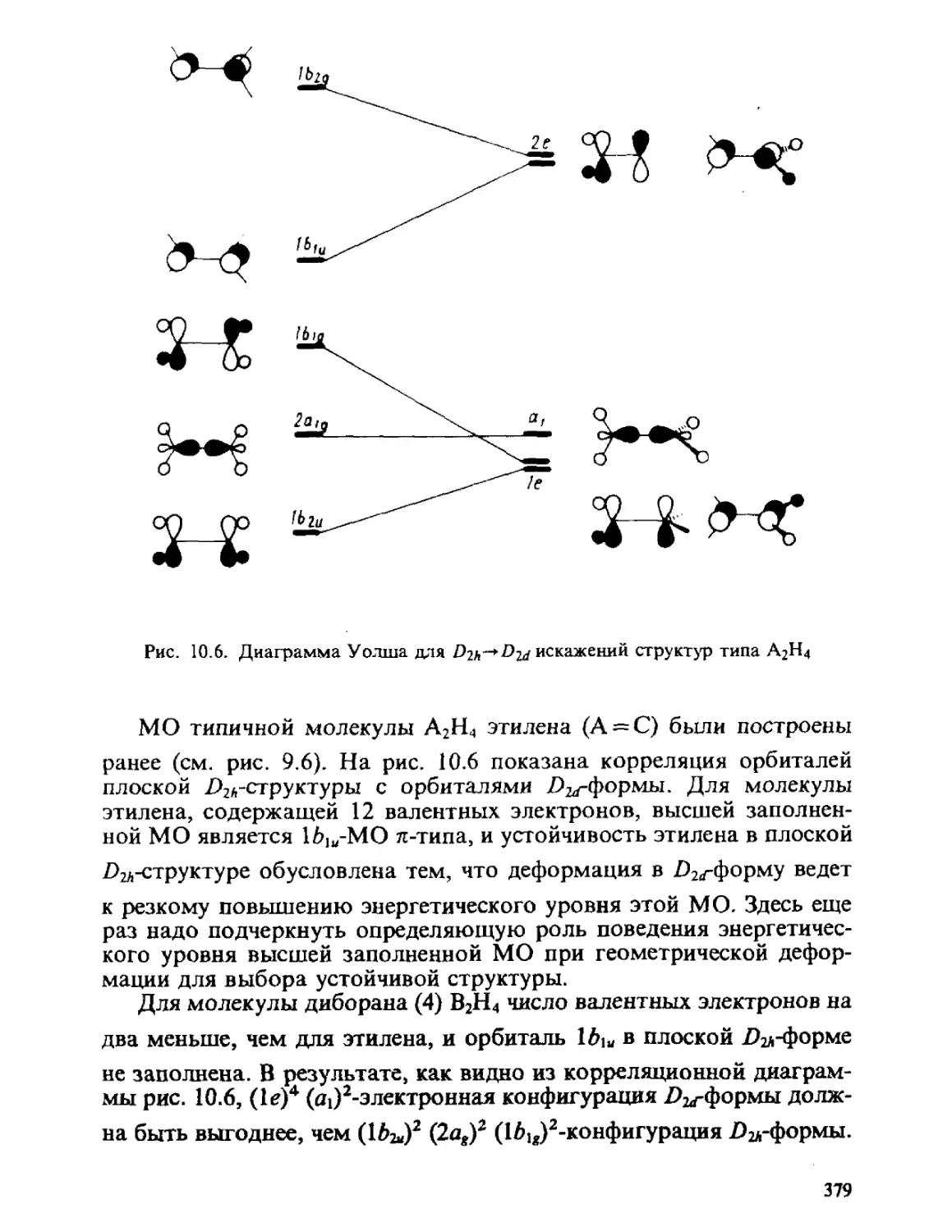
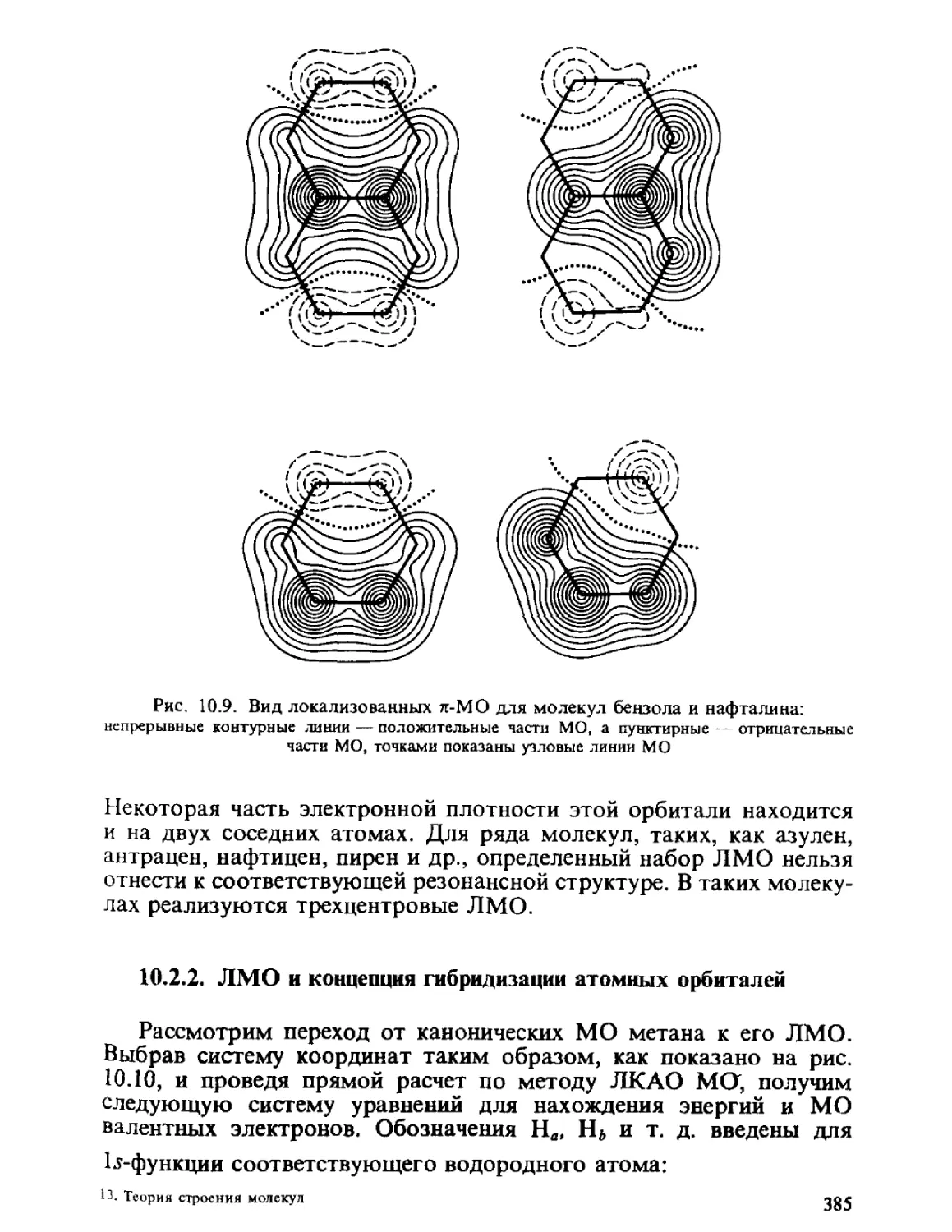
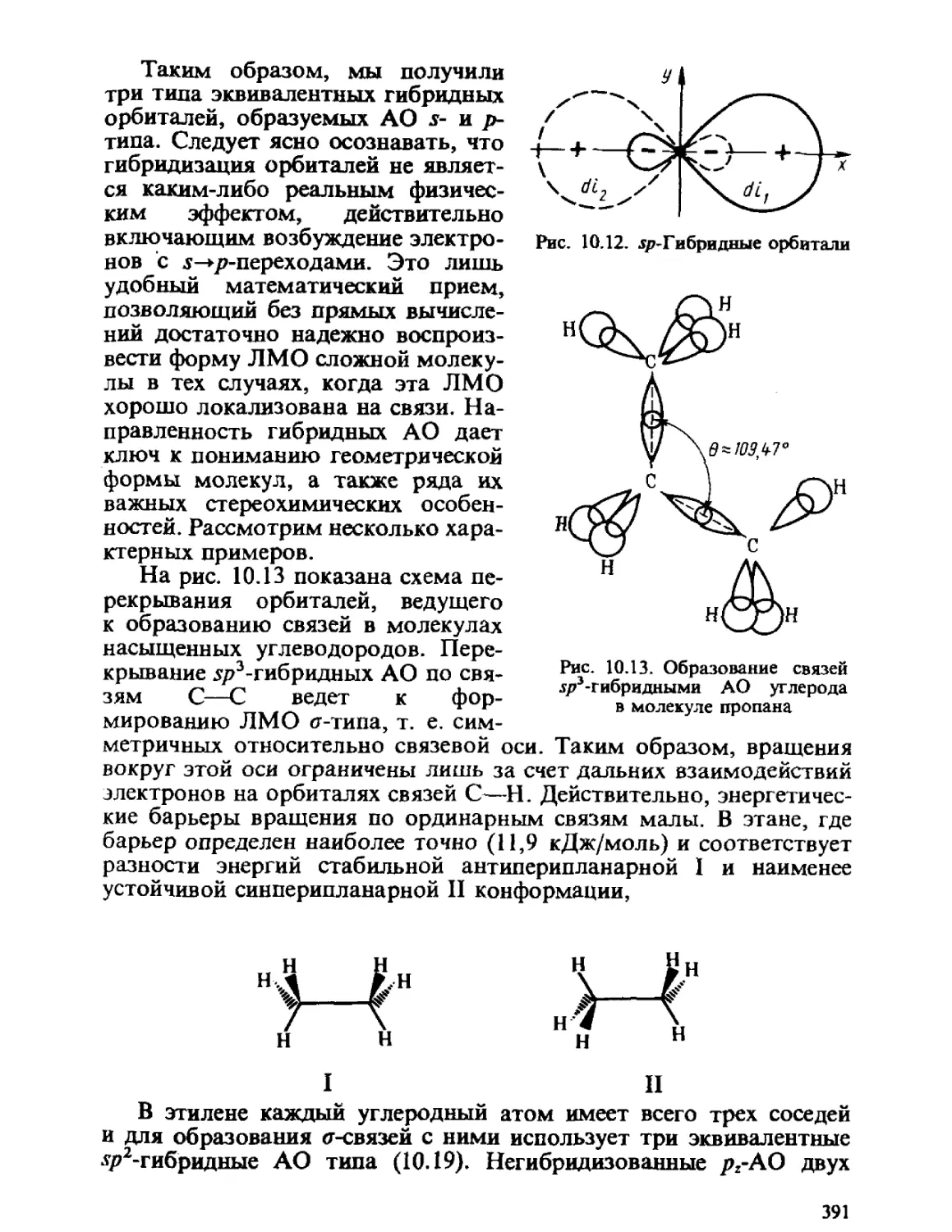
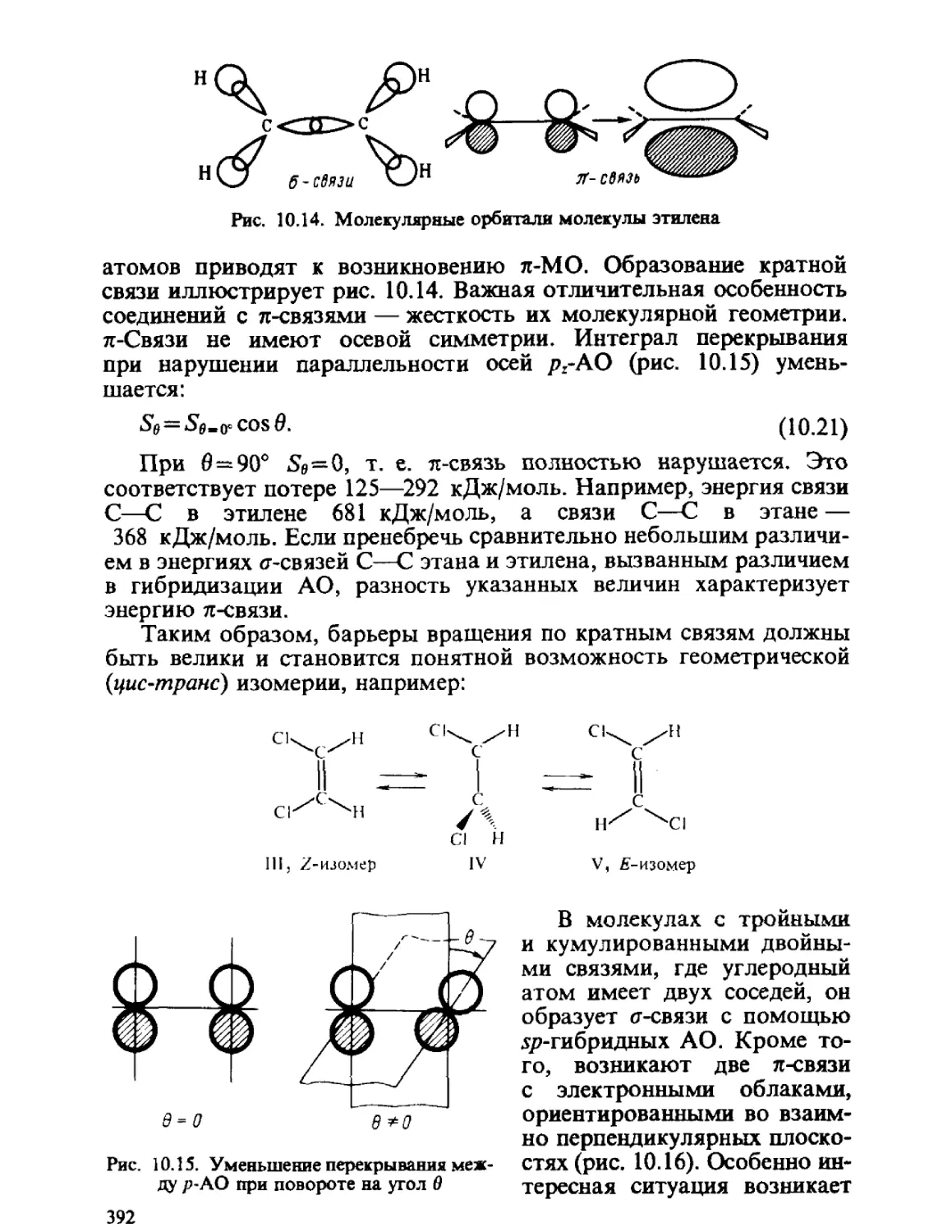
/































































































































































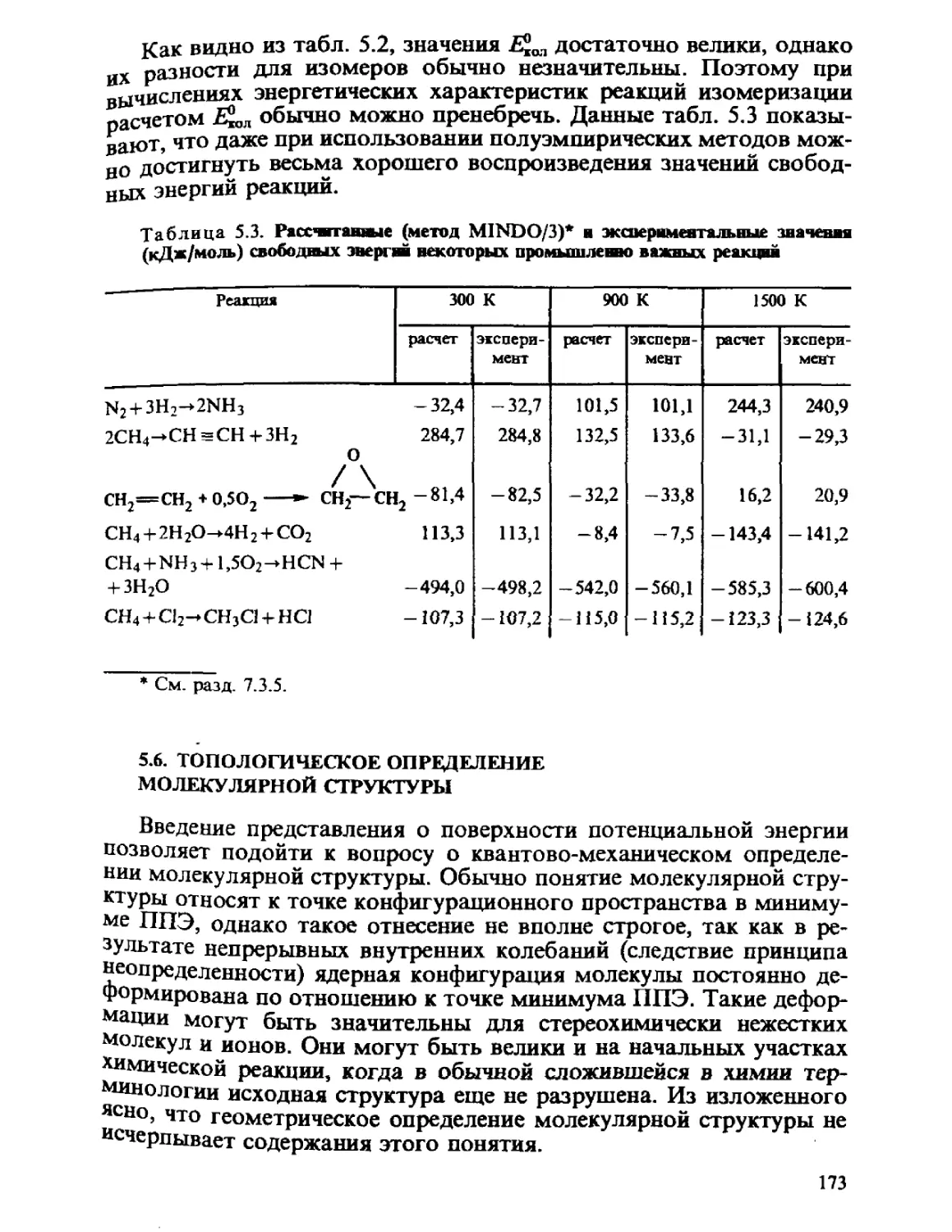





































































































































































































































































































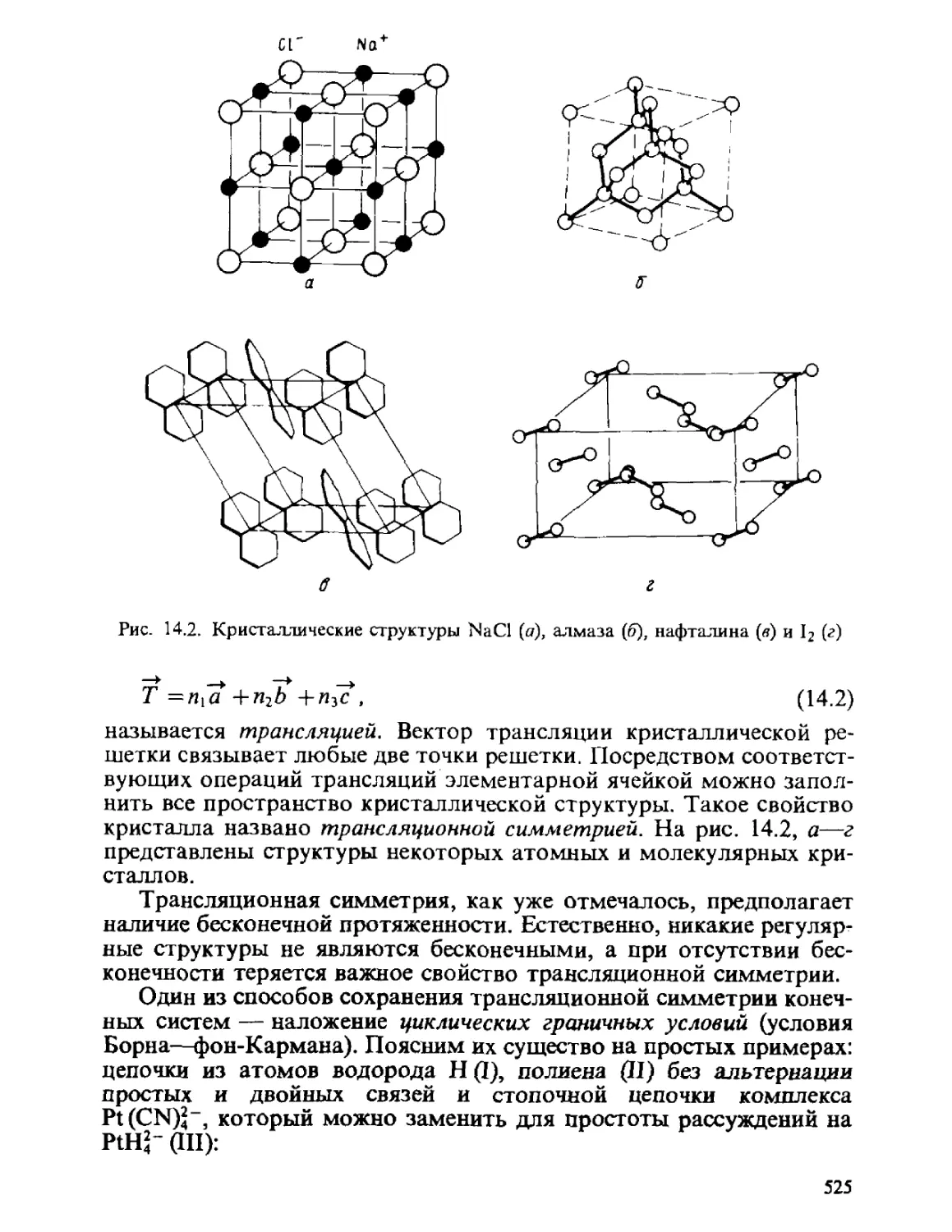
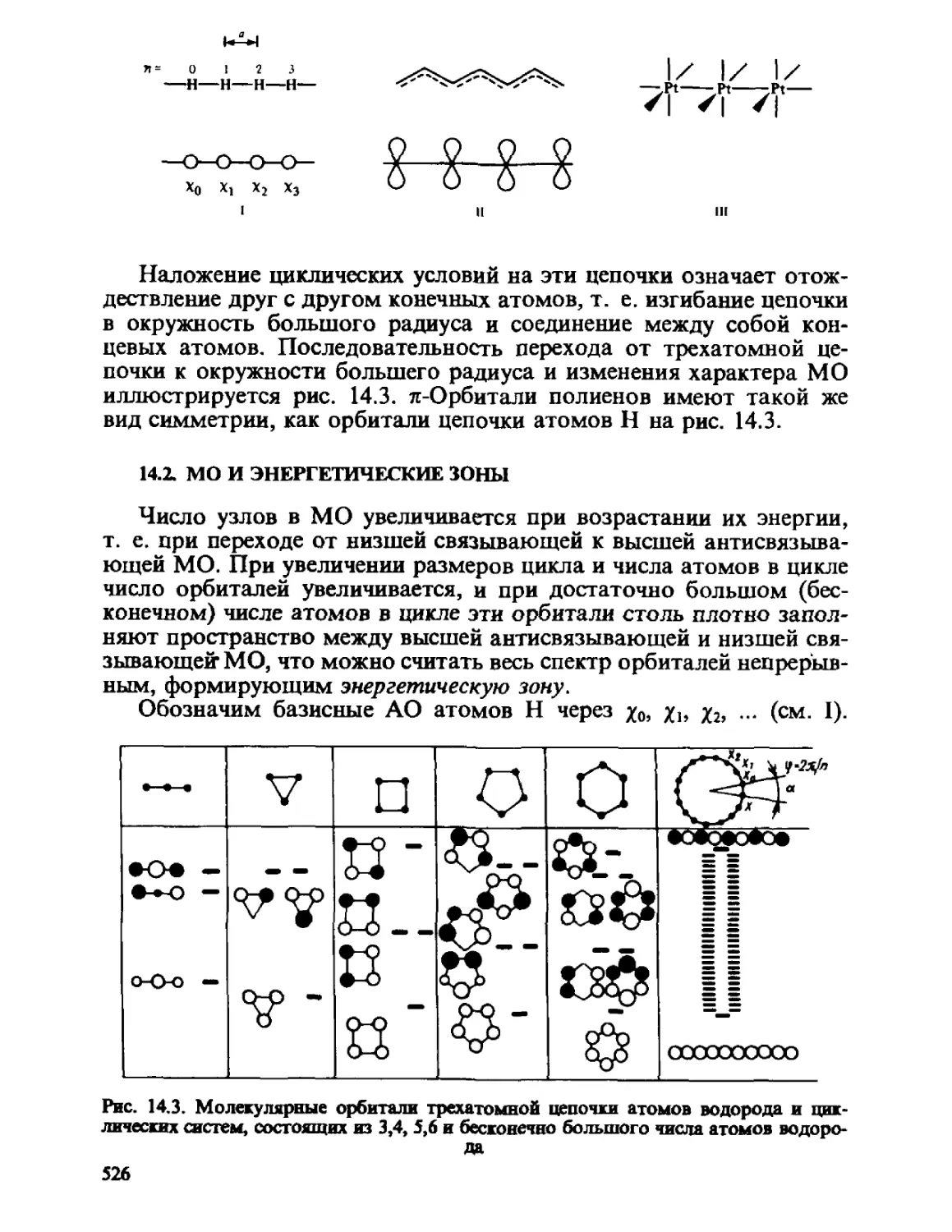


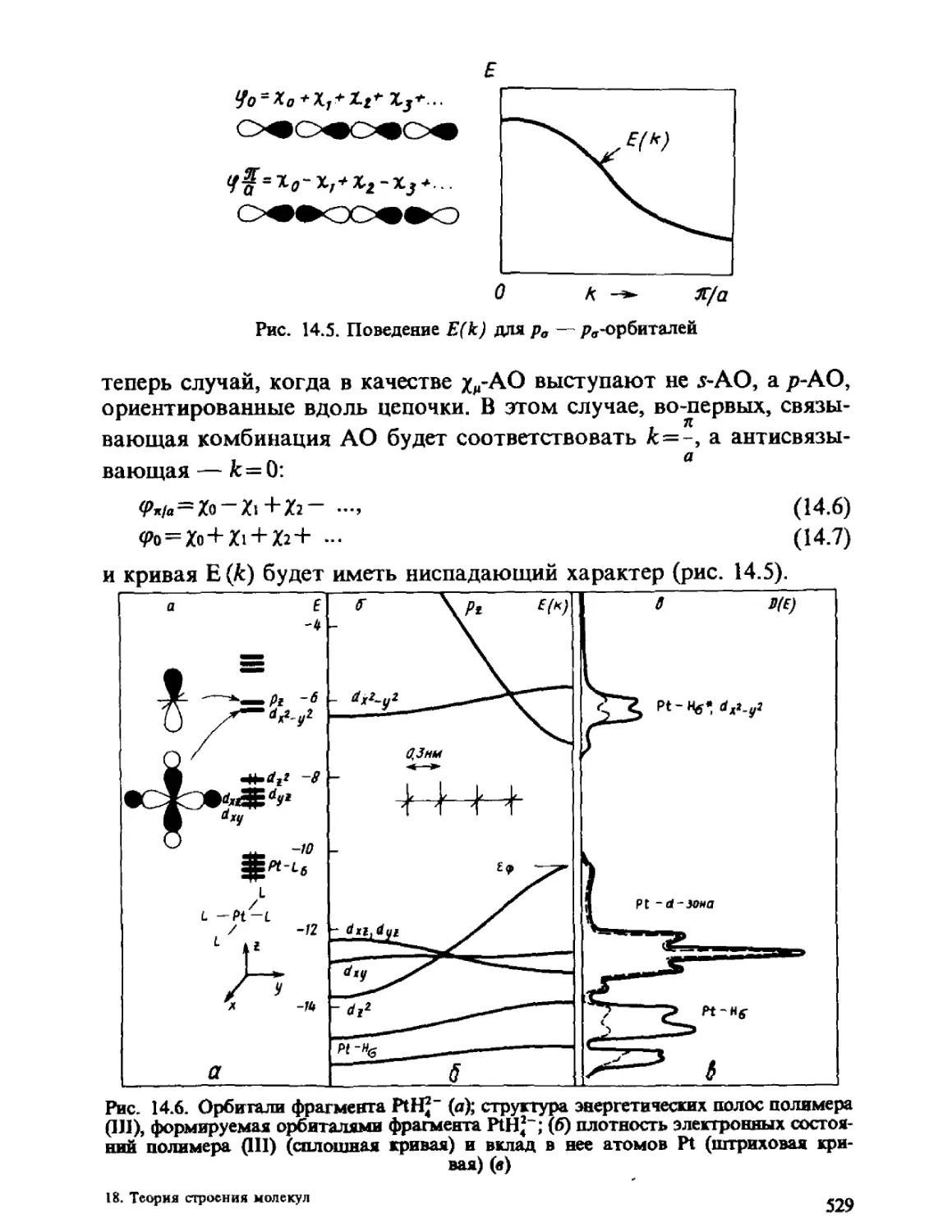















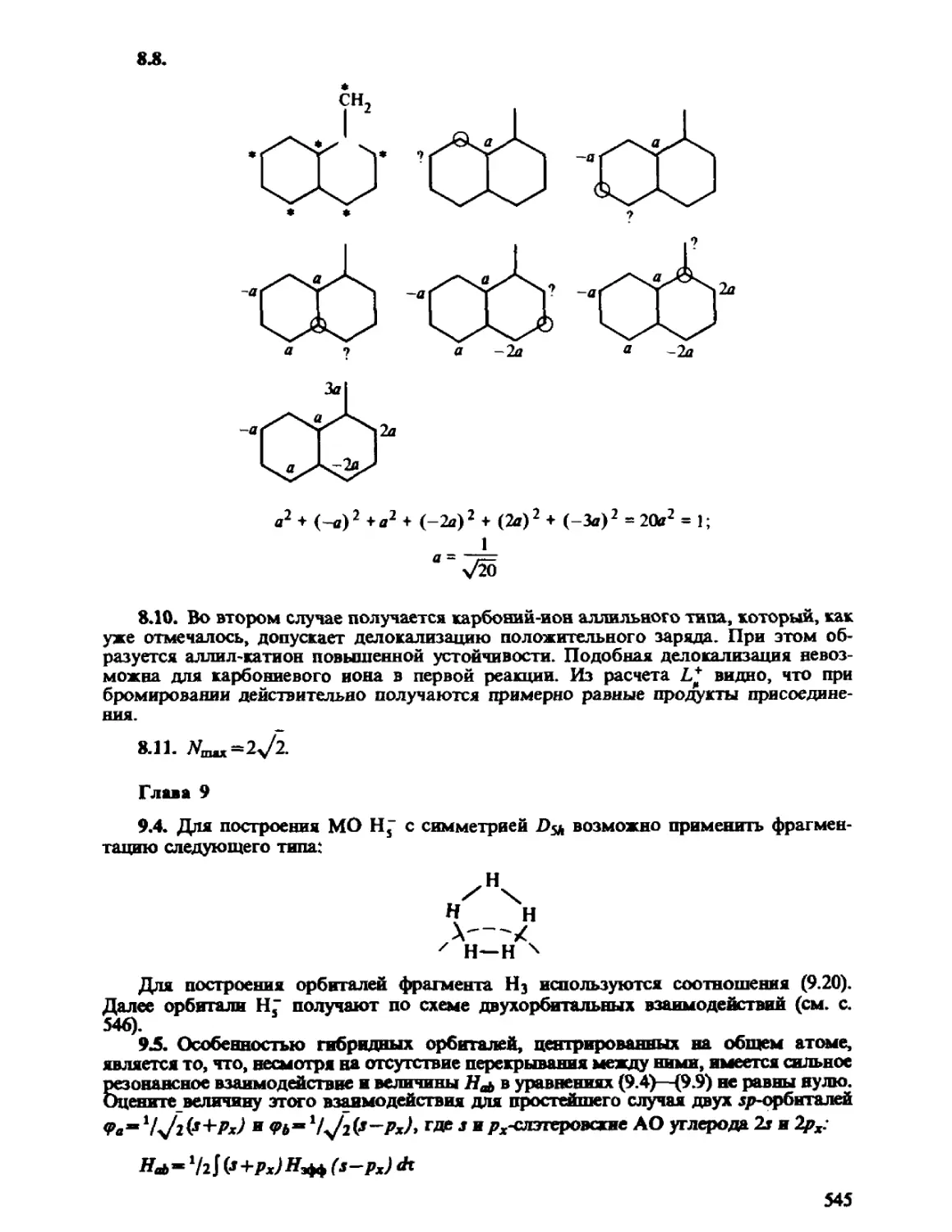
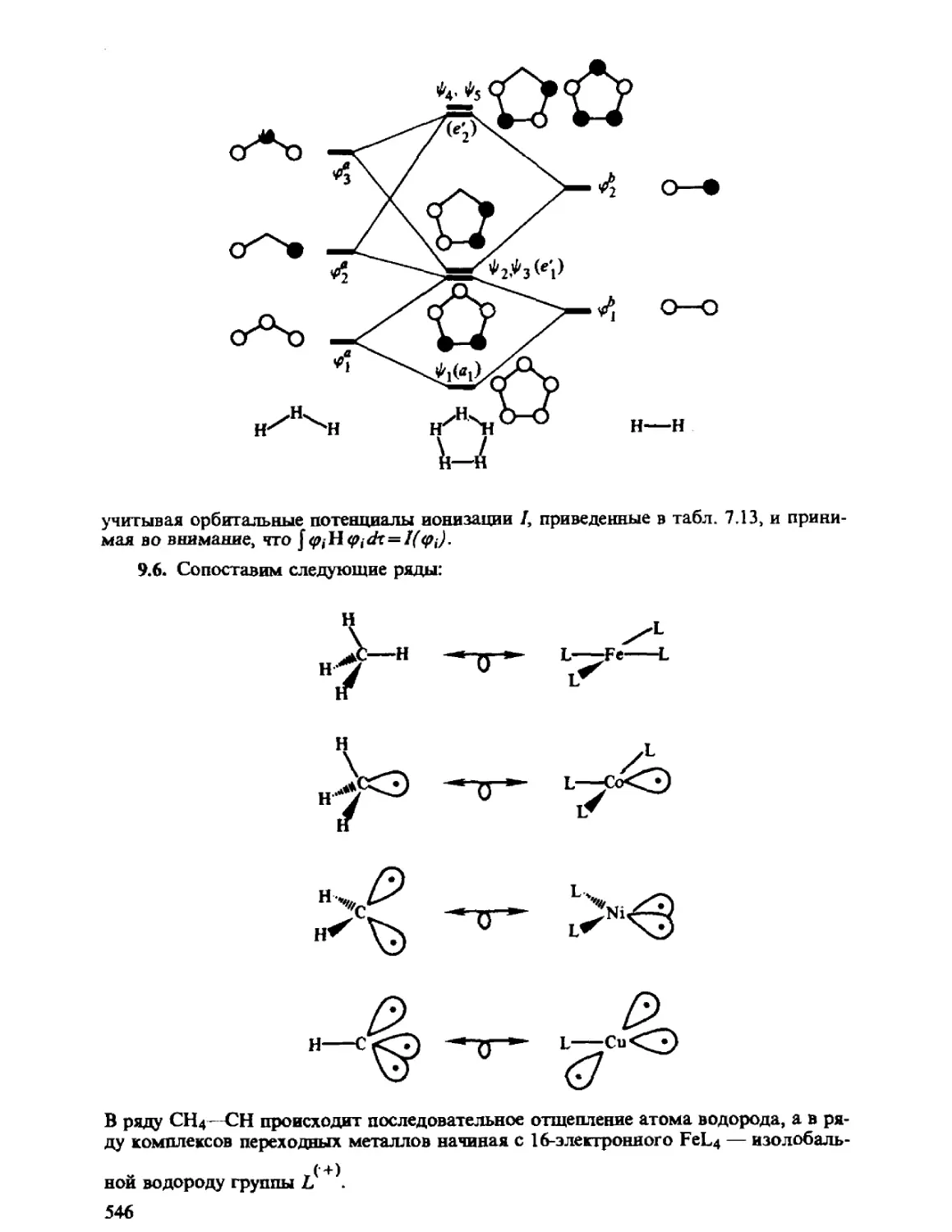

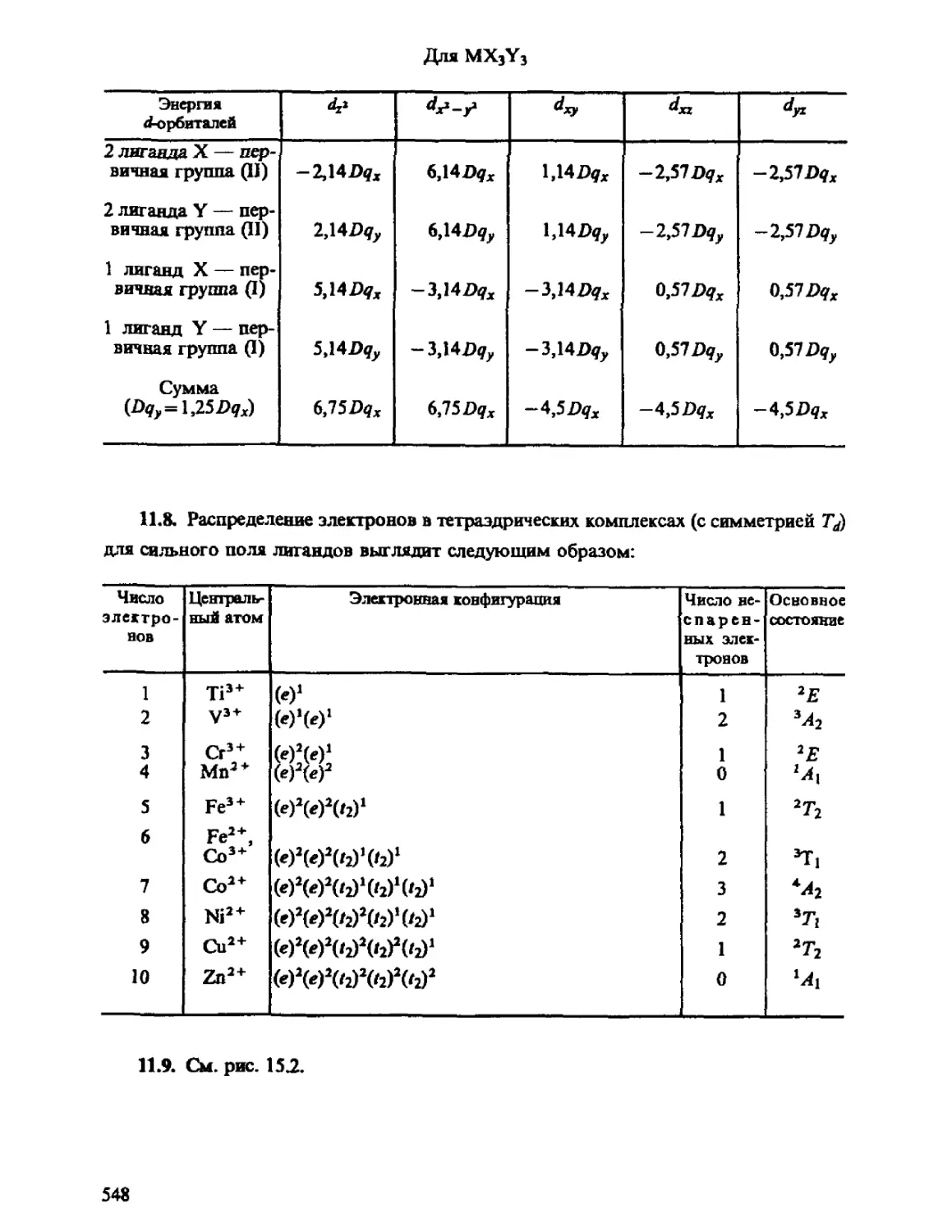

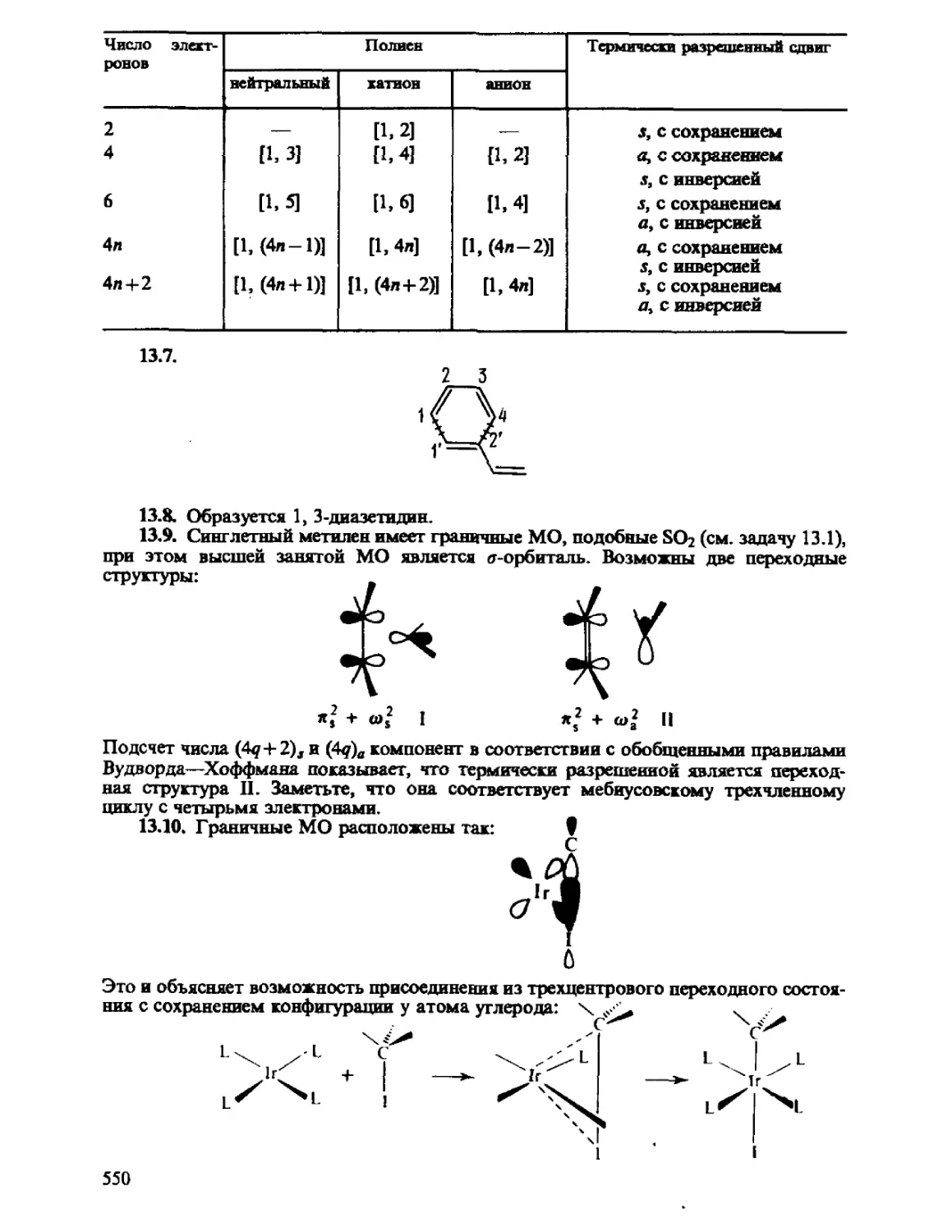








Автор: Минкин В.И. Симкин Б.Я. Миняев Р.М.
Теги: физика квантовая химия издательство феникс строение атомов серия учебники и учебные пособия
ISBN: 5-222-00106-7
Год: 1997




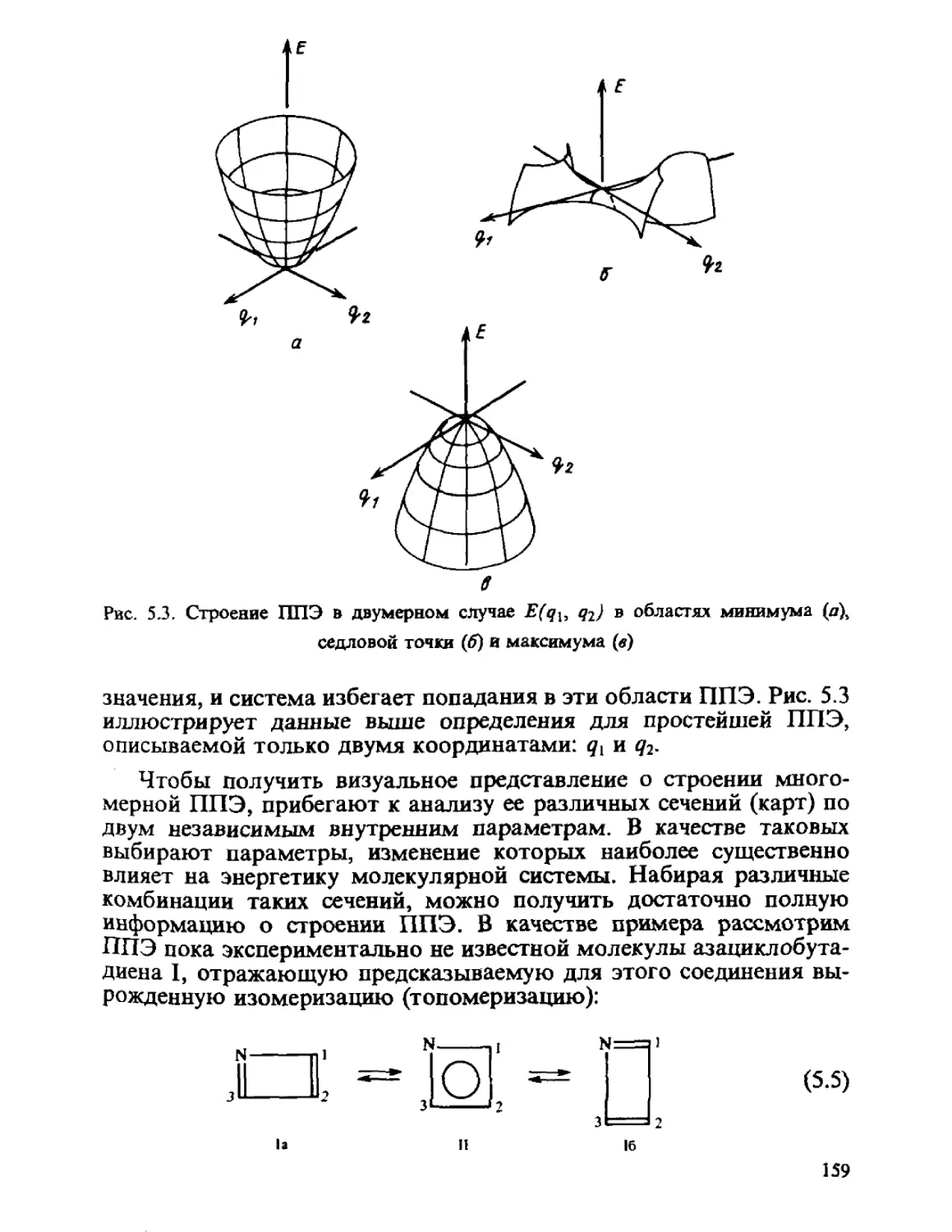
Текст
БК 22.3 Я 72
Т38
Рецензенты: кафедра квантовой химии Санкт-Петербургского го-
государственного университета (зав. кафедрой — проф. А.В.Тулуб)
и проф. М.В.Базилевский (Научно-исследовательский физико-хи-
физико-химический институт им. Л.Я.Карпова)
Минкин В.И., Симкин Б.Я., Миняев P.M.
38 Теория строения молекул./Серия «Учебники и учебные
пособия». Ростов-на-Дону: «Феникс», 1997 — 560 с.
По мере развития и углубления научных знаний меняются характер и
содержание теоретических курсов «Строение атомов и молекул», «Стро-
«Строение вещества», «Квантовая химия», меняются подходы к их преподава-
преподаванию и требования к их усвоению. Кроме овладения основами теории хи-
химической связи, общего ознакомления с формальным аппаратом и тер-
терминологией (что успешно решено имеющимися учебными пособиями)
возникает необходимость более тесного знакомства с конкретными рас-
расчетными схемами, критического понимания их реальных возможностей и
ограничений. Пособие предназначено для студентов химических факуль-
факультетов университетов, химико-технологических институтов, аспирантов и
преподавателей.
ISBN 5-222-00106-7 ББК 22.3 Я 72
В.И.Минкин, Б.Я.Симкин,
P.M.Миняев, 1997
Оформление: изд-во «Феникс», 1997
ПРЕДИСЛОВИЕ
Глубокое понимание основ теории строения атомов и молекул,
природы химической связи и движущих причин химической реакции
стало отправным пунктом при изучении практически любого физи-
физико-химического курса в университетах и химико-технологических
вузах.
По мере развития и углубления научных знаний меняются харак-
характер и содержание теоретических курсов «Строение атомов и моле-
молекул», «Строение вещества», «Квантовая химия», меняются подходы
к их преподаванию и требования к их усвоению. Кроме овладения
основами теории химической связи, общего ознакомления с фор-
формальным аппаратом и терминологией (что успешно решено име-
имеющимися учебными пособиями*) возникает необходимость более
тесного знакомства с конкретными расчетными схемами, критичес-
критического понимания их реальных возможностей и ограничений. От
студента требуется не только усвоение общих идей и принципов
теории строения атомов и молекул, но и их активное применение,
приложение к актуальным, в том числе еще не полностью решен-
решенным, задачам теоретический химии.
Все это и побудило нас к написанию данного учебного пособия
«Теория строения молекул», в котором авторы опирались на
собственный опыт преподавания в Ростовском университете. Стре-
Стремясь сделать пособие достаточно полным и независимым от других
учебников (что удобно для изучающего), мы изложили в гл. 1—4
общие вопросы теории строения атомов и молекул. Гл. 5 и 6, хотя
и основаны во многом на новом материале, также градационны для
учебников по структуре молекул и химической связи. Остальная же
часть книги не имеет аналогий, в ней дается подробный анализ
современных расчетных методов квантовой химин и их приложений
к проблемам структуры молекул и механизмов химических реакций.
Особое внимание уделено концептуальной стороне современной
теории строения и реакционной способности, развитию новых пред-
представлений и правил (сохранение орбитальной симметрии, концеп-
концепция ароматичности, правило полярности и др.).
"Карапетьшщ М. X., Драки С. Н. Строение вещества. — М.: Высшая школа,
1978; Краевое К. С. Молекулы и химическая связь. — М.: Высшая школа, 1977.
Пособие содержит достаточно большое количество задач, снаб-
снабженных ответами и подробными указаниями по их решению. Каж-
Каждая глава заканчивается списком рекомендуемой для углубленного
изучения литературы.
Второе издание книги существенно переработано и дополнено.
Описание расчетных методов квантовой химии менее детализирова-
детализировано, все рассматриваемые методы сведены в одну общую главу.
Введены новые главы, посвященные анализу поверхностей потенци-
потенциальной энергии молекул, а также описанию свойств симметрии
молекулярных систем, теории орбитальных взаимодействий, струк-
структурно нежестких молекул и молекулярного представления зонной
теории твердых тел. При переработке были учтены полезные советы
и замечения, сделанные при ознакомлении с первым изданием книги
М. В. Базилевским, И. Б. Берсукером, ?. Г. Ковалевым, Р. Хоф-
фманном. Пользуемся возможностью выразить им свою искрен-
искреннюю благодарность. Особенно признательны мы также нашим
сотрудникам М. Е. Клейкому и М. Н. Глуховцеву, которые прочи-
прочитали всю рукопись второго издания и сделали ряд ценных рекомен-
рекомендаций.
Мы надеемся, что книга окажется полезной для тех студентов
и аспирантов, специализация которых требует углубленного пони-
понимания современной теории химического строения, умения прило-
приложить ее методы на практике.
Авторы
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
— сродство к электрону атома X
до — радиус первой воровской орбиты
В — магнитная нндукщи
с — скорость света
ci(l — коэффициент в разложении МО по АО
Де — энергия диссоциации
DE — Энергия
Е
р
Е — полная энергия системы
е — заряд электрона
Ffi — индекс свободной валентности
Fp, — матричный элемент оператора Фока
g — фактор Ланде
Н — оператор Гамильтона
Щ — остовный интеграл
/х — потенциал ионизации атома X
i,j, k,l — индексы МО
J, Л — квантовые числа полного момента и его проекции для многоэлек-
многоэлектронного атома в приближении Рассела—Саундерса
J\j — кулоновскин интеграл
Ку — обменный интеграл
^б — константа Больцмана
L, Lz — квантовые числа полного орбитального момента и его проекции
для многоэлектронного атома в приближении Рассела—Саундер-
Рассела—Саундерса
L~ — энергия анионной локализации для положения /i
L* — энергия катионной локализации для положения ц
I — орбитальное квантовое число водородоподобного атома
М — масса ядра
M Ms — магнитные орбитальное и спиновое квантовые числа многоэлект-
многоэлектронного атома
"is — магнитные орбитальное и спиновое квантовые числа водородопо-
водородоподобного атома
те — масса электрона
я — главное квантовое число водородоподобного атома
Р» Pz — операторы импульса н проекции импульса
Р/№ — порядок связи ^—v
— заряд на атоме р
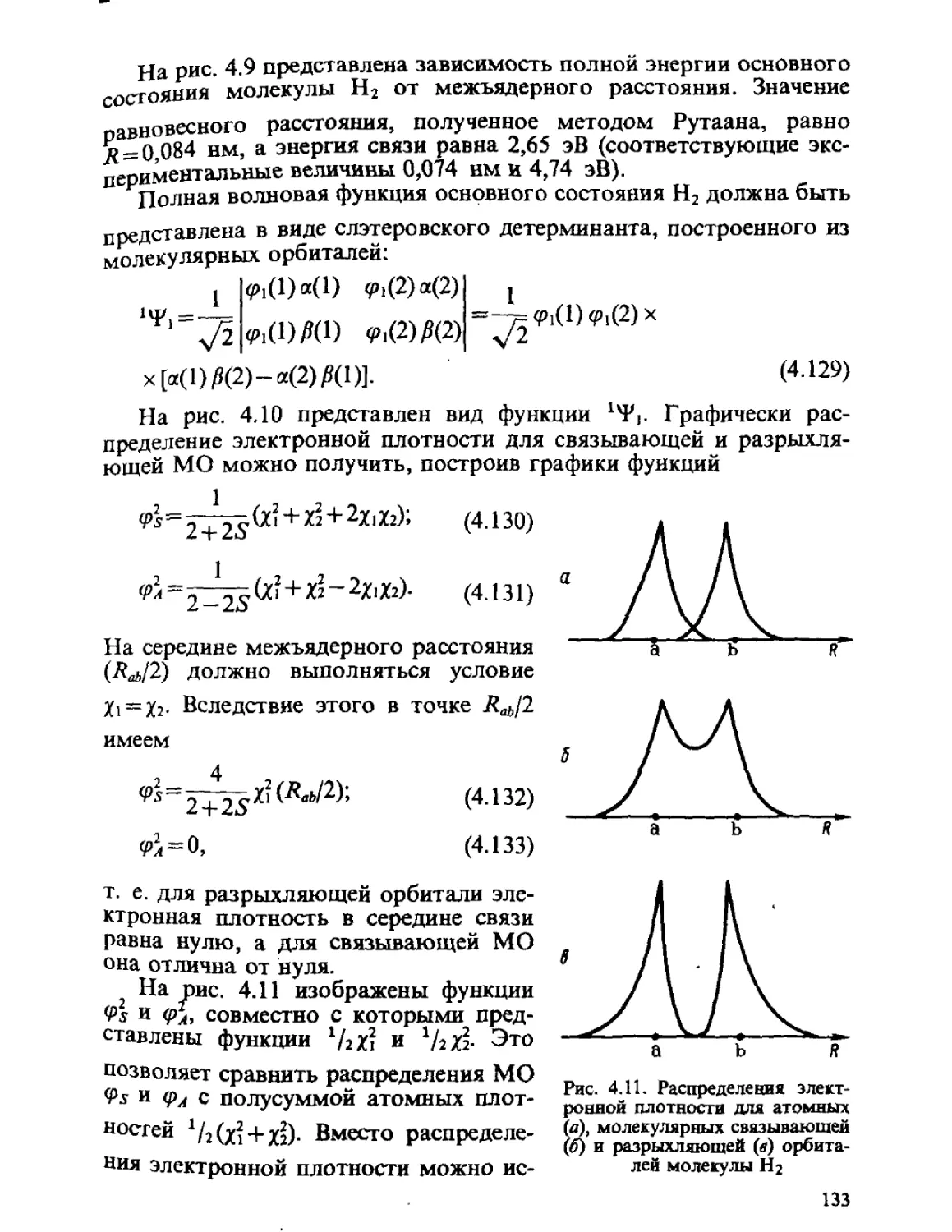
(т) — радиальная часть волновод функции водородоподобного атома
S — спиновое квантовое число многоэлеггронного атома
S/iy — интеграл перекрывания между АО ty и х?
s — оптовое квантовое число водородоподобного атома
Т — оператор кинетической энергии
V — оператор потенциальной энергии
Yim (в, (р) — угловая часть волновой функции водородоподобного атома
Z — заряд ядра
a, ft — спиновые волновые функции
ай — кулоновские интегралы в методе Хюккеля и РМХ
0М — магнетон Бора
/?р, — резонансный интеграл в полуэмпирическях методах
у — гиромагнитное отношение
Ущь ур, — кулоновские интегралы в полуэмпиряческих методах
A=V2 — оператор Лапласа
5 — символ Кронекера
?,- — энергия орбнталн
^ — орбитальная экспонента
(Л}=? — среднее значение оператора
р, v — индексы АО
{ру()м) — двухэлектронный четырехцентровый интеграл
/Г — дипольный момент молекулы *
(р{ — молекулярная орбиталь (МО)
Хц — атомная орбиталь (АО)
X5 — одноэлектронная спиновая функция
Y — полная волновая функция многоэлектронной системы
— постоянная экранирования
п* — эффективное главное квантовое^шсло
E — я-связевая энергия "^
п — ff-связевая энергия
— энергия спин-орбитального взаимодействия
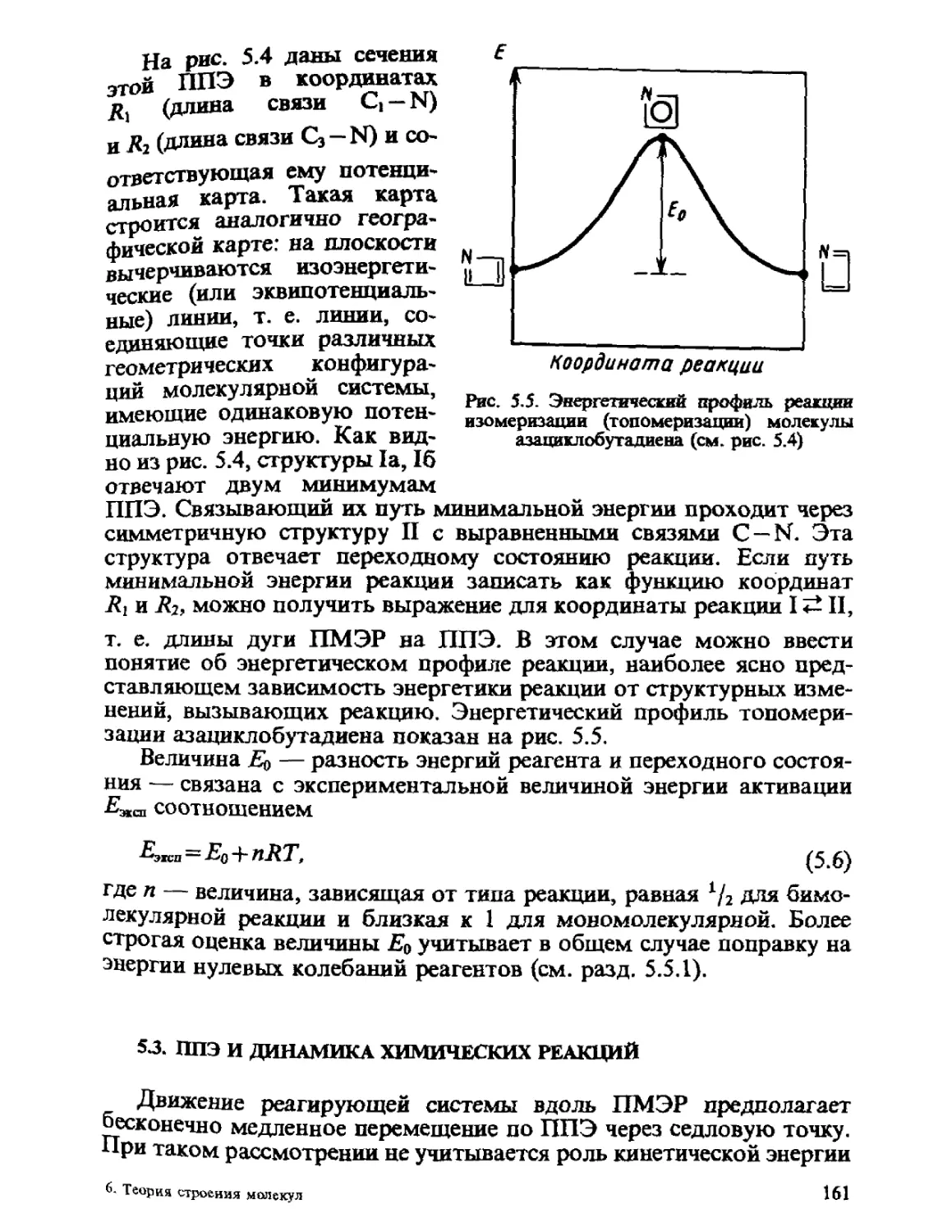
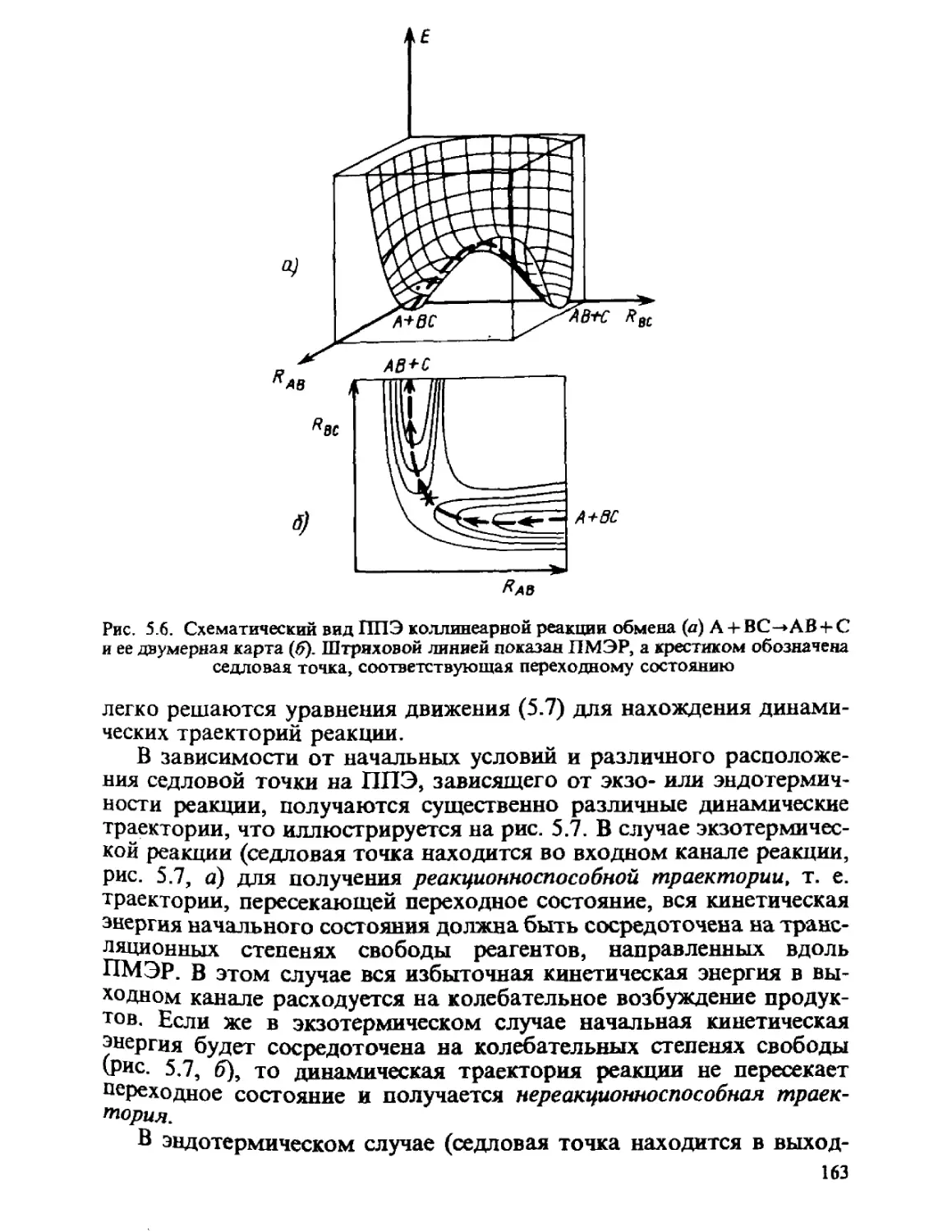
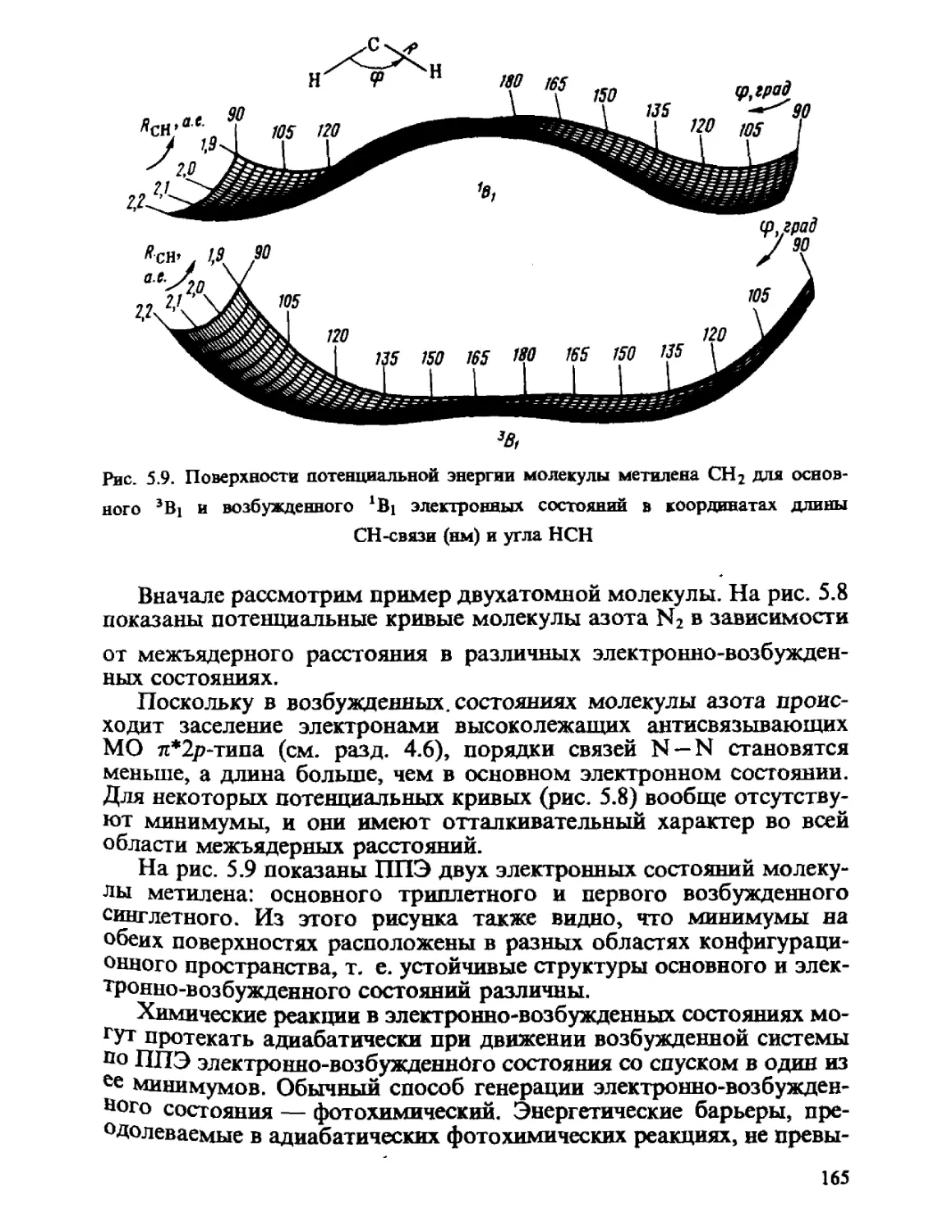
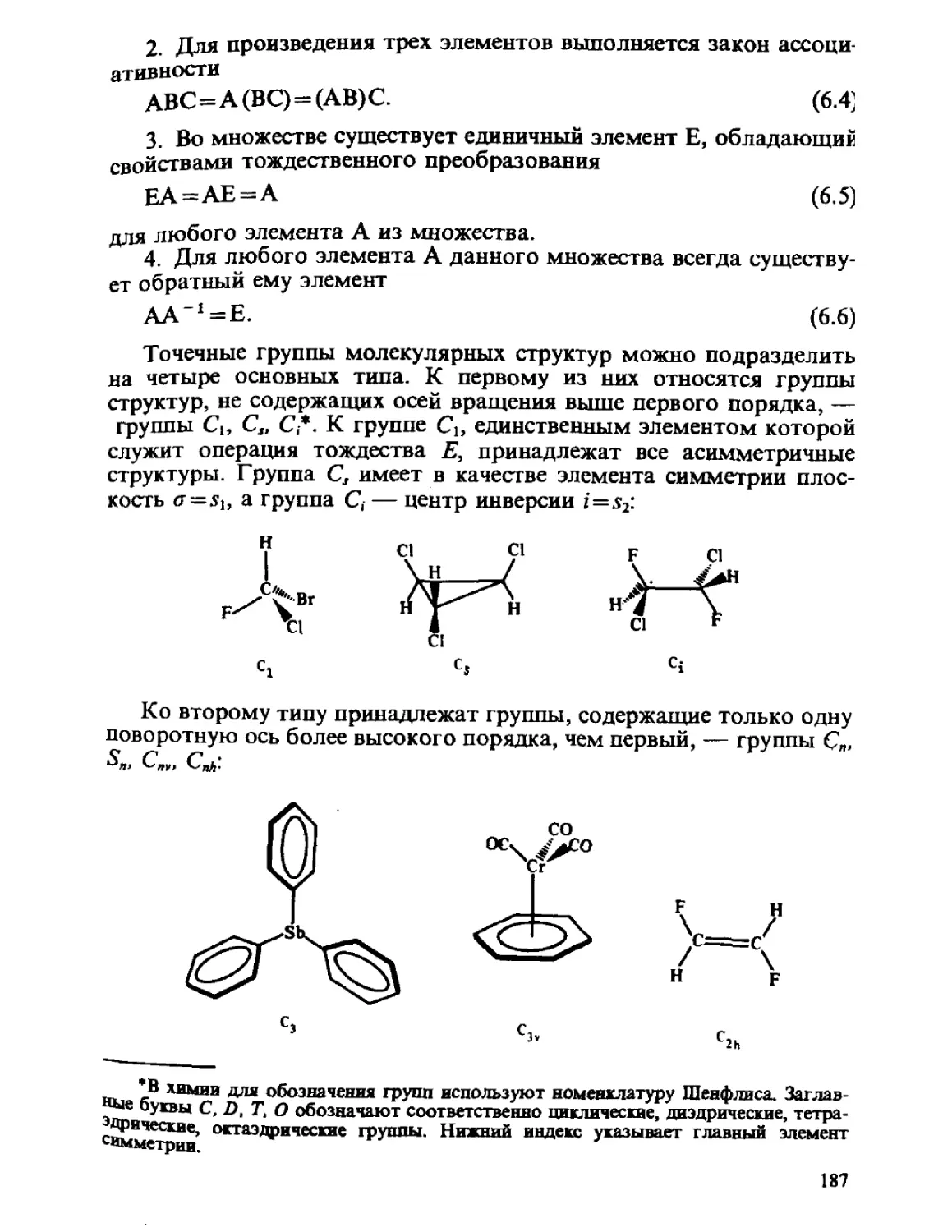
ГЛАВА 1
ОСНОВНЫЕ ПОЛОЖЕНИЯ
КВАНТОВОЙ МЕХАНИКИ
Общий принцип природы состоит в том, что свойства вещества
определяются его составом и строением. Из многих известных видов
элементарных частиц, образующих материю, химия, объектами
изучения которой являются атомы, молекулы, их ионы и радикалы,
в основном оперирует ядрами и электронами. Таким образом, самые
различные химические проявления вещества — его реакционная спо-
способность, пространственное строение молекул, наиболее важные
физические свойства атомов, молекул и их ансамблей — определя-
определяются движением ядер и электронов и физическими законами, описы-
описывающими взаимодействие ядер и электронов между собой.
Уже на рубеже XIX и XX столетий было осознано, что поведение
и свойства электронов, ядер и других микрочастиц не укладываются
в рамки стройных закономерностей хорошо сформированного уже
к тому времени раздела науки — классической механики. Экспери-
Эксперименты по изучению свойств ядер, электронов, атомов показали, что
эти частицы проявляют волновые свойства и, следовательно, свой-
свойства вещества несравненно сложнее и многообразнее. Не только
представления классической механики, но и ее язык (математичес-
(математический аппарат) оказались недостаточными для описания и осмысле-
осмысления новых результатов.
Работами М. Планка, Н. Бора, Л. де Бройля, Э. Шрёдингера
и других выдающихся ученых была создана квантовая механика —
теория движения микрочастиц, включающая в себя классическую
механику как частный случай. Квантовая теория, являющаяся осно-
основой теории строения и свойств атомов молекул, обобщила законы
движения ценой почти полного отказа от привычных классических
представлений.
1.1. ПОСТУЛАТЫ КВАНТОВОЙ МЕХАНИКИ
Вся квантовая механика строится на нескольких основных поло-
положениях, которые не вытекают из какой-либо строгой теории и не
имеют логических доказательств, а отражают огромный экспери-
7
ментальный опыт, сконцентрированный в определенной математи-
математической форме, и научную интуицию творцов этой науки.
Постулат I. Любое состояние системы полностью описывается
некоторой функцией 4Y?i, qi, ..., qnt t) от координат всех образу-
образующих систему частиц и времени, называемой функцией состояния
системы или ее волновой функцией*.
Обобщенная координата q является совокупностью пространст-
пространственных координат (в декартовой системе координат — х, у, z)
и проекции спина частицы.
Величина fV\2dt определяет вероятность нахождения системы
в элементе объема dz. Функция состояния системы должна удовлет-
удовлетворять следующим условиям: 1) однозначности, конечности и не-
непрерывности во всем пространстве переменных; 2) квадратичной
интегрируемости по всему пространству (или условию нормиров-
нормировки**):
A.1)
где ?* — функция, комплексно сопряженная с Ч1. Условие A.1)
отражает тот факт, что вероятность найти систему во всем прост-
пространстве равна единице, а
Задача 1.1. Какие из следующих функций отвечают требованиям, предъяв-
предъявляемым к функциям состояния, и в какой области изменения аргумента: е , е ,
п -дг* . -л2
хе ,sinxe ?
Постулат П. Каждой динамической переменной (координата,
импульс, энергия и т. д.) ставится в соответствие линейный само-
самосопряженный оператор. Все функциональные отношения между ве-
величинами классической механики в квантовой механике заменяются
отношениями между операторами.
Введем определение оператора. Оператор L есть закон, по кото-
которому одной функции / ставится в соответствие другая функция g.
Оператор определяет, какое действие должно быть произведено над
функцией/, чтобы перевести ее в функцию g:
g. A.2)
Оператор L называют линейным, если для любых функций
/ и/2 и любых чисел а, и а2 выполняется соотношение
¦Функцию ? часто называют волновой функцией системы вследствие того, что
она удовлетворяет уравнению Шрёдингера, имеющему аналогию с волновыми урав-
уравнениями классической механики.
¦¦Здесь и в дальнейшем отсутствие пределов интегрированвя означает, что
интегрирование ведется по всему пространству.
8
Оператор L является самосопряженным, или эрмитовым, если для
любых функций /и ? справедливо соотношение
[
где L* получается из L изменением знака перед мнимой частью.
Суммой операторов L, и L2 называют оператор, результат действия
которого равен сумме результатов действия слагаемых, т. е.
L^Lj+Lj, A.5)
если для любой функции выполняется
L/^W+L/. A.6)
Оператор L является произведением операторов Lj и 1^ слева
b^UU, A.7)
если для любой функции/выполняется условие
L/=L,(W). A.8)
Операторы L, и 1^ являются коммутирующими, т. е.
|Llf LJ=1^-1^1,1= 0, A.9)
если для любой функции/
UfLtf^UCLJ). A.10)
- - -
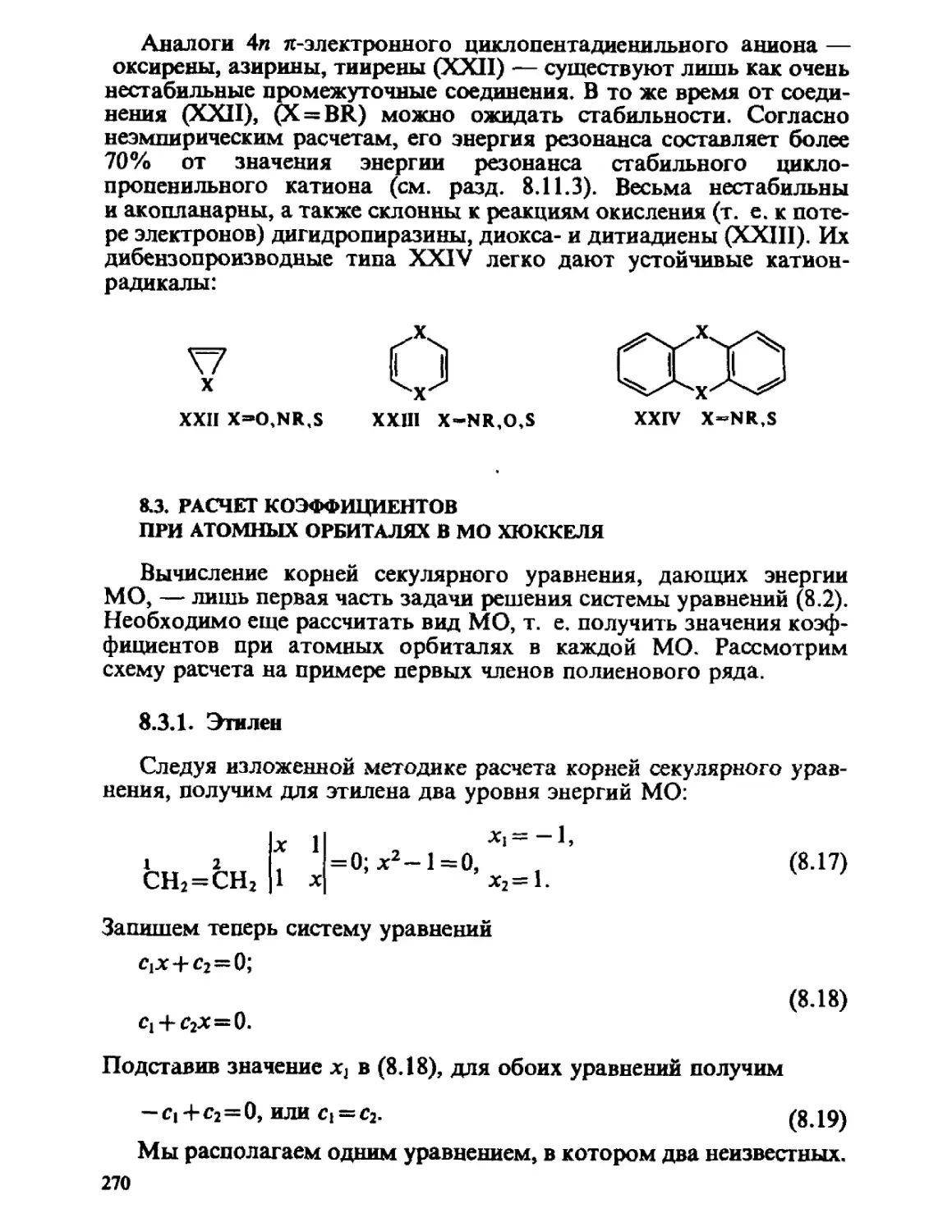
Задача 1.2. Какие из операторов линейны: а) —; 6) —; в) cosx; г) V2;
d d
Задача 1.3. Проверить самосопряженность операторов — н i—.
dy dy
Задача 1.4, Доказать» что произведение двух линейных операторов
А и В является линейным оператором.
Рассмотрим операторы основных физических величин. Подобно
тому как в классической механике свойства системы могут быть
выражены заданием координат и импульсов всех частиц, так
и в квантовой механике операторы различных физических величин
задаются через операторы координат и импульсов. Оператор коор-
координаты есть просто координата, и его действие на любую функцию
заключается в умножении ее на г*, т.е.
Tf-Tf. AU)
Оператор импульса ]? определяется через операторы его проек-
проекции, например на декартовы оси координат:
а?' <ш>
^ A-13)
Так, например,
^ A.15)
Функция от любых динамических переменных f(p,q) заменяется
на оператор f (р, q), который получается из классического выраже-
выражения этой функции заменой р и q на отвечающие имьоператоры р и q:
* <1Л6>
Например, оператор кинетической энергии электрона легко полу-
получить, заменяя в классическом выражении
=^+^+^
компоненты импульса. рх, ру ирг соответствующими операторами из
2 д2 82
A.18)
или вводя обозначение Л — оператора Лапласа:
(Оператор V (набла) был впервые введен в физику Максвеллом
и назван так по аналогии с восточным музыкальным инструментом,
имеющим сходную треугольную форму.) Выражение A.18) для
оператора Т принимает форму
ю
Т=^Д- A.20)
Полная энергия Е классической системы равна сумме кинетичес-
кинетической Т и потенциальной V энергий. Аналогично, в квантовой меха-
механике оператор полной энергии Н—? (оператор Гамильтона, или
гамильтониан системы) есть сумма операторов Т кинетической
и V потенциальной энергий:
Потенциальная энергия W—V(q, t) есть функция только коор-
координат и времени, вследствие чего оператор V выражается через
операторы координат по тем же формулам, что и потенциальная
энергия в классической механике, т. е.
V= V(qt t). A-22)
Из правил построения операторов динамических переменных
видно, что квантовая механика принципиально нуждается в клас-
классической для своего построения и обоснования.
Рассмотрим, для каких операторов квантовой механики выпол-
выполняется условие A.9), т. е. какие операторы коммутируют между
собой. Заметим, что [х, у]=0; [р*, р,]=0 и т. д.
Операторы импульса р и координаты г не являются коммутиру-
коммутирующими. Легко проверить, что для них выполняются соотношения
[р*. х]=р,х-хрж- -1*й;
to. У] = Р>У-УР>= -*'*• A.23)
[р_т, z]=pz2-zpz=;-ih;
ах 1"|
Задача 1.5. Найти коммутатор | —, е j.
Id?'
Отметим, что две физические величины могут быть одновремен-
одновременно измерены только в том случае, если их операторы коммутируют
между собой (доказательство этого утверждения см. на с. 16). От-
Отсутствие коммутации операторов риг между собой и отражает то
обстоятельство, что координата и импульс одной и той же частицы
не могут быть одновременно измерены с любой наперед заданной
степенью точности. Таким образом, соотношения A.23) являются
другой математической формой принципа неопределенности (см.
разд. 1.2).
Постулат Ш. Функция состояния должна удовлетворять урав-
уравнению
11
H (p. q, t) 4f (q, 0 = ihj4 (q, t). A.24)
Это уравнение не может быть выведено, оно постулировано
Э. Шрёдингером A926) и известно как уравнение Шрёдингера*.
В обычных задачах структурной химии и молекулярной фи-
физики, при интерпретации реакционной способности и физических
свойств молекул важны только так называемые стационарные
состояния системы, т. е. состояния, не зависящие от времени.
При их описании считается, что гамильтониан системы явно
не зависит от времени. Волновую функцию Y можно представить
тогда в виде произведения координатной *F (q) и временной
Ф (t) частей:
4(q,t)=4(q)<S>(t). A.25)
Подставляя A.25) в A.24) и разделяя переменные, получим
Левая часть уравнения A.26) не зависит от времени, а правая — от
координат, вследствие чего каждая из частей должна быть равна
константе Е, которая определяет полную энергию системы:
0-27)
^ A.28)
Выражение A.27) называют уравнением Шрёдингера для стаци-
стационарного состояния. Это линейное дифференциальное уравнение
второго порядка в частных производных эллиптического типа. Фун-
Функция Ч? (q) является собственной функцией оператора Н, а Е
— собственным значением. Из теории уравнений типа A.27) извест-
известно, что линейный самосопряженный оператор, каким и является Н,
*Эрвин Шрёдингер (Е. Schrodinger) A887—1961) — австрийский физик, один из
основателей новой эпохи в физике, связанной с созданием в 20-х гг. нашего столетия
квантовой механики. Э. Шрёдингером сформулированы основные математические
положения, относящиеся к введенной им функции ?, разработаны многие разделы
квантовой механики, в частности теория возмущений. Свои главные работы по
квантовой механике он опубликовал в 1925—1926 гг.; в 1928 г. был избран почетным
членом АН СССР; в 1929 г, ему была присуждена Нобелевская премия по физике.
Э. Шрёдингер разработал ряд разделов статистической механики, общей теории
относительности, космологии. В книге Э. Шрёдингера «Что такое жизнь с точки
зрения физика?» A945) впервые была обоснована мысль о молекулярной природе
наследственности и возможности физико-химического истолкования жизни.
12
всегда имеет полную систему собственных функций1*. Каждому
собственному значению Et соответствует собственная функция
^ifq). Бели одно собственное значение Et соответствует одновре-
одновременно нескольким собственным функциям Ч^ (»„—i+l, i+2, ...,
i+m), то состояние называется вырожденным с кратностью вырож-
вырождения, равной т. Любая линейная комбинация функций, соответст-
соответствующих вырожденному состоянию, также будет удовлетворять ура-
уравнению A.27) с тем же самым собственным числом Ех.
Задача 1.6. Показать, что если Ч^ и Т2 — две собственные функции опера-
оператора Н, соответствующие различным собственным значениям Е\ н Е%, то их
любая линейная комбинация не будет являться собственной функцией этого
оператора Н.
Функции *Р, и *Pj, относящиеся к различным собственным значе-
значениям ?j и ?/, ортогональны, т. е. выполняются соотношения
A.29)
Система собственных функций i-го вырожденного состояния не
обязательно ортогональна, однако всегда можно найти такие их
линейные комбинации, которые будут ортогональны. В даль-
дальнейшем будем считать, что система собственных функций операто-
оператора Н ортонормирована. Условие одновременной ортогональности
и нормированное™ функций ^?( (i=l, 2 ..., оо) записывается следу-
следующим образом:
I
A.30)
где Ьи — символ Кронекера, определяемый следующим образом:
[О, если 1Ф}%
1, если i=j.
¦
•Систему функций У) (/=1, 2, ..., со) называют полной, если любую функцию
00
g можно разложить в ряд по функциям/^: g= ? с$ь где
Если при этом выполняется условие If^fjdt—Stj дл
этой системы, то ее называют ортонормированной (о символе 5у см. далее).
Если при этом выполняется условие If^fjdt—Stj для любых функций f\ и fj из
13
Аналогично, для любого оператора L функции (р, и числа Lt (*=1,
2, ...), удовлетворяющие уравнению
A.31)
называют соответственно собственными функциями и собственными
числами оператора L. Если L — самосопряженный (эрмитов) опера-
оператор, то для него, так же как и для Н, справедливы все вышеприве-
вышеприведенные утверждения: система функций <р, является полной; <р, и q>j,
соответствующие различным собственным числам L, и Lh ортогона-
ортогональны.
Задача 1.7. Найти собственные функции и собственные значения оператора
h2 #
— — на [0, а]
Ъп chr
Если есть два различных оператора Li и Ьг, то собственные
функции одного оператора отличны от собственных функций друго-
другого оператора. Но имеется весьма важное исключение из этого
правила, которое приводится без доказательства: если два операто-
оператора Li и Ьг коммутируют между сдбой, т. е. [L1i,2] = 0) то можно
выбрать систему базисных функций так, чтобы они являлись
собственными функциями как Ьь так и L2. Таким образом, если
какой-либо оператор L коммутирует с Н, то система волновых
функций Y,- оператора Н будет также системой собственных функ-
функций оператора L.
Постулат IV. Единственно возможными значениями, которые
могут быть получены при измерении динамической переменной L,
являются собственные значения L операторного уравнения
V?^L%. . A.32)
Постулат V. Среднее значение физической величины X, имеющей
квантово-механический оператор X, в состоянии Ч? определяется
соотношением
к <А> = | Ч*Х?ах= <^ ЩЧГ>\ A.33)
обозначение <^F |Я| ^> введено П. Дираком.
Исходя из A.33) среднее значение полной энергии системы в со-
состоянии Y равно
JU ¦ Л. 11IUI = \ I 1ХД| X /. ll.J*tl
14
Дипольный момент системы
A.35)
где ft =?е,1^ — оператор дипольного момента системы; е(
соответственно заряд и радиус-вектор г-и частицы.
Пусть набор ортонормированных функций % (/=1, 2, ..., оо)
образует полную систему собственных функций оператора Н, т. е.
HVi=Et%. A.36)
Разложим Y в ряд по функциям этой системы:
A.37)
где с,= I^VA. Подставим A.37) в A.34) и, учитывая ортонор-
мироваыность системы, получим
?~ У У с*с (^УАЩ^у— У У clcjE&i^ У |с,|2Д. • П 38)
i-lj-l i-1 >-l i-1
Аналогично, для любого оператора L, у которого система со-
собственных функций ?, совпадает с системой собственных функций
оператора Н, т. е. ?, являются решениями уравнения
среднее значение L равно
со
A.40)
со оо со
Выражения A.38) и A.40) аналогичны определению статистического
среднего по результатам измерений физической величины при усло-
условии, что рх раз было получено значение Lx (Ех), р2 раз — значение
L% (E2) и т. д. Действительно, в этом случае
?2>? A.41)
где N — полное число измерений (N—Yj>,)f pJN — вероятность
того, что в результате отдельного измерения будет получено значе-
значение ?,. Легко показать, что для коэффициентов с, в A.37) выполняет-
выполняется соотношение
15
A.42)
t
означающее условие нормированное™ *F при разложении по ор-
тонормированному базисному набору. Тогда с учетом A.42) воз-
возможна следующая интерпретация |с,|. эта величина есть вероят-
вероятность того, что в результате отдельного измерения наблюдаемой
величины L будет получено значение Ц, отвечающее собственной
функции 4EV Если *F совпадает с одной из функций Ч?ь тогда
&=ЕЬ L=L,. A.43)
Отсюда следуют два важных вывода: 1) в квантовой механике
физическая величина имеет определенное значение в данном состоянии
? только в том случае, когда волновая функция, описывающая
состояние системы, является собственной функцией оператора, соответ-
соответствующего данной физической величине; 2) если два оператора (в
вашем случае Н и L) имеют одинаковую систему собственных функций,
то они могут одновременно иметь определенные значения, т. е. быть
одновременно измеримыми с любой наперед заданной точностью.
Задача 1.8. Вычислить средние значения следующих величин: а) г; б) г;
в) г1; г) г, если волновая функция системы имеет вид
Покажем, что если две физические величины L и М одновремен-
одновременно могут иметь определенные значения, то их операторы
L и М коммутируют. Математически утверждение, что физические
величины L я М одновременно имеют определенные значения, как
следует из ранее изложенного, выражается тем, что операторы
L и М имеют одинаковую систему собственных функций:
L?,=ZA MM^Jlf^. A.44)
Умножая слева первое из этих уравнений на оператор М, а второе
на Ь и вычитая из первого полученного уравнения второе, при этом
учитывая, что L, и Mt являются числами, которые можно перестав-
переставлять, получим
(ML-JM^^MLiMt-MiLd^i^O. A.45)
Аналогично для любой функции имеем
O, A46)
00
где Ч* разложена в ряд *F=
16
Равенство A.46), по определению A.10), выражает свойство ком-
коммутации операторов L и М:
[М, L] = (ML-LM) = 0. A-47)
Задача 1.9. Можно ли одновременно измерить с любой степенью точности
скорость и потенциальную энергию частицы, движущейся в сфервчески-симмет-
ричном потенциальном поле V(r) =*aV2 + (br2 +e ), где a, b и с — константы;
v — скорость частицы?
Постулат VI. Если система может находиться в состояниях,
описываемых волновыми функциями *?\ и ?2, то она может нахо-
находиться и в состоянии
A.48)
где С( и С2 — произвольные константы, которые при условии ор-
тонормированности *?\ и ?2 находят из соотношения (см. примеча-
примечание на с. 13)
О; — I T X • иХ.
J
A.49)
Этот постулат известен под названием принципа суперпозиции. Из
постулата V следует, что функция Ч? описывает такое состояние, при
котором система находится либо в состоянии *Fj с вероятностью,
равной С], либо в состоянии ?2 с вероятностью С\.
Постулат VH. Волновая функция системы частиц с полуцелым
спином (в частности, электронов) должна быть антисимметрична
относительно перестановки координат любых двух частиц:
Ч(Я1> 9г» -» Яи •••» Qj> •— Я^= -^(Яь Ягу —» 4j> —» Яи —> Ял)- A.50)
Антисимметрия волновой функции электронов была постулирована
В. Паули A925).
1JL СООТНОШЕНИЯ НЕОПРЕДЕЛЕННОСТЕЙ
В классической механике микрочастиц движение систем с л сте-
степенями свободы полностью характеризуется заданием п значений
импульсов и п значений координат для определенного момента
времени. При этом принимается, что все 2п значений динамических
переменных могут быть определены с любой нужной степенью
точности. Экспериментальные исследования свойств микрочастиц
(атомы, электроны, ядра и др.) показали, что точность этого опре-
определения ограничена. Действительно, пусть с помощью микроскопа
17
определено положение микрочастицы. Ясно, что неопределенность
этого измерения связана с длиной волны используемого света
Последняя может быть как угодно малой, и, следовательно, коор-
координата х в принципе определяется с любой точностью. Однако
использование очень коротковолнового света приведет к заметному
изменению импульса наблюдаемой частицы и, как следствие, к не-
неопределенности его величины
В результате неопределенности координаты и импульсы связаны
соотношением
AxApxh. A.51)
Путем аналогичных рассуждений о рассеянии частиц можно связать
неопределенности измерения энергии и времени регистрации
AEAtxh. A-52)
Таким образом, точность одновременного определения двух
канонически сопряженных величин регулируется^ принципом неоп-
неопределенности, что было впервые^ установлено В. Гейзенбергом
A927), который писал: «Никогда нельзя одновременно точно знать
оба параметра, решающим образом определяющие движение такой
мельчайшей частицы: ее место и ее скорость. Никогда нельзя одно-
одновременно знать, где она находится, как быстро и в каком направле-
направлении движется. Если ставят эксперимент, который точно показывает,
где она находится в данный момент, то движение нарушается
в такой степени, что частицу после этого нельзя даже снова найти.
И наоборот, при точном измерении скорости картина места полно-
полностью смазывается».
Другими словами, динамические переменные, характеризующие
систему, могут быть разделены на две (взаимно дополнительные)
группы: 1) пространственные координаты и время (q и /); 2) импуль-
импульсы и энергия (р и Е), причем невозможно определить одновременно
переменные из разных групп с любой желаемой степенью точности.
Это связано не с ограниченной разрешающей способностью прибо-
приборов и техники эксперимента, а отражает фундаментальный закон
природы. Его математическая формулировка дается соотношениями
A54)
18
Соотношения A.53) и A.54) называют соотношениями неопределен-
неопределенностей. Они являются одним из самых фундаментальных следствий
постулатов квантовой механики, определяя пределы применимости
классической механики.
Действие соотношений неопределенностей проявляется во всем
устройстве микромира. С его помощью легко ответить, например,
на не вполне ясный с точки зрения классической механики вопрос
о том, почему электрон в атоме не падает на притягивающее его
ядро, ведь, двигаясь по орбите, электрон должен терять энергию за
счет излучения. Действительно, если бы электрон упал на ядро, то
его положение было бы известно с точностью, соответствующей
размеру ядра, т. е. ~Ю~13 см; следовательно, Д#~10~13 см. Со-
Соответственно неопределенность импульса, вычисленная из A.53),
равна
Д/>~~= 1,0545913¦1{Г27-1013 = 1,0545913 ИГ14 г-см/с,
а неопределенность кинетической энергии электрона
V # 1 _ 1,05459132-10-54
ж~2~2Д2~10-26-2-
= 6,104-10" 2 эрг=3,811О10 эВ.
Такое значение кинетической энергии значительно превышает энер-
энергию электронов в атоме, которая, например, для атома водорода
равна 13,6 эВ. Электрон, обладающий такой энергией, покинет
атом.
Соотношения неопределенностей свидетельствуют об отсутст-
отсутствии классического детерминизма в микромире, основное положение
которого заключается в том, что «если мы точно знаем настоящее,
то сможем вычислить и будущее». Однако в этом утверждении, как
отметил В. Гейзенберг, ошибочен не вывод, а предпосылка, так как
в соответствии с соотношением неопределенности мы никогда не
сможем точно знать настоящее.
13. ВАРИАЦИОННЫЙ МЕТОД
Точное решение стационарного уравнения Шрёдингера A.27)
возможно только для простейших систем (атом водорода, молеку-
молекулярный ион водорода, гармонический осциллятор и т. д.). Боль-
Большинство задач квантовой химии и механики решается с помощью
приближенных методов. Наиболее важными подходами к получе-
получению приближенных решений являются вариационный метод и те-
теория возмущений. Вариационный метод основывается на следующей
теореме.
Теорема. Если самое низкое собственное значение гамильтониана
системы Н равно Eua^f\ — точная волновая функция этого состо-
19
яния, то для любой произвольной нормированной функции ? выполня-
выполняется соотношение
WEl. A,55)
Действительно, произвольная функция *Р может быть представ-
представлена в виде ряда ортонормированных собственных функций любого
эрмитова оператора, например Н:
A.56)
Если Ч* нормирована, то
Г
или
00 00
V/r-Vr2-! (i 571
f-1 i-l
Подставляя в A.55) вместо *F разложение A.56) и учитывая A.38),
получим
со со со со
?= X A Cj CjEjoij~ X CjCjE^ X я А- _^ v1*38/
Умножая обе части уравнения A.57) на J?j и вычитая полученное
выражение из A.58), имеем
Так как с\ всегда положительно или равно нулю и по условию
Ех является наименьшим собственным числом оператора.Н, т. е.
то ?>Ej и, следовательно,
;= Jy*
Приближенную функцию, подставляемую в A.55), называют
обычно пробной волновой функцией. Чем лучше пробная волновая
функция аппроксимирует точную, тем ближе значение энергии,
полученное с помощью этой пробной функции, к истинному значе-
значению. Для придания гибкости пробной функции в нее удобно ввести
неизвестные варьируемые параметры си съ .,., сп. Величины си с2,...,
С находят из условий
20
= 0, /=1,2, ..., п. A.60)
Задача 1.10. С помощью вариационного метода найти с в функции
-а* 1 , 1
е (в атомных единицах), Н = — V —.
2 г
1.4. ВАРИАЦИОННЫЙ МЕТОД РИТЦА
В вариационном методе Ритца пробная волновая функция берет-
берется в виде линейной комбинации независимых функций
A.61)
где с\9 с2,... — варьируемые параметры. Подставляя функцию A.61)
в выражение для энергии A.55) и считая, что ? не нормирована,
a q>i и q>j не ортогональны, получим
Е Е C*CJ I <^*
?=-LJ =-^ , A.62)
'• j * i
где Hij=$(p*H(pjdT — матричные элементы гамильтониана Н,
а ?у-= J<p* (jO/rfr — матрица интегралов перекрывания функций. Пере-
Перепишем A.62) в другом виде:
ЕЕ^я*-?ЕЕ<^А,=о. A.63)
i J i J
Условиями минимума энергии, вычисленной с помощью выражения
A.62), являются уравнения A.60). Дифференцируя A.63) по с*,
получим
яр
-Т* ZllbCjSu-EZcjStj+ZcjH^O. A.64)
oct i j j j
Из условий A.60) следует, что
OCf { .
тогда A.64) превращается в систему уравнений
21
М=О, " A.66)
j J
которую можно записать в более удобном виде:
?*УГ#<,-?^~0. 0.67)
Система однородных уравнений A.67) имеет нетривиальные ре-
решения только тогда, когда ее детерминант равен нулю, т. е.
A.68)
Это уравнение называют секулярным или вековым, из его решения
находят п корней Elt Е2,..., Е„. Наименьшее Д соответствует энергии
основного состояния, остальные корни представляют собой значе-
значения энергии возбужденных состояний.
Для нахождения волновой функции основного состояния необ-
необходимо наименьший корень уравнения A.68) подставить в систему
уравнений A.67) и найти коэффициенты ct. Таким способом можно
найти и волновые функции возбужденных состояний. Следует по-
помнить, однако, что в общем случае вариационная теорема и, как
следствие, вариационный принцип позволяют корректно опреде-
определить только низшее энергетическое состояние. Кроме того, укажем,
что волновая функция, оптимальная для энергии, не обязательно
оптимальна для расчета других свойствjjsaHTOBO-механической си-
системы.
1.5. ТЕОРИЯ ВОЗМУЩЕНИЙ
Другим важнейшим приближенным методом решения уравнения
Шредингера является теория возмущений. В ее основе лежит идея
нахождения волновых функций и энергетических уравнений исследу-
исследуемой сложной системы с гамильтонианом Н исходя из соответству-
соответствующих данных, известных для более простой системы (систем) с опе-
оператором Гамильтона JT . В этом случае необходимо представить
оператор Н в виде
Н=Н@)+ЛН'>
где Я — параметр; ЯН' — так называемое возмущение оператора
H*°\ которое должно быть по отношению к нему достаточно ма-
малым. Оператор Н* выбирается таким образом, чтобы для него
были известны ряды его собственных значений ?^ и собственные
функции Ч^, т. е. решена задача
22
Уравнение Шрёдингера A.27) для искомой системы с операто-
оператором A.69) запишется в виде
^*.. A.71)
Будем считать, что в ряду Д, (п= 1, 2,...) нет одинаковых значений,
т. е. все Е„ невырождены. Так как ?„ и Е„ являются функциями Я, то
можно предположить справедливость их разложения в ряды вида
A.73)
где Е ит — поправки т-то порядка соответственно к энергии
и волновой функции. Подставляя A.72) и A.73) в A.71), получим
+ к
... . 0-74)
Чтобы уравнение A.74) удовлетворялось при различных Я, коэф-
коэффициенты при X в одной степени по обе стороны уравнения должны
быть равны:
A.75)
A-76)
(Н<0) - ?<0>) ^?J = Щ> У® + ?(') ?<0 _ H'^i1J. (I -77)
Систему уравнений A.75) — A.77) называют системой рекур-
рекуррентных формул теории возмущений Рэлея-—Шрёдингера, так как
аналогичные уравнения возникают при использовании введенного
еще Рэлеем метода расчета колебаний струны.
Уравнение A.75) не что иное, как уравнение A.70), решения
которого известны. Для решения уравнения A.76) воспользуемся
представлением функции Ч^ в виде разложения в ряд по невоз-
невозмущенным ортонормированным функциям
A.78)
Подставляя A.78) в A.76), умножая слева обе части полученного
уравнения на Ч*0^ и интегрируя, имеем
О=4° - J ЧфнчР их. A -79)
Таким образом мы получили энергию возмущения первого по-
порядка:
23
Аналогичной подстановкой и умножением на *Р?}* можно опреде-
определить коэффициенты ст (тФп):
где
jC-JVfflTffdT. С1-82)
Коэффициент ся легко получить из условия нормировки 4V Это
условие дает ся=0, и, следовательно, функция Ч1?' имеет вид
?™ ч«о) A.83)
Соотношение A.83) иллюстрирует условия применимости те-
теории возмущений
т. е. матричные элементы оператора Н должны быть малы по
сравнению с разностями невозмущенных уровней энергии.
Разлагая в уравнении A.77) функцию^К?1 и поступая аналогично
A.79) — A.81), можно найти поправыНйгорого порядка к энергии
и волновой функции. С учетом поправок первого и второго поряд-
порядков Ея и ?я примут такой вид:
2
.... A.85)
,12 V Г V НктНтп НьнНкп 1 ^/ц
На практике почти всегда выбирают параметр Я= 1, т. е.
A-87)
, A.88)
... . A.89)
Использование теории возмущений особенно эффективно при
решении качественных задач, когда требуется определить, напри-
например, как скажется геометрическая деформация или замена одного
структурного фрагмента молекулы другим на энергетических уров-
24
нях и волновых функциях молекулы. Кроме того, при помощи
теории возмущений принципиально возможно изучать процессы,
зависящие от времени, в отличие от вариационного подхода, приме-
применимого только для стационарных состояний.
При использовании теории возмущений ценным оказывается
применение теории групп (см. гл. 6). Анализ симметрии позволяет
отобрать равные нулю интегралы. Например, таким способом мож-
можно установить, равна ли нулю поправка первого порядка к энергии
и какие коэффициенты в разложении первого порядка для волновой
функции или в разложении второго порядка для энергии оказыва-
оказываются равными нулю. Подобные данные фактически составляют
основу подхода Бэйдера—Пирсона (см. разд. 5.7) или эффекта
Яна—Геллера второго порядка, определяющего форму симметрич-
симметричных молекул.
ГЛАВА 2
ОДНОЭЛЕКТРОННЫЕ АТОМЫ
Хотя из всех атомов периодической системы только водород
и его изотопы относятся к одноэлектронным атомам, квантово-
механическое рассмотрение систем этого типа имеет фундаменталь-
фундаментальное значение. Это объясняется тем, что для атомов и ионов с одним
электроном (так называемых водородоподобных атомов) может
быть точно решено уравнение Шредингера, а полученные решения
служат основой для изучения всех более сложных задач о многоэле-
многоэлектронных атомах и даже молекулах.
2.1. РЕШЕНИЕ УРАВНЕНИЯ ШРЁДИНГЕРА
ДЛЯ АТОМА ВОДОРОДА
Потенциальная энергия V (г) одноэлектронного атома является
энергией кулоновского взаимодействия ядра с зарядом Ze и элект-
электрона (заряд -е):
Будем рассматривать движение электрона вокруг ядра, учитывая
при этом, что ядро несколько смещается относительно центра масс
системы. Тогда в оператор кинетической энергии следует включить
приведенную массу Ма. Без учета спинового момента электрона
гамильтониан водородоподобного атома приобретает вид
V2+V(r).
B.2)
25
Если использовать вместо приве-
приведенной массы массу электрона т„
то погрешность составит всего
0,05%, поэтому в дальнейшем за-
е л меним Мл на т*.
/ I В связи с тем, что кулоновский
'—I *-у потенциал сферически-симметри-
! чен (потенциал центральных сил),
т. е. зависит только от расстояния
между взаимодействующими ча-
Рис. 2Л. Wmiocn, декартовы* спщами, задачу целесообразно ре-
и сферических координат шать в сферических координатах,
связь которых с декартовыми ко-
координатами ясна из рис. 2.1. Соотношения между сферическими
и декартовыми координатами имеют следующий вид:
x~rsin$cos(p; 0<г<оо;
v=rsin#sina>; О^0^я; »
B.3)
z=rcos9; 0^<p^2n;
dv—dxdydz—г2 sin в dOdcpdr,
где dv — элемент объема.
Переход к сферическим координатам создает возможность раз-
разделения переменных в уравнении Шредингера, чего нельзя сделать
при записи этого уравнения в декартовых координатах.
В сферических координатах оператор Лапласа A.19) принимает
вид
„, 1 д ( ,д\ 1 д /. лд\ 1 д2
v = {* ?{в
Задача 2.1. Получить выражение оператора Лапласа A.19) в сферических
координатах.
Заменив в B.2) М„ на те и подставив B.2) и B.4) в уравнение
Шредингера A.27), получим
г*дг\ дг^г^втвдву дв J i2 sin2 в д<р2
Дифференциальное уравнение в частных производных B.5) можно
решить с помощью разделения переменных:
26
У (г, О, q>)=R (r)B (в) Ф (9). B.6)
г2
Подставляя B.6) в B.5) и умножая на , получаем
д ( лд&\ 1
50 у дв) Ф$т
дгФ
Sin а об V 00 / Ф Sin 0 0<р
Левая часть равенства B.7) зависит только от переменной г, а пра-
правая — от переменных в и (р. Но обе части, зависящие от разных
переменных, могут быть равны друг другу только в том случае,
если значения этих частей равны некоторому постоянному числу С.
Таким образом, из B.7) для Я получается уравнение
= 0. B.8)
Аналогично можно разделить переменные в и (р в правой части
уравнения B.7), приведя его к виду
1 д ( . де\ 1 1 ?2Ф „
дв I sin. в Ф д(р
Фд<р2"
Правая часть уравнения B.9) зависит только от ф, а левая — от 0;
следовательно, каждая из них равна постоянной, которую обозна-
обозначим т2. Выбор положительного числа для константы диктуется
тем, что функция Ф отвечает физическим требованиям только тогда,
когда константа положительна.
Легко получить два уравнения
B.10)
+ Csin2e=m2,
( лдв\
\ дв)
которые можно переписать в виде
Ф=0; B.11)
27
l d / ae
im~0S\ ~дв
sin
Итак, мы разделили переменные в я (p.
2,1.1. Решение Ф-уравнення
Решением уравнения B.11), как в этом легко убедиться прямой
подстановкой, будет функция
Ф=Ае±ыГ B.13)
Из условия однозначности волновой функции следует
ф(ф=0)=Ф(<р в 2я) B.14)
или
А—Ае ;е —L U-^J
Используя формулу Эйлера для комплексных чисел, получим
выражение B.15) в виде
cosB7cw)±ismBjtm) = l. * B.16)
Это равенство возможно лишь при условии
т=0, ±1, ±2,... . B.17)
Таким образом, т может принимать только целочисленные зна-
значения. Константа А находится из условия нормированности фу-
функции Ф:
2* 2х
= 1- B.18)
Окончательно функция Ф имеет вид
. 1 +*м
B.19)
|ф*ф^=^2 \
2.1.2. Решение в-уравнеаня. Полиномы Лежаыдра
Уравнение B.12) хорошо известно в теории дифференциальных
уравнений. Оно имеет конечное решение только в случае выполне-
выполнения условий
С=1A+1), /=0, 1, 2, .., B.20)
B.21)
28
при этом решениями являются так называемые функции или поли-
полиномы Лежандра. Нормированные функции 0 имеют вид
функции Р^ (cos в) называют присоединенными полиномами Лежа-
Лежандра и определяют следующим образом:
0)=—[l-(cos0J]w/2
2'/!
(dcosd)
i+м
(cos2 0-1I. B.23)
Присоединенные полиномы Лежандра B.23) связаны с полиномами
Лежандра B.24)
1 d'
P,(cos 0)=-^ - (cos2 0-1)'
'л (dcosQI
B.24)
соотношением
Py*(cos0)=(l-cos20)w/2
JP?(cos0)=P/(cose).
9)
W
B.25)
Задача 2.2. Получить четыре первых присоединенных полинома Лежандра,
воспользовавшись уравнением B.23).
В табл. 2.1 представлен вид функций Qi,m(B) для некоторых
значений /.
Таблица 2.1. Вед фуякщш ®i,m@)
0
1
Функция
~2
1 /-
~~2
~2
1
2
Функция
©2,0=-
4
©2. ±1 =
©2, ±2 =
2
4
29
Задача 2.3. Присоединенные полиномы Лежандра являются ортогональ-
ортогональными функциями, т. е.
Го, если 1ФГ,
, если 1=1.
!
Проверьте ортогональность трех первых полиномов Лежандра (см. табл. 2.1).
Произведение функций 0@) и Ф(ф) представляет собой угловую
часть волновой функции
B.26)
Функции Yfo называются шаровыми функциями или сферическими
гармониками. Подставляя в B.26) выражения для ЩВ) и ЩВ) из
B.22) и B.19), запишем угловую часть в общем виде:
cos б) Г; О»)
/_л i j 'ж——/ —/-1-1 П I 7 n
2.1.3. Решение Л-уравнения. Полиномы Лягерра
Перепишем уравнение B.8) в другом виде, подставив вместо
С соотношение B.20):
d*R 2dR V2E TZ 1A+1)
dr2+r dr+l
где
Я=0, B.28)
^o=—г B.29)
Это уравнение подробно исследовано в математической физике,
и его решение необходимо искать в виде ряда
B.30)
где введено обозначение
>Е<0-
Подставляя B.30) и B.31) в B.28), получим
30
bO+D^+t (J+l) (Hi-l)bjrJ+l-2+
B-32)
Выражение в квадратных скобках должно обращаться в нуль при
всех значениях г. Это возможно только в случае равенства нулю
суммы коэффициентов при одинаковых степенях г. Собирая коэф-
коэффициенты при J* , получим рекуррентное соотношение
Функция B.30) должна быть конечной для любых г, т. е. ряд
bj r должен сходиться- Сравним этот ряд с хорошо известным
разложением функции е *"\
е =1+2^+...+ -1Е^г+-^г+.... B.34)
Отношение двух соседних членов этого ряда при больших j равно
B5)
Отношение двух соседних членов ряда B.30) при больших j также
равно
oo .
2цг
Таким образом, ряд ??,У близок к функции e2ftr, что позволяет
записать функцию B.30) в виде
l*' = riJ'. B-37)
При г-+со функция B.37) стремится к бесконечности по экспоненци-
экспоненциальному закону. Для того чтобы удовлетворить условию конеч-
конечности волновой функции яри любых г, необходимо оборвать ряд,
т. е. для некоторого j должно выполняться условие bj+2=0 или
1,У=0, 1, 2, ... . B.38)
31
Обозначив —
7+/+1=л, л = 1,2, ... , B.39)
получим связь между / и и:
/<л-1, где /=0, 1, 2,..., л~1. B.40)
Подставляя ц из B.31) в B.38), получим выражение для полной
энергии атома водорода
которое полностью совпадает с формулой Бора.
Величину л называют главным квантовым числом, так как она,
согласно B.41), определяет энергию водородоподобного атома.
С учетом нормировки функция B.30), которую называют ради-
радиальной частью волновой функции, записывается следующим образом:
1 Zr
(iZr) B42)
Функция m^i (г) представляет собой так называемый присоединен-
присоединенный полином Лягерра, который связан с полиномом Лягерра
f) следующим дифференциальным соотношением (формула
Родрига):
dk
—L,(r), B.43)
drk
где
Ц,(г)=е' — (re~r). B.44)
dr"
Полиномы Лягерра с различными ли/ ортогональны между собой,
что определяет ортогональность радиальных функций.
Получим вид функции Яа(г) для п — 1 и /=0:
=_2 (*
32
Задача 2.4. Получить вид функций Rni(r) для и=2,3 и показать их ортонор-
мированность.
В табл. 2.2 приведены функции R^fr) для различных ли/.
Таблица 2.2. Нормированные функции
0
Zr
2у/гW
Zr
_y/2/ 2Zr t
W V 3
8
27^6
<»o
/
Z2, АТОМНЫЕ ОРБИТАЛИ
Полная волновая функция водородоподобного атома зависит от
трех квантовых чисел: п, 1шт. Целочисленные значения и взаимо-
взаимосвязь их обусловлены требованиями конечности и непрерывности
волновой функции:
bbn(Q)Q>m(<p)=Rml(r)Yim(Q,<p). B.46)
Теория строения ыолекул "
Таким образом, появление дискретных квантовых чисел автомати-
автоматически следует из математических условий, налагаемых на волновую
функцию.
По предложению Малликена волновую функцию B.46), соответ-
соответствующую определенному набору квантовых чисел п, I и /и, принято
называть атомной орбиталью (АО)*. Этим подчеркивается как
определенная аналогия с боровскими орбитами — траекториями
движения электрона вокруг ядра, так и различие в трактовке клас-
классического понятия орбиты и орбитали, в которую вкладывается
квантово-механическое вероятностное понимание.
Физический смысл главного квантового числа п ясен из рассмот-
рассмотрения решения для радиальной части волновой функции и формулы
для энергии водородоподобного атома B.41). Смысл же квантовых
чисел / и т будет выяснен позже. При классификации электронных
состояний атома для каждого квантового числа / приняты следу-
следующие буквенные обозначения:
/....О 12 3 4 5
Обозначение .... s p d f g h
Функцию с /=0 называют 5-функцией, с /=1—р-функцией
и т. д.
Первые четыре буквенных обозначения имеют происхождение,
связанное с названиями спектральных линий, обнаруживаемых
в атомных спектрах: s, p,d,f— первые буквы английских слов sharp
(резкий), principal (главный), diffuse (диффузный), fine (тонкий). Так
были названы наиболее давно и подробно изученные серии линий
в спектре атома водорода.
Вероятность нахождения электрона в пространстве между значе-
значениями г и r+dr, согласно B.3), равна
п2п
Я
п2п
\Valm(r,9t<p)\2r2sm6drd0d(p =
о о
г2dr \\[Ybn@, <P)?sin
о о
r2 dr=PJr) dr. B.47)
•Термин «орбиталь» используют для произвольной одноэлектронной волновой
функции. В случае атома водорода одноэлектронная волновая функция совпадает
с полной волновой функцией системы. Однако очевидно, что это не так для много-
многоэлектронных систем.
34
i г j
Рис. 2.2. Вид радиальных функций атома водорода
Функцию Pnifr), определяющую вероятность нахождения электрона
на расстоянии г от ядра, называют радиальной функцией распределе-
распределения. На рис. 2.2 и 2.3 показаны радиальная функция Rni(r) и ради-
радиальная функция распределения Pni(r) для некоторых наборов кван-
квантовых чисел ли/. Точки, в которых радиальная часть обращается
в нуль, называют узловыми точкамщшш просто узлами. Аналогич-
Аналогично, поверхность, в каждой точке которой радиальная часть обраща-
обращается в нуль, называют узловой поверхностью. Из рис. 2.2 видно, что
радиальные функции Is, 1р и 3d не имеют узловых точек (т. е. не
пересекают ось г); функции Is и Ър имеют одну узловую точку,
a 3s — две узловые точки. Легко заметить закономерность, соглас-
согласно которой число узлов радиальной части равно п — 1— 1.
Вероятность нахождения электрона в какой-либо точке прост-
35
/ г
si*.
aPlr)
0,2
oj
аР(г)
0,2
о л
1 2 3
5 S 7 8 9г/ао
ранства определяется не только
значением г, но также и величина-
величинами углов в и ф и, следовательно,
зависит как от радиальной R*(r)9
так и от угловой Y^fB, <p) частей
атомной орбитали. Рассмотрим
более подробно сферические гар-
гармоники Yb,@, cp). Функции B.27)
— комплексные, что ясно из вида
Ф-функций B.19). Между тем в бо-
большинстве случаев удобнее рабо-
работать с действительными функция-
функциями. Так как функции 1^/0, <p)
и Yi-mF, <p) отвечают вырожден-
вырожден5 В 7 8 9г/п
Рис. 2.3. Радиальные функции рас-
предеяения атома водорода
ному состоянию, можно восполь-
воспользоваться свойством, согласно ко-
™рому ИХ линейная комбинация
также является решением уравне-
уравнения Шрсдингера с тем же собствен-
собственным значением (см. с. 13). Функции Y'^ и У? будут решениями
уравнения B.9):
B.48)
Тогда вместо функции B.27) получим два набора действительных
решений:
(~1 B/-ь1) (/— М)Пх/2 ., _ fcos|m|9,
YU0. <Р)Ч — —— TTTTT^T I *Г (cos в) i _._ , , _ B.49)
2тс
[sin \m\ <р,
где /=0, 1, 2, ... л-1, /п=0, 1, 2, ... . Для перехода от B.27) к B.49)
необходимо воспользоваться формулами Эйлера
sin тер =
cos ту—¦
7Х
+е
Для удобства и без потери общности можно считать, что от-
отрицательным значениям т соответствуют функции с sin]m\<p, а поло-
положительным — cos|m|<p.
В табл. 2.3 представлены угловые функции B.49) для некоторых
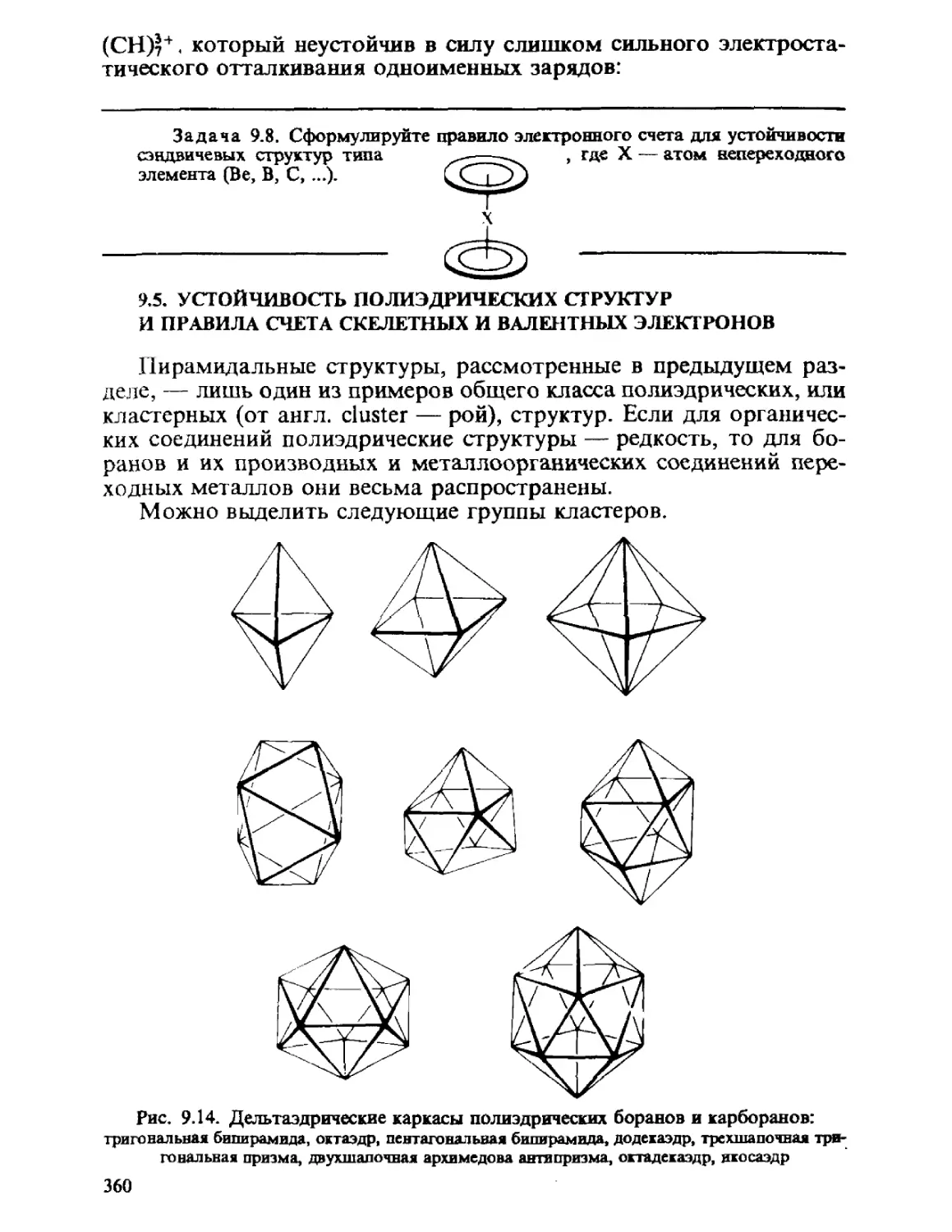
значений / и т.
36
Таблица 2.3. Угловые части волновой фушивш атома водорода
1
0
1
1
1
2
2
2
2
2
m
0
0
-1
1
0
1
— 1
2
-2
1
2.-\j 2.V.
2^2л
^—C cos2 5-1)
4^7t
—= sin 25 sin ^
— 51П (/ COS 4-Ц>
~$&Tl U Sill ^tp
Ayjit
Линейная
1
Ф
1
1
Jl l
1
/V.
-{'2
Ф
КОМ
i ¦
I-
2 +
2-
У,-,)
,-,)
У2-2)
Обозначение
Pz
Py
Px
4.
Отнесение сферических гармоник и их линейных комбинаций
к орбиталям, ориентированным в декартовых осях, понятно из
соотношений B.3) между сферическими и декартовыми координата-
координатами, откуда
. z
cos0=-;
г
• л • У
г
sin0cos^)=-.
г
37
z
*У
Рис. 2.4. Диаграмма угловых частей волновой функции атома водорода
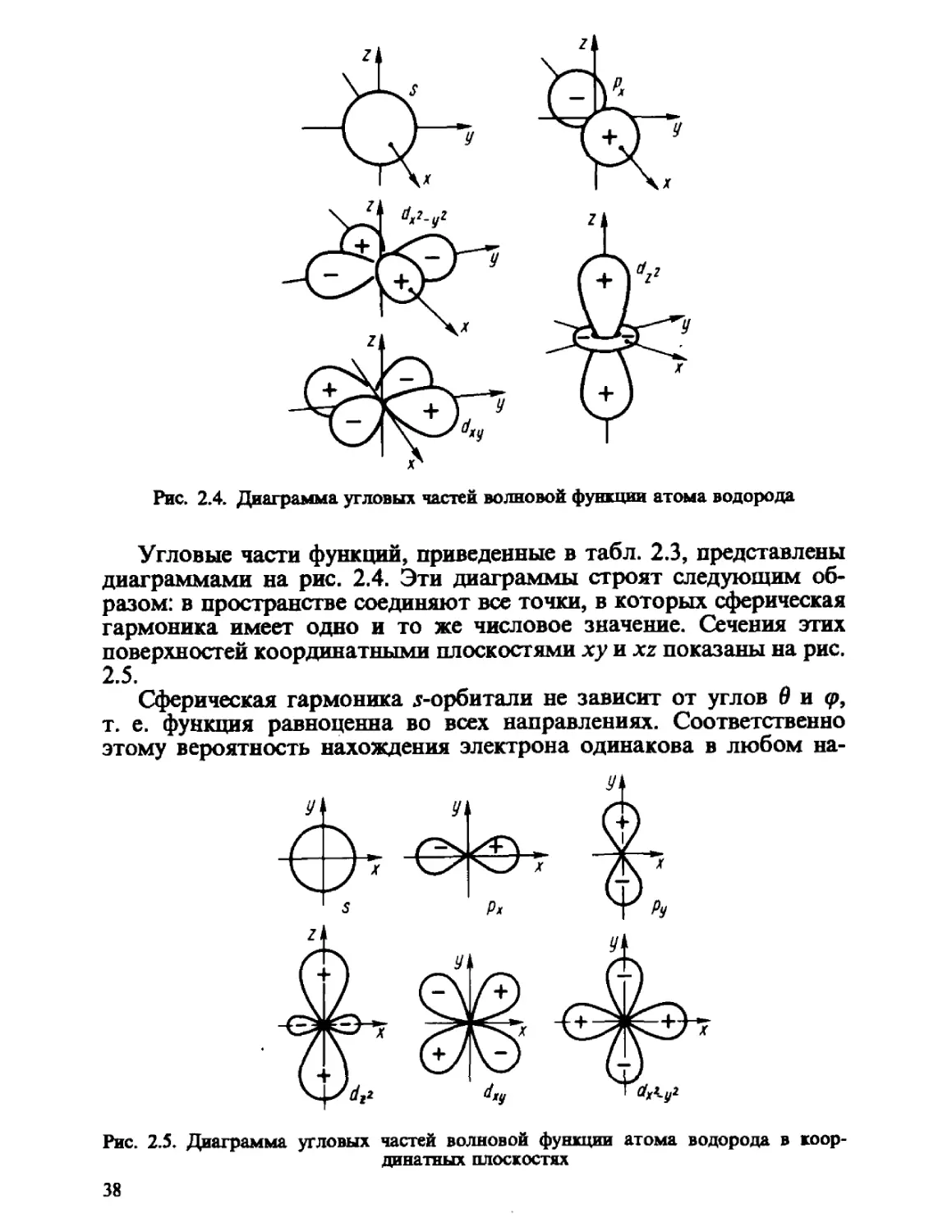
Угловые части функций, приведенные в табл. 2.3, представлены
диаграммами на рис. 2.4. Эти диаграммы строят следующим об-
образом: в пространстве соединяют все точки, в которых сферическая
гармоника имеет одно и то же числовое значение. Сечения этих
поверхностей координатными плоскостями ху и xz показаны на рис.
2.5.
Сферическая гармоника j-орбитали не зависит от углов В и <р,
т. е. функция равноценна во всех направлениях. Соответственно
этому вероятность нахождения электрона одинакова в любом на-
Рис. 2.5. Диаграмма угловых частей волновой функции атома водорода в коор-
координатных плоскостях
38
правлении. ^-Орбиталь имеет форму объемной косинусоиды, вытя-
вытянутой вдоль оси z, знак этой орбитали в некоторой точке простран-
пространства определяется знаком cos в. рг-Орбиталь равна нулю в любой
точке плоскости ху @ = 90°), которая, следовательно, является уз-
узловой плоскостью этой орбитали. Орбитали рх, ру имеют в качестве
узловых плоскостей соответственно плоскости yz и xz. Орбитали d^,
dxz, dyz и dxi_yi имеют две взаимно перпендикулярные узловые плос-
плоскости, a s-ia. ф-орбитали совсем не имеют узловых плоскостей.
Общее число узлов и узловых плоскостей любой АО равно л— 1, где
п — главное квантовое число.
Рассмотрим полную функцию водородоподобного атома. Дей-
Действительные нормированные волновые функции для некоторых зна-
значений п, I и т приведены в табл. 2.4.
Таблица 2.4. Волновые функции водородооодобвого Атома
т
Обозначе-
Обозначение
Функция
Zr
О
О
О
О
Zr
Zr
О
2pz
-I re ^cos0
Zr
-1
^1 — j re
re sin в cos 9
Zr
Zr
о
о
о
3s
1 fZ\3/2/ Zr Z2r2\
Zr
;h
39
Продолжение табл. 2.4
л
3
3
3
3
3
3
3
/
1
1
2
2
2
2
2
т
1
-1
0
1
-1
2
-2
Обозначе-
Обозначение
*,
34*
3</2 2
3^
Функция
Zr
у/2 /Zy/2f Zr\ ~^Z
=( — 1 16 ire sin в cos q>
Zly/nW/ \ "о/
Zr
=(—1 (б Ire sin0sm<p
ZX-s/n^w \ °o/
Zr
1 (гХ12г*ш~3ао(Зсош*0 П
— i i re \j\л)я и if
ZXy/lnyuQ/
Zr
n/2 /ZV/2 ~щ .
=j —J rv siii^cosficos^)
Zr
81^X^0/
Zr
1 /Z\y2 3Oq .
Zr
1 /ZV/2 , ЗДо .
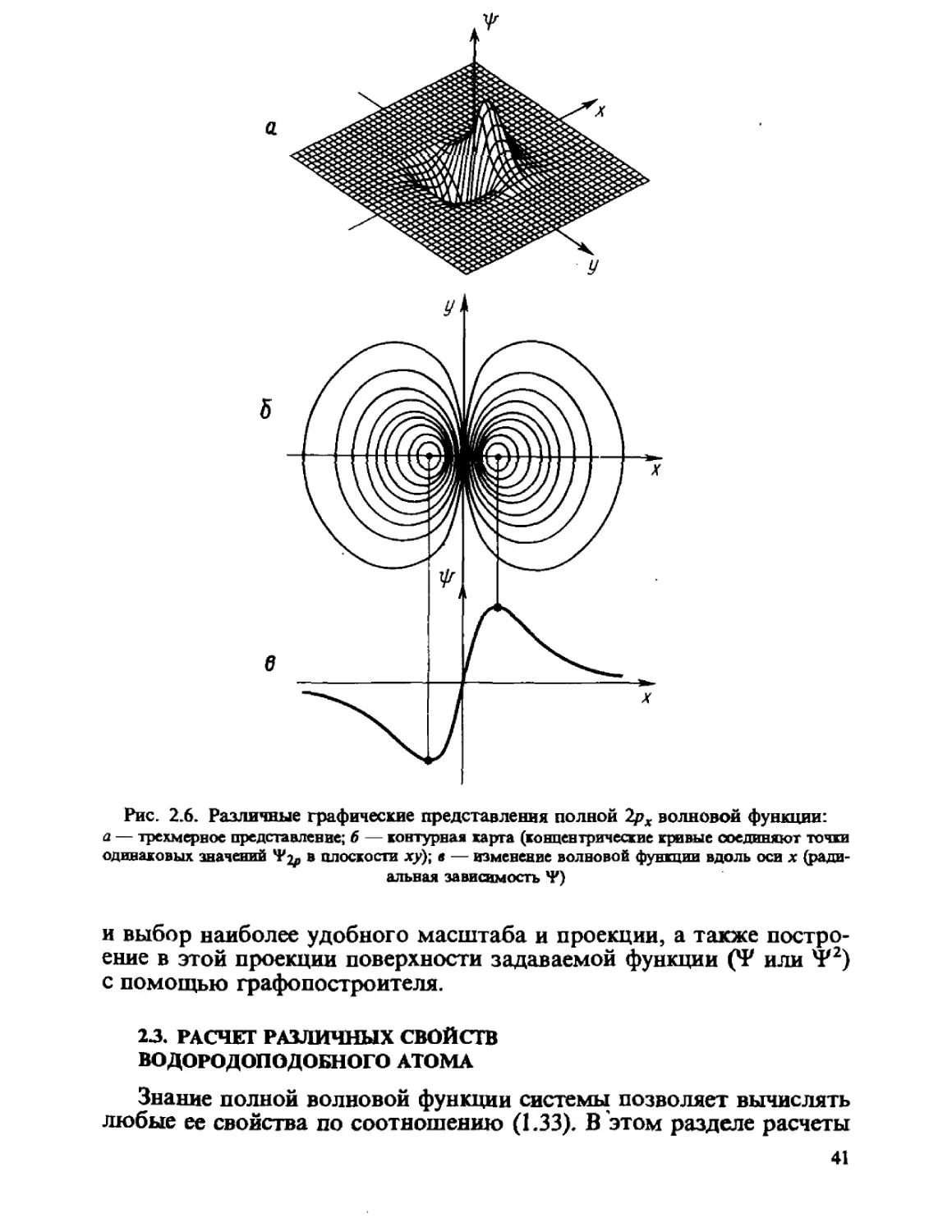
Графическое изображение полной волновой функции затрудни-
затруднительно, так как она зависит от трех переменных: г, 0 и q>. В связи
с этим используются ее различные диаграммные представления.
Один из способов заключается в комбинировании двух зависимо-
зависимостей для угловой и для радиальной частей. Например, такой вид
2/?х-орбитали показан на рис. 2.6. Не обладая достаточной нагляд-
наглядностью, он содержит все необходимые данные для оценки значений
Ч? (г, в, (р) в любой точке пространства.
Лучшим способом представления полной волновой функции яв-
являются пространственные контурные карты Ч* и W2 от двух пере-
переменных (при одной фиксированной). На рис. 2.6 даны зависимости
*? и ?2 от координат хну (при z=0). Для построения используют
электронные вычислительные машины и созданы специальные про-
программы. Последние включают не только вычисления функции, но
40
в
Рис. 2.6. Различные графические представления полной 2рх волновой функции:
й — трехмерное представление; б — контурная карта (концентрические кривые соединяют точки
одинаковых значений Ч*^ в плоскости ху); в — изменение волновой функции вдоль оси х (ради-
(радиальная зависимость *Р)
и выбор наиболее удобного масштаба и проекции, а также постро-
построение в этой проекции поверхности задаваемой функции Q? или ?2)
с помощью графопостроителя.
23. РАСЧЕТ РАЗЛИЧНЫХ СВОЙСТВ
ВОДОРОДОПОДОБНОГО АТОМА
Знание полной волновой функции системы позволяет вычислять
любые ее свойства по соотношению A.33). В этом разделе расчеты
41
подобного типа иллюстрируют вычислением некоторых важных
характеристик водородоподобного атома.
Найдем среднее расстояние между ядром и электроном в основ-
основном состоянии атома водорода. Волновая функция этого состояния
имеет вид (ал. табл. 2.4)
Используя определение средней величины A.33), найдем
0 B.50)
2жп
Интеграл типа
B.51)
a
о
часто встречается в квантово-механических расчетах. Учитывая
B.51), получаем
- 3
Таким образом, среднее расстояние электрона от ядра в основном
состоянии атома водорода равно полутора радиусам первой бо-
ровской орбиты. В общем виде среднее расстояние между электро-
ном и ядром для различных п и / водородоподобного атома опреде-
определяется формулой
/G+1Л
~} B53)
Задача 15. Проверить справедливость формулы B.53) для 2s- и 2р-функций
атома водорода (Z= 1) (см. табл. 2.4).
Задача 2.6. Определить среднее значение г2 в основном состоянии атома
водорода.
42
Интересно сравнить среднее расстояние Г между электроном
и ядром в основном состоянии атома водорода с наиболее вероят-
вероятным положением электрона в атоме. Плотность вероятности нахож-
нахождения электрона от ядра на расстоянии г в основном Ь-состоянии
равна, согласно B.47),
2
Максимальное значение этой функции, соответствующее наиболее
вероятному положению электрона, может быть найдено из ее экст-
экстремума:
dP(r) 2r "Z 2r2 "Z Л
Наиболее вероятное расстояние электрона от ядра точно совпадает
с радиусом первой боровской орбиты.
Несовпадение наиболее вероятного и среднего расстояний элект-
электрона от ядра легко понять из рис. 2.3. Плотность вероятности,
определяемая функцией распределения Р(г), не является симмет-
симметричной функцией относительно своего максимума. Существует до-
достаточно большая вероятность найти электрон на расстояниях,
больших 2flo> в связи с чем среднее расстояние всегда будет превы-
превышать наиболее вероятное.
Задача 2.7. Показать, что lj-функция атома водорода (см. табл. 2.4) не
является собственной функцией операторов Т и V в отдельности, а есть собствен-
собственная функция суммы Т -(- V.
Задача 2.8. Для основного состояния атома водорода найти <Т> и <V>
и показать, что их сумма равна полной энергии Е. Проверить, что
1
Е=— <T)=~<V>. Эти равенства выполняются для любых многоэлектронных
2
систем, их называют теоремой вириала*.
"Квантово-механическая теорема вириала (от лат. vires — силы) — полный ана-
аналог подобной теоремы в классической механике, за исключением того, что в клас-
классической механике среднее берется по времени, а не по состоянию системы. В клас-
классической механике эта теорема была введена еще Клаузнусом. В квантовой механике
ее впервые доказали М. Борн, В. Гейзенберг и П. Иордан A925). Теорема вириала
выполняется только для точных решений. Отклонение от этой теоремы является
одним из основных тестов для проверки точности решения. О теореме вириала см.
также гл. 5.
43
2.4. СПЕКТР ВОДОРОДОПОДОБНОГО АТОМА.
ПРАВИЛА ОТБОРА
Поглощение и испускание света, а следовательно, и спектраль-
спектральные переходы атома подчиняются правилу частот Бора
hv=Ek-Eh B.56)
где Ек и Ег — уровни энергии, соответствующие состояниям с вол-
волновыми функциями Ч?к и *Fj.
Для водородоподобного атома, согласно B.41), частота кванта
поглощаемого света в результате перехода из состояния с главным
квантовым числом п — 1 в более высокоэнергетическое состояние
с главным квантовым числом п определяется соотношением
п
B.57)
На рис. 2.7 представлены возможные значения энергетических уров-
уровней атома водорода (Z= 1).
Спектры атомов характеризуются не только значениями энергий
поглощаемых или излучаемых квантов света, т. е. их частотами, но
и вероятностями этих процессов. Последние определяют интенсив-
интенсивности наблюдаемых полос поглощения (испускания). Вероятность
электронного перехода (сила осциллятора) из состояния хР,=хРя4я^г,
В, (р) в Yjt=4V/w (r, В, (р) зависит линейно от энергии перехода
и квадратично от величины дипольного момента перехода
Ды (формула Малликена—Рике):
B.58)
коэффициент пропорциональности, зависящий от выбора
а Т?н определяется
где К
?,эв
системы единиц,
как
п-2
-13.6
Ряс. 2.7. Потенциальная энер-
энергия атома водорода и его энер-
энергетические уровни
B.59)
где г ^ — радиус-вектор ^-го электрона
в атоме. _+
Переходы, для которых D д,=0, на-
называют запрещенными в дипольном
приближении. Большая часть возмож-
возможных переходов в атоме запрещена,
в связи с чем в спектроскопии важное
значение имеют правила отбора для
разрешенных переходов.
44
Отметим, что переходы, запрещенные в д и по льном приближе-
приближении, могут иногда проявляться в атомных спектрах с очень низкой
интенсивностью, если они разрешены в более высоких приближени-
приближениях (квадрупольном, октупольном и т. д.). Вероятность таких пере-
переходов по сравнению с разрешенными в дипольном приближении
весьма мала и обнаружить соответствующие им линии можно
только с помощью высокочувствительных спектральных методов.
Правила отбора для разрешенных спектральных переходов во-
водородного атома могут быть выведены из рассмотрения интегралов
\ пЬп'>
шп,; B.60)
которые определяют вероятность поглощения (испускания) свето-
световой волны, поляризованной соответственно по осям х, у, г.
Собственные функции водородоподобного атома пблучены
в сферических координатах, поэтому целесообразно интегралы
B.60) также вычислять в этих координатах. Рассчитаем сначала
последний из интегралов B.60). Учитывая, что z = rcos0, его можно
записать в виде
r2dr \Вш(В)В(т(в) cos dsin 6de jV^V-'dp. B.61)
0 0 0
Интеграл по <р не равен нулю только при т~т\ откуда правило
отбора по квантовому числу т имеет вид:
Д/и = 0. B.62)
Подставляя т—т' в интеграл по 6, получим
ж
I'
(В) cos О sin в сЮ. B.63)
О
Учитывая, что ©-функции представляют собой присоединенные
полиномы Лежандра B.23), перепишем B.63) в виде
I ^ (у) У-Р/"' (у) dy, B 64^
-1
где у=cos в.
Для присоединенных полиномов Лежандра существует рекур-
рекуррентное соотношение
=0. B.65)
45
Используя B.65), представим B.64) как
Присоединенные полиномы Лежандра ортогональны4 (см. задачу
2.3), поэтому интеграл B.66) отличен от нуля только при выполне-
выполнении соотношения /=/'+1 или /=/' —1, что приводит к правилу
отбора по орбитальному числу
Д/=±1. - B.67)
Оценка интеграла по г в B.61) намного труднее. Отметим толь-
только, что для уровней с различными значениями главного квантового
числа п не существует каких-либо ограничений в спектральных
переходах.
Правила отбора B.62) и B.67) справедливы только при поглоще-
поглощении света, поляризованного по оси z. Расчет величин
Dnint. nbn и -bjrtw. nim приводит к правилам отбора для циркулярно
поляризованного света, т. е. поляризованного вдоль осей х и у:
Д/=±1;Дж=±1. B-68)
Выделение правил отбора для поглощения света, поляризован-
поляризованного в различных направлениях, имеет смысл только в том случае,
когда одно из направлений пространства, например ось г, задано
условиями эксперимента. Такая ситуация реализуется, например,
при изучении спектров атомов в магнитном поле (эффект Зеемана)
или электрическом (эффект Штарка), где направление поля связыва-
связывается с направлением оси z. В обычных экспериментах все направле-
направления в пространстве неразличимы и единственным правилом отбора
является требование А/= +1.
Таким образом, в спектрах одноэлектронных атомов проявля-
проявляются переходы из ^-состояния только в /^-состояние, из р-состоя-
ния — в s- и ^-состояния, из ^/-состояния — в р- и /-состояния.
Остальные переходы относятся к запрещенным и не регистрируют-
регистрируются в спектрах указанных атомов.
Важным следствием правил отбора является то, что водородо-
подобные атомы не из всех возбужденных состояний могут перейти
в основное состояние за короткое время. Например, переход из
состояния с волновой функцией ?200 в основное состояние Ч'юо за-
запрещен правилом отбора B.67). Переход Ч'гоо-^Ч'юо может произой-
произойти только за счет внешнего воздействия или вследствие безызлуча-
тельной дезактивации, которая возникает в результате столкнове-
столкновений атомов. Долгоживущие возбужденные состояния называют ме-
тастабильными.
46
2.5. УГЛОВЫЕ МОМЕНТЫ АТОМА
Понятие углового момента (момента импульса) особенно важно
при классификации состояний атомных и молекулярных систем.
Угловые моменты дают возможность прояснить физический смысл
квантовых чисел / и т.
2.5.1. Операторы квадрата и проекции углового момента
В классической механике момент импульса отдельной частицы
определяется как
L-У хр ~
г j к
х у z
Рх Ру Pz
B.69)
где г* — радиус-вектор частицы; рх, ру, pz — проекции ее импульса
на координатные оси. Используя выражение B.69) и правила
преобразования классических соотношений к виду квантово-меха-
нических операторов, получим операторы компонент углового
момента:
B-7o)
Квадрат углового момента L2 можно выразить через операторы
проекций импульса:
B-71)
Выражения B.70) и B.71) в сферических координатах имеют вид
/Я Я
Ьх=-Щ sin<p~T-ctg0cos<jp
\ си
/я
Ly=-Mcos(p—-ctgflsinpJ;
\ ov u(pj
B.72)
т2вд<р2_
47
Используя B.70), легко доказать коммутационные соотношения
B.73)
н
=0; , B.74)
L2LX-LXL2 = O.
Задача 2.9. Доказать коммутационные соотношения B.73) я B.74).
В соответствии с одним из основных законов квантовой меха-
механики соотношения B.73) и B.74) показывают, что нельзя одновре-
одновременно измерить две компоненты углового момента, т. е. нельзя
с любой заданной степенью точности определить направление век-
вектора углового момента в пространстве. В то же время можно
одновременно измерить одну из компонент углового момента и ве-
величину его квадрата и, следовательно, знать вместе со значением
одной из проекций скалярную величину углового момента.
2.5.2. Физический смысл квантовых чисел / и т
Найдем собственные значения L2. Для этого необходимо решить
уравнение
?2^=L2^ ' B.75)
Операторы L2 и Lr коммутируют с гамильтонианом Н, т. е.
?2Н - Ш>=0; ?ZH - HL,=0. B.76)
Задача 2.10. Доказать, что операторы L2, HhU подчиняются коммутаци-
коммутационным соотношениям B.76).
Если два оператора коммутируют, то можно выбрать систему
базисных функций так, чтобы они являлись собственными функци-
функциями обоих операторов (см. разд. 1.1). Следовательно, найденные
при решении уравнения Шредингера собственные функции операто-
оператора Н (см. табл. 2.4) являются собственными функциями операторов
L2 и Lz. Используя выражение оператора L2 B.72), запишем уравне-
уравнение B.75) в явном виде:
48
B.77)
Разделяя переменные, получим
sin20 1 д { . ^д®\ L2sin20 I д2Ф
\ дв
r-^ Sin0 Б } + 2 = ~л Т2- B-78>
Часть равенства, содержащая функцию Ф, совпадает с B.11). Следо-
Следовательно, правая и левая части уравнения B.78) могут быть прирав-
приравнены к mz. Тогда для ©-функции имеем
д ( дВ\ /L2 m2
Сравнивая B.79) с B.12), легко заметим, что они совпадают, если
С=^. B.80)
Но уравнение B.12) имеет конечные решения только при выпол-
выполнении условия B.20); следовательно, функция 0 будет решением
B.79) при
¦ B-82>
Таким образом, квантовое число /, называемое обычно орбиталь-
орбитальным квантовым числом, определяет значение углового момента. Так
как орбитальное квантовое число принимает лишь целочисленные
значения, то величина углового момента атома также принимает
дискретные значения, т. е. квантуется. Для состояний с /=0 E-
функции) угловой момент равен нулю. Это объясняется сферичес-
сферической симметрией ^-орбитали, т. е. независимостью формы орбитали
от углов в и <р.
Найдем теперь собственные значения оператора Lr. Для этого
необходимо решить уравнение
ЪЯ=Ь?У B.83)
или
Подставляя Ч? из B.6) и Ф из B.19) в B.84), получим
Lz=mk B.85)
Квантовое число т—0, ±1, ±2,... характеризует значение проекции
углового момента на выбранную ось. В конкретном физическом
49
эксперименте такая ось задается, например, направлением прило-
приложенного поля.
Отметим, что собственное значение оператора абсолютной вели-
величины момента B.81) всегда больше максимального значения (АО его
проекции на любую выбранную ось. Действительно, при равенстве
полного углового момента одной из его проекций Ь=1^ две оста-
остальные проекции должны точно быть равны нулю. Это означало бы,
что все три компоненты углового момента могут быть одновремен-
одновременно точно измерены, что противоречит коммутационным соотноше-
соотношениям B.73) и, следовательно, принципу неопределенности Гейзен-
берга.
2.5.3. Магнитный орбитальный момент атома
С угловыми механическими моментами атомов связаны их маг-
магнитные моменты. Выражение для магнитного момента электрона
можно получить с помощью квантово-механического формализма,
однако можно воспользоваться более наглядными классическими
аналогиями.
Электрон, движущийся по замкнутой орбите, создает магнитное
поле. На расстояниях, больших по сравнению с размерами орбиты,
создаваемое поле можно вычислять как поле магнитного диполя:
где / — величина тока; П — площадь орбиты.
Для круговой орбиты с радиусом г магнитный момент равен (в
электромагнитных единицах)
t-—- B.87)
С
и
Через определенную точку орбиты электрон проходит -— раз в се-
кунду (V — скорость электрона). Так как заряд электрона равен — е,
то сила этого кругового тока равна
ev
1 B88)
Из соотношений B.87) и B.88), учитывая что скаляр углового
момента L=mevr, следует
emevr e
е
где у=- гиромагнитное отношение.
Так как, согласно B.82), L=hy/l(t+\), то
50
^ B90)
eh
Величина -— = /?M—9,2741 • 10 24 ДжДс представляет собой атом-
2тес
ную единицу магнитного момента, называемую магнетоном Бора.
Величина
y=i B-91)
в уравнении B.89) имеет физический смысл отношения величин
магнитного момента к механическому.
Проекция вектора орбитального момента jt на выбранное на-
направление, например ось z, связана с проекцией углового магнит-
магнитного момента с помощью гиромагнитного отношения
\lz = - yL2 =-ymh=- mfiM. B.92)
Наличие магнитного момента атома, связанного с орбитальным
движением электрона, обусловливает его взаимодействие с магнит-
магнитным полем. Энергия такого взаимодействия согласно классической
электродинамике равна
E=-fB* =- |/Г ||i*| cos в, B.93)
где В — вектор индукции магнитного поля; 9 — угол между /Г*
и В . Приняв за ось z направление поля В , получим
!/Г | cos в=fiz = - тр», B.94)
откуда
Е=три\В*\. B.95)
Уровни энергии, определенные выражением B.95), отстоят друг от
друга на величину fiM \B |, не зависящую от / и т.
Итак, в магнитном поле энергия атома водорода зависит не
только от главного квантового числа и, но и от магнитного кван-
квантового числа т (последнее и получило отсюда свое название).
2.5.4. Спин электрона
Экспериментальные данные (опыты Штерна — Герлаха, тонкая
структура спектров щелочных металлов и др.) привели к выводу
о том, что нельзя описать движение электрона только с помощью
классических координат и импульса. Необходимо ввести понятие
о собственном угловом моменте количества движения электрона
и о собственном магнитном моменте электрона. В 1925 г. Г. Гаудс-
мит и С. Уленбек предположили, что электрон обладает собствен-
51
ным моментом количества движения S, который не связан с его
орбитальным движением. Полный момент количества движения
электрона равен
Собственный момент электрона раньше пытались отождест-
отождествить с моментом импульса, возникающим вследствие его враще-
вращения вокруг своей оси. Отсюда принятое для обозначения
собственного момента количества движения электрона название
«спин» (от англ. spin — верчение), хотя такая аналогия неос-
неосновательна потому, что электрон не является классической
частицей.
Поскольку спин не имеет классического аналога, отсутствует
и соответствующее ему классическое соотношение, выраженное че-
через координаты и импульс. В связи с этим невозможно получить
в явном виде оператор спинового момента, пользуясь правилами
написания квантово-механических операторов.
Однако возможно все же определить функциональное отноше-
отношение между операторами квадрата собственного углового момента
S и его проекциями Sx, Sy, Sr, которые вводятся/по аналогии
с соответствующими операторами углового момента L2, Lx, Lyi 1^:
S2=S2+S2 + S2. B.97)
Эти операторы удовлетворяют перестановочным соотношениям
B.98), аналогичным B.73):
[SyiS2]=ihSx; B.98)
Оператор S2 коммутирует с операторами проекции спина
[S2, SJ=[S2, SJ- [S2, SJ=0. B.99)
Согласно данным эксперимента, имеются только две возможные
спиновые ориентации электрона в магнитном поле, вследствие чего
для каждого электрона можно иметь только две собственные функ-
функции операторов S2 и Sr. Эти функции обозначаются символами
а и р и удовлетворяют соотношениям
<*=-(-+Atfa,
(
B.100)
V; S0/rV5+1) /ЦB+1W
Спиновые функции предполагаются ортонормированными:
52
O. B-101)
Из уравнений B.100) следует, что |5^=^-й, а проекции вектора
спинового углового момента на направление оси z равны ±г&
По аналогии с орбитальным магнитным моментом вводится
спиновый магнитный момент электрона
М,= УД B.102)
Для согласия с опытом спиновое гиромагнитное отношение
с
уя необходимо принять равным 2 (в единицах -— — у), а не 1, как для
орбитального магнитного момента (ср. B.89)). Тогда
Bлоз)
Дирак показал, что множитель 2 возникает из релятивистского
рассмотрения электрона. Недавние теоретические и эксперимен-
экспериментальные исследования показывают, что множитель в B.103) немно-
немного больше 2 и равен 2,0023.
Аналогично B.92) для проекции спинового магнитного момента
принимается
&z = 2mspM=±pM, B.104)
где ms — спиновое магнитное квантовое число, которое, как видно
из B.104), принимает значение только ±1/2. Из выражения B.104)
наиболее четко видна связь множителя 2 с экспериментом. В опыте
Штерна—Герлаха расщепление пучков на экране при выходе из
магнитного поля таково, каким оно было бы при собственном
магнитном моменте электрона, имеющем проекцию на ось z, рав-
равную ±рм.
ГЛАВА 3
МНОГОЭЛЕКТРОННЫЕ АТОМЫ
В гл. 2 указано, что атом водорода и водородоподобные ионы
являются единственными атомарными системами, для которых мо-
могут быть получены точные волновые функции путем прямого реше-
решения уравнения Шрё'дингера. Уже для следующего за водородом
элемента периодической системы — гелия — на этом пути возника-
возникают непреодолимые трудности. Смысл их становится понятным из
53
рассмотрения оператора полной энергии ато-
атома гелия, в котором в поле ядра с зарядом
+ 2 находится два электрона (рис. 3.1):
Рис. 3.1. Координаты
электронов в атоме ге- Основное отличие гамильтониана C.1) от
ли* гамильтониана атома водорода B.2) заклю-
заключается в том, что оператор потенциальной энергии включает не
только члены, описывающие притяжение электронов к ядру, но
и член межэлектронного отталкивания. Его величина зависит от
координат обоих электронов (r12 = |F*2— ?! i), что не позволяет раз-
разделить переменные в любой координатной системе. По этой причи-
причине точное аналитическое решение уравнения Шрёдингера с гамиль-
гамильтонианом C.1) невозможно.
Для более сложных атомов с несколькими электронами не-
необходимо учесть энергию отталкивания всех электронов. Гами-
Гамильтониан многоэлектронного атома с п электронами и зарядом
ядра Z имеет вид
ft2 я я 7р1 я я в*-
^Г^-Е^г+^Т- C2)
€ 1—1 i»l ' I < j У
Здесь первый член — оператор кинетической энергии электронов;
второй — оператор потенциальной энергии взаимодействия п элек-
электронов с ядром; третий — оператор энергии межэлектронного от-
отталкивания.
Для атомов с двумя (и более) электронами волновые функции
могут быть получены лишь с помощью тех или иных приближенных
методов.
3.1. МЕТОД САМОСОГЛАСОВАННОГО ПОЛЯ ХАРТРИ
Одним из наиболее эффективных методов решения задач кван-
квантовой химии является метод самосогласованного поля, предложен-
предложенный в 1927 г. Хартри*. Идея этого метода заключается в том, что
взаимодействие каждого электрона в атоме со всеми остальными
заменяется взаимодействием с усредненным полем, создаваемым
ядром и остальными электронами. Это позволяет заменить в урав-
уравнении C.2) потенциал типа —, зависящий от координат двух элект-
г
¦Хартри Дуглас Рэйнер A897—1958) — известный английский физик-теоретик,
один из создателей метода квантово-механяческого расчета многоэлектронных
атомов- Работал также в области математической физики и вычислительной техники.
54
ронов, выражением, описывающим межэлектронное взаимодейст-
взаимодействие как функцию координат каждого отдельного электрона.
Рассмотрим схему метода Хартри более подробно. Полная вол-
волновая функция атома в этом методе записывается в виде произведе-
произведения волновых функций отдельных электронов:
^=^,A)^B) ... Чя(п)*. C.3)
Форма этого соотношения предполагает независимость движения
каждого электрона в атоме от всех остальных. Согласно вари-
вариационному принципу A.48), энергия системы Е, вычисленная с при-
приближенной функцией C.3), будет всегда выше истинного значения
энергии Ех:
и п
C.4)
:, ... dxn.
i > j r*J
Вынесем в выражении C.4) суммирование по i за знак интеграла:
1-1
В выражении C.5) первые два члена в фигурных скобках зависят
только от координат i-ro электрона, а третий член зависит одновре-
одновременно от координат /-го и j-ro электронов. Это позволяет нам
записать C.5) в следующем виде:
Вследствие ортонормированности функций ^i(i) все интегралы
в круглых скобках равны 1; следовательно,
*3десь и далее волновые функции полагаются действительными, вместо коор-
координат q\, qj, —, qn Для сокращения записи использованы индексы 1, 2, .... п.
55
2 ..
или
л 1 я и
xi = X, •",i"ЬX
• 1 2
где введены обозначения
,f У *ь
Интеграл if,, называемый остовным, представляет собой сумму
кинетической энергии электрона на орбитали % и потенциальной
энергии его притяжения к ядру.
Интеграл Уф называемый кулоновским, представляет собой сред-
среднюю энергию электростатического отталкивания электронов, нахо-
находящихся на орбиталях ?, и Ч*,. Неизвестные функции ?, находят из
минимума полной энергии C.6) при дополнительном условии ор-
тонормированности функций:
J^^A = V (ЗЛО)
Для этого составляется новая функция (функционал)*:
л л
/ j 1-1
где коэффициенты е,у называют множителями Лагранжа.
Равенство нулю первой вариации EФ — необходимое условие
экстремальности, из которого находят функции *Р,-:
^^ C.12)
Проводя варьирование в C.6) по функциям ?„ из выражения
C.12) получим
¦Более строгий вывод уравнений Хартри и Хартри—Фока см.: Фок В. А. Начала
квантовой механики. — М.: Наука, 1976.
56
dTt=O. C.13)
В выражении C.13) левая часть равна нулю для любых вариаций
всех S^i (i=l,2,...) только в том случае, если равны нулю одновре-
одновременно коэффициенты при всех <5*Р„ т. е. справедливы уравнения
v ) C.14)
/=1, 2, ... .
Уравнения C,14) впервые были получены Хартри и названы его
именем. Такие уравнения называют также одноэлектронными урав-
уравнениями. Из уравнений типа C.14) следует, что е, G=1, 2, ...)
описывает энергию электрона на /-й орбитали атома с гамильтониа-
гамильтонианом Хартри, представленным в фигурных скобках в уравнении
C.14). Гамильтониан Хартри для /-го электрона отличается от
точного гамильтониана /-го электрона в атоме [/-е члены в C.2)]
заменой электростатического взаимодействия электронов [послед-
[последний член в C.2)] эффективным потенциалом
C.15)
который представляет собой усредненное электростатическое вза-
взаимодействие /-го электрона со всеми остальными электронами.
е2
Поясним это подробнее. Найдем среднее значение — по волновой
ги
функции /-го электрона, которое, согласно A.34), равно
- (среднее по j
Проводя суммирование этих средних величин по всем/ получим
^ C.15). Вернемся к уравнениям C.14), умножим каждое из
них слева на 4?t(i) и проинтегрируем полученное соотношение по
координатам /-го электрона по всему пространству. Тогда с учетом
обозначений C.8) и C.9) получим выражение орбитальных энергий
через остовный и кулоновские интегралы:
7у. C.16)
51
Учитывая C.16), выражение для полной энергии C.7) можно запи
сать в другом виде:
1
Каждое из уравнений системы C.14) содержит координаты одно-
одного электрона, но, чтобы его составить, нужно знать заранее потен-
потенциал РэффО^, который зависит от искомых функций *?j(j) (j^V-
Преодолеть эту трудность можно лишь использовав метод последо-
последовательных приближений. В качестве начальных волновых функций
4?j берут какие-либо пробные орбитали ?}0), например орбитали
водородоподобного атома. С исходным набором функций Ч']0* рас-
рассчитываются интегралы C.8) и C.9), а затем решаются уравнения
C.14) для каждого /. Найденные таким образом функции первого
приближения YJ1' используют для нахождения соответствующих
энергий межэлектронного взаимодействия. Обычно новые величины
энергий сильно отличаются от первоначальных, что связано с нето-
неточностью исходных функций HffK В связи с этим находят функции
следующего приближения Hff* и т. д. Критерием получения до-
достаточно хороших Ч', является совпадение с заданной точностью
величин C.15), рассчитанных для 4f"; и Ч//я+|;, т. е. потенциалы
C.15) должны быть согласованы с функциями У,. Это требование
обусловливает название метода самосогласованного поля (ССП).
Идею метода ССП широко используют в квантовой химии. Пример
проведения самосогласования будет приведен в гл. 8.
Потенциал C.15) в общем случае не является сферически-сим-
сферически-симметричным, т. е. зависит от углов в и (р. Учет несферичности
потенциала — достаточно сложная задача, а полученные поправки
не приводят к существенному улучшению конечного результата.
В связи с этим используют обычно усредненное по всем направлени-
направлениям (9 и ц>) потенциальное поле, т. е. потенциал C.15) заменяется
сферически-симметричным потенциалом (так называемая аппрок-
аппроксимация центрального поля):
C.18)
где интегрирование в отличие от C.15) ведется еще и по угловым
переменным 0,- и <р{ i-го электрона (но не по rt).
В приближении C.18) волновая функция многоэлектронного ато-
атома сохраняет вид водородоподобной функции
C.19)
58
который позволяет классифицировать атомные орбитали Хартри
по типу функций s, p, dn т. д., как и в одноэлектронном атоме.
Таким образом, для нахождения решений уравнений Хартри
C.14) необходимо найти только радиальную функцию Ri(r). Функ-
Функции R((r) должны быть решениями уравнения
r dr \_Ooe:
<<=°> C.20)
аналогично B.28). Уравнения C.20) получены из B.28) путем добав-
добавления потенциала C.18). Эти интегродифференциальные уравнения
значительно сложнее, чем уравнение для водородоподобного атома,
и их решают численным интегрированием. В связи с этим волновая
функция получается не в аналитической
форме, а в виде таблиц числовых значе-
значений радиальной функции (или других фу-
функций на ее основе) от координат элект-
электронов. На рис. 3.2 приведено полученное
по методу Хартри решение уравнения
C.20) для ls-и 2^-электронов атома Be,
Р(г) вычисляют по соотношению B.47).
Из сравнения рис. 2.3 и 3.2 можно заме-
заметить, что радиальное распределение, по-
полученное по методу Хартри, качественно
аналогично распределению в водородо-
подобном атоме.
Если просуммировать электронные
плотности дважды заполненных уровней
с данным орбитальным квантовым чис-
числом / по всем возможным /я, то получим
выражение для функции ?2 (г, в, ср):
Рис. 3.2. Функция P(r)t полу-
полученная по методу Хартри для
Is- и 2г-электронов атома бе-
бериллия
(г, в, «,)=2
C.21)
Воспользовавшись свойством сферических гармоник
\yuo,<p)\
2-
2/-Н
C.22)
получим
59
- C23)
Таким образом, распределение электронной плотности в атоме,
в котором полностью заполнены все орбитали с данным /, является
сферически-симметричным (не зависит от координат В и q>).
Задача 3.1. Проверить соотношение C.22) для s-, р-, {/-функций атома
водорода.
3.2. ПРИНЦИП ПАУЛИ И ОПРЕДЕЛИТЕЛИ СЛЭТЕРА
Волновая функция многоэлектронного атома, представленная
в виде произведения одноэлектронных функций по типу C.3), не
удовлетворяет принципу Паули. Действительно, для двухэлектрон-
ной системы функция C.3) примет вид
C.24)
Операция перестановки электронов ведет к новой функции
?" = ^B)^A), C.25)
причем ?' ф У. Антисимметричную волновую функцию можно по-
получить в виде линейной комбинации У и ?":
Н'=^'~^"=ЧР1A)Т2B)-Ч'1B)^2A). C.26)
Перестановка электронов в C.26) меняет знак ? на обратный, что
и является условием антисимметричности. Выражение для ? C.26)
удобно записать в виде определителя второго порядка:
C.27)
Здесь и далее под 4ft(i) понимается функция, зависящая как от
пространственных rh так и от спиновых переменных fff i-ro электро-
электрона (см. разд. 2.5.4):
WJi) =4t(?h ад. *= ± 1- C.28)
Функцию C.28) называют спин-орбиталью. Так как гамильтониан
системы C.1) явно не зависит от спиновых переменных, спин-ор-
биталь Ч*,- с хорошей степенью точности можно представить в виде
произведения функций, зависящих отдельно от пространственных
и спиновых переменных, т. е. 4?t(i) -^ifrt) Xj( в0, и, вводя обозна-
обозначения #(+1,) = а(%> и $( — 1)=Р(г), получим
60
^j; C.29)
%. C.30)
В неограниченном методе Хартри—Фока (см. разд. 4.3.4) про-
пространственные функции электронов со спицами аи/? различны.
Задача 3.2. Какие из следующих функций являются полностью симметрич-
симметричными или антисимметричными:
Д1)B)(ОB)
C.31)
)lA)g()g()A)][(l)ta-^(l)aB)];
г) ri2exp[-a{Vi+r2J]?
Постройте из функций, не обладающих симметрией, симметричные и антисим-
антисимметричные комбинации.
Д. Слэтер*, обобщая определение C.27), показал, что единствен-
единственной возможной формой построения полностью антисимметричной
волновой функции л-электронной системы из независимых ортонор-
мированных спин-орбиталей отдельных электронов является опре-
определитель л-го порядка, который называют определителем Слэтера:
-Чг
где (л!) — нормировочный множитель.
Задача 3.3. Показать, что при условии ортонормированности всех Ч*,- нор-
1
мировочньш множитель в C.32) равен —=.
j
Перестановке двух электронов соответствует лерестановка двух
столбцов определителя C.32), в результате чего он меняет свой
знак, но не меняет значения. Спин-орбиталь зависит от четырех
квантовых чисел: п, I, т, т, (пространственная часть зависит от п, I,
т, а. спиновая — от ms). Если в системе какие-либо два электрона
будут иметь одинаковый набор четырех квантовых чисел, то им
*Джон Слэтер A903—1977) — американский физик, один из наиболее авторитет-
авторитетных специалистов в области квантовой теории электронных оболочек атомов и моле-
молекул, теории твердого тела.
61
+ -t- +
+ + + + -
a 5 8 г д е
Рис. 3.3. Графическое представле-
представление распределеяня электронов для
различных состояний атома гелия:
а — основного; 6 — * — возбужденных
будут соответствовать одинаковые
пространственные и спиновые фун-
функции. В этом случае две строки де-
детерминанта C.32) окажутся тожде-
тождественными. Определители такого
типа равны нулю. Заметим, что два
электрона могут иметь одинаковые
пространственные части, если их
спиновые функции отличны, т. е. электроны имеют противополож-
противоположные спины. Два электрона системы, отличающиеся только спинами,
считаются спаренными и описываются функциями C.29) и C.30).
Система, состоящая только из спаренных электронов, называется
системой с замкнутыми оболочками (закрытыми оболочками). Та-
Такая система, содержащая четное число электронов, описывается
одним слэтеровским определителем:
•Vi
4>п(г2п) рBп)
C.33)
Неспаренные электроны образуют незамкнутые оболочки. Системы
с нечетным числом электронов являются системами с незамкну-
незамкнутыми (открытыми) оболочками. В этом случае волновую функцию
в общем виде более корректно представлять не одним детерминан-
детерминантом, а в виде линейной комбинации слэтеровских определителей
(см. разд. 4.3.4).
3.2.1. Двухэлектронная система
Примером двухэлектронной системы служит атом гелия. Рас-
Рассмотрим возможные распределения двух электронов между Is- и 2s-
АО атома гелия. Различные варианты распределения электронов по
орбиталям атома гелия показаны на рис. 3.3, где электрон со
спиновой функцией а обозначен стрелкой, направленной вверх, а со
спиновой функцией р — вниз; запишем для каждого состояния слэ-
теровский определитель:
аA)оB)
№Ж2)
-4%
V2
62
L
аB)
aB)
«D
где функции Ч
Функции Ч*а и ?, имеют симметричную пространственную часть
и антисимметричную спиновую, а функции ^и^, — антисиммет-
антисимметричную пространственную и симметричную спиновую части. Функ-
Функции ?г и Ч'а определяют тождественные состояния, и, следователь-
следовательно, полная волновая функция является их линейной комбинацией,
которая, например, равна
1
Эта функция также имеет антисимметричную пространственную
и симметричную спиновую части.
Задача 3.4. Написать возможные определители Слэтера для всех вероят-
вероятных спиновых состояний атома лития.
Таким образом, для возбужденных состояний б — д двухэлект-
ронной системы можно записать четыре спиновые функции, три из
которых симметричны и отвечают триплетному состоянию (полный
спин системы равен 1), а одна антисимметрична и определяет
синглетное состояние (полный спин системы равен 0):
симметричные функции
63
антисимметричная функция —у= [а( 1) /?B)—/?A) аB)].
3.3. МЕТОД ХАРТРИ—ФОКА
В. А. Фок* усовершенствовал метод Хартри, представив пол-
полную волновую функцию атома вместо C.3) в виде слэтеровского
определителя C.32). Пространственные орбитали определяются из
условия минимума полной энергии системы с помощью вариацион-
вариационного принципа.
Рассмотрим вначале подробнее выражение для полной энергии
атома:
где оператор Н имеет вид C.2), а полная волновая функция является
определителем Слэтера C.33) (для простоты будем считать, что
электронная оболочка атома замкнута и содержит 2п электронов).
Подставляя в уравнение C.34) выражения из C.2) и C.33) и проводя
интегрирование по пространственным и спиновым переменным,
получим формулу для полной энергии атома:
C.35)
где Kjj — обменный интеграл:
C36)
Выражение C.35) отличается от формулы полной энергии атома
в методе Хартри C.7) появлением под знаком суммы обменных
интегралов (сумму Y21 &и называют обменной энергией). Это обус-
обусловлено учетом требования антисимметричности волновой функ-
функции C.33), что является принципиальным отличием метода Харт-
Хартри—Фока от метода Хартри. Поясним физический смысл обменной
энергии.
При учете принципа Паули два электрона с параллельными
спинами не могут находиться в одной точке пространства. Следова-
*Фок Владимир Александрович A898—1977) — выдающийся советский физик-
теоретик, академик. Развил и обобщил метод Хартри для расчета стационарных
состояний атомов и молекул, для описания данных по рассеянию электронов атома-
атомами, фотоэффекту и другим свойствам, определяемым электронными оболочками
атомов и молекул. Метод Хартри—Фока (метод самосогласованного поля), лежа-
лежащий в основе всех практических методов расчета электронных оболочек атомов
и молекул, является основным в современной квантовой химии.
64
тельно, среднее расстояние между электронами в этом случае боль
nje, а электростатическая энергия отталкивания меньше на вел»
чину, соответствующую обменной энергии.
Интегралы Кц и Зи в атомах всегда положительны. Из C.S
и C36) видно, что
Задача 3.5. Если ввести обозначение
OAkl)'\\x^)%{Y>r-1XlV^)^l2)dzl dr2, C.3!
то /у- (и\ц) и Ки*= (ij\ij). , C.3!
Покажите, что для кулоновсхих /,у и обменных Ку интегралов выполняютс
соотношения
Kij^J-ф C.4(
1
¦ C.4
hih^fii- C.4:
Применим вариационный принцип для нахождения орбитале
Ч*,-. Вывод уравнений Хартри—Фока проводится аналогично вывод
уравнений Хартри. Орбитали ЧР, по условию считаем ортонормирс
ванными, поэтому минимизация полной энергии Е C.34) должн
проводиться при учете условия ортонормированности. Для этог
составляется новая функция (функционал)
i j i=l
где ги — множители Лагранжа.
Полная энергия Е C.35) достигает минимума по отношение
к орбиталям ?j при условии обращения первой вариации &.
в нуль или
л п
-ау- J ?i J W% А = 0.
1-1
Подставляя в C.44) выражение для интегралов Зц и Ки из C.S
и C.36) и проводя в них варьирование по 4^, получим
3- Теория строения молекул
Это равенство выполняется при любых 54h только если выражение
в фигурных скобках обращается в нуль:
J-
(i=l, 2, ..., л).
C.45)
Систему уравнений C.45) называют уравнениями Хартри—Фока,
которые отличаются от уравнений Хартри C.14) появлением обмен-
обменного члена [второй член в круглых скобках в C.45)]. Таким образом,
частным случаем уравнений Хартри—Фока C.45) при пренебреже-
пренебрежении в них обменным членом являются уравнения Хартри C.14).
Решение уравнений Хартри—Фока проводят таким же образом, как
и уравнений Хартри, т. е. численно. Полученные функции Ч*, пред-
представляют в виде таблиц. Например, в табл. 3.1 представлена ради-
радиальная функция распределения 15- и 2?-электронов для атома берил-
бериллия.
Таблица 3.1. Радиальная функция Ъу/пг R(r) распределения электронов, полу-
полученная do методу Хартрн—Фока для атоме Be
г(а.с.)
0,05
0,1
0,2
0,3
и
0,601
0,988
1,345
1,385
Ъ
0,126
0,205
0,265
0,243
г(а.е.)
0,5
0,6
0,8
1,0
Is
1,111
0,931
0,612
0,381
2г
0,085
-0,016
-0,214
-0,384
г(а.е.)
1,5
2,0
2,5
3,0
1j
0,102
0,025
0,006
0,002
Ъ
-0,638
-0,685
-0,617
-0,504
На рис. 3.4 дано сравнение функции Р(т) (л = 3, /==1), вычислен-
вычисленной из уравнений Хартри и Хартри—Фока. Легко заметить, что
метод Хартри ведет к недостаточно точному радиальному рас-
распределению электронной плотности для валентных электронов.
Вычисленные с помощью метода
Хартри—Фока электронные плотно-
плотности атомов являются достаточно точ-
точными и хорошо совпадают с экспери-
экспериментальными величинами. Напри-
Например, на рис. 3.5 приводится сравнение
радиальной электронной плотности
вычисленной по методу Хартри—Фо-
Хартри—Фока для атома аргона с эксперимен-
экспериментальными данными, полученными
с помощью метода дифракции элект-
электронов.
0,'
-0,1
-0.J
J ч 5 6 7 8 9г/а0
Рве. 3.4. Сравнение решении ура-
уравнений Хартри A) с решением
уравнений Хартри -Фока B) для
валентного Зр-электрона атома
натрия [по оси ординат отложена
функция Р(г}]
66
Вернемся к уравнениям
C.45). Умножим обе части каж-
каждого из этих уравнений на
4*i слева и проинтегрируем по
всему пространству, что ведет к
Яц+
1-1
C46)
В выражении C.46) е„ как и в ме-
методе Хартри, соответствует
полной энергии электрона, на-
находящегося на орбитали *F(-. Ис-
Используя формулу C.46), выра-
выражение для полной энергии системы C.35) можно переписать в виде
Рис. 3.5. Электронная плотность в атоме
аргона в зависимости от г. Сплошная
линия соответствует экспериментальным
данным, пунктирная — решению уравне-
уравнении Хартри-—Фока
C.47)
Если при отрыве электрона с орбитали 4ft не происходит изменения
волновых функций % (j^i)j то е, можно приравнивать к потенциалу
ионизации 1-й орбитали. Этот результат известен под названием
теоремы Купманса. Неизменность волновых функций при иониза-
ионизации фактически означает отсутствие эффектов электронной реор-
реорганизации. В действительности это предположение строго не выпол-
выполняется. Более правильно потенциал ионизации следует вычислять
как разность полных энергий нейтрального атома и иона, причем
обе величины следует получить с помощью метода Хартри—4>ока.
Уравнения ССП должны быть решены отдельно для нейтрального
атома и иона. Однако потенциалы ионизации, вычисленные при
помощи теоремы Купманса, нередко оказываются хорошим при-
приближением к истинным потенциалам ионизации и поэтому из-за
простоты вычислений используются как оценки.
3.4. ПРИБЛИЖЕННЫЕ АНАЛИТИЧЕСКИЕ ФУНКЦИИ
АТОМНЫХ ОРБИТ АЛЕЙ
Атомные орбитали (АО) Хартри—Фока вычислены для подав-
подавляющего большинства атомов и ионов. Работа с ними чрезвычайно
сложна и неудобна из-за того, что АО Хартри—Фока не могут быть
получены в аналитической форме и представляют собой числовые
таблицы. В связи с этим предложено несколько аналитических
аппроксимаций АО, являющихся приближениями к функциям Харт-
Хартри—Фока.
67
3.4.1. Атомные орбнталн Слэтера — Зенера
Наиболее распространенными и удобными приближенными
функциями являются АО Слэтера—Зенера:
"*~1 " " '¦ - C.48)
где #л=B?)я+1 [Bл)!]~ —нормировочный множитель;
?== - орбитальная экспонента; п* и S3KP — постоянные чис-
п
ла, определяемые в согласии с изложенными ниже правилами.
Форма волновой функции C.48), предложенная Слэтером, тесно
связана с видом атомных орбиталей водородоподобного атома
B.46). Функции C.48) являются решениями уравнения B.28) для
радиальной части гамильтониана водородоподобного атома B.2),
в котором оператор потенциальной энергии имеет вид
п*(п*-\)
5
При больших значениях г вторым членом можно пренебречь, тогда
\(г)с*-е2 . C.50)
г
Из этой формулы ясен физический смысл константы S^p, определя-
определяющей величину экранирования заряда ядра электронами. Собствен-
Собственные значения гамильтониана водородоподобного атома с потенциа-
потенциалом C.49) равны
или (в эВ)
?=-13,6
(п*)
Формула C.51) совпадает с формулой B.41) для водородоподоб-
водородоподобного атома при условии определения (Z—S3ip) как эффективного
заряда ядра и и* как эффективного главного квантового числа.
Константы экранирования S3xp для элементов второго периода до
атома фтора были вычислены Зенером. Слэтер аппроксимировал
Sxp и п* для всех элементов таблицы Д. И. Менделеева так, чтобы
68
эти значения хорошо согласовывались с расчетами Зенера и э
дериментальными данными по рентгеновской спектроскопии атов
Правила Слэтера для определения п* и 5^ сводятся к pjj
положений:
1) значение п* связано с главным квантовым числом следующ
образом:
л 1234 5 б
п* 1 2 3 3,7 4 4,2
2) постоянные экранирования ?ЭЖр находят, распределяя орбаг
ли по группам: (Is\Bs2p), C*3/0, C<0, Ds4p), Dd), D/), Es5p) и т.
Все орбитали одной группы имеют одинаковую радиальную фу!
цию. Значение S3tp для данной орбитали является суммой вклад*
вносимых отдельными электронами, т. е. вклад в 5^ равен:
а) нулю для любых электронов, расположенных во внешв
группах;
б) 0,35 от каждого электрона в той же группе, кроме рассмг
риваемого; в группе Ц вместо этого значения берется 0,30;
в) для s- и р-электронов по 0,85 от каждого электрона с главш
квантовым числом на единицу меньше, чем у рассматриваемо
электрона, и по 1,00 от всех электронов, расположенных ej
глубже;
г) для d- и/-электронов по 1,00 от каждого электрона, pi
положенного во внутренних группах.
Суммируя правила а) — г), запишем формулы для вычислен
констант экранирования:
(Ь)=0,30(Ь-1);
B)B 85(
t. д.
При вычислении констант экранирования для данной электронн
конфигурации необходимо вместо Is, 2s, 2р и т. д. в правой час
равенств подставить числа электронов на данной орбитали.
Рассмотрим в качестве примера расчет констант экранирован
S3ip для разных заполненных орбиталей атома углерода в ochobhi
состоянии. Разобьем все электроны на группы (Is), Bs2p) в соотв<
ствии с п. 2. Тогда для 1$-орбитали константа экранирования рав
6-0,30 = 5,70, а для ls-орбитали — 6-3 @,35)-2@,85) = 3,25.
Таким образом, валентные электроны углерода вследствие :
ранирования их внутренними электронами и межэлектронного с
талкивания испытывают значительно более слабое притяжение к у
РУ, чем это можно было ожидать исходя из величины заряда ядр
Отличие орбиталей Слэтера—Зенера от водородоподобных за-
заключается в том, что от присоединенного полинома Лягерра оста-
оставлен только его главный член. Вследствие этого орбитали Слэте-
Слэтера—Зеыера с одинаковыми / и т, но различными п не ортогональны
друг к другу и не имеют узлов в радиальной части, в то время как
водородоподобные АО имеют л—/— 1 узлов.
Задача 3.6. Вычислить первый потенциал ионизации атомов натрия в ка-
калия.
Задача 3.7. Найти первые потенциалы ионизации для элементов серы, селе-
селена и теллура, используя правила Слэтера, и полученные результаты сравнить
с экспериментальными данными.
3.4.2. Двухэкспоненциальные и гауссовские АО
Радиальные функции слэтеровского типа C.48) недостаточно
точно описывают поведение АО Хартри—Фока на небольших рас-
расстояниях от ядра. Этот недостаток можно устранить, если исполь-
использовать для аппроксимации каждой АО Хартри—Фока по крайней
мере две слэтеровские функции с разными орбитальными экспонен-
экспонентами:
„•-1 -<
C.52)
Двухэкспоненциальные функции C.52) дают хорошее приближение
к функциям Хартри—Фока почти во всей области изменения г. Эти
функции называют дубль-зета-функциями, а базисный набор, по-
построенный на них, — дубль-зета-базисом (DZ). Из рис. 3.6, на
DZ
0,д
г,о г /а0
Рис. 3.6. Сравнение радиальных частей различных АО 3^-волновой функции атома
железа: X—Ф — Хартри—Фожа; DZ — дубль-эета (см. разд. 4.3.5): S — Слэтера
70
котором представлены радиальные части различных АО, видно, что
дубль-зета АО дают хорошее соответствие с функциями Хартри—.
фока. Слэтеровские функции являются диффузными, размазанными
по всему пространству.
При расчетах волновых функций молекул широкое распрост-
распространение получили гауссовские функции типа
Gmkm = NMr'Xexp(-ar2) YJB. Я>)> C.53)
где #„ — нормировочный множитель; а — варьируемый параметр.
Одна слэтеровская АО аппроксимируется обычно несколькими
гауссовскими функциями. Таким образом, базис гауссовских функ-
функций всегда больше базиса слэтеровских АО. Однако это компен-
компенсируется тем, что интегралы различного типа, включающие гаус-
гауссовские функции, вычисляются значительно легче (подробнее см.
разд. 4.3.5).
3.5. ЭНЕРГЕТИЧЕСКИЕ УРОВНИ
МНОГОЭЛЕКТРОННЫХ АТОМОВ
Хартри-фоковские расчеты атомов и анализ атомных спектров
показывают, что орбитальные энергии б, зависят не только от
главного квантового числа п и заряда ядра Z, но и от орбитального
квантового числа /. Если бы экранирование ядра внутренними элек-
электронами было полным, то энергетические уровни внешних электро-
электронов были бы идентичны уровням атома водорода. Отклонение от
уровней атома водорода является непосредственной мерой влияния
неполного экранирования (так называемый эффект проникновения).
Все уровни атома лития расположены ниже соответствующих уров-
уровней атома водорода, причем сдвиг их тем меньше, чем больше
угловые моменты соответствующих орбиталей, т. е. ^-уровень сдви-
сдвигается сильнее ^-уровня, /7-уровень — сильнее ^-уровня и т. д. Энер-
Энергии орбиталей уменьшаются с возрастанием Z. Понижение энергии
орбитали уменьшается с ростом главного квантового числа п. Рас-
Расщепление уровней с данным п возникает из-за межэлектронного
отталкивания. В пределе при Z-* оо орбитали внутренних электро-
электронов с данными л снова становятся вырожденными по /, так как
межэлектронное взаимодействие становится незначительным по
сравнению с электронно-ядерным взаимодействием.
Задача 3.8. Найти первый потенциал ионизации и потенциал ионизации
Зй?-электронов атомов железа, никеля и цивка из формулы C.51) и сравнить
полученные значения с опытными величинами /(Fe)=7,89; /(Ni) = 7,69:
/(Zn)=9,39 эВ.
71
3.5.1. Принцип построения
периодической системы элементов Д. И. Менделеева
Порядок заполнения электронами отдельных уровней в атоме
определяется следующими правилами, совокупность которых назы-
называется принципом построения:
1) заполнение атомных орбиталей электронами происходит
в порядке увеличения энергии орбиталей, за исключением атомов
переходных металлов, у которых вначале заполняются более высо-
высокие j-орбитали, а ^-орбитали остаются вакантными;
2) согласно принципу Паули на каждой АО, характеризующейся
квантовыми числами п, I, т, может находиться не более двух
электронов;
3) АО с одинаковыми / и п заполняются так, чтобы суммарный
спин электронов был максимален, т. е. заполняется максимальное
число орбиталей с разными т (правило Хунда). На рис. 3.7 показа-
показаны электронные конфигурации некоторых атомов. Каждая ячейка
соответствует атомной орбитали, положение которой (в порядке
увеличения энергии) в определенной группе определяется квантовы-
квантовыми числами п, I, m и порядком энергетических уровней (правило 1).
В каждую ячейку можно поместить не больше двух электронов.
Аналогичным образом можно построить электронные конфигура-
конфигурации всех атомов периодической таблицы.
Из приведенной на форзацах книги таблицы Д. И. Менделеева
видно, что электроны в ^-оболочке появляются впервые в пятом
элементе — боре, ^-оболочка начинает заполняться у скандия
(Z=2l), а электроны в/-оболочке появляются у церия (Z=58).
Момент появления в атоме электронов с данным / описывается
формулой
Z= 0,17B/+1K. C.54)
Для /=1, 2 и 3 эта формула дает после округления до ближайших
целых чисел правильные значения: 5, 21 и 58. Для 1=4 формула
C.54) приводит к Z— \2А. Это означает, что ^-электроны должны
впервые появиться лишь в 124-м элементе. Наиболее точные рас-
Li
N
Fe
tl
t
5
/r-2
tl
tl
s
t
' X
r
f
n-3
tl
tv
л
tl
tl
tl
tl
tl
tl
tl
tl
p,
tl
t
t
c/72
Рис. З.7. Распределение электронов по оболочкам в атомах лития, азота и железа
72
четы электронных конфигураций гипотетических тяжелых атомон
до методу Хартри—Фока, которые проведены вплоть до элемента
с z= 172, показывают, что застройка ^-оболочки начинается у эле
мента с Z= 125.
3.5.2. Потенциалы ионизации и сродство к электрону
Потенциалом ионизации атома X (/х) называют минимальное
значение энергии, необходимое для удаления электрона из атома X,
т. е.
Второй, третий и т. д. потенциалы ионизации определяются как
энергии, необходимые для удаления электрона от однозарядного
(X ), двухзарядного (Х2+) положительного иона атома Хит. д.
Основные экспериментальные методы определения потенциалов
ионизации основаны на нахождении предела сходимости спектраль-
спектральных линий в атомных спектрах или применении метода фотоэлект-
фотоэлектронной спектроскопии. Для вычисления потенциала ионизации ато-
атома следует рассчитать его энергию до и после ионизации и взять их
разность. Такая процедура получила сокращенное название ДССП,
если расчет проводится методом Хартри—Фока. Более простой
путь расчета /х заключается в использовании теоремы Купманса.
В табл. 3.2 приведены экспериментальные и теоретически опре-
определяемые по теореме Купманса (так называемые вертикальные
потенциалы ионизации) и методу АССП значения потенциалов
ионизации некоторых элементов.
Таблица 3.2. Вычисленные ¦ экспериментальные значения потенциалов иониза-
ионизация элементов второго и третьего периодов, эВ
Атом
Li
Be
В
С
N
0
F
Ne
Расчет
ДЯссп
5,34
8,04
7,93
10,79
13,96
11,89
15,72
19,84
теорема
Купманса
5,34
8,42
8,43
11,79
15,44
17,19
19,86
23,14
Экспери-
Эксперимент
5,39
9,32
8,30
11,26
14,53
13,62
17,42
21,56
Атом
Na
Mg
Al
Si
P
s
a
Ar
Расчет
Д^ссп
4,95
6,61
5,51
7,66
10,04
9,03
11,80
14,78
теорема
Купмавса
4,96
6,88
5,71
8,08
10,66
11,90
13,78
16,08
Экспери-
Эксперимент
5,14
7,65
5,98
8,15
10,49
10,36
12,97
15,76
Из табл. 3.2 видно, что зачастую значения /х, полученные с по-
помощью теоремы Купманса, лучше согласуются с эксперименталь-
экспериментальными данными, чем величины, определенные по методу ЛССП.
Причина этого кроется, вероятно, в неучете корреляционной энер-
энергии в методе Хартри—Фока (см. разд. 4.4).
73
1
Зависимость потенциалов ионизации от атомного номера имеет
четко выраженный периодический характер.
1. В каждой группе периодической системы потенциал иониза-
ионизации понижается с увеличением атомного номера, что связано с уве-
увеличением размера атома, в то время как тип электронной кон-
конфигурации сохраняется. Исключения составляют элементы, следу-
следующие за лантаноидами. Это является следствием так называемого
лантаноидного сжатия, которое возникает из-за увеличения заряда
ядра без появления более удаленных электронных уровней.
2. В каждом периоде величина /х возрастает слева направо,
наибольший потенциал ионизации имеют атомы инертных газов.
В этой закономерности заметны некоторые отступления, например
he>h, Л><Л*» Л«в>/л1, h<Ip- Причина отступлений: особая устой-
устойчивость замкнутых оболочек Is2, Is2 атома Be и Isz2s22p63sz атома
Mg, а также большая устойчивость конфигураций с максимальной
мультиплетностью: Is22s22p3 атома N и \sz2s22p6Zsz2p5 атома
Р в соответствии с правилом Хунда.
Сродством к электрону атома X (Ах) называют энергию, кото-
которая выделяется при присоединении к нейтральному атому X элект-
электрона с образованием отрицательного иона, т. е.
. C.56)
Экспериментальные методы определения сродства к электрону
весьма сложны, поэтому значения Ах определены не для всех эле-
элементов таблицы Д. И. Менделеева. Впрочем, и расчет этого свойст-
свойства представляет сложную задачу, хотя формальная схема проста:
требуется взять разность энергий нейтрального атома и отрица-
отрицательного иона. Расчет энергии последнего вносит основную ошибку.
Так, например, для атомов галогенов имеем: ЛР(эксп.) = 3,45 эВ,
ЛР(расч.)=1,36 эВ; ^4а(эксп.) = 3,61 эВ; Ла(расч.) = 2,58 эВ;
ABl (эксп.) = 3,36 эВ; АВт(р&сч.) = 2у5^ эВ. Для более точного расчета
сродства к электрону следует использовать более строгие теории,
выходящие за рамки приближения метода Хартри—Фока.
3.6. КВАНТОВЫЕ ЧИСЛА МНОГОЭЛЕКТРОННОГО АТОМА
В случае замкнутой электронной оболочки существует единст-
единственный способ размещения электронов по орбиталям, например для
атома Ne:
Ne 1,'2,'2,' MtJltllH
74
В атомах с не полностью заполненными оболочками имеются
уже несколько вариантов записи определителей Слэтера элект-
электронных конфигураций. Например, для атома углерода возникают
следующие конфигурации:
С Is2 2s2 2p2
и
н
б)
н
t
в)
u
t
t
и т.д.
Часть этих конфигураций может иметь одинаковую энергию,
образуя вырожденный набор состояний. Конфигурация с наимень-
наименьшей энергией соответствует основному состоянию атома, оста-
остальные конфигурации относятся к возбужденным состояниям. Расчет
энергии каждой конфигурации можно произвести с помощью мето-
метода Слэтера. Например, для конфигураций а, б к в атома углерода
слэтеровские детерминанты принимают соответственно следующий
вид:
Ча= \\sct \Spisa2sp 1рхч, 2pJ\,
= \lsa
a
2pzp\,
2pya\.
C.57)
Функция из C.57), для которой средняя энергия атома углерода,
вычисленная согласно A.34), имеет наименьшее значение, определя-
определяет электронную конфигурацию основного состояния. При этом
необходимо следить, чтобы используемые функции были собствен-
собственными функциями операторов S2 и L2 (см. разд. 3.6 и 3.7). Расчеты
такого типа достаточно трудоемки, однако они выполнены в насто-
настоящее время для всех наиболее существенных электронных конфигу-
конфигураций атомов периодической системы, в том числе и для еще не
синтезированных сверхтяжелых элементов. Анализ этих результа-
результатов позволяет перейти к формулировке квантовых чисел многоэлек-
многоэлектронных атомов.
3.6.1. Полные орбитальные и спиновые квантовые числа
При сильном межэлектронном взаимодействии, превышающем
энергию спин-орбитального взаимодействия (см. разд. 3.6.2), кван-
квантовые числа отдельных электронов теряют физический смысл из-за
их неразличимости, т. е. нельзя каждому электрону приписать со-
собственный угловой и спиновый моменты. Имеет смысл в этом
случае говорить лишь о полных орбитальном и спиновом моментах
совокупности электронов. Для них действительны следующие соот-
соотношения:
75
, C-58)
где L и S — соответственно полное орбитальное и полное спиновое
квантовые числа.
Полное орбитальное квантовое число L определяется через зна-
значения орбитальных квантовых чисел I,отдельных электронов. L мо-
может принимать только целые положительные значения и значение,
равное нулю. Замкнутые оболочки Д р6, rf10 и т. д. имеют полный
момент L, равный нулю, и для вычисления L различных элект-
электронных систем необходимо рассматривать электроны только в не-
незаполненных оболочках. Для двух электронов с орбитальными
квантовыми числами /t и 1г квантовое число L принимает значения
-1; (h-h+\),\h~h\, C.59)
всего 2/2 + 1 значение при I2<h или 2/,-f 1 при Д </2.
Если имеется три электрона с //#0, то сложение моментов может
быть произведено последовательным сложением вначале /, для двух
электронов и потом сложением каждого из полученных значений
результирующего L с /3. Например, для двух /7-электронов с /, = /2 = 1
L может принимать значения 2, 1 и 0; для двух р- и ^-электронов
сА = 1 и/2-2 L = 3, 2, 1.
В общем случае, когда число электронов в незамкнутых оболоч-
оболочках больше двух, сначала находят результирующий орбитальный
момент каждой оболочки и затем последующим сложением находят
результирующий орбитальный момент всего атома. Если Lxn L2 —
результирующие моменты двух различных оболочек, то
?=Li + L2, L{ + L2-h -, Li-Li, C.6O)
когда L, >
Аналогично буквенным обозначениям орбиталей в атоме водо-
водорода состояния с различными L обозначают следующим образом:
L ... 0 1 2 3 4 5
Обозначение ... S P D F G Н
Полное спиновое квантовое число S находится по тем же прави-
правилам. Для замкнутых оболочек S—0 и значения полного спина
любой электронной системы определяются спинами электронов
лишь в незамкнутых оболочках. Квантовое число S полного спина
для оболочек, заполненных не более чем наполовину, может прини-
принимать следующие дискретные значения:
76
N N N 1
где N — число электронов в незамкнутой оболочке.
В зависимости от того, является ли N четным или нечетным,
значения S C.61) соответственно будут целыми или полуцелыми.
Аналогично проекциям орбитального т и спинового njs момен-
моментов электрона в водородоподобном атоме для многоэлектронных
систем вводятся проекции полного орбитального ML и полного
спинового Ms моментов, которые могут принимать дискретный ряд
значений:
ML=L, L— 1, ..., — L+l, — L, всего 2L+1 значений;
C.62)
MS=S, 5—1, ..., —5+1, — 5, всего 25+1 значений.
3.6.2. Спин-орбитальное взаимодействие.
Квантовое число полного момента
Токи, связанные с орбитальным движением электрона и с его
спином, взаимодействуют друг с другом. Каждый из этих токов
создает магнитное поле, которое воздействует на другой ток. Вза-
Взаимодействие магнитных полей, создаваемых токами, обусловливает
зависимость орбитального и спинового моментов количества дви-
движения совокупности электронов, его называют спин-орбитальным
взаимодействием или спин-орбитальной связью. Энергия спин-ор-
спин-орбитального взаимодействия много меньше разности энергетических
уровней электронов, но, несмотря на это, она оказывает существен-
существенное влияние на стационарные состояния атома. Это влияние приво-
приводит к снятию вырождения состояний с одним и тем же квантовым
числом орбитального движения. Подобное снятие вырождения слу-
служит основной причиной появления тонкой структуры атомных спек-
спектров (см. разд. 3.9) в отсутствие внешних полей. Строгое рассмотре-
рассмотрение спин-орбитального взаимодействия возможно при решении ре-
релятивистского уравнения Дирака. Однако полуклассический подход
позволяет выявить наиболее важные детали этого эффекта.
Вследствие относительности движения можно рассматривать не
движение электрона вокруг ядра, а ядра вокруг электрона с той же
скоростью if. Движущийся положительный заряд ядра создает
в месте нахождения электрона магнитное поле 2?, равное
C.63)
где"? — напряженность электрического поля у электрона, которую
находят по обычной формуле
77
^ C.64)
г3
Энергия взаимодействия магнитного поля C.63) со спиновым маг-
магнитным моментом электрона jts определяет энергию спин-орби-
спин-орбитального взаимодействия:
= -В /?,= ^ . C-65)
Поскольку, согласно B.103),
-?, —5и!?=А C.66)
тс т
а также учитывая, что вследствие релятивистских эффектов в выра-
выражение для энергии C.65) необходимо ввести множитель 1/2, получим
Ze* -~ (
так как (г х~р ) — L согласно B.69).
Для более общего случая некулоновского поля напряженность
выражается через потенциал V(r) как
г
Тогда выражение для энергии спин-орбитального взаимодействия
C.67) перепишется в виде
Л - 3-f (ti). C.69)
г or
Гамильтониан спин-орбитального взаимодействия записывается
следующим образом:
HL-5-U-T-^^- (ЗЛ0)
2mzcz r дг
где L — оператор орбитального момента с компонентами B.70),
S — оператор спина электрона (см. разд. 2.5.4). Для п электронов
соотношение C,70) легко обобщается:
c2 ~ rt
где AjfrJ имеет вид
Ь Z (LSJ 1 AM (L *s 0-
r,- drf
78
Полный оператор гамильтониана для атома складывается из
операторов C.2) и C.71):
C.73)
Точное вычисление энергии спин-орбитального взаимодействия мо-
можно провести только отыскав собственные функции и собственные
значения оператора C.73). Такая процедура достаточно трудоемка,
однако, поскольку величина энергии спин-орбитального взаимодей-
взаимодействия мала по сравнению с разностью энергий соседних уровней
Ei и Ei+\ гамильтониана Н C.2), это позволяет использовать теорию
возмущений. Например, для атомов второго периода энергия спин-
орбитального взаимодействия равна 10~2—10~3 эВ, а расстояние
между уровнями 2 — 10 эВ.
Энергия спин-орбитального взаимодействия в первом порядке
теории возмущений рассчитывается по формуле A.80), которая
в нашем случае имеет вид
%Щ. C-74)
где Ч* — собственная функция оператора Н.
Используя очевидное соотношение
(L+$J=L2+S2+2L'S, C.75)
перепишем C.74):
Wtt i>]W. C-76)
Таким образом, энергия спин-орбитального взаимодействия зави-
зависит не только от угловых моментов L и 5, но и от их суммы.
В связи с этим возникает необходимость введения полного углового
момента атома J, являющегося векторной суммой полного ор-
орбитального и спинового моментов:
C.77)
—»¦ —> —>
причем величины \J |, так же как и \L \, \S |, квантуются:
C'78>
где / — квантовое число полного углового момента атома
и принимает положительные целые или полуцелые значения:
/=Z,+S,L+S-1, ..., |L-S| + 1, \L-S\. C.79)
При L>S число возможных значений / равно 25+1, при L<S
S2L1
Проекция полного момента J на ось z принимает дискретный
ряд значений:
79
Mj=Jt 7-1,/-2,...., -/+1, -J, C.80)
т. е. всего 2J+ 1 значений. _^
Введем операторы полного момента J и его проекции. Операто-
Операторы J и Jx связаны с ? и S простыми соотношениями
г==1-»г "г" Э2.
Функция *F является собственной функцией операторов L2, S2, Н,
а также J2 (см. ниже). С учетом C.77) и C.78) можно записать
C.82)
Задача 3.9. Показать, что операторы Н C.2), L2, S2, J2 обладают оди-
одинаковой системой собственных функций.
Тогда выражение для энергии спин-орбитального взаимодейст-
взаимодействия C.76) можно представить как
^ C.83)
C.84)
i
где А — константа для данного электронного состояния, которая
зависит от L и S, но не зависит от /.
Задача ЗЛО. Найти энергию спин-орбитального взаимодействия для элект-
электрона, находящегося на 2р-АО атома водорода.
Следует указать, что гамильтониан C.73), включающий спин-
орбитальное взаимодействие, не коммутирует с операторами L2
и S2- В этом случае с полным гамильтонианом коммутируют
только операторы J2 и J2:
Использованные выше способы введения квантовых чисел
L и S и квантового числа полного углового момента / правомерны
для случая сравнительно слабого спин-орбитального взаимодейст-
взаимодействия, когда в первом приближении можно пользоваться представле-
представлениями о полном орбитальном и спиновом угловых моментах. Это
приближение называют связью Рассела—Саундерса или LS-связью.
80
Оно справедливо для относительно легких атомов с Z^30, когдг
взаимодействие спинового момента электрона с его орбитальньш
1 моментом меньше взаимодействия орбитальных и спиновых мо
ментов электронов между собой.
При увеличении атомного номера (при Z>30) приближение
Рассела~-Саундерса становится неприменимым и необходимо те
перь вначале суммировать спиновый и орбитальный моменты каж
дого электрона:
а полученные полные моменты каждого электрона суммироват!
между собой. Такая схема сложения моментов называется jj-связью
3.7. ТЕРМЫ МНОГОЭЛЕКТРОННЫХ АТОМОВ
На основании экспериментальных данных по атомной спектро
скопии и теоретических расчетов энергий различных состояний бы-
были выработаны общие правила классификации атомных состояний
и их энергетической последовательности. Определенное энергети-
энергетическое состояние атома называют атомным термом.
3.7.1. Термы в приближении 7,5-связи
В этом случае классификация термов осуществляется в соответ-
соответствии с величинами орбитального, спинового и полного моментов
атома. Терм обозначают следующим образом: + Lj. Слева вверх)
записывается мультиплетность состояния, определяемая величиной
полного спина 5 и показывающая число возможных значений про-
проекции полного спинового момента. В зависимости от спина состоя-
состояния называются следующим образом:
5= 0, 25+1 — 1 синглетное;
5= 72, 25+1 -2 дублетное;
5=1,25+1 = 3 триплетное;
5= 7э, 25+1 =4 квартетное.
Для данной электронной конфигурации атома может существовать
несколько термов. Порядок расположения термов по энергии опре-
определяется эмпирическими правилами Хунда:
I. Терм основного состояния (наинизший по энергии терм) все-
всегда имеет самое высокое значение спиновой мультиплетности.
II. Если несколько термов имеют одинаковую мультнплетность,
то наиболее стабильным будет тот, который имеет максимальное L.
Ш- Для конфигураций с не более чем наполовину заполненными
оболочками самым стабильным является терм с минимальным
81
значением /; если оболочка заполнена более чем наполовину, то
самым стабильным будет терм с максимальным значением /.
Правила Хунда применимы только для определения терма ос-
основного состояния.
Для определения терма основного состояния атомов удобно
пользоваться такой последовательностью правил.
1. Записывается электронная конфигурация незаполненных обо-
оболочек атома.
2. Электроны распределяются по соответствующим ячейкам
так, чтобы в соответствии с первым и вторым правилами Хунда
получить максимальные значения S и L, т. е. составляется кон-
конфигурация с максимальным числом неспаренных спинов и мак-
максимально возможным числом электронов в ячейках с наибольшими
значениями т.
3. Квантовые числа т неспаренных электронов суммируются
и дают ML. Полученное значение ML определяет величину кван-
квантового числа L.
4. По числу неспаренных электронов определяется мультиплет-
ность терма.
5. В соответствии с третьим правилом Хунда находится кван-
квантовое число /.
Воспользовавшись этими правилами, определим термы основ-
основных состояний некоторых атомов.
Атом кислорода:
I) 2\
2)
1 0 -1
m
НИИ
3) ML= 1; р-состояние;
4) 5=1; следовательно, мультиплетностъ равна 3;
5) возможные значения /=2,1,0. Оболочка заполнена более чем
наполовину; следовательно, выбирается максимальное значение
/=2. В итоге терм основного состояния атома кислорода — 3Р2-
Атом хрома:
1) ЗА*1;
4s
2)
t
f
t
t
t
t
m
I 0-1-2
0
3) ML=05 5-состояние;
4) мультиплетность равна 7;
82
5) имеется единственное значение J—3. Итак, терм основногс
состояния атома хрома — 1S3.
Описанные правила позволяют определить только терм основ-
основного состояния. Для определения всех возможных термов даннок
электронной конфигурации можно воспользоваться процедурой, па
нятной из приведенного ниже примера определения термов кон-
конфигурации р2, например атома углерода. Составим таблицу всеэ
возможных микросостояний (способов отнесения электронов раз
личным квантовым числам), определяемых различными ML и Ms:
ML=ml-\-m2\ ть т2= 1, 0, — 1;
ML=2, 1,0, -1, -2;
-\, 0, -1
Таблица 3.3. Возможные микросостояння конфигураций р1
М,
м<
~2
+
0
о
-1
+
-1
+
о
-Jr -1
о
1 1
! 0
о о
-1 I
-1 0
-1
\ О
/о
1 -I
-I 0
-I
Каждая клетка (табл. 3.3) соответствует состоянию с определен-
определенными ML и Ms, причем данным значениям ML и Ms могут соответ-
соответствовать несколько микросостояний с различными /и, и mSi. В табл.
3.3 не введены 15 тождественных микросостояний. Например, для
Ms— 0 и ML~2 кроме указанного микросостояния 1 1 возможно
- +
и тождественное 1 1. Например, для ML=2 и Ms=\ существует
единственный набор т( и mSi, т.е. ml~m2=\ и mSl =mS2=\/2. Для
ML~0 и Ms~0 существует, однако, три возможных микросостоя-
микросостояния:
83
2) /w, = 1, /и2= -1, msl = l/2, w4 = - V2;
3) m, = -1, m2-1, msi = V2, *яч= - Vi-
Из табл. 3.3 необходимо удалить наборы, не подчиняющиеся
принципу Паули% В нашем случае таких наборов шесть (они пере-
перечеркнуты). Далее выбирается любое микросостояние, причем для
удобства обычно* берут набор, соответствующий максимальному
Ms и ML- В нашем случае это состояние с Ms*=\ и ML—\. Таким
величинам проекции соответствуют L— 1 и S= 1 или терм 3Р. Для
этого терма должно быть девять наборов /я, и mSi. Действительно,
,= 1,0, -1;
?=1,0, -1.
Выберем соответствующие наборы и заключим их в прямоуголь-
прямоугольные рамки. Если в клетке имеется два (или более) набора, то
произвольно выбирается любой. Далее выбирается другой набор
mi и тзр например соответствующий ML—2 и Ms=0. Тогда L=2,
5=0, что дает терм 1D, которому удовлетворяют пять микрососто-
микросостояний:
=2, 1,0, -1, -2;
Ms=0.
Заключим их в овальные рамки. После выделения микросостояний,
принадлежащих термам ЪР и 1Df остается один набор: ML = 0,
Ms—0 и L = 0, ?=0. Он отвечает терму J*S.
Правила Хунда позволяют определить устойчивость получен-
полученных термов 3P>LD>1S (знак > означает большую устойчивость
терма, т. е. более низкую энергию). Терм 3Р девятикратно вырож-
вырожден (трижды по S и трижды по JLV терм lD пятикратно вырожден
(по орбитальному числу), а терм \S является невырожденным.
Для терма 3Р возможны значения /=2, 1, 0. Согласно третьему
правилу Хунда, энергетическая устойчивость термов имеет следу-
следующий порядок: 3P0>*Pi>3P2. Термом основного состояния являет-
является терм 3Р0- Термы 1D и 15 имеют соответственно 7=2 и J— 1, т. е.
записываются как 1D2 и 1SQ. В табл. 3.4 приведены возможные
термы для ряда конфигураций эквивалентных (имеющих одинако-
одинаковые п и I) и неэквивалентных электронов.
84
Таблица 3.4. Термы различных конфигураций эквивалентных и неэквивалент-
неэквивалентных электронов
Эквивалентные электроны
Неэквивалентные электроны
конфигурация
термы
конфигурация
термы
1
47s
1p.3p
13
2Р
3Р, *Z>, lS
*S, 2D, 2P
2D
, ЪР, 1Gt lD,
ss
sp
sd
PP
, XD,
Задача 3.11. Найти термы основного состояния атомов Mg, Р, S и Q.
Отметим, что термы конфигурации, содержащей и эквивалент-
эквивалентных электронов, совпадают с термами конфигураций, в которой не
хватает п электронов до закрытой оболочки. Так, термы конфигура-
конфигураций р2 и р*, dx и d9 одинаковы. Закрытые оболочки всегда имеют
один терм 15.
При определении термов конфигураций, содержащих как эк-
эквивалентные, так и неэквивалентные электроны, необходимо найти
сначала возможные термы эквивалентных электронов. Затем берут-
берутся возможные комбинации величин L и S и определяются нужные
термы.
Проиллюстрируем нахождение термов на примере конфи-
конфигурации 2s2p3. Один s-электрон имеет терм 2S. Конфигурации ръ
соответствуют термы AS, 2D, 2P (см. табл. 3.4). Комбинируя значе-
значения L и S этих термов, получим термы конфигурации sp3 (в порядке
возрастания энергии):
SS>3D>3P>3S> lD> lP.
Величины разностей энергий термов
обычно сравнимы с энергиями химичес-
химических связей и химических реакций. Так,
энергии 1D и lS термов атома углерода
с конфигурацией Is22s22p2 выше терма
основного состояния ЪР соответственно
на 105 и 235 кДж/моль. Терм 5S кон-
конфигурации \s22s2p3 лежит выше терма ЪР
приблизительно на 402 кДж/моль. Рас-
Расположение термов и их относительные
энергии для атома кислорода показаны
на рис. 3.8. Энергию, необходимую для
перевода атома из одной электронной
конфигурации в другую, например s2p2
в .sp , называют энергией промотирова-
(подробнее см. гл. 10).
1,97 ЭВ
Ж"
о
г*
Г
III
Рис. 3.8. Энергетические
уровня \я2Ъ22р -электронной
конфигурации атома кислоро-
кислорода
85
Задача 3.12. Найти все возможные термы атома кислорода.
3.7.2. Энергетические уровни в приближении .//-связи
Нахождение термов в приближении jj-свхж осуществляется на-
наиболее просто в случае неэквивалентных электронов, например для
конфигурации s^p . Квантовые числа электронов равны: /, = О,
S\ — xl-b следовательно» y"i = х/2; /2— 1, s2=ll2 ^Н—11ъ 3/г (возможные
значения jt изменяются от суммы (li+sj до разности \l,—л,|). Путем
комбинации j\ и j2 получим допустимые значения У, также изменя-
ющиеся от суммы (j\ +j2) до разности [/\ — J2\. В нашем случае при
j\~xli и h-^lt имеем /=1, 0; при jl = i/2 и ]2 = Ъ12 допустимые
значения J=2, 1. Итак, энергетические уровни для конфигурации
svpl можно классифицировать так:
Перейдем к более сложному случаю эквивалентных электронов,
например конфигурации рг. Так же как и в случае LS-связч, возмож-
возможны 15 микросостояний, классифицируемых, однако, по другим при-
признакам. Квантовые числа электронов одинаковы, т. е. /i=/2 = l
и Si=s2=1/2. Возможные значения^ и j2: j\ =j2 =:э/2, lj2. Комбинируя
значения j\ и j2 для определения допустимых значений квантового
числа /, следует помнить о принципе Паули, не допускающем
существования в атоме двух электронов с одинаковыми квантовы-
квантовыми числами. Например, если^ =л= Х1ъ то электроны должны иметь
обязательно противоположные спины, или, другими словами, до-
допустимое значение /=0; значение J—1 приводит к противоречию
с принципом Паули. Аналогично, для _/t ==У2=3ii допустимые значе-
значения /=2, 0 (хотя формально /может принимать значения 3, 2, 1, 0).
В общем виде для конфигурации/2 разрешенные значения J изменя-
изменяются по закону 2/— 1, 2/— 3, 2/—5, ..., 0. В случае комбинации j\ = x/2
и к—ъ\г разрешены все варианты от суммы (jv+ji) до разности
l/i—Уг|- Тогда имеем J—2, 1. Итак, запишем энергетические уровни
для конфигурации р2:
[ШШШИ0Ш
86
3=0
L$- связь jj-сбязь
Рис. 3.9. Соответствие термов в приближениях LS- и j/'-связей
Для того чтобы расположить определенные термы по энергии,
следует провести корреляцию с соответствующими термами в при-
приближении LS-связи, для которых справедливы правила Хунда (по-
(подобных правил для^'-схемы не существует). Корреляцию необходи-
необходимо проводить по квантовому числу 7, которое является хорошим
квантовым числом для LS-и j/'-схем. Ранее были определены воз-
возможные термы и их последовательность для конфигурации р2 в при-
приближении 1Л-связи. На рис. 3.9 приведена корреляционная диаграм-
диаграмма, связывающая термы, определенные по разным схемам взаимо-
взаимодействия. При движении слева направо возрастает отношение энер-
энергии спин-орбитального взаимодействия к энергии межэлектронного
отталкивания.
3.8. МАГНИТНЫЕ МОМЕНТЫ МНОГОЭЛЕКТРОННОГО АТОМА
По аналогии с орбитальным и спиновым моментами одноэлект-
ронного атома B.90) и B.102) для многоэлектронного атома также
вводятся понятия орбитального и спинового магнитных моментов:
&L\ = ylLl=jL(L+l)pm C.85)
I? d = 2у|?| = 2y/S(S+l) ft,- C.86)
Полный магнитный момент fi j складывается из спинового и ор-
орбитального магнитных моментов:
1?j=~Hl+Jis C.87)
или
—*
?/= -{li?L|cos(L7)+|?dcosE7)} L C-88)
Ml
87
Используя формулу скалярного произведения и соотношения
(ls7) = ~{J(J+l)+S(S+l)-L(L + \)} C-89)
и 2
*7* * C.90)
перепишем C.88) в виде
где
g 2J(J+1)
называют фактором Ланде.
Таким образом, нами введены все квантовые числа многоэлект-
многоэлектронного атома.
Задача 3.13. Получить соотношения C.89), C.90).
Задача 3.14. Показать, что из соотношений C.88)—C.90) вытекают фор-
формулы C.91) и C.92).
3.9. СПЕКТРЫ МНОГОЭЛЕКТРОННОГО АТОМА
Как и в атоме водорода, в многоэлектронном атоме переход
с электронного уровня Е„ характеризующегося волновой функцией
Ч?ь на уровень Ек с функцией *Р* считается разрешенным, если
отличен от нуля матричный элемент:
M^fZ^^'*- C-93)
Точные вычисления матричных элементов C.93) показывают, что
для многоэлектронных атомов в приближении Ь5"-связи разреше-
разрешены переходы только между термами одинаковой мультиплет-
ности, т. е. изменение полного спина системы должно быть равно
нулю:
AS=0. C-94)
Квантовые числа LvlJ при электронных переходах должны менять-
меняться не более чем на единицу, т. е.
Д/. = 0, ±1; C-95)
ЛУ-0, ±1, C-96)
причем переход из состояния с Z,=0 в состояние с L—Q запрещен,
I
p
•p/2
p
tA
tt'ZS
Рис. ЗЛО. Тонкая структура спектральных линяй атомов натрия и лития
откуда вытекает также условие запрещенности перехода с уровня
с /=0 на уровень с 7=0.
По мере увеличения спин-орбитального взаимодействия запре-
запрещенные правилами C.95) и C.96) электронные переходы могут
появиться в спектрах атомов, однако обычно с весьма малой по
сравнению с разрешенными переходами интенсивностью.
В присутствии внешнего поля энергия уровня зависит от кван-
квантового числа Mj. Правила отбора в этом случае дополняются
соотношением
•=0, ±1. C.97)
При рассмотрении спектральной картины электронных переходов
атомов щелочных металлов, получаемой на спектрографах с высокой
разрешающей способностью, можно обнаружить, что каждая спект-
спектральная линия расщепляется на две близко стоящие друг к Другу
линии*. Этот эффект, известный в атомной спектроскопии как
проявление дублетной структуры спектров щелочных металлов, стал
одним из важнейших экспериментальных оснований введения пред-
представлений об электронном спине. Происхождение трех линий, регист-
регистрируемых на ранних приборах с недостаточным разрешением, легко
объяснить в рамках одноэлектронной модели атома (рис. ЗЛО).
Причиной дублетной структуры спектров атомов щелочных ме-
металлов является спин-орбитальное взаимодействие (см. разд. 3.6.2),
которое расщепляет каждый уровень cL=l и S=Lj2 на два уровня
с J=lj2 и 3/2, тогда как 5-уровни (L = 0) не связаны спин-орбиталь-
спин-орбитальным взаимодействием (рис. 3.10).
*Правило Л апорта: разрешены переходы между термами разной четности. Чет-
Четность терма определяется суммой /, отдельных электронов (?/i). Если сумма ?/,- чет-
i i
ная, то терм четный (g), если сумма нечетная, то терм нечетный (и).
89
ЗЛО. МНОГОЭЛЕКТРОННЫЙ АТОМ В МАГНИТНОМ ПОЛЕ
Рассмотренные энергетические состояния атома (термы) зависят
от квантовых чисел L, S и У, но не зависят от Mj, т. е. терм 27+1
кратно вырожден по J.
В магнитном поле вырождение снимается и энергия терма зави-
зависит также от квантового числа проекции полного момента Mj.
Энергия взаимодействия магнитного момента Jij с магнитным
полем индукции В в классическом случае определяется как
Еп=-?\B=yg7li. C.98)
Здесь использована формула C.91) для ц j. Направим вектор В
вдоль оси z в выбранной системе координат и перейдем в C.98)
к квантово-механическим операторам. Соответствующий гамиль-
гамильтониан
, C 99)
так как оператор магнитной индукции В =В .
Полный оператор Гамильтона для атома с учетом спин-ор-
спин-орбитального взаимодействия C.71) и взаимодействия с магнитным
полем C.99) запишется в виде суммы
Нполн=Н+Н0О + Нвз, C.100)
где Н находится из формулы C.2),
Если магнитное поле невелико, т. е. энергия его взаимодействия
со спиновым и орбитальным магнитными моментами меньше, чем
энергия их взаимодействия друг с другом, то связь LS сохраняется.
В этом случае для нахождения средних значений оператора Н„олн мо-
можно использовать теорию возмущений 1-го порядка, которая при-
приводит к выражению для энергии взаимодействия магнитного мо-
мента fij с полем В :
C.101)
Так как, согласно C.80),
(J2> = Mjh, C.102)
то C.101) можно переписать в виде
. C.103)
Задача 3.15. Вычислить расщепление 5/)-терма атома хрома в магнитном
поле напряженностью 1000 Гс.
Полная энергия электронного уровня равна
90
. C.104)
Из C-104) видно, что магнитное поле снимает вырождение по А/Л
причем величина расщепления пропорциональна приложенному по-
полю. Каждый уровень с квантовым числом J расщепляется магнит-
магнитным полем на BJ+1) подуровней.
На рис. 3.11 показано, как с учетом все более и более тонких
эффектов, т. е. по мере усложнения исходного гамильтониана C.2),
усложняется расщепление уровней атома.
Задача 3.16. Нарисовать диаграмму расщепления всех уровней атома Ti
в слабом магнитном поле.
Задача 3.17. Сколько полос дает на экране пучок атомов Sc, проходя через
слабое неоднородное магнитное поле (опыт Штерна—Герлаха)?
без электрон-
значе- ногрвзаипо-Н
действия
н
СЧ)
М
О
3.10.1. Эффекты Зеемана и Пашен а—Бака
При помещении атома в слабое
магнитное поле его уровни расще-
расщепляются, величина расщепления ка-
каждого уровня определяется форму-
формулой C.104). Этот эффект обуслов-
обусловливает дополнительное расщепление
спектральных линий атома в маг-
магнитном поле, называемое эффектом
Зеемана.
Расщепление уровней, а следова-
следовательно, и спектральных линий зави-
зависит от квантового числа проекции
магнитного момента Мь которое
может принимать B/+1)
ний. Схема расщепления уровней те-
термов 251/2, Рщу 2Ръп атома щелоч-
щелочного металла в магнитном поле по-
показана на рис. З.П. На этом же ри-
рисунке даны разрешенные правилами
отбора электронные переходы, при-
приводящие к наблюдаемым экспериментально десяти спектральным
линиям.
Рис. 3.11. Схематические уровни
(прJ -электронной конфигурации
в приближении LS-связи с учетом
различных эффектов взаимодейст-
взаимодействия в атоме
Задача 3.18. Показать расчетом правильность отнесения полос на рис. 3.12.
В частном случае, когда для одного из участвующих в элект-
91
Z2/3
-Г
ронных переходах термов отсут-
ствует спин-орбитальное вза-
имодействие и S=0, фактор Ла-
нде g~ 1 и, следовательно, рас-
расстояние между расщепленными
уровнями для всех термов оди-
одинаково. В этом случае, называ-
называемом нормальным эффектом Зе-
емана, каждая линия расщепля-
расщепляется всегда на три линии (зеема-
(зеемановский триплет).
Происхождение названий
«аномальный» и «нормальный»
эффекты Зеемана относится к пе-
периоду, когда представления об
электронном спине еще не были введены в квантовую механику.
Поскольку зеемановский триплет может быть объяснен и в рамках
описания с набором обычных квантовых чисел (п, /, т), обуслов-
обусловливающий его эффект был признан «нормальным». А более слож-
сложный, хотя и более общий, случай (SV0) получил вначале наименова-
наименование аномального эффекта.
Рис. 3.12. Расщепление термов и спект-
спектральных линий атома натрия в слабом
магнитном поле
Задача 3.19. Для каких переходов в атоме углерода в слабом магнитном
поле будут наблюдаться нормальный и аномальный эффекты Зеемана?
В сильных магнитных полях, когда величина р„В сравнима
с энергией спин-орбитального взаимодействия, взаимодействие
магнитного поля с орбитальным и спиновым магнитными момен-
моментами каждого электрона становится больше, чем взаимодействие
спинового и орбитального магнитных моментов между собой.
В этом случае связь LS нарушается и энергия взаимодействия
с магнитным полем подчиняется соотношению
1
о
-J
иг
¦4/2
уг
-1/2
1/2
т
-1/2
Рис. 3.13. Расщепление спектральных
линий перехода Л5-^2Р в сильном маг-
магнитном поле
92
C.105)
Эффект расщепления спектраль-
спектральных линий в сильном магнитном
поле называют эффектом Паше-
на—Бака. При этом на расщеп-
расщепление в магнитном поле по
ML накладывается мультиплет-
ное расщепление по Mj. Напри-
Например, терм %S расщепляется на
два уровня с Ms= lU и Ms= —1/>
Терм 2Р расщепляется на 6 уров-
ней. Учитывая правило отбора AMS=O, получим 6 возможных
переходов. Спектрально проявляются только три линии, так как
расщепление уровней по Ms одинаково для всех ML (рис. 3.13).
* При действии на атом внешнего электрического поля также
наблюдается эффект расщепления линий, этот эффект назван эффек-
эффектом Штарка.
Задача 3.20. Рассчитать значение индукции В магнитного поля, взаимодей-
взаимодействие которого с орбитальным моментом Mj=1fi нарушает спин-орбитальное
взаимодействие, и сравнить это поле с максимально достижимыми в настоящее
время с помощью сверхпроводящих магнитов полями Капицы (—100 000 Гс).
Задача 3.21. Доказать, что в слабом магнитном поле переход между тер-
термами Х2?2 и 1ir3 дает три линии.
Задача 3.22. Найти, как изменяется спектральная картина переходов в ато-
атоме Li при перемещении его из слабого магнитного поля в сильное.
Расщепление спектральных линий во внешних электромагнит-
электромагнитных полях является общим эффектом, на основе которого действу-
действуют ЯМР- и ЭПР-спектрометры.
3.10.2. Сверхтонкое взаимодействие
Имеется несколько эффектов, которые также могут быть вклю-
включены в атомный гамильтониан. Конечные размеры ядра и эффекты,
которые дают малые поправки в энергию, связанные с его движени-
движением, не приняты во внимание. Кроме того, имеются релятивистские
эффекты, связанные с взаимодействием спинов электронов между
собой (спин-спиновое взаимодействие). Можно также учесть реляти-
релятивистскую зависимость массы электрона от скорости, которая суще-
существенна только для внутренних электронов тяжелых атомов.
Если ядро имеет ненулевой спин, то существует взаимодействие
между ядерным спиновым магнитным моментом, спиновым и ор-
орбитальным магнитным моментом электрона, которое ведет к так
называемой сверхтонкой структуре атомных спектров. Полный уг-
угловой момент F атома есть сумма полного момента всех электро-
нов J и спинового момента ядра / :
?=7+7. (злоб)
Для примера рассмотрим основное состояние атома водорода.
Спин протона I— /2 и /= 1/2; следовательно, квантовое число F мо-
может быть равно 0 или 1. Переход F =1->0 дает линию 1420 мГц
(Я=21 см), соответствующая энергия перехода 5,9 10~б эВ. Это
знаменитая линия, испускаемая водородом в космическом прост-
пространстве. С открытия этой линии Ивеном и Парселлом A951) ведет
93
начало радиоастрономия. Частота сверхтонкого расщепления ос-
основного состояния водорода является, вероятно, одной из наиболее
точно измеренных физических констант: 1420405751, 786 ±0,010 Гц.
Козман У. Введение в квантовую химию. — М.: ИЛ, 1960. Гл. IV, IX.
Маррел Дж., Кеттл С, Теддер Дж. Теория валентности. — М.: Мир, 1980.
Гл. I—V.
Эйрюг Г., Уолтер Дж., Кимбал Дж. Квантовая химия. — М.: ИЛ, 1948. Гл. III,
VI, VII, ГХ.
Более глубокое и детальное изложение аппарата квантовой мехаяихи имеется в учебниках:
Блохмвце» А. И. Основы квантовой механики. — М.: Наука, 1976.
Заградшк Р., Полах Р. Основы квантовой химии. — М.: Мир, 1979.
Лаядау Л. Д., Лвфшяц Е. М. Квантовая механика. — М.: Наука, 1975.
Цюлмке К. Квантовая химия. — М.: Мир, 1976. Т. 1. Гл. 2—7.
ГЛАВА 4
ТЕОРИЯ ХИМИЧЕСКОЙ СВЯЗИ
Квантовая механика позволяет описать электронное строение
и спектры атомов. Она дает также ответы на основные вопросы
теории химического строения: а) почему атомы отдельных элемен-
элементов соединяются в молекулу, т. е. почему устойчивы одни молеку-
молекулы и неустойчивы другие; б) в каком порядке могут объединяться
атомы, т. е. каково химическое и пространственное строение моле-
молекул, каковы свойства химических связей.
Оператор гамильтона молекулы с N ядрами и п электронами
содержит члены кинетической энергии электронов, потенциальной
энергии притяжения электронов к ядрам, а также члены, обуслов-
обусловливающие межэлектронное отталкивание. Кроме того, по сравне-
сравнению с гамильтонианом атома добавляется член электростатичес-
электростатического отталкивания ядер и их кинетической энергии:
N я
где индексы а и /? принадлежат атомным ядрам, а индексы i и j от-
относятся к электронам. Введены обозначения Д^=|Ла—R$ |,
Rb=Vi-R J и ru=]?t-?}.
Так как гамильтониан молекулы D.1) зависит не только от
координат электронов, но и от ядерных координат, полная волно-
94
вая функция системы должна содержать как электронные (г), так
и ядерные (R) координаты: Ч?(г, К). Это значительно усложняет
задачу математического поиска волновой функции. Поэтому в конк-
конкретных расчетах молекулярных свойств стремятся обычно к раз-
раздельному рассмотрению движения ядер и электронов.
4.1. ПРИБЛИЖЕНИЕ БОРНА—ОППЕНГЕЙМЕРА
Вид гамильтониана D.1) существенно усложнен по сравнению
с гамильтонианом многоэлектронного атома C.2) главным обра-
образом из-за наличия члена кинетической энергии ядер. Однако масса
ядра значительно превышает массу электрона (даже масса легчай-
легчайшего ядра протона в 1836 раз больше массы электрона). Соответст-
Соответственно скорость движения ядер мала по сравнению со скоростью
движения электронов. В результате медленно движущиеся ядра
образуют электростатическое поле, в котором с намного большей
скоростью движутся электроны, успевающие почти мгновенно под-
подстроиться к любому изменению координат ядер. Поэтому в первом
приближении можно считать ядра атомов фиксированными и рас-
рассматривать только движение электронов. На языке квантовой меха-
механики такое приближение эквивалентно допущению, что полная вол-
волновая функция молекулы *F(r, R) может быть выражена в виде
произведения электронной ?э(г, R) и ядерной ?„G?) функций:
D2)
Координаты ядер R входят в ?э(г, R) в качестве параметров.
Рассмотрим условия, при которых справедливо допущение D.2).
Запишем уравнение Шрёдингера для молекулы с гамильтонианом
D.1) и волновой функцией D.2):
х ?,(rf R) Чя( R) = Е?э (г, R) 4>J(R)9 D.3)
n n ZZ^e
где VM = ? Yj —б энергия отталкивания ядер;
V«= — J] J] — энергия притяжения электронов к ядрам;
R
a
л и
^ээ=Х J] энергия отталкивания электронов.
j > j Ги
Обозначим
95
н,= -!j- ? v?+ve+v»+VM; D.4|
H -__ V J-V2
¦"Я— л /j w '«¦
Электронная функция ^э(г, i?) определяется как собственная функ-
функция оператора Нэ:
D.6)
где ?, — электронная энергия, обусловленная движением п эле*
ктронов в поле N ядер молекулы, плюс энергия взаимодейств
между ядрами Vm. Величину Еэ называют адиабатическим
ктронным термом молекулы или адиабатическим потенциалом,
Учитывая, что
перепишем уравнение D.3) в виде
N 1 рр- N \
-.tp- V — v ч* v ч* — — V
Пренебрегая выражением в первых круглых скобках
0 D-9)
и используя соотношения D.4), D.6), получим
~1** I ^^ЧГ-+уАУ.-СТ,Ч',=0. D.10)
Разделив все члены уравнения D,10) на Ч*э и принимая во внима-
внимание D.6), получим уравнение для определения ?„:
№+?э) ?,=??,. D.11)
Условие D.9) означает, что электронная волновая функция
*F3 должна быть настолько медленно меняющейся функцией ядер-
ядерных координат R, что можно пренебречь ее первой и второй произ-
96
водными по этим координатам. М. Борн* и Р. Оппенгеймер**
A927) впервые показали, что электронные волновые функции обыч-
обычно подчиняются этому условию с требуемой степенью точности.
Приближение D.2) является весьма существенным для квантовой
химии, его называют приближением Борна—Оппенгепмера или про-
простым адиабатическим приближением. В этом приближении полная
энергия молекулы представляет собой сумму электронной энергии,
вычисленной при фиксированной конфигурации ядер, и колебатель-
колебательно-вращательной энергии ядер:
D.12)
Естественно, возникает вопрос, насколько оправданно исполь-
использование приближения Борна—Оппенгеймера в квантово-химических
расчетах и каковы при этом ошибки. Чтобы ответить на него, будем
следовать рассуждениям Борна, который в 1951 г. дал новое обо-
обоснование адиабатического приближения.
Предположим, что уравнение Шрёдингера D.6) для электронов
при фиксированных ядрах решено, т. е. известны собственные функ-
функции *F?(r, R) и собственные значения энергии Щ(гш R), соответст-
соответствующие данной конфигурации ядер R.
Тогда, для того чтобы решить уравнение
D.13)
с гамильтонианом Н D.1), представим 4*(rt R) в виде ряда:
г, Л). D.14)
где ?" (R) и 4?}(r, R) — ортонормированные волновые функции
ядер и электронов соответственно в состоянии / для данной кон-
конфигурации ядер R. Это означает, что выполняется условие
fi7
f (r, R) 4} (rt R) dr=Sir, <4Л5>
где интегрирование проводится по координатам всех электронов.
Подставим функцию D.14) в уравнение D.13). Учитывая правила
дифференцирования D.7), получим уравнение
¦Макс Борн A882—1970) — выдающийся немецкий фвзик, один из создателей
квантовой механики. Лауреат Нобелевской премии по физике A954). Иностранный
член АН СССР с 1943 г. Ввел в квантовую механику статистическую интерпретацию
волновой функции и вместе с Н. Винером — понятие оператора.
"Роберт Оппенгеймер A904—1967) — американский физик, специалист в об-
области квантовой механики и теории атомного ядра. Был одним из руководителей
Работ в США по созданию атомной бомбы. В 1953 г. отстранен от за!
ч __- ^ ^^н- р ^ -ш~ ш т ^^ _ _ - ^_
Постов за выступление против развертывания работ по водородной бомбе.
Теория строения молекул 97
. R)(
^
Поскольку оператор Нэ не включает операции дифференцировг
по R, после умножения уравнения D.16) слева на функцию *?f(r,
интегрирования по электронным координатам г и учета уравне!
D.6) получим систему уравнений
=h 2, 3, ... ,DЛ'
где оператор Лу определен как
}?! D.18)
Оператор Ад- зависит от ядерных координат и определяет так
называемое вибронное взаимодействие ядер и электронов.
Выражение D.17) представляет собой бесконечную систему заце-
зацепляющихся интегродифференциальных уравнений и является точ-
точным, так как учитывает связь электронного и ядерного движений.
Если в системе зацепляющихся уравнений D.17) приравнять все
недиагональные элементы матрицы Ав- нулю, то уравнение D.17)
распадается на систему независимых уравнений
DЛ9)
/=1,2,3,....
Система уравнений D.19) отличается от D.11) на величину
и приближение, основанное на решении системы уравнений D.19),
называют адиабатическим в отличие от приближения Борна—Оп-
Борна—Оппенгеймера. Таким образом, с точки зрения теории возмущения
Шрёдингера приближение Борна—Оппенгеймера является нулевым
приближением к точному решению уравнения D.17), учет адиабати-
адиабатической поправки Лг отвечает учету возмущения первого порядка,
98
учет недиагональных элементов Л//-, отвечающих взаимодействию
различных электронных состояний, характеризует поправку более
высокого порядка. Применение адиабатического приближения свя-
связано с малостью возмущающего вклада Лд-. Для случая устойчивых
Многоатомных молекул существует простой критерий применимо-
применимости адиабатического приближения:
«1,
D-20)
где v — наибольшая из частот малых колебаний ядер вблизи точки
равновесия, ^и^, — энергии двух соседних электронных состоя-
состояний. Критерий D.20) обычно выполняется для многих молекул,
вследствие этого расчеты различных физических характеристик мо-
молекул, в подавляющем большинстве основанные на простом ади-
адиабатическом приближении или приближении Борна—Оппенгейме-
ра, позволяют получить данные, хорошо согласующиеся с экспери-
экспериментальными результатами. Ошибка, вносимая при использовании
такого приближения, намного меньше ошибок, вносимых другими
приближениями. Численная проверка надежности приближения Бо-
Борна—Оппенгеймера осуществлена только для самых малых моле-
молекул. Так, в табл. 4.1 приведены энергии диссоциации основных
состояний молекул Н2, HD и D2, рассчитанные в различных прибли-
приближениях и сопоставленные с экспериментом.
Таблица 4.1. Сравнение эксиернмеятальных ¦ тео(
днссоцнащш молекул Н2, HD ¦ D2 в см' '
тальных ¦ теоретических
A см-'^ЦГ2 кДж/i
зна
моль)
энергии
Приближение
Борна—Оппенгеймера
Адиабатическое
Неадиабатическое
Эксперимент
Н2
36112,2
36118,0
36114,7
36113,6
HD
36401,5
36405,7
36402,9
36400,5
D2
36745,6
36748,3
36746,2
36744,2
Из данных табл. 4Л видно, что результаты, полученные в при-
приближении Борна—Оппенгеймера, достаточно хорошо согласуются
с экспериментальными величинами и адиабатическая поправка уме-
уменьшается с ростом массы ядер: для Н2 она равна 0,016%, а для
ЕJ — 0,007%. Естественно ожидать, что для молекул, содержащих
более тяжелые ядра, приближение Борна—Оппенгеймера будет вы-
выполняться с достаточной для квантово-химических расчетов точ-
точностью. Действительно, для большинства задач теории строения
и реакционной способности молекул приближение Борна—Оппен-
99
геймера выполняется достаточно хорошо, кроме некоторых особых
случаев (см. разд. 5.7). Это позволяет ограничиваться решением
только одного электронного уравнения D.6). Поправки для возбуж-
возбужденных электронных состояний значительнее, но обычно ими также
можно пренебречь по сравнению с неточностями, обусловленными
приближенным решением электронного уравнения Шрёдингера
D.6).
Задача 4.1. Показать, что включение в потенциальную энергию члена от-
отталкивания ядер не изменяет электронную волновую функцию, а приводит)
только к изменению полной энергии. ,
42. МЕТОД ВАЛЕНТНЫХ СВЯЗЕЙ. \
РАСЧЕТ МОЛЕКУЛЫ ВОДОРОДА МЕТОДОМ ВАЛЕНТНЫХ СВЯЗЕЙ
Приближение Борна—Оппенгеймера позволяет использовать
для расчета электронной волновой функции гамильтониан типа
D.4), который может быть введен в вариационный интеграл A.55).
Следующий вопрос, который необходимо рассмотреть, связан с вы-
выяснением того, какие идеи следует положить в основу поиска формы
волновой функции и по какому принципу она может быть постро-
построена.
Один из эффективных подходов, получивший впоследствии на-
наименование метода валентных связей,
был предложен в 1927 г. В. Гейтлером
и Ф. Лондоном*, которые выполнили рас-
расчет для молекулы водорода. Физическая
идея предложенного ими подхода основа-
основана на предположении о том, что при об-
образовании молекулы из атомов последние
в значительной степени сохраняют лсвою
электронную конфигурацию, а силы свя-
связывания между атомами обусловлены об-
обменом электронов между ними в резуль-
результате спаривания спинов двух электронов,
находящихся на атомных орбиталях. Это
означает, что молекулярную волновую функцию необходимо стро-
строить исходя из волновых функций отдельных атомов, причем вблизи
ядер волновая функция молекулы должна быть чрезвычайно близка
к атомной орбитали.
Рис. 4.1. Обозначение рас-
расстояний между ядрами (а, Ь)
и электронами A, 2) в моле-
молекуле водорода
¦Вальтер Гейтлер (род. в 1904 г.) — немецкий физик, известен своими работами
по теории химической связи, последнее время работал в области квантовой теории
излучения и космических лучей. Фриц Лондон A900—1954) — немецкий физик, автор
ряда фундаментальных работ по теории химической связи, теории межмолекулярных
сил н сверхпроводимости. Ф. Лондону принадлежит идея спин-валентности.
100
Рассмотрим схему метода на примере молекулы водорода. Опе-
Оператор гамильтониана для Н2 в приближении Борна—Оппенгеймера
имеет вид
е2 е2 е2 е2 е2 е2
D.21)
где все обозначения понятны из рис. 4.1.
В соответствии с идеей метода волновую функцию молекулы
составляют из волновых функций атомов. В основном состоянии
для двух невзаимодействующих атомов водорода а и b волновую
функцию запишем как
% = <?, A)^B), D.22)
где ср — функция ls-АО водорода (см. табл. 2.4). Ввиду нераз-
неразличимости электронов эквивалентной является и другая форма
записи:
У2=<РаB)<РьA). D.23)
Линейная комбинация функций ^i и Т2 была использована
Гейтлером и Лондоном в качестве пробной функции для расчета
молекулы водорода:
Ч>=с1Ч1 + с2Ч>2 = с1<ра0)(РьB) + с2(раB)срЛ1)- D.24)
Вид пробной функции D.24) определяет возможность примене-
применения вариационного метода Ритца для вычисления энергии молекулы
и коэффициентов сх и с2. Система уравнений A.67) в данном случае
примет вид
D.25)
сх (Н21 - ES2l) + с2(Н22 - ES22) = О,
где
Я,, - Я22 = JJ <ра A) срь B) Н<ра( 1) <рь B) dh dr2; D.26)
^2; D.27)
=l; D.28)
= ^ab. D.29)
Отметим, что равенство Н12 — Н21 выполняется вследствие эрмито-
вости оператора гамильтониана, Sab — интеграл перекрывания
Функций <ра и щ:
D.30)
101
Окулярное уравнение A.68) с учетом D.26) —D.29) станов:
достаточно простым:
Н\\— .. „
= 0,
Н\\ — Е— +(H\j — ES>d>).
Решение D.32) дает два значения энергии:
А
D.34)
Вычисления показывают, что ES<EA; следовательно, Es — энергия
основного состояния молекулы водорода, ЕА — энергия первого !
возбужденного состояния (смысл индексов S и А будет понятен из
дальнейших выводов).
Вычислим теперь коэффициенты сх и с2 из уравнения D.25) для
Es и ЕА. Для Es имеем
или, принимая во внимание D.33),
D3б)
Итак, получены отношения коэффициентов, но не их числовые
значения. Такая ситуация является обычной при решении системы
однородных уравнений A.67). Числовые значения коэффициентов
получают, используя нормирован ноет ь полной волновой функции:
С? JJ [<Р*
а B) Щ (I)]
D.37)
Окончательный вид функции основного состояния атома водорода
[ (О » B) + ^ B) <Рь A)]. {4 39)
102
диалогично можно получить волновую функцию возбужденного
состояния:
[A)^B)ФB)A)] D40)
Теперь становится понятным смысл индексов S я А, которые
означают, что функции D.39) и D.40) соответственно симметричны
и антисимметричны относительно перестановок координат электро-
электронов.
Построим сшш-ороитали, соответствующие функциям *F5 и *?А.
Для этого необходимо эти функции умножить на спиновые функ-
функции, причем такой симметрии, чтобы произведение пространствен-
пространственной функции на спиновую было антисимметрично по отношению
к перестановке координат электронов. Функцию 4*s необходимо
умножить на антисимметричную спиновую функцию, а ^А — на
симметричную. В разд. 3.2.1 были рассмотрены возможные спино-
спиновые функции двухэлектронной системы. Эти результаты справед-
справедливы также и для молекулы водорода. Окончательно спин-орбита-
ли основного и возбужденного состояний молекулы водорода в ме-
методе Гейтлера—Лондона имеют вид
у/2
-«BHA)]; D.41)
D.42)
Основное состояние является синглетным, а возбужденное — три-
плетным. Для триплетного состояния имеются три спиновые функ-
функции и соответственно три сшщ-орбитали, которые отвечают одной
энергии.
Вычислим теперь энергии основного и возбужденного состояний
молекулы водорода, используя явный вид функций ?5 и *?л. Для
этого необходимо вычислить матричные элементы Нп и Н12. Опера-
Оператор Гамильтона молекулы водорода можно записать в виде суммы:
H=H,A)+HbB)+Heft; D.43)
103
e2 e2 e2 e2
"^ D.44)
D45)
t
Функции <ра и <рь — 1.У-А0 атома водорода, которые являются со-
собственными функциями операторов Нв и Н$, т. е.
D.46)
где Ен — энергия изолированного атома водорода.
Вычислим Нц1
D.47)
где / — кулоновский интеграл, определяемый как
/= JJ <ра A) Фй B) ЯаЬ сра A) ^ B) A, rfr2. D.48)
Аналогичным образом можно оценить матричный элемент Я]2, где
?Ha- D.49)
Определяя обменный интеграл iT как
ЛГ= Я <Р* Р) ^ A) НвА фд A) фь B) Л, Jt2, D.50)
получим
Я12 = ЛГ+2?„й. D.51)
Подставляя D.47) и D.51) в выражение для энергий D.33) и D.34),
получим
й D-52)
D.53)
Зависимость энергии Es и Д< от межъядерного расстояния Л,,* для
молекулы Н2 показана на рис. 4.2. В табл. 4.2 приведены числовые
значения интегралов /, К, а также энергии состояний Es и ЕА в зави-
зависимости от межъядерного расстояния R^.
104
Метод Гейтлера—Лондона
предсказывает, что энергетическая
кривая, соответствующая синглет-
вому состоянию, имеет минимум,
в то время как триплетное состоя-
состояние отвечает отталкиванию атомов
водорода. Следовательно, в три-
длетном состоянии молекула водо-
водорода не может существовать,
а в синглетном состоянии ix? моле-
молекула устойчива.
Минимум энергии на кривой
Es(R<a) достигается при R^
= 0,086 нм. Экспериментальное
значение равновесного расстояния
в молекуле водорода А=0,074 нм
удовлетворительно согласуется с рассчитанным значением. Энергия
связи (глубина минимума), однако, оказывается равной 3,14 эВ, что
заметно ниже экспериментального значения энергии связи в молеку-
молекуле, равной 4,747 эВ.
Таблица 4.2. Велн^мны интегралов J, К и энергий основного (Es) и возбужденного
(Ел) состояний молекулы водорода в методе Геятлера—Лондона
эксп.
Рис. 4.2. Кривые полной энергии для
различных состояний молекулы во-
водорода
JWoo
0
0,5
1.0
1.5
2,0
2,5
3,0
4,0
J, эВ
00
22,1067
2,6067
-0,2830
-0,5211
-0,3456
-0,1891
-0,0449
К,ъЪ
со
18,0919
-2,4407
-4,3985
-3,2693
-1,9795
-0,9877
-0,2762
Es. эВ
00
-6,2978
-27,1147
-30,2779
-30,0303
-29,1310
-28,3447
-27,5201
Еа.эВ
00
24,4604
-8,0324
-18,5286
-23,0210
-25,1420
-26,1923
-26,9705
Задача 4.2. Построить волновую функцию основного состояния молекул
N2 и LiH по методу ВС.
4.2.1. Уточненные расчеты молекулы водорода
по методу валентных связей
Дальнейшие улучшения волновой функции молекулы водорода
основывались на простых физических идеях. При сближении двух
атомов водорода электроны начинают притягиваться одновремен-
одновременно к двум ядрам, вследствие этого их орбитали сжимаются. Сжатие
орбиталей можно учесть при изменении показателей экспонент
¦ 105
О
I
Рис. 4.3. Зависимость конста-
константы экранирования ос от
межъядерного расстояния для
волновой функции Уанга в мо-
молекуле водорода
атомных орбиталей <рл и щ. Для изо-
изолированного атома Н^ра—СеГл, для
атома Н в молекуле Н2 можно использо-
использовать функцию (рв—Се^*, где а — неко-
некоторый вариационный параметр, выби-
выбираемый из условия минимума полной
энергии молекулы. Аналогично выбира-
выбирается атомная функция для атома Н».
Полная волновая функция строится так
же, как в методе Гейтлера—Лондона.
Тогда, проводя аналогичные вычисле-
вычисления, мы получим, что энергии ЕБл Ел за-
зависят от параметра и. Параметр а для
основного состояния Н2 находится из
условия минимума Е:
dEs(a)
да
=0.
Такое уточнение впервые предложил С. Уанг A928). Он выполнил
свои расчеты лишь для положения равновесия. Н. Розен A931)
обобщил эти расчеты для произвольного межъядерного расстояния.
На рис. 4.3 изображена кривая, найденная Розеном для а как
функции от Rob.
На малых расстояниях, когда два ядра Н сливаются с образова-
образованием атома Не, экспонента а должна приближаться к слэтеровской
экспоненте атома Не. При Да-> оо экспонента стремится к экспонен-
экспоненте изолированного атома водорода: а->1. При равновесном межъя-
межъядерном расстоянии (Rab — 0,0743 hmj a —1,166, а энергия связи равна
3,76 эВ, что значительно точнее прежнего значения 3,14 эВ. Атом,
находясь близко от другого атома, будет поляризовать его, вследст-
вследствие чего атомная орбиталь не будет симметричной относительно
ядра. Этот эффект можно включить в АО, заменив сферически-
симметричную АО поляризованной орбиталью вида
<pa~Xxe
-ага
D-54)
где ось х направлена от атома а к атому Ъ. Аналогичный вид имеет
и функция (рь. Параметры Яь Хъ так же как и а, определяются из
условия минимума энергии. Расчеты с функцией D.54) дают энер-
энергию связи 4,02 эВ.
При сближении атомов есть вероятность того, что оба электрона
окажутся вблизи одного и того же ядра. В этом случае волновая
функция системы имеет вид
106
где
D.55)
D.56)
<рь
~ Се
~*1'ь
о
Если обозначить ранее рассмотренную Рис. 4.4. Зависвмость Я3 от
фуНКЦИЮ, имеющую ЧИСТО КОВаленТНЫЙ R*b ДЛ* молекулы водорода
характер, через *?„„, то полную волно-
волновую функцию можно представить в виде (С. Вейнбаум, 1933)
Энергия, вычисленная с такой волновой функцией, зависит от Хь
Л2, А3, а, <*i и Дл- Расчеты дают Яэ— х/4- Это свидетельствует о том,
что при равновесном расстоянии доля ионного характера связи
значительно меньше доли ковалентного. Зависимость А3 от R^ пока-
показана на рис. 4.4.
Г. Джеймс и А. Кулидж A933) показали, что без включения
в волновую функцию члена г"]2 лучшее значение энергии связи не
может превысить величину 4,27 эВ. Ими была предложена пробная
волновая функция молекулы Н2 в виде ряда
D.57)
i, >. к. I. m
где ^ и ц — эллиптические координаты электронов*. Используя
13-членную функцию этого ряда, Джеймс и Кулидж вычислили
энергию связи, отличающуюся всего на 0,05 эВ от эксперименталь-
экспериментальной величины. Дальнейшие улучшения волновой функции связаны
с учетом большого числа членов в разложении функции по степеням
гп. Наиболее точные расчеты молекул Н2 проведены В. Колосом**
и С. С. Дж. Рутааном*** A960) и В. Колосом и Л. Вольниевичем
Обозначения Д.ь Ял я т. д. см. на ряс. 4.1.
** Влодзимеж Колос (род. в 1936 г.) — польский физик-теоретик, основные
работы посвящены теории химической связи и методам расчета двухатомных моле-
молеку.
*** С С. Дж. Рутаан (род. в 1928 г.) — американский физик, ученик Р. Мал-
. Впервые в методе Хартри—Фока применил для разложения МО идею
линейной комбинации атомных орбиталей и получил уравнения, носящие его имя,
107
кул.
A965) соответственно с 50- и 100-членными функциями типа D.57).
Полученная в результате расчетов теоретическая величина энергии
диссоциации молекулы Н2 JDTeop = 36117,4 см оказалась даже ниже
наиболее точного известного экспериментального значения
А«сп=C6113±0,03) см. Это расхождение между теорией и экс-
экспериментом, противоречащее вариационному принципу (см. разд.
1.3), было необъяснимым в течение нескольких лет. Оно даже дало
толчок для предположений о неполной адекватности самой теории
квантовой механики или о наличии какого-то нового неизвестного
эффекта, не учитываемого в гамильтониане молекулы Н2. Решение
загадки оказалось прозаическим. Г. Герцберг* A969) выполнил
исключительно трудоемкие и весьма точные измерения и показал,
что ошибка в старых измерениях энергии диссоциации была в дейст-
действительности значительно больше, чем предполагалось ранее. Полу-
Полученное им новое значение энергии диссоциации лежало в пределах
36118,3—36116,3 см, причем большее значение является предпоч-
предпочтительным. Согласие с теоретическими расчетами оказалось пре-
превосходным.
Метод ВС можно использовать и для расчета многоатомных
молекул, но такие расчеты не получили широкого распространения.
В последнее время развивается новый подход — обобщенный метод
валентных связей, который синтезирует идеи двух методов: ВС
и молекулярных орбиталей.
4.3. МЕТОД МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ (МО)
4.3.1. Общие положения. Аналогия с теорией многоэлектронного
атома
В 1927—1929 гг. Ф. Хунд**, Дж. Леннард—Джонс и Р. С. Мал-
ликен*** развили идею нового подхода к поиску волновой функции
молекулы, известного под названием метода молекулярных орбита-
лей (МО).
Если в методе ВС волновая функция молекулы строится исходя
из комбинаций волновых функций, образующих молекулу атомов,
которые являются основой всех неэмгшрических и полуэмпирических методов
МО.
*Герхард Герцберг (род. в 1904 г.) — канадский физик, крупнейший специалист
по спектроскопии молекул и радикалов. За работы в этой области в 1971 г. удостоен
Нобелевской премии по химии, хотя известен больше как физик и астрофизик.
**Фридрих Хунд (род. в 1896 г.) — немецкий физик, один из создателей метода
молекулярных орбиталей. Известен работами по квантовой теории атомов, молекул,
спектроскопии, по строению периодической системы.
***Роберт Сэндерсон Малликен A896 — 1986) — американский химик, нобе-
нобелевский лауреат по химии 1966 г. Один из создателей метода МО. Автор фундамен-
фундаментальных работ по теории симметрии молекул и молекулярных спектров.
108
в методе МО полная волновая функция молекулы строится из
функций, описывающих поведение отдельных электронов в поле,
создаваемом остальными электронами и всеми атомными ядрами,
которые образуют молекулярный остов. Тем самым исходная идея
метода МО примыкает к концепции АО с той разницей, что в от-
отличие от последних МО являются многоцентровыми орбиталями.
Значение этой аналогии состоит в возможности перенесения в те-
теорию МО всех основных положений теорий и методов многоэлект-
многоэлектронного атома.
Подобно атомным орбиталям, МО представляет собой одно-
электронную функцию (т. е. зависящую явно от координат только
одного электрона), которая включает пространственную и спино-
спиновую компоненты — спин-орбшпалы
D.58)
Каждая МО характеризуется своим значением энергии, и в соот-
соответствии с принципом построения все электроны молекулы рас-
располагаются попарно (одной пространственной функции отвечают
две спиновые Xi( + l) = a(l) и #,(—1)=/?A)) на МО, заполняя их
в порядке повышения энергии.
Полная волновая функция молекулы, содержащей 2л электронов
на п попарно заполненных МО, описывается слэтеровским опреде-
определителем
-1/2
<pn(?2tt)pBn)
D.59)
учитывающим требование антисимметрии волновой функции во
отношению к перестановке любой пары электронов.
Полная энергия молекулы с волновой функцией D.59) определя-
определяется обычным соотношением
где гамильтониан молекулы берется в приближении Борна—Оппен-
геймера в виде D.4). Как было показано ранее в C.47), полная
энергия системы электронов с волновой функцией D.59) может быть
записана как
D.60)
109
Задача 4.3. Пусть *F из D.59) является хартри-фоковской волновой фу
ей для замкнутой оболочки молекулы. Получить выражение D.60) для по.
энергии молекулы.
Последний член в D.60), отсутствующий в выражении полной;
энергии атома C.35 и 3.47), описывает электростатическую энергию
отталкивания положительно заряженных ядер атомов. ,
Таким образом, аппарат теории многоэлектронного атома легко,
переносится на случай молекулы в приближении МО и основная
задача метода МО вырисовывается как нахождение пространствен-'
ных функций МО <ph на основе которых составляется выражение,
D.60).
43.2. Приближение линейных комбинаций ;
атомных орбнталей (МО ЛКАО) ;
В принципе для нахождения одноэлектронных функций МО мо-
можно использовать метод Хартри—Фока и получить таблицы их
числовых значений подобно тому, как это делается для атома.'
Такой путь ведет к лучшим возможным значениям молекулярных
волновых функций и применяется для некоторых двухатомных мо-
молекул. Его недостатком кроме отсутствия решения в аналитической*
форме являются большие математические трудности, которые
в случае атомов частично устраняются наличием центральной сим-
симметрии системы.
Для молекул соображения симметрии также весьма важны, МО
должна характеризоваться теми же элементами симметрии, какими
обладает сама молекула (конфигурация ее атомных ядер). Однако
эти соображения лишь ограничивают, но отнюдь не определяют
вид волновых функций МО. Из различных приближений, которые
можно применить для построения МО, наилучшим (хотя и не
единственно возможным) является приближение ЛКАО. В этом
приближении каждая МО записывается в виде линейной комбина-
комбинации АО атомов, образующих молекулу, т. е.
N
9>.= ? ад.- D.61)
Форма уравнения D.61) имеет два существенных достоинства.
Во-первых, при «движении» электрона по МО, когда он находится
вблизи ядра атома v, его поведение и его волновая функция должны
«совпадать» с соответствующими характеристиками в атоме. Это
требование хорошо обеспечивается разложением D.61). Во-вторых,
форма уравнения D.61) особенно удачна потому, что для нахожде-
нахождения неизвестных коэффициентов сы может быть применен вариаци-
вариационный метод Ритца (см. разд. 1.4).
по
Приближение ЛКАО для поиска вида МО <pt и представление
полной волновой функции молекулы в виде слэтеровского опреде-
определителя D.59) ведет в рамках метода Хартри—Фока с использовани-
использованием гамильтониана D.4) к уравнениям, полученным впервые
в 1951 г. Рутааном. Эти уравнения являются приближением к урав-
уравнениям Хартри—Фока и лежат в основе почти всех современных
неэмпирических методов расчета сложных молекулярных систем.
Они служат также исходными для развития всех основных полуэм-
полуэмпирических теорий метода МО.
4.3.3. Уравнения Рутаана
Рассмотрим систему с замкнутой оболочкой. МО молекулы
представим в виде ЛКАО D.61). Функции АО выбирают по одному
из типов, описанных в разд. 3.4.2. Коэффициенты разложения с„ не-
необходимо найти исходя из условия минимума полной энергии моле-
молекулы D.60) с учетом ортонормированное™ всех МО <pt. Повторяя
рассуждения, полностью аналогичные сделанным при выводе урав-
уравнений Хартри, и применяя вариационный принцип Ритца, придем
к уравнениям Рутаана
сы (Fp—е, Sp) ~ 0; fi = 1, 2, ..., N, D.62)
v=l
где введены обозначения для матричных элементов:
D.63)
занятые
гдеНГ^-
2т* «-1
j X е
D.64)
D.65)
*2 Z2
v | Ха)={J Xll(]) Zv(l) гГ21 пB) ХоB) dix dx2. D.66)
Суммирование в D.63) ведется только по занятым МО. Матрицу
Sp называют матрицей интегралов перекрывания, так как ее соот-
соответствующие элементы говорят о степени пространственного пере-
перекрывания АО Xit и Ху 0*v I to) — интегралы межэлектронного взаимо-
взаимодействия.
Задача 4.4. Вывести уравнения Рутаана D.62) из выражения для полной
энергии D.60), используя вариационный принцип Ритца.
111
Уравнения D.62) аналогичны уравнениям A-67) в методе
но здесь матричные элементы F^ зависят от коэффициентов
Поэтому уравнения Рутаана D.62) являются нелинейными однород*
ными уравнениями относительно неизвестных величин cjx. Bi
обозначение
где суммирование ведется по всем занятым электронами МО.
Рь называют матрицей порядков связей между АО хл и Ха> Матр!
iy перепишем в новом виде:
Систему нелинейных однородных уравнений D.62) можно свести
к системе линейных однородных уравнений с помощью процедур»
самосогласования. I
Общая схема самосогласования состоит в следующем: задаются
начальными коэффициентами cj?, с помощью котррых вычисляют
.FJJ. Считая, что на этом этапе 1JJ не зависит от cin получают^
систему уравнений
? *Г*Р-«ЛЛ=0, /i=l, 2, ...f N, D.69)
являющуюся линейной и однородной. Эта система D.69) имеет
нетривиальные решения при условии равенства ее детерминанта
нулю:
|1*?-в5у=0. D.70);
Из уравнения D.70) находят корни 40)- Подставляя ?J0) в D.69),
вычисляют коэффициенты с$. Затем найденные с$ снова подставля-
подставляют в Fm вычисляют е\1} и с$ и т. д. Эту процедуру повторяют до тех
пор, пока полная энергия D.60) или матрица порядков связей D.67) '
для двух последовательных вычислений (итераций) не будут со-
совпадать с заданной точностью, т. е.
D.71)
ИЛИ
для всех fi и v.
Количество собственных значений е, и соответствующих им
собственных функций <pi равно порядку определителя D.70), т. е.
числу базисных функций N в разложении ЛКАО D.61).
112
Слэтеровский определитель D.59), определяющий полную вол-
волновую функцию системы, строится из п занятых (n = N/2) электро-
электронами МО. В минимизации полной энергии молекулы участвуют
только занятые МО и, так как матричные элементы F^ зависят
только от Рь„ а порядок связи рассчитывается из волновых функций
только занятых орбиталей, только они могут рассматриваться как
физически определенные. Незанятые МО, получаемые из уравнений
Рутаана, не участвуют в минимизации полной энергии системы,
поэтому их соответствие истинным энергетическим уровням моле-
молекулы не вполне определенно. Такие уровни называют виртуаль-
виртуальными.
В соответствии с идеями вариационного принципа чем ближе
к полному набору базис разложения МО по АО D.61), т. е. чем
больше число базисных функций N, тем более точные решения для
МО могут быть получены. С этой точки зрения в наиболее точных
расчетах стремятся к увеличению базиса. Однако эта тенденция
встречает серьезные ограничения. Для того чтобы провести расчеты
по схеме Рутаана, надо вычислить в первую очередь все члены,
входящие в матричные элементы F^. Основная трудность, опреде-
определяющая требуемое для расчета время работы ЭВМ и, следователь-
следовательно, стоимость расчета, связана с вычислением интегралов (р\\Ха).
Подсчитано, что число р одноэлектронных интегралов типа
S^ и Ир? связано с размером базиса N соотношением p=-N(N + \),
а число двухэлектронных интегралов типа (jiv \ Ха) равно
q=-p(p+l), D.73)
т. е. общее число интегралов можно оценить по формуле
-g- D-74)
Таким образом, с ростом размера базиса разложения N число
интегралов резко возрастает. В табл. 4.3 приведено число интег-
интегралов, которое необходимо вычислять в расчетах методом МО
молекул при использовании минимального базиса (см. разд. 4.3.5).
Орбитали Хм> Ху> Хх, Ха могут принадлежать разным атомам
в молекуле, т. е. могут находиться на разных центрах. Точное
вычисление (с точностью 10 ~5) четырехцентрового интеграла,
включающего, например, 3^-функции фосфора и lj-функции водо-
водорода, занимает ~ 10 с работы ЭВМ типа ЕС-1060 B млн. операций
в 1 с). Для расчета пятиатомной молекулы требуется рассчитать
~ 1000 таких интегралов, что потребует ~3 ч непрерывной работы
ЭВМ этого типа.
из
Таблица 4.3. Общее <нело
свмоста от базвса
Молежула
н2
сн4
Бензол QH$
NC—С—CN
У
JL
О
NC— С— CN
тетрациано-
хмнодиметан
одмолектронвых и
N
2
9
36
84
Базис
тип АО
lJ
1j-( Ъ-, 1р-
АО углеро-
углерода, Ь-АО
водорода
Is-, 2s-, 2р-
АО углеро-
углерода, Ь-АО
водорода
\s-, 2s-, 2р-
АО углеро-
углерода и азота,
1 j-AO водо-
водорода
двухэлектронвых ватегралов в завв-
Одеоэлежт-
ронные ин-
интегралы р
3
45
666
3570
Двухэлект-
ровные инте-
интегралы q
6
1035
222Ш
6374235
Общее число
9
1080
222777
6377805
Таким образом, важнейшая задача в расчетах по методу Рутаа-
на — нахождение наиболее удачного компромисса между размером
базиса и числом подлежащих расчету интегралов. Основные исполь-
используемые способы выбора базиса рассматриваются в разд. 4.3.5.
4.Э.4. Уравнения Рутааяа для открытых оболочек
Для многоэлектронных систем, полный спин которых отличен
от нуля, уравнения Хартри—Фока и Рутаана в представленной
ранее форме для замкнутых оболочек не могут быть применимы.
Поэтому необходимо обобщить теорию Хартри—Фока на системы
с открытыми электронными оболочками, т. е. на системы, в кото-
которых отдельные МО содержат по одному электрону. Предположим,
что в такой системе т МО заняты двумя электронами и п МО
заняты одним электроном. Согласно правилу Хунда, волновая фун-
функция основного состояния должна иметь максимальную мультиц-
летность и, следовательно, ее можно записать в виде следующего
слэтеровского определителя:
"+1^огр=!ф1ф1<Р2ф - <Рт<Рт<Рт+\фт + 2 - фт+п\- D.75)
Мультиплетность этой функции равна л + 1, т. е. число электронов
114
Открытая
Замкнутая
•н-
Ъ Т
ОХФ
НХФ
рис. 4.5. Схема электронного распределения по МО открытых оболочек в ограни-
ченном (ОХФ) и неограниченном (НХФ) методах Хартри—Фока:
D — дублетная; Г — триплетная конфигурации
со спиновой функцией а на л больше числа электронов со спиновой
функцией /?.
Волновые функции подобного типа, записанные в виде одного
слэтеровского определителя D.75), обладают существенным недо-
недостатком: спаренные электроны с разными спиновыми функциями
а и р описываются одной и той же пространственной частью
сшш-орбитали (одной МО), что иллюстрирует рис. 4.5. Но число
а-электронов в нашем случае больше числа /^-электронов, поэтому
электроны с а-спином в дважды заполненных МО будут испыты-
испытывать большее отталкивание от неспаренных а-электронов, чем элек-
электроны с ^-спином в дважды заполненной МО. Этот эффект должен
быть отражен определенными различиями в пространственных
функциях а- и ^-электронов в заполненных МО. Задавать одну
и ту же пространственную часть для а- и /?-электронов — значит
налагать не вполне оправданное ограничение на волновую функ-
функцию, а следовательно, и на пространственное распределение элект-
электронов. Чтобы снять отмеченное ограничение, необходимо задать
для а- и ^-электронов различные формы пространственных функ-
функций ср*, <р"ъ ..., р«+я и <pl cpi ..., (р^ где <р\ф(р\\ (?\Ф(р\ и т. д. (рис.
4.5). Тогда полная волновая функция рассматриваемой системы
примет вид
Напомним, что здесь для аппроксимации МО у* и
приближение ЛКАО D.61):
используется
N
N
115
Волновая функция n+1*Forp является частным случаем функции
и, как следует из вариационной теоремы, использование
должно вести к понижению полной энергии системы, т. е.
D.77)
В этом равенстве считается, что "+Vorp и "+1Ч/неогр нормированы.
Таким образом, "+V^rp — более точная волновая функция, чем
л+ ?огр. Не вдаваясь в детальное рассмотрение, отметим, что
"+ ^ в общем случае не является собственной функцией спиново-
спинового оператора S2 и, следовательно, не соответствует точно чистым
синглетным, дублетным, триплетным и т. д. электронным состоя-
состояниям. Для устранения примешивания состояний высшей мультип-
летности развиты различные варианты методов проектирования
требуемой спиновой компоненты. В то же время R Ч'деогр является
собственной функцией Sz с собственным числом, равным nh. Проце-
Процедура минимизации функционала D,77) с волновой функцией
я+ ^ полностью подобна минимизации с " ^огр. Метод, ос-
осогр.
нованный на использовании ?огр, называют ограниченным методом
Хартри—Фока, а на ^Рнеогр — неограниченным методом Хартри—
Фока. Не будем повторять вывод уравнений Рутаана для открытых
оболочек, а запишем окончательные уравнения, которые представ-
представляют собой систему взаимозацепляемых уравнений:
V-l
N
D.78)
В этих уравнениях матричные элементы /Jy и i^v имеют вид
D.79)
Ха
Ха
где
U6
а матричные элементы полной матрицы плотности
Из выражений D.80) можно найти спиновую матрицу плотности:
псшш_ р« _р% D.81)
л la -~rbi г*¦*'
Отличие уравнений Рутаана для открытых оболочек D.78) от урав-
уравнений для закрытых оболочек D.62) заключается в том, что система
D.78) содержит в два раза больше уравнений BN, где N — базис
ЛКАО), чем система D.62). Таким образом, снятие ограничения на
волновую функцию "+ 4Vp и превращение ее в "+ Тнеогр приводит
к увеличению порядка системы уравнений D.78) по сравнению
с D.62).
Уравнения D.78) принципиально ничем не отличаются от урав-
уравнений D.62), и ясно, что решение их проводится одинаковым итера-
итеративным путем.
4.3.5. Выбор базисных атомных функций
Выбор базисных атомных функций в разложении ЛКАО D.62)
является важной задачей, так как именно им определяется, насколь-
насколько точно ряд ЛКАО аппроксимирует молекулярную орбиталь.Хар-
три—Фока. Этот ряд должен достаточно быстро сходиться, т. е.
малое число атомных орбиталей должно аппроксимировать МО
с требуемой точностью. Существует три основных критерия для
выбора базисных функций:
" 1. Базисные функции должны давать в основном хорошее при-
приближение к истинной волновой функции (например, возле ядер и на
больших расстояниях от них).
2. Базисные функции должны допускать аналитическое вычисле-
вычисление нужных интегралов.
3. Полное число базисных функций не должно быть очень боль-
большим.
Рассмотрим это более подробно. Любую непрерывную функцию
можно разложить по полному набору функций. Полиномы Лягерра,
Лежандра и многие другие образуют полную систему функций.
Например, непрерывную функцию одной переменной f(x) можно
разложить в ряд по полиномам Лягерра:
00
D.82)
Функцию нескольких переменных также можно разложить по каж-
каждому ее аргументу в такой же ряд:
117
f(x, y, z) = YS^c™*b*№Ln(y)Lk(z)' D.83)
к
m н
Естественно, возникает желание использовать такие ряды в раз*
ложении молекулярной орбитали. Трудность состоит в том, что не
для всякой функции эти ряды быстро сходятся, т. е. небольшое
количество членов хорошо аппроксимирует функцию/(*,). Оказы-
Оказывается, что для разложения волновой функции в такой ряд необ-
необходимо брать большое число членов A000 и более), чтобы получить,
достаточно хорошие результаты. Такое увеличение базиса приводит
к увеличению времени счета.
Наиболее быстро сходящиеся ряды строятся на базисе атомных
орбиталей, т. е. физическая идея ЛКАО оказалась также и лучшей
с математической точки зрения. Лучшими атомными функциями
являются самосогласованные атомные орбитали, вычисленные*
Э. Клементи и Р. Ватсоном путем решения уравнений Хартри—'
Фока для атомов. Однако эти функции получаются не в аналитичес-
аналитическом виде, а в табличном. Проводить расчеты с функциями, заданными
в числовом виде, крайне трудно и неудобно. Поэтому нерационально
использовать АО Хартри—^Фока в расчетах по методу Рутаана,
являющегося приближением к уравнениям Хартри—Фока. В качестве
базиса АО можно использовать слэтеровские орбитали. Однако из
рис. 3.6 видно, что слэтеровская АО плохо описывает хартри-фоковс-
кую АО вблизи ядра. В то же время две слэтеровские АО достаточно
хорошо аппроксимируют точную хартри-фоковскую функцию атома,'
в связи с чем был предложен весьма распространенный дубль-зета-
базис (DZ), где каждая атомная функция аппроксимируется двумя'
слэтеровскими функциями с разными экспонентами. Например, для
углерода выбираются экспоненты, представленные ниже: l
АО U W Ъ 2У 2р rf
ехр 5,2309 7,9690 1,1678 1,8203 1,2507 2,7263
Расчеты по методу Рутаана можно разделить на два класса:
расчеты с минимальным атомным базисом и с расширенным. Ми-^
нимальный базисный ряд состоит только из АО внутренних и валент-
валентных оболочек свободных атомов, расширенный базис включает до-^
полнительно атомные орбитали, не занятые в основном состоянии.
Расчеты с минимальным базисом, без сомнения, легче, однако
расширенный базис дает более точные результаты. Как уже указы-
указывалось в расчетах по методу Рутаана, основная сложность заключа-
заключается в вычислении интегралов {ру\Хо), вычисления которых на,
слэтеровских функциях чрезвычайно сложны и трудоемки. Для
упрощения расчетов Бойс A950) использовал набор гауссовского
типа для аппроксимации каждой слэтеровской АО:
Gnbn=NH(a) r"-1exp(-ar2) Y^O, q>), D.84)
118
где Nn(ct) — нормировочный множитель; а — варьируемый пара-
параметр.
Преимущество такой замены заключается в том, что произведе-
произведение любых двух гауссовских функций с центрами на атомах
а и b представляет собой новую гауссовскую функцию с центром
в некоторой точке с. В связи с этим вычисление четырехцентрового
интеграла по гауссовским функциям (GaGb \ GeGJ) сводится к вычис-
вычислению двухцентрового (Gc \ GJ) интеграла, который вычисляется зна-
значительно проще. Основной недостаток гауссовских функций в том,
что они плохо отражают поведение хартри-фоковских АО. Для
аппроксимации АО Хартри—Фока с достаточной точностью необ-
необходимо брать большее число гауссовских АО, чем слэтеровских.
Несмотря на уменьшение как числа интегралов, так и сложности
их расчета, порядок алгебраических уравнений D.62) резко возраста-
возрастает при переходе к GTO (гауссовский тип орбиталей), что ведет
к увеличению времени расчета. Для преодоления этих трудностей
некоторые GTO группируют вместе (контрактируют или сжимают)
и затем работают с одной функцией.
Контрактированная орбиталь Xv определяется через исходный
базисный набор б^, называемый примитивным, с помощью очевид-
очевидного соотношения
^G^, D.85)
где Nv — нормировочный множитель.
- Например, примитивный базис A1$, 1р, \dfbs, \р\ состоящий из
одиннадцати ^-функций, семи ^-функций и одной rf-функции для
элементов второго периода и шести ^-функций и одной /^-функции
для водородных атомов, можно контрастировать до базиса Es, Ар,
ld/3s, \p). Последний включает пять сжатых гауссовских s-орбита-
лей, четыре ^-орбитали и одну */-орбиталь для атомов второго
периода и три 5-GTO и одну p-GTO для атомов водорода. Прими-
Примитивные (основные) базисные ряды принято заключать в круглые
скобки, а сжатые — в квадратные. Два описанных выше базиса
можно кратко представить с помощью следующей символики: A1,
7, 1/6, 1) и [5, 4, 1/3, 1].
Существует несколько стандартных способов сжатия базисов,
причем наиболее распространенными следует считать схемы STO-
NG и M-NPG. Первая схема — минимальный базис орбиталей,
в котором N гауссовских функций использованы для аппроксима-
аппроксимации одной слэтеровской. В большинстве случаев ограничивают
#=3, так как при дальнейшем увеличении точность результатов
расчета растет очень медленно.
119
Базис STO (слэтеровский тип орбит ал ей) типа DZ аппроксими-
аппроксимируется расщепленными полиномами функций гауссовского типа Af.
NPG. Каждая внутренняя АО заменяется М орбиталями GTO,
валентные 2s- и 2р-орбитали представляются двумя наборами — N
и Р функций с разными значениями экспонент. Например, базис
4-31G описывает каждую внутреннюю (Is) орбиталь четырьмя
GTO, валентную Is и 2р АО — тремя GTO с одной экспонентоЙ
и одной GTO с другой экспонентоЙ. Важно отметить, что если
в случае минимального базиса NG-типа при больших значениях
N не достигается уровень точности минимального базиса STO,
применение валентно-расщепленных базисов GTO M-NPG позволя-
позволяет превзойти уровень слэтеровского DZ-базиса. i
Для улучшения описания молекулярных орбиталей на больших'
расстояниях от ядра часто используют базисные ряды с включением j
поляризационных функций. Широко распространены базисы 6*31G*
и 6-31G**. Одна звездочка означает добавление поляризационной
d-GTO к каждой /^-функции, а две звездочки — добавление, кроме
того, p-GTO к ls-орбиталям атомов водорода*
4.4, ЭЛЕКТРОННАЯ КОРРЕЛЯЦИЯ
При хорошо выбранном базисе метод ССП МО ЛКАО Рутаана
дает очень близкое приближение к методу Хартри—Фока, с помо-
помощью которого полная энергия атомов и молекул оценивается с точ-
точностью до 0,3%. Вследствие того, что полные энергии имеют весь-
весьма значительные величины, а задачи физики и химии требуют
знания не столько полной энергии, сколько различий в энергиях
сравниваемых систем, с этой ошибкой метода Хартри—Фока при-
приходится считаться.
Так, энергия диссоциации молекулы Н2 на атомы, вычисленная
как разность полной энергии молекулы и суммы энергий изолиро-
изолированных атомов Bx13,545 эВ) и равная 2,65 эВ, значительно ниже
экспериментального значения D,75 эВ). Кроме того, расчет непра-
неправильно предсказывает энергию состояний, в которые диссоциирует
молекула Нг. Такая же картина наблюдается и для многих других
г о мо ядерных молекул. В табл. 4.4 приведены рассчитанные и экс-
экспериментальные значения энергии диссоциации некоторых молекул
первого периода.
'Использование базисов GTO рассмотренных типов широко распространено
в современной практике расчетов, особенно органических молекул и их взаимодейст-
взаимодействий. Базисы GTO включены в весьма быстродействующие и эффективные програм-
программы GAUSSIAN-70, GAUSSIAN-76, GAUSSIAN-80, GAUSSIAN-85, разработанные
группой Попла и представленные в фонд обмена программ QCPE (Quantum
Chemistry Program Exchange).
120
Таблица 4.4. Энергия диссоциация двухатомных молекул
Молекула
н2
Li2
N2
F2
Энергия диссоциации, эВ
теоретическая
2,65
0,15
1,19
-0,30
экспериментальная
4,75
1,05
9,90
1,68
Рассчитанная энергия диссоциации всегда меньше экспери-
экспериментального значения, а молекула F2, согласно предсказаниям
метода Хартри—Фока, должна быть вообще нестабильна, что,
конечно, противоречит эксперименту. Отмеченные недостатки ме-
метода Хартри—Фока связаны с тем, что истинный кулоновский
потенциал взаимодействия электронов У— заменяется усреднен-
ным потенциалом у qj. .__
__ у _
t.jrv
Эта замена приводит к тому, что в приближении Хартри—Фока
не учитывается скоррелированность движения электронов в атоме
и молекуле в каждый момент времени (а не в среднем), вследствие
которой электроны на отдельных орбиталях стремятся находиться
как можно дальше друг от друга, т. е. существует кулоновская
корреляция электронов. Разность между точной (нерелятивистс-
(нерелятивистской) энергией и энергией, полученной методом Хартри—Фока, назы-
называют энергией корреляции:
¦^Ъорр = -Сточи — Ех—ф • D.86)
Точная нерелятивистская энергия молекулы получается при ре-
решении уравнения Шрёдингера с гамильтонианом, не включающим
релятивистские члены (спин-орбитальное, спин-спиновое и другие
взаимодействия). Значения корреляционной энергии для некоторых
атомов и молекул приведены в табл. 4.5.
Таблица 4.5. Корреляционные энергии
Система
Н"
Не
Li
LiH
н2
Явдрр. ЭВ
-1,08
-1,1461
-1,2319
-1,96
-1,06
Система
СН4
NH3
н2о
н2со
Бензол
^корр. эВ
-8,0378
-9,8255
-9,7840
-16,2661
-46,7957
121
'X-cp расчет с минимальным базисом
орбиглалеи
1
а
в
f
f
х-ср расиет о расширенным Базисом
орвиталёи
х-ср предел
почти полное KB
точная нереляглибастская энергия
эксперимент
4
f
Рис. 4.6. Энергия электронной корреляции*.
а — согласно определению; б — оценка в {фактических расчетах; в — экспериментальная жор-
реляциоввая энергия
а
Определение D.86) страдает тем недостатком, что точные реше*г
иия уравнения Шрёдингера нельзя получить даже для трехатомнош
системы, поэтому в практических расчетах придерживаются нескоч
лько иной оценки, которая поясняется схемой на рис. 4.6. ..*•
Энергия электронной корреляции составляет небольшую долюк
полной энергии молекулярной системы @,5% для молекулы Н2О).>
Однако энергия связывания в молекулах — также малая часть пол-
полной энергии (для Н2О также 0,5% полной энергии молекулы). Кроме
того, в общем случае энергия электронной корреляции — неадди-н
тивиая величина и зависит от геометрии системы. Наиболее упот-j
ребительными в настоящее время способами учета энергии элект-а
ронной корреляции являются метод конфигурационного взаимодей-'
ствия (KB) и метод теории возмущений Мёллера—Плессета (МП).
4.4.1. Метод конфигурационного взаимодействия
Полную волновую функцию в этом методе записывают в виде
линейной комбинации слэтеровских определителей, отвечающих
различным электронным конфигурациям:
^Уь D.87)
где М — число учитываемых конфигураций.
122
Электронные конфигурации
4\ (слэтеровские детерминанты)
определяются как различные спо-
способы размещения электронов по
всем орбиталям. Конфигурация
Ч>0 соответствует конфигурации ос-
основного состояния, а остальные —
возбужденным состояниям. Не все
из них являются истинными возбу-
возбужденными состояниями [см. D.137)
и D.138)], однако это несуществен-
несущественно для дальнейших расчетов. Кон-
Конфигурации, возникающие при воз-
возбуждении одного электрона из ос-
основного состояния, называют одно-
однократно возбужденными. При воз-
возбуждении двух электронов возни-
возникают двукратно возбужденные кон-
конфигурации и т. д.
Коэффициенты Ak можно най-
найти, воспользовавшись вариацион-
вариационным методом. При этом выраже-
выражение для полной волновой функции
D.87) подставляют в формулу
C.34) и проводят минимизацию по
коэффициентам Ак и с? в разложе-
разложении МО ЛКАО. В результате полу-
получаются уравнения, из которых на-
находят коэффициенты Aki и полные
энергии электронных конфигураций
7 R/a0
Рис. 4.7. Сравнение с экспериментом
кривых полное энергии молекул Li2
и F2, полученных в приближении Ха-
ртри—Фока (ХФ) и Хартрн—Фока
с учетом конфигурационного взаимо-
взаимодействия (KB) в зависимости от
межъядерного расстояния. Горизон-
Горизонтальная пунктирная линяя показыва-
показывает хартри-фоковскую полную энер-
энергию свободных атомов
Е,:
= 0' D.88)
л.
где Нш~ J ^ITF/<it; <5w — символ Кронекера; Н — полный гамиль-
гамильтониан молекулы. Весь расчет с учетом KB выполняется следу-
следующим образом: проводят самосогласованное решение уравнений
Рутаана, затем полученные коэффициенты Сщ и энергии е, использу-
используют для вычисления матричных элементов Ни (матрицы KB). После
этого решают систему D.88) и находят коэффициенты Аи и энергии
Et (см. разд. 4.5.3).
Приведем кривые потенциальной энергии молекул 1л2 и F2,
рассчитанные методом Рутаана с учетом и без учета KB (рис. 4.7).
123
Введение конфигурационного взаимодействия приводит к значи-
значительному понижению энергии основного состояния. Энергии дис-
диссоциации молекул Н2, Li2, F2 приведены в табл. 4.6, из которой
видно, что с учетом KB энергия диссоциации увеличивается. '
Таблица 4.6. Рассчитанные н экспериментальные значения энергии днссоцнашн
двухатомных молекул .
Соединение
н2
Li2
F2
Энергия диссоциации, эВ
Х-Ф
3,64
0,17
-1,37
X—ФсКВ
4,13
0,46
0,54
экспериментальная
4,75 !
1,05
1,68
ошибку. Можно считать, что корреляционные эффекты вносят опре-
определяющий вклад в связь F — F в молекуле F2.
4.4.2. Метод многоконфигурациоЕшого
взаимодействия (МКВ)
При более строгом рассмотрении коэффициенты Ам D.87) сами
должны зависеть от коэффициентов сщ в разложении МО ЛКАО.
Поэтому нужно решать систему взаимозацепляемых уравнений Ру-
таана
= 0,
D.89)
в которых матричные элементы FMV зависят не только от коэффици-
коэффициентов ст но и от Akh и систему
ы [Ни (cj - БАЦ - 0,
D.90)
в которой матричные элементы Нм зависят от ciVL.
Уравнения D.89) и D.90) должны решаться самосогласованно.
Сначала задают начальные cjj? и А$, вычисляют соответствующие
Fftt и Ны и находят собственные значения е, (энергия i-й МО) и ?i
(энергия /-й электронной конфигурации) из секулярных уравнений:
0; D.91)
D.92)
124
Затем определяют новые коэффициенты с$ и Л$, и вся процедура
повторяется до самосогласования по cf(l и Аы или по соответст-
соответствующим энергиям. Такие расчеты в связи со сложностью вычисле-
вычислений могут быть проведены только на мощных ЭВМ и для малых
молекул. Поэтому обычно применяют более простой метод КВ.
Для многоатомных молекул понижение энергии различных состоя-
состояний при расчетах по KB или МКВ на 2—3 эВ является обычным.
Расчеты с учетом KB и даже МКВ широко распространены для
учета энергии корреляции. Главная трудность KB — слабая сходи-
сходимость ряда D.87), поэтому необходимо учитывать большое число
конфигураций. Многие из них вносят малый вклад в энергию основ-
основного состояния (~10~3 эВ), но пренебречь ими нельзя, так как их
число довольно значительно (иногда 103— 104). Поэтому полный
вклад всех этих конфигураций может быть ~2—3 эВ. Так, конкрет-
конкретные расчеты по МКВ показали, что для получения правильной
величины и знака дипольного момента СО необходимо учесть 5000
конфигураций в разложении D.87). Развиты различные схемы, уско-
ускоряющие сходимость ряда D.87), из которых наиболее эффективно,
но и трудоемко так называемое приближение связанных элект-
электронных пар (СЕРА).
4.4.3. Метод теории возмущений
Трудоемкость метода KB привела к более широкому исполь-
использованию методов теории возмущений для учета электронной кор-
корреляции. Эти методы проще поддаются алгоритмизации, их ре-
реализация не требует особенно больших затрат машинного времени,
и, кроме того, они позволяют учесть влияние различных порядков
теории возмущений на те или иные рассчитываемые свойства. Осо-
Особенное распространение получил метод теории возмущений Мел-
лера—Плессета, вычислительная схема которого включена в про-
программы типа GAUSSIAN и достаточно эффективна для практичес-
практических расчетов.
Основная идея теории Меллера—Плессета заключается в пред-
представлении решения полной многоэлектронной задачи в виде воз-
возмущения хартри-фоковского решения. Полный гамильтониан
Н; (учитывающий энергию корреляции электронов) складывается из
двух частей: хартри-фоковского гамильтониана Г и его возмущения.
Для оценки последнего применяется теория возмущений Рэлея—
Шрёдингера (см. разд. 1.5):
D93)
где Н — правильный (с учетом корреляционной энергии) гамиль-
гамильтониан, а Я — параметр возмущения. Если А —0, то H^F и хартри-
фоковская волновая функция является решением для Ня. При X — 1
125
Нд=Н. Искомая волновая функция и энергия разлагаются в ряд па
степеням Я:
D.94)
В практических расчетах параметр Я принимается равным 1,
Р = *Р0, т. е. хартри-фоков
состояния. Считают также, что
а *Р = *Р0, т. е. хартри-фоковскои волновой функции основного
,?,, D.95)j
i
где е, — одноэлектронные уровни, определяемые по соотношении!
C.46); rii — числа заполнения электронами указанных уровней:
Таким образом, в методе теории возмущений Меллера—Плессета
энергия в первом порядке возмущений является хартри-фоковскои ¦
энергией, вычисляемой согласно D.60). Выражения для Ч*A), т*, ?* ,
... можно получить через характеристики хартри-фоковских орбита?
лей. Отметим, что они существенно усложняются с ростом порядка
возмущения. Практически разложение доводят не выше чем до
четвертого порядка.
Основной вклад в энергию корреляции ввиду действия теоремы
Бриллюэна вносит член второго порядка ?Г. Он описывает
смешивание волновой функции т со слэтеровскими детерминан-
детерминантами, отличающимися от W заменой двух занятых МО двумя
виртуальными (вакантными) МО. Второй порядок теории воз-
возмущений Меллера—Плессета обозначают обычно МП2. Сокраще-
Сокращение МП2/6-ЗЮ* означает, что расчет произведен в базисе 6-3IG*,
а корреляция учтена в рамках теории возмущений второго
порядка.
Третий порядок теории МП, так же как и второй, описывает
только двойные замены МО. Только в четвертом порядке теории,
который лишь в последнее время стал доступен для практических
расчетов, появляются однократные, двойные, тройные и четверные
замены занятых МО на вакантные.
4.5, РАСЧЕТ МОЛЕКУЛЫ ВОДОРОДА ПО МЕТОДУ МО ЛКАО
4.5.1. Основное состояние молекулы водорода
Проиллюстрируем применение метода Рутаана на примере мо-
молекулы Н2. Для построения молекулярных орбиталей будем пользо-
пользоваться минимальным базисом из lj-АО атомов водорода:
D.97)
где а и Ь — индексы атомов водорода; Д, и Rh — расстояния элект-
электронов до соответствующих ядер.
Уравнения Рутаана D.62) для базиса D.97) имеют вид
D.98)
Вследствие эрмитовости матриц F^ и S^ и симметрии задачи вы-
выполняются равенства:
^12=^21; ^11=^22; sn=s2l=s. D.99)
Уравнения Рутаана, как было показано выше, решаются итера-
итеративным путем, но в рассматриваемом случае из-за простоты задачи
и ее симметрии уже первая итерация ведет к самосогласованному
решению. Уравнения D.98) имеют нетривиальные решения только
в том случае, если детерминант системы равен нулю:
h~e Fl2-sS
где е — энергия МО молекулы водорода. Раскрывая детерминант
D.100), получим квадратное уравнение
корнями которого являются значения
?i=-
?,=
J^S~- D-102)
Подставляя значения D.101) в D.98) вместо е, получим уравнения
Для нахождения коэффициентов с\ и с2 в соответствующей МО:
127
D.103)
Fu+Fu\
Проведя простые преобразования, запишем систему уравнений
в виде
D.104)
n-FnH^iFu-FnS)^.
Отсюда
D.105)
D.106)
Из условия нормировки МО (р\
получим
с?B+25)=1; Cl=v^' D!07}
Второе решение получаем, подставляя в D.98) вместо е выражение
D.102). Это решение имеет вид
ci = -^ D.108)
Соответствующая орбиталь
Ф2=с1(х1-х2). D.109)
Условие нормировки q>2 приводит к выражению
Окончательно молекулярные орбитали молекулы водорода запи-
запишутся в виде
у——
() D.112)
128
где (ps — молекулярная орбиталь,
симметричная относительно переста-
перестановки ядер, а <рА — антисимметрич-
антисимметричная МО.
Следует отметить, что в рассмат-
рассматриваемом простом случае явный вид
МО q>s И <рА МОЖНО было легко полу-
ЧИТЬ ПРЯМО ИЗ требований Симметрии
молекулы. Выражения для энергии
МО D.101) и D.102) удобно преоб-
преобразовать к следующему виду:
ftuj_ 4 8 Энергетическая днаграм-
ма <*Р"зованм связывающей
рода
Fx2-FnS
D.113)
D.114)
Считая в первом приближении атомные орбитали ортогональными,
получим
\ D.П5)
?a=Fu-F12. D.116)
Из уравнений D.113) и D.114) видно, что атомные уровни энер-
энергии водорода, приближенно равные Fu при i?->co, расщепляются
так, что один из новых уровней оказывается выше, а второй —
ниже первоначального уровня. Величина расщепления определяет-
определяется значениями Fn и интеграла перекрывания 5, что иллюстрирует
рис. 4.8. Величина матричного элемента F12 отрицательна, поэтому
более низкой энергией обладает уровень es. Благодаря знамена-
знаменателю, равному A+5), расщепление несимметрично относительно
FUi причем
Fl2-FuS Fl2-FuS
1-5
1+5
раз-
разМолекулярную орбиталь <ps называют связывающей, МО <рА
рыхляющей или антисвязывающеп.
4.5.2. Волновая функция и энергия основного состояния Н2
Найдем матричные элементы i^ и Fn, которые потребуются для
вычисления полной энергии молекулы. Согласно D.68), получим
5. Теория строения молекул 129
D. И'
D.
Должны выполняться следующие очевидные равенства:
A1 22)=B2
( )(
A1 12W
B2 12)=(
) к
21)=П2|И)=B1|11);
21)Н21|22)=A2|22).
D.119)
Из условий симметрии задачи вытекают соотношения
A1|11) = B2|22),(Н|12)=B2|21). D.L
Можно вычислить матрицу порядков связи по D.67), используз
значения коэффициентов с1 и с2в занятой двумя электронами моле
кулярной орбитали q>\.
1
T+s'
D.123
Учитывая D.119) — D.121), раскроем суммы в выражениях D.П7)
и D.118):
D.122)
12I
и Нц находят из следующих формул:
¦ j
D.123)^
где Гц — одноцентровый инте^ал кинетической энергии; V\\
одноцентровый интеграл притяжения к ядру; У\х — двухцентро!
интеграл притяжения к ядру; Тп — двухцентровый интеграл
130
тической энергии; V12 и V\2 — двухцентровые интегралы притяже-
притяжения к ядру- .
Подставляя в явном виде выражения для Х\ и Хгиз D.97) и прово-
проводя необходимые вычисления, запишем в аналитической форме окон-
окончательный вид всех одноцентровых и двухцентровых интегралов:
D.125)
(П\П)=~е2;
(П|22)=—
S^ab
где в последнем выражении
D.126)
Числовые значения этих величин в зависимости от межъядер-
межъядерного расстояния Rab приведены в табл. 4.7.
Таблица 4.7. Значения интегралов для молекулы водорода как функции межъя-
Дерного расстояния
Та
-к
A1122)
A1112)
02|12)
0
0,10
0,15
0,20
1,0000
0,8584
0,7252
0,5864
0,5000
0,3066
0,1952
0,1128
1,0000
0,7293
0,5837
0,4725
1,0000
0,7957
0,5576
0,4060
0,6250
0,5545
0,4903
0,4260
0,6250
0,5070
0,4054
0,3080
0,6250
0,4366
0,2968
0,1841
131
0,25
0,30
0,35
0,50
0,70
0,4583
0,3485
0,2592
0,0965
0,0222
Туг
0,0581
0,0249
0,0063
0,0078
0,0038
'К,
0,3905
0,3300
0,2845
0,1999
0,1428
0,2873
0,1991
0,1359
0,0404
0,0073
(И 122)
Продолжение табл. 4.J,
A2| 12)
0,3684
0,3198
0,2799
0,1996
0,1428
(И 112)
0,2256
0,1607
0,1121
0,0349
0,0065
0,0585
0,0308
0,0037
0,0002
Задача 4.5. Получить выражения для Ти> Vu, Sn и (И | П) в выражении D.125),
Показать, что S\2~+0 при
Электронную энергию молекулы Н2 в основном состоянии*
можно вычислить по формуле C.35), которая в данном случае
примет вид
1
+ 2A1112)
}
D.127)
Полная энергия молекулы есть сумма электронной энергии Ео и
энергии ядерного отталкивания Уя
яя
D.128)
f,
ае
0
-0,6
J Л
15
\
\\
|д\
V >^
-
-
-
¦
*- щ ¦
^
/^
0
-10
-го
-зв
8ff/a0
Рис. 4.9. Кривые полной энер-
энергии молекулы Н2 в зависимости
от межъядерного расстояния
R для основного (*?*), три-
плетного C^^), синглетного
С1^^ я Д&яжды возбужденного
синглетного (*?*)
'М состоявий
Рис. 4.10. Трехмерное представление элегг-
ронной плотности в основном состоянии моле-
молекулы И 2
132
На рис. 4.9 представлена зависимость полной энергии основного
остояния молекулы Н2 от межъядерного расстояния. Значение
новесвого расстояния, полученное методом Рутаана, равно
j? = 0,084 нм, а энергия связи равна 2,65 эВ (соответствующие экс-
экспериментальные величины 0,074 нм и 4,74 эВ).
Полная волновая функция основного состояния Н2 должна быть
представлена в виде слэтеровского детерминанта, построенного из
молекулярных орбиталей:
D.129)
На рис. 4.10 представлен вид функции 1хР,. Графически рас-
распределение электронной плотности для связывающей и разрыхля-
разрыхляющей МО можно получить, построив графики функций
1
2-25
D.130)
D.131)
На середине межъядерного расстояния
(Robfe) должно выполняться условие
Xi —X2- Вследствие этого в точке
имеем
4
D.132)
D.133)
т. е. для разрыхляющей орбитали эле-
электронная плотность в середине связи
равна нулю, а для связывающей МО
она отлична от нуля.
На рис. 4.11 изображены функции
*Ps и Фа> совместно с которыми пред-
представлены функции 11гх] и 72*1- Это
позволяет сравнить распределения МО
9s и (рА с полусуммой атомных плот-
плотностей
а
а
Вместо распределе-
распределения электронной плотности можно ис-
Рис. 4.11. Распределения элект-
электронной плотности для атомных
(а), молекулярных связывающей
(б) и разрыхляющей (в) орбита-
лей молекулы Н
133
005
Рис. 4.12. Контурные диаграм-
диаграммы связывающей {a\s) <ps и раз-
разрыхляющей (a* \s) <рл молеку-
молекулярных орбиталей молекулы Н2
пользовать контурные диаграммы, ко-
которые изображают линии равных зна-
значений <р2. На рис. 4.12 представлены
контурные диаграммы <рл- и (ps~ состо-
состояний. Рис. 4.11 и 4.12 свидетельствуют
о «накачке» электронной плотности ор-
битали <ps в межъядерное пространст-
пространство. В то же время орбиталь <рА харак-
характеризуется пониженной электронной
плотностью между ядрами.
Следует отметить, что кривая энер-
энергии основного состояния lxFt при
Д^-юо стремится не к энергии двух1
изолированных атомов водорода^
а к более высокому значению энергии. (
Эта ошибка возникает из представле-
представления полной волновой функции в виде
одного слэтеровского определителя
Хх?ъ пространственную часть которогоJ
в выражении D.129) можно записать в виде произведения:
D.134)^
В этом выражении слагаемые Xi(l)XiB) и XiWXiQ) соответствую'Г
ионным структурам, когда два электрона находятся либо у ядра а,
либо у ядра b (сравните с соответствующей ситуацией в методе ВС):
Н~+Нъ и На+Щ. Следовательно, при Д^-юо вероятность дис-
диссоциации молекулы Н2 на два нейтральных атома водорода или их
ионы в методе МО одинакова, что, очевидно, неверно даже из!.
простых химических соображений. Полученный результат отражает^
известный недостаток однодетерминантной волновой функции ме-*
тода МО — значительную переоценку вклада ионных структур.
Задача 4.6. Какой из методов, МО или ВС, является наиболее пригодным
для изучения реакций диссоциации? *
4.5.3. Волновые функции
возбужденных состояний молекулы Н2
Волновые функции возбужденных состояний молекулы Н2 мож-
можно записать в виде слэтеровских детерминантов:
1
<РгB)аB)
4f
D.135)
«B)
ф2B)аB)
D.136)
D.137)
D.138)
1Ш _
I ft —
D.139)
Их получают из детерминанта основного состояния заменой в нем
столбца или строчки соответствующей виртуальной спин-орбита-
лью D.58). Собственные функции оператора Н ?2, % и *?6 должны
быть также собственными функциями операторов S2 и Sr Дейст-
Действительно, нетрудно показать, что
С Зт о 1ш С Ш в Ш П> (А. \AC\\
&z х2 — Эг Х6 — 3^4— °z^5 — <J» \<*Л<*\))
S 3vP-, = ft34' • Ч 3vP — — h*W (A 1411
'-'г * 2 « * 2> k-»r *¦ 3 «аз- ^t.tTiy
Эти равенства легко проверить, пользуясь формулами B.100). На-
Например, докажем D.141):
z.
Аналогично доказывается, что 1х?ь Зх?2,
собственными функциями оператора S2:
и 1хР6 являются
135
D.142)
S2 3?2=2# 3?2; S2 34S2tf 3?
Однако Ч*4 и Ч*5 не являются собственными функциями S2 и, следо-
следовательно, не являются решениями уравнения Шрёдингера. Можно
построить их линейные комбинации, которые, как нетрудно прове-
проверить, есть собственные функции S2 и Sz:
B)-92A)^B)] х
D143)
х [«AHB)-«BHA)]. <4144)
Для этих функций выполняются соотношения
S23^4=^3^4; S23?4=0; S21^5-S214'5=0, D.145)
которые показывают, что 3*F4 — функция триплетного, а 1х?5 —
синглетного возбужденных состояний.
Задача 4.7. Доказать соотношения D.142) и D.145).
Состояния 3Ч/2, 3?з и 3xF4 имеют одинаковую электронную энер-
энергию, так как из формул D.135)—D.139) видно, что все они имеют
одинаковые пространственные части, а гамильтониан Н не зависит
от спиновых переменных:
H\\—H\2
-Л:12, D.146)
= I
где Н — гамильтониан молекулы водорода D.21); Jl2 и Кп — куло-
новский и обменный интегралы, которые легко вычислить, исполь-
используя C.9) и C.36) и выражения для (р D.111)—D.112);
1 -A1122)-A2112) ; D.147)
2
D.148)
136
функции 3xF2, 3Ч*з и 3*F4 соответствуют возбужденному триплетному
состоянию. Электронная энергия синглетных однократно возбуж-
возбужденного lvP5 и дважды возбужденного 1х?6 состояний запишется
соответственно в форме
;^ <4149>
2(ЯМ-Я12)
—\-s +/22> DЛ50)
где
^E^l DЛ51)
На рис. 4.9 представлены кривые различных состояний мо-
молекулы Н2. Энергия перехода из основного состояния в возбу-
возбужденное lxF5
1Е0~1Е5 = ПА эВ
достаточно хорошо согласуется с соответствующей эксперимен-
экспериментальной величиной, равной 13,2 эВ.
Из рис. 4.9 видно, что молекула водорода Н2 при возбуждении
может перейти только в состояние lxF5AJ]J), так как только кривая
энергии этого состояния имеет минимум. Остальные кривые воз-
возбужденных состояний не имеют минимумов; следовательно, моле-
молекула Н2 при переводе в эти состояния должна диссоциировать на
два свободных атома водорода.
4.5.4. Конфигурационное взаимодействие
Рассмотрим, как учитывается KB для молекулы Н2. В разд. 4.5.2
были получены различные электронные конфигурации H20Fj — *F6).
Поэтому в D.87) М=6. Согласно теореме Бриллюэыа, матричные
элементы HUf #13, Яи, #i5 равны нулю. Кроме того, с учетом
различий в спиновой симметрии все остальные недиагональные
элементы равны нулю, кроме Н1б. Секулярное уравнение D.88)
примет вид
137
Hn-E
0 J
0
0
0
Я.б
0
0
0
0
0
0
Г 0
Н„-Е
0 l
0
0
0
0
0
U Е*
4 ~~ Ё*
0
0
0
0
0
0
Н55-Е
0
я,
0
0
0
0
я
-?
= 0. D.152)
Четыре решения этого уравнения очевидны: Ei—Нц /7=2, 3, 4, 5).
Два остальных решения можно определить из уравнения
Нц—Е Я16
=0; D.153)
Я,
-Е
, Н *% <к,
#« =
D.154)
Яп
D.155)
16»
где Ei — энергия основного состояния молекулы Н2 с учетом КВ.
Вычисляя Я6б, Я]б и Яц (не путать с матричными элементами
HMV в матрице F^), получим понижение энергии диссоциации до
Х>е=3,23 эВ. Кроме того, расчет правильно предсказывает атомные
состояния, в которых диссоциирует молекула Н2 при R-+ оо (Н-+Н)
(рис. 4.13, 4.14).
46. МО ГОМОЯДЕРНЫХ ДВУХАТОМНЫХ МОЛЕКУЛ
Как следует из рассмотренного подробного расчета молекулы
водорода, основные характеристики молекулы определяются харак-
характером ее молекулярных орбиталей. Вычисление последних составля-
138
at
0,5
'0,5
Рис. 4.14. Трехмерное представление элект-
электронной плотности в возбужденном триплетном
CZ) молекулы Н
2 k В 8 RJaQ
Рис. 4.13. Кривые полной энер-
энергии молекулы Нг в различных
состояниях, полученные с уче-
учетом конфигурационного взаимо-
взаимодействия
ет главную задачу любого квантово-механического расчета по ме-
методу МО.
Для гомоядерной двухатомной молекулы, составленной из эле-
элементов второго периода, ограничивая базис валентными Is-, 2s-
и 2/?-орбиталями, искомые МО в форме ЛКАО можно записать
в виде
(p = ct\sa
D.156)
где а и b — обозначения ядер.
Задача, таким образом, сводится к решению секулярного уравне-
уравнения D.70) десятого порядка и соответствующей системы 10 алгебра-
алгебраических уравнений D.62). В результате будут получены 10 (по числу
базисных АО) различных МО молекулы. Число заполненных МО
определяется числом электронов в молекуле. Расчеты показывают,
что в каждой МО гомоядерной двухатомной молекулы несколько
(обычно два) коэффициентов cifl велики, остальные или равны нулю,
или практически неотличимы от него. Для того чтобы атомные
орбитали входили в МО с большим вкладом, необходимо выполне-
выполнение следующих условий: 1) энергии, соответствующие АО, должны
быть сравнимы по величине; 2) АО должны иметь отличное от нуля
перекрывание, т. е. они должны обладать одинаковыми свойствами
симметрии относительно оси молекулы.
Так, j-AO могут комбинировать с s-, ^-функциями, но не с ру-,
Pz'AO, Действительно, перекрывание s- и ^-функций отлично от
нуля, в то время как s- и /у-функции не перекрываются.
139
Задача 4.8. Найти разрешенные и запрещенные комбинации следующих
АО: s, рх, ру, pz, dxy, dx d d d
Так, например, для основного состояния молекулы Li2 в резуль-
результате расчета по методу Рутаана в минимальном базисе АО слэте-
ровского типа D.156) получаются следующие МО (в порядке воз-
возрастания энергии):
р, = 0,7048(Ue + lsb) + 0,0095B.sa+2sb) + 0,0003B/?^+2pxb);
q>2=0,7054A5. - lsb) + 0,0209Bsa-bb) + 0,0032B^+2pxb);
Легко заметить, что сходные атомные орбитали комбинируются
и дают наибольший вклад в МО. Это позволяет, не проводя точ-
точного расчета, рассмотреть основные качественные закономерности
электронного строения двухатомных молекул первого периода: Н2,
Не2, Li2, Be2, B2, C2, N2 и т. д. и их ионов.
Рассмотрим классификацию и обозначения МО гомоядерных
молекул. При этом принимаются во внимание:
1) симметрия орбитали относительно оси молекулы;
2) симметрия относительно плоскости, перпендикулярной оси
молекулы;
3) тип АО, в которые переходят МО при увеличении межъядер-
межъядерного расстояния;
4) симметрия МО относительно центра инверсии.
Молекулярные орбитали, симметричные относительно отраже-
отражения в плоскости молекулы, называют сг-орбиталями, антисиммет-
антисимметричные относительно такого отражения орбитали называют гс-ор-
биталями. Молекулярные орбитали двухатомных молекул харак-
характеризуются квантовым числом А, аналогичным квантовому числу
mt в атоме, которое определяет величину проекции орбитального
момента электрона на молекулярную ось. Для <т-орбиталей Я = 0,
для тс-орбиталей А=*1. Аналогично, имеются орбитали с Я = 2, 3, ...,
называемые соответственно 5, ф,.... Молекулярные орбитали с ХфО
являются дважды вырожденными, т. е. проекция момента на ось
молекулы может быть равна ±Ак
Все МО характеризуются индексами g (gerade — четный)
и и (ungerade — нечетный). Эти обозначения зависят от того, ин-
инвариантна МО (g) или меняет знак (и) при инверсии относительно
центра симметрии.
Для обозначения антисвязывающих или разрыхляющих орбита-
лей (к которым относятся орбитали, антисимметричные относите-
относительно плоскости, перпендикулярной оси молекулы) используют обы-
140
Объединенные
атомы
Разъединенные
атомы
Рис. 4.15. Корреляционная диаграмма
молекулярных орбиталей гомоядерных
двухатомных молекул
чНо символы <т*, я*, 5* и т. д.
Четность и нечетность МО лежат
в основе правил отбора разрешен-
разрешенных электронных переходов (см.
B.60)). Возможны переходы u+-+g
и невозможны переходы и *-* и
Молекулярная орбиталь опре-
определяется также типом АО, из ко-
которых она построена и в которые
она переходит при увеличении
межъядерного расстояния. На-
Например, запись els означает, что
данная МО с-типа построена из
ls-АО и переходит в них при
Л^-* оо (разъединенные атомы).
Иногда полезно знать, в какую.
АО переходит данная МО при
уменьшении межъядерного рас-
расстояния до нуля, т. е. при слия-
слиянии ядер. Такое состояние моле-
молекулы называют состоянием объединенного атома.
Диаграмму, показывающую, в какие состояния объединенного
и разъединенных атомов переходит данная МО при изменении
межъядерного расстояния, называют корреляционной диаграммой
(реже используют термин «диаграммы соответствия»). Корреляци-
Корреляционная диаграмма МО гомоядерной молекулы показана на рис. 4.15.
Диаграмма построена на основании точных теоретических расчетов
энергии МО Гомоядерных молекул в зависимости от межъядерного
расстояния ЯЛ. Эти расчеты совместно с данными молекулярной
спектроскопии дают следующий порядок МО для молекул, постро-
построенных из атомов первого и второго периодов:
D.157)
Орбитали ag2p и вырожденные nzMu2p имеют близкие энергии
и могут меняться местами в зависимости от конкретной молекулы.
Аналогично можно получить энергетическую последовательность
МО, построенных из АО с большими квантовыми числами. Поря-
Порядок расположения МО для этих молекул может быть отличным.
При построении диаграммы необходимо учитывать правило непере-
непересечения Вигнера—Неймана, согласно которому уровни одинаковой
симметрии, например а, л,..., g, и,.... не пересекаются. Абсолютные
значения энергий МО меняются в зависимости от молекулы, поэто-
МУ корреляционные диаграммы могут быть использованы только
og\s <
141
6*is
6fs
•Ь- 7f2p
Рис. 4.16. Образование связываю-
связывающих и разрыхляющих молекуляр-
молекулярных орбиталей из АО. Пунктиром
показаны узловые плоскости, зачер-
зачернены отрицательные угловые части
в качественных целях. С помо
этих диаграмм можно судить об
менении орбитальной энергии п
изменении межъядерного расст
ния. Видно, что энергия связыв
щих орбиталей повышается с уме;
шением межъядерного рассто
в то время как энергия разр
ющих орбиталей понижается. Кро
того, связывающие орбитали ко
лируют с состоянием объединение)]
атома, имеющим то же кванто
число, что и данная МО. Разр;
ющие МО коррелируют с объедн
ненным атомом, у которого кван
вое число на одну или две един
больше.
Рассмотрим конкретные случ
построения МО гомоядерных мо,
кул. На рис. 4.16 представлен _
МО, построенных из \s-9 2px- и 2pi
АО. Отметим, что разрыхляю
МО имеет на одну узловую пл
кость больше по сравнению с соот^
ветствующей связывающей МО.
Следует кратко остановиться и на классификации электронных
термов гомоядерных двухатомных молекул. Одним из основных
принципов классификации атомных термов была классификация по
значениям полного орбитального момента. Очевидно, что в молекуч
лах подобная классификация невозможна, так как поле ядер не
обладает центральной симметрией и, следовательно, полный ор-
орбитальный момент может не сохраниться. Однако аксиальная сим-
симметрия поля ядер обусловливает сохранение проекции полногЪ
орбитального момента на ось молекулы, что позволяет классифици-
классифицировать электронные термы по значениям этой проекции. Абсолют-
Абсолютную величину проекции полного орбитального момента на ось
молекулы принято обозначать буквой Л; числовым значениям
Л ставят в соответствие большие греческие буквы. Так, Л = 0, I,
2 отвечают соответственно ?-, П-, А-термам.
Каждое электронное состояние молекулы характеризуется пол-
полным спином S всех электронов и связанной с ним мультшлет-
ностью 25+1. Терм ЪУ обозначает состояние с А=0, 5=1. Вов
электронные термы с Л#0 двукратно вырождены. Для У-термо»
следует различать случаи ?+ и ?~, для первого волноваяфункци*
не меняется при отражении в любой плоскости, содержащей ос*
молекулы, для второго — меняет знак. Так же как и для МО*
142
- / V <*'
Рис. 4.17. Контурная карта разностной
электронной плотности для молекулы
Н2) определяемая как разность элект-
электронных плотностей молекулы и свобод-
свободных атомов
вводится классификация по чет-
четности (g) и нечетности (и) волно-
волновых функций, например 3?«,
i?+ и т. д.
4.6.1. Электронные конфигу-
конфигурации гомоадерных молекул
Для получения электронной
конфигурации двухатомной мо-
молекулы необходимо разместить
соответствующее число электро-
электронов на МО с учетом принципа
Паули. Рассмотрим элект-
электронные конфигурации молекул
элементов первого и второго пе-
периодов.
Молекула Н2 имеет два
электрона на а* 1 s-МО, та-
таким образом, конфигурация
основного состояния этой
молекулы (a isJ. Два элект-
электрона на связывающей МО
образуют простую связь.
Энергия диссоциации Н2 ра-
равна Dt — 4,15 эВ, равновес- л 1О __
г Рис. 4.18. Контурная карта разностной элект-
ное расстояние Re=0,074 нм. ронной плотности Не2
Молекула Не2 с конфигурацией (a isJ (a* isJ имеет два связыва-
связывающих и два разрыхляющих электрона, которые в сумме не дают
связи. Действительно, молекула Не2 неустойчива и не наблюдалась
в основном состоянии. В то же время если один из электронов МО
0"* b возбудить на связывающую орбиталь, например а 2г, то
в молекуле Не2 окажется три связывающих и один разрыхляющий
электрон. Возбужденные состояния Не2 могут быть стабильны
и спектроскопически наблюдались. Ион молекулы гелия Не2+ устой-
устойчив, энергия диссоциации De—3 эВ, равновесное расстояние
-^=0,108 нм. Эти величины близки аналогичным характеристикам
Н2. Для стабильной молекулы Н2 электронная плотность возрастает
в межъядерном пространстве (рис. 4.17 и 4.18). Для неустойчивой
Здесь и далее индексы gnu опущены, так как их использование наиболее важно
ри рассмотрении электронных переходов между термами.
143
молекулы Не2 межъядерное п
Рис. 4.19. Контурная карта разностной
электронной плотности молекулы и
в состоянии
странство характеризуется noi
жеыной электронной плои
стью.
Молекула 1Л2. Конфигург
основного состояния
(<г isJ (а* UJ (о- 2sJ
имеет два избыточных связыва-
связывающих электрона, что ведет к
разованию ординарной связ!
Молекула Li2 стабильна, энер!
диссоциации De=l,09 эВ, равж
весное расстояние Л—0,267
Связь в Li2 удлинена по срав)
нию с Н2 @,074 нм) из-за зффе|й
та экранирования ядра внутрен-
внутренними электронами. При образо-г?
вании молекулы Li2 1^-АО лития
перекрываются незначительно,
поэтому энергия alj-MO близка к энергии U-AO. Электронна»;
конфигурация Li2 часто записывается в виде КК (a 2sJ; эта запись
[КК обозначает (a* IsJ] подчеркивает, что при образовании молеку-
молекулы общая энергия внутренних электронов меняется незначительно.
Действительно, можно сказать, что сумма распределений связыва-
связывающей и разрыхляющей МО практически совпадает с простым нало-
наложением атомных плотностей. На рис. 4.19 представлена карта раз-
разностной электронной плотности для молекулы Li2, основное от-
отличие которой от аналогичной карты Н2 заключается в появлении
узловых плоскостей.
Молекула Ве2 с электронной конфигурацией основного состоя-
состояния КК (о 2sJ (a* 2sJ не имеет избытка связывающих электронов.
Эта молекула экспериментально не обнаружена. Возбужденные со-
состояния этой молекулы могут наблюдаться спектроскопически.
Молекула В* Конфигурация основного состояния КК (в 2sJ (с*
2sJ (я 2рJ говорит о стабильности этой молекулы. я-Молекуляр-
ные орбитали являются вырожденными nz2p=nj2p, вследствие чего
электроны должны расположиться так, чтобы мультиплетность со-
состояния была максимальна (правило Хунда). В связи с этим кон-
конфигурация основного состояния будет КК (a 2sJ (a* 2sJ (nz 2pI {щ
2рI, причем электроны на nz~ и я,-МО имеют параллельные спины.
Однако экспериментально обнаруженный парамагнетизм молекулы
144
N
N,
У
НИЛ
ч
\
ч
ч
ч
3:/,y
15
16
17
18
13
20
36
37
38
403
Ц-10
/,
a J,3B
Рис. 4.20. Корреляционная диаграмма молекулярных орбиталей (а) и фотоэлектрон-
фотоэлектронный спектр молекулы N2 (б)
Интенсивность
219Jhm
-t
239,7 нм
-i
В2 поставил под сомнение выводы качественного подхода. Прове-
Проведенные по методу Хартри—Фока расчеты привели к выводу о том,
что основным состоянием В2 является терм 3?~. Однако более
точные вычисления показали, что минимум энергии достигается
в состоянии 5J^T, возникающем из электронной конфигурации
КК (о Ъ)г {а* Ъ)х (я ад2 (а 2рI.
Молекула Сг имеет конфигурацию с четырьмя избыточными
связывающими электронами КК {a 2sJ (сг* ЪJ (л 2р)\ вследствие
чего образуется двойная связь. Как отмечалось, энергии п 2р~
и а 2р-МО близки по величине. В связи с этим для С2 возможна
конфигурация КК (<т 2sJ (a* 2sJ (л 2р)ъ{а2р)х. Для этой конфигура-
конфигурации возможно триплетное состояние. Эксперимент показывает, что
молекула С2 диамагнитна, что свидетельствует в пользу первой
конфигурации п B/?L.
Молекула N2. Конфигурация КК (<т 2sJ (с* ЪJ (п 2рL (с 2рJ
обладает шестью избыточными связывающими электронами, что
согласуется с существованием тройной :N = N:-cbh3h. На рис. 4.20
представлены корреляционная диаграмма молекулярных уровней
145
молекулы азота и ее фотоэлектронный* спектр. Из этого рисунка^
видно, что качественные представления о молекулярных орбиталяд
молекулы достаточно хорошо согласуются с опытными данными. ]
Молекула О2. Конфигурация КК (a 2sJ (а* 2s)z {а 2рJ (я 2pf (тс1»"
2р)г является конфигурацией основного состояния молекулы О^
Спектроскопически обнаружено, что в молекулах О2 и F2 МО а2р,
лежит ниже МО п2р. Четыре связывающих электрона образуют*
двойную связь. Электроны на МО я* 2р имеют параллельные спины,
согласно правилу Хунда, в связи с чем у молекулы О2 основное*
состояние является триплетным. Подобную электронную структуру^
имеет молекула S2. Триплетность основного состояния молекулы^
О2 означает, что эта молекула обладает магнитным моментом,
связанным со спином, т. е. молекулярный кислород представляет"!
собой парамагнетик. Действительно, жидкий кислород притягивает-,
ся к полюсам магнита точно так же, как притягиваются железные1
опилки. Жидкий кислород — одна из немногих жидкостей с таким'
удивительным свойством. В первые годы развития теории молекул*
наблюдалось своеобразное соперничество между методом молеку-*
лярных орбиталей и методом валентных схем. Метод МО очен*
просто объясняет парамагнетизм молекулы О2. Метод Гайтлера—*
Лондона не приводит к такому же предсказанию сколь-нибудь*
простым и ясным путем.
Молекулы F2 и Ne2 имеют электронные конфигурации соответст-
соответственно ^
КК (a 2sJ (а* ЪJ (а 2рJ (п 2р)+ (я* 2р)*
пКК(<т 2sY (а* ЪУ (с 2рУ (п 2pf (я* 2pf (а* 2рJ.
В молекуле F2 имеется ординарная связь, Ne2 не обнаружен.
Можно ввести приближенное числовое определение кратности
связи, введенное Г. Герцбергом, которое согласуется с химичес-
химическими представлениями о характере связей в гомоядерных молеку-
молекулах по формуле
'"Методы фотоэлектронной и рентгеноэлектронной спектроскопии основаны и»
измерении энергетического спектра электронов, выбитых из вещества при бомбарм
дировке его потоком фотонов или заряженных частиц. Энергии выбитых электрово»
?^лн связаны с энергией соответствующих оболочек атомов или молекул в исследу-'
емом веществе (без учета малых поправок на изменения колебательной и вращатель-
вращательной энергии при ионизации) соотношением Exm=ky—Iai где hv — энергия бомбар-
бомбардирующих фотонов; 1а — адиабатический потенциал ионизации электрона в молеку-
молекуле или атоме. Детектируя Еаа и зная энергию квантов hv, определяют /а.
146
p=
n-n*
D.158)
где P — кратность связи; nun* — число электронов на соответст-
соответственно связывающих и разрыхляющих МО.
В табл. 4.8 приведены энергии диссоциации, равновесные рассто-
расстояния и кратности связей гомоядерных молекул первого периода;
здесь энергия диссоциации увеличивается, а равновесное расстояние
уменьшается при увеличении порядка связи. Критерием того, связы-
связывающей или разрыхляющей является данная МО, может служить
энергия диссоциации. Например, высшая занятая МО в N2 является
связывающей. Действительно, энергия диссоциации N2 больше
энергии диссоциации N2+. Для О2 высшая МО разрыхляющая, что
также видно из сравнения энергии диссоциации О2 и О2+. Отметим,
что полная энергия молекулы не может служить критерием связыва-
связывающих или разрыхляющих свойств МО; как те, так и другие умень-
уменьшают полную энергию молекулы.
Таблица 4.8. Свойства основного состояния гомоядерных молекул
Молехула
н2
Не*
Не2
Li2
Ве2
в2
Кратность
связи
V2
1
Чг
0
1
0
1
De, эВ
2,8
4,75
3,0
—
1.1
—
3,0
Rg, нм
0,106
0,174
0,108
—
0,267
—
0,159
Молекула
с2
щ
N2
о2
F2
Ne2
Кратность
связи
2
272
3
2У2
2
1
0
De, эВ
6,4
8,9
9,9
6,6
5,2
1,4
—
Re, нм
0,124
0,112
о,по
0,112
0,121
0,142
—
Задача 4.9. Какая из пары молекул имеет большую энергию диссоциации:
a) Li2 или Li2+; б) С2 или С2+; в) О2 или О"; г) N2+ или N~; д) F2 или F+?
Задача 4.10. Записать волновые функции основного состояния молекулы
F2 в методе МО и ВС.
4.7. МО ГЕТЕРОЯДЕРНЫХ ДВУХАТОМНЫХ МОЛЕКУЛ
Для двухатомных молекул, состоящих из различных атомов
(гетероядерные молекулы), МО несимметричны. В связывающих
МО коэффициенты при АО более электроотрицательного атома
больше. Для антисвязывающих МО вклад более электроотрица-
147
тельного атома меньше. При класси-
классификации МО обычно не указывают
АО, из которых они образованы, а со*
храняют только тип (а, тс, ...) МО и ее
порядковый номер внутри данного ти-
типа. Например, расчет молекулы HF
с минимальным слэтеровским базисом
дает следующие МО в порядке возра-
возрастания энергии:
l«7=0,997(bF)+0,013BjF)+
+0,002B/wrF)- 0,031Ask);
2<т= - 0,240A *F)+0,955ByF)+
Ъа = 0,076A sF) - 0,429B^F) +
+ 0,717B/><xF)+0,520(l5H);
, Bpzn?).
Рис. 4.21. Контурная карта раз-
разностной электронной плотности Из карты электронной дифференци-
гетероядерной молеку^гш HF альной плотности молекулы HF, пред-
в состоянии *? ставленной на рис. 4.21, видно, что при
объединении атомов Н и F в молекулу
электронная плотность из периферийных областей переходит в об-
область между ядрами, что объясняет устойчивость молекулы.
В гетероядерных молекулах отнесение орбитали к связывающе-
связывающему типу наиболее тесно определяется по соответствующей ей карте
дифференциальной плотности, т. е. если на связи электронная плот-
плотность по сравнению с плотностью свободных атомов увеличивает-
увеличивается, то орбиталь связывающая, при уменьшении плотности — ор-
биталь разрыхляющая. Корреляционная диаграмма для гетероя-
дерной двухатомной молекулы строится аналогично корреляцион-
корреляционной диаграмме гомоядерной молекулы на основании расчетов
и спектральных данных.
В табл. 4.9 совместно с экспериментальными данными приведе-
приведены вычисленные значения дипольного момента и некоторые другие
характеристики гетероядерных молекул.
Наиболее точно теоретические расчеты воспроизводят экспери-
экспериментальные данные по равновесным расстояниям. Для получения
правильной геометрической структуры возможно использование
метода Рутаана даже с минимальным базисом. В то же время
глубина потенциальной ямы (энергия диссоциации), а также диполь-
ный момент и некоторые другие физические характеристики весьма
чувствительны к размерам базиса, а также к корреляционным по-
поправкам. Иллюстрацией этого являются многочисленные расчеты
дипольного момента молекулы СО.
148
Таблица 4.9. Сравнение рассчитанных и экспериментальных характеристик гете-
роядерных молекул
.—
Молекула
LiF
LiH
HCl
HF
CO
Re.
расчет
2,941
3,034
2,409
1,6%
2,08
a. e.
экспери-
эксперимент
2,955
3,015
2,410
1,733
2,132
расчет
4,08
1,49
3,48
4,38
7,84
, эВ
экспери-
эксперимент
5,95
2,52
4,62
6,12
11,23
расчет
6,30
5,89
—
1,83
-0,08
D
экспери-
эксперимент
6,28
5,88
—
1,82
-0,11
Потенция.":
эВ
расчет
13,02
7,02
11,84
14,54
14,98
экспери-
эксперимент
„
6,50
12,74
15,77
14,01
Тип расчета ц, D
Минимальный базис ... —0,464
Расширенный базис ... 0,361
Метод Хартри—Фока, предел ... 0,280
Метод Хартри—Фока, +138 двукратно возбужденных
конфигураций... 0,173
Метод Хартри—Фока +138 двукратно возбужденных
и 68 однократно возбужденных конфигураций ... —0,08
(отрицательный знак соответствует направлению дипольного момента от кисло-
кислорода к углероду).
Правильный знак дипольного момента удалось получить лишь
с учетом 138 двукратно возбужденных и 68 однократно возбужден-
возбужденных конфигураций. Направление дипольного момента молекулы
отвечает структуре С~ — О+, что находится в противоречии с элект-
электроотрицательностью атомов, но согласуется со структурой
:~С = О+: с тройной связью.
4.8. ТЕОРЕМА ГЕЛЬМАНА—ФЕЙНМАНА
Гамильтониан любой системы, не зависящий явно от времени,
всегда содержит какие-либо параметры. Например, в гамильтониан
молекулы типа D.1) как параметры входят межъядерные расстоя-
расстояния. В качестве параметров можно рассматривать заряды ядер Z,,
массу электрона met постоянную Планка h и другие величины. От
этих параметров будут зависеть собственные числа Е„ и собствен-
собственные волновые функции ?„ гамильтониана:
Н(А) Чщ(к) =Еп(Х) УЩ(Х). D.159)
Существует соотношение, показывающее, как изменяется собствен-
собственное число Еп при изменении параметра к
149
дЕн(к)
~~дк~
ОА
D. И
Соотношение D.160) впервые было выведено независимо друг
друга Г. Гельманом и Р. Фейнманом, его называют теоремой
льмана—Фейнмана*. Теорема доказывается достаточно просто,
определения A.34) имеем
дк
где последнее слагаемое ввиду эрмитовости оператора Н моз
преобразовать:
(к)Щк)
дк
дк
Поскольку
дЕп(к)
и
имеем
дк
3VJX)
Последняя производная равна нулю в силу нормированности
и теорема D.160) доказана. Теорема Гельмана—Фейнмана позв<
ет связать квантово-механический и классический подходы при
смотрении сил в молекулах. Для двухатомной системы из тео]
D.160) можно получить еще две важные теоремы.
*Г. Гельман A906—1943) — немецкий физик, автор известных работ в
квантовой химии. После захвата власти нацистами в Германии Г. Гельман:
в Советский Союз, где в 1936 г. опубликовал монографию «Квантовая
в которой впервые была выведена теорема D.16).
Р. Фейнман (род. в 1918 г.) — выдающийся американский физик. За ф>
тальный вклад в квантовую теорию поля Р. Фейнман в 1965 г. был у;
Нобелевской премии. Работа Р. Феймана «Силы в молекуле», в которой он:
Ф от Г. Гельмана доказал теорему D.160), была его дипломной работой.
150
Задача 4.1}. Найти средние значения Тш V для водородоподобяого атома
в состояния с главным квантовым числом п, используя теорему Гельмана—
фейнмана (в качестве параметров выбрать число протонов в ядре Z и массу
электрона).
Задача 4.12. Колебательный потенциал в молекулах часто аппроксимиру-
аппроксимируют потенциалом Морзе:
—овг
)
u(r)=A(e -2*
Уровни энергии частицы, движущейся в таком потенциале, определяются фор-
формулой
ah
Ея
Найти средние значения кинетической и потенциальной энергии такой частицы.
4.8.1. Теорема вириала и природа химической связи
Рассматривая межатомное расстояние R как параметр, можно
вывести соотношение
Из теоремы Гельмана—Фейнмана следует, что
D-164)
ИЛИ
dE
- = 0. D.165)
Формулу D.165) называют теоремой вириала для двухатомной мо-
молекулы. Так как полная энергия системы Е есть сумма средних
значений кинетической <7> и потенциальной {V) энергий, можно
переписать D.165) в виде
dF
_ = 0. DЛ66)
В частности, для равновесных положений ядер можно из^ D.165)
и D.166) получить соотношения
-E; : D.167)
>DЛ68)
которые должны выполняться для точных решений любых кван-
тово-механических систем, состоящих из заряженных частиц (для
атома см. задачу 2.8).
151
0,5
*- о
-ко
н.
1
5 6 7 R/an
Рис. 4.22. Зависимость полной Е, кине-
кинетической Т и потенциальной V энергий
от межъядерного расстояния R в моле-
молекуле Нг
Теорема вириала устанавли-^
вает определенные соотношения,
между полной, кинетической
и потенциальной энергиями, ко-
которые должны выполняться]
в любых расчетах молекулярных]
систем. i
Рассмотрим влияние требо-
требований теоремы вириала на об-
образование химической связи
в молекуле водорода Н2.
Из представленного на рт
4.22 вида зависимостей полно]
Е, потенциальной V и кинетичес
кой Т энергий от межъядерно;
расстояния R в Н2 можно заметить, что химическая связь (пониже-
(понижение полной энергии молекулярной системы по сравнению с энергией
свободных атомов) возникает уже на больших межъядерных рассто-
расстояниях R, причем понижение полной энергии Е на больших R полно-:
стью обусловлено понижением кинетической энергии Т. При при-*!
ближении к равновесному состоянию теорема вириала требует вы
полнения соотношения D.167) или D.168), поэтому при приближе-
приближении к Re потенциальная энергия резко понижается, а кинетическая
повышается. Повышение кинетической энергии Т при сближении^
ядер до равновесного расстояния обусловливается перетеканием;
электронной плотности из внешних областей на связь, что видно из;
разностных карт электронной плотности двухатомных молекул (cmJ
рис. 4.17, 4.19 и 4.21). Повышение электронного заряда на связи,*
в свою очередь, приводит к понижению потенциальной энергии V.
Заметим, что все эти процессы происходят одновременно. Te-j
орема вириала выступает здесь в роли регулятора, поддерживая вс^|
время необходимое равновесие между Г и К, не допуская неог**
раниченного увеличения или уменьшения одной из величин. :Щ
4.8.2. Электростатическая теорема -
i
Р. Фейнман и Г. Гельман применили теорему D.160) к изучению^
сил, действующих в молекуле, выбрав в качестве параметров ядер-;
—»
ные координаты R а(ха, ул, za).
Оператор кинетической энергии электронов не зависит явно от^
межъядерного расстояния Л^, поэтому для молекулы теорему Гель-,
мана—Фейнмана D.160) можно записать в форме *
152
8E - (Я .-Г)
Л? ¦* .у ,э^+ I ЗД пэ . D.169)
гле о(?)=х?**? — электронная плотность.
д &Е
Производная _- описывает силу, действующую на ядро в моле-
молекуле. Ее компонента по одной из осей координат равна
Д.- -г. [p(f)PZ;AdV+ 2 «iT- D.170)
Из формул D.169) и D.170) следует, что силу, действующую на ядро
а, можно рассматривать чисто электростатически как сумму элект-
электростатического взаимодействия ядра а с электронным облаком
плотности р(?) и с другими ядрами с зарядами Zp [что отражено во
втором члене уравнения D.170)]. Поэтому теорему Гельмана—
фейнмана, записанную в форме D.169), называют электростати-
электростатической теоремой.
Электростатическая оценка силы, действующей на ядро, воз-
возможна только в приближении Борна—Оппенгеймера. Быстрое дви-
движение электронов в молекуле позволяет им «мгновенно» реагиро-
реагировать на любое изменение в ядерной конфигурации. В результате
этого ядра как бы «чувствуют» не отдельные электроны, а тольхо
общее электронное облако, распределенное в пространстве с плот-
плотностью р(?).
Необходимо отметить, что ошибка в силе, вычисленной по
формуле D.169), существенно зависит от точности вычисления элек-
электронной плотности.
Для двухатомной молекулы все пространство можно разделить
на связывающую область, в которой электронная плотность создает
силы, действующие на ядра по направлению друг к другу, и на
антисвязывающую область, в которой силы, действующие на ядра,
стремятся их раздвинуть. Граничная поверхность, разделяющая
связывающую и антисвязывающую области, определяется усло-
условием
Исходя из формулы D.169), легко показать, что эта поверхность для
гомоядерной двухатомной молекулы имеет вид двухплоскостного
гиперболоида. Области связывания и антисвязывания на плоскости
Для некоторых молекул показаны на рис. 4.23 и 4.24, из которых
видно, что связывающая область концентрируется в районе между
ядрами. Заряд в этом районе стремится приблизить ядра друг
к другу. Электронный заряд, находящийся вне ядер, стремится
Удалить ядра друг от друга, и, естественно, этот район является
153
9-yal
Рис. 4.23. Связыва-
Связывающая в антисвязыва-
ющая (заштрихован-
(заштрихованная) области двух-
атомной молекулы АВ
Рис. 4.24. Связывающая и антисвяз!
(заштрихованная) области молекул NaQ
и НО E)
антисвязывающим. Можно сравнить эти рисунки с контур
диаграммами дифференциальной электронной плотности, пр:
денными на рис. 4.21. При сближении атомов на равновесное рф
стояние часть электронной плотности из несвязывающих областей
переходит в связывающую область. Электронный заряд распредели-;
ется в обеих областях так, чтобы силы, стремящиеся сблизить
и растолкнуть ядра, были одинаковы. д
Литература
Коулсои Ч. Валентность. — М.: Мир, 1965. Гл. 4—б.
Козмал У. Введение в квантовую химию. — М.: ИЛ, 1960. Гл. 10, П.
Маррел Дж., Кеттл С, Теддер Д». Теория валентности. — М.: Мир, 1968.
фудзшшга С. Метод молекулярных орбиталей. — М.: Мир, 1983. Гл. 2, 7, 9.
Фларра Р. Квантовая химия. — М.: Мир, 1985. Гл. 9—П.
Dewar M. J. S., Kelemen J. — J. Chem. Education. 1971, 48, № 8, 494—501. .
Подробное рассмотрение молекулы водорода в рамках теории МО JIKAOj
и детальное рассмотрение роли кинетической энергии в образовании химическое
связи двухатомных молекул. .
Fdnberg M. J., Koedenbeig К. - J. Chem. Physics, 1971, 54 D), 1495—1511;
Wilson С. W. Jr., Goddard W. A. — Theor. Chim. Acta, 1972, 26C), 195—230.
ГЛАВА 5
ПОВЕРХНОСТИ
ПОТЕНЦИАЛЬНОЙ ЭНЕРГИИ (ППЭ) МОЛЕКУЛ
5.1. ОБЩЕЕ ПОНЯТИЕ
Приближение Борна—Оппенгеймера (см. разд. 4.1), позволя-
позволяющее разделить волновую функцию молекулы на ее электронную
и ядерную части, лежит в основе фундаментального понятия о по-
поверхности потенциальной энергии (ППЭ) молекулы. На этом поня-
понятии базируются все современные представления о свойствах отдель-
154
ных молекул или их совокуп-
совокупностей, которые зависят от
геометрических характеристик
системы. Таким образом, по-
вятие о ППЭ является цент-
центральным в спектроскопии, ки-
кинетике, структурной теории.
Поверхность потенциальной
энергии можно определить как
непрерывную функцию потен-
потенциальной энергии молекуляр-
молекулярной системы (т. е. ее полной
энергии за вычетом кинетичес-
кинетической энергии ядер) от всех неза-
независимых геометрических коор-
координат этой системы E(q)
E(q)=E(qu q2, ?э, •», Яън-б)-
E.1)
Рис. 5.1. Кривая потенциальной энергии
основного состояния двухатомной молеку-
молекулы A); отталкивательная кривая взаимо-
взаимодействия двух атомов, не образующих мо-
молекулу B):
ге — равновесная длина связи, DQ—De — энер-
энергия нулевого колебания
В качестве координат q мо-
можно выбрать любой ряд геоме-
геометрических параметров молеку-
молекулы, независимых от ее абсо-
абсолютного положения и ориен-
ориентации в пространстве. Часто
наиболее удобны внутренние координаты молекулы: длины связей
или другие межъядерные расстояния, валентные, торсионные углы,
но в принципе выбор координат определяется спецификой реша-
решаемой задачи. Общее число независимых внутренних геометрических
координат молекулы равно CN— 6), где N — число атомных ядер,
равное для линейной молекулы QN— 5).
Уже для трехатомной молекулы выражению E.1) соответствует
функция в четырехмерном пространстве, поэтому более точно назы-
называть эту функцию не поверхностью, а гиперповерхностью потенци-
потенциальной энергии, однако для простоты обычно сохраняют термин
ППЭ. Хотя ППЭ E.1) является чисто теоретической конструкцией,
многие ее параметры могут быть найдены (поставлены в соответст-
соответствие) экспериментально при помощи спектральных и кинетических
исследований, использовании метода молекулярных пучков, изуче-
изучении термодинамических, объемных свойств вещества и др.
В простейшем случае двухатомной системы (JV=2, молекула
линейна, 3#—5 = 1) единственной координатой, описывающей
ППЭ, служит межатомное расстояние г. ППЭ в данном случае
представляет собой просто одномерную кривую (рис. 5.1). При
больших г состояние системы соответствует разделенным атомам
(диссоциации двухатомной молекулы на атомы). На участке от
155
ппэ
Область
конфигурацией -
д ного проотрамстба
Рис. 5.2. Схематический вид двумерного сечения ППЭ в координатах q\ и q2 при
постоянной третьей координате qi = <Д0)
г =оо до г-ге энергетическая кривая имеет так называемый аттрак-^
тивный характер, т. е. описывает взаимное притяжение атомов^
Точке ге соответствует минимум энергии на потенциальной кривой.
и эта точка отвечает устойчивой геометрической конфигурации^
молекулы. На участке от г-ге до г=0 кривая имеет отталкиватель-1
ный характер. Точка г=0 отвечает состоянию объединенного атома;1
когда ядра атомов, составляющих молекулу, сливаются в одно*
ядро. Разность значений энергии при г=ге и г=оо дает энергию'
диссоциации (разрыва) связи (De). Истинная энергия диссоциации!
двухатомной молекулы Do меньше De на величину энергии нулевого,,
колебания (см. разд. 5.5.1). Функция потенциальной энергии двух-^
атомной молекулы (рис. 5.1) отвечает эмпирическому уравнению^
Морзе г
где а — параметр. .^
Не все потенциальные кривые двухатомных систем имеют при-=
тягивающий характер (см. рис. 5.1, кривая 1). Некоторые атомы или
ионы во всей области изменения г отталкиваются друг от друга,
и в этом случае потенциальная кривая на всем участке изменения
г имеет отталкивательный характер (рис. 5.1, кривая 2). Такой же
тип энергетической кривой может реализоваться и для устойчивой
молекулы при переходе в одно из ее высших электронных состоянии
156
(см. разД- 5-4)" ППЭ тРехатомной системы Е=Е (qu q2, Яг) уже
(
нельзя представить графически, ее расчет и представление ведутся
обычно методом сечений. Для ряда фиксированных значений коор-
координаты Чъ (Рис- 5-2) в плоскости двух других переменных в виде
сетки с заданным шагом вычисляют соответствующие значения
потенциальной энергии. Такие вычисления проводят для всех точек
сетки разбиения конфигурационного пространства изменения пере-
переменных q. Если область варьирования каждой координаты q{ огра-
ограничить всего 10 точками, то для трех переменных qlt q2, q3 нужно
вычислить значения потенциальной энергии системы в 103 точках.
В общем случае для 3N—6 независимых геометрических параметров
число точек конфигурационного пространства, в которых необходи-
необходимо провести вычисление потенциальной энергии, возрастает до
10ЗЛГ6. Даже для сравнительно малых систем, таких, например, как
содержащие всего 9 ядер (молекула фурана или система
B), это число достигает астрономической величины 1021.
Предположим, что имеется сверхмощная ЭВМ, производящая вы-
вычисление энергии в одной точке E(qu q2i ..., qi\) со скоростью 10 с,
даже и в этом (пока нереалистичном) случае полный расчет для
достаточно ограниченной сетки координат q займет 1020 с, или
~ 1012 лет, что больше времени существования Земли.
5.2. ПУТИ И ЭНЕРГЕТИКА ХИМИЧЕСКОЙ РЕАКЦИИ
Приведенный пример показывает, насколько велики расчетные
трудности, возникающие при вычислениях поверхностей потенци-
потенциальной энергии. Однако для большинства практических целей нет
необходимости знать функцию E.1) в полном объеме. Достаточно
располагать сведениями лишь об определенных участках ППЭ,
прежде всего соответствующих минимумам и седловым точкам.
Поиск этих областей связан с нахождением так называемых крити-
критических или стационарных точек ППЭ.
К стационарным точкам любой функции f(q) относятся такие
точки конфигурационного пространства, в которых значения всех
первых производных функции по каждому независимому перемен-
переменному qf обращаются в нуль. В стационарных точках ППЭ
дЕ дЕ дЕ
я~вя~=--5 = 0- E-3)
Соотношения E.3) означают, что внутренние силы, действующие на
Данную систему в некоторых областях пространства, полностью
157
скомпенсированы и равны нулю. При этом условии система нахо-)
дится в устойчивом по отношению к смещениям из найденного {
положения состоянии, т. е. в минимуме ППЭ, если все значения >
кривизны в точке экстремума функции E.1) положительны. Чтобы
убедиться в этом, надо исследовать матрицу вторых производных'
потенциальной энергии (гессиан): '
д2Е
Всегда возможно эквивалентное преобразование этой матрицы к
кому виду, в котором она будет диагональна. Условием минимума^
функции E.1) будут положительные значения всех диагональньщ
элементов fu (силовых констант) таких матриц. Только области^
минимумов ППЭ соответствуют устойчивым молекулярным струк- *
турам. Самый глубокий (глобальный) минимум характеризует тер- >
модинамически наиболее устойчивую форму. Остальным локаль- ]
ным минимумам ППЭ соответствуют энергетически более богатые \
изомеры или другие образования. Все другие точки ППЭ, не при-
принадлежащие к ее глобальному или локальным минимумам, соответ-'
ствуют неустойчивым геометрическим конфигурациям молекуляр-*5
ной системы. В отсутствие внешних сил молекулярная система;
релаксирует из такой точки, т. е. самопроизвольно подстраивается
к геометрической конфигурации ближайшего минимума ППЭ..
В рамках представления о ППЭ такая релаксация означает движе-
движение изображающей точки, подобное скатыванию тяжелого шарика
или капли воды со склона некой реальной поверхности в находящу-
находящуюся на ней яму. Все точки ППЭ, из которых исследуемая молеку-
молекулярная система «скатывается» в данный минимум, формируют об-
область (бассейн) данного минимума. Таким образом, вся ППЭ делит-
делится на области отдельных минимумов.
В терминах ППЭ химическая реакция означает переход молеку-
молекулярной системы из одного минимума в другой. Путь минимальной
энергии для такого перехода есть движение по дну долины ППЭ,
связывающей два ее минимума через точку перевала, называемую
седловой точкой. Диагонализованная матрица силовых постоянных
E.4) имеет в этой точке одно-единственное отрицательное значение*
(отрицательную кривизну). Изображающая точка скатывается
вдоль направления отрицательной кривизны в один или другой'
минимум по пути минимальной энергии реакции (ПМЭР). Геомет-
Геометрическую конфигурацию молекулярной системы, соответствующую
седловой точке на П*МЭР, называют переходным состоянием. Эне-
Энергия переходного состояния относительно исходного минимума
ППЭ определяет энергию активации и, следовательно, скорость
химической реакции.
Области ППЭ, где в силовых постоянных системы содержится
два (или более) отрицательных значения, не имеют химического
158
Рве. 53. Строение ППЭ в двумерном случае E(qh цг) в областях минимума (а\
седловой точки (б) и максимума (в)
значения, и система избегает попадания в эти области ППЭ. Рис. 5.3
иллюстрирует данные выше определения для простейшей ППЭ,
описываемой только двумя координатами: qx и д2.
Чтобы получить визуальное представление о строении много-
многомерной ППЭ, прибегают к анализу ее различных сечений (карт) по
двум независимым внутренним параметрам. В качестве таковых
выбирают параметры, изменение которых наиболее существенно
влияет на энергетику молекулярной системы. Набирая различные
комбинации таких сечений, можно получить достаточно полную
информацию о строении ППЭ. В качестве примера рассмотрим
ППЭ пока экспериментально не известной молекулы азациклобута-
диена I, отражающую предсказываемую для этого соединения вы-
вырожденную изомеризацию (топомеризацию):
N-
N
Jo
N:
E.5)
1а
II
16
159
Рис. 5.4. Поверхность потенциальной энергии молекулы азациклобутадиена (а) и се
двумерная контурная карта (б) в координатах Ri (длина NCt-связи) и Л2 (длина
МСз-связи), полученная при помощи полуэмпирического расчета:
жирной линией обозначен путь реакции изомеризации 1а^=Чб; штрихпунктирвой линией показав*
сепаратрисса, разделяющая области двух минимумов; точка на ППЭ(д) и на карте (б) соответ-
соответствует структуре (II) переходного состояния; числа в разрывах линий на карте (б) — отяоеительг
ные энергии уровней в кДж/моль
160
Координата реакции
Рис. 5.5. Энергетический профиль реакции
изомеризации (топомеризации) молекулы
азациклобутадиена (см. рис. 5.4)
На рис. 5.4 даны сечения
этой ППЭ в координатах
д (длина связи C,-N)
ЯЛ2 (длина связи Q-N) и со-
соответствующая ему потенци-
потенциальная карта. Такая карта
строится аналогично геогра-
географической карте: на плоскости
вычерчиваются изоэнергети-
ческие (или эквипотенциаль-
эквипотенциальные) линии, т. е. линии, со-
соединяющие точки различных
геометрических конфигура-
конфигураций молекулярной системы,
имеющие одинаковую потен-
потенциальную энергию. Как вид-
видно из рис. 5.4, структуры 1а, 16
отвечают двум минимумам
ППЭ Связывающий их путь минимальной энергии проходит через
симметричную структуру II с выравненными связями C-N. Эта
структура отвечает переходному состоянию реакции. Если путь
минимальной энергии реакции записать как функцию координат
Лг и Л2, можно получить выражение для координаты реакции I <-11,
т е. длины дуги ПМЭР на ЛПЭ. В этом случае можно ввести
понятие об энергетическом профиле реакции, наиболее ясно пред-
представляющем зависимость энергетики реакции от структурных изме-
изменений, вызывающих реакцию. Энергетический профиль топомери-
защш азациклобутадиена показан на рис. 5.5.
Величина Ео — разность энергий реагента и переходного состоя-
состояния — связана с экспериментальной величиной энергии активации
соотношением
п=Е0+пЯТ, E.6)
где п — величина, зависящая от типа реакции, равная i/2 для бимо-
бимолекулярной реакции и близкая к 1 для мовомолекулярной. Более
строгая оценка величины Ео учитывает в общем случае поправку на
энергии нулевых колебаний реагентов (см. разд. 5.5.1).
53. ППЭ И ДИНАМИКА ХИМИЧЕСКИХ РЕАКЦИЙ
Движение реагирующей системы вдоль ПМЭР предполагает
бесконечно медленное перемещение по ППЭ через седловую точку.
При таком рассмотрении не учитывается роль кинетической энергии
6. Теория строения молекул
161
системы (колебательного возбуждения молекулы, перерасщ
ния энергии между различными колебаниями, вращения молекул
т. е. всех тех динамических эффектов, которые вынуждают реал
ющую молекулу к деформации, определяемой координатной
ции. С учетом члена кинетической энергии полная энергия сие
всегда превышает уровень, задаваемый ППЭ, а динамические
тории могут сильно отличаться от ПМЭР и зависят от началь]
условий (импульсов).
Обычный способ расчета динамических траекторий состоит в
шении классических уравнений движения для каждого атома
. дН . дН
dpi
где H=T+E(q) —классический гамильтониан; Т—кинетич
энергия системы; E(q)—потенциал E.1); р, — обобщенный
пульс, соответствующий координате qh а <?, и pi — первые произ]
ные по времени.
Для решения уравнений движения E.7) необходимо знать
ную ППЭ системы E.1), а не только характеристики ее крит
точек. Обычно используют аппроксимацию аналитическими
циями, наиболее близко отражающими характер данной ППЭ~ Па-
Параметры, характеризующие начальное состояние системы (коорг
динаты, импульсы), задаются в зависимости от типа задачи. Значе-
Значение динамических расчетов состоит в том, что они существенна
расширяют представление о внутреннем механизме реакции, связык
вают эти представления с реальными условиями протекания
мических превращений.
В качестве общего примера рассмотрим простейшую
мическую реакцию обмена между атомом А и двухатомной
молекулой ВС: i
А + ВС-АВ + С E.
Такие реакции могут быть как термонейтральными, например!
E.9), так и эндо- и экзотермическими. К последнему типу othoci
реакция E.10), идущая со значительным выделением теплоты:
Н+Н2-Н2+Н E.9)
F + H2-FH+H E.K
При коллинеарном сближении реагентов в реакции E.8) всего
две внутренние координаты: межъядерные расстояния'
-Кав и Лвс определяют ППЭ, что допускает достаточно строгие ее!
расчеты для различных типов реакций обмена E.8). Схематический
вид таких ППЭ показан на рис. 5.6. Подобные ППЭ хорошо аппрож^
симируются аналитическими функциями, после чего достаточно^
162 J
АВ+С
- А+дС
Рис. 5.6. Схематический вид ППЭ коллинеарной реакции обмена (а) А + ВС-^АВ + С
и ее двумерная карта E). Штриховой линией показан ПМЭР, а крестиком обозначена
седловая точка, соответствующая переходному состоянию
легко решаются уравнения движения E.7) для нахождения динами-
динамических траекторий реакции.
В зависимости от начальных условий и различного расположе-
расположения седловой точки на ППЭ, зависящего от экзо- или эндотермич-
ности реакции, получаются существенно различные динамические
траектории, что иллюстрируется на рис. 5.7. В случае экзотермичес-
экзотермической реакции (седловая точка находится во входном канале реакции,
рис. 5.7, а) для получения реакционноспособной траектории, т. е.
траектории, пересекающей переходное состояние, вся кинетическая
энергия начального состояния должна быть сосредоточена на транс-
трансляционных степенях свободы реагентов, направленных вдоль
ПМЭР. В этом случае вся избыточная кинетическая энергия в вы-
выходном канале расходуется на колебательное возбуждение продук-
продуктов. Если же в экзотермическом случае начальная кинетическая
энергия будет сосредоточена на колебательных степенях свободы
фис. 5.7, б), то динамическая траектория реакции не пересекает
переходное состояние и получается нереакционноспособная траек-
траектория.
В эндотермическом случае (седловая точка находится в выход-
163
Рис. S.7. Схематический вид дина-
динамических траекторий, полученных
для экзо- (а, б) и эндотермических
(в, г) реакций обмена
ном канале реакции) ситуация
ратная, т. е. реакционноспособ!
траектория получается, если на*
вая кинетическая энергия
лена на колебательных степенях
боды (рис. 5.7, г), а не на транс
онных (рис. 5.7, в).
Значение расчетов динамичс
траекторий реакций состоит не toj
ко в том, что они увязывают тео)
тические представления о мехг
мах реакций с реальными услов]
их протекания, но и в зна*
более глубоком понимании механиз-
механизмов. Такие траектории регистрируй
ются, например, в экспериментах noi
столкновениям в пересекающихся
молекулярных пучках, при селективном лазерном возбуждении ко-
колебательно-вращательных состояний молекул.
5.4. ППЭ ЭЛЕКТРОННО-ВОЗБУЖДЕННЫХ СОСТОЯНИЙ
Все энергетические поверхности, получаемые при решении урав-
уравнения D.6), не соответствующие низшему энергетическому, т. е.
основному состоянию, являются поверхностями электронно-воз-
электронно-возбужденных состояний.
74
12
10
в
2
О
0.12
0,20
0,28
нм
Рис. 5.8. Схема потенциальных кривых для системы N2
164
Ф,град
2,2
Рис. 5.9. Поверхности потенциальной энергии молекулы метилена СН2 для основ-
основного 3Bi и возбужденного *Bi электронных состояний в координатах длины
СН-связи (нм) и угла НСН
Вначале рассмотрим пример двухатомной молекулы. На рис. 5.8
показаны потенциальные кривые молекулы азота N2 в зависимости
от межъядерного расстояния в различных электронно-возбужден-
электронно-возбужденных состояниях.
Поскольку в возбужденных, состояниях молекулы азота проис-
происходит заселение электронами высоколежащих антисвязывающих
МО я*2р-типа (см. разд. 4.6), порядки связей N—N становятся
меньше, а длина больше, чем в основном электронном состоянии.
Для некоторых потенциальных кривых (рис. 5.8) вообще отсутству-
отсутствуют минимумы, и они имеют отталкивательный характер во всей
области межъядерных расстояний.
На рис. 5.9 показаны ППЭ двух электронных состояний молеку-
молекулы метилена: основного триплетного и первого возбужденного
синглетного. Из этого рисунка также видно, что минимумы на
обеих поверхностях расположены в разных областях конфигураци-
конфигурационного пространства, т. е. устойчивые структуры основного и элек-
электронно-возбужденного состояний различны.
Химические реакции в электронно-возбужденных состояниях мо-
могут протекать адиабатически при движении возбужденной системы
во ППЭ электронно-возбужденного состояния со спуском в один из
ее минимумов. Обычный способ генерации электронно-возбужден-
электронно-возбужденного состояния — фотохимический. Энергетические барьеры, пре-
преодолеваемые в адиабатических фотохимических реакциях, не превы-
165
шают 30 кДж/моль, так как времена жизни электронно-возбуждс
ных состояний молекулы очень малы: 1(Г3—10 "9 с. Другой р
пространенный вариант фотохимической реакции — неадиабат
ческая реакция — связан с переходами между ППЭ различных эл<
тронных состояний.
5,5. КОЛЕБАНИЯ МОЛЕКУЛ
Ядерная волновая функция ?Я(Л), получаемая из решения
нения D.11), зависит от 3N переменных R(xb yXi z,, x2, y2, z2, •¦-,
yN, zN\ из которых только 3iV—6 координат относятся к внутренний
координатам, а остальные шесть координат описывают постуш
тельное и вращательное движения молекулы как целой. Операт<
Ня можно представить в виде
ня=нг
+н
EЛ1]
где
нг=-|31>
зависит только от внутренних координат qa и является операторе
кинетической энергии молекулы во внутренних координатах; ко:
фициенты аар есть функции от qa. Остальные члены Н"
*.„ и Н, относятся к движению молекулы как целого и взаимо-
взаимодействию этого движения с колебаниями. Для малых колебаний
влияние вращательного и поступательного движения молекулы hi
ее колебания мало и в достаточно хорошем приближении им можвх
пренебречь. Это означает, что последним членом в уравнении E.11J
можно пренебречь и ядерную функцию можно представить
произведение функций, каждая из которых зависит только от коле
бательных, поступательных и вращательных координат (степене
свободы), причем только колебательная функция зависит от виз
ренних координат:
,вращ
В этом случае уравнения для каждого из этих движений peiuai
ся отдельно и электронная энергия Ё как функция от внутреш
координат входит только в колебательное уравнение:
кол
_ г^Р
166
Полная энергия молекулы — это сумма отдельных ее компо-
компонент:
Рассмотрим более подробно уравнение для колебаний молекулы,
которое можно записать в виде
Яъ
Pdqadq?
Ъ^Ън-д-^е"*"*. E.15)
В окрестности точки минимума q0 функцию ?(q) можно разложить
в ряд Тейлора:
I ЗЛГ-6 / jJ? \
Е>(Я) = Е*Ш + 2 L (^^Jo(?«-^fe.-?S)+ ». E.16)
Линейный по (ga—q0) член в разложении E.16) исчезает, потому что
№1
№1* \
точка q0 — точка минимума, где все производные I dqa) равны
нулю.
Без потери общности можно сдвинуть начало системы коор-
координат в точку q = qQ и E=E*(qQ). Тогда разложение E.16) примет вид
1 ЗЛГ-6
2
Если матрица
62Е
не является вырожденной (ее детер-
минант отличен от нуля), можно ввести такие новые координаты Qa,
в которых отличными от нулевых будут только диагональные
(дгЕ\
члены —=). Такие координаты Qa называют нормальными. С их
использованием разложение E.17) запишется в виде
5.5.1. Гармонические колебания молекул
Переходя в формуле E.15) к новым координатам Qa и учитывая,
что при малых колебаниях около положения равновесия (в окре-
окрестности точки минимума на ППЭ) можно считать коэффициенты
167
аар постоянными при q=qo, получим новое уравнение для
колебаний ядер
-2N-6 / h2 д2 \
[3N-6 /
2М.dQ, 2
В этом уравнении вместо E(Q) стоит разложение E.18), в кот<
ром оставлен только первый квадратичный по Qa член. Ма (диаг<
нальные элементы матрицы aap(Q)) называют приведенной массойш
г (дЕ\ - - *
а/я=1 ——2 1 — силовой постоянной а-го колебания E.4).
Вид уравнения E.19) позволяет искать волновую функций
,жол
(Q) в виде произведения функций ? ' (Qa), каждая из которых
зависит только от одной переменной Qa: ;
При подстановке функции ^^(Q) в этом виде в уравнение E.19)'
последнее распадается на 3W—6 независимых уравнений: :
^2Ма'д^+Га^а) "«'^~ * **(Q*S' E.21)
««1,2, ..., 3JV-6.
Уравнение E.21) представляет собой уравнения состояний гармони- 3
ческого осциллятора. Собственные значения ?|^л гармонического*
осциллятора выражают формулой
где va—(fa/MJ —частоты нормальных колебаний; па — колебав
тельные квантовые числа, принимающие значения 0, 1, 2, ....
Собственные функции гармонического осциллятора имеют вид
1
l\nJ i*(nju
1/2
E.23)
где А=
-; Яя — полином Чебышева—Эрмита степени па от-
относительно Qa. Приняв x=P1J2Qta можно записать для л=0, 1, 2:
= 1; Я, fo> = 2х; Я2Гдг; =4х2 -2.
168
Собственные функции гармони-
гармонического осциллятора E.23) орто-
нормированы, т. е.
E.24)
тал АП —,
ц/кол XV
* па * «я
На рис. 5.10 представлены ко-
колебательные уровни и им соот-
соответствующие графики волновых
функций молекулы НС1 в гармо-
гармоническом приближении. Нижний
уровень, соответствующий кван-
квантовому числу ла = 0, является ну-
нулевым колебательным уровнем.
Соответствующая ему энергия
представляет собой энергию ну-
нулевого колебания.
Колебательные уровни мно-
многоатомной молекулы определяют
в результате решения уравнения
E.19). Подставляя в E.19) выра-
выражение для волновой функции
у^°л(па) в форме произведения
E.20) и учитывая достигаемое
при этом разделение координат,
а также условие ортонормиро-
ванности E.24), получим форму-
формулу для полной колебательной
энергии молекулы
3N-6
г*ол V1 /
9
8
I
" 6
3
2
1 К
Г Т Г 1 t I Г^
3210 123
Рис. 5.10. Потенциальная кривая
и волновые функции для модели гар-
гармонического осциллятора молекулы
НС1:
штриховой линией показаны истинный (нега-
(негармонический) потенциал и ему соответству-
соответствующее энергетические колебательные уровня
молекулы НО,4 нормальная координата соот-
соответствует растяжению связи Н — С1
E.25)
Каждому колебательному уровню энергии многоатомной моле-
молекулы отвечает свой набор колебательных квантовых чисел па от-
отдельных нормальных колебаний. Нижний уровень, для которого все
па~0, называют нулевым колебательным уровнем многоатомной
молекулы, а энергию, ему соответствующую, — энергией нулевых
колебаний молекулы:
1 3N-6
^В2 Е hVa' E-26>
0-1
На рис. 5.11 схематически показаны колебательные уровни дву-
двумерного гармонического осциллятора.
169
Рис. 5.11. Колебательные энергетические уровни двумерного гармонического
лятора:
А A3 ЗА
Таким образом, для того чтобы решить задачу о движении
необходимо, во-первых, рассчитать (в приближении Борна—Ошк
геймера) ППЭ соответствующего электронного состояния в okj
стности минимума, во-вторых, аппроксимировать эту ППЭ в okj
стности минимума • параболоидом (гармоническим потенциалов
и только после этого решать задачу E.21) о колебаниях
в молекуле. Несмотря на столь многие допущения, к котор!
приходится прибегать в таких расчетах, вычисленные указ;
способом частоты (энергии) колебаний для низших пяти-i
колебательных уровней достаточно хорошо совпадают с э)
периментальными значениями. Рис. 5.10 показывает близ]
соответствие вычисленного гармонического и найденного Э1
периментально ангармонического колебательного потенциала
молекулы Н—С1. В табл. 5.1 представлены рассчитанные в raj
моническом приближении с использованием метода ЛКАО М<
Рутаана частоты колебаний для четырехатомной молекулы фор!
альдегида СН2—О, значения которых сопоставлены с эксперимс
тальными.
170
Таблица 5.1. Экспериментальные ¦ теоретически рассчитанные (в гармоническом
пяблвженни) частоты колебаний молекулы формальдегида. НеэмиирнческнЙ расчет
использованием базиса 6-31G* и с учетом корреляционных поправок во втором
порядке теории возмущении Меллера—Плессета
сн2,
СН2,
сн2,
сн2,
Тип холебания
симметричное рас-
растяжение
растяжение
ножничное
асимметричное
растяжение
веерное
крутильное
Симметрия
ВИЯ
*i
жолеба-
Теоретическое значе-
значение, см~*
3019
1786
1567
3064
1249
1194
Экспериментальное
значение, см
2783
1746
1500
2843
1249
1167
5.5.2. Расчет термодинамических функции
Зная геометрические характеристики молекул и рассчитав, как
описано в предыдущем разделе, частоты нормальных колебаний,
можно рассчитать термодинамические характеристики молекул
и образованных ими систем при требуемой температуре. Наиболее
важные из них — энтальпия и энтропия. Вычисление этих функций
производят на основе известных соотношений статистической меха-
механики.
В приближении жесткой молекулы (барьеры внутреннего враще-
вращения и инверсии существенно превышают кТ) можно выделить,
пренебрегая ангармоничностью колебаний, вклады отдельных сте-
степеней свободы — поступательного, вращательного, колебательного
движений молекулы и электронного в энтропию S и энтальпию Н:
E.27)
-
(nVAVBVc)
E.28)
E.29)
E.30)
E.31)
E-32)
171
Здесь п — число молей вещества; iV^ — постоянная Авогадро; М
масса молекулы; р — давление; S — число симметрии; W* — вьц
жденность электронного состояния (практически всегда
hva
h2
%п1акТ
i—T^t гДе h — один из главных компонент
кТ
тов момента инерции; va — частота нормального колебания.
Далее члены в выражении E.28) имеют вид
ЯПОСТ=Явр=3/2 RT; E.3:
u
vj(eu*-l).
E.34)
При Х=0 соотношение E.34) переходит, очевидно, в E.26), т.
представляет энергию нулевых колебаний молекулы.
Рассчитав при требуемой температуре величины S и Н, можна
вычислить свободные энергии G, пользуясь известным соотношени-
соотношением термодинамики
G=H-TS, E.35|
и перейти к расчету связанных с ними констант равновесия, кои*
стант скорости (с привлечением представлений теории абсолютны»
скоростей реакций), изотопных эффектов и пр. 4
В табл. 5.2 приведены данные неэмпирических расчетов, позв1
ляющие оценить значения температурных поправок в энтальпии
некоторых простых молекул и энергии их нулевых колебаний. Р
считанные полуэмпирическим методом MIND0/3 (см. разд. 7.3.
значения свободных энергий при температурах от 300 до 1500 К
шести важных для промышленности реакций сопоставлены с эю
периментальными данными.
Таблица 5.2. Рассчитанные (ab initio, метод Хартря—Фока, базис орбяталеф;
3-21G) теоретические поправки на изменение эвтальшш и энергия нулевых колебаний
(кДж/моль)
Молекула
СО2
CHF3
НСООН
NF3
CH3CN
CH3NC
#C00 К) — Я@)
расчет
7,1
9,2
8,8
9,6
9,2
10,0
эксперимент
9,6
8,3
9,6
9,6
10,5
пд
расчет
27,6
65,2
84,1
26,4
115,5
113,8
эксперимент
30,1
65,2
85,4
26,8
115,1
115,1
172
Как видно из табл. 5.2, значения ?°al достаточно велики, однако
иХ разности для изомеров обычно незначительны. Поэтому при
вычислениях энергетических характеристик реакций изомеризации
расчетом ?2™ обычно можно пренебречь. Данные табл. 5.3 показы-
показывают, что даже при использовании полуэмпирических методов мож-
можно достигнуть весьма хорошего воспроизведения значений свобод-
свободных энергий реакций.
Таблица 5.3. Расс-игганные (метод MINDO/3)* ¦ экспериментальные значения
(кДх/моль) свободных энергий некоторых промышленво важных реакций
. — —
—
2CH4-CH
сн2=сн2
Реакция
2NH3
.сн+зн2
/
+ 0,5О2 ^ СН
СН4+2Н2О->4Н2 + СО2
CH4+NH3
+ ЗН2О
сн4+а2ч
+ 1,5O2->HCN +
снэа+на
0
' \
2—си
300
расчет
-32,4
284,7
[2 -81,4
113,3
-494,0
-107,3
К
экспери-
эксперимент
-32,7
284,8
-82,5
113,1
-498,2
-107,2
900
расчет
101,5
132,5
-32,2
-8,4
-542,0
-115,0
К
экспери-
эксперимент
101,1
133,6
-33,8
-7,5
-560,1
-115,2
1500 К
расчет
244,3
-31,1
16,2
-143,4
-585,3
-123,3
экспери-
эксперимент
240,9
-29,3
20,9
-141,2
-600,4
-124,6
* См. разд. 7.3.5.
5.6. ТОПОЛОГИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
МОЛЕКУЛЯРНОЙ СТРУКТУРЫ
Введение представления о поверхности потенциальной энергии
позволяет подойти к вопросу о квантово-механическом определе-
определении молекулярной структуры. Обычно понятие молекулярной стру-
структуры относят к точке конфигурационного пространства в миниму-
минимуме ППЭ, однако такое отнесение не вполне строгое, так как в ре-
результате непрерывных внутренних колебаний (следствие принципа
неопределенности) ядерная конфигурация молекулы постоянно де-
деформирована по отношению к точке минимума ППЭ. Такие дефор-
деформации могут быть значительны для стереохимически нежестких
молекул и ионов. Они могут быть велики и на начальных участках
химической реакции, когда в обычной сложившейся в химии тер-
терминологии исходная структура еще не разрушена. Из изложенного
ясно, что геометрическое определение молекулярной структуры не
исчерпывает содержания этого понятия.
173
Рис. 5.12. Контурная карта распределения электронной плотности р(?) {а) и
векторов градиента V/>(?) для 2-пропилкатиона [СН3СНСН3] в равновесной;
рии (б), рассчитанные методом аЬ initio в базисе орбиталей STO-3G:
положения седловых критических точек обозначены черными кружками; свяэевые пути —
тории, соединяющие соседние ядра через разделяющую их критическую точку; жирными
жми, проходящими через крвтичессве точки перпендикулярно связевым путям, отделены
ны атомных групп СНэ И СН
Квантово-химическая теория молекулярной структуры (Р.
дер) основывается на топологических свойствах распределения эл<
тронной плотности
и поля вектора ее градиента Vp(?, ~q\ где"^ — координата то
в которой вычислена р для заданной ядерной конфигурации
пе — число электронов в данной молекуле или ионе.
Поле вектор-градиента электронной плотности представл
траекториями Vp(r), как показано на рис. 5.12.
Образуемые этими траекториями градиентные пути подразд
ются на два типа. К первому относятся пути, связанные с прохо
нием через локальные максимумы функции р(?, ~q), которые нахо-j;
дятся в положении атомных ядер. Область пространства, ограниГ
ченная градиентными путями, идущими от бесконечности к д
ному ядру, составляет бассейн соответствующего атома в молеку.
Каждая пара соседних атомных бассейнов отделена друг от др;
узловой поверхностью, на линии пересечения которой с плоек
молекулы лежит седловая критическая точка второго порядка
отрицательных значения кривизны). Объединение нескольких с
них атомных бассейнов также создает изолированные басе©
например групп СН2, СН3, свойства которых определяются pao-j
пределением электронной плотности в общем бассейне. Ц
Второй тип градиентных путей определяет распределение связей I
в молекулярной системе. Он включает два градиентных пути, связи*)
вающих критическую точку, которая находится между каждой Ш*
рой соседних атомных бассейнов, с соответствующими им ядрами*
174
1
н
в
н
н
д
и
Рис. 5.13. Молекулярные графы, определяемые топологическими свойствами распределения
электронной плотности, генерируемые ЭВМ по данным незмпирических расчетов поля
градиента Vp(?, ~4 )'•
а — метав; б — катион циклопропенил СзНз+; в — тетраэдран С4Н4; г — бензол; д — диборав
В2Н6; е — этилен в равновесной геометрии, угол НСН = 115,6°; ж — этилен, угол НСН = 85,6в,
энергия относительно равновесной формы 188 хДж/моль; з — этилен, НСН-58,8"; энергия от-
относительно равновесной формы 790 кДж/моль
Сложение этих двух путей дает линию в пространстве — связевый
путь, вдоль которого электронная плотность сохраняет максималь-
максимальное значение. Существование связевого пути между какой-либо
парой атомов в молекуле является необходимым и достаточным
условием существования между этой парой атомов химической
связи. Полная сеть связевых путей для данной ядерной конфигура-
41111 Q(q) определяет молекулярный граф.
На рис. 5.13 показаны молекулярные графы для отдельных
молекул в их равновесных геометриях. Как правило, такие графы
близко соответствуют обычным структурным формулам. Однако
в Ряде случаев они фиксируют важные особенности. Так, для струк-
175
тур с малыми циклами (рис. 5.13, б, в) связевые пути изогнуты,
свидетельствует в пользу концепции изогнутых (банановых) cbj
в этих соединениях. Проведенные расчеты показали, что неболы
а в ряде случаев и весьма значительные структурные вариации
отражаются на характере молекулярного графа. Действитс
(рис. 5.13, е—з), даже очень большие деформации угла НСН в
не, связанные с огромными энергетическими возбуждениями,
приводят к изменению молекулярного графа, т. е. к нарушен!
исходной структуры. Это позволяет выделить область ядерно!
пространства Q'(q) на ППЭ E(q), которую можно связывать с
стоянной молекулярной структурой. Само понятие «молекуляр!
структура» определяется как эквивалентный класс молекулярн
графов, т. е. одно и то же число связевых путей связывает одни й
же ядра в каждом молекулярном графе, принадлежащем данн<
молекулярной структуре.
Основываясь на развитом выше понимании молекулярю
структуры, можно подразделить все конфигурационное npocrpai
во ядерных координат Q(q) на конечное число структурных облг
тей, каждой из которых отвечает своя молекулярная структура,
границах раздела таких областей происходит скачкообразное из!
нение структурного типа, т. е. переход от одной молекулярш
формы к другой.
Значение топологического подхода к определению понятия
лекулярной структуры состоит главным образом в том, что
подвел под это важнейшее понятие химии строгое физическое
снование, внеся важное дополнение к его трактовке в рамках nj
ставлений о ППЭ.
5.7. ВИБРОННЫЕ ВЗАИМОДЕЙСТВИЯ.
ЭФФЕКТЫ ЯНА—ГЕЛЛЕРА
Приближение Борна—Оппенгеймера (адиабатическое приближе-
приближение) становится неудовлетворительным при сближении поверхно*
стей потенциальной энергии различных электронных состояний мэ*
лекулярной системы, когда разность между ними становится срав-
сравнимой с колебательным квантом, т. е. соотношение D.20) не выпол-
выполняется. В области сближения, касания или пересечения ППЭ проис-
происходит смешивание электронных состояний вследствие сильного вза-
взаимодействия электронного и ядерного движений. Такие взаимодей-
взаимодействия называют вибронными. С точки зрения классической мехаг
ники, в этой области сближения ППЭ скорость движения ядер
приближается к скорости движения электронов. Квантово-механЯ-
чески это означает, что в областях пересечения или сближения ППЭ
нельзя пренебрегать оператором кинетической энергии ядер и необ:
холимо решать общее электронно-ядерное уравнение D.17), где по
крайней мере некоторые из диагональных элементов Лй< отличны от
176
я з этом случае получают состояния объединенной электронно-
верной системы, которые называют еибронными состояниями.
Как правило, пересечение (касание) потенциальных поверхностей
происходит в достаточно узких областях внутренних координат.
Можно сохранить представление об адиабатических поверхностях,
введя специальное рассмотрение только для этих областей кон-
конфигурационного пространства. Такое рассмотрение основано на
анализе так называемых эффектов Яна—Теллера первого и второго
порядков. В наиболее важных с точки зрения структурной химии
случаях пересечения или сближения поверхностей основного и пер-
первого возбужденного электронных состояний эффекты Яна—Теллера
определяют характер искажений, которые испытывает молекуляр-
молекулярная система.
5.7.1. Теорема Яна—Теллера
Рассмотрим некоторую ядерную конфигурацию Qo, определя-
определяющую молекулярную систему с заданным числом ядер и электро-
электронов, для которой известны электронные состояния *?0, ?ь ...,
?* и соответствующие им собственные значения Ео, Еи ..., Ек. Важно
ответить на вопрос, как изменятся энергия и волновая функция
основного электронного состояния при небольшом смещении систе-
системы из точки Qo в результате движения вдоль координаты Qt.
Энергию системы можно представить в виде разложения в ряд
Тейлора подобно E.16). Однако поскольку в общем случае ядерная
конфигурация Qa не обязательно принадлежит точке минимума,
необходимо ввести и линейный член по Qh так как первая производ-
производная энергии по этой координате не равна нулю. Если Qt соответству-
соответствует координате нормального колебания, можно отбросить недиэго-
нальные члены:
Гамильтониан системы, претерпевающей деформацию, можно
записать аналогичным образом:
г\
E.37)
где Hq — исходный гамильтониан; Н — потенциальная энергия,
включающая члены электрон-ядерных и ядерных взаимодействий.
Рассматривая гамильтониан E.37) как гамильтониан возмущенной
системы [см. A.69)] и воспользовавшись теорией возмущений, полу-
полупим следующее выражение:
177
Xih*
Здесь Eq — энергия основного электронного состояния неде(
мированной (Qo) системы; второй и третий члены — попрг
к этой величине соответственно в первом и втором порядках тео]
возмущений.
Волновая функция возмущаемой искажением системы в ochoi
ном (низшем) электронном состоянии записывается в соответсп
с A.86) следующим образом:
,;=%+!
Член возмущения первого порядка в E.38) характеризует из]
нение энергии системы, вызываемое смещением ядер из исхода
конфигурации Qo при сохранении соответствующего QQ электрон»
го распределения Yq- Если этот член имеет отрицательное знач<
то это означает, что рассматриваемая исходная геометричеа
конфигурация Qo не является устойчивой.
__ ii
Привлекая теоретико-групповое рассмотрение, можно показать,
что интеграл | то(^7Т I ^о^х не равен нулю только в следующих.
двух случаях. Первый — когда *Р0 не является вырожденной функ-
цией и имеет симметрию типа А, В с учетом того, что —- преоб-
Q
разуется по тем же представлениям, что и Qh тогда последняя
...
должна иметь симметрию ах. Это ясно из вычисления прямого
произведения (см. разд. 6.7). Данный случай не представляет сдеци-
ального интереса и относится к произвольной точке на склоне ППЭ^
Второй случай, когда функция % орбитально вырождена, напри*
мер для систем с симметрией Qh- или Л^-типа. Искажение Qi тогдв
должно или иметь симметрию п\ (этот вариант не имеет принципи*
ального значения, так как деформация симметрии а\ не отражается
178
на точечной группе симметрии системы), или обладать симметрией,
обеспечивающей снятие электронного вырождения *F0. Г. Ян
и э Теллер* A937) впервые показали, что для любой молекуляр-
молекулярной системы с вырожденным электронным состоянием в нелиней-
нелинейной симметричной ядерной конфигурации Qo всегда существуют
такие деформации Qh для которых интеграл ЧУ idQi) ^о^т не
равен нулю. Этот результат, известный как теорема Яна—Геллера,
был получен посредством перебора всех точечных групп симметрии
для нелинейных молекул. Предложена (И. Б. Берсукер, 1966) следу-
следующая более общая формулировка теоремы Яна—Теллера: если
адиабатический потенциал нелинейной многоатомной системы
E(Q), являющейся формальным решением электронного уравнения
Шрёдингера, имеет несколько пересекающихся ветвей, то в точках
пересечения QOi всегда найдутся такие ядерные смещения Qh для
которых производные потенциальной энергии по этим координатам
в точках пересечения отличны от нуля и, следовательно, потенциал
E(Q) не имеет минимума.
Случай вырождения электронного состояния — не что иное, как
пересечение адиабатических потенциальных поверхностей. Поведе-
Поведение потенциала, отражающее существование вибронных взаимодей-
взаимодействий, получило название эффекта Яна—Теллера первого порядка.
Проявления этого эффекта характерны для высокосимметричных
молекулярных систем с неполным электронным заполнением связы-
связывающих или несвязывающих орбиталей. Типичными примерами
таких систем являются молекулы и ионы координационных соеди-
соединений металлов, в которых высокая симметрия обусловлена коор-
координационным полиэдром. Продолжим рассмотрение структурного
аспекта эффекта Яна—Теллера первого порядка в разд. 11.5.
5.7.2. Псевдоэффект Яна—Теллера
и эффект Яна—Теллера второго порядка
Сложное выражение в фигурных скобках в члене второго поряд-
порядка теории возмущений E.39), служащее коэффициентом при квад-
квадратичном члене Qf, можно рассматривать как силовую постоянную
Для деформации в направлении Q{.
*Эдвард Теллер (род. 1908 г.) — немецкий физик, после прихода к власти наци-
нацистов эмигрировал в США, где его называют «отцом водородной бомбы». Автор ряда
Фундаментальных исследований в области квантовой механики, квантовой химии, •
в частности в области теории химических и особенно термоядерных реакций. Идея
теоремы Яна—Теллера, по словам самого автора, принадлежит Л. Д. Ландау, вы-
высказавшему ее еще в 1934 г.
Герман Ян (род. 1907 г.) — английский физик, автор важных работ по примене-
применению теории групп в квантовой механике.
179
E.40|
где./оо= *F0 [ -г^> 1 % dt характеризует возвращающую силу, дейст*
J \dQiJo j
вующую на систему при ее смещении из точки Qo, происходящее
без изменения исходного электронного распределения. Члены
вид которых определен в E.39), соответствуют энергетическим из*
менениям, связанным с перераспределением электронной плотностЦ
в системе при ее геометрической деформации Qt. Эти члены называд
ют релаксационными. *
ЧленУ^ всегда отличен от нуля и имеет положительное значение/
Учитывая, что I — 1, как отмечалось, имеет ту же симметрию, чтд ¦
координата Q, условием неравенства членов fOm нулю служит нали* j
чие в прямом произведении rvo х Г^т х Ге. полностью симметрии-
ного представления а\ (см. разд. 6.7), т. е.
Это означает, что только те возбужденные электронные состоя?
ния *?„ примешиваются к основному в ходе деформации системы*
которые обладают для этого подходящими свойствами симметрии.
Вклад членов f^ всегда отрицателен (понижение энергии). Эт^
следует из того, что Ет>Е0 и знаменатели в последнем слагаемом
уравнения E.39) имеют отрицательное значение. Как правило, на-
наибольший стабилизирующий вклад вносит ближайшее к основномуf
возбужденное электронное состояние (минимальная энергетическая
щель EQ—Em), если, конечно, его свойства симметрии отвечают
условию E.41).
Если вклад релаксационных членов в выражении E.40) превыша-
превышает по абсолютной величине силовую постоянную /оо, суммарны^
эффект будет состоять в понижении энергии основного состояния
*F0 в результате деформации Q{ из исходного положения Qq. Это#
эффект, определяемый как эффект Яна—Теллера второго порядка*
имеет тем большую величину, чем более сближены подходящие по
симметрии взаимодействующие электронные состояния. В боль-
большинстве случаев решающую роль играет взаимодействие меясдЗГ
основным и низшим электронно-возбужденным состояниями оди?
наковой мультиплетности.
В уравнении E.38) функции ?<ь Ч„ представляют собой элект-
180
с'
т
е'
т
¦4*
BHj NH3 CLFj
Рис. 5.14. Типы искажений плоских /?зл-структур молекулы АХ3, определяемые про-
проявлением эффекта Яна—Теллера второго порядка:
а — симметрии нормальных колебаний молежул АХз в ^ЗА-Ф°РМС; 6 — схематическое представ-
представление орбитального строения молежул ВНз, NH3, CIF3. Показаны гранитные валентные ор*
битали, стрелками указан перенос электронной плотности между граничными орбиталями
в случае их смешивания, достигаемого при деформации
ронные состояния, т. е. выражены слэтеровскими детерминантами,
построенными из функций МО, а Ео, Ет — энергии этих состояний.
Перейдя к орбитальному базису, можно прийти к соотношениям
E.38), содержащим те же интегралы, в которых функции состояний
заменены функциями орбиталей, а энергии состояний — орбиталь-
орбитальными энергиями. Наиболее важным является взаимодействие меж-
между граничными МО — высшей заполненной щ и низшей свободной
(р\. Тип энергетически благоприятной деформации определяется из
соображений симметрии аналогично E.41):
Если существует молекулярное колебание Qh симметрия которо-
которого соответствует симметрии переходной электронной плотности
(ра(рт, то структура с данной ядерной конфигурацией будет неустой-
неустойчивой. Примером, показывающим принцип анализа, основанного
на рассмотрении эффекта Яна—Теллера второго порядка, является
плоская молекулярная система АХ3. На рис. 5.14 показаны типы
нормальных колебаний плоской треугольной молекулы АХ3 в сим-
симметрии D3k и характер электронного заполнения орбиталей трех
Различных молекул этого типа.
Для молекул типа ВХ3, где X=H,R, F и др., симметрия переход-
181
ной электронной плотности при взаимодействии граничных орб]
лей е'ха2=е". Колебание такой симметрии для плоских молекуЦ
АХ3 отсутствует (рис. 5.14, а), и, следовательно, D3*-CTpyi
устойчива. Для NX3(X=H, R, F, ...) переходная электронная
ность (а2 х а\ ~аг) имеет свойства симметрии нормального коле
ния а'2у ведущего к пирамидальному искажению. В случае
свойства симметрии переходной электронной плотности (а\ х е'=<
соответствуют е'-колебанию. Вызываемые им деформации
как следует из рис. 5.14, а, к формированию Т-образной стр}
интергалогенного соединения с различающимися длинами свя:
С1—F. Как будет показано в гл. 10, при более подробном раса
рении структурных характеристик молекул соединений непереход
ных элементов предсказания, основанные на анализе эффекта
Теллера второго порядка, совпадают с выводами других Teoj
ческих методов и экспериментальными результатами.
Особым случаем эффекта Яна—Теллера второго порядка
ется псевдоэффект Яна—Теллера. Этот термин применяют
систем, в которых отсутствует вырождение электронных
однако сохраняется орбитальное вырождение. Пример подобной
системы — квадратная структура циклобутадиена в синглетном
электронном состоянии. Вырожденная ег-МО циклобутадиена
полнена (см. разд. 8.1.2) только двумя электронами, но при у
двухэлектронных членов электронные состояния циклобутадиена
полученные при различных заполнениях, невырождены. В этом:
и других подобных случаях энергетическая щель между основным
и низшим электронным состоянием, как правило, особенно мала
и деформации энергетически благоприятны. В случае квадратно^
формы смешивание низшего синглетного электронного состояний!
5^-типа с ближайшим 1А ^-состоянием достигается в соответствие
с E.41) колебанием 6^-симметрии, ведущим к прямоугольной струм*
туре: \
III, А
4А
'гь
Рис. 5.15 иллюстрирует характер преобразований формы ади-
адиабатических потенциалов в областях их пересечения и сближения^
вызываемых проявлениями эффектов Яна—Теллера. Эти преобра-.
зования можно трактовать как избежание пересечения (отталкива-
(отталкивание) адиабатических поверхностей потенциальной энергии при их
182
Рис. 5.15. Проявления эффектов Яна—Геллера:
а первого поряди при пересечении адиабатических потенциалов в точке (^(вырождение
электронного состояния); б — второго порядка (псевдоэффекта) при сближении адиабатических
потенциалов в точке Qo (избежание пересечения электронных термов одинаковой симметрии).
Пунктирными линиями изображены поверхности без учета взаимодействия, сплошными — с
учетом вибронного взаимодействия электронных уровней
сближении. Такое отталкивательное взаимодействие возникает, как
было показано, лишь в случае определенных симметрийных от-
отношений между взаимодействующими электронными^ состояниями.
Если их симметрия одинакова, то интеграл J ?тЙ ?„<&¦ в силу
теоретико-групповых соображений отличен от нуля. Его величина
тем больше и, следовательно, тем больше раздвижение уровней при
их взаимодействии, чем больше перекрывание j *?„*?„ dz между вза-
взаимодействующими состояниями. Этот принцип анализируется
в разд. 9.2 на более простом примере двухорбитального взаимодей-
взаимодействия.
Таким образом, можно сформулировать правило непересечения
для электронных состояний одинаковой симметрии, обобщающее
введенное выше правило непересечения энергетических уровней для
орбиталей одинаковой симметрии (см. разд. 4.6).
Литература
Берсукер И. Б. Эффект Яна—Геллера в вибронные взаимодействия в современ-
современной химии. — М.: Наука, 1987. Гл. 1, 2.
Мннкяи В. И., Сшмквн Б. Я., Мшшев Р. М Квантовая химия органических со-
соединений. — М.: Химия. 1986. Гл. 1.
Салем Л. Электроны в химических реакциях. — М.: Мир, 1985. Гл. 2, 5.
ГЛАВА 6
СИММЕТРИЯ МОЛЕКУЛЯРНЫХ СИСТЕМ
Понятие симметрии играет важную роль во всех
науках. Свойствами симметрии обладают структуры многих м<
кул, ионов, образуемых ими реагирующих систем. Симметрия в
новых функций точно соответствует свойствам симметрии ядер:
конфигураций, и именно сферическая симметрия водородопод
ного атома является причиной наличия одной s-, трех р-, пяти
семи /-орбиталей и т. д., вырождения уровней я-МО в лине;
молекулах, структурных искажений, вызываемых эффектом Яна
Теллера первого порядка, и пр. Зная свойства симметрии волно
функций различных электронных состояний, можно, не при
к прямым расчетам, определить возможность переходов от одно
состояния в другое и получить тем самым представление о ^
тере спектров молекул. По этим свойствам можно судить также
условиях (пространственной ориентации, типе возбуждения), в
торых возможны или невозможны реакции между отдельными мо-
молекулами. Во всех случаях получаемая информация имеет качест-
качественный характер, однако она имеет принципиальное значение для
целей классификации и выработки основных принципов.
Математической основой теории симметрии является теории
групп, детальное рассмотрение которой выходит за пределы задач,
поставленных в настоящей книге. В данной главе будут представ-
представлены лишь некоторые главные понятия, используемые в последу-
последующих разделах, нашедшие широкое приложение в структурно!
химии.
6.1. ОПЕРАЦИИ СИММЕТРИИ ]
Молекулы могут обладать различными элементами симметрии
(оси, плоскости, центры инверсии). Операцией симметрии молеку-
молекулярной системы называют такое ее движение относительно соответ- j
ствующего элемента симметрии, которое переводит молекулярную j
систему в новое положение, физически тождественное первоначал*^
ному. Возможны следующие операции симметрии. !
1. Поворот относительно оси симметрии (например, oz) на угол i
q>—c9(oz). Обычно используемое обозначение для этой операции —;
ся, где n=2nj(p. Повороту относительно оси второго порядка (^>=iO
соответствует операция с2 и т. д.
2. Отражение в зеркальной плоскости (например, ху) а^ Приня-
Принятые обозначения для этой операции симметрии — ак (плоскост*
симметрии перпендикулярна оси с„), <х, (плоскость симметрии пр<>"
184
cz
6
/
%
/
\
PI
/
/
Рис. 6Л. Элементы симметрии молекул воды {а), бензола (б) и аллеаа (в)
ходит через ось с„) и ad (плоскость симметрии делит пополам угол
между двумя осями с2, перпендикулярными главной оси симметрии
3. Инверсия относительно некоторой точки x — ix.
4. Тождественное преобразование Е, оставляющее неизменным
положение молекулы.
Все остальные операции симметрии представляют различные
комбинации указанных выше операций. Особое значение имеет
операция зеркально-поворотного преобразования Sn-> включающая
последовательно поворот по оси ся и отражение в плоскости ah. Если
учесть, что Si = aky то можно сформулировать симметрийное усло-
условие для существования энантиомерных форм молекулы, т. е. прояв-
185
ления оптической активности. Таким условием является от
в структуре молекулы любых зеркально-поворотных осей
2, ...).
Рассмотрим результаты последовательного применения некс
рых операций симметрии к молекуле бензола (рис. 6.1). Пр]
два раза последовательно операцию io, вернемся к исходному
янию. Символически это записывается как результат умножения:
Легко проверить равенство
которое означает, что последовательное применение двух one
отражения эквивалентно трехкратному повороту на угол 2я/6. Та
ким образом, последовательное применение операций симметр]
эквивалентно другой операции симметрии на этой же структуре.
6.2. ТОЧЕЧНЫЕ ГРУППЫ СИММЕТРИИ
Каждая молекула может быть охарактеризована присущими
структуре операциями симметрии. Полный набор этих операций,
включающий обязательно тождественную операцию ?, состав]
группу. Например, для молекулы воды I группу симметрии образ]
ют операции Et cf, <*?> с? (см. рис. 6.1):
о
н
F,
F S F
М
III
Те же самые операции симметрии могут быть выполнены
молекулах совершенно других классов, например тетрафторида
ры II, фенантрена III. При их осуществлении одна точка в кажд<
молекуле (центр масс) не изменяет своего положения. Поэто;
группы симметрии молекул называют точечными группами. Моле-*
кулы I—III принадлежат к одной точечной группе С2* (см. далее).
Можно сформулировать математические требования, которым^
должно удовлетворять множество элементов А, В, С, ..., составля-
составляющих группу, в частности операции симметрии.
1. Произведение двух элементов множества дает также один из
элементов множества
АВ=С.
F.3)
186
2 Для произведения трех элементов выполняется закон ассоци-
ассоциативности
АВС=А(ВС)-(АВ)С. F.4]
3. Во множестве существует единичный элемент Е, обладающий
свойствами тождественного преобразования
ЕА-АЕ=А F.5)
для любого элемента А из множества.
4. Для любого элемента А данного множества всегда существу-
существует обратный ему элемент
'=E. F.6)
Точечные группы молекулярных структур можно подразделить
на четыре основных типа. К первому из них относятся группы
структур, не содержащих осей вращения выше первого порядка, —
группы Си Сл, С,*. К группе Сь единственным элементом которой
служит операция тождества Е, принадлежат все асимметричные
структуры. Группа С, имеет в качестве элемента симметрии плос-
плоскость a—Si, а группа С, — центр инверсии i=s2:
н
Л-
F C1
Cl F
Ко второму типу принадлежат группы, содержащие только одну
поворотную ось более высокого порядка, чем первый, — группы С„,
И
н
У
Л
h
В химии для обозначения групп используют номенклатуру Шенфлиса. Заглав-
Заглавные буквы С, D, Т. О обозначают соответственно циклические, диэдрические, тетра-
Дрические, октаэдрические группы. Нижний индекс указывает главный элемент
симметрии.
187
Третий тип составляют диэдрические группы, содержащие п
нуго ось л-го порядка и п осей второго порядка, перпендикуляр]
главной оси, — группы Dnt D*h,
*F
H
Н
F
н
D3d
Четвертый тип, к которому принадлежат группы высшей
метрии, включает группы, содержащие несколько поворотных
более высоких порядков, чем второй, — группы Та, Т, Тк (гетр;
рические); Oht О (октаэдрические), /, Д (нкосаэдрические),
(сферические):
и
н
N g
С
NC-
Fe CN
С CN
N
Ъ
Процедура отнесения некоторой структуры к определенной то-
точечной группе состоит в выявлении типа группы и дальнейшей
спецификации операций симметрии, выполняемых на этой струк-
структуре (см. указания к задаче 6.3).
Задача 6.1. Множество состонт нз двух элементов: единичного Е н элемен-
элемента А. Ввести в этом множестве операцию умножения так, чтобы оно стало
группой.
Задача 6.2. Множество состоит из четырех элементов: Е, А, В, С; Е —
единичный элемент. Ввести операцию группового умножения.
Задача 6.3. Определить точечные группы симметрии следующих молекуле
188
н-°\
Cl
6)
в)
Нч
н
н
¦
м
g
о го
Н
н)
ж>
Н
\
W
л)
Н
н
ч
О
63. ТАБЛИЦЫ ХАРАКТЕРОВ
ТОЧЕЧНЫХ ГРУПП СИММЕТРИИ
Каждому элементу симметрии точечной группы можно сопоста-
сопоставить матрицу, выбранную таким образом, чтобы операции между
отдельными матрицами удовлетворяли требованиям F.3) — F.6) и,
следовательно, соответствовали операциям симметрии. Набор мат-
матриц для всех операций симметрии образует представление группы Г.
Существует бесконечно большое число таких наборов, связанных
друг с другом эквивалентными преобразованиями (приводимые
представления). Особое значение имеют неприводимые представле-
представления, к которым относятся такие матричные представления, которые
не приводятся эквивалентным преобразованием к блок-диагональ-
блок-диагональному виду.
Все необходимые сведения о свойствах определенной группы
симметрии содержатся в наборах матриц, образующих неприводи-
неприводимые представления группы. ЗИгу информацию можно представить
в наиболее сжатой форме, вводя определение характеров элементов
группы. Характером %(R) операции симметрии R точечной группы
189
симметрии, которому соответствует матрица Щ, называют
(Trace) или сумму диагональных элементов этой матрицы:
т
Характеры (следы) матриц остаются постоянными при
эквивалентных преобразованиях. Они определяют результаты
ствия операций симметрии на базисные функции молекулы, npi
лежащей данной группе симметрии.
Рассмотрим, например, какие наборы характеров (неприво;
мые представления) соответствуют двум связывающим МО moj
кулы воды, образованным 2pz- и 2ру-АО кислорода и ls-АО атов
водорода (см. разд. 10.1.1):
Применяя операции симметрии, определенные на рис. 6.1, а,
получим
и соответствующее неприводимое представление, т. е. ряд хара|^
теровA, 1, 1, 1). у
Аналогично, для орбитали ?2 ->
V
и неприводимое представление A, — 1, — 1, 1). В общем виде можно
записать
R4=x(*)\? F.8)
где оператор R обозначает операцию симметрии, a x(R) — характер
/-го неприводимого представления. .
Полные наборы неприводимых представлений групп содержат
таблицы характеров. Таблица характеров группы С2„ дана в табл.
6.1.
190
Таблица 6.1.
-&"
Ai
А2
Bj
в2
Е
1
1
1
1
Таблща
с2
1
1
-1
-1
характеров
(Г
1
-1
1
-1
грушял Сю
<
1
-1
-1
1
уЛ у2 JL
JC, J?j», XZ
у, Rx, yz
Принятые обозначения неприводимых представлений приведены
в первой колонке; симметричные по отношению к главной поворот-
поворотной оси обозначают через А, а антисимметричные — В. В случае
диэдрнческой группы Dtk символ А присваивают лишь неприводи-
неприводимым представлениям, симметричным по отношению ко всем трем
осям С2. Нижние индексы соответствуют симметрии (индекс 1) или
антисимметрии (индекс 2) по отношению к плоскости, проходящей
через главную ось. Полносимметричное представление всегда обо-
обозначают как А\. Из табл. 6.1 следует, что функции 4*i и Ч*2 молекулы
воды преобразуют по неприводимым представлениям ах и Ь2* точеч-
точечной группы С2у.
В точечных группах, включающих центр инверсии, добавляются
индексы g (симметрия по отношению к центру инверсии) и и (ан-
(антисимметрия). Если необходимо указать свойства симметрии по
отношению к плоскости <гА, добавляют верхний индекс — штрих
(симметричный) или два штриха (антисимметричный). Вырожден-
Вырожденные неприводимые представления обозначают буквами Е (двойное
вырождение) и Т (тройное вырождение) (см. разд. 6.5).
Последняя колонка в табл. 6.1 характеров включает некоторые
важные функции, которые преобразуются по неприводимым пред-
представлениям данной группы. Символы х, у, z представляют коор-
координаты; Rxt Ry, Rx — вращения относительно соответствующих
осей.
Задача 6.4. Покажите на примере группы Cj* что рад^-АО преобразуется
по тем же неприводимым представлениям группы, что и соответствующие
координаты.
"Обозначение Р. Малликена для неприводимых представлений. Для орбиталей
обычно применяются обозначения со строчными буквами а, Ь, е, I. Для состояний
используют прописные буквы А, В, Е, Т.
191
Таблица 6.2. Характеры неприводимых аредставлений некоторых точечных
групи симметрии
А
В
Ay
Ay
Clh
Ag
А„
BS
в„
c2v
Ах
Аг
Вх
в2
Alg
Big
B2g
Eg
Aiu
A2u
Biu
Blu
Eu
Ei
1
I
1
1
E
1
1
1
1
E
1
1
1
1
2
1
1
1
1
2
E
1
1
E
1
1
2C4
1
1
j
-1
0
1
1
-1
-1
0
c2
1
1
— 1
-1
1
1
-1
-1
Сг
1
1
1
1
— 2
1
1
1
1
-2
1
-1
1
-1
0
1
-1
1
-1
0
1
-1
I
1
-1
1
-1
1
— 1
и
w
1
-1
1
-1
2c;
l
-l
-l
l
0
1
-1
-1
1
0
л
°k
1
-1
-1
1
a
V
1
-1
-1
1
i
1
1
1
1
2
-1
-1
-1
-1
-2
z,
t>y>
Rz
R*.Ry
Tf Jy 15
Пх, Лу, Л,
x.
2S4
1
1
-1
-1
0
-1
-1
1
1
0
y>z
Rz
z
Ry
z
Rz
X, /?j
y.R»
oh
1
1
1
1
-2
-1
-1
-1
-1
2
2av
1
-1
1
-1
0
-1
1
-1
1
0
x2, y2, z2, xy
yz.xz
Bee
функции
У2, г2, ху
xz,yz
f
x2, y2, z2
2od
1
_ 1
-1
1
0
J
1
1
-1
0
xy
xz
yz
Rz
xy
Rx, Ry xz, yz
z
х.У
1
1
j
Ч> ''I
: V '
, ,- "''
¦ г
z2
192
Продолжение табл. 6.2
Dbh
A2u
Big
B\u
Biz
Вы
E 2Сб *<?* <?t* 3C2
ш2С3
1
1
I
1
I
1
1
1
2
2
2
2
^
i -i
i -i
¦i -i
i -i
i
i
i
1
i
1
i
-2
-2
2
1 1
1 1
-1 -1
-1 -1
1 -1
1 -1
-1 I
-1 I
0
0
0
0
0
0
0
0 -
1 1
-1 -1
-1 -1
1 1
-1 1
1 -1
1 -1
-1 1
о о
о о
о о
о о
1
-1
1
-1
1
-1
1
-1
-1
1
-I
1
1
-1
1
-1
-1
1
-1
1
1
-1
-1
1
1
-1 Rz
1 z
-1
1
_ J
1
-1
2 (Rx,Ry) (xz. yz)
~2 (x,.y)
2 f jc* —y*.xy)
-2
с*
At
A2
E
E
1
1
2
2C3
1
1
-1
1
-1
0
2
Rz
(x. y) (Rx,
Ry)
X2
(x2-y2,
+y\ Z2
xy) (xz, yz)
<*g
Big
B2g
Big
Au
Вы
Blu
Въи
E
1
1
1
1
1
1
1
1
C2(z)
1
1
-1
-1
1
1
-1
-1
C2(y)
1
-1
1
-1
1
-1
1
-1
C2(x)
1
-1
-1
1
1
-1
-1
1
i
1
1
1
1
-1
-1
-1
1
1
-1
-1
-1
-1
1
1
c(x.z)
1
-1
1
-1
-1
1
-1
1
a(y.z)
1
-1
-1
1
-1
1
1
-1
Rz
Ry
Rx
z
У
X
x2 v2 z2
xy
xz
yz
'¦ Теория строения молекул
193
Продол
2С3
»*
га*
Е'
Е
I ]
1 ]
2 -]
1 1
1 1
2 -1
I 1
I -1
I 0
L 1
I -1
I 0
1
1
2
-1
-1
2
1
1
-1
-1
-1
1
1
-1
0
-1
1
0
Rz
(х.У)
i
(R*. R,)
%\
oh
A*
*ш
Tig
T*
AXU
А-ы
?-
Tlm
E
1
1
2
3
3
1
1
2
3
3
8C3
1
1
-1
0
0
1
1
-1
0
0
3C2
1
1
2
-1
-1
1
1
2
-1
-1
6C4
1
-1
0
1
-1
1
-1
0
1
— 1
I
1
-1
0
-1
1
1
-1
0
-I
1
i
1
1
2
3
3
-1
-1
-2
-3
-3
8*
1
1
-1
3
0
-1
-1
I
0
0
1
1
2
-1
-1
-1
-1
-2
1
1
ад,
l
-l
0
l
-l
-l
l
0
-1
1
<"<
1
-1
0
-1
1
-1
1
0
1
-1
¦m
f Лх, llj, K.ZJ
(xy,xz.yxt
(x. y, z)
Ihd
1
1
I
1
2
1
1
-1
-1
0
1
1
1
1
-2
1
-1
1
-1
0
1
-1
—1
1
0
194
Td
————
Ax
л2
E
Tx
T2
Б
^ i ¦ "
1
1
2
3
3
8C3
1
1
-1
0
0
1
1
2
-1
1
654
1
-I
0
1
-1
<*<
1
-I
0
-1
f*. 7. ^
Продолжение табл 62
fлу, xz, yz)
В табл. 6.2 приведены характеры некоторых часто встречающих-
встречающихся точечных групп симметрии. Более полные таблицы имеются
в приводимых в конце главы источниках.
6.4. СИММЕТРИЗОВАМНЫЕ ОРБИТ АЛИ
Рассмотрим, как используется знание характеров групп для по-
построения симметризованных орбиталей молекул, т. е. орбиталей,
преобразующихся по неприводимым представлениям точечной
группы молекулы. Возьмем в качестве примера построение симмет-
симметризованных я-МО молекулы на-
нафталина из pz-KO атомов углерода.
Эта молекула обладает симметри-
симметрией 1>2А. Однако для простоты ис-
исключим из рассмотрения плос-
плоскость ah, которая является обяза-
обязательным элементом для всех я-со-
пряженных систем. В этом случае
молекуле можно приписать С2,-
симметрию (рис. 6.2).
В соответствии с F.8) можно,
опуская условие нормировки, запи-
записать следующее выражение для
симметризованной МО q>(J) /-го не-
неприводимого представления:
F.9)
д
где Р — одна из орбиталей перво-
первоначального базиса. Суммирование
происходит по всем операциям
симметрии R данной точечной ^ _, -
Группы Элементы симметрии моле-
молекул нафталина в точечной группе С2г
195
Пользуясь табл. 6.1 и выбрав орбиталь Рх атома углеро;
найдем симметризованную я-МО, преобразующуюся по неправо;
мому представлению Ах:
=Й? ЕРх
Так как ЕРХ = Рх; С2РХ = Р5; о?Рх = Р8 и а?Рх = Р4, то после норми]
ки приходим к симметризованной МО ^-типа:
\
Следуя той же схеме и выбрав для рассмотрения операций симмс
рии орбиталь Р2, найдем следующую МО, преобразующуюся
неприводимому представлению Ах:
1
<P2(Ol) — X (Р2 + Р3 + Pb + Pz)-
2
Преобразование F.9) с выбором в качестве начальных Р3, Р4>
Р8 АО будет давать те же самые симметризованные МО ах-п
Однако при задании начальной АО Р9 получим третью и после
нюю я-МО нафталина, преобразующуюся по неприводимым пред*
ставлениям точечной группы С2„: J
Г
Последовательно подставляя в формулу F.8) все
Рк (k~ I -i- \0) и варьируя все неприводимые представления (Q
ной группы Сщ после нормирования получим весь набор симмет-
ризованных я-МО молекулы нафталина. Они представлены в TadJ
6.3.
6.5. ВЫРОЖДЕННЫЕ ПРЕДСТАВЛЕНИЯ ,
Каждая точечная группа симметрии, содержащая оси вращениж]
выше второго порядка, имеет вырожденные представления, кото-|
рые, согласно Малликену, обозначают: Е (следует отличать :w
обозначения тождественного преобразования) — для двукратно и^
рожденного представления; Т — для трехкратно вырожденнвйв
представления. Примеры таких групп даны в серии табл. 6.2. В эти
группах операции симметрии сведены в классы операций, имеют*
196
1
Таблица 6.3. Симметрвзованные я-МО молекулы нафталана* преобразующее-
преобразующееся по неприводимым представлениям группы C2v
<Р\
=
,=— (Р9 + Л0)
1
-
1
-
1
((>6 2
1
2
/2
2
в2
1
2
1
Задача 6.5. Получить все симметризованные я-МО молекулы нафталина.
Задача 6.6. Получить симметризованные комбинация 5-АО молекулы Н4.
Четыре атома водорода расположены в вершинах прямоугольника с симметрией
одинаковые характеры во всех неприводимых представлениях. Так,
в точечных группах С3» А операции Сэ и 0ъ — СъСг объединены
в общий класс операций и обозначаются 2С3, так же как три
сти плоскости в группе Сз» и две оу плоскости в группе D3h представля-
ются классами операции Ъа0 и 2av соответственно.
Для вырожденных представлений сохраняются все правила, ко-
197
торые используют в случае невырожденных представлений. Q)
применение соотношений F.8), F.9) для произведения с их
шью симметризованных орбиталей имеет важную специфику,
смотрим, например, орбитали, образуемые в молекулах NH3,
комбинациями трех s-AO атомов водорода. Из трех базисных
можно, очевидно, получить не более трех симметризованных
биталей:
Выбрав в качестве пробной орбиталь Su следуя процс
описанной в предыдущем разделе, получим для орбитали агтипа !
<p(al)=Sl
т. е. в нормированном виде
з
Проблема возникает при генерировании симметризованных
преобразующихся по вырожденному ^-представлению. На
процесс последовательно с sb s2, SyAO, получаем для двукра
вырожденного представления три различающихся орбитали, т.
на одну больше, чем возможно:
(Sx) q
(S2)(p2(e)=2S2-S3~Sb -j
(S3) q>3(e)=2S3~Sl-S2.
Выход из этого положения состоит в применении процедуры,
известной под названием ортогонализации Шмидта. Конечный ре-
результат состоит в том, что орбиталь q>i(e) и орбиталь
получаемая при определенной комбинации орбиталей (р\(е),
и (ръ(е)у удовлетворяют требованию взаимной ортогональн щ
и соответствуют свойствам двукратно вырожденного представле*
ния Е. Также любая линейная комбинация симметризованных пп*
биталей ф\(е) и <Р\(е) является подходящим решением*.
*Квантово-химический расчет может быть выполнен как в .
так и в несимметризованном базисах. В последнем случае симметрия воссганаадяЛ*
ется в конце расчета.
198
Задача 6.7. Проверить отсутствие ортогональности орбиталей <р\(е),
ту(е), Фъ(е) ш ортогональность орбиталей q>\(e), <pi'(e). Рассчитайте нормиро-
нормировочные множители для этих орбиталей.
6J6. ПРЯМОЕ ПРОИЗВЕДЕНИЕ
Для многих задач теории строения молекул важно знать, каковы
свойства симметрии произведения двух функций fA, fB, если извест-
известны свойства симметрии каждой из них. В теоретико-групповых
терминах это означает, что необходимо найти матричное представ-
представление VD данного произведения функций исходя из неприводимых
представлений ГА и Гв последних, т. е. вычислить прямое произ-
произведение:
ГО=ГА хГв-
Характер представления, являющегося прямым произведением двух
представлений, равен произведению характеров исходных представ-
представлений:
Определим теперь симметрию волновой функции Ч^Ч^, где
% и *Р2 — две МО молекулы воды (или любой другой молекулы
типа АН^), преобразующиеся, как было показано ранее, по пред-
представлениям ах и Ь2 группы С2„:
Ьг
Е
1
1
1
Сг
1
2
-1
а"
в
1
-1
J
1
I
1
F.12)
Из данных F.12) следует, что а, хЬ2=Ь2, т. е. если в F.10) одно
из представлений, например ГА, является полносимметричным, то
прямое произведение Го имеет такое же представление, как Гв.
Рассмотрим прямое произведение b\ x Ь2. Сравнивая результат
F.12) с таблицей характеров группы С^ (см. табл. 6.2), видим, что
199
bi
b2
fcix/>2=02
E
1
1
1
Сг
-1
-1
1
of
1
-1
-1
-1
1
-1
Получим теперь таким же способом представления для npj
произведений ЬххЬи а2ха2, b2xb2. Во всех случаях, как легко у(
диться, результатом будет полносимметричяое представление
Этот результат важен при определении свойств симметрии эл<
ронных состояний, т. е. волновых функций, строящихся из ф
отдельных МО: Va(?iJW2 •¦¦ (<Р«) (ф«) ¦ Прямое произвел*
представлений функций ц>„ всех заполненных электронами МО дас
представление Г*. В силу свойств ассоциативности F.4) ясно,
представление Г* будет определяться прямым произведе!
Г,я х Г9 , так как парные произведения — типа Г91 х Г9.=а\.
m
Задача 6.8. Определите функции состояний ? = (q>if ... (tpn) (<pm) для ма1
кулы (иона) C2w симметрии с МО <р„ и ц>т соответственно симметрии Ь\ и b
случаев заполнения к, 1=0, 1, 2.
Прямое произведение двух представлений F.10) не обязательна]
является неприводимым представлением. Рассмотрим, например
прямые произведения некоторых представлений группы C4v:
c4v
Ах
А2
Вх
Вг
Е
BixE^E
АгхЕх.Вг=Е
ЕхЕ=Е?
Е
1
I
1
1
2
2
2
4
1
1
1
1
-2 '
-2
-2
4
2С4
1
1
-1
-1
0
0
0
0
2<ту
1
-1
1
— 1
0
0
0
0
1
— 1
-1
1
0
0
0
0
F.U
200
Если прямые произведения ВххЕ, А2хЕхВ2 дают неприводи-
неприводимые представления, то Ё1 является приводимым представлением.
Хак как характеры — не что иное, как след матриц, то к ним
и к представлениям в целом применим закон сложения:
Г*=ГД1+Г,2 + Г,3 + ... +Г,Л. F.15)
причем сумма размерностей представлений ГВ1, Г*2... ГВк должна
быть равна размерности исходного представления Гв.
Исходя из F.15), можно, пользуясь таблицей характеров F.14),
отыскать подходящее разложение прямого произведения Е2 на не-
неприводимые представления:
ЕхЕ=Е2=Ах +А2+В1
В табл. 6.4 дана сводка общих правил для определения неприво-
неприводимых представлений прямых произведений.
Таблица 6.4. Правила умвоження неиршюднмых представлений
Общие правила:
А хА = А; ВхВ=А; Ах В=В; А х Е-Е; Вх Е=Е;
АхТ=Т; ВхТ^Т;
g*g=g; uxu=g; иху=и;
Ax E{ = E\\ AxE2-E2;
Правила для индексов представлений А или В:
1х1=1;2х2 = 1; 1x2^2,
кроме групп D2 и D2b где 1 х 2=3; 2 х 3 -1; 1 х 3 = 2.
Правила для дважды вырожденных представлений:
Для групп С3, СЗЛ, C3vt D2, D3h, DM, Q, C6v, D&, D&. S6, O. Ok, T. Td, Tk
E\ x E2—B\
«ля групп Q, C4v, C4h, Dn, Db S4
Правила для трижды вырожденных представлений для групп Td, О, Of,:
Тх х
201
Знание свойств симметрии прямых произведений очень noj
для качественного анализа многих задач теории строения молеку!
требующих оценки интегралов вида
или
где М — любой квантово-механический оператор; <pt —
МО или электронных состояний.
00
Подобно тому как интегралы J f(x) dx не равны нулю
— 00
в том случае, если функция f(x)—четная, т. е. f(x)=f(~:
интегралы F.16), F.17) не равны нулю лишь если подынтег]
ному выражению отвечает полносимметричное (а^) неприводимс
представление группы симметрии молекулы или приводимое
ставление, содержащее полносимметричное неприводимое предел
вление.
Для интегралов F.16) неравенство нулю будет достигаться,
волновые функции (р, и q>j принадлежат к одному и тому же непршкм)
димому представлению группы симметрии молекулы. Интегралы]
F.17) отличны от нуля в случае, если выполняется соотношение
Г,хГуэГ*. F. И
т. е. в прямом произведении представлений Г, и Гр по котор!
преобразуются функции (рг и <р„ содержится представление, по К(
рому преобразуется оператор М.
Многим важным квантово-мехаиическим операторам соответ-
соответствуют полносимметричные представления в любой группе симме
рии. Такими свойствами обладают оператор Гамильтона Н и
производные (—- }, ( —-=¦) (см. разд. 5.7.2). Следовательно, в этом*
\oQJ \dQ»v ¦•**
случае, чтобы интеграл F.17) был отличен от нуля, функции
(pi и %¦ должны принадлежать к одному и тому же неприводимому
представлению группы или в прямом произведении их представлен
ний Г, х Гу должно содержаться полносимметричное представлением|
Литература
Дьюар М. Теория молекулярных орбиталей в органической химии. — М/. Ммр»
1972.
Подробное рассмотрение метода ППП в его применения к расчету свойств осдоиян
состояний сопряженных молекул. (,
Дьюар М., Дотерт» Р. Теория возмущений молекулярных орбиталей в органе*
ческой химии. — М.: Мир, 1977. Гл. 3.
202 j
Подробное обсуждение сопряженных систем с качественных позиций теории возмущений.
число полезных упражнений.
Костяке» Р. Р-> Беспало» В. Я. Основы теоретической органической химии. —
Л.: ИЗД-во ЛГУ, 1982. Гл. 3.
Основные особенности строения «-электронных систем рассмотрены в рамках методов МОХ
лЛЛП.
Роберте Дж. Расчеты по методу молекулярных орбит. — М.: Мир, 1963.
Ясное описание метода Хюккеля и иллюстрация его на детально разобранных примерах.
Стрештвазср Э. Теория молекулярных орбит для химиков-органиков. — М.:
Мир, 1965.
Хорошее и полное изложение метода Хюккеля и его приложений к различным проблемам
Фларрм Р. Группы симметрии. Теория и химические приложения. — М.: Мир,
1983.
Учебное пособие по теории групп и ее применению в химии и квантовой механике.
Рассмотрены группы симметрии нежестких молекул.
Хохштрассер Р. Молекулярные аспекты симметрии. — М.: Мир, 1968.
Полное математическое изложение основ и аппарата теории групп, иллюстрируемое много-
многочисленными хороню подобранными примерами. Особое в лмание уделено электронным спект-
спектрам.
Cotton F. A- Chemical Applications of Group Theory. New York: J. Wiley. 1971.
Используя в самой сжатой форме необходимый математический аппарат, автор — извест-
известный специалист в области химии металлоорганических и комплексных соединений — просто
и наглядно излагает применение теории групп в задачах построения гибридных орбиталей,
симметризованиых МО комплексов сопряженных органических молекул, а также анализа колеба-
колебательных спагтров.
ГЛАВА 7
РАСЧЕТНЫЕ МЕТОДЫ КВАНТОВОЙ ХИМИИ
7.1. ОБЩАЯ ХАРАКТЕРИСТИКА
Все расчеты многоатомных молекул основаны на приближенных
решениях уравнения Шрёдингера D.3). Практика предъявляет два
главных требования к уровню приближения и выбору расчетной
схемы. Это, во-первых, достаточное соответствие результатов рас-
расчета результатам эксперимента и, во-вторых, достаточная эконо-
экономичность расчетов, т. е. разумные затраты времени при выполне-
выполнении их на быстродействующих ЭВМ. Из двух основных теорий
химической связи — метода валентных связей и метода молекуляр-
молекулярных орбиталей — последний имеет значительные преимущества
при реализации на ЭВМ. Поэтому все основные расчетные методы
современной квантовой химии используют приближение МО
в форме схемы ЛКАО МО Хартри—Фока—Рутаана (см. разд.
^-3.3). В рамках этой схемы возможны как дополнительные усовер-
усовершенствования расчетной модели (учет эффектов электронной кор-
203
реляции), так и значительные упрощения, позволяющие на нескс
ко порядков снижать затраты времени ЭВМ.
Вопрос о том, какой уровень приближения следует выбрать
решения той или иной задачи, решается в прямой зависимости
характера последней. Большая часть задач теоретической
носит качественный характер, и ответы на них могут быть получс
при помощи весьма простых расчетов, воспроизводящих лишь
мые главные свойства волновых функций (узловые характерш
и порядок энергетических уровней граничных МО). Другая
задач требует точной количественной оценки какого-либо одн<
или нескольких структурных и физических параметров (теши
образования, электронного спектра поглощения и т. д.), тогда
остальные свойства молекулы могут быть оценены менее то*
Обе эти группы задач, как правило, целесообразно решать, испоЗ
зуя так называемые полуэмпирические методы квантовой
в которых вычисления ряда интегралов в уравнениях D.62) зам<
ются подстановкой эмпирических параметров, а большей част
этих интегралов вообще пренебрегают. Некоторые совремеш
полуэмпирические методы обладают достаточно большой гиб|
стью, т. е. параметризованы таким образом, что позволяют с xoj
шей точностью предсказывать целый ряд свойств основных и в<
бужденных электронных состояний молекул при довольно мг
затратах машинного времени.
Неэмпирические (от лат. ab initio — сначала) расчеты основаны
точном решении уравнений D.3), которые не включают никак]
экспериментальных параметров, кроме фундаментальных физичс
постоянных. Затраты времени ЭВМ при выполнении неэмпиричес:
расчетов даже с использованием минимального базиса АО значи
но выше (см. табл. 4.3), чем для полуэмпирических методов, и быст^
растут с увеличением размеров базиса АО (см. разд. 4.3.3). Благодаря
быстрому прогрессу технических возможностей современных ЭВ$$
область приложений неэмпирических расчетов непрерывно расширя-
расширяется. В современной теоретической химии неэмпирические расчеты
молекул становятся постепенно общедоступными.
Одно из особенно важных достоинств неэмпирических схем ра№
чета состоит в том, что легко установить их строгую иерархикс
основанную на сопоставлении получаемых с их помощью значени»
полной энергии. Свойство вариационности неэмпирических методов^
дает возможность, улучшая постепенно форму искомой волноври
функции, приближаться к результату, достигаемому вначале в xajrr*
ри-фоковском пределе, а затем к результату, достигаемому точный
решением уравнения Шредингера. Поскольку каждый шаг на этот*'
пути сопряжен с быстрым нарастанием затрат машинного времена
исследователь останавливается на расчетной схеме той или инощ
степени сложности, обеспечивающей должный компромисс между
желаемым уровнем точности решения и затратами машинного вр&
мени. Практика расчетов выработала определенные критерии для
204
бора подходящих при решении различных задач приближений
Эмпирических расчетов, а также соответствующих полуэмпири-
ческих методов.
7.2. НЕЭМПИРИЧЕСКИЕ МЕТОДЫ
Неэмпирические методы расчета необходимы для следующих
главных целей: во-первых, для выяснения, насколько хорошо уста-
установленные экспериментальные факты описываются теорией, т. е.
для установления пределов теории; во-вторых, для расчета свойств
молекулярных структур или эффектов, которые затруднительно или
невозможно (переходные состояния реакций) определить экспери-
экспериментально; в-третьих, для обоснования и развития различных полу-
полуэмпирических схем расчета.
При использовании метода Хартри—Фока—Рутаана главной
характеристикой расчетной модели является выбор базиса орбита-
лей. Чем полнее этот базис, тем точнее воспроизводится полная
энергия молекулы. Однако способность расчета предсказывать дру-
другие молекулярные свойства не всегда монотонно зависит от выбран-
выбранного базиса и учета корреляционных эффектов. Наглядный при-
пример — расчет дипольного момента (р) молекулы воды: при мини-
минимальном базисе ^=1,82 D, в расширенном почти до хартри-фоковс-
кого предела базисе /i=2,57 D, в расширенном базисе с наложением
конфигурационного взаимодействия /i=l,99D, в эксперименте
/j=l,85 D. Аналогичные примеры можно найти и для некоторых
других характеристик. Важно знать, какие ряды базисных орбита-
лей следует использовать для получения надежных результатов
в расчетах различных характеристик мрлекул.
7.2.1. Базисные ряды атомных орбиталей
Простейший уровень неэмпирических (а также полуэмпиричес-
полуэмпирических всевалентных расчетов) — использование минимального базиса
АО (см. разд. 4.3.5). Существенное улучшение точности расчета
достигается при переходе к базису дубль-зета (DZ). Важно от-
отметить, что, если в случае минимального базиса NG-типа даже
при больших значениях N не достигается уровень точности ми-
минимального базиса STO, применение валентно-расщепленных ба-
базисов GTO-M-NPG позволяет превзойти уровень слэтеровского
DZ-базиса.
Распространенными базисами GTO, соответствующими расши-
расширенным базисам STO с включением поляризационных функций,
являются базисы типа 6-31G* и 6-31G** (одна звездочка означает
Добавление поляризационной d-GTO к каждой /ьфункции, две звез-
звездочки — добавление, кроме того, p-GTO к l-s-орбиталям атомов
водорода). Переход от минимального базиса типа STO-3G к рас-
Щи базису с включением поляризационной функции типа
205
6-31G* приводит к ~10—20-кратному увеличению затрат
ного времени, необходимого для расчета.
Таблица 7.1. Палые
Сгружтур*
STO-3G
78,30618
78,30160
77,96889
77,30344
4-31G
79,И593
78,11151
78,85466
78,32567
6-31G*
-79,22876
79,22240
-78,97031
78,45215
6-31G**
79,23823
79,23321
-78,98786
78,49100
Приведенные в табл. 7.1 данные расчетов полных энер]
этана в устойчивой ?>w н заслоненной Dy, конформациях, а
для его гипотетических мостиковых структурных изомеров с
метрией Dy, и /)« дают представление о том, насколько уто*
результаты неэмпирических расчетов при постепенном pacumj
базиса.
Переход от минимального базиса к расширенному приво;
н
н-
С—Н
1 ¦*!
II, я,
III, D2h
к понижению полной энергии даже для таких простых структур'
примерно на 1 а. е. энергии, т. е. ~2500 кДж/моль. Однако в хж*
мни важны не абсолютные, а относительные значения. Как видно из
данных табл. 7.1, порядок относительной устойчивости изомер©*
этана не зависит от базиса, а относительные энергии двух конфор^
меров D-sd и />э*, определяющие высоту барьера вращения по связИ
С—С, заключены в пределах 11,5—17 кДж/моль (см. разд. 1Z1). *
Минимальный базис STO-3G достаточно удовлетворителен длЯ
воспроизведения молекулярной геометрии (ошибки в длинах связей*
0,003 нм и углов ~4°) большинства структур с замкнутыми элекЙ
ронными оболочками. Однако для реакций, в которых продукты в»
сохраняют все типы связей исходных реагентов, требуется исполь-
использование по крайней мере DZ-базисов типа 4-31G. Такой же базяф
орбиталей необходим и для достаточно хорошего воспроизведений
колебательного спектра молекул, а для описания барьеров врапФ*
няя относительно простых связей, включающих гетероатом, и б**
рьеров пирамидальной инверсии (см. разд. 12.4.1) требуется обяза-'
206
ильное добавление поляризационных функций, т. е. применение
пасяшренного базиса.
В табл. 7.2 сформулированы некоторые выработанные, главным
образом практикой расчетов, соответствия между рассчитываемым
Свойством и необходимым уровнем базиса АО. Эти соответствия
характеризуют тгжний предел требований и не гарантируют того,
чходля правильного описания того или иного свойства не потребу-
потребуется в конкретном случае более высокий уровень теории.
Таблица 7J2. Ti
рядов АО, рскомещдуемые для коррекпого опаса-
Свойства молекулярной
системы
Молекулярная геоме-
геометрия
Силовые постоянные
Вращательные барье-
барьеры
Инверсионные барье-
барьеры
Энергии реакций
Взаимодействие ио-
иов и диполей; систе-
системы с Н-связью
Слабые межмолеку-
межмолекулярные взаимодейст-
взаимодействия
—
Минимальный
DZ
Минимальный
DZ+P
DZ,
DZ+P
Минимальный; П7.
rfJM-iiiMjYCtfH мн
Комыевтарви в всжлючения
За исключением расчетов диэдральных
уГЛОП И Геометрии ПИрЯМИЛАШ>НЬ1Х
структур, где необходимо использо-
использовать поляризационные функции
Приемлемый уровень — базис типа 3-
21G. Переход к расширенному базису
6-31G* не дает заметных улучшений
Исключение составляют молекулы
с осью вращения, включающей два ге-
тероатома, например Н2О2 и др., для
которых требуется базис DZ+P
Учет эффектов электронной корреля-
корреляции практически не улучшает резуль-
результат расчета
Для реакций, в которых электронные
пары связей в реагентах сохраняются
в продуктах (переходных состояниях).
В противном случае необходим учет
электронной корреляции
Для расчетов анионов и их взаимодей-
взаимодействий обязательно включение поляри-
поляризационной добавки
Должны быть включены поляризаци-
поляризационные диффузные функции. Необхо-
Необходим учет энергии корреляции электро-
электронов
207
7.2.2. Энергия электронном корреляции
В разд. 4.4 было дано определение энергии электро-
корреляции и рассмотрены методы ее количественной oi
Учет корреляционной энергии имеет важное значение при
делении формы потенциальных кривых и поверхностей,
вычисленные длины связей в методе Хартри—Фока окг
короче экспериментальных значений, а следовательно, кс
тельные частоты оказываются завышенными, что видно
данных табл. 7.3. Учет корреляционных эффектов npj
к существенно лучшему согласию с экспериментальными
чениями.
Таблица 7.3. Влияние учета энергия электронной корреляции на
значения частот колебании toe (см'1)
Молекула
LiH
ВеН
ВН
СН
NH
ОН
HF
NaH
MgH
А1Н
SiH
PH
SH
на
Расчет
Хартри—Фока
1428,2
2145,0
2484,6
3044,1
3546,5
4054,5
4476,2
1184,2
1583,6
1730,8
2126,2
2487,6
2839,9
3141,1
Расчет с учетом эвер-
гии корреляции
1401,5
2064,6
2352,1
2841,7
3269,3
3743,6
4169,3
1172,3
1492,3
1691,7
2034,7
2365,9
2676,4
2977,2
1405,7 Я
2060,8 Щ
2366,9 Ш
2858,5 '«
3282,1 Ш
3739,9 -"Ш
4138,7 %Щ
1172,2 Щ
1497,0 1
1682,6 I
2041,8 1
2380,0 ~Ш
2697,0 Щ
2991,0 Щ
Вклад энергии корреляции в вычисленные барьеры внутрешк
вращения и инверсии, как правило, незначителен. Так, доля энер1
корреляции в барьерах вращения этана и инверсии аюмиака
ляет всего ~2 кДж/моль соответственно при величинах барьеров]
12,4 и 24Д кДж/моль. Хотя иногда учет корреляционной энер!
может существенно влиять на рассчитываемые барьеры инверси*. •
Вклад энергии корреляции отражается и на относительной термодщ
намической стабильности изомеров.
Учет энергии электронной корреляции при теоретическом
зе химических реакций становится совершенно необходимым, когд*|
в ходе реакций происходит разрушение электронных пар свяэе#!
исходных структур. Прежде всего это относится к реакциям раз-
разрыва связей, так как расчет в рамках приближения Хартри—Фощ
может приводить к неправильным диссоциированным состояниям.
К числу таких реакций принадлежат превращения, протекающие
208
структуры бирадикального типа, а также реакции, запрещен-
запрещение правилами СОхранения орбитальной симметрии (см. гл. 13).
нЫ зависимость результатов расчета радикальных процессов от точ-
ости метода иллюстрируется неэмпирическими расчетами реакций
радикального замещения:
F + CH2=CH2-CHF=CH2+H
Как видно из данных, представленных в табл. 7.4, удовлет-
удовлетворительного согласия с экспериментальными значениями теплот
реакций удалось достигнуть только при учете электронной кор-
корреляции по теории возмущений Меллера—Плессета не менее чем
в третьем порядке. При этом ХФ-решение также должно быть
достаточно точным, необходимо использование расширенного ба-
базиса АО с добавлением поляризационных функций.
Таблица 7.4. Сопоставление рассчитанных ш экспериментальных значений теп-
теплот радикальных реакция (кДж/моль)
Метод
HF/3-21G
MP2/3-21G
MP3/3-21G
MP4/3-21G
HF/6-31G*
MP2/6-31G*
MP3/6-31G*
MP4/6-31G*
*r/2hva)
Эксперимент
F + C2H4->CH2H3F+H
76,9
-17,6
9,7
2,5
43,3
-75,8
-35,3
-45,4
-18,9
-46,4±8,4
F+CH4->CH3F+H
115,5
30,6
53,4
47,9
71,8
-35,7
-2,5
-5,5
-15,2
-23,6±8,4
а)
Разность энергий нулевых колебаний. Расчетная величина теплового эффекта
получается суммированием этой разности и представленных в таблице разностей
полных энергий реагентов и продуктов реакций.
Другой важный случай, при котором обязательно требуется учет
энергии корреляции электронов, — расчет структур с вырожден-
вырожденными или псевдовырожденными электронными конфигурациями.
Типичный пример — молекула циклобутадиена. Без учета элект-
электронной корреляции расчеты даже в расширенных базисах приводят
к неправильному выводу о предпочтительности триплетной элект-
электронной конфигурации и квадратной, а не прямоугольной геометрии.
209
Учет корреляционных эффектов обязателен в расчетах
кулярных взаимодействий, так как важный эффект дисперсионж
притяжения молекулярных систем с замкнутыми электро]
оболочками имеет чисто корреляционную природу.
7.3. ПОЛУЭМГШРИЧЕСКИЕ МЕТОДЫ РАСЧЕТА
Уже в 1929 г. П. Дирак* писал: «Основные физические зако]
необходимые для построения математической теории большей
сти физики и всей химии, полностью известны, трудность toj
в том, что точное применение этих законов приводит к
сложным уравнениям. Следовательно, желательно развить при(
женные практические методы, которые могут объяснить п
особенности сложных атомных систем без привлечения
сложных расчетов».
При использовании неэмпирических методов расчета осно]
затраты времени ЭВМ, обычно ~70% всего требуемого для no
го расчета времени, направлены на вычисление интегралов межэл<
тронного взаимодействия {ру\1а). По мере увеличения разме]
молекулы число таких интегралов возрастает примерно nponopi
онально N*, где N— размер базиса АО (см. табл. 4.3). Соотв(
венно этому растут время и стоимость расчета.
Принципиально возможны два различных подхода, напрг
ных на уменьшение вычислительных трудностей. Первый из
связан с пренебрежением части интегралов, входящих в выраяк
D.63), и сохранении во всем остальном схемы неэмпирическ*
расчета. Этот подход реализован в методе PRDDO (Рг
Retention of Diatomic Differential Overlap), в котором сохраняют
одно-, двух- и трехцентровые кулоновские интегралы и ^
и двухцентровые обменные интегралы. Фактически из всех JV* инт
ралов остаются JV3 интегралов. Метод PRDDO, предложен!
У. Липскомом** с сотрудниками в 1973 г., по точности лишь
много уступает ab initio расчетам в базисе STO-3G.
Другой подход, оказавшийся более перспективным, основан
замене большей части интегралов параметрами, взятыми из
перимента (потенциалами ионизации атомов в орбитальных вале!
ных состояниях и др.), и использовании различных приближе!
выражений, включающих эти параметры, для оценки интеграле
Основанные на этом подходе методы называются полуэмпиричс
кими.
*Поль Адриен Морис Дирак A902—1984) — выдающийся английский
теоретик, лауреат Нобелевской премии по физике 1933 г., создатель релятивис
теории движения электрона. П. Дирак предсказал существование позитрона.
**Уильям Нан Липском (W. N. Lipscomb) (род. 1919 г.) — американский
ный, широко известен своими работами в области исследования структуры "
дородов и изучения химической связи в них. Лауреат Нобелевской премии по
1976 г.
210
«7 3.1. Основные требования к полу эмпирическим методам
Почти все полуэмпирические методы, применяемые для расчета
молекул, являются методами валентного приближения, т. е. в от-
отличие от неэмпирических методов они учитывают только валентные
электроны и атомные орбитали валентных оболочек. Влияние нева-
невалентных (остовных) электронов неявно учитывается в эмпирических
параметрах.
Результаты полуэмпирических расчетов не могут передавать
достаточно точно одновременно все физические и химические
свойства молекул, так как подгонка параметров производится по
одному, реже по нескольким свойствам. В связи с этим возникают
различные параметризации методов, призванные удовлетворитель-
удовлетворительно описывать определенное свойство или группу свойств.
Основные требования, предъявляемые к упрощенным теорети-
теоретическим моделям квантовой химии, формулируются следующим об-
образом.
1. Полуэмпирические методы должны быть достаточно просты,
чтобы их можно было применять с помощью современных ЭВМ
для расчета больших молекул (число центров более 100, число
базисных функций более 250).
2. В методах, ориентированных на количественные оценки, не-
необходимо сохранить в гамильтониане основные взаимодействия,
например кулоновское отталкивание электронов, притяжение их
к ядрам и т. д.
3. Результаты расчетов должны быть легко интерпретируемы,
а также должны позволять построение качественных моделей и кон-
концепций, не включенных заранее в вычислительную схему.
4. Полуэмпирические методы должны по возможности с помо-
помощью параметризации компенсировать недостатки метода Хартри—
Фока, который не учитывает, например, электронную корреляцию,
энергию нулевых колебаний и т. д.
5. Результаты расчетов полуэмпирическими методами должны
быть инвариантны по отношению к ортогональным преобразовани-
преобразованиям атомных орбиталей (см. разд. 7.3.3).
7.3.2. Приближение нулевого
дифференциального перекрывания
Число интегралов кулоновского отталкивания электронов мож-
можно резко сократить, используя приближение нулевого дифференци-
дифференциального перекрывания (НДП), введенное впервые Р. Парром A952).
Это приближение, сыгравшее важную роль в становлении и раз-
развитии полуэмпирических методов, основано на том, что многие
интегралы кулоновского отталкивания электронов близки к нулю,
особенно те, которые включают в себя функции типа хД1)ХуA)
с #. Интегралы, содержащие произведения XnO-)X»W> как прави-
211
ло, существенно больше по значению. Поэтому предлагается
щение типа
т. е. предполагается, что орбитали Хщ и х* практически не nej
ваются в пространстве и
Интегралы перекрывания АО, входящих в уравнения
и участвующие в нормировке молекулярных орбиталей, также ш
гаются равными нулю для
Несколько нарушают стройность и логическую последовзд
ность приближения НДП остовные интегралы Я™, которые
быть равны" нулю, если справедливо равенство G.2). Оказ]
однако, что если положить их равными нулю и сохранить ш
вательность приближения G.2), то результаты расчетов весьма
удовлетворительны. В связи с этим интегралы Н^ считаются otj
ными от нуля и рассматриваются как варьируемые параме
определяющие параметризацию.
Приближение G.1) превращает четырехмерный массив
ралов (jUv | ко) в двумерный. Вместе с сокращением числа подле
щих расчету интегралов уменьшается также и время вычисле
одного интеграла, так как двухцентровые интегралы счит*
намного проще трех- и четырехцентровых. Для N= 10 (число
ных функций, относящихся к разным центрам) необходимо bi
лить только 45 двухцентровых интегралов, в то время как
число интегралов (ц\\Ха) равно при этом базисе 4595. ^
Приближение G.2) справедливо в базисе ортогональных ai
ных орбиталей. Переход от неортогонального базиса #, G=1, 2» Л
N) к ортогональному можно осуществить преобразованием
<=1
предложенным П. Левдином, где
Матрица S~ г определяется из условия
Задача 7.1. Получить ортогональный набор функций для #=2.
212
В табл. 7.5 приведены интегралы отталкивания молекулы эти
рассчитанные с ортогонализованными (ОАО) и неортогонали:
ванными АО. Легко заметить, что переход к ортогонализованно
базису почти не меняет интегралы типа (jifi | цц) и (цр | vv), в то вре
как интегралы (цр \ v<r), (jjuj \ /х<т) и (jut \ fiv) становятся очень близки
к нулю, что соответствует приближению НДП. Отметим, что инт
ралы по молекулярным орбиталям почти не изменяются при пе
ходе к ортогонализованному базису. Близость интеграл
/ и К в приближении НДП к их значениям в обычном баз]
в какой-то мере оправдывает это приближение.
Таблица 7.5. Значения интегралов перекрывания и двухэлектронвых интегра.
для этилена в различных базисах АО (pz) и соответствующих базисах МО
Базис
АО
МО
Интегралы
Si 2
(И |Н)
A1112)
A1|22)
A2|12)
Jn
Jn
Hi
Слэтеровсхие ор-
батали
0,277
16,93
9,26
3,58
1,08
13,08
13,01
13,44
4,16
НДП
0
16,93
0
0
13,09
13,09
13,09
3,84
ОАО
0
17,29
8,98
-0,09
-0,12
13,08
13,01
13,44
4,16
Если приближение НДП используется для всех пар атомн
орбиталей, то уравнения Рутаана D.62) примут вид
где элементы матрицы Фока записываются следующим образом
-
Отмечалось, что приближения, вводимые в метод Рутаана,
Должны отражаться на инвариантности полной волновой функи
к ортогональным преобразованиям базиса атомных орбиталей. Р
смотрим влияние приближения НДП на инвариантность полуз
Лирических методов к ортогональным преобразованиям базиса.
7.3.3. Инвариантность полуэмпирических методов
Уравнения Рутаана D.62) инвариантны к ортогональным nj
разованиям молекулярных орбиталей. Если имеются молекуля]
орбитали
N
(pi~ ? Ср
то ортогональным преобразованием можно перейти к другим
зисным функциям
причем
N
ч
где а^ — несингулярная квадратная матрица (ее определитель a$j
равен нулю).
Вычисления методом Рутаана с функциями G.9) и G.10) должнц;
приводить к одинаковым орбитальным энергиям и соответственен
к одинаковой полной энергии. Вместе с тем коэффициенты разложен
ния МО в ЛКАО ср и с'ш будут отличаться. Рассмотрим, каки|
преобразования могут описываться соотношением G.11). -Щ
1. Преобразование, смешивающее орбитали одного атоШ
с одинаковыми п и / и приводящее к их линейной комбинации^
Например, можно смешать 2px-t 2ру-, 2р.-АО или пять З^-функвд^'
Наиболее важным примером такого преобразования служит
поворот декартовой системы координат, выбранной для олрё;
деления АО.
2. Преобразования, которые позволяют смешивать любьц
атомные орбитали одного атома. Если смешиваются функции с рази-
ными /, то получаются гибридные АО. Например, если смешаем 2^i ¦
Хрх-ш 2ру-, 2рг-АО, то получим ?р3-гибридные АО. В этом случае*
Хн — гибридные базисные функции. ''*,
3. Преобразования, позволяющие смешивать функции разный
атомных центров. Эти трансформации ведут к неатомному базису.?
Примером такого преобразования может служить переход к лока* \
лизованным МО (см. разд. 10.2.1). .* ¦
Уравнения Рутаана инвариантны к рассмотренным преобразовав
ниям. Введение приближения G.2) нарушает инвариантность урв*^
нений. Рассмотрим условия, которые необходимо наложить на fflg
тегралы 0^1 vv), H^ и Н^ с тем чтобы восстановить инвариант*
214
ость Мы займемся только преобразованиями типа 1 и 2, преоб-
преобразования последнего вида рассматриваться не будут.
Дифференциальное перекрывание %цХ* может быть двухатом-
jtj^ и одноатомным в зависимости от того, принадлежат ор-
битали Хи и X* °ДНОМУ или разным атомам. Очевидно, что преоб-
преобразования типа 1 и 2 переводят двухатомное дифференциальное
перекрывание х?Ху в ДРУгое перекрывание %Л*- ^С1Ш мы пренеб-
пренебрегаем двухатомным дифференциальным перекрыванием для всех
пар атомов, т. е.
то отсюда следует, что и
Другими словами, пренебрежение двухатомным дифференциаль-
дифференциальным перекрыванием сохраняет инвариантность к преобразованиям
типа 1 и 2. Метод, в котором пренебрегают только двухатомным
дифференциальным перекрыванием, называют методом NDDO
(Neglecting of Diatomic Differential Overlap) или ПДДП (пренебреже-
(пренебрежение двухатомным дифференциальным перекрыванием).
Случай дифференциального перекрывания орбиталей, принад-
принадлежащих одному атому, более сложен. Рассмотрим двухцентровый
интеграл {рхру | .г), где рх и ру — орбитали одного атома. В прибли-
приближении НДП этот интеграл обращается в нуль. Повернем декартову
систему координат на угол 9 относительно оси z. Тогда можем
записать
G.12)
ру= —pxsin v+ру cos в.
В новой системе координат интеграл ipxpy\s2) отличен от нуля.
Действительно,
s2) = — cos в sin 6 (pi j s2) + cos 9 sin 6(p] \ s2) +
+ (cos20-sin2e)(pxPy\s2) = cos6sin6 [(py\s2)-(pl\s2)l G.13)
При получении окончательного вида было учтено, что функции
Рх и ру ортогональны. Из G.13) видно, что равенство обращается
в нуль только при 0 = 0,90°, т. е. оно несправедливо при любом в.
Итак, интегралы кулоновского отталкивания неинвариантны от-
относительно преобразований системы координат в том случае, если
°ни включают в себя перекрывание двух АО одного атома. Анало-
Аналогично можно показать, что они неинвариантны к гибридизацион-
ны изменениям.
215
Выход из создавшейся ситуации был предложен Д. Пошк
с сотр. в 1965 г., которые разработали метод CNDO (Comj
Neglecting of Differential Overlap) или ППДП (полное пренеб{
дифференциальным перекрыванием). Этот метод является наибов
разработанным полуэмпирическим методом, включающим как
так и я-электроны.
7.3.4. Метод CNDO
В методе CNDO учитываются только валентные элй
атомов, внутренние электроны включаются в неполяризовг
остов. Приближение НДП принимается для всех пар атомных
биталей, в том числе и для принадлежащих одному атому,
восстановления нарушаемой при этом инвариантности необхо;
предположить, что
(p)\s2)^(pl\^). G.1
В трехмерном случае имеем
(Л I *) = (p) \s2) = (p]\s1) = (рг | s2). G.1
Это соответствует предположению, что ^-функция имеет сферш
кую симметрию, аналогичную s-орбитали. Интеграл (рг\<
должен быть равен интегралу (J2\s2), где У — сферическая
биталь, имеющая ту же радиальную часть, что и р-АО.
Рассмотрение преобразований второго типа приводит к
бованию
(J2\s2)=(s2\s2). at!
Таким образом, для сохранения инвариантности при введении с|
ближения НДП необходимо выполнение условия
Истинное отталкивание между орбиталями заменяется cj
отталкиванием между электронами атомов А и В, уАВ вычис
со слэтеровскими j-функциями соответствующих атомов:
С учетом приближения G.17) матричные элементы
записать в виде
P+ ? РввУав,
B»*A
G.8) моз
'Джон А. Подл (род. 1925 г.) — американский физик, известный своими^
тами в области создания полуэмпирических методов квантовой химии
CNDO, INDO), а также серии программ GAUSSIAN для неэмпирических
216
F^Hr — Pry** /xeA, veB, G.19)
где ^вв — полная плотность валентных электронов на атоме В:
^- G.20)
Теперь рассмотрим вычисление матричных элементов оператора
остова [здесь и далее используется система атомных единиц (см.
приложение 1)]:
H=~V2-?VB, G.21)
1 в
где VB — потенциал, созданный ядром и внутренними электронами.
Диагональные элементы Нт можно представить в виде
G.22)
Величина Um является характеристикой орбитали ц атома А и пред-
представляет собой энергию электрона, находящегося на орбитали
\i свободного атома А. Ее значение обычно получают полуэм-
полуэмпирически, используя результаты спектральных исследований
атомов.
Интегралы (ji | VB| д) полагают равными Vab, т. е. считается, что
взаимодействие любого валентного электрона атома А с остовом
атома В одинаково. Отметим, что VAB может быть не равно VBA.
Интегралы Н^ для /х, veA обращаются в нуль. Для /xeA, veB
интегралы Н^ в приближении НДП обращаются в нуль. Однако,
положив их равными нулю, можно потерять основной вклад в кова-
лентную составляющую связи атомов. Поэтому, нарушая последо-
последовательность теории, для Hp, не будем применять приближение
НДП.
Запишем Ям„ в виде
G.23)
в
где второй член отражает взаимодействие электронного облака Хц X*
с атомом С. Суммой обычно пренебрегают, так как члены (p. | Vc | v)
217
относятся к трехцентровым взаимодействиям, которые малы,
тавшийся член считают эмпирическим параметром и ш
резонансным интегралом fi^. В методе CNDO предполагается,
величины Д„ пропорциональны интегралу перекрывания SMV (i
ближение Малликена):
Д»=А»—Рав Sfty,
где /?ав — параметр, характеризующий атомы А и В и не завис
от типа взаимодействующих орбиталей. Различные параметр!
метода CNDO отличаются в основном выбором параметра Дю.
Таким образом, инвариантность метода достигается ценой
каза от индивидуальности орбиталей и сведения электронного
пределения атомов к сферически-симметричному. Различие oj
лей будет проявляться в интегралах перекрывания и величинах UM
Суммируя сделанные приближения, матричные элементы G.1
можно представить в виде
G.
Диагональные элементы F^ можно переписать в виде, более у;
ном для анализа:
Здесь QB — заряд атома В:
— Оъ Уав — кулоновская энергия взаимодействия заряда атов
В с электроном атома А, для нейтрального атома В этот член ра
нулю. Величина (ZByAB— ^ав)> по предложению М. Гепперт-Майе$
и А. Скляра, названа интегралом проникновения.
Интеграл проникновения — это энергия кулоновского взаимо-,
действия (без обменных эффектов) электрона атома А с нейтраль-
нейтральным атомом В. Вычисления показывают, что эта энергия достаточ-
*Марня Гепперт-Майер A906—1972 г.) — выдающийся американский
лауреат Нобелевской премии по физике A963); основные работы сделаны в
строения ядра. Ею совместно со Скляром выполнен первый расчет методом
органической молекулы — бензола A938).
218
о мала, и интегралами проникновения обычно пренебрегают. От-
в иМ только, что для заряженных систем величины интегралов
ооникновения достаточно значительны и пренебрегать ими не
всегда корректно.
7.3.4.1. ПАРАМЕТРИЗАЦИЯ CNDO/2
Существует несколько параметризаций метода CNDO, исполь-
используемых для расчетов различных характеристик. Главные из них —
параметризации CND0/2 для расчетов основных и CNDO/S для
расчетов возбужденных электронных состояний.
В методе CNDO параметры $& зависят только от типа соседних
атомов, а не от орбиталей и находятся как полусумма величин:
4 G.28)
Величины /?д подбирают так, чтобы рассчитанные методом CNDO
разности орбитальных энергий (?,— ?,) и коэффициенты разложения
МО в ЛКАО наилучшим образом совпадали с результатами неэм-
неэмпирических вычислений, проведенных с теми же базисными функци-
функциями. Приведем используемые в методе CNDO значения ^:
Атом... Н Li Be В С N О F
-^, эВ ... 9 9 13 17 21 25 31 39
Величину Кдв оценивают по формуле
G.29)
Так как (ZB}>ab— Ую) — интеграл проникновения, то оценка G.29)
соответствует пренебрежению интегралами проникновения. Значе-
Значение Um можно найти из выражения, описывающего энергетику
потери электрона атомной орбиталью:
1,=Е+ -?= - Um- (ZA-1) Vaa, G.30)
где Е+ — энергия катиона атома А; Е — энергия нейтрального
атома; ZA — заряд остова атома А.
Потенциалы ионизации /м находятся из спектроскопических дан-
данных и известны для большинства элементов таблицы Д. И. Мен-
Менделеева. В методе CNDO, как и в других всевалентных методах,
валентными состояниями являются негибридизированные s-t p~,
а-состояния. В то же время в молекуле заведомо найдутся АО,
которые обладают избыточной электронной плотностью по сравне-
сравнению со свободным атомом. Поэтому для таких АО U^ нужно
находить из формулы
219
-Л=^ + 2аУаа,
где Ац — сродство к электрону орбитали р. Чтобы иметь воз
ность одновременно удовлетворительно описывать как потерю,
и приобретение орбиталями электронов, естественно ус
оценки G.30) и G.31), т. е.
f Za—Ьм-
¦•и
Числовые значения -
ода приведены ниже (эВ):
для различных атомов второго ш
Атом....
Н
Li
Be В
N
7,176 3,106 5,946 9,594 14,051 19,316 25,390 32,272
— 1,258 2,563 4,001 5,572 7,275 9,11 11,080"
С учетом G.29) и G.3) диагональные матричные элементы G,
приобретают вид
(hAJ UPZ)~
-ZA)-~(Р,щ-\))уАА
Параметризация метода CNDO, в которой V^s, и ?/да находят \
G.29) и G.30), является наиболее распространенной, ее наз*
CNDO/2. Недиагональные матричные элементы F^ в схс
CNDO/2 находят по формуле
1 ~ ¦ . G.33
Полная энергия в методе CNDO/2 может быть записана в
суммы атомных ?"А и двухатомных ЕАВ членов:
где
А А< В
и ?дв можно представить как
220
fie A veB
veB
G-36)
Областью, где метод CNDO/2 дает наиболее надежные резуль-
результаты, является расчет электронного распределения и свойств, кото-
которые им определяются, например дшюльных моментов. Длины ва-
валентных связей в методе CNDO/2 занижены в среднем на 10%.
Особенно чувствительно занижены длины гипервалентных связей,
таких, как аксиальные связи в тригонально-бипирамидальных
структурах переходных состояний ^2-реакций. Это обстоятельство
отражает известный недостаток метода, сильно сказывающийся на
характере получаемых при его помощи ППЭ, — переоценку (при-
(примерно на 20%) ковалентного связывания. Этот и ряд других недо-
недостатков метода (недооценка энергии сопряжения и переоценка
устойчивости мостиковых структур) вызваны приближением НДП
и трудно устраняются репараметризацией.
Так, например, в отличие от неэмпирических методов расчета,
которые в полном согласии с экспериментальными данными пред-
предсказывают устойчивость протонированного бензола в форме <т-ком-
плекса V, расчет по методу CNDO/2 отдает значительное предпоч-
предпочтение мостиковому изомеру VI, в котором протон связан с обоими
атомами углерода я-связи:
STO ЗГ.
4 31G
CNDO/2
V
0
0
184кДж/моль
116
86
0
VI
кДж/моль
кДж/моль
С другой стороны, описание при помощи метода CNDO/2 реак-
реакций, в которых движущей силой выступает не ковалентное связыва-
связывание, а электростатические взаимодействия, является достаточно
точным, так как этот вид взаимодействий регулируется характером
электронного распределения. С этим связаны хорошее воспроиз-
воспроизведение методом CNDO/2 систем с водородной связью и реакций
протонирования гетероатомных соединений, где ход реакции опре-
определяется видом электростатического потенциала. Так, рис. 7.1 пока-
221
Рис. 7.1. Сравнение карт электростатического потенциала молекулы 2-формш
ноадетамида, полученных в расчетах ab initio (базис 4-31G) (а) и CNDO/2 (б):
электростатический потенциал V(r) в точке пространства с координатой i\ вычисляется
где />{/¦) — электронная плотность молекулы в точке с координатой г; za, Ra — заряд ядра
с координатой Я?; Ь(г —R а) — дельта-функция Дирака; Л^ — число атомов в молекуле ^'
зывает на примере модельного пептида, насколько удачно метсш
CNDO/2 передает картину электростатического потенциала.
Таблица 7.6.
Соединение
LiH
ВеН
СН
ОН
FH
СО
Н2О
NH3
HCN
Метанол
Формальдегид
Фтороформ
Нитробензол
Дниольные моменты молекул,
CNDO/2
-6,16
-0,67
1,87
1,78
1,86
-0,64
2,10
1,97
2,48
1,94
1,98
1,66
5,33
D
ab initio
-6,002
-0,282
1,773
1,780
1,942
-0,077
1,997
1,760
2,110
—
1,006
—
*
Эксперимент \\
-5,882
— ¦ ф
1,460
1,660
1,819
-0,112 ..&
1,846 .,
1,468 !'f
2,986 '
1,690 ¦-«'
2,339 ..".*
4,280 ;1
222
В табл. 7.6 проведено сравнение с экспериментом результатов
асчетов дипольных моментов некоторых молекул методами
CKDO/2 и ab initio. Результаты расчетов по методу CNDO/2 и ab
jnxtio близки и согласуются с экспериментальными значениями.
Следует отметить, что точность расчетов дипольных моментов
в значительной степени зависит от правильного выбора молекуляр-
молекулярной геометрии. В отсутствие точных геометрических данных можно
использовать усредненные для ряда соединений валентные углы
и длины связей.
7.3.4.2. РАСЧЕТЫ ЭЛЕКТРОННЫХ СПЕКТРОВ.
ПАРАМЕТРИЗАЦИЯ CNDO/S
Расчеты электронных спектров в параметризации CNDO/2 с уче-
учетом однократно возбужденных конфигураций приводят к плохому
согласию с наблюдаемыми величинами энергий переходов. Расхож-
Расхождение составляет ~ 1,5—2 эВ A2000—16000 см). Такая ошибка
в вычислении энергий перехода делает непригодным метод CNDO/2
для интерпретации электронных спектров, в то время как геомет-
геометрическая структура возбужденных состояний (пример СН2) может
описываться вполне удовлетворительно. Этот результат — прямое
следствие параметризации резонансных интегралов Д„ под свойства
основных состояний молекул. Для изучения электронных спектров
Дель Бене и Джаффе предложили спектроскопическую парамет-
параметризацию метода CNDO. Этот вариант метода назван CNDO/S.
Резонансные интегралы в схеме CNDO/S разделяются на две
группы /?JV и /?Jv, соответственно отвечающие связыванию о и п АО.
Вычисляют же их по формулам
G.37)
G.38)
где К(К<\) —параметр, уменьшающий /?* по сравнению с
Обычно К= 0,585.
Числовые значения /?д для некоторых атомов приведены в табл.
7.7, там же имеются значения одноцентровых кулоновских интег-
интегралов отталкивания удд. Двухцентровые интегралы находят по
формулам Матага—Нишимото или Оно [см. ниже соотношения
G.66) и G.67)]. Отметим, что параметризация метода CNDO/S
близка к параметризации метода ППП, применяемой для расчета
свойств электронно-возбужденных состояний (см. разд. 7.5.1.1).
Аналогична и схема расчета волновых функций возбужденных со-
состояний и энергий спектральных переходов. В методе CNDO/S
Рассматриваются только однократно возбужденные состояния. Точ-
223
ность метода CNDO/S в определении энергий спектральных перех<
дов п—я*-типа несколько хуже соответствующей точности мстгм
ППП. Преимуществом метода CNDO/S по сравнению с мето;
ППП является его способность рассчитывать энергии п—я*-ш
дов. В табл. 7.8 иллюстрируется точность метода CNDO/S.
Таблица 7.7.
Значешн
Атом
Н
С
N
О
Таблица 7.8.
Соединение
Бензол
Пиридин
Фуран
Фурфурол
Расчеты
Уаа Для векоторых атомов
«.Л
-12,0
-17,0
-26,0
-45,0
спектров методом
Расчет
энергияj
ходлЕ,
4,7
5,2
6,9
4.3
4,8
5,4
7,1
5,2
5,8
7,3
3,2
4,9
5,7
аере-
эВ
сила осцил-
осциллятора/
0,000
0,000
0,593
0,000
0,017
0,002
0,473
0,078
0,009
0,368
0,000
0,158
0,002
CNDO/S
i
УАА,эВ
12,85
11,11
12,01
13,00
Эксперимент
энергия пере-
перехода Е, эВ
4,7
6,1
6,9
4,3
4,8
6,2
7,0
5,9
6,5
7,4
3,5
4,6
6,2
сила осцил-
осциллятора /
0,001
0,100
0,690
0,016
0,030
0,200
1,300
0,12
0,08
—
—
—
—
ПрироЗР
Р**"Л
я—id
*-*¦
я—**
/1-Я**
я—&
я—жЛ
Я—JCT
я—я*
*-*¦
я-**
я-**
t • 1
Существует еще целый ряд параметризаций метода CNDO,
чей которых является точное описание того или иного отдельн<
свойства молекулы. Так, имеются параметризации для изуч<
силовых констант, инверсионных барьеров, барьеров вращс
теплот образования углеводородов и т. д.
7.3.5. Методы INDO, MINDO, MNDO.
7.3.5.1. ПАРАМЕТРИЗАЦИЯ МЕТОДА INDO
Метод INDO (Intermediate Neglecting of Differential Overlap),
ЧПДП (частичное пренебрежение дифференциальным перекр!
ем), занимает промежуточное по сложности и времени вычисли
положение между методами NDDO и CNDO.
224
Недостаток метода CNDO — пренебрежение отличием в куло-
отталкивании электронов с параллельными и антипарал-
рпьными спинами. Это отличие особенно велико для электронов
одного атома, в этом случае двухэлектронный обменный интеграл
(uv\ Mv) М> vei^ определяет разницу в энергии взаимодействия элект-
оонов в синглетном и триплетном состояниях. В методе CNDO эти
интегралы полагают равными нулю, вследствие чего этот метод не
может даже качественно воспроизвести правило Гунда, согласно
которому два электрона на различных орбиталях одного атома
отталкиваются слабее в случае параллельности их спинов. Метод
CNDO плохо работает в случае триплетных состояний, свободных
радикалов, т. е. для молекулярных систем с достаточно большой
обменной энергией.
В методе INDO в значительной мере устраняются эти недостатки
сохранением только одноцентровых обменных интегралов (fiv\fiv).
Матричные элементы оператора Фока в методе INDO для закрытой
оболочки в пренебрежении интегралами проникновения имеют вид
ХеА; G-39)
а с ! d
у - Рав *V—^ */
I
3 1
P(\)P
Метод INDO имеет преимущества перед CNDO при расчете
электронной структуры молекул с открытыми оболочками (*S#0).
Для закрытых оболочек (см. разд. 4.3.3) результаты расчетов мето-
методом CNDO более предпочтительны и требуют меньших затрат
машинного времени.
Методы CNDO и INDO плохо воспроизводят теплоты атомиза-
ции и орбитальные энергии, в связи с этим они неприменимы для
изучения относительной устойчивости различных молекулярных
структур, а следовательно, для построения поверхностей потенци-
потенциальной энергии и изучения механизмов реакций.
73.5.2. МЕТОД MINDO/3
М. Дьюар* модифицировал метод INDO применительно к рас-
чету свойств основных состояний молекул, введя эмпирическую
*Майкл Дж. С. Дьюар (род. 1918 г.) — крупнейший американский химик, вне-
внешни значительный вклад в разработку ряда теоретических проблем органической
имин. Бескомпромиссный сторонник применения полуэмпирических методов рас-
расчета молекул.
8- Теория строения молекул 225
оценку некоторых кулоновских, а также остовных интегралов с
чтобы получить правильные значения теплот образования и п
рии молекул. Этот метод назван MINDO (Modified INDO),
МЧПДП (модификация частичного пренебрежения диффере
ным перекрыванием).
Параметры метода MINDO отличаются от схемы INDO
нием ряда эмпирических зависимостей для интегралов
энергии отталкивания остовов, выбираемых так, чтобы д<
наилучшего согласия рассчитанных и эксперимента]
ных значений теплот образования и геометрических
рактеристик широкого класса стандартных соединены]
Двухэлектронные кулоновские интегралы удв вычисляют
формуле
Резонансный интеграл /^ рассчитывают в приближении
ликена с добавлением эмпирического безразмерного мноз
Gab, характеризующего типы рассматриваемых атомов (связь
где А, В — типы атомов; ц, v — индексы орбиталей;
Gab — параметр, зависящий от типа атомов А и В.
Энергию отталкивания остовов оценивают по формуле
где адв — эмпирический параметр, характеризующий типы
триваемых атомов.
Форма выражения G.42) определена тем, что при малых
атомных расстояниях энергия отталкивания остовов хорошо
проксимируется взаимодействием точечных зарядов:
=1 Т.-
A<B ^AB
а при больших Лав — выражением
А <В
Существуют различные параметризации метода MINDO,
ибольшее распространение получила параметризация MINDO/3^
226
Таблица 7.9. Теплоты образования м геометрические характеристик* некоторых
молекул
Молекул*
¦
СНз-СНз
н2с=сн2
нс=сн
н2с=с=сн2
[
4
1
4 3
<>
1
ДЯ/, кДж/моль
расчетная
-83,1
80,6
242,7
176,4
244,0
143,2
-33,6
эксперимен-
экспериментальная
-84,8
52,0
228,0
192,7
298,5
145,3
-34,8
Геометрия, нм в град
Углы и связи
се
сн
Z.CCH
се
сн
LCCH
се
сн
ее
сн
Z.HCC
с=с
с-с
сн
Z.HCC
¦ . о
CN
С2С3
со
С2С3
С3С4
расчетная
0,1486
0,1108
112,8°
0,1308
0,1098
124,8°
0,1191
0,1076
0,1311
0,1099
118,4°
0,1344
0,1466
0,1109
115,7°
148,3°
0,1335
0,1407
0,1406
0,1343
0,1367
0,1455
эксперимен-
экспериментальная
0,1532
0,1107
111,1°
0,1336
0,1103
121,6°
0,1205
0,1059
0,1308
0,1087
118,2°
0,1340
0,1476
0,1100
117,6°
136,9°
0,1340
0,1395
0,1394
0,1371
0,1357
0,1440
В табл. 7.9 представлены вычисленные методом MINDO/3 и со-
сопоставленные с экспериментальными данными теплоты образова-
образования* и геометрические характеристики некоторых молекул. В схеме
* Теплота образования молекулы ?Лг связана с полной энергией молекулы
формулой
N
t-1
гДе &Hf(i) — теплота образования i-го атома в кДж/моль; ?щ — электронная энер-
энергия молекулы; Е^ — энергия отталкивания остовов; E(i) —электронная энергия
'-го свободного атома; N — число атомов в молекуле; AH/(i) и E(i) — табличные
значения.
227
метода MINDO/З предусмотрена оптимизация геометрии moj
лы с помощью градиентного спуска в энергетический мш
Метод достаточно хорошо воспроизводит кривизну ППЭ в облг
минимумов, что позволяет с удовлетворительной точностью pi
сматривать силовые постоянные и частоты нормальных колебг
молекул. В связи с этим метод MINDO/З широко применяют
построения поверхностей потенциальной энергии и исследовг
механизмов реакции. В то же время метод обладает некотор!
недостатками, которые сужают область его применения.
1. Валентные углы, получаемые из расчета, завышены в ере;
на 6—8°. Особенно преувеличены значения углов при дво!
связях, например в случае 1,3-бутадиена угол ССС получается
ным 131°, тогда как экспериментальное значение 123,6°. Накоплен
таких ошибок в соединениях типа VIII приводит к тому, что расстоЗ!
r = н, сосн3
X = О, NH
VJI
VIII
яние X ... X увеличивается на 0,1—0,15 нм. В связи с этим не только
резко увеличивается барьер миграции VII ^ VIII, но может менять*
ся и механизм реакции.
2. Сильно переоценивается стабильность малых циклов, особен-
особенно содержащих два соседних гетероатома.
3. Как и в методе CNDO/2, переоценивается устойчивость
трехцентровых двухэлектронных связей и сильно занижается их
длина (табл. 7.10).
Таблица 7.10. Длины аксиальных ¦ экваториальных связей (им), pacoum
в переходных состояниях (X) реакций Н~ + СН4 и F +CH3F
Метод, базис
орбиталей
CNDO/2
MINDO/3
PRDDO
STO-3G
3-21G
4-31G
6-31G*
Н .
снакс
0,113
0,1140
0,154
0,1481
0,1702
0,1730
0,1715
.. СНз ... Н
СИ»,
0,121
0,1225
0,110
0,1090
0,1061
0,1059
0,1062
F
CF«
0,1439
—
—
—
0,1776
0,1830
0,1820
... СНз ••• F
CF«,
0,1158 ;
—
—-
—
0,1062
0,1060
0,1061
4. Не воспроизводится энергия стабилизации таких димеров,
(Н2ОJ, СН3ОН, ..., NH3 и ДР> и> следовательно, метод непригоден
228
описания систем с водородной связью. Этот недостаток, одна-
ДЛЯустраняется модификациями параметров.
к0' ? Метод не дает точных результатов при описании соединений,
ержащих соседние гетероатомы с неподеленными парами
С°ектронов, борорганических соединений. Будучи параметри-
ванным по эталонным органическим соединениям с фик-
Зиоованной валентностью образующих их атомов, метод, как
°оавило, недооценивает устойчивость неклассических каркасных
и полиэдрических структур. В качестве примера для сравнения
оезультатов, получаемых различными методами, рассмотрим
реакцию 5лг2-нуклеофильного замещения у тетраэдрического
атома углерода. Реакции SN2 сопровождаются обращением
конфигурации у тетраэдрического атома углерода (вальденовское
обращение, 1895 г.). Этот результат — основа прочно уста-
установившихся взглядов о концертном синхронном механизме,
связанном с тыловым подходом нуклеофила Y к рвущейся
связи С—X и с возникновением тригонально-бипирамидального
состояния:
R
XI
Энергетическая предпочтительность такого пути реакции по сра-
сравнению с любыми другими возможностями подтверждается всеми
как полуэмпирическими CNDO/2, INDO, MIND0/3, так и неэм-
неэмпирическими расчетами.
Особый интерес вызывает структура переходного состояния X.
Как полуэмпирические, так и неэмпирические методы расчета свиде-
свидетельствуют о существенном удлинении аксиальных связей по срав-
сравнению с экваториальным. В силу ограничений, связанных с пренеб-
пренебрежением дифференциальным перекрыванием (см. разд. 7.3.2), полу-
полуэмпирические методы CNDO/2 и MINDO/3 сильно недооценивают
этот эффект. В полной мере его величина воспроизводится в неэм-
неэмпирических расчетах с применением расширенного базиса орбита-
леи. Результаты некоторых важных расчетов представлены в табл.
Как успехи, так и недостатки метода MINDO/3 стимулировали
Работы по дальнейшему совершенствованию полуэмпирических ме-
методов. М. Дьюар с сотр. A977) предложили метод MNDO.
229
7.3.5.3. МЕТОДЫ MNDO И AMI
Метод MNDO (Modified Neglecting of Diatomic Overlap) осн<
на более строгом приближении NDDO (Neglecting of Diat
Differential Overlap), или МПДП, в котором учитываются
ралы межэлектронного отталкивания, включающие одноцещ
перекрывания. Интегралы (/i/i|w) между любыми орбит*
атомах А и В рассчитываются для соответствующих
X? и Zv» a ые аппроксимируются соответствующими инте!
5-орбиталей. Важным преимуществом метода MNDO по сравнс
с MINDO/З является отказ от параметризации резонансного
рала Рр по связевому типу G.41) и переход к соотношению
о ¦ ¦ ¦ G;
В результате, хотя количество разных типов интегралов меэ
ронного взаимодействия в методе MNDO значительно возрг
общее число параметров для элементов, включающих второй ш
од, сокращается со 102 до 41. В отличие от метода MIND 0/3
MNDO легко параметризовать для элементов низших периоде
Затраты времени ЭВМ при использовании метода MNDO возрас
ют по сравнению с методом MINDO/З в ~ 1,5 раза. Хотя в мето/
MNDO и преодолеваются некоторые недостатки метода MIND0/
тем не менее он также непригоден для описания систем с водо]
ной связью, кроме того, метод MNDO по сравнению со схеме
MINDO/З имеет тенденцию слишком завышать рассчитанные
чины активационных барьеров реакций. С целью попытки ус
ния этих недостатков метода MNDO после нескольких лет инт
сивных усилий Дьюар с сотр. A985) предложили новый пол]
лирический метод АМ-1 (Austin Model 1), основанный, так же
и MNDO-метод, на приближении NDDO. В методе АМ-1 6i
модифицирована функция, описывающая отталкивание octoi
и соответственно введены новые параметры.
Таблица 7.11. Средние отклонения расчетных длин, полученных методами
MNDO, MINDO/З и АМ-1, от экспериментальных значений для 200 ее
содержащих атомы С, Н, О, N
Метод
MINDO/3
MNDO
АМ-1
Теплота образо-
образования, кДж/моль
46
27
24
Длина связи, им
0,0022
0,0014
0,0012
Валентный угол,
град
5,6°
2,8°
2,Г
Колебател!
частоты
5%
Как видно из табл. 7.11, составленной на основании
приводимых Дьюаром для более чем 200 соединений, в мете
АМ-1 и MNDO достигается лучшее согласие с экспериментом,
в методе MINDO/З, и в то же время метод АМ-1 воспроизвел
230
ородную связь и дает лучшие результаты для активационных
Параметров, чем метод MNDO.
Прежде чем перейти к рассмотрению более простых методов
оасчета, проведем сравнение затрат машинного времени, необходи-
необходимого для расчета молекулы пропана различными всевалевтными
методами. В табл. 7.12 приведено число интегралов кулоновского
взаимодействия, необходимых для расчета молекулы пропана мето-
методом Рутаана, методами NDDO, CNDO, INDO.
Таблица 7.12. Число интегралов кулововского взаимодействия в молекуле
пропана, возникающих в различных методах расчета
Интегралы
Одноцентров ые
Двухцентроэые
Трех- и четырехцент-
ровые
Общее число
Относительное время
расчета
Метод
CNDO
11
55
0
66
1
Метод
INDO (MINDO)
26
55
0
81
1,2
Метод
NDDO
(MNDO, AM-1)
173
568
0
741
11
Метод Рутаава
(минимальный ба-
базис STO)
368
6652
31206
38226
1
580
В этой же таблице приведено также относительное время расчета
различными методами, если считать, что время расчета пропорци-
пропорционально числу интегралов (это заведомо заниженная оценка, так как
с ростом базиса увеличивается и время диагонализации матриц,
и число циклов, необходимых для проведения самосогласования).
Для более крупных молекулярных систем разница во времени счета
становится еще значительнее.
7.3.6. Расширенный метод Хюккеля (РМХ)
В зарубежной литературе РМХ называют ЕНТ или ЕНМО
(Extended Huckel Theory или Extended Huckel МО). Рассмотренные
выше полуэмпирические методы (NDDO, INDO, MINDO/3,
MNDO) основаны на достаточно строгих теоретических схемах,
учитывающих разные типы электронных взаимодействий. Эти ме-
методы ориентированы на воспроизведение количественных харак-
характеристик молекул. Другая группа полуэмпирических методов имеет
Целью получение результатов преимущественно качественного ха-
характера, значение которых состоит не в числовом выражении,
а в определении последовательностей и формы МО. В этих методах
ренебрегают всеми интегралами кулоновского отталкивания
стремятся компенсировать грубость этого приближения соответ-
соответствующей параметризацией остовных и резонансных интегралов,
ренебрежение интегралами кулоновского отталкивания можно
231
в какой-то степени оправдать, если отталкивание ядер компещ
ется усредненным отталкиванием электронов:
cobi
G3
Если все (ду|Я<т)=О, то матричные элементы
с матричными элементами остова Н^:
F —И
Уравнения Рутаана G.62) примут вид
O, /=1, 2, З,... ;
¦О- G-
Остовные интегралы Н^ не зависят от порядков связи
а следовательно, отпадает необходимость в проведении npoi
самосогласования, которая в основном лимитирует время и
вычислений в полуэмпирических методах. Для нахождения энер!
е, и коэффициентов civ достаточно один раз решить уравнения GJ
и G.49).
Уравнения G.48), G.49) лежат в основе РМХ, введенного в л]
тику Р. Хоффманом* A963). В этом методе МО строятся
линейные комбинации валентных АО (STO) всех атомов. Ин1
ралы перекрывания S^ вычисляют исходя из задаваемой геомс
молекулы. Экспоненты радиальных частей могут выбираться?'
различных источников (см. разд. 3.4). Поскольку интегралы Sp,
висят от расстояния между атомными центрами и их относит
ориентации, в РМХ в явном виде включается зависимость
гетических характеристик и электронных распределений от мо
лярной геометрии. Матричные элементы Н^ напротив, не вычи
ются, а заменяются эмпирическими параметрами или аппрокс!
руются специально подобранными соотношениями, включаки
эти параметры. Так, диагональные матричные элементы Нт noj
ют равными потенциалам ионизации соответствующих валенп
электронов, взятых с обратным знаком:
Нт= —/д. G»
Для вычисления недиагональных матричных элементов Щ
ложено несколько формул:
*Роальд Хоффмая (род. 1937 г.) — американский химик, внесший
вклад в теоретическую органическую химию, химию металлооргааическнх и
динациояных соединений. Совместно с Р. Вудвордом сформулировал
хранения орбитальной симметрии в химических реакциях. Лауреат Не
премии по химии 1931 г. Иностранный член Российской АН.
232
формула Вольфсбсрга — Гельмголыда
= 0,5 К (H
^
Гяе обычно К= 1,75;
2) формула Кьюсака
Я^=0,5 B - |5У ) 5^ Г#*,+Л
3) формула Бальхаузена — Грея
G.51)
G.52)
^ %. G.53)
Ионизационные потенциалы валентных электронов /^ выбирают
из экспериментальных данных. Для некоторых элементов Пери-
Периодической системы Д. И. Менделеева ионизационные потенциалы
валентных электронов приведены в табл. 7.13.
Таблица 7.13. Ионизационные иотенцналы валентных состояний электронов для
некоторых атомов второго ¦ третьего иернодов (эВ)
Атом
Н
Li
Be
В
С
N
j-оболоч-
ка
13,60
5,34
8,41
13,45
21,43
27,50
/>-оболоч-
ка
—
—
8,43
11,42
14,49
</-оболоч-
ка
—
—
—
—
—
Атом
О
F
Si
Р
S
j-оболоч-
га
35,30
39,00
14,68
20,20
20,08
—
/>-оболоч-
ка
17,28
18 ДО
8,08
12,49
13,32
—
^-оболоч-
^-оболочка
1,79
2,50
2,01
——
Полная энергия молекулы с заполненной электронной оболоч-
оболочкой вычисляется как удвоенная сумма энергий занятых МО:
эан
G.54)
1=1
Задача 7.2. Показать, что полная энергия в РМХ может быть вычислена по
формуле Е=2
зан
зал
Несмотря на сделанные при выводе основных уравнений РМХ
рьезные упрощающие допущения, метод имеет вполне определен-
определение обоснование (Г. Аллен, Ч. Коулсон и сотр.)
все м°лекул, имеющих равномерное распределение зарядов по
1отм атомам, т. е. для молекул, атомы которых несильно отлича-
я по электроотрицательности (обычно принимают различие не
233
с:
60'
wo" то" wo" 60' loo* шо* /во*
н-В-нуго/! F-0-Fyeo/r
Рис. 7.2. Сравнение расчетов полной энергии молекулы ВН2 и F2O в зависимс
валентного угла по РМХ и методу ab initio:
разность эл«троотрицательностей Дх = 0.1| * Д™ F и О Az=0,5
более 1,4 по шкале Полинга), матричные элементы можно
ставить в виде
const S,
ftV
G.
В этом случае энергия электростатического отталкивания
[третий член в формуле D.60)] примерно равна энергии межэл*
ронного взаимодействия [второй член в D.60)] и полная энер!
молекулы D.60) хорошо аппроксимируется суммой орбитальи
энергий всех электронов. Тогда кривая полной энергии молекул
D.60) почти «параллельна» кривой удвоенной суммы орбитальр1
энергий G.54), что иллюстрирует рис. 7.2. Следовательно, отнг
тельные энергии (например, конформационные энергии) ДО^
удовлетворительно соответствовать значениям Е, вычя*-па<
в согласии с формулой G.54).
Если же в молекуле атомы сильно отличаются по электро<
тельности и распределение электронного заряда неравномерно,
энергии межэлектронного и межъядерного взаимодействия ре
отличаются, а полную энергию нельзя аппроксимировать формуд
G.54); в этом случае РМХ часто дает неверные результаты (рис. П
Для молекул Н2О, Li2O, UOH и FOH, у которых разно!
234
-89,6 \-
-89,1 \-
«V.
4
Л-3.8
-*тооотрицательностей свя- ._ _ | x77 }-3.3
3 между собой атомов
1,4, результаты расче-
РМХ значительно отлича-
^7ся от величин, предсказы-
предсказываемых методом ab imtio, од-
однако для остальных молекул
согласие приемлемое.
Вследствие неучета в яв-
явном виде межэлектронного
и межъядерного взаимодейст-
взаимодействия РМХ плохо предсказыва-
предсказывает равновесные длины связей
даже для молекул с равномер-
равномерным распределением заряда
по атомам, поэтому для оп-
оптимизации длин связей РМХ
практически не применяется.
Для молекул с неравно-
неравномерным распределением заря-
заряда РМХ приводит к сущест-
существенным ошибкам и в опреде-
определении геометрической конфи-
конфигурации. Так, для молекулы
аммиака РМХ предсказывает
в качестве устойчивой не пирамидальную, а плоскую форму. В то
же время РМХ, несомненно, весьма быстрое и экономное средство
для оценки относительной устойчивости конформеров молекул со
слабо поляризованными связями. Так, РМХ правильно воспроиз-
воспроизводит относительные энергии этана и циклогексана, зигзагообраз-
зигзагообразную структуру полимерной цепи полиэтилена. Кроме того,
чрезвычайно важной положительной стороной РМХ является до-
довольно точное воспроизведение им относительного порядка энер-
энергетических уровней и строения МО молекул со слабой поляризацией
связей, что иллюстрирует рис. 7.4. Это делает РМХ важным сред-
средством качественного анализа структурных эффектов, основанного
на изучении МО молекул, их реакции на геометрическое искажение
и введение заместителей.
100Л МО* 180*
Li-O- Li угол
Рис. 7.3. Сравнение расчетов полной энер-
энергии молекул УгО в зависимости от валент-
валентного угла по РМХ и методу ab initio. Раз-
Разность электроотрицательностей Li и О
Задача 7.3. Рассчитать по РМХ электронное строение гипотетической мо-
молекулы НН*, где Н* — атом «водорода» с потенциалом ионизации 20 эВ и ор-
орбитальной экспонентом \J2(RH_H*=0,l им).
Табл. 7.14 позволяет судить об области применимости различ-
различных полуэмпирических всевалентных ССП МО для изучения физи-
к°-химических свойств молекул.
235
7a ab initio
7 a PMX
2е ab initio
2е РМХ
V
Рис. 7.4. Сравнение связывающих молекулярных орбиталей Та и 2е этильного
она (а) и заслоненного этана (б), полученных методами ab initio и РМХ (пу»
обозначены области отрицательных значений МО)
7.4. АНАЛИЗ ЭЛЕКТРОННЫХ ЗАСЕЛЕННОСТЕЙ
АТОМОВ И СВЯЗЕЙ В МОЛЕКУЛАХ
Электронная плотность р (?) — функция, характеризующая
роятность электронного распределения, — определяется таким
разом, что р (?) d'f — вероятность нахождения электрона в мг
элементе объема d~? близ точки ~г'. Эта функция нормирО1
так, что
-н
I
-СО
G-
где п — полное число электронов в молекуле.
236
Таблица 7.14. Сравнительная характеристика всевалентных полуэмшцшческнх
методов
Метод
CNDO/2
CNDO/S
INDO
MINDO/3
MNDO
АМ-1
Параметризуемое
свойство
Электронная
плотность
Спектр
Спиновые
плотности
Потенциал
атом-атомного вза-
взаимодействия
Теплоты обра-
образования
Теплоты обра-
образования
Хорошо воспроизводи-
воспроизводимые свойства
Дипольные мо-
моменты, длины связей,
валентные углы, си-
силовые константы,
ЯМР корреляции
Спектр
Спиновые плот-
плотности, константы
СТВ, геометрия
Теплоты образо-
образования, потенциалы
ионизации. Длины
связей
Теплоты образо-
образования, геометрия
Теплоты образо-
образования, геометрия
Плохо воспроизводимые
свойства
Теплоты образо-
образования, потенциал ио-
ионизации, электронное
сродство, спектр
Теплоты образо-
образования, геометрия
Теплоты образо-
образования, потенциалы
ионизации, элект-
электронное сродство,
спектр
Спектр, водород-
водородная связь
Спектр, водород-
водородная связь
Спектр
Для анализа электронных распределений в рамках принятых в хи-
химии понятий (атомы, связи) необходимо выбрать схему разбиения
электронной плотности на отдельные вклады атомов, связей. По-
Поскольку эти понятия в молекуле теряют строгий смысл (см. разд.
5.6), разложение G.56) на компоненты не является однозначным.
Наиболее удобная схема такого разложения предложена Маллике-
ном, ее называют анализом заселенностей по Малликену.
Рассмотрим нормированную МО <р( двухатомной молекулы АВ:
ьХь, где деА, veB.
Возведем <р, в квадрат и проинтегрируем по всему пространству:
с
Умножив обе части этого равенства на п (i)-e число электронов на
i-Й МО [п (i) = 0,1 или 2], получаем соотношение
G.57)
в котором выражение первой скобки представляет собой элект-
электронную плотность на атоме А, вносимую i-й МО, а выражение во
второй скобке — на атоме В.
237
Величину n(iJciVicblS^ называют заселенностью перекрывt
Этот член не учитывается в методах, использующих приближед
НДП.
В общем случае многоатомной молекулы с N базисными
биталями полная электронная заселенность атомной орбитали
принадлежащей атому А (орбитальная заселенность), дается вы|
жением
зал
i-l ye A
Для полной электронной заселенности атома А (полная ато!
заселенность) имеем
i-l \jieA (teAv^A
Для характеристики связывания между двумя АО /ihv, npi
лежащих соответственно атомам А и В, и между двумя ато!
А и В Малликен ввел определение межорбитальной заселенной
связи
зан
ы
e A, v e В,
G.60)
и полной заселенности связи
i-I fteA veB
Полная заселенность связи характеризует прочность связывания
между двумя атомами: А и В.
Полный электронный заряд молекулы G.56) можно составит*1
теперь как сумму полных орбитальных или атомных заселенностей;; *
,= л. G.62)
Задача 7.4. Доказать соотношения G.58), G.59).
'J
Соотношения G.58) — G.59) независимы от характера исполь-*
зованного в расчетах приближения и в равной степени справедливы
для всех как полуэмпирических, так и неэмпирических методов <
квантовой химии.
238
7.5 <т, гс-ПРИБЛИЖЕНИЕ, ПОЛУЭМПИРИЧЕСКИЕ
Я-ЭЛЕКТРОННЫЕ МЕТОДЫ
Самостоятельную область квантовой химии представляет те
ооия электронного строения соединений с кратными связями (нена
сышенные системы)- НРИ Расчете ненасыщенных систем обычн'
используют <т, тг-приближение.
В ненасыщенных соединениях многие химические и физически
свойства определяются небольшой частью электронов, заселяющи;
тг-орбитали, так называемыми л-электронами. Соединения с крат
ными связями обладают повышенной реакционной способностью
большой поляризуемостью, характеризуются сильным влияниел
донорных и акцепторных заместителей на электронную структур:
и спектры поглощения. Электронные спектры поглощения сопря
женных молекул в видимой и ближней УФ-области обусловлень
возбуждениями я-электронов.
Ненасыщенные органические молекулы чаще всего плоские. По-
Поэтому все атомные орбитали валентной оболочки можно разбить нг
о- и л, причем первые симметричны, а вторые антисимметричны
относительно отражения в плоскости молекулы. Например, у моле-
молекулы этилена есть десять а-АО и две 27Г-АО.
<7, я-Приближение заключается в том, что вместо решения
вариационной задачи для всех валентных электронов решаются
уравнения только для я-электронов, а-АО считаются непо-
дяризованными и включаются в ядерный остов. Другими
словами, предполагают, что я-электроны движутся в потен-
потенциальном поле, создаваемом ядрами и а-электронами. Эта
плодотворная идея принадлежит Э. Хюккедю A931). Так,
для этилена вместо 12 электронов рассматриваются только
два я-электрона. Для антрацена, например, упрощения еще
более значительны: вместо 66 атомных орбиталей необходимо
рассмотреть только 14.
Одно из обоснований а, я-приближения состоит в том, что
энергетические уровни сг-орбиталей расположены значительно ниже
уровней я-орбиталей. Однако это общее положение имеет немало
исключений, выявленных как расчетным, так и экспериментальным
(фотоэлектронные спектры) путем. Как показывает рис. 7.5, даже
в молекуле бензола энергетические уровни я-МО «прослоены» уров-
уровнями ст-орбиталей.
Тем не менее для многих задач с, я-приближение является
достаточно надежным, так как различная симметрия &, я-орбнта-
леи, определяемая наличием плоскости симметрии, препятствует их
взаимодействию. П. Ликос и Р. Парр показали, что, используя
вариационный принцип, можно дать достаточно строгое обоснова-
обоснование отдельному рассмотрению а- и я-электронов.
239
Рис. 7.5. Диаграмма расположений энергетических уровней МО мол
нафталина и цитозина
7.5.1. Метод Партера—Парра—Попла (ШШ)
В <х, л>приближении уравнения метода CNDO значите
щаются. Каждый атом будет иметь только одну орбита!
индексы ft и v совпадают с обозначениями А и В. Т
в пренебрежении интегралами проникновения и перекры]
но записать в виде
I Р
¦**)IV
« ' It
где I)t(~~ Um) — потенциал ионизации валентного ее
электрона.
240
Уравнения G.63) были предложены Дж. Поплом A953). Метод
Попла, чаще его называют методом Паризера—Парра—Попла
AШП),—это весьма точный полуэмпирический метод*. Вначале
метод был параметризован для воспроизведения электронных спек-
спектров молекул. Позднее Дьюар A965) предложил новую парамет-
параметризацию для расчета свойств молекул в основных электронных
состояниях.
7.5.1.1. ПАРАМЕТРИЗАЦИЯ ДЛЯ РАСЧЕТА
ЭЛЕКТРОННО-ВОЗБУЖДЕННЫХ СОСТОЯНИЙ
Как видно из уравнений G.63), в методе ППП имеется три
основных параметра. Величины /д представляют энергии электро-
электронов на атомных 2pz-, Зрг-орбиталях. Значения /„ для $/72-гибридизо-
ванных атомов, поставляющих в я-систему один или два электрона,
представлены в табл. 7.15. Там же даны значения величин сродства
к электрону Ай для атомов в соответствующих валентных состояни-
состояниях, необходимые для определения у^ по соотношению G.64).
Таблица 7,15. Ионизационные иотенцналы (I^J и сродство к электрону
п-орбнталея валентных состояний атомов второго н третьего периодов
Атом
С
N
6
S
О
• •
S
С1
р
Валентное состояние
trtrtr л
tr2 tr tr к
tr2 tr2 tr %
tr2 tr2 tr n
trtrtr n2
tr2 tr tr %2
tr2 tr tr %2
sp2p2p2
sp2P2P2
V, эВ
11,16
14,12
17,70
12,70
28,72
34,12
22,97
26,37
40,70
A^, эВ
0,03
1,78
2,47
2,76
11,96
15,30
11,05
11,05
18,52
"'Для атомов, вносящих 2 я-электроаа, приведены значения второго потенциала
ионизации.
Одноцентровые кулоновские интегралы уда=(/^|/*/*) можно вы-
вычислить, пользуясь АО хартри-фоковского типа (при использовании
STO-функций получаются сильно завышенные значения). Более
практичным является использование соотношения
*Иногда подразделяют метод Паризера—Парра—Попла на два метода: метод
Попла и метод Паризера—Парра. Под методом Попла понимают уравнения Рута-
ана в приближении НДП и я-электронном приближении. Метод Паризера—Парра —
метод конфигурационного взаимодействия (см. далее).
241
/5
10
A
a? 0,2 0,3 Ob 0.5R,hM
Рис. 7.6. Зависимость двухцентро-
вого жулоновского интеграла отта-
отталкивания у от межъядерного рас-
расстояния R:
Т — теоретическая кривая; / — кривая
Матага—Нишимото; 2 — кривая Оно;
1/Jt — кривая
Ar G.64)
Формула G.64) справедлива для ато-
атома ц, вносящего в я-электронную си-
систему молекулы один я-электрон.
Для атома, вносящего два тс-электро-
на, значение ут вычисляется как
/?)-/!,1), G.65)
/JB) и /j!* — соответственно 2-й
и 1-й потенциалы ионизации.
Двухцентровые кулоновские ин-
интегралы у^ должны зависеть от меж-
межатомного расстояния Гр так, чтобы
удовлетворять граничным условиям:
при г^ = 0,
где /J(
— при г^-юо.
Первое условие не требует пояснений, второе же вытекает из того
факта, что при больших межатомных расстояниях отталкивание
двух пространственно распределенных зарядов должно аппрокси-
аппроксимироваться отталкиванием точечных зарядов. Наиболее часто ис-
используют аналитические соотношения Матага—Нишимото
1
Уму=г +а G.66)
или Оно
где
Clint ^"™ "
G.67)
G.68)
На рис. 7.6 даны зависимости кулоновских интегралов у^ от
Гр для различных оценок.
Резонансный интеграл Д„, является основным «подгоночным»
параметром. Опыт расчетов показывает, что величину р^ не удается
параметризовать так, чтобы одновременно удовлетворительно опи-
описывать свойства основного и возбужденных состояний, например
242
теплоты образования и спектра поглощения. Поэтому существуют
специальные параметризации /?,,„ ориентированные на расчет спект-
спектральных свойств молекул. Наиболее часто применяются и дают
неплохие результаты следующие формулы для оценки р^:
*V/ G.69)
G.70)
в-11
/V— _ _
те
где РцУ — я-электронный порядок связи между атомами ц и v,
константы Aq и Ах подбирают так, чтобы лучше всего воспроизвести
спектральные свойства. В табл. 7.16 приведены значения Ао, А\ для
связей С—X, применяемые в расчетах электронных спектров погло-
поглощения.
Соотношение G.70), полученное Линдербергом, интересно тем,
что оно не содержит эмпирических параметров.
Таблица 7.16. Величины Аоя А{ для расчета спектров поглощения с использова-
использованием соотношения G.69)
Связь
с-с
C-N
С-О
C-S
2,04
2,24
2,44
0,73
-0,51
-0,53
-0,56
-0,32
Предложенный Паризером и Парром метод конфигурационного
взаимодействия для расчета свойств электронно-возбужденных со-
состояний состоит в следующем. Запишем волновую функцию воз-
возбужденных синглетных и триплетных состояний, возникающих при
переходе электрона с орбитали <р, на орбиталь щ\
1 "'" "¦ " "'" ... фяBй)}±
1,3
G.71)
Знаки « + » и « — » употребляются соответственно для синглетной
и триплетной конфигурации. Подставляя функции G.71) в выраже-
выражение для полной энергии A.34), можно вычислить энергию возбуж-
возбужденного состояния заданной мультиплетности. Разность полной
энергии основного и возбужденного состояний определяет энергию
перехода и может быть сравнена с экспериментально наблюда-
наблюдаемыми энергетическими характеристиками.
Единственная конфигурация, представленная уравнением G.71),
243
в общем случае является плохим приближением к возбужденному
состоянию. Как правило, одна конфигурация удовлетворительно
описывает только свойства низших синглетного и триплетного
уровней. Чаще всего волновую функцию возбужденного состояния
записывают в виде линейной комбинации нескольких спин-опреде-
спин-определенных конфигураций типа G.71) данной мультиплетности:
W G.72)
i. *
Коэффициенты Т^к можно найти, используя вариационный
принцип (метод KB, см. разд. 4.4.1). Орбитали <р( являются хартри-
фоковскими, поэтому матричные элементы <Ф^А|Н*| Ф/-/Х где Н* —
7г-электронный гамильтониан молекулы, например, для синглета
могут быть записаны в виде
+2 (jk\li). G.73)
В случае триплетных возбуждений последний член в G.73) отсут-
отсутствует.
В приближении НДП интегралы,(ij|kl) имеют вид
(ij\kl)=Ytci*ci*c*c*yi»- G-74)
Вычитая из G.73) энергию основного состояния Ео и принимая
во внимание G.74), матричные элементы Н^к^ можно записать
в виде
#;_*, ,w/=S(J 5Ы (гк - Bi) + ? [2 A —у) cw c^ cvi cvk -
- См Ср с* cv/] yMV, *" G.75)
где /— постоянная, равная нулю для синглетных и единице для
триплетных возбуждений.
Матрица, построенная из элементов G.75), называется матрицей
конфигурационного взаимодействия. Ее диагонализация дает воз-
возможность получить энергии спектральных переходов и коэффициен-
коэффициенты Т^к. Интенсивность перехода можно вычислить, зная квадрат
дипольного момента перехода D •
/=1,085- lO^vli?!2, G.76)
где
G.77)
G.78)
244
Электронный переход может быть разрешен не для всех состав-
составляющих дипольного момента. Возможны ситуации, когда одна из
проекций дипольного момента перехода, например на ось х, равна
нулю. Если ориентировать молекулы определенным образом (на-
(например, под действием электрического или магнитного поля, в кри-
кристаллах и т. д.), то поглощение света будет существенно зависеть от
его поляризации. Таким образом, возникает еще одна эксперимен-
экспериментальная характеристика полосы поглощения — поляризация пере-
перехода. Количественной мерой поляризации перехода служит угол а,
определенный из соотношения
Dx
ctga=-. G.79)
Важной характеристикой возбужденной молекулы является рас-
распределение электронной плотности. Элементы матрицы плотности
Рр для возбужденного состояния вычисляют как
G.80)
i, к
где Р^ — элементы матрицы плотности основного состояния.
Многочисленными исследованиями показано, что для удовлет-
удовлетворительного расчета спектров достаточно в разложении G.72)
учесть 20—25 однократно возбужденных конфигураций. Увеличение
числа учитываемых конфигураций, а также включение двукратно
возбужденных конфигураций оказывают влияние на интенсивности
переходов и распределение электронной плотности, но не на энер-
энергетические характеристики.
Отметим также, что синглетные и триплетные состояния имеют
различное значение энергии корреляции, в связи с чем с помощью
одного набора резонансных интегралов ft нельзя одновременно
одинаково удовлетворительно вычислить энергии синглетных и три-
плетных уровней. Поэтому для расчета триплетных возбуждений
используют чаще всего специально откалиброванные значения /?.
Рис. 7.7 иллюстрирует точность теоретической оценки энергии
и интенсивности переходов в молекулах анилина, фенола и гидрохи-
гидрохинона. Представленные спектральные кривые содержат кроме синг-
лет-синглетных переходов также синглет-триплетное поглощение,
запрещенное правилами отбора по спину, но проявляющееся с ма-
малой интенсивностью в растворителях, содержащих тяжелые атомы,
например атомы иода, которые увеличивают эффекты спин-орби-
спин-орбитального взаимодействия. Положение вертикальных прямых опре-
определяет рассчитанные энергии переходов, высота прямых пропорцио-
пропорциональна вычисленной интенсивности поглощения.
Как видно из рис. 7.7, метод ППП дает возможность весьма
удовлетворительно описать экспериментальную кривую поглоще-
245
он
-2
100 250 500Л,НМ ZOO 250 500Анм 200 250 500 А,ИМ
а 1Г в
Рис. 7.7. Спектры поглощения молекул анилина (а), фенола (б) и гидрохинона (в)
ния. Точность оценки энергий синглет-синглетных переходов со-
составляет 0,1—0,2 эВ C—5%), совпадение для интенсивностей пере-
переходов обычно хуже, ошибка может составлять 40—50%.
7.5.1.2. ПАРАМЕТРИЗАЦИЯ МЕТОДА 1ШП
ДЛЯ РАСЧЕТА СВОЙСТВ ОСНОВНЫХ СОСТОЯНИЙ
Как указывалось выше, не удается в рамках одного набора
параметров метода ППП с одинаково хорошей точностью воспро-
воспроизвести свойства основного и возбужденного состояний. В связи
с этим Дьюар с сотр. разработали параметризацию метода ППП,
специально приспособленную для расчета свойств основных состоя-
состояний, в первую очередь теплоты атомизации* органических молекул.
Точность метода Дьюара в предсказании энергий основных состоя-
состояний сопряженных молекул составляет 0,1—0,2%, или ~ 12—
20 кДж/моль, что вполне достаточно для решения большинства
задач. Схематично изложим параметризацию Дьюара. Энергия свя-
связи молекулы (?св) записывается в виде суммы:
F — р _1_/г
л-'СВ -^(Г-СВ ' -'-'Л-СВ»
где Еа.сь — энергия сг-связей;
G.81)
энергия я-связывания.
•Теплота атомизации ДЯ — это энергия, необходимая для превращения молеку-
молекулы в изолированные атомы. Для нахождения экспериментальных значений ДЯ
используют данные по теплотам сгорания. Экспериментальные значения АН от-
относятся к 298 К, расчет ведется при 0 К, поэтому для сравнения экспериментальные
величины следует экстраполировать к 0 К.
246
Энергию всех «х-связей в молекуле можно представить как
G.82)
л* х-н
где Nx — число связей С—X (Х = С, N, О, S); NX—h — число связей
X—Н; Ес-х и ЕХ—н — соответственно энергия связей С—X и X—Н.
я-Связевую энергию ?"ячъ определяют как разность между п-
электронной энергией молекулы и энергиями я-электронов изолиро-
изолированных атомов.
Энергию связи молекулы Еа можно сравнить с теплотой атоми-
задии молекул из атомов в газовой фазе, приняв ряд упрощающих
т
предположений. Теплоту атомизации АН при температуре Т запи-
запишем как
G.83)
где v, — частота нормальных колебаний; ^O^Av, — нулевая коле-
колебательная энергия; Ср — теплоемкость при постоянном давлении.
Величины АН (Г=298 К) известны для некоторых углеводородов
и гетероатомных систем, в то время как надежные данные от-
относительно v, и Ср обычно отсутствуют. Положение, однако, не
безнадежно, так как известно, что для насыщенных молекул тепло-
теплоты образования можно с хорошей точностью вычислять по ад-
аддитивной схеме. Если считать, что ситуация не меняется для со-
сопряженных соединений, т. е. энергию <т-связей можно считать ад-
аддитивной функцией, то в этом случае энергии <г-связей можно
подобрать так, чтобы включить поправку на температуру. Нулевую
колебательную энергию включают обычно в величину EK.a. Это
дает возможность записать теплоту атомизации в виде
?-н- G.84)
Величины энергий X—Н-связей считают постоянными, и их
значения подобраны Дьюаром по ряду стандартных соединений
(см. табл. 7.17).
Если значения энергий и длин связей X—Н можно считать
постоянными для различных молекул, то значения энергий связей
С—X существенно меняются от одной молекулы к другой. В связи
с этим энергии <т-связей С—X предложено оценивать с помощью
потенциала Морзе G.85) с эмпирическими параметрами Do, flo и г0:
247
-Do, G.85)
где г — межатомное расстояние С—X в молекуле, нм; Оо — по-
постоянная Морзе, нм; Do — энергия связи при равновесной длине
го, эВ.
Таблица 7.17. Энергии связей X—Н в методе Дьюара
Связь
С—Н
0-Н
S—Н
N—Н
N—Н
Энергия, эВ
4,4375
4,4390
3,5998
4,3753
4,2018
Значения Do, г0 и а0 Для связей С—X приведены в табл. 7.18.
Таблица 7.18. Параметры потенциала Морзе для связей С—X
Связь
С-~С
С—N
С—О
С—S
А),эВ
3,9408
3,3463
3,9987
3,2783
го, нм
0,1512
0,1448
0,1395
0,1802
20,022
19,209
17,870
19,663
Параметры р^ в методе Дьюара определяют по уравнению
=Klt,Sltv, G.86)
где S^ — интеграл перекрывания рг-АО, а константы подбираются
так, чтобы лучше воспроизвести экспериментальные данные по
теплотам образования модельных соединений.
В табл. 7.19 приведены рассчитанные значения теплот атомиза-
ции сопряженных углеводоров совместно с имеющимися экспери-
экспериментальными значениями. За исключением последних четырех
соединений, разница между теоретическими и наблюдаемыми
величинами составляет ~0,03 эВ (ошибка ~0,03%). Совпадение
для i/wc-бутадиена, азулена, аценафтилена и флуорантена зна-
значительно хуже: рассчитанные значения теплот атомизации
превышают экспериментальные величины в среднем на 0,4 эВ
@,4%).
248
Таблица 7.19. Теплоты атомязацвн сопряженных углеводородов
Соединение
Формула
ЛЯ.эВ
рассчи-
рассчитанная
экспери-
менталь-
ментальная
Ошибка
эВ
Бензол
Нафталин
Антрацен
Дифенил
Стирол
Стильбен
транс- бутадиен
vuc-бутадиен
Азулен
Аценафтилен
Флуорантен
57,16
90,61
123,89
109,75
75,91
128,53
42,05
42,08
89,46
104,86
57Д6
90,61
123,93
109,76
75,83
128,48
42,05
41,95
89,19
104,32
138,67
138,11
0
о
-0,04
-0,01
0,08
0,05
0
0,13
0,27
0,54
0,56
0
0
0,03
0,01
0,10
0,04
0
0,31
0,30
0,52
0,40
Столь значительное отличие (ошибка возросла в 10 раз) может
быть объяснено наличием в этих молекулах пространственных пре-
препятствий (уыс-бутадиен) или напряженных циклов (аз у лен, аценаф-
аценафтилен, флуорантен). Вследствие этих эффектов молекулы становятся
менее стабильными, поэтому рассчитанные значения теплот атоми-
зации превышают наблюдаемые. Учесть напряжения циклов и про-
пространственные затруднения в методе ППП невозможно, эта ела-
24S
бость я-электронных теорий преодолевается во всевалентных мето-
методах, например в методах MINDO/3, MNDO, ab initio.
Теплоты атомизации некоторых сопряженных молекул, содер-
содержащих гетероатомы N, О, представлены в табл. 7.20.
Точность расчетов теплот атомизации для гетероатомных моле-
молекул существенно ниже по сравнению с сопряженными углеводорода-
углеводородами, хотя и остается достаточно высокой (средняя ошибка в энергии
0,15 эВ). В большинстве случаев отличие рассчитанных и экспери-
экспериментальных значений находят в пределах ошибки эксперимента.
Отметим, что многие термохимические данные по теплотам сгора-
сгорания сопряженных систем были получены ~30 лет назад, когда
методика измерения была не столь точна, как в настоящее время.
Таблица 7.20. Теплоты атомюацин молекул
Соединение
Пиридин
Хинолин
Пиримидин
Пиррол
Индол
Фуран
Бензальдегид
Формула
[
СО
о
н
СХ>
н
К
с гетероатомамв
ДЯ, эВ
рассчи-
рассчитанная
51,87
85,32
46,86
44,77
78,44
41,56
68,51
экспери-
менталь-
ментальная
51,79
85,18
46,99
44,77
78,06
41,52
68,27
Ошибка
эВ
0,08
0,14
-0,13
0
0,38
0,04
0,24
%
0,15
0,16
0,28
0
0,49
0,10
0,35
Вычисление геометрической структуры молекулы в основном
состоянии можно проводить, находя минимум полной энергии в за-
зависимости от длин связей и валентных углов. Существует второй
250
путь, более простой, но менее строгий. Считая, что валентные углы
в сопряженных системах близки к 120 или 108° (в пятичленныя
циклах)*, можно вычислить длины связей, используя предложенную
Ч. Коулсоном и А. Голебиевским зависимость между л-элект-
ровным порядком связи [см. соотношение (8.72)] и ее длиной:
= Л — ВР,
к».
А и В — эмпирические константы.
Константы А и В в уравнении G.87) и константы
G.86) сведены в табл. 7.21.
Таблица 7.21. Параметры уравнений G.86), G.87)
G.87]
в уравнении
Связь
С—С
С—N
С-О
С—S
А, нм
0,1512
0,1448
0,1395
0,1802
В, нм
0,0174
0,0178
0,0165
0,0229
/»,эВ
6,9270
6,9612
9,2246
15,7265
В тех случаях, когда были проведены расчеты геометрии с пря-
прямой минимизацией энергии и по соотношению порядок связи —
длина связи G.87), рассчитанные двумя способами значения от-
отличались в среднем на 0,0001—0,0002 нм.
Таблица 7.22. Сравнение рассчитанных и эксиернментальных длни связей со-
иряженных молекул
Соединение
Длина связи, нм
рассчитанная
эксперимент аль-
ная
1—2
2—3
3—4
1—5
1—2
2—3
0,1397
0,1428
0,1373
0,1423
0,1403
0,1338
0,1512
0,1397
0,1421
0,1363
0,1415
0,1418
*Рентгеноструктурные исследования подтверждают, что отклонения не превы-
превышают 3—4°.
251
Продолжение табл. 722
Соединение
Длина связи, км
рцсоштянн&я
эксперименталь-
экспериментальная
1—2
2—3
1—2
2—3
3—4
1—2
2—3
0,1338
0,1333
0,1364
0,1351
0,1450
0,1256
0,1471
0,1348
0,1388
0,1334
0,1371
0,1354
0,1440
0,1222
0,1477
0,1322
Ошибка в вычислении (см. табл. 7.22) составляет 0,001 нм, экс-
экспериментальная точность — 0,001—0,004 нм. Соотношение (.87)
может быть использовано для расчета геометрического строения
возбужденных состояний. Для этого необходимо порядок связи
в возбужденном состоянии подставить в уравнение G.87). Экспери-
Экспериментальные данные по геометрии возбужденных состояний крайне
трудно получить. В то же время знание их необходимо для ис-
исследования механизмов дезактивации электронного возбуждения,
определения реакционной способности, которая обычно крайне от-
отличается от основного.
7.5.2. Метод Хюккеля
Несамосогласованным вариантом метода ППП, в котором пре-
пренебрегают интегралами (р.х \ ко), является метод молекулярных ор-
биталей Хюккеля (МОХ), предложенный Э. Хюккелем еще
в 1931 г. В этом методе остовные интегралы Н^ не вычисляются
точно, а рассматриваются как параметры, обозначаемые
Нт=а^; Н^ = рр,. G.88)
Здесь ар — кулоновский интеграл, достаточно хорошей оценкой
которого является потенциал ионизации электрона, находящегося
на АО изолированного атома.
Кроме того, вводится еще одно приближение
S^S^ G.89)
и уравнения G.48) сводятся к системе
G.90)
252
которая имеет нетривиальные решения при равенстве нулю детер-
детерминанта:
Aiv=i
= 0. G.91)
Обычно в методе Хюккеля полагают
если атомы ц и v соединены ковалснтной связью,
[О в противном случае.
Полная энергия молекулы есть сумма энергий орбиталей с уче-
учетом их заполненности [сравните с соотношением G.54)]:
G.92)
где щ — число заполнения МО электронами (л, = 2, 1 или 0).
Метод МОХ — простейший расчетный качественный метод ква-
квантовой химии. Метод имел и продолжает иметь важное значение
в процессе внедрения понятий и языка квантовой химии во все
основные представления о строении, свойствах и реакциях химичес-
химических соединений. Детальный обзор приложений метода МОХ дан
в следующей главе.
* * *
Мы рассмотрели основные полуэмпирические методы расчета
молекул в порядке их упрощения. Последовательность же их ис-
исторического возникновения прямо противоположна: 1931 г. —ме-
—метод МОХ, 1953 г. — метод ППП, 1965 г. — метод CNDO. Это
неудивительно, так как численная реализация методов требует при-
применения ЭВМ, совершенствование которых стимулирует развитие
тех или иных методов расчета. На схеме приведена иерархия основ-
основных современных методов квантовой химии.
Литература
Губанов В. А., Жуков В. П., Литинский А. О. Полуэмпирические методы моле-
молекулярных орбиталей в квантовой химии. — М.: Наука, 1976.
Монография, посвященная теории всевалентных полуэмпнрических методов. Приведены
алгоритмы и программы методов CNDO и INDO в различных параметризациях, написанные на
языке Алгол- 60 применительно к ЭВМ БЭСМ-6.
Дьюар М. Теория молекулярных орбиталей в органической химии. — М.: Мир,
1972.
Обширная монография, в которой отражено развитие полуэмпирических теорий квантовой
химии и их применение к расчету свойств основных состояний органических систем.
Жидомиров Г. М., Багатурышц А. А., Абронин И. А. Прикладная квантовая
химия. — М.: Химия, 1979.
В монографии подробно рассматриваются хвавтово-хнмичеогае методы расчета и их приме-
применение к изучению химических реакций.
Минска В. И., Снмкин Б. Я., Миняев Р. М. Квантовая химия органических со-
соединений. Механизмы реакций. — М.: Химия, 1986.
253
s
:
ю
о.
U
Уравнение Шрёдингера
I
Мулътиконфи-
гурационный
метод (МКВ)
Метол
Хартри-Фока
4KB
Метод
Хартри-Фока
Методы не ССП
Т
ЛКАО приближение
I
I
Метод Рутаана
Методы ССП
Методы,
пренебрегающие
всеми интегралами
PRDDO
AMI
MNDO
NDDO
MINDO
INDO
РМХ
1
CINDO
ст,тс-приолижение
1
. тг -приб/
о, тг -приближение
МОХ
ППП
Монография посвящена детальному анализу применения различных квантово-химических
методов а изучению механизмов важнейших органических реакций.
Полуэмпирические методы расчета электронной структуры. — М.: Мир, 1980. Т.
Двухтомная монография оосгавлева из статей ведущих специалистов в соответствующих
областях квантовой химии. Приведены основные современные методы расчета и их применение
к решению конкретных проблем химии.
Pople J. A-, Beveridge D. L. Approximate Molecular Orbital Theory. — N. Y. Me
Graw-НШ, 1970.
Четкое и ясное изложение теории и приложений основных полуэмпирщесхих методов
CNDO и INDO.
Кларк Т. Компьютерная химия. — М.: Мир, 1990.
Практическое руководство по реалвэацив расчетных методов квантовой химии с исполь-
использованием ЭВМ.
254
ГЛАВА 8
СВОЙСТВА я-СОПРЯЖЕННЬК МОЛЕКУЛ
Первым методом молекулярных орбиталей, который можно
было использовать для расчета свойств многоатомных молекул,
явился метод МО Хюккеля*. Уравнения G.89), G.90) были постро-
построены на ассоциациях с формализмом теории Слэтера и Блоха для
описания поведения электронов в металле. Однако основной идеей
метода, не вытекающей из какой-либо теории, является введение
заимствованного из строения двухатомных молекул представления
о возможности раздельного рассмотрения а- и я-электронов.
Легкость расчетов по методу Хюккеля привела к тому, что
внедрение языка и понятий квантовой теории в органическую хи-
химию в основном велось на основе изучения свойств я-сопряженных
молекул. Вероятно, и в настоящее время теория сопряженных соеди-
соединений является наиболее разработанной областью квантовой хи-
химии. В данной главе ставится цель объяснить, как и в какой мере
с помощью расчетов можно изучать и прогнозировать свойства
7Г-сопряженных систем. Будем использовать, как правило, метод
МОХ, так как его применение не требует ЭВМ и вполне оказывается
достаточным иметь микрокалькулятор. Метод МОХ позволяет осу-
осуществить расчеты в полной мере и пояснить принципы, на которых
основаны и расчеты более сложными методами ССП МО.
8.1. РАСЧЕТ ЭНЕРГИЙ МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
В МЕТОДЕ МОХ. АННУ ЛЕНЫ
Наиболее яркой демонстрацией успеха метода МОХ, его описа-
описательных и предсказательных возможностей является расчет энер-
энергетических уровней и относительной устойчивости сопряженных
моноциклических полиенов (аннулены), методы получения и химия
которых хорошо разработаны. Для этих соединений принято обо-
обозначение [Л^-аннулен, где N — число атомов в цикле. Например,
бензол — [6]-аннулен, циклооктатетраен — [8]-аннулен.
Рассмотрим, продвигаясь от низших аннуленов к высшим, схему
расчета энергетических уровней (энергий МО) в методе Хюккеля,
а также выводы, которые можно сделать на основании таких рас-
расчетов.
¦Эрих Хюккель A896—1980) — немецкий физик, большинство работ которого
связано с решением химических проблем. Совместно с П. Дебаем и Л. Онзагером
им разработана теория сильных электролитов (теория Дебая—Хюккеля). Ему при-
принадлежит одна из наиболее плодотворных идей в теории строения сопряженных
систем — идея а, ^-приближения. Э. Хюккель сформулировал правило ароматич-
ароматичности 4л-1-2, предложил метод расчета я-электронных систем, носящих его имя.
255
8.1Л. Система цнклопропеымла
Рассмотрим получение и решение уравнений метода МОХ на
конкретном примере расчета я-энергетических уровней циклопропе-
нильной системы. Устойчивыми являются катионы, производные
которых синтезируются отнятием гидрид-иона от молекул легкодо-
легкодоступных циклопропанов:
3 + Ph3CH
Три гибридные орбитали углеродных атомов цикла образуют
систему с-связей молекулы, а три pz-орбитали служат базисом
тс-МО циклопропенильной системы.
Пронумеруем /?г-орбитали отдельных атомов углерода и будем
искать МО в виде их линейной комбинации типа
где Хь Х2> Хз — атомные /^-орбитали, например слэ-
теровского типа.
Задача состоит в нахождении коэффициентов
ЛГ с^ атомных орбиталей и энергии Д молекулярных
орбиталей, определяемых решением системы линейных однородных
уравнений
ц-1
Соответствующее секулярное уравнение записывается как
Нц—ESu H\2~ESn Hn
Hn-ESn Н2Ъ
3l i ( H32—ES32 Hi3
Учитывая приближения метода Хюккеля (см. гл. 7), можно
переписать уравнение (8.3) в виде
=0.
(8.3)
а-Е
р а-Е р
р р а-Е
= 0,
(8-4)
где а и р — соответственно кулоновский и резонансный интегралы
метода МОХ, определенные выше.
256
Каждую строку (или колонку) определителя разделим на /? и вве-
введем обозначение
a-E_
p -
Тогда секулярное уравнение примет вид
х 1 1
= 0.
(8.5)
(8.6)
1 х 1
1 1 х
Раскрывая определитель (8.6), получаем характеристическое ура-
уравнение
Это уравнение имеет, очевидно, три действительных корня:
Xj = —2; х2=хъ= 1;
они с учетом (8.5) соответствуют трем энергетическим уровням:
Величины кулоновского а- и резонансного ^-интегралов отрица-
отрицательны. Это означает, что уровень Ех соответствует связывающей
МО, энергия которой ниже, чем а, т. е. энергии электрона на pz-AO
изолированного углерода 5р2-гибридизованного атома, которая
принимается за условный нулевой уровень. Вырожденные уровни
Е2 и Еъ, наоборот, лежат выше нулевого уровня и принадлежат
антисвязывающим МО (рис. 8.1).
Заполнение энергетических уровней электронами происходит л о
обычному принципу стремления системы к минимальной энергии
и максимально возможной мультиплетности.
ы-р
of
-Н-
-н-
¦?•
Рис. 8.1. Энергетические уровни п-МО циклопропенильной системы и их заполнение
электронами в катионе, радикале и анионе
9. Теория строения молекул 257
Радикал циклопропенила интересен с точки зрения теории. Тре-
Третий электрон с равным основанием может быть помещен на любую
из двух вырожденных антисвязывающих МО, и, согласно теореме
Яна—Геллера (см. гл. 5), симметрия ядерной конфигурации молеку-
молекулы должна быть нарушена.
Расположение третьего я-электрона на антисвязывающей МО
приводит к тому, что я-электронная энергия циклопропенильного
радикала (СНK оказывается выше, чем я-электронная энергия цик-
циклопропенильного катиона (CH)J". По этой причине одноэлектронное
электрохимическое восстановление устойчивого катиона трифенил-
циклопропенила (\l)(Rl=R2:=Ri = Ph) требует более высокого по-
потенциала, чем для других карбокатионов.
В циклопропенильном анионе четвертый электрон, заселяющий
я-МО, занимает в соответствии с правилом максимальной мультип-
летности вторую вырожденную МО. Анион, следовательно, должен
иметь триплетное бирадикальное основное состояние. Это состоя-
состояние крайне не стабильно, так как два электрона находятся на
антисвязывающих орбиталях. Действительно, производные циклоп-
ропена (I) должны быть отнесены к самым слабым СН-кислотам
(т. е. они не склонны отщеплять протон с образованием циклоп-
циклопропенильного аниона) и не образуют металлоорганических соедине-
соединений. Константа равновесия реакции депротонирования триметил-
циклопропена оценивается величиной 105 (рКв=65):
т. е. это самая слабая из всех кислота, для которых измерены
константы ионизации, например для метана рКа=59.
8.1.2. Циклобутаднен
Циклобутадиен (IV) — одно из самых интригующих органичес-
органических соединений. Это простейшая циклическая сопряженная система
с четным числом я-электронов, однако электронное и пространст-
пространственное строение циклобутадиена до недавнего времени составляло
предмет оживленной теоретической дискуссии:
о
IV
258
Первые попытки осуществить синтез циклобутадиена были
предприняты еще Кекуле, однако решение этой задачи стало воз-
возможным лишь сто лет спустя, в 1969 г.
Рассмотрим поэтапно расчет энергии МО циклобутадиена (V).
1. Выделим систему />Г-АО, которые должны быть включены
в линейные комбинации с образованием МО, и пронумеруем их
начиная с любой АО. Остальные р- и 5-орбитали атомов углерода
образуют <7-остов молекулы.
2. Запишем секулярный определитель подо-
подобно (8.6), расположив х по диагонали; далее
в соответствии с приближениями МОХ присво-
присвоим значение 1 тем недиагональным матричным
элементам, которые соответствуют соседним
атомам (например, , первому и второму),
и нуль — несоседним (например, первому и третьему):
1 0 1
х
1 X
0 1
1 О
1
X
1
О
1
х
= 0.
(8.7)
3. Раскроем определитель по элементам любой строки или ко-
колонки и получим характеристическое уравнение для молекулы цик-
циклобутадиена
х4-4х2 = х2(хг-4)=0,
корнями которого будут д:, = —2, х2~х3 = 0, хА = 2. Отрицательные
значения корней, согласно определению (8 .'5), соответствуют связы-
связывающим МО: нулевые — несвязывающим, положительные — анти-
связывающим. В соответствии с правилом Хунда четыре л-электро-
на циклобутадиена размещаются на энергетических уровнях, как
показано на рис. 8.2.
Таким образом, расчет показывает, что циклобутадиен имеет
триплетное основное состояние и является би-
радикалом. Кроме того, тот факт, что два из
четырех я-электронов циклобутадиена, находя-
находящиеся на несвязывающих МО, не вносят вкла-
вклада в энергию я-связывания, говорит о том, что
нельзя ожидать от молекулы термодинамичес-
термодинамической стабильности. Следовательно, циклобута-
циклобутадиен должен быть малоустойчивым и высоко-
реакционноспособным (в силу его бирадикаль-
ного характера) соединением.
Осознание этих положений теории не могло
не наложить отпечатка на выбор средств экс-
экспериментального обнаружения циклобутадие-
на. Вместо того чтобы бесконечно разнообра-
В ,
Of-2fi
Cf
ar+fi
+
-N-
Рис. 8.2. Диаграмма
энергетических уров-
25S
зить и искать новые изощренные синтетические схемы, которые
должны приводить к циклобутадиену, исследователи обратились
к технике изучения сверхбыстрых реакций методами регистрации
короткоживущих промежуточных соединений (интермедиатов).
Особенно эффективными оказались два метода. Первый из
них — импульсный вакуум-термолиз, который отличается от обыч-
обычного пиролиза тем, что образец подвергается действию лишь крат-
кратковременного A0~3 —10~2 с) высокотемпературного теплового им-
импульса. Второй — фотолиз (т. е. облучение, обычно УФ-светом)
при низких температурах в матрице инертного газа.
С помощью низкотемпературного фотолиза удается генериро-
генерировать циклобутадиен из довольно простого органического соедине-
соединения а-пирона (VI):
Av(>251hm) ///\^0 Av (УФ)
*^ // J Г *-
8-20K
+ C0,
VIH
В этой реакции промежуточный бицикло- [2, 2, 0]-пиран-2-он
(УП) устойчив и может быть выделен. Хедайя A969) применил
метод импульсного вакуум-термолиза к изучению реакции разложе-
разложения (УП) до циклобутадиена. В этих опытах ячейка с образцом
подсоединялась непосредственно к масс-спектрометру. Начиная
с 400°С прибор мог регистрировать ионы с mje — 52 (С4Н4), количест-
количество которых достигало максимума при 800°С (давление 10~4 мм рт.
ст.). Время жизни (период полупревращения) циклобутадиена при
этих условиях составляет 10~3-ь10 с. Циклобутадиен распадает-
распадается на две молекулы ацетилена, а также димеризуется. Однако при
замораживании в аргоновых, ксеноновых, азотных матрицах до
8—60 К эти реакции ингибируются и создается возможность ис-
исследования некоторых физических свойств, в частности ИК-спек-
тров. Их анализ дает важнейшую информацию о геометрическом
строении молекулы циклобутадиена.
Хотя результаты ранних исследований A972) ИК-спектров цик-
циклобутадиена были интерпретированы как доказательство квадрат-
квадратной конфигурации углеродного остова молекулы, впоследствии бы-
было показано A978), что наличие в ИК-спектре циклобутадиена
четырех полос скелетных колебаний четырехчленного цикла с часто-
частотами ниже 2000 см~1 может быть совместимо только с прямо-
прямоугольной структурой молекулы. Несколько позже A980) этот вывод
был подтвержден экспериментами по матричной изоляции двух
изомерных дидейтерозамещенных производных циклобутадиена
(IX) и (X), полученных при фотолизе 5,6-dfr и 3,6-^-а-пирона соот-
соответственно:
260
Ранее ошибочно предполагали, что в результате указанных реакций
образуется один и тот же продукт, имеющий квадратную структуру
(XI).
Результаты большинства неэмпирических расчетов, выполнен-
выполненных в достаточно расширенных базисах с учетом энергии элект-
электронной корреляции, также свидетельствуют в пользу того, что
в основном синглетном состоянии циклобутадиен имеет равновес-
равновесную конфигурацию симметрии D-& (см. задачу 8.2). Выводы же об
энергии низшего синглетного и триплетного состояний структуры
симметрии DAh были противоречивыми. Наиболее строгие неэм-
неэмпирические расчеты с учетом конфигурационного взаимодействия
в зависимости от базиса приводят к разности в 30,5 —
58,2 кДж/моль между Тг и ^-состояниями структуры D^h симмет-
симметрии в пользу ^-состояния. Этот вывод расчетов получил подтверж-
подтверждение и в эксперименте. С использованием техники флеш-фотолиза
установлено, что Тх-состояние пералкилированного циклобутадиена
имеет энергию, на 50 кДж/моль более высокую по сравнению с эне-
энергией основного синглетного состояния.
Приведем наиболее точные расчетные данные по геометрии (нм)
циклобутадиена:
0,1571 им
0,1336 нм
Характерно, что длина связи С—С в циклобутадиене даже превы-
превышает обычные значения (длина связи С—С в этане равна 0,1537 нм),
что объясняется стремлением к уменьшению дестабилизирующего
эффекта сопряжения. Молекула циклобутадиена может выступать
как те-донор, искажаясь при этом в структуру симметрии D^. Веро-
Вероятно, именно этим обстоятельством объясняется тот факт, что
выдвинутое еще в 1969 г. У. Лонге-Хигпшсом и Л. Оргелом пред-
261
положение о возможности фиксации циклобутадиена бирадикалами
оказалось правильным. В качестве б ир а пикала можно использовать
ион переходного металла, например Ni +:
XI
XII
В настоящее время известно уже большое число я-комплексов на
основе циклобутадиена, например димерный я-комплекс (XI) или
комплекс незамещенного циклобутадиена (XII). В отличие от само-
самого циклобутадиена этот комплекс высоко стабилен и охотно вступа-
вступает в типичные реакции электрофильного ароматического замещения
(дейтерирования, ацилирования по Фриделю—Крафтсу, меркуриро-
вания и т. д.) без изменения принципиальной структуры.
8.1.3. Циклопентадиеиил. Аналитическое решение
и графическое определение энергий МО аннуленов
Циклопентадиен легко реагирует с металлическим калием, давая
соль (XIII):
Эта реакция указывает на исключительно высокую стабильность
циклопентадиенильного аниона и, следовательно, высокую кис-
кислотность циклопентадиена, характеризующуюся, по современным
оценкам, рКаа\5. Стабильность циклопентадиенильного аниона
объясняется строением его я-электронной оболочки.
Расчет энергетических уровней циклопентадиенильной системы
можно выполнить, следуя схеме, намеченной при рассмотрении
циклобутадиена, т. е. начиная с записи и раскрытия секулярного
детерминанта:
X
1
0
0
1
1
X
1
0
0
0
1
X
1
0
0
0
1
X
1
1
0
0
1
X
= 0.
(8.8)
262
Однако для аннулеыов нетрудно найти аналитическое решение
в рамках метода Хюккеля. Действительно, в общем виде систему
линейных однородных уравнений для [JVJ-аннулена можно записать
следующим способом:
В этом можно убедиться, рассматривая определители (8.7) и (8.8).
Решения (8.9) будем искать в виде
с,= сеЮ1. (8.10)
Тогда из первого уравнения системы (8.9) имеем
х=-еа-ет"-1', (8-И)
из второго уравнения —
х=-е'-е'.
откуда
Ш i8(N-l) iQ -Ю
е +е —е +е .
В результате
или
eJ^-k, к=], 2, ..., N. (8.14)
N
Тогда для энергетических уровней циклических полиенов, включа-
включающих N атомов, выполняется общее соотношение
jcjfc=-2cos-^fc, к=1, 2, ..., N. (8.15)
А. Фрост и Б. Мусулин предложили простой н полезный графи-
графический образ уравнения (8.15), показанный на рис. 8.3. Правильный
Л^-угольник, отвечающий молекуле или иону аннулена, вписывается
в окружность, имеющую радиус, равный 2, таким образом, что одна
из вершин TV-угольника лежит на расстоянии радиуса от горизон-
горизонтального диаметра. Последний соответствует нулевому уровню:
х=0, Е—а. Расстояние от него до уровней пересечения окружности
с вершинами TV-угольника определяет, согласно уравнению (8.15),
значения его корней.
Как видно из рис. 8.3, система циклопентадиенила имеет три
263
1,618
связывающих уровня, на кото-
которых можно разместить шесть
я-электронов (число, отвеча-
отвечающее аниону). В нейтральной
системе с пятью я-электрона-
ми одна из связывающих МО
будет недозаполнена и, следо-
следовательно, радикал циклопен-
тадиенила будет менее устой-
устойчивым, чем анион. Но так как
его электроны занимают свя-
связывающие МО, он способен
к существованию, а его пен-
тафенилзамещенные произ-
производные являются стабильны-
стабильными радикалами.
Значительный интерес
представляет катион цикло-
пентадиенила. Теория пред-
предсказывает для него триплет-
ное основное состояние, так
Р как две вырожденные связыва-
связывающие МО должны заселяться
электронами попарно. Поскольку при этом остаются еще две вакан-
вакансии для электронов на связывающих МО, такой катион будет мало
устойчив.
Действительно, существование незамещенного катиона цикло-
пентадиенила
Рис. 8.3. Графическое определение энерге-
энергетических уровней анвуленов в единицах
а—с
+ SbF,
Br H
(SbF5Br)
было доказано лишь недавно. Реакция проводилась в специально
сконструированной ячейке спектрометра ЭПР; спектр ЭПР регист-
регистрировался при температуре жидкого азота в матрице пентаф-
торида сурьмы. Спектр отвечает триплетной электронной кон-
конфигурации катиона с плоской геометрической структурой. Неболь-
Небольшие геометрические искажения могут вести к снятию орбиталь-
орбитального вырождения и устойчивой синглетной электронной кон-
конфигурации. Так, пентафенилзамещенный катион циклопентади-
енила диамагнитен и не обнаруживает в растворах и матрицах
сигнала ЭПР.
264
8.1.4. Бензол. Правило Хюккеля Dл+ 2)
Проблема электронного и пространственного строения молеку-
молекулы бензола хорошо известна. Особая термическая устойчивость
бензола и его производных, стремление молекул этих соединений
сохранять в различного рода химических превращениях неизменной
свою главную структурную единицу — шестичленное сопряженное
кольцо — привели к выделению этих соединений в самостоятель-
самостоятельный, широко разветвленный класс ароматических соединений. Со-
Сопряженные циклические углеводороды и гетероциклические соедине-
соединения, характеризующиеся свойствами, подобными бензолу (термоди-
(термодинамической стабильностью и склонностью к реакциям замещения,
но не присоединения или расщепления), названы бензоидными, а со-
соединения, не обладающие этими свойствами, — небензоидными. На-
Наконец, еще более общее и концептуально важное понятие органичес-
органической химии — ароматичность — также выведено из анализа свойств
бензола и его аналогов.
С точки зрения метода МОХ указанные ранее свойства бензола
определяются строением его электронной оболочки. Шесть я-элект-
ронов бензола размещены на трех связывающих глубоко располо-
расположенных МО (рис. 8.3). Повышенная стабильность соединений с п-
электронным (ароматическим) секстетом давно была известна хи-
химикам, однако именно Э. Хюккель дал ему ясное теоретическое
объяснение: высокая устойчивость циклических сопряженных поли-
енов (аннуленов) объясняется особенностями строения их я-элект-
ронной оболочки и присуща лишь тем из них, которые имеют
полностью замкнутую электронную оболочку, содержащую мак-
максимальное число электронов на связывающих МО и не содержащую
электронов на МО других типов.
Из этого определения понятна устойчивость аннуленов с я-элек-
тронным секстетом (бензол, циклопентадиенильный анион), к аро-
ароматическим соединениям следует также отнести и аннулены, для
которых указанные требования выполнены.
Из формулы (8.15) и рис. 8.3 вытекает, что общее число электро-
электронов, которое можно поместить на связывающие МО аннулена,
складывается из двух электронов в любом аннулене, занимающих
нижнюю связывающую МО, и электронов на попарно вырожден-
вырожденных связывающих МО. Число электронов на каждой паре этих
уровней равно 4, а на л уровнях — 4л. Отсюда замкнутыми элект-
электронными оболочками, в которых все электроны находятся на связы-
связывающих уровнях, и, следовательно, ароматическими свойствами
должны обладать все аннулены или их ионы с общим числом
я-электронов (N), подчиняющимся условию
ЛГ=4л+2;л = 0, 1,2,.... (8.16)
Выражение (8.16) и есть широко известное правило Хюккеля
4л+2, которое объясняет повышенную стабильность бензола (л=1),
265
катиона циклопропенила (л = 0), аниона циклопентадиенила (п —
Кроме того, для и=2, 3, 4, ... правило Хюккеля предсказывает
устойчивость [10]-аннулена, [18]-аннулена, соответственно моноани-
моноанионов [9]-аннулена, [13]-аннулена, монокатионов [7]-аннулена и т. д.
8.1.5. Циклогептатриенил
Уже в первых своих работах Хюккель указал, что в соответствии
с (8.16) катион семичленного циклического сопряженного соедине-
соединения должен отличаться повышенной стабильностью. Это заключе-
заключение теории длительное время не привлекало внимания химиков-
органиков до тех пор, пока В. Деринг A954) не установил, что
хорошо известное солеподобное соединение, получаемое нагревани-
нагреванием продукта присоединения брома к циклогептатриену, имеет стру-
структуру циклогептатриенилбромида (тропилийбромида):
Вг + НВг
XTV
Эта работа, в которой определение структуры и направление син-
синтетического поиска вытекали из выводов теории, сыграла важную
роль в популяризации и дальнейшем широком применении метода
МОХ.
Ни радикал, ни анион циклогептатриенила не удовлетворяют
правилу Хюккеля и, следовательно, неустойчивы. Из диаграммы
энергетических уровней (рис. 8.3) видно, что дополнительные элект-
электроны должны занять антисвязывающие орбитали.
8.1.6. Циклооктатетраен
Теория МОХ предсказывает, что молекула циклооктатетраена
в соответствии с расположением энергетических уровней (см. рис.
8.3) должна иметь на высших МО два неспаренных электрона и,
подобно циклобутадиену, должна быть высокореакционноспособ-
ной и относительно неустойчивой. Тем не менее циклооктатетраен
был еще в 1913 г. синтезирован Вилыптеттером:
Причина кажущегося несоответствия теории и эксперимента
вполне ясна. В отличие от низших аннуленов и их ионов циклоок-
266
татетраен устойчив в виде неплоской конформации типа XIII а,
которая претерпевает быстрые обратимые изомеризации типа «са-
«сама в себя» по двум различным механизмам:
XV а XV в
Первый из них (с константой скорости fci«10 с 1 при 0°С)
обусловлен обычной инверсией кольца, подобной инверсии
шестичленного насыщенного кольца циклогексана. Второй меха-
механизм обусловлен валентной таутомерией: связь а удлиняется, тогда
как связь b укорачивается. В этом превращении плоская конфор-
мация циклооктатетраена является, вероятно, переходным состоя-
состоянием. Валентная таутомеризация XV a-*XW б протекает с более
высокой скоростью, чем инверсия: А:2«70 с при 0°С. К рассмот-
рассмотренным процессам изомеризации добавляется еще внутримолеку-
внутримолекулярная циклизация конформации XV б в цикло-[4, 2, 0]-октатриен-2,
4, 7 (XV с). Эта реакция, имеющая низкую константу равновесия
(~ 10 ~4), относится к типу перициклических реакций (см. гл. 12).
По ряду свойств, прежде всего по устойчивости (теплота об-
образования, атомизация) и спектрам ядерного магнитного резонанса,
высшие аннулены четко разделяют на две группы: аннулены с 4л+ 2
и 4и электронами.
Соединения, относящиеся к первой группе (известны [10]-, [14]-,
[18]-, [22]-, [30]-аннулены), стабильны. Для них характерно сильное
дезэкранирование внешних протонов в результате парамагнитных
кольцевых токов, тогда как внутренние протоны, наоборот, сильно
экранированы и находятся в спектре в области высоких полей.
Именно такими свойствами обладают производные бензола и кон-
конденсированных ароматических углеводородов. 4л-Аннулены менее
устойчивы и характеризуются прямо противоположными эффек-
эффектами в спектрах ЯМР (известны [12]-, [16]-, [20]-, [24]-аннулены).
Хотя по пространственным условиям только для [30]-аннулена до-
достижима ненапряженная плоская конфигурация, уже для [18]-ан-
нулена реализуется структура, близкая к плоской, что объясняется
стремлением к ароматической стабилизации.
267
8.2. ПРАВИЛО An + 2 И ЭЛЕКТРОННОЕ СТРОЕНИЕ
СОПРЯЖЕННЫХ ЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ.
АНТИАРОМАТИЧНОСТЬ
Правило 4л+2 определяет число я-электронов в цикле, соответ-
соответствующее его наиболее выгодной электронной структуре. Оно по-
позволяет ориентироваться в вопросах относительной стабильности
сопряженных карбо- и гетероциклических молекул и ионов, при
оценке их некоторых химических и физических свойств.
Так, можно было ожидать, что в отличие от самого циклобута-
диена его дикатион, имеющий в электронной оболочке всего два
электрона, будет устойчив (см. рис. 8.3). Действительно, Г. Ола
удалось получить устойчивые соли производных этого дикатиона:
(SbF5ci>:
Устойчивым является также дианион циклооктатетраена, в элект-
электронной оболочке которого десять я-электронов (п—2). Он имеет
плоскую геометрическую конфигурацию, характерную для арома-
ароматической системы. Следовательно, не пространственные факторы,
а строение электронной оболочки обусловливает акопланарность
молекулы циклооктатетраена. Интересно, что хотя четыре электро-
электрона дианиона XVI находятся на двух вырожденных несвязывающих
МО, тем не менее замкнутость его я-электронной оболочки обес-
обеспечивает достаточную стабильность XVI:
О
- v ., Х-О, S, NH, Se, Те
XVI XVII
Высокая ароматичность в химическом понимании, т. е. склон-
склонность к реакциям электрофильного замещения в ядре гетероцик-
гетероциклических аналогов циклопентадиенильного аниона (XVII) (фуран,
тиофен, пиррол, селенофен, теллурофен), объясняется тем, что 2рг-
электроны неподеленной пары гетероатома входят в л-систему,
дополняя ее до устойчивого секстета. Также понятна высокая тер-
термическая устойчивость азонина (XVIII) (X=NH), в котором ре-
реализована 10я-электронная оболочка. Его не полностью сопряжен-
сопряженный карбоциклический аналог i/мс-циклононатетраен, напротив, ве-
ведет себя как полиен и быстро полимеризуется уже при слабом
нагревании.
268
В молекулах, в которых сопряженный цикл связан с другой
циклической или нециклической сопряженной системой, правило
4л+2 позволяет легко выявить направление поляризации общего
электронного облака и, следовательно, направление дипольного
момента. Укажем, например, на калицен (XIX):
XIX а
XIX б
Это соединение отличается удивительно высокой для углеводоро-
углеводородов величиной дипольного момента F,3D для гексафенилзамещен-
ного). Если бы распределение электронов в молекуле калицена
правильно отображалось структурой XIX а, его дипольный момент
был бы близок к нулю. Столь большое значение дипольного момен-
момента определяется смещением электронной плотности с трехчленного
кольца, стремящегося оставить на орбиталях лишь два л-электрона,
на пятичленное, охотно приобретающее шестой электрон, недоста-
недостающий до секстета. Подобная поляризация объясняет высокие ди-
польные моменты илидов, например циклопентадиенилиденовых
производных (XX), шестичленных гетероциклов (XXI) и др.:
XX
- CH2Ph
fi-9,7D
Если наличие в сопряженном цикле Dи + 2) я-электронов способ-
способствует стабилизации циклической структуры, то подобные системы
с 4л электронами, наоборот, дестабилизированы по сравнению с не-
нециклическими структурами, имеющими то же количество гс-элект-
ронов. Как уже отмечалось, циклобутадиен крайне реакционно-
способен и склонен спонтанно распадаться на две молекулы ацети-
ацетилена; циклооктатетраен вообще не существует в виде плоской кон-
конфигурации, и даже неплоские [4л]-аннулены малоустойчивы и реак-
ционноспособны.
Р. Бреслоу предложил термин «антиароматичность» для этого
вида электронной дестабилизации сопряженных органических со-
соединений. Далее будет дано более строгое и общее, чем просто
принадлежность к классу [4/2]-аннуленов и их аналогов, определение
антиароматичности. Однако наличие An л-электронов в сопряжен-
сопряженном цикле — важный признак антиароматичности.
269
Аналоги 4л я-электронного циклопентадиенильного аниона —
оксирены, азирины, тиирены (XXII) — существуют лишь как очень
нестабильные промежуточные соединения. В то же время от соеди-
соединения (XXII), (X = BR) можно ожидать стабильности. Согласно
неэмпирическим расчетам, его энергия резонанса составляет более
70% от значения энергии резонанса стабильного цикло-
пропенильного катиона (см. разд. 8.11.3). Весьма нестабильны
и акопланарны, а также склонны к реакциям окисления (т. е. к поте-
потере электронов) дигидропиразины, диокса- и дитиадиены (XXIII). Их
дибензопроизводные типа XXIV легко дают устойчивые катион-
радикалы:
7 О
X'
XXII X=»O,NR,S ХХШ X-NR,O,S XXIV X~NR,S
8.3. РАСЧЕТ КОЭФФИЦИЕНТОВ
ПРИ АТОМНЫХ ОРБИТАЛЯХ В МО ХЮККЕЛЯ
Вычисление корней секулярного уравнения, дающих энергии
МО, — лишь первая часть задачи решения системы уравнений (8.2).
Необходимо еще рассчитать вид МО, т. е. получить значения коэф-
коэффициентов при атомных орбиталях в каждой МО. Рассмотрим
схему расчета на примере первых членов полиенового ряда.
8.3.1. Этилен
Следуя изложенной методике расчета корней секулярного урав-
уравнения, получим для этилена два уровня энергий МО:
СН2=СН2
X 1
1 X
у _- 1
= 0;*2-l=0, (8.17)
Запишем теперь систему уравнений
(8.18)
Подставив значение хг в (8.18), для обоих уравнений получим
-с,+с2=0, или с, = с2. (8.19)
Мы располагаем одним уравнением, в котором два неизвестных.
270
Дополнительное уравнение дает условие нормировки:
Совместное решение уравнений (8.19) и (8.20) дает
1
(8.20)
(8.21)
Таким образом, связывающая МО с энергетическим уровнем
P имеет вид
(8.22)
где Х\ и Ъ — 2р_т-АО двух углеродных атомов этилена.
Для второго энергетического уровня подстановка х2 ~ 1 в уравне-
уравнение (8.18) дает
ci = -c2, (8.23)
что совместно с (8.20) приводит к антисвязывающей МО:
Можно различным образом представить орбитали 4*i и *?2-
На рис. 8.4 показано пространственное распределение элект-
электронной плотности этилена, т. е. значение функций *Р и *F2 в плоско-
Рис. 8.4. Вид МО этилена:
а — радиальные часта; б — квадраты радиальных частей; в — пространственное распределение;
г — схематическое изображение двумя л-^гАО углерода
271
сти, перпендикулярной плоскости молекулы (а, б), и часто употреб-
употребляемый для качественных сравнений вид МО этилена (в, г).
8.3.2. Аллил
Аллильная система является простейшим нечетным полиеном,
т. е. полиеном, включающим нечетное число я-орбиталей (формула
XXV):
Секулярный детерминант для системы XXV представлен в виде
X
1
0
1
1
0
1
X
= 0.
Характеристическое уравнение имеет вид
(8.25)
его корни: *,= —у/5; х2=0; хъ~
Система уравнений для нахождения коэффициента АО в МО
аллила, соответствующая секулярному уравнению (8.25), записыва-
записывается следующим образом:
Вычисляя коэффициенты, получим МО аллила (начиная с нижней
связывающей МО):
2
(826)
ш 1
272
Рис. 8.5. Вид МО аллила:
а — радиальные части; б — пространственное распределение; в — схематическое изображение
тремя pz-AO углерода
На рис. 8.5 МО аллила представлены в графической форме.
8.3.3. Бутадиен
Молекула бутадиена XXX является простейшим диеном.
Многие особенности реакционной способности диенов (см. гл,
13) могут быть поняты и предсказаны исходя из формы их МО. Дл«
бутадиена МО можно рассчитать по той же схеме, что и для аллила,
однако целесообразно применить упрощающий расчеты способ,
который учитывает симметричность ядерной конфигурации бутади-
бутадиена:
XXVIa
Г\
XXVI6
Устойчивой конформацией бутадиена является трансоидная кон-
формация XXVI а, которая выгоднее цисоидной XXVI б на
2,3 ккал/моль (9,6 кДж/моль). Однако метод МОХ не различает
структуры XXVI а и XXVI б, так как он по своему существу
является топологическим, т. е. учитывает только последователь-
27:
ность атомов, которые образуют химические связи, но не их точное
геометрическое расположение.
В общем случае, когда молекула имеет элементы симметрии,
расчет, а также классификацию, МО следует проводить на основе
аппарата теории групп (см. гл. 6). Воспользуемся упрощенным
подходом, введенным Ч. Коулсоном, который особенно полезен
для плоских сопряженных молекул. Так как в методе Хюккеля
важна только топология связывания, то молекулу бутадиена можно
представить в виде
12 3 4
что, естественно, не отражает истинного геометрического строения.
Учитывая симметрию, следует ожидать лишь следующих соотно-
соотношений между коэффициентами АО и МО бутадиена:
с, = с4г с2=с$ — симметричная МО E), (8.27)
сг = — с4, с2 = — с3 — антисимметричная МО (А). (8.28)
Применим эти соотношения к системе уравнений типа (8.2) бутади-
бутадиена:
Ci*+c2 = 0;
Ci + c2x+c3=0;
с2+съх+с4=0ш>
сг+сАх=0.
X
1
0
0
1
X
1
0
0
1
X
1
0
0
1
X
=0.
(8.29)
При условии (8.27) система четырех уравнений сводится к систе-
системе двух уравнений:
откуда
X 1
jc1= -1,618; х2=0,618.
Из условия (8.28) получаем
—1)=0;
х 1
1 х-\
=0;
274
лгэ = 1,618; дг4= —0,618.
Теперь можно легко определить коэффициенты при соответству-
соответствующих МО бутадиена:
?,=<*+ 1,618ft ?i=a+0,618ft ?,=a-0,618ft ?,=a- 1,618ft
У, = 0,372 Xi + 0,602 xi+0,602 Хъ + 0,372 *4, S;
Т2=0,602х1 + 0,372Л-0,372Л-0,602Л, A; (8.30)
% - 0,602 xi - 0,372 xi - 0,372 Хъ+0,602 ^, 5;
Рис. 8.6 иллюстрирует представленные данные.
н
Рис. 8.6. х-Молехулярные орбнтали бутадиена:
а — радиальное распределение вдоль молежулы; б — схематическое иэобра
прострашл пенное распределение
Pz-AO; в —
275
Задача 8.1. Применяя классификацию МО по типу А я S, рассчитать МО
аллильной системы и молекулы циклобутадиена в квадратной конфигурации.
8.3.4. Коэффициенты при АО в векторной и матричной формах.
Уравнения метода Хюккеля
как задача на собственные значения
Запишем коэффициенты при АО в различных МО бутадиена
в виде матриц-столбцов:
(8.31)
Представим матрицу, отвечающую определителю (8.29), в виде
суммы двух матриц: матрицы А и единичной матрицы / [учтено
соотношение (8.5)]:
(8.32)
=А-е/. (8.33)
Умножим матрицу А на вектор-столбец с{:
276
/0,372<х+0,602А
/ 0,602а + 0,974/7 \
i 0,602*+0,974? J
\0,372а+0,602/?/
= (a+l,618/f) АШ (8.34)
В более компактном виде
Act = *ici- (8.35)
Таким образом, уравнения метода Хюккеля сводятся к задаче на
собственные значения матрицы А, названной Н. Хэмом и К. Рю-
денбергом топологической:
=е,с,, (8.36)
Таким образом, изучение свойств топологической матрицы А по-
позволяет исследовать особенности сопряженных систем в рамках
модели Хюккеля. Отсюда вытекает связь метода МОХ с математи-
математической теорией графов, обращение к которой позволило получить
множество обобщений для класса сопряженных молекул.
Векторы с, (8.31) рассматривают как собственные векторы мат-
матрицы А, соответствующие собственным значениям е,. Так как
с]с^\, (8.37)
то, следовательно,
clAci=Bit (8.38)
где символ «т» означает транспонирование.
Построим квадратную матрицу с из векторов:
(8.39)
Матричное произведение с Ас равно
'ос+1,6180 0 0 0
0 а + 0,6180 0 0
0 0 а-1,6180 0 •¦ (8'40)
0 0 0 а-
Итак, матрицу А можно привести к диагональному виду в ре-
результате ортогонального преобразования при помощи матрицы с.
Видно, что диагональные элементы диагонализованной матрицы
А (8.40) являются корнями секулярного уравнения (8.29). Отсюда
ясен алгоритм нахождения решений уравнений метода Хюккеля
с помощью ЭВМ. Сначала необходимо составить матрицу А, кото-
которая отличается от секулярного уравнения тем, что в ней опущены
неизвестные значения орбитальных энергий ?,. Затем эту матрицу
277
приводят к диагональному виду с помощью некоторого унитарного
преобразования. Тогда диагональные элементы диагонализованной
матрицы являются корнями секулярного уравнения, столбцы мат-
матриц унитарного преобразования — коэффициентами МО ЛКАО.
8.4. ВЫСШИЕ ПОЛИЕНЫ
Для энергетических уровней линейных полиенов с общей форму-
формулой cNHN+2> так же как и для аннуленов, в методе МОХ можно
найти общее решение. Запишем уравнения, обобщающие уравнения
(8.29), для случая бутадиена (iV=4):
С2+С!х+С4==0:
Будем искать решение в виде
Из первого уравнения (8.41) имеем
С 20
—-
sin0
= -2cos0. (8.43)
Подставляя (8.42) и (8.43) в уравнение (8.41) и проводя несложные
тригонометрические преобразования, получим
(),
- sin N9 cos в+cos N6 sin 9+2 cos в sin NB=
=cosJ/V0sm0+siniV0cos0=sin(iV+1H=0. С8-44)
Из решения уравнения (8.44) получим возможные значения 0
^. Ь=1, 2, ..., N, (8.45)
и, следовательно, энергетические уровни полиенов можно найти из
общего соотношения
пк
пк - (8.46)
—.
Нормируя множитель с в выражении (8.42) и используя (8.45), легко
278
получить общее соотношение для коэффициентов при АО в МО
полнена:
(8.47)
] Л lj -^j . .., i»>
где к — индекс МО; /х — индекс АО.
Задача 8.2. Получите нормировочный множитель с в выражении (8.42).
Соотношения (8.46) представляют интерес для анализа возмож-
возможности получения органических соединений с высокой электронной
проводимостью. У сопряженного полнена можно ожидать появле-
появления металлической проводимости, если энергетическая щель между
заполненными и незаполненными МО будет близка к нулю. Из
формулы (8.46) действительно следует, что для четного полнена при
/ Ns
N-юо различие в энергиях между высшей заполненной МО ( к—-
и низшей свободной
К-)
стремится к нулю (рис. 8.7). Экс-
Экспериментально показано, что величина энергетической щели при
N-* оо в полиене стремится к определенному предельному значению
~2 эВ. Такое несоответствие объясняется недостатками как метода
МОХ, так и той модели (полная выравненность всех связей), для
которой справедливо соотношение (8.46).
Заметим еще, что для нечетного полнена, для которого значение
N+1 в формуле (8.46) будет четным числом, всегда возможна
величина целого числа к=—-—. При этом среди значении уравне-
xN+l
ния (8.46) будет присутствовать величина —- = 0, т. е. для любого
о
N=2
N=3
¦~tt6t8
. -OfiW
¦ +0J&18
+ 1,6 W
a
2
-2
2эВ
П =oo
Рис. 8.7. Зависимость молекулярных уровней энергии от длины полнена при TV-* со
(JV — четное число):
а — расчет МОХ; б — эксперимент
279
N=2 (этилен)
К 618
-1618
N=4 (бутадиен)
-I
Ы=5'(пентадцен)
Рис. 8.8. Графическая иллюстрация орбитальных энергий линейных полиенов
нечетного полнена существует один несвязывающий уровень, что
показано на рис. 8.7 (подробнее см. разд. 8.9). По аналогии с ан-
нуленами можно и формулу (8.46) для орбитальных энергий поли-
полиенов CNHff+i интерпретировать графически. Действительно, если
первую половину окружности радиусом 2/? разделить на N+ 1 рав-
равных дуг (рис. 8.8), то ординаты точек деления, за исключением
лежащих на вертикальном диаметре, соответствуют орбитальным
энергиям.
8.5. ЦИКЛИЧЕСКИЕ ПОЛИЕНЫ,
ИМЕЮЩИЕ ВИД КОЛЕЦ МЕБИУСА
Представим себе плоскую линейную систему базисных р-АО
(рис. 8.9). При замыкании этой системы в цикл без внутреннего
изгибания цепи все орбитали сохраняют свою фазу по отношению
280
Рис. 8.9. Исходная линейная система базисных />-орбиталей (а); циклическая хюк-
келевская система орбиталей (б); циклическая мебиусовская система орбиталей (в):
стрелка указывает инверсию фазы в базисной системе орбиталей
к соседним, т. е. интеграл перекрывания между любыми соседними
орбиталями имеет положительный знак (рис. 8.9, б^ Это один из
примеров та? называемой хюккелевской циклической системы ор-
орбиталей. шменно так выбраны исходные системы базисных ор-
орбиталей при рассмотрении аннуленов и линейных полиенов.
Если по пространственным причинам, создаваемым, например,
подбором в соответствующих местах полиеновой цепи объемистых
заместителей, добиться закручивания цепи так, что один конец цепи
развернется на 180° относительно другого, а затем замкнуть оба
конца в кольцо, получим так называемую мебиусовскую* цикличес-
циклическую систему орбиталей (рис. 8.9, в).
Можно сделать дальнейшее обобщение и охарактеризовать цик-
циклический полиен как систему хюккелевского типа, если общее число
узлов (инверсий фазы в базисной системе орбиталей) четное @, 2, 4,
...), или мебиусовского типа, если число таких узлов нечетное A,3,
5, ...).
Впервые общее решение для циклических полиенов, имеющих
вид колец Мебиуса, получил Е. Хейльброннер A964). Составим
систему линейных однородных уравнений для определения МО
и орбитальных энергий полнена мебиусовского типа. Будем пола-
полагать, что узел возникает один раз между первым и N-м. атомами
углерода. Тогда по аналогии с (8.9) имеем
(8.48)
Система (8.48) отличается от (8.9) изменением знака на обратный
перед cN в первом и перед с^ в последнем уравнении. Это связано
с тем, что между соответствующими атомами внефазное перекры-
*Август Фердинанд Мебиус A790—1868) — один из крупнейших геометров
XIX в. Его важнейший вклад в топологию — открытие односторонних поверхно-
поверхностей, примером которых служит известный лист Мебиуса. Аналогия с последним
и является основанием для введенного обозначения.
281
вание АО обусловливает замену
резонансного интеграла р на
—р. Будем искать решение в ви-
виде (8.10). Тогда из первого урав-
уравнения системы (8.48) имеем
-1,618
Рис. 8.10. Графический расчет энергий
МО мебиусовских циклических полиенов
х-~е +е
из второго уравнения —
Ш -В
х= —е —е .
В результате
№
'_ 1
ИЛИ
= пBк+\), 9=
N
-_1 о JV—1
¦ —1, X,, ..., IV — 1.
(8.49)
Тогда для полиенов мебиусовского типа получаем простое выраже-
выражение для корней характеристического уравнения:
хк— —2 cos
N
-,*=1,2, ...,N-.
(8.50)
Это выражение очень близко к соотношению (8.15), полученному
для хюккелевских циклических полиенов. Сходство и различие осо-
особенно ясно проявляются при графическом изображении энергети-
энергетических уровней мебиусовских систем. По аналогии со способом
Фроста и Мусулина следует вписать правильный ЛГ-угольник в ок-
окружность радиусом 2р, Однако ориентация iV-угольника должна
быть иной (рис. 8.10).
Задача 8.3. Составьте секулярное уравнение для мебиусовского циклобута-
днена и рассчитайте энергии МО и их вид. Проверьте, что при выборе базисной
системы орбиталей с двумя инверсиями для базисных АО получают те же
решения, что и представленные для хюккелевского цвклобутадиеяа.
Пользуясь рис. 8.10, можно убедиться, что электронные обо-
оболочки циклических полиенов мебиусовского типа замкнуты при
условии
(8.51)
которое для них можно условно назвать правилом ароматичности.
Обобщая (8.16) и (8.51), сформулируем правила ароматичности для
циклических полиенов (табл. 8.1).
282
Таблица 8.1. Прамла ароматячносгш для щпслическжх напевов
Число эяежтровов
4/1+2
4л
Тип системы
хюккелевак&я
Ароматическая
Антиароматическая
мебиусовская
Антиароматяческая
Ароматическая
Условия устойчивости циклических полиенов находят широкое
применение при анализе реакционной способности органических
соединения, в первую очередь их перициклических реакций (см. гл.
в) ^\
8.6. МОЛЕКУЛЫ С ГЕТЕРОАТОМАМИ
Рассмотрим в качестве примера молекулу формальдегида XXVII
2 1
XXVII
В общем виде секулярный детерминант для этой молекулы
представлен при обычных допущениях метода Хюккеля как
R «
Р12 «2 —
Из трех параметров этого уравнения аь fin и а2 известен лишь
последний — кулоновский интеграл ^р2-углеродного атома а. Значе-
Значение резонансного интеграла для связи С=О можно выразить через
резонансный интеграл связи С = С:
Рп=Рсо=Кр. (8.53)
Подобным образом запишем выражение для кулоновского интег-
интеграла кислорода:
*1 = <*о=«+Л0- (8.54)
В соотношениях (8.53), (8.54) h и К — безразмерные параметры.
Подставив <Xi и /}12 из указанных соотношений в (8.52) и поделив
каждую колонку на /?, получим
а~Е
1
+ А К
%-Е
==о.
(8.55)
283
Используя обозначение (8.5), имеем
= 0. (8-56)
К
К х
Таким образом, задача расчета соединений с гетероатомами
сводится к задаче определения параметров Л для кулоновского
и К для резонансного интегралов гетероатома и связей, которые он
образует.
Поскольку метод МОХ — полуэмпирический, общий способ
оценки К и h состоит в следующем. Если требуется провести расчет
каких-то конкретных характеристик, например дипольных момен-
моментов группы соединений, включающих, допустим, .ур2-гибридизован-
ный азот в системе сопряженных связей, проводят расчеты несколь-
нескольких стандартных соединений, варьируя параметры, и выбирают
затем те из них, которые лучше воспроизводят экспериментальные
значения. С этими параметрами рассчитывают затем остальные
соединения.
Такой способ оценки приводит неизбежно к большому числу
разных значений параметров, которые могут зависеть от харак-
характеристики, используемой для их выбора. Однако для стандартиза-
стандартизации можно воспользоваться обоснованными общими соображени-
соображениями об относительных значениях параметров.
Так как величина кулоновского интеграла симбатна электроот-
электроотрицательности атома, можно ожидать существования связи между
ними. Для атомов, вносящих в 7г-систему один электрон, пред-
предложена следующая корреляция между электроотрицательностями
Полинга* и А:
(8.57)
Если атом вносит в я-систему два электрона, например азот в ани-
анилинах, кислород в фуране и др., его эффективная электроотрицате-
электроотрицательность будет выше, так как электроны находятся в поле двухполо-
жительного атомного остова и притягиваются к нему сильнее.
В этом случае значение А должно быть увеличено.
Значения К для резонансных интегралов подбирают эмпиричес-
эмпирически. Хорошим ориентиром для такого подбора служит ожидаемая
пропорциональность между резонансными интегралами и интег-
интегралами перекрывания:
^сх=PcxlРос = Sod Sec- (8.58)
Это соотношение автоматически учитывает различие в интег-
интегралах перекрывания для связей, образованных гетероатомами
X и X. Его используют также для расчета резонансных интегралов
¦Элепроотрицательности элементов по Подвигу приведены в таблице Менделе-
Менделеева на форзацах книги.
284
углерод-углеродных связей, длины которых отличаются от арома-
ароматической связи С—С (/=0,1397 нм). Так, для формально простой
связи — С—С= в бутадиене (/=0,146 нм) К=0,9, а двойной связи
бутадиена @,134 нм) ЛГ=1,1.
Задача 8.4. Рассчитать полную энергию прямоугольной структуры цикло-
бутадиена, приняв для двойной и одинарной связи резонансные интегралы
Р соответственно равными 1,1 и 0,9, и сравнить с энергией центросимметричяой
структуры.
Таблица 8 2. Параметры гетероатомов н образуемых ниш связей, применяемые
в методе МОХ
Атом
С
N
N
N +
6
*х
0
0,5
1,5
2,0
1,0
Связь
С"'О
*- V"ap
=С-С =
>с«с<
-N=C<
-N+=C<
O = C<
К
1
0,9
1,1
1
i
0,8
0,6
1
1
Атом
6
6:
s
s
•
в
**
F
a
Ax
2,0
2,5
0,4
1,3
-1
3,0
2,0
Связь
-0-6=
-o+=c<
s=c<
1 ~
B-C=
B-N~
F-C=
"* 1 ~
К
0,8
1
1
0,6
0,7
0,6
0,7
0,4
Из многих систем параметров наибольшее распространение
получил набор параметров Стрейтвизера, который может быть
рекомендован для большинства случаев, где расчет по методу
Хюккеля целесообразен. В табл. 8.2 приведены параметры
для наиболее часто встречаемых атомов и их валентных со-
состояний.
Кроме системы Стрейтвизера употребляют также близкую си-
систему параметров Пюльманов и набор Оргела, который часто
используют при вычислении спиновых плотностей.
Из табл. 8.2 выберем параметры для карбонильного кислорода
формальдегида и подставим значения h и К в (8.56):
Корни этого уравнения: хг = — 1,618; х2=0,618.
Диаграмма энергетических уровней молекулы формальдегида
в сравнении с этиленом приведена на рис. 8.11. Из диаграммы
видно, что увеличение электроотрицательности одного из атомов
в сопряженной системе приводит к «проседанию» уровней. При
этом связывающая МО стабилизирована значительно выше (на
0,618)?), чем антисвязывающая @,382Д. Этот эффект является об-
285
щим для любых сопряженных си-
систем с гетероатомами произведен-
произведенных из изоэлектронных им углево-
углеводородов.
Уравнения для расчета коэффи-
коэффициентов при АО формальдегида со-
соответствуют его секулярному дете-
детерминанту (8.59):
С1(дс+1)+с2=0;
a-fi
Of
a+J3
Рис. 8.11. Диаграмма энергетичес-
энергетических уровней молекул этилена
и формальдегида
Их решение приводит к МО
?, = 0,850 *,+ 0,525 *2
?2=0,525*,-0,850 *2,
которые показаны на рис. 8.12. Как уже отмечалось при рассмотре-
рассмотрении МО двухатомных молекул, увеличение электроотрицательности
одного из атомов ведет к вспучиванию МО на его АО в связыва-
связывающей и противоположному эффекту в антисвязывающей МО.
(8.60)
Ряс. 8.12. я-Молекулярные орбитали формальдегида:
а — радиальные части; 6 — схематячесжое вэображевие двумя Р^АО; в — пространственвое
рвспредслсяне
286
Задача 8.5. Получать МО молежулы пиридина, используя ее свойства сим-
симметрии.
8.7. ЭЛЕКТРОННЫЕ ПЛОТНОСТИ, ЗАРЯДЫ, ПОРЯДКИ СВЯЗЕЙ
И ПОЛЯРИЗУЕМОСТИ, МАТРИЦА ПЛОТНОСТИ ПЕРВОГО ПОРЯДКА
При описании результатов расчета волновых функций отдель-
отдельных молекул и (в целях сопоставления их с экспериментальными
характеристиками оказывается полезным ввести ряд понятий, опре-
определяемых значениями коэффициентов АО в МО и энергиями по-
последних.
Электронная плотность. Полная электронная плотность, создава-
создаваемая я-электронами сопряженной молекулы, равна в приближении
ЛКАО МО:
— 00 —00 —00
••со
(8.61)
i \ и J
— 00 —00
где л, — число электронов на ьй МО; N — общее число я-электро-
нов в молекуле. Коэффициенты с^ при АО предполагаются дейст-
действительными. При условии ортонормированности АО х? и Zv, прини-
принимаемом в методе МОХ, двойная сумма в (8.61) обращается в нуль
и это выражение сводится к сумме:
?*cJ,+2>4+«.+5>i<i=tf- (8.62)
i ; i
Каждую сумму в выражении (8.62) можно приближенно рас-
рассматривать как вклад электронов, находящихся на орбитали /1-го
атома, в полную электронную плотность. Соответственно выра-
выражение
**.в2>4 (8-63)
I
называют электронной плотностью на ц~м атоме (физически элект-
электронная плотность — это величина еР№, где е — заряд электрона).
Очевидно, что существует тесная связь определения (8.63) с элек-
287
тронной заселенностью атомной орбитали G.4), введенной Мал-
ликеном.
Рассчитаем электронные плотности на атомах в аллильном ка-
катионе, радикале и анионе, воспользовавшись рассчитанными для
этой системы МО [см. (8.26)]:
катион (л, =2): ?11 = Рзз = 2A/2J-0,5; Р22=2 Г-^V —1;
радикал (nt=2, и2 = 1): Ри = Рп = 2 - + —7=] =1;
/v w u;
анион (и, =1^ =
Pv> = 2\—=\ =
Определение электронной плотности (8.63) имеет лишь прибли-
приближенный физический смысл. Действительно, хотя на рис. 8.4, 8.5, 8.6,
8.12 полная электронная плотность Q?2 для связывающих МО)
имеет максимумы в районе ядер атомов, она весьма значительна
и в межъядерной области, что и обеспечивает связывание. Тем не
менее выражение (8.63), общее для всех методов НДП, пренебрега-
пренебрегающих перекрыванием, является полезным и наглядным приближе-
приближением.
Заряд атома определяется выражением
q»=nr-Piv (8.64)
где г}р — число электронов, вносимых атомом ^ в общую я-систему;
^м=1 для углерода, пиридинового азота, карбонильного кислорода
и т/м=2 для пиррольного и анилинового азота, фуранового, феноль-
ного, оксониевого кислорода и пр.
Заряды на атомах для производных аллильной системы опреде-
определяют по рассчитанным ранее величинам:
катион ^1 = ^3 = 0,5,^2 = 0;
радикал ql=q2=q:i = 0;
анион Ч\—Чг~— 0»5, q2 = 0.
Для молекулы формальдегида с помощью соотношений (8.63)
и (8.64) легко вычислить электронные плотности и соответствующие
заряды на атомах 1 (кислород) и 2 (углерод):
Рп-1,445; Р22=0,555; qx= -0,445; q2= +0,445.
288
Ясно, что при изменении чисел заполнения МО —* и, соотноше-
соотношение (8.63) позволяет оценить перераспределение я-электронной
плотности при электронном переходе. Так, для п =п*-возбужден-
=п*-возбужденного состояния молекулы формальдегида щ = 1, пг= 1 имеем:
^1 = 1,0;^= 1,0; Й«0;й=0.
В результате возбуждения атом кислорода, имевший в основном
состоянии большую электронную плотность, теряет ее, а атом
углерода — приобретает. Подобные перераспределения элект-
электронной плотИости при электронных переходах весьма важны для
интерпретации реакционной способности молекул в ходе фотохими-
фотохимических реакций.
Порядок связи. Вследствие отмеченной значительной элект-
электронной плотности в пространстве между связанными атомами в мо-
молекуле необходимо ввести в метод МОХ выражение для ее харак-
характеристики. Ч. Коулсон* A939) предложил использовать в этих це-
целях величину так называемого порядка подвижной связи (или про-
просто порядка связи) между атомами р. и v:
'-С^С«-. (8.65)
i
Эти члены возникают как коэффициенты при интегралах перекры-
перекрывания в двойной сумме (8.61) и вследствие ортонормированности
АО исчезают вместе с интегралами перекрывания. Выражение (8.65)
предполагает все же, что, если произведение с^с* достаточно велико
(и положительно), существует значительная вероятность найти
электрон в i-й МО в пространстве между атомами у. и v. Структура
определения (8.65) тесно связана с межорбитальной заселенностью
Малликена G.4).
Смысл порядка связи и его соответствие со сложившимися
в химии представлениями о степени связности иллюстрируются
подсчетом величины Р12 этилена. Воспользовавшись коэффициен-
коэффициентами связывающей МО (8.22) этилена, имеем
P,2=2-L-L=i
12 фф
Поскольку это порядок лишь я-связи, для определения общего
порядка связи его следует суммировать с порядком с-связи, прини-
принимаемым в МОХ для любых связей равным единице. Общий порядок
связи равен двум.
В случае этилена имеем максимально возможный для любой
системы порядок л;-евязи. Для делокализованных систем с большим
¦Чарльз Коулсон A910—1973) — английский математик, доктор теологии. Ши-
Широко известен своими работами по теории химической связи.
10. Теория строения молекул
числом центров порядки связей имеют более низкие значения. Так,
например, для аллильных производных:
катион Л2=Лэ=2--«-^==0,707; Р,3=2--=-;
радикал Р12 = Р23 = 2-—-=—= = 0,707;
Лэ=2-~0
22
анион Р12 = Р23 = 2
-2П 2
— Z,-- — Z
22.
Рассчитанные порядки связей для соседних атомов указывают на
их полную выравненность в катионе, радикале и анионе. Об этом же
свидетельствуют приведенные ранее величины электронных плот-
плотностей и зарядов.
Имеется еще один аспект сделанного расчета порядков связей.
В аллильном катионе между концевыми атомами существует значи-
значительное связывание Р13=- указывающее на тенденцию к цикличес-
циклической циклопропенильной Dи + 2)-системе:
В радикале это связывание отсутствует, а в анионе порядок связи —
отрицательный, что соответствует антисвязыванию и невы-
невыгодности перехода к циклической анти ароматической системе
циклопропенильного аниона.
Другая интерпретация Рт н /%,. Можно связать понятия элект-
электронной плотности и порядка связи с полной энергией молекулы Е.
В вариационном методе Ритца энергия определяется выражени-
выражением A.62):
iftr
290
В хюккелевском приближении для отдельной нормированной МО
это выражение можно записать так:
. (8.66)
Двойное суммирование проведено только по соседним атомам.
В методе МОХ, где не учитывается отталкивание между элект-
электронами на различных МО (так же как и двух электронов на одной),
полная энергия есть просто сумма энергий отдельных электронов:
|Л- (8.67)
С учетом (8.66)
(8.68)
Рассмотрим, как изменится величина полной энергии с измене-
изменением кулоновского интеграла ам. В первом порядке теории воз-
возмущений
?
ЗЧ(Н?)®Ь
Полная энергия является минимумом по отношению к любому
способу варьирования коэффициентов при АО, а так как элект-
электронные плотности представляют собой функции этих коэффициен-
коэффициентов, все члены суммы в уравнении (8.69) не отличаются от нуля. Из
(8.68) следует, что
дЕ
Р (8-70)
(8.71)
Подобным образом можно показать, что
1 дЕ
(8-72)
Итак, соотношения (8.68), (8.70), (8.72) устанавливают связь
между энергией молекулы и электронными плотностями на атомах
и порядками связей.
Атом-атомные и связь-связевые поляризуемости. Изменение куло-
кулоновского интеграла на атоме \l отразится на величине полной
291
энергии так же, как возмущение второго порядка, так как изменение
электронной плотности на атоме ц повлечет за собой перерас-
перераспределение электронных плотностей на других атомах. Степень
такого перераспределения характеризует величины атом-атомных
поляризуемостей:
д2Е др„дрт
(8.73)
Подобным образом определяются связь-связевые поляризуемости:
дР*> 1 д2Е
, I*~™— г -я яя = пь, ^. (8.74)
дрь 2 дрх op K J
Атом-атомные поляризуемости, как и связь-связевые, можно
представить в виде зависимости от коэффициентов разложения МО
по АО. Предполагая, как и ранее, что коэффициенты при АО
действительные, можно получить соотношение
заисвоб
**= ~4S Z Тг-Б • (8.75)
Для самополяризуемости это выражение выглядит как
заясвоб /Л *Л
-^ (8.76)
и является, следовательно, всегда отрицательной величиной. В рам-
рамках метода МОХ при определении энергий МО (?,, Е,) в единицах
(- ), и
атом-атомные поляризуемости получаются в единицах (- ), и так
как р — отрицательная величина, то самополяризуемости в этих
единицах всегда положительны. Это понятно, так как из (8.73) имеем
Ар1Щ=п1ЩАаA, (8.77)
т. е. увеличение кулоновского интеграла на атоме /г должно приво-
приводить к увеличению электронной плотности на этом атоме.
Матрица плотности первого порядка имеет следующий вид:
(8.78)
Например, для молекулы бутадиена в рамках метода Хюккеяя эта
матрица такова:
292
1 0,896 0 -0,446'
0,896 1 0,446 0
Р=! 0 0,446 1 0,896
-0,446 0 0,896 1
Задача-З.б. Рассчитайте элементы матрицы плотности (8.79), используя
МО (8.30)./
Матрица плотности первого порядка позволяет вычислить лю-
любое одноэлектронное свойство того состояния системы, к которому
она относится. Если обозначить это свойство Л, то его среднее
значение равно следу произведения матриц Р и Л:
(8.80)
р v
В качестве примера рассмотрим вычисление энергии молекулы бу-
бутадиена в приближении МОХ. Умножая матрицу А (8.32) на матри-
матрицу Р (8.79) и суммируя диагональные элементы, получим
'а 0 0 0\ / 1 0,896 0 -0,446'
0 а 0 0 I / 0,896 1 0,446 0
?=<А>=Тг J I I
0 0 р а/ \- 0,446 0 0,896 1
= а + 0,8960 + а + 0,896/?+0,446 р+а + 0,8960+0,4460 + а +
+ 0,8960=4а+4,4760.
Это значение с точностью до погрешности вычисления совпада-
совпадает с энергией бутадиена, вычисленной по соотношению (8.67):
8.8. РЕШЕНИЕ УРАВНЕНИЙ МЕТОДА ППП
ДЛЯ МОЛЕКУЛЫ ФОРМАЛЬДЕГИДА.
СОПОСТАВЛЕНИЕ С ХЮККЕЛЕВСКИМ РАСЧЕТОМ
На примере молекулы формальдегида легко проиллюстриро-
проиллюстрировать проведение расчета на основе метода, учитывающего куло-
новское отталкивание электронов и, как следствие, требующего
проведения итерационной процедуры самосогласования. Кроме то-
того, этот расчет позволит сравнить полученные результаты с дан-
данными метода МОХ. Процедуру самосогласования для молекулы
формальдегида можно провести без использования ЭВМ, так как
необходимо диагонализовывать матрицу второго порядка. Чтобы
293
не затруднять расчет, проведем его с фиксированной длиной связи
С = О, для которой примем, что кулоновский интеграл
уп~8,583 эВ, а резонансный интеграл ^12= —2Д6 эВ. Остальные
параметры равны: уц= 15,227; 722= 11,16; ии= —17,697, «22— — И,16
(нумерация атомов показана на структуре XXVII). Положим внача-
вначале Pn = p22=l, тогда, согласно G.63), имеем в нулевом приближе-
приближении:
FYl= - 17,697 + Va" I' 15,227= -10,084 эВ;
-Fi2=—2,16 —V2-1 -8,583= -6,452 эВ;
,Р22=-11,16+V21 11,13= -5,595 эВ.
= 0;
Теперь необходимо диагонализовать матрицу D.70), которая в на-
нашем случае примет вид
Fn-E Fl2
l2 F22-
Подставляя числовые значения, получим:
10,084 -в -6,452
-6,452 - 5,595 -в
?2 + 15,678е+14,795 = 0;
е,= -14,67 эВ; ?2=-1,01 эВ.
Затем найдем коэффициенты С^ и, следовательно, вид МО. Для
этого необходимо подставить значения энергии в уравнения D.69)
и решить систему линейных однородных уравнений. Найдем коэф-
коэффициенты МО ЛКАО для низшего энергетического уровня ?i (вто-
(второй индекс у коэффициентов опустим):
'(- 10,084 +14,67) с,- 6,452 с2 = 0
14,67) с2 = 0.
Отсюда с2 — 0,711 сх. Используя условие нормировки cf + ^= 1, полу-
получим значения с, =0,815; с2 — 0,579. Для второго значения энергии
( — 1,01) также можно найти коэффициенты в разложении МО по
АО. Однако в данном случае можно сразу записать, что с{ — 0,579;
с2=—0,815. Тем не менее вид этой МО не влияет на процедуру
самосогласования, так как вторая МО является незанятой (вирту-
(виртуальной).
Вычислим теперь значения элементов матрицы плотности (8.65)
в первом приближении:
294
Рп=*2-0,815-0,815 = 1,
Pi2=2'0,5790,815 = 0,
Р22 = 20,5790,579 = 0,
С полученными значениями Р^ч необходимо найти новые матричные
элементы F^:
~l)-8,583=-10,402 эВ;
JF12--^16-1/20,9448,583=-6,213 эВ;
F21=- 11,16 + 1IJ-0,671 ¦ 11,13 + A,329-1) 8583 =-4,603 эВ.
Диагонализуем новую матрицу:
-10,402-е -6,213
-6,213 -4,603-?
?!=- 14,36 эВ; ?2=-0,65 эВ.
Для низшего значения энергии
п = 1,423; Р,2 = 0,906; Р22 = 0,
-0;
Ясно, что в процессе самосогласования коэффициенты cifl для ва-
вакантных орбиталей можно не вычислять, так как они не влияют на
матрицу Р^ и, следовательно, на значения матричных элементов
оператора Фока. Продолжая самосогласование, получим данные,
которые сведены в табл. 8.3.
Таблица 8.3. Изменения е,-, Рцу и иолной энергии Ел молекулы формальдегида
в процессе самосогласования
Номер итерации
1
2
3
4
5
-14,67
-1,0086
-14,3588
-0,6466
-14,197
-0,615
-14,178
-0,613
-14,1260
-0,6164
PflV
1,3286
0,6714
0,9444
1,4230
0,5770
0,9061
1,4332
0,5668
0,9014
1,4586
0,5408
0,8882
1,4672
0,5326
0,8839
En
-40,4483
-40,5439
-40,5462
-40,5503
-40,5526
|А* /ivl max
0,3286
0,0944
0,0102
0,0260
0,0086
295
Продолжение табл. 8. 3
Номер итерации
6
7
S
9
Ч
-14,1082
-0,6142
-14,1020
-0,6124
-14,0990
-0,6114
-14,0978
-0,6130
1,4706
0,5294
0,8824
1,4718
0,5282
0,8817
1,4724
0,5276
0,88В
1,4725
0,5275
0,8813
-40,5564
-40,5568
-40,5570
-40,5571
0,0032
0,0012
0,0006
0,0001
— максимальная разность между элементами матрицы плотности л-й
и (л—1)-й итерации.
Процедуру самосогласования проводят до тех пор, пока jAP^l „,„
не станет меньше заданного числа S. Обычно 6 принимают равным
10~4 —10~5. В некоторых случаях сходимость может достигаться
более медленно, но обычно 15—20 итераций бывает достаточно для
достижения точности 10. Иногда элементы матрицы плотности
осциллируют или даже не стремятся к пределу (отсутствие сходимо-
сходимости). В этих случаях применяют специальные процедуры ускорения
и достижения сходимости.
Хюккелевский расчет молекулы формальдегида (см. разд. 8.6)
привел к значениям элементов матрицы плотности Рп = 1,445;
Рп~0,892; Р22 = 0,555, которые находятся между значениями Р^,
соответствующими третьей и четвертой итерациям метода ППП
(е=10~2). Такая ситуация сохраняется обычно и в расчетах более
сложных молекул. В связи с этим иногда в качестве начальных
Р^ берут хюккелевские значения. Такое приближение сокращает
количество итераций на две-три и вряд ли существенно для ускоре-
ускорения сходимости.
8.9. СВОЙСТВА АЛЫЕРНАНТНЫХ УГЛЕВОДОРОДОВ
Еще в 1940 г. Ч. Коулсон и Дж. Рашбрук ввели важное деление
сопряженных углеводородов на два класса — альтернантные угле-
углеводороды (АУ) и неальтернантные углеводороды (НАУ) — и иссле-
исследовали их свойства.
К АУ относят сопряженные системы, не содержащие нечетно-
членных циклов. Это позволяет подразделить все атомы (орбитали)
296
АУ на два типа: «со звездочкой» и «без звездочки» — таким
образом, чтобы атомы различных типов обязательно чередовались.
АУ делятся по типу числа атомов на четные и нечетные. Для
первых из них, очевидно, число атомов со звездочкой равно числу
атомов без звездочки, для вторых превышает число последних на
единицу.
Таким образом, все четные аннулены и полиены относят к АУ.
Структуры XXVIII — XXXII представляют примеры некоторых
АУ и НУ:
сн
сн2
XXVIII
АУ
XXIX
АУ
'П
XXX А
XXXII НУ
Определение АУ является топологическим и позволяет устано-
установить для них ряд ценных закономерностей и соотношений между
энергетическим уровнями, коэффициентами разложения и их функ-
функциями, такими, например, как электронная плотность, и др. Прежде
чем сформулировать основные из этих закономерностей, укажем,
что результаты расчетов АУ по методам МОХ и ППП близки
и доказанные в рамках метода МОХ соотношения сохраняют свою
силу и в методе ППП. Выделение АУ в отдельный класс имеет
смысл только в приближении НДП (см. гл. 7). Классификация
углеводородов на АУ и НАУ основывается на теоретических пред-
представлениях, а не на обычной химической классификации. Впервые
нижеприведенные теоремы были сформулированы Коулсоном и
Рашбруком, а позднее более подробно разработаны Коулсоном
и Лонге-Хиггинсом.
1. Теорема парности утверждает, что в любом АУ все МО
разбиваются на симметричные относительно нулевого уровня
\х=0) пары, т. е. каждой связывающей МО с энергией —.х, (в
единицах —-—) соответствует антисвязывающая с энергией х{. След-
Р
ствия этого положения иллюстрированы расчетами, представлен-
представленными графически на рис. 8.3 и 8.7. Между коэффициентами при АО
в таких связывающих (индекс г) и антисвязывающих (индекс/) МО
также существует простое соотношение: коэффициенты в антисвя-
антисвязывающих МО на атомах со звездочкой совпадают по значению
и знаку с коэффициентами АО атомов со звездочкой в связывающих
МО, а на атомах без звездочки меняют знак на обратный:
297
*i--** (8.81)
cli=c& (8.82)
**--** (8.83)
Примером, показывающим справедливость этого вывода, могут
служить рассчитанные ранее МО аллила и бутадиена.
Задача 8.7. Доказать теорему парности.
2. В любом нейтральном АУ полная электронная плотность
в молекуле (или радикале) равномерно распределена по всем ато-
атомам, т. е.
Рт=1- (8.84)
3. Порядок связи в нейтральном углеводороде между атомами
одного класса (со звездой или без) равен нулю. Проверка справед-
справедливости второго и третьего утверждений может быть проведена
путем анализа матрицы плотности первого порядка бутадиена
(8.79). Из утверждения 2 следует, что порядок связи между сосед-
соседними атомами не может быть больше 1. Действительно, из нера-
неравенств Коши—Буняковского следует:
(8.85)
Таким образом, можно заключить, что насыщенность связей
в молекулах альтернантных углеводородов не выше насыщенности
двойной связи в молекуле этилена {р^— 1).
Правила 1—3 справедливы для всех альтернантных углеводоро-
углеводородов. Для формулировки остальных закономерностей необходимо
разбить АУ на четные, т. е. содержащие 2Л^атомов углерода,
и нечетные.
4. Для четных нейтральных АУ атом-атомные поляризуемости
(в единицах 1//?) отрицательны, если атомы относятся к одному
и тому же (со звездой или без звезды) ряду.
Эта теорема служит теоретическим основанием для эксперимен-
экспериментально установленного правила альтернирующей полярности в со-
сопряженных системах, чем они отличаются от насыщенных систем,
где передача электронного эффекта от заместителя монотонно осла-
ослабляется по мере удаления реакционного центра.
298
Таблица 8.4. Атом-атомные поляризуемости бензола ¦ электронные плотности
на атомах в молекуле пиридина (А =0,5)
Атом-атомные поляризуемо-
поляризуемости бензола, 1//J
741= 0,398
Я12= -0,1,57
я,3= О,0О9
ям= -0,102
Номер атома в пири-
пиридине XXXIV
t
2
3
4
Электронная плотность на атомах
пиридина
рассчитанная по
(8.77)
1,199
0,921
1,005
0,961
полученная прямым
расчетом
1,195
0,923
1,005
0,950
В табл. 8.4 приведены атом-атомные поляризуемости, вычислен-
вычисленные для молекулы бензола. Пользуясь этими значениями, можно
рассчитать в первом порядке теории возмущений по формуле (8.77)
распределение зарядов в молекуле пиридина. Последнюю можно
рассматривать как результат замены одного из метиновых звеньев
изоэлектронным sp-гибридизованным атомом азота. Величина Да
в этом случае равна 0,5/? (табл. 8.4):
XXXIII
С одной стороны, можно видеть весьма хорошее совпадение
данных приближенного и точного расчетов. С другой стороны,
полученное альтернирование электронных плотностей на атомах
пиридина хорошо объясняет его химические свойства: нуклеофиль-
ное замещение (аминирование, гидроксилирование) в положения 2,
4 ядра и электрофильное замещение (сульфирование) в положение 3.
5. Число несвязывающих молекулярных орбиталей НСМО (Z)
не меньше N-2T, где N — число атомов углерода; Г — максималь-
максимальное число двойных связей в какой-либо резонансной структуре:
Z^N-2T. (8.86)
Проиллюстрируем правило (8.86) некоторыми примерами (см.
с. 300).
В общем виде удается найти только оценку снизу (8.86). Вывести
формулу для нахождения точного числа НСМО не представляется
возможным. С числом НСМО тесно связана задача об определении
мультиплетности основного состояния альтернантного углеводо-
углеводорода.
299
XXXV
N = 7
т = з
Z~ 1
XXXVI
N = 6
Т = 2
Z = 2
XXXVII
N = 8
Т = 3
Z = 2
XXXVIII
N = 4
Т- 1
Z = 2
6. Полный спин S основного состояния альтернантного углево-
углеводорода определяется соотношением (А. А. Овчинников)
\пА-пА*\
где пА и пА* — соответственно число атомов углерода без звезды
и со звездой.
Так, для систем XXXVI — XXXVIII полный спин должен быть
равен единице и основное состояние должно быть триплетным.
Ясно, что соотношения (8.86) и (8.87) взаимозависимы. Правило
(8.87) открывает возможность для целенаправленного поиска ор-
органических соединений с высокой мультиплетностью основного со-
состояния вплоть до ферромагнетиков (S—>oo). Для этого необходи-
необходимо, чтобы полный спин был пропорционален размеру молекулы.
Вот несколько примеров гипотетической реализации указанной за-
зависимости на макромолекулах углеводородов с периодически вне-
внедренными гетероатомами. Последние не должны приниматься во
внимание при разбиении атомов на два класса:
= o,s
XXXIX
Y = В, N
XL
300
7. Несвязывающая МО нечетного альтернантного углеводорода
состоит только из атомных орбиталей отмеченных звездочкой
атомов (полагают, что последних больше, чем атомов без звездо-
звездочки), коэффициенты при атомных орбиталях не отмеченных звездо-
звездочкой атомов обращаются в нуль.
Если дополнительно к этому утверждению воспользоваться
формулой Лонге-Хиггинса, согласно которому в выражении НСМС
сумма коэффициентов МО ЛКАО для отмеченных атомов, присо-
присоединенных к данному неотмеченному атому, обращается в нуль, тс
можно ? помощью простых рассуждений находить вид НСМО.
Поясним^ эту процедуру на примерах аллильного радикала XXVII]
и более сложного бензильного радикала XLI (см. с. 302):
* *
СН2-СН-СН2
а 0 -а
Пусть коэффициент при первом атоме углерода равен а, тогда
при третьем атоме он равен —а. Из условия нормировки МО имеем
-а=-
что согласуется с (8.26).
Если в бензиле обеспечить коэффициент при метиленовом атоме
углерода а, то для остальных атомов со звездочкой необходимо
положить коэффициенты равными
ляет получить величину а:
а
. Условие нормировки позво-
а
у
В катионе и анионе АУ по сравнению с ра-
радикалом с несвязывающей МО отнимается или
на нее прибавляется один электрон. Таким об-
образом, знание вида этой МО позволяет рас-
рассчитать распределение полной электронной плотности в катионе
и анионе. Для этого от значений рт— 1 на каждом атоме радикала
(8.84) следует отнять (для катиона) или прибавить (для аниона)
значение электронных плотностей на несвязывающей МО (см.
с. 302).
Кроме того, учитывая третье правило, можно без труда найти
по виду НСМО порядок связи между отмеченными атомами в кати-
катионах и анионах нечетных альтернантных систем.
301
XLH анион
Задача 8.8. Определите вид НСМО а-нафтилметила
в. 10. ТЕПЛОТЫ АТОМИЗАЦИИ ПОЛИЕНОВ
Изучение энергетических факторов в ряду линейных полиенов
представляет самостоятельный интерес и вместе с тем лежит в ос-
основе количественных критериев ароматичности (см. разд. 8.11).
Принципиальные результаты были получены Дьюаром с помощью
метода ППП из расчетов классических полиенов, строение которых
описывается одной резонансной структурой. Примером таких поли-
полиенов могут служить полиены (XLIII):
Н2С=СН-(СН = СН)п-СН=СНья = О, 1,2,... .
XLIII
Если сопряжение не изменяется с ростом и, т. е. длины ординар-
ординарной и двойной связей остаются неизменными, то теплота образова-
образования полнена может быть записана в виде суммы:
где ?с-с— энергия двойной связи в полиене; ?С-с — энергия оди-
одинарной связи в полиене; ?С-н ~~ энергия связи С —Н.
Теплота атомизации полнена (XLIII) должна быть линейной
функцией числа звеньев п. Действительно, расчеты по методу ППП
в параметризации Дьюара приводят к выводу о том, что связи
в полиенах локализованы и их энергия практически не меняется
с ростом цепи сопряжения (рис. 8.13).
Энергия связи С—Н равна 4,4375 эВ (см. табл. 7.17), это позво-
позволяет по графику рис. 8.13 определить энергии ?^.с и ЕС-с- Известно,
что энергия чисто ординарной С—С связи равна 3,9408 эВ (см.
табл. 7.18) — это на 0,4 эВ меньше, чем энергия формально ор-
ординарной *ВЯЗи в полиенах. Такое отличие в энергиях вызвано тем,
что в полиенах связи С —С имеют довольно значительный я-поря-
302
док (~0,2 нм) и они короче @,146 нм) по
сравнению с чистыми связями С —С
@,1512 нм). Согласно теоретическим
оценкам, энергия двойных связей ?с-с по-
почти не отличается от энергии связи в эти-
этилене.
Такая ситуация, т. е. линейность АН
в зависимости от л, сохраняется и в дру-
других типах (долиенов. Например, для ради-
радиаленов, т. ^.циклических полиенов общей
формулы С„НЯ, в которых каждый цик-
циклический атом углерода связан с метиле-
новой группой двойной связью, энергия
атомизации может быть записана как
Прямая АН(п) для радиаленов должна иметь наклон, аналогичный
наклону прямой для линейных полиенов, и должна проходить через
начало координат. Это подтверждают данные расчетов, представ-
представленные на рис. 8.13. Соединения XLIV и XLV являются соответст-
соответственно 3- и 6-членными радиаленами:
200
160
120
80
40
0
8 п
Рис. 8.13. Зависимость теп-
теплоты атомизации АН полие-
полиенов от длины цепи сопряже-
сопряжения
II
XLIV
XLV
Отметим, что для неклассических полиенов, т. е. полиенов, для
которых возможно написание более чем одной резонансной струк-
структуры, например для винилогов диметилформамида (XLVI)
(сн=сн)и— СН=
\,
—сн)я=сн-
XL И б
локализация связей не очень сильная, тем не менее и в них с несколь-
несколько меньшей точностью (~8—12 кДж/моль) теплоты образования
можно представить как сумму энергий связей.
303
8.11. ЭНЕРГЕТИЧЕСКИЕ КРИТЕРИИ АРОМАТИЧНОСТИ
Разбиение циклических полиенов на два класса — ароматичес-
ароматических и антиароматических — согласно соотношению (8 Л 6) является
качественным. В то же время необходимы количественные энер-
энергетические оценки степени ароматичности — антиароматичности.
Это привело к разработке некоторых новых понятий.
8.11.1. Хюккелевская энергия резонанса
Самый простой способ оценки ароматичности заключается
в расчете энергии делокализации DE, или энергии резонанса, т. е.
разности между полной электронной энергией молекулы и энергиями
изолированных двойных связей, включенных в состав молекулы. Эта
разность характеризует выигрыш в энергии (стабилизация молеку-
молекулы) за счет делокализации тг-электронов:
(8.90)
Для бензола ^
и
Е = 3Bа+20) = 6а+60;
DE = 20.
Для циклобутадиена
= 4л+40; II И Е = 2B« + 20) = 4с + 40; DE = 0
Для бутадиена
^S^XeS^ Е= 4а + 4,4720; II II Е = 2Bв +20) =4а +40; DE = 0,4720.
Теоретические значения DE могут быть сопоставлены с экс-
экспериментальными значениями энергии резонанса. Наиболее общий
способ их вычисления состоит в сопоставлении теплот атомизации
(т. е. энергии, расходуемой на превращение молекулы в изолиро-
изолированные атомы), полученных из экспериментов по теплотам сгора-
сгорания по аддитивной схеме из усредненных значений энергий связей.
Понятие энергии делокализации может быть применено и к мо-
молекулам с гетероатомами. Например, для аминопиридина
=nh2
ч
с-ес=м-ей, (8.91)
Определение энергии делокализации по (8.90) и соотношениям
типа (8.91) страдает довольно серьезно недостатком, связанным
с тем, что в методе МОХ увеличение размеров я-системы будет
всегда приводить к увеличению DE. Между тем экспериментальные
данные нередко не соответствуют этому результату. Так, в химии
полиаценов хорошо известна закономерность: линейное аннелиро-
вание бензольных колец приводит к понижению стабильности по ¦
304
мере накопления бензольных ядер. Если бензол — соединение с ис-
исключительной термодинамической устойчивостью и инертностью
к реакциям присоединения, то пентацен высокореакционноспособен.
Этот результат нельзя объяснить на основе изменения значений DE.
Кроме того, ряд НАУ, для которых метод МОХ предсказывает
значительные энергии делокализации, — фульвен, фульвален, пен-
тален — недостаточно стабильны и высокореакционноспособны,
так что последние два из них вообще не удается изолировать.
Вместе с ^гем DE для пенталена равна 2,46/f а превышает DE
бензола — %§,
Все эти данные удается достаточно хорошо уложить в общую
схему, основанную на применении энергий делокализации, если
изменить определение DE. Вместо сумм изолированных двойных
связей в качестве структуры сравнения для сопряженной цикличес-
циклической системы Бреслоу предложил принять равный ей по числу п-
электронов и их орбиталей нециклический пол иен. Так, для бензола
и циклобутадиена структурами сравнения могут служить соответст-
соответственно гексатриен и бутадиен. При этом DE бензола — положитель-
положительная величина, свидетельствующая о его стабильности или, в хи-
химической терминологии, ароматичности, a DE циклобутадиена от-
отрицательна, т. е. циклобутадиен дестабилизирован по отношению
к бутадиену:
6,9880 Е = 4в+40 Е= 4а+4,4720
DE=-0,472/f
8.11.2. Дьюаровская энергия резонанса
Идея Р. Бреслоу получила развитие в работах Дьюара. При
расчете ациклических полиенов (см. разд. 8.10) Дьюар установил,
что постоянство энергий и длин связей в них не указывает на
отсутствие сопряжения через простую связь, а свидетельствует то-
только о сопряжении, приводящем к одинаковым энергетическим
изменениям во всех полиенах в расчете на отдельную связь. Расчеты
показывают, что ситуация сохраняется в случае введения в молеку-
молекулу ациклического соединения атомов других элементов (например,
О, N, S). В то же время если рассчитать энергию агомизации
циклической молекулы, например бензола, по аддитивной схеме
-н = 56,29 эВ,
то полученное значение будет отличаться от экспериментального
(Ап = 57,16 эВ) на 0,87 эВ, что составляет «84 кДж/моль.
Согласно Дьюару, энергия резонанса равна разности энергии
305
атомизации данного сопряженного соединения и энергии соответст-
соответствующего классического полнена, рассчитанных с помощью метода
ППП в параметризации Дьюара. Другими словами, энергия резо-
резонанса — это мера стабилизации циклической системы по сравнению
с соответствующим соединением с открытой цепью, которая содер-
содержит то же число сопряженных атомов углерода. Подчеркнем, что
в качестве структуры сравнения не обязательно выбирается полнен
с чистыми двойными и ординарными связями, а выбирают тот
полнен, у которого минимальная полная энергия. Например, энер-
энергию резонанса цнклопропенильного катиона XLVII а вычисляют
относительно делокализованного аллильного катиона XLVII б, но
не локализованной структуры XLVII в:
А Л.
XLVH a
XLVH 6
XLVII в
Соединения, для которых дьюаровская энергия резонанса (ДЭР)
положительна, называют ароматическими. Антиароматические
и неароматические вещества обладают соответственно отрицатель-
отрицательной и нулевой (с точностью 4—8 кДж/моль) энергиями резонанса.
Проверим справедливость ароматического правила Хюккеля
Dл+2) на основе расчета ДЭР. На рис. 8.14 приведены значения
ДЭР для полиметинов C^H« (m — четное число), там же имеются
данные расчета энергий делокализации по методу МОХ.
Методы МОХ и ППП приводят к качественно отличным выво-
выводам. Так, согласно методу Хюккеля, с ростом т разница между
ДЭР аннуленов с т=4л+2 и т=4л стремится к нулю, а значение
ДЭР все время возрастает. В то же
время метод ППП приводит к выводу
о том, что с ростом т ДЭР уменьшает-
уменьшается, причем для /м=18 значение ДЭР
положительно и [18]-аннулен аромати-
ароматичен, в то время как для т=30 ДЭР
отрицательно и [30]-аннулен должен
быть антиароматичен, несмотря на то
что для него выполняется правило
Хюккеля (л=7). Правило Хюккеля
справедливо только для п— 1— 4. Для
больших значений п соединения с 4/1+2
л-электронами не являются аромати-
ароматическими. В табл. 8.5 приведены ДЭР
бензоидных и небензоидных углеводо-
углеводородов, рассчитанные по методам ППП
и МОХ.
Метод МОХ правильно передает
рост ароматичности при аннелирова-
16 18т
Рис. 8.14. Зависимость дьюа-
ровской я хюккелевской энергий
резонанса полиметинов Н
от т
306
Таблица 8.5. Эпергш резонанса беюовдных (Б)
водород*»
адных (НБ) уг
Формула
Энергия резонанса,
эВ
МОХ*
ппп
Бензол (Б)
Нафталин (Б)
Антрацен (Б)
Фенантрен (Б)
Азулен (НБ)
Бутален (НБ)
Фульвен (НБ)
Фульвален (НБ)
0,869
1,599
2,308
1,499
1,460
0,721
0,639
1Д17
0,869
1,323
1,600
1,933
0,169
-0,270
0,047
0,109
* Резонансный интеграл /? в методе МОХ выбирался так, чтобы получить
правильную, т. е. совпадающую с вычисленной по методу ППП, энергию резонанса
бензола (/?= -0,44 эВ).
нии, и его результаты достаточно правильны для бензоидных угле-
углеводородов. В то же время этот метод для небензоидных углеводо-
углеводородов приводит к результатам, даже качественно не согласующимся
с данными метода ППП. Хюккелевский расчет приводит к непра-
неправильному выводу о том, что ДЭР для бензоидных и небензоидных
углеводородов должны быть почти одинаковыми. Данные метода
ППП предсказывают для небензоидных углеводородов очень ма-
малую энергию резонанса, а в некоторых соединениях (бутален, пен-
тален) вычисленная энергия резонанса отрицательна.
Отметим, что ДЭР — не аддитивная величина при увеличении
числа аннелированных бензольных колец. Например, для нафтали-
нафталина ДЭР больше на 50%, но не на 100%, по сравнению с энергией
307
резонанса бензола. Энергия резонанса зависит от характера ан-
нелирования, например антрацен и фенантрен имеют различные
энергии резонанса — соответственно 1,600 и 1,933 эВ.
8.11.3. Энергия резонанса для возбужденных состояний
ДЭР можно определять не только для основных, но и для
возбужденных, в частности триплетных, состояний. ДЭР низшего
триплетного ягс*-состояния циклического углеводорода вычисляет-
вычисляется как разность энергии яя*-состояния циклического углеводорода
и энергии возбужденного тгя*-состояния полнена с тем же числом
углеродных атомов.
Это определение легко использовать, так как наиболее стабиль-
стабильная конформация полиенов в триплетном состоянии имеет одну
внутреннюю С=С-связь, повернутую на 90°, и энергия такого три-
триплета есть просто сумма энергий двух соответствующих свободно-
радикальных цепочек, соединенных «чистой» C(sp )—С(.ур2)-связыо.
Таблица 8.6. ДЭР трлплетных состояний сопряженных углеводородов
Соединение
ДЭР.эВ
Бензол (Б)
Нафталин (Б)
Антрацен (Б)
Азулен (НБ)
Фульвен (НБ)
Циклобутадиен (Б)
Цижлооктатетраея (Б)
Бутален (Б)
308
-0,534
^0,065
0,209
0,169
0,169
0,613
0,769
0,600
Вычисления ДЭР для яя*-состояний полиметинов показали
(Бэйд), что хюккелевское правило ароматичности изменяется в три-
плетном состоянии. Ароматичными будут аннулены с 4п я-электро-
ыами, антиароматичными с 4/1+2 я-электронами. В табл. 8.6 приве-
приведены ДЭР триплетных состоянии сопряженных углеводородов.
В отличие от основного состояния ДЭР для бензоидных углево-
углеводородов меньше, чем для небензоидных. Соединения с 4л+ 2 тг-
электронами имеют отрицательную энергию резонанса. Реакцион-
Реакционная способность в возбужденных состояниях должна существенно
отличаться по сравнению с основным.
8.12. РАСЧЕТ ФИЗИЧЕСКИХ СВОЙСТВ
СОПРЯЖЕННЫХ СОЕДИНЕНИЙ
Целесообразно рассмотреть расчет физических свойств сопря-
сопряженных соединений только в рамках метода Хюккеля, так как это
позволит вам без привлечения мощных ЭВМ понять принципы
вычислений. Метод Хюккеля наиболее пригоден для качественных
относительных сопоставлений, но в общем случае не может претен-
претендовать на. точное воспроизведение тех или иных свойств или харак-
характеристик молекулы. Вместе с тем этот метод может дать полезную
информацию, когда требуется или очень примерная оценка, или
необходимо расположить серию соединений в ряд по убыванию или
возрастанию той или иной физической величины без ее абсолют-
абсолютного определения.
8.12.1. Потенциалы ионизации и сродство к электрону
Процессы ионизации (см. разд. 3.5.2) электрона и его захвата
в рамках я-приближения (т. е. в данном случае предполагают, что
эти процессы происходят на МО тг-типа) можно представить схема-
схемами (рис. 8.15).
Потенциал ионизации, рассчитанный как разность энергии поло-
положительного иона и нейтральной молекулы, равен просто энергии
высшей занятой МО (ВЗМО) в приближении метода МОХ:
#он-?=?взмо, (8.92)
где Свзмо — вертикальный потенциал ионизации, который не учиты-
учитывает того, что при отрыве электрона от молекулы происходит
реорганизация ее ядерной и электронной конфигурации. Это выра-
выражается в том, что распределения энергетических уровней в ионе уже
не соответствуют схеме, представленной на рис. 8.15.
При этом уравнение (8.92) не выполняется и значение
Е?аЖ необходимо вычислять отдельно. Рассчитанные таким образом
потенциалы ионизации называют адиабатическими; они соответ-
соответствуют экспериментальным величинам, получаемым наиболее ча-
309
-Ь- -Н-
бутадиен (Бутадиен)*
Ионизация
•4V -Н-
-н- -н-
бутадиен (бутадиен)'
Захват электрона
Рис. 8.15. Схема заполнения уровней МО электронами в катионе и анионе бутадиена
сто методом фотоионизации или из фотоэлектронных спектров (см.
рис. 4.20).
В методе МОХ процедура оценки адиабатических потенциалов
мало уместна, и целесообразно лишь попытаться получить пред-
представление об их относительных величинах. Действительно, как вид-
видно из рис. 8.16, между значениями потенциалов ионизации углеводо-
углеводородов, найденными экспериментально, и значениями энергии
ВЗМО, рассчитанными по методу МОХ, имеется удовлетворитель-
удовлетворительная корреляция. Подобная корреляция может быть получена и для
ряда гетероатомных систем, однако в этом случае требуется еще
уверенность в том, что отрыв электрона происходит с ВЗМО л-типа,
а не орбитали неподеленной электронной пары, которая не входит
в я-систему.
Согласно (8.92), метод МОХ предсказывает, что потенциалы
ионизации радикалов и анионов нечетных АУ должны быть равны
нулю, что не соответствует дейст-
действительности. Так, для аллильного
радикала /=8Д5 эВ. Неудовлетво-
рительны также оценки метода для
значений энергий сродства к элект-
электрону. Как следует из рис. 8.15, сро-
сродство к электрону А должно быть
равным энергии нижней свободной
МО (НСМО) и для АУ величины
/ и А должны быть равны по вели-
величине, обладая разным знаком.
В действительности сродство к эле-
электрону для большинства сопряжен-
ных систем положительно, а абсо-
лютные значения Аи/ сильно раз-
разнятся.
Все же чем ниже НСМО, тем
больше сродство к электрону. На
рис. 8.17 показаны корреляции экс-
E(83M0),fi
Рис. 8.16. Корреляция потенциала
ионизации и энергии высшей занятой
(ВЗМО) молекулярной орбитали для
ненасыщенных углеводородов
310
периментальных значений сродства
к электрону, полученных главным обра-
образом методом электронного захвата
и энергий НСМО.
Ввиду подобных приблизительных
корреляций можно коррелировать с эне-
энергиями ВЗМО способность сопряжен-
сопряженных систем к окислительным, а с энерги-
энергиями НСМО — восстановительным реак-
реакциям. (
\
8.12.2. Электронные спектры погло-
поглощения
Аз длен
\пирен
Бензол
¦0,6 -0 8 4 0
E(HCM0),fi
Рис. 8.17. Корреляция сродст-
сродства к электрону с энергией низ-
низшей свободной молекулярной
орбиталью (НСМО) для нена-
ненасыщенных углеводородов
В рамках метода Хюккеля энергия
электронного возбуждения, ответствен-
ответственного за появление полос поглощения,
сопоставляется с разностью энергий МО, между которыми осущест-
осуществляется электронный переход:
AE=hv = Em_m+n = en-em. (8.93)
Рис. 8.18 иллюстрирует схему возможных электронных п—п*-
переходов в молекуле бутадиена. Показаны электронные конфигу-
конфигурации различных возбужденных состояний, возникающих в резуль-
результате перехода электрона с одной из занятых в основном состоянии
МО на возбужденную орбиталь, так как обычно в электронных
спектрах органических соединений достаточно интенсивны лишь
одноэлектронные переходы.
В УФ-спектре бутадиена проявляется только один электронный
переход с наименьшим значением АЕ. Энергии, требующиеся для
возбуждения в другие электронные конфигурации, приведенные на
?,*
+ 0,618
-0,518
-1,618
-н-
-н-
й
«S
-fr
+*¦-
Рис. 8.18. Схема возможных электронных я, я*-переходов в молекуле бутадиена
311
рис. 8.18, превышают энергии связей в молекуле, и соответствующее
жесткое УФ-облучение ведет к разрушению молекулы.
Неучет электронного отталкивания и невозможность различения
в рамках метода МОХ возбужденных состояний с различной муль-
типлетностью ограничивают возможности метода лишь приблизи-
приблизительными корреляциями экспериментальных полос электронных
переходов с разностями энергий МО.
В табл. 8.7 разности энергий НСМО и ВЗМО линейных полиенов
сопоставлены с экспериментальными длинами волн и соответст-
соответствующими им энергиями переходов длинноволновых полос погло-
поглощения.
Таблица 8.7. Электронные спектры поглощения поляенов Н(СН = СН)„Н
R
1
2
3
4
162,5
217,0
251,0
304,0
Д?, эВ
7,63
5,71
4,94
4,08
2,00
1,24
0,89
0,70
Значение резонансного интеграла /?, рассчитанное из представ-
представленных данных, составляет —2,62 эВ. Близкие значения получают
из корреляций подобного типа для других соединений, например
для бензоидных углеводородов рассчитанное таким способом зна-
значение р равно —2,36 эВ. Полученные значения называют спектро-
спектроскопическими; они намного больше термохимического значения S
8.12.3. Спектры ЭПР и распределение спиновой плотности в со-
сопряженных молекулах
Сигнал электронного парамагнитного резонанса в молекулах
возникает при наличии в них одного или нескольких неспаренных
электронов, что вызвано зеемановским расщеплением спиновых
состояний электрона в магнитном поле, подобном рассмотренному
ранее (см. гл. 3) для атомов. При наличии одного неспаренного
электрона полный спин равен i/2i что соответствует дублетному
состоянию, т. е. радикалу. Парамагнетизм радикалов обусловлен
почти исключительно спином неспаренного электрона, который
всегда находится на высшей занятой МО.
На рис. 8.19 показано происхождение спектра ЭПР на примере
радикала, который содержит один атом с магнитным ядром, име-
имеющим спин /=72 (например, водород).
При наложении магнитного поля снимается вырождение спино-
спиновых состояний и уровни, отвечающие двум значениям mSi расщепля-
расщепляются. Согласно C.103),
312
ВФо
Взаимодействие
с ядерным спином
Рис. 8.19. Происхождение спектра ЭПР (а) и сигнал ЭПР (б) (производная от интен-
интенсивности поглощения по Н) в радикале с одним атомом, имеющим магнитный
момент ядра /= 7г
. (8.94)
Развертывая магнитное поле В при заданной частоте, добиваются
условия резонанса (8.94) и связанного с ним поглощения микровол-
микроволнового излучения. Порядок значений АЕ в спектрах ЭПР, отлича-
отличающихся высокой чувствительностью, составляет 10 кДж/моль.
В рассматриваемом случае сигнал ЭПР проявится не в виде
одной, а в виде двух линий поглощений, что является результатом
взаимодействия электронного спина со спином магнитного ядра
(протона). К числу магнитных ядер, встречающихся в молекулах
органических соединений, принадлежат Н, 13С, 19F, 31P (I=l/2)
и др., тогда как ядра 12С, 16О немагнитны.
В результате взаимодействия с ядерными спинами происходит
дополнительное расщепление энергетических уровней, причем более
низкоэнергетическими оказываются состояния с противоположны-
противоположными знаками электронного и ядерного спинов. Правила отбора
Дт,= ±1;ДЛ//=0 (8.95)
ограничивают число переходов двумя линиями, расстояние между
которыми названо константой сверхтонкого взаимодействия
(СТВ), выраженной в единицах напряженности магнитного поля
(Гс). Константа СТВ связана со спиновой плотностью прямой
пропорциональностью, известной как соотношение Мак-Коннела.
Для протона
an=Qp«> (8.96)
где Q для связей С^*—Н принимается обычно равной 23 Гс (однако,
согласно теории, константы СТВ и Q имеют отрицательный знак),
а ря в методе МОХ соответствует электронной плотности неспарен-
ного электрона на одном углеродном атоме /х в высшей занятой
313
2,303
1618
1303
що
0,6/8
О
-0,618
-KOQO
-1,303
-2,303
Нейтр.
Анион-радикал Натион-радинал
Рис. 8.20. Заселенность электронами МО в вентральной молекуле, анион- и катион-
радикале нафталина
МО, т. е. cj*. Соотношение (8.96) позволяет связать определяемые
из эксперимента константы СТВ с теоретически рассчитываемыми
значениями спиновой плотности pn^ = <ik — в этом и состоит его
исключительная важность.
Рассмотрим расчет констант СТВ для анион-радикала нафтали-
нафталина (АУ), который легко образуется как при электрохимическом
восстановлении, так и при восстановлении с помощью щелочных
металлов. Рис. 8.20 показывает заселенность МО в анион-радикале,
катион-радикале и нейтральной молекуле нафталина.
Уравнения (8.97), (8.98) характеризуют вид ВЗМО и НСМО
нейтральной молекулы нафталина:
У5 = 0,425х, + 0,263x2 - ОДбЗ/з - 0,425/4 + 0,425*5 + 0,263 *6 -
?6=0,425х, - 0,263х2 - 0,263хз + 0,425х4 - 0,425х5 + 0,263х6 +
+ 0,263x7-0,425x8. (8.98)
МО иона в приближении метода МОХ имеют тот же вид, что и для
нейтральной молекулы. Так, для расчета спиновых плотностей
в анион-радикале можно воспользоваться орбиталью 4*bt откуда
(8.99)
Следовательно, для двух разных типов протонов в анион-радикале
нафталина (при соответствующих углеродных атомах)
0,181 =4,16 Гс; анп = 23 • 0,069-1,59 Гс.
Экспериментальные значения получают из анализа спектра, состо-
состоящего из 25 линий. Действительно, четыре эквивалентных протона
314
типа Ь^ дадут пять линий (число линий от п эквивалентных, пар
равно л+1), каждая из которых расщепится с меньшей константой
СТВ на пять линий при взаимодействии с четырьмя протонами Нп.
Анализ спектра дает
яН1=4,90 Геи аНп= 1,83 Гс. (8Л00)
Так как нафталин относится к альтернантным системам, то, соглас-
согласно теорем^ парности, абсолютные значения коэффициентов при АО
в парных МО равны между собой [см. (8.97), (8.98)]. Это означает,
что плотности неспаренного электрона, равные квадрату коэффици-
коэффициентов при АО ВЗМО и НСМО молекулы, в анион- и катион-
радикале одинаковы. Такое предсказание теории хорошо под-
подтверждается экспериментальными данными. Хотя катион-радикал
нафталина неизвестен, данные, представленные в табл. 8.8 для
других АУ, убедительно иллюстрируют справедливость сделанного
вывода.
Таблица 8.8. Экспериментальные и рассчитанные константы СТВ нон-радикалов
альтернантных углеродов
Ион-радикал
Эксперименталь-
Экспериментальное значение
Структура
Название
аниов-
радикал
катион-
радикал
ац по
(8.96)
1
2
Бутадиен
Бензол
I
2
1 9
4 2
Антрацен
Тетрацен
Перилен
1
2
9
1
2
5
1
2
3
7,62
2,79
3,75
2,74
1,51
5,34
1,55
1Л5
4,25
3,08
0,46
3,53
3,06
1,38
6,53
1,69
1,03
5,06
3,10
0,46
4,10
0,362
0,138
0,167
0,097
0,048
0,193
0,056
0,034
0,147
0,083
0,013
0,108
8,37
3,17
3,84
2,29
1,14
4,44
1,29
0,78
3,38
1,83
0,30
2,48
2 1
Из данных табл. 8.8 следует, что метод МОХ несколько занижа-
занижает рассчитанные спиновые плотности по сравнению с эксперимен-
315
тальными, поскольку данный метод'в своем простейшем варианте
может учесть лишь положительные спиновые плотности, как это
вытекает из самого их определения. Эксперимент и строгая теория
указывают на существование и отрицательных спиновых плотно-
плотностей.
Действительно, строгое определение спиновой плотности — это
разность плотностей электронов с /я, = 1/2 (а-спин) и /я, = — 1/2
(/}~спин); если последняя величина больше, возникают отрицатель-
отрицательные спиновые плотности. Так как
!>;=!, (8.101)
то появление отрицательных спиновых плотностей должно быть
скомпенсировано увеличением положительных.
Мак-Лечлан показал, что более точный расчет, позволяющий
получать и отрицательные спиновые плотности в рамках метода
МОХ, может быть проведен по формуле
Здесь с,* — коэффициент при АО ^-го атома в к-й МО неспаренного
электрона; п^ — атом-атомные поляризуемости, вычисляемые со-
согласно (8.75); X — эмпирически подбираемый параметр, принима-
принимаемый обычно равным 1.
Отрицательные спиновые плотности возникают преимуществен-
преимущественно на тех атомах, для которых простой метод МОХ предсказывает
нулевое или очень малое значение спиновой плотности. Фундамен-
Фундаментальное значение данных спектроскопии ЭПР для теории состоит
в том, что сопоставление этих данных с теоретически ожидаемыми
результатами показывает, что представления о МО (по крайней
мере МО неспаренного электрона) достоверны и их предполагаемая
форма близка к действительности.
8.13. ИНДЕКСЫ РЕАКЦИОННОЙ СПОСОБНОСТИ
я-СОПРЯЖЕННЫХ МОЛЕКУЛ
Описание реакционной способности молекул является в принци-
принципе более сложной задачей, чем расчет физических свойств. В то же
время теоретический прогноз положений равновесия и кинетики
реакции представляет, несомненно, самую важную для химии об-
область соприкосновения с квантовой механикой молекул.
8.13.1. Общие ограничения
Согласно термодинамике и теории переходного состояния, кон-
константы равновесия К и константы скорости реакции к связаны со
свободными энергиями равновесия и активации соотношениями
316
(8.103)
** , (8.104)
Л
где ав— так называемый трансмиссионный коэффициент, характе-
характеризующий вероятность прохождения реакции при достижении пере-
переходного состояния и принимаемый обычно равным 1. Свободные
энергии равновесия и активации связаны известными соотношени-
соотношениями с энтальпией (теплотой) и энтропией равновесия и активации:
AG=AH-TAS. (8.105)
Здесь символы равновесия и активационного процесса опущены
вследствие общности соотношения (8.105).
Теоретическая оценка изменения энтропии при переходе от ре-
реагентов к продуктам равновесия или переходному состоянию, хотя
и возможна в принципе, требует точного вычисления таких харак-
характеристик, как частоты нормальных колебаний, моменты инерции
и др., и является труднодостижимой для достаточно сложной систе-
системы. Поэтому возможности квантово-механического расчета ограни-
ограничиваются вычислениями значений АН. По этой же причине задача
расчета абсолютных констант скоростей и равновесия по уравнени-
уравнениям (8.103), (8.104) малодоступна.
Однако исключительно важной для химической практики явля-
является задача расчета относительных констант равновесия и скорости
в определенном заданном ряду соединений сходного строения (ре-
(реакционной серии). Если можно предсказать, что соединение ^А ре-
реагирует с X быстрее, чем В, а В — быстрее, чем С, или реагент
Y будет атаковать преимущественно ла/?а-положение замещенного
бензола, но не мета- или орто-, то в этих утверждениях содержится
весьма важная информация.
Если бы в изучаемом ряду реакций энтропийные члены AS были
постоянны, задача свелась бы к расчету энтальпий реакций и ак-
активации, которые почти не отличаются от внутренних энергий:
AE=AH-AnRT, (8106)
где Ал — изменение числа молекул при образовании переходного
состояния из реагентов или в равновесии.
Значения АЕ можно вычислить в рамках того или иного прибли-
приближения как разность полных энергий реагентов и продуктов для
исследования равновесия системы. Для изучения кинетики реакции,
очевидно, необходимо вычислить разность полных энергий реаген-
реагентов и переходного состояния.
Известно лишь очень небольшое число реакционных серий,
в которых для каждой реакции значения AS были достаточно
постоянны. Наоборот, для большинства реакций, протекающих
в растворах, значения AS, зависящие в основном от сольватации,
317
варьируют от одной реакции к другой. При этом в растворах
выполняется известное так называемое компенсационное правило,
согласно которому в реакционных сериях наблюдается линейная
зависимость между величинами ЛЯ и AS, причем увеличение AS
влечет за собой увеличение АЯ, и наоборот. Для таких реакционных
серий, названных изокинетическими и изоравновеснъши, несмотря
на осложнения, вызываемые растворителем, свободные энергии
(но не энтальпии!) реакции должны коррелировать с рассчитанным
рядом значений А?.
8.13.2. Приближение изолированной и реагирующей молекулы
В рамках методов Хюккеля и ППП, основанных на <т, я-разделе-
нии, построение поверхности потенциальной энергии реакции нере-
нереализуемо, так как невозможно без дополнительных приближений
описать результаты взаимного движения реагентов и процессы
разрыва — образования «т-связей. Поэтому здесь достаточно лишь
приблизительное представление о минимально энергетическом пути
реакции.
Представим себе некоторую реакцию сопряженной системы, ко-
которая для своего осуществления требует перехода из состояния
А в состояние В, вызываемого движением по реакционной коор-
координате АВ (рис. 8.21). Если реакция идет достаточно медленно, т. е.
требует высокой энергии активации (кривая 7), ее переходное состо-
состояние в общем случае достаточно далеко удалено от исходного
состояния А. Переходное состояние X отличается, таким образом,
как от исходных реагентов А, так и от продуктов реакции В. Для
расчета энергии
AE=EXl-EA (8.107)
в реакции А—>В необходимо представить, какое строение имеет X.
Пусть теперь та же реакция по тому же пути (та же реакционная
координата) идет очень быстро с низкой энергией активации (см.
рис. 8.21, кривая II). Чем ниже энергия активации, тем ближе
переходное состояние Хц к исходному А, тем меньше путь, пройден-
пройденный до переходного состояния по реакционной координате, тем
ближе строение Хп и А.
Это простое рассуждение, иллюстрированное на рис. 8.21, есть
не что иное, как одно из следствий принципа Белла—Эванса—Поля-
ни, известное в теории реакционной способности как правило Хэм-
монда: электронное строение переходного состояния быстро проте-
протекающих реакций близко к строению исходных реагентов. Исходя из
этого можно ожидать, что, располагая данными об электронном
распределении (например, электронными плотностями на атомах)
в исходной изолированной молекуле А, можно предвидеть свойства
переходного состояния.
318
Подход к анализу реакционной
способности, основанный на изуче-
изучении особенностей электронного
распределения в исходном соеди-
соединении, назван приближением изо-
изолированной молекулы или стати-
статическим приближением. Подход,
требующий оценки энергии пере-
переходного состояния реакции, назы-
называют приближением реагирующей
молекулы или приближением лока-
локализации. В пределах каждого из
этих приближений предложены ха-
характеристики электронного рас-
распределения и энергии, которые
координата реакции.
Рис. 8.21. Кривые потенциальной
энергии реакции А-+В:
I н II — реагцин соответственно «^большой
и малой энергиями активации; АЕ — энер-
энергия активации: Д?** — разность энергий
АиВ
коррелируют с экспериментальными данными о реакционной способ-
способности. Эти характеристики называют индексами реакционной способно-
способности (ИРС).
Наиболее важными типами реакций сопряженных ароматичес-
ароматических и гетероциклических соединений являются реакции замещения
(электрофильное, нуклеофильное и радикальное) и реакции присо-
присоединения. Рассмотрим ИРС, применяемые для описания каждого из
этих типов реакций.
8.13.3. Электрофильное замещение
Наиболее распространенная задача теоретического рассмотре-
рассмотрения электрофильного замещения — прогнозирование или объясне-
объяснение направления, по которому предпочтительно протекает замеще-
замещение, т. е. определение наиболее активного по отношению к электро-
электрофилу положения в ароматическом или гетероциклическом ядре.
Приближение изолированной молекулы. Естественно предполо-
предположить, что реакции электрофильного замещения (нитрование, суль-
сульфирование, ионное галогенирование, дейтерирование и пр.) будут
идти предпочтительно по месту наибольшей концентрации элект-
электронной плотности и соответственно наибольшего отрицательного
заряда. Действительно, при приближении электрофильной частицы,
например нитроний-катиона, к атому р. атакуемой молекулы п-
электроны сопряженной системы смещаются в его сторону, так как
данный атом приобретает частичный положительный заряд. Этот
результат соответствует увеличению (по абсолютной величине) ку-
лоновского интеграла атома /х (ДаА<0). Изменение тг-электронной
части энергии атакуемой молекулы при таком раннем переходном
состоянии, когда электрофил не связан тесно с субстратом и измене-
изменения резонансных интегралов, а также связывание между субстратом
и электрофилом несущественны, можно представить в виде раз-
разложения по степеням А
319
ье. 1лг„ „+___
Учитывая соотношения (8.70) и (8.73), получим:
1 -• "•¦ (8.109)
Главным членом в разложении (8.109) является первое слагаемое.
Следовательно, чем больше q^ тем больше стабилизирована it-
система, раннего переходного состояния, тем меньше энергия ак-
активации. Действительно, в соответствии с хорошо известными экс-
экспериментальными данными реакции электрофильного замещения
в случае фенола направлены предпочтительно в орпго- и пара-поло-
жения ядра, в бевзальдегиде — в мета-положение, в азулене — в
положение 1 пятичленного кольца, в индоле — в положение 3 гете-
роцикла. Распределение электронных зарядов в перечисленных мо-
молекулах показано на молекулярных диаграммах (параметры Сгрей-
твизера):
-0,474
Н\ 0,034 0,334
10
1°>02* ir°.°23 -0,173 0,145
-Oj323^ ^Г Г^^О^58
нухп
Задача 8.9. Используя теорию возмущений, объясните правила ориентации
для заместителей первого и второго порядков.
Для каждого отдельного типа реакции (нитрование, галогениро-
вание, дейтерирование) поле, создаваемое атакующей электрофиль-
ной частицей, и, следовательно, значения Лоск различны. Сравнение
значений АЕ для различных типов реакции затруднительно и кор-
корреляции ограничиваются обычно рассмотрением активности от-
отдельных молекул и центров внутри молекулы для одного и того же
типа реакции.
В случае АУ электронные плотности всех атомов равны единице
и первые члены в выражении (8.109) одинаковы для любого атома /i.
320
Различия в реакционных способностях определяются теперь слага-
слагаемыми, включающими самополяризуемости %т. Поскольку самопо-
самополяризуемости, выражаемые в единицах 1//?, всегда отрицательны,
значения АЕ будут меньше (больше по абсолютной величине) для
положения, тр&пт — наибольшая {по абсолютной величине).
Таблица 8.9. Относительные скорости реакции нитрования и протодетрв-
тннрования фенантреяа по различным положениям (do отношению к скорости
нитрования нафталина и протодетрнтинрования в положение 1), сопоставленные
с различными ИРС
Атом, /t
9
1
3
2
4
Положение
1 нафталина
Реакционная
нитрование
1,04
0,77
0,64
0,196
0,168
1,000
способность
протодетрити-
ированне
1,67
0,934
0,399
0,174
0,840
1,000
ИРС
0,442
9,439
0,409
0,403
0,429
0,443
q^ ВЗМО
0,392
0,216
0,198
0,033
0,108
_ *
2,299
2,318
2,454
2,498
2,366
2,299
* Нельзя сравнивать различные молекулы по этому ИРС.
Как видно из табл. 8.9, самополяризуемость различных положе-
положений фенантрена (кроме положения 4) коррелирует со скоростями
реакции нитрования:
ю
;оАо
3 4 5 6
Полезные ИРС, применимые как к АУ, так и к любым другим
типам соединений, были предложены К. Фукуи*. Как известно, хи-
химическое поведение атомов определяется в первую очередь электро-
электронами во внешней орбитали. Естественно предположить, что и для
молекул электронное распределение в ВЗМО и НСМО будет играть
решающую роль для их реакционной способности.
В случае рассматриваемых реакций электрофильного замещения
связывание с электрофилом затронет прежде всего ВЗМО, так как
электроны на этой орбитали наименее прочно удерживаются осто-
*Кенити Фукуи (род. 1918) — японский химик-теоретик. Заложил в 1952 г. ос-
основы теории граничных, орбиталей, впоследствии распространенной для объяснения
реакций мвогоцентровых активированных комплексов, а также каталитических реак-
реакций. Нобелевский лауреат A981).
11. Теория строения молекул 321
вом и обладают наибольшей поляризуемостью. В приближении
МОХ электронные плотности на атомах в ВЭМО приравнивают
к квадратам коэффициентов при соответствующих АО. Таким об-
образом» вводится новый индекс реакционной способности — элект-
электронная плотность в граничной орбитали:
взмо=2cJ, взмо- (8.110)
Как видно из табл, 8.9, ИРС (в частности, п^) коррелируют с реак-
реакционными способностями отдельных положений фенантрена. Сле-
Следует иметь в виду, что значения q^ взмо могут быть использованы
только для рассмотрения относительных активностей отдельных
положений внутри данной молекулы, но не для сравнения реакцион-
реакционной способности разных молекул.
Приближение локализации. В случае позднего переходного состо-
состояния (см. рис. 8.21, кривая У), достигаемого при значительном
смещении по реакционной координате, оно уже достаточно сильно
отличается по своей структуре от исходной конфигурации. Требует-
Требуется определенная модель переходного состояния Хь которая позво-
позволила бы рассчитать энергию активации по формуле (8.107).
Такая модель была предложена Г. Уэландом. Суть ее понятна из
следующей схемы механизма реакции электрофильного замещения:
+ Е'
реагенты
Н-
ff-комплекс
продукты
В общем случае реакция протекает через промежуточное об-
образование метастабильных комплексов щ и п2 и включает образова-
образование ^-комплекса, которое требует больших энергетических затрат.
Образование ^-комплекса сопровождается перегибридизацией АО
субстрата, так как /?2-орбиталь атакуемого атома образует общую
(т-орбиталь с вакантной орбиталью реагента. Приведем схему об-
образования а-комплекса для случая реакции бромирования (атаку-
(атакующий реагент катион брома):
322
На образующуюся с-МО С—Вг переходят два электрона из
я-системы ароматического соединения, так что пять />*-АО <г-комп-
лекса, образующие его сопряженную систему, заполнены только
четырьмя я-электронами.
Как правило, сг-комдлекс неустойчив и имеет энергию, близкую
к энергии истинного переходного состояния. Отсюда следует, что
хорошим приближением к энергии активации является разность
энергий а-комплекса и исходной молекулы, называемая энергией
катионной локализации L+. Для бензола
Dа+ 5,4640Н6«+8Я=- Bх+ 2 5,360)
Поскольку в структуре <т-комплекса всегда на 2я-злектрона меньше,
опускают слагаемое 2а, а также знак минус. Энергию катионной
локализации бензола принимают равной 2,536/?. Очевидно, что для
различных положений молекулы, не столь симметричной, как бен-
бензол, я-системы сг-комплексов и соответственно энергии катионной
локализации для разных положений будут различны. Чем меньше
энергия локализации, тем легче протекают для данного положения
реакции замещения. В табл. 8.9 энергии катионных локализаций
различных положений фенантрена сопоставлены с их реакционной
способностью.
Задача 8.10. Бромирование 1,3-бутадиена приводит к примерно равному
соотношению 1,2-дибромбутена-З и 1,4-дибромбутена-2. Объясните этот резуль-
результат на основе расчета энергий катионной локализации.
Обоснование модели Уэланда требует доказательства двух по-
положений: во-первых, подтверждения реальности сг-комплекса как
возможного интермедиата; во-вторых, доказательства того, что он
очень близок по энергии к истинному переходному состоянию.
Второе положение не является пока строго доказанным, но первое
обосновано четкой идентификацией многих катионных «т-комплек-
сов в спектрах ядерного магнитного резонанса и даже выделением
некоторых особо стабильных соединений этого типа.
8.13.4. Нуклеофилыгое замещение
При нуклеофильном замещении атакующая частица — нукле-
офил, несущий отрицательный заряд, — стремится выбрать такое
положение в молекуле, электронная плотность которого мини-
323
мальна. Смещение электронной плотности с нуклеофила на ата-
атакуемый атом /х соответствует приращению Дам>0. Это в соот-
соответствии с (8.109) означает, что приращение АЕ минимально при
наименьших qM.
Действительно, распределения зарядов в молекулах пиридина
и ^-замещенного бензимидазола на молекулярных диаграммах
XLVIII и XLIX объясняют, почему реакция аминирования амидом
натрия (нуклеофильный агент — анион NH2~) идет в а- и у-положе*
ния пиридинового ядра и положение 2 имидазольного кольца:
-0,005Г0^
-0,020
-0,0241-1,
-0,017
-0,218
XLVIII
XLIX
В случае альтернантных углеводородов электронная плотность
не может быть использована как ИРС. Значения AaJ в уравнении
(8.109) положительны, и предпочтительным центром реагирования
с нуклеофилом будет атом с наибольшей (по абсолютной величине)
самополяризуемостью. Таким образом, для АУ следует ожидать,
что нуклеофильное замещение будет идти в том же направлении,
что и электрофильное. Этот же вывод, согласно теореме парности
для АУ, следует и из анализа электронных плотностей на атомах
в граничной МО. Однако при нуклеофильном замещении проис-
происходит перенос электрона с нуклеофила на низшую свободную МО
субстрата (рис. 8.22).
Механизм реакции при этом однотипен с механизмом электро-
фильного замещения и включает промежуточное образование п-
и сг-комплексов, которые заряжены отрицательно.
О
О* О
Электрофильная
атака
нуклсофильная
атака
Рис. 8.22. Схематическое представление электрофильного и вуклеофильного заме-
замещений в методе МО
324
Анионные (т-комплексы известны и выделены во многих случаях.
Таковы, например, стабильные комплексы Мейзенгеймера, стро-
строение которых установлено с помощью рентгеноструктурного анали-
анализа, например
Используя модель анионного <7-комплекса для описания струк-
структуры переходного состояния, можно определить понятие энергии
анионной локализации L~, которая сопоставима с энергией ак-
активации. Например, для гипотетической реакции аминирования N-
замещенного имидазола
0 ¦-¦« ex. - -. o-
LI LI1
4N
R
LIII
энергию анионной локализации для положения 2 рассчитывают
следующим образом:
- -3,2900
В табл. 8.10 приведены данные расчета зарядов и энергий анион-
анионной локализации для положения 2 различных производных и анало-
аналогов имидазола, сопоставленные с имеющимися данными об их
способности к прямому аминированию (действие амида натрия
в ксилоле). Можно видеть, что имеется четкая корреляция между
теоретическими расчетами и опытом. Только те соединения, кото-
которые имеют в положении 2 больший положительный заряд и мень-
меньшую (по абсолютной величине) энергию анионной локализации,
чем фенантримидазол (L^~ —1,976), способны вступать в изуча-
изучаемую реакцию. Такая корреляция создает благоприятные возмож-
возможности для прогнозирования поведения соединений сходного стро-
строения в реакции аминирования.
325
Таблица 8.10. Корреляцая нуклеофнльиого замещения в положенн
ных я аналогов ямндаэола *
Соедивенве
f\
0,094
0,107
0,137
0,151
0,152
0,170
0,161
0,186
-3,290
-2,182
-1,976
-1,965
-1,940
-1,931
-1,915
-1,838
326
Продолжение табл. 8.10
Соединение
Г
t
I )
in
я2
0,256
0,264
Ц-tf)
-1,761
-1,705
Способность
ж аминирова-
нию
+
Не изучалась
* Расчет по методу МОХ проведен с параметрами Стрейтвизера (см. табл. 8.2).
8.13.5. Реакции радикального замещения
Радикалы, генерируемые в растворах, способны атаковать аро-
ароматическое или гетероароматическое ядро, замещая водородный
атом. Так, например, при термическом разложении перекиси ацети-
ацетила в инертном растворителе возникающие активные метильные
радикалы реагируют с ароматическим углеводородом:
(СН3СООJ —*- 2СН3СОО- -^2СО2+2СН3-
сн,
сн* +
н +
LPV
Предполагаемый <7-комплекс LIV может служить аналогом пере-
переходного состояния реакции гемолитического замещения, и энергии
активации могут быть аппроксимированы значениями энергий ра-
радикальной локализации LM. Для реакции метального радикала с
бензолом L,, рассчитывается как
L» = е
В случае радикального замещения удобно использовать другой,
более простой и наглядный подход к ИРС. Оценим изменение
я-электронной энергии АЕ сопряженной системы, атакуемой ради-
радикалом. Поскольку радикал — электронейтральная частица, его при-
327
ближение к атакуемому в молекуле атому /z слабо отразится на его
кулоновском интеграле и более значительными будут эффекты из-
изменения резонансных интегралов Д„, где атомы v являются соседя-
соседями р. Эти изменения положительны, так как в результате образова-
образования новой связи с радикалом ослабляются (Р^ уменьшаются по
абсолютной величине) связи атома р с атомами v. Разложим АЕ по
степеням /? и ограничим разложение только членами первого поряд-
порядка. С учетом соотношения (8.72) величину АЕ можно представить
в виде
соседи, IX зг соседа, р
АА I 2JVAA. (8.111)
Суммирование ведется только по атомам v, связанным с атомом
/л химическими связями.
Полагая А/?^ одинаковыми для всех связей атома /i, получим
соседн,
д, ц
АЕ=2Ар X JV (8Л12)
Сумма порядков всех связей, примыкающих к данному атому ц,
названа связевым числом этого атома N^ Оно характеризует сте-
степень насыщенности рассматриваемого атома, которая тем больше,
чем больше JVM. Наоборот, способность атома к образованию новых
связей тем больше, чем меньше N^. Для характеристики этой спосо-
способности вводится ИРС, называемый индексом свободной валентно-
валентности:
F^N^-N», (8.113)
где Nma]L — максимально возможное число я-связей (максимально
возможное iVp), являющееся постоянной величиной.
С учетом (8.113) выражение (8.112) можно записать в виде
Л?=const -IF^Ap, (8.114)
откуда следует, что для однотипных реакций гемолитического заме-
замещения приращения АЕ тем меньше, чем меньше энергия активации
и чем больше индекс свободной валентности атакуемого атома.
В качестве iV^ принимают сумму порядков связей центрального
атома бирадикала триметиленметана, который образует тг-связи
с тремя соседями.
Приводим МО триметиленметана:
V6
V6
328
1
1
(8.115)
V6
Расчет показывает, что порядок
связи центрального атома с любым
соседним равен 1/V3- Таким образом,
4=
V3
/„=1,732- I Л*-
соседи, ft
I
I
I
I
(8.116)
Ожидаемая на основании расчетов
по методу МОХ бирадикальная при-
природа триметиленметана подтвержде-
подтверждена экспериментальными наблюдения-
наблюдениями его спектра ЭПР в стеклующемся
растворителе.
Нетрудно рассчитать по (8.116) ин-
индексы свободной валентности некото-
некоторых АУ:
тетрацен
1,2-Венштрацен , „
цДб-Диоензантрацен/ пирен
Антрацен
Флуорантен
Хризен о / Пирен
Фенантрен
''нафталин
бензол
Рис. 8.23. Корреляция относи-
относительных скоростей гомолитичес-
кого замещения метальным ради-
радикалом с индексами свободных ва-
валентностей К^Дши — макси-
максимальный индекс свободной вале-
валентности в молекуле]
0,399
Н2С:
0,732
t
=сн.
=СН-
0,391 0,838
t \
0,404
0,104
0,452
В соответствии со значениями F^ ненасыщенные углеводороды
легко реагируют с радикалами, например легко восстанавливаются
атомарным водородом; для ароматических углеводородов эти реак-
реакции идут труднее; наиболее активным к радикальному замещению
положением нафталина является а-углеродный атом.
Задача 8.11. Обычно полагают, что наибольшее значение 7^,^ = 1,732. Од-
Однако для бирадикала, теоретически получаемого удалением двух атомов водоро-
водорода из терминальных метиленовых групп аллена [НС—С = СН] ' ' , можно полу-
получить большее значение /^тм- Рассчитать ^ для бирадикала аллена.
На рис. 8.23 показана корреляция относительных скоростей ре-
реакции гемолитического замещения метильным радикалом с индек-
индексами свободных валентностей.
329
8.13.6. Реакции присоединения
Реакции присоединения вообще мало характерны для ароматичес-
ароматических и гетероароматических систем, но отдельные соединения склон-
склонны к образованию аддуктов с диенофилами, присоединению озона,
оксида осмия (IV) и др.
Типичный и хорошо экспериментально изученный пример — при-
присоединение малеинового ангидрида к сопряженным углеводородам.
Так, антрацен легко взаимодействует с этим диенофилом по схеме
Теоретически реакция может идти и по другим направлениям:
присоединения по связям 1—2, I—4, 2—3.
В приближении локализации, полагая, что степень образования
новых связей в переходном состоянии достаточно велика, можно
оценить потерю энергии сопряжения (главный вклад в энергию ак-
активации) как разность энергий сопряженных систем соответствую-
соответствующих продуктов присоединения и исходной молекулы антрацена. Дру-
Другой подход может быть основан на приближении изолированной
молекулы. В этом случае справедливы рассуждения, рассмотренные
в предыдущем разделе, и подходящим ИРС должна служить сумма
величин индексов свободной валентности атомов, участвующих в об-
образовании связи с диенофилами. Данные представлены в табл. 8.11.
Таблица 8.11. Корреляция вацраалекня присоединения дкенофкла к антрацену
Направление
присоедине-
присоединения
Сопряженная система переходного состо-
состояния
-пер
9—10
1—2
2—3
3,31
3,22
3,68
3,80
1,040
0,867
0,918
0,816
330
Литература (
Дьюар М. Теория молекулярных орбиталей в органической химии. — М.: Мир,
1972.
Подробное рассмотрение метода ППП и его применения к расчету свойств основных
состояний сопряженных молекул.
Дьюар М., Догертв Р. Теория возмущений молекулярных орбяталей в органи-
органической химии. — М.: Мир, 1977. Гл. 3.
Подробное обсуждение сопряженных систем с качественных позиций теории возмущений.
Большое число полезных упражнений.
Костиков Р. Р., Беспалов В. Я. Основы теоретической органической химии. —
Л.: Изд-во ЛГУ, 1982. Гл. 3.
Основные особенности строения я-электронных систем рассмотрены в рамках методов МОХ
и ППП.
Роберте Дж. Расчеты по методу молекулярных орбит. — М.: Мир, 1963.
Ясное описание метода Хюккеля и иллюстрация его на детально разобранных примерах.
Стрейтвизер Э. Теория молекулярных орбит для химиков-органиков. — М.:
Мир, 1965.
Хорошее и полное изложение метода Хюккеля в его приложений к различным проблемам
органической химии.
ГЛАВА 9
КАЧЕСТВЕННЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
ПРОСТРАНСТВЕННОГО И ЭЛЕКТРОННОГО
СТРОЕНИЯ МОЛЕКУЛ
В химической теории особенно важным является выработка
широких обобщений и создание достаточно простых качественных
приемов определения электронного, пространственного строения
и реакционной способности молекул. Это объясняется тем, что
число известных химических соединений составляет уже несколько
миллионов и растет со скоростью около 100 тыс. в год. Следовате-
Следовательно, получить данные точных количественных расчетов для каж-
каждого соединения просто не представляется возможным. Кроме того,
вместо строгих расчетов важнее получить ответы на вопросы:
а) какова относительная устойчивость или реакционная способ-
способность в данном ряду соединений; б) какой структурный признак
определяет устойчивость (или неустойчивость) данной однотипной
группы молекул; в) каким образом отразится то или иное структур-
структурное искажение на положении энергетических уровней и форме ор-
орбиталей той или иной молекулы; г) реакции какого типа должны
быть характерны для данного соединения и при каких условиях их
легче реализовать?
Ответы на эти и другие подобные вопросы можно получать,
пользуясь теорией валентных связей, основанной на представлениях
о локализованных химических связях, или методом молекулярных
331
орбиталещ основанном на представлении о делокализованных ор-
биталях.
9.1. ПРИНЦИПЫ КАЧЕСТВЕННОЙ ТЕОРИИ
МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
В своей наиболее простой форме качественная теория молеку-
молекулярных орбиталей содержит следующие основные допущения и по-
положения: 1) рассматриваются орбитали только валентных электро-
электронов атомов, образующих данную молекулу; 2) полная энергия мо-
молекулы или иона аппроксимируется суммой орбитальных энергией
валентных электронов:
зал
(9.1)
где л, — число заполнения электронами /-й МО, п( может быть
равно 0,1 или 2; е, — энергия i-й МО; q — ядерные координаты;
3) формы МО и порядок их расположения по энергии качественно
одинаковы для всех молекул общего структурного типа; 4) если при
изменении геометрической формы молекулы увеличивается поло-
положительное перекрывание (перекрывание участков функций АО
с одинаковым знаком) между двумя (или более) АО в составе
определенной МО, то энергетический уровень данной МО понижа-
понижается. Наоборот, если указанные деформации уменьшают положи-
положительное перекрывание или увеличивают отрицательное, соответст-
соответствующая МО дестабилизуется {правило перекрывания).
Первые два допущения те же, что и в расширенном методе
Хюккеля. Характер приближения (9.1) становится более ясным при
сопоставлении с выражением для полной энергии D.60) в методе
ССП МО, в котором учитываются также эффекты кулоновского
и обменного электрон-электронного отталкивания
зал
Eccn(q) = I «A (q) - У„ (q) + VM (q), (9.2)
»¦
где Уы — энергия электростатического отталкивания ядер или ядер-
ядерных остовов (в валентном приближении).
При сравнении (9.1) и (9.2) ясно, что первое соотношение может
выполняться при условии, что энергия электростатического оттал-
отталкивания ядер и энергия межэлектронного взаимодействия взаимно
скомпенсированы. В действительности было найдено, что эта ком-
компенсация не является полной, но сумма указанных взаимодействий
весьма точно равна 1/3?. Таким образом, должно выполняться
аналогичное (9.1) соотношение
а зав
^D)^^Lnizl(q). (9.3)
332 '
Справедливость соотношения (9.3) в области устойчивых геоме-
геометрических конфигураций большого числа разнообразных молекул
проверялась прямыми неэмпирическими расчетами, которые до-
доказали выполнимость соотношения (9.3) с точностью не хуже чем
11%. Такая точность, конечно, вполне удовлетворяет требованиям
качественной теории.
Наличие в уравнениях (9.1), (9.3) двух переменных — орбиталь-
орбитальных энергий е, (q) и чисел заполнения л, — позволяет применить
качественную теорию молекулярных орбиталей для решения двух
различных типов задач: I) для установления зависимости орбиталь-
орбитальных и полных энергий системы от вида геометрических конфигура-
конфигураций образующих ее атомов и выявления геометрии устойчивой
структуры; 2) для нахождения при заданном геометрическом стро-
строении q или симметрии молекулы оптимальной электронной кон-
конфигурации, т. е. числа электронов л„ при которых система устой-
устойчива или обладает необходимыми физическими параметрами (по-
(потенциал ионизации, сродство к электрону, магнитные характеристи-
характеристики и пр.).
Примером успешного решения задач первого типа служит схема
Уолша (см. разд. 10.1), тогда как результатами анализа задач
второго типа являются многочисленные правила электронного сче-
счета, предсказывающие устойчивые соединения разных классов.
Практическое применение качественной теории МО требует по-
построения МО молекулы из орбиталей атомов или орбиталей ее
отдельных фрагментов, что принято называть фрагментным (или
рек инструкционным) анализом (Р. Хоффман). Разделив молекулу
на фрагменты таким образом, чтобы по возможности сохранить
в каждом общие элементы симметрии, определяют их валентные
орбитали и соответствующие энергетические уровни. Если молекула
разделена на несколько фрагментов, можно составить ее МО, после-
последовательно оценив все парные взаимодействия орбиталей фрагмен-
фрагментов, а затем суммируя их тем же способом. В общем виде схема
двухорбитальных взаимодействий уже рассматривалась при анали-
анализе электронного строения молекулы водорода (см. разд. 4.5.1)
и двухатомных молекул (см. разд. 4.6 и 4.7).
9.2. ВЗАИМОДЕЙСТВИЕ ДВУХ ОРБИТАЛЕЙ
Наиболее простой случай представляет молекулярная система
А—В, разделяемая на фрагменты А и В, взаимодействие между
которыми сводится к взаимодействию только двух орбиталей
(ра с энергией еа, принадлежащей фрагменту А, и щ с энергией ?ft,
принадлежащей фрагменту В. Чтобы рассчитать энергии
еа и е'ь орбиталей (ра и (рь полной системы А—В, необходимо, как
было показано выше (см. разд. 4.3.2), решить секулярное уравнение
ззз
Ва —
= 0, (9.4)
где Яад—Еа, Ньь—еь (выберем фрагменты А и В так, чтобы
Наь—Ньа — матричный элемент оператора Нэфф, определяющего
взаимодействие между орбиталями фрагментов; S^ — интеграл пе-,
рекрываыия орбиталей <ра и <рь на заданном расстоянии сближения
фрагментов А и В. Точный вид оператора Нэфф зависит от выбира-
выбираемого приближения, например при использовании выражения G.51),
применяемого в расширенном методе Хюккеля:
#л=|р.Няфф«>4А = 0>5(?. + еь; Safest*. (9.5)
Раскрыв определитель (9.4), получим уравнение
(га - б\) (бь - е\) - (Ил - diSab) 2 = 0, (9.6)
решение которого относительно г\ приводит к следующим соот-
соотношениям:
(9-7)
Соответствующая диаграмма орбитальных взаимодействий по-
показана на рис. 9.1.
Задача 9.1. Получите выражения (9.7), (9.8) из (9.6).
Так как гл<ей, то из (9.7) и (9.8) ясно, что понижение энергетичес-
энергетического уровня орбитали <р'а относительно q>a меньше, чем повышение
уровня орбитали (р'ь относительно <рь, т. е. А&а<&?Ь по абсолютной
величине, где Аеа=ев—еа и Аеь—?Ь—&ь-
Следовательно, если обе орбитали фрагментов <ра и щ заняты
парами электронов, которые переходят на МО системы А—В, то
суммарный эффект взаимодействия есть дестабилизация, получи-
получившая название эффекта четырехэлектронной дестабилизации, —
это не что иное, как обменное отталкивание заполненных элект-
электронных оболочек. Возможность описания его соотношениями (9.7),
(9.8) возникает благодаря учету интеграла перекрывания 5^ между
взаимодействующими орбиталями.
334
в
Рис. 9.1. Образование МО системы А — В (<рв, ф^ из орбиталей фрагментов А и В
в правой части рисунка показано взаимодействие вырожденных орбиталей
Если же только одна из взаимодействующих орбиталей <ра или
<ръ заполнена электронами или если каждая из них содержит только
по одному электрону, оба электрона окажутся в молекуле А—В на
нижней орбитали ср'а. Этот эффект взаимодействия есть стабилиза-
стабилизация системы, его называют двухэлектронной стабилизацией.
Величина орбитального взаимодействия тем больше, чем бо-
больше интеграл перекрывания [см. (9.5)] и чем ближе друг к другу
энергетические уровни еа и еь взаимодействующих орбиталей фра-
фрагментов. Наиболее сильное взаимодействие соответствует случаю
вырожденных (ед — ?$=?<>) орбиталей <ра и (рь (рис. 9.1, б). Раскрытие
детерминанта (9.4) приводит в этом случае к квадратному ура-
уравнению
(ео-^-ГДа-^О. (9.9)
Решение уравнения (9.9), очевидно, идентично задаче расчета
МО молекулы водорода (см. разд. 4.5.1). Энергии стабилизации
нижней орбитали (ра и дестабилизации орбитали ц>ь можно пред-
представить в форме
(9.10)
ab
335
(9.11)
МО молекулы А—В <ра и щ являются линейными комбинациями
орбиталей фрагментов А и В:
<P>N(<pa+X<pb); (9.12)
<pb = N(<pb + fi(pa). (9.13)
Здесь N — нормировочный множитель; X и /i — безразмерные
коэффициенты, характеризующие степень «примешивания» орбита-
ли q>b к (ра и наоборот. Подобно тому, как в связывающие МО
двухатомных гетероядерных молекул наибольший вклад вносят АО
более электроотрицательного атома (т. е. АО с более низким энер-
энергетическим уровнем), а в антисвязывающие — АО менее электроот-
электроотрицательного атома, в выражении (9.12) для невырожденного слу-j
чая (sa<eb) орбиталь (ра локализована главным образом на фрагмен-1
те А, а орбиталь щ — на фрагменте В, т. е. коэффициенты X и ц ме-
меньше 1. Это важное обстоятельство отражено в графической форме
на рис. 9.1.
Задача 9.2. Получите общие выражения для МО молехулы А—В, представ-
представляющие линейные комбинации орбиталей фрагментов с энергетическими уров-
уровнями (9.7), (9.8).
Так как взаимодействия орбиталей попарно аддитивны, то рас-
рассмотренная схема орбитальных взаимодействий легко распростра-
нима на случай взаимодействия трех (и более) орбиталей, принад-
принадлежащих различным фрагментам. Предположим, нас интересуют
энергетические уровни и вид МО треугольной молекулы Н3 с сим-
симметрией C2v. Чтобы применить схему двух орбитальных взаимодей-
взаимодействий, будем искать МО Н3, исходя из орбиталей фрагментов:
молекулы Н2 и атома Н, т. е. осуществив фрагментацию:
н ч н
/\ Л\
н н н н
Вначале построим орбитали Н—Н в соответствии со схемой
двухорбитальных взаимодействий (см. рис. 9.1, б), а затем рассмот-
рассмотрим взаимодействие полученных орбиталей <р1а и ^ с орбиталью
336
атома водорода (рь (рис. 9.2). Только орбиталь <ри имеет отличный
от нуля интеграл перекрывания с орбиталью <рь. Взаимодействие
этих двух орбиталей ведет к формированию орбиталей lczi и Ъх\
молекулы Н3. Орбиталь фъ, фрагмента Н2 переходит в молекулу
Н3 без изменения (орбиталь Ь2 молекулы Н3).
Аналогичный подход с успехом применяется и в более сложных
случаях. Так, например, чтобы построить орбитали плоского мета-
метана (см. разд. 10.1.3), удобно разложить молекулу на два фрагмента:
центральный атом углерода и периметрическую группу Н4, т. е.
Н
н—с—н
н
Орбитали последней получают по схеме двухорбитальных вза-
взаимодействий, исходя из орбиталей двух групп Н2, в которых атомы
водорода удалены друг от друга на соответствующее расстояние.
Задача 9.3. Пользуясь последовательно схемой двухорбитальных взаимо-
взаимодействий, построить МО квадратной молекулы Щ.
9.3. ВЗАИМОДЕЙСТВИЕ
НЕСКОЛЬКИХ ОРБИТАЛЕЙ ФРАГМЕНТОВ
Для молекулярных систем с симметрией, более низкой, чем
в рассмотренных выше случаях, схему орбитальных взаимодейст-
взаимодействий не удается свести к двухорбитальным взаимодействиям фраг-
фрагментов и приходится использовать более общие выражения.
При взаимодействии т орбиталей, принадлежащих фрагменту
А, си орбиталями фрагмента В в системе А—В образуются (т + п)
новых орбиталей. Для нахождения значений энергий орбиталей
и волновых функций системы А—В нужно решать секулярное урав-
уравнение (m + w)-ro порядка и соответствующую ему систему линейных
уравнений для нахождения коэффициентов
=0, /=sit 2, ... . (9.14)
337
o-o
fa,
—H2
A
Рис. 9.2. Формирование МО системы
Из [симметрии C2v из орбиталей молекулы
водорода (<??=*, +X2 и q>l=Xi ~Xi) с энер-
энергиями ^ и tj и из атомной водородной
орбитали ^ с энергией e^J
?i ~~ *•/
При этом в качестве базиса ор-
орбиталей выступают т орбита-
лей фрагмента Аил орбиталей
фрагмента В. Можно, однако,
и избежать необходимости ре-
решать уравнения (9.14), прибег-
прибегнув для нахождения значений
уровней энергии st и коэффици-
коэффициентов сл для МО композитной
системы А—В к выводам те-
теории возмущений (см. разд.
1.5).
Полагая, что при формиро-
формировании молекулы А—В из фраг-
фрагментов геометрия последних
сохраняется неизменной, мож-
можно представить орбитали
щ молекулы А—В в виде
(9.15)
т. е. орбиталь <р?, принадле-
принадлежащая фрагменту А, переходит
в орбиталь (pi системы А—В
путем примешивания орбита-
лей фрагмента В.
Коэффициенты Су вычисля-
вычисляются с точностью до членов
первого порядка по формуле
(9.16), в котором член А'опре-
А'определен соотношением (9.5):
Из (9.16) следует, что орбиталь (р) будет вносить в (pt тем больший
вклад, чем меньше разность энергетических уровней ef—d и чем
больше интеграл перекрывания S^—^(p1\(pf) между орбиталью
<р1 фрагмента А и орбиталью ц>) фрагмента В.
Энергия е, орбитали q>t находится с точностью до членов второго
порядка теории возмущений по формуле
¦ырожд / v р* \ 2 rt
JTcB >6В ?'"
338
В этом выражении второе слагаемое — член первого порядка
теории возмущений — является не равным нулю только при нали-
наличии вырожденных по энергии орбиталей в фрагментах А, В. Обычно
в этом случае рассматривают вначале взаимодействие между вы-
вырожденными орбиталями, образуя их невырожденные комбинации,
а затем переходят к расчетам взаимодействий всех невырожденных
орбиталей.
Проиллюстрируем эту схему случаем взаимодействия двух ор-
орбиталей фрагмента А с одной орбиталью фрагмента В — так назы-
называемое трехорбитальное взаимодействие. В качестве фрагмента
А возьмем молекулу водорода Н2, а в качестве фрагмента В — атом
водорода Н и рассмотрим формирование орбиталей молекулы
Нз в треугольной Сггформе. Орбитали Н3 находят по формулам
(9.15) и (9.16):
(K-e*2)S23
<р2 ф2) = (ра2+ —Г- q>\\ (9.18)
Не рассчитывая числовые значения коэффициентов с(>, определим их
знаки. Учитывая, что величины (К—в) имеют отрицательные значе-
значения, интеграл Su = Ss] имеет положительное значение, a S32 = ?23 = О
(см. рис. 9.2). Тогда МО молекулы Н3 в С2у-форме можно записать
следующим образом:
(9.19)
Уравнения (9.19) можно переписать в базисе атомных орбиталей
X (см. рис. 9.2), учитывая опять только знаки коэффициентов
при АО:
(9.20)
339
Форма орбиталей (9.20) типична для случая взаимодействия
любых трех орбиталей. Орбиталь q>\ является сильно связывающей,
орбиталь ц>г обычно классифицируется как несвязывающая, а ор-
орбиталь <рг — антисвязывающая. Такое трехорбитальное взаимодей-
взаимодействие обусловливает устойчивость трехцентровых четырехэлектрон-
ных связей в гипервалентных соединениях (см. разд. 9.4.5).
Задача 9.4. Построить МО пентагонального иона Н~ (симметрия
Устойчив ли данный ион по отношению к фрагментации Н~
9.4. ПОСТРОЕНИЕ ОРБИТАЛЕЙ СЛОЖНЫХ МОЛЕКУЛ
Пользуясь приемами качественной теории МО, можно получить
достаточно точное представление о строении орбиталей весьма
сложных молекул. При этом особенно удобно строить МО сложных
молекул, выделяя те фрагменты, взаимодействие орбиталей кото-
которых определяет устойчивость (или неустойчивость) системы в це-
целом.
9.4.1. Орбитали связей и групп
Анализ волновых функций и электронных распределений широ-
широкого круга органических и неорганических соединений показывает,
что как для насыщенных, так и для ненасыщенных и сопряженных
органических соединений, а также и для координационных соедине-
соединений металлов делокализованные МО могут быть построены из
небольшого числа локализованных и переносимых (трансферабель-
ных) от одной системы к другой орбиталей отдельных связей
и групп. Для углеводородных систем такими группами являются
СН, СН2, СН3. Например, орбитали группы СН3 в соединениях
СН3+, СН4, СН3~, хотя и повышают свои энергетические уровни
в указанной последовательности, почти не изменяют свою форму.
Эти орбитали почти в неизменном виде (рис. 9.3) входят в МО всех
молекул, содержащих группы СН3(С2Н6, QH5CH3, СЩ> СН3ОН
и т. д.).
Для молекул с гетероатомами хорошими свойствами трансфера-
бельности обладают орбитали групп, изоэлектронных СН, СН2,
СН3. Даже в молекулах и ионах с многоцентровыми связями (боро-
водороды и их производные, кластеры переходных металлов и п~
комплексы) оказывается возможным выделить групповые орбитали
340
Рис. 9.3. Орбитали
углеводородных
фрагментов СИ,
СН2 и СН3:
граничные орбитали,
наиболее важные для
межмолекулярвых
взаимодействий, по-
показаны внутри прямо-
прямоугольников; энергети-
энергетические уровни получе-
получены из расчетов do
РМХ
26
22
8
с* (<*,)
-Ю
-1U
-18
-22
ОО
н — с
у
'сн3
н
И -hi С
и'
¦N
Рис. 9.4. Трехмерное представление граничных МО групп СН, СН2 и СН3, получен-
полученных из неэмпирических расчетов
нескольких основных фрагментов — строительные блоки делокали-
зованных МО огромного и разнообразного круга соединений.
На рис. 9.3 представлены орбитали углеводородных фрагментов
СН„ (п=\, 2, 3), построенные в форме линейных комбинаций
атомных орбиталей углерода Bs, 2рх, 2ру, 2/>г) и водорода (Is). На
рис. 9.4 показаны в трехмерном представлении эти же валентные
орбитали групп СН, СН2 и СН3, полученные из неэмпирических
расчетов. Все орбитали можно разделить на два типа. К первому из
них принадлежат с-орбитали (осн, Осн2> °сн3)> форма которых обес-
обеспечивает аккумулирование электронной плотности в области связей
СН. При образовании молекулы из групп СН эти орбитали слабо
взаимодействуют друг с другом, так как перекрывание между ними
незначительно [см. (9.7), (9.8)]. Такие орбитали относят к связевым.
В отличие от гг-орбиталей значительная часть электронной плот-
плотности в орбиталях я- и л-типов распределена вне области внутри-
групповых связей. На рис. 9.3 эти орбитали заключены в прямо-
прямоугольник. Взаимодействие таких орбиталей определяет возникнове-
возникновение новых связей, стабилизацию или дестабилизацию структуры,
составляемой из нескольких групп. Условия максимального пере-
перекрывания граничных ж- и п-орбиталей групп, зависящие от от-
относительного расположения групп, диктуют результирующую гео-
геометрию системы.
Форму и порядок энергетических уровней орбиталей п, я-типа
фрагментов СН4 и изоэлектронных им легко вывести, рассматривая
взаимодействие гибридных атомных орбиталей, которые не уча-
участвуют в образовании внутригрупповых связей С—Н (рис. 9.5). Из
четырех ^р3-гибридизованных орбиталей атома углерода в группе
СНЭ три расходуются на образование связей С—Н, а остающаяся
342
одна .?рэ-орбиталь л-типа может быть использована на образование
новых связей. В группе СН2 форма граничных орбиталей определя-
определяется взаимодействием двух ^3-орбиталей. Их связывающая ком-
комбинация (9.12) с коэффициентом Х=+\ дает орбиталь /icH2Bai)>
а антисвязывающая — 62-орбиталь я-типа. Порядок энергетических
уровней и форма граничных орбиталей группы СН, определяемая
взаимодействием трех sp* -орбиталей, вытекают из соотношений
(9.17)—(9.20). Легко видеть полное сходство орбиталей, произведен-
произведенных указанным простым способом, с орбиталями, полученными на
основании прямых молекулярно-орбитальных расчетов (рис. 9.4).
Задача 9.5. Гибридные орбитали <ра, щ ортогональны по отношению друг
к другу (см. разд. 10.2.2). Объясните, почему, несмотря на то, что Sab—O,
гибридные орбитали, центрированные на одном атоме, взаимодействуют, что
приводит к расщеплению их уровней, как показано на рис. 9.5.
9.4.2. Молекулярные орбитали этилена и иона метония
Рассмотрим на двух примерах, как можно использовать груп-
групповые орбитали для качественного представления МО, определя-
определяющих характер связывания в сложных молекулах.
Рис. 9.6 дает представление о способе образования валентных
МО этилена из орбиталей двух групп СН2 (см. рис. 9.3). В более
общем смысле рассматривая лишь энергетическую последователь-
последовательность орбиталей, а не конкретные значения энергетических уровней,
по рис. 9.6 можно судить об орбитальном строении молекул класса
А2Н4, так как форма и порядок энергетических уровней орбиталей
АН2 для всех элементов 2-го и 3-го периодов постоянны.
Как видно из рис. 9.6, только подходящие по свойствам симмет-
симметрии групповые орбитали комбинируют друг с другом. Природа
стабилизации плоской структуры С2Н4 связана с взаимодействием
только граничных орбиталей \Ь\ и 1а{ (п- и р-тшпл% ведущим
к образованию связевых асе и ясс-орбиталей этилена. Благодаря
значительному перекрыванию взаимодействие лBд,)- и ^A ^^ор-
^^орбиталей приводит к большому расщеплению их уровней, стабилиза-
стабилизации связывающей комбинации л-орбиталей Bа,) и образованию
связывающей комбинации МО, при этом заполнены электронами
только связывающие комбинации п- и я-орбиталей. Взаимодействие
низших орбиталей lai и \Ь2 приводит к дестабилизации системы,
343
Рис. 9.5. Энергетические уровни и форма граничных орбиталей СН„ (п*=
групп, образованных из орбиталей центрального атома углерода
О
-k
-8
-16
-20
-28 -
н,с
\
\
н
п
2h
групп
сн2
яс. 9.6. Формирование МО молекул А2Н4 (симметрия D^) из орбитадей АН2-
Представленное электронное заполнение МО соответствует молекуле этилена (расчет РМХ)
345
так как электронами заполнены как связывающие, так и антисвязы-
вающие комбинации этих орбиталей. Таким образом, понижение
полной энергии системы (^е*) относительно двух изолированных
фрагментов СН2 обеспечивается только понижением энергии двух
орбиталей: \biu (я-типа) и 2ag (и-типа). Именно заполнение тг-МО
1?1м, обеспечивающее заполнение всех связывающих МО, обуслов-
обусловливает устойчивость (энергетический минимум относительно угла
вращения вокруг связи С = С) плоской 2JА-формы молекулы этилена
в основном электронном состоянии.
Анализ орбитальных взаимодействий становится особенно важ-
важным при установлении причин устойчивости так называемых не-
неклассических органических соединений. К последним относят соеди-
соединения, структуры которых нельзя описать при помощи стандартных
представлений валентности. Простейшим примером может служить
ион метония — протонированный метан CHf, зафиксированный
впервые В. Л. Тальрозе A952). В газовой фазе этот катион, в кото-
котором атом углерода окружен пятью (!) атомами водорода, весьма
устойчив: для отрыва протона необходимо затратить
558 кДж/моль, а для отрыва молекулы водорода 184 кДж/моль.
Причина устойчивости СН5" становится понятной, если рассмот-
рассмотреть его орбитальное строение, исходя из орбиталей фрагментов
СН3+ и Н2, т. е. представить как комплекс 1д, в котором молекула
водорода выступает в качестве донорной, а катион СН^ — в качест-
качестве акцепторной компоненты:
Н 0,1087 0,127
\ Н LJ II 1
rtw\C-*- j ^(^ \
н ё н н /
н н
la I, Cs 1б
Диаграмма орбитальных уровней СН/ (рис. 9.7), построенная
комбинацией орбиталей группы СНЭ (см. рис. 9.3) и Н2, показывает,
что восемь электронов СН5+ в С,-структуре могут быть размещены
на четырех связывающих МО системы, три из которых (типа а)
стабилизируются при взаимодействии орбиталей фрагментов, при-
причем основным стабилизирующим фактором служит двухэлектрон-
ное взаимодействие <т-МО Щ с вакантной 2а.\ МО
Показанный на рис. 9.7 порядок энергетических уровней объяс-
объясняет устойчивость С,-формы / катиона метония, которая, как пока-
346
fa'
Рис. 9.7. Формирование МО катиона метоаия CHs+ в 0,-симметрии из орбиталей
фрагментов СНз+ и Н2
зано строгими расчетами, предпочтительна по сравнению со струк-
структурами более высокой симметрии С4„, />зл. C2v. Допирование элект-
электронной плотности с фрагмента Н2 на СН3+ (<т-+2д, взаимодействие)
приводит к разрыхлению связи Н—Н (это расстояние в молекуле
водорода равно 0,073 нм). В то же время длины локализованных
связей С—Н исходного фрагмента при СН3+ почти не меняются при
образовании СН^, так как орбитали этих связей не участвуют
в связывании с Н2. Приведенные на структурной диаграмме 16
расстояния (нм) получены на высоком уровне приближения метода
ССП МО (базис орбиталей 6-31G*, см. разд. 4.3.5).
9.4.3. Орбитали циклических систем
Циклы являются важными строительными блоками многих ор-
органических, металлоорганических и неорганических структур, по-
поэтому знание формы и относительного порядка энергетических
уровней их орбиталей весьма важно для конструирования орбиталь-
орбитальной картины структуры в целом. При качественном рассмотрении
необходимо знать проще всего последовательность энергетических
уровней и форму (узловые свойства) орбиталей. Для системы одно-
однотипных базисных орбиталей, принадлежащих каждому отдельному
центру циклической системы, можно с этой целью воспользоваться
простым методом Хюккеля (разд. 7.5.2), математический аппарат
которого справедлив, очевидно, не только для я-сопряженных си-
систем (базисные орбитали рх-типа), но и для любых других систем
орбиталей.
347
Энергетические уровни для циклической системы iV-орбиталей
q>u q>2, .., Фх в приближении Хюккеля, выраженные через значения
кулоновского а{На^ и резонансного /?(Я^) интегралов, каждый из
которых имеет отрицательную величину, определяются соотноше-
соотношением (см. гл. 8)
Вид образующихся МО циклической системы, записанных
в форме линейной комбинации базисных орбиталей, дается уравне-
уравнением
Рис. 9.8. Порядок энергетических уровней и формы *Fy МО для хюккелевских цик-
циклических систем. На рисунке показаны знаки Ckj коэффициентов в (9.22)
348
Как указывалось (см. гл. 8), Фростом и Мусулином предложен
удобный графический способ нахождения решения уравнения (9.21)
в зависимости от N (см. рис. 8.3). Этот способ можно использовать
и для нахождения энергетических уровней МО, образованных из
<т-АО.
На рис. 9.8 и 9.9 показаны базисные системы орбиталей цик-
циклических систем, образуемых атомами углерода или другими непе-
непереходными элементами в состоянии зр-гибридизации (см. разд.
10.2.2). Две группы радиальных л/7-орбиталей — ориентированные
внутрь (s/?in) и вне цикла (spout) — образуют, как и цикл ^„-орбита-
лей, хюккелевскую систему. Системы тангенциальных />-орбиталей,
Рис. 9.9. Базисные орбитали для А„ циклических систем:
Л — непереходный элемент в лр-гибридном состоянии (п = 3 + 6), />tang — система орбиталей,
при л = 3, 5 и 6 относящаяся к мебиусовсжой, а при л=4 — к хюкхелевской. Расположение
энергетических уровней соответствующих орбиталей показано в низу рисунка
349
e'
Ptang
A>»'.
Рис. 9.10. МО [3]-аннулена, сформированные из МО трех СН-групп:
три pj-орбитали СН-группы (рл) и три jp-орбитали (spm) составляют хюккелевскую систему
и образуют я= (а, е?) и <т-МО Ц, е'); три р^ fPung) орбитали составляют мебиусовскую систему
и образуют уолшевские а-МО с порядком энергетических уровней, обратным п-МО
оси которых лежат в плоскости рисунка
, Для нечетночленных
циклов имеют нечетное число антисвязывающих взаимодействий
между базисными орбиталями при их любой первоначальной ори-
ориентировке и относятся, следовательно, к мебиусовским системам.
Продемонстрируем теперь рассмотренные выше правила на при-
примере построения МО циклопропенильной системы Д3]-аннулен) из
350
орбиталей трех СН-фрагментов (рис. 9.10). Вид последних показан
на рис. 9.3 и 9.10. Рассмотрим взаимодействие только трех групп
орбиталей фрагментов С—Н (pnt р^^ spm), создающих валентную
зону орбиталей циклопропенильного кольца. Четвертая орбиталь
углеродного атома sp^t отвлечена на образование связи С—Н и не
принимает значительного участия в связывании групп СН в цикл.
Три /^-орбитали СН-фрагментов формируют при взаимодейст-
взаимодействии я-систему орбиталей цикла. Интегралы перекрывания между
всеми парами ^„-орбиталей положительны (число узлов четное или
равно нулю). Эта система принадлежит к хюккелевскому типу, при
котором в трехчленном цикле образуются один связывающий
а"г (<?1+<Р2 + фэ) и двукратно вырожденный антисвязывающий е"~
уровень (<pi — 2ф2 + (рг) и {(р\ — <ръ) [сравните с уравнениями (9.20)
и рис. 9.2].
Три орбитали р^-типа, наоборот, образуют мебиусовскую цик-
циклическую систему базисных орбиталей (так как число отрицатель-
отрицательных интегралов перекрывания между базисными орбиталями нечет-
нечетное, равно 3). При этом волновые функции результирующих <7-МО
цикла формируются таким же образом, но система уровней об-
обращается (рис. 9.10).
Заметим, что из 14 валентных электронов (СНK+ шесть находят-
находятся на орбиталях связей СН, а для остальных восьми электронов, как
видно из диаграммы орбитальных взаимодействий на рис. 9.11,
имеются как раз четыре связывающие МО, образованные из девяти
базисных орбиталей СН-групп.
Наиболее важный вывод при таком анализе состоит в том, что
сохраняется полная аналогия в схеме образования о- и я-делокали-
зованных МО. Необходимо лишь четко определить, к какому ти-
типу — хюккелевскому или мебиусовскому — принадлежит базисная
система орбиталей фрагментов, и воспользоваться для построения
соответствующих МО соотношениями, представленными в графи-
графической форме на рис. 9.8 и 9.9.
9.4.4. Орбитали металлоорганических фрагментов.
Изолобальная аналогия
Атомы переходных элементов используют при образовании свя-
связей не только валентные s- и /7-орбитали, но и близко расположен-
расположенные ^-орбитали. Тем не менее существуют глубокие аналогии в ха-
характере связывания металлоорганических и чисто органических
фрагментов, обусловленные сходной формой тех орбиталей этих
фрагментов, взаимодействие которых определяет природу связыва-
связывания.
351
Q
*
'/7
I/,
ML
Рис. 9.11. Групповые орбитали ML,, металлофрагментов:
M — переходный элемент; L — двухэлехтрониый лиганд (СО, СЦ РХз и т. д.); уровни а\ и ?
для МЬд, а также а,\ и е для ML3 инвертированы по отношению к уровням СИ- и СНг-групп (см.
рис. 9.3) иэ-эа того, что узловые свойства d-AO отличаются от узловых свойств р-АО
Важным шагом в сторону выявления указанных аналогий было
введение концепции электроноэквивалентных групп, т. е. групп,
центральному атому которых недостает одинакового числа элект-
электронов до полного заполнения валентной электронной оболочки.
В этом смысле электроноэквивалентными являются, например,
атом С1, радикал СН3 и Мп2 (СОM, дающие устойчивые димеры С12,
С2НбиМп(СОI0.
Более точное соответствие между валентными возможностями
группировок, образованных непереходными и переходными элемен-
элементами, устанавливает введенное Хоффманном определение изоло-
бальных групп, т. е. групп, для которых число, свойства симметрии,
форма и энергии граничных орбиталей примерно одинаковы.
Чтобы произвести формы орбиталей групп МЦ, можно исполь-
использовать обычную схему составления групповых МО из орбиталей
фрагментов М и Ln. Еще более удобный способ — последователь-
352
ное отщепление двухэлектронных лигандов (СО, NH3, PRO от окта-
октаэдра ML6 и анализ взаимодействий свободных гибридных орбита-
лей фрагментов МЦ_,) подобно тому, как граничные орбитали
групп СН3, СН2, СН производятся элиминированием связей С—Н
метана СН4 (см. рис. 9.5).
Атом переходного металла участвует в образовании связей де-
девятью орбиталями: пять md, одна (m+\)s и три (т+\)р, где
т — Ъ—5 обозначает период. Три d-орбитали аксиально-симметрич-
аксиально-симметричного фрагмента MLn не смешиваются с орбиталями лигандов и об-
образуют низко лежащий, слабо расщепленный уровень, который
близко соответствует /2*-УРовню в октаэдрических комплексах.
я-Орбиталей от п двухэлектронных лигандов, взаимодействуя
с остальными орбиталями центрального атома, дают п связыва-
связывающих и п антисвязывающих (на рис. 9.11 последние не показаны)
МО группы MLn, локализованных на связях М—L. Таким образом,
в валентной зоне остаются F—л) орбиталей группы ML,,, которые
заполнены х—2 F — п) электронами, где х — число валентных элект-
электронов металла.
Указанные F—л)-валентные орбитали группа МЬ„ использует
для связывания с другими лигандами или подобными группами.
Хотя эти групповые орбитали содержат значительные вклады от
^/-орбиталей металла, их форма и свойства симметрии аналогичны
рассмотренным выше орбиталям групп СН, СН2, СН3 (см. рис. 9.3
и 9.5). Сравним группы СН и Со(СОK. Легко видеть сходство
между граничными аг и е-орбиталями этих групп, причем атом
кобальта в Со(СО)з, как и атом углерода в СН, располагают тремя
электронами (для Со лг=9) для их заполнения. Электроны, которые
идут на образование связей между группами при их объединении
в каркасные и полиэдрические структуры, предложено называть
скелетными.
Подобным образом фрагмент Мп(СОM, в котором атом Мп
имеет 7 электронов (cPs2) и граничную орбиталь артипа, изолоба-
лен СНэ, а фрагмент Fe(COL изолобален СН2. Для обозначения
изолобальности введен специальный символ — двойная стрелка
с опущенной вниз петлей:
Мп(СОM -•—g—*"" снз
Fe(COL -• g ¦"" СН2
Со(СОK^ ф — СН
12. Теория строения молекул 353
Таблица 9.1. Изолобальвая аналоги между органическими м металлоорганичес-
кямн грушами ML, (М — переходный металл, в ^-оболочку включаются (m+\)s-
электроны)
снх
СНз
сн2
сн
Координацнонвос число атома М
9
^-ML8
tP-ML-,
8
^-ML7
</*-ML6
d*-MLs
7
<f-ML6
tf-ML4
6
<T-ML5
^-ML4
5
^-ML4
rflo-ML3
В табл. 9.1 устанавливаются общие соотношения изолобаль-
ности металлоорганических фрагментов, включающих группы ML»,
произведенные как от октаэдра (л = 6), так и от конфигураций
с другими координационными числами с органическими группами
СН,.
Концепция легко распространяется на металлоорганические
группы, содержащие не только двух-, но и 4,6-электронные лиганды.
Так, например, циклобутадиен имеет 4я-электрона и заменяет два
обычных двухэлектронных лиганда L, циклопентадиениланион или
бензол эквивалентны трем двухэлектронным лигандам. Отсюда
вытекает, например, следующий ряд изолобальных групп:
S3:
В табл. 9.2 типичные для органических структур атомные группы
сопоставлены с изолобальными и электроноэквивалентными им
группами переходных металлов.
Цепные, циклические, каркасные и кластерные структуры, полу-
получаемые объединением изолобальных групп, также обладают анало-
аналогичными системами валентных орбиталей и, следовательно, сход-
сходными структурными возможностями. Так, Со2(СОN имеет валент-
валентные орбитали, сходные с орбиталями ацетилена С2Н2, орбитали
циклических кластеров Fe3{CO)i2 и Fe3(CO)9 аналогичны МО цик-
циклопропана и циклопропена. Последнее особенно наглядно при срав-
сравнении рис. 9.10 и 9.12.
354
Таблица 9.2. Изолобальные соотвошешн
Органическая группа
СНЭ
СН3+
СН2
СН, N, О+
СН+, С, ВН
СН2+, С+, ВеН
СН3 +
Число ва-
валентных
электро-
электронов
7
6
6
5
4
3
2
Число
скелет-
скелетных элек-
электронов
5
4
4
3
2
1
0
Металлоорганичесхая группа
Mn(COM, Fe<COJ07s-C5H5),
ZD(r,s-CsH5), Мо(СОK(^-С5Н5),
Сг(СОM, Mn(COJfo5-C5H5), PtCl3-*
Fe(COL, Cr(COM, CoCO(^s-C5H5),
Cufa5 - С5Н5), Rh(COL+, Ni(PR3J
Co(COK) Ni(^s-C5H5), W(COJ
(r7s-C5H5), Re(COL, Rh^-Q^)
Fe(COK, Co(n5-C5H5)f Os(COK
Mn(COK, Fe(rj5-C5H5)
Cr(COK, Fe(COJ) Cr(if6-C6H6)
* Плоская Т-форма
>
2s
Рис. 9.12. МО трехчленного циклического кластера Fe(CO)9, сформированные из
орбиталей Fe3(COK фрагментов.
Струггура Fe3(CO)9 иэолобальна (СН)^+; при добавлении двух электронов к Рез(СО>9 заполняет-
заполняется 2а\ ге-подобвая МО и дианион Fe(CO)J~ имеет алектронную структуру, близкую к катиону
(СН),+ (см. рис. 9.10)
355
Значение концепции изолобальности состоит в том, что она
перекидывает прямой мост, связывающий структурные представле-
представления органической химии с усложненными структурами, характер-
характерными для химии переходных элементов. Он позволяет вскрыть
аналогию электронного строения между внешне совершенно раз-
различными классами органических, металлоорганических и неоргани-
неорганических соединений, например увидеть, что 7Г-комплекс II является
металлоорганическим спиропентаном. Соответственно неоргани-
неорганическая структура IV также изолобальна спиропентану III:
О
(COLFe
III
JV
Задача 9.6. Постройте группы, изолобальные СН3, СН2, СН, исходя из
плоского комплекса FeL4, где L — двухэлектронный лиганд (СО, PR3, С1~).
Задача 9.7. Произведите структуры органических соединений, имеющих
орбитальное строение, аналогичное следующим металлоорганическим комплек-
комплексам:
Fe(COL
.a) H2C=±=CH2
б) ферроцену: в)
CH, CH,
(CO)
ж)
(СО)-Со
o(COK
Co(COK
9.4.5. Полиэдрические органические молекулы и ионы
Подавляющее большинство органических соединений имеет цик-
циклическую или цепную структуру. Известно немало и трехмерных
каркасных соединений, таких, как адамантан V или додекаэдран VI:
VI
356
В этих соединениях замкнутый молекулярный каркас образован
сочетанием шести- или пятичленных циклов, причем каждый атом
углерода сохраняет свою нормальную валентность. В последнее
время, однако, стали быстро накапливаться данные о новых неклас-
неклассических полиэдрических структурах органических соединений, со-
содержащих пента- и гексакоординированные атомы углерода и высо-
высоконапряженные трехчленные циклы. Получены устойчивые пирами-
пирамидальные катионы — производные (СН)^ и (СНI+:
н
с
VII
VIH
Существование подобных структур и природу их устойчивости
в принципе невозможно предсказать и объяснить на базе классичес-
классических представлений теории валентности. Однако качественная те-
теория молекулярных орбиталей не встречает трудностей при описа-
описании пирамидальных и других полиэдрических структур. Удобно
осуществить реконструкционный анализ МО соединений VII, VIII
и их аналогов, выделив в качестве фрагментов апикальную (вершин-
(вершинную) группу СН и базальный аннуленовый цикл. Так, структуру
катиона VII можно составить из группы СН+ и циклобутадиеново-
го фрагмента, а катиона VIII — из группы СН2+ и циклопентади-
енильного фрагмента:
Vila
Villa
Орбитали СН-групп в циклах локализованы на этих связях и не
принимают участия в связывании с апикальной группой. Таким
образом, только я-МО и ff-MO аннуленовых циклов (см. рис. 9.8
и 9.9) существенно взаимодействуют с орбиталями апикального
центра. Расчеты показывают, что при сближении апикальной груп-
группы — единичного атома или связи X—Н с базальным циклом —
интегралы перекрывания валентных орбиталей этих групп с я-МО
[л]-аннуленового цикла в 3—5 раз больше, чем с <т-МО. Так как
энергия взаимодействия орбиталей пропорциональна квадрату ин-
357
Ьг
/a,
и
с
н
Рис. 9.13. Формирование МО гшрамидальной структуры катиона (CH)S+ из орбита-
лей фрагментов — циклобутадиена и СН+-группы
теграла перекрывания S^ согласно (9.7) и (9.8), то при качественном
рассмотрении достаточно учесть только взаимодействие с я-МО
базального цикла. Диаграмма орбитальных взаимодействий фраг-
фрагментов представлена на рис. 9.13. Можно видеть, что возникают
четыре связывающих уровня, на которых можно разместить восемь
электронов. Этот вывод является общим для всех пирамидальных
358
структур, образованных добавлением вершинного атома X непере-
непереходного элемента или <х-связи X—Н, X—R к аннуленовому фраг-
фрагменту IX, IXfl. Действительно (см. рис. 9.8), представления симмет-
симметрии низших я-орбиталей циклов любого размера одинаковы: а —
для низшей, е и b — для следующих МО, Следовательно, реализу-
реализуются те же орбитальные взаимодействия и сохраняется принципи-
принципиальная общность орбитальной картины электронного строения.
Таким образом, можно сформулировать правило электронного сче-
счета: пирамидальные структуры, в которых апикальный атом X —
непереходный элемент, устойчивы, если сумма л-электронов анну-
ленового фрагмента и валентных электронов X (в случае X—R
к электронам X добавляется один электрон группы R, участвующий
в образовании cr-связи) не превышает восьми (правило восьми
электронов):
IXa
Пользуясь правилом восьми электронов, легко объяснить устой-
устойчивость катионов VII, VIII, а также ряда их аналогов, например
полученных недавно металлоорганических ионов X, XI:
Пользуясь представлениями об изолобальной аналогии (см.
табл. 9.2) и заменяя апикальные группы X, ХН на соответствующие
металлоорганические фрагменты, содержащие переходные метал-
металлы, можно прогнозировать устойчивые я-комплексы переходных
металлов. Так, циклобутадиеновый комплекс XII — аналог органи-
органического катиона VII, а хромо цен XIII — органического трикатиона
XII
XIII
359
(CH)?4. который неустойчив в силу слишком сильного электроста-
электростатического отталкивания одноименных зарядов:
Задача 9.8. Сформулируйте правило электронного счета для устойчивости
сэндвичевых структур типа ,^-^zzz^-^ > r&? ^ — атом непереходного
элемента (Be, В, С, ...).
9.5. УСТОЙЧИВОСТЬ ПОЛИЭДРИЧЕСКИХ СТРУКТУР
И ПРАВИЛА СЧЕТА СКЕЛЕТНЫХ И ВАЛЕНТНЫХ ЭЛЕКТРОНОВ
Пирамидальные структуры, рассмотренные в предыдущем раз-
разделе, — лишь один из примеров общего класса полиэдрических, или
кластерных (от англ. cluster — рой), структур. Если для органичес-
органических соединений полиэдрические структуры — редкость, то для бо-
боранов и их производных и металлоорганических соединений пере-
переходных металлов они весьма распространены.
Можно выделить следующие группы кластеров.
Рис. 9.14. Дельтаэдрические каркасы полиэдрических боранов и карборанов:
тригональная бипирамида, октаэдр, пентагональвая бипирамида, додекаэдр, трехшапочная трв-
гональвая призма, двухшапочная архимедова антипризма, октадекаэдр, икосаэдр
360
1. Во раны, карбораны и металлокарбораны. Бораны, или боро-
водороды, образуют широкий ряд полиэдрических форм, харак-
характеризующихся наличием в них треугольных граней (дельтаэдры).
Примерами таких структур являются пентаборан В5Н9, облада
ющий пирамидальной конфигурацией скелетных связей В—В XIV
и изоэлектронный катиону (СНM+, пирамидальный карборан XV,
изоэлектронный дикатиону (СН)^ , а также бипирамидальный диа-
нион (ВН)б~ XVI, металлокарборан XVII и многие другие более
сложные структуры, связевые скелеты которых показаны на рис.
9.14.
XIV
н
н
2. Металл о органические кластеры переходных металлов, содер-
содержащие обычно в качестве лигандов циклолентадиенильные или
карбонильные группы (см. табл. 9.2). Относительно простыми при-
примерами таких структур служат кластеры
(coKR
XVIII
Ru(COK
361
3. Кластеры, в каркас которых внедрен изолированный атом.
Наиболее важный тип представляют карбидные кластеры, примера-
примерами которых могут служить следующие соединения:
XX,CFe(CO),5; CRu,(CO)
15
XXIa, CRu6 (CO), ?. XXI6, CFe6 (COJ,'
CNi8<CO)z-
Замена любой вершины в каждом из приведенных устойчивых
кластеров на неизоэлектронную или неизолобальную группу, как
правило, ведет к его разрушению. Это приводит к мысли о сущест-
существовании четкого соответствия между формой полиэдра и числом
связывающих орбиталей, обеспечивающих устойчивость кластер-
кластерной структуры. Именно это представление лежит в основе общих
правил электронного счета, разработанных для каркасных и кла-
кластерных структур бороводородов, карбо- и гетероборанов и рас-
распространенных с учетом представлений об электроноэквивалентных
группах на аналогичные металлоорганические соединения (К. Уэйд,
1971).
Эти правила основаны на счете скелетных электронов, понятие
о которых введено в разд. 9.4.4, размещаемых на эндоциклических
орбиталях скелетных связей каркасных и кластерных соединений,
обеспечивающих связывание атомов каркаса. Число таких электро-
электронов равно (v + jc—2), где v — число валентных электронов централь-
центрального атома непереходного элемента, занимающего одну из вершин;
х — число одноэлектронных лигандов (неподеленная пара рассмат-
рассматривается как воображаемый фантом-лиганд). Для групп, образован-
образованных переходными металлами, число скелетных электронов опреде-
определяется как (у+х —12). При этом устанавливается соотношение элек-
троноэквивалентности с группами непереходных элементов (см.
табл. 9.1). Понятно, что разница в 10е является, по сути, отличием
в требованиях 8- и 18-электронной оболочки для атомов непереход-
непереходных и переходных элементов.
Все полиэдрические структуры, реализуемые для бороводоро-
362
HU.UU
К/1030
V
Рис. 9Л5. Генезис и структурная иерархия для клозо-, нидо- и арахно-структур:
косые черточки означают переход при удалении одной вершины, горизонтальные -- коифор-
мациокные аереходы; стабильные клозо-, нидо- и арахно-бораны соответствуют анионам В„Н^~,
дов, карбо- и гетероборанов и металл о органических соединений,
отличающиеся наличием в них треугольных граней, можно произ-
произвести от так называемых клозо(замкнутых)-структур бипирами-
дального типа, представленных на рис. 9.15. Нидо(гнездовые)-стру-
ктуры получаются из клозо-формы усечением одной вершины (пи-
(пирамиды из бшшрамид). Тем же способом из нидо-структур образу-
образуют арахно(паутинные)ч;труктуры. Переход клозо-нидо-арахно ил-
иллюстрируется общей схемой, представленной на рис. 9.16. Качест-
Качественная теория МО и прямые расчеты клозо-структур приводят
к заключению, что для бороводородов В„Н„ имеется л+1 связыва-
связывающих МО, на которых можно разместить 2л+ 2 скелетных электро-
электрона, т. е. устойчивы клозо-структуры дианионов B«Hj~, например
XVI, л = 6, и изоэлектронных карборанов С2ВД_2НЛ, а также ней-
нейтральных боранов ВчНд+2.
Так же как и группа СН, ВН имеет три орбитали, используемые
для скелетного связывания:
~&
sp
Как видно из рис. 9.16, а, орбитали sp^ формируют только одну
а\ связывающую МО бипирамидального клозо-борана В5Н5) тогда
как орбитали р^% и рк дают пять связывающих МО. В сумме это
составляет шесть, т. е. л + 1, связывающих МО, на которые можно
поместить 2л+2 скелетных электрона. Нетрудно показать, пользу-
пользуясь идентичной техникой реконструкционного анализа, что этот
вывод справедлив для л = 6, 7, 8, ..., т. е. для высших клозо-боранов
(рис. 9.16, б). Аналогичное рассмотрение для нидо-боранов В„Н„
обнаруживает л + 2, а для арахно-боранов В„НД—п + 3 связывающие
МО. Таким образом, можно вывести следующие правила счета
скелетных электронов для устойчивых полиэдрических структур:
Клозо ... 2л+2
Нидо ... 2л+ 4 (9.23)
Арахно ... 2л+ 6
где л — число атомов непереходных элементов, образующих остов
кластерной структуры. Эти правила справедливы не только для
боранов и карборанов, но и кластеров, включающих другие непере-
непереходные элементы. Подсчет скелетных электронов ведется в соответ-
соответствии с данными, приведенными в табл. 9.2 и 9.3.
364
Рис. 9.16. Формирование орбиталей клозо-борана В5Н5 вз орбиталей фрагментов
ВН-вершин: (а) — МО, образованные из .грщ-системы; (б) — МО, образованные вз Ptang- и
показаны тольжо по одной опбитали^
трехчленного цикла и двух
(для каждой пары f'ae" МО
Таблица 9.3. Числа скелетных электронов (v+x — 2) для групп, образуемых ве-
иереходнымя элементами А
Число валент-
валентных элегтро-
НОВ V
1
2
3
4
5
6
7
А
Li, Na
Be, Mg, Zn, Cd, Hg
B, AI, Ga, In, Tl
C, Si, Ge, Sn, Pb
N, P, As, Sb, Bi
0, S, Se, Те
F, Q, Br, I
Тип гластернпй енинини
A(jc«O)
— 1
0
1
2
3
4
5
AH, AL'(x=l)*
0
1
2
3
4
5
6
AH2,AL(;c=2)**
1
2
3
4
5
6
7
* Одноэлектронный лиганд L' = Q, Br и др.
** Двухэлектронный лиганд L = NH3, PR3 и др.
Выполнение правил (9.23) можно проверить на бипирамидаль-
ных структурах XVI, XVII. Для нидо-структур действует правило
Bи + 4). Устойчивыми пирамидальными бороводородами должны
быть тетраанионы ВД1*~, а в случае углеводородов — пирамидаль-
пирамидальный катион (СНM+. Наконец, для арахно-структур бороводородов
устойчивы гексаанионы В„НJ~. Для арахно-структур углеводородов
(СН)„, производимых усечением двух вершин бипирамид, т. е. плос-
плоских циклов, это означает устойчивость дианиона (СНL~, аниона
(СНM~, нейтрального (СНN, катиона (СНO"\ что является одним из
следствий правила Хюккеля Dи + 2) (см. разд. 8.2).
Дополнительные электроны могут вносить не только избыточ-
избыточные заряды (в анионах боранов), но и мостиковые одноэлектронные
или двухэлектронные лиганды, а также атомы или группы, внедрен-
внедренные в структуру кластера, как в соединениях XX—XXII. Серия
устойчивых нейтральных боранов с мостиковыми связями В—Н—В
иллюстрирует переход клозо-нидо-арахно в соответствии с требова-
требованиями правил электронного счета (9.23):
и
н
XXIII
В6Н8(клоэо)
XXIV
В5Н9(нидо)
н
XXIV
В4Н|0(арахно)
366
Задача 9.9. Покажите эквивалентность правила восьми электронов для
пирамидальных органических структур и правила Bл+4) для нидо-структур
кластеров непереходных элементов.
Правила счета скелетных электронов легко распространяются на
металлоорганические соединения, для чего следует воспользоваться
соотношениями табл. 9.4. Предложены и другие общие формули-
формулировки; в частности, правила, позволяющие подсчитывать все ва-
валентные электроны кластера, включая электроны экзоциклических
связей (Д. Мингос). Так, для карбонильных кластеров Мет(СОIч~
(q — 0, 1, 2, ...) характеристическое число валентных электронов Nm,
вычисленное по формуле
N^=mNm+2n + lq, (9.24)
где Nm — число валентных (s-, p-, d-) электронов металла, составля-
составляет обычно 86 для кластеров различной формы вплоть до т^%.
Примером служат стабильные октаэдрические кластеры Co6(CO)i6,
Co6(CO)ff, Co6(CO)?4~, родиевый кластер XXV, изоэлектронный
Co6(CO)i6, кластеры XXI. Для арахно-кластера XXVI #вал = 62, а для
нидо-кластеров XX N^
,h(CO),
e(CO),
(COKF
(COh
XXV
XXVI
Таблица 9.4. Правила счета валентных электронов для кластеров неиереходных
н переходных элементов
Струггура
Клозо-дельтаэдр
Нндо-деяьтаэдр
Арахно-дельтаэдр
Гндриды веоереходных
элементов
4л+2(л>5)ВяН^-
4п+4(пМ)ВиНя+4
4л + 6(л>4)ВяНя+6
Карбонильные кластеры ое-
рсходных металлов
14л+2 XIX
14it+4 XX
14л+6 XXVI
367
При увеличении числа атомов, образующих кластер, структур-
структурные формы становятся особенно многообразными, а правила элект-
электронного счета — все более разветвленными. Можно показать, что
приведенные в табл. 9.4 правила (как и другие, эквивалентные им)
распространяются на случаи более сложных кластеров, производи-
производимых путем связывания через общий атом, общую связь или общую
грань кластеров клозо-, нидо- или арахно-типов.
Задача 9.10. Вывести правила электронного счета для конденсированных
бипирамидальных структур, имеющих общую вершину.
Задача 9.11. Объясните природу известного в pjajry бораяов и .карбораяов
окислительно-восстановительного превращения
+ 2е +2е
Клозо ?» Нидо j* Арахно
~2е -2е
Значение рассмотренных правил электронного счета состоит в том,
что они ставят в прямое соответствие число связывающих МО и тип
полиэдрической структуры независимо от природы образующих ее
вершинных групп. Эти правила топологичны по своей природе и ясно
демонстрируют общность структурных принципов неорганической,
металлоорганической и органической химии, наиболее ясно обнару-
обнаруживаемую при использовании методов качественной теории МО.
Наиболее важными выводами последней в приложении к проблеме
структурной организации молекул являются следующие положения.
1. Структурно однотипные молекулы и ионы обладают качест-
качественно однотипной системой молекулярных орбиталей. Принципи-
Принципиальные отличия в электронном строении определяются в данном
случае различием в числе валентных электронов и, следовательно,
различной электронной заселенностью орбиталей.
2. Основным типом орбитальных взаимодействий фрагментов,
определяющим устойчивость системы, является взаимодействие
граничных и ближайших к ним орбиталей п- и 7г-типов. Взаимодей-
Взаимодействие ниже расположенных связевых орбиталей ст-типа является
отталкивательным, однако в силу обычно малого перекрывания
таких орбиталей, принадлежащих разным фрагментам, этот эффект
сравнительно невелик.
3. Стабильным структурам соответствуют системы, число элек-
электронов в которых не превышает удвоенного числа связывающих
орбиталей. Это положение лежит в основе всех правил электронно-
электронного счета, описывающих устойчивость основных типов структур.
Литература
Albright Т. A., Burden J. К., Wbangbo M. H. Orbital Interactions in Chemistry.
New York: J. Wiley, 1985.
368
Подробное систематическое рассмотрение всех аспектов качественной теории МО, включая
анализ электронного и пространственного строения металлооргаяических и координационных
соединений.
Minkin V. I., Minyaev R. M., Zhdanov Yu. A. Nonclassical Structures of Organic
Compounds. Moscow: Mir, 1987.
Представлены основные положения теории орбитальных взаимодействий и реконструкцион-
ного анализа. Рассмотрено их применение для описания и предсказания различных напряженных,
полиэдрических, каркасных органических структур и структур, связанных с ними изолобальными
аналогиями.
ГЛАВА 10
СТЕРЕОХИМИЯ СОЕДИНЕНИЙ
НЕПЕРЕХОДНЫХ ЭЛЕМЕНТОВ
Важнейшей задачей теоретической стереохимии является описа-
описание и предсказание геометрической структуры молекул и образу-
образуемых ими частиц (ионы, радикалы, комплексы и др.). В принципе
все сведения о молекулярной структуре (длина связей, валентные
и межплоскостные углы) можно получить с помощью прямых
расчетов, например по методу МО (см. гл. IV), проводя серию
вычислений с полным варьированием всех 3JV—6 независимых гео-
геометрических параметров и отыскивая минимумы гиперповерхности
потенциальной энергии. Пример такого подхода — расчет прост-
пространственной структуры молекулы метилена в двух различных элект-
электронных состояниях (см. рис. 5.9).
Результаты этого и других подобных сложных расчетов, выпол-
выполненных с достаточно высокой точностью (см. гл. 7), хорошо воспро-
воспроизводят, а в некоторых случаях даже корректируют данные экс-
эксперимента по геометрической структуре молекул. Не менее важная
задача таких расчетов состоит в проверке и оценке точности полуко-
полуколичественных и качественных стереохимических теорий, непосредст-
непосредственно перебрасывающих мост от структурной формулы химичес-
химического соединения к геометрической форме его молекулы или иона.
В данной главе рассмотрим три различающихся подхода к описа-
описанию геометрической структуры соединений непереходных элемен-
элементов: 1) модели, основанной на представлениях качественной теории
МО; 2) теорию гибридизации атомных орбиталей; 3) теорию от-
отталкивания электронных пар валентных орбиталей. По существу,
все эти подходы никак не связаны друг с другом, и их предсказания
совершенно независимы.
10.1. ДИАГРАММЫ УОЛША
Под термином «диаграммы Уолша» (реже «диаграммы Уол-
ша—Малликена») подразумевают корреляционные зависимости,
описывающие изменения энергетических уровней молекулярных ор-
369
Рис. 10.1. Схема образования МО линейной молекулы АН 2 из орбиталей атома
А и симметризованных линейных комбинаций 5-орбиталей двух атомов Н
биталей структуры при ее различных деформациях. Если известно,
что уровни отдельных МО, например для молекулы АХ2, понижа-
понижаются при переходе от ее линейной D^-k угловой С2у-форме, нетруд-
нетрудно представить, какова должна быть электронная конфигурация
молекулы, т. е. заполнение энергетических уровней электронами,
чтобы стабилизировать эту форму. Диаграммы Уолша можно по-
получить как в результате прямых количественных расчетов МО
молекул в их различных геометриях, так и на основании качествен-
качественной теории МО, рассматривая деформации родоначальной струк-
структуры как возмущения, отражающиеся на энергетических уровнях
и форме отдельных МО. Этот подход наиболее важен для понима-
понимания главных закономерностей, связывающих электронное и про-
пространственное строение молекул. Проследим за его возможностями
на ряде характерных серий молекул.
10.1.1. АН2- и АХг-системы
На рис. 10.1 показана схема образования МО линейной молеку-
молекулы АН2. Учтены лишь валентные s- и р-АО атома А. Удобно
образовать МО всей молекулы из АО центрального атома с пред-
предварительно сформированными линейными комбинациями двух АО
370
H — А — Н
26а
2Ъ2
•О*
•и»
оОо
Рис. 10.2. Диаграмма Уолша для
la,
искажения молекул АН2
водородных атомов. Фактически это означает, что МО молекулы
АН2 образуются из АО атома А и МО молекулы Н2, в которой два
атома Н разнесены на такое же расстояние, как в молекуле АН2.
Как было показано (см. гл. 6,9), при образовании МО сложной
молекулы возможны комбинации лишь тех орбиталей, которые
обладают одинаковыми свойствами симметрии. Для линейной мо-
371
лекулы АН2, как и в случае двухатомных молекул А2, важно рас-
рассмотреть симметрию относительного центра инверсии, лежащего
на молекулярной оси. Орбитали ру и pz антисимметричны по от-
отношению к оси Н—А—Н, поэтому они не комбинируются с ag\s-
и с*Ь-орбиталями водородных атомов. Следовательно, в линейной
молекуле АН2 они являются несвязывающими.
На рис. 10.2 показано, как изменяются форма и энергетические
уровни МО линейной молекулы АН2 при угловом искажении. Эти
изменения можно прогнозировать, пользуясь правилом перекрыва-
перекрывания (см. разд. 9.1) — одним из основных положений качественной
теории МО.
Задача 10.1. Обоснуйте правило перекрывания на основе представлений те-
теории возмущений. При каком условии член возмущения второго порядка при
геометрической деформации является стабилизирующим?
Наиболее значительные изменения при угловой деформации
происходят с уровнями ои и пиу линейной молекулы. Первый из них
дестабилизируется, так как увеличивается отрицательное перекры-
перекрывание водородных орбиталей, второй сильно стабилизируется, так
как становится возможным перекрывание ру-АО атома А (несвязы-
вающая пиу-МО линейной молекулы АН2) с бг^-МО Н2. Этот резуль-
результат можно рассматривать как следствие смешивания орбиталей
одинаковой симметрии типа а\ при возмущении — деформации
линейной структуры:
(Ю.1)
Как и в линейной структуре, pz-AO центрального атома не имеет
подходящих по симметрии партнеров и остается несвязывающей
орбиталью и в угловой молекуле. Орбиталь ag несколько стабилизи-
стабилизируется при угловом искажении.
372
Рассмотрение относительного расположения уровней МО (см.
рис. 10.2) позволяет связать структуру молекулы АН2 с числом
электронов на ее валентных орбиталях. Так, для иона Н*, получа-
получающегося при экзотермической реакции Н2+Н+ и обладающего
всего двумя электронами, теория предсказывает угловую структуру.
Два электрона заполняют МО 1аь уровень которой несколько ниже
уровня в линейной структуре. Этот вывод согласуется с эксперимен-
экспериментом.
Молекула ВеН2 с четырьмя электронами в валентной оболочке
должна иметь линейную структуру. Однако для шестиэлектронных
трехатомных молекул следует ожидать угловой конфигурации свя-
связей вследствие значительной стабилизации уровня 2а,. Действитель-
Действительно, метилен СНг, в согласии с наиболее точными эксперименталь-
экспериментальными данными и неэмпирическими расчетами (см. рис. 5.9), имеет
угловую структуру. Такой же структурой с валентным углом —120°
обладает ион NH}.
Восьмиэлектронные молекулы, в частности Н2О, обладают, в со-
согласии с предсказанием качественной теории МО, угловой кон-
конфигурацией связей. В молекуле Н2О восемь валентных электронов
заполняют все четыре нижние МО (см. рис. 10.2). Такая электронная
конфигурация молекулы сильно отличается от предсказываемой
теорией гибридизации АО. Согласно последней, в молекуле воды
обе неподеленные электронные пары совершенно равноценны. Рас-
Распределение же электронов согласно схеме уровней МО на рис. 10.2
обнаруживает только один несвязывающий уровень, который мо-
может быть занят лишь одной неподеленной электронной парой.
Анализ фотоэлектронного спектра молекулы воды полностью
согласуется с такой электронной структурой. Первый потенциал
ионизации A2,62 эВ) соответствует несвязывающей МО элект-
электронной пары кислорода 1Ь\. Два следующих пика в фотоэлектрон-
фотоэлектронном спектре (/2= 13,8 эВ, /3 = 17,2 эВ) обладают тонкой колебатель-
колебательной структурой, характерной для орбиталей, локализованных на
связях. Они относятся к орбиталям 2аi и\Ьг соответственно. Наибо-
Наиболее низко расположенный уровень МО валентных электронов \ах
(/4 = 32,6 эВ) содержит лишь небольшую примесь 15-АО водород-
водородных атомов и фактически является второй неподеленной элект-
электронной парой, локализованной на 2s-AO кислородного атома. Рас-
Рассмотренные отнесения иллюстрируются на рис. 10,3, на котором
представлен фотоэлектронный спектр воды.
Величина валентного угла Н—А—Н существенно зависит от
электроотрицательности центрального атома. При ее понижении
373
эв
Н20
BAA
И
-/5
энергетический уровень \пи (р)-
орбитали этого атома повыша-
повышается и, следовательно, умень-
уменьшается энергетическая щель
между смешивающимися по
схеме (ЮЛ) орбиталями сим-
симметрии fl]. В соответствии с со-
соотношениями теории орби-
орбитальных взаимодействий (9.7)
и (9.8) это ведет к увеличению
стабилизации 2<2]-орбитали,
т. е. уменьшению валентного
угла (см. рис. 10.2). Действите-
Действительно, приводимые ниже дан-
данные экспериментального опре-
определения геометрии изоэлект-
ронных гидридов, содержащих
в валентной оболочке по 8 эле-
электронов, подтверждают общее
правило: чем ниже электроот-
электроотрицательность центрального
атома, тем меньше валентный угол Н—А—Н:
Н2О H2S H2Se H2Te H2F+ H2O H2N"
-?а, -2$
Рис. 10.3. Фотоэлектронный спектр моле-
молекулы воды и потенциалы ионизации МО
воды (/) и атомов Н и О G^)
104,5е
92°
90,6°
90,3е
118,1е
104,5е
104°
Для молекул типа АХ2, в которых атом X — элемент второго
или третьего периодов, имеющий валентную оболочку .sp-типа,
диаграммы Уолша качественно близки к рассмотренной для случая
АН2. Основные эффекты понятны, если рассматривать только элек-
электроны сг-связей А—X, образуемых /ьорбиталями X вместо s-op-
биталей атома Н в АН2. Определяющим фактором при выборе
линейной или угловой геометрии молекулы является также кривиз-
кривизна энергетического уровня высшей занятой МО 2агтипа. Как прави-
правило, понижение уровня этой орбитали более значительно, чем деста-
дестабилизация орбитали \Ь2 при угловом искажении. В результате моле-
молекулы с двумя электронными парами, как, например, ВеС12, имеют
линейное строение, тогда как для молекул с тремя и четырьмя
электронными парами (CF2, OF2) реализуется угловая структура.
Задача 10.2. Построить полную диаграмму Уолша для молекул типа АХ2
(А и X — элементы второго периода).
374
-8s
Ijiii...
пи-
Рис. 10.4. Диаграмма Уолша для молекул АН3. Корреляция МО плоской D3* и
рамидальной C3v структур
10.1.2. АНг и АХз-системы
Диаграмма Уолша для молекул АН3 показана на рис. 10.4. МО
плоской молекулы АН3 производятся при помощи соотношений
(9.21), (9.22) или по схеме (см. разд. 9.2).
При пирамидализащш интеграл перекрывания между орбиталя-
ми Н3 и рх, ру-АО центрального атома уменьшается, в результате
375
чего орбитали le-типа дестабилизируются, а 2е-типа стабилизиру-
стабилизируются подобно орбиталям Ь2 в молекулах АНг при угловом искаже-
искажении. Пирамидализация ведет к сильному смешиванию орбиталей
2а! и Ъп\ (в 7)зл-форме эти МО имеют различную симметрию, но при
искажении приобретают а{-симметрию). Поведение этих орбиталей
аналогично взаимодействию агорбиталей в молекулах АН2 [см.
A0.1)]. Размещая электроны на МО плоской и пирамидализованной
структур АН3, нетрудно заключить, что для шестиэлектронного
гидрида ВН3 следует ожидать плоской структуры, а для восьмиэлек-
тронной молекулы NH3, где в пирамидальной форме сильно стаби-
стабилизирована высшая заполненная МО 2аь предпочтительна пирами-
пирамидальная форма.
Рассмотрим радикал СН3. В этом радикале дестабилизация двух
полностью заполненных электронами орбиталей \е уравновешена
стабилизацией заполненной лишь одним электроном орбитали 2ах.
Радикал имеет плоскую структуру, но в его инфракрасном спектре
присутствует низкочастотное колебание Я2-симметрии, приводящее
к динамическому переходу между D3h- и С3„-Ф°рмами.
При рассмотрении молекул АХ3 (где X —F, C1, Вг, I), как и в слу-
случае молекул АХ2, можно пользоваться диаграммой Уолша для
молекул АНз, считая все валентные электроны атома А и только по
одному от каждого атома галогена. Таким образом, шестиэлект-
ронные галогениды А1С13, BF3 должны быть плоскими, а восьми-
электронные молекулы и ионы NF3, SnCl3~% SF3+ — пирамидаль-
пирамидальными.
Задача 10.3. Десятиэлектронные молекулы и ионы типа АХ3) C1F3, SeCl~,
XeF3+ имеют плоскую Т-образную структуру. Объясните эти данные, построив
корреляционную диаграмму D^— С2(,-структур.
10.1.3. АН4-системы
Для тетракоординированных систем, простейшими представи-
представителями которых служат гидриды АН4, наиболее важны две принци-
принципиальные геометрические формы: тетраэдрическая и плоская. Удоб-
Удобно построить МО плоской молекулы (см. разд. 9.2), а затем, дефор-
376
мируя плоскую структуру, рассмотреть, как реагируют на эту дефо-
деформацию энергетические уровни и форма отдельных МО:
(Ю.2)
с
Корреляционная диаграмма МО для систем АН4 показана на
рис. 10.5.
Рис. 10.5. Диаграмма Уолша для T^-^D4h искажения молекул АН4
377
Для двух орбиталей 1/2 группы деформация Td-^D^ приводит
к понижению энергетического уровня, так как связана с увеличением
интеграла перекрывания между ls-AO атомов водорода и р-АО
центрального атома. Однако третья орбиталь из этой группы резко
дестабилизируется, так как, по условиям симметрии, рх-АО цент-
центрального атома в 1LА-структуре молекулы АН4 не перекрывается
с орбиталями атомов водорода. Дестабилизация а^-МО в восьми-
электронных гидридах, важнейшим представителем которых явля-
является молекула метана, — это главная причина неустойчивости плос-
плоских конфигураций этих соединений. Точные расчеты дают для моле-
молекулы метана разность энергий между тетраэдрической и плоской
формами ~ 650 кДж/моль. Если учесть, что энергия разрыва связи
С—Н в метане составляет 435 кДж/моль, становится понятной
невозможность внутримолекулярной изомеризации A0.2) для моле-
молекулы метана.
Можно предположить, что плоский гидрид АН4 реализуется
в шестиэлектронной системе, например в случае иона СЬЦ+. Однако
дикатион метана неустойчив и распадается по схеме
CHj+->CH^4-H + . Чтобы стабилизировать плоскую структуру
восьмиэлектронной молекулы АН4, необходимо стабилизировать
а2«-орбиталь, что достигается при ее смешивании с незаполненными
и подходящими по симметрии АО заместителей. Наиболее подхо-
подходящими заместителями являются Li, BH2. Действительно, как пока-
показывают весьма строгие неэмпирические расчеты, например для мо-
молекулы CH2Li2, плоская структура более предпочтительна, чем тет-
раэдрическая.
10.1.4. А2Н4-системы
Диаграммы Уолша — удобное средство для анализа влияния
структурных изменений и в более сложных молекулярных системах,
чем АН„ и АХ„. Рассмотрим, как скажется вращение относительно
центральной связи А—А в молекулах типа А2Н4:
Н\ /" Н\ х^н
ь/ ^н н
378
3-5
h^
/e
Рис. 10.6. Диаграмма Уолша для
искажений структур типа А2Н4
МО типичной молекулы А2Н4 этилена (А = С) были построены
ранее (см. рис. 9.6). На рис. 10.6 показана корреляция орбиталей
плоской /^-структуры с орбиталями Оъгформы. Для молекулы
этилена, содержащей 12 валентных электронов, высшей заполнен-
заполненной МО является \blu-MO я-типа, и устойчивость этилена в плоской
^-структуре обусловлена тем, что деформация в /J(Гформу ведет
к резкому повышению энергетического уровня этой МО. Здесь еще
раз надо подчеркнуть определяющую роль поведения энергетичес-
энергетического уровня высшей заполненной МО при геометрической дефор-
деформации для выбора устойчивой структуры.
Для молекулы диборана D) В2Н4 число валентных электронов на
два меньше, чем для этилена, и орбиталь 1Ь\и в плоской
не заполнена. В результате, как видно из корреляционной диаграм-
диаграммы рис. 10.6, AеL (а^-электронная конфигурация Л^формы долж-
должна быть выгоднее, чем (l^J Ba*J A6иJ-конфигурация Я^-формы.
379
Хотя диборан D) пока не получен экспериментально, имеющиеся
достаточно строгие неэмпирические расчеты с использованием ба-
базиса типа DZ + P свидетельствуют об энергетической предпочти-
предпочтительности D5 кДж/моль) /Эг^гструктуры по сравнению с /^-фор-
/^-формой. Более того, последняя вообще не соответствует точке миниму-
минимума на ППЭ В2Н4. Для В2С14 и В2Вг4 в газовой и жидкой фазах
экспериментально установлена структура /^типа, однако молеку-
молекула B2F4 устойчива в /Эгл-форме. Причина этого состоит, очевидно,
в том, что благодаря сильным л-донорным свойствам фтора сво-
свободная МО \biu вовлекается в общую я-систему молекулы.
Сходная ситуация возникает в случае молекулы гидразина N2H4,
имеющей уже на два электрона больше, чем молекула этилена. Эти
два электрона должны занять антисвязывающую л* МО (lb2g на
рис. 9.6 и 10.6). Из схемы уровней на рис. 10.6 очевидна предпоч-
предпочтительность структуры /J<гтипа. На самом деле показано, что угол
поворота по связи N—N составляет ~90°. Дополнительным стаби-
стабилизирующим фактором служит пирамидализация связей при атоме
азота в этой форме.
Хорошая прогностическая способность анализа геометрии со-
соединений непереходных элементов, основанного на применении диа-
диаграмм Уолша, подтверждена большим объемом данных незмпири-
ческих расчетов (см. гл. 7) зависимостей энергетических уровней МО
от валентных углов для многих десятков молекул типа АХ2, АХ3
и др. Ключ к пониманию правомерности представлений Уолша
состоит в обосновании того, что зависимость суммы орбитальных
вал
энергий валентных электронов Уег от валентного угла имеет мини-
минимум в той же области, что и полная энергия D.60), а производная
д ™ дЕ
— > ?, имеет тот же знак, что —.
со. до
Доказательство этих условий в общем виде невозможно, но для
ряда конкретных молекул и ионов соблюдение этого требования
проверено прямыми расчетами. Исключение часто составляют со-
соединения с ионными связями, тогда как для молекул с равномерным
распределением электронной плотности указанные условия всегда
выполняются.
Поскольку только правильный знак т^Уе, требуется для пред-
дд .
сказания геометрии, многие приближенные методы квантовой хи-
химии и, в частности, весьма упрощенный расширенный метод Хюк-
келя (см. гл. 7) дают весьма удовлетворительные результаты.
380
10.2. ТЕОРИЯ ГИБРИДИ1АЦИИ
В качественной теории МО получаемые в результате прибли-
приближенных решений уравнения Шрёдингера молекулярные орбитали
многоатомных молекул являются в общем случае многоцентро-
многоцентровыми функциями — линейными комбинациями АО нескольких
атомных центров. Такое описание не связано прямо с понятием
химической связи в структурной теории, где связь представляет
собой локальное свойство, относящееся к двум соседним атомам.
Можно преобразовать атомные орбитали таким образом, чтобы
придать им направленность, характерную для конфигурации об-
образуемых данным атомом химических связей, и на основе этих
новых (гибридных) АО подойти к описанию и прогнозированию
геометрии молекул. Представления о гибридизации атомных ор-
биталей были введены в 30-х годах нашего столетия Л. Полингом*.
Понятие о гибридизации орбиталей тесно связано с понятием
о локализованных МО.
10.2.1. Локализованные молекулярные орбитали
Делокализованные по многим центрам молекулы МО, получен-
полученные в результате решения уравнений Рутаана D.62), называемые
каноническими МО, можно преобразовать к другому виду МО,
локализованных на определенных связях (или фрагментах) молеку-
молекулы. Такие МО называют локализованными и обозначают как ЛМО.
Если молекула содержит N электронов, размещенных на n = Nj2
дважды заполненных МО, ее полная волновая функция может быть
записана в виде определителя:
?=¦
<р,B)аB)
срп{2)р{2)
<Pi(N)a(N)
A0.4)
В. Фок еще в 1930 г. в первой статье по теории атома, где
вводилась антисимметризованная волновая функция A0.4), отме-
отметил, что не только МО <ри ... , срп можно использовать в данном
*Лайнус Карл Полинг (род. 1901 г.) — выдающийся американский химик, один
из немногих ученых, которому была дважды присуждена Нобелевская премия
A954 г. — по химии, 1962 г. — премия Мира). Л. Полингу A970) была присуждена
Ленинская премия за укрепление мира между народами. Один из создателей метода
ВС, теории гибридизации, концепции резонанса, электроотрицательности и др. Внес
огромный вклад в создание молекулярной биологии (спиральное строение полипеп-
полипептидной цепи, существование гемоглобина S а т. д.). На русский язык переведены его
книги: «Не бывать войне», «Природа химической связи», «Общая химия» и др.
381
определителе. Значение полной волновой функции ? будет оста-
оставаться неизменным, если подвергнуть канонические МО <pt линей-
линейному ортогональному (унитарному) преобразованию. Последнее
должно отвечать условию
A0.5)
где Л, — новые МО; Ту — элементы матрицы, для которой
/ A0.6)
1
Задача 10.4. Пусть ?=^
V2
q>2B)
1
=-=[^A) р2B)-<р2A) 01B)]. Про-
верить унитарность преобразования и найти новые к
/2 1\
1сГ= .
\з г)
Конечно, существует множество унитарных преобразований, но
нас интересуют такие, которые позволят осуществить идею локали-
локализации МО, сосредоточения их на химических связях или отдельных
участках молекулы. Это возможно сделать, наложив дополнитель-
дополнительные требования. Для МО естественно потребовать (Дж. Леннард-
Джонс, С. Эдмистон, К. Рюденберг), чтобы критерием максималь-
максимальной локализации было условие минимального взаимодействия (от-
(отталкивания) всех электронных пар в молекуле. Для двух электронов
на орбиталях Я,- и Д, это взаимодействие определяется как
А*B) d dil} I A?> A0.7)
а для всех пар электронов — как
Таким образом, условием максимальной локализации является
минимальность выражения A0.8). Локализованные орбитали зани-
занимают наименьший объем в пространстве; следовательно, взаимо-
взаимодействие двух электронов, находящихся на одной и той же ЛМО,
максимально.
Обменное взаимодействие между различными ЛМО должно
быть минимально. Локализованные МО двухатомных гомоядерных
382
локализованное МО
п=2 i п-3
Рис. 10.7. Схематический вид занятых электронами канонических МО и им соот-
соответствующих локализованных МО в гомоядерных двухатомных молекулах:
номер дважды занятых электронами валентных МО обозначен д. Из двух lng и \яи МО показано
только по одной, а также из каждой трех tf,, te- и i^-орбиталей показано по одной
молекул показаны на рис. 10.7, где они сопоставлены каноничес-
каноническими МО (см. разд. 4.6).
Для молекул с двумя валентными электронами вид ЛМО мало
отличается от канонического типа. Для четырехэлектронных моле-
молекул вместо сг-связывающей и сг-антисвязывающей канонических МО
возникают две эквивалентные ЛМО, форма которых соответствует
орбиталям неподеленных электронных пар каждого атома. Они
обозначаются al и al'. Вид ЛМО хорошо объясняет неустойчивость
или вообще отсутствие связи в таких молекулах, как Ве2. Связыва-
Связывающая ЛМО cr-типа возникает в шестиэлектронной молекуле (рис.
10.7 — ab). Электронная конфигурация 10-электронных молекул
в формализме ЛМО представлена двумя несвязывающими орбита-
лями неподеленных электронных пар al и of и тремя эквивалент-
эквивалентными связывающими ЛМО tb. Напомним, что при использовании
канонических МО тройная связь в молекуле азота определяется
C<7вJ A7СцL-электронной конфигурацией, т. е. включает одну а-
383
н
Нч
/V4"
н »
н
н
н
Рис. 10.8. Вид локализованных МО для некоторых структур катиона С4Н7+
и две 7С-СВЯЗИ. Для 14-электронной молекулы F2 имеются только
одна связывающая ЛМО сг-типа и шесть орбиталей неподеленных
электронных пар. Нетрудно усмотреть в этом описании прямую
аналогию с формулами, основанными на классических октетных
представлениях Дж. Льюиса:
:N = N: :F-F:
На рис. 10.8 показаны локализованные МО насыщенных много-
многоатомных структур. Ясно видна локализация МО на отдельных
связях. В отличие от молекул, не содержащих сопряженных кратных
связей, например представленных на рис. 10.8, критерий локализа-
локализации A0.7), как и другие возможные критерии, не приводит к одно-
однозначно определенному набору ЛМО. Для ненасыщенных систем
возможно несколько наборов ЛМО, отвечающих различным резо-
резонансным структурам. Например, на рис. 10.9 приведены наборы
ЛМО ароматических углеводородов, из которого видно, что лока-
локализованная я-орбиталь не центрируется полностью на двух центрах.
384
ч —
Рис. 10.9. Вид локализованных тс-МО для молекул бензола и нафталина:
непрерывные контурные линии — положительные части МО, а пунктирные — отрицательные
части МО, точками показаны узловые линии МО
Некоторая часть электронной плотности этой орбитали находится
и на двух соседних атомах. Для ряда молекул, таких, как азулен,
антрацен, нафтицен, пирен и др., определенный набор ЛМО нельзя
отнести к соответствующей резонансной структуре. В таких молеку-
молекулах реализуются трехцентровые ЛМО.
10.2.2. ЛМО и концепция гибридизации атомных орбиталей
Рассмотрим переход от канонических МО метана к его ЛМО.
Выбрав систему координат таким образом, как показано на рис.
10.10, и проведя прямой расчет по методу ЛКАО МО, получим
следующую систему уравнений для нахождения энергий и МО
валентных электронов. Обозначения Нв, Нь и т. д. введены для
Ь-функции соответствующего водородного атома:
13. Теория строения молекул
' A0.9)
Рис. 10.10. Пространственное
расположение sp3 -гибридных ор-
биталей молекулы метана
Трехкратное вырождение орбита-
лей типа t2 обусловлено тетраэдричес-
кой симметрией молекулы. Данные
фотоэлектронной спектроскопии под-
подтверждают наличие двух энергетичес-
энергетических уровней в молекуле: первый потен-
потенциал ионизации A3,2 эВ) соответству-
У ет отрыву электрона с одной из /2-ор-
биталей, а второй B2,1 эВ) — с орби-
тали *Fj.
Канонические МО полностью дело-
кализованы, как видно из соотноше-
соотношений A0.9), по всем атомам водорода
в молекуле и не центрированы на от-
отдельных связях С—Н. Используя ми-
минимальность соотношения A0.8), можно перейти к ЛМО метана.
Они представлены в табл. 10.1.
Таблица 10.1. Локализованные МО молекулы метана
Атом
С
Hi
н2
Н3
Н4
АО
Ъ
2ру
1Рг
\s
15
и
Is
Al(CHi)
0,4089
0
0,6446
0,2356
0,5758
0
0
0
Я2(СН2)
0,4089
0,5693
-0,3337
-0,2362
0
0,5758
0
0
Яз(СН3)
-0,4089
+0,5693
0,3337
0,2362
0
0
-0,5758
0
А4(СН4)
0,4089
0
0
0,7079
0
0
0
0,5758
Сравнивая ЛМО из табл. 10.1 с каноническими МО A0.9), можно
видеть, что в отличие от последних каждая из ЛМО является только
двухцентровой, т. е. четко локализована на соответствующей связи
С—Н. Этот результат прямо соответствует идее двухцентровой
химической связи. Однако в образовании такой связи между сосед-
386
ними атомами водорода и углерода участвует несколько различных
АО углерода. Удобно заменить набор нескольких АО углерода
одной орбиталью смешанного (или гибридного) типа, построенной
как линейная комбинация тех его АО, которые включаются в соот-
соответствующую ЛМО Х\
А,=4Г<РсЛ+ВЛ/, A0.10)
где
((pe)t=a, (s) +bt (px) +ct (py) +4 (pj. A0.11)
Учитывая ортогональность АО, можно определить вклад каждого
типа АО углерода в его гибридную АО (рс как
17)
Рассчитаем величины аел и хр для А„-ЛМО метана. Они равны
соответственно 0,25 и 0,75. Введя обозначение fi2 — — и проводя
операцию нормировки, перепишем
((pc)i=N (s+upt). A0.13)
Для Я4 № — л/3 и соответствующая гибридная АО
). A0.14)
Такая гибридная АО обозначается как s 4p 4 (дроби означают квад-
квадраты коэффициентов, т. е. вклады АО) или лу? -орбиталь. Графичес-
Графически 5/>3-гибридная орбиталь углеродного атома показана на рис.
10.11. К такому же виду в результате преобразований A0.13), A0.14)
приводятся и ЛМО Л\, Я2, Я3.
Если выделить гибридные орбитали атома углерода из ЛМО
этана, пропана и других насыщенных углеводородов, то в них
вклад /^-функций также будет
примерно в три раза больше
вклада s-функций, т. е. гибрид-
гибридные орбитали углеродных
атомов также отвечают зр3-типу.
Это говорит о возможности пере-
перенесения (трансферабельности) по- s Lp
лученных волновых функций гиб- J
ридных орбиталей углерода от
ОДНОЙ насыщенной молекулы Рис. 10.11. Углеродная *р3-гибридная
v гтг»игг»ы орбиталь. Пунктиром показана узло-
к другой. вая линяя
381
О+оо
10.2,3. Гибридизация и пространственная направленность
химических связей
Как видно из рис. 10.11, гибридная АО отличается от водородо-
подобных АО значительным концентрированием электронной плот-
плотности в определенном направлении пространства. Естественно по-
полагать, что именно в этих направлениях будут возникать химичес-
химические связи при перекрывании электронных облаков орбиталей, об-
образующих связь атомов. В обобщенной форме эта идея известна как
принцип максимального перекрывания: наиболее прочные химические
связи образуются в направлении наибольшего перекрывания ор-
орбиталей атомов. Таким образом, определив относительную ориен-
ориентацию гибридных АО атома, можно установить направление об-
образуемых ими связей. Проследим за тем, как представления о гиб-
гибридизации АО позволяют описать геометрическую форму молекул.
Полезно при этом также рассмотреть, как можно подойти к концеп-
концепции гибридизации с чисто качественных позиций (Л. Полинг,
Дж. Слэтер).
Вернемся опять к молекуле метана СН4. Известно, что эта
молекула имеет тетраэдрическую структуру, все четыре связи С—Н
в ней равноценны, а атом углерода проявляет валентность, равную
четырем. Для этого необходимо перейти от электронной конфигу-
конфигурации основного состояния
:(lsJBsJBPx)lBpy)lBp2H
с двумя неспаренными электронами к возбужденной
Такое возбуждение допускается правилами отбора (см. разд. 3.9)
и требует затраты энергии ~402 кДж/моль, которая компенсирует-
компенсируется затем при образовании двух дополнительных связей С—Н (энер-
(энергия каждой из которых составляет 435 кДж/моль). Однако четыре
связи, которые теперь способен образовать углеродный атом, долж-
должны быть неравноценны, так как три из них осуществляются АО
/кгипа, а одна — 5-типа.
Для того чтобы все связи С—Н были эквивалентны, необходима
эквивалентность атомных орбиталей углерода. Этого можно до-
достигнуть, представив валентные АО углерода не как чистые s-
и /^-функции, а как их линейные комбинации:
tex - ах s+Ьхрх+схру+dxpz,
x + сфу+a\pt.
Гибридные АО должны быть нормированы и ортогональны, т. е.
388
$(aiS+btpx+Cipy+dipz) (ajs+bjpx + cjpy+djpjdz^dij, A0.16)
что, учитывая ортонормированность АО, ведет к следующей систе-
системе уравнений для коэффициентов при АО:
dj^O (/V/), ПО
/,;=!, 2, 3,4.
Система A0.17) содержит четыре нормировочных уравнения
и шесть условий ортогональности, т. е. всего 10 уравнений при 16
неизвестных. Поэтому шесть коэффициентов можно установить
произвольно. Выбор трех из них ведет просто к разным
ориентациям всей системы гибридных функций в пространстве.
Поэтому фактически вполне произвольно можно задать лишь три
параметра.
Оказывается при этом, что существует лишь три способа зада-
задания произвольных параметров, которые приводят к получению
эквивалентных (т. е. содержащих одинаковые вклады s- и />-функ-
ций) гибридных АО.
Первый способ. ax—bi — Ci—d^. Решая уравнения A0.17) при этих
условиях, получим
A0.18)
Орбитали A0.18) относятся к s 4p 4- или 5р3-типу. Их называют
также тетрагональными (te) орбиталями, так как они направлены
(см. рис. 10.10) к вершинам правильного тетраэдра. Гибридные АО
в силу их четко выраженной направленности можно отождествлять
с векторами, ориентация которых в декартовой системе координат
определена значениями коэффициентов при АО рх, ру, pv Углы
между направлениями гибридных АО вычисляют как углы между
векторами. Простой расчет показывает, что угол между осями
любых двух 5/7 -гибридных АО составляет 109°28', как в правильном
тетраэдре:
pos0(/eif te2)=l l~ll~rl l= ~, 0=1О9°28\
389
Задача 10.5. Гибридные функции A0.18) отличаются от соответствующей
гибридной АО, полученной методом МО A0.14). Это связано с различной
ориентацией гибридных орбиталей относительно осей координат. Гибридная
орбиталь A0.14) была направлена вдоль одной из осей (ось г). Показать, что если
на рис, ЮЛ 0 ось г совместить с направлением одной из гибридных орбиталей, то
орбиталь te\ будет иметь вид (ЮЛ 4).
Второй способ. ах =bY = cx\dx =0. Такой способ задания произволь-
произвольно варьируемых параметров приводит к трем эквивалентным гиб-
гибридным АО s р 3- или лр2-типа, называемым также тригональными
(tr) орбиталями:
0
A0.19)
1 yf% -у/б
—2~Рх—
Гибридные 5/?2-орбитали лежат в одной плоскости и направлены
под углом 120° друг к другу. Форма sp2-KO похожа на форму
^3-орбиталей, но sp2-АО более сжаты и вытянуты (рис. 10.12).
Образуемые ими связи должны быть, согласно принципу макси-
максимального перекрывания, более прочными.
Задача 10.6. Рассчитать угол между осями 5^2-гибридных АО и преобразо-
преобразовать орбитали A0.19) так, чтобы одна из них была направлена по оси у.
Третий способ. ax=bu Ci=d\ = Q. Эти условия ведут к двум эк-
Бивалентным гибридным АО s р - или 5/ьтипа, называемым также
диагональными (di) орбиталями, так как они направлены диамет-
диаметрально противоположно друг другу (см. рис. 10.12):
dU^—=
di
—7=
A0-20)
Хъ—Ру,
390
\
Рис. 10.12. sp~Гибридные орбитали
Таким образом, мы получили
три типа эквивалентных гибридных
орбиталей, образуемых АО s- и р-
типа. Следует ясно осознавать, что
гибридизация орбиталей не являет-
является каким-либо реальным физичес-
физическим эффектом, действительно
включающим возбуждение электро-
электронов с 5->/?-переходами. Это лишь
удобный математический прием,
позволяющий без прямых вычисле-
вычислений достаточно надежно воспроиз-
воспроизвести форму ЛМО сложной молеку-
молекулы в тех случаях, когда эта ЛМО
хорошо локализована на связи. На-
Направленность гибридных АО дает
ключ к пониманию геометрической
формы молекул, а также ряда их
важных стереохимических особен-
особенностей. Рассмотрим несколько хара-
характерных примеров.
На рис. 10.13 показана схема пе-
перекрывания орбиталей, ведущего
к образованию связей в молекулах
насыщенных углеводородов. Пере-
Перекрывание 5/73-гибридных АО по свя-
связям С—С ведет к фор-
формированию ЛМО сг-типа, т. е. сим-
симметричных относительно связевой оси. Таким образом, вращения
вокруг этой оси ограничены лишь за счет дальних взаимодействий
электронов на орбиталях связей С—Н. Действительно, энергетичес-
энергетические барьеры вращения по ординарным связям малы. В этане, где
барьер определен наиболее точно A1,9 кДж/моль) и соответствует
разности энергий стабильной антиперипланарной I и наименее
устойчивой синперипланарной II конформации,
Рис. 10.13. Образование связей
л/?3-гибридными АО углерода
в молекуле пропана
>н
н
I II
В этилене каждый углеродный атом имеет всего трех соседей
и для образования G-связей с ними использует три эквивалентные
¦«р2-гибридные АО типа A0.19). Негибридизованные pz-AO двух
391
н
6-связи
Рис. 10.14. Молекулярные орбитали молекулы этилена
атомов приводят к возникновению я-МО. Образование кратной
связи иллюстрирует рис. 10.14. Важная отличительная особенность
соединений с я-связями — жесткость их молекулярной геометрии.
я-Связи не имеют осевой симметрии. Интеграл перекрывания
при нарушении параллельности осей pz~AO (рис. 10.15) умень-
уменьшается:
. A0.21)
При 0 — 90° Se—О, т. е. тг-связь полностью нарушается. Это
соответствует потере 125—292 кДж/моль. Например, энергия связи
С—С в этилене 681 кДж/моль, а связи С—С в этане —
368 кДж/моль. Если пренебречь сравнительно небольшим различи-
различием в энергиях а-связей С—С этана и этилена, вызванным различием
в гибридизации АО, разность указанных величин характеризует
энергию я-связи.
Таким образом, барьеры вращения по кратным связям должны
быть велики и становится понятной возможность геометрической
(цис-транс) изомерии, например:
С1
с
н
си
с
н
си
с
н
ci-
III,
II ^z
Z-изомер
(
а н
IV
"^-
*
v,
11
с
?-изомер
Рис. 10.15. Уменьшение перекрывания меж-
между р-АО при повороте на угол в
392
В молекулах с тройными
и кумулированными двойны-
двойными связями, где углеродный
атом имеет двух соседей, он
образует ег-связи с помощью
лр-гибридных АО. Кроме то-
того, возникают две я-связи
с электронными облаками,
ориентированными во взаим-
взаимно перпендикулярных плоско-
плоскостях (рис. 10.16). Особенно ин-
интересная ситуация возникает
в кумуленах. Простой анализ
структуры связывающих ЛМО,
построенных из гибридных АО,
приводит к выводу о возмож-
возможности оптической изомерии
производных аллена. Как видно
из структур VI, VII, существует
два варианта образования п-
МО в аллене:
Рис. 10.16. а- и тс-молекулярные орбита*
ли молекулы ацетилена
Геометрически они различаются относительным расположением
плоскостей концевых СН2-групп. В первом случае одна из централь-
центральных р-АО углерода остается выключений из сопряжения с ос-
остальными. В структуре VII образуются две ортогональные я-систе-
мы. Общие соображения и конкретные расчеты указывают на выго-
выгодность структуры VII.
При различных заместителях у 5р2-углеродных атомов замещен-
замещенные аллены, если их структура отвечает формуле VII, становятся
зеркальными и могут быть разделены на оптические антиподы:
i
i
/ VIII. A R'IR'
зеркальная плоскость
IX, А Ч
Впервые такие оптически активные аллены были получены в 30-х
годах нашего столетия (К., = СбН5, K2 — i-Ci^i7). Их существование
ясно подтверждает структуру УП. Переход от одного оптически
активного изомера к другому требует перехода через плоскую
структуру. Для аллена, как показывает эксперимент, это требует
затраты 305 кДж/моль.
Задача 10.7. Какой общий вывод о зависимости геометрической структуры
молекул кумуленов СН2(=вС = )„СН2 от числа двойных связей можно сделать на
основании рассмотрения их электронной структуры с позиции гибридизации АО?
393
10.2.4. Неэквивалентные гибридные атомные орбитали
До сих пор мы рассматривали только эквивалентные гибридные
АО, т. е. такие, в которых вклады s- и /^-функций одинаковы.
Использование таких орбиталей при образовании связей приводит
к валентным углам в 109°28', 120° и 180°. Эти значения наиболее
характерны для валентных углов в самых разных соединениях, но
нередко случаются и достаточно заметные отклонения.
Так, в молекуле СН3Вг(Х) три угла Н—С—Н равны по 111,2°,
а каждый угол Н—С—Вг составляет 106°. Следовательно, три связи
СН атом углерода образует гибридными АО одного типа, а четвер-
четвертую (С—Вг) — другого. Следовательно, в молекуле СН3Вг атом
углерода использует неэквивалентные гибридные АО. Согласно
принципу максимального перекрывания, вид этих орбиталей полно-
полностью определяется углами между связями, которые являются экс-
экспериментальным критерием типа гибридных АО, формирующих
связь. Рассмотрим это определение в более общем виде. Как было
показано, любую гибридную АО можно представить в виде
Vt^Nrfs+mpO, A0.22)
где fif характеризует степень смешения s- и р-АО. Направление
максимума электронной плотности орбитали <р,- совпадает с направ-
направлением ориентации р-АО. Пусть имеются две неэквивалентные гиб-
гибридные АО типа A0.13), центрированные на одном атоме и об-
образующие связи с двумя другими, угол между которыми ви:
Вг
Лс
X XI
Гибридные орбитали должны быть ортогональны:
fs+faр^ (s-fpjPj) dx =
,* = 0. N A0.23)
Так как АО s, p% и pj ортонормированы, то первый интеграл равен
единице, а два последующих — нулю. Последний интеграл, соглас-
согласно A0.21), равен cos0y. Следовательно,
' I ' A0.24)
394
Соотношения A0.24) связывают геометрические характеристики мо-
молекулы с типом гибридизации АО, участвующих в образовании ее
связей. Для эквивалентных гибридных АО /i,=yu> и условие A0.24)
сводится к
0
г A0.25)
Нетрудно проверить, что для ц = у/з, л/l и 1 (sp3-, sp2- и sp~
гибридные орбитали) соответствующие углы в равны 109°28', 120°
и 180°. Если гибридные АО представлены в общем виде
(Pi
+ Cjpy + djpx,
то можно показать, что угол 6у определяется условием
a,aj
cos 0,= , = , A0.26)
Задача 10.8. Доказать соотношение A0.26).
Как видно из соотношений A0.24) и A0.26), при углах 01:,<9О0
гибридные орбитали не могут быть представлены в виде дейст-
действительных функций. Возможно, однако, обобщить указанные соот-
соотношения и использовать представление о гибридизации АО для
углов ву<90°, если перейти к комплексной форме представления
гибридных АО. При таком способе рассмотрения линия связи совпа-
совпадает, как и в разобранных выше примерах, с осью гибридной АО.
Другой подход допускает использование гибридных АО в дейст-
действительной форме даже для описания связей, образующих между
395
собой углы меньше чем 90°. В этом случае необходимо допустить,
что область наибольшего перекрывания гибридных орбиталей ле-
лежит вне линии связей. Хорошо изученным примером служит моле-
молекула циклопропана, в которой углы ССС равны 60°. Схема об-
образования связей в этой молекуле показана на структуре XII:
Интересно отметить, что данная схема согласуется с эксперимен-
экспериментальной картиной распределения электронной плотности в молеку-
молекуле циклопропана, полученной в результате рентгеноструктурного
исследования: максимумы электронной плотности в этой молекуле
не находятся на линии связей С—-С
Особый случай — выявление вида гибридных АО, занимаемых
несвязывающей парой электронов. Он иллюстрируется следующей
задачей.
Задача 10.9. Валентный угол НОН в молекуле воды равен 104,5°. Рас-
Рассчитать вид гибридных АО кислорода, образующих связи О—Н и занятых
неподеленными электронными парами.
Необходимо отметить, что фиксируемая соотношениями
A0.24)—A0.26) прямая связь между геометрическим строением
и типом гибридизации АО сохраняет полную силу лишь в том
случае, когда тип гибридизации полностью определяется условием
максимального перекрывания. Для высокосимметричных молекул,
таких, как АН4 и АН3, это требование выполняется. Для менее
симметричных молекул, в которых связи образуются неэквивалент-
неэквивалентными гибридными АО, выигрыш в энергии связи за счет мак-
максимального перекрывания гибридных орбиталей часто недостато-
недостаточен, чтобы компенсировать высокие затраты на промотирование
электронов с низко лежащих 5-орбиталей. Как показывает анализ
данных большого числа достаточно строгих неэмпирических рас-
расчетов, проведенный в последние годы, для большинства молекул,
особенно включающих атомы третьего и низших периодов Пери-
Периодической системы, реализуются (^J-электронные конфигурации
и связывание обусловливается главным образом /г-орбиталями. Во-
Вопреки распространенному мнению представления о гибридизации
вообще не являются необходимыми для правильного предсказания
тетраэдрической геометрии молекулы метана: исключение из базиса
25-АО углерода не изменяет вывода об устойчивости правильной
тетраэдрической конфигурации СН4.
Таким образом, представления о гибридизации служат скорее
средством условного описания, а не теоретического прогнозирова-
прогнозирования геометрической структуры. Тем не менее тесная связь представ-
представлений о гибридизации АО с фундаментальным понятием ЛМО
и универсальность принципа максимального перекрывания являют-
являются обоснованием применения этих представлений.
396
10.3. ТЕОРИЯ ОТТАЛКИВАНИЯ ЭЛЕКТРОННЫХ ПАР
ВАЛЕНТНЫХ ОРБИТАЛЕЙ
Представления о локализованных молекулярных орбиталях
(ЛМО) лежат в основе другого важного теоретического подхода
к описанию геометрической формы молекул — теории отталкива-
отталкивания электронных пар валентных орбиталей (ОЭПВО). Идея этого
подхода была высказана впервые английскими учеными Сиджвиком
и Пауэлом A940) на основании анализа накопленных к тому време-
времени экспериментальных данных о геометрической структуре молекул
(около 350 молекул и ионов). В последнее время значительный
вклад в развитие теории ОЭПВО внесен канадским химиком
Р, Гиллеспи.
10.3.1. Основные положения
Основная идея теории ОЭПВО состоит в следующих поло-
положениях.
1. Конфигурация связей многовалентного атома (или иона) обус-
обусловливается исключительно числом связывающих и несвязывающих
электронных пар в валентной оболочке центрального атома.
2. Ориентация облаков электронных пар валентных орбиталей
определяется максимальным взаимным отталкиванием заполняю-
заполняющих их электронов.
Если бы природа сил взаимного отталкивания электронных пар
имела чисто электростатический характер, эти силы определялись
бы соотношением (г "), где л = 2. Однако кроме электростатичес-
электростатического взаимодействия электронные пары на разных ЛМО испытыва-
испытывают отталкивание еще в силу действия принципа Паули (см. гл. 3),
поэтому в выражении для сил отталкивания
F~-n> A0.27)
г
где г — расстояние между «центрами тяжести» облаков элект-
электронных пар ЛМО, л-кзо.
Задача поиска расположения центров облаков электронных пар,
расталкивающихся в соответствии с A0.27) при равных для всех пар
величинах г, эквивалентна задаче размещения нескольких частиц на
поверхности сферы при их максимальном удалении друг от друга.
Эта задача решается строго для числа частиц от 2 до 12 и дает
следующий результат (табл. 10.2).
В табл. 10.2 в число q входят электронные пары как на связыва-
связывающих ЛМО, так и на несвязывающих, т. е. неподеденные элект-
электронные пары. Гиллеспи ввел для связывающих электронных пар
обозначение X, а для несвязывающих — Е. С учетом этих обозначе-
обозначений можно следующим образом представить геометрическую кон-
397
Таблица 10.2. Конфигурация связей центрального атома А в зависимости от
•тела электронных пар q на его валентных орбиталях
ч
Конфигурация
2 Линейная
3 Равносторонний треугольник
4 Тетраэдр
5 Тригональная бшшрамида
6 Октаэдр
7 Октаэдр с дополнительной верши-
вершиной
Я
Конфигурация
% Квадратная автилризма
9 Треугольная призма с тремя до-
дополнительными вершинами
10 Квадратная антипризма с двумя
дополнительными вершинами
11 Икосаэдр без одной вершины
12 Икосаэдр
фигурацию молекул типа АХ^Е,,. Как видно из данных табл. 10.3,
в рамках теории ОЭПВО для определения топологии связей цент-
центрального атома в молекулах, образованных непереходными элемен-
элементами, необходимо только сосчитать число электронных пар на
связывающих и несвязывающих орбиталях и разместить их на осях
соответствующего многогранника. При этом полезно дополнить
исходные положения теории ОЭПВО правилами, изложенными
в следующем разделе.
Таблица 10.3. Геометрия структуры молекул mua AXmEn без кратных связен
Общее
число
элект-
электронных
пар
Тип АХ„ЕЙ
Конфигурация
Примеры
2
3
АХ2
АХ3
АХ2Е
АХ3Е
АХ2Е2
АХ5
ВеН2, HgCl2
BF3,A1(CH3)
SnCl2
СН4, ВН4"
NH3, H3O+
Н2О, SC12
Pd5
398
Продолжение табл. 103
Общее
число
элект-
электронных
пар
Тип АХ„ЕП
Конфигурация
Примеры
АХ4Е
АХ3Е2
АХ2Е3
АХ4Е2
АХ7
АХ6Е
АХ8
SC14
CIF3
XeF2
SF6
1F3
XeF4
IF7
XeF6
Ba(H2O>;,
10.3.2. Развитие теории ОЭПВО
1. Неподеленная электронная пара занимает больший объем, чем
пара электронов на орбитали, участвующей в образовании ординар-
ординарной связи. Сила отталкивания электронных пар в данной валентной
оболочке понижается в следующем порядке: неподеленная пара —
неподеленная пара (Е — Е) > неподеленная пара — связывающая
пара (Е—X) > связывающая пара — связывающая пара (X—X).
Это допущение вытекает из таких простых аргументов, как то,
что неподеленная электронная пара находится в поле только одного
положительного атомного остова и, следовательно, более диффуз-
на, чем связывающая электронная пара, испытывающая сжатие
полем зарядов двух атомных остовов. Отсюда следует вывод, что
в серии изоэлектронных молекул (т. е. содержащих равное число
399
электронных пар в валентной оболочке) неподеленная пара, заменяя
связывающую электронную пару, стремится занять больше про-
пространства вокруг атома. Это уменьшает валентные углы между
связями, например, в ряду
Н н
|Ю9°28' Тю7°18' |104°30'
/ н ИУ \н ' \н
XIII XIV XV
Аналогичное объяснение имеют отклонения от полностью сим-
симметричной ориентации связей в молекуле типа АХ5Е, AX4E, АХ3Е2.
Поскольку отталкивания электронных пар типа Е — X доминируют
по сравнению с отталкиваниями X—X, валентные углы ХАХ неско-
несколько сжаты:
F 84°30'
XVI XVII XVIII
В прямой связи с данными об эффективных объемах связыва-
связывающих и неподеленных электронных пар находится такое важное
следствие, как положение о том, что в молекулах типа АХ4Е, АХ3Е2,
АХ2Е3, геометрическая форма которых производится от структуры
тригональной бипирамиды, электронные пары всегда занимают
экваториальные положения (см. табл. 10.3):
ах
ах X X
XX XXI
Это объясняется тем, что в структуре XX имеются только два
невыгодных Е—Х-взаимодействия электронных пар, когда их оси
составляют угол 90°. В структуре XXI (топомерной XX) таких
невыгодных взаимодействий три.
2. Объем электронной пары, участвующей в образовании связи,
уменьшается с увеличением электроотрицательности лиганда.
Более электроотрицательный лиганд сильнее притягивает общее
400
электронное облако связывающей пары, что можно представить как
дополнительное сжатие этого облака. Данная электронная пара
будет более удалена от центрального атома и испытывает меньшее
отталкивание со стороны других соседних электронных пар. Все это
поведет к тому, что валентные углы, составляемые связями центра-
центрального атома с наиболее электроотрицательными лигандами, долж-
должны иметь наименьшие значения.
Эффект влияния изменения электроотрицательности лигандов
на валентные углы связей иллюстрируется сравнением молекул NH3
и NF3. Большая электроотрицательность фтора уменьшает размеры
пары на связи N—F, в результате углы FNF составляют всего 102°,
что на 5° меньше, чем углы HNH в аммиаке. Такая же тенденция
наблюдается в ряду Р13 A02°), РВг3 A01,5°), РС13 A00,3°), PF3(97,8°).
Интересный пример — молекула (CH3JPF3. Валентные углы
в этой молекуле уменьшаются в порядке
СРС > CPF > FPF, соответствующем порядку
возрастания электроотрицательности лигандов.
Следует отметить, что, если рассматривать непо-
деленную электронную пару как некий вообража-
воображаемый лиганд (фантом-лиганд) с предельно малой
электроотрицательностью, правила 1 и 2 легко
обобщаются.
3. Две электронные пары двойной связи или три электронные
пары тройной связи занимают больший объем, чем электронная пара
ординарной связи.
Это правило является основным при рассмотрении геометричес-
геометрической структуры молекул, содержащих кратные связи. Как и для
остальных молекул, определение конфигурации связей центрально-
центрального атома основывается на выделении электронных пар <7-связей
и неподеленных электронов. Электроны тг-связей на этой стадии не
учитываются. Так, например, чтобы определить форму молекулы
SO2 (XXIII), необходимо учесть, что из шести электронов в валент-
валентной оболочке серы два расходуются на образование двух тг-связей.
Из оставшихся четырех два образуют с неподеленными
электронами кислородных атомов а-связи, а два остают- т
ся в виде неподеленной пары. Таким образом, необходи- ^S4^
мо учесть относительную ориентацию облаков трех элек- ° °
тронных пар, что в согласии с данными табл. 10.4 ведет ХХШ
к угловой конфигурации.
В табл. 10.5 представлены данные о геометрической структуре
широкого ряда молекул непереходных элементов с кратными связя-
связями. Так как кратная связь содержит более чем одну электронную
пару, ее электронное облако занимает большее пространство, чем
электронная пара ординарной связи. Размер электронного облака
401
двойной связи по сравнению с размером орбитали неподеленной
электронной пары недостаточно определен. Обычно их размеры
принимают равными. Больший размер кратной связи виден из
примеров молекул типа Х2СО и Х2С=СН2, пирамидальных моле-
молекул типа X2SO, тетраэдрическнх молекул типа РОХ3. Из. табл. 10.5
видно, что угол ХСХ всегда меньше 120°, угол XSO больше угла
XSX.
Таблица 10.4. Геометрическая структура молекул непереходных элементов, со-
содержащих кратные связи
Общее число
электронных
пар <гч>рбита-
лей и несвя-
несвязываю щвх
орбиталей
2
3
4
5
6
AIY)
[исло
сг-связеЙ
2
3
2
4
3
2
5
4
6
Число не-
поделен-
ных пар
0
0
1
0
1
2
0
1
0
Конфигура-
Конфигурация связей
Линейная
Треуголь-
Треугольная
Угловая
Тетраэдри-
ческая
Пирами-
Пирамидальная
Угловая
Тригональ-
аая бипира-
мида
Бисфеноид-
вая
Октаэдри-
ческая
Примеры
О=С=О, H-C=N
о о
'6* N
// Ч - г/ V
о о о а
о он
л
F
F ОН
|>V)H
ОИ
ОН
ОН
Важно подчеркнуть, что выводы теории ОЭПВО о геометричес-
геометрическом строении молекул легко экстраполируются на более сложные
молекулы и ионы, чем рассмотренные в табл. 10.3—10.5. В каждом
случае необходимо выделить фрагмент, содержащий центральный
атом, координирующий около себя другие атомы или их группиров-
группировки, и установить число и тип окружающих данный атом элект-
электронных пар. Таким образом, нетрудно определить, например, стру-
структуру молекулы Р4. Каждый атом фосфора в ней имеет три соседа и,
кроме того, сохраняет одну неподеленную электронную пару. Сле-
Следовательно, должна реализоваться тетраэдрическая конфигурация
осей электронных пар, отвечающая молекулярной структуре XXIV:
p—1—p
.Se. Se
\)-7/
о о
Se
/
о
XXV
XXIV
XXVI
Таблица 10.5. Валентные утлы в некоторых молекулах
Молекула
F2CO
CH3COF
С12СО
н2со
(МНзЪСО
(NH2bCS
Угол,
хсх
108,0
ПО
111,3
115,8
118
116
град
схо
126
128; 122
124,3
122,1
121
122
Молекула
Н2С = СН2
H2C = CHF
H2C=CF2
Н2С = СС12
F2C = CH2
F2C = CFCI
Угол,
ХСХ
116,8
115,4
109,3
114
ПО
114
град
хсс
122
123,3; 120,9
125,3
123
125
123
Молекула
F2SO
Br2SO
(CH3JSO
xsx
92,8
96
100
97,3
XSO
106,8
108
107
106,2
Молекула
POF3
poa3
POBr3
PSF3
XPX
101,3
103,3
108
100,3
XPO
113
112
110
113,8
Таким же образом приходим к пирамидальной конфигурации
связей атома селена XXV в полимерном диоксиде селена /С1Л ч
403
угловой структуре полимерной цепи кристалла HF (XXVI). Поло-
Положения теории ОЭПВО полезны при определении строения не только
стабильных молекул и ионов, но также для описания относительной
ориентации групп в метастабильных комплексах и даже переходных
состояниях реакций присоединения, замещения. Так, например,
в полном согласии с данными строгих расчетов предсказывается
тригонально-бипирамидальное строение переходного состояния
в реакции бимолекулярного замещения на хр-углеродном центре:
н
F-C».... + F"
н
F—С—F
F"+ H,,.C-F
H И Н II
V Н
XXVII XXVIII XXIX
Действительно, пять электронных пар, окружающих централь-
центральный атом углерода в переходном состоянии реакции замещения,
обусловливает его структуру XXVIII.
Задача 10.10. Определить геометрическое строение молекул HNO2, HNO3,
I2O5) SOF4, SF3N, ХеОэ, SbBr^, TeO2) A1H3 2 (CH3KN, BH3 • СО.
Задача 10.11. Объясните с помощью основных положений теории ОЭПВО
правило полярности, согласно которому в структурах молекул АХз, АХфЕ,
АХ3Ё2, произведенных от тригональной бипирамиды, более электроотрицатель-
электроотрицательные лиганды всегда занимают аксиальные положения и аксиальные связи удли-
удлинены по сравнению с экваториальными. v S \ s
2- Tic Re
Задача 10.12. Кластер [R^Cls] имеет структуру типа у ч у \
Учитывая, что, согласно Коттону и Полингу, связь Re—Re является четверной
связью, объясните заслоненную конформацвю аннона.
10.3.3. Недостатки теории ОЭПВО
и отклонения от ее предсказаний
Как и всякая приближенная теория, основанная на той или иной
модели, теория ОЭПВО сталкивается с рядом трудностей, предоп-
предопределенных недостатками модели, лежащей в ее основе. Укажем на
некоторые из них.
1. Как было отмечено в предыдущих разделах, теория приложи-
ма к описанию строения молекул только непереходных элементов,
т. е. элементов, не имеющих не полностью заполненные внутренние
электронные оболочки. Дело в том, что наличие таких оболочек,
например fif-электронов в атомах переходных элементов, приводит
к отклонениям от сферической симметрии распределения электро-
электронов остова. Это, в свою очередь, ведет к тому, что распределение
404
облаков электронных пар в пространстве относительно центрально-
центрального атома не подчиняется точно соотношению A0.27). Эти отклоне-
ния особенно заметны при значительном количестве F—9) электрс
нов в ^-оболочках переходных элементов.
2. Участие ^-орбиталей в связях, образуемых элементами низ-
низших периодов, также приводит к отклонениям от ожидаемой на
основании представлений теории ОЭПВО геометрии. Хорошо из-
известным примером являются угловые искажения молекул галогени-
дов щелочноземельных металлов. Эти отклонения иллюстрируются
табл. 10.6.
Таблица 10.6. Ковфигурацм связей в молекулах галогеяндов
щелочноземельных металлов МХ2 (л — линейная, у — угловая
конфигурации)
Be
Mg
Са
Sr
Ва
F
л
У
У
У
У
а
л
л
л
У
У
Вг
л
л
л
л
У
I
л
л
л
л
У
Причины этих отклонений вызваны изменениями в типе ор-
биталей центрального атома, образующих связи с галогенами, пере-
переходом от sp-типа к sd-тшу по мере возрастания порядкового номе-
номера элемента и электроотрицательности лиганда. Теория ОЭПВО
в отличие от представлений ЛМО и теории гибридизации АО не
учитывает прямо тип орбиталей электронных пар, что и не позволя-
позволяет учесть отдельные тонкие различия.
3. В соединениях типа АХ^Е и других с высоким координацион-
координационным числом центрального атома неподеленная электронная пара
является стереохимически инертной и структура соответствует кон-
конфигурации, получаемой без учета электронной пары Е. Так, анионы
SbClf", TeClJ" имеют октаэдрическое строение, хотя они, как и гек-
сафторид ксенона XeF6, содержат в валентной оболочке по семь
электронных пар. Однако XeF6 имеет в согласии с теорией ОЭПВО
структуру неправильного октаэдра (табл. 10.3), тогда как в указан-
указанных анионах все связи равноценны. Другой пример — Cs2 pCeF8] .
Анион этой соли, в котором центральный атом окружен девятью
электронными парами, вопреки ожиданиям теории (см. табл. 10.2)
имеет строение квадратной антипризмы. Причина отмеченных от-
отклонений состоит в том, что одна из валентных электронных пар,
405
а именно ns2, сильно локализована и по своим свойствам резко
отличается от характеристик остальных электронных пар (см. разд.
10.2.4).
4. Большие расхождения с предсказаниями теории ОЭПВО на-
наблюдаются для соединений с высокополярными связями, близкими
к ионному типу. Так, молекула Li2O, относящаяся к типу АХ2Е2,
имеет не угловую, а линейную форму. Последнее понятно из элект-
электростатических соображений, если представить Li2O в форме ионной
структуры Li+02~Li+.
5. В теории ОЭПВО характеристики заместителей X фактически
не принимаются во внимание. Кроме неправильных предсказаний
для ионных соединений, это ведет к неточному предсказанию и для
соединений, в которых X представляет собой л-сопряженную систе-
систему. Так, анионы АХ3Е типа С (CN)i~> С (NO2K~ имеют не ожидаемую
пирамидальную, а плоскую форму вследствие того, что последняя
обеспечивает лучшие условия для включения неподеленной элект-
электронной пары в общую я-систему.
Несмотря на отмеченные недостатки, представления теории
ОЭПВО исключительно полезны и при правильном применении
достаточно надежны для объяснения и предсказания структурных
характеристик молекул и ионов, образованных непереходными эле-
элементами в самых различных валентных состояниях. Теория ОЭПВО
может служить примером простой и эффективной теоретической
концепции, позволяющей предвидеть главные детали молекулярной
структуры без проведения трудоемких расчетов.
Литература
Гнллесии Р. Геометрия молекул. — М.: Мир, 1975.
Монография, посвященная воем аспектам теории ОЭПВО. Содержит обширный системати-
систематизированный экспериментальный материал о структуре молекул.
Коулсон Ч. Валентность. — М.: Мир, 1965. Гл. 8.
Последовательное изложение представлений о гибридизации АО а соотношений между
свойствами связей и типом образующих ее гибридных АО.
Gimarc В. М. Molecular Structure and Bonding. The Qualitative Molecular Orbital
Approach. New York: Academic Press. 1979.
В простой нематематизированной форме дано подробное рассмотрение приложений качест-
качественной теории МО х описанию геометрической структуры молекул. Рассматриваются корреляци-
корреляционные диаграммы Уолша для всех основных типов молекул, проводится значительный экс-
экспериментальный материал, который сопоставляется с предсказаниями качественной теории
и данными количественных расчетов.
Vowles R. S., Stevenson P., Hotter J. — J. Chem. Education, 1987, 24, N 1, 24.
Electron pair repulsion theory — a microcomputer program.
Программа на языке Бейсик, алгоритм которой реализует положения теории ОЭПВО.
Позволяет при задании последовательности атомов в многоатомной молекуле и общего числа
валентных электронов получить набор всех возможных структур с выводом их и другой
информации на дисплей.
406
ГЛАВА 11
СТРОЕНИЕ КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ
11.1. КООРДИНАЦИОННАЯ СВЯЗЬ
К координационным, или комплексным, относят такие соедине-
соединения, в молекулах которых можно выделить центральный атом
(комплексообразователь), связанный с несколькими соседними
группами атомов (лигандами), причем число лигандов превышает
количество связей, которые способен образовать центральный атом
в соответствующем валентном состоянии за счет своих неспаренных
электронов.
Таким образом, образование химической связи в координацион-
координационных соединениях отличается от рассмотренных ранее схем возник-
возникновения ковалентных связей, однако это отличие относится не
столько к природе связи, сколько к способам ее описания. Так,
согласно данному выше определению, следует отнести к координа-
координационной связь между атомами бора и азота в боразане A) ВН3' NH3:
н н
Хотя в результате перекрывания валентных АО бора и азота
образуется обычная cr-связь В—N, оба электрона на отвечающую
этой связи локализованную сг-МО поставляет азот. Следовательно,
бор в боразане имеет более высокое координационное число (КЧ),
т. е. число непосредственно координированных около него атомов
больше, чем его максимальная валентность, равная наибольшему
возможному числу неспаренных электронов в валентных АО, т. е.
трем. Такую связь называют обычно донорно-акцепторной, и она
представляет лишь один возможный тип координационной связи.
Весьма важным примером донорно-акцепторной связи служат во-
водородные связи, при образовании которых водородный атом прояв-
проявляет КЧ большее, чем его валентность, например в симметричном
анионе II или диборане III:
?—и—f ^в\
п
ж ш
н н
401
Соединения IV—IX иллюстрируют некоторые другие многооб-
многообразные примеры структур с координационными связями:
Во всех этих случаях формальные соображения о валентности цент-
центрального атома недостаточны, чтобы объяснить реально возника-
возникающую в координационных соединениях IV—IX структуру связей.
Наибольшее количество координационных соединений образовано
переходными элементами, т. е. элементами с не полностью запол-
заполненными J-оболочками, и лантаноидами, содержащими незапол-
незаполненную /-оболочку. Однако и непереходные элементы способны
к образованию координационных связей.
Наиболее точные неэмпирические расчеты, проведенные для мо-
молекулы SF6, оксианионов типа QO4", указывают на крайне малую
заселенность rf-орбиталей центрального атома в этих соединениях.
408
Следовательно, связи в них также принадлежат к типу координаци-
координационных связей.
Подытожим особенности координационной связи, отличающие
ее от обычной двухцентровой ковалентной связи, в которой каждый
атомный центр участвует в формировании ЛМО связи одной своей
АО, содержащей один неспаренный электрон.
1. Координационная связь имеет донорно-акцепторную приро-
природу, т. е. возникает при перекрывании полностью заселенных элект-
электронами АО лиганда и вакантных АО центрального атома. Локали-
Локализованные координационные связи такого типа реализованы в комп-
комплексах V, VI. К ним относятся также экзоциклические связи В—Н
в III, Со—С в соединении VII и связи Pt—СН3 в я-комплексе VIII.
2. Координационные связи могут быть многоцентровыми, т. е.
электронная пара связывающей МО может быть делокализована
в области, охватывающей не два, а несколько атомных центров.
Таковы трехцентровые связи В—Н—В и Со—С—Со в комплексах
III, VII, многоцентровые связи центрального атома с углеродными
атомами циклопентадиенильного и бензольного колец в я-комплек-
сах VIII—X.
Указанные особенности координационной связи приводят к ко-
колоссальному многообразию структурных типов молекул координа-
координационных соединений, а также кристаллических структур твердых
тел. Природа сил, обусловливающих координационную связь, луч-
лучше и правильнее всего описывается с помощью теории МО. Ввиду
сложности структуры молекул и ионов координационных соедине-
соединений прямые расчеты не всегда возможны или требуют при их
проведении многих упрощающих допущений. Это вызывает особую
необходимость в развитии полуколичественных теоретических пред-
представлений, позволяющих предсказывать устойчивость и свойства
координационных соединений. Кроме качественной теории МО,
в химии координационных соединений получила широкое распрост-
распространение теория кристаллического поля, которая, хотя и основывает-
основывается на упрощенной физической модели строения, позволяет система-
систематически описать многие важные свойства комплексов. Теории
ОЭПВО и гибридизация АО в химии координационных соединений
не нашли столь широкого применения, как в случае соединений
непереходных элементов.
11.2. ТИПЫ КООРДИНАЦИОННЫХ ПОЛИЭДРОВ
Наличие центрального атома, координирующего от двух до
10—12 лигандов (атомов или групп, связанных двух- или многоцен-
многоцентровыми связями с центральным атомом), приводит к высокой
симметрии молекул координационных соединений и координацион-
координационных узлов в твердом теле. Когда роль центрального атома выпол-
выполняет переходный металл, число возможных для него конфигураций
409
связей (координационных полиэдров) возрастает по сравнению с со-
соединениями непереходных элементов. Это объясняется большим
многообразием типов связей, образуемых переходными элементами
вследствие участия в них частично (или полностью) заполняемых
электронами валентных rf-орбиталей. Кроме того, для соединений
переходных металлов структурная жесткость координационного по-
полиэдра обычно не так высока, как для соединений непереходных
элементов, и это допускает возможность существования различных
типов координационных полиэдров для одного и того же валент-
валентного состояния данного центрального атома при варьировании
лигандов и даже типа кристаллической решетки или условий. На-
Например, комплексный анион [Ni (CNM] ~ существует в кристалличес-
кристаллической фазе в тригонально-бипирамидальной XI и квадратно-пирами-
квадратно-пирамидальной XII формах:
CN
L-CN I
NC—N*
CN
XI XII
В табл. 11.1 показаны основные типы координационных полиэд-
полиэдров для КЧ = 2—8, реализуемых в молекулах координационных
и металлоорганических соединений переходных металлов. Приве-
Приведем отдельные примеры соединений каждого типа, содержащих
двухцентровые связи металл—лиганд:
О==С— СЬ—Zn—Co—C=O
f>c
XIII XIV
RN NR
xv, R-трет-С^Н,
Табл. 11.1 содержит также указания на тип гибридизации ор-
битали центрального атома, осуществляющего двухцентровую
связь с лигандом. Схема образования гибридных орбиталей, вклю-
включающих ^-функции, не отличается от рассмотренной для случая
^гибридных орбиталей (см. разд. 10.2.3). Гибридные орбитали,
состав и форма которых определены типом координационного по-
полиэдра, обеспечивают максимальное перекрывание с локализован-
410
Таблица 11.1. Основные координационные иолюдры в комплексных соед!
неннях переходных металлов с КЧ центрального атома 2—8
кч
2
3
4
5
6
7
8
Конфигурация связей цент-
центрального атома
Линейная
Угловая
Треугольная
Пирамидальная
Тетраэдрическая
Плоская
Тригональная бипира-
мида
Квадратная пирамида
Октаэдрическая
Григональная призма
Пентагональная бипира-
мида
Одношапочный октаэдр
Додекаэдр
Квадратная антипризма
Тип гибридизации
sp, dp
ds
sp2, ds2, dp2
*P
sp3. cPs
dsp2
dzisp5, d*sp
dxi-y*sp3, d^sp, d*s
d2sp3
d*spt cPp, ?ръ
cPsp; d*sp2
Пример структуры
[Ag(NH5Jl+;XIII(Zn)
—
Fe[N(SiMe3Jb;Ni(COK
Mo(COK
Ni(COL;[Co(NH3L]2+
[Ni(CNL}2-;V
XI;XIII(Co);Fe(COM
XII; [MnCl5]2-
Cr(CoN;[Co(NH3N}3 +
XIV
[Mo(CNOJs-;[UO2F5]3-
XV
{Mo(CN)eJ*~
[TaF8]3-
ными орбиталями лигандов, ориентированными в направлениях
осей орбиталей центрального атома. Гибридизация s, px,
ру и d^_y2 АО центрального атома дает четыре эквивалентные
гибридные орбитали ?&р2-типа, направленные вдоль диагоналей
квадрата XVI. Октаэдрической координации отвечают шесть эк-
эквивалентных <Р"Бръ гибридных орбиталей XVII. Для КЧ = 5 возмож-
возможны две различные комбинации, одна из которых, XVIII, соответ-
соответствует тригонально-бипирамидальной, а другая, XIX, — квадрат-
квадратно-пирамидальной конфигурации связей. Очевидно, что в отличие
от эквивалентных dsp2- и iPsp*-гибридных орбиталей квадратной
и октаэдрической конфигураций <&/?3-наборы содержат неэквива-
неэквивалентные орбитали. Так, связи, лежащие в экваториальной плоскости
тригональной бипирамиды, могут включать вклады от рх- и ру-АО
центрального атома, тогда как две аксиальные связи — вклад толь-
только от одной р2-АО:
411
XVI dsp2(s.px,p,.,dx2 yi) XVII d1spi{s,px,py,pI, dx2 yi,dzi)
¦
XVIII
XIX
Задача 11.1. Для определения углов между гибридными АО центрального
атома, содержащих вклады s, p, rf-AO x=zas + bp + cd, JI. Полинг выаел A976)
уравнение
cost)— — t
Оцените углы между связями, образуемыми центральным атомом, для
3-, cPsp3- и Л/>3-структур.
На ранней стадии развития теории строения координационных
соединений представления теории гибридизации находили примене-
применение для описания их строения и магнитных свойств. Так, парамаг-
парамагнетизм солей аниона [NiQ4]2~ может служить основанием для
правильного предсказания тетраэдрическои структуры этого аниона
в соответствии со следующей схемой электронного заполнения АО
никеля при образовании координационных связей:
3d
4s
Ni2+ CF)
[NiClJ
2-
[Ni(CNL]
2-
n
u
1^
u
u
n
н
1
^
u
1
^
u
u
u
it
H
H
<v3L
u
1t
412
В то же время соли аниона [Ni(CNLJ2 диамагнитны, т. е.
электронная оболочка комплекса не содержит неспаренных электро-
электронов. В рамках представлений о гибридизации это объясняется за-
заполнением восемью электронами лигандов двух вакансий в Ъй-
о бол очке центрального иона и шести в s- и /^-оболочках. Гиб-
Гибридизация <&р2-типа ведет к геометрической конфигурации плоско-
плоского квадрата. Однако представления о гибридизации лишь с боль-
большой натяжкой могли объяснить, например, парамагнетизм октаэд-
рического комплекса [CoFJ3~ и диамагнетизм октаэдрического ко-
комплекса [Co(NH3N]3 + . При рассмотрении координационных соеди-
соединений с многоцентровыми связями они теряют предсказательную
силу.
С существенными ограничениями при объяснении пространст-
пространственного строения соединений переходных элементов встречается
также теория ОЭПВО. Как и в случае представлений о гибридиза-
гибридизации, эта теория (по самой сути связанная с концепцией Л МО)
неприложима к описанию координационных соединений с много-
многоцентровыми связями. Кроме того, следует вспомнить еще одно
исходное положение теории ОЭПВО, требующее, чтобы элект-
электронное облако внутренних оболочек центрального атома харак-
характеризовалось сферической симметрией. Это условие выполняется,
если все внутренние оболочки заполнены, т. е. для d°, ^-элект-
^-электронных конфигураций центрального атома. Сюда же относится
электронная конфигурация сг, в которой каждый электрон занимает
отдельную d-AO. Во всех остальных случаях электронное облако
внутренних оболочек не имеет сферической симметрии. По этой
причине соотношение A0.27), управляющее ориентацией облаков
электронных пар в валентных орбиталях, выполняется не вполне
точно.
Оценочные расчеты показывают, что, если количество электро-
электронов на d-AO невелико A—3 электрона), нарушения сферической
симметрии незначительны. Поскольку во внешних электронных
оболочках координационных соединений нет неподеленных элект-
электронных пар, геометрическое строение их молекул или координаци-
координационных узлов в кристаллических структурах типа АХ* достаточно
точно отвечает требуемой правилами теории ОЭПВО взаимной
ориентации п связывающих электронных пар (см. табл. 10.3).
Для соединений XI, XIII, других комплексов с КЧ = 2, 3, 4, 6,
приведенных в табл. 10.1, теория ОЭПВО дает достоверные пред-
предсказания о структуре координационного узла. Для комплексов,
в которых число ^-электронов центрального атома велико G—9),
как правило, наблюдаются значительные отклонения от предсказа-
предсказания теории ОЭПВО. Наиболее важные из них: 1) возможность
существования плоских координационных узлов при КЧ, равном
четырем; 2) искажения конфигурации связей тригональной бипира-
413
миды в сторону квадратно-пирамидальной; 3) тетрагональные ис-
искажения октаэдрических структур, связанные с удлинением или
укорочением одной пары связей. Показательным примером являет-
является строение октаэдрического узла СиС16 в кристаллической решетке
СиС12:
С1 CI
4>-о,23О„м
jfi >-0,295 ни
6CI CI
(СиС12)и XX
Чтобы охватить эти отклонения, в теории ОЭПВО вводится
предположение о том, что электронное облако внутренних rf-элект-
ронов принимает эллипсоидальную форму. Тогда можно показать
на электростатических основаниях, что если это облако вытянуто,
например, вдоль одной из осей октаэдра, то связи, проходящие
через эту ось (Си—С15,б в структуре XX), будут удлинены. Однако
заранее нельзя определить, как ориентирован эллипсоид элект-
электронного облака внутренних ^-электронов. Следовательно, теория
в подобных случаях теряет предсказательную силу.
Кроме того, как будет показано, действительная причина многих
геометрических искажений координационного узла, в частности тет-
тетрагонального искажения октаэдрической конфигурации связей, зна-
значительно глубже и вызвана специфическими электронно-колеба-
электронно-колебательными эффектами типа эффекта Яна—Геллера.
Таким образом, в целом теория ОЭПВО в химии координацион-
координационных соединений полезна для примерного суждения о конфигурации
координационных связей. Следует в каждом отдельном случае счи-
считаться с возможностью отклонения истинной структуры от иде-
идеализированной. Теория не может помочь при определении геомет-
геометрии комплексов с многоцентровыми связями. Кроме того, эта
теория не предназначена для описания магнитных и спектральных
свойств координационных соединений. Эту задачу на уровне мо-
модельных представлений призвана выполнить теория кристалличес-
кристаллического поля.
11.3. ТЕОРИЯ КРИСТАЛЛИЧЕСКОГО ПОЛЯ
Из изложенного выше вытекает особая роль б?-электронных обо-
оболочек центрального атома для понимания структуры и свойств
координационных соединений переходных элементов. В 1929 г.
414
Г. Бете* в статье «Расщепление атомных термов в кристаллах»
заложил основы теоретического подхода к описанию распределения
электронов в d- (а также /-) оболочках центрального атома, коор-
координирующего относительно себя группы лигандов в окружениях
различной симметрии. Этот подход, получивший название теории
кристаллического поля (ТКП), включает следующие основные ис-
исходные положения.
1. Природа сил, вызывающих наличие координационных связей
и определяющих устойчивость координационного соединения, сво-
сводится к чисто электростатическому взаимодействию между цент-
центральным ионом-комплексообразователем и лигандами.
2. Лиганды независимо от их реальной структуры рассматрива-
рассматриваются в ТКП как точечные заряды (реже — диполи), характер рас-
расположения которых относительно центрального иона определяет
симметрию действующего на него внешнего электростатического
поля.
3. Электронная конфигурация центрального иона учитывается
в явном виде в рамках квантово-механических представлений.
4. С учетом указанных выше приближений для описания свойств
комплексов привлекается формализм квантовой механики.
Расчет волновых функций центрального иона-комплексообразо-
вателя в рамках ТКП требует решения уравнения Шрёдингера
с гамильтонианом:
H=H0+V, (ИЛ)
где Но — оператор Гамильтона для свободного иона-комплексооб-
разователя; V — оператор, описывающий взаимодействие этого ио-
иона с созданным лигандами электростатическим полем. Вид этих
операторов отвечает выражениям
. (Ц.2)
y^. 01.3)
rr \Rrr -1
где ZxH, — эффективный заряд центрального иона, вычисляемый,
например, по правилам Слэтера (см. разд. 3.4.1); qj — точечный
заряд, характеризующий лиганд, фиксированный в точке R j (Rjt 6jf
VjJt'fifrh ®u фО — электронная координата. Суммирование прово-
проводят по всем Л^ лигандам и п электронам центрального иона.
*Ганс Бете (род. 1906 г.) — немецкий физик-теоретик. Нобелевский лауреат
A967). Известен своими работами по теории ядерных реакций и особенно открытием
источников энергии звезд.
415
Прежде чем перейти к применению выражений A1.1)—(П.З)
в конкретных расчетах, рассмотрим более важные качественные
аспекты ТКП.
11.3.1. Расщепление «^-уровней центрального иона
Широкие различия магнитных и спектральных характеристик
координационных соединений обусловлены проявляющейся в полях
лигандов неэквивалентностью энергетических уровней ^/-электронов
центрального иона. Расщепление этих уровней, пятикратно вырож-
вырожденных в свободном ионе, вызвано изменением симметрии поля
центрального иона при окружении его лигандами.
Характер расщепления можно установить, не проводя точных
расчетов для координационных соединений ионов, имеющих в d-
оболочке один электрон, например Ti3 + , V4+ (основной терм 2D).
В таких ионах отсутствуют усложняющие эффекты взаимодействия
^-электронов.
Обратимся к рис. 2.4, показывающему пространственное рас-
распределение электронной плотности в d-KO водородоподобного ио-
иона. Все эти АО энергетически равноценны и сохраняют свою эк-
эквивалентность также и в гипотетическом сферически-симметричном
поле лигандов, хотя в этом поле их общий уровень поднят в силу
взаимодействия с отрицательно заряженными лигандами над уров-
уровнем в свободном ионе на некоторую величину % Рассмотрим теперь
поведение электрона в ^-оболочке в полях лигандов более низкой
симметрии.
Как видно из рис. 11.1 и 11.2, в октаэдрическом поле лигандов
d-орбитали делятся на две неравноценные группы. К одной из них,
обозначаемой t2g (см. разд. 6.3), принадлежат dxy-, dxz- и dyz-AO. Если
единственный ^/-электрон рассматриваемого центрального иона
Рис. 11.1. Схема располо-
расположения лигандов в октаэдри-
октаэдрическом комплексе
1 J. A-lQDq
Сферическое Октаэдричесмое
поле ло/ге
Рис. 11.2. Расщепление одноэлектрон-
ных {/-состояний атома металла в сфе-
сферическом и октаэдрическом полях ли-
лигандов (?о~20+40 эВ, Д~1-;-3 эВ)
416
t
X -Х-
Рис. 11.3. Представление J2*-AO как линейной комбинации d?-&- и ^-
остается в комплексе на одной из этих орбиталей, он оказывается
более удаленным от расположенных по координатным осям одно-
одноименно заряженных лигандов, чем в том случае, если бы с?1-элект-
с?1-электрон занимал Ф_у- или ф-орбитали (^-орбитали). Эквивалентность
трех /2?-орбиталей в октаэдрическом поле лигандов достаточно
очевидна, тогда как, судя по рис. 2.4, ф- и ф_^-АО имеют весьма
различную форму. Это различие кажущееся, так как истинная
форма ф-АО в декартовой форме d^-py, т. е. fi^1-*1-** (см- табл. 2.3
и 2.4). Следовательно, ф-АО можно представить как линейную
комбинацию Ф_х*- и ф_/-АО, откуда ясно, что ф и d#_y* равноцен-
равноценны по пространственному распределению электронной плотности
(рис. 11.3). Радиальные части полной волновой функции всех d-KO
одинаковы.
Расщепление t-^- и е^-уровней рассматривается как некоторый
эмпирический параметр и обозначается А, или \0Dq:
E(eg)-E(t2g) = 10Dq. A1.4)
Если за нуль отсчета принимается пятикратно вырожденный энер-
энергетический уровень rf-электронов в сферически-симметричном поле
лигандов ёо, можно оценить относительную стабилизацию электро-
электронов на t2g- и дестабилизацию на ^-уровнях в единицах Dq.
Предположим, что все три t-ц- и два е^-уровня ^-электронов
центрального иона заселены электронами. В этом случае сохраняет-
сохраняется, очевидно, сферическое распределение электронной плотности
d -электронов центрального иона и выполняется соотношение, из-
известное как теорема центра тядкести:
A1.5)
14. Теория строения молекул
417
Коэффициенты в левой части уравнения (Н.5) соответствуют коли-
количеству электронов, которые можно разместить на eg- и ^-уровнях.
Используя соотношение A1.4) и введя для энергии стабилизации
и дестабилизации обозначения
получим после несложных преобразований
2 3
x—~A=4Dq, y=-A =
Задача 11.2. Хотя величину lODq целесообразно рассматривать как пара-
параметр, оцениваемый из экспериментальных данных, можно рассчитать ее по
теории возмущений (Шлапп, Пенни). Расчет приводит к выражению
. (П.6)
где д — точечный заряд лиганда; R — расстояние металл—лиганд; <г*> — сред-
средняя величина для четвертой степени расстояния электрона от ядра центрального
иона. Используя радиальные функции слэтеровского типа, оцените величины
</•*> и \QDq для комплекса [TiFg]3", полагая q= — e и R — 0,2 нм.
При тетраэдрической координации центрального иона также
происходит расщепление уровней rf-орбиталей, однако при этом
положение электрона на е-орбиталях d? и ф_/ оказывается более
выгодным, чем на da~t dys-t ^-орбиталях (t2).
Причину этого, а также сближение е- и ^"Уровней в тетраэд-
рическом поле по сравнению с октаэдрическим можно наглядно
объяснить тем, что в тетраэдре лиганды находятся на диагоналях
вписанного в сферу куба, а в октаэдре — на его гранях:
АТСтр=--Аожг. (U.7)
Расщепление уровней ^/-орбиталей в полях другой симметрии
показано на рис. 11.4. Во всех представленных случаях изложенный
ранее качественный подход, основанный на рассмотрении взаимо-
взаимодействий электрона на отдельных rf-орбиталях с зарядами лигандов,
достаточен для получения показанной на рис. 11.4 картины расщеп-
расщепления d-уровней. Относительные значения расщеплений отдельных
уровней можно получить лишь при количественном расчете.
Задача 11.3. Объясните схему расщепления rf-орбиталей центрального иона
в поллх различной симметрии, приведенную на рис. 11.4. В каких полях произой-
произойдет также расщепление /^-уровней центрального иона?
418
[х12м] [мх8]
Иносаздр Атпиаразт Куб
[МХ6] [UX5Y]
Тетраэдр Сферическая дктаздр
симметрия
Отаздр
(транс)
[мх4] ^
Ялзский
квадрат
Рис. 11.4. Расщепление «/-уровней в полях различной симметрии
В табл. 11.2 даны представления симметрии rf-орбиталей для
точечных групп симметрии, отвечающих наиболее важным кон-
конфигурациям координационного узла комплексов, что поясняют обо-
обозначения на рис. 11.4.
Таблица 11.2. Представления симметрии для {/-орбиталей в различных груииях
симметрия
Координа-
Координационное
число
4
5
6
Конфигурация
Тетраэдр
Квадрат
Квадратная пирамида
Тригональная бипярамида
Октаэдр
Группа
симмет-
симметрии
Td
AlA
C4v
oh
Представление симметрии
е
aXg
^t
а\
е*
dx*~y>
е
big
b\
е'
dXy
t2
b2g
e'
d*
h
eg
e
*
dyz
t2
eg
e
en
419
11.3.2. Количественная оценка расщеплешш
Применение методов теории групп позволяет провести расчет
величины расщеплений уровней rf-орбиталей центрального иона
в полях лигандов различной симметрии. Именно этот подход был
применен в работе Г. Бете.
Для случая электронной конфигурации d1 достаточным оказыва-
оказывается более простой расчет, использующий теорию возмущений (см.
разд. 1.5). Потенциал У в выражении A1.1) можно рассматривать как
относительно малое (по сравнению с внутриатомными взаимодейст-
взаимодействиями) возмущение. Тогда, согласно теории возмущения для вырож-
вырожденного случая, поправки е* к энергиям каждой отдельной ^-орбнтали
можно вычислить, решая секулярное уравнение пятого порядка (терм
2 Л пятикратно вырожден, квантовые числа т варьируют от — 2 до 2):
\vMn-5nuti?k\ = 0f A1.8)
где Vmm— матричные элементы возмущения потенциалом лигандов
V [соотношение A1.3)] вычисляются как
N
00 1*1
A1-9)
-00
причем ф*т, \jfm — функции rf-орбиталей в сферических координатах
(см. табл. 2.4).
Хотя вычисления матричных элементов A1.9) достаточно слож-
сложны, тем не менее они могут быть полностью проведены в аналити-
аналитической форме. Путь расчета подробно разобран в монографиях,
посвященных ТКП. Результаты расчетов табулированы.
Таблица 11.3. Отвосвтельвые энергш ?-орбяталей при разлн
циях лжгандов X (в едшацах Dq). Ось х является осью высшего порядка
конфмгура-
Коорди-
национ-
национное число
2
4
5
6
8
Тип ком-
комплекса
МХ2
МХ4
мх5
мх«
мх«
Конфигурация
Пинрйнаа
Тетраэдрическая
Квадратная
Квадратная пирамида
Тригональная бипира-
мида
Ожтаэдраческая
Кубическая
Квадратная антипризма
10Д8
-2,67
-4^8
0,86
7,07
6,00
-5,34
-5,34
628
-2,67
12Д8
9,14
-0,82
6,00
-5,34
-0,89
-6,28
1,78
2,28
-0,86
-0,82
-4,00
3,56
-0,89
1.14
1,67
-5,14
-4,57
-2,72
-4,00
3,56
3,56
1,14
1,78
-5,14
-4,57
-1,72
-4,00
3,56
3,56
420
ABC
Рис. U.S. Иллюстрация трех первичных групп лигаддов ML, ML2 и ML*
Поскольку приближение точечных лигандов для многих случаев
оказывается слишком упрощенным, важно знать не столько аб-
абсолютные значения екз сколько их относительные величины в полях
различной симметрии. Последние удобно выразить в единицах эм-
эмпирического параметра Dq октаэдры ческой конфигурации лигандов.
Данные для наиболее важных конфигураций приведены в табл. 11.3.
Эти данные использованы при построении картины расщепления
^-орбиталей на рис. 11.4. Обратимся к ним позднее, при рассмотре-
рассмотрении вопроса об энергиях стабилизации кристаллическим полем.
Как видно из соотношения A1.9), матричные элементы возмуще-
возмущений уровней ^-орбиталей рассчитываются как аддитивные функции.
Это позволяет предложить весьма полезный способ аддитивного
расчета относительных энергий расщеплений ^-орбиталей при раз-
различных конфигурациях лигандов, составляя полную конфигурацию
всего из трех типов так называемых первичных групп (Кришнамур-
ти, Шаап, 1970). Первичные группы представлены на рис. 11.5.
Таблица 11.4. Оп
энерпш <?орбп-алей для трех
ядов (в едошщх Dq)
помп-
Коорди-
национ-
национное число
1
2
4
6
Тип группы
(А)
(В)
(С) Правильный тетраэдр
(/f«109°28')
(С) Вытянутый тетраэдр (/У=90°)
(С) Сплюснутый тетраэдр
(/У-120е)
1 лигянд ня new *
1 лиганд ня оси z
2 лиганда в плоскости ху
2 лиганда в плоскости ху
Суммарная энергия 6 лигандов
октаэдра
5,14
-2,14
-2,67
0,64
-3,70
5,14
5,14
-2,14
-2,14
6,00
dj-y*
-3,14
6,14
-2,67
-5,14
-1,43
-3,14
-3,14
6,14
6,14
6,00
-3,14
1,14
1,78
-2,64
4,30
-3,14
-3,14
1,14
1.14
-4f00
dxz
0,57
-2,57
1,78
3,57
0,465
0,57
0,57
-2,57
-2,57
-4,00
dyz
0,57
-2,57
1,78
3,57
0,465
0,57
0,57
-2,57
-2,57
-4,00
421
В табл. 11.4 приведены величины относительных энергий d-op-
биталей для первичных групп.
Чтобы вычислить, например, энергии расщепления d-орбиталей
в октаэдрическом поле лигандов, нужно составить октаэдрическую
конфигурацию из двух первичных групп (А) и двух групп (В). Тогда
потенциал в октаэдрическом поле имеет вид
X i
/»
X
XXI
{
» /
х
Изложенный аддитивный способ подсчета относительных рас-
расщеплений особенно полезен, когда необходимо оце-
оценить картину расщеплений в комплексе, имеющем
неэквивалентные лиганды. Рассмотрим, например,
расчет расщепления уровней rf-орбиталей в октаэд-
октаэдрическом комплексе типа XXI.
Конфигурацию XXI можно составить из двух пер-
первичных групп МХ2 типа (В) и двух первичных групп
типа (A) MY. Результаты расчета будут выражены через два эм-
эмпирических параметра: Dqx и Dqyt каждый из которых характеризует
величину расщепления A=IOD<7 в октаэдрических комплексах МХ$
иМУ6:
Энергия rf-орбита-
лей
2 ляганда — пер-
первичная группа (А)
4 лиганда X — пер-
первичная группа (В) -4,28?>^ \2,2%Dqx
Сумма
(Dqy=\,25Dqx)
Симметрия </-орби-
тали (В4к)
dXy
,2SDqx
,VDqx
dXz
\MDqy
-5,\4Dqx
-3,7152)9,
eg
dyi
\,\4Dqy
-5,\4Dqx
-3t7l5Dqx
eg
Задача 11.4. Сопоставьте полученную в данном расчете картину расщепле-
расщепления уровней в транс-дизамещенном октаэдре с расщеплением в октаэдре MX4Y2
на рис. 11.4. Укажите, удлиненные или укороченные связи М—Y имеются в виду
(точечные заряды лигандов X и Y предположите равными).
Задача 11.5. Составьте схемы расщепления уровней d-орбиталей в цис-ок-
таэдре MX4Y2 и MX3Y3, полагая Dqy=\t25Dqx.
11.3.3. Спектрохимический ряд
Представляется важным получить представление о порядке ве-
величин А= lODq. Эти значения могут быть взяты из эксперименталь-
экспериментальных данных о спектрах поглощения октаэдрически координирован-
422
200 300 Ш 500 600
Рис. 11.6. Спектр поглоще-
поглощения (Ti (Н2О)б]3+
ныл ионов, обладающих одним электро- 1де
ном в ^/-оболочке (терм основного состоя- 3,0
ния 2D).
На рис. 11.6 показан спектр поглощения
октаэдрического комплекса (Ti(H2ON]3 + . 2fi
В длинноволновой области имеется полоса
слабой интенсивности с максимумом по-
поглощения 20 400 см. Эта полоса, со-
ответствующая фиолетовой окраске рас-
растворов комплекса, отсутствует в безвод-
безводном сульфате или хлориде трехвалентного
титана. Природа ее связана с расщеп-
расщеплением d-уровней в октаэдрическом поле
и электронным переходом с t2g- на eg-
уровень (см. рис. 11.4). Энергия этого
перехода приравнивается к разности эне-
энергий tig- и е^-уровней, т. е. lODq. При
наличии более чем одного электрона в центральном ионе не-
необходимо внести дополнительные поправки, чтобы получить
величину \0Dq, но порядок этой величины правильно передается
энергией самого длинноволнового спектрального перехода в ко-
комплексе. В общем, величины lODq варьируют для разных це-
центральных ионов и лигандов в интервале 1—4 эВ (8000—
35 000 см).
Следует подчеркнуть малую интенсивность d—^-переходов в ок-
таэдрических комплексах. В октаэдрическом окружении при нали-
наличии центра инверсии сохраняется правило отбора AL= + 1 (см. разд.
3.9). Следовательно, d—^-переходы (AL = 0) запрещены по симмет-
симметрии. Проявление полос d—(/-переходов обусловлено электронно-
колебательными взаимодействиями. В результате непрерывного ко-
колебательного движения атомов в молекулах некоторые из них (при
не полностью симметричных колебаниях) в каждый определенный
момент времени оказываются слабо искаженными по сравнению
с идеальной октаэдрической геометрией. При таких искажениях
становится возможным наблюдать слабоинтенсивные полосы по-
поглощения формально запрещенных электронных переходов.
На основании изучения большого экспериментального матери-
материала по электронным спектрам поглощения комплексов и оценочных
расчетов удалось сформулировать ряд правил о зависимости вели-
величины \0Dq от типа центрального иона и лиганда. Наиболее важное
из них правило спектрохимического ряда.
Величина lODq возрастает слева направо в следующем ряду
наиболее характерных лигандов, причем указанный порядок не
меняется для различных центральных ионов:
423
Г <Br" <SCN" <СГ <NOf <F" <0H~ <H2O<
NCS-<CH3CN<NH3<Py<En<NOf<CN-<CO, ( ' }
где Py — пиридин; En — этилендиамин.
Кроме этого правила отметим еще два достаточно общих поло-
положения.
При одинаковом лигандном окружении наибольшая величина
\0Dq соответствует комплексам центральных ионов с наибольшими
зарядами. Например, XODq для [Со(Н2ОN]3+ равна «20 000 см,
адля[Со(Н2О)б]2+ «9700 см.
Величина lODq для комплексов ионов переходных металлов,
принадлежащих к разным периодам периодической системы, но
имеющих одинаковую электронную оболочку, примерно постоянна
при однотипном лигандном окружении.
Задача 11.6. Как видно из рис. 11.6, полоса поглощения d—^-перехода
в комплексе [Ti (Н2О)б]3 + сильно размыта, что обусловлено электронно-колеба-
электронно-колебательными эффектами, но все же, как ожидается из картины расщепления уровней
в октаэдрическом поле, в спектре водного раствора комплекса ТЮз'бНгО
имеется всего одна полоса электронного перехода. Вели же определить спектр
поглощения этого комплекса в твердой фазе, то обнаруживаются две полосы
d—{/-переходов при 15 000 и 18 300 см. Объясните происхождение этих полос,
отнесите их к определенным электронным переходам, свяжите объяснение с из-
изменением структуры координационного узла комплекса в кристаллическом со-
состоянии по сравнению с состоянием в растворе.
Задача 11.7. Ион [СиСЦ]2" в комплексе Cs2[CuCl4] имеет структуру сплюс-
сплюснутого тетраэдра (/?«120°). Объясните происхождение трех полос поглощения
в спектре этого комплекса в ближней ИК-области (около 9000, 8000 и 5000 см" *).
11.3.4. Комплексы сильного и слабого полей.
ТКП и магнитные свойства комплексов
До сих пор мы ограничивались рассмотрением комплексов толь-
только тех центральных ионов, в rf-оболочке которых содержится только
один электрон. Оценка величины \dDq и относительных энергий
расщеплений ^-орбиталей позволяет подойти к простому, но до-
достаточно надежному описанию распределения нескольких электро-
электронов по ^-орбиталям центрального иона. Ясно, что результат этого
распределения электронов в незамкнутой оболочке определит число
неспаренных электронов в комплексе и, следовательно, его магнит-
магнитные свойства.
При этом, как и в случае многоэлектронных атомов, необходимо
учесть и количественно оценить два главных эффекта, действующих
часто в противоположных направлениях: стремление электронов
424
образовать полностью заполненную оболочку и тенденцию к об-
образованию электронной конфигурации с максимальным числом
неспаренных спинов.
Рассмотрим вначале правильную октаэдрическую конфигура-
конфигурацию лигандов относительно центрального иона. Расщепление уров-
уровней энергии </-орбиталей в этой конфигурации показано на рис. 11.2.
Нетрудно видеть, что для случаев одного, двух и трех электронов
возможен только один вариант размещения электронов, показан-
показанный схематически в табл. 11.5. Для конфигурации ег и сР (как и для
di0) есть только один способ размещения электронов на t2g- и eg-
уровнях, при котором требования максимальной мультиплетности
и максимального количества электронов на низшем энергетическом
уровне удовлетворяются одновременно.
Однако для <t-, d5-, сР-, ^-электронных конфигураций возможен
выбор между двумя способами размещения. Они отличаются тем,
что в первом случае низший ^-уровень заселяется настолько полно,
насколько это допускает общее число электронов. Во втором вари-
варианте определяющим служит требование наибольшего числа неспа-
неспаренных электронов. Ясно, что выбор между этими двумя возмож-
возможностями заполнения электронной оболочки центрального иона бу-
будет в основном зависеть от величины расщепления tlg- и ^-уровней,
т. е. от параметра \0Dq. Если эта величина большая, как для
лигандов, находящихся в правой части спектрохимического ряда, то
центральный ион сформирует электронную оболочку с наибольшим
числом электронов на нижнем ^-уровне. Этот вариант соответству-
соответствует сильному электростатическому полю лигандов, сами
лиганды, создающие такое поле, называют лигандами сильного поля,
а комплексы — низкоспиновыми.
При относительно малых величинах расщеплений, возникающих
в слабых полях лигандов (левая часть спектрохимического ряда),
тенденция к образованию термов с максимальной мультиплетно-
стью оказывается преобладающей. Этот вариант отвечает случаю
слабого поля лигандов, комплексы такого типа называются
высокоспиновыми.
Приведенные в табл. 11.5 теоретически ожидаемые величины
магнитных моментов рассчитаны в предположении, что они имеют
чисто спиновую природу и зависят только от числа неспаренных
электронов по соотношению C.87). Из сравнения с приведенными
экспериментальными величинами для различных лигандных окру-
окружений видно, что, хотя в комплексах центральных ионов с большим
числом ^-электронов и наблюдаются заметные отклонения вследст-
вследствие спин-орбитального взаимодействия, тем не менее по величине
магнитного момента комплекса практически всегда можно уверенно
установить количество неспаренных электронов центрального иона
и отнести комплекс к высокоспиновому или низкоспиновому типу.
425
Таблица 11.5. Распределение электронов в октаэдрнческих комплексах для слабого и сильного поля лнгаядов, рассчитанные
и экспериментальные значения магнитных моментов комплексов
Число
элект-
электронов
1
2
3
4
5
6
7
8
9
10
Централь-
Центральный ион
Ti3+,
v*+
v3+
V3+'
Mn*4
Cr2*
Mn3^
Mn3+,
Fe» +
SP
Ni* +
Cu3 +
Zni +
Слабое поле
электронная конфигурация
w
С,I С,I С,I
WWWWW
WWWWW
wwwww
WWWWW
лигандов
число
неспа-
рен-
в ы х
элект-
электронов
1
2
3
4
5
4
3
2
1
0
теоре-
тичес-
к и й
маг-
нит-
нитный
мо-
момент
Рм
1,73
2,83
3,87
4,90
5,92
4,90
3,87
2,83
1,73
0
эксперимен-
экспериментальный маг*
нитный мо-
я
мент дм
1,65—1,78
2,75—2,85
3,7-4,0
4,75—5,00
5,65—6,10
4,3-5,7
4,3-5,2
2,8—3,5
1,7—2,2
0
Сильное поле лигандов
электронная конфигурация
¦—
—
—
WWW
www
www
У g' \*gS \ g' V g'
wwwww
wwwww
wwwww
ЧНСЛО-
веспа-
p e н -
H Ы X
элект-
электронов
-—
—
—
2
1
0
1
2
1
0
теоре-
тичес-
к и й
маг-
нит-
нитный
мо-
момент
—
—
—
2,83
1,73
0
1,73
2,83
1,73
0
эксперимен-
экспериментальный маг-
магнитный мо-
момент Рм
—
—
—
3,18—3,30
1,8-2,5
0
1,8 2,0
2,8—3,5
1,7—2,2
0
Так, например, для комплексов [FeFJ3 магнитный момент,
определенный экспериментально в растворе, равен ~6,0Д«. Это
согласуется с магнитным моментом, ожидаемым для высокоспино-
высокоспинового комплекса с пятью неспаренными электронами, а также с тем,
что величина A = lODq для фтора, находящегося в левой части
спектрохимического ряда, равна 13 900 см. Для другого комплек-
комплекса железа [Fe(CN)J^~ Л«30 000 см и его магнитный момент
равен «l,6/jM.
Для тетраэдрических комплексов также возможен простой ана-
анализ, аналогичный описанному выше для октаэдрических комплек-
комплексов. Хотя для некоторых конфигурации ^-электронов в тетраэд-
рическом поле лигандов теоретически можно ожидать низкоспино-
низкоспиновые состояния комплексов, однако следует иметь в виду соотноше-
соотношение A1.7). Оно указывает, что тетраэдрическое поле лигандов явля-
является весьма слабым по сравнению с октаэдрическим. Действитель-
Действительно, низкоспиновые тетраэдрические комплексы неизвестны даже для
лигандов, находящихся в самой правой части спектрохимического
ряда A1.10).
Задача 11.8. Определите конфигурации ^-электронов центрального иона
в тетраэдрических комплексах, для которых теоретически возможны низкос-
низкоспиновые состояния.
Тетрагональное искажение октаэдрической конфигурации коор-
координационного узла снимает, как видно из рис. 11.4, вырождение
е^-орбиталей октаэдра. Этот эффект, если величина его достаточна,
чтобы привести к заметному расщеплению ^-уровня, может иметь
особенно интересные магнетохимические следствия для ^-элект-
^-электронной конфигурации. В сильном поле комплексы соответствую-
соответствующих октаэдрически координированных центральных ионов могут
стать низкоспиновыми, т. е. диамагнитными.
Хотя для искаженных октаэдрических комплексов ионов такой
результат еще не был экспериментально замечен, но, по существу,
именно этот эффект объясняет диамагнетизм плоских комплексов
Ni2+, Pd2+, Pt , Rh1 + , Au3 + . Действительно, плоскую конфигура-
конфигурацию можно рассматривать как предельно вытянутый вдоль одной
оси октаэдр. Как видно из рис. 11.4 и данных табл. 11.3, расщепле-
расщепление высших орбиталей типа Ьн и Ь^ в квадратных комплексах
весьма велико. Поэтому до настоящего времени не известны высо-
высокоспиновые плоские комплексы ионов сР.
На примере комплексов с ^-электронной оболочкой централь-
центрального иона отчетливо иллюстрируется значение магнетохимических
данных для решения некоторых стереохимических задач координа-
427
ционных соединений. Для N-замещенных бмс-салицилальдиминатов
никеля XXII возможны либо плоская (наиболее характерная для
ионов rf8), либо тетраэдрическая конфигурация связей центрального
иона. Действительно, если заместитель R при азоте — н-алкильная
группа, комплексы XXII диамагнитны, как и ожидается для плоской
координации XXIII:
\\п
XXIII XXIV
Однако для комплекса XXII [R = C(CH3K] измеренный в рас-
растворе магнитный момент равен 3,3/?м, что свидетельствует о нали-
наличии двух неспаренных электронов центрального иона и, следова-
следовательно, тетраэдрической структуры молекул XXIV. В случаях, когда
R — вторичная алкильная группа (мз0-С3Н7, циклогексил), измерен-
измеренный магнитный момент имеет промежуточное значение между
О и 3,3/JM. Этот результат говорит о конформационном равновесии
в растворе XXIII ll XXIV. Содержание парамагнитного тетраэд-
рического комплекса и наблюдаемый эффективный магнитный мо-
момент в растворе зависят от температуры последнего.
11.3.5. Энергия стабилизации кристаллическим полем
Представления о сильных и слабых полях лигандов, а также
данные об относительных энергиях rf-орбиталей в полях различной
симметрии привлекаются в рамках ТКП для объяснения предпоч-
предпочтительности того или иного типа координации в зависимости от
электронной конфигурации центрального иона. С этой целью вво-
вводится понятие энергии стабилизации кристаллическим полем
(ЭСКП). ЭСКП оценивается по отношению к нерасщепленному
электронному уровню в поле лигандов сферической симметрии
с помощью данных табл. 11.3. Необходимо установить элект-
электронную конфигурацию центрального иона в сильном или слабом
поле лигандов и подсчитать общую энергию электронов.
Результаты таких подсчетов сведены в табл. 11.6.
При использовании результатов табл. 11.6 следует иметь в виду,
что нельзя прямо сравнивать ЭСКП для низкоспиновых и высоко-
428
Таблица 11.6. ЭСКП для комплексов центральных новов с различными элект-
электронными ковфигурацвямя (в едвнвцах Dq) в различных полях лигандов
Центральный ион
элект-
электронная
конфигу-
конфигурация
<Р
d1
<?
<Р
с?
<?
<?
<Г
d°
<?
d10
примеры
Na+, Sc3 +
Ti+, V4+, U4+
Ti2+, V3 +
V2+, Cr3+
CH+, Mn3+
Mn2 + ,Fe3+,Os3 +
Fe2+, Co3+, Ir3+
Co2+, Rh2 +
Ni2+,Pt2+, Au3+
Cu2+, Ag* +
Cu+, Zn2+, Ag+
Октаэдр
слабое
поле
0
-4
-8
-12
-6
0
-4
-8
-12
-6
0
сильное
поле
0
-4
-8
-12
-16
-20
-24
-18
-12
-6
0
Квадрат
слабое
поле
0
-5,14
-10,28
-14,56
-12,28
0
-5,14
-10,28
-14,56
-12,28
0
сильное
поле
0
-5,14
-10,28
-14,56
-19,70
-24,84
-29,12
-26,56
-24,56
-12,28
0
Тетраэдр*
слабое по-
поле
0
-2,67
-5,34
-3,56
-1,78
0
-2,67
-3,56
-3,56
-1,76
0
* Поскольку тетраэдрические комплексы сильных полей неизвестны, данные
расчета для них не приводятся.
спиновых комплексов. В последнем случае нужно учесть, что элект-
электроны центрального иона стремятся занимать отдельные орбитали.
При этом на каждый электрон выигрывается энергия, равная
обменному интегралу Ки (i, j — индексы ?/-орбиталей), частично
компенсирующая или даже превышающая потерю в орбитальной
энергии. Кроме того, необходимо помнить о приближенности ос-
основных положений ТКП, поэтому при пользовании ЭСКП наиболее
важно учитывать не числовые результаты, а тенденции.
Из данных табл. 11.6 ясно, что для if-, ^10-электронных кон-
конфигураций, а также для конфигурации rf5 в случае слабого поля
лигандов ЭСКП равно нулю. Поэтому для комплексов соответству-
соответствующих центральных ионов (см. табл. 11.6) характерны лишь октаэд-
рическая и тетраэдрическая (четыре лиганда) координации, ожида-
ожидаемые на основании теории ОЭПВО. Тетраэдрическую координацию
имеют ионы [FeClJ1-, MnO4", [Cd(NH3L]2+.
Вообще тетраэдрическая координация — относительно редкий
(по сравнению с плоской) случай структуры координационных со-
соединений. Наиболее характерные примеры известны для комплек-
комплексов центральных ионов типа <Г> например [СоСЦ]2 ~. Стабилизация
тетраэдрического координационного узла для ионов понятна из
429
сравнения ЭСКП тетраэдрических систем с октаэдрическими
и плоскими в слабом поле лигандов. Из данных табл. 11.6 вытекает
также особая устойчивость октаэдрической структуры для ионов d6
в сильном поле и плоской для ионов типа сР.
Значения ЭСКП широко привлекаются также для качественной
интерпретации многих свойств координационных соединений: теп-
лот образования, теплот гидратации ионов, констант устойчивости,
реакционной способности.
11.3.6. Более строгое рассмотрение расщепления уровней
для ^"-конфигураций центральных ионов
Если центральные ионы содержат в ^/-оболочке более одного
электрона, необходимо принимать во внимание межэлектронное
взаимодействие, которое в предыдущем рассмотрении явно не учи-
учитывалось. Схема расчета расщеплений энергетических состояний
таких ионов в полях лигандов значительно усложняется и требует
в общем случае теоретико-группового анализа. Отметим наиболее
существенные моменты.
В слабых полях, создаваемых лигандами, можно считать, что
взаимодействие между ^-электронами существенно сильнее, чем их
взаимодействие с заряженными лигандами, которые можно рас-
рассматривать как возмущение. Энергетические состояния системы
^"-электронов можно тогда классифицировать в рамках схемы Рас-
Рассела—Саундерса (см. разд. 3.6.2).
Расчет расщеплений производится так же, как с потенциалом
A1.3), но в качестве волновых функций при вычислениях матричных
элементов A1.9) должны фигурировать функции LS-терма ?(Lf М,
S, Л/у), создаваемые из микросостояннй соответствующего терма
(см. разд. 3.7). Расчеты показывают, что в октаэдрическом и тетра-
эдрическом, а также кубическом электростатических полях лиган-
лигандов термы центрального иона независимо от мультиплетности рас-
расщепляются следующим образом:
A1.11)
Обозначения А, Е, Т и индексы имеют тот же смысл обозначений
степени вырождения и симметрии, что и описанные в разд. 11.3.2.
Прописные буквы используют для классификации симметрии вол-
волновой функции многоэлектронной системы, тогда как строчные а, е,
t применяют для систематизации волновых функций отдельных
430
K2)
7\ \
Co3*
\ <g
\
Co2t
T's
1G)
\
Ni2+ Г
\m
f
u2+
Mn
2+
Fe2'
Рис. 11.7. Расщепление термов основного состояния ионов от Ti3 + до Си* * в октаэд-
рическом поле. Цифра в скобках — степень вырождения уровня
орбиталей. В скобках указана степень вырождения каждого расщеп-
расщепленного уровня.
Термы основных состояний ионов переходных металлов третье-
третьего периода легко устанавливаются до правилам, описанным в разд.
3.7:
D
5D
D
*F 2D
Расположение термов симметрично относительно 5-терма d5-
конфигурации. Диаграммы расщепления уровней термов по схеме
A1.11) также симметричны относительно терма й?5-конфигурации
(рис. 11.7).
Конфигурациям dx и d9 соответствуют эквивалентные диаграм-
диаграммы расщепления, но с обращенным порядком энергетических состо-
состояний Eg и T2g*.
Подобные аргументы справедливы также для сР~ и <Р~, d3- и d1-,
d*- и ^-конфигураций. В случае слабого поля выполняется также
еще одно правило дополнительности: d" и tf~* имеют одинаковые
расщепления, но с обращенным порядком состояний. Таким об-
образом, появляются дополнительные пары: d1 и d*, d2 и d3. В итоге
в случае слабых полей лигандов требуются расчеты только для d1-
и ^-конфигураций. Диаграмма расщепления основного терма d1-
*Подчеркнем, что речь идет не об обращении порядка t^- и ^-орбиталей
^электронов. Для ^-конфигурации в октаэдрическом поле заполнение этих ор-
орбиталей соответствует данным в табл. 11.5. Но волновая функция девятиэлектронной
системы, записанная в виде соответствующего определителя Слэтера, при таком
заполнении относится х представлению Еш (двукратное вырождение).
431
Возрастание dq ВозрастаниеBq
Рис. 11.8. Диаграмма Оргела для ко-
комплексов d2, <Р, а1 и еР
Возрастание D q Возрастание Hq
Рис. 11.9. Диаграмма Оргела для
комплексов ef1, <Л <г и tt*
и ^-конфигураций в слабых октаэдрическом и тетраэдрическом
полях лигандов показана на рис. 11.8, 11.9.
Для тетраэдрического поля лигандов порядок расположения
расщепленных состояний каждого терма обращен по сравнению
с октаэдрическим полем, поэтому диаграммы расщепления на рис.
11.8 и 11.9, называемые диаграммами Оргела, исчерпывают все
возможности для ^—^-конфигураций центральных ионов в тетра-
тетраэдрическом и октаэдрическом полях лигандов. Кроме того, показа-
показано расщепление лишь термов основных состояний, которое, как
можно видеть, возрастает с увеличением силы поля лигандов. В об-
общем случае, пользуясь схемой составления термов многоэлектрон-
многоэлектронных атомов из микросостояний и определив термы возбужденных
состояний, можно затем по правилам A1.11) получить, учитывая
условия дополнительности, полные диаграммы расщеплений. Зна-
Знание их особенно важно для интерпретации электронных спектров
поглощения. Так, из схемы расщепления на рис. 11.8 следует, что
для октаэдрических комплексов №2+(^) в длинноволновой об-
области поглощения возможны три разрешенных правилами отбора
(Д?=0, AL= ± 1) электронных перехода:
3^-37V ъАъ-+*Тъ(Г) и 3A2g^Tlg(P).
Действительно, как видно из рис. 11.10, в области d—d-переходов
октаэдрических комплексов никеля четко проявляются три полосы
поглощения, относимые к названным электронным переходам.
В случае сильных полей лигандов расщепление {/-уровней
многоэлектронных ионов нельзя описать с помощью диаграмм (см.
рис. 11.8 и 11.9). В сильных полях лигандов взаимодействие d-
электронов с лигандами превышает электростатическое взаимодей-
взаимодействие между rf-электронами. Следовательно, /,5-связь нарушается
и каждый rf-электрон выбирает ту орбиталь, на которой его оттал-
432
25000
:, CM"
tfbiiQ WWQ
8000
12
10
8
6
VV"
kOO 600 800 WOO 1200
Рис. 11.10. Молярный коэффициент эк:
стивкции в для некоторых комплексов Ni
(II) в растворе нитрометана:
/-Ni(NH3K(Cio4J;
2 -
кивание от отрицательных заря-
зарядов лигандов минимально. По-
Поскольку на каждой орбитали не
может быть более двух электро-
электронов, схема заполнения ^/-орбита-
лей электронами в сильном поле
лигандов соответствует предста-
представленной в табл. 11.5.
Наиболее общий случай —
так называемые произволь-
произвольные поля лигандов, промежу-
промежуточные между слабыми и силь-
сильными. Методы расчета расщеп-
расщеплений уровней при этом еще бо-
более усложняются. Я. Танабе
и С. Сугано A954) выполнили
расчеты расщеплений в произ-
произвольных полях для различных
йГ-конфигу раций и представили
результаты расчетов в виде диаграмм зависимости расщепления
термов от силы поля (&~lODq). Эти диаграммы приведены во
многих справочниках и монографиях по химии координационных
соединений. Как и диаграммы Оргела, они весьма полезны при
интерпретации спектров поглощения.
11.4. ПРИМЕНЕНИЕ ТЕОРИИ МОЛЕКУЛЯРНЫХ ОРБИТ АЛЕЙ
ДЛЯ ОПИСАНИЯ ЭЛЕКТРОННОГО СТРОЕНИЯ
КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ
Теория кристаллического поля (ТКП) — чрезвычайно ценная
модель описания свойств координационных соединений, которая
основана на рассмотрении орбитальной структуры только одного
центрального атома (иона) координационного соединения и не учи-
учитывает орбитали лиганда.
В связи с этим ТКП в принципе не может учесть ряда важных
эффектов, определяющих природу химической связи в координаци-
координационных соединениях. Так, ТКП неприложима к я-комллексам с мно-
многоцентровыми орбиталями лигандов, в ТКП не укладываются пред-
представления о дативной связи, обусловленной донированием элект-
электронной плотности й?-электронных пар на вакантные орбитали лиган-
лиганда (связь, аналогичная донорно-акцепторной и играющая сущест-
существенную роль при образовании некоторых комплексов сильных по-
полей лигандов, например цианидных). Вообще все характеристики
комплекса, проявляющиеся в поведении лиганда (необычные реак-
реакции координированных лигандов, перераспределение плотности не-
спаренных электронов в парамагнитных комплексах по орбиталям
433
Рис. 11.11. а-Орбиталн лиган-
дов L(NH3, Н2О, РМе3) октаэд-
рического комплекса MLJ+
(*0 1)
лиганда, полосы металл-лигандных пе-
переходов в электронных спектрах и др.),
остаются вне поля зрения ТКП.
Наиболее общий подход к рассмот-
рассмотрению электронной структуры компле-
комплексов связан с расчетами полных волно-
волновых функций комплекса как целого (а
не только центрального иона по мето-
методу МО). В области координационных
соединений обобщения, полученные на
основе метода МО, названы теорией
поля лигандов. Главная особенность ее
состоит в том, что ввиду обычно высо-
высокой симметрии координационного узла
ус=и, 1,...) мо молекулы или иона координацион-
координационного соединения классифицируются по
представлениям симметрии и принципиальную схему их образова-
образования из орбиталей лигандов можно зачастую построить, не проводя
конкретных расчетов. tj
11.4.1. МО координационных соединений с лигандами,
имеющими «т-орбитали
Рассмотрим вначале наиболее простой, но весьма распростра-
распространенный случай комплексов, образуемых лигандами L типа (СНэ)эР,
NH3, Н2О, ОН", Н~, которые используют для связывания с цент-
центральным атомом металла неподеленную гибридную пару электро-
электронов или пару электронов на s-орбитали (гибрид-ион). Начнем рас-
рассмотрение с наиболее характерного типа координации — октаэд-
рического. Координационные связи в комплексе MLj+, где М — пе-
переходный металл, образуются при донировании электронов с а-
Рис. 11. IX Образование «Г-МО комплекса из dx*-y*-AO металла и групп новое
орбитали (о\ — а2+0з—<*а) (?)', отсутствие перекрывания d^-KO металла с орбиталя-
ми лигандов {б)
434
орбиталей лигандов на вакантные 3d-, 4s-, 4/м>рбитали металла
(возьмем атом металла третьего периода). Чтобы рассчитать ва-
валентные МО комплекса, выберем координатные оси, как показано
на рис. 11.11, расположив вдоль них лиганды.
Полный базис валентных АО состоит из 15 орбиталей: девяти —
металла, шести — лигандов. Только такие комбинации лигандных
АО будут обобщаться в форме МО с различными орбиталями
металла, которые преобразуются по одинаковым представлениям
симметрии в точечной группе О* (см. разд. 6.3). Нетрудно подо-
подобрать соответствующие комбинации <т-АО лигандов (называемые
групповыми орбиталями). Они представлены в табл. 11.7.
Таблица 11.7. Валентные орбнталн металла н грушювые орбиталш лигандов
в октаэдрическом комплексе ML*+ (связывающие комбинации)
Металл
4s
4ру
3dz> C^_^_у)
3dsy. V» 3dyz
1
1
1
1
72(О5"'6)
1
— { G\ ff2
J\2
1
2 ' 2 3
Лнганд
( + ff4 + ff5 + ff6)
-а3~(Т4 + 2а5 + 2(т6)
Представле-
Представление симмет-
рки
aig
Рис. 11.12, а иллюстрирует выбор групповой орбитали е^-сим-
метрии, комбинирующейся с г^.^-орбиталью металла. Из рис.
11.12, б ясно, почему любая комбинация с-АО лигандов дает нуле-
нулевое перекрывание с ^-орбиталями металла типа t2g.
На рис. 11.13 дана корреляционная диаграмма МО октаэдричес-
кого комплекса. Она строится исходя из правил качественной те-
435
м
Рис. 11.13. Корреляционная диаграмма МО октаэдрического комплекса ML*+,- по-
построенных в валентных орбиталях металла и групповых орбиталях лиг&ндов: отдель-
отдельно выделены групповые орбитали лигандов Од-симметрии
ории МО (см. гл. 9). Имеются также многочисленные прямые
расчеты электронного строения разнообразных комплексов МЦ+,
вьшолненные в различных вариантах приближений, которые дают
согласующуюся с диаграммой картину энергетических уровней.
Из диаграммы (см. рис. 11.13) следует, что в комплексе, об-
образованном <7-лигандами и центральным атомом переходного ме-
металла, имеется шесть связывающих {aig, tiu, \eg) и три несвязыва-
ющих (*2f) валентных электронных уровня, на которых можно раз-
разместить 18 электронов. Если каждый из шести лигандов октаэд-
октаэдрического комплекса вносит по 2 сг-электрона, это означает, что
436
устойчивой конфигурацией центрального иона или атома будет
6-электронная.
В согласии с ТКП граничными орбиталями комплекса являются
орбитали t^- и 2е,-типа. Однако если /^-орбитали в случае комплек-
комплексов с сг-лигандами действительно представляют собой локализован-
локализованные ^-орбитали центрального атома, то орбитали 2eg делокализова-
ны и содержат вклад орбиталей лиганда (см. рис. 11.13). Поскольку
эти орбитали имеют антисвязывающий характер, их заселение ведет
к разрыхлению связей М—L. Так, для низкоспинового комплекса
[Co(NH3)J3+, в котором 2е^-орбитали не заселены, длины связей
Со—N равны 0,194 нм, тогда как в высокоспиновом комплексе
[Co(NH3)<j]2+, где эти орбитали заполняются, длины связей Со—N
увеличиваются до 0,211 нм.
11.4.2. МО координационных соединений
с лигаидами, имеющими р- и я-орбитали
В более общем случае, чем рассмотренный выше, лиганды L,
например галогены, могут образовывать связи с центральным ио-
ионом также за счет своих р-АО, оси которых ориентированы перпен-
перпендикулярно связям М—L. При этом создается возможность я-связы-
вания двух типов. На рис. 11.14, а — в показаны два вида групповых
я-орбиталей лигандов. Групповые орбитали первого типа способны
а б в
Рис. 11.14. Общая и локальная системы координат для октаэдрического комплекса
MLJ+:
<х-ороитали лигандов направляются по локальным z-осям, орбитали it-типа — по осям х и у (а);
п-орбиталь на основе р-АО металла и р-АО лигандов (б); одна из трех tjg-rpyunobbix. орбиталей
лигандов -
), способная к перекрыванию с dxy-AO (tig) металла (*)
437
Рис. 11.15. t2g-M0 Сг(СО)б, обеспе-
обеспечивающая дативную связь М->С при
донировании электронов с dxy-AO Сг
на я*-МО С==О
образовать тс-МО комплекса при
перекрывании с р-АО металла.
Групповые орбитали лигандов (см.
рис. 11.14, в) способны к перекры-
перекрыванию с t^-AQ металла. Учет п-
связывания приводит к включению
этих орбиталей в комплексах типа
[CoF6]*~ и других в связывающие
и антисвязывающие комбинации
с орбиталями лигандов, тогда как
в комплексах типа [Co(NH3)d3+
/^-орбитали имели несвязыва-
ющий характер. Именно этот эф-
эффект объясняет возможность обра-
образования дативных связей металла
с лигандами, обусловливающих передачу электронной плотности
с заполненных /^-АО металла на вакантные р-, а также я*-орбитали
лиганда.
Другая особенность образования связей центрального атома
металла с лигандами, обладающими /ьорбиталями, состоит в том,
что последние образуют отдельную систему /?я-орбиталей ^-сим-
^-симметрии. Они вместе с сг-орбиталями той же симметрии могут вза-
взаимодействовать с р-АО (tiu) центрального атома металла. Посколь-
Поскольку ст-перекрывание намного больше, чем я-перекрывание, можно
в первом приближении пренебречь я-взаимодействием и относить
/7*-ряд лигандных орбиталей t{„симметрии к несвязывающему типу.
Это дает возможность рассматривать комплексы с заполненными
электронами />-орбиталями (галогены) в рамках схемы (см. рис.
11.13).
Задача 11.9. Составьте групповые орбитали и энергетическую диаграмму
МО квадратного комплекса ML4 типа [PtCJ^2". Учтите 3s- в Зр~АО лиганда.
Наиболее важен эффект обобществления d-AO центрального
атома металла с вакантными л*-орбиталями таких лигандов
(С == О, С s N) в карбонильных и цианидных комплексах. На рис.
11.15 показана структура одной из t^-MO гексакарбонила хрома,
образующейся при перекрывании dxy-AO металла с антисвязыва-
ющими я*-МО оксида углерода. На рис. 11.16 дана корреляционная
диаграмма МО гексакарбонила хрома, построенная с учетом я-
и п*-орбиталей лиганда.
438
Cr
Cr(C0N
(C0)t
Рис. 11.16. Корреляционная диаграмма МО октаэдрического гексакарбонила хрома*,
все 21 валентных МО комплекса, вжлючая 1ц, заполнены электронами
Для большей ясности «т-орбитали различных типов (см. рис.
11.13) даны в виде одного общего уровня. Расщепления, вызыва-
вызываемые взаимодействиями АО металла с я-орбиталями лигандов,
которые можно скомбинировать в групповые орбитали, пользуясь
правилами теории групп, сравнительно малы. Как и в случае лиган-
лигандов с /г-орбиталями, можно рассматривать эти взаимодействия (за
исключением /^—гс-взаимодействия) как второстепенные по сравне-
сравнению с взаимодействиями d—сг-типа. В то же время эффект приме-
примешивания 7г*-орбиталей лиганда к орбиталям металла /^-ряда играет
важную роль, он приводит к увеличению расщепления t2g—е^-уров-
ней. Шесть СО-групп гексакарбонила хрома вносят 12 электронов
неподеленных электронных пар, 24 я-электрона, а атом хрома дает
439
шесть электронов, т. е. всего имеется 42 электрона, заполняющих
21 связывающие МО (см. рис. 11,16).
11.4.3. я-Комплексы м металлоцены
Особенная и чрезвычайно широкая область координационных
соединений металлов представлена я-комплексами — соединения-
соединениями, в которых центральный атом образует многоцентровые связи
с лигандами в результате обобществления своих */-орбиталей с де-
локализованными я-орбиталями лигандов. Примерами я-комплек-
сов служат соединения VIII—X, в которых в качестве лигандов
выступают циклические сопряженные системы. Прототипом я-ком-
плексов являются более простые алкеновые комплексы металлов,
первый представитель которых — хлороплатинат К [С2Н4 ¦ PtCl3] —
в виде кристаллогидрата был получен В. Цейзе еще в 1827 г. при
кипячении в этиловом спирте платинохлористоводороднои кислоты
с последующим добавлением к раствору КС1.
Природа связи в соли Цейзе XXV объясняется так называемой
синергической моделью Дьюара—Чатта—Дункансона A953), близ-
близкой по своей основной идее к описанию связывания в гексакар-
бониле хрома:
XXV XXVI а XXVI б
Электронная плотность с высшей заполненной я-МО этилена
переносится на вакантную (spn) орбиталь металла (прямое донирова-
ние XXVIa/ Упрочнение связи достигается также за счет эффекта
обратного допирования (дативной связи) — частичного переноса
электронов с заполненной б?-орбитали металла на вакантную я*-МО
этилена. Строгие расчеты хорошо согласуются с общей схемой
XXVI формирования связи в я-комплексе XXV и других подобных
ему структурах. Отвлечение электронной плотности со связыва-
связывающей и частичное заселение антисвязывающей орбиталей этилено-
этиленового фрагмента в XXV должно приводить к разрыхлению связи
С=С в комплексе. Действительно, если частота валентного колеба-
колебания этой связи в молекуле этилена равна 1623 см, а длина связи
0,1337' нм, то в соли Цейзе частота колебания связи понижается до
1511 см , а длина связи возрастает до 0,1354 нм. Еще более
значительные изменения происходят в этиленовых я-комплексах:
440
/cc = 0,143 нм
XXVII
Fc(COL
* ^
c = 0,146 нм
XXVIH
Учитывая типы взаимодействий XXVIa, б, определяющих свя-
связывание в я-комплексах, можно подойти к решению вопроса о кон-
формационной предпочтительности я-комплексов. Например, для
железокарбонильного комплекса XXVIII можно представить две
альтернативные конформации, учитывая бисфеноидную геометрию
фрагмента Fe (COL и предпочтительность экваториального положе-
положения алкена:
XXVIlla
XXVIII6
На рис. 11.17 показана диаграмма орбитальных взаимодействий
фрагментов для двух рассмотренных ориентации (орбитали
Fe (СОХ —~ заселенность центрального атома c^s2 = сР — взяты из
рис. 9.12). Поскольку октаэдрическая симметрия уже не сохраняет-
сохраняется, /2*-ряд соответствующим образом расщеплен.
Можно видеть, что взаимодействия, реализующие связь ме-
металл—лиганд, отвечают схеме Дьюара—Чатта-т—Дункансона. Одна-
Однако оба типа связывающих эффектов — прямое и обратное дониро-
вание — возможны только в конформации XXVIII а, тогда как в ко-
конформации XXVIII6, отличающейся поворотом одного из фрагмен-
фрагментов в экваториальной плоскости на 90°, исчезает d-*>n* (Ь2~*п*)-
перекрывание. Это приводит к отсутствию обратного донирования,
ослабляет связывание и дестабилизирует конформацию XXVIIIб.
По данным спектроскопии ЯМР, энергетический барьер для враще-
вращения фрагмента Fe (COL в XXVIlla, связанный с прохождением через
XXVIII6, составляет 42—65 кДж/моль.
Аналогичная схема связывания осуществляется и в я-комплек-
я-комплексах, образуемых более сложными сопряженными алкенами, таких,
как, например, металлкарбонильные комплексы XXIX—XXXII:
Fe(COK Fe(COK Co(COK Сг(СОK
XXIX
XXX
XXXI
хххн
441
r\-), J
я
Рис. 11.17. Реконструкционный анализ МО я-комплексов XXVIII в двух конфор-
мациях с эквивалентным расположением этилена согласно Р. Хоффманну и др.
A978), демонстрирующий предпочтительность конформации XXVIIIa
Рассмотрим схему возникновения связывания фрагментов в ком-
комплексе XXIX. Железотрикарбонильный фрагмент, имеющий сим-
симметрию С3у, обладает тремя валентными орбиталями (см. рис. 9.11),
образующими подходящие по симметрии комбинации с л-орбита-
лями 1,3-бутадиена. Формы этих орбиталей были приведены выше
(см. разд. 9.4.4):
442
I
Рис. 11.18. Реконструкционный анализ МО тс-комплекса С4Нб Fe(C0K XXIX
Рис. 11.18 показывает схему орбитальных взаимодействий фраг-
фрагментов. Можно видеть, что, как и в случае комплексообразования
с этиленом, основной вклад в связывание вносят граничные \па-
и 2я*-М0 алкена, хотя характер их перекрывания с частично запол-
заполненными е- и вакантной агорбиталями Fe(COK несколько отлича-
отличается от схемы XXVI.
443
Задача 11.10. Построить МО п-комплекса XXX из орбиталей фрагментов.
Установите, используя соотношения изолобальности, соответствие этого комп-
комплекса различным органическим структурам.
Особое внимание в ряду ^-комплексов привлекли так называ-
называемые металлоцены, или соединения с сэндвичевой структурой. Пер-
Первым известным металлоценом, полученным в 1951 г. Т. Кили
и П. Посоном, стал ферроцен XXXIII (M-Fe), послуживший прото-
прототипом для многих синтезированных впоследствии его аналогов.
Аналогичное сэндвичевое строение было доказано и для хромоце-
на — дибензолхрома XXXIV и его аналогов. В табл. 11.8 представ-
представлены данные по электронному строению основных соединений этих
типов:
XXXIII
XXXIV
Таблица 11.8. Электронные конфигурация основных состояний я иервые иотеяцн-
алы ионизация G) некоторых металлоценов тниа XXX] 11 н XXXIV
XXXIII, M
Fe
V
Сг
Mn
Ru
Co
Ni
XXXIV, M
Ti
Cr
Mo
w
V*
Электронная конфигурация
Ulf- (aigJ (e4f
*A{g-(e^J(alg)i
3E2g-(e2gK(algI
6Aig~(e2gK(algJ
1Aig—(aigJ(e2g)*'
2E\g— (a\g) 2 (e2g)* (e\g) *
A\%~(a\%) (&2g) (e\t)
Электронная конфигурация
lAlt-(tb)*(*\tJ
1Alg~(e2g)*(algJ
A\g—f^2g) {&lg)
A\g— (eig) (a\g)
I, эВ
6,88
6,78
5,71
6,91
8,51
5,56
6,51
/, эВ
5,50
5,45
5,52
5,40
5,33
* 1, 3, 5, 1', 3', 5'-гексаметильное производное.
444
Объяснение природы связывания в молекуле ферроцена было
дано впервые М. Дяткиной и Е. Шусторовичем A959). Было пока-
показано, что только тг-орбитали лигандов играют существенную роль
в стабилизации сэндвичевой структуры комплекса. Центросиммет-
ричные металлоцены XXXIII, к которым принадлежит ферроцен,
относятся к точечной группе D^, и групповые орбитали двух цик-
лопентадиенильных лигандов можно классифицировать по пред-
представлениям aXg, e\g, eig, аги, eWt е1и (см. разд. 6.2):
Ф
—г
Ф-
[()()] A1.12)
e2g: ^=-y=
е2и: %=
Ф
Представим вид я-МО колец, получаемый по правилам (см.
разд. 8.1.3):
6 6 6 6
6* -6- -6- -6
+ +
f(e2b)
445
efg, С/и
ia
C2g>e2u
Ферроцен
M
4 ибензоц SHoxpOM
'6h
Рис. 11.19. Схема образования МО в двух типах металлоценовых комплексов
XXXIII и XXXIV
Групповые орбитали колец комбинируют с орбиталями металла
соответствующей симметрии:
а\к\ As,
\ 4рх, 4ру.
На рис. 11.19 показаны происхождение и последовательность
валентных энергетических уровней в ферроцене XXXIII (M = Fe),
а также в дибензолхроме XXXIV (М = Сг), получаемые на основа-
основании теории орбитальных взаимодействий и подтвержденные данны-
данными многочисленных полуэмпирических и неэмпирических расчетов.
446
Рис. 11.20. Схематический вид высших занятых связывающих МО молекулы фер-
ферроцена (см. рис. 11.19)
Восемнадцать валентных электронов ферроцена (восемь от цент-
центрального атома d6sz) заполняют все нижние уровни вплоть до
Связи между металлом и кольцами обусловливаются МО alg и elg,
образующимися за счет перекрывания ф, dyl и dxz с л-орбиталями
циклопентадиенильных колец, при этом происходит перенос заряда
с л-МО колец на ф, dy: и 4рх-, 4ру-АО железа и встречный перенос
с ф, Ф-/ и dxy на п*-МО циклопентадиенилов, в результате воз-
возникает распределение
Вид некоторых МО ферроцена показан на рис. 11.20.
Электронное строение других металлоценов XXXIII и XXXIV
описывается той же схемой МО, что и представленная на рис. 11.19.
Это вытекает из данных табл. 11.8, отражающей результаты стро-
строгих расчетов и экспериментальных исследований. Дополнительные
электроны в кобальтоцене и никелецене XXXIII (М = Со, Ni) заселя-
заселяют антисвязывающие МО е\„, что обусловливает парамагнетизм их
молекул. В кобальтоцене ХХХШ верхняя вырожденная е!г-МО
занята одним электроном. Молекула имеет вырожденное основное
электронное состояние (см. табл. 11.8), что приводит к проявлению
эффекта Яна—Теллера (см. разд. 5.7) в этом соединении. Как пока-
показано электронно-дифракционными исследованиями, эффект Яна—
Теллера для этого соединения имеет динамический характер, выра-
выражающийся в очень быстрой флуктуации относительно усредненной
центросимметричной структуры XXXIII.
Тип связывания, реализованный в металлоценах XXXIII,
XXXIV и обусловленный d—л-взаимодействиями, осуществляется
и для более сложных ценовых, так называемых трехпалубных струк-
структур, например стабильных соединений X, XXXV—XXXVII:
447
R В:
XXXV
XXXVI
XXXVII
Аналогичный характер имеют орбитальные взаимодействия,
определяющие устойчивость сэндвичевых комплексов лантаноидов
и актиноидов. Поскольку для этих элементов активными валент-
валентными орбиталями являются орбитали /-типа с квантовым числом
/—3, подходящие по симметрии я-орбитали циклического лиганда
должны обладать уже не одной узловой плоскостью (как ех- и е2-МО
циклопентадиена, комбинирующие с rf-AO металла), а двумя. Это
возможно лишь для циклических полиенов больших размеров, на-
например для циклооктатетраена. Действительно, ураноцен XXXVII,
как и другие быс-(циклооктатетраенилъные) комплексы актиноидов,
весьма устойчив. На рис. 11.21 показан схематический вид одной из
связывающих МО ураноцена — комбинации 5fxyz-AO урана с анти-
связывающей е2-МО циклооктатетраена.
11.4.4. Правило 18 электронов
Анализ корреляционной диаграммы МО типового октаэдричес-
кого комплекса ML6, в котором М — атом переходного металла
(см. рис. 11.13), показывает, что в комплексе имеется девять низ-
колежащих валентных МО (шесть связывающих и три несвязыва-
ющих), которые могут быть заселены 18 электронами. Диаграмма
на рис. 11.13 относится к простейшему случаю лигандов L, образу-
образующих двухцентровые двухэлектронные связи. Как и в случае а-
лигандов с р- и я-орбиталями (например, С1~, СО, ...), именно
двухцентровые двухэлектронные связи М—L определяют общую
стабильность комплекса и можно ограничиться подсчетом электро-
электронов только на орбиталях этих связей. Из ряс. 11.16 вытекает, что
число электронов на таких орбиталях вместе с электронами на
несвязывающих ^/-орбиталях металла также равно 18. Можно, та-
таким образом, подойти к формулировке общего, весьма важного
в химии координационных соединений правила 18 электронов:
448
в устойчивых комплексах переходных металлов ML,
общее число электронов на связях М—L и несвязыва-
ющих электронов металла равно 18.
Это правило можно трактовать как стремление цент-
центрального атома металла иметь замкнутую электронную
оболочку соответствующего атома инертного газа. Мо-
Можно представить и другое общее объяснение, позволя-
позволяющее одновременно предсказать важные исключения из
данного правила. На рис. 11.22, а приведена обобщенная
диаграмма орбитальных взаимодействий валентных р-,
s-t ti-орбиталей (общее число которых равно 9) с и сим-
мстризованными <т-орбиталями лигандов L в комплексе
MLn. Подходящие по симметрии л-орбитали централь-
центрального атома (можно рассматривать их как соответст-
соответствующие cf, /, //-гибридные орбитали) образуют с ор-
биталями лиганда п связывающих и п антисвязывающих
МО, а (9 — п) d-орбиталей металла, имеющих отличную
симметрию, образуют несвязывающий уровень. Очеви-
Очевидно, что на связывающих и несвязывающих уровнях
можно разместить 18 электронов.
При практическом использовании правила 18 электронов прене-
пренебрегают всеми другими орбитальными взаимодействиями, кроме
сг-связывания, и каждый лиганд рассматривается как вносящий два
электрона в валентную оболочку комплекса. Таким образом, объ-
объединяются и обычные cr-лиганды (NR3, PR3, Н2О, СН3~), и о-лиганды
с />->г/-донорным эффектом (С1~, ОН~), и сг-лиганды с г/->л:*-акцеп-
торными свойствами (СО, CNR, CR2, NO+). Кроме того, я-лиганды
рассматриваются аналогичным образом как льюисовские основа-
основания, причем число вносимых ими электронов приравнивается к чис-
числу электронов на связывающих и несвязывающих я-МО. Это число
(ниже выбрана электронейтральная форма) можно оценить соглас-
согласно энергетической диаграмме на рис. 8.3:
Рис, 11.21.
Иллюстра-
Иллюстрация связы-
вающего
взаимодей-
взаимодействия /АО
центрально-
центрального атома
урана с я-
МО двух
(8]-аннуле-
нов в моле-
молекуле урано-
цена
A1-13)
Электронный
счет 2344 5 6 7 8
Для металла подсчитываются все электроны на s-, p~, rf-оболоч-
ках и учитывается общий заряд комплекса.
Нетрудно проверить выполнение правила 18 электронов для
большинства рассмотренных выше я-комплексов, например фер-
ферроцена XXXIII (M = Fe), хромоцена XXXIV (М = Сг). В отличие от
последнего в валентной оболочке рутеноцена в структуре D6*-chm-
15. Теория строения молекул
449
метрии IX = XXXIV (M = Ru) содержалось бы не 18, а 20 электро-
электронов (Ru—Л2, 8 электронов и по 6 электронов от каждого
бензольного кольца). Чтобы иметь в валентной оболочке 18
электронов, структура рутеноцена должна быть искажена таким
образом, чтобы одно из бензольных колец участвовало в связыва-
связывании лишь двумя я-связями. Именно такая структура XXXVIII
найдена для перметильного производного рутеноцена. В отличие от
неполярного симметричного яромоцена IX молекула XXXVIII
характеризуется достаточно высоким дннольным моментом 2,03
D в растворе. Другой аналогичный пример — искажение пятичлен-
ного циклопентадиенового кольца в сэндвичевом вольфрамдикар-
бонильном комплексе XXXIX, позволяющее вывести одну я-связь
(два электрона) из общего связывания. Атом вольфрама вносит
шесть, два карбонильных лиганда — четыре, плоский циклопен-
тадиенильный фрагмент — пять, а деформированный — три элект-
электрона:
н
XXXVIII
XXXIX
Необычная структура карбонила кобальта Со2(СО)8 VII также
объясняется тем, что в ней достигается 18-электронная конфигура-
конфигурация валентной оболочки. Мостиковые карбонильные группы об-
образуют многоцентровые связи, при формальном рассмотрении они
отдают по одному электрону на оболочку каждого атома кобальта.
Диамагнетизм Со2(СО)8 свидетельствует о спаривании электронов
кобальта и образовании связи Со—Со. Действительно, расстояние
Со—Со составляет, по данным рентгеноструктурных исследований,
всего 0,25 им. Интересно отметить, что в растворе структура VII
находится в равновесии с изомерной ей структурой XL, также
согласующейся с правилом 18 электронов, если два электрона на
а-связн Со—Со XLI приписывать одновременно каждому атому
кобальта. Рассуждая аналогичным образом, в комплексе XLJI сле-
следует ожидать наличия двойной связи Rh=Rh:
450
OS3C—Co
XL
XL!
XUI
Задача 11.11. На основании правила 18 электронов предсжажитс
туры: 1) Fez (СО)» 2) (к-С^ЪС^ОО^ 3) Mn3(COI0; 4) (ОЩЪ
Я
Правило 18 электронов имеет немало исключений, и его следует
рассматривать только как один из факторов, способствующих об-
образованию стабильной структуры координационного соединения.
Отклонения от правила связаны часто с пространственными ограни-
ограничениями, не допускающими координации центральным атомом не-
необходимого для заполнения 18-электронной оболочкой числа ли-
гандов. Например, ясно, что ион V3 f (cP) должен координировать
восемь двухэлектронных лигандов, чтобы заполнить валентную
оболочку полностью. Однако пространственные возможности до-
допускают только октаэдрическую координацию. Другие примеры
отклонения от правила 18 электронов можно найти в табл. 11.8, где
встречаются и 15-электронный ванадоцен, и 20-электронный никеле-
цен. Эти соединения в отличие от 18-электронного ферроцена от-
отличаются малой устойчивостью и высокой реакционной способ-
способностью. Тем не менее они способны к существованию и отвечают
минимумам на соответствующих поверхностях потенциальной
энергии.
Особенно важным отклонением от требований правила 18 элект-
электронов являются плоскоквадратные тетракоординированные и плос-
плоские трикоординированные комплексы. Как следует из схемы ор-
орбитальных взаимодействий, приведенной на рис. 11.22, а—в, в обоих
этих случаях pz-AO центрального атома остается отключенной от
связывания с лигандами, так как ее узловая плоскость совпадает
с плоскостью, в которой располагаются оси <т-орбиталей лигандов
L (см. схему XVI <&р2-гибридизации орбиталей центрального атома
в плоскоквадратных комплексах). Из рисунка следует, что при этом
образуется в сумме только восемь связывающих и несвязывающих
орбиталей комплекса, на которых могут разместиться лишь 16
электронов. Следовательно, в случае плоских тетра- и трико-
ординированных структур переходных металлов устой-
устойчивой является 16-электронная конфигурация.
451
ПМ'1
антисОяз.
р SS
шантисбяз.
U несбяз
5нес6яз
U м-1Сдяз
a MLn
L— М— I
\
L
L
Рис. 11.22. Обобщенная диаграмма орбитальных взаимодействий в комплексе МЬ„
(выбрано л = 6), объясняющая правило 18 электронов (а); диаграммы для плоских
комплексов ML4, ML3, объясняющие устойчивость их 16-электронных конфигураций
(б, в)
Действительно, рассмотрим электронную конфигурацию цент-
центрального атома Pt в соли Цейзе XXV. Этиленовый л-лиганд и три
сг-лиганда С1~ дают каждый по два электрона, т. е. всего восемь
электронов, в валентную оболочку. Учитывая заряд комплексного
аниона —1, заряд центрального иона (или число окисления цент-
центрального атома) определяют как — 1— (—3)=+2. Ион Pt2+(dr*) дает
в валентную оболочку восемь электронов. Общая сумма валентных
электронов, определяемая таким образом, равна 16(8 + 8). Шестнад-
Шестнадцать электронов содержатся также в валентных оболочках плоских
трикоординированных комплексов [Fe (SiNMe2K], XXVII, и др.
Задача 11.12. Какая электронная конфигурация (число электронов) должна
быть наиболее устойчивой в случае линейных комплексов переходных металлов
типа ML.2?
452
11.5. ДЕФОРМАЦИИ КООРДИНАЦИОННЫХ ПОЛИЭДРОВ
И ЭФФЕКТЫ ЯНА—ГЕЛЛЕРА
Высокая симметрия координационных полиэдров обусловливает
вырожденность электронных термов для многих комплексных со-
соединений и их структурные деформации, вызванные эффектами
Яна—Геллера. Проявления этих эффектов могут носить как стати-
статический характер — стабилизация структуры пониженной симмет-
симметрии, так и динамический, когда искажение сравнительно мало и при-
приводит к структуре, занимающей неглубокий минимум на ПЙЭ
системы. Такие структуры претерпевают быстрые перегруппировки
между несколькими эквивалентными ядерными конфигурациями,
т. е. находятся в состоянии вырожденного динамического равнове-
равновесия. Статический или динамический характер искажения Яна—Тел-
лера зависит от величины интеграла т01 ^тг т«»"г в выражении
E.38). Если его величина значительна и обеспечивает достаточное
уменьшение энергии молекулы или иона при понижении их симмет-
симметрии, соответствующее структурное искажение имеет статический
характер.
Примером статического эффекта Яна—Теллера служат октаэд-
рические комплексы иона Си2+. Девять ^-электронов центрального
иона помещаются на орбиталях tlg- и ей-типа двумя равноценными
способами (рис. 11.23). Основное электронное состояние комплексов
Си2 + в октаэдрическом окружении @л-симметрия) является дважды
вырожденным — ?^-терм.
Активными для проявления эффекта Яна—Теллера первого по-
порядка являются смещения по типу колебания ^-симметрии:
Eg х Eg=Alg+Eg, — приводящие либо к удлинению, либо к сжатию
октаэдра — так называемое тетрагональное искажение (рис. 11.24).
Направление искажения октаэдрической структуры диктуется
формой е^-колебаний октаэдра,
но оно может быть легко понято ^тЬ "jr- 7J~ ? d 2
j 7J ? d
просто из рассмотрения элект- *г-у2 1 * ~у z
йй
рто и ррен
ростатических взаимодействий. -fy- -Ц- -Jf- -Н" ~Н~
Так, если в электронной конфи- г2% tzg
гурации на рис. 11.23 элект- а 6
ровная дырка будет находиться ^ ,]а д^™* ящожпевве ^
на а^^-ОрОИТали, ЛИГанды новного электронного состояния октаэд-
В положениях I, 2, 3, 4 (СМ. рис. Ричеосого комплекса Cv? + ?Eg):
11.11) будут СИЛЬНее ПрИТЯГИ- а — электронна* конфигурация (t2g)*
ваться К центральному ИОну, ЧТО ^л,_yi)i^l; б — электронная жонфнгура-
приведет к увеличению расстоя- ^ (t2g)^d^(dx,_y^
453
к.-
У\ •
У
Рис. 11.24. Тетрагональное (а) в тригональное (б) насажени» ожтаэдряческих струк-
структур вследствие эффекта Яна—Геллера:
для комплексов с частично заполненными г^орбиталями возможно только тетрагональное
возможны оба типа
искажение; для комплексов с частично заполненными
искажений; триговальное искажение проявляется, например, в комплексе (СО)в
ний М—L5i М—Ц по сравнению с М—L^. Подобное проявление
эффекта Яна—Геллера объясняет тетрагональное искажение в окта-
эдрических окружениях иона меди в кристаллической решетке
(СиС12)д XX и во многих других кристаллических решетках, в кото-
которых ион Си2+ окружен шестью лигандами. Другие примеры стати-
статического эффекта Яна—Геллера также связаны со случаями вырож-
вырождения на е,-орбиталях. Например, структуру координационного уз-
узла высокоспинового комплекса [MnF6]3~ в кристалле MnF3 описы-
описывают как ромбически искаженный вследствие двукратного вырожде-
вырождения (/2;K (^-конфигурации октаэдр с тремя различными длинами
связей Мп—F @,179, 0,191 и 0,209 нм). В т/хшс-ацетилацетонате
марганца две связи Мп—О имеют длину 0Д948 нм, а четыре дру-
другие — 0,20000 нм.
При электронном вырождении, обусловленном заполнением
/^ орбиталей, эффект Яна—Геллера выражен значительно слабее
и носит обычно динамический характер, проявляющийся в упшре-
нии спектральных полос (см. разд. 5.7.1). Так, значительное ушире-
ние полос электронного спектра поглощения в газовой фазе зареги-
зарегистрировано для низкоспинового {<Р) гексакарбонила ванадия
V(CON.
454
Задача 11.13. Объясните, привлекал представление об эффекте Яна—Гел-
Яна—Геллера доя состояний, вырожденных но типу ех, наличие длинноволнового переги-
перегиба в полосе d—^перехода нова [Ti (HjOfc]3* (см. рис 11.6) в области 575 нм.
Задача 11.14. Для каких электронных конфигураций тетраэдрических и ок-
таэдрическях комплексов сильных и слабых полей лигаядов (d1—rf9) можно
ожидать проявления эффекта Яна—Геллера первого порядка?
Берсукер И. Б. Электронное строение и свойства координационных соедине-
соединений. — Л.: Химия, 1986.
Рт.ыа полное углубленное описание освовных положений современной теории строения
координационных соединений и ее приложений к стереохимии, спектроскопии, устойчивости
и реакционной способности.
Дьячков П. Н., Левши А- А. Вибронная теория относительной стабильности
изомеров в неорганических молекулах и комплексах //Итоги науки и техники. Стро-
Строение молекул и химическая связь. Г. 11. — М.: ВИНИТИ, 1987.
Проблема относительной устойчивости изомеров неорганических молекул и комплексов
тнпа MLpCfc рассмотрена как проблема электронно-колебательных взаимодействий с привлече-
привлечением представлений об эффекта» Яна—Геллера.
Оргея Л. Введение в химию переходных металлов. — М.: Мир, 1964.
Последовательное и ясное качественное изложение основ теории кристаллического поля.
Хьмм Дж. Неорганическая химия. Строение вещества и реакционная способ-
способность. — М.: Химия, 1987. Гл. 10, И, 13.
Представлен большой и хорошо систематизированный фактический материал do строению,
изомерии и реакциям координационных и металлоорганических соединений. Изложение базиру-
базируется на использовании теорий кристаллического поля и молекулярных орбиталей.
KristunmnrtBy К., Schaap W. В. — J. Chemical Education, 1969, 46 (N 12), 799;
1970, 47 (N 6), 433.
Подробный раэбор схемы расчета расщепления «/-орбиталей в полях лигандов различной
симметрии и ее применения для объяснения устойчивости и спектральных свойств комплексов
переходных металлов. Для понимания статей достаточно звания материала гл. 1—4, 6, 9—11
настоящей книги.
ГЛАВА 12
СТРУКТУРНО НЕЖЕСТКИЕ МОЛЕКУЛЫ
К структурно иежестким относят такие молекулы (ионы, радика-
радикалы), которые обнаруживают способность к быстрым и обратимым
внутримолекулярным перестройкам, приводящим к динамическому
равновесию нескольких изомерных (топомерных) форм этих моле-
молекул. Поверхности потенциальной энергии (ППЭ) таких молекул
имеют, следовательно, несколько минимумов с одинаковой или
почти одинаковой глубиной, разделенных невысокими перевалами.
455
12.1. ОСНОВНЫЕ ТИПЫ СТРУКТУРНОЙ НЕЖЕСТКОСТИ
Причины структурной нежесткости и формы ее проявления в мо-
молекулярных системах весьма разнообразны. Наиболее общий меха-
механизм связан с внутренним вращением вокруг простых а-
с в язей. Поскольку электронная плотность сг-связи имеет ципинд-
рическую симметрию, барьеры вращения относительно этой связи
возникают в результате взаимодействий между несвязанными ато-
атомами или группами, например между атомами водородов при
различных углеродных атомах молекулы этана:
<
i^
\
нЛ
Н',
н,
1,
A2.1)
. D,
п. а
Устойчивой структурой, отвечающей минимуму ППЭ, является
заторможенная конформация I, которая трехкратно вырождена.
Конформационные переходы между вырожденными изомерами (то-
померами) 1а ^116 ^ 1в связаны с прохождением через неустойчивые
заслоненные конформации И (группа симметрии D3k), служащие
переходными состояниями. На рис. 12.1 показано сечение потенци-
потенциальной поверхности молекулы этана вдоль координаты, характери-
характеризуемой углом вращения <р вокруг связи С—С. Барьеры вращения
вокруг ординарных (простых) связей обычно очень малы. Для моле-
молекулы этана величина АЕ0 равна, по наиболее точным оценкам,
11,9 кДж/моль. Согласно A2.9), это означает, что время жизни
(т—1/&) каждой отдельной устойчивой конформации I составляет
при комнатной температуре всего ^Ю1 с.
В результате внутреннего вращения вокруг ординарных связей
и -
SO ПО 180 240 300 360 ф
Рис. 12.1. Потенциальная функция внутреннего вращения в молекуле этана
456
происходит изменение конформации молекулы (для этана на стадии
переходов 1+±П) без изменения конфигурации связей каждого ато-
атома. Так, на всех стадиях конформационного равновесия A2.1),
включающего взаимопревращения /K<r и /)ЗЛ-форм, сохраняется тет-
раэдрическая конфигурация связей углеродных атомов.
Второй важнейший тип структурной нежесткости — политопные
перегруппировки (термин введен И. Мьюттертизом A970)). К ним
относят низкобарьерные внутримолекулярные превращения, свя-
связанные с изменением конфигурации структурных узлов перегруп-
перегруппировывающейся молекулы в промежуточных или переходном со-
состояниях, происходящие без разрыва связей.
Примером политопной перегруппировки служит инверсия пира-
пирамидальной структуры соединений трехвалентного азота, осуществ-
осуществляющаяся через переходное состояние с плоской треугольной кон-
конфигурацией связей:
н3
l2 A2.2)
W Ш б
Можно заметить, что изменение конфигурации связей у атома
азота происходит не только на стадии переходной плоской формы
IV, но и в результате превращения Ша ^ 1Ш, так как эти структуры
являются зеркальными отображениями и несовместимы друг с дру-
другом при условии различения отдельных атомов водорода (энанти-
отопомеры) или замены их неодинаковыми заместителями (энанти-
омеры).
Зависимость потенциальной энергии молекулы аммиака от ин-
инверсионной координаты (угол пирамидализации) имеет вид, пока-
показанный на рис. 12.12. Величина энергетического барьера равна,
согласно экспериментальным данным, 24,3 кДж/моль. Это означа-
означает, что инверсионные переходы Ша;±Шб происходят при комнат-
комнатной температуре с частотой порядка 108 с~* (время жизни 10~8 с).
Третий тип структурной нежесткости связан с молекулярными
перегруппировками, обусловленными процессами разрыва — обра-
образования химических связей. Быстрые и обратимые перегруппировки
этого типа называют таутомерными*. Одним из ярких примеров
структурной нежесткости, вызванной таутомерными перегруппи-
перегруппировками, является реакция взаимопревращения вырожденных изо-
изомеров (топомеров) бульвалена, регистрируемая методом спектро-
спектроскопии ядерного магнитного резонанса:
•Термин «таутомерия» (от греч. tovto — тот же самый и ряро? — часть) пред-
предложен Лааром A885). Современное понимание таутомерии как равновесной динами-
динамической изомерии ведет начало от работ А. М. Бутлерова.
457
A23)
Каждое отдельное превращение, например Va-»V6, связано
с разрывом одной «т-связи С—С (Ci —Сэ в Va) и образованием
другой <г-связи С—С (С5—С7 в V6), сопровождающимися смещени-
смещениями я-связей (реакции 3,3-сигматропных смещений, см. гл. 13).
В молекуле семибульвалена VI, содержащей на две СН-группы
меньше, чем V, реализуется тот же самый процесс в более простом
виде:
vie
Здесь вырожденная перегруппировка VIa^±VI6 соответствует ста-
стадии Va^V6 перегруппировки бульвалена.
Молекула бульвалена обладает той замечательной особенно-
особенностью, что в результате наличия в каждой структуре трех возможных
путей ее изомеризации, показанных на схеме A2.3), любая из десяти
СН-групп молекулы может в ходе вырожденной перегруппировки
занять любое положение. Это означает, что в перегруппировыва-
перегруппировывающейся молекуле нет ни одной пары углеродных атомов, связь
между которыми сохраняется неизменной. Каждая отдельная груп-
группа СН как бы скользит по поверхности молекулы, оказываясь
в какой-то момент времени связанной с любым состоянием трех
других групп СН. Так как в молекуле бульвалена 10 групп СН
и каждый вырожденный изомер сохраняет ось симметрии третьего
порядка, то представленный на схеме A2.3) динамический процесс
охватывает переходы между 10!/3 A 209 600) отдельными струк-
458
турами. Эти переходы происходят при комнатной температуре
с частотой ~1(Г с, а перегруппировки Vla^*V16 в семибуль-
валене — с частотой ~108 с~х. Соединения, в которых, подобно
V и VI, реализована подвижная система химических связей, относят
к типу соединений с флуктуирующей структурой.
Термодинамические и спектральные свойства структурно не-
нежестких молекул нельзя правильно описать, исходя из точечной
группы симметрии мгновенной формы, т. е. одной из структур,
участвующих в процессе внутримолекулярной перегруппировки.
Общий подход к построению волновой функции нежесткой системы
на основе волновых функций, соответствующих мгновенным равно-
равновесным ядерным конфигурациям, состоит в учете взаимодействия
между отдельными жесткими структурами путем включения нену-
ненулевых матричных элементов гамильтониана. Если эти состояния
вырождены, как в рассмотренных выше случаях, и взаимопревраща-
взаимопревращаются в результате перестановки ядер или инверсии, то указанное
взаимодействие снимает вырождение и приводит к расщеплению
вырожденных в приближении жесткой структуры энергетических
уровней. Пример такого взаимодействия — инверсионное расщеп-
расщепление колебательных энергетических уровней молекулы аммиака
(см. рис. 12.12) будет рассмотрен далее. В общем, спектры структур-
структурно нежестких молекул значительно богаче, чем жестких. Для их
описания и систематизации используются специальные инверсион-
инверсионно-перестановочные группы симметрии.
12.2. МЕТОДЫ ИССЛЕДОВАНИЯ
СТРУКТУРНО НЕЖЕСТКИХ МОЛЕКУЛ
При энергиях возбуждения порядка величин энергетических
барьеров, связывающих отдельные взаимопревращающиеся формы,
структуру нежесткой молекулы можно описывать, используя усред-
усредненное по всем отдельным формам статистическое распределение
атомов. Такое распределение будет зависеть от температуры, и полу-
получаемые на его основе геометрические характеристики могут существен-
существенно отличаться от ядерных конфигураций в минимумах ППЭ. Поэтому
если для определения строения жесткой молекулы достаточно полу-
получить сведения о геометрических характеристиках атомной конфигура-
конфигурации, соответствующей минимуму адиабатического потенциала, то для
структурно нежестких молекул такие сведения необходимо дополнить
данными о высоте энергетических барьеров, связывающих все точки
минимумов ППЭ. С этими величинами, как следует из уравнения
(8.104), прямо связаны времена жизни x—\jk взаимопревращающихся
изомеров или топомеров. В зависимости от времени жизни отдельной
изомерной формы т0 структурная нежесткость может быть обнаружена
одними методами измерения и совсем не фиксироваться другими.
Действительно, отдельные изомерные формы оказываются экспери-
459
ментально неразличимыми, если их времена жизни т0 меньше, чем
характеристическое время т' данного экспериментального метода
(время акта взаимодействия с внешним возмущением, приводящего
к возбуждению системы).
При условии т'<т0 измерения выявляют лишь те структурные
формы, которые отвечают минимумам потенциальной поверхно-
поверхности, так как за время измерения не успевают совершаться переходы
между отдельными изомерами (топомерами). Напротив, если т/>т0,
то система за время измерения успевает перейти из одной формы
в другую и эксперимент фиксирует не отдельные изомерные или
топомерные структуры, а некоторую усредненную картину.
Характеристическое время данного метода измерения определя-
определяется временем жизни возбужденного состояния At = т', образующе-
образующегося при поглощении соответствующего кванта энергии. В спект-
спектральных измерениях информация о величинах At заключена в пара-
параметре естественной (т. е. освобожденной от аппаратурных факто-
факторов) ширины линии Av. Последняя в силу действия принципа неоп-
неопределенности (см. разд, 1,2)
Д?Д/~А/Bтг) A2.4)
не может быть бесконечно малой. Поскольку E—hv, она следующим
образом связана с At:
A v ^ 1 /Bтг At). (I z. j)
Полная форма спектральной линии в любом диапазоне наиболее
точно описывается функцией Лоренца
1
L(cojk-co0)=-
ж
A2.6)
где oH—2nv, ш^ — резонансная частота, отвечающая переходам
между k-ы и_/-м энергетическими уровнями; т — время релаксации,
т. е. время, требуемое для восстановления равновесной заселен-
заселенности энергетических уровней системы.
Максимальная амплитуда функции A2.6), показанной на рис.
12.2, равна т/я. Важным является значение полуширины линии
спектра Асо^ на полувысоте:
2
откуда
460
я
1
т
-С/
A2.7)
'1/2:
ИЛИ
т=
A2.8)
A2.9)
Для жестких структур, где
единственным механизмом ре-
релаксации являются физические
процессы тушения возбужден-
возбужденных состояний, т = Д/=т\ Од-
Однако при наличии химического
обмена — реакции интерконве-
интерконверсии изомерных форм — ме-
механизм релаксации усложняет-
усложняется, и величину т можно опре-
определить как
1 1 1
т т' т0'
Рис. 12.2. Нормированная функция Ло-
Лоренца A2.6):
полуширина спектральной линии на
полувысоте. Значение функции в максимуме
40) =~
я
A2.10)
При малых скоростях химического обмена, когда то»т',
процессы взаимопревращения изомеров не отражаются на
форме линии спектра, но по мере сближения этих величин
спектральная линия должна уширяться. В зависимости от
значения т' быстрые взаимопревращения изомерных форм
могут вызывать значительные уширения линий в одном спе-
спектральном диапазоне и не влиять на линии спектра в другом.
Например, для того чтобы обменные процессы могли отразиться
на форме линии в колебательном спектре (ИК-спектр или
спектр комбинационного рассеяния), они должны протекать
со скоростями, превышающими скорости реакций, лимитируемых
диффузией, т. е. взаимопревращения изомерных форм должны
реализоваться чаще,чем 10 с. Действительно, минимальная
естественная ширина линий в ИК спектре равна примерно
ОД см, откуда, согласно A2.5), время жизни возбужденного
состояния А/=5 101 с, или к=—^1012 с. Методами ко-
ш
лебательной спектроскопии можно регистрировать такие быстрые
процессы, как внутреннее вращение. Так, процесс быстрого
взаимопревращения двух поворотных изомеров 1,2-дихлорэтана —
гош (Vila) и транс (VII6) — отражается в ИК-спектре в уширении
и удвоении каждой из 18 колебательных полос, принадлежащих
отдельным конформерам:
461
A2.11)
В методе ядерного магнитного резонанса минимальная естест-
естественная ширина линии составляет «0,1 (^(Гц). Следовательно,
уширенве спектральных линий, регистрируемое этим методом, по-
позволяет, согласно A2.5), фиксировать обменные процессы с време-
временами жизни ниже 2 с или со скоростями, превышающими 0,5 с.
Для слияния одиночных пиков сигналов, принадлежащих двум вза-
имопревращающимся изомерам или топомерам и разделенных,
например, на 200 Гц (обычный диапазон химических сдвигов в спек-
спектрах ЯМР13С), скорость процесса химического обмена должна быть
равна 103 с. Поскольку скорость реакции является функцией тем-
температуры (8.104), для одного и того же процесса при разных тем-
температурах можно выполнить условия как очень быстрого (т'>т0),
так и очень медленного (т'<т0) обмена, а также найти диапазон
промежуточных скоростей обмена (т'^т0), в котором уширение
спектральной линии максимально. Эта ситуация иллюстрируется на
рис. 12.3, где представлена температурная зависимость спектра
ядерного магнитного резонанса бульвалена V. При низкой тем-
температуре скорость (частота) взаимопревращений топомерных форм
Va^V6^± ... мала по сравнению с разностью резонансных частот
любой пары неэквивалентных ядер углерода. При повышении тем-
температуры раствора скорость перегруппировок A2.5) растет, дости-
достигает и превосходит диапазон химических сдвигов неэквивалентных
ядер 13С. Выше 70°С в «спектре ЯМР13С фиксируется только один
сигнал от ядер 13С. Это означает, что в рамках метода ЯМР 13С все
группы СН в бульвалене при этих температурах эквивалентны и его
структура имеет более высокую симметрию, чем точечная группа
Сэ, характеризующая отдельные топомерные формы Va, V6 и т. д.
В табл. 12.1 приведены характеристические времена различных
физических и физико-химических методов, применяемых при ис-
исследовании динамики структурно нежестких соединений.
В таких методах, как дифракция рентгеновских лучей, электро-
электронов и нейтронов, частота излучения имеет очень высокие значения
и характеристические времена т' составляют «10 ~18 с, что намного
меньше, чем время, требуемое для любой молекулярной перестрой-
перестройки, и даже меньше, чем периоды нормальных колебаний отдельных
связей молекул. Поэтому при помощи данных методов можно
462
Г*0.8тС
7a гс
39 ГС
шгс
59.9*С
С,
J 1 1 1 1
1
JL
I
2,8Ю~6СГ
I /О*с-'
I
I
1.5ЮЧ
22 /О3С*
/40 /20 80 40 О З'н.д.
Рис 123. Спектр ЯМР — 13С бульвалена V в растворе (дяметилформамид) при
различных температурах:
при — 59,9°С в спектре видны сигналы ядер углерода для всех четырех неэквивалентных положе-
положений структуры V; минимальная разность частот различных сигналов (рабочая частота прибора
25 МГц) для ядер Q i С( составляет примерно 8 с'1, максимальная для ядер С#—С, —
примерно 2600 с'1. Указаны температуры растворов и вычисленные скорости перегруппировок
регистрировать лишь мгновенные конфигурации структурно не-
нежесткой молекулы.
Обычные кинетические методы химического исследования пред-
предполагают возможность препаративной* изоляции метастабильной
463
log k(c-*)
;
-16,6
'19
-160
-120
-80
ноль
160-
Препаративмая изоляция
при комнатной температуре
120-
Л.
ЯМР
80-
Флеш
фотолиз
Ультра-
Ультразвук,
поглоще-
поглощение ЗПР
I
1
40-
\
микроволн,
спектра-
Ч сколия
vomis
)
Необратимые
перегруппировки
Равновесная смесь
изомерных форм
Рис. 1X4. Физические методы для изучения процессов структурной нежесткости,
характеризуемых энергетическими барьерами различной величины:
ори разности свободных рвергий отдельных форм А С^>20—25 кДж/моль содержание минор-
минорных форм меньше 10~6 а они обычно ве фиксируются
изомерной формы и наблюдения за ее релаксацией в более устой-
устойчивую структуру. Это означает, что время жизни такой формы
должно соответствовать по крайней мере нескольким минутам или
часам, т. е. энергетический барьер превращений при комнатной
температуре должен быть не ниже 90—100 кДж/моль. На рис. 12.4
показано, какие физические методы могут быть использованы для
изучения процессов структурной нежесткости, характеризующихся
различными энергетическими барьерами.
464
Таблица 12.1. Характеристические времева различных физических методов
Метод
10В
l(Tie
ю-1В
ю-18
10-14-105
И
7
-*10"e
10-*-10
OlO
боле 101
Рентгенография
Нейтронография
Электронография
Фотоэлектронная спектроскопия
УФ-спектроскопия и спектроскопия в видимой области
ИК-спектроскопия и спектроскопия комбинационного
рассеяния
у-спектроскопия (эффект Мессбауэра)
ЭПР-спектроскопия
Поглощение ультразвука
ЯКР-спектроскопия
ЯМР-спектроскопия
Скачок температуры
Обычные кинетические методы
12.3. ЭЛЕКТРОННАЯ ПРИРОДА
СТРУКТУРНОЙ НЕЖЕСТКОСТИ МОЛЕКУЛЫ
Явление структурной нежесткости по своей природе тесно связа-
связано с вибронными эффектами. В случае жесткой молекулы, отвеча-
отвечающей устойчивой структуре, минимум потенциальной поверхности
имеет значительную глубину. Величина А?, отделяющая данный
минимум от ближайшей седловой точки, которая связывает его
с соседним минимумом ГШЭ, превышает 90—100 кДж/моль. При
деформации структуры, т. е. при смещении от равновесного поло-
положения в направлении любой из CiV—6) внутренних координат,
энергия системы резко возрастает. Ближайшее возбужденное элект-
электронное состояние ?] достаточно удалено от основного электронно-
электронного состояния ?о. Эта ситуация представлена на рис, 12.5, о.
В случае структурно нежестких молекул существует по крайней
мере одна из ZN—6 внутренних координат, смещение по которой
в сторону ближайшего минимума ППЭ сопряжено с преодолением
достаточно малого энергетического барьера Д? (рис. 12.5, б). Такие
координаты Qi называют нежесткими координатами. Число коле-
колебательных уровней в минимумах адиабатического потенциала таких
молекул невелико, и для многих структур потенциальная поверх-
поверхность электронно-возбужденного состояния расположена в непо-
непосредственной близости от поверхности основного состояния. Хотя
приближение Борна—Оппенгеймера, позволяющее разделить элект-
электронные и колебательные волновые функции, сохраняет свою силу
для структурно нежестких молекул этого типа, однако эффективная
ядерная конфигурация и зависящие от нее характеристики не опре-
определяются точно точками минимумов, а должны описываться веро-
вероятностным распределением — квадратом ядерной волновой функ-
465
а <г а
Рис. 12.5. Сечение ППЭ вдоль нежесткой координаты Q;.
Ч*о и *?\ — основное и ближайшее возбужденное электронное состояния; заштрихованные участ-
участки — квадраты функции 4*(Q): Qo, Q\ — равновесные ядерные конфигурации: а — структурно
жесткая система; 6 — стереохимически нежесткая система (сближение поверхностей Ч*о и
в — стереохимически и электронно нежесткая система
ции ^l(Q). Если энергия нулевых колебаний изомерных форм
меньше, чем барьер взаимопревращения этих форм, то сохраняется
принципиальная возможность раздельного наблюдения и даже фик-
фиксация этих форм при низких температурах. При более высоких
температурах, когда система переходит на высшие колебательные
уровни, ее ядерная конфигурация полностью делокализована между
двумя изомерными структурами.
Рассмотренный тип структурной нежесткости относят к сте-
реохимической нежесткости. Потенциальные поверхности (рис.
12.5, б) характерны для молекулярных систем с внутренним враще-
вращением (см. рис. 12.1), а также для систем, способных к политопным
и таутомерным перегруппировкам. Отличительной особенностью
последних является узкая энергетическая щель между потенциаль-
потенциальными поверхностями основного и возбужденного электронных со-
состояний в зоне переходной структуры. Ядерные конфигурации, со-
соответствующие данной области (Q\ на рис. 12.5, б), подвержены
эффекту Яна—Геллера второго порядка (см. разд. 5.7.2), который
можно в этом случае рассматривать как главную причину сте-
реохимической нежесткости. Как отмечалось, обычно (но не всегда)
наибольший вклад в вибронное взаимодействие поверхностей сбли-
сближенных электронных состояний ЯРо, Ч^ (рис. 12.5, б) вносит взаимо-
взаимодействие между граничными орбиталями деформируемой при дви-
движении по нежесткой координате молекулы.
Характер этого взаимодействия схематически показан на рис.
466
Рис. 12.6. Взаимодействие (расталкивание) граничных МО в симметричной проме-
промежуточной структуре Q{ при ее деформации по нежесткой координате (?,:
в качестве примера показано взаимодействие а'2 и а-МО для /^-структуры молекулы аммиака
при ее пирамидализации
12.6. Деформация симметричной структуры Q{ (см. рис. 12.5, б)
приводит к появлению перекрывания между высшей заполненной
и низшей свободной орбиталями. Из теории орбитальных взаимо-
взаимодействий (см. разд. 9.2) следует, что такое взаимодействие ведет
к стабилизации связывающего и дестабилизации антисвязывающе-
го уровней, т. е. к двухэлектронной стабилизации. Последняя до-
достигает максимальной величины при некотором значении нежест-
нежесткой координаты Q, — QQi соответствующей ядерной конфигурации
с более низкой, чем Qu симметрией. Примером стереохимической
нежесткости рассматриваемого типа является инверсия молекулы
аммиака III (конфигурации Qo, Q'Q) через стадию высокосимметрич-
высокосимметричной плоской структуры IV (конфигурация Q}).
Если движение по нежесткой координате Qi сопряжено с пересе-
пересечением поверхностей электронных состояний То и %, это приводит
к вырожденному электронному состоянию в точке Qi и проявлению
эффекта Лна—Геллера первого порядка. Когда это пересечение
происходит вблизи точки Qo, соответствующей ядерной конфигура-
конфигурации минимума ППЭ, эффект Яна—Геллера имеет динамический
467
характер (см. разд. 5.7.1). В зависимости от симметрии потенциаль-
потенциальной поверхности существует несколько разделенных невысокими
энергетическими барьерами флуктуирующих структур. Такой тип
структурной нежесткости (см. рис. 12.5, б) называется электронной
нежесткостью. Волновая функция электронно нежестких систем
представляется как вибронная суперпозиция всех п B, 3, ...) вырож-
вырожденных электронных состояний в адиабатическом приближении:
t A2.12)
Из соотношения A2.12) следует, что вклад отдельного элект-
электронного состояния в *F (Q) зависит от колебательных состояний
*?" (Q) • Таким образом, изменяя при варьировании температуры
заселенность колебательных уровней, можно варьировать одновре-
одновременно и ядерную конфигурацию, и электронные свойства молеку-
молекулярной системы. Многочисленные примеры электронной нежест-
нежесткости, сопряженной со стереохимической нежесткостью, найдены
в координационных соединениях переходных металлов с не полно-
полностью заполненными ^-оболочками (см. разд. 11.5).
Интересным случаем электронной и стереохимической нежест-
нежесткости является динамическое равновесие в биядерных комплексах
металлических ионов с переменной валентностью VIII, в которых
осуществляется адиабатический перенос электрона через я-сопря-
женыые мостики:
2+ #~~\ /~^ 3+
(NH3MCo —N^ у— X ^ N Ru(NH3M
Villa A2,13)
(NH3MCo N у X—^ N Ru(NH3M
VII16
X=S, CH*=CH,C*C
Наряду с геометрическими перестройками в этих реакциях резко
перестраивается электронная конфигурация, что четко отражается
изменениями в электронных спектрах поглощения и люминесцен-
люминесценции. Величины энергетических барьеров и скорости взаимопрев-
взаимопревращений Villa ?*VIII6 меняются в широких пределах в зависимо-
зависимости от природы мостиковой группы X.
12.4. ПОЛИТОПНЫЕ ПЕРЕГРУППИРОВКИ
В этом разделе рассматриваются наиболее важные политопные
перегруппировки, связанные с динамическими трансформациями
468
конфигураций центральных атомов в соединениях типа АХ«. Будут
выделены основные моды перегруппировок, т. е. нежесткие коор-
координаты, варьирование которых определяет переход от одной формы
к другой, а также рассмотрены зависимости энергетических барье-
барьеров перегруппировок от структурных факторов.
12.4.1. Пирамидальная инверсия
трнкоордиынрованных структур
Инверсия пирамиды в молекуле аммиака A2.2) — наиболее
изученная (теоретически и экспериментально) политопная перегруп-
перегруппировка. В табл. 12.2 приведены данные о величинах барьеров
пирамидальной инверсии для структур типа АХ3, где А — непере-
непереходный элемент:
A2.14)
Таблица 12.2. Барьеры пирамидальной инверсии для молекул и ионов АХз ио
результатам неэмииртеских расчетов (базисы inua DZ + Р)
Соединение
сн~
SiH;
NH3
РН3
AsH3
SbH3
Барьер инверсии, кДж/моль
8,8
108
23,4 (эксп. 24,3)
146 (эксп. 132)
192
192
Соединение
он+
sh;
SeH3+
NLi3
NF3
PF3
Барьер инверсии, кДж/моль
3,3
134
126
Ал-структура
314
690
Как видно из табл. 12.2, значения барьеров инверсии охватыва-
охватывают чрезвычайно широкий диапазон — от 3—9 кДж/моль для от-
отличающихся исключительной структурной, нежесткостью метилани-
она, катиона гидроксония и молекулы аммиака до 140—
690 кДж/моль в структурно жестких молекулах фосфина,
трифторида азота и трифторида фосфора. Чтобы понять основные
тенденции в изменениях значений барьеров инверсии, воспользуем-
воспользуемся представлениями качественной теории АО и эффектами Яна—
Теллера второго порядка.
Как отмечалось, энергетически наиболее важным является воз-
возникающее при искажении вдоль нежесткой координаты взаимодей-
взаимодействие высшей заполненной и низшей свободной МО (см. рис. 12.6).
Возникающее при таком искажении перекрывание орбиталей
469
а2 и d\ />зА-структуры (см. рис. 10.4) ведет к их смешиванию, причем
энергию стабилизации нижней МО at в пирамидализованной
структуре можно в соответствии с (9.5) и (9.7) представить как
Будем полагать, что изменения в энергии высшей заполненной
МО при пирамидализации плоской структуры определяют общую
тенденцию в изменении полной энергии молекулы, т. е. остальные
энергетические уровни молекулы слабо реагируют на пирамидаль-
пирамидальную деформацию (сравните с диаграммой Уолша на рис. 10.4).
Тогда из соотношения A2.15) следует, что стабилизация пирами-
пирамидальной формы по отношению к плоской будет тем больше и,
следовательно, тем выше будет инверсионный барьер, чем меньше
энергетическая щель между граничными орбиталями в исходной
плоской структуре. Из теории возмущений, например соотношений
(9.7) и (9.17), вытекает, что с уменьшением электроотрицательнвсти
центрального атома А в ряду соединений АХ3 энергетический уро-
уровень высшей связывающей МО будет повышаться, так как эта
орбиталь (см. рис. 12.6) в /)ЗА-форме молекул АХ3 полностью
(Х=Н) или главным образом (Х^Н) локализована на центральном
атоме А. Аналогичные соображения, подтверждаемые прямыми
расчетами, свидетельствуют о том, что варьирование электроот-
электроотрицательности центрального атома слабо отражается на положении
энергетического уровня низшей свободной МО ?>3/гформы. Таким
образом, понижение электроотрицательности центрального атома,
например в ряду NH3—PH3—AsH3 — SbH3, приводит к уменьшению
знаменателя в A2.15) и увеличению А?, т. е. стабилизации пирами-
пирамидальной формы по отношению к плоской, и тем самым к увеличе-
увеличению барьера пирамидальной инверсии A2.14). С этим выводом
согласуются данные табл. 12.2. Аналогичная закономерность — по-
повышение энергетического барьера пирамидальной инверсии в соеди-
соединениях низших периодов — существует и для соединений элементов
VI, IV групп.
Теория орбитальных взаимодействий дает простое объяснение
и для влияния заместителей X на величины барьеров пирамидаль-
пирамидальной инверсии A2.4). Как видно из схемы двухуровневых взаимодей-
взаимодействий на рис. 12.7, я-донорные электроотрицательные заместители
X (такие, как атомы фтора), обладающие низколежащей заполнен-
заполненной МО, должны повышать энергетический уровень высшей связы-
связывающей МО ?>ЗА-формы АХ3 (AX2R, AXRiR2). При этом одновре-
одновременно несколько понижается уровень низшей свободной МО, в зна-
значительной мере локализованной на лигандах X. В итоге энергети-
470
a 5
Рис. 12.7. Диаграмма орбитальных взаимодействий pz fo^-орбитали центрального
атома А в плоских (Dyd молекулах АХз, AX2R, AXRi R2 с/>-орбиталями заместителя
X:
а — Х-элеггроотрицательная, я-донорная группа; б — Х-элеггроотрицатепьная, «-акцепторная
группа
ческая щель между граничными орбиталями уменьшается и барьер
инверсии увеличивается. Молекулы NF3, PF3 обладают жесткой
пирамидальной структурой. Напротив, если X — я-акцепторный
лиганд с низкой электроотрицательностью, уровень высшей заня-
занятой 1Ч1О понижается до такой степени, что плоская форма становит-
становится предпочтительной, как предсказывают расчетные данные для
молекул NH2Li, NHLi2, NLi3, NH2BH2, NH2BeH и др.
12.4.2. Плоская инверсия
дикоординированных структур
Динамический инверсионный процесс может происходить
и в плоских молекулах. Так, в молекуле формальдимина IX переход
между топомерными структурами 1Ха ?? 1X6 может происходить
как в результате движения связи N—Н в плоскости молекулы через
переходную структуру X (плоская инверсия), так и вследствие вра-
вращения по связи C = N (XI):
C=N—H
X
к 90° ^. 1X6
НЧ
C=N
1Ха
\
н
Чн
XI
471
Рассчитанные методом ab initio (базис DZ-типа) относительные
энергии переходных структур X и XI составляют 109 и
238 кДж/моль соответственно. Расчеты, проведенные с учетом элек-
электронной корреляции (МР2/6-31 G**), сохраняют тот же порядок.
Эти расчеты, как и многие другие, проведенные для аналогичных
превращений соединений дикоординированного азота, свидетель-
свидетельствуют в пользу предпочтительности инверсионного механизма.
Экспериментально найденные значения барьеров плоской инвер-
инверсии варьируют в широких пределах: 40—160 кДж/моль. Введение
электроотрицательных заместителей к атому азота резко увеличи-
увеличивает барьер плоской инверсии, подобно тому, как это имеет место
в случае пирамидальной инверсии. Так, в случае диарилметилени-
миновых производных Ar2C = NX частота инверсии при переходе от
заместителя Х~С(?15 к Х=ОСН3 уменьшается в 1014 раз.
При замене иминного азота на более электроположительную
изоэлектронную группу в карбанионе XII барьер инверсии возраста-
возрастает, тогда как введение более электроотрицательного оксониевого
центра приводит к значительному понижению барьера. Этот вывод
следует из данных неэмпирических (базис DZ-типа) расчетов для
следующих простых соединений:
\ -С С N
г/ \ К \
:N
XII 163 кДж/моль IX 109 кДж/моль ХШ 72 кДж/молъ
Как и в случае пирамидальной инверсии A2.14), этот результат,
полностью подтвержденный экспериментальными данными, может
быть объяснен исходя из анализа зависимости расщепления уровней
граничных орбиталей в переходной структуре типа X от электроот-
электроотрицательности инвертирующего центра.
Задача 12.1. Объясните влияние элеггроотрицагельяости атома X в соеди-
соединениях Н2С=ХН на величину энергетического барьера плоской инверсии в со-
соединениях IX, XII, XIII.
12.4.3. Тетраэдрическая инверсия
тетракдорднннроваииых структур
Две основные структуры тетракоординированных центральных
атомов — тетраэдрическая XIV и квадратная XV — связаны друг
с другом инверсионным процессом A2.17):
472
ч
XV. J*/ A2.17)
XlVa ^ \У XIV6
XV6
Если все заместители при центральном атоме различны, тетра-
эдрические структуры XlVa, б представляют энантиомерные
формы. Переходы XlVa z± XVa ^ XIV6 соответствуют начальному
/^искажению исходных Т^форм (механизм тетраэдрического
сжатия), а переходы XlVa<=*XV6 «*XIV6 — начальному ^-иска-
^-искажению при повороте плоскости ЗА4 по отношению к плоскости 1А2
(механизм дигонального твиста). Для тетраэдрических структур
соединений непереходных элементов внутримолекулярные пере-
перегруппировки, связанные с промежуточной плоской квадратной фор-
формой XV, нереализуемы вследствие крайней неустойчивости послед-
последней. Как было показано (см. разд. 10.1.3), главная причина неустой-
неустойчивости плоских тетракоординированных структур непереходных
элементов — наличие высоколежащей несвязывающей МО а2и, за-
заполняемой двумя электронами (см. рис. 10.5). Введение электропо-
электроположительных тг-акцепторных заместителей существенно стабилизи-
стабилизирует плоскую форму (сравните с данными табл. 12.2 для трикоор-
динированиых структур, NLi3). Как показали данные весьма стро-
строгих неэмпирических расчетов с использованием расширенного бази-
базиса орбиталей и с учетом корреляционных поправок, замена двух
атомов водорода в структурно жесткой молекуле метана на атомы
лития приводит к стереохимически и электронно нежесткой молеку-
молекуле дилитиометана. Структуры четырех низших электронных состоя-
состояний этой молекулы как для тетраэдричесхой, так и для плоской
форм имеют почти одинаковые энергии:
Li Li
\/
С
к н
Li Li
\/
С
4%.
ff h
Li Li
\ /
С
/ \
H H
Li
\
/
H
L
С
\
H
8,8CB2) 16,3CBj) 34,7(lA,)
473
В отличие от соединений непереходных элементов тетракоор-
динированные структуры переходных элементов отличаются значи-
значительно большей лабильностью. Пример стереохимической и элект-
электронной нежесткости — интерконверсия плоской (синглетное 1АЖ ос-
основное электронное состояние) и тетраэдрической (триплетное ЪЕ
основное электронное состояние) изомерных форм хелатных комп-
комплексов никеля (см. разд. 11.3.4). Аналогичное конфигурационное
равновесие существует в растворах моно- и бидентатных фосфино-
вых комплексов:
f x
\ /
Ni
^PR
i 1Ч.Ч
XVI XV11
R = Alk, Ar
X = Cl,Br, I
Скорость взаимопревращений между тетраэдрическими XIV
и плоскими XV формами этих комплексов никеля удалось опреде-
определить при помощи метода спектроскопии ЯМР. Частота этих пре-
превращений составляет 105—106 с" при 25°С (энергетические барье-
барьеры 40—50 кДж/моль).
Если для комплексов тетракоординированного никеля (элект-
(электронная конфигурация центрального иона йг8) относительные энергии
плоской и тетраэдрической структур настолько близки, что эти
формы существуют в динамическом равновесии друг с другом, то
для комплексов металлов с заполненной ^-оболочкой (Zn, Cd, Hg,
РЬ) плоская форма энергетически невыгодна. Однако разность энер-
энергий тетраэдрической и плоской форм намного меньше, чем в случае
соединений непереходных элементов. Это объясняется тем, что
\s /Mv \У A2.18)
CR . .
/ \ CR' !
1\ XVIII (К) /\r XVIII (S)
R R
M=Zn,Cd,Hg,Pb; X = O,S,Se
474
L М
/
L
Ч
I
Рис. 12.8. Корреляционная диаграмма МО для плоских D*/, тетраэдрических
комплексов переходных металлов:
L — двухэлектронный лиганд; электронное заполнение в ML4 отвечает </*° — электронной кон-
конфигурации центрального атома
в плоских тетракоординированных структурах соединений rf-элемен-
тов высоко лежащая орбиталь а^-типа. не заполняется электронами
(сравните рис. 10.5 и 12.8).
475
При помощи метода ЯМР-спектроскопии были определены
энергетические барьеры тетраэдрической инверсии для широких
серий бисхелатных комплексов общего типа [см. A2.18)].
Соединения XVIII являются энантиомерами. При помощи спек-
спектров ЯМР процесс энантиомеризации можно наблюдать без пред-
предварительного разделения энантиомеров по изменениям формы ли-
линий сигналов диастереотопных групп R в прохиральных замести-
заместителях CR/R2- Это обусловлено тем, что они обменивают свои
положения в молекуле в результате обращения конфигурации свя-
связей хирального центра М. Для хелатов Zn энергетические барьеры
интерконверсии XVIII (R) ^ XVIII (S) через стадию образования
плоской формы составляют 75—90 кДж/моль, для хелатов Cd —
45—60 кДж/моль, а для хелатов РЬ и Hg —35—45 кДж/моль.
Для аналогичных хелатов Ni они не превышают 20—25 кДж/моль.
Как видно из орбитальной диаграммы на рис. 12.8, причина
относительной неустойчивости плоской структуры для комплексов
*/10-металлов связана с заполнением антисвязывающей 2blg-MO,
которая в тетраэдрической форме трансформируется в более низ-
колежащую МО /2-ряда. Для комплексов Ni(II), для которых 2big-
орбиталь вакантна, можно ожидать большей устойчивости плоской
квадратной структуры по сравнению с тетраэдрической, что и на-
наблюдается в действительности.
Задача 12.2. Покажите, используя представления об эффекте Яна—Геллера
второго порядка, что механизм тетраэдрического сжатия является основной
модой интерконверсии D^h- и 7>структур непереходных и */1О-элементов.
12.4.4. Пентакоординированные структуры.
Псевдовращение Берри
Для соединений непереходных элементов устойчивой пентакоор-
динированной структурой является структура с тригонально-бипи-
рамидальной конфигурацией связей XIX с неравноценными акси-
аксиальными и экваториальными связями. Например, для молекулы PF5
длины связей PF^. и PF3m как показывают данные электроно-
графических исследований, равны соответственно 0,1577
и 0,1534 нм. Тригонально-бипирамидальные структуры XIX реали-
реализуются и для многих соединений переходных элементов, что об-
обнаруживается методами с высоким временным разрешением (см.
табл. 12.1), такими, как рентгенография и электронография, ИК-
спектроскопия. При изучении подобных соединений с помощью
476
методов с меньшим характеристическим временем и аксиальные,
и экваториальные лиганды оказываются полностью равноценными.
Так, в спектрах ЯМР~19? пентафторида фосфора PF5 даже при
температуре — 150°С содержится только сигнал, общий для всех
ядер фтора (дублет за счет спин-спинового взаимодействия с яд-
ядрами ^1Р). В спектрах ЯМР 13С пентакарбонила железа Fe(COM
даже при — 170°С также наблюдается только один общий сигнал от
всех ядер углерода. С точки зрения метода ЯМР структуры указан-
указанных соединений обладают сферической симметрией или по крайней
мере одной осью пятого порядка. Причиной наблюдаемой сте-
реохимической нежесткости является процесс, приводящий к быст-
быстрому в шкале времени метода ЯМР обмену положений экватори-
экваториальных и аксиальных лигандов. Р. Берри A960) высказал пред-
предположение, которое в настоящее время нашло полное подтвержде-
подтверждение в результате анализа колебательных спектров, спектров ЯМР
и квантово-химических расчетов, что такой обмен осуществляется
по следующей схеме:
A2.19)
. С
,
Результат перегруппировки — обмен одной пары аксиальных
лигандов на экваториальные. Поскольку в этой перегруппировке
может участвовать любая пара экваториальных лигандов, при бы-
быстром обмене происходит эффективное усреднение положений всех
лигандов. Политопная перегруппировка A2.19), являющаяся глав-
главным механизмом структурной нежесткости пентакоординирован-
ных структур, названа псевдовращением Берри. Соответствующая
псевдовращению Берри нежесткая координата совпадает в своей
начальной фазе с вырожденным нормальным колебанием е'-сим-
метрии тригонально-бипирамидальных структур:
477
Именно такую симметрию нежесткой координаты для ?Kд-три-
гонально-бипирамидальных структур можно предсказать, применяя
представления об эффекте Яна—Геллера второго порядка и учиты-
учитывая, что последовательность МО для ?3л-структур фосфоранов РН5,
РХ5 (Х-ст-лиганд) соответствует заполнению
(а,J DI- Ыг)г Bа,)г № Bаг)°. A2-20)
Из A2.20) следует, что смешивание граничных МО достигается
при деформации, имеющей симметрию ах хе' = е', т. е. XXI, приво-
приводящей к С4,-структуре.
В табл. 12.3 приведены данные о барьерах политопных перегруп-
перегруппировок A2.19) для ряда тригонально-бипирамидальных структур
непереходных и переходных элементов.
Таблица 12.3. Энергетические барьеры иолнтоиных оерегруииировок структурно
нежестких пентакоординироваяных структур
Соединение
РН5
PF5
PF4NMe2
PF3(NMe2J
AsF5
O = SF4
VF5
Fe(COM
Fe(COLC2H4
[NiCl(PMe3L]+
[Cua5]3-
UF5
t?(C^-Dih), кДж/моль
4—16
12—14
40
45
9—10
20
5—6
-4
45—60
28
8
5
Метод определения
Квантово-химический расчет
(РМХ, CNDO/2, ab initio)
ИК-спектроскопяя
ЯМР-спектроскопня
Я М Р-спектроск опия
ИК-спектроскопия
ЯМ Р-спектроскопия
ИК-спектроскопия
Спин-решеточная релаксация
ЯМР-спектроскопия
ЯМР-спектроскопия
Квантово-химический расчет
Квантово-химический расчет
Как видно из приведенных данных, для комплексов переходных
элементов энергетическая разность между тригонально-бипирами-
дальной и квадратно-пирамидальной формами может достигать
478
предельно малых значений. Найдено, что многие пентакоорди-
нированные структуры, например Fe (COM и Os (PF3M, существуют
в растворах в виде динамического равновесия Х)ЗА- и С4и-форм.
Для ряда пентакоординированных структур переходных элементов,
например Сг(СОM, [Co(CNM]3 , С^-структуры становятся пред-
предпочтительными .
Задача 12.3. Объясните причины стереохимической нежесткости 2>эл-струк-
тур низкоспиновых комплексов переходных металлов с d6- и ^-электронными
конфигурациями.
12.4.5. Структуры с более высокими
координационными числами
Октаэдрические соединения АХ$, образованные непереходными
элементами, относятся к структурно жесткому типу. Это объясняет-
объясняется большой величиной энергетической щели между граничными
орбиталями в молекулах и ионах SF6, PF6", SiFj", CIF^ и др. В то же
время октаэдрические комплексы переходных металлов, а также
некоторых непереходных металлов обнаруживают стереохимичес-
кую нежесткость. Наиболее характерный механизм — политопная
перегруппировка по типу тригонального твиста — состоит в дви-
движении по нежесткой координате, соответствующей поворотам от-
относительно Сэ-оси. Наиболее изученные примеры — трихелатные
комплексы — дикетонаты или трополонаты:
о
ххп
XXIII
XXIVa (R)
XXV
XXIV* (S)
A2.21)
479
Через структуру XXV с призматической конфигурацией октаэдри-
ческая структура XXIVa переходит в энантиомерную или энантиото-
померную (а=в) форму XXIVb. Если в хелатном кольце соединений
XXII, XXIII один из заместителей R содержит диастереотопную
группу, можно, как и в случае интерконверсии тетраэдрических
комплексов XVIII, определить с помощью ЯМР-спектральных иссле-
исследований скорость процессов рацемизации октаэдрических комплек-
комплексов XXIV, не проводя разделения энантиомеров. Комплексы XXII,
XXIII с М = Al, Ga, In, Sc, Mn, Fe обнаруживают стереохимическую
нежесткость, и энергетические барьеры для перегруппировки по типу
тригонального твиста находятся в пределах 45—90 кДж/моль.
Тригональный твист A2.21) — не единственный возможный ме-
механизм политопных перегруппировок октаэдрических структур. На-
Надежная дифференциация различных перегруппировочных мод тре-
требует проведения достаточно строгих квантово-химических расчетов
путей соответствующих перегруппировочных реакций с локализаци-
локализацией структур переходных состояний и промежуточных соединений.
Пока такие расчеты для соединений АХ*, с т^б еще представляют
определенные трудности, поэтому моды политопных перегруппиро-
перегруппировок этих соединений не так строго идентифицированы, как для
соединений с более низкими координационными числами. Однако
экспериментальные исследования показывают, что с увеличением
координационного числа структурная нежесткость соединений АХт
еще более увеличивается, а типы политопных перегруппировок ста-
становятся более многообразными.
12.5. ТУННЕЛЬНЫЙ МЕХАНИЗМ ПРЕВРАЩЕНИЙ
СТРУКТУРНО НЕЖЕСТКИХ МОЛЕКУЛ
Перегруппировки структурно нежестких молекул, как и другие
реакции, характеризуемые низкими энергетическими барьерами,
могут протекать не только через колебательно-возбужденные состо-
состояния, энергия которых превышает высоту активационного барьера,
но и в результате подбарьерного квантово-механического туннели-
роваыия. Условием преобладающего вклада туннелирования в эф-
эффективную скорость реакции, складывающуюся из подбарьерных
(туннельных) и надбарьерных (классических) переходов, является
выполнение неравенства
A2.22)
где Т — температура, при которой осуществляется реакция; АЕ —
энергетический барьер; m — масса частицы; d — значение полуши-
полуширины барьера; к — постоянная Больцмана.
Из A2.22) следует, что вероятность туннелирования возрастает
480
в случае узких барьеров, для частиц с малой массой и с понижением
температуры. Туннелирование — подбарьерное просачивание си-
системы из одного минимума потенциальной поверхности в другой —
особенно характерно для реакций переноса легких частиц — прото-
протона, электрона. В последнем случае туннельные эффекты проявляют-
проявляются даже для очень больших d— 3 -г 30 нм. Однако и в случае более
тяжелых частиц (атомов второго, третьего периодов), положение
которых относительно центра масс изменяется в результате молеку-
молекулярной перегруппировки незначительно, механизм туннелирования
может играть существенную роль. В таких низкобарьерных поли-
топных перегруппировках, как пирамидальная инверсия, псевдов-
псевдовращение Берри, валентная таутомерия циклобутадиена (см. разд.
5.7.2) и др., значения d достаточно малы и условие A2.22) выполня-
выполняется для относительно высоких температур. Оценочные расчеты,
проведенные для пентафторида фосфора PF5> дают следующие зна-
значения констант скоростей для туннельных и классических надба-
рьерных перегруппировок A2.19): при 100 К величина к^~
= 31(Г2с-Л ?О=4-Ш-5 с; при 400 К ^^=10* с, ?м=
= 6 108 с. Характерным проявлением туннелирования является
инверсионное расщепление колебательных уровней двухъямного по-
потенциала молекулы аммиака в результате пирамидальной инверсии
A2.2). Переходы О+-+О~ с частотой 0,794 см и 1 + -*1~ с часто-
частотой 0,365 см относятся к микроволновой области спектра (пер-
(первый мазер).
12.6. ТАУТОМЕРИЯ
К таутомерному типу взаимопревращений структурно нежестких
молекул относятся перегруппировки их изомерных или топомерных
форм, связанные с внутримолекулярными реакциями разрыва —
образования химических связей. Условие достаточно низкого энергети-
энергетического барьера выполняется обычно только в том случае, когда такая
перегруппировка сопряжена с разрывом не более чем одной связи.
Наиболее важный и широко распространенный тип таутомерных
перегруппировок — внутримолекулярные переносы (миграции)
групп. В случае наиболее легкого мигранта — водорода (прото-
(протона) — переходы между отдельными формами совершаются преиму-
преимущественно по туннельному механизму.
Примером может служить вырожденная таутомерия енола ма-
малонового диальдегида XXVI (Ri = R2=H):
м н
сг о (г х) о
Т
XXVIa XXV11 XXVI6
16. Теория строения молекул .
V,
го
*2Л
Рис. 12.9. Туннелирование в симметричном (малоновый альдегид) и несимметри-
несимметричном (замещенный /? — оксиакролеин, R{ т^Лз) потенциалах:
а — энергетические уровни системы; 6 — волновые функция соответствующих состояний
Характеристическое время метода ЯМР-спектроскопии превы-
превышает время жизни топомераых форм XXVI, и в шкале времени
этого метода молекула малонового диальдегида (R! = R2 —H) долж-
должна характеризоваться симметричной Сгу-структурой XXVII. Доста-
Достаточно строгие неэмпирические расчеты с использованием расширен-
расширенного базиса орбиталей и с учетом корреляционных поправок свиде-
свидетельствуют о том, что структура XXVII представляет собой пере-
переходное состояние реакции, имеющее энергию на 20 кДж/моль вы-
выше, чем XXVI. При изучении строения малонового диальдегида
методом микроволновой спектроскопии с более высоким времен-
временным разрешением выявляется устойчивость структуры XXVI, пре-
претерпевающей быстрые обратимые перегруппировки A2.23) с часто-
частотой 6,3 1011 с. Такая высокая скорость протонных миграций
обусловлена эффектами туннелирования, так как оцененная для
барьера в 20 кДж/моль частота переходов XXVIa ^ XXVIб состав-
составляет при комнатной температуре всего 1,5' 109 с.
Вероятность туннелирования протона сквозь барьер двухъям-
ного потенциала сильно зависит от его симметрии. При симметрич-
симметричном профиле ППЭ (рис. 12.9, а, 6) для вырожденных превращений
собственные функции системы делокализованы по двум ямам, тогда
как асимметричный потенциал (невырожденные системы R^Rz)
характеризуется локализацией собственных функций вблизи мини-
минимума. Только при приближении к вершине барьера повышается
вероятность обнаружения протона в обоих минимумах потенциала.
Подбарьерные переходы между двумя минимумами симметричного
потенциала подтверждаются наличием туннельного расщепления
колебательных уровней, которое для малонового альдегида состав-
составляет 23 см~*.
В качестве мигрантов, быстро обменивающих свои положения
в молекуле, могут выступать и объемистые атомные группы, напри-
например металлоорганические фрагменты. Первым примером A956)
482
таутомерии такого вида была пятикратно вырожденная перегруп-
перегруппировка производных циклопентадиена:
XXVIII
A2.24)
= Fe(ff-C6H5)(COJ; Сг(я - С5Н5) (NOJ ; Hg(o-C5H5); Cu[P(C5H5K]
Смещения металл о органических мигрантов по периметру цик-
лопентадиенового кольца происходят при комнатной температуре
с частотой порядка 103 с, и в спектрах ЯМР1!! в этих условиях
все протоны кольца равноценны, т. е. соединения XXVIII харак-
характеризуются усредненной структурой с эффективной осью симмет-
симметрии пятого порядка.
Выше уже рассматривались примеры валентной таутомерии —
внутримолекулярных перегруппировок, в которых обратимые про-
процессы разрыва—образования связей происходят без миграций от-
отдельных групп атомов по молекулярному остову (см. соединения V,
VI). В некоторых случаях, даже при использовании методов с очень
высоким временным разрешением, трудно определить, к какой
таутомерной форме должно быть отнесено соединение, структурная
нежесткость которого вызвана валентной таутомерией. Рассмотрим
в качестве примера валентную- таутомерию [10] аннулен—динор-
карадиен ХХ1Ха^?ХХ1Хб. Для дицианопроизводного (R, = R2 =
= CN), имеющего, по данным рентгеноструктурных исследований
в кристалле, длину связи С! —С2 0,1526 нм, принадлежность к типу
XXIX6 достаточно ясна. Так же очевидно, что незамещенное соеди-
соединение XXIX (R, = R2 = H) с длиной связи С, —С2 0,2235 нм устой-
устойчиво в кристалле в форме XXIXa. Однако для диметильного произ-
производного (R1 = R2 = CH3) выделены две различные кристаллические
формы, для одной из которых связь Ct —С2 имеет длину 0,1770 нм,
а для другой — 0,1826 нм:
A2.25)
ХХ|Ха
A(R1=R2=CN)
/(С,С2) =0,1526 нм
XXIX6
0,177Онм
0,1826 нм
0,2235 нм
483
в
А
С
Сз
Рис. 12.10. Распределение электронной плотности, градиентные пути и молекуляр-
молекулярные графы соединений XXIХа согласно данным неэмпирических расчетов (базис
STO—3G):
показан молекулярный фрагмент, соответствующий цшслопропановому кольцу в XXIX6: а —
градиентные пути, ограничивающие бассейны атомов; 6 — связывающие градиентные пути;
в — седловые точки на связывающих путях (минимумы электронной плотности), определяющие
наличие связи. В первой часта рисунка показаны фрагменты молекулярных графов в области
атомов Cj, С2, С3.
А-Ль
С-Дь
0,1543 нм); B-
нм)
Решить в этом и подобных случаях вопрос о том, на какой
стадии удлинения связи С]-С2 происходит ее разрыв и при каком
расстоянии между этими атомами связь еще существует, можно
только на основании анализа топологических характеристик рас-
распределения электронной плотности в молекулах XXIX (см. разд.
5.6). Как показывает рис. 12.10, связевый путь между атомами Q,
С2 и соответствующая седловая точка еще сохраняются для струк-
484
туры с l(CjC2) =0,1710 нм, тогда как для структуры
с 1(С[С2) =0,1826 нм эти характеристики уже отсутствуют. Следо-
Следовательно, в первой структуре молекулам диметильного производ-
производного соединения XXIX отвечает таутомерная форма XXIX6, а во
второй — XXIXa. В растворах обе формы переходят друг в друга
с очень высокой скоростью.
12.7. ВНУТРЕННЕЕ ВРАЩЕНИЕ
Изучение процессов стереохимической нежесткости началось
с обнаружения Дж. Кемпом и К. Питдером A936) внутреннего
вращения в молекуле этана. Рассчитывая термодинамические функ-
функции этана, эти авторы обнаружили, что теоретическое значение
энтропии отличается от экспериментального. Это расхождение за-
заставило предположить наличие еще одной степени свободы в эта-
этане— свободного внутреннего вращения вокруг связи С—С A2.1),
но разногласия опыта и теории полностью устранить не удалось.
Если допустить, что вращение происходит не свободно, а затор-
моженно, требуя преодоления активационного барьера
~ 12 кДж/моль, достигается полное согласие с экспериментом. Та-
Таким образом, полная энергия молекулы этана зависит от взаимного
расположения водородных атомов метильных групп (см. рис. 12.1).
Значения энергетических барьеров вращения относительно про-
простых связей обычно весьма низкие, некоторые из них представлены
в табл. 12.4. Как следует из данных таблицы, даже применение
минимального базиса орбиталей позволяет достаточно точно восп-
воспроизвести экспериментальные значения барьера вращения.
Таблица 12.4. Рассчитанные в экспериментальные значения энергетических
барьеров вращения вокруг иростых связей (кДж/моль)
Молекула
СН3—СН3
СН3—NH2
СНз—ОН
СН3—SiH3
СН3—РН2
СН3—СНО
ОН—ОН (цис-б&рьер)
(транс-барьер)
ВН3—NH3
STO-3G
12,1
11,8
8,3
5.4
7,9
4,6
38,0
0,4
8,8
6-31 G*
12,5
8.3
6,3
5,9
8,2
4,6
38,5
3,7
7,9
Эксперимент
11,9(ИК-спектр)
8,3 (микроволн.)
4,5 (микроволн.)
7,1 (термодин.)
8,2 (микроволн.)
4,9 (микроволн.)
29,4 (ИК-спеггр)
4,6 (ИК-спектр)
485
При введении заместителей в молекулу этана и метильные груп-
группы соединения (см. табл. 12.4) потенциальная функция вращения
становится асимметричной, число минимумов ППЭ значительно
возрастает и они имеют разную глубину. Поскольку значения
барьеров переходов между отдельными конформационными изо-
изомерами имеют в отсутствие выраженных стерических помех до-
достаточно низкие значения, ППЭ, усложняясь, сохраняют уплощен-
уплощенный рельеф. Примером служит расчет ППЭ внутреннего вращения
молекулы н-бутана XXX, которая имеет два устойчивых гош
(ХХХа)- и транс (ХХХб)-конформера (рис. 12.11).
н
ч
н
н
н,
н
н
н
XXX ХХХа. 0, = 60° ХХХб. в, = 180е
В «-бутане предпочтительным (~5 кДж/моль) является про-
пространственно менее затрудненный транс-ковформер. При наличии
полярных заместителей, как в дихлорэтане VII, или замене отдель-
отдельных cr-связей неподеленными электронными парами более устой-
устойчивыми оказываются конформеры, строение которых удовлетворя-
удовлетворяет так называемому гош-правилу: молекулярная структура наиболее
устойчива в форме с максимальным числом гош-взаимодействий
между электронными парами или полярными связями.
Таблица 12.5. Устойчивые конформацш молекул в соответствии с гош-правнлом
Молекула
N2H4
Р2Н4
Н2О2
H2S2
fch2—он
Стружтура
н
Диэдральный угол, град
теория
90—95
90—100
111
90,6
60
эксперимент
116
123
91,3
486
360'
Данные, представленные
в табл. 12.5, иллюстрируют
гвш-правило. Молекулы на-
наиболее устойчивы в формах
с максимальным числом ско-
скошенных друг относительно
друга (угловых) расположе-
расположений электронных пар или по-
полярных связей.
В циклических молекулах
вращения относительно от-
отдельных ст-связей совершают-
совершаются лишь в интервалах диэд-
ральных узлов, определяемых
размерами цикла. Результа-
Результатом таких согласованных
между собой процессов явля-
является инверсия циклической
формы, которая может фик-
фиксироваться подходящим экс-
экспериментальным методом
в зависимости от величины барьера и характера замещения. В четы-
четырехчленных циклах XXXI инверсионные барьеры, рассчитанные на
основе анализа колебательных (ИК- и комбинационного рассеива-
рассеивания) спектров, имеют предельно низкие значения:
о2
Рис. 12.11. Сечение поверхности потенци-
потенциальной энергии E(Qi, Qj) при ?i = 180° мо-
молекулы н-бутана
XXXIa
XXXII
XXXI6
A2.26)
Х=СН2 О S Se C = O
ДЕ=6Д кДж/моль . . 0,2 3,3 4,5 0,04
Форма адиабатического потенциала таких молекул показана на
рис. 12.12. Нижний колебательный уровень находится выше барьера
инверсии. Это означает, что даже при температуре 0К инверсион-
инверсионный процесс A2.26) «не заморожен» и молекула флуктуирует между
двумя нешюскими формами. Все ее экспериментально наблюда-
наблюдаемые характеристики будут иметь усредненные по этому потенци-
потенциалу значения. Колебания таких систем отличаются очень высокой
степенью ангармонизма. Для сравнения (см. рис. 12.12) показаны
также потенциальные кривые для систем с промежуточными (NH3)
и высокими энергетическими барьерами инверсии.
С увеличением размеров цикла число конформационных изо-
487
Е,нДж/тлъ
JO" 0 30' a 20° 0 20° a 50' 0 SO' ex
Рис. 12.12. Потенциальные функции инверсии и схема расположения нижних колеба-
колебательных уровней в молекулах циклобутана, аммиака и формальдегида:
угол а — угол изверевг
меров, принимающих участие в процессах инверсии колец, быстро
возрастает. В наиболее изученном теоретически и экспериментально
процессе инверсии шестичленного цикла циклогексана, связанном
с коррелированным вращением по шести углерод-углеродным свя-
связям, промежуточным конформером является mewcm-форма, а полу-
кресловидная конформация и конформация ванны отвечают пере-
переходным состояниям инверсионного процесса (рис. 12.13).
К процессам внутреннего вращения относятся и многие низ-
низкобарьерные изомеризации я-комплексных соединений, вызывае-
вызываемые поворотами металлоорганических фрагментов относительно
?, кДж!' MO/ft>
ОО
Координата реакции
Рис. 12.13. Потенциальная кривая инверсии циклогексаяового кольца согласно ква-
нтово-химическям расчетам и экспериментальным данным
488
остальной части молекул. Простым примером является взаимопре-
взаимопревращение двух форм комплекса этилен — Fe(COL (см. разд. 11.4.3).
Барьер для такого процесса достаточно высок — 45 кДж/моль.
Значительно более низкие барьеры зафиксированы, по данным элек-
тронографических и ИК-спектральных исследований, для поворота
колец в сэндвичевых я-комплексах:
A2.27)
XXXIIIi XXX1V ХХХП16
М = Fe C,8 кДж/моль); Ni, Со G.5 кДж/моль)
В хромоцене (дибензолхроме) аналогичный барьер также состав-
составляет 7—8 кДж/моль, а в ураноцене (бис-циклооктатетраенуране) он
падает почти до нуля. Форма потенциала для таких систем подобна
представленной на рис. 12.12 (левая часть), хотя содержит большее
число минимумов.
Рассмотренные типы структурной нежесткости не исчерпывают
все многообразие этих процессов, хотя и представляют наиболее
важные их типы, к которым можно свести большинство превраще-
превращений стереохимически и электронно нежестких молекул. С развитием
техники экспериментальных исследований низкобарьерных пере-
перегруппировок и методов квантовой химии, надежность предсказаний
которых обеспечивается прогрессом ЭВМ, становится ясным, что
структурная нежесткость — одно из самых общих свойств молекул
различных классов.
Литература
Внутреннее вращение молекул/Под ред. В. Дж. Орвилл-Томас— М.: Мир,
1977.
Сборник статей, посвященных экспериментальному в теоретическому изучению проблем
внутреннего вращения и инверсии, конформационной изомерии молекул. Рассмотрены история
вопроса, применения различных физических методов.
Дашевскнй В. Г. Конформационный анализ органических молекул. — М.: Хи-
Химия, 1982.
Награда М. Стереохимия. — М.: Мир, 1984. Гл. 2.
Применение метода ЯМР-спеггросжопии для изучения процессов внутреннего вращения,
инверсии, псевдовращения в органических и элементоорганичесжих соединениях.
Пирсов Р. Правила симметрии в химических реакциях. — М.: Мир, 1979. Гл.
1—3.
Четкий анализ природы проявлений структурной нежесткости, основанный на диаграммах
Уолша и представлениях об эффектах Ява—Геллера.
489
Minkin V. L, Olekhnovich L. P., Zhdanov Yd. A. — Molecular Desigh of
Tautomeric Compounds. — D. Reidel Publ. Dordrecht—Boston—Tokyo—Toronto.
1988.
Русское издание: Мшкяа В. И., Олехяовп Л. П., Ждавов Ю. А. Молекулярный дизайн
таутомервых систем. Иад-во Ростовского университета, 1977.
Классификация и обзор таутомернык превращений.
ГЛАВА 13
СОХРАНЕНИЕ ОРБИТАЛЬНОЙ СИММЕТРИИ
В ХИМИЧЕСКИХ РЕАКЦИЯХ
Расчет поверхностной потенциальной энергии (ППЭ) реакции,
выявление координаты реакции и переходного состояния (седловой
точки ППЭ) представляют собой наиболее прямой и строгий под-
подход к анализу проблемы реакционной способности. Такой подход
сопряжен обычно с весьма большим объемом вычислений. Между
тем во многих случаях не обязательно знать полную ППЭ реакции
и достаточно сведений о структуре ее отдельных участков, опреде-
определяющих способ сближения реагентов и тип переходного состояния
реакции. По этим причинам, а также вследствие потребности в вы-
выработке концепций и правил, способных объяснять реакционную
способность и механизм реакций на основе данных о строении
исходных реагентов, возникает необходимость в создании эффек-
эффективных упрощенных методов рассмотрения указанных задач.
В 1965 г. Р. Вудворд* и Р. Хоффман выдвинули чрезвычайно
плодотворную идею анализа стереохимии и относительной оценки
энергетических барьеров весьма широкого круга, органических реак-
реакций, основанную на сопоставлении свойств симметрии орбиталей
исходных реагентов и продуктов. В наиболее общей форме эта идея
сводится к положению о том, что согласованные (одностадийные)
реакции, в которых заполненные орбитали системы реагирующих
м^ттнгутт и прбитппи \тпш*куп продуктов рннгции ¦¦- ттолтюстыо
соответствуют друг Другу по свойствам симметрии на протяжении
всего пути реакции (коррелируют между собой), протекают в задан-
заданном электронном состоянии легче, чем согласованные реакции,
в которых указанное соответствие нарушается. Несколько упрощая,
¦Роберт Берне Вудворд A917—1979) — американский химик, крупнейший специ-
специалист в области синтетической органический химии, Нобелевский лауреат по химии
A965). Им было проведено более 20-ти сложных направленных синтезов природных
продуктов (например, хинин, резерпин, хлорофилл, тетрациклин и др.), до этого
считавшихся неосуществимыми. Р. Б. Вудворд — иностранный член АН СССР (с
1976).
490
можно сократить формулировку до утверждения Р. Вудворда
и Р. Хоффмана: «...в согласованных реакциях сохраняется орби-
орбитальная симметрия».
Этот принцип носит весьма общий характер и применим не
только к органическим реакциям, но также и к реакциям неор-
неорганических и координационных соединений. Далее рассмотрим от-
отдельные примеры таких реакций. Однако наиболее характерный
класс химических превращений, для которых применение принципа
сохранения орбитальной симметрии имеет первостепенное значе-
значение, — это так называемые перициклические реакции органических
молекул.
13.1. ПЕРИЦИКЛИЧЕСКИЕ РЕАКЦИИ
Прежде чем перейти к анализу следствий и приложениям
принципа сохранения орбитальной симметрии, полезно определить
терминологию и обозначения, а также дать краткое введение в об-
область перициклических реакций.
В самой формулировке принципа отмечено, что его действие
распространяется на согласованные (концертные) реакции. К согла-
согласованным реакциям относят такие, которые протекают в одну
стадию, т. е. без участия промежуточных продуктов (ионов,
радикалов, молекулярных комплексов, других интермедиатов).
Примерами согласованных реакций могут служить бимолекулярное
5^2-замещение в алифатическом ряду, реакции диенового синтеза,
большинство циклоприсоединений, многие другие, рассмотренные
далее. Важной особенностью реакций этого типа является их высо-
высокая стереоспецифичность. В случае многостадийной реакции каж-
каждую ее отдельную стадию, заключенную между двумя локальными
минимумами ППЭ реакции, можно анализировать как согласован-
согласованную реакцию.
Большинство согласованных реакций связано с формированием
циклических переходных состояний, в которых наиболее существен-
существенные изменения характера связывания относятся к атомам и ор-
биталям, образующим замкнутые системы. Такие реакции были
названы Вудвордом и Хоффманом перициклическими. В перицик-
перициклических реакциях орбитали тех связей и групп, которые непосред-
непосредственно принимают участие в процессах разрыва старых и об-
образования новых связей, могут быть объединены единой замкнутой
кривой, которую называют соединяющим циклом (см. формулы
I—V). Узловые свойства таких систем орбиталей, включенных в со-
соединяющие циклы, играют решающую роль при определении харак-
характера протекания реакции.
Выделяют несколько типов перициклических реакций.
491
13.1.1. Реакции цнклопрнсоедннення
Реакции димеризации этилена
н2с=сн2
н2с=сн2
(«2 + *г)
н2с сн2
н2с—сн2
и дейтерообмен
(о1
II
HD + HD
(*2 + о2)
представляют собой наиболее простые примеры реакций цикло-
присоединения. Они классифицируются по типу орбиталей, образу-
образующих связи, и числу электронов на этих орбиталях. Следует заме-
заметить, что обратная циклоприсоединению реакция циклораспада ци-
клобутана на две молекулы этилена описывается как присоединение
двух сг-связей.
Широко известная реакция диенового синтеза (реакция Дильса—
Альдера) — присоединение олефинов к диенам — рассматривается
как циклоприсоединение п2 + т& или п2 + п2 + я2, а обратная реакция
циклораспада — как с2+с2 + 2
(я* +
m
((Г2
Пг)
Для более полного описания реакции вводится дополнительное
определение, характеризующее стереохимию присоединения. Если
связи, образуемые (или рвущиеся) данным компонентом реакции,
например молекулой этилена, лежат по одну сторону молекулярной
плоскости, такое превращение относится к супраповерхностпным
492
и обозначается индексом «s». Все рассмотренные реакции относятся
к супра-супраприсоединениям.
Если же образуемые молекулой связи расположены в переход-
переходной структуре по разные стороны ее молекулярной плоскости, такое
присоединение относится к антараповерхностному типу и обознача-
обозначается индексом «а». Так, если реакция циклодимеризации этилена
осуществляется через переходное состояние типа IV, ее следует
отнести к tcJ +^-присоединению:
С
н
г
CHt
Н2С
CHi
Схема A3.1) дает представление о разделении реакций присо-
присоединения на супра- и антараповерхностные в зависимости от харак-
характера перекрывания орбиталей, участвующих в образовании (раз-
(разрыве) связей. Стрелки показывают направления подхода реагентов
к орбиталям, принимающим участие в образовании переходного
состояния. Если в связывании участвует лишь одна р-орбяталь
молекулы, то для реакции вводят обозначения со0, ш1 или со2
в зависимости от числа электронов на р-орбитали:
й И
A3.1)
s _Jr ш'
Задача 13.1. Установите тип следующих реакций циклоприсоединения:
a)
б)
13.1.2. Электроциклические реакции
Такое название принято для внутримолекулярных реакций цик-
493
лизации полиенов и их гетероатомных аналогов, приводящих к со-
соединениям циклоалкенового и гетероциклического рядов:
A3.2)
Наиболее простой случай — циклизация 1,3-Диенов (л=0). Ог-
Огромный экспериментальный материал, накопленный химиками-ор-
химиками-органиками, в частности при изучении методов синтеза сложных
природных циклоалкенов, показывает, что при проведении реакций
в обычных условиях — при нагревании бутадиенов V — образуют-
образуются почти исключительно транс-циклобутены Via. Если же вести
реакцию при облучении УФ-светом, т. е. через электронно-возбуж-
электронно-возбужденное состояние, единственными продуктами оказываются цис-\щ-
клобутены VI6. Эти результаты являются следствиями двух различ-
различных путей реакции — конротаторного и дисротаторного вращений
концевых групп бутадиена:
¦Ч*
Ч*-
У!а 3t?
конротаторное вращение
дисрота/порное вращение
Электроциклические реакции можно классифицировать в соот-
соответствии с общими обозначениями перициклических реакций, рас-
рассматривая их как внутримолекулярные реакции циклоприсоедине-
ния или циклораспада. Конротаторное и дисротаторное вращения
ведут к перекрыванию различных лопастей ря- и^-орбиталей, кото-
которые подразделяются на супра- и антараповерхностные в соответст-
соответствии со схемой A3.1).
494
Подробное исследование широкого круга электроциклических
реакций типа A3.2) привело к выводу о том, что для полиенов
с четными л, как и для бутадиена (л = 0), в термических условиях
легко протекает коиротаторная циклизация. Для полиенов с нечет-
нечетным числом двойных связей (л нечетное) в этих условиях легче
осуществляются дисротаторные замыкания. Природа такой стерео-
селективности будет выяснена далее.
13.1.3. Сигматропные реакции
Под таким названием объединяется обширная группа согласо-
согласованных реакций, в которых а-связь, сочлененная с одной или не-
несколькими сопряженными л-системами, мигрирует в новое положе-
положение. Типичная сигматропная реакция — важная в органической
химии перегруппировка Коупа:
и + я; + я*
2'
3'
Vita.
VII б
Здесь <7-связь из положения 1,Г смещается в положение 3,3'.
Соответственно реакция классифицируется как [3,3]-сигматропный
сдвиг, так как обозначение [/, j] отвечает смещению сг-связи из
положения 1,Г в положение ij (отсчет начинается от атомов, связан-
связанных мигрирующей связью).
Подробно изучены [l,j]-сигматропные сдвиги атомов водорода
в полиеновых углеводородах, их циклических и гетероатомных ана-
аналогах:
1 "t— (CH=CH)n— СН=С
\,
1
С=СН—(СН=СН)П—
Легкость протекания термической реакции сильно зависит от
числа виниленовых звеньев п и регулируется правилами, рассма-
рассматриваемыми в разд. 13.3.3. Переходные состояния для [1,у]-сигма-
тропных сдвигов водорода имеют циклическую структуру, обес-
обеспечивающую их принадлежность к классу перициклических про-
процессов. В зависимости от характера перекрывания базисных АО
в переходном состоянии (и, следовательно, геометрии последнего)
реакции подразделяются на супра- и антараповерхностные (рис.
13.1).
495
Рис. 13.1. Циклические переходные состояния для согласованных реакций супрапо-
верхностного (а) и антараповерхностного (б) [I, jj-сдвигов водорода в сигматропных
реакциях типа A3.3)
13.2. АНАЛИЗ ПЕРИЦИКЛИЧЕСКИХ РЕАКЦИЙ
С ПОМОЩЬЮ МЕТОДА КОРРЕЛЯЦИОННЫХ ДИАГРАММ
Наиболее распространенный подход к анализу перициклических
реакций связан с методом корреляционных диаграмм Вудворда—
Хоффмана. Общая идея подхода проистекает из аналогий с кор-
корреляционными диаграммами для двухатомных молекул (см. разд.
4.6) и квантово-механическим правилом непересечения уровней оди-
одинаковой симметрии. Различия заключаются в том, что вместо пред-
предполагаемых реакций типа «атом 4- атом->двухатомная молекула-»-
-*¦ объединенный атом» анализируются молекулярные реакции.
13.2.1. Циклоприсоединение
Рассмотрим общую схему построения корреляционной диаграм-
диаграммы химической реакции вначале на примере простейшей реакции
Gг2 + 7Г2)-циклоприсоединения. Целесообразно придерживаться сле-
следующих правил.
н
ОС
б
Рис. 13.2. Плоскости симметрии, относительно которых классифицируются МО
в системе I (а); вид орбиталей и относительные энергетические уровни двойной связи
этилена (б); С—С-связи циклобутана (в)
496
1. Выделить связи и орбитали, принимающие главное участие
в реакции, учесть порядок энергетических уровней выделенных ор-
биталей реагентов и продуктов.
2. Задав путь сближения реагентов или отдельных связей при
внутримолекулярной реакции в направлении формирования ожида-
ожидаемой структуры переходного состояния, классифицировать МО ре-
реагентов и продуктов по свойствам симметрии, присущей выбранной
структуре переходного состояния.
3. Установить соответствие между МО реагентов и продуктов
в рамках выбранной классификации орбиталей по симметрии. При
этом необходимо, чтобы оси или плоскости симметрии ожидаемого
переходного состояния проходили через рвущиеся и образующиеся
в ходе реакции связи. Элементы симметрии, не пересекающие рву-
рвущиеся и образующиеся связи, несущественны для анализа реакции.
В реакции димеризации этилена, в которой сближение двух
реагирующих исходных молекул проходит по пути, соответству-
Рис. 13.3. Корреляционная диаграмма для реакции димеризации двух молекул эти-
этилена в основном электронном состоянии:
а — я-орбитали изолированных молежул этилена; б — <т-орбитали взаимодействующих молекул
этилена. Указаны свойства симметрии относительно плоскостей 1 и 2 на рис. 13.2; в —орбитали
молекулы циклобутана
497
п
Рис. 13.4. Разрешенное
по симметрии фотоинду-
цированное циклоприсое-
динение двух молекул
этилена
ющему конфигурации I, происходит превра-
превращение двух я-связей в две <т-связи, орбитали
которых показаны на рис. 13.2. Две плоско-
плоскости симметрии секут рвущиеся и образую-
образующиеся связи и являются поэтому правильно
выбранными элементами симметрии. По-
Поскольку отдельная я-орбиталь этилена или
(т-орбиталь связи С—С циклобутана не удо-
удовлетворяет требованию симметрии или ан-
антисимметрии по отношению к обеим вы-
выбранным плоскостям симметрии, нужно об-
образовать такие линейные комбинации этих
вырожденных МО, которые обеспечивают
указанное требование.
Рис. 13.3 показывает построение таких
орбиталей, а также классификацию их на
симметричные S и антисимметричные А относительно двух выбран-
выбранных плоскостей симметрии. Сопоставляя МО системы сближенных
молекул этилена с МО циклобутана, можно видеть, что высшая
заполненная Л-орбиталь реагентов не переходит в результате реак-
реакции в высшую заполненную орбиталь циклобутана (не коррелирует
с ней). В то же время наблюдается пересечение уровней заполнен-
заполненных электронами МО реагентов с антисвязывающим уровнем цик-
циклобутана. Таким образом, при условии сохранения орбитальной
симметрии в процессе согласованной химической реакции два запо-
заполненных электронами уровня системы реагирующих молекул этиле-
этилена не могут переходить в два низших энергетических уровня цик-
циклобутана. Данная реакция считается запрещенной по симметрии
в основном электронном состоянии.
Предположим теперь, что реакция димеризации осуществляется
в условиях фотохимического возбуждения системы реагирующих
молекул этилена УФ-светом. Большинство фотохимических реак-
реакций протекает через низшее возбужденное электронное состояние,
соответствующее переносу одного электрона на низший свободный
уровень. Как видно из рис. 13.4, в этом случае имеется полное
соответствие по свойствам симметрии всех заполненных электрона-
электронами орбиталей реагентов и продукта в первом возбужденном элект-
электронном состоянии и не происходит пересечения уровней заполнен-
заполненных и валентных МО реагентов и продуктов. Реакция относится
к разрешенным по симметрии в электронно-возбужденном состоя-
состоянии. Для ее реализации необходимо облучение светом с длиной
волны, соответствующей энергии я->я*-электронного перехода
в этилене.
Примером реакции я2+я2-циклоприсоединения в электронно-
возбужденном состоянии служит изученная на многих примерах
фотохимическая димеризация ненасыщенных соединений в кристал-
498
лическом состоянии. Если кристаллическая упаковка этих соедине-
соединений такова, что соответствует супра-супраориентации и плоскости
двойных связей достаточно сближены @,35—0,42 нм), при облуче-
облучении кристалла происходит твердофазная реакция циклодимериза-
ции, например в случае производных л-дивинилбензола:
Надо иметь в виду, что запрет по симметрии не означает дейст-
действительную невозможность протекания реакции в данном элект-
электронном состоянии. Запрещенные по симметрии реакции требуют
высокой энергии активации или протекают предпочтительно не как
согласованные процессы, а включают промежуточное образование
радикальных или ионных частиц. Так, экспериментально установ-
установлено, что обратная циклодимеризации этилена реакция термолиза
циклобутана включает промежуточное образование дирадикала те-
траметилена:
сн
н2с
сн2-
н2с
сн,
сн2»
Такой механизм термической реакции объясняется пространст-
пространственными затруднениями образованию промежуточной структуры
IV при супра-антарарасположении этиленовых фрагментов, кото-
которые, как будет показано далее, отвечают условию сохранения ор-
орбитальной симметрии.
6 то же время реакции циклоприсоединения, не связанные с про-
пространственными ограничениями и разрешенные по симметрии в ос-
499
\-J
Рис. 13.5. Корреляционная диаграмма для реакции супра-супра (Я4-+-я2)-циклопри-
соединения 1,3-бутадиена и этилена:
орбиталв классафицврованы по симметрии относительно плоскости, перпендикулярной плоско-
плоскости образующихся 0-связей
500
Рис. 13.6. Корреляция вза-
взаимодействующих орбиталей
1,3-бутадиена и этилена при
супра-супра цикл оприсое дине-
нии, ведущем к циклогексеяу,
по данным неэмпирических
(базис STO—3G) расчетов:
заметьте, что уровни МО оди-
одинаковой симметрии не пересека-
пересекаются
¦-/•¦
.S ^—
новном состоянии, требуют, как правило, низких энергий актива-
активации. К числу разрешенных циклоприсоединений относится реакция
(п* + я^-присоединения при геометрии сближения III. На рис. 13.5
показана орбитальная корреляционная диаграмма для реакции цик-
лоприсоединения бутадиена и этилена, а на рис. 13.6 приведены
данные неэмпирического расчета, позволяющего проследить дейст-
действительный характер изменений энергетических уровней основных
взаимодействующих орбиталей в ходе данной реакции.
Задача 13.2. Построить корреляционную диаграмму для согласованной
реакции: 1) (я*+л|)-присоединения бутадиен + бутадиен; 2) (л^ч-л^)-циклодиме-
ризации этилена.
Задача 13.3. Термические реакции (п]+я^-цяхлоприсоединения достаточно
легко протекают в условиях гомогенного катализа ионами переходных металлов,
особенно с ^-электронной оболочкой. Покажите, пользуясь методом корреляцион-
корреляционных диаграмм, что эти реакции являются разрешенными по орбитальной симметрии.
Обобщая рассмотренные выше данные для реакции циклодиме-
ризации этилена и решения задачи 13.2, можно получить общие
правила для разрешенных и запрещенных по симметрии супра-
и антараповерхностных циклоприсоединений полиенов в зависимо-
зависимости от числа их двойных связей или числа электронов. Эти правила
представлены в табл. 13.1.
Таблица 13.1. Разрешенные по орбитальной симметрии реакции циклопрясоеди-
вення в зависимости от общего числа л-электронов (q~ 0, 1, 2,...)
Тип присоединения
Супра, супра
Антара, ангара
Супра, антара
Термическая реакция
4? + 2
Фотохимическая
реакция
4д
501
13.2.2. Электроциклические реакции
Построим корреляционные диаграммы для двух отмеченных
(см. разд. 13.1.2) путей внутримолекулярной циклизации замещен-
замещенных бутадиенов.
Для конротаторного вращения элементом симметрии, относи-
относительно которого следует классифицировать МО исходного бутади-
бутадиена и продукта реакции циклобутена, служит ось симметрии второ-
второго порядка:
н
Вид МО бутадиена известен из разд. 8.3.3. Для циклобутена
необходимо принять во внимание я-орбитали двойной связи и а-
орбитали простой связи, рвущейся при размыкании кольца. Кор-
Корреляционная диаграмма представлена на рис. 13.7.
Как видно из рис. 13.7, заполненные орбитали бутадиена полно-
полностью коррелируют с двумя заполненными орбиталями циклобуте-
а о
Рис. 13.7. Корреляционная диаграмма для конротаторной циклизации бутадиена:
а — МО бутадиена; б — МО циклобутена, играющие главную роль в реакции
502
на. Следовательно, реакция разрешена по симметрии в основном
электронном состоянии.
Построим теперь корреляционную диаграмму для дисротатор-
ного замыкания бутадиена:
В этом случае элементом симметрии служит уже не ось, а плос-
плоскость, и МО классифицируются на симметричные и антисимметрич-
антисимметричные относительно плоскости симметрии, как показано на рис. 13.8.
Высшая занятая МО бутадиена коррелирует при дисротаторном
процессе не с орбиталью основного состояния циклобутена, а с низ-
низшей свободной МО. Процесс запрещен по симметрии в основном
состоянии. Однако легко убедиться, пользуясь корреляционной диа-
диаграммой на рис. 13.8, что первое возбужденное состояние бутадиена
(щJ faI (щI коррелирует с первым возбужденным состоянием ци-
циклобутена (аJ (л;I (я*I. Следовательно, реакция разрешена по сим-
симметрии при фотохимическом пути.
Общие для электроциклических реакций A3.2) правила сохране-
сохранения орбитальной симметрии сформулированы в табл. 13.2.
Рис. 13.8. Корреляционная диаграмма для дисротаторной циклизации бутадиена
503
Таблица 13.2. Разрешенные но орбитальной симметрии электроциклнческне ре-
реакции в зависимости от общего числа я-электровов 07 = 0, 1, 2,...)
Тип lafuruTHTraa
Конротаторный
Дисротаторный
Термическая реакция
Ц
4^+2
Фотохимическая
реакция
4? + 2
13.2.3. Истинные пути электроциклических реакций
Точные расчеты критических участков ППЭ перициклических
реакций обнаруживают, что истинные пути их реакций в дейст-
действительности сложнее и, как правило, менее симметричны, чем при-
принятые в приведенном выше качественном рассмотрении. Проил-
люстрирум это на примере электроциклической реакции бутадиен—
циклобутен, в которой в качестве координаты реакции принимался
угол вращения метиленовых групп.
Прямые расчеты показывают, что вращение метиленовых зве-
звеньев не определяет координату реакции на ее начальном участке.
Если рассматривать реакцию со стороны циклобутена, такой коор-
координатой служит растяжение связи С —С. Расчеты показывают, что
при изменении R от равновесной для циклобутена величины
0,1534 -г- 0,2270 нм метиленовые группы не вращаются. Вращение
начинается лишь при достижении R = 0,207 нм (MINDO/3)
0,2270 нм (ab initio, 4-31 G), причем в процессе вращения метилено-
метиленовых групп величина R уже не меняется (см. рис. 13.9). Именно в этой
стадии реакции возникают отличия в энергетике конротаторного
и дисротаторного каналов (см. рис. 13.10). Только после окончания
вращения метиленовых групп до требуемой структурой 1,3-бутади-
ена величины угла в продолжается растяжение R до равновесного
значения 0,2819 нм в бутадиене. Неплоское переходное состояние
С2-симметрии VHI с диэдральным углом ^ = 54° возникает в зоне
перехода реакционной координаты от растяжения связи С — С к вра-
вращению метиленовых групп:
Н л X, 1*
С
У) к
0
С s
VIH
504
Схематически путь реакции пока-
показан на рис. 13.9, который под-
подчеркивает, что изменения парамет-
параметров R и 0 практически независимы.
Успех правил Вудворда—Хоф-
фмана в объяснении предпочти-
предпочтительности термического конрота-
торного процесса объясняется тем,
что изменение R в указанных пре-
пределах не отражается на относитель-
относительном расположении и симметрии гра-
граничных и близких к ним орбиталей.
Вследствие этого выводы, сделан-
сделанные на основании рассмотрения рав-
равновесных структур, оказываются
справедливыми и для той области
пути реакции, в которой фактически
и различаются разрешенный конро-
таторный и запрещенный дисрота-
торный пути.
Для правильного описания последнего необходим точный учет
эффектов электронной корреляции. Это связано в первую очередь
пересечением орбиталей при
О
Рис. 13.9. Схематический вид син-
синхронного (прямая 2) и несинхрон-
несинхронного (пунктирная 3 и сплошная
1 линии) путей реакции циклобу-
тен — бутадиен:
2 — одновременное изменение R а в
1 — горизонтальный участок — измене-
изменение R, вертикальный — изменение в.
3 — горизонтальные участки Л, верти-
вертикальный — изменение в
С
следовании реакции по дисрота-
торному пути. Так, представ-
представленные на рис. 13.10 данные не-
неэмпирических расчетов показы-
показывают, что учет электронной кор-
корреляции при помощи метода KB
не только более правильно опи-
описывает барьер и энергетику пре-
превращения, но и устраняет де-
дефект хартри-фоковского расчета
для дисротаторного механизма.
Приближение метода Хартри—
Фока дает результат, противо-
противоречащий принципу микроскопи-
микроскопической обратимости, так как те-
термически запрещенная дисрота-
торная реакция приводит
к двум различным электронным
состояниям (разные заполнения
орбиталей) в зависимости от то-
того, идет ли реакция в направле-
направлении от бутадиена к циклобутену
или наоборот.
EJ8
-9
-11
-13
-П
-Т9
-21
i
0* 15* 30
60' 75' 90'
в
Рис. 13.10. ПРМЭ электроцшишческой
реакции бутадиен — циклобутен, рассчи-
рассчитанный в приближениях метода Харт-
ри—Фока и ХФ+КВ:
координата реакции в — угол синхронного
вращения метиленовых звеньев; за ноль энер-
энергии принята энергия — 154 а. е.
505
13.2.4. Обобщенное правило отбора по симметрии
для согласованных перициклических реакций
Рассмотрев все различные виды перициклических реакций как
реакции циклоприсоединения (см. разд, 13Л), Вудворд и Хоффман
сформулировали следующее обобщенное правило отбора для со-
согласованных реакций: перициклическая реакция разрешена по сим-
симметрии в основном электронном состоянии, если общее число
Dq + 2)su Dq)a структурных компонент реакции нечетное. Для фото-
фотохимической реакции, разрешенной по симметрии, общее число от-
отмеченных компонент должно быть четным.
Нетрудно убедиться в том, что обобщенное правило включает
в себя как частный случай правила, представленные для реакций
циклоприсоединения (см. табл. 13.1) и электроциклических реакций.
Так, например, для конротаторного замыкания бутадиена Va
(ttJ + ttJ) имеется одна Dq + 2Х-компонента реакции (<? = 0). Реакция
должна быть разрешена правилами сохранения орбитальной сим-
симметрии. Для дисротаторного замыкания V6 (гс? +тг;) число отмечен-
отмеченных компонент равно двум. Реакция запрещена по симметрии.
Обобщенное правило отбора, весьма удобное для анализа широ-
широкого круга реакций, было сформулировано его авторами чисто
индуктивным путем. Его истинная природа понята на основе анали-
анализа топологии соединяющего цикла, объединяющего орбитали пери-
перициклической системы.
13.3. АРОМАТИЧНОСТЬ И ПРАВИЛА ВУДВОРДА — ХОФФМАНА
ДЛЯ ПЕРИЦИКЛИЧЕСКИХ РЕАКЦИЙ
Главная особенность перициклических реакций — возможность
выделения циклической системы базисных орбиталей в промежу-
промежуточной между исходными реагентами и продуктами молекулярной
конфигурации или комплексе. Если такая циклическая система ор-
орбиталей оказывается изосопряженной с ароматической D#+2)-си-
стемой, как, например, в структуре III (см. разд. 13.1.1), соответст-
соответствующее переходное состояние можно рассматривать как аромати-
ароматическое, т. е. стабилизированное. Такая реакция должна протекать
с малой энергией активации и относится к разрешенным по симмет-
симметрии.
Подобный подход может быть существенно обобщен и рас-
расширен при более подробном анализе топологических характеристик
(узловых свойств) системы базисных орбиталей промежуточного
комплекса в перициклической реакции.
13.3.1. Правила Циммермана для перициклических реакций
Г. Циммерман доказал, что узловые свойства базисного ядра
орбиталей соединяющего цикла перициклической реакции являются
506
доминирующей характери-
характеристикой при оценке стабильно-
стабильности ее переходного комплек-
комплекса. Соотношения типа пред-
представленных в табл. 13.3 выпо-
выполняются для базисных рядов,
составленных из различного
вида орбиталей (рис. 13.11).
Таблица 13.3. Правила Цнмме-
рмаиа для разрешенных иеряцнклв-
ческих реакций
а.
Рис. 13.11. Примеры хюккелевской (а) и ме-
биусовской (б) систем базисных орбиталей
соединяющего цикла перициклическои реак-
реакции:
стрелка указывают инверсии фазы орбиталей
Число узлов я
Четное
Нечетное
Топология
Хюккелевская
Мебиусовская
Число электронов 2/и
термическая
реакция
фотохимическая
реакция
4<?
4q + 2
м
Z=2
Поскольку наличие циклической системы орбиталей — главный
признак перициклическои реакции, создается возможность оцени-
оценивать относительную устойчивость ожидаемых переходных состоя-
состояний различных реакций (их ароматичность или антиароматич-
антиароматичность), учитывая лишь топологию базисных орбиталей соединя-
соединяющего цикла.
Для применения правил (см. табл. 13.3) необходимо в первую
очередь определить соединяющий цикл,
затем выделить его базисные орбитали,
сосчитать число узлов (инверсий фаз ор-
орбиталей) в соединяющем цикле и число
электронов Bт), заселяющих орбитали
связывающего цикла.
На первый взгляд может показаться,
что существует некоторый произвол
в выделении соединяющего цикла op- w w w so
биталей перициклическои реакции. Дей- v ' * •
ствительно, как следует из рис. 13.12,
например, для реакций циклоприсоеди-
нения (см. разд. 13.1.1) можно с рав-
равными основаниями по-разному предста-
представить соединяющий цикл орбиталей.
Легко убедиться также в том, что
независимо от того или иного выбора
соединяющего цикла для реакции
я?)-циклоприсоединения число уз-
1=0 Z=2
Рис. 13.12. Сохранение четно-
четности узловых характеристик со-
соединяющего цикла базисных
орбиталей при различных вы-
выборах их исходной ориентации:
стрелки указывают области инвер-
инверсии знака интеграла перекрывания
507
лов в цикле всегда четное и, поскольку система относится к q
электронной (#=1), реакция относится к термически запрещенным.
С другой стороны, D^+2)-электронная реакция (teJ + яй-циклопри-
соединения, где число узлов четное, относится к термически раз-
разрешенным.
С точки зрения правил Циммермана легко объяснить разрешен-
ность термических конротаторных циклизаций бутадиенов и запре-
щенность дисротаторных реакций (см. разд. 13.1.2). Для этого
достаточно рассмотреть топологию системы базисных орбиталей
соединяющих циклов Va, V6 и соотнести ее с правилами табл. 13.3.
Следует подчеркнуть достоинство действия рассматриваемых
правил, т, е. фактически обобщенной концепции ароматичности для
циклических систем типа Хюккеля и Мебиуса. В отличие от метода
корреляционных диаграмм, требующего наличия элементов сим-
симметрии для переходного состояния реакции, данный подход свобо-
свободен от указанного ограничения.
Задача 13.4. Пользуясь правилами табл. 13.3, объясните, почему в присут-
присутствии в качестве катализаторов переходных металлов запрещенные по симметрии
реакции (/г2 + тф-циклоприсоединения легко протекают без фотохимического воз-
возбуждения. Рассмотрите различные способы выбора системы базисных орбиталей.
13.3.2. Анализ снгматропыых реакций
Ограничимся рассмотрением [1, /]-сигматропных смещений
групп. Для миграций водородных атомов рассмотрение связыва-
связывающих циклов орбиталей (см. рис. 13.1) позволяет отнести суп-
раповерхностные сдвиги к хюккелевским, а антараповерхностные —
к мебиусовским системам. Отсюда вытекают следующие правила
для разрешенных и запрещенных по симметрии реакций (табл. 13.4).
Таблица 13.4. Правила отбора для сяпиатроиных смешений атомов водорода
в полненовых системах типа A3.3)
Число
электронов
2
4
6
4*
Aq+2
нейтральный
—
[1.3]
[1.5]
[1,D^-1I
[1,D?+ 1)]
Полнен
катион
П.21
[1,41
ид
[1,41
[1, D?+2)]
аннон
—
[1,2]
[1,4]
[1, D*-2)]
[1.4*]
Термически раз-
разрешенная мигра-
миграция
s
а
s
а
s
508
Если в качестве мигранта в [1,у]-сигматропной перегруппировке
выступает более сложная, чем атом водорода, группа, создается
возможность более подробного анализа строения переходного со-
состояния реакции по ее стереохимическому результату. Например,
для детально изученных 1,2-сигматропвых миграции алкнльных
групп в карбониевых катионах IX
ь
„mtC"
Г'
IX а
IX б
экспериментально установлено сохранение конфигурации мигриру-
мигрирующего углеродного центра при перегруппировке. Этот результат
легко объясним с точки зрения действия правил Циммермана, так
как в этой системе возможен по пространственным причинам лишь
супраповерхностный сдвиг алкила.
Действительно (см. табл. 13.3), соединяющий цикл базисных
орбиталей при z=0 (рис. 13.13, а), заселенный в катионе двумя
электронами, предпочтительнее, чем мебиусовский двухэлектрон-
ный цикл при z—\ (рис. 13.13, б).
Задача 13.5. Выведите общие правила термически разрешенных путей
[1, /|-сигматропных сдвигов хиральных алкильных групп с сохранением и об-
обращением конфигурации мигрирующего центра.
Я. ''Л,
Рис. 13.13. Сохранение конфигурации (а) и обращение конфигурации (б) мигриру-
мигрирующего углеродного центра аллильной группы при [1, 2]-сигматропиой перегруп-
перегруппировке карбониевого иона
509
13.3.3. Обобщенное правило Вудворда—Хоффмана
и свойства хюккелевских и мебиусовских систем
базисных орбиталеи
Правила ароматичности и правила отбора разрешенных перицн-
клических реакций (см. табл. 13.3) можно сформулировать еще
более общим образом. Отметим вначале, что в соединяющих цик-
циклах базисных орбиталеи инверсии знаков появляются лишь тогда,
когда в состав базиса включаются р-орбитали (или вообще ор-
битали с квантовыми числами /^ 1). При этом можно выделить два
типа узловых плоскостей. Первый относится к межорбитальному
перекрыванию, число отрицательных перекрываний равно Z. Вто-
Второй тип узловых плоскостей относится к внутриорбитальному об-
обращению знака, например для р-орбитали. Число внутриорбиталь-
ных инверсий обозначим N.
Прямое рассмотрение орбитальной структуры соединяющих
циклов (см. рис. 13.11—13.13, а также формулы I, Va, V6)
показывает, что для любого выбранного цикла числа Z и N имеют
одинаковую четность. Так, например, для произвольной цик-
циклической системы базисных орбиталеи хюккелевского типа на
рис. 13.11, а, где Z=0, N=2\ для системы мебиусовского типа
на рис. 13.11, б, где Z=3, N=3. Такое соотношение между
Z и N позволяет предельно упростить формулировку правил
Циммермана. Необходимо лишь учесть, сколько (N) ра-орбита.лей
включено в связывающий цикл перициклической реакции и ско-
сколько Bт) электронов несут все орбитали этого цикла» Тогда
получим:
Термически разрешенные реакции (ароматические
переходные состояния) m+N нечетное.
(П.4)
Фотохимически разрешенные реакции (антиарома-
(антиароматические переходные состояния) m+N четное.
Например, для переходного состояния (см. рис. 13.13, б) т—\,
N—\ и m + N=2, т. е. супраповерхностная миграция алкильной
группы с обращением конфигурации должна быть запрещена по
симметрии.
Пользуясь правилами A3.4), можно прояснить истинный смысл
обобщенного правила Вудворда—Хоффмана (см. разд. 13.2.3). Для
этого разобьем соединяющий цикл орбиталеи на те же структурные
компоненты, что и представленные в схеме A3.1). Существует про-
простое соответствие между супра- и антараповерхностными знаками
и четностью (N) внутриорбитальных инверсий знаков: антарапо-
верхностному присоединению структурного компонента реакции
отвечает нечетное число внутриорбитальных знаковых инверсий,
супраповерхностному — четное. Это положение иллюстрируется
схемой A3.5), которую следует сопоставить со схемой A3.1):
510
ы
-oo- l
6sINr-2)
A3.5)
\ {
Очевидно, что полное число N является суммой по всем струк-
структурным компонентам реакции циклоприсоединения:
A3.6)
где «I — четное, если число электронов в структурном компоненте
2m = 4q (q = 0, I, ...), и нечетное, если 2/я=4^ + 2 (? = 0, 1, ...)• В итоге
устанавливается связь между терминологией Вудворда—Хоффмана
и описанием, основанным на анализе узловых свойств соединяюще-
соединяющего цикла.
Пользуясь табл. 13.5 и соотношениями A3.4) и A3.6), легко
понять, что в основе обобщенных правил Вудворда—Хоффмана
лежат, по существу, представления об узловых ха-
характеристиках взаимно трансформирующихся в результате перици-
клической реакции орбиталей соединяющих циклов реагентов
и продуктов.
Таблица 13.5. Классификация структурных компонент аерицнклнческнх реакция
m
Четное
Нечетное
N
Четное
Нечетное
Четное
Нечетное
m + N
Четное
Нечетное
Четное
Обозначения
Вудворда—Хоффмана
13.4. ПРИМЕНЕНИЕ ТЕОРИИ ВОЗМУЩЕНИЙ
ДЛЯ АНАЛИЗА ПЕРИЦИКЛИЧЕСКИХ РЕАКЦИЙ
Рассмотренные способы анализа согласованных перицикличес-
ких реакций имеют чисто качественный характер. В настоящем
разделе будут рассмотрены возможности упрощенного расчетного
511
подхода к изучениям согласованных реакций, основанного на те-
теории возмущений (см. разд. 1.5).
Применение такого подхода не требует сложных вычислений, но
позволяет количественно охарактеризовать различия между раз-
разрешенными и запрещенными по симметрии согласованными реак-
реакциями, а также вычислить (а не отобрать из нескольких предполага-
предполагаемых) оптимальный путь сближения взаимодействующих молекул,
ведущий к реакции.
В соответствии с основной идеей теории возмущений волновая
функция реагирующей системы строится из волновых функций
исходных (невозмущенных) реагентов. Полная энергия этой систе-
системы складывается из энергий отдельных реагентов и членов воз-
возмущения, составляющих так называемую энергию взаимодействия.
Знак и величина последней определяются конкретным видом
параметра возмущения в выражении типа A.85) для полной энер-
энергии. В общем случае этот член должен включать все виды энер-
энергетических взаимодействий между двумя сближающимися молеку-
молекулами (ионами, радикалами): кулоновские, индукционные, обмен-
обменное отталкивание, перенос заряда, дисперсионные. Конкретный
вид получаемых при этом уравнений зависит также и от особен-
особенностей принятого расчетного приближения (МОХ, ППП, CNDO
и пр.). Рассмотрим наиболее простой. вариант, основанный на
применении МО Хюккеля, — метод межмолекулярных орбиталей
(ММО).
13.4.1. Расчет энергий взаимодействия
по методу межмолекулярных орбиталей
Метод ММО, предложенный Л. Салемом*, удобен для нахожде-
нахождения оптимальных путей реакции сопряженных молекул с хорошо
разделенными системами я- и очМО. Молекулярные орбитали си-
системы взаимодействующих молекул строятся из МО изолирован-
изолированных участников реакции, которые рассчитываются по методу Хюк-
Хюккеля (см. гл. 8). Такие МО называют межмолекулярными орбиталя-
ми. Они образуются при ст-перекрывании МО реагентов (см., напри-
например, структуры I, III). Основной отличительной особенностью ме-
метода Салема является то, что в нем в явном виде учитывается этот
тип перекрывания. Включение интегралов перекрывания позволяет
учесть электронное отталкивание при сближении я-систем с запол-
заполненными электронными оболочками. Важное значение этого эффек-
эффекта становится понятным при рассмотрении образования ММО пу-
¦Лионель Салем (род. 1937 г.) — французский физик-теоретик, известен работа-
работами в области расчетов электронных спектров органических молекул, механизмов
фотохимических реакций и гетерогенного катализа.
512
о
В А АВ В А АВ В
Рас. 13.14. Стабилизация и дестабилизация орбиталей межмолекулярного комплек-
комплекса АВ:
а — образование ММО из вырожденных орбиталей (эффект смешивания первого поряджа) без
учета интеграла перекрывания; 6 — образование ММО из вырожденных орбиталей с учетом
интеграла перекрывания; в — образование ММО из невырожденных орбиталей реагевтов (эф-
(эффект смешивания второго порядка)
тем взаимодействия двух вырожденных МО, принадлежащих от-
отдельным молекулам А и В.
Как видно из рис. 13*14, а, в, эффекты стабилизации и дестабили-
дестабилизации двух ММО комплекса АВ, образуемых при смешивании
вырожденных МО реагентов А и В (возмущение, или смешивание,
первого порядка), равны и противоположны по знаку. Суммарный
эффект, следовательно, равен нулю, что не позволяет отразить
такое физически важное взаимодействие, как отталкивание атомных
и молекулярных систем с полностью заполненными электронными
оболочками*.
Включение интеграла перекрывания между орбиталями
А и В правильно описывает отталкивательное взаимодействие. Как
видно из рис. 13.14, б и из выражений для энергии связывающего
и антисвязывающего уровней D.33), D.34), дестабилизация всегда
больше, чем стабилизация, т. е. суммарный эффект при смешива-
смешивании первого порядка в случае заполненных МО всегда сводится
к отталкиванию.
В случае сопряженных молекул, обладающих системой запол-
заполненных и вакантных МО, наиболее важную роль при взаимодейст-
взаимодействиях между ними играют не эффекты смешивания вырожденных
орбиталей (строгое вырождение — относительно редкий случай),
а эффекты смешивания второго порядка, т. е. взаимодействия меж-
между невырожденными МО.
Пример такого смешивания, в котором участвуют связывающая
МО одной молекулы А и антисвязывающая МО другой молекулы
В, показан на рис. 13.14, в. Связывающий уровень несколько «просе-
«проседает», что ведет к стабилизации МО комплекса АВ. Величина
стабилизирующего эффекта сильно зависит от разности энергий
¦Наиболее ярким примером такого типа взаимодействий, связанного с действи-
действием принципа Паули (см. разд. 3.2), .служит отталкивание атомов инертных газов.
17. Теория строения молекул 513
между взаимодействующими уровнями; она тем больше, чем мень-
меньше эта разность, что ясно из общей формулы A.85).
При взаимодействии двух сопряженных систем А и В нужно
учесть: 1) смешивания всех пар заполненных электронами МО
и 2) смешивания всех пар заполненных МО одной молекулы с ва-
вакантными МО другой. Взаимодействия первого типа (четырехэлект-
ронные) дадут общий дестабилизирующий эффект — отталкивание
А от В. Взаимодействия второго типа будут стабилизировать комп-
комплекс АВ — притяжение А к В.
Из-за пренебрежения кулоновским отталкиванием полный га-
гамильтониан в методе Хюккеля может быть записан в виде суммы:
A3.7)
где h(/) — оператор Гамильтона для отдельной молекулы, участ-
участвующей во взаимодействии с другими;
h = t+v+v\ A3.8)
причем t и v — операторы кинетической и потенциальной энергии
ядер и электронов внутри отдельной молекулы; v' — средний потен-
потенциал, обусловленный приближением одной молекулы к другой.
Принимается, что включение этого члена не нарушает требования
ортогональности внутримолекулярных орбиталей, т. е. МО А и В.
Физический смысл энергии взаимодействия (Jlf) между двумя
МО (i и Г) различных молекул А и В раскрывается при вычислении
матричного элемента hi(:
1 о 1 о г
= -Z fySif + ГSf Sif + Jit»
где *?{, Ч?г, е, и е,- — соответствующие МО молекул А и В и их
энергии. Так как указанные МО представлены в форме ЛКАО,
интегралы в A3.10) раскрываются через интегралы по атомным
орбиталям Хц (молекула А) и х» (молекула В):
A.J&[Jc>]afr? ОЗ.Ю)
И
514
причем ввиду малости указанных выражений достаточно учесть
лишь те атомные центры \л и v, которые сближаются при взаимодей-
взаимодействии А и В.
В силу общих ограничений теории возмущений, на которой
основывается описываемый подход, на величины SMV накладывается
ограничение:
S^0,2. A3.12)
Фактически соотношение A3.12) ограничивает расстояние между
сближающимися атомными центрами до 0,23—0,25 нм, так как на
меньших расстояниях /^/^-перекрывание становится большим, чем
0,2, и возмущения уже нельзя считать малыми.
После учета с помощью общих соотношений типа A.85) и A.86),
адаптированных с учетом сделанных допущений, всех парных вза-
взаимодействий (смешиваний) МО молекул А и В, Салем получил
следующее общее выражение для энергии взаимодействия молекул
А и В с полностью заполненными электронными оболочками:
Первая сумма в A3.13) отражает отталкивание между сближен-
сближенными молекулярными системами с заполненными электронными
оболочками. Второй и третий члены уравнения описывают притя-
притяжение вследствие образования МО при смешивании заполненных
МО одной молекулы со свободными МО другой.
Выражение A3.13) для случая взаимодействия полярных моле-
молекул следует дополнить членом, описывающим кулоновское взаимо-
взаимодействие:
^=rj?ZPZv>V' A3.14)
II. У
где D — диэлектрическая проницаемость среды (в газовой фазе
Z>=1); Z^ Zv — заряды на атомах; у^—двухцентровый кулонов-
ский интеграл, который в приближении МОХ можно принять рав-
равным Г~у.
Соотношения A3.13) и A3.14) можно применять для вычисления
энергий взаимодействия с МО, полученными как по методу МОХ,
так и по методу ППП.
515
13.4.2. Расчет начальных участков пути
химической реакции
Соотношения A3.13) и A3.14) наиболее эффективны для расчета
относительной ориентации взаимодействующих сопряженных моле-
молекул на начальных участках пути химической реакции между ними.
На этих участках, когда реагенты еще достаточно далеки друг от
друга, можно пренебречь деформациями связей молекул, проис-
происходящими непосредственно в зоне реакции. Такие деформации (из-
(изменения длин связей и углов, их разрывы и образование) уже не
могут быть описаны в приближении теории возмущений, опериру-
оперирующей с МО изолированных молекул.
Подход, основанный на теории возмущений, фиксирует внима-
внимание на установлении оптимальной геометрической конфигурации
взаимодействующих молекул при приближении их вдоль пути реак-
реакции к переходному состоянию.
Чем меньше (с учетом знака) энергия взаимодействия (?"*,), тем
выгоднее данная конфигурация. В случае двухцентровой реакции
расчет ?83 для различных взаимных расположений реагирующих
молекул позволяет определить положение предпочтительного ре-
реагирования, например ориентацию замещения (скажем, в 0-, м- или
л-положения монозамещенного бензола при электрофильном заме-
замещении). В случае многоцентровой реакции сравнение величин
?ta для разных переходных конфигураций определяет стереоспеци-
фичкость реакции.
Рассмотрим в качестве примера выбор между двумя возмож-
возможными направлениями реакции циклоприсоединения этилена к бута-
бутадиену:
* +
Поскольку взаимодействующие молекулы принадлежат к типу
альтернантных углеводородов, я-заряды на всех атомах равны ну-
нулю и энергия взаимодействия вычисляется только из соотношения
516
A3.13). Воспользуемся орбиталями бутадиена и этилена (см. гл.
8.3), их энергетическими уровнями в единицах стандартного резо-
резонансного интеграла /?с-с = /?0 и выпишем отдельно члены элект-
электронного отталкивания и притяжения в выражении A3.13). Выделим
при этом вклад взаимодействия между ВЗМО и НСМО, вносимый
в последний член (подчеркнутая сумма в скобках).
Для структуры X—(я? + 7г,)-присоединение энергетический вклад
отдельных членов в энергию взаимодействия равен
A3.15)
Р
A3.16)
+ 0,172 ЙО
@,104^,-0,340 Pupa + 0,276 Р\г)}.
Для структуры XI —(я* + ^-присоединение
1
~р
Задача 13.6. Используя МО этилена и бутадиена (8.3.1) и (8.3.3), проведите
вычисления Ею по формуле A3.13) и проверьте правильность полученных соот-
соотношений A3.15) и A3.16).
Чтобы рассчитать числовые
значения энергий взаимодействия,
необходимо оценить значения резо-
резонансных интегралов р^. Для этого
используется пропорциональность
резонансных интегралов и интегра-
интегралов перекрывания [см. соотноше-
соотношения G.24) и G.85)]:
^*=АУ^. A3.17)
В расчетах с волновыми функциями
метода МОХ рекомендуется испо-
использовать величину К— 3, а с волно-
волновыми функциями метода ГОТП —
величину А"=14; ft принимается
обычно равным — 3 эВ.
Вернемся к соотношениям
A3.15) и A3.16) и, не пытаясь пока
Рис. 13.15. Зависимость энергии вза-
взаимодействия от интегралов перекры-
перекрывания между сближающимися цент-
центрами бутадиена и этилена в конфигу-
конфигурациях типа X:
значения 5MV Bpff2/v)«0,2, что отвечает
сближению до 0,237 вм
517
получить их числовые значения, сравним значения их отдельных
членов. Учитывая A3.17), можно полагать, что при одинаковых
сближениях реагентов в конфигурациях X и XI энергии отталкива-
отталкивания Em будут примерно равны. Сопоставив энергии притяжения,
можно заметить главное отличие между теми же компонентами,
которые обусловлены смешиванием ВЗМО и НСМО. Для конфигу-
конфигурации X перекрестный член в этой части общей суммы получается
с отрицательным знаком и уравновешивает стабилизирующий
вклад остальных членов. Для конфигурации ХЕ все члены в анало-
аналогичном компоненте имеют одинаковые знаки, соответствующие
стабилизации этой структуры.
Подставив A3.17) в A3.16), получим полное выражение для
энергии взаимодействия (в единицах 0) для конфигурации XI:
?„= -lt06(S?r+S3r)+6,12$, -S*. A3.18)
Поскольку интегралы перекрывания между атомными орбиталя-
ми Spy являются прямыми функциями межъядерных расстояний г^
выражение A3.18) служит, по существу, аналитической формой
ППЭ, в которой энергетическая характеристика представлена энер-
энергией взаимодействия. На рис. 13.15 представлен участок ППЭ реак-
реакции бутадиена с этиленом, соответствующий сближению реагентов
в конфигурации типа XI.
По мере сближения реагентов энергия взаимодействия увеличи-
увеличивается. Пунктирная линия обозначает оптимальный путь реак-
реакции — линию наибольшей кривизны энергетической поверхности,
т. е. гребень холма на этой поверхности, вдоль которого проис-
происходит сближение реагирующих молекул в начале пути. Как видно из
рис. 13.15, рассчитанный путь реакции связан с одновременным
и равномерным сближением соответствующих центров. Это и есть
условие согласованной и притом синхронной реакции циклоприсо-
единения. Указанный результат подтвержден строгими ab initio
расчетами ППЭ реакции бутадиена с этиленом.
Задача 13.7. С помощью метода ММО определите оптимальную конфигу-
конфигурацию сближения двух молекул бутадиена на начальных участках пути реакция;
а> /= б> /у v\ в> /= г) /t ^
\_У
Задача 13.8. С помощью теории ММО установите вероятное направление
циклодимеризация иминов:
а) HC=NH Н,С—NH б) H2C^NH H2C—NH
2 II + э- I I
HN=CH2 HN—CH2 H2C = NH H2C—NH
518
13.5. ГРАНИЧНЫЕ ОРБИТАЛИ ВЗАИМОДЕЙСТВУЮЩИХ МОЛЕКУЛ
И ОПТИМАЛЬНЫЙ ПУТЬ ХИМИЧЕСКОЙ РЕАКЦИИ
Из выражений A3.15) и A3.16), полученных в разд. 13.4.2, ясна
особая роль граничных орбиталей при определении предпочтитель-
предпочтительного пути перициклической реакции. Эта роль понятна также при
рассмотрении корреляционных диаграмм (см. рис. L3.3—13.12), из
которых видно, что именно корреляции ВЗМО и НСМО определя-
определяют разрешенность или запрещенность по симметрии той или инок
реакции.
Хотя в общем случае необходимо учитывать корреляцию всея
МО, а при вычислениях Е„ могут быть существенными вклады от
смешивания МО, удаленных от ВЗМО и НСМО, обычно имение
взаимодействия граничных орбиталей реагентов играют определя-
определяющую роль при выборе оптимальной ориентации реагирующих
молекул в начальной стадии химической реакции. Эта идея, которая
принадлежит Фукуи, распространена им не только на перицикличес-
кие, но и на многие другие типы согласованных реакций. Основной
принцип состоит в том, что наиболее легко протекают такие хи-
химические реакции, в которых взаимная ориентация реагентов обес-
обеспечивает наилучшее перекрывание ВЗМО одной молекулы с НСМО
другой. Для выявления оптимального пути реакции необходимо,
зная структуру граничных МО, выделить такой способ сближения
реагентов, который соответствовал бы максимальной величине ин-
интеграла перекрывания между данными МО, а следовательно, их
наибольшему смешиванию в промежуточном комплексе и наиболь-
наибольшему переносу заряда с ВЗМО одной молекулы на НСМО другой.
Подобный подход — удобный облегченный качественный спо-
способ исследования пространственных требований реакции, который
не заменяет, но в некоторых случаях полезно дополняет более общие
методы корреляционных диаграмм и теории возмущений. Рассмот-
Рассмотрим несколько примеров применения метода граничных орбиталей.
Реакции синглетных карбенов с олефинами высокостереоспеци-
фичны. Так, если в реакцию вступает i/мс-олефин
продуктом взаимодействия является циклопропан исключительно
с доорасположением заместителей R относительно плоскости
трехчленного кольца. Для транс-олефина реакция идет в сторону
образования транс-замещенного циклопропана. Огереоспецифич-
ность присоединения синглетного карбена к олефинам указывает на
согласованный характер этой реакции.
519
6,6! ЗВ
7Г*
J g
Ж
Рис. 13.16. Строение и энергетические уровни (метод MINDO/3) граничных МО
метилена в этилена
Казалось бы, сближение реагентов по схеме A3.19), отвечающей
так называемому ff-пути, должно вести к наиболее энергетически
выгодному пути реакции. Однако анализ строения граничных ор-
биталей метилена и этилена (рис. 13.16) обнаруживает интересные
особенности. Как видно из рис. 13.17, при симметричном а-сЬяиже-
нии отсутствует перекрывание между парами граничных МО мети-
метилена и этилена. Следовательно, этот вариант сближения невыгоден.
Наоборот, при подходе метилена к этилену, при котором сохраня-
сохраняется только одна плоскость симметрии — я-путь (рис. 13.17, б),
граничные МО компонент реакции перекрываются в фазе, что ведет
к стабилизации такой конфигурации промежуточного комплекса.
Возможен еще один вариант сближения по с-пути, но с потерей
одной из плоскостей симметрии по сравнению с симметричным
путем (рис. 13.17, в). Можно видеть, что при несимметричном
(Т-пути сближения реагентов осуществляется ненулевое перекрыва-
перекрывание граничных МО. Этот путь реакции предпочтительнее симмет-
симметричного (т-пути (рис. 13.17, а).
Наиболее выгодный путь реакции синглетного метилена с этиле-
этиленом, как можно ожидать из данного рассмотрения и как показыва-
показывают детальные расчеты ППЭ реакции, является промежуточным
между я-путем и несимметричным «г-путем. Результаты расчетов
сечения ППЭ в плоскости трех углеродных атомов показаны на рис.
13.18. Только при весьма тесном сближении молекул этилена с ме-
метиленом, когда связанный с приближениями теории возмущений
метод граничных орбиталей теряет свою силу, метилен занимает
ориентацию, отвечающую ст-пути.
Задача 13.9. Используйте обобщенные правила Вудворда—Хоффмана
и правила Циммермана для выбора конфигурации промежуточного комплекса
реакции синглетного метилена с этиленом.
В случае внутримолекулярных реакций также можно использо-
использовать общую идею подхода: реакция развивается в направлении
520
Г
П+7Т
Jo
a
в
Рис. 13.17. Перекрывание граничных МО метилена и этилена при различных вариан-
вариантах сближения: а — симметричный «r-путь; б — л-путь; в — несимметричный а-путъ
наибольшего перекрывания граничных МО взаимодействующих
фрагментов. Рассматривая электроциклическую реакцию раскрытия
циклобутенового кольца как (тг + о^присоединение нетрудно по
р р р
циклобутенового кольца как (тг + о^-присоединение, нетрудно по-
понять с указанных позиций, почему конротаторная координата реак-
реакции предпочтительна по сравнению с дисротаторной:
Из пары смешиваемых граничных МО наибольший вклад в энер-
энергию взаимодействия вносит та пара ВЗМО и НСМО, энергетические
уровни которых наиболее близки. Поэтому в большинстве случаев
достаточно проанализировать лишь взаимодействие этой пары гра-
граничных МО. Например, один из важнейших в органической химии
стереохимических результатов — вальденовское обращение, т. е.
инверсия конфигурации тетраэдрического углеродного атома в би-
бимолекулярном нуклеофильном замещении, — легко вытекает из
рассмотрения направления максимального перекрывания ВЗМО ну-
клеофила (орбиталь неподеленной пары) и <т*-орбитали рвущейся
связи.
Для сохранения конфигурации тетраэдра необходимо, чтобы
атакующий нуклеофил Nu: подошел со стороны рвущейся связи
(XII) и занял место замещаемой группы (в рассматриваемом приме-
примере). При этом не достигается необходимого для стабилизации ука-
указанной структуры перекрывания граничных МО. Такое перекрыва-
521
6-путь
н
У
О -
0,12 г}нм
Рис. 13.18. Геометрия сближения и сечение ППЭ реакции присоединения синглет-
синглетного метилена к этилену (Дьюар„ метод MIND0/3):
на больших расстояниях домвнирует ориентация, отвечающая я-пути; прв сближении увеличива-
увеличивается вклад несимметричного с-путв; за начало отсчета принята энергия циклопропана
ние обеспечивается подходом нуклеофила с противоположной заме-
замещаемой группе стороны, что ведет к обращению конфигурации
углеродного тетраэдра (XIII) в результате реакции:
Nu
сохранение
¦J
конфигурации
XII
\
WuOD^Ox<f
обращение конфигурации
XIII
Задача 13.10. Пирсон A970) наблюдал сохранение конфигурации углерод-
углеродного атома в реакции окислительного присоединения галогензамещенных ал-
килов к иридиевым комплексам типа IrL4 с плоскоквадратной структурой:
Объясните этот результат с помощью метода граничных МО.
Лятературт
орбитальной симметрии. — М.: Мир, 1971.
принципа сохранения орбитальной симметрии. Основное
уделено методу корреляционных диаграмм, приложения которого пронллюстрнрова-
тяпов органически
Вудворд Р.,Хоффмм Р. Сохра
Оригинальное изложение идей
ны множеством примеров ртэ.
522
Джалкряст Т., Сторр Р. Органические реакции и орбитальная симметрия. — М.:
Мир, 1976.
Более доступное и более наглядное описание применения метода корреляционных диа-
диаграмм, правил Циммермана и метода граничных орбиталей. Подробно рассмотрен очень
широкий жрут перициклическвх реакций разных типов.
Лер Р., Марчанд А. Орбитальная симметрия в вопросах и ответах. — М.: Мир,
Ориентиром ними на химика-органика задачник по практическому приложению теоретичес-
теоретического подхода Вудворда—Хоффмана к анализу першдеклических термических и фотохимических
реакций. Разбираются главным образом примеры из научно-периодических изданий. Задачи
снабжены подробными ответами и указаниями.
Пирсов Р. Правила симметрии в химических реакциях. — М.: Мир, 1979.
В книге дается детальный анализ обоснования и применения правил симметрии к химичес-
химическим реакциям. Книга написана достаточно просто и включает большое количество задач
и упражнений.
Салем JL Электроны в химических реакциях. — М.: Мир, 1985.
Изучаются современные представления электронной теории химических реакций, рассмат-
рассматриваются новейшие тенденция развития основных физических идей в приложении к теории
строения и реакционной способности молекул.
ГЛАВА 14
МЕТОД МО ДЛЯ ТВЕРДОГО ТЕЛА
Твердое тело можно рассматривать как совокупность большого
числа атомов, молекул или ионов (~ 1023 моль ), связанных друг
с другом обычными силами межатомного взаимодействия (см. гл.
4). Свойства твердого тела являются коллективными свойствами
всей совокупности составляющих его частиц. Твердое тело является
в некотором роде «большой молекулой», и подходы к описанию его
свойств принципиально не отличаются от рассмотренных в пре-
предыдущих главах для молекул. Однако большое число атомов, об-
образующих твердое тело, делает невозможным прямое перенесение
на него методов количественного расчета электронных и простран-
пространственных характеристик молекул и требует учета упорядоченности
структуры твердого тела. В данной главе проиллюстрируем основ-
основную схему описания электронного строения твердых тел на примере
атомных и молекулярных кристаллов, включающих бесконечное
число идентичных атомов или молекул, однородно упакованных
в регулярные ряды и плоскости, заполняющие весь объем кристал-
кристалла. В отличие от такого идеального кристалла реальные кристал-
кристаллические тела содержат дефекты кристаллической решетки, наруша-
нарушающие регулярность. Крайним случаем нарушения регулярности
является совсем случайное, хаотическое расположение атомов или
молекул в твердом теле, какое наблюдается в аморфных твердых
телах, как и в жидкостях. В зависимости от степени регулярности
расположения атомов или молекул в твердом теле используют
и различные модели для описания их строения и свойств.
523
Рис. 14.1. Элементарная ячейка про-
пространственной кристаллической ре-
решетки:
т ] и г 2 — векторы трансляции с щ, nj и гц,
соответственно равными 1, 1, 1 и 2, 2, 1
Зонная теория твердых тел ос-
основана на модели идеального бес-
бесконечного кристалла, в которой не
учитываются поверхность образца
и различные дефекты. Она дает
возможность обобщенного объяс-
объяснения многих экспериментально
наблюдаемых свойств твердого
тела в тех случаях, когда отклоне-
отклонения от идеальной структуры ма-
малосущественны и определяющую
роль играет так называемый даль-
дальний порядок.
При учете дефектов, а также
при расчете характеристик реаль-
реального кристалла, для которых важ-
важную роль играет ближний поря-
порядок, т. е, характер взаимодейст-
взаимодействия выделенного атома с ближай-
ближайшими его соседями, прибегают
к супермолекулярным, или кла-
кластерным, моделям твердого тела.
Зонная теория представляет собой приложение одноэлектроннои
модели к кристаллам. Она эквивалентна методу МО для молекул.
Однако молекулярные орбитали идеального кристалла должны
удовлетворять условию так называемой трансляционной симмет-
симметрии, что несколько видоизменяет их характер.
14.1. ТРАНСЛЯЦИОННАЯ СИММЕТРИЯ
В идеальном кристалле всегда можно ввести три вектора транс-
трансляций ~а , b i&~c так, что физические свойства кристалла в некоторой
произвольно выбранной точке г точно воспроизводятся в любой
другой точке г ', удовлетворяющей условию
"?=?' + П|"?-г-л26 +пэ?, A4.1)
где пь пъ щ — произвольные целые числа. Совокупность точек г ,
определяемая выражением A4.1), при различных пь п2 и и3 дает
кристаллическую решетку, которая является регулярным периоди-
периодическим расположением точек в пространстве.
Параллелепипед, имеющий в качестве ребер вектора а , Ь и с ,
называется элементарной ячейкой (см. рис. 14.1). Перемещение всего
кристалла как целого параллельно самому себе, описываемое век-
вектором Т
524
Рис. 14.2. Кристаллические структуры NaCl (а), алмаза (б), нафталина (в) и \2 (г)
+n2b +пъс ,
A4.2)
называется трансляцией. Вектор трансляции кристаллической ре-
решетки связывает любые две точки решетки. Посредством соответст-
соответствующих операций трансляций элементарной ячейкой можно запол-
заполнить все пространство кристаллической структуры. Такое свойство
кристалла названо трансляционной симметрией. На рис. 14.2, а—г
представлены структуры некоторых атомных и молекулярных кри-
кристаллов.
Трансляционная симметрия, как уже отмечалось, предполагает
наличие бесконечной протяженности. Естественно, никакие регуляр-
регулярные структуры не являются бесконечными, а при отсутствии бес-
бесконечности теряется важное свойство трансляционной симметрии.
Один из способов сохранения трансляционной симметрии конеч-
конечных систем — наложение циклических граничных условий (условия
Борна—фон-Кармана). Поясним их существо на простых примерах:
цепочки из атомов водорода Н A), полнена. A1) без альтернации
простых и двойных связей и стопочной цепочки комплекса
Pt(CNL~, который можно заменить для простоты рассуждений на
PtHj" (III):
525
Л = О 1 2 3
—н—н—н—н—
I/ \/ I/
S-H-8
in
Наложение циклических условий на эти цепочки означает отож-
отождествление друг с другом конечных атомов, т. е. изгибание цепочки
в окружность большого радиуса и соединение между собой кон-
концевых атомов. Последовательность перехода от трехатомной це-
цепочки к окружности большего радиуса и изменения характера МО
иллюстрируется рис. 14.3. я-Орбитали полиенов имеют такой же
вид симметрии, как орбитали цепочки атомов Н на рис. 14.3.
14.2. МО И ЭНЕРГЕТИЧЕСКИЕ ЗОНЫ
Число узлов в МО увеличивается при возрастании их энергии,
т. е. при переходе от низшей связывающей к высшей антисвязыва-
антисвязывающеи МО. При увеличении размеров цикла и числа атомов в цикле
число орбиталей увеличивается, и при достаточно большом (бес-
(бесконечном) числе атомов в цикле эти орбитали столь плотно запол-
заполняют пространство между высшей антисвязывающеи и низшей свя-
связывающей* МО, что можно считать весь спектр орбиталей непрерыв-
непрерывным, формирующим энергетическую зону.
Обозначим базисные АО атомов Н через хо> Хъ Хг> ¦¦• (см- I)-
V
П
•О# -
о-О-о —
(XXXXJUUUUO
Рис. 14.3. Молекулярные орбитали трехатомной цепочки атомов водорода и цик-
циклических систем, состоящих из 3,4, 5,6 и бесконечно большого числа атомов водоро-
водорода
526
Молекулярные орбитали <рп всей системы (I) будут являться линей-
линейной комбинацией АО х« (п = 0, 1, 2, ...)• Однако трансляционная
симметрия, т. е. сдвиг (трансляция) всей цепочки (I) на величину,
кратную д, который возвращает систему в эквивалентное исход-
исходному состояние, накладывает на волновую функцию дополнитель-
дополнительное условие периодичности. Будем искать линейные комбинации
АО цепочки (I) в виде симметризованных орбиталей, преобразу-
преобразующихся по неприводимым представлениям группы CN, как это было
описано в гл. 6 (см. разд. 6.4). Все представления группы Си одноме-
одномерны, и их число равно N. При этом волновая функция, принад-
принадлежащая каждому л-му (л = 1, ..., N) неприводимому представлению,
при повороте на угол 2n/N (см. рис. 14.3) умножается на число
ехр[2тс1(л-1)/Л].
Такие комбинации, или симметризованные МО, имеют вид
11 12
/ 7ni— 2я1 2
<Р\=-г\Хъ+е "li + e "хг+-+е " Xn-X A4-3)
J\ J
(ЛГ-1J
2ni— 2m
y/N\ ~ )'
В более общей форме выражения A4.3) можно переписать как
пт
2т—
. л = 0, 1,2 JV-1. A4.4)
Введем обозначения к——, где а — параметр решетки I (см.
а N
также рис. 14.3); к — так называемый волновой вектор, изменя-
п п
ющийся в пределах <к<-.
а а
В этом случае выражение A4,4) можно записать как
1
A4.5)
Молекулярная орбиталь (р„, записанная в виде A4.4) или A4.5),
называется блоховской функцией и обладает трансляционной сим-
527
-oooo-oo-
метрией, т. е. сдвиг всей цепоч-
цепочки на величину, кратную а, не
меняет функцию <р„.
При Дс = 0 получим полно-
полностью связывающую волновую
функцию <ро> соответствующую
самому нижнему энергетичес-
энергетическому уровню — дну энергети-
О -к-**Л/аО -к
Рис. 14.4. Зависимость E(k) и ширины
энергетической зоны от расстояния между
соседними атомами Н
ческой зоны. При к~- имеем
а
полностью антисвязывающую
МО (рф (ф#-1), соответству-
соответствующую самому верхнему энер-
энергетическому уровню, т. е. гра-
границе энергетической зоны.
Из рис. 14.3 можно заме-
заметить, что расстояния между
энергетическими уровнями на
краях зоны меньше, чем в сере-
середине зоны, т. е. плотность
уровней (число уровней на еди-
нлцу энергии) не оди-
одинакова, по всей зоне; у краев
зоны плотность выше, чем в середине зоны. Для описания зависи-
зависимости расстояния уровней от к вводят непрерывную функцию Е (к)
(закон дисперсии), которую называют также структурой полосы.
Введение этой функции оказалось возможным благодаря предполо-
предположению о сплошном (непрерывном) заполнении зоны (полосы) энер-
энергетическими уровнями.
Разность между высшим и низшим уровнями МО называют
шириной зоны (полосы), она характеризует дисперсию энергии. Ши-
Ширина зоны определяется величиной взаимодействия между сосед-
соседними атомами, которая зависит от интеграла перекрывания АО.
Поясним это более подробно. Для двух взаимодействующих вы-
вырожденных уровней энергия связьшающего и разрыхляющего вновь
образуемых уровней определяется формулами (9.10) и (9.11), из
которых ясно видно, что расстояние между этими уровнями увели-
увеличивается с увеличением интеграла перекрывания между ними. Ана-
Аналогичная ситуация возникает и при взаимодействии орбиталей це-
цепочки. Только в этом случае формируются не две МО, а целая
полоса. Ширина этой лолосы определяется степенью взаимодейст-
взаимодействия АО. Так, рис. 14.4 показывает увеличение ширины зоны при
уменьшении расстояния между атомами, т. е. при увеличении пере-
перекрывания между их АО. Отметим, что кривая E(fc) имеет воз-
возрастающий характер при изменении & от 0 до п(а. Рассмотрим
528
О К -^ Ж/а
Рис. 14.5. Поведение Е(к) для ро — р,-орбиталей
теперь случай, когда в качестве х**~АО выступают не 5-АО, a jp-AO,
ориентированные вдоль цепочки. В этом случае, во-первых, связы-
связывающая комбинация АО будет соответствовать ? = -, а антисвязы-
вающая — к= 0:
-, A4.6)
... A4.7)
и кривая Е (к) будет иметь ниспадающий характер (рис. 14.5).
а
Рис. 14.6. Орбигали фрагмента PtHj" (а); структура энергетических полос полимера
(Ш), формируемая орбиталями фрагмента PtHj~; (б) плотность электронных состоя-
состояний полимера (III) (сплошная кривая) и вклад в нее атомов Pt (штриховая кри-
кривая) (в)
18. Теория строения молекул
529
О к -*- Jija 0 BfE) -+
Рис. 14.7. Соотношение между кривыми Е(к) и D(E)
Рассмотрим теперь цепочку, состоящую из фрагментов PtH42
(III). На рис. 14.6, а показаны орбитали фрагмента PtHj", на
рис. 14.6, б показана структура энергетических полос Е(?) для
различных орбиталей фрагментов. Характер поведения Е(к) для
различных орбиталей фрагмента PtHj" существенно различается.
Так, кривые Е(?) для d^_y^, d#- и d^-полос являются возра-
возрастающими, а для dxz- и dyz — убывающими. Во фрагменте PtHj"
электронами заполнены все орбитали вплоть до ^-орбитали; сле-
следовательно, в полимере (III) все энергетические зоны, включая
ф-зону, будут заполнены электронами. Высшая орбиталь в тве-
твердом теле, заполненная электронами, называется уровнем Ферми
(еф). В данном случае уровень Ферми совпадает с верхним краем
ф-зоны (рис. 14.6, б).
Полоса, в которой отсутствуют энергетические уровни между
нижней энергетической зоной, заполненной электронами, и верхней,
не заполненной электронами, называется запрещенной зоной.
14.3. ПЛОТНОСТЬ СОСТОЯНИЙ
Для характеристики распределения энергетических уровней в зо-
зоне вводят понятие плотности состояний D(E), которая определяет-
определяется как число энергетических уровней N(E), находящихся в интерва-
интервале между Е и Е+ dE:
В общем, D(E) обратно пропорциональна тангенсу угла накло-
наклона касательной к кривой Е(к), что иллюстрируется рис. 14.7.
Интегрирование D(E) no dE от самого нижнего уровня до
уровня Ферми — ?F дает полное число занятых МО. Умножая
530
-1k -
антисвязыб:
Pt-H
сбязыванш
Pt-Pt
онтисбязыд сбяэыбонис
Я(Е)
О
COOP
о
COOP
Рис. 14.8. Плотность состояний D(E) и кривые COOP ддя связей Pt-H и Pt-Pt
полимера A11)
это число на два, получаем полное число электронов в данной
энергетической зоне. На рис. 14.8 показана суммарная плотность
состояний для полимера, состоящего из фрагментов (PtH4 2). Пол-
Полная плотность состояний D(E) может быть разложена на ло-
локальные плотности состояний, т. е. на вклады отдельных атомов
или орбиталей в полную плотность состояний, выделением из
всех молекулярных орбиталей, заполненных электронами, соот-
соответствующих членов, относящихся к данному атому или орбитали.
Так, на рис. 14.10 штриховой линией показана плотность состояний
атома марганца.
Химическую связь между двумя атомами, например I и 2,
характеризует произведение 2ciC2Sl2 G.57); суммируя эту величину
по всем занятым МО, получаем малликеновскую заселенность свя-
связи 1—2. Для кристалла предложено ввести (Р. Хоффман) аналогич-
аналогичное понятие — заселенность перекрывания кристаллических орбита-
лей (COOP) (от англ. — crystal orbital overJap population), которую
находят суммированием ic^cJS^ в рамках одной МО для всех
атомов, одинаково расположенных друг по отношению к другу.
Проиллюстрируем это на примере цепочки из атомов водорода.
Если рассмотреть только все 1, 2-взаимодействия, т. е. взаимодей-
взаимодействия между соседними атомами Н, то получим кривую 1, 2 для
COOP на рис. 14.9. Ясно, что на дне энергетической зоны молеку-
молекулярная орбиталь полностью связывающая (все с^>0) и, следова-
531
л
—н-н-н-н—н-н—н-
антисб.
\
>
связке.
а 6
Рис. 14.9. Соотношения между кривыми
~ 0 +
6 COOP
и COOP
тельно, все члены 1, 2-взаимодействия (c^+15№ A+i) являются поло-
положительными и связывающими. К середине зоны связывающий хара-
-*-
-22-
-АнтисОныбание
О Связывание
COOP
Рис. 14.10. Плотность электронных состояний D(E) (штриховая кривая) (а) и вклад
в нее атомов Мп (сплошная кривая) и кривые COOP F) для кристаллической пленки,
состоящей из MP
532
ктер 1, 2-взаимодействий в МО уменьшается до нуля. К вершине
зоны 1,2-взаимодействие становится антисвязьшающим. Для 1, 3-
взаимодействия на рис. 14.9, в получаем кривую COOP (штриховая),
имеющую другой характер, чем кривая COOP для 1, 2-взаимодейст-
2-взаимодействий. Ее амплитуда значительно меньше, так как интеграл перекры-
перекрывания для 1, 3-взаимодействия значительно меньше, чем для
1, 2-взаимодействия.
Таким образом, положительные области кривых COOP харак-
характеризуют связывание, а отрицательные — антисвязывание в твер-
твердом теле. Амплитуда этих кривых зависит от числа состояний
в энергетическом интервале, интеграла перекрывания и соответству-
соответствующих коэффициентов в МО. Интеграл от COOP до уровня Ферми
дает полную малликеновскую заселенность данной связи.
На рис. 14.8 представлены кривые COOP для связей Pt—Н
и Pt—Pt в полимере (III). Из этого рисунка хорошо видны связыва-
связывающие и антисвязывающие области кривой COOP.
До сих пор рассматривалось образование энергетических зон из
орбиталей мономерных единиц в полимерах. Аналогично проис-
происходит образование энергетических зон и в трехмерных кристаллах.
Например, на рис. 14.10 представлены плотность электронных со-
состояний и COOP для пленки, состоящей из Мп2Р2~ со структурой,
показанной в верхней части рис. 14.10.
Литература
Ленин А. А. Введение в квантовую химию твердого тела. — М.: Химия, 1974.
Теория химической связи в твердых телах рассматривается с позиций квантовой химии,
прослеживаются аналогии между химической связью в молекулах и твердых телах.
Р. Хоффман. Строение твердых тел и поверхностей. — М.: Мир, 1990.
Книга посвящена молекулярно-орбитальному представлению проблемы строения и свойств
твердого тела. Показано, как терминология в язык, принятие в физике твердого тела, смыкаются
с концепциями теоретической химии.
УКАЗАНИЯ К РЕШЕНИЮ ЗАДАЧ
Глава 1
-(-*>
1.2. Использовать разложение Тейлора для ехр>>, где у—\— —
1.7. Функцию необходимо искать в виде (p=Asin(Bx+C).
1.8. Воспользоваться формулой B.51).
1.9. Найти коммутатор [v, V(r)\.
Глава 2
2.1. Воспользоваться правилами дифференцирования сложной функции.
2.7. Необходимо показать, что У{^$)фЕ\ Aj); J(ls)^E2(\s); (Ъ)-обозначение
Ij-АО атома водорода.
Глава 3
3.3. Функцию можно переписать в виде
/ ... 4*»], где N — нормировочный множитель; Р —
р
оператор перестановки, применение которого к волновой функции означает обмен
координатами двух электронов и умножение на ±1 в зависимости от четности
перестановки.
3.4. Учесть только Is- и ls-АО лития.
3.5. Кулоновский и обменный интегралы в атомах всегда положительны. Вос-
Воспользоваться очевидным соотношением |xFjA)xF/2)-xFjeA)xFX2)|2 — >0.
3.6. Необходимо вычислить константы экранирования S для нейтрального ато-
атома и катиона и воспользоваться соотношением C.51).
3.8. Чтобы найти потенциал ионизации 3^-электрона, необходимо из энергии
атома без двух 4у-электронов вычесть энергию атома без двух 4л-электронов и одно-
одного 3</-электрона.
3.9. Доказать коммутацию операторов.
1 1
3.10. (г 3>= , где п,1 — квантовые числа,
dL 1
)
3.16. Для конфигурации tf возможны термы lS, XD, lG 3P 3F.
3.19. Основная конфигурация (р2) имеет термы — 1D, iP, iS. При возбуждении
возникает конфигурация неэквивалентных электронов (pd), термы которой 1Р и 3Р.
534
Глава 4
4.1. Доказательство провести, основываясь на приближении Борна—Оппенгей-
мера.
4.2. В молекуле азота предположить, что Is- я 2?-электроны не участвуют
в образовании связи. Для UH учесть только 1 л-АО водорода и 2s-АО атома лития.
4.5. Интегралы Гц, VJ, и A1111) вычисляются в сферической системе координат,
a S\2 — в эллиптической системе.
4.12. В качестве параметра выбрать А.
Глава б
6.4. Рассмотрите угловые части валентных функций р-АО (табл. 2.4) и учтите
соотношения между сферическими и декартовыми координатами rcos0=z и т. д.
6.7. Найти интеграл Jrf
Глава 7
7.1. Необходимо построить матрицу S'1, которая находится из условия
S~lS = I, где / — единичная матрица.
7.2. Учесть формулу G.54), а также то, что ?, = |^*Нф,Л, где <рг-
7.3. Для нахождения 5MV воспользоваться формулой D.125), а для F^ — G.51).
В условия нормировки волновой функции в методе РМХ входит интеграл перекрыва-
перекрывания (в отличие от я-электронных методов). Это приводит к тому, что коэффициенты
Сщ могут быть больше 1»
Глава 8
8.1. Для аллильной системы с\ = +с$; для циклобутадиена Ci = ± Сг ^ + сз=
8.3. В секулярном уравнении для мебиусовского циклобутадиена недиагональ-
недиагональным матричным элементам для орбитален с обращенными фазами приписать значе-
значения — 1.
8.5. с2=
8.7. Построить для АУ секулярный детерминант. Для АУ, содержащего
N атомов, пронумеровать от 1 до п все меченые атомы и от л + 1 до N все
немеченые. Для простоты положить <х1=а2= — =а#=0. Воспользоваться тем,
что при умножении любого ряда детерминанта на —1 детерминант также ум-
умножается на — 1.
8.10. Учесть, что первичной стадией реакции является атака Вг + .
Глава 9
9.1. Рассмотрите соотношение A.85) и представьте матричные элементы
Н^, в базисе АО, полагая, как в расширенном методе Хюккеля, что H^—kS^ (Hm-?
+ Нуу). При вычислении дискриминанта квадратного уравнения (9.6) следует вос-
1/2 1
пользоваться приближенным соотношением A+х) =1н—х— ... . Соотношения
2
535
(9.7), (9.8) можно получить также, пользуясь выражением A.85) для поправки теории
возмущений второго порядка, так как для невырожденного взаимодействия поправ-
поправка первого порядка равна нулю.
9.2. Воспользуйтесь МО двухатомных гомоядерных молекул (см. разд. 4.6) для
реконструкционного анализа МО АХ2. При преобразованиях целесообразно вос-
воспользоваться приближенным соотношением (\—х)~1=\+х+х2+ ,„ . Примером
прямого численного расчета является вычисление коэффициентов при АО в молекуле
формальдегида (см. разд. 8.6 и 8.8).
9.8. Рассмотрите расщепление уровней орбиталей аннуленовых циклов при их
сближении на расстояние, соответствующее сэндвичевой структуре, а затем произ-
произведите диаграмму орбитальных взаимодействий с валентными орбиталями цент-
центрального атома.
9.9. Для пирамидальной нидо-струггуры, включающей п вершин, из 2л+4 ске-
скелетных электронов 2(л— 1) электрона образуют внутряциклячесхие it-связи базаль-
ного цикла, а 2 электрона заняты на экзоциклической связи (в электронной паре)
апикальной группы. Подсчитайте число электронов, связывающих указанные фраг-
фрагменты.
9.10. Рассчитайте образование сдвоенной бипирамиды, используя фрагмен-
фрагментацию
Нидо-структуры имеют высшие занятые МО а\- или е-типа, донирующие элект-
электроны на валентные МО клозо-структур. Для общего вывода правил см.: D. Mingos.
Accounts Chem. Res., v. 17, p. 311—319 A984) Polyhedral Skeletal Electron Pair
Approach.
Глава 10
10,3. Для ответа на вопрос достаточно рассмотреть корреляционную диаграмму
для соответствующих структур АН"
10.5. Орбитали рх, ру и рг преобразуются при повороте системы координат как
единичные векторы.
10.8. Использовать условие ортонормированности A0.17).
10.9. Гибридные АО кислорода, участвующие в образовании ОН-связей, эк-
эквивалентны, т. е. ii\**ni. Кроме того, можно считать эквивалентными орбитали
неподеленных пар. Учесть условие ортогональности и нормировки МО.
10.12. Конфигурация электронных пар вокруг атома Re соответствует квадрат-
квадратной антипризме.
Глава 11
11.5. Конфигурацию комплексов составлять из первичных групп так, чтобы
число групп типа (II) было максимальным.
11.6. В кристаллическом состоянии координационный узел имеет трансоктаэд-
рическое строение jTifl^O^Cld3* 10/)?«20 000 см, IQDq <\QDq^^ согласно
A1.10). Необходимо воспользоваться рве. П.4.
11.7. Вычислить диаграмму расщепления уровней ^-орбиталей в поле сплюс-
сплюснутого тетраэдра, пользуясь аддитивной схемой. Рассмотреть электронные переходы
в конфигурации <г.
536
Глава 12
12.1. Пользуясь теорией орбитальных взаимодействий, постройте из групповых
орбиталей СН2 и ХН(СН) МО молекулы СН2=ХН. Следует учесть, что высшая
занятая МО Дртипа промежуточной С2,,-структуры X, являющаяся орбиталью непо-
деленной пары электронов, при деформации в менее симметричную С-структуру IX
взаимодействует не с низшей свободной я*-МО ^-типа (отсутствие перекрывания
при смещении X—Н), а со следующей свободной <х-МО в]-типа:
Н
Н X:
12.2. Воспользуйтесь орбитальными диаграммами на рис. 10.5 и 12.8 и соот-
соотношением F.18). Рассмотрите формы нормальных колебаний плоской АХ^формы.
12.3. Воспользуйтесь приближением теории кристаллического поля для опреде-
определения порядка энергетических уровней </-орбиталей. Низкоспиновые d16-комплексы
типа Сг(СО)з в Х>зл-форме подвержены действию эффекта Яна—Теллера первого
порядка, а низкоспиновые ^-комплексы типа Fe (СОM — эффекту Яна—Теллера
второго порядка.
Глава 13
13.1. SO2 имеет у атома серы заполненную и вакантную орбитали, пригодные
для связывания с другими атомами:
немо
взмо
537
13.3. Следует учесть вз.дамодействие я-орбиталей двойных связей с dyz-t dXI-
орбиталями иона переходного металла.
13.4. Учесть симметрию </-орбиталей металла.
13.8. Рассчитать МО формальдимина с параметрами Огрейтвизера. При учете
кулоновских членов положить
Я-1; Va~' г-0,22 нм; ycx:=7CNssyNN-
13.10. ВЗМО иридиевого комплекса — ^с-орбиталь.
ОТВЕТЫ И РЕШЕНИЯ ЗАДАЧ
Глава 1
1.1. е при — со<х^0;х е при любом х; sin хе при любом х. Функция е не
отвечает требованию конечности.
1.2. а, б, в, г, е.
d d
1.3. /— — самосопряженный; несамосопряженный оператор.
dy dy
1.4. L(/'/) L(L/'
-ax I \ _( I -ax\ a -ax
—ae ) Vae —n(n-\-\)
x -1 л x x
Если бы комбинация *Fi и 4*2 была собственной функцией Н, то
= Е(с1х^1+с2у?2) = с1^к^1 + с2^2- Линейная комбинация будет являться собственной
функцией Н только при Е[ = ?2.
г-
II ПК Л Я /Г
1.7. ^?л= /- sin —х; Е„= , где п-1, 2, 3, ... оо.
V а а
Bn-lJnBz)" '
2п + \
-;</¦>—
1.9. Можно.
1.10. Е(с)=\е
-J-
1 . 1
г
-сг2
е
Ъс 2л/2 г dE 3
2 ул Л 2
Ve!e.
9п
Глава 2
д дг 3 дв д dtp д
2.1. —= н н . Из B.3) имеем
дх дх дг дхдв дх д<р
538
r=(x2+y2+z2) ; 0=arccos z(x2+y2+z2) \; <p=*aictg-.
dr dO cos0cos<p dtp sirup
Тогда — = sin 0cos tp', — =* ; — =
dx дх г дх rsin0
d 3 1 д 1 3
и — = sin 0 cos (p —I—cos0cos<p sin<p —.
дх dr г 86 rsinB dtp
d2 d2 d2
Аналогичным образом можно получить —; — и —-.
dx1 dy2 dz2
2.2. P°o(cos0)=l; /*(ff)e P\(e)p1(
3 cos2 0-1
2.6. <>
e2 e2
2.8. <T>=—;<V> .
2uq Oq
2.9. Lxhy
г / e2 э2 \ / в2 a a2 \i
-Щ z[ x z )-W x z ,
L V dydz dydxj \ dz2 dx dzdxjJ
г /г d2 d2\ ( d2 d2 Y]
-ft2 \x — +z y-— )~z [z—y~ I .
L \dy dzdy dz2) \ dxdy dxdzj]
/ 3 d\
льно, [ЬХЦ] = — fr [у — ~x — )==— iTiLr.
\ dx dy/
б
Следовательно
\ y/
Аналогичным образом доказываются и другие равенства.
Глава 3
3.2. в, г, д — полностью симметричные,
б — антисимметричная,
а — функция, не обладающая симметрией (функция общего вида).
1
Симметричная комбинация —= [/(l)gB) +/B)g(l)] a(lJB).
У
Антисимметричная комбинация —=[/(l)gB)-X2)g(l)]a(l)aB).
/
у/
3.3. Число возможных перестановок равно л'. Из условия нормировки с учетом
ортонормированности функций *Р,- имеем №п\ — 1; N— (п\)
3.4. Ч
V3!
7=|*1A)с<1) »F2B)aB)
n/3!
539
1
"л
3.5. Докажем, что K^Jij. Так как [*1A)»Р/B)-Ч'лA)Ч'/B)]2->0, то, интегрируя
это неравенство по пространству электронов 1 и 2, получаем, что результирующий
интеграл больше или равен нулю. Это, в свою очередь, дает соотношение
(и\Ю + (кк\в)
Условие Kij<Jij — частный случай при /«/, k=j.
Теперь несложно показать, что (ij\ij) (ik\ik)^(ik\jlJ, а также что
(\) (&\&)(&\уJ- Эт°> в свою очередь, доказывает последнее из неравенств
3.6. /Na = 7,314 эВ; 7К = 4,808 эВ;
3.7.
= 5,138 эВ; ^" = 4,339 эВ
3.8.
Атом
S
Se
Те
Атом
Fe
Ni
Zn
Потенциал ионизации (эВ)
по Слэтеру
15,10
23,21
19,86
¦Л
11,24
13,36
15,65
эксперимент
10,36
9,75
9,01
hd
25,05
28,94
32,44
У] (эхспер.)
7,89
7,69
9,39
3.10. Для атома водорода константа сшш-орбитального взаимодействия
1 1 dV(r) 1 1 d ( е2\ е1 1
__ = { \=
2wVr дг 2n?c2rdr\ r) Infer*'
Для ^-состояния
Воспользовавшись указанием к задаче, имеем
1 ,.
1 1
/~-, L«l, S=~, n=2.
2 2
3.11, Атом .... Mg
Терм .... Sq
5 эВ,
а
540
3.12. lD, ЪР, *5.
3.15. Терму SZ> соответствуют уровни 3Z>4,31>з, si>2» 5&u 5A)- Стабое магнитное
поле (а поле 10* Тс является слабым; см. задачу 3.20) расщепят первые четыре
уровня соответственно на 9,7,5, 3 эквидистантных уровней (терм sZ>o не расщепится)
с расстоянием между ними, равным &?#. Так как L= S= 2, то g=3/2 для всех термов
и расщепление в магнитном поле равно - /fM5=0,9' 10 эВ.
2
3.16. Диаграмма уровней представлена на рис. 15.1.
3.17. Четыре полосы.
3.18. Hv = hvQ+fluB(giMj2—gxMix). Необходимо вычислить выражение в скоб-
скобках для различных переходов и расположить в порядке возрастания энергии.
3.19. Нормальный эффект Зеемана проявится для переходов JPj-¦ *S0 и iPi~*iD2.
Аномальный эффект для атома углерода проявляться не будет.
3.20. EipV2-E2PlBxl,5 10~* эВ, ?вз=^м^З//<1,5 10 эВ;В<\03 Гс.
3.21. Частота перехода равна v=vo+ (М^—Мц), так как ?=1 (см. задачу
3.18). Используя правила отбора AAfL=O, ±1 (что следует из условия неизменности
мультидлетности при переходе терма xDi в терм 1F$), получим три частоты: v0 ,
п
РмВ
V0) vo+ • 15 возможных переходов можно разбить на три группы с соответст-
h
вующей частотой.
3J.Z. В спектре будут наблюдаться три линии.
Глава 4
4.2. X?N2==\Pi\Pz2Px\PxlPy\Py2\~\Pi\PzlPx\PxlPy\Pyl\ —
-\Pz\Pz2Px\PxlPy\Py7\-\Pi\Pi2Px\Px7Py\Pyl\ +
+ \Pz\PzlPx\Px2Py\Pyl\ — \PzlPzlPxlPx2PylPy2\ +
+ \Pz\PzlPxlPxlPy\Py2\—\PzlPz2PxlPxlPylPy2\>
xVuHsso{^Ts\-\hls\}+b\\sTs\-\-c\2s24; а, б, с находят из условия минимума
энергии.
4.4. Полную энергию в методе Рутаана можно представить в виде
Подставляя вместо Pp, выражение D.67) и минимизируя по коэффициентам с^ выра-
выражение для полной энергии, легко получить уравнения Рутаана D.62).
4.6. Метод ВС.
541
4.8.
s
Px
Py
dxy
dx*
*В
разрешенные комбинации
s, px. dj
s, Px, dj
Py, dxy
Py, dxy
s. Px. dx*
dy^
запрещенные комбинации
Py, Pz. dxy, dyz, dxz, dy*-z*
Py, Pz, dxy, dyz, d^, dy*-z*
J. Px, Pz, dyI, dxz, dx*. dy*-.?
$, Px, Pz, dyz. dxz, dj, dyi^xi
Py, Pz, dxy, dyz, dxz, df-z2
s, Px, Py, Pz- dxyf dyr, dxz, dx*-y*
4.9. а) &?г>Е&*; б) D?>D?*; в) D^>I^'; r) J^/k^'; д) Eft
4.10.
x
) (ag2pz) (ag2px) (ng2px) {ng*2px) (ng2py) (n'g2py)\.
=(lPzlP~72\-\Pz\Pz2\)-
dE
4.11. a) — =
д ( Й2 , Zf
— [ v2
dZ\ 2me r
e
r
2/e2
8En
6)
dme
2m?
V2
_•_ ZV
dme~ 2n2tf
п
BE
4.12. —
dA
дИ\
dA
BIA
dA
ah
Тогда
_ AaA
U— —A\ 1 A n+- 1 ;
L J2mA\ V\
к 2/J dA A
( 1\Г о* / IN"]
V VI JlmA\ У]
Глава б
6.3. a) Cs; 6) Ct, в) D3k; г) Td; д) C3v; e) C2v; ж)
к) Dlh; л) ОА; м) C2v; н) C4v; o) 5g.
6.5. См. табл. 6.З.
6.6. (
; з) D4k; и) ZMA;
1
1
Глава 7
7.1. Обозначим через Sy элементы матрицы S, условие G.6) для ЛГ=2 можно
записать в виде
(::)
Отсюда 5П =—,
•»12 ^11
= —-> S22=~, где
Д Д
S\2 S22
-1/2
Обозначая через Sy элементы S , имеем
S22
22
Следовательно, Su =S22l 5j,+5j2=-; 2SuSn- -—-
A A
Теперь можно получить явный вид 5ц и 512; ортогонализованные атомные орбитали
),\ И Яг СООТВетСТВеННО раВНЫ Х\ = Ц>\к\\+<$>2Г&\2\ ^2 = Ф1^12 + 9>2^22-
73. Sii = l; Si2=0,5610; ?| = -22,0288 эВ^?2=0,0038 эВ.
Тогда <pi= 0,3666xi + 0,7472x25 (Рг= -1,1511 Xi + 0,9492x2; Е*=2г{ = -44,0578 эВ.
Глава 8
8.1. Для циклобутадаеяа
1111
Ф1=-Х1+-Х2+-Хэ+-*4;
1111
Учитывая свойства вырожденных МО, построить
алляльной системы см. (8.26).
8.3. Секулярное уравнение имеет вид
1111
-xi--Z2--x3+-
1111
и <рг с узлами на атомах. МО
1
0
1
1
X
1
0
0
1
X
-1
1
0
-1
X
=0,
откуда x\t 2=л/2: *з, 4= —л/2- Найдите вид МО.
543
8.4. Энергия прямоугольной структуры Е=4а+ 8,080, энергия квадратной струк-
структуры равна 4at + 4/f. Уровни энергии прямоугольника ei, 4ва±4Д> «2, э = *±0,04Д.
8.5. xi - -2,107; *2«-1,167; хэ=-1; х4~
<p1=0,521xi+0,419x2+0,361x3+0,343x4+0,361x5+0,419x6;
^=0,571X1+0,191X2-0,349X3-0,598X4-0,349X5+0,191X6;
Ъ=0,500 X2+0,500 хз - 0,500x5 - 0,500 хй
^4 = 0,546X1-0,366X2-0,238X3+0,566X4-0,238X5-0,366X6;
^5=0,500*2-0,500x3 +0,500X5-0,500X6»*
^6=0,323X1-0,393X2+0,437X3-0,452X4+0,437X5-0,393X6.
8.7. Детерминант для АУ запишется в виде
п
N-n
— ?
0
0
A.
A
rl,
0
—«
0
л + 1 •
0...
0...
0...
0
0
—
A. *+i
л-.
8 Al »+!
• *•+'¦ -
• Ai n 0
j t
••• A, N
•» A. V
™ Pn. N
_E
... — S
=0.
п
N-n
Последовательная замена знаков у п строк и N—п столбцов приводит к выражению
det(e)=(-l)"det(-«).
Следовательно, для четного N секулярноё уравнение будет содержать только четные
степени е, а при нечетном N — только нечетные и е, = 0. При этом ?лг_^-_ \) — — ?,. "
коэффициентов МО cift имеем
N
N
1^ у также корни секулярното уравнения, причем
2» -• "Л"
Заменим i на N— (i— I):
ВИДНО, ЧТО —Cjf-(i-\)t ^ И
CN-(i-l). ii=-Сщ
Итак, МО имеют вид
N
544
8.8.
a1 +
+e2 + (-2aJ + {2aJ + (-3eJ = 20te2 = 1;
1
8.10. Во втором случае получается карбоний-ион аллильного типа, который, как
уже отмечалось, допускает делокализацию положительного заряда. При этом об-
образуется аллил-катион повышенной устойчивости. Подобная делокализация невоз-
невозможна для карбониевого иона в первой реакции. Из расчета L* видно, что при
бромировании действительно получаются примерно равные продукты присоедине-
присоединения.
8.11.
возможно применить фрагмен-
фрагменГлава 9
9.4. Для построения МО Н~ с симметрией
тацию следующего типа:
Н
Н
'
н—н
Для построения орбиталей фрагмента Н3 используются соотношения (9.20).
Далее орбятали Н~ получают по схеме двухорбитальных взаимодействий (см. с.
546).
9.5. Особенностью гибридных орбиталей, центрированных на общем атоме,
является то, что, несмотря на отсутствие перекрывания между ними, имеется сильное
онансное взаимодействие и величины Н^, в уравнениях (9.4)—(9.9) не равны нулю,
е величину этого взаимодействия для простейшего случая двух 5/ьорбнталей
1/,/г(л+Рх) *<Рь= 1/<s/i(s-Px)> где s и рх-слугероваснс АО углерода 2s я 2рх:
s-px) dx
545
я
О—О
н—н
учитывая орбитальные потенциалы ионизации /, приведенные в табл. 7.13, и прини-
принимая во внимание, что \<р1?1<р^=1(<р{).
9.6. Сопоставим следующие ряды:
л
т
о
V
?
н—с<Т) - 0 - L—
?
L—с О
В ряду СН4—CH происходит последовательное отщепление атома водорода, а в ря-
ряду комплексов переходных металлов начиная с 16-электронного FeL,4 — изолобаль-
нон водороду группы L
546
Глава 10
10.4. Ai=
10.5. 120°, tri
={ s
tTi=—=( S +
у/А 2- г")
10.7. При четном числе кумулированных связей возможна цис-транс-взоиеряя,
при нечетном — оптическая изомерия.
10.9. ф\ =—-(
sp*;
^ = N/0^3(^-0,5^-0,646^+1,29^) | 2зз
<Р4 = ч/0,3E-0,5^-0,646^-1,291^) Г 5р *
Угол между неподеленными парами равен 115° 15'.
10.10. Угловая, треугольная, пирамидальная (центральный атом — I), бисфено-
ид, тетраэдр, пирамида, октаэдр, угловая, бисфеноид (центральный атом —А1),
тетраэдр.
Глава 11
11.2. <г4>«102 см4; 10Dqx0,5 эВ.
11.3. р- У ровни расщепляются в полях G^, D^h, D3, Cjy
11.4. Удлинение.
11.5.
Для
Энергия
^-орбиталей
4 лиганда X — пер-
первичная группа (II)
2 лиганда Y — пер-
первичная группа A1)
Сумма
(Dqy=l,2SDqx)
-4,2SDqx
-2,UDqy
- 6,955 Dqx
d^_yi
12,28 Dqx
6tlADqy
l9,55Dqx
dxy
2,2SDqx
\,l4Dqy
3,705Dqx
dxl
-2,51Dqx
-2,57Dqy
-5,7832)?*
-2,57Z>^
-2,57 Dqy
-5,183Dqx
547
Для MX3Y3
Энергия
<й>рбвталей
2 лигаяда X — пер-
первичная группа A1)
2 лиганда Y — пер-
первичная группа (II)
1 лиганд X — пер-
первичная группа (I)
1 лиганд Y — пер-
первичная группа (I)
Сумма
(Dqy=\,25Dqx)
-2,l4Dqx
2,\4Dqy
5,HDqx
5,l4Dqy
6,75Dqx
6,l4Dqx
6,\4J)qy
- 3,14 Dqx
-\\4Dqy
b,15Dqx
1,14 Dqx
hl4Dqy
-\^Dqx
-3,\4Dqy
-4,5Dqx
dn
-2,5lDqx
-2,57Dqy
0,57Dqx
0,57Dqy
-4,5Dqx
dyz
-2,57Dqx
-2,51Dqy
0,57Dqx
G,57Dqy
-4,5Dqx
11.8. Распределение электронов в тетраэдрических комплексах (с симметрией TJ)
для сильного поля лигандов выглядит следующим образом:
Число
электро-
электронов
1
2
3
4
5
6
7
8
9
10
Централь-
Центральный атом
Ti3+
v3+
Cr3 +
Mn3 +
Fe3 +
Fe2+,
Co3+
Co2+
Ni2 +
Cu2+
Zn2+
Электронная конфигурация
(еУ
(еJ(еУ
(eJU2
(e)\eJ(t2y
{eJ{eJ{t2y{t2y
(еПеПьШЧ^2
Число не-
спарен-
ных элек-
электронов
1
2
1
0
1
2
3
2
1
0
Основное
состояние
2Е
3А2
2Е
гАу
2т2
*Аг
ъТг
гТ2
ХАХ
11.9. См. рис. 15.2.
548
Металл
s
d*
dx*-f
Pi
Px
Py
dXy
4-
Групповые
ст-типа
1
—
1
2 2 °4
1
1
J2°*
—
—
орбитали лигандов
0
\
/
Групповые орбитала лигандов я-
типа
—
1
2
1
— (Я1у — Ил*}
V2
1
1
1
1
Представле-
Представление группы
} аи
а2и
\
Г
J
}•¦
Глава 13
13.1. а) Разрешено так называемое согласованное линейное (л)+со^)-присоеди-
нение или
или
Ч)
б) (<yj-f 7г^)-присоединение.
13.2. См. рис. 15.3. Обе реакции термически разрешены.
13.5.
549
Число элект-
электронов
2
4
6
4/t
4л+2
нейтральный
—
[1,3]
[1.5]
ГI / ixM I 1 ]
1 Ж - l~fl Ж Ш J
[1,D/1+1)]
Полнен
катион
[1,2]
[1,4]
[1, 6]
[1, 4/1]
[1, D/1 + 2)]
анион
_—
[1,2]
П. 4]
[1, D/1-2)]
[1,4/1]
Термически рачрешентп^й сдвиг
s, с сохранением
д, с сохранением
5, с инверсией
j, с сохранением
в, с инверсией
а, с сохранением
5, с инверсией
s, с сохранением
а, с инверсией
13.7.
13.8. Образуется 1, 3-диазетидин.
13.9. Синглетный метилен имеет граничные МО, подобные SO2 (см. задачу 13.1),
при этом высшей занятой МО является ст-орбиталь. Возможны две переходные
структуры:
I
Подсчет числа D^+2), и Dq)a компонент в соответствии с обобщенными правилами
Вудворда—Хоффмана показывает, что термически разрешенной является переход-
переходная структура II. Заметьте, что она соответствует мебиусовскому трехчленному
циклу с четырьмя электронами.
13.10. Граничные МО расположены так:
Это и объясняет возможность присоединения из трехцентрового переходного состоя-
состояния с сохранением конфигурации у атома углерода: ч *-
V-
Тг
550
ПРИЛОЖЕНИЯ
I. Атомные едшшщд** (а. е)
Величина
Обозначение Значение в СИ
Значение фи-
физической ве-
величины! я. е.
Масса электрона
протона
нейтрона
Дляна боровского радиуса
Время одного оборота по
одной орбите атома водо-
водорода
Энергия ионизации атома
водорода
Скорость света в вакууме
Постоянная Планка
Заряд электрона
Магнетон Бора
Постоянная тонкой структу-
структуры
g-фактор для электрона
Постоянная Ридберга
Постоянная Больцмана
Универсальная газовая по-
постоянная
Постоянная Авогадро
0,9109534-100 кг
мр
1,6726485-10" 27 кг
1,6749543'10" 27 кг
А2
0,52917706-100 м
2Д43985 10
~17
с
h
Н
е
Дм =
eh
4,8509477-108 Дж
299792458 м/с
6,626176 10~3* Джс
1,0545887-10* Джс
1,6021892-10~19 К
9,274078-10* ДжТл
,-1
е2
Ла->=-
2aohc
kE=RjNA
0,0072973506
1,0011596567
1,097373177 107 м'1
1,380662 Ю3 Дж/град
8,31441 Дж/мольград
6,022045 1023 моль
1
1836,15
1838,68
1
1/2
137,036
1
1/27Е
1
1/4
Рекомендуемые согласованные значения фундаментальных физических посто-
постоянных A973).
551
П. Таблица перевода энергетических величин
эрг
эВ
см
эрг
эВ
'1
см
ккал/моль
а. е.
1
1,6022' 102
1,9865-106
6,9477-104
4,3598-10" "
6,2415 10й
1
1,2398 10'4
4,3363-10 "а
27,212
5,0340-1013
8065,5
1
349,75
219474,6
Продолжение табл.
1 эрг
1 эВ
1 см
1 ккал/моль
1 а. е.
ккал/моль
1,4393* 10"
23,061
2,8592-10" 3
1
627,52
а. с.
2,2937 1010
3,6749 10
4,5563 10"8
1,5936 10
1
ОГЛАВЛЕНИЕ
Предисловие 3
Условные обозначения 5
Глава 1. Основные положения квантовой механики 7
1.1. Постулаты квантовой механики 7
1.2. Соотношения неопределенностей 17
1.3. Вариационный метод 19
1.4. Вариационный метод Ритца 2\
1.5. Теория возмущений 22
Глава 2. Одноэлектронные атомы 25
2.1. Решение уравнения Шрёдингера для атома водорода 25
2.1.1. Решение Ф-уравнения 28
2.1.2. Решение ©-уравнения. Полиномы Лежандра 28
2.1.3. Решение /Е-уравнення. Полиномы Лягерра 30
2.2. Атомные орбитали 33
2.3. Расчет различных свойств водородоподобного атома 41
2.4. Спектр водородоподобного атома. Правила отбора 44
2.5. Угловые моменты атома 47
2.5.1. Операторы квадрата и проекции углового момента ...... 47
2.5.2. Физический смысл квантовых чисел / и т 48
2.5.3. Магнитный орбитальный момент атома 50
2.5.4. Спин электрона 51
Глава 3. Многоэлектронные атомы 53
3.1. Метод самосогласованного поля Хартри 54
3.2. Принцип Паули и определители Слэтера 60
3.2.1. Двухэлектронная система 62
3.3. Метод Хартри—Фока 64
3.4. Приближенные аналитические функции атомных орбиталей .... 67
3.4.1. Атомные орбитали Слэтера—Зенера 68
3.4.2. Двухэкспоненциальные и гауссовские АО 70
3.5. Энергетические уровни многоэлектронных атомов 71
3.5.1. Принцип построения периодической системы элементов
Д. И. Менделеева 72
3.5.2. Потенциалы ионизации и сродство к электрону 73
3.6. Квантовые числа многоэлектронного атома 74
3.6.1. Полные орбитальные и спиновые квантовые числа 75
3.6.2. Спин-орбитальное взаимодействие. Квантовое число полного
момента 77
3.7. Термы многоэлектронных атомов 81
3.7.1. Термы в приближении LS-связв 81
3.7.2. Энергетические уровни в приближении jy-связи 86
3.8. Магнитные моменты многоэлектронного атома 87
553
3.9. Спектры многоэлектронного атома 88
ЗЛО. Многоэлектронный атом в магнитном поле 90
3.10.1. Эффекты Зеемана и Пашена—Бака 91
3.10.2. Сверхтонкое взаимодействие 93
Литература 94
Глава 4. Теория химической связи 94
4.1. Приближение Борна—Оппенгеймера 95
4.2. Метод валентных связей. Расчет молекулы водорода методом валент-
валентных связей 100
4.2.1. Уточненные расчеты молекулы водорода по методу валентных
связей 105
4.3. Метод молекулярных орбиталей (МО) 108
4.3.1. Общие положения. Аналогия с теорией многоэлектронного атома 10В
4.3.2. Приближение линейных комбинаций атомных орбиталей
(МО ЛКАО) 110
4.3.3. Уравнения Рутаана 111
4.3.4. Уравнения Рутаана для открытых оболочек 114
4.3.5. Выбор базисных атомных функций 117
4.4. Электронная корреляция 120
4.4.1. Метод конфигурационного взаимодействия 122
4.4.2. Метод многоконфигурационного взаимодействия (МКВ) .... 124
4.4.3. Метод теории возмущений 125
4.5. Расчет молекулы водорода по методу МО ЛКАО 127
4.5.1. Основное состояние молекулы водорода 127
4.5.2. Волновая функция и энергия основного состояния Н2 129
4.5.3. Волновые функции возбужденных состояний молекулы Н2 . . 135
4.5.4. Конфигурационное взаимодействие 137
4.6. МО гомоядерных двухатомных молекул 138
4.6.1. Электронные конфигурации гомоядерных молекул 143
4.7. МО гетероядерных двухатомных молекул 147
4.8. Теорема Гельмана—Фейнмана 149
4.8.1. Теорема вириала и природа химической связи 151
4.8.2. Электростатическая теорема 152
Литература 154
Глава 5. Поверхности потенциальной энергии (Ш1Э) молекул 154
5.1. Общее понятие 154
5.2. Пути и энергетика химической реакции 157
5.3. ППЭ и динамика химических реакций 161
5.4. ППЭ электронно-возбужденных состояний 164
5.5. Колебания молекул 166
5.5.1. Гармонические колебания молекул 167
5.5.2. Расчет термодинамических функций 171
5.6. Топологическое определение молекулярной структуры 173
5.7. Вибронные взаимодействия. Эффекты Яна—Геллера 176
5.7.1. Теорема Яна—Геллера 177
5.7.2. Псевдоэффект Яна—Геллера и эффект Яна—Геллера второго по-
порядка 179
Литература 183
Глава 6. Симметрия молекулярных систем 184
6.1. Операции симметрии 184
6.2. Точечные группы симметрии 186
6.3. Таблицы характеров точечных групп симметрии 189
6.4. Симметризованные орбитали 195
554
6.5. Вырожденные представления 196
6.6. Прямое произведение 199
Литература 202
Глава 7. Расчетные методы квантовой химия 203
7.1. Общая характеристика 203
7.2. Неэмпирические методы 205
7.2.1. Базисные ряды атомных орбиталей 205
7.2.2. Энергия электронной корреляции 208
7.3. Полуэмпирические методы расчета 210
7.3.1. Основные требования к полуэмпирическим методам 211
7.3.2. Приближение нулевого дифференциального перекрывания . . . 211
7.3.3. Инвариантность полуэмпирических методов 214
7.3.4. Метод CNDO 216
7.3.4.1. Параметризация CNDO/2 219
7.3.4.2. Расчеты электронных спектров. Параметризация CNDO/S . . 223
7.3.5. Методы INDO, MINDO, MNDO 224
7.3.5.1. Параметризация метода INDO 224
7.3.5.2. Метод MINDO/3 225
7.3.5.3. Методы MNDO и AMI 230
7.3.6. Расширенный метод Хюккеля (РМХ) 231
7.4. Анализ электронных заселенностей атомов и связей в молекулах . .236
7.5. ег, я-Приближение, полуэмпирические я-электронные методы . . . 239
7.5.1. Метод Паризера—Парра—Попла (ППП) 240
7.5.1.1. Параметризация для расчета электронно-возбужденных состо-
состояний 241
7.5.1.2. Параметризация метода ППП для расчета свойств основных
состояний 246
7.5.2. Метод Хюккеля 252
Литература 253
Глава 8. Свойства л-сооряженных молекул 255
8.1. Расчет энергий молекулярных орбиталей в методе МОХ. Аннулены . 255
8.1.1. Система циклопропенила 256
8.1.2. Циклобутадиен 258
8.1.3. Циклопентадиенил. Аналитическое решение и графическое опреде-
определение энергий МО аннуленов 262
8.1.4. Бензол. Правило Хюккеля Dл+2) 265
8.1.5. Циклогептатриенил , 266
8.1.6. Циклооктатетраен 266
8.2. Правило 4л+2 и электронное строение сопряженных циклических со-
соединений. Антиароматичность 268
8.3. Расчет коэффициентов при атомных орбиталях в МО Хюккеля . . . 270
8.3.1. Этилен 270
8.3.2. Аллил 272
8.3.3. Бутадиен 273
8.3.4. Коэффициенты при АО в векторной и матричной формах. Урав-
Уравнения метода Хюккеля как задача на собственные значения . . 276
8.4. Высшие полиены 278
8.5. Циклические полнены, имеющие вид колец Мебиуса 280
8.6. Молекулы с гетероатомами 283
8.7. Электронные плотности, заряды, порядки связей и поляризуемости,
матрица плотности первого порядка 287
8.8. Решение уравнений метода ППП для молекулы формальдегида. Сопо-
Сопоставление с хюккелевским расчетом 293
8.9. Свойства альтернантных угловодородов 296
8.10. Теплоты атомизации полиенов 302
8.11. Энергетические критерии ароматичности 304
555
8.11.1. Хюккелевская энергия резонанса 304
8.11.2. Дьюаровсхая энергия резонанса 305
8.11.3. Энергия резонанса для возбужденных состояний 308
8.12. Расчет физических свойств сопряженных соединений 309
8.12.1. Потенциалы ионизации и сродство к элеггрону 309
8.12.2. Электронные спектры поглощения 311
8.12.3. Спектры ЭПР и распределение спиновой плотности в сопряженных
молекулах 312
8.13. Индексы реакционной способности я-сопряженных молекул . . . . 316
8.13.1. Общие ограничения 316
8.13.2. Приближение изолированной и реагирующей молекулы . . . . 318
8.13.3. Электрофильное замещение 319
8.13.4. Нуклеофильное замещение 323
8.13.5. Реакции радикального замещения 327
8.13.6. Реакции присоединения 330
Глава 9. Качественные методы определения просгранствеяпого и электрошю-
го строешн молекул 331
9.1. Принципы качественной теории молекулярных орбиталей .... 332
9.2. Взаимодействие двух орбиталей 333
9.3. Взаимодействие нескольких орбиталей фрагментов 337
9.4. Построение орбиталей сложных молекул 340
9.4.1. Орбитали связей и групп 340
9.4.2. Молекулярные орбитали этилена и иона метония 343
9.4.3. Орбитали циклических систем 347
9.4.4. Орбитали металлоорганических фрагментов. Изолобальная анало-
аналогия 351
9.4.5. Полиэдрические органические молекулы и ионы 356
9.5. Устойчивость полиэдрических структур и правила счета скелетных и
валентных электронов 360
Глава 10. Стереохимия соединенна неоереходшх элементов 369
10.1. Диаграммы Уолша 369
10.1.1. АН2- и АХгСистемы 370
10.1.2. АН3- и АХэ-системы 375
10.1.3. АН4-системы 376
10.1.4. А2Н4-системы 378
10.Z Теория гибридизации 381
10.2.1. Локализованные молекулярные орбитали 381
10.2.2. ЛМО и концепция гибридизации атомных орбиталей .... 385
10.2.3. Гибридизация и пространственная направленность химических
связей 388
10.2.4. Неэквивалентные гибридные атомные орбитали 394
10.3. Теория отталкивания электронных пар валентных орбиталей . . . 397
10.3.1. Основные положения 397
103.2. Развитие теории ОЭПВО 399
10.3.3. Недостатки теории ОЭПВО и отклонения от ее предсказаний . 404
Глава И. Строение координационных соедшенй 407
11.1. Координационная связь 407
11.2. Типы координационных полиэдров . 409
11.3. Теория кристаллического поля 414
11.3.1. Расщепление {/-уровней центрального иона 416
11.3.2. Количественная оценка расщеплений 420
11.3.3. Спектрохимический ряд 422
556
11.3.4. Комплексы сильного и слабого полей. ТКП и магнитные свойства
комплексов 424
11.3.5. Энергия стабилизации кристаллическим полем 428
11.3.6. Более строгое рассмотрение расщепления уровней для d -конфигу-
-конфигураций центральных ионов 430
11.4. Применение теории молекулярных орбиталей для описания электрон-
электронного строения координационных соединений 433
11.4.1. МО координационных соединений с лигандами, имеющими о-
орбитали 434
11.4.2. МО координационных соединений с лигандами, имеющими р~ и
я-орбитали 437
11.4.3. я-Комплексы и металлоцены 440
11.4.4. Правило 18 электронов 448
11.5. Деформации координационных полиэдров и эффекты Яна—Геллера 453
Глава 12. Структурно нежесткие молекулы 455
12.1. Основные типы структурной нежесткости 456
12.2. Методы исследования структурно нежестких молекул 459
12.3. Электронная природа структурной нежесткости молекулы 465
12.4. Полигонные перегруппировки 468
12.4.1. Пирамидальная инверсия трикоординированных структур . . . 469
12.4.2. Плоская инверсия дикоординированных структур 471
12.4.3. Тетраэдрическая инверсия тетрахоординированных структур . , 472
12.4.4. Пентакоординированные структуры. Псевдовращение Берри . . 476
12.4.5. Структуры с более высокими координационными числами . . 479
12.5. Туннельный механизм превращений структурно нежестких молекул . 480
12.6. Таутомерия 481
12.7. Внутреннее вращение 485
Глава 13. Сохранение орбитальной симметрия в химических реакциях . . . 490
13.1. Перициклические реакции 491
13.1.1. Реакции циклоприсоединения 492
13.1.2. Электроциклические реакции 493
13.1.3. Сигматропные реакции 495
13.2. Анализ перициклических реакций с помощью метода корреляционных
диаграмм 496
13.2.1. Циклопрнсоединение 496
13.2.2. Электроциклические реакции 502
132.3. Истинные пути электроциклических реакций 504
13.2.4. Обобщенное правило отбора по симметрии для согласованных пе-
перициклических реакций 506
13.3. Ароматичность и правила Вудворда—Хоффмана для перициклических
реакций 506
13.3.1. Правила Циммермана для першщклнческих реакций 506
13.3.2. Анализ сигматропных реакций 508
13.3.3. Обобщенное правило Вудворда—Хоффмана и свойства хкжкелев-
схих и мебиусовских систем базисных орбиталей 510
13.4. Применение теории возмущений для анализа перициклических реак-
реакций 511
13.4.1. Расчет энергий взаимодействия по методу межмолекулярвых ор-
орбиталей 512
13.4.2. Расчет начальных участков пути химической реакции 516
13.5. Граничные орбитали взаимодействующих молекул и оптимальный
путь химической реакции 519
Глава 14. Метод МО для твердого тела 523
14.1. Трансляционная симметрия 524
557
14.2. МО и энергетические зоны 526
14.3. Плотность состояний 530
Указания к решению задач 534
Ответы и решения задач 538
Приложения 551
Учебное издание
В.И.Минкин, Б.Я.Симкин, Р.М.Миияев
ТЕОРИЯ СТРОЕНИЯ МОЛЕКУЛ
Редактор С.С.Трапезникова. Технические редакторы С.В.Светикова,
И.А.Грохочинская. Корректор Г.Н.Буханова.
Лицензия ЛР № 062308 от 24 февраля 1993 г.
Сдано в набор 10.09.97 г. Подписано в печать 14.10.97 г.
Формат 60х881/16- Бумага офетная.
Гарнитура Тайме.
Тираж 5000. Заказ № 3077.
Отпечатано с готового оригинал-макета в ГУИПП «Курск».
305007, г. Курск, ул. Энгельса, 109.