















































































































































































































































































































































































































































































Автор: Беликова Н.А. Гришина Г.В.
Теги: органическая химия химия практикум органический синтез переводная литература издательство мир учебное пособие для студентов
ISBN: 978-5-03-003806-3
Год: 2008




Organisch-chemisches Grundpraktikum
Von
Heinz G O. Becker
Werner Berger
Giinter Domschke
Egon Fanghanel
Jiirgen Faust
Mechthild Fischer
22., vollstandig iiberarbeitete
Von
Rainer Beckert
Egon Fanghanel
Frithjof Gentz
Karl Gewald
Reiner Gluch
Roland Mayer
Klaus Miiller
Dietrich Pavel
und aktualisierte Auflage
WolfD. Habicher
Peter Metz
Hermann Schmidt
Karl Schollberg
Klaus Schwetlick
Erika Seiler
Giinter Zeppenfeld
Dietrich Pavel
Klaus Schwetlick
WILEY-
VCH
WILEY-VCH Verlag GmbH & Co. KGaA
ЛУЧШИЙ
ЗАРУБЕЖНЫЙ
УЧЕБНИК
ft
В двух томах
1
4-е издание
Перевод с немецкого
канд. хим. наук С. В. Грюнера и профессора, д-ра хим. наук 77. Б. Терентъева
Допущено УМ О по классическому университетскому образованию
в качестве учебного пособия для студентов высших учебных заведений,
обучающихся по специальности ВПО 020101.65 — химия
БЯНОМ
Москва 2008
УДК 547
ББК24.2
0 64
Авторы: X. Беккер, Р. Беккерт, В. Бергер, К. Гевальд, Ф. Генц,
Р. Глух, Г. Домшке, Э. Затер, Р. Майер, П. Мец, К. Мюллер,
Д. Пафелъ, Э. Фангхэнелъ, Ю. Фауст, М. Фишер, В. Хабихер,
К. Шветлик, Г. Шмидт, К. Шольберг, Г. Цеппенфелъд
Органикум: В 2-х т. Пер. с нем. 4-е изд. — М.: Мир, 2008. —
064 Т.2-488с.ил.
ISBN 978-5-03-003806-3
В учебном издании, написанном авторским коллективом из Германии,
подробно рассмотрены все особенности работы в практикуме по органической
химии, включая лабораторное оборудование и методики синтеза.
Том 2 включает описание препаративных методов синтеза (методики про-
проведения реакций окисления и гидрирования, реакций карбонильных соедине-
соединений, перегруппировок), методов идентификации органических соединений
(включая разделение смесей, вопросы и упражнения), описание свойств и ме-
методик приготовления реагентов и растворителей, классификацию по токсич-
токсичности важнейших химикатов, перечень методик, вошедших в т. 1 и 2, предмет-
предметный указатель.
Для студентов, аспирантов и преподавателей университетов и химико-тех-
химико-технологических вузов, а также научных сотрудников.
УДК 547
ББК 24.2
Редакция литературы по химии
Originaly published in the German language by
WILEY-VCH Verlag GmbH, Pappelalle 3,
D-69469 Weinheim, Federal Republic of
Germany, under the title «Schwetlick et al.:
Organikum».
ISBN 978-5-03-003806-3 (русск.) © 2004 WILEY-VCH Verlag GmbH, Weinheim
ISBN 978-5-03-003807-0 © перевод на русский язык, оформление,
ISBN 5-527-31148-3 (нем.) издательство «Мир», 2008
ПРЕПАРАТИВНАЯ ЧАСТЬ
(продолжение)
6. ОКИСЛЕНИЕ И ДЕГИДРИРОВАНИЕ
6.1. ОБЩИЕ ЗАКОНОМЕРНОСТИ
Окисление связано с потерей электронов. Оно всегда сопряжено с восстановле-
восстановлением (присоединением электронов). Окислительно-восстановительная
(редокс-) реакция состоит в передаче электронов от восстановителя (донора
электронов, нуклеофильного соединения) окислителю (акцептору электронов,
электрофильному соединению); при этом восстановитель окисляется, а окис-
окислитель восстанавливается, например:
R3NI + Fe3© R3N? + Fe2® [Г.6.1]
Это общее определение можно применить и к органическим реакциям, про-
протекающим с образованием и разрывом ковалентных связей, если ввести поня-
понятие формального числа окисления (степени окисления).
Для нахождения числа окисления следует рассматривать соединение таким образом, как
будто оно состоит из ионов. Электронную пару ковалентной связи относят к более электроот-
электроотрицательному атому. Связанным с углеродом атомам приписывают следующие числа окисле-
окисления: Н (+1), О (-2), ОН (-1), Hal (-1), С-С @), С=С @), С=С @).
Атомы в молекулах простых веществ имеют числа окисления, равные нулю. Сумма чисел
окисления нейтрального соединения должна быть равна нулю, заряженного — соответствую-
соответствующему зарядовому числу. В соответствии с этими правилами можно получить, например, ряды,
представленные в табл. Г.6.2.
Итак, реакцию можно назвать окислительной, если она сопровождается уве-
увеличением числа окисления реакционного центра субстрата.
Однако в органической химии понятие «окисление» не получило широкого
распространения; например, хлорирование алканов или присоединение брома
к этилену не считаются реакциями окисления. Обычно в органической химии
под окислением понимают потерю электронов, отщепление водорода или введе-
введение кислорода. Часто отщепление водорода сопровождается присоединением
кислорода.
Реакции окисления органических соединений большей частью протекают по сложному
механизму, в соответствии с которым окисление субстрата происходит через ряд последова-
Г Препаративная часть (продолжение)
Таблица Г.6.2. Числа окисления атомов углерода в различных соединениях
-4
-3
-2
-1
0
+ 1
+2
+3
+4
сн4
•енз
®СН3, :СН2
СНзОН, СН3С1
СН2=О, СН2С12
НСО2Н, НСС13, HCN
СО2, СОС12, СС14
СНз-СНз
СН2=СН2
СН^СН, СН3СН2ОН
СНзСНО, СНзСНСЬ
СН3СО2Н, СН3СОС1, CH3CN
(СНзЪСНОН
(СНз)зСОН
(СН3JСО
тельных стадий. Важнейшими типами таких элементарных стадий, которые включают пере-
перенос электронов, водорода и кислорода, являются:
— перенос электронов (окисление соединений с п- или я-электронами, например,
фенолов и ароматических аминов, см. разд. Г,6.4; анодное окисление);
— перенос атома водорода (отщепление Н, например, дегидрирование углеводородов и
спиртов, см. разд. Г,6.3; аутоокисление, см. разд. Г,6.2);
— перенос гидрид-иона (отщепление Н", например, реакция Оппенауэра — окисление
первичных и вторичных спиртов, см. разд. Г,7.3.1.2);
— присоединение кислорода (например, озонирование, гидроксилирование, эпоксиди-
рование олефинов, см. разд. Г,4.1.6, Г,4.1.7, Г,6.5.1);
— окислительное присоединение (например, к комплексам металлов побочных групп
периодической системы, см. разд. Г,4.5.1).
С термодинамической точки зрения реакционноспособность соединений в
окислительно-восстановительных реакциях можно оценивать по величинам их
окислительно-восстановительных потенциалов.
Электронный перенос определяется в соответствии с уравнением
= п?Е°
(ArC° — стандартная молярная свободная энтальпия реакции; F— число Фарадея, п — коли-
количество обменивающихся электронов) разностью стандартных потенциалов окислителя и вос-
восстановителя
[Г.6.3]
донор
' акцептор
Реакция протекает самопроизвольно, если разность молярных свободных энтальпий отрица-
отрицательна. На опыте установлено, что скорость реакции тем больше, чем меньше (отрицательнее)
окислительный потенциал донора и чем выше (положительнее) восстановительный потенци-
потенциал акцептора.
Таким образом, соединение окисляется тем легче (т. е. тем сильнее оно как восстанови-
восстановитель), чеМ меньше его стандартный электродный потенциал, и соответственно окислитель тем
сильнее, чем больше его стандартный потенциал.
Стандартные электродные потенциалы принято оценивать по отношению к потенциалу
стандартного водородного электрода (СВЭ) в воде при рН 1 и 25 °С. Например:
Мп2+ + 4Н2О -» МпО4- + 8Н+ + 5е-, Е° = 1,51 В
[Г.6.4]
6. Окисление и дегидрирование
Таблица Г.6.5. Стандартные окислительно-восстановительные потенциалы наиболее
распространенных неорганических окислителей и полярографические потенциалы по-
полуволны окисления некоторых органических соединений
Стандартный
потенциал
F2/HF
S2O827SO42-
Н2О2/Н2О
Се4+/Се3+
МпО47Мп2+
НОС1/С1-
РЬ4+/РЬ2+
С12/С1-
Сг2О72-/Сг3+
Mn(VMn2+
О2/Н2О
Вг2/Вг-
HNO3/NO
СЮ-/С1-
Fe3+/Fe2+
MnO4-/MnO2
ь/i-
S/H2S
н+/н2
Zn2+/Zn
Na+/Na
ia, В
+3,06
+2,01
+ 1,77
+ 1,71
+ 1,51
+ 1,50
+ 1,46
+ 1,36
+ 1,36
+ 1,23
+ 1,23
+ 1,09
+0,96
+0,88
+0,77
+0,58
+0,54
+0,14
0,00
-0,76
-2,71
Стандартный
потенциал8, В
2,3-Дихлор-5,6-дициано-
+1,00
1,4-бензохинон в/2,3-дихлор-
5,6-дицианогидрохинон
Тетрахлорхинон г/тетрахлор- +0,74
гидрохинон
Хинон/гидрохинон
Метилхинон/метилгид-
рохинон
Красный BiopcTepa/N,N-
диметил-л-фенилендиамин
Хинон/гидрохинон (при
РН7)
Антрахинон/9,10-дигид-
роксиантрацен
+0,7
+0,64
+0,34
+0,3
+0,13
Анодный потенциал
полуволны6, В
Бензол
Толуол
л-Ксилол
4-Метокситолуол
Анилин
Нафталин
Фенантрен
4-Хлор-/п/7онс-стильбен
/ираяс-Стильбен
1,1 -Д ифенилэтилен
Антрацен
4-Метокси-/яр«ис-стильбен
Ацетанилид
Триэтиламин
4-Диметиламинотолуол
2,54
2,23
2,01
1,82
1,80
1,78
1,74
1,74
1,72
1,70
1,61
1,41
1,15
1,09
0,86
а В воде при 25 °С. Участвующие в реакции протоны не указаны.
6 В ацетонитриле относительно стандартного водородного электрода.
в 4,5-Дихлор-3,6-диоксо-1,2-дицианоциклогексадиен-1,4.
г Хл оран ил.
Стандартные потенциалы часто встречающихся в органической химии неорганических
окислителей приведены в табл. Г.6.5.
Лишь очень немногие органические субстраты образуют равновесную окис-
окислительно-восстановительную систему. Из данных табл. Г.6.5 следует, например,
что гидрохинон в стандартных условиях окисляется ионом Fe3+, но не может
быть окислен серой. Точно также любой хинон, стоящий в табл. Г.6.5 выше,
способен окислить любой гидрохинон, расположенный ниже.
Поскольку органические соединения редко хорошо растворимы в воде и окислительно-
восстановительные реакции почти никогда не являются обратимыми, то их стандартные по-
потенциалы не всегда можно определить. Вместо этого чаще определяют полярографически или
методом цикловольтаметрии окислительный EOKm(D) [=E(D+/D)] и восстановительный
^восст(А) [=ДА/А~)] потенциалы в органическом апротонном растворителе (обычно в ацетонит-
ацетонитриле) относительно сравнительного электрода (например, насыщенного каломельного электро-
электрода). Если в окислительно-восстановительной реакции разность ЕОКис (D) — iwcr (A) + ЕкуЛ отри-
отрицательна, то такая реакция термодинамически возможна. Все потенциалы при этом должны
быть измерены относительно одного и того же электрода и в одном и том же растворителе, а
8 Г Препаративная часть (продолжение)
если в реакции участвует сильнополярное или несущее заряд вещество, следует учитывать так-
также энергию кулоновского взаимодействия (.Е^л).
В последней колонке табл. Г.6.5 приведены окислительные потенциалы (относительно
стандартного водородного электрода) некоторых органических соединений, из которых следу-
следует, что электронодонорные (или акцепторные) заместители, как и следовало ожидать, умень-
уменьшают (соответственно акцепторные — увеличивают) окислительный потенциал. К сожале-
сожалению, из величин стандартных электродных потенциалов, приведенных в первой колонке табл.
Г.6.5, невозможно оценить величины ArG°, поскольку потенциалы, определенные в ацетонит-
риле, по непонятной пока причине на 0,5-1 В (и более того) выше, чем в воде. Поэтому при
оценке окисляемости субстрата часто приходится ограничиваться анализом других рядов ре-
реакционной способности (в особенности рядов нуклеофильных замещений) (см. разд. Г,2.2.2).
Окислительно-восстановительные потенциалы и полярографические потенциалы полу-
полуволны прямо связаны с энергиями граничных орбиталей соединения. Экзоэнергетический
перенос электронов возможен, если энергия ВЗМО донора электронов (восстановителя) боль-
больше, чем энергия НСМО акцептора (окислителя) (см. разд. В,6).
Отдача электрона субстратом, сопровождающая окисление, происходит тем
легче, чем больше энергия его ВЗМО, (т.е. чем меньше величина E°otMCll донора).
Аналогично эффективность окислителя оказывается тем больше, чем меньше
энергия его НСМО (или чем больше величина Е^^ акцептора).
В общем случае энергия заполненных s-орбиталей значительно ниже, чем
р- и л-орбиталей. Поэтому способность к окислению увеличивается в указанных
ниже рядах следующим образом:
R-H < R-OH < R-NH2
II \ /
—с-с— < —с=с— < с=с
II / \
Поэтому связи С—С и С—Н обычно не окисляются путем потери электрона.
Напротив, окисление с переносом электронов возможно для олефинов, аренов,
спиртов и прежде всего аминов. Наиболее известный пример — окисление
Ы,Ы-диметил-и-фенилендиамина (см. схему [Г.6.48]).
При окислении вещества X, сопровождающемся переносом водорода, опре-
определяющим фактором является энергия диссоциации связи AD#° (см. табл. Г. 1.4).
В общем случае это возможно только при условии, что в реакции Y* + R—Н ->
Y-H + R' (для Y* = С1'; см. схему [ГЛ.13]) AD/f(Y-H)>AD/f(R-H) (см. схему
[В.20]иразд.Г,1.3).
При этом способность к окислению насыщенных углеводородов по механиз-
механизму отщепления водорода увеличивается от первичных к третичным связям С—Н.
Отщепление водорода от группы О—Н в спиртах энергетически невыгодно, по-
поэтому третичные спирты, например mpe/n-бутанол, окисляются с большим тру-
трудом. Напротив, водород легко отщепляется от группы ос-С—Н первичных и вто-
вторичных спиртов и С-Н альдегидов; то же относится и к муравьиной кислоте.
Сравнение энергий диссоциации показывает также, что альдегиды окисляются
легче, чем первичные спирты. Это объясняет, например, тот факт, что из пер-
первичных спиртов трудно получить альдегиды, избежав их дальнейшего окисления
в карбоновые кислоты.
Ароматические соединения и олефины также с большим трудом окисляются
посредством отщепления атома водорода от связи =С—Н (сравните, однако, с
окислительной атакой на двойную связь С=С; разд. Г,4.1.6и Г,6.5.1).
6. Окисление и дегидрирование
В то время как отщепление водорода от насыщенных углеводородов происхо-
происходит относительно легко, связь С—С рвется с трудом, что не согласуется с относи-
относительно низкой энергией диссоциации этой связи (около 290—370 кДж/моль).
Очевидно, центральная связь С—С экранируется от атаки окислителя заместите-
заместителями.
В природе широко распространены процессы ферментативного окисления. Оксидоредук-
тазы катализируют дегидрирование, оксидазы — электронный перенос, диоксигеназы — пере-
перенос О2 (к двойным связям С=С), а гидроксилазы — гидроксилирование связей С—Н кислоро-
кислородом. Дыхательной цепью называют ферментативную систему клеточного дыхания, в процессе
которого водород переносится ступенчато от субстрата к молекулярному кислороду. При этом
активные группы амида никотиновой кислоты (ср. [Г.7.247]) и рибофлавина переносят в про-
промежуточных стадиях атомы водорода (два электрона и два протона), а цитохромы переносят
электроны.
6.2. ОКИСЛЕНИЕ МЕТИЛЬНЫХ И МЕТИЛЕНОВЫХ ГРУПП
Насыщенные углеводороды окисляются с трудом и крайне неселективно. Поэ-
Поэтому какие-либо препаративные методики таких процессов окисления отсут-
отсутствуют.
В промышленности алканы окисляют воздухом в присутствии катализаторов — соедине-
соединений тяжелых металлов (например, пентаоксида ванадия или солей марганца и кобальта); кроме
того, процесс окисления можно проводить с добавлением аммиака (аминоокисление). Воз-
Возможно, реакция протекает через стадию образования пероксидов (аутоокисление, см. обсужде-
обсуждение в разд. Г, 1.5).
В соответствии со схемой [Г.6.6] при окислении алканов могут образовывать-
образовываться спирты, кетоны или кислоты (частично с разрывом С—С-связей):
—сн3 —> —сн2он —- —сно —~ —соон
\ \ \ [Г.6.6]
СН2 —- СНОН —- СО (—- —СООН с расщеплением связи С-С)
Борная кислота препятствует расщеплению связи С—С, что используется для техническо-
технического синтеза вторичных спиртов из «-парафинов (окисление по Башкирову)
ОН
+ О? 1 R' л П'
R
OH
Первоначально образующийся эфир борной кислоты гидролизуется. При низкой степени
превращения (почему?) образуется смесь всех возможных вторичных спиртов, которые можно
использовать, например, в производстве моющих средств (см. схему [Г.2.53]).
В промышленности часто является допустимым образование смеси веществ; так, например,
при окислении бутана кислородом с ацетатом кобальта в качестве катализатора при 165 °С под
давлением получают смесь продуктов с примерным соотношением метилэтилкетон : уксусная
кислота : метил- и этилацетаты 1:15:3. Промышленное окисление циклогексана в присутствии
ацетата кобальта приводит к смеси циклогексанола и циклогексанона (см. табл. Г.4.103).
Способность метильной или метиленовой группы к окислению существенно
повышается, если она находится у двойной связи или ароматического ядра, пос-
поскольку введение в соединение кратной связи уменьшает его окислительный
10
Г Препаративная часть (продолжение)
потенциал и облегчает перенос электрона (см. табл. Г.6.5) или поскольку при
аутоокислении возникает энергетически более устойчивый радикал аллильного
типа. В этом случае реакция идет более селективно, и в определенных условиях
могут быть получены спирты или даже альдегиды.
Следует иметь в виду, что активирование метильной или метиленовой группы олефиновой
двойной связью не всегда удается использовать для получения непредельных карбонильных со-
соединений, так как двойная связь С=С в общем случае быстрее поддается действию кислотных
окислителей — хромовой кислоты и перманганата калия (с гидроксилированием и расщепле-
расщеплением связи С-С; см. разд. Г,4.1.6 и Г,6.5.1), чем алкильная группа. Для селективного окисления
алифатического атома углерода, соседнего с кратной связью, используют аутоокисление или
окисление диоксидом селена (разд. Г,6.2.3) либо некоторыми соединениями иода(Ш).
Например, в промышленности акролеин получают окислением пропена кислородом в га-
газовой фазе при 350-400 °С над катализатором (оксид меди). Акролеин далее через аллиловый
спирт превращают в глицерин (разд. Г,4.1.6). Аутоокислением пропена над катализатором
(соль молибдена) при 200—500 °С и давлении 1 МПа получают акриловую кислоту. Из изобу-
тена аналогичным методом — метакриловую кислоту. Из бутена-2, а также его смеси с буте-
ном-1 окислением кислородом воздуха в присутствии V2C>5 получают ангидрид малеиновой
кислоты (разд. Г,6.5.1); в качестве побочных продуктов образуются уксусная, акриловая, кро-
тоновая и фумаровая кислоты.
Большое техническое значение имеет окисление углеводородов воздухом в присутствии
аммиака. Так, из метана получают синильную кислоту (метод Андрусова), из толуола и других
метиларенов — бензонитрил и его производные, из пропена — акрилонитрил (табл. Г.6.9).
Последняя из названных реакций, которая проводится в газовой фазе при 400—450 °С над ка-
катализатором (В12О3/МоОз), представляет собой в настоящее время наиболее важный метод
синтеза акрилонитрила. Побочными продуктами являются ацетонитрил и синильная кислота.
Этим же методом получают метакрилонитрил из изобутена.
R-СНз
R-CH=O
R-CH=NH
R-CHN
[Г.6.8]
(об аутотермическом дегидрировании см. разд. Г,6.3.1).
Таблица Г.6.9. Нитрилы, получаемые в промышленности окислительным аммонолизом
Конечный продукт
Синильная кислота
Акрилонитрил
Бензонитрил
Фталонитрил
Метакрилонитрил
Исходное
соединение
Метан
Пропен
Толуол
о-Кислол
Изобутен
Применение
В органическом синтезе (см. разд. Г,5.1.8.2,
Г,7.2.1.1); как инсектофунгицид
Получение полиакрилонитрила (ПАН)
Растворитель для набухания ПАН
Получение фталоцианинов
Аналогично метилметакрилату (см. табл. Г.3.37)
6.2.1. ОКИСЛЕНИЕ АЛЮШРЕНОВ ДО АРОМАТИЧЕСКИХ КАРБОНОВЫХ КИСЛОТ
Из-за высокого окислительного потенциала (см. табл. Г.6.5) ароматические сое-
соединения окисляются лишь очень сильными окислителями, такими как перман-
ганат калия, смесь хромовой и серной кислот или нитрат церияAУ)-аммония
при температуре кипения, согласно уравнению [Г.6.6]. (Значение потенциалов в
табл. Г.6.5 получены в ацетонитриле, и для воды они должны быть уменьшены
6. Окисление и дегидрирование 11
по крайней мере на 0,5 В). Реакция вдет по схеме переноса электрона:
Аг-СНз + М"® (Аг-СНз)? + М("-1)® [Г.б.Юа]
(Аг-СНз)? Аг-СН2- + Н® [Г.6.106]
Аг-СН2- + Мл® - Аг-СН2® + М<"-1>® [Г.б.Юв]
[Г.б.Юг]
Ar-CH2®+ Nu-H (илиЫи0) > Ar-Nu + H®
Nu-H = Н2О или СН3СООН
Бензильный радикал очень быстро окисляется дальше, так что в воде или уксусной кисло-
кислоте он регистрируется в форме бензилового спирта или соответственно бензилацетата. Впрочем
этот первый продукт окисления также легко подвергается дальнейшему окислению по схемам
[Г.6.10а]—[Г.6. Юг], так что в воде как нуклеофиле в конце концов образуются кислоты. Одна-
Однако эфиры уксусной кислоты обладают более высоким окислительным потенциалом по сравне-
сравнению с соответствующими спиртами (см. падение величин потенциалов от анилина к ацетами-
ду в табл. Г.6.5), поэтому диацетат дигидрата альдегида далее не окисляется (см. [Г.6.16]).
Алкиларены с более длинными алкильными цепями образуют первоначаль-
первоначально а-арилалкилкетоны, которые (благодаря способности к енолизации) в ре-
результате расщепления С—С-связи также превращаются в карбоновые кислоты.
Арилалкилкетоны могут быть получены также и методом пероксидирования.
В лаборатории для окисления алкиларенов в ароматические карбоновые
кислоты еще и сегодня применяют классические окислители: перманганат
(обычно щелочной раствор), хромовую смесь, хромовую кислоту в уксусной или
серной кислоте, а также кислород в присутствии солей кобальта или марганца,
например:
+ 2KMnO4 [I | + 2MnO2 + 2K0H ГГ6 11 1
СН2-СН3
+ 4КМпО4 (f Т + СО2 + 4МпО2 + 4 КОН [Г.6.116]
\\ \ + Na2Cr207 + 4H2SO4 [| J + Cr2(SO4K [Г.б.Пв]
/Ч^^ О-
Na2SO4 + 5H2O
3 ^
+ 3/2O2 ^^ \\ T + H2O
H3C — НзС^ [Г6Л1Г]
Диметилированные ароматические соединения в присутствии пермангана-
та сразу переходят в соответствующие дикарбоновые кислоты, например о-кси-
лол — во фталевую кислоту, однако при воздействии кислорода в присутствии
катализатора (солей кобальта) они сначала окисляются до монокарбоновых
кислот. Только в более жестких условиях или в присутствии кислот, например,
НВг с катализатором ацетат-бромид кобальта, оказывается возможным окис-
окислить вторую метильную группу (почему?).
12
Г Препаративная часть (продолжение)
Этот факт можно использовать для частичного окисления полиалкилиро-
ванных ароматических соединений. Горячая 30%-ная азотная кислота также
окисляет только одну метильную группу. ор/ло-Заместители в общем затрудняют
окисление; амино- и гидроксигруппы перед окислением следует защищать
(почему, как?).
Более длинные, в том числе разветвленные и ненасыщенные, боковые цепи
аренов при действии перечисленных выше окислителей, как правило, превра-
превращаются в непосредственно связанную с ядром карбоксильную группу. Рассмот-
Рассмотренные окислительные реакции используют в аналитических цепях для иденти-
идентификации алкиларенов, поскольку таким путем можно установить положение
алкильных групп в ядре. Для этих целей, как правило, применяют хромовую
кислоту или перманганат.
Для аналитических целей предпочтительнее окисление перманганатом, так как при ис-
использовании хромовой кислоты труднее очищать небольшие количества образовавшихся кар-
боновых кислот. Кроме того, перманганат в условиях межфазного катализа (см. разд. Г,2.4.2)
позволяет сократить продолжительность реакции. Соединения с группами, чувствительными
к щелочам, необходимо подвергать кислотному окислению, например, в 30%-ном растворе
Na2Cr207 в 50%-ной серной кислоте (примерно 1,5 г анализируемого вещества в 20 мл).
Ф Общая методика препаративного и аналитического получения ароматических карбоновых
кислот из алкиларенов (межфазный катализ) (табл. Г.6.12). В кругл одонную колбу с обрат-
обратным холодильником наливают 70 мл воды, добавляют 0,5 мл аликвата 336 и 0,02 моль карбо-
карбоната натрия; для нитроароматических соединений — 0,02 моль гидрокарбоната натрия; в слу-
случае пиридинов основание не добавляют. Затем вводят 0,015 моль алкиларена или примерно
Таблица Г.6.12. Аренкарбоновые кислоты
Конечный продукт
Бензойная кислота
л-Хлорбензойная кислота
о-Нитробензойная кислота
л-Нитробензойная кислота
Фталевая кислота
Терефталевая кислота
Пиридин-4-карбоновая
кислота6'в (изоникотиновая
кислота)
Пиридинкарбоновая-3
кислота6'г (никотиновая
кислота)
Сахарин
л-Ацетамидобензойная
кислота
Исходное соединение
Толуол,этилбензол
л-Хлортолуол
о-Нитротолуол
о- Нитроэтил бензол
л-Нитротолуол
о-Ксилол
л-Ксилол
4-Метил пиридин
(у-пиколин)
З-Метилпиридин
(Р-пиколин)
Амид о-толуолсуль-
фоновой кислоты
л-Ацетамидотолуолд
Т. пл.,°С
122(вода)
241 (возг.; водный этанол)
148 (вода)
240 (возг.; вода)
191а (вода)
300 (возг.; вода)
311а(вода)
235 (вода)
228-229 (возг.; ацетон)
150—152 (разд.; этанол)
Выход, %
65-75
75
52-60
68
70
80
55
54
62
55
а В запаянном капилляре.
6 При обработке раствор упаривают на треть и конц. соляной кислотой доводят до
изоэлектрической точки (см. разд. Г,7.2.1.1).
¦ рНЗ,6.
г рНЗ,4.
д Берут 0,07 моль КМпО4. Непрореагировавшее исходное соединение выпадает после
фильтрования и его снова вводят в реакцию.
6. Окисление и дегидрирование
13
1,5 г анализируемого углеводорода и 0,05 моль перманганата калия. В препаративном синте-
синтезе это количество перманганата необходимо для окисления одной метальной группы, для
этильной группы берут 0,1 моль КМпО4 (для более длинных и разветвленных цепей соответ-
соответственно еще больше окислителя). Смесь нагревают до обесцвечивания перманганата или
45 мин с обратным холодильником. Затем горячий раствор фильтруют, диоксид марганца
дважды промывают небольшим количеством горячей воды и в случае необходимости обесц-
обесцвечивают раствор гидросульфитом натрия. В заключение смесь подкисляют разбавленной
A:1) серной кислотой, охлаждают и после завершения кристаллизации осадок карбоновой
кислоты отфильтровывают. Перекристаллизовывают из воды или разбавленного спирта.
Выход можно немного повысить, если перед подкислением раствор упарить или маточный
раствор экстрагировать эфиром. Небольшие количества диоксида марганца можно растворить
без фильтрования раствором гидросульфита или щавелевой кислотой. При большем количе-
количестве исходных веществ следует наполовину уменьшить указанный в методике объем воды. Так
как реакция сильно экзотермична, при исходном количестве алкиларена более 0,15 моль пер-
манганат следует вводить порциями или в виде раствора по каплям, уменьшив в соответству-
соответствующей степени количество воды.
Реакция не идет до конца, так как даже введенный в избытке перманганат расходуется на побоч-
побочные процессы. При больших количествах исходных веществ непрореагировавшее исходное соедине-
соединение можно регенерировать экстракцией диоксида марганца и щелочного раствора эфиром.
©Общая методика аутоокисления замещенных в ядре толуолов в соответствующие бензойные
кислоты (табл. Г.6.13). В трехгорлую колбу на 500 мл, снабженную мешалкой (смазывать
только чистым парафиновым маслом!), газоподводящей трубкой, изображенной на
рис. А. 13,а (не использовать резиновые пробки!), водоотделителем и обратным холодильни-
холодильником (шлифы не смазывать, смазка ингибирует реакцию), помещают 0,5 моль перегнанного
производного толуола, 70 мл хлорбензола и 0,3—0,5 г стеарата кобальта (см. часть Е). При
окислении ксилолов и мезитилена загрузку увеличивают до 1 моль, хлорбензол не добавляют.
Через предохранительную промывную склянку, промывалку с щелочным раствором КМпО4 и
осушительную колонку с КОН (рис. АЛ 1) в кипящую смесь пропускают кислород (примерно
30 л/ч; измеритель скорости потока см. разд. А, 1.6). С помощью реле устанавливают темпера-
температуру нагревателя таким образом, чтобы обеспечить слабое равномерное кипение. В ходе реак-
реакции нагревание следует несколько усиливать.
Окисление начинается не позже чем через 2 ч (для инициирования реакции можно доба-
добавить 0,1 г азобисизобутиронитрила); в среднем реакция продолжается 6—10 ч. В случае ксило-
ксилолов реакцию прекращают после того, как в водоотделителе соберется около 5 мл воды. В дру-
других случаях окисление ведут до полного прекращения выделения воды. Если выпадающий
продукт реакции мешает дальнейшему введению газа, то процесс прерывают, осадок отфильт-
отфильтровывают, а фильтрат снова вводят в реакцию.
По окончании реакции смесь оставляют на ночь в холодильнике, осадок отфильтровывают и пе-
перекристаллизовывают. Фильтрат перегоняют на колонке. Выход в расчете на исходное производное то-
толуола составляет около 50%. С помощью перекристаллизации (из толуола) твердого остатка от пере-
перегонки выход можно повысить.
Таблица Г.6.13. Получение замещенных бензойных кислот аутоокислением
Конечный продукт
о-Толуиловая кислота
л-Толуиловая кислота
л-Толуиловая кислота
3,5-Диметилбензойная кислота
л-Хлорбензойная кислота
Монометиловый эфир
терефталевой кислоты
Исходное соединение
о-Ксилол
м- Ксилол
я-Ксилол
Мезитилен
л-Хлортолуол
Метиловый эфир л-толуиловой
кислоты
Т. пл., °С
105 (вода)
111 (вода)
180 (разб. этанол)
170 (возг.; этанол)
240(пропанол)
230 (возг.; вода)
14
Г Препаративная часть (продолжение)
Таблица Г.6.14. Применение промышленно важных карбоновых кислот, получаемых
окислением метиларенов
Кислота
Бензойная кислота
Фталевая кислота (ангидрид)
Изофталевая кислота
Терефталевая кислота
л-Нитробензойная кислота
Никотиновая кислота
Изоникотиновая кислота
Основное применение
Консервирующее средство;
-> сложные эфиры (репелленты, душистые вещества)
-» Октиловый, бутиловый, этиловый эфиры (пластифи-
(пластификаторы)
-> Полиэфирные смолы (алкидные смолы)
-> Антрахинон (красители)
->• Алкидные смолы
-> Пластификаторы
->• Полигликольтерефталат (синтетические волокна:
терилен, гризутен, дакрон, диолен, лавсан)
—> л-Аминобензойная кислота, лекарственные средства
(прокаин [новокаин], анестезин) (табл. Г.7.42)
-» Амид никотиновой кислоты (витамин) -> 3-аминопи-
ридин (фармпрепараты)
-» М,М-Диэтиламид никотиновой кислоты (ницетамид,
аналептик)
-» Инозитолникотинат (гипотензивное средство)
-> Гидразид изоникотиновой кислоты (INH, неотебен,
изониазид, римифон — противотуберкулезное лекар-
лекарственное средство)
Получение замещенных бензолмоно- и -дикарбоновых кислот из толуола и кси-
ксилола аутоокислением в присутствии ацетата кобальта и уксусной кислоты —
НВг: Hay A. S., Blanchard H. S. Canad. J. Chem., 1965, 43, 1306.
Сведения о техническом применении ароматических карбоновых кислот, получаемых в
больших объемах из соответствующих алкиларенов, приведены в табл. Г.6.14. В качестве окис-
окислителя используют воздух в присутствии пентаоксида ванадия или солей кобальта или марган-
марганца, а также азотную кислоту (Бофорс-процесс). В присутствии катализатора Со/Мп-аце-
тат/бромид в уксусной кислоте ксилолы одностадийно окисляются до фталевых кислот
(Амоко-процесс), т. е. упомянутые в табл. Г.6.13 трудноокисляемые толуиловые кислоты
в данном случае в качестве промежуточных продуктов не образуются. Другие трудно-
окисляемые производные толуола, такие как нитротолуол или толунитрил, дальше не окисля-
окисляются.
Никотиновую кислоту в промышленности получают окислением 2-метил-5-этилпириди-
на до пиридин-2,5-дикарбоновой кислоты с последующим селективным декарбоксилирова-
нием 2-карбоксильной группы. Важное подслащивающее вещество неуглеводной природы —
сахарин (натриевая соль имида о-сульфобензойной кислоты) получают окислением о-толуол-
сульфонамида (см. табл. Г.6.12).
or
КМпО4
SO2NH2
соон
SO2NH2
-Н2О
со
т
so2
[Г.6.15]
Часто сахарин применяют в смеси с очень сладкой натриевой солью N-циклогексилсульфона-
мида.
6. Окисление и дегидрирование 15
6.2.2. ОКИСЛЕНИЕ АЛКИЛАРЕНОВ ДО АЛЬДЕГИДОВ И КЕТОНОВ
Превращение метиларенов в альдегиды затруднено, поскольку образующийся
альдегид окисляется легче, чем метильная группа. Поэтому альдегид необходи-
необходимо непрерывно удалять из реакционной смеси, например, переводя его в более
устойчивые производные. В качестве окислителя пригодна хромовая кислота в
ацетангидриде; в этой смеси альдегид стабилизируется в виде диацетата:
Ar-СНз *»/<°*°О*>, АГ-ф [Г.6.16]
ОСОСНз
Примеры получения ароматических альдегидов B- и 4-нитробензальдегидов,
4-бромбензальдегида, 4-формилбензонитрила) окислением соответствующих
метиларенов хромовым ангидридом в уксусном ангидриде с последующим гид-
гидролизом образующегося диацетата: Nishimura Т., Org. Synth., Col. Vol. IV A963),
713; Tsang S. M., Wood E. H., Johnson J. R., Org. Synth., Coll. Vol III A955), 641;
Lieberman S. V., Connor R. Org. Synth., Coll. Vol. II A943), 441.
Ароматические альдегиды могут быть также получены точно дозированным
окислением метиларенов диоксидом марганца (образующимся в результате
диспропорционирования смеси перманганата калия с сульфатом марганца).
Другим селективным окислителем является нитрат церия:
3 + 4Се4®+ Н2О Н3С-/ \-СН=О + 4Се3® + 4 Н® [Г.6.17]
\—/
Ф Получение л-толуилальдегида из n-ксилола [Trahanovsky W. S., Young L. В. J. Org. Chem.,
1966, 31, 2033]. В двугорлой колбе емкостью 500 мл с мешалкой и обратным холодиль-
холодильником смешивают раствор 0,4 моль церийаммонийнитрата в 200 мл 50%-ной уксусной
кислоты с 0,1 моль «-ксилола и нагревают при интенсивном перемешивании 20 мин на кипя-
кипящей водяной бане. (Смесь в процессе реакции должна стать светло-желтой.) После охлажде-
охлаждения реакционную смесь трижды экстрагируют эфиром: эфирный раствор промывают сначала
1,5 н. раствором соды (СОг!), а затем небольшим количеством воды, сушат над MgSO4 и пере-
перегоняют. Т. кип. 206-208 °С; 106 "С A0 мм рт. ст.); выход 68%.
По такой же методике получают о-толуилальдегид из о-оксилола; выход 25%.
Если стоящая у ароматического ядра метильная группа достаточно реакци-
онноспособна, то в качестве селективного окислителя можно использовать и
диоксид селена (см. часть Е). Этот метод применяют прежде всего для окисления
метилзамещенных гетероциклических соединений. Так, этим методом можно
получить альдегиды из 2-метилбензотиазола, 2- или 4-метилпиридина, -хино-
лина, -хиназолина а также 2-метилнафталина.
Получение нафталинкарбальдегида-2 из 2-метилнафталина: Султанов А. С,
Родионов В. М., Шемякин М. М. ЖОХ, 1946, 16, 2073.
Хинолинкарбальдегид-4 из 4-метилхинолина: McDonald S. F. J. Am. Chem.
Soc, 1947, 69, 1219.
Урацилкарбалъдегид-6 из 6-метилурацила: Zee-Cheng К. J., Cheng С. С. J.
Heterocycl. Chem., 1967, 163.
В промышленности бензальдегид получают частичным окислением толуола воздухом на
катализаторе (соли вольфрама и молибдена) при 500-600 "С или в жидкой фазе.
Другим возможным путем получения ароматических альдегидов является хлорирование
метиларенов до бензилидендихлоридов с последующим гидролизом (см. табл. Г. 1.27 и Г.2.68).
16 Г Препаративная часть (продолжение)
Метиленовые группы, активированные соседними арильными остатками, в целом ряде
соединений можно селективно окислить до кетогрупп окислителями, указанными в разд.
Г,6.2.1, например о-нитроэтилбензол в особых условиях:
В промышленности ацетофенон получают окислением этилбензола над ацетатом марганца
при 130 "С, однако здесь одновременно идет побочная реакция (какая?). Подобный метод при-
применяют также для синтеза о- и «-нитроацетофенонов (и далее о- и и-аминоацетофенонов),
которые нельзя получить нитрованием.
Получение о-, м- и п-нитроацетофенонов из 2-, 3- и 4-нитроэтилбензолов
окислением перманганатом калия в присутствии сульфата алюминия: Кочер-
гин П. М., Титкова Р. М., Засосов В. А., Григоровский А. М. Ж. прикл. хим., 1959,
32, 1806.
Синтез флуоренона из флуорена окислением ^гСггСЬ: Huntress Е. Н.,
Hershberg Е. В., Cliff I. S, J. Am. Chem. Soc. 1931, 53, 2720.
В промышленности ацетофенон можно получать окислением этилбензола воздухом при
130 °С в присутствии ацетата марганца, однако при этом протекает побочная реакция (какая?).
Аналогичным образом получают 2- и 4-нитроацетофеноны (для последующего восстановле-
восстановления их в 2- и 4-аминоацетофеноны), которые нельзя получить нитрованием ацетофенона.
6.2.3. ОКИСЛЕНИЕ АКТИВИРОВАННЫХ МЕТИЛЬНЫХ И МЕТИЛЕНОВЫХ ГРУПП
В КАРБОНИЛЬНЫХ СОЕДИНЕНИЯХ
6.2.3.1. Окисление диоксидом селена
Метальные и метиленовые группы, находящиеся рядом с карбонилом, действи-
действием диоксида селена могут быть селективно превращены в карбонильные груп-
группы. При этом с весьма различными выходами образуются а-оксоальдегиды,
например:
НзС-СНО ОНС-СНО
глиоксаль
[Г.6.19]
н3с-сн2-со-сн3 —- н3с-сн2-со-сно + н3с-со-со-снз
17% 1%
Вероятно, при этом енол окисляется селенистой кислотой с выделением эле-
элементного селена:
о
L Se. О О
*° ??С° А^? V° [Г.6.20]
В качестве растворителей используют ксилол, этанол или диоксан. Следы
воды во многих случаях способствуют повышению выхода.
6. Окисление и дегидрирование
17
Таблица Г.6.21. Получение сс-дикарбонильных соединений окислением диоксидом
селена
Продукт реакции
Фенилглиоксаль
«-Бромфенилоксоацетальдегид
«-Этилфенолоксоацетальдегид
2,4,6-Триметилфенолоксоацет-
альдегид
I -Фенилпропандион-1,2
Циклогександион-1,2
Исходное соединение
Ацетофенон
п- Бромацетофенон
л-Этилацетофенон
2,4,6-Триметилацето-
фенон
Пропиофенон
Циклогексанон
Т. кип. (мм рт. ст.), °С
95-97 B5);
т. пл. 91 (гидрат)
пл. 135-142A7);
т. пл. 132—134 (гидрат)
110-114B0);
т. пл. 93—95 (гидрат)
106 D); «д191,5520
103 A2);/7fl191,5334
78 A6); т. пл. 34
Выход, %
65
50
45
60
35
25
О получении а-дикарбонильных соединений через изонитрозокетоны
см. разд. Г,8.2.3.
(Hj) Общая методика получения арилоксоацетальдегидов и 1,2-дикетонов (табл. Г.6.21)
jj Внимание! Селенсодержащие остатки тщательно собрать!
В трехгорлой колбе на 500 мл, снабженной мешалкой, обратным холодильником и термо-
термометром, к 0,25 моль кетона по каплям добавляют раствор 0,25 моль возогнанного диоксида
селена (см. часть Е) в 180 мл диоксана и 12 мл воды. Температура при этом не должна подни-
подниматься выше 20 °С (в случае необходимости колбу охлаждают водой), затем кипятят 6 ч при
перемешивании, для отделения выпавшего в осадок селена еще горячий раствор фильтруют
(не отсасывают!), осадок промывают диоксаном. (В случае необходимости следует отфильтро-
отфильтровать еще раз.) После отгонки растворителя в вакууме остаток перегоняют в вакууме, собирая
основную фракцию в широком интервале температур B0-30 °С), затем еще раз ректифициру-
ректифицируют ее, используя короткую колонку Вигре.
При получении арилоксоацетальдегидов их можно перевести в устойчивые гидраты. Для
этого перегнанный сырой продукт кипятят в 4—6-кратном количестве воды; выпавшие после
охлаждения кристаллы отфильтровывают; в случае необходимости их можно дополнительно
очистить перекристаллизацией из 20%-ного этанола с активированным углем.
Для окисления алкильной группы енолизуемых кетонов до сх-гидроксикето-
нов вместо токсичного оксида селена можно использовать соединения
иода(Ш), например диацетоксииодбензол РЫ(ОССОСН3J-
6.2.3.2. Реакция Вильгеродта
В ходе реакции Вильгеродта арилалкилкетоны действием раствора полисульфи-
полисульфида аммония (как правило, под давлением) превращаются в со-арилалканкарбо-
новые кислоты с сохранением общего числа атомов углерода:
Ar-CO-(CH2)n-CH3
(NH4JSX + НгО
Ar —(CH2)n+i-COOH
[Г.6.22]
18 Г Препаративная часть (продолжение)
В результате карбонильная группа кетона восстанавливается до метиленовой
группы, а метильная окисляется до карбоксильной.
Фактически сначала получают тиоамид (или амид) кислоты, который затем
омыляют. Улучшенный вариант, не требующий применения давления, предло-
предложил Киндлер. Вместо раствора полисульфида используют серу и вторичный
амин (чаще всего морфолин):
Аг-СО-СНз +S^qHR2- Ar-CH2-CS-NR2 + 2Н2°- Ar-CH2-COOH + H2S + HNR2 [Г.6.23]
Метод имеет значение прежде всего для получения арилуксусных кислот из
арилметилкетонов, которые в свою очередь легко образуются в результате
ацилирования по Фриделю — Крафтсу (см. разд. Г,5.1.8.1).
Реакция Вильгеродта начинается с образования енамина (см. разд. Г,7.1.1), который далее
присоединяет серу. Дальнейшее течение реакции нельзя описать одним механизмом для всех
субстратов. Для арил метил кетона этот механизм можно себе представить следующим образом:
\\ + R NH \ + Ц \ / \ и
C-CH3 _ 2 ¦ C=CH2 C=C Ar-C-C
Ar 2 Ar Ar H W \
Д
Q Ar4 NR2
S
4
- Ar-c-C--S - C=C ===^ Ar—CH2-C
H H H SH S
Ф Общая методика получения морфолидов тиокарбоновых кислот (реакция
Вильгеродта—Киндлера) (табл. Г.6.25)
¦ Осторожно! Выделяется сероводород! Работать под тягой!
В круглодонной колбе на 100 мл с обратным холодильником нагревают 6 ч при 135 °С (тем-
(температура бани) смесь 0,2 моль морфолина с 0,1 моль арилалкилкетона и 0,2 моль F,4 г) серы или
с 0,1 моль ароматического альдегида и 0,1 моль серы. Еще теплую реакционную смесь вылива-
выливают в 40 мл горячего этанола. Трением стеклянной палочкой о стенку стакана вызывают крис-
кристаллизацию тиоморфолида; раствор с осадком оставляют на ночь в холодильнике, осадок
отфильтровывают и промывают холодным этанолом. Для отделения от примеси серы рекомен-
рекомендуется перекристаллизация из нитрометана. Для омыления можно использовать неочищенный
препарат.
^у) Методика омыления морфолидов тиокарбоновых кислот (табл. Г.6.26)
JJ Осторожно! Выделяется сероводород! Работать под тягой!
К 0,1 моль неочищенного тиоморфолида приливают раствор 80 г 50%-ного КОН в 140 мл
этанола и кипятят 6 ч с обратным холодильником. Затем этанол как можно более полно отго-
отгоняют, остаток разбавляют водой, фильтруют и сильно подкисляют соляной кислотой (выделе-
(выделение сероводорода!). После охлаждения отфильтровывают выпавшую кислоту. Если кислота
растворима в воде или если она отделяется, образуя маслянистый слой, то ее извлекают тремя
порциями эфира по 100 мл, экстракты сушат сульфатом магния и отгоняют эфир. Кислоту
перекристаллизовывают из воды, при необходимости добавляют активированный уголь.
Выход можно повысить, проводя дополнительные экстракции маточных растворов.
6. Окисление и дегидрирование
19
Таблица Г.6.25. Реакция Вильгеродта — Киндлера
Продукт реакции
Морфолид л-толилтиоуксусной
кислоты
Морфолид B,4-диметилфенил)-
тиоуксусной кислоты
Морфолид (л-метоксифенил)-
тиоуксусной кислоты
Морфолид тиогомовератровой
кислоты
Морфолид нафтил-1 -тиоуксусной
кислоты
Морфолид нафтил-2-тиоуксусной
кислоты
Бисморфолид дитиомалоновой
кислоты
Морфолид тиобензойной кислоты
Морфолид л-метокситиобензойной
кислоты
Морфолид л-диметиламинотио-
бензойной кислоты
Морфолид р-фенилтиопропио-
новой кислоты6
Исходное соединение
«-Метил ацетофе нон
2,4-Диметилацетофенон
л-Метоксиацетофенон
3,4-Диметоксифенил-
метилкетон
Нафтил-1 -метилкетон
Нафтил-2-метилкетон
Аллиловый спирт3
(или ацетона)
Бензальдегид
Анисовый альдегид
л-Диметиламинобенз-
альдегид
Пропиофенон
Т. пл., °С
103 (этанол)
83 (метанол)
71 (метанол)
90 (этанол)
141 (вода)
108 (этанол)
195-197 (бутанол)
143 (этанол)
114 (этанол)
154 (этанол)
Выход, %
60
55
60
60
55
65
50 B0)
70
78
80
а Используют 0,4 моль серы.
6 Не выделяют, реакционную смесь перерабатывают далее (см. табл. Г.6.26).
Таблица Г.6.26. Получение арилуксусных кислот
Продукт реакции
л-Толилуксусная кислота
2,4-Диметилфенилуксусная кислота
л-Метоксифенилуксусная кислота
Гомовератровая кислота
Нафтил-1 -уксусная кислота
Нафтил-2-уксусная кислота
Р-Фенилпропионовая кислота
Исходное соединение
Морфолид л-толилтиоуксусной
кислоты
Морфолид B,4-диметил-
фенил)тиоуксусной кислоты
Морфолид (л-метоксифенил)-
тиоуксусной кислоты
Морфолид тиогомовератровой
кислоты
Морфолид нафтил-1 -
тиоуксусной кислоты
Морфолид нафтил-2-
тиоуксусной кислоты
Морфолид р-фенилтиопропио-
новой кислоты (неочищенный)
Т. пл., 'С
92
105
85
68 (гидрат)"
131
140
47 (лигроинN
Выход, %
80
80
75
60
85
85
40
а Безводный продукт имеет п. пл. 96 °С.
6 Неочищенная кислота перегоняется; т. пл. 169—170 °С B8 мм рт. ст.).
20
Г Препаративная часть (продолжение)
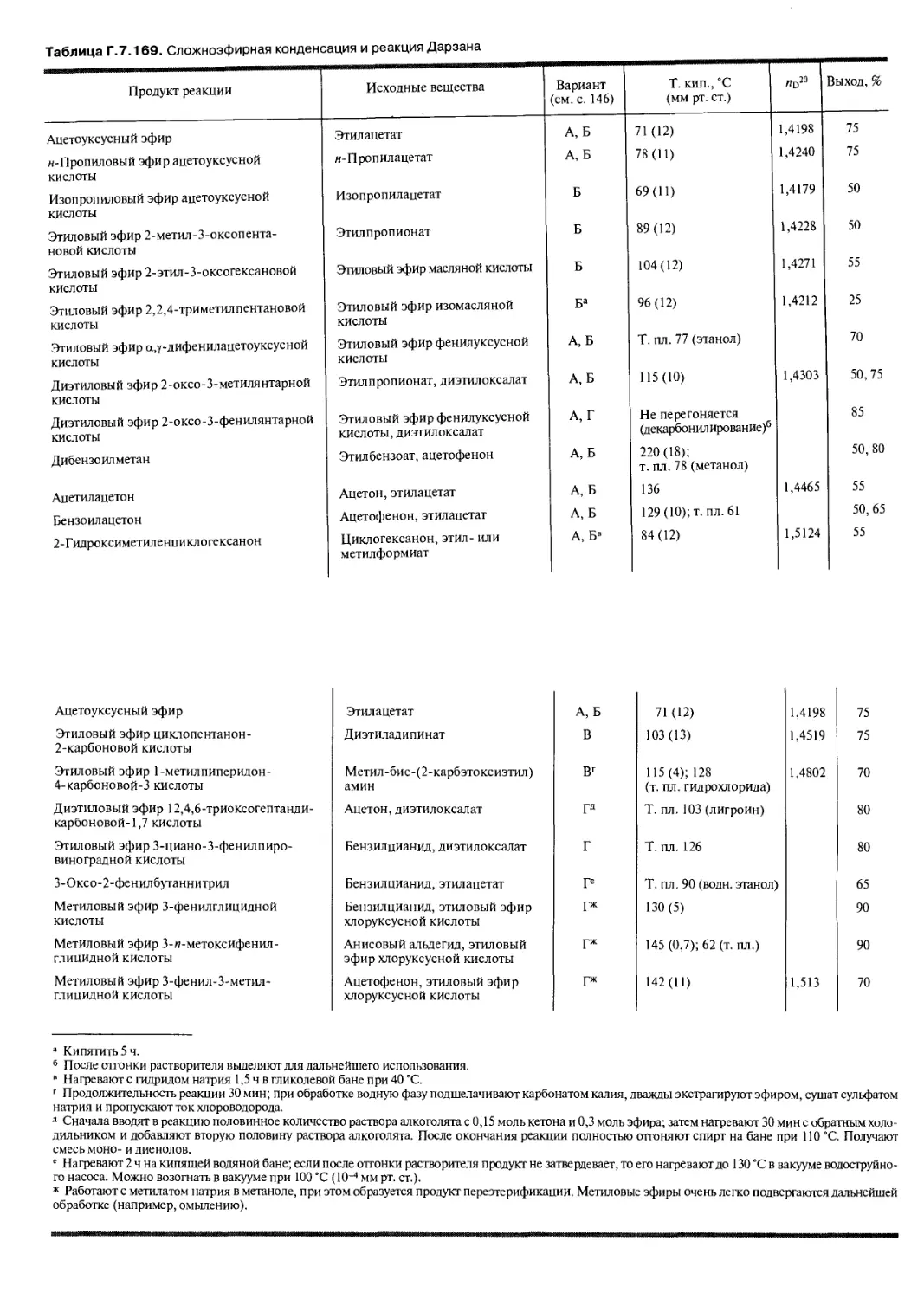
Реакция Вильгеродта—Киндлера входит в ряд синтезов, в ходе которых сера
вводится в структуру органического соединения. Так, в тех же условиях можно
перевести ароматические альдегиды, а также аллиловые спирты в морфолиды
тиокарбоновых кислот (см. табл. Г.6.25).
Многие метил- и хлорметиларены также можно более или менее гладко
окислить серой до производных тиокарбоновых кислот.
Кетоны реагируют с серой и аммиаком с образованием Д3-тиазолинов (реак-
(реакция Азингера):
R'
О
[Г.6.27]
Получение Ai-mua3OAUHoe:AsingerF., Offermanns H. Angew. Chem., 1967,79,953.
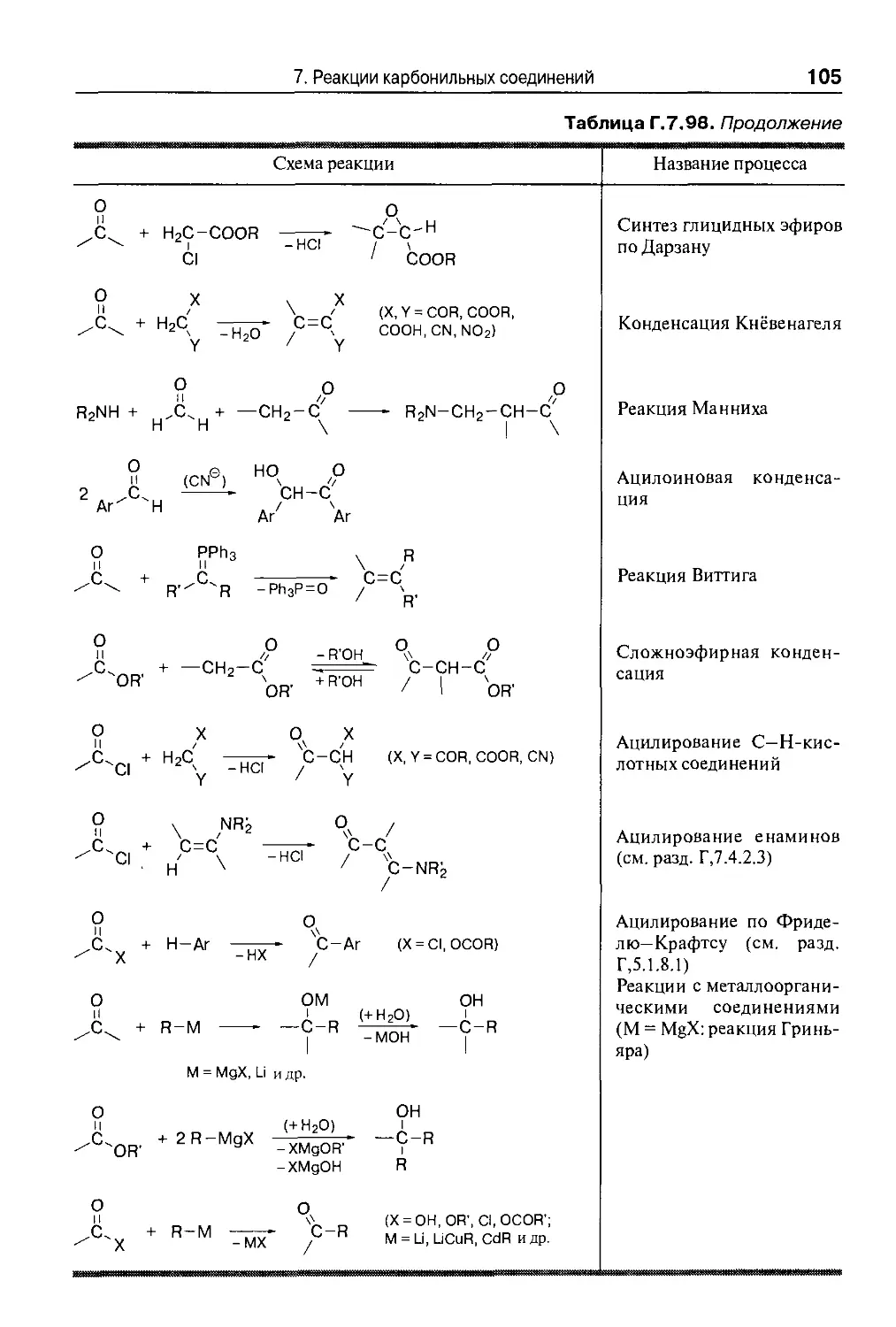
Карбонильные соединения реагируют с С—Н-кислотными нитрилами и се-
серой с образованием 2-аминотиофенов {реакция Гевальда) причем сначала путем
конденсации Кнёвенагеля (разд. Г,7.2.1.4) образуются а-алкилиденнитрилы,
которые затем присоединяют серу, например:
R
R
H COOEt
H CN
COOEt
(амин)
-н2о"
R
R'~hb
H
COOEt
CN
+ s
F
R
1 ?
H
COOEt
CN
[Г.6.28]
R'-'S-^
NH2
Таблица Г.6.29. Получение производных 2-аминотиофенкарбоновой-З кислоты
Конечный продукт
Этиловый эфир 2-амино-4,5-
тетраметилентиофенкарбоно-
вой-3 кислоты
2-Амино-4,5-тетраметилен-
тиофенкарбонитрил-3
Метиловый эфир 2-амино-
4,5-диметилтиофенкарбоно-
вой-3 кислоты
Диэтиловый эфир 2-амино-
4-метилтиофендикарбоно-
вой-3,5 кислоты
Этиловый эфир 2-амино-
4-фенилтиофенкарбоно-
вой-3 кислоты
Этиловый эфир 2-амино-
5-ацетил-4-метилтиофенкарбо-
новой-3 кислоты
Этиловый эфир 2-амино-4-ме-
тил-3-циантиофенкарбоно-
юй-5 кислоты
Исходное вещество
Циклогексанон, этиловый эфир
циануксусной кислоты
Циклогексанон, нитрил малоновой
кислоты
Метилэтилкетон, метиловый эфир
циануксусной кислоты
Этиловый эфир ацетоуксусной
кислоты, этиловый эфир
циануксусной кислоты
Этиловый эфир 2-цианокоричной
кислоты
Ацетилацетон, этиловый эфир
циануксусной кислоты
Этиловый эфир ацетоуксусной
кислоты, нитрил малоновой
кислоты
Т. пл., °С
115
147-148
120-122
108-109
95-96
156-158
210-212
Выход, %
80
78
50
55
60
55
45
6. Окисление и дегидрирование 21
Общая методика получения производных 2-аминотиофенкарбоновых-З кислот (табл. Г.6.29).
К нагретой до 60 "С (температура бани) смеси 30 мл этанола (в случае метиловых эфиров приме-
применяют метанол), 0,1 моль порошкообразной серы, 0,1 моль карбонильного соединения и 0,1 моль
нитрила (или вместо двух последних 0,1 моль илиденнитрила) добавляют по каплям при переме-
перемешивании в течение 10-15 мин 8 мл морфолина (или диэтиламина) и перемешивают еще 1,5 ч
при 60 °С. С циклогексаноном можно работать при комнатной температуре без дополнительно-
дополнительного нагрева. В случае необходимости отфильтровывают нерастворившуюся серу при повышенной
температуре. После этого реакционную смесь оставляют на 1—2 ч до окончания кристаллизации,
затем ставят в воду со льдом. Осадок отфильтровывают, промывают холодным этанолом и пе-
рекристаллизовывают из небольшого количества этанола или нитрометана. Если выход слиш-
слишком низок, маточный раствор перемешивают с водой и перерабатывают.
6.3. ОКИСЛЕНИЕ ПЕРВИЧНЫХ И ВТОРИЧНЫХ СПИРТОВ
И АЛЬДЕГИДОВ
Первичные и вторичные спирты в существенно более мягких условиях реагиру-
реагируют с перечисленными выше (см. окисление метильных и метиленовых групп)
окислителями.
—сн2он —- —сно —- —соон
\ ч [Г.6.30]
снон —- со
Третичные спирты окисляются с трудом, причем процесс сопровождается
разрывом углерод-углеродных связей (см. разд. Г,6.1).
6.3.1. ОКИСЛЕНИЕ ПЕРВИЧНЫХ И ВТОРИЧНЫХ СПИРТОВ
ДО АЛЬДЕГИДОВ И КЕТОНОВ
В качестве окислителей для этих реакций пригодно большое число реагентов.
Используются:
• Соединения xpoMa(VI)
Дихромат калия/серная кислота
Триоксид хрома/серная кислота/ацетон
Комплекс триоксида хрома с пиридином СгО3 • C5H5N
Хлорхромат пиридиния (РСС) C5H5NH+ С1СгО3"
Дихромат пиридиния (PDC) (CsH^NH^C^O?2"
Активированный диметилсульфоксид (DMSO)
DMSO/оксалилхлорид (реакция Сверна) Me2SO/(COClJ
• Галогенсодержащие соединения
Хлорноватистая кислота НСЮ
Периодинан Десса — Мартина (DMP) (см. [Г.6.38])
• Другие соединения
Оксид марганцаA\0 МпО2
Перрутенаттетрапропиламмония (ТРАР) Pr4N+Ru04~
N-оксид N-метилморфолина (NMO)
В лаборатории часто используют хромовую кислоту и ее соединения.
При окислении хромовой кислотой спирт нуклеофильно присоединяется к хромовой кис-
кислоте, при этом отщепляется вода и образуется эфир хромовой кислоты (это первая стадия
22 Г Препаративная часть (продолжение)
реакции, она аналогична образованию сложных эфиров карбоновых кислот; см. разд.
Г,7.1.4.1). Во второй стадии, идущей, вероятно, через циклическое переходное состояние,
а-водород спирта мигрирует к остатку хромата, причем металл переходит из шестивалентного
состояния в четырехвалентное:
R — О R НО
R'-C^Cr^OH - С=О + Сг=О [Г.6.31]
н о R' но
Четырехвалентный хром далее восстанавливается спиртом до трехвалентного, так что суммар-
суммарную реакцию можно записать следующим образом:
3R2CHOH + Na2Cr2O7 + 4H2SO4 3 R2CO + Cr2(SO4K + Na2SO4 + 7 H2O [Г.6.32]
При окислении первичных спиртов смесью дихромата в серной кислоте вы-
выходы альдегидов очень редко превышают 60%. Однако образующиеся альдегиды
можно определенным способом «поймать» (см. разд. Г.6.2.1, реакция [Г.6.16]) и
отогнать из реакционной смеси благодаря их большей летучести, или же прово-
проводить синтез в двухфазной системе и таким образом непрерывно экстрагировать
альдегид (см. последующее изложение).
Получение альдегидов окислением дихроматами в ледяной уксусной кислоте
(общая методика): Bosche H. G. In: Houben-Weyl, Bd. IV/lb, 1975, S. 460.
В качестве селективных окислителей используют комплекс триоксида хрома с
пиридином (СгО3 • 2C4H5N), хлорхромат пиридиния (РСС, C5H5NH+ClCr03~) и
дихромат пиридиния (PDC, [CsHsNH+hC^Oy2"). Эти соединения используют
прежде всего для окисления спиртов, содержащих другие способные окислять-
окисляться группы. Так, например, можно окислить непредельные первичные спирты до
альдегидов, не затрагивая кратную связь С=С.
Окисление вторичных спиртов до кетонов осуществляется еще легче, чем
окисление первичных спиртов. Выходы здесь выше, так как, во-первых, реакци-
реакционная способность вторичных спиртов выше, чем первичных, а во-вторых, обра-
образующиеся кетоны гораздо устойчивее к окислению по сравнению с альдегидами.
Согласно приводимой ниже методике, реакция ведется в двухфазной систе-
системе. Образовавшиеся кетоны извлекаются органическим растворителем и, таким
образом, предохраняются от дальнейшего окисления.
Ф Окисление вторичных спиртов до кетонов смесью дихромата с серной кислотой (общая
методика) (см. табл. Г.6.33)
Внимание/Хромат обладает канцерогенными свойствами. Остерегаться прямо-
прямого контакта. Использовать резиновые перчатки. Хромсодержащие остатки
тщательно собрать для их дальнейшей дезактивации.
В трехгорлую колбу на 500 мл, снабженную мешалкой, капельной воронкой, термометром
и обратным холодильником, наливают раствор 0,2 моль соответствующего спирта в 100 мл эфи-
эфира. При перемешивании добавляют по каплям в течение 15 мин раствор 0,067 моль дихромата
натрия (Na2Cr207 ¦ 2Н2О) и 15 мл серной кислоты в 100 мл воды. Температура реакционной сме-
смеси 25 "С. Перемешивают при этой температуре еще 2 ч, эфирный слой отделяют, водный еще
2 раза встряхивают с 50 мл эфира. Объединенные эфирные вытяжки промывают насыщенным
раствором гидрокарбоната натрия, водой, сушат сульфатом магния или натрия. После отгонки
эфира остаток фракционируют на короткой колонке Вигре. [Brown Н. С, Garg С. P. J. Am. Chetn.
Soc, 1961,83,2952.]
6. Окисление и дегидрирование
23
Таблица Г.6.33. Получение кетонов из вторичных спиртов
Конечный продукт
Циклогексанон
2-Метилциклогексанон
(-)-Ментон
г<ис-Декалинон-2
Изопропилэтилкетон
Пропиофенон
Исходное соединение
Циклогексанол
2-Метилциклогексанол-1
(—)-Ментол
г<ис-Декалинол-2а
2-Метилпентанол-З
1 -Фенилпропанол-1
Т. кип.
(мм рт. ст.)
или т. пл., °С
155
65 B3)
67D)
110A0)
112
93 A1); т. пл. 21
/id20
1,4503
1,4490
1,4536
1,4927
1,3975
1,5270
Выход, %
65
62
70
60
60
65
Можно использовать смесь изомеров, образующуюся при гидрировании; см. табл. Г.4.124.
Методика пригодна для полумикросинтеза: в этом случае удобно применять магнитную
мешалку. Окисление можно использовать и для аналитической характеристики вторичных
спиртов, переводя неочищенный кетон в подходящие производные.
Получение нортрицикланона из нортрицикланола путем окисления хромо-
хромовым ангидридом в ацетоне; MeinwaldJ., Crandall J., Hymnes W. E. Org. Synthesis,
1965, 45, 77.
Особенностью хромсодержащих соединений является их токсичность. Поэ-
Поэтому для селективного окисления первичных и вторичных спиртов соответ-
соответственно до альдегидов и кетонов лучше использовать хлорноватистую кислоту
НОС1, источником которой является щелочной отбеливающий раствор гипо-
хлорита натрия. Этот окислитель менее ядовит и процесс окисления им легко
контролировать. Окисление проводят в двухфазной системе (вода/дихлорметан)
в присутствии стабильного радикала 2,2,6,6-тетраметилпиперидиноксила
(ТЕМПО), КВг и NaHCO3 в качестве буфера.
Этот радикал в процессе реакции образует собственно катализатор — ион 2,2,6,6-тетраме-
тилпиперидин-1-оксила, который дегидрирует спирт с образованием пиперидинола. Послед-
Последний при взаимодействии с НОС1 (в присутствии КВг частично образуется также и НОВг) снова
регенерируется до иона оксопиперидиния.
R ,ОН
¦А
Me
Me
II
О
Me
Me
Me
Me
Me
Me
Me
Me"
N'
i
OH
Me
Me
[Г.6.34]
R1
HOCI
Окисляемый спирт не должен обладать хорошей растворимостью в воде, а
образующееся карбонильное соединение должно хорошо растворяться в дих-
лорметане. По окончании реакции в водной среде должен оставаться небольшой
избыток NaOCl, что можно проверить с помощью крахмальной бумажки, про-
пропитанной КВг (голубое окрашивание). Поскольку реакция сильно экзотермич-
на, то при ее проведении реакционную смесь следует очень хорошо перемеши-
перемешивать и охлаждать, особенно при работе с большими количествами реагентов.
24 Г Препаративная часть (продолжение)
Наличие в структуре субстрата двойной С=С-связи осложняет процесс из-за
присоединения к ней НОС1. Если к двухфазной реакционной смеси добавить
катализатор фазового переноса, например, хлорид метилтриоктиламмония
(аликват 336), то из первичных спиртов (или альдегидов) образуются карбоно-
вые кислоты.
Ф Окисление спиртов и альдегидов гипохлоритом натрия (общая методика) (табл. Г.6.35)
[АпеШР. L, MontanariF., QuiciS., Org. Synth., 1990, 69, 212]
А. Получение альдегидов и кетонов
Процесс проводят ктрехгорлой колбе емкостью 250 мл, снабженной механической мешалкой,
капельной воронкой с насадкой для выравнивания давления и внутренним термометром.
В колбу помещают смесь 0,1 моль соответствующего спирта, 0,16 г A ммоль) 2,2,6,6-тетраме-
тилпиперидиноксила в 40 мл дихлорметана и 1,2 г @,01 моль) КВг в 5 мл воды. Смесь охлажда-
охлаждают на водяной бане смесью соли и льда до —10 °С и при сильном перемешивании в течение
10—15 мин прикапывают ПО мл @,11 моль) 1 М раствора NaOCl с рН 9,5' так, чтобы темпера-
температура смеси не превышала 15 °С. Перемешивают еще 5 мин, при этом в среде должен еще оста-
оставаться избыток NaOCl (проверка иодкрахмальной бумажкой). Органическую фазу отделяют, а
водную — экстрагируют 10 мл дихлорметана. Для удаления катализатора объединенный орга-
органический экстракт промывают 20 мл 10% раствора поваренной соли, содержащей 0,32 г
B ммоль) иодида калия, потом 10%-ным водным раствором тиосульфата натрия и в заключе-
заключение 10 мл 10% раствора бикарбоната натрия и таким же количеством воды. Полученный орга-
органический раствор сушат безводным сульфатом натрия, отгоняют растворитель и остаток пере-
перегоняют (желательно в вакууме) на колонке Вигре. Чистоту продукта проверяют методом ГХ.
Твердые соединения после испарения растворителя перекристаллизовывают.
Б. Получение карбоновых кислот
Для окисления первичных спиртов и альдегидов используют методику А, однако в исходную
реакционную смесь добавляют 1,4 г E ммоль) аликвата 336 в 10 мл воды и в случае спиртов
объем раствора гипохлорита удваивают. После добавления окислителя перемешивают 45 мин.
Полученную двухфазную реакционную смесь встряхивают с 2 н. раствором едкого натра до тех
пор, пока водная фаза не будет иметь рН 12. Водный раствор отделяют и подкисляют 6 н. со-
соляной кислотой. Выпавшую твердую органическую кислоту отделяют и перекристаллизовыва-
перекристаллизовывают. Жидкие кислоты экстрагируют трижды порциями по 20 мл дихлорметана. Экстракт сушат
безводным сульфатом магния, упаривают растворитель и остаток перегоняют на колонке Вигре.
Окисление гипохлоритом натрия можно осуществлять в полумикромасштабе, при этом
количество дихлорметана уменьшают втрое.
Определение содержания гипохлорита в щелочном отбеливающем растворе
В мерной колбе A0 мл) 1 мл отбеливающего раствора разбавляют дистиллированной водой до
метки. Отбирают пипеткой 2 мл полученного раствора в колбу для титрования, добавляют ту-
туда 40 мл дистиллированной воды и титруют 0,1 н. раствором нитрита натрия до появления го-
голубой окраски (по иодкрахмальной бумажке).
[NaOCl] = NaN°2 0,05 моль/л
I^NaOCl
Для приготовления раствора нитрита 100-мл мерной колбе в дистиллированной воде раство-
растворяют 0,345 г нитрита натрия (ч.д.а.) и 2 г бикарбоната натрия.
1 Обычно используемый свежеприготовленный щелочной отбеливатель гипохлорит нат-
натрия имеет молярность 1,8-2,0 и рН около 12,5. При хранении содержание NaOCl падает.
Поэтому раствор следует перед употреблением оттитровать и, добавляя бикарбонат натрия,
довести рН до 9,5 (по индикаторной бумаге).
6. Окисление и дегидрирование
25
Таблица Г.6.35. Окисление спиртов и альдегидов гипохлоритом натрия
Продукты
Гептаналь (энантовый
альдегид)
Октаналь (каприло-
вый альдегид)
Нонаналь (пеларгоно-
вый альдегид)
Деканаль (каприно-
вый альдегид)
Ундеканаль
Бензальдегид
4-Нитробензальдегид
Циклогексанон
Гидрокси-2-этилгек-
санон-3
Гептановая (энанто-
вая) кислота
Ундекановая кислота
Исходные
соединения
Гептанол
Октанол
Нонанол
Деканол
Ундеканол
Бензиловый спирт
4-Нитробензиловый
спирт
Циклогексанол
2-Этилгександиол-1,3
Гептанол
Ундеканол
Т. кип., °С
(мм рт. ст.)
152
72 B0)
81A3)
91A3)
116A8)
64A3)
Т. пл. 106 (эта-
нол/петр. эфир)
155
118A2)
114A3)
168A1)
«о20
1,4279
1,4217
1,4242
1,4580
1,4520
1,5446
1,4503
1,4216
Выход, %
65
72
71
68
65
61
65
73
64
95
55
Дегидрирование первичных и вторичных спиртов можно осуществить с хоро-
хорошими выходами в мягких условиях диметилсульфоксидом (ДМСО) в присутствии
электрофилов («активированный диметилсульфоксид»). В качестве последних
используют дициклогексилкарбодиимид, уксусный ангидрид или оксалилдихло-
рид (в последнем случае это окисление по Сверну).
На промежуточной стадии этой реакции ДМСО взаимодействует с оксалил-
дихлоридом с образованием соли хлорсульфония, сильноэлектрофильный
атом серы которого атакует спиртовый атом кислорода (см. [Г.6.36]), замещая
атом водорода и образуя новую соль. Последняя при взаимодействии с основа-
основанием превращается в илид, расщепляющийся на кетон и молекулу диметил-
сульфида.
©
(H3CJS =
R OH
R H
-o + cicococi
+ ci-i(CH3J -
R
V <-
1 ^
R'
- (CH3JS-
Cl
H
) + S(CH3J
-o-co-
e
X|.ch;
ch3
-co-ci ^o
3 Основание
u®
2 -CO
R
H
- (CH3JS-CI Cl
.0^©^CH3 [Г.
^~^ ©
26
Г Препаративная часть (продолжение)
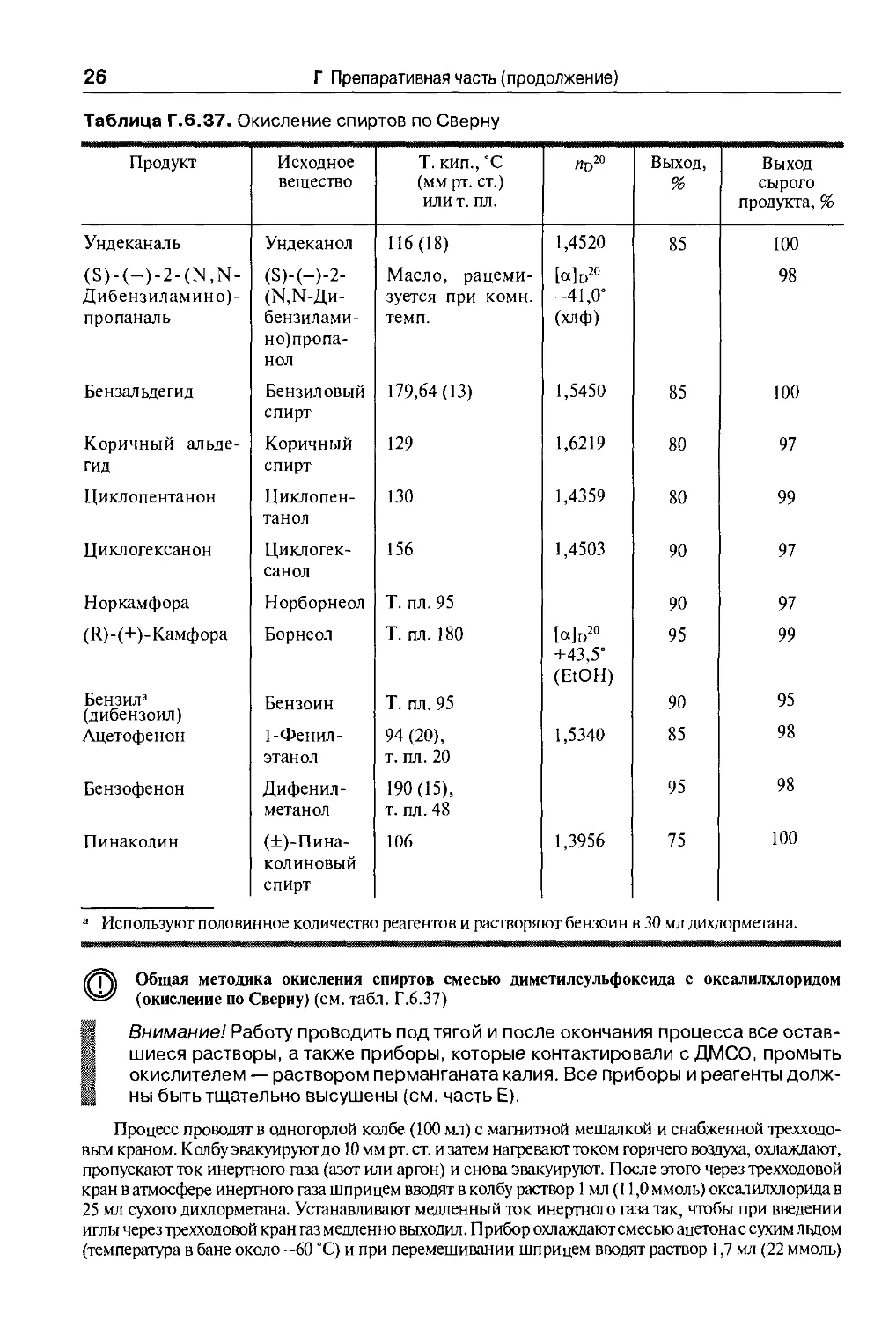
Таблица Г.6.37. Окисление спиртов по Сверну
Продукт
Ундеканаль
(S)-(-)-2-(N,N-
Дибензиламино)-
пропаналь
Бензальдегид
Коричный альде-
альдегид
Циклопентанон
Циклогексанон
Норкамфора
(Я)-(+)-Камфора
Бензила
(дибензоил)
Адетофенон
Бензофенон
Пинаколин
Исходное
вещество
Ундеканол
(S)-(-)-2-
(N.N-Ди-
бензилами-
но)пропа-
нол
Бензиловый
спирт
Коричный
спирт
Циклопен-
танол
Циклогек-
санол
Норборнеол
Борнеол
Бензоин
1-Фенил-
этанол
Дифенил-
метанол
(±)-Пина-
колиновый
спирт
Т. кип., °С
(мм рт. ст.)
или т. пл.
116A8)
Масло, рацеми-
зуется при комн.
темп.
179,64A3)
129
130
156
Т. пл.95
Т. пл.180
Т. пл. 95
94 B0),
т. пл. 20
190A5),
т. пл. 48
106
«D20
1,4520
[а]„м
-41,0°
(хлф)
1,5450
1,6219
1,4359
1,4503
[аЬ20
+43,5°
(ЕЮН)
1,5340
1,3956
Выход,
%
85
85
80
80
90
90
95
90
85
95
75
Выход
сырого
продукта, %
100
98
100
97
99
97
97
99
95
98
98
100
Используют половинное количество реагентов и растворяют бензоин в 30 мл дихлорметана.
\ Общая методика окисления спиртов смесью диметилсульфоксида с оксалилхлоридом
(окисление по Сверну) (см. табл. Г.6.37)
Внимание! Работу проводить под тягой и после окончания процесса все остав-
оставшиеся растворы, а также приборы, которые контактировали с ДМСО, промыть
окислителем — раствором перманганата калия. Все приборы и реагенты долж-
должны быть тщательно высушены (см. часть Е).
Процесс проводят в одногорлой колбе A00 мл) с магнитной мешалкой и снабженной трехходо-
трехходовым краном. Колбу эвакуируют до 10 мм рт. ст. и затем нагревают током горячего воздуха, охлаждают,
пропускают ток инертного газа (азот или аргон) и снова эвакуируют. После этого через трехходовой
кран в атмосфере инертного газа шприцем вводят в колбу раствор 1 мл A1,0ммоль)оксалилхлоридав
25 мл сухого дихлорметана. Устанавливают медленный ток инертного газа так, чтобы при введении
иглы через трехходовой кран газ медленно выходил. Прибор охлаждают смесью ацетона с сухим льдом
(температура в бане около -60 °С) и при перемешивании шприцем вводят раствор 1,7 мл B2 ммоль)
6. Окисление и дегидрирование 27
диметилсульфоксида в 5 мл дихлорметана. Через короткое время в колбу в течение примерно 5 мин
порциями вводят раствор Юммоль окисляемого спиртав 10мл дихлорметана. Через 15 мин добавля-
добавляют порциями 7 мл E0 ммоль) триэтиламина, перемешивают еще 5 мин и, наконец, оставляют смесь
медленно нагреваться до комнатной температуры. Удаляют трехходовой кран, прибавляют
50 мл воды, отделяют органический слой, а водный — экстрагируют 50 мл дихлорметана. Объединен-
Объединенный органический экстракт промывают последовательно 100 мл насыщенного растюра поваренной
соли, 50 мл 1%-ного раствора серной кислоты, 50 мл воды и наконец 50 мл 5%-ного раствора
бикарбоната натрия. Сушат безводным сульфатом магния, удаляют растворитель и остаток перегоня-
перегоняют в вакууме. Получают достаточно чистые соединения (проверить с помощью ИК- и ЯМР-спектрос-
копии). С целью дальнейшей очистки вещества перегоняют повторно или перекристаллизовывают.
Из других специфических реагентов, которые могут быть использованы для селективного
окисления спиртов до альдегидов или кетонов, можно упомянуть следующие:
Так называемый периодинан Десса—Мартина (DMP) — соединение иода(У), получаемое из
о-иодбензойной кислоты. Процесс проходит очень мягко при добавлении пиридина в почти
нейтральной среде. Иод(У) при этом восстанавливается до иода(Ш):
А + О С О + О +2АсОН [Г.6.38]
АсО
ОАс
При этом содержащиеся в структуре субстрата кратные С=С-связи, а также тио-, втор-
амино- и другие функциональные группы не затрагиваются. Реагент может быть использован
для проведения реакций в полумикромасштабе. Однако это соединение при повышенных тем-
температурах взрывается. Наиболее безопасно проводить реакцию в растворе дихлорметана.
Селективным окислителем является также тетрапропиламмонийперрутенат PqN+RuOr
(ТРАР) — соединение рутения(У11) в присутствии морфолин-И-оксида (NMO). Перрутенат
берется лишь в каталитических количествах, поскольку в процессе реакции он все время
реокисляется N-оксидом морфолина, который берется в стехиометрических количествах.
Несомненно, очень важным методом получения альдегидов и кетонов явля-
является каталитическое дегидрирование первичных и вторичных спиртов:
(Си)
- R-CH=O + Н2
[Г.6.39]
В качестве катализаторов при этом используются металлические серебро и
медь, а также хромомедный оксид и оксид цинка. Равновесие реакции дегидри-
дегидрирования (см. [Г.6.39]) устанавливается в интервале температур 300—400 "С.
Процесс дегидрирования является сильно эндотермическим (Ar/T = 70...86 кДж/моль).
Однако, поскольку выделяющийся водород сгорает в токе воздуха, суммарная реакция
R-CH2-OH + 1/2 О2 -» R-CHO + Н2О оказывается сильноэкзотермичной (АцН° =
— 160...—180 кДж/моль). Поэтомутакой процесс также называют окислением или аутотерми-
ческим дегидрированием.
Каталитическое дегидрирование спиртов как лабораторный метод утратил свое значение.
В предыдущих изданиях этой книги была описана общая методика такого процесса. Тем не
менее, дегидрирование спиртов до сих пор является важнейшим методом промышленного по-
получения альдегидов и кетонов. Так получают в огромных количествах формальдегид, ацеталь-
дегид, ацетон, метилэтилкетон и циклогексанон. Промышленная дегидроциклизация бутан-
28
Г Препаративная часть (продолжение)
диола-1,4 над медью при 250 °С позволяет получать у-бутиролактон с высокими выходами.
Аналогичным образом оксидегидрирование этиленгликоля при 300 °С в присутствии ингиби-
ингибиторов — галогенидных соединений — позволяет получать глиоксаль.
Каталитическое дегидрирование спиртов имеет место также в процессе
окисления по Оппенауэру (см. разд. Г,7.3.1.2).
6.3.2. ОКИСЛЕНИЕ ПЕРВИЧНЫХ СПИРТОВ И АЛЬДЕГИДОВ
В КАРБОНОВЫЕ КИСЛОТЫ
Все окислители, способные превращать первичные спирты в альдегиды, могут
быть использованы и для получения карбоновых кислот из спиртов (через
альдегиды) и альдегидов.
При окислении альдегида хромовой кислотой, по-видимому, в качестве промежуточного
вещества образуется эфир хромовой кислоты, как это происходит при окислении спиртов (см.
схемы [Г.6.31]—[Г.6.32]); в данном случае эфир возникает из гидратной формы альдегида и
реагирует далее.
Скорость окисления замещенных бензальдегидов возрастает в следующем
ряду заместителей:
jh-C1
/j-NO2
л-СНзО < и-СНз < Н < и-С1 < льСНз 2 2
При окислении первичных спиртов в кислой среде промежуточно образую-
образующийся альдегид может легко превратиться в ацеталь (см. разд. Г,7.1.2), а образу-
образующаяся кислота — в эфир; это приводит к тому, что часть спирта остается
неокисленной. Поэтому первичные спирты лучше окислять до карбоновых кис-
кислот пиридинийдихроматом (PDC) в диметилформамиде или обрабатывать пер-
манганатом в щелочной среде:
3R-CH2-OH + 4КМпО4
3R-COOH +4МпО2 + 4 КОН + Н2О [Г.6.40]
При этом всегда выход снижается из-за побочной реакции а-окисления про-
промежуточно образовавшегося альдегида, которая приводит к распаду молекулы
путем разрыва связи С—С (см. разд. Г,6.5.3).
Ф Общая методика получения карбоновых кислот из первичных спиртов и олефинов в усло-
условиях межфазного катализа (табл. Г.6.41). В трехгорлой колбе емкостью 1 л с мешалкой
и термометром к смеси 0,1 моль спирта или олефина, 150 мл СН2С12, 250 мл воды и
4 мл аликвата 336 при интенсивном перемешивании порциями добавляют 0,2 моль
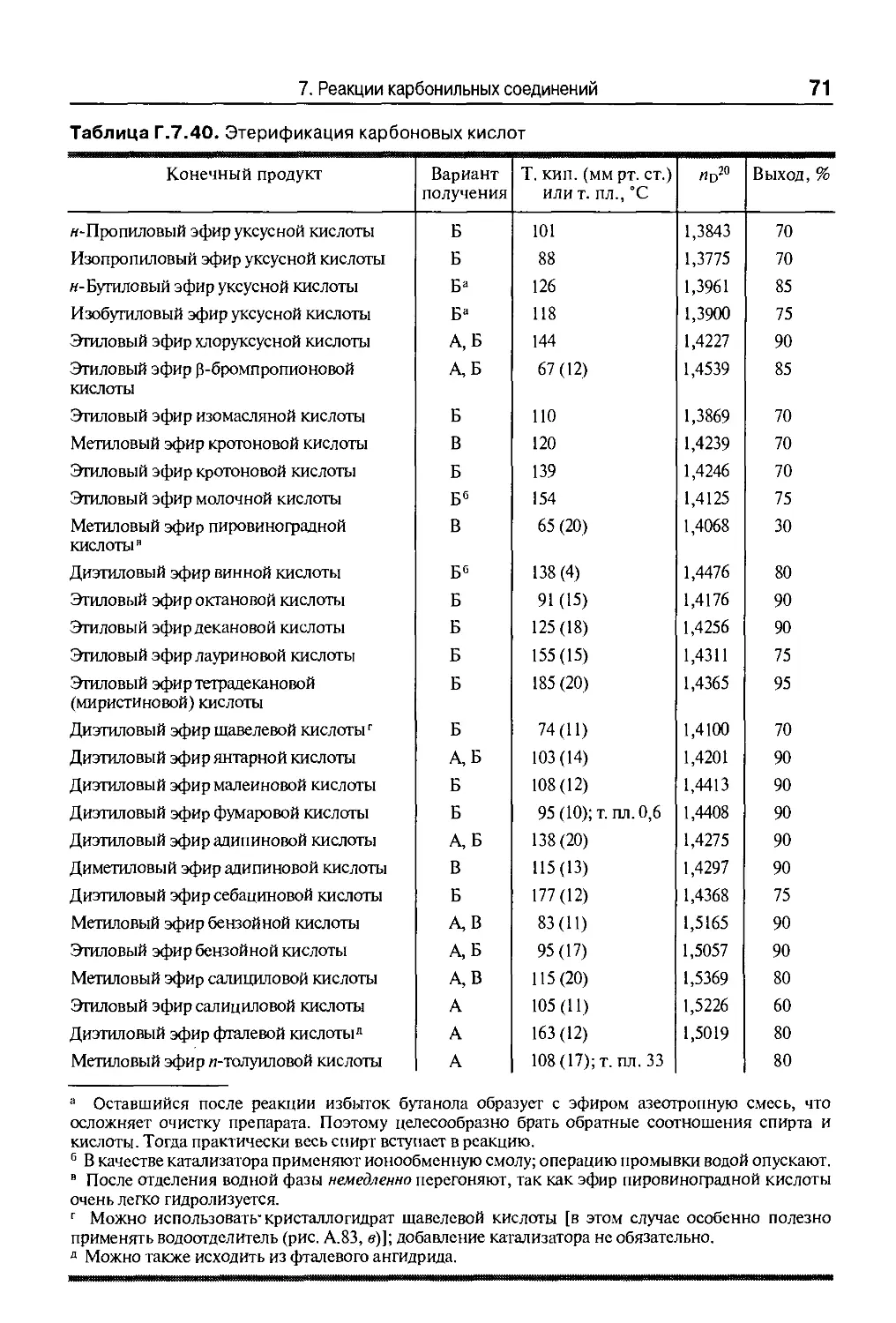
Таблица Г.6.41. Получение карбоновых кислот
Конечный продукт
Гексановая кислота
Октановая кислота
Декановая кислота
Гептановая кислота
Нонановая кислота
Исходное вещество
Гексанол
Октанол
Деканол
Октен-1
Децен-1
Т. кип., °С (мм рт. ст.)
206-208
239-240
129-130A6)
148-150(9)
220-222
115-116A1)
142-143A6)
Выход, %
60
75
55
45
45
6. Окисление и дегидрирование 29
перманганата калия (в случае олефинов 0,25 моль). Охлаждением ледяной водой поддержи-
поддерживают температуру смеси ниже 15 °С при окислении спиртов и ниже 10 °С при окислении оле-
олефинов. Перемешивают реакционную смесь до обесцвечивания перманганата (максимальное
время реакции — 3 ч) и затем растворяют диоксид марганца водным раствором гидросульфи-
гидросульфита. Смесь подкисляют разбавленной серной кислотой и отделяют органический слой. Пос-
Последний сушат небольшим количеством Na2SO4 и перегоняют, лучше всего после отгонки
СН2С12 в ротационном испарителе.
Так как альдегидная группа окисляется легче, чем гидроксильная, то в альдо-
зах, например, можно селективно окислять в мягких условиях только альдегид-
альдегидную группировку. Так, при действии иода в щелочном растворе из D-глюкозы
получают глюконовую кислоту:
СНО СООН
н-с-он н-с-он
H°-fH +,2 + 2OHQ _ H°-?-H + 2,Q+H2O [Г 6 42]
Н-С-ОН Н-С-ОН li.o.t/j
н-с-он н-с-он
СН2ОН СН2ОН
Эту реакцию можно использовать для иодометрического определения Сахаров.
Ионами серебра (в аммиачном растворе; реактив Толленса) или двухввалентной меди (в ви-
виде тартратного комплекса; фелингова жидкость) также можно в щелочной среде селективно
окислять альдегиды в кислоты, причем в ходе реакции указанные ионы восстанавливаются до
металлического серебра и оксида медиA).
Аммиачный раствор нитрата серебра и фелингову жидкость используют для обнаружения
альдегидов; спирты и кетоны их не восстанавливают. Необходимо, однако, иметь в виду, что ке-
тозы восстанавливают фелингову жидкость аналогично альдозам, поскольку в щелочной среде
они легко изомеризуются в альдозы, а частично подвергаются деструкции в низшие альдозы.
Азотной кислотой можно окислить в альдозах альдегидную и первичную
спиртовую группу с образованием гидроксидикарбоновых кислот, например, из
D-галактозы при этом получается слизевая кислота.
СНО СООН
н-с-он н-с-он
но-с-н hno3> но-с-н [Г 6 431
но-с-н но-с-н
н-с-он н-с-он
СН2ОН СООН
(ГП) Получение слизевой кислоты из молочного сахара (окисление азотной кислотой)
jj Осторожно! Выделяются газообразные оксиды азота! Работать под тягой!
0,03 моль молочного сахара растворяют в 120 мл 25%-ной азотной кислоты (d= 1,15), упа-
упаривают на водяной бане до объема 20 мл, добавляют 30 мл воды (образующаяся в качестве по-
побочного продукта сахарная кислота растворима в воде). Оставляют на несколько дней, затем
отсасывают кристаллы кислоты, промывают холодной водой. Выход 30—40%. Для очистки
растворяют в эквивалентном количестве щелочи и осаждают рассчитанным количеством кис-
кислоты. Т. пл. 213 °С (с разложением).
30
Г Препаративная часть (продолжение)
Получение трихлоруксусной кислоты из хлораля (окисление азотной кислотой)
Осторожно! Трихлоруксусная кислота раздражает кожу; работать в резиновых
перчатках!
0,24 моль хлоральгидрата расплавляют в колбе емкостью 250 мл и осторожно добавляют
по каплям 17 мл дымящей азотной кислоты (d = 1,5) (под тягой!). Когда выделение оксидов
азота ослабеет, смесь нагревают до полного прекращения их выделения, затем перегоняют в
вакууме. Т. кип. 102 "С B0 мм рт. ст.); т. пл. 57 °С; выход 55%.
Полигидроксисоединения можно селективно окислять в растворе кислородом на платиновом
катализаторе. Легче всего окисляется первичная гидроксильная группа, образуя в зависимости от
условий альдегидную или карбоксильную группу. Эта реакция важна прежде всего для селективно-
селективного окисления углеводов и их производных, например для получения уроновых кислот.
В промышленности уксусную кислоту в больших объемах получают аутоокислением аце-
тальдегида без катализатора при температуре 50-70 °С. В качестве промежуточного продукта
образуется перуксусная кислота (см. разд. Г, 1.5), которую можно получить в качестве основ-
основного продукта, если процесс проводить при температуре 0 "С и давлении 3 МПа. При действии
60%-ной азотной кислоты из ацетальдегида при 40 °С образуются глиоксаль и некоторые кис-
кислоты. Изобутиральдегид окислением воздухом можно перевести в изомасляную кислоту.
При биохимическом окислении спиртосодержащих растворов (прежде всего в процессах бро-
брожения) культурами Acetobacter aceti и Gluconobacter oxidans получают особые сорта пищевого уксу-
уксуса. Кроме того, в промышленности с помощью штаммов Acetobacter suboxidans осуществляют окис-
окисление D-сорбита в L-сорбозу. Последнюю, предварительно защитив гидроксигруппы образовани-
образованием ацеталей с ацетоном, окисляют далее, например, гипохлоритом натрия до 2-оксогулоновой
кислоты. При подкислении она лактонируется в аскорбиновую кислоту (витамин С).
ОН ОН ОН ^cetobacter °Н
suboxidans)
HO,
ОН ОН ОН
D-сорбит
ОН ОН ОН
L-сорбоза
"ЮН
-OH
L-сорбопираноза
OH
ди-0-изопропилиден-
L-сорбофураноза
ОН ОН О
(H0)
HO OH
OH \=/
[Г.6.43а]
ди-0-изопропилиден-
2-оксоч.-гулоновая кислота
ОН
2-оксо-1_-гулоновая кислота i-аскорбиновая кислота
6.4. ПОЛУЧЕНИЕ ХИНОНОВ ОКИСЛЕНИЕМ
6.4.1. ХИНОНЫ ИЗ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
Синтез хинонов окислением ароматических углеводородов согласно уравнени-
уравнениям [Г.6.10] представляет собой процесс переноса электронов и протекает тем
легче, чем ниже окислительный потенциал E0X{D) арена. Поэтому для окисле-
окисления бензола (,ЕОХ = 2,54 В отн. СВЭ в ацетонитриле) до л-бензохинона необходи-
необходимо использовать сильные окислители — оксид серебра(П) (Ag(II): E° = 2,0 В;
о влиянии растворителей см. разд. Г,6.2.1). Напротив, окислительные потенци-
потенциалы нафталина, антрацена или фенантрена настолько низки (см. табл. Г.6.5), что
6. Окисление и дегидрирование
31
для их окисления достаточно уже хромовой кислоты, пероксида водорода или
кислорода воздуха в присутствии катализатора V2O5; значения ДяG < 0, т. е.
процесс окисления термодинамически возможен.
О
,0
О
л-бензохинон
О
о-бензохинон
[Г.6.44]
При окислении хромовой кислотой в одних и тех же условиях из нафталина,
антрацена и фенантрена получают следующие продукты с указанными выходами:
[Г.6.45]
О О
20% 90% 37%
о-Хиноны обладают большим запасом энергии, чем л-хиноны, поэтому,
например, фенантренхинон легко окисляется в дифеновую кислоту:
[Г.6.46]
НООС СООН
Из нафталина при окислении также получается не только 1,4-нафтохинон,
но и фталевый ангидрид (см. табл. Г.6.47 и разд. Г.6.5.1).
В приведенной ниже методике окисления углеводородов в хиноны использует-
используется большой избыток хромовой кислоты, так как в противном случае в реакционной
смеси остается непрореагировавшее исходное вещество, что затрудняет последую-
последующую очистку. Реакцию следует остановить, как только прореагирует все исходное
вещество, чтобы предотвратить дальнейшее окисление хинона.
(ГП) Общая методика получения хинонов из углеводородов действием триоксвда хрома (табл. Г.6.47)
Ц Осторожно! Оксид xpoMa(VI) — канцероген. Избегать прямых кожных
¦ контактов! Работать в перчатках! Хромсодержащие остатки, растворы и смывы
И посуды следует собирать, но не выливать в раковину!
В трехторлой колбе на 500 мл, снабженной термометром, мешалкой и капельной воронкой
(колба должна сообщаться с внешней атмосферой!), к смеси 0,05 моль исходного вещества (твер-
(твердое вещество следует растереть в порошок) и 90 мл 90%-ной уксусной кислоты при интенсивном
перемешивании в течение 1 ч по каплям добавляют раствор 0,25 моль триоксида хрома в 50 мл
60%-ной уксусной кислоты. Температуру поддерживают в пределах 5-20 °С. Затем перемешива-
перемешивают еще 40—60 мин при температуре 40 "С, чтобы завершить окисление.
Для более точного определения конца реакции перед истечением указанного времени каждые
5 мин берут пробу, разбавляют водой, отсасывают кристаллы, промывают водой. Продукт должен
быть светло-желтым (не зеленым!), запах углеводорода должен исчезнуть. Присутствие исходного ве-
вещества можно также быстро определить, измерив температуру плавления. По окончании реакции
смесь выливают в равное по объему количество воды, продукт отсасывают и перекристаллизовывают.
32
Г Препаративная часть (продолжение)
Таблица Г.6.47. Получение хинонов из ароматических углеводородов
Конечный продукт
Нафтохинон-1,4
2-Метилнафто-
хинон-1,4
Фенантренхинон
Антрахинон
Аценафтохинон
Исходное соединение
Нафталин
2-Метилнафталин
Фенантрен
Антрацен
Аценафтен
Т. пл., °С
124 (гексан)
106(метанол)
207 (этанол или
уксусная кислота)
285 (диоксан)
261 (тетралин)
Выход, %
35
45
60
80
50
Примечания
Защищать от
воздействия света,
легко полимеризуется
Сырой хинон
отмыть содовым
раствором
После добавления
СгО3 нагревать 4 ч
с обратным холо-
холодильником (конец
реакции не опреде-
определяют)
Неочищенный про-
продукт кипятить с тет-
ралином, фильтро-
фильтровать горячим
Антрахинон, важный полупродукт производства красителей (см. разд. Г,5.2.1), в промыш-
промышленности наряду с другими методами (разд. Г,5.1.8.1) получают окислением антрацена триок-
сидом хрома в жидкой фазе при 50—100 "С или воздухом при 370 °С на ванадате железа с высо-
высокой селективностью и хорошим выходом. Нафтохинон-1,4 образуется в качестве побочного
продукта при окислении нафталина до ангидрида фталевой кислоты (см. разд. Г,6.5.1).
6.4.2. ХИНОНЫ ИЗ ЗАМЕЩЕННЫХ АРЕНОВ
Наиболее общим методом получения хинонов является окисление о- или л-ди-
фенолов, аминофенолов и ароматических диаминов. Поскольку эти соединения
обладают очень низкими окислительными потенциалами (см. табл. Г.6.5), про-
процесс переноса электрона термодинамически очень выгоден.
Эти реакции протекают по радикальному механизму. Радикал, возникающий
при отщеплении электрона, заметно стабилизирован за счет мезомерного эф-
эффекта: образуются так называемые семихиноны. Наиболее известный случай —
окисление л-аминодиметиланилина бромом, при этом получают краситель крас-
красный Вюрстера:
NH2
-Вг
Н3С СН3
Н3С СН3
- К )\ ^ [Г.6.48]
Это соединение (катион-радикал) при дальнейшем окислении переходит в
соответствующую хинониммониевую соль, которая в водном растворе очень
быстро гидролизуется до я-бензохинона:
6. Окисление и дегидрирование
33
н,с
-Нв-ВгЭ
+ 2Н2О
-NH3,-H®, -(CH3JNH
[Г.6.49]
Аналогично гидрохинон переходит в я-бензохинон через промежуточный
семихинон, легко обнаруживаемый в щелочной среде1. Эта реакция особенно
легко протекает по механизму аутоокисления кислородом воздуха в присутствии
пентаоксида ванадия:
-Q-O-
юн
юн
+ н-о-о-
+ н2о2 [Г.6.50]
На подобной реакции основан современный промышленный метод получения пероксида
водорода с использованием 2-этил- или 2-т/>е/я-бутилантрагидрохинона2. Образующийся
антрахинон снова гидрируют до антрагидрохинона на никелевом катализаторе при 35 °С.
Принимая два электрона, хиноны вновь превращаются в ароматические со-
соединения: о он
+ 2 еЭ + 2 Н
©
[Г.6.51]
Это придает им свойства окислителей, в ходе реакции они восстанавливаются до
соответствующих гидрохинонов (в кислой среде, например, под действием
диоксида серы). Окислительный потенциал хинонов повышается, если в ядре
находятся электроноакцепторные заместители, так что, например, хлоранил
представляет собой довольно сильный окислитель (см. разд. Г.6.6).
В природе синтез лигнина происходит путем окисления кониферилового спирта, у которо-
которого дегидрогеназа отщепляет электрон и протон. Образовавшийся радикал в последующих реак-
реакциях полимеризуется в макромолекулу со множеством поперечных связей.
1 Поскольку я-бензохинон неустойчив к щелочам, окисление проводят в кислой среде,
где оно протекает через хингидрон. Хингидроны представляют собой интенсивно окрашен-
окрашенные молекулярные соединения из хинонов и гидрохинонов, обычно в молярном соотноше-
соотношении 1 : 1. Их можно получать сливанием водных растворов компонентов, однако устойчивы
они, как правило, лишь в твердом состоянии. (Ознакомьтесь в связи с этим с хингидронным
электродом!)
2 В промышленности пероксид водорода получают также аутоокислением изопропанола
кислородом
Н3С-СНОН-СН3 + О2 Н3С-СО-СН3 + Н2О2
34 Г Препаративная часть (продолжение)
Ф Получение нафтохинона-1,2 [Физер Л. В кн.: Современные методы эксперимента в
органической химии. — М.: Госхимиздат, 1960, с. 240]. (Ср. восстановительное рас-
расщепление азокрасителей по схеме [Г.8.36].)
Получение гидрохлорида 1-аминонафтола-2 восстановительным расщеплением р-нафтол-
оранжа. 0,01 моль р-нафтолоранжа растворяют в 50 мл воды и при 40—50 °С добавляют
0,02 моль дигидрата дитионита натрия. Перемешивают, пока не исчезнет красная окраска и не
выделится желто-розовый осадок 1-аминонафтола-2. Для его коагуляции реакционную смесь
подогревают до начала вспенивания, затем охлаждают в бане со льдом. Осадок отсасывают,
промывают водой и переносят в раствор, приготовленный из 1 мл концентрированной НС1 в
20 мл воды и содержащий ~50 мг хлорида олова(П) в качестве антиоксиданта. Раствор с осадком
слегка подогревают до почти полного растворения, фильтруют через слой активированного
угля, к фильтрату добавляют 4 мл концентрированной НС1. Выпавший гидрохлорид 1-амино-
нафтола-2 растворяют при нагревании, раствор охлаждают в бане со льдом, осадок отсасывают,
промывают холодным раствором 1 мл концентрированной соляной кислоты в 4 мл воды. Гид-
Гидрохлорид необходимо быстро переработать далее, так как он очень неустойчив на воздухе.
Окисление в нафтохинон-1,2. Растворяют при нагревании 0,02 моль гексагидрата хлорида
железа(Ш) в 2 мл концентрированной соляной кислоты и 10 мл воды, охлаждают до комнат-
комнатной температуры и фильтруют полученный раствор. Гидрохлорид 1 -аминонафтола-2 раство-
растворяют при перемешивании в небольшом количестве воды при 35 °С, фильтруют и при переме-
перемешивании вливают в раствор хлорида железа(Ш). Образующийся осадок отфильтровывают,
тщательно промывают водой до нейтральной реакции. Т. пл. 145—147 °С (с разложением);
выход 75%.
Этим же методом из гидрохлорида 1,4-диаминонафталина можно получить
нафтохинон-1,4: ConantJ. В., Freemann S. A. Org. Syntheses, Coll. Vol. I, 383 A941);
Физер Л. В сб.: Синтезы органических препаратов. Сб. 1. — М.: ИЛ, 1949, с. 286;
Сб. 2. - М.: ИЛ, 1949, с. 353.
Получение п-бензохинона из гидрохинона окислением хлоратом натрия в
присутствии оксида ванадияОО см. Ундервуд X., Уолш В. В сб.: Синтезы органи-
органических препаратов. Сб. 2. — М.: ИЛ, 1949, с. 545. То же окислением хромовой
смесью: Влие Е. В сб.: Синтезы органических препаратов Сб. 1. — М.: ИЛ, 1949,
с. 463.
В промышленности и-бензохинон получают сложной экологически неблагоприятной
реакцией окисления анилина МпО2или СгОз в сернокислом растворе (см. разд. Г,6.4.3).
Хиноны широко распространены в природе, являясь, по-видимому, продуктами обмена
веществ в грибах и высших растениях (например, витамин К). Встречаются они и в организ-
организмах животных, где образуются при окислении гидроксифениламинокислот. Ознакомьтесь в
связи с этим, например, с возникновением коричневых и черных пигментов кожи (мелани-
(меланинов) из тирозина или адреналина. Убихинон E-метил-2,3-диметоксибензохинон-1,4 с изоп-
реновым боковым звеном) действует в дыхательной цепи в качестве окислителя. Имеющий
близкое строение пластохинон выполняет аналогичные функции в процессе фотосинтеза.
Реакции хинонов как винилогов карбонильных соединений см. в разд. Г,7.4;
роль хинонов как диенофилов в диеновом синтезе см. в табл. Г.4.99.
6.4.3. ПОЛУЧЕНИЕ ХИНОНИМИНОВ ОКИСЛИТЕЛЬНЫМ СОЧЕТАНИЕМ
Если окислять и-амино-М,М-диметиланилин дихроматом в присутствии
1Ч,г>1-диметаланилина, то указанный в схеме [Г.6.48] катион-радикал оказывает-
оказывается способным присоединять реакционноспособные ароматические соединения.
В следующей стадии окисления продукт реакции стабилизируется путем образо-
6. Окисление и дегидрирование 35
вания связи N—С. Дальнейшее окисление приводит к образованию окрашенной
«хиноидной» системы (сравните ее структуру и мезомерию с кристаллическим
фиолетовым, разд. Г,5.1.8.5):
f V-NH2 + <f ^NMe2 —~ s~ Me2N—/ \—N—^
NH2 + <f ^NMe2 —~ s~ Me2N—/ \—N—^ V-NMe2
\=/ -H , -2e \==/ Л \/
n
A \\ /==\ ©
Me2N-47 N>—N=< V=NMe2 [Г.6.52]
зеленый Биндшедлера
Зеленый Биндшедлера относится к так называемым индаминам (N-фенил-
хинондииминам); незамещенный индамин образуется при окислительном со-
сочетании я-фенилендиамина с анилином (схема [Г.6.53], I). При окислительном
сочетании ароматических аминов и фенолов получаются N-фенилхинонмоно-
имины («индофенолы»); простейший индофенол образуется из я-аминофенола
и фенола (схема [Г.6.53], II).
N'4 ' - [Г.6.53]
i и
Соединения, имеющие структуру типа I или II, часто способны обратимо превращаться в
семихиноны. Способные к мезомерии соли хинониммония, например зеленый Биндшедлера,
можно рассматривать как N-аналоги полиметинов (см. разд. Г,7.2.1.1), что и обусловливает их
глубокую окраску. Этот структурный элемент содержится также в катионных азокрасителях.
(Хинониминные структуры обнаруживаются также в таутомерных формах азокрасителей; см.
разд. Г.8.3.3.)
Индамины и индофенолы не применяются в качестве текстильных красителей, в част-
частности из-за их склонности к гидролизу (см. разд. Г,6.4.2). Однако они являются исходными
продуктами для производства сернистых красителей, которые получают при нагревании с по-
полисульфидами щелочных металлов. Зеленый Биндшедлера реагирует с сероводородом с обра-
образованием метиленового голубого.
Индофеноны и индамины широко применяются как красители в цветной фотографии. На
освещенных местах фотоматериала из галогенида серебра возникают зерна металлического се-
серебра, которые катализируют в растворе цветного проявителя М,М-диалкил-я-фенилендиа-
мина его окислительное сочетание с соединениями, введенными в слои цветной пленки.
Окислителем служит галогенид серебра, желтым компонентом являются анилиды р-оксокар-
боновых кислот, пурпурным — замещенные пиразолиноны-3 (см. разд. Г,7.1.4.2), сине-зеле-
сине-зеленый цвет дают производные 2-ацетамидофенола или а-нафтола, например:
/~\ /~л
E\,H—V %— NH2
-4Ад®, -4НВг
R
При окислительном сочетании анилина получается несколько продуктов.
С хромовой кислотой на холоду образуется в основном смесь так называемых
«олигоиндаминов», в которых в одну цепочку соединяются до восьми молекул
анилина: \
[Г.6.55]
36 Г Препаративная часть (продолжение)
Если одновременно провести гидролиз образовавшихся хинондииминов, то в
качестве основного продукта образуется и-бензохинон, который большей
частью получают именно этим методом (см. разд. Г,6.4.2).
Получение п-бензохинона из анилина: Gattermann L., Wieland Т., Sucrow W.
Die Praxis des organischen Chemikers. — Walter de Gruyter, Berlin, New York,
1982, S. 567.
Дальнейшее окисление приводит к синтезу одного из самых старых синтетических краси-
красителей — анилиновому черному. Он может применяться для окраски хлопковых волокон; при
этом адсорбирующаяся на волокнах соль анилиния окисляется хлоратом или гексацианофер-
ратом(Ш) в присутствии катализатора ванадата аммония. Основой анилинового черного счи-
считают связанные N-мостиками N-фенилзамещенные феназиновые кольца, которые образуют-
образуются при присоединении анилина к промежуточным «олигоиндаминам» [Г.6.55] с последующим
дегидрированием и циклодегидрированием. Применяемые в качестве красителей в пастах для
шариковых ручек, пластмассах, кремах для обуви и типографских красках индулины и нигро-
нигрозины получают окислением анилина при повышенных температурах в виде смеси окрашен-
окрашенных соединений, в которой преобладает сине-черный краситель.
Гидразоны, образующиеся из гетероциклических оксосоединений и имею-
имеющие амидразоновую структуру >N—C=N—NH2, сочетаются при окислении с
ароматическими аминами, фенолами и С-Н-кислотными соединениями в
азокрасители, например:
СН3
'VN_NH2 +
S
Эта реакция дополняет азосочетание (см. разд. Г,8.3.3), так как с ее помощью можно полу-
получать непосредственно как катионные азокрасители (см. схему [Г.6.56]; изобразите вторую ме-
зомерную структуру), так и соединения, которые нельзя получить азосочетанием (напишите
уравнение реакции гидразона 1-метилпиридона-2 с а-нафтолом).
Ф Общая методика получения азокрасителей окислительным азосочетанием (табл. Г.6.57)
[HiinigS., Fritvch К. Н. Liebigs Ann. Chemie, 1957, 609, 143]
А. Фенолы и С—Н-кислотные соединения. К раствору 0,05 моль гидразона N-метилбензоти-
азолона-2 (получение: Riemschneider R., GeorgiS. Monatsh. Chemie, 1960, 91, 623), 0,05 моль вто-
второго компонента и 30 мл воды в 70 мл метанола приливают при перемешивании и охлаждении
до 25—30 °С раствор 0,022 моль гексацианоферрата(Ш) калия в 50 мл воды, 50 мл метанола и
20 мл 25%-ного аммиака. Через 15 мин разбавляют 250 мл воды, осадок отфильтровывают,
промывают водой и после высушивания перекристаллизовывают.
Б. Ароматические амины. К раствору 0,05 моль гидразона Ы-метилбензотиазолона-2 и
0,05 моль второго компонента в 20 мл ледяной уксусной кислоты добавляют на кончике
шпателя CuSO4 и затем при перемешивании — 0,012 моль пероксида водорода в виде
30%-ного раствора. Через 30 мин добавляют 2 г тетрафторобората натрия, растворенного в
небольшом количестве воды и упаривают реакционную смесь наполовину. После охлажде-
охлаждения осадок отфильтровывают, добавляют 7 мл воды, доводят до кипения, снова охлаждают,
отфильтровывают и сушат. Перекристаллизовывают из этилацетата.
6.5. ОКИСЛЕНИЕ С РАСЩЕПЛЕНИЕМ СВЯЗЕЙ С-С
В жестких условиях (высокая температура, большая продолжительность реак-
реакции, избыток окислителя) органические соединения расщепляются с образова-
образованием карбоновых кислот. При полной окислительной деструкции (сжигании) в
качестве конечных продуктов получают диоксид углерода и воду.
Таблица Г.6.57. Реакции окислительного сочетания
Конечный продукт
М'-C-Метилбензотиазолиден-2)-
нафтохинон-1,2-моногидразон-1
М'-C-Метилбензотиазолиден-2)-
хинолиндион-5,8-гидразон-5
Ы'-C-Метилбензотиазолиден-2)-
C-метил-1 -фенилпиразолдион-4,5)-
гидразон-4
Тетрафторборат З-метил-2-
(л-диметиламинофенилазо)-
бензотиазолия
Тетрафторборат 2-
(я-диэтиламинофенилазо)-
3-метилбензотиазолия
Тетрафторборат З-метил-2-
(л-фениламинофенилазо)-
бензотиазолия
Исходное соединение
р-Нафтол
8-Гидроксихинолин
З-Метил-1 -фенилпиразолон-5
Ы^'-Диметиланилин
Ы,Ы'-Диэтил анилин
Дифениламин
Вариант
А
А
А
Б
Б
Б
Т. пл., "С
242—244 (хлорбензол)
253 (монометиловый
эфир гликоля)
258 (ДМФА)
208 (разл.)
199-200 (разл.)
182-183
Я.тах,НМ (lgs)
490 D,42)
(ДМФА)
495 D,55)
(ДМФА)
435 D,46)
(ДМФА)
600 D,80)
(этанол)
595 D,8)
(этанол)
610D,11)
(уксусная кислота)
Выход, %
75
80
75
63
55
60
38 Г Препаративная часть (продолжение)
При 105—120 °С окисление парафинов кислородом воздуха в присутствии марганцевых солей в
качестве катализаторов происходит по обычному механизму аутоокисления (см. схему [Г.1.42]); при
этом промежуточные гидропероксиды переходят в кетоны и далее в гидропероксикетоны. Послед-
Последние снова распадаются на кислоту и альдегид, который окисляется до кислоты в соответствии со схе-
схемой [ГЛ.46]. НОО
-н2о Т R T R [Г.6.58]
А )
В промышленности к-парафины (С»—Сзо) окисляют в жидкой фазе в присутствии катализатора.
Так как все метиленовые группы окисляются кислородом равновероятно (см. разд. Г, 1.5), то продукта-
продуктами реакции является смесь карбоновых кислот с длиной цепи от Q до Сзо. Одновременно образуются
побочные продукты — дикарбоновые кислоты, спирты, кетоны и сложные эфиры (см. разд. Г,6.2).
Важно отметить, что большую часть продуктов реакции составляют карбоновые кислоты с длиной це-
цепи C|2—Qg. Эту фракцию переводят в эфиры и гидрируют на меднохромовом катализаторе в «жирные
спирты», используемые далее в производстве моющих средств (разд. Г,7.3.2). При энергичном окисле-
окислении незамещенных циклопарафинов разрыв кольца в любом месте приводит к образованию одной и
той же дикарбоновой кислоты; например, из циклогексана получается адипиновая кислота:
[Г.6.59]
В ходе этой реакции, имеющей также важное промышленное значение, образуются, кроме того,
продукты более глубокого окисления и деструкции (например, глутаровая и янтарная кислоты).
Функциональные группы (двойные связи, гидроксильные и карбонильные
группы) создают в молекуле центр, предпочтительно подвергающийся атаке
окислителем. Тогда расщепляется преимущественно одна связь С-С, образует-
образуется меньше побочных продуктов и реакция приобретает препаративный интерес.
6.5.1. ОКИСЛЕНИЕ КРАТНЫХ УГЛЕРОД-УГЛЕРОДНЫХ СВЯЗЕЙ
Кратные углерод-углеродные связи очень чувствительны к действию хромовой кис-
кислоты, азотной кислоты или перманганата. Вначале идет присоединение двух гидрок-
сильных групп (^ыс-гидроксилирование, см. схему [Г.4.36]). Далее образовавшийся
диол-1,2 обычно сразу окисляется с расщеплением связи С—С. В условиях межфаз-
межфазного катализа, однако, из олефинов можно получить и диолы-1,2 (гликоли).
Синтез цис-диолов-1,2 окислением олефинов перманганатом калия в щелоч-
щелочной среде: Weber W. P., Shepherd J. P. Tetrahedron Letters, 1972,4907.
Отличным реагентом для осуществления ^ис-гидроксилирования олефинов
является более дорогой и более токсичный тетраоксид осмия. Однако он необхо-
необходим лишь в каталитических количествах, поскольку реокисляется в присутствии
/и/?е/и-бутилгидропероксида или N-оксидов третичных аминов, например триме-
тиламин-N-оксида, М-метилморфолин-М-оксида или пиридин-1Ч-оксида.
Одним из важных методов окисления соединений с кратными связями
является эпоксидирование (см. разд. Г,4.1.6).
В промышленности наряду с эпоксидированием гликоль получают каталитическим окис-
окислением смеси этилена с уксусной кислотой при 170 °С и давлении 2,8 МПа:
Н2С = СН2 + 2АсОН + °* - АсОСН2СН2ОАс + 2Н2°. НОСН2СН2ОН + 2АсОН [Г.6.60]
— П2О
6. Окисление и дегидрирование 39
Обычно при окислении олефинов через промежуточные диолы-1,2 образу-
образуются кислоты или кетоны:
, н но он о о
С=С —С-С-Н .С. + Xv [I.6.61J
/ \ II / \ /- -он
Эта реакция имеет важное значение для синтеза некоторых карбоновых кис-
кислот, например адипиновой кислоты из циклогексена и пеларгоновой и азелаи-
новой кислот из олефиновых кислот (в промышленности — из касторового
масла):
СНз(СН2OСН=СН(СН2OСООН HN°3- СН3(СН2OСООН + НООС(СН2OСООН [Г.6.62]
Примеры получения жирных кислот с нечетным числом углеродных
атомов из алкенов действием перманганата калия приведены в общей методи-
методике к табл. Г.6.41.
Кроме того, эти реакции могут быть использованы для качественного опре-
определения двойной связи (обесцвечивание холодного щелочного раствора пер-
перманганата калия; реакция Вагнера—Байера), а также для определения положе-
положения двойной связи; в последнем случае следует учитывать, что реакция может
сопровождаться миграцией двойной связи. Более подходящим является
расщепление озоном, которое можно использовать и для перпаративного полу-
получения альдегидов, кетонов и кислот (см. разд. Г,4.1.7).
В более жестких условиях расщеплению подвергаются также и ароматические кольца,
прежде всего полициклические соединения. Так, например, фталевый ангидрид в промыш-
промышленности получают окислением нафталина кислородом воздуха над оксидом ванадия(У) при
350-385 "С. При несколько больших температурах D00-500 °С) получают малеиновый ангид-
ангидрид из бензола. Применение фталевого ангидрида см. табл. Г.6.14. Большие количества мале-
инового ангидрида используют в производстве полиэфирных смол.
При окислении хинолина перманганатом калия получают хинолиновую кислоту, которая
может быть декарбоксилирована в никотиновую кислоту (см. табл. Г.6.14).
^СООН ^\ ^СООН
[Г.6.63]
COOH ""
Почему пиридиновое кольцо не подвергается воздействию окислителя?
Ф Получение азелаиновой кислоты из касторового масла (окисление перманганатом)
[HillJ. W., McEwen W. L. Org. Syntheses, Coll. Col. II, 1943, p. 53].
Омыление касторового масла (получениерицинолевой кислоты) (см. разд. Г,7.1.4.3). 100 г кас-
касторового масла 3 ч кипятят с обратным холодильником с раствором 20 г гидроксида калия в
250 мл 95%-ного спирта. Раствор выливают в 600 мл воды, подкисляют разбавленной серной
кислотой F0 мл воды и 20 мл концентрированной серной кислоты). Выделившуюся рицино-
левую кислоту два раза промывают теплой водой, сушат 1 ч при частом встряхивании безвод-
безводным сульфатом магния B0 г), отсасывают от осушителя. Выход неочищенной кислоты 90 г. Ее
следует сразу же использовать для дальнейшего синтеза, так как при стоянии кислота полиме-
ризуется.
Окисление рицинолевой кислоты в азелаиновую кислоту. В трехлитровой трехгорлой колбе,
снабженной мешалкой и термометром, растворяют при нагревании в 2 л воды 0,9 моль A42 г)
перманганата калия. После полного растворения охлаждают до 35 °С и при энергичном пере-
перемешивании сразу добавляют раствор 0,2 моль F0 г) неочищенной рицинолевой кислоты в
40 Г Препаративная часть (продолжение)
400 мл 4%-ного гидроксида калия. При этом температура повышается до -75 °С. Перемешива-
Перемешивание продолжают до момента, когда разбавленная водой проба потеряет окраску перманганата
(примерно полчаса). Реакционную смесь переливают в пятилитровый сосуд и медленно, осто-
осторожно (выделение СОг, вспенивание!) добавляют разбавленную серную кислоту A50 мл воды
и 50 г концентрированной серной кислоты). Для коагуляции диоксида марганца нагревают
15 мин на водяной бане и как можно быстрее отсасывают. Осадок диоксида марганца кипятят
с 500 мл воды для извлечения адсорбированной азелаиновой кислоты, еще раз отсасывают.
Соединенные фильтраты упаривают до объема примерно 1 л и охлаждают в холодильнике.
Осадок азелаиновой кислоты отфильтровывают, промывают небольшим количеством
холодной воды, сушат. Для очистки перекристаллизовывают из кипящей воды (около 15 мл
воды на 1 г неочищенной кислоты). Т. пл. 104—106 °С; выход 35% в расчете на рицинолевую
кислоту.
6.5.2. РАСЩЕПЛЕНИЕ ГЛИКОЛЕЙ
Пользуясь специфическими окислителями, можно селективно расщеплять дио-
лы-1,2 (гликоли) до альдегидов и кетонов. Особенно эффективными реагентами
являются йодная кислота и тетраацетат CBHHua(IV):
_ ' _ /С"°
~С-ОН + РЬ(ОСОСН3L + РЬ(ОСОСН3J+ 2СН3СООН [Г.6.64]
С~ОН \
I с=о
Механизм этой реакции полностью еще не выяснен. Вероятно, гликоль и
тетраацетат свинца сначала образуют сложный эфир, который далее подвергает-
подвергается ионному расщеплению. Как правило, цис-гликолн расщепляются заметно
быстрее, чем /и/?анс-гликоли.
Ф Получение полуацеталя этилового эфира глиоксиловой кислоты1 из диэтилового эфира вин-
винной кислоты действием тетраацетата свинца [Stedehouder P. L. Rec. Trav. Chim. Pays-Bas,
1952,71,831]
у Внимание! Тетраацетат свинцаAУ) — гербицид и весьма токсичен. Избегать
Jj кожных контактов и попадания в рот!
К раствору 1 моль диэтилового эфира винной кислоты в 1 л дихлорметана при энергичном
перемешивании и охлаждении ледяной водой добавляют 1 моль тетраацетата свинца (см. часть
Е), перемешивают 12 ч при комнатной температуре, фильтруют. Медленно, пользуясь 50-сан-
50-сантиметровой колонкой Вигре, в вакууме отгоняют примерно две трети СН2С12 (отгонку прекра-
прекращают, когда проба дистиллята станет давать с конц. аммиаком четкое красное окрашивание:
это означает, что начал перегоняться продукт реакции). После добавления 800 мл абсолютного
спирта оставляют на ночь, фильтруют, твердый осадок промывают небольшим количеством
спирта, большую часть спирта отгоняют через ту же колонку в вакууме. Затем колонку снима-
снимают и остаток перегоняют в вакууме на воздушной бане. Весь собранный дистиллят подвергают
ректификации на колонке. Т. кип. 57—59 °С B2 мм рт. ст.); выход 65%.
Йодная кислота в отличие от тетраацетата свинца растворима в воде и поэтому может быть
использована в качестве реагента для расщепления нерастворимых в органических раствори-
растворителях Сахаров. Она применяется для определения размеров колец гликозидов. Например, при
расщеплении гликозидов альдогексоз пиранозиды образуют в качестве продукта окисления
муравьиную кислоту, а фуранозиды — формальдегид.
1 Этиловый эфир гидроксиэтоксиуксусной кислоты. Об устойчивости полуацеталей
см. разд. Г.7.1.2.
6. Окисление и дегидрирование
41
+ 2НЮ4
МеО О
метил-а-маннопиранозид
-2НЮ3, -Н2О
"Ч
МеО О
ОН
ОН
Л.
[Г.6.65]
+ 2НЮ4
О
. „ -2НЮ3 -Н2О Js.
. ОН ' МеО О'
0
[Г.6.66]
он
метил- а-маннофуранозид
Таким образом удалось показать, что большинство гликозидов имеет шестичленные кольца.
Если периодат использовать в смеси с реагентами, окисляющими олефины до гликолей,
например, с перманганатом калия или тетраоксидом осмия (они берутся только в каталитичес-
каталитических количествах, так как реокисляются периодатом), то возможно осуществить непосредствен-
непосредственное окисление олефинов до альдегидов и кетонов. О получении гликолей, необходимых в каче-
качестве промежуточных соединений, см. разд. Г,4.1.6. При условии коммерческой доступности
генераторов озона, для окисления олефинов можно использовать метод озонирования (см.
разд. Г,4.1.7).
6.5.3. ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ ВТОРИЧНЫХ СПИРТОВ И КЕТОНОВ
При энергичном окислении хромовой смесью или азотной кислотой алифати-
алифатические кетоны и вторичные спирты дают смеси жирных кислот:
о
н он
О
т
о
НО
R'
[Г.6.67]
О
R
НО R"
+ Y
он о
В случае метилкетонов группа СНзСО отщепляется и образует уксусную
кислоту:
сн.
о
НО СН3
[Г.6.68]
Определенное значение имеет окисление алициклических спиртов и кетонов
в дикарбоновые кислоты. Так, например, исходным веществом для промыш-
промышленного получения адипиновой кислоты является циклогексанон.
При галоформной реакции (реакция Эйнхорна) метилкетоны или спирты,
имеющие группировку СН3СНОН, с потерей одного атома углерода превраща-
превращаются в карбоновые кислоты (под действием гипогалогенидов или галогенов в
щелочной среде). В первой стадии спирт окисляется до карбонильного соедине-
соединения, затем происходит полное галогенирование активированной метильной
группы (ср. разд. Г,7.4.2.2). Получающееся тригалогенированное метилкарбо-
нильное соединение сильно поляризовано, поэтому оно очень легко подверга-
42
Г Препаративная часть (продолжение)
ется щелочному гидролизу с образованием соответствующей кислоты и хлоро-
хлороформа или соответственно муравьиной кислоты:
^ЧI * ^^ I i II ^Ч * /*ъ 'II I ^—' |/"\
CI-CI + Н-С-ОЛН.+
СНз
R-CO-СНз + ЗС12
HOIQ
:ю-н
сг + на +
С=О + Н2О
н3с
R-CO-CCI3 + 3HCI
0
[Г.6.69]
R-COOG+ HCCI3
R -CO-CCI3 - R-CO-OH + ICCI3 —
Напишите суммарное уравнение реакции!
Галоформная реакция протекает в очень мягких условиях с высоким выхо-
выходом, так что даже такое неустойчивое соединение, как метилвинилкетон, может
быть превращено в акриловую кислоту. В аналитической химии реакция приме-
применяется для качественного обнаружения групп СН3СО или СН3СНОН. Для этого
действуют иодом в присутствии щелочи, образующийся йодоформ определяют
по цвету, характерному запаху, а также по температуре плавления.
Ф Общая методика окисления метилкетонов гипобромитом (галоформная реакция) (табл.
Г.6.70)
¦ Внимание1. Работать под тягой!
В трехгорлой колбе на 500 мл, снабженной мешалкой, капельной воронкой и термометром
(колба должна сообщаться с атмосферой), при сильном перемешивании и охлаждении к раст-
раствору 1 моль NaOH в 200 мл воды прибавляют по каплям 0,3 моль брома. Температура при этом
не должна подниматься выше 10 °С. Раствор охлаждают до 0 "С и прибавляют по каплям
0,1 моль кетона (температура также не должна превышать 10 °С; твердые кетоны предварительно раст-
растворяют в 100 мл диоксана). Затем смесь перемешивают 1 ч при комнатной температуре. Образовав-
Образовавшийся бромоформ отделяют в делительной воронке или отгоняют с водяным паром; к щелочному
раствору прибавляют 10 г пиросульфита натрия (Na2S2O5) в 150 мл воды, а затем подкисляют концент-
концентрированной соляной кислотой. (Выделяется диоксид серы, работать под тягой!)
Реакционную смесь обрабатывают одним из двух способов: а) отсасывают выделившуюся
кислоту и перекристаллизовывают ее; б) насыщают раствор поваренной солью и экстрагиру-
экстрагируют в течение 8 ч в аппарате для экстрагирования; эфирный раствор сушат сульфатом магния,
отгоняют растворитель, остаток перегоняют.
Таблица Г.6.70. Получение карбоновых кислот из метилкетонов (галоформная реакция)
Продукт реакции
Триметилуксусная кислота
C,C- Диметил акриловая
кислота
Анисовая кислота
Вератровая кислота
и-Хлорбензойная кислота
я-Бромбензойная кислота
а-Нафтойная кислота
(З-Нафтойная кислота
Тиофенкарбоновая-2 кислота
Исходное соединение
Пинаколин
Мезитилоксид
л-Метоксиацетофенон
3,4-Диметоксиацетофенон
л-Хлорацетофен
л-Бромацетофенон
а- Нафтилметилкетон
C-Нафтилметилкетон
2-Ацетилтиофен
Т. пл. или т. кип.,
°С (мм рт. ст.)
Т. кип. 77 B0);
т. пл. 35
Т. кип. 104 B0);
т. пл. 67
184 (вода)
181 (вода)
239(этанол)
254 (вода)
163 (водный этанол)
181 (лигроин)
126 (вода)
Способ
обработки
б
б
а
а
а
а
а
а
а
Выход, %
60
40
80
75
80
90
70
80
90
6. Окисление и дегидрирование 43
Иодоформная проба (общая методика для качественного анализа). Примерно 0,1 г пробы раство-
растворяют в 5 мл диоксана, прибавляют 1 мл 10%-ного раствора гидроксида натрия, затем по кап-
каплям иод в водном растворе иодида калия A г иода, 2 г иодида калия в 10 мл воды). После это-
этого нагревают 2 мин на водяной бане при 60 °С. После исчезновения окраски иода добавляют
еще немного раствора 12/К1 и снова подогревают короткое время. Избыток иода удаляют, до-
добавляя несколько капель 10%-ного раствора гидроксида натрия. Приливают в пробирку воду
и оставляют на 15 мин. Затем фильтруют, сушат и перекристаллизовывают из метанола. Жел-
Желтые кристаллы; т. пл. 121 °С.
Получение адипиновой кислоты из циклогексанола.
Я Осторожно! Выделяются нитрозные газы! Работать под тягой!
ы
В стакане нагревают 0,032 моль 50%-ной азотной кислоты (d = 1,32) и 0,1 г ванадата аммо-
аммония до 90 °С. Из общего количества циклогексанола @,01 моль) вначале прибавляют несколько
капель при перемешивании до начала реакции, затем при охлаждении до 60 "С постепенно при-
прибавляют остальное количество. Через полчаса охлаждают до 0 °С, отсасывают, промывают
ледяной водой, сушат. Выход неочищенного продукта 58—60%; т. пл. 141-145 °С. Для очистки
перекристаллизовывают из концентрированной азотной кислоты, затем из воды. Т. пл. 151 —
152 "С.
6.6. ДЕГИДРИРОВАНИЕ АЛКАНОВ И ЦИКЛОАЛКАНОВ
Если предельные углеводороды нагреть без доступа воздуха выше 500 "С, то они
подвергаются дегидрированию и расщеплению (крекингу), например:
^ Н2С = СЫ-СН3 + Н2 +110кДж/моль 55% 99% [Г.6.71а]
Н3С-СН2-СН3 -650 "С
^^"" Н2С=СН2 + СН4 + ЬЬкДж/моль 45% 1% [Г.6.716]
В то время, как реакция крекинга [Г.6.716] необратима, дегидрирование по
уравнению [Г.6.71а] является обратимым процессом. Поэтому дегидрирование
можно ускорить теми же катализаторами, что и гидрирование, — никелем, плати-
платиной, палладием (см. разд. Г,4.5.2), а с заметно меньшей эффективностью также ок-
оксидами алюминия и хрома. Гидрирование преобладает при низких температурах,
дегидрирование — при высоких (см. также разд. В,2). В промышленности фракции
алифатических углеводородов Сб—Cs подвергают каталитической дегидроциклиза-
дегидроциклизации до аренов (бензол, толуол, ксилолы) (процесс риформинга).
При каталитическом дегидрировании углеводородов трудности нарастают в
следующем ряду: циклоалкены < циклоалканы < алкены < алканы. Алканы можно
дегидрировать с удовлетворительным результатом лишь при 550—600 "С, и едва ли
возможно использовать эту реакцию для препаративных целей; в то же время реак-
реакция с гидроароматическими соединениями (циклоалканами) идет количественно
уже при 300-350°С, и ее можно применять в лаборатории. Как правило, дегидри-
дегидрирование идет вплоть до образования ароматических соединений, а продукты час-
частичного дегидрирования уловить не удается; таким путем получают и гетероарома-
тические соединения, например:
терми-
термически
55%
45%
катали-
каталитически
99%
1%
\ Pd;300-C /Г\ мои
V +2Нг [Г.6.72]
i i
н н
44
Г Препаративная часть (продолжение)
Имеющиеся в молекуле двойные связи существенно облегчают дегидроаро-
матизацию.
Дегидрирование в лаборатории теперь потеряло свое значение. Общая мето-
методика проведения этого процесса и аппаратура были детально описаны в преды-
предыдущих изданиях настоящей книги.
Наряду с каталитическим дегидрированием отщепление водорода от органи-
органической молекулы можно осуществить действием дегидрирующих реагентов,
непосредственно участвующих в реакции, например серой (-> H2S), селеном
(-> H2Se), хинонами (-» гидрохиноны) и другими мягкими окислителями, таки-
такими, как хлорид железа(Ш) или нитробензол.
Дегидрирование серой или селеном методически осуществляется очень
просто нагреванием смеси компонентов; недостатком таких методик является
выделение сероводорода или селеноводорода. Поэтому селен, действующий
только при ЗОО—ЗЗО "С, не играет большой роли в препаративной химии, хотя
образует мало побочных продуктов. Сера реагирует уже при 220—270 °С, давая
серосодержащие побочные продукты, такие, как дитиол-1,2-тионы-3 и произ-
производные тиофена.
Ряд гетероциклических соединений с серой, имеющих важное промышленное значение,
например сернистые красители и фенотиазин, получают дегидрированием с циклизацией и
одновременным включением атома серы.
За немногими исключениями дегидрирование можно применять без всяких ограничений
только в тех случаях, когда продуктами реакции являются ароматические или гетероцикличес-
гетероциклические соединения. Дегидрирование происходит тем легче, чем больше двойных связей в исход-
исходном соединении.
Дегидрирование успешно применяется также при установлении строения терпенов и
стероидов, так как в результате реакции возникают уже известные ароматические системы,
например:
Se; 350 'С
[Г.6.73]
холестерин
метил циклопентенофенантрен
НООС
S;230"C
[Г.6.74]
абиетиновая
кислота
ретен
Из приведенных примеров видно, что при реакциях дегидрирования могут
происходить изменения углеродного скелета и функциональных групп, что
ограничивает препаративное использование дегидрирования, если требуется
однозначное течение процесса. Сравнительно хорошо и однозначно получают-
6. Окисление и дегидрирование
45
ся различные гетероциклические соединения из соответствующих дигидросое-
динений, например:
R
R1
н
-Н
-H2S;160*C R2
TJ) Общая методика дегидрирования серой (табл. Г.6.76)
// N
-R3
[Г.6.75]
Л Внимание! Выделяется сероводород! Работать под тягой!
0,03 моль исходного соединения смешивают с рассчитанным количеством серы и нагре-
нагревают в колбе с воздушным обратным холодильником до ~ 150 °С. Когда начинается выделение
сероводорода, температуру бани постепенно повышают до 250 °С и продолжают нагревание
при 250 "С до прекращения выделения сероводорода. После охлаждения остаток перегоняют
или перекристаллизовывают, добавляя немного активированного угля.
Аналогично дегидрируют А5-пиразолины в пиразолы [Грандберг И. И., Кост А. Н. ЖОХ, 1958,
28, 3071] и Ь?-тиазолины в тиазолы [AsingerF., ThielM. Angew. Chem. 1958, 70, 675].
Хлоранил является дегидрирующим агентом, действующим, как и сера, в довольно мягких
условиях. Его вместе с дегидрируемым соединением нагревают в инертном растворителе (нап-
(например, ксилоле) при 70—120 °С. Хлоранил оказался хорошим дегидрирующим агентом для
гетероциклических соединений. Однако по окончании реакции необходимо отделить не толь-
только гидрохинон, но и непрореагировавший хлоранил.
Дегидрирующее действие хлоранила основано на его превращении в соответствующий
тетрахлоргидрохинон (см. разд. Г,6.4.2):
Таблица Г.6.76. Дегидрирование серой
Конечный продукт
Антрацен
Карбазол
1 -Фенилнафталин
Этиловый эфир 2-ацетамидо-
бензо[Ь]тиофенкарбоно-
вой-3 кислоты6
Фенотиазинг
3 OiiMr-Tfo ar\rfrr\uvr\ii
Исходное соединение
9,10-Дигидроантрацен
1,2,3,4-Тетрагидрокарбазол
3,4-Дигидро-1 -фенилнафталин
Этиловый эфир 2-ацетамидо-4,5-
тетраметилентиофенкарбоно-
вой-3 кислоты6
Дифениламин
Т. кип. или
т. пл., °С
Т. пл.217
(этанол)
Т. пл. 245
(ксилол)а
189A2)
190-191
(этанол)
260A4);
Т. пл.183
(этанол)
Выход, %
60
60
80
85
80
6 Проводить синтез в 15 мл диметилового эфира фталевой кислоты при 220 °С (примерно 4 ч), в
теплую реакционную смесь влить при перемешивании 10 мл этанола.
в Получают кипячением в течение 10 мин 0,1 моль неацетилированного соединения (табл.
Г.6.29) в 50 мл уксусного ангидрида; т. пл. 123 °С (этанол).
г Температура реакции 180—190 °С, добавить 1% иода; продукт реакции перегнать.
Н Н
СГ Y С| [Г.6.77]
¦ О ОН ОН
дигидробензол бензол
Аналогичным образом можно использовать и более сильный дегидрирую-
дегидрирующий агент — 2,3-дихлор-5,6-дицианобензохинон (DDQ) (см. табл. Г.6.5).
46 Г Препаративная часть (продолжение)
6.7. ЛИТЕРАТУРА
Общие сведения о реакциях окисления
HainesA. H.: Methods for the Oxidation of Organic Compounds. Alkanes, Alkenes, Alkynes,
and Arenes. —Academic Press, London 1985.
Haines A. H.\ Methods for the Oxidation of Organic Compounds. Alcohols, Alcohol
Derivatives, Alkyl Halides, Nitroalkanes, Alkyl Azides, Corbonyl Compounds,
Hydroxyarenes, and Aminoarenes. — Academic Press, London 1988.
Hudlicky M., Oxidations in Organic Chemistry. — American Chemical Society, Washington,
DC, 1990.
Получение альдегидов окислением
Bayer О., in: Houben-Weyl. Bd. 7/1 A954), S. 135-191, 332-361.
Offermann H., Prescher G, BornowskiH., u. a. in: Houben-Weyl. Bd. E3 A983), S. 231-349.
Larock R. C, Comprehensive Organic Transformation: A. Guide to Fundamental Group
Preparation. — VCH Publishers 1989. Кар.: Aldehydes and Ketones. 2. Oxidation.
Получение кетонов окислением
Kabbe H. J., in: Houben-Weyl. Bd. 7/2a A973), S. 677-788.
Larock R. С, см. выше.
Получение карбоновых кислот окислением
Henecka H., Ott E., in: Houben-Weyl. Bd. 8 A952), S. 384-418.
Sustmann R., Korth H.-G., in: Houben-Weyl. Bd. E 5/1 A985), S. 199-216.
Реакция Вильгеродта
Brown E. V., Synthesis 1975, 358-375.
Mayer R., in: Organic Chemistry of Sulfur. Hrsg.: S. Oae. — Plenum Press, New York,
London 1977, S. 33-70.
п-Хиноны
Bayer O., in: Houben-Weyl. Bd. 7/3c A979), S. 11-46 (антрахинон).
Grundmann Ch., in: Houben-Weyl. Bd. 7/3b A979), S. 3-89 (о-хиноны).
Ulrich H., RichterR., in: Houben-Weyl. Bd. 7/3a A977), S. 14-647 (л-хиноны).
Получение хинониминов окислением
GmnangerP., in: Houben-Weyl. Bd. 7/3b A979), S. 235-267.
Окисление углеводов
Buttenvorth R. F., Hanessian S., Synthesis 1971, 70-88.
Реакции дегидрирования
Schiller G., in: Houben-Weyl. Bd. 4/2 A955), S. 333-347.
6. Окисление и дегидрирование 47
WimmerK., in: Houben-Weyl. Bd. 4/22 A955), S. 192-205.
StechlH. H, in: Houben-Weyl. Bd. 4/lb A975), S. 873-899 (дегидрирование хинонами).
GolserL., in: Houben-Weyl. Bd. 4/lb A975), S. 963—987 (дегидрирование нитросоедине-
ниями).
Окисление кислородом (см. разд. Г, 1.7)
Heyns К., Paulsen H., in: Neuere Methoden. Bd. 2 A960), S. 208-230.
Карножицкий В. Я. Усп. хим., 1981, 50, 1693-1717.
KropfH., Muller E., WeickmannA., in: Houben-Weyl. Bd. 4/la A981), S. 69-168.
Окисление соединениями металлов
Но Т. L, in: Organic Synthesis by Oxidation with Metal Compuunds. Hrsg.: W. J. Mijs,
С R. H. I. De Jonge. — Plenum Press, New York 1986.
Окисление соединениями свинца
Criegee Я, in: Neuere Methoden. Bd. 1 A949), S. 21-38; Bd. 2 A960), S. 252-267.
Mihailovic M. L., Cekovic Z., Lorenc L., in: Organic Synthesis by Oxidation with Metal
Compounds. Hrsg.: W. J. Mijs, С R. H. I. De Jonge. — Plenum Press, New York 1986,
S. 741-816.
Rothermund G. W., in: Houben-Weyl. Bd. 4/lb A975), S. 167-413.
Окисление соединениями церияA\/)
Matthias G., in: Houben-Weyl. Bd. 4/lb A975), S. 149-166.
Ho T. L, Synthesis 1973, 347-354.
Dudfleld P. J. In: Comprehensive Organic Synthesis. Hrsg.: B. M. Trost I., I. Fleming,
S. V. Ley. — Pergamon, Oxford 1991, Кар. 2.11, S. 345 (хиноны из ароматических
углеводородов).
Ruck К., Kunz H.: J. Prakt. Chem. 1994, 336, 470-471.
Fischer К., Henderson G. N., Synthesis 1985, 641-643 (окисление гидрохинонов в хиноны
Ce(IV)/SiO2).
Broadhurst M. J., Hasall С. H., Tholmas G. J., J. Chem. Soc, Perkin Trans. I 1982, 2239
(окисление диметоксиаренов в хиноны).
Окисление соединениями хрома
Bosche H. G, in: Houben-Weyl. Bd. 4/lb A975), S. 425-464.
Cainelli G, Cardillo G: Chromium Oxidations in Organic Chemistry. — Springer Verlag,
Berlin, Heidelberg, New York, Tokyo 1984.
Luzzio F. A., Ogr. React. 1998, 53, 1—221 (с помощью амино-хромовых(У1)-реагентов).
Piancatellli G., ScettriA., D'Auria M., Synthesis 1982, 245—258 (пиридинийхлорхроматом).
Окисление соединениями марганца
ArndtD., in: Houben-Weyl. Bd. 4/lb A975), S. 465-672.
Arndt D:. Manganese Compounds as Oxidizing Agents in Organic Chemistry. — Open Court
Publishing Company, La Salle 1981.
FatiadiJ., Synthesis 1976, 65-104, 133-167 (окисление MnO2).
Kndlker H.-J., J. Prakt. Chem. 1995, 337, 75-77 (окисление MnO2).
48 Г Препаративная часть (продолжение)
Окисление диметилсульфоксидом
Epstein W. W., Sweat F. W., Chem. Rev. 1967, 67, 247-260.
Martin D., HauthalH. G: Dimethylsulfoxid. — Akademie-Verlag, Berlin 1971.
MancusoA. J., Swern D., Synthesis 1981, 165-185.
Tidwell Т. Т., Synthesis 1990, 857-870.
Tidwell Т. Т., Org. React. 1990, 39, 297-572.
Окисление соединениями галогенов
Buddenberg О., Weickmann A., Zeller K.-P., in: Houven-Weyl. Bd. 4/la A981), S. 435-640.
Окисление гипервалентными соединениями иода
Moriarty R. M., Pakrash О., Org. React. 1999, 54, 273-418.
SpeicherA., Bomm V, Eicher T. J. Prakt. Chem. 338 A996), S. 588-590 (Десса - Мартина перио-
динан)
StangP. J., Zhdankin V, Chem. Rev. 1996, 96, 1123.
Varvoglis A.: The Organic Chemistry of Polycoordinated Iodine. — VCH, New York,
Weinheim, Cambridge 1992.
Varvoglis A.: Hypervalent Iodine in Organic Synthesis. — Academic Press, San Diego 1997.
Wirth Т., Hirt U. H., Synthesis 1999, 1277-1287.
Окисление йодной кислотой
Криге Р. 5 сб.: Новые методы препаративной органической химии. — М.: ИЛ, 1950, с. 13.
Джексон Э. Л. В сб.: Органические реакции. Сб. 2. — М.: ИЛ, 1950, с. 362.
FatiadiA. J., Synthesis 1974, 229-272.
Milewich L, Axelrod L R., Org. Synth. Coll. Vol. 6 A988), 690-691 (KMnO4/NaIO4).
PappeR, AllenD. S., LemiewcR. U., Johnson W. S., J. Org. Chem. 1956,21,478-479 (OsO4/KIO4).
Окисление соединениями серы и селена
Fu P. P., Harvey R. G., Chem. Rev. 1978, 78, 317-361.
KriefA., Hevesi L.:Organoselenium Chemistry I. — Springer. Berlin 1988, S. 76-103.
Ley S. V., in: Organoselenium Chemistry. Hrsg.: R. Liotta. — John Wiley & Sons, New York
1987, S. 163-206.
Rabjohn N., Org. React. 1976, 24, 261-415.
Weickmann A., Zeller K. P., in: Houben-Weyl. Bd. 4/la A981), S. 319-433.
Окисление неорганическими соединениями азота
Buddenberg О., in: Houben-Weyl. Bd. 4/la A981), S. 641-944.
Селективное каталитическое окисление над благородными металлами
Heyns К., Paulsen H., in: Neuere Methoden. Bd. 2 A960), S. 208-230.
Дигидроксилирование олефинов оксидом OsO4 или OsO4/N-оксид амина
Schroder M., Chem. Rev. 1980, 80, 187-213.
Ray R., Matteson D. S., Tetrahedron Letters 1980, 21, 449-450.
Van Rheenen V., Cho D. Y., Hartley W. M., Synth. Coll. Vol. 6 A988), 342-348.
7. Реакции карбонильных соединений 49
7. РЕАКЦИИ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
Карбонильные соединения — важный класс органических соединений. Они
легко получаются, обладают высокой реакционной активностью и поэтому спо-
способны вступать в самые разнообразные реакции.
К типичным карбонильным соединениям относятся альдегиды, кетоны,
карбоновые кислоты, их эфиры, амиды, ангидриды и ацилгалогениды, а также
диоксид углерода. Эти и некоторые другие соединения рассматриваются ниже с
общей точки зрения.
Реакционная способность карбонильной группы определяется ее поляр-
полярностью, связанной с —/-эффектом кислорода, и легкой поляризуемостью:
Таким образом, в карбонильной группе углерод проявляет кислотные
(электрофильные) свойства, кислород — основные (нуклеофильные). Особый
интерес представляют прежде всего реакции нуклеофильных реагентов с угле-
углеродом, так как в общем случае только такие реакции приводят к образованию
определенных соединений:
9
Nul + ,С ^= © А [Г.7.2]
-—-V х Nu v
Здесь Nu| — отрицательно заряженный или нейтральный нуклеофильный
агент, участвующий в реакции своей электронной парой. Продукт присоедине-
присоединения стабилизируется на дальнейших стадиях реакции до конечного продукта.
Скорость реакции [Г.7.2], очевидно, будет тем больше, чем выше нуклео-
фильность основания и чем более электрофилен атом углерода карбонильной
группы.
По возрастающей реакционной способности различные карбонильные сое-
соединения можно расположить примерно в следующий ряд:
.-о .-о .-о .-о .-о .-о .-о
¦--и '-и '-II '-и '-и '--и '-и г„ _,-.
[Г73]
Связанные с карбонильной группой (написанные снизу справа) заместите-
заместители расположены в ряду таким образом, что способность заместителя отдавать
электроны атому углерода карбонильной группы снижается слева направо и тем
самым снижается степень компенсации положительного заряда углеродного
атома1. В наибольшей степени такая способность проявляется в анионе карбо-
новой кислоты. Поэтому такой анион может реагировать только с очень силь-
сильными нуклеофилами, например с алкиллитием с образованием кетонов
(см. [Г.7.215]). Напротив, ацилгалогениды и альдегиды являются в высшей сте-
степени реакционноспособными соединениями. Однако их место в указанном
1 В ряду [Г.7.3] для амида кислоты и ее эфира показаны только мезомерные эффекты,
определяющие свойства таких соединений, а для ацилхлорида — только сильный индукцион-
индукционный эффект, превышающий +Л/-эффект.
50 Г Препаративная часть (продолжение)
выше ряду активности в некоторых реакциях оказывается иным из-за простран-
пространственных факторов.
Углеводородный радикал, связанный с карбонильной группой, также оказы-
оказывает влияние на ее реакционную способность. Группы, проявляющие —/- и
—М-эффекты, повышают реакционную способность карбонильной группы в
реакциях с нуклеофильными агентами и снижают основность кислорода карбо-
карбонильной группы; группы с +/и +М-эффектами снижают реакционную способ-
способность атома углерода и повышают основность кислорода.
Этим обстоятельством объясняется понижение кислотности карбоновых кислот в следую-
следующем ряду: трихлоруксусная > дихлоруксусная > монохлоруксусная > муравьиная > уксусная >
изомасляная > триметилуксусная (пивалиновая).
Влияние заместителей в ароматическом ядре на карбонильную группу можно описать с
помощью уравнения Гаммета [В.69]. Это относится, например, к гидролизу или алкоголизу за-
замещенных бензоилхлоридов или эфиров бензойных кислот, образованию циангидринов заме-
замещенных бензальдегидов и другим реакциям.
Скорость присоединения по карбонильной группе, естественно, тем выше,
чем более нуклеофилен агент, иначе говоря, чем больше его основность. Поэто-
Поэтому, например, эфиры и амиды омыляются гидроксид-ионами гораздо быстрее,
чем слабоосновной водой, а альдегид реагирует с первичными или вторичными
аминами энергичнее, чем со спиртами.
На реакции с участием карбонильных групп в большинстве случаев сильно
влияют катализаторы. Можно заранее предугадать, что все кислотные катализа-
катализаторы будут повышать полярность карбонильной группы, поскольку они могут
реагировать с атомом кислорода карбонильной группы, проявляющим основ-
основные свойства (см. также [Г.5.45] и [Г.5.47]):
©,н _,н „н
101 в 10 10 О)
м +Н -=—*- || —— iff, = |: ©
[Г.7.4]
ю,
м + А1С13
В результате взаимодействия с катализатором (это взаимодействие не обяза-
обязательно должно приводить к образованию настоящей химической связи, как это
показано для большей наглядности на схемах) понижается электронная плот-
плотность на нуклеофильном агенте. Это весьма наглядно показано на представлен-
представленной ниже схеме (где стрелки не обязательно относятся к одновременно протека-
протекающим электронным переходам):
Nul + ^'~Л@ « © %- 1Г.7.5]
-S/ \ Nu" \
С другой стороны, электрофильный катализатор может влиять и на нуклеофильность
агента Nu, вступая с ним во взаимодействие (см. разд. Г,7.1). Такой случай обсуждался выше
на примере электрофильного ароматического замещения, где указывалось на невозможность
ацилирования ароматических аминов по Фриделю — Крафтсу в присутствии хлорида алюми-
алюминия (см. разд. Г,5.1.8.1).
7. Реакции карбонильных соединений 51
В тесном родстве с карбонильной группой находится ряд ее «гетероанало-
гов», в которых кислород карбонильной группы заменен на гетероатом (тиокар-
бонильные соединения, азометины, нитрилы):
.-О5" .-SS~
[Г.7.6]
Реакции гетероаналогов карбонильных соединений рассматриваются в
разд. Г, 8.
Аналогия с карбонильной группой сильнее всего проявляется в реакциях
азометиновой группы. Поскольку, однако, азот менее электрофилен, чем кисло-
кислород, реакционная способность азометиновой группы в нейтральной и щелочной
средах ниже, чем карбонильной группы. В кислой среде, напротив, частичный
положительные заряд на атоме углерода азометиновой группы возрастает вслед-
вследствие сильного —/-эффекта протонированного атома азота.
По тем же причинам, что и азометиновая группа, нитрильная группа также
довольно инертна. Кроме того, тройная связь вообще менее реакционноспособ-
на, чем двойная. Поэтому соединения с нитрильными группами участвуют в
реакциях, характерных для карбонильных соединений, только в более жестких
условиях и в присутствии более эффективных катализаторов.
Нуклеофильные агенты, участвующие в «карбонильных» реакциях, можно
разделить на три группы в зависимости от характера электронной пары, обус-
обусловливающей нуклеофильность. В соответствии с этим принята следующая
классификация реакций карбонильных соединений:
1. Реакции с гетероатомными нуклеофилами.
2. Реакции с углеродными нуклеофилами.
3. Восстановление карбонилсодержащих соединений (реакции с Н-нуклеофилами).
7.1. РЕАКЦИИ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ С
ГЕТЕРОАТОМНЫМИ НУКЛЕОФИЛАМИ
Проще всего рассмотреть вначале реакции карбонильных соединений с нуклео-
нуклеофилами (основаниями Льюиса), имеющими на гетероатоме свободную элект-
электронную пару, например с водой, спиртами, аминами и их аналогами, сероводо-
сероводородом, тиолами и т. д. (в схемах [Г.7.8] и [Г.7.9] они обозначены HNu).
В табл. Г.7.7 приведены важнейшие реакции карбонильных соединений с
гетероатомными нуклеофилами.
Все карбонильные соединения, в том числе их гетероаналоги (нитрилы, азоме-
тины), присоединяют гетероатомные нуклеофилы по одной и той же схеме с обра-
образованием однотипных промежуточных продуктов I (см. схемы [Г.7.8] и [Г.7.9]):
^~Х\ о © I _е _ I _
HNu+xC=O ^^ H-Nu-C-Or == Nu-C-OH
I II
Богатый энергией биполярный ион I (цвиттер-ион) может стабилизировать-
стабилизироваться путем «внутримолекулярной нейтрализации» с образованием продукта II
(схема [Г.7.8]).
Таблица Г.7.7. Важнейшие реакции карбонильных соединений с гетероатомными нуклеофилами
\ -
с=о + н-о-н
/
\
альдегиды,
кетоны
чон
Гидраты
\
С=О + H-O-R
/
Аналогично:
+ H-S-R
\ ,он +roh(h®) \ ,
С — - С
полуацеталь
С"
/ SR
Ацетали
Тиоацетали
H2N-R
Аналогично:
+ NH2-OH
NH2-NH-R
NH2-NH-CO-NH2
\ /0Н
^ С
/ NHR
«альдегидаммиак»
\
C
C=NOH
C=N-NHR
\
C=N-NH-CO-NH2
Азометины (основания Шиффа)
Оксимы
(Замещенные) гидразоны
Семикарбазоны
С=О + H-NR2
/
)н/ОН
С
/ NR2
\
С=С
/ \
Енамины
\
С-
ЯН
0 + IS -О
ONa
\
Р
С р
Бисульфитные соединения
ONa
Таблица Г.7.7. Продолжение
О
+ Н-О-Н
X = галоген, ацилокси
Аналогично:
+ HOR'
-НХ
R-C
ОН
R-C
OR'
HNR2
(R' — также Н)
ынрн —•
+ NH2-NH2
°ч
+ (Na) C-R'
но'
R-/
О
NR2
ОН
R-C
NHOH
NOH
/Р
C
R-C
4
R-CO-O-COR1
Гидролиз ацилгалогенидов и ангидридов карбоновых
кислот
Алкоголиз до сложных эфиров карбоновых кислот
Аминолиз до амидов карбоновых кислот
Образование гидроксамовых кислот
Получение гидразидов карбоновых кислот
Получение ангидридов карбоновых кислот, в т. ч.
смешанных
CI—С + HOR
CI
-HCI
CI-C
HNR2
(R — также Н)
+ H2NR
-на
-на
+ HNR2
-на
-на
RO-C
OR
f
OR
,0
CI—С
NR2
R2N-C/
4
R-N=C=O
Частичный алкоголиз фосгена до эфиров хлоруголь-
ной (хлормуравьиной) кислоты
Алкоголиз фосгена до эфиров угольной кислоты
Аминолиз фосгена до карбамоилхлоридов
Аминолиз фосгена до мочевин
Элиминирование НС1 с образованием изоцианатов
Таблица Г.7.7. Продолжение
о
R-C' + Н-О-Н
OR'
Аналогично:
+ HOR"
+ HNR2'
(R" —также Н)
+ H2N-NH2
+ H2NOH
Р Р
R-C4 + R"—С^ —
OR' OH
R-C^ + H-O-H
(R' —также Н)
0
R-c' + H0R'
OH
Аналогично:
+ HNRj
(R' —также Н)
-C=N + H-O-H ^=^
Аналогично:
+ HCI
+ HOR' -
P
R—С +
OH
— П2
NH
R-C? ==
OH
©
R-C' Cl0
OR'
p
—- R-c'
)H он
p
OR"
<p
NR2'
p
4NH-NH2
P
— R~C\ '
4NHOH
p
R"— &
OR'
p
° OR'
О
- R-<
"nr2
NH2
R4
О
Гидролиз сложных эфиров карбоновых кислот
Алкоголиз (переэтерификация кислотной группы)
Аминолиз до амидов и гидразидов карбоновых кислот
и до гидроксамовых кислот
Ацидолиз сложных эфиров карбоновых кислот
(переэтерификация спиртовой группы)
Гидролиз амидов карбоновых кислот
Этерификация карбоновых кислот
Получение амидов из карбоновых кислот
Получение амидов карбоновых кислот из нитрилов
Получение имидоэфиров присоединением спиртов к
нитрилам
7. Реакции карбонильных соединений 55
В стадии присоединения из тригонального плоского карбонильного соедине-
соединения получается тетраэдрический аддукт (I или II), в котором заместители долж-
должны расположиться более тесно. Поэтому присоединение идет тем труднее, чем
объемистее присоединяемые группы.
Как уже говорилось, реакции присоединения ускоряются кислотами:
HNu +NC=O + Н® ^=^ HNu-C-OH ^=^ Nu-C-OH + Н® [Г 7 9]
I II
Каталитическое действие кислоты необходимо в тем большей степени, чем
менее нуклеофилен реагент. Поэтому, например, сильноосновные соединения
азота (аммиак, амины, гидроксиламин, гидразин и др.) в нейтральной или даже
слабокислой среде легко реагируют с альдегидами и кетонами. Спирты и очень
слабые азотистые основания, такие, как 2,4-динитрофенилгидразин, напротив,
требуют добавления сильной кислоты.
Продукты присоединения II в схемах [Г.7.8] и [Г.7.9] — вещества с относи-
относительно высокой внутренней энергией; они во многих случаях неустойчивы и
легко отщепляют те или иные группы атомов с образованием ненасыщенных
систем {стадия конденсации).
Для аддуктов альдегидов и кетонов можно предложить следующую общую
схему реакции:
_ I _ й _ I ©/*"* -Н2О _!'"'• / © / ^-*—У
Nu-C-OH + Н ^=^ Nu-C-0 . Nu-C© —* Nu=C = Nu-c ГГ7 1П1
| - | V, +H2O \ >. .¦ \ \ U-7.101
II III1 H ¦' |V
Продукт присоединения II протонируется присутствующей в растворе кис-
кислотой (в некоторых условиях самим растворителем). В молекуле существуют два
основных центра. Протонирование центра Nu приводит к обратной реакции (см.
схему [Г.7.9]) и поэтому нас не интересует. Протонирование же гидроксильного
кислорода дает ониевый ион III, который сразу же обратимо стабилизируется
путем отщепления воды и образования карбений-ониевого иона IV с делокали-
зованным положительным зарядом1.
При этом, как обычно (см. разд. Г,2 и Г,3), в результате отщепления протона
или присоединения присутствующего в растворе дополнительного нуклеофила
образуется нейтральный конечный продукт. Различные варианты будут обсуж-
обсуждены ниже (см. [Г.7.11], [Г.7.13], [Г.7.24]).
При реакциях производных карбоновых кислот стадия конденсации в прин-
принципе протекает аналогичным образом. Некоторые особенности будут рассмот-
рассмотрены ниже.
Общую скорость реакции карбонильного соединения может определять как стадия присо-
присоединения [Г.7.8], так и стадия конденсации [Г.7.10]. При реакциях с сильнонуклеофильными
веществами (аммиак, алифатические амины, гидроксиламин) в нейтральной или щелочной
среде присоединение, как правило, идет быстро и скорость процесса определяется дегидрата-
дегидратацией [Г.7.10]. Так как эта стадия всегда катализируется кислотой, добавление последней уско-
ускоряет реакцию. Однако кислота, играющая роль катализатора, взаимодействует также с нукле-
офильным реагентом, превращая его в соль и тем самым блокируя его свободные электронные
1 Вследствие делокализации электронов такие катионы обладают относительно неболь-
небольшой энергией и занимают центральное положение во всех реакциях карбонильных соедине-
соединений, например в реакции Манниха (см. разд. Г,7.2.1.7).
56 Г Препаративная часть (продолжение)
пары. Чем сильнее основание, вступающее в реакцию, тем ниже концентрация кислоты, при
которой происходит блокирование. Образование солей может настолько замедлить реакцию
присоединения [Г.7.8], что эта стадия станет скоростьопределяющей. Поэтому часто бывает
так, что карбонильная реакция при некотором определенном рН идет быстрее, чем в сильно-
сильнокислой или сильнощелочной среде. При этом оптимальном значении рН происходит смена
стадий, определяющих скорость реакции: с одной стороны, стадия дегидратации [Г.7.10] име-
имеет уже достаточно высокую скорость, с другой стороны, имеется достаточно высокая концент-
концентрация свободного непротонированного нуклеофильного компонента. Обычно такое состоя-
состояние наблюдается в области рН, близкой рКа нуклеофильного агента.
Так, скорость реакции фенола (рКа 10,0) с формальдегидом (см. разд. Г,5.1.8.4) действитель-
действительно максимальна при рН 10 и быстро падает при более низких или более высоких значениях рН.
Подобным же образом скорость взаимодействия семикарбазида (рКа 3,6) с фурфуролом и ацето-
ацетоном максимальна при рН 4. Поэтому для превращения карбонильных соединений в семикарба-
зоны наилучшим реагентом является гидрохлорид семикарбазида в присутствии ацетата натрия,
в то время как сам гидрохлорид дает слишком кислую реакцию. В случае гораздо менее основно-
основного 2,4-динитрофенилгидразина уксусная кислота (рКа 4,76) оказывает лишь слабое каталитичес-
каталитическое действие; здесь эффективными катализаторами являются минеральные кислоты.
7.1.1. РЕАКЦИИ АЛЬДЕГИДОВ И КЕТОНОВ С АМИНАМИ
Альдегиды и кетоны легко реагируют с различными азотистыми основаниями
(см. табл. Г.7.7).
Реакция с наиболее нуклеофильными соединениями, например первичны-
первичными и вторичными аминами (рКа 9—11), протекает обычно и без добавления кис-
кислоты. Продукт присоединения II (схема [Г.7.11]) по названным выше причинам,
как правило, очень неустойчив и чаще всего не может быть выделен. Он перехо-
переходит в карбений-иммониевый ион IV, который может образовать устойчивый ко-
конечный продукт разными путями в зависимости от того, имеет атом азота еще
один протон или нет (схема [Г.7.11]).
Из первичных аминов образуются азометины или основания Шиффа, из вто-
вторичных аминов — енамины. (Почему третичные амины не вступают в реакцию?)
В структуре IV отщепление протона от атома азота протекает обычно гораздо
легче, чем от р-углеродного атома (почему?). Поэтому из первичных аминов
не образуются енамины. Если же имеются более благоприятные условия для
элиминирования протона от углеродного атома, например при возникновении
сопряженной системы связей, то с аммиаком и первичными аминами также об-
образуются енамины; например, из аммиака и ацетоуксусного эфира получают
аминокротоновый эфир (схема [Г.7.12]).
\_ \ _ 1е I _<э \_ I _
NH + С=О ==: —N-C-ОГ ^= N-C-OH
/ / ~ А I /I [Г.7.11а]
I и
Первичный амин:
_ I _ я)*) _ I ©,Н -Н2О / _ / я
R-NH-C-OH + Ни := R-NH-C-Q ~ R-NH-C ^=^ R-N=C +H
I I Н +Н2° ^ \ \ [Г.7.116]
II III IV основание
Шиффа
*' Протонирование атома азота, которое на первый взгляд кажется наиболее вероятным,
приводит лишь к образованию исходных соединений.
7. Реакции карбонильных соединений 57
Вторичный амин:
~?Н + Н@-Н2О. А^-Н R2N /
R2N-C-OH — R2N-C^ =^= С = С + Н [Г.7.11в]
| +Н2О,-Н® ^Л / \
II 1У
енамин
О NH2
Н3С-С-СН2-СООС2Н5 + NH3 Н3С-С=СН-СООС2Н5 + Н2О [Г.7.12]
При взаимодействии вторичных аминов с альдегидами типа бензальдегида или формаль-
формальдегида отпадают обе возможности элиминирования протона. В этих условиях к карбений-им-
мониевому иону присоединяется вторая молекула амина с образованием так называемых
«аминалей» (аминоацеталей), например:
Р ЯН + Н@,-Н2О_ %+NHR2 , гг,„,
Ph-C + NHR2 ^^ Ph-CH . Ph-C - Ph-CH Г.7.13
H NR2 +нгО,-H 2
При повышенной температуре аминали альдегидов с а-водородом отщепля-
отщепляют молекулу амина и превращаются в енамины.
Азометины из альдегидов и анилина (анилы) или из бензальдегида и пер-
первичных аминов, а также оксимы, фенилгидразоны, семикарбазоны (см. табл.
Г.7.7), азины1 и другие подобные соединения относительно устойчивы и могут
быть использованы для выделения, очистки и идентификации карбонильных
соединений. (Напишите схемы получения названных соединений! Почему
синтез л-нитро- и 2,4-динитробензилгидразонов требует добавления кис-
кислоты?)
Имины (из альдегидов и аммиака) и азометины (из алифатических альдегидов и первич-
первичных алифатических аминов) легко полимеризуются или дают продукты альдольной конденса-
конденсации (см. разд. Г,7.2.1.3). Так, например, имин ацетальдегида существует в виде циклического
тримера:
ОН
Н3С-СН=О + NH3 Н3С-СН H3C-CH = NH
VlH2
СН [Г.7.14]
HN NH
н 7 7
н3с й н
1 Азины образуются из гидразина и двух молекул карбонильного соединения и имеют
строение
\ /
C=N-N=C
/ \
58
Г Препаративная часть (продолжение)
В случае формальдегида реакция идет еще дальше — аминогруппы реагируют снова с
молекулами альдегида и аммиака с образованием гексаметилентетрамина (уротропина):
Н2
HN'
i
+ 3 НСНО
N
H2CV
n
Н
N
.CH2
IN
CH2OH
-зн2о
[Г.7.15]
Ф Общая методика получения енаминов (табл. Г.7.16). Смесь 1 моль карбонильного соеди-
соединения, 1,2 моль амина и 0,2 г n-толуолсульфоновой кислоты (в случае р-дикарбониль-
ных соединений в качестве катализатора можно добавить 1 мл 85%-ной муравьиной
кислоты) в 200 мл толуола кипятят с обратным холодильником и водоотделителем. При
проведении реакции с газообразными аминами используют эффективный холодильник, а
амин вводят через боковой тубус колбы. Кипячение заканчивают, когда прекратится выделе-
выделение воды. После охлаждения толуольный раствор для удаления толуолсульфоновой кислоты
дважды встряхивают с небольшим количеством воды (муравьиную кислоту можно не отмы-
отмывать), сушат сульфатом магния, отгоняют растворитель и фракционируют в вакууме.
Синтез может быть проведен и в полумикромасштабе. В этом случае используют неболь-
небольшой градуированный водоотделитель (объемом 1-2 мл) или вообще не измеряют объем азеот-
ропно отгоняющейся воды. Объем растворителя относительно взятого количества реагентов
можно при этом увеличить.
Таблица Г.7.16. Получение енаминов
Продукт реакции
Пирролидиноцикло-
пентен-1
Морфолиноцикло-
пентен-1
Пирролидиноцикло-
гексен-1
Морфолиноцикло-
гексан-1
Пиперидиноцикло-
гексен-1
Этиловый эфир
Р-аминокротоновой
кислоты
Этиловый эфир
р-метиламинокротоновой
кислоты
Этиловый эфир
р-диметиламинокрогоновой
кислоты
Этиловый эфир
Р-анилинокротоновой
кислоты
Этиловый эфир
Р-бензиламинокротоновой
кислоты
4-Аминопентен-3-он-2
4- Бензиламинопентен-
З-он-2
Исходные соединения
Циклопентанон,
пирролидин
Циклопентанон, морфолин
Циклогексанон, пирролидин
Циклогексанон, морфолин
Циклогексанон, пиперидин
Ацетоуксусный эфир, аммиак
Ацетоуксусный эфир,
метиламин
Ацетоуксусный эфир,
диметиламин
Ацетоуксусный эфир,
анилин
Ацетоуксусный эфир,
бензил амин
Ацетилацетон, аммиак
Ацетилацетон, бензиламин
Т. кип. (мм рт. ст.)
или т. пл.,°С
85A0)
107A2)
112A2)
119A0)
113A1)
105A5);
т. пл. 18[B5-форма];
т. пл. 32[(?)-форма]а
106 A6)
122A0)
99@,1)
140 @,5)
114A5);т.пл. 39
183 A7); т. пл. 24
По20
1,5150
1,5121
1,5234
1,5132
1,5144
1,5071
1,5227
1,5822
Выход, %
75
75
75
70
75
85
70
80
80
70
80
а При перегонке образуется низкоплавкая модификация, которая при стоянии переходит в
модификацию с более высокой температурой плавления
7. Реакции карбонильных соединений
59
Енамины из альдегидов: Dulou R., Elkik E., Veillard A. Bull. Soc. Chim. France,
1960, 967.
Получение азометинов или енаминов имеет большое значение в органическом
синтезе. Прежде всего описанным здесь методом можно получить азотистые гете-
роциклы, содержащие азометиновые или енаминные группы, например:
H2N
хиноксалины
[Г.7.17а]
о
о
HN
i
Ph
-2Н2О
ч \
N'
[
Ph
1 -фенилпиразолы
[Г.7.176]
о о
NH3
-2Н2О R
N
Н
пирролы
[Г.7.17в]
Эти реакции могут быть также использованы для идентификации 1,2-,
1,3- и 1,4-дикарбонильных соединений.
(Ознакомьтесь с синтезами тиазолов по Ганчу из амидов тиокарбоновых кис-
кислот и а-галогеноальдегидов!)
Продукты конденсации альдегидов с аммиаком или первичными аминами, прежде всего
гексаметилентетрамин, имеют большое техническое значение в качестве ускорителей вулка-
вулканизации и в производстве фенолформальдегидных смол (см. разд. Г,5.1.8.4). Уротропин при-
применяется также для синтеза бризантных взрывчатых веществ (гексоген, октоген). Важное зна-
значение имеют также пластмассы (аминопласты), получаемые взаимодействием формальдегида
с мочевиной или меламином. Сначала образуются так называемые метилольные соединения
(например, метилолмочевина I в [Г.7.18]), из них получаются цепные полимеры III, которые с
новыми молекулами формальдегида дают трехмерные макромолекулы V, например:
H2N-CO-NH2 —- H2N-CO-NH-CH2OH ¦ H2N-CO-NH-CH2-NH-CO-NH2
(—NH-CO-NH-CH2—)
(—NH-CO-N-CH2—)
CH2OH
IV
(— NH-CO-N-CH2—)x [Г.7.18]
CH2
(—NH-CO-N-CH2—)x
V
Семикарбазоны, различные замещенные фенилгидразоны, анилы и многие
оксимы представляют собой хорошо кристаллизующиеся, плохо растворимые в
воде соединения, поэтому они применяются прежде всего для аналитического
определения и выделения альдегидов и кетонов.
60 Г Препаративная часть (продолжение)
Получение семикарбазонов (общая методика для качественного анализа)
1. Спиртовой раствор ацетата семикарбазида (почему применяют ацетат семикарбази-
да, а не гидрохлорид?). Растирают в ступке 1 г гидрохлорида семикарбазида с 1 г без-
безводного ацетата натрия, переносят смесь в колбу, кипятят с 10 мл абсолютного этано-
этанола и фильтруют горячим.
2. К фильтрату добавляют примерно 0,2 г карбонильного соединения, нагревают от 30
до 60 мин на водяной бане, приливают воду до устойчивого помутнения и оставляют
медленно охлаждаться, при этом выкристаллизовывается семикарбазон. Для очистки
его можно еще раз перекристаллизовать из этанола или водного этанола.
Получение 2,4-динитрофенилгидразонов (общая методика для качественного анализа). К 0,4 г
2,4-динитрофенилгидразина добавляют 2 мл концентрированной серной кислоты, а затем при
перемешивании или встряхивании — по каплям 3 мл воды. К теплому раствору приливают
10 мл 95%-ного этилового спирта. Для получения 2,4-динитрофенилгидразона к этому све-
свежеприготовленному раствору, взбалтывая его, прибавляют 1 мл 10—20%-ного спиртового
раствора карбонильного соединения. Гидразон, как правило, выпадает после 5—10-минутного
стояния (в редких случаях приходится оставить раствор на ночь). Выпавший 2,4-динитрофе-
нилгидразон отсасывают, хорошо промывают водой и перекристаллизовывают из этилацета-
та, диоксана, смеси вода — диоксан или этанола.
У динитрофенилгидразонов существуют стереоизомеры, которые плавятся
при различных температурах, что следует учитывать при просмотре литературы.
Фенилгидразоны являются промежуточными продуктами при синтезе индо-
индолов по Фишеру (см. схему [Г.9.44]). Их можно получить также через соли фенил-
диазония (см. схему [Г.8.34]).
сс-Гидроксиальдегиды и а-гидроксикетоны реагируют с фенилгидразином с
образованием сначала (на холоду) фенилгидразонов, а затем (при нагревании и
избытке реагента) озазонов:
\ ^0 \
i + 3H2NNHPh - i + H2NPh + NH3 + Н2О [Г.7.19]
/ "ОН / ^NNHPh
Получение озазонов используется главным образом для разделения и иден-
идентификации Сахаров. (Почему глюкоза, манноза и фруктоза дают один и тот же
озазон?)
Получение озазонов (общая методика для качественного анализа). Встряхивают 0,5 мл фенил-
гидразина с 0,5 мл ледяной уксусной кислоты и 2 мл воды до образования прозрачного раство-
раствора; образуется ацетат фенилгидразина. К полученному раствору добавляют 0,2 г соответствую-
соответствующего сахара, растворенного в 1 мл воды, и нагревают 30 мин на кипящей водяной бане. Уже
через короткое время начинают выпадать озазоны моносахаридов; озазоны дисахаридов обра-
образуются медленнее. Раствор очень медленно охлаждают, осадок отфильтровывают и перекрис-
перекристаллизовывают из воды или этанола.
Температуры плавления озазонов большинства Сахаров находятся в одной узкой области,
так что их идентификация может быть затруднена. Поэтому необходимо дополнительно конт-
контролировать форму полученных кристаллов, для чего капельку полученного раствора с крис-
кристаллами рассматривают под микроскопом.
Микрофотографии типичных кристаллов озазонов: Hassid W. Z, McGready R. M.
Ind. Engng. Chem., Anal. Ed., 1942, 14, 683-686.
Оксимы имеют низкие температуры плавления и поэтому мало пригодны
для идентификации карбонильных соединений. Однако они являются важными
7. Реакции карбонильных соединений
61
исходными веществами в перегруппировке Бекмана (см. разд. Г,9.1.2.4). Образова-
Образование оксимов также часто используют для количественного определения альдеги-
альдегидов и кетонов путем титрования хлороводорода, выделяющегося в ходе реакции:
\
©
С=0 + H3NOH Cl
0
C = NOH + H20 + HCI
[Г.7.20]
Ф Получение (?)-оксима бензальдегида (табл. Г.7.21). К смеси 0,5 моль альдегида, 125 мл
воды, 25 мл этанола, примерно 200 г льда и 0,55 моль гидрохлорида гидроксиламина быстро
при перемешивании прикапывают 50%-ный раствор 1,25 моль едкого натра. Температуру
поддерживают в пределах 25-30 °С, добавляя лед. Перемешивают 1 ч, затем встряхивают с двумя
порциями по 150 мл эфира. Водный слой доводят до рН 6 при 25—30 "С с помощью
концентрированной соляной кислоты и встряхивают с двумя порциями по 400 мл эфира или
дихлорметана. Объединенные экстракты сушат с помощью СаС1г и упаривают в вакууме.
Оставшееся масло кристаллизуют или перегоняют в вакууме. Продукт перекристаллизовывают из
разбавленного этанола.
Таблица Г.7.21. Е-Оксимы бензальдегидов
Продукт
Бензальдегидоксим
З-Хлорбензальдегидоксим
4-Хлорбензальдегидоксим
З-Нитробензальдегидоксим
2-Метоксибензальдегидок-
сим
4-Трифторметилбензальде-
гидоксим
2,5,6-Триметил бензальде-
бензальдегидоксим
Исходное соединение
Бензальдегид
З-Хлорбензальдегид
4-Хлорбензальдегид
З-Нитробензальдегид
2-Метоксибензальдегид
4-Трифторметилбензаль-
дегид
2,5,6-Триметилбензаль-
дегид
Т. пл., °С
35; т. кип. 118°С
A4 мм рт. ст.)
62-64
106-108
119-120
91-93
100-101
125-127
Выход, %
85
65
86
83
84
67
40
Количественное определение реакционноспособных альдегидов или кетонов (оксимное титрова-
титрование). [Houben-Weyl, Bd. II, 1953, S. 458.]
1. Приготовление раствора реактива. 17,5 г гидрохлорида гидроксиламина растворяют в
50 мл воды и приливают 200 мл н-пропанола. В качестве индикатора добавляют 2 мл 0,1%-ного
раствора бромфенолового синего в 20%-ном этаноле. К полученному желтому раствору по кап-
каплям прибавляют 20%-ный водный раствор гидроксида калия до появления сине-зеленой окрас-
окраски. При добавлении одной капли 0,5 н. соляной кислоты к 20 мл этого раствора цвет его должен
стать желто-зеленым, при добавлении одной капли 0,5 н. раствора едкого натра — синим.
2. Проведение титрования. Пробу вещества, содержащую 0,02-0,03 моль карбонильного
соединения, растворяют в 50 мл раствора реактива, колбу закрывают и оставляют на 30 мин.
Появляется желтая окраска. Титруют 1 н. водным раствором гидроксида натрия до перехода
окраски в иссиня-красную.
Переход часто оказывается нечетким, поэтому необходимо делать холостое титрование.
Для этого 50 мл реактива разбавляют водой до объема, примерно равного объему пробы в кон-
конце анализа, и титруют этот раствор 1 н. раствором гидроксида натрия до той же окраски, что и
основную пробу. Затраченное количество раствора гидроксида натрия вычитают из общего
объема, пошедшего на титрование пробы.
3. Расчет. Расчет проводят по формуле
пМ
% карбонильного соединения =
а-10
где а — навеска, г; л — разность объемов 1 н. гидроксида натрия, затраченных на основной и
холостой опыты; М— молекулярная масса карбонильного соединения.
62 Г Препаративная часть (продолжение)
Из уравнения следует, что этот метод может быть использован также для определения мо-
молекулярной (эквивалентной) массы.
Азометины, оксимы, гидразоны и другие аналогичные соединения подвер-
подвергаются гидролизу по реакциям, обратным их образованию (см. схему [Г.7.11]).
Омыление можно рассматривать как катализируемое кислотами присоединение
воды к гетероаналогу карбонильного соединения. Соединение II в схеме [Г.7.22]
идентично соединению II в схеме [Г.7.11а]:
\ NHR \ NHR ^
=^ V =^= V +Н® ГГ7„,
/ noh2 / чон [Г7-221
©
Наряду с циклогексаноноксимом, исходным соединением в синтезе капролактама (см.
разд. Г,9.1.2.4), важное промышленное значение в синтезе полиамидных волокон имеет также
циклододеканоноксим. Ряд оксимов используется в качестве инсектицидов («Бутокарбоксим»
= 2-О-метилкарбамоилоксим 3-метилтиобутанона) или фармпрепаратов («Обидоксимхло-
рид» — 1,Г-(оксидиметилен)-бисD-формилпиридинийхлорид)диоксим — парасимпатиколи-
тик).
7.1.2. РЕАКЦИИ АЛЬДЕГИДОВ И КЕТОНОВ С ВОДОЙ И СПИРТАМИ
При взаимодействии альдегидов и кетонов с водой неустойчивый первичный
продукт присоединения («гидрат») стабилизируется только путем распада на ис-
исходные компоненты:
ХС=О + НОН ^^ V [Г.7.23]
/ х NOH
Равновесие, как правило, сдвинуто влево, в сторону исходных веществ. Этот
сдвиг оказывается тем больше, чем меньше положительный заряд на атоме угле-
углерода карбонильной группы. Поэтому в отличие от кетонов альдегиды частично
гидратированы в водных растворах; в особенности это относится к очень реак-
ционноспособному формальдегиду. Геминальные диолы (гликоли) с двумя
гидроксильными группами у одного атома углерода, как правило, не могут быть
выделены, также как аминогидроксисоединения II (схема [Г.7.11]).
Группы, проявляющие —/- и — Af-эффекты повышают реакционную способность карбо-
карбонильных соединений и тем самым способствуют образованию гидратов. В отдельных случаях
гидраты становятся настолько устойчивыми, что могут быть выделены как индивидуальные
соединения, например гидраты хлораля, глиоксалевой кислоты, мезоксалевой кислоты, нин-
гидрина. (Рассмотрите эти примеры! Какая из кетогрупп нингидрина подвергается гидрата-
гидратации?)
Аналогично обстоит дело с устойчивостью аминогидроксисоединений и полуацеталей
(см. ниже). Так, известен «альдегидаммиак», образованный хлоралем; эфир глиоксалевой
кислоты и хлораль дают устойчивые полуацетали.
Со спиртами альдегиды и кетоны образуют, часто даже без добавления кис-
кислотных катализаторов, полуацетали [соединение 1 на общей схеме (Г.7.24)].
В присутствии сильной кислоты реакция идет далее до ацеталей:
7. Реакции карбонильных соединений 63
\
С=О + ROH ^=^ С - C-OR ^=^ С© ^= С + Н гг 7 0/11
/ / 4 ® / roh / - / 4 [Г.7.24]
Эта реакция сравнима с образованием аминалей (схема [Г.7.13]).
В данном случае не наблюдается стабилизация карбений-оксониевого иона
II с образованием эфира енола, подобная стабилизации при образовании енами-
нов [схема (Г.7.11в)], так как спирт — слишком слабое основание и поэтому не
может вызвать отщепление протона от р-углеродного атома карбонильного сое-
соединения. Эфиры енолов могут быть получены отщеплением спиртов от ацеталей
в кислой среде (см. табл. Г.3.32) или присоединением спиртов к ацетилену
(см. табл. Г.4.50).
Образование ацеталей из карбонильных соединений и одноатомных спиртов в
присутствии безводных минеральных кислот происходит более или менее гладко
только у альдегидов, поскольку в этом случае равновесие заметно смещено вправо.
Кетоны превращаются этим путем в кетали только с малым выходом или вообще
не превращаются. (Объясните этот факт!) Для того чтобы сдвинуть равновесие, не-
необходимо добавить вещества, связывающие воду. Для получения диэтилацеталей
применяют триэтиловый эфир ортомуравьиной кислоты, который представляет
собой легко гидролизующийся ацеталь (эфира муравьиной кислоты):
OEt р
H-c'-OEt + Н20 ^=^ Н-с' +2ЕЮН [Г.7.25]
OEt OEt
При получении диметилацеталей в качестве связывающего воду вещества
можно брать диметилсульфит. Омыление этого очень чувствительного к
действию воды сложного эфира приводит к выделению диоксида серы, в резуль-
результате чего реакция становится необратимой.
Для образования ацеталей из а,р-ненасыщенных карбонильных соединений требуются
особые условия, поскольку эти соединения могут легко присоединять спирт к активированной
двойной связи с образованием ацеталей р-алкоксикарбонильных соединений.
Ф Общая методика получения диэтилацеталей (табл. Г.7.26). К приготовленному при нагре-
нагревании раствору 1 г нитрата аммония в 0,2 моль абсолютного этанола прибавляют
0,2 моль альдегида или кетона и 0,2 моль триэтилового эфира ортомуравьиной кислоты,
тщательно перемешивают и оставляют без доступа влаги воздуха. Продолжительность
реакции для альдегидов составляет 6-8 ч. Для кетонов вместо нитрата аммония используют
0,1 мл концентрированной соляной кислоты и оставляют на 16 ч.
По окончании реакции отфильтровывают выпавшую соль, фильтрат подщелачивают пи-
пиперидином или пирролидином и перегоняют на колонке. Образовавшийся эфир муравьиной
кислоты перегоняется первым. Если температуры кипения ацеталя и этанола близки, то перед
перегонкой необходимо промыть реакционную смесь разбавленным раствором карбоната
натрия и затем высушить карбонатом калия.
Поскольку при равновесных состояниях прямая и обратная реакции всегда
ускоряются одними и теми же катализаторами, то ацетали и кетали под действи-
действием разбавленных минеральных кислот можно легко снова гидролизовать с обра-
образованием исходных веществ (обратная реакция на схеме [Г.7.24]). Гидролизу
особенно подвержены трудно образующиеся ацетали и кетали, которые часто
настолько чувствительны к действию воды, что их можно использовать в хими-
64
Г Препаративная часть (продолжение)
Таблица Г.7.26. Диацетали,
иной кислоты
Продукт реакции
Диэтилацеталь ацетальдегидаа
Диэтилацеталь пропионового
альдегида11
Диэтилацеталь масляного
альдегида
Диэтилацеталь бензальдегида
Диэтилацеталь акролеина
Диэтилацеталь кротонового
альдегида
Диэтилацеталь тиглинового
альдегида
Диэтилацеталь гексанона-2
Диэтилацеталь ацетофенона
Диэтилацеталь циклогексанона
получаемые с помощью триэтилового эфира ортомуравь-
Исходное соединение
Ацетальдегид
Пропионовый альдегид
Масляный альдегид
Бензальдегид
Акролеин
Кротоновый альдегид
Тиглиновый альдегид
B,3-диметилакролеин)
Гексанон-2
Ацетофенон
Циклогексанон
Т. кип., °С
102
123
114
97A2)
123
146
159
69A8)
112A2)
73A3)
«о20
1,3808
1,3897
1,3965
1,4800
1,4012
1,4097
1,4233
1,4109
1,4805
1,4440
Выход, %
65
70
75
95
75
65
79
75
90
95
а Отмыть спирт (см. методику).
ческих реакциях в качестве водоотнимающих веществ. Относительно устойчивы
к гидролизу ацетали формальдегида.
Эфиры енолов также легко подвергаются кислотному гидролизу. Уравнение
реакции аналогично уравнению [Г.7.11] (см. также схему [Г.4.51]). Циклические
эфиры енолов дигидропиран (см. уравнение [Г.9.20]) и алкоксидигидропиран
(см. табл. Г.4.99) дают при этом соответственно 5-гидроксивалериановый альде-
альдегид и глутаровый диальдегид:
Н2О
ЕЮН +
ЕЮ О
О'
О
[Г.7.27]
Наоборот, в щелочной среде ацетали устойчивы (почему?). Своей устойчи-
устойчивостью к действию щелочных и окисляющих реагентов ацетали значительно
превосходят карбонильные соединения, поэтому образование ацеталей исполь-
используют для временного блокирования карбонильной функции. Для этой цели в
последнее время используют преимущественно этиленацетали или этиленкета-
ли (их называют также 1,3-диоксоланами):
\
с=о +
НО.
V
но
,сн2
-сн2
-СН2
+ Н2О
[Г.7.28]
У циклических ацеталей (кеталей) положение равновесия их образования
значительно благоприятнее, чем при реакции карбонильных соединений с одно-
одноатомными спиртами. Кроме того, диоксоланы гораздо устойчивее к гидролизу.
7. Реакции карбонильных соединений
65
Таблица Г.7.29. Получение этиленацеталей (диоксоланов)
Продукт реакции
Этиленацеталь
бензальдегида
Этиленацеталь
л«-нитробензальдегидаа
Этиленацеталь
циклопентанона
Этиленацеталь
циклогексанона
Этиленацеталь
холестен-5-она-З
Этиленацеталь
метилэтил кетона6
Этиленацеталь
3,3-Диметилбутанона-2
Этиленацеталь
мезитилоксида
Этиленацеталь
ацетоуксусного эфира"
Исходное вещество
Бензальдегид
.м-Нитробензальдегид
Циклопентанон
Циклогексанон
Холестен-4-он-З
Метил этилкетон
3,3-Диметилбутанон-2
(пинаколин)
Мезитилоксид
Ацетоуксусный эфир
Т. кип., "С
(мм рт. ст.)
110A4)
Т. пл. 58 (этанол)
57A8)
73A3)
Т. пл.135
[аЬ^-ЗМЧвСНСЬ)
116
147
156
100A7)
ло20
1,5267
2,4481
1,4583
1,4097
1,4236
1,4396
1,4326
Выход, %
90
95
90
90
80
90
90
85
87
a Перегонять с ксилолом; кристаллизуется непосредственно из промытого и упаренного раствора
при охлаждении до 0 °С.
6 Перегонять с дихлорметаном.
в Реакция с хлоридом железа(Ш) должна быть отрицательной.
Получение ацеталей с соответствующим карбонильным соединением можно
использовать также для защиты группы ОН. Примером может служить важный
в промышленности многостадийный синтез L-аскорбиновой кислоты (витами-
(витамина С) из D-глюкозы или L-сорбозы; в его ходе перед стадией окисления четыре
группы ОН защищают превращением в ацетали с ацетоном (см. [Г.6.43а]).
Получение 1,3-диоксоланов проводят обычно с азеотропной отгонкой воды,
образующейся при реакции. Достаточно гладко, аналогично кетонам, реагиру-
реагируют оксокислоты, их эфиры, аминокетоны (в виде гидрохлоридов), гидроксике-
тоны и а-галогенокетоны.
Выбор растворителя, служащего для азеотропной отгонки воды, зависит от
наиболее выгодной в каждом отдельном случае температуры реакции, а также от
температуры кипения превращаемого в ацеталь вещества. Так, ацетон может
быть превращен в диоксолан при использовании в качестве растворителя дихлор-
метана. В этом случае между водоотделителем и реакционной колбой целесо-
целесообразно поставить дефлегматор.
Ф Общая методика получения этиленацеталей (диоксоланов) (табл. Г.7.29). Кипятят с обрат-
обратным холодильником смесь 1 моль кетона или альдегида, 1,2 моль чистого этиленгликоля,
0,1 г л-толуолсульфоновой кислоты или 85%-ной фосфорной кислоты и 150 мл толуола или
ксилола, хлороформа, трихлорэтилена или дихлорметана. Прибор соединен с водоотдели-
водоотделителем; кипячение продолжают до прекращения поступления в последний воды. После этого смесь
охлаждают, промывают разбавленной щелочью и водой, сушат карбонатом калия и перегоняют.
Синтез может быть проведен и в полумикромасштабе (ср. также разд. Г.7.1.1, получение
енаминов).
66 Г Препаративная часть (продолжение)
Ацетали широко распространены в природе. Так, моносахариды существуют в виде внут-
внутренних полуацеталей, которые в зависимости от величины образовавшегося цикла называют
пиранозами или фуранозами (см. разд. Г,6.5.2). В результате образования ацеталя первый угле-
углеродный атом становится асимметрическим и образуются два стереоизомера, например1:
.ОН
н [Г.7.30]
i5 НО '
н-с-он
I 6
ct-D-глюкоза СНгОН p-D-глюкоза
Конфигурацию гидроксильной группы при Q можно определить с помощью борных эфи-
ров (ср. со схемой [Г.2.56]).
При реакции моносахаридов со спиртами в присутствии кислот получаются циклические
ацетали гликозиды, например:
ОН ОН
ОД Й^Д
НООН НООМе l ' ' '
ct-D-метилглюкозид
Если участвующий в образовании ацеталя спирт сам является углеводом, то образуются
ди-, три- и полисахариды. [Ознакомьтесь со структурой сахарозы (тростниковый, свеклович-
свекловичный сахар) и лактозы (молочный сахар). Почему тростниковый сахар является невосстанавли-
вающим веществом и не дает карбонильных реакций? Ознакомьтесь также с получением маль-
мальтозы и целлобиозы осторожным гидролизом крахмала или целлюлозы, а также с техническим
получением глюкозы из крахмала «осахариванием древесины».]
Ацетали низших альдегидов (диметоксиэтан, сольвеном М) применяют, например, в каче-
качестве растворителей целлюлозы. Ацетали ненасыщенных альдегидов (акролеина) являются
фунгицидами и микробиоцидами. Важными синтетическими пластмассами являются ацетали
полимерных спиртов. Промежуточным слоем в небьющемся стекле триплексе является
ацеталь поливинилового спирта и бутиральдегида. Ацетали высших альдегидов благодаря их
высокой устойчивости к щелочам используют в качестве парфюмерной отдушки для мыла.
Диметилацеталь формальдегида применяют в качестве растворителя в реакциях Гриньяра
(см. разд. Г,7.2.2), для депарафинизации минеральных масел, используемых при получении
смазочных материалов, а также в качестве экстрагента при извлечении из натуральных про-
продуктов. Ацетали ароматических альдегидов используются иногда в качестве отдушки, напри-
например, диметилацеталь фенилуксусного альдегида обладает запахом розы.
7.1.3. РЕАКЦИИ АЛЬДЕГИДОВ И КЕТОНОВ С ОБРАЗОВАНИЕМ ТИОАЦЕТАЛЕЙ
И БИСУЛЬФИТНЫХ СОЕДИНЕНИЙ
По аналогии с ацеталями при взаимодействии альдегидов и кетонов с меркапта-
меркаптанами (тиолами) образуются тиоацетали:
х SR
/ NSR
1 Ознакомьтесь в этой связи с явлением мутаротации. Обратите внимание также на то, что
в конформационной формуле глюкозы все группы (ОН, СНгОН) при втором — пятом угле-
углеродных атомах расположены экваториально. В а-глюкозе ОН-группа при атоме С] аксиальна,
в р-глюкозе — экваториальна.
7.Реакции карбонильных соединений 67
Присоединение происходит значительно легче, чем в случае спиртов, что
объясняется большей нуклеофильностью реагента (см. разд. Г,2.2.2). В то же
время гидролиз тиоацеталей идет труднее. С дитиоэтиленгликолем, например,
тиоэтиленацетали (дитиоланы) образуются настолько легко, что отпадает необ-
необходимость в азеотропной отгонке.
Реакция имеет значение для мягкого восстановления кетонов в углеводоро-
углеводороды, которые образуются из дитиоланов при действии водорода, адсорбирован-
адсорбированного на скелетном никеле Ренея.
\с=0 »hs-<*,-ch2-sh ^
с0 х^
Альдегиды и некоторые кетоны дают с концентрированным водным раст-
раствором гидросульфита натрия так называемые бисульфитные соединения, напри-
/ gi S03
Пространственно затрудненные альдегиды и кетоны, а также ароматические
кетоны не реагируют с гидросульфитом. В гидросульфитах щелочных металлов
атом серы обладает наибольшей нуклеофильностью, поэтому образуются натри-
натриевые соли а-гидроксисульфокислот. Как любые соли, эти соединения в общем
случае хорошо растворяются в воде, несколько меньше в концентрированном
«бисульфитном щелоке» или спирте и совсем не растворяются в эфире.
Образование бисульфитных соединений часто используется для очистки или
отделения альдегидов и кетонов (см., например, табл. Г.5.56 и разд. Г,6.2.2).
Расщепление этих соединений происходит достаточно легко при нагревании
с раствором соды или разбавленной кислотой. При этом необходимо учитывать
возможность взаимодействия между карбонильными соединениями и щелоча-
щелочами или кислотами; чтобы избежать этого, образующийся альдегид или кетон
можно сразу отгонять с водяным паром.
7.1.4. РЕАКЦИИ КАРБОНОВЫХ КИСЛОТ И ИХ ПРОИЗВОДНЫХ
С ГЕТЕРОАТОМНЫМИ НУКЛЕОФИЛАМИ
Особенность реакций карбоксильных соединений с основаниями состоит в том,
что в этих случаях никогда не удается выделить первичный продукт присоедине-
присоединения основания к карбонильной группе (соединение II на схемах [Г.7.29],
[Г.7.30]), так как немедленно происходит конденсация, в результате которой сно-
снова возникают производные кислот. Причиной этого является меньшая внутрен-
внутренняя энергия карбоксильных соединений по сравнению с соответствующими
альдегидами и кетонами, так как у производных кислот карбонильная группа
дополнительно стабилизируется способной к мезомерии соседней группой. Это
приводит к снижению их реакционной способности (за исключением ацилгало-
генидов; см. схему [Г.7.3]). Поэтому образовавшийся в процессе присоединения
основания промежуточный продукт с тетраэдрической структурой еще в боль-
большей степени, чем в случае альдегидов и кетонов, стремится перейти в конечный
продукт с низкой внутренней энергией.
68 Г Препаративная часть (продолжение)
Превращения производных карбоновых кислот могут быть изображены в
соответствии с общими схемами [Г.7.8]—[Г.7.10] следующим образом:
Ч> '?|G -н» <1?'л -хе Р
HNul + R-C == R-C-X =^=^ R-C-X == R-C
I II III
Катализируемая добавлением кислоты реакция идет с собразованием анало-
аналогичного промежуточного продукта
Р'-Ле ?Н -н® 97Н -нх Р
HNul + R-C + HU ^=^ R-C-X =^ R-C-X ^=^ R-C [Г.7.35]
Схемы [Г.7.34] и [Г.7.35] не дают точного представления о механизме превращения проме-
промежуточного продукта II в конечный III. Для заместителей с большой основностью (X = NH2,
ОН, OR) здесь в соответствии со схемой [Г.7.9] сначала происходит протонирование II с пос-
последующим отщеплением НХ и образованием карбений-оксониевого иона:
©/ОН О
-^ ^=^ R-c' +H® [Г.7.35а]
Nu
Слабоосновные галогенид-ионы (X = галоген) могут быть элиминированы без предвари-
предварительного протонирования (см. также ряд [Г.2.3]).
Основания, в частности гидроксид-ионы также могут ускорять реакции про-
производных карбоновых кислот, так как в предшествующей равновесной стадии
реагент HNu переходит в более реакционноспособный анион |Nu~ (например,
спирты в алкоголят-анионы; см. схему [Г.2.65]):
НО® + HNu === Н2О + INu® [Г.7.36]
Реакции свободных карбоновых кислот не могут катализироваться основа-
основаниями, так как образуются карбоксилат-анионы, не обладающие карбонильной
активностью (см. ряд [Г.7.3]):
О О!
R-C' + ОН® R-c; В + Н2О [Г.7.37]
ОН (V
Из тетраэдрического промежуточного продукта (I в схемах [Г.7.34] и [Г.7.35]),
разумеется, может отщепляться и HNu, что соответсвтует обратной реакции. Та-
Таким образом, превращения карбоксильных производных являются типичными
обратимыми реакциями. Положение равновесия зависит от того, с какой ско-
скоростью идут обе конкурирующие реакции II —> I или II -> III (схема [Г.7.38]):
Р ?Н Р
R-C4 + HNu ^= R-C-Nu =^= R-C4 + НХ [Г.7.38]
ЧХ X Nu
I II III
Можно предположить, что преимущественно образуются соединения с
меньшей энергией, т. е. стоящие левее в ряду реакционной способности [Г.7.3].
Так, например, сложный эфир действием аминов можно перевести в амид, в
то время как реакция амидов со спиртами протекает значительно труднее.
7. Реакции карбонильных соединений 69
При взаимодействии особенно реакционноспособных ацилхлоридов или ан-
ангидридов кислот с водой, спиртами и аминами равновесие настолько сдвинуто
вправо, что обратной реакции в обычных условиях не наблюдается. Удается лег-
легко и с большим выходом перевести, например, ацилхлорид в сложный эфир,
однако превратить сложный эфир или амид действием хлороводорода в
ацилхлорид нельзя.
Если различия в реакционной способности исходного вещества I и конечно-
конечного продукта III (схема [Г.7.38]) не очень велики (например, между эфирами кар-
боновых кислот, амидами и самими карбоновыми кислотами), то равновесие не
сдвинуто в значительной степени в одну из сторон. Тогда необходимое карбок-
карбоксильное производное можно получить с высокими выходами, сдвигая равнове-
равновесия в нужную сторону обычными методами, т. е. удаляя продукт реакции или
используя большой избыток реагента.
7.1.4.1. Получение сложных эфиров алкоголизом карбоновых кислот и
их производных
Важнейший метод получения сложных эфиров карбоновых кислот — прямая
этерификация свободных кислот {алкоголш карбоновых кислот).
Вследствие малой активности карбонильных групп в карбоновых кислотах
они, как правило, медленно реагируют со спиртами. Этерификацию можно су-
существенно ускорить, добавляя сильные кислоты (серную кислоту, безводный
хлороводород, сульфоновые кислоты, кислые ионообменные смолы):
R--Q-H + R-C//T> ^ R-C-OH /Н;"Нг°- В-/ [Г.7.39]
6 +Н®,+НгО 40R'
6
R'^©^H
(Почему невозможен катализ основаниями?)
Скорость этерификации карбоновой кислоты, как и следовало ожидать, воз-
возрастает вместе с ростом положительного заряда на карбонильном углероде, т. е.
с ростом кислотности. Так, муравьиная, щавелевая, пировиноградная кислоты
реагируют достаточно быстро и без добавления катализатора.
Сильно влияют на этерификацию стерические факторы. С ростом объема
алкильных остатков, связанных с карбоксильной группой, а также спиртовым
гидроксилом, скорость этерификации падает. Поэтому разветвленные у
а-углеродного атома алифатические кислоты, а также о-замещенные аромати-
ароматические кислоты вступают в реакцию медленно и с плохими выходами. В ряду
от первичных к третичным спиртам реакция также затрудняется; кроме того, в
условиях реакции (сильнокислая среда) параллельно возрастает и тенденция к
превращению спиртов в простые эфиры и олефины (см. разд. Г,2.5 и Г,3.1.1.1).
Поэтому в любых случаях эфиры третичных спиртов получаются прямой
этерификацией лишь с очень малыми выходами.
Согласно изложенному, равновесие реакции этерификации [Г.7.39] не очень
благоприятно для получения сложных эфиров. Равновесие можно сдвинуть
вправо, используя 5—10-кратный избыток более дешевого исходного вещества
(им обычно бывает спирт) либо постоянно удаляя из реакционной смеси про-
продукты реакции — воду или сложный эфир.
70 Г Препаративная часть (продолжение)
В простейшем случае образовавшаяся вода связывается добавленной в каче-
качестве катализатора кислотой (например, серной или хлороводородом). В случае
неустойчивых соединений лучше удалять воду азеотропной отгонкой, посколь-
поскольку в этом случае можно обойтись меньшими количествами и менее агрессивных
катализаторов. Выбор растворителя, с которым отгоняется вода, проводят с уче-
учетом температуры кипения наиболее низкокипящего компонента реакционной
смеси. Для получения этиловых, а также пропиловых эфиров пригодны хлоро-
хлороформ или тетрахлоруглерод. Можно использовать также бензол, однако для не-
него соотношение между водой и отгоняемым спиртом менее выгодно (см. табл.
А.82). Высшие спирты, начиная с бутилового, образуют азеотропные смеси с
водой, так что отпадает необходимость в добавлении специального растворите-
растворителя, уносящего при азеотропной отгонке воду.
При так называемой экстрактивной этерификации образовавшийся эфир
удаляют из реакционной смеси с помощью растворителя, который хорошо раст-
растворяет сложный эфир и плохо — воду. Метод особенно удобен для получения
метиловых эфиров (о получении метиловых эфиров действием диазометана см.
табл. Г.8.40), когда сложно осуществить азеотропную этерификацию, поскольку
метиловый спирт отгоняется в столь больших количествах с растворителем, что
в водоотделителе не происходит разделения фаз.
Ф Общая методика этерификации карбоновых кислот (табл. Г.7.40)
А. Связывание воды водоотнимающими средствами. Смешивают 1 моль карбоновой кис-
кислоты (или 0,5 моль дикарбоновой), 5 моль соответствующего абсолютного спирта (если
спирт дороже кислоты, то берут обратное соотношение или лучше выполняют синтез
по варианту Б) и 0,2 моль концентрированной серной кислоты и кипятят без доступа влаги
воздуха с обратным холодильником 5 ч. В случае менее стойких вторичных спиртов лучше не
применять в качестве катализатора серную кислоту, а насытить кипящую смесь хлороводоро-
хлороводородом и увеличить продолжительность кипячения до 10 ч. После этого отгоняют главную массу
избыточного спирта на колонке Вигре длиной 20 см (Осторожно! Не перегревать остаток!) и
выливают остаток в пятикратный объем ледяной воды. Органический слой отделяют, а вод-
водный трижды экстрагируют эфиром. Объединенные органические слои нейтрализуют конце-
концентрированным раствором карбоната натрия, промывают водой до нейтральной реакции,
сушат хлоридом кальция и перегоняют.
Синтез может быть проведен и в полумикромасштабе.
Б. Азеотропная этерификация. Смешивают 1 моль карбоновой кислоты (соответственно
0,5 моль дикарбоновой), 1,75 мольспирта (не обязательно абсолютного), 5 г концентрирован-
концентрированной серной, толуолсульфоновой, нафталинсульфоновой кислоты или ионообменной смолы в
Н-форме, например амберлита IRA-118 (подготовку смолы см. в части Е), и 100 мл хлорофор-
хлороформа или тетрахлоруглерода. Смесь кипятят с обратным холодильником и водоотделителем,
пока не прекратится выделение воды.
При этерификации гидроксикислот, <х,р-ненасыщенных кислот, а также при этерифика-
этерификации вторичными спиртами лучше не применять серную кислоту, чтобы исключить побочные
реакции (какие?). При использовании ионообменной смолы жидкость необходимо переме-
перемешивать, иначе при нагревании может произойти ее выброс из колбы.
По окончании реакции смесь охлаждают, отмывают водой применявшуюся в качестве ка-
катализатора кислоту, промывают водным раствором гидрокарбоната натрия, еще раз водой,
либо отфильтровывают ионообменную смолу. После этого отгоняют растворитель, который
захватывает с собой примесь воды, остаток перекристаллизовывают или перегоняют.
Синтез можно проводить и в полумикромасштабе.
В. Экстрактивная этерификация. Смешивают 1 моль карбоновой кислоты, 3 моль метано-
метанола на каждую карбоксильную группу, 300 мл тетрахлоруглерода (или 1,2-дихлорэтана, или
трихлорэтилена) и 5 мл концентрированной серной кислоты (в случае менее устойчивых ве-
веществ берут 5 г толуолсульфоновой кислоты или ионообменной смолы; см. выше способ Б) и
7. Реакции карбонильных соединений
71
Таблица Г.7.40. Этерификация карбоновых кислот
Конечный продукт
«-Пропиловый эфир уксусной кислоты
Изопропиловый эфир уксусной кислоты
н- Бутиловый эфир уксусной кислоты
Изобутиловый эфир уксусной кислоты
Этиловый эфир хлоруксусной кислоты
Этиловый эфир р-бромпропионовой
кислоты
Этиловый эфир изомасляной кислоты
Метиловый эфир кротоновой кислоты
Этиловый эфир кротоновой кислоты
Этиловый эфир молочной кислоты
Метиловый эфир пировиноградной
кислоты"
Диэтиловый эфир винной кислоты
Этиловый эфир октановой кислоты
Этиловый эфир декановой кислоты
Этиловый эфир лауриновой кислоты
Этиловый эфир тетрадекановой
(миристиновой) кислоты
Диэтиловый эфир щавелевой кислотыг
Диэтиловый эфир янтарной кислоты
Диэтиловый эфир малеиновой кислоты
Диэтиловый эфир фумаровой кислоты
Диэтиловый эфир адипиновой кислоты
Диметиловый эфир адипиновой кислоты
Диэтиловый эфир себациновой кислоты
Метиловый эфир бензойной кислоты
Этиловый эфир бензойной кислоты
Метиловый эфир салициловой кислоты
Этиловый эфир салициловой кислоты
Диэтиловый эфир фталевой кислотыд
Метиловый эфир «-толуиловой кислоты
Вариант
получения
Б
Б
Ба
Б"
А, Б
А, Б
Б
В
Б
Б6
В
Б6
Б
Б
Б
Б
Б
А, Б
Б
Б
А, Б
В
Б
А, В
А, Б
А, В
А
А
А
Т. кип. (мм рт. ст.)
или т. пл., °С
101
88
126
118
144
67A2)
ПО
120
139
154
65 B0)
138D)
91A5)
125A8)
155A5)
185 B0)
74A1)
103 A4)
108 A2)
95 A0); т. пл. 0,6
138B0)
115A3)
177A2)
83A1)
95A7)
115B0)
105A1)
163A2)
108 A7); т. пл. 33
«о20
1,3843
1,3775
1,3961
1,3900
1,4227
1,4539
1,3869
1,4239
1,4246
1,4125
1,4068
1,4476
1,4176
1,4256
1,4311
1,4365
1,4100
1,4201
1,4413
1,4408
1,4275
1,4297
1,4368
1,5165
1,5057
1,5369
1,5226
1,5019
Выход, %
70
70
85
75
90
85
70
70
70
75
30
80
90
90
75
95
70
90
90
90
90
90
75
90
90
80
60
80
80
а Оставшийся после реакции избыток бутанола образует с эфиром азеотропную смесь, что
осложняет очистку препарата. Поэтому целесообразно брать обратные соотношения спирта и
кислоты. Тогда практически весь спирт вступает в реакцию.
6 В качестве катализатора применяют ионообменную смолу; операцию промывки водой опускают.
" После отделения водной фазы немедленно перегоняют, так как эфир пировиноградной кислоты
очень легко гидролизуется.
г Можно использовать1 кристаллогидрат щавелевой кислоты [в этом случае особенно полезно
применять водоотделитель (рис. А.83, в)]; добавление катализатора не обязательно.
д Можно также исходить из фталевого ангидрида.
72 Г Препаративная часть (продолжение)
кипятят 10 ч с обратным холодильником без доступа влаги воздуха. В случае ароматических
карбоновых кислот применяют трехкратное количество катализатора. Обычно образуются два
слоя; слой меньшего объема содержит воду.
После охлаждения органический слой промывают водой, водным раствором гидрокарбо-
гидрокарбоната и снова водой. Растворитель, служивший для экстракции, отгоняют, а остаток перегоня-
перегоняют или перекристаллизовывают.
(Ознакомьтесь по учебникам с циклическими эфирами гидроксикарбоновых
кислот — лактонами и лактидами.)
Прямую этерификацию карбоновых кислот спиртами можно осуществить в
мягких условиях в присутствии М,1Ч'-дициклогексилкарбодиимида (конденси-
(конденсирующий агент DCC) и 4-диметиламинопиридина (DMAP) в качестве катализа-
катализатора ацилирования. DCC пригоден также для получения амидов из карбоновых
кислот и аминов (см. [Г.7.52]). Для этерификации могут быть использованы
также и другие карбодиимиды.
Синтез этил-трет-бутилового эфира фумаровой кислоты из моноэтилфума-
рата и wpem-бутанола: Neises В., Steglich W., Org. Synth. 1985, 63, 183-187.
Для получения сложных эфиров можно использовать также в качестве
исходных веществ эфиры соответствующей кислоты с другими спиртами.
Подобный алкоголю эфиров карбоновых кислот (переэтерификация) может в от-
отличие от обычной этерификации катализироваться как кислотами, так и осно-
основаниями. (Напишите схемы реакций!) В данном случае также имеют место
типичные равновесные превращения.
Если хотят получить высший эфир карбоновой кислоты, то лучше всего
взять в качестве исходного соединения метиловый эфир карбоновой кислоты и
отгонять из равновесной смеси метиловый спирт (см. табл. Г.7.42, искусствен-
искусственное волокно). Кроме этого частного случая спирт, который желательно ввести в
эфир, всегда необходимо добавлять в избытке. Примеры переэтерификации
приведены в табл. Г.7.177 (среди них получение метилового эфира 4-фенилаце-
тоуксусной кислоты из этилового эфира 2-фенилацетилацетоуксусной кислоты;
напишите схему превращения!)
Из-за весьма повышенной карбонильной активности алкоголиз ангидридов
кислот и ацилхлоридов происходит гораздо легче, чем алкоголиз карбоновых кис-
кислот и их сложных эфиров. Однако и в этом случае кислоты и основания оказы-
оказывают ускоряющее действие на реакцию. Их каталитическое влияние особенно
заметно в случае некоторых менее реакционноспособных ангидридов. В этом
можно убедиться следующим образом:
Растворяют 1 мл уксусного ангидрида в 1 мл абсолютного спирта и измеряют температуру
раствора. Затем стеклянной палочкой вносят каплю концентрированной серной кислоты и
наблюдают, как изменяется температура.
В отличие от прямой этерификации эфиры третичных спиртов могут быть по-
получены с помощью ангидридов кислот и ацилгалогенидов. Ряд активности спиртов
(первичный > вторичный > третичный) остается в силе. Так, например, при полу-
получении тре/я-бутилового эфира уксусной кислоты из уксусного ангидрида и трет-
бутилового спирта необходимо добавлять хлорид цинка (ср. табл. Г.7.41).
Ф Общая методика получения эфиров уксусной кислоты из уксусного ангидрида (табл. Г.7.41).
Смесь 1 моль свежеперегнанного уксусного ангидрида и 1 моль соответствующего безвод-
безводного спирта помещают в круглодонную колбу емкостью 500 мл с обратным холодильником,
7. Реакции карбонильных соединений
73
Таблица Г.7.41. Получение эфиров уксусной кислоты из ацетангидрида
Конечный продукт
Гексиловый эфир уксусной
кислоты
Гептиловый эфир уксусной
кислоты
Октиловый эфир уксусной
кислоты
Циклогексиловый эфир
уксусной кислоты
(-)-Ментюювый эфир уксусной
кислоты
т/>е/я-Бутиловый эфир уксусной
кислотыа
О-Ацетиллактонитрилб
Фениловый эфир уксусной
кислоты
л<-Крезиловый эфир уксусной
кислоты
Ацетилсалициловая кислота"
Ацетат холестерина
Пентаацетил-а- О-глюкозаг
Пентаацетил-р-В-глюкозад
Исходное
соединение
Гексанол
Гептанол
Октанол
Циклогексанол
(—)-Ментол
т/>е/я-Бутанол
Ацетальдегид-
циангидрин
Фенол
м- Крезол
Салициловая
кислота
Холестерин
Глюкоза
Глюкоза
Т. кип. (мм рт. ст.)
или т. пл., °С
62A2)
93A4)
98A5)
64A3)
113A9); [аЬ20 -79,4°
96
64A1)
75(8)
99A3)
Т.пл. 136 E0%-ный
водный диоксан)
Т.пл. 115
Т.пл. 114 (этанол);
[a]D20 +111,6°
(в хлороформе)
Т.пл. 135 (этанол);
[a]DM +5,5°
(в хлороформе)
«о20
1,4104
1,4153
1,4204
1,4429
1,4456
1,3862
1,4027
1,5088
1,5004
Выход, %
80
80
80
80
80
55
75
75
75
85
80
50
55
а Вместо серной кислоты в качестве катализатора использовать 0,3 г безводного ZnCb; перед
перегонкой добавить на кончике шпателя КНСО3.
6 Не применять пиридин!
в Взять 1,2 моль ацетангидрида на 1 моль салициловой кислоты; реакция продукта на хлорид
железа(Ш) должна быть отрицательной.
г Перемешивать 0,1 моль моногидрата глюкозы с 0,8 моль уксусного ангидрида в 1,5 моль
пиридина при 0 °С 20 ч, затем вылить на лед.
д Не применять пиридин! Использовать моногидрат глюкозы; 7 моль уксусного ангидрида; только
5 капель концентрированной серной кислоты смешать с 10 мл уксусного ангидрида, эту смесь
добавлять по каплям при перемешивании. В случае повышения температуры выше 100 °С сразу же
охладить!
закрытым хлоркальциевой трубкой, и добавляют 10 капель концентрированной серной кислоты.
После того как пройдет бурная экзотермическая реакция, нагревают еще 2 ч на кипящей водяной
бане. Смесь охлаждают и выливают в 300 мл ледяной воды. Сложные эфиры, выпадающие в виде
твердого осадка, отфильтровывают и перекристаллизовывают. Жидкие эфиры отделяют, водный
слой дважды промывают дихлорметаном или эфиром. Объединенные органические фазы нейт-
нейтрализуют содовым раствором, промывают водой и сушат сульфатом натрия. Затем отгоняют раст-
растворитель и очищают эфир перегонкой или перекристаллизацией.
При малых загрузках, а также в случае дорогих или чувствительных к кислотам спиртов бо-
более целесообразна методика, основанная на катализе основаниями (щелочной катализ):
Нагревают 3 ч с обратным холодильником 10 ммоль свежеперегнанного уксусного ангид-
ангидрида, 10 ммоль соответствующего абсолютного спирта и 12 ммоль сухого пиридина, затем
74 Г Препаративная часть (продолжение)
выливают в ледяную воду; выделяют продукт, как описано выше. Дополнительной операцией,
необходимой для удаления пиридина, является подкисление 10%-ной соляной кислотой и
промывание ею же органического слоя.
При нагревании фталевого ангидрида или ангидридов замещенных фтале-
вых кислот со спиртами, как и следует ожидать, получаются кислые фталевые
эфиры, которые хорошо кристаллизуются и поэтому используются для иденти-
идентификации спиртов (см. разд. Д,2.2). Третичные спирты не вступают в эту реакцию
или дают олефины.
Титруя кислые фталевые эфиры раствором гидроксида натрия, можно легко
определить молекулярную массу соответствующего спирта.
Особый интерес представляют кислые фталевые эфиры рацемических вто-
вторичных спиртов, поскольку они имеют кислотную группу, которая может реаги-
реагировать с оптически активными основаниями (бруцин, хинин и др.). При этом
образуются пары диастереомеров, различающихся по растворимости и другим
физическим свойствам. Эти различия позволяют сравнительно легко отделить
их друг от друга. Последующим омылением выделяют соответствующие энанти-
омерные спирты.
Определение эквивалента спиртов в виде кислых эфиров 3-нитрофталевой кислоты (общая мето-
методика для количественного анализа)
Получение сложного эфира. Смесь 0,3 мл соответствующего спирта, 0,3 г ангидрида 3-нит-
3-нитрофталевой кислоты и 0,5 мл пиридина нагревают 2 ч на кипящей водяной бане, затем выли-
выливают на лед и подксиляют концентрированной соляной кислотой. Кислый фталат отфильтро-
отфильтровывают или экстрагируют хлороформом. Затем его извлекают из органического слоя раство-
раствором карбоната натрия и снова выделяют подкислением.
Определение эквивалентной массы спирта. Точную навеску 0,20—0,25 г чистого кислого
фталата растворяют на холоду в избытке 0,1 н. NaOH (при нагревании возможно омыление!),
избыток щелочи сразу оттитровывают 0,1 н. соляной кислотой.
Расчет ведут по формуле
Навеска (г) 1000
Эквивалент спирта = 193
Объем NaOH (мл) Нормальность NaOH
Для аналитических целей важно, что алкоголиз ацилхлоридов иногда можно
проводить и в водной среде. Спирт можно выделить из водного раствора непос-
непосредственно в виде производного карбоновой кислоты (реакция Шоттена — Бау-
Баумана). Эту реакцию можно проводить в водной среде лишь с такими ацилхлори-
дами, которые трудно растворимы в воде. В этом случае ацилхлорид экстрагиру-
экстрагирует спирт из водной фазы и реагирует с ним в одной фазе (гомогенно). Конкури-
Конкурирующая реакция между ацилхлоридом и водой или добавленным связывающим
кислоту реагентом идет только на границе раздела фаз и поэтому протекает мед-
медленно. Чтобы избежать омыления образовавшегося эфира, следует всегда рабо-
работать в среде, близкой к нейтральной, т. е. прибавляют щелочь по каплям по мере
того, как она расходуется. Надежным способом избежать омыления эфира явля-
является проведение реакции с безводным спиртом в присутствии пиридина, связы-
связывающего кислоту (реакция Эйнхорна).
Для аналитической идентификации спиртов наряду с уже упомянутыми кис-
кислыми фталатами используют в основном эфиры бензойной, я-нитробензойной
и 3,5-динитробензойной кислот.
7. Реакции карбонильных соединений 75
Получение эфиров бензойной кислоты алкоголизом бензоилхлорида (вариант Шоттена — Баумана,
общая методика для качественного анализа). Растворяют в пробирке или суспендируют 0,5 г спирта
в 5 мл воды, прибавляют каплю раствора метилового красного в ацетоне и 1 мл свежеперегнанно-
го бензоилхлорида. Затем по каплям постепенно прибавляют 5 н. КОН. После прибавления каж-
каждой капли пробирку закрывают пробкой и энергично встряхивают, пока желтая окраска индика-
индикатора не перейдет в красную. Прибавление щелочи и встряхивание продолжают до тех пор, пока не
появится устойчивая желтая окраска и не исчезнет запах бензоилхлорида. Образовавшийся слож-
сложный эфир отсасывают, промывают небольшим количеством воды и перекристаллизовывают.
Жидкие сложные эфиры растворяют в диэтиловом эфире, раствор сушат сульфатом натрия и пере-
перегоняют. Однако жидкие эфиры малопригодны для идентификации спиртов.
По аналогичной методике получают и амиды карбоновых кислот. Можно амин растворить
непосредственно в 10-15 мл 2 н. КОН, а бензоилхлорид добавлять отдельными порциями при
встряхивании.
Получение эфиров карбоновых кислот алкоголизом ацилхлоридов (вариант Эйнхорна, общая ме-
методика для качественного анализа). К смеси 0,5 г соответствующего спирта и 3 мл пиридина
постепенно, при охлаждении льдом прибавляют около 2 г ацилхлорида (бензоилхлорида,
л-нитробензоилхлорида, 3,5-динитробензоилхлорида). Затем реакционную смесь нагревают на
кипящей водяной бане, не допуская проникновения влаги воздуха. В случае первичных и вторич-
вторичных спиртов продолжительность нагревания 10 мин, в случае третичных спиртов — 30 мин. Вмес-
Вместо этого можно оставить реакционную смесь на ночь при комнатной температуре. По окончании
реакции смесь выливают в ледяную воду и осторожно подкисляют концентрированной соляной
кислотой. Эфир, часто выделяющийся в виде масла, промывают водным раствором гидрокарбо-
гидрокарбоната или растирают в нем, затем фильтруют и перекристаллизовывают.
Определение скорости этанолиза замещенных бензоилхлоридов (применение
уравнения Гаммета): Органикум. Практикум по органической химии. — М.:
Мир, 1979, т. II, с. 82.
Эфиры карбоновых кислот являются исходными веществами в следующих
важных реакциях: аминолизе, сложноэфирной конденсации (разд. Г,7.2.1.8),
реакции Гриньяра (разд. Г,7.2.2), восстановлении до спиртов (разд. Г,7.3), пиро-
пиролизе (см. разд. Г,3.2).
В химической промышленности сложные эфиры также имеют большое значение. Некото-
Некоторые важные представители перечислены в табл. Г.7.42. Ознакомьтесь с синтезом указанных в
ней соединений по учебнику! Сложные эфиры являются одним из самых распространенных в
природе классов соединений особенно в виде жиров и масел (см. разд. Г,7.1.4.3).
Моющие вещества в большой своей части представляют собой сложные эфиры длинноце-
почечных карбоновых кислот с длинноцепочечными спиртами. Наиболее важными предста-
представителями этих соединений являются пчелиный и другие виды воска, спермацетовое масло и
масло жожоба, а также аналогичные природные масла, широко используемые в косметичес-
косметических композициях.
7.1.4.2. Получение амидов кислот аминолизом карбоновых кислот и
их производных
Аммиак представляет собой довольно сильное основание и поэтому реагирует с
карбоновыми кислотами с образованием солей:
/9
ОН Ъ
/ /9]
R-c' + NH3 ^^ R-c; e NH? [Г.7.43]
Ъ
76
Г Препаративная часть (продолжение)
Таблица Г.7.42. Применение сложных эфиров
Эфир
Применение
Метиловый эфир уксусной кислоты
Этиловый эфир уксусной кислоты
«-Бутиловый и амиловый эфиры уксусной
кислоты
Бензилацетат
Циннамилацетат
Полисольваны (смеси эфиров спиртов Сб—С7
и уксусной и пропионовой кислот)
Эфиры акриловой и метакриловой кислот
Диэтиловый, дибутиловый и 2-этилгексило-
вый эфиры фталевой кислоты
Гликолевый полиэфир терефталевой кисло-
кислоты (получают переэтерификацией диметило-
вого эфира терефталевой кислоты гликолем;
поликонденсацией)
Винилацетат (поливинилацетат)
Алкидные смолы (полиэфиры на основе ан-
ангидридов фталевой и малеиновой кислот и
этиленгликоля или глицерина)
Ацетилцеллюлоза (из целлюлозы и смеси
уксусного ангидрида с уксусной кислотой)
Эфиры 2,4-дихлор- и 2,4,5-трихлорфенок-
сиуксусной кислоты со спиртами Cg—Cm
Этиловые эфиры замещенных циклопро-
панкарбоновых кислот и др. пиретроиды
Аскорбиновая кислота (у-лактон из 2-оксо-
L-гулоновой кислоты
Ацетилсалициловая кислота
Этиловый эфир /т-аминобензойной кислоты
р-Диэтиламиноэтиловый эфир я-аминобен-
зойной кислоты
Растворитель для эфиров целлюлозы (лаков)
Растворитель для лаков и смол
Растворители для нитроцеллюлозы и смол,
экстрагенты пенициллина
Ароматическое вещество
Ароматическое вещество
Экстрагент фенолов (применяется в очистке
сточных вод)
Основа для получения полимеров
Пластификаторы, в т. ч. для ПВХ
Искусственное волокно (полиэстер)
Пластмассы
Пластмассы, сырье для лаков
Пластмассы (например, для негорючей ки-
кинопленки), искусственное волокно (ацетат-
(ацетатный шелк)
Гербициды
Инсектициды (например,
дельтаметрин (см. [Г.7.113])
Витамин С
циперметрин,
Лекарственные средства (ацесал, аспирин,
микристин)
Местный анестетик (бензокаин)
Лекарственные средства (прокаин), солнце-
солнцезащитные препараты
Поэтому для осуществления реакции с кислотой по типу карбонильной реакции
(аммонолиза)
О О
R-c' + NH3 =^=^ R-c' + Н2О [Г.7.44]
ОН NH2
имеются лишь небольшие концентрации свободного аммиака и свободной кислоты,
входящие в равновесие [Г.7.43]. Таким образом, реакция [Г.7.44] идет относительно
медленно, и необходимо постоянно отводить образовавшуюся в реакции воду, нап-
например, нагреванием аммониевой соли кислоты до более высокой температуры.
Вместо аммиака можно использовать мочевину, которая при повышенной
температуре разлагается на аммиак и изоциановую кислоту. Последняя связыва-
7. Реакции карбонильных соединений 77
ет выделяющуюся при реакции воду и переходит в аммиак и углекислоту. (Напи-
(Напишите уравнение реакции и прочитайте о биуретовой реакции.)
Амиды, особенно при действии водоотнимающих веществ (пентаоксид фос-
фосфора, хлороксид фосфора) при повышенных температурах, могут дегидратиро-
дегидратироваться дальше с образованием нитрилов.
При аминолизе карбоновых кислот действием первичных и вторичных ами-
аминов образуются соответственно моно- и дизамещенные амиды, в то время как
третичные амины не образуют амидов (почему?). Сказанное выше верно и для
этой реакции.
Описываемое ниже получение N-метилформанилида из муравьиной кисло-
кислоты и метиланилина представляет собой простейший случай аминолиза, когда
превращение осуществляется просто азеотропной отгонкой воды из реакцион-
реакционной смеси.
Ф Получение N-метилформанилида. В круглодонной колбе емкостью 1 л смешивают 1 моль
N-метиланилина, 1,2 моль 80—90%-ной муравьиной кислоты и 300 мл толуола. Смесь
нагревают с водоотделителем и обратным холодильником. Между колбой и водоотдели-
водоотделителем ставят колонку Вигре высотой 50 см. Когда прекратится отделение воды, отгоняют
толуол и перегоняют в вакууме остаток. Т. кип. 125 °С A3 мм рт. ст.); nD201,5589; выход 95%.
Амиды кислот, как правило, представляют собой хорошо кристаллизующие-
кристаллизующиеся вещества, легко поддающиеся очистке. Поэтому они служат для идентифика-
идентификации как первичных и вторичных аминов (преимущественно в виде ацетат- и
бензамидов, получаемых из соответствующих ангидридов и ацилхлоридов; см.
ниже), так и карбоновых кислот (в виде незамещенных амидов, анилидов, бен-
зиламидов). Кислоты для этой цели выгодно сначала перевести в ацилхлориды
(см. разд. Г,7.1.4.4) и ввести последние в реакцию с аммиаком или анилином.
В ходе качественного анализа, однако, карбоновые кислоты часто оказываются
в водном растворе. В этом случае рекомендуется получать анилиды по следую-
следующей методике.
Получение анилидов из карбоновых кислот и анилина (общая методика для качественного анализа).
Водный раствор карбоновой кислоты нейтрализуют разбавленным раствором гидроксида нат-
натрия, упаривают досуха, сушат остаток при 105 °С. Примерно 0,5 г хорошо растертой высушенной
натриевой соли карбоновой кислоты нагревают с 0,5 мл анилина и 0,2 мл концентрированной
соляной кислоты (или соответствующим количеством гидрохлорида аналина) в течение 45 мин
при 150-160 °С. После охлаждения растирают с водой, фильтруют и перекристаллизовывают из
воды, водного этанола или диоксана.
Аминолиз карбоновых кислот имеет лишь ограниченное препаративное зна-
значение в лаборатории; основным путем получения амидов является аминолиз
ацилхлоридов, ангидридов и сложных эфиров.
Аммонолиз, аминолиз или гидразинолиз эфиров карбоновых кислот (напишите
схемы реакций!) можно проводить в относительно мягких условиях, так как
здесь невозможно образование солей по схеме [Г.7.43] и полностью проявляется
более высокая (например, по сравнению со спиртами) реакционная способ-
способность аминов. Кроме того, сложноэфирная карбонильная группа более реакци-
онноспособна, чем карбонильная группа кислот.
Реакционная способность основания, участвующего в аминолизе (или аммо-
нолизе), растет по мере увеличения его основности, но падает при повышении
78 Г Препаративная часть (продолжение)
степени разветвленное™ амина. Таким образом, максимальной реакционной
способностью обладают первичные амины с неразветвленной цепью. (Поду-
(Подумайте, какой из двух аминов — анилин или бензиламин — более реакционное -
пособен при аминолизе?)
Реакционная способность сложных эфиров при взаимодействии с аммиаком
и аминами изменяется примерно так же, как их реакционная способность в
реакциях с водой (см. разд. Г,7.1.4.3). Так, фениловые эфиры легче поддаются
аммонолизу, чем метиловые, а последние в свою очередь легче, чем этиловые, в
то время как т/>е/л-бутиловые эфиры совершенно инертны по отношению к
аминам. Практически чаще всего применяют легкодоступные метиловые или
этиловые эфиры.
Сложные эфиры, в которых активность карбонильной группы повышена
введением электроноакцепторных групп (например, эфиры циануксусной и
хлоруксусной кислот), вступают в реакцию аммонолиза особенно легко. Эфиры
(J-оксокарбоновых кислот, однако, часто дают смесь амида и эфира C-аминокро-
тоновой кислоты с преобладанием последнего (см. схему [Г.7.12]). Группы
с —/-эффектом, находящиеся в спиртовом компоненте эфира, также повышают
их активность. Подобные, так называемые активированные эфиры, например
цианометиловые, и-нитрофениловые, нашли применение в синтезе пептидов.
При гидразинолизе сложных эфиров гладко образуются гидразиды карбоно-
вых кислот, например из эфира изоникотиновой кислоты образуется гидразид
изо никотиновой кислоты — важный антитуберкулезный препарат (INH, изони-
азид, неотебен, римифон).
Эфиры р-оксокарбоновых кислот конденсируются с фенилгидразином с за-
замыканием кольца, образуя пиразолоны (см. схему [Г.7.59]). По следующей об-
общей методике можно аналогичным образом получить З-аминопиразолоны-5:
NH NH2
0 NH + 2ЕЮН [Г.7.45]
Е/ HN' 2 Аг
Аг
иТ)) Общая методика получения 3-амино-1-арилпиразолонов-5 (табл. Г.7.46)
JJ Внимание! Арилгидразины очень ядовиты и вызывают болезненную сыпь на
jj коже. Тяга!
В трехгорлую колбу емкостью 500 мл, снабженную обратным холодильником (с хлоркальци-
евой трубкой), капельной воронкой и мешалкой, помещают 100 мл абсолютного метанола,
0,1 моль арилгидразина, 0,11 моль моноимида диэтилового эфира малоновой кислоты и 0,5 мл ле-
ледяной уксусной кислоты и оставляют на 2 ч. За это время следует приготовить раствор метилата
натрия из 150 мл абсолютного метанола и 2,5 г натрия (подробнее см. часть Е). Еще теплый раст-
раствор метилата натрия добавляют по каплям при перемешивании в реакционную смесь.
В заключение кипятят 15 мин с обратным холодильником, охлаждают и упаривают досуха на ро-
ротационном испарителе. Остаток растворяют в 400 мл воды, фильтруют и доводят до рН 7 добав-
добавлением ледяной уксусной кислоты при перемешивании. Выпавший продукт отсасывают, сушат и
перекристаллизовывают из ацетонитрила. Средний выход 80%.
7. Реакции карбонильных соединений
79
Таблица Г.7.46. Получение 3-амино-1-арил-Д2-пиразолонов-5 из гидразинов и моно-
имида диэтилового эфира малоновой кислоты
Пиразолиноны
З-Амино-1-фенил-О2-пиразолон-5
З-Амино-1-D-метилфенил)-Б2-пиразолон-5
З-Амино-1 -C-метоксифенил)-О2-пиразолон-5
З-Амино-1 -D-хлорфенил)-О2-пиразолон-5
З-Амино-1-C-хлорфенил)-02-пиразолон-5
З-Амино-1-B-хлорфенил)-02-пиразолон-5
З-Амино-1-D-бромфенил)-О2-пиразолон-5
Исходное вещество
Фенилгидразин
«-Толил гидразин
м- Метокси-фенил гидразин
я-Хлорфенилгидразин
л«-Хлорфенил гидразин
о-Хлорфенилгидразин
я-Бромфенилгидразин
Т. пл., °С
219
182
173
163
190
154
152
Ф Получение цианацетамида и хлорацетамида (аммонолиз реакционноспособного эфира).
[Корсон Б., Скотт Р., Фозе К. В сб.: Синтезы органических препаратов. Сб. 1. — М.: ИЛ,
1948, с. 498.] В стакан емкостью 1 л, снабженный механической мешалкой, помещают
1 моль метилового или этилового эфира циануксусной кислоты; при перемешивании и охлажде-
охлаждении медленно вливают 1,5 моль концентрированного раствора аммиака (d= 0,9). Охлаждением
ледяной водой поддерживают температуру от 30 до 35 °С. После того как весь аммиак прилит,
перемешивают еще 30 мин при той же температуре, затем охлаждают до 0 °С, причем цианацет-
амид выделяется в виде кристаллов. Их отфильтровывают, промывают небольшим количеством
холодного спирта и эфира, перекристаллизовывают из спирта или воды. Т. пл. 118 "С, выход 80%.
Аналогично может быть получен хлорацетамид из этилового эфира хлоруксусной кисло-
кислоты. Синтез проводят при 0 °С, чтобы предотвратить замещение галогена. Т. пл. 116 "С (вода);
выход 80%.
Получение диамида фумаровой кислоты: Моури Д. Т., Бэтлер Дж. М. В сб.:
Синтезы органических препаратов. Сб. 4. — М.: ИЛ, 1963, с. 364.
Ф Получение N-бензиламидов аминолизом эфиров карбоновых кислот (общая методика для
качественного анализа). Нагревают 1 ч с обратным холодильником 0,5 г соответствующе-
соответствующего метилового или этилового эфира, 1,5 мл бензиламина и 0,05 г хлорида аммония,
охлаждают, промывают водой и небольшими порциями разбавленной соляной кислоты. Обыч-
Обычно бензиламид выделяется в виде твердого осадка. Его перекристаллизовывают из водного эта-
этанола или водного ацетона.
Эфиры высших спиртов сначала кипятят 30 мин с обратным холодильником с 3 мл абсо-
абсолютного метилового спирта и 0,05 г натрия и переводят таким образом в метиловые эфиры.
После отгонки спирта проводят синтез по вышеописанной методике.
2,3,4,6-Тетраацетил-О-глюкозу можно получить частичным аминолизом пентаацетил- D-глюкозы.
АсО
АсО
,ОАс
О
ОАс
+ 2H2NCH2Ph|
ОАс
H ?-NHCH2Ph
Н-С-ОАс
АсО-С-Н
Н-С-ОАс
Н-С-ОН
СН2ОАс
-H2NCH2Ph
АсО
АсО
,ОАс
О
ОН
ОАс
[Г.7.47]
Ф Получение 2,3,4,6-тетраацетил-В-глюкозы частичным аминолизом пентаацетил-В-глюкозы.
[Helferich В., Portz W. Chem. Ber., 1953,86,604.] При хорошем охлаждении водой смешива-
смешивают 0,1 моль а- или р-пентаацетил-О-глюкозы (I) с 0,3 моль бензиламина и энергично
перемешивают в течение 10 мин. Из жидкой вначале реакционной смеси постепенно выделяют-
выделяются кристаллы. Тогда сразу же добавляют при перемешивании 75 мл абсолютного эфира, отсасы-
отсасывают кристаллы и дважды промывают их (порциями по 50 мл) абсолютным эфиром.
80 Г Препаративная часть (продолжение)
Кристаллы представляют собой аддукт бензиламина с 2,3,4,6-тетраацетил-0-глюкозой (II),
в то время как эфирный фильтрат содержит N-ацетилбензиламин и избыток бензиламина.
Для очистки аддукт переосаждают, для чего его растворяют в 100 мл сухого хлороформа,
фильтруют и добавляют затем 250 мл абсолютного эфира. Т. пл. 140 °С (с разл.).
Для получения тетраацетилглюкозы (III) можно исходить непосредственно из неочищен-
неочищенного аддукта. Растворяют его в 500 мл хлороформа, фильтруют и дважды извлекают порциями
по 100 мл 5 н. соляной кислоты. Затем органический слой промывают небольшим количест-
количеством водного раствора гидрокарбоната, сушат хлоридом кальция и выпаривают досуха в ваку-
вакууме (в конце наблюдается сильное пенообразование). Оставшийся сиропообразный продукт
оставляют на 24 ч в эксикаторе над пентаоксидом фосфора, затем растирают со 100 мл эфира,
после чего получают первую порцию кристаллического продукта. Маточный раствор упарива-
упаривают, сушат и повторно растирают с эфиром. Повторяя эти операции несколько раз, получают
кристаллическое р-соединение с 85%-ным выходом. Т. пл. 132 °С (смесь ацетон — эфир).
Для описанного в разд. Г,7.1.7.1 разделения (Л,6)-а-фенилэтиламина на оптические анти-
антиподы можно использовать непосредственно сиропообразный продукт, полученный после
упаривания хлороформа.
Аминолиз ангидридов кислот и ацилхлоридов обычно идет очень легко и явля-
является наиболее удобным способом получения амидов. (Напишите схемы реак-
реакций!) Высвобождающийся хлороводород (или карбоновая кислота при ацили-
ровании ангидридами) связывает эквимолярное количество амина, если не при-
применять пиридина или других третичных аминов или щелочей для связывания
кислоты.
В качественном анализе эту реакцию используют для идентификации карбо-
новых кислот.
Получение амидов карбоновых кислот аминолизом ацилхлоридов (общая методика для качествен-
качественного анализа). Растворяют 0,5 г ацилхлорида в 10 мл безводного диоксана (в случае труднора-
труднорастворимых ацихлоридов можно без опасений взять больше диоксана). К раствору по каплям
добавляют 2 г первичного или вторичного амина, растворенного в 10 мл диоксана, и энергич-
энергично встряхивают.
Для получения незамещенных амидов прибавляют избыток концентрированного водного
раствора аммиака.
Через 10 мин выливают в 100 мл ледяной воды, слабо подкисляют разбавленной соляной
кислотой, отфильтровывают и промывают водой до нейтральной реакции. Продукт реакции
перекристаллизовывают из спирта.
В тех же условиях можно проводить реакцию и с ангидридами карбоновых кислот.
Для получения водорастворимых низших алифатических амидов карбоновых кислот в ди-
оксановый раствор пропускают газообразный аммиак, растворитель упаривают в вакууме,
а остаток перекристаллизовывают из бензола или абсолютного этанола.
Получение полиамида (типа найлона) из хлорангидрида себациновой кислоты
и гексаметилендиамина: Sorenson W. R. J. Chem. Educ, 1965, 42, 8.
Образование амидов карбоновых кислот используют также для идентифи-
идентификации аминов. Для этого применяют рассмотренные в разд. Г,7.1.4.1 реакции
Шоттена—Баумана и Эйнхорна. Из-за трудной гидролизуемости амидов
можно при использовании метода Шоттена—Баумана применять избыток
щелочи.
Подобно ацилхлоридам, азиды также дают при аминолизе амиды карбоно-
карбоновых кислот. Исходные азиды получают из гидразидов действием азотистой кис-
кислоты или из ацилхлоридов действием азида натрия.
Возможно осуществлять также аминолиз амидов кислот, при котором проте-
протекает процесс переамидирования (см. [Г.7.56]).
7. Реакции карбонильных соединений 81
Реакции аммонолиза и аминолиза производных карбоновых кислот имеют
большое значение как в лабораторной практике, так и в промышленности.
Выше уже упоминалось о защите аминогруппы от окисления (разд. Г,5.1.3 и
Г,6.2.1) и об идентификации аминов и карбоновых кислот превращением их в
амиды.
Реакции аминолиза лежат в основе многочисленных способов синтеза раз-
разнообразных азотистых гетероциклов (см. [Г.7.45], [Г.7.56] и [Г.7.59]).
1,3-Оксазолины D,5-дигидрооксазолы) образуются взаимодействием карбоновых кислот
или их производных с р-аминоспиртами, например:
fef [r.,481
Благодаря легко протекающему раскрытию их кольца такие соединения используют как
основу для дальнейших синтезов. Кроме того, образование таких оксазолинов представляет
собой удобный метод защиты карбоксильной группы, поскольку при их кислотном гидролизе
цикл расщепляется, освобождая карбоксильную группу (см. разд. В,8.2).
Аналогичным образом при реакции карбоновых кислот и их производных с а,C-диамина-
ми образуются имидазолины, а при взаимодействии с о-фенилендиамином — бензимидазолы
(см. также [Г.7.57]).
NHHVRfrVB^o |Г-74"
Особое значение описанные здесь методы имеют для синтеза пептидов. Для этого реак-
ционноспособное производное аминокислоты, например соответствующий ацилхлорид,
вводят в реакцию со второй аминокислотой, ее эфиром или пептидом. Чтобы реакция шла в
одном направлении, необходимо защитить как аминогруппу аминокислоты, которая высту-
выступает в качестве ацилируюшего компонента, так и карбоксильную группу другой аминокис-
аминокислоты:
/° /Я ° /°
Я
Z-NH-CH-c' + HzN-CH-c/ _нх • Z-NH-CH-C-NH-CH-c/
R X R' OR" R ft OR"
В качестве защитных групп Z и R" в схеме [Г.7.50] можно использовать только такие замес-
заместители, которые после завершения пептидного синтеза могут быть отщеплены без разрыва
пептидной связи.
В частности, для этого непригодны ацетильные и бензоильные группы. Наиболее прием-
приемлемыми являются, например, бензилоксикарбонильная группа, обычно обозначаемая Z, и
т/>е/я-бутшюксикарбонильная группа (Вое). Группа Z вводится ацилированием бензилокси-
карбонилхлоридом (см. разд. Г,7.1.6):
[Г.7.51а]
о
Ph-CH2-O-C-CI -t
4 v '
¦ H2N-CH
R
0
-C— •
R
0
H-C —
82
Г Препаративная часть (продолжение)
Из образовавшегося таким образом уретана (см. разд. Г,7.1.6) бензилоксикарбонильная
группа, как и все О- и N-бензильные группы, может быть легко удалена, например каталити-
каталитическим восстановлением, в результате чего образуются толуол и диоксид углерода:
О
н
- Ph-CH3 + С02 + H2N-CH-C— [Г.7.516]
R
0 0
и и
Ph-CH2-O-C-NH-CH-C— + Н2
R
Часто применяемая mpem-бутилоксикарбонильная группа (СНз)С—О—СО— в очень мяг-
мягких условиях подвергается гидролитическому отщеплению в кислой среде.
В качестве ацилирующего компонента наряду с ацилхлоридами (X = С1 в схеме [Г.7.50])
используют также азиды аминокислот (X = N3) или смешанные ангидриды с моноэфирами
угольной кислоты, получаемые из аминокислот и алкоксикарбонилхлоридов в присутствии
третичных аминов (см. [Г.7.73]). Часто применяют и активированные эфиры, в частности эфи-
ры л-нитрофенола и N-гидроксисукцинимида (схема [Г.7.50], X = я-нитрофенокси).
Наконец, широко используется прямое взаимодействие карбоновых кислот (X = ОН) в
присутствии карбодиимидов, например дициклогексилкарбодиимида (DCC).
R" + / \—N=C=N-/ \
Z-NH-CH-COOH
R
H2N-CH-COOR"
R
/ \
Z-NH-CH-CO-NH-CH-COOR" +
/ У-NH-CO-NH-/ \
Эти реакции используются также и в промышленности, например, в синтезе антигипер-
тензивного препарата эналаприл на стадии получения L-аланил-Ь-пролина и его бензилового
эфира:
СН3
HN'
I
Вое О
N-трет-
COOCH2Ph
бензиловый
-бутокси- эфир L-пролина
карбонил-Ь-аланин
СН3
""I1 if
Вое О COOCH2Ph
СН3
(DCC) ... ,Д^ X ) (CF3COOH) X ,N
9Нз
Н20
(Н®или НОУ)
EtOOC
О СООН
1_-аланил-1_-пролин
н
эналс
О
сн3
II
О
шрил
-N.
COOCH2Ph
[Г.7.53]
—\
к
СООН
Большое распространение получил искусственный подсластитель аспартам — метиловый
эфир Ь-аспарагил-Ь-фенилаланина.
Метод защиты карбоксильной группы широко используется также в производстве полу-
полусинтетических пенициллинов на стадии ацилирования 6-аминопенициллановой кислоты.
См., например, синтез амоксициллина — 6-[амино-D-гидроксифенил)ацетамидо]пеницилла-
новой кислоты.
NH2HCI
н н
-HCI
NH2 H
¦ I Н Н
COOSiMe3
COOSiMe3
НО
N^fT,s
амоксициллин
[Г.7.54]
СООН
7. Реакции карбонильных соединений 83
Лекарственными препаратами являются также некоторые амиды других карбоновых кислот.
Парацетамол D-гидроксиацетанилид) — из 4-аминофенола и уксусного ангидрида —
анальгетик и жаропонижающий препарат, производится в промышленном масштабе.
Никотинамид (применяют в качестве витамина и для лечения пеллагры) в промышленнос-
промышленности получают как амидированием никотиновой кислоты аммиаком, так и частичным гидроли-
гидролизом 3-цианопиридина (разд. Г,7.1.5).
Пантотеновую кислоту (витамин Bs) получают из пантолактона (разд. Г,7.2.1.3) и р-аланина:
НО О
\ / II
[Г.7.55]
H2N
_Н2о
При взаимодействии формамида с этиловым эфиром аланина образуется N-формильное
производное, циклодегидратация которого под действием Р4О10 приводит к соответствующе-
соответствующему 1,3-оксазолу:
Н3С Н3С НзС\ /0Et
CH-COOEt + H2N-CHO *"С'- CH-CO-OEt
H2N -NH4CI HN
сно
Дальнейшая трансформация этого оксазола (по реакции Кондратьевой) приводит к полу-
получению пиридоксина (витамин В^; см. [Г.4.101]).
Реакция ксантогенатов щелочных металлов (см. [Г.7.92]) с 4-метокси-о-фенилендиами-
ном приводит к получению соответствующего 2-меркаптобензимидазола, являющегося ис-
исходным соединением для синтеза лекарственного препарата омепразол (см. [Г.8.5]).
KS ^
C=S I || 4V-SH + ЕЮН + KSH ГГ.7 571
ЕЮ' ^й
Амиды, образующиеся при аминолизе с участием мочевины (так называемые уреиды),
также имеют значение как лекарственные средства. Важнейшие из них — циклические уреиды
малоновой кислоты, являющиеся производными барбитуровой кислоты:
COOEt H2N
> A )=0 + 2Et0H [Г.7.58]
COOEt H2N V
К барбитуратам относятся известные седативные и снотворные средства: фенобарбитал
(лепинал, люминал; R = Ph, R' = Et); гексобарбитал (эвипан, R = циклогексенил-1, R' = СН3,
один NH заменен на N-CH3).
Другую группу важных фармакологических препаратов получают конденсацией эфиров
оксокислот с фенилгидразином, например феназон (антипирин) из ацетоуксусного эфира.
При этом сначала получается фенилгидразон, который путем внутримолекулярного гидрази-
нолиза сложноэфирной группы преобразуется в гетероцикл. Метилирование образовавшего-
образовавшегося пиразолона дает феназон:
СН3 /СНз /СНз
V
+ -roh,-h2o oV oVh oVch3
HN' 2 Ph Ph Ph [Г.7.59]
' З-метил-1- 2,3-диметил-1-
" фенилпиразолинон-5 фенилпиразолинон-5
(феназон)
84 Г Препаративная часть (продолжение)
Производное феназона, имеющее в положении 4 не метальный, а изопропильный ради-
радикал, представляет собой анальгетик пропифеназон.
Замещенные М-арилпиразолоны-5 являются важнейшими представителями «пурпурного
компонента» в цветной фотографии (см. разд. Г,6.4.3); кроме того, они применяются при про-
производстве азокрасителей в качестве компонента реакции азосочетания (см. разд. Г,8.3.3).
Амиды хлоруксусной кислоты применяются в качестве гербицидов многих растений
{алахлор, метолахлор и др.).
Et Me
СО-СН2С1 ^—/ СО-СН2С1
"К \ /-N [Г.7.60]
СН2ОМе \=^ СНСН2ОМе
Et Et Me
алахлор метолахлор
Наконец, полиамиды очень широко используются в качестве синтетических волокон. При
нагревании соли адипиновой кислоты и гексаметилендиамина (так называемая АГ-соль) в
результате поликонденсации получают волокно найлон 66, а при полимеризации е-капролак-
тама (разд. Г,9.1.2.4) образуется найлон б (капрон, перлон).
7.1.4.3. Гидролиз производных карбоновых кислот
Взаимодействие эфиров и амидов карбоновых кислот с водой протекает, как
правило, медленно даже при нагревании, так как эти вещества проявляют низ-
низкую карбонильную активность (см. [Г.7.3]), а вода имеет малую нуклеофиль-
ность. Напротив, в присутствии сильных щелочей или кислот эфиры и амиды
при нагревании легко омыляются.
Механизм катализируемого кислотами гидролиза такой же, как и механизм
катализируемой кислотами этерификации (см. схему [Г.7.39]). Кислотным гид-
гидролизом пользуются только в тех случаях, когда образующаяся кислота неустой-
неустойчива к щелочи (например, в случае галогензамещенных эфиров жирных кислот).
Чаще используют гидролиз, катализируемый гидроксид-ионами (омыле-
(омыление), так как он идет быстрее: гидроксид-ион участвует в реакции как основание
с большой нуклеофильностью и малым объемом и поэтому присоединяется к
сложному эфиру значительно легче, чем вода:
(=>
'?'л ,91
==^ R-C-OR" - R-Q^ |э + HOR' [Г.7.61]
I II III
Кроме того, последняя стадия реакции (II -» III) необратима (почему?), так
что в щелочной среде равновесие постоянно сдвинуто вправо, в сторону гидро-
гидролиза. В то же время из схемы [Г.7.61] видно, что необходимы по меньшей мере
молярные количества щелочи1.
Сложные эфиры в общем случае омыляются тем легче, чем легче они образу-
образуются, т. е. омыление, как и этерификация, сильно зависит от электрофильной
1 С этой точки зрения выражение «реакция, катализируемая основаниями» неверно,
поскольку катализатор принимает участие и необратимо расходуется в реакции.
7.Реакции карбонильных соединений 85
активности карбонильной группы [мерой ее является кислотность образующей-
образующейся кислоты (почему?)] и от пространственных факторов. Так, скорость омыле-
омыления падает в следующих рядах:
Н3С
H3C-COOR > H3C-CH2-COOR > CH-COOR > H3C-C-COOR
НзС' ™3 [Г.7.62]
СНз СН3
R-СООСНз > R-COOCH2CH3 > R-СООСН > R-COOC-CH3
Особенно чувствительны к гидролизу, как следует из сказанного, метиловые
эфиры сильных кислот, например метиловый эфир щавелевой кислоты, кото-
который уже при комнатной температуре разлагается водой.
В то время как щелочной гидролиз эфиров третичных спиртов идет с большим
трудом, катализируемый кислотами гидролиз протекает против ожидания легко. При
этом вначале возникает протонированный сложный эфир, который далее превраща-
превращается в карбоновую кислоту и обладающий меньшей энергией третичный алкил-кати-
он. Последний в зависимости от условий реакции превращается в третичный спирт
(механизм SN1) и(или) изоолефин (механизм Ei) (см. разд. Г,2 и Г,3), например:
R-C-0-С-СНз R-C-O-С-СНз
СН3 Н СН3 [Г.7.63]
О СН3
и Н-)С.©,СНч +Н2О I ел
R-C-OH + 3 С 3 — Н3С-С-ОН + Н®
i i
СН3 СНз
При омылении эфиров малоновой кислоты (их получение см. в разд.
Г,7.2.1.8—Г,7.2.1.10) следует иметь в виду, что первая эфирная группа омыляется
значительно легче, чем вторая (почему?). Таким путем можно легко получать
моноэфиры малоновой кислоты, как это описано в цитируемой ниже работе.
Еще более это различие выражено в замещенных эфирах малоновой кислоты,
у которых вторая эфирная группа омыляется с большим трудом.
Ф Общая методика омыления замещенных диэтиловых эфиров малоновой кислоты (табл.
Г.7.64). В круглодонной колбе емкостью 1 л с обратным холодильником кипятят 4 ч смесь
1 моль соответствующего эфира, 3,5 моль гидроксида калия, 250 мл воды и 500 мл этано-
этанола. После окончания реакции основную массу спирта отгоняют в слабом вакууме. Остаток
(калиевую соль) растворяют в достаточном количестве воды и при хорошем охлаждении льдом
по каплям добавляют концентрированную соляную кислоту до рН 1. Затем 5 раз экстрагируют
эфиром. Для низших малоновых кислот рекомендуется экстракция в перколяторе (см.
рис. А.91). Объединенные эфирные экстракты промывают небольшим количеством насыщен-
насыщенного раствора хлорида натрия и сушат сульфатом магния. Оставшуюся после отгонки эфира ма-
малоновую кислоту перекристаллизовывают из ацетона, этилацетата или метанола. Выход 70—80%.
Получение монометилового и моноэтилового эфиров малоновой кислоты:
Breslow D. S., Baumgarten E., Hauser С. R. J. Am. Chem. Soc, 1944, 66, 1286.
По такой же методике эфиры замещенных циануксусных кислот можно
омылять до соответствующих циануксусных кислот.
Омыление малоновых эфиров, а также эфиров р-оксокислот (омыление и
декарбоксилирование эфиров р-оксокислот называют также кетонным расщеп-
86
Г Препаративная часть (продолжение)
Таблица Г.7.64. Омыление эфиров замещенных малоновых кислот
Продукт реакции
Этил малоновая кислота
Пропилмалоновая кислота
Бутилмалоновая кислота
Изобутилмалоновая кислота
Амилмалоновая кислота
Гексилмалоновая кислота
Аллилмалоновая кислота
Диэтилмалоновая кислота
Циклопропандикарбоновая-1,1
кислота
Циклобутандикарбоновая-1,1
кислота
Исходное вещество
Диэтиловый эфир этилмалоновой
кислоты
Диэтиловый эфир пропилмалоновой
кислоты
Диэтиловый эфир бутилмалоновой
кислоты
Диэтиловый эфир изобутилмалоновой
кислоты
Диэтиловый эфир амилмалоновой
кислоты
Диэтиловый эфир гексилмалоновой
кислоты
Диэтиловый эфир аллилмалоновой
кислоты
Диэтиловый эфир диэтилмалоновой
кислоты
Диэтиловый эфир циклопропандикарбо-
новой-1,1 кислоты
Диэтиловый эфир циклобутандикарбо-
новой-1,1 кислоты
Т. пл., °С
111
96
101
108
82
106
105(толуол)
127
141 (хлороформ)
158 (этилацетат)
лением) создает большие препаративные возможности, так как образующиеся
малоновые кислоты и E-оксокислоты легко декарбоксилируются и таким обра-
образом открывают путь к получению разнообразных кетонов и карбоновых кислот,
например:
НзС-CO-CH-COOR1
R
COOFT
R-CH
COOR'
гидролиз
н3с-со-сн-соон
I
R
гидролиз
СООН
R-CH
COOFT
СООН
R-CH
СООН
-СО2
H3C-CO-CH2-R [Г.7.65а]
R-CH2-COOH [Г.7.656]
Омыление C-оксоэфиров обычно проводят в слабощелочной или слабокислой
среде. В сильнощелочной среде на первое место выступает конкурирующая реакция,
так называемое кислотное расщепление (см. разд. Г,7.2.1.9). Наоборот, эфиры мало-
малоновой кислоты можно без осложнений омылять в щелочной среде (ср. табл. Г.7.64).
Механизм декарбоксилирования этих кислот соответствует схеме [Г.3.52].
Ацетоуксусные кислоты обычно теряют диоксид углерода при температуре ни-
ниже температуры плавления (иногда ниже 100 °С). Малоновые и циануксусные
кислоты устойчивее и могут быть без труда выделены. Они декарбоксилируются
при температуре выше температуры плавления.
Декарбоксилирование каталитически ускоряется кислотами и слабыми осно-
основаниями (аналин, пиридин). В сильнощелочной среде оно идет существенно
труднее, так как в этих условиях практически вся кислота существует в виде анио-
аниона. Стабилизированные анионы могут образовываться и при декарбоксилирова-
нии в сильнощелочной среде.
7. Реакции карбонильных соединений
87
Таблица Г.7.66. Кетонное расщепление эфиров р-оксокарбоновых кислот
Продукт реакции
Метилпропилкетон
Изобутилметилкетон
Метилам ил кетон
Изоамилметилкетон
Диэтилкетон
4-Фенилбутанол-2
Аллилацетон
DL-и-Ментен-Ьон-З
(пиперитонI1
Исходное соединение
а-Этилацетоуксусный эфир
а-Изопропилацетоуксусный эфир
а-Бутилацетоуксусный эфир
а-Изобутилацетоуксусный эфир
Этиловый эфир а-пропионилпропио-
новой кислоты
а-Бензилацетоуксусный эфир
а-Аллилацетоуксусный эфир
2-Изопропил-2(у-оксобутил)-
ацетоуксусный эфир
Т. кип., °С
(мм рт. ст.)
102
119
151
142
102
116A5)
139
116B0)
«в20
1,3902
1,3956
1,4086
1,4078
1,3922
1,5130
1,4388
1,4848
а Синтез проводят по варианту А с 2 моль щелочи. При этом сначала происходит альдольная
конденсация до соответствующего производного циклогексанона (напишите схему реакции;
ср. разд. Г,7.2.1.3).
Ф Общая методика кетонного расщепления эфиров р-оксокислот (табл. Г.7.66)
А. Щелочное расщепление. В круглодонной трехгорлой колбе емкостью 2 л, снабженной
мешалкой и обратным холодильником, перемешивают 4 ч при комнатной температуре
1 моль соответствующего эфира и 1,5 моль NaOH в виде 5%-ного водного раствора. При
этом сложный эфир гидролизуется, а образовавшаяся кислота частично декарбоксилируется. Для
завершения отщепления СОг кипячение с обратным холодильником продолжают еще 6 ч, затем
реакционную смесь охлаждают и многократно извлекают эфиром. Эфирные вытяжки промыва-
промывают водой, сушат хлоридом кальция, отгоняют эфир, остаток очищают перегонкой.
Б. Кислотное расщепление. В круглодонной колбе емкостью 500 мл, снабженной обратным
холодильником, кипятят 0,1 моль C-оксоэфира с 200 мл 20%-ной соляной кислоты, пока взятая
проба после частичной нейтрализации разбавленным раствором NaOH до рН 2—3 не станет да-
давать отрицательную реакцию на р-оксоэфир с хлоридом железа(Ш). На это требуется 3—6 ч.
После этого реакционную смесь охлаждают, многократно извлекают эфиром, промывают
эфирный экстракт водой и сушат хлоридом кальция. После отгонки эфира кетон перегоняют.
Если в табл. Г.7.66 нет особых указаний, одинаково подходят оба варианта. Выход около 70%.
Обе методики пригодны и для синтеза в полумикромасштабе, а также для обнаружения
р-оксоэфиров при качественном анализе.
Получение 5,5-диметшщиклогександиона-1,3 {димедона) из этилового эфира
2,2-диметил-4,5-диоксоциклогексанкарбоновой кислоты: ShrinerR. L., Todd H. R.
Org. Syntheses, Coll. Vol. II, 1943, p. 200.
Получение фенилацетона (бензилметилкетона) кетонным расщеплением
2-ацетил-2-фенилацетонитрила (его получение см. табл. Г.7.169): Julian P. L.,
Oliver J. J. Org. Syntheses, Coll. Vol. II, 1943, p. 391.
Получение фенилацетона из этилового эфира фенилацетилмалоновой кис-
кислоты: Walker H. G., Hauser С. R. J. Am. Chem. Soc, 1946, 68, 1386.
иТ\ Общая методика декарбоксилирования замещенных малоновых кислот (табл. Г.7.67). Соот-
^^ ветствующую малоновую кислоту (получение см. в табл. Г.7.64, можно использовать
неочищенный продукт) помещают в прибор для перегонки и нагревают на бане до 160—
170 °С; при этом интенсивно выделяется диоксид углерода. Реакцию завершают в уме-
умеренном вакууме D-7 кПа; 30-50 мм рт. ст.), образовавшуюся карбоновую кислоту перегоняют в
вакууме. Затем полученный продукт перегоняют еще раз; выход 80-85%.
88
Г Препаративная часть (продолжение)
Препаративное значение реакции выходит далеко за рамки просто получе-
получения кетонов или карбоновых кислот. В сложных синтезах омыление и декарбок-
силирование замещенных эфиров р-оксокарбоновых кислот и эфиров малоно-
малоновой кислоты часто являются важнейшими стадиями. (Напишите схемы двух
последних реакций в табл. Г.7.67!)
Таблица Г.7.67. Декарбоксилирование замещенных малоновых кислот
Продукт реакции
Масляная кислота
Валериановая кислота
Гексановая кислота
4-Метилпентановая кислота
Гептановая кислота
Октановая кислота
Циклобутанкарбоновая
кислотаа
Бутен-З-карбоновая-1 кислота
Исходное соединение
Этилмалоновая кислота
Пропилмалоновая кислота
Бутилмалоновая кислота
Изобутилмалоновая кислота
Амилмалоновая кислота
Гексилмалоновая кислота
Циклобутандикарбоновая-1,1
кислота
Аллилмалоновая кислота
Т. кип., °С
(мм рт. ст.)
162
96 B3)
102A5)
101A3)
114A3)
129A6)
96A5)
91A6)
«D20
1,3980
1,4080
1,4164
1,4140
1,4236
1,4280
1,4430
1,4283
а Циклопропанкарбоновую кислоту нельзя получить этой реакцией из циклопропандикарбоновой
кислоты, так как циклопропановое кольцо по своим свойствам аналогично двойной связи олефинов,
при декарбоксилировании образуется преимущественно бутиролактон.
Из С-алкилированных N-ациламиномалоновых эфиров (см. разд. Г,8.2.3)
получают а-аминокислоты, например из (З-цианэтилацетиламиномалонового
эфира (см. табл. Г.7.285) — глутаминовую кислоту, а также триптофан (соедине-
(соединение II в схеме [Г.7.68] из скатилацетиламиномалонового эфира I в схеме [Г.7.68]
(синтез I см. схему [Г.7.157]):
COOR
CH2-C-COOR
NHAc
СН2-СН-СООН
NHp
[Г.7.68]
Получение триптофана: SnyderH. R., Smith С. W. J. Am. Chem. Soc, 1944, 66,
350; Нош Е. Е., ZambitoA. J., SnyderH. R, TishlerM. J. Am. Chem. Soc, 1945, 67, 38.
Получение глутаминовой кислоты: Albertson N. F., Archer S. J. Am. Chem. Soc,
1945, 67, 2043.
Щелочное омыление сложных эфиров находит также применение для опреде-
определения эквивалентной массы или так называемого числа омыления эфиров (нап-
(например, при количественном анализе жиров). Числом омыления называют количе-
количество гидроксида калия (в мг), которое необходимо для омыления (гидролиза) 1 г
жира или сложного эфира.
7. Реакции карбонильных соединений 89
Определение эквивалентной массы сложного эфира
1. Получение раствора реактива. При нагревании растворяют 3 г гидроксида калия в 15 мл
чистого диэтиленгликоля (не нагревать выше 130 °С!). Охлажденный раствор разбавляют еще
35 мл диэтиленгликоля. Концентрация полученного раствора равна примерно 1 н. Для точно-
точного определения концентрации отбирают пипеткой 5 мл раствора, разбавляют 10 мл дистилли-
дистиллированной воды и титруют 0,1 н. соляной кислотой по фенолфталеину.
2. Омыление. Отбирают пипеткой точно 10 мл оттитрованной щелочи в колбу Эрленмейе-
ра с притертой пробкой, добавляют 0,4—0,6 г сложного эфира (взвешивать на аналитических
весах!), бросают в колбу кипятильники и устанавливают на колбу обратный холодильник,
снабженный трубкой с натронной известью (для исключения доступа СОг). Перемешивают и
нагревают 15 мин при 120—130 °С. Охлаждают до 80 °С, споласкивают холодильник дистилли-
дистиллированной водой и разбавляют 15 мл воды. Непрореагировавшую щелочь оттитровывают 0,1 н.
НС1 по фенолфталеину.
(Для того чтобы оценить, полностью или нет происходит омыление эфира за выбранное
время реакции, следует для параллельной пробы увеличить время реакции вдвое. Если полу-
полученные значения совпадают, то омыление эфира прошло полностью. В противном случае
необходимо увеличивать время реакции еще несколько раз до получения совпадающих резуль-
результатов двух следующих друг за другом проб.)
Аналогично проводят холостой опыт, во время которого сложный эфир не добавляется.
Объем щелочи, затраченной на холостой опыт, вычитают из количества щелочи, использован-
использованной для омыления эфира.
3. Расчет
Е 1000
х=
nN
где х — эквивалентная масса сложного эфира; Е — навеска, г; л — количество использованной
щелочи, мл.; N— нормальность раствора щелочи.
Методы гидролиза сложных эфиров находят применение в промышленности для омыле-
омыления жиров и масел.
Природные жиры и масла представляют собой сложные эфиры высших жирных кислот с
глицерином, причем обычно на молекулу глицерина приходится три молекулы кислоты
(триглицериды). Из последних чаще всего встречается ненасыщенная олеиновая кислота.
В животных жирах наиболее распространены, кроме того, пальмитиновая и стеариновая кис-
кислоты, а в растительных маслах (соевом, арахисовом и других) — линолевая кислота, имеющая
две двойные связи. Для производства масляных красок и лаков важное значение имеют так на-
называемые высыхающие масла (см. разд. Г, 1.5) (например, льняное и китайское тунговое мас-
масла), которые содержат кроме перечисленных ненасыщенные кислоты с тремя двойными
связями (линоленовую и элеостеариновую). Гидролиз триглицеридов проводится либо под
давлением (только водяной или водяной в присутствии основных катализаторов), либо без
давления в присутствии кислых катализаторов, например так называемого реактива Твитчел-
ла (смеси серной кислоты и бензол- или нафталинсульфоновой кислоты, ацилированной по
Фриделю — Крафтсу олеиновой кислотой; сульфоновая кислота действует как эмульгатор).
Омыление щелочами применяют исключительно для получения мыл — щелочных солей жир-
жирных кислот. Получающийся при омылении глицерин также находит разнообразное примене-
применение, (см. разд. Г,4.1.6).
Восстановлением жирных кислот или их эфиров можно получить жирные спирты, кото-
которые перерабатывают в моющие средства (см. рад. Г.7.3.2). Жирные спирты могут быть также
получены омылением спермацета, состоящего из эфиров ненасыщенных жирных кислот с
цетиловым и олеиновым спиртами.
Гидролиз амидов требует, как правило, более жестких условий, чем гидролиз
соответствующих сложных эфиров (почему?), например кипячения в течение
нескольких часов с концентрированными водными растворами кислот или кон-
концентрированными растворами едких щелочей. Условия проведения реакции
90 Г Препаративная часть (продолжение)
должны быть приблизительно такими же, как и при гидролизе нитрилов, поэто-
поэтому в табл. Г.7.80 вместо нитрилов можно поставить соответствующие амиды.
Гидролиз ангидридов кислот и ацилгалогенидов (галогенангидридов кислот),
как и следует ожидать, протекает очень легко. Особенно это относится к низ-
низшим ацилхлоридам. Которые гидролизуются со значительным выделением теп-
тепла, в то время как труднорастворимые в воде высшие и ароматические ацилхло-
риды реагируют с водой лишь медленно. То же можно сказать и об ангидридах
кислот. Во всех случаях гидролиз можно сильно ускорить щелочами или катали-
каталитическими количествами минеральных кислот.
Эта реакция имеет небольшое практическое значение, поскольку ангидриды
кислоты и ацилгалогениды чаще всего получают из кислот. Особым случаем
является получение перкислот из пероксида водорода. Ниже приводится при-
пример такого синтеза.
Ф Получение пербензойной кислоты [Kergomard A., Philibert-Bigou J. Bull. Soc. Chim. France,
1958, 334.] В круглодонной колбе емкостью 1 л, снабженной мешалкой, термометром и
капельной воронкой, охлаждают до 8 °С раствор 1 моль гидроксида натрия в 175 мл во-
воды. При сильном перемешивании прибавляют при температуре 8—10 °С последователь-
последовательно 30%-ный раствор пероксида водорода (содержащий 0,5 моль Н2О2) и 185 мл 96%-ного эта-
этанола, а затем при 3—5 "С по каплям 37 мл бензоилхлорида. Отсасывают на стеклянном фильт-
фильтре, фильтрат помещают в делительную воронку емкостью 1,5 л, в которой находится 100 мл
эфира и 150 г измельченного льда. Подкисляют 10%-ной серной кислотой по метилоранжу,
разбавляют водой до почти полного растворения выпавшего сульфата натрия. Водный раствор
отделяют и еще два раза извлекают эфиром порциями по 50 мл. Осадок со стеклянного фильт-
фильтра растворяют в 500 мл ледяной воды, фильтруют и обрабатывают так же, как описанный вы-
выше фильтрат. Эфирные растворы объединяют, промывают водой и затем 3 раза по 60 мл
40%-ным раствором сульфата аммония, сушат над сульфатом натрия и хранят в холодильнике.
Перед использованием в дальнейшей работе определяют содержание пероксидов. Для это-
этого к 2 мл раствора прибавляют 10 мл 20%-ного раствора иодида калия, подкисляют и через
10 мин титруют 0,05 н. раствором тиосульфата.
Эфирный раствор можно непосредственно использовать для эпоксидирования (см. разд. Г,4.1.6).
7.1.4.4. Ацидолиз карбоновых кислот и их производных
Карбоновые кислоты также способны реагировать с карбонильными соединени-
соединениями в качестве нуклеофильных агентов. Однако их нуклеофильность невелика.
Поэтому в обычно мягких условиях органических реакций карбоксильная
группа не реагирует с карбоновыми кислотами. При высоких температурах мож-
можно добиться осуществления такой реакции; например, уксусная кислота при
700—900 °С образует ацетангидрид, который сразу же превращается в этих усло-
условиях в кетен (см. схему [Г.3.55]):
[Г.7.69]
Значительно легче из соответствующих дикарбоновых кислот образуются
циклические ангидриды с пяти- и шестичленными кольцами. Так, фталевая
Ме-
-t
Me-
( ОН
¦V
VOH
ч
о
Ме-
* -
Ме-
ое
-(-он
©о-н
ч
л
Me—
Me—
я!
^-ОН;
О
! -Н2О
+ Н2О
Ме-
Ме-
j?
Л
О
X
7. Реакции карбонильных соединений 91
кислота при нагревании до 180 °С переходит в ангидрид. (Напишите схемы
аналогичных реакций для малоновой, янтарной и глутаровой кислот. Почему
нельзя перевести в ангидрид фумаровую кислоту?)
Фталевый и малеиновый ангидриды являются важными техническими полупродуктами
(см. разд. Г,6.5.1).
При взаимодействии карбоновых кислот с галогеноводородными кислотами равновесие,
соответствующее схеме [Г.7.69], настолько сильно сдвинуто в сторону исходных веществ, что
получить этим методом ацилгалогениды невозможно.
Иногда ацидолиз довольно легко удается с эфирами карбоновых кислот,
прежде всего при использовании сильной кислоты. Так, например, смесь мети-
метилового эфира акриловой кислоты с муравьиной кислотой превращается в прису-
присутствии следов серной кислоты в смесь метилового эфира муравьиной кислоты и
акриловой кислоты [Rehberg С. Е. Ogr. Syntheses, Coll. Vol. Ill, 1955, p. 33]:
^fL V/°H ^°Н
+ Vqh = ©o-He^Meovo ^ +°ме [Г.7.70]
M \\ О О M \v
о о
Аналогичная реакция находит применение в качественном анализе. При
нагревании эфиров карбоновых кислот с 3,5-динитробензойной кислотой в
присутствии каталитических количеств серной кислоты получаются 3,5-динит-
робернзоаты соответствующих спиртов, входящих в состав эфиров.
Получение 3,5-динитробензоатов ацидолизом эфиров карбоновых кислот (общая методика для ка-
качественного анализа). Смешивают 0,5 мл соответствующего эфира с 0,5 г тщательно измельчен-
измельченной 3,5-динитробензойной кислоты, прибавляют каплю концентрированной серной кислоты и
нагревают с обратным холодильником 30 мин. В случае высококипящих эфиров нагревают до
150 °С. После охлаждения растворяют смесь в 30 мл эфира, нейтрализуют кислоту, встряхивая с
двумя порциями содового раствора, взятого в избытке (осторожно! сильное выделение диокси-
диоксида углерода, вспенивание!), и промывают водой. Остаток после упаривания эфира растворяют в
минимальном количестве горячего спирта, фильтруют и добавляют воды до первого помутне-
помутнения. При охлаждении выкристаллизовывается эфир динитробензойной кислоты.
Легче всего подвергаются ацидолизу, разумеется, ангидриды кислот и ацилгало-
ацилгалогениды.
Взаимодействие ангидрида кислоты с карбоновой кислотой описывается следующими
равновесиями: о о
О R \ R \ ОН
©о-н +
+ V
он
R4
о
о
R —\
О
/
(V —
VOH
о
о
©о-н
R'—/
О
о
R \
о
R' \ О
©О-Н
R \\
О
0 ^=^ //
о R4
/ R^
Л он
©о-н +
э - .о
о
о [Г.7.71]
92 Г Препаративная часть (продолжение)
Сначала образуется смешанный ангидрид, который подвергается ацидолитической атаке
второй молекулой карбоновой кислоты и переходит, таким образом, в симметричный ангид-
ангидрид. Установление равновесия ускоряется каталитическими количествами минеральных кис-
кислот. Реакция служит для получения ангидридов высших карбоновых кислот или ангидридов
дикарбоновых кислот. В последнем случае она протекает особенно легко.
Чтобы получить достаточно высокий выход продукта, карбоновую кислоту (RCOOH в схе-
схеме [Г.7.71]) необходимо постоянно удалять отгонкой из реакционной смеси. Для этого темпе-
температура кипения кислоты должна быть как можно более низкой по сравнению с образующим-
образующимся ангидридом. Поэтому чаще всего в качестве водоотнимающего средства используют
относительно дешевый ацетангидрид.
Ф Получение 3-нитрофталевого ангидрида [Николет Б., Бендер Дж. В сб.: Синтезы орга-
органических препаратов. Сб. 1. — М.: ИЛ, 1949, с. 318]. В круглодонной колбе с обратным
холодильником смешивают 1 моль 3-нитрофталевой кислоты с 2 моль ацетангидрида,
кипятят с обратным холодильником до растворения кислоты. Выливают в стакан, охлаждают
и смешивают с 150 мл эфира, не содержащего спирта. Кристаллическую массу отфильтровы-
отфильтровывают и перекристаллизовывают. Выход 80%; т. пл. 169 °С (ацетон или бензол).
Реакция ацилгалогенидов с карбоновыми кислотами также может привести к образова-
образованию ангидридов (см. также схемы [Г.7.75] и [Г.7.76]):
на ГГТ7О1
[Г.7.72]
И в этом случае первоначально образуется смешанный ангидрид II (схема [Г.7.72]), кото-
который в условиях реакции в соответствии со схемой [Г.7.716] превращается в симметричный ан-
ангидрид. Чтобы обеспечить высокий выход, необходимо удалять образовавшийся хлороводород
нагреванием равновесной смеси. Чаще всего используют ацетилхлорид, который, например,
переводит янтарную кислоту в ангидрид уже при нагревании с обратным холодильником.
В присутствии пиридина или других третичных оснований, связывающих галогеноводо-
род, реакцию кислот с ацилгалогенидами можно проводить в мягких условиях. Этого же мож-
можно добиться, используя щелочные соли карбоновых кислот. Таким путем можно получать и
смешанные ангидриды1.
Смешанные ангидриды аминокисолт и угольной кислоты имеют важное значение в
пептидном синтезе (см. разд. Г,7.1.4.2):
Ac-NH-CH-COOH + CI-CO-OR + NR3 Ac-NH-CH-CO-O-CO-OR + HNR3 Cl® rp 7 731
R R
Ацилгалогениды могут вступать с карбоновыми кислотами в обменную реакцию, которая,
возможно, протекает по схеме
ГО е ОН
к" Ои он г, /
А0
>он
R'4
R-
R'
О®
-fci
©о-н =^
О
о
О
о
©о-н ^=^ ciVo ^^ + [Г.7.74]
0Н R'4 R4 /Cl
Л Л R4
о о
1 Другой путь получения смешанных ангидридов типа CH3COOCOR состоит в действии
кетена на карбоновые кислоты (см. разд. Г,7.1.6). Смешанные ангидриды легко диспропорци-
онируют на два симметричных (часто даже при осторожной перегонке).
7. Реакции карбонильных соединений 93
Эта реакция становится предпочтительной по сравнению с представленной на схеме [Г.7.72],
если образующийся ацилхлорид (R'COCl) более летуч, чем взятый для реакции (RCOC1), и его
можно удалять отгонкой из равновесной смеси, смещая равновесие [Г.7.74] вправо. Это имеет
место при получении низших ацилхлоридов с помощью бензоилхлорида.
Перевести неустойчивые карбоновые кислоты в соответствующие ацилхло-
риды можно также взаимодействием с оксалилдихлоридом (т. кип. 63—64 °С),
который при этом превращается в СОг, СО и НС1. (Напишите схему реакций!)
Таким способом можно получать, например хлорангидриды кислот стероидно-
стероидного ряда без их рацемизации, при этом не затрагиваются функциональные груп-
группы, такие, как эпокси-, кето-, сложноэфирные, а также части макроцикличес-
ких структур, например порфиринов.
Важнейшим и наиболее общим методом получения ацилхлоридов является,
однако, реакция карбоновых кислот с неорганическими соединениями хлора,
например трихлоридом фосфора, пентахлоридом фосфора и тионилхлоридом:
Р /Р
R-C + РС15 - R-C + РОС13 + HCI [Г.7.75а]
ОН CI
3 R-c" + PCI3 - 3 R-c' + H3PO3 [Г.7.756]
ОН CI
<? /Р
R-C + SOCI2 R-C + HCI + SO2 [Г.7.75в]
ОН Ъ
Реакцию с тионилхлоридом аналогично схеме [Г.7.74] можно описать следу-
следующей схемой:
о о
r-c; _/p _/P r-c;
он R Ч -hci_ R^4 ^ а
,Р ~~ Cl-S'-S0 "+НС1 С1-ЦЯ + ,? [Г.7.76]
CI"S, Cl Ъ %
Cl О
I II
Пентахлорид фосфора — наиболее энергично действующий реагент, однако в
нем используется только один атом хлора. Применяют его обычно лишь в тех
случаях, когда не удается получить ацилхлорид действием трихлорида фосфора
или тионилхлорида.
Реакции с трихлоридом фосфора не отвечают полностью стехиометрическому
уравнению [Г.7.756], так как при отщеплении хлороводорода всегда образуется
некоторое количество смешанного ангидрида. Поскольку избыточный трихло-
рид фосфора (т. кип. 75 °С) можно легко отогнать от образовавшегося ацилхло-
рида, то на практике обычно используют небольшой избыток трихлорида
фосфора.
Тионилхлорид (т. кип. 79 °С) является наименее реакционноспособным реа-
реагентом и всегда применяется в избытке. Для получения очень легколетучих
ацилхлоридов (например, ацетилхлорида) этот реактив малопригоден, так как
94
Г Препаративная часть (продолжение)
образовавшийся летучий ацилхлорид в большом количестве уносится выделяю-
выделяющимися газами — диоксидом серы и хлороводородом, а кроме того, затруднено
и отделение от избытка реактива. Реакционноспособность тионилхдорида
повышается в присутствии каталитических количеств диметилформамида.
Реакции между натриевыми солями карбоновых кислот и оксихлоридом фосфора, тионил-
хлоридом или пентахлоридом фосфора используются лишь в отдельных случаях. Их применя-
применяют, например, для синтеза ацетилхлорида особой чистоты или таких ацилхлоридов, которые
не могут быть перегнаны в присутствии хлороводорода.
Для всех описанных синтезов необходимо использовать совершенно сухие
реактивы и аппаратуру.
Таблица Г.7.77. Получение ацилхлоридов
Продукт реакции
Ацетилхлорида
Трихлорацетилхлорид6
Пропионилхлорида
Бутирил хлорид
Изобутирилхлорид"
Стеароилхлорид
Адипоилдихлорид
Себацоилдихлорид
?-2,3 -Диметилакрил оилхлорид
(тиглиноил хлорид)
Фенилацетилхлорид
Циннамоилхлорид6
Бензоил хлорид
л-Метоксибензоилхлорид
л-Толуоилхлорид
и-Хлорбензоилхлорид
.м-Хлорбензоилхлорид
л-Бромбензоил хлорид
л<-Бромбензоилхлорид
л-Нитробензоилхлорид
.м-Нитробензоилхлорид
3,5-Динитробензоилхлорид
а-Нафтоилхлорид
Вариант
получения
А
Б
А
А, Б
А
Б
А, Б, В
Б
Вг
А, Б
Б, В
А, Б
А, Б
А, Б, В
А, Б, В
А, Б
А, Б
А, Б
А, Б, В
А, Б
Б
А, Б
Т. кип. (мм рт. ст.)
или т. пл., °С
51
118
80
102
165 @,4); т. пл. 24
128A8)
166A1)
61 A50)
96A4)
147 A6); т. пл. 36
71(9)
140A4)
95A0)
ПО A5); т. пл. 16
110A5)
120 A5); т. пл. 42
123A5)
154 A5); т. пл. 72
155 A8); т. п.л. 35
196 A2); т. пл. 67
163 A0); т. пл. 26
«о20
1,3898
1,4695
1,4051
1,4126
1,4079
1,4290
1,5336
1,5537
Выход, %
65
80
80
87
80
80
85
80
75
90
80
80
80
80
80
80
80
80
70
80
70
70
а Берут только 90% теоретического количества трихлорида фосфора (почему?).
6 В качестве катализатора берут несколько капель диметилформамида или пиридина.
" Для очистки перегоняют с эффективной колонкой.
7. Реакции карбонильных соединений 95
^П) Общая методика получения ацилхлоридов (табл. Г.7.77)
§1 Внимание! При реакции образуются хлороводород или его смесь с диоксидом
Щ серы! Работать под тягой!
A. Применение трихлорида фосфора. Смешивают в круглодонной колбе 1 моль карбоновой
кислоты с 0,4 моль трихлорида фосфора (в расчете на каждую карбоксильную группу), нес-
несколько раз встряхивают и оставляют на ночь, защитив от влаги воздуха. Можно вместо этого
нагревать 3 ч на водяной бане при 50 "С с обратным холодильником. Затем жидкость деканти-
декантируют с осевшей на дно фосфористой кислоты и фракционируют. Если температура кипения
ацилхлорида ниже 150 °С (в вакууме или при нормальном давлении), то его можно отогнать, не
отделяя от фосфористой кислоты.
Метод также пригоден для полумикросинтезов.
Б. Применение тионилхлорида. Кипятят 1 моль карбоновой кислоты и 1,5 моль тионилхло-
тионилхлорида (на каждую карбоксильную группу) с обратным холодильником, закрытым хлоркальцие-
вой трубкой, до прекращения выделения газа. Затем избыток тионилхлорида отгоняют на
водяной бане; его можно применять для последующих синтезов. Образовавшийся ацилхлорид
перегоняют (в необходимых случаях в вакууме). Если полученный ацилхлорид хотят исполь-
использовать для дальнейших превращений без очистки, то остатки тионилхлорида удаляют нагрева-
нагреванием смеси на водяной бане в вакууме водоструйного насоса. (Нагревание не должно быть
слишком сильным, чтобы не закипел ацилхлорид!)
Синтез можно проводить в полумикромасштабе.
B. Применение оксалилдихлорида. Растворяют (или суспендируют) 0,05 моль соответствую-
соответствующей кислоты в 100 мл сухого толуола или диэтилового эфира, добавляют 0,06 моль оксалил-
оксалилдихлорида и каплю ДМФА и перемешивают 30 мин при комнатной температуре (для дикарбо-
новых кислот 1-2 ч). Удаляют растворитель на ротационном испарителе (температура бани
около 35 °С), и остаток перекристаллизовывают или перегоняют.
Получение фталилдихлорида из ангидрида фталевой кислоты и пентахло-
рида фосфора: Отт Э. В сб.: Синтезы органических препаратов. Сб. 2. — М.:
ИЛ, 1949, с. 547.
7.1.5. ПРИСОЕДИНЕНИЕ ОСНОВАНИЙ К НИТРИЛАМ
Нитрилы представляют собой азотистые аналоги карбонильных соединений и при-
присоединяют основания с образованием азотистых производных карбоновых кислот:
,N1® ,NH
HNul + R—CHN R-C' R-q [Г.7.78]
^—^ YluH Vlu
© ~
При гидролизе (HNu — вода) таким путем образуются имидокарбоновые кис-
кислоты, сразу же превращающиеся в амиды кислот (схема [Г.7.79]; I -» II). Так как
«карбонильная» активность нитрильной группы невелика (см. схему [Г.7.6]), то
омыление нитрилов удается проводить только при высокой концентрации силь-
сильных кислот (например, концентрированной соляной кислотой или 20—75%-ной
серной кислотой) или в 10-50%-ных растворах гидроксидов щелочных металлов:
НОН + R—CEN + Н® ^^ R-c'
R—C=N
R-C
Ъ [Г.7.79]
Таблица Г.7.80. Получение карбоновых кислот омылением нитрилов
Продукт реакции (кислота)
Валериановая
Гексановая
Гептановая
Тридекановая
Янтарная
Глутаровая
Адипиновая
Фенилуксусная
4- Метоксифенилуксусная
3,4-Диметоксифенил уксусная
(гомовератровая)
2,5-Диметоксифенилуксусная
о-Хлорфенилуксусная
л«-Хлорфенилуксусная
я-Хлорфенилуксусная
о-Бромфенилуксусная
л<-Бромфенилуксусная
и-Бромфенилуксусная
2,4-Диметил фенилуксусная
2,5-Диметил фенилуксусная
2,4,6-Триметил фенил уксусная
а-Нафтилуксусная
Исходное соединение3
Валеронитрил
Амилцианид
Гексил цианид
Додецилцианид
Сукцинонитрил
Глутаронитрил
Адипонитрил
Бензилцианид
4-Метоксибензилцианид
3,4-Диметоксибензилцианид
2,5-Диметоксибензилцианид
о-Хлорбензилцианид
л<-Хлорбензилцианид
л-Хлорбензилцианид
о-Бромбензилцианид
л-Бромбензилцианид
и-Бромбензилцианид
2,4-Диметилбензилцианид
2,5-Диметилбензилцианид
2,4,6-Триметилбензилцианид
а- Нафтилацетонитрил
Вариант
А
А
А
А
А
А
А
А
А
А
Б
Б
А
А
Б
А
А
Б
Б
Б
Б
Т. пл., °С
(мм рт. ст.)
Т. кип. 87A5)
Т. кип. 101A6)
Т. кип. 115A1)
43 (водный этанол)
185 (вода)
98; т. кип. 196A0)
152 (уксусная кислота); т. кип. 205 A0)
78 (вода); т. кип. 144A2)
86
68 (гидрат, из воды)
98 (безводная)
124 (вода)
96 (вода)
78 (вода)
106 (вода)
104 (уксусная кислота)
100 (вода)
116 (вода)
106 (вода)
128 (вода)
168 (спирт или лигроин)
133 (вода)
«d20
1,3952
1,4150
1,4236
В качестве исходного соединения можно с равным успехом воспользоваться соответствующим амидом карбоновой кислоты.
7. Реакции карбонильных соединений 97
В условиях, применяемых для гидролиза нитрилов, амиды обычно сразу гид-
ролизуются до карбоновых кислот (см. разд. Г,7.1.4.3). В определенных услови-
условиях, например под действием концентрированной 96%-ной серной кислоты при
комнатной температуре, гидролиз можно остановить и на стадии амида.
Легкость гидролиза возрастает от нитрилов, содержащих группу ON у тре-
третичного углеродного атома, к нитрилам, содержащим ту же группу у первично-
первичного атома углерода. Особенно трудно гидролизуются нитрилы ароматического
ряда с объемистыми заместителями в o/ww-положении (почему?). В случае нит-
нитрилов, даже в жестких условиях с трудом подвергающихся гидролизу, можно
прибегнуть к обходному пути, используя в реакции вместо слабонуклеофиль-
ной воды очень сильно нуклеофильный сероводород. Полученные таким путем
тиоамиды можно затем гладко гидролизовать как в кислой, так и в щелочной
среде. (Напишите схемы реакций!)
Гидролиз нитрилов чаще всего проводят в кислой среде. Так как условия
такого гидролиза (в частности, концентрация кислоты) сильно меняются в
каждом конкретном случае, ниже приведена методика только для щелочного
гидролиза.
Ф Общая методика гидролиза нитрилов (табл. Г.7.80)
А. Легкогидролизуемые нитрилы. Кипятят с обратным холодильником 1 моль нитрила с
2 моль 25%-ного водного раствора гидроксида натрия до прекращения выделения ам-
аммиака D—10 ч; под тягой!). Чтобы предотвратить кристаллизацию в холодильнике твердых
нитрилов, способных к отгонке с водяным паром, добавляют 80 мл спирта (последний отгоня-
отгоняют по окончании реакции).
Б. Трудногидролизуемые нитрилы. В 400 мл моно-, ди- или триэтиленгликоля растворяют
2 моль гидроксида калия и 1 моль нитрила и нагревают с обратным холодильником до прекра-
прекращения выделения аммиака (примерно 5 ч). Затем разбавляют двойным объемом воды.
Обработка. Водный раствор при охлаждении подкисляют 20%-ной серной кислотой, вы-
выпавшую карбоновую кислоту отфильтровывают, промывают водой и перекристаллизовывают.
Жидкие или растворимые в воде кислоты несколько раз извлекают эфиром. После высушива-
высушивания хлоридом кальция эфир отгоняют, а остаток перекристаллизовывают или перегоняют.
Выход 70-95%.
Гидролиз можно провести и в полумикромасштабе.
сс-Гидроксинитрилы можно гидролизовать только в кислой среде. См.,
например, превращение циангидрина бензальдегида в миндальную кислоту:
Корсон Б. Б. и др. В сб.: Синтезы органических препаратов. Сб. 1. — М.: ИЛ,
1949, с. 270.
Частичный гидролиз нитрила на основном ионообменнике: амид никотино-
никотиновой кислоты из 3-цианопиридина: GalatA. J. Am. Chem. Soc, 1948, 70, 394 (вмес-
(вместо указанного в статье ионообменника IRA-400 можно с успехом использовать
вофатит SBW); малоновая кислота из хлоруксусной через циануксусную: Ветер Н.
В сб.: Синтезы органических препаратов. Сб. 2. — М.: ИЛ, 1949, с. 309.
Нитрилы легко могут быть получены синтезом по Кольбе (см. разд. Г,2.6.9), реакциями ци-
анэтилирования, синтезами с циануксусным эфиром и подобными реакциями (см. схемы
[Г.7.140] и [Г.7.184]). Омыление нитрилов обычно является последней стадией этих синтезов и
поэтому имеет большое препаративное и промышленное значение для получения карбоновых
кислот. Гидрокси- и аминокислоты также могут быть получены через циангидрины (см. разд.
Г,7.2.1.1) или синтезом Штреккера (см. схему [Г.7.109]). Примеры синтезов можно найти в цити-
цитированных разделах.
98 Г Препаративная часть (продолжение)
В присутствии безводного хлороводорода спирты присоединяются к нитри-
нитрилам с образованием гидрохлоридов имидоэфиров («иминоэфиров»):
•-—-N A /NH /NH2l
R'-б-Н + R-C=N + H-CI R-c' CIG R-C'' © Cl0 гГ7й11
У ~\^ „. \kR. J 11.7.81J
^ -^ ©p-Rf OR'
h'
Ф Получение диэтилового эфира моноимида малоновой кислоты. В четырехгорлой колбе
емкостью 1 л с мешалкой, обратным холодильником с хлоркальциевой трубкой, ка-
капельной воронкой и внутренним термометром охлаждают до 5 °С смесь 0,85 моль эти-
этилового эфира циануксусной кислоты, 50 мл эфира, 12,5 мл воды и 50 мл 96%-ного спирта.
К этой смеси при перемешивании приливают по каплям при температуре от 0 до 5 °С 0,8 моль
тионилхлорида (выделение SO2! под тягой!). Затем перемешивают еще 2 ч при 10 °С и 4 ч при
комнатной температуре. Оставляют на ночь в холодильнике, охлаждают смесью сухого льда с
метанолом до —20 "С, отфильтровывают осадок и промывают эфиром.
Для выделения имидоэфира порциями при перемешивании вносят полученный гидрохло-
гидрохлорид имидоэфира в смесь 300 мл воды, 100 г льда и 50 г гидрокарбоната натрия, находящуюся в
стакане емкостью 2 л. Выпавший неочищенный продукт отфильтровывают и растворяют в
100 мл бензола. Фильтруют, сушат сульфатом магния, отгоняют в вакууме растворитель и
фракционируют остаток в высоком вакууме. Т. кип. 80 °С B мм рт. ст.); т. пл. 36 °С (петролей-
ный эфир); выход 80%.
Имодоэфиры легко гидролизуются водой до эфиров карбоновых кислот.
Аминолизом получают амидины:
NH u м_и„ NR"
[Г.7.82]
OR- ¦•- NHR" -^
В отдельных случаях амидины получают непосредственной дегидратацией
соответствующим образом замещенных амидов карбоновых кислот, например:
[Г.7.83]
(ГП) Получение 1,8-диазабицикло[5.4.0]ундецена-7 (схема [Г.7.83]). 0,5 моль N-C-aMHHO-
^^ пропил)-Е-капролактама нагревают с обратным холодильником и водоотделителем с
100 мл ксилола и 1 гтолуолсульфоновой кислоты до полного прекращения образования
воды (примерно 24 ч). Растворитель отгоняют в ротационном испарителе, не отмывая от кис-
кислотного катализатора, остаток перегоняют в вакууме. Т. кип. 128 °С A4 мм рт. ст.); выход 85%.
Получение гидрохлорида ацетамидина из ацетонитрила: Dox Л. W. Org.
Syntheses, Coll. Vol. I, 1956, p. 5.
Некоторые амидины используются в качестве растительных пестицидов, например Амит-
раз [Ы-метиламидо-бис(К-2,3-диметиланилидо)формамидин, или М-метил-бисB,4-ксилил-
иминометил)амин].
При алкоголизе имидоэфиров образуются эфиры ортокарбоновых кислот.
Этим путем можно получить, например, триэтиловый эфир ортомуравьиной
кислоты
©
^Нг _ OEt
Н-С? С1° + 2 ЕЮН - H-C^-OEt + NH4CI [Г.7.84]
OEt OEt
7. Реакции карбонильных соединений 99
7.1.6. ПРИСОЕДИНЕНИЕ ОСНОВАНИЙ К
НЕКОТОРЫМ ОСОБЫМ КАРБОНИЛЬНЫМ СОЕДИНЕНИЯМ
Фосген (его можно рассматривать как хлорангидрид угольной кислоты) вступает
в такие же реакции, что и ацилхлориды. Однако в силу наличия связей двух ато-
атомов хлора с карбонильным углеродом продукты реакции фосгена с основаниями
часто также представляют собой активные карбонильные соединения.
Алкоголиз фосгена сначала приводит к эфирам хлоругольной (хлормуравьи-
ной) кислоты (I, схема [Г.7.85]), которые можно выделить в свободном состоя-
состоянии, а затем к диэфиру угольной кислоты II:
с=0 + r-oh -^ cic Р° ГГ7851
Cl OR RO 11./.85J
В пептидном синтезе, например, бензиловый эфир хлоругольной кислоты (см. схему
[Г.7.51 а, б]) применяется для получения N-защищенных аминокислот.
Хлор- или бромциан, будучи гетероаналогами фосгена, при алкоголизе образуют цианаты:
+ N(C2H5K
Ar-OH + Br-CEEN =-z- Ar-O-CEN [Г 7 861
-[HN(C2H5K]®Br° L1./.BOJ
Реакция гладко протекает с большинством фенолов, однако лишь с немногими алифати-
алифатическими спиртами.
Аминолиз фосгена приводит в зависимости от условий реакции через проме-
промежуточный карбамоилхлорид (схема [Г.7.87], I) к производным мочевины II или
с элиминированием НС1 к изоцианатам III:
Cl
\,_„ R"NH2
c=o
Cl
-HCI
о
R-N-C
А С,
[Г.7.87]
При получении изоцианатов в большинстве случаев на гидрохлориды аминов
действуют фосгеном при нагревании. Таким путем можно в значительной степе-
степени подавить образование мочевин и получить продукт с относительно высоким
выходом. Однако для препаративных целей часто удобнее добавлять свободный
амин при охлаждении к избытку фосгена в растворителе (например, толуоле,
ксилоле, хлорбензоле, а-хлорнафталине). При этом образуется смесь карбамо-
илхлорида и гидрохлорида амина. Реакцию заканчивают при более высокой тем-
температуре, при этом происходит полное растворение промежуточных продуктов.
В промышленности диизоцианаты МДИ (дифенилметан-4,4'-диизоцианат), ТДИ B,4- и
2,6-толуоддиизоцианат) и ГДИ (гексаметилендиизоцианат) получают фосгенированием соотве-
соответствующих диаминов и далее перерабатывают в полиуретан.
В принципе аналогичным образом из тиофосгена можно получать изоциана-
ты (горчичные масла). Так как тиосоединения более устойчивы в воде, чем их
кислородные аналоги, с ними часто можно работать в водных средах.
100
Г Препаративная часть (продолжение)
Таблица Г.7.88. Получение изоцианатов фосгенированием аминов
Продукт реакции
Фенилизоцианат
4-Хлорфенилизоцианат
4-Нитрофенилизоцианат
а-Нафтилизоцианат
Исходное соединение
Анилин
и-Хлоранилин
и-Нитроанилин
1 -Аминонафталин
Растворитель
(т. кип., °С)
а-Хлорнафталин
B60)
Толуол A11)
Толуол A11)
Хлорбензол A32)
Т. кип. (ммрт. ст.),
°С
165
81 A0)
Т. пл. 112 (бензол)
145A5)
Выход, %
80
70
50
70
Общая методика получения изоцианатов (табл. Г.7.88)
Внимание! Фосген очень ядовит! Работать только в специально приспособ-
приспособленных помещениях под тягой!
Выходящие через обратный холодильник газы пропускают последовательно через 4
промывные склянки ["ЦПёГЫПёП из которых вторую заполняют 10%-ным водным
раствором гидроксида калия, четвертую — разбавленным вдвое водным концентриро-
концентрированным раствором аммиака, а пустые первую и третью склянки подсоединяют в обрат-
обратном порядке.
В 400 мл сухого растворителя, охлажденного до температуры -7 "С, растворяют 1 моль обез-
обезвоженного фосгена (пропусканием газообразного фосгена до рассчитанного увеличения мас-
массы). Затем по каплям добавляют также при охлаждении раствор 0,75 моль первичного амина
(или 0,4 моль диамина) в небольшом количестве растворителя таким образом, чтобы темпера-
температура только немного поднялась. После добавления амина снимают охлаждение и продолжают
медленно пропускать сухой фосген. Когда повышение температуры прекратится, нагревают в
токе фосгена до 100 °С. После прекращения выделения НС1 подачу газа прекращают и вытес-
вытесняют избыточный фосген током сухого азота.
Растворитель, кипящий при более низкой температуре, чем основной продукт реакции,
отгоняют в вакууме, а остаток фракционируют или перекристаллизовывают.
Растворитель, имеющий более высокую температуру кипения, чем изоцианат, остается в
колбе, а основной продукт отгоняют в вакууме, и в заключение еще раз фракционируют.
Получение п-хлорфеншшзотиоцианата тиофосгенированием «-хлоранили-
на: Dyson G. M. Org. Syntheses, Coll. Vol. I, 1956, p. 165.
Получение фенилцианата из бромцианата и фенола: Martin D., Bauer M. Org.
Syntheses, 1983, 61, 35.
Карбонильные соединения с кумулированными двойными связями присоединяют
основания с образованием карбоксильных производных:
ОН
Х=С=О + INuH
х=с
NuH
х=с
/Р
нх-с
[Г.7.89]
Nu
Nu
В частности, таким путем диоксид углерода при действии щелочей образует гидрокарбона-
гидрокарбонаты, которые при избытке щелочи переходят в карбонаты:
О=С=О
ею-н
ОН + пне н n °
/ + ОН , - п оО /
о=с _— о=с
+ н2о,-оне У
[Г.7.90]
Аналогично при действии аммиака образуется неустойчивая карбаминовая кислота (схема
[Г.7.91], И), которая в избытке аммиака переходит в соответствующую соль аммония III:
7. Реакции карбонильных соединений 101
NH3
INH3 ^^ О=С =^=
! II III
Из аммониевой соли карбаминовой кислоты III по схеме синтеза амидов из аммониевых
солей карбоновых кислот (схема [Г.7.44]) получают мочевину (в присутствии аммиака при 150 °С
и давлении 3,5 МПа C5 атм). Этот метод широко применяется в промышленности.
Сероуглерод, как сернистый аналог диоксида углерода, относительно легко
присоединяет в щелочной среде спирты и амины, давая соли эфиров дитиоуголь-
ной кислоты (ксантогенаты; схема [Г.7.92]) и дитиокарбаматы (схема [Г.7.93]):
7S/©
с_г_с + НПр Na0H- c-r Мя® О-алкилдитиокарбонат fr ? „-,
S-C-S + HOR - S-C Na натрия («ксантогенат») [1-/.Э2\
OR
Q_,~_Q , u NID Na0H- с_ ' м © N-алкилдитиокарбамат [Г.7.93]
S-C-S + H2NR S-C Na натрия
NHR
(Отчетливо уясните себе ход реакции и роль щелочи!) О-Замещенные дитиокар-
бонаты (ксантогенаты) являются исходными веществами для пиролиза эфиров
по Чугаеву (см. разд. Г,3.2). Дитиокарбаматы могут быть использованы для полу-
получения изотиоцианатов (горчичных масел) R—N=C=S. Ознакомьтесь с этими
реакциями по учебнику!
Изотиоцианаты, а также аналогичным образом построенные изоцианаты
(R—N=C=O) и кетены (R—СН=С=О) представляют собой реакционноспособ-
ные карбонильные соединения, легко присоединяющие воду, спирт, амины и
другие нуклеофильные реагенты. На основе общей схемы [Г.7.89] напишите
следующие важные реакции:
Изоциановая кислота + аммиак -» мочевина (по Велеру)
Изоцианаты + вода -> N-замещенные карбаминовые кислоты
(см. схему [Г.9.27])
-> диоксид углерода + амины
Изоцианаты + спирты -> уретаны
Изоцианаты + аммиак (амины) -» N-замещенные мочевины
Изотиоцианаты + аммиак (амины) -> N-замещенные тиомочевины
Кетен + спирты -> эфиры уксусной кислоты
Кетен + аммиак (амины) -> ацетамиды
Кетен + уксусная кислота -> ацетангидрид
Реакции кетенов и изоцианатов часто протекают очень бурно, в то время как
изотиоцианаты несколько менее реакционноспособны. Так, гидролиз послед-
последних до первичных аминов, диоксида углерода и сероводорода удается лишь при
кипячении с концентрированной соляной кислотой; аналогичная реакция с
изоцианатами идет уже при комнатной температуре при действии воды.
Некоторые из перечисленных реакций имеют и промышленное значение. В больших ко-
количествах производится ксантогенат целлюлозы, из которого получают искусственные шелк и
102 Г Препаративная часть (продолжение)
шерсть вискозным методом. Некоторые дитиокарбаматы представляют собой важные ускори-
ускорители вулканизации каучука, а кроме того, применяются в качестве фунгицидов, например
цинковая соль Ы^-диметилдитиокарбаминовой кислоты (цирам), получаемая из диметила-
мина и дисульфида углерода. Для этих же целей служит получаемый окислением этой соли
дисульфид, так называемый тетраметилтиурамдисульфид (тирам).
Производные тиомочевин используются также в качестве лекарственных препаратов, в
частности для лечения проказы, например, тиамбутоксин (М-я-бутоксифенил-М'-л-диметил-
аминофенилтиомочевина).
При присоединении многоатомных спиртов (например, бутандиола-1,4 или сложных и
простых полиэфиров со свободными гидроксильными группами) к диизоцианатам (например,
2- или 4-метил-л«-фенилендиизоцианату или гексаметилендиизоцианату) образуются полиуре-
полиуретаны, которые применяются в качестве синтетических материалов и пенопластов. Присоедине-
Присоединение уксусной кислоты к кетену, получаемому пиролизом той же уксусной кислоты (см. разд.
Г,7.1.4.4 и схему [Г.3.55]) или ацетона, является важным способом получения ацетангидрида.
Уретаны, мочевины и тиомочевины, как правило, хорошо кристаллизуются
и поэтому часто используются для идентификации спиртов и аминов.
Получение N-фенилуретанов присоединением спиртов к фенилизоцианату (общая методика для
качественного анализа). К раствору 0,5 г фенилизоцианата в 10 мл сухого лигроина (т. кип.
80-100 °С) добавляют 0,3-0,5 г соответствующего спирта (предварительно тщательно обезво-
обезвоженного) и 5 мл того же растворителя. По окончании реакции нагревают еще 1—3 ч на кипя-
кипящей водяной бане, фильтруют горячим и охлаждают. Осадок промывают холодным петролей-
ным эфиром и перекристаллизовывают из петролейного эфира или тетрахлоруглерода.
Аналогичным образом из а-нафтилизоцианата получают ос-нафтилуретаны.
Получение замещенных тиомочевин присоединением первичных или вторичных аминов к фенили-
зотиоцианату (общая методика для качественного анализа). Растворяют 0,2 г амина в 5 мл спир-
спирта и прибавляют раствор 0,2 г фенилизотиоцианата в 5 мл спирта. Если реакция не идет при
комнатной температуре, реакционную смесь слегка подогревают в течение 1-2 мин. Если при
последующем охлаждении и трении стеклянной палочкой о стенку стакана кристаллы не вы-
выпадают (это бывает в случае ароматических аминов), снова нагревают в течение 10 мин или
проводят реакцию сначала без растворителя, а затем осаждают продукт 50%-ным водным
спиртом. Тиомочевины перекристаллизовывают из спирта.
7.1.7. ТИОНИРОВАНИЕ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
Карбонильные соединения можно превратить в тиокарбонильные соединения
взаимодействием с пентасульфидом фосфора (P4Si0) в присутствии оснований,
например пиридина или гидрокарбоната натрия:
о s
,,<Х , ^CN , Я' = алкил,арил,ОЙ, NH2, NHR, NR2 [Г.7.94]
R R' R Rf
Таким путем особенно легко превратить ароматические кетоны, эфиры и амиды
карбоновых кислот в соответствующие тиокетоны, тиоэфиры и тиоамиды.
Недавно было найдено, что при реакции анизола с пентаоксидом фосфора с
высоким выходом образуется 2,2-бисD-метоксифенил)-1,3,2,4-дитиадифосфе-
тан-2,4-дисульфид [Г.7.95], названный реактивом Лауссона. С помощью этого
реагента возможно легко осуществлять тионирование карбонильных соедине-
соединений, поскольку он более реакционноспособен, чем P4S[0, и при повышенных
температурах прекрасно растворяется в органических растворителях, что позво-
позволяет проводить тионирование в гомогенной фазе.
7. Реакции карбонильных соединений
103
4PhOMe + P4S10
ОМе
[Г.7.95]
Ниже приведен наиболее вероятный механизм взаимодействия реактива Лауссона с карбо-
карбонильными соединениями:
МеО
МеО
МеО
ЗМеО
о S
11 ч
s ь
P-S
I I
O-C-R'
i
R
ОМе
2 МеО
О
МеО
[Г.7.96]
S44 ,С6Н4ОМе
Р
МеОС6Н4-р
S
p
\
C6H4OMe
(ГП) Общая методика получения амидов тиокарбоновых кислот (табл. Г.7.97)
А. 2,2-бисD-метоксифенил)-1,3,2,4-дитиодифосфетан-2,4-дисульфид (реактив Лауссона).
JJ Осторожно! Выделение сероводорода! Работать под тягой!
В круглодонной колбе (на 250 мл) с обратным холодильником и хлоркальциевой трубкой наг-
нагревают 1 моль анизола с 0,1 моль P4Si0 в течение 6 ч на водяной бане при температуре 155 °С до
получения гомогенного раствора. После охлаждения до комнатной температуры продукт выкрис-
выкристаллизовывается. Его отфильтровывают, промывают смесью эфира с дихлорметаном A:1) и
сушат в вакууме над пентаоксидом фосфора. Выход 80%, т. пл. 227—229 °С. Частично плавится
уже при 214 °С. В эксикаторе над пентаоксидом фосфора продукт может сохраняться до 10 дней.
Б. Получение амидов тиокарбоновых кислот
В круглодонную колбу на 100 мл помещают 10 ммоль соответствующего амида карбоновой
кислоты и 5 ммоль реактива Лауссона в 15 мл диметилового эфира этиленгликоля и получен-
полученный раствор нагревают на водяной бане, в случае первичных амидов — при 80 "С, а в случае
вторичных или третичных амидов — при 100 °С в течение времени, указанного в табл. Г.7.97.
Реакционную массу охлаждают, выливают в 50 мл воды и четырежды экстрагируют порциями
по 25 мл эфира. Объединенные экстракты сушат безводным сульфатом магния и упаривают на
роторном испарителе. Если температура плавления продукта недостаточно высока, то его мож-
можно очистить, растворив в смеси эфир/ацетон A : 1) и пропустив через колонку с силикагелем.
Таблица Г.7.97. Амиды тиокарбоновых кислот
Продукт
Тиоформанилид
Тиобензамид
Тиобензанилид
4-Хлортиоацета-
нилид
Амид тионикоти-
новой кислоты
1М,М-Диметилтио-
формамид
Исходное
соединение
Форманилид
Бензамид
Бензанилид
4-Хлорацетани-
лид
Амид никотино-
никотиновой кислоты
Ы,Ы-Диметил-
формамид
Время
реакции, ч
1
1
8
2
15
3
Т. пл., °С
138-140 (абс.ЕЮН)
115-116 (ЕЮН/Н2О)
97-98 (ЕЮН/Н2О или AcOEt)
141-143 (ЕЮН)
188-190 (PrOH)
Т. кип. 95-97 (при 10мм рт. ст.)
Выход, %
70
68
72
82
68
80
104
Г Препаративная часть (продолжение)
Тиокарбонильные соединения, в особенности амиды тиокарбоновых кислот, находят
практическое применение в качестве фармпрепаратов, растительных пестицидов, вулканиза-
вулканизаторов, ингибиторов коррозии и присадок к смазочным маслам.
Ацетилкоэнзим А, участвующий в биосинтезе жирных кислот, терпенов, а также в цикле синтеза
лимонной кислоты, содержит активную группировку эфира тиокарбоновой кислоты (см. [Г.7.174]).
Тиокарбонильные соединения являются важными исходными продуктами
в синтезе ряда гетероциклов. Напишите реакцию получения 2,4-диметилтиа-
зола из хлорацетона и тиоацетамида.
7.2. РЕАКЦИИ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ С С-НУКЛЕОФИЛАМИ
Карбонильные соединения реагируют с С-нуклеофилами с образованием С—С-связи. Эти
реакции имеют исключительно важное значение в синтезе органических соединений.
В качестве С-нуклеофилов могут выступать:
5- 5+
а) Металлоорганические соединения R—М.
Благодаря +/-эффекту металла связь С—М поляризуется и на атоме углерода
образуется частичный отрицательный заряд.
б) Цианид- и ацетиленид-ионы: eC=N, eC=CR.
Q
в) Илиды)С-Х® (<^)С=Х илены) фосфора (X = PR,) и серы (X = SR2, SOR2).
г) Еноляты)С=С( <->)С—С* альдегидов, кетонов, эфиров и амидов кислот,
а также их азааналогов (енамвды [см. разд. Г,7.4.2.1], а-циано- и а-нитрокарбанионы).
XI е X®
д) Енолы, их эфиры и енамины )С=СС <-»)С—С^ (см. разд. Г,7.4.2).
Соединения (б)—(г) образованы в результате отрыва протона от С—Н-кис-
лотных соединений. Они могут возникать также из этих соединений in situ в про-
процессе реакции под действием оснований.
Таблица
\
с=о +
альдегиды,
кетоны
О
/<Ч +
О
и
/-с\ +
о
11
Ar^ "H
О
1 1
Аг/С"Н
Г.7.98. Реакции карбонильных соединений с С-
н-о-н ^=
н-с=с-н =
^0
—СНг-с' =
+ 0
НзС-С
о
Схема реакции
\ .он
- X
у он
он
^=^ —с-с=с-н
1
1
ОН о \-/
' 1 \ ~Н2° С = 0
Аг Н
у-4 П
-снзсоон н/ Ъ(ЮН
0
Н С-л
+ Н2С-СООН 32НЛ- Х.Л
HNCOPh
Аг N -ph
нуклеофилами
Название процесса
Синтез циангидринов
Этинилирование
Альдольная конденсация
Реакция Перкина
Реакция Эрленмейера
7.Реакции карбонильных соединений
105
Таблица Г.7.98. Продолжение
Схема реакции
Название процесса
-HCI
о
-с-с-н
/
COOR
О
„С^ + H2C-COOR
CI
О х X
" .up' V-r (X,Y = COR, COOR,
^b\ 4 _н,о / 4 cooh.cn, no2)
Y ' Y
о
II
о
if
С
R2NH + U.C.H+ -CH2-C^
О о НП (
О
R2N-CH2-CH-C
OR'
—сн2-с
a/
-Ph3P = C
Ar
\ ,
R'OH_
R
R'
\
чс-сн-с
OR'
9 ,x
+ H2C
CI
> 4C-CH (X, Y = COR, COOR, CN)
О
О
+ c—c
CI. /Л
^C^ + R-M —I
M = MgX, Li идр.
C-NR2
„.асе»
ОМ ОН
I (+Н20) I
?R ?
о
О
OH
(+H20) I
+ 2R-MgX „._„„• —C-R
-XMgOH R
R М _мх Р
(X = OH, OR'.CI, OCOR';
M = Li, LiCuR, CdR идр.
Синтез глицидных эфиров
по Дарзану
Конденсация Кневенагеля
Реакция Манниха
Ацилоиновая конденса-
конденсация
Реакция Виттига
Сложноэфирная конден-
конденсация
Ацилирование С—Н-кис-
лотных соединений
Ацилирование енаминов
(см. разд. Г,7.4.2.3)
Ацилирование по Фриде-
лю—Крафтсу (см. разд.
Г,5.1.8.1)
Реакции с металлооргани-
ческими соединениями
(М = MgX: реакция Гринь-
яра)
106
Г Препаративная часть (продолжение)
Важнейшие реакции С-нуклеофилов с карбонильными соединениями
сведены в табл. Г.7.98.
Некоторые реакции карбонильных соединений уже обсуждались в разд. Г.5 (ацилирова-
ние по Фриделю-Крафтсу, хлорметилирование и родственные превращения).
7.2.1. РЕАКЦИИ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
С С-Н-КИСЛОТНЫМИ ВЕЩЕСТВАМИ
К карбонильной группе способно присоединяться большое число разнообраз-
разнообразных С—Н-кислотных соединений — альдегиды, кетоны, эфиры и амиды кис-
кислот, нитрилы и нитросоединения с атомом водорода в а-положении к функци-
функциональной группе, а также синильная кислота и ацетилен. Эти соединения сами
по себе не обладают основными свойствами, однако в присутствии сильных ос-
оснований способны в ходе равновесной стадии, предшествующей карбонильной
реакции, превращаться в анионы, обладающие достаточной нуклеофильной
активностью, чтобы присоединяться к карбонильной группе.
Положение равновесия зависит от соотношения основностей катализатора-
основания и аниона С-Н-кислотного соединения (табл. Г.7.99).
Катализатор-основание отрывает атом водорода (в виде протона) от ос-поло-
ос-положения карбонильного соединения:
н-с-с"
\
в-н
с-с
\
lOI0
\
с=с
\
\ е,Я
[Г.7.100]
I На 116 Ив
Ацидифицирующее действие карбонильной группы и ее аналогов на соседние с нею ал-
кильные группы объясняется, во-первых, —/-эффектом, повышающим полярность связи
С—Н, во-вторых, возможностью сопряжения с электронной парой, остающейся после отщеп-
отщепления протона, в результате чего стабилизируется анион II (схема [Г.7.100]). Отсюда следует,
что подвижностью обладают только водородные атомы, находящиеся в а-положении. р-Ме-
тильная группа пропионового альдегида уже не может вступать в сопряжение с карбонильной
группой. Водородный атом, стоящий у карбонильного углерода в альдегидах, тоже не спосо-
способен отщепляться в виде протона под действием оснований, поскольку в этом случае не проис-
Таблица Г.7.99. Значения рКа в воде при 25 °С
Вещество
Метан
Бензол
Аммиак
Водород
Трифенилметан
Ацетилен
Ацетонитрил
Метилацетат
Ацетон
Этанол
Метанол
Вода
Диэтиловый эфир малоно-
малоновой кислоты
рК„
-48
-43
-38
~35
-32
-25
-25
-24
-20
16
15,5
15,74
12,9
Вещество
Нитрил малоновой кислоты
Пиперидиний-катион
Ацетоуксусный эфир
Циануксусный эфир
Нитрометан
Фенол
Синильная кислота
Аммоний
Ацетилацетон
Малоновый альдегид
Уксусная кислота
рК„
11,2
11,1
10,8
10,5
10,2
10,0
9,2
9,24
9,0
5,0
4,8
7. Реакции карбонильных соединений 107
ходит удлинения сопряженной системы. Однако этот атом водорода может отщепляться в виде
радикала или аниона (см., например, схемы [Г. 1.46] и [Г.7.238]).
В р-дикарбонильных соединениях (малоновый и ацетоуксусный эфиры, ацетилацетон и т. д.)
сильно проявляется как индукционный эффект в отношении соседней связи С-Н, так и возмож-
возможность делокализации свободной электронной пары в анионе. Поэтому такие соединения облада-
обладают кислотностью, сравнимой с кислотностью фенолов и карбоновых кислот.
Образующийся в реакции [Г.7.100] анион II С—Н-кислотного соединения
может присоединяться к карбонильному соединению таким же образом, как и
ранее рассмотренные основания1 (см. схему [Г.7.8]):
чс'-с + с=о =^== чс-с-с-О10
/ \ / /II"
Пв III IV
[Г.7.101]
О | | О | |
чс-с-с-6~|0 + нв =^ чс-с-с-он + в0
/II" /II
IV V
Образующийся при этом ион алкоголята IV (схема [Г.7.101]) отрывает про-
протон от ранее образовавшегося протонированного основания НВ (схема
[Г.7.100]) или от растворителя, переходя в незаряженное гидроксильное соеди-
соединение V; при этом регенерируется катализатор В". Таким образом, вся реакция
может протекать с каталитическими количествами основания.
Предпосылкой для осуществления последней стадии (IV -» V) является более высокая ос-
основность алкоголят-иона IV по сравнению с В*. Это имеет место, когда в качестве вспомога-
вспомогательного основания используется гидроксид-ион (рКа = 15,7), так как алкоголят-ионы типа IV
являются очень сильными основаниями {рКа » 17-19). Примеры, когда не может осущест-
осуществиться «стадия нейтрализации», рассматриваются позднее. В таких случаях для конденсации
необходимо применять молярные количества основного агента конденсации.
Схема [Г.7.101] является прототипом всех альдольных реакций и аналогич-
аналогичных превращений, катализируемых основаниями. Поскольку все стадии явля-
являются равновесными, образовавшиеся аддукты могут быть снова расщеплены
при действии оснований.
К реакции, протекающей по схеме [Г.7.101], часто примыкает отщепление
молекулы воды с образованием ос,р-ненасыщенных соединений (кротоновая
конденсация). Отщепление воды в подобных случаях протекает очень легко,
так как при этом образуется система сопряженных двойных связей (см. также
разд. Г,3.1.4). Если карбонильным компонентом, реагирующим с С—Н-кислот-
ным соединением, является производное карбоновой кислоты (сложный эфир,
ацилгалогенид, ангидрид), то всегда имеет место стадия конденсации, в ходе
которой происходит отщепление спирта, галогеноводорода или карбоновой
1 Как следует из схемы [Г.7.100], атом кислорода в анионах карбонильных соединений
(енолятах) также имеет нуклеофильные свойства. Однако реакция с ним не может привести к
стабильному продукту, так как образовавшееся соединение полуацетального типа может
стабилизироваться только путем распада на компоненты.
108
Г Препаративная часть (продолжение)
кислоты. В результате образуются анионы (еноляты) р-дикарбонильных соеди-
соединений, обладающие очень малой энергией:
с-с
с=о
оч
чс-сн-с-ог"
/ i г
-НХ
+ НХ
Яч I -©
C-C=C-OI ¦
/ I
с-с-с
/ I Ъ
[Г.7.102]
X = OR, галоген, OCOR
Вследствие своей небольшой основности анионы р-дикарбонильных соединений, как
правило, не способны снова выделить из протонированного катализатора свободное основа-
основание (например, алкоголят). Здесь имеет место один из упоминавшихся случаев, когда необхо-
необходимо использовать молярные количества вспомогательного основания. Если X — галоген или
OCOR (схема [Г.7.102]), то необходим еще один моль конденсирующего вещества.
Реакции С—Н-кислотных веществ с карбонильными соединениями можно ускорить не
только основными, но и кислотными катализаторами или кислотами Льюиса. Кислотный ка-
катализатор повышает карбонильную активность уже известным способом, т. е. катализирует
енолизацию С—Н-кислотного компонента:
н-с-с + н
I ©.он
н-с-с
I \
он
\
с=с
/ \
[Г.7.103]
(Ознакомьтесь по учебнику с кето-енольной таутомерией! См. также разд. Г,7.2.1.8.)
Енол в схеме [Г.7.103] благодаря основным свойствам двойной связи С=С (см. разд. Г,4)
оказывается способным присоединяться как нуклеофильный агент к карбонильной группе:
НО,® | |
с-с-с-он
/ I I
+ н
с-с-с-он
/ I I
[Г.7.104]
Образуется такой же продукт реакции, как и при катализе основаниями (схема [Г.7.101]).
В кислой среде, однако, альдоль немедленно дегидратируется (см. выше):
% ? I
с-с-с-он
/ I I
+ н
-НГ
о н
с-слсто
-Н2О, -YC
+ Н2О, + Не
% /
с-с=с
/ I \
[Г.7.105]
Подобные катализируемые кислотами реакции имеют меньшее значение, чем катализиру-
катализируемые щелочами превращения.
7.2.1.1. Присоединение синильной кислоты к альдегидам и кетонам
В результате присоединения синильной кислоты к альдегидам и кетонам
образуется а-гидроксикарбонитрилы (циангидрины):
/
H-C=N
ОН
R-C-C=N
i
R'
[Г.7.106]
В качестве основных катализаторов в этой реакции используются карбонаты
и цианиды щелочных металлов, аммиак, амины и другие соединения. (Напиши-
(Напишите схему механизма реакции!)
Реакция обратима, поэтому при действии щелочей циангидрины могут рас-
расщепляться на исходные компоненты. Положение равновесия сильно зависит от
7.Реакции карбонильных соединений
109
Таблица Г.7.107. Получение а-гидроксикарбонитрилов (циангидринов) из альдеги-
альдегидов и кетонов
Продукт реакции
Ацетонциангидрин
Циангидрин метилэтилкетона
Циангидрин диэтилкетона
Циангидрин ацетальдегида
Циангидрин бензальдегидаа
Циангидрин циклогексанона
Исходное соединение
Ацетон
Бутанон
Пентанон-3
Ацетальдегид
Бензальдегид
Циклогексанон
Т. кип. (мм рт. ст.)
или т. пл., °С
81A5)
91 B0)
88(8)
95 B0)
Т. пл. 20
126 A8); т. пл. 29
«о20
1,4013
1,4151
1,4251
1,4052
Выход, %
60
50
50
70
70
60
а Циангидрин бензальдегида неустойчив, поэтому неочищенный продукт следует сразу
использовать в дальнейших реакциях (см. разд. Г,7.1.5).
строения карбонильного соединения, причем большое значение имеют как
электронные, так и пространственные факторы. Циангидрины альдегидов
устойчивее, чем циангидрины кетонов. Группы, обладающие —/-эффектом и
находящиеся по соседству с карбонильной группой, облегчают образование
циангидринов. В случае алифатических кетонов получаются плохие выходы, а
чисто ароматические кетоны вообще не вступают в реакцию в этих условиях.
В силу пространственных факторов циангидрины циклогексанона и циклопен-
танона устойчивее, чем циангидрины кетонов с открытой цепью.
(ГП) Общая методика получения а-гидроксикарбонитрилов (циангидринов) (табл. Г.7.107)
Внимание! При реакции образуется свободная синильная кислота! Работать
под тягой в противогазе! Циангидрины также очень ядовиты (почему?). Многие
циангидрины термически нестабильны. Поэтому перед перегонкой их стабили-
стабилизируют добавлением 1-2% концентрированных серной, фосфорной или хлор-
уксусной кислот. В противном случае может произойти разложение со взры-
vl вом. При хранении циангидрины также нужно стабилизировать.
В трехгорлую колбу, снабженную мешалкой, обратным холодильником, капельной ворон-
воронкой и внутренним термометром (см.рис. А.4, г), помещают 1 мольтонкоизмельченного циани-
цианида натрия и 120 мл воды, перемешивают до растворения, после чего смешивают с 1,2 моль кар-
карбонильного соединения. Охлаждают до 0 °С и при интенсивном перемешивании медленно при-
приливают по каплям 0,85 моль 35%-ной серной кислоты с такой скоростью, чтобы температура не
превышала 5 °С. Затем перемешивают еще 15 мин и тотчас отсасывают выделившийся гидро-
гидросульфат натрия (осторожно! синильная кислота!). Слой циангидрина отделяют, осадок на
фильтре промывают дважды 100 мл эфира, водную фазу экстрагируют таким же объемом эфи-
эфира. Органические фазы объединяют, сушат безводным сульфатом натрия, добавляют 1 гхлорук-
сусной кислоты, отгоняют эфир и перегоняют циангидрин в вакууме с короткой колонкой
Вигре (тяга!).
Получение нитрила гликолевой кислоты (формальдегидциангидрина): Горди Р.
В сб.: Синтезы органических препаратов. Сб. 4. — М.: ИЛ, 1953, с. 354.
Гидроксигруппы циангидринов можно ацилировать и алкилировать
(см. также разд. Г,7.2.1.6).
Получение аллиловых эфиров циангидринов из альдегидов, цианида калия и
аллилбромида в условиях межфазного катализа: Mclntosh J. H. Canad. J. Chem.,
1977, 55, 4200.
110
Г Препаративная часть (продолжение)
Циангидрины применяют для синтеза ot-гидроксикислот, причем омыление нитрильной
группы проводят в кислой (почему не в щелочной?) среде (см. также разд. Г,7.1.5; синтез
миндальной и молочной кислот).
Щелочное расщепление циангидринов до альдегидов используют при деструкции альдоз
до ближайшего низшего сахара (расщепление по Волю), например:
СНО
Н-С-ОН
НО-С-Н H2NOH
Н-С-ОН '
Н-С-ОН
СН2ОН
D-глюкоза
CH=NOH
Н-С-ОН
НО-С-Н
Н-С-ОН
Н-С-ОН
СН2ОН
C=N
Н-С-ОАс
i
Ас2О АсО-С-Н CH3ONa
(AcONa)
Н-С-ОАс
Н-С-ОАс
СН2ОАс
СНО
НО-С-Н
н-с-он
н-с-он
СН2ОН
[Г.7.108]
D-арабиноза
Если реакцию между альдегидами и синильной кислотой проводить в прису-
присутствии эквимолярных количеств аммиака или аминов (первичных или вторич-
вторичных), то синильная кислота присоединяется к образующимся иминам, давая
аминонитрилы, кислотный гидролиз которых приводит к а-аминокислотам
(синтез Штреккера):
NR2
R-CH-COOH +
о'?
R-c' + HNR2 + HCN
Vl
NR2
R-C-CN
ь2Н2О, Н
0
I
NH3 [Г.7.109]
При взаимодействии с формальдегидом таким путем цианметилируют
первичные и вторичные амины.
Получение N-метиламиноацетонитрила (саркозинонитрила): Cook A. H.,
CoxS. F. J. Chem. Soc, 1949, 2334.
Этот метод используется в промышленности при производстве нитрилотриуксусной кис-
кислоты [НТК, Трилон A, N(CH2COOHK] и этилендиаминтетрауксусной кислоты (ЭДТК,
Трилон Б, Хелаплекс). Составьте схему их синтеза. НТК комплексует ионы Са и Mg и может
заменять собой фосфаты в моющих веществах.
Трудности в синтезе аминокислот связаны со стадией их отделения от неорганических со-
солей (одинаковый характер растворимости). Растворимость аминокислот минимальна в изо-
электрической точке. Поэтому некоторые труднорастворимые в воде аминокислоты могут
быть выделены из их солевых растворов доведением рН до значения, отвечающего изоэлект-
рической точке (см., например, табл. Г.6.12). В большинстве случаев, однако, аминокислоты
экстрагируют из смесей с неорганическими солями в виде гидрохлоридов, например абсолют-
абсолютным спиртом. Из гидрохлоридов свободные аминокислоты выделяют основаниями в соответ-
соответствующем растворителе или с помощью ионообменных смол. В приводимой ниже методике к
спиртовому раствору гидрохлорида прибавляют диэтил- или трибутиламин, причем гидрохло-
гидрохлорид добавленного более сильного основания остается в растворе, а аминокислота осаждается.
DL-Метионин, незаменимую аминокислоту и добавку к кормам на птицефермах, получают в
промышленности синтезом Бюхерера из р-метилтиопропионового альдегида (см. разд. Г,7.4.1.2):
О
NaCN + (NH4)HCO3
Me'
HN-
NH
H2O + NaOH
1.NaOH
2. H2SO4
CO2 + NH3
[Г.7.110]
7.Реакции карбонильных соединений
111
Таблица Г.7.111. Получение а-аминокислот по Штреккеру
Продукт реакции
DL-Аланин
DL-a-Аминомасляная
кислота
DL-Нормалин
DL-Валин
DL-Метионин
ОЬ-Фенилглицинв
DL-a-Метилаланин
Исходное соединение
Ацетальдегида
Пропионовый альдегид
Масляный альдегид
Изомасляный альдегид
р- Метилтиопропионовый
альдегид
Бензальдегид
Ацетон
Т. пл., °С
295
307 (разл.)
ЗОЗ6 (разл.)
298б (разл.)
281 (разл.)
256
316 (разл.)
Выход, %
50
60
65
14
60
50
55
а Раствор в 100 мл эфира; последний отгоняется при дальнейшей работе.
6 В запаянном капилляре.
в После омыления реакционной смеси добавляют концентрированный аммиак до слабо-
слабощелочной реакции; выпавшую кислоту отфильтровывают. Очистку см. SteigerR E. Org. Syntheses,
Coll. Vol. Ill, 1955, p. 84.
Аналогично из 4-цианобутаналя получают лизин (на промежуточной стадии применяют
гидрирование).
(ГП) Общая методика получения a-аминокислот по Штреккеру (табл. Г.7.111)
Осторожно обращайтесь с цианидами! При подкислении реакционной смеси
выделяется синильная кислота. Работать под тягой! Сосуд для работы под дав-
давлением заполнять только на треть объема, во время реакции обернуть полотен-
полотенцем, перед открыванием охладить! Защитные очки!
В сосуд для работы под давлением вносят насыщенный на холоду раствор 0,55 моль хлори-
хлорида аммония, 100 мл концентрированного раствора аммиака и раствор 0,55 моль цианида нат-
натрия в 50 мл воды. После этого охлаждают ледяной водой и при встряхивании прибавляют по
каплям 0,5 моль альдегида или кетона. В случае ароматических карбонильных соединений до-
добавляют, кроме того, еще 100 мл метилового спирта, чтобы увеличить их растворимость. Сосуд
закрывают и встряхивают на качалке 5 ч при комнатной температуре. Если в реакцию введен
кетон, то вместо этого нагревают 5 ч на водяной бане при 50 "С, часто встряхивая.
После охлаждения сосуд осторожно открывают, переносят содержимое в прибор для пе-
перегонки в вакууме и перегоняют в вакууме водоструйного насоса (~2 кПа) при температуре
водяной бани 30-40 °С. При этом отгоняются аммиак и часть воды. После этого добавляют
300 мл концентрированной соляной кислоты (осторожно! выделяется некоторое количество
свободной синильной кислоты! тяга!) и кипятят 3 ч с обратным холодильником, чтобы гидро-
лизовать аминонитрил. Затем упаривают в вакууме досуха, в конце нагревают на кипящей во-
водяной бане. Горячий остаток дважды экстрагируют 100 мл метанола. Из объединенных вытя-
вытяжек при охлаждении выделяется еще немного хлорида аммония, который отфильтровывают.
После этого добавляют диэтил- или трибутиламин до слабощелочной реакции, выделяя ами-
аминокислоту в свободном виде. Оставляют на ночь в холодильнике, выпавшую аминокислоту
отфильтровывают и промывают метанолом. В случае необходимости можно перекристалли-
перекристаллизовать из водного этанола.
a,a '-Гидразобис(циклогексанкарбонилтрил) и a,a '-гидразобис(изобутиронит-
рил) получают аналогично из циклогексанона или ацетона, сульфата гидразина
и цианида натрия: Оверберджер К., Хуанг Пас-Тунг, Беренбаум М. В сб.: Синтезы
органических препаратов. Сб. 4. — М.: ИЛ, 1953, с. 246.
112 Г Препаративная часть (продолжение)
а-Этил-а-аминомасляная кислота: Штейгер Р. В сб.: Синтезы органических
препаратов. Сб. 3. — М.: ИЛ, 1952, с. 46.
Из альдегидциангидринов реакцией Риттера (первая стадии в уравнении [Г.7.112]) можно
получить а-оксокарбоновые кислоты:
ОН ОН О 0 0
R-CH-CN ,f'B"°H." R-CH-C-NH-f-Bu -^» R-C-C-NH-f-Bu
(H2SO4)
[Г.7.112]
О
н2о
R-C-COOH
-H2N-f-Bu
В промышленности из ацетонциангидрина получают полиметилметакрилат (плексиглас):
ОН
НзС-C-CN H2S°4- Н2С=С-с' °"^Н• Н2С=С-СООСН3 полимеры [Г.7.113а]
СН3 СН3 NH2 3 СН3
из формальдегидциангидрина получают глицин, из ацетальдегидциангидрина — молочную
кислоту. В реакции [Г.7.1136] промежуточно (in situ) образующийся циангидрин ацилируется
хлорангидридом с образованием пиретроидов, например Циперметрина (X = С1) и Дельтамет-
рина (Х= Вг).
О
О CN
-NaCI
[Г.7.1136]
Эти соединения, являющиеся аналогами природных пиретрумов, являются мощными
инсектицидами и производятся в больших количествах (см. [Г.4.83]).
7.2.1.2. Этинилирование карбонильных соединений
Альдегиды и кетоны реагируют с ацетиленовыми соединениями с образовани-
образованием пропаргиловых спиртов C-гидроксиацетиленов).
Этинилирование кетонов проводят обычно в жидком аммиаке в присутствии
амида натрия, который необходимо брать в стехиометрических количествах
(почему?):
е е
NH2 + Н-С=СН ICECH + NH3
О 0 Ю1Э @ ОН [Г.7.114]
R-C + 1С=СН R-C-C=CH R-C-CECH
R R1 R'
I II
Переход! -» II происходит при разложении реакционной смеси водой.
7. Реакции карбонильных соединений
113
Таблица Г.7.115. Этинилирование кетонов
Продукт реакции
2-Метилбутин-3-ол-2
З-Метшшентин-1 -ол-3
1-Этинилциклогексанол
1 -Этинилциклопентанол
2-Фенилбутин-3-ол-2
З-Фенилпентин-1-ол-З
Исходное соединение
Ацетон
Мети л этил кетон
Циклогексанон
Циклопентанон
Ацетофенон
Пропиофенон
Т. кип. (ммрт. ст.),
°С
106
121
78 A5); т. пл. 30
79 A8); т. пл. 27
107 A4); т. пл. 51
107 A0); т. пл. 34
«о20
1,4207
1,4310
1,4805а
1,5302а
Выход, %
60
60
80
40
70
80
Переохлажденный расплав.
Алифатические кетоны вступают в реакцию этинилирования в присутствии
гидроксида калия в качестве катализатора. Альдегиды лучше вводить в реакцию
в присутствии ацетиленида меди, так как указанные основные катализаторы
вызывают побочные реакции (альдольную конденсацию).
При реакциях с низшими кетонами и альдегидами могут образоваться соединения одно-
однократного и двукратного присоединения. Так, из ацетилена и формальдегида можно получить
как пропаргиловый спирт, так и бутиндиол-1,4. Направление реакции можно регулировать,
изменяя стехиометрические соотношения (концентрацию ацетилена) в реакции.
^П) Общая методика этинилирования кетонов (табл. Г.7.115)
,1 Внимание! С жидким аммиаком (т. кип. -34 °С) работать только под тягой! В це-
;| лях безопасности держать противогазы наготове, надеть защитные очки!
У Многие алкинолы могут при перегонке разлагаться со взрывом, прежде всего в
чЛ присутствии основных соединений. Поэтому следует избегать основных осу-
Щ шителей, таких, как карбонат калия; добавьте в перегоняемую смесь немного
й! янтарной кислоты, используйте защитный экран!
Все приборы и реактивы должны быть тщательно высушены (см. часть Е).
Трехгорлую колбу емкостью 1 л, снабженную эффективной мешалкой, газоподводящей
трубкой, термометром и осушительной трубкой, заполненной гидроксидом натрия, от кото-
которой отвод идет непосредственно в тягу, погружают до горла в баню с охлаждающей смесью
метанола и сухого льда. Пропускают быстрый ток аммиака до тех пор, пока в колбе не скон-
сконденсируется 350—400 мл жидкого аммиака. При последующих операциях температуру поддер-
поддерживают от —35 до —40 "С (теперь колбу погружают в охлаждающую смесь неглубоко).
При энергичном перемешивании добавляют 0,1 г нитрата железа(Ш) и пропускают быст-
быстрый ток ацетилена, предварительно пропущенного для удаления паров ацетона (в баллонах
ацетилен растворен в ацетоне, см. часть Е) через две промывные склянки с концентрирован-
концентрированной серной кислотой. Когда серная кислота начнет темнеть и во второй склянке, следует за-
заполнить их заново. Между промывными склянками и реакционной колбой следует установить
приспособление, защищающее от избыточного давления (см. рис. А. 11).
Нарезают тонкими полосками 0,5 г-атом натрия (под слоем сухого бензола) и вносят их в
раствор постепенно, по мере исчезновения появляющегося синего окрашивания. Когда после
114 Г Препаративная часть (продолжение)
введения всего натрия образуется бесцветный или светло-серый раствор или суспензия, пода-
подачу ацетилена прекращают1.
Затем в колбу в течение 30 мин прибавляют по каплям раствор 0,5 моль сухого кетона в
75 мл сухого эфира, снимают охлаждение и перемешивают еще 2 ч. После этого дают испа-
испариться аммиаку (лучше всего оставляют реакционную смесь на ночь). Остаток осторожно раз-
разлагают водой и слабо подксиляют 50%-ной серной кислотой. Затем несколько раз экстрагиру-
экстрагируют эфиром, промывают объединенные вытяжки раствором хлорида натрия, сушат сульфатом
магния и перегоняют над небольшим количеством янтарной кислоты.
Реакция этинилирования представляет значительный интерес для синтеза ненасыщенных
соединений, в частности терпенов, каротиноидов, стероидов. Так, этим путем можно полу-
получить разничные терпеновые спирты (линалоол, гераниол, фарнезол, фитол).
Этинилированием формальдегида в промышленности получают пропин-2-ол-1 (пропарги-
ловый спирт) и бутин-2-диол-1,4. Последний гидрированием до бутандиола-1,4 (см. табл.
Г.4.126) превращают в исходное вещество для синтеза тетрагидрофурана (см. табл. Г.2.61) и у-бу-
тиролактона, а также спиртового компонента для получения полиэфиров и полиуретанов (см.
разд. Г,7.1.6).
В промышленности через 2-метилбутин-3-ол-2 (табл. Г.7.115) получают изопрен — основ-
основной структурный элемент натурального каучука и природных терпенов, который перерабаты-
перерабатывают в 1,4-чис-полиизопрен:
О ОН ОН
н3с-с' + нс=сн —- н3с-с-с=сн -^- н3с-с-сн=сн2 [Г.7.П6]
СН3 СН3 СН3
З-Метилпентин-1-ол-З (метилпентинол, см. табл. Г.7.115) и 1-этинилциклогексилкарба-
мат (этинамат) обладают седативным действием. Этинилэстрадиол, получаемый этинилиро-
этинилированием эстрона, является высокоэффективным синтетическим эстрогеном:
он/-
[Г.7.117]
7.2.1.3. Альдольная конденсация
Адольной или альдольно-кротоновой конденсацией называют взаимодействие
альдегидов и кетонов (карбонильные компоненты) с теми же или другими аль-
альдегидами и кетонами, выполняющими роль С—Н-кислотного (метиленового)
компонента2.
Механизм катализируемой основаниями альдольной конденсации соответ-
соответствует схеме [Г.7.101]. (Напишите схему для пропионового альдегида.) В качест-
качестве оснований используют преимущественно гидроксиды щелочных и щелочно-
1 Синее окрашивание указывает на образование раствора натрия в жидком аммиаке. Со-
Соли железа катализируют его превращение в бесцветный амид натрия. Образование ацетилени-
да натрия происходит очень быстро. Можно вначале получить раствор амида натрия, а потом
вводить ацетилен.
2 В более широком смысле альдольной реакцией называют вообще все реакции между
альдегидами или кетонами и С—Н-кислотными соединениями. Такая классификация право-
правомерна, поскольку во всех этих случаях механизм реакции одинаков.
7. Реакции карбонильных соединений 115
земельных металлов. Указанная в схеме [Г.7.104] реакция, катализируемая кис-
кислотами, не имеет большого значения.
Если работать при низких температурах, то, как правило, нетрудно выделить
простые альдоли. Напротив, альдоли, полученные из ароматических альдеги-
альдегидов, очень легко дегидратируются, так как при этом образуется достаточно
длинная система сопряженных связей. Эта дегидратация особенно легко идет
при кислотном катализе.
Альдегиды очень легко вступают в реакцию в роли карбонильного компо-
компонента, и равновесие [Г.7.101] в этом случае сдвинуто далеко вправо.
Реакционная способность различных альдегидов уже обсуждалась ранее. Наибольшей ре-
реакционной способностью обладает формальдегид (почему?). С очень активными метиленовы-
ми компонентами, например дигидрорезорцином (см. разд. Г,7.4.1.3), он реагирует в водном
растворе даже без катализатора. В противоположность другим альдегидам формальдегид мо-
может давать аддукты, в которых замещены все водородные атомы у а-углерода метиленового
компонента, например:
р р сн2он
3 Н-с' + Н3С-с' НОСН2-С-СН=О [Г.7.118]
Н Н СН2ОН
Это метилольное соединение очень легко вступает в перекрестную реакцию Канниццаро с
образованием пентаэритрита (см. разд. Г,7.3.1.3). Наименее реакционноспособны аромати-
ароматические альдегиды.
При реакции между альдегидом и кетоном последний из-за меньшей карбо-
карбонильной активности всегда играет роль метиленового компонента (реакция
Кляйзена—Шмидта). Кетоны, не имеющие подвижного а-водорода (например,
бензофенон), не реагируют с метиленактивными альдегидами, в этом случае
предпочтительно происходит самоальдолизация альдегида.
Тем не менее оказывается возможным заставить выступить альдегид в качестве метилено-
метиленового компонента, если снизить карбонильную активность альдегида, предварительно переве-
переведя его в основание Шиффа (см. ряд. [Г.7.6]) и превратив последнее, например действием
диизопропиламида лития, в соответствующий анион:
/,NR' q.NR'
R-CH2-c' R-CH'-c' Li [Г.7.119]
чн Vi
Самоконденсация такого аниона идет лишь в малой степени. Реакция с карбонильной
группой других альдегидов или кетонов приводит к образованию N-аналогов альдолей, кото-
которые перегонкой с водяным паром в присутствии щавелевой кислоты можно перевести в
а,р-ненасыщенные альдегиды:
R R Н
I \ / Н
СН С
не с^ +н2о н^-с* / [г.7.120]
R'NHU0H f
Если в кетоне имеются два реакционноспособных центра, как, например, в
ацетоне или бутаноне, то можно получить продукты моно- и диальдолизации.
Если хотят получить моноаддукт, то надо использовать двух- или трехкратный
избыток метиленового компонента. Если в кротоновую конденсацию вводит-
116 Г Препаративная часть (продолжение)
ся несимметричный кетон, то возможно образование двух продуктов реак-
реакции:
^FV [Г.7.121а]
о н у
О
[Г.7.1216]
Катализируемая кислотами реакция с ароматическими альдегидами, как
правило, приводит к конденсации по метиленовой группе [Г.7.1216], в то время
как в щелочной среде атака подвергается преимущественно метильная группа
[Г.7.121а]. Неразветвленные алифатические альдегиды независимо от среды
реагируют преимущественно по метиленовой группе.
Катализируемую основаниями альдольную реакцию со слабо С—Н-кислот-
ными метиленовыми компонентами проще всего провести в двухфазной систе-
системе (ср. межфазный катализ, разд. Г.2.4.2). Примером такой реакции может
служить конденсация ацетонитрила с ароматическими альдегидами, рассмот-
рассмотренная в общей методике ктабл. Г.7.123, вариант Д; ср. схему [Г.7.131].
Равновесие конденсации между одинаковыми или разными кетонами сдви-
сдвинуто в невыгодную сторону, так что, например, диацетоновый спирт D-гидрок-
си-4-метилпентанон-2) можно получить из ацетона с достаточным выходом
только в том случае, если образующийся альдоль постоянно удалять из равно-
равновесной смеси.
Осуществить самоконденсацию ацетона можно также в присутствии силь-
сильной кислоты. (Стадия конденсации сдвигает равновесие в нужном направле-
направлении.) Однако при этом наряду с 4-метилпентен-3-оном-2 (мезитилоксидом)
образуются высшие продукты конденсации: 2,6-диметилгептадиен-2,5-он-4
(форон) и 1,3,5-триметилбензол (мезитилен).
Протонированная карбонильная группа может не только принимать электронную пару
енола (схема [Г.7.104]), но и реагировать с неактивированными алкенами (реакция Принса).
Продукт присоединения может стабилизироваться различными способами. Основными про-
продуктами являются или производные 1,3-диоксана, или р,у-ненасыщенные спирты:
Н
1 /
С-
но-сч нсЛ.
R- R
[Г.7.122]
Реакция Принса используется в промышленности. Присоединением двух молекул фор-
формальдегида к изобутену получают 4,4-диметил-1,3-диоксан, который затем термически разла-
разлагают над фосфатом кальция на изопрен G7%), формальдегид и воду.
7. Реакции карбонильных соединений 117
(ГП) Общие методики проведения альдольно-кротоновых конденсаций (табл. Г.7.123)
А. Конденсация алифатических альдегидов. В трехгорлую колбу емкостью 250 мл, снаб-
снабженную мешалкой, капельной воронкой и внутренним термометром, помещают 1 моль соот-
соответствующего альдегида (используют только свежеперегнанные альдегиды и кетоны) в 75 мл
эфира. При охлаждении водой очень медленно прибавляют 15%-ный раствор 0,02 моль КОН
в метаноле, причем температура в колбе должна поддерживаться в пределах 10—15 "С. Затем
перемешивают еще 1,5 ч при комнатной температуре, нейтрализуют эквимолярным количе-
количеством ледяной кислоты, отделяют осадок ацетата калия, сушат сульфатом натрия и перегоня-
перегоняют при возможно более низкой температуре.
Б. Конденсация алифатических альдегидов (кроме формальдегида) с кетонами. В трехгорлую
колбу емкостью 500 мл, снабженную мешалкой, капельной воронкой, внутренним термомет-
термометром, помещают кетон (свежеперегнанный) и прибавляют 0,03 моль КОН в виде 15%-ного
метанольного раствора. Если кетон имеет только одну реакционноспособную группу (металь-
(метальную или метиленовую), то берут 1 моль, во всех других случаях — 3 моль, если хотят получить
моноаддукт.
При интенсивном перемешивании и охлаждении водой прибавляют из капельной ворон-
воронки (очень медленно, в течение 4—6 ч) 1 моль свежеперегнанного алифатического альдегида в
75 мл эфира (при температуре внутри колбы 10—15 °С), а затем перемешивают еще 1,5 ч при
комнатной температуре. После этого нейтрализуют ледяной уксусной кислотой, сушат суль-
сульфатом натрия и перегоняют.
В. Реакции с формальдегидом. Для получения моноаддукта A:1) суспендируют 1 моль па-
раформа в 5 моль свежеперегнанного метиленового компонента, если последний имеет нес-
несколько реакционноспособных центров, или в 1 моль, если имеется только один такой центр.
К этой смеси добавляют 15%-ный спиртовой раствор КОН до рН 10-11 и реакционную
массу перемешивают в колбе на 500 мл, снабженной мешалкой, обратным холодильником и
внутренним термометром, при температуре 40—45 °С в течение 0,5—1 ч. Время от времени оп-
определяют рН и в случае необходимости добавляют еще раствор щелочи. Затем нейтрализуют
ледяной уксусной кислотой, отфильтровывают выпавшие твердые продукты реакции и про-
промывают водой. В случае жидких продуктов отделяют органический слой и перегоняют.
При соответствующем изменении стехиометрических соотношений можно аналогично
получать продукты а,а-бис(гидроксиметилирования) и а,а,а-трис(гидроксиметилирования).
Синтез может быть проведен в полумикромасштабе. В этом случае работают с магнитной
мешалкой.
Г. Реакции ароматических альдегидов с кетонами
(J Внимание! а,р-Ненасыщенные кетоны часто раздражают кожу и слизистые
jj оболочки. Пораженные места промыть разбавленным спиртом.
В трехгорлую колбу емкостью 1 л, снабженную мешалкой, капельной воронкой и внутрен-
внутренним термометром, помещают раствор 1 моль свежеперегнанных альдегидов и кетона в 200 мл
метилового спирта. Если берут кетоны более чем с одной реакционноспособной метильной
или метиленовой группой и хотят получить продукты моноконденсации, то молярное соотно-
соотношение кетона к альдегиду должно составить 3:1; если хотят получить продукт конденсации
двух молекул альдегида с 1 молекулой кетона, то это соотношение должно быть 0,5 : 1. К раст-
раствору при хорошем перемешивании прибавляют из капельной воронки 0,05 моль гидроксида
калия в виде 15%-ного раствора в метаноле при температуре в колбе 20—25 °С. Реакционную
смесь перемешивают еще 3 ч, нейтрализуют ледяной уксусной кислотой, образовавшиеся
твердые продукты реакции отфильтровывают и промывают водой. В других случаях реакцион-
реакционную смесь разбавляют водой и фильтруют или извлекают эфиром. Эфирные вытяжки промы-
промывают водой, сушат сульфатом натрия и перегоняют.
При получении нитростиролов следует брать 1 моль щелочи, вести реакцию в течение
0,5 ч и после этого вылить реакционную смесь в двойное молярное количество 20%-ной соля-
соляной кислоты.
Д. Конденсация с ацетонитрилом. (Двухфазная реакция; см. работу Gokel G. W., DiBiase S. А.,
Lipisko В. A. Tetrahedron Letters, 1976, 3495.) В круглодонной колбе емкостью 200 мл с мешал-
мешалкой, обратным холодильником и капельной воронкой нагревают смесь 6,6 г @,1 моль) тонко-
Таблица Г.7.123. Альдольно-кротоновые конденсации
Продукт реакции
З-Гидроксибутаналь (ацетальдоль)а
З-Гидрокси-2-метилпентаналь
(пропиональдоль)
Тиглиновый альдегид"
З-Гидрокси-2-этилгексаналь
(бутиральдоль)
4-Гидроксипентанон-2
4- Гидрокси -3 -метилпентанон- 2
4-Гидроксигептанон-2
Бензилиденаиетон
Коричный альдегидг
п- Метоксибензилиденацетон
2,2-Диметил-З-гидроксипропаналь
со-Нитростиролд
Бензилиденацетофеноне (халькон)
Дибензилиденацетон
З-Гидрокси-З-метилбутанон-2
Исходное соединение
Ацетальдегид
Пропионовый альдегид
Ацетальдегид, пропионовый
альдегид
Масляный альдегид
Ацетальдегид, ацетон
Ацетальдегид, бутанон
Масляный альдегид, ацетон
Бензальдегид, ацетон
Бензальдегид, ацетальдегид
Анисовый альдегид, ацетон
Формальдегид, изомасляный
альдегид
Бензальдегид, нитрометан
Бензальдегид, ацетофенон
Бензальдегид, ацетон
Формальдегид, бутанон
Вариант
А
А
А
А
Б
Б
Б
Г
Г
Г
в
г
г
г
в
Т. кип. (мм рт. ст.), °С
83 B0)
85A1)
118
100A0)
60A0)
76A0)
92A2)
140 A6); т. пл. 41
124A6)
185 A8); т. пл. 74
Т. пл. 86 (PhMe/петр. эф)
Т. пл. 58(этанол)
Т. пл. 57 (этанол)
Т. пл. 111 (ацетон, -15 °С)
80A0)
«D20
1,4238е
1,4373е
1,4475
1,44096
1,4265
1,4350
1,4360
1,6195
1,4340
Выход, %
60
60
30
70
60
70
70
60
60
80
80
80
75
70
50
Нитрил и-диметиламинокоричной
кислоты
Нитрил л-диэтиламинокоричной
кислоты
Нитрил я-метоксикоричной кислоты
л-Диметиламинобензальдегид,
ацетонитрил
л-Диэтиламинобензальдегид,
ацетонитрил
Анисовый альдегид,
ацетонитрил
Д
Д
д
Т. пл. 164-166
Т. пл. 97-99
170-190 A8); т. пл. 59-61
55
48
30
а Перегоняют на 20-сантиметровой колонке Вигре. Ацетальдоль при стоянии быстро переходит в димер («паральдоль»), имеющий т. пл. 97 °С (диэти-
ловый эфир):
ОН Hs
ch3-ch-ch/
/
,0-CH
он
\
/
сн2
NCH3
При этом жидкость постепенно становится более вязкой и из нее выделяются кристаллы. Небольшая добавка воды замедляет эту реакцию. При пе-
перегонке в вакууме водоструйного насоса из паральдоля снова образуется мономерный альдоль.
6 Показатель преломления относится к только что полученному соединению.
в Используют по 0,5 моль альдегида, работают в атмосфере азота. После перегонки отделяют воду, образовавшуюся в ходе реакции, сушат хлори-
хлоридом кальция и перегоняют на ректификационной колонке.
г Вместо метанола берут воду, работают в атмосфере азота. В колбу помещают 2 моль бензальдегида и 0,1 моль КОН, прибавляют по каплям
30%-ный водный раствор ацетальдегида. После того как будет прибавлена половина раствора, добавляют еще 0,05 моль КОН в 30 мл воды.
д Молярное соотношение исходных веществ 1:1. Работают ниже +5 °С с эквимолярным количеством щелочи. Через 15 мин медленно приливают
избыток охлажденной во льду разбавленной соляной кислоты.
е Количество метанола увеличивают в три раза; перемешивают 8 ч.
120 Г Препаративная часть (продолжение)
измельченного твердого 85%-ного гидроксида калия, 80 мл очищенного ацетонитрила (см.
часть Е) и 2 мл аликвата 336 (см. разд. Г,2.4.2). Как только смесь закипит, приливают при энер-
энергичном перемешивании раствор 0,1 моль альдегида в 15 мл очищенного ацетонитрила. Затем наг-
нагревают еще 10 мин с обратным холодильником, охлаждают и выливают на 200 г толченого льда.
Смесь дважды встряхивают с дихлорметаном, органическую фазу промывают небольшим
количеством воды, сушат над сульфатом натрия и отгоняют растворитель в вакууме; остаток
перекристаллизовывают из этанола или перегоняют в вакууме. Стереохимию продукта
[(?)- или B)-форма] определяют по спектрам ЯМР 'Н в дейтерохлороформе.
©Получение диацетонового спирта D-гидрокси-4-метилпентанона-2). [ConantJ. В., Turtle N.
In: Amus R. Organische Synthesen. — Vieweg & Sohh, Braunschweig, 1937, S. 192.] В кругло-
донную колбу емкостью 250 мл, снабженную насадкой Сокслета (см. рис. А.88) и эффек-
эффективным обратным холодильником, помещают 1 моль ацетона. Гильзу насадки Сокслета на-
наполняют до полвины объема оксидом бария, прикрыв его сверху ватой. Колбу нагревают на
водяной бане так, чтобы содержимое интенсивно кипело. Конец реакции определяют по прек-
прекращению кипения в колбе при нагревании на кипящей водяной бане (около 30 ч). После этого
перегоняют в вакууме. Т. кип. 73 °С B3 мм рт. ст.); п^20 1,4235; выход 70%.
Псевдоионон из цитраля и ацетона: Рассел А., Кенион Р. В сб.: Синтезы органи-
органических препаратов. Сб. 3. — М.: ИЛ, 1962, с. 380.
Этиловый эфир 3-оксо-А4^°-октагидронафталинкарбоновой-9 кислоты из
этилового эфира 1-C-оксобутил)циклогексанон-2-карбоновой-1 кислоты:
DreidingA. S., Tamasewski A. J. J. Am. Chem. Soc, 1955, 77, 411.
10-Метил-А1'9-октагидронафталиндион-2,5 из 2-метил-2-C-оксобутил)цик-
логександиона-1,3: Назаров И. Н. и др. ЖОХ, 1956, 26, 441.
Пиперин из пипероналя и пиперидина кротоновой кислоты (с помощью меж-
межфазного катализа): SchulzeA., OedigerH. LiebigsAnn. Chemie, 1981, 1725.
При альдольной конденсации прохиральных реагентов, например, альдегида и несиммет-
несимметричного кетона, образуются соединения с двумя асимметрическими атомами углерода, благо-
благодаря чему могут образовываться стереоизомерные формы (два диастереомера и в соответст-
соответствующих энантиомерныхформах) (см. разд. В,7.3.2). Такие диастереомерные кетолы называют
син- (или эритро-) и анти- (или трео-).
О ОН О
о
R'
R"
в» о он о он [Г-7Л241
R'
R" И Н О ОН О ОН
11 5 11 1 анти-кетол
R'^Y^R K^Y В (mpeo)
R" R"
Из ахиральных исходных соединений обе энантиомерные формы обоих диастереомеров
образуются в равных количествах, т. е. в виде рацемической смеси. Однако сами диастерео-
диастереомерные формы образуются не в равных количествах, одна из них преобладает.
Диастереомерность процесса можно понять, если принять во внимание, что образующий-
образующийся при депротонировании основанием (MB) из метиленового компонента енолят может обра-
образовывать стереоизомерные B)- и (?)-формы. Они реагируют с карбонильным компонентом
через шестичленное циклическое (в форме кресла) переходное состояние, в котором металл
М+ координируется с енолятом и карбонильным атомом кислорода.
В случае термодинамически контролируемой реакции (см. разд. В,3.2), т. е. если B)- и
(?)-енолятные формы находятся в равновесии, будет образовываться более термодинамичес-
термодинамически стабильный (?)-енолят. Это имеет место в случае проведения реакции продолжительное
7. Реакции карбонильных соединений
121
время и при высокой температуре. Энергетически более выгодным и менее стерически затруд-
затрудненным переходным состоянием в этом случае будет такое, при котором радикал карбониль-
карбонильного соединения будет занимать экваториальное положение, что приводит к образованию
анти-(трео)-кетопа.
+ мв
R1
+ мв ,. Т
-mf R"
(Zl-енолят
R"
-нв
R
[Г.7.125]
R1
RCHO
О ОН
Н
(Е)-енолят
При кинетическом контроле реакции, при «беспорядочном образовании» енолята преи-
преимущественно реагирует его (Zt-форма, взаимодействие которой с карбонильным компонен-
компонентом приводит к сын- (эритро)-форме кетола. Такое направление процесса характерно в случае
использования сильных оснований, таких, как диизопропиламид лития (ЛДА), пониженной
температуры и короткого времени реакции.
В обоих случаях диастереоселективность увеличивается с увеличением объемов радика-
радикалов R, R', R". Так, например, при реакции бензальдегида (R" = СбН5) с алкилметилкетона-
ми (R = СНз) в присутствии ЛДА преимущественное образование енолята смн-кетола
учеличивается с ростом объема радикала R в ряду R = СН3СН2 < (СН3)гСН < (СН3)зС от 64
до 82 и 98%.
Получение 3-алкил-2-метил-3-оксо-1-фенилпропанола из бензальдегида и
алкилметилкетонов: Heathcock С. Н., Вше С. Т., Kleshick W. A., Pirrung M. С,
Sohn J. E., LampeJ. J. J. Org. Chem., 1980, 45, 1066.
Если при альдольной конденсации исходить из хиральных реагентов, то реакция будет
протекать энантиоселективно, т. е. с преимущественным образованием одного из четырех воз-
возможных стереоизомеров. Для этого в реакции используют, например, предварительно полу-
полученные взаимодействием с бромом и хиральными лигандами B)- и (?)-еноляты или же
закрепленные триметилсилилированием енольные формы в присутствии хиральных катализа-
катализаторов Льюиса, например комплексов титана с хиральными лигандами.
Альдольная реакция с участием ацетальдегида имеет промышленное значение для получе-
получения бутадиена через бутандиол-1,3 (см. разд. Г,7.3.2). Побочным продуктом этого синтеза яв-
является кротоновый альдегид, гидрированием которого получают масляный альдегид и бутанол
(см. табл. Г.3.37). Продукт кротоновой конденсации масляного альдегида также получают в
промышленности и гидрируют до 2-этилгексанола (см. разд. Г,7.3.2). 2-Гидроксиметил-2-ме-
тилпропаналь (табл. Г.7.123) гидрируют до 2,2-диметилпропандиола-1,3, который является
сырьем для синтеза полиэфиров. О получении 1,1,1-трис(гидроксиметил)пропана и пента-
эритрита альдольной конденсацией см. разд. Г,7.3.1.3.
Из мезитилоксида в промышленности получают растворитель лаков изобутилметилкетон,
а также 4-метилпентанол-2.
В первом синтезе антибиотика хлоромицетина (хлорамфеникола) также использовали
альдольную реакцию A949 г.):
122 Г Препаративная часть (продолжение)
(^ У- СНО + СН2-СН2ОН <( )^СН-СН-СН2ОН
NO2 ОН NO2
1. Ацетилирование
/ \ 2 Нитоовзнив / \
<^ У-сн-сн-сн2он :—— о2и—(\ /Ь-сн-сн-сн2оас [г.7.126]
ОН NH2 OAc NHAc
1. Гидролиз
2. +CI2CHCOOCH3
Л\ /У— СН-СН-СН2ОН
ОН NH-COCHCI2
Промышленный синтез пантолактона, необходимого для получения пантотеновой кисло-
кислоты, начинается с альдольной конденсации изомасляного альдегида с формальдегидом:
ОН
Ознакомьтесь также с получением псевдоионона из цитраля!
Используя принцип альдольно-кротоновой конденсации, можно вводить в
реакцию с альдегидами целый ряд соединений, обладающих С—Н-кислот-
ностью. При этом не всегда можно провести четкую границу между этой реакци-
реакцией и реакцией Кнёвенагеля (см. разд. Г,7.2.1.4).
При синтезе Перкина альдегиды взаимодействуют с ангидридами алифатических карбо-
новых кислот, причем образуются а,р-ненасыщенные карбоновые кислоты. В качестве осно-
оснований, служащих конденсирующими средствами, используют щелочные соли карбоновых
кислот или третичные основания (пиридин). Лучше всего идет реакция с ароматическими аль-
альдегидами, она приводит к образованию коричных кисло-»:
<Р /Я
Н3С-С Рп-СН=СН-С +но Рп-СН=СН-СООН
Ph-СНО + р ——- * р 2— [Г.7.128]
н3с-с. -Нг° н3с-с. + н3с-соон
\э чо
Аналогичным образом можно провести конденсацию с бензоиламиноуксусной (гиппуровой)
кислотой. В условиях реакции сначала образуется так называемый азлактон, который и реаги-
реагирует затем с карбонильным соединением:
о о н о
^0Н , ГЛ +R-CH0 R T^o [Г 7 1291
HN^/O -H2O Nss/ "Н2О NW Ll./.l^j
Г
Ph Ph Ph
Образующиеся ненасыщенные азлактоны могут быть омылены до а-оксокислот или
(после предварительного восстановления) до а-аминокислот (синтез аминокислот по Эрлен-
мейеру):
7. Реакции карбонильных соединений 123
н2
[Г.7.130]
о
Выход аминокислот повышается, если вместо гиппуровой кислоты использовать, напри-
например, гидантоин или роданин.
В жестких условиях в качестве метиленового компонента можно использо-
использовать также сложные эфиры карбоновых кислот. Они реагируют с ароматически-
ароматическими альдегидами и кетонами с образованием коричных кислот в присутствии
алкоголятов щелочных металлов в качестве катализаторов. [По отношению к
алифатическим кетонам сложные эфиры ведут себя как карбонильные компо-
компоненты (сложноэфирная конденсация, см. разд. Г,7.2.1.8). Объясните это разли-
различие в поведении сложных эфиров при их взаимодействии с алифатическими и
ароматическими кетонами.]
Ацетонитрил может конденсироваться с кетонами и ароматическими альде-
альдегидами в условиях межфазного катализа в присутствии концентрированного
раствора гидроксида калия; в методике к табл. Г.7.123, вариант Д, описано полу-
получение нитрилов коричных кислот:
Э + H3C-CN —ггг- <л />—CH=CH-CN ГГ 7 1311
-Н2О ^ /J i • • J
Более кислые сложные эфиры а-хлорзамещенных жирных кислот реагируют
в роли метиленового компонента как с альдегидами, так и с кетонами. При этом
сначала образуется хлоргидрин, который в условиях реакции немедленно
отщепляет хлороводород (реакция Дарзана—Кляйзена):
\ ?н Я\
С=О + CH2-COOR ^^ —C-CH-COOR u - —C-CH-COOR [Г.7.132]
/ I |i -HCI /
' Cl ! Cl '
Несколько препаративных примеров этой реакции можно найти в описании
методики сложноэфирной конденсации (см. разд. Г,7.2.1.8).
Полученные таким способом 2,3-эпоксиэфиры (глицидные эфиры) при
омылении декарбоксилируются и перегруппировываются в альдегиды:
О о \
—C-CH-COOR — С-СН-СООН ——— СН-СНО [Г.7.133]
В условиях межфазного катализа из хлорацетонитрила аналогично реакции
[Г.7.132] в присутствии гидроксида натрия можно получить 2,3-эпоксипропио-
нитрилы (см. разд. Г,2.4.2).
124
Г Препаративная часть (продолжение)
Таблица Г.7.134. Получение 2,3-эпоксипропионитрилов (оксиранкарбонитрилов)
[JonczykA., Fedorynski M., Makosza M. Tetrahedron Letters, 1972, 2395]
Продукт реакции
З-Фенилоксиранкар-
бонитрил-2
3,3-Пентаметиленоксиран-
карбонитрил-2
З-Метил-3-фенилоксиран-
карбонитрил-2
3-D- Метоксифенил)окси-
ранкарбонитрил-2
Исходное соединение
Бензальдегид
Циклогексанон
Ацетофенон
Анисовый альдегид
Т. кип., °С (мм рт. ст.)
130-135A4)
104-108 A4)
115-121 F)
160-165A4)
Выход, %
45
50
50
60
(ГП) Общая методика получения 2,3-эпоксипропионитрилов (табл. Г.7.134). Медленно каплям
^-^ при температуре 15—20 "С добавляют 0,2 моль хлорацетонитрила к интенсивно переме-
перемешиваемой смеси 0,22 моль альдегида или кетона, 40 мл 50%-ного гидроксида натрия,
50 мл дихлорметана и 1 г хлорида бензилтриэтиламмония или 1,5 мл аликвата 336. Перемеши-
Перемешивают при этой температуре еще 40 мин, разбавляют 30 мл воды, отделяют органическую
фазу, промывают дважды небольшим количеством воды, сушат сульфатом натрия и перего-
перегоняют.
В случае реакции с кетонами отказываются от дихлорметана и по окончании реакции при-
прибавляют 80 мл эфира.
В качестве других С-Н-кислотных соединений, реагирующих с альдегидами
как метиленовые компоненты, можно назвать, например, а- и y-пиколины, a
также циклопентадиен. Объясните природу С—Н-кислотности этих соедине-
соединений!
Иллюстрацией такого рода синтезов может служить получение кониина — первого алка-
алкалоида, синтезированного Ладенбургом в 1886 г.:
ОНС-СНз
-Н2О
+ 4Н2
СН=СНСН3
С Н 2С Н 2С Н з
[Г.7.135]
Из 2-метилпиридина и формальдегида через промежуточный 2-B-гидроксиэтил)пиридин
в промышленности получают мономер 2-винилпиридин.
При синтезе алкилпиридинов в промышленности промежуточными стадиями также явля-
являются реакции альдольно-кротоновой конденсации, связанные с образованием иминов и дегид-
дегидрированием. Так, из ацетальдегида и аммиака с использованием в качестве катализатора аце-
ацетата аммония в водной фазе при температуре около 250 °С и 100-200 атм образуется в основном
2-метил-5-этилпиридин (МЭП), дегидрированием которого получают 2-метил-5-винилпири-
дин (МВП), используемый при получении синтетических каучуков. С другой стороны, при окис-
окислении МЭП получают никотиновую кислоту (см. разд. Г,6.2.1, табл. Г.6.14). Если взаимодействие
ацетальдегида с аммиаком осуществлять над А12О3 при 450 °С, то образуется смесь
а- и у-пиколина, в тех же условиях в присутствии формальдегида получают пиридин и E-пиколин.
Как показывают эти примеры, альдольно-кротоновая конденсация чрезвы-
чрезвычайно многогранна и имеет важное препаративное значение для создания свя-
связей С-С.
7. Реакции карбонильных соединений 125
7.2.1.4. Реакция Кнёвенагеля
Конденсация Кнёвенагеля в узком смысле является особым случаем альдоль-
но-кротоновой конденсации, когда используемые метиленовые компоненты
имеют особенно большую С—Н-кислотность. К таким соединениям относятся
вещества, в которых метиленовая группа активирована двумя группировками,
как это имеет место в малоновой кислоте, ее моно- и диэфирах, циануксусной
кислоте и ее эфирах, динитриле малоновой кислоты, C-дикетонах и других сое-
соединениях. Вследствие возможности сопряжения двойной связи с р-дикарбо-
нильной системой реакция всегда сопровождается дегидратацией и приводит к
соответствующим ненасыщенным перекрестно сопряженным соединениям,
например:
\ /CN \ /CN
с=о + н2с/ —- с=с' + н2о [Г.7.136]
/ COOR / COOR
Здесь очевидна аналогия с альдольной конденсацией, в результате которой
образуется система двух сопряженных связей (общая методика к табл. Г.7.123;
варианты Г, Д). Поэтому реакцией Кнёвенагеля в широком смысле можно
считать также все виды альдольно-кротоновой конденсации, катализируемые
основаниями.
С более реакционноспособными из названных метиленовых соединений,
прежде всего с циануксусной кислотой и ее эфирами, а также с нитрилом ма-
малоновой кислоты, хорошие выходы дают и альдегиды, и кетоны, а менее реак-
ционноспособные метиленовые соединения гладко реагируют только с арома-
ароматическими альдегидами. В качестве катализаторов используют пиперидин,
ацетат аммония, р-аланин и другие соединения в присутствии ледяной уксус-
уксусной кислоты.
Реакцию некоторых более инертных реагентов, например эфира малоновой
кислоты с кетонами, можно осуществить в смеси тетрагидрофуран—пиридин в
присутствии тетрахлорида титана. Получение этим методом алкилиденмалоновых
эфиров: Lehnert W. Tetrahedron Letters, 1970, 4723; Tetrahedron, 1973, 29, 635.
На практике применяют чаще всего два варианта реакции. По варианту Ко-
упа образующуюся при реакции воду удаляют азеотропной отгонкой. Малоно-
Малоновые кислоты и их моноэфиры в этих условиях реагируют плохо. Для них лучше
применять вариант Кнёвенагеля—Дебнера (см. методику). При этом продукт ре-
реакции декарбоксилируется, в результате чего получаются а,р-ненасыщенные
монокарбоновые кислоты. Этот способ часто гораздо проще, чем классический
синтез Перкина. Кроме того, данный метод имеет еще одно преимущество — его
можно применять и к алифатическим альдегидам; при этом получаются
замещенные акриловые кислоты. (См. получение циннамонитрилов альдоль-
альдольной конденсацией в условиях межфазного катализа, табл. Г.7.123, вариант Д.)
Как можно получить циннамонитрилы методом Кнёвенагеля—Дебнера?
(^J) Общая методика проведения реакции Кнёвенагеля (табл. Г.7.137)
А. Вариант по Коупу. В круглодонной колбе емкостью 500 мл, снабженной водоотделите-
водоотделителем и обратным холодильником, нагревают смесь 0,5 моль метиленовой компоненты (циан-
(циануксусной кислоты, малонового эфира, эфира циануксусной кислоты, нитрила малоновой
Таблица Г.7.137. Конденсация Кнёвенагеля
Продукт реакции
Изопропилиденциануксусная
кислота
Динитрил изопропилиден-
малоновой кислоты
Этиловый эфир З-метил-2-циано-
пентенкарбоновой-2 кислоты
Этиловый эфир циклогексилиден-
циануксусной кислоты
Циклогексилиденциануксусная
кислота^
Циклогексен-1 -ил-1 -ацетонитрил"
Этиловый эфир 3-метил-
2-цианокоричной кислоты
Диэтиловый эфир бутилиден-
малоновой кислоты
Диэтиловый эфир изобутилиден-
малоновой кислоты
Диэтиловый эфир бензилиден-
малоновой кислоты
Этиловый эфир кумаринкарбоно-
вой-3 кислоты
л-Диметиламинокоричная кислота'
Сорбиновая кислота
Исходное соединение
Циануксусная кислота, ацетон
Динитрил малоновой кислоты,
ацетон
Этиловый эфир циануксусной
кислоты, бутанон
Этиловый эфир циануксусной кислоты,
циклогексанон
Циануксусная кислота, циклогексанон
Цианоуксусная кислота, циклогексанон
Этиловый эфир циануксусной кислоты,
ацетофенон
Диэтиловый эфир малоновой кислоты,
масляный альдегид
Диэтиловый эфир малоновой кислоты,
изомасляный альдегид
Диэтиловый эфир малоновой кислоты,
бензальдегид
Диэтиловый эфир малоновой кислоты,
салициловый альдегид
Малоновая кислота, я-диметиламино-
бензальцегид
Малоновая кислота, кротоновый
альдегид
Вариант
А
А
А
А
А
А
А
А
А
А
Ад
Б
Б
Катализатор11
Ал
Ал
Ал
Ам
Ам
Ам
Ам
Пи
Пи
Пи
Пи
Т. кип. (мм рт. ст.),
°С °
Т.гш. 134
(ацетон — бензол)
101 A6)
117A1)
151 (9)
Т.гш. 110(MeNO2)
93A0)
120 B)
144B5)
136 B7)
186 A8); т. пл. 32
Т. пл.94
(этанол — вода)
Т. пл. 216 (разл.)
(этанол)
Т.гш. 134 (вода)
«D20
1,4262
1,4650
1,4950
1,4769
1,5468
1,4425
1,4398
1,5347Г
Выход, %
90
90
85
80
70
75
70
55
90
70
75
75
30
Продукт реакции
Коричная кислота
я-Метоксикоричная кислота
4-Гидрокси-З-метоксикоричная
(феруловая) кислота
.м-Нитрокоричная кислота
3-(Фурил-2)-акриловая кислота
Метиловый эфир 2-цианокорич-
ной кислоты
Нитрил 2-цианокоричной
кислоты
Метиловый эфир 2-циано-
л-метоксикоричной кислоты
Нитрил циннамилиденмалоновой
кислоты
Метиловый эфир циннамилиден-
циануксусной кислоты
Нитрил фурфурилиденмалоновой
кислоты
Исходное соединение
Малоновая кислота, бензальдегид
Малоновая кислота, анисовый альдегид
Малоновая кислота, ванилин
Малоновая кислота, х-нитро-
бензальдегид
Малоновая кислота, фурфурол
Метиловый эфир циануксусной кислоты,
бензальдегид
Нитрил малоновой кислоты,
бензальдегид
Метиловый эфир циануксусной кислоты,
анисовый альдегид
Нитрил малоновой кислоты,
коричный альдегид
Метиловый эфир циануксусной
кислоты, коричный альдегид
Нитрил малоновой кислоты,
фурфурол
Вариант
Б
Б
Б
Б
Б
В
В
В
В
В
В
Катализатора
Таблица Г.7.
Т. кип. (мм рт. ст.),
'С
Т. пл. 136 (вода —
этанол, 3:1)
Т. пл. 172 (этанол —
вода)
Т. пл. 173 (вода)
Т. пл. 203 (этанол)
Т. пл. 140 (гексан)
Т. пл.86
Т. пл.86
Т. пл.96
Т. пл.127
Т. пл.143
Т. пл. 225-228
(нитрометан)
137. Продолжение
«D20
Выход, %
85
50
80
85
85
82
85
85
80
80
80
а Ал — 0,01 моль р-аланина; Ам — 0,05 моль ацетата аммония; Пи — 0,02 моль пиперидина.
6 Промытую и высушенную реакционную смесь упаривают, вьщелившиеся кристаллы отфильтровывают и промывают холодным бензином.
в Промытую и высушенную реакционную смесь перегоняют в вакууме @,5—0,7 мПа). При этом циклогексилиденциануксусная кислота декарбок-
силируется и переходит в циклогексенилацетонитрил, который перегоняется при указанном давлении в интервале 130—140 °С. Дистиллят раство-
растворяют в толуоле, освобождают, как обычно, от кислоты и снова перегоняют в вакууме.
г Переохлажденный расплав.
д Берут только 0,01 моль ледяной уксусной кислоты.
е Для выделения продукта солянокислый раствор, полученный в результате реакции, обрабатывают водным аммиаком.
128 Г Препаративная часть (продолжение)
кислоты), 0,5 моль альдегида или кетона1, 0,01—0,05 моль соответствующего катализатора и
0,1 моль ледяной уксусной кислоты в 150 мл толуола. Реакция заканчивается, когда прекраща-
прекращается отделение воды B—6 ч). Смесь охлаждают, бензольный слой промывают 4 раза неболь-
небольшим количеством полунасыщенного раствора хлорида натрия, сушат сульфатом натрия и от-
отгоняют толуол. Остаток перекристаллизовывают или перегоняют.
Б. Вариант по Кнёвенагелю—Дебнеру. В круглодонной колбе на 500 мл растворяют
1,2 моль малоновой кислоты в ~ 180 мл сухого пиридина и после окончания слабоэкзотерми-
слабоэкзотермической реакции прибавляют 1 моль альдегида и 0,1 моль пиперидина. Затем нагревают на
водяной бане с обратным холодильником до прекращения выделения диоксида углерода,
охлаждают, выливают в смесь льда и концентрированной соляной кислоты, чтобы отмыть
пиридин и пиперидин.
Если карбоновая кислота выделяется при этом в твердом виде, то для завершения кристал-
кристаллизации оставляют на несколько часов в холодильнике, после чего отфильтровывают. Жидкие
продукты реакции извлекают толуолом или эфиром. И в том случае, когда выделяются твер-
твердые продукты реакции, выход часто может быть повышен, если маточный раствор дополни-
дополнительно подвергнуть экстракции. После сушки эфирных или толуольных вытяжек сульфатом
натрия отгоняют растворитель, а остаток перегоняют или перекристаллизовывают.
Подобно малоновой кислоте, можно вводить в реакцию ее моноэфиры, при этом получа-
получаются соответствующие ненасыщенные эфиры.
Оба варианта можно осуществить и в полумикромасштабе. При синтезе по Коупу берут
30-50 мл растворителя и водоотделитель объемом 1-3 мл.
В. Реакция ароматических альдегидов с С—Н-кислотными нитрилами.
1\ Внимание! Бензилиденмалононитрил и прежде всего его замещенные в бензоль-
• ном кольце производные раздражают кожу! Пары раздражают слизистые
i оболочки!
В колбе Эрленмейера емкостью 100 мл растворяют 0,1 моль альдегида и 0,1 моль нитрила
в 30 мл 70%-ного метанола и смешивают с 1,5—2 мл (в случае малононитрила только 1 мл) пи-
пиперидина. Через короткое время начинается экзотермическая реакция. Оставляют на два ча-
часа до окончания кристаллизации, осадок отфильтровывают, промывают небольшим количе-
количеством метанола, охлажденного до 0 °С, и перекристаллизовывают из небольшого количества
этанола.
По типу реакции Кнёвенагеля протекает синтез пирролов по Кнорру. При
этом а-аминокетоны (лучше а-амино-Р-оксоэфиры или а-аминодикетоны)
вводят в реакцию с C-дикарбонильными соединениями, например:
COOR НзС COOR
лх,
[Г.7.138]
Наряду с реакцией Кнёвенагеля при этом одновременно происходит конден-
конденсация кетонной группы с аминной, что приводит к замыканию кольца. а-Ами-
нокетоны могут быть получены восстановлением соответствующих изонитрозо-
кетонов (см. разд. Г,8.2.3).
При реакции триэтилового эфира ортомуравьиной кислоты с С—Н-кислотным и нитрила-
нитрилами получают а-алкоксиметиленнитрилы, из которых взаимодействием с амидинами образу-
образуются пиримидины. Например:
1 Для низших алифатических альдегидов и кетонов (до пентанона) лучше брать 0,6 моль
карбонильного соединения. Жидкие исходные вещества применяют только свежеперегнанные.
7. Реакции карбонильных соединений 129
HC(OEtK +
Чм -
toTT й0~
-сн-с —
NCN
NH2
зС-С
-вон
NH2
N ii^CN
НзС^%Г [Г.7.139]
4-амино-5-циано-
2-метилпиримидин
Они являются исходными соединениями для промышленного получения тиамина (вита-
(витамин Bi).
О синтезе 2-аминотиофенов из а-алкилиденнитрилов и серы, являющемся сочетанием
реакций Кнёвенагеля и Вильгеродта, см. разд. Г,6.2.3.2.
7.2.1.5. Реакция Манниха
Реакцией Манниха называют взаимодействие альдегидов (обычно формальде-
формальдегида) с первичными или вторичными аминами и С—Н-кислотными соединени-
соединениями. Реакция, как правило, осуществляется в кислой среде. Образовавшийся
сначала при обратимом гидролизе [Г.7.140] свободный амин обычным образом
реагирует с формальдегидом:
R2NH2 -=г— R2NH + Н® [Г.7.140]
Н2С=О + HNR2 ^^ Н2С
ОН +н©_н20
-
H2C
NR2 +H2O,-H®
Образовавшийся катион с делокализованным положительным зарядом мож-
можно рассматривать как азотистый аналог формальдегида, вступающий в нормаль-
нормальную катализируемую кислотами реакцию альдольного типа [Г.7.104] с енольной
формой С—Н-кислотного соединения:
°* / Н® Щ /
с-сн ^=^ с=с
/ \ / \
НО / @ НО© | О | @ [Г-7.142]
С=С + H2C-NR2 ^^ C-C-CH2-NR2 =^= C-C-CH2-NR2
/ \ / | / I b
соль основания Манниха
В результате реакции таким образом осуществляется «аминометилирование»
С—Н-кислотного соединения. (Составьте суммарное уравнение реакции!)
Основание Манниха обычно можно получить только в том случае, когда
участвующий в реакции амин обладает большей нуклеофильностью, чем
С—Н-кислотное соединение. В противном случае формальдегид преимущест-
преимущественно реагирует с метиленовым компонентом по типу альдольной реакции. Так,
например, из малонового эфира, формальдегида и диалкиламина нельзя полу-
получить основание Манниха.
Результат реакции в значительной степени определяется кислотностью сре-
среды, так как нуклеофильность С—Н-кислотного соединения и амина в различной
130 Г Препаративная часть (продолжение)
степени зависит от рН. Каждой отдельной реакции Манниха соответствует свое
оптимальное значение рН. Обычно удается добиться наиболее благоприятных
условий, применяя амины в виде гидрохлоридов или солей других кислот. В слу-
случае соединений, обладающих очень слабой С—Н-кислотностью, например
фенола или индола, применяют свободные основания или используют уксусно-
уксуснокислую среду.
Однородные продукты реакции получаются только при использовании вто-
вторичных аминов. При реакции с аммиаком и первичными аминами могут заме-
замещаться все атомы водорода, связанные с азотом. (Напишите уравнения
возможных превращений при взаимодействии ацетофенона, формальдегида и
аммиака.)
При наличии в метиленовом компоненте нескольких реакционноспособных
метальных или метиленовых групп (например, в ацетоне, циклогексаноне) его
всегда берут в избытке (около 4 молей), чтобы подавить образование бис-осно-
ваний Манниха. Как и во всех реакциях альдольного типа, катализируемых кис-
кислотами (см. схему [Г.7.104]), метиленовая группа в кетонах, например в бутано-
не, как правило, более реакционноспособна, чем метильная, что приводит к
образованию преимущественно разветвленных оснований Манниха.
В качестве С—Н-кислотных соединений можно использовать кетоны, альде-
альдегиды, алифатические нитросоединения, синильную кислоту и ацетилен. Кроме
того, по Манниху можно аминоалкилировать ароматические соединения, кото-
которые легко поддаются электрофильному замещению (см. табл. Г.5.2), например
фенолы или гетероциклические соединения (тиофен, пиррол, индол). Из индо-
индола таким образом получают грамин:
CH2N(CH3J
НСНО + HN(CH3J -7Г7Г Г II 7 [Г.7.143]
н
(QJ) Общие методики получения оснований Манниха (табл. Г.7.144)
А. Алифатические кетоны. Смесь 1,5 моль кетона, 0,3 моль формальдегида (в виде 35%-но-
го формалина) и 0,3 моль гидрохлорида амина нагревают 12 ч с обратным холодильником. За-
Затем упаривают в вакууме и гидрохлорид очищают перекристаллизацией. Для получения сво-
свободного основания гидрохлорид при перемешивании и охлаждении вносят в концентрирован-
концентрированный раствор гидроксида калия (при этом температура не должна превышать +5 °С), отделяют
выделившееся основание, сушат его небольшим количеством твердого КОН и перегоняют.
(При реакциях с циклогексаноном амин легко отщепляется от образующегося основания
Манниха, поэтому продукт реакции рекомендуется выделять в виде гидрохлорида.)
Б. Смешанные алкиларилкетоны. Смесь 0,3 моль кетона, 0,5 моль тонкоизмельченного па-
раформальдегида и 0,3 моль гидрохлорида амина в 50 мл абсолютного этанола нагревают до
кипения. Примерно через час прибавляют 0,5 мл концентрированной соляной кислоты; ос-
оставшийся параформальдегид при этом переходит в раствор. Горячую реакционную смесь
фильтруют, фильтрат охлаждают и отделяют выпавший гидрохлорид. Маточный раствор упа-
упаривают в вакууме и твердый остаток растирают с ацетоном. Полученный продукт соединяют с
ранее выделенным гидрохлоридом и перекристаллизовывают или выделяют в виде свободно-
свободного основания, как описано в варианте А.
Получение грамина [Kuhn H., Stein О. Вег., 1937, 70, 567]. Охлажденную льдом смесь 0,05 моль
диметиламина D0-50%-ный водный раствор), 7 г ледяной уксусной кислоты и 0,05 моль фор-
7. Реакции карбонильных соединений
131
Таблица Г.7.144. Получение а-A\1,Р\1-диапкиламинометил)кетонов по Манниху
Продукт реакции
(в виде гидрохлорида)
З-Пипершшно-1 -фенил-
пропанон-1
З-Диметиламино-1-фенил-
пропанон-2
З-Диметиламино-1 -D-ме-
токсифенил)пропанон-1
З-Диметиламино-2-метил-
1 -фенилпропанон-1
5-Пиперидино-1 -фенил-
пентен-1-он-З
4-Пиперидинобутанон-2
4-Морфолинобутанон-2
4-Диметиламино-З-фенил-
бутанон-2
2-Диметиламинометил-
циклогексанон
4-Диметиламинобутанон-2
4-Диэтиламинобутанон-2
4-Диметиламино-З-метил-
буганон-2в
Исходные вещества
Ацетофенон, гидро-
гидрохлорид пиперидина
Ацетофенон, гидро-
гидрохлорид диметиламина
я-Метоксиацетофе-
нон,гидрохлорид
диметиламина
Пропиофенон, гидро-
гидрохлорид диметиламина
Бензальацетон, гидро-
гидрохлорид пиперидина
Ацетон, гидрохлорид
пиперидина
Ацетон, гидрохлорид
морфолина
Фенилацетон, гидро-
гидрохлорид диметиламина
Циклогексанон, гидро-
гидрохлорид диметиламина
Ацетон, гидрохлорид
диметиламина
Ацетон, диэтиламин,
концентрированная
соляная кислота6
Буганон, гидрохлорид
диметиламина
Вариант
(см. с. 130)
Б
Б
Б
Б
Б
А
А
А
А
А
А
А
Т. пл. гидрохлорида,
°С
193 (этанол —
ацетон)
156 (этанол —
ацетон)
181 (этанол)
155 (ацетон)
186(изопропанол)
167 (этанол —
ацетон); т. кип.
основания 101 B0)а
149 (ацетон); т. кип.
основания 116 B0)а
156(ацетон)
158 (этанол — ацетон)
126 (ацетон); т. кип.
основания^ A3)а
77 (ацетон); т. кип.
основания 74 A5)а
Т. кип. основания
58A5)а
Выход, %
75
85
70
60
75
60
60
80
90
60
70
50
а Цифры в скобках соответствуют данлению (мм рт. ст.), которому отвечает данная температура кипения.
6 Эквимолярное количество.
" Гидрохлорид чрезвычайно гидроскопичен, поэтому продукт реакции выделяют в виде свободного
основания.
мальдегида (водный раствор) сразу приливают к 0,049 моль индола. При нагревании образуется
прозрачный раствор, который оставляют при комнатной температуре на несколько часов. Затем
раствор подщелачивают разбавленным раствором гидроксида натрия, выпавший осадок ос-
основания отфильтровывают, промывают водой и сушат в эксикаторе над КОН. Выход 98%;
т. пл. 134 °С (ацетон или гексан).
Псевдопельтьерж из 2-этокси-2,3-Дигидропирана, который предварительно
гидролизуется до глутарового альдегида (см. уравнение [Г.7.27]), метиламина и
ацетондикарбоновой кислоты: Соре А. С, Dryden H. L., Howell С. F. Org. Syntheses,
Coll. Vol. IV, 1963, p. 816. (Имеется перевод 1-го издания: Коуп А., Драйден X.,
Хоуэлл Ч. Синтезы органических препаратов. 9. — М.: ИЛ, 1959, с. 57.)
1-Диэтиламиногептин-2 из гексина-1: Jones E., Marszak J. Bader H. J. Chem.
Soc. (London), 1947, 1578.
Реакцию Манниха в первую очередь используют при синтезе N-замещенных
р-аминокетонов. Эти соединения могут обладать фармакологической актив-
132 Г Препаративная часть (продолжение)
ностью, как, например, анестезирующее средство фаликаин (гидрохлорид 3-пи-
перидино-4'-пропоксипропиофенона).
Реакция Манниха имеет также большое значение при синтезе ряда алкалоидов. Так, нап-
например, тропинон — промежуточный продукт в синтезе атропина — получают двойной конден-
конденсацией по Манниху из янтарного диальдегида, метиламина и ацетондикарбоновой кислоты:
НООС.
ТУСНЗ ^Г \ДгСНз [Г7.145]
НООС НООС
Синтез можно проводить в «физиологических» условиях (комнатная температура, буфер-
буферный раствор).
Кроме того, основания Манниха имеют препаративное значение для синтеза а,р-ненасы-
щенных кетонов (см. разд. Г,3.1.6) и для алкилирования р-дикарбонильных соединений. При-
Примером служит синтез 2-ацетамидо-2-скатилмалонового эфира из грамина и ацетаминомало-
нового эфира:
COOR
CH2-N(CH3J CH2-C-NHCOCH3
N COOR
Н Н
Гидролизом и декарбоксилированием получают триптофан (см. схему [Г.7.68]).
С реакции Манниха начинается также синтез ранитидина — важного блокатора гистамин-
Нг-рецептора.
(CH3JNH + (Н2СО)Х + [ГА .- // \\
\-/^СН,ОН (CH3JNCH2'^^ri'^-CH2OH [Г.7.147]
^~'' "" (ранитидин)
7.2.1.6. Ацилоиновая конденсация и обращение полярности
Сочетание синтеза циангидринов (схема [Г.7.106]) и альдольной конденсации назы-
называют бензоиновой или в более общем плане ацилоиновой конденсацией, в ходе кото-
которой две молекулы ароматического альдегида реагируют между собой в присутствии
каталитических количеств A0—20%) цианида калия. Как и следует ожидать, побочной
реакцией является реакция Канниццаро. Бензоиновая конденсация обратима:
[Г.7.148]
Общая методика ацилоиновой конденсации ароматических альдегидов (табл. Г.7.149)
Внимание! Цианиды щелочных металлов — сильные яды (см. часть Е).
,0
Аг-С' + CN0 =====
Vl
О ОН
и i
===== Аг-С-СН-Аг
IOI0
Аг-С-Н
CN
+ с№
он
==^ Ar-CI0
CN
1
+ Аг-с'
Н
- Аг-с'
Н
НО 101®
Аг-С-С-Аг
| |
NC Н
7. Реакции карбонильных соединений
133
Таблица Г.7.149. Получение ацилоинов
Конечный продукт
Бензоин
2,2'-Фуроин
4,4'-Диметилбензоин
«-Анизоин
Исходное соединение
Бензальдегид
Фурфурол
я-Толуилальдегид
Анисовый альдегид
Т. пл., "С
134
134-136
87-88
111-112
Выход, %
85
60
55
38
Раствор 0,1 моль альдегида и 2 г цианида калия в 30 мл 60%-ного этанола нагревают с обрат-
обратным холодильником. С бензальдегидом реакция заканчивается через 15 мин, с фурфуролом —
через 1 ч. С другими альдегидами нагревают 2 ч, добавляя через 1 ч еще 1 г цианида калия. Если
после охлаждения не выпадают кристаллы, реакционную смесь оставляют на ночь в холодиль-
холодильнике и в случае необходимости встряхивают для инициирования кристаллизации. Осадок
отфильтровывают, промывают водой и перекристаллизовывают из этанола.
Промежуточно образующийся карбанион I (схема [Г.7.148]) не может захва-
захватываться другими электрофильными агентами и использоваться для создания
других связей С—С. Исключением является реакция I с винилогами карбониль-
карбонильных соединений (см. разд. Г,7.4.1.3; реакция Михаэля). Однако если к альдегиду
вместо HCN присоединяется триметилсилилцианид (см. разд. Г,2.7), то образу-
образуются слабые С—Н-кислоты — а-триметилсилилоксинитрилы I (схема [Г.7.150]).
Последние могут депротонироваться диэтиламидом лития до карбанионов II
(схема [Г.7.150]), которые легко реагируют с электрофильными агентами. На
последней стадии реакционный центр в результате гидролиза снова превраща-
превращается в карбонильную группу. С альдегидами и кетонами таким путем получают
ацилоины FVa (схема [Г.7.150]), а с алкилгалогенидами, имеющими нуклео-
фильный заместитель, — кетоны заданной структуры IV6:
,Р
Аг-С
+ Me3SiCN
OSiMe3
Ar-CH
CN
I
OSiMe3
Ar-CI0
CN
OSiMe3
Ar-CI0
CN
0
i-R'-C-R2
M^SiO 101е
Ar-C-C-R2
i i
NC R
hH2O
О ОН
II I
Ar-C-C-R2
R'
[Г.7.150]
IV
OSiMe3
Ar-CIQ
CN
-Br-
OSiMe3
Ar-C-R
CN
нОН
,P
Ar-C
R
IV
134 Г Препаративная часть (продолжение)
Таким путем с помощью временно вводимой группировки можно превра-
превратить электрофильный реакционный центр карбонильной группы в нуклеофиль-
ный. Такой процесс называют обращением полярности, иными словами, обрати-
обратимым изменением полярности реакционного центра (см. разд. В,8.1).
Вместо силилированных производных можно использовать ацилированные циангидрины
(см. табл. Г.7.41) или продукты присоединения циангидринов к виниловым эфирам.
Получение кетонов из ароматических альдегидов и алкилгалогенидов с по-
помощью триметилсилилцианида: Deuchert К., Hertenstein U., Hiinig S., Wehner G.
Chem. Ber., 1979,112, 2045.
По новому варианту алкилирования альдегидов до кетонов вместо высокотоксичного и
дорогого триметилсилилцианида [или вместо упомянутого в конце этой главы сильнопахну-
щего пропандитиола-1,3 (его получение см. разд. Г,7.1.3)] используют легкодоступный бензо-
триазол: Katritzky A. R., LangH., WangZ., ZhangZ., SongH., J. Org. Chem. 1995, 60, 7619.
Такое изменение полярности карбонильной группы алифатических и аромати-
ароматических альдегидов можно осуществить также препаративно и легко с помощью ка-
каталитических количеств @,05—0,1 эквивалента) солей 3-алкил-1,3-тиазолия в при-
присутствии оснований. При этом тиазолиевые соли в атмосфере азота первоначально
депротонируются с образованием илида (З-алкилтиазолий-2-карбениата I
[Г.7.151]), являющегося истинным катализатором. Этот илид I нуклеофильно при-
присоединяется к альдегиду, изменяя таким образом его полярность. Образующийся
при этом аддукт II (замещенный тиазолий-2-метанат) реагирует далее с другой мо-
молекулой альдегида с образованием ацилоина (алифатические ацилоины так полу-
получать легче, чем методом восстановительного сочетания из сложных эфиров; см.
[Г.7.262]). Если в реакцию вводятся а,р-непредельные карбонильные соединения,
то карбанион II (III) предпочтительно присоединяется к их С=С-связи по типу ре-
реакции Михаэля (разд. Г,7.4.1.3) с образованием 1,4-дикарбонильных соединений
(у-дикетонов), эфиров 4-оксокарбоновых кислот, а также 4-оксонитрилов:
г—S /7-S ° л-S 101® г-S ОН
ГС1в -Et3NH@CIS "Г * °* * ?
R1 R1
I
S НО Ю1° /—S lOlfoH О ОН
III
i i i i i
R1
?, . г-SHO R3
R1
OR3 О
ii i и
> I + R2-C-CH-CH2-C-R4
Синтез ацилоинов конденсацией алифатических альдегидов, а также с фурфу-
фурфуролом в присутствии 3-бензил-5-B-гидроксиметил)-4-метил-1,3-тиазолийхло-
рида: StetterH., Kuhlmann H. Org. Synth. 1984, 62, 170.
7. Реакции карбонильных соединений
135
Получение гидроксиметилкетонов перекрестной ацилоиновой конденсацией
с формальдегидом в присутствии 3-этил-1,3-тиазолийбромида: Matsumoto Т.,
Ohishi M., Inoue Sh., J. Org. Chem. 1985, 50, 603.
Обращение полярности в альдегидах можно осуществить также через дитиоацетали (см.
разд. Г,7.1.3). Напишите схему синтеза 1,2-дикарбонильного соединения через следующие
стадии: образование 2-фенилдитиана-1,3 из бензальдегида и пропандитиола-1,3, депротони-
рование бутиллитием, ацилирование ацилхлоридом, гидролиз.
7.2.1.7. Реакции альдегидов и кетонов со сложными эфирами
алкилфосфоновых кислот и алкилиденфосфоранами
7.2.1.7.1. Реакция Хорнера - Вадсворта - Эммонса
Эфиры алкилфосфоновых кислот, которые можно получить реакцией Михаэлиса —
Арбузова (см. разд. Г,2.6.5.2) или из диэтилфосфита и алкилгалогенидов, являются
С—Н-кислотами которые можно депротонировать сильными основаниями.
Однако вследствие большого сродства фосфора к кислороду за альдольным при-
присоединением образовавшегося карбаниона I (схема [Г.7.152]) к карбонильным сое-
соединениям следует элиминирование диэтилфосфата II (схема [Г.7.152]), в результате
чего образуются олефины (реакция Хорнера — Вадсворта — Эммонса):
О
(ЕЮJР
R
СН
R1
NaH
-Н2
IOI0 R
(ЕЮJР-С|
(EtOJP =
R1
R1
Na
©
О
О
и
¦*P(OEtJ
Ce
R''~VR
0IOI P(OEtJ
i
i
—C-C-R
i
R'
IOI0
OjP(OEtJ
' — C-^C-R
I R'
[Г.7.152]
-(EtOJP-O
О
Олефинирование протекает гладко, если депротонированию способствует
заместитель Ri (например, фенильная, карбонильная или нитрильная группа;
R.2 в большинстве случаев — атом водорода). Эта реакция осуществляется в при-
присутствии алкоголятов, а в условиях межфазного катализа даже в присутствии
гидроксида натрия. С ненасыщенными альдегидами образуются диены. Если,
Таблица Г.7.153. Получение стильбенов и 1,4-дифенилбутадиенов-1,3 (реакция
Хорнера - Вадсворта - Эммонса)
Продукт реакции
Стильбен
4-Метилстильбен
4-Хлорстильбен
4-Метоксистильбен
1,4-Дифенилбутадиен-1,3
1-(л-Диметиламинофенил)-
4-фенилбутадиен-1,3
Исходное соединение
Бензальдегид
я-Толуил альдегид
л-Хлорбензальдегид
Анисовый альдегид
Коричный альдегид
и-Д и метиламинокоричн ый
альдегид8
Т. пл., °С
122-124 (этанол)
117 (этанол)
129 (этанол)
136 (этанол)
150-151 (этанол)
168—170 (пропанол)
Выход, %
70
65
65
75
60
50
Растворить в 30 мл толуола.
136 Г Препаративная часть (продолжение)
например, исходить из 1,4-дихлорбутена-2, то можно получить дианион, а из не-
него — триен или (с ненасыщенными альдегидами) полиен. Растворимость или
гидролизуемость отщепляющейся диалкилфосфатной группы может препят-
препятствовать реакции. Если R — карбонильная группа, a R' = Н, то реакция с альде-
альдегидами протекает высокостереоселективно с образованием а,C-непредельных
карбонильных соединений с ^-конфигурацией у кратной связи.
©Общая методика реакции Хорнера — Вадсворта — Эммонса с диэтиловым эфиром бензил-
фосфоновой кислоты (табл. Г.7.153) [Piechucki С, Synthesis 1976, 187]
В трехгорлую колбу емкостью 100 мл, снабженную мешалкой, холодильником и капель-
капельной воронкой, помещают двухфазную смесь 20 мл толуола, 20 мл 50%-ного гидроксида натрия
и 1,5 мл аликвата 336 и при эффективном перемешивании приливают по каплям раствор
25 ммоль диэтилового эфира бензилфосфоновой кислоты и 25 ммоль свежеперегнанного аль-
альдегида в 10 мл толуола. Затем нагревают при перемешивании еще 30 мин при 90 °С с обратным
холодильником. После охлаждения отделяют толуольный слой, промывают 5—10 мл воды,
сушат сульфатом натрия и отгоняют в вакууме растворитель. Остаток перекристаллизовывают.
Отнести стильбены к (?)- или B)-форме можно, определив температуру плавления, и с
помощью спектроскопии ЯМР (в CDC13).
7.2.1.7.2. Реакция Виттига
В реакции Виттига используют соли алкилтрифенилфосфония (см. разд. Г,2.6.5.1).
Их депротонирование гидридом натрия или алкиллитием дает фосфонийилиды I1;
см. схему [Г.7.154] (из формулы II на той же схеме становится более понятным назва-
название «алкилиденфосфораны»). Не являясь ионами, илиды обладают ярко выражен-
выраженным нуклеофильным центром на атоме углерода, который может реагировать с
карбонильными соединениями. Вслед за присоединением к карбонильной группе
происходит элиминирование трифенилфосфиноксида и образование олефина:
[Г.7.154]
В этой реакции двойная связь С=С образуется только на месте карбонильной
группы, изомеризация происходит в виде исключения только в реакциях неко-
некоторых циклических кетонов. Реакция Виттига [Г.7.154] и ее вариант реакция
Хорнера—Вадсворта—Эммонса являются важнейшими методами направленно-
направленного создания двойных связей С=С. В целом эти реакции представляют собой син-
синтезы олефинов из карбонильных соединений и алкилгалогенидов. Напротив,
альдольно-кротоновой конденсацией и реакцией Кневенагеля можно получить
только олефины с электроноакцепторными заместителями.
1 Илидом называют соединение, в котором отрицательно заряженный атом углерода непос-
непосредственно связан с гетероатомом (Р, N, S), имеющим положительный заряд. В названии отра-
отражено то обстоятельство, что гетероатом и атом углерода связаны как атомной связью (ил), так и
ионной (ид). Илиды аммония можно записать только в такой форме, в то время как для фосфо-
фосфора возможна структура с октетом электронов (участие электронов d-орбитали). В этом случае
можно воспользоваться наряду с формой «илида» I (схема [Г.7.154]) также формой «илена» II.
© R
РпзР-СН
R1
0
/C\ + R
-H®
Г3-
© 0,R
Ph3P-CI —
, R'
—C-^C-R1 -
1 R
Ph3P = C
II
- Ph3P = O
R'
\ ,
- c-c
7. Реакции карбонильных соединений 137
Реакционная способность фосфонийилидов в значительной степени зависит
от заместителей у илидного атома углерода и в несколько меньшей степени от
заместителей у атома фосфора. Незамещенный метилентрифенилфосфоран в
высшей степени нуклеофилен и неустойчив. Электроноакцепторные группы R
или R' (например, в солях фенацилфосфония) благоприятствуют депротониро-
ванию до илидов, однако снижают их нуклеофильность. Так, ацилалкилиден-
трифенилфосфораны (R = RCO), которые легко получаются ацилированием
алкилиденфосфоранов ацилхлоридами с промежуточным образованием соотве-
соответствующих фосфониевых солей (напишите реакцию), устойчивы к гидролизу на
холоду и реагируют только с очень активными карбонильными соединениями,
например бензальдегидом. В этом случае мы имеем дело со стабилизированны-
стабилизированными илидами.
Стереоселективность реакции Р-илидов с альдегидами в значительной мере
зависит от того, лабильны илиды или стабильны.
Первой стадией взаимодействия альдегидов с алкилидентрифенилфосфоранами является
[2+2]-циклоприсоединение с образованием оксафосфетана, которое может привести к
получению двух диастереомеров. Это в свою очередь в огромной степени влияет на стереосе-
стереоселективность процесса. Так, лабильные илиды (R' = алкил) образуют, как правило, г<ис-окса-
фосфетаны [Г.7.155], тогда как стабильные илиды (с —Af-заместителями, например, R' = ацил)
преимущественно реагируют с образованием термодинамически более стабильных транс-ок-
сафосфетанов [Г.7.156].
На второй стадии реакции происходит стереоспецифический распад четырехчленного
кольца, при котором образуются трифенилфосфиноксид и олефин. При этом распад цис-ан-
замещенного оксафосфетана дает B)-олефин, тогда как при расщеплении т/гакс-замещенно-
го кольца получают (?)-олефин:
О-РРпз
О PPh3 ,
J, [I у' цис-оксафосфетан (Zl-олефин
П
O-PPh3 ^ /H
r/\. -Ph3P° " [Г-7.156]
гранс-оксафосфетан (Е)-олефин
Ярко выраженная стереоселективность имеет место только в отсутствие ионов лития
(безсолевые условия), поскольку большинство натрийсодержащих оснований, например,
амид натрия, способствуют депротонированию алкилтрифенилфосфониевых солей. В прису-
присутствии ионов лития даже из индивидуальных стереоизомерных оксафосфетанов образуется
смесь стереоизомерных бетаинов 1а и 16 [Г.7.157]), что приводит к образованию смеси B)- и
(?)-олефинов.
(?)-алкены можно получить с высокой стереоселективностью из лабильных илидов по
варианту Шлоссера реакции Виттига в присутствии солей лития. В этом случае промежуточ-
промежуточно образуется литий-бетаин 1, депротонирование которого при действии фениллития дает
новый илид II, протон ирование которого одним эквивалентом НС1 приводит стереоселек-
тивно к получению литий-бетаина 1а. Если же к смеси бетаинов 1а и 16 добавить трет-бут-
лат калия, то литий в 1а замещается на калий, что приводит к образованию /ирянс-оксафосфе-
тана III, стереоселективное расщепление которого дает исключительно (?)-олефин и трифе-
трифенилфосфиноксид.
138 Г Препаративная часть (продолжение)
LiO
R
LiO
©
PPh3
*R'
la
©
PPh3
+ PhLi
-PhH, -Li®
LiO
R
©
PPh3
\
R'
II
+ HCI
-Cle
LiO
У
R
©
P
_J
"r
la
[Г.7.157]
16
(t-BuOK)t O-PPh3 __ XR'
-Li® ,U4 -Ph3PO /^
R R1 R
(ГП) Общая методика получения олефинов по Витгигу (табл. Г.7.158)
А. Получение раствора фосфонийилида. В трехгорлую колбу с насадкой Аншютца циклогек-
саном смывают 0,2 моль гидрида натрия (см. часть Е) и несколько раз промывают н-пентаном.
Колбу снабжают обратным холодильником, мешалкой (при меньших количествах веществ
удобна магнитная мешалка), капельной воронкой с выравнивателем давления и газоподводя-
щей трубкой. Колбу несколько раз вакуумируют и наполняют сухим очищенным азотом. К хо-
холодильнику подсоединяют заполненный диметилсульфоксидом счетчик пузырьков и ток азо-
азота регулируют таким образом, чтобы в 1 мин через счетчик проходило 20-30 пузырьков. Из ка-
капельной воронки добавляют 100 мл тщательно высушенного диметилсульфоксида (см. часть Е)
и смесь перемешивают при 80 °С (температура бани) до прекращения выделения водорода
(около 45 мин). Смесь охлаждают в бане со льдом, добавляют раствор 0,2 моль обезвоженного
галогенида фосфония в 200 мл диметилсульфоксида. Перемешивают 10 мин при комнатной
температуре. Этот раствор используют в следующей стадии.
Б. Конденсации с карбонильными соединениями. К свежеприготовленному раствору илида
добавляют эквимолярное количество карбонильного соединения (твердые вещества — в виде
раствора в диметилсульфоксиде). Реакционную смесь перемешивают и при необходимости
нагревают (см. табл. Г.7.158). По окончании реакции смесь выливают в 300 мл воды, несколь-
несколько раз экстрагируют н-пентаном, объединенные пентановые вытяжки промывают водой,
сушат сульфатом натрия, растворитель отгоняют, а остаток очищают перекристаллизацией,
возгонкой, перегонкой или хроматографически.
Реакция Виттига, и в особенности реакция Хорнера — Вадсворта — Эммонса, применяет-
применяется в промышленности для синтеза витамина А, соответствующей кислоты, C-каротина и
стильбенов (из последних получают затем оптические отбеливатели).
7.2.1.8. Сложноэфирная конденсация
Сложноэфирная конденсация также относится к реакциям типа альдольной
конденсации. При этом образуются р-дикарбонильные соединения из эфиров
карбоновых кислот (в качестве карбонильных компонентов) и следующих
С-Н-кислотных соединений:
Таблица Г.7.158. Получение олефинов по Виттигу
Продукт реакции
Метиленциклогексан
а-Метилстирол
1 -Фенилбутадиен-1,36
1,1 -Дифенилэтен
1,1 -Бис(л-диметил-
аминофенил)этен
9- Винилантрацен
2- Метиленборнан
1,1 -Дифенилпропен-1
Исходные соединения11
Циклогексанон, бромид метилтри-
фенил фосфония
Ацетофенон, бромид метилтрифенил-
фосфония
Коричный альдегид, бромид метилтри-
фенил фосфония
Бензофенон, бромид метилтрифенил-
фосфония
Кетон Михлера, бромид метилтрифенил-
фосфония
Антраценкарбальдегид-9, бромид метил-
трифенилфосфония
(+)-Камфора, бромид метилтрифенил-
фосфония
Бензофенон, бромид этилтрифенил-
фосфония
Т. кип. (мм рт. ст.), °С
Т. пл. 103, /7D201,4516
Т. пл. 162, nD20 1,5360
78A1)
100 A,3); т. пл. 6
Т. пл. 122 (этанол)
Т. пл. 67 (петролейный эфир)
Т. пл. 70 (возг.)
Т. пл.49
Выход, %
85
75
60
80
70
70
70
95
Условия реакции
30 мин, комнатная
температура
1 ч, 65 °С
1 ч, комнатная
температура и
2 ч, 60 "С
1 ч, комнатная
температура
3 ч, 65 °С
10 ч, 65 °С
15 ч, 50 "Ог
3 ч, комнатная
температура; 2 ч,
60°Св
а Вместо бромидов фосфония можно использовать соответствующие иодиды (см. табл. Г.2.87).
6 При перегонке и хранении добавлять для стабилизации гидрохинон.
в После обработки пентановый раствор пропускают через колонку с активным оксидом алюминия, элюируют пентаном.
г Возгоняющийся вначале комплекс время от времени смывают пентаном.
140 Г Препаративная часть (продолжение)
1) эфиров карбоновых кислот (при этом образуются эфиры р-оксокислот):
О
2 R-CH2-COOR' =^= R-CH2-C-CH-COOR' + R'OH [Г.7.159]
R
2) кетонов (при этом образуются р-дикетоны):
О О 0 0
R1-C-OR' + R2-CH2-C-R3 ^=^= R1-C-CH-C-R3 + R'OH [Г.7.160]
R2
3) нитрилов (при этом образуются нитрилы р-оксокислот):
о о
R1-C-OR' + R2-CH2-CN =^^ R1-C-CH-CN + R'OH [Г.7.161]
R2
Вследствие относительно низкой реакционной способности сложноэфир-
ной группы (см. ряд [Г.7.3]) в качестве конденсирующих агентов применяют
сильные основания, преимущественно алкоголяты щелочных металлов. Ход
реакции полностью соответствует схемам [Г.7.100] и [Г.7.102], например:
[Г.7.162]
[Г.7.163]
R0H [Г.7.164]
Поскольку анион IV, образующийся на первой стадии реакции [Г.7.162],
является очень сильным основанием, то равновесие сильно сдвинуто влево. Все
же реакция протекает в соответствии с уравнениями [Г.7.163] и [Г.7.164], так как
в конечном продукте имеется сопряженная ненасыщенная система VI (енолят),
энергия которой относительно мала.
По этой причине сложноэфирная конденсация в присутствии алкоголята в качестве осно-
основания успешно протекает только в том случае, когда соединение, используемое в качестве ме-
тиленового компонента, может дать енолизующийся конечный продукт, т. е. оно должно
иметь не менее двух атомов водорода у а-углеродного атома. Поэтому эфир изомасляной кис-
кислоты в условиях конденсации Кляйзена1 не образует соответствующего C-оксоэфира.
Образующиеся на последней стадии реакции енолы представляют собой бо-
более сильные кислоты, чем спирты (см. табл. Г.7.99). Поэтому алкоголят натрия,
используемый в качестве конденсирующего средства, расходуется на нейтрали-
нейтрализацию и его следует брать по меньшей мере в эквимолярных количествах.
1 Конденсацией Кляйзена называют сложноэфирную конденсацию, осуществляемую с
алкоголятами натрия или калия.
RO0 + H3C-COOR
II
н3с-счх + н2с-с
OR OR
101е о
н3с-с-сн2-сч
OR OR
V
^^ ROH + Н2С-'с/
OR
III IV
^=^ н3с-с-сн2-с
OR 0
V
IOI0 о
=^=^ н3с-с=сн-сч
OR
VI
7. Реакции карбонильных соединений 141
Поскольку все стадии реакции являются равновесными, то выход можно по-
повысить, проводя реакцию не в избытке спирта, а в среде инертного растворите-
растворителя (см. методику на стр. 146, вариант А). Еще лучше удалять образующийся в
ходе реакции спирт из равновесной смеси либо путем отгонки (при необходи-
необходимости в вакууме), либо проводить конденсацию в присутствии щелочных метал-
металлов1 и следовых количеств спирта (см. методику, вариант В).
Выход можно повысить, кроме того, придав необратимый характер первой стадии. Это
возможно при использовании более эффективных, чем алкоголяты, конденсирующих аген-
агентов, например амида натрия2, гидрида натрия, трифенилметилнатрия («тритилнатрия») и
мезитилмагнийбромида.
Эти конденсирующие агенты, которые следует вводить в эквимолекулярных количествах,
из-за исключительно большой основности (см. значения рКа в табл. Г.7.99) переводят практи-
практически весь метиленовый компонент в анион, например:
R-H + Na®NH 2 - RGNa®+ NH3
+ ©/Ph _ + Ph
R-H + Na® IC-Ph ^®Na® + H-C-Ph
i
Ph
[Г.7.165]
CH3
R-H + H3C—<ч h— MgBr - R MgBr + H3C-
CH3 CH3
Этим путем удается осуществить сложноэфирную конденсацию с такими С-Н-кислотны-
ми соединениями, для которых реакция [Г.7.164] не является энергетически выгодной вслед-
вследствие невозможности образования сопряженной ненасыщенной системы VI. (Напишите схе-
схему сложноэфирной конденсации двух молекул эфира изомасляной кислоты с трифенилметил-
натрием.)
Можно было бы предполагать, что в соответствии со схемами [Г.7.162]—[Г.7.164] катион
щелочного металла алкоголята не должен оказывать влияния на ход конденсации по Кляйзе-
ну. Однако это не так; активность алкоголятов щелочных металлов возрастает в ряду Li < Na <
К < Rb < Cs. Это заставляет принять для многих конденсаций по Кляйзену механизм, хотя и
сходный принципиально со схемами [Г.7.162]—[Г.7.164], но предоставляющий катиону
щелочного металла роль центра координации для всех участников реакции, ориентируемых
таким образом в положение, наиболее благоприятное для реакции. Сначала вместо свободно-
свободного карбаниона IV в схеме [Г.7.162] образуется его соединение с катионом щелочного металла,
не проводящее электрического тока и, следовательно, имеющее гомеополярное строение или
являющееся ионной парой. Кроме того, катион металла координационно связывает карбо-
карбонильную группу карбонильного компонента, усиливая тем самым ее поляризацию. Дальней-
Дальнейшие электронные переходы протекают в циклическом комплексе:
1 Реакция в присутствии металлического натрия не всегда приводит к хорошим результатам,
поскольку при этом в значительной степени могут протекать побочные реакции [восстанови-
[восстановительные процессы с образованием а-дикетонов и а-оксоспиртов (ацилоинов); см разд. Г,7.3.3.].
2 Побочно могут образовываться амиды кислот.
142
Г Препаративная часть (продолжение)
+ R'OH
FTO
OR1
[Г.7.166]
Na
R'OH + R
Сложноэфирная конденсация двух одинаковых молекул сложных эфиров
представляет собой важный метод получения эфиров р-оксокислот1. Препара-
Препаративное значение конденсации двух различных сложных эфиров невелико, так
как в общем случае получается смесь различных продуктов реакции (см., одна-
однако, ниже реакции с участием эфиров щавелевой и муравьиной кислот). (Назови-
(Назовите возможные продукты реакции эфира пропионовой кислоты с эфиром уксус-
уксусной кислоты!)
Взаимодействие сложных эфиров с кетонами [Г.7.160] и нитрилами [Г.7.161]
протекает однозначно. В этих условиях сложный эфир выполняет роль карбо-
карбонильного компонента (ср. реакцию Дарзана — Кляйзена между сложными эфи-
рами и альдегидами [Г.7.132], где сложный эфир играет роль метиленового ком-
компонента.
При взаимодействии енолятов ариловых эфиров дизамещенных уксусных
кислот с циклогексаноном с хорошими выходами образуются C-лактоны (напи-
(напишите эти реакции).
Синтез 3,3-диметил-1-оксаспиро[3,5]нонанона-2 из фенилового эфира изомас-
ляной кислоты и циклогексанона: Wedler С, Schick H., Org. Synth. 1998, 75, 116.
Эта реакция тесно связана с синтезом эфиров р-гидроксикарбоновых кислот
из эфиров а-галогенкарбоновых кислот (реакция Реформатского [Г.7.222]).
В предыдущем случае фенолят представляет собой хорошо уходящую группу,
поэтому межмолекулярное взаимодействие промежуточно образующихся
алкоголятов у сложноэфирного карбонильного атома углерода чрезвычайно
выгодно.
Важными в препаративном отношении являются следующие специальные
случаи сложноэфирной конденсации:
1. Конденсация Дикмана. Так называют внутримолекулярную конденсацию
сложных эфиров дикарбоновых кислот с образованием циклических оксоэфи-
ров. (Напишите схемы циклизаций Дикмана, приведенных в табл. Г.7.169, по
1 Другим методом получения эфиров р-оксокислот является расщепление эфиров
а-ацил-р-оксокислот (см. разд. Г,7.2.1.9).
7. Реакции карбонильных соединений 143
варианту В, а также схему получения эфира циклогексанон-2-карбоновой-1
кислоты из эфира пимелиновой кислоты!)
В этой реакции максимальные выходы наблюдаются при образовании пяти-
и шестичленных колец. В случае высших дикарбоновых кислот выходы очень
низки. Эфир янтарной кислоты сначала реагирует по типу обычной межмолеку-
межмолекулярной сложноэфирной конденсации, а на второй стадии в результате внутри-
внутримолекулярной конденсации образуется эфир циклогександион-2,5-дикарбоно-
вой-1,4 кислоты. (Напишите схему реакции!)
2. Сложноэфирная конденсация с участием эфиров щавелевой и муравьиной
кислот. Эти эфиры, не имея а-метиленовой группы, обладают высокой карбо-
карбонильной активностью. (Почему?) Поэтому они однозначно реагируют при сме-
смешанной сложноэфирной конденсации с другими эфирами. (Напишите схемы
реакций эфира щавелевой кислоты с ацетоном и эфиром фенилуксусной кис-
кислоты, конденсации 2 молей бензилцианида с одним молем эфира щавелевой
кислоты, взаимодействия 2 молей эфира щавелевой кислоты с ацетоном. Про-
Продукт последней реакции под действием кислоты отщепляет воду и переходит в
эфир хелидоновой кислоты — производное у-пирона.) Полученные из щавеле-
щавелевого эфира и сложных эфиров или кетонов а-этоксалильные производные при
нагревании > 120 °С отщепляют оксид углерода и превращаются в C-дикарбо-
нильные соединения. Как правило, декарбонилирование происходит особенно
гладко у 3-монозамещенных соединений:
0 0 0 0
t [Г-7.167]
R О R
Этот метод используют для синтеза эфиров алициклических (З-оксокарбо-
новых кислот (например, эфира циклогексанон-2-карбоновой-1 кислоты) и
монозамещенных малоновых эфиров. Последние таким образом могут быть
получены в чистом виде, что при алкилировании малонового эфира в некото-
некоторых случаях невозможно или весьма затруднительно (см. разд. Г,7.4.2.1).
Фенилмалоновый эфир вообще нельзя получить фенилированием малонового
эфира.
Эфироальдегиды и оксоальдегиды, получаемые конденсацией эфиров муравьиной кисло-
кислоты с эфирами карбоновых кислот или кетонами, сильно енолизованы и существуют в виде
а-гидроксиметиленовых соединений, например:
О О ОН О
Д^ + А J^.JL + R"OH rn 1A81
Н OR" |R' H y^ R' [Г.7.168]
R R
а-Гидроксиметиленкетоны, не замещенные в а-положении (R' = Н), очень легко полиме-
ризуются с образованием тримеров, производных бензола (формилуксусный эфир дает эфир
бензолтрикарбоновой-1,3,5 кислоты). Поэтому подобные соединения выделяют только в фор-
форме натриевых солей.
Эфиры угольной кислоты могут вступать в реакцию с кетонами, образуя р-оксоэфиры, и с
нитрилами, образуя эфиры циануксусной кислоты. (Напишите схему реакции фенилуксусно-
го эфира с диэтилкарбонатом.)
Таблица Г.7.169. Сложноэфирная конденсация и реакция Дарзана
Продукт реакции
Ацетоуксусный эфир
н-Пропиловый эфир ацетоуксусной
кислоты
Изопропиловый эфир ацетоуксусной
кислоты
Этиловый эфир 2-метил-З-оксопента-
новой кислоты
Этиловый эфир 2-этил-З-оксогексановой
кислоты
Этиловый эфир 2,2,4-триметилпентановой
кислоты
Этиловый эфир а,у-дифенилацетоуксусной
кислоты
Диэтиловый эфир 2-оксо-З-метилянтарной
кислоты
Диэтиловый эфир 2-оксо-З-фенилянтарной
кислоты
Дибензоилметан
Ацетилацетон
Бензоилацетон
2-Гидроксиметиленциклогексанон
Исходные вещества
Этилацетат
н-Пропилацетат
Изопропилацетат
Этилпропионат
Этиловый эфир масляной кислоты
Этиловый эфир изомасляной
кислоты
Этиловый эфир фенилуксусной
кислоты
Этилпропионат, диэтилоксалат
Этиловый эфир фенилуксусной
кислоты, диэтилоксалат
Этилбензоат, ацетофенон
Ацетон, этилацетат
Ацетофенон, этилацетат
Циклогексанон, этил- или
метилформиат
Вариант
см. с. 146)
А, Б
А, Б
Б
Б
Б
Ба
А, Б
А, Б
А, Г
А, Б
А, Б
А, Б
А, Бв
Т. кип., °С
(мм рт. ст.)
71A2)
78A1)
69A1)
89A2)
104A2)
96A2)
Т. пл. 77 (этанол)
115A0)
Не перегоняется
(декарбонилированиеN
220A8);
т. пл. 78 (метанол)
136
129 A0); т. пл. 61
84A2)
1,4198
1,4240
1,4179
1,4228
1,4271
1,4212
1,4303
1,4465
1,5124
ттштшятття
$ЫХОД, %
75
75
50
50
55
25
70
50, 75
85
50,80
55
50,65
55
Ацетоуксусный эфир
Этиловый эфир циклопентанон-
2-карбоновой кислоты
Этиловый эфир 1-метилпиперидон-
4-карбоновой-З кислоты
Диэтиловый эфир 12,4,6-триоксогептанди-
карбоновой-1,7 кислоты
Этиловый эфир З-циано-3-фенилпиро-
виноградной кислоты
З-Оксо-2-фенилбутаннитрил
Метиловый эфир 3-фенилглицидной
кислоты
Метиловый эфир 3-л-метоксифенил-
глицидной кислоты
Метиловый эфир З-фенил-3-метил-
глицидной кислоты
Этилацетат
Диэтиладипинат
Метил-бис-B-карбэтоксиэтил)
амин
Ацетон, диэтилоксалат
Бензилцианид, диэтилоксалат
Бензилцианид, этилацетат
Бензилцианид, этиловый эфир
хлоруксусной кислоты
Анисовый альдегид, этиловый
эфир хлоруксусной кислоты
Ацетофенон, этиловый эфир
хлоруксусной кислоты
А, Б
В
Вг
Гд
Г
Р
Гж
Гж
рк
71A2)
103A3)
115D); 128
(т. пл. гидрохлорида)
Т. пл. 103 (лигроин)
Т. пл.126
Т. пл. 90 (водн. этанол)
130E)
145 @,7); 62 (т. пл.)
142A1)
1,4198
1,4519
1,4802
1,513
75
75
70
80
80
65
90
90
70
а Кипятить 5 ч.
6 После отгонки растворителя выделяют для дальнейшего использования.
в Нагревают с гидридом натрия 1,5 ч в гликолевой бане при 40 °С.
г Продолжительность реакции 30 мин; при обработке водную фазу подщелачивают карбонатом калия, дважды экстрагируют эфиром, сушат сульфатом
натрия и пропускают ток хлороводорода.
д Сначала вводят в реакцию половинное количество раствора алкоголята с 0,15 моль кетона и 0,3 моль эфира; затем нагревают 30 мин с обратным холо-
холодильником и добавляют вторую половину раствора алкоголята. После окончания реакции полностью отгоняют спирт на бане при 110 °С. Получают
смесь моно- и диенолов.
е Нагревают 2 ч на кипящей водяной бане; если после отгонки растворителя продукт не затвердевает, то его нагревают до 130 °С в вакууме водоструйно-
водоструйного насоса. Можно возогнать в вакууме при 100 "С (Ю-4 мм рт. ст.).
ж Работают с метилатом натрия в метаноле, при этом образуется продукт переэтерификации. Метиловые эфиры очень легко подвергаются дальнейшей
обработке (например, омылению).
146 Г Препаративная часть (продолжение)
Ф Общие методики сложноэфирной конденсации и синтеза глицидных эфиров по Дарзану
(табл. Г.7.169)
jj Осторожно обращайтесь с металлическим натрием! (См. также часть Е.)
A. Реакция с алкоголятом, не содержащим спирта. В трехгорлую колбу на 500 мл с обратным
холодильником, снабженным хлоркальциевой трубкой, капельной воронкой и мешалкой
Гершберга (см. рис. А.6,ж), помещают 0,5 моль освобожденного от корки металлического нат-
натрия, нарезанного крупными кусками, 250 мл сухого толуола и нагревают без перемешивания
до слабого кипения. Используя мотор для мешалки с большим числом оборотов, мешалку
приводят в быстрое вращение и перемешивают до тех пор, пока натрий не превратится в бело-
беловато-серую суспензию; при этом поддерживают слабое нагревание. После образования сус-
суспензии перемешивание прекращают и дают смеси охладиться. Ни в коем случае нельзя пре-
прекращать перемешивание до застывания частиц натрия, так как последние могут вновь соеди-
соединиться в более крупные образования.
К охлажденной суспензии при эффективном перемешивании, а при необходимости и при
охлаждении медленно добавляют 0,5 моль абсолютного спирта1. При этом температура смеси
не должна подниматься выше 85 °С, чтобы избежать повторного расплавления натрия и обра-
образования крупных частиц. В заключение нагревают еще 1 ч при 100 "С, после чего по каплям
добавляют смесь реагентов:
а) При синтезе fi-оксоэфиров карбоновых кислот добавляют 1,5 моль соответствующего
эфира (высушенного над пентаоксидом фосфора и перегнанного), после чего нагре-
нагревают реакционную смесь в течение 15 ч на кипящей водяной бане.
б) При синтезе $-дикетонов при охлаждении водой добавляют смесь 0,5 моль кетона и
1 моль сложного эфира (после предварительного высушивания обоих компонентов над
пентаоксидом фосфора и перегонки), после чего нагревают 4 ч на кипящей водяной бане.
в) При синтезе эфиров оксоянтарной и а-формилкарбоновых кислот (а-гидроксимети-
ленкарбоновых кислот) 0,5 моль диэтилового эфира щавелевой кислоты или
соответственно 0,5 моль эфира муравьиной кислоты смешивают с 0,5 моль карбоно-
вой кислоты и оставляют на ночь при комнатной температуре.
По окончании реакции отгоняют из реакционной смеси фракцию, кипящую ниже 100 °С
(температура бани до 120 °С), остаток охлаждают и к нему добавляют смесь 0,6 моль ледяной
уксусной кислоты со льдом (~33%-ная уксусная кислота). Органический слой отделяют, а вод-
водный несколько раз экстрагируют эфиром. Объединенные эфирные вытяжки тщательно про-
промывают водой и сушат сульфатом натрия. После отгонки растворителя остаток перегоняют
или перекристаллизовывают.
Б. Реакция с гидридом натрия. В литровой трехгорлой колбе, снабженной мешалкой, ка-
капельной воронкой и обратным холодильником с газоотводной трубкой, прибавляют по кап-
каплям при перемешивании к суспензии 0,5 моль гидрида натрия в циклогексане (см. часть Е)
смесь исходных веществ в количествах, указанных в методике А. После этого нагревают в те-
течение 3 ч с обратным холодильником (не пользоваться водяной баней, лучше всего применять
инфракрасный обогреватель). Охлаждают и обрабатывают, как указано в методике А.
B. Циклизация Дикмана с натриевым порошком. Готовят суспензию 0,5 моль металлическо-
металлического натрия в 500 мл толуола, как описано в методике А. К горячей суспензии при энергичном
перемешивании прибавляют по каплям смесь 0,5 моль соответствующего абсолютного эфира
дикарбоновой кислоты и 1 мл абсолютного этилового спирта. По окончании бурной реакции
смесь нагревают в течение 6 ч с обратным холодильником, охлаждают и к ней осторожно до-
добавляют смесь 200 г льда и 0,5 моль концентрированной соляной кислоты. Органический слой
отделяют, водный слой дважды экстрагируют эфиром, объединенные органические фракции
несколько раз промывают небольшим количеством воды и сушат над сульфатом натрия. Раст-
Растворитель отгоняют, остаток перегоняют.
Г. Реакция со спиртовым раствором алкоголята. В трехгорлую колбу на 500 мл с обратным
холодильником, снабженным хлоркальциевой трубкой, капельной воронкой и мешалкой, по-
1 Используют тот спирт, остаток которого содержится в молекуле сложного эфира. Абсо-
лютирование спирта см. в части Е.
7. Реакции карбонильных соединений
147
Таблица Г.7.170. Декарбонилирование оксоянтарной и 2,4-диоксокарбоновых кислот
Продукт реакции
Этиловый эфир 2-оксоци-
клогексан-1 -карбоновой
кислоты
Диэтиловый эфир
фенилмалоновой кислоты
Диэтиловый эфир
метилмалоновой кислоты
Исходное вещество
Этиловый эфир
2-оксоциклогексил-
глиоксалевой кислоты
Диэтиловый эфир 2-оксо-
3-фенилянтарной кислоты
Диэтиловый эфир 2-оксо-
3-метилянтарной кислоты
Т. кип., °С
(мм рт. ст.)
107A2)
151A0)
83A3)
«о20
1,4794
1,4977
1,4126
Выход, %
80
67
95
мещают 0,3 моль металлического натрия и 300 мл абсолютного спирта (продажный абсолют-
абсолютный спирт предварительно обезвоживают, как указано в части Е). После полного растворения
натрия при перемешивании и охлаждении ледяной водой добавляют по каплям смесь, в кото-
которую входит по 0,3 моль обезвоженных исходных веществ.
При получении глицидных эфиров прибавляют смесь 0,2 моль карбонильного компонента и
0,3 моль этилового эфира хлоруксусной кислоты (последний частично расходуется на побоч-
побочную реакцию образования эфира алкоксиуксусной кислоты). Температуру поддерживают на
уровне —10 °С. Реакционную смесь оставляют на ночь при комнатной температуре, нейтрали-
нейтрализуют эквимолярным количеством ледяной уксусной кислоты и выливают в 1 л ледяной воды.
Водный слой несколько раз экстрагируют эфиром (твердый продукт реакции отфильтровыва-
отфильтровывают). Эфирные вытяжки промывают водой и сушат над сульфатом натрия, растворитель
отгоняют, остаток перегоняют или перекристаллизовывают.
Этиловый эфир изовалерилуксусной кислоты из метилизобутилкетона и диэ-
тилкарбоната, этиловый эфир бензоилуксусной кислоты из ацетофенона и диэтил-
карбоната: BrandstromA. Acta Chem. Scand., 1950, 4, 1315.
Этиловый эфир а-цианофенилуксусной кислоты из бензилцианида и диэтил-
карбоната: Wallingford V. Н., Jones D. M., HomeyerA. H. J. Am. Chem. Soc, 1942,
64, 576.
Ф Общая методика декарбонилирования эфиров оксоянтарных и 2,4-диоксокарбоновых кислот
(табл. Г.7.170). В приборе для перегонки медленно нагревают в вакууме D0—50 мм рт. ст.)
соответствующий эфир (можно использовать непосредственно неочищенный продукт
реакции) в присутствии следовых количеств порошкообразного железа и борной кислоты
при температуре бани 140-170 "С. Точнее давление и температуру подбирают таким образом, что-
чтобы декарбонилирование происходило при возможно более низкой температуре без отгонки
исходного продукта. На отщепление оксида углерода указывает увеличение давления. При этом
отгоняется часть декарбонилированного эфира. По прекращении выделения газа температуру ба-
бани постепенно повышают (однако не выше 180 °С) и отгоняют остальной продукт, уменьшая в
случае необходимости давление. Неочищенный продукт еще раз перегоняют в вакууме.
При этом не приходится ожидать конденсации сложноэфирной группы с ме-
тиленактивными соединениями по типу реакции Кнёвенагеля (см. разд.
Г,7.2.1.4). Алкоксиметиленовые соединения, которые могли бы образоваться в
результате подобной реакции, все же можно получить, если исходить из орто-
эфиров карбоновых кислот, например:
OEt
ЕЮ-СН
OEt
COOEt
H2C
COOEt
(Ac2O)
- 2 EtOH
EtO
COOEt
C=C
COOEt
[Г.7.171]
148
Г Препаративная часть (продолжение)
Для проведения этой реакции требуется как относительно высокая С—Н-кис-
С—Н-кислотность метиленового компонента, так и безводная слабокислая среда. (Поче-
(Почему? Сравните реакции ортоэфиров и ацеталей.) Реакция начинается с отщепле-
отщепления спирта от ортоэфира после протонирования последнего.
©Общая методика конденсации ортомуравьиного эфира с метиленактивными соединения-
соединениями (табл. Г.7.172). В колбу емкостью 500 мл, соединенную посредством небольшой
колонки с установкой для перегонки, загружают смесь 0,75 моль ортомуравьиного
эфира, 0,5 моль метиленактивного соединения и 1 моль уксусного ангидрида, нагре-
нагревают в течение 1 ч при температуре бани 140 "С, затем 1 ч при 150 °С. При этом отгоняется
этилацетат. Затем колонку снимают и продукт реакции перегоняют в вакууме.
Этоксигруппа может легко гидролизоваться под действием кислот до (ено-
лизованной) альдегидной группы, а также замещаться на N- или С-основания
(см. разд. Г,7.4.1.5). Для подобных синтезов используют менее реакционноспо-
собные диметиламинометиленовые соединения, получаемые из диметилфор-
мамида (вместо ортоэфира) в присутствии ацетангидрида и РОС13 или SOC12
(ср. реакцию Вильсмейера, разд. Г,5.1.8.3).
В то время как простые альдегиды и кетоны практически полностью существуют в кетон-
ной форме (например, в ацетоне ее содержание составляет до 99,9998%), р-оксоэфиры и
C-дикетоны более или менее енолизованы. Например, в отсутствие растворителя ацетоуксус-
ный эфир содержит при комнатной температуре 7,5% енольной формы, ацетилацетон — 80%.
Кето-енольное равновесие зависит от растворителя. Отношение концентраций таутомеров
равно отношению их растворимостей в данном растворителе. Ацетоуксусный эфир и аце-
ацетилацетон образуют г<мс-еноляты с внутримолекулярными водородными связями. Поэтому
содержание енолов в неполярных растворителях выше, чем в полярных (ацетилацетон еноли-
зован в гексане на 95%, а в ацетонитриле — только на 62%). Все енолы, которые не образуют
внутримолекулярных водородных связей, проявляют обратную зависимость от полярности
растворителя.
Таблица Г.7.172. Получение эфиров а-этоксиметиленкарбоновых кислот конденса-
конденсацией с ортомуравьиным эфиром
Продукт реакции
Этиловый эфир
этоксиметиленциануксусной
кислоты
Этиловый эфир
а-этоксиметиленацетоуксусной
кислоты
Диэтиловый эфир
этоксиметиленмалоновой
кислотыа
Этоксиметиленмалононитрил
Этоксиметиленцианамид8
Исходное вещество
Этиловый эфир
циануксусной кислоты
Этиловый эфир
ацетоуксусной кислоты
Диэтиловый эфир
малоновой кислоты6
Малононитрил
Цианамид
Т. кип., "С
(мм рт. ст.)
173-174A5)
149-151 A5)
159-162A1);
ив20 1,4620
162-163A5);
т. пл. 63—65
57-63@,15)
Выход, %
82
75
55
85
75
а После повторной перегонки.
6 Добавляют 2 г ZnCl2.
" Относительно неустойчив.
7. Реакции карбонильных соединений 149
Енолы р-дикарбонильных соединений дают с хлоридом железа(Ш) окра-
окрашенные соли, имеющие строение хелатных комплексов:
I 12©
0 [Г.7.173]
2CI
Образование комплексов железа используют как качественную реакцию для
обнаружения р-дикарбонильных соединений (см. разд. Д, 1.2.5.2). Реакция про-
протекает уже при содержании 1—2% енолов. Малоновый эфир и его производные
енолизованы в незначительной степени, поэтому они не дают реакции с хлори-
хлоридом железа(Ш). Характерное для хелатного комплекса окрашивание наблюдает-
наблюдается только в спиртовом растворе. В водном растворе помимо этого комплекса
присутствуют простые соли, также окрашенные. Так, например, фенолы не со-
содержащие хелатообразующую группу, дают цветную реакцию с хлоридом желе-
за(Ш) только в водном растворе, что связано с образованием основных солей.
Среди важнейших препаративных методов образования связей С—С, кото-
которые рассматриваются в настоящей главе, сложноэфирная конденсация занима-
занимает особое положение, так как образующиеся р-дикарбонильные соединения и
их аналоги представляют собой вещества с тремя функциональными группами.
Этим объясняется многообразие других соединений, которые могут быть полу-
получены при использовании превращений кетогруппы (восстановление, см. разд.
Г,7.3; образование енаминов, см. схему [Г.7.11в]), атакже реакции метиленовой
группы (присоединение по Михаэлю, разд. Г,7.4.1.3; ацилирование, см. разд.
Г,7.2.1.10; алкилирование, галогенирование, см. разд. Г.7.4.2.1 и Г,7.4.2.2) и
реакции карбоксильной группы (омыление, кетонное расщепление, см.
табл. Г.7.64 и Г.7.66; образование амидов, см. разд. Г,7.1.4.2).
Р-Дикарбонильные соединения часто используют и как исходные вещества
для синтеза гетероциклов.
Техническое значение прежде всего имеет ацетоуксусный эфир, производные которого
(например, пиразолоны) используют для синтеза азокрасителей (см. табл. Г.8.35).
Фенилмалоновый эфир используют при синтезе снотворных средств — барбитуратов
(этилфенилбарбитуровая кислота, фенобарбитал, см. разд. Г,7.1.4.2).
Реакции типа сложноэфирной конденсации протекают также в организмах животных
(цикл жирных кислот, цикл лимонной кислоты). Например, при биосинтезе жирных кислот
концевой фрагмент связанного с белком эфира тиоуксусной кислоты реагирует с аналогично
связанным фрагментом эфира тиомалоновой кислоты с образованием связанного тиоэфира
ацетоуксусной кислоты:
0 0 0 0
HgC-C-S-ACP + H2C-C-S-ACP ¦ H3C-C-CH2-C-S-ACP
I - no AUr
COOH _Co2 [Г.7.174]
ацетил-АСР малонил-АСР ацетоацетил-АСР
(АСР — ацилпереносящий белок)
7.2.1.9. Сложноэфирное и кислотное расщепление р-дикарбонильных соединений
Поскольку все стадии конденсации Кляйзена обратимы (см. схемы [Г.7.162] —
[7.164] и [Г.7.166], то р-оксоэфиры и р-дикетоны можно расщеплять при помо-
помощи спиртового раствора алкоголята (сложноэфирноерасщепление), например:
150 Г Препаративная часть (продолжение)
о он e rioie он о р. о
и I +RO ^-1 с ' " — "
R-C-CH = C-R' R-CLCH=C-R- ^=^= R-C-OR + H2C-C-R'
[Г.7.175]
H2C-C-Rf + ROH
Промежуточный продукт I соответствует соединению V в схеме [Г.7.163].
Как следует из схемы [Г.7.175] и подтверждено экспериментально, енолят реагирует с
неенолизованной карбонильной группой. р-Оксоэфиры под действием алкоголятов енолизу-
ются практически полностью и расщепляются значительно труднее, чем C-дикетоны, кото-
которые, образуя еноляты, сохраняют еще одну реакционноспособную кетонную группу. Енолизу-
ющиеся одним путем C-дикетоны расщепляются преимущественно в одном направлении.
При действии на C-дикетоны или C-оксоэфиры щелочи вместо алкоголятов в
соответствии со схемой [Г.7.175] образуются анион кислоты и кетон или слож-
сложный эфир, который тотчас же омыляется. Анионы кислот вообще не проявляют
карбонильной активности, поэтому обратная реакция с образованием C-дикар-
бонильного соединения невозможна и расщепление идет до конца (кислотное
расщепление; см. табл. Г.7.178).
При кислотном расщеплении р-оксиэфиров в значительной степени осущес-
осуществляется побочная реакция — кетонное расщепление (см. табл. Г.7.64). Оно проис-
происходит в результате гидролиза и декарбоксилирования карбоксильной группы C-ок-
соэфира. Поэтому кислотное расщепление р-оксоэфиров в препаративном отно-
отношении представляет небольшой интерес и предпочтительнее получают соответ-
соответствующие кислоты через замещенные малоновые эфиры (см. схему [Г.7.65]).
Способность р-дикарбонильных соединений к расщеплению при действии ще-
щелочных агентов значительно возрастает при переходе от незамещенных в
а-положении соединений к ос-монозамещенным и к а,а-дизамещенным производ-
производным. Последние уже не способны к енолизации и щелочь не затрачивается на этот
конкурирующий процесс, поэтому сложноэфирное расщепление может осущес-
осуществляться в присутствии каталитических количеств щелочей. Эта реакция является
побочной при диалкилировании р-оксоэфиров, р-дикетонов и малоновых эфиров
(см. разд. Г.7.4.2.1).
ct-Ацил-р-оксоэфиры, получаемые ацилированием эфиров р-оксокислот,
содержат одноверменно группировки р-дикетона и р-оксоэфира. Из сказанного
ясно, что кислотное или сложноэфирное расщепление в этом случае всегда про-
протекает с разрушением р-дикетонной группировки. При действии спиртового
раствора алкоголята (или щелочи) образуется р-оксоэфир, который на холоду
дальше не изменяется и поэтому может быть легко выделен:
О
II
н3с-с-
О
II
-сн-с-
COOR
0
H3C-C-OR
+
О
щелочь II
-R —^ Н3С-С
О
R-C-CH2-COOR
О?
-с^с-
COOR
R
расщепление
эфирной связи
1. + ROH
2. + Н®
Согласно схеме [Г.7.175], вкетоне (р-оксоэфире) обнаруживается та из кетогрупп, которая
была енолизована; поэтому из а-ацил-р-оксоэфира всегда получают тот из двух возможных
7. Реакции карбонильных соединений
151
Таблица Г.7.177. Сложноэфирное расщепление ацилацетоуксусных эфиров
Продукт реакции
Этиловый эфир
бензоилуксусной кислоты
Метиловый эфир
бензоилуксусной кислоты
Метиловый эфир
4-фенилацетоуксусной
кислоты
Этиловый эфир
4-фенилацетоуксусной
кислоты
Этиловый эфир
н-бутирилуксусной кислоты
Этиловый эфир
изобугирилуксусной
кислоты
Исходное вещество
а-Бензоилацетоуксусный
эфир
а-Бензоилацетоуксусный
эфир
а-Фенилацетилацето-
уксусный эфир
а-Фенилацетилацето-
уксусный эфир
а-Бутирилацетоуксусный
эфир
а-Изобутирилацето-
уксусный эфир
Т. кип., °С
(мм рт. ст.)
137D)
122B,5)
125C)
120@,6)
94A5)
85A6)
«о25
1,5254
1,5372
1,5158
(при 20 °С)
1,5011
1,4245
Выход, %
70
70
85
75
90
40
р-оксоэфиров, который имеет большую склонность к енолизации. Обычно это оксоэфир с
большей ацильной группой. Поэтому а-ацилирование ацетоуксусного эфира с последующим
расщеплением открывает большие возможности для синтеза высших р-оксоэфиров, например
бензоилуксусного. (Почему бензоилуксусный эфир нельзя получить при помощи сложно-
эфирной конденсации?)
(ffj\ Общая методика сложноэфирного расщепления ацилацетоуксусных эфиров (табл.
^=^ Г.7.177). Смешивают 1 моль ацилацетоуксусного эфира1 с раствором 1,05 моль КОН в
500 мл этанола или метанола2 и оставляют на ночь. Затем реакционную смесь вылива-
выливают на 3 л льда, к которому добавлено 27 мл концентрированной серной кислоты, и экстраги-
экстрагируют четырьмя порциями по 200 мл эфира. Объединенные эфирные вытяжки промывают
водой почти до нейтральной реакции, сушат сульфатом магния и отгоняют растворитель в
вакууме. Остаток перегоняют в вакууме, используя 25-сантиметровую колонку Вигре.
Кислотное расщепление ос-ацилциклоалканонов представляет препаратив-
препаративный интерес для удлинения цепи карбоновых кислот, так как образующиеся ке-
токислоты можно легко восстановить по Кижнеру—Вольфу (см. разд. Г,7.3.1.6).
(Напишите схемы некоторых реакций, приведенных в табл. Г.7.178.)
Таблица Г.7.178. Кислотное расщепление а-ацилкетонов
Продукт реакции
6-Оксогептановая кислота
7-Оксооктановая кислота
6-Оксооктановая кислота
7-Оксононановая кислота
6-Оксононановая кислота
7-Оксодекановая кислота
Исходное вещество
2-Ацетилциклопентанон
2-Ацетилциклогексанон
2-Пропионилциклопентанон
2-Пропионилциклогексанон
2-Бутирилциклопентанон
2-Бутирилциклогексанон
Т. кип., °С
(мм рт. ст.)
123A)
161 D)
136A,5)
152B)
133@,5)
157B)
Т. пл.,
°С
35
29
52
42
35
Выход, %
55
50
50
70
70
40
1 Получение см. в табл. Г.7.182. Можно применять и неперегнанный продукт реакции.
2 При использовании этанола получают этиловые эфиры; в метаноле в результате пере-
этерификации образуются метиловые эфиры.
152 Г Препаративная часть (продолжение)
Ш Общая методика кислотного расщепления а-ацилкетонов1 (табл. Г.7.178). Смешивают
0,1 моль а-ацилциклогексанона (можно применять и неочищенный продукт) при
100 °С и непрерывном перемешивании с трехкратным молярным количеством горяче-
горячего 60%-ного раствора гидроксида калия и выдерживают еще 15 мин при этой темпера-
температуре. Застывшую реакционную смесь после охлаждения растворяют в 300 мл воды. Затем по
каплям добавляют такое количество концентрированной соляной кислоты, чтобы раствор ос-
оставался слабощелочным, и экстрагируют эфиром. Водный слой сильно подкисляют соляной
кислотой и экстрагируют хлороформом. После отгонки растворителя продукт перегоняют в
высоком вакууме.
Для расщепления а-ацилциклопентанонов (в реакции можно использовать неочищенный
продукт) смесь кипятят 3 ч с 100 мл 5%-ного раствора гидроксида натрия, после чего обраба-
обрабатывают, как описано выше.
7.2.1.10. Реакции ацилхлоридов с $-дикарбонильными соединениями
Подобно сложным эфирам, хлорангидриды (ацилхлориды) и ангидриды кар-
боновых кислот в присутствии основных катализаторов могут реагировать с
С—Н-кислотными соединениями. Механизм реакции аналогичен механизму
сложноэфирной конденсации. Для простейших сложных эфиров и кетонов ре-
реакция не имеет большого значения; в общем случае р-дикарбонильные соеди-
соединения предпочтительнее получать путем сложноэфирной конденсации
(см., однако, разд. Г,7.4.2.3, ацилирование кетонов через енамины).
Реакция ацилирования C-дикарбонильных соединений очень важна в препара-
препаративном отношении. Обычно в реакцию с ацилгалогенидами вводят еноляты метал-
металлов. При этом образуется р-трикарбонильное соединение, обладающее большей
кислотностью, чем исходное дикарбонильное соединение (почему?), поэтому три-
карбонильное соединение передает протон еноляту дикарбонильного соединения:
OQNa® О OGNa® О
2 R-C-CH-COOR + R'-C-CI R-C=C-COOR + R-C-CH2-COOR [Г.7.179]
R'—C=O
+ NaCI
По этой причине необходимо использовать вспомогательное основание,
обычно алкоголят натрия или магния2, в двойном по сравнению с эквивалент-
эквивалентным количестве. (Напишите схему реакции пропионилхлорида с ацетоуксус-
ным или малоновым эфиром.)
Амбидентный анион C-дикарбонильного соединения (см. разд. Г,2.3) в определенных ус-
условиях наряду с С-ацилированием способен и к замещению у атома кислорода енольной груп-
группы (О-ацилированию).
О
R—С-СН—COOR С-ацилирование
COR' [Г.7.180]
R-C-CH-COOR + R'COCI
"NaCI O-COR'
R—C = CH—COOR О-ацилирование
1 HunigS.Kup. Chem. Ber., 1958, 91, 129; 1960, 93, 913.
2 Чаще используют алкоголят магния, так как растворимость магниевых производных
(З-дикарбонильных соединений выше, чем натриевых.
7. Реакции карбонильных соединений 153
Соотношение продуктов О- и С-замещения зависит как от строения ацилирующего аген-
агента и р-дикарбонильного соединения, так и от реакционной среды.
Ацилирование свободных р-дикарбонильных соединений ацилхлоридами в пиридине
также приводит к образованию О-ацилированных продуктов. Ацилирующим агентом в этом
случае служит первоначально образующаяся соль ацилпиридиния I:
Присоединение ацетоуксусного эфира к ацилпиридиниевой соли I аналогично реакции
Манниха, т. е. катализируемой кислотами альдольной реакции с азотистым аналогом карбо-
карбонильного соединения
\+ /
N = C
/ \
Этот первый этап ориентирует реакцию в направлении О-ацилирования. (Составьте суммар-
суммарное уравнение реакции.)
Побочную реакцию — образование сложного эфира из ацилхлорида и спир-
спирта — можно в значительной степени подавить, проводя реакцию при О °С. Аци-
Ацилирование трудно гидролизующимися ацилхлоридами можно проводить даже в
водном растворе NaOH.
Этиловый эфир дибензоилуксусной кислоты из бензоилуксусного эфира и бен-
зоилхлорида: Wright P. E., McEwen W. E. J. Am. Chem. Soc, 1954, 76, 4540-4542.
Этиловый эфир бензоилуксусной кислоты из ацетоуксусного эфира и бензо-
илхлорида в водном растворе (ацилирование с последующим сложноэфирным
расщеплением): StraleyJ. M., Adams А. С. Org. Syntheses, Coll. Vol. IV, 1963, p. 415.
(Имеется перевод 1-го издания: СтрэлиДж.. Адаме А. В сб.: Синтезы органичес-
органических препаратов. 9. — М.: ИЛ, 1959, с. 84.)
Ф Общая методика ацилирования р-дикарбонильных соединений (табл. Г.7.182). В трехгор-
лую колбу на 2 л, снабженную мешалкой (лучше всего конструкции, показанной на
рис. А.6,ж), мощным холодильником с хлоркальциевой трубкой и капельной ворон-
воронкой, помещают 1 моль магниевых стружек, 50 мл абсолютного этанола и 5 мл сухого
тетрахлоруглерода, катализирующего образование этилата магния. Как только реакция нач-
начнет идти с заметной скоростью, добавляют при сильном перемешивании по каплям смесь
1 моль р-дикарбонильного соединения, 100 мл абсолютного этанола и 400 мл абсолютного
эфира так, чтобы смесь энергично кипела. Через несколько часов практически весь магний
растворяется и образуется бесцветное магниевое соединение. Затем, хорошо охлаждая ледя-
ледяной водой, добавляют по каплям раствор 1 моль свежеперегнанного ацилхлорида в 100 мл аб-
абсолютного эфира, еще 1 ч перемешивают при охлаждении и оставляют на ночь. После этого,
охлаждая льдом, прибавляют смесь 400 мл льда и 25 мл концентрированной серной кислоты,
отделяют эфирный слой, водный слой дважды экстрагируют эфиром. Соединенные эфирные
вытяжки промывают водой до почти нейтральной реакции, сушат сульфатом натрия и пере-
перегоняют в вакууме, используя 20-сантиметровую колонку Вигре.
С-Ацильные соединения ацетоуксусного эфира служат исходным материа-
материалом для синтеза высших (З-оксоэфиров, получаемых сложноэфирным расщеп-
расщеплением (см. разд. Г,7.2.1.9). При этом можно использовать ацилированные эфи-
ры без дополнительной очистки.
154
Г Препаративная часть (продолжение)
Таблица Г.7.182. Ацилирование Р-дикарбонильных соединений
Продукт реакции
а-Бензоилацетоуксусный
эфир
а-н-Бутирилацетоуксусный
эфир
а-Изобутирилацетоуксусный
эфир
а-Фенилацетилацетоуксусный
эфир
Ацетилмалоновый эфир
Бензоилмалоновый эфир
Фенилацетилмалоновый
эфир
Исходные вещества •
Ацетоуксусный эфир,
бензоилхлорид
Ацетоуксусный эфир,
н-бутирилхлорид
Ацетоуксусный эфир,
изобутирилхлорид
Ацетоуксусный эфир,
фенил ацетилхлорид
Малоновый эфир, ацетилхлорид
Малоновый эфир, бензоилхлорид
Малоновый эфир,
фенилацетилхлорид
Т. кип., "С
(мм рт. ст.)
175A2)
112A6)
114A5)
156E)
120A1)
148 @,8)
162C)
«D20
1,5390
1,4703
1,4678
1,5134
1,4374
1,5066
Выход, %
75
75
50
85
90
80
90
7.2.1.11. Присоединение С-Н-кислотных соединений к гетерокумуленам
Гешерокумуленами часто называют диоксид углерода (I) и его гетероаналоги: ди-
дисульфид углерода (II), изотиоцианаты (III), карбодиимиды (IV) и изоцианаты
(V) (получение см. разд. Г,7.1.6). Их реакционная способность по отношению к
основаниям (см. разд. Г.7.1.6) в общем случае возрастает слева направо в ряду
[Г.7.183]; она зависит и от природы основания. Реакционная способность арил-
замещенных гетерокумуленов III—V выше, чем соответствующих алкилпроиз-
водных:
О=С=О
s=c=s
N=C=S
N=C=N
IV
[Г.7.183]
С инертным СО2 реагируют только сильные С-основания, например металлоорганические
соединения (см. разд. Г,7.2.2).
Некоторые кетоны могут подвергаться карбоксилированию также в присутствии замещен-
замещенных фенолятов лития. Непрямое карбоксилирование ряда кетонов и нитроалканов осуществля-
осуществляют при помощи метил карбоната магния:
ОСООМе
О
ам
R1
Me
-СО2
-МеОН
О 2Н2О(Н
О
©,
О О
-Мд(ОН)г
ОН
[Г.7.184]
R'
Природные соединения углерода со временем в основном переходят в СОг. В отличие от
природы в промышленности единственными способами возвращения СО2 в естественный
кругооборот являются синтез Кольбе-Шмитта (см. разд. Г,5.1.8.6) и синтез мочевины.
Гетерокумулены II, III и V (схема [Г.7.183]), напротив, реагируют с активны-
активными С—Н-кислотными метиленовыми группами уже в присутствии этилата нат-
7. Реакции карбонильных соединений
155
рия, который не только осуществляет депротонирование, но и расходуется на
образование соли. В случае слабых С—Н-кислот, например кетонов, необходи-
необходимо применять гидрид или амид натрия. Примером служит реакция с циануксус-
ным эфиром:
R,
COOEt
и
С
и
X
+ Н2С
CN
R-NH
C=C
®x' 4CN
R-NH COOEt
С-СН
'/ \
X CN
[Г.7.185]
x = o,s
S
м
С
COOEt
+ Н2С
CN
+ 2ЕКГ '
-2ЕЮН 0
COOEt
с=с
CN
+ 2 Me2SO4
- 2 MeSO?
MeS
COOEt
C=C
MeS
[Г.7.186]
IV
Изоцианаты и изотиоцианаты (I в схеме [Г.7.185]) превращаются в а-акцеп-
торзамещенные карбамиды и тиокарбамиды [Г.7.185, IV), которые можно выде-
выделить и в виде солей. Ендитиолаты [Г.7.186, III], образующиеся из дисульфида уг-
углерода, не могут быть источником получения дитиокислот ввиду лабильности
последних. Можно, однако, дважды алкилировать ендитиолат; при этом образует-
образуется акцепторзамещенный дитиоацеталькетен [Г.7.186, IV], тиометильные группы
которого способны замещаться первичными аминами или другими основаниями
(почему?).
Таблица Г.7.187. Присоединение гетерокумуленов кС-Н-кислотным соединениям
Продукт реакции
Анилид а-ацетилацетоуксусной
кислоты
Анилид а-этоксикарбонил-
ацетоуксусной кислоты
Анилид дициануксусной
кислоты
Анилид этоксикарбонилциан-
тиоуксусной кислоты
Анилид 2-карбамоил-2-циан-
тиоуксусной кислоты
Диметиловый эфир фенил-
карбамоилмалоновой кислоты
Метиловый эфир 2-циан-
3,3-бис(метилтио)пропен-2-
кислоты
Нитрил бис(метилтио)метилен-
малоновой кислоты
Исходное вещество
Ацетилацетон, фенилизоцианат
Ацетоуксусный эфир,
фенилизоцианат
Динитрил малоновой кислоты,
фенилизоцианат
Этиловый эфир
циануксусной кислоты,
фенилизотиоцианат
Цианацетамид,
фенилизотиоцианат
Диметиловый эфир
малоновой кислоты,
фенилизотиоцианат
Метиловый эфир
циануксусной кислоты,
дисульфид углерода
Нитрил малоновой кислоты,
дисульфид углерода
Т. пл., "С
64—66 (этанол)
55-57 (метанол)
170-172 (уксусная
кислота)
114-116 (этанол)
163-165 (этанол)
87-91 (этанол)
87(метанол)
81 (этанол)
Выход, %
60
55
68
78
84
68
75
75
156 Г Препаративная часть (продолжение)
Общая методика присоединения гетерокумуленов к метиленактивным соединениям (табл.
Г.7.187)
Осторожно! Диметилсульфат — сильный яд! Работать под тягой, в защитных
перчатках!
В трехгорлой колбе емкостью 250 мл, снабженной мелашкой и капельной воронкой, растворяют
0,1 моль металлического натрия в 70 мл абсолютного этанола (при работе с дисульфидом углеро-
углерода — 0,2 моль натрия в 140 мл абсолютного этанола; в случае метиловых эфиров используют ме-
метанол). При перемешивании медленно, по каплям, добавляют смесь 0,1 моль гетерокумулена и
0,1 моль производного малоновой кислоты; твердые вещества предварительно растворяют в
10 мл ацетона. Перемешивают при комнатной темпераутре еще 45 мин; при этом в некоторых
случаях выкристаллизовывается твердая соль.
При реакциях с CS2 к смеси медленно добавляют 0,2 моль диметилсульфата. Оставляют на
0,5 ч, затем выливают при перемешивании в четырехкратный объем воды. Кристаллический
осадок отфильтровывают, промывают водой и перекристаллизовывают.
Можно также после выливания реакционной смеси в воду подкислить соляной кислотой
A : 1), отфильтровать осадок и промыть водой. Если продукт растворяется целиком в 0,5 н.
NaOH, его перекристаллизовывают. В противном случае весь продукт суспендируют в доста-
достаточном объеме разбавленного раствора NaOH, фильтруют, фильтрат подкисляют. Выпавший
осадок отсасывают, промывают водой и перекристаллизовывают.
Получаемые таким путем полуфункциональные соединения могут служить исходными ве-
веществами, например для синтеза гетероциклических соединений. Присоединение енаминов к
гетерокумуленам см. в разд. Г,7.4.2.3.
7.2.1.12. Полиметиновая конденсация
Катион соли иммония строения I легко депротонируется, превращаясь в II (схема
[Г.7.188]):
\© R © \© R v. R
N -н_ N N
С._.. 7^® /С-рти© /С^и. [Г.7.188]
Такое поведение характерно и для метальных групп, находящихся в положе-
положениях 2 и 4 к четвертичному атому азота, который входит в гетероароматическую
систему. Возникающие при этом метиленовые группы имеют отчетливо выра-
выраженный нуклеофильный характер (енаминная структура, см. разд. Г,7.1.1 и
Г,7.4.2.3) и склонны к реакциям типа альдольной конденсации. В случае орто-
муравьиного эфира, в молекуле которого в виде ацеталя имеется потенциальная
карбонильная группа, сначала по уравнению [Г.7.171] образуется алкоксимети-
леновое соединение, дающее с другой молекулой метиленового соединения так
называемый триметинцианин [Г.7.189, II]1, например:
СН3 СН3 Н3С
N OEt ^\^N NX.
+ Eto-сн рг- Г |f )=ch{ch=chW' Tl "I
Ъй-зЕюн.-нв 4Asr T UsA^ [г.7.189]
I II (л=1)
I + (EtOJCH-CH2-CH(OEtJ II (n=2)
I + Ph-NH-CH=CH-CH=CH-CH=N-Ph II (n = 3)
1 Три-, пента- и гептаметинцианины (см. схему [Г.7.189]) при отсчете от одного гетеро-
атома до другого (см. табл. А. 126) можно рассматривать как 1,5-, 1,7- и 1,9-дизамещенные
пента-, гепта- и нонаметинцианины соответственно. Аналогично монометиноксонол 1
[Г.7.190] можно отнести к пентаметинам.
7.Реакции карбонильных соединений
157
При конденсации с малоновым альдегидом аналогично получают пентаме-
тинцианины, с глутаконовым альдегидом — гептаметинцианины. Для реакции
применяют не малоустойчивые свободные альдегиды, а более стабильные и бо-
более дешевые ацетали или анилы. Изменяя метиленовые компоненты, можно по-
получать разнообразные полиметины, в том числе и несимметричные. Их структу-
структура лучше всего описывается граничными мезомерными формами. (Напишите
для цианина в [Г.7.189] вторую граничную структуру; см. табл. А. 126. Напишите
также уравнения реакций указанных выше предшественников метинов с
1-этил-2-метил- и 1-этил-4-метилхинолинийбромидами.)
Заряд в полиметинах делокализован по всей цепочке атомов, а результирую-
результирующий заряд определяется гетероатомом; он может быть и отрицательным, как,
например, в полиметиноксонолах (см. табл. А. 126, III).
Монометиноксонол (соединение I в схеме [Г.7.190]) может быть получен из
3-метил-1 -фенилпиразолинона-5 (см. уравнение [Г.7.59]) и ортомуравьиного эфира:
О
Me Me
\_ ©/
N-(CH)n=N —
полиметинцианин
0- ©/
IO-(CH)n=N —
полиметинмероцианин
\© _/
N=(CH)n-N = N^
(Л+3O1
O=(CH)n-N =
О=(СН)П-О|0 =
/
O-(CH)n-N
[Г.7.190]
\
©
n=1,3,5...
(— СН=),атакже
(-CR=),(-N=)
полиметиноксонол
Полиметины общей формулы II [Г.7.190], как и ароматические и полиеновые
соединения, обладают системой сопряженных л-электронов. Сравните УФ-спект-
рывтабл. А.126, а также свойства кристаллического фиолетового1 в разд. Г,5.1.8.5.
Описанные в разд. Г,6.4.3 катионные азокрасители также можно рассматривать
как азополиметины.
Общая методика получения три- и пентаметинцианинов (табл. Г.7.191)
Осторожно! Диметилсульфат — сильный яд! Работать под тягой, в защитных
перчатках!
Смесь 20 ммоль метилгетероароматического соединения с 25 ммоль диметилсульфата нагрева-
нагревают в колбе емкостью 100 мл 30 мин при темпераутре бани 140 "С. После охлаждения добавля-
добавляют 40 мл сухого пиридина и 40 ммоль ортомуравьиного эфира или 30 ммоль тетраацеталя ма-
малонового альдегида (или гидрохлорида дианила малонового диальдегида) и нагревают 40 мин
с обратным холодильником. При перемешивании вносят еще теплую смесь в раствор 4 г иоди-
да калия в 20 мл воды, осаждая полиметин в виде иодида. Отсасывают, промывают холодным
этилацетатом и перекристаллизовывают.
1 Кристаллический фиолетовый — виниленгомолог (см. разд В,5.1 и Г.7.4) нонаметинци-
анина, чем и объясняется его спектральное поведение.
158
Г Препаративная часть (продолжение)
Таблица Г.7.191. Три- и пентаметинцианины
Продукт реакции
1,3-БисC-метилбензо-
тиазолил-2)триметиний-
иодид
1,3-Бис( 1-метилхинолил-
2)триметинийиодид
1,3-БисC-метилбензокса-
золил-2)триметинийиодид
1,5-БисC-метилбензотиа-
золил-2)пентаметиний-
иодид
1,5-БисA-метилхинолил-2)-
пентаметинийиодид6
Исходные вещества
2-Метилбензотиазол,
ортомуравьиный эфир
2-Метилхинолин,
ортомуравьиный эфир
2- Метилбензоксазол,
ортомуравьиный эфир
2-Метилбензотиазол,
тетраэтилацеталь
малонового альдегида
2-Метилхинолин,
тетраэтилацеталь мало-
малонового альдегида
Т. пл., °С
290-293 (ДМФА)
310-312 (вода.
ДМФА, 1:1)
285-288 (пропанол:
вода 2:1)
282-284(пропанол)
240-242 (пропанол)
XmaxOgE)"
569E,13)
614E,20)
491E,15)
662E,13)
720E,11)
Выход, %
70
50
65
50
30
а ^тах — максимум поглощения в длинноволновой области (нм) в ДМФА (диметилформамиде);
е — молярный коэффициент экстинкции.
6 Растворяют KI в 80 мл воды.
Чистоту продукта определяют с помощью тонкослойной хроматографии, например на силу-
фоле; для элюирования используют смесь бутанола, ледяной уксусной кислоты и воды D:1:5
по объему).
Только немногие полиметины нашли применение в качестве красителей в текстильной про-
промышленности, поскольку их светостойкость в обшем случае невелика. Напротив, они широко
используются в фотографии на основе галогенидов серебра в качестве сенсибилизаторов (см. так-
также разд. Г, 1.1). Несенсибилизированный бромид серебра почти нечувствителен к свету с длиной
волны более 500 нм. При нанесении полиметинов на светочувствительный слой галогенидов се-
серебра становится возможным получать изображение не только во всем видимом диапазоне, но и
в инфракрасной области спектра. Как правило, полиметины оказывают сенсибилизирующее
действие в области длин волн, соответствующей их собственному поглощению.
7.2.2. РЕАКЦИИ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ С
МЕТАЛЛООРГАНИЧЕСКИМИ СОЕДИНЕНИЯМИ
С карбонильной группой могут взаимодействовать не только рассматривавшие-
рассматривавшиеся до сих пор нуклеофильные реагенты, но и другие вещества, способные пере-
переносить водородные атомы или алкильные, а также арильные остатки вместе с их
электронными парами (в виде «анионов»). Эти анионы являются особенно
сильными основаниями, однако в ходе реакций они в свободном виде не обра-
образуются, так как ионизация молекул с образованием свободных арильных или
алкильных анионов по схеме [Г. 192] затруднена.
M-R
[Г.7.192]
В подобных ионах отрицательный заряд не может быть внутренне стабилизиро-
стабилизирован. Связь М—R может быть расщеплена только синхронно с реакцией с карбо-
7. Реакции карбонильных соединений 159
нильной группой, причем соответствующее переходное состояние сходно с
переходным состоянием при Б^-реакциях:
Следовательно, расщепление связи М—R не является равновесным процес-
процессом, предшествующим собственно карбонильной реакции, как это обычно име-
имеет место при реакциях С—Н-кислотных соединений.
К реагирующим таким образом соединениям относятся некоторые металло-
органические соединения, алкильные группы которых негативированы +/-эффек-
+/-эффектом металла.
К числу важнейших металлоорганических соединений, используемых в реак-
реакциях с карбонильными соединениями, принадлежат магнийорганические соеди-
соединения — так называемые реактивы Гриньяра (соединения Гриньяра). Важней-
Важнейшим методом их получения служит взаимодействие алкил- или арилгалогенидов
(RX) с металлическим магнием, которое изображают следующей схемой:
R-X + Мд R-Mg-X [Г.7.194]
Обычно эту реакцию проводят в безводном диэтиловом эфире, но пригодны
и другие растворители, не имеющие активных водородных атомов, например
высшие эфиры (дибутиловый эфир, анизол, тетрагидрофуран).
Схема [Г.7.194] представляет гетерогенную реакцию, в которой участвует по-
поверхность металла. Она начинается с переноса электрона от металла к субстрату
RX с образованием анион-радикала, который благодаря слабости связи угле-
углерод—галоген распадается на анион Х~ и радикал R/. Последний реагирует с маг-
магнием, образуя реактив Гриньяра:
R-X + Мд R-X? + Мд® > R- + X0 + Мд® - R-Mg—X [Г.7.194а]
Строение реактива Гриньяра выяснено еще не полностью; оно во многом зависит от кон-
концентрации и растворителя. Важную роль играет так называемое равновесие Шленка:
2 RMgX ^=^ R-Mgf Mg-R =^^ R2Mg + МдХ2 ^^ R-Mg. MgX
"X 'X [Г.7.195]
I II III IV
Нуклеофильный растворитель координационно связывается с атомом магния:
R\ ? Я'
0-Mg—Оч [Г.7.196]
Ry х R'
В эфирных растворах при малых концентрациях предпочтительнее форма I (см. схему
[Г.7.195]), в более основном тетрагидрофуране, по-видимому, преобладает форма II, в то вре-
время как в триэтиламине образуется исключительно форма III. Наконец, в диоксане существует
только диалкилмагний, так как нерастворимый галогенид магния выпадает в осадок.
При дальнейшем описании реакций с участием реактивов Гриньяра для простоты исполь-
используется наиболее часто применяемая форма I (схема [Г.7.195]).
Скорость взаимодействия алкилгалогенидов в реакции (Г.7.194) падает от
иодида к хлориду, однако в случае хлоридов выходы выше, чем в случае броми-
160 Г Препаративная часть (продолжение)
дов и иодидов. Из ароматических галогенопроизводных в реакцию обычно всту-
вступают только бромиды и иодиды.
Связь углерод—магний сильно полярна, при этом атом углерода несет час-
частичный отрицательный заряд (почему?). Поэтому соединения Гриньяра явля-
являются нуклеофильными реагентами, которые легко вступают в реакции с элект-
рофильными агентами. Важнейшими представителями последних являются:
а) соединения, содержащие активный водород;
б) алкилгалогениды;
в) галогениды металлов;
г) соединения с полярными двойными связями (например, карбонильные
соединения).
а) Реактивы Гриньяра реагируют с соединениями, содержащими активный
водород (водой, спиртами, фенолами, карбоновыми кислотами, меркаптана-
меркаптанами, первичными и вторичными аминами, амидами, ацетиленами и другими
С-Н-кислотными соединениями), образуя углеводороды:
R-Mg-Y + Н-Х - R-H + X-Mg-Y [Г.7.197]
Эту реакцию можно использовать для количественного определения актив-
активного водорода, вводя в реакцию метилмагнийиодид и измеряя объем выделив-
выделившегося метана (метод Чугаева—Церевитинова).
Реакция позволяет также получать такие соединения Гриньяра, которые
обычным путем (см. схему [Г.7.194]) получаются с трудом либо совсем не обра-
образуются (пиррол, ацетилен и т. д.):
НС=С-Н + Н3С-МдХ НС=С-МдХ + СН4 [Г.7.198]
К реакциям такого же типа относится взаимодействие фенилуксусной кис-
кислоты с изопропилмагнийхлоридом с образованием реактива Иванова:
С6Н5-СН2-СООН + 2(CH3JCH-MgCI С6Н5-СН-с' +2С3Н8 [Г.7.199]
MgCI OMgCI
б) С алкилгалогенидами соединения Гриньяра дают углеводороды (реакция
аналогична реакции Вюрца):
R-Mg-X + X-R' R-R1 + MgX2 [Г.7.200]
Особенно легко подобным образом реагируют третичные алкилгалогениды,
аллил- и бензилгалогениды (почему?). Эта реакция является нежелательным
побочным процессом при получении соединений Гриньяра по схеме [Г.7.194].
в) Соединения Гриньяра реагируют с галогенидами менее активных, чем маг-
магний, металлов путем обмена галогена на алкильные группы, например:
2RMgX + CdCI2 R2Cd + MgX2 + MgCI2 [Г.7.201]
С галогенидами серебра и меди(П) реакция проходит аномально с выделени-
выделением свободных металлов и образованием углеводородов:
2RMgX + 2AgBr R-R + MgX2 + MgBr2 + 2 Ад [Г.7.202]
7. Реакции карбонильных соединений 161
Реакция по схеме [Г.7.201] имеет значение при получении других металлоор-
ганических соединений. В промышленности таким образом из тетрахлорида
кремния получают алкилхлорсиланы, служащие исходными соединениями при
синтезе силиконов.
г) Реакции реактивов Гриньяра с карбонильными соединениями. В качестве
нуклеофильных реагентов соединения Гриньяра способны присоединяться к
электрофильной карбонильной группе:
\ I
R-Mg—X + С=О R-C-0-Mg— X [Г.7.203]
В реакции часто участвуют 2 моль реагента и 1 моль кетона. Механизм реак-
реакции еще не выяснен во всех деталях. Наиболее наглядной представляется схема
процесса с образованием циклического переходного комплекса. При этом нук-
леофильность магнийорганического соединения в циклическом комплексе
увеличивается за счет второй молекулы реактива Гриньяра:
О .R. -x \ .R
С'^ Мд —С
"л /-; '
О.» < X > Оч + МдХ2
'Мд Мд [Г.7.204]
R(X) R(X)
В формулах I и II стоящая в скобках буква «X» указывает, что вместо RMgX в
переходном комплексе может участвовать MgX2. При этом понижается скорость
реакции, однако подавляются конкурирующие реакции, протекающие по схе-
схемам [Г.7.209] и [Г.7.210].
Образовавшийся алкоголят магния гидролизуют водой:
R-O-МдХ ХМдОН
+ Н2О
или - ROH + или
R-0-Mg-O-R Мд(ОНЬ [Г.7.205]
+ н?о
R-O-Mg-Rf ^— ROH + R'H + Mg(OHJ
Таким методом получают первичные спирты из формальдегида, вторичные
спирты из других альдегидов, третичные спирты из кетонов и карбоновые кис-
кислоты из диоксида углерода.
(Напишите схемы этих превращений!)
Производные карбоновых кислот (сложные эфиры, ангидриды, ацилгалоге-
ниды) сначала реагируют согласно общей схеме [Г.7.204]:
OR"
OR" RJ .R'- 'Hal OR"
R-C +2R'MgHal мл r\ a .- R-C-R' + R'MgHal
C
g л r
°^ OMgHa, [Г.7.206]
Hal
I
162 Г Препаративная часть (продолжение)
Адцукт II можно рассматривать как соль полуацеталя, которая неустойчива
(почему?) и распадается с образованием кетона и алкоголята:
OR" r-
R-C-R1 R-C4 + R"OMgHal [Г.7.207]
OMgHal ЧО
Образовавшийся при этом кетон реагирует по схеме [Г.7.204] со второй
молекулой соединения Гриньяра, образуя третичный спирт.
(Какой конечный продукт получится при реакции с эфиром муравьиной
кислоты?)
В соответствии с рядом активности карбонильных соединений [Г.7.3] кетоны
быстрее реагируют с соединениями Гриньяра, чем сложные эфиры. Поэтому выде-
выделить кетон, образующийся в качестве промежуточного продукта, обычно не удается.
При использовании в качестве карбонильного компонента ацилхлорида при
соблюдении особых условий кетон удается выделить из реакционной смеси
(почему?). Наиболее успешно синтез кетонов проходит с кадмийорганическими
соединениями, так как их реакционная способность достаточна только для
взаимодействия с ацилхлоридами, а кетоны с ними не реагируют.
/° /°
RzCd + 2R-c' 2R-C' + CdCI2 [Г.7.208]
Cl R1
Подобно карбонильной группе, с реактивами Гриньяра реагируют и другие
полярные группировки с кратными связями, например C=N, C=N, C=S, N=O.
(Какие соединения при этом образуются?) Реакции по двойным связям С=С
осуществляются только в тех случаях, если эти связи поляризованы сопряжен-
сопряженной карбонильной группой A,2- или 1,4-присоединение).
Побочные процессы при реакциях Гриньяра наблюдаются в первую очередь в тех случаях,
когда в силу пространственных препятствий невозможно образование циклического переход-
переходного состояния I (схема [Г.7.204]). Если в карбонильном соединении или реактиве Гриньяра
имеются достаточно объемные заместители, то в циклическом комплексе находится место
только для одной молекулы магнийорганического соединения. В этих случаях на карбониль-
карбонильную группу часто переносится не алкильный остаток, а меньший по объему гидридный ион; в
результате карбонильная группа восстанавливается, а соединение Гриньяра превращается в
олефин (гриньяровское восстановление):
-4-^с7- -VH -с-
ti?CPr ~ ^ + -А 1Г.7.209]
Mg \ МдХ
X
В реакции Гриньяра с пространственно затрудненными реагентами в присутствии броми-
бромида магния последний благодаря своему малому объему способен образовать нормальный цик-
циклический переходный комплекс [Г.7.204], что значительно подавляет восстановительный
процесс по схеме [Г.7.209].
В тех случаях, когда пространственно затрудненное соединение Гриньяра не имеет водо-
водородного атома в р-положении, восстановление по схеме [Г.7.209] невозможно. Тогда реакция
протекает с образованием магниевого енолята карбонильного соединения:
I ,9 \ е,ЯМдХ
X-Mg-R + Н-С-С R-H + С-С
I \ / \
7. Реакции карбонильных соединений 163
Подобные соединения Гриньяра можно использовать в качестве сильноосновных конден-
конденсирующих средств при сложноэфирных конденсациях (см. схему [Г.7.165]).
Некоторые указания к проведению реакций Гриньяра. Реакции Гриньяра силь-
сильно мешают вода и спирты (почему?). Необходимо следить за тем, чтобы исполь-
используемый в качестве растворителя эфир был свободен не только от воды, но и от
спиртов. В таком эфире реакция начинается быстро, особенно с низшими
алкилгалогенидами.
Иногда реакция начинается только с большим трудом. В таких случаях к
раствору добавляют несколько капель брома или тетрахлоруглерода и при необ-
необходимости слегка нагревают. Рекомендуется также активировать магний иодом
(непродолжительно нагревать над маленьким пламенем сухие магниевые струж-
стружки и кристаллик иода) или добавлять небольшое количество безводного броми-
бромида магния.
Соединения Гриньяра чувствительны к кислороду. Обычно от окисления
раствор в достаточной степени предохраняет «подушка» паров эфира. Иногда
необходимо работать в атмосфере инетрного газа. (Почему для этого непригоден
диоксид углерода?)
Ф Общая методика получения спиртов и карбоновых кислот с помощью соединений
Гриньяра (табл. Г.7.211)
А. Получение реактива Гриньяра. В трехгорлую колбу емкостью 1 л, снабженную капель-
капельной воронкой, мешалкой и обратным холодильником с хлоркальциевой трубкой, помещают
0,5 моль магниевых стружек и 50 мл абсолютного эфира. При перемешивании прибавляют
примерно 1/20 от общего количества @,5 моль) алкил- или арилгалогенида. Начало реакции
можно установить по легкому помутнению и разогреванию эфира. Если реакция не начинает-
начинается, то добавляют в реакционную смесь 0,5 мл брома или несколько капель тетрахлоруглерода
и слегка подогревают. После начала реакции постепенно прибавляют остальную часть алкил-
или арилгалогенида, растворенного в 125 мл абсолютного эфира. Скорость прибавления регу-
регулируют таким образом, чтобы смесь слегка кипела. Если реакция протекает слишком бурно, то
охлаждают колбу водой. К концу добавления раствора галогенида реакционную смесь нагре-
нагревают на водяной бане, поддерживая умеренное кипение, до полного растворения магния
(примерно 30 мин).
Б. Взаимодействие реактива Гриньяра с альдегидами и кетонами. К раствору реактива
Гриньяра, приготовленному из 0,5 моль галогенида, прибавляют по каплям при перемешива-
перемешивании 0,4 моль карбонильного соединения (сложного эфира берут только 0,2 моль; почему?),
растворенного в равном объеме абсолютного эфира. По окончании прибавления раствора ре-
реакционную смесь нагревают при перемешивании еще 2 ч на водяной бане, охлаждают, гидро-
лизуют, добавляя 50 г размолотого льда, и затем приливают соляную кислоту A : 1) в количе-
количестве, необходимом для полного растворения осадка. При получении третичных спиртов в
этих условиях уже приходится учитывать возможность дегидратации, поэтому соляную кис-
кислоту заменяют насыщенным водным раствором хлорида аммония. Эфирный слой отделяют,
а водный дважды экстрагируют эфиром. Эфирные слои объединяют, промывают насыщен-
насыщенным раствором гидросульфита, раствором гидрокарбоната и небольшим количеством воды.
После сушки сульфатом натрия эфир отгоняют, а остаток фракционируют или перекристал-
лизовывают.
В. Взаимодействие реактива Гриньяра с диоксидом углерода. В охлажденный до —5 °С раст-
раствор реактива Гриньяра пропускают сильный ток сухого диоксида углерода, следя за тем, что-
чтобы температура не поднималась выше 0 °С. По прекращении экзотермической реакции ток ди-
диоксида углерода пропускают еще 1 ч, затем разлагают льдом и соляной кислотой, как описано
в варианте Б, высушивают эфирный слой сульфатом магния и отгоняют растворитель. Остаток
перегоняют в вакууме или перекристаллизовывают из горячей воды, к которой может быть
добавлено немного соляной кислоты.
Таблица Г.7.211. Синтез спиртов и карбоновых кислот по Гриньяру
Продукт реакции
Пентанол-2
Октанол-2
2- Метилпентанол- 3
З-Метил-1 -фенилбутанол-2
2,2,2-Трихлор-1-фенил этанол
1-Фенилпропанол-1
2 - Метилбутанол - 2б
2,3-Диметилбутанол-2
3-Метилпентанол-Зб
1,1 -Дифенил этанол- 1В
1-Фенил-3,4-дигидронафталинг
3-Этилпентанол-Зд
З-Метиллентанол-3
4-Этилгептанол-4
З-Этилгексанол-3
Трифенилкарбинол
Триметилуксусная (пивалиновая)
кислота
Бензойная кислота
Фенилуксусная кислотае
а-Нафтойная кислота
Исходные соединения
Ацетальдегид, пропилмагнийбромид
Ацетальдегид, гексилмагнийбромид
Изомасляный альдегид, этилмагнийбромид
Изомасляный альдегид, бензилмагнийхлорид
Хлоральа, фенилмагнийбромид
Бензальдегид, этилмагнийбромид
Ацетон, этилмагнийбромид
Ацетон, изопропшшагнийхлорид или
изопропилмагнийбромид
Метилэтилкетон, этилмагнийбромид
Ацетофенон, фенилмагнийбромид
а-Тетралон, фенилмагнийбромид
Диэтилкарбонат, этилмагнийбромид
Этилацетат, этилмагнийбромид
Этилпропионат, пропилмагнийбромид
Этилбутират, этилмагнийбромид
Этилбензоат, фенилмагнийбромид
Диоксид углерода,
/лрет-бутилмагнийхлорид
Диоксид углерода, фенилмагнийбромид
Диоксид углерода, бензилмагнийхлорид
Диоксид углерода, а-нафтилмагнийбромид
Т. кип., °С (мм рт. ст.)
119
74 A0)
127
118A5)
145 A2); т. пл. 37
107A5)
102
118
122
155A2);
Т. пл. 90 (эфир)
178A8)
136
122
77A7)
80 D0)
Т. пл. 162 (бензол)
78 B0); т. пл. 35
Т. пл. 122 (вода)
144 A2); т. пл. 76
Т. пл. 160 C0%-ная
уксусная кислота)
1,4053
1,4245
1,4175
1,5091
(при 25 °С)
1,5257
1,4042
1,4176
1,4186
1,4186
1,6297
1,4216
1,4186
1,4439
1,4300
Выход, %
35
45
68
75
70
78
60
70
67
80
60
80
67
58
61
75
63
90
79
80
а См. часть Е.
6 Эфирный раствор не промывают, сушат поташом.
в При перегонке как основной продукт образуется 1,1-дифенилэтилен (см. табл. Г.3.34).
г После отгонки эфира к остатку добавляют 20 мл уксусного ангидрида, нагревают 20 мин на водяной бане, а затем перегоняют.
л На 0,2 моль сложного эфира необходимо 0,75 моль реактива Гриньяра.
е Диоксид углерода вводят при —20 °С.
7. Реакции карбонильных соединений 165
Получение троповой кислоты из реактива Иванова (см. схему [Г.7.199]) и па-
раформа (составьте схему реакции!): Blicke F. К, Raffelson H., Вата В. J. Am.
Chem. Soc, 1952, 74, 253.
Наряду с реактивами Гриньяра все шире применяются литийорганические
соединения. Подобно соединениям Гриньяра их получают из алкил- или арил-
галогенидов и металлического лития:
R—Hal + 2 Li - R—Li + Li—Hal [Г.7.212]
В большинстве случаев литийорганические соединения получают реакцией
обмена бутил- или фениллития, полученных по схеме [Г.7.212], с алкилгалоге-
нидами [Г.7.213] или С—Н-кислотными соединениями [Г.7.214]:
Rf—Hal + R—Li - R1—Li + R-Hal [Г.7.213]
R'—H + R—Li R'—Li + R-H [Г.7.214]
Реакции обмена галогена на металл по схеме [Г.7.213] чаще всего использует-
используется для получения арил- и алкениллитиевых соединений. В этом случае можно
работать при низких температурах (от —60 до —120 °С), при которых, в противо-
противоположность процессам прямого металлирования литием или магнием, не затра-
затрагиваются лабильные заместители, такие, как циано- или нитрогруппы.
По схеме [Г.7.214] депротонироваться могут также очень слабые С—Н-кисло-
ты, такие, как аллил- и бензилсодержащие соединения, 1,3-дитианы (см.
разд. Г.7.2.1.6, с. 135), четвертичные аммониевые и фосфониевые соли (ана-
(аналоги [Г.7.154]), а также олефины и арены, активированные заместителями с
отрицательным индукционным (—/(-эффектом.
Соединения лития, элемента с сильно выраженным электроположительным
характером, более реакционноспособны, чем соединения Гриньяра. Поэтому
они не так просты в обращении, как реактивы Гриньяра. С литийорганически-
ми соединениями необходимо работать при полном отсутствии влаги, кислоро-
кислорода и диоксида углерода в среде инертного газа (лучше всего аргона).
Литийорганические соединения при более высокой реакционной способности
в основном реагируют подобно соединениям Гриньяра (см. с. 160, а—г).
Литийорганические соединения применяют для реакций с карбонильными
соединениями обычно только в тех случаях, когда карбонильные соединения
обладают малой активностью. Так, 2-оксо-1,1-дифенилаценафтен не реагирует
с фенилмагнийбромидом, но без затруднений взаимодействует с фениллитием,
образуя 2-гидрокси-1,1,2-трифенилаценафтен. Литийорганические соединения
позволяют избежать нежелательных реакций восстановления, протекающих с
реактивами Гриньяра (схема [Г.7.209]). Например, 2,2,4,4-тетраметилпентанон-З
и /и/?еот-бутиллитий дают 3-от/7е/и-бутил-2,2,4,4-тетраметилпентанол-3.
Кроме того, взаимодействием литиевых солей карбоновых кислот с литийор-
ганическими соединениями получают кетоны (схема [Г.7.215]; реакция Гилме-
на-Ван-Эсса). Промежуточный продукт III в условиях реакции устойчив и толь-
только при гидролизе переходит в кетон:
>? .+ RU. / . + nu_ \Р" +Нг0 \
\ -RH н Ц, /Ч _2 ион ^-и ГГ 7 21 *51
ОН OLi R' OL\ FT l ' J
166 Г Препаративная часть (продолжение)
Аналогично, с образованием кетонов протекает взаимодействие литийорга-
нических соединений с диоксидом углерода, тогда как реактивы Гриньяра в
этом случае образуют кислоты.
О 1RLi R
RLi + CO2 - R-c' ^^- b=O [Г.7.216]
OLi r'
Получение 4,6-диметилгептен- 1-ола-4 из метилизобутилкетона и аллилли-
тия: Seyferth D., WeinerM. A., Org. Synth. Coll., 1973, Vol. V, 452.
Метилциклогексилкетон из циклогексанкарбоновой кислоты и метиллития:
Ваге Т. М., House H. О., Org. Synth., 1969, 49, 81.
2-Пиридилуксусная кислота из 2-пиридилметиллития и диоксида углерода:
Woodward R. В., Kornfeld E. С, Org. Synth., Coll., 1955, Vol. Ill, 413.
Реакционноспособность магний- и литийорганических соединений может
быть повышена добавлением солей медиA). При этом протекает процесс обме-
обмена металлом, аналогичный реакции [Г.7.201], причем промежуточно образуют-
образуются медьорганические соединения, например алкил- или диалкилкупраты.
RM + СиХ - RCu + MX M = MgX, Li
[Г.7.217]
2RM + CuX - R2CuM + MX X = CI, Br, I,CN
Их можно таким образом выделить в виде индивидуального соединения.
Наиболее важными реагентами являются литийкупраты, получаемые из 2 моль литийорга-
нического соединения и 1 моль иодида медиA). С а,C-непредельными карбонильными соеди-
соединениями они реагируют по схеме 1,4-присоединения, тогда как магний- и литийорганические
соединения образуют при этом только 1,2-аддукты:
OLi
+ (H3cJcuLi г""^ +н2о f^ [Г.7.218]
-H3CCu
С алкил-, алкенил- и арилбромидами, -иодидами и -сульфонатами литийкупраты реагиру-
реагируют по схеме [Г.7.200] с образованием соответствующих углеводородов:
R'X + R2CuLi - R'R + RCu + LiX [Г.7.219]
Хлорангидриды кислот можно таким образом превратить в кетоны:
R'COCI + R2CuLi R'COR + RCu + LiX [Г.7.220]
Получение 3,3-диметилциклогексанона из З-метилциклогексен-2-она и
литийдиметилкупрата: House H. О., WilkimsJ. M., J. Org. Chem., 1976, 41, 4031.
трет-Бутилфенилкетон из бензоилхлорида и литий-тре/и-бутилфенилтио-
купрата: Posner G. И., Whitten С. Е., Org. Synth., 1976, 55, 122.
Аналогично литийкупратам из реактивов Гриньяра и CuBr образуются ал-
килмедьмагнийорганические соединения, так называемые реагенты Норманта.
Они могут стереоспецифически (син-) присоединяться к ацетиленам, а образу-
образующиеся при этом алкенилмедьорганические соединения могут вступать в даль-
дальнейшие разнообразные реакции:
7. Реакции карбонильных соединений 167
RMgBr + CuBr RCuMgBr2
R CuMgBr
RCuMgBr2 + R'C=CH C=C
R'CR=CH2
R'CR=CHI [Г.7.221]
R. H | R'CF^CHR"
*"rsra" R'CR=CHCH=CHR"
Многие из упомянутых выше реакций могут быть осуществлены, исходя из
реактивов Гриньяра в присутствии солей одновалентной меди, таких как CuCl,
CuBr или CuCN.
Получение диэтилового эфира трет-бутилмалоновой кислоты из диэтилизо-
пропилиденмалоната и MeMgl/CuCl: Eliel E. L., Hutchins R. О., KnoeberM., Org.
Synth., 1970, 50, 38.
Этиловый эфир р-циклогексилпропионовой кислоты из этилакрилата и цикло-
гексилмагнийбромида в присутствии CuCl: Liu S.-H., J. Org. Chem., 1977,
42, 3209.
Реакции Гриньяра аналогична и реакция Реформатского, в которой эфиры
а-галогенокислот взаимодействуют с кетонами или альдегидами в присутствии
металлического цинка:
\ Zn I H?O I
С=О + Br-CH2-COOR —C-CH2-COOR —*—¦ —C-CH2-COOR [Г 7 222]
/ 6-ZnBr ОН
Промежуточное цинкорганическое соединение гораздо менее реакционно-
способно, чем магний- или тем более литийорганическое соединение. Поэтому
оно не реагирует с более инертной карбонильной группой сложного эфира и
взаимодействует только с альдегидной или кетонной группами.
Реакцию Реформатского обычного используют для получения ос,р-ненасы-
щенных сложных эфиров, которые легко образуются при дегидратации р-гид-
роксиэфиров (часто уже в ходе синтеза).
Получение этилового эфира $-фенил-$-гидроксипропионовой кислоты из бром-
уксусного эфира и бензальдегида: Hauser Ch. R., Breslow D. S. Org. Syntheses, Coll.
Vol. Ill, 1955, p. 408. (Имеется перевод 1-го издания: Хаузер Ч.. Бресло Д.
В сб. Синтезы органических препаратов. Сб. 3. — М.: ИЛ, 1952, с. 518.)
Этиловые эфиры $-алкил-$-гидроксипропионовых кислот из этилбромацетата
и алифатических альдегидов: Frankenfeld J. W., Werner J. J. J. Org. Chem., 1969,
34, 3689.
7.3. ВОССТАНОВЛЕНИЕ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
Восстановление карбонильных соединений можно осуществлять различными
способами. Важнейшие из них:
— перенос гидрид-ионов с помощью Н-нуклеофилов;
— каталитическое гидрирование элементным водородом;
— перенос электронов от донора электронов.
Реагентами-переносчиками электронов являются алюминий- и борогидри-
ды, а также особые органические соединения, например алкоголяты металлов и
металлоорганические соединения (разд. Г,7.3.1).
168
Г Препаративная часть (продолжение)
Каталитическое гидрирование карбонильных соединений (разд. Г,7.3.2) осу-
осуществляют аналогично и теми же методами, как в случае олефинов (см. разд.
Г.4.5.1 и Г.4.5.2).
Реагенты, являющиеся электронодонорами, представляют собой неблаго-
неблагородные металлы и металлы в низшей валентности (разд. Г,7.3.3).
В табл. Г.7.223 собраны наиболее важные реакции восстановления карбо-
карбонильных соединений.
Таблица Г.7.223. Восстановление карбонильных соединений
с=о +
альдегиды
.0
-ч +
X
-<f*
NR2
\
С=0 +
/Р
—С +
чн
\
с=о +
Г4— f\ л-
О —U +
\
с=о +
\
C=NR
(+Н20) \
нм *'• нс-он
-МОН /
кетоны
2НМ <+^0)- —СН2-ОН
— Мл
-МОН
нм Т^Г- ~<
н
2НМ -1ШГ -CHa-NR2
RN [AI(OR'K] \
нс-он - нс-он •*
R /
с? + н о (Н°в)
чн
/? [AI(ORK] /P
(M = AIH3Li, BH3NaMflp.)
(X = CI,OR)
(X = CI,OR, NR2, 0®
M = AI(O-f-BuKLiM др.)
(M = AIH3Li, BH3NaMflp.)
R
;c=o
R
c' + —сн2-он
OH
н о-сн2-
/ \ /
HN + НСООН HC-N + С02 + Н20
1 1 t- I hill i PI I 1
rl2N NH2 L/M2 +
н2 ^LL \C_OH
t- H2 -^i- HC-NHR
H2O + N2
Восстановление карбо-
карбонильных соединений гид-
гидридами металлов
до спиртов
до альдегидов
до аминов
Восстановление по Меер-
вейну—Понндорфу (окис-
(окисление по Оппенауэру)
Реакция Канниццаро
Реакция Кляйзена—Ти-
щенко
Реакция Лейка рта—Балл аха
Восстановление по Воль-
Вольфу—Кижнеру
Каталитическое восста-
восстановление до спиртов
до аминов
7. Реакции карбонильных соединений
169
Таблица Г.7.223. Продолжение
2Н2
—С + Н2
CI
-НС,
_CH2-NH2
'Я
2 С=0 + Mg
—с' + 4 Na
OR
О
2 —с/ + 4 Na
OR
С=О + 2Zn + 4Н
2H
-Mg2®
+ 3R'OH
-RONa
-3R'ONa
(+2H@)
-2RONa
-2Na®
2 C=O + Ti
\ /
\ /
но-с-с-он
—CH2-OH
HOv
нс-с
/ \
TiO2
CH2 + 2Zn20+ H2O
20
Восстановление нитрилов
до аминов
Восстановление по Розен-
мунду
Восстановление до пина-
колинов
Восстановление по Бу-
во—Блану
Образование ацилоинов
Восстановление по Клем-
менсену
Восстановление по Мак-
Мурри
7.3.1. ВОССТАНОВЛЕНИЕ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ Н-НУКЛЕОФИЛАМИ
7.3.1.1. Восстановление карбонильных соединений алюминий- и борогидридами
Подобно металлоорганическим соединениям (С-нуклеофилам), переносящим
на карбонильную группу свой органический остаток, металлоорганические
гидриды (М—Н) могут также играть роль Н-нуклеофила и отдавать свой атом
водорода вместе с парой электронов на атом углерода карбонильной группы.
При этом перенос гидрид-иона осуществляется как синхронное расщепление
М-Н-связи и завязывание С-Н-связи:
м-н
с=о
м® + н-с-о®
[Г.7.224]
Альдегиды и кетоны таким образом восстанавливаются до алкоголятов.
Гидридные ионы особенно легко переносятся на карбонильные группы от
таких гидридов металлов, как алюмогидрид лития, борогидрид лития или нат-
натрия, например:
I H -
А1Нгч + H-C-OQLi® H-C-O-AI-H Li® [Г.7.225а]
Н
170
Г Препаративная часть (продолжение)
Таким же образом последовательно вступают в реакцию и все остальные ато-
атомы водорода гидрида:
\
LiAIH4 + 4 С=О
,.©
е I
AI(O-C-HL
[Г.7.2256]
Образовавшийся таким образом комплексный алкоголят лития и алюминия
затем подвергается гидролитическому расщеплению:
Li
.©
е I
AI(O-C-HL + 2 Н2О
4 Н-С-ОН + LiAIO2
[Г.7.226]
Если в молекуле субстрата имеются «активные» водородные атомы, то алю-
могидрид лития реагирует в первую очередь с ними, причем выделяется молеку-
молекулярный водород:
4НХ + LiAIH4 - LiAIX4 + 4 Н2 [Г.7.227]
По этой же причине с алюмогидридом лития можно работать только в без-
безводной среде. Отсюда следует, что этот реактив непригоден для восстановления
соединений, не растворяющихся в инертных органических растворителях, нап-
например Сахаров. В таких случаях прекрасно действует борогидрид натрия,
поскольку он разлагается водой лишь медленно.
Восстановление комплексными гидридами имеет важные преимущества по
сравнению с другими методами: оно протекает, как правило, в очень мягких
условиях и с высокими выходами. Поэтому этот метод особенно пригоден для
работы с ценными препаратами и с малыми количествами. Кроме того, он
позволяет гладко восстанавливать малоактивные производные кислот (амиды,
сложные эфиры, сами кислоты).
Из карбоновых кислот, их сложных эфиров или ацилхлоридов обычно обра-
образуются первичные спирты, из амидов и нитрилов — соответствующие амины.
В особых условиях из ацилгалогенидов, а также амидов и нитрилов можно полу-
получить альдегиды. В табл. Г.7.228 указаны количества алюмогидрида лития, необ-
необходимые для восстановления. (Разберите, каким образом определяют это коли-
количество!)
Таблица Г.7.228. Восстановление карбонильных соединений алюмогидридом лития
Карбонильное соединение
Кетон,альдегид
Сложный эфир, ацилхлорид
Карбоновая кислота
Первичный амид (RCONH2)
Вторичный амид (RCONHR)
Третичный амид (RCONR2)
Нитрил
Продукт реакции
Спирт
Спирт
Спирт
Первичный амин
Вторичный амин
Третичный амин
Первичный амин
Количество LiAlH4
на 1 моль
карбонильного
соединения
0,25
0,50
0,75
1,00
0,75
0,50
0,50
7.Реакции карбонильных соединений
171
Таблица Г.7.229. Селективность восстановления комплексными гидридами металлов
(R = алкил, арил)
Соединение
R-COC1
R-CHO, R-COR'
R-COOR'
R-CONR2'
R-NO2
R-CH=CHR'
LiAlH4
в эфире
I
LiAlH[OC(CH3K]3
в тетрагидрофуране
(ТГФ)
1 1 1 1 1+ + +
БисC-метил-
бутил-2)-
боран в ТГФ
1 + 1 + 1 1 +
NaBH4 +
LiClB
джликоле
1 1 1 1 + + +
NaBH,
вЕЮН
1 1 1 1 1 1 + +
Диизобутил-
алюминийгидрид
в гексане
+
Высокой селективности восстановления можно добиться, используя различ-
различные комплексные гидриды и растворители. В табл. Г.7.229 показано, какие соче-
сочетания приводят к восстановлению (+), а какие нет (—).
Кислоты Льюиса могут изменять специфичность реакций. Так, в присут-
присутствии ВРз-эфирата, LiAlH4 и NaBH4 восстанавливают сложные эфиры и лакто-
ны до простых эфиров.
В качестве растворителя при восстановлении алюмогидридом лития чаще всего
используют абсолютный эфир или тетрагидрофуран. Следует иметь в виду, что вос-
восстановление сопровождается значительным выделением тепла. В особых случаях в
качестве растворителей можно применять также пиридин и N-алкилморфолины.
Иногда продажные препараты алюмогидрида лития не полностью растворимы в эфи-
эфире. В этих случаях можно с таким же успехом использовать суспензию в эфире.
Труднорастворимые вещества можно восстанавливать экстракционным ме-
методом. Для этого восстанавливаемое вещество помещают в экстракционную
гильзу непрерывно действующего экстрактора (например, экстрактора Соксле-
та) и извлекают эфиром. В колбе содержится алюмогидрид лития.
Восстановление диизобутилалюминийгидридом можно проводить как в
ТГФ, так и в алканах и циклогексане, а также в толуоле. С его помощью можно
селективно восстанавливать ацетилены до алкенов.
Реакции восстановления борогидридом натрия проводят в воде, водном эта-
этаноле, изопропиловом спирте, ацетонитриле и других подобных растворителях.
Общая методика восстановления алюмогидридом лития (табл. Г.7.230)
Внимание! Осторожно обращайтесь с алюмогидридом лития! При больших заг-
загрузках используйте мешалку с водяной турбиной или взрывобезопасный мотор,
чтобы исключить возможность взрыва гремучей смеси. Очень осторожно разла-
разлагайте гидрид водой! Соблюдайте осторожность при измельчении гидрида!
В колбу Эрленмейера емкостью 200 мл, снабженную магнитной мешалкой, двуро-
двурогой насадкой, капельной воронкой и обратным холодильником с хлоркальциевой трубкой,
помещают алюмогидрид лития в количестве, соответствующем 10%-ному избытку против
необходимого (см. табл. Г.7.230), в 50 мл абсолютного эфира и добавляют по каплям при пос-
постоянном перемешивании раствор 0,05 моль восстанавливаемого соединения в 20 мл абсолют-
абсолютного эфира. При этом эфир должен спокойно кипеть. Закончив добавление эфирного раство-
раствора, перемешивают реакционную смесь 4 ч или кипятят 1 ч с обратным холодильником1.
1 В некоторых случаях выход повышается, если в этот момент еще раз добавить 10%
расчетного количества аланата (алюмогидрида лития) и нагревать при перемешивании еще 1 ч.
172
Г Препаративная часть (продолжение)
Таблица Г.7.230. Восстановление алюмогидридом лития
Продукт реакции
2,2,2-Трихлорэтанол
а-Стирил этанол
а-Фенил этанол
(—)-Ментол и (+)-неоментолб
цис-цис-^-Декалол
DL- Изоборнеол
о-Гидроксибензиловый спирт"
1,2-Бис(гидроксиметил)-
бензол
(З-Фенилэтиламин
Гександиол-1,6
N-Эгиланилин
4-т/>е/я-Бутилциклогексанол
E)-(+)-2-Ы^-Дибензил-
аминопропанол"
Исходное вещество
Хлоральа
Бензальацетон
Ацетофенон
(—)-Ментон
г<ыс-E-Декалон
DL- Камфора
Метил салицилат
Фталевый ангидридг
Бензилцианид
Диметиловый или
диэтиловый эфир
адипиновой кислоты
Ацетанилидг
4-т/>етя-Бутилцикло-
гексанон
Бензиловый эфир (S)-
М,М-дибензилаланина
Т. кип. (мм рт. ст.)
или т. пл., °С; nD25
Т. кип. 56 A3); т. пл. 17
Т. кип. 144 B1); т. пл. 34
Т. кип. 95 A2); т. пл. 20;
яс251,5224
Т. кип. 95-105 A6)
Т. пл. 105 (петрол. эфир)
Т. пл. 212 (в запаянном
капилляре)
Т. пл. 86 (вода)
Т. пл.64
Т. кип. 83A4); nD251,5299
Т. кип. 134 A0); т. пл. 43
Т. кип. 98 A8); nD2S 1,5519
Т. пл.82
Т.пл.41,[<хЬ20 +
92,8° (хлороформ)
Выход, %
50
95
90
80
80
85
60
80
80
80
60
80
75
(МеОН)
а См. часть Е.
6 Смесь содержит ~75% (—)-ментола и -25% (+)-неоментола. Для анализа можно использовать удель-
удельное вращение в этаноле; для (—)-ментола [а]о20 —48,2°, для (+)-неоментола [а]о20 +19,7°.
в Обработку вести, как указано для аминов; осадок гидроксида алюминия кипятить с петролейным
эфиром.
г Прибавлять в виде раствора в сухом тетрагидрофуране.
д Работать в ТГФ при 60 °С. Перед перекристаллизацией 1 ч нагревать при 60 °С и 0,015 мм рт. ст. B Па).
Затем охлаждают колбу ледяной водой и при перемешивании очень осторожно (по кап-
каплям) добавляют ледяную воду до тех пор, пока не прекратится выделение водорода, после чего
приливают 10%-ную серную кислоту до полного растворения образовавшегося осадка гидрок-
гидроксида алюминия. Слои разделяют в делительной воронке, водный слой трижды экстрагируют
эфиром, органическую фазу промывают насыщенным раствором хлорида натрия, сушат суль-
сульфатом натрия и перегоняют.
При получении аминов для разложения используют минимальное количество воды. Отсасывают
выпавший осадок гидроксида алюминия, еще раз взмучивают его с эфиром, снова отфильтровывают
и перегоняют эфирный раствор, предварительно высушив его гидроксидом натрия.
Ф Восстановление 4-трет-бутилцикиогексанона борогидридом натрия. К раствору 0,04 моль
борогидрида натрия в 120 мл изопропанола при комнатной температуре при перемешива-
перемешивании постепенно добавляют 0,1 моль кетона. Для завершения реакции смесь оставляют на
ночь. Затем осторожно прибавляют разбавленную соляную кислоту до полного прекращения вы-
выделения водорода. Полученный раствор 5 раз экстрагируют эфиром, вытяжки сушат сульфатом
натрия и отгоняют растворитель. Осадок обрабатывают фталевым ангидридом по методике полу-
получения моноэфиров 3-нитрофталевой кислоты (см. разд. Г,7.1.4.1), полученный кислый фталат пе-
рекристаллизовывают из смеси этилацетата с пентаном. Фталат разлагают, перегоняя его с водя-
водяным паром из 20%-ного раствора гидроксида натрия. Дистиллят экстрагируют эфиром, эфир отго-
отгоняют. Остаток содержит смесь цис- и т/>анс-4-т/>ет-бутшщиклогексанолов.
7. Реакции карбонильных соединений 173
Изомеры разделяют хроматографически на активированном оксиде алюминия. Для разде-
разделения 1 г смеси изомеров необходимо 30 г оксида алюминия. Элюируют сначала 1 л пентана,
затем 300 мл смеси пентана с диэтиловым эфиром, содержащей 10% эфира. Первая порция
элюата F00-700 мл) содержит основное количество цис-спирта, затем следует промежуточная
фракция (около 300 мл), конечная фракция содержит чистый транс-спирт.
Т. пл. г<"с-4-т/)е/и-бутилциклогексанола 80—81 °С; т. пл. транс-изомера 81-82 °С.
Восстановление ацилхлоридов до альдегидов с помощью три(/и/?е/я-буток-
си)алюмогидрида лития в диглиме, например получение А-нитробензальдегида
из 4-нитробензоилхлорида: Brown H. С, Subba Rao В. С. J. Am. Chem. Soc, 1958,
80, 5377.
Если в приведенной выше методике вместо изопропанола в качестве раство-
растворителя взять метанол, то необходимо увеличить количество восстановителя при-
примерно в 4 раза, так как борогидрид натрия заметно реагирует с метанолом. В ка-
качестве промежуточного продукта при этом образуется триметоксиборогидрид
натрия; являясь пространственно затрудненным реагентом, он восстанавливает
кетоны с высокой степенью стереоселективности.
Несимметричные кетоны прохиральны, при их восстановлении возникает асимметричес-
асимметрический атом углерода (разд. В,7.3.2). Из ахирального исходного соединения образуется рацемичес-
рацемическая смесь равных количеств обоих энантиомеров. Если же кетон уже содержит хиральную груп-
группу, то при его восстановлении образуется преимущественно один из диастереомеров, например:
Н СН3 Н СНз н СН3
О НО* Н Н* ОН
74% 26%
Эти результаты можно объяснить с помощью модели Фелькина-Ана (см. [В.99] и [В. 100]).
Ахиральные кетоны могут быть восстановлены энантиоселективно с использованием хи-
ральных реагентов, в качестве которых лучше попытаться воспользоваться алюмо- или борогид-
ридами с хиральными радикалами. Возможно также проводить восстановление ахиральными
реагентами, но в присутствии хиральных катализаторов. Так, например, использование кетона
борана с хиральным E)-оксазаборалидином 1 в качестве катализатора восстановления позволя-
позволяет получить вторичные спирты с высокой степенью преобладания одного_из энантиомеров:
О A) + ВН3-тгф н0 Н
И илиВН3Ме25 %
Pri СН3 ее > 96% ~ Ph^"CH3 V-N-r'" [Г.7.232]
Me
Натрийцианоборогидрид, благодаря высокой электроотрицательности циа-
ногруппы, обладает меньшей нуклеофильностью, чем натрийборогидрид, всле-
вследствие чего он не способен восстанавливать альдегиды и кетоны при значениях
рН > 5. Более сильноосновные имины, напротив, еще будут восстанавливаться
в кислой среде. Это создает возможность осуществлять прямое восстановитель-
восстановительное аминирование кетонов, селективно переводя их в иммониевые ионы:
\ \ +Н® \ ' (NaBH3CN) \ '
С=О + H2NR —-—-- C=NR C=N© —- HC-N ГГ.7.233]
Получение этилового эфира 2-ацетиламино-2-этоксикарбонил-9-D-имидазо-
лил)-7-азанонановой кислоты из этилового эфира 2-ацетиламино-2-этоксикар-
бонил-6-оксогексановой кислоты и дигидрохлорида гистамина: Могу К., Sugai Т.,
Maeda Y., Okazaki Т., Noguchi Т., Naito #., Tetrahedron, 1985, 41, 5307.
174 Г Препаративная часть (продолжение)
7.3.1.2. Восстановление по Меервейну-Понндорфу-Верлею и
окисление по Оппенауэру
Альдегиды и кетоны можно восстанавливать до спиртов с помощью алкоголятов
магния или алюминия; при этом алкоголят окисляется до соответствующего
карбонильного соединения (см. схему [Г.7.225] восстановление по Меервей-
Меервейну—Понндорфу—Верлею). Реакция идет и со свободным спиртом в присутствии
каталитических количеств алкоголята, так как последний находится в равнове-
равновесии со спиртом.
Алюминий в молекуле алкоголята алюминия (II, схема [Г.7.234]), являясь
кислотой Льюиса, повышает электрофильную активность карбонильной груп-
группы. Одновременно комплексно связанный негативированный атом алюминия
вызывает сдвиг электронов в исходящих от него связях. Вследствие этого а-водо-
родный атом алкоголята (криптооснования) при участии связывающей электрон-
электронной пары переносится к позитивированному карбонильному атому углерода:
R^ ^R l-k ,R' Вч .hU 'R' *<- .Н R1, ^R'
С C-R' R-C C-R1 R-C С
и + i - и i - i + n
° AT ~~~ %'° V ° [Г.7.234]
3 3 3
I II III IV
В отличие от алкоголятов натрия алкоголяты алюминия растворимы в органических раст-
растворителях и перегоняются без разложения. Отсюда следует, что связь между А1 и OR имеет дос-
достаточно сильно выраженный ковалентный характер. Поэтому алкоголяты алюминия далеко не
всегда могут служить источником алкоксигрупп в виде свободных анионов, их основность ма-
мала и они уже не могут енолизовать карбонильные соединения, т. е. они не катализируют аль-
дольную реакцию или катализируют ее только в незначительной степени. Вследствие этого, а
также из-за относительно большой способности к хелатообразованию алкоголяты алюминия
особенно пригодны для восстановления по Меервейну—Понндорфу—Верлею.
Восстановительные свойства у алкоголятов вторичных спиртов выражены
значительно сильнее, чем у первичных; кроме того, они менее склонны к побоч-
побочным реакциям. (Почему нельзя использовать третичные спирты?)
Реакция [Г.7.234] обратима, поэтому для повышения выходов необходимо
постоянно выводить из реакционной среды карбонильное соединение, образо-
образовавшееся из алкоголята алюминия. В связи с этим обычно в качестве восстано-
восстановителя используют изопропиловый спирт, так как образующийся кетон (ацетон)
является самым летучим компонентом системы и может быть отогнан. При
использовании этанола в качестве восстановителя образуется ацетальдегид,
который лучше всего выводить из реакционной смеси током азота.
Основное значение реакции Меервейна—Понндорфа—Верлея состоит в том,
что в ней не затрагиваются двойные углерод-углеродные связи (в том числе и
сопряженные с карбонильной группой), а также нитрогруппы и атомы галогенов.
(Каким образом в технике получают аллиловый спирт из акролеина?)
Восстановление р-дикарбонильных соединений по Меервейну—Понндорфу—Верлею
обычно не удается, так как эти относительно кислые вещества образуют соединения с алюми-
алюминием, которые выпадают в осадок и таким образом выводятся из сферы реакции.
7. Реакции карбонильных соединений
175
Ф Общая методика восстановления кетонов и альдегидов по Меервейну-Понндорфу—Вер-
лею (табл. Г.7.235). В хорошо высушенном приборе для перегонки с 60-сантиметровой
колонкой Вигре (удобно использовать насадку Хана, см. рис. А.77) нагревают 0,2 моль
карбонильного соединения с 0,2 моль изопропилата алюминия (см. часть Е) в виде 1 М раст-
раствора в абсолютном изопропиловом спирте. Температуру нагревательной бани поддерживают
таким образом, чтобы за 1 мин отгонялось примерно 5 капель смеси изопропиловый спирт —
ацетон. Насадку Хана заполняют этанолом. Для контроля за ходом реакции через несколько
часов время от времени отбирают несколько капель дистиллята и встряхивают с 5 мл раство-
раствора 2,4-динитрофенилгидразина @,1 г в 100 мл 2 н. соляной кислоты); о присутствии ацетона
свидетельствует помутнение или образование осадка. Если проба дает отрицательный резуль-
результат, то нагревают еще 15 мин с полным возвратом дистиллята и повторяют пробу. При пов-
повторном отсутствии помутнения большую часть изопропилового спирта отгоняют в слабом ва-
вакууме, к остатку добавляют по 500 г льда на каждый моль взятого для реакции изопропилата
алюминия и гидролизуют 550 мл охлажденной льдом 6 н. серной или соляной кислотой.
Экстрагируют эфиром, эфирный слой промывают один раз водой, сушат сульфатом натрия,
растворитель отгоняют, а осадок перекристаллизовывают или перегоняют.
Ненасыщенные карбонильные соединения не смешивают предварительно с изопропилатом
алюминия, а готовят раствор 0,1 моль такого соединения в 100 мл абсолютного изопропилового
спирта и этот раствор добавляют в течение 6 ч по каплям к кипящему раствору изопропилата,
отгоняя одновременно смесь ацетона с изопропиловым спиртом. Через 1 ч после прекращения
добавления карбонильного соединения проба на ацетон обычно бывает отрицательной.
Метод пригоден и для полумикровосстановления кетонов. При этом целесообразно
использовать тройной молярный избыток изопропилата алюминия. Восстановление обычно
заканчивается через 1 ч.
Кротиаовый спирт (бутен-2-ол-1) из кротонового альдегида: Young W. G.,
Hartung W. Я., Crossley F. S. J. Am. Chem. Soc, 1936, 58, 100.
2-Метилпентен-3-ол-2 из мезитилоксида: Rouve A., Stoll M. Helv. Chim. Acta,
1947, 30, 2216.
Таблица Г.7.235. Получение спиртов восстановлением по Меервейну-Понндор-
фу-Верлею
Продукт реакции
2,2,2-Трихлорэтанол
2,2,2-Трибромэтанол
Коричный спирт
о-Нитробензиловый спирт
и-Нитробензиловый спирт
ж-Нитробензиловый спирт
1-(З-Нитрофенил)этанол-1
4-Фенилбутен-3-ол-2
(-)-Ментол и (+)-неоментола
4-т/>гт-Бутилциклогексанол6
Исходное соединение
Хлораль (см. часть Е)
Бромаль
Коричный альдегид
о-Нитробензальдегид
л-Нитробензальдегид
.м-Нитробензальдегид
м- Нитроацетофенон
Бензилиденацетон
(-)-Ментон
4-трет-Бугилциклогексанон
Т. кип., °С
(мм рт. ст.)
56A3,;т.пл. 17
93 A0); т. пл. 80
(петролейный эфир)
139 A4); т. пл. 34
168 B0); т. пл. 74
185 A2); т. пл. 93
178C); т. пл. 27
Т. пл. 62 (этанол)
125C); т. пл. 39
96A3)
Т. пл. 82-83
Выход, %
80
75
75
90
90
70
60
90
70
а Продолжительность реакции 24 ч. Состав смеси можно определить по удельному вращению:
для (-)-ментола [a]D20 -48,2°, адля (+)-неоментола [а]о20 +19,7° (этанол).
6 Смесь изомеров.
176 Г Препаративная часть (продолжение)
Обратимость реакции [Г.7.234] позволяет также окислять спирт до соответ-
соответствующего карбонильного соединения с помощью кетона или альдегида (окис-
(окисление по Оппенауэру).
При окислении по Оппенауэру невозможно сдвигать равновесие в желатель-
желательную сторону, отгоняя образующийся спирт, так как спирт всегда кипит при
более высокой температуре, чем карбонильное соединение, из которого он об-
образовался. Поэтому целесообразно брать избыток окислителя или подбирать
компоненты реакции таким образом, чтобы образующееся карбонильное соеди-
соединение было самой низкокипящей составной частью реакционной смеси и его
можно было отогнать.
В качестве дегидрирующего средства при реакции Оппенауэра очень часто
используют циклогексанон, кроме того, применяют также коричный и анисо-
анисовый альдегиды.
Обычно окисляемый спирт не превращают заранее в алкоголят алюминия, а
получают последний в результате равновесной реакции с алкоголятом не участ-
участвующего в реакции спирта:
H3Cn R^ H3Cn R^
Н3С-С-О-^[ + НС-ОН =^ Н3С-С-ОН + HC-O-AI_ [Г.7.236]
H3c' 3 r' HgC' r' 3
При этом удобно применять трет-бутилгт алюминия или фенолят алюми-
алюминия (почему?).
Реакция Оппенауэра используется преимущественно для окисления природ-
природных соединений.
Как и при восстановлении по Меервейну—Понндорфу—Верлею, при окисле-
окислении по Оппенауэру не затрагиваются двойные связи, однако возможна изомери-
изомеризация с образованием ос,р-ненасыщенных карбонильных соединений. Приме-
Примером может служить получение Д4-холестенона-3 из холестерина:
[Г.7.237]
©Получение Д4-холестенона-3 из холестерина [Г.7.237]. В колбе емкостью I л, снабженной
обратным холодильником и хлоркальциевой трубкой, растворяют 0,03 моль холестери-
холестерина в 2 моль горячего ацетона, предварительно перегнанного над перманганатом калия,
а затем над гидроксидом калия, и прибавляют раствор 0,05 моль трет-бутилала алюминия (см.
часть Е) в 300 мл абсолютного толуола, не содержащего тиофена. Реакционную смесь кипятят
10 ч с обратным холодильником, дают охладиться и для удаления солей алюминия несколько
раз встряхивают с разбавленной серной кислотой. Толуольный слой промывают водой до
нейтральной реакции промывных вод, сушат над сульфатом натрия и после удаления раство-
растворителя перекристаллизовывают остаток из метанола. Т. пл. 80 °С, выход 85%.
Получение А4-холестенона-3 по Оппенауэру с применением циклогексано-
на: Eastham J. К, Teranishi R. Org. Syntheses, Coll. Vol. IV, 1963, p. 192. (Имеется
перевод 1-го издания: Истхэм Дж., Тераниши Р. В сб.: Синтезы органических
препаратов. Сб. 7. — М.: ИЛ, 1956, с. 75.)
7. Реакции карбонильных соединений 177
7.3.1.3. Реакции Канниццаро и Кляйзена-Тищенко
Диспропорционирование ароматических и неенолизующихся алифатических
альдегидов под действием основных катализаторов (гидроксидов щелочных и
щелочноземельных металлов) с образованием карбоновых кислот и спиртов
называют реакцией Канниццаро. У енолизующихся альдегидов идет только
альдольная реакция, так как ее скорость больше, чем скорость реакции Кан-
Канниццаро.
Механизм реакции Канниццаро сходен с механизмом реакции Меервейна —
Понндорфа. В циклическом переходном состоянии, образованном двумя моле-
молекулами альдегида, гидроксид-анионом и катионом щелочного металла, проис-
происходит перенос атома водорода со связывающими электронами от одной молеку-
молекулы альдегида к другой молекуле. Сначала образуются алкоголят и карбоновая
кислота, которые затем превращаются в спирт и соль карбоновой кислоты:
н
Ar^ i . Н _ „ Ar H
IK§V®|OH " АГ^С-Н + Аг^у°Н ArCH2OH + ArCOONa [Г.7.238]
Na fiH °Na °
[Напишите схему образования а-гидроксифенилуксусной (фенилгликоле-
вой) кислоты из оксофенилацетальдегида (фенилглиоксаля) путем внутримоле-
внутримолекулярной реакции Канниццаро.]
При проведении реакции Канниццаро со смесью альдегида и формальдеги-
формальдегида последний проявляет себя как донор гидридных ионов, окисляясь до муравь-
муравьиной кислоты (перекрестная реакция Канниццаро):
R-CHO + Н2СО -—- R-CH2OH + НСООН [Г.7.239]
Если альдегид R-CHO (схема [Г.7.239]) содержит а-водородный атом, то прежде всего
протекает альдольная реакция. Только при замещении всех атомов водорода в а-положении с
избытком формальдегида идет реакция Канниццаро, например при получении пентаэритрита
из ацетальдегида и формальдегида:
СН2ОН СН2ОН
ЗНСНО + СН3СНО НОСН2-С-СНО —^-^~ НОСН2-С-СН2ОН + НСООН [Г.7.240]
СН2ОН СН2ОН
С реакцией Канниццаро сходна бензильная перегруппировка, в которой вместо атома
водорода мигрирует фенильный остаток со своими связывающими электронами:
Ph ОН Ph OK
Ph-C-C Ph-C-C [Г.7.241]
OK О ОН О
178
Г Препаративная часть (продолжение)
При использовании в качестве катализатора алкоголятов алюминия, кото-
которые являются слишком слабыми основаниями, чтобы катализировать альдоль-
ную реакцию, енолизующиеся алифатические альдегиды также могут реагиро-
реагировать по типу реакции Канниццаро (реакция Кляйзена—Тищенко). Реакция осу-
осуществляется только в отсутствие воды и спиртов (почему?). При этом из двух
молекул альдегида образуется сложный эфир, например из ацетальдегида —
этилацетат:
н
СН3
о?
-
чсн2
[Г.7.242]
-AT
3
Et
Ф Общая методика проведения перекрестной реакции Канниццаро (табл. Г.7.243). В трехгор-
лой колбе, снабженной мешалкой, термометром, обратным холодильником и капель-
капельной воронкой, нагревают до 65 °С смесь 0,2 моль ароматического альдегида, 60 мл
метанола и 0,26 моль формальдегида в виде 30%-ного водного раствора. Затем по каплям при
перемешивании прибавляют раствор 0,6 моль гидроксида калия в 25 мл воды с такой ско-
скоростью, чтобы при охлаждении колбы проточной водой температура внутри колбы поддержива-
поддерживалась в пределах 65—75 °С. По окончании прибавления нагревают еще 40 мин при 70 °С, а затем
кипятят 20 мин с обратным холодильником. После охлаждения добавляют 100 мл воды и отде-
отделившийся маслянистый слой извлекают эфиром. Экстракт промывают водой и сушат сульфа-
сульфатом натрия. После отгонки эфира остаток перегоняют или перекристаллизовывают.
(jff^j) Получение пентаэритрита. В трехгорлой колбе на 1 л, снабженной мешалкой, термомет-
^^ ром, капельной воронкой и обратным холодильником, к смеси 18,5 г оксида кальция и
2,3 моль формальдегида (в виде формалина) прибавляют по каплям раствор 0,5 моль
ацетальдегида в 300 мл воды. В процессе добавления поддерживают температуру около +15 "С,
а затем в течение 1 ч поднимают до 45 °С. Для удаления катализатора пропускают диоксид уг-
углерода до начала растворения образовавшегося осадка карбоната кальция, от избытка диокси-
диоксида углерода освобождаются кипячением, дают смеси охладиться и отсасывают осадок. Фильт-
Таблица Г.7.243. Получение спиртов перекрестной реакцией Канниццаро
Продукт реакции
Бензиловый спирт
л-Метоксибензиловый
спирт
Пиперониловый спирт
о-Хлорбензиловый спирт
.м-Хлорбензиловый спирт
л-Хлорбензиловый спирт
и-Метилбензиловый спирт
Фурфуриловый спирт3
Альдегид
Бензальдегид
Анисовый альдегид
Пиперональ
о-Хлорбензальдегид
.м-Хлорбензальдегид
л-Хлорбензальдегид
л- Метилбензальдегид
Фурфурол
Т. кип., "С
(мм рт. ст.)
98 A4)
136 A2); 23 (т. пл.)
157A6);
56 (т. пл.; вода)
69 (т. пл.; этанол)
105A3)
72 (т. пл.; вода)
118B0);
60 (т. пл.; лигроин)
83 B5)
Лр20
1,5403
1,5535
1,4828
Выход, %
90
90
80
90
70
90
75
60
а Не нагревать до кипения! Перед экстракцией эфиром раствор насытить карбонатом калия,
эфирные вытяжки промыть небольшим количеством насыщенного раствора хлорида натрия.
7. Реакции карбонильных соединений
179
рат под вакуумом упаривают досуха, остаток растворяют в 200 мл горячего этанола, охлаждают
раствор; при этом пентаэритрит выпадает в осадок. Выход 75%. Т. пл. чистого пентаэритрита
(после возгонки в высоком вакууме) 260 °С.
2,2,6,6-Тетра(гидроксиметил)циклогексанол из циклогексанона и формальде-
формальдегида: WittkoffH., Org. Syntheses, Coll. Vol. IV, 1963, p. 907. (Имеется перевод 1-го
издания: Уитткофф Г. В сб.: Синтезы органических препаратов. Сб. 4. — М.:
ИЛ, 1953, с. 458.)
Фуранкарбоновая-2 (пирослизевая) кислота и фурфуриловый спирт из фурфу-
фурфурола: Вильсон В. К. В сб.: Синтезы органических препаратов. Сб. 1. — М.: ИЛ,
1949, с. 351.
Промышленное значение реакция Канниццаро имеет при получении следующих продук-
продуктов: пентаэритрит, который применяется в производстве алкидных смол и взрывчатых ве-
веществ — нитропента (тетранитропентаэритрит); тригидроксинеопентан (метриол; из пропио-
нового альдегида и формальдегида; сложный эфир применяется в качестве пластификатора),
триметилолпропан (из бутиральдегида и формальдегида; в производстве алкидных смол, поли-
полиэфиров, полиуретанов), 1,3-дигидрокси-2,2-диметилпропан (неопентилгликоль; из изобути-
ральдегида и формальдегида производство пластификаторов и полиэфирных смол). Синтез
этилацетата из уксусного альдегида осуществляют в промышленности по Кляйзену—Тищенко.
Превращения типа реакции Канниццаро часто встречаются в физиологических процес-
процессах. Некоторые ферменты способны превращать альдегиды в спирты и кислоты. Так, при мо-
молочнокислом брожении под действием глиоксалазы из метилглиоксаля образуется молочная
кислота.
о н рн
и
О
и
О
[Г.7.244]
7.3.1.4. РеакцияЛейкарта—Валлаха
Реакцией Лейкарта—Валлаха называют восстановительное алкилирование ами-
аминов альдегидами или кетонами в присутствии муравьиной кислоты в качестве
восстановителя. Реакция карбонильного соединения с амином сначала идет
обычным образом по уравнению [Г.7.11а]. Азометиновый (карбений-
иммониевый) катион I (см. схему [Г.7.245]) далее восстанавливается муравьиной
кислотой через циклическое переходное состояние II до амина:
С
©!1
+НСООН
\
—С
Н
О
и
+ С
и
[Г.7.245]
От каталитического аминирования (см. схему [Г.7.256]) реакция Лейкар-
та-Валлаха выгодно отличается тем, что в ней можно использовать вещества,
отравляющие катализатор гидрирования.
Реакцию Лейкарта—Валлаха целесообразно применять для получения тре-
третичных аминов. При получении первичных или вторичных аминов в качестве
побочных продуктов всегда образуются более высокоалкилированные амины.
Высокореакционноспособный формальдегид по большей части приводит к пол-
полностью метилированным аминам.
180
Г Препаративная часть (продолжение)
Служащую восстановителем муравьиную кислоту всегда берут в избытке
B—4 моль на 1 моль карбонильного соединения). При алкилировании формаль-
формальдегидом можно работать в водном растворе, используя формалин и 85%-ную му-
муравьиную кислоту; в случае менее реакционноспособных высших альдегидов,
а в еще большей степени кетонов в присутствии воды выходы резко падают.
Поэтому алкилирование кетонов проводят, как правило, при температурах
150—180 °С, позволяющих удалять воду отгонкой.
В этих условиях из муравьиной кислоты и амина образуется соответствую-
соответствующие формиат аммония или формамид. Последние можно непосредственно
использовать в качестве исходных веществ.
При получении вторичных аминов, особенно при высоких температурах,
образуются формильные производные аминов, поскольку муравьиная кислота
является хорошим формилирующим средством (см. разд. Г,7.1.4.2). В таких
случаях приходится дополнять синтез стадией гидролиза образовавшегося
N-дизамещенного формамида.
Ф Общая методика проведения реакции Лейкарта—Валлаха с альдегидами (табл. Г.7.246).
В круглодонную колбу емкостью 2 л, снабженную обратным холодильником, помеща-
помещают 1 моль соответствующего амина и через холодильник, охлаждая колбу льдом, добав-
добавляют 5 моль муравьиной кислоты (85%-ной при реакциях с формальдегидом, 98%-ной
при реакциях с высшими альдегидами или кетонами).
Затем прибавляют на каждую вводимую алкильную группу по 1,2 моль альдегида (фор-
(формальдегид в виде формалина) и нагревают на водяной бане до прекращения выделения диок-
диоксида углерода (8—12 ч).
Раствор подкисляют концентрированной соляной кислотой до кислой реакции по конго
красному и упаривают досуха на водяной бане в вакууме водоструйного насоса. Остаток раст-
растворяют в небольшом количестве холодной воды, основание выделяют 25%-ным водным раст-
раствором гидроксида натрия и трижды экстрагируют эфиром. Эфирные вытяжки сушат гид-
роксидом калия, отгоняют эфир и перегоняют остаток с помощью 20-сантиметровой колонки
Вигре или перекристаллизовывают его.
Таблица Г.7.246. Получение аминов по Лейкарту-Валлаху
Продукт реакции
Ы,Н-Диметил-и-бугаламин
N, N-Д иметилбензилам ин
N- Метиддициклогексил-
амин6
N-Метилпиперидин
N-Бугилгшперидин
N-Бензилпиперидин
М,М-Диэтилфурфуриламин
Исходные вещества
н-Бутиламин, формальдегид
Бензиламин, формальдегид
Дициклогексиламин,
формальдегид
Пиперидин, формальдегид
Пиперидин, масляный
альдегид
Пиперидин, бензальдегид
Диэтиламин, фурфурол
Т. кип., °С
(мм рт. ст.)
94
78 B6)
153B4)
106
68 B0)
119A3)
74 B4)
«d20
1,3954а
1,4986s
1,4895
1,4464
1,4461
1,5252
1,4630
Выход, %
80
80
65
70
40
40
45
а При25°С.
6 Если продукт при перегонке сильно вспенивается, то его перегоняют при обычном давлении без
колонки. Т. кип. 268 °С G50 мм рт. ст.).
7. Реакции карбонильных соединений 181
а-Фенилэтиламин из ацетофенона и формиата аммония: Ингерсолл А.
В сб.: Синтезы органических препаратов. Сб. 2. — М.: ИЛ, 1949, с. 523.
Амфетамин (СбН5СН2СН(МН2)СНз) и его производные получают из фенилацетона по ре-
реакции Лейкарта—Валлаха. Эти соединения являются лекарственными препаратами — симпа-
томиметиками, но благодаря свойствам привыкания, их использование в качестве доппинг-
препаратов запрещено.
7.3.1.5. Ферментативное восстановление
Карбонильную группу можно мягко восстанавливать (в результате гидридного
переноса) в присутствии ферментов. Используемые для этого оксидоредуктазы
к тому же являются хиральными катализаторами, состоящими из белкового
фрагмента и кофермента (низкомолекулярная активная группа). Одним из ко-
ферментов таких оксидоредуктаз является NADH (гидрированный никотина-
мид-аденин-динуклеотид) или его фосфорилированная форма (NADPH). Его
гидридпереносящим фрагментом является восстановленный никотинамид
[Г.7.247], пиридиновый атом азота которого соединен гликозидной связью с ри-
бозой. Образующийся в процессе реакции NAD+ далее снова восстанавливается
до NADH в результате целого ряда биохимических процессов за счет, например,
добавляемого сахара:
н н
,CONH2 О +н@ ОН ^
Д. Т^ H"W + У [Г.7.247]
i i
R R
Тригональная карбонильная группа (при R * R') представляет собой прохи-
ральную систему, которая взаимодействует с хиральным ферментом преимущест-
преимущественно с той стороны, которая приводит к термодинамически более стабильному
(диастереомерному) переходному состоянию. При этом преимущественно обра-
образуется (за исключением отдельных случаев) только один энантиомерный спирт.
Вместо выделения дорогих ферментов для восстановления, например
эфиров р-оксокарбоновых кислот, используют обычные пекарские дрожжи
A—1,5 кг на 1 моль субстрата). Таким же образом с высоким энантиомерным
выходом могут быть восстановлены эфиры других а- и р-оксокарбоновых
кислот, а-гидроксикетоны и другие подобные соединения (см. указанную ни-
ниже литературу).
Синтез этилового эфира {5)-(—)-3-гидроксимасляной кислоты восстановлени-
восстановлением ацетоуксусного эфира пекарскими дрожжами: Seebach D., Weber R. Я.,
ZugerM. F., Org. Synth. 1985, 63, 1.
См. также: WipfB., KupferE., BertazziR., Leuenberger Y. G. W., Helv. Chim. Acta,
1983,66,485.
7.3.1.6. Восстановление по Кижнеру-Вольфу
Еще один важный метод превращения альдегидов и кетонов в соответствующие
углеводороды — это восстановление по Кижнеру—Вольфу. Если гидразон альде-
альдегида или кетона нагревать до 200 °С в автоклаве в присутствии натрия или алко-
182 Г Препаративная часть (продолжение)
голята натрия, то отщепляется азот и карбонильное соединение превращается в
углеводород:
\ ,2 \
C=N СН2 + N2 [Г.7.248]
\ iN-iNi-i +R0» \j-N-N ^ N 0 +R0H \ н [Г.7.249]
Аналогично происходит а разложение семикарбазонов с образованием углеводородов.
Более новый вариант по Хуангу—Минлону состоит в том, что гидразон полу-
получают из карбонильного соединения и гидразина в высококипящем растворите-
растворителе (этиленгликоле или глицерине) и тотчас, не выделяя, нагревают его до 195 °С;
в этом случае можно работать без давления. Поскольку одновременно из реак-
реакционной смеси отгоняется вода, то вместо дорогого гидразингидрата можно
применять его дешевый 85%-ный раствор, а вместо натрия или алкоголята нат-
натрия использовать гидроксид натрия или калия.
Кетоны и оксокислоты реагируют гладко с высокими выходами. р-Оксоэфи-
ры нельзя восстанавливать таким путем, так как образуются пиразолоны (см.
схему [Г.7.59]). Двойные связи в алкильных остатках изомеризуются и частично
гидрируются (о восстановлении нитрогруппы см. схему [Г.8.9]).
При проведении реакции с альдегидами могут образовываться азины. В этих
случаях лучше применять большой избыток гидразингидрата F—10 моль).
Вариант Хуанга—Минлона позволяет без труда вводить в реакцию большие
количества исходных веществ. В этом, а также в ряде других отношений он пре-
превосходит восстановление по Клемменсену.
(ЦУ) Общая методика восстановления кетонов по Кижнеру—Вольфу (табл. Г.7.250)
j*j Внимание! Соблюдайте осторожность при экстракции эфиром сильнощелоч-
у ного раствора! Защитные очки!
Смешивают 1 моль кетона с 3 моль 85%-ного гидразингидрата (можно применять и более конце-
концентрированные растворы; о концентрировании разбавленных растворов и об определении их конце-
концентрации см. часть Е), прибавляют 4 моль тонкорастертого гидроксида калия (в случае оксокислот —
5 моль) и 1000 мл глицерина (триэтиленгликоля). Смесь кипятят 2 ч с обратным холодильником. За-
Затем соединяют колбу с нисходящим холодильником и медленно отгоняют смесь гидразина и воды,
пока температура не поднимется до 195 "С (внутренний термометр следует защищать металлической
гильзой; почему?). Если нагревают в металлической бане, в которую глубоко погружена колба, то
достаточно измерения температуры в бане. Температуру поддерживают до тех пор, пока продолжа-
продолжается выделение азота (около 4 ч). Окончание выделения газов определяют, погружая время от време-
времени шланг, надетый на форштос холодильника, в воду (осторожно! при охлаждении вода может засо-
саться в холодильник!). Особое внимание следует уделить герметичности прибора и равномерности
нагревания. Легколетучие углеводороды по окончании реакции находятся в основном уже в дистил-
дистилляте. После охлаждения реакционной смеси ее разбавляют равным объемом воды и, если восстанав-
восстанавливалась оксокислота, сильно подкисляют концентрированной соляной кислотой. После этого
несколько раз извлекают эфиром, эфирные вытяжки соединяют с продуктом, отогнанным в ходе
реакции, промывают разбавленной соляной кислотой, затем водой и сушат хлоридом кальция.
В заключение отгоняют эфир и остаток перегоняют или перекристаллизовывают. Выход 80-95%.
Получение 3-(циклогексен-1-ил-1)пропионитрила из л-толуолсульфонилгид-
разона 3-B-оксоциклогексил)пропионитрила: Wittekind R. R., Weissman С,
FarberS., Meltzer R. I. J. Heterocycl. Chem., 1967, 4, 143.
7. Реакции карбонильных соединений
183
Таблица Г.7.250. Восстановление по Кижнеру-Вольфу
Продукт реакции
Этилбензол
Пропил бензол
Бутилбензол
1 -Бром-4-этилбензол
1 -Хлор-4-этилбензол
3,4-Диметокси-1 -этилбензол
4-Метокси-1 -этилбензол
4-Метил-1-этилбензол
4-Фенилмасляная кислота
Ундекандикарбоновая-
1,11 (брассиловая)
Гептановая кислота
Октановая кислота
Нонановая (пеларгоновая)
кислота
Декановая кислота
Исходное соединение
Ацетофенон
Пропиофенон
Бугирофенон
л-Бромацетофенон
л-Хлорацетофенон
Ацетовератрон
4-Метоксиацетофенон
4-Метилацетофенон
З-Бензоилпропионовая кислота
Метилен-бис(дигидрорезорцин)а
6-Оксогептановая кислота
6- (или 7-)Оксооктановая
кислота
6- (или 7-)Оксонановая кислота
7-Оксодекановая кислота
Т. кип., °С
(мм рт. ст.)
136
57B0)
78A0)
94A5)
80A5)
112(9)
90B1)
162
Т. пл.50
Т.пл. 112
(этилацетат)
119 A0); т.пл.-8
132 A6); т.пл. 16
142 A0); т.пл. 1,5
или соотв. 156
146 (8); т. пл. 31
Ло20
1,4959
1,4920
1,4898
1,5488
1,5190
1,5038
1,4950
а Перед реакцией с гидразином под действием щелочи прежде всего происходит расщепление кольца
(см. разд. Г,7.2.1.9), причем образуется 4,8-дикетоундекандикарбоновая-1,11 кислота. Это необходимо
учитывать при определении стехиометрических соотношений.
6 Полиморфные модификации.
Аналогично восстановлению по Кижнеру—Вольфу и Хуангу-Минлону проводят реакцию
по Бамфорду — Стивенсу, в которой монозамещенные гидразоны с электроноакцепторными
заместителями нагревают в этиленгликоле в присутствии алкоголята. Для этой цели хорошо
подходят л-толуолсульфонилгидразоны. В протонных растворителях устанавливается равно-
равновесие (см. схему [Г.7.251]) между гидразонной и азо-формами. После элиминирования суль-
фоната и молекулярного азота образуется карбений-катион, который может вступать в извест-
известные реакции (см. разд. Г,7.3 и Г,7.4). После отщепления протона от соседнего атома углерода
в конце концов образуется олефин; если такой атом водорода отсутствует, то возможны перег-
перегруппировки (см. разд. Г,9):
\ /
R'-C-C
N-NHTs /u©.
R
R-—C-C-H
-N2,-Ts
H
R
R1 = H \
-He
c=c
\© r
C-C-H
/ n
[Г.7.251]
Ts = n-MeQH4SO2—
184
Г Препаративная часть (продолжение)
В апротонных растворителях, например диалкиловых эфирах этиленгликоля, установле-
установление таутомерного равновесия между гидразонной и азо-формами затруднено. Основание
отщепляет связанный с атомом азота протон, а затем после элиминирования молекулярного
азота и сульфината образуется карбен, продукты дальнейших реакций которого можно выде-
выделить с довольно высокими выходами (см. разд. Г,3.3):
N-NHTs
R'-C
_ э
N-N—Ts
R'-c'
п
N=N—Ts
R
О
-N2,-Ts
R._r, . Продукты ГГ 7 2*521
K Ч ппеипашоний 1.1 •'••"¦'J
превращении
Ознакомьтесь с этой реакцией по литературе, приведенной в разд. Г.7.5.
7.3.2. КАТАЛИТИЧЕСКОЕ ГИДРИРОВАНИЕ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
Гетерогенное каталитическое гидрирование С=О-группы протекает аналогично
восстановлению кратных С=С-связей (разд. Г.4.5.2). Водород хемосорбируется
на поверхности катализатора с образованием гидридного комплекса и в такой
форме реагирует с адсорбированным таким же образом карбонильным соедине-
соединением. Альдегиды и кетоны при этом восстанавливаются до спиртов.
С=О + Н2
НС-ОН
[Г.7.253]
Для каталитического гидрирования карбонильных соединений и их анало-
аналогов можно использовать те же катализаторы, что и для гидрирования двойной
связи С=С. (О каталитическом гидрировании см. разд. Г,4.5.2.) В лаборато-
лаборатории применяют прежде всего скелетный никель (никель Ренея), платину и
палладий.
Подобно другим карбонильным реакциям, гидрирование ускоряется кисло-
кислотами. Поэтому в кислой среде благородные металлы являются более эффектив-
эффективными катализаторами, чем в нейтральном или щелочном растворе. При исполь-
использовании скелетного никеля, наоборот, наилучшие результаты дает сильнооснов-
сильноосновный катализатор (например, катализатор Урушибара).
В соответствии с положением в ряду активности карбонильных соединений
[Г.7.3] легче всего гидрируются альдегиды и кетоны. Платина и палладий, одна-
однако, относительно слабо катализируют эту реакцию, так что оказывается возмож-
возможным, например, провести селективное восстановление а,р-ненасыщенных ке-
тонов до насыщенных кетонов (см. табл. Г.4.124). Такие же результаты можно
получить со свободным от щелочи скелетным никелем, дезактивированным
кислотами и метилиодидом. В щелочной среде в присутствии скелетного нике-
никеля, напротив, карбонильная группа легко гидрируется, например, ненасыщен-
ненасыщенные кетоны сразу восстанавливаются в насыщенные спирты.
Нитрилы, азометины, оксимы и подобные соединения легко восстанавли-
восстанавливаются на платине и палладии, в то время как для восстановления на никеле
Ренея, как правило, требуется температура около 100 °С. При гидрировании
нитрилов в качестве побочных продуктов часто образуются вторичные и
третичные амины. Эти побочные реакции протекают через промежуточный
альдимин II (схема [Г.7.254]), который дает с уже образовавшимся первичным
амином III азометин:
7. Реакции карбонильных соединений 185
+ Но +Н?
R-CEN ^ R-CH = NH -~ R-CH2-NH2
-nh3 +ш [Г.7.254]
R-CH = N-CH2-R ^~ (RCH2JNH
IV V
(Образования каких аналогичных продуктов следует ожидать при гидрирова-
гидрировании оснований Шиффа?)
Указанные нежелательные побочные реакции можно подавить. Используя в
качестве катализатора сильноосновный скелетный никель или проводя гидри-
гидрирование в присутствии аммиака.
При каталитическом гидрировании тиолов, тиоэфиров и тиоацеталей отщепляется сера
(в виде сероводорода). На этом основан важный способ превращения кетогрупп в метилено-
вые через дитиоланы (см. схему [Г.7.32]). Галогены также могут быть замещены на водород.
В соответствии с рядом [Г.7.3] можно ожидать, что ацилхлориды должны легко подвергать-
подвергаться каталитическому восстановлению. Действительно, их удается восстановить до альдегидов,
используя частично отравленный палладиевый катализатор, который хотя и способен превра-
превратить ацилхлорид в альдегид, однако не восстанавливает последний (реакция Розенмунда).
Свободные кислоты, эфиры и амиды не затрагиваются в тех условиях, в кото-
которых гидрируются альдегиды, кетоны, нитрилы, основания Шиффа и другие
соединения. Так, из ацетоуксусного эфира легко получить эфир C-гидроксимас-
ляной кислоты.
Для каталитического гидрирования карбоновых кислот и сложных эфиров
наиболее пригоден медно-хромовый катализатор, работающий при высоких
температурах A00—300 °С) и высоком давлении B0—30 МПа; 200—300 атм). Этот
метод применяется преимущественно в промышленности, влаборатории проще
провести восстановление эфиров другими методами [восстановление по
Буво—Блану, см. схему [Г.7.261]; восстановление комплексными гидридами, см.
разд. Г,7.3.1.1].
Ф Общая методика каталитического гидрирования кетонов, альдегидов, нитрилов, оксимов и
азометинов (табл. Г.7.255). (Сведения о безопасных методах работы и необходимых пре-
предосторожностях при каталитическом гидрировании см. в разд. А, 1.8.2 и Г,4.5.2.) Раство-
Растворяют 1 моль соответствующего карбонильного соединения в двойном по объему количестве
метанола, добавляют скелетный никель, приготовленный по Урушибара (см. часть Е) из 30 г
сплава, содержащего 30% никеля, и гидрируют при 10 МПа A00 атм) в автоклаве при встряхи-
встряхивании или перемешивании. В случае простых, малоразветвленных альдегидов и кетонов можно
работать при комнатной температуре, а-третичные альдегиды, кетоны и нитрилы гидрируют
при 90 "С.
После охлаждения и разгерметизации автоклава отфильтровывают катализатор и отгоня-
отгоняют растворитель. Остаток очищают перегонкой или перекристаллизацией. Выход 80—90%.
Небольшие количества можно гидрировать при указанных температурах при нормальном
давлении. При этом целесообразно увеличить количество катализатора.
Рассмотренные реакции гидрирования имеют большое препаративное и промышленное
значение для получения спиртов и аминов. В промышленности, например, этим способом
получают бутанол из кротонового альдегида и 2-этилгексанол из бутиральдоля. Оба спирта в
основном перерабатываются в эфиры (растворители, пластификаторы, см. табл. Г.7.42).
В больших масштабах проводится гидрирование монооксида углерода. На катализаторе из
186
Г Препаративная часть (продолжение)
Таблица Г.7.255. Каталитическое гидрирование карбонильных соединений и их
аналогов
Продукт реакции
Гептанол
Тетрагидрофурфуриловый спирт
Бутанол-2
Циклопентанол
Этиловый эфир р-гидроксимасля-
ной кислоты
а-Фенил этанол
Дифенилкарбинол
4-Фенилбутанол-2
1,2-Дифенилэтиленгликоль
(гидробензоин)
3,3-Диметилбутанол-2
Ментол
Этиловый эфир 4-гидрокси-
1 -метилгшперидинкарбоно-
вой-3 кислоты
D-Сорбит3
Бензил анилин6
Гексаметилендиамин
р-Фенилэтиламин
Этиловый эфир 3-ацетамидопипе-
ридон-2-карбоновой-З кислоты"
Ы-C-Аминопропил)-Е-капро-
лактамг
Исходное соединение
Гептаналь
Фурфурол
Бутанон
Циклопентанон
Ацетоуксусный эфир
Ацетофенон
Бензофенон
Бензилиденацетон
Бензоин
Пинаколин
и-Ментен-1-он-3
Этиловый эфир
1 -метилпиперидон-
4-карбоновой-З кислоты
D-Глюкоза
Бензилиденанилин
Адипонитрил
Бензилцианид
Диэтиловый эфир
1 -ацетамидо-3-цианопропан-
дикарбоновой-1,1 кислоты
М-B-Цианэтил)-
е-капролактам
Т. кип., °С
(мм рт. ст.)
78A0)
80 B0)
100
140
74A1)
94A2)
176 A3); т. пл. 68
(лигроин)
115A3)
Т. пл. 139 (вода)
120
98 A0); т. пл. 36
123D)
Т.пл.~100
173 A0); т. пл. 39
88 A1); т. пл. 40
83A4)
Т. пл. 138 (этанол)
110-120B)
«о20
1,4235
1,4498
1,3971
1,4530
1,4182
1,5211
1,5165
1,4148
1,3742
1,5321
а Гидрируют в смеси этанол — вода при 70 °С; полученный после отгонки растворителя сиропообраз-
сиропообразный остаток хранят в эксикаторе над хлоридом кальция; продукт плохо кристаллизуется, часто только
после внесения затравки.
6 Гидрируют в этилацетате при 20 °С.
" В качестве растворителя применяют этанол! Какой тип реакции имеет место при замыкании кольца,
происходящего спонтанно после гидрирования? — Омыление соляной кислотой приводит к орнитину
(напишите схему реакции!): AlbertsonN. F., Archer S. J. Am. Chem. Soc., 1945,67,2043.
г Выход 50%; если использовать в качестве растворителя метанол, насыщенный аммиаком, выход увеличивается.
оксида цинка и оксида хрома при 300-400 °С и высоком давлении B0 МПа; 200 атм) образует-
образуется метанол. Он используется в основном для производства формальдегида (см. табл. Г.6.40) и
метиламинов, а также в качестве растворителя и антифриза. При повышенной (на 40 °С) тем-
температуре над щелочным катализатором наряду с метанолом получают высшие изоспирты (до
Ci), более всего изобутиловый спирт. Эти спирты также в основном перерабатывают в эфиры.
Каталитическое восстановление жирных кислот и их эфиров (из природных жиров или
продуктов окисления парафинов, см. разд. Г.6.5) приводит к получению высших жирных
спиртов, применяющихся для синтеза моющих средств (сульфатов жирных спиртов). Низшие
спирты (С4—С9), получаемые из жирных кислот — продуктов окисления парафинов, являются
исходными веществами для синтеза эфиров (см. выше).
Восстановлением динитрила адипиновой кислоты получают гексаметилендиамин, приме-
применяемый в качестве аминного компонента в синтезе полиамидов (найлон, см. разд. Г,7.1.4.2).
7. Реакции карбонильных соединений
187
Если гидрирование альдегидов и кетонов проводить в присутствии аммиака,
первичных или вторичных аминов, то вместо спиртов получают соответствую-
соответствующие первичные, вторичные или третичные амины {восстановительное аминиро-
вание)и.
С=0 + NHR,
\
-Н2О /
CH-NR2
R = Н,алкил, арил
[Г.7.256]
Предполагается, что промежуточными продуктами здесь являются азомети-
ны и енамины. И в этом случае необходимо считаться с возможностью побоч-
побочных реакций, упомянутых при рассмотрении гидрирования нитрилов. Поэтому
аминный компонент берут, как правило, в избытке.
Из алифатических альдегидов хорошо поддаются каталитическому восста-
восстановительному аминированию только соединения, содержащие более пяти
углеродных атомов, в то время как низшие альдегиды легко образуют в ходе
реакции продукты конденсации (типа альдолей). В случае алифатических
и ароматических кетонов и ароматических альдегидов реакция идет
гладко.
Таблица Г.7.257. Каталитическое восстановительное аминирование альдегидов и
кетонов
Продукт реакции
Бензиламин
N-Бензилметиламин
N-Бензиланилин
N-Бензил-р-фенилэтил-
амина
Фурфурилам ин
DL-a-Фенил этил амин
DL-2-Амино-1 -фенил-
пропан11
DL-2-Метиламино-
1-фенилпропана
Циклогексиламин
Дициклогексиламин
Исходное соединение
Бензальдегид, аммиак
Бензальдегид, метиламин
Бензальдегид, анилин
Бензальдегид,
р-фенилэтиламин
Фурфурол, аммиак
Ацетофенон, аммиак
Бензилметилкетон,
аммиак
Бензилметилкетон,
метиламин
Циклогексанон, аммиак
Циклогексанон,
циклогексиламин
Т. кип., "С
(мм рт. ст.)
75(8)
82A2)
172 A0); т. пл. 39
170 (9); гидрохлорид:
т. пл. 261
145
70 A0)
92 A2); гидрохлорид:
т. пл. 152
93 A5); гидрохлорид;
т. пл. 140
134
120 A7); т. пл. 20
«о20
1,5424
1,5222
70
1,4886
1,5282
1,5190
1,5123
1,4372
1,4852
Выход, %
80
90
90
50
80
90
80
80
70
а Эти амины лучше всего хранить в виде гидрохлоридов: амин растворяют при охлаждении в избытке
абсолютного этанола, насыщенного хлороводородом (содержание НС1 определяют взвешиванием!),
высаживают соль добавлением абсолютного эфира. Осторожно, вещества ядовиты!
1 Эту реакцию называют также восстановительным алкилированием (аммиака или ами-
аминов). О восстановительном аминировании посредством муравьиной кислоты и аминов (реак-
(реакция Лейкарта-Валлаха) см. разд. Г,7.3.1.4.
188 Г Препаративная часть (продолжение)
Общая методика каталитического восстановительного аминирования альдегидов и кетонов
(табл. Г.7.257)
Внимание! Автоклав не должен иметь медных деталей, соприкасающихся с аммиач-
аммиачным щелочным раствором (многие манометры имеют медные детали!). О мерах пре-
предосторожности при каталитическом гидрировании см. разд. А,1.8.2 и Г.4.5.2.
А. Получение первичных аминов. Растворяют 1 моль карбонильного соединения в 500 мл ме-
метанола, насыщенного аммиаком при 10 °С (т. е. содержащего 5,5 моль аммиака). Вносят ске-
скелетный никель, приготовленный из 30 г сплава, и гидрируют во встряхиваемом автоклаве или
автоклаве с мешалкой при 90 °С и давлении 10 МПа A00 атм).
По окончании поглощения водорода открывают автоклав, отфильтровывают катализатор,
отгоняют избыток аммиака и растворитель. Остаток подкисляют 20%-ной соляной кислотой по
конго красному и извлекают эфиром примеси неосновного характера. Эфирные вытяжки отбра-
отбрасывают. Водный слой при хорошем охлаждении подщелачивают 40%-ным раствором гидрокси-
да натрия и несколько раз экстрагируют эфиром. Эфирные вытяжки сушат гидроксидом калия.
После отгонки растворителя остаток перегоняют на 20-сантиметровой колонке Вигре.
Б. Получение вторичных аминов. Смешивают 1 моль карбонильного соединения с раствором
1 моль первичного амина в 200 мл метанола, гидрируют и обрабатывают, как указано выше.
При обычном химическом синтезе хиральные соединения образуются в виде смеси равных
количеств энантиомеров (D, L; R, S). Напротив, природные соединения такого типа сущест-
существуют в виде определенных индивидуальных энантиомерных веществ. Важно отметить, что би-
биологическое действие (например, запах, вкус, физиологические и фармакологические свой-
свойства) энантиомеров, как правило, резко различаются.
Одной из возможностей получения чистых энантиомеров является разделение синтети-
синтетически полученной рацемической смеси (другим способом является асимметрический синтез,
см. разд. В,7.3.3.2). Рацематы соединений кислотного характера можно разделить с использо-
использованием природных оснований, таких как бруцин(—) и хинин(—), образующих диастереомер-
ные соли. Последние можно уже легко разделить благодаря различию их физических и хими-
химических свойств (разд. В,7.3.3.1).
Примером необычно просто протекающего расщепления рацемата через диастереомеры может
служить получение оптически активных форм а-фенилэтиламина. Из двух его антиподов только
Л(+)-форма образует кристаллический аддукт с 2,3,4,6-тетраацетил-О-глкжозой.
Чистые энантиомеры а-фенилэтиламина можно использовать для разделения других
рацематов вместо упомянутых выше дорогих природных продуктов.
Ф Расщепление рацемического-а-фенилэтиламина на оптические антиподы. [Helferich В.,
Portz V/. Chem. Ber., 1953, 86, 1034.]
В 100 мл эфира растирают 0,15 моль тетраацетил-О-глюкозы (р-форма или сиропооб-
сиропообразная смесь а- и р-форм) и 0,1 моль DL-а-фенилэтиламина с 20 мл эфира. Вскоре начинает-
начинается кристаллизация аддукта Л-(+)-а-фенилэтиламина с тетраацетил-О-глюкозой. Оставляют
на три часа при охлаждении до —78 "С, затем быстро отсасывают и промывают двумя порция-
порциями по 40 мл холодного эфира. Выход 98%.
Для выделения свободного Л-(+)-основания растворяют аддукт в 100 мл хлороформа и
раствор дважды обрабатывают 100 мл 4 н. соляной кислоты. Для отделения последних остат-
остатков тетраацетил-О-глюкозы солянокислый раствор дважды извлекают хлороформом. Затем
подщелачивают 40%-ным раствором гидроксида натрия при хорошем охлаждении, выделивший-
выделившийся амин извлекают эфиром, сушат гидроксидом калия и перегоняют. Т. кип. 70 °С A0 мм рт. ст.);
[a]D19 +35,9° (бензолI.
Тетраацетил-О-глюкозу можно регенерировать из хлороформного раствора после обра-
обработки последнего соляной кислотой, для чего хлороформный раствор сушат хлоридом каль-
кальция и отгоняют хлороформ. Тетраацетил-О-глюкоза получается в виде сиропа, который мож-
можно снова использовать для расщепления амина на антиподы.
?-(-)-а-Фенилэтиламин извлекают из эфирного маточного раствора, оставшегося после отде-
отделения аддукта Л-(+)-формы, обрабатывая эфирный раствор соляной кислотой и далее, как описа-
описано для Л-(+)-основания, и выделяют перегонкой. Т. кип. 70 °С A0 мм рт. ст.); [а]о19 -34,6° (бензолI.
1 Оптические антиподы получаются не совсем чистыми, поэтому (+) и (-)-формы име-
имеют несколько различающиеся углы вращения.
7. Реакции карбонильных соединений 189
Расщепление энантиомеров-а-фенилэтиламина с помощью винной кислоты:
Theilacker W., Winkler H. G. Chem. Ben, 1954, 87, 690.
7.3.3. ВОССТАНОВЛЕНИЕ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ1
НЕБЛАГОРОДНЫМИ МЕТАЛЛАМИ И
МЕТАЛЛАМИ НИЗШЕЙ ВАЛЕНТНОСТИ
В металлах валентные электроны свободно перемещаются («электронный газ»)
и поэтому могут присоединяться к карбонильным соединениям как «нуклео-
фильный реагент»:
+ >c-oie I _0 _ I
л. V ~,© / ~ —с-ог +2н® —с-он
:=О — -C-QI _±_й1е - _^_он [Г.7.258]
/С-^ Г ~ "" Т """ [Г.7.259]
IV V
Результатом этих реакций является восстановление карбонильных соедине-
соединений. При этом сначала путем присоединения одного электрона образуется ани-
анион-радикал I, либо за счет присоединения двух электронов — дианион IV. Ани-
Анион-радикал I может димеризоваться с образованием гликоля III [такая реакция
происходит при восстановлении кетонов металлическим магнием], тогда как
дианион IV, будучи сильным основанием, отщепляет от растворителя ион водо-
водорода и образуют спирт V.
Разумеется, эти окислительно-восстановительные процессы могут протекать
только на поверхности металла. При этом карбонильное соединение более или
менее прочно связывается с поверхностью металла (хемосорбция). По оконча-
окончании переноса электрона хемосорбированная молекула десорбируется. На каж-
каждый перенесенный электрон переходит в раствор соответствующее число атомов
металла в виде катионов:
(|Zn] - Zn2® + 2е0) + С=О + Н® lcf-0-H -^— Н-С-ОН [Г.7.260]
Осуществлять такое восстановление в соответствии с их положением в
электрохимическом ряду напряжений могут только электроотрицательные
неблагородные металлы. Щелочные металлы способны восстанавливать даже
наиболее инертные карбонильные соединения (например, эфиры карбоновых
кислот), а магний и алюминий — только альдегиды и кетоны. Цинк и железо
проявляют восстанавливающие свойства только в кислой среде. (Ср., однако, с
каталитическим гидрированием карбонильных соединений, разд. Г,7.3.2.)
1 Другие методы восстановления карбонильных соединений (комплексными гидридами,
по Меервейну-Понндорфу и др.) будут рассмотрены позже на основе других механизмов ре-
реакций.
190 Г Препаративная часть (продолжение)
Восстановление карбонильных соединений электроотрицательными метал-
металлами, например амальгамированным магнием или алюминием, железом, цин-
цинком и др., может приводить к продуктам как реакции [Г.7.258], так и реакции
[Г.7.259]. Направление, по которому идет реакция, зависит от природы карбо-
карбонильного соединения и от условий проведения реакции (металл, растворитель и
т. д.)- Альдегиды и кетоны восстанавливаются указанными металлами в раство-
растворителях, содержащих активный водород (например, в воде, разбавленных кис-
кислотах и щелочах, спиртах), преимущественно до соответствующих спиртов, а
азометины до аминов. Восстановительное аминирование кетонов удается также
с помощью амальгамы алюминия. Амальгамы магния и алюминия в растворите-
растворителях, не содержащих активный водород (например, в бензоле), восстанавливают
кетоны преимущественно до гликолей (пинаконов). Напишите схему образова-
образования пинакона из ацетона (типа схемы [Г.7.258]; II соответствует в данном случае
пинаколяту магния) и объясните установленную выше зависимость продуктов
реакции от растворителя.
Ф Получение 2,3-диметилбутандиола-2,3 (пинакона). В сухую двугорлую колбу емкостью
1 л, снабженную капельной воронкой и эффективным обратным холодильником с
хлоркальциевой трубкой, помещают 1 г-атом сухих магниевых стружек и 200 мл сухого
толуола (см. часть Е). Из капельной воронки приливают небольшую часть (примерно
25 мл) раствора 0,1 моль сулемы [хлорида ртути(Н)] в 2 моль хорошо обезвоженного ацетона
(см. часть Е). Если реакция не начинается через несколько минут, то подогревают на водяной
бане до тех пор, пока кипение будет происходить без внешнего нагрева за счет начавшейся ре-
реакции. Тогда прекращают нагревание и прибавляют из капельной воронки оставшийся раст-
раствор сулемы в ацетоне возможно быстрее, следя за тем, чтобы не происходило выбросов через
холодильник. Затем приливают раствор еще 1 моль ацетона в 60 мл толуола и нагревают на во-
водяной бане до полного исчезновения магния. Образующийся пинаколят магния заполняет
всю колбу, образуя объемистую пористую массу, поэтому 1-2 раза во время реакции необхо-
необходимо снять холодильник, плотно закрыть колбу пробкой и сильно встряхнуть ее (защитные
очки!), а затем продолжить нагревание.
Для гидролиза магниевой соли по окончании реакции прибавляют через холодильник 60 мл
воды и кипятят еще 1 ч. Затем охлаждают до 50 °С, отсасывают гидроксид магния, кипятят его
с 150 мл толуола и снова отсасывают. Объединенные толуольные фильтраты упаривают до по-
половинного объема, добавляют 70 мл воды и охлаждают в ледяной бане при перемешивании.
При этом выпадает гексагидрат пинакона. Через 1 ч его отфильтровывают и промывают толу-
толуолом. Высушенный на воздухе препарат достаточно чист для дальнейших реакций. В случае
необходимости его можно перекристаллизовывать из воды. Т. пл. 46 °С; выход 40%.
Безводный пинакон можно получить азеотропным обезвоживанием толуолом и перегон-
перегонкой в вакууме. Т. кип. 75 °С A3 мм рт. ст.); т. пл. 43 °С.
Точно таким же образом осуществляется восстановление сложных эфиров
карбоновых кислот или ацилхлоридов действием натрия в спирте (восстановле-
(восстановление по Буво—Блану). В соответствии с общей схемой [Г.7.259] реакция протекает
следующим образом:
р. 0 t
2Na- + R-<1 Ti^r R-8^ »R^§b
i
[Г.7.261]
^R-ch2-qi0
7. Реакции карбонильных соединений
191
Натриевая соль полуацеталя альдегида III, естественно, тут же распадается на
алкоголят и альдегид. Последний восстанавливается таким же образом, в резуль-
результате чего образуется натриевый алкоголят первичного спирта.
На 1 моль эфира требуется 4 г-атома натрия и 2 моль спирта. (Напишите сум-
суммарное уравнение реакции!)
В отсутствие спирта, т. е. при взаимодействии сложного эфира или ацилхлорида с метал-
металлическим натрием, реакция идет по другому пути. Сначала в соответствии со схемой [Г.7.258]
образуется продукт I (схема [Г.7.262]), который через дикетон II восстанавливается до ацило-
ина:
2Na-
/Р
2R-C
2R-C
OR
ГО О
2Na- + R-C-C-R
OR
0IOI CN
i n--1
R-CtC-R
0
eioi ioiq
I I
R-C-C-R
i i
RO OR
I
О О
II II
R-C-C-R
-2RO
QIOI 101®
I I
R-C=C-R
[Г.7.262]
+ 2Na
©
динатриевая соль ацилоина
(Напишите суммарное уравнение реакции, обоснуйте образование дикетона II из первич-
первичного продукта димеризации I и объясните происходящее на последней стадии реакции восста-
восстановление карбонильной группы до динатриевой соли ацилоина!)
Соли ендиолов III при подкислении переходят в свободные ацилоины:
°Ю1 IOI®
i i
R-C=C-R
+ 2Н*
НО ОН
I I
R-C=C-R
О ОН
и i
R-C-CH-R
ацилоин
[Г.7.263]
При восстановлении по Буво—Блану желательно, чтобы используемый спирт
не слишком легко реагировал с натрием, так как в противном случае много нат-
натрия расходуется бесполезно, в больших количествах выделяется газообразный
водород, который не способен восстанавливать эфир. Наиболее пригодны вто-
вторичные спирты, например изомерная смесь трех метилциклогексанолов, полу-
получаемая в промышленности из смеси трех крезолов (каким образом?). В лабора-
лаборатории применяют также изопропанол или циклогексанол.
Восстановление по Буво—Блану дает прекрасные результаты также в приме-
применении к нитрилам. При этом образуются первичные амины.
Ф Общая методика восстановления сложных эфиров и нитрилов по Буво-Блану (табл.
Г.7.264)
Внимание! При работе с натрием и концентрированными щелочами надевать
защитные очки! При разложении реакционной смеси следует проявлять чрез-
чрезвычайную осторожность. Воду добавлять только после того, как все остатки
натрия прореагируют.
Вся аппаратура и реактивы должны быть совершенно сухими. Спирт лучше
всего сушить с помощью магния (см. часть Е), ксилол — над натрием, а эфир
или нитрил — перегонкой в вакууме.
192
Г Препаративная часть (продолжение)
Таблица Г.7.264. Восстановление эфиров и нитрилов по Буво-Блану
Продукт реакции
Октанол-1а
Деканол-1
Додеканол-1
(лауриловый спирт)
Тетрадеканол-1
р-Фенилэтиловый спирт
Этиленкеталь
4- гидрокси бутанона- 2
1,10-Дигидроксидекан
Нониламин"
Ундециламин
Додециламин
Тридециламин
Тетрадециламин
Исходное соединение
Этиловый эфир
октановой кислоты
Этиловый эфир
декановой кислоты
Этиловый эфир
додекановой кислоты
Этиловый эфир
тетрадекановой
(миристиновой) кислоты
Этиловый эфир
фенилуксусной кислоты
Этиленкеталь
ацетоуксусного эфира
Диэтиловый эфир
себациновой кислоты
Октилцианид
Децилцианид
Ундецил цианид
Д одецил циан ид
Тридецилцианид
Т. кип., °С
(мм рт. ст.)
100B0)
112A1)
139A2)
0
172 A6); т. пл. 38
00A0)
1
87A1)
Т. пл.74б
202
115A3)
131 A5); т. пл. 28
160 A4); т. пл. 27
177 A4); т. пл. 40
ло20^40)
1,4300
1,4367
1,5259
1,5315
1,4448
1,4352
1,4403
A,4309)
A,4338)
A,4382)
Выход, %
60
70
8
85
80
60
75
70
80
75
70
75
а В качестве растворителя применяют толуол.
6 После отгонки растворителя перекристаллизовывают из водного этанола.
в Для более полного отделения ксилола объединенные экстракты отмывают 10%-ной соляной кислотой.
Солянокислые вытяжки амина снова встряхивают с эфиром, подщелачивают разбавленной щелочью,
экстрагируют выделившийся амин эфиром, сушат К2СО3 и перегоняют.
Прибор состоит из трехгорлой колбы емкостью 2 л, снабженной мешалкой Гершберга
(см. рис. А.6,ж), эффективным холодильником с хлоркальциевой трубкой и капельной ворон-
воронкой. В колбе нагревают 4,5 г-атома натрия под 800 мл ксилола до плавления (для эфиров ди-
карбоновых кислот берут 1000 мл ксилола на 0,5 моль эфира), добавив на кончике шпателя
стеариновую кислоту в качестве эмульгатора. Когда натрий расплавится, запускают мешалку и
перемешиваютдотехпор и с такой интенсивностью, чтобы натрий диспергировался в тонкую
серую суспензию. Тогда перемешивание прекращают и охлаждают до температуры ниже тем-
температуры плавления натрия. После этого снова при хорошем перемешивании прибавляют из
капельной воронки смесь 1 моль сложного эфира или нитрила (в случае эфира дикарбоновой
кислоты 0,5 моль) с 3,5 моль изопропилового спирта настолько быстро, насколько это позво-
позволяет холодильник. При восстановлении нитрилов смесь необходимо подогревать, чтобы под-
поддерживать частицы натрия в суспендированном состоянии.
Перемешивают еще 15—20 мин и добавляют метанол в количестве, необходимом для пол-
полного разложения натрия. В заключение осторожно добавляют еще 800 мл воды.
После охлаждения разделяют фазы, водный слой экстрагируют эфиром (осторожно — образо-
образование эмульсии), в случае диолов — 5 дней в перколяторе. Органическую фазу и эфирную вытяжку
объединяют, сушат сульфатом натрия и перегоняют с 40-сантиметровой колонкой Вигре.
Другие примеры: МанскеР. В сб.: Синтезы органических препаратов. Сб. 2. —
М.: ИЛ, 1949, с. 170.
7. Реакции карбонильных соединений
193
Аналогичным методом можно восстановить любой жир. Сначала необходимо определить
число омыления, чтобы рассчитать материальный баланс реакции (см. с. 88).
При заключительной перегонке начинают собирать фракцию, кипящую выше 70 °С
A4 мм рт. ст.) (что примерно соответствует спиртам Сб), не разделяя последующие фракции.
Дистиллят растворяют в спирте и разделяют методом газовой хроматографии (см.
рис. Г.7.266).
При действии на альдегиды и кетоны амальгамированного цинка и концент-
концентрированной соляной кислоты происходит их восстановление до углеводородов
(восстановление по Клемменсену):
\
С = О + 2Zn + 4Н
,©
\
СН2 + 2Zn2® + Н20
[Г.7.265]
В ходе этой реакции часто образуются в значительном количестве побочные
продукты, например пинаконы и другие спирты (в соответствии с обычной схе-
схемой восстановления), а также олефины и высокомолекулярные углеводороды.
Кроме того, часто оказывается необходимым проводить реакцию очень длитель-
длительное время, и все же часть карбонильного соединения не восстанавливается.
Тем не менее этот метод в ряде случаев дает хорошие выходы углеводорода,
как это имеет место, например, при восстановлении многих альдегидов и алифа-
алифатических или арилалифатических кетонов, в то время как диарилкетоны, как
правило, реагируют плохо.
а-Оксокислоты часто превращаются лишь в соответствующие а-гидроксикислоты, вос-
восстановление эфиров р-оксокислот возможно, но с невысокими выходами, в то время как у-ок-
сокислоты восстанавливаются с хорошими выходами. В случае труднорастворимых кетонов,
например стероидного ряда, можно для улучшения растворимости прибавлять этиловый
0,34
11,66
14,43
Рис. Г.7.266. Газохроматографическое
разделение спиртов Сб-Cie с четным числом
атомов углерода. Колонка: 3% силикона OV 225 на
хромосорбе W-AW-DMCS; длина 2 м, внутренний
диаметр 3 мм. Программированное изменение тем-
температуры: 2 мин при 110 °С, затем повышение до
240 "С со скоростью 8 °С/мин. Чувствительность
пламенно-ионизационного детектора 30 ГОм;
газ-носитель — азот B,5 л/ч).
а - этанол; б - гексанол; в - октанол; г- деканол;
д - додеканол; е - тетрадеканол; ж - гексадеканол;
э - октадеканол.
16.88
аб в г д е
Время удерживания —
ж
194
Г Препаративная часть (продолжение)
спирт или уксусную кислоту A : 1). Слишком хорошая растворимость кетона в водной фазе,
однако, тоже нежелательна.
Помимо неблагородных металлов восстанавливать карбонильные соедине-
соединения может также целый ряд металлов низшей валентности.
Так, низковалентный титан, вероятно Ti@), образующийся при взаимодей-
взаимодействии TiCl3 с LLAIH4 или калием, например, в ТГФ при реакции с карбонильны-
карбонильными соединениями, возможно, промежуточно образует соединение I (схема
[Г.7.267]), гидролиз которого приводит к диолу II. Нагревание реакционной
смеси вызывает отщепление ТЮг и восстановление субстрата до алкена (сочета-
(сочетание по Мак-Мурри).
2 с=о + л
+Н2О
НО
ч /0Н
II
\ ./
[Г.7.267]
-ТЮ2
Эта реакция имеет большое значение в синтезе алкенов из карбонильных со-
соединений. Если обе карбонильные группы принадлежат одной молекуле, то воз-
возможно образование циклического соединения. При этом выходы не зависят от
размера цикла (внутримолекулярное сочетание по Мак-Мурри).
Синтез 3,3-диметил-1,2-дифенилциклопропена из диметилдибензоилметана
(и других циклопропенов) восстановлением TiCb/LiAlH,»: Baumstark A. L,
McCloskey С. J., Witt К. Е., J. Org. Chem. 1978, 43, 3609.
7.4. РЕАКЦИИ ВИНИЛОГОВ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ И
ДРУГИХ ВИНИЛОГОВЫХ СИСТЕМ
Если двойная связь карбонильной группы (или другого —М-заместителя) соп-
сопряжена с двойной связью С=С, то электрофильные свойства карбонильного
углеродного атома переходят к р-углеродному атому (см. разд. Г,4; олефины с
электроноакцепторными заместителями). В результате нуклеофильный
реагент атакует преимущественно C-положение. Причина этого заключается в
делокализации л-электронов по всей системе двойных связей, о чем сви-
свидетельствуют представленные на схеме [Г.7.268] граничные мезомерные струк-
структуры:
\ /
с=с
/ '-ЛС=О1
\е /
'/4QI
©
с-с.
с^о
[Г.7.268]
Точно также в сопряжении со связью С=С может участвовать неподеленная
электронная пара +А/-заместителя (—NR2, —OR) (см. разд. Г,4; олефины с
электронодонорными заместителями). В этом случае нуклеофильные свойства
+М-заместителя переносятся к р-углеродному атому, который и атакуют в пер-
7. Реакции карбонильных соединений
195
вую очередь электрофильные реагенты. Такие структуры изображены на схеме
[Г.7.269]:
8+
\ 0<
с=с ~
/'-¦ \
\е
/"¦
:;х@
с
\
\5" ..-/
[Г.7.269]
-Х= -01® -ОН, -QR, -NR2
Приведенные на схеме [Г.7.269] структуры представляют собой либо енольные формы
карбонильных соединений, либо их производные (еноляты, эфиры енолов, енамины). (Напи-
(Напишите формулы енолов первичных и вторичных алифатических нитросоединений!) С реакция-
реакциями енолов мы уже сталкивались, например, при обсуждении альдольной конденсации, ката-
катализируемой кислотами, и реакции Манниха.
Если —М- или +М-заместитель связан с одним концевым углеродным ато-
атомом цепи с сопряженными двойными связями, то положительный (или отрица-
Таблица Г.7.270. Реакции винилогов карбонильных соединений и других винилоговых
систем
Y-H + С=С
/ \
Y-C-C-X
Х= —COR, —COOR, —CN и др.
Y= R2N—, RCONR', R0-, RS—, Hal —
Z= —Hal, —SO2R'
OSiMe3
R-C
\ .
C=C
/ \
R'(H)
С
R-C
Cl
\ ,NI
c=c
/ \
M^SiO I o
i i //
- R-C-C-C
R. I \
©
NR2
OH | о
-Me3SiZ i I \
n' i ч
-Cl
7Г C-C-C
0 / | \
+ H20
-R2NH2
\ ^
R-Z + C=C
/ \
©
,NR2
-77- R-C-C-C
:S I I \
+ H2O
-R2NH2®
R1
Vc-c'
/ I \
R-C-C-C
I I \
Присоединение ами-
но-, гидрокси-, суль-
фанилсоединений и
галогеноводородов
Присоединение по
Михаэлю
Алкилирование карбо-
карбонильных соединений
Галогенирование кар-
карбонильных соедине-
соединений
Алкилирование три-
метилсилиловых эфи-
ров енолов
Альдольная реакция
по Мукайаме
Ацилирование
енаминов
Алкилирование
енаминов
196 Г Препаративная часть (продолжение)
тельный) заряд переносится к другому концевому углеродному атому. Подоб-
Подобный перенос полярности в сопряженных системах, а также свойство группиро-
группировок, связанных через виниленовые звенья, проявлять себя таким образом, точно
они связаны друг с другом непосредственно, называют винилогией, а соответ-
соответствующие соединения — винилогами или винилгомологами родоначальника
этого ряда (например, ряда альдегидов, сложных эфиров карбоновых кислот,
аминов); см. также разд. В,5.1.
В табл. Г.7.270 собраны реакции винилогов карбонильных соединений и дру-
других винил оговых систем.
7.4.1. РЕАКЦИИ ВИНИЛОГОВ ЭЛЕКТРОНОАКЦЕПТОРНЫХ СОЕДИНЕНИЙ-
а,р-НЕНАСЫЩЕННЫХ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
Концевой углеродный атом винилога карбонильного соединения благодаря на-
наличию положительного заряда реагирует так же, как и углеродный атом карбо-
карбонильной группы:
HNu + /С^С^=^= /С^.С^=^= /С^С^^=^ /С^°^ [Г.7.271]
I I / н
В результате нуклеофильный реагент избирательно присоединяется к двой-
двойной связи С=С (см. также схему [Г.4.98]).
Винилоги — акцепторы электронов могут быть примерно расположены в
следующий ряд по убывающей активности: а,C-ненасыщенные альдегиды >
а,C-ненасыщенные кетоны >а,C-ненасыщенные нитрилы >а,C-ненасыщенные
эфиры карбоновых кислот >а,р-ненасыщенные амиды карбоновых кислот.
К важным представителям этого класса веществ относятся акролеин, метил-
винилкетон, акрилонитрил1 и эфиры акриловой кислоты. Если в этих соедине-
соединениях часть водородных атомов замещена на алкильные или арильные группы, то
активность подобных соединений всегда ниже, чем соответствующих незаме-
незамещенных соединений. (Сравните со сходными закономерностями для альдеги-
альдегидов, кетонов и карбоновых кислот.)
К винилогам карбонильных соединений, как и к карбонильным соединени-
соединениям, могут присоединяться вещества со свободной электронной парой, например
аммиак, амины, спирты, фенолы, тиолы, некоторые минеральные кислоты, а
также С—Н-кислотные соединения (синильная кислота, альдегиды, кетоны,
Р-дикарбонильные соединения и их аналоги). Реакции соединений первой
группы катализируются как щелочами (которые активируют основание), так и
кислотами (активирующими винилог карбонильного соединения).
Катализ основаниями:
О
Реакции присоединения акрилонитрила называют также цианэтилированием.
7. Реакции карбонильных соединений 197
Катализ кислотами:
№/ ОН _H@Nu4/ ОН Nu/ О
II /н
С—Н-Кислотные соединения должны быть предварительно депротонирова-
ны и переведены таким образом в способные к присоединению анионы. Поэ-
Поэтому такие реакции присоединения как правило, ускоряются основаниями
[Г.7.272].
Катализаторами с основными свойствами чаще всего служат гидроксиды и
алкоголяты щелочных металлов, гидроксид бензилтриметиламмония (тритон В),
а для высокореакционноспособных систем также триэтиламин. Для кислотного
катализа используют серную и уксусную кислоты, трифторид бора и др.
7.4.1.1. Присоединение аминов к винилогам карбонильных соединений
Амины сравнительно легко присоединяются к а,р-ненасыщенным карбониль-
карбонильным соединениям и нитрилам, например:
R2NH + H2C=CH-COOR' R2N-CH2-CH2-COOR' [Г.7.274]
Аммиак и алифатические амины обладают достаточной основностью и поэ-
поэтому присоединяются в мягких условиях даже в отсутствие катализатора. Для
присоединения ароматических аминов, например, требуется температура выше
100 °С и, кроме того, часто присутствие кислотных катализаторов. При реакци-
реакциях с первичными алифатическими аминами в зависимости от стехиометричес-
ких соотношений и температуры можно получить как моно-, так и дипроизвод-
ные. (Напишите схемы этих реакций!) Моноаддукты аммиака можно получить с
удовлетворительным выходом только в специальных условиях.
Ф Общая методика присоединения аминов к винилогам карбонильных соединений (табл.
Г.7.275)
¦ Осторожно! Большинство винилогов карбонильных соединений ядовито или
Л обладает слезоточивым действием. Работать под тягой!
А. Алифатические амины. В трехгорлую колбу емкостью 500 мл, снабженную мешалкой,
капельной воронкой, обратным холодильником и термометром, помещают раствор 1,1 моль
алифатического амина в 150 мл этанола. К этому раствору при перемешивании прибавляют по
каплям 1 моль свежеперегнанного <х,р-ненасыщенного карбонильного соединения, причем
температура реакционной смеси не должна превышать 30 °С. Для получения бис-производно-
го из первичного амина в реакцию вводят 2,5 моль карбонильного соединения.
При получении моноаддуктов акрилонитрила или метилвинилкетона реакционную смесь
оставляют на ночь, при получении моноаддуктов эфиров акриловой кислоты — на 24 ч (при
синтезе бис-аддуктов время удваивают), после чего реакционную смесь перегоняют в вакууме.
Б. Ароматические амины. В круглодонной колбе, снабженной обратным холодильником, в
течение 12 ч кипятят смесь 0,5 моль ароматического амина, 0,5 моль свежеперегнанного
а,р-непредельного соединения в 20 мл уксусной кислоты, после чего реакционную смесь
перегоняют в вакууме.
Присоединение ароматических аминов к а,р-ненасыщенным альдегидам
или кетонам, катализируемое кислотами, используют также при синтезе хино-
Таблица Г.7.275. Присоединение аминов к винилогам карбонильных соединений
Продукт реакции
Этиловый эфир 3-метиламинопропионовой
кислоты
Диэтиловый эфир 4-метил-4-азапентанди-
карбоновой-1,5 кислоты
З-Метиламинопропионитрил
1,5-Дициано-З-метил-З-азапентан
Этиловый эфир 3-пиперидинопропионовой
кислоты
З-Пиперидинопропионитрил
Этиловый эфир 3-бензиламинопропионовой
кислоты
Диэтиловый эфир 4-бензил-4-азапентан-
дикарбоновой-1,5 кислоты
З-Диэтиламинопропионитрил
4-Пиперидинобутанон-2
Этиловый эфир 3-анилинопропионовой кислоты
Метиловый эфир 3-(л-толуидино)пропионовой
кислоты
З-Анилинопропионитрил
3-(«-Анизидино)пропионитрил
Исходные вещества
Метиламин, этилакрилат
Метиламин, этилакрилат
Метиламин, акрилонитрил
Метиламин, акрилонитрил
Пиперидин, этилакрилат
Пиперидин; акрилонитрил
Бензиламин, этилакрилат
Бензиламин, этилакрилат
Диэтиламин. акрилонитрил
Пиперидин, метилвинилкетон
Анилин, этилакрилат
л-Толуидин, метилакрилат
Анилин,акрилонитрил
л-Анизидин, акрилонитрил
Вариант
А
А
А
А
А
А
А
А
А
А
Б
Б
Б
Б
Т. кип., °С (мм рт. ст.)
65A7)
122C)
74 A6)
138 E)
116A7)
115A8)
134 B)
170A)
84A3)
101A1)
146 B)
150 F); 60
(т. пл.; бензол —
петролейный
эфир)
160 F); 49 (т. пл.;
водн. этанол)
221 B1); 64 (т. пл.;
водн.этанол)
«D20
1,4218
(при22°С)
1,4411
1,4342
(при15°С)
1,4606
1,4548
1,4697
1,5060
1,4941
(при 23 °С)
1,4353
1,4630
1,5313
Выход, %
42
80
75
80
80
90
85
80
85
80
50
50
80
70
7.Реакции карбонильных соединений
199
линов по Скраупу (схема [Г.7.276]) или по Дебнеру-Миллеру. а,р-Ненасыщен-
а,р-Ненасыщенные карбонильные соединения при этом часто не вводят в реакцию в готовом
виде, а получают в ходе реакции (например, акролеин из глицерина, кротоно-
вый альдегид из паральдегида). После присоединения амина происходит ката-
катализируемая кислотами реакция альдегидной группы с ароматическим ядром
(см. разд. Г,5.1.8.5), ведущая к образованию 1,2-дигидрохинолина:
9 н он
о
[Г.7.276]
Дигидрохинолиновое производное в заключительной стадии дегидрируется
до хинолина (синтез по Скраупу) или в результате диспропорционирования пе-
переходит в производные тетрагидрохинолина и хинолина (синтез по Дебне-
Дебнеру—Миллеру). (Напишите схему реакции по Дебнеру—Миллеру.)
Для окисления дигидрохинолина в синтезе Скраупа чаще всего используют
нитробензолы, соответствующие применяемому амину. В качестве дегидри-
дегидрирующих средств можно, однако, применять и пентаоксид мышьяка, хлорид
железа(Ш) и другие соединения.
Составьте схемы синтеза 8-гидроксихинолина (антисептик) и 2- и 4-метилхинолинов (из
которых получают полиметиновые красители, см. разд. Г,7.2.1.12).
Ф Общая методика получения хинолинов по Скраупу (табл. Г.7.277). В трехгорлой колбе на
500 мл, снабженной мешалкой, термометром и обратным холодильником, нагревают до
140 "С при перемешивании смесь 0,4 молъ ароматического амина, 1,3 моль безводного гли-
глицерина и 0,47 моль пентаоксида мышьяка. Затем прибавляют через капельную воронку большими
порциями около половины от общего количества A10 г) концентрированной серной кислоты. Ос-
Остальное количество добавляют по каплям после растворения первоначально образующегося осад-
осадка. Смесь нагревают еще 4 ч при 150—155 °С, после охлаждения выливают в 1 л воды и оставляют на
ночь. Затем смесь фильтруют и подщелачивают кислый раствор, прибавляя по каплям при очень
хорошем перемешивании концентрированный раствор гидроксида натрия.
При получении жидких веществ щелочную смесь перегоняют с водяным паром и дистил-
дистиллят несколько раз экстрагируют эфиром. Сушат гидроксидом калия, отгоняют эфир и перего-
перегоняют в вакууме, пользуясь 20-сантиметровой колонкой Вигре.
При получении твердых веществ их отфильтровывают, сушат сырой продукт в вакуум-эк-
вакуум-эксикаторе и осаждают в виде гидрохлорида, пропуская хлороводород через ацетоновый раствор
Таблица Г.7.277.
Продукт реакции
Хинолина
6-Нитрохинолин
1 -Азафенантрен
Синтез хинолинов по Скраупу
Исходное вещество
Ацетанилид6
л-Нитроанилин
2-Нафтиламин
Физические константы
Т. кип. 112 A4ммрт. ст.);
nD201,6218
Т. пл. 151 (водн. этанол)
Т. пл. 93 (лигроин)
Выход, %
50
50
50
а Вместо пентаоксида мышьяка в качестве окислителя применяют нитробензол @,25 моль).
6 В ходе реакции гидролизуется с образованием анилина.
200 Г Препаративная часть (продолжение)
сырого основания. Отфильтрованный осадок растворяют в воде, кипятят с углем, вновь выде-
выделяют свободное основание, как описано выше, отфильтровывают и перекристаллизовывают
из водного этанола.
7.4.1.2. Присоединение воды, галогеноводородов, сероводорода,
спиртов и тиолов к винилогам карбонильных соединений
Присоединение спиртов к активированным двойным связям с акцепторными
заместителями удается осуществить в присутствии кислых или (чаще) щелочных
катализаторов:
ROH + H2C=CH-COOR' RO-CH2-CH2-COORf [Г.7.278]
Таким же образом присоединяется вода, причем образуются либо р-гидрок-
сисоединения, либо соответствующие C,р'-дизамещенные диэтиловые эфиры.
(Составьте схемы реакций!)
При взаимодействии этиленгликоля или глицерина с акрилонитрилом получают аддукты,
используемые в газовой хроматографии в качестве неподвижных фаз. Цианэтиловый эфир
целлюлозы применяется в производстве специальных искусственных волокон.
Присоединение сероводорода и тиолов протекает легче, чем присоединение
воды и спиртов, так как нуклеофильность соединений серы выше. Метантиол
реагирует с акролеином уже без катализатора [прибавляемый ацетат меди(П)
служит ингибитором полимеризации].
Присоединение галогеноводородов приводит к образованию р-галогенкар-
бонильных соединений, что не отвечает правилу Марковникова. (Объясните
этот факт!)
Ф Получение р-метилтиопропионового альдегида присоединением метантиола к акролеину.
[Pierson Е. и др. J. Am. Chem. Soc, 1948, 70, 1450.]
Внимание! Соблюдайте указания о работе с тиолами (см. разд. Г,2.6.6)! Акро-
Акролеин сильно раздражает глаза!
В двугорлой колбе на 500 мл, снабженной газоподводящей трубкой и обратным холодильни-
холодильником с трубкой для отвода газа, осторожно нагревают в медленном токе азота смесь 0,28 моль суль-
сульфата S-метилтиурония1 с 110 мл 5 н. раствора гидроксида натрия. Выделяющийся газообразный
метантиол пропускают последовательно через пустую промывную склянку, затем промывную
склянку с разбавленной серной кислотой A объем концентрированной серной кислоты на 2
объема воды) и осушительную колонку с хлоридом кальция Г~П [Й №щ , после чего направ-
направляют газ в четырехгорлую колбу с газоподводящей трубкой, мешалкой, термометром и обратным хо-
холодильником, снабженным газоотводной трубкой. В этой колбе находятся 0,5 моль свежеперегнанно-
го акролеина и 0,25 г ацетата меди(П). Во время реакции поддерживают температуру около 35—40 °С,
охлаждая колбу в бане со льдом. Примерно через 90 мин сульфат S-метилтиурония полностью раз-
разлагается и реакция заканчивается. Реакционную смесь фракционируют в вакууме, используя корот-
короткую колонку Вигре. Т. кип. 53 °С (при 11 мм рт. ст.); «о20 1,4850; выход 60%.
©Общая методика присоединения галогеноводородов к винилогам карбонильных соедине-
соединений (табл. Г.7.279). Пропускают сухой галогеноводород через 0,2 моль свежеперегнан-
ного винилога карбонильного соединения, защищенного от влаги воздуха и охлаж-
охлажденного до температуры ~—10 °С смесью льда с хлоридом натрия. Скорость пропуска-
1 Получение см. Шильднек П., Унидус У В сб.: Синтезы органических препаратов, Сб. 2. —
М.:ИЛ, 1949, с. 326.
7.Реакции карбонильных соединений
201
Таблица Г.7.279. Присоединение галогеноводородов к винилогам карбонильных сое-
соединений
Продукт реакции
З-Хлорпропионитрил
З-Бромпропионитрил
Этиловый эфир
3-хлорпропионовой
кислоты
Метиловый эфир
3-бро.мпропионовой
кислоты
Этиловый эфир
3-бромпропионовой
кислоты
Метиловый эфир
3-бромизомасляной
кислоты
Исходные вещества
Хлороводород,
акрилонитрил
Бромоводород,
акрилонитрил
Хлороводород,
этилакрилат
Бромоводород,
метилакрилат
Бромоводород,
этилакрилат
Бромоводород,
метил метакрилат
Т. кип., °С
(мм рт. ст.)
87 B0)
92 B5)
80 B9)
65A8)
78A9)
76 B2)
1,4360
1,4789
(при 25 °С)
1,4254
1,4542
1,4569
(при 18 °С)
1,4551
Выход, %
95
90
80
80
90
80
ния газа устанавливают таким образом, чтобы температура в колбе не поднималась выше
-5 °С. После поглощения теоретического количества газа (контроль по массе сосуда!) закры-
закрытую колбу оставляют на ночь при 0 °С. Затем реакционную смесь последовательно промыва-
промывают водой, 10%-ным раствором гидрокарбоната натрия, еще раз водой, сушат сульфатом маг-
магния и перегоняют.
7.4.1.3. Присоединение С-Н-кислотных соединений
к винилогам карбонильных соединений (присоединение по Михаэлю)
Присоединение С—Н-кислотных соединений к винилогам карбонильных сое-
соединений в присутствии основных катализаторов имеет большое преппративное
значение. Особенно гладко эти реакции протекают с р-дикарбонильными сое-
соединениями (почему?), но хорошо идут также с кетонами и нитрилами типа бен-
зилцианида. Часто их называют присоединением по Михаэлю. (В соответствии
со схемой [Г.7.272] составьте уравнение реакции присоединения малонового
эфира к этиловому эфиру акриловой кислоты в присутствии алкоголята натрия!)
При наличии в С—Н-кислотном компоненте нескольких реакционноспо-
собных водородных атомов кроме моноаддукта могут получаться более сложные
продукты присоединения. Моноаддукт обычно удается получить с хорошим вы-
выходом, если брать избыток С—Н-кислотного компонента или проводить реак-
реакцию в разбавленных растворах.
Значительный интерес представляет присоединение альдегидов к винилогам
карбонильных соединений. Альдегидный водородный атом не имеет С—Н-кис-
лотного характера, а углеродный атом карбонильной группы представляет
электрофильный центр; следовательно, для осуществления реакции полярность
последнего необходимо изменить. Такое изменение происходит при реакциях
ароматических альдегидов с цианид-ионом (см. разд. Г,7.2.1.6, ацилоиновая
конденсация).
202
Г Препаративная часть (продолжение)
NC 101®
CN
Ph о ОН
О
[Г.7.280]
О
Промежуточно образующийся карбениевый ион присоединяется к винилогу
карбонильного соединения; при этом регенерируется катализатор (CN~) и с
высоким выходом образуются у-дикарбонильные соединения.
В случае енолизуемых алифатических альцегидов сильноосновный цианид катализирует
альдольную конденсацию. Вместо цианид-иона часто выгодно использовать гетероциклические
цвиттер-ионы, получаемые из гетероциклических четвертичных солей, в особенности 1,3-тиазо-
лов; такой процесс был подробно рассмотрен на схеме [Г.7.151] (см. литературу, цитированную
после общей методики на с. 203).
Вместо винилогов карбонильных соединений часто при присоединении по Михаэлю ис-
используют соответствующие основания Манниха (разд. Г,7.2.1.5). Процесс протекает по меха-
механизму элиминирования - присоединения через винилоги карбонильных соединений или их
аналогов. Так, из грамина (см. схему [Г.7.143]) и бензальдегида описанным выше путем с
CN~ в качестве катализатора получают 3-(бензоилметил)индол:
О
[Г.7.281]
Проведение реакции по Михаэлю часто осложняется тем ,что кроме присоединения од-
одновременно протекает альдольная или кляйзеновская конденсация. Так осуществляется, нап-
например, взаимодействие мезитилоксида с малоновым эфиром в присутствии эквимолярного
количества алкоголята натрия. Эта реакция важна как путь к получению дигидрорезорцинов:
Me
Me Me
нЛ
COOR
COOR
-ROH
H COOR
[Г.7.282]
H COOR
С другой стороны, реакция присоединения по Михаэлю часто следует за кротоновой кон-
конденсацией. Так, а,р-ненасыщенные соединения, образовавшиеся из р-дикарбонильных сое-
соединений и альдегидов в условиях реакции Кнёвенагеля (см. разд. Г.7.2.1.4), часто реагируют с
другой молекулой р-дикарбонильного соединения по типу реакции Михаэля, давая алкили-
ден-бис-р-дикарбонильные соединения:
Н
СН3
COOR'
СН3
COOR'
Н2О
реакция
Кнёвенагеля
[Г.7.283а]
реакция
Михаэля
н' у сн3 н3с
COOR' COOR' R'OOC COOR'
Тенденция к превращениям такого типа особенно сильно выражена у формальдегида.
[Г.7.2836]
7. Реакции карбонильных соединений 203
Побочных реакций, вызываемых основными катализаторами, при взаимодействии с аль-
альдегидами и кетонами можно избежать, если вместо С—Н-кислотного соединения взять соот-
соответствующий енамин (см. разд. Г,7.1.1).
В то время как из енаминов альдегидов обычно образуются устойчивые, перегоняющиеся
без разложения циклобутановые производные, циклобутаны, образующиеся из енаминов ке-
тонов и электрофильных олефинов, при повышенной температуре вновь расщепляются на ис-
исходные вещества, которые затем дают термодинамически выгодные ациклические соединения
(термодинамический контроль); см. схему [Г.7.284]. Образованию соединений с открытой
цепью благоприятствуют полярные апротонные растворители (см. разд. В, 3.3). (Обсудите при-
причины подобного влияния растворителя!)
CN
[Г.7.284]
Общая методика присоединения по Михаэлю (табл. Г.7.285)
Внимание! Многие а,C-ненасыщенные карбонильные соединения ядовиты и
обладают слезоточивым действием! Работать под тягой!
В трехгорлую колбу емкостью 1 л, снабженную мешалкой, термометром, капельной во-
воронкой и обратным холодильником, помещают 1 моль С—Н-кислотного соединения. Добав-
Добавляют раствор катализатора, полученный растворением 0,5 г натрия или 1 г гидроксида калия в
10 мл этанола, после чего по каплям при хорошем перемешивании прибавляют 1,1 моль све-
жеперегнанного а,р-ненасыщенного карбонильного соединения с такой скоростью, чтобы
температура оставалась в пределах 30-40 °С. Для получения ди-, три- и тетрапроизводных бе-
берут соответственно 2, 3 или 4 моль а,р-ненасыщенного соединения на 1 моль С—Н-кислотно-
С—Н-кислотного компонента. Если в С—Н-кислотном компоненте имеется более одного кислотного атома
водорода, а желательно получение моноаддукта, то берут 2 моль С-Н-кислотного компонен-
компонента на 1 моль а,р-ненасыщенного карбонильного соединения. Обязательно нужно следить за
тем, чтобы реакция началась уже после добавления небольшого количества винилога карбо-
карбонильного соединения (это видно по подъему температуры). В противном случае следует увели-
увеличить количество катализатора. После добавления всего количества карбонильного соединения
смесь без перемешивания оставляют на ночь. При выделении твердых продуктов реакции их
отсасывают, промывают водой и перекристаллизовывают. При отсутствии осадка реакцион-
реакционную смесь разбавляют примерно равным количеством дихлорметана или эфира, нейтрализу-
нейтрализуют ледяной уксусной кислотой и промывают водой. После сушки сульфатом магния смесь
перегоняют. При использовании смешивающегося с водой растворителя (табл. Г.7.285) его
отгоняют перед проведением указанных операций.
Дшитрил янтарной кислоты из синильной кислоты и акрилонитрила:
ТерентьевА. П., Хост А. Н. ЖОХ, 1951, 21, 1867.
3-(Индолил-3)пропионитрил из индола и акрилонитрила: Терентьев А. П.,
КостА. Н., Смит В. А. ЖОХ, 1956, 26, 557.
3-B-Оксоциклогексил)пропионитрил из циклогексанона и акрилонитрила:
Bruson H. A., Riener Т. W. J. Am. Chem. Soc, 1942, 64, 2850.
Таблица Г.7.285. Присоединение по Михаэлю
Продукт реакции
БисB-цианэтил)малоновый эфир
Ацетил аминоB-цианэтил)малоновый
эфир2
2-B-Цианэтил)ацетоуксусный эфир
2,2,5,5-ТетракисB-цианэтил)-
циклопентанон
2-B-Цианэтил)-2-этоксикарбонилцикло-
гексанон
а,а-БисB-цианэтил)бензилцианид
М-B-Цианэтил)-Е-капролактамд
5-Ацетил-5-этоксикарбонилнонандион-2,8
З-Изопропил-3-этоксикарбонил-
гептандион-2,6
а-(у-Оксобутил)бензил цианид
9-Гидроксидекалон-2
2-(у-Оксобутил)-2-этоксикарбонилцикло-
гексанон
1,2-Дифенил-1 -цианопентанон-4
Исходные вещества
Малоновый эфир, акрилонитрил
Ацетиламиномалоновый эфир6,
акрилонитрил
Ацетоуксусный эфир, акрилонитрил
Циклопентанон", акрилоншрил
2-Эгоксикарбонилциклогексанон,
акрилонитрил
Бензилцианидг, акрилонитрил
е-Капролактам, акрилонитрил
Ацетоуксусный эфир,
метилвинилкетон
Изопропилацетоуксусный эфир,
метилвинилкетон
Бензилцианид, метилвинилкетон
Циклогексанон, метилвинилкетон
2-Этоксикарбонилциклогексанон,
метилвинилкетон
Бензальацетонж, бензилцианид
Физические константы
Т. пл. 62 °С (этанол)
Т. пл. 94 "С (этанол)
Т. кип. 12ГС B мм рт. ст.); nD2i 1,4446
Т. пл. 176 "С; (ДМФА)
Т. кип. 142 °С @,3 мм рт. ст.); «D251,4700
Т. пл. 70 °С (этанол)
Т. кип. 130 °С A мм рт. ст.)
Т. кип. 160 °С A мм рт. ст.)
Т. кип. 130 °С A мм рт. ст.); ио181,4825
Т. кип. 155 °С B мм рт. ст.)
Т. пл. 148 "С (метилциклогексан)е
Т. кип. 140 °С @,5 мм рт. ст.);
«D251,4730
Т. кип. 184 °С A мм рт. ст.)
Выход, %
90
70
60
95
85
80
55
80
65
60
30
70
80
4-Ацетиламино-4,4-бис(этоксикарбонил)
масляный альдегид3
З-Фенил-2-этоксикарбонилциклогексен-
5-oh-I
Диэтиловый эфир 2-ацетилглутаровой
кислоты
Диэтиловый эфир 2-н-бутил-2-этокси-
карбонилглутаровой кислоты
Ацетиламиномалоновый эфир6,
акролеин
Ацетоуксусный эфир, коричный
альдегид
Ацетоуксусный эфир, этилакрилат
и-Бутилмалоновый эфир, этилакрилат
Т. кип. 162 'С E мм рт. ст.); ло20 1,5635
Т. кип. 135 °С D мм рт. ст.); «D25 1,4398
Т. кип. 112 °С @,1 мм рт. ст.); nD20 1,4416
85
50
80
65
Chlm^^l-Tim П0ЛУЧеНИЯ глУгаминовой ™слоты омылением и орнитина гидрированием и последующим омьиением: Albertson N. К, Archer S. J. Am.
6 В 500 мл этанола.
в В 200 мл толуола.
г В 250 мл этанола.
пя^гТГ^Т РаСПЛаШЫЮТ ПР170 °С; Р««иионную смесь нафевают при 80 'С в течение 3 ч. Получают исходный продуктддя синтеза основания Одигера (см
разд. 1,3.1.1.2), который может без очистки перерабатываться далее. апч^ч^м-
е Возгоняется при 115 °С @,4 мм рт. ст.).
* В 100 мл эфира.
3 Исходный продуктддя получения триптофана: Мое О. A, Warner D. Т. J. Am. Chem. Soc., 1948,70,2763,2765. Без очистки перерабатывается далее.
206 Г Препаративная часть (продолжение)
1-Фенилпентандион-1,4 из бензальдегида и метилвинилкетона: Stetter H.,
Schreckenberg M. Chem. Вег., 1974,107, 2453; Stetter H. Angew. Chem., 1976, 88,695.
4-Оксо-4-(пиридил-3)бутиронитрил из пиридинкарбальдегида-3 и акрило-
нитрила: Stetter H., Kuhlmann H., Lorenz G. Org. Syntheses, 1980, 59, 53.
Ундекандион-2,5 из метилвинилкетона и гептаналя в присутствии хлорида
3-бензил-5-B-гидроксиэтил)-4-метил-1,3-тиазолия: StetterH., Kuhlman H., Haase W.,
Org. Synth., 1987, 65, 26.
Получение метиленбис(дигидрорезорцина). [Stetter H. Angew. Chem., 1955, 67, 769.] Растворяют
0,15 моль дигидрорезорцина в 300 мл воды, прибавляют 0,12 моль водного раствора формаль-
формальдегида и осторожно нагревают до легкого помутнения. После этого оставляют на ночь при
комнатной температуре, осадок отфильтровывают и промывают водой. Выход количествен-
количественный;^ пл. 132 °С.
Аналогичная реакция с 5,5-диметилдигидрорезорцином (димедоном)
используется в качественном и количественном анализе для обнаружения и
количественного определения формальдегида и других альдегидов.
Димедон можно синтезировать кетонным расщеплением соединения, полу-
полученного по схеме [Г.7.282]: Shriner R. L, Todd H. R. Org. Syntheses, Coll. Vol. II,
1943, p. 200.
Присоединение по Михаэлю имеет исключительно большое препаративное
значение, так как этим путем удается удлинять в одну стадию углеродную цепь
на несколько углеродных атомов.
Ниже приводится интересный пример:
НзС
[Г.7.286]
Присоединение метилвинилкетона к 2-метилциклогексанону-1 с последующей циклиза-
циклизацией путем кротоновой конденсации приводит к окталону (соединение I в схеме [Г.7.286]) с
ангулярной метильной группой. Очевидно, что это соединение содержит кольца А и В стеро-
стероидов (см. соединение II в схеме [Г.7.286]). Благодаря большой стереоспецифичности при-
присоединения по Михаэлю оно имеет большое значение при синтезе стероидных соединений.
Все известные синтезы веществ этого класса включают стадию присоединения по Михаэлю.
Взаимодействием ацетоуксусного эфира, аммиака (или первичного амина) и альдегида по
Ганчу получают соединения ряда дигидропиридина:
R H R
Н /-L XOOR1 R'OOC. J^L .COOR1
+ "^Y _ Д jfH 1Г7'287а1
H3C^NH2 СГ^СНз Н3С NH2 СГ^СНз
Н R R
R'OOC^3^.COOR' R'OOC^/L^COOR'
__ у Y ^^^l?il^. T jf [Г.7.2876]
2 H3C N CH3 H3C NT^CHa
H
7. Реакции карбонильных соединений
207
При этом образуются, с одной стороны, р-амино- или р-алкиламинокротоновый эфир
(схема [Г.7.12]), а с другой стороны, в результате реакции Кнёвенагеля — алкилиден- или
аралкилиденацетоуксусный эфир (схема [Г.7.283а]). Далее оба соединения реагируют друг с
другом по типу присоединения по Михаэлю (схема [Г.7.287а]) и дают после циклизации дигид-
ропиридинкарбоновый эфир, который можно легко дегидрировать (например, под действием
оксидов азота) до эфира пиридинкарбоновой кислоты (схема [Г.7.2876]), если в реакции
использовали аммиак.
Упомянутые в качестве промежуточных продуктов соединения можно получить отдельно
и лишь затем ввести их в реакцию для синтеза конечного продукта. Этот принцип замыкания
цикла весьма успешно используют при получении производных пиридина.
Синтез по Ганчу находит промышленное применение, например при получении диэтило-
вого эфира 2,6-диметил-4-(о-нитрофенил)-1,4-дигидропиридиндикарбоновой-3,5 кислоты
(нифедипина), важного препарата для лечения сердечно-сосудистых заболеваний.
Присоединение по Михаэлю енаминов типа [Г.7.287а] к я-бензохинону
(реакция Неницеску) дает интересные в физиологическом отношении 5-гидрок-
сииндолы. Так, из N-монозамещенных эфиров аминофумаровой (или амино-
малеиновой) кислоты и и-бензохинона в присутствии трифторида бора, как
правило, с хорошим выходом получают диэтиловые эфиры 5-гидроксииндолди-
карбоновых-2,3 кислот:
Н COOEt
HN COOEt
i
R
(BF3)
COOEt [Г.7.288]
Ф Общая методика получения диметиловых эфиров N-замещенных 5-гидроксииндолдикар-
боновых-2,3 кислот (табл. Г.7.289). К раствору 0,02 моль N-замещенного диметилово-
го эфира аминофумаровой кислоты в 50 мл абсолютного эфира прибавляют по каплям
при хорошем перемешивании смесь 0,02 моль л-бензохинона и 0,02 моль эфирата трифтори-
трифторида бора в 100 мл абсолютного эфира. Смесь оставляют на ночь. Выпавший осадок отфильтро-
отфильтровывают, промывают водой, растворяют в метаноле, после концентрирования раствора вновь
отфильтровывают осадок и перекристаллизовывают. Если продукт реакции не кристаллизуется,
Таблица Г.7.289. Получение 5-гидроксииндолов из л-бензохинона присоединением
по Михаэлю
Продукт реакции
5-Гидрокси-2,3-диметокси-
карбонил-1 -метилиндол
1 -тл/>«п-Бутил-4-гидрокси-
2,3-диметоксикарбонилиндол
1 -Бензил-5-гидрокси-
2,3-диметоксикарбонилиндол
5-Гидрокси-2,3-диметокси-
карбонил-1 -фенилиндол
Исходное вещество
Диметиловый эфир
N-метиламинофумаровой
кислоты
Диметиловый эфир
М-т/>ет-бутиламинофумаро-
вой кислоты
Диметиловый эфир
N-бензиламинофумаровой
кислоты
Диметиловый эфир
N-фениламинофумаровой
кислоты
Т. пл.,°С
158 (CH2Cl2-CCl4,
1:1)
246 (толуол)
159 (метанол)
208 (дихлорэтан)
Выход, %
80
20
90
60
208 Г Препаративная часть (продолжение)
то эфирный раствор промывают водой, дважды экстрагируют водный слой эфиром, объеди-
объединенные эфирные вытяжки концентируют и фильтруют через сухой активный оксид алюми-
алюминия. Продукт элюируют диоксаном, отгоняют растворитель в вакууме на водяной бане и
остаток перекристаллизовывают.
3-Алкоксикарбонил-5-гидрокси-2-метилиндолы из эфиров C-аминокротоно-
вой кислоты и я-бензохинона: Patrick J. В., Sounders E. К. Tetrahedron Letters,
1979, 42, 4009.
7.4.1.4. Присоединение амидов кислот к винилогам карбонильных соединений
Незамещенные или монозамещенные амиды кислот также могут присоеди-
присоединяться к а,р-ненасыщенным карбонильным соединениям и нитрилам. Эти
реакции осуществляются только в присутствии основных катализаторов. Осо-
Особенно легко в эту реакцию вступают имиды кислот, например фталимид и
сукцинимид, а также амиды сульфоновых кислот, которые под действием
катализатора очень легко переходят в основания, способные к реакциям при-
присоединения (см. схему [Г.8.49]).
Образующиеся продукты присоединения представляют интерес, так как при
омылении амидной группы получаются C-аминоэтильные соединения. Прямым
присоединением аммиака или маноалкиламинов эти соединения можно полу-
получить лишь с трудом. Примером реакции присоединения амидов может служить
удобный лабораторный синтез р-аланина.
^П) Получение р-аланина
^—^ Получение $-фталимидопропионитрила цианэтилированием фталимида. В трехгорлой
колбе емкостью 1 л, снабженной мешалкой, обратным холодильником и термометром,
нагревают до 60 °С на водяной бане смесь 2 моль фталимида, 130 мл диметилформамида и
2,5 моль акрилонитрила. Затем при перемешивании прибавляют за один прием 4 мл 50%-ного
раствора гидроксида калия, что обычно достаточно для начала реакции. Если в течение нес-
нескольких минут температура заметно не повышается, то добавляют еще некоторое количество
раствора гидроксида калия. Необходимое количество гидроксида калия зависит от качества
фталимида, который по возможности не должен содержать примеси фталаминовой кислоты.
После начала реакции температура быстро повышается до ~ 120 °С. Прозрачный, слегка жел-
желтоватый раствор выдерживают еще 20—30 мин при температуре - 120 °С, затем немного охлаж-
охлаждают и выливают в ~ 2 л холодной воды. Это необходимо сделать до начала кристаллизации ре-
реакционной массы. Бесцветные кристаллы отфильтровывают и промывают холодной водой.
Т. пл. 154 "С (этанол); выход 95%.
Омыление $-фталимидопропионитрила до р-аланина. В круглодонной колбе емкостью 3 л в
течение 5 ч кипятят с обратным холодильником смесь 2 моль C-фталимидопропионитрила
(неочищенный продукт предыдущей реакции) и 900 мл 20%-ной соляной кислоты. Образовав-
Образовавшаяся фталевая кислота примерно через 4 ч внезапно выпадает в осадок, вызывая сильные
толчки при кипении (колбу необходимо хорошо закрепить). Горячую реакционную смесь вы-
выливают в стакан и охлаждают при энергичном перемешивании, затем отсасывают выпавшую
фталевую кислоту и тщательно промывают водой. Объединенные фильтраты упаривают досу-
досуха на водяной бане в вакууме водоструйного насоса, после чего сушат еще 1 ч в тех же услови-
условиях. Горячий остаток после сушки смешивают с 150 мл метанола, тщательно перемешивают и
отсасывают. Осадок на фильтре еще дважды обрабатывают таким же образом двумя порциями
метанола по 100 мл. Объединенные экстракты после охлаждения фильтруют и добавляют три-
бутиламин или диэтиламин до слабощелочной реакции. При достижении изоэлектрической
точки аминокислота осаждается. Кристаллы отфильтровывают и промывают метанолом.
Т. пл. 200 °С; выход 80% (в расчете на фталимид).
7. Реакции карбонильных соединений 209
7.4.1.5. Реакции замещения винилогов карбонильных соединений
Группы C1>OCH3>SCH3>>NR21, находящиеся в В-положении винилогов
карбонильных соединений, замещаются основаниями или С—Н-кислотны-
ми соединениями по механизму присоединения—замещения. Так, из р-хлор-
винилальдегидов или В-хлорвинилкетонов (почему эти соединения можно
рассматривать как винилоги ацилхлоридов?) можно получить р-аминови-
нилкарбонильные соединения:
II fi M |_| _i_ Г*11 ~f~* LJ — Г LJ * f1 LJ ГЛI LJ Г M LJ PU Ли Г"Ч_1 Г\ I
пз^ —INn2 + ^1— L-n —L*rl~Un —(Jl *~ n3Li-Nn~Un~tyn-L/r"i~UI
H rCI [Г.7.290]
_I_IC|- H3C —NH—CH = CH —CH=O
При подобных реакциях замещения одновременно реагирует и карбониль-
карбонильная группа; это обстоятельство используется для осуществления многостадий-
многостадийной циклоконденсации. Так, замещенные тиофены получают взаимодействием
Р-хлоркоричного альдегида с эфирами тиогликолевой кислоты:
[Г.7.291]
HS""^COOEt
(Составьте схему синтеза пиразола из р-хлоракролеина и гидразина!)
Другим примером является реакция р-хлорвинилкетонов с енаминами
(винилогами электронодонорных соединений, см. разд. Г,7.4.2), приводящая к
образованию пирилиевых солей:
R3 R3
R \ ^-Н I ..я R ^^ ^s^ R \^^\
К® Л [Г.7.292]
R4 R^O^R4
Аналогично реагируют винилоги гетероаналогов карбонильных соединений,
например иммониевые соединения R—CR=CR— CR=NR2 и нитрилы с соответству-
соответствующими уходящими группами в р-положении. Из нитрилов, таким образом, в ре-
результате внутримолекулярного присоединения образуются гетероароматические
амины; например, из этоксиметиленциануксусного эфира (см. табл. Г.7.172) и гид-
разингидрата получают эфир 3E)-аминопиразолкарбоновой-4 кислоты
COOR H COOR COOR COOR
H2N-NH2 2 N H
1 Последовательность расположения групп в ряду соответствует уменьшению склонности
к замещению.
210
Г Препаративная часть (продолжение)
Таблица Г.7.294. Получение производных 3- или 5-аминопиразолкарбоновой-4 кислоты
Продукт реакции
Этиловый эфир 3- или
5-аминопиразолкарбоновой-
4 кислоты
3- или 5-Аминопиразол-
карбонитрил-4
Этиловый эфир 5-амино-
1 -фенилпиразолкарбоновой-
4 кислоты8
5-Амино-1 -фенилпиразол-
карбонитрил-4
Метиловый эфир 5-амино-
З-метилтио-1-фенилпиразол-
карбоновой-4 кислоты6-"
З-Амино-5-метилтиопира-
золкарбонитрил-4
Исходные вещества
Этиловый эфир этоксимети-
ленциануксусной кислоты,
гидразингидрат
Нитрил этоксиметиленмало-
новой кислоты, гидразин-
гидразингидрат
Этиловый эфир этоксимети-
ленциануксусной кислоты,
фенилгидразин
Нитрил этоксиметиленмало-
новой кислоты, фенилгидра-
фенилгидразин
Метиловый эфир бис-
(метилтио)метиленцианук-
сусной кислоты, фенилгидра-
фенилгидразин
Нитрил бис(метилтио)-
метиленмалоновой кислоты,
гидразингидрат
Т. шт., °С
103 (вода)
175 (вода)
100 (этилацетат)
138 (вода)
114 (петролейный
эфир)
151 (вода)
Выход, %
75
85
70
85
90
90
Образовавшееся после упаривания масло растирают с небольшим количеством толуола.
В качестве растворителя используют метанол.
Выделяется метантиол. Работать под тягой!
Последний при конденсации с формамидом дает важный фармацевтический препарат
4Н-4,5-дигидропиразоло[3,4-с!]пиримидон-4 (аллопуринол, бурмадон), используемый при
лечении артритов.
Ф Общая методика получения производных 3- или 5-аминопиразолкарбоновой-4 кислоты
(табл. Г.7.294). К смеси 20 ммоль нитрила и 20 мл этанола в колбе емкостью 100 мл до-
добавляют порциями при взбалтывании гидразин C0 ммоль 80%-ного гидразингидрата
или 20 ммоль фенилгидразина в 5 мл этанола). Нагревают смесь 1 ч на водяной бане с об-
обратным холодильником, охлаждают, отбирают пробу, разбавляют ее водой. Если при растира-
растирании выпадает продукт реакции, то всю реакционную смесь при перемешивании переносят в
двойной объем воды и через 24 ч отфильтровывают. Если кристаллический осадок не образует-
образуется, то раствор упаривают досуха на водяной бане, растирают остаток с небольшим количеством
воды, продукт отфильтровывают и перекристаллизовывают.
7.4.2. РЕАКЦИИ ВИНИЛОГОВ ЭЛЕКТРОНОДОНОРНЫХ СОЕДИНЕНИЙ
(ЕНОЛЯТОВ, ЕНОЛОВ, ЭФИРОВ ЕНОЛОВ, ЕНАМИНОВ)
Винилоги электронодонорных соединений являются производными енолов
карбонильных соединений:
ОН
Н-С-с
с=с
/ \
с
[Г.7.295]
Их реакционная способность по отношению к большинству электрофиль-
ных агентов соответствует ряду [Г.7.296]:
\ ери
с=с
/ ° \
\ ClOR
= с=с
/ \
\ cJPSiR3
= с=с <
/ \
ч CINR2
: С=С <
/ ° \
\ cjc
: С=С
/ w \
7. Реакции карбонильных соединений 211
[Г.7.296]
Концевой углеродный атом винилога электронодонорного соединения ре-
реагирует так же, как сама электроотрицательная донорная группа, т. е. элект-
рофильные реагенты атакуют преимущественно этот углеродный атом. На
схеме [Г.7.297] это показано на примере важнейших представителей таких
соединений — енаминов (IV в схеме [Г.7.296]):
INR2
[Г.7.297]
Карбонильные соединения могут реагировать с электрофильными реагента-
реагентами с образованием енолов в качестве промежуточных соединений.
•Кето-енольное равновесие [Г.7.295] у простых альдегидов и кетонов смещено в сторону
карбонильного соединения и содержание енольной формы крайне мало (см. разд. Г,7.2.1.8).
Эфиры р-оксокислот и C-дикетоны енолизованы значительно сильнее. Протоны катализиру-
катализируют установление кето-енольного равновесия.
Электрофильные реагенты (Е+) по аналогии со схемой [Г.7.297] атакуют
углеродный атом, находящийся в ос-положении к карбонильной группе:
[Г.7.298]
В результате реакции с электрофильным реагентом енолы постоянно выводятся из равно-
равновесия и процесс может успешно развиваться, даже если равновесие сильно смещено в сторону
карбонильного соединения, см. например, альдольную конденсацию, катализируемую кисло-
кислотами [Г.7.104], галогенирование [Г.7.308], а также взаимодействие С-Н-кислотных соедине-
соединений с азотной кислотой (разд. Г,8.2.3) и с солями диазония (разд. Г,8.3.3 и схема [Г.9.45]).
Значение эфиров енолов [Г.7.296, И] для препаративной химии сравнительно
невелико. Выше упоминались кислотная гидратация и присоединение спиртов,
например, к важнейшему циклическому енолэфиру дигидропирану (см. схему
[Г.9.21]), а также полимеризация.
О-Триметилсшшленолэфиры (Г.7.296, III), которые получают кислотноката-
лизируемым силилированием карбонильных соединений, также могут
взаимодействовать с электрофильными реагентами в р-положении. Такие
силилированные енолы карбонильных соединений можно алкилировать в
ос-положении с помощью SNl-aKTHBHbix алкилгалогенидов в присутствии кис-
кислот Льюиса, например TiCl4. Таким путем можно вводить третичные алкиль-
ные группы в альдегиды, кетоны и сложные эфиры, что не удается с помощью
оснований (почему?).
. п 1. Основание n
I / yj
и р_р ^. |у|сзо|у>1 р_р nv^i у I iyyi4/^ р_р__р ГГ7 2991
I \ / \ -MeaSiCI I \
212 Г Препаративная часть (продолжение)
О-Триметилсилиловые эфиры енолов реагируют с альдегидами и кетонами в
присутствии таких кислот Льюиса, как TiCl4, с образованием аддуктов, гидролиз
которых приводит к альдолям (вариант альдольной конденсации по Мукайаме).
/? \ ,0SiMe3 (new Мез&? I <? +н20(н@, ?Hl Р
R-C + С=С — R-C-C-C J I'- R-C-C-C
\ / \ I I \ "MeaSiOH , | \
Синтез З-гидрокси-3-метил- 1-фенилбутанона-1 из ацетона и О-триметил-
силилового эфира енола ацетофенона: Mukaiyama Т., Narasaka К., Org. Synth.,
1986, 65, 6.
Наивысшей нуклеофильностью в ряду [Г.7.296] обладают еноляты. Как уже
много раз упоминалось, они образуются при депротонировании карбонильных
соединений основаниями (см. [Г.7.100]). С различными нуклеофилами они
реагируют in situ или же в форме натриевых, литиевых или магниевых соедине-
соединений (см. разд. Г.7.2 и последующий текст).
7.4.2.1. Алкилирование карбонильных соединений
Алкилирование карбонильных соединений с активным атомом водорода в а-по-
ложении и других С-Н-кислотных соединений осуществляется при взаимодей-
взаимодействии их анионов (енолятов) с галогеноуглеводородами, сульфатами или тозила-
тами. При этом происходит нуклеофильное замещение атома галогена, причем
анион С—Н-кислотного соединения выполняет роль нуклеофильного реагента
(см. табл. Г.2.4). Особенно легко протекает алкилирование р-дикарбонильных
соединений, которые при взаимодействии с алкоголятами полностью превра-
превращаются в еноляты (см. табл. Г.7.99), например:
R-X +
- ff e
НзС-С-СН-COOR'
IOIQ
H3C-C=CH-COOR'
-X
О
НзС-С-СН-COOR1
I
R [Г.7.301]
O-R
H3C-C=CH-COOR'
Амбидентные анионы (см. разд. Г,2.3) могут претерпевать и О-алкилирование, которое,
однако, протекает в незначительной степени, если соблюдать условия методики к табл.
Г.7.302. О-Алкилированию способствуют апротонные полярные растворители, низкая конце-
концентрация амбидентного аниона, объемистые противоионы, а также малая нуклеофильная
активность алкилирующего агента и жесткость уходящей группы. По этим причинам в случае
вторичных алкилгалогенидов тенденция к О-алкилированию возрастает от иодида к хлориду и
в ряду растворителей: этанол ~ ацетон<ацетонитрил<диметилсульфоксид ~ диметилформа-
мид. Катион оказывает меньшее влияние, хотя в целом натриевые соли способствуют С-алки-
лированию. Иногда продукты О-алкилирования легко перегруппировываются, перенося
алкильную группу к атому углерода карбаниона.
О-Алкилирование подавляется в нейтральной или слабокислой среде. В этом случае для
алкилирования используют уже не алкилгалогениды (почему?), а диазоалканы, ортоэфиры и
соли алкоксония.
Реакционная способность алкилирующих агентов убывает в следующем ря-
ряду параллельно с уменьшением подвижности атома галогена (или кислотного
7. Реакции карбонильных соединений 213
остатка): аллилгалогениды > бензилгалогениды > а-галогенокетоны > диалкил-
сульфаты > алкил-и-толуолсульфонаты > алкилгалогениды. Активность алкил-
галогенидов падает по мере роста объема алкильного остатка, т. е. от метилгало-
генидов к т/ге/я-бутилгалогенидам. Обычно р-дикарбонильное соединение
переводят в натриевое производное действием алкоголята натрия. В результате
побочных реакций при этом из алкилирующего агента образуются простые эфи-
ры и олефины (напишите схемы реакций!). Эти реакции особенно легко проте-
протекают с разветвленными алкилгалогенидами (см. разд. Г,2 и Г,3), что приводит к
резкому снижению выхода. В случае язре/я-бутилгалогенидов вообще не удается
получить приемлемых выходов.
При моноалкилировании р-дикарбонильных соединений часто образуются
диалкилпроизводные, даже если взяты эквимолярные количества реагентов; в
этом случае соответствующая доля карбонильного соединения не вступает в
реакцию. Разделение реакционной смеси (исходное вещество, моно- и диалкил-
диалкилпроизводные) особенно затруднено при работе с низшими продуктами алкили-
рования. В этом случае для выделения чистого моноалкилпроизводного
приходится избирать обратный путь; см., например, получение моноалкилмало-
новых эфиров из эфира 2-оксоянтарной кислоты (схема [Г.7.167]) и алкилиро-
вание енаминов (схема [Г.7.319]).
Полное диалкилирование р-дикарбонильных соединений затрудняется
тем, что кислотность моноалкильных производных меньше, чем незамещен-
незамещенного исходного продукта, а также легкостью сольволитического расщепления
продуктов полного алкилирования в присутствии спиртового раствора алкого-
алкоголята натрия (см. сложноэфирное расщепление, схема [Г.7.175]). (Напишите
схему расщепления дизамещенных малоновых эфиров на эфиры угольной и
диалкилуксусной кислот!) В этих случаях рекомендуется придерживаться
«обратного» порядка проведения реакции, когда к р-дикарбонильному соеди-
соединению по каплям прибавляют алкоголят натрия так, чтобы последний никог-
никогда не был в избытке.
(ГП) Общая методика алкилирования р-дикарбонильных соединений (табл. Г.7.302)
В Внимание! Осторожно обращайтесь с металлическим натрием! (См. часть Е.)
В трехгорлой колбе емкостью 1 л, снабженной мешалкой, капельной воронкой и обратным
холодильником с хлоркальциевой трубкой, готовят раствор алкоголята натрия (см. часть Е) из
1 г-атома металлического натрия и 500 мл абсолютного спирта (во избежание переэтерификации
спирт для получения алкоголята должен быть тем же, что и остаток спирта в сложном эфире).
К еще горячему раствору алкоголята добавляют по каплям при перемешивании 1 моль
р-дикарбонильного соединения и затем 1,05 моль алкилирующего агента с такой скоростью,
чтобы реакционная смесь слегка кипела. Затем смесь при перемешивании нагревают до тех
пор, пока проба не покажет нейтральную реакцию B—16 ч). Основную часть растворителя от-
отгоняют в слабом вакууме при перемешивании (в противном случае кипение сопровождается
толчками из-за осадка соли). Отогнанный спирт совершенно безводен и его можно использо-
использовать для повторных аналогичных синтезов. После охлаждения к остатку добавляют такое коли-
количество ледяной воды, чтобы выпавшая соль растворилась, отделяют органическую фазу в дели-
делительной воронке, а водный слой дважды экстрагируют эфиром. Объединенные органические
вытяжки сушат сульфатом натрия, растворитель отгоняют, а остаток фракционируют на
30-сантиметровой колонке Вигре.
При получении диалкилпроизводных к смеси незамещенного р-дикарбонильного соеди-
соединения и 2 моль алкилирующего компонента при перемешивании прибавляют, защищая от
Таблица Г.7.302. Алкилирование р-дикарбонильных соединений
Продукт реакции
Диэтиловый эфир этилмалоновой кислотыа
Диэтиловый эфир диэтилмалоновой кислоты6
Диэтиловый эфир пропилмалоновой кислоты
Диэтиловый эфир изобугилмалоновой кислоты
Диэтиловый эфир н-бугилмалоновой кислоты
Диэтиловый эфир пентилмалоновой кислоты
Диэтиловый эфир гексилмалоновой кислоты
Диэтиловый эфир аллилмалоновой кислоты
Диэтиловый эфир циклопропандикарбоновой-1,1
кислоты"
Диэтиловый эфир циклобугандикарбоновой-1,1
кислоты"
Триэтиловый эфир этантрикарбоновой-1,1,2
кислоты
Этиловый эфир а-этилацетоуксусной кислотыа
Этиловый эфир а-изопропилацетоуксусной
кислотыа
Этиловый эфир а-бугилацетоуксусной кислоты
Этиловый эфир а-изобугилацетоуксусной
кислоты
Этиловый эфир а-аллилацегоуксусной кислоты
Этиловый эфир а-бензилацетоуксусной кислоты
2-Метилциклогександион-1,3
Исходные вещества
Малоновый эфир, этилбромид
Малоновый эфир, этилбромид
Малоновый эфир, пропилбромид
Малоновый эфир, изобутилбромид
Малоновый эфир, н-бутилбромид
Малоновый эфир, пентилбромид
Малоновый эфир, гексилбромид
Малоновый эфир, аллилбромид
Малоновый эфир, 1,2-дибромэтан
Малоновый эфир, 1,3-дибромпропан
или 1-бром-З-хлорпропан
Малоновый эфир, этиловый эфир
хлоруксусной кислоты
Ацетоуксусный эфир, этилбромид
Ацетоуксусный эфир, изопропилиодид
Ацетоуксусный эфир, бугилбромид
Ацетоуксусный эфир, изобутилбромид
Ацетоуксусный эфир, аллилбромид
Ацетоуксусный эфир, бензилхлорид
Дигидрорезорцин, диметилсульфат
Т. кип., °С (мм рт. ст.)
96A0)
100A2)
108A3)
113A2)
132A7)
135A4)
145A2)
102A0)
106B0)
104A2)
158A5)
80A0)
94A8)
116A6)
120A6)
102A2)
157A4)
120 (т. пл., этанол)
«о30
1,4163
1,4254
1,4197
1,4282
1,4225
1,4259
1,4281
1,4338
1,4335
1,4360
1,4315
1,4194
1,4234
1,4246
1,4242
1,4381
1,4998
Выход, %
85
75
85
80
80
80
80
85
45
45
70
75
75
65
80
85
80
70
а В чистом виде может быть получен только с высокоэффективной колонкой.
6 Применяют обратный порядок (добавляют алкоголят, см. методику).
в Применяют обратный порядок (добавляют алкоголят в течение 2 ч к нагретому до 70 °С раствору). После отгонки спирта перегоняют с водяным паром, дистил-
дистиллят экстрагируют эфиром, эфирные вытяжки обрабатывают, как описано выше. Для реакции берут 1,1 моль 1,2-дибромэтана или 1,05 моль 1,3-дибромпропана.
7. Реакции карбонильных соединений 215
влаги воздуха, 2 моль алкоголята натрия. Можно взять уже готовое моноалкилпроизводное,
смешать его с небольшим избытком алкилирующего компонента и добавлять по каплям раст-
раствор 1 моль алкоголята натрия. Этот метод позволяет получать несимметричные диалкилпроиз-
водные р-дикарбонильных соединений.
Получение этилового эфира а-изопропилацетоуксусной кислоты алкилирова-
нием ацетоуксусного эфира в присутствии трифторида бора: Adams J. Т.,
Levine R., Hauser С. R. Org. Syntheses, Coll. Ill, 1955, p. 405.
Получение этилового эфира $-этокси-[Е\-кротоновой кислоты из ацетоук-
ацетоуксусного эфира и ортомуравьиного эфира: Smissman E. E., Voldeng А. N. J. Org.
Chem., 1964,29,3164.
Монокетоны, сложные эфиры карбоновых кислот, карбоксилат-ионы и нит-
нитрилы можно алкилировать так же, как и C-дикарбонильные соединения. Одна-
Однако в силу их меньшей кислотности они лишь частично переводятся в анионы
действием алкоголята (см. табл. Г.7.99). В этом случае реакционная среда предс-
представляет собой смесь свободных карбонильных соединений и алкоголята, что
способствует протеканию побочных процессов альдольной конденсации и об-
образованию простых эфиров и олефинов. Для подавления таких процессов следу-
следует использовать сильные и по возможности объемистые основания. Для этого
наиболее пригодны диизопропиламид лития (англ. LDA), трет-бутилат калия,
амид натрия и гидроксид калия.
В случае несимметричных кетонов при действии таких реагентов могут обра-
образовываться различные продукты реакции. Так, алкилирование 2-метилциклогек-
санона метилиодидом, например в присутствии диизопропиламида лития, при-
приводит к 2,6-диметилциклогексанону, тогда как та же реакция, но в присутствии
mpem-бутилата калия, протекает с образованием 2,2-дизамещенного изомера.
Такая различная стереоселективность связана с тем, что сильно основный
LDA кинетически контролирует отрыв менее пространственно затрудненного
6-Н-протона метилциклогексанона, тогда как менее основный /я/?ет-бутилат
калия термодинамически контролирует отщепление более кислого 2-Н-про-
тона.
При действии сильно основных амидов возможно также и азотистые ана-
аналоги карбонильных соединений такие, как имины (основания Шиффа) и
гидразоны, превратить в соответствующие енамид-ионы с последующим их
алкилированием (см. [Г.7.119]). Эту реакцию используют прежде всего для
алкилирования а-положения альдегидов, так как прямому их алкилирова-
нию препятствуют процессы альдольной конденсации. Альдегид вначале ре-
реакцией с амином переводится в имин, который далее алкилируется, после че-
чего образовавшийся ос-алкилимин гидролизуется с отщеплением амина:
,.© §.
NR" Ll NR1' R\ NR"
з& ^г н |Г7304]
216 Г Препаративная часть (продолжение)
Образование металленамидов делает возможным проведение энантиоселек-
тивного а-алкилирования прохиральной метиленовой группы карбонильных
соединений. Для этого используют энантиомерные амины или гидразины, нап-
например, E")-1-амино-2-метоксиметилпирролидин {англ. SAMP) или его
(Л)-энантиомер (RAMP), образующие с карбонильным соединением хираль-
ный имин или соответственно гидразон. Прохиральные а-метиленовые группы
в результате реакции с хиральным вспомогательным реагентом становятся диа-
стереотопнымих и при взаимодействии с диизопропиламидом лития алкилиру-
ются диастереоселективно. После удаления из продукта алкилирования хираль-
ного вспомогательного реагента (разд. Г,7.3.2) образуется алкилированное кар-
карбонильное соединение с высоким энантиомерным обогащением.
N' СН2ОСН3
NH2
лрохиральныи
(диастереотоп)
прохиральныи ^^\ прохиральныи
(энантиотоп) Г 1 '(энантиотоп)
Синтез F)-(+)-4-метилгептанона-3 «гидразонным» методом SAMP/RAMP:
Enders D., Kippgardt H., Fey P., Org. Synth., 1987, 65, 183 (с обзором других возмож-
возможностей асимметрического синтеза с помощью метода SAMP/RAMPJ.
Алкилирование ряда слабых С-Н-кислот удается также в условиях межфаз-
межфазного катализа (см. разд. Г.2.4.2), что позволяет избежать металлирования, прове-
проведение которой иногда связано с затруднениями. Во всяком случае при получе-
получении моноалкилсоединений часто не удается избежать побочного образования
диалкилпроизводных.
©Общая методика алкилирования бензилциаиидов в двухфазной системе (табл. Г.7.306)
[Makosza M., Jonc'zyk A. Org. Syntheses, 1976, 55, 91]. В четырехгорлой колбе, снабжен-
снабженной обратным холодильником, мешалкой, капельной воронкой и термометром, наг-
нагревают на водяной бане до 35 °С раствор 30 г гидроксида натрия в 30 мл воды (в дальнейшем
водяная баня используется и для охлаждения). Добавляют 0,5 г хлорида бензилтриэтиламмо-
ния (получение см.: Souto-Bachillerwixp. Org. Syntheses, 1976, 55, 97) и 0,2 моль бензилциани-
да, а затем по каплям при хорошем перемешивании в течение 30 мин 0,2 моль алкилгалогени-
да. Смесь перемешивают еще 2 ч при 35 °С и 30 мин при 40 °С. При работе с незамещенным
бензилцианидом непрореагировавшее соединение переводят в малолетучий нитрил
1 Заместители сами по себе гетеротопны в том случае, если они различаются не структурой,
но топографически, т. е. если внутри молекулы имеют различное химическое окружение.
В ахиральных системах прохиральные группы являются энантиотопными, а в хиральных систе-
системах — диастереотопными. При замене энантиотопного заместителя на другой энантиотопный
радикал образуется энантиомер, а при замене диастереотопного заместителя образуется диасте-
реомер. Диастереотопные заместители можно физически и химически различить, тогда как
энантиотопные — только в хиральных условиях (с помощью хиральных реагентов, хиральных
растворителей или циркулярно поляризованного света; см. также разд. В,7.3.1 и В,7.3.2).
2 SAMP и RAMP получаются из природного E)-пролина или относительно легко доступ-
доступной (Я)-глутаминовой кислоты.
7. Реакции карбонильных соединений
217
Таблица Г.7.306. Алкилирование бензилцианидов в двухфазной системе
Продукт реакции
а-Фенилбутиронитрил
а-Фенилвалеронитрил
а-Фенилизовалеронитрил
1 -Фенилбуген-3-карбонитрил
а-Бензил-а-феншбутиро-
нитрил
Исходные вещества
Бензилцианид, этилбромид
Бензилцианид, пропил-
бромид
Бензилцианид, изопропил-
бромид
Бензилцианид, аллил-
бромид
а-Фенилбутиронитрил,
бензилхлорид
Т. кип., "С
(мм рт. ст.)
103 G)
126A2)
110G)
131A5)
190A2)
«о20
1,5086
1,5063
1,5059
1,5201
1,5593
Выход, %
75
75
5
60
65
а-фенилкоричной кислоты, который легче отделяется от продуктов реакции. Для этого до-
добавляют 2 г бензальдегида, еще 1 ч перемешивают при 25-30 °С, добавляют 25 мл толуола
(или метилендихлорида), отделяют органическую фазу, еще раз экстрагируют водный слой
толуолом (или метиленхлоридом) и промывают объединенную органическую фазу 25 мл во-
воды, разбавленной соляной кислотой и опять водой. После отгонки растворителя остаток
фракционируют в вакууме.
а- Моноалкилирование солей алифатических карбоновых кислот с щелочными
металлами с помощью бутиллития: CregerP. L. J. Am. Chem. Soc, 1970, 92, 1397.
В промышленности алкилированные малоновые эфиры используют прежде всего для по-
получения барбитуратов (снотворных и противоэпилептических препаратов) (см. разд. Г,7.1.4.2
и Г,7.2.1.8). Алкилирование бензилцианида применяется при синтезе петидина — соединения
с морфиноподобным действием, используемого в качестве анальгетика:
-2HCI
CH3
H2SO4, ЕЮН
[Г.7.307]
7.4.2.2. Галогенирование карбонильных соединений
Сравнительно активные С—Н-кислотные соединения, такие, как р-дикарбо-
нильные соединения, а также альдегиды и кетоны, легко галогенируются в ос-по-
ложении к карбонильной группе. Так, при каталитическом действии галогено-
водородов енолы электрофильно присоединяют атом галогена:
ICp-CII +
с=с
/ I \
R1 / R
-на
R1
H О
1 о
-с-с
ГА R
[Г.7.308]
Каталитическое действие могут оказывать и слабые основания (например, ацетат натрия).
В этом случае равновесие между карбонильным соединением и енольной формой заметно
218
Г Препаративная часть (продолжение)
смещено в сторону карбонильного соединения. Однако взаимодействие галогена с енолом в
соответствии со схемой [Г.7.308] сдвигает равновесие и обусловливает возможность заверше-
завершения галогенирования. (Напишите схему реакции!)
Карбоновые кислоты вообще не енолизуются, поэтому с помощью катализа-
катализатора (красного фосфора, трихлорида фосфора) сначала превращают кислоты в
ацилхлорид, который уже способен енолизоваться и в таком виде подвергаться
а-галогенированию:
ЗХ2 + 2Р 2РХ3
Р
3R-CH2-C
ОН
Р
ОН
R-CH2-C
R-CH=C
ОН
R-CH-c"
X X
х2
X
R-CH-c' + НХ
X X
[Г.7.309]
О
R-CH2-COOH
р
R-CH-C + R-CH2-C ит.д.
X
ОН
При хлорировании карбоновых кислот побочно образуются также р-галоге-
нопроизводные, механизм образования которых, по-видимому, включает ради-
радикальные реакции (см. разд. Г,1).
(?ТП) Общая методика получения сс-бромкарбоновых кислот (табл. Г.7.310)
(Все работы вести в хорошо действующем вытяжном шкафу! Об обращении с
бромом см. часть Е.
В трехгорлую колбу (см. рис. А.4,г; сливная трубка капельной воронки должна быть погру-
погружена в жидкость) помещают смесь 0,5 моль соответствующей карбоновой кислоты и 0,15 моль
красного фосфора и при перемешивании по каплям добавляют 0,5 моль безводного брома с та-
такой скоростью, чтобы в холодильнике не появлялись желтые пары брома. При этом температу-
Таблица Г.7.310. Получение а-бромкарбоновых кислот
Продукт реакции
Бромуксусная кислота
а-Бромпропионовая кислота
а-Броммасляная кислота
а-Бромизомасляная кислота
а-Бромвалериановая кислота
2-Бромгексановая кислота
2-Бром-4-метилвалериановая
кислота
Исходные вещества
Уксусная кислота
Пропионовая кислота
Масляная кислота
Изомасляная кислота
Валериановая кислота
Гексановая кислота
4-Метилвалериановая
кислота
Т. кип., "С (мм рт. ст.)
117A5);т.пл.49
95 A2); т. пл. 25
127 B5)
115B4);т.пл.46
(петролейный эфир)
118A2)
137A8)
129A2)
Выход, %
70
70
80
75
80
75
75
7. Реакции карбонильных соединений
219
Таблица Г.7.311. Получение фенацилбромидов
Продукт реакции
Фенацилбромид
л-Бромфенацилбромид
л-Фенилфенацилбромид
Исходное вещество
Ацетофенон
л-Бромацетофенон
и-Фенилацетофенона
а Реакцию ведут в двойном объеме ледяной уксусной кислоты
51;
т. кип.
109
125
Т. пл., °С
128 A0 мм рт. ст.)
Выход, %
60
70
80
ра смеси не должна превышать 40—50 °С. По окончании добавления брома быстро вливают еще
0,5 моль сухого брома и, нагревая смесь на водяной бане при 40 °С, продолжают перемешивание
в течение 48 ч. Затем добавляют 0,5 моль воды, нагревают 5—10 мин при 120—140 "С с обратным
холодильником и перегоняют в вакууме. Выделяющийся в ходе реакции бромоводород погло-
поглощают водой (см. разд. Г, 1.4.2).
Общая методика получения фенацилбромидов (табл. Г.7.311)
Внимание! Соблюдайте осторожность при работе с бромом (см. часть Е)!
Фенацилбромиды раздражают кожу и обладают слезоточивым действием!
В трехгорлую колбу емкостью 500 мл, снабженную мешалкой, капельной воронкой и об-
обратным холодильником, помещают раствор 0,5 моль соответствующего ацетофенона в 100 мл
ледяной уксусной кислоты и несколько капель раствора бромоводорода в ледяной уксусной
кислоте. К этой смеси добавляют по каплям 0,5 моль брома таким образом, чтобы температу-
температура поддерживалась около 20 °С (сначала убедитесь в том, что реакция началась!). После окон-
окончания реакции смесь охлаждают ледяной водой; если не появляются кристаллы, то смесь вы-
выливают в ледяную воду. Твердый осадок отфильтровывают и промывают 50%-ным водным
этанолом до обесцвечивания. Перекристаллизовывают из небольшого количества этанола.
Получение дибутшацеталя бромацетальдегида бромированием паральдеги-
да в н-бутаноле: Baganz Я., Vitz С Chem. Вег., 1953, 86, 395.
Получение бромацетона из ацетона и брома в ледяной уксусной кислоте:
Levene P. A. Org. Syntheses, Coll. Vol. II, 1943, p. 88.
Получение диэтилового эфира броммалоновой кислоты из диэтилового эфира
малоновой кислоты: Palmer С. S., McWherter P. W. Org. Syntheses, Coll. Vol. I,
1941, p. 245.
Получение а-бром-у-бутиролактона из у-бутиролактона, брома и красного
фосфора через а,у-дибромбутирилбромид и а,у-диброммасляную кислоту:
Plieninger К, Chem. Ber., 1950, 83, 265.
Получение 2-хлорциклогексанона хлорированием циклогексанона в воде:
Newman M. S., Farbman M. D., HipsherH. Org. Syntheses, Coll. Vol. Ill, 1955, p. 188.
Получение этилового эфира а-хлорацетоуксусной кислоты из ацетоуксусного
эфира и сульфурилхлорида: Boehme W. R. Org. Syntheses, Coll. Vol. IV, 1963, p. 592.
а-Галогенкарбоновые кислоты, в первую очередь хлоруксусная, и их эфиры
являются промежуточными продуктами в ряде синтезов, например синтезе гли-
цидных эфиров по Дарзану (см. схему [Г.7.132]), получении а-аминокислот по
Фишеру (см. табл. Г.2.85), нитрометана по Кольбе (см. разд. Г,2.6.8) и малоново-
220 Г Препаративная часть (продолжение)
го эфира из циануксусной кислоты. сс-Галогенкетоны и альдегиды используют
при получении тиазолов по Ганчу.
Некоторые а-хлоркарбонильные соединения имеют и промышленное значение. Важней-
Важнейшими из них являются хлоруксусная кислота (ее получают изтрихлорэтилена, см. табл. Г.4.26) и
трихлорацетальдегид (хлораль). Хлоруксусную кислоту используют при получении гербицида
2,4-D (см. разд. Г.2.6.2), малонового эфира (применение см. разд. Г,7.1.4.3), карбоксиметилцел-
люлозы (см. разд. Г,2.6.2), красителей и других веществ, в то время как хлораль применялся глав-
главным образом при синтезе ДДТ и других инсектицидов (см. схему [Г.5.62]). Из 1,3-дибромацето-
на в промышленности получают 1,2,3-трицианопропанол-2 и далее лимонную кислоту.
7.4.2.3. Ацилирование и алкилирование енаминов
Енамины, получение которых рассмотрено в разд. Г,7.1.1, дают с ароматически-
ароматическими ацилгалогенидами в соответствии со схемой [Г.7.297] с хорошими выходами
ацилированные енамины; при этом образуются как соединения с сопряженной
двойной связью (Па в схеме [Г.7.312]), так и несопряженные системы (Пб в
схеме [Г.7.312]). При гидролизе оба соединения превращаются в р-дикарбо-
нильное соединение III:
О
[Г.7.312]
Мб
Образующийся в реакции хлороводород обычно связывают с помощью основа-
основания, например безводного триэтиламина; в противном случае 50% енамина
превращаются в соль. (Почему соляная кислота не взаимодействует с ацилиро-
ванным енамином II?)
В некоторых случаях, например при ацилировании эфирами хлормуравьиной кислоты,
этот метод малопригоден и лучше работать с избытком енамина.
Алифатические ацилхлориды, имеющие подвижный атом водорода в а-по-
ложении, при взаимодействии с енаминами или основаниями сначала превра-
превращаются в кетоны (см. разд. Г,3.1.5).
RV О _ R14
нс-с' Триэтилами" \=с=о [Г.7.313]
R*' Ъ -НС1 #'
(Аналогично из алифатических сульфонилхлоридов образуются сульфены R2C=SO2.)
Затем енамин дает с кетоном производное циклобутанона:
R3 R4 и ^^
?> + н \П"Т~Т о [Г.7.314]
7.Реакции карбонильных соединений
221
При R2= R4= H циклобутаноновое кольцо термически неустойчиво и легко,
например при перегонке, расщепляется с образованием обоих возможных про-
продуктов расщепления:
[Г.7.315]
(Напишите схему расщепления, когда только R2 или только R4=H!)
В случае енаминов циклических кетонов направление расщепления циклобутанонового
кольца зависит от величины цикла кетона. Если в последнем число атомов углерода не более 9,
то образуются ацилиро ванные соединения, а при большей величине цикла преобладает расши-
расширение кольца:
(СН2)Л
СН3
(СН2)П
(СН2)П
[Г.7.316]
(ГП) Общая методика получения р-дикетонов ацилированием енаминов (табл. Г.7.317). В трех-
^=^ гордой колбе емкостью 250 мл, снабженной капельной воронкой, обратным холодиль-
холодильником и мешалкой, растворяют 0,1 моль енамина и 0,12 моль перегнанного над натри-
натрием триэтиламина в 150 мл сухого толуола. Нагревают на водяной бане до 35 °С и добавляют при
этой температуре по каплям 0,12 моль ацилхлорида. Смесь оставляют на 1 ч при 35 °С и на ночь
при комнатной температуре. Добавляют 50 мл 20%-ной соляной кислоты и кипятят 30 мин при
перемешивании с обратным холодильником. Отделяют водный слой, органическую фазу
Таблица Г.7.317. Получение
Продукт реакции
2-Ацетилциклогексанон
2-Пропионилциклогексанон
2-Бутирилциклогексанон
2-Ацетилциклопентанон
2-Бутирилциклопентанон
2-Пропионилциклопентанон
р-дикетонов ацилированием енаминов
Исходные вещества
1 - Морфолиноци клогексен,
ацетилхлорид
1 - Морфолиноциклогексен,
пропионилхлорид
1 - Морфолиноциклогексен,
бутирилхлорид
1-Морфолиноциклопентен,
ацетилхлорид
1 -Морфолиноциклопентен,
бутирилхлорид
1 - Морфолиноциклопентен,
пропионилхлорид
Т. кип., °С
(мм рт. ст.)
112A8)
144A2)
125A5)
78(8)
112A5)
108A5)
Выход, %
50
60
55
60
65
60
222 Г Препаративная часть (продолжение)
промывают водой до нейтральной реакции. Разбавленным раствором гидроксида натрия дово-
доводят рН водного слоя до 5-6 и дважды экстрагируют толуолом. Объединенные толуольные
вытяжки сушат сульфатом натрия. После отгонки растворителя перегоняют в вакууме, собирая
фракцию, кипящую в широком диапазоне. Для кислотного расщепления полученные ацилиро-
ванные кетоны можно использовать без дальнейшей очистки.
Благодаря высокой реакционной способности енамины представляют боль-
большой интерес для препаративной химии.
Реакции ацилирования проводят также с выделенными кетенами. Ацилиро-
вание алифатическими ацилхлоридами сопоставимо с взаимодействием с али-
алифатическими сульфонилхлоридами и триэтиламином (образование сульфенов).
(Напишите схемы этих реакций!)
Вариантом ацилирования енаминов является присоединение изоцианатов и
изотиоцианатов (см. разд. Г,7.1.6 и Г,7.2.1.11), например:
[Г.7.318]
Получение 2-морфолиноциклогексен- 1-карбанилида-1 или -тиокарбанилида-1
из 1-морфолиноциклогексена и фенилизоцианата или фенилизотиоцианата:
HunigS., HiibnerK., BenzingE. Chem. Ber., 1962, 95, 926.
Алкилирование енаминов алкилгалогенидами в соответствии с общей схе-
схемой [Г.7.297] приводит главным образом к моноалкилированным енаминам, ко-
которые можно перевести гидролизом в а-моноалкилированные карбонильные
соединения. (Напишите схему образования этилового эфира 2-оксоциклогек-
силуксусной кислоты из 1-пирролидиноциклогексена-1 и бромэтилацетата!)
Для получения моноалкилпроизводных эфиров (З-оксокарбоновых кислот и
Р-дикетонов также используют реакции их енаминов с алкилгалогенидами или
алкилсульфатами. Образующаяся сначала соль иммония II затем гидролизуется
до р-дикарбонильного соединения III:
©
NR2
R-C=CI
H-FT -
i- o=C=N-
-Ph
NR2 0
1 II
> R-C=C-C-NPh
1
R-
" Vie + H3C-C=t
I
MeOSO2-O-Me + H3C-C=CH-COOEt z-~ H3C-C-CH-COOEt
-MeOSO? l/|e
" [Г.7.319]
О
+ Н2О и
H3C-C-CH-COOEt
-Me2Nhlf
Препаративное значение имеет также алкилирование енаминов электро-
фильными олефинами и ацетиленами, в котором в качестве продуктов реакции
могут получаться производные циклобутана (см. схему [Г.7.284]; ср. с взаимо-
взаимодействием енаминов с л-бензохиноном как винилогом карбонильных соедине-
соединений [Г.7.288]).
Получение а^метилацетоуксусного эфира алкилированием этилового эфи-
эфира р-диметиламинокротоновой кислоты диметилсульфатом: Мистрюков Е. А.
Изв. АН СССР, Сер. хим., 1961, 1512.
7. Реакции карбонильных соединений 223
7.5. ЛИТЕРАТУРА
Получение ацеталей, тиоацеталей; иминов, оксимов, гидразонов и
бисульфитных соединений
Bayer О., in: Houben-Weyl. Bd. 7/1 A954), S. 413-488.
Duni M., Koruntev D., Kovalevi K., Polak L., Kolbah D., in: Houben-Weyl. Bd. E14b/1
A990), S. 434-639 (гидразоны); S. 640-730 (азины).
KlausnerF., u.a., in: Houben-Weyl. Bd. E14a/1 A991), S. 1-783 (ацетали).
Layer R. W., Chem. Rev. 63 A963), 489-510 (получение и реакции иминов).
Meerwein К, in: Houben-Weyl. Bd. 6/3 A965), S. 204-270.
MeskensA. J., Synthesis 1981, 501-522 (ацетали).
PawlenkoS., in: Houben-Weyl. Bd. E14b/1 A990), S. 222-286 (имины).
Rasshofer W., in: Houben-Weyl. Bd. E14a/2 A991), S. 1-819 (О^-ацетали).
UnterhaltB., in: Houben-Weyl. Bd. E14b/1 A990), S. 287-433 (оксимы).
WimmerP., in: Houben-Weyl. Bd. E14a/1 A991), S. 785-836 (О,8-ацетали).
Получение карбоновых кислот, их эфиров, ангидридов, амидов,
гидразидов и ацилхлоридов
Bodanszky М., in: Peptide Chemistry; A Practical Texbook. — Springer-Verlag, New York 1988.
Dopp D., Dopp H., in: Houben-Weyl. Bd. E5/2 A985), S. 934-1183.
Henecka H., u.a. in: Houben-Weyl. Bd. 8 A952), S. 359-680.
Sustmann R., Korth H.-G., u.a. in: Houben-Weyl. Bd. E5/1 A985), S. 193-773.
Получение ортоэфиров карбоновых кислот
Meerwein #., in: Houben-Weyl. Bd. 6/3 A965), S. 295-324.
Simchen G., in: Houben-Weyl. Bd. E5/1 A985), S. 105-122.
De Wolfe R. K, Synthesis 1974, 153-172.
Получение и реакции кетенов
Lacey R. N., Adv. Org. Chem. 2 A960), 213-263.
Quadbeck G., in: Neuere Methoden. Bd. 2 A960), S. 88-107; Angew. Chem. 68 A956),
361-370.
Schaumann E., Scheiblich S., in: Houben-Weyl. Bd. E15/2 A993), S. 2353-2530.
Tidwell Т. Т. Ketenes. — John Wiley & Sons, New York 1994.
Получение и реакции цианатов и изоцианатов
Благонравова А. А., Левкович Г. А. Усп. хим., 1955, 24, 93-119.
Findeisen К., u.a., in: Houben-Weyl. E4 A983), S. 738-834.
Martin D., Bacaloglu R., Organische Synthesen mit Cyansaureestern. — Akademie-Verlag,
Berlin 1980.
Ozaki S., Chem. Rev. 72 A972), 457-496.
Petersen, u.a., in: Houben-Weyl. Bd. 8 A952), S. 119-137.
Sounders J. H., Slocombe R. J., Chem. Rev. 43 A948), 203-218.
224 Г Препаративная часть (продолжение)
Получение и реакции циангидринов, синтез Штреккера
KuritzP., in: Houben-Weyl. Bd. 8 A952), S. 274-285; Nachr. Chem., Tech. Lab. 29 A981),
445-447.
Альдольная конденсация
Bayer О., in: Houben-Weyl. Bd. 7/1 A954), S. 76-92.
Evans D. A., Nelson J. V., Taber T. R., in: Topics in Stereochemistry. Bd. 13. Hrsg. Allinger N. L,
Eliel E. L., Wilen S. H. - Wiley, New York 1982, S. 1-115.
Heathcock С H., in: Modern Synthetic Methods. Bd. 6. Hrsg. R. Scheffold. - VCHA, Basel
1992, S. 1-102.
Heathcock С H, in: Asymmetric Synthesis. Bd. 3. Hrsg. J. D. Morrison. — Academic Press,
New York 1984, S. 111-212.
Mukaiyama Т., Org. React. 28 A982), S. 302-331; Nachr. Chem. Tech. Lab. 29 A981),
555-559.
Nielsen А. Т., Houlihan W. J., Org. React. 16 A968), 1-438.
Wittig G., in: Topics in Current Chemistry. Bd. 67. — Springer-Verlag, Berlin, Heidelberg,
New York 1976, S. 1-14.
Реакция с нитрометаном
LichtenthalerF. W., in: Neuere Methoden. Bd. 4 A966), S. 140-172; Angew. Chem. 76 A964), 84.
Азлактонный синтез
BaltazziE., Ouart. Rev. 9 A955), 150-173.
Картер Г. Е. В сб.: Органические реакции. Сб. 3. — М.: ИЛ. 1951, с. 190-229.
Реакция Перкина
Henecka H, Ott E., in: Houben-Weyl. Bd. 8 A952), S. 442-450.
Джонсон Д. В сб.: Органические реакции. Сб. 1. — М.: ИЛ, 1948, с. 267-344.
Синтез глицидных эфиров по Дарзану
Bayer О., in: Houben-Weyl. Bd. 7/1 A954), S. 326-329.
Ньюмен М. С, Мейджерлеш Б. Д. В сб.: Органические реакции. Сб. 5. — М.: ИЛ, 1951,
с. 319-346.
Реакция Принса
Adams D. R., Bhatnagar S. P., Synthesis 1977, 661-672.
Исагулянц В. И., Чаимова Т. Г., Меликян В. Р., Покровская С. В., Усп. хим., 1968, 37,
61-77.
Конденсация Кнёвенагеля
Jones G., Org. React. 15 A967), 204-599.
7. Реакции карбонильных соединений 225
Реакция с малононитрилом
FatiadiA. J., Synthesis 1978, 165-204; 241-282.
Реакция Манниха
Arend M., Westermann В., Risch N., Angew. Chem. 110 A998), 1096-1122.
Блик Ф. Ф. В сб.: Органические реакции. Сб. 1. — М: ИЛ, 1948, с. 399-454.
Hellmann H., in: Neuere Methoden. Bd. 2 A960), S. 190-207; Angew. Chem. 69 A957),
463-471.
Hellmann H., Opitz G., a-Aminoalkylierung. — Verlag Chemie, Weinheim/Bergstr. 1960.
SchmterR., in: Houben-Weyl. Bd. 11/1 A957), S. 731-795.
Tramontini M., Synthesis 1973, 703-775.
Tramontini M., Angiolini L., Tetrahedron 1990, 1791-1837.
Tramontini M., Angiolini L:. Mannich Bases: Chemistry and Uses. — CRC Press, Boca Raton
1994.
Бензоиновая конденсация
HerlingerH., in: Houben-Weyl. Bd. 7/2a A973), S. 653-671.
Aud В., Бак И. Веб.: Органические реакции. Сб. 4. — М.: ИЛ, 1951, с. 229-269.
Реакция Виттига
Bergelson L. D., Shemyakin M. M., in: Neuere Methoden. Bd. 5 A967), S. 135—155; Angew.
Chem. 76A964), 113-123.
Bestmann H. J., Klein O., in: Houben-Weyl. Bd. 5/lb A972), S. 383-418.
Cadogan J. I. C, Organophosphorus Reagents in Organic Synthesis. — Academic Press,
New York 1966.
Johnson A. W., Ylide Chemistry. —Academic Press, New York, London 1966.
Маркер А. В сб.: Органические реакции. Сб. 14. — М.: Мир, 1967, с. 286—530.
MaryanoffB. Е, ReitzA. В., Chem. Rev. 89 A989), S. 863-927.
Schdllkopf U., in: Neuere Methoden. Bd. 3 A961), S. 72-97; Angew. Chem. 71 A959),
260-273.
TrippettS., Adv. Org. Chem. 1 A960), S. 83-102; Quart. Rev. 17 A963), 406.
Wadsworthp W. S., Org. React. 25 A977), S. 73-253.
Реакции алкилиденфосфоранов
Bestmann H. J., in: Neuere Methoden. Bd. 5 A967), S. 1-52; Angew. Chem. 77 A965), 609,
651,850.
Bestmann H. J., Zimmermann R., Fortschr. Chem. Forsch. 20 A971), 1-141.
Bestmann H. J., Zimmermann R., in: Houben-Weyl. Bd. El A982), S. 616-751.
Flitsch W., SchindlerS. R., Synthesis 1975, 685-700.
Johnson A. W., Ylides and Imines of Prosphorus. — John Wiley & Sons, New York 1993.
Kolodiazhnyi O. /., Phosphorus Ylides. — Wiley-VCH, Weinheim 1999.
Sasse K., in: Houben-Weyl. Bd. 12/1 A963), S. 112-124.
ZbiralE., Synthesis 1974, 775-797.
226 Г Препаративная часть (продолжение)
Сложноэфирные конденсации
Хаузер Ч. Р., Хадсон Б. Е. В сб.: Органические реакции. Сб. 1. — М.: ИЛ, 1948,
с. 344-398.
Henecka Н, in: Houben-Weyl. Bd. 8 A952), S. 560-589.
Конденсация Дикмана
SchaeferJ. P., Bloomfleld J. J., Org. React. 15 A967), 1-203.
Ацилирование карбонильных соединений ацилхлоридами
Henecka H. In: Houben-Weyl. Bd. 8, 1952, S. 610-612.
Hauser С. Л. и др. Org. Reactions, 1954, 8, 59-196.
Полиметины
Berlin L, RiesterO. In: Houben-Weyl. Bd. 5/ld, 1972, S. 227-299.
Реакция Гриньяра
Coates G. E., Wade K., Organometallic Compounds. Vol. I. — Methuen, London 1967.
Иоффе С. Т., Несмеянов А. Н. В сб.: Методы элементоорганической химии. — М.:
АН СССР, 1963.
Kharasch M. S., Reinmuth О., Grignard Reactions of Nonmetallic Substances. — Prentice-
Hall, New York 1954.
NutzelK., in: Houben-Weyl. Bd. 13/2a A973), S. 47-527.
WakefieldB. J., Organometal. Chem. Rev. 1 A966), 131; Усп. хим. 37 A968), 36-60.
Wakefleld В. J., Organomagnesium Methods in Organic Synthesis. — Academic Press,
London San Diego 1995.
Grignard Reagents. Hrsg H. G. Richey, Wiley & Sons, Chichester 2000.
Синтезы с литийорганическими соединениями
GschwendH. W., Rodriguez H. R., Org. React. 26 A979), 1-360.
Jorgenson M. /., Org. React. 18 A970), 1-97.
Mallan J. M., Bebb R. L, Chem. Rev. 96 A969), 693-755.
SchdilkopfU., in: Houben-Weyl. Bd. 13/1 A970), S. 87-253.
WakefieldB. J., Organolithium Methods. — Academic Press, London 1988.
Медьорганические соединения
BahrG., Burba P., in: Houben-Weyl. Bd. 13/1 A970), S. 731-761.
House H. О., Асе. Chem. Res. 9 A976), 59-67.
LishutzB. H., Sengupta S., Org. React. 41 A992), 135-631.
NormantJ. E, Synthesis 1972, 63-80.
NormantJ. E, AlexakisA., Synthesis 1981, 841-870.
Organocopper Reagents. Hrsg.: R. J. K. Taylor. — Oxford University Press, Oxford 1994.
PosnerH. G, Org. React. 19 A972), 1-113; 22 A975), 253-400.
Posner H. G, An Introduction to Synthesis Using Organocopper Reagents. — John Wiley &
Sons, New York 1980.
Taylor R. J. K., Organocopper Reagents. — Oxford University Press, Oxford 1994.
7. Реакции карбонильных соединений 227
Цинкорганические соединения
Erdik Е., Organozinc Reagents in Organic Synthesis. — CRC Press, Boca Raton 1996.
KnochelP., PereaJ. J. A., Tetrahedron 54 A998), 8275-8319.
Knochel P., Jones P., Organozinc Reagents. — Oxford University Press, Oxford 1999.
NiltzelK., in: Houben-Weyl. Bd. 13/2a A973), S. 553-858.
Реакция Реформатского
FiirstnerA., Synthesis 1989, 571-590.
Niitzel K., in: Houben-Weyl. Bd. 13/2a A973), S. 809-838.
Rathke M. iV., Org. React. 22 A975), 423-460.
Синтез кетонов из ацилхлоридов или карбоновых кислот и
металлоорганических соединений
Jorgenson M. /., Org. React. 18 A970), 1-97.
Shirley D. A., Org. React. 8 A954), 28-58.
WinglerF., in: Houben-Weyl. Bd. 7/2a A973), S. 558-603.
Восстановление комплексными гидридами
Браун В. Г. В сб.: Органические реакции. Сб. 6. — М.: ИЛ, 1953, с. 409-460.
Gaylord N. С, Reduction with Complex Metal Hydrides. — Interscience, New York, London
1956.
HajosA., Komplexe Hydride. — Deutscher Verlag der Wissenschaften, Berlin 1966.
HajosA., in: Houben-Weyl. Bd. 4/ld A981), S. 1-486.
Hermann #., in: Neuere Methoden. Bd. 2 A960), 145-154; Angew. Chem. 68 A956), 601-604.
Itsuno S., Org. React. 52 A998), 395-576 (энантиоселективное восстановление
кетонов).
MalekJ., Org. React. 34 A985), 1-317; 36 A988), 1-173.
Рогинская Е. В. Усп. хим., 1952, 21, с. 3-39.
Schenker Т., in: Neuere Methoden. Bd. 4 A966), 173-293; Angew. Chem. 73 A961),
81-107.
Seyden-Penne J., Reductions by the Alumino- and Borohydrides in Organic Synthesis. —
VCH, New York 1991.
Каталитическое гидрирование карбонильных соединений
Adkins H., Org. React. 8 A954), 1-27.
Богословский Б. М., Казакова 3. С. Скелетные катализаторы в органической химии. — М.:
Госхимиздат, 1957.
Schiller G., in: Houben-Weyl. Bd. 4/2 A955), S. 303-312, 318-328.
Tinapp P., in: Houben-Weyl. Bd. 4/lc A980), S. 189-224.
Восстановительное алкилирование аминов
Emerson W. S., Org. React. 4 A948), 174-255.
Клюев М. Б., Хидекель М. Л. Усп. хим., 1980, 49, 28-53.
MollerF., SchrdterR., in: Houben-Weyl. Bd. 11/1 A957), S. 602-648.
228 Г Препаративная часть (продолжение)
Реакция Лейкарта-Валлаха
Богословский Б. М. В сб.: Реакции и методы исследования органических соединений.
Т. 3. - М.: Химия, 1954, с. 253-314.
Moore M. L. Org. Reactions, 1949, 5, 301-330.
MollerF, SchrbterR. In: Houben-Weyl. Bd. XI/1, 1957, S. 648-664.
Реакция Меервейна-Понндорфа-Верлея
УайлдсА. Л. В сб.: Органические реакции. Сб. 2. — М.: ИЛ, 1950, с. 194-243.
Bersin Т. In: Neuere Methoden, Bd. 1, 1949, S. 137-154.
Окисление по Оппенауэру
Bersin Т. In: Neuere Methoden, Bd. 1, 1949, S. 137-154.
Джерасси К. В сб.: Органические реакции. Сб. 6. — М.: ИЛ, 1953, с. 235—300.
Lehmann, In: Houben-Weyl. Bd 4/lb, 1975, S. 905-933.
Реакция Канниццаро
Гейсман Т. А. В сб.: Органические реакции. Сб. 2. — М.: ИЛ, 1950, с. 106-127.
Henecka H., Ott E., in: Houben-Weyl. Bd. 8 A952), S. 455-456.
Реакция Кижнера-Вольфа
AsingerF., Vogel H. #., in: Houben-Weyl. Bd. 5/la A970), S. 251-267, 456-465.
Родионов В. М., Ярцева Н. Г. В сб.: Реакции и методы исследования органических
соединений, т. 1. — М.: Госхимиздат, 1951, с. 7—98.
Shapiro R. #., Org. React. 23 A976), 405-507 (реакция Бэмфорда-Стивенса).
ToddD., Org. React. 4 A948), 378-422.
Восстановление карбонильных соединений неблагородными металлами
BloomfieldJ. J., Owsley D. С, NelkeJ. M, Org. React. 23 A976), 259-403.
Muth H., SauerbierM., in: Houben-Weyl. Bd. 4/lc A980), S. 645-654.
Реакция Клемменсена
Мартин Э. В сб.: Органические реакции. Сб. 1. — М.: ИЛ, 1948, с. 194—296.
VedejsE. Org. Reactions, 1975, 22, 401-422.
AsingerF, VogelH. H. In: Houben-Weyl, Bd. 4/la, 1970, S. 244-250, 450-456.
Реакции восстановления карбонильных соединений
с низковалентным титаном
McmurryJ. E., Chem. Rev. 89 A989), 1513-1524.
LenoirD., Synthesis 1989, 883-897.
FurstnerA., Bogdanovic В., Angew. Chem. 108 A996), 2582-2609.
7. Реакции карбонильных соединений 229
Присоединение аммиака и аминов к
а,$-ненасыщенным карбонильным соединениям
Cromwell N. К, Chem. Rev. 38 A946), 83-137.
MdllerE, in: Houben-Weyl. Bd. 11/1 A957), S. 272-289.
Суминов С. И., КостА. Н. Усп. хим., 1969, 38, 1933-1963.
Синтез хинолинов по Скраупу
Манске Р. X. Ф., Кулка М. В сб.: Органические реакции. Сб. 7. — М.: ИЛ, 1956,
с. 100-145.
Реакция Михаэля
Allen jr. G. R. Org. Reactions, 1973, 20, 337—454 (присоединение енаминов, реакция
Неницеску).
Бергман Э. Д., Гинзбург Д., Паппо Р. В сб.: Органические реакции. Сб. 10. — М: ИЛ,
1963, с. 181-553.
Henecka Н. In: Houben-Weyl. Bd. 8, 1952, S. 590-598.
Little R. D., Masjedizadeh M. R., Wallouist O., McLoughlin J. /., Org. React. 47 A995),
315—552 (внутримолекулярная реакция Михаэля).
Nagata W., Yoshioka M. Org. Reactions, 1977, 25, 255-476 (присоединение синильной
кислоты).
Stetter H. Angew. Chem., 1976, 88, 695—704 (присоединение альдегидов).
Цианэтилирование
Bruson H. A., Org. React. 5 A949), 79-135.
Kurtz P., in: Houben-Weyl. Bd. 8 A952), S. 340-344.
Несмеянов А. Н. и др. Усп. хим., 1967, 36, 1089.
Тереншьев А. П., Kocm A. H. В сб.: Реакции и методы исследования органических сое-
соединений. Т. 2. — М.: Госхимиздат, 1952, с. 47—208.
Алкилирование карбонильных соединений
Соре А. С, u.a. Org. React. 9 A957), 107-331.
Harris Т. М., Harris С. М., Org. React. 17 A969), 155-211.
Henecka H, in: Houben-Weyl. Bd. 8 A952), S. 600-610.
Petragnani N., Yonashiro M., Synthesis 1982, 521-578.
С аминами и солями аммония
BrewsterJ. H, Eliel E. L, Org. React. 7 A953), 99-197.
Hellmann H., Angew. Chem. 65 A953), 473.
Хлорирование и бромирование карбонильных соединений
RoedigA.. in: Houben-Weyl. Bd. 5/4 A960), S. 164-210.
Stroh R., in: Houben-Weyl. Bd. 5/3 A962), S. 611-636.
230 Г Препаративная часть (продолжение)
Получение и реакции металленолятов
Klar G., Kramolowsky R., in: Houben-Weyl. Bd. E15/1 A993), S. 563-597.
Получение и реакции эфиров енолов
Effenberger E, Angew. Chem. 81 A969), 374-391.
Frauenrath H., in: Houben-Weyl. Bd. E15/1 A993), S. 1-349.
Из силиловых эфиров енолов
Brownbridge P., Synthesis 1983, 1-28, 85-104.
Pawlenko S., in: Houben-Weyl. Bd. E15/1 A993), S. 404-462.
Rasmussen J. K., Synthesis 1977, 91-110.
Получение и реакции енаминов
Enamines — Synthesis, Structure and Reactions. Hrsg.: A. G. Cook. — Marcel Dekker,
New York, London 1969.
HickmottP. W^, Tetrahedron 38 A982), 1975-2050; 3363-3446.
Htinig S., Hoch H., Fortschr. Chem. Forsch. 14 A970), 235-293 (ацилирование
енаминов).
RademacherP., in: Houben-Weyl. Bd. E15/1 A993), S. 598-717.
SzmuskoviczJ., Adv. Org. Chem. 4 A963), 1-113.
8. РЕАКЦИИ ДРУГИХ ГЕТЕРОАНАЛОГОВ
КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
Не только кислород карбонильной группы может быть заменен на гетероатомы
(азот, серу); можно представить себе и замещение атома углерода. Таким обра-
образом получаются группировки N=O, S=O, которые не только формально анало-
аналогичны группировке С=О, но и вступают в сходные с ней реакции. Важнейшими
представителями этого типа гетероаналогов карбонильных соединений являют-
являются нитрозо-, нитро- и сульфосоединения, а также их производные.
«Карбонильная активность» нипгрозогруппы, сравнима с карбонильной
активностью альдегидов. Это видно, например из следующих превращений:
+ H2N-R' ^ _R'
1 R -N
[Г.8.1а]
R1
R-
ОН ,N.©,R-
R N [Г.8.16]
IOI0
R1
Ш2С _ || ГГ R 1r1
COR" „Nk X. . 11.8.1В]
R
I
R1
(С какими реакциями альдегидов сходны эти конденсации?)
8. Реакции других гетероаналогов карбонильных соединений 231
По сравнению с нитрозогруппой нитрогруппа обладает меньшей «карбо-
«карбонильной активностью». Это объясняется повышенной склонностью нитрогруп-
нитрогруппы к мезомерии, сравнимой с мезомерией карбоксильной группы или карбок-
силат-аниона. Большая по сравнению с кетонами способность нитросоедине-
ний к восстановлению показывает, что по своей «карбонильной активности»
они стоят примерно между кетонами и карбоновыми кислотами.
Сульфоновые кислоты являются аналогами карбоновых кислот. Из-за боль-
большого числа мезомерных структур их «карбонильная активность» мала:
'-?О1 '-7OI \О\ :'О\ _
R-C' > R-N© > R-C* > R-S*.9' [Г-8-2!
В то время как «карбонильная активность» существенно зависит от вероят-
вероятности мезомерии, кислотность находящегося в а-положении атома водорода
зависит, кроме того, и от индукционного влияния рассматриваемой группы.
Алифатические нитрозосоединения, имеющие атомы водорода в а-положе-
а-положении, изомеризуются в изонитрозосоединения (оксимы); следовательно, нитро-
зогруппа оказывает сильное ацидифицирующее влияние на соседние атомы
водорода:
R N^OI R
V') ~~ ». У-КГ [Г 8 V
R' H R' ОН R'= алкил или Н
(Обсудите, какие нитрозосоединения способны претерпевать реакции [Г.8.1].)
Ацидифицирующее влияние нитрогруппы меньше, чем у нитрозогруппы, но
больше, чем влияние карбонильной группы в альдегидах и кетонах. По этой
причине, в частности, нитросоединения всегда играют роль метиленового ком-
компонента в конденсациях альдегидов и кетонов с нитросоединениями. Подобно
оксимам, первичные и вторичные нитросоединения растворяются в водных
щелочах с образованием солей.
Если свободные енолы простых альдегидов вообще неизвестны, а нитрозо-
нитрозосоединения, имеющие а-атомы водорода, устойчивы только в виде оксимов, то
у нитросоединений, имеющих а-атомы водорода, можно выделить как ацифор-
му (енол), так и нитроформу, подобную кетонной форме карбонильных соеди-
соединений. (Ознакомьтесь по учебнику с таутомерией алифатических нитросоеди-
нитросоединений.)
Сульфогруппа и ее производные ацидифицируют соседние атомы водорода
сильнее, чем карбоксильная группа. Таким образом, С-Н-кислотность падает в
следующем ряду:
I '? I Р I /? I ° I ,?
H-C-n' > H-C-N© > Н-С-с' > H-C-S*0 > Н-С-с' [Г 8 41
I Ou I R I OR I OR
Важнейшие реакции гетероаналогов карбонильных соединений перечисле-
перечислены в табл. [Г.8.5].
232
Г Препаративная часть (продолжение)
Таблица
R-NO2
HO-NO +
и
н
+
©
Ar-NEN
Г.8.5. Важнейшие реакции гетероаналогов
Н®ее
-^— R-NO
Н®ев
——- R-NHOH
Н®ее
—^— R-NH2
Ar-NH2 -
- R-NH2 -
- R2NH
ь R-ОН -
?
ь R2CH-C -
R-CH2-NO2 -
•r R2CH-NO2 -
H2O
н@ее
X9Cu®
Ar-H_
R2CH2|
CC DC
1 1
® II II
•O-Z0 -Z.
1 1
t t
— R-NH-NH-R
Ar-NEN Xu
R-OH
R2N-NO
RO-NO
R2C-/
NO
R-C-NO2
li
N-OH
R2C-NO2
NO
Ar-OH
Ar-NH-NH2
Ar—X
Ar-N=N-Ar
Ar-NH-N=CR2
тттттжжтштжжттттт.
карбонильных соединений
Восстановление
нитросоединений до:
нитрозосоединений
гидроксиламинов
азоксисоединений
азосоединений
гидразосоединений
аминов
Реакции с азотистой
кислотой, ведущие к:
солям диазония
спиртам
нитрозам и нам
нитритам
(эфирам азотистой кислоты)
а-нитрозокарбонильным
соединениям
нитроловым кислотам
псевдонитролам
Реакции солей диазония:
нагревание до фенолов
восстановление до
арилгидразинов
реакция Зандмейера
(образуются галоген- и
псевдогалогенсодержащие
соединения)
азосочетание
(получение азосоединений)
получение гидразонов
8. Реакции других гетероаналогов карбонильных соединений
233
© © +н® © Ar-
H2C-NEN M ¦ CH3
'— -N2
R-C
+ R-cP _
R'
О
l П H .
h "HCI
+ c=c
\ /
c=c
• ICH2 ¦
H®e® H®e
R-SO2CI —'¦—- R-SO2H ——
R'~0H. n <-o on-
RNH2- R-SO2NHR'
— Ar-0-СНз
p
OCH3
CH2-R'
/P
R C\© ©
CH-N=N
2' /^
/ \
— R-SH
Таблица Г.8.5. Продолжение
Реакции диазометана:
получение простых
эфиров фенолов
этерификация карбоновых
кислот
удлинение цепи
карбонильных соединений
диазокетоны из
ацилгалогенидов
1,3-циклоприсоединение
(см. разд. Г, 4.4.4)
образование карбенов и
последующие реакции (см.
разд. Г, 3.3 и Г, 4.4)
Реакции производных
сульфоновых кислот:
восстановление до
сульфиновых кислот и тиолов
образование эфиров
сульфоновых кислот
образование амидов
сульфоновых кислот
8.1. ВОССТАНОВЛЕНИЕ НИТРО- И НИТРОЗОСОЕДИНЕНИЙ
Восстановление нитросоединении можно осуществить под действием небла-
неблагородных металлов (лучше в кислом растворе), путем каталитического гидри-
гидрирования, электролитически, а также некоторыми другими восстановителями.
Механизм восстановления металлами или каталитического гидрирования мо-
молекулярным водородом аналогичен механизму восстановления карбонильных
соединений (см. разд. Г,7.3).
Нитрососдинсние прежде всего восстанавливается до нитрозосоединения:
2eQ+ R-N©
?он
R-N©
Ср)о
+ н
-н2о"
R-N=O
[Г.8.6]
234
Г Препаративная часть (продолжение)
Нитрозосоединение далее превращается в замещенный гидроксиламин
(напишите схему этих превращений!). Вследствие своей более высокой «карбо-
«карбонильной активности» нитрозосоединения гидролизуются быстрее, чем нитросо-
единения, поэтому, как правило, нитрозосоединение не удается уловить во вре-
время восстановления. При восстановлении гидроксиламина металлами в кислых
средах конечным продуктом является первичный амин:
R-NH-OH
_ ©
R-NHTOH2
-Н
-Н2О
© .
R-NH-0
R-NH
Н
R-NH2
[Г.8.7]
[Г.8.8]
В нейтральном или слабокислом растворе, например при взаимодействии
нитросоединения с цинковой пылью в водном растворе хлорида аммония, вос-
восстановление производных гидроксиламина протекает столь медленно, что
таким путем его можно получить препаративно. (Объясните это с помощью схем
[Г.8.7] и [Г.8.8].)
Наконец, в щелочной среде процесс восстановления как нитрозосоединений,
так и производных гидроксиламина замедляется настолько, что решающее зна-
значение приобретает другая конкурирующая реакция. Свободный арилгидрок-
силамин является сильным нуклеофилом и поэтому может легко реагировать с
арилнитрозопроизводными. Это превращение аналогично образованию осно-
оснований Шиффа и приводит к азоксисоединениям, которые могут быть далее вос-
восстановлены до арилазосоединений и, наконец, до арилгидразосоединений.
Ниже приведена общая схема образования промежуточных продуктов при
восстановлении ароматических нитросоединений в различных условиях:
NO2
нитробензол
N0
нитрозобензол
NHOH
фенилгидроксиламин
<HS)
NH2
(НО9)
(Н0)
[Г.8.9]
азоксибензол
азобензол
гидразобензол
Все промежуточные продукты восстановления ароматических нитросоеди-
нитросоединений, за исключением нитрозобензола и устойчивого азобензола, под действи-
действием сильных кислот перегруппировываются. При этом из фенилгидроксиламина
образуется я-аминофенол1, из азоксибензола — и-гидроксиазобензол, а из
гидрозобензола — бензидин. (Напишите схемы образования этих веществ!)
1 л-Аминофенол (родинал) и его производные используются в фотографии как проявите-
проявители. На чем основано их восстанавливающее действие?
8. Реакции других гетероаналогов карбонильных соединений
235
Каталитическое восстановление ароматических нитросоединений проходит
гладко и приводит, как правило, к первичному амину. Вместо молекулярного во-
водорода можно использовать и гидразин, который при этом дегидрируется до азота.
Преимущество восстановления гидразином состоит в селективности данной
реакции. Эта селективность теряется в щелочной среде при повышенной темпе-
температуре (восстановление по Кижнеру—Вольфу; см. разд. Г,7.3.1.6; например
л-нитробензальдегид восстанавливается до ж-толуидина).
Промышленное значение имеет восстановление по Бешану железом в соля-
солянокислой среде (положительными сторонами этого процесса являются деше-
дешевизна металла, небольшой расход кислоты, возможность использования образу-
образующихся оксидов железа в качестве красящих пигментов:
4 PhNO2 + 9 Fe + 4 Н2О
HCI
4PhNH2 + 3Fe3O4
[Г.8.10]
Легкость протекания восстановления оловом в солянокислой среде исполь-
используется при качественном анализе ароматических нитросоединений. Если вос-
восстановление проводить цинком в смеси уксусной кислоты с ацетангидридом, то
сразу образуются ацетилированные амины.
Для восстановления нитросоединений пригодны также сульфиды аммония и
натрия, дитионит натрия. Эти восстановители особенно интересны тем, что с их
помощью можно парциально восстанавливать полинитросоединения (напри-
(например, льдинитробензол до ж-нитроанилина).
Таблица Г.8.11. Восстановление ароматических нитросоединений до первичных
аминов
Амин
Анилин
З-Броманилин
4-Аминофенетол
а-Аминонафталин
4-Хлоранилин
2-Хлоранилин
о-Толуидин
л-Толуидин
З-Хлоранилин
2,4-Диаминотолуол
З-Аминобензофенон
Этиленацеталь 3-ами-
нобензальдегида
Исходное вещество
Нитробензол
З-Нитробромбензол
4-Нитрофенетол
I -Нитронафталин
4- Н итрохлорбензол
2-Нитрохлорбензол
2-Нитротолуол
4-Нитротолуол
3 - Нитрохлорбензол
2,4-Динитротолуол
З-Нитробензофенон
Этиленацеталь 3-ни-
тробензальдегида
Вариант
(см. с. 236)
А, Б
А, Б
А, Б
А, Б
А
А, Б
А, Б
А, Б
А
А, Б
Б
Б
Т. кип., °С
(мм рт. ст.)
69A0)
130A2);
18 (т. пл.)
127(8);
-2 (т. пл.)
160A2);
90 (т. пл.)
130B0);
71 (т. пл., этанол)
115B0)
121 (80)
84 A3); 45 (т. пл.)
113A8)
149 (8); 99 (т. пл.)
87 (т. шт.; этанол)
123@,5)
ло20
1,5863
1,6260
1,5895
1,5728
1,5930
1,5740
Выход, %
90
50
80
90
95
95
70
70
70
80
80
70
236 Г Препаративная часть (продолжение)
©Общая методика каталитического восстановления ароматических нитросоединений
(табл. Г.8.11)
А. Гидрирование молекулярным водородом.
Об общих правилах работы и мерах предосторожности при каталитическом гид-
гидрировании см. разд. Г,4.5.2 и А, 1.8. Ароматические амины и нитросоединения
ядовиты, а иногда и канцерогенны! Они всасываются через органы дыхания и
кожу! Надевайте защитные перчатки!
Во встряхиваемый автоклав или автоклав с мешалкой помещают раствор 1 моль нитросоеди-
нитросоединения в десятикратном объеме растворителя (спирта, диоксана, алкана) и прибавляют скелетный
никель в количестве 1/10 массы нитросоединения. Гидрируют при комнатной температуре и дав-
давлении 100 атм A0 МПа). Реакцию обязательно надо проводить в растворителе, который необхо-
необходим для отвода значительной теплоты реакции A32 ккал/моль; 553 кДж/моль).
Обработка реакционной смеси. После завершения гидрирования катализатор отделяют
фильтрованием, растворитель упаривают и амин перегоняют в вакууме с использованием
колонки Вигре.
Б. Восстановление гидразином. В двугорлой колбе (или круглодонной колбе с насадкой
Аншютца), снабженной обратным холодильником, к 1 моль нитросоединения (или 0,5 моль
динитросоединения), растворенного в десятикратном объеме спирта, прибавляют 2,5 моль
гидразингидрата (80-100%-ного) (о концентрировании водных растворов гидризингидрата и
об определении их концентрации см. часть Е). Смесь нагревают до 30-40 °С и вносят неболь-
небольшими порциями суспензию скелетного никеля в спирте (скелетный никель надо приготовить
в количестве около 5% от массы нитросоединения; при восстановлении динитросоединений
берут вдвое большее количество никеля). Начало реакции можно заметить по выделению азо-
азота. Каждую следующую порцию катализатора прибавляют после того, как выделение газа за-
замедлится. Если после добавления новых порций катализатора выделения газа не происходит,
то реакционную смесь нагревают еще 1 ч с обратным холодильником, отфильтровывают ката-
катализатор, обесцвечивают раствор активированным углем и выделяют амин перегонкой в ваку-
вакууме с небольшим дефлегматором или перекристаллизацией.
Методика пригодна для полумикросинтезов и качественного анализа.
Получение м-нитроанилина парциальным восстановлением .м-динитробен-
зола: Hodgson H. H., WardE. R. J. Chem. Soc, 1949, 1316.
©Восстановление ароматических нитросоединений до первичных аминов оловом в соля-
солянокислой среде (общая методика для качественного анализа). Смесь 0,5 г нитросоединения,
1,5 г мелкогранулированного олова и 8 мл разбавленной A:1) концентрированной соляной
кислоты кипятят 1 ч с обратным холодильником. Затем реакционную смесь охлаждают, жидкость
сливают с нерастворившегося металла, добавляют 5 мл воды и экстрагируют эфиром непрореаги-
ровавшее исходное вещество и примеси неосновного характера. Водный слой быстро выливают в
избыток раствора гидроксида натрия, амин экстрагируют эфиром, экстракт сушат твердым гид-
роксидом калия и отгоняют эфир. Если извлечение эфиром затрудняется выпавшей C-оловянной
кислотой, то амин отгоняют с водяным паром. Для идентификации можно использовать неочи-
неочищенный амин. При восстановлении кислых нитросоединений (например, нитробензойной кис-
кислоты) невозможно выделить продукт реакции извлечением эфиром из щелочного раствора. Обра-
Образующиеся аминокислоты и аминофенолы можно, например, бензоилировать непосредственно в
щелочном растворе с выделением после подкисления и экстрагирования эфиром.
©Определение эквивалентной массы аминов титрованием хлорной кислотой. [Houben-Weyl,
Bd. 2, 1953, S. 661.] Точную навеску 0,5—1 г основания растворяют в 10 мл безводной ледя-
ледяной уксусной кислоты. Добавив несколько капель раствора индикатора, титруют 0,1 н.
раствором хлорной кислоты в уксусной кислоте (см. часть Е) до перехода окраски индика-
индикатора (кристаллический фиолетовый дает переход от синего к зеленому, а а-нафтолбензеин — от жел-
желтого к зеленому). Рассчитывают по формуле
bN
где Е— эквивалентная масса, а — навеска вещества в граммах, b — расход раствора хлорной кис-
кислоты в мл, N— нормальность раствора хлорной кислоты.
8. Реакции других гетероаналогов карбонильных соединений 237
Восстановление ароматических нитросоединений — наиболее распространен-
распространенный метод получения первичных ароматических аминов, поскольку из арилгалоге-
нидов амины, как правило, получить нельзя (см. разд. Г,2.2.1). Алифатические нит-
росоединения труднодоступны; кроме того, алифатические амины в общем случае
легко получать из спиртов или алкилгалогенидов и аммиака (см. разд. Г,2.6.4) либо
каталитическим гидрированием нитрилов (разд. Г,7.3.2).
В промышленности ароматические амины также получают восстановлением нитросоеди-
нитросоединений (разд. Г,5.1.3). Восстановление проводят как каталитически (на меди), так и по Бешану.
Наиболее важным продуктом является анилин, перерабатываемый далее в красители, лекар-
лекарственные препараты (ацетанилид, сульфамидные препараты; см. разд. Г,8.5), ускорители
вулканизации (например, меркаптобензотиазол) и антиоксиданты. О техническом примене-
применении ароматических аминов см. также разд. Г.5.1.3.
До аминов могут быть восстановлены не только нитросоединения, но и нит-
розо- и изонитрозосоединения (оксимы). В связи с большой ролью ацилирован-
ных аминомалоновых эфиров в органическом синтезе (см. разд. Г,7.1.4.3) ниже
описывается получение диэтилового эфира ацетаминомалоновой кислоты.
Ф Получение диэтилового эфира ацетаминомалоновой кислоты. [Shaw К. N. F., Nolan Ch. J.
Org. Chem., 22, 1957, 1668; Zamblto A. J., Howe E. E. In: Org. Syntheses, Coll. Vol. V, 1973,
373.] В трехгорлой колбе с насадкой Аншютца, мешалкой, обратным холодильником и
термометром готовят раствор 0,15 моль аддукта диэтилового эфира изонитрозомалоновой кис-
кислоты и ацетата натрия в 250 мл ледяной уксусной кислоты и 300 мл ацетангидрида. Смесь нагре-
нагревают до 80 °С и постепенно прибавляют 80 г цинковой пыли такими порциями, чтобы темпера-
температура была в пределах 110-115 "С. После окончания добавления цинковой пыли нагревают еще
30 мин при той же температуре. Еще горячий раствор быстро фильтруют (лучше всего через во-
воронку с электрическим обогревом), осадок дважды промывают 70 мл кипящей ледяной уксусной
кислоты (тяга!).
Растворитель удаляют в вакууме (в конце нагревают на кипящей водяной бане). А остаток
кипятят с дихлорметаном. Экстракт промывают насыщенным раствором хлорида натрия, затем
раствором NaHCO3 и сушат сульфатом магния. Растворитель отгоняют, остаток сырого эфира
перекристаллизовывают из изопропанола. Т. пл. 97 °С; выход 80%.
Получение диэтилового эфира ацетаминомалоновой кислоты каталитическим
гидрированием: Vignau M. Bull. Soc. Chim. France. 1952, 638.
8.2. РЕАКЦИИ АЗОТИСТОЙ КИСЛОТЫ
Наиболее важным нитрозосоединением является неустойчивая азотистая кис-
кислота. Ее аналогами в ряду карбонильных соединений являются карбоновые кис-
кислоты. Как можно ожидать на основании этой аналогии, «карбонильная актив-
активность» азотистой кислоты мала, поскольку частичный положительный заряд
атома азота нитрозогруппы в значительной степени компенсируется +А/-эф-
фектом гидроксильной группы (см. разд. Г,7).
Поэтому для ускорения взаимодействия с основаниями необходим кислый
катализ: +NO,e _____
== Н2О + Q=N-O-N=O
© _ _
H-O-N=O
i
H
IV
© _
Н2О + N=0
[Г.8.12]
238 Г Препаративная часть (продолжение)
В соединении II, как и в III и IV (схема [Г.8.12]), реакционная способность к
нуклеофильной атаке повышена. (Почему? Объясните!)
В этих условиях азотистая кислота обладает большей реакционной способ-
способностью, чем карбоновые кислоты. Она реагирует, Например, с С—Н-кислотны-
ми соединениями, давая изонитрозосоединения (оксимы) (см. разд. Г,8.2.3).
Аналогичная же реакция с карбоновыми кислотами не происходит.
Катализируемые кислотами реакции азотистой кислоты далее упрощенно
рассматриваются как идущие через нитрозилкатион III.
Какое из веществ — II, III или IV в схеме [Г.8.12] — выступает в роли нитро-
зирующего средства, зависит от концентрации кислоты в растворе (о механизме
диазотирования см. литературу в разд. Г,8.6)?
8.2.1. РЕАКЦИИ АЗОТИСТОЙ КИСЛОТЫ С АМИНАМИ
Внимание! При реакциях азотистой кислоты с алифатическими аминами и их
производными образуются соединения (нитрозамины, нитрозамиды, диазоалка-
ны), обладающие канцерогенной активностью. Поэтому при работе с этими
веществами необходима крайняя осторожность. Работать только под тягой,
надевать защитные перчатки!
Согласно схеме [Г.8.13], взаимодействие азотистой кислоты с первичными
аминами начинается с образования нитрозосоединения. Так как в III (схема
[Г.8.13]) имеется еще один водородный атом у атома азота, то промежуточный
продукт III перегруппировывается в изонитрозосоединение IV (гидроксидиазо-
соединение):
R-NH2 + N.=0 R-NH2-N=O ~ R-NH-N=O
— н
_ _ _ +н® _ _ © -н2о © _
R-N=N-OH ^^ R-N=N-OH2 - R-N=N
— _н© ~ +н2о
IV V VI
[Г.8.13]
В кислой среде, необходимой для осуществления реакции, гидроксидиазосо-
единение IV присоединяет протон и отщепляет молекулу воды, образуя диазо-
ниевый катион VI. В последнем уже как бы заложена структура молекулярного
азота. Поэтому соли диазония легко отщепляют азот, в частности тогда, когда
диазогруппа не находится в сопряжении со способной к мезомерии системой
или когда катион не может легко перейти в подобную структуру.
В зависимости от структуры амина промежуточно образующийся катион ди-
диазония претерпевает следующие превращения.
Если исходным веществом был алифатический первичный амин (R = алкил), то
отщепление азота происходит уже при температуре <0 °С. Оставшийся карбение-
вый ион стабилизируется обычным образом (см. разд. Г,2.1.1), т. е. путем нуклео-
фильного замещения при взаимодействии с растворителем (чаще всего водой;
схема [Г.8.14]), частично путем депротонирования он переходит в олефин. Перед
этим карбениевый ион может изомеризоваться в энергетически более выгодный
ион, так что, например, из н-пропиламина образуется преимущественно изопро-
пиловый спирт, а в небольших количествах также циклопропан D-5%):
8. Реакции других гетероаналогов карбонильных соединений
239
©
+ Н20
© _
H3C-CH2-CH2-N=N
Н3С СН2
н3с-сн-сн3
©
н3с-сн2-сн2-он
он
н3с-сн-сн3
Н2С=СН-СН3
[Г.8.14]
Аналогично первичные амиды кислот дезаминируются при действии азотис-
азотистой кислоты с образованием карбоновых кислот (метод мягкого гидролиза ами-
амидов кислот).
При взаимодействии эфиров а-аминокислот, а также а-аминокетонов с азо-
азотистой кислотой при низких температурах дезаминирование не происходит, а
отщепляется протон от а-углеродного атома, активированного карбонильной
группой, с образованием а-диазоэфиров или а-диазокетонов.
Данная реакция лучше всего может быть проиллюстрирована на примере
эфира глицина:
10.
~\ © _
C-CH-N34 -ъ
RO' н "Н
^ е © _
C-CH-NEN
C-CH = N=N. —
o'
диазоуксусный эфир
C=CH-NEN
[Г.8.15]
C-CH-N=N
RO
При этом образуется сопряженная система, охватывающая всю молекулу,
происходит значительная делокализация электронов, так что получающееся
диазосоединение достаточно устойчиво.
ос-Диазокетоны и диазомалоновые эфиры (формулы [Г.8.16]) еще более
устойчивы, чем а-диазокарбоновые эфиры (почему?).
°л э © _
C-C-NHN
R' R'
диазокетон
ROOC4 @ _
©IC-NHN
ROOC'
диазомалоновый
эфир
[Г.8.16]
Диазокетоны можно получить и другим путем — действием диазометана на
ацилхлориды (см. разд. Г,8.4.2.2), а также путем «переноса диазогрупп» с
использованием соединений с активной метиленовой группой и реакционнос-
пособных органических азидов. Информацию об этом см. в списке литературы
в конце главы.
Образующиеся при взаимодействии первичных ароматических аминов с азо-
азотистой кислотой (диазотировании) соли диазония — относительно устойчивые
соединения благодаря сопряжению диазониевой группы с ароматическим
ядром. При нагревании эти соединения разлагаются аналогично алкилдиазони-
евым солям (см. разд. Г,8.3.1).
240 Г Препаративная часть (продолжение)
Диазотирование, как правило, проводят в водном растворе, получая азотис-
азотистую кислоту из нитрита натрия и минеральной кислоты. Полученные соли диа-
зония обычно не выделяют, а прямо в растворе перерабатывают дальше.
Поскольку основность ароматических аминов на несколько порядков ниже,
чем основность алифатических аминов, то при реакциях диазотирования осо-
особую важность приобретает активирование азотистой кислоты минеральными
кислотами (см. схему [Г.8.12]). Избыток кислоты необходим, кроме того, для
предотвращения сочетания образовавшейся соли диазония с еще не прореаги-
прореагировавшим свободным амином (образование триазенов; см. схему ([Г.8.33]). В то
же время реагировать с азотистой кислотой может лишь свободный амин, имею-
имеющийся в некотором количестве в равновесии со своей солью.
Первичные ароматические амины, основность которых примерно равна
основности анилина, в общем случае можно диазотировать в разбавленных ми-
минеральных кислотах B,5—3 эквивалента кислоты на моль амина и нитрита нат-
натрия). Менее основные амины требуют более высокой концентрации кислоты.
Например, 2,4,6-тринитроанилин, основность аминогруппы которого пример-
примерно соответствует основности амида карбоновой кислоты, диазотируют в кон-
концентрированной серной кислоте.
Слабоосновные и поэтому трудно диазотируемые амины, а также тетраазо-
тируемые диамины диазотируют в так называемой «нитрозилсерной кислоте».
В большинстве случаев амин растворяют в ледяной уксусной кислоте и прибав-
прибавляют по каплям при энергичном перемешивании к «нитрозилсерной кислоте».
В других случаях применяют вариант Б общей методики (см. ниже). При диазо-
тировании аминов, в молекулах которых имеются кислотные группировки,
например —SO3H, рекомендуется добавлять кислоту к раствору натриевой соли
сложного амина и нитрита натрия (обратный способ диазотирования).
Твердые соли диазония можно получить, проводя диазотирование при помо-
помощи эфиров азотистой кислоты (см. разд. Г,8.2.2) в безводных кислых растворах
(ледяной уксусной кислоте или абсолютном спирте, насыщенных хлороводоро-
дом). Соли диазония в этом случае осаждают, добавляя эфир. В сухом виде они
взрывоопасны (чувствительны к ударам и нагреванию!). Следует избегать выде-
выделения солей диазония в виде хлоридов!
Гораздо удобнее получать устойчивые тетрафторбораты диазония, которые
при диазотировании в 40%-ной тетрафторбороводородной кислоте выпадают
в виде труднорастворимых солей. Последние можно также получать при до-
добавлении раствора тетрафторбората натрия или аммония к раствору соли хло-
хлорида диазония. Если добавлять гексафторфосфат аммония к раствору соли
хлорида диазония, то получают выпадающие в осадок устойчивые гексафтор-
фосфаты диазония.
Получение арилдиазонийтетрафторборатов: Dunker M. F. W., Starkey E. В.,
Jenkins G. L. J. Am. Chem. Soc, 1936, 58, 2308.
(ГП) Общая методика получения растворов диазотированных ароматических аминов
А. Диазотирование нитритом натрия. В колбе или стакане растворяют при добавлении по
каплям (жидкость) или порциями (твердое соединение, предварительно тонко измельченное)
1 моль первичного ароматического амина в 2,5—3 моль разбавленной A:1) концентрирован-
концентрированной соляной кислоты (вместо нее можно использовать разбавленные концентрированные
8. Реакции других гетероаналогов карбонильных соединений 241
бромоводородную A:1) или серную A:2) кислоту1, при этом температура не должна подни-
подниматься выше 50 "С. После этого раствор амина при интенсивном перемешивании быстро ох-
охлаждают до 0 °С смесью льда с поваренной солью и добавляют эквивалентное количество
2,5 М раствора нитрита натрия. Добавление ведут медленно, с такой скоростью, чтобы темпе-
температура не поднималась выше 5 "С. Перед окончанием диазотирования делают пробу на свобод-
свободную азотистую кислоту (синее окрашивание от капли раствора, нанесенной на иодкрахмаль-
ную бумажку). Нитрит прибавляют до тех пор, пока проба не будет положительной через 5 мин
после добавления очередной порции. Поскольку избыток свободной HNO2 может мешать
последующим реакциям, азотистую кислоту разрушают, добавляя небольшое количество
сульфаминовой кислоты. Если при растворении амина в минеральной кислоте из-за слишком
большой концентрации выпадает соль или амин не полностью растворяется в кислоте, то ди-
азотируют при эффективном перемешивании суспензию этой соли. Тонкую суспензию мел-
мелкокристаллической соли получают при интенсивном перемешивании и нагревании выпавшей
соли с последующим быстрым охлаждением (см. выше). Поскольку в гетерогенной системе
реакция идет медленнее, то в этих случаях необходимо эффективное перемешивание.
Методика пригодна и для диазотирования в полумикроколичествах.
Б. Диазотирование «нитрозилсерной кислотой» (нитрозилгидросульфатом). К 10 мл концен-
концентрированной серной кислоты при эффективном перемешивании и охлаждении добавляют
порциями 0,01 моль мелкоизмельченного нитрита натрия так, чтобы температура не поднима-
поднималась выше 10 °С и чтобы не образовывались нитрозные газы. Затем смесь перемешивают
10 мин при 15-20 °С (охлаждая водой) и нагревают до 70 °С. Как только образуется прозрач-
прозрачный раствор, его охлаждают и к нему при перемешивании и охлаждении добавляют 0,01 моль
мелкоизмельченного слабоосновного амина. При добавлении температуру поддерживают в
интервале от 10 до 20 °С, а затем перемешивают еще 3 ч при комнатной температуре. Для раз-
разложения избытка нитрита натрия добавляют немного (на кончике шпателя) мочевины или
сульфаминовой кислоты и оставляют на 30 мин.
Со вторичными аминами азотистая кислота образует нитрозамины:
R." _ _ «. _ _ ©
N-N=0 N-N=0 + ЬГ [Г 8 17]
R® R'
Из N-монозамещенных (вторичных) амидов кислот аналогично образуются
нитрозамиды. (Напишите схему этого процесса для N-метилмочевины! Почему
метилированная аминогруппа реагирует с азотистой кислотой быстрее, чем не-
метилированная?)
N-Нитрозоалкиламины применяются для получения диазоалканов (см.
разд. Г.8.4.1).
(Су) Получение 1Ч-метил-М-нитрозомочевины
Внимание! г\1-Метил-М-нитрозомочевину хранят в темной склянке в холодиль-
холодильном шкафу, так как под действием света и тепла она может взорваться.
Технический метиламин или его гидрохлорид часто содержат диметила-
мин. При действии нитрита натрия он образует очень ядовитый и канцероген-
канцерогенный диметилнитрозамин. Работайте только под тягой!
Раствор 1,5 моль гидрохлорида метиламина и 5 моль мочевины в 400 мл воды нагревают 3 ч
с обратным холодильником. После этого к раствору добавляют 1,6 моль нитрита натрия, смесь
охлаждают до —10 °С и медленно при перемешивании выливают в смесь 600 г льда и НО г кон-
концентрированной серной кислоты, охлаждаемую снаружи смесью льда с поваренной солью.
1 При диазотировании полиаминов количество кислоты и нитрита натрия необходимо
пересчитать в соответствии с числом аминогрупп.
242 Г Препаративная часть (продолжение)
Выпавшее нитрозосоединение отфильтровывают и промывают ледяной водой. Т. пл. 124 °С
(с разложением); выход 80%. Очищают перекристаллизацией из метанола. Продукт без даль-
дальнейшей очистки можно использовать для получения диазометана.
К какому типу относится реакция, которая происходит первоначально при
взаимодействии метиламина и мочевины с образованием метилмочевины?
Почему необходим большой избыток мочевины? Объясните!
Получение N-Memwi-N-нитрозотолуолсульфонамида: De Boer Th. J., Backer H. /.,
Org. Synth., Coll. Vol. IV A963), 943.
Третичные алифатические амины в сильнокислой среде (рН<2) при комнат-
комнатной температуре, как правило, не реагируют или лишь очень медленно реагиру-
реагируют с азотистой кислотой. В слабокислой или нейтральной среде и при повышен-
повышенной температуре происходит дезалкилирование третичных аминов, приводящее
к нитрозаминам. Отщепившийся алкильный остаток окисляется до альдегида.
Бензильные остатки отщепляются легче алкильных групп.
В органическом анализе взаимодействие азотистой кислоты с аминами
используют для того, чтобы отличить первичные алифатические амины от пер-
первичных ароматических аминов. Последние могут диазотироваться и образовав-
образовавшиеся диазосоединения легко обнаруживаются с помощью реакции азосочета-
ния (см. разд. Г,8.3.3). Реакцию с азотистой кислотой применяют также для
отделения и качественного обнаружения вторичных аминов в присутствии пер-
первичных или третичных. Если азотистой кислотой обработать смесь первичного,
вторичного и третичного аминов, то первичные амины дезаминируются, а тре-
третичные остаются без изменения. Образующиеся из вторичных аминов нитроза-
мины имеют желтую окраску, перегоняются с водяным паром и растворимы в
эфире. При нагревании с кислотами они вновь расщепляются на вторичные
амины и азотистую кислоту. Ароматические нитрозамины склонны к перегруп-
перегруппировке в и-нитрозоариламины.
Дезаминирование первичных аминов азотистой кислоты сопровождается
выделением азота, количество которого можно определить по объему; на этом
основан метод Ван-Слайка для количественного определения соединений,
содержащих первичную аминогруппу (алифатических и ароматических аминов,
аминокислот, незамещенных амидов).
8.2.2. РЕАКЦИИ АЗОТИСТОЙ КИСЛОТЫ СО СПИРТАМИ (ЭТЕРИФИКАЦИЯ)
Взаимодействие азотистой кислоты со спиртами аналогично ее взаимодействию
со вторичными аминами. В результате образуются эфиры азотистой кислоты:
©_ © -Н®
R-O-H + N=0 ===== R-O-N=O :=^=^ R-O-N=O
" "" Д +н@ эфир [Г.8.18]
азотистой
кислоты
Как этерификация азотистой кислотой, так и гидролиз ее эфиров протекают
значительно быстрее, чем соответствующие реакции с карбоновыми кислотами.
Эфиры азотистой кислоты часто используются вместо самой азотистой кис-
кислоты в реакциях нитрозирования, например в тех случаях, когда хотят провести
реакцию в неводной среде (см. разд. Г.8.2.1, получение твердых солей диазония)
8. Реакции других гетероаналогов карбонильных соединений 243
или если необходимо проводить реакцию в щелочной среде (см. разд. Г,8.2.3;
нитрозирование С—Н-кислотных соединений).
((Tj) Получение изоамилнитрита
П Осторожно! Вдыхание паров эфиров азотистой кислоты приводит к сильному
Jj расширению периферических сосудов (прилив крови к голове).
В стакане смешивают 1 моль изоамилового спирта с раствором 1,1 моль нитрита натрия в
140 мл воды и при перемешивании охлаждают до 0 °С (смесью льда с поваренной солью). Затем
при эффективном перемешивании, не давая температуре подниматься выше 5 °С, медленно
добавляют из капельной воронки 90 мл концентрированной соляной кислоты. Реакционную
смесь выливают в делительную воронку емкостью 1 л, встряхивают с 400 мл воды, водный слой
отделяют, а органический промывают разбавленным раствором карбоната натрия и затем еще
несколько раз водой. Продукт реакции сушат небольшим количеством хлорида кальция и
перегоняют в вакууме, используя охлаждаемый приемник. Т. кип. 30 °С E0 мм рт. ст.); желтое
маслянистое вещество; выход 75%.
8.2.3. РЕАКЦИИ АЗОТИСТОЙ КИСЛОТЫ С С-Н-КИСЛОТНЫМИ СОЕДИНЕНИЯМИ
С азотистой кислотой могут реагировать и С—Н-кислотные соединения. При
этом осуществляются превращения того же типа, что и при катализируемой кис-
кислотами альдольной конденсации (схема [Г.7.104]). Реакция может происходить
только с реакционноспособными метиленовыми компонентами, имеющими в
а-положении по меньшей мере одну кетонную или нитрогруппу либо две
карбоксильные или сложноэфирные группы:
НО.© I 01 0 1 .0
- C-C-N C-C-N 1.1.6.19]
/ | - -н® / | ~
Если у углеродного атома, при котором оказывается нитрозогруппа, остает-
остается еще хотя бы один атом водорода, то образовавшееся нитрозосоединение тот-
тотчас же превращается в изонитрозосоединение в соответствии со схемой [Г.8.3].
Для соединений с малоактивными метиленовыми компонентами реакция
катализируется сильными основаниями (алкоголятами щелочных металлов).
В этих условиях, естественно, нельзя использовать азотистую кислоту (почему?)
и поэтому применяют ее сложные эфиры. Превращение аналогично сложно-
эфирной конденсации Кляйзена. (Напишите схему реакции!)
©Получение диэтилового эфира изонитрозомалоновой кислоты. К раствору 1 моль свежепе-
регнанного диэтилового эфира малоновой кислоты в 170 мл ледяной уксусной кислоты до-
добавляют в течение 3—4 ч, поддерживая температуру около 0 °С, при энергичном перемеши-
перемешивании раствор 3 моль нитрита натрия в 250 мл воды. После этого реакционную смесь пере-
перемешивают еще 10 ч при комнатной температуре. Образовавшийся изонитрозомалоновый эфир
экстрагируют сначала 400 мл дихлорметана, а затем еще тремя порциями по 100 мл. Объединенные
вытяжки сушат сульфатом магния и встряхивают с 10 г твердого NaHCO3 (осторожно*, выделяется
углекислый газ!). После прекращения выделения газа раствор фильтруют, добавляют 20 г порошко-
порошкообразного безводного ацетата натрия и нагревают 10 мин с обратным холодильником. Затем раст-
раствор фильтруют и упаривают вдвое; к остатку добавляют сухой петролейный эфир до помутнения и
оставляют для кристаллизации на ночь в холодильнике. Кристаллизуется аддукт изонитрозомало-
нового эфира с ацетатом натрия C : 1); выход составляет 75%; т. пл. 88 'С.
1 Аддукт с '/з моль ацетата натрия [Shaw К. N. F., Nolan Ch. J. Org. Chem., 1957, 22, 1966].
244 Г Препаративная часть (продолжение)
В препаративной химии нитрозирование С—Н-кислотных соединений
применяют для получения а-аминокарбонильных соединений (восстановлени-
(восстановлением) и сс-дикарбонильных соединений (гидролизом образовавшихся моноокси-
мов).
Одной из важнейших реакций для синтеза ос-аминокарбоновых кислот (см.
синтез триптофана [Г.7.68], [Г.7.146]) является восстановление и одновремен-
одновременное ацилирование описанного выше изонитрозомалонового эфира. При этом
образуются ацетил- или формиламинодиэтилмалоновый эфир (методику см. в
разд. Г,8.1). (Напишите схемы этих реакций, а также схему получения диацети-
ла из метилэтилкетона.)
Реакции азотистой кислоты с С-Н-кислотными соединениями применяются в аналити-
аналитической химии для идентификации и разделения первичных и вторичных алифатических нит-
росоединений через нитроловые кислоты или соответственно псевдонитролы: бесцветные
нитроловые кислоты растворяются в щелочах с образованием солей темно-красного цвета.
Псевдонитролы, окрашенные в сине-зеленый цвет, солей не образуют.
,14-ОН
R-CH2-NO2 + HNO2 R-C' + Н2О
NO2 [Г.8.20]
нитроловая
кислота
CH-NO2 + HNO2 R.'°4N0 + Н2О [Г.8.21]
R' 2
псевдонитрол
8.3. РЕАКЦИИ СОЛЕЙ ДИАЗОНИЯ
Соли диазония, образующиеся при диазотировании первичных ароматических
аминов, могут реагировать с сохранением группировки N=N или с ее потерей,
сопровождающейся замещением на другую группу.
8.3.1. ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ И ВОССТАНОВЛЕНИЕ
При нагревании или освещении УФ-светом ароматические диазосоединения
теряют элементный азот. В водных средах образовавшийся фенил-катион1 глав-
главным образом дает фенол. Если нагревать растворы, содержащие хлорид- или
бромид-ионы, то в качестве побочных продуктов в небольшом количестве обра-
образуются и галогенопроизводные. Из галогенов гладко вводится только анион
фтора при нагревании тетрафторбората диазония в среде инертного растворите-
растворителя (реакция Шимана):
1 Эта реакция полностью соответствует разложению алифатических диазониевых ионов,
однако из-за большей устойчивости арилдиазониевых ионов она не происходит спонтанно
при комнатной температуре.
8. Реакции других гетероаналогов карбонильных соединений 245
© _
-n2
+ М [Г.8.22]
w F + BF3
О получении из солей диазония других арилгалогенидов см. разд. Г,8.3.2.
При кипячении солей диазония со спиртами наряду с образованием
алкилариловых эфиров наблюдается также конкурентная реакция — восстановле-
восстановление солей диазония до углеводородов. В таких растворителях, как циклические
эфиры (диоксан, тетрагидрофуран и др.), а также в диметилформамиде эта реак-
реакция становится главной (восстановление по Меервейну). При этом в результате
радикального цепного процесса на арильныи остаток переносится атом водорода:
О
• + Су _ (^ун + Гу [Г.8.236]
/Л|_| /"» |_| V©
Г~ О + <^^NEN /~\- + N2 + [О [Г.8.23В]
По сравнению с этой реакцией более старый способ превращения солей диа-
диазония в соответствующие ароматические углеводороды с помощью щелочного
раствора станнита натрия кажется менее целесообразным.
Выходы фенолов при кипячении солей диазония низки. Этим путем фенолы
получают главным образом тогда, когда необходимо синтезировать строго опре-
определенный изомер фенола или когда невозможно получить его иным путем.
Ф Общая методика превращения солей диазония в фенолы (табл. Г.8.24). Раствор соли диа-
диазония, полученный из 0,5 моль амина по методике, описанной в разд. Г,8.2.1 (вариант
А), нагревают на кипящей водяной бане до прекращения выделения азота. Образовав-
Образовавшийся фенол отгоняют с водяным паром до тех пор, пока дистиллят не перестанет давать по-
положительную реакцию с хлоридом железаA II). Дистиллят насыщают хлоридом натрия и фенол
экстрагируют эфиром. Эфирные вытяжки сушат сульфатом магния и фенол выделяют пере-
перегонкой в вакууме.
Методика пригодна и для полумикроколичеств.
Кроме рассмотренного выше восстановления солей диазония до углеводоро-
углеводородов, их можно восстановить также и с сохранением азота в молекуле. При таком
восстановлении водород присоединяется по двойной связи N=N и образуются
1 Реакция инициируется следами ионов металлов в низших степенях окисления или про-
проведением процесса в буферном растворе.
246
Г Препаративная часть (продолжение)
Таблица Г.8.24. Получение фенолов кипячением
Фенол
Фенол
м- Крезол
о-Крезол
л-Крезол
jw-Хлорфенол
л-Хлорфенол
л/-Гидроксибензальдегида
Гваякол6
Амин
Анилин
л«-Толуидин
о-Толуидин
я-Толуидин
jw-Хлоранилин
л-Хлоранилин
л<-Аминобензальдегид
о-Анизидин
эастворов солей диазония
Т. кип.,
°С (мм рт. ст.)
74A0)
86A5)
93B3)
96A5)
55C)
88E)
168A7)
105 B5)
Т. пл., °С
43
(ио20 1,5364)
31
36
32
42
108 (вода)
30
Выход, %
60
60
60
60
65
60
55
50
а После кипячения раствор соли диазония фильтруют горячим, оставшуюся смолу кипятят с водой и из
объединенных фильтратов извлекают эфиром альдегид, который не перегоняется с водяным паром.
6 К раствору соли диазония на холоду прибавляют концентрированную серную кислоту из расчета
300 мл серной кислоты на моль амина и нагревают на металлической бане до 125—130 °С (температура
внутри колбы). При этой температуре гваякол постепенно отгоняется с водяным паром.
арилгидразины. В качестве восстановителей применяют сульфит натрия, цинк в
ледяной уксусной кислоте или дихлорид олова в соляной кислоте (ср., однако,
выше с восстановлением до углеводородов раствором станнита).
Ниже приводится схема восстановления фенилдиазонийхлорида сульфитом натрия до
фенилгидразина:
С/"Е*
СГ + Na2SO3
-NaCI
// ^ © -
SO3Naw
J=N-SO3Na
фенилдиазосульфонат
натрия
[Г.8.25]
H2SO4 + NaHSO4
SO3H
) Общая методика получения арилгидразинов (табл. Г.8.26)
Внимание! Арилгидразины очень ядовиты, попадание на кожу вызывает
болезненные нарывы! Синтез следует проводить только под хорошо действу-
действующей тягой.
Вариант А
1. Приготовление раствора сульфита натрия. В раствор 50 г A,25 моль) гидроксида натрия
в 375 мл воды пропускают SO2 до тех пор, покарН не достигнет значения 6,0-6,5 порН-инди-
каторной бумаге!
2. Восстановление раствора арилдиазонийхлорида. В стакан емкостью 1 л, снабженный ме-
мешалкой и капельной воронкой, помещают раствор сульфита натрия и при перемешивании до-
добавляют порциями охлажденный раствор хлорида диазония, полученный из 0,1 моль амина
(получение см. разд. Г,8.2.1, вариант А, с. 240). После добавления каждой порции проверяют
рН раствора и прикапыванием 30%-ного раствора гидроксида натрия доводят рН до 6,0—6,8.
По окончании прибавления соли арилдиазония доводят рН до 6,5, реакционную смесь, окра-
8. Реакции других гетероаналогов карбонильных соединений
247
Таблица Г.8.26. Получение арилгидразинов восстановлением солей арилдиазония
Гидразин
Фен ил гидразин
л-Толилгидразин
jm-Тол ил гидразин
З-Метоксигидразин
4-Хлорфенилгидразин
З-Хлорфенилгидразин
2-Хлорфенилгидразин
4- Бромфенил гидразин
Исходный амин
Анилин
л-Толуидин
.м-Толуидин
л-Анизидин
4-Хлоранилин
З-Хлоранилин
2-Хлоранилин
4-Броманилин
Вариант
А, Б
А, Б
А, Б
А, Б
А, Б
А, Б
А, Б
А, Б
Т. кип., °С (мм рт. ст.)
или т. пл.. °С
120 A2); т. пл. 19,
nD20 1,6084
Т. пл.54
96B)
105A)
Т. пл. 84,5
89 @,5)
95A)
Т. пл.102
шенную в красный цвет, переносят в круглодонную колбу емкостью 1 л и нагревают с обрат-
обратным холодильником до обесцвечивания B—4 ч). При этом через 40 мин добавлением соляной
кислоты доводят рН горячей смеси до -6,5. После охлаждения реакционную смесь выливают
при перемешивании в концентрированную соляную кислоту (взятую в количестве '/г объема
реакционной смеси); при этом выпадает гидрохлорид гидразина. Смесь охлаждают до О "С и
оставляют на 3 ч при этой температуре; кристаллизация при этом завершается. Осадок от-
отфильтровывают и при необходимости перерабатывают полученный сырой продукт.
3. Очистка. Сырой продукт растворяют или суспендируют в 70 мл воды, подщелачивают
раствором гидроксида натрия и отделившийся гидразин тотчас же экстрагируют хлористым
метиленом D х 30 мл). Растворитель отгоняют на водяной бане при нормальном давлении
(азеотропное высушивание), остаток перегоняют в вакууме или в случае твердого вещества пе-
рекристаллизовывают из петролейного эфира.
Вариант Б
В трехгорлой колбе емкостью 500 мл, снабженной мешалкой, капельной воронкой и тер-
термометром, охлаждают раствор 0,3 моль дигидрата хлорида олова(П) в 70 мл соляной кислоты
(ч. д. а.) до температуры от — 10 до -15 °С (смесь ацетона с сухим льдом). Затем при этой темпе-
температуре прикапывают при перемешивании раствор хлорида диазония, полученный из 0,1 моль
амина с использованием соляной кислоты (ч. д. а.).
Для завершения реакции смесь оставляют на ночь в холодильнике. Затем выпавшую двой-
двойную соль олова отфильтровывают, растворяют или суспендируют в 70 мл воды и добавляют
концентрированный раствор гидроксида натрия до сильнощелочной реакции.
Гидразин экстрагируют эфиром D х 40 мл), объединенный эфирный экстракт сушат суль-
сульфатом магния, растворитель отгоняют, а остаток фракционируют в вакууме или перекристал-
лизовывают из петролейного эфира. Выход около 70%.
Фенилгидразины являются важными реактивами для идентификации альде-
альдегидов, кетонов, Сахаров (см. разд. Г,7.1.1), применяются также в синтезе индолов
по Фишеру (см. разд. Г,9.2). В промышленности их используют в больших коли-
количествах для получения производных пиразолона, которые применяются в каче-
качестве лекарственных препаратов (см. разд. Г,7.1.4.2) или являются компонентами
в производстве красителей (см. разд. Г,8.3.3).
8.3.2. РЕАКЦИЯ ЗАНДМЕЙЕРА
В тех случаях, когда заместитель (например, бром) не может быть введен в аро-
ароматическое ядро вместо диазогруппы при простом кипячении раствора соли ди-
диазония, часто оказывается возможным осуществить эту реакцию при добавле-
добавлении порошкообразной меди или солей медиA) (реакция Зандмейера).
248
Г Препаративная часть (продолжение)
© — га
NHN + Cuu
f\ + ICH0
ее + Си20
Си
,2©
^Л-Cil + е0
Си4
[Г.8.27а]
[Г.8.276]
[Г.8.27в]
При этом в качестве побочных продуктов образуются производные дифени-
ла, что указывает на радикальный характер реакции1 (ср. с образованием ради-
радикалов при окислительно-восстановительных процессах; см. разд. Г,II)-
Ион меди играет роль только переносчика электронов. В связи с этим стано-
становится понятным, почему при введении легко окисляющихся или обратимо
окисляющихся нуклеофильных реагентов катализатор Зандмейера становится
излишним. Так обстоит дело, например, с иодид-ионом, который в известной
мере является собственным катализатором процесса. (В качестве побочного
продукта всегда образуется некоторое количество иода.) То же можно сказать и
об арсенитной группе, при введении которой происходит восстановление кати-
катиона диазония до радикала арилдиазония. Последующая цепная реакция приво-
приводит к образованию ариларсоновой кислоты (происходит необратимое окисле-
окисление арсенитной группы). Приведенные ниже схемы иллюстрируют некоторые
возможности реакций замещения диазогруппы:
-N2
+ Br°;Cu0
+ CN0;Cu0
+ Asof +H2O
+ SO2 +H®+Cu
CEN
[Г.8.28]
-Си*®
B0-«"
Значение этой реакции состоит в том, что с ее помощью можно вводить в
ароматическое ядро при посредстве нитрогруппы с последующим восстановле-
восстановлением и диазотированием такие заместители, которые невозможно ввести пря-
1 Диарилы являются главным продуктом реакции, когда соли диазония действием щело-
щелочей переводят в диазоангидриды (Ar—N=NJO; последние экстрагируют ароматическими
растворителями, в которых они разлагаются. Образующийся арильный радикал вступает в
реакцию с растворителем (получение диарилов арилированием по Гомбергу-Бахману).
8. Реакции других гетероаналогов карбонильных соединений
249
Таблица Г.8.29. Реакция Зандмейера
Продукт реакции
о-Хлортолуол
л<-Хлортолуол
л-Хлортолуол
о-Бромтолуол
ти-Бромтолуол
л-Бромтолуол
о-Хлорнитробензол
ти-Хлорнитробензол
ти-Бромнитробензол
л- Бромнитробензол
Бензонитрил
л-Метилбензонитрил
о-Хлорбензонитрил
л-Хлорбензонитрил
л-Нитробензонитрил
л-Хлориодбензол
Иодбензол
л-Иодтолуол
л-Бромиодбензол
л-Иоданизол
л-Иоднитробензол
Исходное
вещество
о-Толуидин
л/-Толуидин
л-Толуидин
о-Толуидин
ж-Толуидин
л-Толуидин
о-Нитроанилин
л<-Нитроанилин
л<-Нитроанилин
л-Нитроанилин
Анилин
л-Толуидин
о-Хлоранилин
л-Хлоранилин
л-Нитроанилин
л-Хлоранилин
Анилин
л-Толуидин
л-Броманилин
л-Анизидин
л-Нитроанилин
Т. кип., °С (мм рт. ст.)
или т. пл., °С
158
47A7)
44A0)
78 B0)
71A5)
82 C5); 26 (т. пл.)
33 (т. пл., этанол)
45 (т. пл., этанол)
55 (т. пл., этанол)
125 (т. пл., этанол)
70A0)
91 A1); 29 (т. пл.)
43 (т. пл., этанол)
90 (т. пл., этанол)
146 (т. пл., этанол)
56 (т. пл., этанол)
64A0)
133B5); 35 (т. шт.,этанол)
92 (т. пл., этанол)
52 (т. пл., этанол)
174 (т. пл., этанол)
ло20
1,5247
1,5214
1,5221
1,5565
1,5528
1,5289
Выход, %
80
80
80
60
60
60
90
90
90
90
60
60
70
80
75
65
65
80
65
70
75
мым замещением, и в такие положения, в какие они обычно не вводятся. Побоч-
Побочными продуктами могут быть фенолы, диарилы или азосоединения. Обсудите
эти побочные реакции!
©Общая методика получения ароматических хлоридов, бромидов и нитрилов по Зандмейе-
ру (табл. Г.8.29)
• Внимание! При получении нитрилов образуется свободная синильная кислота!
Работать под очень хорошей тягой и в специальном противогазе (дыхательный
фильтр В) (см. часть Е). Цианиды металлов очень ядовиты!
Приготовление медного катализатора. В круглодонной колбе растворяют при нагревании
1 моль сульфата меди (учесть содержание кристаллизационной воды!) в 800 мл воды, к раство-
раствору прибавляют 1,5 моль хлорида (при получении хлоридов) или бромида (при получении бро-
бромидов) натрия. К этому раствору медленно, при перемешивании добавляют раствор 0,5 моль
сульфита натрия в 200 мл воды. Дают смеси охладиться, осадок промывают водой (декантаци-
(декантацией) и растворяют его в 400 мл концентрированной соляной (соответственно бромоводород-
ной) кислоты. Полученный раствор хранят в хорошо закрытом сосуде, так как соль медиA)
чувствительна к кислороду воздуха.
Аналогично можно получить цианид медиA). При этом, однако, сначала проводят восста-
восстановление и лишь затем добавляют цианид натрия. После промывания водой осадок растворя-
растворяют в 600 мл 4,5 М раствора цианида натрия. Осторожней.
Реакция Зандмейера. Реакцию проводят в стакане (осторожно: сильное вспенивание, выде-
выделяется азот!) После прекращения выделения азота реакционную смесь переносят в круглодон-
ную колбу, снабженную насадкой для перегонки с паром. Для получения продуктов, имеющих
высокую т. пл., при перегонке с паром используют колбу (НШ 29/32), с воздушным холодильни-
холодильником, герметично соединенным с двугорлой колбой, снабженной обратным холодильником
250 Г Препаративная часть (продолжение)
Димрота и играющей роль приемника дистиллята. Если вещество затвердевает при перегонке в
воздушном холодильнике, то его расплавляют лабораторным феном, после чего оно попадает в
приемник. Благодаря обратному холодильнику водяной пар конденсируется в приемнике.
Диазотируют 0,75 моль амина по методике, описанной в разд. Г,8.2.1 (вариант А, с. 240).
При этом применяют соляную кислоту при получении хлоридов, бромоводородную — при
получении бромидов и серную — при синтезе нитрилов и иодидов.
При получении хлоридов, бромидов и нитрилов, катализируемом солью одновалентной
меди, раствор соли диазония при 0 "С добавляют при перемешивании в раствор катализатора
(приготовление последнего см. выше). Реакционную смесь перемешивают 1 ч при комнатной
температуре и затем 30 мин на кипящей водяной бане.
Для получения иодидов охлажденный раствор соли диазония прибавляют по каплям в те-
течение 1 ч к водному раствору 1 моль иодида щелочного металла (на 1 г иодида натрия — 3 мл
воды, а на 1 г иодида калия — 5 мл воды). Затем перемешивают 1 ч при комнатной температу-
температуре и нагревают еще 30 мин на водяной бане. После охлаждения реакционную смесь встряхи-
встряхивают с водным раствором сульфита натрия (до исчезновения типичной окраски иода).
Для выделения продукта реакции последний перегоняют с паром. Жидкие вещества
экстрагируют из дистиллята эфиром, объединенные эфирные вытяжки промывают 2 н. раст-
раствором гидроксида натрия (для удаления образовавшихся побочно фенолов) и затем водой, су-
сушат, отгоняют растворитель и перегоняют в вакууме с небольшим дефлегматором. Твердые
продукты реакции отфильтровывают на стеклянном фильтре, промывают 2 н. едким натром и
большим количеством воды, сушат и перекристаллизовывают.
Получение тиосалициловой кислоты из антраниловой кислоты: Аллен К.,
Мак-Кей Д. В сб.: Синтезы органических препаратов. Сб. 2. — М.: ИЛ, 1949,
с. 455 (в методике сочетаются реакция Зандмейера и восстановление дисульфи-
дисульфида; см. разд. Г,8.5).
2,4-Дихлортолуол из 2,4-диаминотолуола: Hodgson H. H., Walker J. J. Chem.
Soc, 1935, 350.
Повышение выхода в реакции Зандмейера за. счет добавления восстанавлива-
восстанавливающего агента: Galli С. Tetrahedron Lett., 1980, 21, 4515.
8.3.3. А30С0ЧЕТАНИЕ. АЗОКРАСИТЕЛИ
Ионы диазония обладают электрофильными свойствами (пониженная элект-
электронная плотность) у концевого атома азота:
" <Q^ [Г.8.30]
Поэтому они способны атаковать ароматические соединения по типу электро-
фильного замещения (азосочетание). Эта реакция вполне аналогична типичным
электрофильным реакциям замещения в ароматическом ряду (нитрованию, га-
логенированию, сульфированию и т. д.).
ст-комплекс азосоединение
[Г.8.31]
Вследствие значительной делокализации положительного заряда ионы диазония не отно-
относятся к числу сильных электрофильных реагентов, и поэтому они могут вступать в реакции
8. Реакции других гетероаналогов карбонильных соединений 251
лишь с сильноосновными ароматическими соединениями. В связи с этим в реакцию азосоче-
тания вступают, как правило, только ароматические амины (сильный +А/-эффект амино-
аминогруппы) и фенолы (сильные +М- и +/-эффекты кислорода в фенолят-ионе, который,
собственно говоря, и вступает в реакцию; см. разд. Г,5.1.2). Лишь в отдельных случаях реакция
идет также с эфирами фенолов или полифенолов либо с полиалкилированными бензолами
(см. ниже).
Поскольку реакционная способность ионов диазония незначительна, они реагируют весь-
весьма селективно (см. также разд. Г,5.1.2), так что образуется почти исключительно продукт пара-
замещения и лишь небольшое количество о/ото-продукта.
В соответствии с теорией —/-и — Л/-заместители в ионе диазония повышают его реакцион-
реакционную способность при взаимодействии с ароматическими соединениями, а группы с +/- и
+М-эффектами понижают ее.
Например, фенилдиазоний-катион способен реагировать с триметиловым эфиром
флороглюцина, а я-нитрофенилдиазоний может вступать в реакцию азосочетания уже с диме-
тиловым эфиром резорцина. Соли 2,4-динитрофенилдиазония гладко сочетаются с анизолом,
и, наконец, 2,4,6-тринитрофенилдиазоний может реагировать даже с мезитиленом.
Каждая реакция сочетания протекает при своем оптимальном значении рН.
В сильнокислой среде реакция, как правило, не идет даже с ароматическими
аминами и фенолами. Концентрация свободного амина в этих случаях из-за об-
образования соли сильно понижена (почему соль диазония не атакует соль аммо-
аммония?). Точно так же концентрация иона фенолята в кислом растворе очень ма-
мала, поскольку диссоциация фенола сильно подавлена. В щелочной среде спо-
способность амина вступать в реакции электрофильного замещения проявляется
наиболее полно, а у фенола эта способность даже сильно повышается вслед-
вследствие образования фенолята, но концентрация иона диазония в этом случае
очень мала, так как в щелочной среде образуется не способный к азосочетанию
диазотат III (схема [Г.8.32]):
/Г~Л © _ +оне /Г\ +оне,-н2о /Г~\ _0
</ Vn=N - (/ V-N=N-OH -¦ <7 >—N=N-01
\=/ +н®, -н2о \=/ +н® \=/ " [Г.8.32]
i и in
При подкислении раствора диазотата можно в результате обратимой реакции получить ди-
азосоединение. Диазотаты могут существовать в двух формах — реакционноспособный
син- (Z) диазотат, находящийся в равновесии с солью диазония, и неактивный анти- (Е) ди-
диазотат. (См. об этом в учебниках.)
Таким образом, наиболее благоприятными условиями для азосочетания с
аминами является слабокислая, а с фенолами — слабощелочная среда.
С первичными или вторичными аминами соли диазония сочетаются в нейт-
нейтральной или очень слабокислой среде преимущественно по атому азота амино-
аминогруппы, являющемуся центром наибольшей электронной плотности. При этом
образуются 1,3-дизамещенные триазены. Такой триазен (осадок канареечного
цвета) образуется, например, в том случае, если при диазотировании анилина
взято слишком мало кислоты:
ytr\ ^ f\ NH_N=N-f\
1,3-дифенилтриаэан
(диазоаминобензол) [1 .o.OJj
252 Г Препаративная часть (продолжение)
Эта реакция, однако, обратима; в частности, обратный процесс наблюдается
уже при такой кислотности раствора, которая не мешает сочетанию в пара-поло-
пара-положение. Поэтому в растворе с соответствующим рН можно изомеризовать триа-
зен в аминоазосоединение. Образующиеся в результате распада диазоаминобен-
зола соединения I и II снова вступают в реакцию в соответствии со схемой
[Г.8.31].
Соли арилдиазония способны к сочетанию и с реакционноспособными али-
алифатическими метиленовыми группами, имеющимися в эфирах, амидах, анили-
дах, нитрилах р-оксокарбоновых кислот, р-дикетонах и енаминах. При этом
образуются арилгидразоны:
R О R О 0 0
С; R' — ° Хн*' ~ R' T ~R' [Г.8.34]
HV"N %'N HN'N
i i i
Ph Ph Ph
В промышленности большое значение имеет также сочетание с С-Н-кислотными пиразо-
лонами, индолами, пиридонами и другими соединениями. Смотрите об этом в учебнике!
(ПУ) Общая методика азосочетания (табл. Г.8.35)
А. Сочетание в слабокислой среде (с аминами). К раствору 0,1 моль азосоставляющей в
эквивалентном количестве 1 н. раствора гидроксида натрия постепенно, при охлаждении до
5—10 °С и перемешивании приливают раствор соли диазония, приготовленный из 0,1 моль
амина (получение см. в разд. Г,8.2.1, вариант А, с. 240). Из кислого окрашенного раствора про-
продукт выделяют нейтрализацией раствором ацетата натрия или карбоната натрия и(или) выса-
высаливанием хлоридом натрия. При высаливании раствор нагревают до 60—80 °С и при переме-
перемешивании медленно (!) смешивают с тонкоизмельченным хлоридом натрия (не более 20 г на
100 мл раствора). В зависимости от растворимости продукта его можно перекристаллизовать
из небольшого количества воды или водного этанола.
Б. Сочетание в кислой среде (с аминами). К раствору 0,01 моль азосоставляющей в 10 мл
10%-ной серной кислоты при перемешивании медленно приливают вязкий коричневый раст-
раствор соли диазония, полученный из 0,01 моль амина диазотированием нитрозилсерной кисло-
кислотой (см. разд. Г,8.2.1, вариант Б, с. 241). Добавлением льда температуру поддерживают около
0 °С. Сочетание начинается уже при смешивании компонентов и завершается примерно через
8 ч. При необходимости продукт выделяют высаливанием, как это описано в варианте А.
В. Сочетание в щелочной среде (с фенолами). К раствору 0,1 моль фенола в 0,2 моль 2 н. раст-
раствора гидроксида натрия (на каждую дополнительную ОН группу в азосоставляющей надо
дополнительно брать эквивалентное количество щелочи) медленно, при перемешивании,
поддерживая температуру 5-10 °С, прибавляют раствор 0,1 моль диазотированного амина (по-
(получение см. в разд. Г,8.2.1, вариант А). рН раствора контролируют при помощи индикаторной
бумажки и в случае необходимости прибавляют карбонат натрия, следя за тем, чтобы раствор
постоянно оставался щелочным. Полноту выпадения красителя повышают, добавляя хлорид
натрия. Для очистки краситель промывают ледяной водой.
Г. Сочетание в буферном растворе ацетата натрия (с С—Н-кислотными соединениями).
К раствору 0,1 моль С—Н-кислотного компонента в 150 моль этанола (в случае цианацетами-
да необходимо 300 мл этанола) и 0,15 моль ацетата натрия в 120 мл 50%-ного этанола медлен-
медленно, по каплям, при перемешивании и охлаждении до 0—5 °С добавляют раствор 0,1 моль
диазотированного амина (получение см. разд. Г.8.2.1, вариант А). Реакционную смесь остав-
оставляют для кристаллизации продукта. Можно также осторожно добавлять небольшими порция-
порциями воду так, чтобы краситель отделялся. Продукт промывают водой и перекристал-
лизовывают.
Таблица Г.8.35. Азокрасители и гидразоны, полученные реакцией азосочетания
Продукт реакции
4'-Диметиламинофенилазобензол-
сульфонат-4 натрия (гелиантин, ме-
метиловый оранжевый)
4'-Диметиламинофенилазобензоат-2
натрия (метиловый красный)
4'-Амино-2'-метил-5'-метокси-4-
нитроазобензол
2-B,6-Дибром-4-нитрофенилазо)-
5-диэтиламиноацетанилид
2'-Бром-4-диаэтиламино-4',6'-ди-
нитроазобензол
5-[БисB-ацетоксиэтил)амино]-
2-B-бром-4,6-динитрофенилазо)-
4-метоксиацетанилид
4-B-Гидроксинафталин-1-азо)бен-
золсульфокислота, динатриевая соль
(р-нафтолоранж)
1-D-Нитрофенилазо)нафтол-2
(пара-красный)
Диазо- и
азокомпоненты
Сульфаниловая кислота,
М,М-диметиланилин
Антраниловая кислота,
N .Ы-диметиланилин
4-Нитроанилин, 5-метил-2-ме-
токсианилин (крезидин)
2,6-Дибром-4-нитроанилин6,
3 -диэтиламиноацетанилид
2-Бром-4,6-динитроанилин6,
М,1Ч-диэтиланилин
2-Бром-4,6-динитроанилин6,
3-[бисB-ацетоксиэтил)амино]-4-
метоксиацетанилид
Сульфаниловая кислота, C-наф-
тол
4-Нитроанилин р-нафтол
Вариант
А
А
А
Б
Б
Б
В
В
Среда — цвет раствора или Х^
(lge)a;T. пл.,°С
Кислая — красный,
щелочная — желтый
Кислая — красный,
щелочная — желтый
Красный; 254 (т. пл.)
^•тах 507 D,40) (хлороформ);
168-172 (т. пл.;ДМФА)
Хтах 552 D,44) (хлороформ);
190-192 (т. пл.;ДМФА)
^-тах 594 D,54) (хлороформ);
146-149 (т. пл.;ДМФА)
Оранжевый
Красный; 246 (т. пл.; толуол)
Выход, %
80
80
85
93
88
93
80
80
Продукт реакции
4-Гидрокси-3-карбокси-4'-нитроазо-
бензол"
2-Гидроксинафтил-1-азобензол
4-[B,4-Диметилфенил)азо]-3-метил-
1 -фенил-Д2-пиразолон-5
2,4-Диоксо-3-(фенилгидразоно)пен-
тан
Этиловый эфир 3-оксо-2-(фенилгид-
разоно)бутановой кислоты
Этиловый эфир и-метоксифенил-
гидразоноциануксусной кислоты
Амид л-метоксифенилгидразоно-
циануксусной кислоты
Диазо- и
азокомпоненты
4-Нитроанилин, салициловая
кислота
Анилин, р-нафтол
2,4-Ксилидин, З-метил-1-фенил-
Д2-пиразолон-5
Анилин, ацетилацетон
Анилин, ацетоуксусный эфир
л-Анизидин, этиловый эфир
циануксусной кислоты
л-Анизидин, цианацетамид
Вариант
В
В
В
Г
Г
Г
г
Таблица Г.8.35.
Среда — цвет раствора или Хтх
(lge)a;T. пл., °С
Кислая — желтый, щелоч-
щелочная — коричневый;
258 (т. пл., уксусная кислота)
Красный; 130 (т. пл.; водный
этанол)
Желтый; 167 (т. пл.)
89 (т. пл.; этанол)
70 (т. пл.; этанол)
78 (т. пл.; этанол)
239—240 (т. шт.; уксусная кис-
кислота)
Продолжение
Выход, %
80
80
90
85
95
75
66
а ^шах длинноволновое поглощение в нм; е — коэффициент молярной экстинкции.
6 Диазотирование «нитрозилсерной кислотой» (см. разд. Г,8.2.1).
в Получается сначала в виде раствора соли щелочного металла и осаждается соляной кислотой.
8. Реакции других гетероаналогов карбонильных соединений 255
Все азосоединения в условиях восстановления нитросоединений превраща-
превращаются в первичные амины:
мо - ми_ми мо. [Г836]
Таким образом, через стадию азосочетания можно ввести аминогруппу в азо-
компонент, получая при этом о- или я-фенилендиамины или аминофенолы, ко-
которые другими путями получить трудно, например, 1-аминонафтол-2 из р-наф-
толоранжа (см. разд. Г,6.4.2).
Азосочетание нашло широкое применение в крупномасштабном производстве азокраси-
телей. Последние составляют примерно половину всех производимых промышленностью
синтетических красителей. (Ознакомьтесь в учебнике с важнейшими типами красителей!)
При взаимодействии солей диазония, полученных из разных ароматических аминов (заме-
(замещенных анилинов, нафтиламинов, других соединений), с разнообразными азокомпонентами
(анилинами, фенолами, нафтиламинами, нафтолами, пиразолонами, их сульфокислотами и
другими производными) получено очень большое число азокрасителей, причем в качестве то-
того или иного компонента реакции все чаще применяют гетероциклические соединения.
Анионные азокрасители благодаря присутствию одной или нескольких сульфогрупп раст-
растворимы в виде солей в воде. Кислотные азокрасители используются при крашении материалов
с основными группами (шерсти, шелка, полиамидных волокон, кож), но могут применяться и
как прямые субстантивные азокрасители для крашения непротравленных волокон клетчатки
(хлопка, льна, регенерированной целлюлозы, бумаги).
Растворимы в воде также и катионные (основные) азокрасители, которые приобретают все
большее значение при окраске полиакрилонитрильного и других волокон.
Малорастворимые в воде дисперсные азокрасители применяются для окраски гидрофоб-
гидрофобных волокон (полиэфирных, триацетилцеллюлозных, а также полиакрилонитрильных и по-
полиамидных). Такие красители адсорбируются волокнами из водных суспензий. Нераствори-
Нерастворимы в воде и так называемые проявляющиеся красители, которые образуются при азосочетании
компонентов непосредственно на волокне. Большое значение имеют красители ряда нафто-
нафтола AS, получаемые изариламидов 3-гидроксинафтойной кислоты (см. разд. Г, 5.1.8.6) и дру-
других ароматических гидроксикарбоновых кислот, гидроксильная группа в которых находится
по соседству с карбоксильной. В крашении текстильных материалов и пластмасс важную
роль играют также металлсодержащие комплексообразующие азокрасители. Азопигменты
(нерастворимые порошкообразные кристаллические красители), как правило, представляют
собой комплексы металлов или их нерастворимые соли.
Светочувствительные соли диазония применяются также для записи информации.
В обычной диазотипии, например, они находятся вместе с фенольными компонентами в моле-
кулярно-дисперсном состоянии в светочувствительном слое. Этот слой имеет кислую реакцию,
поэтому сочетание сначала затруднено. После освещения изображение проявляют, например,
обрабатывая парами аммиака; при этом рН повышается и неразложившаяся соль диазония мо-
может вступать в реакцию азосочетания. Основываясь на этом принципе, можно изготовить мате-
материалы для копирования, обладающие очень высокой разрешающей способностью (например,
диазомикрофильмы).
Исторически известным в химии является антибактериальный сульфамидный фармацев-
фармацевтический препарат белый стрептоцид, в структуру которого входит азогруппа (см. об этом в
учебниках и разд. Г,8.5).
Азокрасители получают также по реакции окислительного сочетания (разд.
Г.6.4.3).
256 Г Препаративная часть (продолжение)
8.4. АЛИФАТИЧЕСКИЕ ДИАЗОСОЕДИНЕНИЯ
8.4.1. ПОЛУЧЕНИЕ ДИАЗОАЛКАНОВ
Диазоалканы нельзя получать диазотированием первичных алифатических ами-
аминов, поскольку в молекулах последних нет активированного а-водородного ато-
атома, так что образовавшийся алкилдиазониевый ион быстрее распадается с выде-
выделением азота, чем депротонирустся до диазоалкана. Поэтому для получения
таких диазосоединений приходится прибегать к обходному пути: нитрозирова-
нию ацилированного первичного алифатического амина и щелочному расщеп-
расщеплению образовавшегося ацилнитрозоалкиламина:
'N=o +2оне в е
R-CH2-N ^ R-CH2-N=N-OI ^^ R-CH-N=N-Q_-H
COR' -R'co° ¦ -нг°
[Г.8.37]
0 0 _ © <3} ©
IR-CH-NEN — R-CH=N=N| = R-CH-NHN
-Ol-^
0
Образующийся промежуточно диазотат распадается до диазоалкана.
Из множества нитрозоамидов для препаративного получения диазоалканов
особенно удобными оказались г>[-алкил-]Ч-нитрозомочевины, N-anioui-N-HHT-
розоуретаны и 1Ч-алкил-1Ч-нитрозотолуолсульфонамиды.
Важнейшим диазоалканом является диазометан. Напишите уравнения реак-
реакций для его получения из перечисленных нитрозоамидов!
По способу, соответствующему схеме [Г.8.37], с удовлетворительными выхода-
выходами можно получать только низшие диазоалканы, так как с ростом длины цепи ал-
кила выходы сильно падают. С хорошим выходом высшие диазоалканы могут быть
получены пиролизом в вакууме литиевых солей тозилгидразонов [Kaufman G. М.
u. a. J. Am. Chem. Soc. 1965, 87, 935-937]. (Напишите эту реакцию!)
((Tj) Получение диазометана из 1Ч-метил-1Ч-нитрозомочевины
Об особенностях работы с N-нитрозо-М-метилмочевиной см. разд. Г.8.2.1.
Внимание! Диазометан (т. кип. -24 °С) взрывоопасен, очень ядовит и канцеро-
канцерогенен. Целесообразно получать его только в растворе. Даже на холоду раство-
растворы хранятся лишь в течение нескольких дней. Лучше всего их использовать
свежеприготовленными. При хранении сосуды с диазометаном нельзя плотно
закрывать (почему?). Все работы с диазометаном необходимо вести за защит-
защитным экраном и под хорошо работающей тягой. См. также Org. Syntheses, 40,
1960, Beilage (приложение).
В колбу Эрленмейера помещают 35 мл холодного 40%-ного раствора гидроксида калия и
100 мл эфира. Затем, постоянно встряхивая колбу, небольшими порциями прибавляют 0,1 моль
N-метил-М-нитрозомочевины. Температура при этом не должна превышать 5 °С. Через
10 мин после внесения последней порции отделяют с помощью делительной воронки эфир-
эфирный раствор диазометана и сушат 3 ч над небольшим количеством твердого гидроксида калия.
Методика пригодна для работы в полумикроколичествах.
Определение содержания диазометана.
а) Гравиметрическое. Добавляют избыток л-бромбензойной кислоты (см. также разд.
Г,8.4.2.1, общую методику метилирования карбоновых кислот). Избыток кислоты удаляют
8. Реакции других гетероаналогов карбонильных соединений 257
встряхиванием с раствором карбоната натрия. Затем взвешивают полученный сложный эфир.
Если необходимо, можно также определить число омыления (см. разд. Г,7.1.4.3, с. 88) и рас-
рассчитать содержание диазометана.
б) Титриметрическое. Аликвотная часть раствора диазометана взаимодействует с 0,2 н.
эфирным раствором бензойной кислоты. Количество непрореагировавшей бензойной кисло-
кислоты определяют обратным титрованием 0,1 н. раствором NaOH [Marshall Е. К., Acree S. F. Вег.
Deut. Chem. Ges. 1910, 43, 2323].
Получение диазометана из 1Ч-метил-1Ч-нитрозотолуолсульфамида: де Боер Т.,
БэкерХ. В сб.: Синтезы органических препаратов. Сб. 8. — М.: ИЛ, 1958, с. 7.
Приведенные способы получения диазометана из 1Ч-метил-М-нитрозомоче-
вины или из М-метил-Тч[-нитрозотолуосульфамида наиболее удобны в лабора-
лабораторных условиях.
8.4.2. РЕАКЦИИ АЛИФАТИЧЕСКИХ ДИАЗОСОЕДИНЕНИЙ
Как видно из схем [Г.8.15] и [Г.8.37], алифатические диазосоединения являются
биполярными, углеродный атом которых представляет собой нуклеофильный
центр, способный подвергаться атаке электрофильными реагентами (например,
протонами, карбонильными соединениями). Эти реакции рассматриваются в
следующих разделах. Кроме того, диазоалканы вступают в реакции [1,3]-цикло-
присоединения с олефинами и ацетиленами (см. разд. Г,4.4.4). Образующиеся из
олефинов Д'-пиразолины легко изомеризуются в А2-пиразолины, которые при
нагревании отщепляют азот и превращаются в производные циклопропана. На-
Напишите уравнения этих реакций! Сами диазоалканы также могут отщеплять N2
при пиролизе, облучении УФ-светом или в присутствии катализаторов (ионов
меди или серебра; см. также разложение а-диазокетонов, разд. Г,9.1.1.3). Реакции
образующихся при этом карбенов обсуждены в разд. Г,3.3 и Г,4.4.1.
8.4.2.1. Реакции алифатических диазосоединений с протонными кислотами
При присоединении протона к нуклеофильному атому углерода алифатических
диазосоединений теряется возможность сопряжения диазогруппы с остальной
частью молекулы. Энергия соответствующего промежуточного соединения нас-
настолько высока, что немедленно происходит выделение азота. При этом образу-
образуется карбениевый ион, который обычно стабилизируется присоединением нук-
леофильного реагента (см. схему [Г.8.38]). В случае других диазоалканов (кроме
диазометана) возможно также образование олефина (см. разд. Г,2.1.1 и Г,3.1.1).
Способность алифатических диазосоединений вступать в реакцию с протонными кисло-
кислотами зависит от их основности. Это свойство диазосоединений, как и следует ожидать, осла-
ослабевает в ряду: диазометан (диазоалканы) > диазоуксусный эфир > диазокетоны > а-диазоди-
карбонильные соединения. Последние устойчивы к ионам гидроксония (алкилирование
диазокетонами см. далее). Скорость реакции алифатических диазосоединений с протонными
кислотами пропорциональна рН среды. Следовательно, определение объема азота, выделяю-
выделяющегося из диазоуксусного эфира, можно использовать для измерения рН.
Получение метиловых эфиров карбоновых кислот и метилфениловых эфи-
ров из диазометана и карбоновых кислот или фенолов по схеме [Г.8.38] имеет
препаративное и аналитическое значение. Реакция протекает количественно и в
258
Г Препаративная часть (продолжение)
очень мягких условиях, так что этим путем можно метилировать, например, и
неустойчивые природные соединения. Поскольку метил-катион при этерифи-
кации не создает стерических препятствий, с диазометаном могут взаимодей-
взаимодействовать и пространственно затрудненные кислоты:
R
\о © ©
1С—N=N1 + Н
\ ©
CH-NSNI
R
IN=NI + ,0-Н
R
[Г.8.38а]
+ НОН
с-н
+ С10
+ R1 —С
+ АЮв
О
О0
R
R
сн-
R
н
-CI
R р
сн-о-с'
r' 4r
R
сн-
-О-Аг
сн-он + н
©
[Г.8.386]
R = Н, алкил, R-CO-, RO-CO-
Кислотность спиртов недостаточна для того, чтобы действием диазометана их можно было
превратить в метиловые эфиры. Если, однако, добавить каталитические количества трифтори-
да бора, то можно осуществить метилирование спиртов:
R-O-H + BF3
F
R-O-B-F
i
F
-N2,BF3
R-O-CH3
[Г.8.39]
Таблица Г.8.40. Простые и сложные метиловые эфиры, полученные метилированием
диазометаном
Эфир
Диметилтерефталат
Метиловый эфир анисовой
кислоты
Метиловый эфир
л-бромбензойной кислоты
Метиловый эфир
л-аминобензойной кислоты
1 -Метоксинафталин
2-Метоксинафталин
л-Нитроанизол
Исходное вещество
Терефталевая кислота
Анисовая кислота
я-Бромбензойная кислота
(водный этанол)
и-Аминобензойная
кислота
а-Нафтол
E-Нафтол
л-Нитрофенол
Физические константы
Т. пл. 142 °С (этанол)
Т. пл. 49 °С (этанол)
Т. пл. 81 "С
Т. пл. 112°С
(водный этанол)
Т. кип. 144 °С
A5 мм рт. ст.); nD201,6225
Т. пл. 72 °С (этанол)
Т. пл. 54 °С (этанол)
Выход, %
80
70
80
50
50
50
65
8. Реакции других гетероаналогов карбонильных соединений
259
Ф Общая методика метилирования карбоновых кислот и фенолов диазометаном (табл.
Г.8.40)
I Внимание! Диазометан ядовит, канцерогенен и взрывоопасен! Работать под
Ц хорошей тягой, за защитным экраном! См. также разд. Г.8.4.1.
В колбе к раствору 0,1 моль алкилируемого соединения в смеси метанол/вода A0:1) добав-
добавляют при перемешивании при комнатной температуре эфирный раствор диазометана, пока не
появится устойчивое слабо-желтое окрашивание; к этому времени должно прекратиться выде-
выделение азота при добавлении новых порций диазометана. (Осторожно'. Прибавление следует
вести медленно, не допуская вспенивания!) Растворитель отгоняют в вакууме на водяной ба-
бане, остаток растворяют в эфире, промывают разбавленными растворами соляной кислоты,
гидроксида натрия и водой, сушат сложный эфир (соответственно фенолят) и после отгонки
растворителя очищают перегонкой или перекристаллизацией.
Методика дает хорошие результаты при работе в полумикроколичествах; ее можно
использовать для качественного анализа.
8.4.2.2. Реакции алифатических диазосоединений
с карбонильными соединениями
Благодаря своей нуклеофильности алифатические диазосоединения могут реа-
реагировать и с карбонильной группой. Однако такая реакция возможна только с
наиболее реакционноспособными карбонильными соединениями.
Наиболее важны реакции диазометана, который гладко реагирует с альдеги-
альдегидами, кетонами, ацилгалогенидами и ангидридами кислот, в то время какдиазо-
уксусный эфир взаимодействует только с альдегидами, но не с кетонами.
Присоединение диазометана к альдегидам и кетонам протекает следующим
путем:
_ © е
NHN-CH2
С=О
? -е
N=N-CH2-C-OI
R'
-N,
© i — я
h2c-c-oi
R1
[Г.8.41а]
I
© I -0
Н2С-С-ОГ
R1
ре
R-CH2-C©
Vr
R
R'-CH2-C0
R-CH2-C
R1
R1 fu /
[Г.8.416]
О
IV
О
внутримо-
внутримолекулярная
Зм-реакция
R1
') ~R означает, что продукт II перегруп-
перегруппировывается с миграцией радикала R.
Как правило, преобладают перегруппировки, ведущие к соединениям типа
III и IV (схема [Г.8.41]). Эта реакция может служить для удлинения углеродной
цепи в кетонах (и для расширения цикла у циклических кетонов). (См. также
разд. Г,9.1.1.3, в котором также приводится механизм перегруппировки.)
260
Г Препаративная часть (продолжение)
Взаимодействие диазометана с ацилгалогенидами и ангидридами карбоно-
вых кислот протекает несколько иначе, поскольку в этом случае отщепление
азота от первичного аддукта I (схема [Г.8.41]) уже не является предпочтительной
реакцией. Отщепляется преимущественно хлороводород или соответственно
карбоновая кислота и образуется относительно устойчивый а-диазокетон (см.
также разд. Г,8.2.1):
_ © е
NHN-CH2
С=О1
_ © I _ с-
NHN-CH2-C-OI
CI
[Г.8.42а]
Н R .
© I I Де
-N-C.-C-^-Or
_ © е Я
NHN-CH-C + HCI
~ ъ
[Г.8.426]
Если реакция проводится не в щелочной среде (в присутствии триэтилами-
на), то отщепляющиеся хлороводород или карбоновая кислота реагируют по
схеме [Г.8.38] еще с одним молем диазосоединения, образуя хлорметан или
соответственно метиловый эфир карбоновой кислоты.
а-Диазокетоны являются важными промежуточными продуктами. Их можно
восстановить до метилкетонов или до а-аминокетонов. Реакция их с галогеново-
дородами приводит к образованию а-галогенкетонов (напишите схему реакции!).
И наконец, диазокетон в присутствии воды, спиртов или аммиака в результа-
результате перегруппировки может быть превращен соответственно в карбоновую кисло-
кислоту, ее эфир или амид. Этот процесс подробно рассматривается в разд. Г,9.1.1.3.
Таблица Г.8.43. Получение диазокетонов и галогенкетонов
Продукт реакции
Бензилдиазометилкетон
Фенилдиазометилкетон
Гептадецилдиазометилкетон
а-Нафтилдиазометилкетон
и-Метоксифениддиазометил-
кетон
Октаметиленбис(диазометил-
кетон)
Бензилхлорметилкетон
Фенилбромметилкетон
Фенилхлорметил кетон
Исходное вещество
Фенацетилхлорид
Бензоилхлорид
Стеароилхлорид
а-Нафтоилхлорид
Анизоилхлорид
Себациноиддихлорид
Бензилдиазометилкетон
Фенилдиазометилкетон
Фенилдиазометилкетон
Т. пл., "С
Маслянистое вещество
49 (со взрывом)
69
56
84
91
Т. кип. 134 A9 мм рт. ст.)
Т. кип. 135 A8 мм рт. ст.);
50 (петролейный эфир)
Т. кип. 140 A4 мм рт. ст.);
59 (петролейный эфир)
Выход, %
80
70
80
80
80
80
80
80
70
8. Реакции других гетероаналогов карбонильных соединений 261
©Общая методика получения диазокетонов и их превращения в галогенкетоны (табл.
Г.8.43)
Внимание! Диазометан взрывоопасен, канцерогенен и ядовит (см. разд.
Г.8.4.1)! Поскольку трение стекла может вызвать взрыв диазометана, необ-
необходимо тщательно смазать мешалку!
Диазокетоны при нагревании разлагаются со взрывом! Работать под хо-
хорошей тягой и за защитным экраном! Диазокетоны следует использовать
для дальнейших превращений сразу после получения без предварительной
очистки.
а-Галогенкетоны — слезоточивые ядовитые вещества!
А. Диазокетоны. В трехгорлую колбу, снабженную эффективной мешалкой, капельной во-
воронкой и термометром, помещают эфирный раствор диазометана, приготовленный из 0,4 моль
N-метил-М-нитрозомочевины по методике, описанной в разд. Г.8.2.1. К этому раствору при
охлаждении до 0 "С и перемешивании добавляют по каплям раствор 0,1 моль ацилхлорида в
100 мл эфира. Взаимодействие протекает очень быстро, с выделением газа. После окончания до-
добавления ацилхлорида реакционную смесь оставляют еще на 1 ч при комнатной температуре.
Поскольку диазокетоны полярны, они труднорастворимы в эфире и могут быть выделены
из раствора при охлаждении до -20 °С (выпадение в осадок и фильтрование с отсасыванием).
Жидкие диазокетоны выделяют осторожным упариванием раствора в вакууме без повышения
температуры. Для определения температуры плавления небольшое количество диазокетона
перекристаллизовывают из эфира. Если далее предполагается получать а-галогенкетоны, то
диазокетоны можно не выделять.
Б. Галогенкетоны. К раствору диазокетона, полученного по приведенной выше методике, при
перемешивании прибавляют по каплям 100 мл концентрированной соляной или бромоводород-
ной кислоты. Сразу же начинается реакция, сопровождающаяся выделением азота. По оконча-
окончании добавления кислоты нагревают 1 ч с обратным холодильником на водяной бане. (Следует
использовать интенсивный обратный холодильник!) После охлаждения реакционную смесь
разбавляют трехкратным объемом воды, эфирный слой отделяют, промывают раствором гидро-
гидрокарбоната натрия, сушат сульфатом магния. Галогенкетоны выделяют перегонкой в вакууме.
8.5. РЕАКЦИИ ПРОИЗВОДНЫХ СУЛЬФОНОВЫХ КИСЛОТ
Сульфоновые кислоты и их производные содержат сернистый аналог карбониль-
карбонильной группы. И все же по своим свойствам они, как правило, более напоминают
серную кислоту и другие неорганические кислоты, чем карбоновые кислоты.
Так, эфиры сульфоновых кислот омыляются с расщеплением связи О—алкил
и в отличие от большинства эфиров карбоновых кислот могут применяться для
алкилирования (см. табл. Г.2.4).
Восстановление производных сульфоновых кислот идет с таким же трудом,
как и восстановление производных карбоновых кислот. Аналогия проявляется и
в том, что здесь тоже легче всего восстанавливаются соответствующие ацилхло-
риды (сульфонилхлориды); таким путем можно получить сульфиновые кислоты
и тиолы или тиофенолы:
RSO2CI + 2 Н RSO2H + HCI [Г.8.44а]
2RSO2H -У-* [r-SO2-S-R -У— R-S-S-r] -^— 2 RSH [Г.8.446]
В соответствующих условиях можно остановить восстановление на стадии
сульфиновой кислоты. Это наиболее важный препаративный метод получения
сульфиновых кислот (другой метод их получения представлен в схеме [Г.8.28]).
262
Г Препаративная часть (продолжение)
H-S-H
сероводород
R-S-H
тиол
R-S-OH
сульфеновая
кислота
R-S-S-R
дисульфид
R-S-R
сульфид
101
ho-s-oh
сернистая
кислота
101
=?= HO-S-OH
II
101
серная
кислота
101
R-S-OH
сульфиновая
кислота
101
R-S-R
сульфоксид
101
R-S-OH
и
101
сульфоновая
кислота
101
R-S-R
и
101
сульфон
[П8.45]
восстановление
В свою очередь сульфиновые кислоты легко окисляются до сульфоновых.
Эти окислительно-восстановительные превращения суммированы на схеме
[Г.8.45]; для сравнения здесь же даны превращения аналогичных неорганичес-
неорганических соединений серы.
При получении тиолов (меркаптанов) в качестве восстановителей использу-
используют неблагородные металлы (например, цинковую пыль) в кислом растворе, а
для синтеза тиофенолов с успехом можно применять также красный фосфор в
присутствии иода. Каталитическое восстановление здесь малопригодно, так как
меркаптаны являются ядами для катализаторов. Кроме восстановления суль-
фохлоридов, тиолы (меркаптаны) можно получать также путем замещения из
алкилгалогенидов (см. разд. Г,2.6.6); для получения тиофенолов этот путь
непригоден.
Тиолы и тиофенолы очень чувствительны к окислителям и при окислении
переходят в дисульфиды. Последние образуются часто уже при контакте с кис-
кислородом воздуха. В связи с этим при получении и последующих превращениях
тиолов чаще всего работают в атмосфере инертного газа (азота, свободного от
кислорода; см. разд.Б). Процесс превращения тиола (тиофенола) в дисульфид
обратим; дисульфиды мягкими восстановителями вновь переводятся в тиолы
(тиофенолы). (О биологическом значении этой реакции на примере системы
цистеин-цистин — посмотрите в учебнике!)
Способность тиолов к окислению используется при очистке технических углеводородов.
При этом нежелательные тиолы окисляют в нейтральные дисульфиды, например с помощью
катализатора фталоцианина кобальта («мерокс-процесс»).
8. Реакции других гетероаналогов карбонильных соединений
263
Сульфоны аналогично сульфоновым кислотам представляют собой очень ус-
устойчивые соединения, восстановить которые до сих пор удавалось только в иск-
исключительных случаях. Принципиально возможно восстановление сульфокси-
дов до тиоэфиров, но этот метод не имеет препаративного значения. Напротив,
обратная реакция — окисление сульфидов (тиоэфиров) до сульфоксидов (напри-
(например, Н2О2, перкислотами и другими окислителями) является важнейшим препа-
препаративным методом получения последних. Тиоэфиры или сульфоксиды окисля-
окисляются до сульфонов приведенными выше окислителями только при повышенной
температуре или такими сильными окислителями, как перманганат калия.
Хорошо кристаллизующиеся сульфоны часто используются для идентифи-
идентификации тиолов и сульфидов. Напишите схему взаимодействия тиола с 2,4-ди-
нитрохлорбензолом с последующим окислением до сульфона в среде ледяной
уксусной кислоты с Н2О2 при повышенной температуре (см. разд. Г,5.2.1 и
Д.2.9.2).
Окисление сульфидов находит применение в химических производствах, например при
синтезе диметилсульфоксида (DMSO, диполярного апротонного растворителя, см. разд. В,3.3
и Г,2.2) и омепразола (важнейшего терапевтического препарата ингибитора Н+/К+-АТРазы):
МеО.
[Г.8.46]
v Общая методика получения тиофенолов (табл. Г.8.47) [ Wagner A. W. Спет. Вег., 1966, 99,
375; см. также Morgenstern J., Mayer R. Z. Chemie, 1970, 10, 449.]
Осторожно1. Тиофенолы имеют исключительно сильный, неприятный и очень
навязчивый запах. Вызывают экзему. Работать под хорошей тягой, лучше все-
всего в особом помещении! К прибору прикасаться только в резиновых перчатках,
для очистки использовать раствор перманганата!
Таблица Г.8.47. Получение тиофенолов восстановлением сульфонилхлоридов
Продукт реакции
Тиофенол
4- Метилтиофенол
2-Метилтиофенол
4-Этилтиофенол
4-й-Пропилтиофенол
4-Изопропилтиофенол
4-н-Бутилтиофенол
4-Хлортиофенол
4- Метокситиофенол
Исходное соединение
Бензолсульфонилхлорид
и-Толуолсульфонилхлорид
о-Толуолсульфонилхлорид
4-Этилбензолсульфонилхлорид
4-н-Пропилбензолсульфонил-
хлорид
4-Изопропилбензолсульфонил-
хлорид
4-н-Бутил бензолсульфонил-
бензолсульфонилхлорид
4-Хлорбензолсульфонилхлорид
4- Метоксибензолсульфонил-
хлорид
Т. кип., °С (мм рт. ст.)
55A2)
Т. пл. 43(водн. этанол)
104 D8); т. пл. 15
102 B3)
106A5)
105 A4); ис20 1,5542
119 A4);/jD20 1,5470
Т. пл. 54 (этанол)
110 A5); ио20 1,5822
Выход. %
80
80
50
60
70
70
60
80
85
264
Г Препаративная часть (продолжение)
Таблица Г.8.48. Получение эфиров п-толуолсульфоновой кислоты из п-толуолсуль-
фонилхлорида и спиртов
Эфир и-толуолсульфоновой
кислоты
Метиловый
Этиловый
я-Пропиловый
н-Бутиловый
н-Ам иловый
н-Гексиловый
я-Гептиловый
н-Октиловый
(-)-Ментиловый
Спирт
Метанол
Этанол
н-Пропанол
н-Бутанол
н-Пентанол
н-Гексанол
н-Гептанол
н-Октанол
(-)-Ментол
Т. кип., °С (мм рт. ст.)
160 A3); т. пл. 29
173 A5); т. пл. 33
140B)
128@,2)
135@,3)
138@,15)
150@,15)
149@,1)
Т. пл.93
(петролейный эфир);
[а] о20 —60° (в хлороформе)
1,4998
1,5044
1,5012
1,4990
1,4966
1,4950
Выход, %
70
60
70
70
70
70
70
70
60
В трехгорлой колбе емкостью 250 мл, снабженной обратным холодильником, капельной
воронкой и мешалкой, при энергичном перемешивании нагревают до кипения смесь 50 мл ле-
ледяной уксусной кислоты, 12,5 г красного фосфора и 0,6 г иода. К смеси по каплям прибавля-
прибавляют 0,15 моль сульфонилхлорида с такой скоростью, чтобы раствор бурно вскипал и появлялись
пары иода. (Твердые сульфонилхлориды вносят через верх холодильника.) Сульфонилхлорид
далее прибавляют с такой скоростью, чтобы реакционная смесь продолжала кипеть, несмотря
на прекращение внешнего нагревания, однако пары иода не должны выходить через верх хо-
холодильника. После прибавления всего количества сульфонилхлорида смесь кипятят еще 2 ч с
обратным холодильником, осторожно добавляют 9 мл воды и кипятят еще 1 ч. Продукт выде-
выделяют перегонкой с водяным паром.
При получении жидких тиофенолов их отделяют в делительной воронке, водную фазу
экстрагируют хлороформом. Органические слои объединяют, сушат сульфатом натрия, раст-
растворитель отгоняют, а тиофенол перегоняют в вакууме. Твердые тиофенолы отфильтровывают
и перекристаллизовывают из водного метанола.
Алкоголиз и аминолиз сульфонилхлоридов, протекающие с образованием слож-
сложных эфиров и амидов сульфоновых кислот соответственно, аналогичны соотве-
соответствующим реакциям ацилгалогенидов. Сульфонилхлориды, однако, как пра-
правило, менее реакционноспособны, чем хлорангидриды карбоновых кислот
(почему? См. разд. Г,8, с. 230). Так, в холодной воде сульфонилхлориды разла-
разлагаются очень медленно, а некоторые из них можно даже перекристаллизовывать
из воды.
Алкоголиз лучше всего проводить в присутствии веществ, связывающих
кислоту. Такими веществами могут служить гидроксид натрия или пиридин
(ср. с алкоголизом ацилгалогенидов; разд. Г,7.1.4.1).
©Общая методика получения алкиловых эфиров л-толуолсульфоновой кислоты (табл. Г.8.48).
К раствору 0,25 моль и-толуолсульфонилхлорида и 0,3 моль абсолютного спирта в 100 мл
хлороформа при 0-3 °С при перемешивании и с защитой от влаги воздуха по каплям при-
прибавляют 0,5 моль абсолютного пиридина. Перемешивание продолжают при той же тем-
температуре еще 30 мин, а в случае спиртов, имеющих более трех атомов углерода, еще 3 ч при ком-
комнатной температуре. Затем прибавляют смесь 200 г льда и 70 мл концентрированной соляной
кислоты, отделяют хлороформный слой, промывают его несколько раз водой, сушат сульфатом
натрия. После отгонки растворителя в вакууме остаток перегоняют с использованием металли-
металлической бани при 0,1—0,3 мм рт. ст., предварительно добавив на кончике шпателя гидрокарбонат
натрия. Твердые эфиры перекристаллизовывают.
8. Реакции других гетероаналогов карбонильных соединений
265
Аминолиз сульфонилхлоридов происходит в принципе так же, как и амино-
лиз ацилгалогенидов.
Сульфонамиды хорошо кристаллизуются и поэтому могут быть использова-
использованы для целей идентификации.
При идентификации аминов с помощью соответствующих сульфонамидов
можно разделять смеси первичных, вторичных и третичных аминов (разделение
по Хинсбергу). Сульфонамиды из первичных аминов растворимы в водных ще-
щелочах с образованием солей, N-дизамещенные сульфонамиды нерастворимы.
Наконец, третичные амины не дают амидов ни с сульфонилхлоридами, ни с
ацилхлоридами.
Кислотный характер аминогруппы монозамещенных сульфонамидов не вызывает удивле-
удивления. Электроноакцепторные заместители вообще уменьшают основность атома азота. Если
аммиак является сравнительно сильным основанием, то обычные амиды карбоновых кислот
способны образовывать соли только с сильными и высококонцентрированными кислотами,
которые в воде немедленно гидролизуются. В водном растворе амиды карбоновых кислот
практически нейтральны. В амидах типа фталимида влияние двух карбонильных групп уже
настолько сильно уменьшает основность (ацидифицирующее влияние), что подобные амиды
растворяются в водных щелочах с образованием солей. (Ср. с ацидифицирующим влиянием
карбонильных групп на а-С—Н-группировки и с высокой кислотностью групп СН В-дикарбо-
нильных соединений.) Влияние сульфонильной группы примерно аналогично влиянию двух
карбонильных групп. Наконец, соединения типа сахарина уже обладают кислотностью карбо-
карбоновых кислот:
O'.'v
NH3
образует
соли в водных
растворах
кислот
,.-О
R NH2
в водных
растворах
нейтральны
,.-о о-. -о..
R^^R " В51г*Нг
'- Г)
R °
образуют соли с NaOH
повышение основности N-атомов
повышение кислотности NH-rpynn
с
IN-H
Со
образуют соли
в растворе
гидрокарбоната натрия
[
[Г.8.49]
(Напишите схемы образования солей со щелочами!)
Сульфонамиды используются также для идентификации сульфоновых кис-
кислот и ароматических углеводородов. Свободные сульфоновые кислоты или их со-
соли со щелочными металлами, которые выпадают в осадок при гидролизе их про-
производных, прежде всего превращают для этого всульфонилхлориды. Наилучшим
образом это превращение удается осуществить с помощью пентахлорида фосфо-
фосфора или тионилхлорида в присутствии диметилформамида. Диметилформамид
значительно повышает реакционную способность тионилхлорида. Сам по себе
тионилхлорид, как и другие используемые при получении ацилхлоридов реаген-
реагенты, дает в применении к сульфоновым кислотам плохие результаты.
Из ароматических углеводородов сульфонилхлориды получают сульфохло-
рированием (см. разд. Г,5.1.4).
Получение сульфонилхлоридов из сульфоновых кислот или их солей со щелочными металлами
(общая методика для качественного анализа). В круглодонной колбочке емкостью 25 мл тща-
тщательно смешивают 1 г безводной сульфоновой кислоты или ее безводной соли со щелочными
металлами с 2 г пентахлорида фосфора. К колбе присоединяют обратный холодильник с хлор-
266
Г Препаративная часть (продолжение)
Таблица Г.8.50. Получение амидов сульфоновых кислот из сульфонилхлоридов
Продукт реакции
л<-Нитробензолсульфонамид
Бензолсульфонамид
л-Толуолсульфонамид
о-Толуолсульфонамид
я-Ацетаминобензолсульфонамид
л-Хлорбензолсульфонамид
л- Метоксибензолсульфонамид
Исходное соединение
.м-Нитробензолсульфонилхлорид
Бензолсульфонилхлорид
л-Толуолсульфонилхлорид
о-Толуолсульфонилхлорид
л-Ацетаминобензолсульфонилхлорид
л-Хлорбензолсульфонилхлорид
и- Метоксибензолсульфонилхлорид
Т. пл., °С
167
153
137
156
218
144
113
кальциевой трубкой и нагревают 30 мин на металлической бане при 120 °С. После охлаждения
приливают 20 мл толуола, смесь вновь нагревают до кипения, а затем охлаждают и фильтруют.
Из фильтрата после отгонки толуола и оксихлорида фосфора в вакууме на водяной бане выде-
выделяют сульфонилхлорид. Неочищенный сульфонилхлорид вполне пригоден для превращения в
сульфонамид.
Ф Общая методика получения амидов сульфоновых кислот (табл. Г.8.50). В трехгорлую кол-
колбу емкостью 1 л, снабженную капельной воронкой, мешалкой, обратным холодильни-
холодильником и термометром, помещают 500 мл концентрированного раствора аммиака и при
60 "С добавляют к нему по каплям при перемешивании 1 моль сульфонилхлорида. Затем пере-
перемешивание продолжают и смесь нагревают на водяной бане до тех пор, пока взятая проба не
будет оставаться прозрачной в разбавленном растворе гидроксида натрия и пока не исчезнет
запах сульфонилхлорида.
После охлаждения сульфонамид отсасывают и очищают перекристаллизацией из воды или
50%-ного этанола. Выход около 80%.
Методика пригодна для получения полумикроколичеств и для аналитических целей.
В этом случае сульфонилхлорид просто нагревают несколько минут до кипения с избытком
аммиака, разбавляют водой и отфильтровывают.
Ы-(п-Толуолсульфонил)антраниловая кислота из w-толуолсульфонилхлорида
и антраниловой кислоты: Scheifele H. J., DeTar D. F. Org. Syntheses. Coll. Vol. IV,
A963), 34.
2-(п-Ацетиламинобензолсульфонамидо)пиридин из я-ацетиламинобензол-
сульфонилхлорида и 2-аминопиридина в диоксане: Crossley M. L, Northey Е. Н.,
HultquistM. E. J. Am. Chem. Soc, 1940, 62, 372.
Разделение смесей аминов через сульфонамиды (разделение по Хинсбергу) (общая методика для ка-
качественного анализа). К 2 г смеси аминов прибавляют 40 мл 10%-ного раствора гидроксида нат-
натрия и затем небольшими порциями 4 г C мл) бензолсульфонилхлорида или 4 г л-толуолсульфо-
нилхлорида. Реакционную смесь непродолжительное время нагревают на водяной бане, пока не
исчезнет запах сульфонилхлорида. Щелочной раствор подкисляют разбавленной соляной
кислотой, осадок отфильтровывают и промывают небольшим количеством холодной воды. Тре-
Третичный амин находится в фильтрате в виде гидрохлорида. Для превращения образующихся ди-
сульфонамидов в моносульфонамиды сухой остаток после фильтрования кипятят 30 мин с раст-
раствором алкоголята натрия, полученным из 2 г натрия и 40 мл абсолютного спирта; затем раствор
разбавляют небольшим количеством воды и отгоняют спирт. Сульфонамид, полученный из
8. Реакции других гетероаналогов карбонильных соединений
267
вторичного амина, отфильтровывают, фильтрат подкисляют разбавленной соляной кислотой и
отфильтровывают выпавший сульфонамид первичного амина. Полученные производные пере-
кристаллизовывают из разбавленного спирта. Третичный амин выделяют из первого кислого
фильтрата подщелачиванием, экстрагируют эфиром и идентифицируют, лучше всего в виде
пикрата (см. разд. Д, 2.1.1.3).
Некоторые амиды л-толуолсульфоновой кислоты, получаемые из л-толуолсульфонилхлорида
(см. разд. Г,5.1.4) относятся к полупродуктам крупнотоннажного органического синтеза. Напри-
Например, натриевая соль N-хлормина (хлорамин Т) — превосходное дезинфицирующее средство.
Амиды сульфоновой кислоты являются эффективными химиотерапевтическими препара-
препаратами при бактериальных инфекциях (сульфамидные препараты). Обычно их получают взаи-
взаимодействием л-ацетамидобензолсульфонилхлорида (см. разд. Г.5.1.4) с определенными
аминами с последующим гидролитическим отщеплением ацетильной группы. Важными
представителями этого класса соединений является сульфаметоксазол
H2N
о о
сульфаметоксазол
.О
[Г.8.51]
силденафил
К амидам сульфокислот относится силденафил (виагра), эффективное средство повышаю-
повышающее половую потенцию.
К важнейшим «оральным» антибиотикам относятся сульфонилмочевины, например
[Г.8.52]
Me
глибенкламид
глимеперид
В качестве важнейших гербицидов используют такие сульфонилмочевины, как хлорсуль-
фурон и его производные
[Г.8.53]
О'
хлорсульфурон
8.6. ЛИТЕРАТУРА
Нитрозо- и нитросоединения
Behnisch R., им., in: Houben-Weyl. Bd. E16d A992), S. 142-405.
BuncelE., NorrisA. R., Russell К. E., Quart. Rev. 22 A968), 123-146.
Cadogan J. I. G., Quart. Rev. 22 A968), 222-251.
268 Г Препаративная часть (продолжение)
Collins С. J., Асе. Chem. Res. 4 A971), 315-322.
Dopp D. О., Fortsch. Chem. Forsch. 55 A975), 49-85.
Feuer H:. The chemistry of The Nitro and Nitroso Groups. Bd. 1-2. — Interscience,
New York 1969-1970.
Kirby G. W., Chem. Soc. Rev. 6 A977), 1.
Urbanski Т., Synthesis 1974, 613-632.
Восстановление нитро- и нитрозосоединений
Bavin R. M. G., Org. Synth., Coll. Vol. 5 A973), S. 30.
Porter H. K., Ogr. React. 20 A973), 455-481.
SchmterR., in: Houben-Weyl. Bd. 11/1 A957), S. 360-494.
Алифатические диазосоединения
BosharM., u.a., in: Houven-Weyl. Bd. E14b A990), S. 961-1257.
BottK., in: Houben-Weyl. Bd. E15 A993), S. 1101.
Burke S. D.; Grieco P. A., Org. React. 26 A979), 361-475.
EistertB., u.a., in: Houben-Weyl. Bd. 10/4 A968), S. 473-893.
Фридман А. Л., Измагилова Г. С, Залесов В. С, Новиков С. С. Усп. хим., 1972, 41, 722-757.
Herrmann W. A., Angew. Chem. 90 A978), 855-868.
Peace В. W., Wulfman D. S., Synthesis 1973, 137-145.
Zollinger H.\ Diazo Chemistry II. — WCH Verlagsgesellschaft, Weinheim 1995.
Regitz M., in: Neuere Methoden. Bd. 5 A970), S. 76; Synthesis 1972, 351-373.
Regitz M., Maas G., Illger W., Dizoaklane. Eigenschaften und Synthesen. — Georg Thieme
Verlag, Stuttgart 1977.
Ried W., MenglerH, Fortschr. Chem. Forsch. 5 A965/66), 1-88.
Диазотирование, соли диазония, азосоединения
Lang-Fugmann S., in: Houben-Weyl. Bd. 16d A992), S. 1-118.
Порай-Кошиц Б. А. Усп. хим., 1970, 39, 608-621.
Putter R., in: Houben-Weyl. Bd. 10/3 A965), S. 1-212.
Roe A., Org. React. 5 A949), 193-288.
Szele I., Zollinger H., in: Topics in Current Chemistry. Bd. 112. — Springer-Verlag, Berlin,
Heidelberg, New York 1983. S. 1-66.
The Chemistry of the Hydrazo, Azo and Azoxy Groups. Hrsg.: S. Patai. Bd. 1—2. — John
Wiley & Sons, London, New York, Sydney 1975.
Ullmanns Encyclopiidie der technischen Chemie. Bd. 10. 4. Aufl. — Verlag Chemie, —
Weinheim 1975. S. 109-132.
Ullmanns Encyclopedia of Industrial Chemistry. Bd. A8. 5. Aufl. — VCH Verlagsgesellschaft,
Weinheim 1987. S. 505-522.
Zollinger H., Chemie der Azofarbstoffe. — Birkhauser Verlag, Basel, Stuttgart 1958.
S. 30-44; Ace. Chem. Res. 6 A973), 335-341; Angew. Chem. 90 A978), 151-160.
Zollinger H., Diazo Chemistry I. — VCH Verlagsgesellschaft, Weinheim 1994.
Реакция Зандмейера, термические превращения солей диазония в растворе,
арилирование
Ambroz Н. В., Kemp Т. J., Chem. Soc. Rev. 8 A979), 353-365.
Cohen Т., DietzA. G., Miser J. R., J. Org. Chem. 42 A977), 2053-3058.
9. Перегруппировки
269
ForcheE., in: Houben-Weyl. Bd. 5/3 A962), S. 213-245.
PfeilE., Angew. Chem. 65 A963), 155-158.
Rondestvedt С S., Org. React. 24 A976), 225-259.
Получение и реакции соединений серы
Block E:. Reactions of Organosulfur Compounds. —Academic Press, New York. 1978.
Houben-Weyl. Bd. 9 A955), S. 3-771.
Organic Chemistry of Sulfur. Hrsg.: S. Oae. — Plenum Press, New York, London 1977.
Sulfur in Organic and Inorganic Chemistry. Bd. 1 A971). Bd. 2 A972). Bd. 3 A972). —
Marcel Dekker, New York.
Topics in Sulfur Chemistry. Bd. 1 A976). Bd. 2 A977). Bd. 3 A978). Bd. 4A979). - Georg
Thieme Verlag, Stuttgart.
9. ПЕРЕГРУППИРОВКИ
Перегруппировками называют реакции, в ходе которых заместитель или группа
переходит от одного атома к другому. При формальной классификации пере-
перегруппировок нумеруют участвующие в перефуппировке атомы так, что атомы
расщепляющейся связи получают номер 1, а атомы по обе стороны от этой
связи — соответственно возрастающие номера; в обозначении типа перефуп-
пировки указывают атомы, между которыми создается новая связь:
[1,2]-Перегруппировка
1R - О
-с
[ 1,2-Н]-Перегруппировка
'Н . п
"С-С-"
1,3]-Перефуппировка
[1,3-Н]-Перегруппировка
vlH
;с-с
0 Iх
.0 <И
;с-с
0 I4
(перегруппировка
винилциклопропан -
циклопентен)
[Г.9.1а]
[Г.9.16]
[Г.9.2]
\ /
С с^ (аллильная [Г9 3а1
С ^ перегруппировка) 1 '
\,Н
(кето-енольная
перегруппировка)
[Г.9.36]
270
Г Препаративная часть (продолжение)
С
н
fig (азометин-енаминная
^ перегруппировка) [Г.9.3в]
[Г.9.3г]
[ 1,5-Н] -Перегруппировка
[2,3]-Перегруппировка
(диенон-фенольная перегруппировка) [Г.9.4]
B,3-перегруппировка
0 е Виттига)
[Г.9.5]
[З,3]-Перегруппировка
(перегруппировка Коупа) [Г.9.6а]
О.
(перегруппировка Кляйзена) [Г.9.66]
У всех представленных перегруппировок всегда происходит разрыв ст-связи и
мигрирующая группа атакует л-центр с образованием новой ст-связи. Такие
перегруппировки называют также сигматропными.
л-Центр может содержать 0, 1 или 2 электрона, т. е. перегруппировки воз-
возможны как у нейтральных соединений (например, л-центры в олефинах, в кар-
бенах, нитренах), у ениевых ионов (например, у карбениевых ионов), так и у
радикалов или анионов (например, карбанионов).
Если рассматривать мигрирующую частицу как внутримолекулярный реа-
реагент, то ее перенос к электронодонорному центру можно классифицировать как
электрофильный, а к электронодефицитному центру — как нуклеофильный.
9.1. [1,2]-ПЕРЕГРУППИР0ВКИ
[1,2]-Перегруппировки известны для любых типов зарядов; впрочем, [1,2]-пе-
регруппировки в ряду радикалов не играют большой роли, т. к. в этом случае
имеют место другие реакции (см. разд. ГД.2). Из электрофильных [1,2]-пере-
группировок следует упомянуть прежде всего реакции илидов (см. разд.
Г,7.2.6.2), например перегруппировку Стивенса [Г.9.7], а также протекающую
9. Перегруппировки
271
аналогично перегруппировку Виттига [Г.9.8]. Перегруппировки как Виттига, так
и Стивенса протекают через радикальные интермедиаты (пары радикалов).
R-CH2-NR3XU
HNaNH2
© I©
R1
Rf
-NH3,-NaX
R-CH-N-R1 - [r-CH-N -R'] CH-NR2
R'
R'
[Г.9.7]
R = RCO, ROOC, Ph
R' = аллил, PhCH 2, Ph2CH, PhCOCH2, метал
+ PhLi
R'
fc) г . Q 1 +H \
R-CH-O-R1 [R-CH-0 -R'J -n— CH-OH
[Г.9.8]
R-CH2-O-R'
-PhH, -I_T
R, R' = алкил, арил, винил
Нуклеофильные [1,2]-перегруппировки — часто встречающиеся и препара-
препаративно важные реакции. Они наиболее вероятны во всех тех случаях, когда в хо-
ходе реакции возникает атом углерода или гетероатом только с 6 электронами
(секстетом электронов), причем неважно, связано ли существование секстета с
наличием заряда или нет. При этом образующийся в реакции сольволиза
первичный карбениевый ион (I) может претерпевать следующие превращения:
R
C=CH2
R Ilia
-H®
(E)
H
©
R-C-CH2 —
P ot
R 1
+ Ye|(SN)
1
;-ch2-y
R V
~H \©
—2— C-CH3 —
R lla
н©
L~z^~ c-ch2-r —
r" не
Г—©* Я = СН2
-H p
H Ilia
+ Y® Y
L-!-^ R-C-CH3
R IVa
Y
^— H-C-CH2-R
R IV6
H
— C=CH-R
"H R 1116
[Г.9.9]
Первоначально образующееся промежуточное соединение с секстетом
электронов I (схема [Г.9.9]) богато энергией и поэтому может сравнительно
неселективно стабилизироваться в результате ряда конкурирующих реакций.
Элиминирование протона (завершение реакции Е1) приводит к Ша, а присое-
присоединение нуклеофила (завершение реакции SN1) — к образованию V; эти
реакции не приводят к перегруппировкам. Возможна также миграция
заместителя (в нашем примере Н или R) от р-атома со своими связывающими
электронами к атому с секстетом электронов, протекающая с образованием
нового промежуточного соединения с секстетом электронов (Па или Пб), кото-
которое окончательно стабилизируется элиминированием Н+ или присоединением
нуклеофила Y~.
272 Г Препаративная часть (продолжение)
В общем случае движущей силой процесса является образование продукта с
низкой энергией из высокоэнергетического промежуточного соединения, на-
например, образование вторичного карбениевого иона II [Г.9.9] из первичного
карбениевого иона I [Г.9.9]. Возможны также так называемые «вырожденные»
перегруппировки, например СН3—СН2+ <-> +СН2—СН3, которые, однако, можно
обнаружить только с помощью искусственных экспериментальных приемов
(например, с помощью изотопов).
В большинстве случаев мигрирующий остаток отрывается от остальной
части молекулы не полностью, а остается в сфере ее влияния (например, в виде
л-комплекса, переходного состояния, аналогичного переходному состоянию в
реакции S^2, или в виде контактной ионной либо радикальной пары). Соответ-
Соответственно при образовании II (схема [Г.9.9]) заместитель большей частью мигри-
мигрирует синхронно от образующегося первичного промежуточного соединения I;
поэтому здесь, как и при ионном элиминировании (см. разд. Г.3.1), сохраняет
силу «принцип четырех центров», а конформационно предпочтительнее проис-
происходит миграция одной из трех углеродных групп, находящихся у р-атома к сс-уг-
леродному атому:
[Г.9.10]
Как показано на схеме [Г.9.10], мигрирующая группа осуществляет атаку с
«лицевой стороны», и, следовательно, миграция сопровождается сохранением
конфигурации, что и было подтверждено экспериментально.
Конечно, если конформация исходного вещества или первичного промежу-
промежуточного продукта I (схема [Г.9.9]) не закреплена, т. е. может легко изменяться, то
вследствие высокой скорости конформационных превращений любой из трех
заместителей может занять способствующее перегруппировке перипланарное
положение (см. схему [Г.9.10]). В такой ситуации мигрирует преимущественно
наиболее нуклеофильная группа и тенденция заместителей к миграции возрас-
возрастает в следующем ряду:
—Н < —СН3 < —СН2СН3 < —СН(СН3J < —С(СН3)з < —Ph [Г.9.11а]
а среди замещенных фенильных групп — в ряду заместителей:
n-NO2 < л-С1 < Н < л-Ph < л-СН3 < л-СН3О [Г.9.116]
Отсюда вытекает следующее правило пространственного эффекта: чем объ-
объемнее заместители у р-атома углерода промежуточного соединения I (схема
[Г.9.9]), тем в большей степени пространственно затруднено тривиальное нукле-
офильное замещение с образованием V (схема [Г.9.9]). Напротив, перегруппи-
перегруппировка объемной, например трет-бугшъной, группы в силу ее высокой склон-
склонности к миграции в соответствии с рядом [Г.9.11а] происходит особенно легко.
Аналогично возможны нуклеофильные перегруппировки и в соединениях с
секстетом электронов у гетероатома:
9. Перегруппировки 273
" _ ~R Rs® © "s
R-C-Y C-Y-R — C=Y-R [Г9Л2]
R R R
Конкретные примеры таких перефуппировок обсуждаются в разд. Г,9.1.2 и
Г.9.1.3.
9.1.1. НУКЛЕОФИЛЬНЫЕ [1 ^-ПЕРЕГРУППИРОВКИ
У АТОМА УГЛЕРОДА
9.1.1.1. Пинаколиновая перегруппировка
Дегидратация диолов-1,2 (ос-гликолей) (I, схема [Г.9.13]) в присутствии кислот-
кислотных катализаторов почти всегда приводит к карбонильному соединению и лишь
в редких случаях образуются сопряженные диены:
R3
[Г.9.13]
Rf 7° з -"@ "' з
©C-C-R3 ^=^ C-C-R3
НО R4 +Н® 6' R4
IV V
Сначала в результате протонирования гидроксильной группы и последую-
последующего отщепления воды образуется карбениевый ион III. Последний стабилизи-
стабилизируется путем миграции группы R1, превращаясь в карбениевый ион IV, который
после отщепления протона от гидроксильной группы превращается в карбо-
карбонильное соединение V. Если заместители R1, R2, R3 и R4 различны, то отщепле-
отщепление гидроксильной группы происходит так, чтобы образовался возможно более
устойчивый катион III. Тенденция к отщеплению возрастает в следующем
ряду:
Н Н R Аг
— СН2ОН < R-C-OH < Ar-C-OH < R-C-OH < Ar-C-ОН [Г.9.14]
(О стабильности катионов см. разд. Г,3.1.4.)
В целом способность заместителей R1, R2, R3, R4 к миграции соответствует
рядам, приведенным в [Г.9.11а, б].
Какие соединения образуются при дегидратации этиленгликоля, глицерина
и 2,3-диметилбутандиола-2,3 (пинакона)?
274
Г Препаративная часть (продолжение)
Таблица Г.9.15. Получение альдегидов и кетонов путем пинаколиновой перегруппи-
перегруппировки
Продукт реакции
Изомасляный альдегид
Циклопентанкарбальдегид
3,3-Диметилбутанон-2
(пинаколин)
Фенилацеталвдегид
2,2-Бис(л-толил)бутанон-2
Исходное соединение
2-Метилпропандиол-1,2
/яра«с-Циклогександиол-1,2
2,3-Диметилбутандиол-2,3
(пинакон)
1 -Фенилэтандиол-1,2
1,1 -Бис(я-толил)-2-метил-
пропандиол-1,2
Т. кип., °С
(мм рт. ст.)
64
137
106
78A0)
47
(т. пл.; этанол)
nD20
1,3730
1,4423
1,3956
1,5254
Выход, %
80
70
70
40
85
Ф Общая методика проведения пинаколиновой перегруппировки (табл. Г.9.15). Синтез про-
проводят в приборе для перегонки с водяным паром (см. разд. А,2.3.4). В перегонной кол-
колбе смешивают 1 моль гликоля и 500 мл 12%-ной серной кислоты и перегоняют с водя-
водяным паром. Отогнанную смесь альдегида или кетона с водой насыщают хлоридом натрия, кар-
карбонильное соединение экстрагируют эфиром. Эфирный слой сушат сульфатом магния или
натрия, упаривают, а затем перегоняют или перекристаллизовывают из этанола.
Не играет роли, каким путем образуется электронодефицитный центр при
нуклеофильной [1,2]-перегруппировке. Поэтому первичные амины при дезами-
нировании азотистой кислотой также претерпевают перегруппировку; из сс-ами-
носпиртов образуются альдегиды или кетоны (перегруппировка Тиффено):
CH2-NH2
ОН
сн2
с-он
¦©
-N2
-ьг
[Г.9.16]
Г0
Эта перегруппировка используется для препаративного получения гомологов
циклических кетонов (реакция Тиффено—Демьянова). При этом вначале проводят
альдольную конденсацию циклических кетонов, имеющих п атомов углерода в
молекуле, с нитрометаном и затем восстанавливают нитрогруппу продукта кон-
конденсации до аминогруппы и далее согласно [Г.9.16] превращают в циклический
кетон с (п + 1) атомами углерода в цикле. Напишите механизм этой реакции!
Выходы циклических кетонов в этом случае выше, чем при аналогичном рас-
расширении цикла кетонов с помощью диазометана (см. разд. Г,9.1.1.3).
Аналогично реагируют эпоксиды, трехчленное кольцо которых при действии
кислот Льюиса расщепляется с образованием электронодефицитного центра,
облегчающего перегруппировку в карбонильные соединения:
Rz R4
0
OBF3 R3
R1-C—C©
R2 R4
-BF3
°s
R2 R4
[Г.9.17]
9. Перегруппировки 275
Для того чтобы предотвратить сольволитическое раскрытие эпоксидного
кольца, реакцию ведут в неполярных растворителях. При раскрытии эпоксид-
эпоксидного кольца под действием кислот Льюиса следует учитывать те же факторы,
которые упоминались выше при обсуждении отщепления гидроксильной груп-
группы от гликолей (пинаколиновая перегруппировка).
Перегруппировка эпоксидов используется и в аналитических целях для
идентификации олефинов, поскольку образующиеся альдегиды и кетоны легко
охарактеризовать в виде производных.
Исходя из рядов [Г.9.11а] и [Г.9.14], подумайте, эпоксиды каких олефинов
перегруппировываются однозначно с образованием соответствующих карбо-
карбонильных соединений, что обеспечивает надежность идентификации.
©Эпоксидирование олефинов и перегруппировка эпоксидов в карбонильные соединения
(общая методика для качественного анализа). [Share/kin I. G., Shwerz H. E. Anal. Chem.,
1961, 33, 635.] К раствору 1 г олефина, растворенного в 5 мл эфира, прибавляют при
комнатной температуре 3 мл 40%-ной перуксусной кислоты, содержащей 5% ацетата натрия.
Оставляют стоять на 20 ч, затем выливают в насыщенный водный раствор карбоната калия,
отделяют эфирный слой, водный несколько раз экстрагируют небольшим количеством
эфира. Объединенные эфирные вытяжки (около 20 мл) сушат 2 ч сульфатом натрия, затем до-
добавляют 2 мл раствора эфирата трифторида бора, встряхивают в течение 5 мин. После этого
промывают 2 мл воды, эфирный слой отделяют, растворитель отгоняют. Остаток растворяют
в 2 н. растворе соляной кислоты в метаноле, прибавляют раствор динитрофенилгидразина и
кипятят. Кристаллизующийся динитрофенилгидразон отфильтровывают и перекристаллизо-
вывают (см. разд. Г,7.1.1).
Камфоленовый альдегид из а-пиненоксида: Royals E. E., Harrell L. L. J. Am.
Chem. Soc, 1955, 77, 3405.
д. 1.1.2. Перегруппировка Вагнера-Меервейна
Перегруппировка Вагнера—Меервейна родственна пинаколиновой перегруппи-
перегруппировке. Ее отличительной чертой является отщепление протона от перегруп-
перегруппировавшегося карбениевого иона с образованием олефина вследствие
стабилизации промежуточной частицы:
R1 н 1
„II.
R2-C-C-R4 @
R3X ~X R3 R4 R2 R3 "H R2 R3 [Г.9.18]
X = галоген, OTos, OH (+ H ®)
Поскольку в ходе этого процесса образуется углеродный скелет исходного соединения
пинаколиновой перегруппировки, ее называют также ретропинаколиновой перегруппи-
перегруппировкой.
2,3-Диметилбутен-2 из 3,3-д,иметилбутанола-2: Whithmore F. С, Rothrock H. S.
J. Am. Chem. Soc, 1933, 55, 1109.
Перегруппировка Вагнера—Меервейна играет решающую роль при реакциях
соединений терпенового ряда, протекающих через ряд последовательных
стадий. Перегруппировке благоприятствует пространственная сближенность
R н
R2-C-C©
R3 R4
R1 R4
-z^-~ c-ch
R2 R3
1
-H®
R1 R4
- c=c
R2 R3
276 Г Препаративная часть (продолжение)
атомов углерода С2 и С6, а также С3 и С5. Так, при дегидратации борнеола1 обра-
образуется камфен:
-—H2°~ -i-—- ~ -'—- -'—— ^-i-— [Г 9 191
Н +Н2О,-Н®
Ознакомьтесь с синтезом камфоры!
Дегидратация тетрагидрофурфурилового спирта до дигидропирана также
сопровождается перегруппировкой Вагнера—Меервейна:
Методика получения 3,4-дигидро-2Н-пирана каталитической дегидратацией фурфурилового
спирта см. в предыдущем русском издании этой книги [Органикум, т. 2. — М.: Мир, 1992].
Являясь циклическим енолоэфиром, 3,4-дигидро-2Н-пиран может присоеди-
присоединять спирты, образуя ацетали (см. разд. Г,4.2.2):
н
+ HOR
О
Этой свойство дигидропирана используют для обратимой защиты спиртовой группы.
9.1.1.3. Перегруппировка Вольфа
Диазокетоны при нагревании или при УФ-облучении отщепляют азот, причем
возникает незаряженный атом углерода с секстетом электронов (карбен). Реак-
Реакция ускоряется в присутствии серебряных катализаторов.
Стабилизация карбена происходит в результате переноса группы R (пере-
(перегруппировка Вольфа) или гидрид-иона с образованием кетена I или соответ-
соответственно а,р-ненасыщенного кетона II:
' Пространственное положение заместителей в подобных бициклических системах ука-
указывается префиксами экзо- и эндо-. В борнеоле гидроксильная группа занимает эндо-положе-
ние (аксиальна), а водород — экзо-положение (экваториален). В изоборнеоле группа ОН
занимает экзо-положение:
9. Перегруппировки 277
© 0 CH2R' CH2R'
O=C-CI —^ O=C = C I
R R
С Cl * C~C, rp q л'л
R' N0 ^2 r' ' \ % Н [".22]
c-ci — c-c и
R H R H
В водной среде кетен сразу присоединяет воду и дает кислоту (схема [Г.9.23],
I). В спиртовых растворах образуются сложные эфиры II, а с аминами — амиды
III (см. также разд. Г,7.1.6).
+ Н2О
R-CH=C=O
HOR1
R-CH2-COOH I
R-CH2-COOR' II [Г.9.23]
R-CH2-CONH2 III
Соотношение между продуктами реакции [Г.9.22] зависит от температуры:
при низких температурах преимущественно образуется а,р-ненасыщенный
кетон, а при нагревании (>50 °С) — производное кислоты. Диазокетоны, не име-
имеющие метиленовой группы рядом с группировкой —CHN2—, дают только кисло-
кислоты и их производные. Например, диазокетоны, образующиеся при взаимодей-
взаимодействии хлорангидридов кислот с диазометаном (см. разд. Г,8.4.2.2) дают только
кислоты и их производные.
Арндт и Эйстерт использовали перегруппировку Вольфа для удлинения угле-
углеродной цепи карбоновых кислот: из хлорангидрида и диазометана получают
диазокетон, в результате перегруппировки (дедиазонирования) которого обра-
образуется карбоновая кислота, имеющая на один атом углерода больше. (Какие
другие методы удлинения цепи карбоновых кислот вы еще знаете?)
Аналогично взаимодействуют с диазометаном кетоны и альдегиды: с выделе-
выделением азота они могут быть превращены в соответствующие карбонильные сое-
соединения, имеющие на одну СНг-группу больше (см. также разд. Г,8.4.2.2).
Метод имеет значение прежде всего для расширения кольца циклических кето-
нов, поскольку при этом образуется единственный продукт перегруппировки:
C=0 - H2CN2 _
(Ср. с реакцией Тиффено [Г.9.14].)
Ф Общая методика получения эфиров карбоновых кислот из диазокетонов перегруппировкой
Вольфа (табл. Г.9.25)
1. Получение катализатора (оксида серебра). К 50 мл 10%-ного раствора нитрата сереб-
серебра прибавляют разбавленный раствор гидроксида натрия до прекращения выделения осадка
Ag20. Осадок отмывают декантацией с водой до нейтральной реакции, затем отфильтровыва-
отфильтровывают и сушат в эксикаторе. Выход около 3 г.
2. Перегруппировка Вольфа. В 300 мл абсолютного этанола растворяют 0,1 моль диазоке-
тона (получение см. в разд. Г,8.4.2.2; можно использовать неочищенные диазокетоны). Раст-
278
Г Препаративная часть (продолжение)
Таблица Г.9.25. Получение сложных эфиров перегруппировкой Вольфа
Продукт реакции
Этиловый эфир гептадекановой
(маргариновой) кислоты
Этиловый эфир нонадекановой
кислоты
Этиловый эфир декандикарбоно-
вой-1,10 кислоты
Этиловый эфир фенилуксусной
кислоты
Этиловый эфир я-метоксифенил-
уксусной кислоты
Этиловый эфир а-нафтилуксус-
ной кислоты
Этиловый эфир дигидрокорич-
ной кислоты
Исходный диазокетон
Диазометилпентадецил-
кетон
Диазометилгептадецил-
кетон
1,10-Бис(диазометил)-
декандион-1,10
Диазометилфенилкетон
Диазометил-л-метокси-
фенилкетон
Диазометил-а-нафтил-
кетон
Бензилдиазометилкетон
Т. кип., °С (мм рт. ст.)
185 E); 28 (т. пл.)
167 @,3); 37 (т. пл.)
193A5); 15 (т. пл.)
100 A0); rtD181,4992
154A7)
179A1)
123 A6); по20 1,4911
Выход, %
60
55
45
35
40
35
35
вор нагревают до 55-60 °С в литровой трехгорлой колбе с обратным холодильником, капель-
капельной воронкой и мешалкой; при перемешивании прибавляют по каплям суспензию 3 г сереб-
серебряного катализатора в 60 мл абсолютного этанола. Содержимое колбы кипятят при перемеши-
перемешивании еще 2 ч, затем в колбу вносят 0,5 г активированного угля, еще раз нагревают до кипения
и фильтруют горячий раствор. Если при охлаждении эфир выделяется в твердом виде, его от-
отфильтровывают и перекристаллизовывают из этанола. Если полученный эфир — жидкость
(или не выпадает при охлаждении), то спирт отгоняют в вакууме и затем эфир перегоняют.
Ф Получение циклогептанона (суберона). [DeBoer Th. /., Backer H. J. Org. Syntheses, Coll. Vol.
IV, 1963, p. 225.] В литровой трехгорлой колбе, снабженной мешалкой, термометром,
капельной воронкой, а также имеющей свободный выход для образующегося в ходе ре-
реакции азота, смешивают 0,5 моль циклогексанона, 0,6 моль N-метил-М-нитрозотолуолсуль-
фонамида, 150 мл этанола и 10 мл воды. Для предотвращения-вспенивания прибавляют нем-
немного силиконового пеногасителя. При перемешивании и охлаждении льдом с солью прибав-
прибавляют из капельной воронки раствор 15 г гидроксида калия в 50 мл 50%-ного этанола с такой
скоростью, чтобы температура в колбе была 10-20 °С. При добавлении щелочи из нитрозоа-
мида образуется диазометан, который сразу же реагирует с циклогексаноном. После оконча-
окончания прибавления щелочи перемешивают еще 30 мин и, не прекращая перемешивания, добав-
добавляют 2 н. соляную кислоту до слабокислой реакции, а затем 300 мл насыщенного раствора тех-
технического гидросульфита натрия. Через несколько минут начинается осаждение бисульфит-
ного соединения суберона. Перемешивают еще 10 ч, затем осадок отфильтровывают и тща-
тщательно промывают эфиром. Бисульфитное соединение разлагают в теплом растворе 125 г
№гСОз ЮНгО в 150 мл воды. Слой кетона отделяют, водный слой 4 раза экстрагируют эфи-
эфиром (порциями по 50 мл). Объединенную органическую фазу сушат сульфатом магния, эфир
отгоняют в вакууме, а остаток фракционируют на 40-сантиметровой колонке Вигре. В первой
фракции содержится непрореагировавший циклогексанон, затем при 65 °С A2 мм рт. ст.) отго-
отгоняется циклогептанон. В остатке остаются кетоны с большим числом атомов углерода (цикло-
октанон и другие). Выход 33%; «о25 1,4600. Высокоочищенный продукт получают ректифика-
ректификацией на колонке (флегмовое число 10 :1).
9.1.2. ПЕРЕГРУППИРОВКИ У АТОМА АЗОТА
При деструкции кислот по Гофману, Курциусу и Лоссену, а также в реакциях
Шмидта и Бекмана перегруппировки происходят через промежуточные соеди-
соединения с секстетом электронов у атома азота (схема [Г.9.26], I и II), которые
9. Перегруппировки
279
имеют характер нитренов или нитрениевых ионов и в свободном виде обычно не
существуют.
Перегруппировка
Гофмана:
Лоссена:
Курциуса:
R-c
4nh2
cf
4NH-OH
4N.-NHN|
0 ©
~R
O=C=N-R
[Г.9.26]
Шмидта:
Бекмана:
R;
. _ ©
C=N-N=N|
R1
C=N-OH
R' ©
L-* C=N
~R © —
—— R'-C=N-R
R'-C
NH-R
R
9.1.2.1. Деструкция по Гофману
При деструкции амидов кислот по Гофману действием гипогалогенитов на ами-
амиды кислот получают первичные амины, имеющие на один атом углерода мень-
меньше, чем исходное вещество. (Эту реакцию не следует путать с реакцией отщеп-
отщепления по Гофману; см. разд. Г,3.1.6.) При этом при определенных условиях в ка-
качестве промежуточного продукта образуется выделяемый галогенамид [Г,9.27],
I; из него после отщепления галогеноводорода и перегруппировки возникает
изоцианат III, сразу же присоединяющий воду. Образующаяся в результате кар-
баминовая кислота IV неустойчива и распадается на диоксид углерода и амин:
„-/
NH2
Вг0,+ОНе
-Н2О
я-/
+ О1-Г
/Р
NHBr
-Вг®, -Н2О
R-C
\
N
~R
O=C=N-R
Ш2О
C-NH-R
СО2 + RNH2
НО
IV
[Г.9.27]
280
Г Препаративная часть (продолжение)
Таблица Г.9.28. Получение аминов деструкцией по Гофману
Продукт реакции
Гидрохлорид метиламина
Гидрохлорид этиламина
Бензиламин
3,4-Диметоксианилин
Антраниловая кислота
Исходный амид
Ацетамид
Пропионамид
Фенилацетамид
3,4-Диметокси-
бензамид
Фталимид
Гипогалогенит
(метод обработки)
NaOBr(a)
NaOBr (a)
NaOBr (б)
NaOCl (б)
B4 мм рт. ст.);
т. пл. 87 (этанол)
NaOBra
Т. кип. и т. пл., °С
Т. пл.227 (этанол)
Т. пл. 108(этанол —
эфир); гигроскопичен
Т. кип. 184
Т. кип. 173
Т. пл. 145 (этанол)
Выход, %
70
70
80
80
60
а Реакционную смесь нейтрализуют соляной кислотой по конго красному. Выпавшую антрамиловую
кислоту перекристаллизопывают из воды при добавлении активированного угля.
Изоцианат является азотистым аналогом кстена, образующегося в ходе
перегруппировки Вольфа.
Если деструкцию по Гофману вести в спиртовом растворе, то образуется
уретан.
В промышленности с помощью деструкции по Гофману из фталимида получают антра-
ниловую кислоту, которая является важным промежуточным веществом в производстве краси-
красителей.
(П)) Общая методика деструкции амидов кислот до аминов по Гофману (табл. Г.9.28)
1. Приготовление раствора гипобромита1. К раствору 6 моль гидроксида натрия в2л воды
добавляют по каплям при 0 °С 1,2 моль брома.
2. Приготовление раствора гипохлорита. 300 г гидроксида калия растворяют в 400 мл во-
воды, охлаждают до 0 °С и смешивают с 1,5 кг измельченного льда, после чего быстро пропуска-
пропускают 85 г хлора.
3. Проведение деструкции по Гофману. В свежеприготовленный охлажденный до —5 °С
раствор гипогалогенита при перемешивании вносят 1 моль амида кислоты2. Если температура
поднимется выше 40 °С, то смесь охлаждают. После перемешивания в течение ночи добавля-
добавляют 20 г сульфита натрия, подкисляют при охлаждении до рН 2, перемешивают еще 15 мин и
снова подщелачивают 50%-ным раствором гидроксида калия.
4. Обработка реакционной смеси.
а) Летучие амины отгоняют с водяным паром, дистиллят насыщают карбонатом калия,
экстрагируют эфиром, экстракт сушат сульфатом натрия и фракционируют. Очень летучие
амины связывают в приемнике соляной кислотой A : 1), дистиллят упаривают и остаток гид-
гидрохлорида амина перекристаллизовывают из этанола.
1 Работать с раствором гипобромита удобнее, но при проведении деструкции по Гофману
растворы гипохлорита обычно дают лучшие выходы.
2 Приготовление из соответствующего галогенангидрида см. разд. Г,7.1.4.2.
9. Перегруппировки 281
б) Если образующийся амин не перегоняется с паром, то реакционную смесь насыщают
карбонатом калия, фильтруют, фильтрат экстрагируют эфиром, эфирный раствор сушат суль-
сульфатом натрия и фракционируют.
Особо летучие амины по данной методике (вариант а) можно получать и п полумикроко-
личествах.
^-Алании из сукцинимида: Кларк X., Бэр Л. В сб.: Синтезы органических
препаратов. Сб. 2. - М.: ИЛ, с. 20, 1949.
9.1.2.2. Деструкция по Курциусу
При деструкции по Курциусу исходным соединением является азид кислоты,
который подвергают термическому разложению:
о О
R-Ce @ ттт* R~cL ~~^~ O=C=N-R [Г.9.29]
Vl-nhni 2 Yj.
Работая в инертном растворителе (например, бензоле), в отличие от деструкции
по Гофману в этом случае можно предотвратить дальнейшее превращение изо-
цианата и выделить его.
Объясните образование дизамещенных мочевин в тех случаях, когда при раз-
разложении азидов не уделено достаточного внимания для защиты от влаги. Какой
продукт реакции образуется при проведении деструкции по Курциусу в спирто-
спиртовом растворе?
В приведенной ниже методике деструкции по Курциусу азид кислоты полу-
получают в водно-ацетоновой среде действием азида натрия на смешанный ангидрид
карбоновой кислоты и моноалкилугольной кислоты. Последний образуется в
реакционной смеси из соответствующей карбоновой кислоты и алкоксикарбо-
нилхлорида (хлоругольного эфира) (см. разд. Г,7.1.4.4). Азиды можно также
получать действием азида натрия на ацилхлориды или действием азотистой
кислоты на гидразиды кислот.
В тех случаях, когда соответствующий азид кислоты заметно разлагается уже
при комнатной или более низкой температуре, получить изоцианат по Курциу-
Курциусу не удается. В этих случаях отщепление азота идет уже при образовании азида
и получающийся изоцианат немедленно реагирует с растворителем (водой).
Ф Общая методика получения изоцианатов из карбоновых кислот деструкцией по Курциусу
(табл. Г.9.30)
Осторожно! Азиды легко взрываются при быстром нагревании или контакте с
серной кислотой! Избегайте их выделения! Работайте в защитных очках! Не
перегоняйте изоцианаты досуха! При разложении азида нагревайте только на
водяной бане.
1. Получение азидов карбоновых кислот. [Weinstock J. J. Org. Chem., 26 A961), 3511.] Втрех-
горлой колбе емкостью 500 мл, снабженной капельной воронкой, мешалкой и термометром,
смешивают 0,085 моль карбоновой кислоты в 150 мл ацетона. Смесь охлаждают до 0 °С льдом
с солью. При этой температуре медленно прибавляют по каплям раствор 0,1 моль триэтилами-
на в 40 мл ацетона, а затем раствор 0,11 моль этоксикарбонилхлорида (этилового эфира хлор-
угольной кислоты) в 40 мл ацетона. Перемешивают 30 мин, а затем также при 0 °С прибавля-
прибавляют по каплям раствор 0,13 моль азида натрия в 30 мл воды. Перемешивают еще 1 ч, после чего
реакционную смесь выливают в 400 мл ледяной воды. Образовавшийся азид трижды экстраги-
282
Г Препаративная часть (продолжение)
Таблица Г.9.30. Получение изоцианатов деструкцией по Курциусу
Продукт реакции
Фенилизоцианат
а-Нафтилизоцианат
р- Нафтилизоцианат
Исходное соединение
Бензойная кислота
а-Нафтойная кислота
р-Нафтойная кислота
Т. кип., °С
(мм рт. ст.)
60 B0)
145A5)
137 A1); 56 (т. пл.)
Выход, %
65
60
70
руют охлажденным до 0 °С толуолом (порциями по 70 мл). Объединенные вытяжки сушат сна-
сначала плавленым сульфатом магния, а затем пентаоксидом фосфора при охлаждении в низко-
низкотемпературном холодильнике или смесью льда с солью.
2. Получение изоцианатов. Трехгорлую колбу, снабженную обратным холодильником и ка-
капельной воронкой, помещают на кипящую водяную баню и по каплям вводят в нее раствор
азида. Перегруппировка происходит с бурным выделением азота. По окончании добавления
нагревают еще 1 ч, растворитель отгоняют в вакууме, а затем перегоняют изоцианат.
9.1.2.3. Реакция Шмидта
Взаимодействие карбонильных соединений с азотистоводородной кислотой в
присутствии сильных кислот сопровождается переносом алкильной группы и
образованием амидов кислот (реакция Шмидта). Собственно перегруппировке
предшествует обычная реакция карбонильной группы (присоединение азотис-
азотистоводородной кислоты и отщепление воды). Реакция с кетонами протекает по
следующей схеме:
s ©
С=О + H-N-NHNI
R' Н
i i ©
R-C-N-NHNI
ОН
R1
-Н2О
©
D = N-NHNI
R1
[Г.9.31]
-N2
©
C = NI
© _
R'-C=N-R
IV
Образовавшийся таким путем карбениевый ион IV реагирует с водой (раст-
(растворителем), давая амид кислоты; с избытком азотистоводородной кислоты он
может образовывать тетразолы:
ОН
*¦-<
© _
R1—C=N-R
N-R
NH-R
_ ©
NH-N=NI
[Г.9.32]
R'-C
N-R
v,
Нитрениевый ион [Г.9.31], III, по-видимому, не является реальным проме-
промежуточным продуктом, поскольку при отщеплении азота от II идет одновремен-
9. Перегруппировки 283
но и миграция группы R. В соответствии со схемой [Г.9.10] следует ожидать, что
мигрировать преимущественно будет заместитель, находящийся в транс-(Е)-
положении по отношению к диазониевой группе:
© _
R. * R'—C=N-R Продукт
C=N-rjl=N| N* [Г.9.33]
R ^- R-C=N-R'
Отщепление воды от I в схеме [Г.9.31] в основном приводит к тому из
?,Z-H3OMepoB II, в котором более объемистый заместитель R находится в
транс-(Е) -положении к диазониевой группе. Поэтому при реакции Шмидта
в случае несимметричных кетонов наблюдается следующий ряд миграцион-
миграционной способности, отличающийся от ряда [Г.9.11а]:
трет-С^Н9 > С6Н6 * мзо-С3Н7 > С2Н5 > СН3 [Г.9.34]
Карбоновые кислоты (R1 = ОН, схема [Г.9.31]) в условиях реакции Шмидта
дают амин, имеющий на один углеродный атом меньше, чем исходная кислота
(ср. с перегруппировкой Курциуса). При этом N-замещенная карбаминовая
кислота, соответствующая амиду V в схеме [Г.9.32], — тот же продукт, который
образуется при деструкции по Гофману [Г.9.27], — немедленно распадается на
амин и диоксид углерода. Из замещенных малоновых кислот таким путем мож-
можно получать а-аминокислоты, поскольку в реакцию вступает лишь одна карбок-
карбоксильная группа. (Напишите схему этой реакции!)
Общая методика проведения реакции Шмидта (табл. Г.9.35)
Внимание! При реакции образуется азотистоводородная кислота — чрезвычайно
ядовитое и взрывчатое вещество. Необходимо работать в хорошо действующем
вытяжном шкафу, за защитным экраном, в защитных очках! См. часть Е.
В трехгорлую колбу на 500 мл, снабженную мешалкой и обратным холодильником с отво-
отводом газа, помещают смесь 0,1 моль карбонильного соединения, 50 мл концентрированной сер-
серной кислоты и 150 мл хлороформа. При комнатной температуре и энергичном перемешивании
в колбу вносят небольшими порциями 0,12 моль азида натрия, следя за тем, чтобы реакция бы-
была не слишком бурной. После внесения всего азида реакционную смесь перемешивают еще 6 ч
при 50 °С (нагревание на водяной бане). Смесь после охлаждения выливают на 400 г толченого
льда, хорошо перемешивают и отделяют хлороформный слой.
Обработка реакционной смеси, а) Амины. Водный слой при охлаждении сильно подщелачи-
подщелачивают концентрированным раствором гидроксида натрия, амин отгоняют с водяным паром в
приемник, содержащий разбавленную соляную кислоту. Упариванием в вакууме получают
гидрохлорид амина. Для получения свободного амина гидрохлорид растворяют в минималь-
минимальном объеме воды и при охлаждении для выделения свободного амина добавляют твердый гид-
роксид натрия. Амин извлекают эфиром, сушат гидроксидом натрия, фракционируют на
30-сантиметровой колонке Вигре.
б) Амиды. Водный слой нейтрализуют концентрированным водным раствором аммиака
при охлаждении. При этом выделяется амид. Твердые амиды отфильтровывают и перекрис-
таллизовывают. Жидкие амиды извлекают хлороформом, сушат сульфатом магния и после
отгонки растворителя фракционируют в вакууме.
Из хлороформного слоя, первоначально отделенного от реакционной смеси, отгонкой
растворителя можно выделить еще небольшое количество амида.
284
Г Препаративная часть (продолжение)
Таблица Г.9.35. Получение аминов и амидов по реакции Шмидта
Продукт реакции
н-Пентиламин
1,4-Диаминобутан
(путресцин)
н-Бутиламин
Анилин
а-Пиперидон
E-валеролактам)
с-Капролактам
Ацетанилид
Пропионанилид
Бути ран ил ид
Бензанилид
N-Ацетил-а-нафтиламин
1,3,4,5-Тетрагидробенз[6]-
азепинон-26
Фенантридон"
Исходное вещество
Гексановая кислота
Адипиновая кислота
Валериановая кислота
Бензойная кислота
Циклопентанон
Циклогексанон
Ацетофенон
Пропиофенон
Бутирофенон
Бензофенон
Метил-а-нафтилкетон
а-Тетралон
Флуоренон
Вариант
а
а
а
аа
б
б
б
б
б
б
б
б
б
Физические константы
Т. кип. 104 °С;л0» 1,4115
Т. кип. 158 "С; т. пл. 27 "С;
т. пл. гидрохлорида 315 °С
(с разл.)
Т. кип. 78 °С; яо2(> 1,4010;
т. пл. гидрохлорида 195 °С
Т. кип. 184 °C;«D201,5863
Т. кип. 137 °С A4 мм рт. ст.);
т. пл. 40 °С
Т.кип. 140°СA2ммрт.ст.);
т. пл. 68 °С
Т. пл. 114 °С (этанол)
Т. пл. 105 °С (водн. этанол)
Т. пл. 96 °С (водн. этанол)
Т. пл. 161 °С (этанол)
Т. пл. 160 °С (этанол)
Т. пл. 141 °С (водн. этанол)
Т. пл. 294 °С (водн. этанол)
Выход, %
70
70
70
60
60
80
97
65
65
80
50
70
90
а При перегонке с паром соляную кислоту в приемник не добавляют, а дистиллят экстрагируют
эфиром.
О
Орнитин из эфира циклопентанон-2-карбоновой-1 кислоты и лизин из
эфира циклогексанон-2-карбоновой-1 кислоты: Adamson D. W. J. Chem. Soc,
1939, 1564.
Пентаметилентетразол (пентетразол) из циклогексанона: Органикум. — М.:
Мир, 1979, т. И, стр. 280.
9.1.2.4. Перегруппировка Бекмана
При обработке оксимов кетонов или альдегидов кислотами (серной кислотой)
или кислотами Льюиса (пентахлоридом фосфора) вначале образуется то же про-
промежуточное соединение, что и при реакции Шмидта (III в [Г.9.31]). В качестве
конечных продуктов образуются амиды карбоновых кислот (перегруппировка
Бекмана):
R'
ЯН +не
R'
© ~R
C = NI —-
© _
R'—C=N
Р
R1—С
NH-R
[Г.9.36]
9. Перегруппировки 285
В этом случае катион II также не появляется как свободная частица: отщеп-
отщепление протонированной гидроксильной группы и перенос заместителя R осуще-
осуществляются из т/?анс-положения одновременно. Промежуточные соединения II и
III существуют в виде ионных пар. Относительная тенденция к перегруппиров-
перегруппировке определяется теми же факторами, что и в случае реакции Шмидта (см. разд.
Г,9.1.2.3). Поэтому, например, из метиларилкетонов образуются преимущест-
преимущественно N-ариламиды уксусной кислоты.
Перегруппировка Бекмана имеет большое промышленное значение как основной способ
производства е-капролактама, полимеризацией которого получают полиамидные волокна и
синтетические материалы.
(ff% Получение е-капролактама из оксима циклогексанона
1. Оксим циклогексанона. В литровой трехгорлой колбе, снабженной мешалкой и ка-
капельной воронкой, растворяют 1,5 моль гидрохлорида гидроксиламина и 1,2 моль кристалли-
кристаллического ацетата натрия в 400 мл воды и нагревают на водяной бане до 60 °С. При перемешива-
перемешивании добавляют по каплям 1 моль циклогексанона, продолжают перемешивание еще 30 мин
при той же температуре, затем охлаждают до 0 "С и выделившийся оксим отфильтровывают.
Водный слой 3 раза экстрагируют эфиром. Твердый оксим сушат в вакуум-эксикаторе, а эфир-
эфирный раствор — сульфатом натрия. Отгоняют эфир, добавляют к остатку твердый оксим и
продукт отгоняют в вакууме. Т. кип. 104 °С при 12 мм рт. ст.; т. пл. 90 °С; выход 70%.
2. z-Капролактам. В стакане емкостью 400 мл при охлаждении и перемешивании (тем-
(температура не выше 20 °С) смешивают 2 моль концентрированной серной кислоты и 1 моль ок-
оксима циклогексанона. Эту смесь прибавляют по каплям к нагретой до 120 "С концентрирован-
концентрированной серной кислоте A,5 моль), помещенной в трехгорлую колбу, снабженную термометром,
мешалкой, капельной воронкой и обратным холодильником. Реакция сильно экзотермична!
Если температура падает ниже 115 °С, то прибавление оксима немедленно прекращают, пока
температура за счет внешнего нагрева снова не повысится до 120 °С (при понижении темпера-
температуры реакция замедляется и если в этот момент прибавить слишком много оксима, то он при
последующем нагревании будет перегруппировываться почти со взрывом!).
После прибавления раствора оксима реакционную смесь нагревают 20 мин при
125—130 °С, а затем охлаждают и выливают на 0,5 кг измельченного льда. Полученную массу
при охлаждении льдом с солью нейтрализуют концентрированным аммиаком (по фенолфта-
фенолфталеину). При нейтрализации температура не должна подниматься выше 20 °С. Е-Капролактам
экстрагируют хлороформом D х 150 мл). Хлороформный раствор промывают водой, сушат
хлоридом кальция, а затем перегоняют в вакууме. Т. кип. 140 °С при 12 мм рт. ст.; т. пл. 68 °С;
выход 80%.
©Полимеризация е-капролактама. В толстостенной пробирке плавят на водяной бане 3 г
очищенного Е-капролактама, к которому добавлена капля серной кислоты. После
этого верхнюю часть пробирки оттягивают на паяльной горелке в тонкий капилляр,
добиваясь того, чтобы пустое пространство над веществом было возможно меньшим. Ампулу
вакуумируют, подсоединив ее с помощью резиновой пробки и стеклянной трубки к водост-
водоструйному насосу, и запаивают под вакуумом. Поликонденсация происходит при 4-часовом наг-
нагревании на металлической бане при 250 °С. После охлаждения содержимое ампулы застывает
в ломкую массу, напоминающую слоновую кость.
Лактамы из циклических кетонов: Olah G. A., Fung A. P. Synthesis, 1979, 537.
9.1.3. ПЕРЕГРУППИРОВКИ У АТОМА КИСЛОРОДА
Соединения с секстетом электронов у атома кислорода RO® (ионы оксения)
формально соответствуют карбенам и нитренам. Уже из положения С, N и О в
периодической системе элементов следует, что энергия ионов оксения выше
286 Г Препаративная часть (продолжение)
энергии карбенов, поэтому их никогда не удается фиксировать как свободные
промежуточные соединения. Перегруппировки у атома кислорода протекают
синхронно; дефицит электронов на атоме кислорода обычно достигается путем
катализируемого кислотами расщепления пероксидов. Наиболее известный
пример такой реакции — синтез фенолов по Хоку:
НзС-С-О-О-Н + Ни Н2О + С-О—(\ /> sp НО-С-О—(ч />
СНз НзС' ^^ -Н° ^ V^
[Г.9.37]
СНз
- о=с + н
Эта реакция имеет большое практическое значение в нефтехимической про-
промышленности для получения фенола и ацетона, однако этим примером и огра-
ограничивается ее применение. В лаборатории перегруппировки пероксидов также
не имеют большого значения, так как их осуществление связано с проблемой
приготовления гидропероксидов и обращения с ними.
Фенол из а,а-диметилбензилгидропероксида: Органикум. — М.: Мир, 1979,
т. 2, с. 283.
Значительно шире используется реакция Байера—Виллигера, в которой
пероксисоединения получают из кетонов или альдегидов и гидропероксикислот
(иногда также из Н2О2) и подвергают перегруппировке in situ:
R4 p R р
С=О + R"—с' HO-C-Q-0-c'
R; io-O-H ft R"
[Г.9.38]
O-R
— н® + о =с'
R» -R"-C0°H R- Я"
Таким путем из линейных кетонов получают эфиры карбоновых кислот, а из
циклоалканонов — лактоны. Тенденция заместителей к миграции здесь соотве-
соответствует рядам [Г.9.11а] и [Г.9.116]. Точно также реагируют и а,р-ненасыщенные
кетоны, причем в этом случае образуются оба продукта миграции как R, так и
ненасыщенного заместителя. В случае альдегидов также мигрируют и R, и Н.
(Напишите уравнения этих реакций!)
(jj| Получение е-капролактона.1 [Fries S. L. J. Am. Chem. Soc, 1949, 71, 2571.] 0,2 моль цикло-
^*^ гексанона и 0,25 моль пербензойной кислоты в 500-600 мл влажного хлороформа остав-
оставляют в темноте при температуре 22-25 "С. Через определенные промежутки времени
содержание пербензойной кислоты определяют тированием, как это описано в разд. Г,7.1.4.3.
Реакция заканчивается приблизительно через 12 ч и поглощение пербензойной кислоты резко
прекращается. Образовавшуюся бензойную кислоту и избыток пербензойной кислоты экстра-
1 Методику получения пербензойной кислоты см. в разд. Г,7.1.4.3. При этом вместо эфира
для экстракции следует применять хлороформ и влажный (мутный) хлороформный раствор
непосредственно использовать далее. Для получения Е-капролактона можно применять и
эфирный раствор пербензойной кислоты, однако выход продуктов в этом случае понижается.
9. Перегруппировки
287
гируют разбавленным раствором гидрокарбоната натрия. Хлороформный слой промывают водой,
сушат сульфатом натрия и перегоняют. Т. кип. 102-104 °С G мм рт. ст.); «о251,4488; выход 71%.
Аналогично из циклопентанона можно получить 8-валеролактон. Эта реакция, однако,
протекает медленнее и для превращения 0,2 моль кетона необходимо около 50 ч. Т. кип.
145-146 °С D0 мм рт. ст.); «D25 1,4352; выход 78%.
К [1,2]-перегруппировкам у электронодефицитного атома кислорода отно-
относится также превращение триалкилборанов (получаемых из олефинов и борана)
в спирты при окислении пероксидом водорода (гидратация против правила
Марковникова), рассмотренное в разд. Г,4.1.8:
R-B + НООН + НО
е
-Н2О
" 0 Л
R-B-O-O-H
B-O-R
Н2О
В-ОН + R-OH
-НО"
R
[Г.9.39]
9.2. [3,3]-ПЕРЕГРУППИРОВКИ
В последние два десятилетия большое значение приобрели синхронные [3,3]-
перегруппировки, отвечающие высоким требованиям к стереохимической нап-
направленности реакций при синтезе природных соединений и фармацевтических
препаратов. Прототипом таких реакций является перегруппировка Коупа:
it
R
R
[Г.9.40]
В исходном соединении один или несколько атомов углерода могут быть за-
заменены на гетероатомы (гетероперегруппировка Коупа).
Такие перегруппировки обычно протекают через перициклическое квази-
квазиароматическое переходное состояние; иными словами, ни на одной стадии ре-
реакции не появляются какие-либо дискретные фрагменты молекул. Это гаранти-
гарантирует очень высокую степень регио- и стереоспецифичности и делает такие пере-
перегруппировки особенно ценными для синтетической органической химии. Как
показано в результате многочисленных экспериментов, переходному состоя-
состоянию отвечает конформация кресла, поскольку в ней 1,1-диаксиальные взаимо-
взаимодействия могут быть минимальными:
конформация
ванны
0,3%
конформация
кресла
99,7%
[Г.9.41]
цис, цис
транс, транс
цис, транс
(Жирными точками обозначены метальные группы.)
288 Г Препаративная часть (продолжение)
Точно так же аналогичное DL-соединение реагирует преимущественно в
конформации кресла, в которой объемные метальные группы находятся в эква-
экваториальном положении (образуется 90% транс,транс-октадиет-2,6 и только
9% цис,цис-окладжна-2,6). (Напишите схемы этих перегруппировок!)
Интересные флуктуирующие структуры наблюдают при вырожденных пе-
перегруппировках Коупа. Быстро протекающая перегруппировка Коупа доказы-
доказывается с помощью ЯМР-спектроскопии для 3,4-гомотрополидена (бицик-
(бициклов, 1,0]октадиен-2,5): при 180 °С к > 103 с~', см. [Г.9.42]. При комнатной
температуре скорость перегруппировки значительно ниже, а при —50 °С пере-
перегруппировка полностью затормаживается. Этот феномен носит название вале-
валентной таутомерии.
[Г.9.42]
Экспериментальное проведение перегруппировки Коупа не представляет ника-
никаких затруднений; здесь требуется только нагревание. В этом отношении особенной
простотой отличается перегруппировка Кляйзена (окса-перегруппировка Коупа), в
которую вступают аллилариловые или аллилвиниловые эфиры, например:
он
[Г.9.43]
COOR' ^^ COOR'
Аллилариловые эфиры легко получаются из аллилбромида и фенолятов
(см. разд. Г,2.6.2). Аллилвиниловые эфиры можно получить путем отщепления
аллилового спирта от диаллилацеталей или спирта от смешанных алкилаллил-
ацеталей (см. также разд. Г,3.1.4; последующую перегруппировку Коупа и пере-
перегруппировку Кляйзена можно проводить без выделения аллиларилового или
аллилвинилового эфира. Аналогично из ортоэфиров карбоновых кислот образу-
образуются (ос-алкоксивинил)аллиловые эфиры (кетенацетали), а из амидацеталей —
(ос-аминовинил)аллиловые эфиры (кетенаминали), которые в результате реак-
реакции Кляйзена перегруппировываются в эфиры или амиды у,5-ненасыщенных
карбоновых кислот соответственно. (Напишите схемы этих реакций!)
Аллилвиниловые эфиры можно получать также по реакции переэтерифика-
ции из алкилвиниловых эфиров и аллилового спирта в присутствии ацетата
ртути или протонных кислот.
Препаративно осуществление перегруппировки Кляйзена очень просто: дос-
достаточно нагреть исходное вещество до температуры, при которой начинается ре-
реакция. Ход реакции контролируют по показателю преломления, температуре
кипения (обычно температура кипения продукта перегруппировки выше, чем ис-
исходного вещества) или с помощью тонкослойной хроматографии. Рекомендуется
проводить реакцию в растворителе, обеспечивающем более равномерный нагрев;
для этой цели особенно удобным оказался 1Ч,М-диметиланилин. В случае неус-
неустойчивых веществ реакцию проводят в вакууме или в атмосфере инертного газа.
9. Перегруппировки
289
©Получение 2-аллилфенола. [ТарбэллД. С. В сб.: Органические реакции. Сб. 2. — М.: ИЛ,
1950, с. 7.] В двугорлой колбе, снабженной обратным холодильником (второе горло
закрыто), нагревают до кипения аллилфениловый эфир. Время от времени из второго
горла отбирают пробы и определяют их показатель преломления. Реакцию считают закончен-
законченной, когда показатель преломления по2Ь достигнет значения 1,54 (обычно для этого требуется
от 5 до 6 ч). Смесь охлаждают, растворяют в двукратном объеме 20%-ного гидроксида натрия
и отделяют образовавшийся в небольшом количестве 2-метил-2,3-дигидробензофуран дважды
экстрагируя его петролейным эфиром (т. кип. 30—60 °С). Щелочной раствор подкисляют и
экстрагируют эфиром, экстракт сушат хлоридом кальция и перегоняют. Т. кип. 103—105 "С
A9 мм рт. ст.); яв24 1,5445; выход 73%.
Из петролейного эфира перегонкой можно выделить 2-метил-2,3-дигидробензофуран.
Т. кип. 86-88 °С A9 мм рт. ст.); nD221,5307.
3-Метилгексен-5-он-2, 4-метилгептен-6-он-2, 2-аллилциклопентанон и 2-ал-
лшщиклогексанон из диаллилкеталей бутанона, пентанона-3, циклопентанона и
циклогексанона: Lorette N. В., Howard W. L. J. Org. Chem., 1961, 26, 3112.
Известна и аналогичная перегруппировке Кляйзена аза-перегруппировка
Коупа. Особый интерес представляет [3,3]-перегруппировка арилгидразонов
альдегидов или кетонов как очень простой путь синтеза индолов (синтез индо-
индолов по Фишеру):
R \-
[Г.9.44]
N ЙН3 -H0,-NH3
Перегруппировке здесь подвергается енамин П. Экспериментами с соедине-
соединениями, меченными 15N, показано, что при замыкании кольца отщепляется тот
атом азота продукта [3,3]-перегрутшировки III, который не находится непосре-
непосредственно у ароматического кольца.
Исходные арилгидразоны I можно получить обычным способом из альдеги-
альдегида или кетона и соответствующего арилгидразина. Фенилгидразоны замещен-
замещенных эфиров пировиноградной кислоты, необходимые для получения 2-этокси-
карбонилиндолов, проще всего получить реакцией Яппа—Клингеманна, при
которой катализируемое основаниями азосочетание и последующее кислотное
расщепление образовавшегося азосоединения приводят непосредственно к
арилгидразону:
R
XOOEt
k/COOEt +HO
(Н0)
-NH3
COOEt
[Г.9.45]
290
Г Препаративная часть (продолжение)
Таблица Г.9.46. Получение индолов по Фишеру
Продукт реакции
5-Метокси-2-этоксикарбонил-
индол
5-Этокси-2-этоксикарбонилиндол
2-Этоксикарбонил-1 Н-бензиндол
2-Этоксикарбонил-ЗН-бензиндол
3-Метил-5-метокси-2-этоксикар-
бонилиндол
3-Метил-5-этокси-2-этоксикар-
бониливдол
З-Метил-2-этоксикарбонил-
1 Н-бензиндол
З-Метил-2-этоксикарбонил-
3 Н-бензиндол
5-Метокси-3-н-пропил-2-этокси-
карбонил индол
3-Фенил-5-этокси-2-этоксикарбо-
нилиндол
Исходные вещества
а-Метилацетоуксусный эфир,
и-анизидин
а-Метилацетоуксусный эфир,
и-фенетидин
а-Метилацетоуксусный эфир,
а-нафтиламин
а-Метилацетоуксусный эфир,
р-нафтиламин
а-Этилацетоуксусный эфир,
и-анизидин
а-Этилацетоуксусный эфир,
л-фенетидин
а-Этилацетоуксусный эфир,
а-нафтиламин
а-Этилацетоуксусный эфир,
E-нафтиламин
а-н-Бутилацетоуксусный эфир,
и-анизидин
а-Бензилацетоуксусный эфир,
и-фенетидин
Т. пл., °С
153 (этанолK
156 (этанол)
170 (этанол с
активированным углем)
161 (петролейный эфир)
147 (этанол)а
167 (этаноле
активированным углем)
176 (этаноле
активированным углем)
176 (этанол с
активированным углем)
106 (этанолK
148 (этанол с
активированным углем)
Предварительно очищают перекристаллизацией из петролейного эфира.
Как изомеризацию арилгидразонов (I в схеме [Г.9.44]) в енамины II, так и об-
образование индольной системы путем элиминирования аммиака катализируют
кислоты. Успех синтеза индолов по Фишеру в значительной степени зависит от
типа и силы кислоты. Пока что здесь не удалось найти общих закономерностей.
В ряде случаев используют полифосфорную кислоту. В одностадийном вариан-
варианте, когда из гидрохлорида арилгидразина, кетона и пиридина в относительно
мягких условиях сразу образуется производное индола, оптимальным катализа-
катализатором часто является образующийся гидрохлорид пиридина, подробнее об этом
см. в указанной ниже литературе.
Ф Общая методика синтеза индолов по Фишеру (получение фенилгидразонов по
Яппу—Клингеманну) (табл. Г.9.46)
11 Осторожно! Ароматические амины — вредные для здоровья вещества! Наф-
Щ тиламины канцерогенны. Во время работы следите за чистотой!
Смешивают охлажденный льдом раствор 0,1 моль а-замещенного ацетоуксусного эфира с
35 мл также охлажденного льдом 50%-ного водного раствора гидроксида калия. Смесь разбавля-
разбавляют 200 мл ледяной воды и затем быстро добавляют при перемешивании раствор соли диазония,
полученный из 0,1 моль амина (см. разд. Г,8.2.1). Перемешивание продолжают еще 5 мин, отде-
отделяют выделяющийся в виде красного масла фенилгидразон, водный слой экстрагируют эфиром.
Соединенные органические фазы сушат сульфатом натрия, растворитель отгоняют. Сырой гид-
разон растворяют в абсолютном этаноле и пропускают сухой газообразный хлороводород до на-
начала выделения хлорида аммония C0-180 мин.). Затем реакционную смесь оставляют на ночь,
выливают в ледяную воду, продукт реакции отфильтровывают или экстрагируют эфиром. Пос-
После удаления растворителя остаток перекристаллизовывают. Выход около 50%.
9. Перегруппировки 291
2,3- Тетраметилениндол, 5-хлор-2,3-тетраметилениндол, 5-метокси-2,3-
тетраметилениндол из циклогексанона и гидрохлорида соответствующего арил-
гидразона в пиридине: Welch W. M. Synthesis, 1977, 645.
Гетероауксин (индолилуксусная-3 кислота) из а-цианэтилацетоуксусного
эфира и анилина: Феофилактов В. В., Семенова Н. К. В сб.: Синтезы органичес-
органических соединений. Сб. 2. 1952, с. 63—69.
Производные индола являются важными природными веществами, особенно триптофан,
входящий в состав белков, и гормон серотонин. Синтез индолов по Фишеру и связанная с ним
реакция Яппа — Клингеманна сыграли большую роль в синтезе подобных природных соеди-
соединений и других биологически активных индолов.
Какие еще методы получения индолов вам известны?
9.3. ЛИТЕРАТУРА
Перегруппировки карбкатионов
BrouwerD. М, Hogeveen H., Prog. Phys. Org. Chim. 9 A972), 179-240.
Kirmse W., in: Topics in Current Chemistry. Bd. 80. — Springer-Verlag Berlin. Heidelberg, New
York 1979, S. 125-311.
Harwood L. M. Polare Umlagerungen. — VCH, Weinheim 1995.
Olah G, SchleyerP. v. R. Carbonium Ions. Bd. 2. — Interscience, New York 1970.
Shubin V. G, in: Topics in Current Chemistry. Bd. 117. — Springer-Verlag, New York 1984,
S. 3269-341.
Перегруппировка Вагнера-Меервейна
Streitwieser A. Chem. Rev., 56 A956), 698.
Перегруппировка Демьянова, реакция Тиффено
Смит П. А. С, БоерД. Р. В сб.: Органические реакции. Сб. 11. — М.: Мир, 1965, с. 167-198.
Пинаколиновая перегруппировка
Collins С. J. Quart. Rev., 14 A960), 357.
Перегруппировка Вольфа. Реакция Арндта-Эйстерта
Бахман В., Струве В. В сб.: Органические реакции. Сб. 1. — М.: ИЛ, 1948.
Непеска #., in: Houben-Weyl. Bd. 8 A952), S. 456-458, 556, 668-669.
Meier H., ZellerK.-R, Angew. Chem. 87 A975), 52.
Ried W., MenglerM., Forschr. Chem. Forsch. 5 A965), 1-88.
Родина Л. Л., Коробицина И. К. Усп. хим., 36 A967), 611-635.
Перегруппировка Стивенса
Pine S. К, Org. React. 18 A970), 403-464.
Stevens Т. S., Watts W. E. Selected Molecular Rearrangements. — Van Nostrand Reinhold,
London, New York 1973, S. 81-116.
Перегруппировка Виттига
Briickner R., Nachr. Chem. Tech. Lab. 38 A990), 1506.
Nakay Т., Mikami K., Chem. Rev. 86 A986), 885.
Nakay Т., Mikami К., Org. React. 46 A994), 105.
SchollkopfU., Angew. Chem. 82 A970), 795.
292 Г Препаративная часть (продолжение)
Электрофильные перегруппировки органических соединений щелочных металлов
Grovenstein E. Angew. Chem. 90 A978), 317.
Деструкция амидов карбоновых кислот по Гофману
Kovacic P., Lowery M. К, Chem. Rev. 70 A970), 639-665.
MollerF., in: Houben-Weyl. Bd. 11/1 A957), S. 854-862.
Уэллис Э. С, ЛэнДж. Ф. В сб.: Органические реакции. Сб. 3. — М: ИЛ, 1951, с. 255-293.
Деструкция по Курциусу
Lwowski W., Angew. Chem. 79 A967), 922.
MollerF., in: Houben-Weyl. Bd. 11/1 A957), S. 862-872.
Смит П. А. С. В сб.: Органические реакции. Сб. 3. — М.: ИЛ, 1951, с. 323-426.
Реакция Лоссена
Bauer L, Exner О. Angew. Chem. 86 A974), 419-428.
Yale H. L. Chem. Rev. 33 A943), 209.
Реакция Шмидта
Колдобский Г. И. и др. Усп. хим., 40 A971), 1790-1813; 47 A978), 2044-2064.
Вольф Г. В сб.: Органические реакции Сб. 3. — М.: ИЛ, 1951, с. 293-321.
Перегруппировка Бекмана
Донарума Л. Г.,Хельд В. 3. Веб.: Органические реакции. Сб. 11. — М.: Мир, 1965, с. 7—166.
Gawley Е. К, Org. React. 35 A988) 1-420.
Кнунянц И. Л., Фабричный Б. П. В сб.: Реакции и методы исследования органических соеди-
соединений. Т. 3. — М.: Химия, 1954, с. 137-251.
MollerF. In: Houben-Weyl, Bd. XI/1, 1957, S. 892-899.
Винник М. К, Захарани Н. Г. Усп. хим. 36 A967) 167-198.
Реакция Хока, окисление по Байеру-Виллигеру
Хасселл Ч. X. В сб.: Органические реакции. Сб. 9. — М.: ИЛ, 1959, с. 82-124.
Hock К, KropfH., Angew. Chem. 69 A957), 313-321.
KropfH., in: Houben-Weyl, Bd. E13 A988), 1085-1094.
Wedemeyer K.-F, in: Houben-Weyl, Bd. 6/lc A976), 117-139.
Перегруппировки Коупа и Кляйзена
BartlettP. D., Tetrahedron 36 A980), 2-72.
Bennett G. В., Synthesis 1977, 589.
BlechertS., Synthesis 1989, 71-81.
Enders D., Knopp M., Schiffers R., Asymmetric [3,3]-Sigmatropic Rearrangements in Organic
Synthesis, Tetrahedron. Asymmetry 7 A996), 184-1882.
Rhoads S. J., Raulins N. R., Org. React. 22 A975), 1-252.
Smith G. G., Kelly F W., Prog. Phys. Org. Chem. 8 A971), 75-234.
ТарбэллД. С. В сб.: Органические реакции. Сб. 2. — М.: ИЛ, 1950, с. 7-60.
WehrliR., BellusD., HansenH.-J., SchmidH., Chimia30 A976), 416.
Winterfeld E., Fortschr. Chem. Forsch. 16 A970), 75.
Синтез индолов по Фишеру
Dopp К, Dopp D., Longer U., GerdingB., in: Houben-Weyl, Bd. E6bi/2a A994), 704-753.
Грандберг И. И., Сорокин В. И. Усп. хим., 43 A974), 266-293.
КитаевЯ. П. Усп. хим., 28 A959), 336-368.
Robinson В., Chem. Rev. 64 A963), 373-401; 69 A969), 227-250.
Robinson В., The Fischer Indole Synthesis. — John Wiley & Sons, New York 1983.
The Chemistry of Indole. — Academic Press, New York, London 1970.
ИДЕНТИФИКАЦИЯ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
1. ПРЕДВАРИТЕЛЬНЫЕ ИСПЫТАНИЯ И
ОБНАРУЖЕНИЕ ФУНКЦИОНАЛЬНЫХ ГРУПП
Идентификацию органических веществ (основные данные о составе исследуе-
исследуемых соединений) можно осуществить с помощью химических реакций, т. е.
простейшими методами, не прибегая к использованию дорогостоящего обору-
оборудования. Из-за неисчерпаемого многообразия органических соединений
невозможно создать стройную схему систематической идентификации, подоб-
подобную существующей в неорганическом качественном анализе.
Прежде чем начать исследование вначале следует установить, имеем ли мы
дело с индивидуальным веществом или смесью веществ. Для решения данного
вопроса лучше всего воспользоваться хроматографическими методами иденти-
идентификации — тонкослойной хроматографией (см. разд. А.2.7.1), газовой хрома-
хроматографией (ГХ, разд. А,2.7.4) и высокоэффективной жидкостной хроматогра-
хроматографией (ВЭЖХ, разд. А,2.7.3). Если анализируемый образец представляет собой
смесь веществ, то вначале следует попытаться разделить их физическими мето-
методами (фракционной перегонкой, перекристаллизацией). Для препаративного
разделения смеси веществ вполне подходят также вышеупомянутые хромато-
графические методы разделения. Но вначале следует попытаться разделить
смесь веществ с помощью химических реакций (особенно в тех случаях, когда
это не требует больших материальных затрат). Обратите внимание на указания,
приведенные в разд. Д,3.
Чистые вещества, а в отдельных случаях также и смеси можно изучать спект-
спектроскопическими методами. Комбинируя их с описываемыми ниже предва-
предварительными испытаниями (зачастую эта информация используется недоста-
недостаточно эффективно), обычно можно сделать важные предварительные выводы о
структуре, а в благоприятных случаях этой информации достаточно для того,
чтобы предложить для изучаемого соединения конкретную структуру.
В разд. Д,2 представлены типичные ИК- и ЯМР-спектры важнейших клас-
классов органических соединений. В сочетании с табл. А. 135, А. 145, А. 148 и А. 153
они должны облегчить интерпретацию спектров неизвестных соединений.
В разд. ДД.2 описаны химические способы обнаружения функциональных
групп. (Органический химический качественный анализ очень полезен с педа-
педагогической точки зрения.) Приведенные здесь предварительные испытания и
294 Д Идентификация органических соединений
методы получения производных позволяют овладеть навыками эксперимен-
экспериментального искусства в области органической химии при работе с небольшими ко-
количествами веществ. (Кроме того, изучение простых реакций часто позволяет
сделать столь же ценные выводы, как и эксперименты на более дорогом и слож-
сложном оборудовании.)
Идентификация неизвестного соединения, с помощью определенных реак-
реакций в силу множества возможных сочетаний последних как никакая другая
практическая работа развивает химическое мышление, закрепляет знание
свойств соединений и таким путем подготавливает к работе в области органи-
органического синтеза.
В предварительных испытаниях сначала выявляют функциональные группы
неизвестного соединения и затем переводят его с помощью соответствующих
реагентов в кристаллическое производное. Если соединение способно подвер-
подвергаться гидролизу, то его сначала гидролизуют на фрагменты, каждый из которых
далее исследуют индивидуально.
Обычно для идентификации достаточно сравнения температур плавления
двух-трех производных неизвестного вещества с данными, собранными в табли-
таблицах (разд. Д,2). Дополнительным доказательством природы вещества может слу-
служить определение молекулярной или эквивалентной массы. Для окончательных
выводов часто проводят еще специальные реакции, описанные для отдельных
веществ в литературе.
Для однозначного доказательства строения вещества достаточно сравнить
его ИК-спектр с ИК-спектром предполагаемого оригинала.
Другие общие указания:
а) Расход вещества для анализа. Если использовать приведенные ниже ме-
методики, то для анализа потребуется не более 5 г вещества. Из этого количества
1—2 г расходуются на предварительные испытания и обнаружение функцио-
функциональных групп, еще 2 г используются для идентификации, остальное является
резервом, позволяющим при необходимости повторить отдельные операции.
Все это позволяет очень экономно расходовать вещество, в особенности при
предварительных испытаниях. Ставя опыты по определению функциональных
групп, следует иметь в виду, что во многих из этих опытов могут быть получе-
получены производные, пригодные для идентификации.
б) Роль контрольных опытов. Для новичка важно с помощью контрольных опы-
опытов приобрести навык в проведении аналитических операций и уверенность в по-
получаемых результатах. С этой целью ставят опыты двух типов. Во-первых, проводят
реакцию в указанных условиях, но в отсутствие анализируемого вещества; это поз-
позволяет обнаружить помехи, вызванные наличием загрязнений в растворителях или
реагентах (например, при цветных реакциях или при окислении раствором пер-
манганата калия в ацетоне; разд. ДД.2.1.2). Во-вторых, если проба, содержащая
анализируемое вещество, дала отрицательный результат, то ставят контрольный
опыт, добавляя такое соединение, которое должно дать ожидаемую реакцию; тем
самым проверяют правильность выполнения анализа. (Например, при отрица-
отрицательном результате на карбонильные соединения добавляют немного ацетона;
если выпадает осадок, то все условия анализа соблюдены.)
в) Подготовка вещества. Анализируемое вещество должно быть чистым.
Поэтому жидкие пробы перегоняют (при необходимости в вакууме). Индивиду-
1. Предварительные испытания и обнаружение функциональных групп 295
альность собранной фракции проверяют газохроматографически. Твердые
вещества после проб на растворимость (см. разд. Д, 1.1.5) перекристаллизовыва-
ют до тех пор, пока вещество не будет иметь постоянную температуру плавле-
плавления, а затем с помощью тонкослойной хроматографии проверяют его чистоту.
Перед проведением всех опытов следует обратить внимание на технику безопасности при
работе с используемыми реактивами и анализируемыми веществами (см. части Е и Ж)!
1.1. ПРЕДВАРИТЕЛЬНЫЕ ИСПЫТАНИЯ
1.1.1. ВНЕШНИЙ ВИД ВЕЩЕСТВА
а) Цвет. Большинство чистых веществ бесцветно. Поэтому в случае окрашен-
окрашенных веществ необходимо проверить, сохраняется ли окраска вещества после
перекристаллизации (перегонки) или она обусловлена примесями.
Окрашены следующие важные классы соединений: нитросоединения, нит-
розосоединения (только в мономерной форме), азосоединения, хиноны. Арома-
Ароматические амины и фенолы, особенно полифункциональные, окрашены в желто-
коричневый цвет; эта окраска обусловлена следовыми количествами продуктов
окисления. Последние, однако, обычно не влияют на реакции аминов и фено-
фенолов, и тщательной очистки можно не проводить.
б) Запах. Характерным запахом обладают определенные классы соединений:
углеводороды терпенового ряда (камфен, карен, пинен), а также циклогексанон,
пинаколин, /и/>ет-бутанол (характерный «терпеновый» запах); низшие спирты;
низшие жирные кислоты (муравьиная и уксусная кислоты имеют очень резкий
запах; пропионовая и следующие — неприятный запах пота); низшие кетоны;
альдегиды; галогеноуглеводороды (дурманяще-сладкий запах); фенолы (запах
«карболки»); эфиры фенолов (анисовый или укропный запах); ароматические
нитросоединения (запах горького миндаля); сложные эфиры алифатических
спиртов (фруктовые запахи); изонитрилы (неприятно-сладкий запах), меркапта-
меркаптаны, тиоэфиры и т. п. (неприятный запах, напоминающий запах сероводорода).
в) Вкус. Упоминаемую в старых работах пробу на вкус ни в коем случае нель-
нельзя рекомендовать, так как значительная часть органических веществ физиологи-
физиологически активна даже в малых дозах.
1.1.2. ОПРЕДЕЛЕНИЕ ФИЗИЧЕСКИХ КОНСТАНТ
Определение физических констант (температур плавления и кипения, показате-
показателя преломления, плотности) и их значение описаны в разд. А,3. Вместе с темпе-
температурой плавления, определяемой с помощью микроскопа с нагреваемым сто-
столиком, следует обращать внимание и на форму кристаллов, а также на возгонку,
кристаллизационной воды и другие явления.
1.1.3. ПРОБА НА ГОРЮЧЕСТЬ И ЗОЛЬНОСТЬ
Несколько капель или несколько кристалликов вещества нагревают и отмечают происходя-
происходящие при этом изменения внешнего вида, цвета, запаха и также появление летучих веществ.
Если вещество горит, надо обратить внимание на цвет пламени; слабо светящее, почти си-
синее пламя указывает на кислородсодержащее соединение (спирт, эфир и т. п.); светящее, жел-
296 Д Идентификация органических соединений
тое, как правило, коптящее пламя — на соединения, содержащие ненасыщенные углеводоро-
углеводороды (ароматические углеводороды, ацетилены и т. п.).
Если после нагревания остается твердый остаток, то его прокаливают до полного окисле-
окисления углеродсодержащих составных частей и анализируют неорганический остаток. Если в
последнем обнаруживают оксиды или карбонаты металла, можно сделать вывод, что исследу-
исследуемое вещество является солью соединения кислотного характера (карбоновой кислоты, фено-
фенола и т. п.). Если остаток содержит сульфид, сульфит или сульфат, то можно предположить, что
исследуемое соединение представляет собой либо бисульфитное соединение альдегида или
кетона, либо соль сульфиновой или сульфоновой кислоты, либо тиолат.
1.1.4. ОБНАРУЖЕНИЕ ЭЛЕМЕНТОВ
Проба Бейлыитейна (обнаружение галогенов). Прокаленную медную проволочку смачивают
исследуемой жидкостью или помещают на нее несколько кристалликов исследуемого вещест-
вещества и вносят в бесцветное пламя газовой горелки. Образующиеся при сгорании летучие галоге-
ниды меди окрашивают пламя в зеленовато-голубой цвет.
Проба очень чувствительна, поэтому отрицательный результат является на-
надежным свидетельством об отсутствии галогенов! Азотсодержащие органичес-
органические вещества часто так же окрашивают пламя, хотя в них нет галогенов.
Наряду с типичными для органических веществ элементами — углеродом,
водородом и кислородом — в них могут содержаться также азот, сера, галогены.
Для обнаружения этих элементов вещество сплавляют с металлическим натри-
натрием, переводя их в растворимую форму:
Na
С, Н, О, N, S, Hal -> Na2S, NaCN, NaHal, NaSCN [Д.1]
Сплавление органических соединений с натрием
Щ Осторожно! Реакции с нитроалканами, органическими азидами, диазоэфирами,
3 диазосоединениями и некоторыми алифатическими полигалогенидами могут
У идти со взрывом. Реакцию и последующее разложение сплава надо проводить за
^ опущенным стеклом тяги и в защитных очках.
5—20 мг вещества помещают в тугоплавкую пробирку для прокаливания. Туда же помеща-
помещают свежеочищенный кусочек натрия длиной около 4 мм так, чтобы он лег чуть выше исследу-
исследуемого вещества. Натрий расплавляют коптящим пламенем горелки; он плавится и горячим1
стекает на вещество. Затем пробирку нагревают до темно-красного каления (часто смесь силь-
сильно обугливается) и еще горячей опускают в маленький стаканчик, в который налито 5 мл
дистиллированной воды. Пробирка лопается, получающийся раствор натриевых солей фильт-
фильтруют и используют для обнаружения гетероэлементов.
Если вещество при смешивании или нагревании с натрием реагирует со взрывом, то посту-
поступают следующим образом: 0,1 г вещества растворяют в 1—2 мл ледяной уксусной кислоты. При-
Прибавляют 0,1 г цинковой пыли и нагревают так, чтобы смесь слабо кипела, пока не растворится
весь цинк. Раствор упаривают досуха и остаток обрабатывают натрием, как описано выше.
Если описываемые ниже реакции обнаружения элементов дают отрицатель-
отрицательный результат, то для надежности операцию повторяют 2—3 раза, добавляя боль-
большее количество натрия.
1 Жидкие вещества рекомендуется вначале обрабатывать натрием без нагревания. Если при
этом происходит выделение водорода, то это указывает на кислый характер соединений — кис-
кислоты, спирты, С-Н-кислоты и т. п.
1. Предварительные испытания и обнаружение функциональных групп 297
Обнаружение азота (проба Лассеня). К 1 мл раствора, полученного после сплавления изу-
изучаемого вещества с натрием, добавляют 0,5 мл водного раствора сульфата железа(П), кипятят
1—2 мин, затем прибавляют 2 капли раствора хлорида железа(Ш), еще раз нагревают и после
охлаждения подкисляют до слабокислой реакции. Если вещество содержит азот, то выпадает
осадок голубой берлинской лазури (или по крайней мере появляется зеленовато-голубая
окраска). Окраску особенно легко заметить, если раствор интенсивно взболтать и нанести нес-
несколько капель на фильтровальную бумагу.
В присутствии серы обнаружение азота может быть затруднено. В таких случаях повторя-
повторяют сплавление с натрием с двойным количеством последнего и проводят определение азота,
добавляя большее количество сульфата железа(Н) (почему?).
Обнаружение серы. К 1-2 мл профильтрованного раствора, полученного после сплавления
с натрием и подкисленного уксусной кислотой, добавляют несколько капель раствора ацетата
свинца. Выпадение черного осадка указывает на присутствие серы. Более чувствительна дру-
другая реакция, когда к 0,5 мл раствора, полученного после сплавления с натрием, добавляют
2 капли водного раствора динатрийпентацианонитрозилферрата(Ш) (нитропруссида натрия).
В присутствии серы появляется фиолетовое окрашивание.
Обнаружение галогенов. Раствор, полученный после сплавления с натрием, подкисляют
концентрированной HNO3 и галогены, как обычно, обнаруживают с помощью нитрата сереб-
серебра. При наличии в веществе азота образовавшуюся синильную кислоту необходимо до прибав-
прибавления нитрата серебра удалить нагреванием на кипящей водяной бане.
Конкретные галогены обнаруживают по известным методикам неорганического анализа.
Бромиды в присутствии хлоридов и иодидов можно обнаружить также с помощью очень
чувствительной эозиновой пробы: 0,5 мл раствора подкисляют несколькими каплями концент-
концентрированной серной кислоты до кислой реакции и добавляют 3—5 капель концентрированного
раствора перманганата. Стаканчик прикрывают бумагой, пропитанной раствором флуоресце-
ина, и нагревают до 40—50 "С. Через 15 мин бумагу помещают в камеру с парами аммиака.
В присутствии брома появляется розовое окрашивание.
Для обнаружения фтора 1 мл раствора упаривают досуха, добавляют 0,5 мл концентриро-
концентрированной серной кислоты, немного дихромата калия и сильно встряхивают. Стенки пробирки
смачиваются. Осторожно нагревают и снова встряхивают: в присутствии фтора повторного
смачивания не происходит.
Фтор можно обнаружить также с помощью циркон-ализариновой пробы: 2 мл раствора под-
подкисляют уксусной кислотой и нагревают до кипения. Несколько капель полученного раствора
наносят на циркон-ализариновую индикаторную бумагу. В присутствии фтора бумага обесц-
обесцвечивается либо окрашивается в желтый цвет.
1.1.5. ОПРЕДЕЛЕНИЕ РАСТВОРИМОСТИ
Определение растворимости имеет большое значение, так как зная раствори-
растворимость, можно сделать определенные выводы относительно полярности молеку-
молекулы и наличия конкретных функциональных групп. Одновременно можно полу-
получить сведения о том, как очистить твердое вещество (подбор растворителя для
перекристаллизации) и удастся ли таким путем разделить смесь.
Целесообразно определять растворимость вещества в нескольких раствори-
растворителях в следующем порядке: вода, эфир, 5%-ный водный раствор гидроксида
натрия, 5%-ный водный раствор гидрокарбоната натрия, 5%-ный раствор соля-
соляной кислоты, концентрированная серная кислота. Далее — в этаноле, толуоле,
ледяной уксусной кислоте, петролейном эфире (чтобы найти растворитель для
перекристаллизации и разделения смесей).
К 0,01—0,1 г исследуемого вещества небольшими порциями добавляют около 3 мл раство-
растворителя, хорошо перемешивая после добавления каждой порции. Если вещество нерастворимо
в воде, то определяют растворимость в разбавленной NaOH, в растворе NaHCO3, HC1. При
смешивании осторожно встряхивают, нерастворившиеся вещества отделяют, а полученный
298 Д Идентификация органических соединений
водный раствор нейтрализуют, наблюдая, происходит ли повторное выделение исходного ве-
вещества. При работе с указанными количествами веществ даже помутнение нейтрализованно-
нейтрализованного фильтрата следует считать признаком присутствия соединений с кислотными или основны-
основными свойствами. При растворении в NaHCO3 необходимо следить, не выделяется ли при этом
диоксид углерода!
Если при комнатной температуре вещество не растворяется, смесь быстро нагревают.
В этом случае необходимо убедиться (особенно после нагревания с кислотами или щелочами),
что не произошел гидролиз или иное необратимое изменение вещества. (Вещество выделяют
и определяют температуру плавления или кипения!)
Выводы, которые можно сделать по растворимости вещества.
а) Растворимость в воде и эфире. По различию растворимости в воде и эфире
все органические соединения можно подразделить на следующие основные
группы:
I. Растворимые в воде, но нерастворимые в эфире.
П. Растворимые в эфире, но нерастворимые в воде.
III. Растворимые и в воде, и в эфире.
IV. Нерастворимые и в воде, и в эфире.
Группа I. Соединения, в которых преобладают полярные группировки: соли,
полиолы, сахара, аминоспирты, гидроксикарбоновые кислоты, ди- и поликар-
боновые кислоты, амиды низших кислот, алифатические аминокислоты, суль-
фоновые кислоты.
Группа II. Соединения, в которых преобладают неполярные группировки: уг-
углеводороды и их галогенопроизводные, простые и сложные эфиры, спирты
более чем с пятью С-атомами, высшие альдегиды и кетоны, высшие оксимы,
средние и высшие карбоновые кислоты, ароматические карбоновые кислоты,
ангидриды карбоновых кислот, лактоны, сложные эфиры, высшие нитрилы и
амиды кислот, фенолы, тиофенолы, высшие амины, хиноны, азосоединения.
Группа III. Соединения, в которых влияние полярных и неполярных группи-
группировок одинаково: низшие алифатические спирты, низшие алифатические аль-
альдегиды и кетоны, низшие алифатические нитрилы, амиды и оксимы, низшие
циклические простые эфиры (тетрагидрофуран, 1,4-диоксан), низшие и сред-
средние карбоновые кислоты, гидрокси- и оксокарбоновые кислоты, дикарбоновые
кислоты, многоатомные фенолы, алифатические амины, пиридин и его гомоло-
гомологи, аминофенолы.
Группа IV. Высококонденсированные углеводороды, амиды высших кислот,
антрахиноны, производные пурина, некоторые аминокислоты (цистин, тиро-
тирозин), сульфаниловая кислота, высшие амины и сульфонамиды, высокомолеку-
высокомолекулярные соединения.
б) Растворимость в основаниях и кислотах. Выполняя эти пробы, всегда необ-
необходимо проверять, не произошло ли с веществом каких-либо изменений. В осо-
особенности однозначно этим реакциям подвержены соединения, относящиеся к
группам II и IV, поскольку в результате солеобразования вещество обычно при-
приобретает способность растворяться в воде. У веществ, относящихся к группам I
и III (т. е. растворимым в воде), следует предварительно определить рН водного
раствора с помощью индикаторной бумаги.
В разбавленной соляной кислоте растворимы алифатические и аромати-
ароматические амины (растворимость последних сильно падает с увеличением числа
1. Предварительные испытания и обнаружение функциональных групп 299
арильных групп: дифениламин почти нерастворим, а трифениламин совсем
нерастворим).
Растворимы и в гидроксиде натрия, и гидрокарбонате натрия сильнокислые
вещества, а именно: карбоновые, сульфоновые и сульфиновые кислоты, неко-
некоторые сильнокислые фенолы (нитрофенолы, 4-гидроксикумарин) и др. Только
в гидроксиде натрия растворимы фенолы, некоторые енолы, имиды, первичные
алифатические нитросоединения, незамещенные или монозамещенные (по
атому азота) арилсульфонамиды, оксимы, тиофенолы, тиолы.
Органические основания при действии щелочей выделяются из своих солей.
При этом они выпадают в кристаллическом виде или в виде маслообразных ве-
веществ. Можно обнаружить их и по запаху. Жирные кислоты, содержащие более
12 атомов углерода, при добавлении щелочей уже не дают прозрачных раство-
растворов; вместо этого образуются типичные опалесцирующие мыла.
Р-Дикарбонильные соединения, образующие соли со спиртовым раствором
гидроксида калия, нельзя нейтрализовать 5%-ным водным раствором гидрокси-
да натрия.
Некоторые вещества растворяются как в кислотах, так и в щелочах. К числу
таких (амфотерных) веществ относятся аминокислоты, аминофенолы, амино-
сульфоновые и аминосульфиновые кислоты и другие соединения.
в) Растворимость в концентрированной серной кислоте. Растворение в кон-
концентрированной серной кислоте часто сопровождается химическими превраще-
превращениями, признаками которых является разогревание, выделение газов и т. д. Про-
Проба с серной кислотой не позволяет отнести вещество к одной из указанных выше
групп, однако она дает возможность сделать другие полезные выводы: ненасы-
ненасыщенные соединения превращаются в водорастворимые эфиры серной кислоты;
кислородсодержащие соединения обычно переходят в раствор с образованием
солей оксония (если органическая группировка содержит не более 9—12 атомов
углерода); спирты этерифицируются или дегидратируются; олефины могут поли-
меризоваться; некоторые углеводороды сульфируются; трефенилкарбинол, фе-
фенолфталеин и родственные соединения обнаруживаются по галохромному
эффекту; иодсодержащие соединения разлагаются с выделением иода.
1.2. ОБНАРУЖЕНИЕ ФУНКЦИОНАЛЬНЫХ ГРУПП
Определение растворимости, обнаружение гетероатомов, определение физичес-
физических констант (температур плавления и кипения, молекулярной массы и т. д.), а
также цвет вещества — все эти данные дают возможность сделать определенные
выводы относительно анализируемого соединения. Для дальнейшего сужения
круга подлежащих рассмотрению типов соединений применяют спектроскопи-
спектроскопические методы.
Другую возможность дает классический путь: испытания при помощи
несложных, выполняемых в очень короткое время реакций, сопровождающихся
такими характерными изменениями, как выпадение осадка, изменение цвета,
образование веществ с характерным запахом, изменение растворимости.
При огромном числе органических соединений высокая вероятность одно-
одновременного присутствия в молекуле нескольких функциональных групп может
300 Д Идентификация органических соединений
затруднять их обнаружение. Несмотря на эти затруднения, можно распознать и
такие соединения, если учитывать влияние всех имеющихся в молекуле функци-
функциональных групп на результаты выполняемой реакции [см. разд. Д,4; вопросы
A.1)-A-5)нас. 358)].
В этой связи следует предостеречь от переоценки значения цветных реакций,
описываемых в литературе как характерных для отдельных соединений: во мно-
многих случаях подобные реакции показывают и другие вещества.
1.2.1. ОБНАРУЖЕНИЕ НЕНАСЫЩЕННЫХ СОЕДИНЕНИЙ
Обнаружение ненасыщенных соединений принципиально возможно с по-
помощью реакций присоединения (см. разд. Г,4). Здесь для этой цели рекоменду-
рекомендуется использовать взаимодействие с бромом в тетрахлорметане или с перманга-
натом (обесцвечивание). Другие данные о наличии ненасыщенных соединений
дает ИК-спектроскопия; кроме того, в спектрах ЯМР 'Н по типичным химичес-
химическим сдвигам можно обнаружить винильные, ацетиленовые и ароматические
протоны.
1.2.1.1. Реакция с бромом
Методику проведения реакции см. в разд. Г,4.1.4.
Тетрахлорметан (тетрахлоруглерод) как растворитель предпочтителен, по-
поскольку он не растворяет бромоводород; поэтому, если бром расходуется на
реакцию замещения, это легко обнаруживается по выделению бромоводорода.
Границы применения: бром присоединяется не ко всем олефинам. Если соеди-
соединение имеет заместители у двойной связи, обладающие —/- или —М-эффектом, то
это либо замедляет реакцию, либо вовсе предотвращает ее. Пространственно зат-
затрудненные олефины присоединяют бром часто лишь в ледяной уксусной кислоте
или воде. Алифатические и ароматические амины также обесцвечивают бром; при
этом может быть сделан ошибочный вывод о присутствии двойной связи.
Ввиду возможности реакций замещения с выделением бромоводорода ана-
аналитическая ценность реакции с бромом не очень высока. Реакции замещения
особенно типичны для енолов, фенолов, метилкетонов, малоновых эфиров.
Легко окисляющиеся соединения, например тиолы, мешают реакции с бромом.
1.2.1.2. Реакция с перманганатом
Эту реакцию всегда следует дополнять реакцией с бромом!
0,1 г вещества растворяют в 2 мл воды или ацетона (ацетон должен быть чистым, не обес-
обесцвечивать перманганат; в противном случае сначала добавляют перманганат до тех пор, пока
он не перестанет обесцвечиваться) и по каплям прибавляют 2%-ный раствор перманганата.
Пробу считают отрицательной, если обесцвечивается не более трех капель.
Границы применения: окисление перманганатом (см. разд. Г,6.2.1) желательно
проводить как дополнение к реакции с бромом. Так, высокосопряженные оле-
олефины реагируют с перманганатом, в то время как бром они присоединяют с тру-
трудом. Естественно, что положительный тест дают и все легко окисляющиеся
1. Предварительные испытания и обнаружение функциональных групп 301
вещества (енолы, фенолы, тиолы, тиоэфиры, амины, альдегиды, эфиры муравь-
муравьиной кислоты, спирты), поэтому их присутствие в качестве основного компо-
компонента или примесей может быть причиной неверных выводов.
1.2.2. ОБНАРУЖЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Большинство приведенных в разд. Г,5 реакций замещения принципиально при-
пригодно для обнаружения аренов. Ценные сведения дают результаты спектраль-
спектральных исследований.
1.2.2.1. Реакция с азотной кислотой
jj Осторожно! Реакция может идти очень бурно; см. разд. Г,5.1.3.
К 0,1 г вещества медленно, при постоянном встряхивании добавляют 3 мл нитрующей
смеси A часть дымящей азотной кислоты плюс 1 часть концентрированной серной кислоты)
и нагревают 5 мин под тягой на водяной бане при 45—50 °С. Затем смесь выливают на 10 г из-
измельченного льда и отделяют выделившееся маслообразное или твердое вещество.
Наличие нитрогруппы в продукте реакции подтверждается ее восстановле-
восстановлением цинком в присутствии хлорида аммония. Образующийся при этом фенил-
гидроксиламин восстанавливает аммиачный раствор нитрата серебра (реактив
Толленса) до металлического серебра.
0,3 г вещества растворяют в 10 мл 50%-ного этанола, прибавляют 0,5 г хлорида аммония и
0,5 г цинковой пыли. Смесь встряхивают и кипятят 2 мин. После охлаждения фильтруют и до-
добавляют реактив Толленса (см. часть Е, с. 381). Выделение металлического серебра указывает
на присутствие нитро- или нитрозогруппы.
Границы применения: см. разд. Г,5.1.3.
1.2.2.2. Реакция с хлороформом и хлоридом алюминия
К 2 мл сухого хлороформа прибавляют 0,1 г вещества, затем осторожно добавляют 0,5 г безвод-
безводного хлорида алюминия (часть его должна оставаться на стенках пробирки). Появление окрас-
окраски (она может быть различной) указывает на присутствие ароматического соединения.
Эта реакция и границы ее применения подробно изучены в работах: Talsky G. Z.
Anal. Chemie, 188 A962), 416; 191 A962), 191; 195 A963), 171.
1.2.3. ОБНАРУЖЕНИЕ ВЕЩЕСТВ,
ЯВЛЯЮЩИХСЯ СИЛЬНЫМИ ВОССТАНОВИТЕЛЯМИ
(РЕАКЦИЯ С АММИАЧНЫМ РАСТВОРОМ СОЛИ СЕРЕБРА)
Сильные восстановители выделяют металлическое серебро из аммиачного раст-
раствора оксида серебра.
0,05 г вещества помещают в чистую пробирку (предварительно ее моют горячей концент-
концентрированной азотной кислотой) и приливают 2-3 мл свежеприготовленного реактива Толлен-
Толленса (см. часть Е, с. 381). Если серебряное зеркало на холоду не образуется, пробирку нагревают
до 60-70 "С.
302 Д Идентификация органических соединений
Положительная реакция свидетельствует о присутствии альдегидов, восста-
восстанавливающих Сахаров, а-дикетонов, а-гидроксикетонов, многоатомных фено-
фенолов, а-нафтолов, аминофенолов, гидразинов, гидроксиламинов, ос-алкокси-
или ct-диалкиламинокетонов и некоторых других соединений. Реакцию дают и
некоторые ароматические амины, например л-фенилендиамин.
1.2.4. ОБНАРУЖЕНИЕ АЛЬДЕГИДОВ И КЕТОНОВ
Альдегиды и кетоны можно обнаружить по характерным полосам поглощения в
ИК-спектрах (см. табл. А.135вт. 1,с. 117). Кроме того, в спектре ЯМР'Н альдегид-
альдегидный протон легко узнать по сильному сдвигу его резонансного сигнала в слабое
поле; карбонильный атом углерода также легко обнаружить в спектре ЯМР 13С.
1.2.4.1. Реакция с динитрофенилгидразином
На присутствие альдегидов и кетонов указывает образование осадка 2,4-динит-
рофенилгидразонов.
Методика: см. разд. Г,7.1.1.
Границы применения: при действии кислого раствора реагента большинство
ацеталей, кеталей, оксимов и азометинов гидролизуется, а образующиеся карбо-
карбонильные соединения выпадают в виде 2,4-динитрофенилгидразонов. Реакция
непригодна для гидроксикетонов (ацилоинов).
Для отличия кетонов от альдегидов можно использовать более легкую окис-
ляемость последних.
1.2.4.2. Реакция с фелинговой жидкостью
0,05 г вещества и 2—3 мл фелинговой жидкости (приготовление см. в части Е, с. 387) нагревают
5 мин на кипящей водяной бане.
Тест на альдегиды и кетоны считают положительным, если выпадает желто-
желтовато-красный оксид медиA).
Границы применения: ароматические альдегиды, как правило, не дают этой
реакции. Реакции мешает присутствие других сильных восстановителей (см.
разд. Д, 1.2.3).
1.2.4.3. Реакция с фуксинсернистой кислотой (реактивом Шиффа)
К двум каплям или 0,05 г вещества прибавляют 2 мл реактива Шиффа (приготовление см.
в части Е, с. 381) и раствор тщательно встряхивают.
Реакцию считают положительной, если раствор окрашивается в розовый или
фиолетовый цвет.
Границы применения: реакцию не дают глиоксаль, сахара, ароматические
гидроксиальдегиды, а,р-ненасыщенные альдегиды. Вещества, легко поглощаю-
поглощающие диоскид серы, могут создать ложное представление о присутствии альдеги-
альдегидов.
1. Предварительные испытания и обнаружение функциональных групп 303
1.2.5. ОБНАРУЖЕНИЕ СПИРТОВ, ФЕНОЛОВ, ЕНОЛОВ
Обратите внимание на характеристические полосы поглощения этих веществ в
ИК-спектре (см. табл. А.135).
Соединения с гидроксильными группами дают с раствором церийаммоний-
нитрата окрашенные комплексы. По реакции с хлоридом железа(Ш) енолы и
фенолы можно отличить от спиртов.
1.2.5.1. Реакция с церийаммонийнитратом
Вещества, растворимые в воде. 0,5 мл раствора реагента (приготовление см. в части Е, с. 390)
разбавляют 3 мл дистиллированной воды и прибавляют 5 капель концентрированного водно-
водного раствора вещества.
Вещества, нерастворимые в воде. К 0,5 мл раствора реагента прибавляют 3 мл диоксана,
затем по каплям воду до получения прозрачного раствора и после этого пять капель концент-
концентрированного раствора анализируемого вещества в диоксане.
В присутствии спиртов появляется красная окраска. Фенолы в водном раст-
растворе дают зеленовато-коричневый или коричневый осадок, а в диоксане — тем-
темно-красное или коричневое окрашивание.
Границы применения: реакция дает однозначный результат в случае соедине-
соединений, содержащих не более 10 атомов углерода; в случае более высокомолекуляр-
высокомолекулярных веществ окрашивание слишком слабое. Многоатомные спирты также мож-
можно обнаружить этим методом, однако из-за окисления окраска может быстро
исчезнуть. Положительную реакцию дают многие амины, а также другие веще-
вещества, легко окисляющиеся с образованием окрашенных продуктов.
1.2.5.2. Реакция с хлоридом железа(Ш)
1 каплю вещества растворяют в 5 мл этанола и прибавляют 1-2 капли 1%-ного водного раст-
раствора хлорида железа(Ш).
При положительной реакции появляется окраска (от кроваво-красной до ва-
васильковой у алифатических енолов и от синей до фиолетовой у фенолов).
Границы применения: положительная реакция указывает на присутствие
фенолов или енолов. Большинство оксимов и гидроксамовых кислот дает крас-
красное окрашивание, гидроксипроизводные хинолина и пиридина — красно-ко-
красно-коричневое, синее или зеленое. Взаимодействие с гидроксипроизводными пяти-
членных ароматических гетероциклов также приводит к окрашиванию в крас-
красноватые оттенки. При реакции с аминокислотами и ацетатами получается
коричневое или красное окрашивание соответственно, с дифениламином —
зеленое. Многие фенолы не дают этой цветной реакции.
1.2.5.3. Реакция с солями меди (II)
Многоатомные спирты образуют с ионами меди(П) комплексы, особенно в ще-
щелочной среде.
5-6 капель вещества растворяют в разбавленной щелочи и добавляют несколько капель
очень разбавленного раствора сульфата меди.
Если осадок гидроксида меди не образуется, то, вероятно, присутствует мно-
многоатомный спирт.
304 Д Идентификация органических соединений
1.2.5.4. Реакция с солянокислым раствором хлорида цинка
(реактивом Лукаса)
Первичные, вторичные и третичные спирты можно отличить друг от друга по раз-
разной скорости замещения гидроксильной группы на хлорид-ион (см. разд. Г,2.5.1).
К 1 мл вещества быстро прибавляют 6 мл реактива Лукаса (приготовление см. в части Е,
с. 381), смесь встряхивают, оставляют на 5 мин и оценивают результат.
Первичные спирты, имеющие до 5 атомов углерода, растворяются. Часто
раствор темнеет, но остается прозрачным.
Вторичные спирты сначала дают прозрачный раствор, однако он быстро
мутнеет, а затем отделяются крошечные капельки хлорида.
В случае третичных спиртов быстро образуются две фазы, одной из которых
является хлорпроизводное.
Границы применения: поскольку проба Лукаса связана с образованием нераст-
нерастворимых хлорпроизводных, она применима лишь для тех спиртов, которые дают
прозрачный раствор в реактиве. Аллиловый спирт ведет себя как вторичный
спирт (почему?).
1.2.5.5. Взаимодействие с реактивом Дениже
Третичные и некоторые вторичные спирты легко дегидратируются концентри-
концентрированной серной кислотой. Образующиеся при этом олефины дают желтые или
красные осадки с ионами ртути.
К 3 мл реактива Дениже (приготовление см. в части Е, с. 381) прибавляют несколько
капель исследуемого вещества и кипятят 1—3 мин.
Третичные спирты образуют осадок желтовато-красных тонов. В случае пер-
первичных и в особенности вторичных спиртов также иногда образуются осадки,
однако последние в большинстве случаев не окрашены. Сложные эфиры тре-
третичных спиртов могут гидролизоваться при действии реагента, а затем уже да-
давать положительную реакцию. Тиофен выпадает в виде комплекса.
1.2.6. ИОДОФОРМНАЯ РЕАКЦИЯ (РЕАКЦИЯ С ГИПОИОДИТОМ НАТРИЯ)
Методика: см. разд. Г,6.5.3.
Границы применения: положительную иодоформную реакцию дают соедине-
соединения следующих типов:
НзС-CO-R
R-CO-CH2-CO-R [Д.2]
H3C-CH(OH)-R
R-CH(OH)-CH2-CH(OH)-R R = Н, алкил, арил
Не дают иодоформной реакции соединения
Н3С-СО-СН2—X X = CN, NO2(COOR) [Д.З]
1. Предварительные испытания и обнаружение функциональных групп 305
1.2.7. ОБНАРУЖЕНИЕ СОЕДИНЕНИЙ, ГИДРОЛИЗУЕМЫХ ЩЕЛОЧАМИ
1.2.7.1. Реакция со спиртовой щелочью (проба Рояна)
0,1 г вещества растворяют в 3 мл спирта, добавляют 3 капли спиртового раствора фенолфтале-
фенолфталеина и 0,1 н. спиртовой раствор гидроксида натрия до появления красного окрашивания. Затем
нагревают 5 мин на водяной бане при 40 °С.
Если красное окрашивание исчезает, реакцию считают положительной. Для
большей надежности пробу повторяют несколько раз, добавляя к одной и той же
смеси новые порции щелочи.
Положительную реакцию дают сложные эфиры, лактоны, ангидриды, легко
гидролизуемые галогенопроизводные, амиды, нитрилы.
Границы применения: свободные кислоты перед выполнением пробы долж-
должны быть нейтрализованы. Пробе могут мешать расщепляющиеся дикетоны
(см. разд. Г,7.2.1.9) или легко осмоляющиеся и диспропорционирующиеся
вещества (см. разд. Г,7.3.1.5).
1.2.7.2. Реакция с гидроксиламином (гидроксамовая проба)
Основой гидроксамовой пробы является аминолиз производных карбоновых
кислот под действием гидроксиламина (см. табл. Г.7.7).
К 0,5 г вещества добавляют I мл 0,5 н. спиртового раствора гидрохлорида гидроксиламина и
0,2 мл 6 н. раствора гидроксида натрия. Смесь нагревают до кипения, охлаждают, прибавляют по
каплям 2 мл 1 н. соляной кислоты. Если смесь становится мутной, добавляют 2 мл этанола. При
прибавлении 1—2 капель 5%-ного водного раствора хлорида железа(Ш) при положительной ре-
реакции появляется окрашивание (от темно-красного до фиолетового). Если окрашивание неус-
неустойчиво, необходимо добавить большее количество хлорида железа(Ш).
Положительную реакцию дают классы органических соединений, перечис-
перечисленные в разд. Д, 1.2.7.1. Из галогенсодержащих соединений реакцию дают толь-
только ацилгалогениды и геминальные тригалогениды.
Границы применения: положительную реакцию дают муравьиная и молочная
кислоты, алифатические нитросоединения. Гидроксамовой реакции не дают:
сложные эфиры угольной и хлоругольной кислот, уретаны, эфиры сульфоновых
и неорганических кислот. Фенолы не мешают реакции.
Аналогично можно обнаружить карбоновые кислоты.
К пробе карбоновой кислоты добавляют 1 мл тионилхлорида, нагревают 10 мин на водя-
водяной бане, избыток тионилхлорида отгоняют в вакууме, остаток обрабатывают гидроксилами-
гидроксиламином, как описано выше.
Границы применения: не удается определить кислоты, образующие при обра-
обработке тионилхлоридом легколетучие ацилхлориды.
1.2.7.3. Реакция с концентрированным раствором гидроксида калия
Амиды и нитрилы карбоновых кислот в общем случае нельзя обнаружить с
помощью пробы Рояна.
К пробе вещества, помещенной в пробирку, добавляют концентрированный раствор гид-
гидроксида калия, края пробирки тщательно очищают от следов щелочи, неплотно закрывают ее
306 Д Идентификация органических соединений
ватой и нагревают до кипения (добавить кипелки!), положив на верх пробирки смоченную
красную лакмусовую бумажку. Ее посинение указывает на присутствие нитрилов или простых
амидов.
Границы применения: положительную реакцию дают также соли летучих ами-
аминов, имиды, гидразиды карбоновых кислот и другие соединения.
1.2.8. ОБНАРУЖЕНИЕ АМИНОВ
Определение растворимости и обнаружение азота при предварительных испы-
испытаний уже позволяют сделать выводы о присутствии или отсутствии аминов.
Первичные амины можно идентифицировать при помощи изонитрильной ре-
реакции. Отличить первичные алифатические амины от ароматических можно с
помощью диазотирования и азосочетания. Первичные, вторичные и третичные
амины разделяют через сульфамиды (реакция Хинсберга, см. разд. Г,8.5).
Следует обращать особое внимание на отнесение частот валентных колеба-
колебаний связи N—Н в ИК-спектрах (см. табл. А. 135).
1.2.8.1. Реакция с хлороформом (изонитрильная проба)
J?f Осторожно! Изонитрилы очень ядовиты! Реакцию проводить под тягой, по окон-
F-j чании работы остатки разлагать концентрированной соляной кислотой.
Небольшое количество анализируемого вещества B-3 капли для жидкостей или «на кон-
кончике шпателя» для твердых веществ) растворяют в 1 мл этанола, прибавляют 2 мл разбавлен-
разбавленной щелочи и несколько капель хлороформа. Смесь быстро нагревают до кипения.
Образование изонитрила обнаруживается по очень сильному неприятному
запаху (проделать холостой опыт!). (Напишите уравнение реакции!)
Границы применения: реакция очень чувствительна, она может давать поло-
положительный результат даже в присутствии следов аминов. Высококипящие ами-
амины образуют изонитрилы с очень небольшим давлением пара, из-за чего их
трудно обнаружить.
1.2.8.2. Реакция с азотистой кислотой
Осторожно! Нитрозамины очень ядовиты и канцерогенны (см. разд. Г.8.2.1).
Остерегайтесь их попадания на кожу! Реакцию проводить под тягой!
Методика: см. разд. Г,8.2.1.
Получаемые из первичных ароматических аминов соли диазония в раство-
растворе сочетаются с р-нафтолом (см. разд. Г,8.3.3). Появление оранжевого или
оранжево-красного окрашивания указывает на присутствие первичного аро-
ароматического амина.
Из вторичных аминов при взаимодействии с азотистой кислотой обычно об-
образуется нерастворимый в воде желтоватый нитрозамин. Низшие алифатичес-
алифатические вторичные амины образуют хорошо растворимые в воде нитрозамины. Тре-
Третичные амины не реагируют (см. разд. Г,8.2.1).
Ы,Ы-Диалкиланилины образуют я-нитрозосоединения (см. также разд.
Г,5.19), которые можно обнаружить по появлению зеленой окраски при подще-
лачивании. Первичные алифатические амины образуют спирты; если углерод-
1. Предварительные испытания и обнаружение функциональных групп 307
ная цепь достаточно велика, то они выделяются в виде масел. Низшие алифати-
алифатические спирты после нейтрализации гидроксидом калия можно выделить
высаливанием, насыщая раствор карбонатом калия.
1.2.8.3. Реакция с нингидрином
К I —2 мг вещества добавляют немного воды, 4—5 капель I %-ного водного раствора нигидрида
и недолго кипятят.
В присутствии аминокислот появляется темно-фиолетовое окрашивание
раствора.
Границы применения: мешают аммиак, первичные амины и их соли; послед-
последние дают похожее окрашивание.
1.2.9. ОБНАРУЖЕНИЕ НИТРО- И НИТРОЗОСОЕДИНЕНИЙ
1.2.9.1. Реакции с цинком и хлоридом аммония
Методика: см. разд. ДД.2.2.1.
Реакция основывается на образовании гидроксиламина, который выделяет
металлическое серебро из реактива Толленса.
Границы применения: мешают вещества, восстанавливающие реактив Толлен-
Толленса (см. разд. Д, 1.2.3).
Первичные и вторичные алифатические нитросоединения могут быть иден-
идентифицированы по методике, изложенной в разд. Д, 1.2.9.3.
1.2.9.2. Реакция аци-нитросоединений с хлоридом железа (III)
Пробу вещества встряхивают с концентрированным раствором гидроксида натрия. Образо-
Образовавшуюся натриевую соль отфильтровывают, растворяют в небольшом количестве воды, до-
добавляют немного эфира и по каплям прибавляют водный раствор хлорида железа(Ш). При
встряхивании эфирный слой приобретает красную или коричнево-красную окраску.
1.2.9.3. Реакция аци-нитросоединений с азотистой кислотой
К пробе вещества добавляют раствор нитрита натрия в 10 н. NaOH. Образующийся осадок
растворяют, добавляя по каплям воду. Затем при охлаждении осторожно по каплям прибавля-
прибавляют разбавленную серную кислоту.
В слабощелочной среде в присутствии первичного нитросоединения появля-
появляется кроваво-красное окрашивание, исчезающее при подкислении. Вторичные
нитросоединения образуют при подкислении окрашенные в интенсивно-синие
или даже зеленые цвета псевдонитролы, которые можно извлечь хлороформом
(ср. также разд. Г,8.2.3).
1.2.10. ОБНАРУЖЕНИЕ ГИДР0ЛИЗУЮЩЕГ0СЯ ГАЛОГЕНА
Несколько капель водного или этанольного раствора галогенсодержащего вещества смешива-
смешивают с 2 мл 2%-ного раствора нитрата серебра в этаноле. Если через 5 мин осадок не образуется,
то нагревают до кипения. Образовавшийся осадок должен сохраняться и после добавления
двух капель азотной кислоты.
308 Д Идентификация органических соединений
Принимая во внимание растворимость газогенсодержащих соединений, их
можно подразделить на следующие классы:
1. Растворимые в воде вещества. Они дают осадок при комнатной температу-
температуре; сюда относятся соли аминов с галогеноводородами, низшие алифатические
ацилгалогениды.
2. Вещества, нерастворимые в воде.
а) Осадок образуется при комнатной температуре: ацилгалогениды, трет-
алкилгалогениды, геминальные алифатические дибромиды, а-галогено-
эфиры, аллилгалогениды, алкилиодиды.
б) Осадок образуется при повышенной температуре: первичные и вторичные
алкилхлориды, вицинальные дибромиды, динитрохлорбензолы.
в) Осадок вообще не образуется: арилгалогениды, винилгалогениды, тетра-
хлоруглерод и другие вещества.
1.2.11. ОБНАРУЖЕНИЕ ТИОЛОВ И ТИОФЕНОЛОВ
Почти все соединения этого типа можно узнать по их очень резкому и очень
неприятному запаху.
Обнаружить их можно также с помощью реакций с растворами солей тяже-
тяжелых металлов или цветных реакций.
1.2.11.1. Реакция с солями тяжелых металлов
Пробу вещества растворяют в небольшом количестве этанола, прибавляют концентрированный
водный раствор соли тяжелого металла [например, ацетата свинца(П), хлорида ртути(П), хлори-
хлорида медиA)].
В присутствии тиолов обычно образуется характерный осадок, который при
нагревании обычно переходит в соответствующий сульфид. Тиолаты свинца и
меди окрашены в желтый цвет, тиолаты ртути(П) не окрашены.
1.2.11.2. Реакция с азотистой кислотой
Пробу вещества растворяют в этаноле и прибавляют твердый нитрит натрия, а затем осторож-
осторожно добавляют разбавленную серную кислоту.
Первичные и вторичные тиолы дают красное окрашивание, третичные тио-
лы и тиофенолы — сначала зеленое, которое затем переходит в красное.
Границы применения: такую же реакцию дают тиоциановая кислота и ее эфи-
ры, а также некоторые ксантогенаты. Меркаптокарбоновые кислоты не дают
интенсивной окраски, а с меркаптокоричной кислотой реакция вообще не про-
происходит.
1.2.11.3. Реакция с динатрийпентацианонитрозилферратом(Ш)
(нитропруссидом натрия)
Пробу вещества растворяют в воде, спирте или диоксане, добавляют 5 капель 2 н. NaOH и 5 ка-
капель водного раствора нитропруссида натрия. Появление фиолетовой окраски указывает на
присутствие тиола.
2. Производные и спектры 309
Проведение описанных выше предварительных испытаний обычно позволя-
позволяет определить, к какому классу относится исследуемое вещество, а также
предоставляет в распоряжение некоторые производные, используемые для
идентификации.
2. ПРОИЗВОДНЫЕ И СПЕКТРЫ
Для идентификации неизвестного соединения используют различные методы:
а) Получение твердых, плавких производных и сравнение их температур
плавления с аутентичным образцом (смешанная проба) или с табличными
данными температуры плавления соответствующих производных.
Для однозначной идентификации для каждого вещества желательно после
предварительных испытаний получить не менее трех различных производных,
температуры плавления которых должны совпадать с температурами плавления
аутентичных образцов.
б) Определение молекулярной массы исследуемого соединения либо исходя
из его эквивалентной массы (определяемой в реакциях омыления или нейтрали-
нейтрализации), либо исходя из количества выпавшего галогенида серебра, либо с по-
помощью обычных прямых физических методов определения молекулярной мас-
массы (см. практикумы по физической химии).
в) Получение производных с последующим определением эквивалентной
массы в реакциях расщепления. Этот метод является ценным дополнением к
идентификации по температурам плавления (сравнение т. пл. образца с таблич-
табличными данными).
г) Для однозначной идентификации вещества используют сравнение
ИК-спектров изучаемого вещества и аутентичного образца (особенно в об-
области «отпечатков пальцев»; см. разд. А,3.5.2). ИК-спектроскопия, а также
спектроскопия ЯМР особенно полезны для идентификации структурных изо-
изомеров (см. разд. А,3.5.2 и А,3.5.3).
2.1. ИДЕНТИФИКАЦИЯ АМИНОВ
Первичные и вторичные амины обычно характеризуют в виде ацильных произ-
производных, третичные — в виде четвертичных оснований. Почти все амины образу-
образуют соли с галогеноводородными кислотами; они особенно рекомендуются для
идентификации третичных аминов.
2.1.1. ПЕРВИЧНЫЕ И ВТОРИЧНЫЕ АМИНЫ
2.1.1.1. Получение бензамидов
Методика: см. разд. Г,7.1.4.1 (она пригодна также и для аминов).
Границы применения: аналогичную реакцию дают спирты, тиолы, фенолы.
Полученные производные следует проверять на содержание азота!
310 Д Идентификация органических соединений
2.1.1.2. Получение бензол- и толуолсульфонамидов и разделение по Хинсбергу
Методика: см. разд. Г,8.5.
Границы применения: разделение хорошо удается лишь в случае аминов, содер-
содержащих не более шести углеродных атомов в цепи. Полученные сульфонамиды
очень устойчивы и гидролизуются с трудом. Так, для получения исходных аминов
сульфонамиды первичных аминов надо кипятить с обратным холодильником с
концентрированной соляной кислотой 24—36 ч, сульфонамиды вторичных ами-
аминов — 10— 12 ч. Удобным методом расщепления сульфонамидов является действие
48%-ной или 30%-ной бромоводородной кислоты в смеси ледяной уксусной кис-
кислоты и фенола. [SnyderH. R. et al. J. Am. Chem. Soc, 74 A952), 2006, 4864.]
Сульфонамиды можно также гидролизовать обработкой хлоридом цинка и
хлороводородом в ледяной уксусной кислоте [Klamann D., Hqfbauer G. Lieb. Ann.
Chemie, 581 A953), 182-197.]
2.1.1.3. Получение пикратов, пикролонатов и стифнатов
0,2 г амина растворяют в 5 мл 95%-ного этанола, добавляют насыщенный раствор пикриновой
(пикролоновой, стифниновой) кислоты в 95%-ном этаноле и доводят до кипения. Выпдающие
при медленном охлаждении кристаллы отфильтровывают и перекристаллизовывают из этанола.
Границы применения: некоторые ароматические углеводороды в описанных
условиях также образуют пикраты, которые, однако, часто не удается перекрис-
перекристаллизовать (ср. разд. Д,2.6.2.4).
Щ Осторожно' Пикраты при нагревании могут взрываться! О пикратах, пикролона-
jfI тах и стифнатах см. Beilstein, Bd. 6, Bd. 24.
2.1.1.4. Получение фенилтиомочевин
Методика: см. разд. Г,7.1.6.
В случае растворимых в воде низкомолекулярных аминов реакцию с фенил-
изотиоцианатом можно проводить тем же способом в водных растворах (оста-
(оставить смесь на ночь).
2.1.1.5. Определение эквивалентной массы
Методика: см. разд. Г,8.1.
Границы применения: метод позволяет определять амины, имеющие рА^ < 14.
Амины с рКь от 9 до 11 (например, пиридин и анилин) можно также титровать
0,1 н. НС1 в водной среде в присутствии метилоранжа.
2.1.2. ТРЕТИЧНЫЕ АМИНЫ
2.1.2.1. Получение пикратов
Получение и границы применения: см. разд. Д,2.2.1.3.
2.1.2.2. Получение метоиодидов и метотозилатов
Методика: см. разд. Г,2.6.4.
Иногда рекомендуется перекристаллизовать соли четвертичных оснований
из смеси дихлорметана с эфиром.
Таблица Д.4. Идентификация аминов (первичных и вторичных)
Амин
Метиламин
Диметиламин
Этиламин
Изопропиламин
т/>е/п-Бутиламин
н-Пропиламин
Диэтиламин
Аллиламин
в/яор-Бутиламин
Изобутиламин
н-Бутиламин
Диизопропиламин
Пирролидин
Изоамиламин
н-Амиламин
Пиперидин
Ди-н-пропиламин
Этилендиамин
1,2-Диаминопропан
Морфолин
Т. кип., "С
-6
7
16
33
46
49
56
56
63
68
78
84
89
96
104
106
109
117
119
130
Т. пл., °С
Т. пл. производных
Бензамид
82
43
71
134
82
жидк.
92
57
41
жидк.
48
жидк.
249
192
75
Бензолсульфонамид
30
52
58
26
36
42
39
70
53
жидк.
жидк.
94
51
168
118
и-Толуолсульфонамид
79
80
63
50
52
60
64
62
78
48
123
жидк.
жидк.
96
360
103
147
Пикрат
211
161
170
150
198
138
74
140
130
151
145
147
112
(желт.)
164
(краен.)
137
138
152
97
233 (ди)
237 (ди)
146
Фенилтиомочевина
113
133
101
102
120
64
34
99
101
82
65
103
69
101
69
187 (ди)
136
Таблица Д.4. Продолжение
Амин
и-Гексиламин
Циклогексиламин
Диизобутиламин
Ди-н-бутиламин
1,5-Диаминопентан
Бензил амин
Анилин
1 -Фенил этиламин
Диизоамиламин
N- Метиланилин
р-Фенил этиламин
о-Толуидин
jw-Толуидин
Ди-н-амиламин
N-Этиланилин
о-Хлоранилин
2,5-Диметил анилин
2,4-Диметиланилин
о-Анизидин
о-Фенетидин
ж-Хлоранилин
Фенил гидразин
л*-Фенетидин
п-Фенетидин
л(-Анизидин
л(-Броманилин
Дибензиламин
1,3-Диаминобутан
Т. кип., °С
130
134
139
159
180
184
184
187
187
196
198
200
203
203
205
209
215
217
225
229
230
243
248
248
251
251
300
159
Т. пл., °С
16
5
19
2
18
27
Т. пл. производных
Бензамид
40
149
135
105
165
120
63
116
146
125
60
99
140
192
66
104
120
168
103
174
120
112
177
Бензолсульфонамид
17
89
56
жидк.
119
88
112
79
69
123
97
130
138
130
89
102
120
154
143
68
/?-Толуолсульфонамид
62
ПО
жидк.
118
103
94
108
114
87
102
233
181
127
164
135
154 (разл.)
157
106
68
81
Пикрат
126
154
121
64
237 (ди)
194
175 (разл.
94
145
169
213
200
72
138
134
171
209
200
177
158
69
169
180
251 (ди)
(разл.)
Фенилтиомочевина
ПО
150
113
86
148
156
154
72
87
135
138
109
89
156
148
152
136
145
124
172
138
148
143
145
о-Броманилин 229
л-Толуидин 200
а-Нафтиламин 300
Индол 254
Дифениламин 302
2-Аминогшридин
л-Анизидин 240
2,4-Дихлоранилин 245
л*-Фенилендиамин 284
л-Броманилин
о-Нитроанилин
л-Хлоранилин 232
Семикарбазид
2,4-Диаминотолуол 292
о-Фенилендиамин 256
Пиперазин 140
р-Нафтиламин 306
л-Нитроанилин
.и-Аминофенол
Бензидин
л-Фенилендиамин 267
(Ди)
л-Нитроанилин
о-Аминофенол
л-Аминофенол
л-Аминобензойная кислота
а После возгонки т. пл. 185 °С (разл.).
6 Диморфизм; т. пл. другой кристаллической формы 119 °С.
32
45
49
52
54
56
58
63
63
66
71
72
96
99
102
104
112
114
122
128
147
147
175
185 (разл.)
187
116
158
160
68
180
165
158
117
240 (ди);
125 (моно)
204
94
192
225
242
301(ди)
196
162
155
174
352 (ди)
203 (моно)
300 (ди)
128 (моно)
199
182 (ди)
234 (ди)
278
120
167
124
95
128
194
134
102
122
191
185
282
102
136
232 (ди)
247 (ди)
139
141
125
212
90
119
157
142
114
172
101
113
192
260 (ди)
173
133
138
157
243 (ди)
266(ди)
191
146
143
129
182
163а
187
182
164
106
184
180
73
208 (разл.)
280
195
113
210 (разл.)
100
146
141
165
152
146
160 (ди)
161
188
158 (разл.)
200
290 (разл.) (ди)
129
156
156
304(ди)
230 (разл.)
145
146
164
314
Д Идентификация органических соединений
н н
в присутствии:
JL
H2C-H2C-NH2
-NH,
б, млн. д.
11,0 10,0 9,0 8Я 7,0 6,0 5,0 4,0 3,0 2,0 1,0 0
Рис. Д.5. Спектр ЯМР 1Н 2-фенилэтиламина в ССЦ.
3600 3400 3200 3000 2в00 2600 2400 2200 2000 1800 1600 1400 1200 ЮОО 800 700 600 600 400
Рис. Д.6. ИК-спектр л-анизидина в КВг (тв.).
10,0
9,0 8,0 7,0 6,0 5,0 4,0 3,0 2,0
Рис. Д.7. Спектр ЯМР 1Н л-фенетидина в ССЦ.
10
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 U0O 1200 ЮОО 800 700 600 500 400
Рис. Д.8. ИК-спектр о-аминобензонитрила в КВг (тв.).
2. Производные и спектры
315
3600 3400 3200 3000 2600 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 500 400
Рис. Д.9. ИК-спектр о-нитроанилина в КВг (тв.).
9,0 8,5 8,0 75 7,0 6,5 6,0 5,5 5,0
Рис. Д. 10. Спектр ЯМР 1Н о-нитроанилина в пердейтерированном ацетоне.
5,5
50
Рис. Д.11. Спектр ЯМР 1Н 2,4-динитроанилина
в пердейтерированном диметилсульфоксиде.
2.1.2.3. Определение эквивалентной массы
Методика: см. разд. Г,8.1.
Границы применения: см. разд. Д,2.1.1.5.
316
Д Идентификация органических соединений
Таблица Д. 12. Идентификация
Амин
Триметиламин
Триэтиламин
Пиридин
а-Пиколин
2,6-Лутидин
р-Пиколин
у-Пиколин
2,4,6-Коллидин
N, N - Д и мети лан ил ин
М,М-Диметил-я-толуидин
М,М-Диэтил анилин
Хинолин
Изохинолин
Хинальдин
Пиримидин
8-Гидроксихинол ин
Трибензиламин
Акридин
Гексаметилентетрамин
(уротропин)
Т.
третичных аминов
кип.,
4
89
116
129
144
144
145
171
194
209
217
237
243
247
124
267
380
345
°С
Т. пл., °С
2
21
76»
95
ПО
280
(разл.)
Т. пл. производных, °С
Пикрат
223
173
167
170
168
150
167
155
163
130
142
203
222
195
156
204
190
208
179
Метоиодид
>355
>230
117
230
233
231
205
102
72а; 133е
159
195
143
184
224
190
Метотозилат
139
150
161
85
126
163
134
205
а Гидрат.
6 Безводное соединение.
в Известны еще три модификации.
100
36Х 3400 3200 3000 2600 2600 2400 2200 2000 1800 1600 1400 1200 1000 600700 600 500 400
Рис. Д. 13. ИК-спектр N-метилморфолина в ССЦ.
°':Г- ™>~СНз
'Ч:нг-сн/
.СН2-СН2.
чсн2-сн/
^
5,5 5,0 и,Ъ 4,0 3.5 3,0 2,5 2,0 1,5
Рис. Д.14. Спектр ЯМР 1Н N-метилморфолина в CDCI3.
1,0
Таблица Д. 15. Идентификация аминокислот
Аминокислота
Антраниловая кислота
jw-Аминобензойная кислота
л-Аминобензойная кислота
Р-Аланин
DL-Пролин
DL-Глугаминовая кислота
L-p-Аспарагин
DL-Треонин
DL-Серин
Глицин
DL-Аргинин
L-Цистин
Температура
разложения, °С
145-147
174
186
200
203
227
227
227
228
232
238
260
Т. пл. производных, °С
Бензамид
182
248
278
120
156
189
145
171
187
230"
181 (ди)
Фенил-
мочевина
181
270
300
168
170
164
178
169
197
160
Rf в системе
фенол —
вода
0,85
0,86
0,81
0,66
0,87
0,31
0,40
0,50
0,36
0,40
0,87
уксусная
кислота —
бутанол — вода
0,37
0,43
0,30
0,19
0,35
0,27
0,26
0,20
0,1
Окраска с
N—CN-индикатором
Золотистая
Зеленовато-коричневая,
при стоянии пурпурно-
коричневая
Зеленовато-коричневая,
при стоянии образуется
красное кольцо
Оранжево-коричневая с
широким оранжевым
кольцом
Серая
Таблица Д. 15. Продолжение
Аминокислота
DL-Фенилаланин
L-Аспарагиновая кислота
DL-Метионин
DL-Триптофан
DL-Изолейцин
DL-Аланин
DL-Норлейцин
DL-Валин
DL-a-Аминомасляная кислота
DL-Тирозин
DL-Лейцин
DL-Лизин
L-Цистеин
Температура
разложения, °С
264
270
281
283
292
295
297
298
307
340
332
Т. пл. производных, °С
Бензамид
188
185
145
193
118
166
123
147
197
141
249
(моно)
Фенил-
мочевина
182
162
120
190
164
170
104
165
196
RF в системе
фенол —
вода
0,85
0,19
0,82
0,76
0,82
0,55
0,88
0,78
0,69
0,59
0,84
0,81
0,57
уксусная
кислота —
бутанол — вода
0,68
0,24
0,55
0,50
0,72
0,38
0,74
0,60
0,45
0,45
0,73
0,14
Окраска с
N—CN-индикатором
Зеленовато-желтая
Серовато-пурпурная с
желтым кольцом
Коричневая с широким
синим кольцом (кольцо
быстро бледнеет)
Светло-синяя
Темно-пурпурная
Пурпурная
Светло-коричневая
Светло-пурпурная с
желтым кольцом
Красно-коричневая, при
стоянии образуется
розовое кольцо
Серая
а Дипроизводное, безводное соединение.
2. Производные и спектры
319
3600 3400 3200 3000 2800 2600 2400 2200 2000 1600 1600 НОО 1200 1000 800 700 600 500 «О
Рис. Д.16. ИК-спектр аланина в КВг (тв.).
2.1.3. АМИНОКИСЛОТЫ
Аминокислоты можно охарактеризовать теми же методами, что и амины и кар-
боновые кислоты. Более пригодными оказываются производные, полученные
реакциями по аминогруппе. Аминокислоты нельзя идентифицировать по
температурам плавления, при нагревании они разлагаются, а их температуры
разложения не характеристичны.
2.1.3.1. Получение бензамидов
I г аминокислоты растворяют в 25 мл воды, содержащей 3 г гидрокарбоната натрия. К раствору
добавляют 1,5 мл бензоилхлорида и смесь встряхивают до завершения реакции, затем фильтру-
фильтруют и подкисляют. Выпавший осадок промывают небольшим количеством холодного эфира (для
удаления бензойной кислоты). Остаток перекристаллизовывают из воды или водного этанола.
2.1.3.2. Получение фенилмочевин
К раствору 0,2 г аминокислоты в 10 мл 2 н. гидроксида натрия добавляют 0,5 г фенилизоциана-
та, встряхивают 2—3 мин и оставляют на 45 мин. Нерастворимую дифенилмочевину, образовав-
образовавшуюся в результате гидролиза, отфильтровывают, фильтрат подкисляют разбавленной соляной
кислотой.
2.1.3.3. Бумажная хроматография
Применяют нисходящую хроматографию (см. разд. А,2.5.4.1). В качестве подвижной фазы
используют насыщенный водой фенол или смесь бутанола, ледяной уксусной кислоты и воды
D:1: 1). Для проявления высушивают хроматограмму 5 мин при 104—110 °С, опрыскивают
N-CN-индикатором (см. часть Е, с. 375) и нагревают 1-2 мин при 105 °С. При этом каждая ами-
аминокислота дает пятно характерной для нее окраски.
2.2. ИДЕНТИФИКАЦИЯ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
2.2.7. АЛЬДЕГИДЫ И КЕТОНЫ
Чаще всего получают фенил-, л-нитрофенил- и 2,4-динитрофенилгидразоны,
а также семикарбазоны и оксимы. Большинство альдегидов и некоторые кетоны
дают при встряхивании с 40%-ным раствором бисульфита натрия кристалличес-
кристаллические аддукты, которые можно использовать для выделения альдегидов и кетонов
(ср. разд. Г,5.1.8.3, Г.6.2.2 и Г.9.1.1.3).
Таблица Д. 17. Идентификация альдегидов
Альдегид
Формальдегид
Ацетальдегид
Пропионовый
Глиоксаль
Акролеин
Изомасляный
н-Масляный
Хлораль
н- Валериановый
Кротоновый
и-Гексаналь
и-Гептаналь
Фурфурол
Гексагидробензальдегид
Янтарный
и-Деканаль
Бензальдегид
5-Метилфурфурол
Фенилацетальдегид
Салициловый
Т. кип., °С (мм рт. ст.)
-19
20
48
50
53
64
75
98
102
102
128
152
162
162
170
170
179
187
194
195
Т. пл., °С
Т. пл. производных, "С
л-Нитрофенил-
гидразон
181
128
124
311 (ди)
151
131
91
131
74
184
73
154
178 (ди)
80
191
130
151
225
2,4-Динитрофенил-
гидразон
166
164
155
327 (ди)
166
187 (Е)
\22(Е)
131
107
195
104
108
212 (Z)
231 (Е)
172
143 (ди)
106
238
210
125
258
Семикарбазон
169а
176
98
273 (ди)
171
125
106г
90
52 (оксим)
215(разл.)
98
73
214
173
188
101
222
210
158
234
Фенилгидразон
32
1006
жидк.
180 (ди)
51"
жидк.
жидк.
56
жидк.
96
124 (ди)
158
147
60
142
Тиофенкарбальдегид-2
л/-Толуиловый
3- Гидроксибутаналь
о-Толуиловый
л-Толуиловый
о-Хлорбензальдегид
Анисовый
Коричный
а-Нафтойный
5-Гидроксиметилфурфурол
о-Метоксибензальдегид
о-Нитробензальдегид
3,4-Диметоксибензальдегид
я-Хлорбензальдегид
Фталевый
л*-Нитробензальдегид
(З-Нафтойный
я-Диметиламинобен-
зальдегид
Ванилин
Антраценкарбальдегид-9
л-Нитробензальдегид
Терефталевый
198
198
60A0)
200
204
212
247
129B0)
292
120@,5)
246
280
215
84 @,8)
150A5)
284
245
12
34
39
44
45
48
56
57
60
74
82
105
107
116
95
157
113
222
200
249
161
195
237
185
208
250
224
244 (разл.)
250
230
186 (разл.)
225
246
281 (ди) (разл.)
242
207
95
194
234
213
252
253 (?)
254
184
253
250 (разл.)
263
270
182
293 (разл.)
270
325
270
265
320
216 (разл.)
233 (разл.)
194
217
234
225
216 (разл.)
215
228
196
219
256 (разл.)
183
227
240 (ди)
246
245
224 (разл.)
229
291
220
>410;
200 (оксим)
139
93
111
121
86
120
168
82
138
94
152
121
127
191(ди)
120
215
148
104
207
159
278 (ди);
154 (моно)
а Указана т. пл. гидрата; для безводного препарата т. пл. 112 °С.
6 Указана т. пл.B)-изомера;т. пл. (?)-изомера57°С.
ь С фенилгидразином в эфире образуется фенилпиразолин.
г Известны также другие модификации.
Таблица Д. 18. Идентификация кетонов
Кетон
Ацетон
Метилэтилкетон
Винилметилкетон
Диацетил
Изопропилметилкегон
Метил-к-пропилкетон
Диэтилкетон
Пинаколин
Хлорацетон
Диизопропилкетон
Бутилметилкетон
Мезитилоксвд
Цихлопентанон
Ацетилацетон
Ди-н-пропилкетон
Циклогексанон
2-Мегилциклогексанон
З-Метилциклогексанон
4- Метилциклогексанон
Гексавдион-2,5
и-Метилацетофенон
Бутирофенон
Т. кип., °С
56
80
81
89
94
102
102
106
119
125
128
130
130
139
144
156
164
167
170
191
226
229
Т. пл., °С
13
и-Нитрофенилщдразон
152
126
330 (да);
230 (моно)
108
117
139
88
133
154
149
(диоксим)
146
132
119
128
210 (да)
192
162
Т. пл. производных, °
2,4-Динитрофенилгидразон
126
117
346 (ди)
123
143
156
126
125
88
108
203
146
209 (моно)
75
162
136
135
134
257 (да)
258
194
С
Фенилщдразон
26
жидк.
261 (да);
134 (моно)
жидк.
жидк.
жидк.
жидк.
142
55
170 (моно)
жидк.
76
45
94
ПО
120 (да)
96
200
Семикарбазон
192
143
141
278 (да)
(разл.);
235 (моно)
116
111
113
140
156
147
1 Cf\
160
125
164
205
185 (моно);
209 (да)
135
166
192
183а
196
224 (да)
209
191
Пропиофенон
Ацетофенон
Фенилацетон
Форон
п- Метоксиацетофенон
Бензилиденацетон
Инданон-1
Бензофенон
Фенацилбромид
я- Бромацетофенон
Метил - р - нафтил кетон
Бензил иденацетофенон
Фенацилхлорид
Бензил фенилкетон
л«-Нитроацетофенон
Флуоренон
Бензил
л-Бромфенацилбромид
Бензоин
Ксантон
DL-Камфора
215
200
213
198
258
262
244 (разл.)
306
256
301
345
244
321
341
347
343
350
возг.
19
20
27
28
38
41
42
48
50
54
54
58
59
60
81
85
95
110
137
174
178
184
143
195
166
235
154
97 (оксимN
89 (оксим)
163
132 (оксим)
269
192 (моно); 290 (ди)
115 (оксим)
161 (оксим)
217
192
248
156
118
232
229
265
232
221
232
262 (разл.)
245
219
204
228
300
189 (моно)
234
164
147
05
85
42
58
35
37
26
76
20
16
27
52
35 (моно);
24 (ди)
58, 108"
52
233
180
199
199 (разл.)
186
198
186 (транс)
247
168
146
208
223 (разл.)
170
157
148
257
245
244 (разл.)
(ли)
206 (разл.)
232 (разл.)
а Для семикарбазона (+)-3-метилциклогексанона наивысшая указанная в литературе температура плавления равна 198 °С.
6 B)-Изомер; т. пл. (?)-изомера 114 °С.
" Данные для двух модификаций.
324 Д Идентификация органических соединений
2.2.1.1. ПОЛУЧЕНИЕ ФЕНИЛГИДРАЗОНОВ
Методика: см. разд. Г,7.1.1.
Для получения фенил- и л-нитрофенилгидразонов вместо серной кислоты в
качестве растворителя используют 50%-ную уксусную кислоту.
2,4-Динитрофенилгидразоны, обычно окрашенные в желто-оранжевые цве-
цвета, как правило, являются хорошо кристаллизующимися соединениями. Из
а,р-ненасыщенных карбонильных соединений образуются производные темно-
красного цвета.
Границы применения: фенилгидразоны низших альдегидов и кетонов в отли-
отличие от 2,4-динитрофенилгидразонов часто оказываются жидкостями и поэтому
непригодны для идентификации. Ацетали, как и оксимы и подобные соедине-
соединения, в условиях реакции также дают соответствующие гидразоны.
2.2.1.2. Получение семикарбазонов
Методика: см. разд. Г,7.1.1.
Все семикарбазоны — твердые вещества; их легко получить в чистом виде с
температурой плавления, совпадающей с табличными значениями.
Границы применения: иногда скорость образования семикарбазонов слишком
мала.
2.2.1.3. Получение димедоновых производных
Получение: Homing E. С, HorningM. G. J. Org. Chem., 11 A946), 95.
Границы применения: димедоновые производные особенно пригодны для
идентификации низших альдегидов. Кетоны реагируют при температуре выше
100 °С в ледяной уксусной кислоте.
2.2.1.4. Определение эквивалентной массы титрованием оксимов
Методика: см. разд. Г,7.1.1.
100
S 80
| 60
I"
[ Т
\
I ' I ' [ '
н
^нлГ
H/v/vV
71 \/ СН3
Г )с~н
»о* ощ
1 ¦ 1 ' 1 ¦ 1
Нз >^3
' SCH3
1 ' 1 ' 1 ' I I
У, см-'1
-1—|—i—|—1_
3600 3400 3200 3000 2600 2600 2400 2200 2000 1600 1600 1400 1200 1000 800 700 600 500 400
Рис. Д. 19. ИК-спектр жидкого изомасляного альдегида (без растворителя).
2. Производные и спектры
325
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 К0О 1200 1000 800 700 600 500 400
Рис. Д.20. ИК-спектр жидкого коричного альдегида (без растворителя).
Э6Ш 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 600 700 600 500 400
Рис. Д.21. ИК-спектр n-диметиламинобензальдегида в КВг (тв.).
У J
интенсивность
уменьшена в 8 раз
-N(CH3J
JL
11,0 10,0 9,0 8,0 7,0 6,0 5,0 4,0 3,0 2,0 1,0
Рис. Д.22. Спектр ЯМР 1Н n-диметиламинобензальдегида в CDCI3.
11,0
10,0 9,0 8,0 7,0 6,0 5,0 4,0 3,0 2,0
Рис. Д.23. Спектр ЯМР 1Н ванилина в CDCI3.
326
Д Идентификация органических соединений
3600 3400 3200 3000 2800 2600 2400 2200 2000 1600 1600 1400 1200 1000 000 700 600 500 400
Рис. Д.24. ИК-спектр жидкого метилизопропилкетона (безрастворителя).
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 600 700 600 500 400
Рис. Д.25. ИК-спектр жидкого ацетофенона (без растворителя).
0
II
с
J
<
.1
P2,c6,.,dc5, *,-v
:::¦¦" <J
(CDCl3
о
>—C —CH.-CHj
-c
J
v
T
<f, млн. д.
220 210 200 190 160 170 160 150 140 130 120 110 И 90 Ю 70 60 50 40 X 20 10
Рис. Д.26. Спектр ЯМР 13С пропиофенона в CDCI3.
3600 3<.0О 3200 3600 3400 3200 3000 2000 1800 1600 1400 1200 1000 600700 600 500 400
б а
Рис. Д.27. ИК-спектр л-аминоацетофенона:
а — в КВг; б — в ССЦ.
2. Производные и спектры
327
3600 3400 3200 3600 3400 3200 3000 2800 2000 1800 1600 1400 1200 1000 800 700 600 500 400
б а
Рис. Д.28. ИК-спектро-аминоацетофенона:
а — твердый в КВг; б — в ССЦ.
2.2.2. ХИНОНЫ
Хиноны обычно узнают по окраске и по чувствительности к щелочам (появле-
(появление окрашивания). С концентрированной серной кислотой образуются сильно-
окрашенные ониевые соединения. В восстановительных условиях хиноны прев-
превращаются в неокрашенные гидрохиноны. При этом в качестве промежуточных
веществ часто образуются зеленые хингидроны.
Хиноны идентифицируют в виде их семикарбазонов или гидрохинондиаце-
татов.
2.2.2.1. Получение семикарбазонов
0,2 г хинона и 0,2 г гидрохлорида семикарбазида нагревают с небольшим количеством воды.
Желтый осадок перекристаллизовывают из воды.
Таблица Д.29. Идентификация хинонов
Хинон
2-Хлорбензохинон-1,4
2-Метилбензохинон-1,4
Бензохинон-1,4
Нафтохинон-1,4
2,6-Дибромбензохинон-1,4
Нафтохинон-1,2
Хинизарин
Фенантренхинон-9,10
Аценафтенхинон
З-Бромфенантренхинон-9,10
Антрахинон-9,10
Хлоранил
Ализарин
Т. пл., "С
57
69
116
126
131
146
201
206
261
268
286
290г
290
Т. пл. производных, °С
Семикарбазон
185а
179а
243 (ди)
2476
225а
1846
220 (моно)
193(моно);271(ди)
242 (моно)
224 (оксим)
Диацетат гидрохинона
70
52
123
128
116
105
200"
183
130
260
245
182
а 4-Монопроизводное.
6 1-Монопроизводное.
в Хинизариндиацетат получают кипячением с ацетангидридом в присутствии небольшого количества
серной кислоты.
г В запаянном капилляре; восстановление приводит к тетрахлоргидрохинону с т. пл. 134 °С.
328
Д Идентификация органических соединений
100
* 80
1 60
«о
|,0
Д 2°
чс-н 1
\
Jl
i.i. t . i
_^ ^ ^
1 | | ч |
V, см~1
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 500 400
Рис. Д.ЗО. ИК-спектр бензохинона-1,4 в КВг (тв.).
220 210 200 190 180 ПО 160 150 140 130 120 110 100 90 60 70 60 50 40 30 20 10 О
Рис. Д. 31. Спектр ЯМР 13С нафтохинона-1,4 в CDCI3.
2.2.2.2. Получение гидрохинондиацетатов
К суспензии 0,5 г хинона в 2,5 мл ацетангидрида добавляют 0,5 г цинковой пыли и 0,1 г по-
порошкообразного безводного ацетата натрия, осторожно нагревают до исчезновения окраски
хинона и после этого кипятят еще 1 мин. Добавляют 2 мл ледяной уксусной кислоты, недол-
недолго нагревают, горячую жидкость декантируют, осадок промывают 3-4 мл горячей ледяной
уксусной кислоты. Соединенные уксуснокислые вытяжки разбавляют небольшим количест-
количеством воды и охлаждают. Перекристаллизовывают из разбавленного этанола или петролейного
эфира.
2.2.3. МОНОСАХАРИДЫ
Наиболее характерные производные простых Сахаров — это озазоны.
2.2.3.1. Получение озазонов
Методика: см. разд. Г,7.1.1.
Границы применения: температуры плавления отдельных озазонов слишком
близки друг к другу, что делает их в этом случае непригодными для идентифика-
идентификации моносахаридов.
Удобными методами идентификации углеводов являются бумажная и тон-
тонкослойная хроматография (см. разд. А,2.5.4.1 и А,2.6.3). В качестве подвижной
фазы при этом рекомендуются смесь бутанола, ледяной уксусной кислоты и
воды D:1:1) или фенол, насыщенный водой. Употребляемые растворители
2. Производные и спектры
329
Таблица Д.32. Идентификация углеводов
Углевод
Рафиноза
D-Рибоза
a-D-Глюкоза
2-Дезокси-0-рибоза
р-Мальтоза
D-Фруктоза
D-Аллоза
a-L-Рамноза
a-D-Ликсоза
DL-Глюкоза
P-L-Рамноза
DL-Ксилоза
p-D-Манноза
DL-Манноза
a-D-Манноза
L-Ксилоза
a-D-Ксилоза
L-Фукоза
p-D-Глюкоза
p-D-Арабиноза
p-L-Арабиноза
DL-Фукоза
DL-Сорбоза
DL-Галактоза
DL-Арабиноза
L-Сорбоза
a-D-Галактоза
Сахароза
L-Аскорбиновая
кислота
Гентиобиоза
Лактоза
р-Целлобиоза
Температура
разложения,
80A19)
87 (95)
90A46)
90
103A60-165)
104
105
105(93)
106-107A01)
112
122-126
129-131
132
132-133
133
144
145
145
148-150
158
160
161
162-163
163 A44)
164
165A59)
167
169-170A85)
190
190-195(86)
201B23)
225
[aW°
+ 105,2
- 21,5 (-23,5)
+ 52,7
+ 2,13
+ 130,4
- 92,4
+ 32,6
+ 8,2
- 14,0
+ 9,1
+ 14,2
+ 14,2
- 18,6
+ 18,8
- 75,9
+ 52,7
+ 104,5
- 43,4
+ 80,2
+ 66,5
- 49,0
+ 8,7
+ 55,3
+ 34,6
Т. пл.
°С
166
205
206
205
190
163
156
210
218
205
160
164
178
210
166
187
170
206
169
162
201
205
162
200
198
Rf в системе
бутанол —
уксусная
кислота —
вода
0,05
0,31
0,18
0,11
0,23
0,37
0,37
0,20
0,28
0,27
0,31
0,20
0,20
0,16
0,14
0,38
0,09
фенол —
вода
0,27
0,59
0,39
0,73
0,36
0,51
0,59
0,45
0,45
0,44
0,63
0,54
0,42
0,44
0,39
0,24
0,38
Для некоторых углеводов указаны две температуры разложения, взятые из разных источников.
безусловно должны быть перегнаны, для контроля одновременно с исследуемой
пробой хроматографируют аутентичный образец. Восстанавливающие сахара
проявляют, опрыскивая анилинфталатом (приготовление см. в части Е, с. 380),
а затем нагревая 10 мин при 105 "С. Невосстанавливающие сахара проявляют
смесью равных частей 0,2%-ного спиртового раствора нафторезорцина и
2%-ного водного раствора трихлоруксусной кислоты с последующим нагрева-
нагреванием до 100 °С (появление окраски).
330
Д Идентификация органических соединений
ot-D-глкжоза й-О-глкжоза
100 90 80 70 60 50 40
Рис. Д.33. Спектр ЯМР 13С D-глюкозы в D2O.
30
2.2.4. АЦЕТАЛИ
Ацетали и кетали идентифицируют с помощью кислотного гидролиза с пос-
последующим раздельным обнаружением карбонильного соединения и спирта
(см. разд. Д,2.5).
Низкомолекулярные ацетали гидролизуются быстро C—5 мин кипячения с
обратным холодильником с 1—2%-ной соляной кислотой), ацетали с большей
молекулярной массой требуют 30-60-минутного кипячения. При гидролизе не-
нерастворимых в воде соединений можно добавить диоксан.
2.2.5. КАРБОНОВЫЕ КИСЛОТЫ
2.2.5.1. Получение п-бром-и п-фенилфенациловых эфиров
Методика: см. разд. Г,2.6.3.
Границы применения: при получении эфира реакционная смесь не должна
иметь щелочной реакции. Большие количества хлорид-ионов мешают из-за
осаждения труднорастворимого и-бромфенацилбромида (т. пл. 117 °С). Затруд-
Затруднено получение производных аминокислот, а также некоторых производных
дикарбоновых и гидроксикарбоновых кислот.
2.2.5.2. Получение амидов карбоновых кислот
1 г карбоновой кислоты кипятят (обратный холодильник, хлоркальциевая трубка) 15-30 мин
с 5 мл тионилхлорида, добавив 1 каплю диметилформамида. Реакционную смесь охлаждают,
выливают в 15 мл охлажденного до 0 °С концентрированного аммиака, осадок отфильтровы-
отфильтровывают и перекристаллизовывают из воды или водного этанола.
Границы применения: этим способом нельзя определить муравьиную кислоту
(почему?); при работе с низкокипящими ацилхлоридами (особенно ацетил- и
оксалилхлоридами) необходимо учитывать их высокую летучесть. В этих случа-
случаях лучше оставить реакционную смесь на несколько часов при комнатной тем-
температуре. Методика не позволяет выделить растворимые в воде амиды. В этих
случаях рекомендуется сначала перевести карбоновую кислоту в метиловый
эфир с помощью диазометана (см. разд. Г,8.4.2.1), а затем провести аммонолиз
концентрированным раствором аммиака.
Таблица Д.37. Идентификация карбоновых кислот
Кислота
Муравьиная
Уксусная
Акриловая
Пропионовая
Изомасляная
н-Масляная
Пировиноградная
Изовалериановая
и-Валериановая
Дихлоруксусная
н-Гексановая
Молочная
Олеиновая
н-Декановая
Левулиновая
Лауриновая
Бромуксусная
Миристиновая
Трихлоруксусная
Хлоруксусная
Пальмитиновая
Тиглиновая
Стеариновая
Кротоновая
Фенилуксусная
Гликолевая
Глутаровая
L-Яблочная
Лимонная (моногидрат)
Т. кип., °С
(мм рт. ст.)
101
118
141
141
155
163
165
176
186
194
205
119A2)
223A0)
269
246
298
208
193A0)
197
187
222A6)
199
189
227
Т. пл.,
°С
8
17
13
14
13
53
14
31
37
45
50
54
57
63
63
65
70
71
78
79
99
100
100
Амид
3
82
85
79
129
115
127
136
106
99
101
79
76
100
108
103
90
105
142
116
141
78
109
161
161
120
94
149 (ди)
138 -
Т. пл. производных, °С
Анилид
48
114
106
103
104
93
104
111
63
94
95
45
69
63
77
162
84
94
134
91
91
95
118
118
96
128 (моно);
223 (ди)
197 (ди)
164
и- Бромфенациловый
эфир
140
86
63
55
63
68
75
72
72
113
67
84
59
81
104
81
68
78
95
89
138
137
179 (ди)
150 (три)
я-Фенилфенациловый
эфир
74
111
165
101
90
97
76
69
71
145
183
77
94
86
90
116
94
106
97
152
204
146
N-Бензиламид
60
61
70
52
87
37
53
42
226
89
89а
89
93
94
95
98
113
122
103
170 (ди)
170
Кислота
Щавелевая (дигидрат)
о-Метоксибензойная
Феноксиуксусная
Пимелиновая
о-Толуиловая
Азелаиновая
jn-Толуиловая
Миндальная
Бензойная
Себациновая
транс- Коричная
Малоновая
Малеиновая
Ацетилсалициловая
л*-Нитробензойная
о-Хлорбензойная
о-Нитробензойная
Бензиловая
Адипиновая
.м-Бромбензойная
л«-Хлорбензойная
Салициловая
а-Нафтойная
л«езо-Винная
2,4-Динитробензойная
Т. кип., °С
(мм рт. ст.)
200
285
300
Т. гот.,
°С
100
101
101
106
107
107
114
120
122
134
133
134
137
143
142
142
147
148
153
156
156
159
162
166
180
Амид
214 (моно);
350 (разя.)
(аи)
128
101
175 (ди)
141
175 (ди)
94
134
127
127 (моно);
210 (ди)
149
121 (моно);
172 (ди)
178 (моно);
181 (аи)"
113Г
143
140
176
155
226 (ди)
155
134
139
202
189 (ди)
203
Таблица Д.37.
Продолжение
Т. пл. производных, °С
Анилид
149 (моно);
252(ди)
74
49
155 (ди)
128
184 (ди)
126
150
165
122 (моно);
201 (ди)
154
132 (моно);
229(ди)
201
137
155
114
156
177
240 (ди)
137
125
136
164
194 (моно)
196
и-Бромфенациловый
эфир
244 (разл.) (ди)
113
137 (ди)
57
131 (ди)
108
119
147 (ди)
146
168 (ди)
134
106
100
152
154
126
117
140
136
204
158
л-Фенилфенациловый
эфир
166 (разл.)
131
146 (разл.)
94
145
136
105
140
183
175
128б
1056
153
123,83е -
140
122
148, 876
155
154
148
N-Бензиламид
128 (моно)
223 (ди)
132
85
153 (ди)
91а
44а
75
105
166 (аи)
106"
142 (ди)
206
102
100
99
156
86
189 (ди)
105а
107"
136
93а
142а
л-Толуиловая 275
р-Нафтойная
Анисовая
Янтарная
3- Гидроксибензойная
3,5-Динитробензойная
Винная (рацемическая)
безводная
л-Гидроксибензойная
З-Нитрофталевая
Фталевая
Никотиновая
Дифеновая
я-Нитробензойная
л-Хлорбензойная
Галловая
Фумаровая
Терефталевая
Изоникотиновая
Изофталевая
л-Нитробензиловый эфир.
Фенациловый эфир.
При получении возможна изомеризация в диамид фумаровой кислоты.
Нерезко; плавление сопровождается перегруппировкой в N-ацетилсалициламид.
В запаянном капилляре.
182
183
184
188
203
206
218
216
219
227
229
232
241
242
258
ЗОО1
300(возг.)
316
348 (возг.)
167
192
167
157 (моно);
268 (ди)
170
180
212 (моно);
240 (ди)
161
174
148 (моно)
121
190 (моно)
201
180
243
309
332 (ди)
154
280
145
167
169
228
157
239
182 (моно);
235 (ди)
195
181 (моно);
233(ди)
170 (моно);
253(ди)
132
181 (моно)
218
194
207
238 (моно);
303 (ди)
313 (моно);
336(ди)
280
153
152
211 (ди)
176
159
205
191
149
153 (ди)
134
126
134
225 (ди)
179 (ди)
165
160
208
1466
154
132
138 (моно)
206 (ди)
142
157а
240, 178б
169 (ди)
182
160
195 (разл.)
198б
191 (диN
133
148 (ди)а
182"
178
185 (ди)а
141
129"
141а
314
265
202а
334
Д Идентификация органических соединений
2.2.5.3. Получение N-бензиламидов карбоновых кислот
Методика: см. разд. Г,7.1.4.2. Получение ацилхлоридов см. в разд. Д,2.2.5.2 и
Г,7.1.4.4.
Избыток тионилхлорида можно не отгонять, а разрушить, добавляя по кап-
каплям безводную муравьиную кислоту, и использовать ацилхлорид в дальнейших
превращениях без очистки.
Границы применения: см. разд. Д,2.2.5.2.
Вместо ацилхлоридов можно применять соответствующие ангидриды карбо-
карбоновых кислот; данная методика может быть использована и для идентификации
ангидридов.
2.2.5.4. Получение анилидов карбоновых кислот
Методика: см. разд. Г,7.1.4.2.
2.2.5.5. Определение эквивалентной массы
Точную навеску очищенной кислоты (около 0,2 г) растворяют в 50—100 мл воды или водного
этанола и титруют 0,1 н. гидроксидом натрия по фенолфталеину:
Эквивалентная масса = ¦
Навеска (г) х 1000
К(мл)№Он
где /V— нормальность.
3600 Э40О 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 5Ш 400
Рис. Д.35. ИК-спектр дигидрокоричной кислоты в КВг (тв.).
-СН-СН2-
А
НвНа
I I
L
10,0
9,0
7,0
6,0
5,0
4,0
3,0
2,0
1,0
Рис. Д.36. Спектр ЯМР 1Н дигидрокоричной кислоты в CDCI3
(спектр высшего порядка группы -СН2-СН2-; система АА'ВВ').
2. Производные и спектры
335
~Счон
13.0
,
J
0
II
ч— Н' 4CH,I|
12.0 Ш
ноосч ,н
^ Г Н' CHj
о
II
I |ЪО:=сГ
сн3
I
S, млн. д.
11,0 10,0 9,0 в,0 7,0 6,0 5,0 4,0 3,0 2,0 1,0 0
Рис. Д.38. Спектр ЯМР 1Н кротоновой кислоты в CDCI3.
3600 3400 3200 3000 2600 2600 2400 2200 2000 И00 1600 1400 1200 1000 800 700 600 500 400
Рис. Д.39. ИК-спектр малеиновой кислоты в КВг (тв.).
3600 3400 3200 3000 2600 2600 2400 2200 2000 1800 1600 1400 1200 1000 600 700 600 500 400
Рис. Д.40. ИК-спектр мапеинового ангидрида в КВг (тв.).
Границы применения: мешает диоксид углерода, в завершающей фазе титро-
титрования его необходимо удалять кипячением. Однако в случае легко декарбокси-
лирующихся веществ проводят определение только при комнатной температу-
температуре. Если кислота малорастворима в воде, то можно попытаться использовать
водный этанол; в этом случае в качестве индикатора лучше взять бромтимоло-
вый синий.
2.2.6. АМИДЫ КАРБОНОВЫХ КИСЛОТ И НИТРИЛЫ
Гидролиз нитрилов и амидов приводит к соответствующим карбоновым кисло-
кислотам. При восстановлении амидов и нитрилов образуются амины.
336
Д Идентификация органических соединений
2.2.6.1. Получениекарбоновыхкислот
Методика щелочного гидролиза: см. разд. Г,7.1.5.
Если карбоновая кислота не выпадает при подкислении, то рекомендуется из
щелочного раствора перевести ее в л-бромфенациловый эфир, как это описано
в разд. Г,2.6.3.
Границы применения: амиды обычно хорошо гидролизуются щелочами, в то
же время гидролиз нитрилов в этих условиях часто происходит слишком мед-
медленно. Поэтому лучше всего гидролизовать нитрилы в кислой среде B0%-ная
соляная кислота, 2 ч).
2.2.6.2. Получение аминов (восстановление по Буво-Блану)
К раствору 1 г нитрила в 20 мл абсолютного этанола при 50—60 °С добавляют небольшими пор-
порциями 1,5 г натрия. После охлаждения осторожно приливают 10 мл концентрированной соля-
соляной кислоты и отгоняют этанол. Остаток подщелачивают 10 мл 50%-ного раствора гидрокси-
да натрия и выделившийся амин отгоняют с водяным паром. Амин лучше всего идентифици-
идентифицировать в водном растворе с помощью бензоилхлорида (см. разд. Д,2.1.1.1).
Границы применения: амиды в этих условиях не восстанавливаются. Получен-
Полученные из нитрилов амины можно непосредственно в спиртовом растворе превра-
превратить в производные фенилтиомочевины (см. разд. Д,2.1.1.4).
10,0
9,0 8,0 7,0 6,0 5,0 4,0 3,0 2,0 1,0
Рис. Д.41. Спектр ЯМР 'Н диметилформамида.
2.2.7. ЭФИРЫ КАРБОНОВЫХ КИСЛОТ
Для идентификации эфиры обычно гидролизуют и каждый из компонентов оп-
определяют в отдельности. Во многих случаях соответствующие производные по-
получают аминолизом или переэтерификацией.
2.2.7.1. Получение карбоновых кислот и спиртов
Смесь 2 г сложного эфира и 20 мл 1 н. раствора гидроксида натрия кипятят с обратным холо-
холодильником до растворения эфира. Часть реакционной смеси используют для идентификации
карбоновой кислоты (см. разд. Д.2.7.2), а из другой части отгоняют воду и спирт (досуха), дис-
дистиллят насыщают карбонатом калия, выделившийся спирт отделяют, сушат MgSO4 и иденти-
идентифицируют, как это описано в разд. Д,2.2.5.
Границы применения: в случае эфиров, образованных нерастворимыми в воде
спиртами, маслянистый слой при гидролизе не исчезает! Кислоты с длинной
2. Производные и спектры
337
9
А ун
СН3-СН,-О/ >>С(
н ^
нэс-сн2-о-
110 10,0 90 6,0
6,0
5,0
Рис. Д.42. Спектр ЯМР 1Н этилового эфира коричной кислоты в ССЦ.
углеводородной цепью образуют мыла. Сложные эфиры, не омыляемые водны-
водными щелочами, гидролизуют в присутствии небольшого количества диоксана или
тетрагидрофурана либо 10%-ным раствором гидроксида калия в этаноле; в этом
случае обычно отказываются от идентификации спирта.
Определение эквивалентной массы: см. разд. Г,7.1.4.3.
Границы применения: с трудом определяются сложные эфиры многоатомных
фенолов, так как в условиях гидролиза последние окисляются (что легко заме-
заметить по появлению окраски и расходу щелочи). Стерически затрудненные слож-
сложные эфиры щелочами не гидролизуются.
2.2.7.2. Получение эфиров 3,5-динитробензойной кислоты
Методика: см. разд. Г,7.1.4.4.
Границы применения: метод пригоден для многих сложных эфиров простого
строения. Однако его нельзя применять в случае сложных эфиров, спиртовая
составляющая которых реагирует с концентрированной серной кислотой (напри-
(например, третичные спирты, легко осмоляющиеся ненасыщенные спирты). Сравни-
Сравнительно высокомолекулярные сложные эфиры реагируют лишь медленно или не
реагируют совсем.
2.2.7.3. Получение амидов карбоновых кислот
Методика: см. разд. Г,7.1.4.2.
Границы применения: методика применима только для метиловых или в лучшем
случае этиловых эфиров. Кроме них в реакцию вступают только так называемые
активированные эфиры (см. разд. Г,2.2.1). Сложные эфиры высших спиртов
необходимо предварительно подвергнуть метанолизу по следующей методике:
0,6—1,0 г эфира кипятят 30 мин с обратным холодильником с 10 мл абсолютного метано-
метанола, в котором предварительно растворено 0,1 г натрия. Избыток метанола упаривают, остаток
непосредственно подвергают аминолизу.
2.3. ИДЕНТИФИКАЦИЯ ПРОСТЫХ ЭФИРОВ
Простые эфиры в общем случае очень устойчивы. Большинство алифатических
простых эфиров растворяется в концентрированной соляной кислоте с образо-
338
Д Идентификация органических соединений
3600 3400 3200 3000 2600 2600 2400 2200 2000 1800 1600 1400 1700 1000 ЯТЮ 700 finfl ЧП 4П0
Рис. Д.43. ИК-спектр жидкого монометилового эфира этиленгликоля (без растворителя).
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 500 400
Рис. Д.44. ИК-спектр жидкого дибензилового эфира (без растворителя).
ванием солей оксония. При разбавлении водой соли распадаются; эту особен-
особенность можно использовать для выделения простых эфиров из смесей. Арилалки-
ловые эфиры образуют оксониевые соли только с концентрированной серной
кислотой, причем частично происходит и сульфирование ароматического ядра.
2.3.1. РАСЩЕПЛЕНИЕ ПРОСТЫХ ЭФИРОВ ИОДОВОДОРОДНОЙ ИЛИ
БРОМОВОДОРОДНОЙ КИСЛОТОЙ
Методика: см. разд. Г,2.5.2.
Выделенные перегонкой алкилиодиды или алкилбромиды идентифицируют
в виде пикратов S-алкилтиомочевин (см. разд. Г,2.6.6).
2.3.2. РАСЩЕПЛЕНИЕ ПРОСТЫХ ЭФИРОВ 3,5-ДИНИТРОБЕНЗОИЛХЛОРИДОМ
В ПРИСУТСТВИИ ХЛОРИДА ЦИНКА
1 г вещества, 0,15 г безводного хлорида цинка и 0,5 г 3,5-динитробензоилхлорида нагревают
1 ч с обратным холодильником. После охлаждения добавляют 10 мл 2 н. раствора карбоната
натрия и нагревают на водяной бане до 90 °С. При стоянии выделяется эфир динитробензой-
ной кислоты; его отфильтровывают, промывают раствором карбоната натрия и водой, а затем
растворяют в 10 мл тетрахлоруглерода. Если раствор мутный, его фильтруют горячим. Если
при охлаждении кристаллы не выделяются, то растворитель упаривают.
Границы применения: эта методика применима лишь для идентификации прос-
простых симметричных алифатических эфиров (почему?). Могут мешать спирты,
амины и другие соединения, поэтому их предварительно необходимо отделить.
2. Производные и спектры
339
2.4. ИДЕНТИФИКАЦИЯ ГАЛОГЕНОУГЛЕВОДОРОДОВ
Проведя испытание согласно разд. Д, 1.2.10, получают сведения о природе гало-
генсодержащего вещества.
Таблица Д.45.
Органический
радикал
Метил
Винил
Этил
Изопропил
н- Пропил
Алл ил
трет-Бутмл
втор'Еупш
Изобутил
к-Бутил
трет-Амил
Изоамил
н-Амил
н-Гексил
Циклогексил
н-Гептил
Бензил
н-Октил
2-Фенилэтил
я-Хлорбензил
о-Бромбензил
Лf-Бpoмбeнзил
л-Бромбензил
л-Нитробензил
Идентификация алкилгапогенидов
Хлорид
-24
-14
12
36
46
46
51
67
68
77
86
100
107
134
142
159
179
184
190
214
110A5)
23 (т. пл.)
50 (т. пл.)
71(т.пл.)
Т. кип.
Бромид
5
16
38
60
71
71
72
90
91
100
108
118
129
157
165
180
198
204
218
51(т.пл.)
31(т.пл.)
41 (т. пл.)
62 (т. пл.)
99 (т. пл.)
(мм рт. ст.), °С
Иодид
43
56
72
89
102
103
98
119
120
130
128
148
156
180
179
204
24.(т. пл.)
225
116A2)
47 (т. пл.)
42 (т. пл.)
73 (т. пл.)
Пикрат S-алкил-
изотиомо-
чевины
224
104
188
196
177
155
160
166
167
180
173
154
157
174
142
188
134
194
222
205
219
Анилид
114
104
104
103
92
114
128
108
109
63
92
108
96
69
146
57
117
57
97
166
Геминальные ди- и тригалогениды (за исключением производных метана)
гидролизуются (см. разд. Г,2.6.1), образующиеся альдегиды и карбоновые кис-
кислоты можно идентифицировать обычными методами. Ароматические фториды
и хлориды можно превратить в производные с помощью реакций нитрования
или сульфохлорирования (см. разд. Д,2.6).
2.4.1. ПОЛУЧЕНИЕ АНИЛИДОВ КАРБОНОВЫХ КИСЛОТ
К 0,4 г магниевой стружки, активированной иодом, добавляют 1,2 г галогенсодержащего ве-
вещества, растворенного в 5 мл абсолютного эфира. По окончании реакции эфирный раствор
декантируют, прибавляют 3—4 г твердого диоксида углерода (происходит образование карбо-
новой кислоты) или 4,5 мл 10%-ного раствора фенилизоцианата в эфире (при этом образуется
анилид). Через 10 мин добавляют 20 г измельченного льда и 1 мл концентрированной соляной
кислоты, перемешивают, эфирный слой отделяют, эфир отгоняют.
В реакцию вступают почти все алифатические алкилгалогениды, а также
арилбромиды и арилиодиды (см. разд. Г,7.2.2). Взаимодействием полученных
Таблица Д.46. Идентификация ароматических галогеноуглеводородов
Галогеноуглеводород
Фгорбензол
Хлорбензол
Бромбензол
2-Хлортолуол
З-Хлортолуол
4-Хлортолуол
1,3-Дихлорбензол
1,2-Дихлорбензол
2-Бромтолуол
З-Бромтолуол
1,4-Диметил-2-хлорбензол
Иодбензол
1,2-Диметил-4-хлорбензол
1,3-Диметил-4-хлорбензол
1,3-Дибромбензол
1,2-Дибромбензол
1-Хлорнафталин
1 -Бромнафталин
4-Бромтолуол
1,4-Дихлорбензол
2-Бромнафталин
2-Хлорнафталин
1,4-Дихлорнафталин
1,4-Дибромбензол
1,5-Дихлорнафталин
Т. кип., °С
85
132
156
159
162
162
173
180
181
183
185
188
195
192
219
219
259
281
185
174
281
265
290
219
Т. пл., "С
7
28
53
59
61
68
89
107
Сульфонамид
Положение
сульфон-
амидной
группы
4
4
4
5
6
2
6
4
5
6
5
5
6
6
4
4
4
2
2
8
8
6
2
3
Т. пл., "С
125
143
162
126
185
143
180
135
146
168
155
207
195
190
176
186
193
165
180
208
126
244
195
204
Нитросоединение
Положение
нитро группы
2,4
2,4
3,5
4,6
2
4,6
4,5
3,5
4,6
5
4
5
6
4
4,5
4,5
4
2
2
1,8
8
2,5
8
Т. пл., °С
52
75
64
91
38
103
ПО
82
103
77
174
63
42
61
114
180
85
47
54
175
92
84
142
Таблица Д.47. Идентификация полигалогеноуглеводородов
Галогеноуглеводород
Дихлорметан
(?)-1,2-Дихлорэтилен
B)-1,2-Дихлорэтилен
Хлороформ
2,2-Дихлорпропан
Тетрахлоруглерод
1,2-Дихлорэтан
Трихлорэтилен
Дибромметан
Тетрахлорэтилен
1,2-Дибромэтан
1,2-Дибромпропан
1,1,2,2-Тетрахлорэтан
Бромформ
Пентахлорэтан
1,3-Дибромпропан
Дииодметан
Бензилидендихлорид
Трихлорметилбензол
Тиролдибромид
Гексахлорэтан
Т. кип., °С
41
48
60
61
70
77
84
87
97
121
132
142
147
151
161
167
180
207
221
74 (т. пл.)
186 (т. пл.)
«d2i)
,4237
,4454
,4486
,4462
,4093
,4630
,4443
,4773
,5419
,5055
1,5379
1,5203
1,4944
1,5977
1,5028
1,5233
1,7405
1,5515
1,5579
</420
1,336
1,257
1,284
1,489
1,093
1,595
1,256
1,464
2,492
1,623
2,179
1,933
1,595
2,887
1,679
1,982
3,321
1,254
1,374
2. Производные и спектры
341
100
=S 80
of
I 60
та
и
с
,&20
н :,-'
С—Н **
м ¦ и
•В ..'• I
г т
1 Н
сн2 г/ у 11
-сн31
СН3-СН2-СН2-Вг
V w
1
^ -1
V, см
3600 3400 3200 3000 2800 2600 2400 2200 2000 1600 1600 U00 1200 1000 800 700 600 500 400
Рис. Д.48. ИК-спектр бромпропана в ССЦ.
100
во
feo
2
(§20
(Аг))с-Н
1 . 1 . Г . 1
н ^
-с-н
н
»v ,
гси3
1 . 1 . , 1
^ ¦¦•¦ * II
, 1 . 1
Па
/СНз
1 .
Г II II \
ьр-с|
f н
1 Н v
1 1 i J. ., 1 ,
1-
-1
см
1 .
3600 3«Ю 3200 3000 2800 2600 2400 2200 2000 1800 1600 К00 1200 1000 800 700 600 500 400
Рис. Д.49. ИК-спектр жидкого о-хлортолуола (без растворителя).
н-
iU"'
1
Г V
'Н
"Н
<
5,40 4,24МЛН.Д.
Q-c-6-Sr ^
Br HB
4C0 МЛН. Д
-J III j*b
1
I ^
LlV\. 4.4
X , AB
l J*B
Д
1' вдв' ,
4.3 4,2
t
Jab ,
i—-L
4;1
rf, млн. д.
7,5
7,0
6,5
6,0
55
5,0
4,5
4,0
3,5
30
Рис. Д.50. Спектр ЯМР 1Н 1,2-дибромэтилбензола в пердейтерированном ацетоне.
НА и Нв магнитно неэквивалентны. Здесь представлен также спектр высшего порядка,
позволяющий определить Д8АВ @,06 м.д.), ЛдхF,09Гц) и Лвх(9,89Гц).
соединений Гриньяра с диметилформамидом можно превратить галогеноугле-
водороды в альдегиды, которые легко идентифицируются в виде 2,4-динитрофе-
нилгидразонов. [SharefkinJ. G., Forschirm A. Anal. Chem., 1963, 35, 1616.]
2.4.2. ПОЛУЧЕНИЕ ПИКРАТОВ S-АЛКИЛИЗОТИОМОЧЕВИН
Методика и определение эквивалентной массы: см. разд. Г,2.6.6.
Границы применения: метод пригоден лишь для алифатических галогеноугле-
водородов.
342 Д Идентификация органических соединений
2.5. ИДЕНТИФИКАЦИЯ СПИРТОВ
Для идентификации спиртов используют эфиры 3,5-динитро-, 4-нитробензой-
ных или 3-нитрофталевой кислот, а также фенил- и нафтилуретаны. Фенолы
могут быть охарактеризованы производными, образующимися взаимодействи-
взаимодействием с ацилгалогенидами и изоцианатами. Многие фенолы дают хорошо кристал-
кристаллизующиеся трибромфенолы.
2.5.1. ПЕРВИЧНЫЕ И ВТОРИЧНЫЕ СПИРТЫ
2.5.1.1. Получение эфиров нитробензойных кислот
Методика: см. разд. Г,7.1.4.1.
Границы применения: реагируют также фенолы, первичные и вторичные ами-
амины, тиолы. Особенно удобно получать эти эфиры в случае растворимых в воде
спиртов, которые часто содержат следовые количества воды (см. также ниже об
уретанах). Для гликолей и полигидроксисоединений выгоднее получать ацетаты
и в особенности бензоаты (см. разд. Г,7.1.4.1). Третичные спирты трудно харак-
характеризовать этим методом.
2.5.1.2. Получение моноэфиров 3-нитрофталевой кислоты
Методика и определение эквивалентной массы: см. разд. Г,7.1.4.1.
Границы применения: третичные спирты чаще реагируют с образованием оле-
финов. Если же спирт действием этилмагнийбромида предварительно превра-
превратить в соответствующий алкоголят, то последующей реакцией с фталевым ан-
ангидридом его можно перевести в полуэфир фталевой кислоты [Fessler W. А.,
Shriner R. L. J. Am. Chem. Soc, 1936, 58, 1384]. Фенолы, первичные и вторичные
амины также реагируют с образованием соответствующих производных.
2.5.1.3. Получение уретанов
Методика: см. разд. Г,7.1.6.
Границы применения: аналогично реагируют фенолы, первичные и вторичные
амины, тиолы. Вода мешает (образуются соответствующие дизамещенные моче-
мочевины), поэтому метод пригоден только для безводных проб. Уретаны третичных
спиртов образуются с трудом.
2.5.2. ТРЕТИЧНЫЕ СПИРТЫ
Третичные спирты для идентификации переводят в соответствующие галогено-
углеводороды, которые затем и определяют.
2.5.2.1. Получение пикратов S-алкилизотиомочевин
Третичный спирт встряхивают с 5—6 объемами концентрированной соляной кислоты. Орга-
Органический слой отделяют, полученный алкилгалогенид идентифицируют в виде пикрата S-ал-
килизотиомочевины (см. разд. Г.2.6.6).
Таблица Д.51. Идентификация спиртов
Спирт
Метанол
Этанол
Изопропанол
/и/>е/я-Бутанол
н-Пропанол
Алл иловый
вжор-Бутанол
Изобутанол
т/>ет-Пентанол
Пентанол-3
н-Бутанол
Пентанол-2
Этиленгликоля
монометиловый
эфир
1-Хлорпропанол-2
2-Хлорэтанол-2
Изопентанол
Этиленгликоля
моноэтиловый
эфир
Пентанол-1
Т. гш.,°С
25
124
135
Т. кип.,
65
78
82
82
97
97
99
108
116
116
118
120
61
127
129
132
138
°С
л-Нитробензоат
96
57
108
116
35
30
25
69
85
17
36
17
56
21
11
3
Т. пл. производных, °С
,5-Динитробензоат
108
93
122
142
40
50
76
86
118
101
63
61
129
62
118а
46
Моноэфир 3-нитро-
фталевой кислоты
153
157
153
142
124
131
179
121
147
103
166
136
Фенилуретан
47
52
90
136
52
70
64
86
44
49
63
113
51
55
67
46
а- Нафтилуретан
124
80
105
80
72
72
75
101
67
68
Таблица Д.51. Продолжение
Спирт
Гексанол-2
Циклопентанол
2-Бромэтанол
2,2,2-Трихлорэтанол
Гексанол-I
Гептанол-2
Циклогексанол
траноЗ-Метилцикло-
гексанол
Фурфуриловый
/яронс-4-Метилцикло-
гексанол
цмс-4-Метил циклогексанол
2,3-Диметилбутандиол-
2,3 (пинакон)
i/wc-3-Метилциклогекса-
нол
1,3-Дихлорпропанол-2
Гептанол-1
Октанол-2
2-Этилгексанол
Пропиленгликоль
Октанол-1
Этиленгликоль
Т. пл.,°С
17
436
Т. кип., °С
140
141
150
151
158
160
161
168
171
171
171
172
173
176
177
180
183
188
195
198
Т. пл. производных, "С
я-Нитробензоат
62
71
7
жидк.
52
63
65
96
48
58
10
28
127 (ди)
17
145 (ди)
3,5-Динитробензоат
38
53
143
60
113
111
81
142
107
99
129
47
32
61
169 (ди)
Моноэфир 3-нитро-
фталевой кислоты
172
124
160
183
127
128
Фенил уретан
132
76
87
42
81
82
46
124
104
215
88
73
60
114
150 (ди)
74
160 (ди)
а-Нафтил уретан
61
87
120
59
54
129
118
130
160
107
129
59
63
59
66
176(ди)
Бензиловый
Бутандиол-1,3
Нонанол-1
Пропандиол-1,3
2-Фенилэтанол
Бутандиол-1,4
Деканол-1
Гераниол
Диэтиленгликоль
Глицерин
Лауриловый
транс-Коричный
L-Ментол
Неопентиловый
Стеариловый
Дифенилметанол
Сорбит
Бензоин
Холестерин
Трифен илметанол
Маннит
D-Борнеол
Пентаэритрит
20
7
18
24
34
43
55
58
69
92
138
149
164
168
205
260
205
208
213
214
219
229
229
230
245
290
264
257
216
113
298
380
84
102
19
119
62
175
30
35
188 (три)
29
78
62
64
132
216Г
123
145"
150г
100е
51
178
57
63
151 (ди)
192 (три)
122
153
74
154
176"
125
123
123
123
162
119
77
123 (ди)
60
81
183
61
81 (ди)
117 (ди)
182
78
91
112
144
80
140
163
168
303
139
134
153 (ди)
65
121
73
142 (ди)
192
80
114
126
100
136
140
160
127
а Т. пл. безводного соединения; т. пл. гидрата 94 °С.
6 Т. пл. безводного соединения; т. пл. гидрата 30 °С.
в Смесь монобензиловых эфиров 3-нитрофталевой кислоты с бензильными группами в положении 1 или 2 имеет т. пл. 151 °С.
г Гексабензоат.
д Бензоат.
е Тетрабензоат.
346
Д Идентификация органических соединений
3600 34.00 3200 3000 2800 2600 2400 2200 2000 1S0O 1600 К00 1200 1000 600 700 600 500 400
Рис. Д.52. ИК-спектр жидкого изопропанола (без растворителя).
11,0
9,0 6,0 7,0 6,0 5,0 4,0 3,0 2,0
Рис. Д.53. Спектр ЯМР 1Н изопропанола в CDCI3.
1,0
Границы применения: вторичные спирты также вступают в эту реакцию,
если вместо концентрированной соляной кислоты взять реактив Лукаса
(см. разд. Д, 1.2.5.4).
2.5.2.2. Определение эквивалентной массы
Методика: см. разд. Г.2.6.6.
Границы применения: для определения эквивалентной массы титрованием
пикраты должны быть тщательно очищены, поскольку присутствие свободной
пикриновой кислоты вносит в результаты погрешность. Более точные результа-
результаты получают при потенциометрическом титровании со стеклянным электродом.
2.5.3. ФЕНОЛЫ
2.5.3.1. Получение бензоатов
Методика: см. разд. Г,7.1.4.1.
Границы применения: аналогично реагируют спирты, тиолы, амины.
2.5.3.2. Получение уретанов
Методика: см. разд. Г,7.1.6.
Границы применения: см. разд. Д,2.5.1.3. сс-Нафтилуретаны обычно образуют-
образуются лучше, чем фенилуретаны. Реакцию катализируют несколько капель сухого
пиридина.
Таблица Д.54. Идентификация фенолов
Фенол
Изоэвгенол
Монометиловый эфир
резорцина
Эвгенол
Карвакрол
Этилсалицилат
о-Бромфенол
л<-Крезол
2,4-Диметилфенол
о-Крезол
л<-Бромфенол
л-Крезол
2,4-Дибромфенол
Фенол
2,4-Дихлорфенол
п-Хлорфенол
Салол
о-Нитрофенол
2,6-Диметилфенол
Тимол
Монометиловый эфир
гидрохинона
л-Бромфенол
2,4,6-Трихлорфенол
2,4,5-Триметил фенол
2,5-Диметилфенол
Т. пл., °С
-17,5
-9,1
1
1
5
12
27
31
32
36
36
42
43
43
42
45
49
51
55
64
67
71
75
Т. кип., °С
(мм рт. ст.)
267
243
253
237
234
194
202
211
192
236
200
238
182
209
217
173A2)
216
203
233
244
236
232
212
Бензоат
103
133
69
83е
87
86
56
165е
138е
88
71
98
71
97
93
80
59
41
103е
87
102
75
63
61
Фенил уретан
118а; 152»
124
101
137
98
124
103
145
115
126
148
111
133
107
144
ПО
166
Производные, т. пл., °С
а-Нафтил уретан
150
129
122
104
129
128
135
142
146
133
166
113
176
160
168
173
Бромпроизводное
946
104г
118я
46
95Г
84Г
179Г
566
108г
95Г
95Г
68
906
117б
79
55
145Г
956
35
796
Арилоксиуксусная
кислота
94
118
100
149
143
102
142
154
108
136
153
101
141
156
158
149
111
154
177
132
118
Таблица Д.54. Продолжение
Фенол
2,3-Диметилфенол
Ванилин
а-Нафгол
2,4,6-Трибромфенол
л«-Нитрофенол
Пирокатехин
Хлоргидрохинон
5-Метилрезорцин
Резорцин
Бромгидрохинон
я-Нитрофенол
2,4-Динитрофенол
я-Гидроксибензальдегид
Пикриновая кислота
р-Нафтол
2,5-Дигидрокситолуол
Пирогаллол
Гидрохинон
Флороглюцин
Т. шт., "С
75
80
94
95
97
105
106
107
ПО
ПО
114
114
115
122
123
125
133
169
218
Т. кип., °С
(мм рт. ст.)
285
280
194G0)
245
263
290
276
285
293
286
Бензоат
58
78Г
257е
81
95
84б
1306
88б
1176
142
132
72
163
107
1206
89Г
204°
173Г
Фенилуретан
173
116
178
168
129
169
154б
1646
148
121
136
158
173Г
224б
Ш
Производные, т. пл., °С
а-Нафтилуретан
152
153
167
160
151
157
Бромпроизводное
160
1056
916
192*
104г
117°
186е
142
118
1816
84
158б
1866
151Г
Арилоксиуксусная
кислота
187
189
192
155
131
194
186
148
198
155
198
а цис-Изомер.
6 Дизамещенное производное.
в /л/>а«с-Изомер.
г Тризамещенное производное.
д Тетразамещенное производное.
е Динитробензоат.
2. Производные и спектры
349
У
ecu
-о-н
Э6С0 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 500 400
Рис. Д.55. ИК-спектр фенола в растворе (комбинированный спектр в СБг и ССЦ).
3600 3400 J200 3000 2800 2600 2400 2200 2000 1600 1600 1400 1200 1000 800 700 600 500 400
Рис. Д.56. ИК-спектр л-нитрофенола в КВг (твердый).
2.5.3.3. Получение бромфенолов
Методика: см. разд. Г,5.1.5.
2.5.3.4. Получение арилоксиуксусных кислот
1 г фенола растворяют в 4 мл 10 н. раствора гидроксида натрия, добавляют 1,25 г монохлорук-
сусной кислоты и 1—2 мл воды для получения гомогенного раствора. Нагревают 1 ч на водяной
бане, после чего охлаждают, разбавляют 10-15 мл воды, затем подкисляют соляной кислотой
до кислой реакции по конго красному. Экстрагируют эфиром E0 мл), эфирный экстракт
встряхивают с 10 мл воды, затем с 25 мл 5%-ного раствора карбоната натрия. Раствор карбона-
карбоната подкисляют разбавленной соляной кислотой (осторожно: вспенивание!), осадок отфильт-
отфильтровывают и перекристаллизовывают из воды.
Границы применения: реакции мешает присутствие электрофильных замести-
заместителей в бензольном кольце.
2.6. ИДЕНТИФИКАЦИЯ УГЛЕВОДОРОДОВ
2.6.1. АЛКАНЫ И ЦИКЛОАЛКАНЫ
Насыщенные углеводороды можно узнать по их химической инертности, т. е.
малой реакционной способности по отношению к обычно употребляемым в ла-
лаборатории реагентам. В простейших случаях их идентификация может быть
проведена путем определения физических констант (температур плавления или
кипения, показателя преломления, плотности, молекулярной рефракции).
350
Д Идентификация органических соединений
Таблица Д.57. Идентификация
Углевоводород
Изопентан
и-Пентан
Циклопентан
2,3-Д иметилбутан
н-Гексан
Циклогексан
н-Гептан
2,2,4-Триметилпентан
Метил циклогексан
2,5 -Д иметилгексан
н-Октан
я-Нонан
транс-п-Ментш
цис-п-Ментш
л-Декан
т^анс-Декалин
цмс-Декалин
алканов и циклоалканов
Т. кип., °С
28
36
50
58
69
80
98
99
101
109
125
151
170
171
174
187
195
«о20
1,3536
1,3574
1,4093
1,3750
1,3750
1,4263
1,3878
1,3914
1,4231
1,3924
1,3890
1,4054
1,4368
1,4431
1,4120
1,4695
1,4810
</420
0,6196
0,6260
0,7450
0,6615
0,6593
0,7786
0,6837
0,6919
0,7694
0,6942
0,7028
0,7176
0,7928
0,8002
0,7300
0,8699
0,8965
100
g со
О 20
р
н
CHj-C-CH2-CH
сн.
сн,
-СН2-|
-сн, -сн3*'
ft-- ^СНз
'1 -сн
!... СНз ЧСНЭ
-с-сн3
СН3
V, см
3600 3400 3200 3000 2800 2600 2100 2200 2000 1800 1600 U0O 1200 1000 600 700 600 500 400
Рис. Д.58. ИК-спектр жидкого 2,2,4-триметилпентана (без растворителя).
[' Расщепление, характерное для /ирет-бутильных и изопропильных групп.]
55 50 45 Ы) 35 30 25 20
Рис. Д.59. Спектр ЯМР 13С 2,2,4-триметилпентана в CDCI3.
2. Производные и спектры
351
2.6.2. АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
Ароматические углеводороды идентифицируют с помощью реакций замещения
в ядре или окисления боковых цепей. Иногда удается также получать пикраты.
2.6.2.1. Получение сульфонамидов
Методика: см. разд. Г,5.1.4 и Г,8.5. См. также разд. Д,2.9.2.
Границы применения: галогентолуолы при сульфохлорировании следует нагревать
10 мин при 50 °С. Полигалогенбензолы требуют более жестких условий A ч при 100°С,
без растворителя). Реакция применима и для ароматических простых эфиров.
2.6.2.2. Получение о-ароилбензойных кислот
Методика: см. разд. Г,5.1.8.1.
Методика позволяет идентифицировать и арилгалогениды. Если ароилбензой-
ная кислота сразу не кристаллизуется, реакционную смесь оставляют на ночь.
Определение эквивалентной массы ароилбензойных кислот проводят по ме-
методике, описанной в разд. Д,2.2.5.5. Найденную эквивалентную массу ароилбен-
зойной кислоты пересчитывают на молекулярную массу неизвестного арена:
Молекулярная масса арена = Эквивалентная масса ароилбензойной кислоты — 148,1 [Д.60]
2.6.2.3. Получение нитропроизводных
Методика: см. разд. Д, 1.2.2.1.
Полученные нитросоединения идентифицируют, как описано в разд. Д,2.7.
Таблица Д.61. Идентификации
Углеводород
Бензол
Толуол
Этилбензол
и-Ксилол
ж-Ксилол
о-Ксилол
Кумол
н-Пропилбензол
Мезитилен
Псевдокумол
я-Цимол
к-Бутил бензол
Дурол
Тетралин
Нафталин
а-Метилнафталин
р- Метилнафталин
Дифенил
Аценафтен
Флуорен
Фенантрен
Антрацен
Т. кип.,
°С
80
ПО
135
138
139
144
151
158
164
169
177
182
193
207
218
241
241
255
278
294
340
351
1 ароматических углеводородов
Т. пл.,
•с
5
13
79
80
34
70
95
114
100
216
Производные, т. пл., °С
Сульфонамвд
148
137
109
147
137
144
107
ПО
141
181
115
155
Ароил бензойная
кислота
128"
138а
122
132
126
178
133
126
212
124
97
264
154
173
168
190
226
198
228
Пикрат
84
88
97
90
91
88
103
97
97
150
141
115
162
84 G9)
143
138
«о20
1,5011
1,4969
1,4959
,4958
,4972
,5054
,4915
,4920
,4994
,5049
,4909
,4898
1,5414
1,6182
а После удаления кристаллизационной воды в вакууме при 100 °С.
352
Д Идентификация органических соединений
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 U00 1200 1000 800700 600 500 400
Рис. Д.62. ИК-спектр жидкого л-цимола (без растворителя).
9,0 8,0 7 0 6.0 5,0 4,0 3,0 2,0
Рис. Д.63. Спектр ЯМР 'Н л-цимола в CDCI3.
2.6.2.4. Получение аддуктов с пикриновой кислотой
Равные количества пикриновой кислоты и анализируемого вещества нагревают до плавления
на водяной бане. После охлаждения аддукт измельчают и перекристаллизовывают. Если
аддукт при перекристаллизации разлагается, то ограничиваются одно- или двукратной про-
промывкой эфиром и сушкой.
Аналогично можно получать стифнаты и пикролонаты.
2.6.2.5. Окисление перманганатом калия или хромовой кислотой
Методика: см. разд. Г,6.2.1.
Границы применения (см. также разд. Г,6.2.1): о-диалкилбензолы можно окис-
окислять лишь в щелочной среде; при действии хромовой кислотой в ледяной уксус-
уксусной кислоте они разлагаются. Некоторые полиядерные арены при окислении
хромовой кислотой превращаются в хиноны (антрацен, фенантрен).
2.6.3. АЛ КЕНЫ И АЛКИНЫ
Во многих случаях идентифицировать ненасыщенные алифатические соедине-
соединения удается с помощью аддуктов с бромом (см. разд. Г,4.1.4), окисления подвой-
ной связи перманганатом калия (см. разд. Г,6.5.1), озонирования и превращения
в альдегиды (см. разд. Г,4.1.7), эпоксидирования и перегруппировки с образова-
образованием кетонов или альдегидов (см. разд. Г,4.1.6). Озонирование и гидрирование
(см. разд. Г,4.5) могут служить для количественного определения олефинов.
2. Производные и спектры
353
Таблица Д.64. Идентификаци!
Углеводород
Пентен-2
Пентин-1
Циклопентадиен
Циклопентен
Диаллил
Гексин-1
Циклогексадиен
Циклогексан
Фенилацетилен
Стирол
(+)-а-Пинен
L-Камфен
D- или L-лимонен
DL-Лимонен
(дипентен)
Инден
транс-Стмлъбен
Т. кип., °С
36
40
42
46
59
70
80
84
140
146
156
160
178
178
180
306; 125 (т. пл.)
1 апкенов и апкинов
</d20
0,651
0,688
0,805
0,774
0,690
0,712
0,840
0,810
0,930
0,925
0,859
0,822
0,841
0,841
0,992
«d20
1,3789
1,4079
1,4470
1,4223
1,4010
1,3989
1,4756
1,4465
1,5524
1,5485
1,4656
1,4621
1,4721
1,4728
1,5710
Дибромпроизводное,
т. пл., °С
73
170
89
104 (тетрабромид)
124(тетрабромид)
237
Другие производные,
т. пл., °С
^.
152(адипиновая
кислота)
98 (пикрат)
94 (пикрат)
2.6.3.1. Превращение в карбонильные соединения
Общая методика: см. разд. Г,9.1.1.1 и Г,7.1.1.
Эпоксидирование можно проводить и с помощью продажной 40%-ной
перуксусной кислоты. [Shareflcin J. G., Shwerz H. Е. Anal. Chem., 33 A961), 635;
см. также разд. Г,4.1.6.]
Границы применения: а,р-ненасыщенные карбоновые кислоты не реагируют.
Олефины с неконцевой двойной связью могут давать изомерные кетоны.
2.6.3.2. Гидратация ацетиленовых производных
Методика: см. разд. Г,4.1.3.
Образующиеся кетоны идентифицируют в виде 2,4-динитрофенилгидразо-
нов. [Shareflcin J. G, Boghosian Е. M. Analyt. Chem., 33 A961), 640.]
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 500 400
Рис. Д.65. ИК-спектр жидкого стирола (без растворителя).
354
Д Идентификация органических соединений
7,8 7,7 7,6 7,5 7,4 7,3 7,2 5,6 5,5 5,4 5,3 5,2 5,1 2,5 2,4 2,3 2,2 2,1 2,0 1,9
Рис. Д.66. Спектр ЯМР 1Н а-метилстирола в CDCI3.
3600 Э400 3200 3000 2800 2600 2400 2200 2000 1S00 1600 1400 1200 1000 800700 600 500 400
Рис. Д.67. ИК-спектр жидкого фенилацетилена (без растворителя).
2.7. ИДЕНТИФИКАЦИЯ НИТРО- И НИТРОЗОСОЕДИНЕНИЙ
Нитро- и нитрозосоединения восстанавливают в кислой среде до соответству-
соответствующих аминов, которые идентифицируют как описано в разд. Д, 2.1.
2.7.1. ПОЛУЧЕНИЕ АМИНОВ ВОССТАНОВЛЕНИЕМ ОЛОВОМ
В СОЛЯНОЙ КИСЛОТЕ
Методика: см. разд. Г,8.1.
Границы применения: после восстановления оловом в солянокислой среде при
подщелачивании выпадают соли оловянных кислот, которые могут адсорбиро-
адсорбировать полученный амин. В таких случаях можно попытаться отогнать амин с
водяным паром. Азокси-, азо- и гидразосоединения также дают при восстанов-
восстановлении соответствующий амин.
2.7.2. ПОЛУЧЕНИЕ АМИНОВ ПРИ ВЗАИМОДЕЙСТВИИ С ГИДРАЗИНГИДРАТОМ
В ПРИСУТСТВИИ СКЕЛЕТНОГО НИКЕЛЯ
Методика: см. разд. Г,8.1.
2.8. ИДЕНТИФИКАЦИЯ ТИОЛОВ И ТИОФЕНОЛОВ
Производные тиолов и тиофенолов получают теми же методами, что и произ-
производные их кислородных аналогов.
2.8.1. ПОЛУЧЕНИЕ 3,5-ДИНИТРОТИОБЕНЗОАТОВ
Методика: см. разд. Г,7.1.4.1.
2. Производные и спектры
355
2.8.2. ПОЛУЧЕНИЕ 2,5-ДИНИТРОФЕНИЛТИОЭФИРОВ И
ИХ ОКИСЛЕНИЕ ДО СУЛЬФОНОВ
Методика: см. разд. Г,5.2.1.
Окисление в сульфоны. 1 г тиоэфира растворяют в минимальном объеме ледяной уксусной
кислоты, по каплям добавляют 4 мл 30%-ного раствора пероксида водорода, а затем нагрева-
нагревают 30 мин на водяной бане с обратным холодильником. Реакционную смесь оставляют на
ночь, прибавляют 20 мл ледяной воды, осадок отфильтровывают и перекристаллизовывают из
2.8.3. ОПРЕДЕЛЕНИЕ ЭКВИВАЛЕНТНОЙ МАССЫ
Точную невеску тиола (около 0,2 г) растворяют в 50-100 мл 20%-ного водного этанола и титру-
титруют 0,1 н. раствором иода в иодиде калия, используя крахмал в качестве индикатора. (Аналогич-
(Аналогично проводят холостой опыт!)
Эквивалентная масса =
Навеска (г)- 1000
[Д.68]
где V\ — объем раствора иода, пошедшего на титрование; N\
этого раствора.
нормальность
Таблица Д.69. Идентификация тиолов
Тиол
Метантиол
Этантиол
Пропантиол-2
Пропантиол-1
2-Метилпропантиол-1
Бутантиол-1
З-Метилбутантиол-1
Пентантиол-1
Этандитиол-1,2
Гексантиол-1
Циклогексантиол
Тиофенол
Пропандитиол-1,3
Гептантиол-1
Фенил метантиол
Октантиол-1
2-Фенилэтантиол-1
.и-Тиокрезол
а-Тионафтол
о-Тиокрезол
л-Тиокрезол
л-Хлортиофенол
л- Бромтиофенол
р-Тионафтол
Т. кип., °С
(мм рт.ст.)
6
36
56
67
88
97
117
126
146
151
159
169
67 A8)
176
194
199
199
200
209
194
195
Т. пл., °С
43
53
15
43
53
74
81
Т. пл. производных, °С
3,5-Динитро-
бензоат
62
62
84
52
64
49
59
40
149
82
120
78
89
2,4-Динитро-
фенилсульфид
128
115
94
81
76
66
95
80
248
74
148
121
194
101
130
98
134
91
176
101
103
123
142
145
2,4-Динитро-
фенилсульфон
189
160
140
128
105
92
83
97
172
161
182
145
155
190
170
190
356
Д Идентификация органических соединений
3600 3400 3200 3000 2600 2600 2Ш 2200 2000 1800 1600 1400 1200 1000 800 700 600 500 400
Рис. Д.70. ИК-спектр о-меркаптобензойной кислоты в КВг (тв.).
Границы применения: аналогично определяются также ксантогенаты калия и
натрия.
Комплексометрическое определение тиолов: Oelsner W., Heubner G., Chem.
Techn., 16 A964), 432.
2.9. ИДЕНТИФИКАЦИЯ СУЛЬФОНОВЫХ КИСЛОТ
Сульфоновые кислоты идентифицируют теми же методами, что и карбоновые
кислоты.
2.9.1. ПОЛУЧЕНИЕ СУЛЬФОНАТОВ S-БЕНЗИЛТИОМОЧЕВИНЫ
0,2 г сульфоновой кислоты растворяют в 2 мл 1 н. раствора гидроксида натрия, добавляют 2 кап-
капли метилового красного и затем по каплям 1 н. гидроксид натрия до перехода окраски индикато-
индикатора. Смесь нагревают на кипящей водяной бане и затем прибавляют горячий раствор 0,5 г хлори-
хлорида S-бензилтиомочевины в 5 мл воды. Смесь охлаждают ледяной водой, при этом выпадает ожи-
ожидаемая соль. При необходимости можно ускорить кристаллизацию, потирая о стенку стеклянной
палочкой. Соединения, содержащие гидрофильные группы, высаливают хлоридом натрия.
Перекристаллизовывают из воды или водного этанола.
2.9.2. ПОЛУЧЕНИЕ СУЛЬФОНАМИДОВ
Методика получения сульфонилхлоридов: см. разд. Г,5.1.4; получение сульфонами-
дов: см. разд. Г,8.5. (Расщепление сульфонамидов см. разд. Д,2.1.1.2.)
Для получения анилидов сульфонилхлорид вводят в реакцию с таким избыт-
избытком анилина, чтобы его хватило также и на связывание образующегося хлорово-
дорода. Продукт реакции осаждают разбавленной соляной кислотой и перек-
перекристаллизовывают.
2.9.3. ОПРЕДЕЛЕНИЕ ЭКВИВАЛЕНТНОЙ МАССЫ
Определение эквивалентной массы проводят, как и в случае карбоновых кислот,
прямым титрованием сульфоновой кислоты 0,1 н. раствором гидроксида натрия
в присутствии фенолфталеина.
Границы применения: при расчете необходимо учитывать, что большинство
сульфоновых кислот содержит кристаллизационную воду. Поскольку большей
частью они гигроскопичны, то их выделение сопряжено с трудностями.
3. Разделение смесей
357
Таблица Д.71. Идентификация сульфоновых кислот
Сульфоновая кислота
Толуолсульфоновая-4
З-Нитробензолсульфо-
новая
2,5-Диметилбензолсульфо-
новая
Бензол сульфоновая
о-Толуолсульфоновая
3,4-Диметилбензолсульфо-
новая
2,4-Диметилбензолсульфо-
новая
4-Хлорбензолсульфоновая
Нафталинсульфоновая-1
Нафталинсульфоновая-2
4-Бромбензолсульфоновая
Нафтол-2-сульфоновая-6
3-Сульфобензойная
2-Сульфобензойная
4-Сульфобензойная
Сульфаниловая
4-Гидроксибензолсульфо-
новая
Т. пл., °С
38
48
48
51
57
64
68
68
91
103
125
133
134
260
290
Кристаллогидрат
Число
молекул
воды
1
2
1
2
2
2
1
2
1
2
3
4
Т. пл., °С
106
86
46
140-150
55
95
67
90
124
98
69
94
Производные, т. пл., °С
Амид
137
167
148
153
156
144
138
144
150
217
166
237
170
(ли)
236
(ДИ)
165
177
Анилид
103
126
112
136
ПО
!04
152
132
119
161
194
252
(ДИ)
200
141
Сульфонат
S-бензилтио-
мочевины
182
146
184
148
170
208
146
175
137
191
170
217
164
206
213
(ДИ)
185
169
3. РАЗДЕЛЕНИЕ СМЕСЕЙ
Химику часто приходится разделять смеси веществ. Эта задача иногда оказыва-
оказывается сложной. В отличие от стандартизованного неорганического анализа разде-
разделение органических веществ из-за богатства их реакционных возможностей
можно осуществлять многими путями. По этой причине невозможно составить
общеупотребительную схему разделения смесей, хотя следует отметить многие
полезные условия в этом направлении (см. приведенную в конце раздела лите-
литературу).
Во многих случаях многокомпонентные смеси легко разделяются с помощью
простых физических приемов [фракционная перегонка или кристаллизация
(см. разд. А,2.3.3 и А,2.2), перегонка с водяным паром, возгонка, хроматография
и др.].
358 Д Идентификация органических соединений
Иногда возможно разделение с помощью химических методов, например,
действием щелочи можно отделить фенолы и карбоновые кислоты от других
веществ. В то же время слабоосновные фенолы чаще всего нерастворимы в
растворах карбоната или бикарбоната натрия. Амины легко отделяются от дру-
других органических соединений путем перевода их в нерастворимые в эфире
соли (гидрохлориды). Относительно сильноосновные алифатические амины
выпадают в осадок в виде амидов угольной кислоты при обработке эфирного
раствора амина углекислым газом (со слабоосновными ароматическими ами-
аминами аналогичная реакция не происходит). Благодаря этому становится воз-
возможным разделение первичного ароматического амина и алифатического
амина химическим путем (в том случае, если такое разделение невозможно
осуществить простой фракционной перегонкой из-за близости т. кип.
аминов).
В некоторых случаях подходит также несложный способ разделения, осно-
основанный на различной растворимости органических веществ в полярных и непо-
неполярных растворителях, в кислотах и основаниях (см. также разд. Д, 1.1.5).
В общем случае с помощью указанных приемов сложные смеси удается раз-
разделить на фракции, каждая из которых даже в наименее благоприятной ситуа-
ситуации содержит лишь 2—3 вещества, подлежащих идентификации.
Для решения подобных задач следует применять преимущественно хрома-
тографические методы, например тонкослойную (см. разд. А,2.7.2), газо-жидко-
стную (см. разд. А,2.7.4) и высокоэффективную жидкостную хроматографию
(см. разд. А,2.7.3).
После хроматографического разделения компоненты смеси характеризуют и
идентифицируют спектральными методами.
Идентификация отдельных компонентов смеси путем перевода их в соответ-
соответствующие производные целесообразно только в тех случаях, когда эти производ-
производные можно легко выделить и в то же время разделение данной смеси веществ
другими методами затруднительно.
Естественно, что и для разделения с помощью производных нельзя дать об-
общеупотребительного рецепта. Метод разделения должен выбираться каждый раз
с учетом результатов предварительных испытаний и обнаруженных функцио-
функциональных групп.
Ниже в разд. Д,4 в качестве примеров приведены некоторые смеси, которые
часто удается разделить, применяя только химические методы.
4. ВОПРОСЫ И УПРАЖНЕНИЯ
1. На что следует обратить внимание при идентификации:
1.1. аминоальдегидов, аминокетонов;
1.2. аминокислот;
1.3. гидроксикарбоновых кислот;
1.4. моноэфиров дикарбоновых кислот;
1.5. эфиров C-оксокарбоновых кислот?
5. Литература 359
2. Опишите ход разделения и идентификации следующих смесей веществ:
2.1. я-бутанол (т. кип. 116 °С) и уксусная кислота (т. кип. 118 °С);
2.2. этанол (т. кип. 78 °С) и метилэтилкетон (т. кип. 80 °С);
2.3. н-амиламин (т. кип. 104 °С) и пиридин (т. кип. 116 °С);
2.4. анилин (т. кип. 183 °С) и бензиламин (т. кип. 184 °С);
2.5. этанол (т. кип. 78 °С), этилиодид (т. кип. 72°) и этилацетат (т. кип. 77 °С);
2.6. пропионовый альдегид (т. кип. 50 °С) и диметилацеталь формальде-
формальдегида (т. кип. 45 °С);
2.7. бензоилхлорид (т. кип. 197 °С) и бензилидендихлорид (т. кип. 205 °С);
2.8. фталимид, антрацен и салициловая кислота;
2.9. монометиловый и диметиловый эфиры гидрохинона;
2.10. стирол (т. кип. 146 °С) ити-ксилол (т. кип. 140 °С);
2.11. нитрометан (т. кип. 101 °С) и ди-н-пропиловый эфир (т. кип. 90 °С);
2.12. а-нафтол и неролин;
2.13. трет-бутанол (т. кип. 83 °С) и этанол (т. кип. 78 °С);
2.14. фенол, бензойная кислота и фенетол;
2.15. бензолсульфохлорид и бензолсульфоновая кислота;
2.16. бутантиол (т. кип. 98 °С) и н-пропанол (т. кип. 98 °С);
2.17. метиловый эфир салициловой кислоты (т. кип. 222 °С) и этил-
бензоат (т. кип. 213 °С);
2.18. бензальдегид (т. кип. 178 °С), бензиновый спирт (т. кип. 205 °С) и
бензойная кислота;
2.19. ди-н-пропиловый эфир (т. кип. 98 °С) и бензол (т. кип. 80 °С);
2.20. DL-фенилаланин, DL-аргинин и p-L-аспарагиновая кислота;
2.21. глюкоза, фруктоза и галактоза.
5. ЛИТЕРАТУРА
Vogel A. I. Practical Organic Chemistry. — Longman Group, London, 1966 (с таблицами
температур плавления).
Hermann С. К. F., Shriner R. L.\ The Systematic Identification of Organic Compounds. —
Wiley-VCH, Weinheim 2003 (с таблицами температур плавления).
Wild F. Characterization of Organic Compounds. — University Press, Cambridge, 1960
(с таблицами температур плавления).
Staudinger H., Kern W., Kammerer H. Anleitung zur organischer qualitativen Analyse. —
Springer-Verlag, Berlin/Heidelberg/ New York, 1968.
Roth H. et al. In: Houben-Weyl, Bd. 2, 1953.
Bauer K. H., Moll H. Die organische Analyse. — Akademische Verlagsgessellshaft Geest &
Portig, Leipzig, 1967.
Feigl F. Spot Tests in Organic Analysis. — Elsevier, Amsterdam, London, New York,
Princeton, 1966.
Veibel S. Analytik organischer Verbindungen. — Akademie-Verlag, Berlin, 1960.
Kaiser R. Quantitative Bestimmung organischer funktioneller Gruppen. Methoden der
Analyse in der Chemie, Bd. 4. — Akademische Verlagsgesellschaft, Frankfurt/M.,
1966.
Untermark W., Schicke W. Schmelzpunkttabellen organischer Verbindungen. — Akademie-
Verlag, Berlin, 1963.
360 Д Идентификация органических соединений
Frankel M. et al. Tables for Identification of Organic Compounds. — The Chemical Rubber
Сотр., Cleveland/Ohio, 1964.
Kemp W. Qualitative Organic Analysis. — McGraw-Hill, London, 1979.
Frei R., Lawrence J. Chemical Derivatization in Analytical Chemistry. — Plenum Press,
New York, 1981, 1982.
Knapp D. Handbook of Analytical Derivatization Reactions. — J. Wiley, New York, 1981.
СВОЙСТВА, ОЧИСТКА И ПРИГОТОВЛЕНИЕ
ВАЖНЕЙШИХ РЕАГЕНТОВ, РАСТВОРИТЕЛЕЙ
И ВСПОМОГАТЕЛЬНЫХ ВЕЩЕСТВ
В данном разделе приведены также сведения о токсичности основных реагентов
(см. часть Ж, Приложение). Дня некоторых веществ указаны симптомы отравления
ими и первая помощь.
АЗОТ И ДРУГИЕ ИНЕРТНЫЕ ГАЗЫ
Для удаления следовых количеств кислорода из инертных газов (азота, аргона,
неона), а также из диоксида углерода применяют специальные катализаторы на
никелевой или медной основе. Подобные методики приведены в соответствую-
соответствующих проспектах предприятий.
Для удаления небольших количеств кислорода азот пропускают через про-
промывную склянку, наполненную раствором 2 г пирогаллола и 6 г гидроксида
калия в 50 мл воды. Затем газ высушивают, пропуская его через колонку с
натронной известью.
А30ТИСТ0В0Д0Р0ДНАЯ КИСЛОТА HN3
Т. кип. 37 "С.
Получение бензольного раствора азотистоводородной кислоты. Работать под
тягой! Суспензию равных по массе количеств азида натрия (ядовит!) и теплой
воды помещают в трехгорлую колбу, снабженную капельной воронкой, термо-
термометром, мешалкой и газоотводной трубкой. На каждые 0,1 моль азида натрия
прибавляют 40 мл толуола, смесь охлаждают до 0 °С и при перемешивании до-
добавляют по каплям эквивалентное количество концентрированной серной кис-
кислоты. Температура при этом не должна превышать 10 °С. Затем реакционную
смесь вновь охлаждают до 0 °С, бензольный слой отделяют и сушат сульфатом
натрия. [Braun J. v. Lieb. Ann. Chem., 490, A931), 125.]
Определение концентрации. 3 мл раствора встряхивают с 30 мл дистиллиро-
дистиллированной воды и титруют 0,1 н. NaOH.
Осторожно! Чистая кислота очень взрывоопасна и ядовита! Как и ее раство-
растворы, имеет невыносимо резкий запах, вызывает головокружение, головные боли,
раздражает кожу, вызывает понижение давления (крови) и расширение сосудов.
362 Е Свойства, очистка и приготовление важнейших реагентов
АЗОТНАЯ КИСЛОТА HNO3
Продажная концентрированная азотная кислота содержит 65—68% HNO3
(d = 1,40 — 1,41); концентрация так называемой «дымящей» азотной кислоты
равна почти 100% (d= 1,52).
Внимание/ Действует разъедающе, огнеопасное вещество! Разлитую азотную
кислоту нельзя собирать легковоспламеняющимся материалом (тряпками, фильт-
фильтровальной бумагой); ее следует разбавить водой и нейтрализовать. Относительно
предосторожностей при работе с азотной кислотой см. также «Нитрозные газы».
АКРИЛОНИТРИЛ CH2=CH-CN
Т. кип. 77 °С; nD20 1,3910; dA20 0,806
Акрилонитрил частично растворим в воде, спирте и эфире. Интервал конце-
концентраций акрилонитрила, в которых происходит смешивание с водой, составляет
3—88%. Азеотроп с водой кипит при 70,6 "С и содержит 85,7% ацетонитрила.
Очистка и сушка. Сушат молекулярными ситами 4А и перегоняют в вакууме
(по возможности при более низкой температуре). В коммерческий продажный
реагент добавляют стабилизатор, предотвращающий полимеризацию (послед-
(последний удаляется при перегонке).
Осторожно! Легковоспламеняющаяся жидкость, ядовит, обладает канцеро-
канцерогенным действием. Образует взрывоопасные смеси с воздухом (от 3 до 17 об. %
акрилонитрила). Акрилонитрил в 30 раз менее токсичен, чем синильная кислота.
АКТИВИРОВАННЫЙ УГОЛЬ
Примеси. Хлорид цинка, соединения серы.
Очистка. Порошкообразный активированный уголь нагревают 2—3 ч на во-
водяной бане с 4-кратным количеством 20%-ной азотной кислоты, отмывают во-
водой до нейтральной реакции, сушат при 100—110 °С.
АЛЮМОГИДРИД ЛИТИЯ (ЛИТИЙАЛЮМИНИЙГИДРИД) LiAIH4
Подходящими растворителями являются диэтиловый эфир, тетрагидрофуран,
N-алкилморфолины. Если литийалюминийгидрид полностью не растворяется, то
работают с его суспензией. Растворители не должны содержать воды и пероксидов!
По окончании восстановления избыток литийалюминийгидрида разрушают,
осторожно приливая небольшими порциями воду. При работе с большими коли-
количествами сначала добавляют этилацетат, пока не прореагирует весь литийалю-
литийалюминийгидрид, а затем добавляют необходимое количество воды для осаждения
гидроксида алюминия.
Осторожно!Легко воспламеняется! Литийалюминийгидрид очень бурно ре-
реагирует с водой, может самовозгораться. Реакции с участием литийалюминий-
литийалюминийгидрида следует проводить лишь при перемешивании со взрывобезопасными
моторами, а выделяющийся водород отводить.
Е Свойства, очистка и приготовление важнейших реагентов 363
АМАЛЬГАМА НАТРИЯ
Амальгама, содержащая 1,2% натрия, при комнатной температуре имеет кон-
консистенцию пасты, а при 50 "С становится жидкой. Амальгамы с более высоким
содержанием натрия при комнатной температуре твердые, их можно измельчить
в порошок.
Получение 2%-ной амальгамы. Под тягой нагревают в тигле 600 г ртути до
30—40 "С и вносят 13 г натрия, нарезанного маленькими кубиками, вводя его
кусочки с помощью длинной острой стеклянной палочки под поверхность рту-
ртути. Реакция сопровождается воспламенением. Чтобы предотвратить разбрызги-
разбрызгивание, тигель накрывают керамической плиткой. После того как амальгама
застынет, ее измельчают в атмосфере азота и сохраняют без доступа воздуха.
О получении амальгамы натрия см. также Физер Л., Физер М. Реагенты для
органического синтеза. Т. 2, с. 366. — М.: «Мир», 1970.
Осторожно! Амальгаму натрия нельзя трогать руками. Она ни в коем случае
не должна соприкасаться с водой.
АМИД НАТРИЯ NaNH2
Амид натрия лучше всего измельчать сухим в ступке. При этом необходимы со-
соответствующие меры предосторожности (защитные очки, тяга, плотные защит-
защитные перчатки)!
Осторожно! С водой амид натрия реагирует со взрывом. Старые препараты
могут детонировать даже при попытке вынуть их из склянки.
Уничтожение отходов. Амид покрывают под тягой слоем толуола или бензи-
бензина и медленно приливают спирт.
АММИАК NH3
Т. кип. -33,5 °С.
Насыщенный при 15 "С водный раствор содержит 35% (или 308 г/л) аммиака
(d= 0,882). Продажный концентрированный раствор аммиака обычно содержит
25% (или 227 г/л) аммиака (</= 0,91).
Сушка. Газообразный аммиак сушат NaOH, КОН или натронной известью.
При повышенных требованиях к отсутствию влаги аммиак дополнительно про-
пропускают над стружками кальция или молекулярным ситом 3 А.
Осторожно!'Аммиак образует взрывоопасные смеси с воздухом A5,5—27 об. %
аммиака). Газообразный аммиак раздражает верхние дыхательные пути и глаза.
При тяжелых поражениях происходит нарушение сердечной деятельности и спаз-
спазмы бронхов, следствием чего может явиться воспаление или отек легких.
Для защиты следует применять дыхательный фильтр К (маркировка зелено-
зеленого цвета).
Первая помощь. Пострадавшего выносят на свежий воздух и дают пить ро-
ромашковый чай или вдыхать пары уксусной кислоты, при угрожающем положе-
положении — кислород, пропущенный через 5—7%-ный раствор уксусной кислоты
364 Е Свойства, очистка и приготовление важнейших реагентов
C0 мин, без искусственного дыхания). При поражении глаз промывать их
15 мин водой, затем 0,9%-ным раствором поваренной соли; следует избегать
химических нейтрализующих средств.
АЦЕТАЛЬДЕГИД (ЭТАНАЛЬ) СН3СНО
Т. кип. 20,8 °С; ио201,3316.
Получение из паральдегида. В перегонный прибор с колонкой помещают па-
ральдегид и каплю концентрированной серной кислоты. При умеренном нагре-
нагревании ниже 35 °С перегоняется ацетальдегид. Его собирают в охлаждаемый
льдом приемник либо непосредственно отгоняют в реакцинную смесь.
Осторожно! Пожароопасен, опасен для здоровья! Образует взрывоопасные
смеси с воздухом D—57 об. % ацетальдегида). Пары ацетальдегида раздражают
слизистые оболочки дыхательных путей, могут вызвать сердцебиение и рас-
расстройство желудка. Предполагается канцерогенное действие.
АЦЕТИЛЕН (ЭТИН) СН=СН
В 100 г ацетона при 13 атм и 15 °С растворяется около 30 л ацетилена.
Чистый ацетилен взрывается уже под давлением 2 атм, поэтому его хранят в
стальных баллонах в ацетоновом растворе. Раствором пропитана пористая мас-
масса (например, кизельгур). Чтобы предотвратить захват ацетона, баллоны при ра-
работе должны стоять вертикально. Технический ацетилен содержит фосфин
(запах!).
Сушка и очистка. Для очистки от фосфина газ пропускают через осушитель-
осушительную колонку с кремнеземом, пропитанным смесью серной кислоты и перман-
ганата калия. Содержащийся в газе ацетон можно удалить активированным
углем. Ацетилен можно сушить, пропуская над пентаоксидом фосфора или
молекулярным ситом ЗА.
Осторожно! Смеси ацетилена с воздухом взрываются при содержании ацети-
ацетилена от 1,5 до 80 об. % ацетилена. Ацетилен нельзя приводить в соприкосновение
с серебром или медью, так как при этом образуются взрывчатые ацетилениды.
Содержащийся в баллонах ацетилен ядовит из-за примеси фосфина.
АЦЕТОН СНзСООСНз
Т. кип. 56,2 "С, «о201,3591, dA20 0,791.
Смешивается со спиртом, эфиром и водой в любых отношениях. Не образу-
образует азеотропной смеси с водой.
Очистка и сушка. Чистота продажного ацетона достаточна почти для лю-
любых целей. Ацетон можно сушить молекулярным ситом ЗА, желательно в ди-
динамическом режиме (см. разд. А, 1.10.2), или оставив приблизительно на 1 ч
над пентаоксидом фосфора, прибавляя время от времени свежий осушитель.
Для менее ответственных целей достаточна сушка хлоридом кальция. После
сушки ацетон необходимо перегнать. Следует иметь в виду, что при сушке
Е Свойства, очистка и приготовление важнейших реагентов 365
основными (и в меньшей степени кислыми) агентами образуются продукты
конденсации.
Осторожно! Легковзрывающаяся жидкость, относится к классу В. Взрыво-
Взрывоопасны смеси с воздухом, содержащие 1,6—15,3 об. % ацетона.
АЦЕТОНИТРИЛ CH3CN
Т. кип. 81,5 "С, nD20 1,3441, d420 0,782.
Смешивается с водой, спиртом и эфиром в любых отношениях. Азеотропная
смесь с водой кипит при 76,7 °С и содержит 84,1% ацетонитрила.
Технический ацетонитрил содержит в качестве примесей акрилонитрил,
амиловый спирт и оксазол.
Очистка и сушка. 1 л ацетонитрила нагревают 1 ч с 0,1 г перманганата калия
при 60 "С и перегоняют затем на эффективной колонке. Первые 10% отгона от-
отбрасывают и затем продолжают перегонку. Сушат молекулярными ситами ЗА.
Осторожно! Пожароопасная жидкость класса В. Ядовит. Особую опасность
представляет имеющаяся в нем примесь свободной синильной кислоты
(см. «Синильная кислота»).
БЕНЗАЛЬДЕГИД С6Н5СНО
Т. кип. 179 °С, 65 "С при 12 мм рт. ст.; nD201,5448.
Бензальдегид летуч с парами воды.
Примеси. Продажный продукт всегда содержит бензойную кислоту (ауто-
окисление, см. схему [Г.1.46]). Перед реакцией бензальдегид необходимо пере-
перегнать в вакууме.
Опасен для здоровья (класс AIII)!
БЕНЗИН
Т. кип. 80-180 "С.
Бензин представляет собой смесь углеводородов (см. также «Лигроин»,
«Петролейный эфир»).
Очистка. См. «н-Гексан».
Внимание! Легковоспламеняющаяся жидкость, относится к классу AI. Смеси бен-
бензина с воздухом взрывоопасны. Бензины не сгорают спокойно, а разбрызгиваются.
БЕН30ИЛПЕР0КСИД С6Н5СО-ОО-СОС6Н5
Т. пл. 107 "С.
Очистка. Пероксид растворяют в небольшом количестве холодного хлоро-
хлороформа и осаждают метанолом. Сырой продажный продукт сушат в вакуум-экси-
вакуум-эксикаторе над пентаоксидом фосфора.
Осторожно! Взрывоопасен! Бензоилпероксид нельзя перекристаллизовы-
вать при нагревании. Температуру плавления определять лишь в исключитель-
исключительных случаях!
366 Е Свойства, очистка и приготовление важнейших реагентов
БЕНЗОЛ С6Н6
Т. кип. 80,1 °С; т. пл. 5,5 °С; nD20 1,5010; d420 0,879.
При 20 °С бензол растворяет 0,06% воды; при той же температуре вода раст-
растворяет 0,07% бензола. Азеотропная смесь с водой кипит при 69,25 °С и содержит
91,17% бензола. О тройной азеотропной смеси с водой и этанолом см. «Этанол».
Примеси. Сырой бензол может содержать до 0,15% тиофена. Тест с изатином
и серной кислотой (индофениновая реакция).
Сушка. Бензол можно осушить азеотропной перегонкой; при этом отбрасы-
отбрасывают первые 10% дистиллята. Для более тщательного обезвоживания применя-
применяют молекулярное сито 4А или натриевую проволоку; натрий добавляют до тех
пор, пока не прекратится выделение водорода.
Удаление тиофена. К 1 л бензола прибавляют 80 мл концентрированной сер-
серной кислоты, энергично перемешивают 30 мин при комнатной температуре, тем-
темный слой кислоты отделяют. Эту операцию повторяют до тех пор, пока кислота
станет окрашиваться лишь слабо. Бензол тщательно отделяют и перегоняют.
Внимание/ Легковоспламеняющаяся жидкость, относится к классу AI.
Ядовит, канцерогенен. Взрывоопасны смеси с воздухом, содержащие 0,8—8,6%
(по объему) бензола.
Бензол является сильным ядом для крови. Он может всасываться и через ко-
кожу. Хронические отравления ведут к поражению печени и нервной системы. Все
работы с бензолом следует проводить под хорошо работающей тягой!
Первая помощь при отравлении ароматическими углеводородами. Снять про-
пропитанную одежду, кожу тщательно промыть водой с мылом. При попадании в
глаза промывать их 10-15 мин проточной водой. Если ароматический углеводо-
углеводород попал в желудок, вызвать рвоту или принять в качестве слабительного пара-
парафиновое масло (из расчета 3 мл на 1 кг веса тела) или раствор 1 столовой ложки
сульфата натрия в 250 мл воды. Ни в коем случае не давать касторовое масло, мо-
молоко или спирт! В тяжелых случаях — дыхание кислородом, консультация врача.
БРОМ Вг2
Т. кип. 58 °С; т. пл. -7,3 °С; rf420 3,14.
Сушка. Встряхивание с концентрированной серной кислотой.
Осторожно/ Бром — очень сильный яд, раздражающий дыхательные пути.
Жидкий бром уже после кратковременного воздействия образует на коже
пузыри, при более длительном действии — болезненные, трудно заживающие
нарывы.
Первая помощь. Кожу промывают спиртом, водой, затем разбавленным раст-
раствором карбоната натрия. При поражении дыхательных путей поступают так же,
как при отравлении хлором.
БРОМОВОДОРОД НВг
Азеотропная смесь с водой кипит при 126 °С; она содержит 47,5% бромоводоро-
да, что соответствует 8,8 М раствору (d= 1,48).
Е Свойства, очистка и приготовление важнейших реагентов
367
Рис. Е.1. Получение бромоводорода.
Получение. В двугорлую колбу прибора, изображенного на рис. Е. 1 помеща-
помещают высушенный (над сульфатом натрия) и перегнанный тетралин и немного
железных опилок. Из капельной воронки медленно прибавляют бром, охлаж-
охлаждая колбу водой; когда реакция замедлится, колбу нагревают на водяной бане
до 30—40 °С. Выделяющийся бромоводород проходит через промывную склян-
склянку с тетралином, где задерживаются пары брома. В ловушке, охлаждаемой до
-60 °С, удерживаются вода, тетралин и остатки брома. Усовершенствованная
аппаратура для получения бромоводорода описана в работе: Hudlicky M. Chem.
Listy, A962), 56, 1442.
Если необходимы большие количества бромоводорода, то следует предпо-
предпочесть метод получения из брома и красного фосфора. [Houben-Weyl, Bd. 5/4,
1960, S. 18.]
Осторожно! Действует разъедающе.
N-БРОМСУКЦИНИМИД
N-Br
\\
О
Т. т. 173 "С.
Получение. 1,62 моль A60 г) сукцинимида растворяют в смеси 1,60 моль F4 г)
NaOH, 300 г измельченного льда и 400 мл воды. К этой смеси за один прием при-
прибавляют при энергичном перемешивании и наружном охлаждении 85 мл брома.
Перемешивание продолжают 1—2 мин, образовавшийся осадок отфильтровыва-
отфильтровывают и промывают водой (охлажденной до 0 °С) до отрицательной реакции на
бромиды. Осадок сушат 8 ч над пентаоксидом фосфора в вакуум-эксикаторе при
0,5 мм рт. ст. или в «пистолете» Фишера при 40 °С и 10—20 мм рт. ст.
Выход 75—81%. Чистота продукта около 97%. [Ziegler К. u. a. Lieb. Ann. Chem.
1942, 551, 109.]
368 Е Свойства, очистка и приготовление важнейших реагентов
грег-БУТИЛАТ АЛЮМИНИЯ [(СН3KСО]3А1
Внимание! Протравленный алюминий должен постоянно находиться под
слоем жидкости, так как на воздухе он мгновенно окисляется.
Получение. 1 моль алюминиевой проволоки, фольги или стружки помещают
в стакан и протравливают 10%-ным раствором NaOH. Как только начнется
бурное выделение водорода, раствор щелочи сливают, алюминий три раза про-
промывают и заливают 20%-ным раствором сулемы. Через минуту жидкость слива-
сливают, образовавшийся осадок промывают водой. После этого трижды промывают
метанолом и дважды абсолютным толуолом. Дают толуолу хорошо стечь, алю-
алюминий переносят в литровую колбу, добавляют 170 г от/>е/п-бутилового спирта
(перегнанного над натрием), нагревают с обратным холодильником (хлоркаль-
циевая трубка) до тех пор, пока потемнение не укажет на начавшуюся реакцию.
Нагревание прекращают и ждут. Если реакция не начнется, прибавляют 0,2 г
сулемы или 2 г изопропилата алюминия. Выделение водорода заканчивается
приблизительно через 15 ч. Прибавляют 500 мл абсолютного толуола, центри-
центрифугируют, упаривают в вакууме. Для удаления следов растворителя сушат в ва-
вакууме 1 ч при 100 °С. Выход 85%. [Schmidt F., Bayer E. In: Houben-Weyl, Bd. 6/2,
1963, S. 16-21.]
трет-Бутилат алюминия следует хранить и использовать без доступа
влаги.
БУТИЛЛИТИИ C4H9Li1
Растворим в углеводородах и эфире.
Получение: Под тягой, за защитным экраном помещают трехгорлую колбу
на 500 мл, снабженную механической мешалкой, обратным холодильником, с
насадкой для подвода азота, термометром и капельной воронкой с выравнива-
выравниванием давления. В колбу помещают 8,6 г литиевой стружки в 200 мл абс. эфира
и затем прибавляют по каплям 68,5 г н-бутилбромида. Сперва прибавляют
лишь 30 капель бромида и охлаждают содержимое колбы до —10 °С. О начале
реакции судят по помутнению реакционной смеси. Как только реакция нач-
начнется, в течение 30 мин прибавляют оставшееся количество бутилбромида,
перемешивают реакционную смесь еще 2 ч приО—10 "С, после чего отфильтро-
отфильтровывают через стеклянную вату.
Определение нормальности бутиллития: 2 мл раствора гидролизуют 10 мл дис-
дистиллированной воды и титруют 0,1 н. раствором соляной кислоты по фенолфта-
фенолфталеину.
Осторожно! Раствор бутиллития пирофорен! Следует избегать попадания
влаги и кислорода воздуха!
1 Предпочтительнее приготовление бутиллития из н-бутилхлорида в гексане (может дол-
долгое время храниться при комнатной температуре без изменения нормальности). Более точно
нормальность бутиллития определяют исходя из его общей (см. выше) и остаточной щелоч-
щелочности, см. Талалаева Т. В., Кочешков К. А., Методы элементоорганической химии. Литий. —
М.: «Наука», 1971, с. 107, 552. — Прим. перев.
Е Свойства, очистка и приготовление важнейших реагентов 369
ВОДОРОД Н2
Очистка и сушка. Чистота водорода из баллонов достаточна для обычных целей.
При проведении особо тонких операций гидрирования водород промывают на-
насыщенным раствором перманганата калия для того, чтобы удалить контактные
яды.
Осторожно! Пожароопасен. Взрывоопасны смеси водорода с воздухом,
содержащие 4—75 об. % водорода. При разгрузке автоклавов водород следует
отводить во внешнюю среду.
н-ГЕКСАН С6Н14
Т. кип. 68,7 "С; яо20 1,3751; d420 0,661. Азеотроп с водой кипит при 61,6 °С и
содержит 94,4% гексана.
Очистка и сушка. Встряхивают с небольшими порциями низкопроцентного
олеума до тех пор, когда кислота станет приобретать лишь слабо-желтую окрас-
окраску. После этого промывают концентрированной серной кислотой, водой,
2%-ным раствором гидроксида натрия и снова водой. Сушат гидроксидом калия
и перегоняют. Для более тщательного обезвоживания применяют натриевую
проволоку, гидрид кальция или молекулярное сито 4А.
Внимание/Легковоспламеняющаяся жидкость, относится к классу AI. Взры-
Взрывоопасны смеси с воздухом, содержащие 1,1—8 об. % гексана.
ГИДРАЗИНГИДРАТ H2NNH2 H2O
Т. кип. 118,5 °С.
Гидразингидрат легко растворим в воде и спирте, нерастворим в эфире.
Гигроскопичен.
Получение 85%-ного гидразингидрата^. 100 г 30%-ного гидразингидрата сме-
смешивают с 200 г ксилола и отгоняют вначале азеотропную смесь ксилола и воды
на колонке Вигре B0 см). После определения концентрации в остатке гидразин
может быть использован без перегонки.
Получение безводного гидразина и 100%-ного гидразингидрата аммоноли-
зом гидразинсульфата описано в работе Fischer H. Org. Syntheses, Coll. Vol. Ill,
1955, S. 515.
Определение содержания гидразина. При титровании кислотой в присутствии
метилоранжа образуется моногидрохлорид.
Осторожно! Гидразингидрат — яд, разъедает кожу и токсичен для крови,
вызывает судороги и поражает сердце; возможно, является канцерогеном.
Первая помощь. Пораженные места кожи промыть разбавленной уксусной
кислотой. При отравлении принимают глюкозу.
Это соответствует 64%-ному раствору чистого гидразина в воде.
370 Е Свойства, очистка и приготовление важнейших реагентов
ГИДРИД НАТРИЯ
Осторожно! Пожароопасен. К гидриду натрия нельзя прикасаться руками.
Его следует защищать от влаги.
Получение. 1 моль натрия и 500 мл сухого циклогексана помещают в автоклав
с магнитной мешалкой (менее пригоден встряхиваемый автоклав). Нагревают с
водородом 12 ч при 200 °С под давлением 200 атм. Продолжая перемешивание,
охлаждают. Полученную суспензию гидрата натрия используют в дальнейшей
работе.
ГЛИКОЛЬ
См. «Этиленгликоль».
ГЛИЦЕРИН (ПРОПАНТРИОЛ-1,2,3) НОСН2-СНОН-СН2ОН
Т. кип. 180 "С при 12,5 мм рт. ст.; т. пл. 20 °С; nD201,4745.
Глицерин гигроскопичен, смешивается в любых отношениях с водой и спир-
спиртом, нерастворим в эфире, бензоле, хлороформе.
Очистка и сушка. Перегонка в вакууме.
ДИМЕТИЛОВЫЙ ЭФИР ДИЭТИЛЕНГЛИКОЛЯ (ДИГЛИМ) (СН3ОСН2СН2JО
Т. кип. 161 °С, 62-63 °С при 15 мм рт. ст.; nD20 1,4073; </420 0,937.
Сушка. 1 л диглима перемешивают 12 ч с 10 г измельченного гидрида каль-
кальция, затем декантируют и перегоняют в вакууме водоструйного насоса (при
12ммрт. ст.).
Внимание!Диглим оказывает тератогенное действие.
ДИМЕТИЛСУЛЬФАТ (CH3JSO4
Т. кип. 76 "С при 15 мм рт. ст.; nD201,3874; d42S 1,321.
Диметилсульфат нерастворим в холодной воде и лишь очень медленно гид-
ролизуется ею.
Очистка. Перегонка в вакууме.
Осторожно! Диметилсульфат очень ядовит. Возможно, обладает канцеро-
канцерогенной активностью. Отравление может произойти как через легкие, так и че-
через кожу. Оно сопровождается появлением язв, судорог и может приводить к
параличу. Поражение легких проявляется лишь через несколько часов. Рабо-
Работать с диметилсульфатом можно только в хорошо действующем вытяжном
шкафу, надевая защитные перчатки.
Первая помощь. Загрязненные диметилсульфатом участки кожи смачивают
разбавленным аммиаком. При попадании на одежду немедленно ее снять.
Е Свойства, очистка и приготовление важнейших реагентов 371
ДИМЕТИЛСУЛЬФОКСИД (ДМСО) CH3SOCH3
Т. кип. 72 "С при 12 мм рт. ст.; т. пл. 18,5 "С; «D201,4783; d,20 1,101.
Примеси. Вода, диметилсульфид, диметилсульфон.
Сушка. Оставляют на сутки над молекулярным ситом 4А или кипятят 2 ч с
обратным холодильником над гидридом кальция. Затем перегоняют в вакууме
водоструйного насоса в токе сухого чистого азота.
ДИМЕТИЛФОРМАМИД (ДМФА) (CH3JNCHO
Т. кип. 153,0 °С; nD2i 1,4269; dA20 0,950.
Диметилформамид смешивается с водой и большинством органических
растворителей в любых отношениях. Кроме того, он растворяет многие соли.
Примеси: амины, аммиак, формальдегид, вода.
Очистка и сушка. Перегоняют смесь 250 мл диметилформамида, 30 мл толуола
и 6 мл воды. Сначала отгоняются толуол, вода, амины, аммиак, затем в вакууме —
диметилформамид.
Чистый диметилформамид имеет найтральную реакцию и не обладает запа-
запахом. Для более тщательной очистки можно нагревать диметилформамид 3—4 ч с
приблизительно 0,5 г гидрида кальция в атмосфере инертного газа (чистого азота
или аргона) в колбе с обратным холодильником. После перегонки в вакууме диме-
диметилформамид хранят в атмосфере инертного газа в темноте; в противном случае
он частично разлагается с образованием диметиламина и формальдегида.
Непрореагировавший гидрид кальция можно еще раз использовать для очист-
очистки диметилформамида; если гидрид не планируют использовать далее, его разла-
разлагают 70%-ным этанолом (оставляют на несколько часов, затем добавляют неболь-
небольшое количество воды и убеждаются, что при этом не происходит выделение газа).
Сушка ДМФА возможна также молекулярными ситами 4А.
Внимание! Диметилформамид не безопасен для здоровья. ДМФА оказывает
тератогенное действие.
1,4-ДИОКСАН
Т. кип. 101 °С; т. пл. 12 °С; nD201,4224; d^ 1,034.
Диоксан смешивается с водой в любых соотношениях. Азеотроп с водой
кипит при 87,8 °С и содержит 81,6% диоксана.
Примеси. Диоксан может содержать уксусную кислоту, воду, этиленацеталь
уксусного альдегида. Относительно образования пероксидов см. «Диэтиловый
эфир».
Очистка и сушка. При перемешивании и взбалтывании осторожно смешива-
смешивают диоксан с 5 об. % концентрированной серной кислоты, затем смесь нагрева-
нагревают с обратным холодильником 2 ч. После охлаждения при энергичном встряхи-
встряхивании добавляют твердый гидроксид калия (обычно внешнее охлаждение не
обязательно), оставляют до разрушения пластиночек КОН; эту операцию пов-
повторяют до тех пор, пока свежий КОН не будет сохранять свою форму в течение
ночи. Диоксан фильтруют, добавляют нарезанный небольшими кусочками
372 Е Свойства, очистка и приготовление важнейших реагентов
натрий и вновь нагревают с обратным холодильником, пока капельки натрия не
перестанут реагировать (останутся блестящими). После перегонки {осторожно*.
Кристаллизующийся диоксан может забить охлаждаемый водой холодильник!)
добавляют натриевую проволоку. Относительно сушки молекулярным ситом 4А
и удаления пероксидов см. «Диэтиловый эфир».
Осторожно!Пожароопасная жидкость класса В. Опасен для здоровья. Взры-
Взрывоопасны смеси с воздухом, содержащие 1,97—25 об. % диоксана. Возможно,
обладает канцерогенной активностью.
ДИОКСИД ПЛАТИНЫ PtO2
Чистый диоксид платины окрашен в коричневый цвет.
Получение. Смесь 2 г платинохлороводородной кислоты, 7 г воды и 20 г чис-
чистого нитрата натрия медленно упаривают в фарфоровой чашке досуха, затем
прокаливают при 400—500 °С (темно-красное каление). Работать под тягой! Ког-
Когда перестанут выделяться оксиды азота, смесь охлаждают. Плав выщелачивают
дистиллированной водой, осадок отфильтровывают и промывают дистиллиро-
дистиллированной водой от нитратов; сушат в эксикаторе.
Осторожно!Способствует горению, действует раздражающе.
ДИОКСИД СЕЛЕНА SeO2
Возгоняется при 315 °С. Гигроскопичен.
Получение из селена. В фарфоровой чашке под тягой нагревают на песчаной
бане 50 мл концентрированной азотной кислоты и осторожно, небольшими
порциями добавляют около 30 г селена. Каждую новую порцию прибавляют
после того, как прореагировала предыдущая. При перемешивании упаривают
досуха, охлаждают и измельчают в порошок.
Активирование диоксида селена. Неочищенный диоксид селена помещают в
фарфоровую чашку, приливают концентрированную азотную кислоту до обра-
образования густой массы. Чашку накрывают перевернутой воронкой и нагревают
на песчаной бане. Сначала испаряются летучие продукты, затем на стенках во-
воронки осаждается возгоняющийся диоксид селена. Скорость возгонки должна
быть такой, чтобы диоксид селена не улетучивался через трубку воронки. Для
возгонки 40 г диоксида селена требуется примерно 2,5 ч. [Синтезы органических
соединений. Сб. 2. - М.: ИЛ, 1952, с. 118.]
Внимание: Диоксид селена — яд, может вызвать поражение кожи. При работе с
диоксидом селена следует избегать вдыхания его паров. Работать в атмосфере аргона!
Первая помощь. Пораженную кожу промывают водой с мылом, а затем
4%-ным раствором бисульфита натрия. При вдыхании диоксида селена следует
вынести пострадавшего на свежий воздух, дать пить молоко, вызвать рвоту.
ДИОКСИД СЕРЫ SO2
Т. кип. -10 "С.
В 100 г воды при 20 °С растворяется 10,6 г диоксида серы.
Внимание!Диоксид серы раздражает слизистые оболочки, однако серьезные
нарушения здоровья вызывают лишь относительно высокие концентрации.
Е Свойства, очистка и приготовление важнейших реагентов 373
Симптомы в этих случаях такие же, что и при отравлении фосгеном. Для защи-
защиты следует использовать дыхательный фильтр Е (желтая маркировка).
Первая помощь: см. «Фосген».
ДИ(грег-БУТИЛ)ПЕРОКСИД
Т. кип. 30 °С при 41 мм рт. ст.
Получение. К смеси 3 моль /яре/и-бутанола и 1 моль 70%-ной серной кислоты
в течение 1,5 ч при энергичном перемешивании и охлаждении до —2 ¦*- —8 "С
добавляют смесь 1 моль 27%-ного пероксида водорода и 4 моль концентриро-
концентрированной серной кислоты. По окончании добавления перемешивают еще 3 ч, ор-
органический слой отделяют, промывают 60 мл воды, 3 раза (по 60 мл) 30%-ным
раствором едкого натра, а затем еще 3 раза (по 15 мл) водой.
После сушки над сульфатом магния пероксид можно непосредственно ис-
использовать для инициирования радикальных реакций.
Внимание! Способствует горению, действует раздражающе на кожу и слизис-
слизистые! Об опасностях при работе с пероксидами см. разд. Г, 1.5.
ДИХЛОРМЕТАН (МЕТИЛЕНХЛОРИД) СН2С12
Т. кип. 40 °С; nD20 1,4246; cf420 1,325.
Азеотропная смесь с водой кипит при 38,1 °С и содержит 98,5% дихлорметана.
Очистка. Промывают кислотой, щелочью и водой, сушат карбонатом калия
и перегоняют. Можно сушить молекулярным ситом 4А.
Внимание! Из-за опасности взрыва нельзя допускать контакта дихлорметана
с натрием. Опасен для здоровья. Дихлорметан вредно действует на нервную сис-
систему; возможно, обладает канцерогенным действием.
1,2-ДИХЛОРЭТАН (ЭТИЛЕНДИХЛОРИД) CICH2CH2CI
Т. кип. 83,7 °С; ио201,4444; </420 1,253.
Азеотропная смесь с водой кипит при 72 °С и содержит 81,5% 1,2-дихлорэтана.
Очистка и сушка. Промывают концентрированной серной кислотой, затем
водой и перегоняют над пентаоксидом фосфора.
Внимание! Пожароопасное вещество класса AI. Яд; возможно, обладает кан-
канцерогенной активностью. Дихлорэтан вызывает расстройство зрения. Дополни-
Дополнительные сведения см. в разд. «Тетрахлорметан». Из-за опасности взрыва нельзя
допускать контакта с натрием. Взрывоопасны смеси с воздухом, содержащие
6,2-15,9 об. % дихлорэтана.
ДИЭТИЛЕНГЛИКОЛЬ (ДИГЛИКОЛЬ) НО(СН2JО(СН2JОН
Т. кип. 244,3 "С, 130 °С при 8 мм рт. ст.; nD2Q 1,4475.
Диэтиленгликоль смешивается с водой.
Примеси. Этиленгликоль, триэтиленгликоль.
374 Е Свойства, очистка и приготовление важнейших реагентов
Очистка. Перегонка в вакууме.
Диэтиленгликоль применяют для заполнения нагревательных бань. Целесо-
Целесообразно поверх налить слой парафинового масла.
Внимание!Относительно ядовитости диэтиленгликоля см. разд. «Этиленгли-
коль».
ДИЭТИЛОВЫЙ ЭФИР (ЭФИР) С2Н5ОС2Н5
Т. кип. 34,6 °С; nD20 1,3527; d^ 0,7193.
При 15 °С эфир растворяет 1,2% воды, а вода при 20 °С растворяет 6,5% эфи-
эфира. Азеотропная смесь с водой содержит 1,26% воды и кипит при 34,15 °С. Про-
Продажный продукт всегда содержит некоторые количества спирта и воды.
Сушка. Абсолютный эфир получают, выдерживая его несколько дней над
хлоридом кальция. Затем хлорид кальция отфильтровывают, а эфир сушат над
натриевой проволокой. Натрий добавляют до тех пор, пока его поверхность не
перестанет терять блеск. При сушке молекулярным ситом 4А для удаления пе-
роксидов рекомендуется добавлять активный оксид алюминия.
Внимание! На свету эфир образует взрывчатые пероксиды (обнаружение пос-
последних см. в разд. Г, 1.5). Гидропероксиды удаляют встряхиванием с раствором
сульфата железа(П) (~ 5 г FeO4 • 7Н2О в 20 мл воды на 1 л эфира).
Эфир следует хранить в темных склянках над едким кали, который переводит
образующиеся гидропероксиды в нерастворимые соли, а кроме того, является и
очень подходящим осушителем.
Осторожно! Легковоспламеняющаяся жидкость, относится к классу AI.
Взрывоопасны смеси с воздухом, содержащие 1,2—51 об. % эфира.
И30ПР0ПИЛАТ АЛЮМИНИЯ [(СН3JСНО]3А11
Т. кип. 130-140 "С при 7 мм рт. ст.; т. пл. 118 "С.
Получение. В литровую колбу с эффективным обратным холодильником,
снабженным хлоркальциевой трубкой, помещают 1 моль алюминиевой проволо-
проволоки или фольги, 300 мл абсолютного изопропилового спирта (продажный изопро-
пиловый спирт перегоняют над натрием, взятым в количестве 5% от массы спир-
спирта) и 0,5 г сулемы (Hg2Cl2), смесь нагревают. Как только смесь закипит, через хо-
холодильник добавляют 2 мл тетрахлорметана и нагревание продолжают, пока не
начнется выделение водорода. В этот момент нагревание прекращают; иногда
колбу приходится даже охлаждать. По окончании бурной реакции кипячение
продолжают до почти полного растворения алюминия F—12 ч). После этого
избыток спирта отгоняют, а остаток перегоняют в вакууме с воздушным холо-
холодильником. Полученный продукт обычно затвердевает лишь через 1—2 дня.
Выход 90-95%.
Для проведения восстановления по Меервейну—Понндорфу—Верлею часто
используют 1 М раствор изопропилата алюминия в абсолютном изопропаноле,
который хранят в тщательно запарафинированной склянке с притертой пробкой.
1 См. также Schmidt F.; Bayer E. in Houben-Weyl. Bd. 6/2 A963) S. 16-21.
Е Свойства, очистка и приготовление важнейших реагентов 375
ИОДОВОДОРОДНАЯ КИСЛОТА HI
Постоянно кипящая смесь имеет т. кип. 126,5 °С, содержит 56,7% иодоводорода,
что соответствует 7,6 М раствору; d= 1,7.
На свету в присутствии кислорода воздуха иодоводородная кислота разлага-
разлагается. Для стабилизации прибавляют 1 г красного фосфора на 1 л кислоты. Для
регенерации содержащую иод кислоту нагревают почти до кипения и прибавля-
прибавляют по каплям 50%-ную фосфористую кислоту до обесцвечивания смеси. После
этого перегоняют.
Внимание!'Первая помощь см. в разд. «Хлороводород».
ИОНООБМЕННЫЕ СМОЛЫ
Ионообменные смолы — это нерастворимые полимеры со способными к диссо-
диссоциации кислотными или основными группами (сульфокислоты, карбоновые
кислоты или аммониевые соли). Противоион, связанный с такой группой, спо-
способен к обмену, например:
Смола-SO3GH® + Na® CMona-SO®Na® + Н®
Относительно различных сортов ионообменных смол и их торговых названий
см. Houben-Weyl, Bd. I/I, A958), S. 528, а также информации химических фирм.
Переведение катионообменной смолы в водородную форму иллюстрируется
ниже на примере сильнокислой сульфофенольной смолы: в хроматографичес-
кую колонку наливают немного воды, затем засыпают 5 г ионообменника.
Колонка должна быть заполнена на три четверти ее высоты. Дают воде стечь до
уровня ионообменной смолы, пропускают через колонку 150 мл чистой 1 н.
соляной кислоты. С помощью крана внизу колонки скорость пропускания уста-
устанавливают около 5 мл/мин. Затем пропускают дистиллированную воду до нейт-
нейтральной реакции вытекающей жидкости. После такой обработки влажный
ионообменник готов к применению.
N-CN-ИНДИКАТОР
Представляет собой смесь двух растворов. Первый раствор: смешивают 50 мл
0,2%-ного раствора нингидрина в абсолютном спирте, 10 мл ледяной уксусной
кислоты и 2 мл 2,4,6-коллидина. Второй раствор: 1%-ный раствор тригидрата
нитрата меди(П).
Для обрызгивания бумажных хроматограмм незадолго до применения
первый и второй растворы смешивают в отношении 25 : 1.
КАЛИИ К
Осторожно! Пожароопасен, действует на кожу разъедающе. Калий может са-
самовозгораться на воздухе; измельчать его можно только под слоем инертного
растворителя (защитные очки!). Остатки следует тотчас же осторожно уничто-
376 Е Свойства, очистка и приготовление важнейших реагентов
жать /и^е/и-бутанолом. Если спирт при этом воспламенится, то сосуд надо закрыть
керамической плиткой. Избегать контакта воды и низших спиртов с калием!
КСИЛОЛ
Т. кип. 136—144 °С (продажный ксилол представляет собой смесь трех
изомерных ксилолов).
Азеотропная смесь с водой кипит при 92 °С и содержит 64,2% ксилола.
Опасен для здоровья. О симптомах отравления см. разд. «Бензол».
ЛИГРОИН
Т. кип. 120—135 °С.
Смесь углеводородов; см. «Бензин», «Петролейный эфир».
Очистка. См. «и-Гексан».
Внимание! Легковоспламеняющаяся жидкость класса AI. Смеси лигроина с
воздухом взрывоопасны.
МЕТАНОЛ СНзОН
Т. кип. 64,7 °С; nD20 1,3286; d420 0,792.
Сушка. К 1 л метанола прибавляют 5 г стружек магния, после окончания
выделения водорода кипятят 2—3 ч с обратным холодильником и перегоняют.
Если в метаноле содержится более 1 % воды, то магний не реагирует. В таком
случае немного магния обрабатывают чистым метанолом и после начала реак-
реакции добавляют эту затравку к основной массе метанола. При этом общее коли-
количество магния несколько увеличивают по сравнению с указанным выше. Путем
динамической сушки молекулярным ситом ЗА (см. разд. А, 1.10.2) содержание
воды можно довести до 0,005%.
Осторожно/Легковоспламеняющаяся жидкость класса В, яд. Взрывоопасны
смеси с воздухом, содержащие 5,5—36,5 об. % метанола.
Метанол вызывает приступы тошноты, нарушает деятельность сердца и
нервной системы, вызывает слепоту.
МЕТИЛЕНХЛОРИД
См. «Дихлорметан».
МОЛЕКУЛЯРНЫЕ СИТА
Молекулярные сита представляют собой синтетические цеолиты. Их применя-
применяют для эффективного обезвоживания жидкостей и газов, а также в качестве
селективных адсобрентов. Молекулярные сита обозначают и применяют в соот-
соответствии с диаметром пор (в ангстремах).
Для сушки растворителей и газов преимущественно применяют молекуляр-
молекулярные сита ЗА и 4А. Методика сушки (и регенерация) приведена в разд. А, 1.10.2.
Молекулярное сито 5А позволяет также удалять из газов примеси (NH3, H2S,
тиолы, следы НС1 и т. д.). Они адсорбируют также «-парафины.
Е Свойства, очистка и приготовление важнейших реагентов 377
Регенерация. Использованное молекулярное сито под тягой встряхивают с
большим количеством воды, чтобы экстрагировать адсорбированный раствори-
растворитель. Во избежание взрыва при прокаливании эта операция особенно необходи-
необходима в случае молекулярных сит, применявшихся для сушки горючих растворите-
растворителей. Воду отделяют и молекулярное сито прокаливают в сушильном шкафу
сначала при 200-250 "С, а затем при 300-350 °С в вакууме (не менее 10~2 кПа,
или 10 мм рт. ст.) до постоянной массы. Охлаждают и только после этого
допускают контакт молекулярного сита с воздухом, пропуская через колонку с
перхлоратом магния.
НАТРИИ
T.m.91,l°C\d=0,91.
Получение суспензии натрия в толуоле или ксилоле: см. разд. Г,7.2.1.8
(«Сложноэфирная конденсация», с. 138).
Осторожно! Пожароопасен, действует разъедающе на кожу. При любых ра-
работах с натрием необходимо надевать защитные очки. Реакционные смеси с ме-
металлическим натрием нельзя нагревать на водяной бане.
Уничтожение отходов. Остатки натрия небольшими порциями вносят в
большой объем метанола.
НАТРИЕВЫЕ АЛКОГОЛЯТЫ
Получение. Необходимое количество натрия помещают в трехгорлую колбу,
снабженную капельной воронкой и обратным холодильником с хлоркальциевой
трубкой. Прибавляют по каплям 10-кратное (по массе) количество соответству-
соответствующего спирта с такой скоростью, чтобы смесь энергично кипела. Не рекоменду-
рекомендуется обратный порядок добавления (натрий к спирту), так как при этом реакция
легче может выйти из-под контроля. В низших спиртах натрий растворяется до-
довольно быстро. В случае высших спиртов необходимо многочасовое перемеши-
перемешивание при 100 "С. Сухой алкоголят можно получить отгонкой спирта из раство-
раствора алкоголята в вакууме. Удобнее, однако, получать алкоголят натрия, добавляя
эквимолярное количество спирта к суспензии натрия в инертном растворителе
(см. также Schmidt F., Bayer E. In: Houben-Weyl, Bd. 6/2 A963), S. 1-70).
НИТРОЗНЫЕ ГАЗЫ
Нитрозными газами называют смеси оксидов азота, которые часто образуются
при работе с азотной кислотой (см. «Азотная кислота»).
Осторожно! Очень ядовиты. Нитрозные газы даже в малых количествах
опасны для здоровья. Они вызывают раздражение дыхательных путей и глаз,
а также тошноту и головную боль. Раздражение проходит через 15—60 мин, одна-
однако через несколько часов неожиданно начинается кашель с пенистыми, ржаво-
красными выделениями, затрудняется дыхание (такое состояние может развить-
развиться даже через двое суток). Это свидетельствует о появлении отека легких, кото-
378 Е Свойства, очистка и приготовление важнейших реагентов
рый вместе с развивающейся метгемоглобинемией крови создает серьезную
угрозу жизни. При отравлении нитрозными газами обязательно надо обратить-
обратиться к врачу.
Для защиты применять дыхательный фильтр В (маркировка серого цвета).
Первая помощь. См. разд. «Фосген».
ОКСАЛИЛХЛОРИД (СОС1J
Т. кип. 63-64 °С; nD20 1,4305; d4201,476.
Оксалилхлорид — бесцветная жидкость с резким запахом. Хорошо раство-
растворим в ароматических углеводородах. Очистка осуществляется перегонкой.
Осторожно! При взаимодействии с водой происходит бурная реакция с вы-
выделением НС1, СО2 и СО. При соприкосновении с металлическими поверхнос-
поверхностями или при действии третичных аминов разлагается с образованием фосгена!
При хранении оксалилхлорида в запаянной ампуле вследствие его частичного
разложения может возникнуть повышенное давление!
ОЛЕУМ
Олеум представляет собой раствор триоксида серы в серной кислоте. При содер-
содержании <40% и 69—70% SO3 олеум — жидкость.
Осторожно/ Олеум действует разъедающе. Ни в коем случае нельзя разбав-
разбавлять олеум водой, а только концентрированной серной кислотой.
ПАЛЛАДИЙ НА АКТИВИРОВАННОМ УГЛЕ (КАТАЛИЗАТОР)
а) Для дегидрирования. Смесь 2,5 г хлорида палладия, 25 мл дистиллированной
воды и 2,1 мл концентрированной соляной кислоты кипятят до растворения
(около 2 ч), раствор охлаждают смесью льда с солью и при перемешивании до-
добавляют 25 мл 40%-ного формалина, 10 г оксида магния (ч.д.а.), 15 г очищенно-
очищенного активированного угля (очистку см. в разд. «Активированный уголь»). Затем
при перемешивании и охлаждении прибавляют раствор 25 г КОН в 25 мл дис-
дистиллированной воды. При этом температура не должна подниматься выше 5 °С.
Катализатор 7 раз промывают дистиллированной водой (с декантацией) и, нако-
наконец, переносят его на стеклянный фильтр и промывают 1 л дистиллированной
горячей воды. Из влажной массы формуют с помощью пресса небольшие цили-
цилиндрики (длиной 3-4 мм) и сушат при 90 °С.
Если пресса нет, то можно поступить так: заполняют тонкую стеклянную
трубку, несколько раз втыкая ее во влажную массу. Подобрав подходящую
палочку, спрессовывают катализатор в трубке, затем осторожно выталкивают,
разрезают и сушат. [Anderson A. G. J. Am. Chem. Soc, 1953, 75, 4985.]
б) Для гидрирования. Смесь 2,5 хлорида палладия, 6 мл концентрированной
соляной кислоты (ч.д.а.) и 15 мл дистиллированной воды кипятят с обратным
холодильником до образования прозрачного раствора (около 2 ч). Раствор раз-
разбавляют 43 мл дистиллированной воды и выливают на 28 г очищенного активи-
Е Свойства, очистка и приготовление важнейших реагентов 379
рованного угля (очистку см. в разд. «Активированный уголь»), находящегося в
плоской фарфоровой чашке. Полученную массу упаривают на водяной бане,
высушивают в сушильном шкафу при 100 "С. Порошкообразную массу хранят в
хорошо закрывающейся склянке. Полученный катализатор можно применять
непосредственно, если не мешает образующийся при гидрировании хлороводо-
род.
В противном случае поступают так: необходимое количество хлорида палла-
палладия на угле (приготовленного, как описано выше) гидрируют в подходящем
растворителе до прекращения поглощения водорода. Отсасывают на стеклян-
стеклянном фильтре, промывают тем же растворителем до отрицательной реакции на
хлороводород; влажный катализатор применяют для гидрирования [Мозинго Р.
Синтезы органических препаратов. Сб. 4. — М.: ИЛ, 1953, с. 409].
ПЕНТАН С5Н12
Т. кип. 36 "С; nD20 1,3577; d420 0,626.
Азеотроп с водой кипит при 34,6 "С и содержит 1,4% воды.
Очистка. См. разд. «н-Гексан».
Внимание!Легковоспламеняющаяся жидкость, относится к классу AI. Взры-
Взрывоопасны смеси с воздухом, содержащие 1,35—8 об. % пентана.
ПЕРОКСИД ВОДОРОДА Н2О2
30%-ный водный раствор пероксида водорода называют пергидролем.
Осторожно! Действует разъедающе! При концентрировании в вакууме раст-
растворы пероксида водорода могут взрываться. Горючие материалы (вата и др.) при
соприкосновении с пергидролем могут загораться.
ПЕТРОЛЕЙНЫЙ ЭФИР
Низкокипящая смесь, состоящая из пентана и гексана, т. кип. 40—70 °С.
Очистка. См. разд. «н-Гесан».
Внимание! Легковоспламеняющаяся жидкость класса AI. Смеси с воздухом
взрывоопасны.
ПИРИДИН C5H5N
Т. кип. 115 °С; nD20 1,5100; </420 0,982.
Пиридин гигроскопичен; он смешивается с водой, спиртом и эфиром в лю-
любых отношениях. Азеотроп с водой кипит при 94 °С и содержит 57% пиридина.
Азеотроп с уксусной кислотой (максимальной концентрации) кипит при
139,7 °С и содержит 65% пиридина.
Очистка и сушка. В большинстве случаев достаточно сушить над КОН
(в течение 2-х недель); перегоняют на хорошей колонке, собирая фракцию
114—116 °С. Можно сушить молекулярным ситом 4А.
380 Е Свойства, очистка и приготовление важнейших реагентов
Очистка и сушка для гидрирования: Пиридин нагревают в колбе с обратным
холодильником и добавляют перманганат калия до тех пор, пока окраска раство-
раствора не перестанет изменяться. Выпавший осадок коричневого цвета отфильтро-
отфильтровывают и пиридин перегоняют. Дистиллят смешивают с равным количеством
ледяной уксусной кислоты, после чего отгоняют примерно 10% от общего
объема смеси (этот отгон отбрасывают). После охлаждения к остатку добавляют
равный объем воды и перегоняют на эффективной ректификационной колонке.
Собирают отогнанный азеотроп (вода—пиридин, состав см. выше) и сушат
азеотропной отгонкой с бензолом (см. «Бензол»).
Осторожно! Легковоспламеняющаяся жидкость класса В. Опасен для здо-
здоровья. Взрывоопасны смеси с воздухом, содержащие 1,8—12,5 об. % пиридина.
Пиридин вызывает на коже экзему. Вдыхание паров пиридина вызывает
тошноту, боли в желудке, заболевание нервной системы.
ПЛАТИНА НА АКТИВИРОВАННОМ УГЛЕ, 10% (КАТАЛИЗАТОР)
Смесь 4,5 г чистого порошкообразного активированного угля, 1,33 г платино-
хлористоводородной кислоты (гексагидрат) и 30 мл воды нейтрализуют раство-
раствором гидрокарбоната натрия, нагревают до 80 °С и медленно, при перемешива-
перемешивании добавляют 3 мл раствора формалина. Одновременным добавлением гидро-
гидрокарбоната натрия поддерживают слабощелочную реакцию. Через 2 ч охлаждают,
осадок отфильтровывают, тщательно промывают и сушат на воздухе.
ПОЛИФОСФОРНАЯ КИСЛОТА
Получение, а) В вакууме водоструйного насоса (~ 12 мм рт. ст.) отгоняют воду из
85%-ной фосфорной кислоты, остаток нагревают 6 ч в вакууме при 150 °С.
Оставшаяся полифосфорная кислота кристаллизуется, б) К 100 мл фосфорной
кислоты (а1 = 1,7) постепенно при охлаждении и перемешивании добавляют
150—210 г Р4ОЮ. Затем смесь нагревают несколько часов на водяной бане. Полу-
Получают полифосфорную кислоту с содержанием Р4О10 от 80 до 84%.
Осторожно! Действует разъедающе!
РАСТВОР ГИДРОСУЛЬФИТА (БИСУЛЬФИТА) НАТРИЯ
Технический гидросульфит — это насыщенный водный раствор гидросульфита
натрия. Обычно его чистота достаточна для синтеза аддуктов с карбонильными
соединениями.
Получение насыщенного раствора гидросульфита натрия: 1 моль гидроксида
натрия растворяют в 150 мл воды, при охлаждении вводят диоксид серы (сернис-
(сернистый газ) до требуемого увеличения массы реакционной смеси или до обесцвечи-
обесцвечивания фенолфталеина.
РАСТВОР ФТАЛАТА АНИЛИНА
0,93 г анилина и 1,66 г фталевой кислоты растворяют в 100 мл насыщенного
водой бутанола.
Е Свойства, очистка и приготовление важнейших реагентов 381
РЕАКТИВ ДЕНИЖЕ
В 100 мл воды растворяют 5 г оксида ртути и 20 мл концентрированной серной
кислоты. Очень ядовит!
РЕАКТИВ ЛУКАСА
Получение. 0,5 моль безводного хлорида цинка при охлаждении растворяют в
0,5 моль концентрированной соляной кислоты.
РЕАКТИВ ТОЛЛЕНСА
Получение. Растворяют 1 г нитрата серебра в 10 мл воды; раствор хранят в темно-
темноте. Непосредственно перед употреблением небольшое количество этого раство-
раствора смешивают с равным объемом раствора 1 г NaOH в 10 мл воды, выпавший
осадок оксида серебра растворяют, осторожно добавляя концентрированный
раствор аммиака.
Осторожно!При стоянии образуется в высшей степени взрывчатое гремучее
серебро, поэтому остатки реактива следует немедленно уничтожать подкисле-
нием.
РЕАКТИВ ШИФФА
Получение. Готовят 0,025%-ный водный раствор фуксина и пропускают через
него сернистый газ до обесцвечивания.
РТУТЬ
d420 13,55; давление пара при 20 °С 1,22 ¦ 10 мм рт. ст. @,16 • 10-3 кПа).
Для очистки ртуть пропускают через фильтр, в острие конуса которого про-
проделано отверстие иглой. Затем каплям ртути дают падать через разбавленную
азотную кислоту и, наконец, несколько раз через воду. Для удаления следов
воды еще раз фильтруют, как указано выше.
Осторожно! Ртуть и ее соли очень ядовиты. Типичные признаки ртутного от-
отравления: сильное слюнотечение, опухоли десен, падение способности к умствен-
умственному сосредоточению. Все работы, в ходе которых переливают ртуть или имеются
другие возможности разлить ее, проводят над ванночкой (например, фотографи-
фотографической), чтобы пролитую ртуть можно было собрать и подвергнуть разложению
(например, конц. HNO3). Если все же ртуть пролили, то ее надо собрать с
помощью специальных «ртутных щипцов». Щели столов или пола, в которые мог-
могла попасть ртуть, следует засыпать порошком серы или иодированным углем.
Мельчайшие капельки ртути собирают, прикасаясь к ним медной проволокой,
предварительно протравленной азотной кислотой и амальгамированной.
Для защиты от ртути следует применять дыхательный фильтр HgP3 (с бело-
голубой маркировкой).
382 Е Свойства, очистка и приготовление важнейших реагентов
Первая помощь при отравлении растворимыми соединениями ртути: съесть
белок (например, сырое яйцо) и вызвать рвоту.
СЕРОВОДОРОД H2S
Т. кип. -0,4 "С.
Сероводород, получаемый в аппарате Киппа из сульфида железа(Н), содер-
содержит значительные количества водорода.
Для сушки сероводород пропускают над хлоридом кальция.
Осторожно! Очень ядовит и пожароопасен. Взрывоопасны смеси с возду-
воздухом, содержащие 4—46 об. % сероводорода.
Отравления небольшими количествами сероводорода проявляются в голо-
головокружении, тошноте и головной боли. В больших концентрациях газ вызывает
мгновенную потерю сознания. Запах можно считать предупредительным сигна-
сигналом только очень короткое время и только при низкой концентрации сероводо-
сероводорода, так как обоняние быстро притупляется. Для защиты следует применять
дыхательные фильтры В (маркировка серого цвета).
Первая помощь. Пострадавшего выносят на свежий воздух и применяют
искусственное дыхание.
СИНИЛЬНАЯ КИСЛОТА (ЦИАНОВОДОРОД) HCN
Т. кип. 25 "С.
Синильная кислота смешивается с водой, спиртом и эфиром в любых соотно-
соотношениях. Давление пара безводной синильной кислоты при 0 °С равно 264 мм рт. ст.
Синильная кислота часто образуется при работе с цианидами.
Осторожно! Синильная кислота пожароопасна и исключительно ядовита.
Смертельная доза равна 50 мг. Синильная кислота парализует внутриклеточное
дыхание, разрушая железосодержащие дыхательные ферменты (образует с ними
комплексные соединения). При вдыхании значительных количеств уже через
несколько секунд наступает внезапная смерть. Для защиты следует применять
дыхательный фильтр В (маркировка серого цвета). Если соединение попало в
организм лишь в малой дозе, то наряду с раздражением (особенно гортани) по-
появляется чувство теплоты, головокружение, шум в ушах, нарушение зрения,
слюнотечение, рвота, сердечные приступы. Пострадавший поправляется очень
медленно, возможны дальнейшие осложнения. Синильная кислота может пос-
поступать в организм и через кожу.
Первая помощь. Все меры помощи необходимо принимать как можно быст-
быстрее! Оказывающий помощь должен позаботиться и о собственной защите
(обычные противогазы не годятся!). Отравленного выносят на свежий воздух.
Если он не потерял сознания, заставляют вдыхать изоамилнитрит (по 3—5 ка-
капель в течение 10 с с интервалами в 2 мин, однако в отсутствие врача не более
5—6 раз, так как имеется опасность чрезмерного понижения кровяного давле-
давления). Пострадавшего надо оставить в лежачем положении.
Попавшие на кожу брызги синильной кислоты необходимо очень тщательно
смыть водой с мылом. Если синильная кислота или цианиды попали в желудок,
Е Свойства, очистка и приготовление важнейших реагентов 383
то немедленно дают выпить рвотное — раствор поваренной соли A столовая
ложка на стакан воды) или суспензию 10 г оксида магния и 2 г сульфата желе-
за(П) в 100 мл воды и вызвать рвоту. Находящемуся без сознания ничего не
вливать! Во всех случаях немедленно вызвать врача.
Уничтожение отходов. К слабощелочному раствору синильной кислоты при-
прибавляют 20%-ный раствор сульфата железа(П) и оставляют на длительное время,
или окисляют пероксидом водорода до изоцианата (при рН 10—11).
СКЕЛЕТНЫЙ НИКЕЛЬ (НИКЕЛЬ РЕНЕЯ)
Получение щелочного высокоактивного скелетного никеля (по Урушибара).
В большой сосуд (объемом 5 л или больше) суспендируют 50 г порошкообразно-
порошкообразного никельалюминиевого сплава, содержащего от 30 до 50% никеля, и 500 мл во-
воды. Прибавляют твердый гидроксид натрия без охлаждения с такой скоростью,
с какой позволяет образующаяся пена.
Осторожно! Бурно протекающая реакция начинается после индукционного
периода, продолжающегося 0,5-1 мин. Смесь начинает бурно кипеть. Когда но-
новые порции гидроксида натрия уже не вызывают заметной реакции (для этого
требуется добавить около 80 г гидроксида натрия), смесь оставляют на 10 мин,
затем выдерживают 30 мин на водяной бане при 70 °С. Никель осаждается в ви-
виде шлама на дно. Водную фазу сливают, катализатор промывают водой 2—3 раза
встряхиванием и декантацией, затем таким же путем 2—3 раза тем же раствори-
растворителем, который планируется применять при гидрировании. Если этот раствори-
растворитель не смешивается с водой, то сначала катализатор промывают растворителем,
смешивающимся как с водой, так и с растворителем для гидрирования.
Катализатор можно хранить некоторое время под слоем растворителя, однако
при этом каталитическая активность никеля падает. Поэтому лучше готовить
катализатор каждый раз заново, непосредственно перед применением [Bilica H. R.,
Atkins H., Org. Syntheses. Coll. Vol. Ill, 1955, p. 176].
Нейтральный скелетный никель получают тщательной промывкой описанно-
описанного выше катализатора. При этом сильно падает активность никеля и получается
катализатор, имеющий степень активности W2.
Дальнейшая инактивация катализатора происходит при его промывке
0,1%-ным раствором уксусной кислоты. Полученный таким путем катализа-
катализатор уже не способен восстанавливать карбонильные группы.
Осторожно! Сухой катализатор самовозгорается. Поэтому фильтры со
скелетным никелем нельзя бросать в корзину для бумаг!
Для уничтожения отходов их сжигают в подготовленном месте; образую-
образующийся при этом оксид никеля собирают для переработки.
СОЛЯНАЯ КИСЛОТА HCI
См. также «Хлороводород». Насыщенная при 15 °С соляная кислота содержит
42,7% хлорводорода. Продажная концентрированная соляная кислота плот-
плотностью 1,184 содержит 37% хлороводорода. Азеотропная смесь с водой кипит
при 110 "С и содержит 20,24% хлороводорода, что соответствует 6,1 М раствору.
384 Е Свойства, очистка и приготовление важнейших реагентов
Осторожно! Концентрированная соляная кислота действует разъедающе,
особенно на глаза и слизистые оболочки.
Первая помощь при поражении глаз: промыть струей воды в течение
-15 мин.
СПИРТЫ
Сушка: см. «Метанол» и «Этанол». Для сушки высших спиртов применяют раст-
раствор метилата магния. Его готовят путем кипячения в течение 2—3 ч
(с обратным холодильником) металлического магния с 10-кратным количест-
количеством метанола (с содержанием воды менее 1%) и с небольшим количеством тет-
рахлорметана. 50 мл полученного таким путем раствора метилата магния добав-
добавляют к 1 л подлежащего сушке спирта, кипятят 2—3 ч, а затем перегоняют.
Обезвоженный таким путем спирт содержит примесь метанола. Для реак-
реакций, в которых метанол может помешать, спирты следует сушить специальными
методами.
СТЕАРАТ КОБАЛЬТА
Получение. 6 г стеариновой кислоты растворяют при 60 °С в 20 мл абсолютного
этанола и нейтрализуют 2 н. NaOH (не содержащим карбоната) по фенолфтале-
фенолфталеину. Полученный гель нагревают, образовавшийся раствор медленно, при эф-
эффективном перемешивании добавляют к горячему раствору 2,8 г СоСЬ • 6Н2О в
20 мл 50%-ного этанола. Осадок тщательно промывают водой, затем спиртом и
ацетоном, отжимают, сушат при 100 °С и измельчают в порошок.
СУЛЬФОЛАН (ТЕТРАМЕТИЛЕНСУЛЬФОН)
о' ЧЪ
Т. кип. 118 °С при 5 мм рт. ст.; т. пл. 28 °С; «D30 1,4820; dA20 1,261.
Очистка. 1 л сульфолана и 10 г гидроксида натрия нагревают несколько часов
в атмосфере азота при 170—180 °С и затем перегоняют в вакууме. Сульфолан
можно очистить также сушкой над молекулярным ситом 13Х с последующей
перегонкой в вакууме. Хранить сульфолан следует в холодильнике.
ТЕТРААЦЕТАТ СВИНЦА(М) (СН3СООLРЬ
Получение. В двухлитровую трехгорлую колбу, снабженную мешалкой и термо-
термометром, помещают 850 мл ледяной уксусной кислоты и 170 мл уксусного ангид-
ангидрида. Смесь нагревают до 40 °С и при энергичном перемешивании к ней добав-
добавляют 0,5 моль C43 г) PD3O4. Температура при этом не должна подниматься выше
65 °С. Затем смесь перемешивают при 60—65 °С до образования прозрачного
раствора. При охлаждении выкристаллизовывается тетраацетат свинца. Его
отсасывают, перекристаллизовывают из ледяной уксусной кислоты и сушат в
вакуум-эксикаторе. Выход около 160 г.
Е Свойства, очистка и приготовление важнейших реагентов 385
Тетраацетат свинца легко гидролизуется с образованием диоксида свинца и
уксусной кислоты, поэтому при кристаллизации и фильтровании его следует
защищать от влаги воздуха. Ядовит.
ТЕТРАГИДРОФУРАН
"о
Т. кип. 65,4 "С; по20 1,4070; d^ 0,887.
Обычно тетрагидрофуран стабилизирован 0,025% 2,6-ди-т/ге/и-бутилкрезо-
ла. Тетрагидрофуран смешивается с водой. Азеотропная смесь с водой кипит
при 63,2 "С и содержит 94,6% тетрагидрофурана.
Очистка. См. «Диоксан». При добавлении серной кислоты необходимо
эффективное охлаждение. Можно сушить тетрагидрофуран молекулярным
ситом 4А.
Тетрагидрофуран хранят над твердым гидроксидом калия.
Осторожно!Легковоспламеняющаяся, действует раздражающе.
В тетрагидрофуране пероксиды образуются существенно быстрее, чем в ди-
этиловом эфире (см. «Диэтиловый эфир»). Тетрагидрофуран, содержащий мно-
много пероксидов, нельзя обрабатывать гидроксидом калия (см. Org. Syntheses, 1966,
46, 105).
ТЕТРАХЛОРМЕТАН (ТЕТРАХЛОРУГЛЕРОД) ССЦ
Т. кип. 76,8 °С; nD201,4603; d420 1,594.
Азеотропная смесь с водой кипит при 66 °С и содержит 95,9% тетрахлорметана.
Тройная азеотропная смесь с водой D,3%) и этанолом (9,7%) кипит при 61,8 °С.
Очистка и сушка. Обычно достаточно перегонки. Вода при этом удаляется в
виде азеотропной смеси (первые части дистиллята отбрасывают). Если к сушке
и очистке предъявляются более высокие требования, то тетрахлорметан кипятят
с обратным холодильником 18 ч с пентаоксидом фосфора и перегоняют на
колонке. Тетрахлорметан можно сушить также молекулярным ситом 4А.
Осторожно! Ядовит. Тетрахлорметан обладает наркотическим действием,
вызывает головную боль, судороги и экзему. Эти симптомы более или менее
сильно проявляются при любых отравлениях хлорированными углеводородами.
В особенности следует помнить, что эти соединения поражают печень и почки.
Возможно, они обладают также канцерогенной активностью. Опасен для окру-
окружающей среды. Тетрахлорметан нельзя сушить натрием (опасность взрыва!).
ТИОНИЛХЛОРИД SOCI2
Т. кип. 79 "С.
Гидролизуется очень легко.
Очистка. Для большинства целей обычно достаточно перегнать продажный
тионилхлорид. Очень чистый, бесцветный продукт получают перегонкой над
хинолином и льняным маслом.
Осоторожно/Тионилхлорид раздражает кожу и слизистые оболочки.
386 Е Свойства, очистка и приготовление важнейших реагентов
ТОЛУОЛ С6Н5СНз
Т. кип. 110,8 "С; «D201,4969; dA20 0,867.
Азеотропная смесь с водой кипит при 84,1 °С и содержит 81,4% толуола.
Сушка. См. разд. «Бензол».
Осторожно! Опасен для здоровья, легковоспламеняющаяся жидкость (клас-
(класса AI). Взрывоопасны смеси с воздухом, содержащие 1,27—7 об. % толуола.
Симптомы отравления: см. «Бензол». Толуол несколько менее токсичен, чем
бензол. Очень вероятно, что толуол вызывает патологию развития плода, т. е.
является тератогеном.
ТРИЭТИЛЕНГЛИКОЛЬ (ТРИГЛИКОЛЬ) НО(СН2JО(СН2JО(СН2)ОН
Т. кип. 287 °С, 165 °С при 14 мм рт. ст.
См. «Диэтиленгликоль».
ТРИХЛОРЭТИЛЕН С12С=СНС1
Т. кип. 87,2 °С; /zD2°l,4778; </420 1,462.
Азеотропная смесь с водой кипит при 73,6 °С и содержит 94,6% трихлорэтилена.
Примеси. В результате самоокисления в трихлорэтилене накапливаются
такие очень ядовитые вещества, как хлороводород, оксид углерода, фосген.
Очистка и сушка. Тщательно встряхивают сначала с карбонатом калия, затем
с водой, сушат над хлоридом кальция и перегоняют на колонке.
Осторожно! Пары могут вызывать наркотическое привыкание. Другие ука-
указания см. в разд. «Тетрахлорметан». Возможно, обладает канцерогенной актив-
активностью.
Трихлорэтилен нельзя сушить над натрием, возможен взрыв!
УКСУСНАЯ КИСЛОТА (ЛЕДЯНАЯ) СН3СООН
Т. кип. 118 °С; т. пл. 16,6 °С; nD201,3720; rf4201,05.
Смешивается с водой во всех отношениях.
Примеси. Следовые количества уксусного альдегида (ацетальдегида), воды.
Очистка и сушка. Для большинства целей достаточно выморозить уксусную
кислоту. Не следует охлаждать слишком сильно, так как при этом в кристалли-
кристаллическую фазу могут перейти также вода и другие примеси. Отсасывают на охлаж-
охлаждаемой воронке, хорошо отжимают. Для более тщательной очистки уксусную
кислоту кипятят 2—6 ч с 2—5%-ным раствором перманганата калия с последую-
последующей фракционированной перегонкой. После этого долго встряхивают с
фосфорным ангидридом и снова фракционируют.
Внимание! Действует разъедающе. Смеси с воздухом взрывчаты, начиная с
4 об. % уксусной кислоты. На коже вызывает образование пузырей.
Первая помощь. Промыть пораженное место большим количеством воды.
При поражении глаз промывать их водой не менее 15 мин.
Е Свойства, очистка и приготовление важнейших реагентов 387
УКСУСНЫЙ АНГИДРИД (АЦЕТАНГИДРИД) (СН3СОJО
Т. кип. 139,6 °С; ио20 1,3904; dA20 1,082.
Гидролизуется теплой водой. При взаимодействии с водными растворами
кислот и щелочей происходит сильное разогревание (иногда это взаимодействие
протекает взрывообразно).
Примеси. Уксусная кислота.
Очистка. Кипячение с безводным ацетатом натрия с последующей перегонкой.
Осторожно!Действует разъедающе. Даже при кратковременном воздействии
сильно поражает кожу.
ФЕЛИНГОВА ЖИДКОСТЬ
Получение. Готовят два раствора: один содержит 1,73 г кристаллогидрата
сульфата меди (гидрат) в 25 мл воды, другой — 8,5 г сегнетовой соли и 2,5 г гид-
роксида натрия в 25 мл воды. Перед употреблением смешивают равные объемы
этих растворов.
ФОРМАЛЬДЕГИД (МЕТАНАЛЬ) НСНО
Т. кип. -21 "С.
Водный раствор, содержащий 30—40% формальдегида и 5—15% метанола,
называют формалином.
Получение сухого газообразного формальдегида. Параформальдегид сушат нес-
несколько дней над фосфорным ангидридом в вакуум-эксикаторе и затем деполи-
меризуют сухой перегонкой. Нагревают до такой степени, чтобы 30 г параформ-
альдегида разлагались примерно за 20 мин.
При проведении реакции Гриньяра следует применять примерно двукрат-
двукратный избыток параформальдегида, так как следы воды в соединительных трубках
и шлангах с реакционным сосудом (эти соединения должны быть максимально
короткими и широкими) вызывают частичную полимеризацию.
Формальдигед разрушает резиновые шланги.
Внимание! Ядовит. На коже формальдегид вызывает экзему. Он поражает
также глаза и дыхательные пути. Возможно, обладает канцерогенной актив-
активностью.
ФОСГЕН СОС12
Т. кип. 7,6 °С.
Фосген хорошо растворяется в бензоле и толуоле, плохо — в холодной воде.
Гидролизуется горячей водой. Фосген имеет своеобразный удушающий запах,
напоминающий запах гниющего сена.
Очистка и сушка. Газообразный фосген пропускают через две соединенные
последовательно промывные склянки, первая из которых наполнена раститель-
растительным маслом (подсолнечным, соевым или хлопковым), а вторая — концентриро-
концентрированной серной кислотой.
388 Е Свойства, очистка и приготовление важнейших реагентов
Осторожно!'Очень ядовит! Фосген — один из наиболее ядовитых газов. Пер-
Первые признаки отравления фосгеном (насморк, затрудненное дыхание, кровавый
кашель) нередко появляются лишь через несколько часов. После того как исчез-
исчезнут указанные первые признаки, может наступить кажущееся временное облег-
облегчение, во время которого, однако, в течение нескольких часов развивается отек
легких, который часто приводит к смерти в результате нарушения сердечной
деятельности.
Фосген может образовываться при пиролизе хлорированных углеводородов.
Осторожно при пользовании огнетушителями с тетрахлорметаном!
Даже при легких отравлениях фосгеном необходимо обратиться к врачу. Для
защиты применяют дыхательный фильтр В (маркировка серого цвета).
Первая помощь. Пострадавшего уложить и транспортировать только в лежа-
лежачем положении. Даже при легком отравлении необходим полный покой. Поле-
Полезен свежий воздух, лучше кислород, однако искусственного дыхания делать нель-
нельзя. Пострадавшего завернуть в одеяло, чтобы предотвратить переохлаждение.
Избыток фосгена поглощают 20%-ным раствором гидроксида натрия.
ХЛОР С12
При 20 °С в 100 г воды растворяется 1,85 г хлора, а в 100 мл тетрахлорметана —
около 17 г хлора. Хлор разрушает резину; шланги быстро становятся ломкими.
При работе с хлором пользуйтесь тефлоновыми шлангами!
Сушка. С помощью концентрированной серной кислоты.
Осторожно! Ядовит. Хлор очень сильно раздражает легкие и слизистые
оболочки. Симптомы отравления хлором сходны с симптомами отравления
фосгеном. Для защиты применяют дыхательный фильтр В (маркировка серо-
серого цвета).
Первая помощь. См. разд. «Фосген». При легком поражении для уменьше-
уменьшения кашля и раздражающего действия вдыхают пары спирта или настоя
ромашки.
ХЛОРАЛЬ (ТРИХЛОРЭТАНАЛЬ) СС13СНО
Т. кип. 98 'С.
Получение из хлоральгидрата. Хлоральгидрат встряхивают с 4-кратным коли-
количеством теплой концентрированной серной кислоты, слой хлораля отделяют и
перегоняют.
Осторожно! Ядовит.
ХЛОРИД АЛЮМИНИЯ А1С13
Возгоняется выше 180 °С.
Очень чувствителен к влаге.
Очистка. Хлорид алюминия должен возгоняться без остатка. Плохие препа-
препараты можно очистить возгонкой в условиях защиты от влаги.
Е Свойства, очистка и приготовление важнейших реагентов
389
Внимание! Хлорид алюминия раздражает кожу и слизистые оболочки. Сухой
препарат реагирует с водой со взрывом.
ХЛОРОВОДОРОД HCI
См. также «Соляная кислота».
Получение. В приборе, собранном в соответствии с рис. Е.2, к смеси тонкоиз-
мельченной поваренной соли и концентрированной соляной кислоты постепенно
приливают концентрированную серную кислоту. (Кончик капельной воронки
целесообразно оттянуть в капилляр.) Хлороводород отбирают через боковой отвод,
сушат концентрированной серной кисло-
кислотой (см. рис. А. 11). Ток хлороводорода регу-
регулируют скоростью подачи серной кислоты.
Осторожно! Действует разъедающе!
Хлороводород поражает легкие и слизис-
слизистые оболочки. Даже 0,05% хлороводорода
в воздухе могут быть смертельными. Для
защиты следует применять дыхательный
фильтр Е (маркировка желтого цвета).
Первая помощь. Пострадавшего выно-
выносят на свежий воздух и оставляют спокой-
спокойно лежать до оказания дальнейшей
медицинской помощи.
концентрированная
серная кислота
хлороводород
соляная кислота
и повареная соль
Рис. Е.2. Получение хлороводорода.
ХЛОРНАЯ КИСЛОТА НСЮ4
Получение 0,1 н. раствора хлорной кислоты в ледяной уксусной кислоте. При ох-
охлаждении льдом и эффективном перемешивании к рассчитанному количеству
уксусного ангидрида постепенно прибавляют столько 70%-ной водной хлорной
кислоты, сколько необходимо для полного связывания воды уксусным ангидри-
ангидридом за счет его превращения в уксусную кислоту. Охлажденную смесь разбавля-
разбавляют чистой ледяной уксусной кислотой до получения 0,1 н. раствора. Раствор не
должен содержать избыток уксусного ангидрида.
Проба на присутствие ацетангидрида. К аликвотной части раствора при пере-
перемешивании добавляют каплю воды и следят за температурой. Если наблюдается
повышение температуры, то воду прибавляют по каплям до тех пор, пока повы-
повышение температуры не прекратится. Исходя из израсходованного количества
воды вычисляют, сколько воды надо добавить ко всему раствору.
Если капля воды не вызывает повышения температуры, то точно таким же
образом следует провести пробу на присутствие в растворе воды, добавляя по
каплям уксусный ангидрид.
Установка титра. Титруют раствором безводного (высушенного при 300 °С)
карбоната натрия (ч.д.а.) в ледяной уксусной кислоте. Индикатор — 0,10%-ный
раствор кристаллического фиолетового в ледяной уксусной кислоте. После
установления титра разбавляют чистой ледяной уксусной кислотой до концент-
концентрации 0,1 н. [Houben-Weyl, Bd. 2, 1953, S. 661.]
Осторожно! Действует разъедающе! Существует опасность возгорания.
390 Е Свойства, очистка и приготовление важнейших реагентов
ХЛОРОФОРМ (ТРИХЛОРМЕТАН) СНС13
Т. кип. 61,2 °С; «D20 1,4455; d4201,4985.
Тройная азеотропная смесь хлороформ — вода — этанол кипит при 55,5 °С и
содержит 3,5% воды и 4% спирта. Продажный хлороформ содержит спирт в
качестве стабилизатора, связывающего образующийся при разложении фосген.
Очистка. Встряхивают с концентрированной серной кислотой, промывают
водой, сушат над хлоридом кальция и перегоняют. Для более полной сушки
можно воспользоваться молекулярным ситом 4А. Удаление значительной при-
примеси фосгена см. в разд. «Фосген».
Внимание/Ввиду опасности взрыва нельзя допускать контакта хлороформа с
натрием.
Опасные свойства тетрахлорметана присущи и хлороформу. Опасен для здо-
здоровья. Возможно, обладает и канцерогенной активностью. Очень вероятно, что
хлороформ оказывает тератогенное действие.
ХЛОРСУЛЬФОНОВАЯ КИСЛОТА CISO3H
Т. кип. 152 "С.
Очень легко гидролизуется!
Очистка. Перегоняют в приборе на шлифах с защитой от влаги воздуха.
Осторожно! С водой хлорсульфоновая кислота реагирует очень бурно. На
кожу и одежду она действует резъедающе даже сильнее, чем олеум.
ЦЕРИЙАММОНИЙНИТРАТ (NH4J[Ce(NO3N]
Получение. 1 г церийаммонийнитрата растворяют в 2,5 мл 2 н. азотной кислоты.
Растворение можно ускорить, слегка подогревая раствор. После охлаждения
реагент готов к употреблению.
ЦИАНИД КАЛИЯ KCN
Почти во всех случаях цианид калия можно заменить более дешевым цианидом
натрия (см. ниже).
Внимание! Очень ядовит. Первая помощь и уничтожение отходов: см.
«Синильная кислота».
ЦИАНИД НАТРИЯ NaCN
NaCN очень гигроскопичен; ниже 35 °С образует устойчивый моногидрат.
Примеси: карбонат и формиат натрия.
Осторожно! Очень ядовит. При работе с цианидом натрия часто выделяется
синильная кислота. Опасности, первая помощь и уничтожение отходов: см.
«Синильная кислота».
Е Свойства, очистка и приготовление важнейших реагентов 391
ЦИАНИД ЦИНКА Zn(CNJ
В противоположность цианидам щелочных металлов цианид цинка плохо раст-
растворим в воде.
Получение. 1 моль цианида натрия, не содержащего примеси карбоната, раст-
растворяют в 60 мл воды, прибавляют насыщенный раствор 0,55 моль хлорида цин-
цинка в 50%-ном спирте. Осадок цианида цинка отсасывают, промывают ледяной
водой, спиртом, эфиром, сушат в эксикаторе.
Внимание! Очень ядовит. Первая помощь и уничтожение отходов: см.
«Синильная кислота».
ЭТАНОЛ (ЭТИЛОВЫЙ СПИРТ) С2Н5ОН
Т. кип. 78,33 °С; nD20 1,3616; d420 0,789.
Этанол смешивается с водой, эфиром, хлороформом и бензолом в любых отно-
отношениях. Азеотропная смесь с водой кипит при 78,17 "С и содержит 96% этанола.
Тройная азеотропная смесь с водой G,4%) и бензолом G4,1%) кипит при 64,85 "С.
Примеси. Синтетический спирт загрязнен уксусным альдегидом и ацетоном,
этиловый спирт, полученный путем брожения, — высшими спиртами (сивуш-
(сивушными маслами). Для денатурации добавляют пиридин, метанол и бензин.
Сушка. В 1 л продажного «абсолютного» спирта растворяют 7 г натрия, прибав-
прибавляют 27,5 г диэтилового эфира фталевой кислоты, кипятят 1 ч с обратным холо-
холодильником, затем перегоняют с небольшой колонкой. Очищенный таким путем
спирт содержит менее 0,05% воды. Путем динамической сушки молекулярным си-
ситом ЗА (см. т. 1, разд. АД.10.2, с. 35) содержание воды можно уменьшить до 0,003%.
Из продажного «абсолютного» этанола следы воды можно удалить и другим спо-
способом. 5 г магния кипятят с обратным холодильником 2—3 ч с 50 мл «абсолютно-
«абсолютного» спирта, к которому добавлен 1 мл тетрахлорметана, затем прибавляют 950 мл
«абсолютного» спирта, кипятят еще 5 ч с обратным холодильником и перегоняют.
Обнаружение воды. Спирт, содержащий более 0,05% воды, осаждает объе-
объемистый белый осадок из бензольного раствора триэтилата алюминия.
Внимание! Легковоспламеняющаяся жидкость класса В. Взрывоопасны
смеси с воздухом, содержащие 2,6—18,9 об. % этанола.
ЭТИЛАЦЕТАТ СН3СООС2Н5
Т. кип. 77,1 "С; nD20 1,3701; d420 0,901.
Этилацетат с водой дает азеотроп с т. кип. 70,4 "С, который содержит 8,1% воды.
Примеси. Вода, спирт и уксусная кислота.
Очистка и сушка. Промывают равным объемом 5%-ного раствора карбоната
натрия, сушат хлоридом кальция и перегоняют. При более высоких требовани-
требованиях к содержанию воды несколько раз (порциями) добавляют фосфорный ангид-
ангидрид, фильтруют и перегоняют, защищая от влаги. С помощью молекулярного
сита 4А содержание воды в этилацетате можно довести до 0,003%.
Внимание! Легковоспламеняющаяся жидкость класса AI. Взрывоопасны
смеси с воздухом, содержащие 2,2—11,4 об. % этилацетата.
392 Е Свойства, очистка и приготовление важнейших реагентов
ЭТИЛЕНГЛИКОЛЬ (ГЛИКОЛЬ, ЭТАНДИОЛ-1,2) НОСН2СН2ОН
Т. кип. 92 °С при 10 мм рт. ст.; ио201,4318; d420 1,113.
Примеси. Диэтиленгликоль, триэтиленгликоль, пропиленгликоль, вода.
Очистка и сушка. Этиленгликоль перегоняют в вакууме, основную фракцию
длительное время сушат сульфатом натрия и вновь перегоняют в вакууме на
хорошей колонке.
Внимание/ Опасен для здоровья, вызывает тошноту и рвоту.
ЭТИЛЕНОКСИД (ОКСИРАН) \^7
Т. кип. 10,7 °С. Этиленоксид поставляется в стальных баллонах.
Осторожно!'Легко воспламеняется, ядовит. Взрывоопасны смеси с воздухом,
содержащие 3—80 об. % этиленоксида. Под действием щелочи полимеризуется
взрывообразно.
При попадании на кожу возникают (часто лишь через несколько часов)
нарывы, затем наступает некроз. Нарывы заживают очень медленно. При вды-
вдыхании даже малых количеств появляются выделения изо рта и носа, длительная
рвота, понос, чувство тяжести в желудке, возбужденное состояние. Высокие
концентрации вызывают наркотическое состояние. Возможны поражения серд-
сердца, печени и почек. Вероятна канцерогенная активность.
Первая помощь. Немедленно снять загрязненную одежду. Основательно про-
промыть кожу. Вдыхать свежий воздух (лучше кислород). Пострадавшего предохра-
предохранять от переохлаждения, транспортировать только в лежачем положении.
ЭФИР
См. «Диэтиловый эфир».
ЛИТЕРАТУРА
Реагенты для органического синтеза
ФизерЛ., Физер М. Реагенты для органического синтеза. Т. I—VII. — М.: «Мир», 1971.
Encyclopedia of Reagents for Organic Synthesis. Bd. 1—8. Ed.: L. Paquette. — John Wiley &
Sons, New York 1995.
Handbook of Reagents for Organic Synthesis. Bd. 1-4. — Wiley — VCH, Weinheim 1999.
Свойства и очистка органических растворителей
Bunge Ж, in: Houben-Weyl, Bd. 1/2 A959), S. 765-868.
LideD.: Handbook of Organic Solvents. - CRC Press, Boca Raton 1995.
Perkin D. D., Armarego D. R., Perrin D. R.: Purification of Laboratory Chemicals. —
Pergamon Press, Oxford 1988.
RiddickJ.A., Bunger W. В., Sakano Т. К:. Organic Solvents. — John Wiley & Sons, New York
1986.
Smallwood I. M:. Handbook of Organic Solvent Properties. —Arnold, London 1996.
ПРИЛОЖЕНИЕ
СВОЙСТВА ОПАСНЫХ РЕАГЕНТОВ
Большинство веществ, с которыми работает химик, являются опасными реаген-
реагентами. Они могут быть:
• взрывоопасными
• пожароопасными
• воспламеняющимися
• ядовитыми (токсичными)
• разъедающими
• раздражающими
• сенсибилизаторами (аллергенами)
• оказывающими канцерогенное действие (канцерогенами)
• вызывающими нарушения репродуктивной системы или патологию
развития плода (тератогенами)
• изменяющими наследственность (мутагенами)
• опасными для окружающей среды (экотоксичными)
Данная систематизация требуется для классификации химикатов по степени
опасности, которая обозначается посредством символов опасности с указанием
вида особой опасности (R-характеристика) и рекомендациями по технике безо-
безопасности при работе с веществами (S-характеристика). (R- и S-характеристики
указаны в конце данного учебникаI.
В табл. П.1 в алфавитном порядке даны важнейшие опасные реагенты,
встречающиеся в методиках данного издания с их символами опасности.
Приведены также летальные дозы (концентрации) токсичных соединений и
параметры максимально допустимых концентраций (мг/м3) этих веществ на
рабочем месте — МАК (нем. — maximale Arbeitsplatz Konzentration) или TRK
(нем. — Technische RichtkonzentrationJ.
Данные об опасности химических веществ постоянно актуализируются; они
приведены здесь по состоянию на апрель 2003 г.
1 В России не принята такая классификация опасных химических веществ. — Прим. перев.
2 См. примечание на с. 394. — Прим. ред.
394 Ж Приложение
В то же время нельзя говорить о том, что те вещества, которые не приведены
в табл. П.1, абсолютно безопасны.
ТОКСИЧНОСТЬ ВАЖНЕЙШИХ ХИМИКАТОВ
Понятие токсичности не является абсолютным, так как степень токсичности
для организма человека зависит от множества взаимосвязанных факторов. Пер-
Первостепенное значение имеют количество токсического вещества, способ его
воздействия (т. е. попало ли оно в организм в процессе дыхания или было прог-
проглочено, или введено инъекцией, или проникло через кожу), физическое состоя-
состояние (пылевидное, грубозернистое, кристаллическое, растворенное, суспендиро-
суспендированное и т. д.). Определенное значение имеет наличие сопутствующих веществ,
которые могут действовать аддитивно или активирующе, и, наконец, сущест-
существенным является психическое и физическое состояние человека, пораженного
токсическим веществом.
Естественно, что результаты действия токсического вещества на организм
определяются тем, сколько времени прошло до обнаружения отравления и ка-
какие были приняты меры. Решающим для сохранения жизни является быстрое и
продуманное вмешательство. Поэтому всегда следует обращать особое внима-
внимание на соответствующие примечания к методикам, описания свойств реагентов
(см. часть Е) и на любые другие указания.
Не меньший вред, чем острые отравления, может приносить и хроническая
токсичность некоторых химикатов. Продолжительный токсический эффект
особенно типичен для канцерогенов, мутагенов, тератогенов, веществ, вызыва-
вызывающих психопатологические и неврологические расстройства, а во всевозраста-
всевозрастающей степени также и для аллергенов. Вещества, обладающие канцерогенной и
мутагенной активностью, способны вызывать соответствующие эффекты спус-
спустя длительное время даже после кратковременного воздействия.
Приведенные в литературе данные о токсичности веществ в большинстве слу-
случаев относятся к определенному виду подопытных животных и специальным ус-
условиям опыта (например, доза смертельная для 50% всех подопытных животных,
получаемая ими в течение 30 сут при внутривенном, подкожном или внутрибрю-
шном введении вещества, растворенного в воде или масле). Во многих случаях
непосредственный перенос полученных таким путем данных на другие виды жи-
животных и на человека затруднителен, а часто вообще невозможен. Однако на
основании этих данных приводятся сведения о степени их опасности.
На основе лабораторного и промышленного опыта, дополненного экспе-
экспериментами на различных видах животных и данными статистики производ-
производственной и клинической медицины, разработаны нормы, предохраняющие от
опасности при работе с важнейшими лабораторными химикатами и продукта-
продуктами химической промышленности (параметры МАК1)- МАК — максимально
1 В России соответствующие величины называются ПДК (предельно допустимые конце-
концентрации). ПДК наиболее распространенных вредных веществ см. в ч. 3 нормативного
документа «Предельно допустимое содержание вредных веществ в воздухе рабочей зоны»,
ГОСТ 12.1.005—88 «Система стандартов безопасности труда. Общие санитарно-гигиеничес-
санитарно-гигиенические требования к воздуху рабочей зоны». — Прим. перев.
Свойства опасных реагентов 395
допустимая концентрация в воздухе на рабочем месте, которая может при
ежедневном 8-часовом рабочем дне и 40-часовой рабочей неделе без видимых
вредных последствий для здоровья человека переноситься неделями, месяца-
месяцами и даже годами.
Потенциальная токсичность, которая свойственна почти каждому химикату,
у высококвалифицированного химика должна вызывать не боязнь при обраще-
обращении с реактивами, а привычку к разумной и обоснованной осторожности. Это
же относится и к тем случаям, когда возникает необходимость в уничтожении
химикатов (см. также указания в частях А и Е).
В табл. П.1 (с. 396-411) приведены характеристики токсичности важнейших
химикатов; используются следующие обозначения:
Таблица П.1. Символы опасности, летальные дозы и МАК-значения опасных веществ
Символы опасности
С разъедающие;
Е взрывоопасные;
F воспламеняющиеся;
F+ легковоспламеняющиеся;
N опасные для окружающей среды;
О пожароопасные;
Т ядовитые;
Т+ очень ядовитые;
Xi раздражающие;
Хп опасные для здоровья.
Легковоспламеняющиеся вещества
AI температура воспламенения ниже 21 °С, не смешивается с водой;
АН температура воспламенения в интервале 21—55 °С, не смешивается с водой;
АШ температура воспламенения 55—100 °С, не смешивается с водой;
В температура воспламенения ниже 21 °С, смешивается с водой при 15 "С.
Категории токсичных реагентов — канцерогенов {к), мутагенов (м) и
реагентов, вызывающих нарушения в репродуктивной системе (р):
1 — токсичны для людей;
2 — токсичны для животных;
3 — токсичность вероятна.
Летальные дозы
ор — летальные дозы для 50% подопытных животных (LD50; для крыс, если не
приводится другого) при пероральном приеме, мг/кг живого веса;
LDLo — низшие летальные дозы при оральном приеме;
инг — летальные концентрации (LC50) в мл/м3 при вдыхании в течение указанного
времени;
LCLo — низшая летальная концентрация при вдыхании.
МАК
TRK. — технические концентрации вредных веществ, которые могут перенести
живые существа;
А — пыль, поступающая через альвеолы;
Е — вдыхаемые фракции.
Таблица П. 1. Символы опасности, летальные дозы и МАК-значения опасных веществ
Вещество
Анилиновая кислота
Азо-бис-изобутиронитрил
Азота оксид
Азотная кислота
Азотной и серной
кислот смесь
Акриламид
Акриловая кислота
Акрилонитрил
Акролеин
Алкоголь см. Этанол
Аллиловый спирт
Аллилхлорид
Алюминия трииэопропилат
Алюминия хлорид
1 -Аминонафгалин
см. а-Нафшламин
2-Аминопирщшн
З-Аминофенол
Аммиака раствор > 25%
Аммиака раствор 10—25%
Аммиака раствор 5-10%
Аммиак безводный
Аммония хлорид
Символ
опасности
Xi
Е,Хп
Т+
о,с
о, с
т
C,N
F,T,N
F,T+,N
T,N
F,T+,N
F
С
Xn,N
C,N
С
Xi
T,N
Xn
R
36
2-11-20/22-52/53
26-34
8-35
8-35
45-46-20/21-25-36/
38-43-48/23/24/24-62
10/20/21/22-35-50
45-11-23/24/25-37/38-41-43-51/53
11-24/25-26-34-50
10-23/24/25-36/37/38-50
11-26-50
11
34
20/22-51/53
34-50
34
36/37/38
10-23-34-50
22-36
S
B)
B-K/9-41-47-61
(l/2-)9-26-28-36/37/39-45
(l/2-J3-26-36-45
(l/2-J3-26-30-36-45
53-45
(l/2-J6-36/37/39-45-62
9-16-53-45-61
23-26-28-36/37/39-38-46-61
(l/2-K6/37/39-38-45-61
A/2-I6-29-33-45-61
B-)8-16
(l/2-O/8-28-45
B-I8-61
(l/2)-26-36/37/39-45-61
(l/2-J6-36/37/39-45-61
(l/2-)9-16-26-36/37/39-45
(l/2-)9-16-26-36/37/39-45-61
22
Категория
токсичности
(к, м, р)
к 2, м 2, р 3
к2
Летальные дозы
ор 11000
ор 700 (мышь)
ор340
ор78
ор26
ор64
ор450
ор 11300
ор3450
ор200
ор924
ор350
ор 1300 (мышь)
МАК,мг/м3
9
5
0,03
7 (ТИС)
0,25
4,8
3
6А
2
35
Вещество
о-Анизидин
л-Анизидин
Анилин
Ацетальдегид
Ацетамид
Ацетангиярид см.
Уксусный ангидрид
Ацетилацетон
Ацетилен
Ацетилхлорид
Ацетон
Ацетона циангидрин
Ацетонитрил
Ацетофенон
Бензальдегид
Бензальхлорид
см. Бензилиденхлорид
Бензиламин
Бензилбензоат
Бензилбромид
Бензилиденхлорид
Бензиловый спирт
Бензилхлорид
Бензоилпероксид
Бензоилхлорид
Символ
опасности
Т
T+.N
T,N
F+,Xn
Xn
Xn
F+
F,C
F,Xi
T+,N
F,Xi
Xn
Xn
С
Xn
Xi
T
Xn
T
E,Xi
С
R
45-23/24/25
26/27/28-33-50
20/21/22-40-48/23/24/25-50
12-36/37-40
40
10-22
5-6-12
11-14-34
11-36-66-67
26/27/28-50/53
11-20/21/22-36
22-36
22
21/22-34
22
36/37/38
22-23-37/38-40-41
20/22
45-22-23-37/38-40-41-48/22
2-36-43
34
S
53-45
(l/2-J8-36/37-45-61
(l/2-J8-36/37-45-61
B-I6-33-36/37
B-K6/37
B-J1-23-24/25
B-)9-16-33
(l/2-)9-16-26-45
B-)9-16-26
(l/2-O/9-27-45-61
(l/2-I6/36/37
B-J6
B-J4
(l/2-J6-36/37/39-45
B-J5
B-K9
(l/2-K6/37-38-45
B-J6
53-45
B-K/7-14-36/37/39
(l/2-J6-45
Категория
токсичности
(к, м, р)
к 2, мЗ
кЗ
кЗ
кЗ
кЗ
к2
Таблица П. 1
Летальные дозы
ор 1505
ор1320
ор250
ор661
ор7000
ор55
ор910
ор5800
ор570
ор815
ор1300
ор1900
ор3249
ор 1230
ор1231
ор7710
ор1900
Продолжение
МАК, мг/м3
0,5 (TRK)
0,51
7,7
90
1200
68
0,1
0,2
5Е
2,8
Таблица П.1. Продолжение
Вещество
>ензол
кнзотрихлорид
:м. Трихлорметилбензол
i-Бензохинон
1,4'-Бис(М,Ы-диметиламиж>)-
Зензофенон см. Михлера кетон
>ифенил (дифенил)
эром
эромбензол
>ромоводородная кислота >40%
эромоводородная кислота 10-40%
зромоводород
Зромоформ
Зромуксусная кислота
5угадиен-1,3
Зуганол-1
Буганол-2
тре/л-Буганол
Буганон
Бугантиол-1
Буген
Бугеналь-2
Бугиламин
н-Бугилацетат
трет- Бугилацетат
Символ
опасности
F,T
T,N
Xi,N
C,T+,N
Xi,N
С
Xi
С
T,N
T,C,N
F+,T
Xn
Xi
F,Xn
F,Xi
F+,
F,T+,N
F,C
F
R
45-11-48/23/24/25
23/25-36/37/38-50
36/37/38-50-53
26-35-50
10-38-51/53
34-37
36/37/38
35-37
23-36/38-51/53
23/24/25-35-50
45-46-12
10-22-37/38-41-67
10-36/37-67
10-20
11-36-66-67
12
11-24/25-26-37/38-41-50-68
11-20/21/22-35
10-66-67
11-66
S
53-45
(l/2-J6-28-45-61
23-60-61
(l/2-O/9-26-45-71
B-N1
(l/2-O/9-26-45
(l/2-O/9-26-45
(l/2-O/9-26-45
(l/2-J8-45-61
(l/2-K6/37/39^15-61
53-45
B-O/9-13-26-37/39-46
B-O/9-13-24/25-26-46
B-)9-16
B-)9-16
B-)9-16-23
(l/2-J6-28-36/37/39-45-61
(l/2-K-16-26-29-36/37/39-45
B-J5
B-I6-23-25-29-33
Категория
токсичности
(к,м,р)
Kl
к2,м2
Летальные дозы
op 930
op 130
op 3280
op 14 (LDLo, человек)
op 2699
инг. 2,4 мг/л, 4 ч
op 933
op 50 (мышь)
op 5480
op 790
op 6480
op 3500
op 2737
op 1500
op 240 (мышь)
op 366
МАК, мг/м3
3,2 (TRK)
0,45
1
0,7
6,7
11 (TRK)
300
300
360
600
1,9
1
15
480
950
Вещество
Бугилхлорид
Буган-2-диол-1,4
Бугарилхлорид
Бутиронитрил
Валериановая кислота
Ванадия пентаоксид
Винилацетат
Водород
Водорода пероксид (раствор) ?20%
Водорода пероксид (раствор) 5-20%
Гваякол
Гексаметилендиамин
Гексан
Гексанол-1
Гексанон-2
а-Гексахлорциклогексан
Гептан
Гептановая кислота
Гептанон-2
Гидразин
Гидразингидрат
Гидразинсульфат
Гидрохинон
Символ
опасности
F
Т
F,C
Т
С
T,N
F
F+
С
Xi
Xn
С
F,Xn,N
Xn
Т
T,N
F,Xn,N
С
Xn
T,N
T,N
T,N
Xn,N
Таблица П.1. Продолжение
11
21-23/25-34-48/22
11-34
10-23/24/25
34-52/53
20/22-37-68-48/23-51/53-63
11
12
34
36/38
22-36/38
21/22-34-37
11-38-48/20-51/53-62-65-67
22
10-48/23-62-67
21-25-40-50/53
11-38-50/53-65-67
34
10-20/22
45-10-23/24/25-34-43-50/53
45-23/24/25-34-43-50/53
45-23/24/25-43-50/53
22-40-41-43-50-68
B-)9-16-29
(l/2-J2-36/37/39-45
(l/2-I6-23-26-36-45
(l/2-L5
(l/2-J6-36-45-61
(l/2-K6/37-38-45-61
B-I6-23-29-33
B-)9-16-33
(l/2-K-28-36/39-45
(l/2-K-28-36/39-45
B-J6
(l/2-J2-36/37-45-60-61
B-)9-16-29-33-36/37-61-62
B-J4/25
(l/2-K6/37
(l/2-J2-36/37-45-60-61
B-)9-16-29-33-60-61-62
(l/2-J6-28-36/37/39-45
B-J4/25
53-45-60-61
53-45-60-61
53-45-60-61
2-J6-36/37/39-61
Категория
токсичности
(к,м,р)
op 104
мЗ, рЗ
рЗ
рЗ
кЗ
к2
к2
кЗ,мЗ
Летальные дозы
ор2670
ор>1000
ор140
ор 600 (мышь)
ор400-500
ор2920
ор 2000 (мышь)
ор725
ор850
ор 28710
ор720
ор2590
ор76
ор7000
ор 1670
орбО
ор129
ор601
ор320
МАК, мг/м3
95,5
0.05А
36
1,4
2,3 Е
180
21
0,5 Е
2100
238
0,13 (TRK)
2Е
Вещество
Диазометан
2,4-Диаминотолуал
1,2-Дибромэтан
Дикетен
Диметиламин
М,М-Диметиланилин
Диметилнитрозамин
Диметилсульфат
Диметилсульфоксид
Диметилформамид
1,4-Диметилциклогексан
1,3-Диншробензол
2,4-Динитротолуол
1,4-Диоксан
Ди-тр«л-бутилпероксид
Дифениламин
1,2-Дихлорбензол
1,3-Дихлорбензол
1,4-Дихлорбензол
Дихлорметан
1,2-Дихлорэтан
Дициклогексиламин
Дициклопентадиен
Диэтиламин
Символ
опасности
Т
T,N
T,N
Xn
F+,Xn
T,N
T+,N
T+
Xi
T
F,Xn,N
T+,N
T,N
F,Xn
O,F
T,N
Xn,N
Xn,N
Xn
Xn
F,T
C,N
F,Xn,N
F,C
R
45
45-20/21-25-36-43-51/53
45023/24/25-36/37/38-51/53
10-20
12-20-37/38-41
23/24/25-40-51/53
45-25-26-48/25-51/53
45-25-26-34-43
36/38
61-20/21-36
11-38-51/53-65-67
26/27/28-33-50/53
45-23/24/25-48/22-51/53-62
11-19-36/37-40-66
7-11
23/24/25-33-50/53
22-36/37/38-50/53
22-51/53
36-50/53
40
45-11-22-36/37/38
22-34-50/53
11-20/22-36/37/38-51/53
11-20/21/22-35
S
53-45
53-45-61
53-45-61
B-K
B-I6-26-39
(l/2-J8-36/37-45-61
53-45-61
53-45
26
53-45
B)9-16-33-61-62
(l/2-J8-36/37-45-60-61
53-45-61
B-)9-16-36/37-46
B-K/7-14-16-36/37/39
(l/2-J8-36/37-45-60-61
B-J3-60-61
B-N1
B-J4/25-46-60-61
B-J3-24/25-36/37
53-45
(l/2-J6-36/37/39-45-60-61
B-K6/37-61
(l/2-K-16-26-29-36/37/39-45
Категория
токсичности
(к, м, р)
к2
к2
к2
кЗ
к2
к2
Р2
кЗ
к 2, м 3, р 3
кЗ
кЗ
к2
Таблица П. 1.
¦шшшвжшяштшштт
Летальные дозы
ор73
орЮ8
ор540
орЮОО
op 1410
op 205
op 14500
op 2800
op 59,5
op 268
op 7120
op 25000
op 2000
op 500
op 580
op<2000
op 1600
op 670
op 373
op 353
op 540
Продолжение
МАК,мг/м3
0,01 (TRK)
0,1 (TRK)
0,8 (TRK)
3,7
25
0,001(TRK)
0,2 (TRK)
160
30
73
5E
300
20
300
350
20 (TRK)
200 (как
мономер)
15
Вещество
Г^Ы-Диэтиланилин
Диэтилацеталь ацетальдегида
Диэтиленгликоль
Диэтилкетон
Дизтиловый эфир
Диэтилоксалат
Диэтилсульфат
Диэтилфталат
Едкое кали см. Калия гидроксид
Едкий натр см. Натрия гидроксид
? Изоамил см. Изопентил
О
м Изобуганол
Изобугилацетат
Изомасляная кислота
Изопентилнитрит
Изопентиловый спирт
Изопрен
Изопропилбензол см. Кумол
Изопропиловый спирт
Иод
Иодоводородная кислота 0,2-10%
Иодоводородная кислота >10%
Калий
Калиягидроксид
Символ
опасности
T,N
F,Xi
Xn
F,Xi
F+,Xn
Xn
T
Xi
F
Xn
F,Xn
F+
F,Xi
Xn,N
С
С
F,C
С
R
23/24/25-33-51/53
11-36/38
22
11-37-66-67
12-19-22-66-67
22-36
45-46-20/21/22-34
10-37/38-41-67
11-66
21/22
11-20/22
12-52/53
11-36-67
20/21-50
34
35
14/15-34
22-35
S
(l/2-J8-37-45-61
B-)9-16-33
B-L6
B-)9-16-25-33
B-)9-16-29-33
B-J3
53-45
24/25
B-O/9-13-26-37/39-46
B-I6-23-25-29-33
B)
B-I6-24-46
B-)9-16-29-33-61
B-O-16-24/25-26
B-J3-25-61
(l/2-)9-26-36/37/39-45
(l/2-)9-26-36/37/39-45
(l/2-M-8-43-45
(l/2-J6-36/37/39-45
Категория
токсичности
(к, м, р)
к 2, мЗ
Таблица П. 1.
Летальные дозы
ор782
ор4570
ор 12565
ор2140
ор 1215
ор400
ор880
ор8600
ор2460
ор 13400
ор280
ор505
ор5045
op 14OO0
ор273
Продолжение
МАК, мг/м3
44
700
1200
0,2 (TRK)
3
300
480
300
500
1,1
Вещество
Калия дихромат
Калия перманганат
Калия фторид
Калия цианид
Кальция гидрид
Кальция карбид
Кальция оксид
Кальция хлорид
J3 е-Капролактам
2,4-Ксилидин
о-, м-, л-Ксилол
о-Крезол
.м-Крезол
л-Крезол
Кротоновый альдегид
Кумол
Кумола гидропероксид
Ледяная уксусная кислота
см. Уксусная кислота
Литий
Литийалюминийгидриц
Метилацетат
Магний
Малеиновая кислота
Символ
опасности
T+,N
O,Xn,N
Т
Т+
F
F
Xi
Xi
Xn
T,N
Xn
T
T
T
F,T+,N
Xn,N
O,T,N
F,C
F
F,Xi
F
Xn
R
49-46-21-25-26-37/38-41-43-50/53
8-22-50/53
23/24/25
26/27/28-32-50/53
15
15
41
36
20/22-36/37/38
23/24/25-33-51/53
10-20/21-38
24/25-34
24/25-34
24/25-34
11-24/25-26-37/38-41-50-68
10-37-51/53-65
7-21/22-23-34-48/20/22-51/53
14/15-34
15
11-36-66-67
15-17
22-36/37/38
S
53-45-6O-6I
B-N0-61
(l/2-J6-45
(l/2-O-28-29-45-60-61
B-O/8-24/25-43
B-)8-43
22-24-26-39
B-J2-24
B)
(l/2-J8-36/37-45-61
B-J5
(l/2-K6/37/39-45
(l/2-K6/37/39^5
(l/2-K6/37/39^5
(l/2-J6-28-36/37/39-45-61
B-J4-37-61-62
(l/2-K/7-14-36/37/39-45-50-61
(l/2-)8-43-45
B-O/8-24/25-43
B-I6-26-29-33
B-O/8-43
B-J6-28-37
Категория
токсичности
(к, м, р)
к 2, м2
мЗ
Таблица П. 1.
Летальные дозы
ор95
ор1090
ор5
орЮОО
op 660
op 467
op 4300
op 121
op 242
op 207
op 240 (мышь)
op 1400
op 382
op 5000
op 708
Продолжение
МАК, мг/м3
0,05 E
0,5
2,5
5E
(какОЧ)
5E
5E
25
440
22
22
22
1
250
250
610
Вещество
Малеиновый ангидрид
Малононитрил (малонодинитрил)
Марганца диоксид
Масляная кислота
Масляный альдегид
Меди сульфат
Мезитила оксид
Метанол
Метантиол
Метилакрилат
Метиламин
N-Метиланилин
2-Метилбуганол-2
Метиленхлорид
см. Дихлорметан
Метилиодид
Метилметакрилат
Метилпентен-З-он-2
Метилпентилкетон
4-Метилпиридин (у-пиколин)
Метилпропилкетон
а-Метилстирол
Метилформиат
Метилциклогексан
Символ
опасности
Хп
T,N
Xn
С
F
Xn,N
Xn
F,T
F+, Xn, N
F,Xn
F+,Xn
T,N
F,Xn
T
F,Xi
Xn
Xn
T
F
Xi,N
F+,Xn
F,Xn,N
R
22-36/37/38-42
23/24/25-50/53
20/22
34
11
22-36/38-50/53
10-20/21/22
11-23/24/25-39/23/24/25
12-20-50/55
11-20/22-36/37/38-43
12-20-37/38-41
23/24/25-33-50/53
11-20
21-23/25-37/38-40
11-37/38-43
10-20/21/22
10-20/22
10-20/22-24-36/37/38
11
10-36/37-51/53
12-20/22-36/37
11-38-51/53-65-67
S
22-28-39
(l/2-J3-27-45-60-61
B-J5
(l/2-J6-36-45
B-)9-29-33
22
B-J5
(l/2-O-16-36/37-45
B-I6-25-60-61
B-)9-25-26-33-36/37-43
B-I6-26-39
(l/2-J8-37-45-60-61
B-)9-16-24/25
(l/2-K6/37-38-45
B-J4-37-48
B-J5
B-J4/25
(l/2-J6-36-45
(l/2-)9-16-33
B-N1
B-)9-16-24-26-33
B-)9-16-33-61-62
Категория
токсичности
(к,м,р)
кЗ
Таблица П. 1.
Летальные дозы
ор400
ор61
ор2940
ор2490
орЗОО
орП20
ор5628
инг. 1,35 мг/л, 4 ч
ор277
oplOO
op 360
op 1000
op 76
op 7872
op 1120
op 1670
op 1290
op 3730
op 4900
op 1500
Продолжение
МАК, мг/м3
0,4
22
0,5 E
врассч. наМп
64
IE
100
260
1
18
12
2
260
2
210
100
238
700
490
120
2000
Вещество
2- Метшшиклогексанол
2- Метилшклогексанон
4-Метокси-2-нитроанилин
Михлера кетон
Морфолин
Муравьиная кислота >90%
Муравьиная кислота 10—90%
Мышьяка триоксвд
Надуксусная кислота
Натрий
Натрия азид
Натрия гидрид
Натрия гидроксид (раствор) >5%
Натрия гидроксид (раствор) 2-5%
Натрия гидроксид
Натрия гипохлорит (раствор)
Натрия дитионит
Натрия дихромат
Натрия карбонат
Натрия нитрит
Натрия перхлорат
Натрия сульфид
Натрия цианид
Нафталин
Символ
опасности
Хп
Хп
Т+
Хп
С
С
с
T+,N
O,C,N
F,C
T+,N
F
С
С
С
С
Хп
O,T+,N
Xi
O,T,N
О,Хп
C,N
T+,N
Xn,N
R
20
10-20
26/27/28-33-52/53
36/37/38-40
10-20/21/22-34
35
34
45-28-34-50/53
7-10-20/21/22-35-50
14/15-34
28-32-50/53
15
35
34
35
31-34
7-22-31
49-46-8-21-25-26-37/38-41-43-50/53
36
8-25-50
9-22
31-34-50
26/27/28-32-50/53
22-50/53
S
B-J4/25
B-J5
(l/2-J8-36/37-45-61
36/37
(l/2-J3-36-45
(l/2-J3-26-45
23-26-36/37/39-45
53-45-60-61
(l/2-K/7-14-36/37/39-45-61
(l/2-M-8-43-45
(l/2-J5-45-60-61
B-O/8-24/25-43
(l/2-J6/36/37/39-45
(l/2-J6/37/39-45
(l/2-J6/37/39-45
(l/2-J8-45-50
B-O/8-26-28-43
53-45-60-61
B-J2-26
(l/2-L5-61
B-J-13-17-46
(l/2-J6-45-61
(l/2-O-28-29-45-60-61
B-K6/37-6O-61
Категория
токсичности
(к, м, р)
кЗ,мЗ
к1
к 2, м2
Таблица П. 1.
Летальные дозы
ор2140
ор 14100
ор6400
ор 1050
орПОО
ор27
ор 500 (LDLo, кролик)
ор4090
ор85
ор1200
ор208
ор6,44
ор>2000
Продолжение
МАК, мг/м3
235
230
70
9,5
0,1 (TRK)
0,2
2
2Е
1,5
0,05 Е
5Т(какСМ)
50
Вещество
а-Нафтиламин
Р-Нафтиламин
а-Нафтол
р-Нафтол
о-, д(-Нитроанилин
л-Нитроанилин
2-Нитроанизол
Нитробензол
Нитрометан
^ а-Нитронафталин
л 2-Нитропропан
2-Нитротолуол
4-Нитротолуол
Нитрующая смесь
см. Азотной и серной кислот смесь
2-Нитрофенол
4-Нитрофенол
Нитрозган
Нонановая кислота
Озон
Оксалилхлорид
Олеум
Олова(И)хлорид
Осмия тетраоксид
Паральдегид
Символ
опасности
Xn,N
T,N
Хп
Xn,N
Т
Т
т
T,N
Хп
Т
Т
T,N
T,N
Хп
Хп
Хп
С
т,с
с
Хп
т+
F
R
22-51/53
45-22-51/53
21/22-37/38-41
20/22-50
23/24/25-33-52/53
23/24/25-33-52/53
45-22
23/24/25-4О-48/23/24-51/53-62
5-10-22
24/25-40
45-10-20/22
23/24/25-33-51/53
23/24/25-33-51/53
22-36/38
20/21/22-23
10-20/22
34
14-23/24/25-34
14-35-37
22-36/37-38
27/27/28-34
И
S
B-J4-61
53-45-61
B-J2-26-37/39
B-J4/25-61
A/2-J8-36/37-45-61
A/2-J8-36/37-45-61
53-45
A/2-J8-36/37-45-61
B-L1
45
53-45
A/2-J8-37-45-61
A/2-J8-37-45-61
26-28
B-J8
B-)9-25-41
A/2-J6-28-36/37/39-45
26-45
A/2-J6-30-45
26
A/2-O/9-26-45
Категория
токсичности
(к, м, р)
к1
к2
кЗ
3
к2
инг. 4,4 ч
Таблица П. 1.
Летальные дозы
ор779
ор 275 (мышь)
ор240
аор1600,лсор535
ор750
ор 1980
ор640
ор940
ор120
ор725
ор891
ор1960
ор328
ор350
орПОО
ор3200
0,2
ор2140
ор700
ор15
Продолжение
МАК, мг/м3
IE
6
5
250
18 (TRK)
0,5 (TRK)
28
310
1
2Е
0,002
Таблица П.1. Продолжение
Вещество
Пентан
Пентанол-1
Пентанон-2
Пентанон-3
Пентилхлорид
Пергипрал см. Водорода перокеид
Пикриновая кислота
Пиперидин
Пиридин
Пирокатехин
Пропанол
Пропанол-2 см. Изопропиловый спирт
Пропен
Пропилацетат
иэо-Пропилацетат
Пропилбензол
Пропилбромид
Пропилхлорид
Пропионовая кислота
Пропионовый альдегид
Резорцин
Ртуть
Ртути ацетат
Ртути оксид
Ртути сульфат
Ргути(И) хлорид
Свинш тетраацетат
Селен
тжтшмштж
Символ
опасности
F,Xn,N
Хп
F
F,Xi
F,Xn
Е,Т
F,T
F,Xn
Xn
F,Xi
F+
F.Xi
F,Xi
Xn,N
Xn
F,Xn
С
F,Xi
Xn,N
T,N
T+,N
T+,N
T+,N
T+,N
T,N
T
шйттттшштттттшшттшш
R
12-51/53-65-66-67
10-20
11
11-37-66-67
11-20/21/22
2-4-23/24/25
11-23/24-34
11-20/21/22
21/22-36/38
11-41-67
12
11-36-66-67
11-36-66-67
10-37-51/53-65
10-20
10-20/21/22
34
11-36/37/38
22-36/38-50
23-33-50/53
26/27/28-33-50/53
26/23/28-33-50/53
26/27/28-33-50/53
28-34-48/24/25-50/53
61-20/22-33-50/53-62
23/25-33-53
мттштштжштштжтжтттт
s
B-)9-16-29-33-61-62
B-J4/25
9-16-33
B-)9-16-25-33
B-)9-29
(l/2-J8-35-37-45
(l/2-I6-26-27-45
B-J6-28
B-J2-26-37
B-O-16-24-26-39
B-)9-16-33
B-I6-26-29-33
B-I6-26-29-33
B-J4-37-61-62
B-)9-24
B-)9-29
(l/2-J3-36-45
B-)9-16-29
B-J6-61
(l/2-O-45-60-61
A/2-I3/28-45-60-61
(l/2-I3-28-45-60-61
(l/2-I3/28-45-61-61
(I/2-K6/37/39-45-60-61
53-45-60-61
(l/2-J0/21-28-45-61
Категория
токсичности
(к, м, р)
рз
Летальные дозы
ор2200
ор3730
ор2140
ор200
ор400
ор891
ор358
ор 1870
ор9370
орЗООО
ор6040
ор>2000
ор2600
орЗЗЮ
ор301
инг. 29 мг/м3
(LCLo,30h)
op 40,9
op 18
op 57
op 1
op 6700
МАК, мг/м3
2950
360
700
700
0,1 E
15
20 E
420
420
31
45 E
0,1
0,01
0,1 E
0,1 E
0,1 E
0,1 E
врасч. raft
0,1 E
Вещество
Стирол
Селена диоксид
Серебра нитрат
Серная кислота > 15%
Серная кислота 5—15%
Сероводород
Сероуглерод
Серы диоксид
Синильная кислота
Соляная кислота >25%
Соляная кислота 10—25%
Сульфаминовая кислота
Сульфаниловая кислота
Сульфурилхлорид
Тетрагидронафталин (тетралин)
Тетрагидрофуран
Тетрагидрофуриловый спирт
Тетралина гидропероксид
Тетрахлсрметан(тетрахлору1жрод)
см. Углерода тетрахлорид
Тиомочевина
Тионилхлорид
Титана тетрахлорид
о-Толуидин
Символ
опасности
Хп
T,N
C,N
С
Xi
F+,T+,N
F,T
T
F+,T+,N
С
Xi
Xi
Xi
С
Xi,N
F,Xi
Xi
O,C,N
Xn,N
С
С
T,N
R
10-20-36/38
23/25-33-50/53
34-50/53
35
36/38
12-26-50
11-36/38-48/23-62-63
23-24
12-26-50/53
34-37
36/37/38
36/38-52/53
36/38-43
14-34-37
19-36/38-51/53
11-19-36/37
36
7-22-34-50/53
22-40-51/53-63
14-20/22-29-35
14-34
45-23/25-36-50
S
B-J3
(l/2-J0/21-28-45-60-61
(l/2-J6-45-6O-61
(l/2-J6-30-45
(l/2-J6-30^*5
(l/2-)9-16-28-36/37-45-61
(l/2-I6-33-36/37-45
(l/2-)9-26-36/37/39-45
(l/2-O/9-16-36/37-3845-60-61
(l/2-J6-45
(l/2-J6-45
B-J6-28-61
B-J4-37
(l/2-J6-45
B-J6-28-61
B-I6-29-33
B-K9
(l/2-K/7-14-26-36/37/39-45-60-61
B-J2-24-36/37-61
(l/2-J6-36/37/39-45
(l/2-O/8-26-36/37/39-45
(l/2-M3-45-61
Категория
токсичности
(к, м, р)
рЗ
кЗ.рЗ
к2
Таблица П.1
Летальные дозы
ор2650
орП73
ор2140
инг. 713, 1ч
ор3188
инг. 2520,1 ч
инг. 120/1 ч
ор3160
ор 12300
ор2860
ор 1650
ор!600
ор 1750
инг. 2700,4 ч
ор670
Продолжение
МАК, мг/м3
86
ОДЕ
0,01 Е
IE
15
16
5
11
8
150
0,5 (TRK)
Вещество
л»-Толуидин
л-Толуидин
Толуол
л-Толуолсульфокислота
1,3,5-Триметилбензол (мезитилен)
1,1,1 -Трихлор-2,2-диD-хлор-
фенил)этан (ДДТ)
Трихлорметилбензол
Трихлоруксусная кислота
* Трихлорэтилен
Триэтиламин
Углерода монооксвд
Углерода тетрахлорид
Уксусный ангидрид
Уксусная кислота >90%
Уксусная кислота 25—90%
Уксусный эфир см. Этилацетат
л-Фенитидин
Фенилгидразин
л-Фенилендиамин
Фенилизоцианат
Фенол
а-Фенилэтиламин
Формальдегид (раствор) >25%
Символ
опасности
T,N
T,N
F,Xn
Xi
Xi,N
T,N
T
C,N
T
F,C
F+,T
T,N
С
С
с
т
T,N
T,N
T+
т
с
т
R
23/24/25-33-50
23/24/25-36-40-43-50
11-20
36/37/38
10-37-51/53
25-40-48/25-50/53
45-22-23-37/38-41
35-50/53
45-36/38-52/53-67
11-20/21/22-35
61-12-23-48/23
23/24/25-40-48/23-52/53-59
10-20/22-34
35
34
23/24/25-33
45-23/24/25-36/38-43-48/23/24/
25-50
23/24/25-43-50/53
10-22-26-34-42
24/25-34
21/22-34
23/24/25-34-40-43
S
A/2-J8-36/37-45-61
A/2-J8-36/37-45-60
B-I6-25-29-33
B-J6-37
B-N1
A/2-J2-36/37-45-60-61
53-45
A/2-J6-36/37/39-45-60-61
53-45-61
A/2-K-16-26-29-36/37/39-45
53-45
A/2-J3-36/37-45-59-61
A/2-J6-36/37/39-45
A/2-J3-26-45
A/2-J3-26-45
A/2-J8-36/37-45
53-45-61
A/2-J8-36/37-45-60-61
23-26-36/37/39-45
A/2-J8-45
A/2-J6-28-36/37/39-45
A/2-J6-36/37/39-45-51
Категория
токсичности
(к, м, р)
кЗ
кЗ
к2
к2
кЗ
к2,мЗ
кЗ
Таблица П. 1.
Летальные дозы
ор974
ор656
орбЗб
инг. 24,4 ч
орбООО
орЗЗОО
ор5650
ор460
LC50, инг.
2,1 мг/л, крысы
ор 2350
ор 1780
орЗЗЮ
ор850
ор188
ор80
ор800
ор317
ор940
oplOO
Продолжение
МАК, мг/м3
9
IE
190
100
IE
0,1 (TRK)
270
4,2
35
65
20
25
22
0,1 Е
0,05
19
0,62
Вещество
Формальдегид (раствор) 5—25%
Фосген
Фосфор, белый
Фосфор, красный
Фссфорилхлорид
Фосфорная кислота
Фосфора(У) оксид
*»¦ Фосфорпентахлорид
** Фосфортрихлорид
Фталевый ангидрид
Фумаровая кислота
Фурфуриловый спирт
Фурфурол
Хлор
Хлораль (-гидрат)
2-, З-Хлоранилин
4-Хлоранилин
Хлорацетонитрил
2-Хлорбензальцегид
Хлорбензол
о-Хлорбензонитрил
Хлордиметиловый эфир
(Хлорметил-метиловый эфир)
Символ
опасности
Хп
Т+
F,T+,C,N
F,N
т+,с
с
с
т+
т+,с
Хп
Xi
Хп
Т
T,N
Т
T,N
T,N
T,N
С
Xn,N
Хп
F,T
R
20/21/22-36/37/38-40-43
26-34
17-26/28-35-50
11-16-50
14-22-26-35-48/23
34
35
14-22-26-34-48/20
14-26/28-35-48/20
22-37/38-41-42/43
36
20/21/22
21-23/25-36/37-40
23-36/37/38-50
25-36/38
23/24/25-33-50/53
45-23/24/25-43-50/53
23/24/25-51/53
34
10-20-51/53
21/22-36
45-11-20/21/22
S
(l/2-J6-36/37-45-51
(l/2-)9-26-36/37/39-45
(l/2-M-26-28-45-61
B-O-43-61
(l/2-O/8-26-36/37/39-45
(l/2-J6-45
(l/2-J2-26-45
(l/2-O/8-26-36/37/39-45
(l/2-O/8-26-36/37/39-45
B-J3-24/25-26-37/39-46
B-J6
B)
A/2-J26-36/37/39-45
(l/2-O/9-45-61
(l/2-J5-45
(l/2-J8-36/37-45-60-61
53-43-60-61
(l/2-L5-61
(l/2-J6-45
B-J4/25-61
B-J3
53-45
Категория
токсичности
(к, м, р)
кЗ
кЗ
к2
Kl
Таблица П. 1.
Летальные дозы
инг. 25/30 мин
(LCLo, человек)
ор 1,4
(LDLo, человек)
ор380
ор 1530
орббО
ор18
ор4020
ор9300
ор177
ор65
инг. 430/30 мин
(LDLo, человек)
ор479
ор 256 (мышь)
орЗЮ
ор220
ор2480
орПОО
ор435
Продолжение
МАК, мг/м3
0,082
0,1 Е
ОДЕ
1,3
1
IE
IE
3
IE
41
20
1,5
0,2 Е (TRK)
46
Таблица П.1. Продолжение
Вещество
1 -Хлор-2,4-динитробензол
Хлороводород
Хлорная кислота
1 -Хлор-2-нитробензол
1 -Хлор-3-нитробензол
1 -Хлор-4-нитробензол
Хлороформ
Хлорсульфоновая кислота
о-Хлортолуол
.м-Хлортолуол
^ п-Хлортолуол
Хлоругольной кислоты этиловый
эфир
Хлоруксусная кислота
д(-Хлорфенол
п-Хлорфенол
Хрома триоксид
Циановодород см.
Синильная кислота
Циклогексан
Циклогексанол
Циклогексанон
Циклогексен
Циклогексиламин
Циклопентадиен см.
Дициклопентадиен
Символ
опасности
T,N
с,т
о, с
т
т
T,N
Хп
С
Xn,N
Xn,N
Xn,N
F,T+
T,N
Xn,N
Xn,N
O,T,C,N
F,Xn,N
Xn
Xn
F,Xn
С
R
23/24/25-33-50/53
23-35
5-8-35
23/24/25-33-52/53
23/24/25-33
23/24/25-33-51/53
22-38-40-48/20/22
14-35-37
20-51/53
20-51/53
20-51/53
11-22-26-34
25-34-50
20/21/22-51/53
20/21/22-51/53
49-8-25-35-43-50/53
11-38-50/53-65-67
20/22-37/38
10-20
11-21/22
10-21/22-34
S
(l/2-J8-36/37-45-60-61
(l/2-)9-26-36/37/39-45
(l/2-J3-26-36-45
28-36/37-45
28-37-45
(l/2-J8-36/37-45-61
B-K6/37
(l/2-J6-45
B-J4/25-61
B-J4/25-61
B-J4/25-61
(l/2-)9-16-26-28-33-36/37/39-45
(l/2-J3-37-45-61
B-J8-61
B-J-28-61
53-45-60-61
B-)9-16-33-60-61-62
B-J4/25
B-J5
16-23-33-36/37
(l/2-K6/37/39-45
Категория
токсичности
(к, м, р)
3
кЗ
Kl
Летальные дозы
ор780
инг. 4746/1 ч
орИОО
ор288
ор470
ор294
ор908
ор3900
ор2100
ор204
ор55
ор570
ор261
ор80
ор 12705
ор2060
ор 1620
ор1940
орЗОО
МАК, мг/м3
8
0,5 Е
50
4,4
4
0,05 Е(ГЖ)
700
200
80
1000
41
Таблица П.1. Продолжение
Вещество
Циклопентанон
Цинка хлорид
Цинка цианид
Щавелевая кислота
Этанол
Эген (этилен)
Этилакрилат
Этиламин
^-Этиланилин
Эгилацетат
Этилбензол
Эгилбромид
Этиленгликоль
Эгиленоксид
Этилметилкетон см. Бутанон
Эгилпропионат
Эгилформиат
Эгилхлорацетат
Эфир см. Диэтиловый эфир
Янтарной кислоты ангидрид
Символ
опасности
Xi
C,N
T+,N
Xn
F
F+
F,Xn
F+,Xi
T
F,Xi
F,Xn
F,Xn
Xn
F+,T
F
F,Xn
T,N
Xi
R
10-36/38
34-50/53
26/27/28-32-50/53
21/22
11
12
11-20/22-36/37/38-43
12-36/37
23/24/25-33
11-36-66-67
11-20
11-20/22-40
22
45-46-12-23-36/37/38
11
11-20/22-36/37
23/24/25-50
36-37
S
B-J3
(l/2-O/8-28-45-6O-61
(l/2-O-28-29-45-60-61
B-J4/25
B-O-16
B-)9-16-33
B-)9-16-33-36/37
B-I6-26-29
(l/2-J8-37-45
B-I6-26-33
B-I6-24/25-29
B-K6/37
B)
53-45
B-I6-23-24-29-33
B-)9-16-24-26-33
(l/2-O/9-45-61
B-J5
Категория
токсичности
(к, м, р)
кЗ
к 2, мЗ
Летальные дозы
ор350
ор7500
ор7060
ор800
ор400
ор334
ор 5620
ор3500
ор 1350
ор4700
ор72
ор 8732
ор 1850
ор235
ор 1510
МАК, мг/м3
690
5E(KaKCN)
IE
1900
20
9,4
1500
440
26
2(TRK)
310
5
412 Ж Приложение
ЛИТЕРАТУРА
Законодательные акты об обращении с химикатами и
пожарной безопасности
См. литературу к части А т. 1.
Свойства опасных веществ;
несчастные случаи в химической лаборатории; первая помощь
Balin R., Fuhrmann G., Legrum, W., Steffen C: Spezielle Toxikologie fur Chemiker. — Teubner,
Munchen 1999.
Bender H. F.: Das Gefahrstoffbuch. Sicherer Umgang mit Gefahrstoffen in der Praxis. — Wiley-VCH,
Weinheim 2002.
Bretherick's Handbook of Reactive Chemical Hazards. Hrsg.: P. G. Urben — Butterworth-Heinemann,
Oxford 1999.
Handbook of Laboratory Safety. Hrsg.: A. K. Furr. — The Chemical Rubber Сотр., Cleveland/Ohio
2000.
Hinweise zur Abfallbeseitigung und Recycling. — Handbuch fur Verwerterbetriebe fur industrielle
Abfalle. Hrsg.: Bundesumweltamt. — Erich Schmidt-Verlag, Berlin.
Hommel G.: Handbuch der gefthrlichen Guter. — Springer-Verlag, Berlin, Heidelberg, New York
1978-2000.
IRPTC — International Register of Potentially Toxic Chemicals. — United Nations, Geneva
1987.
Kuhn R., Birett K.: Merkblatter gefahrlicher Arbeitsstoffe. — Ecomed Verlagsgemeinschaft,
Landsberg/Lech.
List of МАК and BAT Values 2003. Hrsg.: Deutsche Forschungsgemeinschaft. — Wiley-VCH, Weinheim
2003.
Lunn G., Sansone E. В.: Destruction of Hazardous Chemicals in the Laboratory. — John Wiley & Sons,
New York 1994.
Ludewig R., Kappel C, Poelchen W., Regenthal R.: Akute Vergiftungen. — Wissenschaftliche
Verlagsgesellschaft, Stuttgart 1999.
Martinez D.: Immobilisation, Entgiftung und ZerstOrung von Chemikalien. — Verlag Hani Deutsch,
Thun, Frankfurt/Main 1986.
Moeschlin S.: Klinik und Therapie der Vergiftungen. — Georg Thieme Verlag, Stuttgart, New York
1986.
Muller R. K.: Die toxikologisch-chemische Analyse. — Verlag Theodor Steinkopff, Dresden
1976.
Quellmalz E., Wettbacher U., Stormarm R.: Hauptstoffliste, zusammengefuhrte Informationen zu
gefahrlichen Stoffen und Zubereitungen. — WEKA-Fachverlag, Augsburg 1998.
Recycling-Handbuch. Hrsg.: Bundesumweltamt. — Erich Schmidt-Verlag, Berlin.
Reicharg D., Ochterbeck W.: Abfalle aus chemischen Laboratorien und medizinischen Einrichtungen. —
Ecomed Verlagsgesellschaft, Landsberg/Lech 1994.
Richardson M. L., Gangolli S.: DOSE — The Dictionary of Substances and their Effects. Bd. 1-8. — The
Royal Society of Chemistry — Information Services, Cambridge 1992 ff.
Roth L.: Gefahrstoff-Entsorgung. — Ecomed Verlagsgesellschaft, Landsberg/Lech 1995.
Roth L.: Chemie-Ratgeber, Sicherheitsdaten, MAK-Werte. — Ecomed Verlagsgesellschaft,
Landsberg/Lech.
Roth L., Daunderer M.: Giftliste — Ecomed-Verlagsgesellschaft, Landsberg/Lech.
Литература 413
Sorbe G.: Sicherheitstechnische Kenndaten — Ecomed Verlagsgesellschaft, Landsberg/Lech
1983.
Welzbacher U.: Neue Datenblatter fur gefthrliche Arbeitsstoffe nach der GefahrstoffVerordnung. —
WEKA-Fachverlag, Augsburg.
Банкданных
GESTIS-Stoffdatenbank. — Berufsgenossenschaftlicher Institut fur Arbeitssicherheit (BIA), Sankt Augustin.
http://www.hvbg.de/bia/stoffdatenbank.
Hazardous Chemical Database. — University of Akron. USA. http://ull.chemistry. uakron. edu/ erd/
Hazardous Substance Release and Health Effects Database. — Agency for Toxic Substances and Disease
Registry (ATSDR), USA. http://www.atsdr.cdc.gov/hazdat.html.
Merck Sicherheitsdatenblatter auf CD-ROM. — Merck KGaA, Darmstadt.
MSDS-OHS (OHS Material Safety Data Sheets). — MDL Information Systems, USA (Online-Datenbank
liber STN International zuganglich).
ПЕРЕЧЕНЬ МЕТОДИК
Том 1
Общая методика фотохлорирования
ароматических соединений в боко-
боковую цепь 250
Общая методика хлорирования углево-
углеводородов сульфурилхлоридом 253
Общая методика хлорирования заме-
замещенных бензальдоксимов N-хлор-
сукцинимидом 253
Общая методика фотобромирования
алкиларенов в боковую цепь 256
Общая методика бромирования N-бром-
сукцинимидом в аллильное положе-
положение 258
Общая методика получения гидропе-
роксидов из углеводородов 260
Общая методика этерификации спир-
спиртов бромоводородной кислотой
284
Общая методика получения алкилио-
дидов из спирта, иода и красного
фосфора 286
Получение трифенилкарбинола (три-
тилового спирта) 291
Общая методика гидролиза бензили-
дендигалогенидов в концентриро-
концентрированной серной кислоте 292
Общая методика получения простых
эфиров фенола при метилировании
диметилсульфатом 294
Общая методика получения простых
эфиров, спиртов и фенолов с по-
помощью алкилгалогенидов, толуол-
сульфонатов или диметилсульфата
(синтез Вильямсона) 297
Общая методика получения эфиров бен-
бензойной кислоты в условиях межфаз-
межфазного катализа 299
Получение бензилового эфира (>S)-N,N-,hh-
бензилаланина 301
Получение дициклогексилэтиламина
301
Общая методика получения а-амино-
кислот из а-галогенкарбоновых
кислот 302
Кватернизация третичных аминов (об-
(общая методика для качественного
анализа) 302
Общая методика получения алкилтри-
фенилфосфониевых солей 303
Общая методика получения диэтило-
вых эфиров алкилфосфоновых
кислот 304
Общая методика получения симмет-
симметричных диалкил сульфидов 306
Общая методика получения алкил-
тиоцианатов на силикагеле, акти-
активированном тиоцианатом калия
307
Общая методика получения тиолов че-
через S-алкилтиоурониевые соли
309
Общая методика получения алкилфто-
ридов из алкилтозилатов 311
Общая методика получения нитроалка-
нов 312
Получение нитрометана 314
Общая методика получения нитрилов
315
Перечень методик
415
Общая методика триметилсилилирова-
ния амино- и гидроксильных со-
соединений 319
Общая методика дегидратации вторич-
вторичных и третичных спиртов и про-
продуктов альдольной конденсации в
присутствии кислот 337
Общая методика получения простых
эфиров енолов из ацеталей элими-
элиминированием спиртов 337
Общая методика дегидрогалогенирова-
ния алкилгалогенидов с этилди-
циклогексиламином 341
Общая методика дегидрогалогенирова-
ния (детозилирования) с едким
кали в тригликоле 341
Получение дикетена 342
Получение метилвинилкетона 345
Общая методика гидратации ацетиле-
ацетиленовых соединений 361
Общая методика присоединения брома
к олефинам и ацетиленам 364
Качественная реакция на С=С-двой-
ные связи: «бромная проба» 366
Общая методика получения спиртов
оксимеркурированием 367
Общая методика эпоксидирования
олефинов 369
Получение /я/>анс-циклогександиола-
1,2 (/яранс-дигироксилирование
смесью муравьиной кислоты и
пероксида водорода) 369
Получение глицерина из аллилового
спирта 370
Получение цис-циклогександиола-1,2
373
Общая методика получения спиртов
гидроборированием 375
Получение изооктена 378
Катионная полимеризация стирола 378
Общая методика винилирования спир-
спиртов 380
Общая методика присоединения ами-
аминов к эфирам ацетилендикарбоно-
вых кислот 383
Общая методика радикального присое-
присоединения к олефинам 386
Полимеризация полистирола (полиме-
(полимеризация в растворе) 390
Общая методика присоединения дих-
лоркарбена к олефинам 394
Общая методика синтеза 3-D-хлорфе-
нил)-Д2-1,2-оксазолинов и 3-D-хлор-
фенил)-1,2-оксазолов с помощью
1,3-биполярного циклоприсоедине-
ния 398
Общая методика диенового синтеза
401
Общая методика вакер-окисления тер-
терминальных олефинов 404
Получение полиэтилена 406
Получение транс,транс,цис-шклод,о-
декатриена 409
Получение циклооктатетраена из аце-
ацетилена 409
Общая методика каталитического гид-
гидрирования 413
Количественное гидрирование 418
Общая методика нитрования аромати-
ароматических соединений 433
Общая методика сульфохлорирования
аренов 438
Сульфохлорирование (общая методика
для качественного анализа) 441
Получение пиридинсульфоновой-3 кис-
кислоты 441
Получение л-толуосульфоновой кис-
кислоты 442
Получение пикриновой кислоты 442
Общая методика бромирования арома-
ароматических соединений элементным
бромом 445
Бромирование фенолов (общая мето-
методика для качественного анализа)
447
Общая методика бромирования мало-
малоактивных аренов дибромизоциану-
ровой кислотой 447
Общая методика введения тиоцианат-
ной группы 449
Общая методика алкилирования бен-
бензола по Фриделю - Крафтсу 451
Получение трифенилметилхлорида (три-
тилхлорида) 452
Получение бензофенона 453
Общая методика ацилирования ацилх-
лоридами по Фриделю — Крафтсу
457
416
Перечень методик
Общая методика форматирования по
Вильсмейеру 461
Общая методика хлорметилирования
ароматических соединений 465
Синтез 2,2-ди(«-хлорфенил)-1,1,1-
трихлорэтана (ДДТ) 468
Получение кристаллического фиоле-
фиолетового 468
Общая методика карбоксилирования
фенолов 471
Получение NjN-диметил-и-нитрозо-
анилина 472
Получение арил- и алкил-2,4-динитро-
фенилсульфидов (общая методика
для качественного анализа) 476
Получение 2,4-динитрофенилгидра-
зина 476
Получение 2-аминопиридина 476
Получение р-нафтола 477
Получение 3-децилвератрола 482
Общая препаративная методика проведе-
проведения реакции Соногашира 487
Арилирование акриламида (реакция
Хека). Общая методика 492
Том 2
Общая методика препаративного и
аналитического получения арома-
ароматических карбоновых кислот из ал-
киларенов (межфазный катализ)
12
Общая методика аутоокисления заме-
замещенных в ядре толуолов в соответ-
соответствующие бензойные кислоты 13
Получение я-толуилальдегида из и-
ксилола 15
Общая методика получения арилоксоа-
цеталвдегидов и 1,2-дикетонов 17
Общая методика получения морфоли-
дов тиокарбоновых кислот (реак-
(реакция Вильгеродта—Киндлера) 18
Методика омыления морфолидов тио-
тиокарбоновых кислот 18
Окисление вторичных спиртов до ке-
тонов смесью дихромата с серной
кислотой (общая методика) 22
Окисление спиртов и альдегидов гипохло-
ритом натрия (общая методика) 24
Общая методика окисления спиртов
смесью диметилсульфоксида с
оксалилхлоридом (окисление по
Сверну) 26
Общая методика получения карбоно-
карбоновых кислот из первичных спиртов
и олефинов в условиях межфазного
катализа 28
Получение слизевой кислоты из мо-
молочного сахара (окисление азотной
кислотой) 29
Получение трихлоруксусной кислоты
из хлораля (окисление азотной
кислотой) 30
Общая методика получения хинонов
из углеводородов действием триок-
сида хрома 31
Получение нафтохинона-1,2 34
Общая методика получения азокраси-
телей окислительным азосочета-
нием 36
Получение азелаиновой кислоты из
касторового масла (окисление пер-
манганатом) 39
Получение полуацеталя этилового
эфира глиоксиловой кислоты из
диэтилового эфира винной кисло-
кислоты действием тетраацетата свинца
40
Общая методика окисления метилке-
тонов гипобромитом (галоформ-
ная реакция) 42
Иодоформная проба (общая методика
для качественного анализа) 43
Получение адипиновой кислоты из
циклогексанола 43
Общая методика дегидрирования се-
серой 45
Общая методика получения енаминов 58
Получение семикарбазонов (общая мето-
методика для качественного анализа) 60
Получение 2,4-динитрофенилгидразо-
нов (общая методика для качест-
качественного анализа) 60
Перечень методик
417
Получение озазонов (общая методика
для качественного анализа) 60
Получение (?)-оксима бензальдегида
61
Общая методика получения диэтила-
цеталей 63
Общая методика получения этилена-
цеталей (диоксоланов) 65
Общая методика этерификации карбо-
новых кислот 70
Общая методика получения эфиров
уксусной кислоты из уксусного
ангидрида 72
Определение эквивалента спиртов в
виде кислых эфиров 3-нитрофта-
левой кислоты (общая методика
для качественного анализа) 74
Получение эфиров бензойной кислоты
алкоголизом бензоилхлорида (ва-
(вариант Шоттера - Баумана, общая
методика для качественного ана-
анализа) 75
Получение N-метилформанилида 77
Получение эфиров карбоновых кислот
алкоголизом ацилхлоридов (вари-
(вариант Эйнхорна, общая методика для
качественного анализа) 75
Получение анилидов из карбоновых
кислот и анилина (общая методика
для качественного анализа) 77
Общая методика получения 3-амино-
1-арилпиразолонов-5 78
Получение цианацетамида и хлораце-
тамида (аммонолиз реакционнос-
пособного эфира) 79
Получение N-бензиламидов аминоли-
зом эфиров карбоновых кислот
(общая методика для качественно-
качественного анализа) 79
Получение 2,3,4,6-тетраацетил-О-
глюкозы частичным аминолизом
пентаацетил-О-глюкозы 79
Получение амидов карбоновых кислот
аминолизом ацилхлоридов (общая
методика для качественного ана-
анализа) 80
Общая методика омыления замещен-
замещенных диэтиловых эфиров малоно-
малоновой кислоты 85
Общая методика кетонного расщепле-
расщепления эфиров C-оксокислот 87
Общая методика декарбоксилирова-
ния замещенных малоновых кис-
кислот 87
Определение эквивалентной массы
сложного эфира 89
Получение пербензойной кислоты 90
Получение 3,5-динитробензоатов аци-
долизом эфиров карбоновых кис-
кислот (общая методика для качест-
качественного анализа) 91
Получение 3-нитрофталевого ангид-
ангидрида 92
Общая методика получения ацилхло-
ацилхлоридов 95
Общая методика гидролиза нитрилов
97
Получение диэтилового эфира монои-
мида малоновой кислоты 98
Получение 1,8-диазабицикло[5.4.0]ун-
децена-7 98
Общая методика получения изоци-
анатов 100
Получение N-фенилуретанов присо-
присоединением спиртов к фенилизоци-
анату (общая методика для качест-
качественного анализа) 102
Получение замещенных тиомочевин
присоединением первичных или
вторичных аминов к фенилизотио-
цианату (общая методика для каче-
качественного анализа) 102
Общая методика получения амидов тио-
карбоновых кислот 103
Общая методика получения а-гидрокси-
карбонитрилов (циангидринов) 109
Общая методика получения а-амино-
кислот по Штреккеру 111
Общая методика этинилирования ке-
тонов 113
Общие методики проведения альдоль-
но-кротоновых конденсаций 117
Получение диацетонового спирта D-гид-
рокси-4-метилпентанона-2) 120
Общая методика получения 2,3-эпок-
сипропионитрилов 124
Общая методика проведения реакции
Кнёве нагеля 125
418
Перечень методик
Общие методики получения основа-
оснований Манниха 130
Общая методика ацилоиновой конденса-
конденсации ароматических альдегидов 132
Общая методика реакции Хронера —
Вадсворта — Эммонса с диэтило-
вым эфиром бензилфосфоновой
кислоты 136
Общая методика получения олефинов
с помощью реакции Виттига 138
Общие методики сложноэфирной кон-
конденсации и синтеза глицидных
эфиров по Дарзану 146
Общая методика декарбонилирования
эфиров оксоянтарных и 2,4-ди-
оксокарбоновых кислот 147
Общая методика конденсации ортому-
равьиного эфира с метиленактив-
ными соединениями 148
Общая методика сложноэфирного рас-
расщепления ацилацетоуксусных
эфиров 151
Общая методика кислотного расщеп-
расщепления а-ацилкетонов 152
Общая методика ацилирования р-ди-
карбонильных соединений 153
Общая методика присоединения гете-
рокумуленов к метиленактивным
соединениям 156
Общая методика получения три- и
пентаметинцианинов 157
Общая методика получения спиртов и
карбоновых кислот с помощью со-
соединений Гриньяра 163
Общая методика восстановления алю-
могидридом лития 171
Восстановление 4-/ире/и-бутилциклогек-
санона борогидридом натрия 172
Общая методика восстановления кето-
нов и альдегидов по Меервей-
ну-Понндорфу-Верлею 175
Получение Д4-холестенона-3 из холес-
холестерина 176
Общая методика проведения перекре-
перекрестной реакции Канниццаро 178
Получение пентаэритрита 178
Общая методика проведения реакции
Лейкарта-Валлаха с альдегидами
180
Общая методика восстановления кето-
нов по Кижнеру—Вольфу 182
Общая методика каталитического гидри-
гидрирования кетонов, альдегидов, нитри-
нитрилов, оксимов и азометинов 185
Общая методика каталитического вос-
восстановительного аминирования аль-
альдегидов и кетонов 188
Расщепление рацемического-ос-фенилэ-
тиламина на оптические антиподы
188
Получение 2,3-диметилбутандиола-2,3
(пинакона) 190
Общая методика восстановления
сложных эфиров и нитрилов по
Буво-Блану 191
Общая методика присоединения ами-
аминов к винилогам карбонильных
соединений 197
Общая методика получения хинолинов
по Скраупу 199
Получение р-метилтиопропионового
альдегида присоединением метан-
тиола к акролеину 200
Общая методика присоединения гало-
геноводородов к винилогам карбо-
карбонильных соединений 200
Общая методика присоединения по
Михаэлю 203
Получение метиленбис(дигидрорезор-
цина) 206
Общая методика получения диметило-
вых эфиров N-замещенных 5-гид-
роксииндолдикарбоновых-2,3 кис-
кислот 207
Получение р-аланина 208
Общая методика получения производ-
производных 3- или 5-аминопиразолкарбо-
новой-4 кислоты 210
Общая методика алкилирования р-ди-
карбонильных соединений 213
Общая методика алкилирования бензил-
цианидов в двухфазной системе 216
Общая методика получения ос-бромо-
карбоновых кислот 218
Общая методика получения фенацилб-
ромидов 219
Общая методика получения р-дикетонов
ацилированием енаминов 221
Перечень методик
419
Общая методика каталитического вос-
восстановления ароматических нит-
росоединений 236
Восстановление ароматических нитро-
соединений до первичных аминов
оловом в солянокислой среде (об-
(общая методика для качественного
анализа) 236
Определение эквивалентной массы
аминов титрованием хлорной кис-
кислотой 236
Получение диэтилового эфира ацета-
миномалоновой кислоты 237
Общая методика получения растворов
диазотированных ароматических
аминов 240
Получение Ы-метил-Ы-нитрозомоче-
вины 241
Получение изоамилнитрита 243
Получение диэтилового эфира изонит-
розомалоновой кислоты 243
Общая методика превращения солей
диазония в фенолы 245
Общая методика получения арилгид-
разинов 246
Общая методика получения аромати-
ароматических хлоридов, бромидов и нит-
нитрилов по Зандмейеру 249
Общая методика азосочетания 252
Получение диазометана из N-метил-
N-нитрозомочевины 256
Общая методика метилирования кар-
боновых кислот и фенолов диазо-
метаном 259
Общая методика получения диазокето-
нов и их превращения в галогенке-
тоны 261
Общая методика получения тиофено-
лов 263
Общая методика получения алкиловых
эфиров и-толуолсульфоновой кис-
кислоты 264
Общая методика получения амидов
сульфоновых кислот 266
Разделение смесей аминов через суль-
фонамиды (разделение по Хинс-
бергу) (общая методика для качест-
качественного анализа) 266
Общая методика проведения пинако-
линовой перегруппировки 274
Эпоксидирование олефинов и перег-
перегруппировка эпоксидов в карбо-
карбонильные соединения (общая мето-
методика для качественного анализа)
275
Общая методика получения эфиров
карбоновых кислот из диазокето-
нов перегруппировкой Вольфа
277
Получение циклогептанона (суберона)
278
Общая методика деструкции амидов
кислот до аминов по Гофману 280
Общая методика получения изоциана-
тов из карбоновых кислот деструк-
деструкцией по Курциусу 281
Общая методика проведения реакции
Шмидта 283
Получение s-капролактама из оксима
циклогексанона 285
Полимеризация в-капролактама 285
Получение е-капролактона 286
Получение 2-аллилфенола 289
Общая методика синтеза индолов по
Фишеру (получение фенилгидра-
зонов по Яппу-Клингеманну)
290
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адамантан 1:418
Адамса катализаторы 1: 310
Аципиновая кислота 1:418; 2: 38,43, 96, 331
диэтиловый эфир 2: 71, 94
Адреналин 1:407
Адсорбенты 1: 84, 92
Адсорбционная хроматография 1: 86, 97; см.
также Хроматография
Адсорбция 1: 84
Азалактон2:122,123
1-Азафенантрен 2:199, 200
Азид 1: 392, 398
Азии 1: 392; 2: 22, 23, 57
хлорирование 1: 252
Азоксибензол 2: 234
Азометины 1: 300; 2: 51, 56, 57, 62, 185, 222
реакции гидрирования 1:410, 411; 2:184
— с реактивом Гриньяра 2:162
— с аминами 1: 300
Азот
обнаружение в соединениях 2: 296
жидкий 1: 26
маркировка баллонов 1: 29
оксиды, техника безопасности 2:396
очистка 2: 361
Азотистая кислота
реакции 2: 232, 237
— с алифатическими нитросоединениями 2:
306
— с аминами 2: 237, 307
— с СН-кислотами 2: 242
— с первичными ароматическими аминами
2:239
— со спиртами 2: 242
— с тиолами 2: 307
Азотистоводородная кислота 2: 282, 396
Азотистой кислоты эфиры 1: 266, 277, 312;
2: 232, 242
Азотистый иприт 1: 303
Азотная кислота
окислитель 2: 12
— ароматических соединений 2: 301
присоединение к олефинам 1: 355
техника безопасности 2: 362, 396
эфиры 1: 282, 286
Азулен 1:425
Акридин
метилирование 1: 421
производные 2: 316
Акриламид, арилирование 1: 492
Акриловая кислота 1: 338, 356; 2: 10, 90
Акролеин 1: 255, 370
димеризация 1:400
реакции обмена 1:402
D+2)-циклоприсоединение 1: 399,400
|3С-ЯМР 1: 140
Акролеиндиэтилацеталь 1: 343; 2: 63
Активированный комплекс 1: 195
Р-Аланин 2: 82, 208, 281
Алании 1: 302
ИК-спектры2:317
реакции обмена 1: 301
Ализарин 1: 279, 328
Аликват 1: 280
Алкенилацетилен 1: 484,486
Алкенил бензол 1:489
Алкенилгалогениды 1: 483, 486, 489
реакции с металлоорганическими соедине-
соединениями 1: 485, 486
Алкенилтрифлаты 1:483, 486, 489
Алкены см. Олефины
Алкидные смолы1: 371
N-Алкиламинофумаровой кислоты эфир 1:
383
Алкиларены 1: 451,481,484, 489
фотобромирование 1: 256
Предметный указатель
421
фотохлорирование 1: 251
Алкилатбензины 1: 378
Алкилбензолсульфонаты 1: 255, 379,443, 453
Алкилбензолы 1:453
Алкилбораны 1: 374
Алкилбромиды 1: 278, 284, 312
вторичные 1: 311
реакции обмена 1: 298, 303, 311
Б^-реакционная способность 1: 270
Алкилгалогениды 1: 265, 278, 283, 311, 321, 351
гидролиз 1: 265, 283, 290
дегидрогалогенирование 1: 327, 339, 341
масс-спектры 1: 145
нуклеофильное замещение 1: 290
производные 1: 309
реактив Гриньяра 2: 160
реакции обмена 1: 295, 303
хлорирование 1: 254
Алкил-2,4-динитрофенилсульфиды 1:476
Алкиленгалогениды 1: 480, 484,486, 489
Алкилиденфосфораны 2:104, 136
Алкилиодиды 1: 278, 282, 286, 310, 312
восстановление иодоводородом 1: 249
— реакции обмена 1: 296
реакционная способнось 1: 272
карбониевые катионы 1: 146
Алкилирование см. также Фриделя—Крафтса
ал копирование
аммиаки амины 1: 299
ароматические соединения 1:449
карбонильные соединения 1: 278
серосодержащие соединения 1: 305
соединения фосфора 1: 303
Алкилкарбены 1: 349
Алкилнафталины 1: 255
М-Алкил-М-нитрозомочевина 2: 256
1Ч-Алкил-М-нитрозоуретан 2: 256
Алкилпиридин 1: 362
Алкилродамин см. Алкилтиоцианат
Алкилсерная кислота 1: 265, 289, 293, 361, 363
Алкилсульфоновые кислоты 1: 262
Алкилсульфохлориды 1: 261
3-Алкил-1,3-тиазолиясоли2: 134
S-Алкилтиоурониевые соли 1: 289, 309
S-Алкилтиоурония пикраты 1: 309
Алкилтиоцианаты 1: 306, 307
Алкилтриметиламмония гидроксид 1: 343
Алкилтрифенилфосфониевые соли 1: 303
Алкилфенилполигликолевые эфиры 1:453
Алкилфенолы 1: 371, 453
N-Алкилфталимиды 1: 300
Алкилфториды 1: 283, 322
получение из алкилтозилатов 1:311
8н2-реакционная способность 1: 272
Алкилхлориды 1: 278, 285, 293
Алкилхлорсиланы 2: 160
Ал кил цианиды 1:313
Алкильные заместители
стерические эффекты 1: 270
третичные 1: 231
/-эффект 1:206
Алкоголиз
ангидридов карбоновых кислот 2: 52
бензохлорида 2: 75
имидоэфира 2: 98
карбонилгалогенидов 2: 52, 74
карбоновых кислот 2: 69, 70
сложных эфиров 2: 52
фосгена 2: 52
Алкоголяты 1: 294
2-Алкокси-3,4-дигидро-2Н-пиран 2: 64
Алкокситриметилсиланы 1: 319
Аллены 1: 220
Аллилбромид 1: 284
производные 2: 340
реакционная способность 1: 266, 287, 300
Аллилбромирование N-бромсукцинимидом
1:258
Аллилгалогениды 2: 212; см. также Аллилбро-
Аллилбромид, Аллилиодид
реакция с реактивом Гриньяра 2: 160
Аллиламин 1: 255
производные 2:311
Аллилиодид, производные 2: 340
Аллилмалоновая кислота 2: 86, 88
Аллилмалоновой кислоты диэтиловый эфир 2:
86,213
Аллиловый спирт 1: 255, 293, 366; 2: 18
Аллиловый эфир 1: 255
Аллилфениловый эфир 1: 278
4-Аллилфенол 1: 277
2-Аллилфенол 2: 288
Аллилхлорид 1: 255, 364, 366, 387
константа скорости Б^-реакции 1: 270
2-Аллилциклогексанон 2: 289
2-Аллилциклопентанон 2: 289
Аллильный радикал 1: 242
Альдегиды 2: 8
алифатические 2: 134, 167
ароматические
— Вильсмейера синтез 1: 460
— Гаттермана синтез 1:459
— получение из бензилидендигалогенидов 1:
292
— реакция Соммле 1:466
Стиме 1:488
гидратирование 2: 62
получение эпоксидированием 1: 372
— гидратацией ацетиленов 1: 361
— из дигалогенидов 1: 293
422
Предметный указатель
— из озонидов 1: 353,374
— окислением по Оптнауэру 2:175
реакции аутоокисления 1: 259
Дильса—Альдера см. Дильса—Альдера реакция
— Кнёвенагеля 2: 125
— Хорнера—Вадсворта—ЭммонсаЪ: 135
тонкослойная хроматография 1: 68, 76
УФ-спектры 1: 88
хлорирование 1: 252
Альдозы 2: 28, 109
Альдоль 1: 363
Альдольные реакции
кислотный катализ 2:116
конденсация 2: 114 и ел.
реакция Мукайамы 2: 212
стереоселективность 2:120.
Алюминия оксид
адсорбент 1: 84
для обесцвечивания растворов 1: 85
носитель 1: 88
осушитель 1: 36
Алюминия алкоголяты 2: 174, 175, 396
Алюминия хлорид, катализатор реакции Фриде-
ля- Крафтса 1:450
Алюмосиликаты как осушители 1:41
Амбидентные анионы 1: 277
Амбидентные нуклеофилы 1: 215
региоселективность 1: 277
Амидосульфоновая кислота 2: 396
Амил... см. Пентил...
Аминали 2: 57, 63
2-Аминоантрахинон 1: 476, 479
3-Амино-1-арилпиразолинон-5 2:78
4-Аминоацетанилид 1:449
4-Аминоацетофенон 1: 209
З-Аминобензальдегид 2: 246
З-Аминобензойная кислота 1:317
4-Аминобензойная кислота 2:13, 311, 317
З-Аминобензолсульфоновая кислота 1:478
2-Аминобензотиазол 1:448,449
Ы-B-Аминобензотиазолил-6)-ацетамид
1:449
З-Аминобензофенон 2: 235
3-Амино-1-D-бромфенил)-Д2-пиразолинон-5
2:79
р-Аминовинил... 2: 209, 210
4-Амино-2-гидроксибензойная кислота 1: 471
а-Аминокарбонильные соединения 2: 243
р-Аминокарбонильные соединения 2: 208
Аминокарбоновой кислоты гидрохлорид
1:119
Аминокарбоновые кислоты 1: 392
алкилирование N-ациламиномалоновых
эфиров 2: 88
амиды, расщепление по Гофману 2: 280
в пептидном синтезе 2: 80
а-галогенкарбоновые кислоты 1: 302
определение методом тонкослойной хрома-
хроматографии 1: 89
. получение из альдегида и а-аминонитрила
(по реакции Штреккера) 2:97, 109, 111
Аминокетоны 2: 64, 131, 203, 238
Аминокислоты 1: 302
гидрохлорид, ИК-спектры 1: 118, 119
Р-Аминокротоновой кислоты этиловый эфир
1:208; 2: 57, 58, 203
Аминомалеиновой кислоты диметиловый
эфир 1: 383
а-Аминомасляная кислота 1: 302
Аминометилирование ароматических соеди-
соединений 1: 426, 464
2-Амино-6-метилбензотиазол 1: 449
3-Амино-1-D-метилфенил)-Д2-пиразоли-
нон-5 2:79
2-Амино-6-метоксибензотиазол 1:449
1-Амино-2-метоксиметилпирролидин 2: 216
1-Аминонафталин 2: 100, 101, 396
2-Аминонафталинсульфоновая-1 кислота 1:441
1-Аминонафтола-2 гидрохлорид 2: 34
окисление в нафтохинон 2: 34
Аминонитрил 2: 109, ПО
6-Аминопенициллановая кислота 2: 82
4-Аминопентен-3-он-2 2: 58
3E)-Аминопиразолкарбоновая-4 кислота,
этиловый эфир и карбонитрил 2: 210, 211
2-Аминопиридин 1:474, 475, 476; 2: 266
идентификация 2: 314
техника безопасности 2: 396
4-Аминопиридин 1:474
Аминопропановая кислота 1: 302
Ы-C-Аминопропил)-Е-капролактам 2: 98, 99,
185,186
4-Аминосалициловая кислота 1:470, 471
4-Аминосульфонилацетанилид 2: 266
2-Амино-4,5-тетраметилентиофенкарбо-
нитрил-3 2: 20
2-Амино-4,5-тетраметилентиофенкарбо-
новая-3 кислота, этиловый эфир 2: 20
2-Аминотиофенкарбоновой-З кислоты эфир
2:20
2-Аминотиофен 2: 129
а-Аминоуксусная кислота 1: 302
4-Аминофенетол 2: 235
2-Аминофенетол 2: 311, 314
3-Амино-1-фенил-Д2-пиразолинон-5 2: 79
5-Амино-1-фенилпиразолкарбонитрил-4 2:
210,211
5-Амино-1 -фенилпиразолкарбоновой-4
кислоты этиловый эфир 2: 210, 211
2-Амино-1-фенилпропан2: 187
Предметный указатель
423
2-Амино-4-фенилтиофенкарбоновой-3
кислоты этиловый эфир 2: 20
4-Аминофенилтиоцианат 1:449
Аминофенол 1:435,470,471,478, 479; 2: 35, 82,
83,234,255,256,298,314,396
Аминофумаровой кислоты диметиловый эфир
1:383
Аминофумаровой кислоты диэтиловый эфир
1:382
Аминохинолин 1:475
2-Амино-6-хлорбензотиазол 1:449
3-Амино-1-B-хлорфенил)-Д2-пиразолинон-5
2:79
3-Амино-1-C-хлорфенил)-Д2-пиразолинон-5
2:79
3-Амино-1-D-хлорфенил)-Д2-пиразолинон-5
2:79
2-Амино-6-этоксибензотиазол 1: 449
Амины 1: 29
алкилирование аммиака и аминов 1: 266, 299
— азометином 1: 300
— фталимидом (синтез Габриэля) 1: 300
— сульфамидом 1: 301
— уротропином (реакция Делепина) 1: 301
амидометилирование ароматических соеди-
соединений 1:464
ароматические активированные 1: 474
— палладийкатализируемые синтезы арил-
аминов 1:492
— реакции иодирования 1:444
с азотной кислотой 1:471
— роданирование 1:448
— синтез нитрилов 1:313
Вияьсмейера 1: 460
— сульфирование 1: 435
восстановительное аминирование карбониль-
карбонильных соединений, каталитическое 2:185
по Лейкарту—Валлаху 2: 180
нитросоединениями 2: 232, 235
реакция Шмидта 2: 282
восстановление амидами карбоновых кислот
2:170
— гидрирование азометином 2: 168
гетероароматические 2: 209
— синтез по Чичибабину 1: 474
ИК-спектры1:119
кватернизация 1: 266
конденсация Манниха 2: 129
получение из спиртов 1: 302
— из галогенопроизводных 1: 322
— из борорганических соединений 1: 375
— из металлоорганических соединений 1:481
расщепление по Гофману!: 278
реакция восстановления Буво—Блана2:191,336
сушка 1:38
третичные, адсорбционное сродство 1: 84
— кватернизация 1: 302
триметилсилилирование 1: 319
Аммиак 2: 396
в реакциях 1: 382, 383; 2: 58,187
— присоединения 2: 197
— алкилирования 1: 299
Аммония сульфид, восстановитель 2: 234
Аммония формиат 2:180
Аммония хлорид 2: 396
Аммония четвертичные соли 1: 279, 280, 302,
321,322,333,334,366
Аммония ион 1: 203; 2: 106
Аммонолиз
сульфохлоридов 2: 266
эфиров 2: 78
Амоксициллин 2: 82
Ампулы для хранения веществ 1: 159
Амфетамин 1: 203; 2: 181
Анализ органических соединений 2: 293
Анализ структуры кристаллов 1: 156
Анальгетики 2: 82, 83, 217
Ангидрон 1: 36
Анизидин 1: 449; 2: 6, 7, 198, 246, 247, 253, 290,
291,311,314,397
3-(л-Анизидино)пропионитрил, получение 2:198
я-Анизоилхлорид 2: 260, 261
я-Анизоин 2: 133
Анизол 1:440, 446, 458, 462
Анилы 2: 57
Анилин 2: 34, 396
идентификация производных 2: 311
получение 2: 235-237, 284
потенциал окисления 2: 6, 7
реакции замещения 1: 383, 430, 449; 2: 35, 58,
100, 187, 198, 245, 247, 250, 253, 209, 291
УФ-спектры 1: 115
ЯМР13С 1:138
рК 1:203
р-Анилинокротоновой кислоты этиловый
эфир 2: 58
З-Анилинопропионитрил 2: 198
З-Анилинопропионовой кислоты этиловый
эфир 2: 198
Анилинофумаровой кислоты диметиловый
эфир 1: 383
Анисовый альдегид 1: 89, 90, 462; 2: 29, 118,
120,124,136, 144,178,323
Антраниловая кислота 2: 250, 253, 281, 317, 319
Антрахинон 1: 456, 475; 2:13, 14, 32
идентификация 2: 328
электродный потенциал 2: 7, 8
Антрахинонсульфоновая кислота 1: 478, 479
Антрацен 1: 398, 399, 402; 2:45
идентификация 2: 352, 353
424
Предметный указатель
окисление 2: 6,7, 32
роданирование 1:448
УФ-спектр 1:115
Антрацен-9-карбальдегид 2:139
идентификация 2: 323
Арабиноза 2: 329
Аргинин 2: 317, 319
1-Арилпиразолинон-5 2: 83, 84,157
Арил-2,4-динитрофенилсульфид 1:492
Арилалканкарбоновые кислоты 2: 17
Арилалкилиденуксусной кислоты эфир 2: 203,
206
Арилалкиловые кетоны, нитрование 1: 432
Арилалкиловые эфиры 1: 297
Арилацетилен 1: 485,486
Арилборные кислоты 1: 483, 484, 487
Арилгидразин 2: 60, 232, 246, 290, 291
Арилгидразоны 2: 60, 251, 289, 290
Арилдиазония тетрафторборат 2: 239, 240
Ариллитий 1:478,482; 2: 164
Арилонитрил 1: 347, 391, 404
азеотропная водная смесь 2: 362
ИК-спектры 1: 124
осушение 2: 362
реакции обмена 1: 388, 398
Арилфториды, 2: 244
З-Арилциклогексан, 1:491,492
Арин 1:477
какдиенофил 1: 398, 399
реакция Арндта-Эйстерта 2: 276, 277, 291
Ароматические соединения
амидометилирование 1:464
алкилирование по Фриделю—Крафтсу Г. 449,
452, 493
ацилирование по Фриделю—Крафтсу 1: 454,
455, 457, 493
бромирование 1: 445, 447, 448, 493
галогенирование 1: 453
гидрирование 1:423, 424
гидроксиметилирование 1:463
замещение 1:423
— нуклеофильное 1:472,493
— электрофильное 1:425,492,493
ИК-спектр1:119
иодирование 1:493
нитрование 1: 192,431, 432, 433,493; 2: 301
нитрозирование 1: 470, 471
окисление 2: 6, 7,12
— до хинонов 2: 30, 31
получение восстановлением солей диазония
2:244
— каталитическим дегидрированием 2:43
тиоцианатов 1:448,493
синтез Гоша 1:493
сульфонилирование 1:435,441,493
карбоксилирование 1: 468
хлорирование 1: 493
формилирование 1:463
— по Гаттерману 1: 459, 493
— по Вильсмейеру 1:460, 461,493
хлорметилирование 1:465,493
хлорсульфонилирование 1: 438, 439, 493
Ароматичные (душистые) вещества 1: 316; 2:
14,66
Асимметрический атом 1: 220, 221
Асимметрический катализ 1: 227
Аскорбиновая кислота 2: 30, 64, 65, 75, 329
Аспарагин 2: 19
Аспарагиновая кислота 2: 317, 319
Аспартам 2: 82
Аспирин 1:470; 2: 76
Атомная единица массы 1:143
Атомная масса 1: 143
Атропин 2: 132
Аутокисление 1: 259; 2: 8, 9
Аценафтен 1:418; 2: 32, 351
Аценафтохинон 2: 32, 327
Ацетанилид 1: 440; 2: 172, 199, 236, 284
Ацетали 1: 231, 337; 2:64, 288
Ацеталвдегид 1: 361, 362, 382 , 404; 2: 364
Ацетальдегида этиленацеталь 1: 381
Ацетальдегидциангидрины 2:112
Ацетамид 1:203, 319
Ацетамидина гидрохлорид 2: 98
2-Ацетамидобензо(Ь)тиофенкарбоновая-3
кислота 2: 45
4-Ацетамидобензойная кислота 2:12
4-Ацетамидобензолсульфонилхлорид 1:440
З-Ацетамидопиперидон-2-карбоновой-З кис-
кислоты этиловый эфир 2: 186
Ацетат аммония, катализатор 2:125
а-Ацетиламино-3,4-дигидроксикоричная кис-
кислота, гидрирование 1:407
Ацетиламиномалоновой кислоты диэтиловый
эфир 2: 132, 204, 236, 237, 244
АцетиламиноB-цианэтил)малоновый эфир 2:
88; см. также (З-Цианэтилацетиламино-
маловый эфир
2-Ацетиламино-2-этоксикарбонил-9-D-ими-
дазолил)-7-азанонановой кислоты этило-
этиловый эфир 2: 173
2-Ацетиламино-2-этоксикарбонил-6-оксо-
гексановой кислоты этиловый эфир 2:173
Ацетилацетон 2: 20, 58,144,156, 253
а-Ацетилацетоуксусной кислоты анилид 2:155
2-Ацетилгексановой кислоты этиловый эфир
2:87,213,290
Ацетилен 1:29, 350, 366
винилирование циклооктатетраеном 1: 409
галогенирование 1: 362
Предметный указатель
425
получение при элиминировании 1: 323
под давлением 1: 380
реакции обмена 1: 380,409
сочетание с арил- (и алкенил-)-галогенидами
1:485
— с реактивом Гриньяра 2: 160, 166
— с солями меди 1: 485
ЯМР13С 1:140
р/С 1: 204
Ацетилендикарбоновая кислота 1: 343
Ацетиленкарбоновая кислота, декарбоксили-
рование 2: 86
2-Ацетил-2-изопропил-5-оксогексановой
кислоты этиловый эфир 2: 87
О-Ацетиллактонитрил 2: 73
Ацетилмалоновой кислоты диэтиловый эфир
2:154
2-Ацетил-З-метилбутановой кислоты этило-
этиловый эфир 2: 87, 204, 213
N-Ацетил-а-нафтиламин 2: 284
Ацетилнитрат 1: 431
Ацетилсалициловая кислота 1:470; 2: 73, 83
алкилирование 2: 217
ацилирование 2: 151, 152
производные 2: 331
Ацетшшторид 1: 342,458
Ацетилцеллюлоза 2: 76
2-Ацетилциклогексанон 2: 151, 221
2-Ацетилциклопентанон 2: 151,221
2-Ацетоксициклопропанкарбоновой кислоты
эфир 1: 395
Ацетон 1: 259
получение 2: 28, 289
техника безопасности 2: 364, 396
реакции 2:11,109,114, 118, 126, 130,131,144,
164,189,210,219
Ацетондикарбоновая кислота 2: 131
Ацетон итрил 1: 317
азеотропный раствор 2: 365
в качестве растворителя 1: 85, 116
дегидратация 1:40
рКа 1: 204
Ацетонциангидрин 1:109, 337, 338
Ацетофенон 1: 458; 2: 16, 17, 26, 63, 114, 118,
124, 126, 129, 131, 139, 144, 147, 164, 172,
181,182,185,187,219,284,324
производные 2: 324
Ацетофенондиэтилацеталь 1: 338
Ацилирование
ароматических соединений 1: 455
по Фриделю-Крафтсу формилгалогенидами
1: 382; см. также Фриделя—Крафтса
ацилироваание
Ацилоин2:133,134, 135, 191
Ацилоиновая конденсация 2: 104,132
Байера—Виллигера окисление 2: 286, 292
Бакелиты (фенолформальдегидные полиме-
полимеры) 1:467
Балластный газ 1: 31
Бамфорда—Стивенса реакция 2: 183
Бани для нагревания 1: 22
Барбитуровая кислота 2: 83, 150, 217
Бартона правило 1: 332
Батохромный сдвиг 1:111
Башкирова окисление 2: 9
Бекмана перегруппировка 2: 61, 284, 292
Бенз[е]индол-2-карбоновой кислоты этило-
этиловый эфир 2: 290
Бензальбромид 1: 257, 292
Бензалвдегид 1: 255, 291, 292; 2: 15, 25
диэтилацеталь 2: 64, 65
оксим 1:220, 253; 2:61
очистка 2: 365
производные 2: 320
— УФ-спектр 1:115
реакции 1: 252, 431 434; 2: 19, 61, 63, 65, 109,
111, 118, 122, 124, 127, 133, 136, 145, 164,
167,178, 180,187
циангидрин 2: 97, 109
Бензальхлорид 1: 251, 255, 292; 2: 340
Бензамид2:104,309,317
Бензанилид2: 103, 284
Бензгидроксимоилхлорид 1: 254, 397
Бензидин 2: 234, 311
1-Бензил-5-гидрокси-2,3-диметоксикарбо-
нилиндол 2: 207
3-Бензил-5-B-гидроксиэтил)-4-метил-1,3-ти-
азолия хлорид 2: 134, 206
N-Бензил-р-фенилэтиламин 2:187
N-Бензиланилин 2: 186, 187
N-Бензилацетамид 2: 80
N-Бензилметиламин 2:187
S-Бензилтиомочевины сульфонат 2: 356
S-Бензилтиоурония хлорид 1: 309
р-Бензил-а-фенилбутиронитрил 2: 217
Бензиламин2:187,280
идентификация 2: 312
реакции 1: 383; 2: 58, 79, 179, 198
З-Бензиламинопропионовой кислоты этило-
этиловый эфир 2: 198
4-Бензиламинопентен-3-он-2 2: 58
N-Бензиламинофумаровая кислота
диметиловый эфир 2: 207
диэтиловый эфир 1: 383
Р-Бензиламинокротоновой кислоты этиловый
эфир 2: 58
Бензилбромид 1: 257, 303, 304, 308, 311, 314; 2:
340
Бензилвиниловый эфир 1: 381
Бензилгалогениды 2: 160, 212
426
Предметный указатель
Бензилдиазометилкетон 2: 260, 278
Бензилиденацетон 2: 118
производные 2: 323
реакции 1:414; 2:, 131, 172, 175, 186,204
Бензилиденацетофенон 2: 118
производные 2: 323
реакции 1:414
Бензилиденмалоновая кислоты диэтиловый
эфир 2: 127
Бензилиденмалононитрил 2:127
Бензилиодид, производные 1: 310; 2: 340
Бензилкатион в масс-спектрометрии 1: 146
Бензилмагнийхлорид 2:164
Бензилметилкетон см. Фенилацетон
Бензилнитрит 1: 314
Бензиловая кислота, производные 2: 332
Бензиловый спирт 1: 255, 464; 2: 178
производные 2: 345
реакции 1:381; 2: 26
Бензилоксикарбонильная группа 1: 231; 2: 81
N-Бензилпиперидин 2: 180
1-Бензилтио-2-фенилэтан 1: 387
Бензилтиоцианат 1: 308
Бензилтриметиламмония гидроксид2: 197
Бензилтрифенилфосфония бромид 1: 303
Бензилтриэтиламмония хлорид 1: 394; 2: 124,
216
Бензилфенилкетон, производные 2: 323
Бензилфениловый эфир 1:297
Бензилфосфоновой кислоты диэтиловый
эфир 1:305; 2: 135
Бензилхлорид 1: 251, 253, 255, 465
производные 2: 340
реакции 1: 270, 299, 303, 308, 316; 2: 214
Бензилхлорметилкетон 2: 216
Бензилцеллюлоза 1: 255, 299
Бензилцианид 1: 255, 316
реакции 1: 434; 2: 97, 186, 145, 147, 172, 204,
217
Бензилцианиды, алкилирование 2: 217
Бензильная перегруппировка 2: 177
Бензильный радикал 1: 240
Бензимидазол 2: 81
Бензин 1: 477; 2: 365
2-Бензоилтиофен 1:459
сс-Бензоилацетоуксусной кислоты этиловый
эфир 2: 151, 154
(З-Бензоилакриловая кислота 1:459
Бензоиламиноуксусная кислота 2: 122
Бензоилацетон 2:144
Бензоилмалоновой кислоты диэтиловый эфир
2:154
Бензоилпероксид 1: 240, 253, 390; 2: 365
техника безопасности 2: 365
З-Бензоилпропионовая кислота!: 229,459;2:183
З-Бензоилпропионовой кислоты диэтиламид,
ретросинтетический анализ 1: 230
Бензоилуксусной кислоты метиловый эфир 2:
153
Бензоилхлорид 1: 255; 2: 50, 74, 76, 90, 94, 154,
260
Бензоин 2: 26, 133, 186
производные 2: 323, 345
Бензоиновая конденсация 2: 132, 225
Бензойная кислота 1: 203, 418; 2: 11, 12, 14,163
аллиловый эфир 1: 299
бензиловый эфир 1: 299
бутиловый эфир 1: 299
замещенные 1: 209; 2: 12, 14
идентификация 2: 332
метиловый эфир 1:434; 2: 71
получение 2: 11, 12, 163
реакции 1:432, 446, 447; 2: 94, 282, 284
хлорангидрид см. Бензоилхлорид
этиловый эфир 1: 209; 2: 71,145, 164
эфиры1:299;2:50, 75
Бензол 1: 131, 203, 230, 240, 241, 418, 425, 433,
439, 446, 452,458, 459, 465; 2:43, 366
азеотропные смеси 1: 75; 2: 366
алкилирование Фриделя—Крафтса 1: 452
металлирование 1:480
окисление 2: 39
присоединение хлора 1: 385
производные 2: 352
растворитель 1: 85
спектрЯМР|3С1: 139
сульфирование 1: 435
техника безопасности 2: 366
Бензол-1,3,5-трикарбоновой кислоты эфиры
2:143
Бензол-1,3-дисульфоновая кислота 1: 437
Бензолдиазониевая соль 2: 60, 489, 246
Бензолсульфамид 2: 266, 310, 657
Бензолсульфокислота 1:435; 2: 357
Бензолсульфоновой кислоты циклогексило-
вый эфир 1:327
Бензолсульфохлорид 1:439; 2: 263, 266
Бензонитрил 1: 139, 398, 485; 2: 9, 249
Бензофенон 1:453; 2: 26
производные 1: 323,483; 2: 186, 139, 284
Бетаины 2:137
Биарилы см. Диарилы
БисB-аминоэтил)амин 1: 371
5-[БисB-ацетоксиэтил)амино]-2-B-бром-4,6-
динитрофенилазо)-4-метоксиацетанилид
2:253
1,2-Бис(гидроксиметил)бензол 2:172
2,2-Бис(гидроксиметил)пропандиол-1,3 см.
Пентаэритрит
2,2-Бис(я-гидроксифенил)пропан см. Бисфенол А
Предметный указатель
427
1,10-Бис(диазометил)декаццион-1,10 2: 261,
277
Бис(дибромметил)бензол 1: 257, 292, 293
4,4'-Бис(диметиламино)бензофенон 1: 468
2,2'-Бис(дифенилфосфино)-1,1'-динафтил
(BINAP) 1: 493
1,2-Бис(дифенилфосфино)пропан (dppp) 1:489
1,2-Бис(дифенилфосфино)этан (dppe) 1:489
1,1-Бис(л-диметиламинофенил)этилен 2: 139
1,3-БисC-метилбензоксазолил-2)тримети-
нийиодид2: 158
1,3-БисC-метилбензотиазолил-2)тримети-
нийиодид2: 158
1,5-БисC-метилбензотиазолил-2)пентамети-
нийиодид2: 158
БисC-метилбутил-2)боран см. Дисиамилбо-
ран
Бис(метилтио)метиленмалононитрил 2: 155
1,3-БисA-метилхинолил-2)триметинийиодид
2:158
1,5-БисA-метилхинолил-2)пентаметинийио-
дид2: 158
2,2-БисD-метоксифенил)-1,3,2,4-дитиади-
фосфетан-2,4-дисульфид см. Лоусона реа-
реагент
1,4-Бис(тиоцианатометил)бензол 1: 308
3,3-Бис(л-толил)бутанон-2 2: 274
Бисульфитсодержащие соединения 2: 52, 66
Бисфенол А1: 371, 468
Бис(C-хлорэтил)амин 1: 303
БисB-цианэтил)малоновый эфир 2: 204
БисA,5-циклооктадиен)никель 1: 486
Биуретовая реакция 2: 77
Бифенил 1:458; 2: 351
техника безопасности 2: 398
Бицикло[2.2.1]гептен-2 см. Ноборнен
Бицикло[2.2.1]гептен-5-карбоновая-2 кислота
1:402
Бицикло[2.2.1]гептанол-2 см. Норборнеол
Бицикло[2.2.1]гептил, соединения 1: 212
Бицикло[2.2.2]октен-2 1: 402
Бицикло[2.2.2]октен-5-дикарбоновая-2,3 кис-
кислота, ангидрид 1:402
Бицикло[3.2.1]октан 1:402
Бицикло[4.1.0]гептадиен 1: 393
Бицикло[4.1.0]гептан 1: 396
Бицикло[4.4.0]декан 1:402
Бицикло[5.1.0]октадиен-2,5 2:288
Блана реакция 1:464
Блокирование функциональных групп см. За-
Защитные группы
Боденштейна принцип 1: 201
Бора трифторид 1: 372; 2: 215
катализатор реакции Фриделя—Крафтса 1:
450
— присоединения Михаэля 2: 207
Боран 1: 358, 374
Борная кислота, эфир 1: 287, 483; 2:9
Борнеол 2: 276
производные 2: 345
Борорганические соединения 1: 374, 388, 483,
487
Бофорс-процесс 2: 14
Брассиловая кислота (ундекандикарбоновая-
1,11 кислота) 2:183
Бредта правило 1: 347
Бренстеда основания и кислоты 1: 202
Бром 1: 240
как защитная группа при ароматическом за-
замещении 1: 430
реакции 1: 356, 364,445; 2: 218, 300
техника безопасности 2: 366, 398
1-Бромалканы см. Алкилбромиды
Бромаль 2:175
4-Броманизол 1:446
Броманилин 2: 249, 312, 313
Бромароматические соединения 1:445; 2: 247
Бромацетальдегид, дибутилацеталь 2: 219
Бромацетальдегид, диэтилацеталь 1: 343
Бромацетон 2: 219
Бромацетофенон 1:447, 458; 2: 17,42, 181, 219,
323
4-Бромбензальдегид2: 15
Бромбензилбромид 1: 257, 316; 2: 339
Бромбензилиодид, производные 2: 339
2-Бромбензилтиоцианат 1: 308
Бромбензилхлорид, производные 2: 339
Бромбензилцианид 1: 316; 2:96
3D)-Бромбензоилхлорид 2: 94
З-Бромбензойная кислота 1:446,447; 2:94, 332
4-Бромбензойная кислота 2:42, 94, 258
4-Бромбензойной кислоты метиловый эфир 2:
258
Бромбензол 1: 54,434,446
производные 2: 340
техника безопасности 2: 398
4-Бромбензолсульфоновая кислота, произ-
производные 2: 357
4-Бромбензонитрил 1: 484
1-Бромбутан см. Бутилбромид
2-Бромбутан еж. втор- Бутилбромид
2-Бромбутановая кислота 1: 302; 2: 218
4-Бром-/я/>ет-бутилбензол 1:446
а-Бром-у-бутиролактон 2: 219
а-Бромвалериановая кислота 1: 302; 2: 218
2-Бромгексановая кислота 2: 218
Бромгидрохинон, производные 2: 347
2-Бром-1-дейтеро-1,2-дифенилэтан 1: 331
1-Бромдекан см. Децилбромид
2-Бром-4,6-динитроанилин 2: 645
428
Предметный указатель
1-Бром-3,5-динитробензол 1: 447
6-Бром-2,4-динитротолуол 1: 447
2'-Бром-4-диэтиламино-4',6'-динитроазобен-
зол 2: 253
2-Бромизобутановая кислота 2: 218
З-Бромизобутановой кислоты метиловый
эфир 2: 201
Бромирование 1: 239, 263
ароматических соединений 1:493
N-бромсукцинимидом 1: 239, 263
фотобромирование 1: 256
Броммезитилен 1:446
а-Бромкарбоновые кислоты 2: 218
2-Бром-4-метилпентановая кислота 2: 218
2-Бром-4-нитротолуол 1:446
Броммалоновой кислоты диэтиловый эфир 2:219
Бромметилбутанкарбоновая кислота 1: 302
Бромметилирование 1:466
Бромметилнафталин 1: 259,308, 446
Бромметилпропан см. Изобутилбромид
Бромметилфенилкетон 2: 260
1-Бромнафталин 1:485, 487; 2: 341
1-Бром-З-нитробензол 1:446,447; 2: 235
1-Бром-4-нитробензол 1: 434; 2:642
Бромоводород 1: 75, 256; 2: 366
азеотроп с водой 1:256; 2:366
значение рКа1: 203
присоединение к олефинам ионное 1: 355
радикальное 1: 384
расщепление эфиров 1: 289; 2: 720
реакции 1:387; 2: 598
техника безопасности 2: 366, 396
энергия связи 1: 240
Бромоформ 2: 341, 396
1-Бромпентанкарбоновая кислота 1: 302
1 -Бромпропен-2 см. Аллилбромид
З-Бромпропил-1 -фосфорная кислота 1: 305
З-Бромпропионитрил 2: 201
2-Бромпропионовая кислота 1: 302; 2: 218
метиловый эфир 2: 201
этиловый эфир 2: 201
N-Бромсукцинимид 1: 259, 264; 2: 367
техника безопасности 2: 367
4-Бромтиофенол, производные 2: 356
2C)D)-Бромтолуол 1: 257; 2: 249
производные 2: 340
Бромуксусная кислота 2: 218
производные 2: 331
техника безопасности 2: 396
этиловый эфир 1: 311; 2: 222
З-Бромфенантрен-9,10-хинон, производные
2:327
9-Бромфенантрен 1:485
4-Бромфенациловый эфир 1: 299; 2: 323
4-Бромфенацилбромид 1: 299; 2: 219, 323
4-Бромфенилгидразин 2: 79, 247
4-Бромфенилглиоксаль2:17
2C)D)-Бромфенилуксусная кислота 2:96
1-Бром-2-фенилэтан см. 2-Фенилэтилбромид
D-Бромфенокси)триметилсилан 1: 319
Бромфенолы 1: 446; 2: 347
1-Бром-З-хлорпропан 1: 387; 2: 213
З-Бромциклогексен 1:259
2-Бромэтанол, производные 2: 343
1-Бромэтилбензол2:182
B-Бромэтил)фосфоновой кислоты диэтило-
диэтиловый эфир 1: 305
Брэгга уравнение 1: 156
Буво—Блана восстановление 2:169, 191, 336
Буна 1:379, 391,453
Бунзена горелка 1: 22
Бутадиен- 1,3-карбонитрил 1: 347
Бутадиен-1,3 1: 366, 379, 391, 392, 399, 408
реакции 1: 357, 402, 408, 422
техника безопасности 2: 396
Бутан 1: 216
Бутаналь 1: 387
Бутан-1,3-диамин, производные 2: 311
Бутандиол-1,3 2:121,343
Бутандиол-1,4 1: 288, 414,418; 2: 343
Бутановая (масляная) кислота 1:492
производные 2: 331
реакции 2: 94, 219
техника безопасности 2: 396
этиловый эфир 2: 164
Бутанол-1 1:293, 418; 2: 186
производные 2: 343
как растворитель 1: 88
реакции 1: 296, 319, 381; 2: 71, 264
техника безопасности 2: 396
Бутанол-2 1: 293, 362; 2: 185
производные 2: 342
реакции 1: 335, 452
техника безопасности 2: 396
трет-Ъутгяоп 1: 326, 362; 2: 5
производные 2: 342
реакции 1: 378, 452; 2: 477
техника безопасности 2: 398
энергия связи 1: 240
Бутанон
диаллилацеталь 2: 289
реакции 2:109, 118, 126,131,185
реакция Манниха 2: 130
техника безопасности 2: 396
Бутантиол-1 1:310
производные 2: 356
техника безопасности 2: 396
Бутантиол-2 1: 310
Бутен-1 1:343, 361, 379; 2:10, 396
Бутен-2 1: 335, 344, 361, 379, 395, 409; 2:10
Предметный указатель
429
Бутен-2-аль см. Кротоновый альдегид
Бутен-2-ол-1 2: 175
Бутен-З-карбоновая-1 кислота 1:492
Бутен-З-он-2 см. Метилвинилкетон
Бугиламин, 2: 180, 284, 311, 396
трет-Бутиламин 1, 383
производные 2: 311
Ы-/и/>ет-Бутиламинофумаровой кислоты ди-
метиловый эфир 1: 383; 2: 207
2-/я/>ет-Бутилантрагидрохинон 441
Бутилацетат 1: 347
а-Бутилацетоуксусной кислоты этиловый
эфир 2: 214
Бутилбензол 1: 261, 430, 439,446, 452; 2: 182
4-Бутилбензолсульфохлорид 1:439; 2: 263
Бутилбромид 1: 284
производные 2: 340
реакции 1:296,299,303,305,310,316,452; 2:213
Бутилвиниловый эфир 1: 381
1-тре/я-Бутил-5-гидроксииндолдикарбоно-
вой кислоты диэтиловый эфир 2: 207
тре/я-Бутилгидропероксид 1: 370
/я/>ет-Бутилгипобромит 1: 257
/я/ге/я-Бутилгипохлорит 1: 252, 364
Бутилиденмалоновой кислоты диэтиловый
эфир 2: 126
/я/;е/я-Бутилизотиоцианат 1: 308
Бутилиодид 1:286,310
производные 2: 340
«-т/>ет-Бутилкаликсарен 1:463
Бутилкаучук 1:379
Бутиллитий 2: 368
химические свойства 1:480, 482; 2: 164
/я/>е/и-Бутилмагнийхлорид 2: 164
Бутилмалоновая кислота 2: 86, 88
Бутилмалоновой кислоты диэтиловый эфир 2:
86,167, 204,213
Бутилметилкетон, производные 1: 338; 2: 322
Бутилметиловый эфир 1: 289, 296, 362
Бутилнафталин 1:453
Бутилнафталинсульфонат 1: 453
Бутиловый спирт см. Бутанол-1
т/>ет-Бутилоксикарбонильная группа 1: 231,
237
/n/jem-Бутилпероксид см. Ц,п-трет-6угил-
пероксид
N-Бутилпиперидин 2: 180
3-т/>ет-Бутил-2,2,4,4-тетраметилпентанол-3
2:165
4-Бутилтиофенол 2: 263
mpem-Бутилтиоцианат 1: 308
Бутилтрифенилфосфония бромид 1: 303
/я/>ет-Бутилфенол 1: 362,463
Бутилфосфоновой кислоты диэтиловый эфир
1:305
Бутилхлорид 1: 284
производные 2: 340
реакции 1: 308, 452
техника безопасности 2: 396
/я/ретя-Бутилциклогексан 1: 218
4-/я/)е/я-Бутилциклогексанол2: 172, 175
4-т/>ет-Бутилциклогексанон 2: 172, 175
/я/;е/я-Бутильная группа 1: 357
Бутилэтиловый эфир 1: 296
Бутин-2-диол-1,4 1:418; 2: 114
техника безопасности 2: 396
реакции 1:414
Бутиральдегид
диэтилацеталь 2: 63
производные 2: 320
реакции 2: 63, 111,118,126, 180
техника безопасности 2: 396
Бутиральдоль см. 2-Этил-З-гидроксигексаналь
Бутиранилид 2: 284
Бутирилхлорид 2: 94, 154, 221, 396
2-Бутирилциклогексанон 2:151, 221
2-Бутирилциклопентанон 2:151, 221
Бутирилянтарной кислоты диэтиловый эфир
1:386
у-Бутиролактон 1:418,459; 2: 28,114, 219
Бутиронитрил 1:316; 2: 396
Бутирофенол 1:457
производные 2: 324
реакции 2:182, 284
Бутоксибензол 1: 296
Бутокситриметилсилан 1: 319
N-л-Бутоксифенил-М'-л-диметиланилино-
тиокарбамид 2: 102
М-«-Бутоксифенил-М'-я-диметиламинофе-
нилтиомочевина см. Тиамбутоксин
Бюхерера синтез 2: 110
Бюхнера воронка 1:47, 162
Вагнера—Байера реакция 2: 39
Вагнера—Меервейна перегруппировка 2: 275,
291
Вакер-окисление 1: 404
Вакуум 1: 31
Вакуумметр компрессионный 1: 33
Вакуумный форштос 1: 57, 162
Валентная таутомерия 2: 288
Валентные колебания 1: 112, 117, 125
Валериановая кислота 2: 88, 96, 218, 284
производные 2: 331
Валериановый альдегид, производные 2: 320
Валеролактам 2: 284
Валеролактон 2: 286
Валеронитрил 1: 317; 2:97
430
Предметный указатель
Валин 2: 111,318
Вальденовское обращение 1: 269, 285
Ванадия пентаоксид
как катализатор окисления 2: 9
техника безопасности 2: 399
Ванилин 1:90, 295; 2: 127, 325
производные 2: 321, 348
Вератрилхлорид см. 3,4-Диметоксибензилхло-
рид
Вератровая C,4-диметоксибензойная) кислота
2:42
Вератровый альдегид C,4-диметоксибензаль-
дегид) 1: 295
Вератрол A,2-диметоксибензол) 1: 458, 462,
465,482
ВЗМО — НСМО взаимодействия 1: 213
Взрывчатые вещества 1: 287; 2: 178
Вигре колонка 1: 168
Вильгеродта реакция 2: 17,46
Вильгеродта—Киндлера реакция 2: 18
Вильсмейера синтез 1: 426, 461
Вильштеттера расщепление 1: 344
Вильямсона синтез эфиров 1: 265, 291, 295
Винилацетат 1: 362, 390
получение из этилена 1:404
реакции 1: 386, 390, 395, 398
техника безопасности 2: 399
Винилкетон 1: 344
9-Винилантрацен 2: 139
Винилбромид, производные 2: 339
Винилгалогенид, SN-реакции 1: 270
Винилидендихлорид 1: 366, 391
Винилиодид, производные 2: 339
Винилирование 1: 421
Виниловый эфир 1: 121, 380; 2: 288
Винилогия 1: 208; 2:194
2-Винилпиридин 2: 124
2-Винилтиофен 1: 343
Винилхлорид 1: 340, 362, 366, 391, 404
производные 2: 339
Винилциклопропан 2: 269
Винилциклопропан — циклопентен перегруп-
перегруппировка 2: 269
Винильная группа 1: 208
Винная кислота 1: 223, 225
производные 2: 331
Винной кислоты диэтиловый эфир 1: 227,
369; 2: 40, 71
Витамин А 2:138
Витамин Bi 2: 129
Витамин В5 2:83
Витамин В6 1:403; 2: 83
ВитаминС 2:30,65,76
Витамин Е 1: 454
Виттига перегруппировка 2: 271, 291
Виттига реакция 1: 303; 2: 104, 136, 138,
225
Вода
значение рКа 1: 203
растворитель 1: 50, 85, 116, 200, 326
реакция присоединения к олефинам 1: 355,
359,420
к <х,р-непредельным соединениям 2: 200
— с реактивами Гринъяра 2: 160
собственные колебания молекулы в ИК. 1:119
температура кипения, зависимость от давле-
давления 1: 54
энтальпия диссоциации связи 1: 240
Водоотделитель 1: 76
Водород
гидрирование карбонильных соединений 2:
184
— кратных С=С-связей 1: 353, 408,410,424
— нитросоединений 2: 234
очистка и сушка 2: 369
при оксосинтезе 1: 353, 407
техника безопасности 2: 369
характеристика баллона для сжатого газа 1:31
энтальпия диссоциации связи 1: 240
Водородная связь 1: 200
Водоструйный насос 1: 32, 34
Водяная баня 1: 22
Воздушная баня 2: 22
Воздушный холодильник 1: 10, 11, 162
Вольфа перегруппировка 2: 276, 291
Вольфа—Кижнера восстановление 2: 168, 181,
182,228,234
Восстановительная конденсация Мукайамы 1:
288; 2: 288
Восстановительные процессы 2: 5
Восстановительный потенциал см. Электрод-
Электродный потенциал
Восстановление 1:191
алкиновдоалкенов2:172
двойной С=С-связи 1:410, 413
карбонильных соединений 2:168, 227
неблагородными металлами 2: 189, 191,
228
ферментивное 2: 181
Гриньяра 2: 162
каталитическое гидрирование 2: 184,
185,227
комплексными гидридами 2: 169, 172,
227
литийалюминийгидридом 2:171
натрия борогидридом 2: 171
по Буво—БлануЪ: 190
по Канниццаро 2: 177,178, 228
по Клемменсену!: 192, 228
по Лейкарту-Валлаху 2: 179, 180, 228
Предметный указатель
431
по Меервейну—Понндорфу—Верлею!: 174,
228
по Вольфу-Кижнеру 2: 181, 182, 228
нитросоединений 2: 232
— гидразином 2: 235
— каталитическое 2: 234
— с оловом/НС12: 236
радикальное 1: 239, 262
сульфохлоридов 2: 263
сульфонов2:262
сульфокислот 2: 261
сульфоксидов 2: 261
Вуда сплав 1: 24
Вудворда-Гофмана правило 1: 392, 420
Вулыра склянка 1: 34, 38
Вюрстера красный 2: 7, 32
Высокопольный сдвиг при 13С-ЯМР 1: 138
Высшая занятая молекулярная орбиталь
(ВЗМО) 1: 212, 249, 383
значение при циклоприсоединении 1: 392,
400
энергия 1:213, 353
Вюрца реакция 1: 484; 2: 160
Вюрца—Фиттига реакция 1: 484
Вязкость 1: 52
Габриэля синтез 1: 300
Газ-носитель при газовой хроматографии 1: 73
Газовая хроматография (ГХ) 1: 85, 96, 165
Газы 1:19, 36, 37,40
Галактоза, идентификация 2: 329
Галловая кислота 1: 295
производные 1: 295; 2: 331
N-Галогенамины 1: 264
а-Галогенкетоны 2: 65, 219, 260
Галогенгидрины 1: 364
Галогензамещенные углеводороды, алифати-
алифатические
идентификация 2: 295, 339
— производные 2: 339
— растворимость 2: 297
— физические константы 2: 341
как растворитель 1: 50
получение из алифатических диазосоедине-
ний и галогеноводорода 2: 258
борорганических соединений 1: 374
спиртов и галогеноводорода 1: 282, 284
спиртов и неорганических галогенидов
1:284
— при галогенировании насыщенных углево-
углеводородов 1: 245, 249, 253, 259
присоединении галогенов к алкенам и
алкинам 1: 364, 383
галогенововорода к алкенам 1: 359,
384
полигалогеналканов к алкенам 1: 384
расщеплении простых эфиров галогено-
водородом 1:289
обмене галогенов (Финкелыитейна реак-
реакция) 1: 310, 322
химические свойства, алкилирование Фриде-
ля—Крафтса 1:449
— гидролиз 1: 290
— радикальное восстановление 1: 262
Галогензамещенные углеводороды, аромати-
ароматические
получение из солей диазония (по Зандмейеру)
2: 244, 248
— при галогенировании ароматических сое-
соединений 1: 443,445, 447
производные 2: 340
реакции, ацилирование Фриделя—Крафтса 1:
456
— взаимодействие с аминами 1: 492
с магнием 1:479; 2:159
с медью (Ульмана реакция) 1: 485
с литием 1:479,484; 2: 166
с литийкупратами 1: 485; 2:166
с олефинами (Хека реакция) 1: 490
с металлоорганическими соединениями
1:484
— SN 1:272
Галогензамещенные углеводороды, жирно-
ароматические
для тонкослойной хроматографии 1: 99
идентификация 2: 339
получение при хлорметилировании аромати-
ароматических соединений 1:426, 464, 465
— при галогенировании алкилзамещенных
ароматических соединений 1: 251, 253
химические свойства, гидролиз 1: 291
Галогениды металлов, реакции с металлоорга-
металлоорганическими соединениями 1: 483; 2: 160
Галогенирование
алкенов 1: 383
ароматических соединений Г. 425, 443, 445,
447
в боковую цепь в ароматических соединени-
соединениях 1: 444
в ядро ароматических соединений 1: 443
карбонильных соединений 2: 217
радикальное 1: 239, 249, 263
Галогенкарбоновая кислота 2: 218
реакции 1: 302; 2: 219
— аммонолиз 1: 302
— синтез нитрилов 1:313
эфиры 2: 84, 167, 200
Галогеноводороды 1:282
432
Предметный указатель
присоединение к олефинам 1: 355, 359
— к <х,р-непредельным карбонильным соеди-
соединениям 2: 200
Галогентриалкилсилан 1: 322
Галогены 2: 296
идентификация по гидролизу 2: 307
присоединение к олефинам и ацетиленам 1:
355, 420
соединения 1: 239, 262; 2:48
Галоформ 1: 384
Галоформная реакция 2:42
Гаммексан см. у-Гексахлорциклогексан
Гаммета уравнение 1: 209, 234
Гаттермана синтез 1:426, 459
Гаттермана—Адамса синтез 1:459
Гаттермана-Коха синтез 1:426, 459
Гваякол 1:381; 2:246
техника безопасности 2: 399
Гевальда реакция 2: 20
Гексаалкилдистаннан 1: 488
Гексагидробензальдегид, производные 2: 320
1,2,3,5,6,7-Гексагидронафталин 1: 399
Гексагидросалициловой кислоты метиловый
эфир 1: 414
Гексадиен-1,5 2:354
Гексаметилдисилоксан 1: 319
Гексаметилендиамин 1: 366; 2: 80, 84,185, 186
техника безопасности 2: 399
Гексаметилендиизоцианат 2: 99, 102
Гексаметилентетрамин (уротропин) 1: 301,
466; 2: 58
производные 2: 316
Гексаметилфосфорной кислоты триамид как
растворитель 1: 200, 274
Гексан
азеотроп с водой 2: 369
как растворитель 1: 85, 116
очистка и сушка 2: 369
техника безопасности 2: 369, 396
физические константы 2: 349
Гексаналь, производные 2: 320
Гександиол-1,6 2:172
Гександитиол-1,6 1:310
Гексаннитрил 1:316; 2: 96
Гексановая кислота 2: 28, 88, 96, 218, 219
производные 2: 331
Гексанол-1
производные 2: 343
реакции 1: 296; 2: 28, 73, 264
техника безопасности 2: 399
Гексанол-2, производные 2: 343
Гексанон-2 1: 362; 2: 63
техника безопасности 2: 399
Гексанон-2, диэтилацеталь 2: 63
Гексантиол-1 1:310
производные 2: 356
Гексахлорэтан 2: 341
Гексахлорциклогексан 1: 340, 319, 386; 2: 396
Гексахлорциклопентадиен 1: 255, 340
Гексен-1 1:340,344,365
Гексен-5-ил-1, соединения, радикальная цик-
циклизация 1: 388
Гексен-2-ол-1 1:369
Гексен-5-он-2 2:87
Гексиламин 1: 344
производные 2: 311
Гексилбромид 1: 284
производные 2: 340
реакции 1: 299, 314, 316, 340; 2: 213
Гексилиодид 1: 286
производные 2: 340
реакции 1: 308, 314
Гексилмагнийбромид 2:164
Гексилмалоновая кислота 2: 86, 88
Гексилмалоновой кислоты диэтиловый эфир
2:86,213
Гексилметиловый эфир 1: 296
Гексилхлорид 1: 316
производные 2: 340
Гексин-1 1: 342, 362; 2: 131, 354
Гексобарбитал 2: 83
Гелиотропин 1: 374
Гентиобиоза 2: 329
Гептадекановой кислоты этиловый эфир 2:
278
Гептаметинцианин2: 157
Гептан 2: 349
техника безопасности 2: 399
Гептаналь 2: 25
производные 2: 320
реакции 1: 386; 2: 185, 206
Гептанол-1 2:25,73,185,264
производные 2: 343
Гептаннитрил 1:316; 2:96
Гептановая кислота 2: 25, 28, 88, 96,183
техника безопасности 2: 399
Гептаноилянтарной кислоты этиловый эфир
1:386
Гептанол-2, производные 2: 343
Гептанон-2 1:362
техника безопасности 2: 399
Гептантиол-1, производные 2: 356
Гептантрикарбоновой-1,3,3 кислоты триэти-
ловый эфир 2: 204
Гептен-1 1:340,365
Гептен-6-ил-1, соединения, радикальная цик-
циклизация 1: 388
Гептилбромид 1: 284, 340
производные 2: 340
Гептилиодид, производные 2: 340
Предметный указатель
433
Гептилхлорид, производные 2: 340
Гептин-1 1:362
Гераниол 2:114
производные 2: 343
Гербициды 2: 76, 84
Гесса тепловой закон 1:195
Гетероароматические соединения см. Гетеро-
циклы
Гетероауксин 2: 291
Гетерокумулены, взаимодействие с СН-кис-
лотами 2:155
Гетеролиз 1: 191
Гетероциклы
идентификация 1: 115
получение 2-аминобензтиазола тиоцианиро-
ванием 1:449
— аминопиразола 2: 210
— 2-аминотиофена из а-алкилиденнитрилов
2:20
— 3-амино-1-фенилпиразолона-5 из эфиров
моноимидомалоновой кислоты и фенил-
гидразина 2: 78
— 5-гидроксииндолов по Неницеску!: 207
— индолов по Фишеру 2: 290
— оксазолинов и оксазолов 1: 398
— 1-фенилпиразолов из 1,2-дикетонов и фе-
нилгидразинов 2: 59
— пиразолов из пиразолинов 2:45
— пирролов из 1,2-дикетонов и аммиака 2:
59
— тиазолов из тиазолинов 2:45
— тропинона по реакции Манниха 2: 132
— хиноксалинов из 1,2-дикетонов и о-фени-
лендиаминов 2: 59
при [2+2]-циклоприсоединении 1: 396
при [3+2]-циклоприсоединении 1: 397
при дегидрировании 2:43, 45
при внутримолекулярном арилировании
Ы^-арилалкиламиносоединений 1: 493
— хинолинов по Дебнеру—Миллеру 2: 198
по Скраупу!: 198, 229
реакции, аминоэтилирование 1:464
— ацилирование Фридепя—Крафтса 1:456
— Гаттермана синтез 1: 459
— замещение 1:493
— тиоцианирование 1: 448
Гиббса энергия 1: 193, 196
Гиббса—Гельмгольца уравнение 1: 195
Гидантоин2: 123
Гидразиды 2: 52
Гидразин 2: 369
реакции 2: 182,235
— с эфирами карбоновых кислот или хлоран-
гидридами 2: 52, 78
— с р-хлоракролеином 2: 209
— с Ni Ренея как восстановителем 2: 235
сульфат 2: 111,369
техника безопасности 2: 369, 396
энергия разрыва связи 1: 240
Гидразингидрат 2: 210, 369
техника безопасности 2: 369, 399
Гидразинолиз
N-алкилфталимидов 1: 300
сложных эфиров 2: 78
Гидразины 2: 78, 346
Гидразобензол 2: 234
а,а'-Гидразобис(изобутиронитрил) 2: 111
а,а'-Гидразобис(циклогексан-1-карбонитрил)
2:111
Гидразоны 2:51,57,222,232,251,252, 289, 290,
320
восстановление по Кижнеру—Вольфу 2: 181,
182
гидролиз 2: 62
окислительное сочетание 2: 36
синтез индолов по Фишеру!: 290
хлорирование 1: 252
хиральные 2: 216
Гидразосоединения 2: 232
E-Гидридное элиминирование 1:404
Гидридные комплексы 1: 407
Гидриды
комплексные 2: 169, 227
металлов 1:262; 2: 157, 169
Гидрирование 1: 195, 353
каталитическое гетерогенное 1:410,413; 2:234
— гомогенное 1: 407, 423
катализаторы, 2: 378
а,р-Гидроалкоксиэлиминирование (р-элими-
нирование спиртов) 1: 323
а-Гидро-ю-гидрокситриоксиэтилен см. Три-
этиленгликоль
Гидробензоин 2:185
Гидроборирование 1: 353, 374, 375,421
Гидроксамовые кислоты 2: 52, 305
4-Гидроксиазобензол 2: 234
а-Гидроксиальдегиды 2:60
Гидроксиантрахинон 1: 479
4-Гидроксиацетанилид 2: 83
Гидроксибензальдегид 1: 349; 2: 246
производные 2: 347
2-Гидроксибензиловый спирт 2: 172
4-Гидроксибензойная кислота 1: 295, 470
идентификация, производные 2: 331
З-Гидроксибензойной кислоты производные
2:331
3-Гидрокси-1-метилиндолдикарбоновой-2,3
кислоты диметиловый эфир 2: 207
4-Гидроксибензолсульфоновая кислота 2: 357
434
Предметный указатель
4-Гидроксибутанон-2 1: 179
этиленацеталь 2: 192
З-Гидроксибутаналь 1:336; 2: 118
производные 2: 323
Гидроксибутановой кислоты этиловый эфир
2: 181,185
б-Гидроксивалериановый альдегид 2: 64
4-Гидроксигептанон-2 2: 118
9-Гидроксидекалинон-2 2: 204
5-Гидроксииндолдикарбоновой-2,3 кислоты
диметиловый эфир 2: 207
5-Гидроксииндолы 2: 207
а-Гидроксикарбонитрил 2:97, 109
Гидроксикарбоновые кислоты 2: ПО, 167, 192,
298
Гидроксикетоны 1: 456; 2: 60, 65
Гидроксикислоты см. Гидроксикарбоновые
кислоты
4-Гидроксикумарин 2: 298
Гидроксилазы 2: 9
Гидроксиламин 1: 89; 2: 61, 232, 305
Гидроксилирование 1:353,267, 370,420; 2:6, 9,
38
Гидроксильная группа 1: 271, 296
2-Гидрокси-4-метилацетофенон 1: 456
Гидроксиметилбутанон-2 2:118
2-Гидроксиметил-6,6-диметилбицикло[3.1.1]геп-
танси. Пинанол-10
Гидроксиметиленциклогексанон 2:144
Гидроксиметилирование 1: 426,463
Гидроксиметилкетоны 2:135
5-Гидрокси-1-метилиндолдикарбоновой-2,3
кислоты алкиловый эфир 2:207
5-Гидрокси-2-метилиндолкарбоновой-3 кис-
кислоты алкиловый эфир 2: 207
З-Гидрокси-2-метилпентаналь 2: 118
4-Гидрокси-4-метилпентанон-2 1: 336: 2:116,
118
З-Гидрокси-З-метилпентанон-2 1:362
4-Гидрокси-4-метилпентанон-2 1:336; 2: 116,
118
4-Гидрокси-3-метилпентанон-2 2: 118
4-Гидрокси-1-метилпиперидинкарбоновая-3
кислота 2: 185
2-Гидроксиметил-2-метилиропаналь2:118, 121
3-Гидрокси-3-метил-1-фенилбутанон-1 2: 212
Гидроксиметилфурфураль 2: 322
4-Гидрокси-З-метоксибензальдегид см. Вани-
Ванилин
4-Гидрокси-З-метоксикоричная кислота 2:126
Гидроксимоилхлорид 1: 398
З-Гидроксинафталинкарбоновая-2 кислота 1:
470
4-B-Гидроксинафталин-1-азо)бензолсульфо-
новая кислота 2: 253
2-Гидрокси-5-D-нитрофенилазо)бензойная
кислота 2: 253
4-Гидроксипентанон-2 2:118
Р-Гидроксипропионитрил 1: 124, 371
Гидроксисоединения, триметилсилилирова-
ние 1:318
N-Гидроксисукцинимид 2: 82
3-Гидрокси-2,6,6-триметил[3.1.1]гептан см.
Пинанол-3
(Гидроксифенил)аминокислоты 2: 34
5-Гидрокси-1-фенилиндолдикарбоновой-2,3
кислоты метиловый эфир 2: 207
р-Гидрокси-р-фенилпропионовой кислоты
этиловый эфир 2: 167
Гидроксифенилтиоцианат 1:449
а-Гидроксифенилуксусная кислота см. Мин-
Миндальная кислота
8-Гидроксихинолин 2: 37, 198
производные 2: 316
A-Гидроксициклогексил)метилкетон 1: 362
A-Гидроксициклопентил)метилкетон 1: 362
A-Гидроксиэтил)бензол 1: 337
2-B-Гидроксиэтил)пиридин 2: 124
Гидроксиэфиры 1:119
Гидролиз см. также Омыление
алкилгалогенидов, алкилсульфонатов 1: 265,
282, 290, 293
N-алкилфталимидов 1: 300
амидов карбоновых кислот 2: 52, 84, 89
ангидридов карбоновых кислот 2: 52, 89
галогенангидридов карбоновых кислот 2:52,89
галогенидов 2: 92
древесины 2: 66
металлоорганических соединений 2: 501
нитрилов 2: 95, 97
хлорсиланов 1:319
карбоновых кислот 2: 52, 84, 86, 89, 151
Гидропероксиды 1: 259, 260; 2: 38, 286
а-Гидропероксикумол 1:453
Гидроформ ил ирование см. Оксосинтез
Гидрохинон 1:295, 470; 2: 33, 34
как ингибитор радикальных реакций 1: 260
техника безопасности 2: 399
Гидрохинона производные 1: 295; 2: 327, 347
Гиперконъюгация см. Сверхсопряжение
Гиппуровая кислота 2:122
Гипсохромный сдвиг 1:112
Гистамин, дигидрохлорид 2: 132, 173
Глибенкламид 2: 267
Гликозиды 2:40, 65
Гликолевая баня 1: 24
Гликолевая кислота, производные 2:109, 331
Гликоли см. Диолы-1,2, Этиленгликоль
Глимепирид 2: 267
Глиоксалаза 2:178
Предметный указатель
435
Глиоксаль 1:363; 2: 15,320
Глиоксиловая кислота 2:40, 62
Глифосат 1: 303
Глифталь1: 371
Глицерин 1: 293, 366, 370, 371; 2: 396
какрастворительдля масс-спектрометрии 1:143
производные 2: 343
химические свойства 2: 198
Глицерина тринитрат 1: 286, 371
Глицериновый альдегид 1: 221
Глицидные эфиры B,3-эпоксиэфиры) 2: 123
Глицин 1:302; 2: 111,317
сложный эфир 2: 238
Глутаминовая кислота 2: 88, 204, 317
Глутаровая кислота 2: 96, 331
Глутаровой кислоты ангидрид 2: 90
Глутаровый альдегид 2: 64
Глутаронитрил 1:316; 2: 96
Глюкоза 2: 28,60, 65, 73, 185, 329
Глкжоновая кислота 2: 28
Гомберга—Бахмана арилирование 2: 248
Гомолиз 1: 241
3,4-Гомотропилиден 2: 288
Горчичные масла см. Изотиоцианаты
Гофмана перегруппировка амидов карбоновых
кислот 2: 278
Гофмана элиминирование 1: 273, 323, 329, 343,
344,350
Градиентное элюирование 1: 87
Грамин2:130,132, 202
Граничные орбитали 1: 212, 249, 353, 392
Граничные структуры 1: 207
Гремучий газ 1: 195
Гриньяра соединения 1: 478; 2: 159, 163
химические свойства 1: 481, 489; 2: 104, 159,
163, 225
трансметалл ирование 1:388,483,488; 2:160
Губена—Геша синтез 1:426, 459, 460
2,4-Д см. 2,4-Дихлорфеноксиуксусная кислота
Дарзана—Кляйзена реакция 2: 104, 123, 146, 224
ДБА см. Дибензилиденацетон
ДБН см. 1,5-Диазабицикло[4.3.0]нонен-5
ДБУ см. 1,8-Диазабицикло[5.4.0]ундецен-7
Двойной резонанс в ЯМР 1:133
ДДТ 1: 362; 2:448, 467
техника безопасности 2: 399
Дебнера-Миллера синтез хинолинов 2: 198
Дегидратация 1: 334
Дегидрирование 1: 195; 2: 6, 27, 44, 46
Дегидрит, осушитель 1: 36
Дегидробензол 1:477
Дегидробромирование 1: 343
Дегидрогалогенирование 1: 323, 339, 340
Дегидроциклизация 2: 28
Дезинфицирующие средства 1: 302
1-Дезокси-2,3,4,6-тетра-О-ацетил-1-B-циан-
этил)-а-О-глюкопираноза 1: 388
2-Дезокси-О-рибоза 2: 329
3-Дезокси-1,2:5,6-ди-0-изопропилиден-а-0-
рибогексофураноза 1: 263
Дезоксибензоин см. Бензилфенилкетон
Децен-1 1:340,365,377,405
Децин-1 1:342
Декалин (декагидронафталин) 1: 220, 418
реакции 1: 261
физические константы 2: 349
Декалинол-2 1:414; 2: 23, 172
Декалинон-2 2: 23, 172
Декан 2: 349
Деканаль 2: 25
производные 2: 320
Декандикарбоновой-1,10 кислоты диэтило-
вый эфир 2: 278
Декановая кислота 2: 28, 183
производные 2: 331
Декановой кислоты этиловый эфир 2: 71, 192
Деканол-1 1: 376; 2: 25, 28, 192
производные 2: 343
Деканол-2 2: 367
Деканон-2 2:405
Декантация 1: 46
Декарбоксилирование 2: 86, 125
Декарбонилирование 2: 143, 147
Делительная воронка 1: 80, 162
Демьянова перегруппировка 2: 291
Десса—Мартина периодинан 2: 27
Детектор по теплопроводности 1:97
Детозилирование 1: 340, 342
Дефлегматор 1: 72
Деформационные колебания в ИК 1: 117
З-Децил-1,2-диметоксибензол C-децилверат-
рол) 1:482
Децилбромид 1:284,316,340,482
2,4-Ди-/и/>ет-бутилфенол 1: 453
2,6-Ди-т/>ею-бутил-4-метилфенол 1:453
1,Г-Ди(дифенилфосфино)ферроцен (dppf) 1:
493
1,5-Диазабицикло[4.3.0]нон-5-ен (ДБН) 1:
328,339
1,8-Диазабицикло[5.4.0]ундецен-7 (ДБУ) 1:
328, 339; 2:98
Диазоалканы 1: 397; 2: 239, 256, 257
техника безопасности 2: 238
Диазоаминобензол 2: 251
Диазоангидрид 2: 248
а-Диазодикарбонильные соединения 2: 257
а-Диазокарбоновых кислот эфиры 2: 239
436
Предметный указатель
а-Диазокетоны 2: 239, 257
получение из аминокетонов 2: 239
— из хлорангидридов кислот 2: 232, 260
— превращения до эфиров карбоновых кис-
кислот 2: 277
разложение 1: 349
химические свойства 2: 260, 277
Диазомалоновой кислоты эфиры 2: 239
Диазометан 2: 256
реакции с карбоновыми кислотами и фенола-
фенолами 2: 232, 258
техника безопасности 2: 400
фотолиз 1: 349
Диазометилгептадецилкетон 2: 260, 278
Диазометил-«-метоксифенилкетон 2: 260, 278
Диазометилфенилкетон 2: 260, 278
Диазометил-а-нафтилкетон 2: 260, 278
Диазометилпентадецилкетон 2: 278
Диазония соли 1:119; 2: 238, 240, 244, 268
для арилирования 1:486, 489
получение 2: 232, 240, 245
химические свойства 2:244-246
Диазосоединения алифатические 1: 392; 2:256,
267
реакции 1:395, 464; 2: 257, 259
Диазотирование 1: 272; 2: 240
Диазоуксусной кислоты эфиры 1: 395; 2: 239,
257
М,М-Диалкил-4-нитрозоанилин 1:474
М,М-Диалкил-л-фенилидендиамин 2: 35
а-Диалкиламинометилкетоны 2:130
Диалкилбораны 1: 374; 2:171
Диалкилдихлорсиланы 1: 319
Диалкилкупраты 2: 166
Диалкиловые эфиры 1: 288
Диалкилсульфаты 1: 360
гидролиз 1: 282, 290, 293
реакции с аммиаком 1: 299
— с карбоновыми кислотами 1: 298
— с гидросульфидом натрия 1: 305
Диалкилсульфиды см. Сульфиды
Диалкилсульфоны 2: 262
Диалкилфталаты, неподвижная фаза в 1: 99
Диаллилацеталь 2: 289
1,4-Диаминобутан 2: 284
2,4-Диаминотолуол 2: 235, 250
производные 2: 311
техника безопасности 2:400
1,2- (или 1,4)-Диамины, ароматические 2: 255
Диан см. Бисфенол А
о-Дианизидин 1: 89
Диарилдихлорсиланы 1: 319
Диарилсульфиды см. Сульфиды
Диарилы 1:485,488; 2: 248
Диастереомеры 1: 220, 223
Диастереоселективный синтез см. Стереосе-
лективный синтез
Диацетоновый спирт см. 4-Гидрокси-4-метил-
пентанон-2
М,1ч[-Дибензилаланина бензиловый эфир 1:
301
Дибензиламин, производные 2: 311
2-(ТЧ,Ы-Дибензиламино)пропаналь 2: 26
2-(М,М-Дибензиламино)пропанол 2: 26, 172
Дибензилиденацетон 1: 414, 493; 2: 118
Дибензиловый эфир, ИК-спектр 2: 339
Дибензилсульфид 1: 306
Дибензоил 2: 26, 320
Дибензоилметан 2:144
Дибензоилпероксид см. Бензоилпероксид
Дибензоилуксусной кислоты этиловый эфир
2:153
Диборан 1: 374, 375
1,2-Дибромалканы 1: 365
Дибромбензол 1:446
производные 2: 341
2,6-Дибромбензохинон-1,4 2: 327
1,4-Дибромбутан 1:284,290,316
1,2-Дибромгексан 1:342,365
1,2-Дибромгептан 1: 365
1,2-Дибромдекан 1:342,365
1,2-Дибромдодекан 1:342,365
Дибромизоциануровая кислота 1: 444, 447
а,р-Дибромкоричной кислоты этиловый эфир
1:343
а,у-Диброммасляная кислота 2: 219
2,6-Дибром-4-нитроанилин 1:447; 2: 253
2-B,6-Дибром-4-нитрофенилазо)диэтилами-
ноацетанилид 2: 253
1,2-Дибромоктан 1: 342, 365
4,5-Дибромпентин-1 1:364
1,3-Дибромпропан 1: 284, 305, 316, 386; 2: 341
1,2-Дибромпропан 2: 341
2,3-Дибромпропаналь, диэтилацеталь 1: 343
1,2-Дибромстильбен 1: 343, 365
1,2-Дибромстирол 1: 365
Дибромтрифенилфосфоран 1:463
1,2-Дибром-1 -фенилэтан 1: 365
2,4-Дибромфенол 1:446
производные 2: 347
1,2-Дибромциклогексан 1: 342, 359, 365
1,2-Дибромэтан 1: 383; 2: 341
реакции 1: 305, 310, 316
техника безопасности 2: 400
Дибромянтарная кислота 1: 343, 359, 365
Дибутиламин, производные 2: 311
1,2-Дибутилбензол 1:490
Дибутиловый эфир, растворитель для реакции
Гриньяра!: 159
Ди-т/кти-бутилпероксид 1: 240; 2: 373
Предметный указатель
437
техника безопасности 2: 373, 400
Дибутилсульфид 1: 306
1,2-Дивинилоксиэтан 1: 381
Дигалогениды
вицинальные 1: 351, 364
геминальные 1: 291, 321
Дигалогенкарбены 1: 351
Дигалогенметан, дегидрогалогенирование 1:
348
Дигалогенуксусная кислота, для получения
карбенов 1: 348
9,10-Дигидроантрацен 2:45
Дигидрокоричная кислота см. З-Фенилпропа-
новая кислота
1,4-Дигидроксиантрахинон 1: 457
9,10-Дигидроксиантрацен 2: 7
2,4-Дигидроксибензальдегид 1: 462
2,4-Дигидроксибензойная кислота 1:470
1,10-Дигидроксидекан2: 192
2,2'-Дигидроксидиэтилсульфид 1: 306
1,2-Дигидроксипропан 1: 371
1,3-Дигидроксипропан 2: 343
вмч-Дигидроксисоединения 1: 271
2,5-Дигидрокситерефталевая кислота 1:470
2,5-Дигидрокситолуол 2: 347
Ь-р-C,4-Дигидроксифенил)-а-аланин 1:407
4,5-Дигидрооксазол 2: 81
4,5-Дигидропиразолинон-5 см. Пиразолоны
4,5-Дигидро-4Н-пиразоло[3,4-ё]пирими-
динон-4 2:210
3,4-Дигидро-2Н-пиран 2: 64, 275
3,4-Дигидро-2Н-пиран-2(илиЗ)-карбальдегид
1:400
1,4-Дигидропиридин 2: 206
1,4-Дигидропиридиндикарбоновой-3,5 кисло-
кислоты эфир 2: 206
Дигидрорезорцин см. Циклогександион-1,3
1,2-Дигидрохинолин2:198
г<ис-9,10-Дигидро-9,10-этаноантраценди-
карбоновой-11,12 кислоты ангидрид 1:
402
Дигликоль см. Диэтиленгликоль
Диглим, техника безопасности 2: 396
Диеновый синтез см. Дильса—Альдера реакция
Диенон-фенольная перегруппировка 2: 270
Диенофилы 1: 398
Диены 2:135
Диизобутен 1: 378
Диизобутилалюминийгидрид 2: 171
Диизобутиламин 2: 311
Диизопентиламин 2: 311
Диизопропиламин 2: 311
Диизопропилкетон 2: 322
Диизопропиловый эфир 1: 289
Диизопропилфторфосфат 1: 305
Диизоцианат 1:435; 2:99,102
Дииодметан 1: 396
а-Дикарбонильные соединения 2: 17, 59, 243
Р-Дикарбонильные соединения
идентификация, качественная реакция 2:148
получение ацилированием карбонильных со-
соединений 2: 151
— гидролизом ацилированных енаминов 2:
219,221
—декарбоксилированием эфиров оксоянтар-
ной кислоты и эфиров 2,4-диоксокарбо-
новых кислот 2: 142,147
— сложноэфирной конденсацией 2:139,143
— сложноэфирным расщеплением ацилирован-
ацилированных р-дикарбонильных соединений 2:151
реакции, алкилирование 212, 223
— ацилирование 2: 104, 151,152
— кето-енольное равновесие 2: 148, 211
— кислотное расщепление 2: 86, 149,151
— Кнёвенагеля 2: 125
— присоединение винилогичных карбониль-
карбонильных соединений 2: 201
— расщепление кетонов 2: 87, 149
— сложноэфирное расщепление 2:149, 151
— с альдегидами 2: 202
— с аминами 2: 58
— с солями даизония 2: 251
— с фенилгидразином 2: 59
— с хлорангидридами карбоновых кислот 2:151
хелатные комплексы 1: 279; 2: 148
рКа значение 2: 106
у-Дикарбонильные соединения 2: 58, 134, 202
Дикарбоновые кислоты 2: 38
растворимость 2: 298
Дикетен 1: 342, 397
1,4-Дикетоны2: 134
1,2-Дикетоны2:17
1,3-Дикетоны см. р-Дикарбонильные соедине-
соединения
Дикмана конденсация 2: 142, 225
Дильса-Альдера реакция 1: 192, 353, 392, 398,
401
Димедон 2: 87, 206
производные 2: 320
3,3-Диметилакриловая кислота 2: 42
2,3-Диметилакриловая кислота 2: 94
2,3-Диметилакролеин 2: 63
Диметиламин 1: 383; 2: 58, 131
производные 2: 311
техника безопасности 2: 400
Диметиламин, гидрохлорид 2: 131
4'-Диметиламиноазобензолкарбоновая-2 кис-
кислота 2: 253
4'-Диметиламиноазобензолсульфоновая-4
кислота 2: 253
438
Предметный указатель
4-Диметиламинобензальдегид 1: 89, 420; 2: 19,
118, 126
идентификация, производные 2: 322
- ИК-спектр 2: 325
-спектр 'НЯМР2: 325
4-Диметиламинобутанон-2 2:131
4-Диметиламинобутанон-2, гидрохлорид 1:
344
Диметиламинокоричная кислота 2: 126
нитрил 2: 118
этиловый эфир 2: 58, 222
4-Диметиламинокоричный альдегид 2: 136
М,1^-Диметиламиномалеиновой кислоты ди-
метиловый эфир 1: 383
4-Диметиламино-3-метилбутанон-2 2: 131
B-Диметиламино-5-метил фенил )дифенилме-
танол 1:483
З-Диметиламино-2-метил-1 -фенилпропа-
нон-1 2: 131
2-Диметиламинометилциклогексанон 2: 131
3-Диметиламино-1-D-метоксифенил)про-
панон-1 2: 131
4-(N,N-Димeтилaминo)пиpидин 2: 72
4-Диметиламинотиобензойной кислоты мор-
фолид2: 19
М,М-Диметиламино-л-толуидин 1: 483
4-Диметиламино-3-фенилбутанон-2 2:131
3-Диметиламино-1-фенилпропанон-1 2: 131
4-Диметиламинофенилтиоцианат 1: 449
1-D-Диметиламинофенил)-4-фенилбутади-
ен-1,3 2: 136
КЫ-Диметиланилин 1:215; 2:34, 316
реакции 1: 339,449, 462, 467, 472; 2: 36, 253
техника безопасности 2:400
Диметиланилин, производные 2: 311
Диметилацеталь 2: 63
2,4-Диметилацетон 1: 458; 2:19
М,М-Диметилбензиламин 2:180
а,<х-Диметилбензилгалогениды 1: 211
а,а-Диметилбензилгидропероксид 1: 259, 261,
370,391; 2:286
2,4- (и 2,5-)Диметилбензилхлорид 1: 316, 465
2,4- (и 2,5-)Диметилбензилцианид 1:316; 2:96
4,4'-Диметилбензоин 2: 133
3,5-Диметилбензойная кислота 2:13
2,4- (и 2,5-)Диметилбензолсульфоновая кис-
кислота, производные 2: 357
2,3-Диметилбутан 2: 349
2,3-Диметилбутандиол-2,3 (пинакол) 2: 190,
273
производные 2: 344
2,3-Диметилбутанол-2 1:336; 2:164
3,3-Диметилбуганол-2 1: 335; 2: 185, 275
3,3-Диметилбутанон-2, этиленацеталь 2: 65
2,3-Диметилбутен-2 1: 335, 336
М,М-Диметилбутиламин 2: 180
4,6-Диметилгептен-1-ол-4 2: 166
2,6-Диметилгептадиен-2,5-он-4 2: 116
Диметилдибензоилметан 2: 194
5,5-Диметилдигидрорезорцин см. Димедон
4,4-Диметил-1,3-диоксан 2:117
2,2-Диметил-4,5-диоксоциклогексанкарбоно-
вой кислоты этиловый эфир 2: 87
Ы,Ы-Диметилдитиокарбаминовая кислота 2:
101
3,3-Диметил-1,2-дифенилциклопропен 2: 194
Диметилнитрозамин, техника безопасности 2:
400
1Ч,М-Диметил-3-нитроанилин 1:435
М,М-Диметил-4-нитрозоанилин 1:472
2,6-Диметил-4-B-нитрофенил)-1,4-дигидро-
пиридиндикарбоновой-3,5 кислоты диэ-
тиловый эфир 2: 207
Диметиловый эфир 1: 203, 272, 289
Диметиловый эфир ацетилендикарбоновой
кислоты 1: 383
3,3-Диметил-1-оксаспиро[3.5]нонанон-2 2:
142
Диметилоксосульфония метилид 1:473
2,2-Диметилпропандиол-1,3 2:121,179
2,2-Диметилпропанол 1: 284
1,1 -Диметилпропилбромид 1: 284
Диметилртуть, энергия связи 1: 240
Диметилсульфат 1: 289; 2: 370
реакции 1: 295, 316; 2: 158, 215, 222
техника безопасности 2: 370,400
Диметилсульфид, значение рКа 1: 203
Диметилсульфит как влагосвязывающий агент
2:63
Диметилсульфоксид 1:473; 2: 253, 371
как окислитель 2: 25, 48
как растворитель 1: 200, 274
техника безопасности 2: 371,400
1Ч,М-Диметилтиоформамид 2:103
2,4-Диметилфенилтиоуксусная кислота, мор-
фолид 2:19
М,М-Диметил-л-толуидин, производные 2:
316
М,М-Диметил-л-фенилидендиамин, окисли-
окислительный потенциал 2:1
2,3-Диметил-1-фенилпиразолинон-5 2:83
2,4- (и 2,5-)Диметилфенилуксусная кислота 2:
19,96
Диметилфенол, производные 2: 347
Диметилформамид
Вильсмейера синтез 1:460
как растворитель 1: 200, 274
очистка и осушение 2: 371
спектр'НЯМР2: 336
техника безопасности 2: 371, 400
Предметный указатель
439
5,5-Диметилциклогександион-1,3 см. Диме-
дон
Диметилциклогексан 1:414; 2: 396
2,2- (и 2,6)Диметилциклогексанон 2: 215
3,3-Диметилциклогексанон 2:166
1,2-Диметилциклопропан 1: 219
2,3-Диметилциклопропанкарбоновой кисло-
кислоты этиловый эфир 1: 395
2,3-Диметокси-5-метил-1,4-бензохинон 2: 34
3,4-Диметоксианилин 2: 281
3,4-Диметоксиацетофенон 1:458; 2:42,182
2,4-Диметоксибензальдегид 1:462
3,4-Диметоксибензалвдегид 1: 295
производные 2: 321
3,4-Диметоксибензамид 2:281
2,4-Диметоксибензилгалогениды, SN-pearaiHH
1:270
Диметоксибензилхлорид 1:316, 465
2,5- (и 3,4-)Диметоксибензилцианид 1: 316; 2:
96
4,4'-Диметоксибензофенон Г. 458
C,4-Диметоксифенил)метилкетон 2: 19
2,5- (и 3,4-)Диметоксифенилуксусная кислота
2:96
1,2-Диметоксиэтан как растворитель 1: 278
Димрота холодильник 1: 11, 162
Динатрийпентацианонитрозилферрат(Ш) 2:
296, 308
2,4-Динитроанилин 2: 315
3,5-Динитробензоилхлорид 2: 94
Динитробензойная кислота 1: 433; 2: 91, 94,
337
как реагент для идентификации 1: 435
производные 2: 331, 337
1,3-Динитробензол 1: 431, 434, 435, 444. 447; 2:
236
техника безопасности 2: 400
2,2'-Динитробифенил 1: 485
2,4-Динитронафтол-1 1:436
2,4-Динитронафтол-1 -сульфоновая-7 кислота
1:433,436
2,4-Динитротолуол 1:433, 435, 447; 2: 235
техника безопасности 2: 400
2,6-Динитротолуол 1:435
2,4-Динитрофенилгидразин 1: 89, 474, 475; 2:
60
как реагент для идентификации 1: 435
2,4-Динитрофенилгидразоны 1: 114; 2: 56, 57,
60
2,4-Динитрофенилсульфиды 1:476
2,4-Динитрофенол 1:474
производные 2: 347
1,4-Диоксан 1:289
азеотроп с водой 2: 371
как растворитель 1: 50, 200; 2: 159
очистка 2: 371
растворимость 2: 298
техника безопасности 2: 371, 400
1,3-Диоксан, производные 2: 116
Диоксетаны 1:391, 397
Диоксид углерода 1: 29
как гетерокумулен 2:154
реакции с ароматическими соединениями 1:
468
— с СН-кислотами 2:154
— с литийорганическими соединенями 2:165
— с соединенями Гриньяра 2: 161, 163
твердый 1:12, 25; 2: 339
Диоксираны 1: 367
2,4-Диоксокарбоновых кислот этиловый эфир
2:143, 147
1,3-Диоксоланы 2: 64
Диоктилфталат см. Фталевой кислоты диокти-
ловый эфир
Диолы-1,2 1: 287, 367, 373; 2: 38, 273
1,3-Диполи 1:353
Диполь-дипольные силы 1: 199
1,3-Диполярное присоединение 1: 391, 397, 398
Диполярофил 1: 397
Дипропиламин, производные 2: 311
Дипропилкетон, производные 2: 324
Дипропилсульфид 1: 306
Диродан 1:448
Дисиамилборан 1: 375, 376; 2: 171
Диспропорционирование радикалов 1: 243
Дистилляция 1: 52,165 см. также Перегонка
азеотропная 1: 75
вакуумная 1: 59
в атмосфере инертного газа 1: 58
простая 1: 56
фракционная 1: 63,165
Дисульфид углерода см. Сероуглерод
Дисульфиды 2: 262
Дитиоацетали 2: 155, 222
Дитиогликоль 1: 310
Дитиокарбаматы 2: 101
Дитиоланы 2: 66, 184
Дитиомалоновая кислота, бисморфолид 2: 19
Дитиоугольная кислота 2:101
Дитиоугольной кислоты О,8-диалкиловый
эфир 1: 345
пиролиз 1: 345
радикальное восстановление 1: 239, 262
Дифенил см. Бифенил
Дифениламин 1:435
производные 2: 311
реакции 2: 37, 45
техника безопасности 2:400
2,2-Дифенилаценафтенон-1 2: 165
Дифенилацетилен Г. 365,485
440
Предметный указатель
Дифенилацетилхлорид 1: 340, 343
а.у-Дифенилацетоуксусной кислоты этиловый
эфир 2:144
1,4-Дифенилбутадиен-1,3 2:136
1,4-Дифенилбутадиин 1:487
Дифенил-2,2'-дикарбальдегид 1: 374
Дифенил-2,2'-дикарбоновая кислота 2: 31, 333
Дифенилкетен 1: 340
Дифенилметан, производные 1: 353
Дифенилметанол 2:185
производные 2: 343
Дифенилметилвиниловый эфир 1: 345
Дифенилметилгалогениды, Бм-реакции 1:
270
З-Дифеншшетилен-6-трифенилметилцикло-
гексадиен-1,4 1:241
1,5-Дифенилпентанон-З 1:414
Дифенилпикрилгидразил (ДФПГ) 1:244
1,3-Дифенилпропанон-1 1:414
1,1 -Дифенилпропен-1 2: 139
1,3-Дифенилтриазин 2: 251
2,6-Дифенил-4-B,4,6-трифенилпириди-
но)фенолят1:113
а,а-Дифенил-2-фурилметанол 1:483
1,4-Дифенилциклогексен-1 при [4+2]-цик-
лоприсоединении 1:400
1,1-Дифенилэтанол-1 1:336; 2:164
1,2-Дифенилэтанон-1 см. Бензилфенилкетон
1,1-Дифенилэтен 1: 336, 394; 2:139
1,2-Дифенилэтиленгликоль2:185
Дифеновая кислота см. Дифенил-2,2'-дикар-
боновая кислота
Дифеновый альдегид см. Дифенил-2,2'-ди-
карбальдегид
2,4-Дихлоранилин, производные 2: 311
2,4-Дихлорбензальдегид 1: 292
2,4-Дихлорбензилидендибромид 1: 257, 292
2,4-Дихлорбензилидендихлорид 1: 292
1,2- (и 1,3)Дихлорбензол 1:489
производные 2: 340
техника безопасности 2: 400
1,4-Дихлорбензол 1:448
производные 2: 341
техника безопасности 2: 400
7,7-Дихлорбицикло[4.1.0]гептан 1: 262, 394
1,4-Дихлорбутан 1: 289, 290
1,4- (и 2,3-, и 2,4-)Дихлорбуген-2 1: 357, 366
1,1-Дихлор-2-гексилциклопропан 1: 394
1,2-Дихлоргидрин см. 2,3-Дихлорпропанол-1
1,1-Дихлор-2,2-диметилциклопропан 1: 394
Дихлордифенилметан 1: 452
1,1-Дихлордифенилциклопропан 1: 394
2,3-Дихлор-5,6-дициано-1,4-бензохинон
(ДДХJ:6,45
Дихлоркарбен 1: 281, 348, 394
Дихлорметан см. Метилендихлорид
2,4-Дихлор-2-метилбутан 1: 340
1,1 -Дихлор-2-метил-2-фенилциклопропан 1:
262, 395
1,4- (и 1,5-)Дихлорнафталин, производные 2:
341
Дихлоро[1,2-бис(дифенилфосфино)про-
пан]никель(П) 1:489
Дихлоро[ 1,2-бис(дифенилфосфино)этан]ни-
кель(Н) 1:489
1,3-Дихлорпропан 1: 316
2,2-Дихлорпропан 2: 341
2,3-Дихлорпропанол-1 1: 366
2,4-Дихлортолуол 1: 257; 2: 250
Дихлоруксусная кислота 2: 50
производные 2: 331
1,1-Дихлор-2-фенилциклопропан 1: 394
2,4-Дихлорфеноксиуксусная кислота 1: 298,
340, 448; 2: 219
2,4-Дихлорфенол 1: 340, 347,448
1,1-Дихлорциклопропан 1: 394
1,2-Дихлорэтан 1: 340, 366; 2: 341, 373
техника безопасности 2: 373,400
1,1 -Дихлорэтилен см. Винилидендихлорид
1,2-Дихлорэтилен 2: 341
Дихромат 2: 22
Дицианодисульфан см. Диродан
4,4'-Дицианодифенил 1:485
Дицианоуксусной кислоты анилид 2:156
Дициклогексиламин 1: 301; 2:180, 187
техника безопасности 2:400
Дициклогексилкарбодиимид 2: 72, 82
Дициклогексилэтиламин 1: 301, 329, 339
Дициклопентадиен 1:402
Диэлектрическая проницаемость 1: 200, 282
Диэтаноламин 1: 370
Диэтиламин 1: 319; 2: 131, 180, 198
техника безопасности 2:400
З-Диэтиламиноацетанилид 2: 253
4-Диэтиламинобензальдегид 1:462
4-Диэтиламинобутанон-2, гидрохлорид 1: 344
1-Диэтиламиногептин-2 2:131
З-Диэтиламинопропионитрил 2: 198
2-D-Диэтиламинофенилазо)-3-метилбензти-
азолия тетрафторборат 2: 36
Ы^-Диэтиланилин 1: 339, 384; 2: 36, 253
производные 2: 316
техника безопасности 2:401
Диэтилацеталь 2:63
Диэтиленгликоль 1: 24, 370, 371, 373
производные 2: 343
техника безопасности 2: 373,401
Диэтиленгликоля динитрат 1:287
Диэтиленгликоля моноалкиловые эфиры 1:371
Диэтилкарбонат 2:147
Предметный указатель
441
Диэтилкетон 2: 87,109, 289
производные 2: 322
техника безопасности 2:401
Диэтилкетона циангидрин 2:109
Диэтилмалоновая кислота 2: 86
Диэтилмалоновой кислоты диэтиловый эфир
2:86,213
ДиэтилD-нитрофенил)тиофосфат 1: 305
Диэтиловые эфиры алкилфосфоновых кислот
1:304
Диэтиловый эфир B-ацетоксиэтил)малоно-
вой кислоты 1: 386
Диэтиловый эфир 1: 25, 289, 325, 349, 362; 2:
374 см. также Эфир
как растворитель 1: 85, 116, 200, 276
какэкстрагент1: 81
температура кипения, зависимость от давле-
давления 1: 54
техника безопасности 2: 374, 401
Диэтиловый эфир ацетилендикарбоновой
кислоты 1:383, 401
Диэтилсульфат 1: 294, 316
техника безопасности 2: 401
Диэтилсульфид 1: 306
Диэтилфосфит, реакции с алкилгалогенидами
1:305; 2: 135
М,М-Диэтилфурфуриламин 2:180
ДМАП см. 4-(М,>)-Диметиламино)пиридин
Додеканол-1 2: 192
Додекантиол-1 1:310
Додеканон-2 1:405
Додеканнитрил 1: 316; 2:192
Додекановой кислоты этиловый эфир 2: 192
Додецен-1 1:340,365,405
Додециламин 2:192
Додецилбромид 1: 284, 316, 340
Додециловый спирт 1: 54
Додецилтиол 1: 310
Додецилхлорид 1: 310, 316
Додецин-1 1: 342
Доза, токсичная, летальная 2: 395
Донор электронной пары 1: 192, 204; см.
также Электронодонорные группы
ДОФА1:407
Драгендорфа реактив 1: 89
Древесное масло китайское 2: 89
Дрожжи как восстановитель 2:181
Душистые вещества 1: 316; 2:14, 66
Дъюара сосуд 1: 26, 31
Е1 см. Элиминирование мономолекулярное
Е2 см. Элиминирование бимолекулярное
Енамины 1: 208; 2: 62, 185, 194
получение 1: 353, 382; 2: 52, 56, 57, 209, 229
— алкилирование 2:219,222
— ацилирование 2 195,219,221,229
— из р-дикарбонильных соединений 2:57,203
реакции, присоединение изоцианатов 2: 221
— присоединение Михаэля 2: 203
— с образованием солей пирилия 2: 209
— с солями диазония 2: 251
— с электрофилами 2: 210
Енол, простые эфиры 1: 277, 337, 353, 379; 2:
64,194,211,230,288
Енол, сложные эфиры 1: 353
Енолы 1:353; 2:143,148,211
идентификация 2: 302
Еноляты2:106,210
Железа оксид как адсорбент 1: 84
Железо(Ш), хлорид
какдегидрирующийагент2:44, 198
как катализатор 1:456
как реагент-индикатор для гидроксисоедине-
ний 2: 303
комплексы с E-дикарбонильными соедине-
соединениями 2: 148
Железо
для восстановления карбонильных соедине-
соединений 2:189
нитросоединений 2: 234
как катализатор Фриделя—Крафтса 1: 456
Жесткие кислоты и основания см. Кислоты,
Основания
Жидкостная хроматография 1: 86, 92, 94; см.
также Хроматография высокоэффектив-
высокоэффективная (ВЭЖХ) 1: 94, 165
Жирные кислоты 1: 255; 2: 38, 41, 104, 186, 295
Жирных кислот эфиры 1: 255; 2:186
Жиры природные 1: 259; 2: 89
Заместители
второго рода 1:428
первого рода 1:428
константы Гаммета 1: 209
нуклеофугные 1: 272
эффекты 1: 204, 209,269
Заместительная номенклатура 1:179
Замещение ароматическое 1:492
катализируемое металлами 1:479
нуклеофильное 1:472
электрофильное 1:425,492
Замещение у насыщенного атома углерода
бимолекулярное 1:268
мономолекулярное 1: 267
нуклеофильное 1:198, 265, 321
радикальное 1: 245
442
Предметный указатель
Замещения приоритет
в номенклатуре 1: 180
в стереоизомерии 1: 222
Замещения реакции 1: 192
Зандмейера реакция 2: 232, 247, 248, 268
Зарин см. Метилфторфосфоновой кислоты
диизопропиловый эфир
Защитные группы 1: 231, 236
для аминогруппы 1: 301; 2: 82
для гидроксильной группы 1: 296, 319; 2: 64,
276
для карбонильной группы 2: 64
для карбоксильной группы 1: 301, 319; 2: 82
Змеевиковый холодильник 1:11
Зоман см. Метилфторфосфоновой кислоты
пинаконовый эфир
Ибупрофен 1:318
Иванова реагент 2: 160, 164
Идентификация органических соединений 2:
293
получение производных 2: 308
спектроскопическими методами 1: 108, 149;
2:253
хроматографический метод 1: 88
Измеритель скорости потока 1: 20
Изоамилметилкетон 2: 87
Изоамилнитрит 2: 243, 343,401
Изоамиловый спирт 2: 243
Изоборнеол 2: 172, 274, 276
Изобутан 1: 204, 378
Изобутанальдегид 2: 30, 111, 126, 164, 178, 273,
320, 324
Изобутанол см. Изобутиловый спирт
Изобутен 1: 326, 359, 364, 378; 2:10
реакции 1: 373, 394, 452; 2: 9,117
Изобутиламин, производные 2: 311
Изобутилбромид 1: 284; 2: 213, 340
Изобутилвиниловый эфир 1: 381
Изобутилиденмалоновой кислоты диэтило-
выйэфир2:126
Изобутилиодид 2: 340
Изобутилмалоновая кислота 2: 86, 88
Изобутилмалоновой кислоты диэтиловый
эфир 2: 86, 213
Изобутилметилкетон 1: 414; 2: 87, 121, 147
Изобутиловый альдегид 2:179
Изобутиловый спирт 2:185, 396
производные 2: 342
Изобутилхлорид 2: 339
Изобутирилхлорид 2: 94,154
Изовалериановая кислота, производные 2: 331
Изолейцин2:317
Изомасляная кислота 2: 30, 50, 94, 218
производные 2: 331
техника безопасности 2: 401
Изомасляной кислоты сложные эфиры 2: 71,
140, 142, 144
Изониазид2: 13, 78
Изоникотиновая кислота 2: 12, 13, 78, 331
Изонитрилы см. Изоцианиды
Изонитроалканы см. Азотистой кислоты
эфиры
Изонитрозомалоновой кислоты диэтиловый
эфир 2: 236, 243, 244
Изонитрозосоединения 2: 230, 236
Изооктан 1: 378
Изооктен 1: 378, 379
Изопентан 2: 349
Изопентилхлорид, производные 2: 340
Изопентиламин, производные 2: 311
Изопентилбромид, производные 2: 340
Изопентилиодид, производные 2: 340
Изопентилметилкетон см. Изомилметилкетон
Изопентилнитрит 2: 401; см. также Изоамил-
Изоамилнитрит
Изопентиловый спирт см. Изоамиловый спирт
Изопинокамфеол 1: 376
Изопрен 1:340; 2: 114
3-Изопропил-2,6-диоксогептан-3-карбоно-
вой кислоты этиловый эфир 2: 204
Изопропиламин 2: 311
Изопропилбензол см. Кумол
4-Изопропилбензолсульфохлорид 1: 439; 2:
264
Изопропилбромид 1: 284, 303, 450; 2: 217
прозводные 2: 340
Изопропилиденацетон см. 4-Метилпентен-З-
он-2
Изопропилиденглюкофураноза 1: 262
Изопропилиденмалоновой кислоты диэтило-
диэтиловый эфир 2: 166
Изопропилиденмалононитрил 2: 126
Изопропилиденциануксусная кислота 2:126
Изопропилиодид 1: 286, 312
производные 2: 340
Изопропилмагнийхлорид 2:160
Изопропилметилкетон 2: 326
производные 2: 322
Изопропилнитрит 1: 312
Изопропиловый спирт 1: 363
ИК-спектр 2: 343
спектрЯМР'Н2:343
получение 1:293; 2: 238
производные 2: 342
реакции 1:452; 2: 71
техника безопасности 2: 401
4-Изопропилтиофенол 2: 263
Предметный указатель
443
Изопропилтрифенилфосфонийбромид 1: 303
Изопропилхлорид 1: 357, 452
производные 2: 340
Изопулегол см. л-Ментен-8-ол-З
Изосафрол 1: 374
Изотиоцианаты 1: 306; 2: 99,100
ИК-спектр 1:119
как гетерокумулены 2: 154
реакции с аминами 2:101
— с енаминами 2: 221
— с СН-кислотами 2: 155
Изотопный эффект 1:427
Изофитол 1:453
Изофталевая кислота 2: 13
производные 2: 331
Изофталевый альдегид 1: 293
Изохинолин 1:473
производные 2: 316
Изоцианаты 2: 52, 99, 101, 223, 281
ИК-спектр 1:119
как гетерокумулены 1: 395; 2: 154
расщепление по Гофману!: 278
реакции с аммиаком и аминами 2:101
— с водой 2: 101
— с СН-кислотами 2:155
— с енаминами 2: 221
— со спиртами 2: 101
циклоприсоединение 1: 395
Изоцианиды 1:119, 266, 277, 313, 349
идентификация 2: 295
Изоциановая кислота 2:101
Изоэвгенол 1: 373
производные 2: 347
Изоэлектрическая точка 2:109
И К см. Инфракрасная спектроскопия
Илиды2:104, 134, 136
Имидазол 1:425
Имидазолин2:81
Имидоэфиры (иминоэфиры) 2: 52, 98
Имины (основания Шиффа) 2: 215, 223
Ингибиторы 1: 245, 391
Индамины 2: 35
Инданон-1, производные 2: 324
Индантрон (индантреновый синий RS) 1:476
Инден 1: 379
Инден-кумароновые смолы 1: 379
Индол 1:430, 462; 2: 203, 290
производные 1: 362; 2: 311
3-(Индолил-3)пропионитрил 2: 203
Индолилуксусная-3 кислота 2: 291
Индолкарбальдегид-3 1: 362
Индофенол 2: 35
Индукционный эффект 1: 206, 265, 270
Индулины 2: 36
Инсектициды 1: 305, 340, 384, 394, 467
Инфракрасная спектроскопия 1: 100, 108, 110,
116, 117,165; 2: 308
Иод 2: 27, 396
ингибитор радикальных реакций 1: 246
как защитная группа при ароматическом за-
замещении 1:430
присоединение к олефинам 1: 355, 363
реакции с металлоароматическими соедине-
соединениями 1:482
соединения гипервалентные 2: 27, 48
энергия диссоциации связи 1: 251
1 -Иодалканы см. Алкилиодиды
Иоданизол 1:492
Иодбензойная кислота 2: 26, 27
Иодбензол 1: 492; 2: 340
Иодид (ион), в реакциях Sn2 1: 275
Иодирование ароматических соединений 1:
493
1-Иоднафталин 1:487, 492
Йодная кислота 2: 40, 47, 48
4-Иоднитробензол 1: 487
Иодоводород 2: 396
восстановление 1: 282
присоединение к олефинам 1: 355
расщепление простых эфиров 1: 289; 2: 337
техника безопасности 2: 396
энтальпия диссоциации связи 1:240
рА значение 1: 203
2-Иодоктан 1:286, 314
Йодоформ 2:42
Иодоформная проба 2:42, 304
2-Иодпиридинол-З 1: 485
4-Иодтолуол 1: 487
Иодуксусной кислоты этиловый эфир 1:311
Ионное произведение воды 1: 202
Ионные пары 1: 276, 280
Ион-парная экстракция 1: 280
Ионы 1: 277
Искусственное волокно 2: 101
Кадмийорганические соединения 2: 162
Калий 2: 396
Калийорганические соединения 1:480
Каликсарен 1: 463
Калия О-алкилдитиокарбонат 1: 345
Калия /и/>ет-бутилат 1: 330, 480; 2: 214
Калия гексацианоферрат как окислитель 2:
36
Калия гидроксид 2: 396
Калия дихромат 2: 396
Калия иодид, проба на пероксиды 1: 260
Калия ксантогенат 1: 345
Калия перманганат
444
Предметный указатель
гидроксилирование олефинов 1: 373
для обнаружения непредельных соединений
2:300
для окисления алкилзамещенных аромати-
ароматических соединений 2: 12, 352
как окислитель 2:10, 28
как реагент при тонкослойной хроматогра-
хроматографии 1: 89
техника безопасности 2:402
Калия персульфат 1: 391
Калия роданид 1: 308
Калия соли, комплексование с краун-эфира-
ми 1: 279
Калия тиоцианат 1: 308
Калия фторид 2: 402
Калия цианид 2:402
Кальция-алюминия силикат, осушитель 1: 40
Кальция гидрид 1: 36, 88
техника безопасности 2: 402
Кальция карбид 2: 402
Кальция оксид 1: 36
техника безопасности 2:402
Кальция сульфат 1: 36, 88
Кальция хлорид 1: 36,40
техника безопасности 2:402
Камфен 2: 275, 295, 355
Камфоленовый альдегид 2: 275
Камфора 2: 26, 139, 172, 275
производные 2: 324
Канна—Ингольда—Прелога правило 1: 222
Канниццаро реакция 2:132, 168, 177,178, 228
Капиллярные колонки 1: 96
Е-Капролактам 1: 192, 261, 418; 2: 83, 204, 285
техника безопасности 2: 402
Е-Капролактон 2: 286
Капрон 2: 84
Карбазол 1:469; 2:45
Карбазолкарбоновая-1 кислота 1:469
Карбаминовая кислота 2: 100
2-Карбамоил-2-циантиоуксусной кислоты
анилид 2:156
Карбамоилхлориды 2: 52, 99
Карбанионы2: 104,106, 133, 141, 194, 212, 270;
см. также Еноляты
Карбениевые ионы 1: 267, 324, 335, 356, 377,
449; 2: 238, 270, 273
Карбеноиды 1: 393
Карбены 1: 348, 351, 391, 436, 464; 2: 276
Карбиламин (изонитрил) 1: 349
Карбитолы 1: 371
Карбодиимиды 2: 154
Карбоксиметилцеллюлоза 1: 198; 2: 219
Карбонилирования реакция 1:407, 488
Карбонильная группа 2:49
Карбонильные соединения 2: 230, 274; см.
также Альдегиды, Кетоны
алкилирование 2: 211, 229
ацилирование 2: 139, 151, 226
винилоги 2: 196
восстановление 2:169, 189, 227
галогенирование 2: 217, 229
гидрирование, каталитическое 2: 51, 227
енольные формы 2:148, 194
замещение 2: 208
ИК-спектры 1: 119
образование гидрата 2: 51, 62
присоединение альдегидов 2: 202
— амидов карбоновых кислот 2:208
— аминов 2:197,227
— галогеноводородов 2: 201
— СН-кислот2:201
реакция с азотистоводородной кислотой 2:
282
— с СН-кислотами 2:104
— с металлоорганическими соединениями 2:
158
— с нуклеофилами 2: 51, 104
тионирование 2:102
УФ-вид.-спектры 1: 114
этинилирование 2:112
Карбоновые кислоты 2: 13, 217
как растворитель 1: 200
идентификация 1: 299; 2: 330
— ИК-спектр1: 119
— определение эквивалентной массы 2: 331
— растворимость 2: 297
— УФ/вид.-спектр 1:114
получение 2: 9, 37,46, 222
— из бензилгалогенидов 1:466
— из геминальных тригалогенидов 1: 291
— из литийорганических соединений 2: 165
— из соединений Гриньяра!: 161, 163, 339
— окислением алканов 2: 9, 36
алкенов 2: 38
— — алкилзамещенных ароматических
соединений 2: 9, 12
альдегидов 1: 296; 2: 28
первичных спиртов 2: 28
— окислительным расщеплением вторичных
спиртов и кетонов 2:41
— перегруппировкой Вольфа 2: 276
— при галоформной реакции (реакция Эйн-
хорна) 2:41
— при гидролизе амидов карбоновых кислот
2:89
нитрилов 2: 94
эфиров карбоновых кислот 2: 84,
336
— при декарбоксилировании малоновых кис-
кислот 2: 87
Предметный указатель
445
— реакцией Вильгеродта 2:17
— реакцией Кнёвенагеля!: 125
реакции, алкоголю 2: 52,69
— аминолиз 2: 52, 75
— ацидолиз 2:90
— бромирование 2: 218
— винилирование 1: 379
— декарбоксилирование 2: 86
— метилирование диазометаном 2: 258
— с диазометаном 2: 232
— с комплексными гидридами 2:171
— с нуклеофилами 2: 67
— с соединениями Гриньяра 2:160
— хлорирование 2: 217
-Шмидта2: 282
— этерификация 2: 52, 70
Карбоновых кислот азиды 2: 82, 281
Карбоновых кислот амиды
N-бензиламиды 2: 79, 331
получение 2: 75
— аминолизом азидов карбоновых кислот 2:
80
карбоновых кислот 2: 75
хлорангидридов и ангидридов карбоно-
карбоновых кислот 2: 52, 78, 80, 330
эфиров карбоновых кислот 2: 78, 79, 337
— Бекмана перегруппировка 2: 284, 292
— Вольфа перегруппировка 2: 276, 291
— гидролизом нитрилов 2: 89, 95, 97
— из соединений Гриньяра 2: 339
Карбоновых кислот ангидриды
идентификация, ИК-спектры 1: 119
— растворимость 2: 298
— тонкослойная хроматография 1: 89
получение 2: 222
— при ацидолизе ангидридов карбоновых
кислот 2: 52,91
галогенангидридов карбоновых кис-
кислот 2: 52, 92
карбоновых кислот 2: 90
химические свойства, алкоголиз 2: 52, 72
аминолиз 2: 52, 80
ацидолиз 2: 52, 91
взаимодействие с соединениями Гринья-
гидролиз 2: 52, 89
Карбоновых кислот анилиды 2: 75, 331, 339
Карбоновых кислот галогенангидриды
получение из карбоновых кислот 2: 92, 94,
330
— из трихлорметилароматических соедине-
соединений 1:291
хим. свойства, алкоголиз 2: 52, 72
аминолиз 2: 52, 80
ацидолиз 2: 52, 92
гидролиз 2: 52, 89
превращение до диазокетонов 2: 232
до кетенов 1: 340
с комплексными гидридами 2: 171
с соединениями Гриньяра 2: 159,162
Карбоновых кислот гидразиды 2: 52, 78
Карбоновых кислот метиловые эфиры 2: 70,
258
Карбоновых кислот нитрофениловый эфир 1:
299; 2:78
Карбоновых кислот производные
— получение 2: 277
хим. свойства, алкоголиз 2: 69
— — гидролиз 2: 52, 84
с нуклеофилами 2: 67
Карбоновых кислот сложные эфиры 1: 362
активированные 1: 270; 2: 78, 82
— алифатические, а-галогенирование 2:
217
идентификация 2:296
— масс-спектры 1:149
— определение эквивалентной массы 2: 88
— растворимость 2: 298
— тонкослойная хроматография 1: 89, 99
— гидролиз 2: 336
— ИК-спектры 1: 119
— УФ/вид.-спектры 1:114
как растворитель 1: 50
<х,р-ненасащенные, получение 2:167
— реакционная 2: 196
<х,у-ненасыщенные, получение 2: 288
осушение 1:40
получение алкилированием карбоновых кис-
кислот 1: 265, 298, 399, 321
— ацидолизом сложных эфиров 2: 52
— изгалогенидов и ангидридов карбоновых
кислот 2: 52, 72
— из карбоновых кислот 2: 69, 70, 232, 258
— из кетенов и спиртов 2: 101, 277
— из сложных эфиров карбоновых кислот
(переэтерификация) 2: 52, 72
— по Байеру—Виллигеру 2: 286
— перегруппировкой Вольна диазокетонов 2:
276
сродство к адсорбенту 1: 84
химические свойства, алкоголиз 2: 52, 72, 337
аминолиз 2: 52, 78
ацидолиз 2: 52,90
восстановление 1: 262
комплексными гидридами 2:171
по Буво—Блану 2: 191
гидразинолиз 2: 52, 78
гидролиз 2: 52
¦ пиролиз 1: 345
сложноэфирная конденсация 2: 139
446
Предметный указатель
с соединениями Гринъяра 2:161, 163
Карбоновых кислогт фенациловый эфир 1:
299
Карбоновых кислот фениловьгй эфир 1: 119
Карбоновых кислот хлорангидриды 2: 94, 222
винилоги 2:210
химические свойства, алкоголиз 2: 72
аминолиз 2: 80
восстановление до альдегидов 2:167,184
комплексными гидридами 2: 170
до спиртов 2: 190
с диазометаном 2: 277
с СН-кислотами 2:151
с металлоорганическими соединениями
2:161,166
Карвакрол, производные 2: 347
р-Каротин1:113;2:139
Каротиноиды 2:114
Катализаторы 1: 200, 201, 336, 368, 403, 409,
410, 454,456; 2: 184,383
Адамса 1:310
на неметаллической подложке 1: 341
на основе благородных металлов 1:410; 2: 48
Катарометр 1: 97
Катионообменная смола 1:443
Каучук 1:259; 2: 114
Кеталь см. Ацеталь
Кетен 1: 338, 348, 350; 2: 101, 223
взаимодействие с енаминами 2: 220
— с нуклеофилами 2:101
получение 1: 323; 2: 276
— из ацетона 1: 348
циклодимеризация 1: 395
Кетенаминаль 2: 288
Кетенацеталь 2: 288
Кетендиэтилацеталь 1: 343
Кето-енольная таутомерия 2: 107, 148, 211, 269
Кетокислоты см. Оксокарбоновые кислоты
Кетонное расщепление р-дикарбонильных со-
соединений 2: 87, 149
Кетоны
идентификация 2: 302, 317
-ИК-спектр 1:119
— определение количественное 2: 61
— растворимость 2: 297
— тонкослойная хроматография 1: 89, 99
— УФ/вид.-спектр 1:114
а,р-непредельные 1: 392
— получение альдольной реакцией 2:114, 117
из а-ацетоксикетонов 1: 346
из оснований Манниха 1: 344
окислением вторичных спиртов 2: 22
перегруппировкой Лоль$ядиазокетонов
2:276
— реакционная способность 2:196
получение 2: 36, 114,117
— гидролизом азометинов 2: 62
— гидратацией алкинов 1: 362
— из альдегидов 2: 133
— из аминоспиртов реакцией Тиффено 2:
274
— из ароматических соединений ацилирова-
нием по Фриделю-Крафтсу 1: 454,457
— из бисульфитов 2: 66, 277
— из геминальных дигалогенидов 1: 453
— из кетонов с диазометаном 2: 258
— из озонидов при восстановительном рас-
расщеплении 1: 353, 373
— из производных карбоновых кислот и ме-
таллорганическими соединениями 2: 161,
166
— из эпоксидов с кислотами Льюиса 1: 372
— каталитическим дегидрированием вторич-
вторичных спиртов 2: 26
— окислением 2: 46
алканов 2: 8
вторичных спиртов 2: 21, 22
по Оппенауэру!: 168, 173, 176, 227
соединений с активной метиленовой
группой 2; 15
— при ацилоиновой конденсации 2:132
— при расщеплении гликолей 2:40
— расщеплением р-дикарбонильных соеди-
соединений 2: 86, 87
— реакцией Губена-Хеша 1: 459
Стиле 1:488
реакции альдольные 2:114, 117
— алкилирования 2: 210
— восстановления 2: 169, 173, 189
— гидратации 2:62
— гидрирования 2: 184, 185
— Кнёвенагеля!: 125
— Манниха!: 130
— Меервейна—Понндорфа—Верлея 2: 174
— образования ацеталей 2: 62
— окислительного расщепления 2:41
— присоединение, винилоги карбонильных
соединений 2:196
синильной кислоты 2:108
— с аминосоединениями 2: 56
— с диазометаном 2: 274
— с комплексными гидридами 2:169,171
— сложноэфирная конденсация 2:139
— с нуклеофилами 2: 51
— с соединениями Гриньяра 2:161,163, 165
— с эфирами муравьиной кислоты 2: 143
— удлинения цепи 2: 258
— этинилирования 2:112
циклические, расширение цикла 2: 258,
276
Предметный указатель
447
Кинетический контроль реакций 1: 199
Кислород
как ингибитор радикальных реакций 1: 245,
250
как окислитель 1: 259, 263; 2: 9,12, 47
присоединение к олефинам 2: 7
характеристика баллонадля сжатого газа 1: 29
Кислотное расщепление р-дикарбонильных
соединений 2: 86, 149, 151
Кислотно-основные реакции 1: 202
Кислотность 1: 204
СН-2:106, 125, 230
СН-Кислотные соединения
алкилирование 1: 266; 2: 210, 212
ацилирование 2:151, 152
галогенирование 2: 217
присоединение карбонильных соединений 2:
104, 140
реакция Манниха!: 129, 130
— с азотистой кислотой 2: 243
— с винилогами карбонильных соединений 2:
196, 201
— с соединениями Гиньяра 2: 160
— с гетерокумуленами 2:154, 155
тиоцианирование 1: 448
Кислоты 1:202, 209; 2: 106
жесткие и мягкие 1: 204, 214
Клаузиуса—Клапейрона уравнение 1: 53
Клемменсена восстановление 2: 193, 227
Кляйзена конденсация 2: 140, 202
Кляйзена насадка 1: 57, 162
Кляйзена-Тищенко реакция 1: 362; 2: 567, 575
Кляйзена—Шмидта реакция 2: 115
Кнёвенагеля реакция 2: 20, 104, 121, 203, 223
Кнёвенагеля—Дебнера реакция 2: 125
Кнорра синтез пирролов 2:126
Кобальта стеарат 2: 13, 2: 384
Колбы 1:10, 14, 42,47, 55, 67, 162
Колебания молекул 1:111,117
Коллидин, производные 2: 316
Коллодий см. Целлюлоза динитрат
Колонки 1: 67, 68
Кольбе синтез нитрилов 1: 266, 313; 2:97
Кольбе—Шмидта синтез 1:426, 469
71-Комплексы 1: 355, 407
ст- Комплексы 1: 427
Комплексный катализ 1:404
Комплексон см. Этилендиаминтетрауксусная
кислота
Комплексы с переносом заряда 1: 212, 355
Компрессионный вакуумметр 1: 33
Кониин2: 124
Конифериловый спирт 2: 33
Конкурирующие реакции 1:198, 356
Константа взаимодействия 1: 129, 132,140
13С-'Н 1:139
Константа равновесия 1: 193
Константа скорости 1: 195, 197, 201, 209, 246
Конфигурация 1: 221
Конформация 1: 217, 218, 331
Конформер1: 216
Коричной кислоты
амид 1:492
хлорангидрид см. Циннамоилхлорид
этиловый эфир 2: 337
эфиры 2: 122
Коричный альдегид 1: 462; 2: 26, 118, 126, 136,
139,175,204,325
производные 2: 321
Коричный спирт 2: 26,175
производные 2: 343
Коупа вариант реакции Кнёвенагеля 527
Коупа перегруппировка 1: 192; 2: 270, 288,
292
Коупа элиминирование 1: 350
Кофеин 1: 93
Коэффициенты поглощения в УФ 1: ПО,
111,116
Кран-насадка 1: 72
Красители 1: 479; 2: 13, 219, 236, 247, 255
Кратные связи 1:414
18-Краун-6 1:279
Краун-эфиры 1: 276, 279, 311
Крахмал 1:84; 2: 65
ж-Крезилацетат 1: 257
Крезилметиловый эфир 1: 295
Крезол 1: 295; 2: 73, 245
производные 2: 347
техника безопасности 2: 402
Крекинг-процесс 1: 241
Кремния тетрахлорид 2: 160
Кривая кипения 1: 59
Кривая плавления 1: 101
Кривые равновесия 1: 64
Криостат 1: 26
Криптанды 1: 276
Кристаллизация 1:45, 52
Кристаллический фиолетовый 1:468
Кросс-сочетание, катализируемое переходны-
переходными металлами 1:486
Кротиловый спирт 2: 175
Кретоновая кислота 2:9, 335
производные 2: 331
Кротоновой кислоты метиловый эфир 2: 71
Кротоновой кислоты пиперидин 2:120
Кротоновой кислоты этиловый эфир 2: 71
Кротоновый альдегид 1: 336, 338, 418; 2: 121,
185, 198
производные 2: 320
реакции 2: 63, 126, 175
448
Предметный указатель
техника безопасности 2: 402
Кротоновый альдегид, диэтилацеталь 2: 63
Ксантогенаты 2:101
Ксантогеновой кислоты алкиловые эфиры см.
Дитиоугольной кислоты О,5-диалкило-
вый эфир
Ксантон, производные 2: 323
2,4-Ксилидин (аминодиметилбензол) 2: 253
Ксилоза 2: 329
Ксилол 1: 450,453; 2: 376, 396, 416
производные 2: 352
реакции 1: 252, 257, 414,458,465; 2:12,13, 15
техника безопасности 2: 402
Кулона силы 1:199
Кумаринкарбоновой-3 кислоты этиловый
эфир 2:126
Кумилгалогениды 1:211
Кумилгидропероксид см. а,а-Диметилбензил-
гидропероксид
Кумол 1: 259, 261,439,452,453
производные 2: 352
техника безопасности 2:402
Купраты 2:166
Курциуса расщепление 2: 278, 281, 291
Лаки 1: 378; 2: 89, 76
Лактамы 2: 285
Лактоза 2: 29, 66, 327
Лактоны 1: 89; 2: 286, 298
Ламберта-Беразакон 1: ПО, 124
Лассеня проба 2: 296
Латекс 2: 391
Лауриловый спирт 2: 192
производные 2: 343
Лауриновая кислота, производные 2: 331
Лауриновой кислоты этиловый эфир 2:71
Лауссона реактив 2: 102
ЛД см. Летальная доза
ЛДА см. Литийдиизопропиламид
Левулиновая кислота, производные 2: 331
Лейкарта—Валлаха реакция 2:168, 179, 227
Лейцин 1:302; 2: 318
Летальная доза 2: 395
Летучесть 1: 53
Либиха холодильник 1: 11,162
Лигнин 2: 33
Лигроин 2: 396
Лизин2:111,317
а-О-Ликсоза2:329
Лимонен 2: 355
Лимонная кислота 2: 219
производные 2: 331
Лимонной кислоты цикл 2:104, 149
Линалоол2:114
Линдемана—Адамса правило 1: 137
Линдлара катализатор 1:411
Линейная комбинация атомных орбиталей см.
ЛКАО
Линейное отношение свободных энергий 1:
196,210
Линолевая кислота 2:89
Линоленовая кислота 2: 89
Литий 2: 396
Литийалюминийгидрид 2:169, 362
как восстаноситель 2:169,172
техника безопасности 2: 362/402
Литийдиизопропиламид (ЛДА) 2:115,121, 215
Литийорганические соединения 1: 479, 484; 2:
165, 226; см. также Органические соеди-
соединения щелочных металлов
Лития купраты 1:485; 2:166
ЛКАО 1:213
Лоссена кислотная перегруппировка 2: 278,
292
Лукаса реактив 2: 304, 381
2,6-Лутидин, производные 2: 316
Льняное масло 1:259; 2:89
Льюиса кислоты 1: 192, 204
катализаторы реакции Дильса—Альдера 1: 401
— SN-реакций 1: 274
электрофильные реагенты 1: 355
Льюиса основания 1: 192, 204
Магний
восстановление карбонильных соединений 2:
190
реакция с галогенсодержащими соединения-
соединениями 1:478; 2: 159,168
техника безопасности 2:402
Магнийорганические соединения см. Гриньяра
соединения
Магния диалкил 1:159
Магния перхлорат как осушитель 1:41
Магния сульфат как осушитель 1: 36, 41
Магнитная мешалка 1: 17,163
Мак-Лафферти перегруппировка 1: 147
Мак-Мури сочетание 2: 169,194
Максимальная концентрация вредных ве-
веществ на рабочем месте см. ПДК
Малеиновая кислота 1: 102, 219
ИК-спектр 2: 335
производные 2: 331
реакции 1: 359,414
техника безопасности 2:402
Малеиновой кислоты диэтиловый эфир 1: 386;
2:71
Предметный указатель
449
Малеиновый ангидрид 2: 10, 39, 76, 90
ИК-спектр 2: 335
какдиенофил 1: 398
реакции 1:365,402, 459
техника безопасности 2: 403
Малоновая кислота 1: 318, 348; 2:86,97,125,126
производные 2: 85-87, 331
Малоновой кислоты диметиловый эфир 2:156
Малоновой кислоты диэтиловый эфир 2:106
получение из хлоруксусной кислоты 2: 219
реакции 1: 386; 2:126, 148, 154, 204, 213, 219
Малоновой кислоты монометиловый эфир 2:
86
Малоновой кислоты моноэтиловый эфир 2: 86
Малоновой кислоты сложные эфиры 2:148
алкилированные 2: 85, 147, 213, 217
реакция Кнёвенагеля 2: 125
— с мезитилоксидом 2: 202
Малоновый альдегид 2:106,157,158
Малононитрил 2:106
реакции 2: 20, 126,148, 156
техника безопасности 2: 403
р-Мальтоза 2: 329
Маннит, производные 2: 345
Манниха основания 1: 344; 2: 202
Манниха реакция 1:426; 2: 104, 129, 130, 225
Манноза 2: 60, 329
Марганца соединения для окисления 2: 47
Маргариновой кислоты этиловый эфир 2: 278
Марковникова правило 1: 357, 385
Марциуса желтый см. 2,4-Динитронафтол-1
Масла 1:259,418, 493; 2: 89
Масс-спектрометрия 1:141, 143, 144,146
Меди ацетиленид 1:485
МедиA) соли 1:485, 486
Меди(И) соли 2: 28, 303
Меди сульфат 1: 36
техника безопасности 2:403
МедиA) фенилацетиленид 1:485
МедиA) цианид 1:485
Медь
как катализатор дегидрирования 2: 26
Ульмана реакция 1:485
Медьорганические соединения 1: 485; 2: 166,
226
Меди-хрома оксид как катализатор дегидри-
дегидрирования 2: 27
Меервейна—Понндорфа—Верлея восстановле-
восстановление 2: 168, 174
Межфазные катализаторы 1: 298, 338, 343
Межфазный катализ 1: 201, 290, 295, 298, 311,
320, 394; 2: 12, 28, 38,120,124, 135, 216
Мезитилоксид см. 4-Метилпентен-3-он-2
Мезитилен 2: 116, 251
реакции 1:446, 465; 2:13
Мезитилмагнийбромид 2: 141
Мезоксалевая кислота 2:62
Мезомерия 1: 207, 270,427
Меланин 2: 34
л-Ментан 2: 349
я-Ментен-2 1: 332, 334, 342, 345
л-Ментен-3 1: 334, 345
я-Ментен-1-он-З 2:87, 185
и-Мент-3-илтозилат 1: 334, 342
Ментол 1: 345; 2: 175, 172, 185
производные 2: 343
реакции 2: 23, 73, 264
Ментон 2: 23
реакции 2:175, 172
Меркаптали см. Дитиоацетали
Меркаптаны см. Тиолы
1-Меркапто-2-финилэтан 1: 310
2-Меркаптобензимидазол 2: 83
2-Меркаптобензойная кислота 2: 356
2-Меркаптобутан 1: 310
Меркурирование ароматических соединений
1:426
Меры защиты от опасных веществ 1: 159, 237;
2:396
Метакриловая кислота 2: 10
Метакриловой кислоты амид 1: 337
Метакриловой кислоты метиловый эфир 1:
338, 391; 2: 10, 201
техника безопасности 2:403
Метакрилонитрил 2:10
Металлиловый спирт 1:492
Металлилхлорид 1: 364
Металлирование ароматических соединений
1:426,479
opmo-Металлирование направленное (DoM) 1:
481
Металлокомплексный катализ 1:403,423,486,489
Металлоорганические соединения 1: 386, 479,
483, 484; 2: 158, 226
Металлохелаты р-дикарбонильных соедине-
соединений 1: 279
Метан 1: 203, 240; 2: 6, 106
реакции 2:9, 255
Метанол 2: 6
азеотроп с тетрахлорметаном 1: 75
величина р/С 1:203
идентификация, производные 2: 342
как растворитель 1: 85, 116
осушение 2: 376
получение 2: 185
— карбонилированием 1: 407
реакции 1:296; 2: 71, 264
техника безопасности 2: 376,403
энергия диссоциации связи 1: 272
Метантиол 1: 203; 2: 200
450
Предметный указатель
производные 2: 356
техника безопасности 2:403
Метастабильные ионы в масс-спектроскопии
1:147
Метатезис 1:409, 410
4-Метил-4-азагептандинитрил 2: 197
4-Метил-4-азагептановой кислоты диэтило-
выйэфир2:144, 197
Метиламин 1: 203; 2: 132
производные 2: 311
реакции 1: 382; 2: 58, 131, 187, 197
техника безопасности 2:403
Метиламина гидрохлорид 2: 281
N-Метиламиноацетонитрил 2: 110
л-Метиламинобензалвдегид 1: 461
р-Метиламинокротоновой кислоты этиловый
эфир 2: 58
3-Метиламинопропионитрил2:197
З-Метиламинопропионовой кислоты этило-
этиловый эфир 2: 197
2-Метиламино-1-фенилпропан 2:187
N-Метиламинофумаровой кислоты димети-
ловый эфир 2: 207
N-Метиламинофумаровой кислоты диэтило-
выйэфир 1: 383
Метиламины 1:119; 2:186
N-Метиланилин 2: 75
производные 2: 311
техника безопасности 2: 403
Метилароматические соединения
бромирование 1: 256, 259
окисление 2:10,12, 15
хлорирование 1: 251, 253
4-Метилацетанилид2: 12
а-Метилацетоуксусной кислоты этиловый
эфир см. 2-Метил-З-оксобутановой кис-
кислоты этиловый эфир
4-Метилацетофенон 1:458; 2:19, 183
производные 2: 324
4-Метилбензальдегид 2: 15, 178
2-М.етилбензилбромид 1: 257, 308
4-Метилбензилбромид 1: 308
4-Метилбензшювыйспирт2: 178
Метилбензилтиоцианат 1: 308
2-Метилбензилхлорид 1: 252, 308
4-Метилбензилхлорид 1: 308, 465
3-Метилбензо[е]индолкарбоновой-2 кислоты
этиловый эфир 2: 290
3-МетилбензоИиндолкарбоновой-2 кислоты
этиловый эфир 2: 290
4-Метилбензойная кислота 2: 94
2-Метилбензоксазол 2:158
4-Метилбензонитрил 2: 250
2-Метилбензотиазол 2:15, 158
Ы'-C-Метилбензотиазолиден-2)-C-метил-1-
фенилпиразолдион-4,5)гидразон-4 2:
37
Ы'-C-Метилбензотиазолиден-2)нафтохинон-
1,2-моногидразон-1 2:37
Ы-Метилбензотиазолона-2 гидразон 2: 36
Метилбензо-1,4-хинон 2: 327
Ы-МетилбисB,4-ксилилиминометил)амин 2:
98
2-Метил-1,1 -бис(«-толил)пропандиол-1,2 2:
273
Метилбромид 1: 257
идентификация, производные 2: 340
— масс-спектр 1: 144
— 'Н-химический сдвиг 1: 130
реакции 1: 303
энергия диссоциации связи 1: 272
2-Метилбутанол-2 1: 336; 2: 164, 329
производные 2: 342
техника безопасности 2:403
З-Метилбутантиол-1 2:356
2-Метилбутен-1 1:329,336
2-Метилбутен-2 1: 364
получение 1: 329, 336
химические свойства 1: 377
З-Метилбутен-1 1: 336
З-Метилбутен-З-он-2 1:336
Метил-/и/>е/я-бутиловый эфир 1: 289, 362
2-Метилбутин-3-ол-2 2: 114
Метилвинилкетон 2: 196
идентификация 2: 322
получение 1: 344
реакции 1: 337; 2: 198, 204, 203, 204
— галоформная 2:41
— димеризация 1: 399
— как диенофил 1: 399
Метилвиниловый эфир 1: 138
3-Метилгексен-5-он-2 2: 289
4-Метилгептанон-З 2:216
4-Метилгептен-6-он-2 2: 289
1-Метилгептилиодид 1: 286, 312
1-Метилгептилнитрит 1: 312
Метилгидросульфат 1: 287
Метилглиоксаль 2: 179
3-Метил-4-диметиламинобутанон-2 2:131
2-Метил-З-диметиламинопропиофенон 2:131
3-Метил-2-D-диметиламинофенилазо)бензо-
тиазолия тетрафторборат 2: 36
3-Метил-4-B,4-диметилфенилазо)-1-фенил-
А2-пиразолинон-5 2: 253
N-Метилдициклогексиламин 2: 180
Метилен 1: 349, 350
и,я'-Метиленбис(фенилизоцианат) 1: 466; 2:
100, 102
2-Метиленборнан 2:139
Метилендианилин 1:466; 2:100
Предметный указатель
451
Метиленбромид 2: 341
Метилениодид 2: 341
Метиленхлорид 1: 255; 2: 6
идентификация, физические константы 2:
341,373
как растворитель 1:85,116
как экстрагент 1:81
очистка и осушение 2: 373
техника безопасности 2: 373, 403
Метиленовые группы 1: 90; 2: 9, 15
Метилентрифенилфосфоран 2: 137
Метиленциклогексан 1: 346; 2: 138
Метилиодид 1: 272, 275, 287, 296, 303, 305
производные 2: 340
техника безопасности 2: 403
•Н-химический сдвиг 1: 130
Метилирование 1: 343
Метилкетон 1: 89; 2: 41
4-Метилкоричной кислоты амид 1:492
Метилмалоновой кислоты диэтиловый эфир
2:147
Метил-а-маннопиранозид 2:40
Метил-а-маннофуранозид 2:41
N-Метилморфолин-М-оксид 1: 373; 2: 21, 27
N-Метил морфолин 2: 316
1-Метилнафталин 1: 259, 428
производные 2: 351
2-Метилнафталин 1: 259, 446; 2:15, 32
производные 2: 351
Метил-а-нафтилкетон 1:458; 2: 19, 42, 284
Метил-C-нафтилкетон 1: 458; 2: 19, 42
производные 2: 324
2-Метилнафто-1,4-хинон 2: 32
М-Метил-М-нитрозотолуолсульфамид 2: 242
N-Метил-М-нитрозокарбамид 2: 241, 256
Метиловый красный 2: 253
Метиловый оранжевый 2: 253
Метиловый синий 2: 35
Метиловый спирт см. Метанол
Метиловый эфир (сложный) 1: 119; 2: 258
2-Метил-З-оксобутановой кислоты этиловый
эфир 2: 222, 289
5-Метил-З-оксогексановой кислоты этиловый
эфир 2:147
2-Метил-З-оксопентановой кислоты этило-
этиловый эфир 2: 87, 144
4-Метил-З-оксоггентановой кислоты этило-
этиловый эфир 2: 151
1 -Метил-4-оксопиперидонкарбоновой-З кис-
кислоты этиловый эфир 1: 185; 2: 144
З-Метил-2-оксоянтарной кислоты диэтило-
диэтиловый эфир 2: 144, 147
Ю-Метил-Д'-'-октагидронафталиндион-г^г: 120
1|3(или 3)-Метил-бр-пропионилоксиметил-
2,7-диоксабицикло[3.2.0]гептен-3 1: 397
4-Метилпентановая кислота 2: 88, 218
2-Метилпентанол-З 1: 376; 2: 164
4-Метилпентанол-2 1: 376; 2: 121
4-Метилпентен-2 1: 376
4-Метилпентен-3-ол-2 2: 175
4-Метилпентен-3-он-2 1:336; 2: 116
производные 2: 322
реакции 1:414; 2:175, 202
техника безопасности 2: 403
4-Метилпентен-3-он-2-он, этиленацеталь 2:
65
З-Метилпент-1-ин-З-ол 1:362; 2: 114
Метилпентилкетон 2: 87
техника безопасности 2: 403
Метилпентиловый эфир 1: 296
N-Метилпиперидин 2: 180
Метилпиридин см. Пиколин
1-Метилпиридон-2 1:475
1 -Метилпиридона-2 гидразон 2: 36
N-Метилпирролидон 1:418
2-Метилпропандиол-1,2 1:373; 2: 274
2-Метилпропантиол-1 1:310
производные 2: 356
2-Метилпропанол-2 см. тре/я-Бутанол
2-Метилпропен см. Изобутен
Метилпропилкетон 2: 87
производные 2: 322
техника безопасности 2: 403
5-Метилрезорцин, производные 2: 347
Метилсерная кислота см. Метилгидросульфат
4-Метилстильбен 2: 136
а-Метилстирол 1: 394; 2:139
'Н-ЯМР-спектр 2: 355
Метилсульфат кислый 1: 287
З-Метилтиобутанон-2-О-метилкарбамоилок-
сим 2:62
р-Метилтиопропаналь 2: 111, 200
2D)-Метилтиофенол 2: 263
Метилтозилат см. и-Толуолсульфокислоты ме-
метиловый эфир
4-Метилтолан 1:487
N-Метил-М-триметилсилилформамид 1:
319
Метилтрифенилфосфонийбромид 1: 303; 2:
138
Метилтрифенилфосфонийиодид 1: 303
6-Метилурацил 2: 15
3-Метил-2-D-фениламинофенилазо)бензоти-
озолия тетрафторборат 2: 36
3-Метил-1-фенилбутанол-2 2: 164
Метилфениловый эфир см. Анизол
З-Метил-З-фенилоксиран-2-карбонитрил 2:
124
3-Метил-1-фенилпиразолинон-5 2: 37, 83,
157,253
452
Предметный указатель
2-Метил-З-фенилпропионовый альдегид 1:492
N-Метилформамид 1: 319
N-Метилформанилид 2: 75
Метилфосфоновой кислоты диизопропило-
вый эфир 1: 305
Метилфосфоновой кислоты фторидхолино-
вый эфир 1: 305
Метилфторид 1:130, 272
Метилфторфосфоновой кислоты
диизопропиловый эфир (зарин) 1: 304
Метилфторфосфоновой кислоты пинаконо-
вый эфир (зоман) 1: 304
5-Метилфурфураль, производные 2: 320
Метилхиназолин 2: 15
Метилхинолин 2:15, 158, 198
производные 2: 316
Метилхинон/Метилгидрохинон, электродный
потенциал 2: 7
Метилхлорид 1:130, 255, 272, 285; 2: 6
производные 2: 340
Метилцеллюлоза 1: 255, 298
Метилциклогексан 1: 218, 261, 414; 2: 349
техника безопасности 2:403
2-Метилциклогександион-1,3 2:214
2-Метилциклогексанол 1: 414; 2: 23
техника безопасности 2: 404
3D)-Метилциклогексанол, производные 2:343
2-Метилциклогексанон-1 2:23
производные 2: 324
техника безопасности 2:404
химические свойства 2: 206, 215, 216
3D)-Метилциклогексанон-1 2: 324
1-Метилциклогексен-1 1:347
З-Метилциклогексен-2-он 2:166
1-Метилциклогексилацетат, пиролиз 1: 347
1-Метилциклогексилгидропероксид 1: 261
Метилциклопентенофенантрен 2:44
Метальный радикал 1: 250; 2: 6
Метионин2: ПО, 111,317
Метоиодиды 1: 302; 2: 310
4-Метоксиацетофенон1:458;2:19,42,131,182
производные 2: 324
2-Метоксибензальдегид 2: 61
производные 2: 320
2-Метоксибензальдегидоксим 2: 61
2-Метоксибензгидроксимоилхлорид 1: 254
2-Метоксибензилгалогениды 1: 270
4-Метоксибензилиденацетон 2:118
4-Метоксибензиловыйспирт2: 178
4-Метоксибензилтиоцианат 1: 308
4-Метоксибензилхлорид 1: 308, 316, 466
4-Метоксибензилцианид 1:316; 2:96
4-Метоксибензоилхлорид 2: 94
2-Метоксибензойная кислота, 2: 331
4-Метоксибензойная кислота 1: 295; 2: 94
Метоксибензол см. Анизол
4-Метоксибензолсульфамид 2: 266
4-Метоксибензолсульфохлорид 1: 439; 2: 264,
266
З-Метоксибутанол 1: 338
3-Метоксибутилацетат 1: 338
Метоксигруппа 1: 290
5-Метоксииндолкарбоновой-2 кислоты эти-
этиловый эфир 2: 290
4-Метоксикарбонил-1 -метилпиридиния ио-
дид 1:113
4-Метоксикоричная кислота 2: 126
4-Метоксикоричной кислоты амид 1: 492
5-Метокси-3-метилиндолкарбоновой-2 кис-
кислоты этиловый эфир 2: 290
1B)-Метоксинафталин 1:462
2D)-Метоксинафталин-1-карбальдегид 1:462
4-Метокси-2-нитроанилин 1: 435
техника безопасности 2: 404
5-Метокси-3-пропилиндол-2-карбоновой
кислоты этиловый эфир 2: 290
4-Метоксистильбен 2:136
р-Метоксистирол 1: 338
5-Метокси-2,3-тетраметилениндол2: 190
4-Метокситиобензойной кислоты морфолид
2:19
4-Метокситиофенол 2: 264
B-Метоксифенил)виниловый эфир 1: 381
З-Метоксифенилгидразин 2: 79, 247
4-Метоксифенилгидразоноцианацетамид 2:
253
4-Метоксифенилгидразоноциануксусной
кислоты этиловый эфир 2: 253
3-D-Метоксифенил)оксиран-2-карбонитрил
2:124
4-Метоксифенилтиоуксусной кислоты мор-
морфолид 2: 19
4-Метоксифенилуксусная кислота 2: 19, 96
4-Метоксифенилуксусной кислоты этиловый
эфир 2: 278
4-Метоксициннамонитрил 2:118
Метотозилаты 1: 302; 2: 310
Метриол2:179
Мешалок типы 1: 17
Миндальная кислота 2:110
производные 2: 331
Миристиновая кислота, производные 2: 331
Миристиновой кислоты этиловый эфир 2: 71,
192
Михаэлиса—Арбузова реакция 1: 303, 305; 2:135
Михаэля присоединение 2: 133, 201, 203,
229
Михлера кетон 1:468; 2:139
техника безопасности 2: 404
Мииунобу реакция 1:287, 321
Предметный указатель
453
Молекулярная масса 1: 144
Молекулярная рефракция 1: 107
Молекулярность 1:197, 267
Молекулярные ионы в масс-спектрометрии 1:
141
Молекулярные колебания 1:86,86
Молекулярные орбитали (МО) 1: 213 см.
также ВЗМО, НСМО
Молекулярный пик в масс-спектрометрии 1:
144
Молочная кислота 2: 179
идентификация, производные 2: 331
получение 2: ПО, 112
Молочной кислоты этиловый эфир 2: 71
Молочнокислое брожение 2:179
Монооксид углерода 1: 29, 119, 407,488
техника безопасности 2: 408
Моноперфталевая кислота 1: 367, 372
Моносахариды 2: 66, 328
Монохроматор 1:108
Морфолин 2: 58
Вильгеродта—Киндлера реакция 2: 18
производные 2: 311
техника безопасности 2:404
Морфолина гидрохлорид 2:131
Морфолин-Ы-оксид 2: 27
4-Морфолинобутанон-2 2: 131
1-Морфолиноциклогексен-1 2: 58, 221, 222
2-Морфолиноциклогексен-1 -карбанилид 2:
222
2-Морфолиноциклогексен-1-тиокарбани-
лид-1 2:222
1-Морфолиноциклопентен-1 2:58,221
Мочевина 2: 99
получение 2: 52, 101, 155,317
реакции 2: 75, 242
Моющие средства 1:362,443; 2: 89, ПО, 186
получение 1: 261; 2: 10, 38
МС см. Масс-спектрометрия
МТБЭ см. Метил-/я/?ет-бутиловый эфир
Мукайамы восстановительная конденсация 1:
288
Муравьиная кислота 1: 75; 2: 40, 50
производные 2: 331
Муравьиной кислоты этиловый эфир 2: 143,
144,396
Мыла 2: 89
Мягкие кислоты см. Кислоты
Мягкие основания см. Основания
Найлон 1:14, 418; 2: 84; см. также Перлон
Напряжение цикла 1: 212
Напряженность поля при ЯМР 1:127
Насадочная колонка 1:67
Натра едкого раствор см. Натрия гидроксид
Натрий 2: 296, 377
как осушитель 1: 37,40
суспензии, получение 2:146
техника безопасности 2: 377, 404
Натрия N-алкилдитиокарбамат 2: 101
Натрия 0-алкилдитиокарбонат 2:101
Натрия азид 2: 283, 404
Натрия алкоголяты 2: 377
Натрия амальгама 2: 377
Натрия амид 1: 477
химические свойства, агент сложноэфирной
конденсации 2: 141
аминирование пиридином 1:474
инициатор для анионной полимериза-
полимеризации 1: 378
уничтожение отходов 2: 377
Натрия борогидрид 2: 169, 173
Натрия гидрид 1: 144
агент сложноэфирной конденсации 2: 141
техника безопасности 2: 404
Натрия гидроксид 2: 404
Натрия гидросульфид 1: 305
Натрия гипоиодит 2: 304
Натрия гипохлорит 2: 24
техника безопасности 2:404
Натрия дихромат 2:404
Натрия дитионит, восстановитель 2: 234
техника безопасности 2:404
Натрия р-нафталинсульфонат 1:478
Натрия нитропруссид 1: 89; 2: 296, 308
Натрия перхлорат 2: 404
Натрия сульфат 1: 36, 37, 40
Натрия сульфид 1: 305, 306; 2: 234
техника безопасности 2:404
Натрия сульфит 2: 246
Натрия цианид 2: 390
синтез нитрилов по Кольбе 1: 313
— Штреккера 2: 111
техника безопасности 2: 390,404
Натрия цианоборогидрид 2: 173
Натронная известь 1: 36, 40
Натуральный каучук 2: 114
Нафталин 1:418,425
ацилирование по Фриделю-Крафтсу 1:458
производные 2: 351
окисление 2:30, 39
окислительный потенциал 2: 6
реакции 1:434, 458; 2: 32
техника безопасности 2:404
УФ/вид.-спектр 1: 115
Нафталинкарбальдегид 2: 321
Нафталинсульфоновые кислоты 1:436,477
идентификация 2: 357
454
Предметный указатель
3-A-Нафтол)акриламид 1: 492
а-Нафтиламин 2: 235, 290
производные 2: 311
техника безопасности 2: 405
р-Нафтиламин 2:199, 290
производные 2: 311
техника безопасности 2:405
М-(Нафтил-1)ацетамид см. N-Ацетил-а-
нафтиламин
а-Нафтилацетонитрил 1: 316
Нафтилизоцианат 2: 100, 282
а-Нафтилмагнийбромид 2:164
Нафтил-1-метилфосфоновой кислоты диэти-
ловыйэфир1: 305
Нафтилтиоуксусной кислоты морфолид 2:19
Нафтилуксусная кислота 2:19
Нафтил-1-уксусной кислоты этиловый эфир
2:278
Нафтил- 1-уретаны 2: 342
1-A-Нафтил)-2-фенилацетилен 1: 487
1-B-Нафтил)-2-фенилацетилен 1:487
а-Нафтоилхлорид 2: 94, 260
а-Нафтойная кислота 2:42, 94, 164, 282
р-Нафтойная кислота
получение 2:42
реакции 2: 282
производные, идентификация 2: 331
а-Нафтойный альдегид см. Нафталин-1-
карбальдегид
(З-Нафтойный альдегид см. Нафталин-2-
карбальдегид
а-Нафтол 1: 418,443, 477, 478; 2: 35, 258
производные 2: 347
техника безопасности 2:405
р-Нафтол 1:443,477, 478
производные 2: 347
реакции 1: 295,414,418,470; 2: 35, 37, 253, 258
техника безопасности 2: 405
Нафтол желтый S см. 2,4-Динитронафтол-1-
сульфоновая-7 кислота
р-Нафтолоранж 2: 34, 253
Нафтол AS пигмент 1:471; 2: 255
Нафтол-2-сульфоновая-1 кислота 1:439
Нафгол-2-сульфоновая-6 кислота 2: 357
а-Нафтонитрил 1: 485
Нафтохинон-1,2 2:34,328
производные 2: 327
Нафтохинон-1,4 2: 32
Неницеску реакция 2: 207
Неоментол 2:175,172
Неопентанол, производные 2: 343
Неопентилгликоль 2:179
Нернста закон 1: 77
Неролин 1: 295
Нефедипин 2: 207
Нигрозин 2: 36
Никелевые комплексы
катализатор кросс-сочетания 1:486, 488, 492
реакции с арилгалогенидами и сульфонатами
1:479,486
Никель, катализатор гидрирования 1: 410; 2:
184
Николя призма 1: 107
Никотиновая кислота 2: 11, 12, 14, 39, 124
производные 2: 331
Никотиновой кислоты амид 2: 9, 14, 97
Никотинонитрил см. Пиридин-3-карбонит-
рил
Нингидрин 1: 89; 2: 62, 307
Нитрены 1:349, 351,391
Нитрилоксид 1: 392, 398
Нитрилотриуксусная кислота (НТК, трилон
А) 2: НО
Нитрилы 1:277,466
идентификация 2: 305, 335
— ИК-спектр 1: 119
— растворимость 2: 298
а,р-ненасыщенные
— получение конденсацией Кнёвенагеля!: 126
пиролизом сложных эфиров 1: 346
присоединением синильной кислоты к
ацетиленам 1: 363
присоединение аминов 2: 197
— реакционная способность 2: 196
получение 1:313, 322; 2: 97
— из амидов карбоновых кислот 2: 75
— из диазониевых солей (реакция Зандмейе-
ра) 2: 248
— из сульфокислот 1:477
— из 2,3-эпоксипропионитрилов 2: 124
— окислением углеводородов в присутствии
аммиака 2: 10
— цианэтилированием 2: 196, 208, 229
химические свойства, алкилирование 2: 214
альдольная реакция 2:118
восстановление комплексными гидри-
гидридами 2: 171
по Буво—Блану 2:191
гидрирование 1: 316,410; 2:168,184, 185
гидролиз 1: 316; 2: 52, 89, 95
присоединение нуклеофилов 2: 95
сложноэфирная конденсация 2:111, 142
реакции с карбонильными соединения-
соединениями и серой 2: 20
— со спиртами 2: 52, 98
с соединениями Гриньяра 2: 162
Нитриты см. Азотистой кислоты эфиры
Нитроалканы 1: 266, 279, 312; 2: 244
2-Нитроанизол 1: 295; 2: 396
4-Нитроанизол 1:435; 2: 258
Предметный указатель
455
Нитроанилин 1: 112, 210, 435
идентификация, производные 2: 311
-спектр'НЯМР2: 315
-ИК-спектр2:311
реакции 2: 100, 199, 250, 255
техника безопасности 2: 405
2D)-Нитроацетофенон 2: 16
З-Нитроацетофенон 1: 433; 2: 16, 175
производные 2: 324
2-Нитробензальдегид2: 15, 175
производные 2: 320
З-Нитробензальдегид 1: 434
производные 2: 320
реакции 2: 61, 65,126,175
4-Нитробензальдегид 1: 292; 2: 15, 25, 175
производные 2: 320
З-Нитробензальдегида этиленацеталь 2: 65,
235
З-Нитробензальдегидоксим 2: 61
4-Нитробензилбромид 1: 257, 299
производные 2: 340
4-Нитробензилидендибромид 1:257,292
4-Нитробензилидендихлорид 1: 252
2C)-Нитробензиловый спирт 2: 175
4-Нитробензиловыйспирт2:25, 175
4-Нитробензиловый эфир 1: 299
4-Нитробензилхлорид 1: 299
производные 2: 340
4-Нитробензилцианид 360
З-Нитробензоилхлорид 1:459; 2: 94
4-Нитробензошшторид 2: 94
реакции 1:456; 2: 75, 173
Нитробензойная кислота 1:434; 2:11, 12, 14,94
производные 2: 331
З-Нитробензойной кислоты метиловый эфир
1:434
4-Нитробензойной кислоты эфиры 2: 341
Нитробензол 1:54, 215,434
как дегидрирующий агент 2:44, 198
техника безопасности 2:405
реакции 1: 432, 434, 438, 446, 447, 450, 473; 2:
234
4-Нитробензолдиазония ион 2: 251
З-Нитробензолсульфамид 2: 266
З-Нитробензолсульфоновая кислота, произ-
производные 2: 357
З-Нитробензолсульфохлорид 1:438; 2: 266
4-Нитробензонитрил 2: 250
З-Нитробензофенон 1: 459; 2: 235
4-Нитробензофенон 1: 456
Нитрование
алифатических соединений 1: 239, 261
ароматических соединений Г. 425, 431, 433,
435,493
4-Нитровератрол 1:434
1-Нитрогексан 1: 314
Нитроглицерин см. Глицеринтринитрат
Нитрогруппа
как гетероаналог карбонильной группы 2:230
поглощение в видимой и УФ-области 1: 114
4-Нитродифениламин 1:435
Нитрозамины 1: 471; 2: 238, 241
техника безопасности 2: 238
Нитрозилсерная кислота 2:131
Нитрозирование
ароматических соединений 1:426
радикальное 1:261
Нитрозные газы 2: 377
Нитрозоамиды 2: 238, 241
Нитрозобензол 2: 234
Нитрозогруппа
как гетероаналог карбонильной группы 2: 230
поглощение в УФ/вид.-области 1: 114
а-Нитрозокарбонильные соединения 2: 232
Нитрозосоединения алифатические 1: 261; 2:
230
идентификация 2: 306
получение 2: 243
химические свойства 2: 267
восстановление 2: 232
гидрирование 1: 410
с соединениями Гриньяра 2: 162
Нитрозосоединения ароматические
получение восстановлением ароматических
нитросоединений 2: 232
— нитрозированием ароматических соедине-
соединений 1:471
— перегруппировкой ароматических N-нит-
розоаминов 1:471
4-Нитрозофенол 1:474
З-Нитрокоричная кислота 2: 126
Нитроловая кислота 2: 232, 244
Нитрометан 1:155, 220, 261, 313; 2: 106, 118
техника безопасности 2: 405
М-A-Нитронафтил-2)ацетамид 1:434
1-Нитронафталин 1:434; 2: 235
техника безопасности 2: 405
Нитрооктан 1:314
Нитропента см. Пентаэритриттетранитрат
4-Нитропиридин 1:433
4-Нитропиридин-Ы-оксид 1: 433
2-Нитропропан 1: 261, 314
техника безопасности 2: 405
Нитропропаны 1: 261
Нитропруссид натрия см. Динатрийпентациа-
нонитрозилферратA II)
Нитросоединения алифатические 1: 277
идентификация 2: 306
— растворимость в щелочах 2: 298
получение 1: 321
456
Предметный указатель
— алкилированием нитритов щелочных ме-
металлов 1: 277, 312
— нитрованием углеводородов 1: 261
химические свойства, радикальное восста-
восстановление 1: 262
реакция Манниха 2: 130
Нитросоединения ароматические
идентификация 2: 306
-ИК1:119
получение из сульфокислот 1: 433, 441
— нитрованием ароматических соединений
1:431,433,433
химические свойства 2: 267
восстановление 2: 232, 233, 234, 236
комплексными гидридами 2:171
каталитическое гидрирование 1: 411; 2:
234
нуклеофильное замещение 1: 473, 475
ш-Нитростирол 2:118
4-Нитротолан 1:487
Нитротолуол 1: 434, 435
реакции 1: 252, 257, 434, 446; 2:11, 12, 235
техника безопасности 1: 405
4-Нитрофенетол 1: 296, 435; 2: 235
1-D-Нитрофенилазо)нафтол-1 2:253
4-Нитрофенилгидразоны 2: 57
4-Нитрофенилизоцианат2:100
4-Нитрофениловый эфир 2: 78
1-C-Нитрофенил)этанол-1 2:175
Нитрофенолы 1:435; 2: 82
идентификация, производные 2: 347
- ИК-спектр 2: 347
получение 1:434
реакции I: 295, 296; 2: 258
растворимость в щелочах 2: 298
техника безопасности 2:405
З-Нитрофталевая кислота 1:434; 2: 92
производные 2: 331
З-Нитрофталевой кислоты ангидрид 2: 74,
92
З-Нитрофталевой кислоты моноэфир 2: 74,
342
6-Нитрохинолин 2:199
4-Нитрохинолин-Ы-оксид 1:434
Нитроцеллюлоза 1:287; см. также Целлюлозы
тринитрат
Нитроцеллюлозныйлак 1: 287
Нитроэтан1:261;2:396
Нитрующая смесь 1:433; 2: 396
Новолаки 1:467
Номенклатура химических соединений 1:
179
Нонадекановой кислоты этиловый эфир 2:
278
Нонан 2: 349
Нонаналь 2: 25
Нонаннитрил 1:316; 2:192
Нонановая кислота 2: 28, 39, 183
техника безопасности 2: 405
Нонанол 2: 25
производные 2: 343
Нониламин 2: 192
Норборнанол-2 1: 376, 377; 2: 26
Норборнен 1: 332, 367, 376, 395
5-Норборненкарбоновая-2 кислота 1: 402
Норборнеол см. Норборнанол-2
Норвалин1:302;2:111
Норкамфора 2: 26
Норкарадиен см. Бицикло[4.1.0]гептадиен
Норкаран см. Бицикло[4.1.0]гептан
Норлейцин1:302;2:318
Нортрицикланол 2: 23
Носители для хроматографических колонок 1:
98
НСМО 1:213, 249, 353
Нуклеофилы 1:192, 204, 213, 274
амбидентные 1: 215, 277
жесткие и мягкие 1: 275
Нуклеофильность 1: 204, 274
Нутч-фильтр 1: 46
Ньюмана проекции 1:217
Область «отпечатков пальцев» в ИК-спектрах
1:123
Оборудование для перегонки в вакууме 1: 57
Обратный холодильник 1: 10
Обращенная фаза 1: 87
Обращение реакционной способности 1: 229;
2:132, 202
Оверхаузера ядерный эффект 1: 136
Одноэлектронная молекулярная орбиталь см.
ЧЗМО
Озазон 2: 60
Озон 1: 398; 2: 396
Озониды 1: 373
Озонирование 1: 353, 373, 421
Окисление 2: 5, 45
биохимическое 1: 438
молекулярным кислородом 1: 259, 264
по Оппенауэру!: 174, 176, 228
тиолов и сульфидов 2: 262
ферментативное 2: 9
Окисления потенциал 2: 6
Окисления число 2: 5
Окислительное дегидрирование 2: 28
Окислительное присоединение 1:406,478,486,
490; 2:6
Окислительное сочетание 2: 34, 36
Предметный указатель
457
Окраска веществ 2: 295
Оксазол 1: 398,402; 2: 82
1,3-Оксазолин 1:231
Д2-1,2-Оксазолины 1: 354, 398
Оксалилхлорид 2:92, 95
техника безопасности 2: 405
Оксафосфетан 2: 137
Оксациклобутан см. Оксетан
Оксетан 1:391
Оксид азота 1: 398
1,1'-(Оксидиметилен)-бисD-формилпириди-
нийхлорид)диоксим 2: 62
Оксидный катализатор 1:410
Оксиды азота, техника безопасности 2: 396
Оксимеркурирование 1: 353, 366, 420
Оксимы 2: 51, 57
идентификация, ИК 1: 119
— растворимость 2: 297
получение 2: 61, 222
химические свойства, восстановление 2:236
гидрирование 2: 184
перегруппировка Бекмана 2: 284
хлорирование 1: 252
Оксиранкарбонитрил 2: 124
Оксираны 1: 367
р-Оксоальдегиды 2: 143
а-Оксоальдегиды, получение 2:16
1-C-Оксобутил)циклогексанкарбоновой-1
кислоты этиловый эфир 2: 120
5-Оксовалеронитрил 2: 111
З-Оксогексановой кислоты этиловый эфир 2:
151
6-Оксогептановая кислота 2: 151, 183
7-Оксодекановая кислота 2: 151, 183
5-Оксо-2,3-дифенилгексановой кислоты нит-
нитрил 2: 204
Оксокарбоновые кислоты 1: 347; 2: 86, 112,
192
Оксокарбоновых кислот амиды 2: 251
Оксокарбоновых кислот анилиды 2: 35
Оксокарбоновых кислот нитрилы 2: 140
Оксокарбоновых кислот эфиры
получение 2:134, 139, 143
химические свойства, алкилирование 2: 212
ацилирование 2:152
восстановление 2: 192
ферментативное 2: 181
енолизация 2: 148, 211
кетонное расщепление 2: 87
омыление 2: 86
получение амидов 2: 149
сочетание с солями диазония 2: 251
Оксонолы (красители) 1: 114
6G)-Оксононановая кислота 2: 151, 183
3-Оксо-Д4|0-октагидронафталинкарбоновой-
9 кислоты этиловый эфир 2:120
6G)-Оксооктановая кислота 2: 151, 183
4-Оксо-4-(пиридил-3)бутиронитрил 2: 203
Оксосинтез 1: 353,407
Оксоспирты 1: 119
Оксофенилацетальдегид см. Фенилглиоксаль
З-Оксо-2-фенилбутиронитрил 2: 144
З-Оксо-2-фенилгидразономасляной кислоты
этиловый эфир 2: 253
З-Оксо-2-фенилкарбамоилбутановой кислоты
этиловый эфир 2:156
Оксофенилуксусная кислота см. Фенилглиок-
силовая кислота
2-Оксо-6-фенилциклогексен-3-карбоновой-1
кислоты этиловый эфир 2: 204
2-Оксо-З-фенилянтарной кислоты диэтило-
выйэфир2:144,147
2-Оксоциклогексанкарбоновой-1 кислоты
этиловый эфир 2: 144, 147, 204
B-Оксоциклогексил-1 )глиоксиловой кислоты
этиловый эфир 2: 147
B-Оксоциклогексил-1)уксусной кислоты эти-
этиловый эфир 2: 222
3-B-Оксоциклогексил-1)пропионитрил 2: 203
3-B-Оксоциклогексил-1)пропионитрила
и-толуолсульфогидразон 2:183
2-Оксоциклопентанкарбоновой-1 кислоты
этиловый эфир 2: 144
Оксоянтарной кислоты диэтиловый эфир 2:
143, 144, 147, 213
Октадиен-2,6 2:288
1,4,4а,5,8,8а,9а, Юа-Октагидро-1,4;5,8-диэта-
ноантрахинон 1:402
Октакозан 1: 54
Октан 2: 349
Октаналь 2: 25
Октановая кислота 2: 28, 88,183
Октановое число 1: 379
Октановой кислоты этиловый эфир 2: 71, 192
Октанол-1 1: 376; 2: 25, 28, 73, 192, 264
Октанол-2 1: 367; 2: 164
производные 2: 343
Октанон-2 1:362,405
B5)-(+)-Октантиол 1: 288
Октантиол-1, производные 2: 356
Октен-1 1: 328, 340, 365, 386, 394,405; 2: 28
Октилбромид 1: 314, 316, 328, 340
производные 2: 340
Октилнитрит 1: 313
Октилхлорид, производные 2: 340
Октин-1 1:342,362
Олеиновая кислота 2: 38
производные 2: 331
Олеум, техника безопасности 2: 378,405
Олефины
458
Предметный указатель
идентификация 2: 300, 354
-ИК 1:119
— масс-спектроскопия 1:146
— окисление 2: 300
— производные 2: 354
— тонкослойная хроматография 1:89
— УФ/вид.-поглощение 1:113
— физические константы 2: 354
— эпоксидирование 2: 275
получение 1: 282, 350
— дегидратация спиртов 1: 323, 334, 336, 337
— дегидрирование алканов 2:43
— диеновый синтез (Дильса-Альдера реак-
реакция) 1: 392
— из эфиров серной кислоты 1: 328
сульфокислот 1: 327, 340
— Вагнера—Меервейна перегруппировка 2:275
— Виттига реакция 2:136,138
— Мак-Мурри сочетание 2:194
— образование при масс-спектроскопии 1:
145
— при пиролизе ацетатов 1: 346
ксантогенатов (Чу гаев) 1: 345
— при циклоолигомеризации 1:407
— Хорнера—Вадсворта—Эммонса реакция 2:
135
— элиминирование 1:323
спиртов из эфиров 1: 334
галогеноводородов из алкилгалогенидов
1: 339, 340, 343
Гофмана 1: 343, 344
химические свойства 1: 353, 419; 2:194
алкилирование ароматических соедине-
соединений 1:444
восстановление комплексными гидри-
гидридами 2: 169
гидратирование 1: 359
— — гидрирование 1: 424
— — щдроборирование 1:374
окисление 2: 6, 38
до карбоновых кислот 2:28
оксимеркурирование 1:366
основность 1: 355
полимеризация 1: 378
анионная 1: 378
радикальная 1: 389
присоединение 1: 352, 383-388
при металло- и металлокомплексном
катализе 1:403
бензонитрилов 1: 398
брома 1: 364
— карбенов 1:393
галогенов 1: 363
протонных кислот 1: 359
гипогалогенных кислот 1:364
реакции с енаминами 2: 203, 222
с металлокомплексами 1:403,490
сочетания с алкенил- и арилсоедине-
ниями (Хека реакция) 2: 86
циклоолигомеризация 1:407
эпоксидирование 1:367,369
Олигомеризация 1: 353, 377
Олигомеры 1: 391,405; 2: 36
Олова тетрахлорид как катализатор Фриде-
ля— Крафтса 1:450
Олова(П) хлорид 2: 396
Оловоорганические соединения 1: 262, 386,
483, 488
Омыление, см. также Гидролиз
амиды карбоновых кислот 2: 335
N-ациламиномалонаты 2: 88
в качественном анализе 2: 305
замещенных диэтиловых эфиров малоновой
кислоты 2: 85
нитрилов 2:96, 97, 335
эфиров сульфокислот 2: 261
Оппенауэра окисление 2:168, 174,176, 228
Оптическая активность 1:107, 221
Оптическая плотность (экстинкция) 1:85,90
Оптическая спектроскопия 1:108
Орбитали см. ВЗМО, НСМО, ЧЗМО
Орбитальная симметрия, сохранение 1: 393,
420
Органические бораны 1: 374, 388
Органические соединения щелочных метал-
металлов 1:480; 2: 291
Орнитин 2: 204
Ортокарбоновых кислот эфиры 2: 98, 147, 223,
288
Ортомуравьиной кислоты триалкиловый эфир
2:156
Ортомуравьиной кислоты триэтиловый эфир
2:63,98,147,158,214
Осмия тетраоксид 1: 372
техника безопасности 2:405
Основания 1:202, 233
жесткие, мягкие 1:204, 214
Основное состояние 1: ПО
Основной пик в масс-спектроскопии 1: 144,
149
Основность 1: 202
и нуклеофильность 1: 204
Основные колебания в ИК 1:117
Осушители 1:40
Отбеливатель (белильный раствор) 2: 24
Отгонка растворителей 1:61
Отходы и их уничтожение 1:160
Охлаждаемая ловушка 1:31, 35
Охлаждение 1: 22, 25
Охрана здоровья 1:158
Предметный указатель
459
Палладий как катализатор гидрирования 1:
409, 410; 2: 184
Палладия(П) соли 1:428, 486,491
Палладия комплексы 1:404,478,486
Пальмитиновая кислота 2: 89
производные 2:331
ПАН см. Полиакрилонитрил 1:401; 2: 9
Пантотеновая кислота 2: 82
Паракрасный (л-нитроанилиновый красный)
2:253
Паральдегид 2: 198, 219, 362
техника безопасности 2:405
Паралвдоль2: 152
Паратион см. Диэтил-D-нитрофенил)тиофос-
фат
Парафиновое масло для нагревательной бани
1:102
Парафины твердые как неподвижная фаза для
газовой хроматографии 1: 99
Парафины см. Алифатические углеводороды
Парацетамол 2: 82
Пастера пипетка 1: 39
Патентная классификация 1:172
Патентные описания 1:171, 172
Паттерне—Бюхи реакция 1: 397
ПВА см. Поливинилацетат
ПВХ см. Поливинилхлорид
ПДК (предельно допустимая концентрация) 2:
395
Пеларгоновая кислота 2:182,183
Пенициллин 2: 82
Пентаацетилглюкоза 2:73, 79, 80
Пентабромтолуол 1:448
Пентадиен-1,3 1: 111,344
Пентаметилентетразол 2: 284
Пентаметинцианин 2: 158
Пентан 1: 84, 85,137, 255; 2: 349
Пентандиамин-1,5, производные 2: 311
Пентандион-2,4 см. Ацетилацетон
Пентанол-1 1: 286, 287, 296; 2: 264
производные, 2: 343
Пентанол-2 1: 336; 2: 164
производные 2: 343
mpem-Пентанол см. 2-Метилбутанол-2
Пентанол-3, производные 2: 343
Пентанолы 1: 255, 293, 294
Пентанон-2 см. Метилпропилкетон
Пентанон-3 см. Диэтилкетон
Пентанон-3, диаллилацеталь 2: 289,290
Пентантиол-1, производные 2: 355
Пентахлорэтан 2: 341
Пентаэритрит 1: 363; 2: 114, 115, 121, 176, 178,
179
производные 2: 343
Пентаэритриттетранитрат 1: 287; 2:178,179
Пентен-1 1:336,360,361
Пентен-2 1: 336, 360, 361; 2: 354
Пентен-1-ин-4 1:364
Пентетразол см. Пентаметилентетразол
Пентиламин 2: 284, 311
Пентилбромид 1: 284, 285, 316; 2: 213
производные 2: 340
/и/>етя-Пентилгидропероксид 1: 370
Пентилиодид, производные 2: 340
Пентиллитий 1:406
Пентилмалоновая кислота 2: 86, 88
Пентилмалоновой кислоты диэтиловый эфир
2:86,213
/и/>е/я-Пентиловый спирт см. 2-Метилбута-
нол-2
Пентиловый эфир 1: 255
Пентилхлорид 1: 255, 286, 287, 316,406
производные 2: 340
Пентин-1 2:354
Пептиды 2: 80, 81
Пербензойная кислота 1: 367, 372; 2: 90, 286
Пербунан 1: 391
Первольфрамовая кислота для эпоксидирова-
ния 1: 367
Пергидроаценафтен 1:418,419
Переалкилирования реакции 1:453
Перегонка
в глубоком вакууме 1: 62; см. также Дистил-
Дистилляция
с водяным паром 1: 72, 73
Переметаллирование 1: 483, 484, 486; 2: 160,
166, 167
Перенос протона 2: 6
Переносчик цепи 1: 244
Переэтерификация 1: 282, 288
по Вшьямсону 1: 265, 291, 295
спиртов 1: 265, 288, 325
— диазометаном 2: 258
— диметилсульфатом 1: 295
Перипланарная (конформация) 1: 331
Перициклические реакции 1:192
Перкина реакция 2:104, 122, 125, 224
Перколяция 1:81
Перколяторы 1: 82
Перлон 2:84; см. также Найлон
Перманганат см. Калия перманганат
Перокисление 1: 239, 259
Пероксид водорода 1:240, 241; 2:33,90
техника безопасности 2: 399
Пероксиды 1: 161, 259, 386, 391
Пероксисоединения 1: 259, 391; 2: 286
Перуксусная кислота 1: 367; 2: 30
Петидин 2: 217
Петролейный эфир 1: 85; 2: 379
техника безопасности 2: 379
460
Предметный указатель
Пивалиновая кислота 1:203; 2:50,106,164
Пиколины (метилпиридины) 2:11,12, 15,124,
316
Пикраты2:310, 351
Пикрилхлорид 1: 473
Пикриновая кислота 1:433,436,441, 474
производные 2: 347
реакции 1:435; 2: 309, 351
техника безопасности 2: 406
Пикролонаты 2: 310, 351
Пикролоновая кислота 2: 310, 351
Пикте—Трутона правило 1:65
Пимелиновая кислота, производные 2: 331
Пинакол (пиканон) см. 2,3-Диметилбутан-
диол-2,3
Пинаколин (пинаколон) 2: 26, 42, 65, 273
Пинаколиновая перегруппировка 2: 273, 291
Пинаколиловый спирт, превращения 2: 26
Пинаколы 2:192, 273
Пинанол 1: 376
а-Пинен 1: 376; 2: 354
Р-Пинен 1:376
а-Пиненоксид 2: 275
Пиперазин, производные 2: 311
Пиперидин 1: 319,414; 2: 58,125, 180,197
техника безопасности 2:406
Пиперидингидрохлорид 2:131
Пиперидиний-ион 2:106
4-Пиперидинобутанон-2 2: 131, 198
З-Пиперидинопропановой кислоты этиловый
эфир 2:197
З-Пиперидинопропионитрил 2:197
1-Пиперидиноциклогексен-1 2:58
а-Пиперидон 2: 284
Пиперилен см. Пентадиен-1,3
Пиперин 2: 120
Пиперитон см. я-Ментен-1-он-З
Пиперональ2: 120,178
Пиперониловый спирт 2: 178
Пиразолиноны-5 см. Пиразолоны
Д2-Пиразолины
получение 2: 257
реакции 2:45
Д'-Пиразолины 1: 397; 2:257
Пиразолоны 2: 35, 78, 83,149,157, 247
Пиразолы 2:45, 59, 209
Пиранозы 2:66
Пирекс (стекло) 1:7
Пиретроиды 1: 395
Пиретрум 1: 395
2-Пиридилметиллитий 2:166
2-Пиридилуксусная кислота 2:166
Пиридин 1:425
азеотропная смесь 2: 379
идентификация, производные 2:316
— растворимость 2:298
— УФ/вид.-поглощение 1:115
как растворитель 1: 85
очистка и осушение 2: 379
получение 1: 362; 2:125
химические свойства, замещение нуклеофи-
льное 1:474
электрофильное 1:428
превращения 1:414,441, 474
техника безопасности 2: 379, 406
Пиридиндикарбоновая-2,3 кислота 2: 39
Пиридиндикарбоновой-3,5 кислоты эфиры 2:
206
Пиридинкарбальдегид-3 2: 206
Пиридинкарбонитрил-3 2: 97
Пиридинкарбоновая-3 кислота см. Никотино-
Никотиновая кислота
Пиридин-4-карбоновая кислота см. Изонико-
тиновая кислота
Пиридин-Ы-оксид 1: 430
Пиридинсульфоновая-3 кислота 1:441
Пиридиния дихромат 2: 22, 28
Пиридиния хлорхромат 2: 22
Пиридоксин 1:42; 2: 83
Пирилий-катион 1:425
Пирилия соли 2: 209
Пиримидин, производные 2:129, 316
Пировиноградная кислота 2: 69
производные 2: 331
Пировиноградной кислоты метиловый эфир 2:
71
Пирогаллол, производные 2: 347
Пиролюзит (диоксид марганца) 2: 15,21
техника безопасности 2: 403
Пирослизевая кислота 2:179
Пиррол 1:425; 2:43, 126
химические свойства, карбоксилирование 1:
469
электрофильное замещение 1:428,430
Гаттермана синтез 1:459
реакции с соединениями Гриньяра 2:160
Пирролкарбоновая-2 кислота 1:469
Пирролидин 2: 43, 58
техника безопасности 2: 311
1-Пирролидиноциклогексен-1 2:58,222
1-Пирролидиноциклопентен-1 2: 58
Пирролы 2: 59
Пистолет для осушения 1: 38
Пламенно-ионизационный газоанализатор 1:97
Планка уравнение 1: 242
Пластификаторы 1: 255; 2: 13, 14, 76, 185, 186,
178
Пластохинон 2: 34
Платина как катализатор гидрирования 1:410;
2:184, 396
Предметный указатель
461
Платины диоксид 2: 396
Плотность 1: 100, 107
ПМР-спектроскопия 1: 129
Поверхностно-активные вещества 1: 371, 453
Поглотительная колонка 1: 36
Подвижная фаза в хроматографии 1: 86
Полиакрилонитрил 1: 391; 2: 10
Полиамидное волокно 1:435; 2: 62, 83
Полиамиды 1: 366, 418; 2: 83
Полибутадиен 1: 391
Поливинилацетат 1: 362, 390
Поливиниловый спирт 1: 362
Поливинилхлорид 1: 340, 362, 390
Полигалогеналканы, радикальное присоеди-
присоединение к олефинам 1: 384
Полигалогенметаны 1: 384
Полигидроксисоединения 1: 287
Полигликоли 1:97, 99, 276
Полигликоль, простые эфиры 1: 270, 279
Полиены 1:113
Полиизобутен 1: 379
Поликарбоновые кислоты 2: 297
Полимеризация 1: 353, 376, 422
атактическая 1:405
суспензии 1: 391
изотактическая 1:405
радикальная 1: 383, 389
синдиотактическая 1:405
этиленов при низком давлении 1: 405
Полиметакриловой кислоты метиловый эфир
1:338; 2: 111
Полиметилметакрилат (оргстекло) 1: 338, 391;
2:111
Полиметин 2: 35, 156, 226
Полиметиновая конденсация 2: 156
Полиметиновые красители 1: 113
Полинитрофенолы 1:433
Полиолы 2: 297
Полипропилен 1:405
Полисахариды 2: 66
Полисилоксаны 1: 319
Полистирол 1: 390, 391, 443,453
Политерефталевая кислота, гликолевый эфир
2:14,76
Политетраметиленгликоль 1: 289, 418
Политетрафторэтилен 1: 255, 312, 390
Политрибромстирол 1:448
Полиуретаны 1: 366, 418, 435; 2:101, 114
Полифосфорная кислота 2: 396
Полихлорбензол 1:448
Полихлорпентан 1: 255
Полиэтилен 1: 338, 391, 406
Полиэтиленгликоль 1: 24,143, 370, 371
Полиэтиленгликоля моноалкиловый эфир 1:
371
Полиэфиры
простые 2: 101
сложные 1:371, 391, 418; 2: 13,75, 101, 114
— как стационарная фаза при газовой хрома-
хроматографии 1: 97
-волокно 1:371; 2: 13, 75
— термостабильные 1:418
Полуацеталь 2:62,161
Полумикроэкстракция 1: 79
Полями соотношение 1:196
Поляризатор 1:107
Поляризованный свет 1: 100,107, 222
Поляризуемость 1: 204, 206, 276, 357
Поляриметр 1:107, 221
Полярность растворителей 1: 200
Последовательные реакции 1:197
Постоянная магнитного экранирования 1:
128
Потенциал полуволны см. Окисления потенциал
Превращения с переносом заряда 1:111
Преломление света 1:100, 105
Прилежаева реакция 1: 368
Принса реакция 2: 116, 224
Присоединение
катализированное комплексами 1:406
нуклеофильное (AdN) 1: 352, 379
окислительное 1:407, 478,485, 490
против правила Марковникова 1: 374; 2: 199,
200, 287
радикальное (AdR) 1: 243, 352, 383, 421
электрофильное (Adg) 1: 352
Производная (дифференциальная) спектрос-
спектроскопия 1:116
Прокаин 2:14, 76
Пролин2:317
Пропан 1: 240
Пропан-1,2-диамин 2: 311
Пропандиол-1,3 2: 343
Пропандитиол-1,3 1:310; 2:134
производные 2: 356
Пропанол-1 1:381; 2:71, 264
производные 2: 342
техника безопасности 2:406
Пропанол-2 см. Изопропиловый спирт
Пропантиол, производные 2: 356
Пропаргиловые спирты 1: 362; 2: 113 см.
также Пропин-2-ол-1
Пропаргилового альдегида диэтилацеталь см.
Пропиналя диэтилацеталъ
Пропен 1: 360, 364, 378; 2: 9
энтальпия диссоциации связей 1: 240
величина рКа 1: 203
замещение радикальное 1: 245
метатезис 1:410
окисление 2:10
462
Предметный указатель
с гидропероксидами 1:170
техника безопасности 2: 406
хлорирование 1: 255
Пропеноксид 1: 370
Пропиламин 2: 238
производные 2: 311
Пропилбензол 1:439; 2: 182
производные 2: 352
техника безопасности 2: 406
4-Пропилбензолсульфохлорид 1:439; 2: 263
Пропилбромид 1: 284; 2: 341
производные 2: 340
реакции 1: 306, 316,452; 2: 213, 217
техника безопасности 2: 406
Пропилвиниловый эфир 1: 381
Пропилен см. Пропен
Пропиленгликоль см. 1,2-Дигидроксипропан
Пропиленкарбонат как растворитель 1: 275
Пропилиодид 1: 286
производные 2: 340
Пропилмагнийбромид 2:164
Пропилмалоновая кислота 2: 86, 88
Пропилмалоновой кислоты диэтиловый эфир
2:86,213
З-Пропилоксиранметанол 1: 369
4-Пропилтиофенол 2: 263
Пропилхлорид 1: 270,452
производные 2: 340
техника безопасности 2: 406
Пропиналя диэтилацеталь 1: 343
Пропин-2-ол-1 2: 114; см. также Пропарги-
ловые спирты
Пропионамид 2: 281
Пропионанилид 2: 284
Пропионилхлорид 1:457; 2: 94, 152, 221
2-Пропионилциклогексанон 2: 151, 221
2-Пропионилциклопентанон 2:151, 221
Пропионитрил 1: 316
Пропионовая кислота 1: 249; 2: 94, 218
производные 2: 331
техника безопасности 2:406
Пропионовой кислоты этиловый эфир 2:144,164
техника безопасности 2:406
Пропионовый альдегид 2:63,111,118,179
производные 2: 320
техника безопасности 2:406
Пропионового альдегида диэтилацеталь 2: 63
Пропиофенон 1:457,459; 2: 22, 326
производные 2: 324
реакции 2: 17, 19,114, 131,182, 284
Пропиофенона енолят 1: 278
Пропифеназон 2: 84
Пропоксибензол 1: 296
Протона акцепторы 1: 202
Протонные кислоты
как электрофильные реагенты 1:355
присоединение к олефинам 1: 359, 420
Прохиральность 1: 223; 2: 216
Псевдогалогенные соединения 2: 232
Псевдокумол 2: 351
Псевдонитрилы 2: 232, 244, 307
Псевдопельтьерин 1: 344; 2:131
Пурин, производные 2: 298
Пурпурный проявляющий краситель 2: 83
Путресцин 2: 284
Радикалы
время жизни 1: 243,244
изомеризация 1: 243
получение 1:191, 240, 388
реакционная способность 1: 248
селективность радикальных реакциий 1: 247
Радикальная цепная реакция 1: 244, 263
Радикально-функциональная номенклатура 1:
179
Радиолиз 2: 329
Разделительная колонка 1: 92, 98
Разотерм (стекло) 1: 7
Раймера—Тимана синтез 1: 349
Рамноза 2:329
Ранитидин2: 132
Распределительная хроматография 1: 86, 98
Растворимость 2: 297
Растворители 1:49, 61, 66; 2: 185, 396
влияние на реакционную способность 1: 199,
233, 272, 325
для восстановления комплексными гидрида-
гидридами 2: 171
— каталитического гидрирования 1:412
— металлирования 1:480
— масс-спектроскопии 1: 143
— реакции Гриньяра 2: 159
Фриделя—Крафтса 1: 457
Лека 1:491
неполярные 1: 200
очистка 2:396
осушение 1: 37
отгонка 1: 61
полярные, апротонные 1: 200, 272
протонные 1: 200, 272, 326
Растительные масла 2: 89
Рауля закон 1: 53
Раффиноза 2: 329
Рацемат 1: 221, 223, 227; 2: 188
Рашига кольца 1: 68
Реагента индукция 1: 226
Реагента контроль 1: 227
Реакции
Предметный указатель
463
константы 1: 209, 325
скорость 1: 197
— нуклеофильного замещения 1: 267, 277
— реакций карбонильной группы 2:49
— электрофильного ароматического замеще-
замещения 1:427
— элиминирования 1: 325
тепловой эффект 1:192
энергия 1:193
энтропия 1:195
Реакционная способность
алкилгалогениды
— при реакции Гриньяра 2:159
— при нуклеофильном замещении 1: 270
ароматические соединения 1:427
винилогичные электронодонорные соедине-
соединения 2: 210
влияние растворителя 1: 200
диазония ионы 2: 250
гетерокумулены 2:154
карбонильные соединения 2: 158,114,184
литийорганические соединения 2:165
нуклеофильные реагенты 1: 474
— при нуклеофильном замещении 1: 274
— радикальном восстановлении 1: 262
замещении 1: 245
— хлорировании 1: 250
— электрофильном присоединении 1: 355
производные карбоновых кислот 2:68,90,140
радикалы 1: 243
теория возмущений 1: 212
С—Н-связей 1: 249
Региоселективность
при 1,3-диполярном присоединении 1: 397
— перегруппировке Коупа 2: 287
Резолы 1:467
р-Резорциловая кислота см. 2,4-Дигидрокси-
бензойная кислота
Резорцин 1: 297, 415,462,470
производные 2: 348
техника безопасности 2:406
Резорцина диметиловый эфир 1: 295, 462; 2:
251
Резорцина монометиловый эфир 1: 295
производные 2: 347
Рекомбинация радикалов 1:243
Ректификация 1: 53, 62,64, 71, 165
Ренея катализаторы 1:410; 2:184, 383
Рентгеноструктурный анализ 1:156
Реппе синтезы 1: 380
Ретен 2:44
Ретро-дильс-альдеровское расщепление при
масс-спектроскопии 1:147
Ретросинтез 1: 229
Реформатского синтез 2:167, 227
Рефрактометрия 1:106
О-Рибоза2:329
Рибофлавин 2:9
Риттера реакция 2:112
Рицинолевая кислота 2: 39
Роданиды см. Тиоцианаты
Роданин2:123
Роданирование ароматических соединений 1:
426, 448
Родинал 2: 234
Розенмунда восстановление 1: 411; 2: 169,
184
Розенмунда—Брауна реакция 1:485
Рояна проба 2: 305
Ртути(И) ацетат 1: 366; 2:406
Ртути оксид 2:406
Ртути(П) хлорид 2: 407
Ртуть 2: 381,406
Ртутьорганические соединения 1: 388
Салициловая кислота 1:469,471; 2: 73, 253
производные 2: 332
Салициловой кислоты метиловый эфир 1:414;
2:71
Салициловой кислоты фениловый эфир
(салол) 2: 347
Салициловой кислоты этиловый эфир 2:71
производные 2: 347
Салициловый альдегид 2:126,320
Саркозин, нитрил (метиламиноуксусной кис-
кислоты нитрил) 2:109
Сахар 1:84, 222; 2: 66,181,297
озазоны 2: 60, 328
Сахарин 1:443; 2:12,13, 82
Сахарометр 1:108
Сверка окисление 2: 25
Свинец 2:47
Свинца тетраацетат 2: 40; 384
техника безопасности 2: 384,407
Свободный спад индукции (ССИ) 1:135
Себациновая кислота A,8-октандикарбоновая
кислота) 2: 94, 331
Себациновой кислоты дихлорангидрид 2: 80,
94
Себациновой кислоты диэтиловый эфир 2:71,
192
Селективность при радикальном замещении 1:
245
Селен 2:44,407
Селена диоксид 2: 16, 47, 372
техника безопасности 2: 372,407
Семикарбазид 2: 59, 311
Семикарбазоны 1: 114; 2: 51, 56, 57, 320, 327
464
Предметный указатель
Сера 2:18, 44,47, 296
СеребраA) соли как окислители 2: 29
Серебра галогениды 2: 160
Серебра нитрат 2:407
Серебра нитрит 1: 279, 312
Серебра оксид 1: 291
Серебра цианид 1:315
Серебро как катализатор дегидрирования 2:
27
Серебряное зеркало 2: 301
Серии 2: 317
Серная кислота
как осушитель 1: 36, 39,40
присоединение к олефинам 1: 355, 360
растворимость органических соединений в
концентрированной серной кислоте 2:
298
техника безопасности 2: 382, 407
Сернистые красители 2: 35
Серной кислоты эфиры 1: 282, 287, 353, 360
как алкилирующий агент 1: 293; 2: 212
Сероводород 1: 203; 2: 382
присоединение к а,C-непредельным карбо-
карбонильным соединениям 2: 200
— радикальное, к олефинам 1: 384
техника безопасности 2: 382, 407
Серотонин 2: 290
Сероуглерод 1: 85; 2: 101, 155
техника безопасности 1: 25; 2:407
Серы соединения 2: 268
алкилирование 1: 305
диоксид 1:29,437
— техника безопасности 2:407
триоксид 1: 287, 437
Сиккативы 1: 260
Силандиол 1: 319
Силденафил 2: 267
Силаны 1:262, 318
Силикагель 1:40, 84, 87, 94, 97
Силиконы 1: 9, 24, 97, 102, 255, 319
Силиловый простой эфир 1: 319; 2: 121, 210,
229
Симакс (стекло) 1: 7
Симметрии условия
при переносе электрона 1:112
— циклоприсоединении 1: 392
Симмонса— Смита реагенты 1: 395
Синглетное состояние карбенов 1: 349,
Синглетное состояние кислорода 1: 391, 397
Синильная кислота 1: 203; 2:10
присоединение к альдегидам и кетонам 2:108
— к винилогам карбонильных соединений 2:
196
— к олефинам и ацетиленам 1:420
реакция Манниха 2:130
техника безопасности 2: 382, 407
Синклинальная (конформация) 1: 217
Синперипланарная (конформация) 1: 217, 331
Синтеза планирование 1: 228
Синтон 1: 228
Сквален 1:97
Скелетный никель (катализатор) 1:410, 413
Скраупа синтез хинолина 2:198, 229
Слизевая кислота 2: 28
Сложноэфирная конденсация 2: 104, 139, 143,
149
Сложноэфирное расщепление р-дикарбо-
нильных соединений 2: 149, 151
Сложные эфиры 1: 346
Смазочные масла 1: 379
Смеси
азеотропные 1: 64, 75
разделение 2: 293
Снемиуса закон преломления 1: 106
Сокслетта экстрактор 1: 79
Соли, растворимость 1: 49; 2: 297
Сольватация 1:199, 272
Сольватохромный сдвиг 1: 112
Сольволиз 1: 270, 272
Соляная кислота 2: 383, 407
Соммле реакция 1: 466
Соногашира реакция 1:486, 487
Сорбиновая кислота 1: 338; 2:126
Сорбит 2: 30, 185, 343
Сорбоза 2: 30, 65, 329
Сочетание
окислительное 2: 34, 36
солей диазония 2: 250, 252
Спектроскопия
инфракрасная 1: 100, 108, 116, 166
масс- 1: 141
микроволновая 1: 108
производная 1:116
ренгеновская 1:108
электронная 1:111
ЯМР 1:100, 126, 166
Спермацетовое масло 2: 89
Спин 1: 57
Спин-спиновое взаимодействие 1: 131
Спирты
алкилирование ароматических соединений 1:
449
арилирование 1:496
восстановление по Мейервейну—Понндорфу—Вер-
лею2:168
дегидратация 1: 334, 337
ИК-спектры 1:117
дегидрирование 1: 335, 338; 2: 26
— с реактивом Гриньяра 2:159
нуклеофильное замещение 1: 281
Предметный указатель
465
окисление до кетонов 2: 22
— по Оппенауэру!: 175
получение, гидратация спиртов 1: 353, 359,
420
— гидроборирование 1: 353, 374, 421
— гидролиз алкилгалогенидов и алкилсуль-
фатов 1:291, 293
эфиров карбоновых кислот 2: 336
— гидроксилирование 1: 367, 257, 258,420
— оксимеркурирование 1: 288, 367, 306
— оксосинтез 1: 353, 407
— реакции карбонилсодержащих соедине-
соединений с литийорганическими 1: 481
с магнийорганическими 2:104,159,
163
Каннищаро 2:168, 176, 178
— гидрирование каталитическое, реакция Бу-
во—Блана!: 168
с гидридными комплексами 2:169,172
с неблагородными металлами 2: 188
радикальное восстановление 1: 262
реакции присоединения <х,р-непредельных
карбонильных соединений 2: 199
олефинов 1: 384
триметилсилилирование 1: 319
хлорирование 1: 254
элиминирование по Чугаеву 1: 350
этерификация бромоводородной кислотой 1:
284
— винилирование 1: 380
— галогеноводородной кислотой 1: 266, 281
— Мицунобу реакция 1: 288
— осушение 1: 39
— растворитель 1:49, 200, 327
— реакции обмена 1: 297
Сродство к электрону 1:213
Стандартный электродный потенциал 2:6
Станнаны 1: 262
Стационарности принцип по Боденштепну 1:
197
Стеариловый спирт 2: 343
Стеариновая кислота 2: 89, 94, 331
Стеароилхлорид 2: 94, 260
Стереоизомеры 1: 216,
Стереоселективные синтезы 1: 226
алкилирование карбонильных соединений 2:
216
альдольная реакция 2: 120
гидрирование 1:407
— спиртов 1: 376; 2:173
— эпоксидов 1: 369
Виттига реакция 2: 137
Стерические эффекты 1: 211
Стероиды 2: 114, 206
Стефена—Кастро сочетание 1: 485
Стивенса перегруппировка 2: 270, 291
Стилпа реакция 1: 485
Стильбен 1: 331, 394; 2: 6, 136, 355
Стирол 1:327, 379,391,453
идентификация 2: 354, 355
получение 1: 338
реакции 1: 365, 367, 369, 370, 376, 379, 386,
390,394,398,400,414,462
техника безопасности 2:407
Стирола дибромид 1: 342; 2: 341
Стифнаты
аминов 2: 310
углеводородов 2: 351
Стифниновая кислота 1: 435; 2: 309, 351
Суберон 2: 278
Субстрата индукция 1: 226
Сузуки реакция 1: 487
Сукцинальдегид 2: 154, 320
Сукцинимид2:208,281
Сукцинонитрил 1: 316; 2:96, 203
Сульфаметоксазол 2: 266
Сульфамиды см. Сульфаниловой кислоты
амиды, Сульфокислот амиды
Сульфаниловая кислота 2: 253, 266, 298, 357
техника безопасности 2:407
Сульфаниловой кислоты амиды 2: 236, 266
Сульфеновые кислоты 2: 262
Сульфены 1: 340
Сульфидные катализаторы 1: 410
Сульфиды
идентификация 2: 285
получение восстановлением сульфоксидов 2:
262
— из алкилгалогенидов и сульфида натрия 1:
266,305
металлоорганических соединений и се-
серы 1:482
тиолов и активированных ароматичес-
ароматических соединений 1:476
— присоединением тиолов к а,р-непредель-
ным карбонильным соединениям 2:
199
окисление до сульфоксидов 2: 262
Сульфиновая кислота 1:443; 2: 248, 260, 298
2C)D)-Сульфобензойная кислота 2: 357
2-Сульфобензойной кислоты имид см. Сахарин
Сульфогруппа как гетероаналог карбонильной
группы 2:262
Сульфокислоты 2: 260, 265, 297, 356
получение окислением тиолов и сульфино-
вых кислот 2: 260
— сульфированием ароматических соедине-
соединений 1:435,441
— сульфоокисление алифатических углево-
углеводородов 1: 261
466
Предметный указатель
превращения до сульфохлоридов 2: 265
Сульфокислот алкиловые эфиры 1: 294
взаимодействие с анионами карбоновых кис-
кислот 1: 298
восстановление 1: 239, 262
как алкилирующие агенты 1: 272; 2: 260
Сульфокислот амиды (сульфамиды) 1: 300,
443; 2: 236, 264
идентификация 2: 298, 351, 357
получение 2: 208, 232, 266
Сульфокислот хлорангидриды
получение из сульфокислот 2: 265
— хлорсульфированием ароматических угле-
углеводородов 1:438, 439
— сульфохлорированием алифатических уг-
углеводородов 1: 261
химические свойства, алкоголиз 2: 264
аминолиз 2: 265, 266
восстановление 2: 233, 263
гидролиз 1: 261
Сульфоксиды 1:119; 2: 262
Сульфолан см. Тетраметиленсульфон
Сульфониевые соединения 1: 266
Сульфонилмочевина 2: 268
Сульфонирование ароматических соединений
1:425,435,493
Сульфоны 2: 262
Сульфоокисление 1: 239, 261
Сульфохлориды см. Сульфокислот хлорангид-
хлорангидриды
Сульфохлорирование 1: 239, 261
Сульфурилхлорид 1: 253; 2:407
Супероснования 1: 480
Сурьмы пентахлорид (катализатор) 1:449
Сухой лед см. Диоксид углерода твердый
Сушка 1: 35, 38, 62
Таллия гидроксид 1:488
Таммана правило 1: 52
Тарелка теоретическая 1: 65, 70, 95, 98
Твист-конформация 1: 218
Твитчела реактив 2: 89
ТДИ см. Толуилендиизоцианат
Теклю горелка 1: 22
Телоген 1: 390
Теломер 1: 389
Температура
плавления 1:100,101, 103
кипения 1: 54, 105
ТЕМПО 1: 244; 2: 23
Терефталевая кислота 1: 371; 2:13,14, 258
производные 2:333
Терефталевой кислоты диметиловый эфир 2:
76, 258
Терефталевой кислоты монометиловый эфир
2:13
Терефталевый альдегид 1: 292; 2: 321
Термодинамика реакций 1:193
Термодинамический контроль 1: 199
Термолиз 1: 241, 383
Термометр 1:103,162
Термостат 1: 24
Терпеновые спирты 2:114
Терпены 1:369; 2: 104, 114
Тетрааминоэтилен 1: 352
2,3,4,5-Тетраацетил-О-глюкоза 2: 79
1,3,4,5-Тетрагидробенз[Ь]азепинон-2 2: 284
1,2,3,4-Тетрагидрокарбазол 2:45
1,2,3,4-Тетрагидронафтил-1-гидропероксид
(тетралингидропероксид) 1: 261; 2:407
Тетрагидронафталин см. Тетралин
Тетрагидротиофен-1,1 -диоксид 1: 201
Тетрагидрофуран 1: 289,418
идентификация, растворимость 2: 298
как растворитель 1: 85,200, 276; 2:159
получение 1: 289; 2:114
превращения 1: 290, 385
очистка и осушение 2: 385
Тетрагидрофуриловый спирт 2: 276,407
Тетрадеканол-1 2:193
Тетрадекановой кислоты нитрил 2:192
Тетрадекановой кислоты этиловый эфир 2: 71,
192
Тетрадециламин 2:192
2,2,6,6-Тетракис(гидроксиметил)циклогекса-
нол 2:179
Тетракис(трифенилфосфин) палладий 1: 487,
490
Тетракозан 1: 54
Тетралин 1: 262, 418; 2: 351, 407
Тетралина гидропероксид 1: 262; 2:407
а-Тетралон 1:418,459; 2:164, 284
Тетраметилбензол 1:453
2,3-Тетраметилениндол 2: 291
Тетраметиленсульфон (сульфолан) как раст-
растворитель 1: 311; 2: 384
2,2,6,6-Тетраметилпиперидин-1-оксил см.
ТЕМПО
Тетраметилсилан 1:128,130
Тетраметилтиурамдисульфид 2: 102
Тетраметилэтилен см. 2,3-Диметилбутен-2
N,N,N',N'-TeTpaMeTH л эти лен диамин
(ТМДА) 1:480
Тетрапропиламмония перрутенат 2: 27
Тетрапропилен 1: 379
Тетрафторолефины 1: 384, 385
Тетрафторэтилен 1: 391
Предметный указатель
467
а,а,а,о-Тетрахлоралканы 1: 390
1,2,4,5-Тетрахлорбензол 1:479
Тетрахлоргидрохинон 2: 7,45
Тетрахлорметан см. Тетрахлоруглерод
1,1,1,3-Тетрахлорнонан 1: 387
1,1,1,3-Тетрахлороктан 1:387
Тетрахлоруглерод 1: 255, 390; 2: 6
азеотроп с метанолом или водой 1: 75
идентификация, физические константы 2:
340
как растворитель 1: 85, 116
— переносчик воды 1: 75
— экстрагент 1: 81
химические свойства, хлорирующий агент 1:
252, 384, 386,453
осушение и очистка 1: 41; 2: 385
техника безопасности 2: 385, 408
Тетрахлорхинон см. Хлоранил
1,1,2,2-Тетрахлорэтан 1: 340, 366; 2: 340
Тетрахлорэтилен 2: 340
Тетрацианэтилен 1: 352
Тетрозы 1: 222
Тефлон см. Политетрафторэтилен
Д3-Тиазолин 2:45
Тиазолы 2:154
Тиамбутоксин 2: 102
Тиамин 2:129
Тиглиновая кислота 2: 94, 330
Тиглиновый алвдегид 2:64, 118
Тимол, производные 2: 347
Тиоамиды2: 18
Тиоацетали 2: 51, 66
Тиобензамид 2: 103
Тиобензанилид 2: 103
Тиобензойной кислоты морфолид 2: 19
Тиогликолевая кислота 1: 288, 310
Тиогликолевой кислоты эфиры 2: 209
Тиогликоль 2: 66
Тиогомовератровой кислоты морфолид 2: 19
Тиодигликоль 1: 306
Тиокарбонильные соединения
взаимодействие с реактивами Гринъяра 2: 162
получение 2: 103
УФ/вид.-поглощение 1: 114
Тиокарбоновых кислот амиды 2: 59, 103, 155
Тиокарбоновых кислот морфолиды 2:18
Тиокарбоновых кислот эфиры 2:103
Тиокетоны 2:102
Тиокрезол 2: 356
Тиолкарбоновые кислоты, радикальное при-
присоединение к олефинам 1: 384
Тиолы (меркантаны) 1:265; 2:66,310; см. так-
жеТиофенолы
идентификация 1: 119, 309; 2: 295, 298, 308,
355, 356
получение 1: 321
— из алкилгалогенидов и гидросульфидов ще-
щелочных металлов 1: 305
металлоорганических соединений и се-
серы 1:482
— при восстановлении сульфохлоридов 2:261
гидролизе S-алкилтиоурониевых солей
1:309
пиролизе ксантогенатов 1: 345
реакции, арилирование 1:493
— присоединение винилогов карбонильных
соединений 2: 196,200
к олефинам, радикальное 1: 384
— с азотистой кислотой 2: 308
— с алкилгалогенидами 1: 306
— с реактивами Гриньяра 2:160
— с солями тяжелых металлов 2: 308
Тиомочевина 1: 307-410; 2: 102,407
Тионафтол, производные 2: 356
Тионикотиновой кислоты амид 2: 103
Тионилхлорид 1: 285, 287; 2: 93, 95,98
техника безопасности 2: 385,407
Тионирование карбонильных соединений 2:
102
Тиосалициловая кислота 2: 250
Тиосемикарбазон 1: 114
Тиофен 1:425,462,483
ацилирование по Фриделю-Крафтсу 1:459
синтез Гаттермана 1: 460
электрофильное замещение 1:428,430
Тиофенкарбальдегид-2 1:462; 2: 321
Тиофенкарбоновая-2 кислота 2:42
Тиофенол 1: 203, 387; 2: 263, 356
Тиофенолы см. также Тиолы
идентификация 2: 308, 354
— ИК1:119
— определение химического эквивалента 2:
355
— растворимость 2: 298
получение 1:443, 2: 263
реакции, с азотистой кислотой 2: 308
— с солями тяжелых металлов 2: 308
2-Тиофентиол 1:483
Тиофены замещенные 2: 209
Тиоформанилид 2: 103
Тиофосген 2: 99,101
1B)-Тиоцианатометилнафталин 1: 307
Тиоцианаты 1: 307, 321, 426,448
Тиоцианирование см. Роданирование
Тиоэтиленацеталь 2: 66
Тиоэфиры см. Сульфиды
Тирам 2: 102
Тирозин 2: 34, 298, 318
ТитанаAУ) изопропилат, катализатор 1: 368
Титана комплексы 1: 368
468
Предметный указатель
ТитанаAУ) сульфат 1: 260
Титана тетрахлорид 1:450; 2: 211, 407
Титана трихлорид 1: 405; 2:194
Титана соединения в низшей валентности 2:
194
Тиурониевые соли 1: 308
Тиффено реакция 2: 274, 291
ТМС см. Тетраметилсилан
Тобиаса кислота 1:441
Тозилат см. я-Толоулсульфокислоты алкило-
вый эфир
Токоферол 1:454
Токсическая доза 2: 393, 394
Токсичность химикатов 2: 396
Толил- см. также Толуил-
Толилгидразин 2: 79, 247
я-Толилуксусная кислота 2:19
Толленса реактив 2: 29, 301, 381
Толуальдегид см. Толуиловый альдегид
Толуидин1:477;2:235, 310
превращения 1:449; 2: 198, 246, 247, 249
техника безопасности 2:407,408
3-(я-Толуидино)пропионовой кислоты этило-
этиловый эфир 2: 198
2,4- и B,6-)Толуилендиизоцианат 2: 99
Толуиловая кислота 2: 13, 94, 332, 333
Толуиловой кислоты метиловый эфир 2:13, 71
Толуиловый альдегид 1:460; 2:15,133, 135, 321
я-Толуоилхлорид 2:94
Толуол 2:10
азеотроп с водой 1: 75; 2: 386
идентификация, производные 2: 351
значение рА; 1:203
окислительный потенциал 2: 6
получение 1:451
сушка 2: 386
техника безопасности 2: 386, 408
химические свойства, в присутствии аммиака
2:10
метилирование 1:451
окисление 2: 12
превращения 1: 251, 253, 257, 416, 434,
439, 442, 458, 465; 2: 12
радикальное замещение 1: 246
хлорирование 1: 253
энергия диссоциации связи 1: 240
я-Толуолсульфамиды 2:12, 266, 310
Толуолсульфокислота 1:442; 2: 357, 408
я-Толуолсульфокислоты алкиловый эфир 1:
265, 272, 278, 311,450; 2: 212
получение 2: 264
превращения 1: 262, 297, 310
я-Толуолсульфокислоты эфиры 1: 272, 296; 2:
264
я-Толуолсульфокислоты N-хлорамид 2:267
Толуолсульфонаты см. я-Толуолсульфокисло-
я-Толуолсульфокислоты алкиловый эфир
Ы-(я-Толуолсульфонил)антраниловая кисло-
кислота 2: 266
я-Толуолсульфонилгидразон 2: 182,256
Толуолсульфохлорид 1: 365, 443; 2: 263, 264,
266
Толуолы замещенные 1:489; 2: 12, 15
Тонкослойная хроматография 1: 88, 165
Треоза 1: 223
Треонин 2: 317
7>ео-форма 1: 224
Триазены 1,3-дизамещенные 2: 240, 251
Триалкилалюминия соединения в синтезе
Циглера—Натты 1:405
Триалкилбораны 1: 374; 2: 287
Триалкилгалогенсилан 1: 322
1,3,5-Триалкилгексагидро-1,3,5-триазин 1:
464
Триалкилоловохлорид 1:488
Триалкилсилилазид 1: 266
Триалкилсилиламин 1: 266
Триалкилсилилцианид 1: 266
Триалкилсилильная группа 1: 231
Триалкилфосфиноксид 1: 304
Трибензиламин, производные 2: 316
Трибромацетальдегид см. Бромаль
1,2,3-Трибромпропан 1: 365
2,4,6-Трибромфенол, производные 2: 348
Трибромэтанол 2: 175
Три(«-бутил)олова гидрид 1: 263, 264, 386,489
Тригалогениды геминальные 1: 291
Тригалогенметан 1: 348
Тригалогенуксусная кислота 1: 348
2,4,6-Тригидроксибензойная кислота 1:470
3,4,5-Тригидроксибензойная кислота 1: 295
Тригидроксинеопентан 2: 178
Тригликоль см. Триэтиленгликоль
Триглицерид 2: 89
Тридекановая кислота 2: 96
Тридециламин 2: 192
Тридецилбромид 1: 314
Тридецилцианид 1: 314; 2:192
Триизопропилфосфит 1: 304
Трикрезилфосфат для тонкослойной хрома-
хроматографии 1: 99
Три(о-толил)фосфин 1:488,492
Трилон А см. Нитрилотриуксусная кислота
Трилон Б см. Этилендиаминтетрауксусная
кислота
Триметиламин 1: 344; 2: 316
2,4,6-Триметилацетофенон 2:17
2,5,6-Триметилбензальдегид 2: 61
2,4,6-Триметилбензгидроксимоилхлорид 1:
254
Предметный указатель
469
2,4,6-Триметилбензилхлорид 1:316,465
2,4,6-Триметилбензилцианид 1: 316; 2: 96
1,3,5-Триметилбензол 1:446,451; 2: 116, 408
2,3,5-Триметилгидрохинон 1:454
2,4,4-Триметилпентен 1: 330
2,4,4-Триметилпентанол-2 1: 330
2,2,4-Триметилпентан 2: 350
2,2,4-Триметил-З-оксопентановой кислоты
этиловый эфир 2: 144
Триметилсиланол 1: 320
Триметилсилиламин 1:319
N-Триметилсилилацетамид 1: 319
N-Триметилсилилдиэтиламин 1: 319
а-Триметилсилилоксинитрил 2: 133
N-Триметилсилилпиперидин 1: 319
Триметилсилилцианид 1: 319; 2: 133
Триметилуксусная кислота 2:42, 50, 164
2,4,6-Триметилфенилглиоксаль 2: 17
2,4,6-Триметилфенилуксусная кислота 2: 96
2,4,5-Триметилфенол, производные 2: 347
Триметинцианин 2: 156
3,4,5-Триметоксибензойная кислота 1: 295
1,3,5-Тринитробензол 1:432, 435
2,4,6-Тринитротолуол 1:435
2,4,6-Тринитрофенол см. Пикриновая кислота
2,4,6-Триоксо-1,7-гептандикарбоновой кис-
кислоты диэтиловый эфир 2:145
Триплетное состояние карбенов 1: 349, 393
Триптофан 2: 88,132,291,317
Трис(дибензилиденацетон)дипалладий 1:488,492
Тритиловый спирт см. Трифенилметанол
Тритилхлорид см. Трифенилметилхлорид
Тритон Б 2: 197
Тритретбутилфосфин 1:491
1,2,2-Трифенилаценафтен-1-ол 2: 165
Трифенилметан 2: 106
Трифенилметановый краситель 1:459,468
Трифенилметанол 1: 291,453; 2: 164, 345
Трифенилметилгалогениды 1: 270
Трифенилметилнатрий 2: 141
Трифенилметиловый эфир 1: 298
Трифенилметилхлорид 1: 291, 298, 452
Трифенилметильный радикал 1:240,244
Трифенилфосфан см. Трифенилфосфин
Трифенилфосфин 1: 288, 303,487, 490
Трифенилфосфиноксид 2:136
Трифлаты см. Трифторметансульфоновой
кислоты эфиры
Трифторметансульфоновая кислота как ката-
катализатор Фриделя—Крафтса 1:456
Трифторметансульфоновой кислоты эфиры 1:
272, 278,486
4-Трифторметилбензальдегид 2: 61
4-Трифторметилбензгидроксимоилхлорид 1:
254
Трифторэтанол 1: 278
1,1,1 -Трихлор-2,2-диD-метоксифенил)этан 1:
467
1,1,1 -Трихлор-2,2-диD-хлорфенил)этан см.
дат
2,2,2-Трихлор-1-фенилэтанол 2:164
Трихлорацетальдегид см. Хлораль
Трихлорацетилхлорид 2: 94
1,2,4-Трихлорбензол 1: 340
Трихлорметан см. Хлороформ
Трихлорметилароматические соединения 1:
291
Трихлорметилбензол 1: 251,255; 2: 340, 408
Трихлоруксусная кислота 1: 401; 2: 30, 94, 331,
408
2,4,5-Трихлорфеноксиуксусная кислота 2: 76
2,4,5-Трихлорфенол 1:448, 479
2,4,6-Трихлорфенол, производные 2: 344
Трихлорэтан 1: 366
2,2,2-Трихлорэтанол 2:172, 175, 344
Трихлорэтилен 1: 340 , 366; 2: 220, 340
техника безопасности 2: 386,408
1,2,3-Трицианопропанол-2 2:220
Тридеканонитрил 1: 317; 2: 96, 192
Триэтаноламин 1: 370
Триэтиламин 1:342; 2:159, 316,408
Триэтиленгликоль 1: 24, 54, 371; 2: 386
Триэтилфосфит 1: 304
1,3,3-Триэтоксибутан 1: 337
Тропилия катион 1: 146,425
Тропинон2: 132
Троповая кислота (З-гидрокси-2-феншшро-
пионовая кислота) 2: 165
Тростниковый сахар 2: 66, 329
ТСХ см. Тонкослойная хроматография
ТЭБАХ см. Бензилтриэтиламмония хлорид
Углеводороды 1: 29, 240
идентификация 2: 349
— ИК-спектры 1: 119
— тонкослойная хроматография 1: 89
как растворитель 1:49, 200
получение, радикальное восстановление 1:
262
химические свойства, дегидрирование 2: 43,
46
хлорирование 1: 249
Углеводороды алифатические
идентификация 2: 349
— спектр ЯМР13С 1:137
— масс-спектрометрия 1:145
— физические константы 2: 349
получение взаимодействием соединений Гринь-
470
Предметный указатель
яра и галогенсодержащих соединений 2:
160
и соединений с активным атомом
водорода 2:160
— восстановлением альдегидов и кетонов по
Клемменсену!: 192
через дитиолан 2: 66,184
через гидразон 2:181
двойной связи С=С 1:409, 413
карбоновых кислот по Кольбе 1: 242
хлорметильной группы 1:466
— из алкилиодидов 1: 249
— из борорганических соединений 1: 374
— полимеризацией 1: 383, 405
— циклоолигомеризацией 1:407
химические свойства, аммоксидирование 2:7
— каталитическое дегидрирование 2:43
— хлорирование 1: 255
— окисление 1: 260; 2: 36
Углеводороды арилалифатические
получение 1:449, 481,483,489
химические свойства, окисление 2:12
хлорирование сульфурилхлоридом 1:253
фотохимическое 1: 251
фотобромирование 1: 256
Углеводороды ароматические
идентификация 2: 351
— производные 2: 351, 352
— УФ/вид.-спектр 1: 110
получение каталитическим дегидрированием
2:43.
Углеводороды непредельные см. Ацетилены,
Олефины
Углеводы 1: 46, 84, 369; см. также Сахара
Угольной кислоты эфиры 1: 345; 2: 52, 98, 164
Удерживания время 1:95, 96
Удерживаемый объем 1:97
Удлинение цепи с диазометаном 2: 232, 258
Уксусная кислота 1: 203, 363; 2: 5, 50, 101
азеотроп с толуолом 1: 75
идентификация 1:115; 2: 295, 331
как растворитель 1: 50, 85
очистка и осушение 2: 386
получение 1: 260, 407; 2: 9, 30
превращения 2:94, 218
техника безопасности 2: 386,408
Уксусной кислоты ангидрид см. Уксусный ан-
ангидрид
Уксусной кислоты N-ариламиды 2: 285
Уксусной кислоты бензиламид 2:79
Уксусной кислоты замещенные 1: 211
Уксусной кислоты эфиры 1: 346, 363; 2: 72, 101
бутиловый 2: 71-73, 76, 398
виниловый см. Винилацетат
гексиловый 2:73
гептиловый 2: 73
3-дибромметилфениловый 1: 257
— превращения, гидролиз 1: 294
изобутиловый 2: 71
изопропиловый 2: 71,144
л»-крезиловый 2: 73
метиловый 2: 73
метиловый 2: 9, 76, 106
2-оксоалкиловый 1: 346
октиловый 2: 73
пентиловый 2: 76
пропиловый 2: 71, 144
фениловый 2: 73
[3-D-хлорфенил)-Д2-1,2-оксазолин-5-ил]овый
1:398
циклогексиловый 2: 73
этиловый 1:75, 85; 2:76, 391
— получение 2: 9, 178, 391
— превращения 2: 144,164
— техника безопасности 2:391,411
Уксусный ангидрид (ацетангидрид) 1: 338,362
Ускоритель вулканизации 1: 310; 2: 102
Фаза в хроматографии
неподвижная (стационарная) 1: 86
подвижная 1: 85
Фактор разделения C при экстракции 1: 78
Фарнезол2:114
Фелингова жидкость 2: 28, 302, 387
Фелькина—Ана модель 1: 226; 2:173
Феназон 2: 82
Фенантрен 2: 6, 30, 351
Фенантренхинон 2: 32, 327
Фенантридон 2: 284
Фенацетилмалоновой кислоты диэтиловый
эфир 2:154
Фенацетилхлорид 2: 94, 154, 260
Фенацилбромиды 1: 299; 2: 219, 324
Фенациловый эфир (сложный) 1: 270, 299
Фенацилфосфния соли 2:137
Фенацилхлорид 1:271; 2: 324
Фенетидин 1:449; 2: 290, 311, 408
Фенетол 1: 296, 462
1-Фенилазонафтол-2 2:253
Фенилаланин 2: 318
N-Фениламинофумаровой кислоты димети-
ловый эфир 2: 207
Фенилацетальдегид 2: 274, 320
Фенилацетальдегида диметилацеталь 2:66
превращения 1: 337, 414
Фенилацетамид 2:281
Фенилацетилен 1: 342, 365, 398,487; 2: 354, 355
Фенилацетон 1:478; 2: 87,131, 181, 187, 324
Предметный указатель
471
4-Фенилацетоуксусной кислоты эфиры 2:151
4-Фенилацетофенон 1: 342; 2: 219
1-Фенилбутен-3-карбонитрил-2 2:217
2-Фенилбуган-3-ол-2 2:114
4-Фенилбутен-3-ол-2 2:172
1-Фенилбутадиен-1,3 2: 139
2-Фенилбутадиен-1,3 1:400
4-Фенилбутанол-2 2: 185
4-Фенилбутанон-2 1: 414; 2: 87
2-Фенилбутан см. Бутилбензол
а-Фенилбутиронитрил 2: 217
а-Фенилвалеронитрил 2: 217
Фенилвинилкетон 1: 338
Фенилвиниловый эфир 1: 381
Фенилгидразин 2: 59, 60, 79, 209, 246, 311
техника безопасности 2: 408
З-Фенилгидразинопентандион-2,4 2:253
Фенилгидразоны 2:57,290, 320
Фенилгидроксиламин 2: 234, 301
Фенилгликолевая кислота см. Миндальная
кислота
Фенилглиоксаль 2:14,177
З-Фенилглицидной кислоты метиловый эфир
2:144
Фенилглицин 2: 111
Фениддиазония соли см. Бензолдиазония соли
1-Фенил-3,4-дигидронафталин2:45, 164
Фенилендиамины 1:435; 2: 80, 82, 311,408
а-Фенилизовалеронитрил 2:217
Фенилизотиоцианат 2: 102
Фенилизоцианат 2: 100, 101, 156, 222, 282,
408
N-Фенилкарбамоилмалоновой кислоты диме-
тиловый эфир 2: 156
Фениллитий 1: 378; 2:165
Фенилмагнийбромид 2:164
Фенилмалоновой кислоты диэтиловый эфир
2:143, 147
у-Фенилмасляная кислота 1:458; 2:183
Фенилметантиол 1: 310, 386; 2: 356
Фенилмочевина 2: 317
1-Фенилнафталин 2:45
Фенилнитрометан 1: 312
3-Фенил-1,2-оксазол 1: 398
3-Фенил-Д2-1,2-оксазолин 1: 398
З-Фенилоксиранкарбонитрил-2 2:124
З-Фенилпентин-1-ол-З 2:114
1-Фенилпентандион-1,4 2:203
1-Фенил-5-пиперидинопентен-1-он-3 2:131
1-Фенил-3-пиперидинопропанон-1 1: 338; 2:
131
1-Фенилпиразол 2: 60
1-Фенилпропандион-1,2 2:17
1-Фенилпропанол-1 2:22,164
З-Фенилпропановая кислота 1:414; 2:19, 331
З-Фенилпропановой кислоты этиловый эфир
2:278
Фенилпропиоловая кислота 1: 343
Фенил(тиенил-2)кетон 1:458
Фенилтиомочевина 2: 310
З-Фенилтиопропановой кислоты морфолид 2:
19
З-Фенилтиопропиононитрил 1: 386
Фенилуксусная кислота 1: 316; 2: 94, 96, 160,
164,331
Фенилуксусной кислоты хлорангидрид 2: 94
Фенилуксусной кислоты этиловый эфир 2:
142-144,192,278
Фенилуретаны 2: 102, 343
4-Фенилфенацилбромид 1: 299; 2:219
4-Фенилфенациловый эфир 1: 299; 2: 330
1-Фенил-2-фенилтиоэтан 1:386
Фенилфосфоновой кислоты диэтиловый эфир
1:478
2-Фенилфуро[3,2-Ь]пиридин 1:485
N-Фенилхинонимин 2: 35
Фенильный радикал 1: 243
1-Фенилэтандиол-1,2 1:370; 2: 274
2-Фенилэтантиол-1 1:310; 2: 356
1-Фенилэтанол 1: 367; 2: 26, 172
2-Фенилэтанол 1: 375; 2:192, 343
1-Фенилэтиламин 1:225; 2:79,181,187, 188, 311
2-Фенилэтиламин 1: 316; 2: 172, 185, 187, 311,
314
2-Фенилэтилбромид 1: 284; 2: 340
2-Фенилэтилиодид, производные 2: 340
1-Фенилэтилхлорид 1: 252, 253, 327
2-Фенилэтилхлорид 2: 340
Фенобарбитал (веронал) 2: 83
Феноксиуксусная кислота 1: 298; 2: 331
Фенол 1:203,418,453; 2:106
идентификация 1: 130; 2: 347
как растворитель 1: 277
получение 1: 259,443,448,473; 2: 245, 276
превращения 1: 295, 296, 314, 381, 446, 449,
466,470; 2: 56, 72
техника безопасности 2:408
Фенола простые эфиры 1:295; 2: 232, 258
химические свойства, расщепление эфиров 1:
289
синтез Гаттермана 1:459
синтез Вильсмейера 1:460
Фенола сложные эфиры 1:456
Феноддисульфоновая-2,4 кислота 1:436
Фенолкарбоновые кислоты 1:469
Фенолформальдегидные полимеры 1:453,466
Фенолы
идентификация, ИК 1:118
— качественная реакция 2:148, 303
— производные 2: 343
472
Предметный указатель
— растворимость 2: 288
— тонкослойная хроматография 1:90
получение гидролизом галогенароматичес-
ких соединений 1: 448, 473, 477
— действием расплавленной щелочи на аро-
ароматические сульфокислот 1:443,478
— из металлароматических соединений 1:
481,483
— из растворов солей диазония 2: 232, 244,
245
— перегруппировкой арилгидропероксидов
1: 259; 2: 286
реакции, аминометилирование 1:464
— арилирование 1: 492
— бромирование 1:446
— иодирование 1:444
— карбоксилирование 1: 469
— метилирование диазометаном 2: 258
— нитрование 1:433
— нитрозирование 1: 470
— присоединение винилогов карбонильных
соединений 2: 196
нуклеофилов, ацетилен 2: 379
— роданирование 1:448
— синтез Вильсмейера 1:460
— синтез Гаттермана 1: 459
— с соединениями Гриньяра 2: 160
— триметилсилилирование 1: 318
— этерификация 1:295; 2: 232
Феноляты 1: 216, 277, 294
Фенотиазин 2: 45
Фенэтиламин см. Фенилэтиламин
Феруловая кислота 2: 126
Фильтрование 1: 46
Фильтр-пипетка 1: 39
Финкельштейна реакция 1: 266, 282, 310
Фитол 2: 114
Фишера проекции 1: 221
Фишера синтез
аминокислот 1: 302
индолов 2: 247, 289, 292
Флегмовое число 1:65, 71
Флеш-хроматография 1: 93
Флороглюцин 1:470; 2: 347
Флотореагент 1: 302
Флуорен2: 15,352
Флуоренон 2:15, 284, 324
Формалин см. Формальдегид
Формальдегид 1:463, 466; 2:117, 124
идентификация, производные 2: 320
получение 2: 28,41, 185
реакции 2:118,135, 130, 178, 180
— Кажиццаро 2: 176,178
— с соединениями Гриньяра 2:161
техника безопасности 2: 387, 408
Формальдегида циангидрин 2: 109, 111
Формамидомалоновой кислоты диэтиловый
эфир 2: 244
4-Формилбензонитрил 2: 15
2*-Формилдифенилкарбоновая-2 кислота 1:
374
Формилуксусной кислоты эфир 2: 143
Формилфторид 1:459
Формилхлорид 1:459
Формимидхлорид 1:459
Форон2: 116,323
Фосген 1: 30, 252; 2: 5, 100, 99, 387
очистка и осушение 2: 387
техника безопасности 2:409
Фосфан см. Фосфин
Фосфиналкены см. Алкилиденфосфораны
Фосфинистой кислоты эфиры 1: 305
Фосфины 1: 303, 487,491,492
Фосфонистой кислоты диэфиры 1: 305
Фосфония илиды 2: 136, 138; см. также Алки-
Алкилиденфосфораны
Фосфоновой кислоты фторангидриды 1: 305
М-(Фосфонометил)глицин 1: 302
Фосфор белый 1: 158; 2:408
Фосфор красный 1: 286; 2:409
Фосфора пентаоксид 1: 36; 2:409
Фосфора пентасульфид 2:102
Фосфора пентахлорид 1: 252, 284; 2: 93, 94
техника безопасности 2:409
Фосфора соединения 1: 303
Фосфора трихлорид 1: 284; 2: 93, 94, 409
Фосфора хлориды 1: 285; 2: 93
Фосфористой кислоты эфиры 1: 285, 305
Фосфорная кислота 2:409
Фосфорномолибденовая кислота 1: 90
Фосфорорганические соединения 1: 303, 305,
383; 2:135
Фотобромирование 1: 256
Фотолиз 1:242, 383
Фотонитрозирование 1: 262
Фотохлорирование 1: 254
Фотоэлектронные спектры 1:212
Фрагментация 1:143, 145, 324
Фриделя—Крафтса алкилирование 1: 199, 266,
426, 430, 449,452
Фриделя—Крафтса ацилирование 1: 382, 426,
454, 457, 458
Фриса перегруппировка 1:456
Фронтальные орбитали см. Граничные орби-
тали
Фруктоза 2: 60, 329
Фталевая кислота 1: 300; 2: 12, 13, 331
Фталевой кислоты ангидрид 1:456; 2: 13
получение 2: 32, 39, 90
превращения 1:459; 2:73,94,171
Предметный указатель
473
техника безопасности 2:409
Фталевой кислоты эфиры 1: 97, 147; 2: 73
диалкиловый 1:99
дибутиловый 1: 54; 2: 76
диоктиловый 1: 147
диэтиловый 2:71, 76
Фалевый альдегид 1: 293; 2: 321
Фталимид 1: 231, 300; 2: 208, 281
р-Фталимидопропионитрил 2: 208
Фталоиддихлорид 2: 94
Фталонитрил 2: 10
Фталоцианины 2:10
1-Фтор-2,4-динитробензол 1: 474
Фтор 1: 240, 362
Фторбензол 1: 115; 2: 244, 340
Фториды 1:271, 276, 311,322
Фтороводородная кислота 1: 158
Фтороводород 1: 203, 240, 282, 355
Фторхлоруглеводороды (фреоны) 1: 312
Фукоза 2: 329
Фумаровая кислота 1: 219; 2: 9
бромирование 1: 359, 365
идентификация 1: 102; 2: 331
техника безопасности 1: 92; 2: 409
Фумаровой кислоты диамид 2: 79
Фумаровой кислоты эфиры
т/>е/я-бутилэтиловый 2: 72
диметиловый 1: 390
диэтиловый 2: 71
моноэтиловый 2: 72
Функциональная группа 2: 293, 298
3-(Фурил-2)акриловая кислота 2: 126
Фуран 1: 399, 401, 425, 456,459,482
Фуранкарбоновая-2 кислота 2: 179
Фуранозы 2: 66
2,2'-Фуроин 2: 133
Фурфуриламин 2: 187
Фурфурилиденмалононитрил 2: 126
Фурфуриловый спирт 2: 178, 343, 409
Фурфурол 2: 320
реакции 2: 56, 187,133, 126, 134, 178, 180
техника безопасности 2:409
Халкон1:414;2:118
Характеристические группы (номенклатура) 1:
179
Характеристические полосы в ИК-спектрос-
копии 1: 119
Характеристические фрагменты в масс-спект-
роскопии 1: 149
Хека реакция 1:490
Хелатный комплекс 2:148
Хелидоновой кислоты эфиры 2:142
Хемилюминесценция 1: 397
Хемосорбция 1: 201, 410; 2: 184, 189
Химический сдвиг 1: 127,130, 135, 138
Хинсберга расслоение 2: 264, 266, 305
Хиналвдин B-метилхинолин) 2: 316
Хинизарин A,4-гидроксиантрахинон) 1: 457
Хинин 1: 225
Хиноксалины 2:59
Хинолин 1: 54, 339, 473; 2: 39, 198, 229, 316
Хинон см. й-Бензохинон
Хинонимины 2: 35, 46
Хиноны 1: 456, 458; 2: 31, 32, 45, 46, 294, 298,
327
Хиральность 1: 107, 220, 223
Хлор 1: 29; 2: 388
техника безопасности 2:409
химические свойства 1: 386
гомолиз 1: 191
присоединение к олефинам 1: 355, 363,
384
радикальное замещение 1: 249
электрофильное замещение в аромати-
ароматических соединениях 1: 443
энергия связи 1: 240
(З-Хлоракролеин 2: 209
Хлоралканы 1: 255; см. также Алкилхлориды
Хлораль1:362;2:62
идентификация, производные 2: 320
получение из гидрата 1: 468
превращения 1:468; 2: 30,175, 172,164
техника безопасности 2: 409
Хлоральгидрат 1:468; 2: 62, 409
Хлорамин 1: 364; 2: 266
N-Хлорамины 1: 254
Хлорамфеникол (левомицетин) 2: 121
Хлоранил (тетрахлор-л-бензохинон) 2: 6, 45,
328
Хлоранилин 1: 435; 2: 235, 311,409
превращения 1: 449; 2: 100, 245, 247, 250
Хлорантрахинон 1: 478
Хлорарены 1: 443; 2: 248
Хлорацетамид 2: 79
4-Хлорацетанилид 2: 103
Хлорацетон, производные 2: 322
Хлорацетонитрил 1: 270, 305; 2:124
техника безопасности 2:409
а-Хлорацетоуксусной кислоты этиловый эфир
2:219
4-Хлорацетофенон 1:458; 2:42, 182
2-Хлорбензальдегид 1: 292; 2: 178, 320,409
З-Хлорбензальдегид 2: 61, 178
4-Хлорбензальдегид 1: 292; 2: 61, 136, 178, 320
Хлорбензальдегида оксим 2: 61
Хлорбензгидроксимоилхлорид 1: 254, 398
Хлорбензилбромид 1: 257, 259, 316; 2: 340
474
Предметный указатель
4-Хлорбензилидендибромид 1: 257, 292
Хлорбензилидендихлорид 1: 252, 292
2C)D)-Хлорбензиловыйспирт2:178
2-Хлорбензилхлорид 1: 252, 253, 316
4-Хлорбензилхлорид 1: 252, 253, 316; 2: 340
2C)D)-Хлорбензилцианид 1: 316; 2: 96
3D)-Хлорбензоилхлорид 2: 94
2-Хлорбензойная кислота 2: 331
З-Хлорбензойная кислота 2:94, 331
4-Хлорбензойная кислота 2:12,13,42, 94, 331
Хлорбензол 1: 54,447
техника безопасности 2: 407
химические свойства, гидролиз 1: 439, 458,
468,473,477
4-Хлорбензолсульфонамид 2: 266
4-Хлорбензолсульфоновая кислота 2: 357
4-Хлорбензолсульфохлорид 1:439; 2:264,266
2-Хлорбензонитрил 2: 250,409
4-Хлорбензонитрил 2: 250
2-Хлор-1,4-бензохинон 2: 327
7-Хлорбицикло[4.1.0]гептан 1: 262
2-Хлорбутадиен-1,3 1:251,366
4-Хлорбутанон-2 1:338
Хлорбутан см. Бутилхлорид
р-Хлорвинилальдегид 2: 209
Хлоргидрин 1: 298; 2: 525
получение 1: 364, 366
1-Хлоргидрин см. З-Хлорпропандиол-1,2
Хлоргидрохинон 2: 347
4"-Хлор-Г,2-диметилбензол 2: 340
4-Хлор-1,3-диметилбензол 2:340
Хлордиметиловый эфир 1:464; 2: 396
1-Хлор-2,4-динитробензол 1:474,475; 2: 410
Хлордифторметан 1: 312
2-Хлор-1,4-диэтилбензол 2: 340
4-Хлориодбензол 1:487
Хлорирование ароматических соединений 1:
239, 249, 263,493
Хлоркарбен 1: 348
а-Хлоркарбонильные соединения 2: 219
а-Хлоркарбоновая кислота, эфир 2:122
р-Хлоркоричный альдегид 2: 209
Хлоркрезол 1:448
Хлорметан 1: 139
2-Хлорметил-4-нитрофенол 1:466
Хлорметилирование ароматических соедине-
соединений 1:426,465
1-Хлорметилнафталин 1: 305, 308, 316,466
2-Хлорметилтиофен 1:466
Хлорметилфенилкетон 2:260
1-Хлор-2-метил-2-фенилциклопропан 1: 262
Хлормуравьиная кислота 2: 52, 82, 98, 220
1B)-Хлорнафталин 2: 341
1-Хлор-2-нитробензол 1: 473, 485; 2: 235, 250,
410
1-Хлор-З-нитробензол 2: 235, 250,410
1-Хлор-4-нитробензол 1:435,473; 2: 235, 410
2-Хлор-4-нитротолуол 1:435
C-Хлор-4-нитрофенил)диметилтионфосфат
1:305
Хлороводород 1: 203, 240, 355; 2: 201, 389,410
Хлоропрен 1: 363, 366
Хлороформ 1: 75, 255; 2: 390
идентификация, физические константы 2:
341
как растворитель 1: 84, 116
получение 2: 41
превращения 1: 389, 394; 2: 301
техника безопасности 2: 390, 410
Хлорпентаны 1: 255
1-Хлорпропанол-2, производные 2: 343
З-Хлорпропандиол-1,2 1: 366
З-Хлорпропановая кислота 1: 338
З-Хлорпропановой кислоты этиловый эфир 2:
201
Р-Хлорпропионовый альдегид, ацеталь 1: 343
З-Хлорпропиононитрил 2:201
4-Хлорстильбен 2: 136
N-Хлорсукцинимид 1: 252, 254
Хлорсульфонирование 1:438, 439, 443, 493
Хлорсульфоновая кислота 1:437; 2:410
Хлорсульфурон 2: 267
5-Хлор-2,3-тетраметилениндол 2: 290
4-Хлрртиоацетанилид 2:103
4-Хлортиоацетанилид 2:103
Хлортион см. C-Хлор-4-нитрофенил)диме-
тилтиофосфат
4-Хлортиофенол 2: 264, 356
4-Хлортолан 1:487
Хлортолуол 2: 249, 340, 341
превращения 1: 252, 253, 257, 259,477
техника безопасности 2: 410
Хлортриметилсилан 1: 318
1-Хлор-2,4,6-тринитробензол 1:473
Хлортрифенилметан см. Трифенилметилхло-
рид
Хлоруксусная кислота 1:298,302, 310,340,366;
2:50,219,331,410
Хлоруксусной кислоты этиловый эфир 1: 270;
2:71,78,79,144,213,411
3-D-Хлорфенил)-1,2-оксазол 1: 398
3-D-Хлорфенил)-5-фенил-Д2-1,2-оксазол 1:
398
3-D-{лорфенил)-5-фенил-А2-1,2-оксазолин 1:
398
2C)D)-Хлорфенилгидразин 2: 79, 247
4-Хлорфенилизоцианат 2:100
4-Хлорфенилтиоцианат 2:101
2C)D)-Хлорфенилуксусная кислота 2: 96
1-Хлор-2-фенилэтан см. 2-Фенилэтилхлорид
Предметный указатель
475
1-Хлор-1-фенилэтансл«. 1-Фенилэтилхлорид
3D)-Хлорфенол 1:447; 2: 245, 347, 410
Хлорциклогексан см. Циклогексилхлорид
2-Хлорциклогексанол 1: 288
2-Хлорциклогексанон 2: 219
1-Хлор-2,3-эпоксипропан 1: 255, 366, 371
2-Хлорэтанол 1:17, 125,179, 306; 2: 343
1-Хлор-4-этилбензол 2:182
2-A-Хлорэтил)тиофен 1: 343
Холестен-4-он-З 2:65, 176
Холестен-5-она-З зтиленацеталь 2:65
Р-Холестанол 1:414
Холестерол (холестерин) 1: 414; 2: 44, 175, 343
Холестерина ацетат 2: 73
Холин 1: 305
Холиновый эфир 1: 304
Холинэстераза 1: 305
Холостой опыт 2: 294
Хорнера—Вадсворта—Эммонса реакция 1: 304;
2:135
Хризантемовой кислоты эфиры 1: 394
Хрома соединения 2: 21,47
Хроматриоксид2: 21, 31,410
Хроматограмма 1:91, 95
Хроматография 1:85, 86,92,97,164; 2: 317, 328,
329
Хромовая кислота 2: 21
Хромовой кислоты ангидрид см. Хрома триок-
сид
Хромовая смесь (окислитель) 2: 10
Хромофор 1: ПО, 114
Хэммонда постулат 1: 196
Хюккеля правило 1:425
Целлобиоза 2: 65, 329
Целлозольв (моноалкиловые эфиры этилен-
гликоля, растворители) 1: 371
Целлофан 1: 371
Целлулоид 1: 286, 289
Целлюлоза 1: 85; 2: 65
Целлюлозы динитрат 1: 287
Целлюлозы ксантогенат 2: 101
Целлюлозы простые эфиры 1: 298
Целлюлозы тринитрат 1:286
Цеолиты 2: 396
Церевитинова метод 2:160
ЦерияAУ) соединения 2:46
ЦерияAУ)-аммония нитрат 2:10, 15, 302, 390
2-Циан-3,3-бис(метилтио)пропен-2-овой
кислоты метиловый эфир 2:156, 210
2-Циан-3-этоксипропен-2-овой кислоты эти-
этиловый эфир 2: 148,210
Цианамид 2:148
Цианаты 2: 223
Цианацетамид 2: 79, 156, 253
Циангидрин 2: 50, 104, 108, 223
Циангидриналлиловый эфир 2: 109
Цианид цинка 2: 391,411
Цианиды см. Нитрилы
Цианин 1: 114
Циановодород см. Синильная кислота
2-Циано-2-фенилтиокарбамоилуксусной кис-
кислоты этиловый эфир 2:156
а-Цианокоричной кислоты метиловый эфир
2:126
а-Цианокоричной кислоты этиловый эфир 2:
20
а-Циано-р-метилкоричной кислоты этило-
этиловый эфир 2:126
Цианометиловые сложные эфиры 2: 78
2-Циано-3-метилпентен-2-овой кислоты эти-
этиловый эфир 2:126
Цианометилфосфоновой кислоты диэтило-
вый эфир 1:305
а-Циано-4-метоксикоричной кислоты мети-
метиловый эфир 2: 126
З-Цианопиридин см. Пиридинкарбонитрил-3
З-Циано-3-фенилпировиноградной кислоты
этиловый эфир 2: 144
а-Цианофенилуксусная кислота, этиловый
эфир 2: 147
Циануксусная кислота 1:318; 2:86,97,125,126
Циануксусной кислоты эфиры 2: 125
замещенные, гидролиз 2: 86
метиловый 2: 20, 79,126, 156
получение 2: 143
химические свойства, аминолиз 2: 78
этиловый 2: 20, 79, 97, 106, 126,148, 156, 253
Циануровая кислота, превращения 1: 447
р-Цианэтилацетиламиномалоновый эфир 2:
204; см. также АцетиламиноJ-циан-
этил)малоновый эфир
2-B-Цианэтил)ацетоуксусной кислоты этило-
этиловый эфир 2: 204, 290
1-B-Цианэтил)-2-оксоциклогексанкарбоно-
вая кислота, этиловый эфир 2: 204
Ы-B-Цианэтил)-е-капролактам 2: 204, 125
Цианэтилирование 2: 196, 208, 229
Циглера—Натты полимеризация этилена 1:
405
Цикламат 2:15
Циклизация 1: 353, 388
Циклоалканы 2: 349
Циклоалкены 2:194
Циклобутан 1: 395; 2: 203
Циклобутандикарбоновая-1,1 кислота 2:86, 88
Циклобутанирование 1: 212
Циклобутанкарбоновая кислота 2: 88
476
Предметный указатель
Циклобутанон, производные 2: 220
Циклобугилтриметиламмония гидроксид 1:333
Циклогексен-З-карбальдегид-1 1:402
Циклогексадиен-1,3 1: 392, 398,402; 2: 354
Циклогексан 1:417
идентификация 2: 349
как растворитель 1: 84,116
конформация 1: 218
превращения 1: 253, 261; 2: 38
техника безопасности 2: 410
Циклогександиол-1,2 1: 369, 373; 2: 273
Циклогександион-1,2 2:17
Циклогександион-1,3 (дигидрорезорцин) 1:
414; 2:114, 203,214
Циклогександионы-1,3 2:202
Циклогексанкарбоновая кислота 1: 418; 2: 165
Циклогексанол 1:418; 2: 9, 410
производные 2: 343
превращения 1: 319, 336, 381; 2: 25, 26,43, 73
Циклогексанон 2: 28, 410
получение 2: 9, 22, 25, 26
идентификация 1:119; 2:324
реакции 2:17,20,58,63,65,109,111,114, 124,
126, 138, 142, 176, 178, 187, 204, 203, 219,
285, 286
— как дегидрирующий агент 2: 175
— Манниха!: 130
— до адипиновой кислоты 2:41
Циклогексанона производные
диаллилацеталь 2: 289
диэтилацеталь 1: 337; 2: 63
оксим1:192,468
— Бекмана перегруппировка 1:192; 2:284, 285
— получение из циклогексана 1: 262
из циклогексанона 2: 285
циангидрин 2:109
этиленацеталь 2: 65
Циклогексантиол, производные 2: 356
Циклогексен 1: 327; 2:410
идентификация 2: 354
получение 1: 336, 342
химические свойства 1: 259, 365, 369, 373
арилирование 1:491
бромирование 1: 359
гидроксилирование 1: 373
окисление 2: 38
цис- Циклогексен-4-дикарбоновой-1,2 кисло-
кислоты ангидрид 1:402
Циклогексилацеталъдегида диметилацеталь 1:414
Циклогексилбромид 1: 284, 342
Циклогексилвиниловый эфир 1: 381
Циклогексилзамещенные соединения при ра-
радикальной циклизации 1: 388
Циклогексилиденциануксусной кислоты эти-
этиловый эфир 2:126
Циклогексилиодид 1: 286; 2: 340
Циклогексилметилкетон 2:165
Циклогексиловый спирт см. Циклогексанол
Циклогексилокситриметилсилан 1: 319
Циклогексилтриметиламмония гидроксид 1:
333
Циклогексилхлорид 1: 253
Циклогептанон 1:119; 2:278
Циклогептатриен 1: 393
Циклогептатриенильный катион 1:425
Циклогептилтриметиламмония гидроксид 1:333
Циклогексилбензол 1:452
р-Циклогексилпропановой кислоты этиловый
эфир 2:166
Циклододекан 1:418
Циклододеканол 1:418
Циклододеканон 1:408, 418
Циклододеканона оксим 1: 418
Циклододекатриен 1: 373, 408, 418
1,5-Циклооктадиен (cod) 1:485
Циклооктатетраен 1: 344,408
Циклоолигомеризация 1: 407
Циклопентадиен 1: 392, 398; 2: 354, 411
химические свойства 1:400, 402; 2: 124
Циклопентадиенилирование 1:425
Циклопентан 2: 349
Циклопентанкарбальдегид 2: 273
Циклопентанол 1: 336; 2: 26, 185, 343
Циклопентанон 1:119; 2: 26, 322,410
химические свойства 1: 398; 2:58,65,185, 204,
284,287
Циклопентанона диаллилацеталь 2: 289
Циклопентанона этиленацеталь 2: 65
Циклопентаны хлорированные 1: 340
Циклопентен 1: 369; 2: 269, 354
Циклопентилзамещенные соединения при ра-
радикальной циклизации 1: 388
Циклопентилтриметиламмония гидроксид 1:333
Циклоприсоединение 1: 215, 353, 382,
391-395,398,401,422
Циклопропандикарбоновая-1,1 кислота 2:86,213
Циклопропанирование 1:212
Циклопропаны 1: 349, 353, 394; 2: 257
Циклопропенильный катион 1:425
Циклофосфамид 1: 303
Цимат (диметиддитиокарбамат цинка) 2:101
я-Цимол 2: 352, 354
Цинк для восстановления карбонильных сое-
соединений 2: 189
Цинка оксид как катализатор дегидрирования
2:26
Цинка хлорид 2: 337
как катализатор Фриделя-Крафтса 1:449,456
Цинкорганические соединения 1: 395; 2: 166
Циннамилиденмалононитрил 2: 127
Предметный указатель
477
Циннамилиденциануксусной кислоты мети-
метиловый эфир 2: 127
Циннамоилхлорид 2:94
Ципрофлоксацин 1:474
Циркон-ализариновая проба 2: 297
Цистеин 2: 262, 317
Цистин 2: 262, 298
Цитохромы 1: 371; 2: 8
Цитраль2: 118
ЧЗМО (частично занятая молекулярная орби-
таль) 1:192, 249, 383
Чичибабина синтез 1:474
Шаровой конденсатор 1:11
Шарппесса—Кацуки эпоксидирование 1: 227,
369,420
Шимана реакция 2: 244
Шиффа основания 2: 51, 56,184
Шленка равновесие 2: 159
Шлифов типы 1: 9, 15
Шлоссера модификация реакции Виттига 2: 137
Шмидта реакция 2: 279, 282, 283, 292
Шоттена—Баумана реакция 2: 75
Штреккера синтез 2: 97, 109,111, 224
Щавелевая кислота 2: 69, 331,411
Щавелевой кислоты диметиловый эфир 2: 85
Щавелевой кислоты диэтиловый эфир 2: 71,
144,396
Эбуллиометр 1: 105
Эванса соотношение 1: 196
Эвгенол, производные 2: 347
ЭДТА см. Этилендиаминтетрауксусная кислота
Эйнхорна реакция 2:41, 74
Эйринга соотношение 1:81
Экзотермические реакции 1:196
Экстракция 1:45, 78-82
Эластомеры 1: 373, 379, 390
Электродный потенциал 2: 6
Электронная поляризуемость 1: 107
Электронные колебания 1: ПО
Электронные состояния 1:111
Электронные орбитальные переходы 1:111
Электронный перенос 1: 191
Электроноакцепторные группы 1: 111, 204,
353,423, 427; 2:49
Электронодонорные группы 1: 111, 204, 269,
353,427; 2:49,196, 210
Электрофилы 1:191, 204, 215
Электрофугная группа 1: 141
Элементарные реакции 1: 197
Элементы, обнаружение 2: 295
Элеостеариновая кислота 2: 89
Элиминирование 1: 192, 298, 323, 350
анти- 1: 331
бимолекулярное 1: 325, 327
восстановительное 1:406,490
ионное 1: 323
мономолекулярное 1: 324
по Зайцеву 1: 328
син-1:331, 345
стереоэлектронные условия 1: 331
стерическая характеристика 1: 331
<х,а-1: 348
а,р-, воды 1: 323
— галогеноводородов 1: 323
— триалкиламинов 1: 323
Элюат 1: 87
Элюент при тонкослойной хроматографии 1:88
Эмульгатор 1: 371
Эналаприл 2: 82
Энантиомерный избыток 1: 227
Энантиомеры 1:87, 220,225; 2:172,187,216
Энантиоселективный синтез см. Сгереоселек-
тивный синтез
Энантиотопия 1: 223; 2: 216
Энергия
активации 1: 195
внутренняя 1: 193
диссоциации связи 1: 195, 240
— гетеролитической 1: 271
ионизации 1:212
свободная см. Гиббса энергия
испарения 1:53,64
Эозина проба 2: 296
Эпилокс 1: 371
Эпихлоргидрин см. 1-Хлор-2,3-эпоксипропан
Эпоксидирование алкенов 1: 227, 353, 369, 391
Эпоксидные смолы 1: 255, 366, 371,467
Эпоксиды 1: 367, 420
из олефинов и перкислот 1:369; 2:274
из олефинов и кислорода 1: 253
получение из хлоргидринов 1: 370
химические свойства 1: 372; 2: 275
2,3-Эпоксипропанол 1: 371
2,3-Эпоксипропилхлорид см. 1-Хлор-2,3-
эпоксипропан
2,3-Эпоксипропионитрил 2: 124
1,2-Эпоксициклогексан 1: 367, 369
1,2-Эпоксициклопентан 1: 369
1,2-Эпоксиэтилбензол 1: 369
Эритроза 1:222
Эритро-форма 1: 223
478
Предметный указатель
Эрленмейера синтез аминокислот 2:104
Эрленмейера колба 1: 10, 162
Эстроген 2:114
Эстрон2: 114
Эталонное топливо 1: 378
Этан 1:139, 203, 240, 245, 255; 2: 6
Этандитиол-1,2 2:66,356
Этанол 2:6, 391,411
азеотроп с тетрахлорметаном или водой 1: 75
значение рА 2: 106
идентификация, производные 2: 342
как растворитель 1: 84, 116
осушение 2: 391
получение 1: 293
химические свойства 1:192,296,381; 2:71,264
ЯМР'Н 1:133
Этаноламин 1: 370, 371
Этантиол 1: 203; 2: 356
Этантрикарбоновой-1,1,2 кислоты триэтило-
выйэфир2:213
Этен см. Этилен
Этерификация
азеотропная, экстрактивная 1: 282; 2: 69, 70
спиртов с диазометаном 2: 232, 258
— с неорганическими кислотами 1: 265, 282,
284; 2: 242
карбоновых кислот 2: 69, 70
Этиламин2:311,411
Этиламина гидрохлорид 2: 281
N-Этиланилин 2:172, 311, 411
2-Этилантрахинон 2: 33
Этилацетат см. Уксусной кислоты этиловый эфир
4-Этилацетофенон 2: 17
Этил бензол 1:453; 2:411
идентификация, производные 2: 352
получение 1:414; 2: 182
реакции 1: 252, 253, 439; 2:10, 12, 15
4-Этилбензолсульфохлорид 1:439; 2: 263
Этилбромид 1: 284, 305; 2:411
идентификация, производные 2: 340
превращения 1: 296, 306; 2: 213, 217
Этилвиниловый эфир 1: 381,402
2-Этилгександиол-1,3 2:25
2-Этилгексанол-1 2: 7,121,185, 343
З-Этилгексанол-3 2:164
Этилгексиловый эфир 1: 296
4-Этилгептанол-4 2: 164
2-Этил-З-гидроксигексаналь 2: 118, 185
2-Этил-1-гидроксигексанон-3 1: 305
Этилгидросульфат 1: 286
1-Этил-3,4-диметоксибензол 2:182
Этилен 1: 203, 355, 359, 366, 378, 383, 390
получение 1: 325, 409
реакции, бромирование 1:192
— металлирование 1:480
— с кислородом 1: 370
техника безопасности 2: 411
спектр ЯМР13С 1:138, 139
Этиленацеталь 2: 64
Этиленгликоль 1: 24, 371
идентификация, производные 2: 343
как растворитель 1: 278
получение 1: 293, 370; 2: 38
превращения 1: 381; 2:40, 64
очистка и осушение 2: 392
техника безопасности 2: 392, 411
Этиленгликоля эфиры 1: 370
диметиловый 1: 276
моноалкиловый 1: 371
монометиловый 2: 339, 343
моноэтиловый 2: 343
Этиленгликоля динитрат 1: 286
Этилендиамин 1: 366; 2: 311
Этилендиаминтетрауксусная кислота 2:109
Этилендибромид см. 1,2-Дибромэтан
Этилендихлорид см. 1,2-Дихлорэтан
Этиленоксид 1: 298, 370, 379; 2: 392,411
Этиленхлоргидрин см. 2-Хлорэтанол
Этиленциангидрин см. р-Гидроксипропио-
нитрил
Этилизопропилкетон 2:22
Этилизопропиловый эфир 1: 349
Этилиодид 1:134, 286, 305; 2: 340
реакции 1:296,300,305
Этилмагнийбромид 2: 164
Этилмалоновая кислота 2: 86, 88
Этилмалоновой кислоты диэтиловый эфир 2:
86,213
М-Этил-М-метиламид бензолсульфоновой
кислоты 1: 300
а-Этил-а-метилбензилгидропероксид 1: 261
I-Этил-4-метилбензол 2: 183
Этилметилкетон 1: 84, 363; 2: 28, 322, 411
получение 2: 9
реакции 2: 15, 20, 65,114, 164
Этилметилкетона этиленацеталь 2: 65
Этилметилкетона циангидрин 2:109
5-Этил-2-метилпиридин 2:124
1-Этил-2D)-метилхинолиния бромид 2: 157
1-Этил-4-метоксибензол 2: 182
1-Этил-2C)D)-нитробензол 2: 15
З-Этилпентанол-3 2:164
Этилпропиловый эфир 1: 349
Этилтрифенилфосфония бромид 1: 303; 2: 139
З-Этил-1,3-тиазолия бромид 2:135
4-Этилтиофенол 2: 263
Этилфенилбарбитуровая кислота 2: 82, 149
4-Этилфенилглиоксаль 2:17
Этилфосфоновой кислоты диэтиловый эфир
1:305
Предметный указатель
479
Этилхлорид 1: 255, 284, 305, 363; 2: 340
Этилцеллюлоза 1: 255, 298, 363
Этинамат2: 114
Этинилирование 2:102, 112
1-Этинилциклогексанол 1: 362; 2:114
1-Этинилциклогексилкарбамат см. Этинамат
1 -Этинилциклопентанол 1: 362; 2: 114
Этинилэстрадиол 2:114
2-Эгокси-3,4-дигидро-2Н-пиран 1:402; 2:64,131
р-Этокси-[Е]-кротоновой кислоты этиловый
эфир 2: 214
5-Этокси-3-метилиндолкарбоновой-2 кисло-
кислоты этиловый эфир 2: 290
5-Этокси-3-фенилиндолкарбоновой-2 кисло-
кислоты этиловый эфир 2: 291
л-Этоксибензальдегид 1:462
Этоксибензол 1: 296
1-Этоксибутадиен-1,3 1:400
2-Этоксибутадиен-1,3 1:337
2-Этоксигексен-1 1: 337
5-Этоксииндолкарбоновой-2 кислоты этило-
этиловый эфир 2:290
2-Этоксикарбонилиндол см. Индолкарбоно-
вой-2 кислоты этиловый эфир
а-Этоксиметиленацетоуксусный эфир 2:148
а-Этоксиметиленкарбоноюй кислоты эфиры
2:148
Этоксиметиленмалоновой кислоты диэтило-
вый эфир 2:148
Этоксиметиленмалононитрил 2:148, 210
Этоксиметиленцианамид 2:148
а-Этоксистирол 1: 337
2-Этоксициклогексен-3-карбальдегид-11:400
1-Этоксициклогексен 1: 337
Эфедрин 1:459
Эфир 1: 289,466; см. также Диэтиловый эфир
для тонкослойной хроматографии 1: 99
идентификация 2: 337
— растворимость 2: 297
— масс-спектрометрия 1: 145
— расщепление 1: 289; 2:337
как растворитель 1:49, 200
нагревание 1: 25
осушение 1:40; 2: 374
пероксиды 1: 260; 2: 379
получение 1: 293, 320
— алкилированием спиртов и фенолов 1: 282,
295, 325; 2: 258
— дегидратацией спиртов 1: 282, 325
— кислотной этерификацией спиртов 1: 288
— присоединением спиртов к а,р-ненасыщен-
ным карбонильным соединениям 2:199
к олефинам 1: 360
химические свойства, замещение нуклеофи-
льное 1: 282
расщепление 1: 265, 288, 320
элиминирование спиртов 1: 333
Эфироальдегиды 2:143
Эфиры алкилфосфоновых кислот 2: 135
Эфиры см. Карбоновых кислот эфиры, Сульфо-
новых кислот эфиры, Алкилсульфаты и т. д.
Эффект поля 1: 206
Яблочная кислота, производные 2: 331
Ядерные колебания 1:110
Ядерный спин 1:126
Ядерный эффект Оверхаузера 1: 135
Ядохимикаты для защиты растений 1: 255,
298, 305, 342,467; 2: 9, 98, 103, 267
ЯМР-спектроскопия 1: 100, 126,135, 165
Янтарная кислота 1: 414; 2: 38, 96, 333
Янтарной кислоты ангидрид 1:459; 2: 90, 411
Янтарной кислоты диэтиловый эфир 2:71
BINAP см. 2,2'-Бис(дифенилфосфино)-1,Г-
бинафтил
ВМВ см. Дисиамилборан
Вос-группа см. /n/jem-Бутилоксикарбонильная
группа
DFP см. Диизопропилфторфосфат
DCC см. Дициклогексилкарбодиимид
DoM см. орто-Металлирование направленное
dppf см. 1,1 '-Ди(дифенилфосфино)ферроцен
MPLC см. Жидкостная хроматография сред-
среднего давления
NOE см. Оверхаузера ядерный эффект
NTA см. Нитрилотриуксусная кислота
ORTEP-изображение структуры молекулы 1:
158
Rf — характеристический параметр в тонко-
тонкослойной хроматографии 1: 91
SAMP см. 1-Амино-2-метоксиметилпирроли-
дин
SnI см. Замещение нуклеофильное мономоле-
мономолекулярное
Sn2 cm. Замещение нуклеофильное бимолеку-
бимолекулярное
SNR-MexaHH3M реакций 1:478
ОГЛАВЛЕНИЕ
!f\
ПРЕПАРАТИВНАЯ ЧАСТЬ (ПРОДОЛЖЕНИЕ) 5
6. Окисление и дегидрирование 5
6.1. Общие закономерности 5
6.2. Окисление метальных и метиленовых групп 9
6.2.1. Окисление алкиларенов до ароматических карбоновых кислот ... 10
6.2.2. Окисление алкиларенов до альдегидов и кетонов 15
6.2.3. Окисление активированных метильных и метиленовых групп в
карбонильных соединениях 16
6.2.3.1. Окисление диоксидом селена 16
6.2.3.2. Реакция Вильгеродта 17
6.3. Окисление первичных и вторичных спиртов и альдегидов 21
6.3.1. Окисление первичных и вторичных спиртов до альдегидов и
кетонов 21
6.3.2. Окисление первичных спиртов и альдегидов в карбоновые
кислоты 28
6.4. Получение хинонов окислением 30
6.4.1. Хиноны из ароматических углеводородов 30
6.4.2. Хиноны из замещенных аренов 32
6.4.3. Получение хинониминов окислительным сочетанием 34
6.5. Окисление с расщеплением связей С—С 36
6.5.1. Окисление кратных углерод-углеродных связей 38
6.5.2. Расщепление гликолей 40
6.5.3. Окислительное расщепление вторичных спиртов и кетонов 41
6.6. Дегидрирование алканов и циклоалканов 43
6.7. Литература 46
7. Реакции карбонильных соединений 49
7.1. Реакции карбонильных соединений с гетероатомными нуклеофилами 51
7.1.1. Реакции альдегидов и кетонов с аминами 56
7.1.2. Реакции альдегидов и кетонов с водой и спиртами 62
7.1.3. Реакции альдегидов и кетонов с образованием тиоацеталей
и бисульфитных соединений 66
7.1.4. Реакции карбоновых кислот и их производных с гетероатомными
нуклеофилами .. .• 67
Оглавление 481
7.1.4.1. Получение сложных эфиров алкоголизом карбоновых
кислот и их производных 69
7.1.4.2. Получение амидов кислот аминолизом карбоновых кислот
и их производных 75
7.1.4.3. Гидролиз производных карбоновых кислот 84
7.1.4.4. Ацидолиз карбоновых кислот и их производных 90
7.1.5. Присоединение оснований к нитрилам 95
7.1.6. Присоединение оснований к некоторым особым карбонильным
соединениям 99
7.1.7. Тионирование карбонильных соединений 102
7.2. Реакции карбонильных соединений с С-нуклеофилами 104
7.2.1. Реакции карбонильных соединений с С-Н-кислотными
веществами 106
7.2.1.1. Присоединение синильной кислоты к альдегидам и
кетонам 108
7.2.1.2. Этинилирование карбонильных соединений 112
7.2.1.3. Альдольная конденсация 114
7.2.1.4. Реакция Кнёвенагеля 125
7.2.1.5. Реакция Манниха 129
7.2.1.6. Ацилоиновая конденсация и обращение полярности .... 132
7.2.1.7. Реакции альдегидов и кетонов со сложными эфирами
алкилфосфоновых кислот и алкилиденфосфоранами ... 135
7.2.1.7.1. РеакцияХорнера — Вадсворта — Эммонса .... 135
7.2.1.7.2. Реакция Виттига 136
7.2.1.8. Сложноэфирная конденсация 138
7.2.1.9. Сложноэфирное и кислотное расщепление
(З-дикарбонильных соединений 149
7.2.1.10. Реакции ацилхлоридов с р-дикарбонильными
соединениями 152
7.2.1.11. Присоединение С—Н-кислотных соединений к
гетерокумуленам 154
7.2.1.12. Полиметиновая кислота 156
7.2.2. Реакции карбонильных соединений с металлоорганическими
соединениями 158
7.3. Восстановление карбонильных соединений 167
7.3.1. Восстановление карбонильных соединений Н-нуклеофилами .. 169
7.3.1.1. Восстановление карбонильных соединений алюминий-
и борогидридами 169
7.3.1.2. Восстановление по Меервейну-Понндорфу-Верлею и
окисление по Оппенауэру 174
7.3.1.3. Реакции Канниццаро и Кляйзена-Тищенко 177
7.3.1.4. Реакция Лейкарта-Валлаха 179
7.3.1.5. Ферментативное восстановление 181
7.3.1.6. Восстановление по Кижнеру-Вольфу 181
7.3.2. Каталитическое гидрирование карбонильных соединений 184
7.3.3. Восстановление карбонильных соединений неблагородными
металлами и металлами низшей валентности 189
7.4. Реакции винилогов карбонильных соединений и других
винилоговых систем 194
482 Оглавление
7.4.1. Реакции винилогов электроноацепторных соединений —
а,р-ненасыщенных карбонильных соединений 196
7.4.1.1. Присоединение аминов к винилогам карбонильных
соединений 197
7.4.1.2. Присоединение воды, галогеноводородов, сероводо-
сероводорода, спиртов и тиолов к винилогам карбонильных
соединений 200
7.4.1.3. Присоединение С—Н-кислотных соединений к винилогам
карбонильных соединений (присоединение по Михаэлю) 201
7.4.1.4. Присоединение амидов кислот к винилогам карбонильных
соединений 208
7.4.1.5. Реакции замещения винилогов карбонильных соединений 209
7.4.2. Реакции винилогов электронодонорных соединений
(енолятов, енолов, эфиров енолов, енаминов) 210
7.4.2.1. Алкилирование карбонильных соединений 212
7.4.2.2. Галогенирование карбонильных соединений 217
7.4.2.3. Ацилирование и алкилирование енаминов 220
7.5. Литература 223
8. Реакции других гетероаналогов карбонильных соединений 230
8.1. Восстановление нитро- и нитрозосоединений 233
8.2. Реакции азотистой кислоты 237
8.2.1. Реакции азотистой кислоты с аминами 238
8.2.2. Реакции азотистой кислоты со спиртами (этерификация) 242
8.2.3. Реакции азотистой кислоты с С—Н-кислотными соединениями .. .243
8.3. Реакции солей диазония 244
8.3.1. Термическое разложение и восстановление 244
8.3.2. Реакция Зандмейера 247
8.3.3. Азосочетание. Азокрасители 250
8.4. Алифатические диазосоединения 256
8.4.1. Получение диазоалканов 256
8.4.2. Реакции алифатических диазосоединений 257
8.4.2.1. Реакции алифатических диазосоединений с протонными
кислотами 257
8.4.2.2. Реакции алифатических диазосоединений с карбонильными
соединениями 259
8.5. Реакции производных сульфоновых кислот 261
8.6. Литература 267
9. Перегруппировки 269
9.1. [1,2]-Перегруппировки 270
9.1.1. Нуклеофильные [1,2]-перегруппировки у атома углерода 273
9.1.1.1. Пинаколиновая перегруппировка 273
9.1.1.2. Перегруппировка Вагнера-Меервейна 275
9.1.1.3. Перегруппировка Вольфа 276
9.1.2. Перегруппировки у атома азота 278
9.1.2.1. Деструкция по Гофману 279
9.1.2.2. Деструкция по Курциусу 281
9.1.2.3. Реакция Шмидта 282
9.1.2.4. Перегруппировка Бекмана 284
9.1.3. Перегруппировки у атома кислорода 285
Оглавление 483
9.2. [З,3]-Перегруппировки 287
9.3. Литература 291
ИДЕНТИФИКАЦИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ 293
Предварительные испытания и обнаружение функциональных групп 293
1.1. Предварительные испытания 295
1.1.1. Внешний вид вещества 295
1.1.2. Определение физических констант 295
1.1.3. Проба на горючесть и зольность 295
1.1.4. Обнаружение элементов 296
1.1.5. Определение растворимости 297
1.2. Обнаружение функциональных групп 299
1.2.1. Обнаружение ненасыщенных соединений 300
1.2.1.1. Реакция с бромом 300
1.2.1.2. Реакция с перманганатом 300
1.2.2. Обнаружение ароматических соединений 301
1.2.2.1. Реакция с азотной кислотой 301
1.2.2.2. Реакция с хлороформом и хлоридом алюминия 301
1.2.3. Обнаружение веществ, являющихся сильными восстановителями
(реакция с аммиачным раствором соли серебра) 301
1.2.4. Обнаружение альдегидов и кетонов 302
1.2.4.1. Реакция с динитрофенилгидразином 302
1.2.4.2. Реакция с фелинговой жидкостью 302
1.2.4.3. Реакция с фуксинсернистой кислотой (реактивом Шиффа) 302
1.2.5. Обнаружение спиртов, фенолов, енолов 303
1.2.5.1. Реакция с церийаммонийнитратом 303
1.2.5.2. Реакция с хлоридом железа(Ш) 303
1.2.5.3. Реакция с солями меди(П) 303
1.2.5.4. Реакция с солянокислым раствором хлорида цинка
(реактив Лукаса) 304
1.2.5.5. Взаимодействие с реактивом Дениже 304
1.2.6. Иодоформная реакция (реакция с гипоиодитом натрия) 304
1.2.7. Обнаружение соединений, гидролизуемых щелочами 305
1.2.7.1. Реакция со спиртовой щелочью (проба Рояна) 305
1.2.7.2. Реакция с гидроксиламином (гидроксамовая проба) 305
1.2.7.3. Реакция с концентрированным раствором гидроксида
калия 305
1.2.8. Обнаружение аминов 306
1.2.8.1. Реакция с хлороформом (изонитрильная проба) 306
1.2.8.2. Реакция с азотистой кислотой 306
1.2.8.3. Реакция с нингидрином 307
1.2.9. Обнаружение нитро- и нитрозосоединений 307
1.2.9.1. Реакции с цинком и хлоридом аммония 307
1.2.9.2. Реакция аци-нитросоединений с хлоридом железа(Ш) .. 307
1.2.9.3. Реакция аци-нитросоединений с азотистой кислотой .. 307
1.2.10. Обнаружение гидролизующегося галогена 307
1.2.11. Обнаружение тиолов и тиофенолов 308
1.2.11.1. Реакция с солями тяжелых металлов 308
1.2.11.2. Реакция с азотистой кислотой 308
484 Оглавление
1.2.11.3. Реакция с динатрийпентацианонитрозилферратом(Ш)
(нитропруссидом натрия) 308
2. Производные и спектры 309
2.1. Идентификация аминов 309
2.1.1. Первичные и вторичные амины 309
2.1.1.1. Получение бензамидов 309
2.1.1.2. Получение бензол- и толуолсульфонамидов и разделение
по Хинсбергу 310
2.1.1.3. Получение пикратов, пикролонатов и стифнатов 310
2.1.1.4. Получение фенилтиомочевин 310
2.1.1.5. Определение эквивалентной массы 310
2.1.2. Третичные амины 310
2.1.2.1. Получение пикратов 310
2.1.2.2. Получение метоиодидов и метотозилатов 310
2.1.2.3. Определение эквивалентной массы 315
2.1.3. Аминокислоты 319
2.1.3.1. Получение бензамидов 319
2.1.3.2. Получение фенилмочевин 319
2.1.3.3. Бумажная хроматография 319
2.2. Идентификация карбонильных соединений 319
2.2.1. Альдегиды и кетоны 319
2.2.1.1. Получение фенилгидразонов 324 !
2.2.1.2. Получение семикарбазонов 324
2.2.1.3. Получение димедоновых производных 324 \
2.2.1.4. Определение эквивалентной массы титрованием оксимов 324 ~
2.2.2. Хиноны 327
2.2.2.1. Получение семикарбазонов 327
2.2.2.2. Получение гидрохинондиацетатов 328
2.2.3. Моносахариды 328
2.2.3.1. Получение озазонов 328
2.2.4. Ацетали 330
2.2.5. Карбоновые кислоты 330
2.2.5.1. Получение и-бром- и «-фенилфенациловых эфиров ... 330
2.2.5.2. Получение амидов карбоновых кислот 330
2.2.5.3. Получение N-бензиламидов карбоновых кислот 334
2.2.5.4. Получение анилидов карбоновых кислот 334
2.2.5.5. Определение эквивалентной массы 334
2.2.6. Амиды карбоновых кислот и нитрилы 335
2.2.6.1. Получение карбоновых кислот 336
2.2.6.2. Получение аминов (восстановление по Буво-Блану) 336
2.2.7. Эфиры карбоновых кислот 336
2.2.7.1. Получение карбоновых кислот и спиртов 336
2.2.7.2. Получение эфиров 3,5-динитробензойной кислоты 337
2.2.7.3. Получение амидов карбоновых кислот 337
2.3. Идентификация простых эфиров 337
2.3.1. Расщепление простых эфиров иодоводородной или
бромоводородной кислотой 338
2.3.2. Расщепление простых эфиров 3,5-динитробензоилхлоридом
в присутствии хлорида цинка 338
Оглавление 485
2.4. Идентификация галогеноуглеводородов 339
2.4.1. Получение анилидов карбоновых кислот 339
2.4.2. Получение пикратов S-алкилизотиомочевин 341
2.5. Идентификация спиртов 342
2.5.1. Первичные и вторичные спирты 342
2.5.1.1. Получение эфиров нитробензойных кислот 342
2.5.1.2. Получение моноэфиров 3-нитрофталевой кислоты 342
2.5.1.3. Получение уретанов 342
2.5.2. Третичные спирты 342
2.5.2.1. Получение пикратов S-алкилизотиомочевин 342
2.5.2.2. Определение эквивалентной массы 346
2.5.3. Фенолы 346
2.5.3.1. Получение бензоатов 346
2.5.3.2. Получение уретанов 346
2.5.3.3. Получение бромфенолов 349
2.5.3.4. Получение арилоксиуксусных кислот 349
2.6. Идентификация углеводородов 349
2.6.1. Алканы и циклоалканы 349
2.6.2. Ароматические углеводороды 351
2.6.2.1. Получение сульфонамидов 351
2.6.2.2. Получение о-ароилбензойных кислот 351
2.6.2.3. Получение нитропроизводных 351
2.6.2.4. Получение аддуктов с пикриновой кислотой 352
2.6.2.5. Окисление перманганатом калия или хромовой кислотой .. 352
2.6.3. Алкены и алкины 352
2.6.3.1. Превращение в карбонильные соединения 353
2.6.3.2. Гидратация ацетиленовых производных 353
2.7. Идентификация нитро- и нитрозосоединений 354
2.7.1. Получение аминов восстановлением оловом в соляной кислоте 354
2.7.2. Получение аминов при взаимодействии с гидразингидратом
в присутствии скелетного никеля 354
2.8. Идентификация тиолов и тиофенолов 354
2.8.1. Получение 3,5-динитротиобензоатов 354
2.8.2. Получение 2,5-динитрофенилтиоэфиров и их окисление
до сульфонов 355
2.8.3. Определение эквивалентной массы 355
2.9. Идентификация сульфоновых кислот 356
2.9.1. Получение сульфонатов S-бензилтиомочевины 356
2.9.2. Получение сульфонамидов 356
2.9.3. Определение эквивалентной массы 356
3. Разделение смесей 357
4. Вопросы и упражнения 358
5. Литература 359
L СВОЙСТВА, ОЧИСТКА И ПРИГОТОВЛЕНИЕ
ВАЖНЕЙШИХ РЕАГЕНТОВ, РАСТВОРИТЕЛЕЙ
И ВСПОМОГАТЕЛЬНЫХ ВЕЩЕСТВ 361
Азот и другие инертные газы 361
Азотистоводородная кислота HN3 361
486 Оглавление
Азотная кислота HNO3 362
Акрилонитрил CH2=CH-CN 362
Активированный уголь 362
Алюмогидрид лития (литийалюминийгидрид) LiAlH4 362
Амальгама натрия 363
Амид натрия NaNH2 363
Аммиак NH3 363
Ацетальдегид (этаналь) СН3СНО 364
Ацетилен (этин) СН^СН 364
Ацетон СНзСООСНз 364
Ацетонитрил CH3CN 365
Бензальдегид С6Н5СНО 365
Бензин 365
Бензоилпероксид QH5CO-OO-COC6H5 365
Бензол С6Нб 366
Бром Вг2 366
Бромоводород НВг 366
N-Бромсукцинимид 367
трет-Ъуттат алюминия [(СН3)зСО]зА1 368
Бутиллитий C4H9Li 368
Водород Н2 369
н-Гексан QH14 369
Гидразингидрат H2NNH2 • Н2О 369
Гидрид натрия 370
Гликоль 370
Глицерин (пропантриол-1,2,3) НОСН2-СНОН-СН2ОН 370
Диметиловый эфир диэтиленгликоля (диглим) (СН3ОСН2СН2JО 370
Диметилсульфат (CH3JSO4 370
Диметилсульфоксид (ДМСО) CH3SOCH3 371
Диметилформамид (ДМФА) (CH3JNCHO 371
1,4-Диоксан 371
Диоксид платины PtO2 372
Диоксид селена SeO2 372
Диоксид серы SO2 372
Ди(т/>еот-бутил)пероксид 373
Дихлорметан (метиленхлорид) СН2С12 373
1,2-Дихлорэтан (этилендихлорид) С1СН2СН2С1 373
Диэтиленгликоль (дигликоль) НО(СН2JО(СН2JОН 373
Диэтиловый эфир (эфир) С2Н5ОС2Н5 374
Изопропилат алюминия [(СН3JСНО]зА1 374
Иодоводородная кислота HI 375
Ионообменные смолы 375
N-CN-Индикатор 375
Калий К 375
Ксилол 376
Оглавление 487
Лигроин 376
Метанол СНзОН 376
Метиленхлорид 376
Молекулярные сита 376
Натрий 377
Натриевые алкоголяты 377
Нитрозные газы 377
Оксалилхлорид (СОС1J 378
Олеум 378
Палладий на активированном угле (катализатор) 378
Пентан С5Н12 379
Пероксид водорода Н2О2 379
Петролейный эфир 379
Пиридин C5H5N 379
Платина на активированном угле, 10% (катализатор) 380
Полифосфорная кислота 380
Раствор гидросульфита (бисульфита) натрия 380
Раствор фталата анилина 380
Реактив Дениже 381
Реактив Лукаса 381
Реактив Толленса 381
Реактив Шиффа 381
Ртуть 381
Сероводород H2S 382
Синильная кислота (циановодород) HCN 382
Скелетный никель (никель Ренея) 383
Соляная кислота НС1 383
Спирты 384
Стеарат кобальта 384
Сульфолан (тетраметиленсульфон) 384
Тетраацетат свинцаAУ) (СН3СООLРЬ 384
Тетрагидрофуран 385
Тетрахлорметан (тетрахлоруглерод) ССЦ 385
Тионилхлорид SOC12 385
Толуол С6Н5СН3 386
Триэтиленгликоль (тригликоль) НО(СН2JО(СН2JО(СН2)ОН 386
Трихлорэтилен С12С=СНС1 386
Уксусная кислота (ледяная) СНзСООН 386
Уксусный ангидрид (ацетангидрид) (СН3СОJО 387
Фелингова жидкость 387
Формальдегид (метаналь) НСНО 387
Фосген СОС12 387
Хлор С12 388
Хлораль (трихлорэтаналь) СС13СНО 388
Хлорид алюминия А1С13 388
488 Оглавление
Хлороводород HCl 389
Хлорная кислота НСЮ4 389
Хлороформ (трихлорметан) СНСЬ 390
Хлорсульфоновая кислота CISO3H 390
Церийаммонийнитрат (NH4J[Ce(NO3N] 390
Цианид калия KCN 390
Цианид натрия NaCN 390
Цианид цинка Zn(CNJ 391
Этанол (этиловый спирт) С2Н5ОН 391
ЭтилацетатСНзСООС2Н5 391
Этиленгликоль (гликоль, этандиол-1,2) НОСН2СН2ОН 392
Этиленоксид(оксиран) 392
Эфир 392
Литература 392
.о ПРИЛОЖЕНИЕ.
СВОЙСТВА ОПАСНЫХ РЕАГЕНТОВ 393
Токсичность важнейших химикатов 394
Литература 412
Перечень методик 414
Предметный указатель 420
[ЛУЧШИЙ
ЗАРУБЕЖНЫЙ
УЧЕБНИК
Учебное издание
Беккер Хейнц, Беккерт Райнер, Бергер Вернер и др.
ОРГАНИКУМ
В двух томах
Зав. редакцией канд. хим. наук Т. И. Почкаева. Ведущий редактор И. С. Беленькая
Художник М.М.Иванов. Технический редактор Е. В. Денюкова
Оригинал-макет подготовлен А. А. Пудовым, Е. В. Денюковой
Подписано к печати 17.09.07. Формат 70 х 100/16. Бумага офсетная. Печать офсетная.
Бум. л. 15,25. Печ. л. 30,50. Изд. № 3/9916. Тираж 2000 экз. Зак. 12234.
Издательство «МИР»
Министерства культуры и массовых коммуникаций РФ.
107996, ГСП-6, Москва, 1-й Рижский пер., 2.
Диапозитивы изготовлены в издательстве «Мир»
OiiK"m:iHi> с гмкжмх .шншшгшвон в ОАО < Illlk "Южный Урал»
4MHHHI, г. Opi'iioypr. мор. Гпгшолииа. 4.