhCH,Cl 3. Н2О, Н +
58 %
EtCOCH2COMe
1. NaNH2> «.NH,
2. Mel 3. H2O, H +
Me
EtCOCH2COEt
89 %
+ MeCHCOCH2COMe
11 %
Однако, если метиленовая группа дополнительно активирована, на-
пример фенильным заместителем, по ней может проходить алкили-
рование. В общем легкость алкилирования следует порядку
PhCH2> СН3 > СН2.
Ви
1. NaNH2, >k.NH, I
PhCH2COCH2COMe --------------PhCHCOCH2COMe
2. BuBr
77 %
Чтобы избежать самоконденсации /3-кетоальдегидов, при их
алкилировании исходят из мононатриевой соли, которую предва-
рительно переводят в дианион действием одного моля амида ще-
лочного металла, а затем алкилируют:
194
Na +
О О
К - 'I knh2
/С л. 'С. —.ггг
Нзсх чс н *NH3
I
н
о о 2"
I1'
- С С K+Na+ LBuBr „
Н2С С Н 2. н2о, н +
н
---*- ВиСН2СОСН2СНО 72 %
Аналогичные дианионы могут быть получены из /2-кетоэфиров,
/3-кетосульфидов и различных амидов и имидов, например:
Амид натрия оказывается достаточно сильным основанием,
способным генерировать поликарбанионы из поликарбонильных
соединений. Последние, как и дианионы, затем могут быть алкили-
рованы по боковой метильной или метиленовой группе:
ООО
II II II
PhCCH2CCH2CCH3
NH?
PhCH2Cl
О О О р-
Ph С С СН,
I I
н н
PhCOCH2COCH2COCH2CH2Ph
3.4. РЕАКЦИИ НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
б АРОМАТИЧЕСКОМ РЯДУ
Жидкий аммиак относится к числу растворителей, которые ис-
пользуются для проведения реакций, разнообразных по механизму
и традиционно относимых к ароматическому нуклеофильному за-
мещению.
В первую очередь это касается реакций галогенароматических
соединений (один-два атома галогена). Такие реакции протекают
через промежуточное образование дегидробензолов (аринов), и для
их осуществления требуется сильное основание; амиды натрия и
калия в жидком аммиаке используются для этой цели наиболее
часто:
195
ОМе
knh2
>k.NH3
ОМе
K.NH2
ОМе
Самое интересное заключается в том, что входящая группа не
всегда занимает положение, освобождаемое уходящей группой. В
приведенном примере образование ж-аминоанизола в качестве
единственного продукта объясняется тем, что возникший интер-
медиат несимметричен и метоксигруппа направляет входящую
группу не в орто-, а в мета- положение. В случае образования сим-
метричного интермедиата продукт реакции представляет собой
смесь с почти равным соотношением изомеров, что было изящно
продемонстрировано на примере реакции 1-14С-хлоробензола с
амидом калия:
В синтетическом плане реакции с промежуточным образова-
нием дегидробензолов важны, прежде всего, потому, что позволяют
вовлечь в процессы нуклеофильного замещения субстраты, не
реагирующие по другим механизмам. Так, например, в системе
амид натрия - жидкий аммиак арилгалогениды становятся способ-
ными алкилировать еноляты. Эта реакция расширяет спектр соеди-
нений, которые можно синтезировать на основе малонового эфира
(или сходных соединений). Амид металла как основание выполняет
здесь две функции - отрывает протон от органического субстрата,
образуя нуклеофил, и генерирует дегидробензол:
NaNH,
PhBr x —
CH(COOEt)2
CH(COOEt)2
PhCH(COOEt)2
н2о, н
PhCH(COOH)2
- е
Аналогичным образом удается осуществить арилирование
дианионов:
RCOCHCOCH, + PhBr NH2----------- RCOCH,COCH,Ph
2. Н2О, Н +
196
Многие важные реакции нуклеофильного ароматического заме-
щения протекают через промежуточное образование анионных
cr-комплексов (механизм SnAt). Обычно такие реакции реализу-
ются лишь для субстратов, имеющих в орто- и иара-положениях по
отношению к уходящей группе активирующие электроноакцептор-
ные группы. Высокая эффективность жидкого аммиака как среды
для проведения реакций этого типа была продемонстрирована не-
давно на примере нитрогалоген- и полифторароматических соеди-
нений:
Кинетическое изучение реакций на примере полифторарома-
тических соединений позволило установить, что взаимодействие их
с алкоголятами металлов в жидком аммиаке протекает с существен-
но большей скоростью, чем в спиртах.
Большая группа реакций в ароматическом ряду протекает по
механизму радикального нуклеофильного замещения (5^1). И в
этом случае в качестве растворителя часто используется жидкий
аммиак. Такие реакции проходят в мягких условиях и при использо-
вании неактивированных арилгалогенидов. Установлено, что круг
субстратов достаточно широк: PhHal, PhjS, РЬг1 + , РЬзБ + , РЬгО,
РЬгБе, PhOP(O)(OR)2 и PhNMe3 + . Реакция катализируется сольва-
тированными в аммиаке электронами или инициируется облуче-
нием и далее проходит по свободнорадикальному механизму:
АгХ ——— [Агх] ’ ------------
Ar’ + Nu" ----[ArNu] ’ + АгХ --------
где Nu - нуклеофильный реагент.
Ar’ + X"
ArNu + [ Агх] ’
7 Заказ № 137
197
Аммиак оказался наиболее подходящим растворителем для
реакций типа S/wl благодаря следующим обстоятельствам. Он
хорошо растворяет реагенты различной природы - неполярные
органические соединения и соли, образованные щелочными метал-
лами и анионными нуклеофилами. Для фотоинициируемых реак-
ций важным оказывается то, что аммиак прозрачен для ультрафио-
летового света; для реакций, инициируемых сольватированными
электронами, он удобен тем, что не взаимодействуют с последними.
Поскольку большинство нуклеофилов, успешно введенных в аро-
матические реакции типа SrnI , представляют собой анионы высо-
кой основности, особое значение приобретает слабая кислотность
аммиака.
Наиболее изученными нуклеофилами в реакциях арилирования,
протекающих по механизму SrnI , являются еноляты кетонов.
к
ж-NHj
ОН
I
сн2снсн3
^-^ОМе
6,9 : 1
В отсутствие калия и при облучении раствора близким УФ све-
том образуются те же продукты, но в другом соотношении.
Две важные особенности механизма 5^1 заключаются в сле-
дующем: во-первых, требуемый заместитель может быть введен в
желаемое положение ароматического фрагмента (занимаемое в
исходном субстрате уходящей группой) и, во-вторых, в реакцию
могут вступать даже субстраты, имеющие электронодонорные за-
местители. Так, реакция о-галогеноанизолов с амид-ионами в жид-
ком аммиаке протекает по механизму аринового типа, приводя к
образованию л^-анизидина, а при инициировании реакции металли-
ческим калием механизм ее изменяется, и в результате образуется
только о-анизидин:
NH7
ОМе
NH7
ОМе
198
ОМе
к
ж. NH3'
ОМе
Ряд конденсированных и неконденсированных галогенозаме-
щенных полициклических ароматических соединений вовлекается
в реакции по механизму SrnI , давая во многих случаях более высо-
кие выходы, чем соответствующие производные бензола.
1-Хлоро- и 1-иодонафталины реагируют с цианометил-анио-
ном, енолятом ацетона, а также с 2-оксиэтилсульфид-ионом, обра-
зуя продукты замещения с высокими выходами. Реакция с карбани-
онными нуклеофилами может также быть промотирована металли-
ческим калием, но в этих случаях наблюдается и частичное вос-
становление ароматического кольца:
3.5. РЕАКЦИИ ЭЛИМИНИРОВАНИЯ
Амид натрия в жидком аммиаке является одним из оснований,
которые применяются для дегидрогалогенирования алкилгалоге-
нидов. Все закономерности, характерные для реакции элиминиро-
вания, при этом сохраняются. Чаще это сильное основание исполь-
зуют для получения соединений с тройной связью. В этом случае в
роли галогенидов выступают 1,1- и 1,2-дигалогениды, а. также ви-
нилгалогениды:
NaNH,
PhCHBrCH2Br --------PhC^CH
NaNH, NHdCl
MeC = CHCH,OH ------MeC=CCH,ONa ----------—-
| 2 x.NH3 2
Cl ---- MeC = CCH2OH
199
В тех случаях, когда возможно образование терминального
алкина и алкина с внутренней тройной связью, преимущественно
образуется первый, так как он при взаимодействии с амидом натрия
дает соль, сдвигая равновесие в сторону ее образования. Результат
реакции в целом оказывается иной, чем при использовании таких
оснований, как НО” и RO-, когда наблюдается образование алки-
нов с внутренней тройной связью.
3.6. РЕАКЦИИ ПРИСОЕДИНЕНИЯ
Соли очень слабых кислот, легко получаемые в жидком аммиа-
ке, могут быть использованы в реакциях присоединения к некото-
рым ненасыщенным соединениям. Процесс присоединения чаще
всего включает атаку карбанионов на электрофильный атом углеро-
да, который связан с кислородом (как в карбонильных соединениях,
эпоксидах и в диоксиде углерода), либо с азотом (как в нитрилах).
Наиболее важными являются реакции присоединения ацети-
ленид-аниона к карбонильным соединениям с образованием спир-
тов ацетиленового ряда - алкинолов:
М _ R,CO НтО
RC=CH —--------- RC^CM ------------ R,CC = CR »
ж-NHj 2 ।
------ R2C(OH)C = CR om
M = Li, K, Na; R = H, Aik, Ar
Карбонильный компонент может содержать как альдегидную,
так и кетонную группировку. Этим методом пользуются при син-
тезе вторичных алкинолов из альдегидов (начиная с пропионового)
и третичных из кетонов, особенно стерически затрудненных (пента-
метил- и гексаметилацетон), которые не удается ввести в реакцию
Фаворского (представляющую собой альтернативный способ син-
теза алкинолов):
PrCHO b HC=CNa> жЫНз НС^ССН(ОН)Рг
2. Н2о, Н +
Ме2СО BuC = CNa_^ BuC=CC(OH)Me9
ж.МН) 2
znpem-BuCPr-изо HC~CNa> ж^нз НС = СС(ОН)Ви-ирет
II 2. н2о, н+ |
О Рг-изо
200
Для превращения кетонов в алкинолы чаще используют
ацетиленид натрия, хотя иногда применение ацетиленида лития
дает определенные преимущества, особенно в случае «^-непре-
дельных кетонов:
HC=CLi
«.NHj
Me
Me
CH = CHC(OH)C = CH
Me Me
Me
3.7. ПРОМЫШЛЕННОЕ ПРИМЕНЕНИЕ ЖИДКОГО АММИАКА
Кажущимся недостатком жидкого аммиака как растворителя
является низкая температура кипения. Однако применение его во
многих случаях обеспечивает столь высокую скорость реакции, что
позволяет вести процесс при температуре, не превышающей тем-
пературу кипения. Аммиак также используется при более высоких
температурах (комнатной и выше), и хотя это осложняет процесс
(из-за необходимости применять автоклав), однако не создает
непреодолимых препятствий. Заметим также, что теплота испаре-
ния аммиака велика, поэтому, работая с ним, можно использовать
емкости, сообщающиеся с атмосферой. При этом быстрого испаре-
ния аммиака не происходит, и в большинстве случаев можно
обойтись без наружного охлаждения системы. Немаловажным об-
стоятельством является также то, что аммиак можно удалить при
небольшом нагревании. Это облегчает процесс его регенерации, де-
лает возможным повторное использование. Неудивительно поэто-
му, что многие исследователи рассматривают аммиак как перспек-
тивный нетрадиционный растворитель для экологически чистых и
малоотходных технологий.
В нашей стране в качестве растворителя жидкий аммиак исполь-
зуется предприятиями витаминной промышленности при получе-
нии промежуточных продуктов в производстве витаминов А и Е.
Например, из гексагидропсевдоионона и ацетилена в среде жидкого
аммиака в присутствии метанольного раствора гидроксида кальция
при температуре -5 ч-+30 °C и давлении 28-35 атм получают
3,7,11-триметилдодецин-1-ол-3 - промежуточный продукт синтеза
витамина Е. Доля возвращенного в цикл аммиака составляет 90 %.
201
Me
CH = CHC(OH)C = CH
Me Me
Me
Аналогично 3-метилпентен-4-ин-1-ол-3 - промежуточный про-
дукт синтеза витамина А - получают конденсацией метилвинил-
кетона с ацетиленидом кальция в среде жидкого аммиака при тем-
пературе не выше -50 °C:
СЩ
_ । 3
СН9 = СН-С-СНЧ нс = сн, Са^ СН, = СН-С-С = СН
2 II 3 >k,N Hj 2 I
о он
Пример использования жидкого аммиака для синтеза лекарст-
венных препаратов стероидного ряда приведен выше (см. 3.2.1).
Нет причин сомневаться, что с развитием современных техно-
логий аммиак будет находить все большее применение в промыш-
ленных и лабораторных процессах.
3.8. ЭКСПЕРИМЕНТАЛЬНЫЕ РАБОТЫ
Особенности работы с жидким аммиаком, амидами металлов
и ацетиленовыми баллонами. Аммиак выпускается в сжиженном
состоянии в баллонах желтого цвета с надписью черными буквами
„аммиак11. Он содержит примесь 5 • 10-5 весовых частей воды
(азеотропной) и обычно хранится при комнатной температуре под
давлением не более 30 атм. При отборе жидкого аммиака баллон
устанавливают так, чтобы запирающий вентиль был направлен
вниз, и жидкость выпускают через игольчатый вентиль в сосуд
Дьюара. Отбор производят на улице в противогазе.
Жидкий аммиак квалификации „ч“ может быть дополнительно
очищен растворением в нем металлического натрия и последующей
отгонкой в охлаждаемый до -70 °C реакционный сосуд непосредст-
венно перед проведением эксперимента. При такой операции одно-
временно удаляется коллоидное железо, которое контролирует ре-
акцию металлов (особенно калия и натрия, в значительно меньшей
степени - лития) с аммиаком. Поэтому при использовании аммиака
в процессах восстановления желательно, чтобы он был предвари-
202
тельно перегнан. Даже когда используются такие доноры протонов,
как вода, рекомендуется проводить осушение аммиака и используе-
мых растворителей, так как это позволяет точно контролировать
количество донора протонов.
Аммиак раздражает дыхательные пути, а попадая на открытые
кожные покровы, вызывает сильные ожоги. Работа с жидким ам-
миаком должна проводиться в хорошо действующем вытяжном
шкафу. Лицо работающего должно быть защищено экраном, на
руки следует надеть перчатки. Реакционную колбу с жидким ам-
миаком рекомендуется предохранить мешком из плотной ткани
(брезент).
Амиды щелочных металлов относятся к взрывоопасным ве-
ществам. Твердый амид металла при добавлении воды разлагается
со взрывом. На воздухе амиды металлов поглощают влагу, окисля-
ются, разлагаются и становятся взрывоопасными. Метод получения
амидов щелочных металлов с применением жидкого аммиака наи-
более безопасен и удобен. Тем не менее соблюдение мер предосто-
рожности обязательно. Рабочие смеси, содержащие амиды метал-
лов, запрещается нагревать на водяной бане. Посуду с остатками
амидов следует обработать в тот же день. Недопустимо оставлять
реакционные смеси с амидом металла более чем на 1-2 дня.
Ацетилен выпускается в баллонах белого цвета с надписью
красными буквами „ацетилен". Сжатый ацетилен легко взрывается,
поэтому, чтобы уменьшить опасность взрыва, баллон заполняют
пористой массой (пемзой, активированным углем и т. п.). Пористый
наполнитель пропитывают ацетоном, в котором растворяется аце-
тилен. Максимальное давление в баллоне 20-25 атм. При исполь-
зовании баллон обычно находится в вертикальном положении, в
5-10 м от рабочего места. Подвод газа осуществляется через
толстостенный вакуумный шланг либо по системе стальных трубок.
Работа с ацетиленом проводится в хорошо действующем вытяж-
ном шкафу, в котором не должны находиться включенные электро-
нагревательные приборы с открытым обогревом и газовые горелки.
Отвод газа из системы следует направлять непосредственно в канал
вытяжного шкафа.
К выполнению экспериментальных работ следует приступать
после ознакомления с инструкциями по технике безопасности при
работе с жидким аммиаком, щелочными металлами и их амидами, а
также с ацетиленовыми баллонами.
203
Получение амидов натрия и лития в жидком аммиаке
М + NH, ---------- MNH,
3 жМ3 2
Реакцию проводят в трехгорлой колбе вместимостью 500 мл,
снабженной механической мешалкой и воронкой с уравнительной
трубкой. В третье горло колбы помещают стеклянную трубку дли-
ной 10-30 см для предотвращения выплескивания реакционной
смеси при энергичном перемешивании и во избежание разбрыз-
гивания аммиака при его испарении. Через боковой тубус в колбу
наливают необходимый объем жидкого аммиака и при перемеши-
вании в нее бросают 1-3 кусочка металлического натрия (лития)
величиной с горошину. После появления голубой окраски, харак-
терной для растворов щелочных металлов в жидком аммиаке, при-
бавляют несколько кристаллов нитрата железа (II) в виде гидрата
(около 0,1 г), а затем небольшими кусочками заданное количество
свеженарезанного металлического натрия (лития). О превращении
натрия в амид судят по исчезновению голубой окраски раствора и
образованию суспензии серого цвета. Суспензия амида лития бес-
цветна. Реакция обычно занимает 20-30 мин.
2,3-Дифенилпропионовая кислота
Ph
1. NaNH2, жМ, I
PhCH,COOH ----------------3----PhCH,CHCOOH
2. PhCH2CI 3. Н2О, Н+ 2
Реактивы
Натрий металлический 1,3 г
Фенилуксусная кислота 3,7 г
Бензилхлорид 3,4 г
Аммиак жидкий 200 мл
Эфир сухой 115 мл
К суспензии амида натрия, приготовленной из 1,3 г металли-
ческого натрия и 200 мл аммиака, прибавляют 3,7 г (0,027 моль)
фенилуксусной кислоты и образовавшуюся суспензию темно-зе-
леного цвета перемешивают в течение 20 мин. Затем быстро при-
ливают 3,4 г (0,027 моль) бензилхлорида в 15 мл сухого эфира и
смесь перемешивают еще 2 ч. Далее из реакционной смеси аммиак
204
осторожно выпаривают почти досуха на водяной бане (сначала вода
должна иметь комнатную температуру, затем 30-40 °C). К остатку
приливают 50 мл эфира и вновь выпаривают все досуха; такую
процедуру повторяют еще раз. Полученный остаток растворяют в
100 мл воды и раствор промывают тремя порциями эфира по 100 мл.
Водный раствор фильтруют через складчатый фильтр, чтобы осво-
бодиться от механических примесей, и фильтрат подкисляют хло-
роводородной кислотой. Образовавшаяся бесцветная маслянистая
жидкость после охлаждения в течение нескольких минут в бане со
льдом превращается в кристаллическую массу. Если маслянистая
жидкость не затвердевает, ее экстрагируют эфиром, объединенные
эфирные экстракты сушат сульфатом натрия и остаток после испа-
рения эфира вновь пытаются закристаллизовать. Кристаллы от-
фильтровывают, методом ТСХ контролируют присутствие в них
фенилуксусной кислоты. В случае необходимости кристаллы на
фильтре промывают тремя порциями горячей воды (50-60 °C) по 20
мл (растворимость в воде фенилуксусной кислоты заметно больше,
чем 2,3-дифенилпропионовой кислоты). После высушивания
кристаллов получают 5,2 г (85 %) 2,3-дифенилпропионовой кис-
лоты, т. пл. 92-93,5 °C. Ее перекристаллизовывают из петролейного
эфира (т. кип. 70-110 °C) и получают препарат с температурой
плавления 95,5-96,5 °C.
Хроматография: элюент - хлороформ и этилацетат, 3:1; Rf 0,75.
Спектральные характеристики даны на рис. 3.1.
1,1-Дифенилпентан
Ph
1. NaNH2, *NH3
СгЦ ---------------;—
/ 2. BuBr
Ph
Реактивы
Натрий металлический
Дифенилметан
Бутилбромид
Аммиак жидкий
Эфир сухой
Ph
СН(СН2)3СН3
Ph
1,6 г
10,1 г
9,6 г
150 мл
70 мл
К суспензии амида натрия, приготовленной из 1,6 г металли-
ческого натрия и 150 мл жидкого аммиака, при перемешивании
205
Рис. 3.I. Спектральные характеристики
2,3-дифенилпропионовой кислоты:
а - спектр ПМР* в ССЦ (<5, м.д.: 2,95 д.д, J = 14и7Гц(1Н,
СН2), 3,37 д.д, J = 14 и 8 Гц (1Н, СН2), 3,81 д.д, J = 9 и 7 Гц
(1Н, СН), 7, 28 м (ЮН, Ph));
б - ИК спектр* в ССЦ (1720 (С=0), 3200 см”1, ш. (О-Н));
в - УФ спектр* в этаноле (Лмакс., 1g е: 282 тс, 3,08)
206
добавляют по каплям раствор 10,1 г (0,06 моль) дифенилметана в
20 мл сухого эфира. При этом цвет реакционной смеси из серого
становится темно-красным. Образовавшуюся суспензию переме-
шивают в течение 15 мин, а затем к ней прибавляют по каплям
раствор 9,6 г (0,07 моль) бутилбромида в 20 мл сухого эфира; цвет
смеси вновь меняется на серый. Суспензию оставляют на ночь для
испарения аммиака (или осторожно упаривают аммиак на водяной
бане). К реакционной смеси прибавляют 30 мл эфира и затем с осо-
бой осторожностью по каплям и при перемешивании 30 мл воды.
Эфирный слой отделяют, из водного продукт экстрагируют эфи-
ром. Объединенные эфирные растворы сушат сульфатом магния,
эфир удаляют, остаток перегоняют в вакууме. При этом отбирают
фракцию с т. кип. 160-161 °C при 3 мм рт. ст. Получают 12,4 г
(92 %) 1,1-дифенилпентана, Ид6 1,5501.
Хроматография: элюент - хлороформ и петролейный эфир, 2:1;
Rf 0,5. Спектральные характеристики даны на рис. 3.2.
1,1,2-Трифенилэтан
Ph
сн2
Ph
1. Na NH2, >k.NH3
2 PhCH2Cl
Ph
CHCH2Ph
Ph
Реактивы
Натрий металлический
Дифенилметан
Бензилхлорид
Аммиак жидкий
Эфир сухой
1,3 г
8,4 г
6,3 г
200 мл
95 мл
К суспензии амида натрия в жидком аммиаке, приготовленной
из 1,3 г (0,056 моль) металлического натрия и 200 мл жидкого ам-
миака, при перемешивании прибавляют по каплям раствор 8,4 г
(0,05 моль) дифенилметана в 20 мл сухого эфира. Раствор изменяет
цвет на темно-красный. Через 10 мин в колбу прибавляют раствор
6,3 г (0,05 моль) бензилхлорида в 25 мл сухого эфира. Вскоре на-
блюдается обесцвечивание раствора. Реакционную смесь переме-
шивают в течение 0,5 ч, добавляют в нее 50 мл сухого эфира и остав-
ляют на ночь для испарения аммиака (или осторожно упаривают
207
3000 ' 2000 ' 1600 ' 1200 ' Ъсм1
Рис. 3.2. Спектральные характеристики
1,1 -дифенилпентана:
а - спектр ПМР* в ССЦ (<5, м.д.: 0,89 т, J = 7 Гц (ЗН, СН3),
1,32 м (6Н, ЗСН2), 2,03 м (2Н, СН2), 3,86 т, J = 7,5 Гц
(1Н, СН), 7,16 м (ЮН, Ph)); 6 - ИК спектр* в СС1д;
в - УФ спектр* в этаноле (ЛмаКс., нм, 1g £' 255 тс, 3,83).
208
аммиак на водяной бане). Холодную реакционную смесь разлагают,
приливая осторожно при перемешивании 50 мл воды. Эфирный
слой отделяют, из водного проводят экстракцию эфиром. Из эфир-
ных растворов после высушивания сульфатом натрия удаляют раст-
воритель. Остаток кристаллизуется. После перекристаллизации из
спирта получают 10,1 г (78 %) 1,1,2-трифенилэтана, т. пл. 50-54 °C.
Дополнительная очистка может быть проведена сублимацией при
100 °C и 2 мм рт. ст.
Хроматография: элюент - петролейный эфир и хлороформ, 3:1;
Rf 0,1. Спектральные характеристики приведены на рис. 3.3.
Гептин-2-ол-1
1. LiNH,, жДН,
нс=ссн,он ----------------—СН,(СН,),С=ССН,ОН
2 2. BuBr 3. Н2О, Н+ 3 23 2
Реактивы
Литий металлический 1,4 г
Пропаргиловый спирт 5,6 г
Бутилбромид 13,7 г
Аммиак жидкий 200 мл
К энергично перемешиваемой суспензии амида лития, приго-
товленной из 1,4 г (0,2 моль) металлического лития и 200 мл жид-
кого аммиака, прибавляют 5,6 г (0,1 моль) пропаргилового спирта,
перегнанного в небольшом вакууме. Далее в течение 1,5 ч прибав-
ляют по каплям 13,7 г (0,1 моль) бутилбромида, после чего аммиаку
дают испариться, снабдив открытое горло колбы газоотводной
трубкой, наполненной гранулами натронной извести (обычно колбу
оставляют на ночь). Трубка должна располагаться на уровне дна
колбы или ниже его, с тем чтобы после удаления аммиака в колбе
осталась защищающая атмосфера этого газа. Удалить аммиак мож-
но и быстрее - выпариванием на водяной бане (сначала вода должна
иметь комнатную температуру, затем 30-40 °C). К твердому ос-
татку осторожно при перемешивании прибавляют 50 мл воды.
После того как масса растворится, органические вещества из-
влекают экстракцией эфиром (5 раз). Объединенные экстракты
сушат сульфатом магния, эфир удаляют. После перегонки остатка
из колбы с дефлегматором образуется 8,4 г (75 %) гептин-2-ола-1,
т. кип. 83 °C при 12 мм рт. ст., 1,4550. Продукт индивидуален по
данным ГЖХ. Спектральные характеристики даны на рис. 3.4.
209
Гексин-1
1. Na , ж-NH,
HCHCH -------------------► CH,(CH,kC = CH
2. BuBr 3. H20 J 2 3
Реактивы
Натрий металлический 9,7 г
Бутилбромид 54,8 г
Ацетилен (из баллона)
Аммиак жидкий 300 мл
Трехгорлую колбу вместимостью 500 мл снабжают механичес-
кой мешалкой, пропущенной через короткую стеклянную муфту, и
длинной трубкой для ввода газа, которая должна быть погружена в
жидкий аммиак. Трубка соединена с пустым барбатером и барба-
тером, наполненным концентрированной серной кислотой. Остав-
шийся боковой тубус колбы используют сначала для введения
аммиака и натрия, а позже в него устанавливают капельную ворон-
ку. В колбу через боковой тубус наливают 300 мл жидкого аммиака.
В аммиак при перемешивании пропускают быстрый ток ацетилена
(примерно 5 пузырьков в секунду), высушенного при прохождении
через склянку с серной кислотой. Насыщение аммиака ацетиленом
продолжается 5 мин. Затем ток ацетилена уменьшают, в получен-
ный раствор через боковой тубус прибавляют 9,7 г (0,42 моль)
мелко нарезанного (размером с горошину) металлического натрия с
такой скоростью, чтобы раствор не окрашивался надолго в голубой
цвет (если это произойдет, рекомендуется увеличить ток ацетиле-
на). Обычно прибавление натрия занимает 10—15 мин.
Когда весь натрий будет прибавлен, пропускание газа прекра-
щают и в течение 30 мин из капельной воронки при эффективном
перемешивании прибавляют 54,8 г (0,4 моль) бутилбромида. Пере-
мешивание продолжают в течение 1,5 ч. Затем в реакционную смесь
вводят по каплям (во избежание сильного разогревания) 50 мл
25 %-ного водного раствора аммиака и далее 100-150 мл воды.
Когда ледяная корка на наружных стенках колбы начнет оттаивать,
ее легко можно удалить. Холодное содержимое колбы переносят в
делительную воронку и отделяют нижний водный слой. Углеводо-
родный слой промывают последовательно водой, 5 %-ной хлоро-
водородной кислотой (с проверкой кислотности водного слоя), сно-
ва водой, 10 %-ным раствором карбоната натрия и опять водой.
210
fOO
i
60-
20-
0 "L
3500
2500
Рис. 3.3. Спектральные характеристики
1,1 -дифенилэтана:
a - спектр ПМР* в ССЦ (<5, м.д.: 3,33 д, J = 8 Гц
(2Н, СН2), 4,21 т, J = 8 Гц (1Н, СН), 7,1 м (15Н, Ph));
б - ИК спектр* в ССЦ; в - УФ спектр* в метаноле
(2Макс., нм, 1g £: 261 тс, 3,01; 298, 2,96)
—I—I--1-1-1--1-1-
1600 1200
211
Промытый углеводород высушивают хлоридом кальция и фракцио-
нируют, применяя колонку эффективностью 5-10 теоретических
тарелок. Выход гексина-1 составляет 18,1 г (55 %), т. кип. 70-72 °C,
«о 1,3990. Спектральные характеристики даны на рис. 3.5.
1-Этинилциклогексанол
нс=сн
1. Na, )K.NH3
Реактивы
Натрий металлический
Циклогексанон
Ацетилен (из баллона)
Аммиак жидкий
Эфир сухой
/--ч С = СН
\' ОН
5,7 г
24,5 г
300 мл
50 мл
Трехгорлую колбу вместимостью 500 мл снабжают механи-
ческой мешалкой и длинной трубкой для ввода ацетилена, которая
должна быть погружена в жидкий аммиак. Трубка соединена с
пустым барбатером и барбатером, наполненным концентрирован-
ной серной кислотой. Третий тубус колбы закрыт трубкой, запол-
ненной сухой ватой. В колбу наливают 300 мл жидкого аммиака,
прибавляют 1-2 кусочка металлического натрия для обезвоживания
аммиака, а затем в течение 15 мин вносят небольшими порциями
5,7 г (0,25 моль) металлического натрия. Раствор приобретает
голубую окраску. Далее при несильном перемешивании начинают
пропускать достаточно быстрый ток ацетилена до тех пор, пока весь
натрий не превратится в ацетиленид натрия, что будет заметно по
образованию молочно-белой суспензии. Эта операция занимает 1 ч.
Газовводную трубку заменяют капельной воронкой и через нее в
реакционную колбу прибавляют по каплям в течение 30 мин раст-
вор 24,5 г (0,25 моль) циклогексанона в 50 мл эфира. Для завер-
шения реакции смесь перемешивают еще 3 ч, затем прибавляют
50 мл эфира и дают испариться аммиаку (для этого рекомендуется
оставить реакционную смесь на ночь). После испарения аммиака
остаток должен находиться под слоем эфира. Реакционную смесь
при перемешивании и охлаждении осторожно разлагают водой,
добавляя ее по каплям до полного или почти полного растворения
осадка. Эфирный слой отделяют, из водного слоя экстрагируют
212
продукт эфиром, объединенные вытяжки промывают насыщенным
раствором бисульфита натрия, затем насыщенным раствором хло-
рида натрия и сушат сульфатом магния. Эфир удаляют, а остаток
перегоняют над небольшим количеством янтарной кислоты. До-
бавка последней необходима, так как некоторые алкинолы могут
взрывоподобно разлагаться при перегонке, особенно в присутствии
веществ основного характера (перегонку следует вести за за-
щитным экраном). Собирают фракцию с т. кип. 79-81 °C при
Рис. 3.4. Спектральные характеристики гептин-2-ола-1:
а - спектр ПМР* в СС14 (<5, м.д.: 0,93 т, J = 8 Гц (ЗН, СН3), 1,43 м (4Н, 2СН2), 1,95 с
(Ш, ОН), 2,31 м (2Н, СН2), 4,12 т, J = 1 Гц (2Н, СН2О));
б - ИК спектр* в тонком слое (2220 и 2280 см-1 (С=С), 3340 см-1 (О-Н))
213
21 мм рт. ст., «о20 1,4823. Выход 1-этинилциклогексанола составля-
ет 6 г (19,4 %). Чистоту продукта (отсутствие примеси циклогек-
санона) контролируют методом ГЖХ или пробой с 2,4-динитрофе-
нилгидразином. Спектральные характеристики даны на рис. 3.6.
2-Нитроанизол
Рис. 3.5. Спектральные характеристики гексина-1:
а - спектр ПМР* в СС14 (<5, м.д.: 0,93 м, (ЗН, СН3), 1,42 м (4Н, 2СН2), 1,73 м (1Н, СН=),
2,11 м (2Н, СН2= С)); б - ИК спектр в тонком слое (3320 см-1 (С-Н))
214
Реактивы
Натрий металлический 1,3 г
2-Хлоро-1-нитробензол 3,1 г
Метанол 1,8 г
Аммиак жидкий 200 мл
В круглодонную колбу вместимостью 500 мл, снабженную
механической мешалкой и воздушным холодильником, наливают
200 мл жидкого аммиака и прибавляют 1-2 кусочка металлического
натрия величиной с горошину (для обезвоживания аммиака). Затем
вносят небольшими порциями 1,3 г (0,056 моль) натрия и при-
бавляют 1,8 г (0,056 моль) метанола. После того как исчезнет синяя
окраска, характерная для раствора натрия в аммиаке, в колбу вносят
3,1 г (0,019 моль) 2-хлоро-1-нитробензола. Реакционную смесь
перемешивают в течение 4 ч и оставляют до полного испарения
аммиака. К остатку прибавляют 100 мл воды и продукт реакции
экстрагируют эфиром. Экстракт сушат сульфатом магния, раство-
ритель удаляют. Остаток перегоняют, собирая фракцию с т. кип.
148-150 °C при 15 мм рт. ст. Выход составляет 2,7 г (90 %). Хрома-
тография: элюент - хлороформ и этилацетат, 10:1; RfG,l. Спект-
ральные характеристики приведены на рис. 3.7.
4-Нитрофенетол
C1-Z Э— NO2 C2115°.N-->- c2hso-Zno2
V // 1 Ж-NH, 2 э V // L
Реактивы
Натрий металлический 1,3 г
Этанол 2,6 г
4-Хлоро-1 -нитробензол 3 г
Аммиак жидкий 200 мл
В круглодонную колбу вместимостью 500 мл, снабженную
механической мешалкой и воздушным холодильником, наливают
200 мл жидкого аммиака и прибавляют 1-2 кусочка металлического
натрия величиной с горошину. Затем вносят 1,3 г (0,056 моль)
натрия небольшими порциями и 2,6 г (0,057 моль) этанола. После
того как исчезнет синяя окраска, характерная для растворов натрия
в аммиаке, в колбу вносят 3 г (0,019 моль) 4-хлоро-1 -нитробензола.
215
Реакционную смесь перемешивают 5 ч и оставляют до полного
испарения аммиака. К остатку прибавляют 100 мл воды и продукт
реакции экстрагируют эфиром. Объединенные эфирные растворы
сушат хлоридом кальция, эфир удаляют в вакууме. Остаток пере-
Рис. 3.6. Спектральные характеристики 1-этинилциклогексанола:
а - спектр ПМР* в ССЦ (<5, м.д.: 1,64 м (10 Н, 5СН2), 2,77 с (1Н, СН), 4,34 с (1Н, ОН));
б - ИК спектр в тонком слое (3300 см-1 (= С-Н))
216
кристаллизовывают из этанола. Получают 2,9 г (92 %) 4-нитрофене-
тола; т. пл. 59-60 °C. Хроматография: элюент - хлороформ и этил-
ацетат, 10:1;7?/0,8. Спектральные характеристики даны на рис. 3.8.
5,8-Дигидро-1-нафтол
ОН
ОН
Li , EtOH
;k.NHj
Реактивы
Литий металлический
1-Нафтол
Этанол сухой
Аммиак жидкий
2,1 г
10,8г
13,8 г
200 мл
В круглодонную колбу вместимостью 500 мл, снабженную
механической мешалкой, помещают 10,8 г (0,075 моль) 1-нафтола.
К перемешиваемому содержимому колбы достаточно быстро (во
избежание образования твердой корки) прибавляют 200 мл жидко-
го аммиака. После того как нафтол-1 растворится, вносят 2,1 г
(0,3 моль) металлического лития вначале небольшими кусочками,
чтобы не допустить интенсивного кипения аммиака, затем не-
сколько быстрее. Далее раствор перемешивают в течение 20 мин, а
затем, не прекращая перемешивания, прибавляют по каплям 13,8 г
(0,3 моль) сухого этанола (если к концу прибавления произойдет
вспенивание, то интенсивность перемешивания следует умень-
шить). Продолжая перемешивание, дают испариться аммиаку, по-
давая в колбу струю воздуха. Остаток растворяют в 0,1 л воды, про-
мывают эфиром, эфирные растворы отбрасывают. Водный слой
подкисляют и продукт реакции экстрагируют из него эфиром.
Эфирные экстракты промывают водой, высушивают сульфатом
натрия, эфир упаривают. Получают 10,6 г (97 %) неочищенного
5,8-дигидро-1-нафтола; т. пл. 69-72 °C. Соединение неустойчиво и
после получения переводится гидрированием в 5,6,7,8-тетрагидро-
1-нафтол (см. 1.8).
217
Рис. 3.7. Спектральные характеристики
2-нитроанизола:
а - спектр ПМР* в ССЦ (<5, м.д.: 3,95 с (ЗН,
ОСН3), 7,02 д.т, J = 7 и 1 Гц (1Н, Н4 аром.),
7,16 д.д, J = 8,5 н 2 Гц (1Н, Н6 аром.) 7,53 д.т,
J = 8,5 и 2 Гц (Н5 аром.), 7,74 д.д, J = 7 и 2 Гц (IН,
НЗ аром)); б - ИК спектр в ССЦ (1000 см-1
(С-О), 1340и 1520 см-1 (NO2)); в — УФ спектр в
метаноле (2макс., нм, 1g е: 258, 4,11; 321, 3,98)
218
Рис. 3.8. Спектральные характеристики
4-нитрофенетола:
а - спектр ПМР* в СС1д (<5, м.д.: 1,48 т, J = 8 Гц (ЗН,
СН3), 4,17 кв, J = 8 Гц (2Н, СН2), 6,99 м (2Н, Н3-6),
8,15м (2Н, Н3-5)); б - ИК спектр* в СС14 (1120 см~1
(С—О), 1330 и 1500 см“1 (NO2)); в — УФ спектр*
в этаноле (2Макс., нм, 1g е: 230, 3,94; 313, 4,09)
219
Глава 4. ЛИТИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
В ОРГАНИЧЕСКОМ СИНТЕЗЕ
Органические соединения лития начали широко входить в
практику органического синтеза более 30 лет назад. В настоящее
время они продолжают интенсивно исследоваться. Создаются так-
же новые литийорганические реагенты, обладающие уникальными
свойствами, разрабатываются методы синтеза на их основе, изуча-
ются механизмы реакций.
Литийорганические соединения выгодно отличаются от реакти-
вов Гриньяра, часто используемых в синтезе, так же как и от других
металлорганических соединений. Они, как правило, более реакци-
онноспособны, и вследствие этого конечные продукты получаются
с высокими выходами. Выделелять продукты проще, так как боль-
шинство литиевых солей хорошо растворимо в воде. Они менее,
чем магнийорганические реактивы, склонны к реакциям восста-
новления и сопряженного присоединения. Немаловажным факто-
ром является также то, что при синтезе литийорганических соеди-
нений меньше трудностей возникает с выбором растворителей.
Обладая не очень сильно поляризованными связями, эти соеди-
нения хорошо растворяются как в слабополярых (типа простых
эфиров), так и в неполярных (типа углеводородов) растворителях,
чем выгодно отличаются от более реакционноспособных натрий-
органических соединений, которые вследствие солеобразного стро-
ения не растворяются в указанных растворителях, и от магнийорга-
нических соединений, которые требуют более полярных раствори-
телей. Возможность применения углеводородных растворителей
особенно ценна для' промышленной наработки литийорганических
соединений (многие из них благодаря этому вполне доступны) и для
использования их в синтезе практически важных соединений, в
частности лекарственных препаратов.
220
В настоящее время литийорганические соединения приобрели
вполне самостоятельное значение, и с каждым годом применение
их расширяется.
4.1. СТРОЕНИЕ ЛИТИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Понимание свойств литийорганических соединений основано
на знании структуры и характера связи углерод-литий. В литий-
органических соединениях, в противоположность органическим
производным других щелочных металлов, связь углерод-литий
оказывается ионной лишь в таких соединениях, карбанионная часть
которых резонансно стабилизирована, например сопряжением с
ненасыщенными группами:
Н
,С Li+ Zf 'b=^CH2 Li+
H2C''-~'CH2 ViV
В общем случае связь углерод-литий в литийорганических
соединениях является полярной ковалентной:
RCT~ —Ыст +
Однако рассматриваемая связь, вероятно, относится к более
сложному типу. Дело в том, что мономерные литийорганические
соединения оказываются координационно ненасыщенными и фор-
мула RLi в общем не отражает их структуры. Действительно, целый
ряд данных (в том числе данные ЯМР на ядрах 'Н, 13С и 7Li) указы-
вает на то, что литийорганические соединения существуют в виде
олигомеров или полимеров. Степень ассоциации зависит от при-
роды радикала и растворителя, а также от температуры и концент-
рации раствора. Метиллитий тетрамерен в углеводородных раст-
ворителях. Многие стерически незатрудненные алкиллитиевые
соединения, такие как бутиллитий и октиллитий, в неполярных
растворителях типа бензола и циклогексана гексамерны, в то время
как стерически затрудненные, такие как изопропиллитий и трет-
бутиллитий, - тетрамерны (табл. 4.1).
Важно отметить, что степень ассоциации алкиллитиевых соеди-
нений в растворах остается постоянной в большом интервале кон-
центраций и даже очень разбавленные растворы содержат ассоциа-
ты. Олигомеры, таким образом, оказываются достаточно стабиль-
ными. Рентгеноструктурный анализ метиллития и этиллития
8 Заказ № 137
221
обнаруживает, что они яв-
ляются тетрамерами даже
в твердом состоянии, а
Таблица 4.1. Степень ассоциации
некоторых литийорганических
соединений в различных растворителях
Соединение Растворитель Степень ассоциации
MeLi Эфир Тетрамер
BuLi Углеводороды Гексамер
Эфир Тетрамер
ТГФ Димер
BuLi-ТМЭДА Углеводороды Мономер
mpem-BuLi Углеводороды Тетрамер
ТГФ Димер
PhLi Эфир Димер
PhCH2Li Бензол Димер
ТГФ Мономер
CioHvLi Эфир Дцмер
изучение методом масс-
спектроскопии этиллития,
игрет-бутиллития и три-
метилсилиллития показы-
вает, что даже в газовой
фазе присутствуют тетра-
мерные и гексамерные час-
тицы.
Тем не менее степень
ассоциации зависит от при-
роды растворителя. Как
правило, она понижается в
растворителях эфирного
типа. Например, бутилли-
тий в бензоле гексамерен, в
эфире - тетрамерен, а в
ТГФ - димерен. Это связа-
но с характерной способ-
ностью литийорганических соединений координироваться с моле-
кулами, которые являются донорами электронов. Поэтому не
удивительно, что степень ассоциации еще более эффективно, чем
простые эфиры, снижают третичные амины, из которых часто ис-
пользуют тетраметилдиаминоэтан (ТМЭДА) и 1,4-диазобицикло-
[2,2,2]октан (ДАБЦО):
Me /СН2~СН2\ Me И
N
I I
Me Me
ТМЭДА
ДАБЦО
Координируясь с литийорганическими соединениями по атому
лития, эти бидентатные лиганды деполимеризуют ассоциаты. Так,
бутиллитий в гексане в присутствии ТМЭДА существует как мо-
номер:
(BuLi)6 + ТМЭДА
Bu Li Li Ви
✓ X
Me^N" 'jQ^-Me
Me CH2- CH2 Me
222
Удалось даже выделить твердые комплексы, имеющие опре-
деленную стехиометрию: MeLi и Et2<3 состава 1:1, BuLi и ТМЭДА
1:1, BuLi и ДАБЦО 4:1, PhLi и ДАБЦО 4:1. Помимо комплексов с
эфирами и аминами литийорганические соединения образуют
также комплексы с галогенидами и алкоксидами металлов. По-
следнее обстоятельство важно потому, что эти вещества часто
присутствуют в растворах литийорганических соединений и влия-
ют на их реакционную способность. Так, одна или две молекулы
бромида лития могут замещать молекулу метиллития в тетрамере, а
с фениллитием бромид лития образует комплекс состава 1:1, воз-
можно, замещая одну молекулу в его димере.
Способность литийорганических соединений образовывать
ассоциаты и комплексы обусловлена характером связей литий-уг-
лерод. Эта преимущественно ковалентная связь может соединять
атом углерода с одним атомом металла (простая о-связь), с двумя
атомами металлов (трехцентровая двухэлектронная связь). Простая
о-связь реализуется, по видимому, в мономерных соединениях
лития, трехцентровая - в димерах. Так, например, предполагается,
что в димере фениллития мономеры удерживаются двумя трех-
центровыми связями (рис. 4.1). Четырехцентровая связь реали-
зуется, вероятно, в тетрамерах. Рентгеноструктурные исследования
показали, что тетрамеры имеют тетраэдрическую структуру, при-
чем атомы лития находятся в вершинах тетраэдра, а алкильные
группы - над центром каждой из его граней. Это можно видеть на
примере структуры метиллития (рис. 4.2). Каждый атом лития
посредством четырехцентровой двухэлектронной связи одновре-
менно соединен с тремя метильными группами, и каждая метильная
группа одновременно связана с тремя атомами лития:
Me Me Li^|^Li
Li
Четырехцентровые связи удерживают вместе и мономеры в
гексамерах, что было показано рентгеноструктурным анализом
кристаллического комплекса циклогексилллитий • бензол, 6:2. Сое-
динение представляет собой структуру с ядром, содержащим ис-
кривленный октаэдр из атомов металла. Предполагаемая структура
гексамера бутиллития представлена на рис. 4.3.
223
Рис. 4.1. Предпо- Рис. 4.2. Модель тетрамера
латаемая структура в кристаллической структуре
димера фенил- метиллития
Рис. 4.3. Предполагаемая
структура гексамера
н-бутиллития
лития
Степень ассоциации литийорганических соединений в раство-
рах оказывает влияние, иногда решающее, на их реакционную
способность. Как правило, последняя тем больше, чем ниже степень
ассоциации. Поэтому, добиваясь ускорения реакций, важно иметь
способы разрушения ассоциатов. Одним из таких способов явля-
ется применение донорных растворителей эфирного типа. Послед-
ние не только понижают степень ассоциации, но и эффективно
сольватируют катион лития, что приводит к увеличению карбани-
онного характера соединения. Еще более эффективным способом
разрушения ассоциатов оказывается добавление мощных хелати-
рующих оснований типа ТМЭДА и ДАБЦО. В литературе имеется
много примеров изменения реакционной способности литийорга-
нических соединений в зависимости от растворителя или от при-
сутствия хелатирующих оснований. Например, бутиллитий в угле-
водородном растворителе дает продукты присоединения по крат-
ной связи PI1CH2CN, а в эфире замещает водород в «-положении
этого нитрила (металлирует). Бензол практически инертен по отно-
шению к некомплексованному бутиллитию, но легко металлиру-
ется комплексом BuLi • ДАБЦО.
Разрушение ассоциатов - не единственный путь увеличения
реакционной способности литийорганических соединений. Альтер-
нативный путь заключается в использовании смешанных литийор-
ганических - натрий(или калий)органических соединений:
224
ОМе
Na[LiPh2]
Et2O
ОМе
Напротив, реагенты, образованные взаимодействием литийор-
ганического и медьорганического соединений, обладают понижен-
ной реакционной способностью, но вместе с тем и увеличенной
селективностью.
Открытие способов модификации свойств литийорганических
соединений в значительной степени изменило спектр химических
превращений, характерных для этих соединений.
4.2. МЕТОДЫ СИНТЕЗА ЛИТИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
В противоположность магнийорганическим соединениям, кото-
рые имеют общий способ получения, литийорганические соедине-
ния могут быть синтезированы различными путями. Это позволяет
переходить к соединениям любой структуры и получать их с вы-
соким выходом. Здесь заложена одна из причин, по которым тра-
диционно используемые магнийорганические соединения все чаще
стали заменяться литийорганическими.
4.2.1. Замещение галогена литием („прямой" синтез)
Наиболее широко применяемый метод синтеза простейших ал-
кил- и ариллитиевых соединений аналогичен методу получения
реактивов Гриньяра и основан на взаимодействии лития и соот-
ветствующего органического галогенида:
RHal + 2 Li ------RLi + LiHal
Именно этот путь используют в промышленности для полу-
чения метиллития, этиллития, фениллития и, что особенно важно,
бутиллития, который применяется наиболее часто:
Ме! + 2 Li -----► MeLi + Lil
BuBr + 2 Li -----► BuLi + LiBr
Br + 2 Li
Li + LiBr
Механизм этой реакции исследован менее, чем механизм подоб-
ной реакции Гриньяра, но, по-видимому, у них много общего. На
225
первой стадии предполагают перенос одного электрона с поверх-
ности атома лития к алкилгалогениду с образованием анион-ради-
кала. Участие интермедиата такого типа подтверждается, в част-
ности, тем, что реакция алкилгалогенидов с литиевыми солями
ароматических анион-радикалов приводит к образованию литийал-
килов с высокими выходами. После передачи электрона или синх-
ронно с ней происходит разрыв в анион-радикале связи R-Hal с
образованием радикала R и аниона галогена. Образовавшийся ради-
кал R может далее принять второй электрон и превратиться в карба-
нион с одновременным переходом катиона лития в раствор:
RHal + Li -------► RHaL + Li +
RHal* --------► R' + Hal
Hal" + Li+ -------- LiHal
R* + 2 Li -------RLi + Li+
Реакционная способность алкилгалогенида при взаимодействии
с литием уменьшается в ряду RI > RBr > RC1, при этом алкилгало-
гениды более реакционноспособны, чем арилгалогениды. Чаще все-
го литийорганические соединения получают из хлоридов или бро-
мидов, так как иодиды имеют тенденцию давать больше продуктов
сочетания (по типу реакции Вюрца):
RLi + RI ------► R-R + Lil
Исключение составляет метиллитий, который удобно получать
из метилиодида.
К реакции сочетания особую склонность проявляют активные
бензил- и алкилгалогениды. Поэтому для получения бензил- и ал-
лиллития часто применяют в качестве исходных соединений не
галогениды, а простые бензиловые и аллиловые эфиры:
ArCH2OR + 2 Li -------- ArCH2Li + LiOR
CH2 = CHCH2OPh + 2 Li ------- CH2 = CHCH2Li + LiOPh
Хотя выход продуктов в реакции алкилгалогенидов с литием
высок (80-90 %), природа растворителя, его чистота, а также ряд
других экспериментальных деталей могут оказаться весьма сущест-
венными. Например, иногда для уменьшения роли реакции сочета-
ния синтез проводят при низкой температуре (0 -г -50 °C), а для
достижения оптимальной скорости реакции используют литий,
содержащий следы натрия, а также вводят в реакцию примерно
226
20 %-ный избыток лития. Выбор растворителя зависит от многих
факторов, однако большим преимушеством реакций такого типа
является возможность применения углеводородных растворителей
(бензол, пентан и т. д.). Лишь метиллитий и фениллитий ограни-
ченно растворимы в углеводородах, и для их получения требуется
использовать эфир (в котором они достаточно стабильны). Бутил-
литий удобнее получать в гексане, особенно в промышленном
производстве. Виниллитий обычно получают, используя в качестве
растворителя ТГФ.
4.2.2. Реакция металлирования
Большое разнообразие литийорганических соединений доступ-
но для применения в органическом синтезе благодаря легкости, с
которой простейшие литийалкилы могут быть превращены в новые
литийорганические соединения. Основным методом, используе-
мым с этой целью, является металлирование (литиирование), т. е.
замещение водорода металлом (литием):
R1 Li + R2H
R*--H
> I
Li--R2
R*Li + R2H
Реакция металлирования включает атаку аниона или полярной
группы R1 на атом водорода субстрата R2H и протекает, возможно,
через четырехцентровое переходное состояние. Это подтвержда-
ется тем, что эффекты резонанса заместителей в R2 не оказывают
заметного влияния на направление атаки. Так, если R2 - арил, такие
заместители, как ОМе и CF3, направляют атаку в ортео-положение, а
изопропил - в мета- (преимущественно) и пара-. Именно такого
результата можно было ожидать при учете только индукционных
эффектов.
Реакция металлирования может рассматриваться как взаимо-
действие кислоты и соли более слабой кислоты, поэтому для ее
протекания важнейшее значение имеет С-Н-кислотность субстрата
R2H. Равновесие обычно сдвинуто в сторону образования более
слабой кислоты. Так как алканы имеют рКа ~ 45-50, то алифа-
тические литийорганические соединения оказываются пригодными
для металлирования С-Н кислот с рКа <35. Подходящую кислот-
ность, таким образом, имеют многие органические соединения
(см. 3.3). Это и обусловливает широкую область применения рас-
227
сматриваемой реакции. В качестве металлирующего агента чаще
других используют BuLi, главным образом, благодаря его доступ-
ности.
Литиирование алифатических соединений протекает наиболее
легко, если образовавшийся карбанион мезомерно стабилизирован,
например:
СНЭ = СНСН. —BuLi » CH9 = CHCH9Li
2 3 ТМЭДА
РЬСЩ ' »- PhCH.Li-ТМЭДА юо%
3 ТМЭДА 2
Н Li
По той же причине легко проходит литиирование алкинов-1 (у
ацетилена $Ка ~ 25). Однако в реакциях с производными ацетилена
нередко наблюдается их полиметаллирование (получать моноли-
тиевые производные удобнее реакцией ацетилена с литием в жид-
ком аммиаке (см. 3.3.1)):
НС^СН -PhL' - LiC = CLi
НС = ССН3 BuL1 (1 зкв) » LiC = CCH3
Bu Li (4 экв.)
HC = CCH3 ------------ LiC = CCLi3
Возможно также, что взаимное возмущение электронов валент-
ной оболочки серы и неподеленной пары электронов углеродного
атома обеспечивает стабилизацию аниона, легко получаемого из
1,3-дитиана (рКа ~ 31):
Bu Li
ТГФ
до 9 5 %
Такие литиевые соединения представляют интерес, так как исход-
ный 1,3-дитиан - дитиоацеталь карбонильного соединения - пред-
ставляет собой маскированную карбонильную группу и, следо-
вательно, может выступать как нуклеофильный эквивалент этой
группы.
В системах, металлирование которых описано выше, активация
С-Н связей обусловлена мезомерной стабилизацией карбанионной
228
части литийорганического соединения. Электроноакцепторные
группы лишь за счет индуктивного эффекта не могут обеспечить
достаточной стабилизации образующегося карбаниона. К тому же
соединения, содержащие у одного атома углерода литий и элект-
роноакцепторную группу (х), оказываются нестабильными и,
возникая, склонны давать продукты элиминирования, перегруп-
пировки ит. д.:
RCFLX------RCHX -----------:—*- [rCH:"] -----продукты
2 । - LiX u
Li
Лишь в некоторых случаях при очень низких температурах
такие соединения оказываются стабильными и их удается охарак-
теризовать:
Аг2С = СНС1
Bu Li
ТГФ, -45 4--100 °C
Li
Аг2С = с'
"С1
1. со2 _
2. Н +
Аг2С = С(С1)СООН 97 %
Особый интерес реакция металлирования представляет для по-
лучения литийарилов. Ароматические углеводороды обычно явля-
ются более сильными кислотами, чем алифатические (у бензола
рКа ~ 43), поэтому доступный бутиллитий оказывается подходя-
щим реагентом для синтеза литийарилов. Однако реакция самого
бензола протекает очень медленно, и для ее ускорения используют
ТМЭДА или ДАБЦО, а также mpem-бутоксид калия, превращаю-
щий литийорганический реагент в более реакционноспособное
соединение калия:
PhH -------——► PhLi 90 %
тдет-BuOK
Многие замещенные ароматические соединения взаимодейст-
вуют с бутиллитием без осложнений. При этом характерной особен-
ностью реакции является то, что металлирование осуществляется в
орто-положение по отношению к группировкам, содержащим гете-
роатом, таким как OR, NR2, СН2ОН, CH2NR2, CONHR, CONR2
и др.:
Bu Li
Et2O
65 %, орто- 99,8 %
229
CH2NMe2
Bu Li
CH2NMe2
Et2O
> 92 %
По-видимому, на первом этапе реакции такого типа происходит
координация электронодефицитного металлирующего агента с не-
поделенной электронной парой гетероатома субстрата (с одновре-
менной деполимеризацией литийорганического соединения). По-
следующая протофильная атака карбанионного центра на соседний
водородный атом приводит к образованию металлированного про-
Наиболее сильными активирующими и орто-ориентирующими
группами являются азотсодержащие группы типа амидной и суль-
фамидной. В целом относительная ориентирующая способность за-
местителей уменьшается в следующем ряду: SO2NR2, SONR->
> CONR2 > CH2NR2 > OR > OAr >NHAr > NRAr > NR2. Поэтому,
если в ароматическом кольце присутствуют две группы, способные
к координации, конечный продукт реакции содержит металл в ор-
то-положении по отношению к заместителю с большей ррто-ори-
ентирующей способностью:
Ph Li
Et2O
75 %
230
Электроноакцепторные группы (С=О, CF3, F, Cl, I), не способ-
ные координироваться с металлирующим агентом, также проявля-
ют орто-ориентирующий эффект, возможно, благодаря способнос-
ти увеличивать кислотность соседних протонов:
В ароматических гетероциклах литийорганическое соединение
координируется с гетероатомом, в результате чего металлирование
проходит по а-положению:
трет -Bu Li
Et2O
98 %
Ви Li
Et2O
70 %
Помимо строения субстрата на скорость реакции металлирова-
ния влияет природа группы R металлирующего агента. Так, ско-
рость реакции между трифенилметаном и RLi уменьшается в ряду
PhCH? > аллил > Bu > Ph > винил > Me. Последовательность, одна-
ко, может нарушаться, если условия реакции изменяют степень
ассоциации литийорганического соединения. Поэтому в более раз-
бавленном растворе (не 0,1 М, как выше, а 0,01 М) относительная
активность соединений RLi другая: Ви > аллил > > винил > Ph >
> Me. Уменьшение степени ассоциации приводит также к тому, что
emop-бутиллитий реагирует в 24 раза быстрее, чем бутиллитий.
В последние годы границы применимости реакции металли-
рования были расширены введением в практику органического
синтеза сильных оснований, обладающих слабонуклеофильными
свойствами. Таковыми оказались пространственно затрудненные
амиды лития. Наибольшее распространение получили диизопро-
пиламид лития (ДИПАЛ), дициклогексиламид лития, ди(триметил-
силил)амид лития и др. Получают сверхсильные основания чаще
всего действием бутиллития на соответствующий амин и исполь-
зуют их без выделения (in situ):
i«o-Pr2NH —-uL-- »• «3o-Pr2NLi ~ 100 %
231
(Me3Si)2NH
Bu Li
(Me3 Si)2NLi
88 %
Пространственное экранирование атома азота в этих соедине-
ниях приводит к резкому ослаблению нуклеофильных свойств, в то
время как их основность, обусловленная низкой N-Н кислотностью
(рКа вторичных аминов равен примерно 30), очень высока. Это
важно в тех случаях, когда исходное соединение может подвер-
гаться атаке как основаниями, так и нуклеофилами, что на практике
встречается довольно часто:
q____pj 1. R Li - нуклеофил
/2 2. RLi - основание
4LiR
Применение амидов лития для металлирования органических
соединений позволяет избежать конкурентной реакции замещения,
например обмена галогена на металл:
R Br R Br R Li
\ z w дипал X / BuL1 »
/ \ ТГФ,-78 °C / \ Et2O / \
Br Li Br Н Br н
Не менее важно, что удается избежать и реакций присоединения
к группам NO2, СНО, СООН, COOEt, C=N, CONH2 и CONMe2, что
позволяет получать литийорганические соединения, содержащие в
«-положении эти группировки:
Ме
LiN(SiMe3)2
-78 °C
CHjCOOEt
LiCH2COOEt
> 94 %
Me3SiCH2COOBu-/n^e/n Me3SiCHCOOBu-/n^e/n
Как видно из приведенных примеров, реакции металлирования
под действием таких супероснований, как амиды лития, протекают
даже при очень низких температурах. Депротонирование диал-
килформамидов оказалось, по существу, мгновенным даже при
-100 °C:
232
о
II
hcnr2
дипал
-100 °C
О
II
LiCNR2 ~Ю0%
В целом использование оснований типа ДИПАЛ позволяет
осуществить переход к ct-гетерозамещенным литийорганическим
соединениям. Это расширяет возможности органического синтеза,
так как предоставляет в распоряжение химика нуклеофильные син-
тоны, содержащие функциональные группы.
4.2.3. Замещение галогена литием
с помощью литийорганического соединения
Если галогеносодержащее соединение не удается превратить в
литийорганическое производное непосредственно в реакции с
литием, то его вводят во взаимодействие с доступным литийорга-
ническим соединением:
R'Hal + R2Li г- — - R’Li + R2Hal
Реакция представляет собой чрезвычайно удобный метод полу-
чения литийорганических соединений. Наиболее легко она проте-
кает с иодидами и бромидами, труднее с хлоридами и практически
не идет с фторидами, т. е. I > Br > Cl » F. Реакция обратима, при
этом положение равновесия благоприятствует образованию такого
литийорганического соединения, карбанионная часть которого
имеет наиболее эффективно делокализованный отрицательный
заряд (т. е. с литием оказывается связанным тот радикал, который
образует более устойчивый карбанион, а с галогеном - тот, для
которого RH - более слабая кислота).
Изучение равновесия показало, что устойчивость карбанионов
уменьшается в ряду винил > фенил > циклопропил > этил > про-
пил > изобутил > неопентил > циклобутил > циклопентил. Обычно
R2 = алкил, чаще всего бутил (хотя и не всегда), a R1 = арил:
Вг
Bu Li
PhCH3
95 %
233
Реакция обмена галоген-литий - одна из самых быстрых хими-
ческих реакций, и проведение ее при низких температурах позво-
ляет не затрагивать присутствующие в соединении функциональ-
ные группы, которые легко реагируют с карбанионами. С помощью
этой реакции можно осуществить синтез ряда органических соеди-
нений:
Вг
/
МеэС = С
2 \
СООН
Bu Li
ТГФ, -1 00 °C
Li
/
Ме7С = С
z \
COOLi
Н2О, н +
234
Рассматриваемая реакция замещения обычно осложняется кон-
курирующими процессами сочетания и металлирования. Реакции
сочетания удается избежать, контролируя экспериментальные ус-
ловия (скорость смешения реагентов, температуру и т. д.). Реакция
металлирования, казалось бы, должна быть серьезным препятст-
вием для использования рассматриваемого метода, так как можно
ожидать, что индуктивный эффект галогена будет активировать к
металлированию орто-протоны. Тем не менее обычно удается по-
добрать условия, приводящие исключительно к замещению гало-
гена на литий, так как реакция обмена проходит со значительно
большей скоростью, чем металлирование, которое связано с пере-
носом протона между углеродными атомами и поэтому обычно
протекает медленно. Проведение реакции при низких температурах
подавляет конкурирующий процесс. Иногда удается получить лишь
продукт металлирования или лишь продукт обмена галогена на ли-
тий, применяя в качестве исходного различные литийорганические
Механизм процесса обмена галогена на металл окончательно не
выяснен; однако большинство имеющихся данных свидетельствует
о гомолитическом пути реакции:
R’Hal + R2Li [R1*, Hal, Li, R2’] R*Li + R2Hal
клетка растворителя
В пользу этого механизма свидетельствует получение продук-
тов сочетания и диспропорционирования из R1’ hR2‘, а также
наблюдение химически индуцированной динамической поляриза-
ции ядер.
235
4.2.4. Получение из других металлорганических соединений
Многие органические соединения металлов и металлоидов (оло-
ва, ртути, бора, кремния, свинца, сурьмы и селена) способны рас-
щепляться литийалкилами и металлическим литием с образованием
литийорганических соединений:
R'M + R2Li RJLi + R2M
RM + Li RLi + M
Эти реакции не находят столь широкого применения, как мето-
ды, описанные выше, но они бывают полезны для частных случаев.
Например, их нередко используют, чтобы синтезировать литий-
органические соединения, не содержащие галогенида лития, или те,
которые трудно или невозможно приготовить иным путем. Так,
винильные, аллильные, бензильные и циклопропильные литийорга-
нические соединения часто получают на основе соединений олова:
(CH2=CH)4Sn —4 CH2=CHLi 74 %
Ph Li
Ph.SnCH.Ph ——PhCH.Li 90 %
3 2 Et.O 2
Таблица 4.2. Применимость различных методов получения
литийорганических соединений
Литийорганическое соединение RHal + Li RH + R'Li RHal+ R'Li RM + R' Li
/\/Li - -
VLi - + - +
PhCH2Li - + - +
перв.-RLi + -
втор.-RLi + - -
трет.-RLi + - -
^Li + - + +
ArLi + - +
236
Метод переметаллирования был применен также для синтеза
о-дилитийбензола из о-фениленртути:
Рассмотренные методы получения литийорганических соеди-
нений, по существу, имеют много общего. Все они представляют
собой реакции замещения литием галогена, водорода или другого
металла. Возможность применения их в синтезе некоторых соеди-
нений показана в табл. 4.2.
4.3. ЛИТИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
В ОРГАНИЧЕСКОМ СИНТЕЗЕ
При конструировании органических молекул встает задача
поставить в соответствие возникающим нуклеофильным синтонам
синтетические эквиваленты, т. е. реальные химические реагенты.
Возможности органического синтеза во многом зависят от доступ-
ности таких реагентов. Их разнообразие позволяет успешно решать
проблемы, связанные с региоселективностью реакций, их стереохи-
мией, преодолевать низкую реакционную способность и т. д.
Широкое использование литийорганических соединений связа-
но прежде всего с тем, что они являются удобными синтетическими
эквивалентами синтонов нуклеофильного типа. При этом во многих
случаях они имеют преимущества перед более реакционноспо-
собными натрийорганическими и менее реакционноспособными
магний-, кадмий- или ртутьорганическими соединениями. В целом
литийорганические соединения применяются сейчас столь же час-
то, как и магнийорганические. Выбор конкретного металлоргани-
ческого соединения зависит, прежде всего, от целей синтеза.
Как основания, литийорганические соединения способны легко
отрывать протон от групп ОН, NH, SH и, что особенно важно, от
групп CH, СН2 и СНз. Однако отрыв протонов от последних обычно
происходит медленно, что позволяет проводить важнейшие реак-
ции, в которых литийорганическое соединение реагирует преиму-
щественно в качестве нуклеофила, а не сильного основания.
В табл. 4.3 перечислены важнейшие нуклеофильные синтоны,
каждому из которых может быть поставлено в соответствие литий-
237
Таблица 4.3. Синтетические эквиваленты нуклеофильных синтонов
Синтон Синтетический эквивалент Синтон Синтетический эквивалент
Aik- AlkLi conr2 LiCONR2
r2c=ch- R2C=CHLi Li
RC=C RC=CLi RCHCHO
Аг~ ArLi RCHCHO
Hal | yal Li |
R-CH" R-CHLi R]CHC=O I r'chc=o
Me3SiCH2“ Me3SiCH2Li R2 R2
A д о /—S\ < XHLi 4—sz - "° RCHC OR2 Li I //0 r'chc OR2
‘C R /—S\ < /CRLi x—sz RCHCOO Li I RCHCOOLi
органическое соединение, причем для некоторых из них последнее
является единственным доступным синтетическим эквивалентом.
Реакции литийорганических соединений, в результате которых
удается встроить в молекулу нуклеофильный синтон, можно раз-
бить на два типа - реакции присоединения и реакции замещения. К
первому типу относятся многочисленные примеры взаимодействия
литийорганических реагентов с соединениями, содержащими крат-
ные связи (С=С, C=N, С=О, C=S), ко второму - реакции алкили-
рования. В обоих типах реакций литийорганическое соединение
выступает в качестве нуклеофила, а сами реакции относятся к
конструктивным, т. е. приводящим к образованию новых углерод-
углеродных связей и создающим скелет молекулы.
4.3.1. Присоединение к кратным связям углерод-углерод
Присоединение литийорганических соединений к кратным С-С
связям имеет ограниченное применение, так как происходит от-
носительно легко лишь в том случае, если в алкене присутствует
238
анион-стабилизирующая группа, например триалкил- или триарил-
силильная:
Bu Li
Ph3SiCH=CH2 -------► Ph3SiC6H13 67 %
Внутримолекулярная координация в некоторых случаях, веро-
ятно, может облегчить процесс присоединения:
сн2=снсн2сн2 » сн2~снсн2сн2 ------------------►
OR изо-Рг Li ►OR
----► M3o-PrCH2CHCH2CH2OR
Li
Бутиллитий присоединяется к 1,1 -дифенилэтилену и к дифенил-
ацетилену:
BuLi н2о
Ph2C = CH2 ------► Ph2CCH2Bu ——► Ph2CHCH2Bu
Li
Ph Ph Ph Ph
BuLi \ / H2O \ /
PhC^CPh ----------- C-C --------------*- C = C
/ \ / \
Li Bu H Bu
RLi
Наиболее важна рассматриваемая реакция для химии сопря-
женных диенов и стиролов, так как она является первым этапом
реакции полимеризации:
Нэ с н
пт: \ /
сн2=с-сн=сн7 » с=с
2 I 2 / \
С LiH2C CH2R
Возникающее новое литийорганическое соединение в свою
очередь присоединяется к диену. Повторение процесса приводит к
формированию полимера.
Литийорганические соединения могут присоединяться и по
кратным связям ароматических систем. Этот процесс проходит
легче с более реакционноспособными аренами, например с антра-
ценом:
EtLi
циклогексан
Et
98 %
239
4.3.2. Присоединение к кратным связям углерод-азот
Присоединение литийорганических соединений по двойным и
тройным связям углерод-азот часто сопровождается различными
побочными реакциями, в первую очередь а-депротонированием.
Наибольшее значение имеет присоединение к нитрилам, позволяю-
щее получить кетоны с удовлетворительными выходами:
CHjLi v Н3О+
C5HhCN -----—*C5HhC = NH ——* С5НИС = О 60 %
СН3 сн3
Ароматические азотсодержащие гетероциклы, формально име-
ющие двойную связь C-N, также способны присоединять литий-
органические соединения. В этом случае чаще наблюдается 1,2-
присоединение:
RLi
нагрев
-LiH
Реакция проходит по механизму присоединения - отщепления, и
образующийся аддукт удается выделить. При его нагревании от-
щепляется LiH и получается продукт алкилирования, а при исполь-
зовании ариллитиевых реагентов - продукт арилирования.
Гладко проходит присоединение литийорганических соедине-
ний к кратным связям C=N иминов. Если имин относительно ус-
тойчив, эта реакция дает возможность получать вторичные амины с
хорошим выходом:
Ph2C = NPh PrL- -> Ph2CNHPh 77 %
Pr
4.3.3. Присоединение к кратным связям углерод-кислород
Реакция литийорганических соединений с диоксидом углерода
является одним из наиболее важных методов получения карбоно-
вых кислот. Реакция почти универсальна, и поэтому возникающие
карбоновые кислоты часто используются для характеристики ли-
тийорганических соединений. Кислоты получаются с высокими
выходами не только из простых алкил- и ариллитиевых соединений,
240
но и из таких нереакционноспособных, как этиниллитий и трифе-
нилметиллитий, а также из стерически затрудненных реагентов, в
частности пентахлорфениллития:
Ph3CLi ! С°2 » Ph.CCOOH 82 % 2. Н3О+ 3
u- ► | || 60 % 2,СО2 З.Н3О+ Ч^соон
Одной из реакций, которые очень эффективны при использо-
вании алкиллитиевых реагентов, но плохо идут с реактивом Гринь-
яра, является синтез кетонов из карбоновых кислот. Литийоргани-
ческое соединение взаимодействует с солью карбоновой кислоты,
образуя в качестве промежуточного продукта реакции дилитиевую
соль. Последняя оказывается стабильной при комнатной темпера-
туре и лишь при гидролизе реакционной смеси дает кетон; наиболее
часто реакция используется для получения метилкетонов:
Li О О Li О
EtLi \ / Нт О H+ И
Bu С ООН --------► Ви С-Et —► BuCEt 78 %
Et2O
СООН
Ph Me
1. MeLi
2. H2O, H +
Ph Me
90 %
Процессом, осложняющим реакцию, является образование по-
бочного продукта - соответствующего третичного спирта. Избе-
жать этого удается использованием определенных условий, в том
числе добавлением после смешения реагентов триметилсилил-
хлорида, взаимодействующего с избытком литийорганического со-
единения:
НО . , СООН LMeLi г НО . . СОМе
\/ \jZ \/ 2. Me3SiCl \/\/\/
। 3. Н2О ।
Me Me 91 %
Одним из важных методов образования углерод-углеродной
связи является присоединение литийорганических соединений по
карбонильной группе альдегидов и кетонов, что представляет собой
общий метод синтеза первичных, вторичных и третичных спиртов:
241
Bu Li
Рг
Me
CH2NHPh
CH(OH)Ph
75 %
I
Pr
CH(OH)Ph
95 %
Преимущество литийорганических соединений перед реактивами
Гриньяра состоит в том, что они более активны и менее склонны к
реакциям восстановления и енолизации, а также менее чувствитель-
ны к стерическому влиянию. Поэтому выходы соответствующих
спиртов обычно высокие, даже для стерически затрудненных кар-
бонильного и литийорганического соединений:
im-PrLi
трет -Bu 2 С (О Н) Рг-изо
ni/?«n-Bu2CO ----
mpem-BuLi
трет-Ви^СОН
88 %
81 %
Одно из наиболее важных различий в реакционной способности
литийорганических реагентов и реактивов Гриньяра заключается в
разном направлении присоединения их к a, fl -непредельным кар-
бонильным соединениям. Первые взаимодействуют почти исклю-
чительно с карбонильной группой, давая продукты 1,2-присоедине-
ния, в то время как реактив Гриньяра имеет тенденцию к сопря-
женному 1,4-присоединению:
I, 2-
С-С-С-0 + М8 —R8
с=с-с-он
/ I
Rx V I
-с-с-с=о
Вероятно, это различие связано с тем, что литийорганические
соединения меньше по размеру и на их реакции в меньшей степени
влияют пространственные препятствия:
242
Ph Li II.О Ph2CC=CCPh2 ——» OLi OLi - Ph2CC=CCPh2 OH OH
PhCC=CCPh~ II II О О Ph Ph Ph Ph
PhMgX_ 1 1 H30 + PhC=C-C=CPh —J— 1 1 ► PhCCHCHCPh
II II II
OMgX OMgX о о
Если желательно получить продукт 1,4-присоединения с помощью
литийорганического реагента, то его следует предварительно пре-
вратить в диалкилкупрат лития.
Реакции производных кислот - хлороангидридов, эфиров и
амидов приводят, как правило, к образованию третичных спиртов:
R1 R1 R1
\ I \ p2j j
С = О + R2Li ---- R2— с-0" Li+ ----«- С = О »
/ I -LiX /
X R1 х + R1 R2
---- R2— С-OLi --°— R2-C-OH
'2 R2
X = Cl, ОС OR3, NR 2
Промежуточно образующийся кетон удается выделить лишь в реак-
циях литийорганических соединений с амидами, так как группа
NR2, в противоположность группам Hal и OCOR, является плохо
уходящей, вследствие чего промежуточный аддукт оказывается
стабильным и лишь при гидролизе дает кетон. Эта реакция имеет
практическое значение для производных формамида, из которых
чаще употребляются N-метилформанилид и ДМФА. Продуктами
реакций оказываются альдегиды:
Cl /L^OCH2OMe || 1 Bu Li • ТМЭДА J 2. ДМФА Cl Cl A. OCH2OMe у f| H20, H4 * L. JL -NHMe, CHNMe2 OLi x,OCH2OMe 85 % чсно
243
Как было отмечено выше, использование сверхсильных осно-
ваний типа ДИПАЛ позволяет нацело превращать сложные эфиры,
кислоты и кетоны в литийорганические производные, которые по
существу представляют собой сопряженные основания карбонил-
содержащего соединения - еноляты лития:
С=С-О" Li+
Это увеличило набор применяемых в синтезе нуклеофильных
синтонов (см. табл. 4.3) и позволило успешно и зачастую регио-
селективно осуществлять многие превращения на их основе. Еноля-
ты лития вступают в те же реакции, что и другие литийоргани-
ческие соединения. И хотя они являются амбидентными нуклео-
филами (т. е. могут выступать как О- и С-нуклеофилы), наиболее
типичные пути их превращений связаны с С-нуклеофильностью.
Нижеприведенная реакция подтверждает это положение:
ОН
l.CH3CHLiCOOLi
2. Н3О+
снсо2н
сн3
63 %
Введение енолятов лития в органический синтез значительно
расширило область применения реакций с использованием ацето-
уксусного и малонового эфиров, реакций альдольной конденсации,
конденсаций Клайзена, Реформатского, Дарзана, Штоббе, Кневена-
геля, Перкина, а также позволило избежать многих проблем, тради-
ционно осложняющих химию карбонильных соединений: самокон-
денсации, последующей конденсации конечных продуктов, низкой
хемоселективности, снижения реакционной способности из-за сте-
рических препятствий. Эти сложности удается обойти, превращая
карбонилсодержащее соединение действием супероснования в ал-
коголят лития, проводя реакцию при возможно более низких темпе-
ратурах и т. п. Таким путем можно, например, достаточно селек-
тивно провести конденсацию эфиров уксусной и пропионовой
кислот в обоих вариантах:
О
MeCOOEt -Д—л-»- CH2COOEt - SlSQOEt». EtCCH2COOEt
244
ЛИПАЛ — СН о COOEt
EtCOOEt - CHCOOEt ----- ►
™ 0
снз II
----- CH3CCHCOOEt
сн3
4.3.4. Реакции замещения при атоме углерода
Одной из наиболее характерных реакций литийорганических
соединений является реакция нуклеофильного замещения:
R*Li + R2X ---------- R!R2 + LiX
Реакция приводит к образованию новой углерод-углеродной связи
и относится к большой и важной группе реакций алкилирования.
В качестве алкилирующего реагента чаще используют галоге-
ниды, но на практике, однако, эти реакции нередко сопровожда-
ются значительными осложнениями, чаще всего связанными с
конкурентно протекающими процессами обмена галогена и водо-
рода на литий. К тому же алкилгалогениды имеют тенденцию к
дегидрогалогенированию. Тем не менее реакции алкилирования
применяются довольно часто, особенно при использовании в ка-
честве субстрата реакционноспособных бензил- и аллилгалогени-
дов, метил ио дида и других галогенидов:
трет -Bu Li + PhCH2Cl
Me j Si Cl
трет -Bu С H 2 Ph
75 %
50 %
Описать взаимодействие литийорганических соединений с ал-
килгалогенидами в рамках единого механизма не представляется
возможным. В зависимости от строения субстрата и условий реак-
ции механизм может быть гетеролитическим 5д-2 типа или ради-
кальным. Так, реакции аллил- и бензиллития со вторичными алкил-
бромидами протекают с хорошими выходами продуктов и с высо-
кой степенью обращения конфигурации атома углерода, у которого
происходит замещение атома брома:
245
Et Et
I Sn2 I
PhCH2Li + Mei...C\ ---------► PhCH2~C....."Me
tX Br Xi
выход 5 8 % инверсия 100%
Однако имеются данные, указывающие на то, что в реакции
простых литийорганических соединений с алкилгалогенидами
участвуют радикалы. Промежуточно образующиеся радикалы были
обнаружены методами ЭПР и химической поляризации ядер.
Процесс может включать одноэлектронный перенос от карбаниона
к галогеноалкилу с образованием алкильного радикала (R1') и
анион-радикала (R2X')- Последний легко распадается на ион гало-
гена и алкильный радикал. Алкильные радикалы объединяются, да-
вая желаемый продукт, а также продукты диспропорционирования
и симметричные продукты сочетания.
Неактивированные винил- и арилгалогениды имеют достаточно
низкую реакционную способность для проведения алкилирования с
помощью литийорганических соединений. Если алкилирование и
идет, то не как нуклеофильное замещение, а по механизму присое-
динения - элиминирования или через промежуточное образование
дегидробензола:
Продукты сочетания с арилгалогенидами могут также возни-
кать путем первоначального замещения галогена металлом с по-
следующим арилированием образованного алкилгалогенида. Так,
если бутиллитий добавляют к раствору 1-бромонафталина и реак-
ционную смесь через 20 мин обрабатывают диоксидом углерода, то
получают нафтойную-1 кислоту, однако, если смесь кипятят 36 ч, то
получают 1-бутилнафталин:
246
Bu
75 %
Если не удается получить высокий выход продуктов замеще-
ния, то вместо литийорганических реагентов используют купраты
лития:
Ph2Cu Li
PhCH=CHBr -----------► PhCH=CHPh 90 %
Кроме галогенов замещению с участием литийорганических
соединений может подвергаться и ряд других групп. Среди реакций
этого типа широкое распространение получили лишь реакции с ал-
килсульфатами (в частности с диметил сульфатом):
(Me3Si)2CBrLi (Me3Si)2CBrMe 88 %
Как можно было заметить из вышеприведенных примеров,
строение литийорганических соединений не имеет большого зна-
чения для протекания реакций алкилирования. Арил- и виниллитие-
вые реагенты удается алкилировать алкилиодидами и бромидами.
Тем не менее делокализация отрицательного заряда карбаниона
облегчает ход реакции. Этот эффект, в частности, наблюдается при
использовании литийорганических производных 1,3-дитиана, ста-
билизация отрицательного заряда за счет атомов серы в котором
отмечена выше. Такие соединения легко алкилируются, при этом
после гидролиза появляется маскированная образованием дитио-
ацеталя карбонильная группа. В целом открытие данной реакции
позволило превращать доступный электрофильный синтон
RC+ Н(ОН), соответствующий карбонильному соединению, в недо-
ступный ранее нуклеофильный синтон, RC_=O (обращение поляр-
ности синтона). На этом основан общий метод синтеза альдегидов и
кетонов путем алкилирования их простейших представителей:
247
PhCHO
H
Ph
Bu Li
ТГФ
85-90 %
H2O
Hg2+
Pr-изо
o=c
\
Ph
К рассматриваемому типу реакций можно отнести и раскрытие
кольца циклических простых эфиров, особенно легко протекающее
с эпоксидами. Реакция сопровождается инверсией конфигурации
менее пространственно затрудненного атома углерода и протекает
по механизму внутримолекулярного S,y2 замещения:
Ph Li
Ph-CH- СН2
О
PhCH2CHPh
ОН
70 %
Трудности при реакции алкилирования часто удается преодо-
леть использованием енолятов лития. При алкилировании несим-
метричного кетона обычно встает вопрос о направлении реакции.
Использование енолятов лития позволяет избирательно алкилиро-
вать желаемое положение. Региоселективность может быть достиг-
нута двумя путями. Первый состоит в том, что из исходного кетона
предварительно действием подходящего реагента получают енол-
ацетат или силиловый эфир. Последние образуются в виде смеси
изомеров, в которой один из изомеров обычно преобладает. Он без
труда может быть выделен, а затем обработкой 2 моль метиллития
превращен в соответствующий енолят лития. Последующее алки-
лирование енолята позволяет получить а-алкилкетон определенно-
го строения (из более замещенного енолята получают более заме-
щенный кетон, из менее замещенного енолята - менее замещенный
кетон):
248
Me3Si Cl
ГЛИМ
Me Li
ГЛИМ
RI
Me Li
глим
Второй способ алкилирования использует особенности еноли-
зации кетонов под действием оснований типа ДИПАЛ. Из двух
возможных енолятов в этом случае образуется так называемый
кинетический енолят (наименее замещенный). Алкилирование это-
го енолята позволяет получить менее замещенный а-алкилкетон:
Me
ДИПАЛ
ТГФ, -78 °C
RI
О
98 %
Особенности алкилирования Д-дикарбонильных соединений
(дикетонов, кетоальдегидов и кетоэфиров) через промежуточное
образование дианионов под действием амидов металлов в жидком
аммиаке отмечены выше (см. 3.3.2). Недавно те же реакции стали
осуществлять в эфире с использованием в качестве основания
ДИПАЛ:
MeCOCH2COOEt
ДИПАЛ
-78 °C
---RCH.CCHCOOEt
2П
О
CH.CCHCOOEt — "а1*-
2П
+ о
-—«- RCH,CCH,COOEt
2П 2
О
Этот метод применяется при алкилировании второго по кислотнос-
ти положения.
Карбоновые кислоты в реакциях с сильными основаниями типа
ДИПАЛ образуют дианионы, которые в действительности имеют
249
структуры енолята RCH=C(O _)2- Алкилирование дианиона прохо-
дит по более нуклеофильному центру и представляет собой общий
метод синтеза кислот, альтернативный синтезу на основе малоно-
вого эфира:
о- R
Ме2СНСООН Д-ПАЛ *• Ме2С=С 1--На1------и- Ме.ССООН
2 2 \ 2. н2о, н+ 2
О“
4.3.5. Протонирование литийорганических соединений
Литийорганические соединения протонируются даже слабыми
донорами протонов. О протонировании С-Н кислотами, протекаю-
щем при металлировании, было сказано выше. Еще легче литий-
органические соединения реагируют с N-H, S-Н и О-Н кислотами.
RLi + Н+ ---------- RH + Li+
Применение кислот, меченных изотопами водорода (обычно
используют тяжелую воду, дейтерометанол или дейтероуксусную
кислоту), приводит к замене лития на дейтерий или тритий. Это
превосходный метод получения соединений, меченных изотопами
водорода в определенном положении. Степень обогащения обычно
высокая (75-95 %). Таким же путем можно установить положение
лития в молекуле:
PhCH2SiMe3 —tL-*- PhCH(Li)SiMe3
d2o
PhCH(D)SiMe3
85 %
ОМе
ОМе
ОМе
1. Bu Li
2. D2O
ОМе
94 %
ОМе
ОМе
Реакции литийорганических соединений co спиртами, тиолами
и аминами представляют собой удобный способ получения алко-
ксидов лития, тиолятов и амидов. Продукты реакции могут быть
выделены, но могут быть использованы in situ, так как образую-
щиеся одновременно углеводороды не мешают дальнейшему тече-
250
нию реакции. Синтез алкоксидов используется для получения
сложных эфиров из спиртов, которые не выдерживают кислых
условий, например эфиров третичных спиртов:
r'oh —» r'oli R-—1- R2COOR1
ТГФ
Реакции бутиллития со вторичными аминами, приводящие к
синтезу амидов лития, протекают быстро и практически полностью
уже при низкой температуре. Именно эта реакция используется для
получения ДИПАЛ (см. 4.2.2).
4.3.6. Получение реакционноспособных интермедиатов
Нестабильные высокореакционноспособные интермедиаты -
карбены, дегидробензолы, илиды фосфора - получаются с исполь-
зованием литийорганических соединений элиминированием гало-
генидов лития, что является энергетически выгодным процессом.
Карбены образуются из а-галогенолитийорганических соедине-
ний, которые получают металлированием или замещением галогена
литием. а-Галогенолитийорганические соединения (обычно ста-
бильны при низких температурах -70 -г-100 °C) вступают в типич-
ные для карбенов реакции, по-видимому, без промежуточного
образования свободного карбена. Так, взаимодействие с алкенами
является общим методом получения соединений циклопропанового
ряда:
г ™ Ме2СН = СНМе2
СН7С17 + RLi n — LiCHCL—[=СНС1] —-----------------
* -RH —L1C1
Н С1
Ме^^^ Me
Me Me
Как отмечалось выше, орто-галогенозамещенные литийарилы
могут быть получены из галогенобензолов реакцией орто-метал-
лирования или из орто-дигалогенобензолов в результате замеще-
ния галогена на литий:
251
Соединения типа о-фторофениллития также стабильны лишь
при низких температурах, но есть основания полагать, что их реак-
ции протекают через промежуточное образование дегидробензола:
Ph Li
В противоположность карбенам и дегидробензолам илиды от-
носительно стабильны. Наибольшее значение в органическом син-
тезе приобрели илиды фосфора, которые могут быть вовлечены в
реакцию с карбонильными соединениями (реакция Виттига):
PhjP + - RLi
RCH2Hal ----—*- RCH2PPh3Hal —
-------- RCH = PPh3 ------ RCH —PPh3
+ r2\ /R2
R’CH-PPh, + .C=O ------------► R’CH=C + Ph,P=O
3 R3/ V 3
Литийорганические соединения (чаще бутиллитий) участвуют
на стадии депротонирования фосфониевой соли, поэтому они могут
быть заменены другими сильными основаниями. Свойства илидов,
однако, зависят от природы основания. При синтезе с использо-
ванием литийорганических соединений илиды иногда проявляют
специфичные свойства, так как могут существовать в виде металли-
рованного соединения или комплекса с галогенидом лития:
RCH-PPh3 RCH-PPh3- LiHal
Li
4.3.7. Синтез других металлорганических соединений
Литийорганические соединения наряду с магнийорганическими
часто используются для получения других металлорганических
соединений. В основе таких превращений лежит взаимодействие
252
литийорганических соединений с производными (чаще галогенида-
ми) другого, менее электроположительного металла. Наибольшее
значение реакция имеет для получения производных бора, алюми-
ния и меди. Так, при взаимодействии с солями одновалентной меди
первоначально образуется медьорганическое соединение, оно
нестабильно и часто не может быть выделено, однако с избытком
литийорганического соединения легко дает смешанные литийорга-
ническое-металлорганическое соединение:
MeLi + Cui ---
CH3CH=CHLi + Cui
[MeCu] Me2CuLi
(CH3CH=CH)2CuLi
Образовавшиеся смешанные купраты реально могут иметь различ-
ную стехиометрию R^CuLi, RjCujLi, ЯзСиЫг и RvCusLij.
Полученные комплексы, как и литийорганические соединения,
являются синтетическими эквивалентами карбанионов. Однако та-
кие комплексы в значительной степени отличаются по своей нук-
леофильности, основности, способности к комплексообразованию,
благодаря чему удается кардинально изменить реакционную спо-
собность литийорганических соединений и селективность их реак-
ций с электрофилами. Так, было обнаружено, что алкилкупраты
лития почти полностью лишены способности реагировать, как ли-
тийалкилы, с карбонильной группой. Единственным исключением
являются галогеноангидриды кислот, взаимодействие с которыми
проходит исключительно с образованием кетонов, при этом другие
карбонильные группы в молекуле не затрагиваются:
Me2CuLi + СН3(СН2)4СО(СН2)4СОС1 _ 78 О(, -
------► СН3(СН2)4СО(СН2)4СОСН3 95 %
Me2CuLi + 1(СН2)10СОС1 —^7— 1(СН2)10СОСН3 91 %
Будучи инертными по отношению к карбонильной группе, куп-
ратные комплексы проявляют повышенную склонность к присое-
динению по двойной связи, сопряженной с карбонилом:
Me2CuLi +
97 %
9 Заказ № 137
253
Присоединение к а, Д-ненасыщенным карбонильным соединениям
проходит, таким образом, в положение 1,4-, что позволяет селек-
тивно ввести почти любой фрагмент в положение 3 карбонильного
соединения.
Купратные реагенты при определенных условиях легко вступа-
ют и в реакцию нуклеофильного замещения с R-Х, где X = Hal, Ts,
Ас. Примечательно также, что по отношению к купратам электро-
филами являются даже винил- и арилгалогениды. Такие реакции
идут обычно с сохранением конфигурации двойной связи:
Н Вг Н Me
\ / \ /
Me, Си Li + С—С ► С=С 81 %
2 / \ / \
Вг Н Me Н
Ph
\
(CH2 = C)2CuLi + PhBr — *- С=СН, 85 % / 1
Me Me
Интенсивные исследования в области купратных комплексов
лития, проводимые в настоящее время, расширяют синтетические
возможности литийорганических соединений, из которых они лег-
ко получаются.
4.3.8. Разрыв связи кислород-углерод простых эфиров
Разрыв связей С-0 простых эфиров редко применяется в син-
тезе. Однако литийорганические соединения часто готовят и ис-
пользуют в виде растворов в эфире, и поэтому желательно знать,
как быстро происходит этот разрыв и какие продукты при этом
образуются.
Скорость разрыва связи С-0 простого эфира зависит как от его
строения, так и от строения литийорганического соединения. Обыч-
но растворы в ТГФ менее стабильны, чем в диэтиловом эфире. Для
последнего порядок уменьшения стабильности литийорганических
реагентов следующий: MeLi > PhLi > BuLi > EtLi > PrLi > изо-BuLi >
> изо-PrLi > втор-BuLi > трет-BuLi. Так, уменьшение концент-
рации раствора метиллития в эфире от 0,54 М до 0,14 М происходит
при комнатной температуре в течение 1 года; концентрация фенил-
лития падает от 0,4 М до 0,2 М через 12 дней при кипении его эфир-
ного раствора; концентрация бутиллития в эфирном растворе
254
падает до половины своей первоначальной величины через 153 ч
при 25 °C; 0,14 М раствор /ире/и-бутиллития полностью разлагается
через 30 мин при комнатной температуре. В литературе сообщалось
также об отрицательном температурном коэффициенте для реакций
между эфирами и литийорганическими соединениями, так что со-
хранение эфирных растворов и проведение реакций при низких
температурах не всегда желательно.
Показано, что расщепление диэтилового эфира в присутствии
литийорганического соединения приводит к образованию этилена и
этилата лития, а расщепление ТГФ дает енолят ацетальдегида и
этилен. Интенсивное изучение механизма разрыва связей простых
эфиров показало, что он меняется в зависимости от природы реаги-
рующих соединений и даже для одного и того же эфира возможна
реализация альтернативных механизмов. Так, например, для рас-
щепления диэтилового эфира под действием литийорганического
соединения постулированы механизмы, включающие /?- и даже
а',Д -элиминирование:
Et2O
RLi
о п
н-СН2- сн2- OEt
н
I
Н2С-CH-OEt
Li
.О.
Н, с-нс сн,
L?\ fcH2
н 2
н2с=сн2
+
Li OEt
В двух последних случаях разрыву связи С-0 предшествует «-ме-
таллирование эфира. Аналогичный процесс наблюдается для ТГФ:
RLi
-RH
CH, CH,
II 2 + II 2
CH, CH
2 \
O" Li+
Так как литийорганические соединения реагируют с эфирами,
влагой и кислородом, встает задача определения концентрации их
растворов непосредственно перед работой. Титрование является
вполне подходящим для этого методом и дает достаточно точные
255
результаты. Обычно используют простое ацидометрическое титро-
вание, особенно если в синтезе употребляют избыток литийоргани-
ческого соединения. В других случаях, в том числе при исследо-
вании бутиллития, рекомендуется применять метод двойного тит-
рования. Для этого вначале определяется общая щелочность (RLi +
+ LiOR + LiOH + LijO), что достигается добавлением 5-10 мл
отфильтрованного раствора литийорганического соединения к
10 мл воды, чтобы полностью гидролизовать литийорганическое
соединение и алкоголят лития. Титрование стандартной кислотой с
фенолфталеином в качестве индикатора позволяет определить об-
щую щелочность. Далее к 5-10 мл анализируемого раствора добав-
ляют 1 мл свежеперегнанного бензилхлорида в 5 мл сухого эфира.
Происходящие при этом реакции можно выразить рядом урав-
нений:
RLi + PhCH2Cl --------► RCH2Ph + LiCl
RLi + PhCH2Cl — PhCH2Li + RC1
PhCH2Li + PhCH2Cl -------► PhCH2CH2Ph + LiCl
По разности результатов титрования определяют концентрацию
литийорганического соединения.
Использование бензилхлорида дает во многих случаях адекват-
ные результаты, но отмечены и случаи их занижения, возможно,
из-за неполного протекания реакции бензилхлорида с литийорга-
ническим соединением. Вместо бензилхлорида можно применять
1,2-дибромоэтан (особенно для анализа метил- и фениллития),
1,1,2-трибромоэтан, а также аллилбромид (для бутиллития).
4.4. ПРОМЫШЛЕННОЕ ПРИМЕНЕНИЕ
ЛИТИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Как было отмечено выше, одним из преимуществ литийоргани-
ческих соединений по сравнению с магнийорганическими является
возможность использовать для их получения углеводородные раст-
ворители. Это позволяет исключить особо пожароопасные раство-
рители типа эфира. Поэтому многие литийорганические соедине-
ния получают в промышленности, и они вполне доступны.
В свою очередь, промышленные процессы синтеза практически
важных соединений часто включают использование литийоргани-
ческих соединений. Сюда надо отнести, прежде всего, процессы
256
получения полистирола, полиизопрена и других важнейших поли-
меров на основе диенов, которые относительно легко присоединя-
ют литийорганические соединения:
PhCH=CH, BuLi»- (-CH-CH,-)n
z гексан | z п
Ph
Полимеризация этилена протекает лишь под давлением
650-1000 атм, а в присутствии ТМЭДА - при 10-70 атм и приводит
к образованию полиэтилена:
СН2 = СН2 BuLi > C4H9CH2CH2Li -".-Ь1?~СНг »
н,о
-----C4H9(CH2CH2)n+1Li » С4Н9(СН2СН2)П+1Н
Литийорганические соединения используются и во многих дру-
гих промышленных процессах. Без сомнения, число их будет уве-
личиваться по мере того, как промышленный органический синтез
будет осваивать современные технологии, основанные на примене-
нии высокореакционноспособных соединений.
4.5. ЭКСПЕРИМЕНТАЛЬНЫЕ РАБОТЫ
Особенности работы с литийорганическими соединениями. В
отличие от многих реагентов, используемых в органическом синте-
зе, литийорганические при соприкосновении с влажным воздухом,
кислородом и водой самовоспламеняются. Поэтому работа с ними
требует особой осторожности и внимания. Опасность увеличива-
ется при использовании концентрированных растворов. Все работы
с применением литийорганических соединений осуществляются в
токе инертного газа - аргона или азота, высушенного и освобож-
денного от кислорода. Вся стеклянная посуда тщательно высуши-
вается при температуре 120-150 °C.
Растворы литийорганических соединений стараются употреб-
лять немедленно после получения, а при необходимости сохраняют
в герметичной посуде в атмосфере азота или аргона, лучше при
низкой температуре (от 0 до-15 °C).
При получении литийорганических соединений используют
растворители углеводородного или эфирного типа. Уже это обстоя-
257
тельство обусловливает необходимость работать под тягой и вдали
от открытого огня, но основное требование к растворителям заклю-
чается в том, что они должны быть свободны от воды и пероксидов.
С этой целью углеводородные растворители сушат и перегоняют
над натрием, эфиры освобождают от пероксидов обычными спо-
собами и дополнительно перегоняют над натрием, а затем над алю-
могидридом лития.
В случае загорания растворов литийорганических соединений
пламя следует тушить сухим песком, покрывалом. Использование
пенных и углекислотных огнетушителей, воды или тетрахлорида
углерода не допускается.
Обычно получение литийорганических соединений включает
стадию фильтрования их в токе инертного газа. Шлам, содержащий
непрореагировавший литий, должен быть немедленно уничтожен
осторожным растворением в спирте.
Приступать к выполнению работ следует после ознакомления с
инструкциями по технике безопасности при работе с щелочными
металлами и литийорганическими соединениями.
Получение литийорганических реагентов
а) Раствор фениллития в эфире
PhBr --------- PhLi + LiBr
Et2O
Реактивы
Литий металлический 2,3 г
Бромобензол 23,5 г
Эфир сухой 100 мл
Аргон (из баллона)
Установка (рис. 4.4) состоит из колбы (круглодонной или
плоскодонной) вместимостью 300 мл, снабженной двурогой на-
садкой. В один из тубусов помещают капельную воронку с уравни-
тельной трубкой, верхнее ее отверстие снабжают трубкой для ввода
газа, через которую начинают пропускать медленный ток аргона.
Через другое отверстие насадки вносят в колбу 70 мл сухого эфира и
с помощью воронки для сыпучих веществ - 2,3 г (0,33 моль) хорошо
измельченного лития. Для измельчения литий предварительно рас-
258
плющивают, например ударом молотка по
куску, положенному между двумя метал-
лическими пластинами и смазанному пара-
финовой смазкой. Расплющенные пластинки
лития толщиной 1 мм промывают от смазки
петролейным эфиром и разрезают ножница-
ми на узкие полоски непосредственно над
воронкой.
Воронку для сыпучих веществ убирают,
снабжают прибор обратным холодильником,
пускают в ход мешалку и прибавляют к реак-
ционной смеси 1-2 мл бромобензола для то-
го, чтобы инициировать реакцию (если реак-
ция не начинается, смесь осторожно нагре-
вают). Остальное количество бромобензола
разбавляют 30 мл сухого эфира и получен-
ный эфирный раствор прибавляют в колбу с
такой скоростью, чтобы обеспечить слабое
Рис. 4.4. Установка для
получения фениллития
кипение. Прибавление эфирного раствора занимает около 1 ч.
Затем реакционную смесь нагревают 1 ч, поддерживая слабое
кипение эфира, охлаждают и отделяют от мелких кусочков непро-
реагировавшего лития. С этой целью раствор сифонируют под дав-
лением аргона в предварительно заполненные аргоном мерный
цилиндр или плоскодонную колбу с делениями через трубку, в ко-
торую впаян стеклянный фильтр или помещена стекловата. Остатки
металлического лития осторожно нейтрализуют метанолом. Выход
фениллития составляет 90-95 %.
Эфирный раствор, полученный описанным способом, содержит
фениллитий и бромид лития (PhLi • LiBr). Последний не мешает
дальнейшему использованию реагента.
Для установления концентрации полученного эфирного раст-
вора фениллития с помощью тщательно высушенной пипетки отби-
рают 3-5 мл этого раствора, помещают в колбу, содержащую 10 мл
эфира. Добавляют 10 мл дистиллированной воды, гидролизуют об-
разец и титруют продукт гидролиза стандартным раствором кисло-
ты, используя фенолфталеин в качестве индикатора. Предпочти-
тельно получение 1,3 М раствора фениллития.
259
б) Раствор метиллития в эфире
Mel У' - Me Li + Lil
Et2O
Реактивы
Литий металлический 3,5 г
Метилиодид 32,4 г
Эфир сухой 110 мл
Аргон (из баллона)
Эфирный раствор метиллития получают так же, как это описано
для фениллития. После внесения в реакционную колбу 60 мл сухого
эфира и 3,5 г (0,5 моль) лития в нее при перемешивании добавляют
1-2 мл метилиодида. После начала реакции, что заметно по по-
мутнению раствора, равномерно прибавляют оставшийся метил-
иодид, растворенный в 50 мл сухого эфира, с такой скоростью,
чтобы все время поддерживалось умеренное кипение эфира. По
окончании прибавления смесь нагревают 1 ч, затем мешалку оста-
навливают и раствору дают охладиться. После отстаивания эфир-
ный раствор сифонируют под давлением аргона в сосуд с деле-
ниями. Выход метиллития составляет 90 %. Концентрацию полу-
ченного раствора устанавливают ацидометрическим титрованием
аналогично эфирному раствору фениллития. Предпочтительно по-
лучение 1,1 М раствора метиллития.
в) Раствор бутиллития в эфире
BuBr -----——► BuLi + LiBr
Et2O
Реактивы
Литий металлический 4,2 г
Бутилбромид 34,3 г
Эфир сухой 150 мл
Аргон (из баллона)
Процедура приготовления эфирного раствора бутиллития ана-
логична описанной для фениллития. После внесения в реакцион-
ную колбу 100 мл сухого эфира и 4,2 г (0,6 моль) металлического
лития в нее при перемешивании добавляют 20 капель раствора
34,3 г (0,25 моль) бутилбромида в 50 мл сухого эфира. После того
как реакция началась, что можно заметить по помутнению реак-
ционной смеси и появлению блестящих пятен на кусочках лития,
260
реакционную смесь охлаждают до -35 °C и по каплям при интен-
сивном перемешивании в течение 30 мин прибавляют оставшийся
раствор бутилбромида в эфире. Перемешивание продолжают еше
1-2 ч, сначала при охлаждении, а затем постепенно поднимая
температуру до 0 °C. Далее реакционной смеси дают отстояться при
0 °C и спустя некоторое время сифонируют ее под давлением
аргона в круглодонную реакционную колбу вместимостью 250 мл
через трубку, в которую впаян стеклянный фильтр или помещена
стекловата. Остатки металлического лития осторожно нейтрали-
зуют метанолом. Выход бутиллития составляет 75-85 %.
Для определения концентрации полученного раствора исполь-
зуют ацидометрическое двойное титрование. Предпочтительно по-
лучение 1,3 М раствора бутиллития.
г) Раствор бутиллития в гексане
BuCl ----BuLi + LiCl
гексан
Реактивы
Литий металлический 2,4 г
Бутилхлорид 12 г
Гексан сухой 80 мл
Аргон (из баллона)
Работу проводят в установке, изображенной на рисунке 4.4. За-
полнение колбы растворителем и внесение лития проводится в тех
же условиях, что и при получении фениллития. В колбу помещают
50 мл сухого гексана и 2,1 г (0,3 моль) лития, нагревают смесь при
перемешивании до 30-35 °C, приливают 2-3 мл бутилхлорида.
Реакция обычно начинается сразу после прибавления первой пор-
ции бутилхлорида, что заметно по разогреванию и помутнению
раствора. Остальное количество бутилхлорида разбавляют 30 мл
сухого гексана и прибавляют этот раствор к реакционной смеси в
течение 1 ч с такой скоростью, чтобы температура смеси была
50-65 °C. После этого реакционную смесь перемешивают 2 ч при
45-50 °C, вносят в нее 0,3 г свеженарезанного лития и перемеши-
вают еще 1 ч при той же температуре. Далее раствору дают отсто-
яться и спустя некоторое время сифонируют его под давлением
аргона в колбу с помощью трубки, в которую впаян стеклянный
фильтр или помещена стекловата. Остатки металлического лития
261
осторожно нейтрализуют метанолом. Выход бутиллития состав-
ляет 75-85 %.
Для определения концентрации полученного раствора исполь-
зуют ацидометрическое двойное титрование. Для этого берут али-
квотную часть раствора бутиллития (2 мл), гидролизуют ее 10 мл
дистиллированной воды и продукт гидролиза титруют стандарт-
ным раствором кислоты с использованием в качестве индикатора
фенолфталеина. Вновь берут аликвотную часть раствора бутилли-
тия, вливают раствор, содержащий 1 мл тщательно очищенного
бензилхлорида в 5 мл гексана. Смесь перемешивают несколько ми-
нут и гидролизуют 10 мл дистиллированной воды. Затем титруют,
как описано выше. Разность результатов титрования соответствует
концентрации исследуемого раствора бутиллития. Предпочтитель-
но получение 1,3 М раствора бутиллития.
1,13-Трифенилпропен-2-ол-1
(дифенилстирилметанол)
1. PhLi.EtjO
PhCH = CHCOPh -----------7-
2. Н2О, Н +
Реактивы
Фениллитий
Бензальацетофенон
Эфир сухой 40 мл
Аргон (из баллона)
Работу проводят в токе аргона в установке, изображенной на
рис. 4.4. К эфирному раствору, содержащему 0,12 моль фениллития,
медленно прибавляют раствор 20,8 г (0,1 моль) бензальацетофенона
в 40 мл сухого эфира. Реакционную смесь выдерживают в течение
нескольких часов при комнатной температуре. Затем раствор разла-
гают разбавленной уксусной кислотой, продукт реакции экстраги-
руют эфиром. Экстракт промывают сначала раствором бикарбоната
натрия до слабощелочной реакции, затем водой, сушат сульфатом
натрия, эфир испаряют. Затвердевший остаток перекристаллизо-
вывают из декана или петролейного эфира. Получают 25,5 г (89 %)
продукта; т. пл. 108-111 °C. Хроматография: элюент - гексан и
хлороформ, 1:3; Rf 0,5. Спектральные характеристики представле-
ны на рис. 4.5.
PhCH=CHC(OH)Ph2
0,12 моль
20,8 г
262
Из маточного раствора через некоторое время выкристаллизо-
вывается небольшое количество 1,3,3-трифенилпропанбна-1 - про-
дукта присоединения фениллития по двойной связи С=С бен-
зальацетофенона. Т. пл. 90-91 °C. Спектр ПМР* в СС1д (<5, м. д.:
3,82 д, J = 8 Гц (2Н, СН2), 4,75 т, J = 8 Гц (1Н, СН), 7,29 м (13Н, Ph,
И3"5 аром.), 7,97 м (2Н, Н2-6 аром.)).
1,1,2-Трифенилэтанол-1
PhCH2COOEt I. Ph Li, Et2O PhCH2C(OH)Ph2
2. Н2О, Н +
Реактивы Фениллитий Этилфенилацетат Эфир сухой Аргон (из баллона) 0,12 моль 8,4 г 30 мл
Работу проводят в токе аргона в установке, изображенной на
рис. 4.4. К перемешиваемому эфирному раствору фениллития,
содержащему 0,12 моль фениллития, добавляют 8,4 г (0,05 моль)
этилфенилацетата, растворенного в 30 мл сухого эфира. Для за-
вершения реакции смесь нагревают и кипятят в течение 1 ч. После
охлаждения в ледяной бане реакционную смесь осторожно разла-
гают водой. Продукт реакции экстрагируют эфиром. Объединен-
ные эфирные экстракты сушат сульфатом магния, эфир удаляют.
Остаток перегоняют в вакууме при 10 мм рт. ст. Получают 0,5 г
(6 %) исходного этилфенилацетата (т. кип. 135-140 °C) и 9,6 г
(70%) 1,1,2-трифенилэтанола (т. кип. 222 °C). Последний затвер-
девает и его кристаллизуют из гексана или петролейного эфира.
Т. пл. 87-88 °C. Хроматография: элюент - хлороформ, Rf 0,5.
Спектральные характеристики приведены на рис. 4.6.
263
0,06 моль
9,3 г
7,3 г
30 мл
Реактивы
Фениллитий
4-Бромоанизол
Бензофенон
Эфир сухой
Аргон (из баллона)
Работу проводят в токе аргона в установке, изображенной на
рис. 4.4. К 1,3 М эфирному раствору, содержащему 0,06 моль
фениллития, добавляют 9,3 г (0,05 моль) 4-бромоанизола. В про-
цессе прибавления реакционная смесь слабо разогревается. Ее вы-
держивают в течение 24 ч при комнатной температуре. По истече-
нии этого срока прибавляют по каплям 7,3 г (0,04 моль) бензофе-
нона, растворенного в 30 мл сухого эфира. Для завершения реакции
смесь нагревают 1 ч, затем охлаждают и осторожно разлагают во-
дой. Эфирный слой отделяют, из водного продукт экстрагируют
эфиром. Объединенные эфирные растворы промывают водой до
нейтральной реакции, сушат. Эфир удаляют. Остаток при охлажде-
нии затвердевает. После кристаллизации из этанола получают 11,1г
(75 %) бесцветных кристаллов; т. пл. 127-128 °C. Возможна допол-
нительная очистка сублимацией при температуре 150 °C и
давлении 5 мм рт. ст. Хроматография: элюент - хлороформ и пет-
ролейный эфир, 2:1; Rf 0,6. Спектральные характеристики даны
на рис. 4.7.
ОМе
2,6-Диметоксибензальдегид
ОМе
Li /Ме
u l.PhNL
^СНО
ОМе
СНО
Ph Li
Et2O
ОМе
Реактивы
Фениллитий
1,3-Диметоксибензол
N-Метилформанилид
Эфир сухой
Аргон (из баллона)
Работу проводят в токе аргона в установке, изображенной на
рис. 4.4. В колбу помещают 13,8 г (0,1 моль) 1,3-диметоксибензола
2. Н2О, Н
ОМе
0,12 моль
13,8 г
10,8г
40 мл
ОМе
264
265
266
------г--- j ,------------! I I I I
8 7 6 5 4
267
и 1,3 М эфирный раствор, содержащий 0,12 моль фениллития. Реак-
ционную смесь выдерживают в течение 60 ч при комнатной тем-
пературе. При этом в виде больших бесцветных кристаллов выде-
ляется литиевое производное 1,3-диметоксибензола. К смеси по
каплям прибавляют раствор 10,8 г (0,08 моль) N-метилформани-
лида в 40 мл сухого эфира. В результате саморазогревания смесь
закипает. Через 30 мин после того, как кипение прекратится, смесь
выливают в разбавленную серную кислоту, взятую в избытке.
Эфирный слой отделяют и сушат, эфир удаляют. Остаток пере-
гоняют при давлении 13 мм рт. ст. до тех пор, пока температура
паров не достигнет 150 °C. Остаток после перегонки кристалли-
зуют из циклогексана или из большого количества воды и получают
7,3 г (55 %) альдегида в виде бесцветных иглообразных кристаллов;
т. пл. 98-99 °C. Дополнительная очистка может быть проведена
сублимацией при температуре 100 °C и давлении 2 мм рт. ст. Хро-
матография: элюент - хлороформ и этилацетат, 5:1; Л/0,5. Спект-
ральные характеристики представлены на рис. 4.8.
2,6-Диметокситолуол
Me2SO4
Реактивы
Фениллитий 0,12 моль
1,3-Диметоксибензол 13,8г
Диметилсульфат 16,4г
Аргон (из баллона)
Осторожно! Диметилсульфат - сильный яд. Все работы с ним,
включая мытье посуды, производить только под тягой.
Работу проводят в токе аргона в установке, изображенной на
рис. 4.4. К 1,3 М раствору, содержащему 0,12 моль фениллития,
прибавляют 13,8 г (0,1 моль) 1,3-диметоксибензола и раствор остав-
ляют на 60 ч при комнатной температуре. При этом в виде больших
бесцветных кристаллов выделяется литиевое производное 1,3-ди-
метоксибензола. По истечении указанного времени к раствору при-
268
Рис. 4.8. Спектральные
характеристики 2,6-диме-
токсибензальдегида:
а - спектр ПМР* в ССЦ (д, м.д.:
3,76 с (6Н, ОСНз), 6,55 д, J = 8 Гц
(2Н, Н3-5 аром.), 7,43 т, J = 8 Гц
(IH, Н4аром.), 10,5 с (IH, НС=О));
б - ИК спектр* в тонком слое
(1260 см-' (С-О), 1680 см-'
(С=О)); в - УФ спектр* в метаноле
(Лмакс., нм, 1g е: 276,4,12; 330,3,66)
269
бавляют при перемешивании 16,4 г (0,13 моль) диметилсульфата.
Для завершения реакции смесь кипятят в течение 1 ч, снабдив колбу
обратным холодильником. После охлаждения смесь выливают на
лед и продукт реакции экстрагируют эфиром. Объединенный
эфирный раствор сушат сульфатом натрия, эфир удаляют и остаток
перегоняют в вакууме. Собирают фракцию с температурой кипения
выше 110 °C при 20 мм рт. ст., выход на этой стадии составляет 14 г.
На холоду дистиллят кристаллизуется. Его промывают холодным
пентаном и кристаллизуют из пентана. Получают 8,8 г (58 %) бес-
цветных кристаллов; т. пл. 39-40 °C. Хроматография: элюент -
хлороформ, Rf 0,8. Спектральные характеристики приведены на
рис. 4.9.
Дифенил(2,6-диметоксифенил)метанол
Реактивы
ОМе
J\Li
^ЮМе
Фениллитий
1,3-Диметоксибензол
Бензофенон
Эфир сухой
Аргон (из баллона)
1. Ph2CO
2. Н2О
C(OH)Ph2
ОМе
0,12 моль
13,8 г
14,5 г
50 мл
Работу проводят в токе инертного газа в установке, изобра-
женной на рис. 4.4. К 1,3 М эфирному раствору, содержащему
0,12 моль фениллития и помещенному в колбу вместимостью
200 мл, быстро прибавляют раствор 13,8 г (0,1 моль) 1,3-диметокси-
бензола в 20 мл эфира. Смесь выдерживают при комнатной темпе-
ратуре в течение 60 ч. За это время образовавшийся 2,6-диметокси-
фениллитий кристаллизуется. Далее к перемешиваемой суспензии
прибавляют по каплям раствор 14,5 г (0,08 моль) бензофенона в
30 мл сухого эфира, после чего реакционную смесь нагревают и
кипятят в течение 1 ч, затем охлаждают и добавляют 30 мл воды.
Эфирный слой отделяют, из водного слоя делают экстракцию
270
Рис. 4.9. Спектральные характеристики
2,6-диметокситолуола:
а - спектр ПМР* в ССЦ (<5, м.д.: 2,03 с (ЗН, СН3), 3,78 с
(6Н, ОСН3), 6,53 д, J = 8 Гц (2Н, Н3-5 аром.), 7,06 т,
J = 8 Гц (IH, Н4 аром.)); б- ИК спектр* в СС1д
(1260 см-1 (С-О)); в - УФ спектр* в метаноле
(2макс., нм, 1g е: 222,4,20; 270, 3,04)
271
эфиром. Объединенный эфирный раствор сушат сульфатом магния,
эфир удаляют, остаток кристаллизуют из водного этанола. Полу-
чают 18 г (70 %) дифенил(2,6-диметоксифенил)метанола, т. пл.
134-135 °C. Хроматография: элюент - этилацетат и петролейный
эфир, 1:2; Я/0,4. Спектральные характеристики даны на рис. 4.10.
4-Оксиацетофенон
НО—V У- соон
l.CH3Li
2. (CH3)3SiCl
3. Н2О
но—С у-сосн3
Реактивы
Метиллитий
4-Оксибензойная кислота
Триметилсилилхлорид
Тетрагидрофуран сухой
0,2 моль
5,5 г
10 мл
20 мл
Аргон (из баллона)
В реакционную колбу, предварительно заполненную аргоном,
вносят 5,5 г (0,04 моль) 4-оксибензойной кислоты и 20 мл сухого
ТГФ. Раствор с помощью ледяной бани охлаждают до 0 °C и, энер-
гично перемешивая, быстро (менее 1 мин) приливают 1,1 М эфир-
ный раствор, содержащий 0,2 моль метиллития. Перемешивание
продолжают в течение 2 ч при температуре 0 °C, а затем, не прекра-
щая его, быстро добавляют в реакционную смесь 10 мл (0,08 моль)
свежеперегнанного триметилсилилхлорида. Охлаждающую баню
убирают, реакционной смеси позволяют нагреться до комнатной
температуры, после чего в нее прибавляют 30 мл 1 н. хлороводо-
родной кислоты и перемешивают в течение 0,5 ч. В делительной
воронке водный слой отделяют от органического, из водного слоя
делают экстракцию эфиром. Объединенные эфирные слои промы-
вают 40 мл воды и сушат сульфатом магния. Эфир удаляют. Анализ
остатка методом ГЖХ обнаруживает менее 5 % примеси 1 -метил-
1-(4-оксифенил)этанола. 4-Оксиацетофенон очищают перекристал-
лизацией из эфира. Т. пл. 108,5-109,5 °C. Выход 4,6 г (85 %). Хро-
матография: элюент - хлороформ и этилацетат, 1:1; Rf 0,5. Спект-
ральные характеристики даны на рис. 4.11.
272
Рис. 4.10. Спектральные характеристики
дифеиил(2,6-диметоксифеиил)метанола:
а - спектр ПМР* в СС1д (<5, м.д.: 3,39 с (6Н, ОСН3),
5,84 с (1Н, ОН), 6,55 д, J = 8,5 Гц (2Н, Н3-5 аром.),
7,16 м (ПН, Ph н Н4 аром.)); б-ИК спектр* в ССЦ
(1250 см~> (С-О), 3520 см”1 (О-Н)); в - УФ спектр*
в этаноле (2макс., нм, 1g е: 278, 3,28)
273
Рис. 4.11. Спектральные характеристики
4-оксиацетофенона:
а - спектр ПМР* в ССЦ (<5, м. д.: 2,57 с
(ЗН, СН3), 6,95 м (2Н, Н3-5 аром.), 7,9 м
(2Н, Н2-6 аром.), 8,35 уш. с (1Н, ОН));
б-ИК спектр в КВг (1650 см-1 (С=О).
3150 см-1 (О-Н)); в- УФ спектр в метаноле
(Амакс., нм, 1g е: 219,5, 4,04; 275,4,2)
274
Транс-4-нитро-4'-метоксистильбен
no2 L no2
Реактивы
Бутиллитий 0,026 моль
Трифенилфосфин 10,5 г
4-Нитробензилхлорид 6,8 г
4-Метоксибензальдегид 3,2 г
Бензол сухой 40 мл
Аргон (из баллона)
Получение хлорида 4-нитробензилтрифенилфосфина. К раство-
ру 10,5 г (0,04 моль) трифенилфосфина в 20 мл сухого бензола при-
бавляют 6,8 г (0,04 моль) 4-нитробензилхлорида, смесь нагревают и
кипятят в течение 3 ч. После охлаждения выпадает осадок, который
отделяют фильтрованием, промывают небольшим количеством хо-
лодного бензола. Получают 6,3 г фосфониевой соли. Фильтрат упа-
ривают и получают дополнительно 4,8 г соли. Суммарный выход
составляет 11,1г (64 %), т. пл. 275-278 °C (из смеси CCI4 и петро-
лейного эфира).
Получение транс-4-нитро-4'-метоксистильбена. 1,3 М раст-
вор, содержащий 0,026 моль бутиллития в гексане, добавляют в ат-
мосфере аргона к суспензии 8,6 г (0,02 моль) 4-нитробензилтри-
фенилфосфинхлорида в 20 мл бензола и смесь перемешивают в
течение 2 ч. О том, что образовался илид, судят по исчезновению
осадка соли фосфония и образованию окрашенного в красно-оран-
жевый цвет раствора. Затем прибавляют 3,2 г (0,024 моль) 4-мето-
ксибензойного альдегида (анисового альдегида) и смесь переме-
шивают при комнатной температуре в течение 4 ч, после чего ее
275
разбавляют петролейным эфиром. При этом выпадает окрашенный
в темный цвет осадок, который отфильтровывают и кристаллизуют
из этанола. Получают 4,5 г (89 %) /пранс-4-нитро-4'-метоксис-
тильбена в виде окрашенных в желтый цвет кристаллов; т. пл.
131-132,5 °C.
7ранс-4-нитро-4'-метоксистильбен можно получить также с ис-
пользованием эфирных растворов бутиллития или метиллития. В
этом случае после завершения реакции с 4-метоксибензальдегидом
к смеси прибавляют 40 мл эфира, осадок оксида трифенилфосфина
отфильтровывают и промывают на фильтре эфиром. Эфирный
фильтрат промывают водой и сушат сульфатом натрия. После
удаления эфира маслообразный остаток растирают с небольшим
количеством холодного спирта, фильтруют и сушат. Получают 2,7 г
(53 %) кристаллов; т. пл. 133-134 °C (этанол). Спектральные харак-
теристики приведены на рис. 4.12.
М,1\-Диметиламид 1-оксициклогексанкарбоновой кислоты
uso-PnNLi
НС ------—
NMe2
Реактивы
//О
LiC^
NMe2
i-O°
2. NH4C1
НО CONMe2
Бутиллитий Диизопропиламин М,М-Диметилформамид Циклогексанон Эфир сухой ТГФ сухой Аргон (из баллона) 0,25 моль 20,4 г 14,6 г 19,6г 80 мл 20 мл
Трехгорлую колбу вместимостью 250 мл снабжают низкотем-
пературным термометром, капельной воронкой с уравнительной
трубкой, в верхнее отверстие которой вставляют трубку для ввода
инертного газа (аргона) и механической мешалкой. Эфирный раст-
вор, содержащий 0,25 моль бутиллития, охлаждают до -35 °C и по
каплям при интенсивном перемешивании в атмосфере аргона при-
бавляют к нему 20,4 г (0,2 моль) диизопропиламина с такой ско-
ростью, чтобы температура реакционной смеси не превышала
-30 °C. Реакция экзотермична, поэтому необходимо внешнее
276
Рис. 4.12. Спектральные характеристики
транс-4-нитро-4' -метоксистильбена:
а - спектр ПМР* в СС1д (<5, м.д.: 3,83 с (ЗН,
ОСНз), 6,9 д, J = 9 Гц (2Н, Н3 .5’ аром.), 6,98 д,
J = 16 Гц и 7,20 д, J = 18 Гц (2fi СН=СН), 7,47 д,
J = 9 Гц (2Н, Н2/' аром.), 7,57 д, J = 9 Гц (2Н,
Н2-6 аром.), 8,18 д, J = 9 Гц (2Н, Н3-5 аром.)); б-
ИК спектр* в КВг (1180 см~' (С-О), 1330 и
1520 см-1 (NO2)); в - УФ спектр* в этаноле
(Лмакс., нм, 1g е: 248, 4,03; 275, 3,95; 377, 4,40))
277
охлаждение. После прибавления всего диизопропиламина переме-
шивание продолжают 10 мин при -30 °C и получают раствор, со-
держащий около 0,2 моль диизопропиламида лития. Этот раствор
охлаждают в токе аргона до -70 °C. В отдельной колбе охлаждают
Рис. 4.13. Спектральные характеристики N,N-диметаламида
1 -оксицнклогексанкарбоновой кислоты:
а - спектр ПМР* в CDCI3 (<5, м. д.: 1,63 м (10 Н, СН2), 1,83 с (6Н, СН3), 3,79 с (1Н, ОН));
б- ИК спектр в КВг (1610 см-' (С=О))
278
до температуры -78 °C раствор 14,6 г (0,2 моль) диметилформамида
и 19,6 г (0,2 моль) циклогексанона в 100 мл смеси сухого эфира и
ТГФ (4:1). Затем раствор сифонируют под давлением аргона в
колбу с раствором диизопропиламида лития. Реакционную смесь
выдерживают при температуре -78 °C в течение 6 ч, после чего тем-
пературу медленно поднимают до 20 °C и реакционную смесь нейт-
рализуют водным раствором хлорида аммония. Слои разделяют, из
водного слоя делают экстракцию эфиром. Объединенный эфирный
раствор промывают насыщенным раствором хлорида натрия и су-
шат сульфатом магния. Эфир испаряют в вакууме, от маслянистого
остатка отгоняют непрореагировавший циклогексанон. Затвердев-
ший бесцветный остаток перекристаллизовывают из циклогексана.
Получают 15,8 г (45 %) вещества в виде бесцветных игл; т. пл.
105 °C. Спектральные характеристики приведены на рис. 4.13.
279
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
К главе 1
Богословский Б. М., Казакова 3. С. Скелетные катализаторы, их свойства и
применение в органической химии. М.: Изд-во иностр, лит-ра, 1957. 144 с.
Вейганд К, Хильгетаг Г. Методы эксперимента в органической химии / Пер.
с нем. М.: Химия, 1968. 944 с.
Жермен Д. Каталитические превращения углеводородов / Пер. с англ. М.:
Мир, 1972. 308 с.
Новые пути органического синтеза. Практическое использование переход-
ных металлов: Пер. с англ. / X. М. Колхаун, Д. Холтон, Д. Томпсон, М. М. Твигг.
М.: Химия, 1989. 400 с.
Мозеттиг Э., Мозинго Р. Каталитическое восстановление хлорангидридов
кислот в альдегиды И Органические реакции / Пер. с англ. М.: Изд-во иностр, лит.,
1951. Сб. 4. С. 337-357.
Современные методы органического синтеза / Под ред. Б. В. Иоффе. Л.:
Изд-во Ленингр. ун-та, 1980. 232 с.
Carruthers W. Some modem methods of organic syhthesis. 3rd ed. Cambr. Univ.
Press, 1986. 526 p.
Evans R. F. Reduction methods // Modem reactions in organic synthesis / Ed.
C. J. Timmons. L.: Reinhold Co, 1970. P 3-53.
HudlickyM. Reduction in organic chemistry. Chichester: Ellis Horwood Ltd, 1986.
309 p.
Komarewsky И I., Riesz С. H„ Morritz F. L. Catalytic reactions // Technique of
organic chemistry / Ed. A. Weissberger. N.Y., L.: Interscience Publishers Inc, 1956.
Vol. 2. P. 1-255.
К главе 2
Браун В. Г. Восстановление литийалюминий гидридом И Органические реак-
ции / Пер. с англ. М.: Изд-во иностр, лит-ры, 1953. Сб. 6. С. 409-478.
Гейлорд Н. Восстановление комплексными гидридами металлов / Пер. с англ.
М.: Изд-во иностр, лит-ры, 1959. 912 с.
Минович В., Михайлович М. Алюмогидрид лития и его применение в органи-
ческой химии / Пер. с англ. М.: Изд-во иностр, лит-ры, 1957. 257 с.
Хайош А. Комплексные гидриды в органической химии / Пер. с нем. Л.:
Химия, 1971. 623 с.
280
Candlin J. P., Rennie R. A. C. // Elucidation of organic structures by physical and
chemical methods. 2nd ed. I Eds K. W. Bentley, G. W. Kirby. N.Y.: Wiley, 1973.
P. 1-76. (Weisberger Techniques of Chemistry. Vol. 4.)
Carruthers W. Some modem methods of organic synthesis. 3rd ed. Cambr. Univ.
Press, 1986. 526 p.
House H. O. Modem synthetic reactions. Menlo Park: Benjamin, 1972. 852 p.
Hudlicky M. Reduction in organic chemistry. Chichester: Ellis Horwood Wtd,
1986. 309 p.
Reduction/ Ed. R. L. Augustine. N.Y.: Marcel Dekker, 1979. 242 p.
К главе 3
Неводные растворители: Пер. с англ. / Под ред. Т. Виддингтона. М.: Химия,
1971.372 с.
Реутов О. А., Белецкая И. П., Бутин К. П. СН-Кислоты. М.: Наука, 1980.
248 с.
Caine D. Reduction and related reactions of a,/?-unsaturated carbonyl compounds
with metals in liquid ammonia // Organic reactions. N.Y.: Wiley, 1976. Vol. 23.
P. 1-253.
Carruthers W. Some modem methods of organic synthesis. 3rd. ed. Cambr. Univ.
Press, 1986. 526 p.
House H. O. Modem synthetic reactions. Menlo Park: Benjamin, 1972. 852 p.
Smith H Organic reaction in liquid ammonia: Chemistry in non-aqueous ionizing
solvents. N.Y.: Wiley, 1968. Vol. 1, № 2. 343 p.
К главе 4
Бейтс P., Огле К. Химия карбоанионов / Пер. с англ. Л.: Химия, 1987. 112 с.
Гильман Г. Реакция металлирования при помощи литийорганических соеди-
нений // Органические реакции / Пер. с англ. М.: Изд-во иностр, лит-ры, 1956.
Сб. 8. С. 333-391.
Талалаева Т. В., Кочетков К А. Методы элементоргаиической химии. Литий,
натрий, калий, рубидий, цезий. М.: Наука, 1971. 1192 с.
Уэйкфилд Б. Методы синтеза с использованием литийорганических
соединений: Пер. с англ. / Под ред. И. П. Белецкой. М.: Мир, 1991. 183 с.
Gschwend Н. W.. Rodriquez Н. R. Heteroatom-facilitated lithiations // Organic
reactions / Ed. Board. N.Y.: Wiley, 1979. Vol. 26.
NegishiE. Ofganometallics in organic synthesis N.Y.: Wiley, 1980. Vol. 1.
Posner G. H. An introduction to synthesis using organocopper reagents, N.Y.:
Wiley, 1980. 140 p.
Seebach D., Griss К. H. New application of organometallic reagents in organic
synthesis. Amsterdam: Elsvier, 1976.
Wakefield B. J. The chemistry of organolithium compounds. Oxford: Pergamon
Press, 1974. 333 p.
281
Учебное издание
Репинская Ирина Бернгардовна
Шварцберг Марк Самуилович
ИЗБРАННЫЕ МЕТОДЫ СИНТЕЗА
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Редактор издательства Н.В. Кораблина
Художник В.Н. Морошкин
Корректоры Н.В. Кораблина, Е.А. Мордвинова
Компьютерная верстка Т. В. Велигжанина
ЛР№ 020618 от 13.08.97
Подписано в печать 19.04.2000. Формат 60 х 84/16. Гарнитура Таймс. Печать офсетная.
Бумага офсетная. Усл. печ. л. 16. Уч.-изд. л. 24. Тираж 500 экз. Заказ № 137
Издательство Новосибирского университета
630058, г. Новосибирск, ул. Русская, 35
Отпечатано в типографии Сибирского издательско-полиграфического
н книготоргового предприятия „Наука" РАН
630077, г. Новосибирск, ул. Станиславского, 25