/












































































































































































































































































































































































































































































































































































Текст
А.ФЕЙЫ1
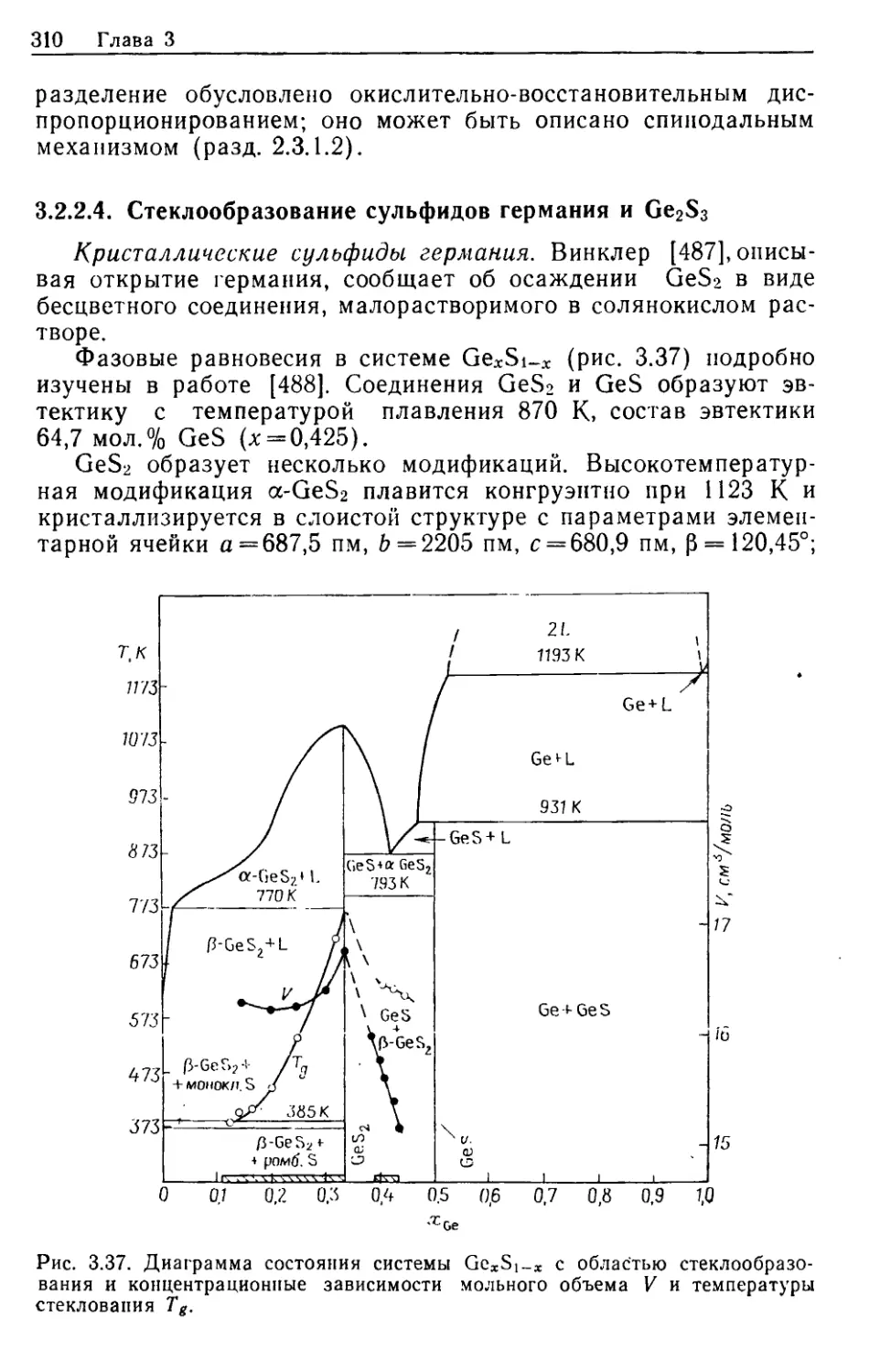
АМОРФНЫЕ
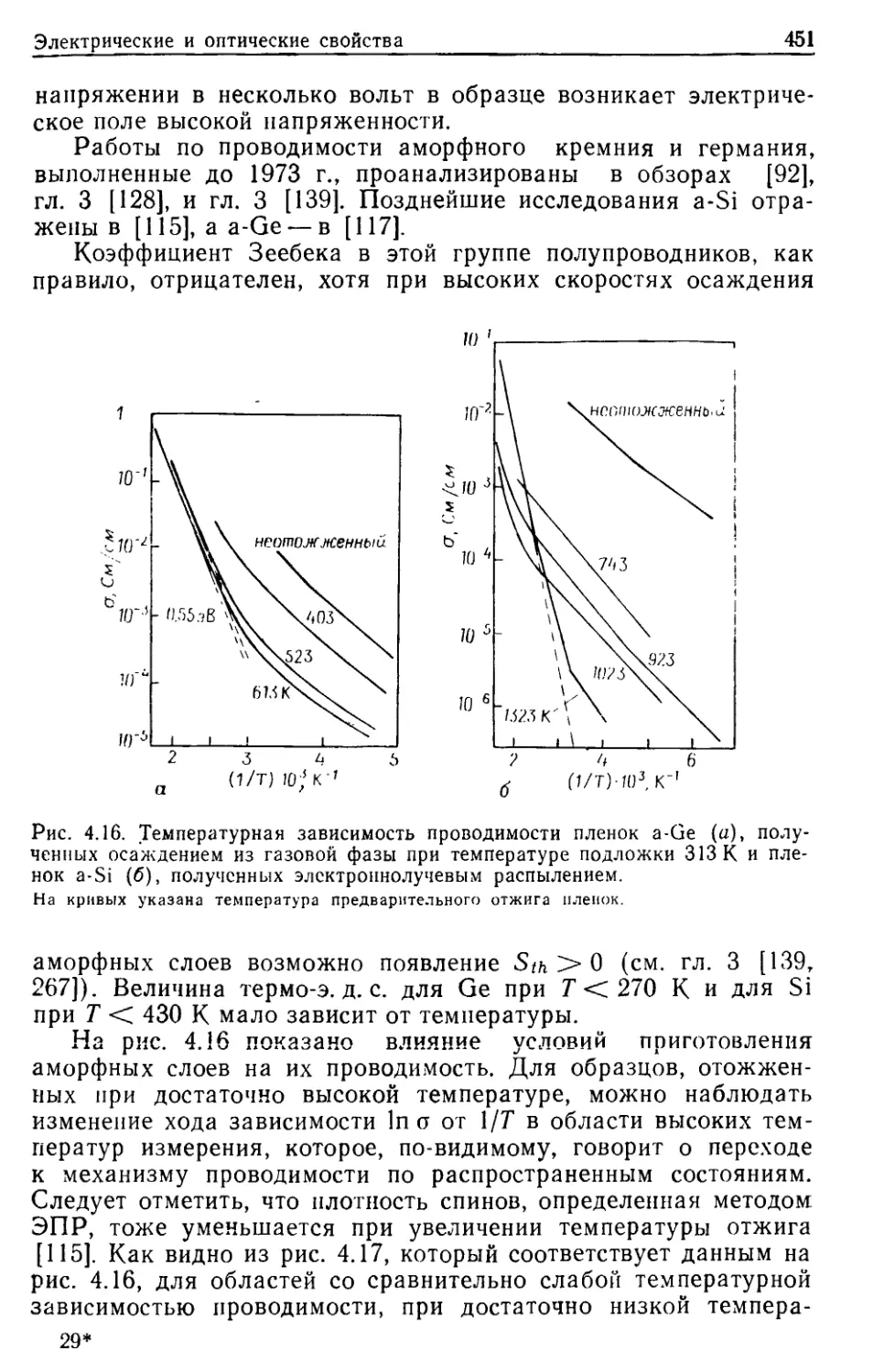
И СТЕКЛООБРАЗНЫЕ
НЕОРГАНИЧЕСКИЕ
ТВЕШЫЕ
ТЕЛА
AMORPHE
UND GLASARTIGE
ANORGANISCHE
FESTKORPER
von
Prof. Dr. Rer. Nat. Habil. Adalbert Feltz
Jena
Akademie-Verlag- Berlin, 1983
А.ФЕЛЬЦ
АМОРФНЫЕ
И СТЕКЛООБРАЗНЫЕ
НЕОРГАНИЧЕСКИЕ
ТВЕРДЫЕ
ТЕЛА
Пер. с немецкого
канд. хим. наук Г. 3. Виноградовой,
канд. физ.-мат. наук А. В. Колобова
и канд. хим. наук И. Б. Куценка
под редакцией
академика И. В. Тананаева
и д-ра хим. наук С. А. Дембовского
Москва «Мир» 1986
ББК 24.1
Ф40
УДК 54-161.6
Фельц А.
Ф40 Аморфные и стеклообразные неорганические твердые
тела: Пер. с нем.— М.: Мир, 1986.— 558 с., ил.
Автор монографии — профессор Фельц является одним из наиболее крупных
специалистов ГДР, ученым с мировым именем. В книге рассматриваются вопросы
теории аморфного и стеклообразного состояния и последние достижения в обла-
сти практического применения аморфных материалов и стекол. Автор приводит
огромный фактический материал по конкретным системам (области стеклообра-
зования, электрические и оптические свойства) и их применению в новых отрас-
лях техники.
Предназначена для химиков-неорганнков, технологов, работающих с некри-
сталлическими материалами, физнкохимиков.
1802000006-319
Ф041(01)-86
84—86, ч. 1
ББК 24.1
Редакция литературы по химии
© Akademie-Verlag - Berlin 1983
© перевод на русский язык, «Мир», 1986
Предисловие редакторов
перевода
Твердые аморфные (некристаллические) и стеклообразные
материалы вызывают постоянно возрастающий научный интерес
и находят все более широкое практическое применение. В по-
следнее время сферы применения стекол уже не ограничиваются
традиционными областями в качестве оптических сред различ-
ного целевого назначения; значительно расширился и круг не-
кристаллических веществ. По ряду свойств эти материалы пре-
восходят кристаллы и поэтому заменяют последние даже
в традиционных областях их применения; к таким новым мате-
риалам относятся, например, аморфные магнетики, аморфный
гидрогенизированный кремний и др. Появились и новые, ранее
неизвестные технологии, позволяющие получить традиционные
материалы — стекла с улучшенными свойствами, например кос-
мическая технология.
Вниманию советского читателя предлагается книга видного
ученого ГДР, профессора Йенского университета А. Фельца.
Это первая выходящая на русском языке монография, посвя-
щенная конденсированному некристаллическому состоянию
практически всех типов неорганических веществ,— от традици-
онных оксидных стекол, стеклообразных полупроводников до
аморфных металлов, получивших широкое развитие в последние
годы. Ранее в русском переводе вышло несколько книг, которые
можно считать предшественниками данной, в частности книга
Г. Роусона *, коллективная монография под редакцией М. Брод-
ски **. Однако рассматриваемые в них вопросы относились лишь
к определенным классам аморфных материалов. Сейчас в миро-
вой науке усиливается интерес к самой проблеме некристалли-
ческого состояния, о чем свидетельствует и выход в свет в тече-
ние последних 2—3 лет сразу нескольких монографий:
Elliott S. R., Physics of Amorphous Materials, London—New
York, Longman, 1983, 374 p.; Paul A., Chemistry of Glasses,
London, Chapman and Hall, 1982, 293 p.; Zallen R., The Physics
of Amorphous Solids, New York, Chichester, Brisbane, J. Wiley
* Роисон Г. Неорганические стеклообразующие системы. Пер. с англ.—
М.: Мир, 1970.
** Аморфные полупроводники. Пер. с англ./Под ред. М. Бродски.— М.:
Мир, 1982.
6
Предисловие редакторов перевода
and Sons, 1983, 297 р. и др. Труд А. Фельца занимает видное
место в этом ряду и будет интересен широкому кругу читате-
лей: физикам, химикам, технологам — как теоретикам, так и
экспериментаторам.
Книга состоит из пяти глав, первая из которых является
кратким введением. Вторая глава, очень большая по объему,
затрагивает методы получения аморфных (в том числе стекло-
образных) веществ, термодинамику стеклообразного состояния,
кинетические аспекты стеклообразования и аморфизации (в том
числе критические скорости охлаждения расплавов), структуру
и химическую связь. Затрагиваются вопросы применимости зон-
ной теории и представлений о дефектах к некристаллическому
состоянию вещества. Эта область интенсивно развивается в на-
стоящее время и поэтому может быть адекватно и на современ-
ном уровне отражена только активно работающим в данном
направлении специалистом. Хорошо освещены традиционные
представления пяти-десятилетней давности, но некоторые на-
правления последних лет, в частности работы советских авто-
ров (например, модель квазимолекулярных дефектов или тео-
рия автолокализации электронных пар, на которые мы даем
соответствующие ссылки), к сожалению, оказались незатро-
нутыми.
Третья глава посвящена рассмотрению конкретных аморф-
ных и стеклообразующих систем, начиная от элементов типа
a-Si, a-Ge и бинарных стекол типа SiCb, В2О3 и др. и кончая не-
которыми четырехкомпонентными системами. Большое внима-
ние уделено тройным халькогенидным стеклообразным полупро-
водникам, которые интенсивно исследовались в лаборатории
профессора Фельца; однако и другие группы аморфных мате-
риалов освещены достаточно полно.
Пожалуй, наиболее сложная для автора часть — гл. 4, по-
священная рассмотрению электрического транспорта (ионного
и электронного) и оптических свойств некристаллических ве-
ществ, а также ряду эффектов, таких, как фотолюминесценция,
фотоструктурныс превращения, пьезооптический эффект, фото-
кристаллизация и т. п.
Заключительная часть книги дает представление о некото-
рых современных аспектах применения аморфных материалов и
стекол — вопрос весьма актуальный, поскольку некристалличе-
ские материалы благодаря широчайшему диапазону свойств за-
нимают в настоящее время все большее место в ряде практи-
ческих применений, о чем говорилось выше.
Следует учесть, что при написании данной монографии ав-
тор ставил целью осветить в целом проблему некристалличе-
ского состояния, не останавливаясь на частных вопросах. Воз-
можно, такой подход не удовлетворит «узких» специалистов, но
для них книга может оказаться ориентиром, а иногда и пер-
Предисловие редакторов перевода 7
воначальным пособием в других областях, тем более что в ней
затронуто и много граничных проблем.
За время, прошедшее с момента публикации книги в ГДР,
в советской периодической литературе опубликованы две по-
ложительные рецензии на нее *. В одной из них указывалось
на недостаточное цитирование советских авторов и источников,
особенно последних лет. Мы полностью разделяем это мнение
и при подготовке русского издания сочли необходимым допол-
нить литературу ссылками на труды советских ученых, отра-
жающие, на наш взгляд, основные направления научных ис-
следований в данной области, проводимые в СССР, а также
затрагивающие ряд приоритетных вопросов. В дополнительную
литературу включены также монографии зарубежных авторов,
переведенные на русский язык, и работы, которые появились
уже после написания автором монографии и могут представ-
лять интерес для советского читателя.
Перевод выполнен канд. хим. наук Виноградовой Г. 3. (гл. 3),
канд. физ.-мат. наук Колобовым А. В. (гл. 4 и 5) и канд. хим.
наук Куценком И. Б. (предисловие, гл. 1 и 2).
И. Тананаев
С. Дембовский
От редакции
Выражаем искреннюю благодарность профессору Б. Т. Коло-
мийцу за помощь, оказанную при редактировании гл. 4 («Элек-
трические и оптические свойства стекол и аморфных тел»).
* Физ. и хим. стекла, 1984, т. 10, № 6, с. 744; Изв. АН СССР, сер.
неорг. матер., 1985, т. 21, № 4, с. 685.
Моей семье и коллегам
Предисловие
Наши знания о некристаллическом состоянии неорганиче-
ских твердых тел существенно расширились и углубились за по-
следние два с половиной десятилетия. Примерно до середины
50-х годов об аморфных неорганических веществах, по суще-
ству, сообщалось лишь в самой общей, описательной форме.
Уже несколько тысячелетий человек использовал стекло
в своей повседневной жизни. Однако только со времен
О. Шотта и Э. Аббе началось систематическое изучение стекла.
С тех пор были найдены многочисленные эмпирические соотно-
шения, связывающие состав стекла с его свойствами. В послед-
ние десятилетия исследования направлены на создание новых
марок стекол, а также на выявление объективных корреляций
между структурой и свойствами с целью создания более глубо-
ких теоретических основ стеклообразного состояния.
Значительные успехи достигнуты в термодинамической ин-
терпретации стеклообразного состояния и в кинетическом опи-
сании стеклообразования и процессов упорядочения, которые мо-
гут протекать в стекле. Материальная основа аморфных ве-
ществ и стеклообразующих систем существенно расширилась
за счет включения в нее металлов и сплавов, а также много-
численных пеоксидных комбинаций элементов, прежде всего
халькогенидных стекол. Решающий успех в развитии теорети-
ческих аспектов достигнут благодаря привлечению аппарата
физики твердого тела при изучении стеклообразных и аморф-
ных веществ. Аморфные модификации некоторых простых ве-
ществ, таких, как селен, кремний и германий, и стеклообразо-
ватели на основе бинарных соединений определенной стехио-
метрии, например кварцевое стекло, впервые досконально изу-
чены методами физики твердого тела. При проведении этих
исследований стало возможным гораздо более надежно выяс-
нить природу химических связей и типы структур в конденси-
рованных системах и четче выделить черты, характерные для
кристаллического состояния.
Благодаря этим исследованиям были также открыты новые
физические явления и необычные свойства веществ. Извест-
ные ранее соотношения, касающиеся прежде всего структуры
и затрагивающие теорию химической связи, впервые получили
удовлетворительное научное обоснование. Речь идет в основном
Предисловие 9
о качественных закономерностях. Нельзя не отметить, что на
достигнутом к настоящему времени уровне развития теории не-
обходимо еще с осторожностью подходить к результатам ко-
личественных расчетов, даже в тех случаях, когда они нахо-
дятся в удовлетворительном согласии с экспериментом.
На каждой стадии развития знаний о стекле очень сущест-
венно, что стекла с определенной комбинацией свойств потен-
циально могут иметь практическое применение. Например, раз-
работка оксидных стекол с электронной проводимостью была
откликом на потребность в связи с развитием техники телеви-
зионной записи в конце 50-х годов; в 1963 г. открыты бистабиль-
ные проводящие состояния в халькогенидных стеклах (так назы-
ваемый эффект переключения Овшинского); большие надежды
связаны с перспективой создания недорогих солнечных батарей
на основе аморфного кремния (1975 г.).
Однако эти исследования не затрагивают традиционных на-
правлений изучения стекол. Результаты современных исследо-
ваний химии стекла нашли свое воплощение при решении тех-
нических задач путем создания таких важнейших новых мате-
риалов, как элементы волоконной оптики, прозрачная керамика,
фотохромные стекла, лазерные стекла или широко используе-
мые оптические стекла со специальными свойствами. При этом,
несомненно, одновременно происходило дальнейшее развитие
теоретических основ науки о стекле. Решающий прогресс в тео-
ретическом обосновании достигнут в настоящее время в рамках
физики и химии твердого тела. На регулярно проходящих засе- -
даниях Международного конгресса по стеклу и Международной
конференции по аморфным и жидким полупроводникам обсу-
ждаются различные аспекты этой проблемы. Стремление объ-
единить оба направления (теоретическое и практическое) ха-
рактерно для публикаций в Journal of Non-Crystallin Solids и
докладов на международных конференциях «Физика некристал-
лических твердых тел».
Те же цели легли в основу настоящей книги. В ней пред-
принята попытка дать обзор и изложить необходимые вводные
представления о нынешнем состоянии теоретической физики и
химии аморфных и стеклообразных твердых фаз в форме, до-
ступной и интересной и для физиков, и для химиков, занимаю-
щихся изучением твердых тел, а также для специалистов в об-
ласти исследования стекла. При этом важно было продемон-
стрировать многообразие веществ, имеющих некристаллическую
структуру, и их различные свойства. Следует иметь в виду, что
речь идет о первом опыте систематического обсуждения аморф-
ных и стеклообразных неорганических веществ в терминах хи-
мии твердого тела. Автор стремился к общедоступному пред-
ставлению прежде всего новейших результатов твердофазных
исследований в данной области, избегая формально-теоретиче-
10 Предисловие
ской проработки определенных соотношений, а также широкого
обсуждения обычных фактов из области химии стекла, которые
в последние годы собраны во многих монографиях.
Несмотря на принятое ограничение затронуть лишь наиболее
характерные соединения-стеклообразователи и основные стекло-
образующие системы, в книге представлена относительно пол-
ная библиография. Цитируемая литература отражает подъем,
который претерпели физика и химия твердых некристалличе-
ских систем в недалеком прошлом. Однако приводимые цитаты
не претендуют на полноту. Они подобраны в соответствии с на-
учными интересами автора и в связи с этим не свободны от
субъективизма.
Книга должна обратить внимание читателя на то, что для
всестороннего описания неорганических твердофазных систем
нельзя обойтись без исследований твердых веществ с неупоря-
доченным строением. Твердое аморфное и стеклообразное со-
стояния в общем тесно связаны со структурой расплава; однако
они имеют и собственные варианты реализации принципов по-
строения неорганических соединений, которые выходят за рамки
наших знаний о соответствующих кристаллических формах.
Пусть же данная книга подтолкнет заинтересовавшихся хими-
ков устремить свои усилия в этом направлении, генерируя но-
вые идеи, которые способствовали бы развитию неорганической
химии.
В течение последних 10 лет коллектив отдела научных ис-
следований по неорганической химии твердого тела института
химии Университета им. Фридриха Шиллера работал в на-
правлении, соответствующем теме книги; результаты работы
были оформлены в виде ряда публикаций. Существенное уча-
стие в написании некоторых разделов книги, в особенности свя-
занных с термохимическими аспектами, а также при выявле-
нии корреляций между структурой и свойствами, принял
доц. д-р Д. Линке. Кроме того, выражаю свою благодарность
д-ру Буркхардту, д-ру Капсу, д-ру Ширмайстеру, д-ру Канту,
д-ру Фойгту, д-ру Людвигу.
За работу с библиографией благодарю мою супругу Урсулу
Фельц, за тщательную подготовку рукописи — Хайке Мюллер
и за выполнение рисунков — Штефи Этцольд.
Выражаю благодарность коллективу Akademie-Verlag за
помощь при редактировании рукописи и за превосходное офор-
мление книги.
Адальберт Фельц
Иена, октябрь 1981
Принятые обозначения
А — поглощающая способность
а—длина связи; средняя длина перескока
д0 — постоянная решетки
аь — активность
а, b — постоянные уравнения Ван-дер-Ваальса
аКрит — критическое расстояние при делокализации электронов
В — пьезооптическая постоянная
С —- емкость
С\, С2 — фотоупругие постоянные
Ср — мольная теплоемкость; теплоемкость при постоянном давлении
£) —диэлектрический сдвиг; коэффициент диффузии
d — толщина слоя
Е — модуль упругости; энергия
— индуцированное полем понижение потенциального барьера
локализованных электронов
^Е * — энергия активации испарения в открытом вакууме
EAfi — сродство к электрону
Ед, Ев — энергия локализованных состояний в области края зоны про-
водимости и валентной зоны
Ев—экспериментально определенная энергия связи
Еь — энергия связи, получаемая путем расчетов
Ес — энергия края зоны проводимости в кристаллических полупро-
водниках; энергия края подвижности в зоне проводимости
некристаллических полупроводников
Eq — энергия активации диффузии; энергия деформации кристалли-
ческой решетки
Ер—энергия Ферми
Ер,п> Е?, р — энергия квазиуровней Ферми электронов и дырок
ЕУ} — энергия активации вязкого течения
Ер—энергия связи электронов /(-оболочки
Ев — энергия образования равновесной концентрации вакансий
^EiOk — область локализованных состояний на границах зон
Eoni — оптическая ширина запрещенной зоны
Ер —энергия поляризации решетки
Ер — энергия поляронного состояния; энергия поляризации
Ео — энергия активации проводимости
Еу — энергия края валентной зоны в кристаллических полупровод-
никах; энергия края подвижности в валентной зоне некри-
сталлических полупроводников
12 Принятые обозначения
в — заряд электрона р — напряженность поля ^крит — критическая напряженность поля при отклонении от закона Ома f', — фокусные расстояния fe — коэффициент рассеяния электронов fl — коэффициент активности fm> fn ~~ атомные амплитуды при рентгеновском рассеянии f N — атомные амплитуды при нейтронном рассеянии Q — модуль сдвига; модуль упругости при кручении; свободная энергия Q — свободная энергия смешения д(7^ — свободная энергия активации вязкого течения дбд—свободная энергия образования вакансий дОпл — мольная свободная энергия плавления — - удельная свободная энергия гомогенного зародышеобразова- ния AgKp — удельная свободная энергия гетерогенного зародышеобразо- вания Д^гпах ~ максимальная свободная энергия гомогенного зародышеобра- зования Ag’max — максимальная свободная энергия гетерогенного зародышеоб- разования gm (И — функция электронной плотности
Д£об, Afi’non — объемная и поверхностная составляющие AgKp
Н — напряженность магнитного поля; энтальпия Д//д — стандартная мольная энтальпия атомизации ДЯЛ — энтальпия активации вязкого течения Д//кр — энтальпия кристаллизации — стандартная мольная энтальпия реакции Д//£—мольная энтальпия образования вакансий Д#пл — мольная энтальпия плавления ДЯиСП — энтальпия испарения h — постоянная Планка / — интенсивность; спин ядра /д —энергия ионизации; плотность потока фотонов, падающего на образец Iv — скорость стационарного зародышеобразования t — скорость нестационарного зародышеобразования J — полуширина зоны / — плотность тока k—постоянная Больцмана; волновой вектор; константа поглоще- ния 1 — средняя длина свободного пробега Af — молекулярная масса; молярная масса Д4 — средняя молекулярная масса
Принятые обозначения
13
М * — комплексный электрический модуль
те — масса электрона
/ИЭфф — эффективная масса электрона
ДГ — концентрация атомов или структурных групп
МА — число Лвогадро
N (£) — плотность электронных состояний
N (W) — плотность фононных состояний
ДГС> Д/у — эффективная плотность состояний на краях зоны проводимо-
сти и валентной зоны
Nt—плотность состояний центров захвата
П — концентрация электронов
Д/2 — избыточная концентрация электронов при возбуждении излу-
чением
По — концентрация связей
Пс — концентрация гомоатомных связей
Псг — показатель преломления при Z=643,85 нм
nD — дрейфовая концентрация носителей
пА— концентрация донорных и акцепторных центров
Пе — показатель преломления при Х=546,07 нм
п — показатель преломления при Х=479,99 нм
Г
Р — удельная поляризация
рм— молярная поляризация
P(F) — функция распределения Ферми—Дирака
Р — составляющая относительной дисперсии
Л/*
р—давление; концентрация дырок
Др — избыточная концентрация дырок при возбуждении излучением
q — скорость охлаждения; скорость нагревания
//крит — критическая скорость охлаждения
Р— газовая постоянная: отражательная способность при много-
кратном отражении; сопротивление
Рн — постоянная Холла
Рм — молярная рефракция
Рт — скорость молекулярного потока при испарении в равновесных
условиях
Р*т — скорость молекулярного потока при испарении в открытом
вакууме
— скорость конденсации при испарении в открытом вакууме
^?исп — скорость испарения в открытом вакууме
Г — отражательная способность при однократном отражении
Гкрит — критический радиус зародыша
Гтя — вектор расстояния между парой атомов
S — энтропия
Se — напряжение при растяжении или сжатии
д5^ — энтропия активации вязкого течения
14 Принятые обозначения
SG — касательное напряжение сдвига; напряжение на срез
А5стекл — изменение энтропии при переходе стекло—расплав
ДЗконф — изменение конфигурационной энтропии
— хроматическая аберрация
Д5ПЛ — мольная энтропия плавления
Sth — термоэлектродвижущая сила; коэффициент Зеебека
Т — коэффициент пропускания; температура
То—коэффициент пропускания при бесконечно малом поглощении
Tew—дилатометрическая температура размягчения стекла
Tg — температура стеклования
Tgt min—температура образования идеального стекла Тг
Гкрит — критическая температура
Тм — температура проседания
Тпл —’температура плавления
Тz — температура затвердевания (замораживания)
tD—время дрейфа
дб/ — термо-э. д. с.
UА—энергия кулоновского взаимодействия
£/эфф — эффективная энергия корреляции электронов
UЕР — энергия корреляции электронов
U (г) — периодический потенциал кристаллической решетки
U — линейная скорость роста кристаллов
й —средняя скорость
V — мольный объем
Vn—диапазон колебаний потенциальной энергии локализованных
электронных центров
У* — активационный объем
Vj — свободный объем
Укрит— критический объем пустого пространства
Укр—объемная доля кристаллов в процессе кристаллизации
V—скорость 1еплового потока
Va, Vl — объемы структурных элементов или вакансий, участвующих
в вязком течении
О/, Vtr — скорости распространения продольных и поперечных звуковых
волн
НУ — кулоновское притяжение между положительно и отрицательно
заряженными дефектными центрами
W D —колебание потенциальной энергии поляронных состояний в не-
кристаллических твердых телах
X ~~ электроотрицательность
X, Xit у — содержание в мольных долях
Хе — содержание вакансий в мольных долях
Z—сумма по состояниям
Z — заряд иона
20 — число структурных элементов на поверхности критических за-
родышей
Zt — координационное число
Принятые обозначения
15
а — коэффициент поглощения; силовая постоянная упругой дефор-
мации кристаллической решетки; коэффициент конденсации
по Кнудсену; угловая деформация тела при упругом напря-
жении сдвига; коэффициент термического расширения; коэф-
фициент линейного термического расширения стекла
арасп — коэффициент линейного термического расширения расплава
Да — изменение коэффициента термического расширения при пере-
ходе стекло—расплав
Р — поляризуемость кристаллической решетки; постоянная Пула—
Френкеля; квантовый выход
у— температурный коэффициент изменения расстояния между
краями зон или краями подвижности
у' — коэффициент перекрывания в теории свободного объема
Д — параметр асимметричности расщепления связывающих и анти-
связывающих молекулярных орбиталей
fgd — внутреннее трение; диэлектрические потери
е — диэлектрическая проницаемость е0Сг
Со — диэлектрическая проницаемость вакуума
ег — относительная диэлектрическая проницаемость
е*—комплексная диэлектрическая проницаемость
е7 — действительная часть диэлектрической проницаемости
Е," — мнимая часть диэлектрической проницаемости
es —статическая диэлектрическая проницаемость
Соо — оптическая диэлектрическая проницаемость
Ew —диэлектрическая проницаемость ниже области частот элек-
тронной и атомной поляризации
Т] — динамическая вязкость; коэффициент внутреннего трения
— к. п. д. солнечных элементов
— температура Дебая
0 — деформация; краевой угол; угол брэгговского отражения
х — адиабатическая сжимаемость
Дх— изменение сжимаемости при переходе стекло—расплав
X — логарифмический декремент; длина волны
р, — коэффициент Пуассона; подвижность
Цд — дрейфовая подвижность
[Хм — холловская подвижность
p,z — химический потенциал
р,п> Цр —подвижность электронов и дырок
V — частота
V — волновое число
— число Аббе
— частота присоединения структурных элементов к поверхности
критических зародышей
yw — скорость присоединения структурных элементов при росте
кристаллов
£ — обратная величина радиуса экранирования
р — плотность
16 Принятые обозначения
р (г) — функция распределения плотности
а — удельная электрическая проводимость
OD — электрическая проводимость, рассчитанная из значений коэф-
фициента диффузии
— фотопроводимость
Т — время релаксации; среднее время жизни
Т/ — индукционный период при нестационарном зародышеобразо-
вании
тя, р — время релаксации фотопроводимости
— время релаксации проводимости
ф (г) — межмолекулярный потенциал
ф(г) — волновая функция Блоха
СО — круговая частота
«Искусство изготовления стекла — одно из самых
прекрасных и благородных видов искусств; и те чу-
деса, которые хранит оно в себе, и те краски, кото-
рые с его помощью можно получить, оказываются
настолько красивы и редки, что они будят в нас
желание постичь основы этого искусства и открыть
все его самые сокровенные тайны.»
Ходискье де Бланкур
1. Введение
Учение об элементах Аристотеля — земле, воде, воздухе и
огне,— которое возникло на базе древнегреческой натурфилосо-
фии, до сих пор можно обнаружить в подходе к общей клас-
сификации веществ по их агрегатному состоянию. Наряду
с твердым, жидким и газообразным состояниями современная
наука выделяет еще четвертое состояние вещества — плазму.
Четыре состояния вещества как бы соответствуют системе Ари-
стотеля.
По результатам структурных исследований, проведенных
в XX в., можно предложить другой подход при рассмотрении
вещества, который лишь в незначительной степени связан с его
внешним видом. В основе такого подхода лежит отделение кри-
сталлических твердых тел с преимущественно упорядоченной
структурой от веществ с разупорядоченпым строением.
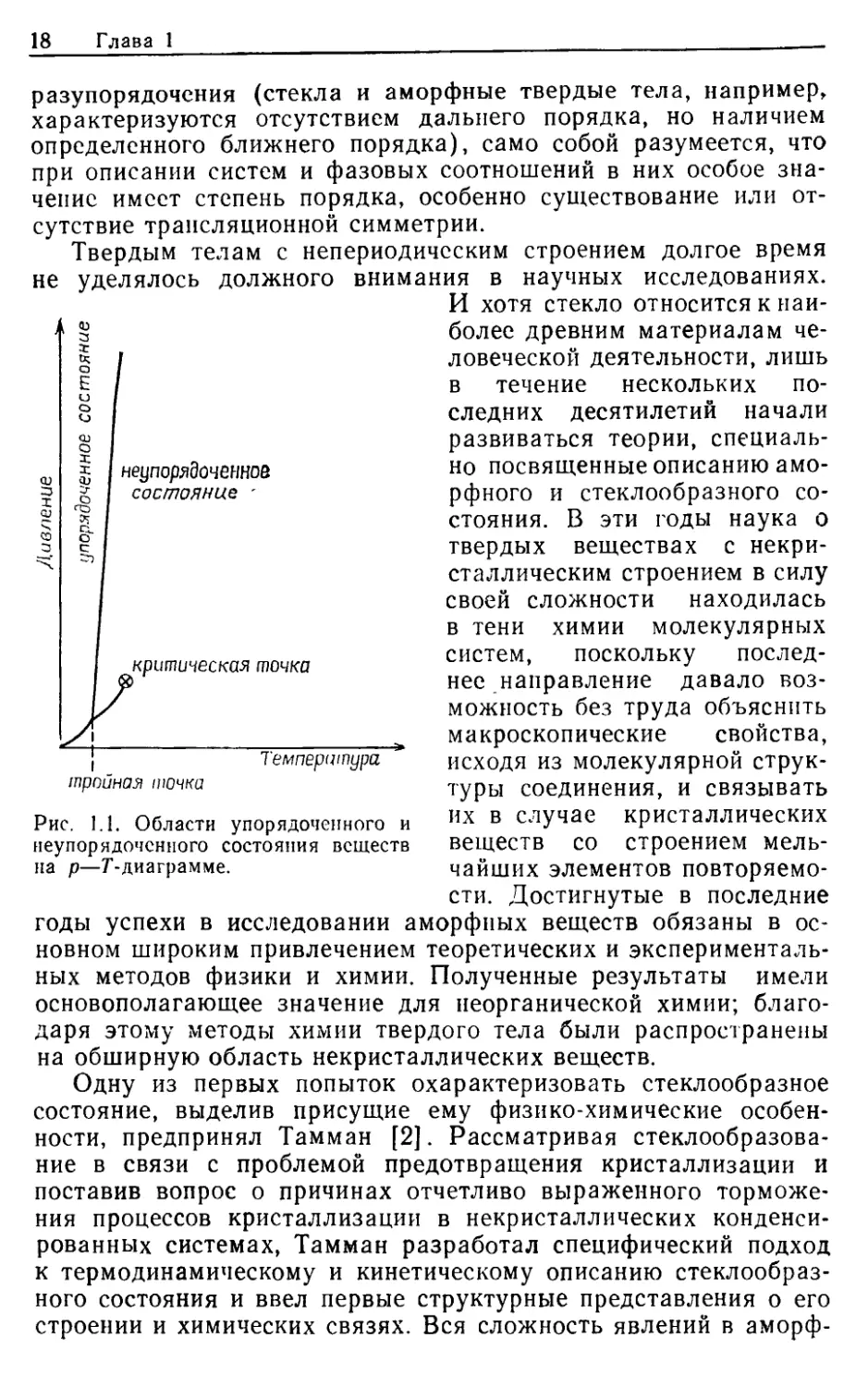
Обратим внимание на рис. 1.1, на котором приведена диа-
грамма температура—давление для простого вещества или чи-
стого соединения в области давлений от 0 до 40 МПа и в обла-
сти температур до нескольких тысяч кельвинов. Очевидно, что
поле веществ с разупорядоченными структурами заметно пре-
восходит поле кристаллических фаз. Аморфные твердые тела и
стекла при повышении давления и температуры непрерывно пе-
реходят в расплавленное состояние (такой переход возможен,
если в достаточной степени замедлен процесс спонтанной кри-
сталлизации), а расплавы — выше критической температуры —
в газообразное состояние. В то же время перейти границу
между упорядоченным кристаллическим состоянием и одним из
неупорядоченных агрегатных состояний практически невоз-
можно без дискретного изменения определенных параметров со-
стояния, например объема, энтальпии или энтропии.
На это различие еще раз недавно указал Гинье [1]. По-
скольку реальные конденсированные системы попадают в об-
ласть между состоянием идеального упорядочения и полного
2 Заказ Хе 413
18 Глава 1
разупорядочсния (стекла и аморфные твердые тела, например,
характеризуются отсутствием дальнего порядка, но наличием
определенного ближнего порядка), само собой разумеется, что
при описании систем и фазовых соотношений в них особое зна-
чение имеет степень порядка, особенно существование или от-
сутствие трансляционной симметрии.
Твердым телам с непериодическим строением долгое время
не уделялось должного внимания в научных исследованиях.
Рис. 1.1. Области упорядоченного и
неупорядоченного состояния веществ
на р—Т-диаграмме.
И хотя стекло относится к наи-
более древним материалам че-
ловеческой деятельности, лишь
в течение нескольких по-
следних десятилетий начали
развиваться теории, специаль-
но посвященные описанию амо-
рфного и стеклообразного со-
стояния. В эти годы наука о
твердых веществах с некри-
сталлическим строением в силу
своей сложности находилась
в тени химии молекулярных
систем, поскольку послед-
нее направление давало воз-
можность без труда объяснить
макроскопические свойства,
исходя из молекулярной струк-
туры соединения, и связывать
их в случае кристаллических
веществ со строением мель-
чайших элементов повторяемо-
сти. Достигнутые в последние
годы успехи в исследовании аморфных веществ обязаны в ос-
новном широким привлечением теоретических и эксперименталь-
ных методов физики и химии. Полученные результаты имели
основополагающее значение для неорганической химии; благо-
даря этому методы химии твердого тела были распространены
на обширную область некристаллических веществ.
Одну из первых попыток охарактеризовать стеклообразное
состояние, выделив присущие ему физико-химические особен-
ности, предпринял Тамман [2]. Рассматривая стеклообразова-
ние в связи с проблемой предотвращения кристаллизации и
поставив вопрос о причинах отчетливо выраженного торможе-
ния процессов кристаллизации в некристаллических конденси-
рованных системах, Тамман разработал специфический подход
к термодинамическому и кинетическому описанию стеклообраз-
ного состояния и ввел первые структурные представления о его
строении и химических связях. Вся сложность явлений в аморф-
Введение
19
ных веществах в их полном объеме стала ясной после обнару-
жения микрогетерогенной структуры стекла во многих стекло-
образующих системах, имеющих практическое значение.
В разработке основ химии и физики стекла приняли участие
многие ученые. Результаты исследований, касающихся прежде
всего свойств стекла и попыток их интерпретации, описаны
в многочисленных работах, например Стевелса [3, 4], Стен-
ворта [5], Морея [6], Эйтеля [7], Роусона [8], Вейла и Марбо
[9], Бартенева [10], Джонса [11], Дайя [12], Голловея [13],
Доремуса [14], Мазурина [15], а также в недавних работах
Шольца [16] и Фогеля [17].
В монографиях, посвященных стеклам, налицо, с одной сто-
роны, тесная связь этого направления развития науки с прак-
тическими задачами производства стекла. При этом отчетливо
прослеживается стремление прогнозировать различные свойства
синтезируемых стекол; например, для данного состава смеси
рекомендуются определенные условия плавления. С другой сто-
роны, бросается в глаза преимущественно описательный харак-
тер изложения. Приводимые соотношения носят в основном эм-
пирический характер. Сказанное в значительной мере относится
к привлекаемым представлениям из теории химической связи,
которые излагаются на довольно низком уровне (например,
[18, 19]) без убедительного теоретического обоснования.
Заметный толчок к развитию науки о стекле дало сущест-
венное расширение круга стеклообразующих систем и аморфных
твердых тел, начавшееся два с половиной десятилетия назад.
Коломиец и Горюнова [20] в 1955 г. обнаружили, что не-
оксидные стекла, в частности халькогенидные стекла с преиму-
щественно ковалентными связями, характеризуются электрон-
ной проводимостью. Позже эта же особенность замечена Мюл-
лером [21], Борисовой [22] и Дембовским [23] при исследова-
нии многих систем. Хилтон [24] указал на применение таких
стекол в качестве прозрачной среды для ИК-излучения. При-
мерно в это же время Стенворт и сотр. [25] сообщили об об-
наружении электронной проводимости оксидных стекол, кото-
рые содержат катионы переходных металлов в различных
степенях окисления. Электронная проводимость стекол, в основ-
ном с полярными химическими связями, отмечена также в ра-
ботах Маккензи [26]. Синтез стекол с металлическими свойст-
вами и их изучение выделились в самостоятельную область со-
временного материаловедения. Впервые об изготовлении
тонких пленок различных металлических сплавов сообщили
в 1960 г. Дувез и сотр. [27].
Из многообразия аморфных и стеклообразных твердых тел,
свойства которых определяются совершенно разной структурой
и различными химическими связями, со всей очевидностью вы-
текает необходимость разработки теории химической связи
2*
20 Глава 1
в стеклообразующих системах, основанной на квантовохимиче-
ском подходе. Толчком для развития теоретических представ-
лений для стеклообразного состояния послужила выдвинутая
Моттом проблема о границах применимости зонной модели
кристаллического твердого тела. Иоффе и Регель [28] ука-
зали граничное условие для некристаллических, аморфных и
жидких полупроводников. Займан [29] в 1961 г. объяснил ме-
таллическую проводимость расплавленных металлов. Примерно
в это же время были проведены первые физические исследова-
ния аморфного германия. Здесь прежде всего необходимо упо-
мянуть заслуги Таука [30] и Григоровичи [31]. В 1965 г.
в Праге состоялась первая международная конференция по
аморфным и жидким полупроводникам. Вторая такая конферен-
ция была проведена двумя годами позже в Бухаресте. В даль-
нейшем физические исследования некристаллических твердых
тел стимулировались использованием аморфного и стеклообраз-
ного селена в электрофотографии. Пристальное внимание при-
обрело изучение бистабильных проводящих состояний аморф-
ных халькогенидных слоев; прогнозируется применение эффекта
переключения Овшинского [32] для работы конкретных техни-
ческих устройств.
Результаты применения методов физики твердого тела
(вместе с необходимым математическим аппаратом) к аморф-
ным системам изложены во многих обзорных трудах [33—35].
Насущные проблемы выразительно изложены в статьях Мотта
[36, 37].
В предлагаемой читателю книге сделана попытка дать все-
сторонний обзор наиболее существенных представлений о строе-
нии и свойствах аморфных веществ. При этом автор стремился
составить по возможности единое представление о состоянии
теории неорганических аморфных веществ и стекол. Изложение
материала в книге проводится с позиций дедуктивного подхода.
Прежде всего приводятся сведения об образовании и получении
аморфных твердых тел и стекол, а затем уже излагается фено-
менологическая термодинамическая теория, описывается кине-
тика процесса стеклообразования, а также дается структурная
интерпретация стеклообразного состояния и затрагиваются
проблемы химической связи в аморфных фазах. При теорети-
ческом рассмотрении привлекаются конкретные системы, тем
самым обеспечивается внутренняя целостность и законченность
изложения.
Большое место в книге отведено описанию стеклообразую-
щих неорганических систем; не включен материал, содержа-
щийся в монографиях по химии стекла и относящийся к техниче-
ски важным стеклообразующим системам. Структуры твердых
тел с непериодическим строением обсуждаются на основе
периодической системы элементов. Из сопоставления различных
Введение
21
кристаллических и аморфных (или стеклообразных) модифика-
ций элементов и их соединений следует, что наши знания об
основных неорганических веществах в конденсированном со-
стоянии могут быть значительно расширены при изучении
структуры жидкости, замороженной в стеклообразном состоя-
нии. Поэтому вопрос о структурных причинах и особенностях
химических связей, вызывающих у некоторых элементов и
многих бинарных соединений ярко выраженную склонность
к стеклообразованию, вырастает в принципиальный. Правиль-
ное решение этой проблемы позволит описать свойства тройных
и некоторых четверных стеклообразующих систем. В связи
с этим автор старался по возможности полнее описать извест-
ные корреляции структуры и свойств в стеклообразующих си-
стемах, а также определить цели дальнейших исследований и
их направления.
До сих пор в литературе по химии стекла доминировали эм-
пирические соотношения между содержанием определенного
компонента и интересующим свойством стекол. Именно таковы
большинство работ, описывающих оптические свойства стекол,
которым посвящена особая глава книги (где также обсужда-
ются явления переноса заряда в аморфных веществах). Про-
блема локализации и делокализации электронных состояний
в аморфных и стеклообразных веществах вызывает наибольший
интерес у исследователей; в этом направлении в последние
годы наблюдаются заметные успехи, в частности развиты новые
представления о химии конденсированного состояния. Известно,
что критерии локализации и делокализации электронных состоя-
ний имеют основополагающее значение для теоретического опи-
сания химических связей в молекулярных системах. Рассмотре-
ние различных свойств, вызванных структурными особенностями
молекул, привычно для каждого химика. В то же время анало-
гичные корреляции в макромолекулярных твердых веществах до
сих пор составляют предмет физики твердого тела. Пришло
время использовать и в химии твердого тела те преимущества,
которые открывает применение зонной модели для интерпрета-
ции экспериментальных данных.
Книга заканчивается кратким обзором некоторых нетради-
ционных областей применения стекол и аморфных слоев.
Литература
1. Guinier A., Proc. 5th Int. Symp. High Purity Mat. in Science and Tech-
nology, Dresden, 1980, p. 55.
2. Tammann G., Der Glaszustand, Verl. L. Voss, Leipzig, 1933.
3. Stevels J. M., The physical properties of glases, Rec. Trev. Chim. Pays-Bas,
60, 85, 62, 17 (1943).
4. Stevels J. M., The structure and the physical properties of glass, in
S. Fliigge, Handbuch der Physik, Bd. XIII, Springer-Verlag, Berlin, 1962,
S. 510.
22 Глава 1
5. Stanworth J. E., Physical properties of glass, Oxford, Clarendon Press,
1950.
6. Morey G. W., The properties of glass, Reinhold, New York, 1954.
7. Eitel W., Silicate Science, Academic Press, New York, 1965, 1976.
8. Rawson H., Inorganic glass-forming systems, Academic Press, New York,
1967.
9. Weyl W. A., The constitution of glasses. A dynamic interpretation, Inter-
science Publishers, New York, 1962—1967.
10. Bartenev G. M., The structure and mechanical properties of inorganic glas-
ses, Wolters-Nordhoff Publ., Groningen, 1970.
11. Jones G. O., Glass, Chapman and Hall Ltd., London, 1971.
12. Introduction to glass science, Pye L. D., Stevens H. J., La Course W. C.
(Eds.), Plenum Press, New York, 1972.
13. Holloway D. G., The physical properties of glass, Wykeham Publications
Ltd., London, 1973.
14. Doremus R. H., Glass science, John Wiley and Sons, New York, London,
Sidney, Toronto, 1973.
15. Мазурин О. В., Стрельцина М. В., Швайко-Швайковская Т. П. Свойства
стекол и стеклообразующих расплавов.— Л.: Наука, т. 1, 1973, т. 2, 1975.
16. Scholze Н., Gias, Natur, Structur und Eigenschaften, Springer-Verlag, Ber-
lin, Heidelberg, New York, 1977.
17. Vogel W., Glaschemie, VEB Verlag Deutscher Verlag fur Grundstoffindust-
rie, Leipzig, 1979.
18. Wondratschek H., Strukturchcmie des Glases, Glashdtten-Handbuch, 40, 1
(1965).
19. Scholze H., Stand des Wissens uber Struktur und Geftige von Gias, Glas-
techn. Ber., 42, 36 (1969).
20. Kolomlec В. T., Vitreous Semiconductors, Phys. Status Solidi, 7, 359
(1964).
21. Мюллер P. Л., Химия твердого тела и стеклообразного состояния.—Л.:
ЛГУ, 1965, с. 9.
22. Борисова С. У. Химия стеклообразных полупроводников.— Л.: ЛГУ, 1972.
23. Дембовский С. А.— Ж. физ. хим. стекла, 1978, т. 4, с. 522.
24. Hilton A. R., J. Non-Cryst. Solids, 2, 26 (1970).
25. Denton Е. Р., Rawson Н., Stanworth J. Е., Nature (London), 172, 1030
(1954).
26. Mackenzie J. D., Semiconducting Oxide Glasses, Mod. Aspects of the Vi-
tieous State, 3, 126 (1965).
27. Klement W., Willens R. H., Duwez D., Nature (London), 187, 869 (1960).
28. Joffe A. F., Regel A. R., Prog. Semicond., 4, 237 (1960).
29. Ziman J. M., Phil. Mag., 6, 1013 (1961); Ziman J. M., Prinzipien der Fest-
korpertheorie, Akademie-Verlag, Berlin, 1974.
30. Tauc J.. Abraham A., Pajasova L„ Grigorovici R., Vancu A., Proc. Conf.
Phys. Non-Cryst. Solids, North-Holland, Delft, 1964, p. 606.
31. Grigorovici R.. Croitoru N., Devenyi A., Teleman E., Proc. Int. Conf. Semi-
cond. Phys., Paris, 1964, p. 423; Phys. Status Solidi, 16, K143 (1966).
32. Ovshinsky S. R., Symmetricel Current Controlling Device, пат. CHIA,
3 271 581* (1966).
33. Mott N, F„ Davis E. A., Electronic Processes in Non-Crystalline Materials,
Clarendon Press, Oxford, 1971, Second Edition, Oxford, 1979.
34. Electronic and Structural Properties of Amorphous Semiconductors, LeCom-
ber P. G., Mort J. (Eds.), Akademic Press, London, 1973.
35. Аморфные полупроводники: Пер. с англ./Под ред. М. Бродски.— М.: Мир,
1982.
36. Miott N. F., Electrons in Glass, Rev. Modern Phys., 50, 203 (1978).
37. Mott N. F., The origin of some ideas on non-crystalline materials, J. Non-
Cryst. Solids, 28, 147 (1978).
2. Аморфное и стеклообразное
состояние неорганических
веществ
«Аморфными» в смысле «бесформенными» или «принимаю-
щими любую форму» называют такие вещества, которые ха-
рактеризуются следующими особенностями: 1) отсутствием за-
висимости свойств от направления; 2) возникновением при
разломе и расколе поверхностей произвольной формы, чаще
всего раковистого излома; 3) отсутствием кристаллических об-
ластей как в компактном, так и в дисперсном состоянии.
Аморфные вещества, как газы и жидкости, изотропны. При их
характеристике прежде всего следует отмечать, что это некри-
сталлические вещества.
С помощью методов дифракции рентгеновского излучения,
нейтронов и электронов можно строго разделить так называе-
мые «рентгеноаморфные» и кристаллические вещества; при
этом понятно, что между этими состояниями вещества сущест-
вует непрерывный переход. Чем меньше области с периодиче-
ским строением, тем сильнее идет рассеяние на множестве пло-
скостей кристаллической решетки. Вместо острых дискретных
дифракционных максимумов на дифрактограммах кристалли-
ческих веществ наблюдаются только размытые интерференцион-
ные кольца, что указывает на нестатистический характер рас-
пределения величин межатомных расстояний; следовательно,
и для аморфного состояния можно говорить о существовании
определенной степени порядка. При представлении результатов
дифракционных экспериментов в виде зависимостей интенсив-
ности полос на дифрактограммах от угла рассеяния использу-
ются функции распределения плотности и функции парного рас-
пределения, которые позволяют делать более определенные вы-
воды о наличии порядка в стекле.
Путем определения равновесных положений, которые зани-
мают химически связанные атомы, установлено, что расстояния
между соответствующими парами атомов в аморфных веществах
статистически имеют такую же величину, что и в кристалличе-
ских соединениях, причем значения, меньшие этой величины,
практически не встречаются. Отсюда следует, что аморфные
вещества, как и кристаллические, характеризуются наличием
областей с ближним порядком. Эти области часто соответ-
24 Глава 2
ствуют структурным полиэдрам кристаллических веществ или
по крайней мере находятся по отношению к ним в структурном
родстве. Направленный характер химических связей позволяет
ожидать, что ближний порядок существует в непосредственном
окружении любого атома (во 2-й и 3-й координационных сфе-
рах), хотя бы в ослабленной форме. С увеличением расстояния
быстро возрастает многообразие структурных конфигураций
из-за изменения длины химических связей, и прежде всего ва-
лентных углов, что приводит к изменению взаимной ориентации
структурных элементов в результате частичного поворота во-
круг осей вдоль химических связей. Таким образом, как бы
осуществляется переход в континуум парных межатомных рас-
стояний. В связи с отмеченными особенностями аморфных тел,
отличающих их от кристаллических, для аморфного состояния
отсутствует дальний порядок. Понятия «аморфный» и «некри-
сталлический» описывают одно и то же состояние вещества и
используются в одинаковом смысле.
Стекла представляют собой некристаллические или аморф-
ные вещества. Понятие стеклообразное вещество применяют
к твердым телам, которые либо получены из расплавов, либо
другими методами, причем в последнем случае образуются ком-
пактные агрегаты в форме тонких пленок, нитей или волокон,
характеризующиеся макроскопическими размерами, по крайней
мере в двух или в одном направлении, и температурным диа-
пазоном размягчения.
Стеклам, как и кристаллам, присущи такие свойства, как
твердость и способность сохранить форму.
В стекловарении, имеющем более чем четырехтысячелетнюю
историю, при получении стекол для различных целей полностью
оправдали себя высокотемпературные методы: смеси неоргани-
ческих веществ путем нагревания переводят в расплав, который
далее в процессе охлаждения затвердевает. Этим объясняется
выделение в определении стеклообразного состояния вещества
способа получения таких веществ из расплава. Опираясь на
определение, данное Американским обществом по исследова-
нию материалов, в 1971 г. была принята следующая формули-
ровка [1]: «Стекло — это неорганический продукт плавления,
который в основном затвердевает без кристаллизации».
Никоим образом не отрицая стеклообразного характера
очень многих органических веществ, имеющих важные сферы
технического применения, невозможно не отметить, что за два
прошедших десятилетия появилось еще много новых методов
получения аморфных и стеклообразных модификаций простых
веществ и соединений не из расплава. Благодаря этим успехам
удалось существенно расширить сами возможности реализации
этих состояний, что потребовало заметного развития соответ-
ствующих теоретических представлений.
Аморфное и стеклообразное состояние 25
В частности, широко изучены аморфные и стеклообразные
тонкие слои. При этом установлено, что отличия от компактных
стекол часто заключаются лишь в различном соотношении объ-
емных и поверхностных свойств и что резкое разграничение
между аморфными пленками и компактными стеклами никоим
образом не обоснованно.
Точка зрения, принятая в данной книге, соответствует боль-
шинству последних публикаций: целесообразно отнести к стек-
лообразным аморфные вещества (независимо от способа их по-
лучения), характеризующиеся наличием типичного для стекол
температурного интервала размягчения.
В более ранней литературе, например при описании некото-
рых модификаций фосфора, мышьяка и селена, понятия
«аморфный» и «стеклообразный» имели один смысл. Этому не
противоречит утверждение, что понятие «аморфное твердое со-
стояние» вещества шире по сравнению с понятием «стеклооб-
разное состояние». Стекла всегда аморфны, но не все аморф-
ные вещества — стекла. Высокодисперсные порошки, например
многие продукты осаждения из водных растворов (гидроксиды,
сульфиды, селениды), промежуточные продукты некоторых
твердофазных реакций, а также термически неустойчивые гели
могут быть аморфными, но их, безусловно, нельзя назвать
стеклами.
2.1. Методы получения аморфных твердых тел
и стекол
Аморфное и стеклообразное вещество обладает по сравне-
нию с кристаллической фазой того же брутто-состава более
высоким запасом энергии. После плавления кристаллического
твердого тела области, характеризующиеся наличием ближнего
порядка, продолжают существовать в качестве структурных мо-
тивов расплава. Однако часто при переходе в жидкое состояние
происходят структурные изменения уже в 1-й координационной
сфере. При растворении, а также при более высоких темпера-
турах, прежде всего в газовой фазе, в результате разложения
образуются, как правило, более мелкие структурные фрагменты.
Дальний порядок исчезает уже при плавлении.
Аморфные (стеклообразные) твердые вещества можно полу-
чить из жидкого агрегатного состояния (из расплавов или рас-
творов) или из газовой фазы в том случае, если удастся вос-
препятствовать процессам зародышеобразования и кристаллиза-
ции. При этом более высокая свободная энергия расплава, рас-
твора или газовой фазы, по крайней мере частично, сохраняется
и при затвердевании. В этом смысле говорят о процессе замо-
раживания. В то же время и кристаллические твердые тела
26 Глава 2
Рис. 2.1. Способы получения стеклообразных твердых тел из расплавов или
растворов, газовой фазы, а также из кристаллического состояния (по Оуэну).
при подводе к ним энергии могут быть переведены в аморфное
(стеклообразное) состояние, минуя жидкую и газовую фазы.
Секрист и Маккензи опубликовали обзор необычных методов по-
лучения некристаллических твердых тел [2]. Для наглядности
различные варианты синтеза в соответствии с предложением
Роя [3] и Оуэна можно систематизировать (рис. 2.1).
Стекла могут быть получены путем достаточно быстрого ох-
лаждения расплавов (/) или из растворов путем высушивания
гелей (2). Процессы осаждения из растворов часто ведут к об-
разованию аморфных осадков (5), как, например, в случае
красного аморфного селена, сульфидов мышьяка, сурьмы, гер-
мания, олова и многих других металлов или оксидов металлов.
Электролитическим осаждением при высоких плотностях тока
(4) получают аморфные слои, например, аморфного германия.
Таким путем впервые получены металлические стекла. Так на-
зываемая «взрывчатая» сурьма получена также при электроли-
тическом осаждении из растворов. Если исходить из газовой
фазы, то необходимо прежде всего назвать различные варианты
термического испарения и конденсации в высоком вакууме (5),
а также катодное распыление (6) и осаждение аморфных слоев
Аморфное и стеклообразное состояние 27
в тлеющем разряде (7). В некоторых специальных случаях
практическое значение имеет химическое осаждение из газовой
фазы (S); например, при образовании кварцевого стекла или
аморфных слоев SiO2 используется гидролиз SiCU или пиролиз
смесей SiCh/Os. Пленки кварцевого стекла или слои SiOx
также можно получить путем непосредственного окисления по-
верхности монокристаллов кремния (9). Кристаллические твер-
дые тела переходят в аморфное состояние под действием удар-
ной волны (10) или интенсивного нейтронного или ионного
облучения (11). Аморфные продукты часто образуются в резуль-
тате химического разложения веществ в твердой фазе (12), на-
пример при обезвоживании каолинита. Необходимо отметить,
что и при механической обработке кристаллических твердых
тел, например путем шлифования и полирования, часто возни-
кают сильно разупорядоченные аморфные поверхностные слон,
что необходимо учитывать при внедрении технологических про-
цессов в производство.
В рамках настоящей книги мы ограничимся в основном та-
кими стеклообразными и аморфными твердыми фазами, кото-
рые могут быть получены в компактной форме или в форме
тонких слоев толщиной до нескольких микрометров и состав
которых одинаков по всей толщине образца, а структура по
меньшей мере близка к гомогенной и изотропной, так что су-
ществует возможность проводить корреляции между структурой
и свойствами. Прерывистые стрелки (рис. 2.1) указывают на
возможность перехода аморфных слоев и пленок путем после-
дующего отжига в компактную форму, близкую к стеклам, по-
лучаемым из расплава.
2.1.1. Получение стекол из расплава
Задача состоит в том, чтобы в процессе охлаждения сохра-
нить гомогенное и изотропное состояние расплава, т. е. вос-
препятствовать процессам кристаллизации и зародышеобразо-
вания. На практике охлаждение стеклообразующих расплавов
часто сочетается с процессом придания формы стеклу. В обла-
сти температур ниже линии ликвидуса максимальные скорости
образования зародышей и скорости роста кристаллов прихо-
дятся на разные температуры (рис. 2.25). Таким образом, для
образования аморфной твердой фазы скорость охлаждения
должна быть настолько велика, чтобы не допустить перекры-
вания обеих кривых (имеется в виду зависимость скорости от
температуры) в критической области (т. е. там, где конкури-
руют кристаллизация и образование аморфной фазы). По-
скольку процессы зародышеобразования и кристаллизации носят
статистический характер, необходимо введение дополнительных
условий.
28 Глава 2
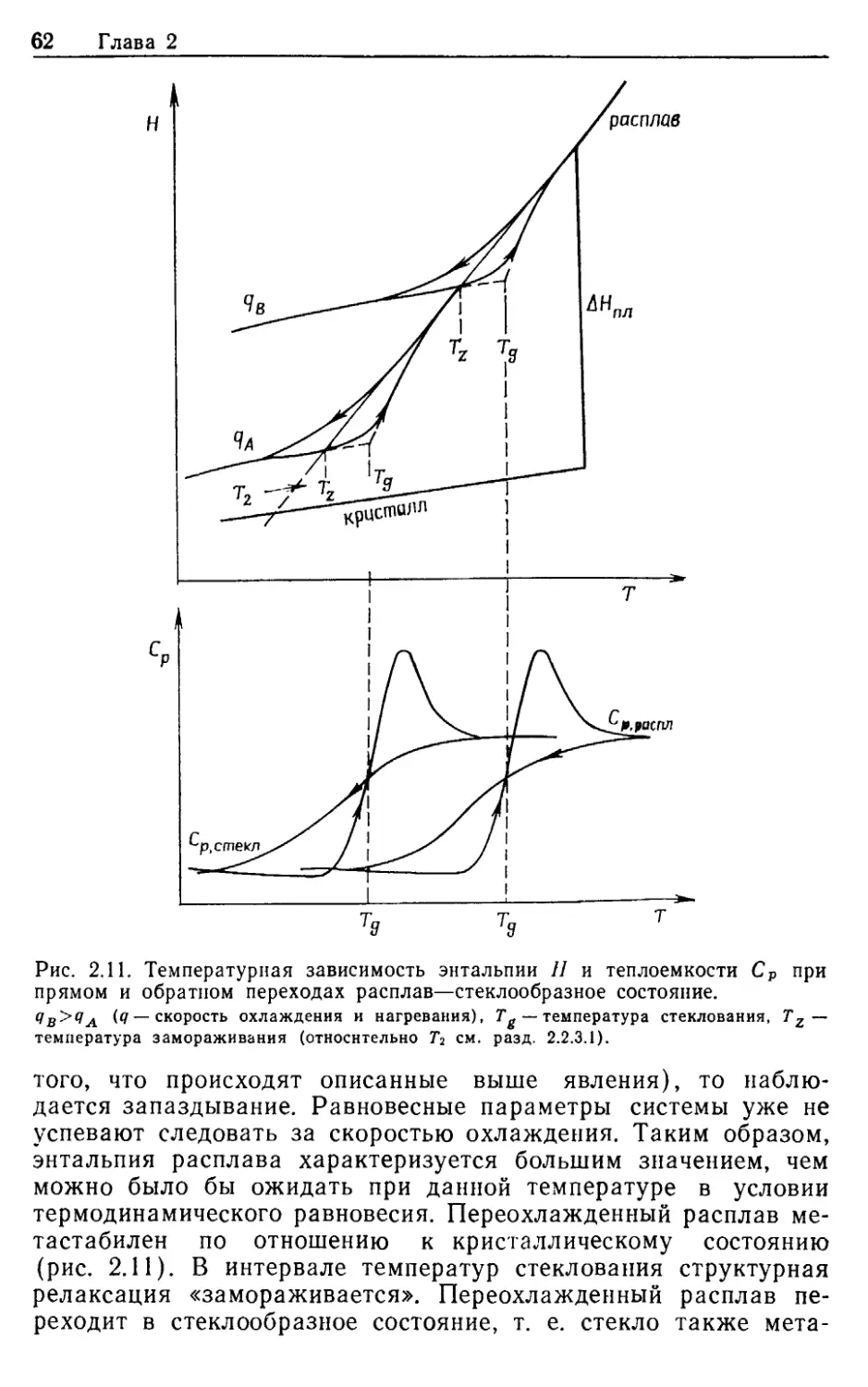
Тернбал [4] в качестве условия для стеклообразования на-
зывает полное отсутствие центров кристаллизации. Критическая
скорость охлаждения в этом случае зависит от количества рас-
плавленной исходной смеси. В других работах [5] стекло оп-
ределяется как продукт плавления, в котором концентрация
центров кристаллизации не превышает одного центра на 1 см3.
Ульманом [6] в качестве критического предела предложена
объемная доля центров кристаллизации, равная 10-6. Таким
образом, стеклообразование из расплава требует таких усло-
вий, которые позволяют замедлить скорость возникновения за-
родышей и скорость роста кристаллов. Опытным путем уста-
новлено, что склонность к возникновению стеклообразного со-
стояния при затвердевании расплавов проявляется тем сильнее,
чем выше вязкость жидкой фазы вблизи температур ликвидуса
и чем резче вязкость возрастает с понижением температуры.
В области температур стеклования осуществляется переход от
пластичного бесформенного переохлажденного расплава в твер-
дое стеклообразное состояние, характеризующееся упругим со-
хранением формы. В термодинамическом смысле уже переох-
лажденный расплав мстастабилен по отношению к кристалли-
ческому состоянию. В стекле же метастабильное состояние
переохлажденного расплава «замораживается».
2.1.1.1. Оксидные стекла
Стекла, нашедшие практическое применение в качестве упа-
ковочных и строительных материалов, в быту, электротехнике
и оптике, производятся в большом ассортименте на крупных
предприятиях путем обработки расплавов, в основном на воз-
духе. Технологические приемы достигли высокого уровня раз-
вития и подробно описаны в монографиях и учебниках по тех-
нологии стекла [7—9].
В научных лабораториях при получении стекол применяют
различного типа печи с прямым и косвенным нагревом. Часто
возникающие экспериментальные затруднения связаны с выбо-
ром подходящего материала для тиглей, особенно если хими-
ческая устойчивость к расплаву таких традиционно исполь-
зуемых в этих целях металлов, как платина, уже недостаточна.
Кроме того, некоторые проблемы обусловлены летучестью от-
дельных компонентов расплава, что приводит к продуктам
с концентрационным градиентом. Надежное достижение равно-
весия в расплаве, например путем удаления определенных ле-
тучих компонентов смеси (рафинирование), имеет существен-
ное значение для воспроизводимого получения гомогенных сте-
кол. Необходимо также обращать внимание на возможное
включение легирующих компонентов в матрицу стекла из-за
частичного растворения в ней материала тигля или внешних
загрязнений (из окружающего пространства).
Аморфное и стеклообразное состояние
29
Для важнейших стсклообразующих оксидов и BeF2
в табл. 2.1 приведены некоторые необходимые характеристики.
Таблица 2.1. Некоторые свойства стеклообразующих оксидов и BeF2
Вещество Температура плавления Г1Л> К II Температура стеклования к Энтальпия плавления Д"пл- кДж/моль Вязкость Г] п₽и Гпл> дПа • с
SiO2 1995 1495 8 ю7
GeO2 1389 853 17,2 7 • 105
В20з 723 550 23 105
р205 853 537 71 5 • 10е
As2O3 585 420 18,7 106
BeF2 823 523 41,2 >106
Комбинация различных стеклообразующих веществ, а также
добавление одного или нескольких оксидных компонентов
к стеклообразующей матрице привели к большому набору сте-
кол, имеющих практическое значение. Поскольку в стеклооб-
разующий расплав могут быть введены не только оксиды ще-
лочных и щелочноземельных элементов, но и практически все
оксиды переходных металлов, а также оксиды элементов глав-
ных подгрупп периодической системы (А120з, Оа2Оз, Sb2Os,
Т12О, PbO, Bi2O3, ТеО2) и даже сульфаты, хлориды и многие
фториды, число возможных комбинаций чрезвычайно велико,
и в настоящее время, как и в дальнейшем, создаются и будут
создаваться новые стекла. По-видимому, эта обширная область
исследований требует отдельного рассмотрения. На соответст-
вующие работы указывалось во введении (см. литературу
в гл. 1 [1—5]).
В 1954 г. Роусон, Стенворт и др. (см. литературу к гл. 1
[25] и гл. 3 [П]) впервые сообщили об оксидных стеклах, об-
ладающих электронной проводимостью. Такие стекла содержат
катионы переходных металлов в средних степенях окисления;
например, наряду с Ti(IV) имеются ионы Ti(III) или наряду
с V(V)—большое или даже преобладающее количество V(IV).
Расплавы такого сорта крайне склонны к окислению. Их при-
готовление требует тщательного удаления влаги, кислорода воз-
духа, СО2 и других примесей, действующих как окислители и
восстановители.
На рис. 2.2 приведена схема кварцевого реактора (труба
длиной 600 мм и шириной 70 мм). Через верхнюю 2 и нижнюю
10 пришлифованные крышки в реактор введены устройства для
перемешивания. Шлифы прибора смазывают вакуумной смаз-
30 Глава 2
Рис. 2.2. Реактор для плавления склонных к окислению оксидных стекол [12].
1 — устройство для перемешивания, 2 — верхняя пришлифованная стеклянная крышка,
3—керамическая трубка, 4—платиновая крышка, 5 — керамическая крышка, 6 — кера-
мический держатель, 7 — керамический стержень, 8 — кварцевый цилиндр, 9 — латунный
стержень, 10 — латунный конус, 11 — стальной стержень.
кой; прибор проверяют на герметичность и после этого запол-
няют чистым аргоном. Во избежании термического разложе-
ния вакуумной смазки в шлифах предусмотрено водяное охла-
ждение. С помощью устройства 1 расплав можно перемешивать
через отверстие в крышке тигля, а также поднимать крышку
тигля перед разливкой расплава. Платинородиевый тигель
(90% Pt, 10% Rh) на 50 мл нагревается индукционно и фик-
сируется в реакторе двумя, приваренными сбоку цилиндриче-
скими кронштейнами; тигель можно переворачивать. С по-
мощью вводимого снизу стержня 7 тигель можно наклонять и
Аморфное и стеклообразное состояние
31
выливать его содержимое во вдвигаемую и охлаждаемую водой
форму. Таким способом удается воспроизводить условия плав-
ления и отливки склонных к окислению стеклообразующих рас-
плавов, содержащих катионы переходных металлов в средних
степенях окисления.
2.1.1.2. Халькогенидные стекла
В последние два с половиной десятилетия в литературе по-
явилось огромное количество работ, посвященных новым «не-
оксидным» сте'клам. Интерес к этому направлению обнару-
жился после исследований стеклообразного селена, а также
после установления в 50-х годах Фрерихсом [13] того факта,
что стекла на основе сульфида мышьяка оптически прозрачны
в ИК-области; Горюнова и Коломиец [14] (см. также в гл. 1
работу [20]) в 1955 г. при изучении системы Tl2Se-SbsSe3—
Tl2Se-As2Se3 открыли полупроводниковое стекло TlAsSe2. Не-
оксидные (не содержащие кислорода) стекла — это прежде
всего сульфиды, селениды и теллуриды элементов IV и V глав-
ных подгрупп периодической системы (главным образом Si, Ge,
Р, As, Sb) и их комбинации между собой, а также с галоге-
нами, халькогенами и халькогенидами тяжелых металлов (Hg,
Ga, In, Tl, Sn, Pb). По сравнению с оксидными стекла, не со-
держащие кислорода, характеризуются худшими механическими
свойствами и пониженной термической устойчивостью, но в то
же время они имеют более высокие коэффициент термического
расширения, термический коэффициент показателя преломле-
ния и относительную упругооптическую постоянную. При
этом прозрачность для инфракрасных лучей заметно сдвинута
в область более длинных волн (т. е. в дальнюю ИК-область),
что объясняется большими атомными массами и меньшими си-
ловыми постоянными связей [15, 16]. Уменьшение средней энер-
гии связи, которая наблюдается с ростом атомной массы эле-
ментов одной группы периодической системы, приводит к тому,
что по сравнению с оксидами в нсоксидных стеклах сужается
запрещенная зона. Почти все халькогенидные стекла (за не-
сколькими исключениями, например желтое стекло GeS2 и крас-
ное стекло As2S3) поглощают свет во всей видимой области
спектра; внешне они представляют собой непрозрачные мате-
риалы черного цвета. Под действием термического возбужде-
ния носителей электрического заряда у халькогенидных стекол
появляется значительная электрическая проводимость. Напри-
мер, известны стекла с удельной электрической проводимостью
10~3 Ом^'См-1 при 298 К. Таким образом, халькогенидные
стекла образуют вторую группу стеклообразных веществ, ха-
рактеризующихся полупроводниковыми свойствами (оксидные
стекла с электронной проводимостью составляют первую
32 Глава 2
группу). Из-за высокой отражательной способности они имеют
сильный характерный блеск.
Халькогенидные стекла получают в тиглях из графита и
стеклографита или в ампулах из тугоплавких оксидов, напри-
мер из кварца. Сравнительно высокая энергия решетки при
относительно низких температурах плавления халькогенидов
приводит к тому, что взаимодействие с кварцем при работе
в кварцевых ампулах идет в существенно меньшей степени,
чем в случае оксидных расплавов. Заметное давление паров
над халькогенидными расплавами и главное их склонность
к взаимодействию с кислородом при повышенных температурах
делают необходимым проводить работу в герметичных прибо-
рах и в бескислородной атмосфере. Поэтому халькогенидные
стекла получают плавлением соответствующих смесей простых
веществ в запаянных под высоким вакуумом кварцевых ампу-
лах. Для гомогенизации расплава ампулы в печах поворачи-
вают. Кроме того, печи время от времени наклоняют. Быстрое
охлаждение (закалка) расплавов осуществляется путем быст-
рого погружения ампул в холодную жидкость или путем охла-
ждения вне печи на воздухе или в другом температурном ре-
жиме. Из-за относительно высокого коэффициента термиче-
ского расширения образцы имеют предрасположенность
к внутренним напряжениям, поэтому перед их механической об-
работкой необходимо проводить отжиг в области температур
стеклования с последующим медленным охлаждением до ком-
натной температуры.
Таблица 2.2. Некоторые свойства неоксидных стеклообразующих веществ
Вещество Температура плавления Гпл> К Температура стеклования к Энтальпия плавления Д^ПЛ’ кДж/моль Вязкость Г) при Тпл, дПа с
Se 493 305 6,23+0,17 22,1
P4Se4 603 455 — ”—-
AS2S3 592 447 28,7+1,3 ю5,1
As2Se3 648 445 40,8+1,3
As2Te3 685 380 55,8+1,5
OeS2 1123 765
OeSe2 1013 665 40
CdAs2 894 545
CdSiAs2 760
CdOeP2 1073 715
CdOeAs2 943 655
Аморфное и стеклообразное состояние 33
В табл. 2.2 приведены характеристики основных стеклооб-
разующих халькогенидов. Здесь же помещены данные о стекло-
образном селене и некоторых других стеклообразующих соеди-
нениях, которые относятся к группе пниктидов. Видно, что лишь
As2S3 и, пожалуй, еще As2Se3 имеют при температуре плавле-
ния вязкость, сравнимую с вязкостью оксидов (см. табл. 2.1).
Другие соединения в области переохлаждения характеризуются
значительно более высокой тенденцией к кристаллизации.
2.1.1.3. Металлические стекла*
В 1960 г. Клемент, Уилленс и Дувез (см. в гл. 1 работу
[27]) впервые описали стеклообразные металлические сплавы,
полученные быстрым охлаждением расплавов. Применяя не-
обычайно высокие скорости охлаждения, удалось получить тон-
кие стеклообразные пленки состава Au3Si. С тех пор синтези-
ровано множество стеклообразных сплавов переходных метал-
лов (Т), главным образом с неметаллами или полуметаллами
(М), причем состав этих сплавов часто находится в области
соединений от ЪМ до Т3М. Кроме того, стали известны и дру-
гие типы металлических стекол (разд. 3.1.1.1).
Получение металлических стекол потребовало разработки
технических приемов, позволяющих достичь скоростей охлажде-
ния ~ 1010 К/с. Число неорганических веществ, которые в ре-
зультате такого охлаждения удалось перевести в аморфное со-
стояние, увеличилось во много раз. В частности, применение
этих новых методов работы заметно ускорило изучение метал-
лических стекол, имеющих важное практическое использование.
По данному вопросу написаны обстоятельные обзоры [17, 18].
Метод распылительной закалки под давлением (splat quen-
ching) или метод выстреливания расплава (gun technique) ос-
нован на действии ударной волны (рис. 2.3, а), которая распро-
страняется в вакуумированную часть реактора, внезапно раз-
рывая диафрагму из тонкой фольги. Под давлением ударной
волны расплав «выстреливается» через небольшое отверстие на
охлажденную подложку, напрямер на медную пластину. Этот
метод впервые применили Дувез с сотр. [17]; позже описаны
и другие его варианты [19].
В методе поршня и наковальни (piston-and-anvil method)
капли расплава, находящиеся в свободном падении или выстре-
ливаемые под действием ударной волны, попадают в зазор
между неподвижно закрепленной наковальней и движущимся
поршнем. Последний «бьет» со скоростью от 2 до 3 м/с и рас-
плющивает каплю в тонкую пластинку (рис. 2.3,6) [20].
* В литературе встречаются два термина — металлические стекла и
аморфные металлы. Более правильно употреблять второй термин.— Прим,
ред.
3 Заказ № 413
Аг или Не
(давление')
диафрагма из
полиэтилена-
вакуум
жидкая капля
2-3 м/с
наковальня поршень
струя
металлического
расплава
подложка
у пленка Q
==х стеклообразная пластинка
а 6
струя металлического расплава
лента
выпуклая
поверхность лецта
г
в
ж
проволока
з
д
е
Рис. 2.3. Схемы различных методов производства металлических стекол.
Аморфное и стеклообразное состояние
35
Преимущество метода спиннингирования расплава (melt-
spinning) состоит в возможности непрерывного изготовления
тонкой фольги в виде ленты [21]. Известно несколько модифи-
каций метода. Расплав выдавливается высоким давлением газа
через сопло и попадает на охлажденную быстро движущуюся
поверхность, например на вращающийся диск (рис. 2.3, в), вра-
щающийся цилиндр (рис. 2.3, г), или прокатывается между
двумя быстро вращающимися роликами (roller quenching)
(рис. 2.3, д). При спиинингировании расплава в центрифуге
(centrifugal spinning) струя расплава направляется на внутрен-
нюю поверхность быстро вращающегося колеса (рис. 2.3, е)
[23]. Центробежные силы обеспечивают надежный термический
контакт, а изогнутая поверхность—быстрое удаление ленты
с подложки. В литературе [18] описаны также установки
(рис. 2.3, ж), в которых твердые тела в форме стержней рас-
плавляются исключительно в месте контакта с вращающимся
диском или роликом (melt extraction), что дает возможность
отказаться от плавления в тигле и выдавливания через сопло.
Проволоку можно получать, используя установку (рис. 2.3, з),
в которой тонкая струя расплава попадает непосредственно
в закалочную жидкость, и далее поступает вместе с пей в охла-
ждаемую направляющую трубку (метод струйной закалки
аморфных сплавов; free jet spinning of wires) [21]. На такой
установке можно производить металлические стекла в виде
проволоки и нитей непрерывно со скоростью до 2 км/мин.
2.1.1.4. Управление скоростью охлаждения
стеклообразующих расплавов
В табл. 2.3 приведены наиболее часто используемые значе-
ния скоростей охлаждения при получении неорганических сте-
кол из расплавов.
Технические стекла и оптические стекла с целью достиже-
ния гомогенности состава и устранения внутренних напряже-
ний необходимо очень медленно охлаждать. Для этого исполь-
зуют специально сконструированные печи, позволяющие выдер-
живать определенный режим охлаждения. Даже при темпера-
турах выше температурного диапазона стеклования охлаждение
должно вестись медленно, чтобы избежать возникновения
в расплаве конвекционных потоков и гидродинамических не-
стабильностей. В результате таких процессов может, например,
происходить неконтролируемый перенос поверхностных слоев
расплава в его объем, что приводит к изменению состава, по-
явлению оптически негомогенных областей (например, свильно-
стей). Такие стеклообразные материалы непригодны для оп-
тики. Чтобы наилучшим образом обеспечить одинаковую тер-
мическую обработку всех областей стекла, необходимо прово-
36 Глава 2
Таблица 2.3. Скорости охлаждения стеклообразующих расплавов
Скорость охлаждения q, К/с Термическая обработка
io-5 ~2 • 10-4 Ю-3—10-2 Отжиг больших зеркал телескопов Отжиг оптических стекол Отжиг обычных стекол
1—2 Закаливание на воздухе халькогенидных расплавов массой 10—20 г в кварцевых ампулах с толщиной стенок 2 мм
8-10 Закаливание в воду при температуре 273 К
35 Закаливание в воду 1 г расплава в ампулах с толщиной стенок 0,5 мм
-180 Закаливание в воду 0,015 г расплава в тонкостенной ампуле [23]
~103 105—106 Метод разбрызгивания расплава Метод спиннингирования расплава
106—107 Метод поршня и наковальни
106—10'° Метод распылительной закалки под действием ударной волны
дить как можно более медленное охлаждение и ниже темпе-
ратур стеклования. При такой термообработке в материале воз-
никают лишь небольшие температурные градиенты от центра
образца к его поверхности. К техническим и оптическим стек-
лам должны предъявляться особенно жесткие требования в от-
ношении устойчивости расплавов к процессам рекристалли-
зации.
При изучении тенденции к стеклообразованию в различных
системах используют высокие скорости охлаждения. При этом
расплавы берут в таких малых количествах, что без особого
ущерба можно пренебречь возможностями возникновения пере-
напряжений в стекле. Перед разливкой и закалкой в данном
случае стремятся как можно более сильно переохладить рас-
плав. Достаточно высокая вязкость расплава и использование
емкостей соответствующей геометрии способствуют торможе-
нию конвекционных потоков. Возникшие напряжения могут
быть в значительной степени сняты путем заключительного от-
жига. При работе с халькогенидными расплавами в запаянных
кварцевых ампулах необходимо иметь в виду относительно вы-
сокие давления паров над ними. При быстром охлаждении та-
ких расплавов от слишком высоких температур резкое пони-
жение давления над расплавом нередко вызывает их вскипа-
ние, в результате чего получают вспененные или с большим
количеством пузырей продукты.
Аморфное и стеклообразное состояние
37
Рис. 2.4. Изменение скорости охлаждения расплава халькогенидных стекло-
образующих расплавов при различных условиях.
Если расплав массой несколько десятков граммов охла-
ждать на воздухе, например вылив его в предварительно нена-
гретую форму или вынув ампулу с халькогенидным стеклооб-
разующим расплавом из печи, то можно заметить, что веще-
ство по краю охлаждается с большей скоростью, чем во
внутренней области, и образец в целом уже нельзя охарактери-
зовать единой термической историей. В этом случае приводится
средняя интегральная скорость охлаждения, которая зависит от
теплоемкости и теплопроводности системы, т. е. как от массы
и средней молярной массы исходной расплавленной смеси, так
и от геометрических параметров емкости, в которую выливают
расплав (например, для халькогенидных стекол — от формы и
толщины стенок кварцевого сосуда).
В кварцевой ампуле с карманом для термопары (рис. 2.4)
можно определить скорости охлаждения халькогенидных рас-
плавов. На рис. 2.4 также приведены кривые охлаждения рас-
плава (образцы массой 10—20 г) в ампулах с толщиной стенок
~2 мм при условии закаливания на воздухе (кривая 7) и
в ледяную воду (кривые 2— исходные расплавы имели разную
температуру).
Поскольку для процесса замораживания определяющим па-
раметром является скорость, с которой образец проходит об-
ласть переохлаждения, т. е. температурный интервал между
температурой плавления ТПл и температурой стеклования TR,
понятно, насколько важна для сопоставления результатов на-
дежность установления скорости охлаждения расплава. На
рис. 2.4 выделена область температур Т= 7'^±50 К. Вблизи
38 Глава 2
этой температуры ( — 300 К) скорость охлаждения при закали-
вании на воздухе составляет 2 К/с (кривая 1), а при закалива-
нии в ледяную воду 8—10 К/с (кривые 2). При работе с образ-
цами массой 1 г в небольших достаточно тонкостенных
( — 0,5 мм) ампулах достижимы скорости охлаждения 35 К/с.
Аналогичные величины приводят Корне и Россье [24]. При за-
каливании в охлажденный концентрированный раствор едкого
натра или в ртуть удается повысить скорости охлаждения еще
в 2—3 раза.
Дальнейшее повышение скорости охлаждения может быть
достигнуто путем уменьшения массы исходной расплавленной
смеси, а также путем улучшения способов отвода тепла за счет
отказа от использования сосудов для плавления, например пу-
тем приведения в непосредственный контакт расплава с охла-
ждающей жидкостью или с охлажденной подложкой.
В методе распыления расплава (spray cooling) образец
плавят в кварцевой трубке, в нижней части которой находится
отверстие размером —100 мкм [25]. Под действием внешнего
давления расплав распыляется, проходя через отверстие, и по-
падает в интенсивно перемешиваемое силиконовое масло. Та-
ким образом, получают стеклянные бусинки диаметром 100—
300 мкм.
Наиболее высокие скорости охлаждения достигнуты с по-
мощью приспособлений, описанных в разд. 2.1.1.3. По данным
Девиса и Халла [26], при закаливании металлических распла-
вов в слоях толщиной 0,15 мкм температура уменьшалась со
скоростью 1010 К/с. Такого рода данные получены на основании
изучения гидродинамических структур, которые образуются
в расплаве в условиях сильной неравновесности во время зака-
ливания [18]. Серджент и Рой, используя описанную технику
[27], сумели перевести в стеклообразное состояние оксиды
V2O5, ТеО2, WO3, МоО3. Совсем недавно Нассо [444] сообщил
о получении стеклообразных форм LiNbO3, КТаО3, УзЕе5О12,
Li4CdK(SO4)4, а также множества других оксидных фаз путем
закаливания расплавов со скоростями охлаждения — 107 К/с.
2.1.2. Получение аморфных твердых материалов
из растворов
Растворенные вещества можно перевести в твердое состоя-
ние путем выпаривания растворителя, изменения температуры
или добавления осадителей, например таких жидкостей, которые
понижают растворимость благодаря изменению полярности рас-
творителя. Реакции осаждения лежат также в основе катод-
ного и анодного выделения веществ.
Если изменения условий происходят настолько быстро, что
образуются сильно пересыщенные растворы, то времени на об-
Аморфное и стеклообразное состояние 39
разование и рост зародышей кристаллов оказывается недоста-
точно, и таким образом получают аморфные вещества. При
этом часто промежуточной стадией является образование кол-
лоидных растворов. Увеличение размеров коллоидных частиц,
обусловленное процессами коагуляции, часто заторможено
(с этим, например, связаны определенные трудности, возникаю-
щие при фильтровании продуктов осаждения). В определенных
случаях затвердению предшествует образование геля, который
далее непрерывно может быть переведен в твердый аморфный
материал. Причины, приводящие к выпадению из растворов не-
кристаллических осадков, весьма многочисленны. Наряду с по-
лучением аморфных форм вещества лишь путем быстрого до-
бавления осадителя или электролизом токами высокой плотно-
сти известно множество химических соединений, которые оса-
ждаются из растворов преимущественно в некристаллическом
виде (разд. 3.2.4.2).
2.1.2.1. Стекла, получаемые гомогенным осаждением гелей
Уже давно известно, что совместное осаждение веществ,
например различных оксидов, гидроксидов и др., весьма удобно
для дальнейшего исследования протекающих твердофазных хи-
мических реакций. Компоненты находятся в продукте осажде-
ния в очень тесном контакте. Часто вследствие образования
промежуточных соединений и смешанных фаз возникает такое
распределение частиц по размеру, что диффузионные пути от-
дельных частиц при протекании твердофазных реакций сильно
сокращаются, благодаря чему превращения протекают в тече-
ние короткого времени и при существенно более низких тем-
пературах.
Рой [28] обратил внимание на то, что эти явления можно
использовать при получении стекол. Дислих [29], подробно
излагая результаты исследования гелеобразования при гидро-
лизе этанольных растворов Si(OC2H5)4, Д1(ОС4Нэ)з, NaOCHg
и КОС2Н5 показал, что при ступенчатом термическом разложе-
нии образовавшегося геля получается стекло. Для этого ока-
залось достаточным нагревание геля до температуры ~800 К
(т. е. до области температур стеклования). Свойства получен-
ных из геля стекол совпадают со свойствами продуктов, полу-
ченных из расплава. Таким же путем получены боросиликат-
ные стекла. Формование стекла проводят прессованием в об-
ласти температур стеклования Tg. При этом удается избежать
попадания в температурную область, где скорость кристалли-
зации максимальна или где происходит процесс разделения фаз
(разд. 2.3.1), и тем самым создать относительно благоприятные
условия для получения стеклообразного твердого тела. Недавно
для многих систем появились сведения о стеклообразных фазах,
возникновение которых идет путем отвердения гелей [427—429].
40 Глава 2
2.1.2.2. Аморфные осадки
Образование аморфных осадков широко распространено
в химии водных растворов. Чаще всего выпадение таких осадков
рассматривают как мешающий фактор, так что все внимание ис-
следователей направлено на то, чтобы найти условия, при ко-
торых получаются хорошо фильтруемые, кристаллические
осадки стехиометрического состава. Кремневая кислота, мно-
гие гидроксиды металлов и большое число основных солей об-
разуют аморфные осадки, не имеющие определенного хими-
ческого состава. Из-за сильно развитой поверхности аморфных
осадков другие находящиеся в растворе ионы адсорбируются
или хемосорбируются ими и легко соосаждаются. Это обстоя-
тельство часто мешает разделению веществ в аналитических
целях.
В то же время известны соединения стехиометрического со-
става, например сульфиды As2S3, As2S5, Sb2S3, Sb2S5, GeS2, при
осаждении из водных растворов образующие аморфные формы.
Благодаря малой растворимости и строго стехиометрическому
составу эти вещества можно использовать для гравиметриче-
ского определения мышьяка, сурьмы и германия. As2Ss и Sb2Ss,
а также M0S3, МоБез, WS3, WSes, V2S5 до настоящего времени
известны лишь в виде аморфных модификаций. Ge2S3 и Ge3Se3
осаждаются из растворов соответствующих тио- и селеногипо-
дигерманатов в виде аморфных осадков. Эти же соединения
можно получить в стеклообразном состоянии и из расплавов
(разд. 3.2.2.4, 3.2.2.5 и 3 2.4).
2.1.2.3. Электролическое осаждение аморфных слоев
Защита поверхностей металлов путем анодного окисления
(анодирования) основана на образовании плотно прилегающих,
как правило аморфных, оксидных слоев, которые предотвра-
щают протекание других окислительных процессов. Толщина
оксидного слоя составляет десятые доли микрометра. Большое
практическое значение имеет анодирование алюминия. Про-
зрачные аморфные пленки ТагО5 устойчивы до температуры
923 К, пока не начинается их кристаллизация [30].
При электролизе токами большой плотности на катоде впер-
вые получены аморфные сплавы фосфора с никелем и кобаль-
том [31].
Таук [32] сообщил об осаждении путем катодного восста-
новления аморфных слоев германия при электролизе раствора
GeCl4 в гликоле. Однако получаемое аморфное вещество
сильно загрязнено вследствие взаимодействия с компонентами
раствора. Так называемая взрывчатая сурьма, полученная при
электролизе кислых растворов SbCl3 и содержащая еще зна-
Аморфное и стеклообразное состояние 41
чительные количества связанного хлора, представляет собой
скорее дихлорид сурьмы. Для получения аморфных веществ
высокой чистоты электрохимические методы мало пригодны.
2.1.3. Получение аморфных слоев из газовой фазы
При получении аморфных и стеклообразных слоев путем кон-
денсации твердого вещества из паровой фазы на подходящую
подложку наиболее часто используются испарение, катодное
распыление, разложение в тлеющем разряде и реакции различ-
ных газообразных компонентов, приводящие к твердым продук-
там. В описанных методиках условия получения аморфных ве-
ществ варьируют очень широко, и поэтому даже в однокомпо-
нентных системах известен довольно многочисленный набор
различных аморфных структур. Часто причиной такого много-
образия является влияние примесей, которые встраиваются
в структуру твердого тела совершенно невоспроизводимым об-
разом. Поэтому чистота слоев и аналитический контроль со-
става имеют особое значение. К сожалению, этому вопросу, не
всегда уделяется достаточное внимание.
При изучении многокомпонентных систем необходимо про-
водить тщательный контроль гомогенности слоев в вертикаль-
ном и горизонтальном направлениях. Сведения о механизме их
образования ограниченны. Лишь для некоторых простых си-
стем, например для некоторых простых веществ и ряда соеди-
нений, получены данные о кинетике испарения и механизме кон-
денсации [33—35]. Однако даже в этих случаях с помощью
масс-спектрометрического анализа в паровой фазе найдены
различные частицы, так что такие результаты передают, пожа-
луй, лишь самую общую картину явления.
Системы, расплавы которых обнаруживают тенденцию к за-
твердеванию в виде стекла, часто проявляют склонность к об-
разованию структур с непериодическим строением и при оса-
ждении из газовой фазы. Химический состав получаемых таким
образом стеклообразных фаз часто практически совпадал
с составами стекол при образовании их из расплавов. По-
скольку при получении аморфных или стеклообразных веществ
из паровой фазы решающее значение имеет подавление зароды-
шеобразования, весьма полезным оказывается использование
охлаждаемой подложки. Чем выше в каждой конкретной си-
стеме склонность к кристаллизации, тем при более низких тем-
пературах необходимо проводить осаждение из газовой фазы.
Благодаря более высоким значениям энтропии пара осаждение
из газовой фазы в экстремальных условиях (например, кон-
денсация на охлаждаемые жидким гелием подложки) открыло
новые пути синтеза стеклообразных и аморфных форм таких
42 Глава 2
веществ, которые никаким другим способом не удавалось пе-
ревести в это состояние, например Mg, Zr, Hf, Nb, Ta, Mo, W,
Re, Cd.
2.1.3.1. Методы термического испарения в вакууме
Испарение является статистическим процессом, который
можно описать в рамках кинетической теории газов. Состояние
равновесия характеризуется: 1) упругими столкновениями ча-
стиц на поверхности раздела между конденсированной и газо-
вой фазами (обратимая передача теплоты) и 2) выделением
в газовую фазу и осаждением из газовой фазы частиц на по-
верхности раздела фаз (обратимая передача работы).
Испарение в условиях равновесия (соотношение Кнудсена).
При испарении вещества в ячейке Кнудсена процесс протекает
в равновесных условиях. Отверстие в испарителе пренебрежимо
мало по сравнению с общей поверхностью испарения. Скорость
эффузии Rm идеального газа можно выразить уравнением, из-
вестным из кинетической теории газов:
Rm = mU/4v = (M/2txRT)'12 р (2.1)
где М — молекулярная масса; й — средняя скорость, соответ-
ствующая распределению Максвелла—Больцмана, т. е. й =
— (SRT/лМуь, a (mlv)-MplRT. Определяя уменьшение массы
образца во времени, можно вычислить давление насыщен-
ного пара. В многокомпонентных системах уравнение (2.1)
описывает парциальные скорости эффузии компонентов Rw;,
и, следовательно, из него можно определить парциальные дав-
ления компонентов р,. Таким образом, в общем случае со-
став пара меняется со временем.
Испарение в вакууме (соотношение Ленгмюра). Чтобы
иметь возможность испарить в течение данного времени боль-
шее количество вещества, используют емкости с большими
отверстиями, например прямо или косвенно нагреваемые от-
крытые тигли или лодочки. В этих условиях система не рав-
новесна. Процесс испарения определяется скоростью, с кото-
рой происходит разрушение поверхности твердых или жидких
веществ, т. е. определяющими факторами этого процесса ста-
новятся его кинетика и перенос тепловой энергии к месту, где
происходит реакция. Несмотря на это, для описания такого
типа испарения также используют уравнение (2.1), но в не-
сколько измененном виде: в уравнение вводят множитель а =
^R'JRm, который по очевидным соображениям должен быть
всегда меньше 1:
/?*т = а(М/2.т/?7’)'',р
(2.2)
Аморфное и стеклообразное состояние 43
где М— средняя молекулярная масса частиц в потоке пара,
т. е. при инконгруэнтном испарении и при различных давле-
ниях паров продуктов разложения неизбежны явления фрак-
ционного разделения. Из температурной зависимости скорости
свободного испарения в вакуум
С = Аехр(-ДЕ7/?7') (2.3)
можно рассчитать энергию активации испарения Д£*, ко-
торая, хотя и не совпадает с энтальпией испарения, все же
представляет собой некий интегральный параметр, характе-
ризующий реакцию разрушения поверхности испаряющегося
вещества.
При достаточно низкой температуре подложки и большом
расстоянии между испарителем и подложкой удается избежать
обратного испарения уже осажденных слоев, т. е. из экспери-
ментальной скорости испарения /?* с учитывая направление
испарения вещества, можно рассчитать скорость конденсации
• Если рассчитанная скорость конденсации согласуется
с экспериментальным значением, то можно говорить о том, что
весь процесс описан данной моделью.
. Чем ниже температура подложки, тем больше опасность
внедрения примесей в осажденные слои, например из окружаю-
щего испарительную установку пространства или из оставшихся
в ней после откачки газов (прежде всего из-за попадания в нее
насосного масла). Кроме того, при таких условиях на под-
ложке осаждаются весьма рыхлые и пористые слои, которые
впоследствии быстро изменяются при контакте с воздухом.
На практике конденсацию стараются вести как можно более
медленно, поддерживая как можно более высокую температуру
подложки. Определенные ограничения метода связаны с темпе-
ратурой стеклования данного материала и давлением пара над
стеклообразной пленкой. Скорости испарения 0,1—0,5 нм/с при
температуре подложки 298 К позволяют, например, в случае
испарения Ge4Se5Te обеспечивать достаточную подвижность ча-
стиц на поверхности для того, чтобы образовывались относи-
тельно плотные пленки. Свойства таких пленок практически не
отличаются от свойств компактных образцов стекла, что сви-
детельствует о сходстве их структурного строения (разд. 3.3.3.2
и 4.1.3.6). При получении аморфных слоев германия аналогич-
ные скорости осаждения оптимальны при температуре под-
ложки 470 К.
В работе [36] описаны различные приборы для получения
аморфных препаратов из паровой фазы. На рис. 2.5, а приве-
дена схема конструкции прибора, который был использован
для изучения испарения стеклообразного Ge2Se3 и поликри-
сталлических смесей GcSe и GeSc2. На вращающемся диске
Рис. 2.5. Общая схема установки для осаждения аморфных слоев методом
термического испарения в вакууме (а) и схема вращающего держателя под-
ложек и мишеней (б) [37].
1—танталовая лодочка, 2 — исходный материал, 3 — предварительно нагретый медный
блок, в который помещается танталовая лодочка с исходным материалом, 4 — токонод-
воды и датчик температуры, 5 — поворачивающаяся заслонка для регулировки потока
пара, 6 — кварцевый пьезодатчик, 7 — зафиксированный блок со сменными держателями
подложек 8, 9 — вращающийся диск с тонкослойными мишенями для нижнего контакта
10, слоя халькогенидного стекла 11 и верхнего контакта 12, /3 — система терморе!j.ni-
ровки подложки, 14— держатель подложек, 15 — образцы тонких слоев для химического
анализа, 16 — арретир для диска с мишенями.
Рис. 2.6. Зависимость (а) логарифма скорости испарения /?*сп (/), скорости
_< *
конденсации лконд (2) и скорости осаждения /?^(5), измеренной с помощью
кварцевого пьезодатчика, от обратной температуры [уравнение (2.3)] при испа-
рении переохлажденного расплава ОегЗез и эквимолярной смеси GcSe/GeSe2
{4} и сравнение (б) энтальпии испарения Д//Исп и энтальпии активации
Д£* кристаллических и стеклообразных веществ (хотя ДНстекл < ДТ/кр»
Д^стекл -•'> А^кр) ’
Аморфное и стеклообразное состояние 45
(рис. 2.5, б) расположены различные мишени-подложки. При
вращении диска осуществляется получение сразу нескольких
аморфных образцов за один рабочий цикл (вакуумирование);
причем можно параллельно изготовить несколько одинаковых
образцов, часть из которых использовать для химического ана-
лиза [37].
Построив графическую зависимость скорости испарения
и конденсации от обратной температуры (рис. 2.6, а), можно
прийти на первый взгляд к удивительному выводу. Хотя гомо-
генное стекло состава Ge2Se3 (в опыте использовались образцы
со средним диаметром частиц 0,3—0,7 мм) обладает значи-
тельно более высоким запасом энергии по сравнению со смесью
кристаллических фаз GeSe/GeSe2, энергия активации свобод-
ного испарения переохлажденного расплава (Тё которого 608 К)
в два раза выше, чем энергия активации испарения поликри-
сталлической смеси [38]. При температуре — 730 К переохла-
жденный расплав кристаллизуется. В условиях равновесного
испарения в области температур <730 К переохлажденный
расплав безусловно характеризовался бы меньшей величиной
энтальпии испарения, а энтальпия сублимации поликристалли-
ческого образца была бы выше. В неравновесных же условиях
свободного испарения скорость испарения определяется кине-
тикой процессов разрушения и перестройки поверхности,
а также скоростью подвода тепловой энергии, необходимой
для протекания испарения (рис. 2.6, б).
Предполагается, что в стекле, полученном из переохлажден-
ного расплава, имеются обширные области с совершенной про-
странственной сеткой из структурных элементов Ge2See/2
(разд. 3.2.2.5), в то время как кристаллический образец пред-
ставляет собой гетерогенную смесь соединений GeSe + GeSc2,
причем оба селенида имеют слоистую кристаллическую ре-
шетку. В результате масс-спектрометрического исследования
установлено, что в паровой фазе в основном содержатся моле-
кулы GeSe и Se2 [38]:
GezSe3 кристаллизация^ GeSe
Ge2Se3 -> GeSe + Se2
GeSe/GeSe2 —> GeSe + Se2
Скорость испарения /?*, определяется скоростью самой
медленной стадии сложного процесса, непосредственно пред-
шествующего переходу частиц в газовую фазу. Энергия акти-
вации свободного испарения часто существенно превосходит
энтальпию испарения и энтальпию сублимации, если в жидком
и стеклообразном состоянии вещество имеет макромолекуляр-
ную (полимерную) структуру (например, аморфный мышьяк,
46 Глава 2
красный аморфный фосфор или стеклообразный и моноклин-
ный AS2O3) [39]. Свободное испарение стеклообразного Аз2Оз ха-
рактеризуется, например, величиной а = 2’2«10-6 [40]. Исходя
из этого, Гутцов [41] сделал вывод, что в условиях, которые
описываются уравнением Ленгмюра (10~5<а< 10_9), для стек-
лообразного состояния характерны очень низкие скорости испа-
рения.
Масс-спектрометрическим анализом состава пара и химиче-
ским анализом осажденных пленок с разной толщиной слоя
установлено, что только при испарении переохлажденного рас-
плава Ge2Se3 (т. е. ниже температуры кристаллизации) можно
достичь независимости состава от времени. В остальных слу-
чаях пар и осажденные слои обогащаются соединением GeSe,
т. е. при разложении структурных групп Ge2Se6/2 в испаряю-
щемся образце частично остается GeSe2. Различия в кинетике
испарения GeSe и GcSe2 в значительной степени сглаживаются
при использовании стеклообразного Ge2Se3. Для многокомпо-
нентных систем при испарении в вакууме более предпочти-
тельно использовать расплавы или стекла, чем смеси кристал-
лических фаз, поскольку первые обладают относительно более
однородной поверхностью. Если удасться подобрать условия
проведения испарения таким образом, что состав пара сохра-
няется неизменным во времени, то гомогенные слои образуются
даже в том случае, когда состав шихты в испарителе несколько
отличается от исходного. При этом необходимо остановить про-
цесс испарения до того момента, когда изменения в составе
шихты начнут оказывать заметное влияние на состав пара. На-
пример, фракционное разделение соединений можно сущест-
венно уменьшить путем соответствующего подбора массы ве-
щества, находящегося в испарителе.
В этом плане испарение под действием лазерного излуче-
ния или направленного электронного излучения вряд ли имеет
какие-либо преимущества перед термическим испарением. Од-
нако при использовании этих методов удается испарять веще-
ство из определенного малого объема образца, достигая высо-
ких локальных температур испарения. Существенно и то, что
в этом случае отпадает проблема выбора материала испари-
тельной камеры, а также его влияния на изучаемую систему.
Использование же методов мгновенного испарения позволяет
заметно уменьшить вероятность фракционного разделения.
Мгновенное испарение и плазменное испарение. При мгно-
венном испарении маленькие частицы вещества попадают
(чаще всего путем свободного падения) в предварительно на-
гретый до высоких температур реактор, в котором они очень
быстро испаряются. В результате в газовой фазе появляется
набор фрагментов молекул, осаждение которых на подложке
Аморфное и стеклообразное состояние
47
приводит к образованию аморфных сильно разупорядоченных
слоев. Из-за быстрого испарения мелкие твердые частички
увлекаются потоком пара и встраиваются в аморфный слой.
Чтобы избавиться от подоб-
ных явлений и обеспечить
образование слоев только
из атомарных и молекуляр-
ных компонентов пара, вну-
три испарителя располага-
ют отражающие перего-
родки. Поток пара, натал-
киваясь на перегородки, мно-
гократно меняет направле-
ние своего движения, что при
водит к увеличению време-
ни пребывания вещества в
зоне высокой температуры
и к более полному его испа-
рению. На рис. 2.7 показа-
но устройство испарителя
в виде U-образной кварце-
вой трубки с косвенным
обогревом, внутри которой
предусмотрено наличие бес-
порядочно расположенных
отражающих перегородок.
Такая конструкция исполь-
зована для мгновенного ис-
парения халькогенидных
стекол [42].
В методе плазменного
испарения (plasma-jet eva-
poration) для испарения ча-
Рис. 2.7. Установка для проведения
мгновенного испарения [42].
1 — гранулированное вещество и дозатор, 2 —
защитный тепловой экран, 3 — охлаждаемая
трубка, 4 — U-образная кварцевая трубка с пе-
регородками 5,6 — обмотка нагревателя, 7 —
защитный тепловой экран, 8 — охлаждаемый
защитный экран, 9 — охлаждаемый блок с под-
ложками 10.
стиц вещества используют
высокотемпературную плазму (после испарения вещество попа-
дает в плазму) [43]. При этом достигаются как полное испаре-
ние вещества, так и высокая скорость потока пара (20 км/с);
поток распространяется строго прямолинейно. Если к расплаву,
испаряющемуся в вакууме, приложено электрическое поле высо-
кой напряженности, то наблюдается эмиссия ионизированных
частиц [44].
2.1.3.2. Получение аморфных слоев методом катодного
распыления
При получении аморфных слоев методом катодного распы-
ления (sputtering) используют газовый разряд, который
48 Глава 2
осуществляют в атмосфере инертного газа — чаще всего ар-
гона— при пониженном давлении (10—10~2 Па) [45—47].
Ионы Аг+ ускоряются в поле и с огромной скоростью на-
правляются на катод, который покрыт испаряемым веществом.
Под действием потока ионов с поверхности мишени отрываются
атомы и небольшие фрагменты молекул. Это происходит как
за счет термического испарения, так и за счет непосредствен-
ной передачи импульса. В зависимости от напряженности поля,
в котором ускоряются ионы Аг+, последние приобретают энер-
гию в сотни—тысячи электронвольт, что намного превосходит
энергию связи атомов в твердых телах [36]. Явления фракци-
онного разделения в этом методе проявляются в гораздо мень-
шей степени, чем в многокомпонентных системах при термиче-
ском испарении.
Удаление атомов с поверхности катода основано на высоком
термическом возбуждении атомов решетки и в основном лока-
лизуется в непосредственной близости от места попадания
ионов Аг+ Из-за высокой плотности энергии явления фракцион-
ного разделения в значительной степени подавлены. Однако
различие парциальных давлений отдельных компонентов
нельзя совсем игнорировать и в рамках данного метода. С ро-
стом энергии ионов Аг+ сильнее проявляется «ударный» меха-
низм данного метода. Это означает, что с ростом приложенного
напряжения уменьшается фракционное разделение компо-
нентов.
Оторвавшееся с катода вещество более чем па 99 % состоит
из нейтральных частиц. Они осаждаются за границей зоны га-
зового разряда, например на соответствующим образом распо-
ложенной подложке. При надлежащем охлаждении можно по-
лучить аморфные слои со сравнительно однородным брутто-со-
ставом даже из сложных смесей веществ.
На рис. 2.8 показаны конструкции типичных установок
с двумя и тремя электродами. При наложении постоянного по-
тенциала 1 —15 кВ при давлении газа 1—10 Па между элек-
тродами возникает плазма (рис. 2.8, а). Осаждаемый на аноде
слой вещества может одновременно подвергаться воздействию
электронов, обладающих высокой энергией. В установках
с тремя электродами (рис. 2.8, б) электроны улавливаются
специальным анодом. Выделение ионов Аг+ из зоны газового
разряда и в этом случае происходит с помощью постоянного
электрического поля напряжением 1—2 кВ (остаточное давле-
ние газа 5-Ю-2—1 Па). В магнитном поле, создаваемом маг-
нитами, расположенными вне установки, контакт плазмы с ми-
шенью (подложкой) ухудшается.
При катодном распылении непроводящих (высокоомных)
веществ на мишени скапливался бы положительный заряд
и весь процесс заканчивался бы в течение короткого времени.
Аморфное и стеклообразное состояние
49
Таким образом, метод катодного распыления при работе с ди-
электрическими материалами требует периодического подвода
положительных и отрицательных зарядов, что может быть до-
стигнуто при наложении переменного напряжения. Для этого
наиболее эффективно применение переменного напряжения
с частотой ~ 10 МГц (рис. 2.8, б). За исключением металличе-
ских стекол, все аморфные пленки в основном относятся к до-
статочно высокоомным материалам, поэтому для их получения
Рис. 2.8. Установки для катодного распыления с двумя (а) и тремя (б) элек-
тродами.
1— ввод газа, 2 — к насосу, 3 — катод, 4 — мишень, 5 — анод. 6 — осаждаемой слой, 7 —
«затемненное» катодное пространство. 8 — область положи[ельного заряда, 9 — вспомога-
тельный анод. 10 — магнитная катушка возбуждения и фокусировки плазмы.
10
7
необходимо применение катодного распыления в высокочастот-
ном поле.
Аморфные слои халькогенидных материалов сложного со-
става, например Aso,3oSio,i2Gco.ioTeo,48, теперь можно воспроиз-
водимо получать методом катодного распыления; это явилось
сильным стимулирующим фактором для исследования элек-
трических свойств этих веществ. Методы катодного распыления
как раз и предназначены для получения аморфных слоев в си-
стемах с несколькими компонентами, давления паров которых
заметно отличаются друг от друга. В аморфных слоях Si и Ge,
полученных методом катодного распыления, обнаружены за-
метные количества аргона, который встраивается в структуру
синтезируемого материала (разд. 3.1.2.3). Если к слоям предъ-
являются очень высокие требования по чистоте, то данные
методы вряд ли применимы. Необходимость поддержания пони-
женного давления газа сопряжена с внесением в продукт
4 Заказ № 413
50 Глава 2
загрязнений из-за встраивания в его структуру примесей из
окружающего пространства, что существенно ограничивает сте-
пень чистоты осаждаемых слоев.
2.1.3.3. Осаждение аморфных слоев в тлеющем разряде
Получение аморфных слоев путем разложения определен-
ных газообразных веществ или газовых смесей в тлеющем раз-
ряде также относится к методам плазмохимии, которые на-
шли применение в промышленности [48—50]. Плазма — это
высокоионизованный газ, свойства которого определяются раз-
личной подвижностью ионов и электронов. В плазме, получен-
Рис. 2.9. Осаждение аморфных слоев путем разложения соответствующего
газа или газовой смеси (например, SiH4) в тлеющем разряде, создаваемом
катушкой индуктивности (а) или конденсатором (б).
1 — катушка индуктивности, 2 — терморегулируемый держатель подложек.
ной за счет нетермического возбуждения, электронная темпера-
тура может быть на четыре порядка выше температуры ионов
и нейтральных частиц.
Тлеющий разряд возникает в некотором замкнутом объеме
или трубке в потоке газа при пониженном давлении (~10 Па),
если на электроды прикладывают напряжение в сотни вольт.
Концентрация электронов и ионов в газовой фазе при тлеющем
разряде составляет ~ 1010 см-3. Энергия электронов 1 —10 эВ
примерно в 30—300 раз превосходит среднюю термическую
энергию ионов и нейтральных молекул. Из-за высокой подвиж-
ности электронов большинство химических связей разры-
вается, благодаря чему химические реакции протекают при
сравнительно более низких температурах. На рис. 2.9 приве-
дены схемы экспериментальных установок, в которых тлеющий
разряд возникает за счет энергии индукционной катушки или
за счет энергии, запасенной в конденсаторе. Чаще всего рабо-
Аморфное и стеклообразное состояние 51
тают в области 10—20 Вт и в диапазоне частот 1 — 100 МГц.
Структура и свойства осажденных слоев зависят от многих па-
раметров, например, температуры подложки, диаметра газораз-
рядной трубки, относительного расположения подложки и ин-
дукционной катушки (или обкладок конденсатора). Суще-
ственно наличие контакта между плазмой и осаждаемым
слоем.
В литературе описаны многочисленные примеры получения
с помощью тлеющего разряда металлических слоев (Sn и РЬ
из Sn(C2H5)4 и РЬ(С2Нб)4 [51]) и плотных аморфных слоев
органических полимеров [50]. Штерлинг [52, 53] применил этот
метод для осаждения аморфных слоев Si и Ge из SiH4 и GeH4.
Полученные слои содержат заметные количества водорода (на-
личие фрагментов Si—Н и Ge—Н позволяет рассматривать их
как высокомолекулярные сетчатые полициклосиланы и поли-
циклогерманы). Для воспроизводимого получения такого сорта
слоев необходимо строгое соблюдение всех условий экспери-
мента. Интерес к синтезам в тлеющем разряде заметно возрос
после удачного получения аморфных слоев кремния, в которых
был реализован п—р-переход (разд. 4.1.3.5 и 5.1.3). При этом
в SiH4 вводились в несколько приемов очень малые добавки
таких газообразных веществ, как РН3 или В2Нв.
Смеси N2O с газообразным SiH4 или О2 с Si(OC2H5)4 дают
в тлеющем разряде тонкие стеклообразные слои SiO2.
Из В(ОСН.з)3 был получен ВгО.з, а из смесей SiH4 и NH3 —
аморфный Si3N4. Сообщалось также об осаждении аморфных
слоев AI2O3 с высокой электрической прочностью на пробой из
смеси А12С1б и О2. Эксперименты с такими системами — при-
меры получения аморфных слоев методом химического оса-
ждения из паровой фазы.
2.1.3.4. Химическое осаждение из паровой фазы
В методе химического осаждения из паровой фазы исполь-
зуют спонтанно протекающее взаимодействие между различ-
ными газами или реакцию между газообразными молекулами
на твердой поверхности (например, на стенке сосуда или па
подложке). Такие методы вызывают повышенный интерес
в связи с выращиванием монокристаллов (газофазная эпитак-
сия) и могут быть использованы также для получения аморф-
ных веществ. Среди наиболее известных примеров применения
этого метода необходимо упомянуть о получении высокочистого
SiO2 путем гидролиза SiCl4 в пламени гремучего газа и путем
пиролиза газообразной смеси SiCl4-!-O2. Оба названных
приема позволяют проводить осаждение практически не содер-
жащих воду форм кварцевого стекла очень высокой степени
чистоты.
4*
52 Глава 2
2.1.4. Перевод кристаллических веществ
в аморфное состояние
Перевод кристаллических твердых тел в аморфное состояние
нетермическими способами основан на достаточно сильном
воздействии на них внешних сил. При этом атомы могут поки-
дать свои равновесные позиции, например за счет получения
извне импульса энергии. В ходе твердофазных реакций при
определенных условиях часто происходит распад структуры
исходных веществ без построения новой кристаллической струк-
туры с периодическим расположением атомов. При этом мо-
гут быть получены аморфные фазы, хотя бы в виде промежу-
точных продуктов. В качестве примеров можно привести ре-
акции термического обезвоживания каолинита или получения
аморфного KFe3O3(SO4)2 при нагревании ярозита
KFe3(OH)6(SO4)2 до 500К [430].
2.1.4.1. Получение аморфных тел путем
механической обработки
О механической аморфизации кварца путем его обработки
в вибрационной мельнице сообщалось в работе Шредера
и сотр. [431], которые подробно изучили этот процесс. Иссле-
дования методом ЭПР-спектроскопии позволили сделать вывод
о высокой концентрации разорванных химических связей в по-
лучаемых материалах и как следствие о повышенной реакцион-
ной активности их [432, 433]. Так, каолин при интенсивном из-
мельчении переходит в аморфное состояние [434]. Отмечалось
[436] также влияние механического активирования сухой смеси
компонентов цементной пасты на схватывание цемента.
При любом механическом воздействии на твердые мате-
риалы необходимо учитывать, что первоначально возникающая
в поверхностных слоях сильная разупорядоченность распро-
страняется в глубь образца и приводит к полной аморфизации
структуры. Протяженный разупорядоченный слой Бейлби при
механическом полировании или при притирке подшипников
и зубчатых профилей возникает как следствие воздействия
узко локализованной энергии. Связанные с этим явления опи-
сываются в рамках специальной области трибохимии (механо-
химии), имеющей особое значение для техники [436].
2.1.4.2. Получение аморфных твердых тел путем облучения
При облучении твердых тел быстрыми нейтронами или под
действием сильно ускоренных ионов нарушается периодичность
расположения атомов в узлах кристаллической решетки. При
этом не наблюдается промежуточной стадии перехода твер-
Аморфное и стеклообразное состояние 53;
дого тела в изотропное расплавленное состояние. Например,
при воздействии потока 1,5-1020 нейтрон/см2 плотность кварца
уменьшается на 15 % и достигает значения 2,26 г/см3. Показа-
тель преломления составляет 1,467. Такие же значения полу-
чают, если исходят из тридимита и кристобалита [54, 55]. Та-
ким образом, речь идет о существовании твердого аморфного
изотропного материала, основные характеристики которого
мало отличаются от свойств кварцевого стекла (плотность
кварцевого стекла 2,205 г/см3, показатель преломления 1,457).
Структурные исследования также обнаружили сходство
в структуре ближнего порядка кварцевого стекла и аморфного-
материала [56]. При малой дозе облучения после дополни-
тельного отжига аморфной фазы плотность вновь возрастает,
и происходит обратное превращение в а-кварц. Если же дозы
облучения велики, то после отжига при 1373 К происходит об-
разование кварцевого стекла, т. е. плотность в этом случае по-
нижается еще больше. Такие же превращения в стеклообраз-
ное состояние известны также для берилла BesACSieOie.
Глубина проникновения заряженных частиц ограничивается
лишь приповерхностной областью. Так, например, при бомбар-
дировке монокристаллов Ge ионами кислорода с энергией
105 эВ при дозе облучения 1015 ион/см2 получены аморфные
слои Ge толщиной 60 нм [57].
2.1.4.3. Получение аморфных веществ под действием
ударной волны
Де Карли и Джамайзон [58] экспериментально показали,,
что в определенных областях монокристалла кварца можно
получить аморфные формы кварца при действии на него удар-
ной волны всего в 360 кбар и максимальной температурой
фронта волны 873 К. Полученный продукт имеет плотность
2,22 г/см3 и показатель преломления 1,46, что близко к соответ-
ствующим свойствам кварцевого стекла. Передача ударного
импульса осуществляется с помощью металлических пластин.
Наилучшие результаты получены при времени действия удар-
ной волны 10-5 с. Температура плавления а-кварца составляет
1673 К, и из-за высокой вязкости линейная скорость распро-
странения плавления при этой температуре составляет
10-8 см/мин [59]. Поэтому в данном случае следует отказаться
от общепринятого механизма плавления. Далее необходимо
подчеркнуть, что аморфный образец SiO2, полученный под дей-
ствием ударной волны в 600 кбар, имеет примерно такую же
плотность, что и продукт, полученный в эксперименте, когда
сила ударной волны составляла 360 кбар. Согласно этим ре-
зультатам, а также как показало изучение той части монокри-
сталла кварца, который остался в неизменном кристаллическом
54 Глава 2
состоянии, аморфное вещество, подобное кварцевому стеклу,
в термодинамическом смысле представляет собой определенную
модификацию SiO2 (разд. 3.2.2.1). Машимо и сотр. [438] опи-
сали характерные особенности (прежде всего структурные ха-
рактеристики) аморфного SiO2, полученного ударным сжатием.
2.1.4.4. Реакции кристаллических твердых тел
при образовании стекла
Большой практический интерес имеют исследования, посвя-
щенные, например, окислительной обработке поверхности мо-
нокристалла Si в потоке газообразного кислорода или водяных
паров при высоких температурах. Согласно кинетическим ис-
следованиям роста аморфного слоя SiO2, происходящие при
этом процессы можно объяснить в рамках лимитируемых диф-
фузией реакций поверхностных слоев монокристалла Si с О2
или Н2О. Из значений скоростей этих реакций рассчитаны
коэффициенты диффузии О2 и Н2О в кварцевом стекле, кото-
рые хорошо согласуются с данными, полученными другими ме-
тодами [60]. Такие же поверхностные слои удается точно также
нанести, например, в кислородной плазме при существенно бо-
лее низких температурах и, следовательно, в относительно бо-
лее мягких условиях [50].
2.2. Термодинамическое описание
стеклообразного состояния
Описанные методы получения стекол (разд. 2.1) позволяют
сделать вывод о близости стекла к изотропным жидкостям
(или расплавам) и пару. Жидкости в свою очередь можно рас-
сматривать как сконденсировавшийся газ либо как сильно
разупорядоченпое твердое тело. Из этого следует, что при опи-
сании стеклообразного состояния целесообразно исходить из
теории жидкостей.
Если принимать во внимание плотность упаковки частиц, то
жидкости гораздо больше похожи на твердые тела, чем на газ.
Однако постоянный обмен позициями между атомами и струк-
турными элементами, происходящий в жидкостях, ставит их
в один ряд с газами. При описании поведения жидкостей ис-
пользуют решеточные и ячеечные модели и принимают во вни-
мание тот факт, что жидкости характеризуются, как правило,
большим по сравнению с кристаллами объемом, так называе-
мым «свободным объемом», в котором происходит статистиче-
ское изменение расположения элементов структуры. Эти пред-
ставления были впервые введены Френкелем [62] и Эйрингом
и сотр. [63]. Позже Элфри с сотр. [64] указали на примени-
Аморфное и стеклообразное состояние 55*
мость теории свободного объема к объяснению процессов за-
твердевания стеклообразных материалов.
Вблизи температуры затвердевания, а также в области пе-
реохлаждения (по крайней мере для малых областей) еще бо-
лее правомочно проводить сравнение жидкости с кристалличе-
ским состоянием. При этих условиях все еще имеют место про-
цессы перемещения частиц с одной позиции на другую, и это
препятствует затвердеванию со стабилизацией формы. Гут-
цов с сотр. [65] предложили модель, описывающую процессы
ассоциации, которые ведут к увеличению упорядоченных об-
ластей. С ростом температуры структура жидкости все более
приближается к структуре газа, а при некоторой критической
температуре наблюдается непрерывный переход из жидкого со-
стояния в парообразное. Качественные связи между основными
параметрами состояния в этом случае весьма наглядно сле-
дуют из уравнения Ван-дер-Ваальса (2.4) или в более общем
виде из уравнения Клаузиуса (2.5):
pV = RT + [b-(a/RT)-]p (2.4}
pV = JRr + 73XK(rl/)rz/ (2.5)
Первое слагаемое связано с вкладом кинетической энергии,
второе — с энергией взаимодействия между атомами и другими
структурными элементами, удаленными друг от друга на рас-
стояние гц. Можно переписать уравнение (2.5), введя функ-
цию радиального распределения р(г) [66]:
оо
pV = RT-(NAl&)\-^Lp{r)^r3dr (2.6)
о г
Это соотношение проясняет физический смысл функции распре-
деления р(г) и потенциала межмолекулярного взаимодейст-
вия <р(г) [67].
При описании процессов текучести и переноса в качестве
параметра используют также время релаксации — временной
интервал, по прошествии которого положение атома или группы
атомов, изменившееся в результате возмущения в момент вре-
мени t, вновь описывается среднестатистическими параметрами,
т. е. возмущение затухает в е раз, и поведение всей системы
приближается к среднестатистическому.
Предложено [68] описывать такие явления с помощью коэф-
фициента автокорреляции ,
г = 7- (/)F(/ + t) (2.7)
который представляет собой скалярное произведение силы F (/)
в момент времени t на ту же силу через время т. Если z = 0,.
•56 Глава 2
то частица «больше не помнит» о том состоянии движения,
в котором она находилась в момент времени t.
Такой подход лежит в основе статистико-термодинамических
расчетов, применимых лишь к простейшим системам, например
к ансамблям атомов инертных газов, включающим несколько
сотен частиц [69]. В приводимых расчетах времена релаксации
т^Ю-8 с до сих пор, как правило, не использовались, и по-
этому во всех практически важных случаях для описания жид-
кого состояния применяли макроскопические феноменологиче-
ские модели.
2.2.1. Релаксационные процессы в жидких
и стеклообразных веществах
В настоящем разделе основные закономерности релаксаци-
онных явлений рассмотрены на примере механической релакса-
ции. Из дидактических соображений особенности структурной
релаксации (независимо от того, пойдет ли речь о растягиваю-
щем напряжении или о напряжении сжатия) обсуждаются
в тесной связи с термической релаксацией. Затем представ-
лены температурная зависимость вязкости и различные мо-
дели, применяемые для ее феноменологического описания. В за-
ключение читатель сможет кратко ознакомиться с методами
корреляционной спектроскопии.
2.2.1.1. Механическая релаксация
Под действием внешних сил стекла, как и любые твердые
тела, подвержены упругой деформации. Например, под дейст-
вием касательного напряжения сдвига или напряжения на срез
Sg = K/F наблюдается угловая деформация (рис. 2.10).
Рис. 2.10. Воздействие на твердое
тело касательного напряжения SG =
=K/F.
Стеклообразное тело под действием силы, вызывающей де-
формацию, меняет свою форму настолько, насколько это воз-
можно в условиях упругой деформации; при снятии внеш-
него напряжения тело вновь приобретает первоначальную
форму. Для модуля упругости при кручении или для модуля
Аморфное и стеклообразное состояние
57
сдвига G справедливо а= (1/G)Sg. Модуль сдвига G связан
с модулем Юнга Е= (l/&l)SE и адиабатической сжимаемостью х:
Е = 2О(1 + р), х = 3(1-2ц)/Е (2.8а)
SE — напряжение сжатия или растягивающее напряжение, на-
правленное перпендикулярно к поверхности, ар — коэффици-
ент Пуассона, p=At//d: А///. Коэффициент Пуассона можно
относительно легко определить, например, путем измерения ско-
рости распространения продольных и поперечных звуковых волн
Vi и vtr в образце
ц = 0,5 1-----------
~ vtl
(2.86)
G можно рассчитать, зная плотность образца р и скорость рас-»
пространения поперечных звуковых волн G = pv2tr.
Зависимость упругого напряжения от времени можно выра-
зить производной
= -^---твердое тело, подчиняющееся закону Гука (2.9)
В случае пластической деформации стеклообразных твердых
тел, т. е. при начавшемся вязком течении, касательное напря-
жение сдвига SG пропорционально градиенту скорости, с ко-
торой слои вещества сдвигаются относительно друг друга в на-
правлении х (рис. 2.10); связывает эти параметры динамиче-
ская вязкость
du Sq
=----— ньютоновская жидкость 10)
Объединение уравнений (2.9) и (2.10) приводит к выраже-
нию для скорости деформации d^i/dt, откуда можно получить
дифференциальное уравнение для вязкоэластичной текучести
(уравнения Максвелла)
d^ 1 dSO I Sq . uSq i/d Sq /су i i \
~dT~~G dt 1 rj ’ ~dT~ ~di v\!G ' '
Изменение во времени напряжения сдвига dScldt пропор-
ционально скорости деформации d$ldt. В случае возникнове-
ния текучести замедление изменения напряжения во времени
тем сильнее, чем больше величина SG и чем меньше отношение
г]/О. Последнее отношение имеет смысл времени релаксации,
в течение которого тело реагирует па внешнее воздействие,
т. е. в течение которого в теле ослабевает напряжение сдвига:
ц —От; т| = (3/5х)т (для ц = 0,25) (2.12)
58 Глава 2
При постоянной деформации (dft/dt = Q)
dSdt So =S'O ехр(-//т) (2.13)
т. е. при t = x Sc—S'cJe. При строго постоянном изменении
деформации во времени (d$!dt = const) из уравнения (2.11)
можно получить уравнение (2.10), т. е. реализуется случаи
стационарного течения ньютоновской жидкости.
Из уравнения (2.11) следует, что существенное значение
для определения агрегатного состояния вещества (стекло или
переохлажденная жидкость) имеет период времени, в течение
которого проводится исследование. Если время наблюдения го-
раздо меньше времени релаксации (А/<Ч), то членом Sg/t
можно пренебречь, и практически речь идет о твердом теле,
подчиняющемся закону Гука. Если тело с достаточной скоро-
стью ударяется о поверхность жидкости (например, воды), то,
даже пренебрегая влиянием поверхностного натяжения, такое
воздействие было бы вполне эквивалентно столкновению с бе-
тоном. Если же А/^>т, то в стеклах и даже в кристаллических
твердых телах возникает пластическая деформация.
В этом смысле уместно обратить внимание на текст из
книги Рихтера, гл. 5, стих 5 (песня Дебора): «...горы разли-
лись перед человеком...». Действительно, речь здесь должна
идти о неземной по длительности жизни, поскольку требуется
слишком большой промежуток времени, чтобы суметь охватить
произошедшие события. Так называемое число Дебора DN опре-
деляется как отношение времени релаксации т к времени
наблюдения А/ [70]. В области £W;>1 расплавы и жидкости
характеризуются свойствами твердых тел, и они могут рас-
сматриваться как стекла, в области £)/V<Cl постоянство формы
веществ отсутствует. Переходная область характеризуется зна-
чениями DN ~ 1.
Дагласом [71] предложено следующее определение стекло-
образного состояния: «Стекло — это жидкость, время релакса-
ции которой нельзя определить».
Каузман [72] формулирует более точно: «В стеклообразном
состоянии время релаксации любой степени свободы слишком
велико в сравнении с длительностью эксперимента. Поэтому
температура стеклования существенно зависит от природы, из-
меряемого свойства, времени эксперимента, а также от того,
какой временной промежуток принят за длительный».
Таким образом, остается открытым вопрос, что понимают
под утверждением «нельзя определить», какие методы исполь-
зуют, как долго действует, например, напряжение сдвига и с ка-
кой точностью устанавливают начавшуюся деформацию тела
или ослабление приложенного напряжения.
Аморфное и стеклообразное состояние 59
При исследованиях стеклообразных тел наиболее часто на
них действуют некоторой периодической нагрузкой '0' = -00 cos со/.
При этом измеряют, например, затухание колебаний крутиль-
ного маятника или используют резонансные устройства для
определения логарифмического декремента затухания Л, из ко-
торого рассчитывают время релаксации т:
Х = л/сот (2.14)
Зная модуль упругости при кручении G или сжимаемость х, по
уравнению (2.12) легко определить вязкость.
При переходе к комплексным переменным Sg, G и О в урав-
нении (2.11) надо заменить соответствующими комплексными
функциями S*, G* и О*: -
S*G = 6*0*, G*= ^~rG, = (2.15)
С учетом сделанных преобразований можно решить дифферен-
циальное уравнение (2.11), выделяя в G* действительную
и мнимую части:
G* = G'4-iG" = G+i , G (2.16}
Из (2.16) можно получить выражение для тангенса угла
потерь:
tgd = G"/G' (2.17)
tg 6 = л/л называют также внутренним трением. Эту величину
нельзя путать с коэффициентом внутреннего трения — вязко-
стью Т). /Ji
Измеряя X или tg д в зависимости от частоты со, можно
рассчитать по уравнению (2.16) функции G'\G и G"IG и
определить время релаксации т. График зависимости G'fG
от 1gшт представляет собой S-образную кривую с точкой
перегиба при (от=1 (при этом значении аргумента Gf/G = ’/2).
Зависимость G"fG от 1g сот имеет вид куполообразной кривой
с максимумом при G"/G = 1/2 и (от = 1. Это означает, что, когда
при частотном измерении осуществляется определяемый вре-
менем релаксации т переход в область жидкого течения, на
кривых G'lG и G"IG от 1g (от возникает перегиб или экстре-
мум соответственно. При частоте 10-1 Гц этому условию отве-
чает время релаксации 1,6 с, откуда в типичном случае (напри-
мер, для As2S3 6 = 6,24 ГПа) т)= 10й дПа-с.
Чтобы определить внутреннее трение в широком диапазоне
времен релаксации, соответствующие измерения проводят при
различных частотах и температурах. При этом установлено, что
в общем случае поведение стекол отличается от поведения тел.
‘60 Глава 2
для которых выполняются уравнения Максвелла. Например,
при высоких значениях скорости напряжения сдвига стекла
обладают малой вязкостью [73], а при очень низкой скорости
сдвига (10-8 см/с) обнаружена некоторая пороговая величина
начала сдвига [74]. Таким образом, вязкоэластические свойства
изменяются не строго линейно, и, следовательно, в уравнение
(2.13) входят дополнительные нелинейные члены. Для интер-
претации этих явлений при описании закономерностей внут-
реннего трения в стеклах и их расплавах вводят функцию рас-
пределения времен релаксации [75, 76], которая зависит от
температуры. Дебаст и Джилард [76] предложили вместо урав-
нения (2.13) следующее:
S0 = Sg ехр(—t/x)b (2.18)
В области //т^ЮО b сравнимо с 1, т. е. здесь работает про-
стая модель х^аксвелла. В области, где вязкость достигает
значений 1015 дПа-с, b ~0,5 [77, 78]. Для интервала 10-3<
<(//т)<10 4 Ья'13. С уменьшением величины b кривые
G'lG и G"IG от 1g шт все более сглаживаются, и при £’ = 0,1
точка перегиба на первой кривой и максимум на второй кри-
вой практически исчезают [79]. При переохлаждении расплава
до области температур стеклования (г) = 10134-1015 дПа-с) ре-
лаксационные явления удается достаточно точно описать с тер-
модинамических позиций, используя представления о времени
релаксации, и лишь в области температур стеклования имеет
смысл вводить коэффициент автокорреляции (2.7).
В температурную зависимость времени релаксации различ-
ные релаксационные процессы входят с различным весом, так
что привычное для описания температурной зависимости хими-
ческих превращений уравнение Аррениуса
т = т0 ехр [£П//?Г] (2.19)
применимо здесь лишь для узкого температурного интервала.
Выше температур стеклования измерение механической ре-
лаксации требует особых экспериментальных ухищрений из-за
деформации исследуемых образцов под действием гравитаци-
онного поля. Для этого применяют, например, ультразвуковые
методы [80]. Корсаро [81] сообщил об использовании акустиче-
ского дилатометра. На практике часто более просто измерить
вязкость и по уравнению (2.12) рассчитать время релаксации.
При изучении распространения электромагнитных волн при
частотах v>10 2 Гц в образцах при температурах стеклова-
ния обнаруживаются свойства упругого твердого тела. В стек-
лообразных соединениях процессы течения и обмена позициями
частиц в этих условиях практически больше не идут, и со свя-
Аморфное и стеклообразное состояние
61
занными с этими явлениями эффектами можно не считаться.
Однако показано, что уже следы примесей в кварцевом стекле
(например, присутствие в нем ОН-групп или оксидов щелоч-
ных металлов) приводят к затуханию колебаний в стекле при
температурах гораздо ниже области стеклования. Поэтому при
изучении используемых на практике стекол сложного химиче-
ского состава получен широкий спектр значений внутреннего
трения в зависимости от температуры. Во многих работах по-
лученные результаты объяснены с позиций структурной химии.
Структурный каркас стекла жесткий, и в целом образец ведет
себя как твердое тело, подчиняясь закону Гука. В то же время
внутри стекла имеют место локальные перескоки частиц с од-
ной позиции на другую; например, ионы щелочных металлов
перемещаются в различные позиции, разделенные потенциаль-
ными барьерами (разд. 4.1.1). К таким же явлениям можно
отнести обмен позициями между мостиковыми и немостиковыми
кислородными ионами [82]. В таких случаях Е^ в уравнении
(2.19) надо заменить энергией активации того процесса, кото-
рый протекает в системе. Бартенев и Щеглова [439] сообщили
о новом механизме релаксации при T<ZTg.
Механическая релаксация в стеклах первоначально описана
в работах Рётгера [83], а затем появились более подробные ра-
боты [84—86]. Работа Переза и сотр. [448] — уже фундамен-
тальное исследование вязкоэластической текучести в области
температур стеклования. В этой работе измерения были прове-
дены с помощью низкочастотного крутильного маятника. Изу-
чение аналогичных явлений проведено также на металлических
стеклах [87].
2.2.1.2. Термическая и структурная релаксация
При охлаждении стеклообразующих расплавов система
в области переохлаждния находится в термическом
равновесии. При этом распределение внутренней энер-
гии и энтальпии по степеням свободы системы успевает
следовать за скоростью охлаждения q = dT:dt. Число и тип
разорванных химических связей, а также размеры неразрушив-
шихся в результате процессов течения структурных элементов,
их поступательное, колебательное и вращательное движение
заметно меняются с температурой. В общем с изменением тем-
пературы в расплаве происходят структурные изменения. Для
понятного и наглядного описания этих явлений вводится тер-
мин структурной релаксации.
Если скорость установления нового состояния равновесия
в системе при любом бесконечно малом понижении темпера-
туры становится меньше, чем скорость охлаждения (за счет
62 Глава 2
Рис. 2.11. Температурная зависимость энтальпии II и теплоемкости Ср при
прямом и обратном переходах расплав—стеклообразное состояние.
<7В><7А (<7— скорость охлаждения и нагревания), Tg — температура стеклования, Tz —
температура замораживания (относительно Т2 см. разд. 2.2.3.1).
того, что происходят описанные выше явления), то наблю-
дается запаздывание. Равновесные параметры системы уже не
успевают следовать за скоростью охлаждения. Таким образом,
энтальпия расплава характеризуется большим значением, чем
можно было бы ожидать при данной температуре в условии
термодинамического равновесия. Переохлажденный расплав ме-
тастабилен по отношению к кристаллическому состоянию
(рис. 2.11). В интервале температур стеклования структурная
релаксация «замораживается». Переохлажденный расплав пе-
реходит в стеклообразное состояние, т. е. стекло также мета-
Аморфное и стеклообразное состояние
63
стабильно по отношению к переохлажденному расплаву. Гово-
рят, что стекло — это замороженное состояние переохлажден-
ного расплава.
Сравнение процесса стеклообразования с процесом замора-
живания широко использовалось Симоном [89]; продолжая ту
же линию, Хаазе [88] предложил следующее определение стек-
лообразного состояния: «Стекло — это замороженная переох-
лажденная жидкость».
Согласно Гутцову, некристаллическое твердое тело ниже
температуры стеклования независимо от способа его получе-
ния можно рассматривать «как кинетически замороженную
термодинамическую систему».
Процесс стеклообразования можно описать соотношением,
аналогичным дифференциальному уравнению (2.11):
d ts.ll х-. dT ts.H iCt a(\\
' dt ~Cp dt т ' }
Если кривую охлаждения разбить на ряд последовательных
интервалов, внутри которых температура все время постоянна
(т. е. dT/dt = 0), а при переходе от одного интервала к другому
меняется скачкообразно, то для этих бесконечно малых обла-
стей выполняется следующее соотношение:
-^- =------(2.21)
Причем в этих условиях температурную зависимость т можно
описать с помощью уравнения (2.19) или других более подхо-
дящих соотношений (разд. 2.2.1.3).
С понижением температуры время релаксации увеличи-
вается, и в зависимости от скорости охлаждения процесс замо-
раживания начинается при различных температурах. Чем выше
скорость охлаждения, тем выше температура, при которой на-
чинается стеклование данного материала (рис. 2.11).
В связи с этим становится понятной причина различий
в структурном строении стекол, имеющих одинаковый состав,
но разную термическую предысторию. В зависимости от терми-
ческой предыстории при данной температуре фиксируется раз-
личная структурная конфигурация. Предложено [90] темпера-
туру, при которой замораживается структура жидкости, назы-
вать фиктивной температурой стеклования (принят термин
«фиктивная температура Тула»). Девис и Джонс [91] опреде-
ляют ее физический смысл как параметр внутреннего порядка.
Описание стеклообразного состояния требует наряду с указа-
нием таких параметров состояния, как температура и давле-
ние, еще по крайней мере одного дополнительного параметра.
Релаксация, с которой осуществляется переход из переохла-
64 Глава 2
жденного расплава в стеклообразное состояние и обратно из
неравновесной стеклообразной фазы в равновесное расплавлен-
ное состояние, находит свое выражение в петле гистерезиса,
которая наблюдается для одной и той же системы при охла-
ждении и нагревании с одинаковой скоростью (рис. 2.И)-?
На рис. 2.11 приведен также ход температурной зависимости
теплоемкости. Приняв Н = Н(Т, /), проведя дифференциро-
вание
и используя соотношения (2.19) и (2.21) для -^~^т==т
удалось получить следующее выражение для разности между
- о / dH X „
наблюдаемой теплоемкостью 1-^-1 =СР и теплоемкостью
стекла = Сп [92]:
\ J t р, стекл * *
\НТ
Ср Ср, стекл — | _ I ехР [ EpjRT g\ (2.23)
I Ч I то. Tg
(знак минус — нагревание, плюс — охлаждение).
Считая, что зависимостью (Ср — Ср, стекл) и &HTg от скоро-
сти нагревания или охлаждения q можно в первом приближении
пренебречь (рис. 2.11), удается получить следующую функцио-
нальную зависимость между q и Tg:
• а^>т\ Ч = [-£п/ет8] (2.24)
а 1/у ix
Эта зависимость соответствует уравнению Бартенева [93,
94]. Она была подтверждена в ряде экспериментальных иссле-
дований. График зависимости In | q | от 1/Т представляет собой
прямую, из наклона которой можно определить энергию акти-
вации вязкого течения в области стеклования. Рассчитанная
таким способом величина энергии активации находится в хоро-
шем согласии с данными, полученными другими методами, на-
пример для As2Se3 [92, 95], В2О3 и других стекол [92]. Мазурин
[79] приводит полный обзор по данному вопросу.
Структурная релаксация стеклообразующих расплавов
и стекол оказывает влияние также па другие свойства материа-
лов, например на плотность, мольный объем или молекулярную
рефракцию. Так, для стеклообразного В2О3 обнаружена
зависимость показателя преломления от времени отжига
(рис. 2.12) [96]. Показатель преломления образца стекла,
отожженного при 583,5 К (т. е. при Tz>Tg) до равновесного со-
Аморфное и стеклообразное состояние 65
стояния, при 298К составляет nD= 1,45337. Последующие от-
жиг стекла при температурах <583,5 К и закалка до комнат-
ной температуры приводят к представленным на рисунке зави-
симостям nD = f(t). Чем выше температура термообработки,
тем меньше, как и следовало ожидать, оптическая плотность
и тем быстрее идет установление нового конечного значения nD
(рис. 2.12, а). Если образец, отжигавшийся в течение 14 ч при
Рис. 2.12. Зависимость от времени показателя преломления
образца
стеклообразного В2О3 после закаливания от 583 К и последующего отжига
при соответствующих температурах (а), а также образца, отожженного в те-
чение 14 ч при 498,6 К, а затем нагретого до 543 К (б) [96].
498,6 К, дополнительно нагреть до 543 К, то при 298 К n,D =
— 1,4597 сохраняет свое значение. Однако, как видно из
рис. 2.12, б, вначале наблюдается уменьшение и лишь затем
быстрое установление значения показателя преломления, соот-
ветствующего температуре 543 К. Из этого следует, что в дан-
ном случае для описания явлений, происходящих в стекле при
изменении температуры и времени, необходимо иметь в виду
протекание по крайней мере двух релаксационных процессов:
первый из них связан с изменением температуры (быстрый
процесс), а второй (медленный процесс), очевидно, отвечает
структурной релаксации. При высокой надежности метода ре-
фрактометрии выявление такого рода закономерностей для
стекол очень важно в практических целях [97].
2.2.1.3. Температурная зависимость вязкости
Для описания температурной зависимости вязкости служит
уравнение
ц = ц0 ехр [ЕП/ЯТ] (2.25)
5 Заказ № 413
66 Глава 2
которое получено из (2.19) и (2.12). Оно адекватно описывает
изменение вязкости в широком интервале температур (т. е. при
этих температурах сохраняется постоянство Etl) лишь для не-
ассоциированных жидкостей с шарообразными частицами и не-
направленными химическими связями.
Однако в стеклообразующих расплавах имеется сильное
взаимодействие между соседними структурными элементами.
С ростом температуры размеры хаотически расположенных упо-
рядоченных областей умень-
Рис. 2.13, Температурная зависимость
вязкости для некоторых стеклообразу-
ющих соединений.
шаются, что связано с возра-
станием числа разрушенных
связей. При этом энергия
активации вязкого течения
уменьшается [98—100].
Тем не менее в области
высоких температур, т. е.
при достаточно низкой
вязкости (E^^kT), можно
применять уравнение Арре-
ниуса (2.25) [101]. Согласно
Эйрингу [102], и в области
очень высокой вязкости
(E^kT) уравнение (2.25)
также приближенно выпол-
няется. В переходной обла-
сти (на рис. 2.13 это заметно для ВзО.з) Еп наиболее сильно
меняется с температурой.
Согласно Френкелю [62], в основе вязкого течения распла-
вов лежат термически активируемые перескоки молекулярных
структурных элементов в соседние микропустоты. При доста-
точно высоких температурах и относительно низких давлениях
количество и размеры таких микропустот в расплаве весьма ве-
лики; при более низких температурах и более высоких давле-
ниях они только начинают образовываться. «Теория свободного
объема» исходит из того, что молекулы или другие агрегаты
частиц вследствие термических флуктуаций могут перераспре-
деляться таким образом, что свободное пространство между
ними будет сосредоточено в одном месте с образованием
«дырки», объем которой близок к объему структурного эле-
мента, участвующего в перескоках. Вероятность образования
дырок и вероятность отрыва одной частицы от своих соседей
для занятия соседней микропустоты определяют возможность
протекания двух различных этапов вязкого течения.
Поэтому логично в энергии активации вязкого течения Е^
выделить два вклада: энергию активации образования вакан-
сий Еь (так называемый вклад свободного объема расплава)
и энергию преодоления потенциального барьера Еп при пере-
Аморфное и стеклообразное состояние 67
движении определенного структурного элемента из одной пози-
ции в другую [103]. Согласно Стенворту [104], для низких тем-
ператур справедливо уравнение
Л = A exp [EL/RT~\ exp [Д^/ЯТ] (2.26)
В области высоких температур El близка нулю, поскольку
в этих условиях количество микропустот достаточно велико,
и приведенное выше уравнение сводится к уравнению (2.25).
Например, для расплава В2О3 в области температур Tg
энергия активации составляет 326 кДж/моль, а при более
высоких температурах — лишь 57 кДж/моль. Разность
в 269 кДж/моль почти точно соответствует энтальпии испаре-
ния В2О3 (А/7ИСц = 274 кДж/моль). Такого рода сравнения яв-
ляются вескими аргументами в пользу того, чтобы применить
к стеклообразующим расплавам рекомендацию Каузмана [72]
об оценке энергии образования вакансий в неассоциированных
жидкостях, исходя из мольной энтальпии испарения. Энталь-
пия испарения — это мера энергии сцепления упорядоченных
областей в жидкости. Поэтому, видимо, физически вполне
обоснованно сопоставлять энергию образования вакансий
с мольной энтальпией испарения. Правда, еще Френкель [62]
указывал на то, что для большинства расплавов энергия обра-
зования пустот намного меньше энтальпии испарения и ее ве-
личина ближе мольной энтальпии плавления.
На основании рассмотрения одной из таких моделей Эйринг
[63, 102], исходя из уравнения теории «абсолютных скоростей
химических реакций»
•п = (ЛГЛЛ/У)exp [Mj'JRT] = (^ЛЯ/V)exp [-AS*//?] exp [Д^'/ЛГ]
(2-27)
вывел следующее соотношение:
т| = (Na/V)'1' (2ЛТ/Р ДЯ„СП) (2nmkT)'h exp [&ErjRT] (2.28)
где Ад —число Авогадро, 7 —мольный объем, АЯИсп — моль-
ная энтальпия испарения, 0 — вероятность перехода, т—-масса
структурного элемента. Вейман [105] получил аналогичное
уравнение, применив аппарат статистической термодинамики
и не используя теорию абсолютных скоростей химических ре-
акций.
Другое направление описания текучести стеклообразующих
расплавов [106, 107] сконцентрировано в значительной степени
на рассмотрении структурно-химических представлений и не-
которых аспектов теории химической связи. Согласно развивае-
мой Мюллером для стекол теории валентности, вязкое течение
неорганических стекол основано на активируемом переносе ко-
валентных мостиковых связей в каркасе структуры стекла.
5*
68 Глава 2
Роль мостиковых связей при рассмотрении вязкого течения под-
черкивается в многочисленных работах Немилова [108]. Мюл-
лер и Немилов интерпретируют ДО* в уравнении (2.27) как
свободную энергию образования связи AGJ. Именно такая
энергия требуется для разрыва химических связей и возник-
новения вязкого течения. Эти модельные представления доста-
точно хорошо описывают вязкое течение в случае стекол с трех-
мерной сетчатой структурой, в области температур стеклования
(разд. 2.4.3.1). Однако для стекол с более низкой степенью
связности структурного каркаса ДО* намного меньше энер-
гии связи. Мольный объем V = V*y связывают с акти-
вационным объемом V* через коэффициент у = ехр [—ДО* (Т)/
RT], который отражает изменение конфигурации структуры
в окрестности положения, где происходит перескок структурных
элементов:
Т) = (P^h/v") exp {[ДО" + ДО'Л (Г)]/СТ} (2.29)
(dhG*/dT)p = —S*— энтропия активации при любой тем-
пературе. Предполагается [109], что для многих силикатных
стекол активационный объем V* совпадает с объемом, необ-
ходимым для размещения кислородных ионов, для халь-
когенидных стекол — с атомным объемом соответствующих
халькогенов. В настоящее время известны различные точки зре-
ния относительно размеров частиц, участвующих в процессе
течения. Так, для силикатных стекол согласно работе [109]
принимается, что в этом процессе участвуют ионы кислорода,
а согласно работам [НО, 111] 1—2 тетраэдра [SiCh]. Это об-
стоятельство дало основание утверждать [112], что «стеклооб-
разующий расплав при вязком течении движется как еди-
ное целое, и наличие отдельных структурных элементов,
участвующих в течении, не следует принимать во внимание».
Образование вакансий из-за чисто феноменологического по-
нимания этого процесса как процесса возникновения свобод-
ного объема тесно связано с изменением внутреннего порядка
в расплаве, т. е. с изменением энтропии.
Уильямс, Лендел и Ферри [113] ввели эмпирические урав-
нения, непосредственно связывающие вязкость со свободным
объемом:
Л = A'exp [В/V)], Vf — Vo • 3 Да(7’ — То), напр. T0 = Tg (2.30)
где Vf — избыточный объем расплава по сравнению с объемом
твердого тела, полученным экстраполяцией к соответствующей
температуре; Да — разность коэффициентов линейного расши-
рения. Ниже области стеклования объемное расширение обус-
ловлено возрастающей ангармоничностью колебаний решетки.
Свободный объем, необходимый для перемещения частиц из од-
Аморфное и стеклообразное состояние 69
ной позиции в другую, при этих температурах еще не достиг-
нут, и поэтому вязкое течение не наблюдается в течение всего
эксперимента. Выше температуры Tg на кривой V(T) наблю-
дается крутой подъем, и в этих условиях с ростом темпера-
туры свободный объем составляет большую часть расплава.
Образующиеся в расплаве «пустоты» имеют размеры, большие
некоторого минимального объема Ук₽ит, который требуется для
структурных элементов, участвующих в процессе течения.
Возможность такого перераспределения свободного объема
следует из потенциала Леннарда—Джонса, который лежит
в основе клеточной модели Тернбала и Коэна [114]. В соответ-
ствии с этой моделью появление «пустоты» с частотой v =
= ехр [у'Ккрит/V/] в непосредственном окружении частицы, за-
фиксированной с помощью своих ближайших соседей, можно
ожидать с достаточной вероятностью лишь в области темпера-
тур стеклования. При этих температурах частица может поки-
нуть свою «клетку» со скоростью, близкой скорости, определяе-
мой кинетической теорией газов v= (3kT/m)l/2. При этом путь
частицы равен а. Эту величину часто приравнивают к среднему
диаметру частицы. Используя уравнение Стокса—Эйнштейна
D kT/3nar\, с учетом геометрического фактора jcr^l/б из урав-
нения D = gavv имеем
1] = [2 (ЗтЬТ)'/г/Зла] ехр [у>критМ] (2.31)
У; можно экспериментально определить как разность между
объемом расплава Граси и величиной объема стекла УСтекл экс-
траполированного к высоким температурам (рис. 2.16, б).
Кфит — критический объем пустоты, у' — коэффициент пере-
крывания. Вообще говоря, принятые здесь как участники
в процессе течения структурные элементы вызывают опреде-
ленные сомнения, особенно потому, что именно их размеры
определяют критический объем пустот [115, 116]. Такого рода
модели используются также в связи с изучением вязкости пе-
реохлажденных металлических расплавов [117].
Другой путь объяснения эмпирического уравнения (2.30)
предложен Адамом и Гиббсом [118]. В рамках статистико-тер-
модинамического подхода температурная зависимость релакса-
ционных процессов в стеклообразующих расплавах объяснена
ими изменением размеров упорядоченных областей, которые
к тому же могут кооперативно менять свою структурную кон-
фигурацию. Свободная энергия активации АСКОнф, характери-
зующая в такой упорядоченной области процесс перехода
к иному расположению структурных единиц, связана с конфи-
гурационной энтропией расплава
т) = т)0 ехр [АОконф/Г А5К0Нф] (2.32)
70 Глава 2
Учитывая, что ДSкoнф=ДC^>[(7, — То)/Т] и что разность моль-
ной теплоемкостей расплава и стекла ДСР примерно
постоянна (разд. 2.2.3.1), можно получить соотношение, ана-
логичное эмпирическому уравнению (2.30). При затвердевании
стекла Д£Конф стремится к очень малой величине, так что
Л5конф~0. В уравнении (2.30) этому соответствует случай,
когда Т ж Tg, т. е. вязкость быстро достигает чрезвычайно вы-
соких значений при Т^Т8, что неизбежна приводит к возник-
новению упругого твердого тела.
Комбинацией уравнений Коэна—Тернбала (2.31) и Эйринга
(2.28) получено [119—121]
Л = А'Т ехр [у' (VKPHT/Kf) + (Д£Я/7?Г)] (2.33)
Варьируя четыре параметра этого уравнения, можно добиться
относительно хорошего согласования с экспериментальными
данными. В зависимости от того, какой из процессов является
определяющим — образование свободного объема (высокий ко-
эффициент термического расширения расплава) или термиче-
ская активация перемещения частиц из одной позиции в дру-
гую,— в уравнении (2.33) доминирует первый или второй член.
Встречающиеся в литературе [122—124] уравнения для опи-
сания температурной зависимости вязкости стекол и их рас-
плавов далеко не исчерпываются приведенными выше. Все они
содержат 3—4 параметра, которые нужно определять экспери-
ментально. В зависимости от типа стеклообразующей системы
и исследуемого температурного интервала эти уравнения с раз-
ной степенью точности описывают экспериментальные кривые.
Очень часто достигается не намного более близкое совпадение
с экспериментом, чем при использовании ранее предложенных
эмпирических соотношений. Уравнение Уильямса—Лендела—
Ферри (2.30) соответствует эмпирическому соотношению Фо-
геля—Фульчера—Таммана [125, 126]:
1пп = Л" + В7(Т-7’о) (2.34)
В оба уравнения входят три экспериментально определяемые
коэффициента. Различные модели, применяемые для описания
температурной зависимости вязкого течения, дают представле-
ние о физическом смысле этих параметров. Однако еще рано
говорить о построении теоретического фундамента для количе-
ственного предсказания температурной зависимости вязкости.
Для измерения вязкости в настоящее время имеется в рас-
поряжении множество различных методов (см. литературу
к гл. 1 [16, 17]). При этом определение температурной зави-
симости в широком температурном интервале представляет со-
бой весьма сложную задачу. Для определения коэффициентов
А", В' и То в уравнении Фогеля—Фульчера—Таммана (2.34)
Аморфное и стеклообразное состояние
71
требуется по меньшей мере три пары значений ц (Т). Для удов-
летворительного описания переходной области от переохла-
жденного расплава к стеклообразному состоянию и обратно слу-
жат определенные фиксированные точки:
1g 11 = 13,0 Температура стеклования Tg
1g т) = 7,6 Температура размягчения,
или температура Литтлтона Те
1g Л = 4,22 Температура проседания Тм
Для установления этих точек разработаны определенные ме-
тоды и условия измерений, которые подробно описаны, напри-
мер, в монографиях Шольце и Фогеля (см. литературу к гл. 1
[16, 17]). Один из новых методов описан в работе [127]. Для
определения очень высоких значений вязкости можно восполь-
зоваться методикой Ли и Ульмана [128].
2.2.1.4. Структурная релаксация и
корреляционная спектроскопия
Явления структурной релаксации в стеклообразующих расплавах выше
температуры размягчения ТЕ в области, где время релаксации т<10-6 с,
можно изучать ультразвуковыми методами [80, 81]. В области температур
ниже температуры стеклования, где 102-Ь103 с, возможно проводить ис-
следования зависимости различных свойств от времени отжига или измерять
механическое затухание колебаний. В промежуточной температурной обла-
сти особенно эффективна корреляционная спектроскопия.
Обусловленные изменениями температуры флуктуации плотности и пока-
зателя преломления характеризуются тем же механизмом, что и структурная
релаксация. Поскольку такого сорта флуктуации вызывают рассеяние света,
экспериментальная регистрация зависимости интенсивности рассеянного света
от времени содержит информацию о кинетике структурной релаксации.
Обычно проводят измерение интенсивности рассеянного под определенным
углом света луча лазера и определяют автокорреляционную функцию путем
регистрации рассеянного импульса в определенные моменты времени [(ура-
внение (2.7)]. Автокорреляционная функция пропорциональна квадрату ре-
лаксационной функции (2.18) [131]. Такими методами было проведено пря-
мое изучение релаксационных явлений в стеклообразующих расплавах
вплоть до температур стеклования, т. е. в наиболее важной для процесса
стеклования температурной области. Соответствующие данные приведены
для As2Sc3, В2О3 и других стекол [132].
В связи с применением стекол для передачи информации с целью умень-
шения потерь, связанных с затуханием колебаний, в высшей степени важны
знания о флуктуациях плотности стекол. Для оценки пригодности стекол
для этих практических целей введена новая функция — произведение Tg на
адиабатическую сжимаемость х [133] ф = 7’йх.
2.2.2. Температура стеклования
Поскольку температура стеклования — один из важнейших
параметров, характеризующих процесс перехода в стеклооб-
разное состояние, экспериментальное определение этой величины
72 Глава 2
представляет как практический, так и теоретический ин-
терес. Установлены и обоснованы многочисленные корреляции
между Tg стекол и другими термодинамическими свойствами.
Попытки связать тенденцию к стеклообразованию в различ-
ных системах с реальным переходом в стеклообразное состоя-
ние предпринимались неоднократно.
2.2.2.1. Методы определения температуры стеклования
Температурные зависимости многих свойств стекол претер-
певают при температуре Тё характерные изменения, поэтому
в распоряжении исследователей имеется много методов изме-
рения этого параметра. По определению температуре Tg соот-
ветствует вязкость ~ 1013 дПа «с. На графике зависимости 1g т]
от температуры в области вязкости 1012—1014 дПа-с наблю-
дается точка перегиба. В зависимости от скорости, с которой
охлаждают стекло, эта реперная точка на температурной зави-
симости вязкости может отвечать различным температурам
(разд. 2.2.1.1). Зная модуль сдвига G, по уравнению (2.12)
можно рассчитать время релаксации, отвечающее вязкости т| =
= 1013 дПа -с.
Из табл. 2.4 видно, что для измерения Tg необходимо ис-
пользовать такие скорости нагревания и охлаждения, чтобы ин-
тервал температур стеклования был пройден за несколько ми-
нут. В целях сравнения между собой результатов различных
измерений Tg наряду с договоренностью о скорости нагревания
(обычно измерения проводят при нагревании образцов) необ-
ходима также стандартизация предварительной термической
обработки образцов.
Таблица 2.4. Значения модуля сдвига G и времени релаксации т
для некоторых стеклообразных соединений в области температур
стеклования (ч= Ю13 дПа-с)
Параметр Se AS2S3 As2Se3 Ge2S3 Oe2Se3 GeS2 B2O3 SiO2 GeO2
G, ГПа 3,74 6,24 6,87 10,46 8,86 6,06 6,8 30,7 18,7
т, с 270 160 145 97 113 165 147 32,6 53,4
Обычно для опытов используют предварительно отожжен-
ные образцы. После синтеза стекла его еще раз нагревают до
температур выше Tg и охлаждают сначала до температуры на
100 К ниже Tg со скоростью 1 К/мин, а затем быстрее. Ско-
рость нагревания, которую, как правило, используют при изме-
рении Tg, составляет примерно 5 К/мин. При экспериментах
Аморфное и стеклообразное состояние
73
следует по возможности ориентироваться на эти условия. По-
грешность измерения Tg в редких случаях составляет менее
±2 К. Поскольку скорости нагревания и охлаждения q [урав-
нение (2.24)] связаны логарифмической зависимостью с Тё, ма-
лые отклонения условий измерения практически не влияют на
получаемый результат.
Измерение объемного (термического) расширения удобнее
всего проводить бесконтактным интерферометрическим методом.
Интерференционные полосы, возникающие при отражении от
плоскопараллельной поверхности исследуемого образца и от
Рис. 2.14. Интерференционный ди-
латометр для бесконтактного из-
мерения расширения.
1 — образец, 2 — кольцевой ограничи-
тель, 3 — клиновидная стеклянная
крышка, 4 — датчик температуры, 5 —
печь с линейным нагревом, 6 — полу-
прозрачное зеркало, 7 — источник мо-
нохроматического света, 8 — спектро-
метр, 9 — усилитель, 10 — самописец.
клиновидной прикрывающей крышки, смещаются при нагрева-
нии печи (схема измерительной установки приведена на
рис. 2.14). Это смещение полос обусловлено различным терми-
ческим расширением образца и кварцевого кольца и вызван-
ного этим изменением воздушного клина.
Зарегистрированное с помощью спектрометра число интер-
ференционных полос р является мерой линейного расширения
74 Глава 2
рл = 2Д/; далее можно рассчитать коэффициент линейного рас-
ширения стекла:
«ДГ = (дХ/2/0 ДГ) + «кварц (2.35)
На рис. 2.15 схематически
висимость изменения объема
представлена температурная за-
стеклообразного образца, а на
Рис. 2.15. Температурная зависимость изменения объема в области стекло-
вания.
qA и 9в — скорости охлаждения, q\ и q? — скорости нагревания, причем qд^<71 и
Каждой скорости нагревания qt и q2 соответствуют температуры стеклования Tg{ и Tg2.
Каждой скорости охлаждения qA и qB (qqА) соответствуют температурам замора-
живания Т7А и Т 7 в. TEW — дилатометрическая температура размягчения, — тем-
пература, при которой происходит изменение формы образца под действием собственного
веса (Т2 и Tg, mln — см. в разд. 2.2.3.1).
рис. 2.16, а приведены две типичные кривые, полученные для
двух образцов стекла состава GesSe? [115].
Ход зависимости термического расширения обстоятельно
изучен Рётгером [134, 135]. При скорости охлаждения qA
уменьшение объема происходит в соответствии с кривой, изоб-
раженной на рис. 2.15. При нагревании со скоростью q^qA
наблюдается гистерезис, связанный с протеканием релаксаци-
онных процессов (см. рис. 2.11). Температуру стеклования Tg\
можно получить путем проведения касательных к эксперимен-
тальной кривой. Лишь при температуре TEW (т. е. при темпе-
ратуре дилатометрического размягчения, когда т) = 1011>3дПа-с)
зависимость выходит на ту кривую, которая отвечает равно-
весному состоянию переохлажденного расплава. При темпе-
ратуре Ту! происходит изменение формы образца под дсйст-
Аморфное и стеклообразное состояние
75
Рис. 2.16. Измерение термического расширения двух образцов стекла Ge3Se7
с помощью интерферометра.
а — изменение числа интерференционных полос р='2\Ц\ [уравнение (2.35)] с температурой;
б — изменение объема V стекла (/) и расплава (2) от температуры для T>TEW.
вием собственного веса. При экстраполяции отрезка между точ-
ками TF,W и Тут в область низких температур до пересечения
с кривой термического расширения стекла можно получить тем-
пературу замораживания Tza, характерную для скорости охла-
ждения qA. Чем выше скорость нагревания образца в процессе
измерения, тем выше определяемая таким способом темпера-
тура Т g, например, для скорости </2 определяется Tgz>Tg\.
Из сказанного следует, что вся термическая предыстория об-
разца «входит» в экспериментально определяемую величину
температуры стеклования; это типично для всех неравновес-
ных систем.
76 Глава 2
Рис. 2.17. ДТА-кривые стеклообразного As?Se3 в зависимости от скорости
охлаждения расплава [137].
/ — </=0,5 К/с, 2 —<7=15 К/с, 3 —<7 = 20 К/с. Скорость нагревания 3 К мни.
На рис. 2.15 изображено также поведение стеклообразую-
щего расплава, закаленного с гораздо более высокой скоро-
стью. Температура замораживания TZB в этом случае нахо-
дится при более высокой температуре, то же можно сказать
и о Tg (для этого случая на рис. 2.15 значение Tg не нанесено).
При этом предполагается, что скорость нагревания q~^qB-
Если нагревание проводить со скоростью qi, как и предыдущем
примере, г. е. q\<qB, то кривая зависимости изменения объ-
ема от температуры пройдет через минимум, прежде чем выйти
в равновесную область.
На рис. 2.15 и 2.16 отчетливо видно увеличение свободного
объема выше температуры Tg. Естественно, что возрастание
подвижности частиц, например ионной проводимости стекол
сложного состава, также используют при определении Tg [136].
Дифференциально-термический анализ (ДТА) и дифферен-
циальная сканирующая калориметрия (ДСК). Как показывает
сравнение рис. 2.11 и 2.15, между температурными зависимо-
стями объема и энтальпии можно выделить определенные ана-
Аморфное и стеклообразное состояние
77
Рис. 2.18. Изменение теплоемкости Ср в области температур стеклования
для стеклообразного селена в зависимости от времени отжига при 300,4 К
(скорость нагревания одинакова. 20 К/мин) [138].
логии. Используя основные принципы измерения, лежащие
в основе ДТА и ДСК, можно относительно просто определить
температуру стеклования и изучать последующую кристаллиза-
цию стекол.
На рис. 2.17 приведены некоторые экспериментальные кри-
вые ДТА для стеклообразного As2Se3 [137], а на рис. 2.18 пред-
ставлены данные по исследованию изменения теплоемкости
стеклообразного селена в зависимости от времени отжига [138].
Возрастание теплоемкости при переходе стекла в состояние
переохлажденного расплава проявляется на кривых ДТА и ДСК
78 Глава 2
в виде отрицательного (эндотермического) отклонения от ос-
новной линии. Температура стеклования определяется как
точка пересечения касательных к двум участкам кривой
(рис. 2.17). Возникающий в результате релаксационных про-
цессов гистерезис ведет к появлению заметного теплового эф-
фекта на кривой ДТА (кривая 1), который проявляется тем от-
четливее, чем сильнее отличается скорость нагревания образца,
используемая при измерении, от скорости предшествующего
Рис. 2.19. ДТА-кривые стеклообразного As2Se3, полученного различными ме-
тодами.
1 — 62 мг гранулированного стекла, полученного из расплава, 2 — 130 мг порошкообраз- *
ного AS2SC3, полученного осаждением из пара, 3 — 90 мг аморфного слоя, осажденного
на поверхности датчика температуры.
охлаждения. Этот тепловой эффект тем больше, чем медленнее
проводилось предварительное охлаждение стекла и чем дольше
оно отжигалось перед измерением. Аналогичное влияние ока-
зывает увеличение скорости нагревания при измерении.
На температурной зависимости теплоемкости (рис. 2.18) этот
релаксационный эффект находит свое выражение в возраста-
нии Ср и стремлении ее производной по температуре к беско-
нечности в точке Tg (разд. 2.2.3.1, рис. 2.11).
Если уменьшить скорость нагревания образца по сравнению
со скоростью его предварительного охлаждения, то на кривой
ДТА возникает обратный (экзотермический) тепловой эффект
(рис. 2.17, кривые 2 и 3). При приближении к Tg система
с уменьшением объема переходит в переохлажденный расплав,
и теплота выделяется (ср. с кривой qB на рис. 2.15 для слу-
чая 71<7в). Такого рода кривые ДТА были аналитически опи-
Аморфное и стеклообразное состояние 79
саны Мойниханом и Маседо [139] в рамках общей теории ре-
лаксации.
На рис. 2.19 приведены кривые ДТА для стеклообразного
As2Ses, полученного из расплава, и аморфного слоя As2Se3, ко-
торый был осажден непосредственно на поверхности датчика
температуры, используемого при измерениях [140]. Можно за-
метить, что аморфный слой As2Se3, получаемый путем напыле-
ния, характеризуется такой же температурой стеклования Tg,
что и образец, полученный из расплава. Различия в Tg между
двумя образцами ~ 10 К вполне допустимы с учетом высоких
скоростей закалки. Согласно полученным при этом данным,
аморфный слой As2Se3 с полной определенностью можно рас-
сматривать как стеклообразный материал.
2.2.2.2. Температура стеклования и термическое расширение
Коэффициент термического расширения стекла а часто при-
мерно соответствует коэффициенту расширения кристалла того
же состава. В области температур Tg ангармоничность колеба-
ний решетки так велика, что упругие силы в решетке практи-
чески больше не действуют. Чем больше коэффициент термиче-
ского расширения, тем ниже Tg-, предложено [141] следующее
уравнение (А—коэффициент пропорциональности):
aTs — K • 2|л/3 (1 - 2ц) (2.36)
Как правило, для стекол ц = 0,24-0,33 (наиболее низкое
значение (ц = 0,17) для кварцевого стекла, а наиболее высо-
кие ц = 0,394-0,42— для металлических стекол; разд. 2.4.3.5).
Таким образом, можно заранее сказать, что произведения aTg
по меньшей мере должны сближаться.
В табл. 2.5 представлены коэффициенты термического рас-
ширения типичных стеклообразующих соединений, а также про-
изведения коэффициентов расширения на температуры стекло-
вания. Если коэффициенты термического расширения для край-
них членов приводимого ряда (кварцевое стекло и селен) отли-
чаются друг от друга в 80 раз, то произведения aTg для этих
фаз различаются лишь в 15 раз. Если же SiO2 исключить из
рассмотрения, то aTg различаются в 3 раза. Используя коэф-
фициенты Пуассона, приводимые в работах Куркяна и сотр.
[61] и Линке [327, 343, 351], можно получить представленные
в табл. 2.5 значения коэффициента пропорциональности К из
уравнения (2.36). Для всех стеклообразных фаз, кроме квар-
цевого стекла, значения А близки между собой. Этот вывод
подтверждается результатами исследований Коэпена [141] раз-
личных оксидных стекол и органических полимеров, а также
данными для халькогенидных стекол, т. е. веществ с различной
степенью связности стеклообразного каркаса [437].
80 Глава 2
Таблица 2.5. Взаимосвязь а (300—500 К) и Tg для некоторых
стеклообразных соединений
Параметр SiO2 BeF2 ОеО2 GeSj OejSj GeSej
а • 106, К’1 0,5 6,8 7,7 12 10,9+0,6 16
aTg • 103 0,75 3,56 6,57 9,18 6,93 10,6
а, 0,17 0,214 0,244
К • 103 4,37 26,3 21,8
Параметр Ge?Se3 BjO3 AS2S3 As2S 63 Se
a * 10е, К-' 12,2+0,3 15 23,9+1,3 21,6+1,4 40
aTg ♦ 103 7,48 8,25 10,87 9,61 12,2
И 0,258 0,273 0,305 0,297 0,329
к • io3 21,0 20,6 20,85 19,7 18,5
Выше температуры Tg разрыв химических связей приводит
к высокой концентрации вакансий xl = NlI (Na + Nl) , имеющих
размер Vl\ при этом она значительно превышает концентра-
цию вакансий в стекле (здесь NA — число атомов или струк-
турных групп размером vA). Связанное с этим процессом из-
менение свободной энергии = — Т&SL характери-
зуется, с одной стороны, возрастанием внутренней энергии при
постоянном давлении АЯЬ в результате увеличения свободного
объема системы = а с другой — изменением энтропии
которое, помимо небольшого вклада, связанного с измене-
нием колебательной энтропии, определяется энтропией смеше-
ния Nl вакансий и NA атомов или структурных групп. В лите-
ратуре используются’ два способа описания конфигурационной
энтропии [142, 143].
Если предположить, что вакансии и атомы (структурные
группы) при смешении образуют идеальный раствор, т. е. ~
^vL, то выражение для конфигурационной энтропии имеет вид
АЗК0Нф — 7? [(А^l/Nл) In %£ -|- In Хд\
(2.37)
откуда в равновесных условиях (д&G/dNL = 0) следует
Nl = Улехр[—XHJRT] и (NL/NA]xxL при NL < (NA + NL)
(2.38)
Если принять во внимание, что размеры вакансий и соот-
Аморфное и стеклообразное состояние
81
ветствующих частиц (структурных групп или структурных эле-
ментов, участвующих в процессе течения) различны, то
Nl = (UaVaM ехр [-bHL/RTl V = Nava [1 + ехр (-ДЯ£//?Г)]
(2.39)
Для коэффициента расширения расплава арлСп = а+Асх
с учетом, что VA = NAvA, можно привести следующее соотно-
шение:
арзспл = ехр =
1 хт In хт
=°~т-&г (2-40)
Концентрацию вакансий xL,g при температуре Tg можно
определить также из произведения Да-Т^ (^а~ Ут^).
Да —а —а —________1
- Чраспл u - Т 1 I „ —
1 8 1 -Г XL, g
/ М, \
=(-^-]^P(-^L/RTg) (2.41)
V К-1 £ '
Согласно этой модели, концентрация вакансий xllig при Tg
оказывает влияние на свойства стекол. В связи с этим находит
свое объяснение эмпирическое правило Симха и Бойера [144]
ДаТg ъ const (2.42)
Используя коэффициенты расширения образца, имеющего
форму куба, можно получить, что константа из уравнения (2.42)
составляет -~0,116. Это соответствует мольной доле вакансий
Xl^3,6% [144, 145]. Согласно данным [426], это правило
удовлетворительно выполняется для стекол в системах As—S—I
и As—S—Т1, хотя они и имеют относительно большие разли-
чия в значениях Тg. По данным Коэнена [141], для некристал-
лических тел с цепочечной структурой, например для органи-
ческих полимеров или боратных стекол, содержащих оксиды
щелочных металлов, характерны существенно иные значения
произведения ДаТ^, чем для алюмоборосиликатпых и других
стекол, имеющих трехмерную сетчатую структуру. Изучение
халькогенидных стекол с различной структурой сетчатого
каркаса позволило сделать вывод [437], что концентрация ва-
кансий не единственный фактор, определяющий свойства за-
твердевающего стеклообразующего расплава. Для полного опи-
сания явления необходимо также использовать сведения
о структуре стекла.
Из уравнений (2.41) и (2.42) следует, что Tg и ДНг связаны
корреляционной зависимостью, a &HL определяется в высоко-
6 Заказ Д’» 413
82 Глава 2
вязком расплаве работой, затраченной на образование вакансий
и свободного объема. Эти закономерности справедливы также
для стеклообразующих затвердевающих расплавов металличе-
ских сплавов [117].
На взаимосвязь между AHL и энтальпией испарения Л//„сп
указывалось ранее (разд. 2.2.1.3). Дональд и Девис [146] про-
вели корреляцию между T# и энтальпией сублимации для мно-
жества металлических стекол.
Рис. 2.20. Изменение количества вакансий (в отн.единицах) с температурой
при переходе расплава в кристаллическое и стеклообразное состояние.
Если уравнение (2.38) переписать, подставив в него хг,=
=Nl/(Na + Nl), то xl= [exp(&HTJRT) + I]-1; соответствующая
температурная зависимость концентрации вакансий представ-
лена на рис. 2.20. При температуре затвердевания проис-
ходит скачкообразное изменение концентрации вакансий. Затем
в кристаллическом состоянии с понижением температуры наб-
людается дальнейшее постепенное уменьшение этой величины.
При достаточно низких температурах в кристаллах существует
вполне определенная концентрация точечных дефектов, которая
связана со скоростью охлаждения образца (здесь имеются
в виду чисто кинетические причины их сохранения). В области
переохлажденного расплава ход температурной зависимости
концентрации вакансий отвечает равновесной кривой вплоть до
температуры Tz. Начиная с температуры замораживания Tz
вакансии в определенной концентрации «замораживаются»
в структуре стекла. Связанный с Tz остаточный свободный
объем оказывается уже недостаточным для процесса обмена
Аморфное и стеклообразное состояние
83
позициями между различными частицами. Выше температуры Tz
(и Tg) наблюдается более крутой рост xL с температурой, что
отвечает (в соответствии с имеющимися модельными представ-
лениями) увеличению свободного объема.
В этом смысле можно провести аналогию между стеклооб-
разным состоянием и метастабильным состоянием сильно пе-
ресыщенного твердого раствора, характеризующегося наличием
дефектов Френкеля [147].
2.2.2.3. Температура стеклования и теплоемкость
Возникновение вакансий при переходе стекла в состояние
переохлажденного расплава повышает теплосодержание си-
стемы, что приводит к скачкообразному росту мольной теп-
лоемкости (рис. 2.11). По аналогии с тепловым расширением
мольную теплоемкость можно разложить на два вклада:
Ср> стекл — вклад стеклообразного состояния, связанный в основ-
Рис. 2.21. Температурная зависимость мольной теплоемкости Ср некоторых
стеклообразных соединений.
ном с фононным спектром упругих твердых тел, и ДСР— вклад
процесса образования вакансий при переходе в переохлажден-
ный расплав:
Ср = Ср, СТекл + L ( qy ) = Ср, СТекл + ДСр (2.43)
Комбинируя уравнения (2.43) и (2.39) при условии, что xL~
^Nl/Na, получаем
, vA ( ЬНГ \2 Г Д/7, I vA
ДСр = /?-^(^-) exp [--------=/? —d-xUlnxz.)* (2.44)
На рис. 2.21 приведены зависимости СР(Т) [148]. Видно,
что уменьшение теплоемкости Ср в области температур стекло-
6*
84 Глава 2
вания значительно. В отдельных случаях ДСР составляет поло-
вину теплоемкости расплава.
Аналогично теплоемкости Ср и термическому расширению а
величина сжимаемости х также характеризуется скачкообраз-
ным изменением на Ах в области температур стеклования.
2.2.2.4. Температура стеклования и температура плавления
Температуры стеклования Tg и плавления Тпл связаны эм-
пирическим правилом, сформулированным Каузманом [72],
в соответствии с которым-в стеклообразующих системах в ши-
роком интервале температур (100—2000 К) и для многих клас-
сов соединений при скоростях закалки 10_|4-102 К/с приведен-
ная температура стеклования TgfT^ примерно постоянна
Г&/Тпл«2/з (2.45)
Гутцову [111, 149] удалось свести правило Каузмана к теории
свободных объемов. Он смог показать, что отношение Тё1Тпл
описывается следующим приближенным уравнением:
^/7'пл = ’/2 + /? (2.46)
где F — функция замороженной вязкости. При скоростях охла-
ждения ^^lO2 К/с (ц^Ю13 дПа-с) для стеклообразных ве-
ществ F примерно постоянно и равно 0,14-0,15 (рис. 2.22).
Стеклообразующие оксидные системы сложного химического
состава также укладываются в простую линейную зависимость
[151]. В этом случае вместо температуры плавления ГПл
в уравнение (2.46) следует подставить температуру ликвидуса
TL (т. е. Тцл для каждого состава). Правило выполняется
также для стеклообразных органических полимеров [151]. При-
менимость правила к металлическим стсклам всесторонне про-
верялась Дональдом и Девисом [146]. Исходя из уравнения
Линдеманна, Капно [440] вывел соотношение А1У2/30^ /Тпл —
= const (где А1 — молекулярная масса, V — мольный объем,
— температура Дебая).
Путем разложения в ряд уравнения (2.37) Гутцову удалось
показать, что для конфигурационной энтропии во всей области
переохлаждения 0<.Т<СТпл выполняется соотношение
АЗконф 3R exp[-&HL/RT] (2.47)
Для случая неассоциированных жидкостей применимо приб-
лиженное соотношение А5конФ=А//Пл/Гпл~#. Из уравнения
(2.37) тогда следует, что Хь = 0,35. Это означает, что при плав-
лении соответствующего твердого тела количество вакансий
составляет 35 % числа занятых и незанятых узлов структуры
Аморфное и стеклообразное состояние
85
500 1000 1500~ 2000
Т К
Рис. 2.22. Корреляция между температурой стеклования Те и температурой
плавления ГПл (наклон прямой соответствует правилу Каузмана [уравнение
(2.45)].
(ЛД + ЛД). Гутцов [152] провел исследование применимости
этих сильно упрощенных модельных представлений к стеклооб-
разующим расплавам. Даже эта простая теория позволяет
установить взаимосвязь между конфигурационной энтропией
и правилом Каузмана.
Используя правило Трутона для оценки \HL и вводя тем-
пературу Го = 7,пл/2, при которой принимается, что =
можно получить температурную зависимость конфигурационной
энтропии в температурном интервале Тпл~Т0:
ДЗкопф~ДЗпл(277Гпл- 1)
(2.48)
откуда при температуре T = Tg с учетом правила Каузмана
следует
Д.$стекл/ДЗпл /з
(2.49)
Изменение энтропии Д5Стекл при превращении переохлажден-
ного расплава в стекло составляет ~7з изменения энтропии
при кристаллизации расплава Д5Пл при температуре Гпл. От-
ношение обеих величин, следовательно, остается примерно по-
стоянным.
Поскольку в соответствии с уравнением (2.24) Tg связана
со скоростью охлаждения q экспоненциальной зависимостью,
аналогичную закономерность можно ожидать и для ДЗстекл,
86 Глава 2
т. е. для изменения степени порядка замороженного при охла-
ждении расплава. На рис. 2.23 приведены полученные Гутцо-
вым [111] кривые, которые иллюстрируют изменение конфигу-
рационной энтропии затвердевания в зависимости от некоторого
tg q[K/c]
Рис. 2.23. Отношение изменений конфигурационных энтропий Д$Стекл и А5Пл
затвердевания стеклообразных и кристаллических тел из расплава в зависи-
мости от скорости охлаждения и некоторого параметра b (значения приве-
дены у кривых) [111].
Заштрихованное поле соответствует веществам с отчетливой тенденцией к стеклообразо-
ванию.
параметра Ь, который, как следует из теории, для типичных
стеклообразующих расплавов принимает значение от 3,5 до
4,5. Из рис. 2.23 видно, что с ростом скорости охлаждения q
в стекле резко возрастает степень разупорядочения. Правило
Каузмана применимо к системам с «нормальной» тенденцией
к стеклообразованию при скоростях охлаждения 1О-2<<7<
<10 К/с.
2.2.2.5. Тенденция к стеклообразованию
Температуру стеклования в определенном смысле можно
рассматривать как меру термической стабильности стекла. Чем
выше Tgi тем в более широких температурных границах суще-
ствует стекло без изменения формы. Выше температуры
стеклования в общем случае начинается кристаллизация. Нод
тенденцией расплавов к стеклообразованию понимают «силу
инерции», которая сохраняет стеклообразующий расплав при
определенных сравнимых условиях в жидком состоянии, за-
медляя и не давая проявляться процессам кристаллизации
(разд. 2.3.3).
Исходя из физического смысла температуры стеклования
как параметра, используемого для характеристики стеклообраз-
ного состояния, неоднократно предпринимались попытки свя-
Аморфное и стеклообразное состояние
87
зать тенденцию к образованию стекла с этим легко определяе-
мым экспериментальным параметром — температурой стекло-
вания.
Вещества, расплавы которых не удается перевести в стек-
лообразное состояние при скоростях охлаждения
<10К/с, часто все-таки образуют стекла при использовании
других препаративных методов (разд. 2.1.1.4). Найдено, что
для таких веществ отношение Тg/T 1[Л принимает более низкие
значения, что можно рассматривать как выражение меньшей
тенденции к стеклообразованию. Однако во многих случаях
и в этих системах достигаются значения TgjT^, отвечающие
правилу Каузмана. При этом необходимо отметить, что при
крайне высоких скоростях охлаждения Tg повышаются и, сле-
довательно, как указывалось в разд. 2.2.2.4, «замораживаются»
состояния с более высокими значениями конфигурационной
энтропии, так что экспериментально измеренное отношение
Тg(Tn^ = 2/3 или отношение ASctckji/ASu.^ = 1/3 в действительно-
сти принимает более низкие значения, если эти величины
(Тв/Тпл или АЗСтекл/А5пл) пересчитать к условиям, где приме-
нимо правило Каузмана. Применительно к металлическим
стеклам более высокие значения отношения Тне обнару-
жены, несмотря на то что здесь часю вместо температуры Tg
используют температуру кристаллизации. Поэтому объяснимо
критическое отношение к данным Дональда и Девиса. Сопо-
ставление этих данных с полученными в других работах тре-
бует приведения к единой скорости охлаждения или уточнения
правила Каузмана для более высоких скоростей охлаждения,
используемых при получении металлических стекол. Отклоне-
ние к более низким значениям отношения 7,^/ТПл<2/з четко от-
ражает уменьшение тенденции к стеклообразованию в рассмат-
риваемых системах. В этих случаях стекла часто получают пу-
тем конденсации паров на охлажденные до низких температур
подложки.
Другое полуэмпирическое правило для оценки тенденции
к стеклообразованию сформулировано Коэном и Тернбалом
[154]. Они ввели приведенную температуру плавления
Т red == йен (2.50)
и высказали предположение, что тенденция к стеклообразова-
нию увеличивается с уменьшением Tred. Чем выше энтальпия
испарения при Гпл и чем ниже температура плавления или
температура ликвидуса, тем сильнее выражена тенденция рас-
плава к затвердеванию с образованием стекла. Если принять,
что А//„СП»А//Л, то более низкая TrCd одновременно означает,
что А5Конф [уравнение (2.47)] мало, т. е. вещества, для кото-
рых конфигурационная энтропия мало меняется в точке плав-
ления, склонны к образованию стекла при затвердевании
88 Глава 2
расплава. В таких расплавах сильное обменное взаимодействие,
характерное для кристаллических твердых тел, по-прежнему
существует в большей или меньшей степени, и при их образо-
вании разрушается малое количество химических связей, т. е.
речь идет о расплавах с высокой степенью ассоциации струк-
турных групп.
Применение соотношения (2.50) наиболее эффективно в ря-
дах, аналогичных по составу стеклообразных соединений. На-
пример, на его основе находит простое объяснение большая
тенденция к стеклообразованию у веществ вблизи области глу-
бокой эвтектики на диаграмме состояния. Эта тенденция осо-
бенно заметно проявляется у металлических стекол
(разд. 3.1.1.1).
Маркус и Тернбал [155] предложили в качестве дополни-
тельного критерия тенденции к стеклообразованию растворов
в расплавах использовать отклонение измеренной температуры
ликвидуса TL раствора от ожидаемого значения; исходя
из модели идеальных растворов:
ar TLJi-TL ьнЖ, ,
TL,td TL,ld ’ L',d ан*—я ln(l — хв) гД,
где A//kv— мольная энтальпия плавления растворителя, Кк —
температура плавления растворителя, хв— мольная доля рас-
творенного компонента.
Возрастание отношения \Т/TL> id отражает увеличение взаи-
модействия растворенного компонента В с растворителем А.
Тенденция к стеклообразованию также возрастает в этом на-
правлении. Дональд и Девис [146] применили это соотношение
в упрощенном виде к металлическим стеклам, в которых от-
сутствуют промежуточные фазы интерметаллидов. При этом,
опираясь на правило Вегарда, они использовали следующее вы-
ражение: TL, id = Xfj!t„ 4- ХвТпл. Найдено, что при отношении
АТ/Тl, id > 0,2 и при скоростях охлаждения q < 107 К/с ме-
таллические системы, у которых в области составов от А3В до
А5В есть эвтектика, в подавляющем большинстве случаев
склонны к стеклообразованию.
Хруби [156] ввел параметр Kg, характеризующий тенденцию
к стеклообразованию, который связывает температуру стекло-
вания Tg с температурой плавления Тпл и температурой начи-
нающейся кристаллизации в области переохлажденного рас-
плава Ткр:
KG = (Ткр “ Тё)/(Тпл - Ткр) (2.52)
Чем больше разность Ткр—Tg и чем меньше температурный ин-
тервал Тпл—Ткр, тем сильнее заторможены процессы зародыше-
Аморфное и стеклообразное состояние 89
образования и кристаллизации и тем, следовательно, выше
тенденция к стеклообразованию в данной области составов
системы. Все входящие в выражение (2.52) величины легко
определяются экспериментально методами ДТА и ДСК.
2.2.3. Термодинамические функции и стеклообразование
Как следует из рис. 2.11 и 2.15, существует принципиальное
различие в температурной зависимости функций состояния V
и Н при затвердевании кристаллических и стеклообразных фаз
из расплава. На зависимостях объема и энтальпии происходят
скачкообразные изменения этих функций при переходе из рас-
плава в кристаллическое состояние:
V = (-2Я-) ; H^G-T ; S = — ;
X др )т \дТ )р \дТ )р'
<2-53а’
Первые производные свободной энергии претерпевают раз-
рыв, а вторые производные — коэффициент термического рас-
ширения а, сжимаемость % и мольная теплоемкость Ср при
температуре ТПл стремятся к бесконечности:
_ 1 / d2G X. _ 1 / d2G X с _ т ( d2G X
а — V \др дТ J ’ л~ V \ др2 )т' Ьр~ 1 \дТ2 )р
(2.536)
По классификации Эрепфеста, такого рода переходы называют
фазовыми переходами 1-го рода.
В отличие от описанного случая при температуре замора-
живания Tz переохлажденного расплава функции V, Н и S
меняются нескачкообразно. Лишь для вторых производных сво-
бодной энергии G, т. е. для величин а, х и СР, наблюдается
скачкообразное изменение (рис. 2.11 для Ср). И хотя на пер-
вый взгляд кажется, что переход переохлажденного расплава
в стеклообразное состояние и обратный переход можно рас-
сматривать как фазовый переход второго рода, возникает су-
щественное возражение против такого подхода, поскольку стек-
лообразование из-за температурной зависимости процессов ре-
лаксации подчиняется кинетическим закономерностям и не мо-
жет быть описано в рамках термодинамических представлений.
Стекла — это замороженные неравновесные системы. По своей
сути переход из расплава в стеклообразное состояние не отно-
сится к фазовым переходам 2-го рода, поскольку последний
предполагает равновесное существование обеих фаз при каждой
температуре в области перехода. При затвердевании же стекла
происходит переход из равновесного в неравновесное состояние.
90 Глава 2
2.2.3.1. Парадокс Каузмана
Каузман [72] обратил внимание на то обстоятельство, что
в основе перехода в стеклообразное состояние лежат термоди-
намические предпосылки.
Если предположить, что время проведения эксперимента на-
столько велико, что в системе устанавливается равновесие, то,
построив зависимости Н (Т) и V(Т) для переохлажденных рас-
плавов (рис. 2.11 и 2.15), можно мысленно продлить соответст-
вующие кривые в область низких температур до их пересече-
ния с соответствующими кривыми Н(Г) и V (Т) для твердых
фаз. В точке пересечения 7grmin имеем АН = ЯЖ——О,
откуда
д(//ж-//кр)
дТ
= \СР = Т
д (Зж SKp)
дТ
(2.54)
т. е. АЗ = 0. При еще более низких температурах AS оказалось
бы отрицательной, т. е. 5ж<Зкр. Вряд ли можно себе предста-
вить, что энтропия расплава могла бы существенно сблизиться
с энтропией кристаллической фазы. Поэтому из термодинами-
ческих соображений разности //ж—Я1ф и Зж—SKp должны, на-
чиная с определенной температуры, стремиться к некоторому
постоянному положительному значению или в крайнем случае
к нулю. В области стеклования происходит именно такой пе-
реход. Мольная теплоемкость переохлажденного расплава
уменьшается па АСР. Аналогичные изменения происходят с ко-
эффициентом термического расширения (Аа) и сжимаемостью
(Ах). Причем мольная теплоемкость, коэффициент термического
расширения и сжимаемость стеклообразных фаз лишь незна-
чительно отличаются от соответствующих параметров кристал-
лических твердых тел. Замороженный переохлажденный рас-
плав характеризуется значениями Ср, а и х, которые тем ближе
Таблица 2.6. Коэффициенты расширения и мольная теплоемкость
некоторых кристаллических и стеклообразных соединений
Соединение “стекл • |0'!- К“‘ акр’10’’ К- Ср, стекл’ Дж/(моль-К) ср, кр* Дж/(моль’К)
Se 40 74 (_Lc), -17,9 ( Ц с) 25,63 (300 К) 24,52 (300 К)
AS2S3 20 — 23,0 (300 К) —
As2Se3 21 — 25,0 (300 К) 24,6 (300 К)
В2О3 15,4 — 7,83 (169,4 К) 7,52(170 К)
S1O2 0,5 14,2 (_1_с), 8,7 ( [| с) 14,58 (298 К) 14,92 (298 К)
Аморфное и стеклообразное состояние 91
к соответствующим свойствам кристаллов, чем медленнее про-
водилось- охлаждение, т. е. избыточные величины ДСР, Да и Дх
связаны с процессом релаксации и проходят через нуль при
переходе в стеклообразное состояние (при температурах ниже
Tg) (разд. 2.2.1.3).
Чтобы избежать физически бессмысленной ситуации, когда
5ж<5Кр, необходимо допустить, что переход в стеклообразное
состояние термодинамически неизбежен. Это предположение
на первый взгляд плохо гармонирует с объяснением процесса
стеклования в рамках релаксационных явлений.
Объяснение кажущегося противоречия предложено в рабо-
тах Гиббса и сотр. Так, удалось показать [157], что в переох-
лажденных расплавах, в которых существуют структурные эле-
менты или области с достаточной асимметрией и малой
подвижностью, при некоторой температуре T2^Tg должен
происходить фазовый переход 2-го рода. Полагают, что, даже
если достижение равновесия кинетически не заторможено, все
равно требуется достаточно длительное время для установления
равновесия в системе. Температура Т2 имеет те же особенно-
сти, что и температура стеклования. Ее нижняя граница соот-
ветствует значению Т#, min на рис. 2.11 и 2.15. Эксперименталь-
ное определение этой точки, которую называют температурой
перехода в идеальное стекло, потребовало бы бесконечно дол-
гого времени.
В согласии с экспериментальными фактами находятся ре-
зультаты расчетов, проведенных в рамках молекулярно-стати-
стической модели. В соответствии с этими расчетами темпера-
тура фазового перехода 2-го рода Т2 хорошо согласуете^ & ве-
личиной Tg, если принять, что эксперимент проводится доста-
точно длительное время. Такое совпадение свидетельствует
о том, что оба процесса, протекающие при температурах Tg
и Т2, близки по своей природе. Другим результатом этого чисто
термодинамического рассмотрения является вывод об относи-
тельно значительном уменьшении числа микросостояний системы
при приближении к температуре фазового перехода 2-го рода.
Поэтому неизбежно должно наступить существенное изменение
кинетических свойств системы и, следовательно, времени про-
текания релаксационных процессов. Происходящие в кристал-
лах с изменением температуры переходы порядок беспоря-
док точно так же относятся к фазовым переходам первого или
более высоких порядков [158].
В этом смысле переход в стеклообразное состояние является
обязательным процессом с термодинамической точки зрения.
Однако в условиях реального эксперимента это превращение
регулируется кинетическими факторами, например заморажи-
ванием релаксационных процессов. Таким образом, парадокс
Каузмаиа представляет собой мнимую проблему. Существенно
92 Глава 2
новым вкладом является утверждение факта, что стеклообра-
зование обусловлено термодинамическими законами, а релак-
сационные процессы можно рассматривать всего лишь как про-
изводные (второстепенные) явления.
Возрастание времени релаксации, по-видимому, является
следствием уменьшения конфигурационной энтропии в области
температур стеклования [118]. Размеры кооперативных обла-
стей, в которых могут происходить изменения конфигураций
структуры, возрастают с понижением температуры. Растущий
дефицит структурных конфигураций — причина постоянного по-
нижения конфигурационной энтропии Д5КОнф, которая при не-
которой определенной температуре Г2 (температура фазового
перехода 2-го рода) стала бы равной нулю, если бы можно
было наблюдать за пей. Такой подход позволяет не только ка-
чественно интерпретировать эмпирические уравнения (2.30) и
(2.34), но и полуколичественно объяснить хорошее совпадение
входящих в них коэффициентов с экспериментальными дан-
ными. Сообразно с этим температура Т2, при которой Л5КОнф = 0,
во многих стеклообразующих системах должна лежать при-
мерно на 50 К ниже температуры стеклования Tg (разд. 2.2.1.3).
В этом случае говорят об идеальном аморфном состоянии твер-
дого тела, которое находится во внутреннем равновесии. Од-
нако, поскольку T2 = Tg, min можно достичь лишь за бесконечно
длинный промежуток времени, это состояние остается, таким
образом, неким реально недостижимым состоянием. При Тg>
>Т2 переход в реальное стеклообразное состояние можно на-
блюдать экспериментально. Он происходит с образованием за-
мороженной структуры жидкости, которая характеризуется не-
которой остаточной конфигурационной энтропией. Считается,
что реальное состояние стекла может совсем немного отли-
чаться от «равновесного состояния» идеального стекла при тем-
пературе Т2, если проводить достаточно медленное охлаждение.
Несмотря на то что энтропийная модель Гиббса неодно-
кратно признавалась как лучшая теоретическая концепция для
объяснения процесса стеклования и даже была использована
Коэном [159] для интерпретации вязкости переохлажденных
металлических расплавов, она не свободна от некоторых проти-
воречий. Равенство нулю Д5Копф означает, что стекло в обла-
сти температур 0<Т<Т2 можно рассматривать как состояние
с одинаковой степенью порядка. Однако наш опыт говорит
о том, что в зависимости от термической предыстории стекло
может находиться в различных состояниях.
Гольдштейн [160] указывал, что разность мольных теплоем-
костей ДСР переохлажденного расплава и стекла содержит раз-
личные вклады, которые не могут быть сведены лишь к изме-
нению конфигурационной энтропии. Речь идет, например, о том,
что при описании данного конфигурационного состояния также
Аморфное и стеклообразное состояние 93
необходимо учитывать изменение частот и ангармоничности
колебаний. Переход 2-го рода в этой связи может быть обо-
снован даже при отсутствии вклада от конфигурационной
энтропии. Согласно молекулярно-статистическихМ моделям
Канно [441], последнее обстоятельство тесно связано с транс-
ляционным движением, которое при температурах >7'2, но ниже
температуры стеклования регулируется кинетическими пара-
метрами.
2.2.3.2. Уравнение Пригожина—Дефея
Из различного наклона кривых, описывающих температур-
ную зависимость объема и энтальпии (рис. 2.11, 2.15 и 2.16),
а также из изменения сжимаемости образца следует характери-
стическое уравнение Эренфеста для фазовых переходов 2-го
рода [161]:
П » ДС? Дх/ZV (Да)2 = 1; = = (2'55)
Экспериментально было найдено, что это соотношение не
выполняется для стекол: dTg/dp<AK/ha. Согласно представле-
ниям Пригожина и Дефея [162], уравнение (2.55) вытекает из
положений термодинамики необратимых процессов. В системе
с заданным брутто-составом, в которой протекает п независи-
мых реакций, существует п различных внутренних параметров,
определяющих состояние системы. В таком случае свободная
энергия зависит как от внешних параметров р, Т и х, так и
от степени протекания каждой из этих реакций. В заморожен-
ном состоянии им соответствуют п параметров порядка, кото-
рым для наглядности можно придать смысл степени превраще-
ния в ходе различных процессов. В то же время замораживание
различных протекающих независимо друг от друга изменений
структурных конфигураций происходит при различных темпе-
ратурах, и весь процесс можно описать введением некоторой
фиктивной температуры Тула [90]. Из сказанного становится
понятной корреляция внутренних параметров системы со спект-
ром времен релаксации, о существовании которого упомина-
лось в разд 2.2 1.1
По мнению Пригожина и Дефея, уравнение (2.55) справед-
ливо в случае, когда для описания системы достаточно одного
параметра порядка наряду с р, Т и концентрацией х. Если не-
обходимо использовать несколько параметров порядка (как
в случае перехода в стеклообразное состояние), то выполняется
неравенство Девиса и Джонса [91]
П = ЬСР \k/TV (Да)2 > 1 (2.56)
Гупта и Мойнихан [163] определили, что для стеклообразного
В2Оз П = 4,7, а для стеклообразного селена П = 2,0. Из изме-
94 Глава 2
рений AV(AT), AT (Др), АЯ (AT) получены по уравнению (2.18)
разные значения функций релаксации [164]. Существование
нескольких параметров порядка рассмотрено на примере стек-
лообразного В20з[164] в рамках нелинейной модели структур-
ной релаксации. Проведено обобщение модели с учетом напря-
женности электрического поля в качестве дополнительного
внешнего параметра [442].
2.2.3.3. Термические свойства систем при низких
температурах
Еще Тамман (см. литературу к гл. 1 [2]) установил, что
мольная теплоемкость стеклообразных и кристаллических мо-
дификаций одних и тех же веществ почти одинакова. Поскольку
температуры замораживания Тг и стеклования Тё находятся
всегда выше так называемой температуры перехода в идеаль-
ное стекло Т2, структура стекла при температуре Tg характе-
ризуется небольшим значением конфигурационной энтропии
Д5Конф. Следовательно, при абсолютном нуле энтропия стекла
на эту небольшую величину должна отличаться от нуля, если
предположить, что мольная теплоемкость имеет нормальный
ход температурной зависимости
Tg
So, стекл = А^конф, == j ^Ср d In Т (2.57)
В этом уравнении АСР соответствует изменению мольной теп-
лоемкости в области стеклования.
Согласно теории Дебая, температурная зависимость Су
в твердых телах выражается соотношением
Cv = 97? 4
X3
ехр (х — 1)
dx
Т ехр [©gr/r) — 1]
(2.58)
где Qgr — определяемая максимальной частотой колебаний
Vgr характеристическая температура Дебая, равная B = hxSr/k,
a x = hvlkT. При высоких температурах (7’>0gr) Cv равна
классическому значению 37? (рис. 2.21), а при низких темпера-
турах (7<c0g?.) Су—>0 причем вблизи абсолютного нуля функ-
ция Су приближенно описывается законом Дебая, называемым
законом Г3 (Су = аТ3). Для металлов, у которых заметный вклад
в теплоемкость вносит электронный газ, выполняется соотно-
шение Су = аТ3 + ЬТ. Связь между Су и более доступной и
удобной для измерений Ср дается выражением
Ср - Су = TV и2/и
(2.59)
Аморфное и стеклообразное состояние
95
Из общего уравнения следует, что
т
Д5кол = 5т-5г-.о= J Cpd\nT (2.60)
г=о
Колебательная энтропия при приближении к температуре абсо-
лютного нуля стремится к нулю.
На первый взгляд теория Дебая, описывающая температур-
ную зависимость мольной теплоемкости в области низких темпе-
ратур, должна была бы быть справедливой и для стеклообраз-
ных твердых тел. Ниже температуры 1 К длины волн домини-
рующих в спектре фононов больше, чем 104 пм, что существенно
превышает размер флуктуаций плотности и даже микроскопи-
ческих дефектов, возникающих из-за апериодического сочленения
структурных групп. В связи с этим можно ожидать, что для ди-
электрических стекол справедлив закон Дебая Т3. Измерения за-
тухания ультразвуковых воли и калориметрические исследова-
ния привели, однако, к удивительному результату: закон Дебая
нс выполняется вовсе или выполняется только в узких темпера-
турных диапазонах. Подробный обзор по этому вопросу дан
Йсклем и сотр. [165].
Для кристаллического селена закон Т3 выполняется в обла-
сти от 8 до ГО К, для кристаллических модификаций SiO2 —
только в области 1—2 К [166, 167]. Температурная зависимость
мольной теплоемкости стеклообразного селена проходит через
максимум при 4 К, причем при этой температуре теплоемкость
в 10 раз больше значения теплоемкости кристаллического се-
лена [166]. В кварцевом стекле широкий максимум наблюдается
при 50 К, при температуре 5 К существует еще один менее рез-
кий подъем теплоемкости [165, 168, 169]. Существование такого
рода особенностей на кривых теплоемкости характерно и для
других стеклообразных соединений, например BeF2 [170, 171],
GeO2, ZnP2C>6, В2О3 и Аз20з [170], причем эти эффекты весьма
чувствительны к термической предыстории образца. Наличие
таких эффектов объясняют термически активируемой релакса-
цией определенных структурных дефектов, например при су-
ществовании двух различных равновесных позиций мостиковых
кислородных атомов.
К обсуждаемым эффектам можно отнести такой температур-
ный ход теплоемкости стекол, когда до температуры ~2 К вы-
полняется закон Дебая Т3, а начиная с 1 К происходит сильное
отклонение теплоемкости в сторону положительных значений.
Цеттер и Поль [172] установили, что при низких температурах
в этих случаях теплоемкость описывается линейным уравнением
Cv =АТ. Эта низкотемпературная область особенно подробно
изучена в работах Дрансфельда и сотр. [173]. Удельная тепло-
емкость кварцевого стекла, например, при 30 мК примерно
96 Глава 2
в 1000 раз выше, чем у кристаллического SiO2 (рис. 2.24), а теп-
лопроводность заметно меньше и пропорциональна Т2. В отличие
от кристаллического состояния в кварцевом стекле затухание
Рис. 2.24. Удельные теплоемкости Ср SiO2
при низких температурах [173].
1 — кварц, 2 — кварцевое стекло.
ультразвуковых волн зна-
чительно и даже возраста-
ет с уменьшением темпе-
ратуры.
В качестве наиболее
интересного факта необ-
ходимо упомянуть о су-
ществовании зависимости
затухания ультразвуко-
вых волн от амплитуды в
области сверхнизких тем-
ператур, а также о зави-
симости этого явления от
состава. Аналогичные эф-
фекты обнаружены при
изучении распростране-'
ния электромагнитных
волн. На основании сово-
купности этих явлений
можно заключить о нали-
чии двух вкладов в теп-
лоемкость, отвечающих
двум видам дефектов,
физическая природа ко-
торых до сих пор не уста-
новлена.
Универсальность этих
характерных для стекол
температурных эффек-
тов позволяет предположить, что в данном случае речь идет
о свойствах, которые присущи лишь стеклообразному состоянию
и которые выделяют его среди других состояний вещества. Ста-
новится понятным, что небольшая часть конфигурационной
энтропии стеклообразного расплава, которая замораживается
в твердом теле при его затвердевании, оказывает влияние
на определенные свойства стекла лишь при низких температу-
рах.
2.3. Фазовые переходы и стеклообразование
Образование стекла из переохлажденных расплавов или оса-
ждение аморфных веществ из газовой фазы связано с подавле-
нием процессов кристаллизации. При достаточно низких скоро-
Аморфное и стеклообразное состояние 97
стих кондесации и относительно высоких температурах под-
ложки или при скорости охлаждения расплава ниже критиче-
ской, а также при проведении отжига стекол выше температур
стеклования возникают кристаллические фазы различного хи-
мического состава.
Расстекловывание прежде всего рассматривается как нега-
тивное явление, поскольку оно мешает изготовлению стекла.
При проведении же направленной кристаллизации с целью по-
лучения стеклокерамических материалов со специальными свой-
ствами необходимо использовать знания о закономерностях про-
текания в стеклообразующих системах процессов упорядочения.
Теоретические соотношения, описывающие процессы кристал-
лизации, также служат основой при определении условий от-
сутствия фазовых превращений в переохлажденных стеклооб-
разующих расплавах. Таким образом, изучение процессов
кристаллизации позволяет характеризовать стеклообразное со-
стояние и тенденцию к образованию стеклообразных фаз в раз-
личных системах с новой стороны. В этой связи на первый план
выдвигаются кинетические аспекты стеклообразования.
2.3.1. Выделение фаз из стеклообразующих расплавов
Переохлажденные расплавы стеклообразующих соединений
переходят при отжиге в кристаллические модификации этих ве-
ществ. При наличии нескольких полиморфных модификаций
сначала, как правило, образуется фаза, характеризующаяся
большим запасом энергии. Фазовый переход происходит без
изменения состава и может быть описан с помощью законов
кристаллизации.
Из расплавов, брутто-состав которых находится вне обла-
сти гомогенности определенного химического соединения, в пер-
вую очередь кристаллизуется фаза другого состава. В этом слу-
чае фазовый переход можно точно так же свести к двум одно-
временно протекающим процессам — образованию зародышей
кристаллов и их росту из гомогенного расплава.
Часто в сложных системах метастабильный гомогенный пе-
реохлажденный расплав не устойчив ниже определенной тем-
пературы, и в нем перед выделением кристаллических фаз про-
текают процессы ликвации. Эти процессы характеризуются
тем, что из расплавов выделяется, например, жидкая фаза
в форме отдельных каплевидных областей. Матричная и капле-
видные области имеют разный химический состав. Законы заро-
дышеобразования и роста в данном случае можно применять
к процессам образования этих расслаивающихся областей. Кри-
сталлизация обычно следует за этими процессами. Чаще всего
первоначально кристаллизация протекает в каплевидных обла-
стях или на их границе с матричной областью.
7 Заказ № 413
98 Глава 2
Кроме того, в системах, для которых характерна ликвация
в жидком состоянии, можно наблюдать существование таких
областей сложного состава, где процесс разделения фаз про-
текает без образования зародышей и, следовательно, без за-
медления.
2.3.1.1. Зародышеобразование и рост кристаллов
Тамман предложил (см. литературу к гл. 1 [2]) описывать
кристаллизацию жидкостей или выпадение кристаллов из рас-
творов, используя модели процессов зародышеобразования и
связанных с ними процессов роста кристаллов. Выведенные
исходя из упрощенных модельных представлений теоретиче-
ские положения в различной форме применены к реальным си-
стемам и подвергнуты экспериментальной проверке. Обобщен-
ные результаты проведенных исследований приведены в мно-
гочисленных публикациях [174—180, 453].
Гомогенное стационарное зародышеобразование. Для кон-
груэнтно плавящихся соединений скорость стационарного за-
родышеобразования можно выразить числом способных к росту
кристаллических центров, которые образуются в единице объ-
ема в единицу времени в переохлажденном расплаве за счет
термических флуктуаций:
Iv = NKTzovk (2.61)
Здесь NK — концентрация зародышей критического размера
в 1 см3 расплава в условиях равновесия; Г — коэффициент
Зельдовича, отражающий неравновесное состояние системы;
20 — коэффициент, характеризующий количество структурных
элементов на поверхности зародыша критического размера;
zqvk — скорость, с которой частицы и структурные группы пе-
реходят из переохлажденного расплава на поверхность заро-
дыша критического размера, что приводит к его росту.
Движущая сила процесса кристаллизации определяется раз-
ностью свободных энергий расплава 6расп и кристалла GK:
&Gl4) = GK—Gpacn- Она возникает лишь в области переохлажде-
ния (7’<7’пл) и действует после некоторой временной задержки.
Причина этого состоит в том, что при такого рода фазовых пе-
реходах процесс развивается не сразу во всем объеме. Субми-
кроскопические зародыши обладают большим запасом энергии,
чем макроскопический кристалл. Из-за относительно большого
числа частиц, которые находятся на поверхности зародышей,
температура плавления последних понижена, причем тем
больше, чем меньше размеры кристаллических зародышей. Для
того чтобы учесть эти обстоятельства, обычно рассматривают
раздельно разность свободных энергий одних и тех же ансамб-
Аморфное и стеклообразное состояние
99
лей частиц в расплаве и в объеме зародыша (AgO6) и разность
свободных энергий ансамблей частиц в расплаве и на поверх-
ности зародыша (Л^пов). Таким образом, переходят от моль-
ных к удельным величинам. Agnos действует в обратном направ-
лении по сравнению с движущей силой Agoo, которая соответ-
ствует веществу, находящемуся в объеме кристаллического за-
родыша. Так что
AgKp== Ag06 Н- А^пов (2.62)
Если зародыши имеют сферическую форму
AgKp = — (4л/3) г3 Ago6 + 4лг2 Ag’noe (2.63)
На графике зависимости AgKp от г имеется максимум при
Гкрит= 2 А^пов/А^об", В ЭТОЙ ТОЧКе Agmax = 16л Ag пов /3Ago<j. Тер-
модинамически в области гсгкрит должен происходить распад
зародышей, и лишь для г>гкРит вероятность дальнейшего роста
зародышей становится преобладающей. гкрит называют критиче-
ским радиусом зародыша, a Agmax— максимальной свободной
энергией зародышеобразования, которую необходимо затратить,
чтобы произошел фазовый переход.
Если для оценки Ag06 использовать приближение Хоффма-
на [181], а для А^пов — приближение Тернбала [182]
Дг„0 = -^!-ГДГ; = (2.64)
’ * ПЛ
ТО
. 1блр3//плт^л . 2₽к’^,л
Agmax 3NAT2(bT)2 ’ Гкрит — ду.
(2.65)
где АЯПл — мольная энтальпия плавления, А7’ = ТПл—Т— пере-
охлаждение, р — коэффициент, который для стеклообразных
соединений различного типа равен 0,32 [209]; для металлов
этот коэффициент равен 0,46. Можно заметить, что при малом
переохлаждении Agmax велико, и также велико значение Гкрит*
Таким образом, термодинамическая вероятность образования
способных к росту зародышей очень мала при температурах
чуть ниже точки плавления, и, следовательно, для протекания
этого процесса требуется значительное время. И Agmax и гкрит,
однако, уменьшаются при понижении температуры, т. е. с уве-
личением переохлаждения. Равновесная концентрация зароды-
шей NK критического размера возрастает по следующему)
закону. )
AK = Mexp[—Agmax/ZeT) (2.66
Константа скорости vK в уравнении (2.61) может быть связана
с коэффициентом диффузии в поверхностном слое DK = a2VK>
7 *
100 Глава 2
если принять, что путь частицы, находящейся на поверхности
зародыша, равен ес среднему диаметру а. Используя уравнение
Стокса—Эйнштейна D = kT/3m]a и соотношение а'А = М/рМ а,
можно получить выражение для скорости гомогенного зародыше-
образования
/об = Г 4=2- ex Pl-Д g^JkT]
(2.67)
Рис. 2.25. Температурная зависимость ско-
рости гомогенного зародышеобразования /Об
и скорости зародышеобразования /об на
поверхности переохлажденного расплава,
а также скорости роста кристаллов и для
стекла состава (Na2O)2i,25(CaO)2i.25(SiO2)57,5
[184].
Тошев и Гутцов [183] приводят
множителя Г:
После подстановки в не-
го уравнения (2.25) полу-
чаем
I -р NRTp
7об= 1 ~ —
ЗлЛщо
X
X [exp— (Ag max + E^)/k Г]
(2.68)
Это уравнение спра-
ведливо в приближении,
что перенос вещества че-
рез границу между по-
верхностью зародыша и
расплавом определяется
тем же механизмом, что
и перенос в объеме пере-
охлажденного расплава
{DK = D).
следующее выражение для
г = (1/Sfx) VА^тах/Зл/гГ
(2.69)
т. е. Г зависит от температуры. gK — число структурных эле-
ментов в кристаллическом зародыше.
При низких температурах, т. е. при сильном переохлажде-
нии, значение /Об невелико, так как оказывает существенное
влияние повышение Если переохлаждение мало, то /Об
также уменьшается из-за высоких значений Agmax- И лишь
в области среднего переохлаждения можно ожидать макси-
мальной скорости зародышеобразования. На рис. 2.25 приве-
дена экспериментально полученная кривая скорости зародыше-
образования в стекле Na2O—СаО—SiO2 [184].
Нестационарное зародышеобразование. Как уже говорилось
(разд. 2.2.1), при описании любых процессов при нагревании
или охлаждении высоковязкого переохлажденного расплава не-
Аморфное и стеклообразное состояние
101
(2.70)
^/Об не за-
сл еду ющее
обходимо считаться с некоторым отставанием во времени. На-
чиная с определенных температур это относится и к установ-
лению стационарной скорости зародышеобразования, т. е. вме-
сто уравнения (2.68) необходимо использовать
/об. е =/об ехр (—
Это означает, что только через время t > 5тг /Об, t
висит от времени. Тошев и Гутцов [183] вывели
уравнение для индукционного периода тг:
А£повт1а ^Л^пл
= 1,6
(2.71)
Рис. 2.26. Образование зародыша
К из расплава S на инородной
подложке (0 — угол смачивания).
(ЛГ)2
еще
более приближенные вы-
[185] использовал следую-
(где гкрит«4а)
(2.72)
Впоследствии предложены
ражения для ту. Например, Хиллиг
щее соотношение:
_ ~ ~ (Гкрит)2 _ Зл2Л1г]
ту-«л — RT(y
и время релаксации в уравнении (2.12), пропорцио-
вязкости, а поскольку вязкость экспоненциально возра-
понижением температуры, индукционный период может
Тг, как
нально
стает с
характеризоваться чрезвычайно большими значениями. В таких
условиях зародышеобразование практически невозможно наблю-
дать. Для кварцевого стекла по оценке индукционный период
при 1573 К составляет Тг = 5-104 с [186], а экспериментально
найдено Ti =3 • 105-4-106 с [187].
Гетерогенное зародышеобразование. В общем случае на го-
могенное зародышеобразование в объеме переохлажденного рас-
плава накладывается гетерогенное зародышеобразование, кото-
рое инициируется неконтролируемым присутствием инородных
частиц и чаще всего протекает на поверхности или в местах
контакта расплава со стенками тигля. Гетерогенное зародыше-
образование в значительной мере определяется степенью сма-
чивания расплавом зародышей и чужеродной подложки
(рис. 2.26). Его можно описать путем введения дополнитель-
ного параметра — угла смачивания 0, который может меняться
от 0 = 0° (полное смачивание) до 9=180° (отсутствие смачи-
вания).
Если обозначить свободные энергии поверхностей раздела
соответствующих фаз как Д§’ПОв(5, Л), Д§’ПОв(5, U), Д§’пов(Л,
102 Глава 2
(7*), то с помощью уравнения Юнга для углов смачивания мо-
жно записать следующее соотношение для изменения мольной
свободной энергии процесса гетерогенного зародышеобразования:
AgKp = — ЛГ2 (1 — COS2 0) AgnoB (S, Д') cos 0 -4-
4- 2лг2 (1 — cos 0) AgnOB (S, К) — (л/3) г3 (2 + cos 0) (1 — cos 0)2 Ago6
(2.73)
Радиус критического зародыша равен гкрит = 2 Agn0B(S, K)/Ag06,
а для максимальной свободной энергии зародышеобразования
А ' 4л пов ($, К) .
Agmax = -----------------(2 + COS 0) (1 — COS 0)2
3 Д£об
bg™ = Ag™ (2.74)
Если подложка не смачивается зародышем (0=180°), то
Agmax — Agmax, и в этом случае гетерогенное зародышеобразо-
вание не оказывает никакого влияния на гомогенное зародыше-
образование. Предполагается [443], что гетерогенные частицы
и поверхности раздела практически не влияют на скорость го-
могенного зародышеобразования, если углы смачивания превы-
шают 0>1ОО°. Напротив, в случае сильного смачивания макси-
мальное изменение свободной энергии гомогенного зародышеоб-
разования сильно уменьшается, и это ускоряет процесс кри-
сталлизации из раствора.
В уравнение (2.62) можно ввести еще один дополнительный
член: AGz — изменение мольной свободной энергии упругой де-
формации. Эта величина оказывает такое же тормозящее влия-
ние на скорость зародышеобразования, как и АСПов- Поскольку
AGz приходится в основном на поверхность, процессы зароды-
шеобразования по энергетическим причинам заканчиваются
преимущественно на поверхности даже в отсутствие гетероген-
ных частиц (рис. 2.25). При этом часто трудно отличить гомо-
генное зародышеобразование от гетерогенного.
Скорость кристаллизации. Исходной информацией для рас-
чета скорости кристаллизации является скорость /ог>, с которой
в единицу времени в единице объема возникают в случае ста-
ционарного процесса способные к росту зародыши. Последую-
щие этапы кристаллизации можно описать, опираясь на законы
роста кристаллов во времени. Линейная скорость роста кри-
сталлов может быть определена как разность скорости переноса
* Буквы 5, К и U обозначают расплав, зародыш и подложку соответ-
ственно.— Прим, перев.
Аморфное и стеклообразное состояние
103
вещества avw из расплава в направлении к поверхности заро-
дыша или кристалла и скорости движения частиц в противопо-
ложном направлении (последняя связана со свободной энергией
плавления):
и = avuz[l — ехр(—AG[I;I//?r)] Q (2.75)
Существенное значение имеет количество активных центров
на поверхности зародыша, поскольку осаждение вещества идет
преимущественно на этих центрах. При этом могут происходить
поверхностная диффузия частиц осаждаемого вещества (диф-
фузия Фольмера) и их переход в энергетически более выгод-
ные позиции.
Возможны различные механизмы роста кристаллов. Напри-
мер, в рамках обычной модели предполагается, что все пози-
ции на поверхности зародыша эквивалентны и каждая из них
может служить местом дальнейшего роста кристалла. Рассмат-
риваются также модель роста двумерного зародыша или мо-
дель присоединения молекул вещества к грани винтовой дисло-
кации. В зависимости от того, какой из механизмов роста до-
минирует, Q принимает различные значения или описывается
различными зависимостями.
Принимая, что vw=Dw/a2, с учетом уравнения Стокса—
Эйнштейна D = kTf3nr\a и приближенной зависимости АбКр~
~ АТ/пл ДТ’/Т’пл уравнение (2.75) можно переписать:
“ = -ехр(-д^дШГГ„;,)] (2'76>
откуда для малых переохлаждений следует, что линейная ско-
рость роста кристаллов прямо пропорциональна переохла-
ждению:
,, QA/У пл
U~ 3na\NAT пл
АГ
(2.77)
В основе уравнения (2.76), как и в случае зародышеобразова-
ния, лежит предположение, что механизм переноса вещества
через фазовую границу поверхность кристалла — переохлажден-
ный расплав практически не отличается от механизма переме-
щения частиц из одной позиции в другую в объеме расплава
(DW = D).
Ниже температуры плавления Тил скорость кристаллиза-
ции и быстро достигает высоких значений. Уже при малых
переохлаждениях она проходит через максимум (такой же макси-
мум характерен и для температурной зависимости /об) и пони-
жается до крайне малой величины в области температур стек-
лования Tg (рис. 2.25). Положение максимума может быть оп-
ределено по уравнению (2.76) путем учета температурной
104 Глава 2
зависимости вязкости [разд. 2.2.1.3, уравнение (2.34)]. Для этого
первую производную скорости роста кристалла по температуре
приравнивают к нулю. По данным Шольце и сотр. [188], между
вязкостью вблизи температуры плавления и вязкостью рас-
плава в области максимальной скорости кристаллизации су-
ществует следующее приближенное соотношение:
ln'lrma;,-Innr,,., » I (2-78)
На рис. 2.25 изображена температурная зависимость /Об и и
для силикатного стекла. Видно, что в области малых переохла-
ждений скорость кристаллизации достигает максимума, в то
время как скорость зародышеобразования все еще очень мала.
В то же время при температурах, характеризующихся макси-
мальной частотой образования зародышей, скорость роста кри-
сталлов очень мала. Область перекрывания обеих кривых ки-
нетически наиболее благоприятна для фазового перехода пе-
реохлажденного расплава в кристаллическое состояние. Таким
образом, низкие значения /Об и и нс единственное условие, бла-
гоприятное для образования стекла из переохлажденного рас-
плава. Уменьшение области перекрывания обеих кривых играет
существенную роль для подавления процесса кристаллизации.
Законы протекания кристаллизации во времени. Аврами
[189] составил и решил дифференциальное уравнение, описы-
вающее зависимость изменения объемной доли закристаллизо-
вавшегося вещества Укр во времени (см. также [190]):
lnln[l/(l -Укр)] = (п+ 1) In £ -J- In (F/o6u~) (2.79)
При 72 = 3 уравнение описывает рост кристаллов сферической
формы, при п = 2 — образование плоских кристаллов; при
п = 1 — возникновение игольчатых кристаллов. F — коэффи-
циент, учитывающий форму кристалла. В случае образования
сферических кристаллов справедливо следующее приближенное
соотношение:
УКР = 1 — ехр (—FIo6u3t4) ж (л/3) (если УкР мала) (2.80)
Можно подставить выражения (2.67) для /Об и (2.76) для и
в (2.80) для описания зависимости изменения объемной доли
закристаллизовавшегося переохлажденного расплава от тем-
пературы и времени.
2.3.1.2. Явления разделения фаз
Во многих системах сложного химического состава отклоне-
ние от состояния равновесия сопровождается распадом гомо-
генного расплава на две жидкие фазы. В переохлажденных
расплавах такого рода процесс идет значительно чаще, чем
Аморфное и стеклообразное состояние
105
кристаллизация расплава. Это имеет как энергетические, так и
энтропийные причины.
Из термодинамических соотношений, связывающих свобод-
ную энергию, энтропию и энтальпию, следует, что преимущест-
венное взаимодействие между одинаковыми частицами часто
приводит к распаду расплавов выше температуры плавления
или температуры ликвидуса на две равновесные жидкие фазы.
На диаграмме состояния системы в этом случае возникает об-
ласть расслаивания, которая исчезает лишь выше определенной
критической температуры ТКрит. Эта температура может быть
определена экспериментально по правилу Кайете—Матиаса пу-
тем изучения равновесных концентраций обеих фаз при раз-
личных температурах. С понижением температуры тенденция
к расслаиванию увеличивается из-за все уменьшающегося
вклада энтропии смешения в величину свободной энергии.
В стеклообразующих расплавах, которые выше температуры
ликвидуса представляют собой гомогенные и равновесные жид-
кости и характеризуются явно выраженной склонностью к пе-
реохлаждению, также можно ожидать протекания процессов
расслаивания, по крайней мере в определенных областях со-
ставов. Если критическая температура лежит ниже линии лик-
видуса данной системы, говорят о существовании скрытой об-
ласти расслаивания. Субликвидусное расслаивание можно об-
наружить на диаграмме состояния по точке перегиба на кри-
вой ликвидуса. Если тенденция к расслаиванию проявляется
только ниже температуры стеклования, то разделения фаз не
наблюдается из-за того, что в этом диапазоне температур про-
цессы перемещения частиц в значительной степени блоки-
рованы.
Области микронеоднородности в стекле впервые обнару-
жены в начале 50-х годов благодаря электронно-микроскопиче-
ским исследованиям [191—194]. Позже, в основном усилиями
Фогеля и сотр. [195, 196], на многих системах со всей очевид-
ностью установлено, что это явление весьма широко распро-
странено в стеклообразных системах. В промышленном произ-
водстве стекла изучение процессов разделения фаз привело как
к совершенствованию технологии, так и к созданию совершенно
новых материалов [197, 198]. Наряду с электронной микроско-
пией особое значение для изучения микроликвации имеют ме-
тоды исследования рассеяния света и малоугловое рассеяние
рентгеновских лучей. Успехи в теоретическом описании про-
цессов ликвации в большой степени связаны с работами Кана
[199] и Хиллерта [200]. В многочисленных обзорных статьях
собраны разнообразные сведения о явлении разделения фаз
в переохлажденных стеклообразующих расплавах [201—203].
Существенные закономерности приведены также в одной из ра-
бот Ульмана [204].
106 Глава 2
Рис. 2.27. Изменение свободной энергии смешения G от состава (х2 — мольные
доли) в квазибинарной системе с расслаиванием (а) при температурах Т=Т\
(б), Г=Г2<7’1 (в) и Т=Т3<Т2 (г).
Типичными бинарными системами с отчетливо выраженной
тенденцией к ликвации являются системы SiO2—В2О3 [205],
SiO2—Р2О5, В2О3—Р2О5 (см. литературу к гл. 1 [17]), А12О3—
SiO2 [206], РЬО—В2О3 [207], а также многие другие системы
на основе оксидов металлов.
Термодинамика процессов разделения фаз. На рис. 2.27 при-
ведены результаты применения критериев устойчивости Гиббса
для бинарных смесей к стеклообразующим расплавам, в кото-
рых процессы расслаивания начинаются уже выше температуры
ликвидуса [108].
При температуре Т=Т\ система гомогенна и однофазна.
Свободная энергия смешения выражается соотношением
G = (1 — х2) ц? + W2 + (1 — x<i)RT In а\ + XiRT In а2 (2.81)
Аморфное и стеклообразное состояние 107
Таким образом, кривая G(x2) расположена ниже пунктирной
прямой, которая соединяет значения ц? и ц” на рис. 2.27,6
(0<а< 1). Кривая G(x2) тем круче уходит вниз от прямой, чем
больше коэффициент активности отклоняется от 1 в сторону
более низких значений (f<l). Если f> 1, кривая имеет более
пологий вид. Рис. 2.27,6 соответствует ходу кривой при />1.
Во всей области смешения кривая вогнута к оси абсцисс:
г > 0 (2.82)
Речь идет, таким образом, о гомогенной равновесной фазе.
В общем случае условиями устойчивого равновесия являются
положительные вторые частные производные функций состоя-
ния по одной из их характеристических экстенсивных перемен-
ных (V, S, л') и отрицательные вторые производные по интен-
сивным переменным (р, 7).
При температуре Т = Т2<1\ критерии устойчивости (2.82)
нс выполняются в области составов между двумя точками пе-
региба 1 и 2, где 62(7/6х2 = 0 (рис. 2.27, в). В этой области
фаза самопроизвольно распадается на две даже в результате
бесконечно малого колебания концентрации. Превращение про-
исходит мгновенно и протекает без образования зародышей.
Его называют спинодальны-м распадом. Равновесный состав
обеих сосуществующих фаз отвечает, разумеется, обоим мини-
мумам на привой G(x). Уравнение
(дО/дх{3))Р, т = (дО/дХ(4])р> т (2.83)
отвечает одинаковому наклону касательных в точках 3 и 4.
Лишь при данных составах система устойчива. Между точками
3 и 1 и 2 и 4 выполняется условие (2.82). В этих областях мо-
жет существовать гомогенная фаза, однако по отношению
к смеси двух фаз составов 3 и 4 эта фаза метастабильна. Здесь
процесс разделения фаз проходит стадию зародышеобразова-
ния с последующим ростом областей расслаивания.
Таким образом, стабильное существование только одной_го-
могенной фазы возможно лишь в областях, где кривая G(x)
вогнута в сторону оси х. Между точками 3 и 4 кривая выпукла,
и стабильность нарушается. Здесь существуют две метаста-
бильные области составов (между точками 3 и 1 и 2 и 4),
а также одна лабильная область (между точками 1 и 2). Если
рассмотреть процессы расслаивания при других температурах,
то в результате соединения набора точек 3 и 4 можно получить
равновесную кривую зависимости состава сосуществующих жид-
ких фаз от температуры (рис. 2.27, п). Точки перегиба 1 и 2
образуют спинодаль (штриховая линия на рис. 2.27, а), которая
разделяет стабильную область от нестабильной.
108 Глава 2
При температуре Т=Т3<Т2 в равновесии с гомогенной жид-
кой фазой находится кристаллическая фаза 5 (рис. 2.27, г).
В зависимости от кинетики установления равновесия между
этими состояниями кристаллическая твердая фаза будет иметь
различную морфологию.
Кинетика процессов разделения фаз. Если исходить из со-
става у (рис. 2.27), то при охлаждении расплава даже с отно-
сительно высокой скоростью перед началом кристаллизации
происходит самопроизвольный распад гомогенной смеси на две
жидкие взаимно проникающие фазы. При этом размеры обла-
стей расслаивания тем меньше, чем выше скорость охлаждения.
В случае субликвидусного расслаивания также можно ожидать
в области температуры стеклования Tg образования взаимно
проникающих структур. Это происходит при условии, что про-
цессы переноса вещества в области переохлаждения при дан-
ной скорости охлаждения еще успевают следовать за образо-
ванием фаз с составами, соответствующими спииодали. На
электронно-микроскопических снимках стекла в этом случае
можно обнаружить соответствующие микроструктурированные
области.
Возможно также, что перед затвердеванием расплава
в стекло или в процессе последующего отжига стекла
в одной из двух фаз начнутся процессы зародышеобразования,
которые могут привести к кристаллизации (например, к выпа-
дению фазы 5).
Кроме того, необходимо обратить внимание на то, что со-
ставы, отвечающие спинодали, не являются равновесными. Сле-
довательно, двухфазная система должна стремиться перейти
в равновесное состояние, отвечающее положениям минимумов
на кривой G(x2) (точки 3 и 4). Такой переход произойдет на-
столько глубоко, насколько позволит кинетика процессов пе-
реноса вещества в расплаве и между обеими жидкими фазами
при данных температурах. На пути к равновесному состоянию,
т. е. при подходе к точкам 3 и 4 (а на рис. 2.27, г и при при-
ближении к точке 5), могут начаться процессы зародышеобра-
зования и кристаллизации.
Если исходить из расплава состава г, спинодалыюго рас-
пада не происходит. В метастабильной области законы
термодинамики требуют в случае распада двух жидких фаз
стадии зародышеобразования, вслед за которой должен на-
чаться процесс роста, лимитируемый диффузией. Если опере-
дить эти процессы, применяя высокие скорости охлаждения, то
можно получить гомогенные стекла. В случае если охлаждение
ведется достаточно медленно или если проводится дополнитель-
ный отжиг стекла при температурах T^Tg, то образуются до-
статочно высокие концентрации (1012—1016 см-3) каплевидных
Аморфное и стеклообразное состояние
109
ликвационных областей, которые можно обнаружить на элек-
тронно-микроскопическом снимке. Увеличение размера областей
ликваций в зависимости от температуры и времени исследо-
вано во многих работах [204] (см. литературу к гл. 1 [17]. По-
сле образования микроликвационных фаз процесс обмена ве-
ществ с матрицей часто затормаживается, так что при более
низкой температуре вновь наступает пересыщение, которое мо-
жет привести ко вторичной ликвации как в каплях, так и
в матрице. Известны даже примеры многостадийного протека-
ния таких ликвационных процессов.
Вместо микроликвации при охлаждении расплава состава г
(рис. 2.27) может происходить непосредственная кристалли-
зация. В этом случае определяющее значение имеет макси-
мальная свободная энергия зародышеобразования, поскольку
система, стабилизируясь, переходит в состояние с большей сте-
пенью порядка. Agmax- зависит от свободной энергии поверхно-
сти раздела Agn0B [уравнение (2.65)], а последняя в случае об-
разования каплевидных некристаллических зародышей несом-
ненно меньше, чем в случае образования кристаллических
центров. Измерения, проведенные Хеммелом [210], показали,
что свободная энергия поверхности раздела между расплавом и
капельной фазой для изучавшихся силикатных стекол более
чем на порядок величины меньше, чём между расплавом и кри-
сталлическим зародышем. Отсюда понятно, что процессы лик-
вации имеют бесспорное преимущество перед кристаллиза-
цией, а в областях со сложным химическим составом и в обла-
сти расслаивания они предшествуют процессам расстекловыва-
ния.
В заключение этого краткого обсуждения необходимо ска-
зать о том, что часто невозможно точно установить истинный
механизм микрофазиого разделения. Без дополнительных ис-
следований трудно различить, протекает ли спинодальный рас-
пад переохлажденного расплава или идет процесс зародышеоб-
разования. Каплевидные ликвационные области растут за счет
диффузионных процессов, и благодаря тенденции к уменьше-
нию поверхностного натяжения возникает движущая сила их
коалесценции. Таким образом, может образовываться вторичная
упорядоченная структура [211—213]. В то же время обнару-
жено, что возникшая при спинодальном распаде микрострук-
тура впоследствии может превращаться в отдельные каплевид-
ные ликвационные области [214]. Описаны также примеры,
показывающие, что в зависимости от термической предысто-
рии образца возникает та или иная микроструктура [215,
216].
Механизм ранних стадий процесса разделения фаз часто
бывает замаскирован вторичными процессами, идущими в стек-
лообразующих расплавах. Поэтому экспериментальная проверка
НО Глава 2
теоретических положений сопряжена с существенными трудно-
стями. Можно констатировать, что наши знания о собственно
процессах образования фаз далеки от совершенства.
2.3.2. Критическая скорость охлаждения и тенденция
к стеклообразованию
В соответствии с современными представлениями стекло
представляет собой замороженный переохлажденный расплав,
имеющий гомогенное (в смысле изотропности жидкости) или
микрогстсрогенное строение (если в системе в процессе охла-
ждения произошло разделение фаз). В последнем случае обе
фазы жидкие или — при температурах ниже температуры стек-
лования— стеклообразные. Таким образом, в понятие стекло-
образное состояние включаются как гомогенные, так и микро-
гетерогенные структуры. Различие этих структур проявляется
лишь при переходе в кристаллическое состояние, хотя процесс
разделения фаз можно рассматривать по отношению к гомо-
генному расплаву как первую стадию стабилизации структуры
с более высокой степенью порядка.
Ряд эмпирических соображений, используемых для описа-
ния тенденции к стеклообразованию соединений и сложных си-
стем, приведен в разд. 2.2.2.5. Другой подход к объяснению
этого важнейшего для возникновения аморфных твердых тел
свойства связан с привлечением закономерностей, в основе
которых лежит кинетика образования кристаллических зароды-
шей и их роста. Согласно уравнению (2.67), частота образо-
вания гомогенных зародышей тем меньше, чем выше вязкость
и чем выше максимальная свободная энергия зародышеобразо-
вания. Линейная скорость роста кристаллов, согласно уравне-
нию (2.76), имеет такую же зависимость от вязкости. Она
больше у тех веществ, у которых выше энтальпия плавления.
Наиболее старое определение понятия склонность к стекло-
образованию принадлежит Дитцелю [217]. В качестве пример-
ной меры тенденции вещества к стеклообразованию им предло-
жено использовать обратную линейную скорость кристалли-
зации.
Более удобной и лучше подходящей к кинетическому описа-
нию процессов стеклообразования, по-видимому, является кри-
тическая скорость охлаждения gKpilT, под которой понимают
минимальную скорость охлаждения, когда еще происходит об-
разование стекла. Поскольку совсем непросто обнаружить су-
ществование закристаллизованных областей в стекле, при их
небольшом содержании (это во многом зависит от метода ис-
следования) для частично закристаллизованных твердых тел
необходимо каждый раз проводить более детальные исследо-
вания.
Аморфное и стеклообразное состояние
111
Серджент и Рой |218] сформулировали полуэмпирическое
приближенное правило. Они исходили из простой предпосылки
о том, что <7кРит прямо пропорционально Тпл и обратно про-
порционально времени релаксации т, т. е. вязкости г| [уравне-
ние (2.12)] при ТпЛ:
<7крит = (2.84)
Значение коэффициента с = 2,0-10-6, правда, получено из дан-
ных для NaCl, что кажется странным, поскольку ^Крит для
этого вещества не удалось до сих пор определить ни одним
экспериментальным методом. Для целого ряда различных окси-
дов на базе данного приближения проведена оценка критиче-
ской скорости охлаждения, но достаточно хорошего совпадения
с экспериментальными данными достигнуто не было.
В работах [4, 219] для оценки тенденции к стеклообразо-
вавию выбрана скорость образования зародышей. Она должна
быть ниже критической скорости охлаждения. По уравнению
(2.67) рассчитано, например, критическое время /Крит, в тече-
ние которого может образоваться 1 зародыш в объеме 1 см3.
Тогда для интервала времени Д/, за который в процессе охла-
ждения проходится область максимальной скорости зародыше-
образования, ДОЛЖНО ВЫПОЛНЯТЬСЯ уСЛОВИе Д/^/крпт-
Ульман [6] для описания процесса объемной кристаллиза-
ции обратился к классическому уравнению (2.80). Объемная
доля закристаллизовавшегося расплава Укр, равная 10-6, рас-
сматривается как предельно определяемая. Комбинацией урав-
нений (2.80), (2.76), (2.67) и (2.65) получено соотношение,
позволяющее приближенно рассчитать время t превращения
расплава (УКр=Ю 4 об. %) в кристаллическое состояние в за-
висимости от температуры:
, 9,3т)4 Г а9ИКр Г 16лр3Д//пХл 1/Г1 угпТТ
/ = -1Г-{-ГоТу-[еХР 3^/?ГЗ(ДГ)2 J/H-cxP (~^пл АГЖГПЛ)]}
(2.85)
Для расчета необходимо знать температурную зависимость
вязкости в области переохлаждения.
На рис. 2.28 приведены так называемые ТТГ-кривые (от
англ. Time—Temperature—Transformation) для SiO2 [6], Те, Ge,
а также Ni (в качестве примера типичного металла) и одного
металлического сплава [220]. Можно заметить, что при малых
переохлаждениях заранее заданная объемная доля закристал-
лизовавшегося расплава образуется в течение достаточно дли-
тельного времени. При большом переохлаждении скорость кри-
сталлизации также мала. Наиболее короткое время, в течение
112 Глава 2
Рис. 2.28. ТТГ-кривые.
Кривые используют для определения временных интервалов, достаточных для кристал-
лизации при данном переохлаждении Д7'=7'пл — Т 10-4 об. % расплава. TN—темпера-
тура «передней» части кривой.
которого кристаллизуется 10-6 доля расплава, отвечает тем-
пературе Ту (выступающая часть Т7Т-кривой; индекс (7V) от
нем. die Nase — нос). Критическую скорость охлаждения мо-
жно определить с помощью касательной к /ТТ-кривой в точке
Ту. Таким образом,
<7кРит = (Т пл — TN)/tN (2.86)
Значения критической скорости охлаждения, получаемые из
ТТТ-кривых для SiO2, Ni и Pc^Siis, по крайней мере грубо
совпадают с результатами эксперимента (расхождения состав-
ляют меньше одного порядка величины), однако в случае Те и
особенно Gc расчет приводит к величинам, далеким от экспе-
риментальных условий синтеза стеклообразных и аморфных
фаз этих веществ.
В работе [209] принято во внимание нестационарное зароды-
шеобразование в высоковязких переохлажденных расплавах
Аморфное и стеклообразное состояние
113
[уравнение (2.70)], которое не учитывалось в подходе Ульмана.
Используя уравнение (2.72), можно найти соотношение, связы-
вающее скорость зародышеобразования /Оо, t и скорость охла-
ждения <7=(7’пл—7,)/Д^=Д7,/Д/:
об, t — Iоб ехр
Зл2Мт]д *1
RT ДГр J
(2.87)
Число зародышей Пк в объеме v:
t
nK = v\ /об> t dt
о
(2.88)
определяется скоростью охлаждения q = \TII\t, причем крити-
ческая скорость охлаждения в этом случае отвечает частоте
появления одного зародыша в объеме 1 см3.
В работе [221] по аналогии с работой Ульмана приняли
в качестве критерия склонности к стеклообразованию время,
которое при данной скорости охлаждения проходит до момента
достижения температуры стеклования Tg. В качестве предель-
ного значения предложено время, за которое кристаллизуется
объемная доля расплава, равная 10-4.
В табл. 2.7 приведены критические скорости охлаждения
<7крит ряда стеклообразующих соединений и других веществ,
определенные различными методами. Поскольку приведенные
данные трудно сопоставимы, можно сделать вывод, что пока не
выработано удовлетворительного способа оценки тенденции
к стеклообразованию в различных системах. Такое же замечание
относится и к ТТГ-кривым (рис. 2.28). Положение кривых на
Таблица 2.7. Критические скорости охлаждения веществ
Соединение Чкриг (К/с) по различным данным
1218] 10'1 [209] [221]
SiO2 4 2 • 10-4 ю-> 1,4 • 10-3
GeO2 — 7 • IO’2 10-" 1,2 • 10-3
В2О3 — — 10-16 5,2 • 10-8
р2о5 — — Ю-2Э 1,2- 10-6
As2O3 — — 107 1,3 • 104
BeF2 — — 10-3 9,5 • IO"5
Pb 108 — 1010 4,7 • 108
8 Заказ № 413
114 Глава 2
плоскости температура—время лишь в первом приближении
совпадает с экспериментальными данными.
Теоретические разработки проблемы описания образования
некристаллических форм твердых тел при конденсации из га-
зовой фазы находятся лишь в начальной фазе своего развития.
Первые работы, в которых обосновываются кинетические кри-
терии, пригодные для описания осаждения кристаллических или
аморфных (стеклообразных) форм вещества, приведены в ра-
боте Аврамова и Гутцова [445].
2.4. Структура и химическая связь в стеклах
и аморфных телах
Применение термодинамических и кинетических теорий для
макроскопического описания явления стеклообразования вовсе
не достаточно для моделирования процессов, идущих на мик-
роскопическом уровне. Весьма полезным в этом плане оказы-
вается учет геометрических и энергетических соотношений
между отдельными атомами и структурными группами в си-
стеме. Тенденция к образованию аморфных или стеклообраз-
ных твердых фаз меняется в различных системах в широких
пределах. Поэтому уместно поставить вопрос, какие структур-
ные соотношения и какие типы химических связей характерны
для отчетливо выраженной тенденции к стеклообразованию.
В данном разделе обсуждаются прежде всего наиболее ча-
сто применяемые методы структурных исследований стекол и
продемонстрированы на некоторых примерах возможности этих
методов. Результаты структурных исследований дают основание
считать, что структура стекла состоит из областей, характе-
ризующихся ближним порядком, причем эти области беспоря-
дочно сочленяются друг с другом. Это представляет собой ос-
новную особенность аморфного и стеклообразного состояний
твердых тел.
Для обычных стеклообразующих соединений, т. е. так на-
зываемых основных стеклообразователей, целесообразным ока-
залось введение модели идеальной структуры стекла в качестве
некоторого предельного состояния системы. Структура реаль-
ного стекла, предусматривающая непериодическое сочленение
областей с ближним порядком, отличается от идеального слу-
чая наличием определенных точечных дефектов, т. е. собствен-
ным разупорядочением. Такого рода модельные представления
в значительной мере способствовали пониманию стеклообраз-
ного состояния с точки зрения химии твердого тела.
Результаты измерения макроскопических свойств в зависи-
мости от состава часто могут быть объяснены со структурно-
химических позиций в случае, если есть сведения о типе струк-
турных групп в стекле. Однако выводы о структуре материала
Аморфное и стеклообразное состояние 115
могут быть сделаны лишь на основании комбинаций различных
методов исследования. Характерные изменения на зависимостях
определенных свойств рассматриваются как выражение струк-
турных изменений в данной системе, но однозначное объяснение
явлений без использования других методов вряд ли возможно.
2.4.1. Основные методы структурных исследований
некристаллических твердых тел
Отсутствие трансляционной структуры кристаллической ре-
шетки означает, что структуру твердого тела нельзя больше
свести к мельчайшим элементам повторяемости, элементарным
ячейкам. Можно лишь высказывать предположения вероятно-
стного характера о распределении частиц в окрестности лю-
бого данного атома. Данные о наиболее часто встречающихся
расстояниях между двумя отдельными атомами в образце
стекла можно получить с помощью дифракционных методов,
которые позволяют построить представления о ближнем по-
рядке, хотя вполне определенная картина возникает лишь при
исследовании гомоатомпых систем. Кроме того, на этой основе
возможно делать выводы о ближайшем окружении атомов, т. е.
о второй, третьей, а при определенных условиях и о более да-
леких координационных сферах, а также о способах сочлене-
ния областей ближайшего порядка.
Для расшифровки структур в гетероатомных стеклообраз-
ных системах оказываются в высшей степени полезными моле-
кулярно-спектроскопические методы, в особенности ИК-спек-
троскопия и спектроскопия КР. Получаемая таким образом
структурная информация относится прежде всего к самим об-
ластям ближнего порядка и их симметрии.
Третья группа методов использует исключительно те свой-
ства атомов, на которые влияет их химическое окружение, т. е.
вид, число и расположение партнеров по химической связи, ок-
ружающих центральный атом. К этим методам относятся фо-
тоэлектронная спектроскопия, ядерный магнитный резонанс,
мёссбауэровская спектроскопия, а для определенных катионов
переходных металлов — УФ-спектроскопия и спектроскопия
в видимой части спектра, а также другие физические методы
исследования твердых тел.
В последнее время возрастает поток информации о струк-
турном строении стекол, полученных на основании применения
химико-препаративных и химико-аналитических методов.
2.4.1.1. Дифракционные методы
Дифракция рентгеновских лучей. Аморфные твердые тела,
рассеивая монохроматическое рентгеновское излучение, дают
такую же дифракционную картину, как жидкости и газы. Эта
116 Глава 2
картина может быть описана с помощью функции распределе-
ния интенсивности рассеянных лучей в зависимости от перемен-
ной s = 4nsin0/Z. Дифракционная картина при интерференции
лучей, рассеянных в определенном направлении всеми атомами,
и содержит информацию о распределении расстояний между
рассеивающими частицами в данном веществе. Теория вопроса
изложена во многих научных трудах [222—224].
Функция интенсивности описывается формулой Дебая
/ = £ Е f™ СО f п (s) s'"5'"1'' (2.89)
т п
где гтп — модуль вектора расстояния между атомами т и п,
fm и fn — амплитуды рассеяния атомов т и п. Если принять,
что в образце имеется N одинаковых атомов с одинаковым ок-
ружением, уравнение (2.89) упрощается:
у__,/Vf2 s*1' тп
Sr тп
т
(2.90)
Откуда, принимая во внимание, что (sin srnin)/(srmn) -+
->• 1 и вводя функцию плотности р(г), имеем
[00
1 + f 4№р (г)dr
о sr
(2.91)
р(г)—число атомов на расстоянии г от данного атома,
4nr2p(r)rfr — число атомов, содержащихся в сферическом слое
толщиной dr на расстоянии г от центрального атома. Учитывая
плотность исследуемого образца р0, имеем:
I = + j 4лг2[р(г) — р0]
I о
Sin sr J I f . 9 sin sr .
------dr -\- \ 4nrp0--------dr
sr J r sr
(2.92)
При измерении больших углов в области значений s вторым
интегралом можно пренебречь. Применение интегральной тео-
ремы Фурье приводит к следующему выражению для функции
радиального распределения плотности (ФРР), которая может
быть рассчитана из интегрального выражения в пределах из-
меренных значений s:
оо
4лг2р (г) = 4лг2р0 4- J s (-щ-2-----------1) sin sr ds (2.93)
Далее не обсуждаются погрешности, которые касаются стро-
гости полученных структурных выводов, имея ввиду ограниче-
ние пределов интегрирования (эффекты обрыва ряда), и по-
Аморфное и стеклообразное состоянне
117
11)0 200 300 /'00 500 600
г, пм
Рис. 2.29. Функция радиального распределения для стеклообразного селена
[225].
правки, которые следует производить по измеренным кривым
интенсивности.
На рис. 2.29 приведена функция радиального распределе-
ния плотности для стеклообразного селена [225]. Кривая
усредненной рассеивающей плотности 4лг2р0 описывает гипо-
тетический случай некоторого непрерывного распределения
плотности в пространстве, при котором имеется равная вероят-
ность всех расстояний между точечными рассеивающими ча-
стицами. Отклонение ФРР от кривой усредненной рассеиваю-
щей плотности и дает информацию о структуре вещества. Мо-
жно заметить, что расстояния между двумя атомами, которые
были бы меньше длины химической связи, не встречаются. Ин-
тегрирование площади первого максимума дает число частиц
в первой координационной сфере и позволяет высказать опре-
деленные соображения о структуре ближнего порядка. В дан-
ном случае речь идет о двух атомах. На основании этого можно
сделать вывод о цепочечном сочленении атомов в стеклообраз-
ном селене и, следовательно, о сходстве ближнего порядка
118 Глава 2
в этом веществе со структурой кристаллического селена. Вто-
рой максимум может быть интерпретирован также еще отно-
сительно определенно. Длина связи между одним атомом се-
лена и четырьмя соседними, которые в кристаллическом селене
принадлежат трем цепочкам селена, окружающим централь-
ную цепочку (разд. 3.1.3.2), в стеклообразном селене увеличи-
вается из-за вандерваальсова взаимодействия и случайно со-
впадает с расстоянием до двух следующих атомов в централь-
ной цепочке. Таким образом, площадь под вторым максимумом
соответствует шести атомам. Однозначность отнесения других
максимумов к определенному расположению атомов уменьша-
ется из-за многообразия похожих конфигураций. Поэтому с ро-
стом расстояния кривая ФРР все больше приближается к усред-
ненной кривой.
Для гетероатомных систем с .V одинаковыми структурными
элементами из уравнения (2.89) следует
т =#= п
' = N£ f2„ + 2 Е Ы- S'"rr""'- (2.94)
р т п
Первая сумма относится ко всем атомам одного структурного
элемента, вторая соответствует любой паре атомов в образце.
Снова можно перейти к бесконечному распределению атомов
в окружении данного атома. Принимая, что число атомов т
и п в сферическом слое радиуса г и толщиной dr равны ат
и ап, можно определить взвешенную функцию плотности
4nr2pm(r)dr= X amfm- Тогда с учетом вышеприведенного урав-
нения
/ = М
Е +£ М 4л,*2р^ (и
_ т О
sin sr
sr
dr
(2.95)
В этом выражении функции р™(г) и fm зависят от s, и по этой
причине нельзя произвести фурье-преобразование.
Таким образом, для веществ, состоящих из атомов различ-
ного типа, требуется введение некоторого приближения. Оно
может быть найдено на основании очень близких зависимостей
атомных амплитуд от s в случае, если порядковые номера ато-
мов не сильно отличаются друг от друга. Для всех атомов
считают, что атомная амплитуда — величина, кратная фактору
рассеяния свободного электрона. Тогда коэффициент пропор-
циональности имеет смысл числа эффективных электронов атома
fm(s) = Kmfe(s) (2.96)
а функция распределения атомной плотности pm(r) может быть
заменена функцией электронной плотности gm(r)‘-
Pm (г) = fegт (Г) (2.97)
Аморфное и стеклообразное состояние 119
Вводя величину средней электронной плотности gQ и пренебре-
гая рассеянием на малых углах, получаем
оо
4---£ й = We J 1л Л(И - go] Г2 ЛЛ dr (2.98)
п От
Откуда, применяя интегральную теорему Фурье, имеем
°° I __ у £"
EV' 2r f N
Kmgm (Г) = 4nr2go ), Km + — J «--T2--- Sin Sr ds
m m 0 / e
(2.99)
В рамках принятого приближения функцию радиального
распределения (ФРР) удобно представлять графически в виде
функции е^г от расстояния г.
На рис. 2.30 представлена ФРР кварцевого стекла, пост-
роенная по экспериментальным данным (2 и 3) [226, 227] и
рассчитанная из статистической модели сетчатой структуры
Белла и Дина (/) [228]. Для сравнения приведены более ста-
рые данные (4) [229]. Хотя последние и характеризуются низ-
кой разрешающей способностью, в свое время они послужили
экспериментальным подтверждением развитой Захариасеном
[230] сетчатой модели структуры стекла.
Положение первого максимума (161 пм) соответствует длине
связи Si—О, а площадь, ограниченная его ветвями,— электрон-
ной плотности области ближнего порядка состава [SiCX]. Такие
структурные элементы составляют основу структуры стекла и
имеют тетраэдрическую симметрию. Второй максимум можно
с большой вероятностью отнести к расстоянию между атомами
О—О соседних тетраэдров. Отсюда следует, что величины ва-
лентных углов с вершинами на мостиковых кислородных ато-
мах находятся в интервале 120—180° с максимумом распреде-
ления на 144° [227].
Обычно строят модель структуры вещества и варьируют
конфигурации структурных групп до тех пор, пока не добива-
ются хорошего совпадения ФРР модели и экспериментально
полученной ФРР. При этом необходимо обратить внимание
на то, что многообразие структурных конфигураций, незначи-
тельно отличающихся друг от друга, быстро возрастает с уве-
личением расстояния г, и, таким образом, не существует кри-
терия однозначности расшифровки структуры обширных обла-
стей при достигнутой к настоящему времени разрешающей спо-
собности метода.
Здесь нельзя не заметить, что интерпретация ФРР квар-
цевого стекла опирается на дополнительную информацию. Вы-
вод о существовании связей Si—Si и О—О можно сделать,
Рис. 2.30. Функция радиального распределения для кварцевого стекла.
1 — расчетные данные (226] по модели Белла и Дина [228], 2 и 3 — экспериментальные
данные [226, 227], 4 — [229].
Аморфное и стеклообразное состояние 121
опираясь на простые химические обоснования. В гетероатомных
системах, например в металлических стеклах, даже в случае
присутствия в них лишь двух различных сортов атомов А и В
для определения функций парного распределения А—А, А—В
и В—В требуются три независимых дифракционных экспери-
мента [231]. Дифракция нейтронов и, в определенных преде-
лах, дифракция электронов — методы, существенно дополняю-
щие метод дифракции рентгеновских лучей.
Нейтронография. Дифракция нейтронов происходит на яд-
рах атомов, чьи геометрические размеры намного меньше длины
волны нейтронов. Получаемая дифракционная картина ха-
рактеризуется сферической симметрией. Это означает, что ам-
плитуды атомного рассеяния Ду не зависят от угла рассеяния,
что упрощает обработку. Обычно используют нейтроны с дли-
ной волны 0,07 нм или тепловые нейтроны с длиной волны
0,12 нм. Сечение рассеяния относительно мало. Следовательно,
требуются образцы большего размера, чем при съемке кривых
рентгеновского рассеяния. Размеры в несколько сантиметров
вполне достаточны. Кроме того, время съемки кривых нейтрон-
ного рассеяния относительно велико.
Вообще говоря, амплитуды атомного рассеяния слабо рас-
тут с увеличением порядкового номера элемента (по закону
Z1'3). Изменения этих величин от элемента к элементу, а также
разность их для различных изотопов одного элемента выра-
жены весьма резко, так что вклад различных сортов ядер
в интенсивность рассеяния, например в результате изменения
изотопного состава образца, легко можно обнаружить. Такого
рода исследования весьма дорогостоящи; в настоящее время
известно лишь несколько примеров [232]. Комбинированные
эксперименты по изучению рассеяния рентгеновских лучей и
нейтронов проведены на аморфном сплаве Coes^Pie.e- Разре-
шающая способность нейтронографического метода повышается
при использовании поляризованного излучения. Из трех неза-
висимых экспериментов, например съемка кривых рентгенов-
ского и нейтронного рассеяния, а также получение дифракци-
онной картины с помощью поляризованных нейтронов, можно
определить парциальные интерференционные функции для рас-
стояний между парами атомов Со—Со, Со—Р, Р—Р (рис. 2.31)
[233].
Применение методов нейтронной дифракции, в том числе и
для исследования аморфных и стеклообразных твердых тел,
рассмотрено в обзорах [234, 235]. Сообщается [449] об изме-
рениях дифракции нейтронов с целью изучения некристалличе-
ских магнитных твердых тел.
Электронография. Электроны при взаимодействии с веще-
ством рассеиваются существенно сильнее, чем рентгеновские
122 Глава 2
Рис. 2.31. Парциальные функции дифференциального распределения плотности
для аморфного Со8злР1б.б [233].
лучи. Поэтому применение методов электронной дифракции ог-
раничено исследованием тонких пленок толщиной 0,01—0,1 мкм.
Зависимость интенсивности рассеяния от угла дифракции силь-
нее, чем в случае рентгеновских лучей. В то время как ампли-
туды атомного рассеяния для рентгеновских лучей пропорцио-
нальны примерно квадрату порядкового номера элемента Z2,
для электронного рассеяния на элементах с маленькими по-
рядковыми номерами соблюдается примерная пропорциональ-
ность Z,/3. Благодаря этому с помощью электронной дифрак-
ции можно с высокой чувствительностью обнаруживать наряду
с положением тяжелых атомов положение легких атомов, что
исключено в случае рентгеновских методов.
Данный метод был применен многократно для изучения
структуры аморфных пленок (гл. 3).
Метод ПТ СР С (метод протяженной тонкой структуры рент-
геновского спектра)*. Сайерс, Литл и Стерн [236] открыли
новый метод изучения структуры аморфных твердых тел. Им
удалось интерпретировать давно известную тонкую структуру
края К-полосы поглощения рентгеновского спектра, соотнося
ее со структурой окружения данного атома [236, 237]. Если
ввести обозначения: а — коэффициент поглощения, х— тол-
щина образца, то произведение ах = 1п(/0//) отражает периоди-
* В англоязычной литературе известен как метод EXAFS (Extended
X-ray Absorption fine Structure).
Аморфное и стеклообразное состояние
123
ческие изменения на высокоэнергетической стороне края по-
лосы поглощения, которые связаны с непосредственным окру-
жением поглощающего атома. Речь идет об особого рода диф-
ракции электронов, которая характеризуется тем, что фотоэлек-
трон, обладающий энергией E = hv—Ек (Ек — энергия связи
электрона Л-уровня), испускается атомом, который предвари-
тельно поглотил энергию.
Первоначальный поток
фотоэлектронов взаимо-
действует со вторичной
волной, вызванной рас-
сеянием на соседних ато-
мах. В соответствии с
волновым числом фото-
электрона происходят из-
менение дипольного мо-
мента в матрице и, сле-
довательно, изменение
вероятности перехода
электрона. Такой подход
объясняет явление осцил-
ляции коэффициента по-
глощения.
На рис. 2.32 приведе-
ны спектры ПТСРС для
стеклообразного GeO3
и аморфного GeSe, при-
чем для последнего по-
глощение как Ge, так и
Se [236]. Точкой отсчета
принята энергия £ = 0
(EK=hx)\ подобные
спектры могут быть сняты
Аналитическое выражение
коэффициента рентгеновского поглощения можно получить с по-
мощью фурье-преобразования. Таким методом можно определить
отдельно для каждого сорта атомов длины химических связей
и координационное число первой координационной сферы.
В стеклообразных As2Se3 и GeSe2 координационное число селена
равно ~2, а координационные числа As и Ge 3 и 4 [239]. Метод
оказался особенно подходящим для изучения мало исследован-
ных структурных соотношений в металлических стеклах [240].
Применение спектроскопии ПТСРС к изучению стекол на основе
силиката натрия недавно привело к достойному внимания ре-
зультату— оказалось, что и для ионов Na+ существует опреде-
ленный ближний порядок [446].
Энергия фотоэлектронов, эВ
Рис. 2.32. Спектр ПТСРС стеклообразного
GeOs (/), аморфного GeSe (поглощающий
атом Ge) (2) и аморфного GeSe (погло-
щающий атом Sc) (3) [236].
отдельно для каждого сорта атомов,
для описания осциллирующего хода
124 Глава 2
2.4.1.2. Инфракрасная спектроскопия и спектроскопия
комбинационного рассеяния (рамановская)
Оба метода позволяют изучать колебательный спектр ис-
следуемого вещества. Поглощение электромагнитного излучения
в ИК-области, как известно, основано на возбуждении перехо-
дов между колебательными уровнями молекулы (кристалла),
что связано с изменением дипольного момента. В КР-спектро-
скопии для возбуждения системы используют более коротко-
волновое излучение. В современных измерительных установках
применяют лазерное излучение в видимой области спектра.
Частота первичного светового потока меняется как в сторону
своего уменьшения на величину частоты возбужденных излуче-
нием колебаний (стоксовский сдвиг), так и в сторону своего
увеличения вследствие взаимодействия с термически возбужден-
ной системой атомов исследуемого вещества (антистоксовский
сдвиг). Причиной эффекта Рамана является изменение поляри-
зуемости в процессе колебания. Поскольку механизмы возбу-
ждения в этих методах различны, возникают различные правила
отбора, и оба метода дополняют друг друга.
Число колебательных степеней свободы молекулы состав-
ляет F = 3N—6. Колебания молекул различных типов симмет-
рии можно описать, привлекая теорию групп. Сопоставление
теоретического описания с экспериментально получаемыми спек-
трами позволяет сделать вывод в пользу той или иной струк-
туры молекулы. Макромолекулярные твердые тела, например
кристаллы, как правило, характеризуются спектрами, струк-
тура которых определяется периодичностью кристаллической
решетки и симметрией кристалла. Как уже отмечалось, стекла
занимают в известном смысле промежуточное положение между
кристаллическими твердыми телами с макромолекулярной
структурой и веществами молекулярного строения. При отне-
сении полос в колебательных спектрах стекол и аморфных
веществ необходимо учитывать как теорию колебаний кри-
сталлической решетки твердых тел, так и молекулярные мо-
дели.
Решеточная модель колебательных спектров некристалличе-
ских твердых веществ. Согласно классической теории Борна
и Кармиана, периодическую решетку кристалла можно рассмат-
ривать как бесконечно большую дискретную систему, состоя-
щую из nN точек определенной массы. Здесь п — число атомов
в самом маленьком элементе повторяемости, который нельзя
смешивать с обычной элементарной ячейкой. Последняя уста-
навливается в соответствии с определенными правилами, и ее
симметрия, так называемая фактор-группа, всегда изоморфна
кристаллографической точечной группе. В таком кристалле су-
Аморфное и стеклообразное состояние
125
ществует 3nN—3 типов простейших (нормальных) колебаний,
которые можно привести в соответствие определенным наборам
частот, так называемым ветвям нормальных колебаний, состоя-
щим из N отдельных колебаний. В рамках приближения упру-
гих сил движение атомов в кристаллической решетке можно
описать как распространение не связанных между собой плос-
ких волн. В такой волне идентичные атомы различных эле-
в
Рис. 2.33. Схематическое представление колебаний решетки, состоящей из
цепочек атомов двух разных сортов [247].
а — оптическая и акустическая ветви; б — распределение плотности состояний колебаний;
в — ИК-поглощение в заштрихованной области.
ментарных ячеек, т. е. частицы п примитивных подрешеток, ко-
леблются с одинаковой частотой и с одним и тем же фазовым
сдвигом, который определяется длиной волны X или волновым
вектором k = 2л/Х.
Для решетки, состоящей из п — 2 различных атомов, реше-
ние уравнений движения в случае рассмотрения лишь продоль-
ных колебаний приводит к изображенному на рис. 2.33, а поло-
жению дисперсионных ветвей. Очевидно, что налицо имеются
две ветви нормальных колебаний, которые разделены запрещен-
ной зоной. Частоты при k = Q (Х = оо) называются предель-
ными частотами. На нижней ветви для k = 0 также ш = 0.
В этом случае все атомы решетки движутся в одной фазе с оди-
наковой амплитудой в одном направлении, и, таким образом,
речь идет о поступательном движении. В области k > 0 фазо-
вый сдвиг от атома к атому не равен точно нулю. Возникают
упругие волны низкой частоты, длины которых велики по срав-
нению с размерами элементарной ячейки. Поэтому эта ветвь
нормальных колебаний называется акустической ветвью.
На верхней ветви волновому вектору k — 0 отвечает самая
высокая в решетке собственная частота. Она может быть опре-
делена исходя из масс атомов и силовых постоянных. В этом
126 Глава 2
случае атомы разного сорта в элементарной ячейке колеблются
точно в противофазе, причем центр тяжести каждой ячейки на-
ходится в состоянии покоя, а все ячейки жестко связаны друг
с другом, так как Х = оо, т. е. обе примитивные подрешетки
различных сортов атомов колеблются относительно друг друга.
В кристаллах полярных веществ возбуждение таких колебаний
связано с временным изменением дипольного момента. Ветвь
нормальных колебаний называется в этом случае оптической
ветвью, а соответствующие собственные колебания — оптиче-
скими колебаниями.
На рис. 2.33,6 приведен ход кривой плотности состояний
N(а) для описания распределения частот колебаний. Видно, что
плотность состояний на краях полос особенно высока, а затем
круто опускается к нулю.
Если допустить пространственное движение атомов в трех
направлениях, т. е. включить также в рассмотрение и попереч-
ные волны, то появляются две другие акустические ветви
с N—1 колебаниями. Для k = 0 вновь существует поступа-
тельное движение в одном из двух других направлений. Всего
имеются три акустические ветви — одна продольная и две по-
перечные с 3(jV—1) нормальными колебаниями. Аналогично
утраивается также количество оптических ветвей, из которых
3(п—1) состоят из ЗА(п—1) колебаний, причем N (п—1) ко-
лебаний продольных, a 2N(п—1) поперечных. Таким образом,
колебательное движение в кристаллической решетке можно
описать 3N(n—1) + ЗА— 3 = 3nN — 3 нормальными колеба-
ниями.
Например, для кристаллического вещества, состоящего из
двух сортов различных атомов, получаются три акустические
и три оптические ветви. В случае кубической симметрии, напри-
мер в решетке типа NaCl, они вырождаются. Наименьшим эле-
ментом повторяемости в этом случае является ромбоэдриче-
ская ячейка (п = 2). Решающее значение имеет правило отбора,
согласно которому электромагнитным излучением возбуждаются
только такие колебания, при которых трансляционно эквивалент-
ные частицы колеблются в одной фазе. Это означает, что в ИК-
спектре можно наблюдать предельные частоты оптических ве1-
вей. В ИК-спектре NaCl существует только одна интенсивная
полоса поглощения (рис. 2.33,в). Полосы в КР-спектре точно
так же отвечают предельным частотам оптических ветвей. Воз-
буждение колебаний в акустической ветви можно изучать с по-
мощью бриллюэновского рассеяния.
В наименьшем элементе повторяемости а-кварца имеются
три формульные единицы SiO2. Поэтому в данном случае
имеются 3 акустические и 24 оптические ветви (п = 9), так что
при k — 0 следует ожидать наличие полос поглощения в ИК-
и КР-спектрах.
Аморфное и стеклообразное состояние
127
Фактор-группе D3 соответствуют типы колебаний: 4/1JKP),
4/12(ИК)^-З/ЦИК+КР), т. е. всего в ИК- и КР-спектрах может
быть обнаружено 12 полос, 8 из которых — в обоих спектрах
с примерно одинаковыми волновыми числами.
В табл. 2.8 [241] приведены наблюдаемые колебательные
полосы а-кварца с их соответствующим отнесением. Здесь при-
ведены также соответствующие рассчитанные [242] и экспери-
ментальные [243] значения частот для кварцевого стекла. Рас-
пределение частот для пространственной сетки, которая состоит
из тетраэдров [SiO4], рассчитано Беллом и сотр. [244]. Нельзя
не заметить, что имеются различия в значениях для стекло-
образного и кристаллического SiO2; однако описание колеба-
тельного спектра кварцевого стекла с помощью кристалличе-
ской модели, судя по этому сравнению, оказывается обосно-
ванным. Кажется, будто в кварцевом стекле существует
«расшатанная» вплоть до исчезновения дальнего порядка кри-
сталлическая решетка, в которой имеются тетраэдрические
Таблица 2.8. Колебательные полосы в а-кварце и кварцевом стекле
Тип симметрии колебаний V, см-1
а-кварц кварцевое стекло
расчет. экспер.
At 207 273 275
At 356 385 377
464 485 490
At 1084 1152 1100
Аг 364 308 ?
Аг 495 503 ?
Аг 778 772 800
Аг 1080 1199 1190
Е 394 282
Е 401 374 370
Е 450 480 490
Е 509 589 610
Е 795 672 660
Е 807 792 792
Е 1072 948 950
Е 1231 1134 1060
Е 1235
128 Глава 2
структурные элементы [SiCh] в виде неизменных областей
ближнего порядка.
Наблюдаемые в средней ИК-области основные линии позво-
ляют выявить далеко иду-
щую аналогию. Из сравне-
ния приведенных на рис. 2.34
спектров можно сделать
определенный вывод о том,
что в кварцевом стекле
не существует областей,
родственных ни структуре
стишовита, ни структуре
коэсита [245, 246]. Спектр
в значительной степени соот-
ветствует кварцу, тридимиту
и кристобалиту. Структура
кварцевого стекла состоит
из тетраэдрических областей
с ближним порядком, ос-
новной структурный мотив
которых элементы [SiO;].
Большие различия между
кристаллическим и стекло-
образным SiOa проявляют-
ся в длинноволновой части
спектра. Правило отбо-
ра, которое ограничивает
возбуждение колебаний
в области предельных ча-
стот, связано с трансляци-
онной симметрией кристал-
лической решетки. Если ре-
шетка разрушается, все ко-
лебания, связанные с изме-
нениями дипольного момен-
та, становятся оптически ак-
тивными. Уширение полос
Рис. 2.34. ИК-спектры кварцевого стекла поглощения в кварцевом
и различных кристаллических модифи- стекле — проявление этого
каций SiO2 [246]. эффекта. Возникающие при
нарушении периодичности
решетки эффекты наиболее отчетливо видны в далекой инфра-
красной области спектра. На рис. 2.35 приведен ход зависимости
tgd = aX/2^n от частоты (где a — коэффициент поглощения,
п — показатель преломления; см. разд. 4.2.1) для кварца и
кварцевого стекла с различными добавками [247]. В кварцевом
стекле вся полоса длинноволновых колебаний (ср. с рис. 2.33, в)
Аморфное и стеклообразное состояние
129
оптически активна. Из-за малых изменений дипольного момента
поглощение относительно мало. Небольшие добавки НгО или
NaaO могут заметно влиять на поглощение кварцевого стекла
как раз в длинноволновой части спектра.
Молекулярная модель колебательных спектров стекол.
На примере кристаллических и стеклообразных форм S1O2
показано, что предельные
частоты оптически актив-
ных колебаний, интерпре-
тированные в рамках ре-
шеточной модели, нахо-
дятся в тесной взаимосвя-
зи с колебательными спек-
трами изолированных тет-
раэдрических анионов
SiO4- или нейтральных
частиц с такой же струк-
турой и типом химической
связи (табл. 2.9).
Можно заметить, что
оба валентных колебания
vs и v(; симметрии Аг
и Fz в спектре а-кварца
и кварцевого стекла дей-
ствительно хорошо со-
Рис. 2.35. Поглощение (в единицах диэлек-
трических потерь tg б) кварца и кварцевого
стекла, а также патриево-силикатного стек-
ла в микроволновой области [247].
гласуются с соответству-
щими полосами анионов
SiO4- и молекул SiF^. Ме-
жду деформационными
колебаниями типов
симметрии F2 и Е связанных структурных групп [SiCh] и коле-
баниями анионов SiO4~ также существует хорошее соответствие.
Как и следовало ожидать, в а-кварце и кварцевом стекле про-
исходит частичное расщепление вырожденных полос, что опре-
деляется законами симметрии.
Из тесной взаимосвязи между колебательными спектрами
молекул или комплексных анионов и спектрами стеклообразных
твердых тел, содержащих те же или аналогичные структурные
группы в виде областей ближнего порядка, вытекает правомер-
ность применения молекулярных моделей для описания колеба-
тельных спектров аморфных фаз. Экспериментальные спектры
отражают внутренние колебания структурных групп, которые
можно представить в виде следующей модели: один централь-
ный атом с высоким координационным числом, окруженный
большим или меньшим числом лигандов. Если энергию связи
9 Заказ № 413
130 Глава 2
Таблица 2.9. Колебательные полосы структурных единиц [SiX4]
симметрии Тл
Тип симметрии и тип колебаний соответствующей структурной группы V, см-1
а-кварц кварцевое стекло sioj- S1F,
ик [241| ик [2481 КР |249| [250|
A2v^(SiX) (ИК + КР) 11611 1078J С* 11841 1098) 10681 ? с. 1087) 1055 сл. 1050 1010
(SIX) (КР) 800 сл. 804 сл. 815 ср. 800 ср. 800 800
/’26rf(XSiX) (ИК + КР) 695 сл. 695оч.сл. 605 с. 625 390
Edd (XSiX) (КР) 5101 ? с. 455) 468 475 4901 _ ?оч. с. 435) 500 268
стеклообразного вещества можно было бы разделить на две
составляющие: энергию связи областей ближнего порядка
и энергию взаимодействия между этими областями, то величина
первой составляющей будет заметно превосходить величину вто-
рой. Взаимодействие нормальных колебаний таких областей
ближнего порядка в стеклах более слабое по сравнению с взаи-
модействием для кристаллического состояния из-за отсутствия
периодичности решетки. Совершенно естественно в рамках дан-
ной модели привлечение методов ИК- и КР-спектроскопии к ис-
следованию стекол для определения структуры ближнего по-
рядка.
Молекулярная модель была применена для интерпретации
спектров халькогенидных стекол [251, 252]. В последней работе
сумели расшифровать структуру ближнего порядка для многих
стеклообразующих соединений и производных стеклообразую-
щих систем. Сравнением спектров кристаллических и стекло-
образных форм AS2S3, As2Se3, As2Te3, GeS2, GeSe2, а также
аморфного GeTe2 и изучением спектров молекул галогенидов
As(III) и Ge(IV) удалось расшифровать строение областей
ближнего порядка в этих стеклах [253] (разд. 3.2.2.6 и 3.3.5.2).
В аморфных Si, Ge и As молекулярные структурные элементы
выделить не удалось. В этих веществах атомы имеют в среднем
одинаковые координационные числа, и поэтому молекулярная
модель не применима.
Если массы атомов, связанные между собой в областях
ближнего порядка стекла, различаются в два раза и более или
если кратность связи между атомами в первой координационной
сфере велика, существует еще одно приближение молекулярной
Аморфное и стеклообразное состояние 131
модели. Согласно правилу Губо, нормальные колебания в этих
случаях могут быть сведены к характеристическим валентным
и деформационным колебаниям, отличительной особенностью
которых является то, что периодическое движение в основном
выпадает на долю более легких атомов. Это неоднократно ис-
пользовалось в молекулярной спектроскопии для интерпретации
определенных химических связей и характеристических групп,
например связей О—Н или Si—Н, а также при обнаружении
оксидов в халькогенидных стеклах (4.2.1.2).
В заключение необходимо указать на то, что концепция
Тарте [254], согласно которой в рамках молекулярной модели
рассматриваются отдельные, независимые колебания, была рас-
пространена на молекулярно-спектроскопическое исследование
координации катионов в стеклах. Обзорные работы, в которых
обсуждаются результаты изучения оксидных стекол, опублико-
ваны Вонгом и Энжеллом [241] и Найротом [255]. Боратные
стекла особенно подробно изучал Коньедук [256]. Гриском
[257] опубликовал обзор о значении молекулярно-спектроскопи-
ческих методов исследования структуры боратных стекол.-Гали-
нер [258] заложил основы модельных расчетов для стекол типа
кварцевого стекла: SiO2, GeO2, BeF2. При этом он исходил из
общей решеточной модели и включил в рассмотрение возбу-
жденные колебания, инициируемые дефектами. Кроме того, им
использовалась аналогия с молекулярными моделями.
2.4.1.3. Другие методы исследования твердых тел
Фотоэлектронная спектроскопия. Примерно такой же универ-
сальностью, как и описанные ранее методы, характеризуется
фотоэлектронная спектроскопия (метод ESCA; от англ. Electron
Spectroscopy for Chemical Analysis) [259]. В зависимости от
типа возбуждения различают рентгеновскую фотоэлектронную
спектроскопию (РФС) и ультрафиолетовую фотоэлектронную
спектроскопию (УФФС).
В РФС исследуемый образец, находящийся в высоком ва-
кууме, облучается характеристическим рентгеновским излуче-
нием. При этом проводят измерение кинетической энергии испу-
скаемых электронов Ek-\-^s = hv — Еъ. Таким образом, может
быть определена энергия связи Еъ электронов определенного
энергетического уровня для любых атомов. Фл— работа выхода
материала спектрометра, учет которой важен при исследовании
диэлектриков. Например, этот параметр можно учесть при гра-
дуировке системы, которая заключается в том, что па образец
напыляют слой углерода и электронный уровень 1s углерода
с энергией 285,0 эВ используют в качестве реперной величины.
Энергия связи электронов, находящихся на определенном уровне
132 Глава 2
атома, зависит, например, от состояния окисления. На ее вели-
чину влияет также характер химической связи данного атома
с соседними: изменение химического окружения приводит к из-
менению положения данного энергетического уровня, что можно
обнаружить по спектру. Кроме того, метод фотоэлектронной
спектроскопии позволяет сделать вывод о распределении состоя-
ний в валентной зоне твердых тел.
Исследования с использованием фотоэлектронной спектро-
скопии описываются в многочисленных монографиях и обзор-
ных статьях [260—262]. Возможность обнаружения различия
между мостиковыми и немостиковыми кислородными атомами
способствует существенному расширению и углублению пред-
ставлений о структуре силикатных, фосфатных и алюмосиликат-
ных стекол [447] (разд. 2.4.2.6). Применение фотоэлектронной
спектроскопии для исследования халькогенидных стекол
описано, например, в работе [263].
Спектроскопия ядерного магнитного резонанса (ЯМР). Энер-
гетический уровень атомных ядер с магнитным моментом
ц = как известно, расщепляется в магнитном поле на
214- 1 эквивалентных термов с разностью энергий Л£ = gnl^H
(где Н — напряженность магнитного поля, ga— ядерный g-фак-
тор, — ядерный магнетон, а / — спин ядра). В области вы-
сокочастотных полей (мегагерцевая область) определенные ре-
зонансные частоты вызывают переходы между уровнями Зее-
мана. Экранирование внешнего магнитного поля окружением
данного атома проявляется в виде химического сдвига, измеряе-
мого относительно принятых эталонных веществ. Сопоставление
напряженности магнитного поля, при которой возникает ядер-
ный резонанс при измерениях на постоянной частоте, позволяет
сделать выводы (после градуировки) о химическом окружении
данного атома и типе взаимодействия между ними и соседними
атомами.
ЯМР-спектроскопия применена для изучения структуры сте-
кол, содержащих следующие элементы: В, Н, Li, Be, О, F, Na,
Al, Si, P, Se, Sc, Cd, Sn, Те, V, Cs, T1 и Pb. Брэй [264] приво-
дит подробный обзор применения ЯМР-спектроскопии. Брэй
и сотр. [265—268] продемонстрировали работоспособность ме-
тода при изучении боратных стекол. Обнаружив изменения ко-
ординации между кислородом и бором и учитывая строение со-
ответствующих кристаллических соединений, они сделали важ-
ные структурные выводы. При выяснении структуры силикатных
стекол весьма полезным оказался метод ЯМР-спектроскопии на
ядрах ^Si. В комбинации с химическими методами анализа
анионов ЯМР-спектроскопия дает надежную информацию
о структуре стекла [269—271].
Аморфное и стеклообразное состояние
133
Мёссбауэровская (у-резонансная) спектроскопия. Эффект
Мёссбауэра основан на резонансном поглощении у-квантов оп-
ределенными ядрами, которые переходят при этом в возбужден-
ные состояния. Здесь речь идет об изомерных переходах, причем
энергетический уровень возбужденного состояния отличается от
основного состояния на величину Е = 1044-105 эВ. Среднее
время жизни изомера составляет 10-7 > т > 10-10 с, и поэтому
ширина спектральных линий Г мала. Для наиболее часто при-
меняемых «мёссбауэровских» ядер она составляет 10-6—10~9 эВ.
или соответствует скорости движения источника v порядка не-
скольких миллиметров в секунду согласно уравнению v=Vc)E.
Источниками излучения служат радиоактивные изотопы
с подходящими для проведения экспериментальных исследова-
ний периодами полураспада. При их распаде происходит необ-
ходимый для измерения изомерный переход. Так, в результате
К-захвата в изотопе 57Со (период полураспада 270 сут) возникает
возбужденный изомер 57Fe, причем энергия перехода из воз-
бужденного состояния в основное составляет 14,4 кэВ. Анало-
гично в ходе изомерного превращения 119?nSn (период полурас-
пада 245 сут) в 119Sn возникает энергетическое состояние, отли-
чающееся на 24,0 кэВ от основного состояния. Исходным явля-
ется состояние, характеризующееся энергией 89 кэВ.
Если ядра атомов в исследованном веществе имеют то же
самое химическое окружение, что и в источнике у-излучения,
то происходит резонансное поглощение у-квантов в отсутствие
движения источника. При неодинаковом химическом окруже-
нии источник должен двигаться относительно поглотителя,
чтобы, используя доплеровский эффект, добиться резонансного
поглощения у-лучей. Таким образом, как и в случае ЯМР-спек-
троскопии, информацию о структуре окружения данного ядра
можно получить, исследуя химический сдвиг.
Влияние химической связи на положение энергетических
уровней ядер происходит в связи с конечными и различными
размерами ядра в основном и возбужденном состояниях и в связи
с ненулевой плотностью вероятности нахождения электронов
(особенно s-электронов) на ядре. Структурные конфигурации,
которые создают локальные электрические поля несферической
симметрии, вызывают у ядер со спином / > */2 квадрупольное
расщепление. Так, для lb9Sn с 1=^1^ в возбужденном состоянии
происходит расщепление на 2 компоненты. Такое расщепление
характерно, например, для соединений Sn(II) из-за асимметрич-
ного строения элементов структуры.
На рис. 2.36 приведены мёссбауэровские спектры стекол си-
стемы SnS—GeS—GeS2 и для сопоставления некоторых кри-
сталлических соединений с известной структурой. Какие же
структурые выводы можно делать с помощью мёссбауэровской
спектроскопии? В данном случае очевидно, что в стеклах
134 Глава 2
I I____I--L___1___I---1___1____1-----
Q 7 2 3 4 5 G 7 8
<тм, мм/с
Рис. 2.36. Мёссбауэровские спектры
стекол (SnS2)2,5(GeS)6o(GeS2)37,5 (/),
(SnS)2o(GeS)5o(GeS2)so (2) и jSnSJsj-
(GeS) 3s(GeS2)3o (3), а также других ве-
ществ, взятых для сравнения [272].
существует ближний порядок, аналогичный структуре SnGeSs,
в которой атомы Sn(II) окружены пятью атомами S. Такие
области ближнего порядка имеют конфигурацию тетрагональной
пирамиды [272]. Примене-
ние мёссбауэровской спек-
троскопии для изучения сте-
кол, содержащих Sb, Sn и
Fe, подробно дано в обзорах
[273, 274]. Кроме того,
мёссбауэровский эффект
можно получить на ядрах
61Ni, 67Zn, 125Те, 1271, изотопах
Os, Ir, W, Re, Hg и ряда
редкоземельных элементов.
Ультрафиолетовая и ви-
димая спектроскопия. Элек-
тронные спектры в видимой
и ближней УФ-областях
используют для изучения
координации кислородных
атомов в стеклах, содержа-
щих переходные металлы.
При этом часто параллель-
но проводят исследования
магнитных свойств этих же
стекол. При интерпретации
спектров опираются на пред-
ставления теории кри-
сталлического поля и тео-
рии поля лигандов. Литера-
тура по данному вопросу
собрана в обзоре [276] (ко-
торый, к сожалению, опуб-
ликован сравнительно дав-
но). Заметного прогресса
в изучении стекол этими методами достигли Гиттер и сотр.
[277]. О структурной интерпретации спектров электронного
поглощения стеклообразных соединений в УФ-области см.
в разд. 4.2.1.3.
2.4.1.4. Химические методы изучения строения стекол
В последние годы благодаря применению некоторых приемов
эксперимента из полимерной химии достигнут существенный
прогресс в выяснении структурных особенностей неорганических
стекол. Под действием определенных растворителей, в данном
случае при добавлении подходящих реагентов, иногда в мягких
Аморфное и стеклообразное состояние
135
условиях, удается перевести в раствор отдельные части молекул
или даже целые структурные единицы каркаса стекла и, про-
ведя аналитические исследования, сделать выводы об их
структуре. В основе такого подхода лежит предпосылка
о том, что молекулярная структура стекла может сохраняться
в неизменном виде при переведении в растворимое состояние.
Если при растворении идет химическая реакция, то необходимо
знать структурно-химическую взаимосвязь между конечным про-
дуктом взаимодействия и компонентами стекла. Если же из-
вестны условия, при которых гарантируется выполнение при-
нятой предпосылки, то химические методы исследования пре-
восходят физические методы в том плане, что они приводят
к однозначным выводам о строении больших по размеру струк-
турных областей.
Наиболее ярким примером таких исследований является изу-
чение стекол на основе фосфатов щелочных металлов. Тило
и сотр. [278] и Ван Везер [279] химическим путем установили
структуру этих материалов. Стеклообразный NaPO3 или стекло
с соотношением Na:Ps=Cl,25 растворяли в воде в строго кон-
тролируемых условиях. Под действием ацетона из водного рас-
твора в осадок выпадают полифосфаты. Различные анионы вы-
деляют путем фракционного осаждения с использованием
определенных катионов. Например, с помощью раствора
[Со(ЫНз)б]С13 в ацетоне выделяют пента- и гексациклофосфат-
иоиы (РО3)э~ и (Р03)о~. Такого сорта фрагменты составляют
0,5—0,8 % в структуре стеклообразного NaPO3, и до сих пор
они препаративно получены лишь описанным выше способом
[281]. Если иметь в распоряжении достаточно большой выбор
поли- и циклофосфатов для проведения сравнительного анализа,
то можно методом бумажной хроматографии качественно опре-
делить присутствие тех или иных молекулярных фрагментов
в стекле и в растворе. Количественный анализ может быть
проведен фотометрически или с помощью радиохимических ме-
тодов [282].
Строение анионов в стсклообразующих силикатных распла-
вах подробно исследовано Массоном [283] и Балтой и др.
[284]. На основании экспериментально установленной зависи-
мости термодинамической активности оксидов в силикатных
расплавах от их мольной доли построены модели распределе-
ния анионов в расплаве. В качестве таких моделей рассмат-
ривается, например, исключительно линейное сочленение
структурных элементов типа [S1O4] или возможное разветвле-
ние цепей. Эксперименты по растворению, проводимые для
подтверждения имеющегося в стекле распределения анионов,
должны исключать разрыв мостиковых связей Si—О—Si.
В чтой связи наиболее эффективными оказались два метода.
136 Глава 2
При взаимодействии с (CH3)3SiCl, (CH3)3SiNR2 или
[(CH3)3Si]2N—СОСН3 анионы, присутствующие в кристалли-
ческих силикатах и стеклах, можно перевести практически без
побочных реакций в относительно устойчивые триметилсилило-
вые эфиры [285—287]. Их разделение проводят с помощью
газовой хроматографии. Комбинация этого метода с методом
ЯМР-спектроскопии на ядрах 29Si позволила достигнуть значи-
тельного прогресса в изучении протекающей в водной среде
реакции конденсации кремневых кислот [269—271].
Второй метод основан на прямом растворении образцов. Си-
ликаты с относительно низкой степенью полимеризации раство-
ряются достаточно быстро в НС1 или H2SO4. При оптимизации
условий, при которых в процессе растворения происходит ми-
нимальное количество изменений, обращают внимание на то,
чтобы исходные силикаты имели определенную структуру анио-
нов, и учитывают количество образовавшихся побочных про-
дуктов. При рНЗ разбавленные растворы силикатов устойчивы
в течение нескольких часов и даже суток [288]. Об изучении
таких растворов методами бумажной хроматографии сообща-
лось в работах Викера и Хоббела [289]. Разработан кинетиче-
ский метод количественного определения различных силикатов
в водных растворах [290]. В рамках этого метода на основании
изучения скорости образования ортокремневой кислоты в про-
цессе реакции гидролиза делают выводы о типе конденсирован-
ных силикатов и их концентрации в растворе. При этом коли-
чество образующейся ортокремневой кислоты определяют фото-
метрически по содержанию иопов SiMo^Olo’, образующихся при
добавлении молибдатов.
Для определения структуры неоксидных стекол химические
методы до настоящего времени применялись лишь в отдельных
случаях. При взаимодействии стеклообразных Ge2S3 и Ge2Se3
с Na2S и Na2Se в водных или метанольных растворах при
298 К получены соединения Na6Ge2Se и Na6Ge2Se6. Строение их
анионов установлено методами молекулярной спектроскопии
[291] и препаративными методами — путем взаимодействия по-
лученных соединений с бромом; в результате образуются
Na3GeS3Br и Na3GeSe3Br [292]. В соединениях обнаружена
связь Ge—Ge, и, следовательно, их можно рассматривать как
гексатио- и гексаселеногиподигерманаты. В ходе этой химиче-
ской реакции области ближнего порядка Ge2S6/2 и Ge2Se6/9 вы-
свобождаются из трехмерного каркаса и переводятся в отрица-
тельные шестизарядные ионы, которые могут быть выделены
с помощью соответствующих катионов. В таких стеклах изуче-
ние функции радиального распределения не даст однозначного
заключения даже о строении первой координационной сферы
(разд. 3.2.2.4).
Аморфное и стеклообразное состояние 137
Необходимо заметить, что в ходе этих исследований, на-
правленных на определение строения стекол, был открыт ряд
новых неорганических соединений. Такие соединения удалось
получить относительно простым способом, когда в качестве ис-
ходных веществ при их синтезе использовали стеклообразные
формы веществ.
О том, с какой осторожностью необходимо подходить к хи-
мическим реакциям, используемым для расщепления структуры
стекла в процессе растворения, показывает пример со стекло-
образным селеном. Растворение стеклообразного селена в CS2,
проведенное Бриглебом [293] примерно пять десятилетий назад,
служило экспериментальным подтверждением положения о том,
что стекло в значительной степени состоит из молекул See. При
этом не обращалось внимания на то, что деполимеризация
в присутствии растворителей может сопровождаться замыканием
цепей Se8 с соответствующей конфигурацией в кольцеобразную
структуру (разд. 3.1.3.2).
2.4.2. Структурные теории стеклообразования
Согласно результатам исследований стекол методами,
описанными в разд. 2.4.1, структура стеклообразных и аморф-
ных твердых веществ может быть описана на основании имею-
щихся на сегодняшний день сведений о структуре ближнего
порядка. Склонность атомов к образованию тех или иных типов
химических связей приводит к возникновению структурных эле-
ментов определенной симметрии, которые часто совпадают с об-
ластями ближнего порядка. О существовании аналогичных об-
ластей ближнего порядка часто уже известно из исследований
соответствующих кристаллических твердых фаз. Даже если воз-
можность такого сравнения отсутствует, в некристаллических
твердых телах, вообще говоря, могут быть обнаружены области
ближнего порядка и способы их сочленения.
В настоящее время актуальны в основном два круга вопро-
сов, которые и должны составлять предмет современных ис-
следований структуры стекла. Во-первых, до сих пор не сущест-
вует надежных количественных данных, подтверждающих тео-
ретические соображения о том или ином типе сочленения
структурных элементов между собой. Выводы, получаемые
с помощью имеющихся в распоряжении методов изучения струк-
туры, часто не однозначны. Нередко для одного и того же стек-
лообразующего соединения существуют различные сетчатые
модели с непрерывной и беспорядочной ориентацией областей
ближнего порядка. Сетчатые модели стекла могут, однако, дать
определенное представление о некотором усредненном по-
рядке в системе. Особое положение занимают кластерные мо-
дели.
138 Глава 2
Второй круг вопросов, с одной стороны, связан с возмож-
ностью некоторой нерегулярности соединения структурных
групп, происходящей без изменения их строения. Появление
структурных групп, связанных друг с другом общими ребрами,
можно рассматривать как некоторое топологическое разупоря-
дочение пространственной структуры, в которой области ближ-
него порядка соединяются в основном своими вершинами. Во-
вторых, необходимо принимать во внимание конституционное
разупорядочение в самих областях ближнего порядка. К таким
типам дефектов структуры относятся разрушение структурных
группировок или разрыв связей в процессе вязкого течения.
Оба типа разупорядочения по своей сути представляют со-
бой точечные дефекты и могут быть интерпретированы в случае
их гомогенного распределения как собственное разупорядоче-
ние предельного идеального состояния данной структуры стекла.
Однокомпонентные стекла, например на основе простых ве-
ществ или соединений с определенной стехиометрией, характе-
ризуются определенным видом областей ближнего порядка. В них
можно выделить следующие основные типы разупорядочения.
а) колебания плотности как проявление замороженных тер-
мических флуктуаций;
б) геометрическое разупорядочение как проявление отсутст-
вия дальнего порядка;
в) собственное разупорядочение в областях ближнего по-
рядка: топологическое разупорядочение и структурное разупоря-
дочение областей ближнего порядка.
2.4.2.1. Прототипы стеклообразующих соединений
Основные виды областей ближнего порядка и их симмет-
рия для типичных стеклообразующих веществ стехиометриче-
ского состава приведены в табл. 2.10. Такие соединения назы-
вают стеклообразователями. Координационные числа элементов
указаны в первой колонке при соответствующем химическом
символе элемента справа вверху в квадратных скобках. Мер-
ность макромолекулярного, практически бесконечного сочлене-
ния отдельных структурных единиц приведена в виде соответ-
ствующего обозначения перед формульным типом.
За исключением BeF2 типичными стеклообразователями яв-
ляются оксиды и халькогениды. Атомы халькогенов участвуют
в образовании двух химических связей, т. е. это мостиковые
лиганды, принадлежащие сразу двум структурным единицам.
Сочленение структурных элементов может продолжаться, и при
этом возникают полимерные макромолекулярные цепи (напри-
мер, стеклообразные селен и сера, которые образуются при за-
твердевании). Несмотря на то что такие цепи изогнуты под
разными углами и при макроскопическом подходе нельзя об-
Аморфное и стеклообразное состояние
139
Таблица 2.10. Виды областей ближнего порядка типичных
стеклообразователей
Формульный тип и'степень связности стеклообразного каркаса Структурная единица Группа симметрии идеальной структуры Стеклообразователь
i х'2> хх2/2 С2у Se, S
~ mJ31xJ2J мх3/2 ^3v AS2O3, AS2S3, АЗгЗбз, As2Te3
00 mJ31xJ2Ix2 хмх3/2 ^3v P2O5
~ Mj3IXj21 мх3/2 D3h B2O3
~ mhix'2' 3 МХ4/2 Td SiO2> BeF2, GeO2, GeSj, GeSe2, GeTe2
СО Mj4IXi2* М2Хб/2 &3d Ge2Sa, Ge2Se3
наружить определенного направления их расположения, все же
говорят об образовании одномерной сетки. Такой структуре
приписывают степень связности стеклообразного каркаса, рав-
ную 1.
Двумерная бесконечная сетчатая структура характерна для
халькогенидов мышьяка, а также для Р2Об и В2О3. И в этом
случае по данным рентгеновского исследования можно выделить
лишь слоистую структуру. Эти слои сильно деформированы,
так что макроскопические свойства изотропны. Степень связно-
сти стеклообразного каркаса в этих веществах равна 2.
Пространственная сетчатая структура с трехмерным соедине-
нием областей ближнего порядка существует в стеклах и аморф-
ных веществах с общей формулой МХ2. Области ближнего
порядка имеют симметрию типа Та, как и молекулы СН4, SiF4
или GeCU (разд. 2.4.1.2). Пространственная структура с обла-
стями ближнего порядка, аналогичными молекулам С2Нс или
Ge2Cl6, впервые для стеклообразующих соединений обнаружена
у Ge2S3 и Ge2Se3 (разд. 3.2.2.4 и 3.2.2.5). Составлен [454] пере-
чень различных типов ближнего упорядочения с включением
в него и характерных структур металлических стекол.
2.4.2.2. Сеточная модель стеклообразователей
На основании существования ближнего порядка в совокуп-
ности с однородным строением стеклообразных однокомпонент-
ных систем вполне оправдан вывод об образовании простран-
ственных структурных сеток. Для понимания структурно-хими-
140 Глава 2
ческих предпосылок, необходимых для реализации стеклообраз-
ного и аморфного состояния, уместно задать вопрос об
общности этого явления.
Ответ на этот вопрос применительно к стеклообразующим
оксидам дан около полувека назад в виде гипотезы Захариасена
[230], которая противопоставлялась предложенной ранее
(1921 г.) кристаллитной теории Лебедева (последняя нашла
экспериментальное подтверждение в работе Ренделла и сотр.
[294], где рентгенографически обнаружено наличие кристобали-
топодобных областей в стекле). К тому времени было известно,
что теплоемкости стеклообразных и кристаллических форм ве-
щества практически не отличаются друг от друга. Исходя из
этого, сделан вывод об идентичности областей ближнего по-
рядка. Отсутствие дальнего порядка, в чем заключается глав-
ная отличительная особенность стеклообразных и аморфных
фаз, согласно Захариасену, связано в оксидах МОХ с определен-
ными структурно-химическими условиями. Остановимся на этих
условиях.
Каждый атом кислорода имеет координационное число, рав-
ное 2, и, следовательно, может быть связан не более чем
с двумя атомами М. Согласно данным, приведенным в табл. 2.10,
число атомов кислорода, окружающих атом М, не должно быть
большим. Желательно, чтобы это число было равно 3 или 4.
Области ближнего порядка состоят из координационных поли-
эдров МОХ, имеющих общие вершины, а не ребра и грани. Для
образования трехмерной пространственной сетки необходимо,
чтобы по крайней мере три вершины полиэдра были общими
с соседними полиэдрами.
Эти правила без дальнейшей доработки могут быть пере-
несены на случай неоксидных стеклообразующих соединений.
В отдельных исключительных случаях, однако, возможны неко-
торые отклонения от этих правил. Так, например, в сульфидах
и селенидах мышьяка и германия (As2S3, As2Se3, GeS2, GeSe2)
в построении каркаса стекла принимают также участие струк-
турные группы, соединенные своими ребрами (разд. 3.2.1.4,
3.2.1.5, 3.2.2.4, 3.2.2.5). При этом необходимо обратить внима-
ние на то, что такого типа отклонения, которые называют топо-
логическим разупорядочением, вызывают уменьшение степени
связности стеклообразного каркаса. В аморфных формах моно-
халькогенидов германия, как и в ряде оксидных стекол слож-
ного состава, атомы халькогена, по крайней мере частично,
имеют координационное число 3. Это означает, что и правило
о том, что координационное число мостиковых атомов должно
быть равно 2, не всегда выполняется (ср. также со стеклообраз-
ным мышьяком, разд. 3.1.2.5).
В итоге можно сказать, что правила Захариасена, сформу-
лированные первоначально в виде гипотезы, оказались вполне
Аморфное и стеклообразное состояние
141
справедливыми и позже подтверждены структурными исследо-
ваниями. В 1935 г. Уоррен и сотр. [229] установили, что полу-
ченные ими рентгенограммы кварцевого стекла лучше всего
интерпретируются в рамках модели непрерывной беспорядочной
сетки тетраэдров [SiO4]. Поэтому основополагающая и сохра-
нившаяся до сегодняшнего дня в почти неизменном виде струк-
турная теория стекла связана с именами Захариасена и Уор-
рена.
Правила Захариасена отражают, собственно говоря, лишь
геометрические и стерические предпосылки, которые должны
быть выполнены для обеспечения возможности образования
Рис. 2.37. Структура ближнего порядка и искажение валентных и диэдри-
ческнх углов в кварцевом стекле (а) и стеклообразном селене (б).
большого числа структурно примерно эквивалентных конфигу-
раций. Одинаковая вероятность образования этих конфигура-
ций— причина появления твердых тел, не имеющих дальнего
порядка. В кварцевом стекле многообразие структурно экви-
валентных конфигураций обусловлено весьма широкой вариа-
бельностью валентных углов между связями мостикового кис-
лородного атома и возможностью свободного вращения вокруг
связи Si—О, т. е. конформационной статистикой. Углы и осо-
бенно длины химических связей между элементами внутри тет-
раэдров, напротив, остаются в основном неизменными. На
рис. 2.37 схематически показаны возможности изменения кон-
формации для двух связанных между собой тетраэдров [SiO4] -
Другие сочленения таких тетраэдров ведут к образованию про-
странственной сетки с беспорядочным непрерывным распределе-
нием всех возможных конфигураций. Таким образом, как уже
говорилось, многообразие конфигураций обусловлено конфор-
мационной статистикой, определяющей образование тех или
иных типов связей между структурными группами.
Белл и Дин [228] предложили модель структуры такого
рода для кварцевого стекла. Само собой разумеется, что в за-
висимости от величины или функции распределения валентного
угла а между связями мостикового кислородного атома су-
ществует сколь угодно большое количество таких моделей
142 Глава 2
решетки. В случае SiO2 валентные углы между связями кисло-
рода, согласно экспериментальным данным, меняются от 120
до 180° с максимумом при 150° [228]. Такого рода структуру
данного стеклообразователя называют идеальной. В этом
смысле и следует понимать сравнение структуры кварцевого
стекла с одной из кристаллических модификаций (рис. 2.38).
На рис. 2.37 изображен фрагмент структуры ближнего по-
рядка для цепочки атомов Se в
Рис. 2.38. Проекция на плоскость фраг-
мента непрерывной беспорядочной сет-
чатой структуры кварцевого стекла.
стеклообразном селене. Длины
химических связей и углы
в группировке SeSe2/2 со-
впадают с соответствующи-
ми величинами в кристал-
лическом селене. Атомы /,
2 и 3 находятся в одной
плоскости. Таким образом,
многообразие конфигураций
цепи обусловлено лишь зна-
чениями диэдрических углов
ср между четвертым атомом
селена и тремя другими
на плоскости или между
плоскостями, в которых на-
ходятся атомы /, 2, 3 и 2,
3, 4. По данным Малорана
и Диксмайера [295], все
диэдрические углы (0—90°)
одинаково вероятны, т. е.
в цепи происходит беспрепятственное вращение вокруг
связи Se—Se. Результаты последующих исследований
методом фотоэлектронной спектроскопии показали, что из-за
образования гибридных связей между атомами селена диэдри-
ческпй угол сохраняется постоянным с небольшими откло-
нениями (разд. 3.1.3.2). Таким образом, все возможные кон-
фигурации ограничиваются в основном двумя альтернативными
положениями четвертого атома Se относительно трех других,
находящихся в одной плоскости. Поскольку вероятность этих
конфигураций определяется статистическими законами, следует
вновь обратиться к конформационной статистике. Необходимо
указать, какие структурные группы или конформацион-
ные положения описывает конформационная статистика. Про-
блема однозначного определения взаимного расположения ато-
мов за пределами первой и второй координационных сфер
встает особенно остро, если учитывать тот факт, что от нахо-
ждения четвертого атома селена в цис- или транс-положении
зависит возникновение кольцевых молекул See. Другими сло-
вами, конфигурация цепи влияет на образование или исчезно-
вение молекул Seg. Кольцевые молекулы вовсе не обязательно
Аморфное и стеклообразное состояние 143
возникают даже в случае ^wc-конфигурации (рис. 3.20). Если
для стеклообразного селена учесть, что на кривой ФРР макси-
мум нечетко выражен, то вряд ли возможно сделать определен-
ные выводы о доле молекул See в структуре этого вещества.
Между тем в настоящее время принято считать, что в стекло-
образном селене присутствует заметное количество молекул
Seg [293]. Поэтому, естественно, применимость представлений
о сетчатой структуре к строению стеклообразного селена под-
вергается оправданному сомнению.
Пример со стеклообразным селеном показывает, как тяжело
экспериментально установить строение областей стекла, в кото-
рых возможно статистически случайное распределение раз-
личных типов организации структуры. (Из этого распределения
вытекает изотропность стеклообразных и аморфных твердых
тел.) Если же наблюдается частичное предпочтение определен-
ной конфигурации (на рис. 2.37, например, тетраэдры ориенти-
рованы таким же образом, как в а-кварце, p-кварце, тридимите
или кристобалите), то это сразу приводит к применимости мо-
дели сетчатой структуры кварцевого стекла к большим струк-
турным областям, имеющим сходное строение с кристалличе-
скими модификациями [297, 298]. Предельным случаем таких
структурных представлений является кристаллитная модель,
пригодность которой для описания большинства стеклообразных
веществ по современным понятиям невелика. В кварцевом
стекле кристаллиты играют определенную роль лишь в случае
неполного расплавления кристалла кварца. Однако, как пока-
зали недавние исследования [299], кристаллиты не типичны для
стеклообразного состояния. По мнению Моззи и Уоррена [227],
для больших областей стеклообразного образца нельзя прини-
мать существование сетчатой структуры, характеризующейся
предпочтением одной определенной конфигурации тетраэдриче-
ских структурных групп. Результаты модельных статистических
расчетов для структурной сетки кварцевого стекла [228] нахо-
дятся в хорошем согласии с экспериментальными данными.
Сетчатые модели с непрерывным беспорядочным сочлене-
нием областей ближнего порядка до сих пор являются лучшим
приближением для описания структуры аморфных Si и Ge.
После основополагающей работы Полка [300] они неоднократно
так или иначе совершенствовались для наилучшего согласова-
ния с имеющимися экспериментальными данными (разд. 3.1.2.3).
Грифе и Девис [301] с успехом перенесли модель Полка на
аморфный As (разд. 3.1.2.5). В металлических стеклах общей
формулы А4В [302] также существуют области ближнего по-
рядка с расположением атомов меньшего размера В, в основном
в пустотах, вокруг которых располагаются 8—9 соседних ато-
мов. В этом смысле существует известная аналогия со струк-
турной теорией жидкостей Бернала, согласно которой в жидкости
144 Глава 2
существует плотная статистическая упаковка сферических
частиц А (разд. 3.1.1).
Подробный обзор кристаллических прототипов стеклообра-
зующих соединений и аморфных полупроводниковых твердых
тел опубликован Моссом [303]. Весьма исчерпывающую обра-
ботку литературных данных, прежде всего результатов изучения
колебательных спектров, провели Коннел и Луковски [304].
Дальнейшее развитие принципов топологии сетчатой структуры
стеклообразующих веществ содержится в работах Филлипса
[305]. Исходя из теоремы Каузмана (разд. 2.2.3.1), опираясь
на тот факт, что дифракционный максимум на кривых рассея-
ния на малых углах характеризуется значительной интенсив-
ностью, а также используя данные молекулярно-спектроскопи-
ческих исследований, в этих работах выявлен некоторый
средний порядок в структуре сеток (прежде всего для халькоге-
нидных стекол), который распространяется на области 150—
300 пм [450]. Райт и сотр. [451] на основании геометрических
преобразований провели систематическую классификацию струк-
тур аморфных твердых тел.
2.4.2.3. Кластерные модели
Кластерные модели вытекают из очевидного физического
представления о том, что при охлаждении расплава или в про-
цессе образования аморфных веществ другим путем могут воз-
Рис. 2.39. Кластер, состоящий
из 20 атомов Si или Gc с осью
симметрии 5-го порядка, не
может быть растущим зароды-
шем.
никать низкомолекулярные струк-
турные конфигурации, что сопровож-
дается определенным выигрышем
в энергии. Из-за того что такие об-
разования характеризуются особой
симметрией, они не могут служить
ни зародышами кристаллов, пи цен-
трами роста твердых аморфных
фаз. Такая ситуация имеет место
в случае структур, имеющих, напри-
мер, ось симметрии 5-го порядка,
когда атомы или структурные груп-
пы с тетраэдрическими связями
соединяются между собой, обра-
зуя заслоненные конформации (рис.
2.39). В случае плоских пятиуголь-
ных циклов с углами 108° дефор-
мация тетраэдрических углов относительно мала и энергетиче-
ские характеристики таких кластеров, вероятно, сопоставимы
со способными к росту кластерами, состоящими из шестичлен-
ных колец. Каждый атом может образовывать радиальные
связи с другими атомами. Однако процесс увеличения кластера
Аморфное и стеклообразное состояние
145
сопровождается его сильным искажением, и поэтому непрерыв-
ное заполнение пространства в одном направлении идет путем
пристраивания других додекаэдров. Присоединение шестичлен-
ных колец в форме ванны также ведет к структуре с симмет-
рией 5-го порядка.
Если между двумя атомами Si или Ge (рис. 2.39) можно
было бы встроить атом кислорода, то возник бы кластер, ха-
рактерный для аморфных форм SiO2 и GeO2, с осью симмет-
рии 5-го порядка. Тилтон [306] назвал такого рода структур-
ные образования нитронами. Структура, построенная из вит-
ронов, должна быть дискретна, поскольку при встраивании
в сетку компактных аморфных твердых тел витроны должны
присоединяться каким-то другим способом к беспорядочно ори-
ентированным структурным элементам. Эта модель получила
дальнейшее развитие в работе Робинсона [307]. Опираясь на
структурную теорию Бернала для одноатомной жидкости [308],
было принято, что диаметр цилиндрических кластеров равен
— 3 нм. Эти кластеры образуют почти плотную упаковку и
соединяются друг с другом химическими связями. Позиции
кластерной модели несколько пошатнулись после того, как
было установлено, что зарегистрированные в ходе электронно-
микроскопических исследований области негомогенности
в стекле имеют размеры того же порядка, что и области, неза-
твердевшие после плавления. Поэтому в более поздних работах
все больше утверждается мнение, что различие между рассчи-
танными и экспериментально найденными кривыми радиального
распределения указывают на то, что модель непрерывной беспо-
рядочной сетки лучше соответствует строению кварцевого
стекла, чем кластерная модель [309].
Для кластера из атомов Ge, изображенного на рис. 3.39,
предложено название «аморфон». В работе [310] достигнуто
хорошее согласование расчетной функции радиального распре-
деления с экспериментальной ФРР для аморфного германия
(см. литературу к гл. 1 [31]). В рамках модели непрерывной
статистической сетки Полка удалось достичь еще лучшего сов-
падения (разд. 3.1.2.3). Однако кластерная модель все еще ис-
пользуется в литературе в качестве альтернативы сетчатой мо-
дели аморфных тел [311].
2.4.2.4. Химическая связь и стеклообразование
Структурно-химические предпосылки стеклообразования
представляют собой объединение энергетических и стерических
факторов. Характер химических связей в веществе определяет,
в какой степени склонность к стеклообразованию ведет к воз-
никновению структурных групп с достаточной асимметрией или
порождает такие свойства, которые создают стерические пред-
10 Заказ № 413
146 Глава 2
посылки для большого числа различных конформаций. Исходя
из этих требований, для возникновения стеклообразных фаз
наиболее подходят структурные мотивы, представляющие собой
области ближнего порядка с невысокими координационными
числами (3 или 4). Для октаэдрических структурных групп
(координационное число 6) изменение валентных углов между
связями мостикового кислородного атома в значительной сте-
пени ограниченно. Поэтому соединения типа TiO2 или SnO2 не
образуют стекол. Решающее значение при обсуждении даже
самой возможности возникновения стекол имеет существование
данного вещества в различных структурных конфигурациях, ко-
торые энергетически либо равнозначны, либо очень слабо от-
личаются. Соединения, в которых характер химической связи
определяет образование областей ближнего порядка с такого
рода геометрическими свойствами, отличаются отчетливой тен-
денцией к стеклообразованию.
В этой связи необходимо упомянуть о достаточной жест-
кости структурных групп. Таким образом, стеклообразователи
характеризуются направленными химическими связями в об-
ластях ближнего порядка. Значительная доля ковалентности
связи — надежный критерий существования для данного веще-
ства стеклообразного состояния.
Если асимметрия молекулярных групп выражена достаточно
четко, то стеклообразование возможно даже в случае органиче-
ских жидкостей, например для изопентана, изопропилбензола
или метанола [312]. Сохранение формы стеклообразных твердых
тел связано в этих веществах с существованием вандервааль-
сового взаимодействия или, как в случае спиртов, с наличием
водородных связей. Даже вода застывает при достаточно высо-
ких скоростях охлаждения в виде стеклообразной фазы (Т(,=
= 135 К) [312, 313].
В металлических стеклах, например, общей формулы А4В
ковалентные связи стабилизируют структуру жидкости Бер-
нала. Речь идет о ковалентных связях между атомами В мень-
шего размера, находящимися в пустотах структуры, и окру-
жающими их 8—9 атомами А. Стеклообразующие оксиды отли-
чаются от оксидов металлов с гетерополярными связями более
высокой степенью ковалентности химических связей. При пе-
реходе к халькогенидным стеклам полярность связи еще умень-
шается, и в стеклообразных селене и мышьяке, как и в аморф-
ных германии и кремнии, структуру определяют гомеополярные
связи.
В литературе описаны многие попытки свести стеклообразо-
вание к определенному параметру, характеризующему химиче-
скую связь. При этом не принимались во внимание некоторые
количественные, имеющие ясный смысл стерические факторы.
Поэтому все эти подходы оказались несостоятельными; они
Аморфное и стеклообразное состояние
147
пригодны лишь для классификации определенных групп стекло-
образующих соединений.
Смекал [314] обратил внимание на роль смешанных хи-
мических связей в связи с вопросами стсклообразования. Стен-
ворт [315] ввел критерий разности электроотрицательности
компонентов. Для стеклообразующих оксидов бора, кремния,
фосфора, германия и мышьяка эта разность составляет 1,8—
2,1. Для оксида мышьяка и SnO2 разность электроотрицатель-
ностей, однако, приходятся примерно на тот же интервал зна-
чения. Для BeF2 степень ионности связи оценивается 85%, для
As2S3— лишь 5%, хотя оба соединения и являются отличными
стеклообразователями. Сан [316] предпринял попытки прове-
сти корреляцию склонности к стеклообразованию оксидов и
энергии связи. Однако при этом не удалось отчетливо разгра-
ничить различные оксиды. Подход Роусона (см. литературу
к гл. 1 [8]), состоящий в проведении корреляции между энер-
гиями одинарных валентных связей и температурой плавления
вещества, также не оказался эффективным.
Для стеклообразующих оксидных систем прочно укорени-
лась концепция напряженности поля Дитцеля [317]. Дитцель
в грубом приближении записал выражение для энергии взаимо-
действия Uл в виде UA = ZiZ2e2/a; тогда для напряженности
поля можно получить F = Z\Z2ela2. Если ограничиться только ок-
сидами, то заряд аниона z2 и заряд электрона е постоянны.
Таким образом, мерой напряженности поля является отношение
г/а2 (где z — заряд катиона, а — длина связи в ангстремах).
Для типичных стеклообразующих оксидов среднее значение
этой величины составляет 1—2.
Винтер [318] обратила внимание на известную взаимо-
связь между стеклообразованием и числом р-электронов внеш-
ней электронной оболочки. Соединения, склонные к стеклооб-
разованию, содержат элементы, у которых имеется 2—4 р-элек-
трона. Однако если в качестве примера рассмотреть В2О3 и
BeF2, для которых этот критерий выполняется лишь для мо-
стиковых атомов, а также кислород и теллур, то надежность
предлагаемого критерия окажется весьма сомнительной. Более
современные представления о собственном структурном разупо-
рядочении учитывают роль неподеленных пар р-электронов
в общем ансамбле структурных конфигураций стекла
(разд. 2.5.2).
2.4.2.5. Структура идеального стекла и собственное
разупорядочение
Непрерывное беспорядочное соединение структурных групп
в изменяющемся (с точки зрения возможных пространственных
конфигураций), но жестком после образования всех связей
10*
148 Глава 2
структурном каркасе называют идеальной структурой данного
некристаллического твердого тела. Если рассмотреть приведен-
ные в табл. 2.10 соединения, расплавы которых в процессе ох-
лаждения переходят в стеклообразное состояние, то возникает
вопрос о причинах наблюдающейся подвижности структуры
выше температуры стеклования. Какие из химических связей
разрушаются и перераспределяются при переходе стекла в со-
стояние переохлажденной жидкости и, наоборот, какие связи
фиксируются в процессе замораживания неравновесного стекло-
образного твердого тела? Поскольку текучесть, характерная
для жидкостей, требует достаточной деполимеризации, замо-
роженные в стеклообразном состоянии термические флуктуации
плотности непосредственно отражают элементарные стадии вяз-
кого течения.
Феноменологическое описание процессов течения переохла-
жденных расплавов дано в разд. 2.2.1.3. Механистические пред-
ставления развиты слабо, так как их трудно проверить экспе-
риментально. Лишь теория активированного переключения кова-
лентных связей Мюллера [ 106] представляет собой плодотвор-
ный подход к структурно-химическому анализу проблемы.
Процессы течения не могут быть сведены лишь к умень-
шению степени связности стеклообразного каркаса, например,
путем «разматывания» цепей при образовании колец или к пе-
реходу в такие состояния, которые характеризуются кратко-
временным возникновением соединенных ребрами структурных
групп. В идеальной сетчатой структуре именно топологическое
разупорядочение понижает степень связности стеклообразного
каркаса. Но такому разупорядочению сопутствует одновремен-
ное образование более крупных жестких структурных групп,
что ограничивает возможность их различных сочленений. Так,
доля соединенных ребрами тетраэдрических групп в структу-
рах GeS2 и GeSe2 составляет 50%, поэтому эти вещества имеют
слоистую решетку. Тенденция к кристаллизации их расплавов
в отличие от GeO2 и SiO2 существенно повышается (разд. 3.2.2.4
и 3.2.2.5). В SiS2, в котором тетраэдры [SiS4] связаны между
собой исключительно ребрами, существуют жесткие линейные
макромолекулы. Совершенно понятно, что у этого соединения
стеклообразование не обнаружено.
Если стремиться описать явления вязкого течения с точки
зрения энергетических аспектов теории химической связи, не-
обходимо иметь в виду наличие определенных равновесных кон-
центраций разрушенных связей, т. е. изменений, происходя-
щих в областях ближнего порядка.
Разрушение связей происходит в системах, включающих
атомы с неподеленными электронными парами. Часто при раз-
рыве связи электронная пара остается на одном из фрагмен-
тов, т. е. происходит гетеролитический разрыв связи. При этом
Аморфное и стеклообразное состояние
149
возникают один положительно заряженный и один отрица-
тельно заряженный дефектные центры. Необходимая для раз-
рыва связи энергия отчасти компенсируется за счет существова-
ния неподеленной электронной пары у атомов, находящихся
в ближайшем окружении. Таким образом, число химических
связей в целом не меняется. Наличие р-электронов у мостиковых
Рис. 2.40. Структурное разупорядочение в кварцевом стекле.
кислородных атомов или атомов халькогенов обеспечивает ста-
билизацию такой структуры (рис. 2.40, 2.41). Следовательно,
в переохлажденном расплаве должны существовать положи-
тельные и отрицательные дефектные центры, концентрация ко-
торых пс, согласно уравнению (2.102, б), определяется свободной
2SeSe^2
е е
SeSe# + SeSe^
Рис. 2.41. Структурное разупоря-
дочение в стеклообразном селене.
энергией реакции образования дефектов (разд. 2.5.2). Концен-
трация дефектных центров, отвечающая температуре T=Tg,
замораживается в процессе затвердевания стекла. Такого сорта
центры присутствуют также и в стеклообразном состоянии ве-
щества, поскольку совершенно невероятно, чтобы все разорван-
ные химические связи при затвердевании переохлажденного
расплава образовались бы вновь и чтобы структура идеального
стекла оставалась бы бездефектной. Таким образом, в гомоген-
ных стеклах стехиометрического состава наряду с колебаниями
плотности и имеющимся, согласно определению идеального
стеклообразного состояния, топологическим разупорядочением
150 Глава 2
различного вида существует еще и структурное разупорядоче-
ние. Последнее проявляется в виде положительно и отрицательно
заряженных дефектных центров. При этом, как и в случае то-
чечных дефектов в кристаллах, доминируют дефекты, реакции
образования которых характеризуются наименьшим изменением
свободной энергии.
Для кварцевого стекла и стеклообразного селена наиболее
вероятны точечные дефекты (рис. 2.40 и 2.41). В определен-
ных местах каркаса стекла вследствие термических колебаний
связи Si—О поляризуются так сильно, что энергетически вы-
годным становится образование новых химических связей с со-
седним мостиковым кислородным атомом. Таким способом воз-
никают новые структурные группы [О(SiO3/3)3] + с положи-
тельным зарядом на атоме кислорода и SiO3/2O~ с отрицатель-
ным зарядом на атоме кислорода. Луковски [319] в результате
изучения ПК и КР-спектров кварцевого стекла с определен-
ностью установил наличие таких центров и оценил их концен-
трацию величиной 1019 см“3.
В рамках такого подхода точка зрения Смекала [314]
о смешанных химических связях может рассматриваться как
выражение тенденции к возникновению изомерии связи. К об-
разованию таких связей часто склонны элементы главных под-
групп правой половины периодической системы элементов. Не
имеет решающего значения, что связи Si—О характеризуются
степенью ионности 50%, так как и в аморфном селене сущест-
вуют дефектные центры примерно с той же концентрацией
(разд. 3.1.3.2). Предлагаемое на рис. 2.41 уравнение, описы-
вающее равновесный процесс в расплавленном селене, соответ-
ствует протеканию в неметаллических системах реакций дис-
социации, например 2H20z± Н3О+ + ОН_, ЗВг2 Вгз+Вгз или
312 13 +1 з . PC13F2 и PBr3F2 известны как ковалентные соеди-
нения (тригонально-бипирамидальные молекулы), а также
в виде своих гетерополярных модификаций [РС14]+ [PF6] ~ и
[РВг4]+ [PFa]_. В работах Кольдица и сотр. [321J описаны
и многие другие соединения этого типа.
Собственное разупорядочение в стсклообразующих систе-
мах, которое характеризуется возникновением положительно
и отрицательно заряженных дефектных центров, подробно об-
суждается в разд. 2.5.2. Из качественных соображений, касаю-
щихся лишь структуры и химических связей в условиях вяз-
кого течения, оказывается вполне понятным существование
собственного разупорядочения в стеклообразующих системах.
Число и вид ковалентных связей остаются неизменными как
в неполярных, так и в полярных формах веществ.
Аморфное и стеклообразное состояние 151
В качестве другой возможности образования дефектных
центров как одного из типов собственного разупорядочения не-
обходимо рассмотреть возникновение связей элемент—элемент
в гетероатомной системе. При этом с термодинамических по-
зиций удобно использовать среднюю энергию связи Ев.
Энергию связи можно найти либо по методу Мюллера [322]
(находят отношение стандартной энергии атомизации &НА дан-
ного соединения к числу связей, содержащихся в одной формуль-
ной единице вещества), либо в случае неизвестной структуры
или неоднозначно определенных координационных чисел из
разности элсктроотрицательностей X по Полингу [323]:
Ем_х =0,5(£м_м + Ех_х) + 100 (Хм - XJ - 6,5 (Хм - Хх)<
Для указанных элементов М и X значения /?м-м и fx-x
получают _ как частные от деления мольных стандартных
энергий атомизации на число связей, приходящихся на один
атом. Энтальпии атомизации ХНА^ приведены в литературе
[323—326]. Для соединений, например, приведенных в табл. 2.10,
при расчетах методом Мюллера необходимо учитывать стан-
дартные мольные энтальпии реакций их образования ХНц,
а также энтальпии кристаллизации стеклообразных веществ
Д/Др. Термохимические свойства в стеклообразующих системах
подробно изучены в работе [327]. В соответствии с уравнением
АЯЛ = £ Xi ЬНА, i-XHR + АЯкр (2.100)
для величины энтальпии атомизации стеклообразных Se,
As2S3 и GeSe2 получено
Se ХН А = 4-227 + 0 — 3 — +224 кДж/моль
Aso>4So,6 АЯЛ =+289 + 31 — 3 =+317 кДж моль
Geo,33Seo,67 АЯЛ ==+227 + 30 — 6 =+301 кДж/моль
В пересчете на формулу GeSe4/2 энтальпия атомизации
равна A/+t = 903 кДж/моль, а средняя энергия связи £'oe-se=
= 0,75-301 = 226 — 225 кДж/моль. Приведенные в табл. 2.11 зна-
чения энергий связи рассчитаны таким же способом (погреш-
ность оценивается в 10 кДж/моль, поэтому значения округ-
лены). А//кР оценивалась по формуле А//кр = 0,4 ХНп:1 (где
А//пл — мольная энтальпия плавления).
Если для оценки термохимической стабильности связей
М—X использовать уравнение
2М—X - М—М + X—X (2.101)
например, —35 кДж/моль + 2Ge—Se Z2 Ge—Ge + Se—Se
(2GeSe4/2 r: Ge2See/j + Se2/j)
или —15 кДж/моль + 2As—Те д: As—As + Те — Те
(2AsTe3/2 ZT As2Te4/2 + Te2/j)
152 Глава 2
Таблица 2.11. Средние мольные энергии связи Ев [327]
Связь Eg, кДж/моль Связь Eg , кДж/МОЛЬ
С/5 1 С/5 280 As —Те 205
Se—Se 225 Sb-S 260
Те—Те 195 Sb-Se 230
S—Se 255 Sb-Те 195
Se—Те 220 Ge—S 265
Р-Р 225 Ge—Se 225
As—As 200 Ge—Те 200
Sb—Sb 175 Si—Те 220
Si—Si 225 P—0 (для P4O6) 425
Ge—Ge 185 As -0 335
P-S 270 SI—0 465
P-Se 240 Ge—0 355
As—S 260 BO 520
As —Se 230
то во всех случаях получается, что связь М.—X энергетически
выгоднее эквивалентного количества связей М—М и X—X.
При этом в первом приближении энтропийными вкладами мо-
жно пренебречь, и при расчете равновесия свободные энергии
образования связей можно заменить на энтальпии образования
связей. Кроме того, необходимо обратить внимание на то, что
в уравнения термохимического баланса (2.101) должна вхо-
дить энтальпия при температуре стеклования. В расчетах же
используются соответствующие значения для 298 К. Таким об-
разом, такие расчеты дают лишь грубую оценку.
Структурные модели, в основе которых лежит одинаковое
статистическое распределение всех возможных связей, не учи-
тывают различий в энергиях связей. Они часто привлекаются
для интерпретации экспериментальных кривых радиального рас-
пределения, однако, вероятно, нс соответствуют действитель-
ности из-за того, что существуют равновесия типа (2.101). Если
отвлечься от неточности, которую вносит пренебрежение энтро-
пийным вкладом, то из экспериментальных данных с энергети-
ческих позиций можно рассчитать склонность к образованию
химических связей для приведенных в табл. 2.10 областей
ближнего порядка. Правда, значения энергии связи в селени-
дах и теллуридах, как показывают примеры, относительно не-
велики, и поэтому при температуре плавления GeSe2 (1013 К)
Аморфное и стеклообразное состояние 153
и даже вблизи температуры стеклования (665 К) нужно счи-
таться с частичным образованием связей Ge—Ge и Se—Se.
Для числа связей в 1 см3 п0 можно записать общее урав-
нение
л0 = (р/М) £ XiNAZi/2 (2.102а)
где М — средняя молярная масса атомов, xt— мольная доля
соответствующего компонента, Na — число Авогадро, Zi — со-
ответствующее координационное число, р — плотность. Если,
например, для расплава GeSes в рамках указанных прибли-
жений принять AGC = 35 кДж/моль, то, согласно уравнению за-
кона действующих масс для температуры 7, = 7,^ = 665 К, можно
получить концентрацию связей Ge—Ge и Se—Se в стеклооб-
разном состоянии:
пс = л0ехр(-АОс/2/?Г) (при лс<п0) (2.1026)
Она составляет 2-1021 на 1 см3. Это означает, что примерно
4% общего числа всех связей составляют связи М—М и X—X.
Если же учесть энтропийный вклад, то концентрация гомоядер-
ных связей скорее всего еще больше увеличится. Такое же
влияние оказывает уменьшение энтальпии реакции, происходя-
щее с ростом температуры. Поэтому проведенная оценка — это
нижний предел концентрации гомоядерных связей. Таким об-
разом, в стеклообразующих расплавах веществ, характеризую-
щихся средними и низкими энергиями связи, наряду с образо-
ванием положительно и отрицательно заряженных дефектных
центров необходимо считаться с описанными выше дефектами
в областях ближнего порядка структуры идеального стекла.
2.4.2.6. О структуре сложных стеклообразующих систем
Многочисленные стеклообразующие системы появляются
в результате всевозможных комбинаций различных стеклооб-
разователей между собой, а также при добавках к стеклам
других оксидов или халькогенидов. В таких системах реали-
зуется широкий набор структур, и поэтому вряд ли удасться
провести их систематическое обсуждение, как в случае простых
стеклообразующихся соединений.
Прежде всего необходимо указать на то, что даже веще-
ства с постоянным составом и определенной структурой при
плавлении могут перейти в смесь различных соединений. По-
этому даже в этом простейшем случае необходимо считаться
с особенностями, характерными для смешанных фаз.
В метафосфате NaPO3 имеется, как и в волластоните CaSiOs,
бесконечная пространственная цепочка тетраэдрических струк-
турных групп с периодом повторяемости, равным 3. В расплаве
154 Глава 2
происходит существенная перегруппировка структурного кар-
каса и при застывании в стеклообразную фазу обнаруживаются
циклофосфаты, содержащие до 7 атомов фосфора в цикле.
В силикатах с цепочечной структурой наряду с циклическими
анионами содержатся также дву- и трехмерные сетчатые
структурные группы, а также низкомолекулярные образова-
ния [283].
Добавки оксидов металлов к стеклообразующим оксидам
Рис. 2.42. Схематическое
изображение структурной
сетки в присутствии моди-
фикатора.
или халькогенидов металлов к типич-
ным халькогенида м-стеклообразовате-
лям модифицируют структуру сетки.
Оксидные или халькогенидные ионы
нуклеофильно разрывают мостико-
вые связи с локализацией отрицатель-
ного заряда на концевых атомах кис-
лорода или халькогена, которые преи-
мущественно располагаются вокруг
введенных ионов металла. Их взаи-
модействие обусловлено в основном
электростатическими силами; таким
образом, с введением добавки сетка
разрушается и степень связности стек-
лообразного каркаса постепенно по-
нижается. Ионы кислорода или халь-
когена на концах цепей, взаимодей-
ствующие с ионами металлов, на-
зываются немостиковыми атомами.
На рис. 2.42 схематически изображе-
на модифицированная добавками
структурная сетка.
Основываясь на вышеизложенном, стеклообразователи мо-
жно называть также сеткообразователями, а компоненты мо-
дифицирующих добавок — модификаторами сеток. Согласно
концепции Дитцеля о напряженности ноля, модификаторы дол-
жны иметь параметр напряженности поля 0,1—0,4.
Введение модификаторов оказывает существенное влияние
на свойства стекла. Из-за появления в структуре стекла ме-
нее направленных связей каркас ослабевает и наблюдается тен-
денция к образованию более плотной упаковки. Мольный объем
при этом уменьшается. Температура стеклования падает из-за
уменьшения степени связности стеклообразного каркаса. От-
рицательно заряженные немостиковые атомы повышают поля-
ризуемость и, следовательно, ангармоничность колебаний ре-
шетки. Коэффициент термического расширения возрастает. Чем
выше в стекле содержание модификатора, тем больше концен-
трация немостиковых атомов и тем больше нехватка в спо-
собных к координации лигандах. Кроме того, скопление отри-
Аморфное и стеклообразное состояние
155
цательного заряда на сеткообразователе способствует большей
мобильности неподеленных электронных пар и поляризации
связи.
Если бы существовал такой катион, который можно было
бы добавлять в стекло в небольших концентрациях, практиче-
ски не вызывая изменения структуры и химических связей, и
спектр поглощения которого был бы достаточно чувствителен
к влиянию донорных свойств оксидных лигандов, то можно
было бы установить новую систему параметров, которая позво-
лила бы характеризовать стекла в рамках кислотно-основной
теории. Спектроскопический переход ’So-->-3^i 6$2-электронов
катионов Т1+, РЬ2+ или Bi3+ (например, в свободном ионе РЬ2+
он наблюдается в области 60 700 см1) тем сильнее сдвигается
в сторону длинных волн, чем выше поляризуемость атомов кис-
лорода в стекле. В СаО соответствующая полоса лежит в об-
ласти 29 700 см-1, и, таким образом, сдвиг составляет
31 000 см-1. Даффи и Инфрем [328] ввели определение опти-
ческого параметра основности
^Pb (П) = (^РЬ2+ (свободный) ^РЬ2+ (С1-екло))/ЗЮ00
Apb(ii),cao= 1- Параметр основности стекол всегда меньше 1.
С ростом содержания модификатора некоторая ковалент-
ность связей металл—кислород и металл—халькоген начинает
играть все более важную роль. Поэтому структуру и свойства
стекла определяют соответствующие координационные поли-
эдры (как области ближнего порядка) совместно с оставшимися
анионами (разд. 2.4.1.3).
Существование стекол с высоким содержанием оксидов или
халькогенидов металлов можно объяснить достаточно просто.
По данным Трепа и Тевелса [329], в стекле (Na2O) ^(КгО) 15-
(CaO)i5(BaO) 15(8102)40 с общей формулой M3Si2O7, начиная
лишь с 40 % SiO2, связанная сетка из силикатных ионов больше
не образуется. Это означает, что за образование стабильной
структуры стекла становятся ответственными координационные
полиэдры металл—кислород. Меньшая степень ковалентности
связи М—О и связанная с этим относительно высокая теку-
честь расплавов — причины ярко выраженной склонности к кри-
сталлизации. Этой тенденции противодействует рост конфигу-
рационной энтропии, обусловленной присутствием нескольких
оксидов металлов, характеризующихся различными координа-
ционными числами и разными длинами связей.
При введении модификаторов склонность к стеклообразо-
ванию, как правило, возрастает из-за расширения возможностей
для возникновения различных структурных конфигураций, при-
чем в области эвтектических составов часто стеклообразование
наиболее вероятно. Температура ликвидуса падает. Также
ведет себя приведенная температура [уравнение (2.59)]. В то же
156 Глава 2
время даже малые добавки посторонних веществ могут за-
метно уменьшать вязкость сеткообразователя, что в первую оче-
редь сильно облегчает протекание процессов кристаллизации
и тем самым ограничивает склонность к стеклообразованию
(разд. 2.3.2).
Распределение в сетке немостиковых атомов, а следова-
тельно, и катионов часто происходит неравномерно, так как
может оказаться более выгодным, чтобы структурный каркас
сеткообразователя в определенных областях было бы в основ-
ном разрушен, а в других, наоборот, остался бы неизменным.
(Такая структурная организация может следовать из соотно-
шения энергетических и энтропийных факторов.) При этом уже
в расплаве происходит разделение на две жидкие фазы или
возникает скрытая область расслаивания (разд. 2.3.1.2), и на
кривой температуры ликвидуса появляется точка перегиба.
Лишь при дальнейшем добавлении модификаторов при попада-
нии в область низкоплавкой эвтектики, где в равновесии с го-
могенным расплавом находятся несколько кристаллических фаз,
в полной мере проявляется повышенная склонность к стеклооб-
разованию и происходит образование гомогенного стекла. По-
явление в стеклообразующих системах областей расслаивания
вблизи низкоплавкой эвтектики, однако, также допустимо.
2.4.2.7. Возможные критерии для структурно-химического
объяснения процесса разделения фаз
Правило simila similibus solvuntur (подобное растворяет по-
добное) испытано временем. Тем не менее нелегко сказать, ка-
кие факторы определяют «подобие» смешивающихся друг с дру-
гом веществ и какие структурные соотношения и параметры
химической связи ведут к описанному в разд. 2.3.1.2 явлению
расслаивания [343].
В неполярных низкомолекулярных веществах взаимодейст-
вие обычно описывается с помощью так называемой «удель-
ной энергии сцепления» А//ИСп/^. (Это нашло свое выражение
при описании образования гомогенных смесей.) Гильдебранд
и др. [330] использовали «параметр растворимости» А =
= (Д/7исп/Г)1/2. Здесь ДТ/исп — мольная энтальпия испарения,
V — мольный объем формульной единицы. Неполярные жид-
кости с близким значением L должны смешиваться в любых
соотношениях. В случае большой разности значений L следует
ожидать расслаивания. В полярных веществах надо учитывать
также силы притяжения между неодинаковыми атомами. Для
такого случая параметр растворимости L предложено модифи-
цировать [331].
Чаще всего ожидаемая смешиваемость, следующая из рас-
четных значений L для веществ, которые предполагается иссле-
Аморфное и стеклообразное состояние
157
довать в качестве компонентов стеклообразующих систем, не
совпадает с экспериментальными данными. Структурные группы
в стеклообразующих расплавах сильно ассоциированы, так что
испарение сопровождается деполимеризацией, а также разложе-
нием. Энтальпия испарения А//исп в этом случае уже не совпа-
дает с энергией сцепления в расплавах стекол.
При оценке «подобия» расплавов при температурах вблизи
температуры плавления более подходит энтропия плавления
ASu.-i. Предлагается [332—333] использовать для такой оценки
отношение энтальпии плавления к энтальпии атомизации
АЯПл/АЯа, которое характеризует изменение энергии сцепле-
ния. Если при плавлении происходит изменение координацион-
ного числа и, следовательно, типа связи, то получаются отно-
сительно большие значения как А5ПЛ, так и А//Пл/А//а.
Сопоставив эти параметры для многих халькогенидов и со-
единений на их основе, Линке [335] пришел к следующим вы-
водам.
Если вещества в кристаллическом состоянии «подобны» друг
другу и А5ПЛ отличаются мало, то подобие сохраняется и для
расплавов, т. е. должна быть полная смешиваемость. Примеры:
Sb2S3—Sn2Se3, AS2S3—As2Se3, Sg-—P4S10, SiO2—GeO2.
Если вещества имеют близкие А5ПЛ, но существенно раз-
личаются в кристаллическом состоянии по другим свойствам,
например поляризуемостью связей или степенью связности кар-
каса, то различия в основном сохраняются и при переходе
в расплавленное состояние и смешиваемости вряд ли можно
ожидать. Примеры: Sn—SnS, Sg—T1S2, TI—TI2Se.
Если вещества в кристаллическом состоянии не имеют сход-
ства друг с другом и А5ПЛ сильно отличаются, то при плавле-
нии может происходить как сближение свойств веществ (на-
пример, в системах Ge—GeTe, Те—As2Te3, где наступает их пол-
ная смешиваемость), так и усиление различий и образование
областей несмешиваемости (системы SiO2—В20з, SiO2—Р2О5,
S8—Sb2S3 Pb—PbSe, Sb—SnS).
2.4.3. Корреляции структура—свойства
Изучение взаимосвязей между макроскопическими свойст-
вами и определенными структурными параметрами и характе-
ристиками химических связей представляет собой испытанный
принцип классификации химических соединений. При этом
даже в гомологических рядах, в которых происходит очевид-
ное изменение структуры и химических связей, трудно устано-
вить взаимосвязь с определенными свойствами для данной
группы веществ. Обычно существует несколько факторов, ко-
торые часто воздействуют на поведение системы в противопо-
ложных направлениях. Четкое структурно-химическое толко-
158 Глава 2
вание изменения свойств в рядах веществ возможно лишь на
основе предшествующего выяснения структуры. Если выявлены
соотношения между структурой и свойствами (например, пра-
вило Вегарда для твердых растворов), то непрерывное изме-
нение плотности можно, наоборот, рассматривать как указание
на образование твердых растворов. Отклонения от идеальных
законов смешения указывают на структурные изменения в си-
стеме. В чем конкретно состоят эти изменения, нельзя опреде-
лить без применения других методов исследования.
В стсклообразующих системах изучение свойств в рядах
образцов с определенным изменением состава представляет со-
бой эффективное средство для описания различных явлений.
Набор образцов для сравнения надо готовить из исходного ма-
териала, представляющего собой стеклообразующис соедине-
ния с известной структурой. В таком случае изменение свойств
в известных пределах может быть интерпретировано на осно-
вании ясных предпосылок о структурно-химическом поведении
атомов и групп, участвующих в построении системы.
Поскольку корреляции между структурой и свойствами мо-
жно проводить неограниченно, далее остановимся на таких
свойствах, как мольный объем, вязкость, температура стекло-
вания Tg, коэффициент термического расширения, упругие по-
стоянные, и покажем их связь со структурой стекла. Соотно-
шения между структурой стекла и проводимостью или диэлек-
трической проницаемостью рассмотрены в гл. 3.
2.4.3.1. Зависимость мольного объема от структуры
Плотность стекол можно определить с достаточной надеж-
ностью различными методами, например пикнометрически. Она
зависит от скорости охлаждения в области температур стекло-
вания. Образцы халькогенидных стекол, охлаждающиеся со
скоростью 100 К/мин, имеют плотность, на 1—2 % меньшую,
чем в случае охлаждения со скоростью 3—4 К/ч. На это не-
обходимо обращать внимание при сравнении результатов экс-
периментов.
Чтобы исключить влияние массы образца, от плотности пе-
реходят к мольному объему. При этом одному и тому же ве-
ществу можно приписать различные формулы в зависимости от
того, какие формулы наиболее показательны при проведении
сравнения образцов в данном ряду. Например, для соединения
GeSe2 можно вести расчет как на состав области ближнего
порядка GeSe4/2, так и на 1 моль Geo,3sSeo,67. В сложных стек-
лообразующих системах средний мольный_объем V=M/p можно
связать со средней молярной массой Л7 = £ хдМ,-, где М,-—
молярные массы определенных структурных единиц или атом-
ные массы компонентов, Хг— соответствующие им мольные
доли.
Аморфное и стеклообразное состояние
159
Из общих положений термодинамики растворов следует,
что разложение среднего объема на отдельные вклады в соот-
ветствии с формулой V — Е XjVi имеет мало смысла. Идеального
поведения системы все равно можно ожидать лишь тогда,
когда компоненты имеют близкие мольные объемы, их структур-
ные элементы похожи по геометрии и энтальпия смешения пре-
небрежимо мала.
В определенных предельных случаях структурно-химическая
интерпретация изменения среднего мольного объема тем не ме-
нее возможна при правильном выборе состава сравниваемых
образцов. Если атомы, из которых состоит система, имеют при-
мерно равные эффективные радиусы, то наблюдаемые, обычно
отрицательные, энтальпии смешения в бинарных системах мо-
жно объяснить некими структурными особенностями. В много-
компонентных системах весьма полезно попарное взаимное за-
мещение определенных атомов со сравнимыми структурно-хими-
ческими свойствами при постоянстве мольных долей всех
остальных компонентов, например, с целью проверки изоморф-
ности замещения. И наконец, если в вещества вводить катионы,
имеющие относительно небольшие эффективные радиусы, так,
чтобы они попадали в пустоты между крупными частицами, оп-
ределяющими объем системы (в данном случае анионы и ка-
тионы), то рекомендуется вести расчеты по формуле V1{ —
= V/Y,xR,i (где Хн — мольная доля крупных атомов или ионов,
которые определяют заполнение пространства).
Близкое расположение в периодической системе и относи-
тельно небольшая разность электроотрицателыюстей позволяют
считать, что компоненты стеклообразующих систем GexSei-.v
и AsxSei_x имеют примерно сравнимые эффективные радиусы.
При введении в селен германия и мышьяка структура из цепо-
чечной переходит в сетчатую с образованием структурных еди-
ниц GeSe4/2 и AsSe3/2. Поэтому средний мольный объем умень-
шается (отрицательная энтальпия смещения). В ряду стекол
системы As—Se при составе АзгБез (х=0,4) имеется минимум
на зависимости мольного объема от состава (рис. 3.34). В си-
стеме Ge.vSei-x средний мольный объем уменьшается лишь до
состава GeSe4 (х = 0,2), а затем вновь возрастает (рис. 3.40).
В этой системе на стеклообразующее соединение GeSe2 прихо-
дится максимум мольного объема. Трехмерное сочленение за-
фиксированных тетраэдрических структурных групп приводит
к образованию относительно рыхлой структуры (рис. 2.38).
Модификаторы уменьшают средний мольный объем из-за
нарушения сетчатой структуры и появления немостиковых ато-
мов, которые образуют ненаправленные полярные связи с катио-
нами. Структура меняется в сторону более плотной упаковки.
160 Глава 2
На рис. 2.43 приведен пример попарного взаимного заме-
щения атомов Ge и Sn, а также Se и Те в стеклах ряда
Ge0,4-xSnxSeo,6-yTey [336]. Состав строго соответствует общей
формуле М2Хз, т. е. при х = 0 и y=Q реализуется трехмерная
структурная сетка стекла GesSea. Если при постоянной кон-
центрации Ge и Sn (х = const) атомы Se замещать на Те, то
зависимость среднего мольного объема от у имеет вид прямой
Рис. 2.43. Изменение среднего мольного объема V и температуры стеклования
Тg от состава.
а-стекла Ge0>4Se0 6_уТеу (/ и2) и As0 4Se06_yTey (3); б — стекла Ge0>4_xSnxSe0 5Те01
(^)> Ge0 4_xSnxSe0 45Te0il5 (5) и Ge0 4_xSnxSe0i4Tc0 2 (6).
с положительным наклоном. На рис. 2.43, а приведен пример
такой зависимости при х = 0. Возрастание мольного объема
можно объяснить различием объемов атомов Те и Se. Оче-
видно, Те может изоморфно замещать атомы Se в структуре
стеклообразного GeaSes с сохранением координационного числа 2.
Для сравнения па этом же рисунке приведено изменение моль-
ного объема стекол ряда Aso^Seo.e-yTe^. Несмотря на то что
в этих стеклах наблюдается двумерное сочленение областей
ближнего порядка, средний мольный объем здесь меньше, что
также может указывать на структурный эффект. При трехмер-
ном сочленении областей ближнего порядка с направленными
связями степень эффективного заполнения пространства меньше,
а объем пустых мест между структурными группами больше.
На рис. 2.43, б приведена зависимость мольного объема от
содержания Sn при постоянных^ концентрациях Se и Те. При
замещении Ge на Sn значение V в рядах стекол (г/=0,1; 0,15;
0,2) понижается. Если для стекол Geo^-xSnxSeo.e-j/Tey принять
строение, соответствующее формуле (SnSei_zTez)x(GeSe]_z-
Tez)i/5_x(GeSe2--2zTe2z)i/5 с z=5#/3, то и в этом случае нужно
ожидать роста V. При кристаллизации в системах SnSc—GeSe
Аморфное и стеклообразное состояние
161
и SnTe—GeTe имеет место образование твердых растворов без
пустот в их кристаллической решетке [337]. При образовании
стекла из расплавов в случае модифицированной молекулами
GeX и SnX сетки GeX2 тем более должно было происходить
изоморфное замещение обоих монохалькогенидов. Различие эф-
фективных радиусов Ge и Sn должно проявляться_в росте сред-
него мольного объема. Наблюдаемое падение V показывает,
что GeSe и SnSe, а также GeTe и SnTe не могут изоморфно
замещать друг друга в стекле. Необходимое для образования
структурных единиц Ge2X62 количественное соотношение халь-
когенидов GeX и GeX2 устанавливается «автоматически». Халь-
когениды олова (II), выполняющие функцию модификаторов,
образуют собственные структурные полиэдры. Это объясняет
уменьшение среднего мольного объема стекла, а также пони-
жение температуры стеклования [336]. Поэтому структуру сте-
кол можно передать формулой (SnSei-2Te2)x(Ge2Se3—ЗгТвзг) 1/5—х"
(GeSe2_2zTe22)x с z = 5f//3 (разд. 3.3.6.4).
Примеры пренебрежимо малой деформации сетки при вве-
дении в стекло небольших по размеру катионов можно найти
в системах КгО—SiO2—TiO2 [338] и К2О—SiO2—VO2 [339].
Если, например, приписать частицам сравнимых размеров (т. е.
ионам К+ и О2-) средний мольный объем и пересчитать его по
приведенному уравнению к 1 молю вещества, то для всех сте-
кол 7я=12,7 см3/моль (±2%) [339].
2.4.3.2. Зависимость вязкости от структуры
Опираясь на «валентную теорию стекла» Мюллера [106,
107], предпринимались неоднократные попытки, в том числе
Немиловым [108, 340], провести корреляцию между структурой
стекла и его вязкостью.
Основу такого подхо-
да составляет уравнение
(2.29) (разд. 2.2.1.2). Бы-
ла изучена зависимость
свободной энергии акти-
вации вязкого течения
AG* от состава в обла-
сти температуры размяг-
чения (рис. 2.13). Из
этой зависимости рассчи-
Рис. 2.44. Зависимость энтро-
пии активации вязкого течения
от состава (в мольных долях х)
для стеклообразующих рас-
плавов в системах GexSei-x
(/) и AsxSei_x (2) [340].
И Заказ № 413
162 Глава 2
тана энтропия активации S* = (dG*/dT) р и построен график
зависимости S* от состава.
п
На рис. 2.44 приведен участок зависимости энтропии акти-
вации от состава для области стсклообразования в системах
GexSei-x и AsxSei-x.
До сих пор для хода зависимости от состава не дано
исчерпывающего структурного толкования. Обращают на себя
внимание три особенности: 1) существенное различие в пони-
жении в области малых добавок As и Ge; 2) «ступенька»
между 10 и 15 мол. %; 3) существование минимума в окрест-
ности составов AsSe3 и GeSe4.
Относительно недавно выполненные измерения вязкости
в силикатных стеклах сопровождаются структурными объясне-
ниями [341]. При этом исходили также из представлений
Мюллера; было обнаружено, что 1g г] обратно пропорционален
концентрации модификатора. Между свободной энергии акти-
вации и параметром напряженности поля Дитцеля существует
линейная зависимость.
2.4.3.3. Температура стеклования и структура стекол
При изучении влияния структуры на температуру стеклова-
ния Tg необходимо учитывать описанные в разд. 2.2.2 требо-
вания по стандартизации термической предыстории стекол и
условий проведения измерений.
В самом общем виде можно сказать, что Tg тем выше, чем
прочнее связь между подвижными структурными элементами.
Как уже отмечалось в разд. 2.2.1.3, 2.2.2 и 2.2.3, энергетика
взаимодействия в процессе вязкого течения в значительной сте-
пени определяется энтропийными факторами.
В кристаллических изоструктурных веществах имеет место
известная симбатность между энергией решетки и температурой
плавления. Это объясняется одинаковой природой химических
связей, аналогичными принципами соединения структурных
групп, совпадением координационных чисел, т. с. единым ближ-
ним порядком. Можно ожидать, что и в стеклах должны су-
ществовать аналогичные корреляции, несмотря на то что в об-
ласти температуры стеклования происходят разрыв или пере-
группировка наименее прочных связей. Линке [327, 343] пред-
ложил в качестве параметра использовать мольную энтальпию
атомизации (разд. 2.4.2.5).
Мольная энтальпия атомизации \На стеклообразных твер-
дых тел с преобладающими ковалентными связями может быть
рассчитана по уравнению (2.100) из значения Ь-Нал элемен-
тов, стандартной энтальпии реакции образования А/Л? и эн-
Аморфное и стеклообразное состояние 163
тальпии кристаллизации А7/кр. В входит энтальпия обра-
зования кристаллических соединений, имеющихся в системе,
а также энтальпия смешения, которой чаще всего пренебре-
гают. Таким образом, ^НА = ^ Xi\HA, i+ |АЯД|— |АЯКр|. Как
видно из приведенных ранее расчетов, АЯкр составляет всего
несколько процентов в общем энергетическом балансе. С энер-
гетических позиций также ясно, что определяющее значение
для образования химических связей в стеклах имеет именно
ближний порядок, а не отсутствующий дальний порядок, ко-
торому соответствует член А//Кр- Поэтому очевидно, что свой-
ства стекол в основном тесно связаны со структурой ближнего
порядка. Даже энтальпия образования соединений, имеющихся
в данной системе, составляет в случае халькогенидных стекол
5—15% АЯа- Следовательно, в нулевом приближении для оце-
нок можно использовать средние мольные энтальпии атомиза-
ции кристаллических элементов.
В случае оксидных стекол А Яд целесообразно рассчиты-
вать из данных для бинарных оксидов, а не из данных для эле-
ментов. Это объясняется высокой степенью полярности связи
в оксидах и, следовательно, относительно высокими вкладами
энтальпии образования оксидов в &НА. Поэтому полезно ис-
пользовать обычный для химии стекла способ записи состава
стекла как смеси различных оксидов. Значения стандартной
мольной энтальпии атомизации отдельных оксидов, как пра-
вило, можно найти в справочниках [342]. Ее можно рассчи-
тать на основании данных по стандартной мольной энтальпии
образования оксида, стандартной мольной энтальпии ато-
мизации данного металла и энергии диссоциации молеку-
лярного кислорода. АЯКр в случае оксидных стекол составляет
лишь 1 % энтальпии атомизации, и ей можно пренебречь.
входящая в уравнение (2.100), представляет собой комбинацию
стандартных мольных энтальпий образования оксидных компо-
нентов, участвующих в образовании тройных или четверных со-
единений, с учетом энтальпии их смешения.
В работе [343] собраны и сведены в таблицы значения А Яд
для стеклообразующих соединений и ряда многокомпонентных
систем. На рис. 2.45 нанесена зависимость Tg от средней моль-
ной энтальпии атомизации АНа (здесь для расчета АЯа исполь-
зовалась стандартная мольная энтальпия реакции АЯД бинар-
ных соединений, а для оксидных стекол АЯД соответствующих
тройных фаз).
Наиболее высокие значения Tg характерны для стекол
с трехмерным сетчатым каркасом. К ним относятся кристалли-
зующиеся в алмазоподобной халькопиритной структуре соеди-
нения CdGeAs2, CdGeP2, а также кварцевое стекло и GeO2. Во
всех названных веществах все атомы имеют тетраэдрическое
окружение. В SiO2 и GeO2, как и в GeS2 и GeSe2, кроме этого
11*
164 Глава 2
т3>к
1500
1300
700 -
-1--------------1_____________I_____________i_
200 300 ЦОО 500
А Нд , кДлг/моль
I
600
Рис. 2.45. Зависимости температуры стеклования Tg от энтальпии атомизации
А#а [327, 343].
тетраэдры соединены друг с другом через атомы халькогенов.
Следует отметить, что в этот же ряд веществ попадает и GcTeg,
который известен в настоящее время лишь в стеклообразном
состоянии. В твердых телах, имеющих трехмерную сетчатую
структуру, процессам плавления, стеклообразования и вязкого
течения должно сопутствовать сильное возбуждение связей,
и, следовательно, эти процессы зависят от их прочности. Так,
Аморфное и стеклообразное состояние
165
энергия активации вязкого течения расплава GeSe2, равная
241+5 кДж/моль, хорошо совпадает со средней энергией связи
£oe-se=225 кДж/моль [344] (табл. 2.11). В то же время в бо-
гатых селеном расплавах энергия активации составляет 40—
50 кДж/моль, что намного ниже энергии связи Se—Sc.
Соединения со средней степенью связности стеклообразного
каркаса находятся вблизи прямой 2оо (рис. 2.45). В эту группу
входят стеклообразующие соединения с двумерным бесконеч-
ным сочленением областей ближнего порядка. Следует заме-
тить, что стеклообразный BeF2, которому приписывают струк-
туру, аналогичную кварцевому стеклу, также попадает в эту
группу веществ. Для образования двумерной сетчатой струк-
туры потребовалось бы, чтобы ~50 % тетраэдрических групп
[BeF-i] соединились между собой ребрами, что не подтвержда-
ется структурными исследованиями (разд. 3.2.3.1). При объяс-
нении этого факта прежде всего необходимо учесть меньшую
энергию связей, образуемых мостиковыми атомами галогенов.
Именно этим стеклообразный BeF2 качественно отличается от
оксидных и халькогенидных стекол.
Стсклообразующие оксиды, модифицированные различными
компонентами, попадают в область между прямыми 2оо и Зоо.
Степень связности стеклообразного каркаса этих соединений
не может быть указана однозначно, поскольку при высокой кон-
центрации модификатора кислородные полиэдры, включающие
ионы металлов, сами участвуют в построении структурного кар-
каса стекла (разд. 2.4.2.6). Хотя в стеклообразном NaPO3 осу-
ществляется одномерное сочленение структурных групп [POJ,
это стекло в связи с указанными причинами попадает в группу
веществ с двумерной сеткой.
Вблизи прямой loo находятся такие стекла, основной струк-
турный мотив которых—атомные цепочки. Кроме халькоге-
нов серы, селена и теллура к таким веществам относятся AsXI
и GeXI2 (где X = S, Se) (разд. 3.3.2) и НРО3.
Влияние структуры стекла на температуру стеклования
весьма выразительно продемонстрировано на рис. 2.46 [327,
343, 344].
В работе Дембовского [345] эю направление исследований
получило дальнейшее развитие: температура стеклования Tg
связывается со средним координационным числом Z = Y.XiZi.
При этом обращается внимание, что в ряду халькогенидных
стекол приблизительно выполняется соотношение Тg/Z~423±
±10 К. Беркее [346] провел корреляцию между Tg и числом
связей XiZil2, приходящихся на 1 моль вещества АХВ]_Х. При
этом он указал на отклонение в сторону более высоких коор-
динационных чисел в системах AsxSei-x при малых мольных до-
лях х<0,15.
Рис. 2.46. Зависимости температуры стеклования Tg в типично халькогенидных стеклообразующих системах от энтальпии
атомизации (по данным Линка).
Аморфное и стеклообразное состояние 167
2.4.3.4. Зависимость термического расширения
от структуры
Определение коэффициента линейного расширения а может
быть проведено с помощью изображенного на рис. 2.14 дилато-
метра. В ходе непрерывного измерения а, проводимого при на-
гревании образца, необходимо строго следить за тем, чтобы
температура образца успевала изменяться вслед за изменением
температуры нагревателя. Поэтому необходимо соответствую-
щим образом размещать датчик температуры и обеспечивать
достаточно большую изотермическую зону. Другой источник
возможных ошибок при определении а связан с измерением
длины твердого образца /0 [уравнение (2.35)]. Весьма полезна
проверка надежности дилатометра по стандартному образцу
с известным коэффициентом термического расширения (напри-
мер, титановый стандарт).
Термическое расширение образца следует из ангармонич-
ности колебаний решетки. В выражение для потенциальной
энергии V(x)=bx2—сх3 входят коэффициент b — упругая со-
ставляющая возвращающего усилия (гармонические колеба-
ния) и коэффициент с — мера ангармоничности. Именно второй
член выражения обусловливает расширение. Для температур-
ной зависимости среднего смещения — величины, пропорцио-
нальной коэффициенту термического расширения,— можно
записать (k — постоянная Больцмана)
а - dx/dT = 3kc/4b2 (2.103)
Ослабление упругих сил решетки (из-за уменьшения сте-
пени связности стеклообразного каркаса) позволяет ожидать
относительное увеличение вклада слагаемого, отвечающего за
ангармоничность системы. Поэтому с уменьшением b и увели-
чением с коэффициент термического расширения должен воз-
растать.
Учет других членов разложения в ряд приведенного выше
выражения для потенциальной энергии позволяет определить
температурную зависимость а. которой в широком температур-
ном диапазоне нельзя пренебрегать. С целью проведения срав-
нения в связи с обсуждением структуры стекла весьма полезно
стандартизировать условия измерения коэффициента термиче-
ского расширения, например, определив хотя бы температурный
интервал на 100—200 К ниже Тё. Последнее требование не-
выполнено для данных, приведенных в табл. 2.5 и 2.6. Из прак-
тических соображений удлинение х приведено к единому ин-
тервалу температур, не связанному с Tg. Этот интервал темпе-
ратур включает и комнатную температуру.
Как следует из табл. 2.5, наиболее низкие значения коэф-
фициентов термического расширения имеют стеклообразующие
168 Глава 2
соединения с трехмерной сеткой областей ближнего порядка.
Далее по мере нарастания а следуют стекла с двумерным сочле-
нением структурных групп. А стеклообразный селен выделяется
наибольшим значением а. Добавление к сеткообразователям,
например к стеклообразующим веществам, которые приведены
в табл. 2.10, модификаторов приводит к увеличению коэффи-
циента термического расширения, что вполне согласуется с при-
веденными выше представлениями.
Рис. 2.47. Коэффициент термического расширения а стекол [350].
1- (PbSe)a(GeSe)0>7_a(GeSe2)03 (область/), 2 - (PbSe)e(GeSe)0>e5_e(GeSe2)0i35 (об-
ласть //), 3 — (PbSe)a(GeSe)0i5_0t5a(GeSe2)0i5_0i5a (область ///), 4 — (PbSe)a(GeSe)a-
(GeSe2)(_2a (область IV)
Последнее справедливо лишь в тех случаях, когда введе-
ние модификатора не вызывает никаких других изменений
структуры, кроме разрыва сетки за счет образования более
полярных связей. Если же добавки ионов металлов ведут к по-
вышению координационного числа сеткообразователей, как
в случае боратных стекол, то вначале происходит уменьшение
коэффициента термического расширения.
Добавки В20з, А120з и TiO2 могут существенно компенси-
ровать эффект разрыхления структуры, возникающий при вве-
дении модификаторов [347]. Так, добавление ТЮ2 в кварцевое
стекло ведет даже к образованию стекол с отрицательным рас-
ширением [348, 349]. Использование таких эффектов состав-
ляет основу для получения стекол с исчезающе малым терми-
ческим расширением.
Аморфное и стеклообразное состояние 169
Измерение коэффициента термического расширения — один
из методов обнаружения изменений структуры стекла. Такие
измерения проведены для стекол системы PbSe—GeSe—GeSe2
[350] (рис. 2.47). Коэффициент а образцов I и II группы, т. е.
(PbSc)x(GeSe)oi7-x(GeSe2)o>3 и (PbSe)x(GeSe)0,65-x(GeSc2)0135
(разд. 3.3.6.4), проходит через минимум при х = 0,3, а именно
для состава с эквимолекулярным соотношением компонентов
GcSc и GeSe2. Коэффициенты термического расширения образ-
цов III группы (PbSe)a(Ge2Se3)i-a резко уменьшаются. Ни
мольные объемы, ни температуры стеклования не имеют такого
хода зависимости от состава. Лишь кривые удельной электро-
проводности характеризуются такой же прерывистостью при
соотношении фаз GeSe/GeSez=l [350].
2.4.3.5. Структура стекла и упругие постоянные
Упругие свойства стекол исследуются преимущественно аку-
стическими методами. Перенос энергии при распространении
звуковых волн происходит, как и в кристаллических твердых
телах, в форме продольных и поперечных колебаний. Прочность
химических связей, плотность упаковки, координационные чи-
сла, степень связности структуры стекла должны оказы-
вать такое же влияние на распространение волн, какое они
оказывают на температуру стеклования. Соотношение между
скоростями продольного и поперечного распространения волны
Vi и vtr, модулем упругости Е, модулем сдвига G и сжимае-
мостью х, а также коэффициентом Пуассона ц уже приводи-
лось в разд. 2.2.1.1 [уравнения (2.8а) и (2.86)]. Из граничных
значений ц (0<ц<0,5) следует, что EI2>G'>EI3.
На рис. 2.48 изображена зависимость модуля упругости от
средней мольной энтальпии атомизации кНА для некоторых
важных стеклообразующих соединений и систем. Данные для
халькогенидных стекол взяты из работ [327, 343, 351]. Данные
для Se имеются также в работе [352], а для Аэ25з и As2Se3 —
в работе [353]. Можно заметить, что в соответствии с умень-
шающимся относительным удлинением М/l модуль упругости
Е= (//А/) • Se закономерно возрастает в рядах AsxSi-x, AsxSei-x
и GexSe<-x с увеличением степени связности стеклообразного
каркаса. Халькогенидные стекла в системах с большим поряд-
ком имеют близкие значения Е [например, для Geo^Aso.sSeo.s
£=17,6 ГПа (ц = 0,28), для Ge0 iSi012AS0зТе0 48 £ = 21,8 ГПа
(ц = 0,27)] [354].
Для соединений GeSe2, GeO2 и SiO2 с трехмерным сочлене-
нием тетраэдрических областей ближнего порядка наблюдается,
как и для температуры стеклования (рис. 2.45), примерно ли-
нейная зависимость £ от кН а. Возрастание энергии связи при-
водит к повышению модуля упругости. Например, у GeO2 он
170 Глава 2
Рис. 2.48. Зависимость модуля упругости стекол от энтальпии атомизации.
равен 43,3 ГПа, а у кварцевого стекла 72 ГПа. Примечательно,
что стеклообразный В20з имеет относительно низкий модуль
упругости (17,5 ГПа). Объяснение, что степень связности стек-
лообразного каркаса В2О3 понижена по сравнению с структурой
стеклообразного SiO2, вряд ли можно признать удовлетвори-
тельным, особенно если учесть, что GeSe2 и As2Se3 имеют близ-
кие модули упругости Е.
Введение оксидов щелочных металлов в виде модификато-
ров в кварцевое стекло, как и следовало ожидать, понижает
модуль упругости [80] (см. литературу к гл. 3 [55]). На
рис. 2.48 это показано на примере калиево-силикатного стекла.
В то же время добавки оксидов щелочноземельных металлов
(также модификаторов) вызывают заметное повышение Е. Это
связано с увеличением кулоновского вклада в энергию решетки
Аморфное и стеклообразное состояние 171
[356]. Для стекла состава 0,5MgO-0,5CaO-SiO2 модуль упру-
гости, например, равен 99,1 ГПа. Введение модификаторов
в стеклообразный В2О3 приводит также к увеличению модуля
упругости, что может быть объяснено образованием областей
ближнего порядка с тетраэдрической координацией атомов
бора. Так, для боратного стекла, содержащего 28,5 мол. %
Na2O, £ = 56,5 ГПа [357]. Аналогичное поведение наблюдается
у стекол системы Na2O—GeO2. Здесь необходимо иметь в виду
структурно-химические свойства четырехвалентного германия,
который склонен к образованию октаэдрических полиэдров
[GeOe]. Натрий-германатиое стекло, содержащее 18 мол. %
Na2O, имеет модуль упругости £ = 67,5 ГПа (рис. 2.48).
По сравнению с модулем упругости коэффициент Пуассона
ц меняется с изменением структуры в обратном направлении.
Он уменьшается с возрастанием степени связности стеклооб-
разного каркаса. Из всех стекол самый низкий коэффициент
Пуассона имеет кварцевое стекло (ц = 0,17). Согласно опреде-
лению коэффициента Пуассона как отношения относительного
поперечного сжатия к относительному продольному удлинению,
низкое ц кварцевого стекла означает, что напряжение при растя-
жении образца приводит к малому поперечному сжатию из-за
того, что структурные группы [SiO4] со всех сторон соединены
с такими же структурными элементами. Низкие значения ц отве-
чают высокой упругости. Самые высокие значения для стекол
характерны для стеклообразных металлов (р = 0,39-4-0,42)
[358—360].
Металлические стекла поэтому имеют относительно высокие
значения модуля упругости. Как видно из рис. 2.48, для группы
сплавов (Pdi-xNi.T)o>8B5Sio,i65 с 0<х<0,6 модуль упругости Е
меняется от 87,8 до 110 ГПа; это намного выше, чем значения
Е для кварцевого стекла [361]. Аналогичная зависимость Е от
состава характерна для стекол (Pdi_.TNi.T)oi8Po,2, (Pdi-xFex)o,8Po,2,
(Pti-xNix)o,75Po,25- Указанные факты объясняются, вероятно, вы-
сокой плотностью упаковки элементов кристаллической решетки
металлов. Значения Tg, напротив, меняются в этих рядах до-
статочно слабо. В упругие свойства стскол свой вклад вносит
не только энергия связи и степень связности стеклообразного
каркаса, но и степень заполнения пространства. На наличие
такого влияния указывают относительно высокие значения мо-
дуля упругости для группы стекол PbS—GeS—GeS2. Можно
предположить, что неожиданное понижение модуля упругости
в случае стеклообразного В2О3 также объясняется меньшей
степенью заполнения пространства в этом стекле по сравнению
с кварцевым стеклом. Для обоих стскол расчет дает примерно
одинаковый средний мольный объем VR, приходящийся на оди-
наковое количество кислородных частиц: для кварцевого стекла
VR= 13,5 см3/моль, для стеклообразного В2О3 VR — 12,7 см3/моль.
172 Глава 2
2.5. Электронные состояния в некристаллических
твердых материалах
Реализация тех или иных структур аморфных твердых тел
и стекол обусловлена общими термодинамическими принципами
равновесия и устойчивости системы (когда система стремится
к минимуму свободной энергии) и кинетическими факторами.
Последние ответственны в основном за процессы образования
стекла. Структура стекла определяется взаимодействием элек-
тронов на внешних орбиталях атомов, участвующих в образо-
вании данного вещества. При этом значительная часть энергии
взаимодействия расходуется на создание областей ближнего
порядка, структура которых очень близка отдельным фраг-
ментам кристаллической структуры. Беспорядочное сочленение
областей ближнего порядка, которое в известной степени сбли-
жает некристаллические твердые тела с изотропными жидко-
стями и газами, вносит ничтожный вклад в общий энергетиче-
ский баланс системы. Поэтому вполне логично при теорети-
ческом описании химических связей в стеклообразном
состоянии исходить из моделей кристаллических твердых тел.
Основой описания коллективного взаимодействия в конденсиро-
ванных системах является зонная модель электронной струк-
туры.
Если мысленно представить себе тело, состоящее из Na
атомов, находящихся па очень большом расстоянии друг от
друга, то система по отношению к каждому электронному терму
будет Л/л-кратно вырождена. При сближении атомов вырожде-
ние уровней снимается и возникает зонная электронная струк-
тура вне зависимости от того, образуется кристаллическая ре-
шетка или нет.
Описание электронных состояний в кристаллических твер-
дых телах в рамках зонной модели Блоха, Пайерлса и Виль-
сона позволяет объяснить существование квазисвободных элек-
тронов в металлах и существование запрещенной зоны между
валентной, зоной и зоной проводимости r изоляторах и полу-
проводниках. Образование делокализованных электронных со-
стояний, согласно данной теории, связано с трансляционной ин-
вариантностью решетки, т. е. с периодическим расположением
элементарных ячеек и, следовательно, наличием дальнего по-
рядка. В приближении Блоха волновая функция, описывающая
движение электрона в периодическом поле кристаллической ре-
шетки, представляет собой линейную комбинацию NA однотип-
ных волновых функций электронов, локализованных на кристал-
лографически эквивалентных атомах. В результате обменного
взаимодействия возникает зона из NA энергетических уровней»
которые могут быть заполнены парами электронов. Плотность
Аморфное и стеклообразное состояние
173
вероятности, характеризуемая данной волновой функцией, оди-
накова в каждой ячейке кристаллической решетки. Таким обра-
зом, этот подход изначально предусматривает делокализацию
электронов. С учетом симметрии кристаллической решетки и
свойств симметрии атомных функций из спектра дискретных
энергетических уровней атома получается электронная струк-
тура в виде разрешенных и запрещенных энергетических зон.
В приближении Бриллюэна зонная структура возникает из
условия брэгговского рассеяния электронных блоховских волн
на семействе кристаллографических плоскостей.
В аморфных твердых телах и стеклах вследствие отсутст-
вия кристаллографической периодичности не существует набора
плоскостей кристаллической решетки. Стекла не дают брэг-
говских отражений (разд. 2.4.1.1). Поэтому может возникнуть
вопрос, правильно ли использовать концепцию зонной модели
в качестве основы для интерпретации электронных состояний
в некристаллических твердых телах. Однако наш опыт говорит
о том, что стекла близки по ряду свойств соответствующим кри-
сталлическим соединениям; например, они оптически прозрачны
в определенном интервале длин волн (оксидные стекла — в ви-
димой части спектра, халькогенидные — в ИК-области). Халь-
когенидные стекла проявляют полупроводниковые свойства,
а в аморфных металлах даже в отсутствие дальнего порядка
сохраняется металлическая проводимость. Учитывая, что раз-
личие между кристаллическим и аморфным состояниями в энер-
гетическом отношении относительно невелико, приведенные
факты вряд ли могут быть объяснены, если не принять приме-
нимость основных положений зонной модели для описания бес-
порядочного сочленения областей ближнего порядка.
Но как в таком случае можно представить себе связь между
распределением энергии и пространственной структурой в си-
стеме, с одной стороны, и непериодическим строением — с дру-
гой? Каким образом ближний порядок влияет на структуру зон?
Бесспорно, что высокая степень разупорядочения в аморфных
твердых телах и стеклах должна приводить к значительной
концентрации локализованных электронных состояний. В связи
с этим возникает вопрос о критериях локализации электронов
и о том, каким образом можно описать переход из делокализо-
ванных в локализованные электронные состояния.
В связи со стремлением применить данную теорию для ин-
терпретации структуры и свойств некристаллических твердых
тел зонная модель должна претерпеть определенные уточнения
по сравнению с первоначальным вариантом. Границы приме-
нимости зонной модели к кристаллическим твердым телам к на-
стоящему времени вполне определились. Постановка вопроса
о критериях локализации электронов связана с наблюдаемым
переходом от металлов к изоляторам. В последнее десятилетие
174 Глава 2
в этом направлении достигнуты заметные успехи, связанные
с развитием идей Мотта (в 1977 г. Мотт, Андерсон и Ван Флэк
стали лауреатами Нобелевской премии по физике).
2.5.1. Зонная модель и локализация электронов
в твердых телах
В этом разделе прежде всего будут сформулированны ос-
новные положения зонной модели кристаллических твердых тел.
При определенных условиях даже в кристаллическом состоянии
в рамках зонной модели возможно появление локализованных
электронов. Сильно легированные и частично компенсирован-
ные полупроводники могут служить простейшими модельными
системами, пригодными для описания твердых тел со стати-
стическими флуктуациями потенциальной энергии взаимодейст-
вия атомов. Перенос основных положений теории на аморфные
твердые тела и стекла обоснован, в соответствии с экспери-
ментальными фактами, развитием качественных представле-
ний о характере перехода от делокализованных электронных
состояний в энергетических зонах к локализованным состояниям
на краях зон и возникновении дефектных центров в запрещенной
зоне.
2.5.1.1. Основные положения зонной модели
Зонная модель возникла в результате применения метода
МО Хунда и Малликена для описания гигантской молекулы
кристалла. Было изучено движение электрона в строго перио-
дическом поле всех Na атомов. При этом считалось, что роль
всех остальных электронов сводится исключительно к экрани-
рованию полей атомного остова. В основе зонной модели лежат
два основных постулата: 1) в рамках адиабатического при-
ближения не учитывается совместное движение атомных ядер;
2) при сближении электронов пренебрегается межэлектронным
взаимодействием.
Волновая функция Блоха получается линейной комбинацией
одинаковых атомных функций:
ф(г) =—±=-u(r)elkr или в одномерном случае
yNA
ф (х) = !_ и (х) eikx (2.104)
Nna
Волновая функция представляет собой плоскую волну, ампли-
туда которой модулируется периодическим потенциалом кри-
сталлической решетки н(г) или w(x). Для волнового вектора
Аморфное и стеклообразное состояние
175
k из условия периодичности кристаллической решетки в обла-
сти значений х=±а0^л/2 получаем
k = (2л/а0)(n/NA) (п = 0, ±1, ±2, .... ±NA/2),
--— <k< —
aQ а0
(2.105)
где а0 — постоянная решетки. Из решения уравнения Шредин-
гера в рамках теории возмущений для одномерного случая вы-
текает выражение для энергии
Е = EAt -J- С 4- J cos ka0 (2.106)
Рис. 2.49. Функция E(k) энергетической зоны делокализованных электронных
состояний в соответствии с уравнением (2.106).
Отклонение от параболы обусловлено влиянием периодического поля кристаллической
решетки на свободный электронный газ.
где ЕА1— энергия невозмущенного атомного терма, С — энер-
гетический вклад кулоновского притяжения электрона в поле
атомного остова. В соответствии с приведенными [уравнение
(2.105)] пределами значений волнового вектора k все энерге-
тические уровни сосредоточены в энергетической зоне вблизи
EAt + C (ширина зоны 2/) (рис. 2.49). Ширина зоны тем
больше, чем больше перекрывание волновых функций, чем
меньше эффективный заряд ядра 73фф и чем меньше расстояние
«о. Для й = 0 и ±nfa на границе зоны возникают отраженные
волновые функции. Они отвечают стоячим плоским электрон-
ным волнам. Всего имеется NA состояний, которые могут за-
полняться парами электронов в соответствии со статистикой
Ферми. Плотность состояний N(£) по определению — количе-
ство заполненных электронных состояний, приходящихся на еди-
ницу объема и единицу энергии. На краях зон N(Е) макси-
мальна, в середине зоны — минимальна (рис. 2.49). Это осо-
бенность одномерной решетки, в трехмерной решетке картина
176 Глава 2
те
при & = 0, имеем
(2.108)
(2.109)
от параметра /.
обратная. На краях зоны плотность состояний в совершенном
кристалле круто падает до нуля.
Чтобы понятия классической теории электронного газа мо-
жно было бы распространить дальше, необходимо принять сле-
дующее приближение. Зависимость E(k) (рис. 2.49) аппрокси-
мируют уравнением параболы, что в известных пределах вполне
справедливо на краях зоны. Согласно модели потенциальной
ямы Зоммерфельда, кинетическую энергию свободного элек-
тронного газа с учетом волновой природы электронов и гра-
ничного условия 2а0 = пД (п2 — число пробегов) можно пред-
ставить в следующем виде:
/j2n2 ^2^2
Е =-----— = , а для кривизны кривой Е (k) имеем
8maQ Zme
d2E Ъ2
4k=— (2.107)
Объединяя это уравнение с уравнением (2.106)
1 / ^эфф — J ^о/h
Отсюда следует выражение для подвижности
И = (е/Шэфф) (//и) = eJall/h2v
Масса электрона на краях зоны зависит
определяющего ширину зоны. Поэтому массу электрона лучше
заменить некоторой эффективной массой. Большая ширина
зоны обусловливает значительную кривизну на краях зоны и,
следовательно, небольшие эффективные массы электронов.
Точно так же подвижность ц зависит не только от длины сред-
него свободного пробега / и скорости v, но и от ширины зоны.
Из модели трехмерной потенциальной ямы Зоммерфельда
можно весьма просто вывести следующее уравнение для зави-
симости плотности состояния N (Е):
N (£) = (л/4) (8те/Л2)3/2 Е11 (2.110)
Таким образом, в рамках принятого приближения (шР можно
заменить на /лЭфф, которую принято считать практически не
зависящей от энергии) плотность состояний внутри зон кри-
сталлических твердых тел меняется пропорционально корню
квадратному из энергии.
Заполнение того или иного состояния определяется принци-
пом Паули и происходит в соответствии с функцией распреде-
ления Ферми—Дирака Р(Е) = {exp [ (Е — Ее)/kT]+ I}-1. Таким
образом, для концентрации электронов в единице объема ме-
Аморфное и стеклообразное состояние
177
таллов и их скорости в области уровня Ферми получаем
п = | Р (Е) N (Е) dE = -2- |
/ 8/Лэфф£ \3/г
7 Т у л5 J
_______E'^dE__________
exp [(£ - Ep)!kT] + 1 “
(2.1Н)
Vf — (/r/Шэфф) (Зл2п)'/з; Vf « 108 см/с
Точно так же может быть вычислен интеграл в выражении
(2.111) в случае полупроводников и изоляторов. Однако из-за
существования запрещенной зоны в таких материалах при не
слишком высоких температурах занятой оказывается лишь не-
большая часть состояний выше границы зоны проводимости Не-
соответственно лишь небольшая часть состояний ниже границы
валентной зоны оказывается незаполненной. Так что критерий
вырождения статистики Ферми не выполняется: поскольку раз-
ность Е—ЕР велика, слагаемым +1 в знаменателе выражения
можно пренебречь. Это означает, что для расчетов можно ис-
пользовать статистику Больцмана. Если дополнительно ввести
в качестве точки отсчета для кинетической энергии электронов
в зоне проводимости энергию границы зоны проводимости Ес
и заменить Е на разность Е—Ес, то
« = ^-)’Л j (е - ЕсГ’ехр [-(Е - Ef)/W] dE (2.112)
сс
п = 2 (2nm3^kTlh^!1 exp ( —= N с ехР С ~~ ьт^
\ КI / X /v« 4
(2.112а)
Аналогичное уравнение можно записать для концентрации
незаполненных электронных состояний (дырок) р -у верхней
границы валентной зоны с потенциальной энергией Ev:
р = 2 (2лтэфф^Т//г2),/2ехр (—Ер ) = Nv exp ( —
(2.1126)
Здесь и Nv — эффективная плотность состояний вблизи
границ соответствующих зон. Принимая, что кривизна диспер-
сионной кривой E(k) (ширина зон) вблизи границ валентной
зоны и зоны проводимости почти одинакова, можно найти, что
эффективные массы электронов и дырок примерно равны. Из ус-
ловия п=р (собственный полупроводник) следует, что Ес—Ef =
= Ef—Ev, и тогда EF= (Ec-\-Ev)l2, т. е. уровень Ферми
12 Заказ № 413
178 Глава 2
находится точно в середине между валентной зоной и зоной
проводимости:
( Е ____Е \
n = p = (Nc/Nv)'hexV[----ТлгЧ <2Л13)
Это же соотношение можно получить, применяя закон дейст-
вующих масс к процессу образования пар электронов и дырок
в соответствии со схемой, изображенной на рис. 2.50, а. В ре-
Рис. 2.50. Зависимость плотности состояния N(£) в области валентной зоны
и зоны проводимости для собственного полупроводника (а). Для наглядности
приведены энергетические состояния в полупроводнике n-типа (б).
зультате устанавливающегося через запрещенную зону шириной
(Ес—Ev) =RT \п К равновесия между n-электронами и р-дыр-
ками возникают Nv занятых и Nc незанятых состояний:
пр = NCNу ехр ( —
(2.114)
Приравнивая скорость образования g к скорости рекомби-
нации w~rnp электронов и дырок, получаем тот же результат,
поскольку np = glr — const. •
Разупорядочение в реальных кристаллах приводит к тому,
что внутри запрещенной зоны ниже Ес и выше Ev появляется
определенная плотность локализованных электронных состоя-
ний, которые обладают донорными и акцепторными свойствами.
К аналогичным последствиям ведет введение соответствующих
легирующих примесей. Как показано на рис. 2.50, б, в случае
существования nD донорных центров уровень Ферми при до-
статочном числе донорных состояний находится в середине
между Ес и Ed'
п = (поМс/2)'1г ехр(
2kT
(2.115а)
Аморфное и стеклообразное состояние
179
Для образования дырок в валентной зоне при заполнении
акцепторных уровней по аналогии с (2.115а) имеем
(£ _£ ч
----Л2^Г V ) (2.1156)
Лишь в случае практически полного удаления доноров или
при введении акцепторов (создание примесных центров) уро-
вень Ферми вновь смещается в направлении середины-запре-
щенной зоны.
2.5.1.2. Границы применимости зонной модели
кристаллических твердых тел
Учет межэлектронного взаимодействия, которое не рассмат-
ривается в одноэлектронном приближении, позволил Мотту
сформулировать критерии локализации электронов и тем са-
мым определить факторы, вызывающие при определенных ус-
ловиях исчезновение делокализации электронных состояний
в кристаллах. Рассмотрев соотношение между шириной зоны,
подвижностью и средней длиной свободного пробега электронов
и используя уравнение (2.109), ~Иоффе и Регель ,указали на
ограничения зонной модели. И наконец, Андерсон вывел ус-
ловие, которое описывает переход от зонной структуры к лока-
лизованным состояниям под действием статистических флуктуа-
ций потенциальной энергии взаимодействия атомов.
Выход за рамки второго приближения, лежащего в основе
зонной модели — введение в рассмотрение смещения окру-
жающих .элементов кристаллической решетки при описании
электронных состояний,— привел к формулировке условий ло-
кализации заряда и к открытию так называемого полярошюго
эффекта.
Переход Мотта. Согласно зонной модели Блоха—Вильсона,
при расширении кристаллического твердого тела вследствие
бесконечной делокализации электронных волновых функций сле-
дует ожидать конечной ширины запрещенной зоны и при до-
статочно больших межатомных расстояниях. При наличии ва-
лентных электронов должна возникать металлическая прово-
димость. Ширина разрешенных зон крайне мала, эффективные
массы электронов на границах зон очень велики, но тем не ме-
нее электронные волны могут распространяться беспрепятст-
венно.
Такая ситуация физически абсурдна, выводы зонной модели
ошибочны. Уже Вигнер [363] указывал на то, что электрон-
ный газ при определенной плотности состояний «кристалли-
зуется» из-за межэлектронного взаимодействия (кулоновское
взаимодействие е2/Г]2 больше, чем выигрыш энергии в результате
12*
180 Глава 2
образования зоны шириной 2J). Мотт [364—366] обратил
внимание на то, что при образовании полосы электронных со-
стояний из Na однократно занятых волновых функций атомов
в периодическом поле происходит попарное заселение электрон-
ных состояний. При этом действует межэлектронное отталкива-
ние Ulp—так называемая энергия корреляции электронов. Эта
энергия как раз равна разности между энергией ионизации 1А
и сродством к электрону EAff (например, для натрия £7Гр = 5эВ):
ULP = IA-EAff (2.116)
Рис. 2.51. Зависимости электропроводности от расстояния между центрами
локализации электронов в решетке.
Это приводит к возрастанию энергии системы по сравнению
с состоянием одиночно локализованных электронов. В зонной
модели таким взаимодействием пренебрегают, и поэтому полу-
чается, что даже при очень больших расстояниях между ато-
мами все еще существует металлическая проводимость. В дей-
ствительности же в одновалентных и двухвалентных металлах
электропроводность падает до нуля уже при конечных межатом-
ных расстояниях и при приближении температуры к абсолют-
ному нулю (рис. 2.51). В случае двухвалентного металла такое
уменьшение электропроводности происходит в результате исчез-
новения перекрывания зон и связанного с этим возникновения
запрещенной зоны. В случае одновалентных металлов этот эф-
фект определяется положительной величиной энергии корреля-
ции электронов при переходе металл—изолятор.
Согласно Мотту [367, 368], такой переход сопровождается
скачкообразным изменением проводимости (переход I рода).
При оценке расстояния, при котором происходит переход от ло-
кализованных к делокализованным электронным состояниям,
исходят из кулоновского потенциала V(r)=—е2/г, который опи-
сывает взаимодействие электронов, находящихся на соседних
центрах. При этом учитывают эффект экранирования со сто-
роны соседних электронов, который проявляется тем сильнее,
Аморфное и стеклообразное состояние 181
чем меньше постоянная решетки а0- Эффект экранирования,
который учитывают, вводя в выражение для потенциала мно-
житель ехр(—£г), имеет ту же природу, что и в случае взаи-
модействия в разбавленных растворах электролитов, которое
описывается теорией Дебая—Хюккеля
V(r) = (—ег/г)ехр(—?г), у = (2.ц7>
Если радиус экранирования меньше радиуса электрон-
ной орбиты водородного атома г0=А2/те2, спаривания электронов
не происходит из-за сильного кулоновского отталкивания со
стороны соседних электронов. Отсюда следует, что расстояние
а0, до которого могут сближаться атомы в периодическом поле
кристаллической решетки, чтобы произошла делокализация
электронов, имеет некоторое предельное значение, примерно
равное учетверенному боровскому радиусу
йо = п~'/з ^4го (2.118)
Эти соотношения неоднократно проверялись во многих экс-
периментальных работах, например при изучении переходов
изолятор—металл в оксидах и халькогенидах переходных ме-
таллов [369]. В большинстве случаев такой переход, иниции-
руемый повышением температуры или давления, связан с фа-
зовым превращением, т. е. с изменением симметрии кристалли-
ческой решетки. Поэтому переход Мотта в чистом виде может
быть обнаружен пе сразу. Примером может служить ряд твер-
дых растворов (Vi-xCr.v)2О3 [370]; правда, здесь атомы V(III),
введение которых определяет возникновение носителей заряда,
распределены статистически в кристаллической решетке, и, сле-
довательно, их расположение не периодично. Аналогичные
ограничения характерны для перехода к проводимости по при-
месной зоне в сильно легированных монокристаллах Si и Ge.
При достаточно небольшом среднем расстоянии между донор-
ными центрами (рис. 2.50, б) возникновение делокализованных
электронных уровней вполне объяснимо (рис. 2.52). В этом
случае проводимость вблизи- абсолютного нуля высока, хотя
периодического расположения центров не существует. В ряде
работ [365, 368, 371, 372] обнаружено хорошее согласие пре-
дельного значения а0 при переходе к делокализованным состоя-
ниям со значением а0 из уравнения (2.118).
Критерий Иоффе—Регеля. В уравнение для подвижности
(2.109) можно ввести выражение средней скорости электронов
ц= (2^7,//7гЭфф)1^ (статистика Больцмана):
ц = (е/аоМ)(7/26Т)'/2
(2.119)
182 Глава 2
Если принять в качестве нижнего предела ширины зоны
J^kT и допустить по физически вполне понятным соображе-
ниям, что средняя длина свободного пробега в условиях обра-
зования делокализованных электронных состояний имеет тот
же порядок величины, что и постоянная решетки (а0^/), то
подвижность р = 1ч-5 см2/(В-с). При более низких значениях
подвижности применение зонной модели становится невозмож-
зона проводимости
|| Н1ЦЩ I
N(E)
зона проводимости
_t ® ® л. ® 4?
4® Iе 4е М(Е)
валентная зона
валентная зона
зона проводимости
5 э- е -е - ё 0-ее
I® |® j® 4®
зона проводимости
валентная зона
валентная зона
г
Рис. 2.52. Схематическое представление перехода Андерсона в легированном
и частично компенсированном кремнии.
а —Пр=»1015 атом. Р/см3, локализация электронов при низкой температуре; б — частичная
компенсация алюминием, V'o/2/>35; в — nD>3-1015 атом. Р/см3, проводимость по при-
месной зоне; г — частичная компенсация, Е0/2/<3-е-5.
ным, и понятие длины среднего свободного пробега теряет
всякий физический смысл.
Первыми на такое ограничение зонной модели указали
Иоффе и Регель (см. литературу к гл. 1 [28]). Вследствие
рассеяния на колебаниях кристаллической решетки, сущест-
вования дефектов строения последней и наличия примесей
в ней волновая функция Блоха (2.104), которая обычно рас-
сматривается в бесконечном пространстве, в реальном кри-
сталле ограничена некоторым объемом /3, связанным со
средней длиной свободного пробега. Процессы рассеяния обус-
ловливают неопределенность волнового вектора k. \k пропор-
ционально /*, однако \kjk не может быть больше 1. Отсюда сле-
.дует
/6>1, fc = 2n/Z
(2.120)
Аморфное и стеклообразное состояние 183
При превышении этого граничного значения должна про-ч
исходить локализация электронов. Такую ситуацию следует
ожидать в случае, когда перекрывание мало и полосы узки, или
в области низкой плотности состояний на границах зон в не-
кристаллических твердых телах, где средняя длина свободного»
пробега заметно уменьшается в связи с высокой степенью раз-
упорядочения.
Переход Андерсона. Андерсон в своей теоретической работе
[375] изучил проблему трехмерного периодического располо-
жения атомов, потенциальная энергия которых статистически
распределена в области потенциала Уо и которые определяют
ширину зоны 2/ в случае одинаковых потенциальных энергий,
при некотором среднем значении. Ему удалось определить кри-
тическое предельное отношение
V0/2J < 3 5 (2.121)-
которое зависит от типа кристаллической решетки и в первую
очередь от координационного числа. Андерсон показал, что
статистическое разупорядочение в трехмерной решетке в опре-
деленных пределах не противоречит образованию делокализо-
ванных электронных состояний. За этими пределами происхо-
дит локализация электронов. Работа Андерсона стала осново-
полагающей в теории некристаллических полупроводников.
Согласно Андерсону [376], переход изолятор—металл реа-
лизуется, например, в легированных и частично компенсиро-
ванных монокристаллах Si и Ge. Экспериментальные данные
для этих веществ получены в работе [425]. Как можно заме-
тить из схемы (рис. 2.52), умеренное легирование монокристалла
Si донорными примесями, например введение Р в количестве
~ 1015 атом/см3, ведет при низких температурах к заполнению
донорных центров электронами.
Вблизи температуры абсолютного нуля такие легированные
монокристаллы представляют собой изоляторы. Образование
так называемых примесных зон не происходит из-за недостаточ-
ной плотности электронных состояний и межэлектронного
отталкивания, которое доминирует в данном случае (рис. 2.52, а)..
Частичная компенсация донорных примесей за счет введения
акцепторов, например атомов А1, приводит к появлению свобод-
ных мест донорных уровней, причем межэлектронное от-
талкивание уменьшается. По соседству возникают положи-
тельно и отрицательно заряженные центры (рис. 2.52, б), куло-
новское притяжение которых вызывает случайные флуктуации
потенциальной энергии центров обоих знаков вследствие их
нерегулярного распределения в кристаллической решетке.. Пе-
ренос заряда между двумя соседними состояниями на до-
норных уровнях определяется прыжковым механизмом.
184 Глава 2
(разд. 4.1.2 и 4.1.3), на который пе оказывает влияние меж-
электронное отталкивание. Поэтому при компенсации донор-
ных примесей проводимость возрастает. Несмотря на это, кри-
сталл при абсолютном нуле по-прежнему является изолято-
ром, поскольку критическое отношение Андерсона Уо/2/, оче-
видно, остается превышенным. Если концентрации примесных
донорных атомов По>3-1018 см*3, то и при температуре абсо-
лютного нуля существует некоторый уровень проводимости
(рис. 2.52, в). В этом случае говорят о металлической проводи-
мости кристалла, имея ввиду примесную зонную проводимость.
Она сохраняется и в случае введения некоторого количества
акцепторной примеси (рис. 2.52, г), сравнимого с количеством
этой примеси для случая, изображенного на рис. 2.52, б. Таким
образом, при более высокой плотности состояний предельное
для перехода Андерсона критическое значение не достигается,
и даже, как показывает пример, в случае случайных флуктуаций
потенциальной энергии атомов возможна делокализация элект-
ронов [374, 375].
Таким же путем на базе исходной решетки кристаллического
твердого тела может возникать разупорядоченная система ато-
мов с одинаковым окружением или по крайней мере с оди-
наковым ближним порядком, что является характерной особен-
ностью аморфных и стеклообразных твердых тел. Геометриче-
скому разупорядочению стекал и аморфных веществ соответст-
вует различная потенциальная энергия отдельных структурных
групп. Это аналогично случайному разбросу энергетических
уровней доноров и акцепторов в легированных и частично ком-
пенсированных полупроводниках.
Основной вывод, следующий из вышесказанного, состоит
в том, что образование делокализованных состояний в полупро-
водниках физически вполне реально даже в отсутствие даль-
него порядка.
Поляронные состояния. В неполярных кристаллах, таких,
как Si или Ge, электрон, находящийся на своем донорном
уровне, характеризуется слабой локализацией в простран-
стве. Так, простая оценка [362] показывает, что для Ge (е= 16)
3/4 заряда s-электрона распределяется в окрестности 925 ато-
мов Ge. Подобные рассуждения не применимы к глубоко ле-
жащим центрам с сильно связанными электронами, в особенно-
сти в решетках полярных веществ, например к F-центрам,
а также к узкозонным полупроводникам, например к оксидам
переходных металлов. Если ограничиться локализацией элек-
тронов определенного уровня лишь вблизи нескольких соседних
атомов, то нельзя больше пренебрегать искажением, которое
возникает в решетке в связи с существованием данного заряда.
Волновая функция электрона взаимодействует с фононами кри-
Аморфное и стеклообразное состояние
185
сталлической решетки, что приводит к стабилизации локализо-
ванного электронного состояния. Таким образом, от одного
центра к другому заряд переносит с собой поляризационные
искажения решетки. Исходя из этого было введено представ-
ление о новой квазичастице — поляроне. Энергетический уро-
вень полярона назвали поляронным состоянием [379—380].
Мотт и Гарни [381] получили выражение для понижения
потенциальной энергии локализованных электронов при воз-
никновении поляронного состояния, используя различие между
Рис. 2.53. Поляронпое состояние [381] (а) и модель конфигурационных коор-
динат [379] (см. также литературу к гл. 1 [33]) (б).
оптической диэлектрической проницаемостью 8<х> (жесткая ре-
шетка) и статической (или низкочастотной) диэлектрической
проницаемостью es (последняя «учитывает» колебания ре-
шетки). После введения радиуса гр, сравнимого с постоянной ре-
шетки, получено следующее выражение для понижения энергии:
VP(rp)=- -7-(-£---------г-) при г<Гр <2121>
Гр \ ЕОО S /
а для спуска в возникшую потенциальную яму (рис. 2.53, а)
= -----ПРИ г>гр (2.122)
г \ еоо e-S /
Согласно модели конфигурационных координат [379] (см.
также литературу к гл. 1 [33]), при образовании поляронного
состояния наблюдается выигрыш энергии (рис. 2.53, б). Общая
энергия Гобщ равна сумме деформационной энергии £‘деф =
= возникающей из существования упругих сил кристал-
лической решетки, и поляризационной энергии £‘Пол = Р7-
186 Глава 2
В минимуме функции
£общ == ~2~ aq
(2.123)
конфигурационная координата принимает значение q0 = fi/a, из
которого следует выражение для понижения энергии электрона
в поляронном состоянии
р2 _ е2 / i________________LA
2а ~ гр \ еж е5 )
(2.124)
На такую величину понижается энергия локализованных элек-
тронов при взаимодействии с фонопами решетки. Это пониже-
ние энергии тем сильнее, чем больше поляризуемость £ элемен-
тов решетки вблизи данного состояния и чем меньше постоян-
ная а (связана с упругой деформацией решетки).
Если выигрыш в энергии за счет расщепления электронных
состояний одного энергетического уровня меньше, чем пониже-
ние энергии при образовании поляронных состояний, то в си-
стеме происходит локализация электронов. Таким образом, не
всегда можно исходить из адиабатического приближения, лежа-
щего в основе зонной модели.
2.5.1.3. Зонная модель некристаллических твердых тел
При определенных условиях (разд. 2.5.1.2) образование зон-
ной структуры нарушается уже в кристаллической решетке. В то
же время пример проводимости по примесной зоне показывает,
что и при случайном распределении центров локализации по
эквивалентным узлам решетки при достаточной плотности со-
стояний возможно существование делокализованных электрон-
ных состояний. Согласно Андерсону, даже статистические флук-
туации потенциальной энергии таких центров в определенных
пределах совместимы с образованием энергетических зон.
По мнению Мотта [368, 382, 383] и Коэна [384], в усло-
виях существующего в стеклах и аморфных твердых телах
ближнего порядка также возможно возникновение энергети-
ческих зон. В областях пониженной плотности состояний (ма-
лая степень перекрывания), например на границах зон, проис-
ходит, согласно критерию Андерсона (2.121), переход к локали-
зованным состояниям. Уже Губанов [385] и Банья [386]
высказывали предположения, что зонная модель некристалличе-
ских твердых тел также характеризуется наличием локализо-
ванных электронных состояний; это приводит к размытию гра-
ниц зон, а следовательно, плотность состояний не уменьшается
так резко, как в кристаллах (рис. 2.50, а). В связи с геометри-
Аморфное и стеклообразное состояние
187
ческим разупорядочением, которое имеет место в сетчатых
структурах аморфных тел и стеклообразующих соединений,,
и связанными с этим флуктуациями потенциальной энергии гра-
ницы зон оказываются не такими резкими. Могут происходить
локальные сдвиги границ, которые создают некоторую плотность
состояний внутри запрещенной зоны. Поэтому в некристалличе-
ских твердых телах энергетический спектр характеризуется нали-
чием так называемых «хвостов» локализованных состояний,
проникающих в запрещенную зону (рис. 2.54, а).
зона проводимости
валентная зона
I край
р \ подвижности ..
I
।
V
iO ’ ю° Ю’ ю2
6
а
Рис. 2.54. Плотность состояний N(E) в некристаллических полупроводниках
(а) и зависимость края подвижности от энергии (б).
Ес' и £г' —границы зон в кристаллическом состоянии.
Границы между делокализованными и локализованными
электронными состояниями обозначены символами Ес и Еу.
В случае заполнения состояний выше Ес при абсолютном нуле
следует ожидать некоторого уровня проводимости. Напротив,
при заполнении состояний ниже Ес вещество является изоля-
тором. Эти же рассуждения относятся к уровню Еу. В области
энергий Ес и Еу подвижность ц меняется скачкообразно
(рис. 2.54, б). Ранее введенные определения подвижности [урав-
нения (2.109) и (2.119)] в области локализации состояний
теряют свой смысл. Необходимо дать новое определение под-
вижности с учетом прыжкового механизма переноса электронов
(разд. 4.1.2 и 4.1.3).
Коэн, Фриче и Овшинский [387] ввели в теорию некристал-
лических полупроводников понятие края подвижности.
На рис. 2.54, а представлена типичная зависимость плотно-
сти состояния от энергии для идеальных аморфных и стекло-
образных твердых тел (определение идеальной структуры дано
в разд. 2.4.2.5). В таком виде можно было бы описать энер-
гетическую структуру валентной зоны и структуру зоны;
188 Глава 2
проводимости, лежащей выше щели подвижности, например
в случае существования непрерывной беспорядочной сетки со-
единенных друг с другом атомов Si и Ge, все связи которых
насыщены. Приведенная схема также относится к стеклообра-
зующим оксидам и халькогенидам. При этом необходимо обра-
тить внимание на то, что валентная зона образуется из неподе-
ленных электронных пар мостиковых атомов.
Структуру стекол и аморфных твердых тел нельзя пол-
ностью описать с помощью представлений о геометрическом
разупорядочении беспорядочной непрерывной сетки связанных
друг с другом областей ближнего порядка с учетом их топо-
логического разупорядочения (разд. 2.4.2). В стеклах и аморф-
ных телах возможны различные типы структурного разупоря-
дочсния областей ближнего порядка (разд. 2.4.2.5). Оборван-
ные химические связи и модифицированные формы типичных
структурных групп могут давать целый спектр различных де-
фектных центров, частота возникновения которых определяется
свободной энергией реакции образования каждого из таких
центров. Возникающие дефекты могут быть как донорными,
так и акцепторными, и следует ожидать их определенного рас-
пределения в области щели подвижности. Таким образом, пе-
реход к реальному стеклу требует внесения соответствующих
изменений в зонную модель, приведенную на рис. 2.54, а.
Многие свойства, такие, как высокая концентрация спинов
в аморфных Si и Ge или люминесцентные свойства (разд. 4.2.2),
но прежде всего различные явления переноса заряда
(разд. 4.1.3), указывают на высокую плотность локализован-
ных состояний в щели подвижности [388—390]. На основании
проведенных измерений установлено, что эта величина состав-
ляет ~ 1017—1020 см~3-эВ-1. В работе [391] обращается вни-
мание на то, что электропроводность стеклообразных полупро-
водников, как правило, слабо зависит от незначительных коле-
баний состава или присутствия небольших количеств примесей.
При введении добавок в количестве, меньшем чем 1019 см-3,
обычно не меняется ни величина проводимости, ни ее темпера-
турная зависимость. Фриче [392] приводит важный аргумент
в пользу того, что уровень Ферми находится примерно в сере-
дине щели подвижности. Из-за высокой плотности локализован-
ных состояний энергия Ферми должна быть «закреплена», как
и в легированных и компенсированных кристаллических полу-
проводниках.
Эти экспериментальные факты использованы Моттом, Коэ-
ном, Фриче и Овшинским при создании модели локализован-
ных состояний в запрещенной зоне (модель Мотта—КФО).
Слабая чувствительность проводимости аморфных полупровод-
ников к наличию примесей может быть объяснена тем, что
в стеклах все валентные электроны участвуют в образовании
Аморфное и стеклообразное состояние
189
связи [368]. В то время как, например, кристаллическая ре-
шетка Si «навязывает» координационное число 4 вводимым
в качестве донорной примеси атомам Р, в аморфных и стекло-
образных полупроводниках в этом случае связывающему со-
стоянию с самым низким уровнем энергии может отвечать
координационное число 3. Такие центры больше не являются
донорами электронов.
В рамках модели КФО [387] приведенные соображения по-
лучили дальнейшее развитие. Структурное разупорядочеиие
областей ближнего порядка
существенно повышает плот-
ность локализованных со-
стояний в области хвостов,
выходящих из валентной
зоны или зоны проводимо-
сти. Это приводит к тому,
что локализованные состоя-
ния распространяются да-
леко в глубь щели подвиж-
ности, так что в середине
зоны происходит их частич-
ное перекрывание. Посколь-
ку в этом случае незапол-
ненные энергетические цент-
Рис. 2.55. Плотность состояний заря
женных дефектных центров в щели по-
движности согласно модели КФО.
ры, выходящие из зоны про-
водимости, занимают более
низкие уровни энергии,
чем попарно заполненные
центры, выходящие из валентной зоны, то следует ожидать
перераспределения электронов. Это и имеет место (рис. 2.55)
в области уровня Ферми. Здесь создается соответствующая кон-
центрация положительно и отрицательно заряженных центров,
которые из-за сильного электростатического взаимодействия
вызывают заметные флуктуации потенциальной энергии и за-
крепляют уровень Ферми.
Модель КФО дает объяснение многим явлениям, но и ста-
вит новые вопросы. Высокой концентрации положительно и
отрицательно заряженных центров должна соответствовать вы-
сокая концентрация неспарснных электронов. Попарное запол-
нение уровней с образованием двукратно заряженных положи-
тельных и отрицательных центров, вообще говоря, не должно
осуществляться из-за межэлектронного отталкивания и положи-
тельной величины энергии корреляции электронов ULP
(разд. 2.5.1.2, переход Мотта). Однако исследования спектров
ЭПР [393] со всей очевидностью свидетельствуют о том, что
концентрация спинов в халькогенидных стеклах <П015 см-3. По
данным Бишопа и сотр. [394], лишь под действием облучения
190 Глава 2
концентрация фотоиндуцированных неспаренных электронов до-
стигает 1017—10г§ см 3 и возникает полоса оптического погло-
щения, обнаруженная при низких температурах (разд. 4.2.2.2).
В аморфных Si и Ge, напротив, концентрация неспаренных
электронов в зависимости от способа получения достигает
1020 см-3. Наличие однократно заряженных положительных и
отрицательных дефектных центров заставляет ожидать замет-
ную проводимость при низких температурах. Как и в случае
легированных и частично компенсированных полупроводников,
проводимость можно описать с помощью прыжкового меха-
низма. В халькогенидных стеклах вклад такой проводимости
обнаружен не был, в то время как в аморфном Ge он заметен.
Все эти данные свидетельствуют о том, что в случае лока-
лизованных электронных состояний в щели подвижности речь
идет о наличии более или менее дискретных энергетических уров-
ней, а не о непрерывном распределении, которое предполагает
модель КФО.
Объяснение этого различия стало возможным в связи со
структурно-химической интерпретацией наличия дефектных цен-
тров с позиций теории химической связи. Это привело к. углуб-
лению представлений о структурном строении стекол и аморф-
ных твердых тел.
2.5.2. Собственное разупорядочение и заряженные
дефектные центры в некристаллических системах
Наиболее существенный вклад в развитие излагаемых здесь
представлений также внес Андерсон. В одной из своих работ
{395] он указал на то, что энергия корреляции электронов
ULP должна быть всегда положительной, если исходить из се
определения как меры межэлектронного взаимодействия в слу-
чае заполнения эквивалентных электронных состояний в изоли-
рованной системе (разд. 2.5.1.2). Если учесть поляризуемость
окружающих электроны участков кристаллической решетки, то
оказывается, что энергия корреляции электронов может прини-
мать и отрицательные значения. Это дало основание ввести
в рассмотрение некоторую эффективную энергию корреляции
t/эФФ, которая по смыслу аналогична энергии стабилизации по-
ляронных состояний (разд. 2.5.1.2):
U эфф = U lp — Р2/2а (2.125)
Если эффективная энергия корреляции отрицательна и по
абсолютной величине больше, чем kT, то в конденсированной
системе должно происходить заполнение одинаковых локализо-
ванных состояний, причем таким образом, что половина центров
останется незанятой и приобретет тем самым положительный
заряд, в то время как другая половина вследствие ее заполне-
Аморфное и стеклообразное состояние 191
ния парами электронов будет иметь отрицательный заряд. Ши-
роко распространенное в конденсированных системах явление
диамагнетизма находит при этом вполне однозначное объясне-
ние. Линейный ход молекулярной теплоемкости стекол в обла-
сти низких температур (разд. 2.2.3.3) также может быть объяс-
нен с этих позиций.
Стрит и Мотт [396] использовали эти представления для
интерпретации экспериментальных данных, которые касаются
обнаружения локализованных электронных состояний в щели
подвижности некристаллических твердых тел. Они построили
модель заряженных ненасыщенных связей (модель СМ).
Кастнер, Адлер и Фриче [397] рассматривают связывающие
орбитали атомов, участвующих в образовании структуры, и вза-
имодействие между ними в рамках схемы двухцентровых моле-
кулярных орбиталей. Соотнесение определенных энергий с раз-
личными заполненными состояниями позволило оценить, какие
конфигурации энергетически более предпочтительны. С построе-
нием модели пар с переменной валентностью были впервые
сформулированы понятные химические представления о струк-
туре дефектных центров, оказывающих существенное влияние
на многие свойства стекол и аморфных твердых тел (модель
КАФ).
Обе модели с точностью до отдельных деталей описывают
один и тот же эффект. Главное, что их объединяет,— единство
подхода к сути явления. Наиболее существенные положения
обеих моделей в обобщенном виде можно назвать «моделью
заряженных дефектных центров*.
В заключение необходимо отметить, что структурное разупо-
рядочение в некристаллических системах можно поставить
в один ряд с такими известными явлениями, как диспропор-
ционирование и самоокисление — самовосстановление [398]. Та-
кие аналогии можно распространить и на случаи изомерии
связи — существование ковалентной и гетерополярной форм
связи в одном и том же соединении.
2.5.2. !. Различие между веществами, связанное со знаком
эффективной энергии корреляции электронов
Возникновение отрицательных значений эффективной энер-
гии корреляции электронов £/Эфф(Р2/2а > Ulp) обусловлено
главным образом взаимодействием незанятых положительно за-
ряженных центров, образующихся при гетеролитическом раз-
рыве химических связей, со свободной неподеленной парой элек-
тронов соседних атомов. На существование такого взаимодейст-
вия указывалось в разд. 2.4.2.5 при описании образования
различных типов связей (рис. 2.40 и 2.41). Халькогены и стекло-
образующие халькогениды и оксиды, имеющие неподеленную
192 Глава 2
пару р-электронов у мостикового атома, в принципе в состоянии
оказывать такое стабилизирующее влияние на структуру ве-
щества, что приводит к отрицательным величинам £/Эфф. Эту
особенность надо учитывать не только в случае стеклообразую-
щих, но и в любых конденсированных системах, в состав ко-
торых входят атомы со свободной парой р-электронов.
Необходимо провести разграничение между этими вещест-
вами и соединениями, состоящими из атомов, все валентные
электроны которых участвуют в образовании ковалентных свя-
зей. Ко второй группе веществ относятся, например, алмазо-
подобные полупроводники, такие, как Si и Ge, соединения
AinBv или CdGeAs2. В таких веществах описанного выше взаи-
модействия нет, и, следовательно, энергия корреляции положи-
тельна.
Промежуточное положение занимают пниктиды сурьма,
мышьяк, фосфор, а также соединения AIVBVI, например моно-
халькогениды германия, кристаллическая структура которых
характеризуется наличием связевых состояний всех р-орби-
талей. Атомы имеют, правда, еще одну пару s-электронов,
которая из-за более сильного взаимодействия с ядром (более
слабая нуклеофильность) способна сравнительно слабо взаимо-
действовать с незанятым положительно заряженным центром.
В зависимости от условий корреляционная энергия здесь может
принимать как положительные, так и отрицательные значения.
Влияние знака энергии корреляции электронов на стати-
стику заполнения определенных типов центров в щели подвиж-
ности изучено Адлером и Иоффе [399, 400].
В приведенной энергетической схеме (рис. 2.56) No локали-
зованных состояний с энергией То можно описать набором сле-
дующих параметров:
Hi Ei gi
0 0 1
1 To 2
2 2T 0 + U эфф 1
Здесь Hi — число электронов, занимающих данное состояние.
Если состояние не занято электронами, то Е = 0; если состоя-
ние занято одним электроном, Е = То', если состояние занято
двумя электронами, Е = 2То+^эФФ‘, g—вырождение. Из на-
бора этих параметров с учетом соотношения Ut=Ei — tiiEpi
для суммы по состояниям Z= [£ gi exp (—Ut/kT)} получаем
i
(EF— энергия Ферми)
Z = {1 + 2 exp [- (To - EF)/kT] + exp [- (2T0 +
+ £/эфф - 2EF)/fc7’]}’'" (2.126)
Аморфное и стеклообразное состояние
193
Рис. 2.56. Энергетическая диаграмма положения дефектных центров с поло-
жительной (а, б) и отрицательной (в, г) эффективной энергией корреляции
электронов в зависимости от заполненности состояний [399, 400].
Стрелки соответствуют оптическим переходам.
Рассчитав среднее число электронов, приходящихся на один
дефектный центр n = N/No, по уравнению n(EF, Т) =
= (kT/N0)(d\n Z/dEF)T, получаем при условии | С^фф | kT
для (/эфф<0 ЕГ = ТО-----^-^эфф---^Пп(2п-1 — 1) (2.127а)
для иЭфф > 0 Ef = То — ЛТ1п[2(п— 1)/(2 — п)], 0 < п < 1
(2.1276)
£г = 7’о + ^Эфф + /гГ1п[2(п- 1)/(2-п)], 1 < п < 2
При отрицательной энергии корреляции электронов измене-
ние п, например, в пределах !/з < п < 5/з практически не при-
водит к изменению EF, так что энергия Ферми оказывается дей-
ствительно «закрепленной». Другие аргументы в пользу по-
стоянства значения энергии Ферми приводятся в работах Мотта
и сотр. [377, 401]. В случае положительной энергии корреляции
можно отметить обратную картину: при изменении п в пределах
2/з < п < 7з наблюдается изменение положения уровня Ферми
в зависимости от степени занятости состояний. Если концентра-
ция электронов п (выражена по отношению к концентрации
13 Заказ № 413
194 Глава 2
дефектных центров) достигает 1, состояния с энергией То за-
няты одним электроном, а для п > 1 происходит скачок на
величину t/дфф в связи с частичным заполнением центров двумя
электронами (рис. 2.56,6).
При отрицательных значениях энергии корреляции ситуация
иная. В связи с тем что электроны располагаются в основном
парами, отрицательно заряженные центры с энергией То—£эфф
всегда заполнены, а состояния с энергией То являются носите-
лями положительного заряда из-за того, что они остаются не-
занятыми. В области О < п < 2 (исключая /г = О и п = 2) по-
ложение уровня Ферми не меняется, изменяются лишь площади
пиков (рис. 2.56, в, г).
Тип проводимости зависит от положения уровней То и (Афф-
В халькогенидных стеклах обычно доминирует р-проводимость
(разд. 4.1.3.1). Электронной проводимости прыжкового типа
между локализованными состояниями в области уровня Ферми
обнаружено не было. Отсутствие парамагнитных центров легко
объяснимо. Под действием облучения, частота которого при-
мерно соответствует ширине щели подвижности, при низких тем-
пературах возникает соответствующая концентрация неспарен-
ных спинов. Эти спины исчезают при отжиге или под дейст-
вием более длинноволнового излучения (разд. 4.2.2). Спектр
люминесценции халькогенидных стекол характеризуется явно
выраженным стоксовским сдвигом, что дает основание прийти
к заключению о сильном электрон-фононовом взаимодействии
[уравнения (2.124) и (2.125)].
Проводимость в аморфных веществах типа германия
(£эфф>0) при комнатной температуре и ниже, напротив, мо-
жет быть описана с помощью модели прыжкового механизма
перемещения электронов из одного локализованного электрон-
ного состояния в другое в области энергии Ферми. Такие полу-
проводники относятся в основном к полупроводникам м-типа. Как
и следовало ожидать, в них обнаружена высокая концентрация
парамагнитных центров. В спектре веществ этого класса при
облучении нс обнаруживаются люминесцентные полосы в обла-
сти запрещенной зоны.
2.5.2.2. Модель заряженных дефектных центров
Основываясь на гипотезе Андерсона [395], Стрит и Мотт
при построении своей модели (модель СМ) исходили из вполне
понятного положения о том, что в халькогенидных стеклах
гетсролитический разрыв химической связи с образованием по-
ложительно и отрицательно заряженных дефектов энергетически
более выгоден, чем гомолитический разрыв связи, который ве-
дет к возникновению центров с неспаренными спинами. При-
чина этого вполне очевидна из схем, приведенных на рис. 2.40
Аморфное и стеклообразное состояние
195
Рис. 2.57. Образование заряженных дефектных центров D+ и D~ в результате
гетеролитического разрыва связей на примере селена (а) и образование мета-
стабильного нейтрального центра DQ путем возбуждения электронов (б).
и 2.41; имеет место взаимодействие с парой р-электронов со-
седнего мостикового атома халькогена. Данная концепция до-
норно-акцепторного взаимодействия, сопровождающегося поля-
ризацией и упругой деформацией окружающей решетки, есть не
что иное, как интерпретация отрицательной энергии корреляции
в теории химической связи.
Описанное явление проиллюстрировано на рис. 2.57 на при-
мере селена. В результате такого взаимодействия образуется
связывающее состояние, аналогичное F-цснтру или полярону.
Существованием пары s-электронов пренебрегают. Происходя-
щее в результате взаимодействия с р-электронами расщепление
терма позволяет двукратно занятому связывающему состоянию
опуститься глубоко внутрь валентной зоны неподеленных пар
р-электронов. В то же время антисвязывающее состояние имеет
электронную конфигурацию, которая описывается £)+-термом.
Последний располагается в запрещенной зоне ниже зоны про-
водимости (рис. 2.57,а).
Терм D~ относится к такому состоянию, когда дефектный
центр имеет полностью заполненную валентную электронную
оболочку; такое состояние поэтому не образует никаких допол-
нительных связей. Поскольку энергия корреляции Ulp положи-
тельна, терм поднимается из валентной зоны в запрещенную
13*
196 Глава 2
зону. Предполагается, что поляризация решетки в окрестности
£)--центра пренебрежимо мала. Тем сильнее поляризационный
эффект, создаваемый £)+-центром. Антисвязывающее состоя-
ние гораздо сильнее дестабилизирует систему [из-за наличия
члена р2/2а в уравнении (2.125)], чем состояние D~. Разность
между энергетическими уровнями центров D+ и D~ в точности
отвечает отрицательной энергии корреляции (Афф-
Если оторвать, например, с помощью оптического возбужде-
ния один электрон от центра D~ и перевести его в зону прово-
димости, то образуется нейтральный центр D°. Этот центр точно
так же стремится прореагировать с парой соседних р-электро-
нов (рис. 2.57,6). Антисвязывающее состояние центра D* за-
нято одним электроном, взаимодействие в целом ослаблено,
поэтому относящийся к нему терм находится ниже, примерно
в середине между центрами D+ и D~. Аналогичное расположе-
ние термов вытекает из возбуждения электрона валентной зоны
и перевода его в центр D+. Поэтому стоксовский сдвиг спектров
люминесценции находит свое естественное объяснение в раз-
личной поляризации окружающей терм решетки, если исходить
из существования одного центра D~ и одного центра D°. Связы-
вающие состояния D° нестабильны, поскольку энергия корреля-
ции в такой системе отрицательна. Поэтому состояние D° обна-
руживаются лишь при низкой температуре. Эти состояния могут
быть, например, заморожены и зафиксированы в виде фото-
индуцированного сигнала в спектре ЭПР или по появлению
соответствующей полосы поглощения (разд. 4.2.2).
Если записать реакцию самопроизвольного образования цен-
тров D+ и D~ из центров D° в виде двухстадийного процесса
и для энергетической характеристики каждой из этих стадий
использовать энергию ионизации /д:
D-_.£)o + e + /x(1) (а)
D° ->D+ + e+IA(2) (б)
2D^-D+ + D- _[/л (1) _ /л (2)] (в) (2.128)
то, так как константа равновесия К > 1 (самопроизвольный
процесс), получаем 1А (1) > 1А (2). Такая последовательность
реакций ионизации совершенно нс характерна для изолирован-
ных атомарных или молекулярных систем (например, для иони-
зации частиц в вакууме). Существование двухстадийного про-
цесса типа (2.128) — типичная особенность определенных кон-
денсированных систем; она обусловлена сильным взаимодейст-
вием данного связывающего состояния со своим окружением
(разд. 2.5.2.3). Энергия ионизации 2-й стадии реакции /д(2)
уменьшается из-за энергетической стабилизации центров D+
в результате их взаимодействия с соседними атомами Se на-
столько сильно, что /д(2) <7д(/). Отсюда следует важный вы-
Аморфное и стеклообразное состояние
197
вод для явления переноса заряда: отщепление двух электронов
путем двукратного возбуждения центра D~ энергетически более
выгодно, чем генерация электронов путем однократного воз-
буждения различных центров (разд. 4.1.3.5, рис. 4.21) [401 —
404].
Для оценки положения равновесия в системах с ковалент-
ными связями, в которых образуется некоторое количество по-
ложительно и отрицательно заряженных дефектных центров,
можно привлечь модель циклического процесса, например для
стеклообразного селена:
a) 6SeTB — 6Ser + 6\НА
(A/7A — энтальпия атомизации)
б) Ser - Se+ + e 4- IA (IA — энергия ионизации)
в) Ser + е {Е’АЦ — сродство к электрону)
г) Se+ + 3Ser -Se+, -3£j (Е^~, — энергия связи Se —Se)
д) Se^ + Ser -*Se2?r-£r
е) Se4') t. + Se<r г — Se^Se^T Т) - ±\HA-W (W — кулоновское притяжение)
ж) 6SeTB - s*4+se2; „ + 4 ЬНЛ - 3£#+-£; + !a - ЕЛ11 - F
3) 68етв = 8е+8е27„+/л-£л„ (2.129)
Разность энтальпий кристаллического и стеклообразного со-
стояния вносит относительно небольшой вклад в гораздо боль-
шую величину энтальпии атомизации АНЛ (разд. 2.4.2.5) (для
Se 225 кДж/моль), и поэтому этой разностью можно пре-
небречь. Реакции разупорядочения можно также записать для
образования точечных дефектов в кристаллическом селене. Ре-
акции (2.129г, д) отвечают образованию центров D+ и D~ в ре-
зультате взаимодействия ковалентных связей с ближайшими
соседними атомами селена. Реакция (2.129е) описывает образо-
вание идеального разбавленного твердого раствора (затвердев-
шего в виде стекла). W учитывает кулоновское притяжение.
В первом приближении А//д ~ Ёь ~ Еь и W « 0 (для 1018 цен-
тров D+ и D~ в селене, диэлектрическая проницаемость кото-
рого ег = 6,15, в случае беспорядочного распределения W =
= 0,03 эВ), тогда в правой части уравнения (2.129ж) остается
198 Глава 2
лишь разность 1а— Еац, что точно соответствует энергии кор-
реляции Ulp [уравнение (2.116)].
Применяя закон действующих масс по аналогии с (2.102),
получаем
[ОЧ [О~] = njexp(-Uip/RT); [d4, [D-]-«no (2.130)
где По = jVap/Л! — концентрация структурных групп, участвую-
щих в рассматриваемом равновесии. Например, для равновесия,
записанного на рис. 2.41, эти группы имеют состав SeSe2/2
(в этом случае М — молярная масса данного структурного эле-
мента). Пренебрегая энтропийным членом (AG° = АН0 = U lp),
можно получить, что концентрация дефектных центров в основ-
ном определяется положительным значением энергии корреля-
ции Ulp. При температуре T = Tg в структуре стекла фикси-
руется равновесная концентрация таких центров.
Проведение количественной оценки Ulp по уравнению
(2.129) невозможно, так как величины различных вкладов
в Ulp по отдельности не доступны. В основу уравнения
(2.129з) положено слишком грубое приближение. Если принять
для свободных атомов селена /^ = 9,752 эВ, а £^// = 2,0201 эВ,
то разность обеих величин при ег = 6,15 (селен) выражается
слишком малым числом, и, следовательно, концентрация де-
фектов окажется слишком малой. Поэтому расчет ведут от
обратного—из экспериментально найденной концентрации цен-
тров находят U lp- Если в стеклообразном селене [£)+] =
= [Z)~] = 1018 см-3, то из уравнения (2.130) Ulp ~ 0,5 эВ.
Для AS2S3 и As2Se3 Ulp = 0,7 4- 0,8 эВ [405].
Кастнер, Адлер и Фриче [397] назвали центры D+ и D~
парами с переменной валентностью (Valence Alternation Pairs).
В модели КАФ халькогенидные атомы обозначаются буквой С,
элементы главной подгруппы V группы—Р, элементы
IV группы — Т. Координационное число приводится в виде ин-
декса справа внизу, заряд (+, — или 0) — справа вверху.
Для халькогенов, например для селена, можно предложить
схему возможных связывающих состояний (рис. 2.58). Такая
схема учитывает строение валентной электронной оболочки дан-
ного атома и взаимодействие р-орбиталей с соответствующими
валентными электронами атомов селена в 1-й координационной
сфере. Еъ — сопровождающее образование ковалентных связей
понижение энергии. Такая энергия выделяется при заполнении
связывающих состояний р-электронами данного атома (второй
электрон принадлежит партнеру по связи). А — энергия, обус-
ловленная асимметрическим расщеплением терма (влияние так
называемого интеграла перекрывания [406]). Ulp— энергия
корреляции, которая входит в виде положительного вклада
в терм состояния С i (центр D~). Данная модель отличается от
подхода Стрита и Мотта [396], в модели которых, согласно
Аморфное и стеклообразное состояние
199
Конфигурация
центра
Заполнение р орбитали
центральный * атом
атом & лиганда
Энергия данной
конфигурации
Д
-2£Ь
-s\
-З£д
Рис. 2.58. Схема энергетических термов для описания возможных связываю-
щих состояний атомов Se в конденсированном агрегатном состоянии [397].
— 2 + Д
рекомендациям Андерсона, учитывалась поляризация окружаю-
щих данный дефектный центр участков решетки [уравнение
(2.125)]. Кроме этого, в модели КАФ принимается, что взаимо-
действие между центром С Г и центром С? настолько сильное,
что в центре Сз образуются три одинаковые ковалентные связи.
При таком взаимодействии учитывается энергетический вклад
Р2/2а, ответственный за отрицательное значение энергии корре-
ляции электронов.
Для реакции (2.129ж) можно записать
2С^С3( +C^ + ULP (2.131а)
Поскольку ULp положительна, равновесие сильно сдвинуто
влево. Реакция, (2.128в) соответствующая реакции
2С° - С+ + Cf + (ULP - 2Д) (2.1316)
напротив, экзотермическая, так как 2Д>(Др.
200 Глава 2
Из равновесий (2.1316) и (2.128в) следует, что если раз-
ность Ulp — 2Д отрицательна, то в основном происходит по-
парное заполнение электронных состояний, так как в этом слу-
чае константа равновесия >1. Если Ulp> 2Д, то равновесие
сдвинуто в сторону образования однократно занятых центров
с неспаренным спином. Эффективная энергия корреляции элек-
тронов U3$$ = Ulp — 2Д определяет положение равновесия
в системе. Из этого следует соотношение между энергией поля-
ризации, равной, согласно Андерсону, ЕР = [}2/2а [уравнение
(2.125^]', и возникающей под действием так называемого инте-
грала перекрывания асимметрией расщепления на связываю-
щую и антисвязывающую молекулярную орбиталь; эти соотно-
шения отражают взаимосвязь между положительно заряженным
центром и парой р-электронов Д = 02/4а.
Согласно проведенным оценкам [405, 407], в селенсодержа-
щих стеклах Д « 1 эВ. В халькогенидных стеклах центры Сз
практически не встречаются, так как 2Д > Ulp', следовательно,
^эфф < 0. Таким образом хорошо объясняется отсутствие пара-
магнитных свойств у халькогенидных стекол.
В заключение необходимо также отметить, что кулоновское
взаимодействие между акцепторами и донорами может привести
в кристаллических полупроводниках к образованию связанных
пар [408]. Равновесия (2.131а) и (2.1316) при этом могут быть
еще больше сдвинуты в сторону существования положительно
и отрицательно заряженных дефектных центров. Правда, энтро-
пия смешения в области температуры замораживания противо-
действует этому процессу. По оценке [405] можно считать, что
концентрация статистически распределенных пар с переменной
валентностью сравнима с концентрацией сближенных пар
с переменной валентностью. В энергетическом балансе
равновесий (2.131а) и (2.1316) можно дополнительно учесть
кулоновское притяжение центров W [уравнение (2.129)]. Во
всяком случае, отсутствие зависимости спектров люминесцен-
ции от воздействия внешних электрических и магнитных полей,
которое было обнаружено экспериментально, объясняют сущест-
вованием таких дипольных центров.
2.5.2.3. Ковалентные и полярные связывающие состояния
в конденсированных химических системах
и окислительно-восстановительное
диспропорционирование
Галогены и межгалогенные соединения. Концепция образования некото-
рого количества положительно и отрицательно заряженных ионов в рас-
плавленном селене (рис. 2.41 и 2.57) основана на многочисленных эксперимен-
тальных данных и теоретически обоснована в предыдущих разделах. Этот
Аморфное и стеклообразное состояние
201
процесс можно описать также с помощью реакции самоокисления — самовос-
становления
л^еж -— п/6$е4^сольв “Ь n/eSe2> сольв (2.132)
Индекс «сольв» указывает на взаимодействие с атомами растворителя
(в данном случае с тем же селеном) с образованием ковалентных связей.
Аналогичное равновесие уже давно известно, например, из результатов
измерения ионной проводимости расплавов иода [409, 410]. Иод плавится
в атмосфере собственных паров при 386,7 К; удельная проводимость при
413 К составляет 1,7-10—4 (Ом-см)-1. Это заставляет предположить, что
в расплаве имеет место равновесие диссоциации [411]
312, ж — сольв + Ь, сольв + LP (2-133)
Более слабые межмолекулярные силы приводят в данном случае к доста-
точной подвижности. Таким образом, уже из ионной природы проводимости
следует вывод о наличии дефектных центров. Из-за отчетливо выраженных
электрофильных свойств ион 1+ склонен к сольватации. Его координацион-
ное число равно 2. Известны, например, многочисленные соединения, в со-
став которых входит комплексный катион [1(руг)2]+. Протекание процесса
самоокисления — самовосстановления в расплавленном иоде обусловлено,
как и в случае расплавленного селена, сильным донорно-акцепторным взаи-
модействием. Катионы 1з+ существуют, например, в солеобразном соедине-
нии ЬДАЮЦ]- [412], а также в растворе безводной H2SO4. По данным КР
и спектроскопии ядерного квадрупольного резонанса установлено, что этот
катион имеет нелинейную конфигурацию с валентным углом 97°. Связанный
с этим выигрыш энергии более чем достаточен, чтобы компенсировать меж-
электронное отталкивание Ui.p, которое возникает в результате гетеролити-
ческого разрыва связи в ионе 1~. В этом случае также (7Эфф<0. Следует
заметить, что ион I- образует хотя и слабую, но весьма стабильную связь
с молекулой 12. Отрицательный заряд в образующейся частице распреде-
ляется здесь между тремя атомами. Поэтому Ui.p значительно понижена,
и равновесие сильнее сдвинуто вправо, чем в случае селена (Ulp<_Ulp).
В ту же сторону сдвигает равновесие взаимодействие между ионом I-
и молекулой Ь, способствуя образованию полярных связывающих состояний.
В химии водных растворов ион 1^" уже давно известен. Его существование
ставит вопрос, насколько подобные образования участвуют в стабилизации
центров D~ или центров в расплаве Se. До сих пор такими взаимо-
действиями пренебрегали (разд. 2.5.2.2). Ион линеен. В этой частице
всего имеется 16 р-электронов и 3 пары s-электронов. Это означает, что
с учетом двух ковалентных связей на каждый атом приходится более
8 электронов. Из 9 р-орбиталей три используются на образование двух о-
связей. Не привлекая к рассмотрению d-орбитали, можно предположить
существование линейной трехцентровой четырехэлектронной связи (рис.
2.56,6), что ведет к некоторому понижению терма на величину Еь . Остав-
шиеся 6-2=12 электронов занимают в атомах иода попарно несвязываю-
щис р-орбитали. Из четырех электронов, занимающих молекулярные орби-
тали, лишь два связывающие. Поэтому связи слабее, чем в нормальной двух-
центровой двухэлектронной связи (Еь <.ЕЬ). Энергия связи в молекуле иода
Еь и небольшая энергия взаимодействия, которая высвобождается при обра-
зовании иона I-, в сумме дают энергию ‘2ЕЬ обеих связей трехцеитровой
четырехэлсктронной системы, которая понижается на величину ULP. Оба
электрона на несвязывающей орбитали в основном находятся (рис. 2.59)
202 Глава 2
Конфигурация
центра
Заполнение р-ор8итали Энергия данной,
конфигурации
~ЕЬ
~ЕЬ+ А
1Р
^ь-<р
4ЦЦЦМ1-
в
8~ 8+ 6 '
I — I — I
d
Рис. 2.59. Схема энергетических термов для описания связывающих состояний
галогенов [398].
Такие соотношения можно ожидать на основании существующего самоокисления—само-
восстановления в расплаве иода.
в области обоих концевых атомов I. Поэтому эти атомы характеризуются
некоторой избыточной электронной плотностью (распределение отрицатель-
ного заряда отсюда ULP <ZUlp) Описание валентных связей в ионе
требует преобразования валентной оболочки центрального атома иода
в гибридную $рМ-орбиталь. При этом в химическую связь включается пара
s-электронов, что кажется весьма проблематичным из-за относительно боль-
шого различия энергий по сравнению с р-орбиталями у тяжелых атомов.
Для пар с переменной валентностью (VAP) в галогенах действительна
такая же схема энергетических термов (рис. 2.59) [398], что и в случае,
изображенном на рис. 2.58.
Положение равновесия реакции (2.133) описывается уравнением
3/У? ZZ Н% + Я2- + U'LP - 2ЕЬ + Еь (2.134)
По сравнению с однократно занятым состоянием 1$ = Н2 образование поло-
Аморфное и стеклообразное состояние 203
жительно и отрицательно заряженных центров, как и в случае селена, об-
легченно из-за отрицательной величины эффективной энергии корреляции
+ + (2.135)
Центры МОГУТ также соединяться между собой таким образом, что
из однократно занятых антисвязывающих состояний возникает расщепленная
полоса (рис. 2.59, в). Обусловленное этим понижение энергии может на-
столько повлиять на энергетический баланс —(2Еъ' + 2\— U'lp), что со-
стояние окажется стабильным [452].
Темно-голубое соединение включения иода с кристаллическим «-декст-
рином или другие соединения, в структуре которых предполагаются каналы
с близкими размерами, содержат цепочки иода, имеющие в поперечнике
размер ~306 пм (для молекулы 12 266 им; реакция иода с крахмалом [414]).
Длина цепи зависит от типа катиона, который встраивается в решетку
вместе с нолииодидпым анионом. Совместно с катионом Cd2+, например,
существуют ионы 15_, которые взаимодействуют между собой [153]. В этом
случае состояние Н'} реализуется аналогично состоянию С3 в расплавах
теллура. Вдоль цепи иода, связь которой можно описать, используя
представления о рй?2г-гибридизации, существует металлическая проводимость.
Согласно модели одномерного электронного газа Куна [415], структура цепи
интерпретирована с учетом всех 7 валентных электронов иода. Модель заря-
женных дефектных центров позволяет дать однозначное объяснение сущест-
вованию полииодид-анионов. Образующаяся из однократно занятых анти-
связывающих молекулярных орбиталей центров //2° полоса заполнена напо-
ловину и поэтому может принимать другие электроны. Отсюда понятно, что
могут возникнуть полииодидные ионы с различной длиной цепи.
Собственная проводимость брома, согласно очень старым данным, на
несколько порядков меньше, чем проводимость иода [416]. В сильнокислых
растворителях удалось обнаружить катионы Вг3!" [417]. Катионы С1^~ сущест-
вуют в Cl3+[AsF6]~ [418].
Как видно из табл. 2.12, в ряду межгалогенпых соединений самоокисле-
ние— самовосстановление идет еще сильнее. О термодинамических свойст-
вах таких систем известно еще достаточно мало. Майер и Гутман [419]
предприняли попытки изучить явление самоокисления—самовосстановления,
исходя из донорных свойств молекул указанных жидкостей.
Таблица 2.12. Самоокисление — самовосстановление галогенов
и некоторых межгалогенных соединений [420]
Соедине- ние ГПЛ’ К Т’кип- К О, Ом-1 • СМ-1 Равновесие диссоциации
С12 172 238,5 ю-'6 (203 К) 2С12 Cl^ + CI-
Вг2 265,9 331,9 7,9 • 10-14 (267,6 К) 3Br2 Z7: Вг^ + Вг^
h 386,7 457,5 1,7 • 10-4 (413 К) 31, = 13+ + 13-
а-1С1 300,3 373 4,4 • 10-3 3ICIZ2 12С1+ + 1СГГ
IFs 282,7 371 1,6 . ю-5 2IF5 - IF+ + IFf"
BrF3 281,9 400,7 8,1 • Ю-3 (Г11Л) 3BrF3zzBr2F5!' + BrFf
Явления самоокисления—самовосстановления в полярных жидкостях и
системах с изомерией связи. Обсуждавшиеся выше явления самоокисления—
самовосстановления в галогенах и межгалогенных соединениях характерны
204 Глава 2
также для многих полярных растворителей. Ионное произведение воды —
общеизвестное понятие. Отщепление протона от молекулы воды по модели
заряженных дефектных центров облегчено тем, что ионы Н3О+ или Н9О4
весьма устойчивы. Число связей в общем балансе остается неизменным.
Поэтому образование некоторого количества ионов Н3О+ и ОН- может рас-
сматриваться как структурное разупорядочение воды.
В табл. 2.13 приведены некоторые данные для наиболее распространен-
ных полярных растворителей. Как и в случае воды, для всех растворителей
равновесие заметно сдвинуто в сторону образования недиссоциировапных
молекул. При реакции диссоциации в виде двухстадийного процесса [ура-
внения (2.128)] можно найти, что изменение свободной энергии для первой
стадии отщепления заряженной частицы, например протона, меньше, чем
изменение свободной энергии второй стадии (AGi<AG2).
Таблица 2.13. Диспропорционирование некоторых полярных растворителей
Раство- ритель Г..,К пл Т'ки.рК пл, кДж/моль е о, Ом ’1 - см-1 Реакция диссоциации
Н2О 273,1 373,1 6,01 81,7 4 • 10-8 (291 К) 21 ГО:/ 1ДН3О++О11-
NH3 239,8 195,4 5,65 22 5 • 10-11 (240 К) 2NH3- — NH4+ 4- NHT
СНзСООН 391,2 289,8 11,72 6 5 • 10-7 (298 К) 2СН3СООН П 7ZCH3CO2H.+ + + CH3COO-
HF 292,6 190,1 4,58 83,6 1,4 • 10 5 2HF zt H2F+ + + hf2-
so2 262,9 200,4 7,36 12,35 4 • IO-8 (263 К) 3SO2 /_* — S2O5+ + SO^-
Условие AG,<AG2— общий критерий протекания процессов, в которых
частицы в средних степенях окисления диспропорционируют с образованием,
например, более положительно и более отрицательно заряженных состояний.
Типичные примеры таких явлений известны в литературе, например реакции
переноса ионов.
Многие галогениды элементов V группы главной подгруппы образуют
наряду с ковалентными модификациями также полярные формы с ионными
связями. Кольдиц [321, 421] назвал такие процессы с переменой валентно-
сти изомерией связи. Так, для хлорида и бромида тропилия С7Н7Х описано
существование и жидкой ковалентной формы, и твердой солеобразной формы
[С7Н7]+Х_ [422]. РС15, например, существует в твердом состоянии в виде
[РСЦ]+(РС1б]_, а в СС14 растворяется в виде молекул с тригонально-бипира-
мидальной структурой. В таких полярных растворителях, как CH3CN, встре-
чаются одновременно как катионы [РС14]+, так и анионы [РС16]_. При взаи-
модействии РС15 или РВг5 с AsF3 получены соединения [РС14]+ [PF6]— и
[PBr4]+ [PFe]- [423, 424]. В ковалентном состоянии эти вещества известны как
PCI2F3 и РВг2Рз. PCI4F переходит самопроизвольно в ходе медленно проте-
кающего процесса в [РС14]+ [PC14F2]— [150].
Такие системы можно отнести к системам с диссоциативным равновесием,
Аморфное и стеклообразное состояние
205
поскольку число ковалентных связей в обоих состояниях остается неизменным:
2PXS ZT [РХ4] + + [РХе]- (2.136)
Связанное с гетеролитическим разрывом связи Р—X межэлектронное
отталкивание ULp действует лишь условно, так как происходит изменение
электронного состояния, и отрицательный заряд распределяется на весь
большой комплексный анион, который стабилизируется за счет перехода
в октаэдрическую конфигурацию:
Одновременно стабилизируется и катион путем внутренней перегруппировки
в тетраэдрическую частицу. Без сомнения, кулоновское взаимодействие и
связанная с превращением (2.136) энтальпия кристаллизации совместно обу-
словливают в этих случаях большую энергетическую выгодность полярного
состояния связей. Таким образом, равновесие смещено в сторону образова-
ния заряженных центров и отвечающих им структурных групп.
Влияние растворителя иногда оказывается вполне достаточным (как
и для РС15), чтобы сместить положение равновесия в обратную сторону.
При этом с особой ясностью видно, что образование полярных и ковалент-
ных форм в химических системах зависит от типа межмолекулярного взаи-
модействия.
Если по аналогии с уравнением (2.128) записать двухстадийный процесс
для превращения PCls в [РС14]+ [РС16]“ при добавлении CH3CN к раствору
хлорида фосфора (V) в СС14:
a) [PClfi]-zr PC1S + Cl- + AGi
6) PCI5z=[PC14]+ + C!- + AG2
b) 2PC15 - [PCI4]+ + [PC16]- - (AGj - AG2) (2.137)
то по аналогии с потенциалом ионизации в уравнении (2.128) AG!>AG2
при условии самопроизвольного протекания превращения (константа равно-
весия реакции А>1).
Окислительно-восстановительное диспропорционирование. Реакции дис-
пропорционирования характеризуются самопроизвольным переходом веще-
ства из средней степени окисления (Redox) в более низкую (Red) и более
высокую (Ох) степень окисления:
a) Red zz Redox + п{е + AG®
б) Redox zi Ox + n2e + AG®
в) 2Redox Red + Ox + (n2 — n{)e — (AG?- AG®) (2.138)
Условие AG® >AG® означает для стандартных электрохимических по-
тенциалов, что Eoi>Eo2- Если это требование выполняется, реакция диспро-
порционирования протекает самопроизвольно. В случае когда £02>£оь
стабильна средняя степень окисления Redox. Это можно подтвердить с по-
мощью взятых из справочника значений стандартных электрохимических
потенциалов. Например, для ЕеТв/Ее2+ Е0} = — 0,44 В, а для Fe2+/Fe3+ £02 =
= +0,77 В или для SnTB/Sn2+ EOi =—0,24 В, а для Sn27Sn44- Е02=+0,15 В.
Железо и олово в степени окисления +2 устойчивы в водных растворах;
диспропорционирование не идет.
206 Глава 2
Для меди характерно другое соотношение: для CuTB/Cu+ Z?oi = O,52 В,
а для Cu+/Cu2+ £02=0,18 В. Ионы Си+ в водном растворе неустойчивы. Они
диспропорционируют с осаждением металлической меди и образованием
ионов Си2+. Для стабилизации средней степени окисления в данном случае
можно использовать комплексообразование, например связывать CiT в виде
[CuCN)4]3-. Ионы Cu(I) имеют заполненную валентную электронную обо-
лочку d10, а ионы Си (II)—один нсспарснный электрон. Согласно концепции
Андерсона, можно было бы ожидать противоположных эффектов. На самом
деле этот пример подчеркивает роль химического окружения — поляризуе-
мость среды — для возникновения определенных электронных состояний
в локализованных центрах.
Другие типичные примеры можно найти в химии одновалентной ртути
или соединений марганца средних степеней окисления (например, диспропор-
ционирование МпО42- на МпО2 и МпОч-). Можно также рассмотреть оки-
слительно-восстановительные свойства Н2О2.
Тесная внутренняя связь диспропорционирования с самоокислением —
самовосстановлением халькогенов и галогенов проступает особенно явно,
если рассмотреть процесс диспропорционирования галогена, например хлора,
в воде:
a) CI-г? '/2С12 + е £01 = 1,39В
б) 72С12 +2Н2О —HOCI+ Н3О+ + е £02= 1,59В
в) С12 + 2Н2О — НОС1 + С1- + Н3О+ (2.139)
Зная разность £Oi — £о2=О,О59 lg 7G, можно найти константу равновесия
суммарной реакции (2.139) (К.,= 10-3’39). Таким образом, С12 находится
в водном растворе преимущественно в виде гидролизовапной молекулы.
Комбинируя уравнение (2.139) с уравнением диссоциации хлорноватистой
кислоты (Дп = Ю“7’25) с учетом ионного произведения воды, получаем урав-
нение реакции
С12 + 2ОН-СЮ' +С1' + Н2О (2.140)
константа равновесия которой Кь = К^Кг>К~~ = 1017’3. С12 диспропорционнруст
в щелочной среде полностью на СЮ- и С1_. При добавлении основания
поляризуемость водного раствора резко возрастает, и, таким образом, вы-
игрыш энергии благодаря стабилизации состояния С1+ путем образования
соединения с ионами кислорода сдвигает, положение равновесия в сторону
продуктов диспропорционирования хлора. Другими словами, можно сказать,
что эффективная энергия корреляции электронов принимает отрицательное
значение лишь при достаточной поляризуемости окружающей среды. В спль-
нокислых растворах направление процесса (2.139) определяет положитель-
ное значение ULP, и поэтому в этих условиях устойчива средняя степень
окисления, в данном случае незаряженное состояние.
Для одинаковых концентраций Cl2, НОС1 и С1_ из закона действующих
масс
= [НОС1] [Н3О+] [С1-] = 10_3,39
следует, например, при содержании С12 с=0,1 моль/л ([С12] = [НОС1] = [CI_] —
= 0,05 моль/л), что Ks=0,05 [Н3О+]. Согласно модели заряженных дефектных
центров, таким образом, получается, что при pH 2,1 в 0,1 М водном рас-
творе С12 концентрация различных форм хлора одинакова, £7Эфф = 0 и ULP —
= р2/2а.
Такие примеры известны и в органической химии, например диспропор-
ционирование альдегидов. Только в щелочной среде протекает реакция Кан-
ницарро с образованием спирта и соли соответствующей карбоновой кислоты.
Аморфное и стеклообразное состояние
207
Литература
1. DIN 1259, Gias. Begriffe fiir Glasarten, Juli, 1971.
2. Secrist D. R., Mackenzie J. D., Modern Aspects of the Vitreous State,
Vol. 3, 149 (1956).
3. Roy R., J. Non-Cryst. Solids, 3, 33 (1970).
4. Turnbull D., Contemp. Phys., 10, 473 (1969).
5. Jackson K- A., in: Nucleation Phenomena. Amer. Chem. Soc., Washington,
1966, S. 37.
6. Uhlmann D. R., J. Non-Cryst. Solids, 7, 337 (1972).
7. Salmahg H., Die Glasfabrikation, Springer-Verlag, Berlin Gottingen, Hei-
delberg, 1957.
8. Технология стекла/Под рсд. И. И. Китайгородского,—3-е изд.,— М., 1961.
9. Jebsen-Marwedel Н., Glastechnische Fabrikationsfehler, Springer-Verlag,
1959.
10. Tooley F. H., Handbook of Glass Manufacture, V. II, Ogden Publ. Comp.,
1961.'
11. Baynton P. L., Rawson H., Stanworth J. E., Nature (London), 178, 910
(1956).
12. Wollenhaupt R., Dissertation A., Fridrich-Schiller-Universitat, Jena, 1979.
13. Frerichs R., Phys. Rev., 78, 643 (1950).
14. Горюнова H. А., Коломиец Б. T.— Ж. техн, физ., 1955, т. 25, с. 984; Изв.
АН СССР, сер. физ., 1956, т. 20, с. 1496.
15. Hillon A. R., Jones С. Е., Brau М., Phys. Chem. Glass., 7, 105 (1966).
16. Feliz A., Linke D., Voigt B., Z. Chem., 20, 81 (1980).
17. Giintherodt H.-J., Festkorperprobleme (Adv. Solid State Phys.), XVII, 25
(1977).
18. Handrich K, Kobe S., Amorphe Ferro- und Ferrimagnelica, Akademic-
Verlag, Berlin, 1980.
19. Duwez P., Willens R. H., Trans. Met. Soc. AIME, 227, 362 (1963).
20. Pietrokowsky P., Rev. Sci. Instr., 34, 445 (1963).
21. Kavesh S., Proc. 1976 ASM Seminar on Metallic Glasses, Niagara Falls,
N. Y., 1976; ASM, Metals Park, Ohio, 1977.
22. Chen H. S., Polk D. E., J. Non-Cryst. Solids, 15, 174 (1974).
23. Chen H. S., Miller С. E., Mater. Res. Bull., 11, 49 (1976).
24. Cornet J., Rossier D., J. Non-Cryst. Solids, 12, 61 (1973).
25. De Neufville J. P., J. Non-Cryst. Solids, 8—10, 85 (1972).
26. Davies H. A., Hull J. B., J. Mater. Sci., 9, 707 (1974).
27. Sarjeant P. T., Roy R., J. Amer. Ceram. Soc., 50, 500 (1967).
28. Roy R., J. Amer. Ceram. Soc., 52, 344 (1969).
29. Dislich H., Angew. Chem., 83, 428 (1971).
30. Young L., Anodic. Oxide Films, Academic Press, London, New York, 1961.
31. Brenner A., Couch D. E., Williams E. K, J. Res. Nat. Bur. Stand., 44,
109 (1950).
32. Tauc J., Abraham A., Zallen R., Slade M., J. Non-Cryst. Solids. 4. 279
(1970).
33. Zinsmeister G., Vacuum, 16, 529 (1965); Thin Solid Films, 2, 497 (1968);
4, 363 (1979); 7, 51 (1971); Vakuum-Tcchnik, 22, 85 (1973).
34. Shevchik M. J., J. Non-Cryst. Solids, 12, 141 (1973).
35. Gutzow J., Avramov I., J. Non-Cryst. Solids, 16, 128 (1974). Thin Solid
Films, 85, 203 (1981).
36. Maissel L. I., Physics of Thin Films, New York, 3, 61 (1966).
37. Kaps Ch., Dissertation A, Univ. Jena, 1978.
38. Feltz A., Raps Ch., Thin Solid Films, 70, 117, 175 (1980).
39. Somorjai G. A., Lester J. E., Solid State Chem., 4, 1 (1967).
40. Bestul A B., Blackburn D. H., J. Chem. Phys., 33, 1274 (I960).
41. Gutzow I., Z. Phys. Chem. (Frankfurt/Main), 81, 195 (1972).
42. Kahnt H., Dissertation, Univ. Jena, 1981.
208 Глава 2
43. Davis S., Fischer M., Giessen В. C., Polk D., Proc. 3rd I nt. Conf, on Ra-
pidly Quenched Metals, Brighton, 1978; Giintherodt H.-J. et al., Electrons
in Disordered Metals and at Metallic Surfaces, Phariseau P., Gyorffy B. L.,
(Eds.), Plenum Publ. Corp., 1979.
44. Clampitt R„ Scott M. G., Aithen K. L., Gowland L., ibid.
45. Holland L., Electronic Components, 1970, 11.
46. Koenig H. R., Maissel L. I., IBM J. Res. Dev., 14, 168 (1970).
47. Stuart P. R., Vacuum, 19, 507.
48. Hollahan J. R., Bell A. T., Technique and Application of Plasma Chemistry,
Wiley, New York, 1974.
49. McTaggert F. K-, Plasma Chemistry in Electrical Discharges, Elsevier,
New York, 1967.
50. Drost H., Plasmachemie, Akademie-Verlag, Berlin, 1978.
51. Бузин В. В., Литвичко С. В., Пистолкорс В. А.— Укр. хим. жур., 1969,
т. 35, с. 987.
52. Sterling Н. F., Swann R. С. G., Solid State Electron, 8, 653 (1965).
53. Chittick R. C., Alexander J. H„ Sterling H. R., J. Electrochem. Soc., 116,
77 (1969).
54. Primak IF., Phys. Rev., HO, 1240 (1958).
55. Wittels M., Sherril F. A., Phys. Rev., 93, 1117 (1954).
56. Simon I., J. Amer. Ceram. Soc., 40, 150 (1957).
57. Parsons J. R., Phil. Mag., 12, 1159 (1965).
58. De Carli P. S., Jamieson J. C., J. Chem. Phys., 31, 1675 (1959).
59. Ainslie N. G., Mackenzie J. D., Turnbull D., J. Phys. Chem., 65, 1718
(1961).
60. Doremus R. H., Proc. Xlth Inter. Congr. Glass., Prag 1977, V. I, 263.
61. Kurkjian C. R., Krause J. T., J. Amer. Ceram. Soc., 49, 134 (1966).
62. Френкель Я. И., Кинетическая теория жидкостей.— М.: Изд-во АН СССР,
1959, с. 108, 207.
63. Glastone S., Laidler К. J., Eyring Н., Die Thcorie der Ratenprozesse,
McGraw-Hill, Book Co., New York, 1941.
64. Alfrey T., Goldfinger G., Mark H., J. AppL Phys., 14, 701 (1943).
65. Gutzow I., Konstantinow I., Kaischew R., Cominun. Dept. Chem. Bulg.
Acad. Sci., 45, 433 (1972).
66. Pryde J. A., The Liquid Statr, Hutchinson, 1966.
67. Douglas R. W., J. Non-Cryst. Solids, 14, 1 (1974).
68. Kirkwood J. G., J. Chem. Phys., 14, 180 (1946).
69. Macdonald I. R., Singer K-, Quart. Rev., 24, 238 (1970).
70. Stevels J. M., J. Non-Cryst. Solids, 26, 307 (1971).
71. Douglas R. W., Non-Cryst. Solids, Frechette V. D. (Ed.), Wiley and Sons
Inc., New York/London, 1960, 374.
72. Kauzmann W., Chem. Rev., 43, 219 (1948).
73. Ei J. H., Uhlmann D. R., J. Non-Cryst. Solids, 3, 127 (1970).
74. Cornelisse Ch. W., Visser T. J. M., Stein H. M., Stevels U. M., J. Non-
Cryst. Solids, 1, 150 (1969).
75. Rotger H., Silikattechnik, 10, 524 (1959).
76. De Bast J., Gilard P., Rech C. R., Trav. Cent. Techn. Sci. Ind. Beige du
Verre № 32 (1965), N 36 (1969).
77. De Bast J., Gilard P., Phys. Chem. Glass, 4, 117 (1963).
78. Kurkjien C. R., Phys. Chem. Glass, 4, 128 (1963).
79. Mazurin О. V., Proc. 11th Inter. Congr. Glass, Prag 1977, Survey Papers I,
129; J. Non-Cryst. Solids, 25, 129 (1977).
80. Deeg E., Glastechn. Ber., 31, 85, 124, 229 (1958).
81. Corsaro R. D., Phys. Chem. Glass, 17, 13 (1976); J. Amer. Ceram. Soc.,
59, 115 (1976).
82. Stevels J. M., Non-Cryst. Solids, 4th Inter. Conf. The Physics of Non-Cryst.
Solids 1976, Frischat G. H. (Ed.), Trans. Tech. Publication, 1977, p. 18.
83. Rotger H., Glastechn. Ber., 19, 192 (1941); J. Non-Cryst. Solids, 14, 201
(1974).
Аморфное и стеклообразное состояние
209
84. Van Ass Н. М. J. М.. Stevels J. М., J. Non-Cryst. Solids, 14, 27, 131
(1974).
85. Shelby J. E., Day D. E., J. Amer. Ceram. Soc., 52, 169 (1969); Verste-
gen E. H., Day D. E., J. Non-Cryst. Solids, 14, 142 (1974).
86. Taylor T. D., Rindone G. E., J. Non-Cryst. Solids, 14, 157 (1974).
87. Tyagi S., Lord A. E., J. Non-Cryst. Solids, 30, 273 (1979).
88. Haase R., Thermodynamik der Mischphasen, Springer Verlag, Berlin, 1956,
S. 306.
89. Simon F., Ergebn. exakt. Naturwissenschaften, 9, 222 (1930).
90. Tool A. Q., J. Amer. Ceram. Soc., 29, 240 (1946).
91. Davies R. D., Jones G. O., Adv. Physics, 2, 370 (1953).
92. Moynihan С. T., Eastreal A. J., Wilder J., Tucker J., J. Phys. Chem., 78,
2673 (1974).
93. Бартенев Г. M.— ДАН СССР, 1951, т. 76, с. 227.
94. Bartenev G. М., Sanditov D. S., Rasumovskaja /. W., Lukjanov I. A.,
Silikattechnik, 23, 155 (1972).
95. Коломиец Б. T., Любин В. M., Шило В. П.— Изв. АН СССР, Неорган.
материалы, 1971, т. 7, с. 858.
96. Macedo Р. В., Napolitano A., J. Amer. Ceram. Soc., 53, 148 (1970); J. Res.
Nat. Bur. Stand., 71Л, 231 (1967).
97. Lillie H. R., Ritland H. N., J. Amer. Ceram. Soc., 37, 466 (1954).
98. HolzmUller №., Z. Phys. Chem., 303, 163 (1954).
99. Jenckel E., Z. Phys. Chem., A184, 309 (1939).
100. Litovitz T. A., Macedo P. B., Physics of Non-Cryst. Solids, North-Holland
Publ. Co., Amsterdam, 1965, p. 229.
101. Andrade E. N., Phil. Mag., 17, 497, 698 (1934).
102. Eyring H., J. Chem. Phys., 4, 283 (1936).
103. Douglas R. W., J. Soc. Glass Tcchnol., 33, 138 (1949).
104. Stanworth J. E., J. Soc. Glass Tcchnol., 32, 20 (1948).
105. Weymann H. D., Kolloid-Z. Z. Polym., 181, 131 (1962).
106. Мюллер P. Л.— Ж. нрикл. химии, 1955, т. 28, с. 1077.
107. Мюллер Р. Л. В сб.: Стеклообразное состояние.— М.: Изд-во АН СССР,
1960, с. 61.
108. Немилое С. В.— Физ. тв. тела, 1964, т. 6, с. 1375.
109. Немилое С. В.— Успехи реологии полимеров.--М.: Химия, 1970, с. 241.
110. Kumar S., Phys. Chem. Glass, 4, 106 (1963).
111. Лущов И.— Физ. химия стекла, 1975, т. 1, с. 431.
112. Sasek L., Proc. 11th Int. Congr. Glass, Preg 1977, V. 1, 203.
113. Williams M. L., Landel R. F., Ferry J. D., J. Amer. Ceram. Soc., 77, 3701
(1955); J. Phys. Chem., 59, 95 (1955).
114. Turnbull D., Cohen M. H., J. Chem. Phys., 34, 120 (1961).
115. Feltz A., Z. anorg. allg. Chem., 412, 20 (1975).
116. Haward R. N., J. Macromol. Sci.-Revs. Macromol. Chem., C4(2), 191
(1970).
117. Ramachandrarao P., Cantor B., Cahn R. W., J. Non-Cryst. Solids, 24, 109
(1977); J. Mater. Sci.. 12, 2488 (1977).
118. Adam G., Gibbs J. H., J. Chem. Phys., 43, 139 (1965).
119. Litovitz T. A., Macedo P. B., J. Chem. Phys., 42, 245 (1965).
120. Tweer H., Simmons J. H., Macedo P. B., J. Chem. Phys., 54, 1952 (1971).
121. Simmons J. H., Macedo P. B., J. Res. Nat. Bur. Stand., A75, 175 (1971).
122. Oldekop W., Glastechn. Ber., 30, 8 (1957).
123. Holzmiiller W., Z. physik. Chem., 203, 163 (1954).
124. Boesch L., Napolitano A., Macedo P. B., J. Amer. Ceram. Soc., 53, 148
(1970).
125. Vogel H., Physik. Z„ 22, 645 (1921).
126. Tammann G., Hesse G., Z. anorg. allg. Chem., 156, 245 (1926).
127. Gulati S. T., Fontana E. H., Plummer A., Phys. Chem. Glass, 17, 114
(1976).
128. Li J. H„ Uhlmann D. R., J. Non-Cryst. Solids, 1, 339 (1969).
14 Заказ № 413
210 Глава 2
129. Висаго J. A., Dardy И. D., Corsaro R. D., J. Appl. Phys., 46, 741 (1975).
130. Demoulin C., Montrose C. J., Ost rowsky N., Phys. Rev., A9, 1740 (1974).
131. Lai С. C., Macedo P. B., Montrose C. J., J. Amer. Ceram. Soc., 58, 121
(1975).
132. Moynihan С. T., Macedo P. B., Montrose C. J., Gupta P. K-, De Bolt M. A.,
Dill J. F., Dom В. E., Drake P. W., Eastell A. J., Eltermann P. B., Moel-
ler P. P., Sasate H., Wilder J. A., Technical Report 5, N00014-75-C-8056,
NR 22-007, Vitreous State Lab., Catholic Univ. America, Washington.
133. Laberge N. L., Gupta P. K., Macedo P. B., J. Non-Cryst. Solids, 17, 61
(1975).
134. Rotger H., Silikattechnik, 20, 404 (1969).
135. Rotger H., Amorphous Materials, Proc. 3rd Int. Conf. Phys. Non-Cryst.
Solids, Douglas R. W., Ellis B. (Eds.), Sheffield, 1970, Wiley J., London,
1972, p. 125.
136. Meissner B., Meissnerova H., Sasek L., Glastechn. Ber., 46, 240 (1973).
137. Ludwig W., J. Therm. Anal., 8, 75 (1975).
138. Stephens R. B., J. Non-Cryst. Solids, 20, 75 (1976).
139. Moynihan С. T., Macedo P. B., J. Phys. Chem., 75, 3379 (1971).
140. Ludwig W., Proc. 6th Int. Conf. Thermal Analysis, Birkhauscr-Verlag,
Stuttgart, 1980, p. 293.
141. Coenen M., Glastechn. Ber., 50, 74 (1977).
142. Hirai N., Eyring H., J. Appl. Phys., 29, 810 (1958); J. Polym. Sci., 37,
51 (1959).
143. Frenkel J. L, Kinetic Theory of Liquids, Dover Publ., New York, 1955,
p. 93.
144. Simha R., Boyer R. F., J. Chem. Phys., 37, 1003 (1962).
145. Eisenberg A., Saito S., J. Chem. Phys., 45, 1673 (1966).
146. Donald 1. W., Davies H. A., J. Non-Cryst. Solids, 30, 77 (1978).
147. Eckstein B., Glastechn. Ber., 36, 323 (1963).
148. Angell C. A., J. Amer. Ceram. Soc., 51, 117 (1968).
149. Gutzow I„ Proc. 3rd Int. Conf. Phys. Non-Cryst. Solids, Douglas R. W.,
Ellis B. (Eds.), Sheffield 1970, J. Wiley, London, 1972, p. 159.
150. Kolditz L, Z. anorg. allg. Chem., 293, 147 (1957).
151. Sakka S., Mackenzie J. D., J. Non-Cryst. Solids, 6, 145 (1971).
152. Gutzow 1., Commun. Dept. Chem. Bulg. Akad. Sc., 11, 764 (1978).
153. Saenger W., Angew. Chem., 92, 343 (1980).
154. Cohen M. H., Turnbull D., Nature, 189, 131 (1961).
155. Marcus M., Turnbull D., Natur. Sc. Eng., 23, 211 (1976).
156. Hruby A., Czech. J. Phys., B22, 1187 (1972).
157. Gibbs H., Di Marzio E. A., J. Chem. Phys., 28, 373 (1958).
158. Eckstein B., Zeitschr. Krist., 128, 273 (1969); J. Non-Cryst. Solids, 4,
366 (1970).
159. Chen H. S., J. Non-Cryst. Solids, 22, 135 (1976).
160. Goldstein M., J. Chem. Phys., 64, 4767 (1976); 67, 2246 (1977).
161. Базаров И. П. Термодинамика.— М.: Физматгиз, 1961.
162. Пригожин И., Дефей Р. Химическая термодинамика: Пер. с англ.— Но-
восибирск: Наука, 1966.
163. Gupta К., Moynihan С. Т., J. Chem. Phys. Phys., 65 (10), 4136 (1976).
164. Moynihan С. T., Gupta Р. К., J. Non-Cryst. Solids, 29, 143 (1978);
Hsish H. S.-L, J. Amer. Ceram. Soc., 60, 485 (1977).
165. Jackie J., Piche L., Arnold W., Hunklinger S., J. Non-cryst. Solids, 20,
365 (1976).
166. Meissner M., Wobig D., The Physics of Selenium and Tellurium, Grosse P.
(Ed.), Springer Verlag, Berlin, 1979, 68.
167. Leadbetter A. J., Phys. Chem. Glass, 9, 1 (1968).
168. Anderson O. L., Bommel H. E., J. Amer. Ceram. Soc., 38, 125 (1955).
169. Anderson O. L., J. Phys. Chem. Solids, 12, 41 (1959).
170. Krause I. T., Kurkjian C. R., J. Amer. Ceram. Soc., 51, 226 (1968).
171. Leadbetter A. J., Wycherley К. E., Phys. Chem. Glass, 12, 41 (1971).
Аморфное и стеклообразное состояние
211
172. Zeller R С., Pohl R. О., Phys. Rev., В4, 2029 (1971).
173. Hunklinger S., Sussner H., Dransfeld К., Festkorperprobleme XVI, 267
(1976); Hunklinger S., Festkorperprobleme, XVII, 1 (1977); Dransfeld K.,
Hunklinger S., Proc. 7th Int. Conf. Amorph. Liqu. Semicond. Edinburgh
1977, University Edinburgh, p. 155.
174. Thakur R. L., de Sarkar В. K., Basak В. C., Bibliography on glass-cera-
mics (1961 —1970), Calcutta, Central Glass and Ceramic Research Institute,
1974.
175. Hench L. L., Freiman S. W. (Eds.), Advances in Nucleation and Crystal-
lisation in Glasses, Columbus (Ohio), The American Ceramic Society, 1971.
176. Porai-Koshils E. A. (Ed.), The Structure of Glass, New York, Consultants
Bureau, 1958—1973, V. 1—8.
177. Bergeron С. C., General Aspects of the Crystallisation of Glass in Intro-
duction to Glass Science, Plenum Press, New York, 1972.
178. Hammel J. J., Nucleation in Glass, in: Advances in Nucleation and Crystal-
lisation in Glasses, Columbus "(Dhio), The American Ceramic Society, 1971.
179. Prins J. A. (Ed.), Physics of Non-Cryst. Solids, Proc. Inter. Conf. Delft,
1964, North-Holland Publ. Comp., Amsterdam, 1965.
180. Turnbull D., Thermodynamics and Kinetics of Formation of the Glass
State and Initial Devitrification, Proc. Inter. Conf. Denft, 1964, North-
Holland Publ. Comp., Amsterdam, 1965.
181. Hoffmann J. D., J. Chem. Phys., 29, 1192 (1958).
182. Turnbull D., J. AppL Phys., 21, 1022 (1950).
183. Toschev S., Gulzow L, Kristall und Technik, 7, 43 (1972).
184. Strnad Z., Douglas R. W., Phys. Chem. Glass, 14, 33 (1973).
185. Hillig W. B., Proc. 6th Inter. Symp. React. Solids, Schenectedy 1968,
Wiley-Intersciencc, New York, 1968, p. 639.
186. Uhlmann D. R., in: Kinetics of Reactions in Ionic Systems, Grey T. J.,
Frechette V. D. (Eds.), Plenum Press, New York, 1969, p. 172.
187. Леко В. К., Комарова Л. А.— Изв. АН СССР, неорган. материалы, 1975,
т. 11, с. 1753.
188. Kumm К.-А., Scholze Н„ Tonind.-Ztg., 93, 332, 360 (1969).
189. Awrami М., J. Chem. Phys., 7, 1103 (1939); 8, 212 (1940); 9, 177 (1941).
190. Freiman S. W., Hench L. L., J. Amer. Ceram. Soc., 52, 111 (1969).
191. Slayter G., J. Amer. Ceram. Soc. Bull., 31, 276 (1952).
192. Prebus A. E., Michener G. W., Ind. Engng. Chem., 16, 147 (1954).
193. Шетобский В. Л.— Стекло и керамика, 1954, т. 11, с. 19.
194. Oberlies F., Naturwissenschaften, 43, 224 (1956).
195. Vogel W., Gerth К., Glastechn. Ber., 31, 15 (1958); Silikattechnik, 9, 353,
495 (1958).
196. Vogel W., Struktur. u. Kristallisation der Glaser, VEB Deutscher VerL
fur Grundstoffindustric, Leipzig, 1965.
197. Stookey S. D., Glastechn. Ber., 32K, 1 (1959).
198. Heidenreich E., Ehrl R., Vogel W., Proc. Xlth Inter. Congr. Glass, Prag,
V. 2, p. 1 (1977).
199. Cahn J. W., Acta metall., 9, p. 795 (1961); 10, 179 (1962).
200. Hillert AL, Acta metall., 9, 525 (1961).
201. Zarzycki J., Discuss. Farad. Soc., 50, 122 (1970).
202. Charles R. J., Bull. Amer. Ceram. Soc., 52, 673 (1973).
203. James P. F., J. Mater. Sci., 10, 1802 (1975).
204. Uhlmann D. R., Koibeck A. G., Phys. Chem. Glass, 17, 146 (1976).
205. Charles R. J., Wagstatt F. E., J. Amer. Ceram. Soc., 51, 146 (1968).
206. Me Dowell J. F., Beall G. H., J. Amer. Ceram. Soc., 52, 17 (1969).
207. Simmons J. H., J. Amer. Ceram. Soc., 56, 284 (1973).
208. Cahn J. W., Charles R. J., Phys. Chem. Glass, 6, 181 (1965).
209. Vreeswijk J. C. A., Grossink R. G., Stevels J. M., J. Non-Cryst. Solids, 16,
15 (1974).
210. Hammel J. J., J. Chem. Phys., 46, 2234 (1967).
14*
212 Глава 2
211. Srinivasan G. R., Tweer J., Macedo P. B., Sarkar A., Haller W.t J. Non-
Cryst. Solids, 6, 221 (1971).
212. Seward T. P., Uhlmann D. F., Turnbull D., J. Amer. Ceram. Soc., 51,
634, 278 (1968).
213. James P. F„ McMillan P. IF., Phys. Chem. Glass, 11, 59 (1970).
214. Burnett D. G., Douglas R. IF., Phys. Chem. Glass, 11, 125 (1970).
215. Elmer T. H., Carrier M. E., Carrier G. B., Korda E. J., J. Amer. Ceram.
Soc., 53, 171 (1970).
216. Reiss H., Ber. Bunsenges. Phys. Chem., 79, 943 (1975).
217. Dieizel A., Glastechn. Ber., 22, 41, 81, 212 (1948).
218. Sargeant P. T., Roy R., Mat. Res. Bull., 3, 265 (1968).
219. Gesling R. J. H., Stein H. N., Stevels J. M.. Phys. Chem. Glass, 7, 185
(1967).
220. Davies H. A., Phys. Chem. Glass, 17, 159 (1976).
221. Ruckenstein E., Him S. K., J. Chem. Soc., 72, 764 (1976).
222. Debye D„ Ann. Phys., 46, 809 (1915).
223. Klug H. P., Alexander L. E., X-ray Diffraction Procedures, J. Wiley, New
York, 1974.
224. Warren В. E., X-ray Diffraction Reeding, Addison-Wesley, 1969.
225. Krebs H., Steffen R., Z. anorg. allg. Chem., 327, 224 (1964).
226. Zickert K., Steil H., Gelk G., Herms G., Becherer G., Kristall und Technik,
14, 1147 (1979).
227. Mozzi R. L„ Warren В. E., J. Appl. Cryst., 2, 164 (1969).
228. Bell R. J., Dean P., Phil. Mag., 25, 1381 (1972).
229. Warren В. E„ Krutter H., Morningstar O., J. Amer. Ceram. Soc., 19, 202
(1936).
230. Zachariasen IF. H., J. Amer. Chem. Soc., 54, 3841 (1932).
231. Keating D. T., J. Appl. Phys., 39, 923 (1963).
232. Enderby J. E., North D. M., Egelstaff P. A., Phil. Mag., 14, 961 (1966).
233. Sadoc J. F., Dixmier J., Mater. Sc. Eng., 23, 187 (1976).
234. Wagner C. N. J., J. Non-Cryst. Solids, 31, 1 (1978).
235. Steeb S.. Lamparter P., Z. Metellk., 70, 660 (1979).
236. Sayers D. E., Lytle F. IF., Stern E. A., J. Non-Cryst. Solids, 8—10, 401
(1972).
237. Stern E. A., Phys. Rev,. B10, 3027 (1974).
238. Lytle F. IF., Sayers D. E., Stern E. A., Phys. Rev., Bl 1, 4825, 4836 (1975).
239. Sayers D. E„ Lytle F. IF., Stern E. A., Proc. 5th Int. Conf. Amorph. Li-
quid Sernicond., Garmisch-Partenkirchen, 1973, Taylor-Francis, London,
1974, p. 403.
240. Hayes T. M., J. Non-Cryst. Solids, 31, 57 (1978).
241. Wong J., Angell C. A., Appl. Spectr. Rev., 4, 155 (1971).
242. Bock J, Su G.-J., J. Amer. Ceram. Soc., 53, 69 (1970).
243. Simon J., Modern Aspects of the Vitreous State, Butterworth, London,
1960, p. 138.
244. Bell R. J., Bird N. F., Deen P„ J. Ph vs. C (Solids State Phvs.). 1. 299
(1968).
245. Lyon R. J. P., Nature, 196, 266 (1962).
246. Zarzycki J., Proc. Int. Conf. Phys. Non-Cryst. Solids, Delft, 1964,
Prins J. A. (Ed.), North-Holland Publ. Comp., Amsterdam, 1965, 525.
247. Amrhein E.-M., Glastechn. Ber., 43, 1 (1970).
248. Ferraro J. R., Manghnani M. H., Quattrochi A., Phys. Chem. Glass, 13,
116 (1972).
249. Hass M., J. Phys. Chem. Solids, 31, 415 (1970).
250. Накамото К. Инфракрасные спектры неорганических координационных
соединений: Пер. с англ.— М.: Мир, 1966.
251. Austin 1. G., Garbett Е. С., Phil. Mag., 23, 17 (1971).
252. Lucovcky G., Martin R., J. Non-Cryst. Solids, 8—10, 185 (1972).
253. Lucovsky G., Proc. 5th. Int. Conf. Amorph. Liquid Sernicond., Garmisch-
Partenkirchen 1973, Taylor-Francis, London, 1974, V. 2, p. 1099.
Аморфное и стеклообразное состояние
213
254. Tarte Р., Proc. Int. Conf. Phys. Non-Cryst. Solids, Delft, 1964, Prins J. A.
(Ed.), North-Holland Publ. Comp., Amsterdam, 1965, 549.
255. Neuroth N., Glastechn. Ber., 41, 243 (1968).
256. Konijnendijk W. L., Philips Res. Repts., Suppl. 1 (1975); Konijnendijk W. L.,
Stevels J. M., J. Non-Cryst. Solids, 18, 307 (1975).
257. Griscom D. L., Mat. Sci. Res., 12, 11 (1977).
258. Galeener F. L., Phys. Rev., В19, 4292 (1979); Galeener F. L., Sen P. N.,
Phys. Rev., 17, 1928 (1978).
259. Siegbahn K. et al., ESCA (Atomic, Molecular and Solid State Structure
Studies by Means of Electron Spectroscopy), Uppsala, 1967; ESCA Ap-
plied to Free Molecules, Amsterdam, London, 1969.
260. Nordling C., Angew. Chem., 84, 144 (1972).
261. Meisel A., Leonhardt G., Z. Chem., 18, 294 (1978).
262. Shirley D. A., Electron Spectroscopy, North-Holland Publ. Comp., Am-
sterdam, 1972.
263. Hatt B. A., Savage J. A., Bosnell J. R., Waghorne R. M., Proc. 5th Int.
Conf. Amorph. Liquid Seinicond., Garmisch-Partenkirchen 1973, Taylor-
Francis, London, 1974, v. 1, p. 412.
264. Bray P. J, in: Glaschemie, Vogel W. (Hrsg.), VEB Verlag Deutscher
Verlag fur Grundstoifindustrie, Leipzig, 1979; The Physics of Non-Cryst.
Solids, 4th Inter. Conf. Clausthal-Zellerfeid 1976, Fi ischatt G. 11. (Ed.),
Trans. Tech. Publications, 65.
265. Bray P. J., Edwards J. 0., O’Keefe J. G., Ross V. F., Tatsuzaki L, J.
Chem. Phys., 35, 435 (1961).
266. Bray P. J., O’Keefe J. G., Phys. Chem. Glass, 4, 37 (1963).
267. Kriz H. M., Bray P. J., J. Non-Cryst. Solids, 6, 27 (1971).
268. Kim K. S., Bray P. J., Phys. Chem. Glass, 15, 47 (1974).
269. Hoebbel D., Garzo G., Engelhardt G., Janeke H., Franke P., Wieker W.,
Z. anorg. allg. Chem., 424, 115 (1976).
270. Engelhardt G., Altenburg W., Hoebbel D., Wieker IT., Z. anorg. allg.
Chem., 437, 249 (1977).
271. Hoebbel D„ Garzo C., Engelhardt G., Z. anorg. allg. Chem., 450, 5 (1979).
272. Feltz A., Schlenzig E., Arnold D., Z. anorg. allg. Chem., 403, 243 (1974).
273. Taneje S. P., Kimball C. W., Sheffer J. С., цит по C. A., 1974, 43320 n.
274. Wong J., Angell C. A., Appl. Spectr. Rev., 4, 97 (1970).
275. Herber R. IL, Progr. Inorg. Chem., 8, 1 (1967).
276. Juza R.. Seidel H., Tiedermann J., Angew. Chem., 78, 41 (1966).
277. Gitter M., Vogel W., Proc. 1st Otto-Schott-Kolloquium, Jena, 1978, Wiss.
Zeitschr. Friedrich-Schiller-Universitat, Mathem.-Naturwiss. Reihe, 28, 307
(1979).
278. Thilo E., Schulz G., Wichmann E.-M., Z. anorg. allg. Chem., 272, 182
(1953).
279. van Wazer J. R., J. Amer. Chem. Soc., 72, 644 (1952).
280. Westman A. E. R., Beatty R., J. Amer. Ceram. Soc., 49, 63 (1966).
281. Thilo E., Schiilke LL, Z; anorg. allg. Chem., 341, 293 (1965).
282. Wieker W., Z. Elektrochem., Ber. Bunsenges. Phys. Chem., 64, 1047 (1960).
283. Masson C. R., Proc. Xlth Int. Congr. Glass., Prag. 1977, Survey Papers,
V. 1, p. 3.
284. Balta P., Balta E., Ceausescu V., J. Non-Cryst. Solids, 20, 323 (1976).
285. Lentz C. IT., Inorg. Chem., 3, 574 (1964).
286. Kolb К. E., Hansen K. W., J. Amer. Ceram. Soc., 48, 439 (1965).
287. Masson C. R„ J. Chem. Soc., Al970, 2683: 1971, 686.
288. Wieker W., Z. anorg. allg. Chem., 360, 307 (1968).
289. Wieker W., Hoebbel D., Z. anorg. allg. Chem., 366, 139 (1969).
290. Thilo E., Wieker W., Stade H., Z. anorg. allg. Chem., 340, 261 (1965).
291. Feltz A., Pfaff G., Z. anorg. allg. Chem., 442, 41 (1978).
292. Feltz A., Pfaff G., Z. anorg. allg. Chem., 467, 211 (1980).
293. Briegleb G., Z. phys. Chem., A144, 321 (1929).
214 Глава 2
294. Randall J. T., Rooksby H. P., Cooper В. S., Z. Krist., 75, 196 (1930).
295. Malaurent J. C., Dixmier J., Proc. Conf. Structure of Non-Cryst. Mat.,
Cambridge, 1976, p. 49.
296. Robertson J., Phil. Mag., 34, 13 (1976).
297. Cartz L., Z. Kristallogr., 120, 241 (1964).
298. Konnerl J., Karie J., Trans. АСА, 10, 29 (1974).
299. Seward T. P„ Uhlmann D. R., Proc. 3rd Intr. Conf. Phys. Non-Cryst.
Solids, 1970, J. Wiley, London, 1972, p. 327.
300. Polk D. E., J. Non-Cryst. Solids, 5, 365 (1971).
301. Greaves G. W., Davis E. A., Phil. Mag., 29, 1201 (1974).
302. Polk D. E., Acta Metall., 20, 485 (1972).
303. Moss S. C„ Proc. 5th Int. Conf. Amorph. Liquid Semicond., Garmisch-
Partenkirchen 1973, Taylor-Francis, London, 1974, V. 1, p. 17.
304. Connell G. A. N„ Lucovsky G., J. Non-Cryst. Solids, 31, 123 (1978).
305. Phillips J. C., J. Non-Cryst. Solids, 34, 153 (1979).
306. Tilton L. W., J. Res. Nat. Bur. Stand., 59, 139 (1957).
307. Robinson IL A., J. Phys. Chem. Solids, 26, 209 (1965).
308. Bernal J. D„ Nature, 183, 141 (1959); 185, 68 (1960).
309. Stell H., Dissertation A, Wilh.-Pieck-Univ., Rostock, 1979.
310. Grigorovici R., Thin Solid Films, 9, 1 (1971).
311. Goodman С. H. L., Proc. Conf. Structure of Non-Cryst. Mat., Cambridge,
1976, p. 197.
312. Suga H., Seki S., J. Non-Cryst. Solids, 16, 171 (1974).
313. Sugisaki M., Suga H., Seki S., Bull. Chem. Soc. Japan, 41, 2591 (1968);
Physics of Ice, Plenum Press, New York, 1969, p. 329.
314. Stnekal A., Glastechn. Ber., 22, 278 (1949).
315. Stanworth J. E., J. Soc. Glass Tcchnol., 32, 366 (1948).
316. Sun K. IL, J. Amer. Ceram. Soc., 30, 277 (1947).
317. Dietzel A., Z. Elektrochem., 48, 9 (1942).
318. Winter A., J. Amer. Ceram. Soc., 40, 54 (1957).
319. Lucovsky G., Phil. Mag., 39, 513 (1979).
320. lander G., Die Chemie in wasserahnlichen Losungsmitteln, Springer-Ver-
lag, 1949, S. 192—208.
321. Kolditz L., Advanc. Inorg. Chem. Radiochem., 7, 1 (1965).
322. Мюллер P. Л. Химия твердого тела/Под ред. Борисовой С. У.— Л.: ЛГУ,
1965, с. 9.
323. Pauling L., Die Natur dor Chemischen Bindung, Verlag Chemie; Weinheim,
1962, S. 94.
324. Урусов В. С. Энергетическая кристаллохимия.— М.: Наука, 1975.
325. Honig R. Е., Kramer D. A., RCA-Rev., 30, 285 (1969).
326. Кондратьев В. II. Энергия химической связи, потенциалы ионизации и
сродство к электрону.— М.: Наука, 1974.
327. Linke D., Proc. Xlth Int. Congr. Glass., Prag 1977, V. I, p. 149.
328. Duffy J. A., Infram M. D., J. Non-Cryst Solids, 21, 373 (1976).
329. Trap H. L L., Stevels I. M„ Glastechn. Вег.. 32K, 31 (1959).
330. Hildebrand I. H., Scott R. L., The Solubility of Nonelectrolytes, Dover
Publ. Inc., New York, 1964.
331. Mott B. W., Phil. Mag., 8, Ser. 2, 259 (1957).
332. Жузе В. П., Шелеч А. И.— Физ. тв. тела, 1965, т. 7, с. 1775.
333. Meyers М. В., Felty Е. I., J. Electrochem. Soc., 117, 818 (1970).
334. Blachnik R., Hablitationsschrift, Univ. Glausthal, BRD, 1972.
335. Linke D., Proc. Int. Conf. Amorph. Semicond., Balatonfiired 1976, Verl.
Ungar. Akad. Wiss., Budapest, 1977.
336. Feltz A., Kunzel B., Linke L, Z. anorg. allg. Chem., 437, 237 (1977).
337. Krebs H., Langner D., Z. anorg. allg. Chem., 334, 37 (1964).
338. Rao В. V., Phys. Chem. Glass., 4, 22 (1963).
339. Feltz A., Linke L, Z. anorg. allg. Chem., 415, 156 (1975).
340. Немилое С. B.— Ж- прикл. химия, 1964, т. 37, с. 293; Изв. АН СССР, сер.
физ., 1964, т. 28, с. 1283.
Аморфное и стеклообразное состояние
215
341. Sasek L., Proc. Xlth Int. Congr. Glass., Prag 1977, V. 1, p. 203.
342. Аппен А. А., Глушкова В. Б., Каялова С. С.— Изв. АН СССР, неорган.
материалы, 1965, т. 1, с. 576.
343. Linke D., Dissertation В, Friedrich-Schiller-Univ., Jene, 1978.
344. Linke D., Proc. Amorph. Sernicond., Pardubice, 1978, p. 57.
345. Dembovskij S. A., Phys. Chem. Glass., 10, 73 (1969).
346. Berkes J. S., Non-Cryst. Solids, 4th Int. Conf. The Physics of Non-Cryst.
Solids, 1976, Frischat G. H. (Ed.), Thans Tech. Publications, 1977, p. 405.
347. Scholze H., Gias, Natur., Struktur und Eigenschaften, Springer-Verlag,
Berlin, Heiderberg, New York, 1977. S. 147.
348. Schultz P. C., Smyth IL T., Amorphous Materials, Proc. 3rd Inter. Conf.
Phys. Non-Cryst. Solids, Sheffield 1970, Wilcy-lnterscience, London, 1972,
p. 453.
349. Павлова Г. А., Аматуни А. Н,— Изв. АН СССР, Неорган. материалы,
1976, т. 11, с. 1443.
350. Feltz A., Senf L., Z. anorg. allg. Chem., 444, 195 (1978).
351. Linke D., Eberhardt G., Proc. 1st. Otto-Schott-Kolloquium, Jena 1978,
Wiss. Zeitschr. Friedrich-Schiller-Universitat, Mathem.-Naturwiss. Reihc,
28, 339 (1979).
352. Soga N., Kunugi M., Ota R., J. Phys. Chem. Solids, 34, 2143 (1977).
353. Thompson J. C., Bailey К. E., J. Non-Cryst., Solids, 27, 161 (1978).
354. Farley J. AL, Saunders G. A., J. Non-Cryst. Solids, 18, 417 (1975).
355. Kozlovakaja E. L, The Structure of Glass., V. 1—8, Porai-Koshits E. A.
(Ed.), Consultants Bureau, New York, 1958, V. 2, p. 299.
356. Soga N., Yamanaka H., Hisamoto C., Kunugi AL, J. Non-Cryst. Solids,
22, 67 (1976).
357. Fanderlik 1., Skrivan M., The Structure of Glass, V. 1—8, Porai-Ko-
shits E. A. (Ed.), Consultants Bureau, New York, 1958, V .6, p. 145.
358. Dutoil M., Chen H. S., Appl. Phys. Lett., 23, 357 (1973).
359. Aladdin R., Masumoto T., Mater. Sci. Eng., 9, 153 (1972).
360. Leamy H. J., Chen H. S„ Wang T. T., Metallurg. Trans., 3, 699 (1972).
361. Chen H. S., Krause J. T., Coleman E., J. Non-Cryst. Solids, 18, 157
(1975).
362. Spenke E., Elektron. Halbleiter, Springer-Verlag, 1965.
363. Wigner E.y Trans. Faraday Soc., 34, 678 (1938).
364. Mott N. F.y Proc. Phys. Soc. (London), A62, 416 (1949).
365. Mott N. F., Canad. J. Phys., 34, 1356 (1956).
366. Mott N. F., Rev. Mod. Phys., 40, 677 (1968).
367. Mott N. F., Phil. Mag., 6, 287 (1961).
368. Mott N. F., Advan. Phys., 16, 49 (1967).
369. Mott N. F., Phil. Mag., 20, 1 (1969).
370. Me Whan D. B., Reimeika J. P., Phys. Rev., B2, 3734 (1970).
371. Mott N. F., Twose W. D., Advan. Phys., 10, 107 (1961).
372. Alexander M. N., Holcomb D. F., Rev. Mod. Phys., 40, 815 (1968).
373. Mott N. F., Contemp. Phys., 10, 125 (1969).
374. Mott N. F., Festkorperprobleme, IX, 21 (1969).
375. Anderson P. W., Phys. Rev., 109, 1492 (1958).
376. Anderson P. W., Rev. Modern. Phys., 50, 191 (1978).
377. Mott N. F., Phil. Mag., 34, 1101 (1976).
378. Mott N. F.. Davis E. A., Phil. Mag., 17, 1269 (1968).
379. Gerthsen P., Kauer E., Reik H. G., Festkorperprobleme, IX, 1 (1966).
380. Bosman A. L, Daal H. J., Advan. Phys., 19, 1 (1970).
381. Mott N. F., Gurney R. W., Electronic Processes in Ionic Crystals, Claren-
don Press, Oxford, 1940, p. 116.
382. Mott N. F., Phil. Mag., 19, 835 (1969).
383. Mott N. F., Allgainer R. S., Physica Status Solidi, 21, 343 (1967).
384. Cohen M. H.y J. Non-Cryst. Solids, 2, 432 (1970); 4, 391 (1970).
385. Gubanov A. Quantum Electron Theory of Amorphous Conductors, Con-
sultants Bureau, New York, 1965.
216 Глава 2
386. Banyai L., Physique des Semiconducteurs, Hulin M. (Ed.): Dunod, Paris,
417.
387. Cohen M., Fritzsche H., Ovshinsky S., Phys. Rev. Lett., 22, 1065 (1969).
388. Spear W. E., Proc. 5th Int. Conf. Amorph. Liquid Scmicond., Garmish-
Partenkirchen 1973, Taylor-Fracis, London, 1974, V. 1, p. 1.
389. Brcnig W., Proc. 5th int. Conf. Amorph. Liquid Semicond., Garmish-Per-
tenkirchen 1973, Taylor-Fracis, London, 1974, V. 1, p. 31.
390. Fritzsche H., Proc. 7th Int. Conf. Amorph. Liquid Semicond., Edinburgh,
1977, University Edinburgh, p. 3.
391. Андриеш A. M., Коломиец Б. T.— Физ. тв. тела, 1965, т. 6, с. 2652.
392. Fritzsche Н., in: Electronic and Structural Properties of Amorphous Semi-
conductors, P. G. Le Comber, J. Mort, Academic Press, London, 1973,
p. 56.
393. Agarwal S. G., Phys. Rev., B7, 685 (1973).
394. Bishop S. G., Strom U., Taylor P. C., Phys. Rev. Lett., 34, 1346 (1975);
.. 36, 543 (1976).
395/ Anderson P. W., Phys. Rev. Lett., 34, 953 (1975).
396. Street R. A., Mott N. F„ Phys. Rev. Lett., 35, 1293 (1975).
397. Kastner M., Adler D., Fritzsche H., Phys. Rev. Lett., 37, 1504 (1976).
398. Feltz A., J. Non-Cryst. Solids, 45, 355 (1981).
399Г. Yotfa E. L, Adler D., Phys. Rev., В12, 2260 (1975).
400. Adler D., Yoffa E. F., Phys. Rev. Lett., 36, 1197 (1976); Canad. J. Chem.,
55, 1920 (1977).
401. Mott N. F., Davis E. A., Phil. Mag., 32, 961 (1975).
402. Mott N. F., Street R. A., Phil. Mag., 36, 33 (1977).
403. Mott N. F., Rev. Phys. Appl., 12, 619 (1977).
404. Mott N. F., Non-Cryst. Solids, 4th Int. Conf. The Physics of Non-Cryst.
Solids 1976, G. H. Frischat (Ed.), Trans. Tech. Publications, 1977, p. 3.
405. Kastner M., Fritzche H., Phil. Mag., 37, 199 (1978).
406. Cumber C. W. N., Wave Mechanics for Chemists, Heinemann Educational
Books Ltd., London, 1966.
407, Kastner M., J. Non-Cryst. Solids, 31, 223 (1978).
408. Kastner M., Hudgens S. J., Phil. Mag., 37, 665 (1978).
409. Rabinowitsch M., 1. physikal. Chem., 119, 59 (1926).
410. Plotnikov IF. A., Fialkov J. A., Tschalig IF. P., Z. physikal. Chem., Al72,
304 (1935).
411. Andrieth L. F., Kleinberg J. L., Non-Aqueous Solvents, J. Wiley, New York,
1953, p. 251.
412. Merryman D. J., Edwards P. A., Corbett J. D., McCarley R. E., Chem.
Comm., 1972, 779.
413. Gillespie R. J., Morton M., Sowa J. M., Advan. Raman. Spectr., 1, 530
(1972).
414. Cramer F., Chem. Ber., 84, 855 (1951); Naturwisscnschaften, 39, 256
(1952).
415. Kuhn H., Z. Electrochemie, 53, 165 (1949).
416. Exner F., Ann. d. Phys., 15, 412 (1882).
417. Gillespie R., Passmore J., Adv. Inorg. Chem. Radiochem., 17, 54 (1975).
418. Gillespie R. J.. Morton M. J., Inorg. Chem., 9, 811 (1970).
419. Mayer V., Gutmann V., Advanc. Inorg. Chem. Radiochem., 17, 189 (1975).
420. Landold-Bornstein, Eigenschaftcn dcr Materie in ihren Aggregatzustanden,
7. Teil, Elektr. Eigenschaften II, Springer-Verlag, 1960.
421. Kolditz L., Halogen Chemistry, V. 2, V. Gutmann (Ed.), Academic Press,
London. 1967, p. 115.
422. von Eggers W., Krauch H., Angew. Chem., 68, 661 (1956).
423. Kolditz L., Z. anorg. allg. Chem., 284, 144 (1956).
424. Kolditz L., Feltz A., Z. anorg. allg. Chem., 293, 155 (1957).
425. Fritzsche H., Phys. Rev., 115, 386 (1959); 119, 1899 (1960); 125, 1552
(1962).
Аморфное и стеклообразное состояние 217
426. Suzuki S., Sakamuya Н., Imaoka М., Yogyo-Kyokai-Shi, 79, 299 (1971).
427. Furukawa Т., White W. В., J. Non-Cryst. Solids, 38, 39, 87 (1980).
428. Puyane R., James P. F„ Rawson H., J. Non-Cryst. Solids, 41, 105 (1980).
429. Sakka S., Kamiya K., J. Non-Cryst. Solids, 42, 403 (1980).
430. Martin A., T. anorg. allg. Chem., в печати.
431. Schrader R., Dusdorf IT., Kristall u. Technik, 1, 59 (1966).
432. Schrader R., Wissing R„ Kobsch H., Z. anorg. allg. Chem., 305, 191
(1965).
433. Voland U., Schrader R., Schneider H., Z. anorg. allg. Chem., 368, 317
(1969).
434. Schrader R., Kutzer H.-J., Hoffmann B., Tonind.-Ztg., 94, 410 (1970).
435. Knape G., Rolling W., Schumann H., Silikattechnik, 18, 388 (1967).
436. Хайнике Г. Трибохимия: Пер. с англ.— М.: Мир, 1987.
437. Feltz A., Fink G., неопубликованные данные.
438. Mashimo Т., Nishii К., Some Т., Sawaoka A., Phys. Chem. Minerals, 5,
367 (1980).
439. Bartenev G. M., Scheglova N. N., J. Non-Cryst. Solids, 37, 285 (1980).
440. Kanno H., J. Non-Cryst. Solids, 44, 409 (1981).
441. Kanno H., J. Non-Cryst. Solids, 37, 203 (1980).
442. Lesikar A. V., Moynihan С. T., J. Chem. Phys., 73, 1932 (1980).
443. Yinnon H., Uhlmann D. R., J. Non-Cryst. Solids, 44, 37 (1981).
444. Nassau K-, J. Non-Cryst. Solids, 42, 423 (1980).
445. Avramov J., Gutzow I., Materials Chemistry, 5, 315 (1980).
446. Greaves G. N., Proc. IX. Inter. Conf. Amorph. Liqu. Semicond., Grenoble,
1981, J. de Physique Collogue C4, Suppl., № 10, 42, 225 (1981).
447. Bruckner R., Chun H.-U., Goretzki H., Sammet M., J. Non-Cryst. Solids,
42, 49 (1980).
448. Perez J., Duperray P., Lefevre D., J. Non-Cryst. Solids, 44, 113 (1981).
449. Wright A. C., J. Non-Cryst. Solids, 40, 325 (1980).
450. Phillips J. C., J. Non-Cryst. Solids, 43, 37 (1981).
451. Wright A. C., Etherington G., Desa J. A. E., Sinclair R. N., Con-
nell G. A. N., Mikkelsen J. C., J. Non-Cryst. Solids, 49, 63 (1982).
452. Perlstein J. H., Angew. Chem., 89, 534 (1977).
453. Gutzov J., Contemp. Phys., 21, 121 (1980).
454. Messmer R. P„ Wong J., J. Non-Cryst. Solids, 45, 1 (1981).
Дополнительная литература *
Аппен А. А. Химия стекла.— Л.: Химия, 1974, 351 с.
Андреев Н. С., Мазурин О. В., Порай-Кошиц Е. А. и др. Явления ликва-
ции в стеклах.— Л.: Наука, 1974, 219 с.
Бонч-Бруевич В. Л., Звягин И. FL, Кайпер Р. и др. Электронная теория не-
упорядоченных полупроводников.— М.: Наука, 1981, 383 с.
Бонч-Бруевич В. Л. Вопросы электронной теории неупорядоченных полупро-
водников.— Усп. физ. наук, 1983, т. 140, № 4, с. 583—637.
Беленький В. 3. Геометрико-вероятностные модели кристаллизации: феноме-
нологический подход.— М.: Наука, 1980, 84 с.
Губанов А. М. Квантово-электронная теория аморфных проводников,— М.-Л.:
Изд-во АН СССР, 1963, 250 с.
Dembovsky S. A., Chechetkina Е. A. Defects and glass formation.— Non.-Cryst.
Sol., 1984, 64, p. 95—111.
Dembovsky S. A., Chechetkina E. A. To the classification of amorphous solids
and the calculation of chitical cooling rates for amorphous metals and glasses.
Mat. Res. Bull., 1982, v. 17, N 12, p. 1531 — 1538.
Закис Ю. P. Дефекты в стеклообразном состоянии вещества.— Рига: Зинантне,
1984, 203 с.
Представлена редактором С. А. Дембовским.
218 Глава 2
Займан Дж. Модели беспорядка. Теоретическая физика однородно неупоря-
доченных систем,— М.: Мир, 1982, 591 с.
Skuj L. N.. Silin A. R. Model for the non-bridging oxygen center in fised
silica.— Phys. Stat. Sol.(a), 1982, 70, p. 43—49.
Ковнеристый Ю. К., Осипов Э. К., Трофимова Е. А. Физико-химические
основы создания аморфных металлических сплавов.— М.: Наука, 1983, 145 с.
Кобеко П. П. Аморфные вещества,— М.-Л.: Изд-во АН СССР, 1952, с. 431.
Кокорина В. Ф. Влияние химической связи на стеклообразование и свойства
стекол.— В кн.: Стеклообразное состояние. Л.: Наука, 1971, с. 87—92.
Клингер М. И., Карпов В. Т. Автолокализация электронных пар в неупоря-
доченных системах.— ЖЭТФ, 1982, т. 82, № 5, с. 1687—1703.
Клингер М. И. Автолокализнроваппые состояния электронов и дырок.— Усп.
физ. наук, 1985, т. 146, № 1, с. 105—142.
Калинина А. М., Фокин В. М., Филлипович В. Н. Индукционный период за-
рождения кристаллов в стекле Li2O-2SiO2 и его температурная зависимость.—
Физ. и хим. стекла. 1977, т. 3, № 2, с. 122—129.
Колмогоров А. Н. К статистической теории кристаллизации металлов.— Изв.
АН СССР, сер. матем. 1937, № 3, с. 355.
Лившиц И. М., Г редескул А. С., Пастур Л. А.— Введение в теорию неупо-
рядоченных систем.— М.: Наука, 1982, 358 с.
Мухин Е. Я., Гуткина И. Г. Кристаллизация стекол и методы ее предупре-
ждения.— М.: Оборонгиз, 1960, 125 с.
Мазурин О. В. Стеклование и стабилизация неорганических стекол.— Л.:
Наука, 1978, 62 с.
Милюков Е. М., Касымова С. С. Нссмешивающиеся расплавы и стекла.—
Ташкент: Фан, 1981, 176 с.
Мазурин О. В., Стрельцина М. В., Швайко-Швайковская Т. П. Свойства сте-
кол и стеклообразующих расплавов: Справочник в 4-х т./Под рсд. Неми-
лова С. В. Т. I, 1973, 444 с.; т. II, 1975, 632, с.; Т. III, ч. 1, 1977, 586 с.;
Т. III. ч. 2, 1979, 486 с; Т. IV, ч. 1, 1980, 462 с; Т. IV, ч. 2, 1981, 375 с.
Немилое С. В. Энергетика и свойства стеклообразных и кристаллических
тел.— В кн.: Стеклообразное состояние, Л.: Наука, 1971, с. 10—15.
Немилое С. В. Взаимосвязь между скоростью распространения звука, массой
и энергией химического взаимодействия.— ДАН СССР, 1968, т. 181, с. 1427—
1429.
Немилое С. В. Валентно-конфигурационная теория вязкого течения пере-
охлажденных жидкостей и ее экспериментальное обоснование. Физ. и хим.
стекла, 1978, т. 4,— № 2, с. 129—148.
Немилое С В. Развитие представлений о характере внутренних изменений
систем при переходе стекло—жидкость.— Физ. и хим. стекла, 1980, т. 6, № 3,
с. 257—268.
Пинскер Г. 3. Определение решеточных закономерностей в аморфной струк-
туре.— Физ. и хим. стекла, 1980, т. 6, № 5, с. 521—524.
Пинскер Г. 3. О правильном расположении атомов в области ближнего по-
рядка в аморфных телах.— В кн.: Физические методы исследования неоргани-
ческих материалов, 1981, с. 117—131.
Порай-Кошиц Е. А. Развитие структурных исследований стеклообразных ве-
ществ в течение последнего пятилетия.— В со.: Стеклообразное состояние,
Л.: Наука, 1983, с. 5--10.
Попов И. А. Новая модель дефектов в халькогенидных стеклообразных полу-
проводниках.— Письма Ж.ЭТФ, 1980, т. 31, № 8, с. 437—460.
Попов Н. А. Квазимолекулярные дефекты и их связь с вязкостью полупро-
водниковых халькодегидных расплавов,—физ. и техн, полупроводников, 1982,
т. 16, с. 344—347.
Регель А. Р., Глазов В. М. Периодический закон п физические свойства элек-
тронных расплавов.— М.: Наука, 1978, 307 с.
Rawson И. Properties and application of glass. Ams. Oxf., N-Y, 1980, p. 316.
Силинь A. P. Структура и механизм образования простейших собственных
дефектов в стеклообразном кремнеземе.— В кн.: Электронные процессы и
Аморфное и стеклообразное состояние 219
структура дефектов в стеклообразующих системах Рига: Изд-во ЛГУ, 1982,
с. 3—10.
Салли И. В. Кристаллизация при сверхбольших скоростях охлаждения.—
Киев: Наукова думка, 1972, 136 с.
Сандитов Д. С., Бартенев Г. М. Физические свойства неупорядоченных струк-
тур.— Новосибирск: Наука, 1982, 259 с.
Тарасов В. В. Проблемы физики стекла.— М.: Стройиздат, 1979, 255 с.
Фокин В. М., Филипович В. Н., Калинина А. М. Стационарная и нестацио-
нарные скорости зарождения кристаллов 2Na2OCaO-3SiO2 в стекле того же
состава.— Физ. и хим. стекла, 1980, т. 6, № 2, с. 148—152.
Фокин В. М., Филипович В. Н., Калинина А. М. Исследование влияния пред-
варительной термообработки стекла Li2O-2SiO2 на зарождение в нем кри-
сталлов.— Физ. и хим. стекла, 1977, т. 3, № 2, с. 129—136.
Чернов А. А. Физика кристаллизации.— М.: Знание, 1983, 64 с.
Шульц М. М. О химическом строении стеклообразующих расплавов и сте-
кол.— В сб.: Стеклообразное состояние, 1983, Л.: Наука, с. 5—10.
Эфрос А. Л. Физика и геометрия беспорядка.— М.: Наука, 1982, 175 с.
Шульц М. М., Борисова И. В., Кожина Е. Л. Термодинамики тугоплавких
оксидных систем в свете критериев устойчивости равновесия.— В кн.: Химия
силикатов и оксидов. Л.: Наука, 1982, с. 3.
Paul A. Chemistry of Glasses. London: Chapman and Hall, 1982, 293 p.
Шульц M. M. Кислотно-основная концепция в применении к оксидным рас-
плавам и стеклам и учение Д. И. Менделеева о стеклообразном состоянии.—
Физ. и хим. стекла 1984, т. 10, № 2, с. 129—138.
3. Аморфные
и стеклообразные
системы
Как было показано в предыдущей главе, твердые тела, су-
ществующие в стеклообразном или аморфном состоянии, харак-
теризуются особой структурой и типом связи. Поэтому понятно,
что при образовании таких веществ может не быть построена
кристаллическая решетка. Элементы периодической системы су-
ществуют в виде многочисленных некристаллических модифи-
каций. При взаимодействии элементов образуются стеклообраз-
ные соединения, что ведет к огромному разнообразию систем
с непериодически организованной структурой. В этой главе из-
ложены основные взаимосвязи между макроскопическими свой-
ствами и структурой этих веществ.
К обсуждению некристаллических модификаций элементов
привлекаются данные по металлическим стеклам, которые по
составу представляют собой сплавы или интерметаллические
фазы. Далее следуют данные по двойным и тройным соедине-
ниям в соответствующих стеклообразующих системах. Когда
в качестве стеклообразователей выступают оксиды и фториды,
то не наблюдается больших различий между стеклообразовате-
лем и производными от него стеклами. При обсуждении этих
систем приводятся ссылки на обзорные статьи и монографии
(см. литературу к гл. 1 [3-17] ).
3.1. Некристаллические модификации элементов.
Сплавы
Давно известны аморфные модификации некоторых элемен-
тов, например бора, фосфора, мышьяка, сурьмы и селена. Зна-
чительно позже была исследована связь между их структурой
и свойствами. Применение новых препаративных методов [кон-
денсация паров на сильно охлажденных подложках в глубоком
вакууме, катодное распыление или очень быстрая закалка рас-
плава (разд. 2.1.1.3, 2.1.1.4 и 2.1.3)] привело к значительному
расширению известного набора элементов, которые могут су-
ществовать в стеклообразном или аморфном состоянии.
При осаждении паров металлов на подложках, охлаждаемых
жидким гелием, получены в стеклообразном состоянии Bi, Ga
Аморфные и стеклообразные системы
221
и сплав олова с 10 % Си [1]. Zn, Hf, V, Nb, Та, Сг, Mo, W, Re,
Fe, Ni и Al в стеклообразном состоянии были получены позже.
На рис. 3.1 [2] показано, какие из элементов периодической
1 — испарение и конденсация иа охлаждаемых подложках; 2 — химическое осаждение;
3 — осаждение при очень большой скорости охлаждения; 4 — электролитическое осажде-
ние; 5 — плазменное испарение; 6 — в чистом виде.
системы известны к настоящему времени в стеклообразном или
аморфном состоянии. Поскольку даже от ничтожных (следо-
вых) количеств примесей часто зависит образование аморфных
веществ (иногда эти примеси вызывают зародышеобразование
и инициируют кристаллизацию), всегда нужно обращать особое
внимание на чистоту веществ при таких работах.
3.1.1. Металлические стекла *
После того как был предложен метод разбрызгивания рас-
плава с мгновенным его охлаждением (splat quenching) (см.
литературу к гл. 1 [27] ) и этот метод был использован для
получения стеклообразного AusSi, было разработано очень
много непрерывных процессов получения металлических стекол
в виде проволоки и тонкой ленты. Основные методы приведены
на рис. 2.3.
Сообщается (см. литературу к гл. 2 [220]) о получении
в присутствии следовых количеств кислорода стеклообразной
никелевой фольги при охлаждении со скоростью до 1010 К/с.
Для большинства изученных сплавов эвтектического состава ис-
пользовались скорости охлаждения от 106 К/с, в то время как
* См. примечание к разд. 2.1.1.3.
222 Глава 3
для получения стекол в системах Pd—Си—Si, Pd—Ni—Р или
Pt—Ni—Р оказалась достаточной даже скорость охлаждения
102 К/с [4]. При этом можно получить компактные образцы
диаметром 1—3 мм.
В некоторых обзорных работах [2, 5—8] (см. также литера-
туру к гл. 2 [17, 18] ) сообщается о большом разнообразии раз-
личных сочетаний элементов, расплавы которых при таких
условиях охлаждения образуют стекла или аморфные вещества
(при испарении и конденсации на охлаждаемых подложках,
а также при катодном распылении). В монографии [784] из-
ложены новейшие результаты исследований аморфных ме-
таллов.
3.1.1.1. Классификация металлических стекол
Большая группа сплавов, затвердевающих с образованием
стекол, включает сплавы переходных металлов (Т) с такими
элементами (М), как Be, В, С, Si, Ge, Sn, Р, As, Sb или Те;
состав этих сплавов ле-
Рис. 3.2. Низкоплавкие эвтектики в двой-
ных системах Au—Si (/), Со—Р (2) и
Pd—Si (3).
ведены составы таких стекол наряду
жит в области от Т5М до
Т3М, в большинстве слу-
чаев вблизи Т4М. Наи-
большую стеклообразую-
щую способность, как
правило, имеют сплавы,
состав которых соответст-
вует на диаграмме со-
стояния низкоплавкой
эвтектике, например в си-
стемах Au—Si и Pd—Si —
это составы Au8iSii9
(7пл = 643К) и Pd82Sii8
(Тпл=943 К) [9] или
в системе Fe—В — со-
став Fe83Bi7 (ГПл =
= 1400 К) [10].
На рис. 3.2 приводят-
ся фазовые диаграммы
систем Au—Si, Со—Р
и Pd—Si. В табл. 3.2 при-
с тройными и четверными
системами, произведенными от двойных эвтектик. В тройных си-
стемах по эвтектическим линиям часто образуются ряды заме-
щения, а для четверных стекол возможности изменения состава
еще больше. Известны также многочисленные примеры, когда
стеклообразование не связано с эвтектическими составами. При
определенных условиях охлаждения конгруэнтно плавящиеся
Аморфные и стеклообразные системы
223
Таблица 3.1. Металлические стекла а
Двойные стекла Тройные стекла Четверные стекла
Au81SiI9 (290) (гл. 1 [27]) Au73Ge27 (290) (гл. 2 [146]) Pd82SiI8 (670) [14] Pd8iPi9 (615) (гл. 2 [146]) Ni8OP2o (607) (гл. 2 [361]) Ni7gP25 [20] Со7аР25 [22] Fe83P17 (520) [23] Fe83B17 (630) [23] (674) [24] Ti80Si20 (867) (гл. 2 [146]) Ni60Nb40 (970) (гл. 2 [146]) Cu57Zr43 (740) [23] Cu5oZr5O (680) Od67Co33 (250) [26] Au77Ge14Si9 (295) [15] Pd77Au5SiI8 (650) [16] Pd77Cu6Si17 (635) [17] Pd80Si16Sb3 (650) [18] Pd6oFe2OP2o (617) (гл. 2 [361]) Ni80Si8B12 (670) Ni40Pd40P2()(587) (гл.2) [361]) Ni22Pd62Si16 (690) [19] Ni75Si8Bi7 (777) (гл. 2 [146]) Ni7sPi5Bio [21] Co75Si15B10 (780) (гл. 2 [146]) Fe83B2Pi5 (600) [23] Fe81Cr2Bi7 (759) Fe80P13C7 (700) [23, 25] Fe79SiioBn (813) Fe75PI5CI0 [27] Ti4oNi4oSi2o (>1000) Ti62Ni30B8 (775) Ti72Fei2Sie (850) Ni75P16B6AI3 (691 [23]) Ni68Cr(oSi 10B12 (772) NigeNbioSiioB^ (849) Fc75P15C6A14 (713 [26]) Fe75P16B6Al3 (693 [26]) Fe40Ni40PI4B6 (663 [26])
а В круглых скобках приведены значения Тй в кельвинах.
интерметаллические фазы также могут переходить в стеклооб-
разное состояние. Правда, в этих случаях температуры плав-
ления этих фаз значительно ниже, чем у чистых компонентов.
К этой группе относятся металлические стекла, образованные
переходными металлами побочных подгрупп I и VIII групп пе-
риодической системы, с другими элементами, например Ni3Nb2,
Си32г2 или Rh9Taii [11].
Еще одну группу составляют металлические стекла, в состав
которых входят актиноиды и лантаноиды, например и7Еез,
П7Сг3 [8], Gd2Co [12] или La7Al3 и Се7А1з [8].
224 Глава 3
Стеклообразное состояние известно и для металлических
сплавов на основе элементов главных подгрупп, например
Ca7Mg3 [8].
Для описания стеклообразующей способности в системах на
основе сплавов были привлечены различные теоретические кон-
цепции (см. гл. 2) [5, 6]. Приведенная температура стеклования
Tg = TglT-nn [разд. 2.2.2.4, уравнение (2.45)] для металлических
стекол лежит в интервале 0,45 < 7^ < 0,67; оценка скорости
охлаждения из теории зародышеобразования для этих условий
дает величину до 106 К/с. В группе стекол типа Т4М повышение
концентрации М в узких пределах, при котором сохраняется
стеклообразование, приводит к незначительному возрастанию
Tg. При этом образуется определенный ближний порядок,
что обусловливает возрастание вязкости. Понижение темпе-
ратуры ликвидуса ТПл приводит к реализации условия 7g->0,45.
Стеклообразующая способность наиболее велика в области
низкотемпературной эвтектики (см. разд. 2.2.2.5 и 2.3.2). Ее
можно также выразить через критическую скорость охлажде-
ния ^крит-
Такие системы характеризуются сильным взаимодействием
между атомами Т и М, относительно высокими значениями эн-
тальпии плавления и низкими температурами кристаллизации.
Стеклообразующую способность можно также характеризовать
как разность между величиной температуры ликвидуса, рас-
считанной для идеальной смеси, и экспериментально измерен-
ной температурой [разд. 2.2.2.5, уравнение (2.51)]; она также
может применяться для поиска сочетаний элементов, от кото-
рых можно ожидать стеклообразования. При соблюдении реаль-
ных пределов применимости этого способа прогнозирования
были обнаружены многие металлические стекла [13] (см. также
литературу к гл. 2 [146] ).
3.1.1.2. Структура металлических стекол
Попытки представить структуру металлических стекол на
основе кристаллоподобных областей, связанных между собой
через неупорядоченные промежуточные слои не привели к со-
гласию с результатами экспериментов по рентгеновской дифрак-
ции [28].
Плотность металлических стекол, как правило, только на
2 % меньше, чем для кристаллических фаз. Кривые радиаль-
ного распределения в значительной степени схожи с кривыми
радиального распределения для расплавов.
На рис. 3.3 приведены функции радиального распределения
для аморфного и жидкого кобальта, а также для стеклообраз-
ного сплава Со78Р22 [29]. Как следует из рис. 3.4, кривые ра-
Аморфные и стеклообразные системы
225
диального распределения для металлических стекол различных
составов в значительной степени совпадают (см. литературу
к гл. 2 [234] ), что позволяет сделать вывод о близости структур
и типов связи.
Были опробованы модели, основанные на представлениях
о беспорядочной плотной упаковке твердых шаров [30, 31]. Они
базируются на структурной модели одноатомной жидкости Бер-
налла (см. литературу к
гл. 2 [308]). Среднее ко-
ординационое число ле-
жит в интервале от 11 до
13. Пример подобной мо-
дели — модель Беннета
(см. литературу к гл. 2
[18]), согласно которой
к исходному кластеру из
трех атомов присоединя-
ются остальные атомы
таким способом, что в об-
разовавшейся структуре
соблюдается тройной кон-
такт [32]. В поверхност-
ном слое предпочитают-
ся позиции, расположен-
ные ближе к центру тя-
жести кластера.
Жидкость Берналла
может быть построена из
структурных полиэдров
Рис. 3.3. Кривые радиального распределения
(КРР) для аморфного Со (/), аморфного
Со78Р22 (2) и расплава Со (3) [34].
(рис. 3.5), причем груп-
пировки в—д встречают-
ся только в аморфных
структурах. Доля таких
группировок составляет 21 %. Согласно работам [33] (см. также
литературу к гл. 2 [302]), атомы М легко встраиваются в пустоты
таких полиэдров, тем самым стабилизируя структуру расплава,
что объясняет частое образование относительно низкоплавкой
эвтектики вблизи состава Т4М и повышенную склонность к стек-
лообразованию в этой области [29]. Опыты по рентгеновской
дифракции показали, что статистическое распределение расстоя-
ний в парах Т—Т, М—М и Т—М не соблюдается. Приведен-
ные на рис. 2.31 парциальные функции радиального распреде-
ления показывают, что окружение атомов Со и Р в аморфном
Со4Р различно (см. литературу к гл. 2 [233]). Относительно
большие расстояния Р—Р не подтверждают существования со-
ответствующих связей. Координационное число атомов Р со-
ставляет ~9.
15 Заказ № 413
Рис. 3.4. Кривые радиального распределения металлических стекол различ-
ного состава (см. литературу к гл. 2 [234]).
Рис. 3.5. Структурные полиэдры берналловой жидкости.
Аморфные и стеклообразные системы 227
Эффективные радиусы М-компонентов оказываются слишком
велики для заполнения тетраэдрических пустот, хотя они и со-
гласуются с экспериментальными результатами, полученными
с помощью кривой радиального распределения [34]. Стекла
в системе Ni—Nb проявляют исключительную термостойкость
(табл. 3.1). Компоненты этой системы относятся к типичным
металлам, и область стеклообразования простирается от 23
до 66 мол. % Nb [35]. Этот пример показывает, что нельзя
объяснить стеклообразующую способность только заполнением
пустот в структуре металлического расплава атомами М. Ока-
залось, что для стекол системы Pd—Si—Си парциальные атом-
ные объемы Si и Си зависят от состава [36]. Чем меньше за-
полнение пустот в неправильной упаковке атомов металличе-
ского расплава, тем более вероятно образование связей
и обусловленное этим формирование определенных областей
ближнего порядка, необходимое для образования структуры сте-
кол. Новое объяснение структуры таких стекол приводится в ра-
боте [37] (см. также (784) )•
Исследование стеклообразного Pd4Ge методом протяженной
тонкой структуры рентгеновского поглощения (ПТСРП) пока-
зало, что существуют упорядоченные области, в которых коор-
динационное число атомов Ge составляет от 8 до 9; эта вели-
чина хорошо соответствует большим размерам пустот в жидко-
сти Берналла, но не соответствует концепции заполнения
тетраэдрических пустот [38]. Расстояния Ge—Ge в этой системе
также отклоняются от статистических значений.
Представления теории химической связи еще только разви-
ваются для прогнозирования предпочтительного взаимодействия
в системах Т—М. Нагель и Таук [39] обратили внимание, что
в оптимальных для стеклообразования составов минимумы
электронной плотности и энергии Ферми совпадают, т. е. изо-
тропное жидкое состояние обладает более низкой энергией,
чем кристаллическое. Сильное понижение температуры плавле-
ния эвтектического состава могло бы служить выражением осо-
бой устойчивости структуры жидкости, а переход к области
составов с повышенной кристаллизационной способностью ин-
терпретируется подобно тому, как последовательность фаз
Юма—Розери в соответствующих системах.
3.1.1.3. Свойства металлических стекол
Электропроводность металлов, как известно, ограничивается
процессами рассеяния на дефектах кристаллической решетки
и на фононах. В кристаллах преобладает последний процесс.
Благодаря большой подвижности электронов в области энергии
Ферми [разд. 2.5.1.1, уравнение (2.111)] средняя длина свобод-
ного пробега носителей заряда может составлять 10~6—10-1 см,
15*
228 Глава 3
а при температурах жидкого гелия даже достигать 10 см. Элек-
тропроводность металлов последовательно возрастает при пони-
жении температуры. Из-за нарушения периодичности строения
кристаллической решетки и большой степени неупорядоченно-
сти в металлических стеклах средняя длина свободного пробега
носителей составляет только 10-7 см. Электропроводность ме-
таллических стекол ниже, чем у металлов, и температурный
коэффициент для них также значительно ниже.
Близость расплавов металлов и стеклообразного состояния
проявляется также при исследовании их электрических свойств
[8, 40]. Иногда в зависимости от состава можно наблюдать
положительный или отрицательный температурный коэффициент
электропроводности (и сопротивления) (например, в системе
Pd0>5_o,5xNio,5-хРх) [41]. Такое же, как у металлических стекол,
сильное уменьшение длины свободного пробега встречается
только у сверхпроводников II рода. Известны сверхпроводящие
стекла с температурой перехода в сверхпроводящее состояние
~4,7 К [42, 43]. Коэффициент Холла таких стекол в 100 раз
больше, чем у металлов в кристаллическом состоянии из-за
пониженного сопротивления.
Губанов теоретически предсказал наличие ферромагнетизма
у аморфных тел [44], что экспериментально подтвердилось
на стеклах системы Au—Со [145]. Ранее сообщалось о ферро-
магнетизме в аморфных пленках системы Со—Р, полученных
электролитическим путем (см. литературу к гл. 2 [31] ). В стек-
лах, образованных с участием редкоземельных элементов, на-
личие магнитного момента связано со строением глубоко лежа-
щих 4/-уровней; их магнитные свойства, следовательно, не
зависят от структуры. Для переходных металлов З^-ряда маг-
нитные свойства значительно меняются при переходе от стекло-
образного к кристаллическому состоянию. В стеклах на основе
З^-переходных металлов коэрцитивная сила мала; если в их
составе присутствуют редкоземельные металлы с заполненными
f-орбиталями, то для многих свойств стекол наблюдается зна-
чительный анизотропный эффект.
Малая коэрцитивная сила, подвижность границ между доме-
нами, высокая магнитная проницаемость и низкая электропро-
водность в сочетании с высокой коррозионной стойкостью — все
эти положительные качества выдвигают металлические стекла
(FegoBao, Fe82Bi2Si6, Fe^PuBe, Регэ^ддРнВбЗцг или РеувМогВго)
как перспективные материалы для использования в электро-
технике (например, в качестве материала сердечников транс-
форматоров) [46, 47]. Особенно высокая коррозионная стойкость
характерна для стеклообразных сплавов Fe и Ni, содержащих
до 8 % Сг, Мо или W (см. литературу к гл. 2 [48] ). Часто
в аморфных пленках наблюдается магнитная анизотропия в на-
правлении большего размера; так, в аморфных пленках сплавов
Аморфные и стеклообразные системы
229
редкоземельных элементов с переходными металлами обра-
зуются так называемые магнитные пузыри, имеющие прибли-
зительно одинаковые размеры ( — 10 нм) [49]. Использование
топких пленок таких материалов в электронно-вычислитель-
ной технике позволяет достигать плотности записи информации
109 бит/см2.
Для металлических стекол прочность на растяжение состав-
ляет 7ю—7б теоретического значения, что значительно больше,
чем у кристаллических металлов, близких по составу [50]. Во
многих работах [19, 23, 51, 52] (см. также литературу к гл. 2
[22, 361] ) приводятся данные по механическим свойствам
и упругим постоянным металлических стекол.
В отличие от оксидных и халькогенидных стекол металличе-
ские стекла в области Т <zTg проявляют некоторую пластич-
ность, но в противоположность кристаллическим металлам по-
сле механической обработки их твердость не возрастает. Не все
металлические стекла ковки, они часто хрупки при Т намного
ниже температуры кристаллизации. Структурная релаксация
(разд. 2.2.1.2) влияет также на многие другие свойства металли-
ческих стекол; она исследована в работах [53, 54].
При температурах выше температуры стеклования, как пра-
вило, металлические стекла претерпевают быструю кристалли-
зацию. В системах, где образуются металлические стекла,
также изучается кинетика кристаллизации в связи с фазовым
разделением [16, 55, 56].
3.1.2. Элементы — неметаллы в аморфном состоянии
3.1.2.1. Бор
Модификации. Чистый бор в виде простого вещества впер-
вые получен в 50-х годах. В нормальных условиях термодина-
мически стабильна ^-ромбоэдрическая модификация бора. Она
выделяется при кристаллизации из расплава. Крупные моно-
кристаллы такой модификации бора (длина 30 см, толщина
1 см) получены в 1958 г. методом зонной плавки; их чистота
составила 99,99 % [57].
а-Ромбоэдрическую модификацию получают путем пиролиза
чистых галогенидов бора в атмосфере Н2 на раскаленной танта-
ловой проволоке в интервале температуры 1173—1773 К [58,
59]. Желто-красные кристаллы a-модификации обнаруживаются
в виде вкраплений в слое аморфного (стекловидного и не-
прозрачного) бора черного цвета. Сообщается [60] об одновре-
менном выделении тетрагональной модификации II. Примеси
СН4, SiBr4, N2, РВг3 или А1Вг3 в газовой смеси способствуют
230 Глава 3
образованию ^-ромбоэдрической модификации, если содержание
С, Si, N, Р или А1 не превышает следовых количеств; если же
концентрация примеси велика, то образуется тетрагональная
модификация I. Эта модификация была получена в 1943 г.
Лаубснгайером и сотр. [61], однако первоначально ее иденти-
фицировали не как модификацию бора, а как соединения
включения (В^^ВгСг или (612)462^. При дальнейшем воз-
растании концентрации атомов углерода происходит переход
Рис. 3.6. Структура а-ромбоэдрической модификации бора.
а — икосаэдр В12; б — упаковка икосаэдров В,2 в ромбоэдрической элементарной ячейке;
в — трехцентровые двухэлектронные связи между икосаэдрами в слоях, перпендикуляр-
ных направлению (111), которые заканчиваются атомами В в е-позициях.
к структурам карбидов бора, например Bi3C2. В работе [62]
описано получение боридов А1В2, А1Вю, AIB12 и А1(В12)2С4.
Наряду с пиролизом метод разливания расплава на охла-
ждаемой металлической пластине также привел к получению
чистого аморфного бора [63]. Аморфный бор используется
в ядерных реакторах для поглощения нейтронов.
Плотность ^-ромбоэдрической модификации бора 2,35 г/см3,
а-ромбоэдрической 2,46 г/см3. Низкое давление пара (0,05 Па
при 2187 К), высокая температура плавления (—2450 К)
и большая энтальпия сублимации (554 кДж/моль), а также
значительная твердость (И по шкале Мосса; ср. для ал-
маза 15) обусловлены прочными химическими связями во всех
трех кристаллографических направлениях.
Структура. Характерной структурной единицей в обеих мо-
дификациях бора с известной структурой является икосаэдр
(рис. 3.6, а). Каждый атом бора связан с 5 такими же атомами,
образующими правильный пятиугольник. Этот икосаэдр можно
представить как пятиугольную антипризму, над основаниями
которой находятся по одному атому бора, или как две соединен-
Аморфные и стеклообразные системы
231
ные друг с другом тригональные антипризмы (октаэдры) раз-
личных размеров.
В а-ромбоэдрическом боре икосаэдры Bi2 соединены друг
с другом близко к кубической гранецентрнрованной упаковке
(ромбоэдрический угол 58,06°, а0 = 505,7 пм) (рис. 3.6,6). Вза-
имодействие в слое, перпендикулярном направлению (111), осу-
Рис. 3.7. Структурная единица В84, об-
разующая p-ромбоэдрическую модифи-
кацию бора.
ществляется через трехцентровую двухэлектронную связь
(рис. 3.6, в), в которой участвуют атомы бора, находящиеся
в вершинах большей тригональной антипризмы (так называе-
мые экваториальные или 2-вершины), посредством 4 электронов
икосаэдра (6-2/3 = 4). Взаи-
модействие между слоями
осуществляется за счет нор-
мальной ковалентной двух-
элсктронной связи в напра-
влении трансляции ромбо-
эдрической ячейки; в этой
связи участвуют атомы бо-
ра меньшей тригональной
антипризмы (так называе-
мые ромбоэдрические или
r-вершины), а также 6 элек-
тронов икосаэдра. Из 36 ва-
лентных электронов (12-3 =
= 36) 26 остаются на 13
связывающих молекуляр-
ных орбиталях (как следу-
ет это для В12-икосаэдра из
теории молекулярных орби-
талей) [64].
Структурной единице ^-ромбоэдрической модификации
(рис. 3.7) соответствует полиэдр с 60 вершинами, который
можно представить как обычный футбольный мяч со швами.
Его поверхность образована 12 правильными пятиугольниками
и 20 правильными шестиугольниками со всеми одинаковыми
длинами ребер. Помимо 60 атомов бора в вершинах внутри
полиэдров находятся еще 24 атома. 12 из них дополняют пяти-
угольники в радиальных направлениях до пятигранных пира-
мид. Со своей стороны, они связывают вершины икосаэдра Bi2
с его центром. Такие группировки В84 занимают вершины ромбо-
эдрической элементарной ячейки с <2=1014,5 пм и а = 65°17',
в которых помещается по крайней мере 21 атом В, из них
20 атомов находятся в пустотах над шестиугольниками группи-
ровки В84 и, вероятно, один атом бора в центре ячейки [65, 66].
Описание кристаллической структуры тетрагональной моди-
фикации I, содержащей примесные атомы, можно найти в учеб-
никах по кристаллографии. В элементарной ячейке (Bi2)4B2N2
232 Глава 3
в трехмерную пространственную сетку связываются 4 икосаэдра
В12 с двумя атомами азота в тетраэдрической конфигурации
и двумя атомами бора [67].
Кривая радиального распределения аморфного бора хорошо
согласуется с кривыми радиального распределения кристалли-
ческих модификаций [68]. Согласно кривой радиального рас-
пределения аморфного бора, межатомные расстояния в нем
определяются размерами элементарных ячеек. Перед нами на-
лицо типичный пример вещества, для которого обсуждались воз-
можности метода рентгеновской дифракции (разд. 2.4.1.1). Со-
гласно представлениям современной химии бора, можно при-
нять, что в образовании аморфного бора принимают участие
различные фрагменты икосаэдров Bi2.
Свойства. Размеры элементарной ячейки модификаций бора,
обусловленные сложной структурой, позволяют ожидать совпа-
дения свойств кристаллических модификаций и аморфного бора.
Модельные представления, разработанные для описания струк-
туры и типа связи в аморфных и стеклообразных твердых те-
лах, могут быть перенесены на кристаллические модификации
бора. Электрические свойства, электропроводность и термо-
э. д. с. p-ромбоэдрического и аморфного бора интерпретиру-
ются в различных работах, исходя из модельных представлений
[69—71].
Электропроводность в области низких температур подчи-
няется закону Мотта (пропорциональна Т разд. 4.1.3.1);
она обусловлена скачками электронов между локализованными
состояниями в области энергии Ферми. Плотность состояний
в центре зоны составляет ~1018 см-3. Выше 600 К кривая
электропроводности достигает области собственной проводимо-
сти. Энергия активации электропроводности составляет для
аморфного бора 0,65 эВ, для ^-ромбоэдрического бора 0,70 эВ.
Исходя из предположения, что уровень Ферми закреплен на
середине запрещенной зоны, из этих измерений можно оценить
ширину зоны; она составляет ~1,3—1,4 эВ. Край поглощения
как для аморфного, так и для ^-ромбоэдрического бора в об-
ласти 0,5 < hv <0,9 эВ характеризуется экспоненциальным
ходом.
3.1.2.2. Углерод
Кристаллические модификации. Элементарный углерод из-
вестен в основном в виде многочисленных форм графита, кото-
рые характеризуются различной чистотой и степенью дисперсно-
сти. Плотность чистого графита 2,266 г/см3. Плотность алмаза
значительно больше (3,514 г/см3). Устойчивость графита при
Аморфные и стеклообразные системы 233
300 К и нормальном давлении на 0,29 кДж/моль выше, чем
алмаза. Энтальпия атомизации графита составляет при 300 К
71,8 кДж/моль, температура плавления при нормальном дав-
лении ~4100 К [72].
В графите атомы углерода образуют правильные шестичлен-
ные кольца, которые связаны в плоские слои. Каждый атом
находится от других трех атомов на расстоянии 141,3 пм. Угол
между углерод-углеродными связями составляет 120°. Структуру
графита можно описать как двумерный каркас из сг-связей, об-
разованный за счет взаимодействия 5р2-гибридных орбиталей.
Оставшийся электрон углеродного атома характеризуется ко-
нечной плотностью вероятности (л-электроны) только выше
и ниже плоскости, в которой атомы связаны в шестичленные
кольца (как пчелиные соты). Он делокализован, что приводит
к высокой металлической проводимости графита. Параллельно
слоям проводимость составляет —2 См/см, перпендикулярно
слоям — только 10~4 См/см. Слои располагаются в гексагональ-
ной последовательности АВАВ на расстоянии 335,4 пм, а могут
также располагаться в последовательности АВСАВС или в од-
ном из множества сочетаний этих двух пограничных случаев
(ромбоэдрический графит). Холл [73] сообщает о различных
формах графита, в которых реализуется один из возможных
вариантов статистического распределения позиций слоев отно-
сительно друг друга.
Из-за относительно больших расстояний между слоями проч-
ность связи между ними мала, что объясняет низкую микро-
твердость графита. Внутри слоев прочность связи очень велика.
Прочность па растяжение графитовых слоев составляет 100 ГПа
(модуль упругости ~ 1 ТПа). В монокристалле толщиной в во-
лос (так называемые «усы» углерода) прочность на растяжение
достигает 20 ГПа (модуль упругости 700 ГПа) [74].
В решетке алмаза валентное состояние атома углерода реа-
лизуется с помощью четырех симметричных эквивалентных
5р3-гибридных орбиталей. Их взаимодействие приводит к обра-
зованию трехмерного каркаса о-связей, что обусловливает не-
обыкновенную твердость этой модификации углерода. Меж-
атомное расстояние в алмазе составляет 154,45 пм, валентный
угол 109°28/. Алмаз — это изолятор с шириной запрещенной
зоны 5,3 эВ. Его структуру можно описать как кубическую
гранецентрированную решетку, в которой центры чередующихся
«восьмушек» куба заняты четырьмя атомами углерода, или как
две кубические гранецентрированные решетки, смещенные одна
к другой на ’/4 диагонали куба. Эту решетку можно представить
(рис. 3.8) как трехмерную сетку циклогексановых колец в форме
«кресла». Последовательность АВС участков решетки, содержа-
щих атомы углерода (тип цинковой обманки, рис. 3.8, а), можно
представить также в виде последовательности АВ (тип вюр-
234 Глава 3
Рис. 3.8. Структуры сфалерита (цинковой обманки) (а) и вюрцита (б).
а — при идентичности атомов 1 и 2 получается решетка алмаза; б- при идентичности
атомов 1 и 2 получается структура алмаза с гексагональной решеткой.
цита, рис. 3.8,6) или одним из разнообразных сочетаний обоих
типов.
В решетке вюрцита часть циклогексановых колец располо-
жена в форме «ванны». Для SiC описано более 140 вариантов
упаковки такого типа [75]. Известна также аналогичная вюр-
циту модификация, так называемый гексагональный алмаз [76,
77]. Предполагают, что такого рода топологическое нарушение
порядка также существует в аморфных алмазных пленках,
полученных специальным способом [78]. Известна еще одна
бесцветная модификация углерода — карбин [79, 80]. Атомы
углерода в пей связаны в прочные линейные цепи, которые раз-
личаются двумя формами, одна из них с кумулятивными двой-
ными связями, вторая с чередующимися простыми и тройными:
=С==С=С=С=С=С=
—С=С-С=С-С=С—
Карбин образуется в аморфном состоянии при окислении С2Н2
в присутствии CuCi или из поливинилиденхлорида при обра-
ботке Na/NH3 (кристаллическая форма карбина образуется при
нагревании в вакууме). Его также получают путем сублимации
пиролитического графита [81]. Эти формы углерода можно
также получить из графитовых участков гнейсов Nordlinger
Ries [82]. Новые исследования показали, что карбин геохими-
чески широко распространен, но вместе с графитом встречается
редко.
Аморфный и стеклообразный углерод. Многочисленные виды
так называемого аморфного углерода, например древесный
уголь или сажа, представляют собой микрокристаллические
Аморфные и стеклообразные системы
235
формы графита. Аморфные пленки углерода, осаждаемые с при-
менением различных методов из пара, состоят, как показывают
структурные исследования, из алмазоподобных и графитоподоб-
ных областей ближнего порядка, причем последний тип явно
преобладает [83, 84]. Статистическое распределение не соблю-
дается. Исследования методом дифракции электронов указы-
вают на существование локальных областей упорядочения раз-
мером 1 нм со структурой, аналогичной графиту, и наличие
нарушений в последовательности слоев. Эти области структуры
связаны через такие же конфигурации атомов, как в решетке
алмаза и его многочисленных разновидностей. Для описания
структуры аморфных слоев углерода подходит кристаллитная
модель (разд. 2.4.2.2).
Спектр поглощения аморфного углерода [85], и его элек-
тропроводность можно объяснить на основе этой модели [86].
Пленки аморфного углерода имеют темный цвет, а их электро-
проводность анизотропна, например 30 См/см в плоскости
слоя и 10-4 См/см перпендикулярно плоскости. Измерения
методом ЭПР дают сообразно с ожидаемой положительной
энергией корреляции плотность спинов 3-1019 см-3 [87]. В КР-
спектре аморфного углерода проявляется одна полоса 1500 см-1
[88], в спектре монокристаллов графита — полоса 1575 см ’,
у алмаза— 1332 см 1. Эти данные показывают, что в аморфных
слоях углерода преобладают области, аналогичные графиту.
Стеклообразный углерод — технический продукт, который по-
лучают пиролизом трехмерных сетчатых органических полиме-
ров, например продуктов конденсации фенола и фурана [90].
При карбонизации нс образуется жидкой фазы. Процесс графи-
тизации контролируется кинетически. Кристаллиты графита при
нагревании до 3100 К остаются маленькими, например, их
размеры ограничены 490 нм параллельно плоскости слоев
и 310 нм перпендикулярно им [89]. В стеклообразном графите
расстояние между слоями 344 пм (по сравнению с 335 пм
в упорядоченном графите). Пакеты слоев связаны в открытую
трехмерную сетку из атомов углерода, что приводит к изотроп-
ности свойств, большой твердости и аналогичной обычным стек-
лам хрупкости на излом. Плотность составляет только 1,5 г/см3.
Проницаемость стеклографита для Не мала; она такая же, как
у силикатных стекол, и составляет 10-’1 см2/с. Характеристиче-
ский размер пор составляет 136 нм. Высокая коррозионная
стойкость объясняется тем, что только малое число пор откры-
вается наружу. Удельная электропроводность стеклографита
равна 0,02 См/см [74]. Исследование структуры подтверждает,
что стеклографит построен из микрокристаллических графито-
подобных домепов, которые уложены в области, характеризую-
щиеся трехмерным каркасом связей атомов углерода и отсутст-
вием периодичности [91].
236 Глава 3
3.1.2.3. Кремний, германий и производные системы
Кристаллические модификации. Эти элементы (Si I и Ge I)
кристаллизуются в решетке алмаза; межатомные расстояния
составляют 235 и 245 пм, плотности 2,328 и 5,323 г/см3, а опти-
ческая ширина запрещенной зоны достигает величин 1,12
и 0,78 эВ соответственно.
Удельная электропроводность кремния под давлением 15 ГПа
такая же, как у А1. При этом Si ведет себя как металл; его
структура соответствует решетке белого олова. Эта модифика-
ция, существующая под высоким давлением, известна как Si II.
Ее плотность выше на 23%, а межатомное расстояние при-
мерно на 6 % больше, чем у Si I. Аналогичная модификация
германия (Ge II) существует под давлением 12 ГПа [92, 93].
Объемное сжатие Ge II составляет 21 %, и среднее межатомное
расстояние (в сочетании с возрастанием координационного
числа до 6) возрастает примерно на 8 %. И Sill, и Ge II не-
устойчивы при нормальном давлении.
При снятии давления Si II переходит в другую модификацию
Si III с гранецентрированной кубической структурой (а —
= 663,6 ±0,005 пм, плотность 2,55 г/см3); элементарная ячейка
содержит 16 атомов. В такой структуре координационное число
атома кремния опять равно 4; углы тетраэдра составляют 99
и 108°, межатомные расстояния в направлении тригональной
оси укорочены до 230 ± 1 пм, остальные связи удлинены до
239 ± 1 пм. Возрастание плотности связано с уменьшением рас-
стояний до вторых соседей до 345 пм по сравнению с 384 пм
в Si I (остаточное действие давления). В решетке алмаза каж-
дый атом участвует в образовании 12 различных шестичленных
колец (рис. 3.8,а); в Si II существует такая конфигурация ато-
мов, что наряду с уменьшением числа шестичленных колец по-
являются также восьмичленные кольца. Удельная электропро-
водность Si III в 10—100 раз меньше, чем у Si II. При нагре-
вании до 500—900 К образуется модификация со структурой
вюрцита (рис. 3.8,6). Плотность ее составляет 2,33 г/см3,
а удельная электропроводность, равная 10~4 См/см, резко убы-
вает при возрастании температуры (как у Si I).
Из фазы высокого давления Ge II при снятии давления обра-
зуется тетрагональная фаза Ge III (а = 593 ± 1 пм, с —
= 698 ± 1 пм); элементарная ячейка содержит 12 атомов. Плот-
ность Ge III 5,91 г/см3 (на 11 % больше, чем у Ge I). Меж-
атомные расстояния 248—249 пм немного увеличены, тетраэдри-
ческий угод значительно искажен. Расстояние до вторых соседей
составляет 381 пм по сравнению с 400 пм в кубическом герма-
нии. В Ge III наряду с шестичленными кольцами существуют
пяти- и семичлснные.
Аморфные и стеклообразные системы
237
Использование высоких давлений, начиная с достаточно низ-
ких температур, приводит к образованию такой же модифика-
ции Ge, как Si III. Эту модификацию обозначили Ge IV. Еще
одна модификация германия, алло-Ge, получена по топотактиче-
ской твердофазной реакции из Li7Gei2 (Li извлекают при обра-
ботке водой или бензофеноном) [807].
Структура расплава. Кремний плавится при температуре
1686 К, германий — при 1231,6 К. Электропроводность при пере-
ходе в расплав обоих металлов намного превышает (для Si
в 30 раз, для Ge в 15 раз) электропроводности типичных рас-
плавленных металлов (104 См/см) (см. литературу к гл. 1
[28] ). Оба элемента характеризуются аномальным поведением
плотности при температуре плавления. Расплав этих элементов
занимает мольный объем на 8 % меньше, чем кристаллическое
вещество, и, как и у воды, температура плавления понижается
на —3 К при повышении давления на 100 МПа.
Кривая радиального распределения расплава Ge показала,
что координационное число Ge в расплаве составляет ~6,
а ближний порядок в расплаве совпадает со структурой белого
олова [94, 95]. Для того чтобы фиксировать при комнатной
температуре изотропное состояние расплава, требуется очень
высокая скорость охлаждения. Аморфные пленки Ge получают
из расплава при скорости охлаждения 1010 К/с (см. литературу
к гл. 2 [26] ), однако по своей структуре они не обнаруживают
ожидаемого сходства с расплавом. Кристаллизация происходит
лишь при 965 К. Исследования этих пленок методом дифракции
электронов показали, что они аналогичны по структуре аморф-
ному германию, получаемому различными способами осаждения
из газовой фазы, электролизом или ионной имплантацией.
Аморфные формы кремния и германия. Аморфные кремний
и германий получают путем испарения и конденсации элемен-
тов в глубоком вакууме или катодного распыления в аргоновой
плазме. Использованные методы в зависимости от условий экс-
перимента (скорости осаждения, температуры подложки, по-
ковке) позволяют получать аморфные слои, сильно отличаю-
щиеся по свойствам. Обзор литературных данных и своих работ,
отчасти противоречивых, приводит Биненсток [96].
Свойства аморфных пленок германия, полученных в сверх-
высоком вакууме, описаны в работе [97]. Воспроизводимость
свойств слоев тем лучше, чем ниже остаточное давление,
меньше скорость осаждения и выше температура подложки.
Очевидно, образованию определенных метастабильных аморф-
ных структур способствует достаточная подвижность атомов
на поверхности. Температура подложки ограничена температу-
рой кристаллизации аморфного вещества. Критическая темпе-
238 Глава 3
ратура нуклеации для поверхностной кристаллизации состав-
ляет для чистого аморфного Ge 653 К [98], для аморфного Si
770—870 К. Для энтальпии кристаллизации аморфного Ge
в литературе приводятся два несовпадающих значения: 11
и 16 кДж/моль. Температуры стеклования для аморфных моди-
фикаций обоих элементов не обнаружены.
Для осаждения аморфных слоев Si и Ge необходимо еще
одно условие—поток пара и плоскость подложки должны об-
разовать прямой угол, т. е. возможно большее расстояние между
испарителем и подложкой. Угол падения сильно влияет на раз-
мер пустот, образующихся при росте слоев. Методом малоугло-
вого рассеяния электронов установлены размеры пустот (от 70
до 2000 пм), например доказано образование пустот, вытянутых
в направлении, перпендикулярном плоскости слоев [99]. Число
и форма пустот сильно зависят от условий получения. Этим
объясняются различия в плотности, переменная концентрация
неспаренных электронов и оборванных связей, являющихся де-
фектными центрами и локализующихся на внутренней поверх-
ности таких пустот. Этим же объясняется чувствительность
к загрязнениям, например к загрязнению кислородом; примеси
влияют на плотность спинов в спектре ЭПР, а также на элек-
трические транспортные свойства. Слои германия, полученные
методом катодного распыления при температуре подложки
625 К, включают 1,8 ат. % Аг [96, 100].
Высокие температуры осаждения, низкие скорости и исклю-
чительно высокая чистота, что можно осуществить практически
только путем испарения в сверхвысоком вакууме, приводят при
соблюдении геометрических факторов в расположении испари-
теля и подложки к получению аморфных слоев Si и Ge, свой-
ства которых при последующей обработке не будут существенно
меняться.
Аморфные слои Ge можно получить также методом электро-
литического осаждения, например, из раствора GeCU в пропи-
ленгликоле [101] (см. также литературу к гл. 2 [32] ).
Был испытан еще один способ получения определенных
форм аморфных Ge и Si. В основе его лежит соображение
о том, что неспаренные электроны оборванных связей либо
связываются с определенными атомами примесей, либо всту-
пают в химическое взаимодействие, и при этом высокая плот-
ность локализованных состояний, чувствительная к различ-
ным влияниям, резко понижается [102]. Во многих работах при-
водятся данные по испарению Si под небольшим давлением кис-
лорода. В работе [103] вводили несколько процентов Н2 при
катодном распылении Si в аргоновой плазме.
Особенно успешным оказался примененный впервые Спиром
с сотр. [104, 105] метод разложения SiFU в тлеющем разряде.
В продуктах реакции содержится 5—50 мол. % водорода, а вме-
Аморфные и стеклообразные системы
239
сто чистого кремния — макромолекулы сетчатого полимера поли-
циклосилана. Состав таких пленок зависит от типа разряда,
его мощности, парциального давления и расхода SiH4, темпе-
ратуры подложки. В общем содержание водорода уменьшается
при повышении температуры подложки; обычно используют тем-
пературы 470—670 К.
Ограничение влияния локализованных состояний на пере-
нос заряда обеспечивается путем насыщения разорванных свя-
зей, плотность которых высока в чистом аморфном Si. Электро-
проводность таких слоев можно модифицировать посредством
легирования подходящими донорами и акцепторами. Если до-
бавлять к находящемуся под действием тлеющего разряда по-
току SiH4 последовательно один за другим РН3 и В2Нб, то
можно осуществить таким путем р, n-переход в аморфных слоях
SiHx, что вызвало значительный интерес в связи с возможностью
потенциального применения, например, в солнечных батареях
большой площади [109, НО] (разд. 4.1.3.5 и 5.1.3).
Работы в этой области, особенно в направлении препаратив-
ной химии, в последние годы развивались особенно бурно [111].
С целью улучшения стабильности приборов при одновременном
повышении к. п.д. подробно изучались также возможности
частичного или полного замещения атомов Н на другие при-
месные атомы, например О или F [112—114]. Подробные сведе-
ния об условиях получения и свойствах таких слоев приведены
в работе [785].
Структура аморфных кремния и германия. Сопоставление
структуры и свойств кристаллических элементов выявило полез-
ную роль аморфных германия и кремния как модельных ве-
ществ, исследование которых должно привести к основным
успехам в понимании аморфных и стеклообразных полупровод-
ников. Поэтому большое число работ посвящено исследованию
структуры аморфных германия и кремния.
С помощью методов дифракции рентгеновских лучей, элек-
тронов и нейтронов для аморфных элементов Si и Ge, получен-
ных с помощью испарения, катодного распыления или электро-
литического осаждения, построены кривые радиального распре-
деления, ход которых выявляет относительно единую картину
[1 16—118, 807] (см. также литературу к гл. 2 [303]). Даже
бомбардировка кубической модификации Ge ионами германия
высокой энергии приводит к получению аморфной формы, кото-
рая мало отличается по структуре от аморфных слоев. Отсюда
можно сделать вывод, что в аморфных модификациях обоих
элементов существует в некотором приближении одна и та же
метастабильная равновесная конфигурация атомов.
На рис. 3.9, а приведены кривые радиального распределения
(КРР) для кристаллического и аморфного Si [119]. Первые
Рис. 3.9. КРР кристаллического и аморфного Si (а) [119]
Сплошная кривая — экспериментальные данные (см. литературу к
и аморфного Ge (б).
гл. 2 [304]).
Аморфные и стеклообразные системы
241
и вторые максимумы г1 = г0 = 235 пм и г2 = (8/з)1/,*235 =
= 384 пм совпадают для обеих форм. Что касается координа-
ционного числа, то для первой координационной сферы оно
равно 4, а для второй 12. Различия наблюдаются в дальнейшем
ходе кривых. Третий максимум на кривой аморфного кремния
практически отсутствует. На рис. 3.9,6 изображены аналогич-
ные экспериментальные результаты для аморфного Ge [120].
Для сравнения приведены кривые, рассчитанные по различным
моделям неупорядоченной сетки [121 —123]. КРР для аморф-
ных слоев Ge, полученных катодным распылением или электро-
литическим осаждением, хороню согласуются [117]. Максимумы
первой (Г1 = 246 ± 1 пм) и второй (г2 = 400 ± 4 пм) координа-
ционных сфер хорошо согласуются с максимумами кристалли-
ческого Ge.
Кристаллитныс модели в отличие от случая аморфного угле-
рода нс дают удовлетворительного сходства с эксперименталь-
ными данными ио дифракции. Для интерпретации эксперимен-
тальных данных неоднократно привлекались также кластерные
модели [124, 125] (см. также литературу к гл. 2 [310]).
Модель непрерывной неупорядоченной трехмерной сетки
использована Полком (см. литературу к гл. 2 [300]). При соблю-
дении обязательного условия континуума эта модель строится
из 440 атомов; сделан соответствующий расчет. Модель пред-
ставляет собой пространственно-разветвленную структуру
с почти постоянными межатомными расстояниями Si—Si, сред-
ним отклонением валентного угла ±10° (максимальное отклоне-
ние ±20°) и произвольным диэдрическим углом. В этой струк-
туре имеются 6- и 5-членные кольца, взятые в отношении
4:1; плотность такого твердого тела достигает 97 ± 2 % плот-
ности соответствующей кристаллической кубической модифика-
ции [126]. Экспериментальная кривая рассеяния аморфного Si
и рассчитанная из нес КРР удовлетворительно согласуются
в отношении положения максимумов с кривой, рассчитанной
по модели Полка. Межатомные расстояния, соответствующие
3-й координационной сфере, сильно уменьшаются по сравнению
с кристаллической модификацией из-за участия более плоских
пятичленных колец в аморфной структуре. Модель была рас-
ширена до 519 атомов и уточнена путем оптимизации в направ-
лении лучшей подгонки к поверхности [127].
Из неоднозначности кривых радиального распределения
в области больших расстояний следуют различные возможности
при интерпретации. Непрерывная беспорядочная сетка из 5-,
6- и 7-членных колец, взятых в отношении 4:9:10, описывает
экспериментальную КРР аморфного Ge, и приближение, пока-
занное на рис. 3.9,6 (кривая /) [122, 128]. Когда беспорядоч-
ная сетка составлена только из шестичленных колец, то в ней
оказывается очень мало крупных нарушений структуры
16 Заказ № 413
242 Глава 3
(кривая 2) [121]. На рис. 3.9, б (кривая 3) приведен также ход
кривой для сеточной структуры, построенной из многих колец
с нечетным числом членов [123]. Предполагается [129], что
чем меньше статистические различия в составе колец, тем боль-
шую роль играет распределение диэдрических углов. Конфигу-
рация «кресла» более вероятна, чем конфигурация «ванны».
Обнаружено также, что кривая радиального распределения кри-
сталлической модификации Ge III прекрасно согласуется
с усредненной экспериментальной кривой аморфного Ge [130].
Один из трех максимумов на КРР кубической модификации,
Рис. 3.10. Вакансии в решетке кристаллического Si или Ge с широкими коль-
цами и разорванными связями (а) и широкие кольца в аморфном Si или
Ge (б).
отвечающий увеличенному межатомному расстоянию, не прояв-
ляется у этой кристаллической модификации (Ge III). Когда
специфическая структурная информация о веществе, извлекае-
мая из КРР, ограничена, то для описания аморфных твер-
дых тел в рамках этого метода измерения также приме-
нимы модели кристалла с достаточно большой элементарной
ячейкой (разд. 3.1.2.1). Эти модели нельзя смешивать с кри-
сталличными моделями. Например, модель кубического кри-
сталла из 64 атомов (тетраэдрическое окружение) с некоторым
отклонением от правильной структуры привела к успешной ин-
терпретации дифракционных кривых [131]. Поэтому в рассмот-
рение включены также модели аморфного германия, основанные
на структуре алмаза с высокой концентрацией в решетке моно-
и дивакансий [132] (рис. 3.10). Модель с винтовыми дис-
локациями, характеризующаяся плотностью 1014 см-2, также по-
строена с нарушениями дальнего порядка, и КРР для этой
аморфной структуры хорошо согласуются с экспериментальной
кривой [133].
Из рис. 3.10 видно, что одна вакансия в решетке соответст-
вует четырем разорванным связям с одновременным возникно-
Аморфные и стеклообразные системы 243
вением относительно больших колец. Модели неупорядоченной
сетки предполагают кольца меньшего размера и допускают
изолированные оборванные связи. Они допускают большое раз-
нообразие структурных конфигураций возможных изолирован-
ных центров, имеющих неспарениый электрон.
В работах [786, 787] описаны исследования структуры водо-
рода, содержащегося в аморфном кремнии.
Свойства аморфных слоев Si и Ge. Свойства обоих элемен-
тов в аморфном состоянии определяются высокой плотностью
дефектных центров в запрещенной зоне [134]. Исследова-
ния методом ЭПР очень чистых слоев, полученных методом
вакуумного испарения, показали [135], что они содержат в 1 см3
от 1019 до 1020 парамагнитных центров, т. е. на 103 атомов Si
приходится одна оборванная связь. Такие дефекты появляются
на внутренних поверхностях пустот, образовавшихся при полу-
чении пленок; они обладают высокой реакционной способностью
и ответственны, например, за загрязнение атомами примесей
во время получения или в дальнейшем.
Аморфные слои Si, полученные разложением SiH4 в тлею-
щем разряде, характеризуются на 3—4 порядка меньшей плот-
ностью спинов. Дефектные центры в таких слоях заняты связями
Si—Н, и интенсивность сигнала ЭПР ниже предела обнаруже-
ния. Нагревание до температур ниже температуры кристалли-
зации приводит к значительному выделению водорода. Напро-
тив, аморфные слои, полученные испарением в высоком ва-
кууме, можно насыщать водородом благодаря наличию
оборванных связей. За протеканием таких твердофазных реак-
ций удобно наблюдать с помощью методов ЭПР и ПК-спектро-
скопии [135, 136]. Процессы старения аморфных слоев и влия-
ние примесей исследуются во многих работах.
Проводимость аморфных слоев Si и Ge в области от низких
температур до комнатной осуществляется за счет перескоков
электронов вблизи уровня Ферми, где проводимость подчиня-
ется правилу Мотта (разд. 4.1.3.1) и пропорциональна Т ’А
Механизм переноса зарядов зависит от плотности дефектных
центров, т. с. от условий получения; последующая термообра-
ботка не оказывает существенного влияния [137—140]. Лишь
при повышенных температурах осуществляются переходы ме-
жду зонами. Энергия активации проводимости в этой обла-
сти температур составляет для аморфного Ge 0,55 эВ, для
аморфного Si 0,75 эВ [138]. Для слоев Si, полученных
разложением SiH4 в тлеющем разряде, при комнатной
температуре проводимость меньше на 6 порядков; найденная
из температурной зависимости электропроводности энергия ак-
тивации Еа составляет 0,75 эВ. Предполагая, что уровень
Ферми закреплен вблизи середины запрещенной зоны, можно
16*
244 Глава 3
оценить ширину щели подвижности (рис. 2.54) между валент-
ной зоной и зоной проводимости; для аморфного Ge она состав-
ляет 1,10 эВ, для аморфного кремния 1,50 эВ, т. е. значительно
выше соответствующих значенй, полученных для кристалли-
ческих Ge и Si (0,78 и 1,12 эВ соответственно).
Аморфные системы Si—С, Si—Ge и Ge—Sn. Аморфные слои
в этих системах можно получить теми же методами, что и для
Si и Ge. Дифракционные, оптические, электрические и спектро-
скопические исследования дают согласующиеся результаты
о тетраэдрическом ближнем порядке. При этом предполагается
наличие наряду с гетеросвязями связей между одинаковыми
атомами и соответственно более высокой степени разупорядоче-
ния структуры, чем для чистых элементов. Например, для много-
численных кристаллических модификаций SiC принято сущест-
вование только связей Si—С [141], а в аморфной форме они
отчасти замещаются связями Si—Si и С—С. Геометрическая и
топологическая неупорядоченность накладывается на разупоря-
доченность по составу [142]. Это подтверждают исследования
методом КР-спектроскопии, которые обнаруживают присутствие
графитоподобных областей [143]. Осаждение в тлеющем раз-
ряде из смесей SiH4 и С2Н4 приводит к получению относительно
однородных слоев, фотолюминесценция которых в видимой об-
ласти спектра представляет практический интерес [146].
Сообщается об аморфных слоях Ge—Si [144] и аморфных
фазах Gei_xSrix (0<х<0,5) [145]; в образующихся структурах
около половины связей образовано одинаковыми атомами. Срав-
нение с аморфными модификациями соединений AinBv обнару-
живает хорошее соответствие в КРР [147].
Аморфные соединения AInBv и AnBVI. Особенно хорошее
соответствие КРР обнаружено для аморфных слоев Ge, GaAs
и GaP, а также для GeSn, InSb и GaSb. Соединение колец,
имеющих нечетное число членов, с образованием непрерывной
беспорядочной сетки, построенной по модели Полка, осуществля-
ется путем возникновения связи между одинаковыми атомами.
Особенно отчетливое структурное сходство аморфных GeSi и
GaP обнаруживается при сравнении кривых рентгеновской диф-
ракции. На основе известных структурно-химических представ-
лений связи Ga—Ga и Р—Р приемлемы, и, напротив, малая
энергия связи Ga—Р в общем энергетическом балансе должна
играть незначительную роль. Электронная структура атомов
в таких «неправильных -связях» должна отличаться от струк-
туры остальных атомов матрицы. КР-спектры свидетельствуют
о присутствии таких «неправильных» связей [148]. В то же
время показано, что моделирование структуры аморфного Ge
никак не связано с образованием колец из нечетного числа
Аморфные и стеклообразные системы 245
членов. Наилучшее приближение к КРР для аморфных со-
единений AniBv получено с помощью модели непрерывной
сетки только с использованием колец с четным числом членов
[149]. Сведения об оптических и электрических свойствах
аморфных слоев GaP и GaAs приводятся во многих работах
[150—152]. Выполнен обзор [153] по аморфным пленкам для
соединений AnBVI, например ZnSe, CdTe и ZnTe. В этой группе
соединений также сохраняется тетраэдрический ближний по-
рядок.
Соединения AnBlvCy и производные стеклообразующие
системы. Физические свойства тройных полупроводников типа
AnBIVCJ в твердом состоянии широко исследованы [154]. Они
кристаллизуются в решетке халькопирита CuFeS2 и принад-
лежат к структурной группе алмаза. Путем охлаждения рас-
плавов со скоростью 7 — 50 К/с соединения CdGeAs2 и CdGeP2
получают в виде компактных стекол (с Тя 660 и 720 К) [155].
По температуре стеклования стеклообразного CdSiAs2 имеются
несовпадающие данные 760 К [156] и 703 К [163]. Исследо-
ваны также свойства по разрезам CdGe(AsxPi-x)2 [157],
CdSixGei_xAs2 [158], CdSnxGei-xAs2 [159] и CdPbxGe(_xAs2
[160]. Эти тройные стекла существуют в более или менее про-
тяженных областях стеклообразования (простирающихся до
соединений CdAs2 и CdP2). CdAs2 и CdP2 получены в стекло-
образном состоянии [161]. Их ближний порядок в кристалли-
ческом состоянии также характеризуется тетраэдрическими
группировками. Наряду с [CdAs4] имеются группировки
[AsAs2Cd2] со связями As—As; как и в соединениях с решет-
кой типа алмаза, здесь на каждый атом приходится в среднем
4 валентных электрона (правило Гримма—Зоммерфельда).
CdAs2 образует стекла в относительно широких границах в со-
ответствии с общей формулой CdMxAs2, где M = Ge, Si, Tl, In,
Al, Sb, Mg или Ga [162]. На рис. 3.11 показана область стек-
лообразования в системе Cd—Ge—As [163]. Следует отметить,
что эти стекла обладают большей плотностью, чем кристаллы
того же состава; например, плотность стеклообразного CdGeAs?
5,72 г/см3, а для кристаллического вещества 5,61 г/см3
[161].
Исследование структуры тройных стекол с помощью кривых
радиального распределения представляет собой вследствие
большого разнообразия возможных атомных связей еще более
трудную задачу, чем для аморфных форм соединений AinBv.
В работе [164] из различных моделей структуры выбрали
структуру беспорядочной объемной сетки, в которой преобла-
дают кольца с четным числом членов, где, как и в кристалли-
ческой решетке, отсутствуют «неправильные» связи.
246 Глава 3
Рис. 3.11. Область стеклообразования в системе Cd—Ge—As [163].
1 — границы области стеклообразования при скорости охлаждения 102 К<с, 2 — границы
области стеклообразования при скорости охлаждения 1—10 К/с.
3.1.2.4. Фосфор
Кристаллические модификации. Белый фосфор состоит из
тетраэдрических молекул Р4, которые сохраняются и при тем-
пературах выше температуры плавления. Тпл 317,2 К, плот-
ность 1,82 г/см3, легко растворяется в CS2, расстояние Р—Р
221 пм, валентный угол 60° (отсюда и нестабильность этой
модификации). Выше 453 К происходит полимеризация, и по
твердотельной реакции, которая контролируется кинетически,
через промежуточную стадию образования аморфного продукта
образуется фиолетово-красный фосфор Гитторфа. Кристаллы
получают только после длительного нагревания до 770 К-
Фосфор Гитторфа содержит 84 атома в моноклинной элемен-
тарной ячейке (р = 2,36 г/см3), а = 919 пм, 6 = 913 пм, с =
= 225,5 пм, р= 106,1°) [165]. Средние межатомные расстояния
составляют 221,9 и 346 пм, средний угол связи 100,9°.
На рис. 3.12 приведена структура фосфора Гитторфа (по
Кребсу); она построена из двух группировок Р8 и Р9, в одной
из них, Р8, атомы расположены, как в реальгаре. Обе группи-
ровки чередуются, соединяясь через два линейно связанных
атома фосфора, так что получаются бесконечные длинные
трубки с пятиугольным сечением (рис. 3.12,а и б). Трубки уло-
Аморфные и стеклообразные системы
247
жены в слои, параллельные друг другу. Два таких слоя соеди-
няются друг с другом через атомы групп Р9, так что оси тру-
бок образуют угол 89,6° (рис. 3.12, в). Соединение двух таких
двойных слоев осуществляется без прямого контакта и обуслов-
лено только вандерваальсовыми силами. Благодаря этому фос-
фор Гитторфа расщепляется, как слюда. Образование такой
чрезвычайно сложной структуры можно объяснить, очевидно,
стремлением каждого атома реализовать нормальный валент-
Рис. 3.12. Кристаллическая структура фосфора Гитторфа [165].
ный угол — 100°. Выше 870 К происходит разрушение полимер-
ной структуры с образованием молекул Р4. Для температуры
плавления фосфора Гитторфа приводится значение 893 К.
Вплоть до температуры 823 К термодинамически стабилен
черны?! фосфор. Он образуется при нагревании белого фосфора
до 470 К под давлением от 1,2 ГПа или при нормальном давле-
нии при нагревании от 500 до 640 К в присутствии Hg как
катализатора и затравки кристаллов черного фосфора. Плот-
ность черного фосфора 2,69 г/см3, ромбическая элементарная
ячейка, а = 331,4 пм, 6 = 437,6 пм, с= 1048 пм (параметры соот-
ветствуют слоистой решетке) [166], образуется из решетки
NaCl путем замещения позиций идентичными участками при-
митивной кубической решетки (рис. 3.13, а). Получаются «гоф-
рированные» двойные слои, когда позиции атомов в направ-
248 Глава 3
Рис. 3.13. Кристаллическая структура двух модификаций фосфора.
а — ромбический черный фосфор: ri = 222,4 пм, г2==331,4 пм, г3 = 224,4 пм, г4 = 359,2 пм. а. =
=96°34', <р2=102°9'; слои расположены в направлении (100); б — ромбоэдрический фосфор:
П=213 пм, г2=327 пм, ф=.104°30'; слои расположены в направлении (111).
лении [100] решетки NaCl попарно связаны и для углов cpi и
q>2 допускаются отклонения от прямого угла. Слои состоят из
связанных друг с другом шестичленных колец. Ваидерваальсово
расстояние г4 между сдвоенными слоями больше, чем расстоя-
ния внутри двойных слоев. Кристаллы черного фосфора такие
же чешуйчатые на вид, как и графит. Черный фосфор обла-
дает металлической проводимостью. Его проводимость при ком-
натной температуре равна 0,5 См/см, ширина запрещенной
зоны ~0,33 эВ.
Под давлением >83 кбар из черного фосфора образуется
еще одна кристаллическая модификация, аналогичная по струк-
туре ромбоэдрическому мышьяку [167]. Ее также можно пред-
ставить производной решетки NaCl, если обратить внимание
на идентичные участки, образованные примитивной кубической
решеткой. Здесь позиции атомов в направлении [111] попарно
связаны в двойные слои, так что образуются 3 короткие связи
с расстоянием Г] и 3 длинные с расстоянием г2- Валентный угол
внутри слоя 104,5°. Эта структура представляет собой двумерно
связанные шестичленные кольца, аналогичные циклогексану.
Повышение давления до 11,1 ГПа приводит к обратимому
превращению ромбоэдрического фосфора в примитивную куби-
ческую решетку с одинаковыми межатомными расстояниями
237,7 пм (Г1 = Г2) по всем трем направлениям и валентным уг-
Аморфные и стеклообразные системы
249
лом 90° [167]. Фосфор в этих условиях существует в виде мо-
дификации с высокосимметричной структурой типа а-полония.
Аморфный фосфор. Аморфный красный фосфор получают
путем нагревания белого фосфора при 667 К в течение трех
суток. Для этой модификации спята и интерпретирована кривая
радиального распределения [168, 180]. Первый максимум г\
находится на расстоянии 224—231 пм, ему отвечает координа-
ционное число 3, что характеризует ближний порядок для эле-
ментарного фосфора. Второй максимум при 348 пм (362 пм
[180]) определяет расстояние до вторых соседей. Найденное
для второй координационной сферы координационное число 7,7
интерпретируется Кребсом и Грубером таким образом, что
в координационной сфере данного атома находится заметно
большее число атомов, чем в черном фосфоре. Обнаружено,
что красный аморфный фосфор, полученный из белого фос-
фора, состоит из различных возможных структурных фраг-
ментов, среди которых имеются и трубчатые группировки
фиолетового фосфора, что было установлено при структурном
анализе кристаллических полифосфидов металлов [169]. Най-
денные для соединения HgPbPi4 средние расстояния Р—Р,
равные 430, 520 и 600 пм [170], соответствуют с хорошим
приближением выраженным максимумам на КРР аморфного
красного фосфора с г3 = 428 пм, г4 = 518 пм и г5 = 590 пм. Иссле-
дование кривой радиального распределения аморфного фос-
фора, полученного путем химической транспортной реакции
в водородной плазме низкого давления, привело к выводу, что
его структура близка к структуре черного фосфора [171] (ri =
= 232 пм, гг = 352 пм).
В аморфном фосфоре, содержащем водород, при его полу-
чении образуются в качестве побочных продуктов высококон-
дснсированные полифосфины. Они образуются при освещении
Р2Н4 или при осаждении аморфного фосфора в процессе раз-
ложения РН3 в тлеющем разряде.
Из смеси паров фосфора и азота в тлеющем разряде оса-
ждают аморфный нитрид фосфора P3N5, структура которого
неизвестна [172].
3.1.2.5. Мышьяк
Кристаллические модификации. У мышьяка наряду с очень
неустойчивой желтой модификацией, состоящей из молекул As4
(плотность 2,07 г/см3), и по структуре, аналогичной белому
фосфору, существует ромбическая модификация (плотность
5,54 г/см3), слоистая структура которой аналогична черному
фосфору (рис. 3.13, а); параметры элементарной ячейки: а =
= 365 пм, Ь = 447 пм, с= 1100 пм [173], межатомные расстоя-
ния в двойных слоях 248 и 249 пм, валентные углы 94,1 и
50 Глава 3
98,5°, а расстояние между слоями 356 пм. Энергия активации,
рассчитанная по температурной зависимости проводимости,
0,15 эВ. Ромбическую модификацию получают при нагревании
до 370 К ромбоэдрического мышьяка в присутствии ртути в ка-
честве катализатора. При 520 К происходит превращение
в ромбоэдрическую модификацию (плотность 5,72 г/см3), устой-
чивую при нормальных условиях; параметры элементарной
ячейки: <2=413,1 пм, а = 54°10' (рис. 3.13, б), межатомное рас-
стояние в двойных слоях 251 пм, валентный угол 97°. Во вто-
рой координационной сфере вокруг выбранного атома находится
6 атомов на расстоянии 376 пм; они принадлежат к тому же
двойному слою, что и выбранный атом. Расстояние между
слоями (между соседними атомами) г2 = 315 пм. Ромбоэдриче-
ский мышьяк обладает металлической проводимостью при
300К (о = 3-104 См/см). При высоком давлении и низкой тем-
пературе элементарные As, Sb и Bi переходят в сверхпроводя-
щую форму, структуру которой можно описать как плотнейшую
кубическую упаковку [174].
Аморфный мышьяк. Компактный аморфный мышьяк можно
получить сублимацией кристаллического As в холодный конец
запаянной вакуумированной кварцевой трубки или разложением
AsH3 [175, 176]. При этом получают материал с различной
плотностью. Склонность ромбоэдрического мышьяка быстро
окисляться с поверхности при комнатной температуре сильно
уменьшается в аморфном состоянии. В технике стеклообраз-
ный, или аморфный, мышьяк получают сублимацией при низ-
ком парциальном давлении Н2. Плотность аморфного мышьяка
обычно достигает 4,7 г/см3. Остаточное содержание водорода
в нем ниже предела обнаружения метода ИК-спектроскопии,
а содержание металлических примесей ^10~4%.
Аморфные слои As получают различными способами (ваку-
умное испарение, катодное распыление, разложение AsH3
в тлеющем разряде). Как и для аморфных слоев Si и Ge, свой-
ства аморфного мышьяка зависят от условий получения,
а также от того, контактировали ли образец в момент получе-
ния с воздухом и присутствовали ли в нем по какой-нибудь
причине другие примеси [176]. Для получения аморфных слоев
As также с успехом используется осаждение на подогретых под-
ложках, например при 400 К в тлеющем разряде. Свойства
аморфного мышьяка близки к свойствам компактных материа-
лов. Загрязнение водородом, явно вызванное экзотермическим
разложением AsH3, в аморфном As ниже, чем в аморфных
слоях Si, полученных осаждением из смеси SiH4 с аргоном
в тлеющем разряде.
Температура кристаллизации компактного аморфного
мышьяка ~600 К; для аморфной пленки этот параметр ниже
Аморфные и стеклообразные системы
251
г, пм
Рис. 3.14. Сравнения КРР двух образцов аморфного мышьяка с отличающейся
плотностью (/ и 2) (см. литературу к гл. 2 [225]) с кривой, рассчитанной по
модели непрерывной беспорядочной сетки (3) [180].
и составляет 520 К. Энтальпия кристаллизации 5,98±
±0,13 кДж/моль.
Структура аморфного мышьяка. Методами рентгеновской и
нейтронной дифракции получены КРР атомной плотности со
следующими максимумами (в скобках указано координацион-
ное число):
1-й максимум 2-й максимум 3-й максимум 4-й максимум Литература
249 пм (3,0) 378 пм (10 ) 462 пм (6,3) 576 пм (19) 225
245 пм (3,2) 375 пм (10,2) 475 пм (4,7) 565 пм (17) 178
На рис. 3.14 приведены КРР (см. литературу к гл. 2 [225])
для двух образцов аморфного As с несколько различающейся
плотностью.
Сравнение расстояний, соответствующих 1-му и 2-му макси-
мумам, с межатомными расстояниями и расстояниями до
252 Глава 3
вторых соседей в кристаллическом мышьяке свидетельствует
о большом сходстве ближнего порядка кристаллического и
аморфного состояний. Межатомные расстояния внутри первой
координационной сферы в аморфном мышьяке несколько уко-
рочены по сравнению с ромбоэдрической модификацией и
больше соответствуют ромбическому мышьяку. В аморфном
мышьяке 2-й максимум совпадает с расстоянием до шести вто-
рых соседей внутри двойного слоя ромбического мышьяка.
С этим коррелирует еще по крайней мере положение трех бо-
лее дальних атомов. Таким образом, приходят к заключению,
что расстояние 315 пм отвечает несколько уширенному вандер-
ваальсовому расстоянию [179], причем на этом расстоянии
находятся, согласно КРР, только немногие атомы. Следует за-
метить, что если исходить из ромбического мышьяка, то
удается удовлетворительно интерпретировать эксперименталь-
ные результаты [178]. Кристаллитные модели для этой цели
не привлекаются, потому что на кривой малоуглового рассея-
ния аморфного мышьяка имеется острый максимум дифракции,
что характерно для веществ со слоистой структурой.
Предложена (см. литературу к гл. 2 [301]) модель непре-
рывной сетки с трехмерной беспорядочной увязанностью струк-
турных единиц [AsAs3/3 ]; она близка модели Полка (см. лите-
ратуру к гл. 2 [300]), описывающей аморфные Si и Ge. Сетча-
тая структура, построенная из 533 атомов As, приводит к пони-
женной на 10—20 % плотности по сравнению с кристалличе-
скими формами As; такая структура хорошо согласуется с экс-
периментальными данными. В сетчатой модельной структуре
имеются многочисленные пустоты различных размеров; она
построена из колец, включающих различное число членов (4—
15 атомов As). Чаще всего встречаются 5-, 7- и 6-члепные
кольца. Отклонения валентных углов от среднего значения со-
ставляют ~ 10°, распределение диэдрических углов примерно
равномерное, с предпочтением конфигурации «кресла» по срав-
нению с «ванной». На кривой малоуглового рассеяния макси-
мум, наблюдаемый вблизи 100 пм, на К-шкале соответствует
расстоянию 600 700 пм (2л/&). Предложено (см. литературу
к гл. 2 [301]) объяснение обязательного образования слоев,
исходя из петлеобразной структуры непрерывной беспорядоч-
ной сетки. Однако при этом не считается обязательным образо-
вание связи, подобной связи в слоях. Эта модель уточнена пу-
тем просчета на ЭВМ с учетом условия минимизации энергии
(180]. При этом получена плотность 4,7 г/см3. Результирующая
КРР и соответствующие экспериментальные кривые приведены
на рис. 3.14. Следует заметить, что модель беспорядочной сетки,
состоящей из колец с четным числом членов, также позволяет
удовлетворительно описать экспериментальные данные [176].
В ИК-спектре аморфного мышьяка присутствуют две основ-
Аморфные и стеклообразные системы
253
Энергия данной
кошриа/рицгш
-3Р£Ь
~2р~Ь
7 с + I!
^Р'-О^ PJLP
-^sp£b+ £Hb
3spEb+ spULP* ЕНЬ
~2spEb* spULP* EHb
Рис. 3.15. Энергетические уровни в аморфном мышьяке и различные дефект-
ные центры по модели пар с переменной валентностью (см. литературу к гл. 2
[405]).
ные полосы ~230 и ~ 130 см-1, которые обусловлены валент-
ными и деформационными колебаниями структурных единиц
[AsAs3/3] [181]. В КР-спектре наблюдаются полоса ~220 см-1
и вторая широкая полоса 60—150 см-1 [182].
Собственные дефекты аморфного мышьяка. Кроме геометри-
ческих и топологических дефектов, характеризующих сетчатую
структуру, для аморфного мышьяка рассматривают еще соб-
ственные дефекты. Исходя из положения мышьяка в перио-
дической системе [он находится между элементами с тетраэд-
рическими связями (Si или Ge) и халькогенами], можно су-
дить о знаке эффективной корреляционной энергии (разд. 2.5.2),
однако нет никаких доказательств образования нейтральных де-
фектных центров с неспаренными электронами (разорванные
гомосвязи) или более устойчивых положительно или отрица-
тельно заряженных дефектов (пары с переменной валент-
ностью). Устойчивость элементов главной подгруппы V группы
периодической системы можно оценить по аналогии со схемой,
приведенной на рис. 2.58 и 2.59 [176, 183] (см. также литера-
туру к гл. 2 [405]). На рис. 3.15 приводятся конфигурации и
соответствующие энергии по модели КАФ (разд. 2.5.2.2). Из
нормальной конфигурации [AsAs3/3] (рР°3), атомов As с разор-
ванной гомосвязью образуется отрицательно заряженный
мостиковый атом рР~ (псевдохалькогенный атом) и четы-
рехкоординированный положительно заряженный центр As+As4,3
254 Глава 3
(spP+), для чего необходимо участие s-электронных пар и энер-
гия гибридизации Ень. Следует различать р-орбитали (индекс
р) и гибридные 5р3-орбитали связи (индекс sp). Для реакции
2AsAs3/3 7Z AsAs4/3 + AsAs2/3-f- Д (3.1)
2pF^~spPf + рРГ + EHb + pVLP - 4 (spE„ - pEb)
наблюдается эндотермический эффект, pEb=2,l эВ (табл. 2.11),
spEb= (4/3) -р£б = 2,8 эВ (см. литературу к гл. 2 [323]), Ень =
= 7,5 эВ [рассчитана для расщепления терма s—р в As по из-
мерениям методом рентгеноэлектронной спектроскопии (РЭС)
[184] и pULP=0,7 эВ, т. е. равновесие практически полностью
сдвинуто влево (&Hbzx +5,7 эВ).
Следует принять во внимание, что валентный угол в при-
родном мышьяке (см. данные по кристаллической структуре),
исходя из топологии сетки, во многих случаях может расши-
ряться приблизительно до тетраэдрического угла, слсдова-
Г) О
тельно, исходное состояние 5Ргз уже проявляет заметную не-
устойчивость (EHb-\-SpULp). Для приведенных выше численных
значений реакция
2spP» X + /Г + PULP - 2spULP - Еиь + 2,^ - 2рЕь (3.2)
экзотермическая. Достаточно небольшого искажения угла для
того, чтобы в аморфном мышьяке оказалось возможным образо-
вание положительно и отрицательно заряженных дефектных
центров. По сравнению с нейтральными дефектами с неспарен-
ными электронами пары с переменной валентностью, конечно,
нестабильны. Реакция
2рР» S spP+ + „Р2- + pULP + Ень + 2/ь - A spEb (3.3)
эндотермическая (энергия ~ 1,2 эВ). Поэтому парамагнитные
центры рР? в аморфном As должны быть стабильными. Квап-
товомеханические расчеты позволяют обосновать такое пред-
сказание [185].
Если для реакции (3.3) принять во внимание обусловленную
искажением дестабилизацию термов на определенных ато-
мах сетки (например, частичный переход в направлении к со-
стоянию spPz), то это приведет к большей стабильности пар
с переменной валентностью. Чем больше склонность атомов
к гибридизации, тем больше это способствует образованию за-
ряженных дефектных центров. Поэтому в аморфном фосфоре
можно ожидать в пределах собственного разупорядочения боль-
шей относительной доли заряженных дефектных центров, в то
Аморфные и стеклообразные системы
255
время как в аморфной сурьме преобладают функции р-связей
с неспарепными электронами [186].
Свойства аморфного мышьяка. Благодаря значительной раз-
ности энергий s- и p-связей пара s-электронов мышьяка нахо-
дится, как у халькогена (например, селена), ниже края ва-
лентной зоны, построенной из p-состояний. В халькогенах не-
поделенные пары р-электронов образуют валентную зону.
У мышьяка эти пары участвуют в образовании связей, когда
нет свободных пар электронов. Что касается оптического погло-
щения в области запрещенной зоны или щели подвижности, то
ожидаются соответствующие переходы между связывающими и
антисвязывающими p-состояниями. Последние образуют зону
проводимости.
Найденная из оптических измерений ширина запрещенной
зоны компактного аморфного мышьяка составляет 1,4 эВ [176,
187], для аморфных слоев мышьяка 1,1 эВ. В спектрах погло-
щения ромбоэдрического мышьяка в той же области наблюда-
ется минимум. В кристаллическом состоянии из-за взаимодей-
ствия двойных слоев между собой (г2 = 315 пм) наблюдается
перекрывание валентной зоны и зоны проводимости; в резуль-
тате этого происходит делокализация электронов, что приводит
к возрастанию коэффициента поглощения на низких частотах
[188]. В аморфном As перекрывания зон практически не про-
исходит (г2 = 378 пм), чем и обусловлена типичная для изоля-
тора и полупроводника ширина запрещенной зоны (рис. 4.42, б).
Проводимость компактного аморфного As при комнатной
температуре на 13 порядков ниже, чем у ромбоэдрического As
(о=10-9 См/см). Энергия активации проводимости составляет
0,81 эВ, что несколько больше, чем половина оптической ши-
рины запрещенной зоны. При температуре немного ниже 298 К
проводимость компактного аморфного мышьяка мало зависит
от температуры, а энергия активации проводимости составляет
0,50 эВ [189]. Таким образом, при понижении температуры ме-
няется механизм переноса зарядов. В области малых энергий
активации перенос заряда осуществляется в области локализо-
ванных состояний у края зоны.
По спектру ЭПР в аморфном мышьяке можно обнаружить
намного более низкую концентрацию оборванных гомосвязей,
чем в аморфных Si или Ge. При 4,2 К концентрация этих свя-
зей меняется от 1015 до 1017 см-3. При повышении температуры
(энергия активации 10—30 мэВ) она возрастает и достигает
при 300 К Ю18 см 3. Это можно объяснить, рассмотрев темпе-
ратурную зависимость равновесия (3.3) [190, 191]. Появление
полос люминесценции при освещении аморфного мышьяка и
наблюдение по аналогии с халькогенидными стеклами стоксова
сдвига также приводят к заключению о существовании в аморф-
ном мышьяке заряженных дефектных центров.
256 Глава 3
Сообщается также о модифицировании свойств аморфного
мышьяка с помощью направленного легирования [176] и при-
водятся данные по кинетике кристаллизации аморфного
As [192].
3.1.2.6. Сурьма
При нормальных условиях структура сурьмы аналогична
ромбоэдрическому мышьяку (рис. 3.13, б); г\ = 290,8 пм, г2 =
= 335,5 пм. Валентный угол <р = 95°35', т. е. близок к углу 90°,
характерному для p-связей. Под давлением 85 кбар сурьма пе-
реходит в состояние, для которого характерны только р-связи.
Этот переход приводит к выравниванию межатомных расстоя-
ний: Г1 = г2 = 296 пм, ф = 90°.
Желтая сурьма состоит из аморфной сетки атомов Sb, в ко-
торой часть связей занята водородом [193]. Эту модификацию
сурьмы получают пропусканием кислорода в жидкий SbH3 при
183 К. При более высокой температуре, особенно под дейст-
вием света, образуется черная монотропная модификация
сурьмы, также аморфная. Таким образом, желтая сурьма соот-
ветствует не ранее описанным желтому мышьяку или белому
фосфору, а слабоокрашенным высококонденсированным полиар-
синам или полифосфинам с различным содержанием водорода.
КРР расплавленной сурьмы свидетельствует, что при плавле-
нии происходит уменьшение валентного угла до 90° (гл. 2 [225]).
Среднее координационное число ~6, т. е. соответствует ближ-
нему порядку с примитивной кубической решеткой [194]. Плот-
ности и вязкости расплава см. [195].
Проводимость аморфных слоев сурьмы, полученных путем
катодного распыления, может быть описана процессом переско-
ка электронов и подчиняется правилу Мотта (закон [196].
Исходя из температурной зависимости проводимости, прихо-
дят к выводу о преобладании нейтральных дефектов рРъ с не-
спаренными электронами в аморфной сурьме [186]. Кристал-
лизация аморфных слоев сурьмы происходит при комнатной
температуре.
Так называемую взрывчатую аморфную сурьму получают
электролизом кислых галогенидных растворов Sb(III) при вы-
сокой плотности тока (разд. 2.1.2.3). При электролизе происхо-
дят восстановление до низших галогенидов и спонтанная кри-
сталлизация с сильным экзоэффектом, протекающая с испаре-
нием значительного количества SbX3 (Х = Cl, Br, I) [197].
3.1.3. Стеклообразование халькогенов
Структурная химия элементов серы, селена и теллура ха-
рактеризуется образованием ковалентных связей каждого атома
с двумя соседними. При этом образуются цепочечные поли-
Аморфные и стеклообразные системы 257
меры с различными степенями полимеризации, для которых
благодаря искажению валентного угла (отклонение от 180°) су-
ществует большое многообразие различных структурных кон-
фигураций, например кольцевые молекулы, число членов в ко-
торых также не подчиняется никаким ограничениям. В более
тяжелом гомологе, теллуре, взаимодействия между цепочками
оказывают возрастающее влияние на образовавшуюся струк-
туру, и в а-полонии, наконец, достигается трехмерная система
эквивалентных ортогональных связей с координационным чис-
лом 6 для каждого атома. Гибридизация s- и p-связей умень-
шается при возрастании атомной массы в данной группе пе-
риодической системы. Структуру а-полония определяют исклю-
чительно свойства симметрии р-орбиталей.
З.1.З.1. Сера
Сведения об аллотропных модификациях серы приведены
в обзорных статьях [198—201], поэтому об этом говорится лишь
кратко. Характеристика и непостоянство параметров связей
S—S обсуждаются в работе [202], а современное состояние
вопроса приводится в обзоре [788].
Наряду с серой S8, для которой до сих пор известны три
различные кристаллические структуры, можно указать еще 15
кристаллических модификаций с известной структурой; в со-
вокупности же известны 19 четко охарактеризованных модифи-
каций, большинство из которых построено как молекулярные
структуры из колец серы, например S6, S7, S9, Si0, Sn, S!2, Si8
и S2o. При исследованиях расплавов серы обнаружены кольца
с еще большим числом составляющих атомов (до S34) [203,
789]. Смесь низкомолекулярных форм серы, не включая S8, на-
зывают также л-серой.
Полимерная сера, называемая также пластической или ц-
серой, получается при охлаждении расплава, нагретого выше
432,5 К; полученный плав далее обрабатывают CS2 и таким
образом извлекают сохранившиеся части кольцевых молекул.
Пластическая сера имеет волокнистую структуру и при рас-
тяжении кристаллизуется [204]. Предполагают, что спираль-
ные цепи состоят из ~ 10 атомов серы с тремя периодами иден-
тичности. Расстояние S—S 206,6 пм, валентный угол S—S—S
106°, диэдричсский угол S—S—S—S 85,3°. Для достаточно круп-
ных кольцевых молекул серы, например S12, Si8 и S2o, также
определены параметры связи. Плотность пластической серы
2,01 г/см3. Полимерная сера всегда содержит обрывки кольце-
вых молекул.
При 388,3 К (температура плавления) равновесный рас-
плав состоит из 0,6 % S8, 2,8 % S7, 1,5 % Sx (х<12) и 95,1 °/о
S8 [789]. При 423 К доля колец серы с числом членов, отли-
17 Заказ № 413
258 Глава 3
чающихся от 8, достигает 10% [788]. При 432,5 К вязкость
расплава скачкообразно возрастает более чем на 5 порядков;
расчетным путем найдено, что это соответствует увеличению
средней длины цепи до 105 атомов серы. При дальнейшем по-
вышении температуры наблюдаются следующие изменения: при
533 К содержание Su возрастает до 55%, при 460 К вязкость
расплава максимальна (932 дПа-с), а при более высокой тем-
пературе снова убывает; при 717,7 К сера кипит (рис. 3.19).
В жидкой сере находятся в равновесии многочисленные моле-
кулярные структуры и полимерные цепи, причем их распределе-
ние зависит от температуры. При охлаждении расплава серы
в зависимости от начальной температуры и приложенной ско-
рости охлаждения получают пластическую серу, состоящую из
различных молекулярных структур. Если резко охлаждать рас-
плав ниже 243 К, то получается полимерная сера в стеклооб-
разной форме. Температура стеклования серы, как и следовало
ожидать, сильно зависит от предварительной термообработки
и скорости охлаждения и лежит в интервале 230—250 К. При
высоком давлении получают модификацию серы с металличе-
ской проводимостью, которая при температуре ЮК переходит
в сверхпроводящее состояние [206].
Система S—Se. В разное время в литературе сообщалось
об образовании смешанных колец SenSs-n, например SeS7,
Se2S6, Se3S5 и Se2S5 или Se2Si0 [207—211]. Стабильная моди-
фикация селена построена из винтовых цепочечных структур,
так что возникает вопрос: при каком содержании селена про-
исходит спонтанная полимеризация кольцевых молекул? На
диаграмме состояния составу Se4S6 отвечает слабо выраженная
эвтектика [212, 213]. Согласно рентгенографическому иссле-
дованию [214], сплавы состава Se5,iS2;9, Se3,3S4,7 и Se4i7S3j3 кри-
сталлизуются в структуре известной модификации серы S8; их
можно также описать как твердые растворы на основе един-
ственного компонента общей формулы SeTtSe-n-
Богатые Se расплавы SexSi-Y характеризуются высокой вяз-
костью и в широком интервале составов при затвердевании об-
разуют стекла. При возрастании содержания серы Tg линейно
убывает и при х = 0,7 достигает 273 К [215]. В работе [216]
рассматриваются явления переноса заряда в стеклах системы
S—Se.
Система S—Те. В литературе сообщается о многочисленных
попытках получить соединения в. системе S—Те. TeS2 устойчив
при температурах <253 К [217]. При взаимодействии H2S
с ТеС14 получают с низким выходом темно-красные кристаллы,
которые, согласно рентгенографическому исследованию, пред-
ставляют собой твердые растворы Se, TeS7 и Te2S6 [218].
Аморфные и стеклообразные системы
259
3.1.3.2. Селен
На структуре и свойствах селена весьма часто останавли-
ваются во многих монографиях и обзорных статьях (см., на-
пример, [219—221]). Этот элемент существует в нескольких ал-
лотропных модификациях, из которых термодинамически ста-
билен вплоть до температуры плавления серый тригональный
селен. Обзор некристаллических модификаций селена приво-
дится в работе [205].
Рис. 3.16. Структура кристаллических модификаций Se.
а — тригональный Sc: ri=237,3 пм, г2=342,6 пм; б — трамс-конфигурация селеновых цепей:
О — валентный угол, ф — диэдрический угол; в — структура «короны» молекул Ses в мо-
ноклинных а- и Р-модификациях селена.
Кристаллические модификации. Селен образует три мета-
стабильные моноклинные модификации красного цвета; а- и
0-формы известны уже давно. В работе [790] сообщается
о у-модификации. Эти модификации состоят из молекул Se8.,
которые напоминают корону (рис. 3.16,в). Межатомные рас-
стояния отличаются лишь на немного [222, 790]. Расстояние
Se—Sc составляет 232 или 233—234 пм, для а-модификации
валентный угол 105,9°, диэдрический угол = 101,0° (рис. 3.16, б).
В моноклинной элементарной ячейке а- и 0-селена содер-
жатся 4 молекулы Se8, в у-селене— 8. а-Моноклинный селен
имеет следующие параметры элементарной ячейки: а = 905,4 пм,
6 = 908,3 пм, с= 1160 пм, 0 = 90,81°; плотность 4,39 г/см3. Моно-
клинные модификации селена получают из растворов в CS2.
При медленной кристаллизации получают а-моиоклинный се-
лен, при быстром испарении раствора—0-модификацию. а-Мо-
ноклиниый селен плавится при температуре 417 К и спонтанно
превращается в тригональный селен; 0-модификация ведет себя
подобным образом, но выше 373 К. Как видно из рис. 3.16, в,
кольца Se8 можно рассматривать как ^«^-конфигурацию четы-
17*
260 Глава 3
рсх атомов Se. При фазовом переходе образуется транс-конфи-
гурация в результате разрыва кольца (рис. 3.16,6).
Структура тригонального селена приведена на рис. 3.16,а
[223]. Винтовые цепи с периодом идентичности 3 связаны
в кристаллическую решетку тригональной сингонии с парамет-
рами элементарной ячейки: а = 436,8 пм, с = 495,8 пм. Кратчай-
шее расстояние Se—Se и = 237,3 пм, расстояние до вторых со-
седей внутри селеновой цепи 372 пм, а валентный угол 103,1°.
Диэдрический угол ср, характеризующий степень закручивания
цепи, достигает 100,6°, приближаясь к величине угла в моле-
куле Sc8. Расстояние г2 выбранного атома до четырех вторых
соседей (рис. 3.16,а), принадлежащих к трем спиралям селена,
составляет 342,6 пм. Такая структура связана с кубической ре-
шеткой а-полония, имеющей 6 одинаковых длин связей.
Плотность тригонального селена 4,819 г/см3 (298 К). Коэф-
фициент термического расширения и молекулярная теплоем-
кость приведены в табл. 2.6. Энтальпия плавления 6,23±
±0,17 кДж/моль [224]. Для удельной проводимости в литера-
туре приводятся различные значения: 10-5—10-6 См/см [220]
и 10~10 См/см [225] (при 298 К). При высоких давлениях Se
и Те образуют кубическую решетку, аналогичную сс-полонию,
причем происходит переход в сверхпроводящее состояние; тем-
пература перехода зависит от давления (6,7 К при 13 ГПа и
3,3 К при 5,6 ГПа) [226, 227].
Аморфный селен. Красный аморфный селен (плотность
4,27 г/см3) получают восстановлением подкисленных растворов
селенитов:
H2SeO3 + N2H4 - Se + ЗН2О + N2 (3.4)
H2SeO3 + 2SO2 + H2O - Se ± 2H2SO4 (3.5)
или охлаждением паров Se до 85 К. Структурных данных для
красного селена до сих пор мало; согласно новейшим исследо-
ваниям, растворимость этой модификации в CS2 нельзя рас-
сматривать в качестве доказательства молекулярного строения.
Энтальпия перехода красного аморфного селена в тригональ-
ный составляет при 298 К 12,5± 1,7 кДж/моль. Эта величина
в 2,5 раза больше, чем изменение энтальпии при рекристалли-
зации стеклообразного селена. Можно было бы думать, что бо-
лее высокое содержание энергии в полимерном селене обус-
ловлено большим содержанием напряженных конфигураций,
например цис-группировок с искаженными диэдрическими уг-
лами, что позволяет участкам кольцевых молекул Se8 свободно
вступать во взаимодействие с растворителем. Из химии серы
давно известно об образовании («развертывании») цепочек серы
при разрыве колец S8.
Аморфные и стеклообразные системы
261
Рис. 3.17. Колебательные спектры различных модификаций Se.
а — ИК-спектры красного и черного аморфного селена [2301; б — ИК-спектр стеклообраз-
ного и кристаллического моноклинного a-Se; в ~ КР-спектр стеклообразного и a-Se; г —
КР-спектр тригонального и стеклообразного селена [233].
Если нагревать красный аморфный селен, то уже при 310 К
наблюдается эндотермический эффект, и образуется черный
аморфный селен. Энтальпия перехода — 0,4 кДж/моль [228,
229]. Структура черного аморфного селена до сих пор неиз-
вестна. ИК-спектры обеих аморфных модификаций (рис. 3.17,а)
очень близки [230].
Методом вакуумного испарения получают аморфные слои
Se, которые были подробно изучены в связи с их широким при-
менением в электрофотографии. Аморфные слои Se, полученные
путем вакуумного испарения, полностью растворяются в CSs-
При небольшой толщине красные слои прозрачны.
Состав паровой фазы селена очень сложен. Наряду с мо-
лекулами Se8, Se2 и Se7 при температуре —600 К преобладают
группировки Se6 и Se5 [231]. Концентрация последних вплоть
до 1000 К сохраняется неизменной. При этом с повышением
262 Глава 3
г пм
Рис. 3.18. Дифференциальные кривые радиального распределения компактного
стеклообразного селена (а) и аморфных слоев Se, осажденных при темпера-
туре подложки 298 К (б) и 77 К (е) [232].
температуры растет доля молекул Se2 и 8ез за счет более круп-
ных молекул. В зависимости от условий испарения и конденса-
ции (температура и скорость испарения, температура под-
ложки) можно получить аморфные слои с различной структу-
рой. При этом необходимо исключать загрязнение примесями.
Следы примесей могут вызвать замедление или ускорение кри-
сталлизации во всех аморфных формах Se; образование мак-
рокристаллических включений, как правило, мешает проведе-
нию измерений.
На рис. 3.18 приведены дифференциальные КРР (см. также
рис. 2.29) для компактного стеклообразного селена и для двух
аморфных слоев Se, полученных при различных температурах
подложки [232]. Во всех трех случаях кривые чрезвычайно по-
хожи, что позволяет сделать вывод, как и для аморфных Si
и Ge, о не зависящем от условий получения равновесной кон-
Аморфные и стеклообразные системы 263
фигурации пространственном расположении областей ближнего
порядка в указанных аморфных структурах. На рис. 3.17,6 и в
для сравнения приведены ЙК-спектры стеклообразного и кри-
сталлического красного a-Se и КР-спектры стеклообразного се-
лена и обеих кристаллических модификаций [233]. Согласно
приведенным экспериментальным данным (рис. 3.17 и 3.18),
очевидно значительное сходство ближнего порядка всех форм
селена. В то же время весьма близкое сходство КРР свиде-
тельствует о том, что дифракционными методами нельзя уло-
вить тонкие структурные различия.
Близость ЙК-спектров стеклообразного и моноклинного
a-Se (рис. 3.17,6) рассматривается как подтверждение предпо-
ложения о присутствии в стеклообразном Se значительного ко-
личества молекул Se8 (см. разд. 2.4.1.4) [234] (см. также лите-
ратуру к гл. 2 [293]); такое предположение возникло при изу-
чении растворения этих форм селена. Только недавно обратили
внимание на то, что присутствие сегментов цепей в ц«с-кон-
фигурации с немного искаженными углами может вызвать
в спектре такие же колебания. Для изолированных кольцевых
и цепочечных молекул Se можно ожидать одинаковую частоту
валентных колебаний [235]. Вследствие взаимодействия между
соседними цепями частота колебаний в тригональном селене
понижается с 256 до 237 см-1; в стеклообразном селене из-за
отсутствия таких взаимодействий характеристическая частота
для молекул Se8 возрастает. Однако нельзя без других дока-
зательств считать наблюдаемое сходство в спектрах как до-
казательство существования молекул Sc8.
Расплав селена. Тригональный Se плавится при 490 К. При
этом плотность скачкообразно меняется от 4,70 до 4,01 г/см3,
а при дальнейшем повышении температуры монотонно убывает
[236]. На температурной зависимости проводимости при тем-
пературе плавления наблюдается скачок (см. литературу к гл. 1
[28]). Изменение проводимости при плавлении сильно зависит
от удельной проводимости предшествующей твердой фазы, а по-
следняя в свою очередь — от содержания примесей. Для чи-
стого селена при плавлении проводимость убывает примерно на
полпорядка. Удельная проводимость расплава селена при
490 К 4-10~9 См/см [225]; преобладает р-тип проводимости.
Термическая энергия активации электропроводности равна
2,31 эВ. Только при температурах >1570 К в области давлений
выше критического селен приобретает металлическую прово-
димость [237].
Структуру расплава селена часто рассматривают при изу-
чении структуры и типа связи расплава серы, хотя эти эле-
менты сильно различаются по термодинамической устойчиво-
сти своих структурных форм. Для селена стабильная форма —
264 Глава 3
Рис. 3.19. Температурные зависимости вязкости т| и степени полимеризации Р
расплавов серы и селена [238, 239].
цепочечная структура тригонального селена, а образующиеся
при высоких температурах расплава молекулы Se8 метаста-
бильны. Напротив, для серы метастабильной формой считается
цепочечная структура.
На рис. 3.19 приведены температурные зависимости вязко-
сти ц и степени полимеризации Р (выраженной в структур-
ных единицах из 8 атомов) для расплавов серы и селена
[238, 239].
Начало полимеризации кольцевых молекул в расплаве се-
ры при 432,5 К связано с возрастанием вязкости на 5 поряд-
ков. Выше этой температуры превращения вязкость вновь убы-
вает, при этом уменьшается среднее число структурных единиц
серы (Р), осуществляющих текучесть. Вязкость селена при
температуре плавления составляет ~25 дПа-с, а при темпе-
ратуре кипения 958 К понижается до 10-2 дПа-с.
Если интерпретировать расплавы серы по аналогии с рас-
плавами селена, то средняя длина цепей в области переохла-
ждения должна возрастать. В области температур затвердева-
ния стеклообразного селена (303—313 К) кажущаяся энергия
активации вязкого течения возрастает в 10 раз, и при пониже-
нии температуры текучесть убывает [240]. Температура по-
лимеризации расплава селена, аналогичная температуре превра-
щения серы, лежит ниже температуры плавления в области
более высокой вязкости и составляет 350 К [238, 239].
Такого же, как у серы, снижения вязкости у селена не
наблюдается. Для расчета структурного равновесия кольца
цепи в расплаве Se учитывалось, что при охлаждении рас-
плава от температуры 490 К в зависимости от скорости охла-
Аморфные и стеклообразные системы
265
ждения до 40 % стекла растворяется в CS2 (гл. 2 [293]).
При охлаждении расплава от 700 К доля растворенного стекла
составляет только 18%. При этом предполагалось, что селен,
растворимый в CS2, в стекле существует в виде молекул See.
При охлаждении возможен также разрыв структурных единиц
Se8 с образованием конфигураций, соответствующих фрагмен-
там цепей. Поэтому общеизвестные и распространенные пред-
ставления о структуре распла-
ва Se могут вызывать неко-
торые сомнения и нуждаются
в проверке.
Против предположения о
такой большой доле молекул
Se8 в расплаве Se и стеклооб-
разном Se, полученном при
охлаждении расплава, свиде-
тельствуют и эксперименталь-
ные данные. Например, кон-
центрация молекул Se8 в рас-
плаве, нагретом до 495 К, по
сравнению с расплавом, на-
гретым до 675 К, должна ме-
няться в 2 раза (см. литера-
туру к гл. 2 [293]. Однако на
ИК-спектрах соответствую-
щим образом полученных сте-
Рис. 3.20. Области с цис- и транс-
конфигурациями в цепочечной струк-
туре стеклообразного селена.
кол не наблюдается практиче-
ски никаких различий [241]. ИК-Спектр стекла после экстрак-
ции в CS2 также почти не меняется [230]. Установлено, что
стеклообразный селен растворяется в CS2 только после облу-
чения светом с энергией >2,3 эВ [242]. В цитируемой выше
работе (см. литературу к гл. 2 [293]) об этом ничего не гово-
рится. В работе [242] молекулярные спектры компактного
стеклообразного селена интерпретируются, исходя из предпо-
ложения о цепочечной структуре, которая характеризуется не-
правильной последовательностью цис- и транс-конфигураций.
Участок такой цепи изображен на рис. 3.20.
Стеклообразный селен можно достаточно просто получить
в виде относительно больших компактных образцов путем за-
калки расплава (критическая скорость охлаждения Ркрит^
^20 К/мин *) при условии достаточно низкой концентрации
примесей, вызывающих кристаллизацию. Температура стекло-
вания такого материала 303,2 К. Плотность при 298 К
4,285 г/см3, коэффициент термического расширения (КТР)
а<т§ =4,0-10 5 К-1 и a>Tg = 15-10"5 К-1, модуль упругости
£=10,2 ТПа, модуль сдвига G = 3,7 ТПа, модуль сжимаемости
266
Глава 3
х=1,04 ТПа-1 [220]. Соотношения давление—объем—темпера-
тура в жидком и стеклообразном селене подробно изучаются.
При повышении давления до 200 МПа Tg возрастает до 318 К
[243].
Для выяснения структуры стеклообразного селена во мно-
гих работах привлекаются методы рентгеновской и нейтронной
дифракции [232, 244—247, 249—255] (см. также литературу
к гл. 2 [225, 295]). Независимо от условий получения для
стекла и расплава селена наблюдается большое сходство
в ближнем порядке. На КРР (рис. 2.29 и 3.18,а) проявляется
максимум при 232 пм, площадь которого соответствует коор-
динационному числу 2, т. е. каждый атом Se связан с двумя
ближайшими атомами, находящимися примерно на таком же
расстоянии, как в кристаллических модификациях. Второй мак-
симум при 372 пм соответствует расстоянию до двух вторых
соседей в цепочках или кольцах кристаллических модификаций
Se (рис. 3.16), но его площадь соответствует нс двум, а шести
атомам. Очевидно, расстояние до четырех атомов Se, которые
принадлежат в тригональном селене трем соседним цепям,
возрастает от 342,6 пм до вандерваальсова расстояния 372 пм.
Расстояние (372 пм) случайно совпадает с расстоянием до
вторых соседей в селеновой цепочке.
Что касается других максимумов, то на КРР выражены
только относительно нерезкие максимумы вблизи 570 и 720 пм.
Если обратиться к рис. 3.18, максимум при 570 пм нельзя
скоррелировать ни с одним расстоянием любой кристалличе-
ской модификации Se. При 500 и 525 пм на КРР аморф-
ных слоев Se (298 К) не наблюдается максимумов. Кри-
сталлитные модели в этом случае неприемлемы. Модели па
основе деформированных колец Se8 [132], сочетания молекуляр-
ных структурных единиц Se8 и колец с большим числом членов
с селеновыми цепочками [245, 247] (гл. 2 [225]) или структуры
на основе молекул See [244, 256] частично согласуются с экс-
периментальными кривыми в области больших расстояний.
Во многих работах приводятся структурные модели, где пре-
обладают селеновые цепочки [246, 249—251, 254, 255]. Разли-
чия между кольцевыми структурами Se8 и цепочками из селе-
новых атомов должны проявляться в четвертой координацион-
ной сфере. Отсутствие максимума на расстояниях 500—525 пм
позволяет принять модель Полка (гл. 2 [300]), основанную
на цепочечной структуре, в которой диэдрический угол мо-
жет принимать любые значения [253—255] (см. также литера-
туру к гл. 2 [295]). КРР, рассчитанная на основе цепочки,
построенной из 200 атомов, довольно хорошо согласуется с экс-
периментом [253] (см. также литературу к гл. 2 [295]). Энер-
гетические расчеты рентгеновских и УФ-фотоэлектропных спек-
тров показали, что в цепочках стеклообразного селена диэдри-
Аморфные и стеклообразные системы 267
ческий угол сильно изменен (см. литературу к гл. 2 [296]).
Исходя из степени sp-гибридизации валентных орбиталей, ди-
эдрическому углу 102° соответствует минимальная энергия, что
также должно влиять на структуру расплава. Предполагается
статистическое распределение цис- и тра«с-конфигураций, обра-
зованных третьим и четвертым атомами Se (рис. 2.38) со всеми
другими атомами цепочки, так что образуется нерегулярная
последовательность конфигураций (участок такой цепочки при-
веден на рис. 3.20). Экспериментальная КРР стеклообразного
Se хорошо совпадает с кривой, рассчитанной по этой модели
[254).
С этими данными по структуре стеклообразного селена рас-
ходятся расчеты зонной структуры, в которых удовлетвори-
тельно интерпретированы экспериментальные результаты, по-
лученные методом рептгеноэлектронной спектроскопии
[258, 259].
В разд. 2.4.2.5 и 2.5.2 сообщается о собственных дефектах
в расплаве селена и типе точечных дефектов, которые возни-
кают в одномерных структурных единицах, из которых состоит
расплав селена. Дефекты влияют прежде всего на электриче-
ские свойства стеклообразного Se, и их можно обнаружить из
спектров фотолюминесценции, из плотности спинов, индуциро-
ванных облучением при низких температурах, а также при
появлении соответствующих полос поглощения (разд. 4.2.2.2).
В литературе приводятся также квантовохимические рас-
четы дефектных состояний для стеклообразного селена [260].
Свойства стеклообразного селена. Очищенный компактный
стеклообразный селен при комнатной температуре является вы-
сокоомным полупроводником (о^Ю-16 См/см). Квазистатиче-
ская диэлектрическая проницаемость этого материала 6,15±0,06
[261]. Расчет термической энергии активации из данных по
проводимости и термо-э. д. с., дает приблизительно совпадаю-
щие результаты 1,0 и 1,15 эВ. В широком интервале темпера-
тур для стеклообразного Se и расплава наблюдается линейная
зависимость Igo от 1/Т [262, 263]. По измерениям дрейфо-
вой подвижности найдены подвижность электронов 6• 10—з см2/
(В-с) и подвижность дырок Ю-1 см2/(В-с) [262, 264, 265, 394].
Установлено, что термическая энергия активации подвижности
мала, она составляет 0,16—0,3 эВ. В стеклообразном селене
доминирует дырочный тип проводимости (разд. 4.1.3.4). Пере-
нос заряда путем скачков электронов между локализованными
состояниями в области энергии Ферми, который описывается
правилом Мотта (зависимость Т '4), не был обнаружен. В от-
личие от аморфного мышьяка в аморфном селене иод дейст-
вием давления механизм переноса заряда не меняется [266].
На проводимость стеклообразного селена сильно влияют
268 Глава 3
определенные примеси, например оксиды, галогениды, щелоч-
ные и тяжелые металлы (разд. 4.1.3.5).
Оптическая ширина запрещенной зоны стеклообразного се-
лена 2 эВ; сдвинута в коротковолновую область по сравне-
нию с величиной для тригонального селена; она несколько
меньше удвоенной термической энергии активации проводимо-
сти [267]. В работе [268] приводится значение оптической ши-
рины запрещенной зоны для аморфных слоев Se.
Изучены также время релаксации стеклообразного селена
(разд. 2.2.1.2 и 2.2.2.1) [269] (см. также литературу к гл. 2
[138]) и кинетика кристаллизации селена (разд. 2.3.1.1)
[270, 271].
3.1.3.3. Теллур
Теллур кристаллизуется в том же структурном типе, что и
тригональный селен (рис. 3.16, а) [223]. Спиральные цепи с рас-
стояниями Те—Те 287,8 пм образуют кристаллическую решетку
с параметрами элементарной ячейки: а = 445,1 пм, с = 592,6 пм;
валентный угол 0= 101,8°, диэдрический угол <р, образованный
плоскостью, в которой лежат 3 атома Те, и четвертым атомом
~ 100°. Каждый атом Те, кроме двух атомов, с которыми он
связан внутри цепочки, связан еще с 4 атомами, находящимися
на расстоянии 349 пм и принадлежащими трем соседним це-
почкам. Связь между цепочками только в ~2 раза слабее, чем
связь внутри цепей. Плотность теллура 6,25 г/см3 (298 К),
удельная электропроводность при комнатной температуре
— 2 См/см. Электрические и оптические измерения приводят
к значениям для ширины запрещенной зоны (0,33 эВ) [267].
Температура плавления теллура при нормальном давлении
725 К, температура кипения 1663 К.
При температуре плавления плотность резко уменьшается
от 6,21 до 5,770 г/см3 [226, 272]. Как при более высоких тем-
пературах, так и при понижении температуры в области пере-
охлажденного расплава плотность уменьшается, так что на кри-
вой температурной зависимости плотности можно заметить ано-
малию. Удельная проводимость теллура в отличие от селена
при переходе в расплав, несмотря на понижение плотности, воз-
растает от — 180 до 1800 См/см (см. литературу к гл. 1 [28,
33]). Температурный коэффициент электропроводности Те по-
ложительный; для расплава Те энергия активации проводимо-
сти 0,5 эВ [273]. Проводимость Те возрастает при повышении
температуры, достигая максимального значения —3000 См/см
при 1173 К, а при дальнейшем повышении температуры убы-
вает.
Структура расплавленного Те изучена методами дифрак-
ции нейтронов [274] и рентгеновской дифракции [275]. При
Аморфные и стеклообразные системы 269
температуре 870 К первый и второй максимумы на КРР соот-
ветствуют расстоянию 297 и 381 пм, а их площади отвечают
координационному числу 3. Третий отчетливый максимум соот-
ветствует расстоянию 452 пм. Сравнение межатомных расстоя-
ний показало, что ближний порядок в жидком теллуре выше
температуры плавления отличается от ближнего порядка в кри-
сталлическом состоянии по координации. В кристаллической ре-
щетке имеются два более коротких и четыре более длинных
межатомных расстояния, а в расплаве — три ближайших и три
более дальних атома. Аномалию, наблюдаемую на температур-
ной зависимости плотности в области переохлажденного рас-
плава, можно объяснить частичной перестройкой структуры
жидкости в направлении большего координационного соответ-
ствия кристаллической решетке.
Структура расплавленного теллура близка к структуре ром-
боэдрического мышьяка. Три p-связи определяют взаимодейст-
вие с тремя ближайшими и тремя более дальними атомами.
Таким способом образуются локальные слоистые структурные
области, которые связаны с участками соседних слоев. Валент-
ное состояние большинства атомов Те в расплаве можно опи-
сать с помощью С° или D° (по аналогии с состояниями
атомов в расплавленных халькогенах см. разд. 2.4.2.5 и
2.5.2, рис. 2.58). Валентное состояние s2p4 атомов в кристалли-
ческом Те, которое ответственно за образование двух ковалент-
ных связей с преобладающим р-характером и поэтому за це-
почечную структуру, переходит в расплаве в состояние s2p3
с тремя ковалентными p-связями, а один электрон переходит
на антисвязывающий уровень центров ТеТе3. . Электрон, отще-
пившийся от атома Тс, переходит с антисвязывающего уровня
в зону проводимости; он в значительной степени делокализован,
что обеспечивает большую величину электропроводности. При
температурах ~1970 К первый максимум на КРР при 307 пм
примерно соответствует координационному числу 6, в то время
как второй максимум значительно ослаблен. При высоких тем-
пературах межатомные расстояния выравниваются и структура
расплава приближается к структуре а-полония.
Структурные изменения, обусловленные плавлением теллура,
выражаются в значительно более высоком значении энтропии
плавления по сравнению с этим же параметром для селена
[энтропия плавления для Те 24,3 Дж/(моль 1 • К 1), для Se
12,6 Дж/(моль1-К’)] [274]. Исследование методом нейтрон-
ной спектроскопии показало, что колебательный спектр рас-
плава совершенно отличается от спектра кристаллического
теллура [276].
Вследствие частичной делокализации валентных электронов
вязкость расплава Те настолько понижена, что переход в стек-
270 Глава 3
лообразное состояние может достигаться только при исклю-
чительно высоких скоростях охлаждения. Чтобы получить тон-
кие аморфные слои Те, требуются скорости охлаждения
~1010К/с (см. литературу к гл. 2 [26]). Тё аморфного Те
285 К (см. литературу к гл. 2 [146]). Аморфные слои также
можно получить конденсацией паров теллура на подложки, ох-
лажденные до 77 К. Найденная путем экстраполяции к ком-
натной температуре проводимость этих слоев составляет
~10-4 См/см, термическая энергия активации проводимости
0,44 эВ [267]. Как показали измерения, происходящие в этих
слоях при изменении температуры, процессы обратимы только
до температуры ~200 К. Выше этой температуры происходит
кристаллизация. В интервале температур 77—170 К перенос
заряда осуществляется электронами по прыжковому механизму
в соответствии с правилом Мотта (закон Г-1''4; разд. 4.1.3.1).
Энергия активации проводимости 0,046 эВ [277].
Система Те—Se. Твердые растворы обоих элементов при
плавлении характеризуются почти идеальным поведением.
В системе Se—Те существует непрерывный ряд твердых рас-
творов [278—280]; сообщается об отклонении этих твердых рас-
творов от закона Вегарда [281].
Если к расплаву Те прибавить Se, то максимум плотности
в системе TexSei_x смещается в область высоких температур
[236]. Аномальному ходу температурной зависимости плотности
соответствуют изменения электропроводности [221, 231]. В об-
ласти температур, где ближний порядок в расплаве больше
соответствует селену, чем теллуру, наблюдается более высокая
энергия активации электропроводности.
Расплав состава Teo,5Seo,5 вплоть до температуры 973 К, при
которой происходит изменение структуры, характеризуется
почти такой же энергией активации вязкого течения, как и рас-
плавленный селен (для Teoi5SeOi5 En = 51,9 кДж/моль, для Se
Еп = 58,2 кДж/моль) [283, 284]. Считают, что расплавы, богатые
Se, состоят из спиральных цепочек, в которых атомы Se ста-
тистически замещены на Те, с кольцами Te2Se6 [275, 285, 288].
Компактные образцы стекол состава TcxSci_x (масса 10—
50 г) можно получить в ампулах из кварцевого стекла путем
закалки расплава в воде. Таким образом получают стекла
с содержанием Те от 0 до 15 ат.%. Стекла с содержанием Те до
~40 моль.% получают охлаждением расплава путем его раз-
брызгивания (spray cooling) [286]. Методом катодного распы-
ления получают аморфные слои с содержанием Те до ~67 ат.%.
Для состава Seo,5Teo>5 по сравнению с чистыми компонентами
Тё повышается до 330 К. Эти стекла по своим свойствам в значи-
тельной степени близки к стеклообразному селену. В работе [287]
изучены релаксационные явления в стеклах системы Se—Те.
Аморфные и стеклообразные системы
271
3.2. Двойные стеклообразующие соединения
и системы *
3.2.1. Триоксид бора и системы на основе оксидов
и халькогенидов фосфора, мышьяка и сурьмы
3.2.1.1. Триоксид бора
Модификации. В2О3 был относительно поздно получен в кри-
сталлическом состоянии путем дегидратации НВО2 [289]. Мо-
нокристаллы получали посредством медленного охлаждения
расплава под давлением от 1 до (максимально) 2 ГПа [290,
291]. Это соединение плавится при температуре 748 К, энтро-
пия плавления 30 Дж/(моль-К). Согласно диаграмме состоя-
ния, температура плавления значительно возрастает при повы-
Рис. 3.21. Цепочки ^ВО2
в структуре LiBO2 дают пред-
ставление о структуре кристал-
лического В2О3.
шении давления. В области повышенных температур плавления
благодаря низкой вязкости реализуются благоприятные усло-
вия для зародышеобразования и роста кристаллов В2О3.
Исследование структуры показало, что В2О3 кристаллизу-
ется в гексагональной решетке с параметрами элементарной
ячейки: « = 433, 6 пм, с = 834,0 пм; состав элементарной ячейки
выражается 3 формульными единицами. Плотность кристалли-
ческого В2О3 2,56 г/см3. Кристаллическая решетка В2О3 пред-
ставляет собой относительно разрыхленную пространственную
сетку, состоящую из связанных друг с другом тригональных
структурных единиц BO3/j среднее межатомное расстояние
В—О 137,2 пм. Структуру В2О3, подобно структуре LiBO2, мо-
жно представить в виде бесконечных плоских лент или цепей.
В цепочках ВО2 атомы кислорода несут отрицательный заряд
(рис. 3.21). В кристаллическом В2О3 эти атомы кислорода
* Сведения о стеклообразовании и фазовых равновесиях в 25 двойных
и более чем в 150 тройных халькогенидных системах приводятся в книге:
Виноградова Г. 3. Стеклообразование и фазовые равновесия в халькогенид-
ных системах.—М.: Наука, 1984, 174 с,—Прим, перев.
272 Глава 3
играют роль мостиковых лигандов, так как связывают парал-
лельно расположенные ленты одного слоя с соседним слоем,
в котором ленты расположены почти перпендикулярно первому
слою, в трехмерную пространственную сетку [291].
Под давлением >20 кбар В2О3 переходит в другую кристал-
лическую модификацию со значительно большей плотностью
(р = 3,11 г/см3) [292, 293]. Эта модификация кристаллизуется
в ромбической решетке, причем атомы бора имеют четверную
координацию, а атомы кислорода — отчасти тройную.
Расплав В2О3 получают обезвоживанием Н3ВО3 при посте-
пенном повышении температуры. При очень медленном охла-
ждении расплав затвердевает в стекло, в котором обнаружи-
вается некоторое количество воды в виде связанных ОН-групп.
Плотность стеклообразной фазы зависит от содержания ОН-
групп и может меняться от 1,81 до 1,86 г/см3; температура стек-
лования 530 К.
Структура стеклообразного В20з. Изменение плотности при
переходе от кристаллического В2О3 к стеклообразному и относи-
тельно большая величина энтропии плавления говорят о том,
что переход в стеклообразное состояние сопровождается суще-
ственным изменением структуры. Кристаллизация обычно свя-
зана со значительной перегруппировкой структурных единиц,
в случае В2О3 она сильно замедляется вследствие высокой вяз-
кости расплава. Однако по сравнению с кремнеземом вязкость
В2О3 значительно ниже. При сравнительно близких средних
значениях энергии связи Si—О и В—О (444 и 460 кДж/моль)
вязкости обоих расплавов различаются, например, при темпе-
ратуре 1533 К в 10-11’6 раз [294]. Структура стеклообразного
В20з исследована в работах [295] (см. также литературу
к гл. 2 [257]).
Исследования методом ЯМР свидетельствуют о существова-
нии в стеклообразном В20з тригональных плоских структурных
единиц [ВО3] [296—298]. При этом исключается присутствие
в стекле большой концентрации тетраэдрически координирован-
ных атомов бора. КР-Спектр стеклообразного В2О3 характе-
ризуется присутствием интенсивной полосы vj при ~806 см-1.
Она соответствует симметричному валентному колебанию струк-
турных групп ВОз/2. Антисимметричное валентное колебание
v3 и в ИК-, и в КР-спектрах проявляется как группа полос
в интервале 1250—1430 см-1. Деформационные колебания V4 =
= 740 см-1 также наблюдаются в ИК- и в КР-спекграх. Дефор-
мационные колебания V2, изгибающие плоскости [ВО3], напро-
тив, можно ожидать только в ближней области ИК-спектра.
Уже простой анализ расщепления колебательного спектра по-
зволяет предполагать существование в стекле бороксоловых ко-
лец [299]; согласно новейшим исследованиям, считается на-
Аморфные и стеклообразные системы
273
дежно установленным, что именно наличие этих колец в стекле
определяет его структуру [808].
Два первых максимума на КРР стеклообразного В2О3 мо-
жно удовлетворительно интерпретировать на основе предпо-
ложения о тригональных плоских структурных единицах [ВОз]
с кратчайшими межатомными расстояниями В—О 137 пм и
О—О 237 пм. Для описания структуры стеклообразного В2О3
привлекают представления о непрерывной беспорядочной сетке,
состоящей из связанных друг с другом структурных группиро-
вок BOs/j (рис. 3.22,а) (см. литературу к гл. 2 [229]). С по-
Рис. 3.22. Проекция сетки стекла В2О3 со статистическим распределением
структурных единиц ВО3,г (а) и предпочтительным образованием бороксоло-
вых колец (б).
мощью КРР можно также обнаружить в стеклообразном В2О3
на расстоянии ~600 пм значительное структурирование. Как
показано в работе [301], дальнейший ход КРР можно хорошо
описать на основе беспорядочной трехмерной сетки из свя-
занных между собой бороксоловых колец, как схематически
показывает проекция на плоскость (рис. 3.22,6). Установлено
существование структурного мотива, например, как в кристал-
лическом соединении Cs2O-9B2O3 [302]. Экспериментальную
КРР можно также интерпретировать исходя из представлений
о беспорядочной сетке из структурных групп ВОз/2 [303]. С по-
мощью структурной модели (рис. 3.23), предложенной для
аморфного мышьяка (см. литературу к гл. 2 [301]), которая
основана на тригонально-пирамидальных структурных единицах
[AsAs3], в работе [303] рассчитана КРР сетчатой структуры,
состоящей из 533 атомов В и 866 атомов О (при этом струк-
турные единицы заменены па [ВОз]). Новые расчеты, приве-
денные в работе [791], основаны на статистической модели.
18 Заказ № 413
274 Глава 3
Лучше всего описывает результаты экспериментов непрерыв-
ная беспорядочная сетка из бороксоловых колец [792], что
также подтверждается новыми измерениями дифракции нейтро-
нов [809].
В разд. 2.2.1.3 сообщалось о вязком течении расплава В2О3.
Температурная зависимость вязкости В2О3 приводится в работе
[793]. Значительное различие энергий активации вязкого тече-
ния расплавов SiO2 и В2О3 (710 и 310 кДж/моль) при примерно
Рис. 3.23. Модель структуры стеклооб-
разного В2О3 — производная от струк-
туры аморфного мышьяка [303].
одинаковых энергиях связи Si—О и В—О (табл. 2.11) позво-
ляет сделать вывод, что механизм перемещения связан с из-
менением координационного числа атомов бора. Принимая во
внимание сильнокислотные свойства координационно-ненасы-
щенного бора, можно предположить частичное образование
трехсвязанных атомов кислорода и четырехсвязанных атомов
Рис. 3.24. Механизм текучести расплава В2О3.
бора. Путем перемещения структурных групп такие конфшу-
рации должны вновь образовывать связи В—О—В с обычным
координационным числом 3 для атома В и 2 для кислорода
(рис. 3.24). Благодаря скольжению структурных группировок
энергия, необходимая для разрыва связей, значительно пони-
жается. Подобный механизм вязкого течения обсуждался для
расплавов стеклообразного кремнезема. Такому механизму спо-
собствует в случае стеклообразного В2О3 кислотный (по Льюису)
характер структурных группировок [ВО3] при одновременном
присутствии основных электронных пар на мостиковых атомах
кислорода.
Аморфные и стеклообразные системы
275
3.2.1.2. Оксиды фосфора, мышьяка, сурьмы
Оксиды, фосфора. Р4Ое кристаллизуется в структуре уротро-
пина (рис. 3.25); он плавится при температуре 296,9 К с образо-
ванием бесцветной жидкости, которая выше температуры 483 К
диспропорционирует с образованием красного фосфора и оксида
среднего состава РОг. Описаны также производные Р40б оксиды
фосфора Р4О7, Р4О8 и Р4О9 [304, 794]. Конечный продукт окис-
ления — оксид фосфора (V) Р4Ою, для которого наряду с моди-
фикацией молекулярной структуры известны еще две полимер-
ные и одна стеклообразная модификации.
Н4О6 или As4o5 р^°ю
стеклообразный Р205
Рис. 3.25. Структура Р4О6, Лз40б, Р4Ою и стеклообразного Р2О5.
Для оксида мышьяка As4Os параметры решетки приведены в скобках.
В Р4О10 по сравнению с Р4О8 каждый из четырех атомов Р
связан еще с одним атомом О (рис. 3.25). В ромбоэдрической
элементарной ячейке Р4Ою с параметрами п = 744 пм, а = 87°
содержатся 2 формульные единицы. Плотность Р4Ою 2,30 г/см3.
Р4Ою плавится под давлением собственного пара при темпера-
туре 693 К и возгоняется при 632 К при нормальном давлении.
Это соединение всегда образуется при сублимации различных
оксидных форм фосфора (V), но из-за своей явно выраженной
гигроскопичности оно устойчиво только при полном отсутст-
вии влаги. Твердый P4Oin медленно превращается под влия-
нием постоянно присутствующих следов воды в высокомолеку-
лярную модификацию, образованную трехмерной сеткой струк-
турных группировок О = РО3/2.
Это превращение сильно ускоряется при повышении темпе-
ратуры. Соответственно расплав Р4Ою быстро полимеризуется
с образованием стекла, из которого при температуре 700 К вы-
деляется кристаллическая модификация со структурой прост-
ранственной сетки. Эта модификация кристаллизуется в ромби-
ческой сингонии с параметрами элементарной ячейки а =
= 1630 пм, b = 814 пм, с = 526 пм; в элементарной ячейке содер-
18*
276 Глава 3
жится 8 формульных единиц Р2О5; плотность этой модифика-
ции 2,72 г/см3 [305]. При нагревании этой модификации при
393 К в течение месяца образуется другая модификация с бо-
лее низкой текучестью. Она характеризуется слоистой струк-
турой с параметрами элементарной ячейки а = 923 пм, 6 = 718 пм,
с = 494 пм; элементарная ячейка включает 4 формульные еди-
ницы Р2О5 [306]. Высоковязкие расплавы затвердевают с обра-
дованием стекла плотностью 2,37 г/см3 при комнатной темпе-
ратуре.
Стеклообразный Р2О5, как и его кристаллические модифи-
кации, сильно гигроскопичен. Его степень полимеризации, тем-
пература стеклования и другие свойства в основном определя-
ются содержанием ОН-групп. ИК-Спектр стеклообразного Р2О5
подтверждает симметрию C3V структурной единицы О = РО3/2.
Изучен [308] также КР-спектр стеклообразного Р2О5 и по со-
поставлению со спектром POF3 сделано отнесение полос. Двой-
ной связи Р = О соответствует полоса поглощения ~ 1390 см ’.
Оксид мышьяка(111). Одна из модификаций оксида мышья-
ка (III)—арсенолит, состоящая из молекул AS4O6, кристалли-
зуется в кубической решетке (межатомные расстояния и ва-
лентные углы приведены на рис. 3.25 в скобках) [309]. В эле-
ментарной ячейке с параметром а = 1107,4 пм содержится
8 формульных единиц As4Oe; плотность этой модификации
3,86 г/см3. Выше 240 К арсенолит превращается в клаудетит —
высокомолекулярную модификацию, характеризующуюся слои-
стой решеткой. Этот фазовый переход контролируется кинети-
чески; вообще, он происходит только при повышенных темпера-
турах, и ему особенно благоприятствуют условия гидротермаль-
ного синтеза. Клаудетит I кристаллизуется в моноклинной
структуре с параметрами а = 525 пм, 6 = 129,9 пм, с = 453 пм,
р =93,88°; в элементарной ячейке содержатся 4 формульные
единицы Аз20з; плотность 4,23 г/см3. В клаудетите 1 тригональ-
ные структурные единицы AsO3/2 с атомом As в вершине свя-
заны через общие вершины (атомы О) в сильно гофрированные
слои (рис. 3.26,а). Межатомные расстояния As—О для клау-
детита I составляют 172—181 пм (среднее значение 177,2 пм).
Значения углов As—О—As лежат в интервале 123,3—134,6°,
а для углов О—As—О — в интервале 89,7—103,3°. Расстояния
до вторых соседей As—О больше 280 пм, расстояния О—О
250—275 пм. Все расстояния между гофрированными слоями
превышают 300 пм [310]. Клаудетит I плавится при 585 К.
Сообщается [307] еще об одной модификации — клаудетите II
с чередующимся ориентированием структурных групп AsO 3/2
в слоях.
Стеклообразную модификацию As2O3 получают как охла-
ждением расплава, так и конденсацией паров As4Oo выше
температуры 583 К [311]. Плотность стекла 3,75 г/см3. Вовлаж-
Аморфные и стеклообразные системы
277
ной атмосфере стеклообразный AS2O3 кристаллизуется уже при
комнатной температуре. В зависимости от условий получения
стеклообразного As2O3 температура стеклования меняется от
423 до 473 К. Вязкость переохлажденного расплава As2O3 в ин-
тервале температур 600—700 К сравнима с вязкостью расплава
В2О3 [312].
В работе |313] КРР стеклообразного As2O3 в соответствии
с ранее полученными результатами интерпретируются на ос-
нове модели беспорядочной непрерывной сетки из структурных.
арсенолит
Рис. 3.26. Слоистая структура клаудетита (а) и области структуры (б), оди-
наковые по расположению структурных единиц, для арсенолита, клаудетита
и стеклообразного As2O3 [316].
группировок AsO , производной от структуры клаудетита..
Согласующиеся результаты получены при исследовании струк-
туры стеклообразного As2O3 методом протяженной тонкой
структуры рентгеновского поглощения (ПТСРП) [314]. Колеба-
тельные спектры позволяют обнаружить как структурные об-
ласти, подобные клаудетиту, так и фрагменты As4Oe, в то
время как молекулы As4Oe не обнаружены [315]. Обе кристал-
лические модификации As2O3 отличаются друг от друга конфи-
гурацией структурных группировок AsO3/2. В модификации,,
состоящей из молекул As4Oe, диэдрические углы, образованные
последовательностью связей As—О, примерно сохраняют свою
величину и направление вращения, а в клаудетите, напротив,
углы различаются как по величине, так и по знаку. В работе
[316] доказано, что обе модификации можно представить на
основе непрерывной беспорядочной сетки без других допуще-
ний (рис. 3.26, б), и КР-спектр стеклообразного As2O3 можно
объяснить без допущения о наличии изолированных молекуляр-
ных структурных единиц As4O6 в структуре стекла. Уже сооб-
щалось об аналогичной особенности в стеклообразном селене.
Машинный расчет зависимости плотности состояний от водно-
278 Глава 3
вого числа в стеклообразном AS2O3 подтверждает появление
валентных углов, известных для кристаллических модифика-
ций, которые характеризуют средний порядок [797].
Оксид мышьяка(У). AS2O5 получают в аморфном состоянии
путем обезвоживания мышьяковой кислоты под действием
Р2О5, однако при нагревании выше температуры 873 К полу-
чаемый оксид разлагается на As2O3 и О2. Монокристаллы
AS2O.5 успешно выращивают в автоклавах под повышенным дав-
лением кислорода, где безводный AS2O5 в течение нескольких
недель нагревают при температуре выше 873 К. Структура
AS2O5 представляет собой трехмерную пространственную сетку
Рис. 3.27. Ленточная структура
Sb2Os (валентинит).
•октаэдров [AsOe] и тетраэдров [AsCU], которые связаны через
общий атом кислорода, расположенный в вершине [317]; струк-
туру As2O5 можно описать формулой [AsO6/2] [AsO4/2] или
JoAs^As^O^1. Тетраэдрические позиции атомов As в AS2O5
могут замещаться без появления вакансий атомами фосфора,
октаэдрические позиции — атомами сурьмы [318].
Оксиды сурьмы. Оксид сурьмы (III), как и аналогичное со-
единение мышьяка, существует в виде кубической Sb4Oe (се-
нармонит, рис. 3.25, а) и ромбической модификаций (валенти-
нит) с ленточной струкгурой, построенной из двойных цепей
(рис. 3.27). Валентные углы при атомах сурьмы составляют
81, 93 и 99°. Фазовый переход из одной модификации в дру-
гую происходит при 879 К [319], температура плавления
Sb2O3 929 К.
Sb2O3 может образовать гомогенное стекло только при усло-
вии применения исключительно высокой скорости охлаждения.
Снята КРР для стекла, содержащего 95 мол. % Sb2O3
и 5 мол. % В2О3. С экспериментальной кривой удовлетвори-
тельно согласуется расчет в предположении структуры, произ-
водной от валентинита [320].
Аморфные и стеклообразные системы 279
Для кислородных соединений сурьмы (III, V) описано мно-
жество различных модификаций (см. обзор [321]).
Кристаллический Sb2Os, как и As20.5, получают при повы-
шенном давлении кислорода [322]. По структуре Sb2O5 пред-
ставляет собой трехмерный каркас, состоящий из октаэдров,,
связанных по вершинам и ребрам, и относится к блочной
структуре, производной от структуры типа ReO3 или рутила.
Sb2O5 изоструктурен с B-Nb2Os‘, структуру Sb2Os можно опи-
сать формулой [SbO6/3][SbO6/2] или doSb^O^1 [O3pV
3.2.1.3. Сульфиды и селениды фосфора*
Химия соединений в системах Р—S и Р—Se характери-
зуется выраженной тенденцией к образованию молекулярных
структур. Средняя мольная энергия связей Р—Р 225 кДж/моль,
Р—S 270 кДж/моль и Р—Se 240 кДж/моль (табл. 2.11). По-
скольку эти величины не сильно отличаются друг от друга, то
в многочисленных сульфидах и селенидах фосфора связи Р—Р
существуют наряду со связями Р—S и Р—Se. В системе Р—Те
соединения до сих пор не обнаружены.
Соединения в системах Р—S и Р—Se. Давно известные со-
единения P4S[o, P4S7 и P4S3 согласно диаграмме состояния,
плавятся конгруэнтно, в то время как плавление P4S9, P4S5
и P4S4 инконгруэнтно [323, 324]. Молекулярная структура не-
которых соединений приведена на рис. 3.28.
Фазовая диаграмма системы Р—Se изучена со стороны, бо-
гатой Р, до инконгруэнтно плавящегося соединения P4Se3 [325]’
и подтверждена методами рентгеноструктурного анализа [326],.
свидетельствующего об изоструктурности P4Se3 и P4S3, и ко-
лебательной спектроскопии [327]. Согласно работам [300, 328],.
на диаграмме состояния проявляются помимо P4Se3 еще кон-
груэнтно плавящиеся соединения P4Sei0 и P4Se4. Кристаллиза-
ция расплавов, содержащих меньше 50 мол. % Р, сильно за-
торможена, поэтому селениды фосфора, аналогичные богатым
серой соединениям P4SKJ, P4S9, P4S7 и P4S;), едва ли можно рас-
сматривать как достоверные. Стеклообразный P4Se4 может от-
носительно легко переходить в кристаллическое состояние в ре-
зультате кратковременного нагревания до 583 К и последую-
щего отжига [324]. Это соединение окрашено в коричневый
цвет; оно испытывает фазовый переход при 573 К и плавится
* Взаимодействие Те с различными модификациями фосфора (белый,
красный, черный) изучено в работе [Майсашвили Н. Г., Виноградова Г. 3.,
Тимофеева Н. В., Лужная И. П.— ЖНХ, 1980, т. 25, с. 680]. Показано, что
в системе Рбелый — Тс. образуется неустойчивое соединение ориентировоч-
ного состава РТе.— Прим, персе.
280 Глава 3
при 603 К. Масс-спектроскопическое исследование [329] свиде-
тельствует о том, что структура P4Se4 аналогична P4S4
(рис. 3.28). P4S4 также имеет другую модификацию, аналогич-
ную по структуре реальгару [795]. О колебательных спектрах
P4S5 и Р4§7 сообщается в работе [796]. P4Sc5 до настоящего
времени нс обнаружен на диаграмме состояния, его нужно по-
лучать путем взаимодействия Р48ез с Вг2 [330].
Характерная для сульфидов и селенидов фосфора клеточ-
ная структура является производной от молекулы белого фос-
P4S4. P4Se4
S
II
s
p^s
^10
Рис. 3.28. Структуры некоторых соединений фосфора.
фора Р4. Даже в соединении P4S? сохраняется еще одна связь
Р—Р, и только в случае P4Sio образуется структура, аналогич-
ная уротропину или адамантину; также построен и Р4Ою.
Стекла в системах Р—S и P—Se. В системе PxSi-x в ин-
тервале составов 0,05<1аг<С 0,25 образуются термически мало-
устойчивые стекла желтого цвета [331, 332]*. Они очень не-
устойчивы на воздухе и быстро гидролизуются с выделением
H2S. Рентгенографическое исследование таких стекол показало,
что при малых концентрациях фосфора атомы фосфора обра-
зуют мостики между цепочками серы [333]. При более высоком
содержании фосфора образуются полимерные группировки на
основе молекул P4Sio, так что расплавы кристаллизуются даже
при закалке в воде.
Стекла в системе PxSei-x получают в виде компактных об-
разцов в интервале составов 0<х<0,52 [331, 333, 334]. Они
* О двух областях стеклообразования в системе Р—Se и диаграмме
состояния этой системы см. работу [Ким Е. И., Чернов А. П., Дембов-
ский С. А., Борисова 3. У.— Изв. АН СССР, нсорг. матер., 1976, т. 12,
с. 1021].— Прим, перев.
Аморфные и стеклообразные системы
281
химически устойчивы до состава с х = 0,2 и в этой области со-
ставов подобны стеклам системы As—Se; при большом содер-
жании фосфора их устойчивость на воздухе значительно пони-
жается, о чем свидетельствует выделение НгБе (появление
характерного запаха). Приведенные границы области стекло-
образования относятся к навеске 10 г при условии охлаждения
на воздухе (скорость охлаждения ~2 К/с). Стекла системы
Рис. 3.29. Области стеклообразоваиия и свойства стекол в системах PxSi-x
(2 и 3) и PxSe,_x (/ и 4).
Р—Se в области с х>0,4 окрашены в темно-красный цвет и во
всей области стеклообразоваиия однородны. В литературе не
указывается на расслаивание для стеклообразных фаз системы
Р—Se.
Свойства стекол в системах Р—S и Р—Se. Зависимости
температур стеклования и молярных объемов от состава для
стекол в системах Р—S и Р—Se приведены на рис. 3.29 [331].
Для стекол системы Р—Se в области концентраций 0,1<х<0,4
Tg не зависит от состава. В системе Р—Se для состава х = 0,5
на кривой зависимости Tg от состава имеется максимум, на
кривой зависимости мольного объема от состава — минимум;
это позволяет предположить, что соединение Рд5е4 в стеклооб-
282 Глава 3
разном состоянии характеризуется большой степенью развития
сетчатой структуры. Структура этого соединения до сих пор не
известна.*
Удельная проводимость при комнатной температуре стекол
системы Р—Se составляет 10-15—10 16 См/см, что значительно
ниже удельной проводимости стеклообразного селена. Диэлек-
трическая проницаемость характеризуется подобной концентра-
ционной зависимостью [335].
3.2.1.4. Сульфиды мышьяка и стеклообразование
в системе As—S
Общеизвестно, что AS2S3 осаждается в аморфном состоянии
из подкисленных водных растворов. Образование стекла при за-
твердевании расплава As2S3 впервые описано еще в 1870 г.
[336].
Сульфиды мышьяка. В кристаллическом состоянии AS2S3
встречается в природе в виде аурипигмента. Вероятно, этот ми-
нерал имеет гидротермальное происхождение.
Кристаллы As2S3 получить очень трудно, их получают при
термообработке переохлажденного расплава или посредством
газотранспортных реакций. Для того чтобы AS2S3 закристалли-
зовался при температуре ~530К, требуется 2500 ч [337]. Моно-
кристаллы AS2S3 удается получить методом гидротермального
синтеза [338].
As2S3 кристаллизуется в моноклинной сингонии с парамет-
рами элементарной ячейки а = 1147,5 ±0,5 пм, b =957,7 +
+ 0,4 пм, с = 425,6 + 0,2 пм, р = 90°23' + 5; элементарная ячейка
содержит 4 формульные единицы [339].
Как показывает рис. 3.30, в аурипигменте тригональные
структурные группы [AsS3] связаны через мостиковые атомы
серы в двумерную бесконечную растянутую сетку, состоящую
из двенадцатичленных изогнутых колец. Структурный мотив
аурипигмента аналогичен клаудетиту (рис. 2.26, а). Струк-
туру аурипигмента можно также описать как винтовые цепочки
вдоль оси с из чередующихся атомов As и S, связанные в слои
через дополнительные атомы серы, находящиеся между ато-
мами As из соседних цепочек. Межатомные расстояния As—S
относительно мало различаются (в интервале 224,3—229,2 пм).
2
Формула ooAs^SPl выражает степень разветвления сетки и ко-
ординационные числа атомов. Температура плавления As2S3
сильно зависит от скорости нагревания и составляет ~592К.
Согласно работе [340], на диаграмме состояния системы
As—S имеется второе конгруэнтно плавящееся соединение ре-
* О структуре двух модификаций P4Se4 см. работу [329].— Прим, перев.
Аморфные и стеклообразные системы
283
альгар AS4S4 (температура плавления 594 К). Согласно работе
[341], AS4S4 плавится инконгруэнтно при температуре 578 К,
в то время как в работе [363] в значительной степени подтвер-
ждаются результаты более ранних работ. На рис. 3.32 приве-
дены диаграмма состояния системы As—S (показана область
стеклообразования) и концентрационные зависимости некото-
рых свойств в этой системе. Для AS4S3 обнаружена дистектика.
При большом содержании As температура ликвидуса резко
возрастает. В системе As—S имеется область расслаивания,,
о чем свидетельствует горизонтальный участок на кривой лик-
Рис. 3.30. Слоистая структура (вдоль оси в) кристаллического AS2S3 (а) и.
структура беспорядочной сетки стеклообразного As2S3 (б).
видуса в области составов 0,7<Сх<0,9 при температуре
>1000 К [363].
Реальгар состоит из молекул As4S4; образование таких
группировок можно представить как замыкание цепочек As—S
с ^нс-конфигураций (в отличие от транс-конфигураций) в коль-
цевые структуры (рис. 3.30, a). As2S3 содержит цепочки
с транс-конфигурацией. В реальгаре отсутствуют дополнитель-
ные атомы S, связывающие цепочки, поэтому атомы As насы-
щаются путем образования связей As—As. В молекуле As4S4
атомы As расположены в вершинах тетраэдра, а расположение
атомов серы близко к плоскому квадрату (рис. 3.31) [339,342].
При взаимодействии паров As и S или быстром охлаждении
расплава AS4S4 образуется отличающаяся от а-реальгара р-мо-
дификация [343—346], структура которой приведена на
рис. 3.31 [346].
Структуры а- и р-рсальгара отличаются от аналогичного
сульфида фосфора P4S4 (рис. 3.28), причем для P4S4 известна
также одна модификация, соответствующая реальгару. Соеди-
нения P4S3 и AS4S3, напротив, изоструктурны (рис. 3.31) [347].
As4S3 также встречается в природе в виде минерала димор-
‘284 Глава 3
фита. Наконец, сходство между низшими сульфидами фосфора
и мышьяка особенно заметно проявляется в образовании со-
единения P2As2S3 [348]. В работе [349] описан еще один суль-
фид мышьяка As4S5. Его молекулярная структура приведена
на рис. 3.31. Термодинамические данные для AS2S3, AS4S4
и As4S3 приводятся в работе [363].
AS2S5 до сих пор известен только в аморфном состоянии *;
его получают, например, добавляя по каплям раствор тиоар-
сената в избыток соляной кислоты. Исследования методами
колебательной и фотоэлектронной спектроскопии, а также с по-
мощью КРР указывают на образование структурной единицы
cc-As4S4 (As4Se4) MS4S4
As4S3!As4Se3) AS4S5
Рис. 3.31. Сульфиды мышьяка с молекулярной структурой [346].
S=AsS3/2 [350]. Каждый атом As в такой структурной единице
связан с тремя мостиковыми атомами S, находящимися на
расстоянии 230 пм, и одним концевым атомом S, находящимся
на расстоянии 212 пм. Из сравнения ИК- и КР-спектров P4Sio
и Ge4S4~ со спектрами As2S.5, полученного осаждением из рас-
твора, приходят к выводу об аналогии в их структуре. Сход-
ство в свойствах AS2S5 со стеклообразным селеном
(разд. 3.1.3.2) и стеклообразным AS2O3 (разд. 3.2.1.2) показы-
вает, что определенные конфигурации, которые реализуются
в молекулярной структуре As4Sio, могут также образовывать
полимерную сетку. Нерастворимость и условия реакции оса-
ждения AS2S5 из раствора позволяют предположить для него
макромолекулярное строение.
Сульфидномышьяковые стекла. Стеклообразование в си-
стеме AsxSi-x подробно исследовано [215, 351]. В системе
AsxSi-x для навески в несколько десятков граммов и скорости
охлаждения 2 К/с образование гомогенных стекол происходит
в интервале 0,05<Сх<С0,45 (рис. 3.32).
* О соединении As2Ss, полученном в кристаллическом состоянии при вы-
соком давлении и температуре см. работы [Тимофеева Н. В., Виногра-
дова Г.З., Фекличева Е. М. и др. в кн.: Современные проблемы физической
химии.— М.: Изд-во МГУ. т. 6, с. 234, 1972; Kravchenko Е. Л., Timofeeva К. V.,
Vinogradova G. Z.— J. Mol. Struct., 1980, т. 58, с. 253].— Прим, перев.
Аморфные и стеклообразные системы
285
Рис. 3.32. Диаграмма состояния системы AsxSi-x с областью стеклообразо-
вания (/ — [363]; 2 — [340]) и концентрационные зависимости свойств (темпе-
ратуры стеклования Tg, энергии активации вязкого течения Е^, микротвердо-
сти по Викерсу Ну и молярного объема V) (х— мольные доли).
Для стекла состава Aso,0580,95 температура стеклования со-
ставляет 273 К, т. е. в этой области составов стекла при ком-
натной температуре склонны к спонтанной кристаллизации
с выделением серы. Существующие в расплавах серы обрывки
цепей и кольца связываются с мышьяком с образованием струк-
турных единиц AsS3/2.
Исследования стекол системы AsxSi-x методами рентгенов-
ским и ИК-спектроскопии в сравнении с кристаллическим AS2S3
приводят к выводу о существовании структурных единиц
AsS в стеклах [352, 353]. До состава As2Si0 в стеклах ча-
стично присутствует растворенная сера, которую можно выде-
лить с помощью экстракции. Результаты исследований с по-
мощью колебательной спектроскопии показывают, что вплоть
286 Глава 3
до состава Aso.asSo.es в стеклах должны существовать молекулы
Ss [354, 355]. Чем больше содержание As в стеклах, тем короче
средняя длина цепочек серы между структурными группиров-
ками [AsS3]. Судя по молекулярным спектрам, в стеклах не
реализуется равномерного распределения структурных единиц
AsS3/2 и цепочек серы. Наряду со структурными областями, со-
стоящими из связанных друг с другом структурных единиц
AsS3/2, происходит преимущественное образование более длин-
ных цепочек серы из колец. Однако следует заметить, что ИК-
и КР-спектры стеклообразного As2S5, полученного из расплава,
и аморфного вещества, полученного осаждением из раствора,
очень похожи между собой. Для описания структуры аморф-
ного продукта принимается во внимание структурная единица
S=AsS3/2 [350]. Образование аналогичных структурных груп-
пировок в расплавах AS2S5 до сих пор достоверно не исклю-
чается.
Для состава AS2S3 (х = 0,4) осуществляется такая струк-
тура, в которой структурные единицы связываются друг с дру-
гом непосредственно через атомы серы, каждый из которых об-
разует две связи, причем в отличие от кристаллической моди-
фикации (рис. 3.30, б) образуются неправильные кольца раз-
личной величины [356].
КРР стеклообразного As2S3 нс свидетельствует о наличии
большого числа вторых соседей, которого можно ожидать в со-
ответствии со структурой кристаллической модификации, из
этого следует вывод о значительном гофрировании слоев и об-
разовании пространственной сетчатой структуры в стекле
As2S3 [357].
В то же время наличие выраженного максимума на малых
углах па кривой рентгеновской дифракции свидетельствует
о параллельном расположении фрагментов слоев в малых по
размеру областях. Среднее расстояние между слоями, согласно
такой интерпретации, возрастало бы от 480 пм для кристалли-
ческого As2S3 до 519 пм для стекла [352]. Плотность кристал-
лического As2S3 (р = 3,46 г/см3) отличается от плотности стекло-
образного (р = 3,17 г/см3) на ~8 %.
Па рис. 3.32 представлены концентрационные зависимости
некоторых свойств стекол системы AsxSt_x. На всех представ-
ленных зависимостях (температура стеклования Tg [215], энер-
гия активации вязкого течения [352, 358, 359], твердость по
Викерсу Ну [351], мольный объем V [ЗЫ, 359]) составу х = 0,4
отвечают экстремумы. Как показывают кривые, по макроскопи-
ческим свойствам As2S3 — стеклообразующее соединение в си-
стеме AsxSi-x. Энергия активации вязкого течения для стекол
AsxSi-x достигает и даже превышает среднюю энергию связи
As—S (260 кДж/моль, табл. 2.11), но для расплавов с боль-
Аморфные и стеклообразные системы
287
шим содержанием серы она остается гораздо ниже энергии
связи S—S (280 кДж/моль), что подчеркивает влияние степени
разветвления сетки стекла на вязкое течение и одновременно
указывает на сложность таких процессов в силыюассоцииро-
ванных жидкостях. Результаты новейших прецизионных изме-
рений вязкости стекол системы As—S приведены в работе [810].
Если содержание серы в стеклах системы AsxSi-x меньше,
чем в As2Sa (х> 0,4), то Tg, энергия активации вязкого тече-
ния и микротвердость Hv опять убывают по сравнению
с теми же параметрами для AS2S3, а средний молярный объем
возрастает, что может свидетельствовать как об образовании
энергетически менее устойчивых связей As—As (200 кДж/моль),
например при образовании структурных единиц As2S4/2, так
и о тенденции к образованию молекулярных циклических груп-
пировок (рис. 3.31). Эти стекла (с большим содержанием As)
склонны к расслаиванию [360]. Границы области стеклообра-
зования системы As—S существенно зависят от скорости охла-
ждения и чистоты расплава. В работе [352] стекла получены
до состава реальгара, а в работе [361] область стеклообразо-
вания ограничена составом х^0,45, что примерно соответствует
составу As3S.5. При большем содержании мышьяка нельзя по-
лучить гомогенных стекол из-за микроликвации и выделения
кристаллов J3-As4S4 [362].
Стеклообразный As^S^. Благодаря ярко выраженной стекло-
образующей способности AS2S3 наряду с селеном или В2О3 рас-
сматривается как прототип стеклообразователя. Tg для AS2S3
составляет 463 ± 5 К. Сочленение тригонально-пирамидальных
структурных единиц AsS3/2 осуществляется через цис- и транс-
группировки благодаря возможности изменения диэдрического
угла в цепочках As—S в виде непрерывной беспорядочной про-
странственной сетки, как в стеклообразном AS2O3.
Основные колебания структурных групп AsS3 можно легко
скоррелировать с полосами в ИК- и КР-спектрах AsCl3 (точеч-
ная группа Сзу). Введя некоторый коэффициент, можно эти
спектры совместить друг с другом, откуда следует вывод о зна-
чительном сходстве структуры и типа связи для AsS3/2
и AsCl3 (см. литературу к гл. 2 [352]).
Первый максимум на КРР, полученных методами рентгенов-
ской и нейтронной дифракции, находится на расстоянии 228 пм
и совпадает с межатомным расстоянием As—S в кристалличе-
ском As2S3 [364]. Однако конфигурация во второй координа-
ционной сфере остается невыясненной, например неизвестна
степень сочленения структурных единиц по ребрам и по верши-
нам. В работе [362] на основе КР-спектров стеклообразного
AS2S3 сделан вывод об участии в образовании структуры
288 Глава 3
стекла структурных группировок, сочлененных по ребрам, т. е.
в стеклообразном AS2S3 микроскопические участки трехмерной
сетки плоские; она разрыхляется благодаря наличию областей
с цепочечной структурой (см. рис. 3.30, б). В недавней работе
[811] моделируется структура стеклообразного AS2S3.
Другой вариант сетчатой структуры стеклообразного As2Ss
основан на возможности образования собственных дефектов.
В приложении к разд. 2.4.2.5 и 2.5.2 при обсуждении свойств
материалов для расплава AS2S3 принято следующее уравнение
образования дефектов:
4AsS3/2 - [S (AsS2/2)3]+ + AsS2/2S~ + ULP (3.6)
Гетеролитическое расщепление связи As—S с образованием
отрицательно заряженного концевого атома серы посредством
взаимодействия координационной вакансии на атоме As с мо-
стиковым атомом серы в S (AsS2/2 )2 энергетически выгодно (от-
рицательная эффективная энергия корреляции). Согласно этой
концепции, положительная суммарная энергия ULp определяет
состояние равновесия и в соответствии с уравнением (2.130)
концентрацию заряженных дефектных центров. Приведенное
в разд. 2.4.2.5 равновесие разрыва связей с образованием свя-
зей As—As и S—S:
2AsS3/,- As2S4/2+ S2/2 (3.7)
2 As—S As—As S—S
—2 • 260 —200 —280 кДж/моль
показывает, что связи As—S по сравнению с гомосвязями эле-
мент—элемент только на ~40 кДж/моль более устойчивы
(0,4 эВ}. По уравнению (2.102) и в рамках использованного
там приближения n0 = S xiNaZ i/2)p/M число связей для As2Ss
достигает при температуре стеклования 463 К равновесной кон-
центрации гомосвязей 2,7-1020 см-3. Согласно работам [355,
365], наблюдаемые в КР-спектре стеклообразного As2S3 слабые
полосы —231 и 490 см-1 коррелируют с низкой концентрацией
связей As—As и S—S. Полоса 231 см-1 исчезает при малой
примеси серы в As2S3, в то время как полоса 490 см-1 усили-
вается. Напротив, в стеклах с большим содержанием As, чем
в As2S3 (х>0,4), слабая полоса поглощения 231 см-1 возра-
стает, а полоса 490 см-1 больше не наблюдается.
При наличии связей S—S в стеклообразном As2S3 или
в стеклах с избытком S следует принимать во внимание гетеро-
литический разрыв связей с образованием дефектных центров
[AsS4/2]+ и отрицательно заряженных концевых атомов серы:
AsS3/i+ S2/2-[AsS4/2]+4--Sl/2 (3.8)
Аморфные и стеклообразные системы
289
Спектры люминесценции, без сомнения, указывают на присут-
ствие заряженных дефектных центров в стеклообразном AS2S3
(разд. 4.2.2) [366].
В литературе приводятся многочисленные сведения по ши-
рине запрещенной зоны компактных образцов стеклообразного
AS2S3; при расчетах использовались оптические и электрические
измерения (см. обзор [367]). Рассчитанная по краю оптиче-
ского поглощения ширина запрещенной зоны составляет 2,32—
2,4 эВ. Термическая энергия активации проводимости (1,1 —
1,2 эВ) составляет несколько меньше половины оптической
ширины запрещенной зоны. Для квазистатистической диэлек-
трической проницаемости ег приводится значение 7,9 (298 К).
Аморфные слои AS2S3. В связи с фотохимическими иссле-
дованиями возник интерес к аморфным слоям трисульфида
мышьяка, полученным испарением. Структура и свойства таких
пленок значительно отличаются от компактного стекла, причем
термообработка слоев дает возможность узнать, какая струк-
тура зафиксирована при охлаждении расплава AS2S3. На кри-
вых рентгеновской дифракции пленок трисульфида мышьяка
обнаруживается более интенсивный и острый первый максимум
(чем для компактного стекла), что было неоднозначно истолко-
вано. В работах [364, 368] сделано предположение о более
сильно выраженной в пленках слоистой структуре по сравнению
с компактным стеклом, а в работе [369] — о существовании
молекулярных структурных единиц As4Se, размер которых обус-
ловливает наблюдаемый максимум на дифракционной кривой.
Исследования методом электронной дифракции еще раньше
показали, что в паре над расплавом AS2S3 существуют моле-
кулы As4S6 [370]. Согласно диаграмме состояния [340], AS2S3
испаряется конгруэнтно, и образование молекул As4Se приводит
к мысли о деполимеризации расплава с образованием струк-
туры, аналогичной арсенолиту и уротропину.
Однако новые измерения давления пара ставят под сомне-
ние присутствие молекул As4Se в паре над As2Ss [571]. Иссле-
дования КР-спектров свидетельствуют об отсутствии в паре
молекул As4S4 и присутствии AS4S3 [372]. В масс-спектрах
ионы [As4Se]+ не обнаружены, но идентифицированы осколки
[As4Ss]+; последние считают продуктами распада молекул
As4S6 [373]. Но As4Ss — соединение с молекулярным строением,
так что образование таких структурных единиц может проис-
ходить в результате разложения паров в процессе испарения
A.S2S3. Новые масс-спектрометрические исследования по-преж-
нему подтверждают предположение о присутствии молекул
As4S6 в паровой фазе [374].
В работе [375] путем анализа кривых рентгеновской ди-
фракции аморфных слоев AS2S3, полученных испарением
19 Заказ № 413
290 Глава 3
в вакууме, сделан вывод о возрастании разупорядочения в таких
слоях по сравнению с компактным стеклом. При этом высказы-
вается мнение, что пленки As2S3 состоят из смеси молекулярных
сульфидов мышьяка, которые во время конденсации полиме-
ризуются и приобретают слоистое строение. Согласно работе
[376], в парах доминируют молекулы AS4S4, и об участии таких
структурных групп в полимеризации можно судить, привлекая
результаты и рентгеновской, и нейтронной дифракции.
3.2.1.5. Селениды мышьяка и стеклообразование
в системе As—Se
Селениды мышьяка. Впервые об исследовании диаграммы
состояния системы As—Se сообщалось в работе [377]. Позже
диаграмма состояния подробно изучена в работе [215]; эти
данные были подтверждены [363]. На рис. 3.33 приведена диа-
Рис. 3.33. Диаграмма со-
стояния системы AsxSct-x
с областью стеклообразова-
ния и концентрационные за-
висимости Тё и модуля
сдвига G (мольной доли х).
грамма состояния системы As—Sc, а также зависимости Tg
и модуля сдвига G от состава стекол [378]/
As2Se3 кристаллизуется в слоистой решетке, изоструктурной
As2S3 (рис. 3.30), с параметрами элементарной ячейки: а =
= 1205,3 пм, 6=989,0 нм, с = 427,7 пм, р=90°28' [379—381].
Межатомные расстояния As—Se в спиральных цепях вдоль
оси с составляют 236—237 пм, т. е. значительно короче, чем
Аморфные и стеклообразные системы
291
для связей между атомами Se и As, объединяющих эти спи-
рали в слои; эти межатомные расстояния As—Se составляют
247 пм. As2S3 и As2Se3, как и следовало ожидать, изоморфны.
Исследования методами рентгеновской и нейтронной дифрак-
ции свидетельствуют об образовании в области 0<х<0,5
твердых растворов As2 (Sei-xSx)3 [382], при этом установлено
отличающееся от статистического распределение атомов S и Se
в позициях атомов халькогенов в кристаллической решетке.
Атомы S предпочтительно замещают атомы Se в цепочках
As—Se с более короткими межатомными расстояниями As—Se.
As2Se3 плавится при 648 К. Плотность кристаллического
As2Se3 составляет 4,825 г/см3, т. е. только на —4 % превышает
плотность стекла того же состава (для стекла р = 4,60 г/см3).
В системе AsxSei-x существует вторая кристаллическая
фаза As4Se4, аналогичная а-реальгару (рис. 3.31) [381, 383].
Это соединение плавится инконгруэнтно при температуре 550 К.
Фазу As4Se3, соответствующую диморфиту As4Se3, можно полу-
чить сублимацией аморфного образца [384].
Мышьяковоселенидные стекла. Приведенная на рис. 3.33
область стеклообразования системы As—Se простирается до
большего содержания As по сравнению с системой As—S.
На способность системы As—Se к стеклообразованию благопри-
ятно влияет заметное разложение селенидов с большим содер-
жанием As при инконгруэнтном плавлении As4Se4. При исполь-
зовании веществ высокой чистоты и навески 10—50 г в квар-
цевых ампулах при охлаждении на воздухе (скорость охлажде-
ния 7^4 К/с в области температуры стеклования) удается
получить в стеклообразном состоянии в виде компактных образ-
цов составы, содержащие до 70 ат. % As [385].
В отличие от системы As—S стекла системы As—Se могут
кристаллизоваться при отжиге. Только в области эвтектики в ин-
тервале 0,15<С%<СО,23 для осуществления кристаллизации не-
обходимо применение давления [386]. Для As2Se3 Tg 447 ± 4 К;
стеклообразный As2Se3 кристаллизуется при скорости нагрева-
ния 2 К/мин, т. е. это может происходить при записи ДТА-кри-
вых. Кинетика кристаллизации стеклообразного As2Se3 изучена
в работе [387]. Молекулярная теплоемкость кристаллического
As2Se3 при 400 К составляет 126,3 Дж/(моль-К) [363], а для
стеклообразного As2Se3 125 Дж/(моль-К) [387], т. е. эти вели-
чины близки. При температурах >Tg теплоемкость переохла-
жденного расплава составляет 200 Дж/(моль-К) (разд. 2.2.3.1,
табл. 2.6).
Исследования методами рентгеновской и нейтронной ди-
фракции показали, что структура стекол системы AsxSef-x
в области составов х^0,4 сравнима со структурой мышьяково-
сульфидных стекол аналогичного состава [364]. КРР для
19*
292 Глава 3
компактного стекла и расплава As2Se3 очень близки [388, 389].
Соответствующий кратчайшему межатомному расстоянию пер-
вый максимум на кривой радиального распределения стекла
находится на расстоянии 243 пм; эта величина совпадает со
средним межатомным расстоянием As—Se в кристаллическом
As2Se3. Площадь под первым максимумом соответствует сред-
нему координационному числу 2,4, что совпадает с ожидае-
мым значением (0,4-3 + 0,6-2). На кривых малоуглового рент-
геновского рассеяния стеклообразного As2Se3 наблюдается
отчетливый максимум, откуда сделан вывод о наличии опре-
деленных параллельно ориентированных микрообластей
в сильно гофрированных слоях. Исследования стекла As2Se3
методом рентгеноэлектронной спектроскопии также свидетель-
ствуют о двумерной увязанности структурных единиц AsSe,/2
[390].
Частоты основных колебаний структурных групп [AsSe3]
почти совпадают для кристаллического и стеклообразного
As2Se3 [391]; отнесение полос можно сделать по ИК- и КР-
спектрам AsBr3 (см. литературу к гл. 2 [252]). Колебательная
спектроскопия также подтверждает сходство между структу-
рами селенидов и сульфидов мышьяка (путем умножения на
коэффициент 0,71 можно перейти от одних спектров к другим)
[392]. На концентрационной зависимости действительной части
диэлектрической проницаемости стекол системы AsxSef-x также
отражаются образование структурных единиц AsSe4/j и их тип
колебаний.
На рис. 3.34 приведены зависимости диэлектрической про-
ницаемости и молярного объема от содержания As для стекол
системы AsxSei-x. Диэлектрическая проницаемость стеклооб-
разного As2Se3 составляет 10,2 ±0,1. На рис. 3.34 приведена
также зависимость молярной поляризации Рм от состава; пу-
тем вычитания из нее молярной рефракции, рассчитанной из
показателя преломления и учитывающей электронную поляри-
зацию, получают зависимость от состава, соответствую-
щую вкладу в диэлектрическую поляризацию колебаний, актив-
ных в ИК-области.
Используя приведенную в работе [393] силу осциллятора
можно из хода зависимости диэлектрической проницаемости от
состава рассчитать в общих чертах частоты основных колеба-
ний структурных групп [AsSe3l [385].
Изменение наклона кривой вблизи состава AsSe3 позволяет
предположить, что в области х<0,25 мостиковые связи
As—Se—As совсем не встречаются или встречаются в неболь-
шой концентрации.
Предполагают, что в стекле состава AsSe3 ближний порядок
определяется преимущественно образованием связей As—Se—
Аморфные и стеклообразные системы
293
Рис. 3.34. Зависимость свойств стекол от состава в системе As—Se.
er—диэлектрическая проницаемость, V — мольный объем, Рм — молярная поляризация,
Ру^Рм ~ где молярная рефракция; ДРГ коррелирует с данными, получен-
ными из ИК-спектров для стекол системы As^Se^* [393].
—Se—As и только в области О,25<Сх<СО,4 происходит образо-
вание сетчатой структуры с характерными для As2Se3 мости-
ковыми связями двухвалентных атомов Se между структур-
ными группировками. Такая степень упорядочения придает не-
кристаллическому AsSe3 признаки стеклообразующего соеди-
нения с определенной структурой (разд. 3.2.4).
As2Se3, как и As2S3 в системе As—S (рис. 3.32), характери-
зуется экстремумами на кривых зависимости свойств от со-
става (рис. 3.33 и 3.34). По аналогии с уравнением (3.6) для
объяснения разупорядочения можно привлечь к рассмотрению
уравнение:
4AsSe3/2 - [Se (AsSe2/2)3] + + AsSe2/Se~ + U LP (3.9)
294 Глава 3
В расплавах с большим избытком Se, вероятно, сохраняется не-
изменной характерная для стеклообразного Se дефектная
структура (разд. 2.4.2.5, 2.5.2 и 4.1.3.5) [394]. Соответствую-
щая уравнению (3.8) реакция образования дефектов, например
2AsSeSe2/2^ [AsSe4/J4 + AsSe2/2Se“ (3.10)
т. е. замещение [SeSe3/2]+ центрами [AsSe//2]+, должна сопро-
вождаться повышением энергии за счет вовлечения s-электрон-
ных пар, что энергетически еще менее выгодно, чем в системе
As—S. Напротив, можно описать разрыв связей As—Se [по
аналогии с уравнением (3.7)]:
2As—Sez2As—As-j-Sc—Se (3.11)
—2 ♦ 230 - -200 —225 кДж/моль
Связи As—Se всего лишь на 35 кДж/моль более стабильны,
и расчет количества гомосвязей по уравнению (2.102) в общем
случае, если по= (2 XiNAzil2)pM, дает пс = 4-1020 см-3 при
447 К. Классификация дефектов, существующих в стеклообраз-
ном As2Se3, теоретически обоснована в работе [798].
В КР-спектрах стекол, богатых мышьяком (х>0,4), наблю-
даются две новые полосы при 220 и 155 см-1, которые также
присутствуют в спектре аморфного мышьяка [355]. Очевидно,
происходит нарушение взаимного расположения некоторых ато-
мов As, поэтому образуются фрагменты структуры аморфного
As, которые связываются в стекле мостиковыми атомами Se.
Тот факт, что соединение As4Se4 в стеклообразном состоянии на
зависимостях свойство—состав (рис. 3.34) почти не прояв-
ляется, позволяет предположить его разложение в расплаве.
Соответствует ли минимум на составе As3Se2 определенной
структурной единице, нельзя обнаружить, если исследовать
одни макроскопические свойства.
Оптическая ширина запрещенной зоны стеклообразных
сплавов уменьшается при добавлении мышьяка к селену [для
стеклообразного As2Se3 оптическая ширина запрещенной зоны
составляет 1,73 эВ (300 К)]. При дальнейшем возрастании со-
держания As ширина запрещенной зоны опять возрастает [395,
396]. Стеклу As2Se3 отвечает минимум на кривой зависимости
ширины запрещенной зоны от состава стекол, как и на кривой
молярного объема. В работе [267] приводится другое значение
ширины запрещенной зоны для стеклообразного As2Se3, равное
1,8 эВ (для кристаллического As2Se3 2,0 эВ [397]).
Удельная проводимость стеклообразного As2Se3 при ком-
натной температуре равна 3-1013 См/см; энергия актива-
ции проводимости 0,91 эВ, что примерно соответствует половине
оптической ширины запрещенной зоны. Как при повышении,
Аморфные и стеклообразные системы
295
так и при понижении содержания As по сравнению с As2Se3 на
Ах = ±0,1 удельная проводимость убывает больше, чем на пол-
порядка, что в основном связано с изменением подвижности
[395] (см. разд. 4.1.3.1). В стеклах системы As—Se наблюдается
только p-тип проводимости. Порядок подвижности носителей
заряда составляет при 298 К 10 6 см2/(В-с). Электрические
свойства стеклообразного As2Se3 изучены в работах [398, 399].
Аморфные слои селенидов мышьяка. Путем испарения
в вакууме можно получить все составы системы As.vSei_Y (0<С
<х<С1) в аморфном состоянии. Структура аморфных слоев
системы AsxSei-x, согласно исследованиям методом рентгенов-
ской дифракции, значительно отличается от структуры ком-
пактных стекол, полученных из расплавов (как и в случае
аморфных слоев на основе сульфидов мышьяка). Масс-спек-
трометрическое исследование свидетельствует об инконгруэнт-
ном испарении селенидов мышьяка. При испарении As2Se3
и As2Se,5 наряду с молекулами As4Se3 и небольшим количеством
As4Se4 в паре присутствуют фрагменты As3Se и As2Se2,
а также As4 и Se2 в сравнимых количествах. As4Se4 в газовой
фазе образует молекулы As4Se3, Se2 и As4 [400].
3.2.1.6. As2Te3 и стеклообразование в системе As—Те
Согласно диаграмме состояния, в системе AsxTei-.v имеется
одно кристаллическое соединение As2Te3 [401] (рис. 3.35)*.
По сравнению с системой As—Se стеклообразующая способ-
ность в системе As—Те сильно понижена, она несколько возра-
стает вблизи состава 50 ат. % As. Аналогичное реальгару со-
единение в системе As—Те до сих пор не обнаружено. As2Te3
удается получить в стеклообразном состоянии при использова-
нии скорости охлаждения ^180 К/с, что коренным образом от-
личает эту систему от системы As—S.
Кристаллический As2Te3 и по своей структуре отличен от
As2Se3 и As2S3 [402, 403]. В As2Te3 атомы Те образуют струк-
туру, близкую плотнейшей гексагональной упаковке, в пусто-
тах которой атомы As с координационными числами 6 и 3 рас-
положены таким образом, что образуются ленты, в которых
координационные полиэдры [AsTc6] и [AsTe3] связаны друг
с другом. При этом каждый атом Те окружен 3 или 2 атомами
As. Межатомные расстояния As—Те составляют 272—286 пм,
кратчайшие расстояния между одинаковыми атомами Те—Те
и As—As 405 и 401 пм.
* Несколько отличающаяся диаграмма состояния приводится в работе
[Дембовский С. А., Кириленко И. А., Хворостенко А. С.— ЖНХ, 1968, т. 13,
с. 1462].— Прим, перев.
296 Глава 3
Рис. 3.35. Диаграмма состояния системы AsxTei-x с областью стеклообра-
зования.
Обозначенные на рис. 3.35 границы области стеклообразо-
вания 0,48<х<0,58 определены для скорости охлаждения
q^2 К/с [404] [хотя, согласно другим данным (см. литературу
к гл. 2 [24]), необходима скорость охлаждения >10 К/с].
При использовании скорости охлаждения >180 К/с удается
получить стекла в области составов 0,2<х<0,7. Согласно раз-
личным данным [406, 407] (см. также литературу к гл. 2 [24]),
температура стеклования колеблется от 379 до 389 К. Плот-
ность стеклообразного As2Te3 5,53 г/см3, кристаллического
As2Te3 6,23 г/см3; понижение плотности при переходе из кри-
сталлического состояния в стеклообразное для As2Te3 замет-
нее, чем для соответствующих сульфида As2S3 и селенида
As2Se3, что приводит к выводу о более существенном структур-
ном различии между стеклом и кристаллами для Лэ2Тез.
Исследования стеклообразного As2Te3 методом рентгенов-
ской дифракции показали, что его структуру можно предста-
вить в виде непрерывной беспорядочной сетки, в которой пре-
обладают структурные единицы AsTe3/2 , т. е. структура анало-
гична стеклообразным As2S3 и As2Se3 [408, -409]. Исследование
расплава Аз2Тез методом дифракции нейтронов свидетельствуют
о совпадении КРР стекла и расплава [410]. Тот факт, что первая
координационная сфера, соответствующая первому максимуму
на КРР стеклообразного Аз2Тез, находится на расстоянии
266 пм и, следовательно, межатомное расстояние As—Те
Аморфные и стеклообразные системы
297
в стеклообразном As2Te3 значительно укорочено по сравнению
с кристаллической модификацией, указывает на усиление ко-
валентной связи в стекле. Структура стеклообразного As2Te3
изучена также методом колебательной спектроскопии (см. ли-
тературу к гл. 2 [252, 353]). Частоты антисимметричных ва-
лентных колебаний структурных групп [AsX3] (X = S, Se, Те)
в стеклах AS2X3 линейно коррелируют с соответствующими ча-
стотами молекулярных соединений AsCI3, AsBr3 и Ash
(рис. 3.45). Стеклообразный А5гТе3 также подчиняется такой
корреляции в ряду соединений-гомологов. Кинетика кристалли-
зации стеклообразного As2Te3 изучена в работе [411].
Для того чтобы описать процесс разупорядочения As2Te3
[уравнение (2.9)], принимаются во внимание дефектные центры
[Te(AsTe2/2 )з]+ и концевые отрицательно заряженные атомы
Те в группировках AsTe2/2 Те- и главное очень сильное наруше-
ние связи As—Те. Расчет по уравнению (2.102) при использо-
вании величин, приведенных в табл. 2.11, дает для связи As—Те
по сравнению с сочетанием связей As—As и Те—Те выигрыш
в энергии только 15 кДж/моль (т. е. связь As—Те более устой-
чива). Поэтому необходимо учитывать значительное образова-
ние собственных дефектов в стеклообразном As2Te3 (~10%).
В области составов с х>0,4 должны образовываться
в большом количестве связи As—As, и уже известная для си-
стемы As—Se склонность к образованию структурных конфигу-
раций аморфного мышьяка [355] усиливается при переходе
к системе As—Те. Стеклообразные фазы в системе As—Те
(рис. 3.35), несомненно, включают фрагменты структуры
аморфного мышьяка. О структурных единицах таких стекол
мало известно.
Кристаллический As2Te3 обладает при комнатной темпера-
туре относительно высокой проводимостью (0,4 См/см; энер-
гия активации проводимости 0,03 эВ). Предполагается ме-
ханизм проводимости ио дефектам. Ширина запрещенной зоны
для Аэ2Тез указана только для температуры 500 К (0,45 эВ;
значение определено из собственной проводимости) [412]. Про-
водимость стеклообразного As2Te3 неоднократно исследовалась
[407, 413, 414]. Она составляет при комнатной темпера-
туре ~5-10-5 См/см с энергией активации £а=0,42 эВ, кото-
рая при понижении температуры незначительно изменяется.
Стеклообразный As2Te3 обладает p-типом проводимости. Для
стекла состава AsTe (х- 0,5) при низких температурах наблю-
дается изменение наклона кривой температурной зависимости
проводимости в направлении значительно более низкой энергии
активации [396]. Оптическая ширина запрещенной зоны стек-
лообразного As2Te3 составляет 0,87 эВ [413].
При испарении As2Te3 полностью диссоциирует с отщепле-
нием молекул As4 и со значительным обогащением остатка Те
298 Глава 3
[415]. Методом катодного распыления получают аморфные слои
As2Te3, для которых оптическая ширина запрещенной зоны до-
вольно хорошо совпадает с оптической шириной запрещенной
зоны компактных стекол [412, 416, 417]. Термическая энергия
активации электропроводности для аморфных слоев As2Te3
почти не меняется по сравнению с этим параметром для ком-
пактного стекла, но меняется абсолютная величина электро-
проводности на постоянном токе. Отжиг при комнатной
температуре приводит к тому, что проводимость аморфных
слоев As2Te3 достигает величины, характерной для компакт-
ного стекла. Первоначальная плотность спинов, существующая
в аморфных слоях, при отжиге также исчезает [418].
3.2.1.7. Аморфные сульфиды и селениды сурьмы
Структура Sb2S3 и Sb2Se3 характеризуется цепями, которые
связаны друг с другом с образованием лент (рис. 3.36).
На диаграмме состояния системы SbxSi-x по обе стороны
от состава Sb2S3 расположены области расслаивания [419].
В системе SbxSei-x наличие точки перегиба на кривой ликви-
Рис. 3.36. Структуры Sb2S3
и SbaS3 построенные из двой-
ных лент.
дуса в области х<0,4 указывает на скрытую область расслаи-
вания; в области 0,54 <х<0,85 также образуются две жидкие
фазы [420]. Аморфные соединения Sb2S3 и Sb2Se5, получаемые
осаждением из водных растворов (разд. 3.2.4.2), на диаграм-
мах состояния не проявляются.*
При использовании обычных скоростей охлаждения в си-
стемах Sb—S и Sb—Se не удается получить стекол. Однако
электрические и оптические свойства аморфных пленок Sb2S3
* Разложение аморфного SbjSs при высоком давлении (30—70 кбар)
в интервале температур 190—200 °C с образованием кристаллической фазы
состава SbjS^ описано в работе [Тимофеева И. В., Виноградова Г. 3., Апо-
лонова В. И. и др. в сб.: Аморфные полупроводники 78, Pardibice, CSSR,
1978, р. 92—95].— Прим, перев.
Аморфные и стеклообразные системы
299
и Sb2Se3 изучены подробно (например, [421]). Исследование
расплава Sb2Se3 методом рентгеновской дифракции приводит
к выводу о сходстве ближнего порядка в расплаве и кристал-
лическом веществе [422]. По способу закалки путем разбрыз-
гивания (разд. 2.1.1.3, табл. 2.3) Sb2Se3 получен из расплава
в виде тонкой стеклообразной пленки; охарактеризованы неко-
торые свойства этой пленки [423]. Стекло Sb2S3 в виде компакт-
ных образцов массой 1,5—2 г получено путем закалки в ледя-
ной воде (7^250 К/с) [424]. Согласно работе [546], в стекло-
образных Sb2S3 и Sb2Se3 существует ближний порядок, сходный
с халькогенидами мышьяка.
3.2.2. Оксидные и халькогенидные системы кремния
и германия
Наряду со стеклообразным кремнеземом и производными
от него силикатными стеклами возросло значение стеклообра-
зующих систем с GeO2 и недавно полученных стекол на основе
халькогенидов германия. Получение стекол на основе сульфи-
дов и селенидов кремния не представляет интереса из-за их вы-
раженной склонности к гидролизу.
3.2.2.1. Стеклообразный кремнезем и аморфная система SiO2_a
Диаграмма состояния системы SixOi-x показывает, что
в интервале составов 0о<0,33 эта система является гетеро-
генной, а почти во всей области концентраций 0,33<х<1,0 при
температурах >1973 К обнаружена область расслаивания
[425]. Только путем осаждения из газовой фазы при условиях,
когда происходит затвердевание из неравновесного состояния,
в этой области удается получать аморфные гомогенные твер-
дые фазы SiO2-a. Диоксид кремния в широкой области темпе-
ратур и давлений — единственное термодинамически стабиль-
ное соединение системы Si—О. Сочетание обоих элементов, наи-
более распространенных в земной коре, привлекает к SiO2
с давних пор пристальное внимание и с теоретической, и с прак-
тической стороны.
Диоксид кремния. Известны три модификации SiO2, ста-
бильные при нормальном давлении: кристобалит, тридимид
и кварц. Кроме того, описано много других модификаций SiO2,
часть которых получают при высоких давлениях и температу-
рах [426]. Наиболее известны из них коэзит и стишовит. За ис-
ключением двух последних модификаций высокого давления,
в которых диоксид кремния обладает структурой, аналогичной
рутилу, в остальных модификациях структурные группировки
представляют собой тетраэдры [SiO4]. Эти тетраэдры обычно
300 Глава 3
связаны вершинами, в которых находятся атомы О, в простран-
ственную сетку; иногда они связаны по ребрам, как в моди-
фикации, аналогичной SiS2. Благодаря возможности изменения
валентного угла мостикового атома кислорода существует мно-
гообразие различных конфигураций с различной ориентацией
тетраэдров относительно друг друга, причем модификации SiO2
значительно отличаются одна от другой.
а-Кварц при 846 К переходит в р-кварц, который при 1143 К
превращается в р-тридимит, а он в свою очередь при 1743 К —
в р-кристобалит. Метастабильный р-тридимит в интервале тем-
ператур 393—443 К переходит в a-модификацию, а р-кристоба-
лит в интервале температур 473—548 К-—в а-кристобалит.
р-Кристобалит плавится при 1983 К. Все фазовые переходы ки-
нетически заторможены, и, следовательно, их катализируют
следы примесей. Валентный угол атома кислорода составляет
в а-кварце 143°, в а-кристобалите 150° и в р-кристобалите 152°.
SiO2 разлагается при испарении с отщеплением кислорода
и образованием в газовой фазе устойчивых молекул SiO. Ме-
тодом матричной изоляции показано, что молекулы SiO2, изо-
структурные СО2, метастабильны в газовой фазе [427]. Устой-
чивость SiO2 в твердом состоянии обусловлена в основном
энергией решетки, которая выделяется в результате образова-
ния трехмерно связанных макромолекул, состоящих из тетра-
эдрических структурных единиц SiO4, Энтальпия плавления
SiO2 (~8 кДж/моль) очень мала по сравнению с энергией об-
разования (энтальпия атомизации SiO2 1857 кДж/моль).
Используя представления теории химической связи, с еди-
ных позиций можно описать с хорошим приближением как
кристаллические модификации, так и стеклообразный кремне-
зем, полученный охлаждением расплава.
Химическая связь в структурных группировках SiO4/ осу-
ществляется за счет $р3-гибридных функций Si, взаимодей-
ствующих с функциями мостикового атома кислорода, степень
гибридизации которого определяется валентным углом и изме-
няется от sp до sp2. Речь идет о ковалентных связях, которые
поляризованы в соответствии с разностью электроотрицатель-
ностей атомов, участвующих в связи. Согласно Полингу, при-
нимают, что доля ионности связи Si—О составляет 50 %. В этом
случае аргументация в пользу ионных радиусов не отражает
реального положения вещей, и от такого подхода следует от-
казаться. Для SiO2 независимо от агрегатного состояния соблю-
дается тетраэдрическая симметрия с валентным углом атома Si,
равным 109,5°. Межатомное расстояние Si—О составляет
161,5 пм. Постоянство межатомного расстояния Si—О подтвер-
ждено при структурных исследованиях 314 различных веществ,
в том числе и многочисленных силикатов.
Аморфные и стеклообразные системы 301
Получение стеклообразного кремнезема. На практике стек-
лообразный кремнезем получают различными методами, по-
этому партии кремнезема имеют различное содержание приме-
сей, что значительно влияет на свойства.
Из кристаллов горного хрусталя при нагревании в электри-
ческой печи в вакууме или в инертной атмосфере получают
плавленое кварцевое стекло, не содержащее ОН-групп; содер-
жание примесей оксидов металлов составляет =С0,01 %. Плав-
ление кристаллов кварца в пламени гремучего газа (смесь Н2
и О2) приводит к содержанию ОН-группы в стекле, равному
=СЗ-10-2%. Синтетическое кварцевое стекло получают паро-
фазным гидролизом SiCl4; это стекло почти не содержит окси-
дов металлов, но из-за использования смеси Н2 и О2 содержа-
ние ОН-группы возрастает до 0,12%, и кроме этого в стекле
содержится 0,01 % С1. Разложение SiCk с последующим оса-
ждением SiO2 в пламени плазменной горелки приводит к по-
лучению кварцевого стекла, не содержащего воды, а содержа-
ние в нем С1 может быть еще выше, например 2,4-10-2 %.
Влияние термообработки на плотность кварцевого стекла по-
зволяет предположить значительные различия в содержании
ОН-групп [429]. Понятно, что время релаксации кварцевого
стекла также зависит от содержания ОН-групп [436].
В полупроводниковой технике значение кварцевого стекла
все возрастает в качестве материала для изготовления диффу-
зионных масок, пассивирующих и изолирующих слоев. Их
получают путем термического окисления (разд. 2.1.4.3) [430],
химического осаждения аморфных слоев SiO2 из газовой
фазы на подложки (разд. 2.1.3.4) или путем катодного
распыления (разд. 2.1.3.1 и 2.1.3.2) [431, 432]. В производ-
стве световодов для осаждения SiO2 используют пиролиз смесей
SiCl4/O2.
Структура стеклообразного кремнезема. Относительно проч-
ные тетраэдрические структурные группировки SiO4/2 образуют
в кристаллических модификациях SiO2 четко выраженную про-
странственную структуру. Плотность кварца 2.65, кристобалита
2,32, стишовита 4,35 г/см3. В основе стишовита лежит струк-
тура, близкая к плотнейшей гексагональной упаковке атомов
кислорода. Плотность стеклообразного кремнезема в зависимо-
сти от термической предыстории меняется от 2,20 до 2,22 г/см3.
Большая доля пустот в структуре SiO2 позволяет ожидать зна-
чительной растворимости и подвижности атомов примесей. Экс-
периментально найденная растворимость Не или Ne в решетке
р-кристобалита удовлетворительно совпадает с рассчитанной
концентрацией вакансий (2,34-1022 см3) [433].
Строение кварцевого стекла по данным рентгеновской ди-
фракции обсуждается в разд. 2.4.1.1 (рис. 2.30), а также
302 Глава 3
в разд. 2.4.2.2 (рис. 2.37 и 2.38). Модель непрерывной беспо-
рядочной сетки, состоящей из трехмерных структурных группи-
ровок SiO4/ с произвольным значением диэдрического угла
оказалась наилучшим приближением к экспериментальной КРР
кварцевого стекла (гл. 2 {226—229, 399]). Однако снова выска-
зываются представления о большей степени упорядочения
микрообластей в кварцевом стекле. Методом электронной мик-
роскопии высокого разрешения недавно обнаружены кристал-
литоподобные области среднего размера 170 пм [434]. Из по-
луэмпирического описания химической связи в SiO2 [435] на
основе концепции ионности предложена модель областей сред-
него порядка, состоящих из случайно ориентированных мета-
стабильных субмикрокристаллитов, беспорядочно связанных
друг с другом. Методом молекулярной спектроскопии показано
(см. разд. 2.4.1.2, табл. 2.8 и 2.9, рис. 2.34), что ближний по-
рядок для кристаллического и стеклообразного SiO2 совпадает.
Для понимания процесса структурной релаксации в SiO2 при-
влекают КР-спектроскопию, причем в зависимости от термиче-
ской предыстории образца в КР-спектре исследуют интенсив-
ности определенных полос [436].
В последние годы во многих работах методом рентгено-
электронной спектроскопии (РЭС) изучена электронная струк-
тура кварцевого стекла. В работе [437] представлены экс-
периментальные результаты по расчету зонной структуры SiO2..
Ширина валентной зоны составляет ~ 10—11,5 эВ; она обра-
зована в основном р-орбиталями атома кислорода. Верхняя
граница оптической ширины валентной зоны оценивается из
данных ио фотопроводимости, она составляет 9,0 эВ. Оптиче-
ская ширина запрещенной зоны а-кварца на 0,4 эВ больше, чем
у кварцевого стекла. В соответствии с этим для кварцевого
стекла следует ожидать прозрачности далеко в вакуумной
УФ-области спектра (край поглощения соответствует ~8,3 эВ
или ~ 151 нм). Однако на практике пропускание обычно ограни-
чено длиной волны 250 нм вследствие рассеяния излучения
и поглощения на дефектных центрах, которые могут действовать
как окрашенные центры. В очень чистом кварцевом стекле до
200 нм достигается пропускание, близкое к 100 %.
Дефектные центры в стеклообразном кремнеземе. Многооб-
разие встречающихся в кварцевых стеклах дефектных центров
широко изучено оптическими и спектроскопическими методами.
Наряду с различного вида примесями и нарушениями струк-
туры, порождаемыми излучением высоких энергий (такие
центры проявляют характеристический спектр ЭПР), в очень
чистом кварцевом стекле имеются собственные дефекты, ко-
торые обусловлены частичным изменением состава структур-
ных единиц SiO4. (разд. 2.4.2.5, рис. 2.40) [438].
72
Аморфные и стеклообразные системы
303
Представления, что так называемые Е+-центры, возникаю-
щие при расщеплении определенных Si—О-связей, есть, как
правило, точечные дефекты с наименьшей свободной энталь-
пией образования, оказались ошибочными. Стеклообразный
кремнезем, как и стеклообразный As2S3, диамагнитен, и спек-
тры люминесценции обоих соединений можно единообразно
описать [439]. Обозначим энергию $р3-гибридных связей в тет-
раэдрической структурной группировке как Е' а энергию
связей трехсвязанного атома О через Еь, тогда (рис. 2.40)
4SiO./l= [O(SiO1;,)3]+ + SiO1; р-
-I6£i -3£6-9£; -3E‘b + ULP
Реакция описывает равновесное состояние, в котором обра-
зование групп SiO4/i сопровождается экзотермической энталь-
пией образования (ЗЕь—Eb)+ULp (разд. 2.4.2.5). Число о-свя-
зей во время реакции образования дефектов не меняется
(разд. 2.5.2.3). Этим связям отвечает основная часть энергии
связи. Энергетический расчет для связи Si—О, исходя из сред-
ней молярной энтальпии атомизации (619 кДж/моль), дает
значение 4,8 эВ/моль. В предположении, что Еь~Еь, при об-
разовании заряженных дефектных центров энергия затрачи-
вается только па межэлектронное отталкивание ULP (энер-
гия корреляции электрона), и это гораздо меньше, чем при
образовании предполагаемых в работе [440] центров [SiO32]+
и SiO3/2 О~, для которых требуется определенная энергия вслед-
ствие гетеролитического разрыва связи. Методом молекулярной
спектроскопии [319] экспериментально подтверждается, что
концентрация центров С+ и С~ [уравнение (3.11)] в кварцевом
стекле близка к ~3-1019 смД t/LP = 2,0 эВ [уравнение (2.130)].
Транспортные свойства аморфных пленок SiO2, например
подвижность дырок, которая составляет 20—40 см2/(В-с), можно
удовлетворительно объяснить в предположении присутствия
таких заряженных центров [441]. Пары с переменной валент-
ностью, преобладающие в теоретическом анализе, рассматри-
ваются в этом случае только как диполи [442]. Облучение
светом приводит к появлению двух сигналов ЭПР, а также
двух полос поглощения с энергией 2 и 5,8 эВ, причем последняя
соответствует Е'-центру [443]:
SiO„ О- + [о (SiO./a)3J h —- SiO,; о; + о (SiO./2)2 + lSiO,/a (/?')- (3-12)
Сильный стоксов сдвиг от 5,8 до 2,7 эВ, который в спектре
люминесценции характеризует Е'-центр, и низкий уровень вто-
рой полосы (2,0—1,8 эВ) не противоречат ситуации, приведен-
ной в уравнении (3.12).
304 Глава 3
О собственных дефектах другого рода в аморфном SiC>2,
которые происходят от точечных дефектов и относятся к группе
макроскопических нарушений структуры, сообщается в работе
[444]. Речь идет об ориентированных областях с более высоким
упорядочением, которые образуются при топохимическом взаи-
модействии кремниевой основы в процессе получения аморф-
ных слоев SiO2 посредством окисления. Такие дефекты назы-
вают канальными. Изучена диффузия кислорода в кварцевое
стекло [445, 446].
Посторонние примеси, например оксиды металлов (ИагО),
изменяют картину собственных дефектных центров в кварцевом
стекле:
SiO3/O- + [О (SiO3,2)3]4 + Na2O - 2SiO3/2O- + 2Na+ (3.13)
Таким же образом под действием воды происходит разложение
центров С+ с образованием групп Si—ОН [447—449]. Под дей-
ствием Нг только при температурах >773 К возникают связи
Si—Н и Si—ОН [449, 450]. Различия в спектрах ЭПР кварце-
вых стекол, свободных от воды, и содержащих ОН-группы, об-
суждаются в работе [451].
Свойства стеклообразного кремнезема. Стеклообразный
кремнезем принадлежит, как известно, к неорганическим мате-
риалам, исключительно устойчивым в химическом и механиче-
ском отношении. Температура стеклования Тё, при которой
вязкость достигает 1013 дПа-с, составляет 1495 К, но его можно
подвергать пластической обработке только при температурах
вблизи 2000 К.
Модуль упругости 72 ГПа кварцевого стекла очень высок
(рис. 2.48), коэффициент сжимаемости х при комнатной темпе-
ратуре составляет 0,027 ГПа-1, а коэффициент Пуассона ц =
— 0,176 относительно низок [уравнение (2.8а) в разд. 2.2.1.1].
В отличие от большинства твердых тел и также от а-кварца
при повышении давления коэффициент сжимаемости х квар-
цевого стекла сначала возрастает до 0,036 ГПа-1 и только при
давлении >4 ГПа х убывает, т. е. под действием гидростати-
ческого давления кварцевое стекло легче меняет форму; этот
эффект, несомненно, связан с разрыхленной структурой квар-
цевого стекла. На кривой температурной зависимости сжимае-
мости при температуре 60 К наблюдается максимум; коэффи-’
циент термического расширения (КТР) кварцевого стекла при
низких температурах отрицателен, в области температур
—60 К он характеризуется минимумом. При 200 К КТР кварце-
вого стекла равен нулю, и только после 400 К КТР почти не за-
висит от температуры и составляет 2-Ю-6 К-1. В работе [452]
предлагается модель, интерпретирующая эти аномалии квар-
цевого стекла.
Аморфные и стеклообразные системы
305
Существует обширная литература, посвященная явлениям
релаксации кварцевого стекла [453]; много работ посвящено
изменению свойств кварцевого стекла под действием давления
или облучения [454, 455].
Оксиды кремния с пониженным содержанием кислорода.
Эти вещества образуются как метастабильные продукты поли-
меризации при химическом осаждении из газовой фазы, содер-
жащей SiO, смеси SiO/O2 или из смешанных паров Si/SiO. При
этом получают оптически однородные пленки. Только при до-
статочно высокотемпературном отжиге происходит, согласно
диаграмме состояния, распад низших оксидов на Si и SiO2.
Исходя из оптических спектров аморфных слоев SiO2-a, из-
меренных до энергии 26 эВ, приходят к выводу, что структура
таких слоев основана па непрерывном неправильном распре-
делении структурных единиц, в которых осуществляются все
возможные комбинации тетраэдрической координации атомов
Si с другими атомами Si и/или с атомами О. При этом у ато-
мов О координационное число равно 2 [456]. Формула струк-
турной единицы [Si (SiyO4-y) ] (0<t/<4) основана на предпо-
ложении о непрерывном переходе между структурой аморф-
ного кремния и кварцевого стекла под давлением. Эта струк-
турная модель подтверждена методом рентгеноэлектронной
спектроскопии (РЭС) и полуэмпирическими расчетами [457,
458], а в работе [459] показано, что экспериментальная КРР
согласуется с рассчитанной по модели, основанной на стати-
стическом распределении структурных группировок [SiSi4],
[SiSi3O], [SiSi2O2], [SiSiO3] и [SiO4]. С этими результатами со-
гласуются и данные дифракции электронов [460].
Нитрид кремния Si^N^ [461] также получают в аморфном
состоянии, например путем химического осаждения из газовой
фазы, содержащей NH3, SiCl4 и Н2 (последний в качестве
газа-носителя) [462]. В связи с эффектами накопления заряда
в аморфных слоях Si3N4 для этого материала также были раз-
виты представления о заряженных дефектных центрах [463], КР-
и ИК-спектры различных форм Si3N4 приведены в работе [799].
3.2.2.2. Халькогениды кремния
Кристаллические соединения. Как известно, переход от SiO2
к SiS2 связан с образованием связанных по ребрам тетраэдри-
ческих структурных единиц SiS4/2. SiS2 состоит из макромоле-
кул, строение которых подобно спиранам. В такой структуре
невозможно свободное вращение вокруг мостиковой связи
халькогена, поэтому необходимое для стеклообразоваиия разно-
образие структурных конфигураций не может осуществиться,
20 Заказ № 413
305 Глава 3
и склонности к стеклообразованию у этого вещества уже
не наблюдается. Напротив, для селенида кремния при таких же
одномерных цепях реализуются различные структурные конфи-
гурации.
В SiS2 межатомное расстояние Si—S составляет 214 пм.
Напряженные четырехчленные кольца состоят из чередующихся
атомов Si и S, валентный угол при атоме S составляет 80°, при
атоме Si — 100°. Заметная склонность этого соединения к гид-
ролизу объясняется тем, что энергия связи Si—S на — 30%
ниже, чем энергия связи Si—О.
При испарении SiS2 и SiSe2, как и SiO2, разлагаются. Сооб-
щается [464] о соединениях SiS и SiSe.
В системе Si—Те установлено существование только соеди-
нения Si2Te3 [465—467]. Оно кристаллизуется в тригональной
сингонии с параметрами элементарной ячейки а = 743,0 пм, с =
= 1348,2 пм; в виде монокристаллов это соединение получают
путем выращивания из газовой фазы и по методу Бриджмена.
Рентгеноструктурный анализ показал, что Si2Te3 характери-
зуется слоистой структурой, в которой структурные группи-
ровки Si2Te6/2 с попарно связанными друг с другом атомами
Si соединяются через общие атомы Те [468]. Межатомное рас-
стояние Si—Si —230 пм. Сведения об ИК- и КР-спектрах Si2Te3
приведены в работе [469]. В соединении КбВ12Те6 обнаружены'
шестизарядные отрицательно заряженные структурные группи-
ровки с межатомным расстоянием Si—Si, равным 240 пм [470].
Стеклообразование в системе Si—Те. В системе SixTei _х
в интервале составов 0,15<7х<Д23 из навесок — 20 г полу-
чают компактные стекла путем закалки ампул с расплавом
в воду (скорость охлаждения — 8 К/с) [471]. При скорости
охлаждения 180 К/с область стеклообразования лежит в интер-
вале 0,10<7х<0,22 [472], при скорости охлаждения 250 К/с —
в интервале 0,10<х<0,275 [473]. В литературе приводятся
и другие границы области стеклообразования, например
0,15<х<0,20 [474].
Изучена диаграмма состояния системы Si—Те [466]. Соеди-
нение Si2Te3 (х = 0,4) плавится инконгруэнтно при 1165 К. Эв-
тектика между Те и Si2Te3 обладает относительно низкой тем-
пературой плавления (682 К) и отвечает ориентировочному
составу х = 0,17ч-0,18, подобно эвтектике в системе Ge—Тё.
В этой области составов вязкость расплавов довольно значи-
тельна, поэтому в данных условиях возможно затвердевание
расплава с образованием стекол.
Несмотря на свою большую устойчивость по сравнению
с соединениями в системах Si—S и Si—Se, темно-красные кри-
сталлы Si2Te3 (край поглощения — 2,0 эВ) все же медленно
гидролизуется, причем на поверхности кристаллов в результате
Аморфные и стеклообразные системы
307
окисления образуется черный налет Те. Стекла, богатые Те,
относительно устойчивы химически и на воздухе сохраняются
сколько угодно долго.
КРР стекол системы Si—Те интерпретируют в предположе-
нии координационных чисел 4 для Si и 2 для Те [475]. Ts этих
стекол лежат в интервале 373—443 К и возрастают при увели-
чении содержания Si. Электропроводимость этих стекол при
комнатной температуре составляет ~4-10-5 См/см.
Испарение соединений в системе Si—Те изучено в работе
[467]. SiTe, по-видимому, существует только в газовой фазе.
3.2.2.3. Диоксид германия и стеклообразование
в системе GeO—GeO2
Кристаллические модификации. Устойчивая при нормаль-
ных условиях модификация GeO2 по структуре аналогична ру-
тилу. Межатомные расстояния Ge—О в октаэдрах [GeOe] со-
ставляют 187—191 пм, причем октаэдры расположены таким
образом, что кислород занимает положение, близкое к плот-
нейшей гексагональной упаковке. Эта модификация GeO2 кри-
сталлизуется в тетрагональной сингонии с параметрами а =
= 439,5 пм, с=286,0 пм; плотность 6,277 г/см3.
При 1322 К эта модификация GeO2 медленно переходит
в высокотемпературную модификацию, устойчивую до темпера-
туры плавления 1389 К. Структура высокотемпературной мо-
дификации является производной от структуры р-кварца. Эта
модификация в метастабилыюй области температур (<1293 К)
превращается в модификацию, соответствующую но структуре
а-кварцу, которая при комнатной температуре устойчива в те-
чение длительного времени. Среднее межатомное расстояние
Ge—О (165 пм) и валентный угол (106,3—113,1°) при атоме
Ge в структурных группах [GeO-i] свидетельствуют об отклоне-
нии от тетраэдрической симметрии. Плотность a-GeO2 4,28 г/см3.
a.-GeO2 растворим в воде с образованием коллоидного рас-
твора (при 298 К растворяется 4,53 г/л), что можно рассмат-
ривать как признак координационной ненасыщенности атома
Ge в соответствующей структурной единице.
Стеклообразный GeO2. Высоковязкий расплав GeO2 при от-
носительно медленном охлаждении затвердевает в стекло; плот-
ность стекла 3,62 ± 0,02 г/см3, температура стеклования 823 К.
При 973 К происходит кристаллизация с образованием a-GeO2;
энтальпия плавления 17,2 кДж/моль (см. табл. 2.1) [476], а эн-
тальпия кристаллизации 12,2 кДж/моль [477].
Структура стеклообразного GeO2 аналогична структуре
кварцевого стекла: структурные единицы GeO 4/г связаны в трех-
мерную беспорядочную сетку. Среднее межатомное расстояние
20*
308 Глава 3
Ge—О 174 пм, средний валентный угол при мостиковом атоме
кислорода 133° [478, 479]. Результаты сравнительного исследо-
вания стеклообразных GeO2 и ZnCl2 методом ПТСРП приве-
дены в работе [800]. ИК-спектры стекла GeO2 хорошо согла-
суются со структурной моделью беспорядочной сетки [480].
При модификации структуры стеклообразного GeO2 путем
добавок оксидов металлов, например Na2O, с целью изменения
сетчатой структуры сначала происходит увеличение плотности
с достижением максимума, а затем ее уменьшение; этот факт
рассматривается в литературе как указание на изменение ко-
ординационного числа от 4 до 6. При исследовании структуры
ближнего порядка стеклообразного GeO2 и системы
Na2O—GeO2 при изменении давления до 1,5 ГПа обнаружена
взаимосвязь между плотностью, вязкостью и особенностями
КР-спектров, однако такой вывод подвергается сомнению [481].
Новые исследования стекол системы Na2O—GeO2 методами
молекулярной спектроскопии и ПТСРП показали, что атомы
Ge в этой системе все-таки имеют координацию 4 и 6 [801, 802].
Структура дефектов в стеклообразном GeO2 изучена термо-
люминесцентным методом [482]. Значение оптической ширины
запрещенной зоны из данных по фотопроводимости составляет
~5 эВ [483]. Облучение при низких температурах приводит,
как и в кварцевом стекле, к возникновению двух раздельных
сигналов ЭПР.
Предполагается, что реакция образования собственных де-
фектов в стекле GeO2 описывается уравнением, аналогичным
уравнению (3.11). Исследования структуры стеклообразного
GeO2 методом КР-спектроскопии дают плотность дефектных
центров, равную 1019 см-3, откуда можно рассчитать энергию
корреляции ULp таким же образом, как и для SiO2. Получают
Ulp=1,2 эВ.
Стеклообразование в системе GeO—GeO2. Расплав GeO2
в атмосфере, не содержащей кислорода, отщепляет кислород;
причем при повышении температуры доля выделенного кисло-
рода увеличивается. При выдержке расплава GcO2 в азоте при
температуре 1673 К в нем образуется — Ю1й дефектных цент-
ров, которые имеют характеристическую полосу поглощения
245 нм. По интенсивности этой полосы оценивают концентрацию
дефектных центров. Продолжительная выдержка GeO2 при
1423 К на воздухе приводит к уменьшению концентрации де-
фектов до ~ 1015 см3. В работе [477] изучено влияние плотно-
сти дефектных центров на кинетику кристаллизации и на ход
плавления GeO2.
Можно себе представить, как в результате удаления
атома О мостиковой связи из двух фрагментов GeO3/ , имею-
щих по одному неспаренному электрону, могут образовы-
Аморфные и стеклообразные системы 309
ваться либо структурные единицы Ge2O6/2 со связью Ge—Ge,
либо положительно или отрицательно заряженные дефектные
центры [O(GeO3, )2] + и GeO“ . В дефектных центрах германий
двухвалентен. Оценка с использованием энергий связи, полу-
ченных из энтальпий атомизации (табл. 2.11), дает
2GeO4/2-Ge2O6/2 + 72O2 (3.14)
—8 • 353 —6 • 353 —249,2 кДж/моль
-185
4GeO</2- [GeO3/2]- + [О (GeO3/2)3]+ + '/2О2 (3.15)
—16-353 —3- 351 —12-353 --249,2 кДж/моль
Отсюда энергетический баланс для (3.14) составляет
272 кДж/моль, для (3.15) 104 кДж/моль, т. е. образование за-
ряженных центров предпочтительнее. Следует заметить, что
расплавы GeO2 с пониженным содержанием кислорода дают
сигнал ЭПР, откуда делается вывод о наличии дефектных
центров GeO3/ [504]. Измерения вязкости показали, что GeO
как компонент стеклообразоваиия проявляет в расплаве GeO2
совершенно иные структурно-химические свойства, чем ти-
пичный оксид-модификатор (например, Na2O) [484]. Если
добавление Na2O приводит к резкому уменьшению энер-
гии активации вязкого течения, то в системе GeO2_« (при воз-
растании а) происходит, напротив, лишь постепенное линейное
уменьшение этого параметра.
Путем сплавления смесей Ge/GeO2 в графитовых тиглях
в атмосфере аргона и медленного охлаждения расплава удается
получить стекла вплоть до состава GeOi,s; в условиях очень
больших скоростей охлаждения (рис. 2.3, д) получают гомо-
генные стекла до состава GeOi,6 [485]. Аморфные слои с еще
более высоким содержанием Ge получают путем вакуумного
испарения. GeOi,6 соответствует формуле (GeO)2(GeO2)3, но
только до состава (GeO) i (GeO2)3 можно представить структуру
стекла, согласно уравнению (3.15), как образование заряжен-
ных центров [GeO 3/ ]~[O(GeO3/2) 3|+. При большом содержании
Ge часть мостиковых атомов кислорода должна иметь по три
связи и поэтому нести положительный заряд. Предполагают,
что система перестраивается и частично образуются, согласно
уравнению (3.14), структурные единицы Ge2O«/2: CieOi.6 =
= (Ое2Об/г)2 (GeO*/,).
В области концентраций GeO—GeOi,85 в стеклах, подверг-
нутых отжигу при 673 К, наблюдается фазовое разделение,
о чем свидетельствует их почернение. Выделяется фаза аморф-
ного германия (размеры вкраплений 5—10 пм) [486]. Фазовое
310 Глава 3
разделение обусловлено окислительно-восстановительным дис-
пропорционированием; оно может быть описано спинодальным
механизмом (разд. 2.3.1.2).
3.2.2.4. Стеклообразование сульфидов германия и Ge2S3
Кристаллические сульфиды германия. Винклер [487], описы-
вая открытие германия, сообщает об осаждении GeS2 в виде
бесцветного соединения, малорастворимого в солянокислом рас-
творе.
Фазовые равновесия в системе GexSi-x (рис. 3.37) подробно
изучены в работе [488]. Соединения GeS? и GeS образуют эв-
тектику с температурой плавления 870 К, состав эвтектики
64,7 мол. % GeS (х = 0,425).
GeS2 образует несколько модификаций. Высокотемператур-
ная модификация a-GeS2 плавится конгруэнтно при 1123 К и
кристаллизируется в слоистой структуре с параметрами элемен-
тарной ячейки a = 687,5 пм, Ь = 2205 пм, с = 680,9 пм, |3 = 120,45°;
Рис. 3.37. Диаграмма состояния системы GexS^x с областью стеклообразо-
вания и концентрационные зависимости мольного объема V и температуры
стеклования Tg.
Аморфные и стеклообразные системы
311
плотность a-GeS2 2,935 г/см3 [489]. Искаженные тетраэдрические
структурные группировки [GeSi] характеризуются межатом-
ными расстояниями Ge—S 217—229 пм, и половина из них свя-
зана друг с другом через общие атомы S в вершинах, а поло-
вина— ребрами. Углы тетраэдров в структурных группировках
[GeS',] составляют 99,8—117,6°, валентные углы при атомах S
99,8—102,8°. Структура a-GeS2 занимает промежуточное поло-
жение между структурным типом SiS2, где связь осуществля-
ется по ребрам, и пространственной сеткой, как в решетке
кварца и низкотемпературной модификации |3-GeS2.
Структура p-GeS2 представляет собой трехмерную простран-
ственную сетку, состоящую из структурных групп [GeS4], свя-
занных по вершинам; плотность (3-GeS2 2,99 г/см3 [490, 491];
параметры элементарной ячейки а=687,5 пм, 6=2255 пм, с =
= 680,9 пм, [3 = 120,45°, межатомные расстояния Ge—S 219—
223 пм, валентные углы при мостиковых атомах S (82,1 —102,2°)
меньше, чем для мостиковых атомов О в любой из модифика-
ций SiO2, так что по конфигурации тетраэдрические структур-
ные группы [GeS4] не идентичны структурным группам моди-
фикаций кремнезема.
Из фаз высокого давления при температуре 873 К и давле-
ниях ^300 МПа стабилен y-GeS2 [488]; другую модификацию
получают при температуре 1173 К и давлении 3 ГПа [491]. Ука-
занные модификации не идентичны. Получены также еще фазы
высокого давления при температуре 673 К и давлении 1 ГПа,
а также при температуре 773 К и давлении 7 ГПа [492].
GeS — вещество цвета от серого до черного с металлическим
блеском, которое при растирании образует порошок цвета от
темно-коричневого до красного; плавится инконгруэнтно при
931 ±4 К. По структуре отвечает структурному типу черного
фосфора (рис. 3.13, а) [493].
Уточнение структуры GeS [494] привело к параметрам эле-
ментарной ячейки а = 1047,0 пм, 6=429,7 пм, с = 364,1 пм; три
межатомных расстояния Ge—S в структурных группах [GeS.?]
равны 244,1 пм; расстояние до вторых соседей (атомов серы)
составляют 327 пм внутри гофрированных слоев и 327,8 пм до
атома S соседнего слоя.
Стеклообразный GeS2 и стекла в системе GeS2—сера. Об-
ласть стеклообразования в системе GexSi-x приведена на
рис. 3.37 [495]. Германий взаимодействует с серой с образова-
нием GeS2, а при добавлении к GeS2 серы структурные еди-
ницы GeS4/ связывают в пространственную сетку цепочки
серы, находящиеся в расплаве. Из стекол в порошке в области
составов GeS4_5 (0,17 < х < 0,20) можно экстрагировать Se се-
роуглеродом, что по аналогии с системой As—S свидетельствует
о присутствии в стеклах различных структур. Очевидно,
312
Глава 3
в стеклах системы GexSi-x сосуществуют структурные обла-
сти, богатые серой и GeS2. Точка перегиба на кривой ликви-
дуса указывает на склонность к расслаиванию в этой области
составов [488].
Температура стеклования Tg, как и микротвердость Hv (по
Викерсу), в системе GexSi-x возрастает при увеличении содер-
жания Ge, а коэффициент термического расширения а убывает
от 9- 10~5 К-1 для стеклообразной серы до 1,2- 10“5 для стекло-
образного GeS2 [496]. На рис. 3.37 представлены зависимости
Tg и среднего молярного объема от состава. Температура
стеклования GeS2, равная 768 К, является самой высокой среди
всех известных до сих пор халькогенидных стекол. В отличие
от системы As—S молярный объем стекол в системе Ge—S
при введении Ge не убывает, а возрастает и достигает макси-
мума для состава GeS2. Средний молярный объем стеклообраз-
ного GeS2 на 10 % больше, чем в случае стеклообразного As2S3,
что указывает на менее плотную упаковку для GeS2. Плотность
стеклообразного GeS2 2,717 г/см3. Зависимость упругих постоян-
ных стеклообразного GeS2 от температуры и давления приве-
дена в работе (табл. 2.4) [497].
Расплав GeS2 характеризуется значительной кристаллиза-
ционной способностью. Она существенно зависит от содержа,-
ния примесей, особенно от присутствия GeS в малых количест-
вах. Критическая скорость охлаждения ^Крит для GeS2 в обла-
сти температуры стеклования составляет 17 К/с (закалка 10 г
расплава в воду) [498]. В то же время расплав состава GeS2,oe
можно перевести в стеклообразное состояние при навеске 50—
100 г охлаждением на воздухе (7«рит~3 К/с), а состав GeSi.w
закалкой 2 г расплава в воду (<7крит~40 К/с). Только при бо-
лее высокой концентрации GeS, когда относительно высокая
температура ликвидуса, характерная для поля GeS2, достаточно
понижается, а вязкость возрастает, что соответствует сильно
пониженной первичной кристаллизации, в области составов
0,392 < х < 0,435 или (GeS) y(GeS2)i-y, где 0,45 < у < 0,70,
опять складываются благоприятные условия для стеклообразо-
вания (^Крит^8 К/с). Вблизи эвтек1ики GeS/GeS2 существует
вторая область с выраженной склонностью к стеклообразо-
ванию.
Структура золотисто-желтого стекла GeS2, которое хорошо
поддается обработке, согласно исследованиям методами рентге-
новской дифракции [552] и молекулярной спектроскопии [499],
характеризуется тетраэдрическими структурными единицами
GeS4/2 , которые через мостиковые атомы серы связаны в трех-
мерную пространственную сетку. Среднее межатомное расстоя-
ние Ge—S, найденное из первого максимума на КРР стеклооб-
разного GeS2, составляет 224 пм и соответствует в хорошем
приближении длинам связей в кристаллических модификациях
I Аморфные и стеклообразные системы
313
GeS2. Структурная интерпретация остальных максимумов позво-
ляет прийти к выводу, о том, что стекло GeS2 построено из
структурных единиц [GeS/,], связанных по вершинам и ребрам
[500]. В таком смысле трактуется появление двух полос в об-
ласти симметричных валентных колебаний тетраэдрических
структурных группировок в КР-спектре a-GeS2 и стеклообраз-
ного GeS2 [501, 502, 540].
Участие связанных по ребрам структурных групп в образо-
вании стеклообразного GeS2 приводит к уменьшению средней
степени разветвления сетки. В этой связи следует отметить ли-
нейную корреляцию между Ts стекол МХ2 и энтальпией атоми-
зации (рис. 2.45, разд. 2.4.3.3.), которая значительно отличается
от зависимости для стекол As2X3 с двумерной сетчатой струк-
турой. При температуре Tg в течение нормированного времени
наблюдения происходит пластическая деформация (вязкое те-
чение) стекла за счет энергии, которая необходима для разрыва
достаточного числа связей (разд. 2.2.2). Очевидно, в таком
процессе уменьшение средней степени разветвления сетки
стекла из-за того, что тетраэдрические группировки частично
связаны по ребрам, мало влияет до тех пор, пока вообще
сохраняется трехмерный каркас связей.
Оптическая ширина запрещенной зоны стеклообразного GeS2
составляет ~2,7 эВ.
Собственные дефекты стеклообразного GeS2. Другие ис-
кажения сетчатой структуры вызваны возможностями образова-
ния собственных дефектов. По аналогии с разд. 2.4.2.5 и 2.5.2
можно рассмотреть поведение стехиометрического расплава
GeS2; для него, как и в случае кварцевого стекла и других стек-
лообразных халькогенидов, исходя из энергетических соображе-
ний, принимают во внимание следующие реакции образования
дефектных центров:
4GeS,/2 - GeS3/ + [S (GeS3/2)3] + ULP (3.16)
Исходя из предположения, что для группировок, стоящих в ле-
вой и правой частях уравнения (3.16), энергии связи Ge—S
примерно одинаковы, получают, что выигрыш при образовании
группировок GeS4, против образования заряженных дефект-
ных центров равен только энергии корреляции Ulp- Из сравне-
ния с другими халькогенидными стеклами ULp оценивают 0,8—
1 эВ, откуда плотность заряженных центров равна ~1018 см-3.
Далее следует заметить, что GeS2 при температуре плавле-
ния характеризуется большим давлением пара продуктов раз-
ложения, причем склонность к отщеплению халькогена усили-
вается по сравнению с расплавом GeO2. Даже в запаянных под
вакуумом кварцевых ампулах существующий свободный объем
314 Глава 3
занимает паровая фаза, характеризующаяся более высоким со-
держанием S, чем расплав, т. е. расплав GeS2 обедняется
серой.
По аналогии с уравнением (3.15) можно оценить энергетиче-
ский баланс реакции:
40eS./l^ [| OeS,,,]- + [S (OeS.Jjp + '/2S2 (3.17)
-16.265 -3-265 -12-265 —212,7
Откуда получается, что образование группировок GeS4/ энер-
гетически более выгодно (энтальпия образования GeS4,2
52 кДж/моль). Согласно (3.17), при температуре плавления
GeS2 (1123 К) в расплаве должно существовать ~ 1019—
— 1020 см-3 дефектных центров; такая концентрация, по-види-
мому, физически вполне возможна, и принятое приближение
(пренебрежение энтропийными эффектами) оправдывается
в истинном смысле.
Разрыв связей Ge—S с образованием связей Ge—Ge и S—S
требует затраты энергии 65 кДж/моль:
2Ge—S — Ge—Ge + S—S (3.18)
—2 • 265 —185 -280 кДж/моль
Откуда по уравнению (2.102) для стеклообразного GeS2 при
рассчитана плотность гомосвязей, составляющая 3 -1020 см-3.
Стекла на основе сульфидов германия принадлежат к не-
многим халькогенидным стеклам, для которых в твердом со-
стоянии при отсутствии освещения, при комнатной температуре
в спектрах ЭПР обнаруживается сигнал; отсюда в их структуре
предполагается присутствие центров с неспаренными электро-
нами [504]. Для стеклообразного GeS2 плотность неспаренных
электронов составляет 1016 см-3 [503]. Она, как оказалось, за-
висит от условий получения стекла, уменьшаясь вследствие за-
грязнения примесями из стенок кварцевой ампулы при возра-
стании времени контакта расплава со стенкой [504]. Природа
эффекта стабилизации, который благоприятствует образованию
таких парамагнитных центров против образования заряженных
центров, до сих пор не ясна. В работе [590] сильно размытый
край поглощения стеклообразного GeS2 объясняют очень высо-
кой степенью геометрического разупорядочения. Из частичного
перекрывания плотности состояний положительно и отрица-
тельно заряженных центров должно следовать такое замеще-
ние дефектных центров, которое энергетически более предпочти-
тельно. В области составов с х<0,33 к характеристической для
стеклообразного GeS2 резонансной линии прибавляется добавоч-
ное поглощение, по-видимому, вызываемое неспаренным элек-
троном на цепочках серы различной длины [505]. В области со-
ставов с х>0,33 в спектре, например стекла Ge2S3, проявляется
Аморфные и стеклообразные системы
315
сигнал ЭПР, точно такой же, как в спектре стеклообразного
GeS2; отжиг стекла полностью устраняет этот сигнал, а под
действием УФ-облучения он возобновляется [504].
Сильно выраженная кристаллизационная способность высо-
кочистого расплава GeS2 резко снижается при добавлении ми-
нимального количества серы, что приводит к выводу о частич-
ной диссоциации расплава при высоких температурах
(>1123 К), причем зародышеобразованию сильно способствует
образование Ge (II), т. е. связей Ge—Ge, или радикалов
tGeS,,,.
Стекла в системе GeS—GeS2. О структуре стекол в системе
(GeS) y(GeS2)i-y в области составов 0,45 < у < 0,70 долгое
время не было единого мнения. Предполагалось [495], что ком-
понент GeS выполняет функцию модификатора стеклообразной
сетки, при этом межатомные расстояния Ge—S в стеклах дол-
жны быть не меньше, чем в кристаллическом GeS. Однако по-
ложение первого максимума на КРР стекол системы GeS—GeS2
не допускает такого объяснения [552]. Предположение [506]
о том, что при возрастании мольной доли у среднее координа-
ционное число атома S также растет до 4, приписываемого
аморфному GeS, следует отклонить, исходя из положений тео-
рии химической связи. Для стекол системы GeS—GeS2 хоро-
шее согласие с площадью первого максимума на КРР дает
расчет, если принять координационное число Ge, равное 4, S,
равное 2, и статистическое распределение связей Ge—Ge, Ge—-S
и S—S, т. e. при отрицании предпочтительного образования свя-
зей Ge—S, несмотря на это, в работе [552] указывается па не-
однозначность КРР. ИК- и КР-спектры стекол системы GeS—
GeS2 также не удается интерпретировать с позиций вышеизло-
женного предположения о структуре. Использование модели,
предложенной для аморфных пленок SixOi_x [456], т. е. предпо-
ложения о статистическом распределении структурных единиц
[(SsGe)nGeS4-n], где 0 О < 4 [507], также не привело к удов-
летворительному объяснению результатов спектроскопических
исследований.
В работах [508, 509], исходя из эквивалентности атомов Ge
в рентгеноэлектронном спектре стеклообразного Ge2S.% прихо-
дят к выводу о наличии высокой степени химического упорядо-
чения в стекле этого состава. Энергетически более выгодны
связи Ge—S, г. е. равновесие (3.18) должно быть сдвинуто
в сторону образования связей Ge—S, а связи Ge—Ge образу-
ются только в случае недостатка халькогена (х>0,33). По-
этому едва ли можно ожидать, что связи S—S могут существо-
вать наряду со связями Ge—Ge. Из этого следует, что стекла
вплоть до эквимолярного содержания GeS и GeS2 (0,33 < х <
<0,40), что соответствует формуле (Ge2S »/2 )2(GeS </2 )i-z (где
316 Глава 3
z=(3x—1)/(1—2х)], состоят из структурных единиц GeS4/2
и Ge2S6// В стеклах в основном сохраняется структура про-
странственной сетки. Тетраэдры как бы «вдвигаются» друг
в друга в возрастающем количестве (рис. 3.38), и это объяс-
няет уменьшение среднего молярного объема (рис. 3.37).
В структуре стекла состава ОегЗз преобладают структурные
единицы Ge9Se/ , которые обязательно содержат связи
Ge—Ge; соединение в пространственную сетку осуществляется
через двухвалентные атомы серы. На концентрационной зави-
Рис. 3.38. Взаимодействие в расплаве GeS с GeS2 или GeSe с GeSe2 с образо-
ванием структурных группировок Gc?Xtj,2 и их диспропорционирование при
кристаллизации.
симости Tg составу ОегЗз отвечает скачок [510]. Например,
в кристаллической решетке GaS существуют аналогичные
структурные группы Ga2 So/ , объединенные через атомы S, об-
разующие три связи, в слоистую решетку [511, 512].
В области 0,40 < х < 0,4285, т. е. до границы области стек-
лообразования, при интерпретации структуры используют тот
же подход и в соответствии с формулой (Ge3S e/Jz(Ge2S e/)i-z
[где z=(5x — 2)/(1—2х)] предполагают образование структур-
ных единиц Ge„Se. При этом остается неясным, до какого
числа членов продолжается соответствующий мотив. Кроме
того, принимают во внимание также разложение структурных
групп Ge S (2п+2)/2 на структурные единицы с большим и мень-
шим п, т. е. образование структурных единиц с соответственно
большим числом связей Ge—Ge, например
2QenS(2n + 2)/2<—Ge$4/2 4“ ^С2П— 1$4п/2 (3.19)
Для составов с большим п (х—>-0,5), где быстро возрастает
число структурных изомеров, образуемых атомами Ge, возможно
Аморфные и стеклообразные системы
317
бесконечное разнообразие структурных единиц. Предполагают,
что в структурных единицах и фрагментах структуры таких
стекол наряду с координационными числами 4 для Ge и 2 для
S встречается координация 3 для обоих атомов, как в аморф-
ных модификациях фосфора и мышьяка. Исследования аморф-
ных GeS и GeSe методами ПТСРП и рентгеноэлектронной спек-
троскопии (ЭСХА) показали, что для атомов германия и халь-
когена координационное число равно 3 [803].
В области существования компактных стекол, получаемых
из расплава, вполне вероятны структурные единицы
Ge nS (2п+2)/2 ’ это подтверждается положениями максимумов на
КРР, и прежде всего данными ИК- и КР-спектроскопии. Моле-
кулярные спектры можно свободно интерпретировать на основе
таких моделей [353, 513].
Стеклообразный Ge2S3. Структура стеклообразного Ge2S3
представляет собой трехмерную сетку, составленную из струк-
турных единиц Ge2S6/ ; вещество проявляет существенные при-
знаки индивидуального соединения. Взаимодействие стекла
GesS,? с раствором Na2S в метаноле, есть реакция, которую мо-
жно рассматривать как доказательство состава этого стекла
[514] (см. также литературу к гл. 2 [291]):
Ge2S3 + 3Na2S С-Нз°-+ Na6Ge2S6 • 4СН3ОН (3.20)
По аналогии с образованием тиогерманатов (IV) из GeS2 и
растворимых сульфидов под действием нуклеофильного расщеп-
ления связей мостиковых атомов S из структуры стекла выде-
ляются группировки [Gc2S6] и превращаются в шестизарядные
отрицательные анионы. Их состав определяют из сравнения
данных для гексатиогиподигерманата натрия NaeGe2S6 с моле-
4 —
кулярными спектрами Si2Cl6 и P2Se , а также по реакции взаи-
модействия с бромом, в результате которой образуется соеди-
нение NaaGeSgBr (см. литературу к гл. 2 [292]):
Na6Ge2S6 + Вг2 - 2Na3GeS3Br (3.21)
Если прибавлять по каплям водный или метанольный рас-
твор Na6Ge2S6 в соляную кислоту, то выделяется похожий на
AS2S3 золотистый, труднорастворимый осадок аморфного соеди-
нения GeaSa.
Раньше приводились сведения об аморфном сублимате со-
става GeaS.3, образующемся при инконгруэнтном испарении GeS2
[517, 518]. Предпринимались, но безуспешно многочисленные
попытки выделить кристаллическое вещество этого состава. При
отжиге стеклообразного или аморфного Ge2Ss в соответствии
с фазовой диаграммой происходит разложение, которое может
рассматриваться как диспропорционирование, и образуется
318 Глава 3
гетерогенная смесь GeS и GeS2 (рис. 3.38). Плавление этой
смеси, напротив, приводит к взаимодействию с образованием
в расплаве и стеклообразном состоянии характеристических
структурных группировок Ge nS (2п+2)/2. Из эквимолярной смеси
GeS и GeS2 получают Ge2S3 в виде гомогенного стекла, которое
на основе своей структуры ближнего порядка рассматривается
как индивидуальное соединение; оно может существовать только
в виде непериодически связанных структурных групп
(разд. 3.2.4).
3.2.2.5. Стеклообразование селенидов германия и Ge2Se3
Диаграмма состояния системы GexSei-x (рис. 3.39) неодно-
кратно приводилась в литературе [519—524]. Средний состав
эвтектики отвечает л' = 0,43 и выражается формулой (GeSe)yX
(GeSe>)i-y (где у = 0,673). Температура плавления эвтектики
846 К [524].
Кристаллические селениды германия. GeSe3— соединение
желтого цвета, которое плавится при 1013 К и по структуре
Рис. 3.39. Диаграмма состояния системы GcxSei_x с областью стеклообразо-
вания и зависимость Tg от состава.
Аморфные и стеклообразные системы 319
аналогично слоистой модификации a-GeS2 с параметрами эле-
ментарной ячейки a = 701,7 пм, Ь = 1679,7 пм, 1183,2 пм,
Р= 90,66° (р = 4,39 г/см3) [525]. Угол тетраэдра в структурной
группировке GeSe4/2 составляет 94,5—116,0, межатомные рас-
стояния 233,7—236,9 пм, а валентный угол Ge—Se—Ge 80,2—
100,1° Сообщается [492] и о других кристаллических модифи-
кациях, полученных под давлением.
GeSe — вещество черного цвета с сильным блеском, струк-
тура которого, как и GeS, является производной от структур-
ного типа черного фосфора [526, 527]. Параметры элементарной
ячейки а = 438,8 пм, b = 1082,5 пм, с = 383,3 пм соответствуют
межатомным расстояниям Ge—Se 257,4 пм и дважды 256,4 пм,
а три другие расстояния 331,6—336,7 пм.
Согласно данным работы [524], GeSe существует в узкой
области гомогенности, которая при 873 К достигает хж ±0,005.
При отклонении от стехиометрии температура обратимого фазо-
вого перехода |3-GeSe a-GeSe меняется в интервале 876—
900 К. GeSe плавится инконгруэнтно при 924 К, а при 934 К
наблюдается второй эффект, обусловленный монотектикой. Со-
гласно другим данным [528], превращение [J- в a-GeSe происхо-
дит при 924 К, а инконгруэнтное плавление наблюдается при
943 К. Структура высокотемпературной модификации a-GeSe
соответствует искаженному структурному типу NaCl.
Стеклообразный GeSe2 и стеклообразование в системе Ge—
Se. В системе GexSei_x расплавы образуют стекла в относи-
тельно широкой области составов, начиная от состава селена.
В системе GexSe[_x, как и в аналогичной системе Ge—S, при ус-
ловиях навеска 20—30 г, скорость охлаждения ~2 К/с в интер-
вале температуры стеклования проявляются две области стек-
лообразования в интервалах составов 0 < х < 0,33 и 0,388 <
< х < 0,417 [529, 530]. При закалке расплава (навеска
~0,5 г) в маленьких тонкостенных кварцевых ампулах в воду
(скорость охлаждения ~30—35 К/с) удается получить стекла
в единой области в интервале составов 0 < х < 0,42. На
рис. 3.39 приведены границы области стеклообразования в си-
стеме Ge—Se. Приводимые в литературе противоречивые дан-
ные о границах стеклообразования в системе Ge—Se, возможно,
обусловлены влиянием примесей. В работе [531] показано, что
высокочпстый расплав GeSe2 (навеска 20 г) при охлаждении на
воздухе образует стекло, не содержащее кристаллических вклю-
чений. Ранее сообщалось о стекле состава Ge2Se,3 [532]. О стек-
лообразоваиии в системе GeSe—GeSe2 также сообщалось в ра-
боте [533].
Методами рентгеновской и нейтронной дифракции показано,
что структура стеклообразного GeSe2 аналогична стеклу GeS2
{534—536]. Среднее межатомное расстояние Ge—Se (236,5 пм),
320 Глава 3
найденное из первого максимума КРР, и среднее координацион-
ное число 2,62 (0,33-4 + 0,67-2) для стеклообразного GeSe?
свидетельствуют о сходстве с ближним порядком, характерным
для кристаллического GeSe2. Исследование расплава GeSe? при
1044 К методом дифракции нейтронов показало, что по сравне-
нию со стеклом его структура в основном неизменна [389].
Сравнение частот валентных колебаний, встречающихся
в ИК,- и КР-спектрах этих стекол, с колебательным спектром
GeBr.; также позволяет предположить тетраэдрические струк-
турные группы [538, 539] (см. также литературу к гл. 2 [253]).
Симметричное валентное колебание проявляется в спектре стек-
лообразного GeSes, как и в случае стекла GeS2, в виде вырож-
денной полосы, что получило противоречивое толкование в ли-
тературе [538, 540—543]. Расщепление полосы с несколько
меньшей разностью волновых чисел также наблюдается в кри-
сталлическом GeSe?, что в этом случае объясняют присутствием
структурных группировок GeS^ , связанных по вершинам и
ребрам. Вытекающее из этого предположение о средней степени
разветвления сетки стекла приводит к обязательному парал-
лельному ориентированию структурных группировок в микрооб-
ластях с двумерными структурными единицами и может объяс-
нить аномально острый первый максимум на дифракционной
кривой [389, 501, 540]. В то же время следует заметить, что для
стекла GeSe? выполняется корреляция между Tg и энтальпией
атомизации, характерная для стекол с трехмерной сеткой
(рис. 2.45); следовательно, доля структурных областей, состоя-
щих из связанных по ребрам структурных группировок, не на-
столько велика, чтобы ее учитывать.
В связи с изложенным в разд. 2.4.2.5 и 2.5.2 и по аналогии
со стеклообразующими халькогенидами мышьяка и стеклами
общей формулы МХ2 (SiO2, GeO2, GeS2) для стеклообразного
GeSe2 также можно ожидать разупорядочения с образованием
заряженных дефектных центров. Реакцию образования таких
центров можно записать по аналогии с уравнением (3.16):
4GeSe4/. GeSe,,.Se- + [Se (GeSe,/2)3] + + f/,p (3.22)
Из-за относительно низкого давления пара селена над рас-
плавом GeSe2 образование дефектных центров по аналогии
с уравнением (3.17) намного менее вероятно, чем в случае
стекла GeS2. Также следует очень внимательно отнестись к раз-
рыву связи Ge—Se, аналогичному уравнению (3.18)
(разд. 2.4.2.5) (см. литературу к гл. 2 [107]). Германиевоселе-
нидные стекла не обнаруживают сигнала в ЭПР-спектрах.
Свойства стекол в системе Ge—Se. На рис. 3.40 представ-
лены концентрационные зависимости среднего мольного объема
Аморфные и стеклообразные системы
321
^Ge
Рис. 3.40. Концентрационные зависимости мольного объема V, диэлектриче-
ской проницаемости еГ) молярной поляризации Рм, молярной рефракции Rm
и АР=РМ—Rm для стекол системы GexSei-x.
АР' — отклонение молярной поляризации АР от линейности.
V, квазистатистической диэлектрической проницаемости ег и
молярной поляризации P3f=(er—1)/ для стекол системы
Ge—Se. Найдя разность Рм и молярной рефракции RM (из по-
казателя преломления), можно построить зависимость АР от
х по аналогии с тем, как это сделано для стекол системы
AsxSei-x; функцию АР(х) интерпретируют как вклад валент-
21 Заказ № 413
322 Глава 3
ных колебаний в действительную часть диэлектрической прони-
цаемости (рис. 3.34).
Известно, что стеклообразующее соединение GeSe?, как и
в случае GeS?, характеризуется максимумом на концентраци-
онных зависимостях мольного объема и Tg (рис. 3.39). Темпе-
ратура стеклования GeSe2 достигает относительно высокой для
халькогенидных стекол величины 665 К, а плотность стеклооб-
разного GeSe2 (4,22 г/см3) только на 4 % меньше плотности
кристаллической модификации. В области состава GeSe2 кон-
центрационные кривые диэлектрических свойств стекол претер-
певают разрыв.
Следует отметить наличие явного структурно-чувствитель-
ного вклада в диэлектрическую проницаемость, который в стек-
лах системы Ge.rSei_x проявляется в области х =0,2 в виде по-
ложительного отклонения кривой АР от линейной зависимости
[385]. В отличие от стеклообразного Se, для которого колебания
вносят сравнительно малый вклад в молярную поляризацию
(ег = 6,15, /г2 = 5,95), при увеличении концентрации структур-
ных единиц GeSe4/ АР линейно возрастает. При этом появля-
ется еще дополнительный вклад, который отчетливо обусловлен
характеристической степенью разветвленности структурных еди-
ниц и наиболее значительно проявляется для состава GeSe4.
Зависимость ДР'(х) получают вычитанием из кривой ДР (х)
значений, соответствующих линейной зависимости. Точно для
состава х = 0,2 наблюдается экстремум.
На концентрационной зависимости мольных объемов для
стекол системы Ge—Se этому составу (х = 0,2) отвечает мини-
мум [530, 544, 545], на зависимости коэффициента термиче-
ского расширения КТР от состава для х = 0,2 наблюдается пе-
региб [545]. На кривой Tg— х при х = 0,2 также обнаружива-
ется изменение наклона (рис. 3.39). Резкое возрастание темпе-
ратуры, соответствующей вязкости ~1013 дПа-c, при добавле-
нии Ge к Se можно объяснить разветвлением сетки стекла за
счет структурных группировок GeSe4/ (см. литературу к гл. 2
[340]). В рамках теории свободного объема структурной еди-
ницей текучести в системе Ge—Se является атом Se, т. е. теку-
честь — это результат скольжения и сдвига подобных частиц,
или разрыва и нового образования соответствующих связей
[547, 548] (см. также литературу к гл. 2 [115]). На зависимости
кинематической вязкости от состава расплава системы Ge—Se
для х = 0,33 обнаружен максимум, который при повышении тем-
пературы становится более плоским [659]. Второй уширенный
максимум на кривой вязкости соответствует х = 0,2. Он прояв-
ляется тем отчетливее, чем ниже температура расплава. Осно-
вываясь на измерениях плотности расплава системы Ge—Se и
аномалии в температурной зависимости плотности вблизи со-
Аморфные и стеклообразные системы 323
става GeSe?, сделан вывод [550] об изменении структуры ближ-
него порядка, начиная с температуры ~1073 К, связанной
с большим заполнением пространства. Исследованы также за-
висимость упругих постоянных для стекол системы Ge—Se от
состава и от давления [545]. Для стеклообразного GeSe2 мо-
дуль упругости равен 27,5 ГПа, а модуль сдвига 11 ГПа; оптш
ческая ширина запрещенной зоны составляет 2,25 эВ [564],
а удельная проводимость стекла GeSe2 лежит в интервале от
10~15 до 10-16 См/см при 298 К; энергия активации проводимости
Еа составляет ~2 эВ.
Судя по ходу кривых, приведенных на рис. 3.40, наряду
с GeSe2 стекло состава GeSe4 также проявляет себя как инди-
видуальное соединение. Исследование стекол системы Ge—Se
методом КР-спектроскопии [513] показало, что в отличие от
стекол системы Ge—S аналогичного состава непосредственная
связь структурных группировок GeSe4/2 через мостиковые атомы
Se нарушается, пока это допускает содержание Ge. Согласно
КРР, структуру стекла состава GeSe4 можно описать состоящей
из тетраэдров GeSe4, которые связаны мостиковыми связями
Se—Se в пространственную сетку [562]. Валентный угол при
атоме Se позволяет реализовать, как и в стеклообразном GeSe2>
плотнейшую упаковку тетраэдрических структурных группиро-
вок. Вполне допустимо было бы, что определенные крутиль-
ные колебания взаимосвязанных с атомами германия мостико-
вых группировок Se—Se вносят дополнительный вклад в мо-
лярную поляризацию.
Стекла системы GeSe—GeSe2 и Ge^Se^. Структура стскол си-
стемы (GeSe) y(GeSez)i-y в области 0,419 < у < 0,60 исследо-
вана методом рентгеноэлектронной спектроскопии; исходя из
координации атомов Ge, сделан вывод о структуре пространствен-
ной сетки общей формулы Ge nSe (2п+2}/2 [509]. На концентра-
ционной зависимости вязкости расплавов составу Ge2Se,3 отве-
чает размытый максимум [549]. Согласно уравнению [2.101],
связи Ge—Se должны быть энергетически более выгодными, чем
связи Ge—Ge и Se—Se (причем последние встречаются в этих
стеклах только в незначительном количестве). Составу с п~2
или у = 0,5 соответствует образование структурных единиц
Ge2Se6/ , которые образуют трехмерный каркас связей и через
мостиковые двухкоординированные атомы Se объединяются
в непрерывную неупорядоченную сетку. В структуре GaSe
структурные группы Ga2 See/ связаны через трехкоординиро-
ванные атомы Se в слоистую решетку [551].
Как доказательство возможности образования таких струк-
турных групп, рассматривается образование соединения
21*
324 Глава 3
NacGezSes при взаимодействии стеклообразного Ge2Se3 с NazSe
в СНзОН [см. уравнение (3.20)]:
Ge2Se3 + 3Na2Se -СНз°Н > Na6Qe2Se6 • 4СН3ОН (3.23)
Исследования этого вещества методом молекулярной спек-
троскопии показали, что оно содержит отрицательно заряжен-
ные ионы Ge2See~, которые образуются при взаимодействии
с Na2Se из структурных группировок стекла Ge2Se6/2 [514] (см.
также литературу к гл. 2 [291]). При взаимодействии с бромом
связи Ge—Ge расщепляются, и образуется соединение
Na3GeSe3Br красного цвета (см. литературу к гл. 2 [292]):
NacGe2Se6 -|- Вг2 — 2Na3GeSe3Br (3.24)
Если водный или метанольный раствор Na6Ge2Se6 добавлять
по каплям в соляную кислоту, то выпадает труднорастворимый
осадок оранжево-желтого цвета, отвечающий аморфному соеди-
нению Ge2Se,3 с формально трехвалентным германием.
Представления о структуре стеклообразного Ge2Se3 на основе
изучения ИК- и КР-спектров [513] также предполагают сущест-
вование структурных единиц Ge2Se6/ . Ge2Sea, как и GezS3,
имеет свою собственную структуру ближнего порядка, поэтому
его можно считать индивидуальным соединением, существую-
щим, очевидно, только в аморфном или стеклообразном состоя-
нии. При отжиге стеклообразного Ge2Se,3 происходит его разло-
жение (рис. 3.38) с образованием в соответствии с фазовой
диаграммой гетерогенной смеси GeSe и GeSe2. Плавление экви-
молярной смеси GeSe и GeSe2 приводит к их взаимодействию,
и при переходе в стеклообразное состояние «фиксируется» ха-
рактерная для GezSe3 структура расплава.
Структуру стекол (GeSe) у (GeSe2) в интервале концентра-
ций 0 <Z у < 0,5 можно описать как пространственную сетку
связанных друг с другом структурных групп GeSe^ и Ge2Se6?.
Уменьшение среднего мольного объема можно объяснить, как
и для аналогичных стекол системы Ge—S, предположением
о взаимном проникновении тетраэдрических групп (рис. 3.38).
Температура стеклования также уменьшается при повышении
содержания структурных единиц Ge„Se^ . Как показывают
данные, приведенные на рис. 3.41, переход от стекла GeS2 (или
GeSe2) к стеклам Ge2S3 (или Ge2Se3) обозначает не только по-
нижение Тё, но одновременно и выравнивание Tg. Отсюда сле-
дует, что в стеклах GeS2 и GeSe2 вязкое течение в интервале
температур стеклования связано со скольжением и сдвигом свя-
зей Ge—S и Ge—Se (рис. 2.40); таким образом, очевидно, раз-
личие в энергиях связей (Ge—S и Ge—Se) приводит к разно-
сти в Tg на ~100 К. Небольшое различие в Tg (~20 К) для
Аморфные и стеклообразные системы
325
стеклообразных соединений Ge2S3 и Ge2Se3 позволяет предполо-
жить разрыв связей Ge—Ge. Поскольку энергия связи Ge—Ge
равна всего 185 кДж/моль, связи Ge—Ge — наиболее слабые
места в сетке стекла. В предположении гетеролитичсского рас-
щепления связей принимают во внимание взаимодействие пустой
орбитали на атоме Ge со степенью окисления +4 и свободной
электронной пары соседнего мостикового атома Se, так что
происходит образование положительно заряженного центра
Энергия связи, кДж/моль
Ge-S 265
Ge-Se 230
Ge-Ge: 185
Температура сштиния Тд,к
GeS2- 765 6e2S3: 636
GeSe2: 663 Ge2Se3: 613
Рис. 3.41. Образование собственных дефектов в стеклообразных соединениях
Ge2$3 и Ge2Se3 и механизм вязкого течения.
[Se(GeSe3/ )3]+, в то время как на атоме Ge со степенью окис-
ления + 2 получается отрицательно заряженный центр
30e2Ses/2zr [Se3/GeSe(Ge2Se5/2)2]+ + [| GeSe,,,]- (3.25)
Согласно этому уравнению реакции образования дефектных
центров (при написании уравнения использовали аналогию со
стеклообразным Ge2S3), связь Ge—Ge (185 кДж/моль) разры-
вается и появляются новые связи Ge—Se (230 кДж/моль).
Энергия корреляции Ulp, равная ~50 кДж/моль, уменьшается
таким образом на 45 кДж/моль, так что для этого случая необ-
ходимо считаться с образованием дефектных центров.
Вязкое течение расплавов Ge2S3 и Ge2Se3 можно предста-
вить как процесс распада этих структур и их образования. При
охлаждении расплава при температуре стеклования (или замер-
зания) фиксируются имеющаяся равновесная концентрация по-
ложительно и отрицательно заряженных точечных дефектов и
характеристическое разупорядочение сетки стекла, состоящей
из структурных единиц Ge2,Se6^ или Ge2S6^ . О равновесном
состоянии системы Ge—Se до сих пор еще мало что известно.
326 Глава 3
Можно предположить, что содержание разупорядоченных струк-
турных областей составляет от нескольких процентов до не-
скольких десятков процентов. Доказательства можно отыскать
в результатах структурных исследований. Даже если обратиться
к данным по проводимости, то вполне оправдан вывод, что со-
держание областей с разупорядоченной структурой в стекле
Ge2Ses [в соответствии с уравнением (3.25)] относительно ве-
лико (рис. 4.22).
Структуру стекол (GeSe) v (GeSe?) i-y в области у > 0,5
с большой долей вероятности можно описать в предположении
координационного числа 3 для обоих атомов, т. е. по аналогии
с аморфным мышьяком [803, 804]. По данным ИК- и КР-спек-
троскопии сделан вывод о существовании в стеклах структур-
ных единиц типа GenSe(2n+2)/2 для п > 2 [353, 513]; однако
в другой работе [553] при анализе КР-спектров найдено дока-
зательство того, что большая часть GeSe не участвует в обра-
зовании сложных структурных единиц сетки стекла, а в струк-
туру стекла включаются характерные для кристаллического
GeSe структурные единицы GeSe3/j .
Аморфные слои на основе селенидов германия. Структура
и свойства армофных слоев системы GexSei-x или (GeSe)yX
X(GeSe2)i-y хорошо изучены. При испарении GeSe2 диссоции-
рует на молекулы GeSe и Se2 [554, 555]. При испарении Ge2Ses
в условиях вакуума и последующей конденсации происходит
сильное изменение состава слоя (от у = 0,5 до у = 0,8 4-0,9)
(см. литературу к гл. 2 [38]). GeSe испаряется без разложения
[556, 557].
Исследования структуры аморфных слоев GeSe2 методом
ПТСРП показали, что она близка структуре компактного стекла
GeSe2 (см. литературу к гл. 2 [239]). Измерения аморфных
слоев GeSe?, полученных методом испарения с помощью ди-
фракции электронов, также подтверждают характерный для
GeSe2 ближний порядок [558]. Для интерпретации структуры
аморфных слоев GeSe2 привлекается модель со статистическим
распределением связей Ge—Ge, Ge—Se и Se—Se, которая хо-
рошо подходит для аморфных слоев GeSe. При этом атомы Ge
всегда имеют координационное число 4, атомы Se — координа-
ционное число 2. Среднее межатомное расстояние Ge—Se, най-
денное из положения первого максимума на КРР для аморф-
ных слоев GeSe, составляет 238 пм; оно значительно укорочено
по сравнению с самым коротким расстоянием Ge—Se в кри-
сталлическом GeSe, но приблизительно совпадает с парамет-
ром, найденным для GeSe2 (237 пм). По ИК-спектру можно об-
наружить различия в структуре ближнего порядка между кри-
сталлическим и аморфным GeSe [559]. Однако исследования
структуры аморфных слоев GeSe методом ПТСРП указывают,
Аморфные и стеклообразные системы
327
что предложенная ковалентная модель со статистическим рас-
пределением связей не подходит [560]. В работе [561] приве-
дены аргументы из теории химической связи в пользу того, что
укорачивание межатомного расстояния в аморфном GeSe, воз-
можно, вызвано образованием структурных группировок GeSe.^
с координационным числом 3 для Ge и Se.
3.2.2.6. Стеклообразование в системе Ge—Те, GeTe4 и GeTeg
Кристаллический GeTe. Диаграмма состояния Ge—Те. На
диаграмме состояния системы Ge—Те известно одно индивиду-
альное кристаллическое соединение GeTe [565]. Оно плавится
конгруэнтно при 977 К [566] и кристаллизуется (a-GeTe)
в структурном типе ромбоэдрического мышьяка (искаженная
структура NaCl с параметрами элементарной ячейки а =
= 596 пм, а = 88,27°) [567]. В GeTe существуют 3 коротких
межатомные расстояния Ge—Те, равные 300 пм, внутри двой-
ных гофрированных слоев и 3 длинных расстояния между сло-
ями (рис. 3.13).
а-GeTe имеет область гомогенности, охватывающую область
составов от Geo.99.5Te до Geo,9i5Te. При повышении температуры
происходит выравнивание отличающихся межатомных расстоя-
ний, причем угол описанной выше элементарной ячейки прибли-
жается к 90° (ромбоэдрический угол составляет 60°). При тем-
пературе 703 К со стороны Ge и при 673 К со стороны области
гомогенности, богатой Те, образуется Р-GeTe, характеризую-
щийся неискаженной структурой NaCl. ^-Модификация харак-
теризуется областью гомогенности от Gei.ossTe до Ое0,92зТе и
устойчива вплоть до температуры плавления. В интервале от
Ge0,97Te до Ge0,93iTc ниже 638 К проявляется еще одна модифи-
кация у-GeTe, структура которой аналогична GeS (структур-
ный тип черного фосфора) [566, 568]. у-Модификацию также
получают при кристаллизации GeTe под давлением.
На рис. 3.42 приведена диаграмма состояния системы
Ge—Те [472, 566, 568, 569]. GeTe образует с теллуром эвтек-
тику, сс состав GcsTei; (х = 0,15), температура плавления 648 К.
По другим данным [569] эвтектике отвечает состав с % = 0,18, и
точно на этот состав приходится максимальная стеклообразую-
щая способность (критическая скорость охлаждения ^крИт»
«2 К/с).
Стекла и аморфные слои в системе Ge—Те. При скорости
охлаждения 180 К/с в системе GexTei-x область стеклообразо-
вания расположена в интервале 0,12 < % < 0,22; при использо-
вании метода растекания расплава по охлаждаемой плоскости
(splat quenching) и достигаемой скорости охлаждения ~105 К/с
(разд. 2.1.1.3.) области стеклообразования отвечают границы
328 Глава 3
Рис. 3.42. Диаграмма состояния системы GexTei-x; стеклообразование наблю-
дается для состава Ge3Tei7 (х=0,15).
0,10 < х < 0,25. В работе [286] найдены близкие границы об-
ласти стеклообразования для образцов, полученных методом
разбрызгивания мелких капель расплава (spray cooling)
(разд. 2.1.1.4). Методом катодного распыления получены слои
в интервале 0,05 <С х < 1. На диаграммах состояния систем,
содержащих Те, эвтектика часто расположена вблизи ~20 ат. %
второго компонента. Во многих случаях такие расплавы при
охлаждении переходят в стеклообразное состояние [569, 570].
Уже достаточно малой добавки третьего компонента для того,
чтобы в области эвтектического состава склонность расплава
к кристаллизации еще больше понизилась. Области стеклообра-
зования тройных систем Ge—Те—I (разд. 3.3.2), Ge—Те—Se
(разд. 3.3.3.2), Ge—Те—Р (разд. 3.3.5.5), Ge—Те—As
(разд. 3.3.5.4) или Ge—Те—Si (разд. 3.3.4.4) тесно связаны
с узкой эвтектической областью двойной системы Ge—Те. В си-
стеме Ge—Те—Hg также обнаруживается узкая область стек-
лообразования [771].
Исследования структуры стекол системы Ge—Те методами
дифракции рентгеновских лучей и нейтронов показали, что она
отличается от структуры кристаллического GeTe; результаты
этих исследований можно объяснить, используя различные
структурные модели [572, 560]. Первый максимум на КРР рас-
положен на расстоянии 265 пм (дифракция нейтронов) или
275 пм (дифракция рентгеновских лучей), а второй —на рас-
стоянии 420 пм. Сумма ковалентных радиусов (Ge 122 пм, Те
Аморфные и стеклообразные системы
329
132 пм) довольно хорошо совпадает со средним межатомным
расстоянием, найденным по первому максимуму, что указывает
на относительно большую долю ковалентности связи Ge—Те
в стеклах. В работе [561] предлагается учитывать предположе-
ние о ближнем порядке в стеклах системы Ge—Те, аналогичном
ближнему порядку в кристаллическом GeTe. Укорачивание
длины связи Ge—Те можно объяснить, например, уменьшением
взаимодействия между слоями. При сравнении кристаллов GeTe
с аморфными слоями оказалось, что рост степени ковалентно-
сти связи Ge—Те в аморфных слоях экспериментально проявля-
GeTe5
' Те
Те^ \ Те
Те
Те
ХТе
GeTej
GeTe5
в
Рис, 3,43. Различные типы структуры ближнего порядка для стекол системы
Ge—Те в области эвтектики.
ется как понижение на ~ 1 эВ энергии связи электронов Ge
ЗсР/2-уровня в рентгеноэлектронном спектре (ЭСХА), а также
как сдвиг Ка-рентгеновского края поглощения Ge [573]. Резуль-
таты исследования стекла состава Geo,i?Teo,83 методом рентге-
новской дифракции можно интерпретировать как с помощью
моделей, где для Ge принято координационное число 4, а для
Те 2, так и с помощью представлений о структуре, в которой
атомы Ge находятся в окружении 3 атомов Те, а один из этих
атомов Те одновременно окружен двумя другими атомами Те
(рис. 3.43, а) [574]. Неупорядоченная сетка, построенная из та-
ких структурных единиц, характеризуется средним координаци-
онным числом z = £ лдгг-= (0,167 • 3 + 0,167 • 3 + 0,67 • 2) =2,33.
Состав GeTe., примерно соответствует эвтектике. Происходящее
в расплаве при ~673 К повышение среднего координационного
числа от 2,43 до 3,25 [575] позволяет предположить, что при
повышении температуры атомы Те (рис. 3.43, б) также перехо-
дят в состояние с координационным числом 3, характерным для
теллура в жидком состоянии: z= (0,167 • 4 + 0,167 • 3 + 0,67 • 3) =
-3,17.
Метастабильные соединения GeTe± и GeTez. Плотность
стекла состава Оео.пэТеожэ 5,26 г/см3, а плотность расплава при
330 Глава 3
Рис. 3.44. Проводимость при 298 К (/—3) и оптическая ширина запрещенной
зоны Ь’опт (а=104 см1) (Г—3') для аморфных слоев по разрезу. GexTej-ySe,,
для у=\ (1 и Г), у = 0,5 (2 и 2') и у = 0 (3 и 3') (см. литературу к гл. 2 [25]
и гл. 4 [144]).
673 К 5,87 г/см3. Точно так же в этом температурном интервале
проводимость при переходе от стекла к расплаву возрастает
на 1,5 порядка и достигает 3,4- 104 См/см [576].
Существование кристаллической фазы состава GeTe4 рас-
сматривают как веское доказательство того, что в расплаве
в области переохлаждения или в стекле вблизи эвтектического
состава преобладают структурные единицы, изображенные на
рис. 3.43, в [577]. Термообработка стекла состава GeTe4 при
453 К приводит к кристаллизации фазы, рентгенограмма кото-
рой проиндицирована в кубической сингонии с 8 формульными
единицами в элементарной ячейке. При повышении темпера-
туры происходит разложение этой фазы на GeTe и Те в соот-
ветствии с диаграммой состояния. Обнаружено [578], что
в аморфных пленках состава х 0,2 кристаллиты Те продол-
жают расти, а в пленках с х > 0,3 они разрушаются, что также
Аморфные и стеклообразные системы
331
объясняется образованием соединения GeTe4. Здесь явно идет
речь о метастабильном соединении. Несмотря на то что до сих
пор для этого соединения не определены параметры, существо-
вание соединения GeTe4 является указанием на возможность
существования стеклообразного соединения GeSe4.
На рис. 3.44 приведены концентрационные зависимости
свойств стекол и аморфных веществ в системе Ge.rTei-x (см.
литературу к гл. 2 [25] и гл. 4 [144]). На концентрационной
зависимости Tg для состава GeTe2 при температуре 498 К на-
блюдается отчетливый мак-
симум. Этот состав свобод-
но подчиняется корреляции
между температурой стек-
лования и энтальпией ато-
мизации, характерной для
трехмерно связанной сетки
стекла (рис. 2.45), что по-
зволяет прийти к выводу о
том, что этот состав близок
по структуре к другим стек-
лообразователям общей
формулы МХ2. Оптическая
ширина запрещенной зоны
характеризуется максиму-
мом для состава GeTe2, а
удельная проводимость —
минимумом. Зависимости
Рис. 3.45. Частоты валентных колебаний
V3 структурных группировок GcX4/2 и
GeY4 (/), а также AsXSj,2 и AsY.i (2),
где X*=S, Se, Те, Y=C1, Вг, I.
удельной проводимости и оптической ширины запрещенной зоны
от состава в системе Gex(Tei_ySey) i-x, где i/ = 0; 0,5; 1
(рис. 3.44), свидетельствуют об изоструктурном замещении Те
па Se (см. также рис. 2.43, а).
Близость ИК- и КР-спектров аморфного GeTe2 и Geh,
а также подобные закономерности для пар GeSe2—GeBr4 и
GeS2—GeCI4 трактуются (см. литературу к гл. 2 [253]) в пользу
существования гомологического ряда, в котором частное от де-
ления частот определенных полос поглощения является посто-
янной величиной. На рис. 3.45 приведены соотношения частот
для полос поглощения т.з(772), обусловленных структурными
единицами Gc\z (X = S, Se, Те) и молекулярными соедине-
ниями GeY4(Y = Cl, Вг, I), а также соотношения частот для
полос поглощения ¥з(£) стекол со структурными единицами
AsX и молекулярных соединений! AsY3.
Согласно этим результатам, едва ли можно сомневаться
в существовании соединения GeTe2 в аморфном состоянии.
В кристаллическом состоянии, как это следует также из диа-
граммы состояния, оно до сих пор не получено, при рекристал-
332 Глава 3
лизации происходит его разложение на кристаллический GeTe
и теллур.
Результаты исследования аморфных слоев GeTe2 меюдом
дифракции электронов согласуются с предположением о струк-
туре пространственной сетки, состоящей из структурных единиц
GeTe4/, связанных по вершинам и ребрам; для интерпретации
использована модель статистически равновероятного распреде-
ления связей Ge—Ge, Ge—Те и Те—Те [580]. Сделан вывод
[581] о координационном числе 3 для Ge и Те в аморфных
слоях GeTe2. Результаты исследований структуры этих слоев
методом дифракции нейтронов также подтверждают координа-
ционные числа 3 для Ge и Те [583]. Как и в других стеклооб-
разующих системах, в стеклах системы Ge—Те возможен широ-
кий спектр разнообразных дефектов.
На основании многочисленных экспериментальных результа-
тов еще не удалось установить, реализуется ли для состава
Ge2Te3 (х = 0,4) образование структурных единиц Ge2Te...
Существование таких структурных группировок в виде отрица-
тельных шестизарядных анионов [Ge2Tee]6- в соединении
KeGe2Te6 обнаружено методом рентгеноструктурного анализа
[582].
Выполнены сравнительные исследования аморфных и кри-
сталлических слоев состава GeTe [584, 585]. Эти исследования
свидетельствуют о структурных различиях между обоими со-
стояниями, но не позволяют решить, какой структурный мотив
преобладает в аморфной модификации. Исследования расплава
GeTe методом дифракции нейтронов показали, что в жидком
состоянии реализуется искаженная структурная группировка —
одна из известных для кристаллического и аморфного состоя-
ния [410].
3.2.3. Стеклообразующие фториды
Из стеклообразующих галогенидов уже давно известны ZnCl2
и BeF2. Стеклообразный ZnCl2 изучен методом дифракции ней-
тронов [812]. Лишь недавно изучена возможность применения
ZrF4, HfF4, AIF3 и PbF2 в качестве компонентов стеклообразова-
телей [805]. Недавно открыты стекла на основе CaF2 с A1F3
и другими фторидами щелочноземельных металлов [373].
З.2.З.1. Фторид бериллия
BeF2 по структуре близок SiO2. Для BeF2 установлено су-
ществование модификаций, аналогичных кварцу, тридимиту,
кристобалиту и коэзиту. Благодаря ослаблению кулоновского
Аморфные и стеклообразные системы
333
взаимодействия химических связей температура плавления
BeF2 значительно понижена (823 К по сравнению с 1983 К для
S1O2). Температура стеклования чистого BeF2 составляет 523 К
[586]. Коэффициент термического расширения стеклообразного
BeF2 достигает 6,8- 10-6 К-1, диэлектрическая проницаемость на
частоте 106 Гц составляет 9,7, плотность 1,982 г/см3. Эти свой-
ства относятся к стеклообразному BeF2, очищенному дистилля-
цией под пониженным давлением в атмосфере HF/H2 при ис-
пользовании графитового тигля при температуре 1123 К. Как
правило, BeF2 получают путем термического разложения
(NFLhBeFi: при соответствующей температуре расплава XH4F
улетучивается.
Работа с расплавами BcF2 требует преодоления особых экс-
периментальных затруднений, связанных с гигроскопичностью
BeF2 и взаимодействием со многими материалами, приме-
няемыми для изготовления тиглей. Из-за токсичности работы
с BeFa проводятся в герметичных установках. Несмотря на это,
в некоторых областях техники стекла на основе BeF2 приобре-
тают все большее значение. Область прозрачности стеклообраз-
ного BeF2 простирается от 150 до 4000 нм. Имея необыкновен-
ное сочетание значений показателя преломления (п^= 1,27) и
числа Аббе (vd= 107), стеклообразный BeF2 занимает исклю-
чительное положение на диаграмме n,v (рис. 4.51).
Благодаря очень низкой нелинейной составляющей показа-
теля преломления стекла на основе BeF2 исключительно подхо-
дят для использования в лазерной технике [587]. В системах на
основе BeF2 и фторидов щелочных и щелочноземельных метал-
лов, PbF2, AIF3, а также фторидов редкоземельных металлов об-
наружено множество новых стеклообразных фаз (см. литера-
туру к гл. 1 [17]).
Структура стеклообразного BeF2 характеризуется трехмер-
ным каркасом из структурных группировок BeF^, связанных
через общие вершины. КРР стеклообразного BeFo, полученная
методом рентгеновской дифракции, характеризуется максиму-
мами на расстояниях 150, 255 и 490 пм, что соответствует меж-
атомным расстояниям Be—F в структурных группировках
[BcFa] (в модификации ВеРг, аналогичной кристобалиту, меж-
атомное расстояние Be—F составляет 150 пм), расстояниям
F—F внутри тетраэдра и расстоянию Be—F во второй коорди-
национной сфере [588]. Согласно КРР, в каркасе стекла BeF?
присутствуют в значительном количестве структурные группи-
ровки, связанные по ребрам. Исследования этого стекла мето-
дом дифракции нейтронов выявили наряду с отчетливым мак-
симумом, соответствующим межатомным расстояниям Be—F
154 пм и расстояниям F—F 256 пм, также слабо выраженный
максимум для расстояний Be—Be 309 пм [589]. Средний валент-
334 Глава 3
ный угол при мостиковом атоме F в стекле BeF2 составляет
146°. В литературе также описан колебательный спектр BeF2
(см. литературу к гл. 2 [244]).
3.2.3.2. Стекла на основе ZrF4
Кристаллический ZrF4 представляет собой трехмерную про-
странственную сетку из структурных группировок [ZrFs], гео-
метрический образ которых передается через тетрагональную
антипризму. Структурные единицы ZrF связаны по вершинам
через общие атомы F. Этот структурный признак является глав-
ны,м и выполняется в многочисленных неорганических стеклооб-
разующих системах, поэтому ZrF4 может использоваться по
крайней мере как один из компонентов стеклообразующей си-
стемы.
Обнаружены многочисленные тройные и четверные фторид-
ные стекла, в которых ZrF4 служит стеклообразователем, на-
пример в системах BaF2—LaFs—ZrF4 [592, 593], BaF2—NdFs—
ZrF4 и BaF2—ThF4— ZrF4 [594].
3.2.4. Двойные некристаллические соединения
Как можно обнаружить из сопоставления соединений, при-
веденных в табл. 3.2, наряду со стеклообразующими или аморф-
ными соединениями, которым соответствуют кристаллические
модификации, существует множество веществ определенного
состава, которые в равновесных условиях не проявляются на
диаграммах состояния. К последней группе относятся соеди-
нения, которые получают осаждением в виде аморфных осад-
ков из расплава в виде стекла или из газовой фазы в виде
аморфных пленок; не следует путать эти соединения с вещест-
вами, которые образуются только в результате определенных
химических реакций.
3.2.4.1. Стеклообразующие соединения и структура расплава
Обсуждаемые выше двойные системы характеризуются об-
разованием определенных типов стеклообразующих соединений
(табл. 2.10), в кристаллическом состоянии структура которых
описывается несколькими типами решеток: SiO2, GeO2, GeS2,
GeSe2, As2S3, As2Te3. Исследования структуры стекла позволяют
приблизиться к структуре расплава, так как между ними на-
блюдается значительное соответствие. Только кристаллический
As2Te3 по своему структурному мотиву не укладывается в об-
щую закономерность. Соединепия-стсклообразователи обнару-
живают максимум или скрытый максимум на кривой темпе-
ратуры ликвидуса и, таким образом, проявляются на диа-
Аморфные и стеклообразные системы
335
Таблица 3.2. Двойные неорганические некристаллические соединения
Соединение Получается путем осаждения из раствора, разложе- ния тио- или селеносолей Известно в кристалли- ческом состоянии Проявля- е1ся на диаграмме состояния Известно в виде компактного стекла Известно в виде аморфного слоя
GeS2 + + + + +
GeSe2 + 4- 4- + +
GeSe4 — — + +
GeTe2 — — - 4-
GeTe4 — + — + +
As2S3 + + + + +
As2Se3 + + + +
AsSe3 — — + 4*
As2Te3 — 4" 4- + —
Sb2S3 + 4“ + 4- +
Sb2Se3 + + + + +
Sb2S5 + — — — —
Sb2Se5 — — — —
As2S5 + — — + +
Ge2S3 + — — + +
Oe2Se3 + — — + +
v2s5 + — — — —
MoS3 + — — — —
MoSe3 4- — — — —
ws3 + — — — —
WSe3 + — — — —
Re2S7 H2S 4" RC2O7 — — — —
RuS2 RuCJ3 4- NH4HS 4* + — —
Fc2S3 FeCI3 + Na»S • . 9H2O
336 Глава 3
грамме состояния. Об образовании в расплаве определенной
структуры ближнего порядка можно судить исходя из концен-
трационных зависимостей свойств стекол и взаимных корреля-
ций, а также корреляций с другими параметрами в определен-
ных областях составов, причем такое состояние упорядочения
не обязательно должно проявляться на кривых ликвидуса и/или
солидуса, и тогда гомогенные кристаллические фазы не обра-
зуются. Очевидно, исключением является GeTe4, который про-
является как метастабильное соединение перед тем, как насту-
пает его диссоциация с образованием гетерогенной смеси GeTe
и Те. Из-за недостаточной возможности сравнения с кристалли-
ческими модификациями суждения о структуре часто нена-
дежны, как, например, для стекол AsSe3 и GeSe4. Структурная
единица GeTe4/2, вероятно, все-таки соответствует эксперимен-
тальным данным для расплава GeTe2 или аморфного слоя та-
кого же состава, хотя диаграмма состояния не дает никаких
указаний на существование такого соединения.
Особый интерес представляют выводы о структуре жидко-
сти в области эвтектического состава. Как раз в этой области
стеклообразующая способность часто максимальна (разд. 2.2.2.5
и 2.3.2). Как стеклообразующие соединения GeSe4 и GeTe4, так
и Ge2S3 и Ge2Se3 по составу близки к соответствующим эвтек-
тикам. Однако области эвтектических составов вообще рассмат-
риваются прямо как отрицание определенных соединений.
В то же время постоянство температуры плавления эвтекти-
ческой смеси можно связать с изотермическим процессом проте-
кания твердофазной реакции, в результате чего компоненты ге-
терогенной смеси образуют новое соединение с беспорядочным
расположением структурных единиц в расплаве. Это означает,
что при непериодической упаковке структурных групп, учитывая
соображения симметрии и энергетический баланс, может осу-
ществляться относительно высокая термодинамическая ста-
бильность. Поэтому система пребывает в таком состоянии до
относительно низких температур. Только ниже температуры
плавления эвтектики происходит распад на две или несколько
различных кристаллических фаз. Таким образом, наличие низ-
коплавких эвтектик можно понимать как указание на образо-
вание относительно упорядоченной структуры жидкости, состоя-
щей из определенных структурных единиц. В таком случае эв-
тектическая температура для соединения, существующего
в жидком состоянии, является характерной температурой разло-
жения. Низкотемпературные эвтектики металлических систем,
склонных к стеклообразованию (рис. 3.2), также приводят к вы-
воду об особой структуре жидкости (подобной соединению)
(разд. 3.1.1.2).
Особенно проблематичной является интерпретация струк-
туры в тех случаях, когда отправной точкой служит корреля-
Аморфные и стеклообразные системы
337
ция между составом и каким-нибудь свойством или сравнение
с аналогичным веществом, а прямые методы, например моле-
кулярная спектроскопия или дифракционные методы, дают не-
однозначные результаты. Такая ситуация, очевидно, существует
для стеклообразного As2S5 (разд. 3.2.1.4.). Это соединение, как
и Sb2S3, получают в виде аморфного осадка, и оно принадлежит
вместе с многочисленными сульфидами и селенидами к давно
известным «простым» неорганическим соединениям.
3.2.4.2. Аморфные соединения
К аморфным соединениям, которые получают только осаж-
дением H2S или из растворов тио- или селеносолей, а также
разложением соответствующих солей аммония, принадлежат
наряду с Sb2Ss и Sb2Se5 многочисленные халькогениды переход-
ных металлов (табл. 3.2).
Согласно более ранним предположениям, эти вещества яв-
ляются индивидуальными соединениями. КРР, полученные ме-
тодом рентгеновской дифракции, для MoS3, MoSe3 и WS3 при-
водят к предположению о структурных единицах, в которых 3
атома Мо или W, как в металлических кластерах, связаны
с атомами халькогена [595]. Аналогичные исследования с вы-
сокой разрешающей способностью с привлечением рентгено-
электронной спектроскопии допускают, что по структуре ближ-
него порядка эти соединения относятся к ряду кристаллических
халькогенидов МХ3 (M = Ti, Zr, Hf, Th, Nb, Ta; X = S, Se, Те)
[596]. В этих соединениях атомы М окружены 6 атомами X,
образующими тригональную призму, и структурные единицы
связаны через 2 атома халькогена в жесткие колонны: ооМХ .
Между призматическими колоннами действуют другие связи
[597]. Аморфная структура, очевидно, является результатом сто-
хастического распределения дисульфидных группировок в коор-
динационных полиэдрах МХб/2 [806]. Эти соединения благодаря
аморфной структуре обладают большой поверхностью и вызы-
вают интерес в связи с возможным использованием в качестве
материала катода в электрохимических Li-ячейках с неводными
растворителями. Группировка MoS3 обратимо связывает до
3,8 атома Li, что обеспечивает накопление высокой плотности
энергии [598].
Изучена структура аморфного V2S5 [599]. Следует отметить
образование аморфного RuS2 из RuCI3 и NH4HS в метоксиэти-
ловом эфире; в этом соединении рутений двухвалентен.
Выше 623 К RuS2 кристаллизуется с образованием фазы со
структурой пирита [600]. Аморфный сульфид железа(III) изу-
чен методом ПТСРП [601].
22 Заказ № 413
338 Глава 3
3.3. Тройные стеклообразующие халькогенидные
системы
Число стеклообразующих систем очень велико из-за разно-
образия возможных сочетаний элементов. Изучены оксид-халь-
когенидные стекла и халькогенид-галогенидные стекла, в лите-
ратуре также указывается на использование оксид-фторидных
стекол в волоконной оптике [602—604]. .
Тройные халькогенидные стекла по составу разделяются на
три группы: 1) стекла, два компонента которых халькогены;
2) стекла, образованные халькогеном и двумя элементами од-
ной группы периодической системы; 3) стекла, включающие
элементы IV, V, VI групп периодической системы. Ниже в изло-
жении материала преобладает описательный характер. Из-за
структурно-химической сложности таких систем макроскопиче-
ские свойства рассматриваются только в малой степени, в част-
ности в связи со строением микрообластей.
3.3.1. Оксид-халькогенидные стекла
Из-за слабо выраженного кристаллохимического сродства
между оксидами и сульфидами оксид-халькогенидные стекла,
вообще говоря, не имеют большого значения. В пирометаллур-
гических процессах нашли применение для практических целей
разделения многие оксид-халькогенидные системы с обширными
областями расслаивания; при экономической оценке способа
разделения нередко в качестве критерия рассматривают оста-
точную растворимость одной фазы в другой.
Несмотря на свое практическое значение, следует признать,
что оксид-халькогенидные системы к настоящему времени еще
недостаточно систематически изучены. Какие высокосимметрич-
ные структуры здесь возможны, показывает, например, полиа-
нион SnioO/^o; этот крупный тетраэдрический анион построен
из 10 тетраэдров (SnSi), причем 4 октаэдрические пустоты за-
няты ионами кислорода [605].
К оксид-халькогенидным системам принадлежат также
стекла типа рубиновых; эти стекла готовят путем гомогенного
растворения в основном стекле, например, нескольких десятых
долей процента CdS или CdSe и последующего отжига, при ко-
тором происходит выделение мелкодисперсной фазы, причем
край поглощения стекол сдвигается из УФ- в видимую область
спектра и устанавливается определенная характеристическая
его величина (наведенная окраска стекла) [606] (см. также ли-
тературу к гл. 1 [9]).
В системах Na?S—В2О3 и NaaS—S1O2 возможно стеклообра-
зование вплоть до составов с содержанием до ~40—50 мол. %
Аморфные и стеклообразные системы 339
Na2S [607]. Некоторые стекла бывают окрашены, что связано
с образованием различных полисульфидных ионов [608].
Исследовано [609] влияние добавок оксидов на свойства
халькогенидных стекол; например, Sb2O3 и РЬО вводили
в стекла As2S3—As2Se3. В обзорной работе [610] приводятся
данные для других систем, например As2S3—As2Se3 с HgO или
CuO, As2Se3—GeSe2—HgO, As2Ses—Sb2Se3—БЬгОз, As2Se3—
As2Te.3—CuO и As2Se3—As2Te3—ЗЬгО.з. Изучены также электри-
ческие свойства стекол в системах As2Se3—As2O3, As2Se3—SeO2,
As2Se3—CuO, As2Se3—CdO и As2Se3—V2O5 [611, 612].
3.3.2. Галогенсодержащие халькогенидные стекла
Из 18 систем, которые образуются из сочетаний трех различ-
ных мышьяковохалькогенидных и германиевохалькогенидных
систем с хлором, бромом и иодом, стеклообразование изучено
только в системах мышьяк—сера—галоген и в системах, содер-
жащих иод. Халькогениды сурьмы с галогенами также обра-
зуют стекла. Интерес к системе Sb—S—I вызван, очевидно, сег-
нетоэлектрическими свойствами соединения SbSI.
При замещении атомов халькогена на галоген средняя сте-
пень разветвления сетки стекла уменьшается, поэтому темпера-
тура стеклования значительно понижается. Некоторые из этих
расплавов затвердевают в стекло только при температурах
ниже комнатной. В этих системах образуются обширные обла-
сти стеклообразования.
Граница области стеклооб разования Tg,K Литература
As—S—С1 <10 ат. % CI —'410 613
As—S—Br <50 ат. % Вг -213 614
As—S—I <30 ат. % I -256 615
Ge—S—I <50 ат. % I <298 616
As—Se—I <40 ат. % 1 <298 617 (см. также литерату-
Ge—Se—J <50 ат. % I <298 618 ру к гл. 1 [22])
В соединениях AsXY(AsSI) и GeXY2(GeSI2) группировки
—Asl— и —Geb—, имеющие две связи, формально можно рас-
сматривать как псевдохалькогены, что позволяет без особых
усилий объяснить образование обширных областей стеклообра-
зования.
3.3.2.1. Стекла в системах As—Те—I и Ge—Те—I
На рис. 3.46 приведены границы областей стеклообразова-
ния в этих тройных системах, определенные для скорости
охлаждения ~ 10 К/с. Известно, что добавки иода замедляюще
22*
340 Глава 3
действуют на кристаллизацию. При изучении стеклообразова-
ния в тройных системах исходили из двойных систем As—Те
(разд. 3.2.1.6) и Ge—Те (разд. 3.2.2.6) и в составы с повышенной
стеклообразующей способностью вводили иод как третий компо-
нент. В системе Ge—Те—I Tg составляет ~393 К [619]. В си-
стеме As—Те—I при содержании >30 мол. % I Tg <373 К
[620].
Изучение стекол системы As—Те—I обусловлено в основном
их электрическими свойствами. Для стекол, обогащенных Те и
с малым содержанием I, удельная проводимость достигает ЗХ
ХЮ-3 См/см при 298 К, при этом термическая энергия актива-
ции проводимости составляет 0,36 эВ [614, 620]. Коэффициент
Зеебека 4,3 мВ/К [621].
Давно известно, что в стеклах системы As—Те—I наблюда-
ется нелинейная зависимость силы тока от напряжения, и при
высоких напряжениях эти стекла характеризуются эффектом
переключения. При этом наблюдается переход из высокоомного
полупроводникового состояния в низкоомное (разд. 4.1.3.7) [614,
621].
Аморфные и стеклообразные системы
341'
Удельная проводимость стекол системы Ge—Те—I, даже при
большом содержании Те, ниже, чем в системе As—Те—I (по-
рядка ~10-6 См/см при комнатной температуре; ZFCT = 0,95 эВ).
3.3.2.2. SbSI
SbSI— это высокоомный сегнетоэлектрик и полупроводник;
температура фазового перехода 292,3 К. Диэлектрическая про-
ницаемость при температуре немного выше температуры пере-
хода, близка к ~ 104. Диаграмма состояния системы Sb2S3—
Sbl3 приведена в работе [622]. Соединение SbSI находится вне
области стеклообразоваиия [623]. Сообщается также о стекло-
образовании в четвертой системе Ge—Sb—S—I [624], Тё стекла
состава SbSI • GeS2 равно 523 К.
3.3.3. Тройные халькогенидные стекла,
содержащие два халькогена
На основе рассмотренных в разд. 3.2 двойных сульфидных,,
селенидных и теллуридных систем можно составить следующие
тройные системы:
P—S—Se
As—S—Se
Si—S—Se
Ge—S—Se
P—S—Те
As—S—Те
Si—S—Те
Ge—S—Те
Р—Se—Те
As—Se—Те
Si—Se—Те
Ge—Se—Те
Стекла, содержащие Si или P, еще мало изучены из-за их
химической неустойчивости. Фазовые равновесия в системе
Р—S—Se изучены по нескольким разрезам [625]. В то же
время тройные системы на основе халькогенидов мышьяка и
германия исследованы подробно.
3.3.3.1. Стеклообразование и свойства в системах
As—S—Se, As—S—Те и As—Se—Те
Области стеклообразоваиия. На рис. 3.47 приведены границы
областей стеклообразоваиия в системах As—Se—S [215] и
As—Se—Те [626], полученные для скоростей охлаждения
~ 10 К/с. Исследованы также стекла в системе As—S—Те
[627]. Область стеклообразоваиия в этой системе из-за наличия
расслаивания ограничена интервалом составов с большим со-
держанием As2S3.
Структура стекол. Предполагают, что структура стекол
в указанных системах образована структурными единицами, су-
ществующими в двойных системах As — халькоген
-342 Глава 3
Рис. 3.47. Области стеклообразования в системах As—S—Se и As—Se—Те.
(разд. 3.2.1.4—3.2.1.6). Температура стеклования в указанных
системах возрастает при повышении содержания As, поскольку
степень разветвления сетки стекла в результате образования
структурных единиц AsSe3. и AsTe3/2 увеличивается; на разре-
зах (As2Ses) 7/(As2S3)i-y и (As2Tes) 7/(As2Se3)i-y Tg достигает
максимума, а при дальнейшем возрастании концентрации As
снова убывает [626, 628], т. е. стекла, составы которых распо-
ложены вдоль штриховой линии (рис. 3.47) характеризуются
самыми высокими температурами стеклования Tg. В работе
[629] приводятся концентрационные зависимости Tg и коэффи-
циента термического расширения для стекол системы As—S—
Те.
Исходя из широких областей изоморфного замещения в си-
стемах из двух халькогенов, например в системах SxSei-x
(разд. 3.1.3.1.) и Te.rSei-x (разд. 3.1.3.3.), можно допустить, что
атомы халькогенов образуют цепочки с распределением, близ-
ким к статистическому. В системе As2S;;—As2Se,3 образуется не-
прерывный ряд твердых растворов [630]. Исследования моно-
кристаллов состава As2(SxSei-x)з (где 0 < х < 0,5) методами
дифракции рентгеновских лучей и нейтронов показало, что об-
разование твердых растворов происходит посредством предпоч-
Аморфные и стеклообразные системы
343
Рис. 3.48. Отклонения от аддитивности
мольных объемов по разрезам (As2S3)i-y-
(As2Se3)y (/), (Ge2S3)i-!/(Ge2Se3)!/ (2) (см.
литературу к гл. 2 [343]), (As2Se3)i-y-
(As2Tcj)y (<?) (см. литературу к гл. 1 [22]).
тителыюго замещения атомов Se атомами S в цепочках As—Se
и характеризуется укорочением межатомных расстояний, т. е.
взаимодействие между атомами отличается от статистического
смешения. С этим предположением согласуется наблюдаемое
для стекол системы (As2S.3)i-y(As2Se.3) у небольшое отрицатель-
ное отклонение концентрационной зависимости мольного объ-
ема от аддитивности (см. литературу к гл. 2 [243]).
В системе (Аз2Тез) у (As2Ses)i-y, согласно диаграмме состоя-
ния, образуются ограни-
ченные твердые растворы
на основе двойных соеди-
нений, кристаллизую-
щихся в разных струк-
турных типах. В средней
части диаграммы взаим-
ная растворимость компо-
нентов отсутствует [631].
Расплавы и стекла в этой
системе гомогенны, в них
не наблюдается расслаи-
вание. Концентрацион-
ная зависимость моль-
ных объемов стекол этой
системы, рассчитанных
из данных по плотности,
свидетельствует о поло-
жительном отклонении от
аддитивности, что типич-
но для систем, где отсут-
ствует смешение (см. литературу к гл. 2 [343]). Приведен-
ная на рис. 3.48 кривая показывает, что в стеклах,
структурные группировки AsSe3/ и AsTe3/ , по крайней мере
частично, образуют друг с другом более крупные ассоциаты,
и возможно образование структурных группировок со смешан-
ными атомами халькогенов.
В ИК-спектрах отражения стекол системы (As2S.4)i-yX
X (As2Se3)y присутствуют характеристические полосы колеба-
ний As—S (310 см-1) и As—Se (220 см-1) (см. литературу
к гл. 2 [253]). Эти полосы относятся к антисимметричным ва-
лентным колебаниям структурных группировок AsX^..
Для обеих полос сила осциллятора линейно зависит от мольной
доли компонента. В ИК-спектрах отражения стекол системы
(Аэ2Тез) у(As2Ses) t-y наблюдаются полосы 184 и 218 см-1 (см.
литературу к гл. 2 [253]).
Свойства стекол. В литературе приведены температурные за-
висимости вязкости стекол и расплавов систем As—Se—Те1
344
Глава 3
[626], As2Te3—As2Se3 (см. литературу к гл. 1 [20]) и As2Ss—
As2Se3 [637], а также концентрационная зависимость упругих
свойств стекол системы As2Ss—As2Se3 [633] и коэффициента
термического расширения стекол в системе As—Se—Те [626].
Большое внимание привлекли оптические и особенно элек-
трические свойства этих стекол. Изучена (см. также литературу
к гл. 1 [20]) [634] зависимость края оптического поглощения от
состава для стекол в системах As2S3—As2Se3 и As2Se3—As2Te3.
При введении As2Se3 в As2S3 происходит сдвиг края поглощения
Рис. 3.49. Концентрационные зависимости энергии активации проводимости
£’а, диэлектрической проницаемости ег, молярной поляризации Ру и мольного
объема V для стекол в системах (As2Se3) у (As2S3) 1_у и (As2Te3) у (As2Se3) i-y.
из видимой области в сторону длинных волн, а при добавлении
As2Te3 к стеклообразному As2Se3 наблюдается максимум фото-
проводимости для состава (As2Te3)o75(As2Se3)o25, отвечающий
— 1 эВ.
Изучена удельная проводимость стекол в системах
Aso.4So,6-xSex и Aso.4Seo.6-xTex [628, 634, 635], а также удельная
проводимость на постоянном и на переменном токе для стекол
системы As—Se—Те [636, 637].
На рис. 3.49 приводятся концентрационные зависимости тер-
мической энергии активации Еа, диэлектрической проницаемо-
сти ег, молярной поляризации Ру и среднего мольного объема
V для стекол в системах (As2Se;) 7/(As2S3)i-y и (Аз2Тез)т/Х
X(As2Se3)i-y [261].
Известно, что при введении тяжелых халькогенов ширина
запрещенной зоны уменьшается. При изменении состава стекол
в системе As—Se—Те удельная проводимость на постоянном
Аморфные и стеклообразные системы 345-
токе меняется на 11 порядков. Стекла, обогащенные Те, пред-
ставляют особый интерес как модельные вещества для изуче-
ния основных процессов переноса заряда в халькогенидных
стеклах. Удельная проводимость этих стекол измерена и в обла-
сти низких температур (см. разд. 4.1.3.1.).
В стеклах Se—Те и As—Те в области низких температур об-
наружен переход к малым энергиям активации проводимости
[638], что интерпретируется как изменение механизма переноса
заряда от зонной модели к прыжковому механизму. Аномалии,
обнаруженные для эффекта Холла, интерпретируются таким
же образом [639]. В литературе приводятся также результаты
измерений термо-э. д. с. [640]. Согласно измерениям дрейфовой
подвижности в стеклах системы As—Se—Те приблизительно
на порядок больше, чем цп [641].
3.3.3.2. Стеклообразование и свойства стекол в системах
Ge—S—Se, Ge—S—Те и Ge—Se—Те
Стекла в системе Ge—S—Se. Расширение области стеклооб-
разования в системе Ge—S—Se относительно просто объяснить,
исходя из соображений о структуре и типе связи (разд. 3.2.2.4.
и 3.2.2.5). В структуре этих стекол можно ожидать значитель-
ного изоморфного замещения. В структуре стекла группы GeS4,2
и GeSe4/ или такие группы, в которых произошел обмен атома
одного халькогена на другой халькоген, должны связываться
через цепочки различной длины из атомов халькогенов, а в си-
стеме (GeS?) y(GeSe2)i-y структурные группы непосредственно
соединяются друг с другом через собственный атом халькогена,
образуя пространственную сетку.
Проведено отнесение характеристических полос в И Коспек-
трах для структурных группировок: GeS4/ 367 см-1 (vs, F2) и
160 см-1 (v?, Е)', GeSe4 254 см"1 (v.3, Ft) и 100 см"1 (v?, Е)
[642]. Для структурных единиц, содержащих различные атомы
халькогена в первой координационной сфере, антисимметричное
валентное колебание дает характеристические полосы для свя-
зей Ge—S и Ge—Se. Сила осциллятора линейно зависит от
мольной доли соответствующей структурной единицы. Из одних
молекулярных спектров нельзя заключить, осуществляется ли
статистическое распределение связей Ge—S и Ge—Se или же
присутствуют структурные единицы GeS4/2 и GeSe42, т. е. на-
сколько предпочтительно образование смешанных структурных
единиц.
346 Глава 3
В области стеклообразования при избытке S или Se отме-
чено, что стекло состава GeSe4 обладает относительно высо-
кой степенью упорядочения, в то время как стекло состава GeS4
микронеоднородно: цепи S различной длины сосуществуют со
структурными областями, в которых несколько структурных
единиц GeS связано друг с другом (разд. 3.2.2.3.). В ряду
(GeS4) у (GeSei) i-y склонность к расслаиванию должна убывать
при уменьшении мольной доли у, в этом направлении наблюда-
ется переход к большей структурной однородности тройных
стекол.
Возрастание содержания Ge в стеклах больше, чем это соот-
ветствует составу Ge(S, Se)2, усиливает склонность к кристал-
лизации расплава. Это наблюдение относится к обеим областям
стеклообразования в двойных системах GexSi-x и GexSei-x; для
тройной системы в области х >• 0,33, очевидно, также должны
существовать стекла. Однако это предположение до настоящего
времени еще не получило экспериментального подтверждения,
так что вопрос о том, существует ли в центре фазового тре-
угольника системы Ge—S—Se область стеклообразования, со-
единяющая две области стеклообразования двойных систем,
остается открытым; эта область должна простираться прибли-
зительно до линии составов (06283)2/(0686.3)1-2/. Для этих сте-
кол вполне обоснованно предположение о структуре, представ-
ляющей собой трехмерную пространственную сетку, состоящую
из структурных единиц Ge2Sc/2 и Ge2Se6/2 или из соответствую-
щих структурных единиц, содержащих атомы различных халь-
когенов (разд. 3.2.2.4 и 3.2.2.5). Зависимость средних мольных
объемов от состава для стекол системы (GeaSs) v (GezSea) 1—у об-
наруживает небольшое положительное отклонение от аддитив-
ности (см. литературу к гл. 2 [343]). Для этих же стекол тем-
пература стеклования составов Ge2S,3 и Ge2Se3 отличается
только приблизительно на 20°С и почти линейно зависит от со-
става.
Стекла системы Ge—S—Те. Диаграмма состояния тройной
системы Ge—S—Те изучена в работе [643]. В системах GeS —
GeTe и GeSa—GeTe наблюдается простое эвтектическое взаи-
модействие, а составу (GeTe)0,328(GeS)0.10(GeS2)0,572 отвечает эв-
тектика с температурой плавления 807 К. В этой области со-
ставов получены гомогенные стекла [644]. Уточнение границ
области стеклообразования в этой системе еще не закончено.*
* Границы стеклообразования в системе Ge—S—Те приведены в работах
[Maneglier-Lacordaire S., Besancon Р„ Rivet L, Flahaut J.— J. Non-Cryst. So-
lids, 1975, t. 18, 439; Алексеева И. В., Образцова А. А., Борисова 3. У„
Фунтикова В. А.— Физ. и хим. стекла, 1978, т. 4, с. 490; Маковская 3. Г.,
Жуков Э. Г.— Изв. АН СССР, неорг. матер., 1980, т. 16, с. 251].— Прим, перев.
Аморфные и стеклообразные системы
347'
Рис. 3.50. Границы области стеклообразования в системе Ge—Se—Те.
/ — [286], 2 — [652], 3 — [649]. £ —эвтектики в двойных системах GeTe—Те и GeSe—GeSe2:
£i и £2 — эвтектики в тройной системе.
Стекла системы Ge—Se—Те. Диаграмма состояния тройной
системы Ge—Se—-Те изучена в работе [645]. Разрез GeSe2—
GeTe характеризуется простым эвтектическим взаимодействием.
GeSe и GeTe в широкой области концентраций образуют твер-
дые растворы, причем каждый из компонентов растворяется
в кристаллической решетке другого, а в средней области кон-
центраций ниже эвтектики при понижении температуры раство-
римость компонентов отсутствует [646].
В отличие от системы GeS2—Те, где в расплаве наблюдается
гомогенная растворимость и в области, богатой Те, располо-
жена эвтектика, в системе GeSe2—Те наблюдается большая
область расслаивания [645, 647, 648]. Тройная эвтектика Е}
(температура плавления 633 К) в области, богатой Те, соответст-
вует составу Geo,i9Seo,o9Teo,729. Вторая эвтектика £2 отвечает со-
ставу Geo44Seo,4Teo,i6 или (GeTe)o,364(GeSe)o,364(GeSe2)o,i72 и пла-
вится при температуре 751 К; состав эвтектики расположен
в треугольнике, образованном тремя указанными двойными со-
единениями. Обе эвтектики можно соединить друг с другом ли-
нией минимальных температур ликвидуса. Область стеклообра-
зования расположена примерно вдоль этой эвтектической линии
'348 Глава 3
(рис. 3.50). Вторая область стеклообразоваиия расположена со
стороны, богатой халькогенами. Две области стеклообразоваиия
разделяются псевдобинарным разрезом GeSe2—Те, причем при
большом содержании GeSe2 сплавы склонны к кристаллизации,
а в интервале 28—86 мол. % Те наблюдается расслаивание
с образованием двух жидких фаз.
Область стеклообразоваиия в системе Ge—Se—Те приве-
дена на рис. 3.50 [286, 649]. Показаны эвтектики Е двух двой-
ных систем GeTe—Те и GeSe—GeSe2, а также эвтектики и
Е2 тройной системы. Границы области стеклообразоваиия опре-
делены для навесок расплава в 1 г при охлаждении в ледяной
воде (скорость охлаждения ~35 К/с) [286, 652]. Массивные
•образцы (навеска 10 г) можно получить при скорости охлаж-
дения ~2 К/с (ампулы вынимают из печи и охлаждают на воз-
духе) [649, 650].
Термические свойства стекол в системе Ge—Se—Те подробно
описаны в литературе; приводятся данные по температурам
стеклования и кристаллизации стекол [286]. В разд. 3.3.2.6 при-
ведены результаты исследования свойств стекол в системе
Gex(Tet-?/Se2/)i-x, причем на основе этих экспериментальных ре-
зультатов приходят к заключению о существовании структур-
ных единиц GeTe4/2 в аморфных слоях состава GeTes- Кроме
того, следует принять, как и в системе Ge—Se, что связанные
друг с другом атомы халькогена оказывают более существен-
ное влияние на структурообразование только в области соста-
вов х С 0,33, т. е. за разрезом (GeTe2) у (GeSe) i-y (рис. 3.50)
находятся стекла, в структуре которых преобладают структур-
ные единицы GeTe^ и GeSe4^ или структурные единицы, со-
держащие различные атомы халькогенов. Состояние упорядоче-
ния, проявляющееся в стеклах составов GeSei и GeTe4
(разд. 3.3.2.5 и 3.3.2.6), приводит по разрезу (GeTe4) у (GeSei) i-y
к расширению границ растворимости одного компонента в дру-
гом (рис. 3.50). В центре фазового треугольника (рис. 3.50)
взаимная растворимость компонентов отсутствует — в системе
присутствует несколько кристаллических фаз.
В связи с наблюдаемым эффектом переключения (разд. 4.1.3.7)
исследованы зародышеобразование и кристаллизация стекол
как для массивных образцов, так и для аморфных слоев в си-
стеме Ge—Se—Те в области составов с высоким содержанием
халькогенов, особенно в области эвтектики GeTe/Te, например
для составов Geo,2Seo,4Teo,4 [653] или Geo.15Seo.o4Teo.8i [652]. При
обсуждении переноса заряда в сильных электрических полях
сообщается (разд. 4.1.3.6) об аморфных слоях системы Ge—
Se—Те, полученных испарением в вакууме.
Аморфные и стеклообразные системы
349
3.3.4. Тройные халькогенидные стекла с двумя различными
элементами V и IV групп периодической системы
Исходя из описанных в разд. 3.2 двойных сульфидных, се-
ленидных и теллуридных систем, можно обсудить следующие
тройные системы:
Р—As—S
Р—Sb—S
As—Sb—S
Si—Ge—S
P—As—Se
P—Sb—Se
As—Sb—Se
Si—Ge—Se
P—As—Те
P—Sb-Те
As—Sb—Те
Si—Ge—Те
Из-за слабо выраженной стеклообразующей способности си-
стемы Р—As—Те, Р—Sb—Те и As—Sb—Те не имеют практиче-
ского значения, а сплавы, образующиеся в системах Si—Ge—S
и Si—Ge—Se, имеют ярко выраженную склонность к гидролизу
и поэтому также не представляют интереса.
3.3.4.1. Стекла в системах Р—As—S и Р—As—Se
Для этих систем подробно исследована взаимосвязь струк-
туры и свойств стекол; сообщается и о стеклообразовании
в этих системах [248, 563]. Согласно работе [257], стекла полу-
чают из расплава вплоть до состава ?2As2Se; исследована за-
висимость Tg стекол от состава по разрезам (Po.sAso.sJxSi-x и
(Po.sAso.r,) Sei-x, а также в системах P4Se4—As2Ses и P4Se.3—
As2Se3. Приведены данные [177] по удельной проводимости,
плотности и микротвердости стекол в системе Р—As—Se.
«Клеточная» структура сульфидов и селенидов фосфора и
мышьяка (рис. 3.28 и 3.31) позволяет предположить образова-
ние смешанных фаз. Например, из расплава выделено в си-
стеме P4S3—As2S,3 соединение P2AS2S3 [348]. По структуре это
соединение соответствует молекуле P4S3, но содержит связь
As—As, в то время как связь Р—Р не обнаружена (разд. 3.2.1.3,
3.2.1.4).
3.3.4.2. Стекла в системе Р—Sb—Se
Высокое давление пара расплава Р—S при температуре
плавления Sb^Ss ограничивает возможности получения стекол
в системе Р—Sb—S с относительно низким содержанием Sb.
При этом не достигается существенного повышения термиче-
ской и химической устойчивости фосфорносульфидных стекол.
Стеклообразование и свойства стекол в системе Р—Sb—Se
приведены в работе [405]. Как следует из рис. 3.51, гомогенные
стекла образуются в интервале составов от разреза P4Seio—
Sb2Se3 до ~25 мол.% Р (скорость охлаждения ~2 К/с). В об-
350 Глава 3
Рис. 3.51. Области стеклообразоваиия в системах Р—Sb—Se [405] и Si—Ge—Те
[471, 529].
ласти, богатой Р, при охлаждении расплава на стенке ампулы
осаждаются из газовой фазы низшие селениды фосфора, так что
приведенные границы области стеклообразоваиия могут быть
не вполне достоверны. На диаграмме указаны продукты пер-
вичной кристаллизации. Область стеклообразоваиия в системе
Р—Sb—Se значительно меньше, чем в родственной системе
As—Sb—Se.
Аморфные и стеклообразные системы
351
3.3.4.3. Стекла в системах As—Sb—S и As—Sb—Se
Наибольшая протяженность области стеклообразования в си-
стеме As—Sb—S наблюдается по разрезу (Sb2Ss) y(As2S3)i-y
(г/ гС 0,62) [515], вдоль которого проходит и примерная гра-
ница области стеклообразования. При увеличении содержания
Sb2S3 температура стеклования немного повышается, а коэффи-
циент термического расширения уменьшается.
В системе As—Sb—Se свойства стекол изучены в основном
по разрезу (Sb2Se3) у (As2Se3) i-y. Граница области стеклообра-
зования отвечает составам с г/= 0,45 4-0,5 [516]. Термическая
энергия активации проводимости составляет 0,93 эВ для As2Se3
и 0,72 эВ для стекла состава (As, Sb)2Ses. Эти величины соот-
ветствуют значениям оптической ширины запрещенной зоны,
найденным из измерений края поглощения. Tg в системе
(Sb2Se3) у (As2Se.?) i-y не зависит от состава [562].
3.3.4.4. Стекла в системах Si—Ge—Те
В системе Si—Ge—Те область стеклообразования (рис. 3.51),
как и можно было ожидать, находится между двумя областями
стеклообразования в двойных системах Si—Те и Ge—Те
(разд. 3.2.22 и 3.2.2.6) [471, 529]. Исходя из свойств эвтектиче-
ских расплавов многочисленных систем с Те в области
с -80 мол. % Те, можно предположить, что стеклообразованию
способствуют относительно низкие температуры ликвидуса вдоль
эвтектической линии, соединяющей обе двойные системы. Tg
стекол понижается при добавлении Ge к стеклам системы
Si—Те.
3.3.5. Тройные халькогенидные стекла, образованные
элементами IV, V и VI групп периодической системы
Сочетание описанных в разд. 3.2.1.3—3.2.1.7 двойных халь-
когенидных систем, содержащих элемент V группы, с двойными
халькогенидными системами кремния и германия (разд. 3.2.2.2,
3.2.2.4—3.2.2.6) приводит к ряду тройных систем, причем в не-
которых из них в широких областях концентраций образуются
устойчивые стекла:
Р s Si—P—S Se Si-P—Se Те Si—P—Те
Si As Si—As—S Si—As—Se Si—As—Те
Sb Si—Sb—S Si—Sb—Se Si—Sb—Те
Р Ge—P—S Ge—P—Se Ge—P—Те
Ge As Ge—As—S Ge—As—Se Ge—As—Те
Sb Ge—Sb—S Ge—Sb—Se Ge—Sb—Те
352 Глава 3
Сплавление сульфидов и селенидов кремния с сульфидами
или селенидами фосфора и мышьяка опять-таки приводит к по-
лучению продуктов реакции, сильно склонных к гидролизу.
Предполагают, что большие различия в температурах плавле-
ния двойных соединений неблагоприятны для стеклообразова-
ния. Данные об областях стеклообразоваиия в системах Si—
Sb—S и Si—Sb—Se приводятся в работе [651]. Расплавы в си-
стемах Si—Sb—Те и Ge—Sb—Те при обычных условиях охлаж-
дения не образуют стекол.
З.З.5.1. Стеклообразование в системах Ge—Р—S и Ge—Р—Se
Области стеклообразоваиия * в системах Ge—Р—S и Ge—
Р—Se приведены на рис. 3.52, а и б. К изучению этих систем
обратились в связи с поиском новых стекол, прозрачных в ИК-
области и обладающих возможно большей термостойкостью
[651]. Относительно высокие значения средних молярных энер-
гий связи, которые характерны для двойных соединений в этих
системах, дают возможность ожидать высоких температур стек-
лования. Для стекол системы Ge—Р—S на рис. 3.52, а выде-
лена область, где стекол достигает 753 К.
Расширение областей стеклообразоваиия, о чем сообщалось
для систем Ge—Р—S [651] и Ge—Р—Se [654] (см. также ли-
тературу к гл. 1 [22]), не подтверждено. Над расплавами, со-
держание Р в которых выше, чем для составов, лежащих на
разрезах GeSs—P4S3 или GeSe2—Р4$ез, в запаянных под вакуу-
мом кварцевых ампулах развивается значительное давление
пара низших халькогенидов фосфора. При охлаждении эти пары
осаждаются на затвердевшем образце и на стенках ампулы, и
при вскрытии ампулы происходит самопроизвольное воспламе-
нение, поэтому в этой области составов получение гомогенных
стекол заданного состава невозможно. Фазовые равновесия
в системе Ge—Р—S изучены по разрезам GeSz—P4S3, GeS?—
P4S7 и GeSz—P4S10 [655]. Тройные соединения в системе не об-
наружены.
Определены границы области стеклообразоваиия в системе
Ge—Р—Se (навеска 10 г, скорость охлаждения ~2 К/с [656]
(см. также литературу к гл. 2 [343]) (рис. 3.52, б). Линия раз-
реза GeSez—P4Se3 близка к границе образования гомогенных
стекол.
Мольные объемы стекол в системе Ge—Р—Se подчиняются
зависимости, обнаруженной в системах PxSe]_x и GexSei_x
* Уточнение границ области стеклообразоваиия в системе Ge—Р—S
[Виноградова Г. 3., Майсашвили Н. Г.— ЖНХ, 1979, т. 24, с. 1116] пока-
зало, что область стеклообразоваиия значительно меньше, чем по данным
работы [651].— Прим, перев.
354 Глава 3
(рис. 3.29 и 3.40). Концентрационная зависимость мольных объ-
емов проходит через минимум, расположенный между соста-
вами GeSe4 и P4Se4 [656]. Зависимость Ts от состава в тройной
системе образуют некоторую искривленную поверхность, причем
температура стеклования меняется от низких значений для сте-
кол с большим содержанием Se в системах GexSei-x и PxSei-x
до высоких значений при увеличении содержания в стекле
структурных единиц GeSe4/i . Изучены ИК-спектры пропуска-
ния стекол в системе Ge—Р—Se [651, 656, 657]. Эти стекла яв-
ляются высокоомными полупроводниками. Измерения удельной
проводимости стекол системы Ge—Р—Se показали, что термиче-
ская энергия активации проводимости лежит в интервале 0,8—
0,95 эВ.
3.3.5.2. Стеклообразование и свойства стекол в системах
Ge—As—S и Ge—As—Se
Области стеклообразования. Стеклообразованию в этих си-
стемах посвящены многочисленные работы, причем, как пра-
вило, различные результаты не согласуются друг с другом; по-
видимому, эксперименты проводились со стеклами, получен-
ными в различных условиях; кроме того, не всегда достаточно
строго учитывается влияние примесей. Стекла системы Ge—
As—Se представляют интерес для использования в ИК-оптике
(разд. 4.2.1.2 и 5.2), а также в связи с исследованиями фотоин-
дуцированных структурных превращений (разд. 4.2.4.1 и 5.3.1).
Области стеклообразования в системах Ge—As—S и Ge—
As—Se приведены на рис. 3.53, а и б. Нанесенные на рисунках
линии связывают области стеклообразования в соответствую-
щих двойных системах. В этих системах обнаружены опреде-
ленные фазы, тройные соединения GeAsS и GeAsSe [658] * и
GeAs4Se [659]. Приведенные на рис. 3.53, а границы области
стеклообразования [660] не были подтверждены. Правда, в ра-
боте [661] по разрезу As2SsGen получены большей частью гомо-
генные стекла, однако в области 5—10 мол.% Ge наблюдается
расслаивание. Границы области стеклообразования (рис.
3.53, а) [662] соответствуют охлаждению расплава на воздухе
(навеска 50 г, скорость охлаждения ~2 К/с).
Расплавы в системе Ge—As—Se в аналогичных условиях
образуют стекла; концентрационные границы области стекло-
образования приведены на рис. 3.53, б. Данные различных ра-
бот для этой системы лучше согласуются между собой [663—
* О соединении GeAsS см. работу [658], о GeAsSe см. работу [Виногра-
дова Г. 3., Дембовский С. А., Лужная Н. П.— ЖНХ, 1968, т. 13, с. 1445].—
Прим, перев.
Ge
Рис. 3.53. Области стеклообразования в системах Ge—As—S [662] (о) и
Ge—As—Se [664] (б).
356 Глава 3
666]. На рис. 3.53,6 приведены границы области стеклообразо-
вания согласно работе [664].
Структура стекол. В системах Ge—As—S и Ge—As—Se для
стекол разных составов можно ожидать реализации совершенно
различных структур. Относительная ясность в отношении струк-
тур имеется для области А. Для стекол в двойных системах из-
вестны характерные структурные единицы; поэтому строение
стекол в области S—As2S3—GeS2 или Se—As2Ses—GeSe2 можно
описать как смесь структурных единиц AsX3/2 и GeX4/ , связан-
ных или непосредственно друг с другом, или через цепочки из
атомов халькогенов. Согласно отнесению частот в ИК- и КР-
спектрах, стекла по псевдобинарным разрезам (As2Ss)yX
X(GeS2)i-!/ и (AsSes) y(GeSe2)i-2/ должны быть построены из
тригональных и тетраэдрических структурных группировок
[642, 668]. Расчет по валентно-энергетической модели приводит
к валентным колебаниям типа Ai и F2 или Е [669]. Волновые
числа для экспериментально наблюдаемых и рассчитанных по-
лос приведены в табл. 3.3.
Таблица 3.3. Полосы поглощения стекол в системах (As2Ss) v (GeS2)-y
и (As2Se)w(GeS2)i-i/ [669]
Структурная единица Симметрия Правило отбора Волновое число, см-*
рассчнт. экспер.
ик КР
GeS,, */2 Aj КР V1 342 345
Т2 ик, КР v3 367 367 374, 408
GeSe,, 4/2 А, КР Vi 200 201, 219
а2 ик, КР V3 248 254 248, 267
AsS3/2 Ai КР vi 345 345
Е ик, КР v3 310 310 310
AsSe3/2 Ai КР vt 230 239
Е и к, КР v3 217 217 225
Область В ограничена соединениями As2S.3, AS4S4, GeS и
GeS2 или As2Se3—As4Se4—GeSe—GeSe2. Даже для псевдо-
бинарных систем на основе халькогенидов мышьяка и герма-
ния в литературе существует противоречивая интерпретация
экспериментальных данных. В тройных системах необходимо
дополнительно принимать во внимание реакции окисления-вос-
становления, тип и состояние равновесия которых едва ли мо-
жно сейчас обсуждать. С химических позиций вполне возможно
восстановление соединений мышьяка соединениями германия
Аморфные и стеклообразные системы 357
низкой степени окисления. Степень протекания таких реакций и
структурный состав равновесного расплава в настоящее время
еще невыяснены.
Исходя из КР-спектров стекол состава As2Se.3-n, даже и pH
относительно маленьких значениях п можно прийти к выводу
о наличии структурных единиц AsAsz , характерных для
аморфного мышьяка [355]. Почти наверняка не будет ошибкой
предположение, что протеканию таких процессов способствуют
добавки GeS или GeSe.
При введении в расплав стекол состава As2Sa—As4S4 или
As2Se.3—As4Se4 добавок SnS или SnSe в мессбауэровском спек-
тре получаемых стеклообразных фаз проявляется лишь четы-
рехвалептное олово. Только при более высокой концентрации
олова наряду со Sn(IV) одновременно появляется и Sn(II)
[670, 671].
В области В GeS и GeSe также должны действовать как
компоненты стеклообразования, т. е. они будут отбирать халь-
коген у структурных единиц AsS или AsSe„ для того, чтобы
/2 /2
образовать структурные единицы GeS.. или GeSe,, . Такой
/2 /2
процесс обязательно приводит к образованию структурных еди-
ниц, богатых As. В этой связи для стекол системы AsxSei-.x
следует указать на наличие выраженного минимума для состава
As,-,Se2 на концентрационной зависимости молярной поляриза-
ции (рис. 3.34). Такие выводы о структуре стекол по разрезу
As2SesGen сделаны в работе [661]. Исследование этих стекол
методом КР-спектроскопии приводит к таким же результатам
[672].
В заключение следует заметить, что мышьяк также образует
связь с германием. В системе Ge—As—Se известны стабильные
стекла, составы которых относительно далеки от области В.
В области С образование стабильной сетки стекла можно до-
пустить только в том случае, если реализуются связи Ge—As.
Следует заметить, что, по данным рентгеновской дифракции,
для стекла состава GeAs2Se2 среднее координационное число
( — 2,5) совпадает с этим же параметром для стекла Ge.'.Se?.
Для стекла состава Ge^AsaSe, среднее координационное число
составляет лишь 2,2. При 893 К в расплаве еще сохраняется
такой ближний порядок, и только при — 1043 К происходит пе-
реход к более высокому координационному числу. Для соеди-
нений GeAsS и GeAsSe в стеклообразном состоянии расчет
среднего координационного числа по первому максимуму на
КРР дает 3,6—3,7 [675]. Кристаллы GeAsSe по структуре соот-
ветствуют структурному типу красной модификации Hgl2 [667].
Свойства стекол и расплавов. Результаты измерений вязко-
сти в системе Ge—As—Se приведены в работах [676, 677]. Тем-
358 Глава 3
пературная зависимость плотности для расплава, состав кото-
рого расположен в области С (рис. 3.53, б), проходит через
минимум в области 970—1070 К [678]. В области более низких
температур при повышении температуры наблюдается резкое
возрастание удельной проводимости до 103 См/см [666]. В рас-
плаве для составов Ge9As6Se5 и GenAs6Se3 при ~ 1023 К
происходит изменение структуры, связанное со скачкообразным
переходом к металлической проводимости. В этой же области
температур также проявляется аномалия плотности. При повы-
шении координационного числа структурные единицы перегруп-
пировываются, так что происходит перекрывание зон. Вязкость
таких расплавов с металлической проводимостью понижается
на 3—4 порядка.
Аморфные и стеклообразные системы
359
Рис. 3.54. Концентрационные зависимости мольного объема V, мольной ре-
фракции и сжимаемости х для стекол в системах (As2S3) y(Ge2S3) t-y и
(As2Se3);; (Ge2Se3) !-v (п), а также диэлектрической проницаемости ег в систе-
мах As2Se3—Ge2Se3 и Tg в системах As2S3—Ge2S3, As2S3—GeS2 и As2Se3—
GeSe2 (6).
Концентрационные зависимости среднего мольного объема
для стекол систем Ge—As—S и Ge—As—Se изучены во многих
работах [660, 661, 679]. В работе [679] для стекол системы
Ge—As—Se приводятся подробные данные о показателе пре-
ломления, твердости по Викерсу, упругим постоянным, а также
температурам стеклования Tg и вязкости в интервале Tg. Там
же приводятся значения коэффициента термического расшире-
ния. Следует отметить, что температура стеклования в области
360 Глава 3
стеклообразного соединения GeAsSe (область С) достигает от-
носительно большого значения (673 К). Стеклообразное состоя-
ние, очевидно, значительно стабилизируется при образовании
связей Ge—As. Упругие постоянные стекол системы Ge—As—Se
приводятся в работе [680].
На рис. 3.54, а представлены концентрационные зависимо-
сти средних мольных объемов, молярной рефракции и объемной
сжимаемости стекол по разрезам Ge2S3—As2S3 и Ge2Se3—As2Se3.
Приведены также мольные объемы стекол в зависимости от
состава по разрезу GeS2—As2S3. Резкое снижение мольного объ-
ема при переходе от стеклообразного GeS2 к стеклу As2S3 по-
нятно с позиций структурно-химического подхода. Отклонения
от линейности но разрезам Ge2X3—As2X3 интерпретируются
в предположении реакций окисления-восстановления, в резуль-
тате которых из структурных групп Ge2X,7 образуются
GeX4/ , например
GC2S3 -J- AS2S3 z; 2GeSe2 4- 72AS4S4 (3.26)
бОегбсз 4~ 3As2Se3 Д2 10GeSe2 4~ 2As3Sc2 (3.27)
Уменьшение объема при переходе от стеклообразного As2X3
к составам, обедненным халькогеном (рис. 3.32, 3.34), компен-
сируется приростом среднего мольного объема вследствие ча-
стичного образования GcX2 за счет Ge2X3 (рис. 3.37, 3.40). Для
зависимостей мольного объема и сжимаемости от сооава
можно ожидать положительного отклонения от линейности, для
молярной рефракции — отрицательного.
Из зависимости Tg от состава по разрезам Ge2S3—As2S3,
Ge2Se3—As2Se3 и GeS2—As2S3 (рис. 3.54, б) можно сделать
вывод, что GeS2 или GeSe2 частично снова образуются в рас-
плаве. Максимальная Tg отвечает составам
5Ge,S3 4- AS2S3 8GeS2 4- 2GeAsS (3.28а)
5Ge2Se3 4- As2Se3 7Д80eSe2 4- 2GeAsSe (3.286)
Только при еще более высоком содержании Ge2X3 в струк-
турных группировках GcoX., , очевидно, сохраняются связи
Ge—Ge и Tg уменьшается до величин, характерных для стек-
лообразных Ge2S3 и Ge2Se3.
3.3.5.3. Стеклообразование и свойства стекол
в системах Ge—Sb—S и Ge—Sb—Se
Области стеклообразования в системах Ge—Sb—S
и Ge—Sb—Se намного меньше, чем в аналогичных системах
Ge—As—S и Ge—As—Se. Вероятно, связи Ge—Sb не обра-
зуются. Большие области расслаивания в системе Sb—S влияют
на плавление в широкой области концентраций системы
Аморфные и стеклообразные системы
361
Ge—Sb—S. Стекла в системе Ge—Sb—Se используются
в ИК-оптике (разд. 5.2).
Области стеклообразования и структура. На рис. 3.55, а
приведены границы области гомогенных стекол в системе
Ge—Sb—S, которые соответствуют навеске 10 г и скорости охла-
ждения — 2 К/с [681].
По разрезу Sb2S3—GeS стеклообразование мало распростра-
нено; оно связано с областью стеклообразования в частной си-
стеме GeS—GeS2, а не со стеклами в системе GeS2—S. Только
при достаточно большом содержании Sb2S3 подавляется склон-
ность GeS2 к кристаллизации и гомогенные стекла можно по-
лучить лишь в ограниченной области системы Sb2S3—GeS2—S.
Большая часть области стеклообразования расположена в ча-
стной системе, образованной тремя двойными сульфидами
(GeS—GeS2—Sb2S3); область стеклообразования сильно вытя-
нута в направлении соединения Sb2S3.
В системе Ge—Sb—S обнаружены две области расслаива-
ния, которые соответствуют двум областям расслаивания в си-
стеме Sb—S. При большом содержании S в частной системе
GcS2—Sb2S3—S стеклообразные фазы сосуществуют с верхним
слоем, богатым серой, в частной системе GeS—Sb2S3- -Sb —
с нижним кристаллическим слоем, сильно обогащенным сурь-
мой. В областях, переходных к зоне однородных стекол, мето-
дами оптической и электронной микроскопии обнаружено рас-
слаивание.
Па рис. 3.55, с5 приведена область стеклообразования в си-
стеме Ge- Sb- Se по данным работ [682—684]. При исполь-
зовании малых навесок и охлаждения расплава в воде область
стеклообразования удалось расширить [685]; расслаивания
в системе не наблюдалось. В области Sb2Sc3—Se диаграммы со-
стояния системы Sb—Sc наблюдается склонность к расслаи-
ванию, однако она значительно меньше, чем в системе Sb—S.
В системе Sb2Se3—GcSe2—Se при охлаждении расплавов
стекла образуются в более широкой области, чем в аналогич-
ной сульфидной системе. Соединения Sb2Se3 и GcSe2 образуют
эвтектику Е\ [686]. На разрезе Sb2Se3—GeSe также находится
эвтектика £2; она образована соединениями Sb2Se3 и Ge4Sb2Se?
(последнее соединение плавится по перитектической реакции
при 723 К)- На третьей стороне фазового треугольника пссвдо-
тройной системы также расположена эвтектика f3, ее состав
отвечает мольному соотношению GeSe : GeSe2 = 0,673. В системе
GcSc/GeSe2 образование стекол, по-видимому, происходит
в расплаве путем взаимодействия компонентов; в результате
возникают структурные единицы Gen Se(2n+2)/2 со связями
Ge—Ge; пространственная сетка из этих структурных единиц
обусловливает высокую вязкость расплава (разд. 3.2.2.5). Стек-
Рис. 3.55,а. Область стеклообразоваиия в системе Ge—Sb—S [681]. б. Область
стеклообразоваиия в системе Ge—Sb—Se [684].
Аморфные и стеклообразные системы 363
лообразованию способствует добавление Sb2Se3. Ограничение
областей стеклообразования разрезами Sb2S3—GeS или
Sb2Se3—GeSe показывает, что проявляющиеся в аналогичных
мышьяковых стеклах реакции окисления-восстановления в си-
стемах Sb—Ge—S и Sb—Ge—Se усиливаются. Стекло состава
Sb2S3-Ge2S3 кристаллизуется при 360 °C с выделением сурьмы,
о чем свидетельствует рентгеновский анализ:
Sb2S3 + 3Ge2S3 - 6GeS2 + 2Sb - 94 кДж/моль (3.29)
При обсуждении структуры стекол следует учитывать на-
ряду со структурными единицами GeS4/2 и Ge2S6/2 структуры
гомологического ряда Оеп$(2п+2)/2 с более высоким содержа-
нием Ge или структурные единицы GeS3/3, а также характер-
ные для Sb2S3 структурные единицы со структурой двойных
слоев и группировки SbS3/2. Кроме того, в присутствии ука-
занных структурных единиц может происходить частичное вос-
становление Sb2S3:
Sb2S3 + 2GeS — Sb2S2 + Ge2S3 (3.30)
В системе Ge—Sb—Se, по-видимому, для области стеклооб-
разования можно предложить аналогичную оценку, хотя тер-
мохимическая оценка реакции окисления-восстановления [урав-
нение (3.30)] свидетельствует о малой вероятности ее протека-
ния. Соединение Ge4Sb2Se7, вероятно, следует рассматривать
как селенид двухвалентного германия и трехвалентной сурьмы.
Свойства стекол. Для разреза (Sb2S3)y(Ge2S3)i~y системы
Ge—Sb—S на рис. 3.56 приведены зависимости мольных объ-
емов от состава [681]. Более медленное охлаждение образцов
приводит одновременно к уменьшению ошибки измерений на 1 —
1,5 % [687]. Для стекол системы Ge—Sb—Se наблюдается
небольшое положительное отклонение мольных объемов от
аддитивности (рис. 3.48) [684]. Положительное отклонение
мольного объема для разрезов Ge2S3—Sb2S3 и Ge2Se3—Sb2Se3
значительно меньше, чем для аналогичных разрезов As2S3—Ge2S3
и As2Se3—Ge2Se3 (рис. 3.54), что может быть вызвано образо-
ванием структурных единиц GeS4/2 или GeSe^ , занимающих
больший объем. Вполне вероятно, что для систем, содержащих
Sb, по крайней мере в случае сульфидных стекол, в расплаве
также частично протекают реакции окисления-восстановления.
Для стекол по разрезам Sb2S3—G2S3 и Sb2Se3—Ge2Se3
(рис. 3.56) наблюдается значительное положительное отклоне-
ние концентрационной зависимости от аддитивности.
Для стекол в системе Ge—Sb—S температуры стеклования
меняются в интервале 473—623 К [681, 688], микротвердость
Рис. 3.56. Концентрационные зависимости модуля упругости Е, молярной ре-
фракции RM и мольного объема V стекол в системах Ge2Se3—Sb2Se3 (/) и
Ge2S3—Sb2S3 (2 и 3).
1 и 2 - данные получены при скорости охлаждения 3—4 К/ч, 3— 100 К/мин.
по Викерсу 150—300 кг/мм2. Для обоих свойств наблюдается
линейная зависимость от энтальпии атомизации стекол. Для
области стеклообразоваиия в системе Ge—Sb- -Se также про-
ведены детальные измерения температуры стеклования [684,
689, 690]. В области устойчивого стеклообразоваиия системы
Ge—Sb—Se, где охлаждением расплава получают образцы сте-
кол массой до нескольких килограммов, например для состава
(Sb2Se3)3(Ge2Se3)7, Tg достигает — 563 К. В работе [691] при-
водятся данные по термическому расширению стекол.
Для многих составов стекол в системах Ge—Sb—S [688.
692] и Ge—Sb—Se [693, 694] изучена зависимость оптического
поглощения от состава, а для некоторых образцов — и от тем-
пературы.
Аморфные и стеклообразные системы 365
Приводятся данные для термической энергии активации про-
водимости на постоянном токе для стекол систем Ge—Sb—S
[688] и Ge—Sb—Se [537, 696—697]. Как показали измерения
удельной проводимости расплава в системе Ge—Sb—Se, при по-
вышении температуры происходит переход к металлической
проводимости [179].
3.3.5.4. Стекла в системах Ge—Р—Те и Ge—As—Те
В то время как в системе As—Те удается получить стекла
только при достаточно высокой скорости охлаждения, после
введения Ge в качестве третьего компонента образуется доста-
точно большая область стеклообразования и компактные гомо-
генные стекла становятся доступными уже при значительно более
медленном охлаждении. Из-за большой атомной массы и пони-
женной силовой постоянной связи ожидается, что область про-
зрачности этих стекол должна распространяться в более даль-
нюю ИК-область спектра (рис. 4.37).
Кроме того, стекла системы Ge—As—Те многократно иссле-
довались в связи с тем, что у них обнаружен эффект переклю-
чения. Стекла системы Ge—Р—Те мало привлекают внимания
к настоящему времени, очевидно, из-за незначительной протя-
женности области стеклообразования.
На рис. 3.57 показаны области стеклообразования в систе-
мах Ge—As—Те и Ge—Р—Те, которые соответствуют навеске
10—20 г и скорости охлаждения ~5 К/с вблизи температур
стеклования.*
Согласно работам [698, 699], в системе Ge—As—Те область
стеклообразования состоит из двух частей, что обусловлено су-
ществованием также двух областей стеклообразования в си-
стеме As—Те. Между двумя областями стеклообразования на-
ходятся расплавы, которые, очевидно из-за характерной для
As2Te3 структуры и типа связи, проявляют высокую кристалли-
зационную способность. В работах [700—702] приводятся не-
полные данные по стеклообразованию в этой системе, и их
трудно отнести к надежным. Область стеклообразования в си-
стеме Ge—Р—Те, как и область А (рис. 5.57) в системе
Ge—As—Те, имеет вполне определенную связь с эвтектикой
в системе GeTe—Те.
В образовании стекол области А принимают участие струк-
турные единицы GeTe4/2 и AsTe3/ ', а также цепочки из атомов
* Уточнение состава стекол в системе Ge—Р—Те с помощью химиче-
ского анализа показало, что в состав стекол входит не больше, чем 1 —
2 ат. % Р, т. е. о стеклообразовании в системе Ge—Р—Те можно говорить
лишь условно [Виноградова Г. 3., Майсашвили Н. Г.— ЖНХ, 1979, т. 24,
с. 1116].— Прим, перев.
366 Глава 3
Те, образующих две связи. Структурные исследования стекол
тройной системы Ge—As—Те до настоящего времени не про-
водились. Состав стекол в области В (рис. 5.57) позволяет
предположить участие в структуре ближнего порядка связей
Ge—As, как в аналогичной системе Ge—As—Se.
Измерения температурной зависимости удельной проводимо-
сти свидетельствуют об изменении структуры расплава [702].
Они показывают, что для стекол характерны направленные
связи и низкие координационные числа. В этом случае расплав
имеет достаточно высокую вязкость, а при заданной скорости
охлаждения зародышеобразование и рост кристаллов подав-
ляются.
Для стекол в системе Ge—As—Те Tg лежит в интервале
408—543 К. Область оптической прозрачности этих стекол рас-
пространяется до 20 мкм, потери на отражение из-за высокого
показателя преломления (3,4—3,5) относительно высоки.
Удельная проводимость стекол в системе Ge—As—Те при 298 К
достигает 10~5—10-4 См/см [701, 703]. Для каждой температур-
ной области характерен свой механизм проводимости [704].
Аморфные и стеклообразные системы
367
3.3.5.5. Стекла в системах Si—Р—Те и Si—As—Те
Введение Si вместо Ge позволяет ожидать для тройных сте-
кол, содержащих Те, значительного повышения температуры
стеклования. Изучение таких систем проводилось уже давно
с целью получения стекол, прозрачных в ИК-области спектра
и обладающих повышенной термостойкостью. Из кремнийсодер-
Рис. 3.58. Области стеклообразования в системах Si—As—Те и Р—Si—Те.
жащих халькогенидных стекол химически более устойчивы со-
держащие Те, чем содержащие Se или S.
На рис. 3.58 приведены области стеклообразования, отно-
сящиеся к таким же условиям получения, какие были описаны
в предыдущем разделе для систем Ge—Р—Те и Ge—As—Те
[651]. Сообщается [700] о расширении области стеклообразова-
ния в системе Si—As—Те до состава SigAssTee или Si2AssTe3.
Стекла в системе Si—Р—Те получены до содержания Р~
— 20 мол. % и Si —35 мол. % [651]; максимальное значение
температуры стеклования в этой системе составляет —475 К.
Для стекол системы Si—As—Те Tg достигает 625 К. Приве-
денные в работе [705] температуры размягчения завышены на
50 К, что обусловлено методикой определения. Следует отме-
368 Глава 3
тить значительное расширение области стеклообразоваиия в си-
стеме Si—As—Те по сравнению с системой Ge—As—Те.
В работе [706] впервые для системы As—Si—Те приведены
данные по плотности и электрической проводимости на посто-
янном токе; подробное исследование выполнено в работе [707].
Из температурной зависимости удельной проводимости и термо-
э. д. с. сделан вывод о переносе носителей заряда в соответст-
вии с зонной моделью и вкладе дырок в «прыжковую» прово-
димость.
3.3.6. Тройные халькогенидные стекла
с халькогенидами металлов
в качестве модификатора стеклообразной сетки
По аналогии с большим разнообразием веществ, которые
образуются из сочетаний стеклообразующих оксидов с раз-
личными оксидами металлов, сочетание сульфидов, селенидов
и частично теллуридов мышьяка и германия с соответствую-
щими халькогенидами металлов приводит к образованию мно-
гочисленных новых стекол.
Сообщается |708, 709] о стеклообразовании GeS2 с сульфи-
дами щелочных металлов; исследовано также взаимодействие
GeS2 с As2S3, T12S, Ag2S, ZnS, CdS и PbS. Co всеми сульфи-
дами металлов I группы, кроме T12S и Na2S, обнаружены от-
носительно небольшие области стеклообразоваиия. Из халько-
генидов щелочноземельных металлов только BaS растворяется
до 10 мол. % в расплаве GeS2 с образованием стекла [710].
Относительно большие области стеклообразоваиия описаны
в системах GeS2—La2S3 и GeS2—Ga2S3, а также в тройных си-
стемах с участием этих сульфидов [711]. Халькогениды пере-
ходных металлов ни сами, ни в сочетании с другими компонен-
тами не склонны к стеклообразованию [710, 712]. Их высокие
температуры плавления, а также низкая основность сильно сни-
жают растворимость в расплавах типичных стеклообразующих
халькогенидов. Только для халькогенидов марганца, например
в системе Мп—Ge—Se, обнаружено участие в образовании халь-
когенидных стекол [713].
В литературе подробно описаны Т1-содержащие стекла. Си-
стематические исследования стеклообразоваиия в системах с тя-
желыми металлами, например Ag, Hg, Sn или Pb, в расплавах
халькогенидов германия показали, что при особенно больших
концентрациях халькогениды тяжелых металлов могут уча-
ствовать в стеклообразовании в сочетании со стеклами систем
GeS—GeS2 и GeSe—GeSe2 [714]. До сих пор неясно, почему из
халькогенидов элементов побочной подгруппы I и II групп
Ag2S и халькогениды ртути выпадают из общего правила.
Аморфные и стеклообразные системы
369
3.3.6.1. Натриевотиогерманатные и натриевотиоарсенатные
стекла
Стеклообразование и свойства стскол в этих системах были
изучены в связи с поиском твердых тел с высокой ионной про-
водимостью (твердых электролитов). При повышении содержа-
ния Na2S в стеклах сильно возрастает и их склонность к гид-
ролизу.
Область стеклообразования в системе (Na2S)x(GeS2) i-x об-
наружена для составов 0<х<0,6 [715]. Согласно диаграмме
состояния, в системе Na2S—GcS2 существуют инконгруэнтно
плавящиеся соединения Na4GeS4 и Na2GeS3 и соединение
Na4Gc4Sio, характеризующееся конгруэнтным плавлением [716].
Структура аниопа Ge4Sio такая же, как у P4Sio [717]. Напро-
тив, структура Na2GeS3 полимерная, близкая к цепочечной, что
позволяет объяснить склонность его к стеклообразованию.
Ионная проводимость этих стекол достигает 5 • 10-6 См/см
(298 К) и характеризуется энергией активации 0,44 эВ [715].
Сообщается [719] о сравнительном исследовании систем
Na2S—SiS2, Na2S—GeS2, Na2S—P2S.s и Li2S—GeS2 и о причи-
нах повышенной ионной проводимости оксидных стскол таких
же составов. Li2GeS3 образует стекло, обладающее наибольшей
ионной проводимостью среди некристаллических твердых тел
(4-10 5 См/см при 293 К).
В системе (Na2S)(AsS3/2) i-.v область стеклообразования
значительно меньше, чем в системе Na2S- GeS2; стеклообразо-
вапие существует для составов 0<х<0,1, поэтому стекла
в этой системе характеризуются более низкой удельной прово-
димостью [720]. Расплав As2S.-, уже при небольших добавках
Na2S склонен к кристаллизации. Соединение NasAsS3 кристал-
лизуется в структуре NaC103 (заполненной) [721], а для
NaAsS2 и NaAsSe2 характерна цепочечная структура [722].
3.3.6.2. Халькогенидные стекла, содержащие Си и Ag
О стеклообразовании в системе Си—As—Se много сообща-
лось в связи с исследованиями эффекта переключения [723]
(разд. 4.1.3.7). В системе Cux(As0,4Se0,6) i-.v область стеклооб-
разования распространяется до х = 0,35. Добавление 2,4 мол. %
Си повышает удельную проводимость от 5,6-10-13 до
5,8-10-11 См/см; при этом энергия активации понижается от
0,86 до 0,73 эВ [724]. В этой системе образуются тройные со-
единения Cu3AsSe4 и CuAsSe2, структура которых является
производной от структуры цинковой обманки или вюрцита
[725]. Согласно исследованиям этих стекол методами рентгенов-
ской дифракции и ПТСРП, координационное число Си1 равно
4, а для селена в зависимости от состава меняется от 2 до 4
24 Заказ № 413
370 Глава 3
[726]. В этих стеклах As образует три связи, а в области с низ-
кой концентрацией Se существуют связи As—As. Что касается
Cu-содержащих стекол на основе сульфидов мышьяка и халь-
когенидов германия, то они мало освещены в литературе.
Среди систем с серебром, напротив, преобладают сульфид-
ные системы. В системе Ag—As—S по разрезам AS2S3—Ag
и As2S3—Ag2S обнаружено стеклообразование вплоть до со-
Рис. 3.59. Области стеклообразования в системах Ag2S—GeS—GeS2 [730],
T12S—GeS—GeS2 [743] и Tl2Se—GeSe—GeSe2 [744].
става c 40 мол. % Ag [727], однако, по данным работы [728],
область стеклообразования значительно меньше. Область стек-
лообразования включает соединение AgAsS2 и распространена
до 40 мол. % Ag. Как и следовало ожидать, соединение Ag3AsS3
(прустит) находится вне области стеклообразования.
Для системы Ag2S—GeS—GeS2 в литературе [729, 730] при-
водятся различные данные по границам стеклообразования
(рис. 3.59). Расплав смеси Ag2S/GeS2 не образует стекла при
охлаждении. В аргиродите Ag8GeSe6 впервые был обнаружен
Ge. Ag2GeS3 кристаллизуется в структурном типе вюрцита
[731], что и объясняет отсутствие стеклообразования. Только
в сочетании с GeS расплавы системы Ag2S—GeS2 образуют
стабильные стекла. Стекло состава (Ag2S)4(Ge2S3)3 характери-
Аморфные и стеклообразные системы 371
зуется самым высоким содержанием Ag (~30 мол. %). Напро-
тив, в системе Ag—Ge—Se при обычных условиях охлаждения
расплавов стекла не образуются.
В системе (Ag2S)y(P2S5)i-y в области 0,45<z/<0,55 также
получены стекла с высоким содержанием серебра [732].
Измерения проводимости и чисел переноса показали, что
перенос заряда осуществляется почти исключительно легко
подвижными ионами Ag+ [732, 733]. В системах Ag2S—GeS—
GeS2 и Ag2S—As2S3 удельная проводимость стекол с боль-
шим содержанием серебра при 293 К составляет 3-10-4—
9-10-5 См/см [733].
3.3.6.3. Халькогенидные стекла, содержащие TI и Ga
Системы Т1—S, Т1—Se и Т1—Те. В системе TlxSi-x в интер-
валах концентраций 0,05<х<0,27 и 0,69<х<0,90 в расплаве
существуют области расслаивания [734]. T12S плавится конгру-
энтно; TI4S3, T1S, T12S3 и T12S5 плавятся инконгруэнтно. Послед-
нее соединение уже при 423 К образует гомогенный расплав.
В области от T1S до T1S2,5 при скорости охлаждения ~ 10 К/с
образуются компактные стекла [735].
В системе Т1—Se со стороны, богатой Se, (0<х<0,23)
также обнаружено расслаивание; имеются индивидуальные со-
единения Tl2Se и TISe [736]. В соответствии с формулой
TlT[TllIIX2] (X = S, Se) T1S и TISe можно описать цепочечной
структурой, в которой искаженные тетраэдрические структур-
ные единицы Т11ПХ4/2 связаны аналогично спиранам по реб-
рам (структурный мотив SiS2). Согласно ИК-спектрам стекол
системы TlxSei_.v, полученных охлаждением (скорость охла-
ждения >>103 К/с) расплава по методу поршня и наковальни
(рис. 2.3, б) [737], в области х<0,30 соединение таких струк-
турных группировок осуществляется через цепочки Se. В си-
стеме TlxSei_x компактные стекла в области составов 0<х<
<^0,33 можно получить путем закалки в воду от температуры
1070 К малых количеств расплава в тонкостенных кварцевых
ампулах (скорость охлаждения ~ 10 К/с) [738]. Аморфный TISe
осаждают из раствора по реакции между солью таллия и селе-
носульфатом натрия при 273 К [739].
Аморфные слои TlxTei-x в широкой области концентраций
можно получить осаждением из паровой фазы. Для составов
Т12Те, Т15Те3, Т13Те2, Т1Те, Т12Те3, Т1Те2, Т12Те5, Т1Те3 и Т12Те7
наблюдается минимальная проводимость (10-8 См/см), в то время
как в соседних областях проводимость достигает 102 См/см
[740, 741]. Из большого числа описанных в литературе кристал-
лических фаз соединения Т12Те, Т1Те и Т12Те3 можно рассмат-
ривать как достоверные [742].
24*
372 Глава 3
Стекла в системах Tl2S—GeS—GeS2 и Tl2Se—GeSe—GeSe2.
Если в системы Т1—S и Т1—Se в качестве третьего компонента
вводить германий, то в полученных сплавах в широкой области
концентраций обнаруживаются две фазы [743, 744] (см. также
литературу к гл. 2 [343]), в то время как в системах
T12S—GeS—GeS2 и Tl2Se—GeSe—GeSe2 образуются гомогенные
стекла (рис. 3.59).
Стеклооб разевание в системах Т1—4s—S, Т1—4s—Se
и Т1—4s—Те. Обширная область стеклообразования в системе
Т1—As—S велика [745] и ограничена максимальным содержа-
нием Т1, равным ~55 мол. %; однако, по другим данным [728],
область стеклообразования в этой системе значительно уже.
Гомогенные стекла по разрезу (Tl2S)y(As2S3)1_y, по-видимому,
получаются в интервале 0<#<0,68, т. е. приблизительно до
состава TI4AS2S5 [728, 746], причем это высокоомные стекла
и для некоторых из них 7\,<370 К. Сообщается [747] о соеди-
нениях TlAsS2, TlAsSe2 и TlAsTe2, их свойствах в кристалличе-
ском и стеклообразном состояниях и приводятся [615] данные
о коэффициенте термического расширения и Tg стекол в си-
стеме Т1—As—S.
В соответствии с расширением области стеклообразования
в системе As—Se по сравнению с системой As—S при добавле-
нии Т1 получают стекла, состав которых по разрезу
(Tl2Se)y(As2Se3) 1-,/ меняется в более широких пределах, чем
для соответствующей системы с серой, и распространяется как
в направлении более высокого, так и более низкого содержания
As [745]. Согласно диаграмме состояния, в системе
Tl2Se—As2Se3 имеются тройные индивидуальные соединения
TlAsSe2 и Tl3AsSe3 [748].
Удельная проводимость стекол при повышении содержания
Tl2Se возрастает от 10-13 до ~10-7 См/см (293 К, TlAsSe2)
(см. литературу к гл. 1 [20, 22]). Удельная проводимость
и термо-э. д. с. стекол по разрезу TlaAs2Se3 (0<а<1,99) иссле-
дована в работе [749]; перенос заряда осуществляется за счет
дырок. Согласно данным по удельной проводимости этих сте-
кол, получена более высокая энергия активации, чем при из-
мерениях термо-э. д. с.; но в обоих случаях энергия активации
проводимости линейно уменьшается с температурой до
<0,5 эВ. Изучены оптические свойства стекол в системе
Т1—As—Se [750] (см. также литературу к гл. 1 [33, 22]).
Тё стеклообразного TlAsTe2 составляет 338 К; удельная про-
водимость при комнатной температуре 3-Ю'3 См/см [747].
Энергия активации проводимости равна 0,25 эВ, а в области
температур 165 К выражается еще меньшей величиной [751].
Термо-э. д. с. стеклообразного TlAsTe2 также экспоненциально
зависит от обратной температуры; энергия активации, рассчи-
Аморфные и стеклообразные системы 373
тайная по данным по термо-э. д. с., составляет 0,13 эВ. В от-
личие от большинства других халькогенидных стекол знак ко-
эффициентов Зеебека и Холла для стеклообразного TlAsTe2
один и тот же (положительный). Оптическая ширина запрещен-
ной зоны составляет при 300 К 0,62 эВ.
При исследовании системы TlSe-Sb2Se3—Tl2Se-As2Se3 Го-
рюнова и Коломиец в 1955 г. впервые обнаружили халькоге-
нидные стеклообразные полупроводники (см. литературу
к гл. 2 [14]) и при замещении As2Se3 на As2Te3 по разрезу
Tl2Se-As2Se3—Tl2Se-As2Te3 обнаружено стеклообразование во
всей области составов (см. литературу к гл. 1 [20]). Для сте-
кол состава Tl2Se-As2Te3 определены [752] диэлектрические
потери в интервале частот от 109 до 1014 Гц.
Системы Ga—Ge—S и Ga—Ge—Se. Слоистая решетка со-
единений GaS, GaSe и GaTe характеризуется структурными
единицами Ga2Xd/ со связями Ga—Ga [511, 551, 753]. В ра-
боте [754] исследовано стеклообразование GaS в сочетании
с GeS и GeS2, при этом в расплаве образуются структурные
группировки Ge„S(2rt | 2)/2, например при /2 = 2 (Ge2S6/2). Воз-
можные окислительно-восстановительные реакции приводят
к большому разнообразию структурных единиц в этих стеклах.
GeS2 и Ga2S3 взаимодействуют с образованием простой эвтек-
тики [755].
Описаны фазовые равновесия в тройной системе Ga—Ge—Se
[756] и стеклообразование в этой системе [757].
3.3.6.4. Германиевохалькогенидные стекла,
содержащие Sn и РЬ
Исходя из структуры и типа связи, в стеклах систем
GeS—GeS2 и GeSe—GeSe2 особый интерес приобретает изме-
нение структуры путем постепенного замещения монохалько-
генида германия на SnS или SnSe и PbS или PbSe. Оловотио-
германатные, свинцовотиогерманатные и' свинцовоселеногерма-
натные стекла содержат неожиданно много тяжелых металлов;
в системе Sn—S—GeS—GeS2 получены стекла вплоть до
47,5 мол. %. SnS [508] (см. также литературу к гл. 2 [272]),
а в системах PbS—GeS—GeS2 и PbSe—GeSe—GeSe2 — до
57 мол. % PbS и 54 мол. % PbSe [758] (см. также литературу
к гл. 2 [350]). Области стеклообразования в указанных системах
приведены на рис. 3.60. Очевидно, сходство областей стеклообра-
зования в системах с T12S и Tl2Se и аналогичных систем с PbS
и PbSe (рис. 3.59). В системе SnSe—GeSe—GeSe2 при использо-
вании обычных скоростей охлаждения стеклообразования не об-
наружено. SnS образует с GeS2 тройное соединение SnGeS3, ко-
торое инконгруэнтно плавится при 880 К [759]. Его кристалличе-
374 Глава 3
Рис. 3.60. Области стеклообразоваиия в системах SnS—GeS—GeS2 (см. лите-
ратуру к гл. 2 [272]), PbSe—GeSe—GeSe2 (см. литературу к гл. 2 [350]) и
PbS— GeS—GeS2 [758].
скую структуру можно описать на основе структурных группи-
ровок [GeS^, связанных между собой в цепочки, причем атомы
серы из этих группировок образуют вокруг атомов Sn11 тетра-
гонально-пирамидальные координационные полиэдры с коорди-
национным числом 5 [760, 761]. В системе SnSe—GeSe2 обра-
зуется простая эвтектика [762].
При введении сульфидов и селенидов тяжелых металлов
в смеси GeS—GeS2 и GeSe—GeSe2, а не в GeS2 и GeSe2 удается
проще получить стекла. Это свидетельствует о том, что в рас-
плавах, дающих при охлаждении стекла, имеются структурные
единицы Ge X
п (Лп4-Л)/2
Средние мольные объемы и температуры стеклования при
введении SnS или PbS в стекла системы GeS—GeS2 умень-
шаются, что было показано по разрезам (MS)y(GeS)o,7-y(GeS2)o(3
и (MS)J/(GeS)o,65-j/(GeS2)o,35, где M=Sn, Pb [714, 763].
На рис. 3.61 приведены зависимости средних мольных объемов
и Tg стекол от состава в системе PbSe—GeSe—GeSe2 по разре-
зам [350]. Резкое уменьшение мольного объема и Tg особенно
заметно проявляется на разрезе III. Очевидно, PbSe в раз-
ветвленном каркасе структурных групп [Ge2See] играет роль
Аморфные и стеклообразные системы
375
Рис. 3.61. Концентрационные зависимости среднего мольного объема V (J)
и Tg (2) для стекол по разрезам.
1 — (PbSe)J/(GeSe)0 65_y(GeSe2)0 35, И — (PbSe)y (GeSe)0 7_у (GeSe2)0 3,
Hl-(PbSe)y(Ge2Se3)0i5_0j5j/, IV - (PbSe)y (GeSe)y (GeSe2)1_2y.
модификатора структурной сетки стекла. РЬП занимает пу-
стоты, и только при достаточно высокой концентрации удовлет-
воряются их пространственные требования и заметно возра-
стает их участие в формировании структурных полиэдров.
GeSe2 вносит самый большой вклад в средний мольный
объем. Уменьшение этого параметра по разрезу
(GeSe)y(GeSe2)i-y (рис. 3.40) понятно, так как можно пред-
376 Глава 3
ставить структурные группы Ge2Se6/2H Ge3Se8/2 как взаимопро-
никающие тетраэдры [509, 510]. По разрезу IV этот эффект
еще усиливается из-за того, что PbSe играет роль модифика-
тора, поэтому по разрезам (PbSe)y(GeSe2) i_3y(Ge2Se3)y (где
</^7з) и (PbSe)y(Ge2Se3)2_52/(Ge3Se4)3y-i (где г/^/з) наблю-
дается понижение мольных объемов на ~ 10 %. По разрезам /
и II РЬП замещает германий (формально Ge11) без изменения
содержания селена. При возрастании содержания PbSe повы-
шается доля стеклообразующих структурных группировок, ко-
торые «требуют» возрастания относительного объема. Для раз-
резов / и II кривые проходят через минимум, причем их восхо-
дящая ветвь после минимума может быть обусловлена как объ-
емными «требованиями» РЬ11, так и переходом от Ge3Se8/2
к Ge2See/2 и далее к GeSe4/2- По всем разрезам при добавлении
малых концентраций PbSe температура стеклования убывает,
а в остальной области составов Tg не зависит от концентрации.
Очевидно, в этой области разрыхление структуры, связанное
с разрывом мостиковых связей Ge—Se—Ge, компенсируется
упрочнением структуры, которое вызвано образованием свинцо-
воселеновых координационных полиэдров.
Концентрационные зависимости коэффициента термиче-
ского расширения (КТР) по указанным разрезам приведены на
рис. 2.47. Очевидно, что на границах областей стеклообразова-
ния происходят структурные изменения, которые вызваны из-
менением состава. Наименьшие а имеют стекла GeSe2
и Ge2Se3. В соответствии с модифицирующей функцией PbSe
по разрезам наблюдается приращение а. По разрезам / и II
соответствующие зависимости проходят через максимум, для
состава у = 0,4 (т. е. эквимолярного содержания GeSe и GeSe2)
наблюдается минимум, а при дальнейшем повышении содержа-
ния PbSe вплоть до границы стеклообразования коэффициент
а резко возрастает. Разрез IV также пересекает «долину».
По разрезу III а понижается до «дна долины». По-ви-
димому, снижение коэффициента термического расширения
в средней области составов разрезов / и // можно объяснить
упрочнением структуры. Средний мольный объем в этой обла-
сти возрастает, и соответственно участие свинцовоселеновых
координационных полиэдров в структурном каркасе этих сте-
кол становится значительным.
Кристаллохимия тиосиликата, селеносиликата и тиогерма-
ната РЬП характеризуется различными координационными чис-
лами. В Pb2SiSe4 четко проявляются 3 более короткие связи
РЬ—Se с межатомным расстоянием от 290 до 300 пм, причем
координационное число возрастает до 7 за счет 4 более даль-
них атомов селена. Вторая группа атомов РЬ11 образует 5 свя-
зей РЬ—Se с межатомным расстоянием от 300 до 310 пм [764].
В структуре Pb2GeS4 с координационными числами 6 и 7 для
Аморфные и стеклообразные системы 377
атомов РЬП также встречаются 3 укороченные межатомные
расстояния РЬ—S [765]. Существует и аналогичное соединение
Pb2GeSe4 [766], плавящееся инконгруэнтно при 863 К. В соеди-
нениях PbGeS3 и SnGeS3 для атомов металлов координацион-
ное число равно 5 [760, 761]. Исследование стекол системы
SnS—GeS—GeS2 методом у-резонапсной спектроскопии пока-
зало, что при высокой концентрации атомов Sn11 ближний по-
рядок в стеклах сходен с ближним порядком в кристалличе-
ском SnGeS3 (разд. 2.4.1.3, рис. 2.36). При малом содержании
Sn11 в стеклах наблюдается заметное квадруполыюе расщепле-
ние, что указывает на более низкую координацию и сильную
асимметрию связей.
Результаты структурных исследований подтверждают пред-
положение о том, что взаимодействие PbSe со стеклообразова-
телем приводит к образованию структурных группировок
[PbSe3] путем разрыва мостиковых связей Ge—Se—Ge под дей-
ствием PbSe и одновременного возникновения двух связей
РЬ—Se—Ge и координационной связи с атомом Se из соседней
связи Ge—Se—Ge. При этом возникает возможность реализа-
ции связей с большими межатомными расстояниями РЬ—Se.
В этой области коэффициент термического расширения а воз-
растает. В примыкающей области составов понижение а, воз-
можно, связано с повышением координационного числа до 5
по аналогии со стеклами системы SnS—GeS—GeS2.
Упругие постоянные стекол в этой области повышены
вследствие возрастания среднего числа связей, и поэтому ко-
эффициент термического расширения а понижается, а, достиг-
нув минимума, вновь возрастает при переходе в область раз-
резов / и // благодаря новой структуре и типу связи. Предла-
гаемая структурная модель для стекол системы PbSe—GeSe—
—GeSe2 согласуется с результатами исследования стекол с по-
мощью КРР [767]; согласно этой работе, средняя длина связи
РЬ—Sc составляет 300 пм, а координационное число атомов
PbTI лежит в интервале от 3 до 5.
Следует заметить, что минимум а отвечает мольному соот-
ношению PbSe : GeSe : GcSe2 — 1, откуда следует, что при этом
составе существует соединение с собственной структурой. Пред-
полагают, что при медленном охлаждении расплава образуется
кристаллическая метастабильная фаза с составом, близким
PbGe2Se4. При отжиге в результате монотропного превращения
она распадается с выделением GeSe2 и другого соединения, что
подтверждается рентгенографически.
Результаты исследования электрических свойств стекол си-
стемы PbSe—GeSe—GeSe2 приведены на рис. 3.62. При возра-
стании содержания PbSe в стеклах удельная проводимость
возрастает, а термическая энергия активации убывает. Для
стекол по разрезам / и //, несмотря на значительное изменение
378 Глава 3
Рис. 3.62. Удельная проводимость а при 298 К и энергия активации проводи-
мости Ео для стекол системы PbSe—GeSe—GeSe2 по разрьзам I, II, III, IV
(см. рис. 3.61).
концентраций РЬ11, наблюдаются почти совпадающие значения
о0: по разрезу / lgo0 = 2,0±0,2 и по разрезу // lgOo=l,4±
± 0,2. Следует заметить, что эти разрезы относительно мало
отличаются по содержанию GeSe2 (в пересчете на Se различия
составляют 0,94мол. % Se); именно в этом, по-видимому, и со-
стоит причина близости значений о0-
По обоим разрезам в средней области составов происходит
переход к более уплощенным кривым. По разрезу // перегиб
на кривых и lg<J298 соответствует минимуму на концентра-
ционной зависимости коэффициента термического расширения
Аморфные и стеклообразные системы
379
(рис. 3.62). Обсуждавшееся выше повышение координационного
числа от 3 до 5 должно привести к незначительному возраста-
нию средних межатомных расстояний РЬ—Se в координацион-
ных полиэдрах, что позволяет объяснить нерезкое уменьшение
термической энергии активации проводимости в области с боль-
шим содержанием PbSe.
3.3.6.5. Германиевохалькогенидные стекла, содержащие Hg
Из-за летучести соединений ртути сейчас известны только
отдельные случаи их использования в качестве компонентов
оксидных стеклообразующих расплавов; например, под давле-
нием получены боросиликатные стекла, содержащие до
65 мол. % HgO [768]. Для Hg-содержащих расплавов неоксид-
ных систем, например стеклообразующих халькогенидов
мышьяка и германия, можно ожидать пониженную тенденцию
к разложению из-за более высокой термической стабильности
HgS, HgSe и HgTe по сравнению с HgO.
Согласно работе [769], в системах (HgS)y(As2S3) i-y
и (HgSe)y(As2Se3)i_y стекла удается получить до мольного со-
держания х^0,33 в первой системе и х^0,18 во второй. Сооб-
щается [770] о вхождении Hg в As2Se3 по разрезу
Hgx(As2Se3) 1-х до состава с 0,286. В системе HgS—GeS—
—GeS2 также образуются стекла, но при достижении темпера-
тур плавления взятых компонентов в кварцевых ампулах раз-
вивается все-таки значительное давление паров Hg [710]. В от-
носительно широких концентрационных пределах стекла полу-
чены также в системах HgSe—GeSe—GeSe2 [571],
HgTe—GeSe—GeSe2 и HgTe—GeTe—GeSe2 [771, 714, 763].
Области стеклообразования в этих системах приведены на
рис. 3.63 для скорости охлаждения ~2 К/с.
В системе HgSe—GeSe—GeSe2 получены стекла с содержа-
нием до 50 мол. % HgSe. Область стеклообразования в этой
системе больше, чем в аналогичной системе с селенидом свинца,
и вытянута в направлении стороны HgSe—GeSe. Тот факт, что
из смесей HgSe—GeSe, HgTe—GeSe и HgSe—GeTe в опреде-
ленных концентрационных границах можно при плавлении по-
лучить стекла, указывает на окислительно-восстановительное
равновесие, в результате которого наряду со ртутью, находя-
щейся в низкой степени окисления, частично образуется GeSe2.
Продукты, образующиеся при плавлении таких смесей, и при
правильном температурном режиме совсем не содержат метал-
лической ртути.
В отличие от свинцовоселенидных стекол температура стек-
лования приблизительно линейно убывает при возрастании
концентрации HgSe, например по разрезу (HgSe)J/(GeSe)o,6-J/X
X(GeSe2)o,4 от 597 К (*/ = 0) до 503 К (*/ = 0,45). Концентраци-
380 Глава 3
Рис. 3.63. Области стеклообразования в системах
HgTe—GeSe—GeSe2, HgTe—GeTe—GeSe2 [571, 771].
HgSe—GeSe—GeSe2,
онная зависимость среднего мольного объема примерно такая же.
Как показывает рис. 3.64, в структуре этих стекол преоб-
ладает влияние структурных мотивов германиевоселенидпых
стекол. Для разрезов // в случае стекол, содержащих HgSe
и PbSe, понижение мольного объема для стекол с HgSe на-
много меньше. По разрезу (MSe) ДОегЗез^.э-о.йу введение PbSe
(у =0,05, М = РЬ) вызывает понижение мольного объема ог
71,44 до 15,7 см3/мол, a HgSe в таком же мольном соотноше-
нии (// = 0,05, M = Hg) понижает мольный объем всего до
17,36 см3/моль. Поэтому о модифицирующей роли HgSe едва
ли можно говорить.
В области средних составов кривые, приведенные на
рис. 3.64, характеризуются немонотонным ходом. Стеклообра-
зующая способность, выраженная через уравнение (2.52) как
соответствующий параметр, в этой области составов наиболее
велика. С двух сторон эти кривые характеризуются противопо-
ложной кривизной. При приближении к области средних кон-
центраций HgSe изменение объема с двух сторон возвращается
к одинаковым значениям, а структура «опрокидывается» к дру-
гой конфигурации. Очевидно, в области стеклообразования мо-
Аморфные и стеклообразные системы
381
Рис. 3.64. Концентрационные зависимости среднего мольного объема стекол
в системах HgSe—GeSe—GeSe2 и PbSe - GeSe—GeSe2.
Разрезы: I — (IIgSe)y (GeSe)08_y(GeSe2)0>2, II — (HgSe)y (GeSc)0 7_y (GeSe2)0/J, Ill—
(HgSe)y (GeSe)0 6_,/(GeSe2)(M, IV — (HgSe)y(GeSe)0 55_y(GeSe2)0 45,
V •- (PbSe)y(GeSe)0 7_J/(GeSe2)0 3. ( • ).
гут реализоваться стекла различной структуры. Такой же вы-
вод следует из диэлектрических свойств этих стекол [771].
На рис. 3.65 приводятся данные по измерению проводимо-
сти стекол по разрезам (MSe)y(Ge2Se3)0.5-0.57/, где М = РЬ или
Hg. Энергия активации проводимости стекол в системе
PbSe—Ge2Se3 с ростом содержания PbSe убывает от 1,0 до
0,7 эВ, а удельная проводимость возрастает на 4 порядка, в то
время как для стекол системы HgSe—Ge2Se3 эти же параметры
почти не меняются.
Для разреза /// по концентрационным зависимостям про-
водимости и энергии активации наблюдается согласие с этими
результатами (рис. 3.66). Удельная проводимость убывает
больше, чем на порядок; для составов с 23,75 и 35 мол. % HgSe
она достигает минимума (—1g (J298 = 14,84-14,9). Даже
для стекла состава (HgSe)o,4(GeSe)o,2(GeSe2)o.i или
(HgSe)o.4(Ge2Se3)o,2(GeSe2)o,2 удельная проводимость только
Рис. 3.65. Концентрационные зависимости удельной проводимости а при
298 К и энергии активации проводимости Ед для стекол по разрезам
(MSe) у (ОегБез) 0,5-0,5».
1 — м=РЬ, 2 — M-Hg.
Рис. 3.66. Изменение энергии активации проводимости Ед (/), оптической ши-
рины запрещенной зоны (2) и удельной проводимости Оаэв (<?) Для стекол по
разрезу III (рис. 3,64).
Аморфные и стеклообразные системы
383
немного выше, чем для стекла состава (GeSe)o>6(GeSe2)o,4 (ко-
нечный состав этого разреза). Термическая энергия активации
проводимости этих стекол лежит в интервале 1,0—0,9 эВ. На-
против, для стекла состава (PbSe)o,4(GeSe)o,25(GeSe2)o,35
—1g о = 9,9 и энергия активации £а = 0,67 эВ.
Как показывает рис. 3.66, изменения —1g о2э8 и Еа с соста-
вом обратны. Количественная корреляция, как в случае свин-
цовоселеногерманиевых стекол (см. литературу к гл. 2 [350]),
для стекол системы Hg—Ge—Se не проявляется на зависимо-
стях Igo и £а от состава, что, как и концентрационная зави-
симость мольного объема для стекол системы Hg—Ge—Se, сви-
детельствует о существовании двух областей с различной струк-
турой. Обнаружено также два типа концентрационных кривых
по этому разрезу, в случае энергии активации наблюдается ми-
нимум, а для удельной проводимости — максимум (рис. 3.66).
Зависимость удельной проводимости от концентрации HgSe
и структуры стекол выражена на порядок слабее, чем в стеклах
системы PbSe—GeSe—GeSe2, параметры электрической прово-
димости остаются близкими к величинам, характерным для
германиевоселенидных стекол.
Высокая проводимость HgSe (~103 См/см) и энергия акти-
вации, составляющая 0,1 эВ, обусловлены реализацией в се-
лениде ртути структуры цинковой обманки. Модификация
HgSe, полученная под высоким давлением, соответствует по
структуре красной киновари, а ее проводимость на 6 порядков
ниже [772]. Ближний порядок, существующий в расплаве HgSe,
может быть в основном описан координационными числами 2
или 2 + 4 (см. литературу к гл. 1 [28]). При температуре плав-
ления HgSe, равной 963 К, проводимость убывает до ~ 10 См/см.
Край поглощения или щель подвижности расплава этой системы
отвечают 2,3 эВ.
Предположение о малом координационном числе хорошо
согласуется с относительно малым понижением мольного объ-
ема при добавлении HgSe (рис. 3.64). Низкое координационное
число, например 2 для катионов Hg2+, одновременно означает
и сильное ослабление модифицирующей функции HgSe в гер-
маниевоселенидных стеклах. Образование гомогенных стекол
в области HgSe—GeSe2—Se также согласуется с этим выводом
(рис. 3.70).
3.3.7. Некоторые четверные халькогенидные
стеклообразующие системы
Сочетание 4 элементов дает возможность более широкого
изменения состава материалов, что намного превышает вариа-
ции составов в тройных стеклообразующих системах. До сих
пор не стремились к систематическому изучению четверных
384 Глава 3
систем, и они изучены еще далеко не полностью. При выборе
четверных систем ориентиром служат тройные системы, отли-
чающиеся большими областями стеклообразования и особенно
стабильными стеклами.
В стекла системы Ge—As—Se удалось ввести до 10 мол. %
Sn или РЬ [773]. В работе [774] изучена область стеклообразо-
вания и кристаллизация в системе As2S3GcxTey, а в работах
[775, 776] исследованы термические, электрические и оптические
свойства стекол по разрезу As2GeS3-xTex. Стекла системы
Ge—As—Se—Те также изучены во многих работах. Результаты
измерений проводимости и термо-э. д. с. стекол по разрезу
(Aso,744Geo,256)i-x(Seo,i97Teo,8O3)x приведены в работе [777]. В си-
стеме Ge—Sb—Se стеклообразование сохраняется при замеще-
нии до 25 мол. % Se на Те [778]. Горюнова и Коломиец от-
крыли класс халькогенидных стеклообразных полупроводников
при исследовании разрезов Tl2Se-Sb2Se3—Tl2Se-As2Se3
и Tl2Se-As2Se3—Tl2Se-As2Te3 (разд. 3.3.6.3) (см. также лите-
ратуру к гл. 2 [14]).
Расширенные области стеклообразования также получены
в германиевоселенидных системах, содержащих Sn, РЬ или Hg,
при участии Те как четвертого компонента.
Определенные составы стекол в системе As—Ge—Se—Те
обнаруживают эффект переключения.
3.3.7.1. Стеклообразование и свойства стекол в системах
Ge—Sn—Se—Те и Ge—Pb—Se—-Те
Область стеклообразования для оловосодержащих стскол
приведена на рис. 3.67 (см. литературу к гл. 2 [336]). Взаим-
ное влияние пар элементов—гомологов Ge, Sn и Se, Те изу-
чено по разрезу Geo,4-xSnxSeo,6-yTey. При этом составы изучен-
ных образцов всегда соответствовали формуле М2Х3 (М =
= Ge-l Sn, X = Se + Te).
Принимая во внимание уже обсуждавшееся изменение сред-
них мольных объемов (рис. 2.43) и применяя известные кри-
сталлохимические соотношения, был сделан вывод о том, что
мопоселепиды Ge и Sn в стекле не изоморфны. Образование
из GeSe сетки структурных группировок Gc2Sc6/2 вследствие
взаимодействия с GeSe2 и влияние SnSe как модификатора
стеклообразной сетки подтверждаются полученными результа-
тами (рис. 2.43). Se и Те взаимно замещают друг друга в кар-
касе стекла (рис. 2.43).
Измерения удельной проводимости по такому разрезу дают
следующие результаты:
г/=0,1; 0,02 <х< 0,08; 11,6 > — 1g а298 > 9,3; 0,75 > > 0,63 эВ
#=0,15; 0<х<0,12; 12,4 > — 1g сг298 > 7,0; 0,82 > Еа > 0,49 эВ
#=0,2; 0<х<0,12; 11,3 > — 1g а298 > 6,6; 0,78 > Еа > 0,53 эВ
Аморфные и стеклообразные системы
385
Рис. 3.67. Область стеклообразовании в системе Ge—Sn—Se—Те и три раз-
реза, проходящие через 10, 15 и 20 мол. % Те.
Для всех стекол сщ в уравнении 1g о = 1g о0— EJkT не зависит
от состава; 1g а0 = 1,3 0,2.
Область стеклообразоваиия в системе Ge—РЬ—Se—Те по
разрезам Ge—Se—Тс (рис. 3.50), Ge—РЬю—Se—Те,
Ge—Pbis—Se—Те и Ge—РЬго—Se—Те приведена па рис. 3.68.
В этой системе также проявляется аналогичное соотношение
[77-9]. Следует заметить, что сравнение стекол, содержащих
РЬ и Sn, при идентичных в остальном составах показывает,
что у стекол с Sn проводимость выше. На рис. 3.69 приведены
значения —Igo для четырех образцов M0.f->GexSe?/Tez с тремя
значениями х, у, z, (M = Sn или РЬ).
Несмотря на больший ионный радиус РЬП по сравнению со
Sn, средние мольные объемы стекол, содержащих РЬ, при
идентичных составах примерно одинаковы или даже меньше.
Проводимость Pb-стекол ниже, а энергия активации электропро-
водности несколько выше, хотя для их структуры предпола-
гается более высокая плотность упаковки. На электрических
свойствах SnTe и РЬТе также обнаруживается такое различие.
Ширина запрещенной зоны SnTe составляет 0,26 эВ, а для
РЬТе 0,31 эВ [780].
25 Заказ № 413
Рис, 3.68. Область стеклообразования в системе Ge—Pb—Se—Те и разрезы,
проходящие через 0 (/), 10 (2), 15 (<3) и 20 (4) мол. % РЬ.
Рис. 3.69. Сравнение удельной проводимости а298 и энергии активации прово-
димости для стекол аналогичного состава в системах Ge—Sn—Se—Те (/) и
Ge—Pb—Se—Те (2).
Аморфные и стеклообразные системы
387
3.3.7.2. Стекла в системе Hg—Ge—Se—Те
На рис. 3.70 объединены области стеклообразования, при-
веденные на рис. 3.63, и области стеклообразования систем
HgSe—GeSe2—Se и Ge—Se—Те (рис. 3.50) [571]. Исходя из
ориентации разрезов и обозначенных границ стеклообразова-
Рис. 3.70. Область стеклообразования в системе Hg—Ge—Se—Те с разрезами,
соответствующими рис. 3.63.
ния в этих системах, можно установить положение области со-
ставов, в которой при охлаждении расплавов образуются
стекла.
Смеси HgSe—HgTe—GeSe или HgSe—GeTe—GeSe также
образуют стекла в определенных пределах. Вблизи эвтектики
GeTe/Te при добавлении Hg в качестве третьего компонента
также образуется узкая область стеклообразования.
3.3.7.3. Стекла в системе As—Ge—Si—Те
Как видно из рис. 3.71, где изображена область стеклообра-
зования по разрезу AsxGeySi2Teo,5 [471, 529], внутри тетраэдра
объединены области стеклообразования в ограничивающих
тройных системах Ge—As—Те (рис. 3.57), Ge—Si—Те (рис. 3.51)
и Si—As—Те (рис. 3.58). В работе [781] изучен разрез
AsxGe^TezSioj. Электрические свойства тонких слоев такого
25*
388 Глава 3
состава Asot3Geo,iSio(i2Teo,48 были подробно изучены (разд. 4.1.3.7).
Вблизи этого состава путем термообработки получают стекла
и переохлажденные расплавы без признаков кристаллизации.
Материал не кристаллизуется с поверхности при недельной
термообработке. Такие стекла также очень стабильны по от-
Рис. 3.71. Область стскло-
образования в системе As—
Ge—Si—Те.
ношению к структурным изменениям, что особенно важно для
определенных применений. Температура стеклования этого
состава ~510 К; в системе Si—As—Те она достигает 590 К
(для теллуридных стекол это достаточно высокое значение).
Проводимость стекла состава Aso,3Sio,iGeo,i2Teo,48 при 298 К
составляет 4-Ю-8 См/см, термическая энергия активации про-
водимости £а = 0,68 эВ [751]. Ширина запрещенной зоны, най-
денная из оптических измерений, 1,14 эВ [782, 783].
Литература
I. Bucket W., Hilsch R., Z. Phys., 138, 109 (1954); Hilsch R., Non-Crystalline
Solids, V. D, Frechette (Ed.), Wiley, New York, 1960, p. 348.
2. Takayama S., J. Mater. Sci., 11, 164 (1976).
3. Davies H. A., Lewis B. G., Scripta Metall., 9, 1107 (1975).
4. Chen H. S., Acta Metall., 22, 1505 (1974).
5. Chaudhari P., Turnbull D., Science, 199, 11 (1978).
6. Davies H. A., Phys. Chem. Glass., 17, 159 (1976).
7. Warlimont H., Z. Metallkunde, 69, 212 (1978).
8. GUntherodt H. J., Beck H., Oelhafen P., Ackermann К. P., Liard M., Mid-
ler M., Kiinzi H. U., Rudin H„ Agygman K., Electrons in Disordered Me-
tals and at Metallic Surfaces, Plenum Publ. Corp., 1979, p. 501.
9. Хансен M., Андерко К. Структуры двойных сплавов. Т. 1, 2.— М.: Ме-
таллургиздат, 1964, с. 1488.
10. Davies Н. A., Rev. Chim. Miber., 16, 349 (1979).
11. Ray R., Giessen В. C.. Grant N. J., Scripta Metall., 2, 357 (1968).
Аморфные и стеклообразные системы
389
12. Herd. S. R., Chaudhari Р., Phys. Stat. Solidi (a), 18, 603 (1978).
13. Vi nd Nielsen H. J., J. Non-Cryst. Solids, 33, 285 (1979).
14. Duwez P., Widens R. H., Crewdson R. C., J. Appl. Phys., 36, 2267 (1965).
15. Chen H. S., Turnbull D., J. Chem. Phys., 48, 2560 (1968).
16. Chou С. P., Turnbull D., J. Non-Cryst. Solids, 17, 169 (1975).
17. Chen H. S., Turnbull D., Acta MetalL, 17, 1021 (1971).
18. Marcus M. A., J. Non-Cryst. Solids, 30, 317 (1979).
19. Chen H. S., Krause J. T., Coleman E., Scripta MetalL, 9, 787 (1975).
20. Barmatz M., Chen H. S., Phys. Rev., В 9, 4073 (1974).
21. Yamanchi K., Nakagawa Y., Jap. J. Appl. Phys., 10, 1730 (1971).
22. Anderson O. L., Physical Acoustic, 3 B, 43 (1965).
23. Chen H. S., Proc. Conf. The Structure Non-Cryst. Mat., Cambridge, 1976,
p. 79.
24. Walter J. P., Mat. Sci., Eng., 39, 95 (1979).
25. Duwez P., Lin S. С. H., J. Appl. Phys., 38, 4096 (1967).
26. Polk D. E„ Chen H. S., J. Non-Cryst. Solids, 15, 165 (1974).
27. Rastogi P. K., Duwez P., J. Non-Cryst. Solids, 5, 1 (1970).
28. Cargill G. S., J. Appl. Phys., 41, 12 (1970).
29. Leung P. K., Wright J. G., Phil. Mag., 30, 955 (1974).
30. Cargill G. S„ J. Appl. Phys., 41, 2249 (1970).
31. Finney J. L., Proc. Roy. Soc. (London), A 319, 479 (1970).
32. Bennett C. FL, J. Appl. Phys., 43, 2727 (1972).
33. Polk D. E., Scripta Met., 4, 117 (1970).
34. Cargill G. S., Kirkpatrick S., AJP Conf. Proc. Structure and Exitations of
Amorphous Solids, New York, 1976, p. 339.
35. Ruhl R. C., Giessen В. C., Cohen M., Grant N. J., Acta MetalL, 15, 1693
(1967).
36. Chen H. S„ Park В. K., Acta MetalL, 21, 395 (1973).
37. Cargill G. S., Rapidly Quenched Metals, M. J. T., Cambridge, 1976, p. 293.
38. Hayes T. M„ Allen J. W., Tauc J., Giessen В. C., Hauser J. J., Phys. Rev.
Lett., 40, 1282 (1978).
39. Nagel S. R., Tauc J., Phys. Rev. Lett., 35, 380 (1975).
40. Midler R.. Gilntherodt H. J., KUnzi H. U., Liard M., Muller M., Rudin H.,
Non-Cryst. Solids. 4th Inter. Conf. The Physics Non-Cryst. Solids 1976,
Ed. G. H. Frischat. Trans. Tech. Publications, 1977, p. 212.
41. Boucher B. Y., J. Non-Cryst. Solids, 7, 277 (1972).
42. Johnson W. L., Poon S. J., Duwez P., Phys. Rev., 11 B, 150 (1975); J.
Appl. Phys., 46, 1787 (1975).
43. Togano K., Tachikawa K-, Phys. Lett., 54 A, 205 (1975).
44. Губанов А. И.— Физ. тв. тела, 1960, т. 2, с. 468.
45. Mader S.. Nowick A. S., AppL Phys. Lett., 7, 57 (1965).
46. Egami T., Flanders P. J., Graham C. D., Appl. Phys. Lett., 26, 128 (1975).
47. Fujimori H„ Masumoto T., Obi Y., Kikuchi M., Jap. J. AppL Phys., 13,
1889 (1974).
48. Naka M., Hashimoto K., Masumoto T., J. Non-Cryst. Solids, 29, 61 (1978).
49. Wright J. G., 7th Int. Colloqu. Magnetic Thin Films, Regensburg, 1975.
50. Pampillo C. A., J. Mater. Sci., 10, 1194 (1975).
51. Chen H. S., Leamy H. J., Barmatz M., J. Non-Cryst. Solids, 5, 444 (1971).
52. Soshiroda T., Koiwa M., Masumoto T., J. Non-Cryst. Solids, 22, 173 (1976).
53. Waseda Y., Miller W. A., Physica Status Solidi (a), 49, К 31 (1978).
54. Greer A. L.. Leake J. A., J. Non-Cryst. Solids, 33, 291 (1979).
55. Bagley B. G., Vogel E. M., J. Non-Cryst. Solids, 18, 29 (1979).
56. Marcus M. A., J. Non-Cryst. Solids, 30, 317 (1979).
57. Borchert W., Dietz W., Kolker H., Z. Angew. Phys., 29, 277 (19'TQ).
58. McCarty L. V., Carpenter D. R., J. Electrochem. Soc., 107, 38 (1960).
59. -Amberger E., Dietz W., Z. anorg. allg. Chem., 332, 113 (1964).
60. Amberger E., Angew. Chem., 83, 893 (1971).
61. Laubengayer A. W., Hurd D. T., Newkirk A. E., Hoard J. L., J. Amer.
Chem. Soc., 65, 1924 (1943).
390 Глава 3
62. Wohler F., Liebigs Ann. Chem., 141, 268 (1867).
63. Galasso F., Vaslet R., Pinto J., Appl. Phys. Lett., 8, 131 (1966).
64. Decker B. F., Rasper J. S., Acta Crystallogr., 12, 503 (1959).
65. Hoard J. L., Sullenger D. B., Rennard С. H. L., Hughes R. E., J. Solid
State Chem., 1, 268 (1970).
66. Geist D., Rloss R., Follner H., Acta Crystallogr., В 26, 1800 (1970).
67. Will G., Ploog R., Nature, 251, 406 (1974).
68. Badzian A. R., Mater. Res. Bull., 2, 987 (1967).
69. Borchert W., Dietz W., Herrmann H., Z. angew. Physik, 19, 485 (1965).
70. Werheit H., Festkorperprobleme X, 189 (1970).
71. Berezin A. A., Golikova O. A., Razanin M. M„ Rhomidov T., Mirlin D. N.,
Petrov A. V., Umarov A. S., Zaitsev V. R., J. Non-Cryst. Solids, 16, 237
(1974).
72. Bundy F. P., J. Chem. Phys., 38, 618, 631 (1963).
73. Hall С. E., J. Appl. Phys., 19, 271 (1948).
74. Vohler 0., Reiser P. L., Martina R., Overhoff D., Angew. Chem., 82, 401
(1970).
75. Parthe E., Crystal Chemistry of Tetrahedral Structures, Gordon and
Breach, Science PubL, New York/London, 1964.
76. Hanneman R. E„ Strong H. M., Bundy F. P., Science, 155, 955 (1967).
77. Bundy F. P., Rasper J. S., J. Chem. Phys., 46, 3437 (1967).
78. Aisenberg S., Chabot R., J. Appl. Phys., 42, 2953 (1971).
79. Rasatochkin V. I., Rorchak V. V., Rudrjavcev J. P., Sladkov A. M., Stern-
berg J. E., Carbon, 11, 70 (1973).
80. RoptuaK В. В., Нудрявцев И. P., Сладкое A. M.— Вести. АН СССР, 1978,
№ 1, c. 70.
81. Whittaker A. G., Rintner P. L., Science, 165, 589 (1969).
82. Goresy A. E., Donney G., Science, 161, 363 (1968).
83. Rakinoki J., Ratada R., Haiowa I., I no T., Acta Crystallogr., 13, 171
(1960).
84. Бойко Б. T., Палатник JI. С., Деревянченко A. C.— Д. АН СССР, 1967,
т. 179, c. 316.
85. Grtgorovici R.. Devenyi A., Sheorghiu A., Belli A., J. Non-Cryst. Solids,
8—10, 793 (1972).
86. Hamilton E. M., Cross J. A., Adkins C. J., Proc. 5th Int. Conf. Amorph.
Liquid Semicond., Garmisch-Partenkirchen 1973, Taylor-Francis London,
1974, p. 1225.
87. Wada N., Gaczi P. J., Solin S. A., J. Non-Cryst. Solids, 35/36, 543 (1980).
88. Smith J. E., Bull. Amer. Phys. Soc., 17, 136 (1972).
89. Mildner D. F. R., Carpenter J. M., Bull. Amer. Phys. Soc., 17, 463 (1972).
90. Noda T., Inagaki M„ Yamada S., J. Non-Cryst. Solids, 1, 285 (1969).
91. Stenhouse B. J., Grout P. J., J. Non-Cryst. Solids, 27, 247 (1978).
92. Wentorf R. H., Rasper J. S-, Science, 139, 338 (1963); Jamieson J. C.,
Science, 139, 762 (1963).
93. Rasper J. S., Richards S. M., Acta Crystallogr., 17, 752 (1964).
94. Rrebs H., Lazarev V. B., Winkler L., Z. anorg. allg. Chem., 352, 277
(1967).
95. Isherwood S. P., Orton B. R., J. Non-Cryst. Solids, 8—10, 691 (1972).
96. Bienenstock A., Proc. 5th Int. Conf. Amorph. Liquid Semicond., Garmisch-
Partenkirchen 1973, Taylor-Francis, London, 1974, p. 49.
97. Elliott P. J., Yoffe A. D., ibid., Vol. 2, 1139; Viscor P., Yoffe A. D., J.
Non-Cryst. Solids, 35/36, 409 (1980).
98. Barna A., Barna P. B., Pocza J. F., J. Non-Cryst. Solids, 8—10, 36, (1972).
99. Cargill G. S. Ill, Phys. Rev. Lett., 28, 1372 (1972).
100. Temkin R. J., Connell G. A. N., Paul W., Proc. 5th Int. Conf. Amorph.
Liquid Semicond., Garmisch-Partenkirchen 1973, Taylor-Francis London,
1974, p. 533.
101. Camphausen D. L., Connell G. A. N., Paul W., J. Non-Cryst. Solids, 8—10,
223 (1972).
Аморфные и стеклообразные системы 391
102. Le Comber Р. G., Loveland R. J., Spear W. E., Vaughan R. A., Proc.
5th Int. Conf. Amorph. Liquid Sernicond., Garmisch-Partenkirchen 1973,
Taylor-Francis, London, 1974, p. 245.
103. Connell G. A. N., Pawlik J. R., Phys. Rev., В 13, 787 (1976).
104. Spear W. E., Le Comber P. G., Solid State Commun., 17, 1193 (1975).
105. Spear W. E., Le Comber P. G., Kinmond S., Brodsky M. H., Appl. Phys.
Lett., 28, 105 (1976).
106. Triska A., Demieson D., Fritzsche H., Bull. Amer. Phys. Soc., 20, 392
(1975).
107. Brodsky M. H., Frisch M. A., Ziegler J. F., Lanford W. A., Appl. Phys.
Lett., 30, 561 (1977).
108. Zanzucchi P. J., Wronski C. R., Carlson D. E., J. Appl. Phys., 48, 5227
(1977).
109. Le Comber P G., Spear W. E., Doped Amorphous Semiconductors in
Topics in Appl. Phys. 36 — Amorphous Semiconductors — M. H. Brodsky
(Ed.) p. 251.
110. Carlson D. E., Wronski C. R., ibid., p. 287.
111. Proc. 8th Internal. Conf. Amorph. Liqu. Sernicond. Cambridge (Ma.), 1979,
J. Non-Cryst. Solids, 35, 36.
112. Ching W. У., J. Non-Cryst. Solids, 35/36, 61 (1980).
113. Madaa A., Ovshinsky S. R., J. Non-Cryst. Solids, 35/36, 171 (1980).
114. Knight J. C., Street R. A., Lucovsky G., J. Non-Cryst. Solids, 35/36, 279
(1980).
115. Craczyk J. F., Chaudhari P., Phys. Status Solidi (b), 58, 163 (1973).
116. Craczyk J. F., Chaudhari P., Appl. Phys. Lett., 32, 466 (1978).
117. Shevhik N. J., Paul W., J. Non-Cryst. Solids, 8—10, 381 (1972).
118. Kinney W. J., J. Non-Cryst. Solids, 21, 275 (1976).
119. Moss S. C., Graczyk J. F., Phys. Rev. Lett., 23, 1167 (1969).
120. Temkin R. J., Paul W., Connell G. A. N., Advances Phys., 22, 581 (1973).
121. Connell G. A. N., Temkin R. J., Phys. Rev., В 9, 5323 (1974).
122. Steinhardt P., Alben R., Weaire D., J. Non-Cryst. Solids, 15, 199 (1974).
123. Beeman D., Bobbs B. L., Phys. Rev., В 12, 1399 (1975).
124. Grigorovici R., J. Non-Cryst. Solids, 1, 303 (1969).
125. Grigorovici R., Manaila R., J. Non-Cryst. Solids, 1, 371 (1969).
126. Turnbull D., Polk D. E., J. Non-Cryst. Solids, 8—10, 19 (1972).
127. Polk D. E., Boudreaux D. S., Phys. Rev. Lett., 31, 92 (1973).
128. Graczyk J. F., Chaudhari P., Phys. Rev. Lett., 17, 299 (1975).
129. Temkin R. J., J. Non-Cryst. Solids, 28, 23 (1978).
130. Henderson D.^ Hetman F., Ortenburger I. B., Proc. 5th Int. Conf. Amorph.
Liquid Sernicond., Garmisch-Partenkirchen 1973, Taylor-Francis, London,
1974, p. 991.
131. Henderson D., Herman F., J. Non-Cryst. Solids, 8—10, 359 (1972).
132. Graczyk J. F., Phys. Status Solidi (a), 46, 611 (1978).
133. Koizumi H., Ninomiya T., J. Phys. Soc. Jap., 44, 898 (1978).
134. Davis E. A., Solomon L, in: Amorphous Semiconductors, M. H. Brodsky
(Ed.), Springer-Verlag, 1979. p. 41. 189.
135. Thomas P. A., Brodsky M .H., Kaplan D„ Lepine D., Phys. Rev., В 18,
3059 (1978).
136. Kaplan D., Sol N., Velasco G., Thomas P. A., Appl. Phys. Lett., 33, 440
(1978).
137. Pankove J. L, Lampert M. A., Tarng M. L., Appl. Phys. Lett., 32, 439
(1978).
138. Mell R., Proc. 5th Int. Conf. Amorph. Liquid Sernicond., Garmisch-Parten-
kirchen, 1973, Taylor-Francis London, 1974, p. 203.
139. Beyer W., Stuke J., Proc. 5th Int. Conf. Amorph. Liquid Sernicond., Gar-
misch-Partenkirchen, 1973, Taylor-Francis, London, 1974, p. 251.
140. Voget-Grote U., Stuke J., Wagner H., Proc. Conf. Structure and Excita-
tion Amorph. Solids, Williamsburg, 1976, A. I. P. New York, p. 91.
141. Shaffer P. T. B., Acta Crystallogr, В 25, 477 (1969).
392 Глава 3
142. Fagen Е. A., Proc. 5th Int. Conf. Amorph. Liquid Semicond., Garmisch-
Partenkirchen 1973, Taylor-Francis, London, 1974, p. 601.
143. Tejeda J., Shevchik N. J., Cardona M., Proc. 5th Int. Conf. Amorph. Liquid
Semicond., Garmisch-Partenkirchen 1973, Tavlor-Francis, London, 1974,
p. 557.
144. Lannin J. S., Proc. 5th Int. Conf. Amorph. Liquid Semicond., Garmisch-
Partenkirchen 1973, Taylor-Francis, London, 1974, p. 1245.
145. Connell G. A. N., Temkin R. J., Paul W., Proc. 5th Int. Conf. Amorph.
Liquid Semicond., Garmisch-Partenkirchen 1973, Taylor-Francis, London,
1974, p. 1201.
146. Engemann D., Fischer R., Knecht J., Appl. Phys. Lett., 32, 567 (1978).
147. Shevchik N. J., Paul W., J. Non-Cryst. Solids, 13, 1 (1973/74).
148. Whil M., Cardona M., Tauc J., J. Non-Cryst. Solids, 8—10, 172 (1972).
149. Connell G. A. N., Temkin R. J., Proc. Int. Conf. Tetrahedr. Bonded Amorph.
Semicond. Yorktown Heights, N-Y., 1974, 192.
150. Yates D. A., Penchina С. M., Proc. 5th Int. Conf. Amorph. Liquid. Semi-
cond., Garmisch-Partenkiichen 1973, Taylor-Francis, London, 1974, p. 617.
151. Gheorghiu A., Theye M.-L., Proc. 7th Int. Conf. Amorph. Liquid Semicond.,
Edinburgh 1977, University Edinburgh, p. 462.
152. Paul W., Moustakas T. D., Anderson D. A., Freeman E., Proc. 7th Int.
Conf. Amorph. Liquid. Semicond., Edinburgh 1977, University Edinburgh,
p. 467.
153. Brodie D. E., Proc. 7th Int. Conf. Amorph. Liquid Semicond., Edinburgh
1977, University Edinburgh, p. 472.
154. Smith R. C., J. de Physique, 36, C 3—89 (1975).
155. Вайполин /1. .4., Гопюнова H. А., Османов E. О., Рудь Ю. В.— ДАН
СССР, 1965, т. 160, с. 633.
156. Борисова 3. У., Школьников Е. В., Ярембаш Е. И.— Изв. АН СССР,
нсорг. материалы, 1970, т. 6, с. 383.
157. Борисова 3. У., Горюнова И. А., Кузова И. И., Османов Е. О.,
Рудь Ю. В.— Физ. техн, полупроводников, 1968, т. 2, с. 1518.
158. Аксенов В. В., Петров В. М., Харахорин Ф. Ф.— Изв. АП СССР, нсорг.
материалы, 1972, т. 8, с. 1152.
159. Uemura О., Satow Т., Phys. Status Solidi (а), 40, 303 (1977).
160. Satow Т., Uemura О., Watanabe S., Phvs. Status Solidi (a), 44, 731
(1977).
161. Gorjunova N. A., Kuzmenko G. S., Osmanov E. O., Mater. Sci. Eng., 7,
54 (1971).
162. Cervinka L., Hruby A., Matyas M., Simecek T., Skacha J., Stourac L.,
Tauc J., Vorlicek V., Hoschl P., J. Non-Cryst. Solids, 4. 258 (1970).
163. Hruby A., Houserova J., Czech. J. Phys., В 22, 89, 861 (1972).
164. Popescu M., Manaila R., Grigorovici R., J. Non-Cryst. Solids, 23, 229
(1977).
165. Thum H„ Krebs H., Angew. Chem., 78, 1101 (1966).
166. Brown A., Rundquist S., Acta Crystallogr., 19, 684 (1965).
167. Jamieson J. C., Science, 139, 1291 (1963).
168. Krebs H., Gruber H. U., Z. Naturforsch., A 22, 96 (1967).
169. v. Schnering H. G., Catenation of Phosphorus Atoms in: Homoatomic
Rings, Chains and Macromolecules of AAain-Group Elements, A. L. Rhein-
gold (Ed.), Elsevier, New York, 1977, p. 317.
170. Krebs H., Ludwig T., Z. anorg. allg. Chem., 294, 257 (1958).
171. Beyeler H. U., Veprek S., Phil. Mag., 41, 327 (1980).
172. Osterlag P., Dissertation Sektion Chemie der TU Dresden, 1972.
173. Smith P. M., Leadbetter A. J., Apling A. J., Phil. Mag., 31, 57 (1975).
174. Reale C., Vacuum, 28, 1 (1978).
175. Sldhr H., Z. anorg. allg. Chem., 242, 138 (1939).
176. Greaves G. N., Elliott S. R., Davis E. A., Adv. Phys., 28, 49 (1979).
177. Борисова 3. У., Крылова 3. A.— Жури, прикл. хим., 1967, т. 40. с. 61.
178. Bellisent R., Tourand G., J. de Physique, 37, 1423 (1976); Proc. Vlth Inter.
Аморфные и стеклообразные системы
393
Conf. Amorph. Liqu. Semicond., Leningrad 1975, Structure and Properties
of Non-Cryst. Semicond., p. 160.
179. Krebs H., Festkorperprobleme, IX, 1 (1969).
180. Davis E. A., Elliott S. R., Greaves G. N„ Jones D. P.t Proc. Conf. Struc-
ture of Non-Crystalline Materials, Taylor and Francis, London, 1977,
p. 205.
181. Lucovsky G., Knights J. C., Phys. Rev., В 10, 4324 (1974).
182. Lannin J. S.t Phys. Rev., В 15, 3863 (1977).
183. Elliott S. R., Davis E. A., J. Non-Cryst. Solids, 35, 36, 849 (1980).
184. Ley L., Pollak R. A., Kowalczyk S. P., McFeely R., Shirley D. A., Phys.
Rev., В 8, 641 (1973).
185. Robertson J., цпт. no Elliott S. R., Davis E. A., J. Non-Cryst. Solids, 35,
36. 849 (1980).
186. Elliott S. R., Davis E. A., J. Phys. C, Solid State Phys., 12, 2577 (1979).
187. Greaves G. N., Davis E. A., Phys. Mag., 34, 265 (1976).
188. Raisin C., Leveque G., Robin J., Solid State Commun., 14, 723 (1974).
189. Mytilineou E., Davis E. A., Proc. Vllth Int. Conf. Amorph. Liqu. Semi-
cond., Edinburgh, 1977, p. 632.
190. Taiflor P. C., Friebele E. J., Bishop S. G., Solid. State Commun., 28, 247
(1978).
191. Taylor P. C., Strom U., Bishop S. G., Phys. Rev., 18, 511 (1978).
192. Mytilineou E., Davis E. A., J. Non-Cryst. Solids, 35, 36, 883 (1980).
193. Krebs FL, Angew. Chem., 65, 293 (1953).
194. Aymerich F. M., Delumas A., Phys. Status Solidi (a), 31, 165 (1975).
195. Crawley A. F., Kiff D. R., MetalL Trans., 3, 157 (1972).
196. Hauser J. J., Di Salvo F. J., Hutton R. S., Phil. Mag., 35, 1557 (1977).
197. Delunas A., Aymerich F., Phys. Status Solidi (a), 46, 703 (1978).
198. Schmidt M., Angew. Chem., 85, 474 (1973).
199. Meyer B., Chem. Rev., 76, 367 (1976).
200. Meyer B., Advan. Inorg. Chem. Radiochem., 18, 287 (1976).
201. Meyer B., Sulfur, Energy and Environment, Elsevier Scientific Publishing
Comp., Amsterdam/Oxford/New York, 1977.
202. Steudel R., Angew. Chem., 87, 683 (1975).
203. Steudel R., Mausle H.-J., Angew. Chem., 91, 165 (1979).
204. Donohue J., The Structures of the Elements, Wiley, New York, 1974.
205. Andonov P„ J. Non-Cryst. Solids, 47, 297 (1982).
206. Яковлев E. H.— Письма ЖЭТФ, 1978, т. 28, с. 369, 390.
207. Cooper R., Culka J. V., J. Inorg. Nucl. Chem., 32, 1857 (1970).
208. Meyer B., Spitzer K-, J. Phys. Chem., 76, 2274 (1972).
209. Weiss J., Pupp M., Angew. Chem., 82, 447 (1970).
210. Weiss J., Bachtler W., Z. Naturforsch., В 28, 523 (1973).
211. Calvo C., Gillespie R. J., Vekris J. E., Ng H. N., Acta Crystallography
В 34, 911 (1978).
212. Ringer W. E., Z. anorg. allg. Chem., 32, 183 (1902).
213. Lozana L., Gazz. Chim. Ital., 53, 396 (1923).
214. Weiss J., Z. anorg. allg. Chem., 435, 113 (1977).
215. Myers M. B„ Felty E. J.. Mat. Res. Bull., 2, 535 (1967).
216. Schottmiller J., Tabak M., Lucovsky G., Ward A., J. Non-Cryst. Solids, 4,
80 (1970).
217. Hagemann A. M., J. Amer. Chem. Soc., 41, 339 (1919).
218. Pupp M., Weiss J., Z. anorg. allg. Chem., 440, 31 (1978).
219. Physics of Selenium and Tellurium, W. C. Cooper (Ed.), Pergamon Press,
Inc., Oxford, 1969.
220. Selenium, R. A. Zhigaro, W. C. Cooper (Ed.), Van Nostrand-Reinhold,
New York, 1974.
221. The Physics of Selenium and Tellurium, E. Gerlach, P. Grosse (Eds.),
Springer-Series in Solid-State Science 13, Springer-Verlag, 1979.
222. Unger P., Cherin P., in: Physics of Selenium and Tellurium, W. C. Coo-
per (Ed.), Pergamon Press, Inc., Oxford, 1969, p. 223.
394 Глава 3
223. Кребс Г. Основы кристаллохимии неорганических соединений: Пер.
с англ.— М.: Мир, 1971.
224. Moynihan С. Т., Schnaus U. Е., Mat. Sci. Eng., 6, 227 (1970); Chang S. S.,
Bestul A. B., J. Chem. Thermodynamics, 6, 325 (1974).
225. Eckhart E., Ann. Phvsik, 14, 233 (1954).
226. Wittig L, Phys. Rev' Letters, 15, 159 (1965).
227. Me Cann D. R., Cartz L., J. Chem. Phys., 56, 2552 (1972).
228. Gattow G., Heinrich G., Z. anorg. allg. Chem., 331, 275 (1964).
229. Gobrecht H., Witters G., Wobig D„ J. Phys. Chem. Solids, 31, 2145 (1970).
230. Kawarada M., Nishina У., Sci. Rep. RITU, 26 A, 101 (1976); Jap. J. Appl.
Phys.. 14, 1519 (1975).
231. Cooper W. C., Westbury R. A., in: Selenium, R. A., Zhigaro, N. C. Cooper
(Eds.), Van Nostrand-Reinhold, New York, 1974, p. 87.
232. Kaplow R., Rowe T. A., Averbach B. L., Phys. Rev., 168, 1068 (1968).
233. Zallen R., Lucovsky G., in: Selenium, R. A. Zhigaro, N. C. Cooper (Eds.),
Van Nostrand-Reinhold, New York, 1974, p. 167.
234. Lucowsky G., Mooradian. A., Taylor W., Wright G. B„ Keezer R. C., Solid
State Commun., 5, 113 (1967).
235. Martin R. M., Lucovsky G., Helliwell K., Phys. Rev., В 13, 1383 (1976).
236. Thum H., Ruska J., J. Non-Cryst. Solids, 22, 331 (1976).
237. Hoshino H., Schmutzler R. W., Warren IT. W'., Hensel F., Phil. Mag., 33,
255 (1976).
238. Tobolsky A. V., Eisenberg A., J. Amer. Chem. Soc., 81, 780 (1959).
239. Eisenberg A., Tobolsky A. V., J. Polymer. Sci., 46, 19 (1960); 61, 483
(1962).
240. Cukierman M., Uhlmann D. R., J. Non-Cryst. Solids, 12, 199 (1973).
241. Keezer В. C., Lucovsky G., Martin R. M., Bull. Am. Phys. Soc., 20, 323
(1975).
242. Lucovsky G„ in: The Physics of Selenium and Tellurium, E. Gerlach,
P. Grosse (Eds.), Springer-Series in Solid-State Science 13, Springer-
Verlag. 1979, p. 178.
243. Berg J. I., Siniha R., J. Non-Cryst. Solids, 22, 1 (1976).
244. Hendus H., Z. Phys., 119, 265 (1942).
245. Krebs И., Schultze-Gebhardt F., Acta Crystallogr., 8, 412 (1955).
246. Richter H., Gomtnel G., Z. Naturforsch., A 12, 996 (1957).
247. Андреевский А. И., Набитович И. Д., Волощук Ю. В.— Кристаллография,
1962, т. 7, с. 865.
248. Горюнова Н. А., Коломиец Б. Т.— Журн. техн, физ., 1958, т. 28, с. 1922.
249. Henninger Е. И., Buschert R. С., Heaton L., J. Chem. Phvs., 46, 586
(1967).
250. Breitling G., Richter H., Mater. Res. Bull., 4, 19 (1969).
251. Richter H., J. Non-Ciyst. Solids, 8—10, 388 (1972).
252. Hansen F. Y., Knudsen T. S., J. Chem. Phys., 62, 1556 (1975).
253. Malaurant J. C., Dixtnier J., Phys. Status Solidi (a), 43, К 61 (1977).
254. Long M.. Galison P.. Albert R Connell G A К Phys Rev, В 13 1^21
(1976).
255. Meek P. E., Phil. Mag., 33, 897 (1976).
256. Richter H., Herre F., Z. Naturforsch., A 13, 874 (1958).
257. Blachnik R., Hoppe A., J. Non-Cryst. Solids, 34, 191 (1979).
258. Nielsen P., Proc. 5th. Int. Conf. Amorph. Liquid. Sernicond., Garmisch-
Partenkirchen 1973, Taylor-Francis, London, 1974, p. 639.
259. Vaidyanathan A., Mitra S. S., Tsai Y. F., Phys. Rev., В 21, 2475 (1980).
260. Vanderbilt D., Joannopoulos J. D., Phys. Rev. Lett., 42, 1012 (1979).
261. Feltz A., Aust H., J. Non-Cryst. Solids, 51, 395 (1982).
262. Hartke J. L., Phys. Rev., 125, 1177 (1962).
263. Owen A. E., Contemp. Phys., 11, 227, 257 (1970).
264. Spear W. E., J. Non-Cryst. Solids, 1, 197 (1969).
265. Schottmiller J., Tabak M., Lucovsky G., Ward A., J. Non-Crvst. Solids, 4,
80 (1970).
Аморфные и стеклообразные системы
395
266. Dolezalek F. К., Spear W. Е., J. Non-Cryst. Solids, 4, 97 (1970).
267. Stake J., Festkorperprobleme, IX, 46 (1969).
268. Andiere J. D., Mazieres C., Carballes J. C., J. Non-Cryst. Solids, 27, 411
(1978).
269. Matsuura M., Suzuki K., J. Mater. Sc., 14, 395 (1979).
270. Kawarada M., Nishina Y., Jap. J. Appl. Phys., 16, 1525, 1531 (1977).
271. Andiere J. P., Mazieres C., Carballes J. C., J. Non-Cryst. Solids, 34, 37
(1979).
272. Глазов В. M., Чижевская С. Н„ Глаголева Н. Н. Жидкие полупровод-
ники, М., Наука, 1967, 244 с.
273. Perron J. С., J. Non-Cryst. Solids, 8—10, 272 (1972).
274. Tourand G., Cabane В., Brenil M., J. Non-Cryst. Solids, 8—10, 676
(1972).
275. Hoyer W., Thomas E., Wobst M., Proc. Vlth Internat. Conf. Amorph. Li-
quid Semicond., Strukture and Properties of Non-Crystalline Semicon-
ductors, Leningrad, 1976, Nauka, p. 405.
276. Axmann A., Gissler W., Kollmar A., Springer T., Faraday-Discussion,
1970/71.
277. Phahle A. M., Thin Solid Films, 61, L 21 (1979).
278. Pellini G., Vio G., Atti Reale Accad. Lincei, Rend., 15, 46 (1906).
279. Lanyon H. P. D., Hockings E. P., Phys. Status Sol., 17, К 185 (1966).
280. Grison E., J. Chem. Phys., 19, 1109 (1951).
281. Смородина T. П.— Физ. тв. тела, 1960, т. 2, с. 807.
282. Mahdjuri F., Proc. 5th Int. Conf. Amorph. Liquid Semicond., Garmisch-
Partenkirchen, 1973, Taylor-Francis, London, 1974, p. 1295.
283. Fischer M., Krebs H., Glastedhn. Ber., 47, 42 (1974).
284. Rialland J. F., Perron J. C., Proc. Vlth Int. Conf. Amorph. Liquid Semi-
cond., Structure and Properties of Non-Crystalline Semiconductors, Lenin-
grad 1976, Nauka, p. 371.
285. Ward A. T., J. Phys. Chem., 23, 4110 (1970).
286. Sarrach D. J., De Neufville J. P., Haworth IF. £., J. Non-Cryst. Solids,
22, 245 (1976).
287. Das G. C., Bever M. B., Uhlmann D. R., J. Non-Cryst. Solids, 7, 251
(1972).
288. Bellissent R., Tourand G., J. Non-Cryst. Solids, 35, 36, 1221 (1980).
289. Kracek F. C., Morey G. W., Merwin H. E., Amer. J. Sci., 35 A, 143 (1938).
290. Mackenzie J. D., Claussen W. F., J. Amer. Ceram. Soc., 44, 79 (1961).
291. Gurr G. E., Montgomery P. W., Knutson C. D., Gorres В. T., Acta Crys-
tallogr., В 26, 906 (1970).
292. Dachille F., Roy R., J. Amer. Ceram. Soc., 42, 78 (1959).
293. Prewitt С. T., Shannon R. D., Acta Crystallogr., В 24, 869 (1968).
294. Fajana K., Barber S. W., J. Amer. Chem. Soc., 74, 2761 (1952).
295. Krogh-Moe J., J. Non-Cryst. Solids, 1, 269 (1969).
296. Kritz H. M., Bray P. J., J. Non-Cryst. Solids, 6, 27 (1971).
297. Silver A. H., Bray P. J.. J. Chem. Phys , 29, 984 (1958).
298. Snt/der L. C., Peterson G. E., Kurkjian C. R., J. Chem. Phys., 64, 1569
(1976).
299. Goubeau J., Keller H., Z. anorg. allg. Chem., 272, 303 (1953).
300. Monteil Y., Vincent H.y Bull. Soc. Chim. Fr., 1975, 1029.
301. Mozzi R. L., Warren В. E., J. Appl. Cryst., 3, 251 (1970).
302. Elliott S. R., Phil. Mag., В 37, 435 (1978).
303. Krogh-Moe J., Ihara M., Acta Crystallogr., 23, 427 (1967).
304. Heinz D., Z. anorg. allg. Chem., 336, 137 (1965).
305. de Decker H. C. J., Rec. Trav. Chem. Pays-Bas, 60, 413 (1941).
306. Mac Gillary С. H., de Decker H. C. J., Niiland L. M., Nature, 164, 448
(1949).
307. Pertlik F., Monatsh. Chem., 106, 755 (1975).
308. Galeener F. L., Mikkelsen J. C, Solid State Commun., 30, 505 (1979).
309. Pertlik F., Chech. J. Physics, В 28, 171 (1978).
396 Глава 3
310. Pertlik F., Monatsh. Chem., 109, 277 (1978).
311. Stranski I. N., Wolff G., Z. Naturforsch., A 4, 23 (1949).
312. Katutz J., Stranski I. N., Z. anorg. allg. Chem., 292, 330 (1957).
313. Imaoka M., Hasegawa H., Phys. Chem. Glass, 21, 67 (1980).
314. Garman S. J., Pettifer R. F., Phil. Mag., 40, 345 (1979).
315. Galeener F. L., Lucovsky G., Geils R. H., Phys. Rev., В 19, 4251 (1979).
316. Lucovsky G., Galeener F. L., J. Non-Cryst. Solids, 35, 36, 1209 (1980);
37, 53 (1980).
317. Jansen M., Z. anorg. allg. Chem., 441, 5 (1978); Z. Naturforsch., 34b,
10 (1979).
318. Winkler A., Thilo E., Z. anorg. allg. Chem., 339, 71 (1965); 350, 320
(1967).
319. White W. B„ Dachille F„ Roy R., Z. Kristallogr., 125, 450 (1967).
320. Hasegawa H., Sone M., Imaoka M., Phys. Chem. Glass., 19, 28 (1978).
321. Rumpel H., Berndt W., Adam K., Schwarzmann E., Z. Naturforsch., В 33,
39, (1978).
322. Jansen M., Acta Crystallogr., В 35, 539 (1979); Angew. Chem., 90, 141
(1978).
323. Vincent H., Bull. Soc. Chirn. Fr., 1972, 4517.
324. Blachnik R., Hoppe A., Z. anorg. allg. Chem., 457, 91 (1979).
325. Robinson P. L„ Scott IF. E., Z. anorg. allg. Chem., 210, 57 (1933).
326. Kenlen E., Vos A., Acta Crystallogr., 12, 323- (1959).
327. Maroni V., Schablaske R. V., J. Inorg. Nucl. Chem., 33, 3182 (1971).
328. Monteil Y., Vincent H., J. Inorg. Nucl. Chem., 37, 2053 (1975).
329. Monteil Y., Vincent H., Z. anorg. allg. Chem., 416, 181 (1975).
330. Penney G. J., Sheldrick G. M., J. Chem. Soc. A 1971, No. 1—2, 245.
331. Header F„ Linke D., Z. Chem., 13, 480 (1973).
332. Monteil Y., Vincent H., Z. anorg. allg. Chem., 428, 259 (1977).
333. Soklakov A. J., Zdanov G. S., цит. no Wright A. C., The Structure of
Amorphous Solids by X-Ray and Neutron-Diffraction- in: Hoppe W., Ma-
son R., Advances in Structure Research by Diffraction Methods, Vol. 5,
Verlag, Vieweg, Braunschweig, 1974, p. 70.
334. Борисова 3. У., Касаткин Б. Е., Кам Е. И.— Изв. АН СССР, неорг.
матер., 1973, т. 9, с. 822.
335. Ким Е. И., Гутенев М. С., Викторовский И. В., Байдаков Л. А.— Журн.
прикл. хим., 1975, т. 2, с. 281.
336. Schultz-Sellack С., Ann. Phys. Chem., 139, 182 (1970).
337. Господинов Г. Г., Пашинкин А. С.— Изв. АН СССР, неорг. матер., 1974,
т. 10, с. 2074.
338. Bohac Р., Della Casa A., Gaumann A., Krist. u. Tcchn., 9, 237 (1974).
339. Mullen D. J. E., Nowacki W., Z. Kristallogr., 136, 48 (1972).
340. Jonker W. P., Z. anorg. allg. Chem., 62; 89 (1909).
341. Hruby A., J. Non-Cryst. Solids, 28, 139 (1978)’.
342. Ito T., Morimoto N., Sadanaga R., Acta crystallogr., 5, 775 (1952).
343. Maruno S., Jap. J. Appl. Phys., 7, 1434 (19b8).
344. Strathdee B. A., Pidgeon L. M., Canad. Mining Met. Bull., 54, 883 (1961).
345. Street С. B., J. Inorg. Nucl. Chem., 32, 3769 (1970).
346. Kutoglu A., Z. anorg. allg. Chem., 419, 176 (1976).
347. Whitfield H. J., J. Chem. Soc. (A), 1970, 1800; J. Chem. Soc. Dalton
Trans., 1973, 1737.
348. Leiva A.-M., Fluck E., Muller H., Wallenwein G., Z. anorg. allg. Chem.,
409, 215 (1974).
349. Whitfield H. J., J. Chem. Soc. Dalton Trans., 1973, 1740.
350. Diemann E., Rev. Chim. Miner., 16, 237 (1979).
351. Tsuchihashi S„ Kawamoto Y., Adachi K-, J. Ceram. Assoc. Japan, 76, 101
(1968); J. Non-Cryst. Solids, 5, 286 (1971).
352. Tsuchihashi S., Kawamoto У., Yogyo-Kyokai-Shi, 77, 35 (1969).
Аморфные и стеклообразные системы
397
353. Lucovsky G., Galeener F. L., Geils R. H., Keezer R. C., Proc. Conf. Struc-
ture of Non-Crystalline Materials, Taylor and Francis, London, 1977,
p. 127.
354. Ward A. T., Adv. in Chem., 110, 163 (1972).
355. Ewen P. J. S., Sik M. J., Owen A. E., Proc. Conf. Structure of Non-Crys-
talline Materials, Taylor and Francis, London, 1977, p. 231; Solid Stale
Commun., 33, 1067 (1980).
356. Порай-Кошиц E. А., Вайполин A. A.— Физ. тв. тела, 1963, т. 5, с. 246.
357. Grigorovici R., The Structure of Amorphous Semiconductors. Electronic
and Structural Properties of Amorphous Semiconductors, P. G. Le Comber,
J. Mort (Eds.), Academic Press, London, 1973, p. 219.
358. Виноградова Г. 3., Дембовский С. А., Кузьмина Т. IL, Чернов А. П.,—
ЖНХ, 1967, т. 12, с. 3240.
359. Немилое С. В.— Физ. хим. стекла, 1979, т. 5, с. 398.
360. Мюллер Р. JI., Борисова 3. У., Ильинская О. В,—Жури. прикл. химии,
1971, т. 34, с. 664.
361. Maruno S., Noda М., J. Non-Cryst. Solids, 7, 1 (1972).
362. Bertoluzza A., Fagnano С., Monti Р., Semerano G., J. Non-Cryst. Solids,
29, 49 (1978).
363. Blachnik R., Hoppe A., Wicket U., Z. anorg. allg. Chem., 463, 78 (1980).
364. Leadbetter A. J., Apling A. J., J. Non-Crvst. Solids, 15, 250 (1974).
365. Ewen P. J. S., Owen A. E., J. Non-Cryst. Solids, 35, 36, 1191 (1980).
366. Bosch M. A., Shah J., Phys. Lett, 74 A, 446 (1979); Solid State Commun.,
31, 769 (1979).
367. Bobb L. C., Kramer K., Byer H. H., J. Non-Cryst. Solids, 21, 441 (1976).
368. Apling A. J., Leadbetter A. J., Proc. 5th Int. Conf. Amorph. Liquid Semi-
cond., Garmisch-Partenkirchen 1973, Taylor-Francis, London, 1974, p. 457.
369. de Neufville J. P., Moss S. G., Ovshinsky S. R., J. Non-Cryst. Solids, 13,
191 (1974).
370. Lu C. S., Donohue J., J. Amer. Chem. Soc., 66, 818 (1944).
371. Mills К. C., Thermodynamic Data for Inorganic Sulphides, Selenides and
Tellurides, Butterworth, London, 1974, p. 131.
372. Solin S. A., Papatheodorou G. N., Phys. Rev., В 15, 2084 (1977).
373. Ehrt D„ Erdmann C., Vogel W., Z. Chem., 23, 37 (1983).
374. Janai M., Rudman P. S., Mandelbauin A., J. Non-Cryst. Solids, 27, 67
(1978).
375. Apling A. J., Leadbetter A. J., Wright A. C., J. Non-Cryst. Solids, 23,
369 (1977).
376. Daniel M. F., Leadbetter A. J., Wright A. C., Sinclair R. N., J. Non-Cryst.
Solids, 32, 271 (1979).
377. Дембовский С. А., Дужная H. П.— ЖНХ, 1964, t. 9, c. 389, 660.
378. Чернов А. П., Дембовский С. А., Чистов С. Ф.— Изв. АН СССР, неорг.
матер., 1968, т. 4, с. 1658.
379. Вайполин А. А.— Кристаллография, 1965, т. 10, с. 596.
380. Kitaci М., Asakura N., Yamada S., Jap. J. Appl. Phys., 8, 499 (1968)._
381. Renninger A. L., Averbach B. L., Acta Crystaiiogr., В 29, 1583 (19/3).
382. Smith B. A., Cowlam N., Shamah A. M., Phil. Mag., В 39, 111 (1979).
383. Goldstein P., Paton A., Acta Crystallogr., В 30, 915 (1974).
384. Bastow T. J., Whitefield H. J., J. Chem. Soc. Dalton Trans., 1977, 959.
385. Feltz A., Aust H., J. Non-Cryst. Solids, 55, 179 (1983).
386. Тимофеева H. В., Виноградова Г. 3., Фекличев E. M., Семененко Т. В.,
Дембовский С. А., Калашников Я. А.— ЖНХ, 1970, т. 15, с. 3391.
387. Thornburg D. D., Johnson R. I., J. Non-Cryst. Solids, 17, 2 (1975).
388. Sagara Y., Uemura O., Okuyama S., Satow T., Phys. Status Solidi (a),
31, К 33 (1975).
389. Uemura O., Sagara Y., Muno D., Satow T., J. Non-Cryst. Solids, 30, 155
(1978).
390. Liang K. S., J. Non-Cryst. Solids, 18, 197 (1975).
391. Aral T., Koniya S., Kudo K., J. Non-Cryst. Solids, 18, 289 (1975).
398 Глава 3
392. Zallen R., Slate M. L., Phys. Rev., В 9, 1627 (1974).
393. Vasko A., Kieslich K., Proc. 7th Int. Conf. Amorph. Liquid. Semicond.,
Edinburgh, 1977, University Edinburgh, p. 105.
394. Owen A. E., Spear W. E., Phys. Chem. Glass, 17, 174 (1976).
395. Fisher F. D., Marshall J. M., Owen A. E., Phil. Mag., 33, 261 (1976).
396. Hulls K., McMillan P. W., J. Non-Cryst. Solids, 15, 557 (1974).
397. Althaus H. L., Weiser G., Nagel S., Phvs. Status Solidi (b), 87, 117
(1978).
398. Pfister G., Taylor P. C., J. Non-Cryst. Solids, 35, 36, 793 (1980).
399. Melnyk A. R., J. Non-Cryst. Solids, 35, 36, 837 (1980).
400. Leadbetter A. J., Apling A. J., Daniel M. F., J. Non-Cryst. Solids, 21,
47 (1976).
401. Eifert J. R„ Peretti E. A., J. Mater. Sci., 3, 293 (1968).
402. Spenser Ch. IT., J. Metals, AIME Trans., 203, 144 (1955).
403. Carron G. J., Acta Crystallogr., 16, 338 (1963).
404. Eusner P. R., Durden L. R., Slack L. IL, J. Amer. Ceram. Soc., 55, 43
(1972).
405. Linke D., Hey H., Z. Chem., 16, 413 (1976).
406. Hruby A., Stourac L., Mater. Res. Bull., 6, 465 (1971).
407. Seager С. H„ Quinn R. K-, J. Non-Cryst. Solids, 17, 386 (1975).
408. Cornet J., Rossier D., J. Non-Cryst Solids, 12, 85 (1973).
409. Fitzpatrick J. R., Maghrabi C., Phys. Chem. Glasses, 12, 105 (1971).
410. Uemura O.. Sagara Y., Tsushima M., Kamikawa T., Satow T., J. Non-
Cryst. Solids, 33, 71 (1979).
411. Johnson R. T., Quinn R. K-, Borders J. A., J. Non-Crys. Solids, 15, 289
(1974).
412. Platakis N. S., IBM Technical Report 02.769 (1977).
413. Stourac L„ Abraham A., Hruby A., Zavetova M., J. Non-Cryst. Solids,
8—10, 353 (1972).
414. Brasen D., J. Non-Cryst. Solids, 11, 131 (1972).
415. Northrop D. A., Mat. Res. Bull., 7, 147 (1972).
416. Weiser K., Brodsky M. H., Phys. Rev., В 1, 791 (1970).
417. Rockstad H. K-, J. Non-Cryst. Solids, 2, 192 (1970).
418. Hauser J Di Salvo F. J., Hutton R. S., Phil. Mag., 35, 1557 (1977).
419. Jaeger F. M., van Klooster H. S., Z. anorg. Chem., 78, 246 (1912).
420. Орлова Г. M., Кожина И. И., Короленко В. Г.— Вести. ЛГУ, сер. физ.
и хим., 1973, т. 4, с. 90.
421. Wood С., Н игу ch Z., Shaffer J. С., J. Non-Cryst. Solids, 8—10, 209 (1972);
11, 153 (1972).
422. Satow T., Uemura 0., Akaike S., J. Non-Cryst. Solids, 29, 215 (1978).
423. Brasen D., J. Non-Cryst. Solids, 15, 395 (1974).
424. Мелех Б. T., Маслова С. В., Аблова M. С., Жукова T. Б., Андреева A. A.—
Физ. хим. стекла, 1976, т. 2, с. 189.
425. Johnson R. Е„ Миап A., J. Amer. Ceram. Soc., 51, 430 (1968).
426. Hilbner К., Phys. Status Solidi (a), 40, 487 (1977).
427. Schnockel H., Angew. Chem., 90, 638 (1978).
428. Baur W. H., Acta Crystallogr., В 34, 1751 (1978).
429. Bruckner R., J. Non-Cryst. Solids, 5, 281 (1971).
430. Deal В. E., Grove A. S., J. Appl. Phys., 36, 3770 (1965).
431. Agajanian A. H., Solid-State Techn., 20, 36 (1977).
432. Schnable G. L., Kern W., et al., J. Electrochein. Soc., 122, 1092 (1975).
433. Shackelford J. F., Masaryk J. S., J. Non-Cryst. Solids, 30, 127 (1978).
434. Bando Y., Jshizuka K., J. Non-Cryst. Solids, 33, 375 (1979).
435. Hilbner K., Lehmann A., Phys. Status Solidi (a), 46, 451 (1978).
436. Mikkelsen J. C., Galeener F. L., J. Non-Cryst. Solids, 37, 71 (1980).
437. Griscom D. L., J. Non-Cryst. Solids, 24, 155 (1977).
438. Greaves G. N., Phil. Mag., В 37, 447 (1978).
439. Gee С. M., Kastner M., Phys. Rev. Lett., 42, 1765 (1979).
440. Greaves G. N., J. Non-Cryst. Solids, 32, 295 (1979).
Аморфные и стеклообразные системы
399
441. Lucovsky G., Phil. Mag., 39, 531 (1979).
442. Lucovsky G., J. Non-Cryst. Solids, 35, 36, 825 (1980).
443. Lucovsky G., Phil. Mag., 41, 457 (1980).
444. Revesz A. G., Phys. Status Solidi (a), 57, 235, 657 (1980); 58, 107
(1980).
445. Williams E. L., J. Amer. Ceram. Soc., 48, 190 (1965).
446. Doremus R. H., Proc. Xlth Int. Congr. Glass, Prog., 1977, Vol. I, p. 261.
447. Hartwig С. M., Rahn L. A., J. Chem. Phys., 67, 4260 (1977).
448. Van der Steen G. H. A. M., van den Boom H., J. Non-Cryst. Solids, 23,
279 (1977).
449. Shackelford F., J. Non-Cryst. Solids, 21, 55 (1976).
450. Van der Steen G. H. A. M., Non-Cryst. Solids, 4th Int. Conf. The Physics
of Non-Cryst. Solids, 1976, G. H. Frischat (Ed.), Trans. Tech. Publica-
tions, 1977, p. 486.
451. Stapelbroek M., Griscom D. L., Friebele E. L, Sigel G. H., J. Non-Cryst.
Solids, 32, 313 (1979).
452. Vukcevich M. R., J. Non-Cryst. Solids, 11, 25 (1972).
453. Hsieh S. Y., Montrose C. J., Macedo P. B., J. Non-Cryst. Solids, 6, 37
(1971).
454. Revesz A. G., J. Non-Cryst. Solids, 7, 77 (1972).
455. Gaskell P. H, Johnson D. W., J. Non-Cryst. Solids, 20, 153, 171 (1976).
456. Philipp H. R., J. Non-Cryst. Solids, 8—10, 627 (1972).
457. Hollinger G., Tousset J., Due T. M., AlP-Conf. Proc., 20, 102 (1974).
458. Hiibner K-, Phys. Status Solidi (a), 52, 541 (1979).
459. Temkin R. J., J. Non-Cryst. Solids, 17, 215 (1975).
460. George C. F., D’Antonio P., J. Non-Cryst. Solids, 34, 323 (1979).
461. Fitzer E., Hegen D., Angew. Chem., 91, 316 (1979).
462. Aiyma T., Fukunaga T., Nichara K., Hirai T., Suzuki K., J. Non-Cryst.
Solids, 33, 131 (1979).
463. Kirk С. T., J. Appl. Phys., 50, 4190 (1979).
464. Emons H. H., Hellmold P., Mohlenrich S., Z. Chem., 15, 249 (1975).
465. Vennik J., Callaerts R., C. R. Acad. Sci., 260, 496 (1965).
466. Bailey L. G., J. Phys. Chem. Solids, 27, 1593 (1966).
467. Brebrick R. F., J. Chem. Phys., 49, 2584 (1968).
468. Ploog K-, Stetter W., Nowitzki A., Schonherr E., Mater. Res. Bull., 11,
1147 (1976).
469. Zwick U., Rieder К- H., Z. Physik, В 25, 319 (1976).
470. Dittmar G., Acta Crystallogr., В 34, 2390 (1978).
471. Feltz A., Maul W., Schonfeld L, Z. anorg. allg. Chem., 396, 103 (1973).
472. Cornet J., Proc. Vlth Int. Conf. Amorph. Liquid Semicond., Structure and
Properties of Non-Crystalline Semiconductors, Leningrad, 1976, Nauka,
p. 72.
473. Andreev A. A., Ablova M. S., Melek В. T., Nasredinov F. S., Seregin P. P.,
Turaev E., Proc. 7th Int. Conf. Amorph. Liquid Semicond., Edinburgh 1977,
University Edinburgh, p. 44.
474. Savage J. A., J. Non-Cryst. Solids, 11, 121 (1972).
475. Bartsch G. E. A., Bromme H., Just T., J. Non-Cryst. Solids, 18, 65 (1975).
476. Newns G. R., Hanks R., J. Chem. Soc., (A), 1966, 954.
477. Vergano P. J., Uhlmann D. R., Phys. Chem. Glass, 11, 30, 39 (1970).
478. Lcadbelter A. J., Wright A. C., J. Non-Cryst. Solids, 7, 37 (1972).
479. Bondot P. P., Acta Crystallogr., A 30, 470 (1974).
480. Chen В. T.-K, Su G.-J., Phys. Chem. Glass, 12, 33 (1971).
481. Sharma S. K., Virgo D., Kushiro L, J. Non-Cryst. Solids, 33, 235 (1979).
482. Bohm H., Phys. Chem. Glass, 1 1, 177 (1970).
483. Bohm H. F., J. Non-Cryst. Solids, 7, 192 (1972).
484. de Neufville J. P., Drummond С. H., Turnbull D., Phys. Chem. Glass, 11,
186 (1970).
485. de Neufville J. P., Turnbull D., Discuss Farad. Soc., 50, 182 (1970).
486. Dtummond С. H., Turnbull D., J. Non-Cryst. Solids, 17, 19 (1975).
400 Глава 3
487. Winkler Cl., Ber. Deutsch. Chem. Ges., 19, 210 (1886).
488. Viaene W., Moh G. H., Neucs Jahrb. Mineral., Monatsh., 283, 1970; Neucs
Jahrb. Mineral. Abh., 119, 113 (1973).
489. Ditlmar G., Schafer IL, Acta Crystallogr., В 31, 2060 (1975); В 32, 1188
(1976).
490. Zachariasen W. H., J. Chem. Phys., 4, 618 (1936).
491. Prewitt С. T., Young H. S., Science, 149, 535 (1965).
492. Shimada M„ Dachille F., Inorg. Chem., 16, 2094 (1977).
493. Zachariasen W. H., Phys. Rev., 40, 917 (1932).
494. Bissert G., Hesse K.-F., Acta Crystallogr., В 34, 1322 (1978).
495. Kawamoto Y., Thuchihashi S., J. Amer. Ceram. Soc., 52, 626 (1969).
496. Kawamoto Y., Thuchihashi S., J. Amer. Ceram. Soc., 54, 131, 526 (1971).
497. Ota R., Kunugi M., J. Phys. Chem. Solids, 38, 9 (1977).
498. Voigt B., Z. anorg. allg. Chem., 447, 153 (1978).
499. Lucovsky G., de h’eufville J. P., Galeener F. L., Phys. Rev., В 9, 1591
(1974).
500. Feltz A., Pohle AL, Herms G„ Stell H., J. Non-Cryst. Solids, в печати.
501. Nemanich R. J., Solin S. A., Solid State Commun., 21, 273 (1977).
502. Bridenbaugh P. M., Espinosa G. P., Griffiths J. E., Phillips J. C., Re-
meika J. P., Phys. Rev., В 20, 4140 (1979).
503. Aral K-, Namikawa H., Solid State Commun., 13, 1167 (1973).
504. Voigt B., Feltz A., Schroder B., Z. Chem., 18, 77 (1978).
505. Cerny V., Frumar M., J. Non-Cryst. Solids, 33, 23 (1979).
506. Cervinka L., Hruby A., Proc. 5th Int. Conf. Amorph. Liquid Sernicond.,
Garmisch-Partenkirchen 1973, Taylor-Francis, London, 1974, p. 431.
507. Lucovsky G., Galeener F. L., Keezer R. C., Geils R. H., Six H. A., Phys.
Rev., В 10, 5134 (1974).
508. Feltz A., Voigt B., Schlenzig E., Proc. 5th Int. Conf. Amorph. Liquid Semi-
cond., Garmisch-Partenkirchen 1973, Taylor-Francis, London, 1974, p. 261.
509. Feltz A., Voigt B., Burckhardt W., Senf L., Leonhardt G., Proc. Vlth Int.
Conf. Amorph. Liquid Sernicond. Structure and Properties of Non-Cryst.
Semiconductors, Leningrad, 1976, Nauka, p. 88.
510. Feltz A., Burkhardt W., Senf L., Voigt B., Zickmiiller K-, Z. anorg. allg.
Chem., 435, 172 (1977).
511. Hahn H., Frank G., Z. anorg. allg. Chem., 278, 340 (1955).
512. Kuhn A., Chevy A., Acta Crystallogr., В 32, 983 (1976).
513. Lucovsky G., Nemanich R. J., Galeener F. L., Proc. 7th Conf. Amorph.
Liquid Sernicond., Edinburgh 1977, University Edinburgh, p. 130.
514. Feltz A., Zickmiiller K., Pfaff G„ Proc. 7th Int. Conf. Amorph. Liquid
Sernicond. Edinburgh 1977, University Edinburgh, p. 125.
515. Kawamoto Y., Tsuchihashi S., Yogyo-Kyokai-Shi, 77, 328 (1969); 79 264
(1971).
516. Platakis N. S., Sadagopan V., Gatos H. C., J. Electrochem. Soc., 116,
1436 (1969).
517. Spandau H., Klanberg F„ Z. anorg. allg. Chem., 295, 29i (1958).
518. Stephan O., Dissertation, Braunschweig, 1971.
519. Лю-Цюнь-Хуа, Пашинкин A. C.t Новоселова A. B.— ДАН СССР, 1962,
т. 146, с. 1092.
520. Дембовский С. А., Виноградова Г. 3., Пашинкин А. С.— ЖНХ, 1965,
т. 10, с. 1657.
521. Виноградова Г. 3., Дембовский С. А., Сивкова Н. Б.— ЖНХ, 1968, т. 13,
с. 2029.
522. Карбинов С. Г., Зломанов В. П., Новоселова А. В.— Вести. МГУ, сер.,
9, химия, 1968, т. 9, с. 96.
523. Ross L., Bourgon М„ Canad. J. Chem., 47, 2555 (1969).
524. Quenez P., Khodadad P., Geolin R., Bull. Soc. Chim. France, 1, 117 (1972).
525. Dittmar G„ Schafer H., Acta Crystallogr., В 32, 2726 (1976).
526. Kyriakov D. S., Karakostas T. S., Economou N. A., J. Cryst. Growth., 35,
223 (1976).
Аморфные и стеклообразные системы 401
527. Wiedemeier Н., v. Schnering Н. G., Z. Kristallogr., 148, 295 (1978).
528. Wiedemeier II., Sietners P. A., Z. anorg. allg. Chem., 411, 90 (1974).
529. Feltz A., Biittner H. 1., Lippmann F. I., Maul W7., J. Non-Cryst. Solids,
8—10, 64 (1972).
530. Feltz A., Lippmann F. I., Z. anorg. allg. Chem., 398, 157 (1973).
531. Voigt В., неопубликованные данные.
532. Аветикян Г. Б., Байдаков Л. А., Страхов 3. П.— Изв. АН СССР, неорг.
матер., 1969, т. 5, с. 1667.
533. Пазин А. В., Образцов А. А., Борисова 3. У.— Изв. АН СССР, неорг.
матер., 1972, т. 8, с. 247.
534. Satow Т., Uemura О., Sagara У., J. Jap. Inst. Metals, 37, 1348 (1973).
535. Полтавиев Ю. Г., Познякова В. М.— Изв. АН СССР, неорг. матер., 1973,
т. 9, с. 766.
536. Uemura О., Sagara У., Satow Т., Phys. Status Solidi (а), 32, 91 (1975).
537. Giridhar A., Narasimhan Р. S. L., Mahadevan S., J. Non-Cryst. Solids,
37, 165 (1980).
538. Kumagai N., Shirafuji I., Inuishi Y., J. Phys. Soc. Jap., 42, 1261 (1977).
539. Sen P. N., Thorpe M. F., Phys. Rev., В 15, 4030 (1977).
540. Nemanich R. I., Phys. Rev., В 16, 1655 (1977).
541. Popovic Z. V., Nikolic P. M., Solid State Commun., 27, 561 (1978).
542. Ball G. J., Chamberlain J. M., J. Non-Cryst. Solids, 29, 239 (1978).
543. Tronc P., Bensoussan M., Brenac A., Sebenne C., Phvs. Rev., В 8, 5947
(1973); Solid State Commun., 24, 79 (1977).
544. Azoulay R.. Thibierge II., Brenac A., J. Non-Cryst. Solids, 18, 33 (1975).
545. Ola R., Yamate T., Soga N., Kunugi M., J. Non-Cryst. Solids, 29, 67
(1978).
546. Cervinka L., Hruby A., J. Non-Cryst. Solids, 48, 231 (1982).
547. Laugier A., Chaussemy G., Fornazero J., J. Non-Cryst. Solids, 23, 419
(1977).
548. Chaussemy G., Fornazero J., Laugier A., Rev. Phys. Appl., 12, 687 (1977).
549. Глазов В. M„ Ситулина О. В.— ДАН СССР, 1969, т. 187, с. 799.
550. Ruska I., Thum И., J. Non-Cryst. Solids, 22, 277 (1976).
551. Kuhn A., Chevy A., Chevalier R., Phys. Status Solidi (a), 31, 469 (1975).
552. Rowland S. C., Narasimhan S., Bienenstock A., J. Appl. Phys., 43, 2741
(1972).
553. Kawamura H., Matsumura M., Solid State Commun., 32, 83 (1979).
554. Лю-Цюнь-Хуа, Пашинкин А. С., Новоселова A. B.— ЖНХ, 1962, t. 7,
c 2159
555. Irene E. A., Dissertation-Abstracts, Physic. Chem., 5753-B, В 33 (12, Pt. 1),
753 (1975)
556. Barrow R. F„ Jewons W., Proc. Phys. Soc., 52, 534 (1940).
557. Irene E. A., Wiedemeier H., Z. anorg. allg. Chem., 411, 182 (1975).
558. Uemura O., Sagara Y., Satow T., Phys. Status Solidi (a), 26, 99 (1974).
559. Chamberlain J. M., Sirbegovic S. S., Nikolic P. M., J. Phys. C: Solid State
Phys., 7, L 150 (1974).
560. Betts F„ Bienenstock A., Ovshinsky S. R., J. Non-Cryst. Solids, 4, 554
(1970).
561. Bienenstock A., J. Non-Cryst. Solids, 11, 447 (1973).
562. Malaurent J. C., Dixmier J., J. Non-Cryst. Solids, 35, 36, 1227 (1980).
563. Егорова E. А., Кокорина В. Ф.— Оптико-механ. промышл., 1963, т. 1,
с. 33.
564. Bensoussan М., Rev. Physique Appl., 12, 753 (1977).
565. Klemm W., Frischmuth G., Z. anorg. allg. Chem., 218, 249 (1934).
566. Legendre B., Souleau С., C. R. Acad. Sci., Ser. C, 1977, 315.
567. Schubert K„ Fricke H., Z. Metallkunde, 44, 457 (1953); Z. Naturf., 69,
781 (1951).
568. Карбанов С. Г., Зломанов В. П., Новоселова А. В.— Изв. АН СССР,
неорг. матер., 1969, т. 5, с .1171.
569. Cornet I., Ann. Chim., 10, 239 (1975).
26 Заказ № 413
402 Глава 3
570. Andreev A. A., Ablova М. S., Melek В. Т., Nasredinov F. S., Seregin Р. Р.,
Turaev Е., Proc. 7th. Int. Conf. Amorph. Liquid Semicond., Edinburgh
1975, University Edinburgh, p. 44.
571. Feltz A., Burckhardt IF., Z. anorg. allg. Chem., 461, 35 (1980).
572. Bienenstock A., Betts F., Ovshinsky S. R., J. Non-Cryst. Solids, 2, 347
(1970).
573. Betts F., Bienenstock A., Bates C. W., J. Non-Cryst. Solids, 8—10, 364
(1972).
574. Betts F., Biennenstock A., Keating D. T., de Neufville J. P., J. Non-Cryst.
Solids, 7, 417 (1972).
575. Nicotera E., Corchia M., De Giorgi G., Villa F., Antonini M., J. Non-Cryst.
Solids, 11, 417 (1973).
576. Nakamura Y., Numata M., Hoshino H., Shimogi M., J. Non-Cryst. Solids,
17, 259 (1975).
577. Moore A. G., Maghrabi C., Parker J. M., J. Mater. Science, 13, 1127
(1978).
578. de Neufville A., Gerard P., Devenyi J., J. Non-Cryst. Solids, 22, 77 (1976).
579. Popovic Z. V., Nikolic P. M., Solid-State Commun., 27, 561 (1978).
580. Dove D. B., Chang J., Molnar B., J. Non-Cryst. Solids, 8—10, 376 (1972).
581. Fisher G. B., Tauc J., Proc. 5th Int. Conf. Amorph. Liquid Semicond.,
Garmisch-Partenkirchen 1973, Taylor-Francis, London, 1974, p. 1259.
582. Dittmar G., Z. anorg. allg. Chem., 453, 68 (1978).
583. Pickatd S. J., Sharma Y. P., de Neufville J. P., J. Non-Cryst. Solids, 34,
183 (1979).
584. Fisher G. B., Lindau I., Orlovski B. A., Spicer W. E., Verhelle Y., Proc.
5th Int. Conf. Amorph. Liquid Semicond., Garmisch-Partenkirchen 1973,
Taylor-Francis, London, 1974, p. 621.
585. Ribbihg C. G., Kallne E., Proc. 5th Int. Conf. Amorph. Liquid Semicond.,
Garmisch-Partcnkiichen 1973, Taylor-Francis, London, 1974, p. 823.
586. Baldwin С. M., Mackenzie J. D., J. Non-Cryst. Solids, 31, 441 (1979); 40,
135 (1980).
587. Cline C. F., Weber M. J., Proc. I Int. Otto-Schott-Kolloquium, Wiss.
Zeitschr. der Friedrich-Schiller-Universitat, Mathemat.-Naturwissenschaftl.
Reihe, 28, 351 (1979).
588. Zarzycki J., Phys. Chem. Glass., 12, 97 (1971).
589. Leadbelter A. L, Wright A. C., J. Non-Cryst. Solids, 7, 156 (1972).
590. Watanabe /., Ishikawa M„ Shimizu T., J. Phys. Soc. Jap., 45, 1603 (1978).
591. Burbank B. R., Bensey F. N., U. S. Atomic Energy Commun., K-1280
(1956).
592. Lecoq A., Poulain M., J. Non-Cryst. Solids, 34, 101 (1979).
593. Poulain M., Lucas J., Brun P., Mat. Res. Bull., 10, 243 (1975).
594. Lucas J., Chanthahashini M., Poulain M., J. Non-Cryst. Solids, 27, 273
(1978).
595. Diemann E., Z. anorg. allg. Chem., 432, 127 (1977).
596. Liang K. S., de Neufville J. P., Jacobson A. J., Chianelli R. R., J Non-
Cryst. Solids, 35, 36, 1249 (1980).
597. Furnseth S., Brattas L., Kjekshus A., Acta Chem. Scand., A 29, 623 (1975).
598. Jacobson A. J., Chianelli R. R., Rich S. M., Whittingham M. S., Mat. Res.
Bull., 14, 1437 (1979).
599. Diemann E., Muller A., Z. anorg. allg. Chem., 444, 181 (1978).
600. Passaretti J. D., Collins R. C., Wold A., Mat. Res. Bull., 14, 1167 (1979).
601. Diemann E., Z. anorg. allg. Chem., 461, 201 (1980).
602. Evstropjev K. S., Petrovskij G. T., Chalilev V. D., IX Int. Glaskongr.,
Versailles 1971, Sammelband, p. 485.
603. Chalilev V. D., Wasylak J. P., Igitjanjan J. G., Oganesjav P. H., Pego-
sjan M. A., XI Int. Glaskongr. Prag, 1977, Vol. I, 133.
604. Heidenreich. E., Kittel Th., Gerth K., Ehrt D., Burger H., Vogel W'., DDR-
WP C03c, 219 030.
605. Schiwy W., Krebs B., Angew. Chem., 87, 451 (1975).
Аморфные и стеклообразные системы
403
606. Rehfeld A., Katzschmann R., Silikattechnik, 29, 298 (1978).
607. Hanaka Т., Soga N., Kunugi M., J. Non-Cryst. Solids, 21, 65 (1978).
608. Ahmed A. A., El-Shamy T. M., Sharaf N. A., J. Non-Cryst. Solids, 33,
159 (1976).
609. Шило В. П., Коломиец Б. Т.— Изв. АН СССР, сер. хим., 1964, т. 28,
с. 1285.
610. Коломиец Б. Т. Стеклообразное состояние.—Л.: Наука, 1971, с. 82.
611. Minami Т., Fujikawa N., Hattori М., Tanaka М., Yogyo-Kyokai-Sci, 82,
597 (1974).
612. Minami Т., Hibino М., Tanaka М., J. Non-Cryst. Solids, 15, 141 (1974),
613. Deeg W., Hobashy M., Loutfi R. D., Sprechssal, 100, 757 (1967), 5070.
614. Pearson A. D., N orthover W. R., Dewaid J. F., Peck W. F., Adv. Glass
Techno!. VI. Congr. Glass, Washington D. C., 1962, Plenum Press, New
York, 1962, p. 357.
615. Suzuki S., Sakamura H., Imaoka M., Yogyo-Kyokai-Shi, 79, 301 (1971).
616. Дембовский С. А., Кириленко В. В., Буслаев Ю. А.— Изв. АН СССР,
неорг. матер., 1971, т. 7, с. 328.
617. Munir Z. A., Fuke L. М., Kay Е., J. Non-Cryst. Solids, 12, 435 (1973).
618. Дембовский С. А., Попова Н. 7/.—Изв. АН СССР, неорг. .матер., 1970,
т. 6, с. 138.
619. Feltz А., ВйИпег Z. Chem., 12, 393 (1972).
620. Quinn R. К., Johnson R. T., J. Non-Cryst. Solids, 7, 53 (1972).
621. Eaton D. L., J. Amer. Ceram. Soc., 47, 554 (1964).
622. Рязанцев A. A., Bapexa Л. M., Позукин Б. А., Ляховицкая В. А., Ново-
селова A. В.— Изв. АН СССР, неорг. матер., 1969, т. 5, с. 1206.
623. Туряница И. Д„ Коперлекс Б. М.— Изв. АН СССР, неорг. матер. 1973,
т. 9, с. 851.
624. Turjanitsa I. D., Miholinec I. М„ Koperles В. М., Kopinets I. F., J. Non-
Cryst. Solids, 11, 173 (1972).
625. Montell У., Vincent H., Bull. Soc. Chim. Fr., 1975, 1025.
626. Cornet J., Schneider J., Non-Cryst. Solids, 4th Int. Conf. The Physics of
Non-Cryst. Solids 1976, G. H. Frischat (Ed.), Trans. Tech. Publications,
1977, p. 397.
627. Minami T., Yoshida A., Tanaka M., J. Non-Cryst. Solids, 7, 328 (1972).
628. Образцов А. А., Борисова 3. У.— Изв. АН СССР, неорг. матер., 1970,
т. 6, 1247, 1417.
629. Minami Т., Hattori Н., Tanaka М„ Yogyo-Kyokai-Shi, 78, 101 (1970).
630. Жуков Э. Г., Джапаридзе О. И., Дембовский С. А., Попова Н. П.— Изв.
АН СССР, неорг. матер., 1974, т. 10, с. 1886.
631. Хвороаенко А. С., Дембовский С. А.— ЖНХ, 1970, т. 15, с. 1705.
632. Доморяд И. А., Хижниченко Л. П.— Изв. АН СССР, сер. физ. мат., 1963,
т. 4, с. 99, т. 5, с. 87.
633. Доморяд И. А., Коломиец Б. Т.— Изв. АН СССР, неорг. матер., 1971,
т. 7, с. 1620.
634. Edmond J., Non-Cryst. Solids, 1, 39 (1968).
635. Бийдаков Л. А., Борисова 3. У., Ипатьева В. В.— Вести. ЛГУ, сер. физ.
хим., 1962, с. 90.
636. Owen А. Е., Robertson J. М., J. Non-Cryst. Solids, 2, 40 (1970).
637. Minami Т., Yoshida A., Tanaka М., Yogyo-Kyokai-Shi, 78, 299 (1970).
638. Hulls К-, Me Millan P. IF., J. Non-Cryst. Solids, 15, 357 (1974).
639. Mytilineou E., Roilos M.. Phil. Mag., 37, 387 (1978).
640. Mahadevan S., Rao K. J-, J. Non-Cryst. Solids, 34, 53 (1979).
641. Takahashi T., J. Non-Cryst. Solids, 34, 307 (1979).
642. Lucovsky G., Keezer R. C., Six H. A., Geils R. H., Proc. Vlth Int. Conf.
Amorph. Liquid Sernicond., Structure and Properties of Non-Cryst. Semi-
conductors, Leningrad 1976, Nauka, p. 296.
643. Maneglier-Lacordaire S., Rivet J., Khodadad P., Flahaut J., Bull. Soc.
Chim. Fr., 1973, 1930.
644. Brau M. пат. США, 3 371 211 (1968).
26*
404 Глава 3
645. Bordas S., Clavaguera N., Geli M„ Gasas- Vazquez J., Clavaguera-
Mora M. T., Thermal. Anal., Proc. 5th. ICTA Conf., Kyoto 1977, M. Chi-
haia (Ed.), Samyo Shoppan Boeki Co., Inc., Tokyo, 1977, p. 14.
646. Шелимова Л. E., Абрикосов H. X., Жданова В. В., Чижова С. Н.— Изв.
АН СССР, неорг. матер., 1966, т. 2, с. 2103.
647. Образцов А. А.— Изв. АН СССР, неорг. матер., 1972, т. 8, с. 374.
648. Bordas S., Geli М.. Casas-Vazquez J., Clavaguera N., Clavaguera-
Mora M. T., Rev. Phys. Appl., 12, 677 (1977).
649. Пазин А. В., Образцов А. А., Борисова 3. У.— Изв. AH СССР, нсорг.
матер., 1972, т. 8, с. 247.
650. Noda М., Maruno S., Yogyo-Kyokai-Shi, 82, 56 (1974).
651. Hilton A. R., Jones С. Е., Brau М., Infrared Physics, 4, 213 (1964).
652. Wieder J., Aronson S., J. Non-Cryst. Solids, 33, 405 (1979).
653. Sarrach D. J., de Neufville J. P., Haworth W. L., J. Non-Cryst. Solids,
27, 193 (1978).
654. Радауцан С. И., Маслянко P. А., Ляликова P. IO., Колоскова В. Г.—
Изв. АН СССР, неорг. матер., 1975, т. 11, с. 1508.
655. Виноградова Г. 3., Майсашвили Н. Г.— ЖНХ, 1979, т. 24, с. 1067.
656. Heyder F., Dissertation A, Friedrich-Schillcr-Universitat, Jena, 1978.
657. Ое К., Toyoshima У., Nagai Н., J. Non-Cryst. Solids, 20, 405 (1976).
658. Виноградова 3. Г., Дембовский С. А.— ЖНХ, 1971, т. 16, с. 2036.
659. Pachali К. Е., Ruska У., Thum IL, Inorg. Chem., 15, 991 (1976).
660. Мюллер Р. Л., Орлова Г. М., Тимофеева В. Н.— Вести. ЛГУ, 1962, т. 17,
с. 146.
661. Andreichin R., Nikiforova М., Skordeva Е., Yurukova L., Grigorovich R.,
Manila R., Popescu M., Vancu A., J. Non-Cryst. Solids, 20, 101 (1976).
662. Eberhardt G., Dissertation A, Fricdrich-Shieller-Universitat, Jena, 1979.
663. Айо Л. Г., Кокорина В. Ф.— Оптико-механ. промышл., 1961 (6). с. 48,
1963 (2), с. 36.
664. Немилое С. В.— Журн. приклад, хим., 1964, т. 37, с. 1699.
665. Savage J. A., Nielsen S„ Phys. Chem. Glass., 5, 82 (1964).
666. Krebs H„ Haisty R. W., J. Non-Cryst. Solids, 1, 427 (1969).
667. Hulliger F., Siegrist T., Mat. Res. Bull., 16, 1245 (1981).
668. Nemanich R. J., Solin S. A., Lucovsky G., Proc. Vlth Int. Conf. Amorph.
Liquid Semicond., Structure and Properties of Non-Crystalline Semicon-
ductors, Leningrad, 1976, Nauka, p. 518.
669. Lucovsky G., Nemanich R. J., Solin S. A., Keezer R. C., Solid State Com-
miin., 17, 1567 (1975).
670. Серегин П. П., Васильев Л. H., Пронкин А. А.— Изв. АН СССР, неорг.
матер., 1972, т. 8, с. 376.
671. Бахтияров А. П., Васильев Л. Н.— Изв. АН СССР, неорг. матер., 1975,
т. 11, с. 741.
672. Gichter М., Pilz W., Phys. Status Solidi (b), 93, 641 (1979), 94, К 81
(1979).
673. Krebs H„ Welte H., J. Solid State Chem., 2, 182 (1970).
674. Krebs IL, Ackermann F. Glatechn. Ber., 45, 213 (1972).
675. Полтавцев Ю. Г., Позднякова В. М.— Изв. АН СССР, неорг. матер.,
1974, т. 10, с. 1932.
676. Немилое С. В.— Ж. прикл. химии, 1964, т. 37, с. 1452, 1690.
677. Fischer М., Krebs Н., Glastechn. Вег., 47, 72 (1974).
678. Krebs Н., Ruska J., J. Non-Cryst. Solids, 16, 329 (1974).
679. Webber P. J., Savage J. A., J. Non-Crys. Solids, 20, 271 (1976).
680. Title U., Frischat G. H., Leers К. J., Non-Cryst. Solids, 4th Int. Conf.
The Physics of Ncn-Cryst. Solids 1976, G. H. Frischat (Ed.), Trans. Tech.
Publications, 1977, p. 631.
681. Linke D., BCckel L, Z. anorg. allg. Chem., 419, 97 (1976).
682. Борисова 3. У., Пазин А. В. Химия твердого тела.— Л.: ЛГУ, 1965. с. 98.
683. Brau М. J., Johnson R. Е., Patterson R. J., пат. США 3 360 649 (1967).
684. Linke D„ Heyder F., Z. anorg. allg. Chem., 425, 155 (1976).
Аморфные и стеклообразные системы
405
685. Haisty R. W., Krebs Н., Angew. Chem., 80, 999 (1968); J. Non-Cryst.
Solids, 1, 399 (1969).
686. Орлова Г. M., Кожина И. И., Короленко В. Г.— Вести. ЛГУ, сер. физ.
хим., 1973, № 4 (1), с. 90.
687. Linke D., Eberhardt G., Z. anorg. allg. Chem., 442, 263 (1978).
688. Frumar M„ Ticha H., Bures M., Koudelka L., Z. Chem., 15, 199 (1975).
689. Пазин А. В., Борисова 3. У.— Изв. АН СССР, неорг. матер., 1972, т. 8,
с. 242.
690. Frumar М., Koudelka L., Tomiska Р., Ticha Н., Z. Chem., 12, 25 (1972).
691. Savage J. A., Webber P. J., Pitt A. M., J. Mater. Sc., 13, 859 (1978).
692. Ticha H„ Tichy L., Frumar M., Phys. Status Solidi (a), 54, К 163 (1979).
693. Frumar M., Ticha FL, Klikorka J., Tomiska P., J. Non-Cryst. Solids, 13,
173 (1973).
694. Ticha H., Frumar M., J. Non-Cryst. Solids, 16, 110 (1974).
695. Пазин А. В., Борисова 3. У.— Изв. АН СССР, неорг. матер., 1969, т. 5,
с. 1903.
696. Борисова 3. У. Химия стеклообразных полупроводников.— Л.: ЛГУ, 1972,
с. 145.
697. Пазин А. В.— Изв. АП СССР, пеорг. матер., 1978, т. 14, с. 411.
698. Hilion A. R., Jones С. Е., Brau М., Infrared Physics, 6, 183 (1966).
699. Krebs И., Fischer Р., Discuss. Farad. Soc., 50, 35 (1970).
700. Savage J. A., Nielson S., Phys. Chem. Glass., 7, 56 (1966).
701. Панус В. P., Борисова 3. У.— Жури. прикл. .химии, 1966, т. 39, с. 987;
1967, т. 40, с. 998.
702. Панус В. Р — Вести. ЛГУ, сер. физ. и хим., 1969, т. 24, с. 135.
703. Seager С. Н., Emin D., Quinn R. К., Phys. Rev., В 8, 4746 (1973).
704. Panwar О. S., Radhakrishna Al., Srivastava К. K-, Lakshminarayan K. N.,
J. Non-Cryst. Solids, 33, 411 (1979).
705. Hilton A. R., Brau M., Infrared Physics, 3, 69 (1963).
706. Панус В. P., Борисова 3. У., Алексеева Т. Т.— Жури, прикл. химии,
1968, т. 41, с. 2759.
707. Nagels Р., Callaerts R., Denayer Al., Proc. 5th Int. Conf. Amorph. Liquid
Semicond, Garmisch-Partenkirchen 1973, Taylor-Francis, London, 1974,
p. 867.
708. Imaoka M., Yamazaki T., Seisan Kenkyn, 19, 261 (1967).
709. Plumat E. R„ J. Amer. Ceram. Soc., 51, 499 (1968).
710. Voigt B., Dissertation A, Friedrich-Schiller-Universitat, Jena, 1974.
711. Lozach A. M., Barnier S., Guillard M., Besan^on P., Flahaut J., Ann.
Chim., 9, 127 (1974).
712. Hilton A. R., Texas-Instruments-Report 08-67-18 (1967).
713. Hilton A. R., пат. США, 3 511 672 (1970).
714. Feltz A., Burckhardt W., Senf L., Kunzel B., Proc. VI Int. Conf. Amorph.
Liquid Semicond., Structure and Properties of Non-Crystalline Semicon-
ductors, Leningrad, 1976, Nauka, p. 24. A1P Conf. Proc. Struct. Excit.
Amorph., Solids Williamsburg, Americ. Inst. Phy^ . 1976.
715. Barrau B., Kone A., Ribes M., Souquet J.-L., C. R. Acad. Sci., Ser. C,
287, 43 (1978).
716. Oliver-Fourcade J., Fhillipot E., Ribes M., Maurin .VI., Rev. Chim. Mine-
rale, 9, 757 (1972).
717. Krebs B., Pohl S., Z. Naturforsch., 26b, 853 (1971).
718. Muller A., Cyvin B. N., Cyvin S. J., Spectrochcm. Acta, 32 A, 67 (1976).
719. Ribes M., Barrau B., Souquet J. L., J. Non-Cryst. Solids, 38, 39, 271
(1980).
720. Feltz A., Holand M., неопубликованные данные.
721. Sommer H„ Hoppe R., Z. anorg. allg. Chem., 430, 199 (1977).
722. Eisenmann B., Schafer H., Z. anorg. allg. Chem., 456, 87 (1979).
723. Asahara У., Izumitani T., J. Non-Cryst. Solids, 11, 407 (1973); Jap. J
Appl. Phys., 11, 109 (1972).
406 Глава 3
724. Kolomiec В. Т., Rukljadev Т. Y., Silo V. Р., J. Non-Cryst. Solids, 5, 389
(1971).
725. Саван И., Кожина И. И., Орлова Г. М., Биндер М.— Изв. АН СССР,
несрг. матер., 1969, т. 5, с. 492.
726. Hunter S., Bienenstock A., Proc. VI Int. Conf. Amorph. Liquid Sernicond.,
Structure and Properties of Non-Crystalline Semiconductors, Leningrad,
1976, Nauka, p. 151; Proc. 5th Int. Conf. Amorph. Liquid Sernicond., Gar-
misch-Partenkirchen 1973, Taylor-Francis, London, 1974, p. 79.
727. Maruno S., Noda M., Yamada T., Yogyo-Kyokai-Shi, 81, 445 (1973).
728. Kawamoto Y., Agata M., Tsuchihashi S., Yogyo-Kyokai-Shi, 82, 46 (1974).
729. Kawamoto Y., Nagura N„ Tsuchihashi S., J. Amer. Ceram. Soc., 56, 189
(1973).
730. Feltz .4., Thieme Ch., Z. Chem., 14, 33 (1974).
731. Nagel A., Range K-L, Z. Naturforsch., 33b, 1461 (1978).
732. Kawamoto Y., Nishida M., J. Non-Cryst. Solids, 20, 393 (1976).
733. Kawamoto Y., Nagura N., Tsuchihashi S., J. Amer. Ceram. Soc., 57, 489
(1974).
734. Kabre S., Guittard M., Flahaut J., C. R. Acad. Sci., Ser. C, 278, 1043
(1974).
735. Cervinka L., Hruby A., J. Non-Cryst. Solids, 30, 191 (1978).
736. Tausend A., Wobig D., Z. Phys. Chem. (Frankfurt/M.), 96, 199 (1975).
737. Zirke J., Dromer C., Tausend A., Wobig D., J. Non-Cryst. Solids, 24,
283 (1977).
738. Cervinka L., Hruby A., J. Non-Cryst. Solids, 34, 275 (1979).
739. Mangalam M. J., Rao K. N., Rangarajan N., Siddiqui M. I. A., Suryana-
rayama С. V., Jap. J. Appl. Phys., 8, 1258 (1969).
740. Paesler M., Phys. Rev., В 13, 5578 (1976).
741. Ferrier R. P., Prado J. M., Anseau M. R., J. Non-Cryst. Solids, 8—10, 798
(1972).
742. Robertson J., Phil. Mag., В 39, 501 (1979).
743. Linke D., VIII. Arbeitstagung Elektronenmikroskopie, Berlin, 1975, Kurz-
referate Vortrage, S. 257.
744. Linke D., Gitter M„ Krug F., Z. anorg. allg. Chem., 444, 217 (1978).
745. Flaschen S. S., Pearson A. D., Northover W., J. Amer. Ceram. Soc., 43,
274 (1960).
746. Коломиец Б. T., Шило В. П.— Стекло и керамика, 1963, т. 20, с. 10.
747. Дембовский С. А.— Изв. АН СССР, пеорг. матер., 1968, т. 4, с. 1920;
Виноградова Г. 3., Дембовский С. А.— ЖНХ, 1970, т. 15, с. 1949.
748. Дембовский С. А., Кириленко В. В., Хворостенко А. С.— ЖНХ, 1969,
т. 14, с. 2561.
749. Strunk R., J. Non-Cryst. Solids, 12, 168 (1973).
750. Викторовский И. В., Байдаков Л. А.— Изв. АН СССР, неорг. матер.,
1971, т. 7, с. 508.
751. Nagels Р„ Callaerts R., Denayer М., de Coninck R., J. Non-Cryst. Solids,
4. 295 (1970).
752. Bishop S. G„ Taylor P. C., Mitchell D. L., J. Non-Cryst. Solids, 5, 351
(1971).
753. Julien-Pouzol P. M., Jaulmes S., Guittard M., Atapini F., Acta Crystallogr.,
В 35, 2848 (1979).
754. Feltz A., Krautwald A., Z. Chem., 19, 18 (1979).
755. Loireau-Lozac’h A.-M., Guittard M., Ann. Chim., 101, (1975).
756. Thiebault C, Guen L„ Eholie R., Flahaut J., Bull. Soc. Chim. France,
1975, 967.
757. Ollitraut-Fichet R., Eholie R., Rivet }., Flahaut J., Ann. Chim., 31 (1977).
758. Feltz A., Voigt B., Z. anorg. allg. Chemie, 403, 61 (1974).
759. Feltz A., Ludwig W„ Seiss R., Kristall u. Techn., 13, 405 (1978).
760. Fenner J., Moolz D., Naturwisscnschaft., 61, 127 (1974); Z. anorg. allg.
Chem., 427, 123 (1976).
Аморфные и стеклообразные системы
407
761. Ribes М., Fourcade J. О., Philippol Е., Maurin М., Acta Crystallogr., В 30,
1391 (1974).
762. Lamine М. М., Khodadad Р., С. R. Acad. Sci. Ser. С, 278, 243 (1974).
763. Feltz A., Burckhardt W., Senf L., Voigt B., Proc. Xlth Int. Congr. Glass,
Prag., 1977, Vol. 1, p. 389.
764. Iglesias J. E., Steinfink H., J. Solid State Chem., 6, 93 (1973).
765. Susa K., Steinfink H., 3, 75 (1971).
766. Feltz A., Ludwig W., Senf L., Simon C., Krist. u. Tcchn., 15, 895 (1980).
767. Rowland S. C., Ritland F., Haferburns D., Bienenstock A., Proc. 7th Int.
Conf. Amorph. Liquid Semicond., Edinburgh 1977, University Edinburgh,
p. 134.
768. Strickler D. W., Roy R., J. Mater. Sc., 6, 200 (1971).
769. Ши.ю В. П. Дисс. на соиск. ст. канд. хим. наук, ИОНХ АН СССР, М.,
1967.
770. Дойников Л. И., Борисова 3. У. Химия твердого тела.— Л.: ЛГУ, 1965,
с. 93.
771. Feltz A., Burckhardt W., J. Non-Cryst. Solids, 41, 301 (1980).
772. Kafalas J. A., Gatos H. C., Lavine M. C., Banus M. D., J. Phys. Chem.
Solids, 23, 1541 (1962).
773. Ефимов A. M., Кокорина В. Ф. Стеклообразное состояние.— Л.: Наука,
1965. с. 177.
774. Артамонова Г. И., Ботвинкин О. К.— ЖПХ, 1970, т. 15, с. 3388.
775. Mackowski J. М., Kumurdian Р., Touset J., Thin Solid Films, 34, 55
(1976).
776. Hollinger G„ Kumurdian P., Mackowski] J. M., Petrosa P., Porte L.r
Tran. Minh Due., J. Elektron. Spectr. Rel. Phen., 5, 237 (1974).
777. Green M., Radjy N. A., Phil. Mag., 39, 65 (1979).
778. Bordas S., Sasas-Vazqzez J., Clauaguera N., Clavaguera-Mora M. T.r
Thermochem. Acta, 28, 387 (1979).
779. Feltz A., Kley G., Linke L, J. Non-Cryst. Solids, 33, 299 (1979).
780. Dalven R., Infrared Physics, 9, 141 (1969).
781. Savage J. A., J. Mater. Sc., 7, 64 (1972).
782. Kumeda M„ Jinno Y., Suzuki M., Shimizu T., Jap. J. Appl. Phvs., 15, 201
(1976).
783. Suzuki M., Ohdaira H., Matsumi T., Kumeda M., Shimizu T., Jap. J. AppL
Phys., 16, 221 (1977).
784. Glassy Metals I, Topics in Applied Physics, Vol. 46, 11.-J. Guntherodt,
H. Beck (Eds.), Springer-Verlag Bcrlin/Heidelberg/New York, 1981.
785. Proc. IX, Int. Conf. Amorph. Liquid. Semicond., Grenoble, 1981.
786. Schalke W., Phil. Mag., В 43, 451 (1981).
787. Guttmann L., Phys. Review В 23, 1866 (1981).
788. Steudel R., Topics in Current Chemistry, Vol. 102, Springer-Verlag, 1982,
p. 149.
789. Steudel R., Z. anorg. allg. Chem., 478, 139, 156, 177 (1981).
790 Foss O., Janickis V., J. Chem. Soc. Chem. Comm., 1977, 834
791. Soules T. F„ J. Chem. Phys., 73, 4032 (1980).
792. Bell R. J., Carnevale A., Phil. Mag., В 43, 389 (1981).
793. Phillips J. C.y J. Non-Cryst. Solids, 44, 17 (1981).
794. Jansen M., Vo.ss M., Angew. Chem., 93, 120 (1981).
795. Minshall P. C„ Sheldrick G. M., Acta Crystallogr., В 34, 1326 (1978).
796. Bues W., Somer M., Brockner W., Z. anorg. allg. Chem., 476, 153 (1981).
797. Beeman D., Lynds R., Anderson AL R., J. Non-Cryst. Solids, 42, 61 (1980).
798. Vanderbilt D., Joannopoulos J. D., Phys. Rev., В 23, 2596 (1981).
799. Wada N., Solin S. A., Wong J., Prochazka S., J. Non-Cryst. Solids, 43,
7 (1981).
800. Wong J., Lytle F. №.. J. Non-Cryst. Solids, 37, 273 (1980), 49, 103 (1982)
801. Sakka S., Komiya K., Rev. Chim. Minerale, 16, 293 (1979).
802. Cox A D„ Me Millan P. W., J. Non-Cryst. Solids, 44, 257 (1981).
408 Глава 3
803. Oyanagi Н., Tanaka К.. Hosoya S., Minomora S., Proc. IX Int. Conf.
Amorph. Liquid Semicond., Grenoble 1981, J. de Physique Colloque C4,
Suppl. № 10, 42, 221 (1981).
804. O’Reilly E. P., Robertson J., Kelly M. J., Solid State Commun., 38, 565
(1981).
805. Baldwin С. M., Almeida R. M., Mackenzie J. D., J. Non-Cryst. Solids, 43,
309 (1981).
806. I.iang K- S., Cramer S. P., Johnston D. C., Chang С. H., Jacobson A. J.,
de Neufville J. P„ Chianelli R. R., J. Non-Cryst. Solids, 42, 345 (1980).
807. Etherington G., Wright A. C.. Wenzel J. T., Dore J. C., Clarke J. El.,
Sinclaire R. N.t J. Non-Crvst. Solids, 48, 265 (1982).
808. Windisck С. F.. Risen W'. M., J. Non-Cryst. Solids, 48, 325 (1982).
809. Johnson P. A. K, Wright A. C., Sinclair R. N., J. Non-Crvst. Solids, 50,
281 (1982).
810. Pohl K. D., Briickner R., Phys. Chem. Glass., 22, 150 (1981).
811. Itoh S., Fujiwara T., Okazaki Л1., J. Non-Cryst. Solids, 50, 49 (1982).
812. Desa J. A. E., Wright A. C., Wong J., Sinclair R. N., J. Non-Cryst. Solids,
51, 57 (1982).
Дополнительная литература *
Абдуллаев Г. Б., Абдинов Д. Ш. Физика селена.— Баку: ЭЛМ, 1975, 403 с.
Борисова 3. У. Химия полупроводниковых стскол.— А.: Изд-во ЛГУ, 1984,
327 с.
Ватолин Н. А., Пастухов Э. А. Дифракционные исследования строения вы-
сокотемпературных расплавов.— М.: Наука, 1980, 189 с.
Демкина Л. И. Исследование зависимости свойств стекол от их состава. М.:
Оборонгиз, 1958, 239 с.
Леко В. К., Мазурин О. В. Свойства кварцевого стекла.— Л.: Наука, 1985,
164 с.
Мазурин О. В. и др. Свойства стекол и стеклообразующих расплавов. Спра-
вочник в 4-х т./Под ред. Немилова С .В., Т. I, 1973, 444 с.: Т. II, 1975, 632 с.:
Т. III, ч. 1, 1977, 586 с.; Т. Ill ч. 2, 1979, 486 с.; Т. IV, ч. 1, 1980, 462 с.;
Т. IV, ч. 2, 1981, 375 с.
Полтавцев Ю. Г. Структура полупроводниковых расплавов.— М.: Металлур-
гия, 1984, с. 176.
Пинскер Г. 3. Определение решеточных закономерностей в аморфной струк-
туре.— Физ. и хим. стекла 1980, т. 6, № 5, с. 521—524.
Пинскер Г. 3. Определение параметров структуры стекла SiO2 — Физ. и хим.
стекла, 1980, т. 6, № 6, с. 651—657.
Пинскер Г. 3. Идентификация двух типов a-Se.—ДАН СССР, 979, т. 248,
№ 1, с. 115—118.
Rawson Н. Properties and application of glass. Ams., Oxf„ N.-Y., 1980, 316 p.
Татаринова Л. И. Структура твердых аморфных и жидких веществ.— М.:
Наука, 1983, 151 с.
Силинь А. Р., Трухин А. Н. Точечные дефекты и элементарные возбуждения
в кристаллическом и стеклообразном SiO2—Рига: Зинантне, 1985, 244 с.
* Представлена редактором С. А. Дембовским.
4. Электрические
и оптические свойства
стекол и аморфных тел
Явления переноса заряда и взаимодействия вещества с элек-
тромагнитным излучением дают основную информацию о ха-
рактере связей между атомами и структурными группами в не-
кристаллических твердых телах. Так, в гл. 3 при анализе
корреляций между структурой и свойствами показано, что неор-
ганические вещества можно классифицировать на аморфные
и стеклообразные по типу и порядку величины проводимости
или по области оптической прозрачности.
Увеличение степени разупорядоченности и соответствующее
увеличение концентрации вакансий и других дефектов создают
благоприятные условия для термически активированных струк-
турных перестроек. При этом даже небольшие добавки одноза-
рядных катионов в практически непроводящие стекла способны
существенно снизить их сопротивление. Например, при добавке
к халькогенидным стеклам оксидов или халькогенидов щелоч-
ных металлов возникает характерная для таких составов ион-
ная проводимость.
Из-за сильного кулоновского взаимодействия двух- и более
зарядных катионов с отрицательным зарядом сетки их подвиж-
ность оказывается на несколько порядков ниже подвижности
однозарядных катионов. Эта особенность определяет существо-
вание высокоомных оксидных стекол, прозрачных в видимой
области спектра, а также высокоомных халькогенидных стекол.
Такие материалы обладают запрещенной зоной достаточно ши-
рокой для юго, чтобы термическая генерация носителей была
практически исключена.
Некристаллические твердые тела с электронной проводи-
мостью на основе непроводящих стекол можно получить двумя
основными способами. Во-первых, если в матрицу непроводя-
щего стекла ввести компоненты, дающие электронные состоя-
ния внутри запрещенной зоны стекла (например, если к оксид-
ному стеклу добавить оксиды переходных металлов). Во-вто-
рых, если уменьшить ширину запрещенной зоны пу чем введения
элементов или комбинаций элементов, понижающих энер-
гию и полярность связей в стекле. Первый случай отвечает
оксидным стеклам с характерной электронной проводимостью,
410 Глава 4
второй — полупроводниковым элементам IV и V групп периоди-
ческой системы и халькогенидным стеклам.
В металлических стеклах валентная зона и зона проводи-
мости перекрываются, вследствие чего запрещенная зона исче-
зает и явления переноса заряда описываются так же, как в ме-
таллических расплавах.
4.1. Перенос заряда
в некристаллических твердых телах
В области выполнения закона Ома
j = oF (4.1)
плотность тока / и напряженность электрического поля F свя-
заны между собой коэффициентом о, который называют удель-
ной электропроводностью (электропроводностью, проводимо-
стью). Проводимость можно представить как произведение кон-
центрации носителей заряда на их подвижность ц
о = пер (4.2)
Как было показано в работе [1], проводимость некристал-
лических твердых тел в более или менее широком интервале
температур подчиняется эмпирическому закону Аррениуса
в форме уравнения
о = о0 ехр(—EJkT) (4.3)
Если электропроводность осуществляется по состояниям
в зонах, то ее экспоненциальную температурную зависимость
по уравнению (4.3) определяет в основном температурная за-
висимость концентрации носителей п или р в соответствии
с уравнениями (2.112, 113, 115), а температурной зависимостью
подвижности можно пренебречь. В случае прыжкового меха-
низма переноса по локализованным состояниям, т. е. в усло-
виях примерно постоянной концентрации носителей, основной
вклад дает термическая активация подвижности. Последняя
зависит от вида носителей заряда (электронная или ионная
проводимость) и от причин локализации (разд. 2.5.1.2), кото-
рые, в свою очередь, зависят от состава и структуры стекло-
образного или аморфного твердого тела.
Для некристаллических твердых тел при измерениях на по-
стоянном токе проводимость вначале меняется во времени, и ее
стационарное значение устанавливается не сразу. Измерения
на переменном токе возможны в широком диапазоне частот;
в литературе имеются данные для частот от 10~6 до 1016 Гц.
Из таких измерений можно получить представление о кинетике
процессов переноса и смещения зарядов, а из полученных зна-
Электрические и оптические свойства
411
чений резонансных частот сделать вывод о механизме релак-
сации.
По аналогии с механической релаксацией (разд. 2.2.1.1)
можно записать подобно уравнению (2.11) дифференциальное
уравнение для зависимости электрического смещения D =
= e0F + Р = eoerF = eF (Р—поляризация) от времени
rfF _ 1 dD_________F_
£° dt Er dt Er/о
(4.4)
При сопоставлении величины er/o с диэлектрической проницае-
мостью вакуума е0 из соотношения ег/о = т/Оо получаем посто-
янную времени т, имеющую смысл времени релаксации. Так
же, как в уравнение (2.11), в уравнение (4.4) входят «упругие»
и «неупругие» параметры (параметры переноса). При D =
= const, т. е. (dDIdt) = 0 получаем
F = F'exp(-f/T) (4.5)
Это уравнение означает, что уменьшение напряженности элек-
трического поля в конденсаторе, заполненном высокоомным ве-
ществом (диэлектриком), с течением времени замедляется
из-за замедления процесса перераспределения заряда. Время
релаксации т также можно определить из декремента затуха-
ния X в соответствии с уравнением (2.14); для этого измеряется
уменьшение напряженности электрического поля.
В большинстве случаев уменьшение напряженности поля не
описывается простой экспоненциальной зависимостью, и по-
этому необходимо пользоваться функцией распределения вре-
мен релаксации g(r); соответственно
F (t) — Ff j g (т) ехр (—f/т) dx (4.6а)
о
По аналогии с уравнением (2.18) экспериментальные данные
можно описать эмпирическим выражением
F(t) = F ехр(—t/x)b (4.66)
Величины F, D и бг из уравнения (4.4) можно представить
в комплексной форме как F*, D* и е*. При подстановке в урав-
нение (4.4)
D* = eoe*F*, F* = F0elO)<, 1/е*= (Ют)/(<от - Z) (1/eJ (4.7)
оно имеет решение. Если величину 8W выразить через соотно-
шение
Е = Еоо + Е0Ет (4.8)
412
Глава 4
то можно исключить из рассмотрения которая вплоть до
частот 109 Гц не зависит от частоты и учитывает суммарную
поляризацию (электронную, атомную и ионную).
В работе [2] был введен электрический модуль М* = 1/е*,
разделение которого на действительную и мнимую часть дает
М* = ,,r + J'!,., + мг (4.9а)
и соответственно
м* = м +1 м - м' +<4-9б)
откуда получаем для тангенса угла диэлектрических потерь
tg6 = M'7M' = е'7е' (4.10)
Действительная часть диэлектрической проницаемости е' опи-
сывает «упругую» составляющую реакции среды на влияние пе-
ременного электрического поля и соответствующие потери энер-
гии, мнимая часть е"—«неупругая» составляющая—отвечает
процессам диссипации энергии; они связаны между собой со-
отношением
ст (со) — eowe" = e08'w t g 6 = еое7т (4.11)
Если из величины е", связанной с еш, вычесть величину
о(0)/(080, отвечающую проводимости на постоянном токе, прак-
тически измеряемой в области низких частот, то оставшаяся
часть комплексной диэлектрической проницаемости
8р = е — i (& —а (0)/(ое0) (4.12)
представляет собой диэлектрическую релаксацию, которая
в простейшем случае ансамбля однотипных диполей, находя-
щихся в тепловом равновесии, описана Дебаем [3]:
е’ = е — it = 82 + (е, — е2) -Тт~/<чт — £2 + (ei — £г) X
1 -j- 4С0Т
X Г 1 J 2 2--riMT—1 (4.13)
где 8i = lime' (wt<C1) и 82 = lime' (wt^>1). Здесь т пред-
ставляет собой время релаксации; строго говоря, следует учи-
тывать функцию распределения времен релаксации. Помимо
постоянных молекулярных диполей в электрическую поляриза-
цию могут давать вклад противоположно заряженные дефекты
(не способные взаимно нейтрализоваться), а также простран-
ственные смещения электронов, поляронов и ионов (которые
предполагаются, например, при прыжковом механизме прово-
димости).
Электрические и оптические свойства
413
Для определения истинных параметров проводимости сле-
дует учитывать возможное влияние измерительной схемы (не-
удачные /?С-сочетания могут быть приняты за релаксирующие
объекты в образце), а также контактные явления. При изме-
рениях на постоянном токе с блокирующими контактами можно
получить сопротивление на несколько порядков выше истин-
ного. В случае исследования веществ с ионной проводимостью
следует иметь в виду возможность поляризационных эффектов
и образования объемного заряда в приконтактной области.
В крайне высокоомных веществах даже в случае надежных
омических контактов стационарная проводимость вследствие
диэлектрической релаксации устанавливается лишь через неко-
торое время. При таких измерениях исследуют образец в виде
конденсатора с емкостью С и сопротивлением R, для которого
время релаксации
т = /?С = е0Ег/о (4.14)
Например, при ег = 8 и а=10 12 См-см-1 т = 708 с, а время
установления постоянного значения о составляет 5т(е0 =
= 8,854-10-14 Ф/см). Измерение такого образца при его тол-
щине £ = 1 мм, диаметре контактов 10 мм (площадь контак-
тов 0,79 см2) и приложенном напряжении (/=10 В находится
на пределе возможностей стандартного электрометра с вибро-
преобразователем (чувствительность я? 10-13 А). При этом
для подвижности носителей ц=1 см2-В-1-с-1 в соответствии
с уравнением (4.2) получаем весьма низкую концентрацию но-
сителей, равную 6300 см-3 (сравнить с концентрацией атомов
в твердом теле ~ 1022 см-3).
Таким образом, измерения электропроводности очень нена-
дежны и могут, использоваться как метод оценки структуры вы-
сокоомного твердого тела лишь в сочетании с другими мето-
дами исследования или при установлении корреляций в гомо-
логических рядах. Отметим также, что и в этом случае следует
обязательно исключать поверхностную проводимость, для чего
измерения должны проводиться в высоком вакууме или с ис-
пользованием заземленных кольцевых контактов.
4.1.1. Стекла с ионной проводимостью*
Ионная проводимость стекол описана во многих работах
(см., например, [4—6]). В стеклах, содержащих ионы металла,
электроперенос осуществляется в основном катионами и ведет
к изменению структурной сетки стекла. Числа переноса ионов
щелочных металлов или других однозарядных ионов практи-
* См. также Евстропьев К. К. Диффузионные процессы в стекле.— Л.:
Стройиздат, 1970, 168 с.— Прим, перев.
414 Глава 4
чески равны единице; двухзарядные катионы гораздо менее
подвижны.
При измерениях проводимости на постоянном токе приме-
няют либо неполяризующиеся электроды, например растворы
электролитов, расплавы солей или амальгамы (в этом случае
для преодоления потенциального барьера в граничном слое тре-
буются достаточно высокие напряжения, когда становится за-
Рис. 4.1. Частотные зависимости проводимости и диэлектрической проницае-
мости стекол.
а — (Li2O)s(SiO2)i9 при различных температурах [7]. Л = 5,44 см3, L=-2,01 мм; б — стекло
Na2Si3O7 при 298 К [8].
меткой зависимость проводимости от напряженности электри-
ческого поля), либо блокирующие электроды, например, из
платины (при этом используют достаточно малые плотности
тока, чтобы в условиях высокой плотности поверхностного за-
ряда избежать его заметного истощения). Согласно уравнению
(4.14) в высокоомных веществах велико время установления
стационарной проводимости, поэтому предпочтительней изме-
рения на переменном токе. В последнем случае можно экспери-
ментально определить как действительную, так и мнимую часть
диэлектрической проницаемости, а значит и величину проводи-
мости.
На рис. 4.1 приведены частотные зависимости проводимости
стекол (Li2O)6(SiO2)i9 [7] и NajzSiaO? [8]. В низкочастотной об-
Электрические и оптические свойства
415
ласти значение о(0) отвечает проводимости на постоянном
токе. Изгиб при малых частотах связан с поляризацией элек-
тродов, и только при достаточно низких температурах (и соот-
ветственно при высоких сопротивлениях) этот изгиб практиче-
ски исчезает при частотах от 1 до 0,1 Гц, т. е. непосредственно
проявляется проводимость на постоянном токе. В области ча-
стот, где наблюдается примерное постоянство значений о, кри-
вая е' имеет плечо (рис. 4.1), отвечающее обычному для таких
стекол значению диэлектрической проницаемости 20—30. Кру-
той рост действительной части диэлектрической проницаемости
при низких частотах обусловлен поляризацией электродов. Что
касается о, то после достижения некоторой частоты начинается
ее рост. Повышение температуры образца действует на прово-
димость так же, как увеличение частоты поля.
4.1.1.1. Ионная проводимость
на постоянном токе
Ионную проводимость стекол при измерениях на постоянном
токе можно представить как результат перемещения катионов
по вакансиям, хаотически расположенным в структуре стекла.
Согласно Френкелю [9] ионы выходят из своих положений
в узлах решетки и становятся свободными, «блуждающими»
ионами, одновременно освобождая свои места и создавая, та-
ким образом, условия для вакансионной проводимости. Мюл-
лер [10] рассматривает образование свободных ионов как про-
цесс диссоциации. По Шарлю [11] вследствие неполного запол-
нения пространства в стеклах число возможных мест для ионов
щелочных металлов больше числа имеющихся ионов, следова-
тельно, их перемещение происходит по вакансионному меха-
низму. Сходный-механизм рассматривался в работе [12]. На ос-
нове представлений о флуктуациях энергетических барьеров
и потенциальной энергии в некристаллических веществах в рам-
ках перколяционной модели неоднократно делались попытки
связать процесс протекания заряда в таких веществах с осо-
бенностями их структуры.
Обозначим через N\ число мест в объеме V, которые могут
занимать перемещающиеся катионы, например Na+ или Li+,
а через w — вероятность заполнения этих мест. Тогда концент-
рация носителей заряда равна n = N\w/V. При вероятности за-
полнения вакансий равной 1—w, длине прыжка а и вероятно-
сти термически активированного перескока v (частота переско-
ков) получаем выражение для коэффициента диффузии
D = а2 (1 — w) v = а2 (1 — w) v0 exp (—E^kT) (4.15)
где v0 — средняя частота фононов. С использованием соотно-
шения Нернста—Эйнштейна, связывающего коэффициент
416 Глава 4
диффузии с подвижностью в форме \\. = eD!kT, и уравнения
(4.1) получаем выражение для проводимости
п (1 — w) e2a2vo . „ ne2D
а = —------------ exp (~E0/kT) = (4.16)
Итак, ионная проводимость пропорциональна концентрации
носителей заряда, квадрату длины прыжка иона и вероятности
1 — w, которая характеризует степень разупорядоченности
структуры стекла. Так как величины, входящие в предэкспо-
ненциальпый множитель, зависят в основном от состава стекла
и в обычных условиях мало зависят от температуры, то в со-
ответствии с уравнением (4.16) проводимость экспоненциально
зависит от обратной температуры и, таким образом, рассмот-
ренный «прыжковый» механизм электропроводности хорошо
описывается эмпирическим уравнением (4.3). По наклону зави-
симости Igo от \/Т (или лучше 1g (аТ) от 1/Т) можно опреде-
лить энергию активации ионной проводимости на постоянном
токе. Например, для рассмотренных в работах [7, 8] стекол
(Li2O)6(SiOs) 19 и NasSisO? были получены величины 0,683
и 0,65 эВ соответственно.
При выводе уравнения (4.16) предполагалось, что переме-
щение ионов под действием поля происходит по тому же меха-
низму, что и самодиффузия. Однако, как правило, рассчитанная
из коэффициента диффузии проводимость оказывается за-
ниженной, что учитывается корреляционным коэффициентом f:
oD = fG (4.17)
Величина j<Z 1 означает, что каждый последующий диффузи-
онный шаг зависит от предыдущего. Для стекла Na2ShO7 f =
= 0,5 при 573 К [13].
На проводимость высокоомных стеклообразных оксидов
и халькогенидов оказывает влияние даже небольшое количество
примесей, и особенно примесей щелочных оксидов. Так, при
573 К плавленый кварц с содержанием 4-10~6 % Na+ имеет со-
противление 1013 Ом-см, а при содержании 2-1О’зо/о Na+ —
5• 1 (Г Ом-см [15]. Аналогичное влияние примеси Na2O па про-
водимость было обнаружено в стекле В2Оз [16].
При увеличении концентрации носителей, например в обыч-
ных бинарных щелочносиликатных стеклах, зависимость о от
п в двойном логарифмическом масштабе выражается прямой
(о~/г6); крутизна прямой уменьшается при повышении тем-
пературы. Одновременно с увеличением проводимости при уве-
личении концентрации носителей изменяется сам характер
влияния добавок оксидов щелочных металлов, который объяс-
няют модификацией решетки и изменением подвижности носи-
телей. Другие факторы: изменение длины прыжка, вероятности
Электрические и оптические свойства 417
заполнения вакансий 1—w и средней частоты фононов Vo не
оказывают существенного влияния на величину проводимо-
сти. Энергия активации проводимости Еа уменьшается с уве-
личением относительного содержания в стекле атомов кисло-
рода.
Природа вводимого щелочного катиона практически одина-
ковым образом влияет па проводимость разных стекол. Так, ион
Li+ благодаря малому радиусу может сравнительно легко пере-
мещаться в матрице стекла, однако в то же время он эффек-
тивно удерживается решеткой за счет сильного кулоновского
взаимодействия с отрицательно заряженными кислородными
анионами. Ион К+, имеющий больший радиус, слабее связан
с решеткой. Как правило, при уменьшении радиуса иона ще-
лочного металла проводимость стекол в гомологическом ряду
несколько увеличивается, что имеет место, например, в щелоч-
носиликатных стеклах при переходе от К+ к Li+. Наибольшую
ионную проводимость обеспечивают ионы Ag+. Правда, суще-
ствование соответствующих оксидных стекол в открытой си-
стеме (в отсутствие повышенного давления кислорода) огра-
ничивается сравнительно легкоплавкими системами, например
серебросодержащими фосфатными или боратными стеклами.
В результате изучения влияния природы основного стекло-
образователя на ионную проводимость в смешанных системах
выяснилось, что в ряду силикатных, германатных и фосфатных
стекол при одинаковом содержании NasO проводимость умень-
шается, а при переходе к сульфидным системам — увеличи-
вается. Последнее можно объяснить более слабым кулоновским
взаимодействием ионов щелочных металлов (или ионов се-
ребра) со структурным остовом тиосиликатных, тиофосфатных
или гиогерманатных стекол. Соответствующая большая поля-
ризуемость остова отвечает меньшим потенциальным барьерам
между катионными узлами и вакансиями, чю обусловливает
наблюдаемое уменьшение энергии активации проводимости.
Сильная разупорядоченность и связанная с ней высокая
подвижность катионов позволяет рассматривать стекла с ион-
ной проводимостью как твердые электролиты.
Стекла как твердые электролиты. В последние годы уве-
личился интерес к твердым электролитам в связи с миниатю-
ризацией и связанной с ней потребностью в малогабаритных
энергоемких источниках тока [17, 18]. Среди так называемых
суперионных веществ (это прежде всего соединения серебра
при температуре выше некоторой критической [19—21]) луч-
шими катионными проводниками являются р-оксид алюминия
NaAliiOj/(о1СН3 См-см-1) и Li3N (п = 2-10 •’ См-см-1 при
298 К) [19—21]. Хорошим анионным проводником является
PbSnF4 (а^ 10~3 См-см-1 при 300 К) [22].
27 Заказ № 413
418 Глава 4
Благодаря тому, что стекла изотропны и в них исчезает ха-
рактерная для поликристаллических твердых тел неопределен-
ность, связанная с наличием границ зерен, ионную проводи-
мость в стеклообразных материалах можно оптимизировать
двумя способами. Во-первых, повышением подвижности катио-
нов за счет перехода от оксидных к халькогенидным систе-
мам, содержащим высокие концентрации ионов щелочных ме-
таллов или серебра. Во-вторых, путем разрыхления структуры
оксидных стекол за счет введения в их состав солей с относи-
тельно крупными катионами. Для получения таких нетради-
ционных стекол, например LiNbO3 с о =10“5 См-см-1 при 298 К,
используются высокие скорости охлаждения (см. разд. 2.1.1.3).
В табл. 4.1, составленной по данным Равайна [23] и других ав-
Таблица 4.1. Оксидные и халькогенидные стекла
с ионной проводимостью
Состав о (См/см) при 298 К [23] £o, эВ
AgPO3 4,5 • 10-7 0,52
NaPO3 5,6 • 10-“ 0,82
AgPS3 8 • IO-6 0,41
5 • IO'6®
NaPS3 5,7 • 10-8 0,54
3,9 • IO6® 0,54
Na2SiS3 1,1 • IO"5 0,43
Na20e2Ss 1,0 • io-8 0,54
Na2GeS3 1,0 • 10е 0,51
5,0 • 10 sa 0,51
2,9 • 10-7 0,59
Na6Ge2S7 7,3 • 106
Na2SiO3 2 • IO'7 0,64
Na2GeO3 2,3 • IO8 0,69
A g A s% 2 • 10-7
Ag6Ge7Si3 2,5 . IO6
LieGezSn 4,4 . IO7 0,63
Li2GeS3 4,0 • IO’5 0,51
а Данные взяты из нескольких работ (см. литературу к гл. 3
[719, 732,733]).
торов, представлены стекла, частично рассмотренные ранее
в разд. 3.3.6.1. Из таблицы видно, что кроме Na2SiS3 весьма
Электрические и оптические свойства
419
высокую для стекол проводимость 0 = 4-10~5 См-см-1 при 298 К
обнаруживает также Li2GcS3. Однако некоторые из стекол,
включенных в табл. 4.2, имеют еще большую проводимость.
Вероятно, за счет добавления определенных солей концентра-
ция подвижных катионов в этих стеклах особенно высока,
а структурный остов разрыхлен вследствие равномерного
встраивания крупных анионов.
Таблица 4.2. Стекла нетрадиционных составов с ионной проводимостью
Состав 0 (См/см) при 298 К Eq ,ЭВ
ЫРОз 2 • IO’9 0,7
(ЫРОз)з (LiF)2 2- 10-8 0,63
(ЫРОз)з (LiCI)2 8,9 • 10-8 0,64
(LiPO3)3 (LiBr)2 7,7 • IO-7 0,55
(LiPO3)7 (Li2SO4)3 1,5 • IO-7 [23] 0,60
В2Оз(Ы2О)0;57 6,3 • 10-” 0,73
В20з(1л20)о,57 (LiCI)0,86 3,2 • 10-6 0,46
L i2S i2O5 3,2 • IO 9 0,66
Li2Si2Os (Li2SO4)0,4 Li2O — SiO2 — Li2SO4 (Li/Si<2,2) AgPO3 — AgSO4 L1PO3 — Li2SO4 AgPO3—AgCI . AgPO3 — AgBr AgPO3 — Agl AgPO3(MI2)0,19 (M = Pb или Cd) Ag2MoO4AgI Ag7!4AsO4 Agel4B2O4 1,3 • IO-8 < 10-s (при 363 К) [25] <4 • 10-® [26] <ю-7 < io-< < IO'3 [27] < 10-2 < 10-2 [27] <IO-2 [28] ~ 10-2 [29] ~2 • IO’2 [30] 0,66
В системах AgX—Ag2O—Р2О5 (Х = С1, Вг или I), где обна-
ружены области стеклообразования [31], было установлено уве-
личение проводимости при возрастании радиуса вводимых анио-
нов; при этом могут быть достигнуты величины, близкие или
даже превышающие проводимость кристаллического р-оксида
алюминия.
27*
420 Глава 4
4.1.1.2. Ионная проводимость
на переменном токе
Из рис. 4.1 видно, что по достижении некоторой частоты про-
водимость начинает возрастать с ростом частоты, причем, как
было отмечено в работе [31], для веществ с различным соста-
вом и структурой обычно выполняется соотношение
а (со) ~ соп, п 1
(4.18)
В области высоких частот показатель степени п достигает ти-
пичного значения между 0,8 и 1, при этом в", в соответствии
с уравнением (4,11), фактически не зависит от частоты. В об-
ласти меньших частот е" монотонно уменьшается с повыше-
Рис. 4.2. Схема флуктуаций потенциальных барьеров в стекле, дающих вклад
в действительную часть диэлектрической проницаемости е/.
нием частоты, что можно интерпретировать на той же основе,
что и релаксацию проводимости в режиме постоянного тока.
Для осуществления такой релаксации требуется наличие непре-
рывного перколяционного пути (капала протекания тока)
между электродами. Однако при повышении частоты все боль-
ший вклад в проводимость вносит смещение зарядов на рас-
стояния, меньшие межэлектродного, и при достаточно высоких
часто!ах проводимость преимущественно определяется пере-
скоками ионов между соседними центрами. При дальнейшем
увеличении частоты ноля проводимость, в соответствии с урав-
нением (4. И:, линейно возрастает с частотой.
Очевидно, смещение ионов в структуре стекла с характер-
ными флуктуациями потенциала, изображенными на рис. 4.2,
приводит к скоплению зарядов у наиболее высоких барьеров.
Такое скопление и соответствующее рассасывание заряда дает
вклад в действительную часть комплексной диэлектрической
проницаемости s'. При высоких частотах скопление зарядов
не успевает произойти, и, как видно из рис. 4.1, б, е' умень-
шается (от величины Ei до величины е2 в соответствии с урав-
нением (4.13)).
При сопоставлении частотных зависимостей е" и проводи-
Электрические и оптические свойства
421
мости с использованием уравнения (4.11) получаем соотно-
шение
о (0) = еое7та (4.19)
где Та — время релаксации проводимости [2, 7]. При обработке
экспериментальных данных по уравнению (4.19) выясняется,
что требуется вводить несколько времен релаксации; это со-
гласуется с моделью флуктуирующих потенциальных барьеров
(рис. 4.2).
Энергия активации в уравнении
та = та (0) ехр (£а//г7) (4.20)
отвечает £с по уравнению (4.3), если та понимать как среднее
время релаксации [7]. С другой стороны, в работе [2] пока-
зано, что введение распределения времен релаксации проводи-
мости позволяет получить частотную зависимость е' ио. В ра-
боте [33] описано влияние термической предыстории на вре-
мена релаксации в щслочносиликатных стеклах.
4.1.1.3. Полищелочной эффект
В стеклах, содержащих два типа щелочных металлов, пара-
метры процессов переноса, связанных с подвижностью катио-
нов, т. е. коэффициент диффузии, проводимость, вязкость рас-
Рис. 4.3. Полищелочной эффект в ще.тэчносиликатпых стеклах с парами катио-
нов Cs+—Li+, Cs+—i\a+, Cs* -K+, Cs+— Rb+ (a) [34j и LiT—Na+, Na+—K+,
K+—Ы (6) [35].
плава, нелинейно меняются с составом. Из рис. 4.3, где пред-
ставлены концентрационные зависимости удельного сопротивле-
ния щелочносиликатных стекол, видно, что в области примерно
равных концентраций щелочных металлов наблюдается
422 Глава 4
максимум, тем более резкий, чем сильнее различаются ионные
радиусы катионов. Этот эффект наблюдается также и для коэф-
фициента о0 в уравнении (4.3), и для энергии активации Еа по
уравнению (4.16) [36]. Полищелочной эффект наблюдается и
в других системах, например в щелочноборатных стеклах, при-
чем он усиливается с ростом температуры.
Обзор работ, посвященных интерпретации этого явления,
дается в работах [37, 38]. Щелочной эффект обсчитывался
также статистически [39]. Из того факта, что в районе макси-
мума эффекта коэффициенты диффузии обоих щелочных катио-
нов падают примерно на 40%, причем энергия активации
удваивается, следует, что между катионами существует сильное
взаимодействие, уменьшающее их подвижность. В электродина-
мической теории Хэндриксона и Брэя [40] учитывается взаим-
ное влияние катионов, расположенных вблизи отрицательно за-
ряженного иона кислорода. Таким образом, рассчитывается
энергия диполь-дипольного взаимодействия, повышающего энер-
гию активации подвижности носителей. Эта модель, впоследст-
вии развитая в рамках теории кооперативных термически акти-
вированных процессов в соответствующих стеклах [41], явля-
ется лучшим из существующих теоретическим подходом к по-
ниманию природы полищелочного эффекта.
В связи с рассматриваемым явлением следует отметить, что
в стеклах МгО—А120з—SiO2 при эквимолярном отношении
М1: А1 полищелочной эффект отсутствует [42], и что в работе
[43] было рассмотрено явление релаксации в смешанных ще-
лочносиликатных стеклах.
4.1.1.4. Сверхвысокоомные стекла
Ниже температуры стеклования подвижность двухзарядных
катионов в оксидных стеклах на несколько порядков ниже по-
движности однозарядных, и особенно мала для щелочноземель-
ных ионов [44—46]. Многозарядные катионы могут сущест-
венно блокировать подвижность щелочных ионов; например,
проводимость стекла состава (Na20)o,i (BaO)o.2(Si02)o,7 на
шесть порядков меньше проводимости стекла (Na20)o,3(Si02)o.7.
Стекло Са25!зО8 имеет проводимость 10-13 См • см-1, которая
не зависит от концентрации ионов Na+ при содержании Na2O
от 3,5- 10~3 масс. % до 1,3 мол. % [47]. На рис. 4.4 приведены
другие примеры, взятые из работы Маккензи [48].
Если введение SrO и ВаО в оксидные стекла тоже приводит
к сильному повышению их сопротивления, то в случае MgO
наблюдается более слабое влияние на электропроводность. По-
видимому, в последнем случае сказывается тенденция MgO вхо-
дить в структурную сетку как стеклообразователь, вследствие
Электрические и оптические свойства 423
чего ослабляется его блокирующее действие на щелочные ионы.
Однако такое объяснение сложного эффекта, сходного с поли-
щелочным эффектом, явно упрощено.
Влияние двухзарядных ионов на проводимость натрийсили-
катных стекол и стекол, содержащих катионы высокой валент-
Рис. 4.4. Зависимость сопротивления высокоомных стекол от температуры [48].
1 — Na2O—CaO—SiO2, 2 — SiO2 с 1 • Ю~3 % Na+, 3 — Ca3Si3O8 с 2-10-2% Na+, 4 -
(CaO)0 43(A^Og^gjg(§102)0,42 С 0,1 % Na+, 5 — СаВ^О? с о % Na+.
ности, рассматривалось в работах [46, 49]. Стекла с аномально
низкой диэлектрической проницаемостью (3,5 < ег < 5,4) на
основе В2О3 (80 мол.%) с примерно одинаковым содержанием
AI2O3 и щелочноземельного оксида описаны в работе [50].
4.1.2. Оксидные стекла с электронной проводимостью
В случае кристаллических оксидов переходных металлов
общей формулы МО (решетка типа NaCl), М2О3 (решетка типа
корунда) или МО2 (решетка типа рутила) закономерности из-
менения электрических свойств можно понять при учете взаи-
модействия d-орбиталей, не образующих о-связей, но которые
(самостоятельно или с участием р-электронов) могут образо-
вывать л-связи. При этом чем сильнее вытянуты в пространстве
d-орбитали, тем больше ширина зоны 2/ (см. разд. 2.5.1.1 и
рис. 2.49). Тенденция к образованию делокализованных элек-
тронных состояний, характерных для металлической проводи-
мости, увеличивается для изоструктурных оксидов в горизон-
тальных или вертикальных рядах периодической таблицы при
увеличении атомной массы переходного металла (уменьшение
эффективного заряда ядра и увеличение радиального перекры-
тия волновых функций). ИеОз, например, проявляет металличе-
скую проводимость.
Если двигаться по диагонали таблицы от оксида ванадия
к оксиду платины, то наблюдается скачкообразное изменение
424 Глава 4
свойств с исчезновением металлической проводимости. При
этом мы как бы частично переходим к высокоомным полупро-
водникам, в которых перенос осуществляется в основном прыж-
ковым механизмом с термической активацией подвижности.
Причиной такого перехода металл—диэлектрик является обра-
зование поляронных состояний (разд. 2,5.1.2), которые появля-
ются, когда в результате взаимодействия с решеткой (фонон-
ным спектром) электрон энергетически локализуется на узле,
например на катионе переходного металла. Этот эффект ста-
новится существенным при уменьшении ширины зоны ниже не-
которой критической.
Такие оксиды, как VO2, V2O3 или Т120з, выше некоторой тем-
пературы переходят в состояние с металлической проводимо-
стью. Но если затем понизить их температуру, становится
устойчивой кристаллическая модификация с более низкой сим-
метрией, ведущая себя как полупроводник или даже диэлек-
трик; переход к этой модификации сопровождается скачкооб-
разным уменьшением проводимости на несколько порядков.
В рамках зонной модели такой переход можно интерпретиро-
вать как появление запрещенной зоны при снижении симметрии
кристаллической решетки. Однако зоны оказываются весьма
узкими, а подвижности в условиях сильного искривления зон
малы; поэтому предпочтительным здесь является описание
процессов переноса на основе взаимодействия между локализо-
ванными состояниями. Так, например, спектр поглощения зеле-
ного оксида никеля отвечает поглощению катионами
[Ni(Н2О)б]2+; следовательно, электронные переходы осущест-
вляются только внутри системы уровней d-электронов (спектр
поля лигандов). С другой стороны, в TiO, кристаллизующемся
в решетке NaCl, имеет место металлическая проводимость
с су = 104 См • см-1.
Второй важный фактор, влияющий на электронную прово-
димость в кристаллических оксидах переходных металлов -- об-
менное взаимодействие, которое существенно при наличии
в структурно эквивалентных местах решетки одноименных ка-
тионов в разных степенях окисления. Например, катионные де-
фек1ы могу; на несколько порядков повышать проводимость
NiO, если они обеспечивают образование трехвалентных качио-
нов никеля. В соответствии с этим в ряду Li.-\hJnNi:112vO вве-
дение уже 5-10~3 % Li (х = 5-10 5) приводит к увеличению
проводимости на 10 порядков (при > =0,1 проводимость дости-
гает величины лз 10 См • см ч) [51, 52].
В оксидных стеклах с переходными металлами, обладающих
электронной проводимостью, в ослабленном виде проявляются
закономерности, характерные для кристаллических оксидов пе-
реходных металлов (оксидные стекла с металлической прово-
Электрические и оптические свойства
425
Рис. 4.5. Температурная зависимость электропроводности в стеклах
с электронной проводимостью для двух гомологических рядов состава
(BaO)x(K20)o,25-x(Si02)o,5(M02)o,25 при M=VIV или TiIV по [53].
I, Г - х=0, 2, 2' — х=0,075, 3, 3'-х=0,125, 4, 4' - х=0,15, 5, 5' - №0,175. 6, 6' - х=0,25.
димостью в настоящее время не известны). Поскольку чистые
оксиды переходных металлов не образуют стекол, для получе-
ния стекол с электронной проводимостью обязательно исполь-
зуют также стеклообразующие оксиды и модификаторы. При
этом необходимо обращать внимание на возможность маски-
ровки электронной проводимости ионной, поэтому предпочти-
тельней системы без щелочных металлов или системы, содер-
жащие добавки, ограничивающие подвижность щелочных ионов
(разд. 4.1.1.4). Для иллюстрации на рис. 4.5 изображена тем-
пературная зависимость проводимости в гомологическом ряду
стекол (ВаО)х(КгО)0,25-х(SiCh)о,5(МОг)о,25 (M = VIV или TiIV)
[53]. При х = 0 преобладает ионная проводимость, причем di-
Таблица 4.3. Оксидные стекла с электронной проводимостью
Система Проводимость (См/см) и энергия активации 1ЭВ) Свойства3 Литература
(V2Oa)x(P2O5)j_x IO-4’3; 0,38; х = 0,1 (298 К) (J (=) См. лите- ратуру к гл. 1 [25]
О ( = ) 55
ЯМР 56
IO'4; 0,32; х =0,1 (298 К) Термо-э.д.с. (—) о (=, 57
х = 0,2 а (~) 58
(BaO)x(P2Os)0j4 _ х(^20а)о,б 10-5’5; х = 0,2; а = 4,8 о (=) 59
(GeO2)x(P4O,0)y(V2O5).r 10 -3,2; х =у = = 0,025; z = 0,95 о (--) Термо-э.д.с. (—) Эффект Холла 60
МпО — А120з — SiO2 < ю-“ Поверхностная 61
проводимость ю-9
(P2Og)o,45(FeOa)o,55 10~9-2; (298 К); 0,65; а= 1,24 Термо-э.д.с. (!-), а = 1,24 62
Термо-э.д.с. (-), а = 1,24
PbO—B2O3—Fe2O3 < 10~4’5 (573 К) 63
(МпО)0>5з(В2Оз)0,47 10~7’5 (543 К) 64
(СоО)01з(СаО)0>23(В2О3)047 10* 6’5(623 К); 1 эВ
(BaSiBsOeb-HTiOa-ylr 10-8 (423 К); х = 0,30; у = 0,016 65
(PO2i5)0,47(TiO2 - y)o,53 10~8’3 (573 К); «/ = 0,01 66
а Знак = означает измерения на постоянном токе, а ~ — на пере-
менном.
Электрические и оптические свойства
427
электроны в стеклах с M. = VIV практически не проявляются
(например, при поглощении света). При замене КгО на ВаО
ионная проводимость уменьшается, но при х>0,15 для VO2-
содержащих стекол электропроводность начинает возрастать,
в то время как для оптически прозрачных ТЮг-содержащих
стекол, в которых нет di-электронов, наблюдается дальнейшее
падение проводимости. Итак, электронная проводимость стекол
отчетливо проявляется только после достаточной блокировки
ионной проводимости [54].
Чем выше концентрация катионов с частично заполненными
d-оболочками и соответственно меньше среднее расстояние
между такими катионами, тем легче перемещается заряд. По-
этому для получения стекол с электронной проводимостью ши-
роко используются оксиды титана и ванадия, которые можно
ввести в стекло в высокой концентрации; в таких стеклах на-
ряду с эффективным перекрыванием d-орбиталей может выпол-
няться и условие переменной степени окисления. Небольшая
электронная проводимость наблюдалась также в стеклах, со-
держащих оксиды железа, кобальта или марганца.
Дентон, Роусон и Стенворт впервые доказали электронную
природу проводимости в стеклах системы VO2—V2O5—Р2О5
с величиной проводимости до 10-4 См • см-1 (см. гл. 1 [25]).
В табл. 4.3 приведены наиболее важные оксидные полупровод-
никовые стекла. Стекла других составов рассмотрены в рабо-
тах [48, 70].
В литературе обычно не уделяют должного внимания усло-
виям получения стекол, что затрудняет анализ литературных
данных, в частности обсуждение влияния на проводимость на-
личия катионов с разной степенью окисления. Последняя за-
висит от температуры расплава, от парциального давления
кислорода (оно определяет положение окислительно-восстано-
вительного равновесия) и, прежде всего, от кислотности рас-
плава (причем добавки восстановителей не изменяют сущест-
венно средней степени окисления катионов переходных метал-
лов). Кроме того, для получения гомогенного стекла необхо-
димо установление равновесия в расплаве.
Прежде чем делать какие-либо выводы о структуре стекла
по изменению свойств в зависимости от состава, следует иметь
в виду трудности, часто возникающие при попытках получить
стекло определенного состава. Например, на рис. 4.6 показано,
как сильно меняется исходный состав системы (PO2.5)x(TiO2) i-.r
при переходе в расплав. В данном случае исходные смеси гото-
вились «мокрым» способом (гомогенное осаждение) и затем
обезвоживались продолжительным прогревом при 1100 К; рас-
плав выдерживался 4 ч при 1600 К. Полученные стекла имели
темно-синий цвет и характеризовались малым содержанием ти-
тана. Повысить содержание титана таким способом невозможно,
Рис. 4.6. Содержание кристаллического фосфата титана(IV) в исходной
смеси и состав стекол после синтеза. Внизу показана область стеклообразо-
вания в системе ТЮ2—P2OS.
Рис. 4.7. Стекла в системах VO2—VO2,5—РО2.5 и VOi,5—VO2— PO2,s (7), полу-,
ченные из смесей УОзл/РОг.з и VO2/P2O2,5 (2) [67].
Электрические и оптические свойства
429
так как в расплаве Р2О5 восстанавливается до низших степе-
ней окисления и даже до элементного фосфора.
Еще ярче зависимость средней степени окисления переход-
ного металла в стекле от состава исходной смеси проявляется
в системе (PO2,5)x(VO2±a)i-x [67]. Из рис. 4.7 видно, что в об-
ласти исходных составов х 0,55 независимо от первоначаль-
ной степени окисления ванадия (смеси P2O5/VO2 или P2O5/V2O5)
при затвердевании расплава на воздухе всегда получаются
стекла системы Р2О5—V2O5—VO2, причем содержание VIV
в стекле увеличивается с увеличением концентрации Р2О5
в шихте. При составе исходной смеси х 0,6 получаются
стекла системы Р2О5—VO2—V2O3. Получить ванадий-фосфатное
стекло, содержащее только VIV довольно трудно из-за неизбеж-
ного улетучивания оксидов ванадия при высоких температурах,
а также из-за возможного диспропорционирования VJV на V111
и Vv в расплаве [68].
4.1.2.1. Механизм переноса заряда
По Маккензи [48] вывод об электронном характере проводи-
мости можно сделать на основании четырех независимых на-
блюдений:
1) проводимость на постоянном токе не зависит от времени
(отсутствуют поляризационные эффекты, обусловленные дви-
жением ионов);
2) для стекол с ионной проводимостью, ведущих себя как
твердые электролиты, следует ожидать зависимости разности
потенциалов от разности парциального давления кислорода на
электродах; для стекол с электронной проводимостью такое
поведение не характерно;
3) в высокоомных стеклах с ионной проводимостью сопро-
тивление уменьшается на несколько порядков при введении
в их состав оксидов переходных металлов. При этом о появле-
нии электронной проводимости говорит то, что электропровод-
ность не коррелирует с величиной oD, рассчитанной из измере-
ний диффузии с использованием соотношения Нернста—Эйн-
штейна, причем энергия активации подвижности существенно
ниже энергии активации диффузии;
4) в то время как электропроводность стекол с ионной про-
водимостью уменьшается с увеличением гидростатического дав-
ления, электронная проводимость возрастает вследствие уси-
ления перекрытия электронных орбиталей при увеличении плот-
ности вещества.
В работах [48, 57, 62, 69] показано, что величина термо-
э. д. с. в оксидных стеклах с электронной проводимостью прак-
тически постоянна в широком диапазоне температур. Поэтому
концентрацию носителей в таких стеклах можно считать при-
430 Глава 4
мерно постоянной, и тогда термическая активация проводимо-
сти обусловлена термической активацией подвижности
(разд. 4.1.1.1). Величина и знак термо-э. д. с. меняется при из-
менении средней степени окисления катионов переходных ме-
таллов, причем при реализации низших степеней окисления
электроперенос осуществляется в основном электронами, а выс-
ших — дырками.
Проводимость в рассматриваемом случае можно описать
выражением, отвечающем уравнению (4.16) (разд. 4.1.1.1), ио
в рамках электронной модели. Тогда в выражении n = Nj.wlV
в случае ванадийсодержащих стекол Nj— число узлов в объ-
еме V, занятых атомами ванадия, w — вероятность заполнения
этих узлов электронами, равная концентрации VIV; причем Ni
определяется соотношением NifV = tii!a3, где rii число атомов
ванадия в элементарном «объеме прыжка» а3 {а — длина
прыжка). С использованием уравнения (4.16) получаем
7Z V
о = -^-®(1-а>)ехр(-В„/4Т) (4.21)
где Еа — энергия активации перескока электрона между доста-
точно близкими центрами захвата (ловушками), например ме-
жду атомами Vrv и Vv. В соответствии с поляронной моделью
(см. рис. 2.53) захват заряда обусловлен взаимодействием
электрона с решеткой. Терм электрона в стабилизированном
захваченном состоянии определяется энергией поляризации
ЕР = —р2/2а [уравнение (2.124)]. По Мотту и Остину [72—74]
(см. также гл. 1 [33] и гл. 3. [263]) вероятность прыжка элек-
трона между двумя центрами (например, катионами в различ-
ных степенях окисления) велика, если разность его энергий
в результате такого прыжка U7D (определяется флуктуациями
потенциала в стекле) соизмерима с энергией фононов. С ис-
пользованием соответствующей конфигурационной модели по-
лучаем
1 1
Е,=------+ w (4.22)
и при условии EP^>WD
Е„ = р2/4а + 4-Г° (4.23)
Таким образом, £а тем больше, чем выше поляризуемость ре-
шетки *, характеризуемая коэффициентом 0, и чем ниже ее уп-
ругость (коэффициент а).
Уравнение (4.21) соответствует адиабатическому случаю,
когда за время ~10“12 с, отвечающее существованию локаль-
* Структурной сетки в случае некристаллических твердых тел.— Прим,
ред.
Электрические и оптические свойства
431
ной конфигурации с близкими энергиями соседних узлов *,
возможен многократный переход электрона из одной потенци-
альной ямы в другую. Если же это время мало по сравнению
со временем электронного перехода, то справедливо неадиаба-
тическое приближение, в котором частота прыжков v = v0X
X [exp (—Ea/kT)]p2 учитывает вероятность перескока электрона
Рг (в адиабатическом приближении рг=1). Для р2 Холстейн
[117] вывел следующее выражение:
p, = (hv„rl (л/Е.,кТ)'Ч2 (4.24)
где J есть мера перекрытия волновых функций. Для характери-
стики последней Мотт (см. гл. 1 [33]) ввел фактор перекрытия
е~2са, учитывающий спад волновой функции локализованного
электрона по длине прыжка а при Т >0/2 (0—дебаевская
температура); при этом
о = — w (1 —• w) exp (—2cd) exp (—Ea/kT) (4.25)
Уравнение (4.25) представляет проводимость в виде произведе-
ния диффузионного члена, фактора перекрытия и больцманов-
ского фактора.
При изменении состава стекла чем выше будет концентра-
ция катионов переходных металлов, т. е. чем меньше среднее
расстояние между центрами захвата а, тем выше должна быть
проводимость. С другой стороны, при изменении состава обычно
меняются и другие параметры, определяющие Ео. Следует от-
метить, что на поляризационный параметр Ер при изменении
состава в первую очередь будет влиять состояние кислородных
лигандов в координационных полиэдрах вводимых переходных
металлов. Влияние природы самих вводимых катионов на вели-
чину w(l —w) обсуждалось выше.
Максимум проводимости в железосодержащих оксидных
стеклах, как и следовало ожидать, отвечает примерно равным
концентрациям Fe11 и Fe111 [62, 70]. Однако в ванадийфосфат-
ном стекле (10 мол.-/о Р2О5) ему соответствует отношение
VIV/(VIV + VV) =0,1 [57, 70], а в стеклах системы (ВаО)хХ
X (Р2О5)0.4-х, (V20a)o,6, приведенной в табл. 4.3, он приходится
на х = 0,25 [59]. Аномальное поведение ванадийсодержащих сте-
кол можно объяснить в предположении, что координационные
полиэдры, образованные связями Vv—О, не участвуют или ча-
стично участвуют в переносе заряда [57]. В соответствии с этим
ЯМР таких стекол показывает наличие по крайней мере двух
типов ближнего порядка.
* Такие конфигурации периодически возникают в процессе тепловых
колебаний решетки (сетки).— Прим. ред.
432 Глава 4
В оксидных стеклах с относительно высокой проводимостью
при комнатной температуре возможно измерение электропро-
водности до весьма низких температур [57, 75—77], как это по-
казано, например, на рис. 4.8. При температурах Т <0/2 с по-
нижением температуры Еа постепенно спадает до величины
Wd/2, т. е. энергия фононов слишком мала для поляронной ста-
билизации заряда. При этом потенциальные барьеры, обуслов-
ленные геометрической разупорядоченностью, оказываются
Рис. 4.8. Температурные
зависимости проводимости
ванадийфосфатных стекол
в системе V2O5-a— P2Os—
V2O5 [77].
Содержание V2O5 (мол. %): 1 —
80, 2 — 70, 3 - 60.
слишком высоки для перескоков электронов между соседними
центрами. Мотт [72] (см. также гл. 1 [33]) показал, что в этом
случае вероятны перескоки электронов на расстояния 1а, зна-
чительно большие а (Z>1).* В этом случае появляется в /3
раз больше мест для возможного перескока электрона, и можно
ожидать, что среди них найдутся места с энергиями близкими
к энергии рассматриваемого заполненного локального уровня.
При разности этих энергий WdH? прыжки между соответствую-
щими узлами будут происходить с частотой
v = v0 exp (—2са) exp (—D!l2kT)
дающей известный закон
v = v0 exp [— const (ca^'tWo/kT'1*)] (4.26)
* Этот механизм проводимости называется прыжковым механизмом
с переменной длиной прыжка (variable range hopping).— Прим. ped.
Электрические и оптические свойства
433
и соответственно
о = о0 ехр (—В/т'1*)
Ход экспериментальных кривых на рис. 4.8 в области низких
температур удовлетворительно описывается уравнением (4.26).
Измерения проводимости на переменном токе подтверждают
применимость механизма проводимости посредством прыжков
поляронов для оксидных стекол с электронной проводимостью
[57, 58].
4.1.2.2. Проводимость в рядах оксидных стекол
со сходной структурой
В сложных стеклообразных системах при изменении их со-
става реализуются многообразные формы ближнего порядка и
статистических конфигураций, что затрудняет установление
корреляций между структурой стекла и его свойствами, напри-
мер электропроводностью. Тем не менее в некоторых случаях,
а именно в рядах образцов со сходной структурой, можно опре-
деленными приемами выявить влияние отдельных факторов на
закономерности структурных изменений.
Как было показано при изучении систем КгО—SiO2—МО2
[78], BaO—SiO2—МО2 [79] и BaO—К2О—SiO2—МО2 [53] (Л4 =
= Ti или V), на стекла распространяются в известных преде-
лах кристаллохимические закономерности, касающиеся изотип-
ных и изоморфных соотношений между оксидами Ti (IV) и
V(IV). Так, если в системе ВаО—SiO2—VO2 выдерживать по-
стоянные концентрации SiO2 и VO2, но постепенно замещать
ионы Ва2+ на.2К+ или 2/3La3+, т. е. получать следующие ряды
образцов:
А (ВаО)0,25-у (МгО)у (VOg)o,25 (SiOs)o,5
MZO.= K2O, 0 <у <0,1; MzO = La0,e7O, 0 < у <0,05
В (ВаО)о,зз-у (MzO)y (VOg)o,22 (SiO)0,45
МгО = Ьа0,б7О, 0 <у <0,125;
С (ВаО)0>35_у (МгО)у (V02)o,25 (SiO)0,4
MZO=K2O, 0 < у <0,1; M/) = La0,67O, 0<^< 0,175
то вследствие сравнимого радиуса катионов К+, Ва2+ и La3+
(1,33; 1,35 и 1,15 А соответственно) структурные изменения
должны быть не слишком велики.
Следует отметить, что при синтезе рядов стекол необходимо
исключить действие воздуха и влаги, для чего плавление и за-
твердевание расплава должны проводиться в инертной атмо-
сфере (см., например, аппаратуру на рис. 2.2). По результатам
28 Заказ № 413
434 Глава 4
Рис. 4.9. Зависимость электропроводности при 350 К и энергии
проводимости от состава для рядов Л и С.
активации
химического анализа средняя степень окисления ванадия в та-
ких стеклах составляет 4,00—4,01 [80].
Из измерений плотности следует, что среднее расстояние
между атомами ванадия в рассматриваемых рядах не меняется.
С другой стороны, как видно из рис. 4.9, в рядах А и С прово-
димость изменяется примерно на пять порядков вследствие из-
менения напряженности электрического поля вблизи ванадий-
кислородных полиэдров за счет изменения заряда катиона.
В рассматриваемом диапазоне концентраций ионная проводи-
мость не проявляется. Если для постоянной температуры по-
строить зависимость 1g (аа) от Еа, то из наклона прямой в со-
ответствии с уравнением (4.25) получаем постоянную Больц-
мана.
Химия четырехвалентного ванадия в оксидных системах
в значительной мере определяется его координацией [81].
Обычно ванадий(IV) образует комплексы с координационным
числом 5 или 6 и симметрией C4V. В изоструктурных соедине-
ниях BazTiOSizOy и BazVOSizO; атомы V (Ti) окружены пятью
лигандами кислорода, расположенными в объеме тетрагональ-
ной пирамиды [82]. Из спектров отражения этих стекол сле-
дует, что кроме переходов в поле лигандов, показанных на
рис. 4.10 (слева), имеют место и переходы с переносом заряда.
Этот переход соответствует возбуждению л-электрона оксива-
надиевого (IV) комплекса, отвечающего двойной связи вана-
дий—кислород, на dxy-орбиталь ванадия. Для стеклообразного
BazVOSizCh, полученного из расплава, наблюдается размытый
Электрические и оптические свойства
435
спектр, из которого можно сделать вывод о незначительном
искажении координационного полиэдра ванадия.
В работе [83] показано, что на перенос заряда большое
влияние оказывает сила кристаллического поля в окрестности
координационного полиэдра. Исследование ЭПР-спектров сте-
кол, содержащих VIV, подтвердили координационное число 5
по ванадию при расположении лигандов (ионов кислорода)
в конфигурации тетрагональной пирамиды [84, 85].
В случае комплексов с Vv ближний порядок несколько ме-
няется. В кристаллическом V2O5 группы VO3+ связаны посредст-
вом двух- и трехкоордипированных ионов кислорода таким об-
разом, что получается координационное число 5 по ванадию
{86]. Очевидно, лиганды — анионы кислорода — тем сильнее де-
формируются в поле центрального катиона — ванадия, чем
больше его заряд. При этом при переходе от VIV к Vv энер-
гия связей увеличивается и соответственно уменьшается спо-
собность ионов кислорода к смещению, т. е. затрудняется об-
разование поляронов (см. выше механизм проводимости по-
средством прыжков поляронов), вследствие чего увеличивается
энергия активации электропроводности.
Из-за снижения атомной и ионной поляризации кислорода
уровень 2Ь2 при расщеплении в поле лигандов заглубляется
меньше, чем в рассмотренном выше случае оксиванадиевых(IV)
комплексов (рис. 4.10): можно сказать, что заглубление элек-
тронов на t/.vy-орбитали тем сильнее, чем «мягче» действие кис-
лородных лигандов на центральный катион. Как показано на
рис. 4.10, система нерасщепленных уровней в комплексе Vv бо-
лее дестабилизирована (лежит выше но энергии), чем в ком-
плексе VJV. С другой стороны, большее расщепление уровней
в случае Vv отвечает большей стабилизации Vv в поле лиган-
дов по сравнению с VIV; этот эффект, однако, не перекрывает
эффекта дестабилизации для нерасщепленных уровней.
При переходе носителей заряда с VIV на Vv длина связей
между центральным атомом и атомами лигандов увеличива-
ется, а уровень dxy понижается (рис. 4.10), что можно тракто-
вать как стабилизацию поляронного состояния. Такую стабили-
зацию называют «самозаглублением», причем для него качест-
венно показано, что различие в энергиях уровней еху для VIV
и Vv тем меньше, чем меньше поляризуемость кислородных
ионов в координационной сфере.
Из разности между молярной поляризацией Рм (измерения
диэлектрической проницаемости) и молекулярной рефракцией
Rm (измерения показателя преломления) можно получить ион-
ную поляризацию Pi = (4л/3) • Na Е XiPt, где Xi и pt отвечают
отдельным компонентам стекла. Так как в составе стекла пре-
обладают ионы кислорода, то можно ожидать, что ионная по-
ляризация будет пропорциональна их поляризации ро, которая
28*
436
Глава 4
ванадий (У)
ванадий (IV)
Рис. 4.10. Расщепление в поле лигандов d-уровней атома ванадия с симмет-
рией Civ.
Расщепление больше для VV, чем для VIV; сферическая симметрия приводит к боль-
шей дестабилизации системы уровней Vv.
связана с поляризуемостью р, используемой в конфигурацион-
ной модели, соотношением ро = т$. При $=npPi (/?p = const)
из уравнения (4.23) следует
£ —_у/ (4.27)
4akT 1 2 и ' '
При постоянной температуре зависимость Еа от Pj и зависи-
мость 1g (аа) от Pi, таким образом, графически изображаются
прямыми [82].
На рис. 4.11 представлены данные по электропроводности
ванадийсодержащих оксидных стекол (для удобства проводимость
нормирована по концентрации носителей); такой же характер
зависимости от среднего расстояния между атомами ванадия
демонстрирует и подвижность. В системах VO2—V2O5 [58, 77],
CaO—VO2—V2O5—Р2О5 (или В2О3) [87] и BaO—А12О3—VO2 -
V2O5—В2О3 [88] изменяются не только эти расстояния, но и со-
отношение между VIV и Vv, что также сказывается на величине
проводимости. В рядах А, В и С доминирующее влияние на
Электрические и оптические свойства
437
Рис. 4.11. Зависимость проводимости (нормированной к концентрации носи-
телей заряда) от среднего расстояния между атомами ванадия в различных
стеклообразующих системах.
1 и 2 - VO2-V2O5— Р2О5 [58]. 3 и 4 - CaO-VO2—V2O5— Р2О5 (или В2ОЯ) [87], 5— ВаО-
VO2-V205-Al203-B203 [88], 6 - BaO—VO2—SiO2 [79]; А, В, С - см. с. 433.
проводимость оказывает изменение поляризации оксидных
ионов (рис. 4.11).
Отметим в -заключение, что еще Мунаката [89] указывал на
зависимость механизма проводимости от величины поляризуе-
мости кислородных ионов, а Кеннеди и Маккензи, отмечая раз-
личие в проводимости ванадиевоборатных и ванадиевофосфат-
ных стекол, указывали на различную поляризуемость ионов
кислорода в обоих случаях [87]. Таким образом, анализ корре-
ляций между электропроводностью и поляризуемостью кисло-
родных стекол в изоструктурных рядах подтверждает точку
зрения Мотта [72], согласно которой энергия активации по-
движности определяется в основном поляризацией вблизи ло-
кализованных носителей заряда.
4.1.3. Электропроводность в стеклах и аморфных телах
с малой полярностью связей
Описанные в гл. 3 элементарные аморфные твердые тела
(В, Si, Ge, As и Se) и многочисленные халькогенидные стекла
являются полупроводниками, так как ширина их запрещенной
438 Глава 4
Е
Рис. 4.12. Схема плотности состояний в стеклообразном полупроводнике с не-
полярными и слабополярными связями.
Нейтральные центры у краев зон соответствуют связям между одинаковыми атомами.
Следует различать три вклада в проводимость: 1 — возбуждение в зонные состояния,
2— возбуждения в локализованные состояния в области &Elofl, 3 -- прыжки электронов
в области энергии Ферми.
зоны достаточно мала для термической генерации носителей
тока. В разд. 2.5.1.3 было показано, что разупорядоченность
в аморфных полупроводниках естественно приводит к появле-
нию локализованных электронных состояний у краев зон. На
основе критерия локализации Андерсона (см. уравнение
(2.121)) Мотт [90] ввел понятие порога подвижности, разде-
ляющего локализованные и делокализованные (распространен-
ные) состояния, и соответственно щели подвижности (псевдоза-
прещенная зона), показанную как интервал энергий (Ес — Ev)
на рис. 4.12 (см. также рис. 2.54). Коэн (гл. 2 [384]), однако,
высказал предположение, что падение подвижности при пере-
ходе от распространенных к локализованным состояниям не
столь резкое, и постулировал существование некоторой проме-
жуточной области для делокализованных состояний, располо-
женной выше Ес и ниже Ev.
Собственный беспорядок в аморфных полупроводниках при-
водит к появлению внутри щели подвижности многочисленных
уровней, отвечающих положительно и отрицательно заряжен-
ным дефектным центрам, а также — вследствие существования
гомополярных связей — уровней нейтральных центров
(рис. 4.12). Модель щели с многочисленными максимумами
выше и ниже уровня Ферми, отвечающими зонам доноров и
акцепторов, впервые была предложена Маршаллом и Оуэном
[91]. Вскоре (после 1975 г.) она была подтверждена исследо-
ваниями люминесценции, фотопроводимости и дрейфовой по-
движности, причем удалось установить определенную связь
Электрические и оптические свойства
439
между экспериментально определяемыми локальными уровнями
в зоне и предполагаемой структурно-химической природой соот-
ветствующих центров (разд. 2.5.2).
Процесс электрического транспорта в некристаллических
твердых телах, где макроскопическая проводимость обуслов-
лена термическим возбуждением в веществе, рассматривают
обычно с помощью трех механизмов (см. также рис. 4.12):
1) Перенос носителей между делокализованными состояни-
ями в зоне проводимости (Е>Ес) и в валентной зоне (Е<
<ЕУ); наличие таких состояний обусловлено ближним по-
рядком.
2) Перемещение носителей в хвостах зон (Ес — Ел) и (Ев —
— Еу); существование хвостов ширины AEZoft связано с тополо-
гическим беспорядком.
3) Прыжки носителей между локализованными состояни-
ями, расположенными в зонах вблизи уровня Ферми; эти со-
стояния обусловлены собственными дефектами и посторонними
примесями.
Учет всех трех механизмов дает следующее выражение для
электропроводности ([92, 93]; см. также гл. 1 [33]):
о = Ci exp (—\Ea/kT) + С2 ехр — (ЛЕ, + Л^,)/^Т] + С3 ехр (—
— \W2jkT) (4.28)
Для проводимости по распространенным состояниям, прини-
мая по аналогии с температурной зависимостью ширины опти-
ческой запрещенной зоны (разд. 4.2.1.4)
ЛЕо = Ео-уГ (4.29)
получаем
о, = С, ехр (y/fc) ехр (—Ea/kT) = aOi ехр (—Ea[kT} (4.30)
где ooi = e- М)фф- Цен • ехр (y/k) (Л^эфф — эффективная плотность
состояний на уровне Ес или Ev [см. уравнение (2.112)]). Для
некристаллических твердых тел обычно poi^5 см2«В-1-с-1, что
соответствует средней длине пробега, меньшей межатомного
расстояния (разд. 2.5.1.2). При этом по Коэну (см. гл. 2 [384])
движение носителей описывается как диффузионное — типа
броуновского молекулярного движения, откуда следует
Мо1 = (еа2/6^Т) ve (4.31)
При частоте перескоков электронов ve~1015 с-1, у = 2-?8Х
ХЮ-4 эВ • К-1 (см. [93] и гл. 1 [33]), poi~5 см2 • В-1 • с-1 и
Мэфф^Ю19 см-3 получаем для ooi величину порядка 103 СмХ
Хсм-1.
Если перенос заряда осуществляется по локализованным со-
стояниям в хвостах зон, то проводимость обусловлена прыж-
440 Глава 4
ками электронов * и подвижность можно описать выражением
аналогичным (4.24) с vo = vph/Q, а именно:
ехр(-ДГ,/ЛГ)=(1юехр (~&W,/kT) (4.32)
Тогда при vp/i~10'2 с-1** получаем уменьшение подвижности
при переходе от распространенных к локализованным состоя-
ниям в 103 раз (порог подвижности). В рассматриваемом слу-
чае проводимость описывается уравнением
а2 = е/УтЦог exp (—\WJkT) ey,k exp (—Ei/kT) (4.33)
При a^l нм и е2са^1 в предположении ц02~Ю-2 см2-В ’Х
Хс-1 и Мг«1019 см-3 (плотность состояний в хвостах вблизи
Ес или Ev) получаем для o02=C2ev/ft = e-• рог-е^ значение
<у02~0,5 См-см-1.
Из соотношений Еа > Е{ и Qoi > ^02 следует, что в области
низких температур си < стг, т. е. низкотемпературная проводи-
мость осуществляется в основном по прыжковому механизму,
в то время как в высокотемпературной области преобладает
проводимость по распространенным состояниям. В приближе-
нии линейной зависимости плотности локализованных состоя-
ний в хвостах от энергии можно записать [93]
ог = ок С„2ехр[ - (Е, + ДГ,)/*Т] (4.34)
LbE-lOk.
где С02 = 1—ехр (—AEioh/kT) [1+ (AEiok/kT)]-, в этом случае за-
висимость In а от 1/Т не является линейной. При повышенных
температурах, когда оба механизма проводимости сосущест-
вуют, а2 обычно подавляется и в измерениях проявляется
только Оь
Третье слагаемое в уравнении (4.28) соответствует переско-
кам носителей между локализованными состояниями вблизи
уровня Ферми; этот механизм сходен с механизмом примесной
проводимости в сильно легированных и частично компенсиро-
ванных кристаллических полупроводниках (см. рис. 2.52). Если
вероятность прыжка определить как p = vPhexp (2са — Д^г/бТ),
то по Мотту (гл. 2, [382]) получаем (с использованием соот-
ношения Нернста—Эйнштейна \\,=eDjkT и выражения D =
= (1/ера2) уравнение, сходное с уравнением (4.32):
о3 = ea2vPhN (EF) exp (—2са) ехр(—\W2/kT) =
= о0зехр(—&W2jkT) (4.35)
* Дырок для «хвоста» (Ев—Ev).— Прим. ред.
** и W~kT.—Прим. ред.
Электрические и оптические свойства
441
где Af (EF) — плотность дефектных состояний в районе уровня
Ферми, a N(EF)kT — число электронов, участвующих в прово-
димости.
Множитель ехр (—2са—AWz/kT) принимает максимальное
значение при прыжках между соседними центрами. В случае
механизма с переменной длиной прыжка получаем закон Мотта
Г ’'4 (см. 4.1.2.1) [72, 93] (см. также гл. 1 [33]) *
_ Г 9 /____
6 [8jicN(EF)kT ] т'1* )
(4.36)
где Л = 2,l(c3/kN(EF)]\
Обзор по прыжковой проводимости в некристаллических
твердых телах дается в работе [94]. Следует отметить также,
что проводимость в таких материалах успешно описывается
в рамках поляронного подхода Эмина [95, 96] (см. разд. 2.5.1.2
и 4.1.2.1).
4.1.3.1. Перенос заряда в халькогенидных стеклах
Статическая проводимость. В литературе неоднократно об-
суждалась проводимость халькогенидных стекол, частично рас-
смотренных ранее в гл. 3; при этом отмечалось, что в относи-
тельно широком интервале температур наблюдается линейная
зависимость In о от обратной температуры [92] (см. также гл. 1
[20] и гл. 3 [263, 267, 636]. Так как возможность измерения
электропроводности при низких температурах ограничена ха-
рактерным для стекол высоким сопротивлением, то для таких
измерений наиболее пригодны теллурсодержащие стекла, кото-
рые являются сравнительно низкоомными. Верхняя темпера-
турная граница измерения электропроводности обычно ограни-
чивается температурой стеклования Т#, выше которой, как из-
вестно, начинается размягчение и кристаллизация ** стекол;
однако во многих работах было показано, что при переходе
к расплаву кривая проводимости нс претерпевает разрыва ***
(см., например, гл. 3 [267, 636]).
Из рис. 4.13, на котором представлены данные по электро-
проводности для различных стеклообразующих систем, видно,
* В случае, когда плотность состояний в середине зоны обращается
в нуль («кулоновская щель») зависимость проводимости описывается зако-
ном Г'^2 [Шкловский, В. И., Эфрос /1. Л. Электронные свойства легирован-
ных полупроводников.— М.: Наука, 1979, 416 с.].— Прим, псрев.
** Для типичных стекол заметная кристаллизация происходит значи-
тельно выше Tg, однако максимум скорости зародышеобразования может при-
ближаться к температуре стеклования.— Прим. ред.
*** Если при нагреве удалось избежать кристаллизации.— Прим. ред.
442 Глава 4
что энергия активации проводимости увеличивается при увели-
чении содержания компонентов с меньшей атомной массой. По-
добным образом меняются оптическая ширина запрещенной
зоны и величина щели подвижности. Хотя значения проводи-
мости при обычных температурах сильно различаются для раз-
ных составов, в пределе 1/7' 0 величины о0 сходятся в до-
вольно узкой области.
Рис. 4.13. Температурная зависимость проводимости полупроводниковых сте-
кол и аморфных слоев.
1 ~ GeTe, 2 — Tl2As2SeTe3, 3 — As2Te3> 4 — Asi0Se3Tei2, 5 - - CdGeAs2, 6 — Tl2As2Se3. 7 —
AseSbiSeis, 8 — As2Se3, 9 — Ge2Se3, 10 — As2S3, 11 — Se.
Из рис. 4.14, где сведены данные по проводимости ковалент-
ных некристаллических полупроводников в координатах 1g о
от энергии активации проводимости, соответствующих уравне-
ниям (4.3) или (4.28), видно, что в высокотемпературном пре-
деле су0= 102Н-104 См-см-’1. Такие величины предэкспоненты
характерны для кристаллических полупроводников. Этот факт,
а также то, что величина энергии активации проводимости
в некристаллических полупроводниках, как правило, составляет
половину оптической ширины запрещенной зоны, позволяют
утверждать, что в данном случае проводимость собственная,
а уровень Ферми расположен вблизи середины щели подвиж-
ности.
Собственная проводимость в случае кристаллических полу-
проводников означает, что в соответствующей температурной
Электрические и оптические свойства
443
Рис. 4.14. Связь проводимости при 298 К с энергией активации проводимости
для некристаллических (О) и кристаллических (ф) полупроводников.
области примеси и дефекты решетки не дают вклада в электро-
проводность, причем независимость положения уровня Ферми
от температуры отвечает условию п = р и приблизительного ра-
венства эффективных масс электронов и дырок. Если взять не-
кристаллические полупроводники и среди прямых на рис. 4.13
провести усредненную прямую, то для о = 10-15 См - см-1 она
пересечет ось температур при 140 К. Тогда при энергии акти-
вации Еа = 0,5 эВ и подвижности ц~1 см2-В-1-с-1 получаем
концентрацию носителей порядка 103 см-3, которая отвечает на-
личию в щели подвижности не более 5- 102 доноров или акцеп-
торов. Но, с другой стороны, из эксперимента, особенно из из-
мерений эффекта поля [92, 97], известно, что в стеклах имеется
высокая концентрация разнообразных дефектных центров, обес-
печивающих существование многочисленных максимумов плот-
ности состояний внутри щели подвижности (см. рис. 4.12). По-
этому считают, что в некристаллических полупроводниках не-
зависимость энергии Ферми от температуры возникает как след-
ствие особой природы дефектных центров, обеспечивающих
444 Глава 4
с обеих сторон уровня Ферми высокую плотность донорных и
акцепторных состояний *.
Таким образом, собственная проводимость некристалличе-
ских полупроводников, в отличие от кристаллических, обуслов-
лена не термическим возбуждением переходов носителей между
валентной зоной и зоной проводимости (в этом случае также
имеем EF ~(ЕС— Еу)^, а возбуждением электронов из распро-
страненных состояний вблизи потолка валентной зоны в локали-
зованные состояния вблизи уровня Ферми (тогда Еа в уравне-
нии (4.30) отвечает EF— Ev) или из состояний вблизи уровня
Ферми в зону проводимости (Еа^Ес — EF)\ в зависимости ог
преобладающего процесса можно говорить соответственно о ды-
рочной или электронной проводимости. В случае прыжковой
проводимости по локальным состояниям в хвостах зон в урав-
нение (4.33) подставляется энергия активации Е{, равная ЕА—
Ef или Ef.— Ев (см. рис. 4.12). В халькогенидных стеклообраз-
ных полупроводниках обычно (Ес— EF) > (EF — Ev) и
(Ел — EF) > (Ef — Ев), т. е. уровень Ферми несколько смещен
от середины щели в сторону валентной зоны. С этим связывают
экспериментально наблюдаемое преобладание дырочной прово-
димости в таких материалах.
Стрелки на рис. 4.12 характеризуют эффективную отрица-
тельную корреляционную энергию, о которой говорилось
в разд. 2.5.2.2. Согласно схеме (2.128) IA (1) >/д(2), т. е. дву-
кратная ионизация центров 7?_ пли Z)4- (двухэлектронное воз-
буждение) энергетически более выгодна, чем их одноэлектрон-
ное возбуждение.
Эффект Зеебека. Измерения термо-э. д. с. в халькогенидных
стеклах указывают на дырочный тип их проводимости [92]
(см. также гл. 1 [33] и гл. 3 [267]). Термоэлсктродвижущая
сила (термо-э. д. с., коэффициент Зеебека) Siu определяется как
коэффициент, связывающий напряжение ДУ, возникающее ме-
жду контактами при разнице их температур \Т : SV^Siu • \Т.
Для большинства полупроводников зависимость термо-э.д.с.
от температуры хорошо описывается уравнением
5л=±4(4г + Л) <4-37>
При положительном знаке Stu в случае проводимости по рас-
пространенным состояниям в некристаллических полупроводни-
ках имеем
sn (1) = ~ (~FiTEv - V + 9 <4-38>
* Это общепринятое объяснение явления закрепления или пинпинга ([.’in-
ning) уровня Ферми, которое особенно ярко выражается в характерной
нелегируемости халькогенидных стекол (см. разд. 4.1.3.5).— Прим. ред.
Электрические и оптические свойства
445
Однако экспериментально определяемое из зависимости Sth от
1/Т значение Es оказывается ниже, чем энергия активации,
определяемая по наклону прямой In о в зависимости от обрат-
ной температуры. Это расхождение можно объяснить вкладом
прыжкового механизма проводимо-
сти (см., например, модель прыж-
ков поляронов для интерпретации
стационарной проводимости [93] и
также разд. 4.1.2.1), так как энер-
гия активации прыжков не входит
в выражение для Sth [92, 93] *. Ес-
ли же справедливо равенство Е$ =
= АЕ0 = Еа — уТ [см. уравнение
(4.29)], то из измерений термо-
э. д. с. можно независимым спосо-
бом получить значение температур-
ного коэффициента для оптической
ширины запрещенной зоны.
В случае достаточно низких тем-
ператур, когда проводимость осу-
ществляется прыжками в хвостах
зон, в уравнение (4.38) вместо
Еъ—Ev вводится Ef — Ев:
(4.39)
Из рис. 4.15 видно, что при повы-
шении температуры происходит
переход от прыжковой проводимо-
сти к проводимости по распростра-
ненным состояниям, при этом в со-
ответствии с изменением дрейфо-
вой подвижности и проводимости
Рис. 4.15. Корреляции между
Sth, 1g о и дрейфовой подвиж-
ностью цо при изменении ме-
ханизма проводимости [98].
термо-э. д. с. демонстрирует переход из одного режима в дру-
гой, с различными Es [93, 98] (см. также гл. 1 [33]).
По измерениям Оуэна и Робертсона (см. гл. 3 [636]) для
аморфных As2Se3, 2As2Se3 • As2Te3 и As2Se3 • 2As2Te3 температур-
ная зависимость Sth, соответствующая проводимости ио рас-
пространенным состояниям, проявляется только в расплаве.
При измерении эффекта Зеебека в стеклах часто попадают
в переходную область, причем, если имеет место резкий переход
* Для прыжкового механизма по хвостам зон общий вид уравнения
(4.37) сохраняется, ио при этом ES~EF— ЕА для электронного типа про-
водимости и Es = En — Ef для дырочного.— Прим. ред.
446 Глава 4
от di к 02, то в переходной области наблюдается неожи-
данный рост S(/t с увеличением температуры, как это показано
на рис. 4.15.
Следует ожидать, что при снижении температуры будут по-
следовательно реализовываться механизмы проводимости со все
более низкими энергиями активации. На практике, однако, со-
ответствующие изломы на зависимости In о от 1/7 удается за-
фиксировать далеко не всегда, а только для сравнительно низ-
коомных стекол, в которых возможны измерения проводимости
в довольно широком диапазоне температур. Так, в объемных
стеклах, которые не относятся к сравнительно хорошо прово-
дящим, вклад о3 вообще не наблюдался. Тем не менее, с по-
мощью измерений термо-э. д. с. в общем случае удается выде-
лить энергетические составляющие уравнения (4.28) Ea = EF—
— Ev, Ei=Ef — Ев и AIFi, которые относятся к сп и о2. Соот-
ветствующие данные приведены в табл. 4.4.
Экспериментальные данные для As2Se3 и As2Te3, обработан-
ные в соответствии с обычно применяемой концепцией о двух
составляющих проводимости (0 = 01 + 02), показывают ано-
мально большое для 002 значение предэкспоненциального мно-
жителя (табл. 4.4), причем из измерений термо-э. д. с. известно,
что проводимость в данном случае осуществляется по хвостам
зон. Это противоречие между ожидаемой и наблюдаемой вели-
чиной О02 удается объяснить в рамках модели поляронных
прыжков, которые обеспечивают, по-видимому, перенос заряда
в стеклообразующих системах As—Те, Ge—As—Те и As—Те—I
[96] (см. также гл. 3 [407, 703] и разд. 4.1.2.1). При этом тем-
пературная зависимость проводимости определяется в основном
термической активацией подвижности, а предэкспонента оцени-
вается как Oo2 = Ю2 +- 103 См • см-1 (см. гл. 3 [636]). Тогда
уменьшение наклона зависимости In о от 1/7 при снижении тем-
пературы, которое наблюдается, например, для стекол As2Te2l2
и AsTe (см. гл. 3 [703]) можно рассматривать как переход от
адиабатического к неадиабатическому режиму прыжков поля-
ронов (см. разд. 4.1.2.1). С другой стороны, температурное
поведение проводимости в стеклах состава AsTei.sS*- и в важ-
ном модельном стекле As3,oGei,oSe2,2Te4,8 было объяснено [93]
(см. также гл. 3 [707]) на основе модели Мотта—Девиса (см.
гл. 1 [33]), без привлечения поляронных представлений.
Примером некристаллического полупроводника с отрица-
тельным знаком термо-э. д. с. является аморфный мышьяк. Для
стекол системы GexSei-x, в частности для Ge2Se3, из измерений
дрейфовой подвижности (см. разд. 4.1.3.4) тоже был сделан вы-
вод об электронном типе проводимости [143]. Электронными
проводниками, по-видимому, являются и стекла Ge—Se—Те
[144]. Увеличение содержания висмута в стеклах систем Bi—
Ge—Se и Bi—Ge—Se—Те приводит к увеличению электронного
Электрические и оптические свойства
447
вклада в проводимость по сравнению с дырочным [145, 146].
В случае установления электронного типа проводимости вели-
чины (EF —Еу) и (Ef — Ев) в табл. 4.4 и в уравнениях (4.30,
4.34, 4.38, 4.39) заменяются на (Ес— EF) и (ЕА — EF).
Эффект Холла. В образце, по которому протекает ток плот-
ности /а, под действием внешнего магнитного поля Нн возни-
кает электрическое поле FH. Эти величины связаны между со-
бой коэффициентом Холла (постоянной Холла)
Rh = FhH„Hh = --^- или (4.40)
Знак Rn совпадает со знаком носителей заряда в случае уни-
полярной проводимости (преобладание одного типа носителей),
а из величины Rn можно (при известном коэффициенте рассея-
ния гн) определить концентрацию носителей. Уравнение
Ця = | Rn | о = | гп | р (4.41)
определяет холловскую подвижность рн, которая в гн раз пре-
вышает подвижность в уравнении для проводимости (4.2). Для
кристаллических полупроводников гн немного больше единицы.
В некристаллических полупроводниках длина свободного про-
бега носителей сравнима с межатомным расстоянием, поэтому
обычная интерпретация эффекта Холла здесь неприменима.
Для большинства халькогенидных стекол коэффициент
Холла отрицателен, т. е. наблюдается аномальное несовпадение
знака носителей, определенного из эффектов Холла и Зеебека
[111, 112] (см. также гл. 1 [20]). Исключением является, на-
пример, стекло TlAsTe2, в котором знаки Sth и RH совпадают
(см. гл. 3 [751]). В аморфном мышьяке RH 2> 0, но •< 0
(см. гл. 3 [189]).
Фридман * получил для случая межзонной проводимости со-
отношение
4л еа2 z
Ня — з о, •* А/Эфф ——
(4.42)
где J — интеграл перекрытия между соседними центрами за-
хвата, А.эфф — плотность состояний в области Ес или Еу, z—
координационное число, z — число взаимодействующих центров.
Величина связывается с подвижностью для проводимости ц
следующим образом:
\iHlv = kTIJ
(4-43)
* Строго говоря, это соотношение было получено Фридманом в модели
случайных фаз для кристаллических полупроводников, однако его исполь-
зуют и для некристаллических полупроводников в режиме проводимости
по распространенным состояниям.— Прим. ред.
Таблица 4.4. Проводимость типичных халькогенидных стекол
Стекло o0i, Сч/см !-.o=l:p- Ey, эВ o02, Cm/cm
Se 104 1,13 —
Asa 8 • 10’ 0,81 13
TlAsTe2 1,2 • 103 0,285 1,1
Т1 AsTe( !5Se0,5 2,5 • 103 0,355 33
As2Se3 1.2; 1,06 1,5 • 103
1,5 . Ю4 0,99 2 • 102
As2Te3 1,5 • 103
Ei—Ep — Ер эВ £S, эВ Д1Р|, эВ Литература
— 1,15 — 100, см. литературу к гл. 1 [33]
0,50 0,165 0,280 0,50 0,098 См. литературу к гл. 3 [189] 105 105
0,91 0,60 0,31 См. литературу к гл. 3 [267, 407]
0,78 0,09 См. литературу к гл. 3 [639]
0,42 (ЗЗОК) 0,38 (130К) 0,46 (>230К) 0,44 0,25 0,25 (210К) 0,30 0,17 0,13 См. литературу к гл. 3 [407] 101' 102
Заказ № 413
AsTej >5Si0,25 3200 0,46 90
AsTe! i5Sio,44 3200 0,615 70
AsTe
As6Te0I
AsgTelg
AsTejeGe3
Ge2Se| 5,6 • 103 1,16 0,59
Ge2Se2Tea 9,2 • 102 0,86 0,0023
CdAs2 7,5 • 104 0,67 4,8 • 102
а В этих материалах преобладает электронная проводимость.
0,383 0,515 0,44 0,25 0,19 См. литературу к гл. 3 [707] Там же
См. литературу к [703] гл. 3
0,36 0,32 0,04 См. литературу к [411] гл. 3
0,45 0,27 0,18 См. литературу к [703] гл. 3
0,43 0,89 0,56 0,453 0,24 0,19 См. литературу к [703] 106 114 гл. 3
450 Глава 4
откуда при 1 эВ получаем цн не более 101 см2-В-1«с-1,
т. е. в случае проводимости по делокализованным состояниям
холловская подвижность не превышает !/ю величины подвиж-
ности для проводимости.
Из теории Фридмана следует отрицательный знак Rn в слу-
чае конфигурации из трех взаимодействующих центров, даже
если электропроводность осуществляется дырками; таким обра-
зом, впервые было объяснено несоответствие знаков эффектов
Холла и Зеебека для стеклообразных полупроводников [109].
Холстейн получил такой же вывод в поляронной картине — для
прыжков поляронов в трехцентровой конфигурации [НО].
Теоретически оцененная величина Цн~10”1 см2-В-1-с-1 и
ее независимость от температуры по уравнению (4.42) не про-
тиворечат экспериментальным данным для стекол и расплавов
систем As—Se—Те [Hl], Ge—As—Те [112] и Т1—As—Se—Те
[111, ИЗ] (эти данные приведены также в [92]). В то же время
в стеклообразных халькогенидах мышьяка (см. гл. 3 [639]) и
в стеклах систем As—Те, As—Те—I и Ge—As—Те [92] в обла-
сти низких температур холловская подвижность начинает
уменьшаться. В системе As2Se3—As2Te3 величина prf и ее энер-
гия активации проходят через минимум в области 50 мол. %
(гл. 3 [639]). Для стекол AsTei>5Sex и Aso,3oSio,i2Gco(ioTeo>4i вели-
чина цгг линейно зависит от 1/Т, что, вероятно, связано с пере-
ходом от проводимости по распространенным состояниям к про-
водимости по состояниям в хвостах зон.
4.1.3.2. Электрические свойства
аморфных кремния, германия
и родственных систем
Как было показано в разд. 2.5.2.1, в некристаллических
твердых телах с положительной эффективной корреляционной
энергией преобладают нейтральные дефектные центры с неспа-
ренными электронами, представляющие собой оборванные го-
молитические связи. Однако вследствие разупорядоченности и
связанных с ней флук1уаций потенциальной энергии возможно
образование некоторого количества заряженных центров, что
можно рассматривать как результат перекрытия полос плотно-
сти состояний вблизи уровня Ферми (см. рис. 2.56). Заряжен-
ные дефектные центры, однако, нетипичны для таких материа-
лов.
В данном случае нельзя получить объемные образцы, и элек-
трические свойства изучаются на пленках по методике, изложен-
ной в разд. 2.1.3, причем, помимо чувствительности пленок
к условиям приготовления, при измерениях на тонких пленках
возможен неомический характер проводимости, так как даже при
Электрические и оптические свойства
451
напряжении в несколько вольт в образце возникает электриче-
ское поле высокой напряженности.
Работы по проводимости аморфного кремния и германия,
выполненные до 1973 г., проанализированы в обзорах [92],
гл. 3 [128], и гл. 3 [139]. Позднейшие исследования a-Si отра-
жены в [115], a a-Ge — в [117].
Коэффициент Зеебека в этой группе полупроводников, как
правило, отрицателен, хотя при высоких скоростях осаждения
Рис. 4.16. Температурная зависимость проводимости пленок a-Ge (а), полу-
ченных осаждением из газовой фазы при температуре подложки 313 К и пле-
нок a-Si (б), полученных электроннолучевым распылением.
На кривых указана температура предварительного отжига пленок.
аморфных слоев возможно появление Sth > 0 (см. гл. 3 [139,
267]). Величина термо-э. д. с. для Ge при Т < 270 К и для Si
при Т <С 430 К мало зависит от температуры.
На рис. 4.16 показано влияние условий приготовления
аморфных слоев на их проводимость. Для образцов, отожжен-
ных при достаточно высокой температуре, можно наблюдать
изменение хода зависимости 1п о от 1/7 в области высоких тем-
ператур измерения, которое, по-видимому, говорит о переходе
к механизму проводимости по распространенным состояниям.
Следует отметить, что плотность спинов, определенная методом
ЭПР, тоже уменьшается при увеличении температуры отжига
[115]. Как видно из рис. 4.17, который соответствует данным на
рис. 4.16, для областей со сравнительно слабой температурной
зависимостью проводимости, при достаточно низкой темпера-
29*
452 Глава 4
туре выполняется закон Мотта Т~'1* [см. уравнение (4.36)], свя-
зываемый с прыжками носителей по локализованным состоя-
ниям вблизи уровня Ферми. Однако для слоев, полученных на-
пылением в сверхвысоком вакууме, было показано, что при
толщине слоев, сравнимой с длиной прыжка, наблюдается пе-
реход к зависимости Т~'/з [118].
Девис и Мотт [119] связывают проводимость в a-Ge при
Т 2> 300 К с переносом носителей по делокализованным состоя-
Рис. 4.17. Выполнение закона Мотта
рис. 4.16) [116, 117].
у-'Л в пленках Ge (а) и Si (б) (см.
ниям вблизи дна зоны проводимости (ooi ~ Ю3 См-см-1), при-
чем с повышением температуры предварительного отжига соот-
ветствующие энергетические уровни смещаются к Ес (см. гл. 1
[33]). При Т < 300 К осуществляется механизм прыжковой
проводимости по локализованным состояниям вблизи уровня
Ферми; однако до температур выше 200 К далеко не всегда
реализуется механизм с переменной длиной прыжка, где спра-
ведлив закон Т~'!\
В работе [120] была показана применимость закона Мотта
для a-Si. По [116] перенос заряда в аморфном кремнии при
Т > 500 К осуществляется по распространенным состояниям
(Ео = 0,69 эВ), при 200 <С Т <С 500 К по состояниям в хвостах
зон (Ei=0,15 эВ) и при Т<200 К по состояниям вблизи
уровня Ферми (AU72 = 0,l эВ). По наклону прямой на
рис. 4.17, б для неотожженного образца с использованием урав-
нения (4.36) можно получить концентрацию дефектных цент-
ров, равную 1019 см-3.
Электрические и оптические свойства
453
Что касается легированных слоев кремния и германия, то
особый интерес представляют гидрогенизированные или фтори-
рованные пленки a-Si : Н (или a-Si: F) и a-Ge : Н (или a-Ge :
: F) *, в которых концентрация неспаренных спинов и прыжко-
вая проводимость на уровне Ферми уменьшаются настолько,
что не поддаются измерению обычными методами. Такое пове-
дение объясняют эффективным насыщением оборванных связей
германия или кремния (см. гл. 3 [134]). Так как температур-
ная зависимость проводимости в координатах 1п а от \/Т
спрямляется [121 —124], но имеется различие между энергиями
активации проводимости и термо-э. д. с., то можно сделать вы-
вод о переносе заряда в таких материалах но локализованным
состояниям в хвостах зон.
Электрические свойства аморфного бора сходны со свойст-
вами аморфных германия и кремния (разд. 3.1.2.1). В тетра-
эдрических стеклах CdGeAs2, CdSnAs2, ZnGeP2 и CdSiP2, полу-
чаемых быстрым охлаждением расплава, как и в халькогенид-
ных стеклах, проводимость можно описать на основе двух
составляющих oi и о2 [уравнения (4.28)] [125]. Концентрация
парамагнитных центров в таких стеклах, если они вообще есть,
очень низка [126]. В стеклообразном CdAs2, в котором высо-
котемпературная проводимость осуществляется по локализован-
ным состояниям в хвостах зон, при Т < 180 К наблюдали пере-
нос заряда по локализованным состояниям вблизи уровня
Ферми по механизму с переменной длиной прыжка [114].
4.1.3.3. Динамическая проводимость
Если перенос заряда осуществляется по распространенным
состояниям, расположенным выше Ес или ниже Ev, то прово-
димость не зависит от частоты, по крайней мере до частот
~ 10s Гц. В случае же переноса заряда за счет прыжков ме-
жду локализованными состояниями в хвостах зон (см. область
^Eiok на рис. 4.12) или вблизи уровня Ферми проявляются про-
цессы релаксации, которые можно наблюдать уже при часто-
тах ~ 103 Гц.
Для прыжковой проводимости по состояниям вблизи уровня
Ферми Остин и Мотт [74] получили выражение
а (со) = e2kT [А (EF)]2 [In vpft/(o]4 (4.44)
где N(Ef) —плотность состояний вблизи уровня Ферми, с опи-
сывает спадание волновой функции по длине прыжка [см. урав-
нение (4.24)], a vPh есть фононная частота (~1012н-1013 с-1).
* Мы используем здесь общепринятое обозначение этих материалов.—
Прим. ред.
454 Глава 4
По уравнению (4.44) о (со) должна линейно возрастать с ростом
температуры, а крутизна кривой 1п ст(со) от In со несколько
уменьшается с увеличением частоты. Уравнение (4.44) по ана-
логии с уравнением (4.18) можно представить в виде
о (со) « const о)" « const о0,8 (4.45)
где показатель степени п определяется как t/(ln o)/d(ln со). При
п=1—4/ln (vp/i/o)) и Vph= Ю13 с-1 имеем /2 = 0,84 для со = 102с“1
и /2 = 0,65 для со = 108 с-1. Таким образом, общепринятая вели-
чина /г = 0,8 должна быть справедлива только в области частот
- 104 Гц.
Случай переноса заряда по хвостам зон тоже отвечает тер-
мически активированным прыжкам носителей по локализован-
ным состояниям, и, следовательно, можно ожидать частотную
зависимость вида <о [In (vp^/co)]4. При этом температурная за-
висимость проводимости, как и для проводимости на постоян-
ном токе, имеет активационный характер:
о (со, Г) ~ &Т ехр[— (A^/AjT)] (4.46)
Конкретный механизм проводимости в данном веществе за-
висит как от температуры, так и от частоты. Эксперименталь-
ные данные для аморфных Si и Ge хорошо описываются урав-
нением (4.44) причем полученная таким образом плотность со-
стояний 1019—1020 эВ-1 • см 3 хорошо согласуется с величиной,
определенной из температурной зависимости проводимости
в области справедливости закона Мотта Т_,/4 [127—129]. В ра-
боте [130] проведен критический анализ соответствующих экс-
периментальных данных, включая данные для халькогенидных
стекол. Следует отметить, что в стеклах As2Se3 [131 —133],
As2S3—As2Te3 (см. гл. 3 [636]), As2Te3, AsSeTe2, As32Sii0Ge5Te53
[135] и многих других проводимость описывается уравнением
(4.45) с показателем степени п от 0,7 до 1,0 [92, 93].
Из рис. 4.18, где представлены типичные экспериментальные
данные, видно, что в динамическом режиме проявляется и про-
водимость на постоянном токе, вклад которой тем больше, чем
выше температура и меньше частота. При этом в области низ-
ких температур динамическая составляющая слабо зависит от
температуры. Разделение проводимости на две составляющие
означает реализацию двух различных механизмов переноса за-
ряда, однако следует соблюдать осторожность при интерпре-
тации этих механизмов. Так, например, обработка эксперимен-
тальных данных для халькогенидных стекол по уравнению
(4.44) дает плотность состояний в окрестности уровня Ферми
1018—1021 эВ-1-см-3, однако, как известно, прыжковая прово-
димость в этом классе стекол в статическом режиме не прояв-
ляется.
Электрические и оптические свойства
455
Новые теоретические исследования проводимости на пере-
менном токе с точки зрения прыжкового механизма выполнены
Звягиным [147]. Температурную и частотную зависимость ди-
намической проводимости для халькогенидных стекол можно
Рис. 4.18. Температурная и частотная зависимость проводимости стеклообраз-
ного As2Se3 по 1133].
/ — статическая проводимость, 2 — 5104 Гц, 3- -3-105 Гц, 4 — 3.10е Гц, 5 — 1,4 - 10т Гц.
Штриховые линии соответствуют разности между статической и динамической проводи-
мостью при одной и той же температуре.
объяснить на основе модели специфических дефектов — пар
с переменной валентностью, рассматривая совместные прыжки
двух электронов такой пары через потенциальный барьер [136]
(разд. 4.1.3.5). Очевидно, на динамическую проводимость силь-
ное влияние должны оказывать близко расположенные поло-
жительные и отрицательные дефекты.
4.1.3.4. Дрейфовая подвижность
Измерения дрейфовой подвижности оказываются весьма по-
лезными для определения плотности состояний, прежде всего
в хвостах зон, и для изучения природы дефектов. В таких экс-
периментах па образец (толщиной, например, 50 мкм) нано-
сятся контакты, причем в случае оптического возбуждения но-
сителей один из них делается прозрачным. При облучении
коротким импульсом сильнопоглощаемого света (или при бом-
бардировке электронами) в приконтактной области создается
высокая концентрация носителей, которые при соответствущем
знаке будут распространяться через образец к другому кон-
такту. Если в образце имеется высокая концентрация собствен-
ных носителей, то избыточные носители экранируются и
456 Глава 4
поэтому в эксперименте проявляются только неосновные носи-
тели заряда. Однако в случае некристаллических полупроводни-
ков с характерным высоким сопротивлением (обычно р>
> 107 Ом • см) при времени пролета носителей (transit time)
tD меньшем, чем время диэлектрической релаксации, инжекти-
руемые носители не экранируются, вследствие чего появляется
возможность раздельного определения электронной и дыроч-
ной подвижности. Дрейфовая подвижность определяется как
VD = d/FtD (4.47)
где F — напряженность электрического поля, приложенного
к образцу толщиной tZ; время пролета определяется экспери-
ментально из измерений зависимости дрейфового тока от вре-
мени после приложения возбуждающего импульса (рис. 4.19).
Этот метод (time-of-flight experiments) для некристаллических
полупроводников впервые применил Спир (гл. 3 [264]); см.
также [92, 93] и гл. 1 [33].
За время прохождения заряда носители периодически за-
хватываются на ловушки (и освобождаются с них при возмож-
ности термической активации), и изменение их числа в про-
цессе дрейфа можно выразить как
nD(f) = nD(0)exp(—t/xD) (4.48)
где Тл — среднее время жизни носителей. С захватом носителей
на ловушки можно связать тот факт, что дрейфовая подвиж-
ность, как правило, меньше подвижности ц, входящей в урав-
нение для проводимости. При плотности дрейфующих носите-
лей пр, плотности захваченных носителей nt и условии =
= ц[«п/(пр+п«)1 получаем
Но = Н [1 + WNC)] exp [ЕС - Et)/kT]~l (4.49а)
и соответственно
Но « Н (Nc/Nt) exp [- (Ес - Et)/kT] (4.496)
где Nt — плотность ловушек на локальном уровне Et, a Nc —
эффективная плотность состояний на уровне Ес. Таким обра-
зом, из наклона зависимости In нл от 1/Т можно определить
(Ес — Et) и, следовательно, глубину залегания ловушек, а по
пересечению этой зависимости с осью ординат можно опреде-
лить концентрацию ловушек. Применение этого метода при
различных толщинах образца и температурах дает возможность
исследовать спектр ловушек в образце.
В идеальном случае, когда ловушки отсутствуют, все носи-
тели, «рожденные» светом, одновременно достигают противопо-
ложного контакта, и ток скачком падает до нуля (рис. 4.19, а).
При предполагаемом гауссовом распределении ловушек по
энергиям следует ожидать кривую, показанную на рис. 4.19, б;
Электрические и оптические свойства
457
Рис. 4.19. Зависимость дрейфового тока от времени.
а — идеальный случай; б — случай гауссова распределения; в — дисперсионный транспорт;
г — перестройка кривой (в).
однако и этот случай практически не реализуется, особенно
для низких температур [93, 138, 139]. Реальная ситуация изо-
бражена па рис. 4.19, в — она известна как дисперсионный пе-
ренос. Обработка таких кривых в двойном логарифмическом
масштабе (рис. 4.19, г) дает время tXy которое подставляется
в уравнение (4.47) как время пролета. Полученная таким об-
разом дрейфовая подвижность цд оказывается зависящей от
приложенного поля и толщины образца (см., например,
рис. 4.20). В работах [140—142] обсуждается теория дисперси-
онного переноса.
Измерения дрейфовой подвижности используются и лля вы-
яснения влияния примесей на спектр ловушек; например, в ра-
боте Оуэна и Спира (см. гл. 3 [394]), из которой взят рис. 4.20,
приводятся результаты такого исследования на стеклообразном
селене с небольшими добавками мышьяка.*
Типичные данные по дрейфовой подвижности приведены
в табл. 4.5. Интересно, что по данным [99] дрейфовая подвиж-
ность носителей в слоях AS2S3, полученных при температуре
* О влиянии примесей на подвижность носителей заряда в стеклообраз-
ном селене см. также работу [Коломиец Б. Т., Лебедев Э. А.— Физ. и техн,
полупроводников, 1966, т. 8, с. 1136].— Прим, перев.
458 Глава 4
Рис. 4.20. Зависимость дрейфовой подвижности электронов и дырок в аморф-
ных слоях селена от температуры и напряженности электрического поля [394].
Таблица 4.5. Дрейфовая подвижность в некристаллических твердых телах
Вещества Up («). cm2/B-»c—i Ec~Et, эВ Bp (P) СМ2В-Ч:-' Et - Ev эВ Литература
Se 3—6 . 10-3 (300 K) 0,29— 0,33 1-2X ХЮ-’ 0,13— 0,28 См. литературу к гл. 3, 394
As2Se2,3 1,3 . io-4 0,44 —
As2Seg — — 2 • 10-6 0,53 142, 150; см. так-
же литературу к гл. 3, 395
GeSe2 1,4 • 10-* 4 . 10-2 151
GeSe4 1,0 • io-’ 0,28 3,8- 10-2 0,42
Ge2Se3 7,4 • 10-’ 1,6 - IO-2 143
Se97,3Te2i7 9,6 • 10-4 0,25 i,o-io-‘ 0,25
As6,6Se87,oTe6>4 1,2 • IO"4 0,29 4,0 • 10-3 0,14 См. литературу к гл. 3, 641
SiO2 <io-5 20-40 152; см. литера- туру к гл. 3, 441
Электрические и оптические свойства 459
298 К, почти на порядок выше, чем в слоях, напыленных на
подложку при температуре 458 К, близкой к температуре раз-
мягчения (или стеклования) Tg. Большое различие в подвиж-
ности электронов и дырок в кварцевом стекле говорит о силь-
ной асимметрии плотности состояний в хвостах валентной зоны
и зоны проводимости. Луковским (см. гл. 3 [441]) приведена
непротиворечивая трактовка экспериментальных данных для
SiO2 на основе модели взаимодействующих С~- и С+-центров
(см. рис. 2.40) при их концентрации 3- 1019 см-3. В работе [341]
на основе простых физических представлений связываются про-
цессы дрейфа и рекомбинации носителей.
4.1.3.5. Примеси и электрические свойства
Как впервые показал Коломиец (см., например, гл. 1 [20]),
примеси практически не влияют на электрические свойства по-
лупроводниковых стекол. Хотя на кривых проводимости и энер-
гии активации проводимости наблюдаются экстремумы вблизи
составов, отвечающих по фазовой диаграмме образованию со-
единений [например, при составе As2Se,3 в стеклах системы
As—Se (см. гл. 3 [395]); см. также рис. 3.44], но изменения
в районе этих экстремумов невелики по сравнению с очень
сильным влиянием нестехиометрии на электропроводность
в кристаллических полупроводниках.
Мотт (см. гл. 2 [382]) объясняет характерную нелегируе-
мость стеклообразных полупроводников способностью атомов
в стеклообразующих системах насыщать свои ковалентные
связи уже в расплаве вследствие лабильной структуры послед-
него; эта структура с насыщенными связями сохраняется и
в расплаве вследствие лабильной структуры последнего; эта
структура с насыщенными связями сохраняется и в твердом
стекле. В кристалле же атомы основного вещества образуют
строго определенную решетку, при введении в которую посто-
ронние атомы не всегда способны удовлетворить свои валент-
ные возможности, вследствие чего возникает примесная прово-
димость.
Модифицирование халькогенидных стекол *. Эксперимен-
тально было показано, что некоторые примеси способны сильно
* В данном разделе автор рассматривает введение примесей через рас-
плав, в то время как термин «модифицирование» (или химическая модифи-
кация) относится к так называемому холодному легированию, когда в ре-
зультате высокочастотного сораспыления основного вещества с примесями
переходных металлов резко меняется электропроводность пленок, чего не
удастся добиться введением той же примеси при синтезе массивных стекол,
т. е. через расплав. Случай холодного легирования кратко рассмотрен в сле-
дующем подразделе.— Прим. ред.
Таблица 4.6. Электрические свойства некоторых халькогенидных стекол в зависимости от вводимой нримсси
Стекло о, Cm/cm (300 K) Ea, эВ Содержание примеси о, См/см Ла, эВ Литература
Se IO"18 1,13 0,002% О 10-” 0,75 153
0,05% CI ю-9 0,56 гл. 2 [377]
0,1 мол. % К 10-13
As2Se3 IO '2 0,99 5 мол. % Си 10-8 0,68 154, 155
6 .IO-13 0,86 2,4 мол. % Си 6 . 10-'1 0,61 гл. 3 [724]
10-12 0,9 28,6 мол. % TI 2,5 • IO7 0,48 гл. 3 [749]
0,92 3 мол. % Ga 0,75
0,2 мол. % В 0,75 157
0,6 мол. % In 0,75
0,4 мол. ?6 AI 0,75
As3oSii2GeioTe48 4 • 10-8 0,68 1 мол. % Мп 4 • 10-7 0.5 гл. 3 [782]
(PbSe)y (Oe2Se3)o,5-o,5y (f/ = 0) 6,3 • io-15 «/ = 0,40 1,6 • io-10 0,70 гл. 2 [350]
(PbSe)tz (GeSe)0,65- у
(GeSe2)0,35 (t/ = 0) 1,8 . IO-15 0,97 у = 0,45 1,6 • ю-'° 0,65 гл. 2 [350]
Ge0)4-x SnvSeo|5Teo,i (x — 0) io-'5 0,78 х = 0,08 5 • 10-10 0,63 гл. 2 [336]
Geo,4-xSnvSeO)4Teo,2 (x==0) 2 • 10-13 0,56 х = 0,12 2,5 • 10-7 0,47 106
Электрические и оптические свойства
461
изменять электрические свойства стеклообразных полупровод-
ников. По-видимому, эти примеси действуют на состояния
в щели подвижности, связанные с дефектными центрами, и из
соответствующих экспериментальных зависимостей можно де-
лать определенные предположения о природе дефектов.
В табл. 4.6 приведены данные по влиянию модифицирующих
примесей на электрические свойства халькогенидных стекло-
образных полупроводников.
Для стеклообразного селена собственная разупорядочен-
ность приводит к появлению положительно и отрицательно за-
ряженных дефектов, как это показано на рис. 2.41; эти центры
фиксируют уровень Ферми примерно в середине щели подвиж-
ности. При введении калия или Кг5е собственные центры се-
лена С+ (или структурные единицы SeJ/2 Se+) разрушаются по
реакции
K2Se + Se3/jSe+ 2К+ + Se,/2Se~ + SeSe2/2 (4.50)
в соответствии с которой возрастает концентрация С~-центров
(или структурных единиц Seiy Se~). Если же вводить кислородо-
или хлорсодержащие примеси, например SeO2 или SeCh, или
провести частичное окисление или хлорирование, то из реак-
ций
SeCI2 + Se1/2Se-^ Se3/Se+ + 2Cr (4.51)
или
2SeO2 + SeSe2/a + 2Se, fSe~ 2Se3/2Se+ 4- 4SeI/2O~ (4.52)
следует образование отрицательно заряженных ионов хлора
или концевых ионов кислорода при одновременном возрастании
концентрации С+-центров. Эти процессы изображены на
рис. 4.21.
Если записать скорость генерации носителей в нелегирован-
ном селене как
Ро ~ ехр[-2 (Er- Ev)/W] = ехр[- (lA (1) + 1Л (2))/АТ] (4.53)
а скорость их рекомбинации как
Ря ~ р2 (4.54а)
то из условия динамического равновесия Pg = Pr следует выра-
жение для концентрации дырок
р ~ ехр [—(Ef — Ev)!kT} (4.55а)
462 Глава 4
Рис. 4.21. Парное возбуждение носителей заряда в халькогенидных стеклах
с отрицательной энергией корреляции на примере селена и схема, поясняющая
влияние примесей кислорода, хлора и калия на энергию активации прово-
димости.
На рис. 4.21 (вверху слева) сдвоенная стрелка отвечает преоб-
разованию центра С+ в центр С~ в результате термического
возбуждения двух электронов из валентной зоны, причем в со-
ответствии со схемой (2.128) имеем 2(£f—£v) = IA (1) +IA (2).
Концентрация полученных таким образом Су-центров доста-
точно высока и на ней практически не сказывается изменение
числа носителей за счет их термической генерации.
При легировании селена кислородом или хлором число
Су-центров уменьшается [рис. 4.21 (справа вверху)], и поэтому
следует принимать во внимание термическую генерацию носи-
телей, что приводит по Мотту (см. гл. 2 [377, 403]) к выра-
жению
Ря~р2[СГ] (4.546)
откуда следует
р — ехр [—2 (ЕЛ — Ev)/3/?r]
(4.556)
Электрические и оптические свойства
463
При введении таких добавок компенсация нарушается и следует
ожидать уменьшения энергии активации проводимости па '/з
по сравнению с величиной для нелегированного селена, что
приближенно согласуется с данными табл. 4.6. Оптическая ши-
рина запрещенной зоны селена при этом не меняется.
При модифицировании стеклообразного селена калиехМ уро-
вень Ферми должен смещаться в противоположную сторону
[рис. 4.21 (внизу)] с возможностью появления электронного
типа проводимости. Однако влияние калия на электропровод-
ность селена сравнительно невелико, даже при 0,1 мол. % К
(табл. 4.6). Это связывают с тем, что уровень Ферми в нелеги-
рованном селене смещен от середины щели в сторону валент-
ной зоны. Мотт (см. гл. 3 [237]) из анализа эксперименталь-
ных данных для селена, легированного различными примесями,
оценил величину такого смещения в 0,13 эВ.
В соответствии с рис. 4.21 генерация и рекомбинация носи-
телей в стеклообразном селене сопровождается разрушением и
образованием ковалентных связей. Центры С~ и С+ находятся
в динамическом равновесии, взаимно превращаясь друг в друга
путем двухэлектронных (двухдырочных) возбуждений. Рассмот-
ренные выше примеры легирования показывают тесную связь
между явлениями переноса заряда и изменениями структуры,
когда термическая генерация носителей связывается с обрати-
мыми перестройками собственных дефектов. Такое поведение
характерно для веществ с отрицательной эффективной корре-
ляционной энергией.
Аналогичным образом можно объяснить изменение проводи-
мости при введении 1 мол. % Мп в стекло Aso.3Sio,i2Geo,iTeo,48
(см. гл. 3 [782]). По данным ЭПР при этом образуются тет-
раэдрические структурные единицы МпТе4, которые должны
повышать концентрацию £)~-центров и понижать концентрацию
£>4-центров, т. е. нарушать компенсацию. При введении Мп
ширина щели не меняется. Вследствие сложного состава и
структуры стекла ничего определенного о строении дефектов
в этом случае сказать нельзя.
Атомы меди входят в стеклообразный As2Se3 в виде ионов
Си1, причем тоже в тетраэдрической координации [154, 158];
такие же структурные единицы реализуются и для других при-
месей в АзгЗе.з, включенных в табл. 4.6. Кастнер [159] предпо-
ложил, что соответствующие центры нейтральны и поэтому не
оказывают влияния на равновесие собственных дефектов в рас-
плаве As2Se3. Вхождение СщБе в сетку As2Se3 можно предста-
вить реакцией
7Se (AsSe2/2)2 + Cu2Se - > 2 {Se2/2 AsSeCu [Se (AsSe2/2)2]J (4.56)
464 Глава 4
N(E) N(E)
Рис. 4.22. Нарушение компенсации положительно и отрицательно заряженных
дефектов при введении PbSe в стекло Ge2Se3.
При этом отрицательно заряженные собственные дефекты обра-
зуются лишь при диссоциации:
Se2/2AsSeCu [Se (AsSe2/J2]3 4- Se (AsSe2/2)2
- {Cu [Se (AsSe2/2)2]J+ + Se2/j AsSe~ (4.57)
(см. разд. 3.2.1.5 и уравнение (3.9)), вследствие которой сни-
жается концентрация С+-центров, т. е. [Se(AsSe2/)3]+. Кон-
центрации образованных таким образом дефектов зависят от
соотношения их энтальпий; соответствующие равновесия были
рассмотрены в работе [160] (см. также гл. 2 [390]). Преиму-
щественно электронейтральным вхождением примеси в сегку
стекла можно объяснить слабое, по сравнению с кристаллами,
влияние примесей на электропроводность стекол. Электрические
свойова стекол в системах РЬ—Ge—Se и Ge—Sn—Se—Те,
рассмотренных ранее в разд. 3.3.6.4 и внесенных в табл. 4.6,
объясняются таким же образом.
В стеклах Geo,4Seo,5Teo,i и Geo,4Seo,4Teo,2 (без примесей), ко-
торые относятся к рассмотренному в разд. 3.2.2.5 типу Ge^X.!
(см. рис. 3.41), преобладает электронная проводимость (см.
табл. 4.4 и 4.5). Она связана, вероятно, с возбуждением элек-
тронов Т^-центров в зону проводимости (рис. 4.22), так как
трехкоординированный атом германия с неподеленной электрон-
ной парой, по-видимому, имеет наименьшую в системе энергию
ионизации. Образующийся при этом Т^-дефект «залечивается»
Электрические и оптические свойства 465
за счет взаимодействия с неподеленными парами электронов
атомов селена в соседних цепочках; при этом концентрация
С^-центров меняется незначительно и скомпенсированное со-
стояние сохраняется (аналогичные рассуждения применялись
выше для селена).
Введение в стекла системы Ge—Se—Те примесей SnSe или
SnTe изменяет структуру стекла, как это показано на рис. 4.22
на примере PbSe, вводимого в Ge2Ses; при этом концентрация
С+-центров снижается. Это относится и к стеклам (PbSe)yX
X (Ge2Se3) 0,5-0,5У, представленным на рис. 3.61. Для стекол /г-типа
в системах Bi—Ge—Se и Bi—Ge—Se—Те электронная прово-
димость, по-видимому, сохраняется при введении в них Sn11
или РЬ11 [145, 146].
На рис. 4.22 приведена схема, поясняющая изменения, про-
исходящие с исходным состоянием Ge2Se3 при введении при-
меси PbSe (SnSe, SnTe). Эти примеси встраиваются в струк-
туру стекла с разрывом мостиковых связей селена, т. е. с обра-
зованием центров С~, вследствие чего разрушаются С+-центры.
В то же время концентрация центров Т~ не меняется, и в ре-
зультате компенсация нарушается. Однако, как следует из из-
мерений мольного объема (рис. 2.43 и 3.61), при введении SnX
или PbSe возможно образование структурных единиц типа
Т^-центров; в этом случае концентрация С+-центров должна
повышаться, и можно ожидать перехода к дырочной проводи-
мости.
Заметное уменьшение энергии активации проводимости по-
является только при сравнительно большом содержании SnSe,
SnTe или PbSe, что говорит об относительно высокой концен-
трации положительно и отрицательно заряженных дефектов
в исходном стекле Ge2Se3 (см. гл. 2 [300, 398]). Как уже гово-
рилось в разд. 3.2.2.5 (рис. 3.41), в расплаве Ge2Se3 создаются
особенно благоприятные условия для диссоциации с образова-
нием таких дефектов.
В стеклах (PbSe) w (GeSe) 0.65—w (GeSe2) 0.3.5 при увеличении со-
держания PbSe энергия активации проводимости падает. (При
у —0 можно получить объемные высокоомные образцы, элек-
трические параметры их, как и для стекол GeSe—GeSe2, опре-
деляются экстраполяцией.) При замене GeSe на PbSe ширина
запрещенной зоны, а также предэкспоненциальный множитель
для проводимости на постоянном токе не меняются (см.
рис. 3.62). В стеклах GenSe(2+n)/2 концентрация С+- и Су-цеп-
тров, по-видимому, особенно велика при м>2. При п<2, ве-
роятно, наряду с С+-центрами образуются центры Т+:
3Ge2Se3 + GeSe -[Se(Ge25Мз]+ + [QeSe>/.]' (4.58)
30 Заказ Ns 413
466 Глава 4
Спектры комбинационного рассеяния таких стекол (см. гл. 3
[543]) подтверждают эти предположения.
Модифицирование аморфных пленок халькогенидов. Если
равновесие между собственными и примесными дефектами не
успевает установиться, возможно существенное изменение элек-
трических свойств [160]. Так, при соосаждении или катодном
сораспылении халькогенидов с переходными металлами наблю-
дается как сильное изменение электропроводности, так и
уменьшение энергии активации проводимости [161, 162]. На-
пример, в пленках состава Nix (Оео.згАво^Зео.згТео.зг) i-x послед-
няя изменяется от 0,74 эВ при х = 0 до 0,18 эВ при х = 0,114,
а проводимость при этом увеличивается на 9 порядков, дости-
гая величины 0,2 См • см-1 [163]. Влияние различных примесей
на характеристики переноса в пленках АзгЗВез, прежде всего на
дрейфовую подвижность, описано в работе [164].
Легирование аморфных кремния и германия. В разд. 3.1.2.3
уже частично обсуждались электрические свойства a-Si и a-Ge,
причем отмечалась их существенная зависимость от условий
приготовления. Из исследований ЭПР (гл. 3 [135]) и эффекта
поля [165] можно оценить плотность состояний в щели, связан-
ных с дефектами, величиной 1019—1020 эВ-1 • см-3. В соответст-
вии с положительной эффективной корреляционной энергией,
характерной для таких материалов, эти состояния обладают
песпаренным спином.
Электрические свойства аморфных гидрогенизированных
кремния и германия (a-Si : Н и a-Ge:H), которые получают
разложением S1H4 или GeH4 в тлеющем разряде, тоже сильно
зависят от условий приготовления, прежде всего от темпера-
туры подложки. На рис. 4.23, а сплошной линией показано рас-
пределение плотности состояний в a-Si : Н, которое было полу-
чено из экспериментов по эффекту поля на пленках, осажден-
ных при 520 К [166]; ясно видно разделение спектра на распро-
страненные состояния, локализованные состояния в хвостах
зон и локализованные состояния внутри щели подвижности.
Для a-Ge : Н получается сходная картина [167]. Из рис. 4.23, а
видно, что, в отличие от a-Si и a-Ge (верхняя пунктирная кри-
вая), в гидрогенизированных пленках германия и кремния
плотность -состояний в щели значительно снижена, что, веро-
ятно, обусловлено насыщением оборванных связей атомами во-
дорода.
Предполагается, что уровни дефектных центров в полосе,
прилегающей к зоне проводимости, нейтральны выше уровня
Ферми и отрицательно заряжены при (Ес— Е) <.EF; а уровни
дефектов, прилегающие к валентной зоне, нейтральны ниже
уровня Ферми и положительны выше этого уровня (см. рис.
4.23, а и 2.56). При легировании a-Si : Н фосфором в резуль-
Электрические и оптические свойства
467
тате соосаждения силана с РНз, избыточные электроны за счет
пр атомов фосфора занимают свободные локальные состояния
выше уровня Ферми, который смещается при этом на величину
A£f вверх, вследствие чего проводимость увеличивается. При
введении атомов акцептора, например бора, уровень Ферми
смещается в противоположную сторону.
Ранее, в разд. 2.5.2.1, уже обсуждались причины, по кото-
рым уровень Ферми в некристаллических полупроводниках
Рис. 4.23. Плотность состояний в аморфном кремнии, полученном напыле-
нием в вакууме (—) и осажденном в тлеющем разряде ( — )[166, 167] (а),
и зависимость проводимости а-Si : Н при 298 К от примеси и от уровня леги-
рования (б) [137].
с положительной эффективной корреляционной энергией легче
смещается при легировании, чем в халькогенидных стеклах.
Основным условием легируемости является низкая концентра-
ция собственных дефектных центров, дающих уровни в щели
подвижности. В соответствии с этим Спир и Ле-Комбер [137]
впервые показали возможность эффективного легирования
а-Si : Н и а-Ge : Н, где оборванные связи германия или кремния
пассивированы атомами водорода; на рис. 4.23, б представлены
их экспериментальные данные для a-Si, который вообще изу-
чен наиболее подробно. Библиографию по данному вопросу см.
в гл. 3 [109].
4.1.3.6. Зависимость проводимости
от напряженности электрического поля
Высокое сопротивление некристаллических полупроводников
обеспечивает возможность измерения в них проводимости
в сильных электрических полях без саморазогрева образца,
30*
468 Глава 4
причем для пленок такие поля легко получить эксперимен-
тально. Критерием сильного поля можно принять величину на-
пряженности F > 103 В • см-1, когда обычно наблюдаются от-
клонения от закона Ома.
При интерпретации процессов переноса заряда в сильных
полях следует различать явления, связанные с контактными и
с объемными эффектами. Если ток ограничен контактным со-
противлением, то вольт-амперные характеристики (ВАХ) не за-
висят от толщины образца (за исключением очень тонких об-
разцов, толщина которых соизмерима с дебаевским радиусом
экранирования); этот радиус в некристаллических полупровод-
никах вследствие высокой плотности локализованных состоя-
ний весьма мал, а именно ~5 нм [92]). В случае неблокирую-
щих контактов, которые сравнительно несложно изготовить,
обедненные приконтактные слои, по-видимому, не формируются
благодаря глубокому залеганию уровня Ферми (приблизи-
тельно в середине щели подвижности).
Многочисленные исследования (см., например, [92]), в том
числе на пленках GeiSesTe [169], Aso,3Geo,iSeo,i2Teo,4s [170] и на
стеклах GeAs4Te5 [171], показали, что при обсуждении нелиней-
ности ВАХ можно исключить из рассмотрения контактные эф-
фекты и (в случае отсутствия саморазогрева) следует рассмат-
ривать следующие явления:
токи, ограниченные пространственным зарядом,
эффект Пула—Френкеля,
зависимость подвижности от напряженности поля и
переход электронов из валентной зоны в зону проводимости
или эффект Зенера (его можно ожидать в полупроводниках
с запрещенной зоной не более 1,5 эВ и при 106 В • см~’ —
см. гл. 2 [362]).
Зависимость подвижности от напряженности электрического
поля. Определим критическую напряженность электрического
поля Екрит реализацией условий, когда скорость электрона, раз-
виваемая в поле, т. е. v = pF становится больше скорости теп-
лового движения vth = (ZkTIm*),/2:
г 1 / 3/гТ (Л КПч
^крит > — д/ (4-59>
Тогда для проводимости по делокализованным состояниям при
т*, равной массе покоя электрона, 7 = 300 К и ц = 20 см2Х
ХВ-1-сн (эта величина заведомо превышает соответствующие
подвижности), получаем ЕКрИт > 6 • 105 В • см-1.
При рассмотрении проводимости за счет прыжков по лока-
лизованным состояниям в хвостах зон следует учитывать зави-
симость подвижности от напряженности поля. Барьер AU7i
в уравнении для прыжковой проводимости в хвостах (4.32)
Электрические и оптические свойства 469
в поле понижается на величину eFa!2, где а — расстояние ме-
жду соседними центрами захвата. Отсюда получаем
М-2 (F) — Ц02 ехр [~AU7i/kT + qFa/2kT] — ц2 (0) ехр [eFa/2kT] (4.60)
При учете угловой зависимости изменения барьера AUZi в элек-
трическом поле его снижение записывается как eFa cos 0/2, и
тогда
1 pFa
ц2 (F) = ц2 (0)sh ац, гдеац=—ту- (4.61)
Uu Kl
Пфистер [150] применил это выражение для описания полевой
зависимости дрейфовой подвижности в стеклообразном As2Se.3.
Ток, ограниченный пространственным зарядом. Простран-
ственный (объемный) заряд образуется, например, в вакууме
вблизи эмиттирующего электроны катода или при протекании
тока в высокоомных полупроводниках с инжектирующими элек-
тродами. Это явление было использовано в работах [101, 172—
174] для интерпретации нелинейной ВАХ в аморфных полупро-
водниках. Вид уравнения для плотности тока, ограниченного
пространственным зарядом, в зависимости от напряженности
поля определяется выбранной функцией распределения плотно-
сти локальных уровней, причем в это уравнение обязательно
входит толщина образца. Наблюдаемая на опыте зависимость
(j/F) ~ехр (F/Fo) согласуется с предположением о равномер-
ном распределении локальных состояний. Выражение для Fo =
= еЬМтНТ/ее2 позволяет сравнивать образцы с различной тол-
щиной. Следует отметить, что этот подход широко использу-
ется, но недостаточное внимание, особенно в ранних работах,
уделялось вопросам воспроизводимости.
С другой стороны, против такого механизма возникновения
нелинейности ВАХ в аморфных полупроводниках говорит ха-
рактерная для них высокая концентрация локализованных со-
стояний. При плотности соответствующих уровней Nt^
~ 1018 см-3 ток, ограниченный пространственным зарядом, не
будет заметен на фоне основного тока [169], и поэтому его сле-
дует учитывать лишь в исключительных случаях [92].
На рис. 4.24 приведены ВАХ для пленок Ge4o,8±i,2Se49±i,2X
ХТею±1,о, осажденных из паровой фазы по способу, описанному
в разд. 2.1.3.1. В приведенных здесь координатах ВАХ не за-
висит от толщины образца: такое поведение указывает на реа-
лизацию эффекта Пула—Френкеля.
Эффект Пула—Френкеля для изолированных кулоновских
центров. Френкель [178] на основании более ранних экспери-
ментальных работ Пула [179] связал увеличение проводимости
высокоомных твердых тел во внешнем электрической поле с по-
470 Глава 4
Рис. 4.24. Ход усредненных полевых зависимостей для проводимости пленок
различной толщины.
Рис. 4.25. Потенциальная
яма для изолированного
центра Пула—Френкеля.
нижением энергии ионизации соответствующих центров. Потен-
циальная энергия центра (рис. 4.25) равна
е2
E™=-^r-eFr <4-62>
В одномерном случае барьер ионизации снижается на величину
"Р" г'"
2ер'/г
(4.63)
Электрические и оптические свойства
471
Отношение для вероятностей ионизации в поле и без поля при
условии равенства подвижностей совпадает с отношением со-
ответствующих проводимостей:
>=>=>=“₽ ^)= <<•<*>
При учете углового распределения эммитированного заряда от-
носительно направления поля получаем
6F = -|}F1/2cos,/20 (4.65)
При рассмотрении трехмерного случая Коннелл с сотр. [180]
получил выражение
о (F)/o (0) = 2«п2 (1 + «п — sh «ц — ch ап) (4.66)
Пай [181] отметил связь уравнения (4.66) с теорией Онзагера
(теория диссоциации слабых электролитов в электрическом
поле). Действительно, применив методы статистической термо-
динамики к модели броуновского движения центров, взаимодей-
ствующих между собой по закону Кулона, во внешнем электри-
ческом поле, получаем уравнение
ГА 1 ( ai YZ1 2
o(F)/o(0) = [X /| (/+,), И") <4-67>
которое совпадает с уравнением (4.66) при ai = 2an.
Ширрмейстер [169, 177] дал в асимптотическом приближе-
нии решение уравнения (4.67) в форме
о (F)/a (0) = 2an2 (1 + ац shan — chan) (4л)“'/4 ап’74^11 (4.68)
откуда при an = ^F'/2/kT получаем
In =-^TF'l2+ 1п[(4л)“'/'(-у^)’/‘о(0)] (4.69)
С помощью уравнения (4.69) можно из экспериментальных
данных непосредственно определить константу Пула—Френ-
келя р. Для ее оценки можно использовать также усредненную
величину диэлектрической проницаемости er = 8± 1 (прямые из-
мерения дают значения 7,2±0,7). Обычное значение о(0) = (54-
4-3)10-12 См • см-1, причем для объемного стекла при 298 К
a(0) = 1 • 10-13 См-см-1. Из рис. 4.26 видно, что эксперимен-
тальные данные для образцов различной толщины действи-
тельно ложатся на одну зависимость, описываемую уравне-
нием (4.69) с использованием приведенных выше оценок р и
о(0).
472 Глава 4
Рис. 4.26. Полевые зависимости проводимости пленок ОедБезТе в сопостав-
лении с уравнением (4.68).
В работе [169] показано, что в данном случае концентрация
центров, проявляющихся в эффекте Пула—Френкеля, состав-
ляет 5 • 1016 см-3.
В заключение отметим, что для состава Ge4Se5Te, на основе
которого были приготовлены образцы, представленные на
рис. 4.25 и 4.26, наблюдается сильная зависимость параметров
проводимости от условий приготовления. Если учесть, что
в объемных стеклах такого состава Еа = 0,90±0,01 эВ, Оо =
= (1,3±0,2) • 103 См • см"1 и о = 8- 10"14 См • см-1 при 300 К;
в термически осажденных пленках Еа = 0,82±0,01 эВ, оо =
= (2±1)-102 См • см-1 и о = 5-10"12 См • см-1 при 300 К;
а в пленках, полученных методом взрывного распыления, £о =
= 0,72 эВ и а = 8- 10"11 См • см"1 при 300 К, то очевидна зако-
номерность: чем выше разупорядоченность структуры, тем
меньше энергия активации проводимости и тем выше проводи-
мость. На основе исследований фотопроводимости (см. рис. 4.34)
удалось показать, что эти изменения связаны с уменьшением
щели подвижности [106] (см. также гл. 2 [42]).
Эффект Пула—Френкеля для экранированных кулоновских
центров. В случае пленок, полученных методом взрывного рас-
пыления (см. разд. 2.1.3.1) наблюдается гораздо большая их
однородность и меньшая зависимость от особенностей приготов-
Электрические и оптические свойства
473
ления шихты, чем для пленок, полученных обычным способом
термического осаждения из паровой фазы. Хотя и в этом слу-
чае полевая зависимость проводимости нечувствительна к тол-
щине пленки [106] (см. также гл. 2 [42]), но такие пленки, как
видно из рис. 4.27, отличаются от обычных: кривая для прово-
димости лежит выше и лучше спрямляется в координатах
Igo — F, чем в координатах Igo — F'1*. Такая полевая зависи-
Рис. 4.27. Полевая зависимость проводимости для пленок Ge4Sc5Te, получен-
ных методом термического осаждения (/) и взрывного испарения (2).
мость наблюдалась и для других многокомпонентных халько-
генидных пленок [174, 183, 184], и тогда для описания экспе-
риментальных данных используют эмпирическое выражение
o(F, Т) = о(0, T)exp[ea(T)F/kTj (4.70)
Для интерпретации зависимости (4.70) подходит модель экра-
нированных кулоновских центров. В этой модели уравнение для
потенциальной энергии
z>2 1
£пот= — -д——— ехр(—r/Го) — erfcosO (4.71)
(где го—радиус экранирования) записывается относительно
координаты положения максимума барьера гт в виде
ехр (—г т/r 0) [rm2 + (rmr0)~‘] — Р ----- 4лг0ег = 0 (4.72)
474 Глава 4
Численное решение вышеприведенных уравнений при ег = 8 и
го = 2 нм хорошо согласуется с экспериментальными данными
на рис. 4.27 (светлые точки); соответствующий вывод приведен
в работах [182, 340] (см. также гл. 2 [42]). При условии, что
го= (kT£o&r/e2Nr)1/2 и температура замораживания 280 < Т <
<1270 К получаем концентрацию центров, проявляющихся
в эффекте Пула—Френкеля, равную 2 4-8 • 1018 см-3, т. е. по
меньшей мере на два порядка выше, чем в рассмотренных
в предыдущем разделе обычных пленках.
В области Т < 300 К при снижении температуры все более
существенным становится влияние поля на подвижность носи-
телей в хвостах зон. В этом случае при комбинации уравнений
(4.72) и (4.61) удается вполне удовлетворительно описать экс-
периментальные зависимости [182] (см. также гл. 2 [42]).
Эффект Пула—Френкеля в модели заряженных дефектов.
На основе представлений об отрицательной эффективной кор-
реляционной энергии (см. разд. 2.5.2.1) Адлер и Иоффа (гл. 2
[399, 400]) рассмотрели условия неравновесной проводимости
при высоких напряженностях поля, использовав понятие ква-
зиуровней Ферми по Шокли. Тогда в соответствии с уравнени-
ями (2.126, 2.127) можно записать
Ер, „ = Т,- ~ 1/,фф + t>E = EF + ЛЕ (4.73)
Ер,Р = Т„-~и^-1>Е = Ер~ЬЕ (4.74)
откуда при
on (F) = ецЛэфф, A exp [— (Ес — EF, n)/kT] (4.75)
и
и (Л) = ецр^фф, иехр[— (EFtP — Ev)/kT] (4.76)
получаем выражение для эффекта Пула—Френкеля в прибли-
жении изолированных центров [186]:
а (F) = [оЛ (0) + Ор (0)] ехр [ЬЕ/ЕГ] = о (0) exp (bE/kT)
Этот подход сам по себе не дает указаний о природе цен-
тров, однако если бы проявляющиеся в эффекте Пула—Френ-
келя центры характеризовались одноэлектронным возбужде-
нием, то концентрация получающихся нейтральных центров
~ 1018 см-3 отвечала бы измеримому сигналу ЭПР, который не
наблюдается в действительности в аморфных полупроводни-
ках с отрицательной эффективной корреляционной энергией.
Кроме того, трудно было бы объяснить экранирование центров.
Поскольку неспаренпые спины в халькогенидных стеклообраз-
Электрические и оптические свойства
475
ных полупроводниках практически не реализуются, для таких
веществ была принята модель противоположно заряженных де-
фектов, которые превращаются друг в друга путем двухэлек-
тронных возбуждений (см. разд. 2.5.2.2); этими же центрами
можно объяснить эффект Пула—Френкеля. Таким образом,
возникновение нелинейности на ВАХ в области сильных полей
может служить аргументом в пользу принятой модели дефек-
тов, дающих положительно и отрицательно заряженные лока-
лизованные состояния в щели подвижности.
Рассмотренные в предыдущих подразделах пленки Ge—Se—
Те имеют электронную проводимость [144], однако можно пред-
положить, что центры Т~, изображенные на рис. 3.41 и 4.22,
обратимо преобразуются в С+-центры, обеспечивая таким обра-
зом выполнение предложенной модели эффекта Пула—Френ-
келя.
4.1.3.7. Эффект переключения * *
В высокоомных полупроводниках и диэлектриках при напря-
женности поля выше некоторой критической величины насту-
пает пробой — необратимое разрушение вещества. Механизм
пробоя связан с лавинообразной термо-полевой ионизацией или
инжекцией носителей заряда из контактов, причем в любом
случае происходит сильный разогрев вдоль путей протекания
тока, которые соответствуют «слабым» местам в образце, обра-
зующимся при его изготовлении или последующем легировании
основного вещества материалом контактов.
Для халькогенидных стеклообразных полупроводников при
измерении проводимости на переменном токе, а также в им-
пульсном режиме, можно наблюдать выше некоторой напря-
женности обратимый переход в низкоомное состояние (порого-
вое переключение). При определенных условиях низкоомное со-
стояние сохраняется и после выключения напряжения, и воз-
врат к исходному высокоомному состоянию осуществляется
только после пропускания достаточно сильного и резко спадаю-
щего импульса тока (переключение с памятью). Соответствую-
щие ВАХ приведены на рис. 4.28 (см. также [187] и гл. 3
[614]).
Эффект переключения привлекает особое внимание в связи
с перспективами его использования в ячейках памяти. Начиная
с 1963 г. было оформлено множество соответствующих патен-
тов [188].
* Этот эффект, характерный для халькогенидных стеклообразных полу-
проводников, был открыт в 1963 г. Коломийцем и Лебедевым (см. ссылку
(190]).— Прим. ред.
* См. также Костылев С. А., Шкут В. А. Электронное переключение
в аморфных полупроводниках.— Киев: Паукова думка, 1978, 217 с.— Прим,
перев.
476 Глава 4
Переключение с памятью. В этом случае переход в низко-
омное «включенное» состояние (ON-state) связан с кристалли-
зацией вещества в канале, по которому течет ток. Канал фор-
мируется непосредственно перед моментом переключения за
счет тепловых и, возможно, электронных эффектов (см.
разд. 4.2.4.2). При этом вещество в канале, разогретом выше
температуры стеклования, кристаллизуется, а падение сопро-
тивления связано с тем, что образующиеся микрокристаллики
за счет своей упорядоченности обладают гораздо лучшей про-
водимостью по сравнению со стеклообразной матрицей
(рис. 4.28, а). В соответствии с такой трактовкой переключе-
Q
Рис. 4.28. Вольт-амперная
характеристика пленок
аморфных полупроводников.
а — эффект переключения с па-
мятью, б—пороговое переклю-
чение.
ние с памятью наблюдается в тех халькогенидных стеклах, ко-
торые склонны к кристаллизации и имеют сравнительно низ-
кую температуру стеклования. Условия реализации низкоом-
ного состояния можно описать с помощью уравнений нестацио-
нарной теплопроводности [195, 196].
Обратный переход в высокоомное «выключенное» состояние
(OFF-state) требует импульса тока достаточной мощности,
чтобы расплавить вещество в канале, и достаточно крутого,
чтобы (с учетом геометрии образца и теплофизических свойств
материала) обеспечить быстрое охлаждение расплава со ско-
ростью, большей критической (см. разд. 2.3.2), необходимой
для образования стекла.
При исследовании обратимых переходов «стекло—кристалл»
следует исключать возможность разных условий кристаллиза-
ции вследствие температурных градиентов по образцу, приме-
нять инертные контакты, материал которых при диффузии в ос-
новное вещество не влияет на образование кристаллических за-
родышей, а также учитывать сложное взаимодействие кристал-
лического канала со стеклообразной матрицей, особенно при
его образовании и плавлении. Кристаллические соединения мо-
жно разделить на две группы по сохранению или изменению,
ближнего порядка при плавлении [197, 204]; в состав пленок,
где наблюдается переключение с памятью, могут входить со-
единения обеих групп (например, As2Se.3— см. разд. 3.2.1.5,
Аз2Тез — разд. 3.2.1.6, GeTe4). Наиболее эффективно переклю-
Электрические и оптические свойства
477
чают стекла GenTe85X4 (где X4 = As4, AS2S2, ЗЬгЗг и т. д.), со-
став которых лежит вблизи эвтектики системы GeTe—Те. В та-
ких стеклах в канале образуются кристаллиты теллура, кото-
рые при последующем плавлении входят в структуру стекла
с изменением своей координации (разд. 3.2.2.6). Эффект пере-
ключения с памятью исследовался также в слоях CdAs2 [199]
и неоднократно наблюдался в полупроводниковых оксидных
стеклах n-типа, содержа-
щих переходные метал-
лы (см., например, [200,
201]).
Пороговое переключе-
ние. В слоях толщиной
10 4-н10 5 см, приготов-
ленных на основе стекол,
устойчивых к кристалли-
зации, можно наблюдать
ВАХ типа, изображен-
ного на рис. 4.28, б, при-
чем такие характеристи-
ки безынерционно следу-
ют за каждым импуль-
сом (или периодом) на-
пряжения вплоть до ча-
стот 106 Гц.
Рис. 4.29. Зависимость сопротивления
аморфных слоев от напряжения при раз-
личных температурах [172]. Пороговое на-
пряжение Vth (рис. 4.28,6) показано штри-
ховой линией.
В целях практическо-
го применения фирма
Energy Conversion Devi-
ces разработала оптими-
зированные составы
As3oGeioSii2Te48 и
As35Ge6Sii8Te4o (состав указан в мол. %) (см. разд. 3.3.7.3);
пленки этих составов осаждают методом катодного распыле-
ния. Для пороговых переключателей пригодны и другие со-
ставы при условии стабильности их стеклообразного состояния
(устойчивости к кристаллизации) и при величине щели подвиж-
ности 0,8< (Ес — Еу)< 1,4 эВ [188, 200] (см. также гл. 2 [387]).
Полученные к настоящему времени экспериментальные осо-
бенности эффекта можно суммировать следующим образом:
а) В высокоомном состоянии (OFF-state) проводимость
сильно зависит от температуры (что вообще характерно для
халькогенидных стеклообразных полупроводников), и отчетливо
проявляются отклонения от закона Ома [172, 173, 205, 206].
Последние интерпретировались в рамках теории токов, ограни-
ченных пространственным зарядом [101, 177], и на основе тео-
рии эффекта Пула—Френкеля [170]. Как видно из рис. 4.29,
478 Глава 4
можно выделить две области: при сравнительно небольших на-
пряженностях поля эксперимент описывается в рамках эффекта
Пула—Френкеля для экранированных центров, но по мере при-
ближения к порогу для переключения проводимость все более
отклоняется от ожидаемой по данной модели экспоненциальной
зависимости. Это явление до сих пор не получило удовлетво-
рительного объяснения.
б) Пороговое напряжение Vth (threshold voltage), показан-
ное штриховой линией на рис. 4.29, слабо зависит от темпера-
туры при Т<200К. Коло-
миец с сотр. [207] обнару-
жили, что в слоях толщи-
ной менее 10 мкм Vth не
зависит и от толщины об-
разца, из чего следует су-
ществование некого крити-
ческого напряжения для пе-
реключения.
в) Проводимость в низ-
коомном состоянии (ON-
state) не зависит от площа-
ди контактов. Это указыва-
ет на образование достаточ-
но узкого токопроводящего
1,4
Рис. 4.30. Время задержки td и его ста-
тистический разброс [2101.
канала; принимают, что
площадь канала пропорциональна общему току. Проводи-
мость в ON-state не зависит от температуры и от толщины
пленки; по-видимому, напряжение падает в приконтактной об-
ласти.
г) При исследовании на прямоугольных импульсах выявля-
ется время задержки td, как это показано на рис. 4.30 [202,
208, 209]. При амплитуде импульса чуть выше порогового на-
пряжения время задержки составляет несколько микросекунд
и воспроизводится с некоторым статистическим разбросом при
многочисленных переключениях. При увеличении напряжения
импульса величина td экспоненциально уменьшается, стремясь,
по-видимому, к характерному времени самого переключения
IO"10 с [210].
д) Если очередной импульс подать через время, меньшее
~ 1 мкс (так называемое время восстановления), то переклю-
чение происходит при пониженном значении Vth. По-видимому,
в течение времени восстановления постепенно снижается повы-
шенная концентрация носителей в области бывшего канала и
в барьерном слое вблизи контактов.
Модели, предложенные для объяснения порогового переклю-
чения, условно можно разбить на две группы: тепловые и элек-
тронные. Сторонники теплового механизма сводят переключе-
Электрические и оптические свойства
479
ние к высокотемпературному пробою [211, 212], исходя при этом
из диссипации энергии в канале тока и трактуя переключение
как нестабильность, которая следует из решения уравнений не-
стационарной теплопроводности при определенных граничных
условиях. Расчеты показывают, что в этом случае температура
в канале должна на несколько сот градусов превышать темпе-
ратуру окружающей среды, т. е. материал канала должен на-
ходиться в размягченном состоянии (для большинства пере-
ключающих стекол температура стеклования лежит в пределах
4204-620 К). При этих условиях следует ожидать существен-
ной нестабильности и плохой воспроизводимости при много-
кратном переключении (Попеску справедливо отмечал, что ста-
бильное переключение в этом случае возможно, лишь если
время существования низкоомного состояния не превышает по-
стоянной времени структурной релаксации, которая определя-
ется вязкостью расплава — см. разд. 2.2.1.2). Однако пленки,
приготовленные из особочистых материалов, выдерживают до
10й циклов переключений без заметной формовки и старения
[203]. Кроме того, было показано, что в течение времени за-
держки происходит накопление заряда в слое, причем накоп-
ленная таким образом электрическая энергия тем больше, чем
выше температура [209], что, очевидно, не согласуется с теп-
ловой моделью.
Сегодня можно с уверенностью утверждать, что в основе по-
рогового переключения, по крайней мере для образцов толщи-
ной не более 5 4-10 мкм, лежит электронный механизм — об
этом говорит уже независимость порогового напряжения от
толщины слоя в тонких пленках [207]. В обосновании элек-
тронного механизма переключения существенный вклад внесли
Хэниш с сотр. [213—215]. В одной из работ [213] они показали,
что интенсивное облучение светом из области собственного по-
глощения не меняет порогового напряжения, в то время как ве-
личина порогового тока повышается почти на порядок. Далее,
если в течение времени задержки поменять полярность тре-
угольных импульсов, то время задержки увеличивается [214].
Наконец, если использовать несимметричные контакты: n-Ge
(отрицательный)/графит (положительный), p-Ge (положитель-
ный)/графит (отрицательный) или n-Ge (отрицательный)/p-Ge
(положительный), эффект ведет себя обычным образом, но про-
падает при перемене полярности; он пропадает и при контактах
только п- или только р-типа [215, 216]. Все эти результаты не
укладываются в модель теплового пробоя.
Можно, кроме того, оценить температуру в канале тока. По
Адлеру [217] она повышена всего на 13 К. Наконец, наблюде-
ние порогового переключения на паносекундпых импульсах,
которые могут заметно повысить температуру в канале только
в гипотетических условиях полного отсутствия теплоотвода от
480 Глава 4
образца, тоже свидетельствует в пользу электронного меха-
низма [218, 219].
Низкоомное состояние можно исследовать с помощью нано-
секундных импульсов различной длительности и амплитуды.
При определенных длительностях импульса существует состоя-
ние без тока (blocked ON-state или transient ON-state), кото-
рое характеризуется наличием определенного потенциала под-
держания Vh (рис. 4.31). В другом временном режиме условием
Рис. 4.31. Характеристика низкоомного со-
стояния при измерениях на наносекундных
импульсах.
существования низкоом-
ного состояния является
наличие тока поддержа-
ния: если ток уменьшает-
ся, то скачком восстанав-
ливается высокоомное
состояние, причем в рам-
ках тепловой модели не-
возможно объяснить
столь резкий переход.
Из ВАХ на наносе-
кундных импульсах типа
изображенной на рис.
4.3.1 можно определить
величину удельного со-
противления материала в
низкоомном состоянии
р = 0,08 ±0,02 Ом-см.
[203]. Поскольку время
восстановления зависит
от поперечного сечения
канала тока [217], то исчезновение низкоомного состояния
при снижении тока, вероятно, обусловлено диффузионным
растеканием носителей. Подвижность носителей, оцененная по
коэффициенту диффузии, составляет 10 см2-В-1-с-1, откуда
с учетом величины р получаем концентрацию носителей 7Х
Х1018 см-3. На основании этих оценок и теплофизических рас-
четов можно сделать вывод, что повышение температуры в ка-
нале тока не превышает 60 К [217].
В соответствии с разд. 4.1.3.6 можно предположить, что про-
цессы, предшествующие собственно переключению, обусловлены
неравновесными носителями. По-видимому, упомянутые выше
отклонения на рис. 4.29 при повышенных напряженностях поля
связаны с неравновесной инжекцией носителей из электродов.
При этом будет происходить преимущественное преобразова-
ние в центры одной зарядности: D+ в D~ или D~ в D+— в за-
висимости от положения уровня Ферми в исходном образце.
В сильно неравновесных условиях появляется возможность од-
ноэлектронного возбуждения дефектов с образованием £)°-цен-
Электрические и оптические свойства
481
тров. В результате неравновесного заполнения уровней локали-
зованных состояний происходит смещение уровня Ферми, и низ-
коомное состояние предположительно описывается кривыми,
приведенными на рис. 4.32, б, причем потенциал спадает преи-
мущественно либо в одной, либо в обеих приконтактных обла-
стях.
Рис. 4.32. Схематическое
изображение падения на-
пряжения в слое полупро-
водника в высокоомном (а)
и пизкоомном (б) состоя-
ниях.
По вопросу об электронной природе низкоомного состояния
до сих пор нет единого мнения. Хэниш [220] и Мотт (см. гл. 2
[373]) предложили модель двойной инжекции электронов и ды-
рок (см. также [221]), которая подтверждается в эксперимен-
тах с несимметричными электродами. Не исключено также силь-
ное увеличение числа £)°-центров, вследствие чего уменьшается
рассеяние носителей заряда, и их движение соответствует эф-
фективному переносу по делокализованным состояниям выше
Ес или ниже Еу.
4.1.4. Фотопроводимость
Наличие фотопроводимости, т. е. генерации электронов и
дырок под действием поглощаемых фотонов, послужило для
Коломийца и Горюновой указанием на полупроводниковую при-
роду халькогенидных стекол (см. гл. 2 [14]). Фотопроводимость
измеряется как в объемных образцах, так и на пленках, и вы-
ражается величиной фототока в зависимости от выбранного
параметра: длины волны X (или волнового числа v), интенсив-
ности или плотности потока фотонов 1А (при этом учитывается
31 Заказ № 413
482 Глава 4
только та часть светового потока, которая попадает в образец,
т. е. IA — Iph — Ir, где IR — связанная с показателем преломле-
ния материала интенсивность отраженного от поверхности об-
разца света), температуры или приложенного электрического
поля.
Образцы для измерений готовят в двух вариантах. При ком-
планарной геометрии образца между электродами имеется за-
зор в несколько десятых миллиметра, через который и происхо-
дит облучение. В этом случае получить абсолютное значение
фототока невозможно, измеряется лишь изменение сопротивле-
ния образца, которое зависит от его геометрии. В сандвичевом
варианте вещество расположено между нижним и верхним элек-
тродами, один из которых — через него производят освещение
образца — должен быть прозрачным (обычно это тонкая метал-
лическая пленка или стеклянная подложка, покрытая SnCh).
И в этом случае можно определить лишь относительный фото-
ток. То же относится и к квантовому выходу фотоэффекта.
Измерения можно проводить в двух режимах: статическом
и динамическом, и соответственно различают статическую и ди-
намическую фотопроводимость. В последнем случае применяют
прямоугольные световые импульсы малой частоты (несколько
десятков герц). По характеру кривой спада фототока после вы-
ключения света можно судить о кинетике релаксационных про-
цессов, в частности о кинетике рекомбинации носителей. Спир
[264] использовал сандвичевые структуры также и для измере-
ний на коротких мощных импульсах при исследовании дрейфо-
вой подвижности (разд. 4.1.2.4).
Поток фотонов 1А, который поглощается по толщине образца
с коэффициентом поглощения в соответствии с законом Лам-
берта (—dIAldx) =а • 1А, генерирует в нем носители обеих зна-
ков. При квантовом выходе 0 скорость генерации носителей
-^- = -^£-=Мл (4.77)
Если время диэлектрической релаксации больше времени жизни
избыточных носителей, то устанавливается тепловое равнове-
сие; в противном случае имеет случай релаксационных полу-
проводников, исследованный Ван Розеброком [222].
Процесс, обратный генерации носителей — их рекомбинация.
Она может происходить либо при непосредственной встрече
электрона и дырки, либо после их раздельного захвата на ло-
кализованное состояние — центр рекомбинации. Скорость ре-
комбинации пропорциональна концентрации носителей п = по4-
-|-А« и р=ро+Ар (по и ро — равновесные концентрации носи-
телей), откуда из (4.77) следует
^-=- V <"« + (₽• + М + у М <4-78)
Электрические и оптические свойства 483
При условии п = р и Д/г = Ар получаем
—= ра/а — у (2/г Д/г + (Ап)2) (4.79а)
= ₽а/л - у (2р Др + (Др)2) (4.796)
Рассмотрим два крайних случая. При малой интенсивности
возбуждения и высокой температуре имеем /г, р^>Д/г, Ар. Тогда
изменение концентрации избыточных носителей во врем-ени опи-
сывается уравнением скорости 1-го порядка, и в стационарных
условиях имеем
Д/г = -^-; кр = -^ (4.80)
2\'П г 2ур ' '
Если определить среднее время жизни избыточных носителей
как
=------Ц— , Тр =-------Ц— (4.81)
qnvn^n ’ р qpVpkp v '
(где qn и qp — сечения захвата для электронов и дырок, a vn
и vp — средние тепловые скорости носителей), то получаем
Д п = ра/лт„, Д р = ра/лтр (4.82)
откуда для стационарной фотопроводимости имеем
вph = ера/А (цптп + ЦрТр) (4.83)
При высокой интенсивности возбуждения и низкой темпе-
ратуре преобладают неравновесные носители: /го, ро-САя, Др.
При этом с использованием уравнений (4.79) получаем уравне-
ния второго порядка
Д»-^)7’; ДР = (-^-)'/’
/ ра/л \’/«
®ph — в ” ) (Мп “F Цр)
(4.85)
На рис. 4.33 приведены типичные данные по фотопроводимо-
сти. Экспериментальные зависимости имеют вид кривых с мак-
симумом, причем при снижении температуры в области за мак-
симумом фотопроводимость оказывается выше темновой про-
водимости при той же температуре, а при повышенных темпе-
31*
484 Глава 4
ратурах ее величина меньше, чем величина темновой прово-
димости. По-видимому, в первом случае реализуется прибли-
жение, описываемое уравнением (4.85), а во втором—(4.83).
При достаточно низких температурах кривые фотопроводимо-
сти стремятся к насыщению.
Аналогичные кривые были получены для многих халькоге-
нидных стеклообразных полупроводников: селена [224], As2Se3
Рис. 4.33. Темновая проводимость
и фотопроводимость аморфных
пленок Gei5Te8iSb2S2 [223]. Усло-
вия возбуждения: галогенная
лампа мощностью 100 Вт, Х=
= 1,06 мкм (а = 2,6104 см-1). f~
= 1,36-1017 фотон-см-2-с-1, при-
чем Iph=f (/), 0,If (2), 0,0 If (3),
0,00 If (4).
[125], 2As2Te3-As2Se3 [226] и т. д. В работе [230] исследовался
Аз2Тез, и из факта совпадения энергии активации до и после
максимума (0,37 эв при >. = 0,65 мкм; энергия активации опре-
делялась по наклону зависимости lgoPh от 1/Т) авторы делают
вывод, что рекомбинация в обоих случаях определяется глу-
бокими ловушками, лежащими внутри щели подвижности.
Из кривых спада фототока можно выделить две составляю-
щие. Одна из них отвечает постоянной времени тп или тР~
~10-3Ч-10-4 с, для другой постоянная времени на несколько
порядков больше, достигая десятков секунд [227—229]. По-ви-
димому, кинетика спада проводимости определяется захватом
избыточных носителей на глубокие уровни дефектов D+ и D~
с образованием 7)°-центров, а последующее восстановление
о
-0,2
-0,4
-0,6
to
CD
о-0,8
'"-j.o
-1.2
-1,4
-1,6
-1,8
о беол SeQ6 (аморфные слои)
• GeOi( Sefl6 (компактное стекло)
® ^ео,4 5®о, 55 Te005 (аморфные слои)
(pmpwe мои)
8 70 72 14 76 18 20 22
р-10 3, см~1
Рис. 4.34. Спектральная зависимость фототока для некоторых стекол.
Рис. 4.35. Спектральная зави-
симость фототока для стекол
(PbS)o,2(GeS)o.3(GeS2)o>5 (/) и
(PbSe)o,2(GeSe)o,3(GeSe2)o,5 (2),
измеренная с различными кон-
тактами.
486 Глава 4
равновесных концентраций заряженных центров происходит бо-
лее медленно.
Величины запрещенной зоны, определенны из измерений фо-
топроводимости и из коэффициента поглощения а (£опт), хо-
рошо согласуются между собой (см. гл. 3 [417]). Как известно,
для определения £Опт необходима спектральная зависимость ос.
Сходные спектральные зависимости наблюдаются и для фото-
тока (рис. 4.34 и 4.35). На рис. 4.34 представлены данные для
объемных образцов и пленок того же состава, полученных ме-
тодом взрывного испарения [106, 182]; при этом оказывается,
что ширина запрещенной зоны в последнем случае умень-
шается.
Рис. 4.35, где приведены данные для объемных стекол, ил-
люстрирует влияние микропримесей на фотопроводимость. Ока-
залось, что наиболее сильно влияют оксидные примеси, так что
измерение фотопроводимости может служить методом их об-
наружения [231]. Коломиец с сотр. [332] (см. также гл. 3
[724]) исследовал влияние Си, Ag, Са, In и Т1 на фотопрово-
димость стеклообразного АвгЗез. Имеется также детальный об-
зор Коломийца и Любина [228] по фотоэлектрическим явле-
ниям в халькогенидных стеклах, опубликованный в 1973 г.
4.2. Оптические свойства
Широкое практическое применение стекол обусловлено
главным образом их прозрачностью в видимой области спектра
и сравнительно легким получением оптических сред с задан-
ными значениями показателя преломления и его дисперсии.
Область оптического пропускания, так называемое оптиче-
ское окно, с длинноволновой стороны ограничена поглощением,
связанным с комбинированными и сложными колебаниями
(многофононное поглощение) в группах ближнего порядка. При
дальнейшем увеличении длины волны начинается непрерывный
спектр фононного поглощения, обусловленный неупорядочен-
ностью структуры стекла (отсутствие дальнего порядка). Связь
между составом, колебательным спектром и структурой стекол
обсуждалась ранее в разд. 2.4.1.2.
В области коротких длин волн окно прозрачности ограни-
чено поглощением при переходах электронов из валентной зоны
в зону проводимости. Эта коротковолновая граница пропуска-
ния называется также краем поглощения и отвечает оптической
ширине запрещенной зоны.
При переходе от традиционных оксидных стекол к халько-
генидным оптическое окно смещается в сторону инфракрасной
области спектра (рис. 4.36 и 4.37), соответственно уменьшается
ширина запрещенной зоны и увеличивается электропровод-
ность. Эти изменения связаны с увеличением атомных масс
Рис. 4.36. Зависимость коэффициента поглощения а и показателя преломле-
ния стекла AS2S3 от волнового числа [234].
5еь—-------------------------------------------------
AsTe^i— ------------------------------------<
G ете г 1---------------------------------------1
A s2S е 3i------------------------------------------1
G е 2 S е 3 i—------------------------- t
G e S e 21 J
G 62 S3 ।
A s ZS з' '
GeSz 1---------------------------------------------1
GeOz 1 1
° A !----------------------------------1
B2O3I I
5;ozi----------------------------------------------1
BeFzl-------------------------------------<
ИК видимая УФ
I------------1____________I____________I-----------1----Ml nun in-1-----
2,0 2,5 5,0 3.5 _ 4.0 4,5
Ig V [CM'1]
Рис. 4.37. Окна прозрачности для типичных стекол. Коротковолновая граница
окна определяется значением а=104 см-1, длинноволновая — полосой в соб-
ственном колебательном спектре с наибольшим значением v (например, по-
лоса, отвечающая антисимметричным валентным колебаниям).
488 Глава 4
и уменьшением силовых констант *. В действительности опти-
ческие окна уже изображенных на рис. 4.37 из-за наличия мно-
гофононного поглощения и размытия края поглощения в стек-
лах по сравнению с кристаллами.**
При облучении стекол светом с длиной волны, соответст-
вующей краю поглощения, наблюдается не только рассмотрен-
ная выше фотопроводимость (разд. 4.1.4), но и явление фото-
люминесценции, обусловленное локализованными уровнями де-
фектов в запрещенной зоне, а также фотохимические реакции
и фотоструктурные превращения (разд. 4.2.2 и 4.2.4).
4.2.1. Поглощение и пропускание
Для определения коэффициента поглощения а необходимо
выделить ту часть падающего на образец электромагнитного
излучения /о, которая действительно поглощается в веществе,
т. е. определить величину То = 1о — Ir, где IR — интенсивность
отраженного света. В приближении нормального падения луча
в случае многократного отражения имеем [235, 233]
R = r + Т = (4-86, 4.87)
1 4- r e 1 — r e
I — P~ad
A = (l-r) -S—s-, (4.88)
1 — re
где
(n- l)24-£2
(n 4- 1 )2 4- k2
4nk
(4.89, 4.90)
= 4nk v
a =
Здесь R = Ir/Io — отражение, А = 1аЩ — поглощение, d — тол-
щина образца, a — коэффициент поглощения, г—коэффициент
отражения при однократном отражении и k — постоянная по-
глощения. Величина k практически не влияет на г, если
^0,1, т. е. при или a^v. Для очень малых величин a
(например, а^10~2 см-1) из уравнений (4.86) и (4.87) полу-
* Как видно из рис. 4.37, выход окна из видимой области спектра
обусловлен изменением края поглощения, следовательно в данном случае
силовые константы (они относятся к длинноволновому колебательному спек-
тру) не при чем, а основное значение приобретает ширина запрещенной
зоны, определяемая электронной структурой зон.— Прим., ред.
** Смысл предложения неясен.— По-видимому, имеется в виду, что спек-
тральные характеристики стекол очень чувствительны к условиям их приго-
товления, поэтому, например, выбор а=104 см-1 еще не гарантирует совпа-
дения соответствующих значений v для разных образцов стекла одинакового
состава, и что такая неопределенность особенно сильна при установлении
длинноволновой границы оптического окна.— Прим. ред.
Электрические и оптические свойства
489
чаем выражения для отражения R и пропускания Т = 1тЩ‘.
г = <4-91’ 4-92)
что отвечает /1 ~ 0. Если ad>>l, то из уравнения (4.86) имеем
R = r, т. е. вклад, связанный с многократным отражением, ис-
чезает из-за сильного поглощения света в объеме образца.
Из выражения
можно рассчитать а, если измерить пропускание Т и отраже-
ние R = r или последнюю величину оценить по уравнению (4.89)
при известном показателе преломления п (величиной k можно
пренебречь для /.<10 мкм и а<103 см-1). Так как ехр(—2aJ)
намного меньше, чем ехр(—ad), то уравнение (4.87а) можно
приближенно представить в виде
T = (l -R)- e~ad (4.876)
Если коэффициент отражения неизвестен, то для определения
коэффициента поглощения из данных по пропусканию можно
воспользоваться уравнением
Т~ То ехр (—ad) (4.87в)
предварительно определив То в спектральной области очень ма-
лых а.
Описанным выше способом можно определять коэффициент
поглощения а^10-3 см-1, но если при измерениях исключить
отражение, то удается снизить предел определения а вплоть до
10-5 см-1 [236]. Соответствующие методы: лазерная калоримет-
рия [237], фотоакустическая калориметрия [238] или интерфе-
ренционная калориметрия [239] основаны па непосредственном
измерении световой энергии, поглощаемой в образце. В работе
[240] дается обзор различных методов измерения поглощения
и отражения (см. также [234]).
Стекла с высокой прозрачностью применяются в световодах
(оптических волноводах), в связи с этим постоянный интерес
вызывают вопросы их очистки и оптического контроля [241,
242, 243]. Качество стекол для световодов характеризуют вели-
чиной оптических потерь, измеряемых в дБ/км. Децибелл опре-
деляется выражением
10 1g (/«//) = 4,34<xd = 1 дБ (4.93)
при этом потери 1 дБ/км соответствуют значению а — 2,3X
X Ю-6 см-1. Обычно волноводы работают в видимой области
490 Глава 4
спектра, но в литературе есть сведения и о волноводах для
ИК-области [243].
Верхняя граница определения коэффициента поглощения из
данных по пропусканию обычно отвечает а—102 см-1. В этом
случае для регистрации величины пропускания, равной 1 %
То, необходимы образцы толщиной 0,1 мм, в то время как для
измерений а=104 см-1 требуемая толщина образца ~1 мкм.
Для области а>102 см-1 необходимо либо измерять угловую
зависимость коэффициента отражения с последующей обработ-
кой результатов по уравнению Френеля, либо, в предположе-
нии нормального падения луча, проводить анализ спектров от-
ражения по Крамерсу—Кронигу J244, 245]. При этом путем
разделения уравнения Френеля на действительную и мнимую
часть получаем выражения для пик:
______1 — R________. ___ —2 V/? sin 0
1 +/? — 2 V/? cos 0 ’ ~~ 14-/? —2 V/? sin 0
где е = ,ln^(y)-lnj?fa) dv 95)
я о vi — V
Экспериментальные данные обычно представляют в форме
дисперсии коэффициента поглощения и показателя преломле-
ния (см., например, рис. 4.36). Можно также построить зави-
симость е" = 2п£ и е'=п2— k2 от длины волны X, волнового чи-
сла v или частоты v. Связь этих величин с характеристиками
высокочастотной проводимости дается выражением
<4-96)
откуда с учетом уравнения (4.11) получаем
а (со) = а>/гаХ = госпа (4.97)
(где с — скорость ветра в вакууме). Зависимость па от волно-
вого числа в дальней ИК-области спектра отвечает частотной
зависимости проводимости в СВЧ-диапазоне [248].
4.2.1.1. Многофононное поглощение
В связи с широким применением стекол в качестве оптиче-
ских сред интенсивно исследовалось их остаточное поглощение,
связанное с комбинированными (смешанными) колебаниями
в длинноволновой области спектра (см., например, [246, 234]).
Что касается халькогенидных стекол, то они представляют ин-
Электрические и оптические свойства
491
терес не только как материалы для световодов в ИК-области
спектра, но и для фокусировки мощных лазерных импульсов,
в частности СО2-лазера с длиной волны 10,6 мкм (940 см-1).
На рис. 4.38 приведен типичный для халькогенидных стекол
вид спектра многофононного поглощения, в котором наблю-
даются различные сочетания частоты валентных колебаний
(va=345 см1- для As2S3 и уд =240 см-1 для As2Se3) с частотой
колебаний мостиковой связи (vb = 485 см-1 для As—S—As
и у' =340 см-1 для As—Se—As) — см. табл. 3.3. При сопоста-
влении приведенных спектров отчетливо выявляется изострук-
турность стекол As2S3 и As2Se3. Из-за наличия широкого спек-
тра многофононного поглощения в случае As2Se3 величина а
довольно велика (10-2 см-1) даже при частотах 1500 см-1, хотя
вызывающие этот спектр основные колебания лежат в гораздо
более длинноволновой области (200—500 см4).
Спектр многофононного поглощения в стеклах выражен го-
раздо сильнее, чем в кристаллах, так как вследствие неупоря-
доченности снимаются некоторые запреты в правилах отбора.
Кроме того, следует иметь в виду, что при возрастании поля-
ризуемости (например, при переходе от оксидных стекол
492 Глава 4
к халькогенидным) увеличивающаяся в соответствии с уравне-
нием (2.103) ангармоничность колебаний тоже приводит к воз-
растанию вероятности комбинационных колебаний и колебаний
высших гармоник.
При изучении спектров многофононного поглощения в стек-
лах необходима тщательная очистка от примесей, дающих по-
лосы поглощения в той же области спектра; это относится не
только к исходным материалам, но и к обеспечению таких усло-
вий синтеза, которые исключали бы возможность взаимодей-
ствия вещества с кислородом воздуха и влагой.
4.2.1.2. Влияние примесей на пропускание.
Очистка стекол
Прозрачность оксидных стекол с оптическими окнами в ви-
димой и ИК-области спектра снижается в присутствии примеси
воды, входящей в сетку стекла в виде гидроксильных групп.
Соответствующие полосы валентных колебаний водородных свя-
зей ОН-группы расположены в интервале 3700—3000 см-1. Во-
просы очистки оксидных стекол были рассмотрены ранее на при-
мере кварцевого стекла (см. разд. 3.2.2.1).
В халькогенидных стеклах прозрачность снижают, в основ-
ном, оксидные примеси, которые вызывают появление оксид-
ных полос в области многофононного поглощения, причем та-
ким образом, что при приближении к оптическому окну коэф-
фициент поглощения сильно возрастает. Поглощение за счет
кислородных примесей в области 8—12 мкм ограничивает ис-
пользование этих стекол в системах регистрации теплового из-
лучения.
Влияние кислородных примесей на колебательный спектр
стекол As2Se3 и GeAs2Ses исследовали в работах [249] и [250].
При этом оказалось, что введение в шихту всего 0,005 масс. %
GeO2 вызывает сильное падение пропускания на длине волны
10,6 мкм (943 см-1) за счет интенсивного поглощения в поло-
сах As—О (650 см-1) и Ge—О (780 и 1260 см-1). При больших
интенсивностях излучения СОг-лазера это может привести к пе-
регреву и разрушению оптической среды.
Для очистки халькогенидных стекол от кислородных приме-
сей можно применять дистилляцию в потоке водорода [253,
254]. При этом для того, чтобы исключить попадание в стекло
ионов тугоплавких оксидов вследствие взаимодействия с мате-
риалом ампулы (кварцевое стекло) добавляют небольшие ко-
личества углерода или графитизируют внутренние стенки ам-
пулы (см. гл. 2 [155, 156]). Можно также в качестве геттеров
кислорода добавлять А1 или Zn, т. е. металлы с большой теп-
лотой образования оксидов [249, 257]. Следует отметить, од-
нако, что эти способы часто приводят к появлению полос по-
Электрические и оптические свойства
493
глощения в других областях спектра (например, полосы, отве-
чающей колебаниям связи халькоген—водород) или же про-
зрачность падает вследствие рассеяния на мелкодисперсных
частицах углерода или оксида геттера. Надо также иметь
Рис. 4.39. Ампула для очистки селена или мышьяка дистилляцией с после-
дующей сублимацией.
в виду, что для получения максимального пропускания тре-
буется длительная и трудоемкая очистка исходных материалов
и длительный отжиг предварительно очищенной ампулы из
Рис. 4.40. Спектры стеклообразного селена (а) и спектр пропускания стекла
Ge3As2Se5 в области полос поглощения, обусловленных оксидными приме-
сями (6).
Содержание кислорода (в процентах) в исследуемых образцах селена (в скобках — тол-
щина слоя): / —0 (d=15,5 мм), 2 — 7.4-10-4 (d=15,5 мм), 3— 11,3-Ю-4 (d=13,5 мм), 4 —
70.3-10-4 Щ= 13,6 мм).
кварцевого стекла в высоком вакууме. Кроме того, взвешива-
ние и загрузка компонентов в ампулу должна производиться
в инертной атмосфере, так как даже кратковременный контакт
с кислородом воздуха приводит к окислению поверхности,
прежде всего за счет окисления мышьяка.
494 Глава 4
Для примера рассмотрим методику получения очищенного
стекла Ge3oAs2oSe5o. В кварцевую ампулу, обработанную в рас-
творе HF/HNO3 и отожженную 8 ч при 1173 К в вакууме, за-
гружаются промышленный германий полупроводниковой чи-
стоты и предварительно очищенные селен и мышьяк. Поскольку
исходный промышленный селен полупроводниковой очистки
содержит не только SeO2, но и существенные количества воды
(до 0,015 масс. %), его необходимо чистить от кислородных
примесей. С этой целью исходный селен загружают в ампулу,
изображенную на рис. 4.39, которую далее вакуумируют до
остаточного давления 1,5-10—3 Па. Селен, находящийся в уча-
стке ампулы /, нагревается до 573 К, т. е. выше температуры
плавления; в процессе нагрева вода и SeO2 перегоняются
в участки ампулы <? и 4. Затем суженный участок ампулы
между 2 и 3 перепаивают, и селен перегоняется через пористый
фильтр в участок 2. Мышьяк очищают аналогичным образом
(после заключительной перегонке мышьяка в участке / оста-
ется небольшое количество черного осадка, который, как и кис-
лородные примеси, способен сильно снижать пропускание
стекла). После загрузки компонентов (в инертной атмосфере)
ампула эвакуируется до 1,5-10~3 Па и отпаивается; синтез ве-
дется при 1070 К в течение 8 ч. В заключение расплав охла-
ждается до комнатной температуры со скоростью 3 К/мин.
Для спектроскопического исследования из полученного та-
ким образом стекла нарезаются шайбы, которые полируются
с обеих сторон. Как видно из рис. 4.40, б, полоса кислорода,
связанная с оксидами, практически исчезает в результате очи-
стки. По калибровочной зависимости, получаемой на ряде об-
разцов с известным содержанием GeO2, внесенного в шихту из
предварительно дезоксидированных компонентов, можно оце-
пить содержание кислорода в стеклах, синтезированных в раз-
личных условиях. В случае, отвечающем рис. 4.40, б, содержа-
ние кислорода, определенное с помощью калибровочной зависи-
мости, не превышает 3-10“4 % для стекла, очищенного описан-
ным выше способом. В таком стекле коэффициент поглощения
на 10,6 мкм равен 10 2 см~! (см. гл. 2 116]).
Для кривых на рис. 4.40, а содержание кислорода оценива-
лось по интенсивности полосы поглощения 932 см-1 [258]. По-
лосы 490 и 740 см'1 возникают в результате комбинации частот
собственных колебаний селена 119, 250 и 269 см-1 [259]
(разд. 3.1.3.2).
В работе [260] описан способ определения кислорода в стек-
лах Geo,28Sbo,i2Seo-6 и Geo,33Aso.i2Seo,55 с помощью аннигиляции
позитронов (активационный анализ). Хилтон с сотр. [257, 261]
разработали метод непрерывного получения халькогенидных
стекол высокой чистоты в количествах от 5 до 10 кг.
Электрические и оптические свойства 495
4.2.1.3. Спектр электронного поглощения
(поглощение при больших энергиях) *
Как известно, при достаточно большой частоте электромаг-
нитного излучения, действующего на полупроводник, возможны
переходы электронов между валентной зоной и зоной проводи-
мости. Эксперименты по исследованию дрейфовой подвижности
(разд. 4.1.3.4) и фотопроводимости (разд. 4.1.4), наглядно по-
казывающие возникновение при облучении квазисвободных но-
сителей, подтверждают применимость зонных представлений
и к некристаллическим веществам.
Структура зон Бриллюэна в кристаллах отвечает существо-
ванию областей с минимальным расстоянием между свобод-
ными и занятыми зонными состояниями; соответствующие пе-
реходы отвечают краевому поглощению, которое первым про-
является в спектре межзонных переходов. Вообще различают
прямые и непрямые переходы. Первые происходят с сохране-
нием волнового вектора k (см. гл. 2 [ 106J и рис. 2.49); некото-
рые из них могут быть запрещены требованиями симметрии.
В непрямых переходах волновой вектор изменяется от 0 до
л/2, или обратно. Закон сохранения импульса требует, чтобы
в таких переходах участвовал хотя бы один фонон, вследствие
чего вероятность непрямых переходов меньше вероятности
прямых.
В некристаллических твердых телах правила отбора при
оптических переходах менее жестки, чем в кристаллах, благо-
даря малым длинам свободного пробега носителей и вытекаю-
щей отсюда неопределенности вектора k [см. граничные усло-
вия— уравнение (2.120)]. Однако это утверждение становится
проблематичным для переходов с энергией, выше (£с — Ev),
т. е. для межзонных переходов. С другой стороны, высокая
плотность состояний в зонах для некристаллических твердых
тел дает основания ожидать и здесь достаточно высоких ко-
эффициентов поглощения, соизмеримых с таковыми для кри-
сталлов, хотя в соответствующих спектрах должны проявляться
различия в электронной структуре аморфной и кристаллической
форм вещества.
Из-за больших величин коэффициента поглощения в обла-
сти межзонных переходов измерения проводят на отражение,
определяя спектральную зависимость R. Далее, с помощью со-
отношений (4.94) получают дисперсию показателя преломле-
ния п и постоянной поглощения k. Из рис. 4.41, где приведены
полученные таким способом спектральные зависимости е"=
* Подзаголовок, отсутствующий в оригинале, поясняет, что здесь рас-
сматривается поглощение, которое имеет место при энергиях выше края
(фундаментального) поглощения и соответствует межзонным переходам
электронов.— Прим. ред.
496 Глава 4
а
Рис. 4.41. Спектры электронного поглощения аморфного и кристаллического Si
[262] и аморфного и кристаллического Ge [263, 264].
(.....) — термически напыленный слой, ( — — • — ) — пленка, полученная катодным
распылением при температуре подложки 298 К, (-----) — то же при 623 К.
= 2nk для аморфных Si и Ge и их кристаллических аналогов,
видно, что явно выраженные в кристаллах двойные максимумы
не проявляются в спектрах аморфных форм. Следует отметить,
однако, что в области первого кристаллического максимума
в аморфных германии и кремнии наблюдаются сравнимые по
величине пики поглощения, второй же кристаллический пик
сильно ослаблен. Этот экспериментальный результат можно
объяснить тем, что более низкочастотный пик в спектре кри-
сталла [265] отвечает переходам в направлении [111], кото-
рое, по-видимому, воспроизводится и в аморфном состоянии,
а переходы в направлении [100], отвечающие высокочастотному
кристаллическому пику, не имеют аналогов в аморфном со-
стоянии. Такая трактовка подтверждается особенностями
структуры Si и Ge, рассмотренными в разд. 3.1.2.3*.
* Направление [111] в кристаллических Si и Ge отвечает направлению
вдоль ковалентной связи, а [100] — нет. Тогда сохранение низкочастотного
пика в аморфных Si и Ge можно связать с сохранением ближнего порядка,
который определяется ковалентными связями.— Прим. ред.
Электрические и оптические свойства 497
На рис. 4.42 приведены спектры для аморфной и кристалли-
ческой форм селена и мышьяка, из которых видно, что в дан-
ном случае интенсивность длинноволновой части спектра сильно
ослаблена, а сам спектр размыт [272]. Но в то же время для
спектра аморфного Se (рис. 4.42, а) повторяется основная осо-
бенность кристаллического спектра, а именно разделение ва-
лентной зоны на две составляющие [266, 268]; такой же ре-
зультат следует из фотоэмиссионных измерений [269].
Рис. 4.42. Спектр электронного поглощения кристаллического и стеклообраз-
ного селена [272] и аморфного мышьяка (гл. 3 [176]).
Отсутствие в спектре аморфного селена полосы 2 эВ согла-
суются с расчетами зонной структуры, в соответствии с кото-
рыми эта полоса в кристаллическом селене связывается со
взаимодействием между цепочками [270] (см. также г2 на
рис. 3.16, а)—поскольку в разд. 2.4.1.1 и 3.1.3.2 (рис. 2.29
и 3.18) было показано, что в стеклах это взаимодействие
сильно ослаблено. По-видимому, в стеклообразном селене па-
раллельно с ослаблением межцепочечного взаимодействия уси-
ливаются связи внутри цепочек, что приводит к смещению всего
спектра в коротковолновую область. Аналогичную картину
можно наблюдать при сравнении спектров кристаллического
и аморфного теллура; отмеченные выше особенности прояв-
ляются здесь еще ярче [271, 272] (см. также гл. 3 [267]). Для
аморфного мышьяка ослабление взаимодействия между слоями
по сравнению с кристаллом (см. гл. 3 [176] и рис. 3.13, б), при-
водит не только к сдвигу спектра в коротковолновую область
(см. рис. 4.42,6), но и к потере металлической проводимости.
32 Заказ № 413
498 Глава 4
Из рис. 4.43, а видно хорошее соответствие между спек-
трами кристаллической и аморфной форм As2Se3, если от-
влечься от различий в их интенсивности; то же справедливо
Рис. 4.43. Спектры электронного поглощения кристаллического и стеклооб-
разного As2Se3 [274] (см. литературу к гл. 3 [397]) и SiO2 [276] (см. литературу
к гл. 3 [437]).
и для As2S3. Рис. 4.43, б показывает, насколько хорошим мо-
жет быть соответствие электронной структуры зон в кристал-
лической и аморфной формах одного и того же вещества.
4.2.1.4. Край поглощения
На спектрах, приведенных на рис. 4.41—4.43, край погло-
щения не проявляется, так как соответствующие измерения от-
вечают очень высоким значениям коэффициента поглощения
(так, при п«2,5, 2/г£=1 и л=0,63 мкм или 2 эВ получаем
а = 2-104 см-1). Специальные измерения оптического поглоще-
ния при а<104 см~* дают информацию о плотности состояний
в хвостах зон.
На основании того, что в аморфных полупроводниках удво-
енная величина энергии активации проводимости примерно со-
ответствует частотам, отвечающим а=104 см-1, Штуке предло-
жил определять оптическую ширину запрещенной зоны по этому
значению а. Такие величины £УПт хорошо коррелируют с ре-
зультатами измерений фотопроводимости. Как и следует ожи-
дать, никакого разрыва на спектре поглощения при а=104 см-1
не наблюдается.
Электрические и оптические свойства
499
Оптическая ширина запрещенной зоны. По Тауцу [277]
в области а^Ю4 см-1 оптическое поглощение обусловлено пе-
реходами электронов из делокализованных состояний в валент-
ной зоне (E^EV) в делокализованные состояния в зоне про-
водимости (Е^Ес). В приближении параболической зависи-
мости плотности состояний
висимость коэффициента
поглощения следующего
вида:
а (<о) = Л[^о) — (Ес —
— Eyfflha (4.98)
Рис. 4.44. Край поглощения стеклообраз-
ного Аз25з при 293 и 90 К [280].
где А — некоторая посто-
янная [274] (см. гл. 1 [33]).
Девис и Мотт [278],
принимая линейную за-
висимость плотности со-
стояний в хвостах зон от
энергии, получили для
переходов из локализо-
ванных состояний в хво-
сте валентной зоны в де-
локализованные состоя-
ния в зоне проводимости
(Е^Ес) или из делока-
лизованных состояний
в валентной зоне (E^EV) в локализованные состояния в хво-
сте зоны проводимости уравнения, аналогичные (4.98);
а (<о) = а0 [Агсо — (Ел — Е И)]2/Л(о \Eiok
а(<о) = а0рмо — (Ес — Ев^/Ьм \Eiok (4.99)
где Д£/0^ = Ес — Еа=Ев — Ev (см. рис. 4.12). Возможность пе-
рехода электронов между хвостами зон пока не учитывалась.
Представленная на рис. 4.44 экспериментальная зависи-
мость характерна для некристаллических твердых тел, а также
для кристаллических полупроводников с непрямыми перехо-
дами. До величины [а((о)й(о]1/2 = 60 эВ+* 2-см_1'2 она описы-
вается уравнениями (4.98) или (4.99). Экстраполяцию линей-
ного участка кривой до пересечения с осью абсцисс (йчо =
= ЕС— Еу), как это показано на рис. 4.44, часто используют
для оценки оптической ширины запрещенной зоны (см. гл. 1
[33]). Величины Е0ПГ, определенные таким способом, как пра-
вило, на 0,1—0,2 эВ меньше определенных по способу Штуке
(т. е. для а= 104 см-1).
На рис. 4.45 показана корреляция между усредненной мо-
лярной энергией связи для типичных стеклообразующих соеди-
32*
500 Глава 4
нений, определенную из энтальпии атомизации (см. табл. 2.11)
и оптической шириной запрещенной зоны. Наличие общей кор-
реляции для стекол с самым различным строением указывает
на общие законы формирования их зонной структуры. Дей-
ствительно, во всех оксидных и халькогенидных стеклах ва-
лентная зона образована состояниями электронов неподелен-
ных пар, а полностью насыщенные связевые состояния распо-
ложены гораздо глубже. Отклонение от общей зависимости для
Рис. 4.45. Корреляция между средней энергией химических связей и шириной
оптической зоны (а=101 см~') для стеклообразующих соединений.
В2О3 можно объяснить наличием свободной р2-орбитали, с по-
мощью которой совершается переход с переносом заряда
между кислородными лигандами и центральным атомом Вш,
вследствие чего ширина зоны оказывается меньше ожидаемой.
Отклонение для BeF2 следует из очевидной разницы между
электронной структурой атомов кислорода и халькогенов —
с одной стороны, и атомов галогенов — с другой.
Правило Урбаха и температурная зависимость края погло-
щения. В кристаллических полупроводниках с прямыми разре-
шенными переходами коэффициент поглощения резко спадает
в крае фундаментального поглощения, изменяясь на несколько
порядков при снижении энергии на несколько десятых элек-
тронвольта. В стеклах, а также в щелочногалогенидных кри-
сталлах, CdS и тригональном селене в области а<104 см-1 ко-
эффициент поглощения уменьшается очень медленно: спад до
а= 14-0,1 см-1 растягивается на несколько электронвольт, при-
Электрические и оптические свойства
50Г
чем коэффициент поглощения в этой области экспоненциально
уменьшается с частотой. Естественно попытаться связать такое
поведение с постепенным изменением плотности состояний на
краях и в хвостах зон для некристаллических полупровод-
ников.
Экспоненциальное уменьшение коэффициента поглощения
с уменьшением частоты в области края поглощения впервые
Рис. 4.46. Край поглощения кристаллического и стеклообразного As2Ss в ко-
ординатах, соответствующих правилу Урбаха [уравнения (4.100)] [280, 281] (а)
и край поглощения двух различных типов кварцевого стекла [282] (б).
наблюдал Урбах [279] на щелочногалогенидных кристаллах.
Его можно описать эмпирическим выражением
а = В ехр [у' (ha) — ЕоптУбГ] (4.100}
причем В соответствует коэффициенту поглощения при /ио =
= ЕОпт- На рис. 4.46 в соответствующих координатах приве-
дены зависимости для AS2S3 и кварцевого стекла. При этом ока-
зывается, что ЕОпт, определяемая уравнением (4.100), отвечает
а = 0,1 и 1 см1.
Из рис. 4.44 видно, что положение края поглощения зависит
от температуры. Из температурно-частотных зависимостей а
можно с использованием уравнения (4.100) определить у'
и с ее помощью Еопт. Полученная таким образом оптическая
ширина запрещенной зоны для As2Se3 линейно зависит от тем-
502 Глава 4
пературы вплоть до температур расплава (рис. 4.47). В пред-
положении £Опт = £о11т(0) —уТ из уравнения (4.100) получаем
In а = In В 4- y'y/k + у' [йсй~Е0ПТ (0)]/£Т (4.101)
т. е. на частоте, отвечающей Й(о = £ОПт(0), коэффициент погло-
щения не зависит от температуры. Такие же исследования были
выполнены и для а-Si: Н [342]. Следует отметить, однако,
что определять этим способом ширину оптической запрещенной
зоны в общем случае некорректно, так как температурная за-
Рис. 4.47. Температурная зависимость оптической ширины запрещенной зоны
Вот для стеклообразного и жидкого As2Ses [284].
висимость, соответствующая уравнению (4.100), наблюдается
далеко не всегда.* Например, она выполняется в щелочнога-
логенидных кристаллах уже с 300 К, но во многих стеклах —
только выше температуры стеклования. Для объяснения та-
кого поведения Тауц [285] использовал представление о замо-
раживании флуктуаций потенциального рельефа при темпера-
туре стеклования. Для интерпретации правила Урбаха пред-
ложены и другие модели [286—288]. например Скеттрап [289]
рассмотрел вариант с тепловыми флуктуациями энергии опти-
ческой запрещенной зоны.
Поглощение в области малых а. Как правило, на экспери-
ментальных зависимостях Ina от Аго при a< 1 см-1 появ-
ляется излом, соответствующий переходу к прямой с меньшей
крутизной:
а = С ехр (hti'/Et), где Et > kT)y' (4.102)
* Простая модель урбаховского края предложена в работе [Kolobov А. V.,
Konstantinov О. V., Philos. Mag., 1979, В40, р. 475].— Прим, перев.
Электрические и оптические свойства
503:
На рис. 4.48 видны такие изломы для стекла As2S3 при раз-
личных температурах. Уменьшение наклона должно свидетель-
ствовать о постепенном уменьшении плотности состояний
в хвостах зон.
С другой стороны, было показано, что в особочистых и опти-
чески однородных образцах переход к «плоскому» участку про-
Рис. 4.48. Температурная зависи-
мость края поглощения стеклооб-
разного AsjSj в области малых
коэффициентов поглощения [281].
исходит при гораздо меньших значениях а. Так, Вашко с сотр.
[290] наблюдали урбаховский край на отдельных образцах се-
лена вплоть до 3-10~3 см-1. Банку с сотр. [291] показали, что
«плоский» участок связан с эффектами рассеяния. При этом
центрами рассеяния могут служить свили, посторонние ча-
стицы, примеси и т. д. (см. например [292]), и уменьшение про-
пускания в области малых а только кажется связанным с изме-
нением поглощения.
4.2.2. Фотолюминесценция
Дефектные центры в некристаллических твердых телах го-
раздо сильнее проявляются в спектрах фотолюминесценции,
чем в рассмотренных выше спектрах поглощения. Фотолюми-
несценция наблюдается при облучении светом из области края
поглощения, причем максимум спектра возбуждения отвечает
а—100 см"'. Появление максимума связано, вероятно, с ма-
лыми глубинами проникновения возбуждающего излучения при
больших коэффициентах поглощения. Примечательно, что сам
спектр излучения расположен при гораздо меньших частотах —
при энергиях, соответствующих приблизительно половине опти-
504 Глава 4
ческой ширины запрещенной зоны, т. е. имеет место сильный
стоксов сдвиг.
Такая фотолюминесценция, как отмечалось (разд. 2.5.2.1)
характерна для аморфных полупроводников с отрицательной
эффективной корреляционной энергией; она не наблюдается
в a-Si и a-Ge. В то же время, в слоях a-Si: Н, полученных раз-
ложением силана в тлеющем разряде, т. е. в условиях, когда
оборванные связи кремния «залечиваются» водородом, можно
Рис. 4.49. Спектр фотолюминесценции, спектр возбуждения фотолюминесцен-
ции и коэффициент поглощения а для стеклообразных АвгБез (а) и Se (б)
[294].
наблюдать сходную с описанной выше полосу люминесценции
[293] *.
На рис. 4.49 представлены типичные спектры фотолюминес-
ценции на примере Se и As2Se3. Подробно исследовались также
AS2S3 [295], и системы As—Se [294, 296], Ge—S [297] Ge—Se
[298, 307]. Спектр фотолюминесценции кварцевого стекла
имеет такой же характер [299].
Одна из первых работ по фотолюминесценции халькогенид-
ных стекол была выполнена Коломийцем с сотр. [300], которые
объясняли ее рекомбинацией на глубоких локализованных
уровнях вблизи уровня Ферми (см. также [278]). В модели
Стрита [301] такие уровни отвечают положительно и отрица-
тельно заряженным дефектным центрам, расположенным по
обе стороны уровня Ферми; эти дефекты находятся в термо-
динамическом равновесии в расплаве, но замораживаются ниже
* Люминесценция в a-Si: Н гораздо менее эффективна, чем в халькоге-
нидных стеклах, и характеризуется меньшим стоксовым сдвигом. - Прим,
ред.
Электрические и оптические свойства
505.
температуры стеклования. Попытки связать фотолюминесцен-
цию с электрическими и оптическими свойствами стеклообраз-
ных полупроводников в сочетании с гипотезой Андерсона (см.
гл. 2 [395]) привели к созданию конкретных моделей заря-
женных дефектных центров (см. разд. 2.5.2): модели Стрита—
Мотта (гл. 2 [395]) и модели Кастнера—Адлера—Фрицше
(гл. 2 [357]).
4.2.2.1. Спектр люминесценции с точки зрения модели
заряженных дефектных центров
Тот факт, что максимум спектра фотолюминесценции (ФЛ)
по энергии в два раза меньше максимума спектра возбуждения
(ВФЛ), указывает на сильное электрон-фононное взаимодейст-
вие в люминесцирующих стеклах. Другими словами, потенци-
альная энергия центра люминесценции сильно меняется при
удалении с него электрона вследствие сильной поляризации
и деформации ближайшего атомного окружения. При этом воз-
<7 б Р г-
Рис. 4.50. Схема уровней заряженных и нейтральных дефектов, а также пар^
с переменной валентностью для интерпретации спектров фотолюминесценции
в халькогенидных стеклах.
никает аналогия с известными центрами окрашивания в кри-
сталлах, спектроскопическое проявление которых трактуют на
основе поляронной модели (см. рис. 2.53).
В соответствии с рис. 4.50, а примем, что при поглощении
фотона с энергией £' из области краевого поглощения проис-
ходит перенос электрона от D~ или С~-центра (например,
Se.Se- по рис. 2.57) в зону проводимости (Е^Ес) с резуль-
тирующим образованием нейтрального дефекта D°(SeI/2Sef)
или <7°. К тому же результату приводит возбуждение элек-
трона непосредственно из валентной зоны в зону проводимости
при условии, что дырка из валентной зоны захватывается на
£>~-центр сразу же после своего возникновения. В результате
:'5О6 Глава 4
реакции окружения центра на образование D° (С®) уровень
дефекта смещается из положения А' в положение С', т. е.
почти в середину запрещенной зоны. При возвращении элек-
трона из зоны проводимости на уровень дефекта происходит
рекомбинационное излучение (фотолюминесценция), а пони-
женная по сравнению с энергией возбуждения энергия излуче-
ния (т. е. Ey<ZE'a) связана с описанным выше смещением
уровня дефекта (А'->С')-
Аналогичный процесс можно рассмотреть для оптического
возбуждения дырки (процесс Е" на рис. 4.50, б), которая обра-
зуется при переходе электрона из валентной зоны на ^-уро-
вень или в зону проводимости (Е^ЕС) с последующим захва-
том на £>+-центр. Состояние D° (С®) стабилизируется при
смещении уровня А"-+С", откуда с испусканием рекомбина-
ционного излучения Е" электрон возвращается в валентную
зону.
Для простоты на рис. 4.50 уровень Ферми показан лежа-
щим в середине запрещенной зоны и расстояния Е+ и Е~ при-
няты равными, т. е. Еа = Еа = Еа и Еу = Еу = Еу.
На конфигурационной диаграмме (рис. 4.50, г) рассмотрен-
ные выше оптические переходы в соответствии с принципом
Франка—Кондона изображены вертикальными линиями. Если
для простоты предположить, что форма потенциальной ямы
•одинакова для центра в любом состоянии (D, D° или D+),
т. е. что взаимодействие ближайшего окружения с дефектом
^сводится лишь к смещению его потенциальной ямы без ее из-
менения, то при энергии поляризации Ер разность между энер-
гией возбуждения и энергией излучения, которая наблюдается
в эксперименте (рис. 4.49), отвечает величине 2ЕР, что соот-
ветствует также соотношению С'—А'—А" — С"~2ЕР.
Термически индуцированные переходы между основным
и возбужденным состоянием совершаются, как правило, между
колебательными уровнями с наименьшей энергией — см. стрелку
Ер на рис. 4.50, г. На зонной схеме появляются соответствую-
щие уровни В' и В", которые отстоят от уровней возбужден-
ных дефектов С' и С" на величину поляронного сдвига Ер
(рис. 4.50, в). Рассмотрим, например, процесс термического
возбуждения электрона между валентной зоной (Е^Еу) и ос-
новным колебательным уровнем состояния D+ или С+
[ (SeSe3/)+] — В". Этому процессу соответствует энергия воз-
буждения или ионизации 1а(1)=Ес — Ev—(Е+ + ЕР) (см.
уравнение (2.128) и рис. 4.21,а). Затем следует релаксация
в положение С' или С®, т. е. fSeSe3/ ), и после возбужде-
ния второго электрона дальнейшая стабилизация центра с пе-
Электрические и оптические свойства
507
реходом его в положение В' (D~ или С~, т. е. Se- + SeSe2/).
Для возбуждения второго электрона требуется меньшая энер-
гия 1А(2)=Е~— Ер (см. также разд. 2.5.2.2. и 4.1.3.1). Анало-
гично можно рассмотреть термическое возбуждение электрона
с £)~-центра, т. е. с уровня В~, в зону проводимости (см.
рис. 4.50, в).
Из соотношения IA (1) — 1А (2) = В" — В' — £7Эфф, т. е. из раз-
ности энергий D+- и /)~-цеитров для термических переходов,
определяется величина корреляционной энергии ULP (см.
разд. 2.5.2.2, уравнение (2.131в) и рис. 2.56), которая возни-
кает вследствие межэлектронного отталкивания: Ulp =
= С ~С" = 2ЕР-Е^.
Процессы излучательной и безызлучательной рекомбинации
в халькогенидных стеклах исследовались во многих работах
(см., например, [302—305]). Фрицше с сотр. [306] исследовали
влияние разности электроотрицательностей элементов, входя-
щих в состав стекла, на спектр его люминесценции. Кастнер
и Хадженс (гл. 2 [408]) предложили модель люминесценции
на основе сближенных пар с переменной валентностью
(IVAP), с помощью которой можно объяснить нечувствитель-
ность люминесценции к легированию определенными приме-
сями. Такая нелегируемость впервые установлена Бишопом
с сотр. [394] на примере As2Se3 (интересно, что те же примеси
оказывают сильное влияние на электропроводность — см.
разд. 4.1.3.5). Исследование люминесценции методом времен-
ного разрешения при использовании наносекундных импульсов
позволило выделить два процесса, обусловленных различными
группами состояний [347].
4.2.2.2. Фотоиндуцированное
неравновесное дефектообразование
Экспериментально показано увеличение интенсивности фо-
толюминесценции с понижением температуры.* Это можно объ-
яснить уменьшением вероятности ухода возбужденных элек-
трона или дырки от центра люминесценции, вследствие чего
возрастает вероятность рекомбинации. Кроме того, на опыте
наблюдается так называемая усталость люминесценции (lumi-
nescence fatigue) которая проявляется в снижении интенсив-
ности излучения в процессе непрерывного возбуждения. «Уста-
лость» можно объяснить в рамках поляронной модели, полагая,
что при пониженных температурах способность окружения
* В халькогенидных стеклах фотолюминесценцию вообще можно наблю-
дать лишь при низких температурах, например, при 7<100К для Se или
при Т< 150 К для AS2SC3, поэтому такие измерения обычно относятся к 4 —
77 К.— Прим. ред.
508 Глава 4
центра к релаксации уменьшается, так что дефекты остаются
в метастабильном нейтральном состоянии DQ, из которого они
уже не могут быть возбуждены. В процессе возбуждения па-
раллельно с развитием «усталости» появляется широкая по-
лоса поглощения, которая начинается при энергиях, соответст-
вующих максимуму люминесценции, и продолжается в область
больших энергий [309, 344]. По-видимому, эта полоса фотоин-
дуцированного поглощения отвечает переходам из £)°-центров
на другие центры, энергия которых лежит между А' и С' или
А" и С" (см. рис. 4.50, в).
При низкотемпературном облучении сильнопоглощаемым
светом халькогенидные стекла дают отчетливый ЭПР-сигнал,
который можно трактовать как появление 2)°-центров, парамаг-
нитных вследствие наличия неспаренного электрона; эти центры
можно разрушить либо термически, либо при облучении све-
том из области фотоиндуцированного поглощения [310] (см.
также гл. 2 [397] и гл. 3 [191]). В работе [312] сообщалось
о фотоиндуцированном увеличении проводимости на перемен-
ном токе.
4.2.3. Показатель преломления и дисперсия света
При использовании твердых тел в качестве оптических сред
для систем записи и преобразования информации важное зна-
чение имеет показатель преломления и его зависимость от ча-
стоты, т. с. дисперсия. Теория даст зависимость показателя
преломления п от длины волны X в виде
п - 1 = £ лд7(х2 - X?) (4.103)
i
где If — длины волн, соответствующие собственным частотам
элементарных осцилляторов в среде, Ai— эмпирические кон-
станты или «силы осцилляторов». Для определения параметров
At и X,- требуется прецизионное измерение показателя прелом-
ления в возможно более широкой области спектра; такие из-
мерения очень немногочисленны. Очевидно, на параметры Ai
и X/ и, следовательно, на дисперсию существенно влияет струк-
тура вещества. Поскольку практически важные стекла имеют
сложный состав и структуру, соответствующие корреляции
установить трудно. Поэтому на практике используют введенную
Аббе среднюю дисперсию \п = п\— п2. Для характеристики
дисперсионного поведения стекла в видимой области спектра
измеряют показатель преломления на длинах волн, отвечаю-
щих спектральным линиям кадмия F' (479,99 нм) и С'
(643,85 нм); соответственно получают п\— nF' и п2 = пС'. Вели-
чину nF' — пС' называют главной дисперсией. Для перехода
ж характеристической относительной величине используют нор-
Электрические и оптические свойства
509
мировку на показатель преломления при промежуточной длине
волны, а именно, на длине волны зеленой линии в спектре
ртути е (546,07 нм). Тогда выражение (пР'—nC')f(пе-— 1)
определяет относительную дисперсию, а обратная величина из-
вестна как число Аббе
(пе—— пс) = уе (число Аббе) (4.104)
Рис. 4.51. лв/ув-диаграмма для
некоторых оптических стекол.
На рис. 4.51 представлена «е/\’е-диаграмма для оптических
стекол в видимой области спектра *. Малые числа Аббе гово-
рят о высокой относительной дисперсии, т. е. отвечают сильно-
преломляющим стеклам. Из халькогенидных стекол, область
прозрачности которых обычно лежит в ИК-области спектра,
в диаграмму попадает только желтое стекло GeS2.
На рис. 4.52 приведена аналогичная диаграмма для ближ-
ней ИК- (опорная длина волны Х = 2,0 мкм), а на рис. 4.53 —
для дальней ИК-области спектра (Х=10 мкм), при этом:
«2,0 — 1 «10,0 — 1
V2,0 — ~ ~ > V1Q = — ~
«1,8 — «2,2 «8,0 — «12,0
(4.105, 4.106)
* На рис. 4.51 приведено деление диаграммы Аббе, принятое в ГДР
(где К— крон, F — флинт, S — тяжелый, L — лантан, Р — фосфор. В — ба-
рий). В СССР принято Другое деление диаграммы Аббе. Так, в зависимости
от значений пе и ve стекло LaK называется либо сверхтяжелым кроном
(СТК), либо тяжелым кроном (ТК) и т. д. См. Совместный каталог опти-
ческого стекла СССР/ГДР, 1976.— Прим, перев.
2.0
Ge9
•AgCl
Csl M
*•* BaF,
NaCl •
500 400
MgF2
_l__________I__________l__
300 200 700 V
Рис. 4.52. n/v-диаграмма для оптических сред в области 2 мкм в соответствии
с уравнением (4.105). KRS-5 — твердый раствор (Т1Вг)о,44(ТП)о,5в.
И/о ° Се30Аз1з5е27"Ге30
2,8 As£Sf?3
о □
Ge22As22Se36Te20
° Ge30As13Se377e20
* Ge12,5Sb20Se675
6elOAsA0Se50 Ge10As30Seo60 . Ge125As10Sbt0Se675
0 Ge30As13^e477e17
Ge2OAs1OSe7O ° Ge30As20Se50
Ge125As20Se67>5 6e20As20Se60 Ge33As12Ses5
• о °
о Ge30As15Se55
6eioA5$oSe7o 0 Ge30As10Se60
• о
6eZOSe80
2.31___________1__________________l—
775 150
125
100
75
Рис. 4.53. n/v-диаграмма для оптических сред в области 10 мкм в соответ-
ствии с уравнением (4.106).
Электрические и оптические свойства
511
Область I на рис. 4.52 отвечает стеклам, представленным на
ne/ve— диаграмме (рис. 4.51), а область II отвечает стеклам,
оптические данные для которых взяты из работ [313] (На)
(гл. 2 [351] (Нб)). Для сравнения тут же приведены характе-
ристики для некоторых кубических кристаллов. Оптические па-
Рис. 4.54. Соответствие различных стекол дисперсионной формуле (4.103)
в точках равного наклона дисперсионных кривых dn/d'k——0,1 мкм~1 и dnld)^
~—0,2 мкм-1.
раметры стекол, изображенных на рис. 4.53, взяты из работ
[312—315].
Корреляции, представленные на рис. 4.54, показывают, что
стекла, даже сильно различающиеся по своим оптическим свой-
ствам, в частности по области прозрачности, обнаруживают
внутреннюю взаимосвязь с точки зрения их дисперсии.
4.2.3.1. Нормальная и аномальная
частные дисперсии
Проекционная оптика, как известно, требует сочетания сте-
кол с различными показателями преломления. Однако следует
иметь в виду, что качественное изображение в полихроматиче-
512 Глава 4
ском свете возможно лишь при одинаковой частной дисперсии
на всех участках дисперсионной кривой. В противном случае
у изображения вследствие хроматической аберрации возникает
цветная кайма.
При сочетании призм или линз цветовая компенсация до-
стигается при условии п'х—п' =const(п" — п"). Постоянная
в этом выражении определяется для призм отношением малых
преломляющих углов 0, а для линз — отношением фокусных
расстояний f; таким образом, можно регулировать ахроматизм
соответствующим выбором геометрических параметров на дли-
нах волн F' и С' главной дисперсии:
d' v' Прг — Пс, [' v" nF,— пс,
” ve nF,-nc, I Ve nF, — nc,
(4.107, 4.108)
Однако на длинах волн, отличных от F' и С' вновь возни-
кают хроматические аберрации (так называемый вторичный
спектр), и для их коррекции необходимо ввести относительную
частную дисперсию
Р = —, Ап—,—, например = —-------------— (4.109)
nF, — пс, nF, — nc,
В каталоге стекол, действующем в ГДР и СССР, используются
24 значения относительной частной дисперсии.
По Аббе большие группы стекол описываются эмпириче-
скими закономерностями вида (см. рис. 4.55, а)
PkL’ — CL + bve + CV2e (4.1 10)
где p2F' определяется уравнением (4.109). Уравнение (4.110)
показывает, что существует соответствие между изменением
дисперсии в некоторой области спектра и изменением диспер-
сии во всей рассматриваемой спектральной области. В этом
случае говорят о нормальном поведении или нормальной част-
ной дисперсии.
Вторичный спектр при сочетании нескольких линз можно
охарактеризовать через относительные частные дисперсии
f (4.111)
ve~ ve
где f — фокусное расстояние объектива. Переменными здесь
являются X и соответственно т]А. Как видно из рис. 4.55, б, кор-
рекция ахроматической аберрации для двух длин волн соответ-
ствует нулевой точке с р^' = р'^р,= \. Если p'KF, и p^F, не равны
Электрические и оптические свойства 513
единице, но равны между собой и имеют разные знаки, то полу-
чаем еще одну нулевую точку для третьей длины волны (апо-
хроматическая аберрация). Соответственно подобранные пары
стекол, отличающиеся от «нормальной» прямой на рис. 4.55, а,
применяются в оптических системах с минимальной хромати-
Рис. 4.55. Нормальная прямая (а), соответствующая уравнению (4.110) и
вторичный спектр в соответствии с уравнением (4.111) (б).
ческой аберрацией. При этом комбинируются стекла, резко
различающиеся по положению и интенсивности полос поглоще-
ния в электронном и колебательном спектрах, а, значит, и по
структуре [317]. Различные группы стекол можно выделить
в данном случае следующим образом: если провести «нормаль-
ную» прямую (см. рис. 4.55, а) для силикатных стекол, то бо-
ратные стекла дадут положительные, а фосфатные — отрица-
тельные отклонения от этой прямой [318, 319].
4.2.3.2. Пьезооптический эффект
(фотоупругость)
Отличительной особенностью стеклообразного состояния яв-
ляется то, что твердые стекла, подобно жидкостям, изотропны.
С другой стороны, на основании описанного в разд. 2.4.3.5 раз-
личия в упругих постоянных стекол в зависимости от направ-
ления распространения упругих волн относительно направле-
ния смещения частиц, можно ожидать, что под действием меха-
нических сил оптическая изотропия будет нарушаться.
Действительно, в работе [348] сообщалось о возникновении
двулучепреломления в ориентированном расплаве AsxSi~x.
33 Заказ № 413
514 Глава 4
Как известно, зависимость характеристик распространения
света в среде (изотропной или анизотропной) от направления
описывается индикатрисой. На рис. 4.56 представлена индикат-
риса для первоначально оптически изотропной среды, в которой
под действием одноосного сжатия или растяжения сферическая
симметрия сменяется вращательной эллипсоидной. В результате
появления оптической оси световой луч, падающий перпенди-
кулярно направлению внешней механической силы, разде-
ляется на два луча: обыкновенный и необыкновенный, причем
последний колеблется в плоскости, перпендикулярной направ-
лению силы. В этом случае при повороте анализатора перио-
анализатор
Рис. 4.56. Индикатриса изотропной оптической среды, находящейся под внеш-
ним механическим напряжением.
дически появляются участки с большим или меньшим пропу-
сканием. Разность хода лучей выражается через разность в по-
казателях преломления в направлении, параллельном и перпен-
дикулярном оптической оси, и связана с величиной деформи-
рующего напряжения:
дЛц=п0-п =C|So
п — п = ( с — с A S = BS
II 1 \ 1 2/ о о
(4.112)
где Ci и с2 — упругооптические коэффициенты, а В — упругооп-
тическая постоянная, измеряемая в ТПа-1 (1 ТПа-1 =
= 0,981 см2/кгс=1 брюстер). Будучи разностной величиной, В
не определяет влияние нагрузки на сам показатель преломле-
ния, так что даже при величине В, близкой к нулю, коэффи-
циенты Ct и с2 и, следовательно, Дп и Дп± могут принимать
большие значения.
В оксидных стеклах упругооптическая постоянная В изме-
няется от —2 до +4 ТПа-1, а в халькогенидных, как было по-
казано Линке и др. при изучении около 120 стекол (гл. 2
[351]),— от +20 до —40 ТПа-1 (см. также гл. 2 [16]).
На рис. 4.57 приведены зависимости величины упругооптиче-
Электрические и оптические свойства
515
Рис. 4.57. Зависимость упругооптической постоянной от состава для некото-
рых стекол.
ской постоянной от состава системы, из которых можно сде-
лать вывод, что решающее влияние на величину и знак В ока-
зывает степень увязанпости структурной сетки стекла, а при
одинаковой степени увязанности величина В уменьшается
с увеличением поляризуемости атомов в составе стекла. В стек-
лах с трехмерной сеткой (GeSe2, GeS2, Ge2S3, Ge2Se3, SiO2
и т. д.) В положительна, а в стеклах с двумерной (AS2S3 или
AssSe^) или цепочечной (Se) структурой — отрицательна. Как
видно из рис. 4.57, введение Sb2S3 или Sb2Se3 в стекла с трех-
мерно-увязанной структурой действует на изменение в соответ-
ствующих системах таким же образом, как введение Аз25з или
As2Se3. По Линке (гл. 2 [343], см. также [324]) такое поведение
упругооптической постоянной объясняется следующим образом.
В стеклах с трехмерной структурой одноосная деформация
всегда приводит к большему увеличению оптической плотности
в направлении сжатия. Из этого следует п. >п т. е. В>0.
В стеклах с двумерной или одномерной структурой этот эффект
более чем компенсируется за счет соответствующей ориентации
структурных групп ближнего порядка, что приводит к л
и В < 0. Отмеченное выше уменьшение В при увеличении поляри-
зуемости атомов, составляющих стекло, можно объяснить тем,
33*
516 Глава 4
что при уменьшении энергии связи (увеличении «рыхлости»
структуры) упругая деформация сильнее сказывается на изме-
нении углов, чем на изменении расстояний, что приводит к наи-
большему изменению оптической плотности в направлении, пер-
пендикулярном оси деформации.
В соответствии с этими представлениями силикатные стекла
с высокой степенью увязанности структуры имеют большие по-
ложительные значения В, например +3,5 ТПа1 в SiO2. При
модификации кварцевого стекла другими оксидами степень увя-
занности уменьшается, но ее не удается связать определенным
образом с появлением различных катионно-кислородных поли-
эдров: как видно из рис. 2.48, упругие постоянные силикатных
стекол сложным образом зависят от их состава. Чтобы пони-
зить В, можно вводить в стекло катионы с большой поляризуе-
мостью, например РЬ2+, Т1+ или Bi3+; на такой способ снижения
фотоупругости указывал еще Покельс [320]. Стеклами с мини-
мальной фотоупругостью являются фосфатные, в которых мала
степень увязанности структурной сетки.
Данные по фотоупругости кристаллов подтверждают рас-
смотренную ранее структурную интерпретацию явления. Как
видно из табл. 4.7, в кубических кристаллах Si и Ge, так же
Таблица 4.7. Фотоупругость стеклообразных и кристаллических веществ
Стекло В, ТПа-' An Кристалл В, ТПа-' An
OeS2 +22,1 +2,2 • IO’5 Si +11,8 1,2 • 10-5
Ge2S3 +21,0 +2,1 • 10-5 Ge +24,5 2,4 • IO’5
Ge2S3 +8,5 +0,9 . IO’5 ZnS +0,02
SiO2 +3,5 +0,35 • 10-5 CdS +0,03
Полистирол +6,7 Триг. Se + 1,04
As2S3 -7,8 —0,8 • 10-5 MoS2 —2,3
As2Se3 —39,4 —3,9 • 10-5 Графит —0,5
Se —30,4 —3,0 . io-5 FeCO3 -0,24
(Oe2Se3) — (Sb2S3)2 ±0 0 CaCO3 Be3AI2Si6Ote —0,17 —0,01
как и в стеклах с трехмерной структурой, величина В положи-
тельна. Вюрцит, CdS и тригональный селен (оптически одно-
осные кристаллы с трехмерной структурой) являются анизо-
тропными кристаллами, и эффект по Д/г на три порядка выше,
чем в предыдущем случае, хотя величина В еще положительна.
Электрические и оптические свойства
517
В кристаллах со слоистой структурой, в которых анизотропия
еще сильнее, появляются отрицательные значения В.
Теоретические вопросы фотоупругости рассмотрены в рабо-
тах [321—323]. По Меллеру [322], где приводится количествен-
ное рассмотрение, величина В связывается с поляризуемостью,
однако при этом не привлекаются какие-либо структурные
представления.
4.2.4. Фотоиндуцированные структурные превращения
в стеклах
Электронное возбуждение стекол, как неравновесных систем
с высокой концентрацией замороженных дефектов, может при-
вести к протеканию в них фотохимических реакций, которые,
в свою очередь, способны вызвать необратимое или обратимое
изменение структуры и свойств. К эффектам такого рода отно-
сится давно известная соляризация — потемнение стекла при
интенсивной солнечной засветке.
Фотохромные стекла с широко варьируемыми свойствами
получают при введении примесей галогенидов серебра, меди
или кадмия, молибдата или вольфрама серебра, а также опре-
деленных комбинаций редкоземельных элементов. Практиче-
ский интерес представляют, во-первых, стекла, которые быстро
темнеют, но затем быстро восстанавливают свою прозрачность
(светозащитные пластины и очки, модуляторы света), и, во-
вторых, фотохромные стекла для оптической записи информа-
ции. На параметры таких стекол (квантовый выход, контра-
стность, постоянная времени, соответствущая увеличению или
уменьшению пропускания в два раза) можно эффективно
влиять изменением состава стекла или приданием ему опреде-
ленной структуры, например, путем направленного выделения
микрофаз. Полихромные стекла позволяют, кроме того, полу-
чать видимое изображение распределения температур в иссле-
дуемом объекте [325].
В результате фотохимических реакций могут появляться суб-
микроскопическне выделения металлов (например, серебра),
которые служат центрами кристаллизации. Образующиеся ге-
терогенные микрообласти быстрее растворяются в травителе
(например, в плавиковой кислоте), чем необлученные участку
стекла. Таким образом, получают фотоформы с высоким разре-
шением.
Фотохромные стекла и соответствующие процессы рассмот-
рены во многих работах; для первоначального ознакомления
можно рекомендовать монографии Фогеля (гл. 1 [17])
и Шольце (гл. 1 [16]) вместе с цитированной там литературой.
В халькогенидных стеклах фотоиндуцированное изменение
свойств происходит и без обычных фотохромных добавок.
518 Глава 4
По-видимому, сравнительно малая энергия химических связей
в таких стеклах облегчает протекание фотохимических реакций
и фотоструктурных превращений. Это относится, в частности,
и к фотокристаллизации: в халькогенидных стеклах с мень-
шей, чем в оксидных, энергией активации кристаллизации
фотокристаллизация протекает даже без дополнительного на-
грева.
4.2.4.1. Фотоиндуцированные изменения
края поглощения и показателя преломления
в халькогенидных стеклах
В этих экспериментах, связанных с облучением халькогенид-
ных стекол интенсивным светом из области края поглощения,
следует обращать внимание на возможность процессов окисле-
ния и гидролиза [326, 327]. Соответствующие исследования
обычно выполняются в высоком вакууме на пленках толщиной
несколько микрометров, которые получают термическим испаре-
нием в вакууме или катодным распылением и предварительно
отжигают при температурах вблизи 7\.
При облучении халькогенидных пленок наблюдается сдвиг
в длинноволновую область, причем тем больший, чем ниже тем-
пература, при которой производится облучение [328, 345, 346]
(см. также гл. 3 [369])*. Для As2S3 при 14 К была достигнута
величина сдвига —0,2 эВ. Как видно из рис. 4.58, а, эффект
фотопотемнения обратим: отжиг при температурах вблизи Tg
приводит к восстановлению исходного спектра поглощения.
Обратимость фотоструктурных превращений подтверждается
исследованиями методом рентгеновской дифракции (рис. 4.58,6).
Сдвиг края поглощения сопровождается изменением показа-
теля преломления (для As2S3, например, было получено кпх
»0,1 (см. гл. 3 [369]), что представляет интерес для гологра-
фической записи и храпения информации [330]. Следует отме-
тить, что фотопотемнение в AS2S3 сопровождается также обра-
тимым увеличением объема примерно на 0,4 %, причем в плен-
ках As4Se5Ge. напротив, наблюдается уменьшение объема при
облучении, примерно на 0,2 % [329]. Фотоструктурные превра-
щения детально исследованы в пленках As2Se3 [331] (см. также
гл. 3 [369]), As—Se и Ge—Se [332], Ge—S [333], Ge—As—Se
[334, 335, 338] и Ge—As—S [336] **.
* В этой связи cm. [Ke Tanaka, J. Non-Cryst. Solids, 59/60, 925 (1983)]. —
Прим. ped.
** Большой вклад в исследование фотоструктурных превращений внесли
исследователи ленинградской школы см., например: Kolomiets В. Т., Lyu-
bin. V. М., Mater. Res. Bull., 13, 1343 (1978); Лихолит И. Л. Любин В. М.,
Макаров В. Ф., Федоров В. А.— ФТТ, 1984; т. 26, с. 172; Averianov V. L.,
Kolobov А. V., Kolomiets В. Т., Lyubin V. М., Phys., Stat. Sol., А57, 81
(1980).— Прим. ред.
Электрические и оптические свойства 519
Фотоиндуцированные эффекты в халькогенидах мышьяка
можно трактовать на основе представлений о разрыве связей
As—X с образованием связей As—As и X—X [334], а также
с точки зрения полимеризации изолированных молекулярных
групп, изображенных на рис. 3.31. В работе [337] была пред-
ложена модель взаимного превращения структурных групп
с изменением связей внутри и по краям группы; по-видимому,
Рис. 4.58. Обратимые фотоструктурные превращения в пленках Аз25з [328,
329].
а — сдвиг края поглощения; б — кривая рентгеновской дифракции. X — слой, отожженный
при 443 К,О—после облучения при 290 К. •—после облучения при 77 К.
эта трактовка имеет отношение к изменению валентного взаи-
модействия между соседними атомами, которое проявляется
в различии спектров поглощения красного и тригонального се-
лена и кристаллического и аморфного мышьяка (см.
4.42, б).
В заключение отметим, что обсуждаемый эффект потемне-
ния не следует путать с фотонаведенным поглощением, описан-
ным в разделе 4.2.2.1. Последнее наблюдается только при про-
должительном облучении при 14 К, характеризуется увеличе-
нием поглощения между полосой люминесценции и краем
поглощения, и связано, по-видимому, с неравновесным заполне-
нием дефектных состояний внутри щели подвижности.
520 Глава 4
4.2.4.2. Фотокристаллизация
Можно ожидать, что аморфные слои, кристаллизующиеся
при нагреве около температуры стеклования, способны кри-
сталлизоваться при воздействии света за счет локального разо-
грева, т. е. в результате безызлучательной диссипации энергии
(см. рис. 4.50). Что касается теплового действия света, то в ра-
боте (гл. 3 [572]) установлено, что после воздействия корот-
кого импульса света с крутым задним фронтом на предвари-
тельно закристаллизованную эвтектику GeTe/Te материал мо-
жет перейти в стеклообразное состояние.
В работе [ЮЗ] сообщается о кристаллизации пленки
Gei5Te8iSb2S2 толщиной 2 мкм при облучении в течение 0,045 с,
что соответствует скорости кристаллизации п^4-10-3 см-с-1.
В [104] наблюдали увеличение скорости кристаллизации селена
в 20 раз при освещении, соответствующем созданию 3• 1018 элек-
тронно-дырочных пар на 1 см2 в 1 секунду; при этом «=
= 3-10“5 см-с-1. В данном случае из спектральной зависимо-
сти было установлено, что скорость кристаллизации опреде-
ляется концентрацией носителей заряда, а не поглощенной
энергией.
Другие доказательства того, что электронный вклад оказы-
вается существенным при фотокристаллизации приведены в ра-
ботах [108, 134, 149]. При облучении пленок Gei5Te8iSb2S2
и Se—Те лазерным лучом диаметра 3 мкм, интенсивностью от
10 до 100 мВт и продолжительностью импульса от 1 до 10 мкс
кристаллизация происходит за время, меньшее 5 мкс, что отве-
чает и~ \ см-с-1. В таких опытах концентрация электронов
и дырок на 4 порядка больше, чем в описанных выше опытах
с селеном, на основании чего можно ожидать сильного возбу-
ждения всей системы связей, вплоть до их разрыва. При этом
высокая скорость кристаллизации объясняется увеличением
подвижности структуры, т. е. созданием благоприятных кинети-
ческих условий для зарождения и роста кристаллов.
Фотокристаллизацию можно провести в условиях, когда
размер кристаллических микрообластей обеспечивает плотность
записи 500 линий на миллиметр. Отличие этих областей по ко-
эффициенту поглощения и показателю преломления от стекло-
образной матрицы обеспечивает оптическую запись информа-
ции, которую стирают с помощью лазерных импульсов соответ-
ствующей длины волны и интенсивности с достаточно крутым
задним фронтом. Описанный принцип записи и стирания инфор-
мации был запатентован [156, 168].
Дополнительные сведения по данному вопросу можно найти
в работах: [189], где подробно описаны обратимые фотостиму-
лированные фазовые превращения, а также в [278, 308], по-
Электрические и оптические свойства
521
священных фотокристаллизации аморфного селена, и [339], где
исследовалась фотокристаллизация в пленках системы
GexSei-x*.
Литература
1. Rasch Е., Hinrichsen F. W., Z. Elektrochem. angew. phys. Chem., 14, 41
(1908).
2. Macedo P. B., Moynihan С. T., Bose R., Phys. Chem. Glass., 13, 171
(1972).
3. Debye P., Polar Molecules, Dover, N. Y. ,1945.
4. Stevels J. M., in: Handbuch der Physik, Springer-Verlag, Berlin, 1957,
B. 20, S. 350.
5. Taylor H. E., J. Soc. Glass Technol., 41, 050 T (1957); 43, 124 T (1959).
6. Owen A. E., Electric conduction and dielectric relaxation in glass, in:
Progr. in Ceramic Science, J. E. Burke (ed.), Pergamon Press, 1963, V. 3,
p. 77.
7. Provenzo V., Boesch L. P., Voltera V.. Moynihan С. T.. Macedo P. B.,
J. Amer. Ceram. Soc., 55, 492 (1972).
8. Blank R., Glastechn. Ber., 39, 489 (1966).
9. Frenkel J., Z. Physik, 35, 657 (1926).
10. Мюллер P. Л.— ЖТФ, 1955, t. 25, c. 236.
11. Charles R. J., J. Amer. Ceram. Soc., 45, 105 (1962).
12. Boskcy Z., Lengyel B., Z. physik. Chem. (Leipzig), 241, 36 (1969); J.
Non-Cryst. Solids, 14, 79 (1974)
13. Engel J. R., Totnozawa M., J. Amer. Ceram. Soc., 58, 183 (1975).
14. Frischat G. H., Ionic diffusion in oxide glasses, Aedermannsdorf (Schweiz),
Trans. Techn. Publ., 1975.
15. Owen A. E., Douglas R. W., The electrical properties of vitreous silica,
J. Soc. Glass Technol., 43, 159 (1959).
16. Schtuchukerev W. A., Muller R. L., Z. physik. Chem., 150A, 183 (1975).
17. Rickert H., Angew. Chern., 90, 38 (1978).
18. Holzapfel G., Rickert H., Naturwissenschaft., 64, 53 (1977).
19. Junod P., Hediger H„ Rilchdr B„ Wulischleger J., Phil. Mag., 36, 941
(1977).
20. Sakuma T., Hoshino S., Techn. Rep. ISSP Ser. A No. 1037 (1980).
21. Hayes T. M., Boyce J. B., Beeby J. L., J. Phys. C: Solid State Phys., 11,
2931 (1978).
22. Villenneuve G., Echengut P., Lucat C., Reau J.-M., Hagenmuller P., Phys.
Stat. Solidi (b), 97, 295 (1980).
23. Ravaine D., J. Non-Cryst. Solids, 38, 39, 353 (1980).
24. Barrau B., Ribes M., Maurin M., Rone A., Souquet J.-L., J. Non-Cryst
Solids, 37, 1 (1980).
25. Souquet J.-L., Rone A., Ribes M., J. Non-Cryst. Solids, 38, 39, 307 (1980).
26. Malugani J. P., Wasniewski A., Doreau M., Robert G., C. R. Acad. Sc.
Paris, Ser. C, 287, 455 (1978).
27. Magulani J. P., Wasniewski A., Doreau M., Robert G., Al Rikabi A., Mat.
Res. Bull., 13, 427, 1009 (1978).
28. Minami T., Hambu H., Tanaka M., J. Amer. Ceram. Soc,. 60, 467 (1977).
29. Grant R. J., Ingram M. D., Turner L. D. S., Vincent C. A., J. Phys. Chem.,
82, 2838 (1978).
30. Chiodelli G., Magistris A., Schiraldi A., Elektrochem. Acta, 23, 585 (1978).
* Следует упомянуть также обнаруженный советскими физиками про-
цесс фотолегирования \Костышин М. Т„ Михайловская Е. В., Романенкс*
П. Ф.— Физ. твердого тела, 1966, т. 9, с. 571].— Прим, перев.
522 Глава 4
31. Minatni Т., Tanaka M.t Rev. Chim. Minirale, 16, 283 (1979).
32. Jonscher A. K., Nature, 267, 673 (1974); Phys. Status Solidi (b), 83, 585
(1977); The universal dielectric response, Chelsea Colege, London, 1978.
33. Boesch L. P„ Moynihan С. T., J. Non-Cryst. Solids, 17, 44 (1975).
34. Hakitn R. M., Uhlmann D. R., Phys. Chem. Glass., 8, 174 (1967).
35. Lengyel B., Boskay Z., Z. phis. Chem. (Leipzig), 223, 49 (1963).
36. Terai R., J. Non-Cryst. Solids, 6, 121 (1971).
37. Day D. E., J. Non-Cryst. Solids, 21, 343 (1976).
38. Isard J. 0., J. Non-Cryst. Solids, 1, 235 (1969).
39. Lengyel B., Boskay Z., Varga M., Z. physik. Chem. (Leipzig), 242, 37
(1969); 217, 357 (1961); 223, 186 (1963).
40. Hendrickson J. R., Bray P. J., Phys. Chem. Glass., 13, 43 (1972).
41. Rouse G. B., Gordon J. M., Resin W. M., J. Non-Crust. Solids, 33, 83
(1979).
42. Hayward P. J., Phys. Chem. Glass, 17, 54 (1976).
43. Hamikawa H., J. Non-Cryst. Solids, 14, 88 (1974), 18, 173 (1975).
44. Mackenzie J. D., Bull. Amer. Ceram. Soc., 42, 223 (1963).
45. Petrovskij, Silikaty (Praha), 3, 336 (1959).
46. Мазурин О. В., Браиловская P. В.— Физ. твердого тела, 1960, т. 2, с. 1477.
47. Schwarz М., Mackenzie J. D., J. Amer. Ceram. Soc., 49, 582 (1966).
48. Mackenzie J. D., Mod. Aspects of the vitreous state, 3, 126 (1965).
49. Lengyel B., Boskay Z., Z. physik. Chem. (Leipzig), 222, 183 (1983), 242,
23 (1969).
50. Dunn B., Chen F., Mackenzie J. D., J. Non-Cryst. Solids, 16, 313 (1974).
51. Johnston W. D., J. Chem. Educat., 36, 605 (1959).
52. van Houten S., J. Phys. Chem. Solids, 17, 7 (1960).
53. Feltz A., Linke L, Z. anorg. Chem., 415, 161 (1975).
54. Linke L., Dissertation A., Friedrich-Schiller-Univ., Jena, 1975.
55. Китайгородский H. И., Фролов В. К.— Стекло и керамика, 1960, с. 5.
56. Lansberger F. R., Bray Р. J., J. Chem. Phys., 33, 1757 (1976).
57. Linsley G. S., Owen A. E., Hayatee F. M., J. Non-Cryst. Solids, 4, 208
(1970).
58. Sayer M., Mansingh A., Reyes J. M., Rosenblatt G., J. Appl. Phys., 12,
2857 (1971); Phys. Rev., В 6, 4629 (1972).
59. Munakata M., Kawamura S., Asahara J.. Iwamoto M., J. Ceram. Assoc.
Jap., 67, 74 (1959).
60. Janakirama-Rao В. V., J. Amer. Ceram. Soc., 48, 311 (1965).
61. McMillan P. W„ Adv. Glass Technol., 1, 333 (1962).
62. Hansen K. W., J. Electrochem. Soc., 112, 994 (1965).
63. Hirashima H., Yoshida T., Yogyo-Kyokai-Shi, 85, 434 (1977).
64. Mackenzie J. D., J. Amer. Ceram. Soc., 47, 211 (1964).
65. Takamori T., Sero IL, Tashiro M., J. Amer. Ceram. Soc., 52, 582 (1969).
66. Feltz A., Unger B., Z. Chem., 24, 100 (1984).
67. Feltz A., Unger B.. Z. anorg. allg. Chemie, в печати.
68. Unger В., Dissertation A., Friedrich-Schiller-Univ., Jena, 1977.
69. Jordan B. D., Calvo C., Canad. J. Phys., 55, 436 (1977).
70. Murawski L., Chung С. H, Mackenzie J. D., J. Non-Cryst. Solids, 32,
91 (1979).
71. Aral K., Kumata K., Kadota K-, Yamamoto K-, Namikawa H., Saito S.,
J. Non-Cryst. Solids, 13, 131 (1973).
72. Mott N. F., J. Non-Cryst. Solids, 1, 1 (1968).
73. Austin I. G„ J. Non-Cryst. Solids, 2, 474 (1970).
74. Austin I. G., Mott N. F., Adv. Phys., 18, 45 (1969).
75. Nestler H. H, Kingery W. D., Compl. Rend. VII Congr. Int. Verres, Brus-
sel, 1965, Vol. 1, 1, 106.
76. Schmid A. P., J. Appl. Phys., 39, 3140 (1968).
77. Greaves G. N., J. Non-Cryst. Solids, 11, 427 (1973).
78. Feltz A., Linke L, Z. allg. Chem., 415, 156 (1975).
Электрические и оптические свойства
523
79. Feltz A.. Schtnalifuss S., Linke L, Z. Chem., 15, 118 (1975).
80. Feltz A.. Wollenhaupt R., Z. anorg. allg. Chein., 466, 41 (1980); J. Non-
Cryst. Solids, 35, 36, 1015 (1980).
81. Selbin J., Chem. Rev., 65, 154 (1965).
82. Feltz A., Scmalfuss S., Langbein IL, Tietz M., Z. anorg. allg. Chem., 417,
125 (1975).
83. Feltz A., Z. anorg. allg. Chem., 355, 120 (1967).
84. Toyuki H., Akagi S., Phys. Chem. Glass., 13, 15 (1972).
85. Livage J.. Pineau P., Leroy M. C., Michaud M., Phys. Status Solidi (a),
39, 73 (1977).
86. Абдулаев А. А., Кисловский Л. П.— Кристаллогр., 1970, т. 15, с. 988.
87. Kennedy T. N., Mackenzie J. D., Phys. Chem. Glass., 8, 169 (1967).
88. Paul A., Yee N„ J. Non-Cryst. Solids, 24, 259 (1977).
89. Munakata M., Solid State Electronics, 1, 159 (1960).
90. Davis E. A., Mott N. F., Phil. Mag., 22, 903 (1970).
91. Marshall J. M., Owen A. E., Phil. Mag., 24, 1281 (1971).
92. Fritzsche H., Electronic Properties of Amorphous Semiconductors, in:
Amorphous and Liquid Semiconductors, J. Tauc (ed.), Plenum Press, Lon-
don, 1974.
93. Nagels P., in: Amoiphous Semiconductors, M. H. Brodsky (ed.), Springer-
Verlag, Berlin, Heidelberg, New York, 1979, p. 113.
94. Overhof H., Festkorperprobleme (Advances in Solid State Physics) V. XVI.,
J. Frensch (ed.), Vicveg, Braunschweig, 1976, S. 239.
95. Emin D., in: Electronic and Structural Properties of Amorphous Semi-
conductors, P. G. Lie/Commer J. A\ort (eds.). Academic Press, London,
1973, p. 261; Proc. 7th Int. Conf. Amorph. Liquid Semiconduc., Edinburgh
1977, University Edinburgh, p. 249.
96. Emin D., Seager С. H., Quinn R. K-, Phys. Rev. Lett., 28, 813 (1972).
97. Mahan J. E., Bube R. FL, J. Non-Cryst. Solids, 24, 29 (1977).
98. Mott N. F., Science, 201, 871 (1978).
99. Pfeister G., Morgan M., Phil. Mag., В 41, 191 (1980).
100. Henkels H. W., Maczuk J., J. Appl. Phys., 24, 1056 (1953).
101. Croitoru N., Popescu C., Lazarescu M., J. Non-Cryst. Solids, 4, 493 (1970).
102. Grant A. I., Moustakas T. D., Penney T.. Weiser K-, Proc. 5th Int. Conf.
Amorph. Liquid Sernicond., Garmisch-Partenkirchen 1973, Taylor-Francis,
London, 1974, p. 325.
103. Evans E. J., Helbers J. H., Ovshinsky S. R., J. Non-Cryst. Solids, 2, 334
(1970).
104. Dresner L, Stringfellow G. B., J. Phys. Chem. Solids, 29, 303 (1968).
105. Nagels P., Colson R., van Gool L., Proc. 7th Int. Conf. Amorph. Liquid
Sernicond., Edinburgh, 1977, University Edinburgh, p. 546.
106. Kahnt FL, Feltz A., J. Non-Cryst. Solids, 98, 211 (1982).
107. Holstein T., Ann. Phys. (N. Y.), 8, 343 (1959).
108. Feinleib I., De Deufville L, Moss S, C Ovshinsku S R.. Annl. Phvs.
Lett., 18, 254 (1971).
109. Friedman L., J. Non-Cryst. Solids, 6, 329 (1971).
110. Holstein T„ Phil. Mag., 27, 225 (1973).
111. Male J. C., Brit. J. Appl. Phys., 18, 1543 (1967).
112. Панус В. P., Ксендзов Ю. M., Борисова 3. У.— Изв. АН СССР, неорг.
матер., 1968, т. 4, с. 778.
113. Ивкин Е. Б., Коломиец Б. Т., Лебедев Э. А.— ДАН СССР, физ. сер. 1964,
т. 28, с. 1190.
114. Nagels Р., Colson R., van Gool К, J. Non-Cryst. Solids, 35, 36,427 (1980).
115. Stuke J., Proc. Vlth Int. Conf. Amorph. Liquid Sernicond., В. T. Kolomiets
(ed.), Leningrad, Nauka, 1976, p. 193.
116. Nakashita T„ Hirose M., Osaka Y., Jap. J. Appl. Phys., 17, 985 (1978).
117. Hirose M., Suzuki T„ Yoshifuji S., Osaka Y., Jap. J. Appl. Phys., 13,
40 (1974).
524 Глава 4
118. Viscor Р„ Loffe A. D., J. Non-Cryst. Solids, 35, 36, 409 (1980).
119. Davis E. A., Mott N. F., Phil. Mag., 22, 903 (1970).
120. Grunewald M„ Thomas P., Phys. Status Solidi (b), 94, 125 (1979).
121. Lewis A. J., Pioc. Vlth Int. Conf. Amorph. Liquid Semicond., В. T. Kolo-
miets (ed.), Leningrad, Nauka, 1976, p. 299.
122. Jeffrey F. R., Shanks H. R., Danielson G. C., J. Non-Cryst. Solids, 35, 36,
261 (1980).
123. Van Dong A’., Hai T. G., J. Non-Cryst. Solids, 35, 36, 351 (1980).
124. Beyer W., Stritzker B., J. Non-Cryst. Solids, 35, 36, 321 (1980).
125. Tauc J, Stourac I.., Vorlicek V., Zavetova M., Proc. IXth Int. Conf. Phys.
Semicond., Moscow, 1968, p. 1251.
126. Di Salvo F. J., Menth A., Waszczak J. V., Tauc J., Phys. Rev., В 6, 4574
(1973).
127. Agarwal S. C., Guha S., Narasimhan K. L., J. Non-Cryst. Solids, 18, 429
(1975).
128. Yoshida A., Arizumi T., Shimakawa K., Nitta S., Proc. Vlth Int. Conf.
Amorph. Liquid Semicond., В. T. Kolomiets (ed ), Leningrad, Nauka, 1976,
p. 231.
129. Long A. R., Balkan N., J. Non-Cryst. Solids, 35, 36, 415 (1980).
130. HUI R. M., fonsher A. K-, J. Non-Cryst. Solids, 32, 53 (1979).
131. Lakatos A. !.. Abkowitz M., Phys. Rev., B3, 1791 (1971).
132. Crevecoeur C., de Wit H. J., Solid State Commun., 9, 445 (1971).
133. Ivkin E. B., Kolomiets В. T., J. Non-Cryst. Solids, 3, 41 (1970).
134. Ovshinsky S. R., Fritzsche H., Metallurg. Transact., 2, 641 (1971) IEEE
Transact. Electron. Devices, 20, 91 (1973).
135. Rockstad H. K., Solid State Commun., 9, 2233 (1971).
136. Ellioit S. R., Phil. Mag., 36, 1291 (1977), 37, 553 (1978), 40, 507 (1978);
J. Non-Cryst. Solids, 35, 36, 855 (1980).
137. Spear IT. E., Le Comber P. G., Proc. 7th Int. Conf. Amorph. Liquid Semi-
cond., Edinburgh, 1977, University Edinburgh, p. 309.
138. Pfister G., Scher H., Proc. 7th Int. Conf. Amorph. Liquid Semicond., Edin-
burgh, 1977, University Edinburgh, p. 197.
139. Marshall J. M., Phil. Mag., 24, 959 (1977); Proc. 7th Int. Conf. Amorph.
Liquid Semicond., Edinburgh. 1977, University Edinburgh, p. 541.
140. Marshall J. M., Owen A. E.. Phil. Mag., 24, 1281 (1971).
141. Scher H., Montroll E. W., Phvs. Rev., В 12, 2455 (1975).
142. Pfitster G„ Scher H„ Phys. Rev., В 15. 2062 (1971).
143. Kim Gi IL, Shirafuji J., Jap. J. Appl. Phys., 17, 1789 (1978).
144. De Neufville J. P., 4th Semi-Annual Technical Report, DACH 15-7O-C-O187,
Troy, 1973.
145. Tohge N., Minami T., Tanaka M., J. Non-Cryst. Solids, 37, 23 (1980);
38, 39, 283 (1980).
146. Tohge N., Minami T., Yamamoto Y., Tanaka AL, J. Appl. Phys., 51, 1048
(1980).
147. Zvyagin I. P„ Phvs. Status Solidi (b), 83, 63 (1977); 85, 105, (1978);
97. 143 (1980).
148. Elliott S. R., Solid State Commun., 27, 749 (1978).
149. Ovshinsky S. R., Klose P. H., J. Non-Cryst. Solids, 8—10, 892 (1972).
150. Pfister G., Phil. Mag., 36, 1147 (1977); Proc. 7th Int. Conf. Amorph. Li-
quid Semicond.. Edinburgh, 1977, p. 765.
151. Toth L., Phvs. Statis Solidi (a), 54, К 159 (1979).
152. Hughes R. C., Phys. Rev., В 15, 2012 (1977).
153. Twaddel V. A., Lacourse W. C., Mackenzie J. D., J. Non-Crvst. Solids,
8—10, 831 (1972).
154. Liang K. S„ Bienenstock A., Bates C. W., Phys. Rev., BIO, 1528 (1974).
155. Данилов .4. В., Мюллер P. Л.— Ж. прикл. химии, 1962, г. 35, с. 2012.
156. Ovshinsky S. R., пат ГДР 85100 (1969).
157. Oda Т., Yamada Е., J. Phys. Soc. Jap., 46, 515 (1979).
Электрические и оптические свойства
525
158. Hunter S .Н., Bienenstock A., Hayes Т. Л4., Proc. 7th Int. Conf. Amorph.
Liquid Semicond., Edinburgh, 1977, University Edinburgh, p. 78.
159. Kastner M., Phil. Mag., В 37, 127 (1978).
160. Fritzsche H., Kastner M., Phil. Mag., В 37, 285 (1978).
161. Ovshinsky S. R., Proc. 7th Int. Conf. Amorph. Liquid Semicond., Edin-
burgh, 1977, University Edinburgh, p. 519.
162. Ovshinsky S. R., Adler D., Contemp. Phys., 19, 109 (1978).
163. Flasck R., Izu M., Sapru K-, Anderson T., Ovshinsky S. R., Fritzsche H.,
Proc. 7th Int. Conf. Amorph. Liquid Semicond., Edinburgh, 1977, Univer-
sity Edinburgh, p. 524.
164. Pfister G., Morgan M., Phil. Mag., 41, 209 (1980).
165. Madan A., Le Comber P. G., Soear W. E., J. Non-Cryst. Solids, 20, 239
(1976).
166. Spear W. E., Le Comber P. G., J. Non-Cryst. Solids, 8—10, 727 (1972);
Phil. Mag., 33, 935 (1976).
167. Jones D. L, Spear IT. £., Le Comber P. G., J. Non-Cryst. Solids, 20, 259
(1976).
168. Feinleib J., пат. ГДР 86863 (1971).
169. Schirrmeister F., Dissertation A., Friedrich-Schiller-Univ., Jena, 1979.
170. Reehal H. S., Thomas C. B„ Phil. Mag., В 39, 321 (1979).
171. Wallage A. M., Owen A. E., Robertson J. M., Phil. Mag. В 38, 57 (1978).
172. Walsh P. J., Vogel R., Evans E. J., Phys. Rev., 178, 1274 (1969); J. Non-
Cryst. Solids, 2, 107 (1970); Hall G. E., J. Non-Cryst. Solids, 2, 125
(1970).
173. Davis E. A., Shaw R. F., J. Non-Cryst. Solids, 2, 406 (1970).
174. de Wit H. J., Crevecoeur C., J. Non-Cryst. Solids, 8—10, 787 (1972);
Solid State Electronics, 15, 729 (1972).
175. Rose A., Phys. Rev., 97, 1538 (1955).
176. Feltz A., Scirrmeister F., Kahnt H., J. Non-Cryst. Solids, 35, 36, 865
(1980).
177. Schirrmeister F„ Feltz A., Phys. Status Solidi (a), 57, 591 (1980).
178. Frenkel J., Phys. Rev., 54, 647 (1938).
179. Poole H. H., Phil. Mag., 32, 112 (1916).
180. Connell G. A. N„ Camphausen D. L., Paul W., Phil. Mag., 26, 541 (1972);
J. Non-Cryst. Solids, 8—10, 223 (1972).
181. Pai D. M., J. Appl. Phys., 46, 5122 (1975); Phys. Rev., В 11, 5163 (1975).
182. Kahnt E., Schirrmeister F., Phys. Status Solidi (b), 115, 171 (1983).
183. Marshal! J. M., Miller G. R., Phil. Mag., 27, 1151 (1973).
184. Fagen E. A., Fritzshe H., J. Non-Cryst. Solids, 2, 170 (1970).
185. Poole H. H., Phil. Mag., 34, 195 (1917).
186. Kahnt H., Schirrmeister F., Feltz A., Phys. Status Solidi (a), 57, К 183
(1980).
187. Ovshinsky S. R., J. Non-Cryst. Solids, 2, 99 (1970); Mat. Res. Bull., 5,
681 (1970).
188. Ovshinsky S. R., пат. США 3 271 591.
189. Silptitz P., J. Signal AM. 2. 85 (1974).
190. Коломиец Б. T., Лебедев Э. A.— Радиотехника и электроника, 1963, т. 8,
с. 2097.
191. Stocker Н. J., J. Non-Cryst. Solids, 2, 371 (1970).
192. Sie С. H., J. Non-Cryst. Solids, 4, 548 (1970).
193. Llttecht R., Stevenson H., Sie С. H., Ragnavan K. S., J. Non-Cryst. Solids,
2, 358 (1970).
194. Bagley B. G., Bair H. E., Gent Lecture, 1968.
195. Collins F. AL, J. Non-Cryst. Solids, 2, 496 (1970).
196. Hayes T. M., Thornburg D. D., J. Phys. C: Solid State Phys., 6, 450
(1973).
197. Thornburg D. D., J. Electronic Mat., 2, 3 (1973); J. Non-Cryst. Solids, 11,
113 (1972).
526 Глава 4
198. Evans Е. J., Helbers J. H., Ovshinsky S. R., J. Non-Cryst. Solids, 2,
334 (1970).
199. Thornburg D. D., Johnson R. I., Kloffenstein T. J., J. Non-Cryst. Solids,
20, 225 (1976).
200. Drake C. F., Scanlan I. F., Engel A., Phys. Status Solidi, 32, 193 (1969).
201. Holzinger R., Holzinger 0., Z. angew. Phys., 28, 196 (1970).
202. Ovshinsky S. R., Phys. Rev. Lett., 21, 1450 (1968).
203. Adler D., Henish H. K., Mott N. F., Rev. Modern Phys., 50, 209 (1978).
204. Ovshinsky S. R., Fritzsche H., IEEE Transactions Electron Devices, 91
(1973).
205. Fritzsche IL, Ovshinsky S. R., J. Non-Cryst. Solids, 2, 393 (1970).
206. Walsh P. J., Vezzoli G. C., Appl. Phys. Lett., 25, 28 (1974); Proc. 5th Int.
Conf. Amorph. Liquid Sernicond., Garmisch-Partenkirchen, 1973, Taylor-
Francis, London, 1974, p. 1391.
207. Коломиец Б. T., Лебедев Э. А., Таксами И. А.—Физ. и техн, полупровод-
ников, 1969, т. 3, с. 267.
208. Shanks R. S., J. Non-Cryst. Solids, 2, 504 (1970).
209. Haberland D. R., Solid State Electronics, 13, 207 (1970); Frequenz, 24,
185 (1970).
210. Lee S. H., Henish H. K., Burgess W. D., J. Non-Cryst. Solids, 8—10, 422
(1972).
211. Robertson J. M., Owen A. E., J. Non-Cryst. Solids, 8—10, 439 (1972).
212. Thomas D. I., Male J. C., J. Non-Cryst. Solids, 8—10, 522 (1972).
213. Henish H. K., Smith W. R., Wihl IF., Proc. 5th Int. Conf. Amorph. Liquid
Sernicond., Garmisch-Partenkirchen 1973, Taylor-Francis, London, 1974, p. 567.
214. Pryor R. W., Henish II. K., Appl. Phys. Lett., 18, 324 (1971).
215. Vendura G. J., Henish H. K., J. Non-Cryst. Solids, 11, 105 (1972).
216. Stiegler IL, Haberland D. R., J. Non-Cryst. Solids, 11, 147 (1972).
217. Petersen К. E., Adler D., J. Appl. Phys., 47, 256 (1976).
218. Pryor R. W., Henish IL K., J. Non-Cryst. Solids, 7, 181 (1972); 8—10,
415 (1972); Нотта К- Henish К., Ovshinsky S. R., 35, 36, 1105 (1980).
219. Vezzoli G. C., Walsh P. J., Doremus L. IF., J. Non-Cryst. Solids, 18, 333
(1975).
220. Henish H. K., Sci. Am., 221, 30 (1969).
221. Henish H. K. Fagen E. A., Ovshinsky S. R., J. Non-Cryst. Solids, 4, 538
(1970).
222. van Roosenbroeck W.. J. Non-Cryst. Solids, 12, 232 (1973).
223. Arnoldussen T. C., Bube R. H., Fagen E. A., Holmberg S., J. Appl. Phys.,
43, 1798 (1972).
224. Vautier C., Viger C., J. Non-Cryst. Solids, 23, 287 (1977).
225. Kitao M„ Ikeda H., Hasegawa H., Yamada S., Phys. Status Solidi (a),
45, 107 (1978).
226. Weiser K., Fischer R., Brodsky M. IL, Proc. Xth Int. Conf. Phys. Semi-
cond., Cambridge/Ma. (1970), S. P. Keller, J. C. Hensel, F. Stern (eds.)
(US Atomic Energy Commission), p. 667.
227. Коломиец Б. T., Любин В. М,— ДАН СССР, 1959, т. 129, с. 789; Физ.
твердого тела, 1959, т. 1, с. йнн.
228. Kolomiets В. Т„ Lyubin V. М., Phys. Status Solidi (а), 17, 11 (1973).
229. Fagen Е. A., Fritzsche Н., J. Non-Cryst. Solids, 4, 480 (1970).
230. Main C., Owen A. E., Photoconductivity and Noise in Chalcogenide Glas-
ses, in: Electronic and Structural Properties of Amorphous Semiconductors,
P. G. LeCommer, J. Mort (eds.), Academic Press, London, 1973, p. 527;
Proc. 5th Int. Conf. Amorph. Liquid Sernicond., Garmisch-Partenkirchen
1973, Taylor-Francis, London, 1974, p. 783.
231. McMillan P. W., Shutov S. D., J. Non-Cryst. Solids, 24, 307 (1977).
232. Kolomiets В. T., Bukhliadev J. V., Shilo V. P., J. Non-Crvst. Solids, 5,
402 (1971).
233. Vasko A., Infra-red Radiation, ILIFFE Books Ltd., London, SNTL-Publi-
shcrs of Technical literature, Prague, 1968.
Электрические и оптические свойства 527
234. Treacy D., Taylor Р. С., Optical Properties of Highly Transparent Solids,
S. S. Mitra, B. Bendow (eds.), Plenum Publ. Corp., 1975, p. 261.
235. Bergmann L., Schaffer CL, Lehrbuch der Experimentalphysik, Bd. Ill,
Optik, Atomphysik, 1. Teil, Wellenoptik, W. De Gruyter (ed.), Berlin, 1962.
236. Heitmann W., Appl. Optics, 15, 256 (1976); 14, 3047 (1975).
237. Pinnow D. A.. Rich T. C., Appl. Optics, 12, 984 (1973).
238. Rosencwaig A., Opt. Comm., 7, 305 (1973); Phys. Today, 28, 23 (1975).
239. Skolnik L., Hordvik A., Kahan A., Appl. Phys. Lett., 23, 477 (1973).
240. Hordvik A., Appl. Optics, 16, 2827 (1977).
241. Neuroth N., Silikat Journal (1978), Heft 6, Schott-Sonderdruck.
242. Conradi J., Kapron F. P., Dyment J. C., IEEE Trans. Electron Devices,
25, 180 (1978).
243. Zembutsu S., Fukunishi S., Appl. Optics, 17, 1953 (1978); 18, 393 (1979).
244. Moss T. S., Optical Properties of Semiconductors, Butterworth Publ. Ltd.,
1959.
245. Kortum G., Reflexionsspectroskopie, Grundlagen, Methoden, Anwendungen,
Springer-Verlag, Berlin, 1969.
246. Hass M., Bendow B., Appl. Optics, 16, 2882 (1977).
247. Makland M. S, Mohr R. K-, Howard R. E., Macedo P. B., Moynihan С. T.,
Solid State Commun., 15, 855 (1977).
248. Strom U., Taylor P. C., Phys. Rev., В 16, 5512 (1977).
249. Moynihan С. T., Macedo P. B., Maklad M. S., Mohr R. K-, Howard R. E.,
J. Non-Cryst. Solids, 17, 369 (1975).
250. Ma D. S., Danielson P. S., Moynihan С. T., J. Non-Cryst. Solids, 37, 181
(1980).
251. Savage J. A., Webber P. J., Pitt A. N., Appl. Optics, 16, 2938 (1977).
252. Savage J. A., Nielsen S., Phys. Chem. Glass, 6, 90 (1965).
253. Savage J. A., Nielsen S., Infrared Phys., 5, 195 (1965).
254. Kettlewell B. R., Kinsman В. E., Wilson A. R., Pitt A. M., Webber P. J.,
J. Mater. Sci., 12, 451 (1977).
255. Jones С. E„ Hafner H. C., TI-Report AFAL-TR-68-368 (1969).
256. Minami T., Ben T., Zairyo Kagaki, 20, 1070 (1971).
257. Hilton A. R., J. Non-Cryst. Solids, 2, 28 (1970).
258. Voigt B., Dresler G., Analyt. Chiin., Acta, 127, 87 (1981).
259. Srb I., Vasko A., Czech. J. Phys., В 11, 664 (1961); В 13, 827 (1963).
260. Swincle D. L., Schweikert E. A., Taianta, 22, 84 (1975).
261. Hilton A. R., Hayes D. J., Rechtin M. D., J. Non-Crvst. Solids, 17, 319,
339 (1975).
262. Stuke J., Proc. 8th Int. Conf. Phys. Semicond., Cambridge/Ma. (1970),
S. P. Keller, J. C. Hensel, Stern F. (eds.) (U. S. Atomic Energy Com-
mission), p. 14.
263. Connell G. A. N., Temkin R. J., Paul IF., Adv. Phys., 22, 643 (1973).
264. Donovan T. M., Spicer W. E., Bennett J. M., Ashley E. J., Phys. Rev.,
В 2, 397 (1970).
265. GiceiiWuy D. L„ ilurueke G., Optical Properties and Band Structure of
Semiconductors, Pergamon Press, Oxford, 1968.
266. Mohler E., Stuke J., Zimmerer G., Phys. Status Solidi, 22, К 49 (1967).
267. Tutihasi S., Chen I., Phys. Rev., 158, 623 (1967).
268. Leiga A. G., J. Appl. Phys., 39, 2149 (1968); J. Opt. Soc., Am., 58, 880,
1441 (1968).
269. Shevchik N. J., Cardona M., Tegeda J., Phys. Rev., В 8, 2833 (1973).
270. Treusch J., Sandrock R., Phys. Status Solidi, 16, 487 (1966).
271. Tutihasi S., Roberts G. G., Keezer R. C., Drews R. E., Phvs. Rev., 177,
1043 (1969).
272. Stuke J., J. Non-Crvst. Solids, 4, 1 (1917).
273. Drews R. E., Emerald R. L., Slade M. L„ Zallen R., Solid State Commun.,
10, 293 (1972).
528 Глава 4
274. Connell G. A. JV., in: Amorphous Semiconductors, M. H. Brodsky (ed.),
Springer-Verlag, Berlin, Heidelberg, New York, 1979, p. 73.
275. De Vore 11. B., Phys. Rev., 102, 86 (1956).
276. Philipp H. R., Solid State Commun., 4, 73 (1966).
277. Tauc J., Optical Properties of Solids, F. Abelcs (cd.), North-Holland, Am-
sterdam, 1970.
278. Clement R., Carballes J. C„ de Cremoux B., J. Non-Cryst. Solids, 15, 505
(1974).
279. Urbach F., Phys. Rev., 92, 1324 (1953).
280. Kosek F„ Tauc J., Czech. J. Phys., В 20, 94 (1970).
281. Tauc J., Menth A., J. Non-Cryst. Solids, 8—10, 569 (1972).
282. Kammow I. P., Bagley B. G., Olson C. G., Appl. Phys. Lett., 32, 98
(1978).
283. Edmond J. T., Brit. J. Appl. Phys., 17, 979 (1966).
284. Aral T., Komiya S., Kudo K., J. Non-Cryst. Solids, 18, 295 (1975).
285. Tauc J., Mat. Res. Bull., 5, 721 (1970).
286. Kurik M. V., Phys. Status Solidi (a), 8, 9 (1971).
287. Dow J. D., Redfield D., Phys. Rev., В 5, 594 (1972).
288. Sumi H., Toyozawa K., J. Phys. Soc. Jap., 31, 342 (1971).
289. Skettrup T., Phys. Rev., В 18, 2622 (1978).
290. Vasko A., Lezal D., Srb I., J. Non-Cryst. Solids, 4, 311 (1970).
291. Vancu A., Sladaru St., Grigorovici R., Proc. 5th Int. Conf. Amorph. Liquid
Semicond., Garmisch-Partenkirchen, 1973, Taylor-Francis, London, 1974,
p. 631.
292. Zavetova M., Henrion IF., Phys. Status Solidi (a), 24, К 27, (1974).
293. Street R. A., Phil. Mag., В 37, 35 (1978).
294. Street R. A., Searle T. M., Austin 1. G., Phil. Mag., 30, 1181 (1974).
295. Street R. A., Austin 1. G., Searle R. M., Smith B. A., J. Phys., C 7, 4185
(1974).
296. Bishop S. G., Strom U., Friebele E. J., Taylor P. C., J. Non-Cryst. Solids,
32, 359 (1979).
297. Aral K-, Itoh U., Romine H., Namikawa 11., Proc. Vlth Conf. Amorph.
Liquid Semicond., Structure and Properties of Non-Cryst. Semicond.
В. T. Kolomiets (ed.), Leningrad, Nauka, 1976, p. 322.
298. Street R. A., Biegelsen D. K-, J. Non-Cryst. Solids, 22, 339 (1979).
299. Gee С. M„ Kastner M., J. Non-Cryst. Solids, 35, 36, 927 (1980).
300. Kolomiets В. T., Mamontova T. N.„ Negreskul V. V., Phys. Status Solidi,
27, К 15 (1968); Kolomiets В. T., Mamontova T. N„ Babaev A. A., J.
Non-Cryst. Solids, 4, 289 (1970); 8—10, 1004 (1972).
301. Street R. A., Advanc. Phys., 25, 397 (1976).
302. Mott N. F., Phil. Mag., 36, 413, 979 (1977).
303. Stoneham A. M., Phil. Mag., 36, 383 (1977).
304. Street R. A., Phys. Rev., В 17, 3984 (1978); Solid State Commun., 24, 363
(1977).
305. Fischer R., Festkbrperprobleme XVII (1977).
306. Fritzsche 11., Gaczi P. 1., Kastner M., Phil. Mag., В 37, 593 (1978).
307. Vassiljev V. A., Koos M., Somogyi I. K-, Phil. Mag., В 29, 333 (1979).
308. Gross G., Stephens R. B., Turnbull D., J. Appl. Phys., 48, 1139 (1977).
309. Bishop S. G., Strom U., Optical Properties of Highly Transparent Solids,
B. Bendow, S. S. Mitra (eds.), Plenum, N. Y„ 1975, p. 317.
310. Bishop S. G., Strom U., Taylor P. C., Phys. Rev., В 15, 2278 (1977).
311. Taylor P. C., Strom U., Bishop S. G., Phil. Mag., В 37, 241 (1978).
312. Kocka J., Elliott S. R., Davis E. A., J. Phys. C, Solid State Phys., 12,
2589 (1979).
313. Ajo J,. G., Efimov A. M., Kokorina V. F., J. Non-Cryst. Solids, 27, 299
(1978).
314. Savage J. A., Webber P. J., Pitt A. Appl. Optics, 16, 2938 (1977).
315. Hafner H. C., National Technical Information Service U. S. Department
of Commerce AD-773 174.
Электрические и оптические свойства
529
316. Burckhardt IT., J. Non-Cryst. Solids, 50, 173 (1982).
317. Kas H., Glastechn. Ber., 45, 1 (1972).
318. Brewster G. F., Heusler J. R., Road J. L„ Weidel R. A., Appl. Optics, 5,
1891 (1966).
319. Izumitani T., Nakagawa K-, Mitt, der Hoya Glass Works, Tokyo, Japan.
320. Pockets F., Ann. Phys., 7 ,745 (1902).
321. Weyl W. A., Glastechn. Ber., 30, 269 (1957).
322. Mueller H., Physics 6, 179 (1935); Phys. Rev., 47, 947 (1935); J. Amer.
Ceram. Soc., 21, 27 (1938).
323. Левенберг В. А., Лунтер С. Г.— Физ. хим. стекла, 1976, т. 2, с. 63, 557.
324. Eberhardt G., Dissertation A., Friedrich-Schiller-Univ., Jena, 1979.
325. Smith G. P., Glass Technol., 20, 149 (1979).
326. Berkes J. S., Short J. M., Johnson K. J., Proc. 5th Int. Conf. Amorph.
Liquid Sernicond., Garmisch-Partenkirchen 1973, Taylor-Francis, London,
1974, p. 117.
327. Berkes J. S., Ing S. W., Hillegas W. J., J. Appl. Phys., 42, 4908 (1971).
328. Tanaka K-, Appl. Phys. Lett., 26, 243 (1975).
329. Tanaka K-, AIP Conf. Proc., № 31, 148 (1976).
330. Keneman S. A., Appl. Phys. Lett., 19, 207 (1971).
331. Treacy D. J., Taylor P. C., Klein P. B., Solid State Commun., 32, 423
(1979).
332. Nang T. T., Okuda M., Matsushita T„ J. Non-Cryst. Solids, 33, 311 (1979).
333. Maruno S., Ban S., Asahi Garasu Kogyo Gijutsu Shoreikai Kenkyu Ho-
koko, 31, 71 (1977); Jap. J. Appl. Phys., 19, 97 (1980).
334. Nang T. T., Okuda M., Mastushita T., Phys. Rev., В 19, 947 (1979).
335. Igo T., Toyoshima У., J. Non-Cryst. Solids, 11, 304 (1973); SuppL J. Jap.
Soc. Appl. Phys., 43, 106 (1974), 41, 61 (1971).
336. Ohmachi Y., Igo T., Appl. Phys. Lett., 20, 506 (1972).
337. Tanaka K., Hamanaka H., lizima S., Proc. 7th Int. Conf. Amorph. Liquid
Sernicond., Edinburgh 1977, University, Edinburgh, p. 787.
338. De Neufville J. P., Seguin R., Moss S. C., Ovshinsky S. R., Proc. 5th Conf.
Amorph. Liquid Sernicond., Garmisch-Parkenkirchen 1973, Taylor-Francis,
London, 1974, p. 737.
339. Suzuki A., Matsushita T., Okuda M., Nang T. T., Jap. J. Appl. Phys., 18,
47 (1979).
340. Feltz A., Kahnt H„ Sghirrmeister F., 42, 935 (1981).
341. Scher H., Proc. IX Int. Conf. Amorph. Liquid Cemicond., Grenoble, 1981,
J. de Phisique Collique C4, Suppl, n° 10, 42, 935 (1981).
342. Perrin J., Solomon I., J. Non-Cryst. Solids, 37, 407 (1980).
343. Maurer R. D., J. Non-Cryst. Solids, 42, 197 (1980).
344. Mollot F., Cernogora J., Benoit a la Guillaume C., Phil. Mag., В 42, 643
(1980).
345. Tanaka K„ in: Fundamental Physics of Amorphous Semiconductors,
F. Yonezawa (ed.), Springer-Verlag, 1981.
346. Tanaka K., Solid State Commun., 34, 201 (1980).
347. Higashi G. S., Kastner M., Phys. Rev. Lett., 47, 124 (1981).
348. Pohl K- D., Bruckner R., Phys. Chem. Glass., 23, 23 (1982).
Дополнительная литература*
Бонч-Бруевич В. Л.; Звягин И. П., Кайпер Р. и др. Электронная теория не-
упорядоченных полупроводников.— М.: Наука, 1981, 383 с.
Бонч-Бруевич В. Л. Вопросы электронной теории неупорядоченных полупро-
водников.— Усп. физ. наук, 1983, т. 140, № 4, с. 583—637.
Губанов А. М. Квантово-электронная теория аморфных проводников,— М.-Л.:
Йзд-во АН СССР, 1963, 250 е.
Grinfelds A. U., Silin A. R., Skuja L. N., Plekhanov V. G. Low temperature
* Представлена редактором С. А. Дембовским.
34 Заказ № 413
530 Глава 4
recombination luminescence in amorphous and crystalline SiO2.— Ph. St. Sol.
(a) 1984, 81, К 23.
Dembovsky S. A., Chechetkina E. A. Model photostructural changes in chal-
cogenide glassy semiconductors. Mat. Res. Bull., v. 20, p. 321—328, 1985.
Физико-химические основы производства оптического стекла./Под ред.
Л. И. Демкиной.— Л.: Химия, 1976, 456 с.
Жданов В. Г., Малиновский В. К- Фотоиндуцированное двулучепреломление
и дихроизм в пленках AS2S3 — Письма ЖТФ, 1977, т. 3, № 18, с. 943—946.
Жданов В. Г., Малиновский В. К, Соколов А. П. Фотоиндуцированные из-
менения структуры пленок халькогенидных стеклообразных полупроводни-
ков.— Автометрия, 1981, № 5, с. 3—13.
Займан Дж. Модели беспорядка.— Теоретическая физика однородно неупо-
рядоченных систем.— М.: Мир, 1982, с. 591.
Клингер М. И., Карпов В. Т. Автолокализация электронных пар в неупоря-
доченных системах.— ЖЭТФ, 1982, т. 82, № 5, с. 1687—1703.
Клингер М. И. Автолокализированные состояния электронов и дырок.— Усп.
физ. наук. 1985, т. 146, № 1, с. 105—142.
Лифшиц И. М., Гредескул А. С., Пастур Л. А. Введение в теорию неупоря-
доченных систем.— М.: Наука, 1982, 358 с.
Мюллер Р. Л. Электропроводность стеклообразных веществ.— Л.: Изд-во
ЛГУ, 1968, 251 с.
Мазурин О. В. Электрические свойства стекла (Область слабых полей).— Л.:
Госкомиздат. 1962, 162 с.
Регель А. Р„ Глазов В. М. Физические свойства электронных расплавов,— М.:
Наука, 1980, 268 с.
Шкловский Б. И., Эфрос А. Л. Электронные свойства легированных полу-
проводников.— М:. Наука, 1979, 416 с.
Закис Ю. Р. Дефекты в стеклообразном состоянии вещества.— Рига: Зинантне,
1984, 203 с.
Elliott S. Р. Physics of Amorphous Materials. London—New York: Longman,
1983, 374 p.
Zallen R. The Physics of Amorphous Solids. New York, Chichester, Brisbane:
J. Wiley and Sons, 1983, 297 p.
Гуревич Ю. Я. Особенности термодинамики суперионных проводников.— Усп.
физ. наук, 1982, т. 136, № 4, с. 693—728.
Практическое использование научных результатов —
непреходящий родник, питающий научно-техниче-
ский прогресс и вместе с тем гарантия их досто-
верности.
Ж. Бернал
5. О некоторых применениях
Многочисленные применения стеклообразных материалов
связаны, как известно, с особенностями их свойств. Наряду
с оптической прозрачностью, дисперсией показателя прелом-
ления, химической 'стойкостью и механической (вакуумной)
прочностью таких материалов следует также назвать техноло-
гичность их обработки и возможность получения материалов
с заданными свойствами. Детальное изложение многочисленных
практических аспектов выходит за рамки этой главы.
На основе изложенного можно в общих чертах определить
направления, по которым проводились исследования с целью
практического использования стеклообразных или аморфных
материалов. Прежде всего выделяются исследования аморфного
кремния, а также халькогенидных стекол в связи с попытками
технической реализации преобразователей солнечной энергии.
Широкий поиск в этом направлении сильно стимулировался
интересом к средам, прозрачным в ИК-области спектра, средам
для записи информации, а также их отличительными свойст-
вами, например существованием двух состояний проводимости.
Следует отметить, что представитель этой группы материалов
селен является важнейшим материалом для электрофотогра-
фии. Оксидные стекла с электронной проводимостью также на-
ходят ряд применений в электронике.
Важное практическое значение имеет установление корреля-
ций типа «структура—свойства».
Применение различных методов исследования привело в по-
следние годы к значительным успехам, например в изучении
кварцевого стекла, причем эти исследования стимулируются
также практическими потребностями. Требования микроэлек-
троники, предъявляемые к аморфным слоям SiOa, касаются
не только воспроизводимости свойств; необходимо знать связи
между структурой, морфологией и свойствами некристалличе-
ского оксида кремния. Еще одна важная область практического
34*
532 Глава 5
применения — волноводы с малыми потерями. Некоторые
аспекты практического применения рассмотрены в разд. 2.1.1.3,
3.1.1 и З.2.2.1.
5.1. Использование некоторых стекол и аморфных
слоев с полупроводниковыми свойствами
Стекла с ионной проводимостью первоначально использо-
вались в передающих телевизионных трубках суперортиконного
типа в качестве накопительных пленок. Вследствие своего ко-
роткого срока службы они вскоре были заменены стеклами
с электронной проводимостью [1].
Исследование оптимальных ионных проводников на основе
стекол, рассмотренных в разд. 4.1.1.1, связано с повышенным
интересом к электрохимическим батареям повышенной надеж-
ности. В какой степени стекла окажутся приемлемыми для этой
цели, пока неизвестно.
Халькогенидные стекла нашли применение как резисторы
в некоторых специальных случаях, например в качестве тер-
мисторов в управляющих схемах ядерных реакторов. Сооб-
щается [2], что дозы облучения 1020 нейтронов/см2 не приводят
к заметным изменениям проводимости. Повышенная стабиль-
ность стекол к облучению связывается с наличием в них высо-
кой концентрации дефектов и вакансий.
5.1.1. Применение стекол с ионной проводимостью
В последние годы интенсивно исследуются стекла с ионной
проводимостью с целью применения в качестве твердых элек-
тролитов. Стеклянные электроды с ионной чувствительностью
широко применяются в устройствах для измерения pH. В этой
связи для селективного определения различных катионов
и анионов в различных твердых телах с ионной проводимостью
наряду с другими материалами предложены и стекла.
5.1.1.1. Твердые стеклообразные электролиты
Кривые проводимости на постоянном токе для стекол уже
рассматривались в разд. 4.1.1.1. По возможности высокие зна-
чения удельной проводимости представляют интерес при кон-
струировании электрохимических ячеек (первичные цепи) и ба-
тарей высокой надежности. Кроме того, большое значение при-
дается высоким выходным напряжениям, т. е. свободная энталь-
пия реакции должна иметь по возможности большую отрица-
тельную величину. При этом делается ориентация на щелочные
элементы, такие, как литий и натрий, так как их катионы от-
личаются относительно высокой подвижностью в структурном
О некоторых применениях
533
остове стекла (табл. 4.1 и 4.2). Достаточная стабильность во
времени стекол при непосредственном контакте с этими ще-
лочными металлами вплоть до температур в несколько сотен
градусов отвечает еще одному требованию, которому должны
удовлетворять твердые тела с ионной проводимостью для их
использования в качестве твердых электролитов.
Описано [33] устройство, включающее несколько тысяч
стеклянных капилляров из натрийсиликатного стекла, напол-
ненных натрием. Другие примеры подобных устройств приве-
дены в работе [34].
5.1.1.2. Ионоселективные электроды
Наиболее известны и уже в течение долгого времени при-
меняются стеклянные электроды для измерений pH. Нижняя
часть такого электрода в форме шарика изготовлена из тонкой
пленки кальцийсиликатного стекла, обогащенного Na или Li
(например, 20 % Na2O, 8—10 % СаО, 70 % SiO2); к шарику
припаяна стеклянная трубка. Внутри этой конструкции нахо-
дится раствор с определенной активностью водородных ионов,
в котором помещен электрод сравнения (например, электрод
второго рода — хлорсерсбряный или каломельный — в буфер-
ном растворе). Стеклянный электрод погружают в раствор, pH
которого необходимо измерить. В результате частичного заме-
щения ионов Na+ ионами Н+ образуется стабильный гелевый
слой, потенциал которого в области pH 1—8 количественно
зависит от активности ионов водорода в растворе [35—37].
Халькогенидные стекла на основе As2Sa и As—Se или
As—Se—Те, содержащие до 25 мол. % подходящих металлов,
например Си или РЬ, оказались пригодными для использования
в качестве ионоселективных электродов для определения ионов
Си2+ или РЬ2+.
5.1.2. Применение оксидных стекол с электронной
проводимостью
В настоящее время достигли стадии технической завершен-
ности телевизионные передающие трубки типа суперортикон
с накопительной пленкой из стекла с электронной проводимо-
стью и канальный умножитель вторичных электронов на основе
стекол с поверхностной проводимостью.
5.1.2.1. Накопительные электроды из стекла
в суперортиконах
На внутренней стороне входного окна суперортикона нахо-
дится фотокатод. При проектировании изображения возбу-
жденные электроны ускоряются и попадают на стеклянную
534 Глава 5
пластинку толщиной несколько микрометров (рис. 5.1). На пла-
стине возникает положительное электростатическое изображе-
ние, а находящийся перед ней сеточный электрод, на который
подают небольшое напряжение противоположного знака, за-
хватывает вторичные электроны. Изображение, записанное
в виде рельефа потенциала, проходит сквозь пластину и оказы-
Рис. 5.1. Схема суперортикона с накопительным электродом (2) из стекла
с электронной проводимостью, находящимся перед ним сеточным электродом
(3) и фотокатодом (/).
вает модулирующее действие на считывающий электронный луч
с обратной стороны пластины.
Нетрудно понять, что стекла с ионной проводимостью с од^
ненаправленным перемещением катионов для этой цели исполь-
зоваться не могут. Стекла с электронной проводимостью сво-
бодны от эффекта «старения».
Толщину стеклянной пластинки следует подбирать таким
образом, чтобы ограничить поперечное растекание положитель-
ного заряда за время прохождения его к обратной стороне пла-
стинки. При проводимости 1О-10 См/см оптимальные толщины
составляют ~3 мкм. Для изготовления накопительных элек-
тродов пригодны многие стекла, включенные в табл. 4.3.
5.1.2.2. Канальный умножитель вторичных электронов
Тонкие полые стеклянные волокна толщиной 40—200 мкм
формируют параллельные связки, которые соединяются с пе-
редней и задней сторон (рис. 5.2) с тем, чтобы можно было
подводить напряжение 2,5—3 кВ [3]. При воздействии первич-
ного излучения, например электронного, ионного, УФ- или
рентгеновского излучения на внутренних стенках стеклянных
волокон возбуждаются электроны, которые линейно умно-
жаются в канале с коэффициентом 108—109. Используются, на-
пример, ванадийфосфатные стекла с сопротивлением 107—
10° Ом-см. Возможно также использование стекол, содержа-
О некоторых применениях
535
щих свинец или висмут, на поверхности которых в результате
восстановления устанавливается определенная проводимость.
Стекла, содержащие марганец, имеют повышенную поверхно-
Рнс. 5.2. Схематическое устрой-
ство канального умножителя
вторичных электронов.
стную проводимость после нагревания на воздухе [4]. Такие
канальные пластины нашли применение в преобразователях
рентгеновского изображения, а также в космической технике.
5.1.3. Переключатели типа Ovonic
О переключателях на стеклообразных полупроводниках уже
говорилось в разд. 4.1.3.7. Подробное рассмотрение различных
стеклообразных соединений, в которых возможны высокоомное
и низкоомное состояния, проведено в работе [5]. Центральное
место занимает патент Овшинского (см. литературу к гл. 1
[32]). Типичные конструкции, предложенные Овшинским и др.,
приведены на рис. 5.3.
На рис. 5.3, а изображено устройство, включающее два
электрода, выполненных из графита (стеклографита) в виде
полусфер, покрытых стеклянной пленкой; электроды слегка при-
жаты друг к другу [6]. На рис. 5.3, б показана интегральная
сандвич-структура. Слои наносятся друг на друга катодным
распылением либо фотолитографическим способом с примене-
нием соответствующих масок. На рис. 5.3, в изображен пере-
ключатель на кремниевой шайбе. Часто в электронных пере-
ключателях используются диоды в непосредственном контакте
с подложкой [11].
В работе [6] сообщается, что на площади 6,45 см2 удалось
расположить 2500 переключателей. Переключатели с матрич-
ным х, «/-соединением легко управляются при последовательном
включении диода. Такие элементы RMM-256 (Read Mostly Me-
mory, 16X16 элементов) на основе переключателей с памятью
536 Глава 5
Рис. 5.3. Переключатель типа Ovonic в дискретном (а) и тонкопленочном
интегральном (б и в) исполнении.
в качестве накопителей постоянных производятся фирмой
Energy Conversion Devices. О их использовании в переключа-
тельных цепях сообщалось во многих работах [6—11].
5.1.4. Солнечные элементы на основе слоев
аморфного кремния
Преобразование солнечной энергии с использованием мо-
нокристалла кремния ( р,п-переход) известно с 1954 г. [12]; при
этом получен к. п. д. ~6 %. Впоследствии к. п. д. был повышен
до 19 % [13], а при использовании элементов на основе GaAs —
до 23 % [14].
В слоях гидрогенизированного аморфного кремния (поли-
циклосилановые пленки), полученных разложением силанов
в тлеющем разряде (разд. 2.1.3.3, 3.1.2.3 и 4.1.3.5) путем после-
довательных добавок подходящих газов, таких, как РН3 и В2Нв
(рис. 4.23), также можно получить р,п-переход (см. литературу
к гл. 2 [53] и гл. 3 [104]); причем до сих пор достигнут к. п. д.
~6%. Преимущество слоев на основе аморфного кремния со-
стоит в возможности осуществления непрерывного технологи-
ческого производства элементов большой площади, существенно
более дешевых, чем монокристаллические преобразователи.
К. п. д. определяется соотношением
i\s — jsVs/Is (5.1)
где Vs—фото-э. д. с., js — плотность тока, a Is — интенсивность
солнечного света при нормальном падении (в ясный день со-
ставляет ~100 мВт/см2). Для применения полупроводниковых
элементов в качестве преобразователей солнечной энергии они
должны удовлетворять определенным условиям (см. литера-
туру к гл. 3 [ПО]).
О некоторых применениях
537
1. Коэффициент поглощения в широкой области спектра,
особенно в области максимума интенсивности, должен быть по
возможности высок. В видимой области при толщине пленок
1 мкм а>103 4 см-1.
2. Возбужденные вследствие поглощения электроны
и дырки должны по возможности полностью собираться на
электродах, т. е. диффузионная длина неосновных носителей
должна быть сравнима с толщиной образца.
нелегированный нелегированный
Рнс. 5.4. Несколько вариантов солнечных элементов на основе гидрогенизиро-
ванного аморфного кремния (см. литературу к гл. 3 [НО]).
3. Последнее условие имеет определяющее значение для
достигаемого к. п. д. Возникающая фото-э.д. с. определяет ве-
личину отводимой электрической энергии.
4. Наконец, следует обращать внимание на то, чтобы по-
следовательное сопротивление солнечного элемента (кроме со-
противления нагрузки) было мало, чтобы прибор потреблял
лишь малую долю преобразованной энергии.
Несколько вариантов солнечных элементов на основе аморф-
ного кремния приведено на рис. 5.4. В работе [15] подробно
рассмотрены физические свойства гидрогенизированных слоев
аморфного кремния и некоторые аспекты его использования
для преобразования солнечной энергии. Общие вопросы, касаю-
щиеся материалов, используемых для преобразования солнеч-
ной энергии, рассмотрены в работе [16].
До сих пор не получила объяснения потеря стабильно-
сти системы связей Si—Н при действии достаточно интенсив-
ного электромагнитного излучения. При разработке солнечных
элементов много внимания уделяется исключению попадания
воздуха и их герметизации. Сообщается [17, 19] (см. также ли-
тературу к гл. 3 [113, 114]) о попытках частичной замены свя-
зей Si—Н связями Si—F и Si—О—Si с целью сохранения в те-
чение длительного времени требуемых свойств гидрогенизиро-
ванного кремния, а также целенаправленного изменения
538 Глава 5
свойств. Технологические достижения в производстве солнечных
элементов на основе аморфного кремния обсуждаются в ра-
боте [40].
5.2. Халькогенидные стекла как среды,
прозрачные в ИК-области спектра
Требования, которым должны отвечать стекла с высокой
оптической прозрачностью, рассматривались в разд. 4.2.1.2.
В качестве материалов, прозрачных в ИК-области (8—
12 мкм), могут быть использованы селенидные и теллуридные
стекла. Сульфидные стекла, например AS2S3, а также халько-
генидные стекла, содержащие Si, прозрачны только до
~ 10 мкм. Поскольку расплавы теллуридов, не содержащие се-
лена, могут давать стекла лишь при высоких скоростях охла-
ждения (исключение составляют системы Si—As—Те и
As—Ge—Si—Те), селенидные стекла более предпочтительны.
Промышленное производство халькогенидных стекол для
ИК-оптики существует более 10 лет в СССР, США (Texas
Instruments), а также осуществляется фирмами Servo-Corpo-
ration и British Drug House. Сообщалось [21] об изготовлении
в СССР стекол Ge—As—Se, а также стекол ИКС-29 в виде
монолитов массой 5—13 кг. Фирмой Texas Instruments нала-
жено техническое производство стекол TI20 GeaaAsisSess
и Т11173 Gc2sSbi2Se6o (см. литературу к гл. 4 [261]). Имеются
данные о получении образцов в виде дисков толщиной 10 мм
и диаметром 70 мм, а также в виде пластинок 30x60 см2.
В качестве сред, прозрачных в ИК-области спектра,
халькогенидные стекла используются в термографии, лазерной
технике, инфракрасной фурье-спектроскопии, а также для изго-
товления оптических элементов высокого разрешения для при-
боров ночного видения. В отличие от традиционно используе-
мых щелочногалогенидных и щелочноземельнофторидных сте-
кол, халькогенидные стекла более устойчивы к химическому
воздействию атмосферы. Из халькогенидных стекол можно, на-
пример, изготавливать кюветы для работы с водными раство-
рами (от нейтральных до сильнокислых). В щелочных средах,
однако, стойкость халькогенидных стекол на основе мышьяка
и германия оказывается недостаточной.
Из-за относительно высоких значений показателя прелом-
ления для повышения пропускания необходимо принимать
меры, обеспечивающие уменьшение отражения. Механическая
прочность и термическая стабильность халькогенидных стекол
невелика, а коэффициент термического расширения, темпера-
турный коэффициент показателя преломления и упругооптиче-
ская постоянная их больше, чем у оксидных стекол.
Оптические элементы из монокристаллов Ge—GaAs, а также
О некоторых применениях
539
из халькогенидов цинка и кадмия не имеют этих недостатков.
Они конкурируют с халькогенидными стеклами (см. [22, 23],
а также литературу у гл. 4 [233]). ZnSe и CdTe характери-
зуются на длине волны излучения СОг-лазера (10,6 мкм) ко-
эффициентами поглощения 4-10~4 и 3-Ю-4 см”1. Оба материала
могут с успехом использоваться для передачи и фокусировки
лазерного излучения. В халькогенидных стеклах столь малые
значения коэффициента поглощения не могут быть реализованы
из-за многофононного поглощения (разд. 4.2.1.1).
Однако монокристаллы не позволяют широко варьировать
форму изделия. Поэтому крупногабаритные элементы для ПК-
приборов, например оптические окна, до недавнего времени
изготавливались только на основе халькогенидных стекол. Не-
давно было показано, что возможно получение сантиметровых
поликристаллических пластин из ZnSe площадью несколько
квадратных метров методом химического осаждения из паро-
вой фазы. В ходе реакции обмена между парами цинка и HaSe
при 873 К при пониженном давлении (104 Па) получены скоро-
сти роста 100 мкм/ч. Материал характеризуется высокой про-
зрачностью в области 0,5—18 мкм [23].
Если из соображений коррекции изображения необходимо
«подогнать» показатель преломления и дисперсию под опреде-
ленные величины, пользуются стеклообразными материалами,
так как кристаллические вещества, кроме твердых растворов,
не обладают такой перестраиваемостью свойств.
Конечная цель и обязательное сопоставление достоинств
и недостатков того или иного материала — вот критерии, опре-
деляющие выбор данной оптической среды. Подробные сведения
о кристаллических средах для ИК-области спектра приведены
в работе [24].
5.3. Халькогенидные стекла как среды
для оптической записи информации
Существует три основных процесса в халькогенидных стек-
лах, на основе которых возможна запись информации: фото-
стимулированное изменение поглощения и показатели прелом-
ления, фотокристаллизация и различия между темновой и фо-
топроводимостью, которые помимо других требований необхо-
димы в электрофотографии.
5.3.1. Оптические среды на основе фотоструктурных
превращений
Сдвиг края поглощения при облучении пленок халькогенид-
ных стекол и связанные с ним изменения показателя прелом-
ления и объема описаны в разд. 4.2.4.1. В зависимости от
540 Глава 5
состава материала возможно как потемнение, так и просветление
материала. Этот эффект на примере слоев составов GesoAs^Seis
и GeioAs4oSe5o предложен фирмами Fa. Nippon Telegraph и Te-
lephone Public Corporation как новый принцип записи инфор-
мации [25].
Важное значение имеет использование различий в скоро-
стях травления облученных и необлученных участков пленки.
Пленки стеклообразного Ge—Se растворяются, например, на
облученных участках в разбавленных растворах NaOH или
аммиака со скоростью на 18 % большей, чем было предложено
использовать для селективного травления [26]. Разрешающая
способность получена 3000 линий/мм.
Эффект может быть значительно усилен, если на пленку
халькогенидного стекла (0,03—3 мкм) нанести тонкий метал-
лический слой, преимущественно используются серебро или
медь (толщиной 0,03—0,05 мкм). Облучение полупроводнико-
вого слоя светом из области собственного поглощения через
полупрозрачный металлический слой приводит к фотодиффузии
атомов металла в пленку халькогенидного стекла. Канон [27]
предложил использовать этот процесс для получения фотошаб-
лонов.
5.3.2. Использование фотоиндуцированного перехода
стекло—кристалл (овография)
Описанный в разд. 4.2.4.2 процесс фотокристаллизации
можно использовать в новых типах оптических запоминающих
устройств сверхбольшой емкости [28]. На этой основе может
быть основан копировальный процесс, аналогичный ксеро-
графии.
Как представлено на рис. 5.5, управляемый ЭВМ лазерный
луч записывает информацию на халькогенидном слое, нанесен-
ном на барабан. При этом возможна либо кристаллизация
в облучаемых точках, либо обратный переход в стеклообразное
состояние, если пленка предварительно была закристаллизо-
вана при отжиге. Изменения проводимости позволяют форми-
ровать электростатическое изображение.
Размер точки составляет 1 мкм. При расстоянии между
точками в строке 3 мкм и расстоянии между строками 6 мкм
получаем плотность записи 5-10е бит/см2. С учетом того, что
лазерный луч может быть сканирован по поверхности в 1 м2
получаем емкость записи 5-Ю10 бит. Каждая точка обрабаты-
вается за время порядка нескольких микросекунд. Для записи
приводились значения битовой скорости 2-Ю8 с-1 [29], в случае
использования фазового перехода из поликристаллического со-
стояния в стеклообразное.
О некоторых применениях 541
Предложены также варианты для получения фотографий
(см. литературу к гл. 4 [149]). Пленка стеклообразного селена
толщиной несколько сотен микронов наносится на прозрачную
подложку. При облучении (10-4 Дж/см2) получается позитив-
ное изображение на отражение (контраст 20:1) и негативное —
управляющий сигнал
Рис. 5.5. Схема копировального процесса, основанного на использовании полу-
проводниковых аморфных слоев, в которых возможна фотокристаллизация.
на пропускание. При поглощении одного кванта света обра-
зуется кристаллическая точка, состоящая из 100 атомов селена.
Разрешающая способность ~100 линий/мм. В других случаях,
например при использовании пленок, содержащих теллур, изо-
бражение может быть усилено путем последующей температур-
ной обработки.
5.3.3. Ксерография
Обзор применений аморфных полупроводников был бы не-
полным, если бы не сказать о ксерографии на основе слоев
стеклообразного селена толщиной 50 мкм. Метод восходит
к работе Карлсона [31], предложившего в 1938 г. принцип
«сухого копирования» (ксерография) на основе полупроводника
с фотопроводимостью большой площади. Последовательность
операций ксерокопирования схематически показана на
рис. 5.6 [32].
542 Глава 5
На цилиндрический барабан наносится пленка селена, ко-
торая затем положительно заряжается с помощью коронного
разряда (а). На следующей стадии на селеновую пленку пере-
носится копируемое изображение (б). При поглощении света
в пленке селена «рождается» множество пар электрон—дырка.
Дырки дрейфуют к подложке из алюминия, а электроны — к по-
нагреватель
\QOOOQOOOOQDOQQ.Q.Q QO O OQQQPJjQO ОУ 0
д
Рис. 5.6. Схема процесса ксерокопирования.
а — подача положительного заряда на селеновую пленку; б — перенос изображения; в —
проявление с помощью красителя; г — перенос изображения на бумагу; д — закрепление
изображения на бумаге; е — очистка и снятие остаточного заряда путем засветки.
верхности, где они нейтрализуют положительный заряд. Для
фиксации изображения (Ь) селеновая пленка обрабатывается
красителем, отрицательно заряженные частицы которого за
счет электростатического электричества задерживаются на точ-
ках, несущих положительный заряд. Затем рисунок переносится
на бумагу с помощью второго коронного разряда (г) и закреп-
ляется (д) при нагревании. После очистки селеновой пленки
и снятия остаточного заряда путем засветки (е) селеновый слой
готов к повторному использованию.
О некоторых применениях
543
Подробно ксерокопировальные устройства описаны в рабо-
тах [5, 15, 18, 19].
Для улучшения панхроматичности, прежде всего для повы-
шения чувствительности селена к красному свету, а также для
оптимизации параметров зарядки в селен вводятся определен-
ные добавки. В ксерографии нашел применение также АзгБез
[20]. В качестве высокочувствительных слоев для электрофото-
графии описаны пленки Se—Bi—I [30].
Литература
1. Rawson И., Denton Е. Р., пат. Великобритании, 801 570 (1958); Macken-
zie J. D., Mioff S. Р., пат. США, 3 258 434 (1966).
2. Edmond J. Т., J. Sc. Instr. Ser., 21, 373 (1968).
3. Adams J., Manley B. W., Phillips Techn. Rundschau, 28, 256 (1967).
4. Trap H. J. L., Sprechsaal fiir Keramik, Gias Email. Silikate, 101, 1103
(1968).
5. Bosnell J. R., Physics in Technology, 4, 113 (1973).
6. Neale R. G., J. Non-Cryst. Solids, 2, 558 (1970).
7. Nelson D. L., J. Non-Cryst. Solids, 2, 528 (1970).
8. Fleming G. R., J. Non-Cryst. Solids, 2, 540 (1970).
9. van Laudingham К. E., IEEE Transyct. Electron. Dev., ED-20, 178 (1973).
10. Zingg R. J., Zimmermann R. E., Pohm A. V., IEEE, Transyct. Electron
Dev., ED-20, 188 (1973).
11. Neale R. G., Aseltine .1. A., IEEE Transyct. Electron. Dev., ED-20, 195
(1973).
12. Chapin D. M„ Fuller C. S., Pearson G. L., J. Appl. Phys., 25, 676 (1954).
13. Lindmeyer J., Wrigley С. У., National Science Foundation — 43 090, Wa-
shington, D. C., 1975.
14. James L. W., Moon R. L., Proc. 11th IEEE Photovoltaic Specialists Conf.,
Scottsdate, Arizona, 1975, p. 402.
15. Dessauer J. 11., Clark H. E., Xerography and Related Processes, Focal
Press, N. Y., 1965.
16. Mattox D. M., J. Solid State Chem., 22, 31 (1977).
17. Feldmann B. J., J. Non-Cryst. Solids, 35, 36, 309 (1980).
18. Weigl J. W., Angew. Chem., 89, 386 (1977).
19. Berger S. B., Enck R. C., Scharfe M. E., Springett В. E., in: Physics of
Selenium and Tellurium, E. Gerlach, P. Grosse (eds.), Springer Series
in Solid State Science 13, Springer-Verlag, 1979, p. 256.
20. Bayer E., Kempter K., Kiendl A., Non Cryst. Solids, 4th Int. Conf. The Phy-
sics of Non-Cryst. Solids, 1976, G. H. Frischat (ed.), Trans. Tech. Publ.,
1977, p. 242.
21. Киишцкая E. А., Носов В. Б., Кокорина В. Ф.— Физ. хим. стекла, 1977,
т. 3, с. 624.
22. Lussier F. М., Laser Focus, 12, 47 (1976).
23. Miles Р., Optical Engineering, 15, 451 (1976).
24. Donald 1. W., McMillan O. W„ J. Mater. Sci., 13, 1151 (1978).
25. Nippon Telegraph and Telephone Public Corporation, E. C. L., Technical
Publication, № 67.
26. Nagai H., Yoshikawa A., Toyoshima Y., Ochi O., Mizoshima Y., Appl. Phys.
Lett., 28, 145 (1976).
27. Canon К. К.. пат. ФРГ, 2 201 178 (1972).
28. Feinleib J., Iwasa S., Moss S. C., de Neufville J. P., Ovshinsky S. R., J.
Non-Cryst. Solids, 8—10, 909 (1971).
29. von Gutfeld R. J., Chaudhari P., J. Appl. Phys., 43, 4688 (1972).
544 Глава 5
30. Hillegas W. J., Neyhart J. H., J. Non-Cryst. Solids, 27, 347 (1978).
31. Carlson C. F., Xerography, in: Progress in Photography, P. A. Spenser
(ed.), 1955—1958, Focal Press, London, 1958.
32. Mort J., Proc. 5th Int. Conf. Amorph. Liquid Semicond., Garmisch-Parten-
kirchen, 1973, Taylor-Francis, London, 1974, p. 1361.
33. Levine C. A., Proc. 10 IECEC, 621 (1975).
34. Wicsener K-, Gurche J., Schneider U7., Elektrochemische Stromquellen, Aka-
demie-Verlag, Berlin, 1981.
35. Schwabe K-, pH-Messtechnik, Berlin, 1963.
36. Eisenman G. et al., The Glass Electrode, Interscience Publ., N. Y., 1985.
37. Schwabe K., Physikalische Chemie, Band 2, Eiektrochemie, Akademie-Verlag,
Berlin, 1974.
38. Jasinski R., Trachtenberg L, Rice G., J. Electrochem. Soc., 121, 363 (1974).
39. Owen A. E., J. Non-Cryst. Solids, 35, 36, 999 (1980).
40. Hamakawa T., IX. Int. Conf. Amorph. Liquid Semicond., Grenoble, 1981,
Proc. IX. Int. Conf. Amorph. Liquid Semicond., Grenoble, 1981, J. de Phy-
sique Coiioque C4, Suppl. n° 10, 42, 1131 (1981).
Дополнительная литература *
Алимбарашвили H. А., Бродзели М. И. и др. Новые материалы для оптиче-
ской записи информации.— Тбилиси: Мецниереба, 1983, с. 149.
Панасюк Л. М. Фототермопластические носители на основе слоев аморфных
полупроводников.— В кн.: Структура, физико-химические свойства и приме-
нение некристаллических полупроводников. Кишинев, Штинца, 1980, с. 238.
Петровский Г. Т., Воронков Г. Л. Оптическая технология в космосе.— Л.'.
Машиностроение, 1984. 158 с.
Rawson Н. Properties and application of glass. Ams., Oxf., N-Y., 1980, 316 p.
Физико-химические основы производства оптического стекла/Под ред.
Л. И. Демкиной — Л.: Химия, 1976, 456 с.
Костылев С. А., Шкут В. А. Электронное переключение в полупроводниках.—
Киев: Наукова думка, 1978, 203 с.
* Представлена редактором С. А. Дембовским.
Предметный указатель
Аморфные модификации элементов
221, 229
Аморфные системы 244
Аморфные слои в системе Т1—Те 371
---------Ge—Те 327, 332
---------As—S 289
---------As—Se 79, 295
---- на основе селенидов германия
326
---- получение
------ катодное распыление 47
------ лазерное испарение 46
------ мгновенное испарение 46
------осаждение в тлеющем раз-
ряде 50
------плазменное испарение 46
------ термическое испарение в ва-
кууме 42
------химическое осаждение из па-
ровой фазы 51
Аморфные соединения 337
----AmBv и AnBVI 244
----AnBrvC2v 245
----CdAs2, CdP2 245
Аморфные твердые тела, определение
23
— — — получение
---------осаждением из растворов
38, 40
--------- под действием ударной
волны 53
--------- путем механической обра-
ботки 52
----—------облучения 52
Бериллия фторид, ближний порядок
139
35 Заказ № 413
---- вязкость 29, 66
----коэффициент Пуассона 80
----критическая скорость охлажде-
ния 113
----КТР 80, 333
----показатель преломления 333
---- структура 333
---- температура плавления 29
----— стеклования 29, 164, 166
----ширина запрещенной зоны 487
---- энтальпия атомизации 164, 166
------ плавления 29
Бор аморфный 230, 232
— модификации 229
— свойства 232
Бора оксид, ближний порядок 139,
273
---- время релаксации 72
---- вязкость 29, 274
— — колебательный спектр 272
----коэффициент Пуассона 80
----критическая скорость охлажде-
ния 113
----КТР 80, 90
---- модуль сдвига 72
— упругости 170
•---показатель преломления 65
— — структура 273
-—- температура плавления 29, 85
------ стеклования 164
----теплоемкость 83, 90
----фотоупругость 516
------ ширина запрещенной зоны 500
---- энтальпия атомизации 164
------ плавления 29
Время релаксации 55, 76, 92, 411
Вязкость 29, 32, 64, 66
546
Предметный указатель
— температурная зависимость 65
— энергия активации вязкого тече-
ния 64
— энтропия активации 68, 161
Галогены 202
Германий аморфный 237
— КРР 241
— легирование 466
— модификации 236
— проводимость 451
— спектр электронного поглощения
496
— ТУТ-кривая 112
- фотоулругость 516
Германия диоксид, ближний порядок
307
—— время релаксации 72
—— вязкость 29
— — коэффициент Пуассона 80
----кристаллические модификации
307
----край поглощения 487
----критическая скорость охлажде-
ния 113
----КТР 80, 90
---- модуль сдвига 72
----собственные дефекты 309
- — стеклообразный 307
---- температура плавления 307
------- стеклования 307
----ширина запрещенной зоны 500
---- энтальпия атомизации 164
------- плавления 29, 307
— диселенид, аморфный осадок 335
----ближний порядок 139, 320
----диэлектрическая проницаемость
321
----дрейфовая подвижность 458
---- колебательный спектр 320
------ край поглощения 487
----КТР 80
----собственные дефекты 320
---- структура 123, 319
----температура стеклования 164
----ширина запрещенной зоны 500
---- энергия активации вязкого те-
чения 165
---- энтальпия атомизации 164
— дисульфид, аморфный осадок 335
----ближний порядок 139
---- время релаксации 72
----колебательный спектр 313
----край поглощения 487
— — критическая скорость охлаж-
дения 312
----КТР 80, 312
----микротвердость 312
----модификации 310
---- модуль сдвига 72
---- мольный объем 312
----показатель преломления 510
----собственные дефекты 313
— стеклообразование 310
---- структура 313
— - температура плавления 310
------ стеклования 310
----фотоупругость 516
----ширина запрещенной зоны 500,
313
---- энтальпия атомизации 164, 166
— селенид 319
— сульфид 311
— теллурид 327
Гипотеза Андерсона 190, 194
— Захариасена 140
— Лебедева 140
Дисперсия света 508, 511
Диспропорционирование 204, 205
Дифракция рентгеновских лучей 115
ДСК 76, 77
ДТА 76, 77, 78
Зародышеобразование 98
— гомогенное, стационарное 98
— гетерогенное 101
— нестационарное 100
Зонная модель 172, 174, 179, 186
ИК-слектроскопия 124
Иод, дефектные центры 150, 201
— проводимость 203
Ионоселективные электроды 533
Предметный указатель
547
Канальный умножитель 534
Кварц см. Кремния диоксид
Кластеры 144
Коэффициент поглощения 487, 489,
491, 498, 502
КР-спектроскопия 124
Кремнезем см. Кремния диоксид
Кремний аморфный 237—239
— кластерная модель 144
— легирование 466
— модификации 236
— проводимость 451
— слои a-Si:H 238
— спектр электронного поглощения
496
— электрические свойства 450
Кремния диоксид, ближний порядок
139
---- время релаксации 72
— — вязкость 20
•---дрейфовая подвижность 458
---- колебательный спектр 127
----коэффициент Пуассона 80, 304
----край поглощения 302, 487
------ критическая скорость охлажде-
ния 113
----КТР 80, 304
----модификации 299
----модуль сдвига 72
•-----упругости 304
----сжимаемость 304
— — собственные дефекты 149, 302,
303
— — спектр электронного поглоще-
ния 498
---- температура плавления 29
— ----стеклования 164, 301
----теплоемкость 83, 90
----777-кривая 112
----фотоупругость 516
----ширина запрещенной зоны 500
---- энтальпия атомизации 164
------- - плавления 29
— диселенид 305, 306
— дисульфид 305, 306
— нитрид 305
— оксиды с пониженным содержани-
ем кислорода 305
35*
— теллурид 307
Ксерография 541
Ликвация см. Разделение фаз
Межгалогенные соединения 203
Металлические стекла, классификация
222
----модуль упругости 171
---- получение 33, 34
----• свойства 227
— — структура 224
----777-кривые 111
Мёссбауэровская (у-резонансная)
спектроскопия 132
Микронеоднородность 105
Модель КАФ 191, 198, 199
— Мотта—КФО 188
— Полка 143, 241
— сеточная 139
— Стрита—Мотта 191, 194
Модификаторы стеклообразной сетки
154, 155, 170
Модифицирование аморфных пленок
халькогенидов 466
— селена 463
— халькогенидных стекол 459
Многофононное поглощение 491
Мышьяк аморфный 250
— модификации 249
— проводимость 250, 448
— спектр электронного поглощения
497
— структура 250
Мышьяка оксид(Ш), ближний поря-
док 139
---- вязкость 29
---- испарение 46
----критическая скорость охлажде-
ния 113
----модификации 276, 277
---- структура 277
---- температура плавления 29
—-----стеклования 29, 164
---- энтальпия атомизации 164
------— плавления 29
548
Предметный указатель
— оксид(V) 278
— селенид 291
— триселенид, аморфные слои 79,
295
----ближний порядок 139
---- время релаксации 72
----диэлектрическая проницаемость
293
----дрейфовая подвижность 458
---- колебательный спектр 292
----коэффициент Пуассона 80
----критическая скорость охлажде-
ния 113
----КТР 80, 90
----многофононное поглощение 491
----модуль сдвига 72
------- упругости 170
---- мольный объем 293
----показатель преломления 510
----примеси 460
----проводимость 448
----спектр электронного поглощения
498
---- структура 123
----температура плавления 32, 85,
290
-------стеклования 32, 85, 164
— — теплоемкость 90
----фотоупругость 516
— — ширина запрещенной зоны 294,
500
---- энтальпия атомизации 164
------- плавления 29
---- энтропия активации вязкого те-
чения 161
— сульфид (реальгар) 283, 284
— трисульфид (аурипигмент), аморф-
ные слои 289
----ближний порядок 139
— — время релаксации 72
— — вязкость 32, 66
— — диэлектрическая проницаемость
289
----колебательный спектр 356
----коэффициент Пуассона 80
•---край поглощения 499, 501, 503
----КТР 80, 90
---- микротвердость 285
----многофононное поглощение 491
---- модуль сдвига 72
------ упругости 170
---- мольный объем 285
----показатель преломления 510
— — проводимость 289
—— структура 286
---- температура плавления 32, 85,
285
—-----стеклования 32, 85, 164, 285
----теплоемкость 90
----фотоструктурные превращения
519
----фотоупругость 515, 516
----ширина запрещенной зоны 289,
500
---- энергия активации вязкого те-
чения 285
---- энтальпия атомизации 164
------ плавления 29
— теллурид, ближний порядок 139
---- колебательный спектр 297
----край поглощения 500
----критическая скорость охлажде-
ния 295
----проводимость 297, 460
----собственные дефекты 297
---- структура 296
---- температура плавления 32, 85,
296
----— стеклования 32, 85, 164
----ширина запрещенной зоны 297,
500
---- энтальпия атомизации 164
------ плавления 32
Нейтронография 121
Овография 540
Оксиды стеклообразующие 29
Парадокс Каузмана 90
Переключатели 535
Предметный указатель
549
Переключение пороговое 477
— с памятью 476
Подвижность дрейфовая 455
— зависимость от напряженности 468
Показатель преломления 508, 518
Правило Каузмана 87
— Серджента и Роя 111
— Урбаха 500
Примеси в халькогенидных стеклах
459, 492
Проводимость стекол, зависимость от
напряженности 467
--- ионная 413
— — металлических стекол 227
---на переменном токе 420
------ постоянном токе 415
--- оксидных стекол 423
---прыжковый механизм 410
--- статическая 441
--- стационарная 413
--- частотная зависимость 415
--- электронная 423, 429
ПТСРП (EXAFS) 122
Разделение фаз 104
---- кинетика 108
---- критерии 156
---- механизм 109
Растворение стекол 135—137
Реальгар 283, 284
Релаксация механическая 56
— структурная 61, 64, 71
— термическая 56
Рост кристаллов 98
---- механизм 103
Самоокисление—самовосстановление
203
Селен аморфный 260
— ближний порядок 139
— время релаксации 72
— вязкость 32, 264
— дрейфовая подвижность 458
— колебательный спектр 261
— коэффициент Пуассона 80
— КТР 80, 90, 265
— модификации 259
— модуль сдвига 72
- - очистка 492
— примеси 492
— проводимость 260, 458
— расплав 263
— растворение 137
собственные дефекты 195
— спектр электронного поглощения
497
— стеклообразный 265
— температура плавления 32, 85
стеклования 32, 85, 164
— теплоемкость 77, 90
— фотоунругость 516
-- ширина запрещенной зоны 268
— энергия связи 152
— энтальпия атомизации 164
--- плавления 32
Сера, вязкость 258, 264
модификации 257
Скорость испарения 45
— охлаждения критическая НО, 11S
---стеклообразующих расплавов 36’
— кристаллизации 102
Собственное разупорядочение 147
Солнечные элементы 537
Стекла в оксидных системах Li2O—
В2О3 419
---------Li2O—SiO2 414, 416, 419
---------Li2O—SiO2—Ij2SO4 419
---------Li2O—P2O5 419
---------Na2O-B2O3 416
---------Na2O—SiO2 164, 414, 416,.
418
_________\a,O—GeO2 164, 418
_________Ха О P2O5 164, 418
_________\’a2O—CaO—SiO2 423
_________Na2O—BaO—SiO2 423
_________K2O—SiO2—TiO2 161
_________K2O—SiO2—VO_> 161
_________CaO-Al2O3—SiO2 423
_________CaO -SiO2 422
_________B2O3-PbO—Fe2O3 426
_________A12O3—SiO2—MnO 426
_________GeO—GeO2 317, 310
_________GeO >-P2O5- V2O5 426
_________V;O5—P2O5 426
550_________________________
-------VO2—V2O5—P2o5 427
---TiO2—P2O5 428
--- халькогенидных системах
-------Li2S—GeS2 369, 418
-------Na2S-SiS2 369, 418
-------NTa2S—GeS2 369, 418
-------Na2S—P2S5 418
-------Na2S—As2Ss 369
-------Cu—As—Se 369
-------Ag—Ge—S 370, 418
-------Ag—As—S 370, 371. 418
-------Hg—Ge—Se 379, 383
-------Hg—Ge—Se—Те 379
-------Hg—Ge—Те 379
-------Ga—Ge—S 373
-------Ga—Ge—Sc 373
-------T1 -S 371
-------Tl—Se 371
-------Tl—Ge—S 370
-------Tl—Ge—Se 370
-------Tl—As—S 372
-------Tl—As—Se 372
-------Tl—As—Те 372, 448
-------Tl—As—Sb—Se 384
-------Tl—As—Se—Те 384, 448
-------Si—Те 306
-------Si—Ge—Те 350, 351
-------Si—P Tl 367
-------Si—Ge—As—Те 388
-------Si—As—Те 367, 449
-------Ge—S 311, 315, 317
-------Ge—Se 319, 320, 323
-------Ge—Те 327, 329
-------Ge—Pb—S 373, 379
-------Ge—Pb—Se 373, 375, 381
-------Ge—Pb—Se—Те 384
-------Ge—Sn—S 373, 377
---— — Ge—Sn—Se—Те 385
-------Ge—P—S 352
-------Ge—P—Se 352
-------Ge—P—Те 365, 366
---Ge—As—S 354, 357
---— — Ge—As—Se 354, 492, 494,
510, 518
-------Ge—As—Те 365, 449, 477
-------Ge—Sb—S 360
-------Ge—Sb—Se 360
Предметный указатель
--------Ge—S—Se 345
--------Ge—S—Те 346
--------Ge—Se—Те 347, 449
--------Ge—S—I 339
--------Ge—Se—I 339
--------Ge—Те—I 340
----— P—As—S 349
--------p_As—Se 349
--------P—Sb—Se 349
------As—Sb -S 351
--------As—Sb—Se 351
---- p_ S Se 341
— As -S—Se 341, 343
—-------As— S -Те 341
— As—Se-Te 341, 343
- - — As -S—Cl 339
— - — As S-Br 339
--------As—S—I 339
--------As—Se—I 339
--------As—Те—I 340
--------Sb—S—I 341
— оксид-халькогенидные 338
----прозрачные в ИК-области 487,
538
----с ионной проводимостью 413,
418. 419, 532
— —•с электронной проводимостью
423, 533
---- сверхвысокоомные 422
— щелочносиликатные 421
Стекло, определение 24, 25, 63
— получение из расплава 27
----осаждением гелей 39
Структурное разупорядочение 149
Суперортикон 534
Сурьма, аморфные слои 256
— оксиды 278
— селениды 298, 335
— сульфиды 298, 335
Твердые электролиты 417—419
---- применение 533
Теллур 268
— критическая скорость охлаждения
113
— структура 269
— температура стеклования 164, 166
Предметный указатель
551
— ГГГ-кривые 112
— энтальпия атомизации 164
Температура плавления 29, 32
— стеклования 29, 32, 71
— — зависимость от состава 160
---методы определения 72—79
---связь с термическим расшире-
нием 79
---------теплоемкостью 83
— температурой плавления 85
--------- энтальпией атомизации
164, 166
Теории стеклообразоваиия 140
Теплоемкость 77, 83, 92, 95
Термическое расширение 73, 75, 79,
89, 167
ТТГ-кривые 111, 112
Углерод аморфный 234
— модификации 232
— стеклообразный 235
УФ-спектроскопия 134
Фазовые переходы 96
Фосфор аморфный 249
— модификации 246
Фосфора оксиды 275
---ближний порядок 139
— — вязкость 29
— — критическая скорость охлажде-
ния 113
--- температура плавления 29
•----- стеклования 29
— — энтальпия плавления 29
— селениды 279
— сульфиды 279
Фотоиндуцированные структурные
превращения 517, 518, 539
Фотокристаллизация 520
Фотолюминесценция 503, 505
Фотопроводимость 481
Фотоупругость 513, 516
Фотоэлектронная спектроскопия 131
Химическая связь 145
Ширина запрещенной зоны 498, 499
Щель подвижности 188, 191
Электронное поглощение 495
Электронные состояния 172
Электронография 121
Электрофотография 539
Энергия связи 152
Энтальпия атомизации 164, 166, 170
Эффект Зеебека 444
— переключения см. Переключение
пороговое
— полищелочной 421
— Пула—Френкеля 474
— Холла 447
ЯМР-спектроскопия 132
Оглавление
Предисловие редакторов перевода ..................................... 5
Предисловие.......................................................... 8
Принятые обозначения ................................................ 11
1. Введение ........................................................ 17
Литература ...................................................... 21
2. Аморфное и стеклообразое состояние неорганических веществ . . 23
2.1. Методы получения аморфных твердых тел и стекол........... 25
2.1.1. Получение стекол из расплава.......................... 27
2.1.1.1. Оксидные стекла.............................. 28
2.1.1.2. Халькогенидные стекла........................ 31
2.1.1.3. Металлические стекла......................... 33
2.1.1.4. Управление скоростью охлаждения стеклообра-
зующих расплавов....................................... 35
2,1.2. Получение аморфных твердых материалов из растворов 38
2.1.2.1. Стекла, получаемые гомогенным осаждением ге-
лей ................................................... 39
2.1.2.2. Аморфные осадки......................... 40
2.1.2.3. Электролитическое осаждение аморфных слоев 40
2.1.3. Получение аморфных слоев из газовой фазы........... 41
2.1.3.1. Методы термического испарения в вакууме . . 42
2.1.3.2. Получение аморфных слоев методом катодного 47
распыления ..................................... 50
2.1.3.3. Осаждение аморфных слоев в тлеющем разряде
2.1.3.4. Химическое осаждение из паровой фазы .... 51
2.1.4. Перевод кристаллических веществ в аморфное состояние 52
2.1.4.1. Получение аморфных тел путем механической
обработки ............................................. 52
2.1.4.2. Получение аморфных твердых тел путем облуче-
ния ................................................... 52
2.1.4.3. Получение аморфных веществ под действием
ударной волны ......................................... 53
2.1.4.4. Реакции кристаллических твердых тел при обра-
зовании стекла......................................... 54
2.2. Термодинамическое описание стеклообразного состояния . . 54
2.2.1. Релаксационные процессы в жидких и стеклообразных
веществах ................................................... 56
2.2.1.1. Механическая релаксация ...................... 56
2.2.1.2. Термическая и структурная релаксация .... 61
2.2.1.3. Температурная зависимость вязкости............ 65
2.2.1.4. Структурная релаксация и корреляционная спек-
троскопия ............................................. 71
Содержание
553
2.2.2. Температура стеклования............................
2.2.2.1. Методы определения температуры стеклования
2.2.2.2. Температура стеклования и термическое расши-
рение ..............................................
2.2.2.3. Температура стеклования и теплоемкость . . . .
2.2.2.4. Температура стеклования и температура плавле-
ния ................................................
2.2.2.5. Тенденция к стеклообразованию..............
2.2.3. Термодинамические функции и стеклообразование . . .
2.2.3.1. Парадокс Каузмана..........................
2.2.3.2. Уравнение Пригожина—Дефея..................
2.2.3.3. Термические свойства систем при низких темпе-
ратурах ............................................
2.3. Фазовые переходы и стеклообразование....................
2.3.1. Выделение фаз из стеклообразующих расплавов . . . .
2.3.1.1. Зародышеобразование и рост кристаллов . . .
2.3.1.2. Явления разделения фаз ....................
2.3.2. Критическая скорость охлаждения и тенденция к стек-
лообразованию ...........................................
2.4. Структура и химическая связь в стеклах и аморфных телах
2.4.1. Основные методы структурных исследований некристал-
лических твердых тел.....................................
2.4.1.1. Дифракционные методы.......................
2.4.1.2. Инфракрасная спектроскопия и спектроскопия
комбинационного рассеяния (рамановская) . . .
2.4.1.3. Другие методы исследования твердых тел . . .
2.4.1.4. Химические методы изучения строения стекол . .
2.4.2. Структурные теории стеклообразоваиия...............
2.4.2.1. Прототипы стеклообразующих соединений . . .
2.4.2.2. Сеточная модель стеклообразователей........
2.4.2.3. Кластерные модели .........................
2.4.2.4. Химическая связь и стеклообразование.......
2.4.2.5. Структура идеального стекла и собственное раз-
упорядочение .......................................
2.4.2.6. О структуре сложных стеклообразующих систем
2.4.2.7. Возможные критерии для структурно-химиче-
ского объяснения процесса разделения фаз . .
2.4.3. Корреляции структура—свойства .....................
2.4.3.1. Зависимость мольного объема от структуры . .
2.4.3.2. Зависимость вязкости от структуры..........
2.4.3.3. Температура стеклования и структура стекол . .
2.4.3.4. Зависимость термического расширения от струк-
туры ...............................................
2.4.3.5. Структура стекла и упругие постоянные . . . .
2.5. Электронные состояния в некристаллических твердых мате-
риалах ......................................................
2.5.1. Зонная модель и локализация электронов в твердых
телах ...................................................
2.5.1.1. Основные положения зонной модели...........
2.5.1.2. Границы применимости зонной модели кристал-
лических твердых тел................................
2.5.1.3. Зонная модель некристаллических твердых тел
2.5.2. Собственное разупорядочение и заряженные дефектные
центры в некристаллических системах......................
2.5.2.1. Различие между веществами, связанное со зна-
ком эффективной энергии корреляции электронов
2.5.2.2. Модель заряженных дефектных центров . . •
2.5.2.3. Ковалентные и полярные связывающие состояния
в конденсированных химических системах и оки-
71
72
79
83
84
86
89
90
93
94
96
97
98
104
НО
114
115
115
124
131
134
137
138
139
144
145
147
153
156
157
158
161
162
167
169
172
174
174
179
186
190
191
194
554 Содержание
слительно-восстановительное диспропорциониро-
вание ............................................ 200
Литература ......................................................... 207
3. Аморфные и стеклообразные системы................•.................. 220
3.1. Некристаллические модификации элементов. Сплавы .... 220
3.1.1. Металлические стекла..................................... 221
3.1.1.1. Классификация металлических стекол............. 222
3.1.1.2. Структура металлических стекол.................... 224
3.1.1.3. Свойства металлических стекол..................... 227
3.1.2. Элементы-неметаллы в аморфном состоянии.................. 229
3.1.2.1. Бор .............................................. 229
3.1.2.2. Углерод .......................................... 232
3.1.2.3. Кремний, германий и производные системы . . 236
3.1.2.4. Фосфор ....................................... 246
3.1.2.5. Мышьяк............................................ 249
3.1.2.6. Сурьма ....................................... 256
3.1.3. Стеклообразование халькогенов............................ 256
3.1.3.1. Сера ............................................. 257
3.1.3.2. Селен ............................................ 259
3.1.3.3. Теллур ........................................... 268
3.2. Двойные стеклообразующие соединения и системы............. 271
3.2.1. Триоксид бора и оксидные и халькогенидные системы
фосфора, мышьяка и сурьмы........................................ 271
3.2.1.1. Триоксид бора..................................... 271
З.2.1.2. Оксиды фосфора, мышьяка, сурьмы................... 275
3.2.1.3. Сульфиды и селениды фосфора....................... 279
З.2.1.4. Сульфиды мышьяка и стеклообразование в си-
стеме As—S................................................. 282
3.2.1.5. Селениды мышьяка и стеклообразование в си-
стеме As—Se.............................................. 290
3.2.1.6. As2Te3 и стеклообразование в системе As—Те 295
3.2.1.7. Аморфные сульфиды и селениды сурьмы . . . 298
3.2.2. Оксидные и халькогенидные системы кремния и герма-
ния ............................................................. 299
3.2.2.1. Стеклообразный кремнезем и аморфная система
SixO^x .................................................. 299
3.2.2.2. Халькогениды кремния ............................. 305
3.2.2.3. Диоксид германия и стеклообразование в си-
стеме GeO—GeO2..................................... 307
3.2.2.4. Стеклообразование сульфидов германия и Ge2S3 310
3.2.2.5. Стеклообразование селенидов германия и Ge2Se3 318
3.2.2.6. Стеклообразование в системе Ge—Те, GeTe4 и
GeTe2 ..................................................... 327
3.2.3. Стеклообразующие фториды ................................ 332
3.2.3.1. Фторид бериллия .................................. 332
3.2.3.2. Стекла на основе ZrF4............................. 334
3.2.4. Двойные некристаллические соединения..................... 334
3.2.4.1. Стеклообразующие соединения и структура рас-
плава ..................................................... 334
3.2.4.2. Аморфные соединения............................... 337
3.3. Тройные стеклообразующис халькогенидные системы .... 338
3.3.1. Оксид-халькогенидные стекла ............................ 338
3.3.2. Галогенсодержащие халькогенидные стекла . . . 339
3.3.2.1. Стекла в системах As—Те—I и Gc—Те—I . . . 339
3.3.2.2. SbSI ............................................. 341
35*
Содержание
555
3.3.3. Тройные халькогенидные стекла, содержащие два халь-
когена .................................................... 341
3.3.3.1. Стеклообразование и свойства в системах As—
S—Se, As—S—Те и As—Se—Те....................... 341
3.3.3.2. Стеклообразование и свойства стекол в системах
Ge—S—Se, Ge—S—Те и Ge—Se—Те........................... 345
3.3.4. Тройные халькогенидные стекла с двумя различными
элементами V и IV групп периодической системы . . . 349
3.3.4.1. Стекла в системах Р—As—S и Р—As—Se . . 349
3.3.4.2. Стекла в системе Р—Sb—Se.............. 349
3.3.4.3. Стекла в системах As—Sb—S и As—Sb—Se . . 351
3.3.4.4. Стекла в системе Si—Ge—Те.............. 351
3.3.5. Тройные халькогенидные стекла, образованные элемен-
тами IV, V и VI групп периодической системы .... 351
3.3.5.1. Стеклообразование в системах Ge—Р—S и Ge—
Р—Se.................................................. 352
3.3.5.2. Стеклообразование и свойства стекол в систе-
мах Ge—As—S и Ge—As—Se................................ 354
3.3.5.3. Стеклообразование и свойства стекол в систе-
мах Ge—Sb—S и Ge—Sb—Se................................ 360
3.3.5.4. Стекла в системах Ge—Р—Те и Ge—As—Те . . 365
3.3.5.5. Стекла в системах Si—Р—Те и Si—As—Те . . 367
3.3.6. Тройные халькогенидные стекла с халькогенидами ме-
таллов в качестве модификатора стеклообразной сетки 368
3.3.6.1. Натриевотиогерманатные и натриевотиоарсенат-
ные стекла .................................... 369
3.3.6.2. Халькогенидные стекла, содержащие Си и Ag 369
3.3.6.3. Халькогенидные стекла, содержащие Т1 и Ga 371
3.3.6.4. Германиевохалькогенидные стекла, содержащие
Sn и РЬ............................................... 373
3.3.6.5. Германиевохалькогенидные стекла, содержащие
Hg.................................................... 379
3.3.7. Некоторые четверные халькогенидные стеклообразующие
системы .................................................... 333
3.3.7.1. Стеклообразование и свойства стекол в системах
Ge—Sn—Se—Те и Ge—Pb—Se—Те............................. 384
3.3.7.2. Стекла в системе Hg—Ge—Se—Те................. 387
3.3.7.3. Стекла в системе As—Ge—Si—Те................. 387
Литература .................................................... 388
4. Электрические и оптические свойства стекол и аморфных тел . . 409
4.1. Перенос заряда в некристаллических твердых телах .... 410
4.1.1. Стекла с ионной проводимостью....................... 413
4.1.1.1. Ионная проводимость на постоянном токе . • 415
4.1.1.2. Ионная проводимость на переменном токе . . 420
4.1.1.3. Полищелочной эффект ...................... 421
4.1.1.4. Сверхвысокоомные стекла................... 422
4.1.2. Оксидные стекла с электронной проводимостью . . . 423
4.1.2.1. Механизм переноса заряда .................... 429
4.1.2.2. Проводимость в рядах оксидных стекол со сход-
ной структурой ..................................... 433
4.1.3. Электропроводность в стеклах и аморфных телах
с малой полярностью связей ................................ 437
4.1.3.1. Перенос заряда в халькогенидных стеклах . . 441
4.1.3.2. Электрические свойства аморфных кремния, гер-
мания и родственных систем............................ 450
4.1.3.3. Динамическая проводимость.................... 458
556 Содержание
4.1.3.4. Дрейфовая подвижность ..................... 455
4.1.3.5. Примеси и электрические свойства .......... 459
4.1.3.6. Зависимость проводимости от напряженности
электрического поля ................................. 467
4.1.3.7. Эффект переключения ....................... 475
4.1.4. Фотопроводимость .................................. 481
4.2. Оптические свойства..................................... 486
4.2.1. Поглощение и пропускание........................... 488
4.2.1.1. Многофононное поглощение .................. 490
4.2.1.2. Влияние примесей на пропускание. Очистка сте-
кол ................................................. 492
4.2.1.3. Спектр электронного поглощения............. 495
4.2.1.4. Край поглощения ........................... 498
4.2.2. Фотолюминесценция ................................. 503
4.2.2.1. Спектр люминесценции с точки зрения модели
заряженных дефектных центров......................... 505
4.2.2.2. Фотоиндуцированное неравновесное дефекгооб-
разование ........................................... 507
4.2.3. Показатель преломления и дисперсия света........... 508
4.2.3.1. Нормальная и аномальная частные дисперсии 511
4.2.3.2. Пьезооптический эффект (фотоупругость) . . . 513
4.2.4. Фотоиндуцированныс структурные превращения в стек-
лах ..................................................... 517
4.2.4.1. Фотоиндуцированные изменения края поглощения
и показателя преломления в халькогенидных
стеклах ............................................. 518
4.2.4.2. Фотокристаллизация ........................ 520
Литература................................................... 521
5. О некоторых применениях....................................... 532
5.1. Использование некоторых стекол и аморфных слоев с полу-
проводниковыми свойствами .................................... 532
5.1.1. Применение стекол с ионной проводимостью.......... 532
5.1.1.1. Твердые стеклообразные электролиты........ 532
5.1.1.2. Ионоселективиые электроды.................. 532
5.1.2. Применение оксидных стекол с электронной проводимо-
стью .................................................... 533
5.1.2.1. Накопительные электроды из стекла в суперор-
тиконах .................................... 533
5.1.2.2. Канальный умножитель вторичных электронов 534
5.1.3. Переключатели типа Ovonic.......................... 535
5.1.4. Солнечные элементы на основе слоев аморфного крем-
ния ..................................................... 536
5.2. Халькогенидные стекла как среды, прозрачные в ИК-области
спектра ...................................................... 538
5.3. Халькогенидные стекла как среды для оптической записи ин-
формации ..................................................... 539
5.3.1. Оптические среды на основе фотоструктурных превра-
щении ................................................... 539
5.3.2. Использование фотоиндуцированного перехода стекло—
кристалл (овография) ................................... 540
5.3.3. Ксерография ....................................... 541
Литература .................................................. 543
Предметный указатель............................................. 545