/
































































































































































































































































































Автор: Флид М.Р. Трегер Ю.А.
Теги: физическая химия твердых тел, жидкостей и газов органическая химия химия высокомолекулярных соединений полимеры химия
ISBN: 978-5-89530-019-0
Год: 2008




Текст
\ 'll . 11 I . I ‘u 1(17 )
I<ld 1 и
• I' •
Рецензенты:
Н.Ф. Гретьяков, член-корреспондент РАН, профессор,
Н.Ф. Швец, доктор химических наук, профессор
Флид М.Р., Трегер Ю.А.
<1’72 Винилхлорид: химия и технология. В 2-х кн. Кн. 1. — М.: Калвис,
2008. — 584 с.
ISBN 978-5-89530-019-0 (кн. 1)
ISBN 978-5-89530-017-6 (общ.)
Винилхлорид — один из наиболее крупнотоннажных продуктов органическо-
го синтеза. Сведения о нем изложены в двух книгах.
В первой книге рассмотрены научные и прикладные аспекты способов полу-
чения винилхлорида, приведены основные кинетические и технологические
закономерности процесса его получения гидрохлорированием ацетилена, а также
процессов прямого и окислительного хлорирования этилена и пиролиза дихлор-
этана, являющихся составными частями производства винилхлорида по сбалан-
сированной схеме. Описаны катализаторы, использующиеся в этих процессах.
Приведены разные модификации промышленных технологических процессов
получения винилхлорида и параметры их работы. Рассмотрена сырьевая база для
производства винилхлорида.
Для научных и инженерно-технических работников институтов и предприя-
тий химической промышленности, а также для студентов химико-технологических
факультетов и вузов.
УДК 544.23:547.79 (075)
ББК 24.7я7
ISBN 978-5-89530 019-0 (кн. 1)
ISBN 978-5-89530-017-6 (общ.)
©М.Р. Флид, Ю.А. Трегер, 2008
©14зда1ельство «Калвис», 2008
Оглавление
Предисловие ..........................................................'I
Введение.............................................................. 6
Глава 1. Физико-химические свойства винилхлорида ................... 1(>
Глава 2. Сырьевая база для производства винилхлорида..................37
2.1. Хлор.........................................................37
2.2. Хлороводород.................................................53
2.3. Ацетилен....................................................6-1
2.4. Этилен.......................................................73
Глава 3. Химия и технология получения винилхлорида....................89
3.1. Общие положения..............................................8ч
3.2. Щелочное омыление 1,2-дихлорэтана ...........................95
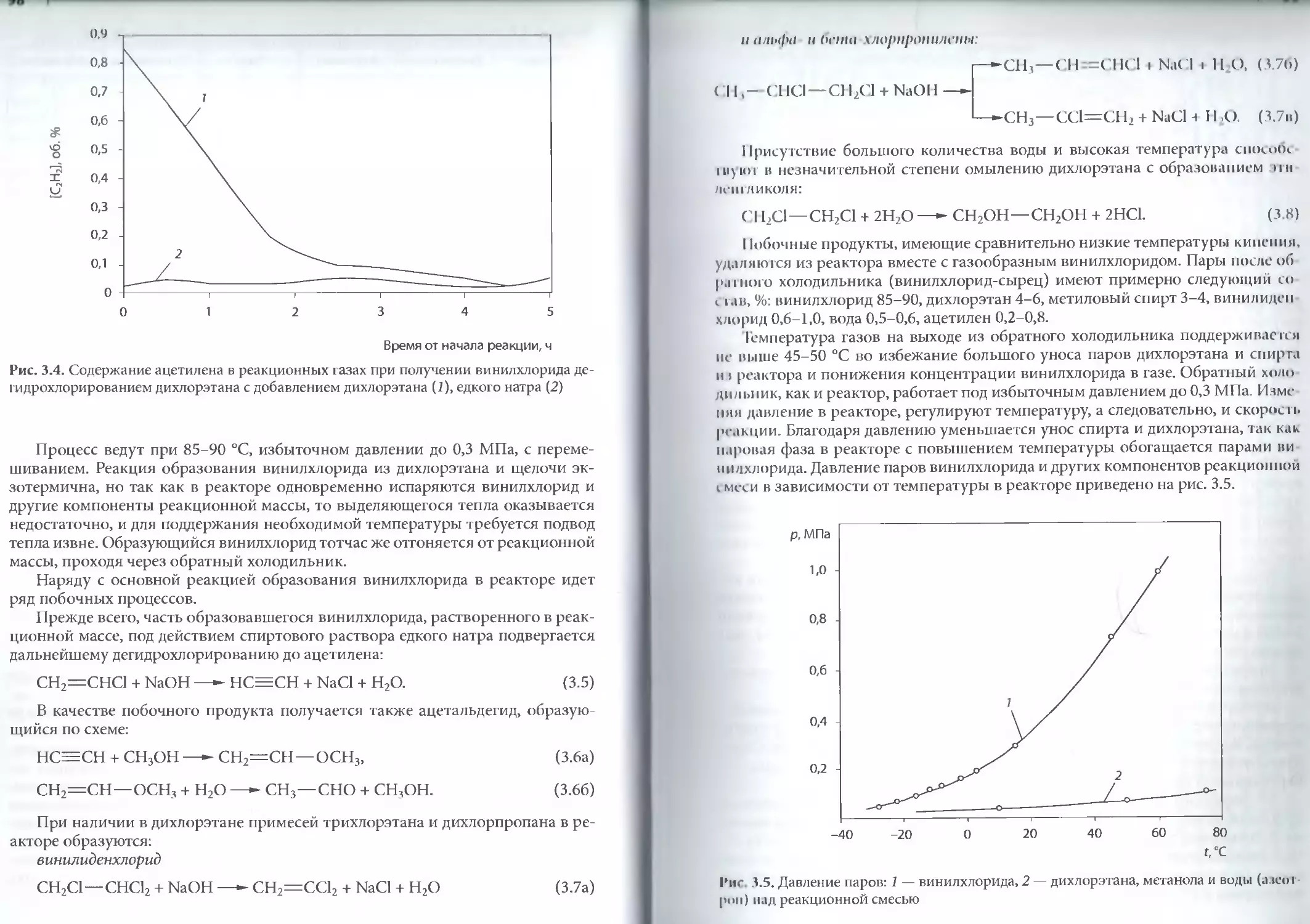
3.3. Газофазное гидрохлорирование ацетилена
в неподвижном слое катализатора ................................ 112
3.4. Гидрохлорирование ацетилена
в псевдоожиженном слое катализатора........................... 155
3.5. Гидрохлорирование ацетилена в присутствии инициаторов...... 169
3.6. Жидкофазное гидрохлорирование ацетилена.................... 182
Глава 4. Получение винилхлорида из этилена по сбалансированной схеме. 197
4.1. Принципы сбалансированности
и их использование в производстве винилхлорида.................. 197
4.2. Основные свойства 1,2-дихлорэтана ..........................203
4.3. Окислительное хлорирование этилена.......................... 2М
4.4. Прямое хлорирование этилена.................................377
4.5. Пиролиз 1,2-дихлорэтана.....................................477
Глава 5. Технологические и экологические аспекты
производства винилхлорида по сбалансированной схеме . 53(1
5.1. Принципиальная технологическая схема
производства винилхлорида сбалансированным методом..............53(1
5.2. Производственные выбросы в окружающую среду................5-18
5.3. Характеристика готового продукта ...........................552
Глава 6. Получение винилхлорида из этана.............................551
CHt .. -' --- CHCI
Предисловие
СН2 — ---- CHCI
В настоящей работе рассматриваются научные и прикладные аспекты син-
теза винилхлорида (ВХ) — полупродукта в производстве поливинилхлорида
(ПВХ). Именно производство ВХ-мономера вносит до 70% добавленной стои-
мости в ПВХ-продукт, отличается существенным разнообразием протекаю-
щих при этом процессов (жидко- и газофазных, гетерогенно-каталитических
и радикально-цепных), технологических решений, конструкций реакторных
узлов. В силу этого создание современного производства винилхлорида требует
детальной химической и инженерно-технологической проработки всех состав-
ляющих его стадий.
Несмотря на то, что некоторые производства винилхлорида в России
эксплуатируются достаточно долго (больше 40 лет), и при этом накоплен
шачительный опыт их работы, до сих пор в отечественной научной литера-
। уре отсутствует обобщающий материал, сочетающий анализ значительного
массива научных исследований с результатами работы промышленных уста-
новок.
Нами рассмотрены основные свойства винилхлорида, сырьевая база (орга-
ническая и минеральная) для его производства, химико-технологические аспек-
ты основных стадий его получения, опыт эксплуатации промышленных произ-
водств. Наряду с большим количеством опубликованных данных, принадлежа-
щих в том числе авторам настоящей книги, последние руководствовались так
же собственным опытом при создании и пуске отечественного производства в
Стерлитамакском АО «Каустик», усовершенствовании и модернизации других
производств винилхлорида в России и Украине.
Отдельное внимание авторами уделено проблеме квалифицированной пе-
реработки отходов производства винилхлорида, определяющей при создании
производства как с экономической, так и с экологической позиций.
Данная книга может оказаться полезной инженерно-техническим работни-
кам промышленных предприятий, аспирантам и сотрудникам научных учреж-
дений, занимающимся вопросами химии и технологии основного органическо-
го синтеза, студентам химических и химико-технологических специальностей
высших учебных заведений.
Авторы с благодарностью примут советы, замечания и дополнения, относя
щиеся к содержательной стороне настоящей работы.
Мы считаем своим долгом вспомнить советских и российских специалистов,
как ныне здравствующих, так и тех, кого уже нет с нами, внесших фундамен-
тальными и прикладными исследованиями огромный вклад в понимание про-
блемы винилхлорида и развитие ее в России: это — П.И. Павлович, С.С. Бобков,
Я.П. Чопоров, Р.М. Флид, М.В. Хрулев, Я.М. Абдрашитов, З.С. Смолян, А.А. Пан-
филов, В.С. Этлис, В.К. Нейман, А.Л. Левин, Б.Л. Каменко, А.Л. Энглин, Э.В. Со-
пни Г.Д Гужнивская, О А. Зайдман, 11.Ф. Кришталь, Л.II Тимашов, АН. Гелы»
штейн, Ю.М. Бакши, Е.И. Гсльперин, ЕС. Яблонский, С.М. Данон и др.
В написание книги значительный вклад внесли ведущие российские спсцна
листы в области хлорорганического синтеза — М.Г Аветьян, которого нс ci.wk
и период подготовки книги к печати, и Л.М. Карташов.
Ан горы глубоко признательны В.Ф. Третьякову, члену-корреспонден i у РА 11
д.х. н., профессору и В.Ф. Швецу, д.х.н., профессору за полезность обсуждения г
ценные замечания, высказанные по существу данной работы.
Авторы выражают глубокую благодарность М.В. Баботиной за большую но
мощь, оказанную при подготовке рукописи к печати.
Выражаем также признательность АО «Саянскхимпласт» — крупнейшем’
производителю ВХ и ПВХ в России — за спонсорскую поддержку настоящен
издания.
:hj j-l.. — .... chci
Введение
CH2 ' - 7. ---- CHCI
Винилхлорид (во всем мире принято сокращение VCM — vinyl chloride
monomer) — уникальный и самый многотоннажный в химической промыш-
ленности продукт комплексной переработки минерального и органического
сырья — поваренной соли и нефти. Объем его мирового производства достиг в
2006 г. 31 млн т. До 98% всего выпускаемого винилхлорида направляется на по-
лучение поливинилхлорида (ПВХ), важнейшего полимерного материала, кото-
рый производится в промышленных масштабах уже более 70 лет. Общая схема
превращений с учетом последующей переработки винилхлорида в поливинил-
хлорид представлена на рис. В.1.
Безальтернативность использования каустической соды в широком спектре
отраслей промышленности указывает на необходимость утилизации соответ-
с гвующего количества хлора. Из ~50 млн т хлора, ежегодно производимого в
мире, до 40% идет на получение винилхлорида и далее — поливинилхлорида,
ч го свидетельствует об определяющем значении данного направления в струк-
। у ре ряда отраслей промышленности [1]. Можно утверждать, что если бы ви-
нилхлорида не было, его следовало бы придумать.
Впервые винилхлорид был получен германским химиком Юстусом фон
Либихом и его учеником — французским химиком Анри-Виктором Реньо
в 1835 г. действием спиртового раствора едкого кали на дихлорэтан. Одно-
временно Реньо обнаружил способность винилхлорида полимеризоваться
Рис. B.I. С'хема, включающая получение винилхлорида и переработку его в поливи-
Ri» Л.ние -----------------
I 7
иод действием света, но нс придал этому шачения ввиду инертное i и пора
«овавшегося соединения к действию разных химических реагентов. В 1858 i
III.Л. Вюрц и Э. Франкленд синтезировали винилхлорид дейс твием на дихлор
хан этилата натрия. В 1902 г. Блитц получил винилхлорид каталитическим
разложением дихлорэтана на пемзе при температуре красного каления, а в
1908 г. Ж.Б. Сандеран — разложением дихлорэтана на обезвоженном гликозе
ме при 370 °C [2].
Реальными же предпосылками промышленного производства винилхлорида
явились открытые практически одновременно в 1912-1913 гг. реакции получс
ния винилхлорида методом гидрохлорирования ацетилена в газовой и жидкой
фазе (Ф. Клатте, Германия) и его фотополимеризации (И.И. Остромыслеискин,
Россия). Сформировавшееся к концу 1920-х годов понимание большого значс
ния IIBX в мировой экономике привело к созданию в 1929 г. в Рейнфельдепе
(1ермания) первого промышленного комплекса ВХ-ПВХ.
По степени прикладного значения, непрерывно возрастающего с 1950 х го
дов до настоящего времени, открытие винилхлорида Ю. Либихом и Л. Реньо
должно быть отнесено к наиболее значительным в истории развития химии.
Выше было отмечено, что роль винилхлорида как одного из ключевых про
дуктов в промышленности органического синтеза определяется потребностью
разных отраслей народного хозяйства в каустической соде и поливинилхлори
де. Подробное рассмотрение вопросов, связанных с каустической содой, не вхо
ди г в задачу данной книги. На поливинилхлориде, его значении и перспективах
мы остановимся подробнее.
Продукты из ПВХ отличаются чрезвычайной долговечностью, стойке»
стью к климатическим условиям, низкой воспламеняемостью и просто гоп
ухода. До 60% производимого ПВХ используется в строительной промыт
лепности, для труб газо- и водопроводов, профилей окон, жалюзи и дверей,
покрытий полов, герметизирующих пленок, водосточных желобов, обоев, ка
больных трубопроводов, а также для облицовки фасадов и потолков. В хими
ческой промышленности на основе ПВХ широко используются резервуары,
емкости, ванны.
Таблица ВЛ. Потребление винилхлорида
Срок эксплуатации Примеры Доля в общем потре- блении ПВХ (%)
До 2 лет Упаковочные, медицинские материалы, канцелярские принадлежности 15
От 2 до 10 лет Напольные покрытия, предметы домашнего хозяйства, канцелярские принадлежности, пластинки,обои, обувь 16
От 10 до 20 лет Машины, мебель, кабельные изоляции 28
Свыше 20 лет Трубы, окна, шторы, кабели II
Книга туммя
1----------------------------------------------------------------
Важной областью применении IIBX являются упаковочные материалы, та-
не, как пленки, бутылки и стаканы. ПВХ используется для кабельных изоля-
ций, чековых и 1сле(|>оппых карточек, термеги шрующих пленок для приборных
щ гкон автомобилей, а также в медицине для изготовления мешков, в которых
ранят кровь, мягких трубок и противоспидных перчаток.
Интенсивно растет использование ПВХ как заменителя дерева. В табл. В.1
|редставлены данные по относительному потреблению ПВХ.
Удачное сочетание высоких потребительских свойств и сравнительно невы-
окой цены способствует росту потребности в поливинилхлориде и в связи с
। им увеличению мощностей по производству винилхлорида.
Данные по динамике мирового производства ПВХ (рис. В.2), мощности
статювок по производству ПВХ (рис. В.З) и по спросу на ПВХ в сопоставлении
мощностями установок по его производству (табл. В.2) свидетельствуют о тен-
1енции к существенному увеличению спроса на данный продукт.
Но данным [26, 27] на начало 2006 г. общая мощность установок по про-
1 нюдству винилхлорида в Китае составила 9,7 млн т/год при годовом объеме
напуска 6,5 млн т. При этом 74% всего винилхлорида выпускалось на основе
карбидного ацетилена. В табл. В.2 приведена сравнительная характеристика
проса (числитель) на ПВХ и мощностей (знаменатель) по производству по-
। и винилхлорида в мире.
Но состоянию на конец 2005 г. основными производителями винилхлорида
ч поливинилхлорида являются США — 7600, Китай — 6500, Япония — 3200,
Германия — 2000 тыс. т/год. При этом наблюдается устойчивая тенденция к ро-
сту выпуска данных продуктов, в среднем на 3,5-4,5% в год.
пт
FTRV
9
1M>W
В табл. В.З представлены данные по производству винилхлорида и ценам на
него в США за последние 50 лет [21,22], которые подтверждают данную тендем
цию. Колебания цен на винилхлорид полностью зависят от стоимости сырья —
этилена и хлора.
В таблице В.4 показан относительный рост потребности в поливинилхлори
де в разных регионах земного шара за 1999 2004 и 2005-2010 гг. Эти показа! ел и
подтверждают возрастание значения ПВХ как индекса экономического состоя
ния региона. Наибольший рост потребности в поливинилхлориде соотноси гея
с ростом валового внутреннего продукта и характерен прежде всего для стран
Восточной Езропы и Юго-Западной Азии.
По уровню потребления полимерных материалов в мире ПВХ занимает
ус гойчивое второе место после полиэтилена. Потребление ПВХ на душу населс
Таблица В.2. Спрос (С) на ПВХ и мощность (М) установок по его производству
Регион планеты С/М, тыс. т Среднегодовые темпы прироста, %
1997 г. 2002 г.
Северная Америка 6627 8318 5,0
7632 9512 4,5
Западная Европа 5472 6411 3,2
5753 6157 1,4
Азия 9023 12164 6,2
8939 13834 9,1
Всего в мире 24374 31 130 5,0
26395 35558 5,4
.......... II I 111>1111 НИ1Д1 । но (11) niuiii л хлорид,! и цены (Ц) n.i neio и ( 111Л н период 19 ’5
tin > II
||1Д II. II.H. t и. долл./г Год II, тыс. т Ц. до л л./г Год II, 1ыс. г И. долл./т
1'1,. ’ It) 220 1991 5031 326 1999 7323 409
I'!(>() 1 t) 260 1992 5374 303 2000 7023 564
907 180 1993 5496 367 2001 6740 421
|9 0 1833 110 1994 6020 465 2002 6995 443
1975 1903 240 1995 5875 520 2003 7334 573
1980 2933 490 1996 6552 446 2004 7609 679
1985 3586 365 1997 6971 495 2005 8018 -
1990 4678 433 1998 7031 343
аблица B.4. Региональная структура, динамика и прогноз развития мирового рынка ПВХ
Регион планеты Рост потребности в год, % Потребление, тыс. т
1999- 2004 гг. 2005-2010 гг. 2004 г. 2010 г.
Северная Америка 0,6 0,8 7322 7650
Западная Европа -0,2 0,8 5621 5680
Восточная и Центральная 8,2 7,6 980 1546
Азия и Океания* 0,6 4,0 5864 7348
Китай 12,5 6,5 6642 10 108
Южная Америка -0,1 3,7 1173 1368
Африка+Ближний Восток 5,1 5,2 1436 1950
Всего в мире 3,0 3,5 29 038 35 650
Включая Японию.
ния в США, Германии, Японии составляет 18-25 кг/год, в России эта величина
не превышает 5 кг/год. Естественно, что уровень производства винилхлорида-
мономера подчиняется аналогичным соотношениям (рис. В.4). Сопоставление
тгих значений указывает, что производство и потребление поливинилхлорида
гесно связано с экономическим состоянием страны. Этот вывод подтверждает-
ся также данными на рисунках В.5 и В.6, иллюстрирующих взаимосвязь между
потреблением ПВХ и ростом ВВП.
Необходимость дальнейшего увеличения выпуска винилхлорида и поливи-
нилхлорида обусловлена также тем, что эти производства являются крупней-
шими потребителями хлора. Производство хлора в мире в 2006 г. составило
49 млн т. На получение ВХ и ПВХ было израсходовано 17,5 млн т или 35,7%.
Таким образом, наличие многотоннажных комплексов ВХ-ПВХ кардинально
Mini I
Рис. B4. Производство винилхлорида в 2005 г. (всего 29 млн т): а — валовое; 6 — на душу
населения
решает проблему утилизации хлора как своего рода побочного продукта в про
изводстве широко используемой в целлюлозно-бумажной, текстильной, алю
миниевой промышленности каустической соды, спрос на которую растет на
3-3,5% ежегодно.
Перечень десяти крупнейших производителей винилхлорида и поливинил
хлорида (рис. В.7) указывает на транснациональный характер развития этого
направления и подтверждает тезис о глобализации развития экономики в XXI в.
Участие России в глобальных процессах, предполагающих, в частности, устой
чивое развитие на рыночной и демократической основе [20], является важным
условием экономических преобразований в стране, и увеличение мощности
комплексов ВХ-ПВХ будет являться одним из индикаторов данного вектора.
Рис. В.5. Потребление ПВХ на душу населения в сравнении с ВВП в 2004 г.
Рис. В.6. Потребление ПВХ и относительный рост ВВП в России
Согласно данным по доле использования мощностей в производствах, вхо-
ящих в комплексы ВХ-ПВХ в мире, этот коэффициент составляет 0,85-0,90
лис. В.8), что свидетельствует о высокой устойчивости этих производств и по-
виляет максимально эффективно и долговечно эксплуатировать промышлен-
ое оборудование.
g
I*
1
3
гм
§
J
Ф
X
о
HJ
X
I
X
5
о
S
fl
1.5
3
2,5
2
1,5
1
0,5
0
Южная Америка
3320
2885
660
1450
Северная и центральная Америка
Западная Европа
2070 2070
* Formosa
Westlake
Gulf
Partners
290
SHIN-ETSU OxyvinilsLP INEOS LG Chem® Yinholit
Рис. B.7. Десять крупнейших производителей ПВХ
I.. .1 ПВХ, млнт ИИВХ, млнт ° ПВХ, % —ВХ, % А Дихлорэтан, %
Рис. В.8. Диаграмма мощностей в производствах, входящих в комплексы ВХ-ПВХ в мире
Необходимо также остановиться на проблеме экологической безопасное ги
использования изделий на основе ПВХ. Достаточно распространено мнение
о том, что ПВХ в ходе его эксплуатации и последующей утилизации выделяет
винилхлорид-мономер, а также высокотоксичные диоксины, и этим наносит се
рьезный ущерб как здоровью людей, так и окружающей среде. Подобного рода
рассуждения основаны на слабом знании предмета и недобросовестной копь
niKiype. Опубликовано дос и точно материалов, доказывающих чрезвычайно
i.iOoc ан । ропотен ное воздействие ПВХ и изделий из пето на окружающую CPC-
у. Убедительные доказательства этого приведены, например, в статье [3|, где
ка iaiio, что винилхлорид из ПВХ и изделий из него не выделяется пи при каких
словиях. С овременные предприятия производят ПВХ с содержанием остаточ-
ого ВХ менее 1 i в 1 т ПВХ. При разложении ПВХ (терморазложении, старении)
сполимеризации не происходит.
Доказано [41, что при неконтролируемом сжигании ПВХ эмиссия диоксинов
окружающую среду не превышает соответствующего значения при сжигании
ис той древесины (6,67 и 3<-28 мкг/т соответственно). На количество образую-
щихся диоксинов при сжигании прежде всего влияют конструкция печей и ра-
очие характеристики процесса, а не наличие или отсутствие ПВХ в горящем
агериале [5].
Материалы на основе ПВХ практически полностью пригодны для повтор-
ого использования (рециклинга). Разработан ряд промышленных методов их
типизации [6-8]. При этом значительное количество оконных профилей, на-
ольных покрытий, кабельной изоляции и других продуктов содержат до 70%
ерсработанного ПВХ [19].
I (одробный анализ достоинств и недостатков ПВХ при использовании его в
азных отраслях промышленности приведен в [9]. Суммируя основные выводы
анпой работы, можно утверждать:
1. ПВХ — один из самых старых из существующих полимеров, второй по
бъему производства и потребления, отличающийся при этом разнообразием
»орм применения и глубиной проникновения в разные сферы человеческой
еягельности.
2. ПВХ, состоящий на 57% из хлора, обладает меньшей энергоемкостью по
равнению с прочими крупнотоннажными термопластами, что с учетом по-
ледних успехов в области переработки отходов ПВХ существенно улучшает его
кобаланс.
3. ПВХ потенциально не более опасен, чем другие полимеры. Из всех по-
имеров он наиболее изучен, следовательно, его применение обещает меньше
еприятных сюрпризов. Однако хлор, сообщающий ПВХ ряд ценных потреби
ельских качеств (пониженная горючесть, возможность высокочастотной свар-
:и медицинских изделий и т.п.), может при неадекватной технологии производ-
тва, неправильном применении изделий или неграмотном обращении с отхо-
,ами ПВХ стать причиной образования токсичных веществ. Определенной ток-
ичностью обладают и некоторые добавки, используемые в композициях ПВХ.
) го заставило (и заставляет) производителей ПВХ в 1000 раз сократить выделе-
|ис ВХ; производителей готовых ПВХ-композиций заменять стабилизаторы на
основе тяжелых металлов на экологически безопасные Ca-Zn (несмотря на то,
по химически связанные металлы не могут переходить в окружающую среду)
ши использовать менее летучие пластификаторы; разработчиков мусоросжи-
а гелей использовать параметры и устройства, исключающие выделение НО и
образование диоксинов (несмотря на ничтожно малый вклад ПВХ в мировой
щоксиновый баланс, отсутствие прямой связи между выбросами диоксинов
ори техногенных катастрофах и присутствием ПВХ, а также доказательств кан-
(ерогенности диоксинов для человека).
I ( ci одни нс । oi новации hiкап. .un.iepn.il пну 11BX, да ее и глобальном м.к
iin.ibc н не существует: в ряде полимерных изделий (таких, как линолеум, мио
гнс Н1дсли>1 медицинского назначения, антогермегики, большинство кабелей,
окна) сю просто нечем заменить; в друшх изделиях (например, упаковочные
пленки. । идрои золяционные мембраны, пленки для интерьера автомобиля, i ру
<>ы для канализации и водоснабжения) ПВХзаменим, но при худшем coin ноше
ппп «цепа качество»; в третьем случае, наиболее редком, ПВХ уступает другим
полимерам, обладающим лучшим набором свойств и/или более высокой тех
политичное п.ю (например, вытеснение ПВХ полиэтилентерефталатом с рынка
<»у । ылок для питьевой воды).
5. Мировой рынок ПВХ уже давно имеет глобальный характер. Несмо гря па
циклический характер ввода новых мощностей, мировое производство ПВХ в
i реднем растет пропорционально росту мирового валового продукта.
Поливинилхлорид еще достаточно долго останется полимерным материл
лом, незаменимым практически во всех отраслях промышленности.
Глава 1.
Физико-химические свойства
винилхлорида
СН2 ... 1 --CHCI
Винилхлорид (СН2=СНС1) впервые был получен А.-В. Реньо в 1835 г. при
взаимодействии спиртового раствора едкого кали с «маслом голландских хи-
миков» (дихлорэтаном) [31], хотя, по некоторым данным [14], это явилось пов-
юрением более ранних, неопубликованных работ Ю. Либиха. Е. Бауманн [32] в
1872 г. обнаружил и описал полимерные соединения, образующиеся в виде бе-
лых хлопьев при продолжительном воздействии солнечного света на винилхло-
рид. Им же была предложена эмпирическая формула этого полимера: (С2Н3С1)„.
Впервые реакция получения винилхлорида путем взаимодействия ацетилена и
хлорида водорода в присутствии хлорида ртути (сулемы) была запатентована в
1ермании в 1912 г. [33], а в 1929 г. также в Германии было создано первое про-
мышленное производство винилхлорида на основе ацетилена.
При нормальных условиях винилхлорид — бесцветный газ с температурой
кипения -13,8 °C и температурой плавления -158,4 °C. Технический продукт —
бесцветная легколетучая жидкость с резким эфирным запахом.
Физические свойства [34-38]
Молекулярная масса 62,499.
Плотность жидкости р = 0,983 г/см3 при -20 °C и р = 0,911 г/см3при 20 °C.
Плотность относительная по воздуху d - 2,17.
Температурный коэффициент объемного расширения в интервале от -13 до
28 °C р = 2,2/(1000 °C).
Таблица 1.1. Давление паров
Г, °C р, кПа 1,°С р, кПа 1, °C р, МПа t,°C р, МПа
-105,6 0,13 53,2 13,33 20,0 0,337 94,0 2,027
-90,8 0,67 -41,3 26,67 30,1 0,448 100,0 2,518
-83,7 1,33 -28,0 53,32 34,0 0,507 120,0 3,719
-75,7 2,67 -20,0 75,84 40,0 0,602 140,0 5,294
-74,0 4,00 -13,8 101,31* 50,45 0,736 150,0 6,237
-66,8 5,33 0 172 61,5 1,013
-61,1 8,00 4,3 203 80,0 1,540
* Атмосферное давление, равное 760 мм рт. ст.
11.ip.iMc । ры ьри i ического cociояния:
Давление, Ml la................5,34
l< Miicp.ii yp.i, °C..... 158,4
1 loiicpx пос гное натяжение:
/. ( 60 зо -20 -10 20 60 100 1 10
н, м11/м ...29,2 23,9 22,3 20,9 16,9 10,8 5,5 1,3
Ко >(|><|>ициен1 преломления n10D = 1,4046; n °р= 1,3700.
I еплопроводность жидкости при 20 °C X = 0,138 Вт/(м-К).
Тепло га испарения:
/,"(............. -60 -13,8 25 60 100 1 10
кДж/моль.... 22,51 20,8 18,64 16,74 13,6 8,24
1а1шмца 1.2. Вязкость динамическая, мкПа-с
Жидкость Пар
1,°С В Г, °C В
-60 420 -60 7,70
-40 334 -20 9,20
-30 303 20 10,71
-20 278 60 12,20
-10 256 100 13,71
20 180 140 15,21
60 130 180 16,71
220 18,21
260 19,70
Таблица 1.3. Теплоемкость
Жидкость Пар
t,°C Ср Дж/(г-К) /,‘С Ср, Дж/(г-К)
-40 1,046 0 0,785
-20 1,146 25 0,858
0 1,247 100 1,000
20 1,351 200 1,172
40 1,448 300 1,301
60 1,556 400 1,397
500 1,473
600 1,540
leiuioia плавления q,,,, = 4711 кДж/моль
leiuiora ci орания qu = 1198,1 кДж/моль.
Теплота полимеризации qIi;iM = 92,11 кДж/моль.
leiuiora образования стандартная АН мн = 37,26 кДж/моль.
Энтропия вещества в стандартном состоянии = 263,96 Дж/(моль-К).
Дипольный момент ре - 4,84-10’30 Кл м (1,45 ± 2% D).
Диэлектрическая проницаемость е = 6,26 при 25 °C.
Винилхлорид — легковоспламеняющийся и токсичный продукт.
Температура вспышки, °C:
в закрытом приборе.....................................-61,1
в открытом приборе.....................................-77,8
Температура самовоспламенения, °C.........................472
Температурный предел взрываемости паров в воздухе, °C.....<-45
Область воспламенения паров (об. %):
в воздухе.............................................3,6-33,0
в кислороде............................................4 70
ПДК паров в воздухе рабочей зоны
производственных помещений................................1 мг/м' (Россия).
Винилхлорид хорошо растворяется в 1,2-дихлорэтане, хлороформе, эфире,
ряде других органических растворителей. Это свойство широко используется
для извлечения винилхлорида из разбавленных газовых смесей. Достаточно
полные данные по растворимости винилхлорида в органических растворителях
получены В.А. Киреевым [39] и З.С. Смоляном [40]. Некоторые из этих данных
представлены в табл. 1.4.
Таблица 1.4. Растворимость винилхлорида в некоторых органических соединениях, на
1 мл растворителя
Растворитель Газ, мл Жидкость, мг
Дихлорэтан (смесь изомеров) 330 921
Тетрахлорэтан 273 762
Тетрахлорид углерода 271 756
Диметилформамид 291 812
Ацетон 366 1021
Циклогексанон 305 851
Анизол 257 717
Этилацетат 318 897
Бутилацетат 300 837
Этилбензол 247 689
Хлорбензол 252 703
о-Ксилол 252 703
Толуол 219 611
м Гептан 165 461
н Октан 166 463
Метанол 150 418
Винилхлорид малорасгворим в воде: при 20 °C рас гворяс п. в 0,25%, а при
25 "(: — О, I I %.
Растворимость (Р) коды в винилхлориде:
Г, °C...... О 10 20 25 30 40 50
1>,%....... 0,042 0,070 0,097 0,110 0,124 0,152 ОДНО
В табл. 1.5 представлены расчетные данные конденсации винилхлорида и i
смеси газов |2].
Спектры инфракрасного поглощения для винилхлорида приведены Дж. Гор
кингтоном, Дж. Томсоном [41] и М. Мамедовым [42].
Частоты колебаний винилхлорида, Гц:
Планарные колебания С—Н растяжение у, = 3030 у2 = 3080 уз = 3130
С—Н растяжение С-—Cl-растяжение С—С1-деформация ( Н2-деформация у4 = 1610 у5 = 724 Уб =395 у7= 1370 у8= 1030 у9= 1280
СН2-кручение Ую = 622
11епланарные колебания С—Н-деформация Ун = 895 у,2 = 940
Таблица 1.5. Конденсация винилхлорида (ВХ) из смеси газов (расчетные данные)
ВХ в газе, об. % Степень конденсации, %, при температуре
-20 °C -25 °C -30 °C -35 °C -40 С
50 - - 25,9 32,9 50,0
60 - - - 53,3 62,3
70 - 33,5 - 71,2 78,9
80 16,4 59,4 75,6 83,3 87,7
90 62,8 81,8 89,1 92,5 94,5
95 82,4 91,4 94,2 96,4 97,4
98 93,2 96,7 98,0 98,6 99,0
Химические свойства
Молекула винилхлорида содержи! дне функциональные группы — атом
хлора и двойную связь. Поэтому все превращения винилхлорида могут быть
ра (делены на реакции, обусловленные наличием атома хлора, и реакции, обу-
словленные наличием двойной связи.
Замещение связи С—CI
Атом галогена, связанный с атомом углерода, находящимся при С=С-свя-
зи, проявляет меньшую активность по сравнению с атомом галогена, находя-
щимся при С—С-связи.
Инертность атома хлора в молекуле винилхлорида может быть объяснена
следующим образом: л-электроны двойной связи взаимодействуют со свобод
ными электронными парами атомов хлора, в результате чего образуется общее
электронное облако.
СН2=СН^С1=^СН2=СН=С1. (1.1)
Смещение электронного облака приводит к тому, что связь С=С приобре-
тает некоторые свойства одинарной связи, а С—С1 — двойной. Данное пред-
положение подтверждается более высоким значением длины связи С—С в ви-
нилхлориде по сравнению с этиленом (138 и 134 пм соответственно). Длина же
связи С—О в винилхлориде (169 пм) ниже, чем в предельных хлоруглеводоро-
дах (176-177 пм).
Смещение электронной плотности в сторону СН2 группы приводит к
уменьшению полярности связи С—С1. Это подтверждается значением диполь-
ного момента, составляющего для винилхлорида 4,84 10 3(’Кл-м (1,45 D), тогда
как для этилхлорида эта величина составляет 6,0-10 30 Кл-м (1,805 D).
Инертность атома хлора в винилхлориде может быть также объяснена с по-
зиции мезомерии. Благодаря сопряжению двойной связи с атомом хлора молеку-
ла винилхлорида может образовать мезомерную структуру |СН2—СН—С1|+,
вследствие чего атом хлора становится малоподвижным.
По указанным причинам отщепление хлорида водорода или замещение ато
ма хлора требует достаточно жестких условий. Например, дегидрохлорирование
винилхлорида с образованием ацетилена протекает либо при высоких (> 500 °C)
температурах, либо под влиянием очень сильных оснований, например, натрия
в среде жидкого аммиака [42]. Основными продуктами при термическом разло-
жении винилхлорида при 525-575 °C являются ацетилен и хлоропрен; при этом
дополнительное введение в систему хлорида водорода снижает выход хлоро-
прена [43]. Горение винилхлорида в воздухе при 510-795 °C приводит к образо-
ванию в основном диоксида углерода и хлороводорода. В продуктах содержатся
также незначительные количества оксида углерода и фосгена [44].
Атом хлора, несмотря на свою низкую подвижность, может быть замещен
нуклеофильными агентами в присутствии палладия и других благородных
металлов [45-51]. В качестве продуктов образуются винилацетат, алкоголяты,
простые и сложные виниловые эфиры. Дж.А. Шебен [52] полагает, что процес-
ki>i npik осдинения и >л иминиронания 11роiскаioi через первоначальное oopa io
ii nine п комплексов
Алкоголя гы при повышенной температуре и под давлением peainpyioi сип
пплхлоридом, образуя соответствующие виниловые эфиры [69]. При 80 100 "(
винилхлорид, реагируя со спиртами, также образует виниловый эфир с иы.хо
дом до 90%:
«0-90 °C
(I1,=СНС1 + RONa--------- СН2=СН—OR + NaCl. (1.2)
кон
Фенолы реагируют значительно труднее и только при более высокой темпе
рагуре. Аналогично можно винилировать тиофенолы, карбоновые кислоты и
некоторые азотистые соединения, например, карбазол [70]. В частности, с i ио
крезолом реакция протекает следующим образом:
c;il2=CHCl + HS—
—сн3 —сн3—
— S—СН=СН2. (1.3)
В отдельных случаях атом хлора может реагировать с солями некоторых ор
ынических кислот с образованием сложных виниловых эфиров [71]. Например,
при нагревании в толуоле винилхлорида и натриевой соли янтарной кислоты
при 100-130 °C получается дивиниловый эфир янтарной кислоты:
СН2—COONa СН2—COO—СН=СН2
2СН2=СНС1+ — +2NaCl. (1.4)
СН2—COONa СН2—COO—СН=СН2
Натриевые и калиевые комплексы винилхлорида могут быть получены при
действии на последний металлических натрия и калия в среде тетрагидрофура
на [72].
При взаимодействии винилхлорида с оксидом углерода в присутствии пере
ходных металлов с низкой валентностью образуется хлорангидрид акриловой
кислоты [52]. Механизм этой реакции может включать окислительное присо
единение переходного металла. Карбонилирование винилхлорида в прису тс
твии этанола как растворителя и восстановленного палладиевого катализатора
приводит к образованию виниловых эфиров [53, 54].
Реакция винилхлорида с бутиллитием и далее с диоксидом углерода в рас г но
ре эфира приводит при низких температурах к образованию а-, [З-ненасыщен 11 ы.ч
органических кислот [55]. При реакции винилхлорида с боранами в растворе
тетрагидрофурана хлор замещается на водород с образованием этилена [56].
Винилхлорид может служить исходным соединением для синтеза реактива
Гриньяра — винилмагнийхлорида [57], который можно использовать для при
соединения винилового аниона к ряду органических функциональных групп
Винилмагниевые соединения могут взаимодействовать с монохлоридом меди
при -60 °C с образованием бутадиена [58]. При взаимодействии винилмагпии
хлоридами ,р ненасыщенными соединениями <><>р.г1уюня у ,б непасыщеппые
соединения [59]. Реакции пипилмагнийхлорида с органическими кислотами ве-
дут к образованию виниловых kciohoii и спиртов |6О, 61].
Виииллитиевые соединения образуются при взаимодействии винилхло-
рида с диспергированным литием, содержащим 2 мае. % Na при 0-10 °C [62].
Виниллитисвый комплекс образуется как промежуточное соединение при син-
тезе виниловых спиртов из альдегидов, винилкетонов из органических кислот,
вииилсульфитов из дисульфитов и монозамещенных алкенов из органических
галоидов [63-65]. Виниллитий может быть также преобразован в винилмедные
соединения, которые используются для стереоселективного введения винило-
вой группы в разные а-, p-ненасыщенные системы [66]. Виииллитиевые реаген-
ты, взаимодействуя с триалкилборанами, превращаются во вторичные спир-
ты [67].
При взаимодействии винилхлорида с фторидом водорода над Сг2О3/А12О3-
катализатором при 380 °C образуется винилфторид [68].
Практически важно восстановление винилхлорида заменой хлора на водо-
род [73]. Наиболее универсальным методом является каталитическое гидри-
рование. Реакция идет предположительно по ионному механизму В зави-
симости от условий процесс может протекать или только с замещением атома
хлора, или с одновременным восстановлением С—Cl-связи и гидрированием
двойной связи. В паровой фазе в присутствии Ni- или Pd-содержащих катали-
заторов винилхлорид восстанавливается до этилена и далее до этана:
Ni
СН2=СНС1 + Н2—► СН2=СН2 + НС1. (1.5)
Многие металлы: натрий, магний, цинк, алюминий, медь в сочетании с до-
норами водорода — водой, кислотами, спиртами, аммиаком и т.д. могут восста-
навливать связи С—С1.
Под действием катализаторов, содержащих медь, кадмий, свинец или олово,
винилхлорид при 400 °C в паровой фазе способен конденсироваться с образо-
ванием хлоропрена [73]:
2СН2=СНС1 —— СН2=СН—СС1=СН2 + на.
Реакции подвойной связи
Полимеризация. Способность к полимеризации — наиболее важное хи-
мическое свойство винилхлорида, определяющее его практическую ценность.
Процесс полимеризации винилхлорида протекает при 40-90 °C и давлении
0,5-1,5 МПа в присутствии инициатора. Винилхлорид полимеризуется по ради-
кальному механизму, состоящему из следующих элементарных актов.
Инициирование — образование первичного активного центра полиме-
ризации из молекулы мономера (М) за счет внешнего импульса (Ru). Внешний
импульс может быть обеспечен химическими инициаторами, активным светом,
радиацией и т.п.:
+ М—- !<,—М*. (1.6)
Jtor.iki является наиболее шергоемким во всей цени и определяющим для
реакции полимеризации в целом (энергия активации 60 80 кДж/моль).
Рос г цепи — присоединение молекул мономера к активному цен тру. . )нер
। ия активации составляет 21-34 кДж/моль, что в несколько раз ниже по сравне
пию с актом инициирования:
Ru—М + иМ—* Ru—М„—М . (1.7)
Обрыв цепи — при столкновении микрорадикала с ингибирующими при
месями, стенкой или с другими радикалами:
Ru—М„—M’ + R— Ru—М„—М—R. (1.8)
Передача цепи — характерный акт процесса полимеризации, при кого
ром за счет столкновения микрорадикала с мономером или полимерной мо
лскулой происходит передача спаренных электронов с возникновением либо
вновь возбужденной молекулы, либо разветвления цепи:
Мх
I.
Ru—М„—М +МХ—М—Му—*RU—М„—М—М . (1.9)
м>.
Акт передачи цепи может приводить к образованию трехмерных и сетчатых
структур полимера.
При полимеризации винилхлорида в основном получаются полимеры со
структурой 1,3-монохлорида и средней молекулярной массой 60-200 тыс.:
hv
иСН2=СНС1 — [—СН2—CHCI—СН2—СНС1—]п12. (1.10)
Окисление. Наличие двойной связи делает винилхлорид достаточно реакци
онноспособным при взаимодействии с такими окислителями, как перманганат
калия, азотная кислота, оксид хрома (СгО3), кислород, озон, перкислоты и др.
При этом в зависимости от ряда факторов образуются разные кислородсодер
жащие соединения [73]. Наиболее важной реакцией окисления является реак
ция с кислородом, поскольку она в той или иной степени влияет на полимери
зацию.
Хорошо очищенный винилхлорид не реагирует с газообразным кислоро
дом при 45 °C в течение недели, а также с кислородом в присутствии воды или
НС1 [74, 75]. Быстрое окисление винилхлорида при контакте его с воздухом
или кислородом инициируется азо-бис-изобутиронитрилом [76, 77], УФ-све
том [78], пероксидами бензола и ацетилбензоила [76], следами ацетальдегида,
ацетилена, хлорида железа [74]. Ряд авторов [74-81] полагают, что при окис
лении винилхлорида кислородом образуются промежуточные пероксидные со
единения, которые остаются после испарения мономера в виде масла [76, 77|,
сиропов [78] или твердого белого осадка [79]. Эти вещества, легко отщепляю
щие под действием воды НС1, выделяют иод из кислых растворов KI и взрыва
ются при трении и нагревании [74-80]. Количество пероксидных соединении
упе/шчиплси» с увеличением акшвшн hi niniiiii.iiopj |77|; при ном скорость
накопления иероскидпых групп совпадает е начальной скоростью поглощения
кислорода [81|.
Установлено, что полипероксиды являются продуктами сополимеризации
винилхлорида с кислородом и имеют предположительное строение (— О —
CHiCHCl—О—)и, где и = 174-26 [77], либо представляют равновесную смесь
двух форм СН2=СС1ООН СН2СНС1—О—О— [80]. Полярографичес-
кое исследование твердых продуктов окисления винилхлорида показало в по
л и пероксиде наличие трех пероксидных групп —О—О—С—О, —О—О и
— О—ОН разной степени устойчивости [75, 81].
Фотоокисление винилхлорида смесью кислорода и хлора в газовой фазе при
100 °C или в растворе тетрахлорэтана при 90 °C приводит к образованию хлор
ацетальдегида с выходом 95% [82].
Е. Санхуэза с соавторами [83] показал, что окисление винилхлорида, содер-
жащего сенсибилизированный атом хлора, приводит к образованию С1СНО
(выход 74%) и СО (выход 25%) при 30%-ной конверсии винилхлорида. При
высоком соотношении кислород: хлор реакция не протекает по цепному меха-
низму. Реакция винилхлорида с триплетным кислородом [О(3р)] дает высокие
выходы СО и хлорацетальдегида [84]. Показано также, что окисление в газовой
фазе при t > 250 °C не приводит к образованию карбонильных соединений С2,
причем винилхлорид является единственным из хлоролефинов, чье окисление
не идет по радикально-цепному механизму.
При взаимодействии винилхлорида с озоном образуются неустойчивые
взрывчатые озониды, превращающиеся в карбонильные соединения или кар-
боновые кислоты по схеме [73]:
Оз
+о, / \ \ /\ /
>С=С< — >с—С< —- с—С —- >С=О + >с+—о—О . (1.11)
о —о о+
Реакция озона с винилхлоридом используется для удаления последнего из
отходящих газов промышленных производств. Продуктами окисления являют-
ся муравьиная кислота и формилхлорид [85, 86].
Абсолютное значение констант скорости реакции озона с хлоролефинами
в растворе СС14 при 25 °C указывает на существенное влияние атомов хлора и
пространственных факторов у двойной связи на скорость ее озонолиза. Так, от-
ношение констант скорости озонолиза перхлорэтилена, винилхлорида и этиле-
на [87]:
^С2С14 : ^c^HjCl: ^с2н4 = 1:1200 : 25 000. (1 12)
Надкислоты окисляют винилхлорид до эпоксидов или гликолей в зависи-
мости от условий реакции [73, 88]:
>С—с< 11« 4)0011-
1« ООП
--- C(OCOR)—с—ОН
I! О
—ГЛИКОЛЬ. (1.11)
Аналогично действует азотная кислота, давая карбонильные соединения
либо с сохранением числа углеродных атомов, либо с деструкцией по двойной
связи. Реакция осложняется побочными процессами нитрования [73].
Перманганат калия дает аналогичные продукты окисления с деструкцией по
двойной связи.
Галоидирование. При галоидировании непредельных углеводородов, в час i
ности, винилхлорида, возникают две возможности: присоединение галогена ио
двойной связи либо замещение водорода хлором с сохранением непредельной
связи. Возможность реализации двух одновременных и конкурирующих про
цессов хлорирования создает проблему выбора оптимальных условий хлорн
рования углеводородов с непредельными связями. Обычно при низких темпе
ратурах протекает реакция аддитивного хлорирования с образованием 1,1,2-
грихлорэтана:
С1,
СН2=СНС1—— СН2С1—СНС12, (1-11)
а при высоких (250-500 °C) — преобладает замещение с образованием ди- и
трихлорэтиленов. В частности, А.И. Субботиным с сотр. [89,90] показано, ч го н
процессе хлорирования винилхлорида при 320-380 °C одновременно образую i
ся продукты присоединения и замещения. При этом основная часть продуктов
образуется путем распада продуктов присоединения.
Хлорирование винилхлорида может протекать как по ионному, так и
по радикальному механизму. В жидкой фазе в присутствии кислот Льюиса
(FeClj) практически количественно образуется 1,1,2-трихлорэтан. Тог же
продукт образуется по радикальному механизму при t < 250 °C, а также при
фотохимическом хлорировании [91-93]. Присутствие небольших (<0,5%) ко
личеств кислорода резко увеличивает скорость радикально цепного хлорн
рования [90].
Хлорирование винилхлорида в водной среде [94] с pH - 7 в присутствии
эмульгаторов (сульфатов, полученных из высших парафиновых углеводородов)
при охлаждении до 16 °C идет с образованием чистого 1,1,2-трихлорэтана без
примеси хлорированных продуктов.
При определенных условиях хлорирование винилхлорида в водной среде
может приводить к образованию монохлорацетальдегида [95].
Некоторые авторы указывают, что жидкофазное хлорирование олефинов и.
в частности, винилхлорида, протекает по молекулярному механизму, вредно
лагающему образование продуктов из молекулярных компонентов, минуя сто
дию диссоциации на ионы или радикалы [ 1145]. Относительный вклад того или
иного направления реакции зависит от условий проведения процесса: темпе
ратуры, концентрации реагентов, полярности среды и т.д. Механизм действия
хлорида железа связан с предварительным образованием комплекса с хлором
и углеводородом. В »гом случае ск<»р<н н> реакции хлорирования записывается
уравнением третьего порядка:
r=k[>C=C<] [С12] [РеС1,]. (1.15)
При »гом увеличение количества атомов хлора в молекуле органического
соединения способствует снижению его реакционной способности.
Винилхлорид может также взаимодействовать с бромом с образованием
I хлор-1,2-дибромэтана при t < 100 °C; широкого промышленного значения
нот процесс не имеет.
Гидрогалоидирование. Реакции гидрогалоидирования винилхлорида про-
текают в основном по гетеролитическому механизму и катализируются хлори-
дами металлов. Реакция может проходить как в жидкой, так и в газовой фазах,
преимущественно в соответствии с правилом Марковникова:
на
СН2=СНС1—► СН3—СНС12. (1.16)
Реакция протекает с выделением тепла (-ДН298 = 72,81 кДж/моль) и значи-
тельным уменьшением энтропии (-S298 = 143,8 Дж/(моль-К)). Равновесный вы-
ход 1,1-дихлорэтана составляет (в числителе — при 0,1 МПа, в знаменателе —
при 1 МПа): 0,93/0,98 при 100 °C, 0,74/0,92 при 150 °C, 0,52/0,83 при 200 °C [97].
Р. Ринкер и В. Коркорен [98] предложили уравнение, удовлетворительно описы-
вающее реакцию гидрохлорирования винилхлорида в газовой фазе:
r=k(Al А11 -А/К), (1.17)
где г — скорость образования 1,1-дихлорэтана;
к — константа скорости;
А, А1, А" — активности соответственно 1,1 дихлорэтана, НС1 и винилхлорида
в газовой фазе;
К — константа равновесия.
Выражение для константы равновесия как функции температуры имеет сле-
дующий вид:
1п К = 8,096 + (14,135/7 — 4,900 In Т + 0,0054 Г — 21,09)/1,987. (1.18)
Наилучшие результаты при газофазном гидрохлорировании винилхлорида
получены в случае применения катализатора ZnCl2 (20%) на цеолите-408 [99].
В отсутствие катализатора гидрохлорирование винилхлорида в газовой фазе
практически не идет из-за высокого активационного барьера (150-170 кДж/
моль).
Скорость жидкофазной реакции гидрохлорирования винилхлорида при
20-25 °C достаточно велика и сочетается с высоким равновесным выходом це-
левого продукта. Термодинамические параметры реакции имеют следующие
значения [97]:
ДН298 = -92,5 кДж/моль;
Д5298 = -239 Дж/(моль-К);
1g Кр = 4870/7-12,5.
В жидкой фале нолмо кно полное превращение исходных peatен гон при гид
рохлорироваиии. Согласно В.Л. Аверьянову 1100] для реакций гидрохлорпро
нация, в частности винилхлорида, типичен кислотный катализ но следующим
двум маршру гам:
- первичное присоединение протона катализатора по двойной связи с обра
зованием иона карбония;
- первичное активирование реагента с образованием более сильного элек i
рофила, способного присоединяться по двойной связи.
Реакция гидрохлорирования винилхлорида существенно ускоряется в при
сутствии кислот Льюиса (А1С13, FeCl3, SnCl4 и др.) за счет образования кислоты,
более сильной, чем хлорид водорода, в результате связывания аниона С1 в ком
плекс с кислотой Льюиса, например:
НС1 + FeCl3 H+FeCl4, (1.19)
либо в воздействии кислоты Льюиса на первоначально образовавшийся
п-комплекс, что ускоряет переход л-комплекса в ион карбония:
НС1 НС1-А1С13
+НС1 | +А1С13 |
> с=с< —— >с=с< [>с=с<] >снс+< + а1сг4 —*
— * >СНС1С< + А1С13. (1.20)
Исследования электропроводности растворов НС1 и FeCl3 в 1,1,2,2-тетра
хлорэтане показывают, что в присутствии соли константа диссоциации хлорида
водорода увеличивается на 7 порядков [278].
Установлено [101, 664], что в присутствии суспендированных А1С13 и FeCI,
скорость реакции гидрохлорирования винилхлорида в 12-15 раз выше соот-
ветствующих значений гомогенного процесса. Хлорид алюминия более активен
по сравнению с хлоридом железа в сопоставимых условиях.
Взаимодействие винилхлорида с бромидом водорода протекает по ради
кально-цепному механизму, в котором наиболее значительны реакции на с га
днях присоединения и передачи цепи:
Вг' + СН2=СНС1 — Вг—СН2—СНСГ (АН = 20,9 кДж/моль),
Вг—Н2—СНСГ + НВг —- Вг—СН2—СН2С1 + Вг (АН = 46,0 кДж/моль).
Гидробромирование отличается от гидрохлорирования винилхлорида экзо
термичностью обеих стадий. Более вероятен радикальный механизм реакции
гидробромирования, а для реакции с участием НС1 такой механизм возможен
только в особых случаях. Гомолитический процесс протекает против правила
Марковникова [102]:
С1—СН=СН2 + НВг—
В темноте FeCI3
Медленно
Пероксиды
Быстро
С1—СНВг—сн2
С1—СН2—СН2—Вг.
I iiiiosaioiidiipottatiuc Реакция приведши пин хлорнотгг негой кислоты к
олефиновым соединениям и, в частности, к винилхлориду, протекает согласно
правилу Марковиикова с последующим отщеплением хлорида водорода и обра-
зованием хлорацегальдегида [73, 103, 104]:
( 112=( 11С1 + НОС1 —► [С1СН2—CHCl(OH)] — С1СН2—СНО + НС1. (1.22)
Выход хлорацетальдегида достигает 75%. О. Эрнст и Г. Ланге [103] отметили
1акже образование небольших количеств 1,1,2-трихлорэтана. В целом наличие
хлора в молекуле винилхлорида существенно замедляет его гипохлорирование
но сравнению как с этиленом, так и с олефинами, содержащими аллильный
хлор. Из-за продолжительной реакции это приводит к образованию побочных
продуктов за счет присоединения хлора по двойной связи.
Винилхлорид способен также взаимодействовать со смесью формальдегида
и хлорида водорода с образованием соответствующих хлоргидринов [105, 106]:
с Н2=СНС1 + СН2О + НС1 —
-СН2ОНСНС1СН2С1
СН2ОНСН2СНС12.
(1.23а)
(1.236)
Присоединение нитрозилхлорида. Особенность реакции хлорида нитрози-
ла с галоидсодержащими олефинами состоит в том, что промежуточно образу-
ющиеся нитрозосоединения не перегруппировываются в оксимы, а окисляются
до нитросоединений. Поэтому продукты реакции представляют смесь нитро- и
полихлоралканов. Обычно реакция идет при t = (-20)з-(-10) °C в хлороформе
или тетрахлориде углерода по гетеролитическому механизму нуклеофильного
присоединения. Наличие электроноакцепторного заместителя в хлорсодержа-
щих мономерах сильно замедляет процесс.
Нитрозилхлорид присоединяется так, что положительная нитрогруппа идет
к наименее гидрогенизованному углероду в соответствии с направлением поля-
ризации [107, 108]. Промежуточное хлорнитрозосоединение реагирует дальше
с избытком хлорида нитрозила с образованием неустойчивого диазонитрата,
разложение которого в присутствии хлорида нитрозила дает хлорид, азот и
диоксид азота. При взаимодействии диоксида азота с исходным непредельным
соединением по гомолитическому механизму образуется нитросоединение, у
которого нитрогруппа связана с наиболее гидрогенизованным углеродом. Од-
новременно с присоединением нитрозилхлорида в значительной степени идет
побочная реакция хлорирования по двойной связи и образуются смолистые ве-
щества за счет полимеризации и окисления.
При взаимодействии винилхлорида с нитрозилхлоридом образуется 1,1-
дихлор-2-нитроэтан и 1,1,2-трихлорэтан [109, ПО]:
СН2=СНС1 + NOC1
NOCI
CH2NO—СНС12 ——
—-СН2С1—СНС12.
CH2NO2—СНС12
(1.24а)
(1.246)
Нитрозохлорирование может активироваться катализаторами Фриделя-
Крафтса, образующими с нитрозилхлоридом комплексы типа NO+[A1C14] .
Присоединение нитрилхлорида. В результате присоединения ин 1 рилхлорн
да подвойной связи образуется хлорин трос оедииение:
>С=С< + C1NO2 —* >CC1C(NO2)<. (1.25)
Винилхлорид в паровой фазе реагирует со смесью диоксида а то га и хлора и
дает при 325 °C 1,2-дихлор-2-нитроэтан.
2СН2=СНС1 + N2O4 + С12 —— 2СН2С1—CHC1NO2. (1.26)
Найдено [111], что смесь N2O4 и хлора действует аналогично нитрилхлориду
в случае непредельных соединений с электроноакцепторными заместителями, а
в случае электронодонорных заместителей ориентации присоединения про in
воположны. Авторы объясняют это тем, что электронодонорные заместители
у двойной связи способствуют протеканию реакции по ионному механизму, а
электроакцепторных — по радикальному. В силу этого галоидолефины, в час i
ности винилхлорид, реагируют по радикальному механизму.
Нитрилхлорид в смеси с винилхлоридом легко взрывается [112].
Диэтилхлорамин присоединяет винилхлорид с выходом 82% [113].
Присоединение сулъфурилхлорида и тионилхлорида. Сульфурилхлорид
(SO2C12) и тионилхлорид (SOC12) активно присоединяются по двойной связи не
предельных соединений, в частности винилхлорида. Согласно Н.К. Кочеткову
[114], сульфурилхлорид при взаимодействии с винилхлоридом в среде тетра
хлорида углерода в присутствии хлорида алюминия дает рф-дихлорэтилсуль-
фанилхлориды. При этом в значительных количествах образуется 1,1,2-три
хлорэтан — продукт аддитивного хлорирования винилхлорида:
СНС12—CH2SO2C1
СН2=СНС1 + SO2C12
А1С1,
СН2С1—СНС12
(1.27а)
(1.276)
Выход рф-дихлорэтилсульфанилхлорида составляет 31,5% от теоретичес
кого.
Более энергично протекает реакция винилхлорида с тионилхлоридом [115].
Принимая, что при растворении А1С13 в тионилхлориде образуется хлортио
нилкатион С1—S+=O, авторами предложен механизм сульфохлорирования.
Медленность реакции с сульфурилхлоридом объясняется малой склонностью
к образованию комплекса (S'O2C1)A1CI4 и меньшей координационной ненасы
щенностью атома серы в S+O2C1, чем в S+OC1. Для уменьшения хлорирующе
го действия в случае присоединения тионилхлорида рекомендуется насытить
смесь реагентов SO2.
Гетеролитическое (электрофильное) присоединение галоидуглеводородов.
Взаимодействием монохлорсодержащих углеводородов с винилхлоридом [116
121] в условиях реакции Фриделя-Крафтса можно получить соединения с CCI
группой:
RC1 + СН2=СНС1 — RCH2CHC12. (1.28)
Мстил и >i нлхлорид нс присоединяю) oi к винилхлориду. При нитровании
до 50 "(' смеси л илхлоридас винилхлоридом и присутствии хлорида алюминия
1,1 -дихлороу гап не обра зуется; в реакционной смеси найдены 1,1-дихлорэтан и
1,1,3 трихлорбу ran [117]
И HiecTHo присоединение разных вторичных [116,117, 119,120] и третичных
11 17, 119, 1211 алкилхлоридов к винилхлориду. Низшие хлоралканы, третбутил
и амилхлориды реагируют с винилхлоридом как в присутствии FeCI3 при 20 °C,
гак и в присутствии А1С13 при -20 °C (выход 70%).
Аддукты винилхлорида с высшими хлоралканами, например, 2-хлор-2,3-ди-
метилбутаном и 2-хлор 2,3,3-триметилбутаном в присутствии FeCl3 при 20 °C
дают продукты дегидрохлорирования с образованием алкенов и димеров. Эти
же хлоралканы с А1С13 при -20 °C образуют с выходом 35-50% соответствую-
щие дихлориды. В присутствии А1С13 выход дихлоридов выше, чем в присутс-
гвии FeCl3. Так, выходы продуктов присоединения изопропилхлорида и трет
бутилхлорида к винилхлориду в присутствии А1С13 составляют 34 и 96%, а в
присутствии FeCl3 — 23 и 77% соответственно [117].
В реакцию с винилхлоридом вступают лучше всех третичные хлоралканы,
хуже — вторичные и практически не реагируют первичные.
С вторичным амилхлоридом винилхлорид дает с высоким выходом 1,1-
дихлор-3,4-диметилпентан.
сн3 сн3 сн3
СН,=СНС1 + CH—СНС1—СН3 FeC1 ► CH—СН—сн2—СНС12. (1.29)
сн3 сн3
Известно присоединение циклогексилхлоридов к винилхлориду [122] с об-
разованием
/---\ К
< BY
N---7 СН2—СНС12 (1.30)
(при R - Н — выход 38%; при R = СН3 — выход 52%). Так же реагирует и метил-
циклопентилхлорид с выходом 20% [122].
При 0-5 °C винилхлорид реагирует с бензолом, образуя смесь 1-хлорэтил-
бензола и 1,1-дифенилэтана [123, 124]; 1,1-замещенные продукты образуются
последовательно взаимодействием первичного аддукта со второй молекулой
промежуточного соединения. Быстрая реакция первичного продукта с молеку
лой толуола дает выход 1,1-дитолилэтана 68%, а анизол (метоксибензол) — вы-
ход 1,1-ди-п-анизилэтана 42% [125,126]. Фенол также реагирует с образованием
п-винилфенола [127]. Конденсация винилхлорида с формальдегидом и хлори-
дом водорода (реакция Принса) дает 3,3-дихлоро-1-пропанол и 2,3-дихлоро-1-
пропанол [106, 128]. Винилхлорид может взаимодействовать с бромоформом в
присутствии пентакарбонила железа [129].
В присутствии хлоридов цинка или олова винилхлорид реагирует с моно-
хлордиметиловым эфиром, давая с удовлетворительным выходом 1,1-дихлор-
3-метоксипропан [130]:
/|>< I
ci |,=( 11( lull ,0(1I2C1 — * Cl I ,OCI 1.( I l;CI ICI2. (1.3 r)
Виимодейсгвием ROCH2C1 (R = CH3, C2HS, н CtH7, н-С|Нч) с винилхлори
дом были получены соединения строения ROCH2CH2CHC12 с выходом 25- 16%
11311. Наибольший выход продукта получен в случае хлорметилового эфира
В реакции с хлорметиловым эфиром наряду с аддуктом (46%) были выделены
СН3СНС12 (5%) и СН3О(СН2)2, СНС1СН2СНС12 (5,5%), а также продукт катион
ной геломеризации с и = 2.
При проведении реакции электрофильного присоединения с хлорангидри
дами карбоновых кислот конечным продуктом реакции в основном является
продукт дегидрохлорирования промежуточного аддукта.
При взаимодействии винилхлорида с ацетилхлоридом в присутствии А1( 13
можно выделить промежуточно образующийся дихлоркетон с выходом 50%
который легко дегидрохлорируется в метилф-хлорвинилкетон [132];
СН3СОС1 + СН2=СНС1 —* СН3СОСН2СНС12. (1.32)
При проведении реакции винилхлорида с хлорангидридами кислот арома ги
ческогоряда образуются арилфф-дихлорэтилкетоны [133], которые значигель
но более устойчивы, чем их алифатические аналоги. Фенил-, п-нитрофенил ,
п-метоксифенил, о-бромфенил-рф-дихлорэтилкетоны получены с выходом 71,
67, 49, 45% соответственно [132].
Дихлорангидриды алифатическмх дикарбоновых кислот реагируют с ни
нилхлоридом в присутствии А1С13 в растворителе (СС14, СН2С12, СН2С1СН2С1)
при температуре от -5 до +20 °C с образованием (выход 55-89%) соответствую
щих тетрахлоркетонов [134, 135]:
С1С(О)(СН2)ИСОС1 + 2СН2=СНС1 —-
—- СНС12СН2С(О)(СН2)„СОСН2СНС12 (и = 3+7). (1.33)
Дихлорангидрид янтарной кислоты с винилхлоридом дает лишь t,e
дихлороксокапроновую кислоту (выход 60%). Увеличение времени, темпера гу-
ры реакции, количества катализатора [134] не приводит к образованию диад
дукта. Оксалилхлорид реагирует с винилхлоридом при t - -10^0 °C с образов»
нием аддукта — 1,1,5,5-тетрахлорпентанола-З (выход 69%); проведение реакции
при 10-20 °C ведет к дегидрохлорированию аддукта с образованием 1,5-дихлор
пентадиен-1,4-она-З (выход 67%) [134, 136].
Гомолитические присоединения галоидуглеводородов. Гомолитические при
соединения хлорсодержащих соединений к винилхлориду с образованием по
вых углерод-углеродных связей протекают по свободно-радикальному мехапиз
му. Присоединение хлорсодержащих соединений к ненасыщенным с разрывом
С—Х-связи соединений и образованием новой С—С-связи протекает по об
щей схеме [73]:
I I I
X—С—С1 + >С< — X—С—с—Cl (X = Н, Br, Cl, I). (1.31)
I I I
К k.i'icl ।не иннцн.пороп тполыуюгся карбонилы мегаллоп с переменной
палеиi ikk n>io и нуклеофильный сокагализагор (изонронанол, димегилформа
мид, а цени in i рил), подавляющие побочную реакцию геломеризации. В услови-
ях ной реакции винилхлорид присоединяется в присутствии пентакарбонила
железа и изопропанола к метилхлороформу, давая с относительно высоким вы-
ходом 1, 1,3,3,- ге грахлорбутан:
СН (СС!3 + СН2СНС1------------- СН3СС12СН2СНС12.
(СН,),СНОН
(1-35)
Аналогично присоединяются к винилхлориду 1,1,1-трихлорпропан и 1,1,1,5-
е грахлорпентан с образованием соответствующих аддуктов и некоторого ко-
личества теломеров.
Присоединением хлороформа к винилхлориду в присутствии хлорного
железа и хлоргидрата диэтиламина в спирте получен 1,1,3,3-тетрахлорпропан
1137]:
FeCI,
СНС13 + СН2=СНС1---------------*- СНС12СН2СНС12.
(C2H5)2NHHC1
(1.36)
Винилхлорид реагирует также с трихлорбромметаном в присутствии тет-
ракарбонила никеля с образованием 1,3,3,3-тетрахлор-1-бромпропана (выход
75%) [138]:
Ni(CO)4
СС13Вг + СН2СНС1--------- СС13-СН2-СНС1Вг.
(1.37)
Та же реакция протекает за счет УФ-инициирования [139].
Присоединение к винилхлориду трихлорметильных производных RCC13,
инициированное пентакарбонилом железа в присутствии изопропанола, при-
водит к соединениям общей формулы RCC12CH2CHC12 с выходом 26-35% от
теоретического [140, 141].
По свободно-радикальному механизму протекает и реакция взаимодейс-
твия винилхлорида с хлоридами проматических диазониев в присутствии солей
меди:
С=С + ArN2Cl —► С1С—CAr + N2.
(1.38)
Эта реакция хлорарилирования или реакция Меервейна обычно прово-
дится в водно-ацетоновой среде при строгом поддержании pH среды. При
взаимодействии винилхлорида с толуолдиазонийхлоридом в присутствии
СпС12 и СаО и t - 5-г 20 °C образуется 1,1 дихлор-2-толуилэтан с выходом
50-70% [142]:
СиС12
СН2=СНС1 + CH3C6H5N2C1 — С6Н5—СН2—СНС12 + N2. (1.39)
Радикальная тсломсри ищия. Геломери лация — эго реакция непредельно
то соединения (например, винилхлорида) с каким либо вещее гном (телогеном),
в результате которой образуется смесь продуктов разной молекулярной массы
(теломеров). Молекулы теломеров построены из нескольких молекул мономера
и содержат концевые группы — фрагменты телогена. В общем виде теломериза
ция может быть выражена схемой:
X—Y + nC=C —X—[—С—С—]„—Y. (1.40)
Телоген Теломеры
Реакция инициируется пероксидами, азосоединениями, соединениями мс
таллов с переменной валентностью, облучением.
В присутствии пероксидных инициаторов винилхлорид теломеризуется
тетрахлоридом углерода с образованием высших теломеров с п - 2<-7 [143—145].
При инициировании теломеризации хлоридом железа в изопропаноле винил
хлорид с тетрахлоридом углерода дает жидкие теломеры с и = 1+3.
Стереохимию радикальной теломеризации винилхлорида исследова
ли спектроскопическими методами. Показано [146, 147], что в теломериза
ции винилхлорида с СС14, инициируемой пероксидом бензоила или системой
FeClj+i-CjHyOH рост цепи до теломеров с и < 10 носит нестереорегулярный ха
рактер. Исследована стереохимия теломеризации винилхлорида хлороформом
с выделением фракций, отвечающих всем низшим теломерам [148]. Примене-
ние методов ИК, ЯКР и ПМР-спектроскопии позволило установить, что тело
мер СС13(СН2СНС1)3Н образуется при 90 °C и пероксидном инициировании в
виде двух диастереомерных рацемических форм с соотношением 1,5+2:1. Был
сделан вывод, что присоединение алифатических радикалов к винилхлориду
протекает с некоторой стереонаправленностью.
Теломеризация винилхлорида трихлорсиланами в присутствии пероксида
бензоила приводит к образованию теломеров H(CHCICH2)„SiCl3 с и - 2 и 3 [149|.
Теломеризация винилхлорида бромтрихлорметаном приводит к соединен и
ям строения СС13(СН2СНС1)„Вг. Идентифицированы теломеры с п = 1+7. Опре-
делены константы передачи цепи [150].
Стабилизация винилхлорида
Использование хлорсодержащих мономеров в производстве соответству-
ющих полимеров и сополимеров неизбежно связано с необходимостью сохра
нения в течение определенного времени качества мономера — технического
продукта. Вместе с тем высокая реакционная активность хлорсодержащих мо
номеров и наличие ряда примесей в техническом продукте делает эту задачу
достаточно сложной.
Для решения этой задачи необходимо рассмотреть возможные превраще
ния в хранящемся хлорсодержащем мономере, важные с учетом изменения ка
чества. Результатом таких превращений могут быть изменение состава техпи
ческого продукта, появление мути, взвесей, осадка, изменение окраски.
I Ьиболсс важной химической реакцией в рассматриваемом аспек те являем
ся реакция взаимодействия хлорсодержащих мономеров с кислородом.
Р шее было указано, ч го хлорсодержащие мономеры энергично взаимодейс-
твуют с кислородом даже в следовых количествах и при комнатной темпера-
туре. Образующиеся при этом полипероксиды линейного или циклического
строения способны инициировать неуправляемый процесс полимеризации.
11оскольку полимеры хлорсодержащих мономеров практически не растворяют-
ся в мономере, их образование приводит к появлению взвесей и осадка, а также
изменению состава технического продукта.
Полипероксиды — вещества малостабильные и разлагаются, особенно в
присутствии воды, с образованием оксидов углерода, хлорида водорода, фор-
мальдегида, глиоксаля, гликолевой кислоты и т.п. Все это также изменяет состав
технического продукта; кроме того, кислые вещества и прежде всего хлорид во-
дорода вызывают коррозию поверхности емкостей с образованием хлоридов
железа. Хлориды железа могут далее катализировать реакцию автоокисления
хлорсодержащих мономеров, а также реакцию дегидрохлорирования хлорсо-
держащих мономеров, особенно низкомолекулярных.
Первая из этих реакций увеличивает количество полипероксидов и последо-
вательно идущих за этим процессов. Вторая — может приводить к образованию
трехмерных структур или соединений с сопряженными двойными связями, да-
ющими окрашивающий мономер, а также повышающих содержание хлорида
водорода с негативными последствиями.
Реакцию автоокисления способны инициировать и такие возможные при-
меси в техническом продукте, как ацетилен и ацетальдегид. Вся эта сложная
система взаимо-последовательно-параллельных реакций в конечном итоге из-
меняет качественный состав технического продукта и приводит к ухудшению
качества получаемого полимера.
Необходимо выделить узловые реакции:
- окисления хлорсодержащего мономера с образованием полипероксидов,
идущие преимущественно по радикальному механизму;
- разложения полипероксидов, катализируемую теплом, светом, влагой и
хлоридами железа с образованием хлорида водорода;
- полимеризации хлорсодержащих мономеров, инициируемую полиперок-
сидами и продуктами их распада;
- кислотного разрушения металла емкостей с образованием хлоридов же-
леза.
Таким образом, теоретически для обеспечения стабильности качественно-
го состава хлорсодержащих мономеров необходимо заблокировать эти узловые
реакции, для чего используют соответствующие химические вещества.
Для блокирования реакции окисления мономера можно использовать анти-
оксиданты (в частности, гидрохинон, фенилнафталамин и др.).
Для блокировки реакции разложения полипероксидов могут быть исполь-
зованы дезактиваторы металлов, образующие прочные комплексы и, в частнос-
ти, оксикислоты, диамины, эфиры фосфорной кислоты и др.
Блокировать реакцию полимеризации мономеров можно ингибиторами
цепных процессов и, в частности, фенолом, ароматическими аминами, дифени-
ламином, тимолами.
Для блокировки pi акции кислотного ра (рушения мешала можно исколь
lonaib акцепторы хлорида водорода и, в частности, алифатические амины
шоксисосдипения. Всгесгвенно, что между этими соединениями пег четкой
разграничения об lacieii действия. Например, фенол может выступить в роли i
акцептора кислорода и ингибитора свободно-радикальных процессов. 1Io.ttom)
для с (абилизации на практике используют как одно-, так и многоком нонен i ны<
стабилизирующие составы.
Для винилхлорида в качестве стабилизатора обычно используется одпоком
понентная система. При этом стабилизатор добавляется в таком количестве
которое обеспечивает необходимую стабильность продукта, но в то же врем»
позволяет использовать стабилизированный мономер непосредственно для но
димеризации, не удаляя стабилизатора.
В производстве винилхлорида прежде всего обеспечиваются условия, ис
ключающие контакт с кислородом и светом. В процессе получения винилхло
рида стабилизаторы не применяются. Стабилизируется уже готовый продукт.
Наиболее простым приемом стабилизации винилхлорида является прокус
какие жидкого мономера через чешуированную щелочь, активный оксид ал«>
миния или их смеси. При этом связываются следовые количества влаги и хло
рида водорода.
В качестве стабилизирующих добавок предлагаются фенол, параамипофе
пол, оксипиридин, ионол, пентаметилфенол, параметоксифенол, бутоксифенол
гидрохинон, пирогаллол, соединения ряда тиазина и, в частности, фенотиазип
соединения ряда карбазидов (в частности, семикарбазид) и ряд других. Эти со
единения добавляются к винилхлориду в количестве от (1ч-10)-10 4 до 100-10
мае. %, чаще в виде спиртового раствора. Стабилизация происходит посредс
том непрерывного дозирования стабилизатора в поток винилхлорида в конце
технологической линии после его ректификации. Использование таких стабк
лизаторов позволяет сохранить свойства технического винилхлорида в течение
нескольких месяцев.
Стабилизируют винилхлорид также органическими соединениями свинца
олова, германия и эфирами пирофосфорной кислоты.
Методы синтеза винилхлорида [34]
Реакция дегидрохлорирования
Винилхлорид получается отщеплением хлорида водорода от 1,2-дихлор
этана.
Дегидрохлорирование может проводиться в жидкой фазе с помощью водно
го или спиртового раствора щелочи:
СН2С1—СН2С1 + КОН —— СН2=СНС1 + КС1 + Н2О. (1.11
Реакция дегидрохлорирования может проводиться и в паровой фне к pi
300-500 °C в присутствии катализаторов, инициаторов или без них:
СН2С1—СН2С1 — СН2=СНС1 + НС1. (1.-12
Реакция гидрохлорирования
Винилхлорид нолучаки гидрохлорироиаиисм ацетилена как в паровой, гак
и жидкой фазе, в присутствии катализаторов — хлоридов ртути, меди и др.:
HgCI2
СН=СН + НС1-----СН2=СНС1. (1.43)
Реакция хлорирования
Винилхлорид можно получать хлорированием этилена или этана в объеме
или на катализаторе при 450-550 °C:
СН2=СН2 + С12 —— СН2=СНС1 + НС1, (1.44а)
СН3—СН3 + 2С12 —— СН2=СНС1 + ЗНС1. (1.446)
Селективность, особенно при хлорировании этана, невысока.
Реакция оксихлорирования
Винилхлорид можно получать окислительным хлорированием этилена или
этана в паровой фазе при 450-550 °C на катализаторе СпС12/носитель (катали-
затор Дикона):
СН2=СН2 + НС1 + 0,5 О2 — СН2=СНС1 + Н2О, (1.45а)
СН3—СН3 + НС1 + О2 —* СН2=СНС1 + 2Н2О. (1.456)
Реакция дехлорирования
Винилхлорид можно получать из 1,1,2-трихлорэтана взаимодействием
с цинком, железом или алюминием в присутствии водяного пара:
СН2С1—СНС12 + Zn —- СН2=СНС1 + ZnCl2. (1.46)
Реакция диспропорционирования
Винилхлорид может быть получен в результате реакции диспропорциони-
рования ацетилена и 1,2-дихлорэтана:
СН=СН + СН2С1—СН2С1 —2СН2=СНС1. (1.47)
Синтез из этилена и 1,2-дихлорэтана в паровой фазе
над безводным сульфатом кальция
СН2С1—СН2С1 + СН2=СН2 —- СН2=СНС1 + СН3—СН2С1. (1.48)
Глава 2.
Сырьевая база
для производства
винилхлорида
СН2 -- сн<
Основными видами сырья для производства винилхлорида являются хлн
или хлорид водорода, а также углеводороды — ацетилен, этилен и, в меньше
с гепени, этан. Масштабы производства этих видов сырья исчисляются со i ням
। ысяч и миллионами тонн в год.
Масштабы производства винилхлорида (31 млн т в год) предполагают шин
чие адекватного количества хлорного и углеводородного сырья, направляемо!
на его получение. Так, на производство винилхлорида расходуется 15-18% все!
производимого этилена и, по некоторым оценкам, 15-17% ацетилена. Вини;
хлорид является также крупнейшим потребителем хлора.
Рассмотрим кратко свойства сырья и основные методы его произволе i и
чтобы лучше понимать всю технологическую цепочку, приводящую к получ<
нию винилхлорида.
2.1. Хлор
Впервые хлор был получен шведским химиком К. Шееле в 1774 г.
В промышленных масштабах хлор начали получать в 1785 г. окислением с<
ляной кислоты оксидом Мп (IV) (метод Вельдона), позднее, во второй полот
не XIX в. — кислородом в присутствии катализатора (метод Дикона). В кош
XIX в. был разработан способ производства хлора электролизом водных ра
творов поваренной соли, первый патент был выдан в 1879 г. российским изо!
ретателям Н. Глухову и Ф. Ващуку [165].
При электролизе поваренной соли одновременно образуется эквивален гш
количество каустической соды (1,13 т на 1 т хлора).
Основные физические и физико-химические свойства хлора приведены
ряде справочников (например [166, 167]). Ниже представлены лишь некогор!
его характеристики.
Физические свойства
Хлор — зеленовато-желтый газ с характерным резким и раздражающи
запахом; в сжиженном состоянии прозрачная жидкость янтарного цис!
1КИП = -34,05 °C, 1ПЛ = -101,6 °C.
Плотность жидкого хлора
/,"(.......... 3-1,05 0 20
р, кг/м'......1558,9 1483,0 1427,3
Температурный коэффициент объемного расширения:
/,°С........-884-30 -254-+5 10-15 20-25
0, (1000 °C) 1 .... 1,409 1,793 1,978 2,030
Таблица 2.1. Давление паров хлора
t,°C р, кПа t,°c р, МПа t,°c р, МПа
-88 5,00 -30 0,122 20 0,671
-80 8,33 -25 0,152 25 0,773
-70 15,73 -20 0,186 30 0,887
-60 28,00 -15 0,226 40 1,165
-55 36,66 -10 0,266 65 2,027
-50 46,66 -5 0,318 84,8 3,040
-45,0 59,32 0 0,371 101,6 4,053
-40,0 74,65 5 0,431 115,2 5,066
-35,0 93,98 10 0,502 127,1 6,080
-34,05 101,31* 15 0,583
Ратм = 760 мм рт. ст.
Параметры критического состояния:
Давление, МПа........7,71
Температура, °C ...... 144,0
Плотность, г/см3.....0,573
Объем, м3/моль.......0,124
Вязкость динамическая
а) жидкости:
t, °C р, мПа-c... . -53,1 . 0,569 -45 0,530 -35,3 0,494 0 0,385 20 0,345 50 0,300 100 0,249
б) пара: t, °C ..0 20 50 100 150 200 250 300 400
р, мкПа-c.. .12,3 13,3 14,7 16,8 18,9 20,9 22,9 24,9 25,7
I loiicpxnoc i ное натяжение:
.. -72 -50 -10 30 0 10 20 >0 100
<i,m!1/m ....33,0 29,2 27,3 25,4 21,7 20,02 IK,34 13,30 1,90
Коэффициент преломления жидкости nD14 = 1,367 nD2(1 = 1,385.
Коэффициент преломления пара h^20 - 1,0008.
Теплоемкость
а) жидкости:
1,пС................-173 -123 -73 -17,78+37,8
Ср, кДж/(кг-К)......0,5950 0,7197 0,9412 0,9881
6) пара:
1,°С.............. 10-38 38-65,5
Ср, кДж/(кг-К)......0,482 0,511
Теплопроводность
а) жидкости при 30 °C: 0,620 Вт/(м-К)
б) пара:
t, °C -40 -28,9 -17,8 -6,7 -4,4
А. Вт/(м-К).. ... 0,00640 0,00692 0,00727 0,00761 0,00796
1, °C 15,6 26,7 37,8 48,9
А, Вт/(м-К)... ... 0,00848 0,00883 0,00917 0,00969
Таблица 2.2. Теплота испарения
t,°C ^ИСП> кДж/кг f,°C 4исп> кДж/кг t,°C *7111. II» кДж/kl
-100 315,70 0 249,55 100 158,69
-80 303,56 20 234,47 120 126,03
-60 290,16 40 218,56 140 54,01
-40 277,18 60 201,81 144,1 0
-20 263,78 80 182,97
Теплота плавления дпл = 6,81 кДж/моль.
Энтропия для идеального газового состояния:
/, К...........293,15 100 500 600 00 81)0 900 1000
S1’, Дж/(моль-1<).. .222,57 233,33 241,31 247,95 253,62 258,57 262,97 266,92
Растворимость в воде:
1,°С................... 10 20 30 40 50 60 70 80 90
Растворимость, г/100г.... 1,01 0,74 0,58 0,47 0,39 0,33 0,28 0,22 0,13
Дипольный момент жидкости в интервале температур от -65 до 18 °C:
ре = 0,767-10 30 Кл-м (0,23 D)
Диэлектрическая проницаемость жидкости:
1,°С....-65,2 -60 -50 -33,2 -20 10 14 77 142
Е.......2,147 2,150 2,100 2,048 2,030 1,970 1,910 1,730 1,540
Эбуллиоскопическая постоянная равна 1,73.
Пожароопасные и токсические свойства
Хлор не горюч, но поддерживает горение многих органических веществ,
с водородом образует смеси, взрывающиеся на солнечном свету.
Предельно допустимые концентрации в воздухе:
- рабочей зоны производственных помещений — 1 мг/м3;
- населенных мест — 0,10 мг/м3 максимальная разовая, 0,03 мг/м3 средне-
суточная (Россия).
Современные методы получения хлора и каустической соды
В настоящее время в мире используются три основных метода электрохи-
мического получения хлора и каустической соды: ртутный, диафрагменный
и мембранный. Наряду с этими основными методами каустическую соду в
небольших количествах получают химическим методом из кальцинирован-
ной соды обменной реакцией с известковым молоком (известковый метод)
или спеканием кальцинированной соды с оксидом железа с последующим
гидролизом водой промежуточного продукта — феррита натрия (ферритный
метод).
Хлор в небольших количествах получают также в качестве побочного про-
дукта при получении натрия и магния электролизом расплавленных солей,
электролизом абгазной соляной кислоты или химическим окислением хлорида
водорода кислородом воздуха (процесс Кель-Хлор).
Электрохимические методы получения хлора, водорода и гидроксида на-
трия основаны на электролитическом разложении раствора поваренной соли:
2 NaCl + 2 Н2О —► 2 NaOH + С12 + Н2. (2.1)
В целях жопомии электроэнергии процесс ведут при rcMitepuiype >лскгро
лига 85-95 °(
Методы разнятся по реакциям на катоде и в прикагодном пространстве, <
также по способам разделения продуктов электролиза.
Анодный процесс по всем методам одинаков и сводится к выделению хлор,
на аноде по реакции:
2СГ—*С12 + 2е. (2.2
В электролизере с ртутным катодом на катоде происходит разряд ионон па
трия с образованием жидкого сплава — амальгамы натрия:
Na+ + е + п Hg—»-Na(Hg)„. (2.3
Амальгама натрия в отдельном аппарате (разлагателе), взаимодействуя с во
дой, образует водород и гидроксид натрия:
Na (Hg)„ + Н2О — NaOH + 0,5 Н2 + и Hg. (2.4
Регенерированная ртуть из разлагателя возвращается в электролизер. II pt
получении щелочи в разлагатель подают обессоленную воду, что позволяет по
лучать раствор гидроксида натрия высокой степени чистоты.
В диафрагменных и мембранных электролизерах на катоде выделяется во
дород, а в прикатодном пространстве образуется раствор гидроксида натрия:
2Н2О + 2 е—► Н2 + 2ОН . (2.5
Методы разнятся способами разделения продуктов электролиза. В диафр.п
менном методе анодная и катодная камеры разделены фильтрующей диафра!
мой. Для предотвращения переноса ионов гидроксида в анодную камеру мере
диафрагму под действием электрического поля создают проток электролиз
через диафрагму из анодной камеры в катодную. Вследствие этого в катили i
всегда присутствует хлорид натрия, а концентрация гидроксида натрия нс нре
вышает 10-11%.
В мембранном методе анодная и катодная камеры разделяются непроточно!
катионообменной мембраной, проницаемой для катионов натрия и непропиц.
емой для анионов гидроксида и хлора. Современные катионообменные мембра
ны позволяют получать раствор гидроксида натрия с концентрацией 32-35*4
Для получения раствора щелочи такого состава в катодную камеру при элект
релизе добавляют обессоленную воду, что позволяет получать раствор гидрок
сида натрия высокой степени чистоты.
Полученный тем или иным методом влажный хлор охлаждают, сушат серии
кислотой, компримируют и направляют потребителям. При необходимое ги и
газообразного получают жидкий хлор. Электролитический водород охлаждаю
компримируют и используют в основном для синтеза хлорида водорода и соля
ной кислоты. В отдельных случаях, при наличии соответствующих потребим
лей, водород сушат на цеолитах и чистят от примеси кислорода. Водород, нону
чаемый в разлагателях ртутных электролизеров, перед выбросом в атмосфер
или подачей потребителям обязательно чистят от ртути [196].
Подготовка рассола, принцип работы и конструкция электролизеров, дот
дение до товарной формы получаемого при электролизе гидроксида натрия
каждом методе имеют сноп особенности Ниже дастся более подробное описи
ине каждого из методов.
Ртутный метод
I |риинициальное устройство электролизера с ртутным катодом показано на
рис. 2.1.
Катод 1 электролизера представляет стальную прямоугольную плиту ши-
риной 1,5-2 м и длиной 10-15 м, установленную с наклоном 10-25 мм на 1 м
длины, по которому тонким слоем течет ртуть. Стенки электролизера стальные
гуммированные, крышка стальная гуммированная или гибкая из специальных
сортов резины. Для увеличения съема продукции с 1 м2 площади пола в ртут-
ном электролизе, с самого начала стремились работать с высокими плотностя-
ми тока. Современные электролизеры с ртутным катодом работают при плот-
ностях тока 10-13 кА/м2, мембранные — при 3-6 кА/м2, диафрагменные — при
1,5-2,5 кА/м2.
Аноды 2 устанавливаются параллельно потоку ртути на расстоянии 3-5 мм
от ее поверхности. Для уменьшения сопротивления электролита и снижения
перенапряжения при выделении хлора электролиз ведут при 80-85 °C. В старых
конструкциях электролизеров в качестве анодов использовался искусственный
графит, обладавший недостаточной химической стойкостью. С появлением ме-
таллооксидных анодов (титан, активированный оксидами благородных метал-
лов) графитовые аноды были вытеснены как в новых, так и в старых конструк-
циях электролизеров.
Концентрированный рассол (содержащий хлорид натрия 300-310 г/л) по-
дают в верхнюю часть электролизера. Из нижней части электролизера выво-
дят обедненный рассол (содержание NaCl 250-270 г/л) вместе с хлором или
раздельно. Амальгама натрия с его концентрацией 0,2-0,4% через гидроза-
твор самотеком поступает в разлагатель 4. Амальгама в разлагателе течет
Рис. 2.1. Принципиальная схема электролизера с ртутным катодом (обозначения в
тексте)
сверху mini противотоком к дсмнпералн «шанпой воде. 11римспенис демп
нерали.юканпой воды обеспечивает получение каустической соды высокой
степени чистоты, пригодной для использования в производствах вискозы
и синтетических волокон. Для ускорения процесса разложения амальгамы
разлагатель шполняется катализатором — кусочками графита или карбида
тигана размером 8-20 мм. Ртуть из нижней части разлагателя насосом 3 воз
вращается в электролизер.
При взаимодействии амальгамы натрия с водой выделяется значительное
количество тепла, вследствие чего температура в верхней части разлагателя
достигает 120 °C. Для уменьшения уноса паров ртути и воды с водородом па
разлагателе установлен обратный холодильник 5, в котором водород охлажда
ют до 30-40 °C. Выходящий из разлагателя каустик с содержанием едко! о натра
45-50% охлаждают до 50-60 °C, фильтруют от мелкодисперсной ртути и о ткачи
вают на склад готовой продукции.
При нормальной эксплуатации производства выход по току в электроли и
рах с ртутным катодом составляет 95-97%. Остальная часть тока (3-5%) расхо
дуется на побочные реакции: выделение кислорода на аноде, выделение водоро
да и частичное восстановление растворенного в анолите хлора на катоде. В силу
этого электролитический хлор всегда содержит примеси кислорода (1-2 об. %)
и водорода (0,4-0,8 об. %).
Удельный расход электроэнергии постоянного тока на получение 1 т NaOl I
в пересчете на 100% составляет 2820-3320 кВт-ч.
Принципиальная блок-схема производства хлора и каустической соды
представлена на рис. 2.2. В качестве исходного сырья используется твердая тех
ническая или выпаренная соль. Основное требование к качеству исходного сы
рья — отсутствие амальгамных ядов (Cr, V, Mo, Ge). Присутствие соединений
этих металлов в соли и рассоле приводит к увеличению доли тока, расходуемого
на выделение водорода, загрязняющего хлор. Повышенное содержание водоро
да в хлоре не только затрудняет переработку хлоргаза, но в отдельных случаях
делает процесс электролиза взрывоопасным (нижний предел взрываемости но
дорода в хлоре 5,8 об. %).
Применение в производстве больших количеств токсичной ртути требуо
соблюдения специальных мер безопасности для защиты обслуживающего пер
сонала и окружающей среды от воздействия ртути. В Европе, США и Японии
значительные усилия были направлены на совершенствование методов очис
тки сточных вод и газовых выбросов от ртути, а также на совершенствование
технологии электролиза. Это позволило снизить выбросы ртути в атмосферу и
водоемы до пренебрежимо малого уровня в сравнении с такими процессами,
как вулканическая деятельность и сжигание топлива. Расход ртути на upon i
водство 1 т хлора снижен с 200-300 (в начале 1970-х годов) до 1-3 г в настоящее
время. Тем не менее, в Японии в середине 1970-х годов был принят закон, защи-
щающий применение ртутного метода производства хлора и каустической соды
в промышленности, и к середине 1986 г последняя установка, работавшая по
этому методу, была закрыта. В настоящее время расходы на природоохранные
мероприятия в ртутном методе производства хлора и каустической соды дос i и
гают 15-20% от общих эксплуатационных расходов.
Твердая соль
Водород Каустик (50%) Хлор
Рис. 2.2. Принципиальная блок-схема производства хлора и каустической соды по ртут-
ному методу
Диафрагменный метод
г хеми wick। роли icpj с iiepiикалыюй фильтрующей диафрагмой предс гаи
лена па рис. 2.3.
Катод >ле|<1роли юра представляет стальную коробку / без крышки и дна, и
которой размещен металлический каркас сложной формы, обтянутый метал
лической сеткой 2. На катодную сетку нанесена асбестовая диафрагма 3, раз
деляющая электролизер на анодную камеру (с внешней стороны диафрагмы) и
катодную камеру (с внутренней стороны диафрагмы, прилегающей к катодной
сетке).
В нижней части катода имеется устройство 4 для отвода из электролизера
элек грощелоков, в верхней части — штуцер для вывода водорода.
Между противолежащими сторонами катодных ячеек размещаются ано
ды 5, закрепляемые на днище 6, служащем для подвода тока к анодам. Корпус
катода электрически изолирован от анодного днища резиновой прокладкои 7.
На корпус катода устанавливается крышка 8 со штуцерами для ввода рассола и
отвода хлора [168].
Выход по току в современных конструкциях диафрагменных электрон и
зеров составляет 94-96%. Часть тока, как и в электролизерах с ртутным каю
дом, расходуется на выделение кислорода на аноде и на восстановление хлора
па катоде (растворенный в анолите хлор вместе с потоком рассола проникает в
катодную камеру через диафрагму). Основные потери тока связаны с перепо
сом гидроксид-ионов в анодную камеру под действием электрического поля, где
они, взаимодействуя с растворенным в анолите хлором, образуют гипохлори г и
хлорат натрия:
2 NaOH + С12 —— NaClO + Н2О + NaCl, (2.6а)
6 NaOH + 3 С12 —- NaClO3 + 5 NaCl + 3 Н2О. (2.6б)
Рис. 2.3. Принципиальная схема электролизера с вертикальной фильтрующей дна
фрагмой
Вода Соль (рассол)
Водород Каустическая сода Хлор
(50%)
Рис. 2.4. Принципиальная блок-схема производства хлора и каустической соды по диа-
фрагменному методу
Если । ипохлори I натрия, попадая н катодную камеру, практически нсетда
полностью восстанавливается па катоде, го хлорат натрия, как более прочное
соединение, па катоде по восстанавливается и загрязняет элекгрощслока. При
нормальной работе электролизера содержание хлората натрия в щелоках со
ставляег 0,2-0,3 г/л.
В отличие от ртутных и мембранных электролизеров, диафра! меппые >лск
тролизеры не могут работать при переменных нагрузках. Как правило, пониже
ние силы тока снижает концентрацию гидроксида натрия в щелоках, по после
дующее повышение нагрузки приводит к скачкообразному возрастанию кон
центрации. Одновременно возрастает и содержание хлората натрия (с 0,2 0,3
до 0,7-1,5 г/л и даже выше).
Напряжение на электролизере и, соответственно, удельный расход элскт
роэнергии на единицу продукции существенно зависят от плотности тока, по
этому рабочую нагрузку на электролизере обычно определяют технико-эконо
мическим расчетом с учетом стоимости электроэнергии в регионе и требуемых
капиталовложений в создание производства заданной мощности
Принципиальная блок-схема производства хлора и каустической соды по
диафрагменному методу представлена на рис. 2.4.
Рассол, полученный методом подземного выщелачивания соли или рас то
рением привозной технической соли, подвергается содово-каустической очис
тке от солей кальция, магния и железа. Ионы кальция осаждают раствором
кальцинированной соды, ионы магния и железа — гидроксидом натрия, содер
жащемся в так называемом обратном рассоле, который осветляют в отстойнике
гравитационного типа, фильтруют и подают на электролиз. Содержание приме
сей ионов кальция и магния в очищенном рассоле составляет не более 5 mi/л,
содержание взвесей — не более 1 мг/л.
Полученные в электролизерах электрощелока, содержащие 125-145 г/л i ид
роксида натрия и 180-210 г/л неразложившегося хлорида натрия, направляю!
на концентрирование для получения товарного продукта. Концентрирование
щелоков осуществляют в многокорпусных выпарных установках с трех-, четы
рехкратным использованием пара. Существуют схемы с прямоточным и проги
воточным движением пара и раствора.
Полученный товарный раствор каустической соды содержит 46-50% i ид
роксида натрия, 1,5-3,5% хлорида натрия, до 0,8% карбоната натрия, до 0,3%
хлората натрия и не более 0,007% железа (содержание примесей дано в иерее че
те на 100%-й продукт).
Мембранный метод
Принципиальное устройство мембранного электролизера показано n.i
рис. 2.5.
В мембранном электролизере анодная 1 и катодная 2 камеры разделены
непроточной катионообменной мембраной 3. В катодной камере параллельно
мембране размещается катод 4, в анодной — анод 5. При электролизе, как и в
электролизерах с диафрагмой, на аноде выделяется хлор, на катоде — водород,
в прикатодном слое образуется раствор щелочи. Под действием электричес
кого поля ионы натрия свободно проходят через катионообменную мембрану
Водород
Хлор
Рис. 2.5. Принципиальное устройство мембранного электролизера
из анодной камеры в катодную, тогда как миграция гидроксид-ионов из ка-
тодной камеры в анодную практически отсутствует, так как катионообменная
мембрана непроницаема для анионов.
В анодную камеру непрерывно поступает концентрированный и выводится
обедненный хлоридом натрия рассол (анолит). Концентрация хлорида натрия
в ходе электролиза снижается с 300-310 до 210-220 г/л. В катодную камеру не-
прерывно подают деминерализованную воду из расчета получения раствора
щелочи с содержанием гидроксида натрия 32-35%, что отвечает оптимальным
условиям работы современных мембран. При электролизе в катодной камере
образуется щелочь высокой чистоты, близкая по качеству ртутной. Так как
мембраны не обладают идеальной селективностью, из анодной камеры в като
лит вследствие концентрационной диффузии проникает незначительное ко-
личество хлорида натрия (до 0,02%), сульфата и хлората натрия (до 0,001%).
В свою очередь, вследствие концентрационной диффузии и миграции под
действием электрического поля 4-6% гидроксид-ионов проникает в анодную
камеру и, взаимодействуя с хлором, образует хлорат натрия, накапливаю-
щийся в рассольно-анолитном цикле. В связи с этим схемы очистки рассола
для мембранного электролиза дополнительно оборудуются узлом химичес-
кого разрушения хлората натрия.
Kaiпонооомсипые мемi>p.i11i.i и и<>i.in/iпн.ин11 и ii.11>< шин <|>11i|»ik»iiиMi p
пых смол с ан тинными сульфо и карОом ильными i руинами о(нц< и <|><'р
мулы R-SO3H и R-COO1I. Современные мембраны мноиь. лпнны, содержа)
смолы как с сульфо , гак и с карбоксильными группами. Опп пьноко ,\п
мичсски стойки и высокоселективны: первые мембраны с активными суль
фогруппами имели выход по току около 85% при концентрации щелочи
15-20% и служили около года; современные мембраны обеспечиваю! выход
но току 95-97% при концентрации щелочи 32-35% и служат не менее четы
рех лет, что весьма существенно, так как стоимость мембран значительна
(-800 долл./м2) [169].
Высокая стоимость мембран диктует необходимость эксплуатировать
элек тролизеры при максимально возможной плотности тока (3-6 кА/м ). Гем
не менее, за счет применения активированных анодов и катодов с низким не
ренапряжением для выделения хлора и водорода, использования высокоэф
фективных мембран с малым электросопротивлением, минимально возмож
кого межэлектродного расстояния, напряжение на электролизерах составил
ег 3,05-3,10 В, а расход электроэнергии на 1 т гидроксида натрия — порядка
2100 кВт-ч, что ниже, чем в диафрагменных электролизерах при существенно
меньших плотностях тока.
Благодаря использованию высоких плотностей тока мембранные электро
лизеры компактнее диафрагменных и требуют для своего размещения мепь
ших производственных площадей. Принципиальная блок схема мембранного
метода представлена на рис. 2.6.
Выходящий из электролизеров анолит с содержанием хлорида натрия
210-220 г/л подвергают сначала вакуумному, а затем полному химическому
обесхлориванию. Во избежание накопления хлората натрия в рассольно-апо
литном цикле перед вакуумным обесхлориванием хлорат натрия разрушают
соляной кислотой. Обесхлорснный анолит донасыщают твердой поваренной
солью (или упаривают), очищают от солей кальция, магния и железа и подави
на узел доочистки от ионов кальция и магния с помощью ионообменных смол.
Далее очищенный рассол вновь подают на электролиз.
Часть обесхлоренного анолита очищают от сульфатов одним из трех мето
дов: физико-химическим, химическим (осаждением хлоридом бария), нано
фильтрацией (через ультрамикропористую перегородку).
Ионообменная очистка рассола — важнейшая стадия подготовки расы»
ла: именно она определяет срок службы мембран. Наряду с ионами кальция,
магния, суммарное содержание которых должно быть не более 0,02 мг/л, в
очищенном рассоле жестко лимитируется содержание стронция, алюминия,
тяжелых металлов (бария, марганца, хрома, никеля), кремнезема, йода. Ка
чество рассола контролируют современными физико-химическими методами
(атомно-абсорбционной, эмиссионной спектроскопией).
Полученный при электролизе 32-35%-ный раствор гидроксида натрия
концентрируют до 48-50% в выпарной установке. Используются аппараты с
принудительной циркуляцией выпариваемого раствора. Материал инвара
тов — никель. Расход пара на концентрирование мембранных щелоков сос i ан
ляет ориентировочно 1,26-1,46 ГДж/т.
Хлор Водород Каустическая сода (50%)
Рис. 2.6. Принципиальная блок-схема мембранного метода производства хлора и кау-
стической соды
Мощности и объемы производства хлора в России и и миро
Ниже приведены данные по производству хлора, n>i<. I, в промышленно
развитых странах, включая Россию и СССР с 1950 по 2005 г.:
1<>ды ..1950 г. 1960 г. 1970 г. 1980 г. 1990 г. 2005 г.
США ... .1890 4210 8860 10 360 13 000 13 800
1срмания .... 220 660 1725 2 990 3 500 -
Франция 82 330 1030 1 240 1 420 -
Англия ....310 560 950 940 1 020 -
Италия 45 292 840 870 1 070 -
Китай - - - - 10 700
Япония 17 620 2 150 2 600 - -
Вос точная Европа ... - - - - - 1 800
( ССР .... 202 506 1 414 2 380 2 884 -
РСФСР, РФ .... 183 424 1 165 1 814 1 958 1 100
В мире всего ... 3 200 8 400 20 000 26 500 37 500 49 900
Крупнейшим производителем хлора и каустической соды являются США,
па долю которых приходится около 26% общего производства хлора. По уровню
мощностей в 2005 2006 гг. к США стремительно приближается Китай. В табл. 2.3
представлены данные о мощностях по производству хлора по регионам и их
доля в мире в 2005 г.
В последнее десятилетие прирост объемов производства хлора и каусгичсс
кой соды в экономически развитых странах (США, Западной Европы, Японии)
практически прекратился. Годовые объемы производства хлора колеблются
около средней величины, следуя за ростом или падением спроса на продукцию
хлорорганического синтеза. Рост объемов производства хлора в мире в основ
пом связан со строительством новых хлорных установок в странах третьего
мира (Китае, Индии, странах Арабского Востока, Африки, Южной Америки,
Океании). Тем не менее, прирост производственных мощностей в США состав
ляет около 2,5% в год [277].
Таблица 2.3. Соотношение мощностей по производству хлора в мире (за 2005 г.)
Регион Мощность, млн т Доля, %
Северная Америка 13,9 25,7
Западная Европа 12,3 22,8
Восточная Европа 2,9 5,4
Китай 12,7 23,5
Япония 4,3 8,0
I (си тральная и Южная Америка 2,1 3,9
Прочие 5,8 10,7
Ikero 54,0 100
Ко >ффициеп г иик)Л1> iob.iiihh ирон шодс шейных мощное гей но хлору в
мире составляет 75-90% и в сильной степени подвержен влиянию конъюн-
ктуры рынка. Гак, в Западной Европе в последнее десятилетие он составлял
85-90%, а в США дос i тпал 95-98%, что было связано с выводом из эксплуата-
ции значительной части мощностей с изношенным оборудованием в преды
дущие годы.
В Западной Европе и США наблюдается тенденция к укрупнению произ-
воле гвенных мощностей. Ниже приведены показатели по США за 1970-1995 гг„
характеризующие эту тенденцию:
Год ..1970 1980 1990 1995
Средняя мощность завода по хлору, т/сут . ...404 489 577 792
Число производителей хлора ... 38 34 26 25
Число заводов, имеющих хлорные установки .., ... 70 64 52 45
Соотношения диафрагменного (Д), ртутного (Р) и мембранного (М) мето-
дов производства хлора в ведущих странах мира, СССР и РФ, в % от общего
объема производства хлора:
Год . .1970 1980 1990
Метод д Р д р д Р М
США 66 28 72 24 78 16 3
Германия 7,6 89 19 78 32 65 0,2
Франция 30 70 35 65 43 57 0,2
Италия 3 92 10 90 10 83 7
СССР 71 29 66,5 33,5 71 29 Нет
РСФСР, РФ 73 27 65 35 65 35 Нет
Год . . 1995 2005
Метод Д Р М Д Р М
США 76 12 9,5 65 5 30
Западная Европа - - 20 45 35
Япония - - 2 - 98
Китай - - 50 - 50
Российская Федераци я . . 64 36 Нет 68 20 12
На рис. 2.7 представлены данные за 2005 г. и прогнозируемые на 2010 г. по
производству хлора в мире разными методами.
2005 г.
Рис. 2.7. Производство хлора в мире ртутным (Р), диафрагменным (Д) и мембрапиыг
(М) методами
2010 г.
2.2. Хлороводород
Хлороводород впервые был получен в 1772 г. английским химики,
Дж. Пристли. Синтез хлороводорода прямым взаимодействием хлора с вод<
родом был впервые осуществлен в 1809 г. французским химиком Л.-Ж. Ген.
ром.
Хлороводород (молекулярная масса 36,461) при нормальных условия
(20 °C и 0,1 МПа) — бесцветный газ с резким запахом, который при атмосфе|
ном давлении и -85,1 °C конденсируется в бесцветную, легкоподвижную жи;
кость. Жидкий НС1 при -114,22 °C переходит в твердое состояние. Твердый IК
существует в двух модификациях: ромбической, стойкой при температуре пил
-174,75 °C, и кубической.
Основные свойства хлороводорода [151]
Плотность газа:
при нормальных условиях, г/см3 .................................. 1,6391
по отношению к воздуху..........................................1,2679
I Указатель преломления:
жидкого НС!...................................................1,256
газообразного НС! при нормальных условиях.... ..................1,0004456
Критические константы:
t,°C...........................................................51,4
р, МПа..........................................................8,258
р, г/см3........................................................0,42
Гемпер.н yp.i,"( :
тройной точки...................................................-I 1-1,24
кипени» при 0,1 МПа .. .......................................-85,1
плавления при 0,1 МПа.........................................-114,22
перехода от ромбической к кубической структуре................-174,74
Теплота, 10 Дж/кмоль:
плавления.....................................................1,993
парообразования...............................................16,19
сублимации при температуре тройной точки......................1,908
образования газа при 25 °C....................................-9,217
Энергия образования газа при 25 °C, 10' Дж/кмоль................9,537
Диэлектрическая постоянная при 25 °C и 0,1 МПа..................1,0046
Коэффициент теплового расширения жидкости при Тпл, °C”1.........0,00278
Таблица 2.4. Плотность хлорида водорода (числитель — жидкости, знаменатель —
пара)
t,°c р, г/см 3 t,°C р, г/см3
-112,4 1,265/- -20,0 0,997/10 3-32,0
-101,5 1,2347/10 3-0,83 0 0,924/10 3-54,0
-90,0 1,204/10"3-1,67 7,5 0,873/-
-85,1 1,187/10 3-2,5 15,85 0,838/-
-80,0 1,178/10”33,2 20,0 0,831/10 3 97,0
-60,0 1,122/10“38,3 40,0 0,697/0,18
-40,0 1,063/10 3-17,0 51,4 0,424/0,424
-30,0 1,031/10 3-23,0
Таблица 2.5. Плотности двух форм НС1
t,°C р, г/см3 t,°C р, г/см3
Ромбическая форма Кубическая форма
-273 1,540 -178,15 1,550
-195,15 1,507 -174,15 1,490
-192,15 1,503 -166,15 1,469
В г.16л. 2.1 и 2.5 представлены .шачепия n/ioniocin 1вердою и жидкою 1И
и его насыщенного пара (151 -153).
Значения мольной теплоты испарения твердого HCI (154):
Г, °C.......... 139,15 -126,0 -120,45 -119,2 117,15
Д1„ 1()7 Дж/кг. 1,970 1,939 1,927 1,917 1,923
Поверхностное натяжение на границе НС1 (ж) - НС1 (п):
t,°C...........-110,0 -101,4 -92,9
р, Н/мм........27,37 26,25 24,72
Удельная теплоемкость хлороводорода при 10 °C и постоянном давлении с<
ставляет 811,82 Дж/(кг.К). Отношение cvlcy = 1,41. В интервале от 0 до 1700 "
удельную теплоемкость хлороводорода можно определить по формуле:
Ср-а + ЬТ, (23
где а = 0,1805; b - 2,687-10 5; Т — температура, К.
Растворимость НС1 в хлорэтанах в интервале температур (-20)4-(+б0) °C и пр
парциальных давлениях НС1 (0,27-^-1)-10 ’ Па находятся в пределах (04-0,08) мо/п
моль растворителя и уменьшается в ряду: дихлорэтан > трихлорэтан > тетр;
хлорэтан > трихлорэтилен [155]. Теплота растворения хлороводорода снижай-
ся в ряду: дихлорэтан > трихлорэтан > трихлорэтилен > тетрахлорэтан.
Более подробная информация о физических и физико-химических слог
ствах хлороводорода содержится в монографии [151].
Существует ряд промышленных методов получения хлороводорода. ( г;
рейшим из них является сульфатный, предложенный И.А. Лоури в 1934 i О
применяется в тех случаях, когда есть потребность в сульфате натрия (или к;
лия) и нет запасов природного сульфата.
Сульфатный метод основан на взаимодействии хлорида натрия с koi щеп
рированной серной кислотой:
2NaCl + H2SO4 —Na2SO4 + 2НС1 - 40,25 кДж. (2.1
Ввиду эндотсрмичности реакции для ее протекания требуется подвод тепл
Взаимодействие хлорида натрия с серной кислотой начинается при 0 °C с выд
пением в газовую фазу почти безводного хлороводорода, но быстро прекращу
ется; при нагревании реакция возобновляется.
В зависимости от температуры реакционной массы образующийся суд,
фат натрия может полностью оставаться в жидкой фазе или частично крис i .и
лизоваться без нее в виде кислых солей: 2 Na2SO4 • 9 H2SO4; Na2SO4 • 2 J1 >S( 1
Na2SO4- H2SO4; 3Na2SO4 • H2SO4.
Во избежание преждевременного затвердевания реакционной массы нео<
ходимо, чтобы температура была достаточно высокой: при применении Na<
500-550 °C, при применении KCI 700-750 °C.
Процесс можно проводить в механических или вращающихся печах |15<
или в реакторах кипящего слоя [157]. Концентрация хлороводорода в реакции!
ной зоне колеблется от 10 до 65% в зависимости от условий ведения процесса
В основе прямого синтеза хлороводорода лс.ын реакция взаимодействия
хлор । с водородом:
П2 + С12^^2НС1 + 184,7 кДж.
(2.9)
Диссоциация НС1 на элементы становится заметной при > 1500 °C. Констан-
ia равновесия реакции диссоциации
Кр = р2на/Рн, Ра2 (2.10)
рассчитывается по уравнению [152]:
IgXp = (9554/Т) — 0,5331gT + 2,42. (2.10а)
Теплота реакции синтеза, кДж, с повышением температуры несколько уве-
личивается:
д,= 172,5 + 0,00461. (2.11)
При адиабатическом сгорании стехиометрической смеси хлора и водорода
при 0 °C теоретическая температура факела пламени равна 2500 °C [158]. Прак-
тически вследствие некоторой диссоциации НС1 температура пламени сни-
жается до 2200-2400 °C [152]. Избыток одного из компонентов газовой смеси
(обычно водорода) несколько понижает температуру горения.
При обычной температуре в отсутствие световых лучей реакция образова-
ния НС1 из элементов идет очень медленно. При нагревании смеси хлора и во-
дорода или под действием яркого света происходит взрыв вследствие цепной
реакции:
кг
С12 + hv Cl + О , (2.12а)
М
СГ + Н2 — НС1 + Н’, (2.126)
Н* + С12 — НС1 + СГ, (2.12в)
СГ + Н2 — НС1 + Н’ (2.12г)
и т.д.
В присутствии кислорода реакция хлора с водородом замедляется [151].
Зависимость скорости фотохимической реакции от концентрации компо-
нентов может быть выражена эмпирической формулой:
4на] [а2]2-[н2]
л ’ [о2}{[н2]+о,1[аJ ’
где к — константа скорости реакции, зависящая от интенсивности возбуждаю-
щего светового луча или теплового импульса.
Определяющей стадией неинициированно!о процесса является рсакци
( Г 1 II.—► IK I ( II’ 11511. Если обозначить константу скорости «гои pi
акции A.,, <i константу диссоциации молекул хлора к2, го констан та скорое и
образования I К Л будет равна = 0,5 к2- к{ и может бы гь вычислена по фор
муле:
к = 118 exp[-35722/(RT)]. (2.1 I
Скорость реакции зависит также от присутствия катализаторов — в/iai н
твердых пористых тел (древесного угля, губчатой платины) и некоторых ми
неральных веществ (кварца, глины и др.).
Абсолютно сухие хлор и водород не взаимодействуют между собой. При
сутствие следов влаги ускоряет реакцию столь интенсивно, что она може
произойти со взрывом. Повышение содержания влаги сверх 5-10 5% не отра
жается на скорости реакции [151].
В производственных условиях осуществляется спокойное, невзрынное го
рение водорода в токе хлора, которое обеспечивается равномерным пост ул
лением хлора и водорода и смешением их только в факеле пламени горелки
Газы сгорают при выходе из горелки, не образуя взрывоопасной смеси в печи
Водород подается в избытке (5-10%), что позволяет полностью использона i
хлор и получать не загрязненную хлором соляную кислоту. Помимо этот
избыток водорода постоянно обеспечивает в печи восстановительную среду
что способствует снижению коррозионного разрушения как стальных, так i
графитовых печей.
Принципиальная схема получения хлороводорода представлена и
рис. 2.8.
Камеры сжигания могут быть выполнены из стали, графита, кварца, от
неупорного кирпича. Каждый из материалов имеет свои достоинства и нс
достатки. Например, стальные печи могут работать под давлением, что уне
личивает концентрацию HCI в газе благодаря устранению подсосов воздуха
При этом, однако, хлорид водорода содержит большое количество желе ы
что ухудшает качество соляной кислоты.
Наибольшее применение в качестве конструкционного материала иолу
чил графит, импрегнированный феноло-формальдегидными смолами (ко
робоном, игуритом). Такое сочетание придает необходимую коррозионную
стойкость и предотвращает загрязнение НС1.
Для синтеза хлороводорода обычно используются печи с восходящим и/п
нисходящим потоком горения. Достоинством аппаратов является тог фак1
что в едином агрегате объединяются как камера сжигания, так и абсорбци
онное устройство. Горелки состоят из двух графитовых трубок: наружной i
внутренней; при этом расположение отверстий в трубках может в.трьиро
ваться с целью обеспечения наиболее эффективного смешения. Мощное i
современных графитовых печей достигает 65 т/сут [151].
Инертные газы
Рис. 2.8. Принципиальная технологическая схема получения хлороводорода из хлора и водорода:
1 — реактор сжигания водорода; 2, 4, 6, 10, 12 — холодильники; 3 — абсорбционная колонна; 5 — водяной скруббер; 7 — сборник 31-33%-
ной соляной кислоты; 8, 14 — циркуляционные насосы; 9 — колонна отпарки хлороводорода; 11 — кипятильник; 13 — сборник азеотропной
соляной кислоты
Характеристики печей синтеза HCI:
Производительность*, г/сут 5-18 15- 20 10 .0 50 6 ч
Диаметр, мм
внутренний 300 450 700 800
наружный .. .400 560 840 910
Высота, мм 3200 5000 8000 9000
Площадь внутреннего сечения, м2 ... . 0,0707 0,159 0,3848 0,5027
Охлаждаемая площадь (внутр.), м2 3,01 7,07 17,6 22,6
Объем, м3 0,226 0,795 3,08 4,52
Производительность на единицу:
охлаждающей площади, т/(сут-м2). ... 2,66 2,83 2,84 2,88
объема, т/(сут-м3) 35,4 25,2 17,2 14,1
* В расчете на 35%-ю соляную кислоту.
Одним из распространенных методов получения хлороводорода являет и
его десорбция из соляной кислоты при ее кипячении в насадочных колоннах
При обычном нагревании смеси НС1 и Н2О эти компоненты невозможно pa i
делить полностью из-за образования азеотропа, содержащего 20,22% НС1 [159|
Увеличение или уменьшение давления изменяет состав смеси, но этого недос r.i
точно для реализации рентабельных процессов разделения под разным давле-
нием хлорида водорода и воды.
Низкая температура кипения хлороводорода при нормальном давлен hi
(-85 °C) делает нецелесообразным процесс обычной ректификации для полу
чения сухого хлороводорода. Однако поскольку давление насыщенных наро
воды над соляной кислотой очень мало, нетрудно получить хлороводород с Со
держанием влаги менее 0,01% при -10 °C и давлении 0,1 МПа.
Процесс десорбции можно осуществить в колоннах отгонного типа с после-
дующим охлаждением паров хлороводорода и воды последовательно в водя ног
и рассольном холодильниках и с возвратом конденсата в колонну. Технолш и
ческая схема процесса представлена на рис. 2.9.
Содержание хлороводорода в газе не менее 99%, влаги не более 0,01 мае. ‘
давление 60-140 кПа.
Для получения хлороводорода из разбавленной соляной кислоты и кисло! i
азеотропной концентрации сочетают ректификацию с использованием подоен
нимающих средств [160,161]. Наиболее приемлемыми являются способы, осне
ванные на применении в качестве водоотнимающих средств концентрирован
ных растворов MgCl2 [162] и СаС12 [163].
Влияние концентрации СаС12 на относительную летучесть НС1 и Н2О нок.
зано на рис. 2.10а, на состав азеотропа — на рис. 2.106. Установлено, что полно
разрушение азеотропа соляной кислоты происходит при достижении концеп
рации СаС12 равной 6,6 мол. % или 30,4 мае. %.
Принципиальная технологическая схема процесса концентрирования сол>
ной кислоты представлена на рис. 2.11.
Соляную (6%-ю) кислоту направляют в стальную футерованную кер.
мическими плитками колонну 1 для концентрирования до содержания ’()'
На эжектор
[СаС12], мол.%
Рис. 2.10. Влияние концентрации СаС12: а — на относительную летучесть смеси 111
и Н2О; б — на состав азеотропа (Ха)
НС1. Эффективность колонны — шесть теоретических тарелок. Из кондеи
сатора колонны отводят дистиллат при pH = 6-=-7, а из кипятильника колон
ны — 20%-ю соляную кислоту. Сюда же поступает 50%-й раствор СаС12. Смс
ситель также защищен слоем полиэбонита и керамических кислотоупорны
плиток.
Смеситель 4 имеет графитовый подогреватель блочного типа для поддс
ржания температуры питающей смеси 107 °C. Пары, образующиеся при смете
IK I (1.11)
Рис. 2.11. Принципиальная технологическая схема процесса концентрирования соля-
ной кислоты:
1, 5,11 — ректификационные колонны; 2, 6, 12 — кипятильники колонны; 3, 8, 10, 13 — сборники;
4 — смеситель; 7 — холодильник; 9 — вакуум-выпарной аппарат
нии горячих растворов, отводят в укрепляющую секцию ректификационной
колонны 5, а смесь подают в верхнюю часть исчерпывающей секции этой же
ректификационной колонны. В кипятильнике 6 колонны 5 поддерживают тем-
пературу 120 °C. Выводимый из холодильника 7 НС1 (газ) имеет температуру
не выше 20 °C и концентрацию не ниже 99,5% НС1. Кубовый остаток, содержа-
щий 0,2% НС1, поступает в приемник 8, а оттуда — в стальной гуммирован-
ный вакуум-выпарной аппарат 9. Упаренный до 50% раствор СаС12 направляют
через сборник 10 в смеситель 4. Выходящую из кипятильника 12 колонны 11
соляную кислоту через сборники 13 и 3 подают на приготовление смеси НС1 с
СаС12 в смеситель 4.
Выход НС1 с концентрацией 99,5% составляет 98%.
Известен также способ получения НС1 [164] ректификацией растворов со-
ляной кислоты в присутствии хлорида магния с концентрацией 46-55%. Соот-
ношение массовых долей растворов 2-ь5:1. Температура процесса 140-180 °C.
При более низких температурах выделяется твердая фаза, а выше 180 °C разла-
гается MgCl2i также с выделением твердой фазы.
По мере развития методов очистки хлороводорода и его вторичного исполь-
зования вышеприведенные классические способы получения НС1 теряют свое
значение.
В п.к нппцес время просматривается общая тенденция создания сбалапи
ровапных по хлору производств, г.е. полное использование хлороводорода, о<
разующсгося в промышленных процессах.
Абгазный хлороводород образуется в значительных количествах при ос)
ществлении следующих процессов:
1. Хлорирование ароматических и алифатических соединений. В этих ре.н
циях, идущих как по ионному, так и по радикальному механизму, образовали
основного продукта всегда сопровождается выделением хлороводорода:
RH + С12 —— RC1 + НС1. (2.1е
Образующиеся продукты имеют весьма широкую область применения.. )т
растворители, дезинфектанты, мономеры для синтеза высокомолекулярных с<
единений, инсектициды, пластификаторы, охлаждающие вещества и др.
2. Дегидрохлорирование, например, получение винилхлорида из дихло|
этана, трихлорэтилена из тетрахлорэтана:
RH—R^l —R=R, + НС1. (2.1 (
3. Конденсация по Фриделю-Крафтсу:
RH + RjCl—— R—Rj + HC1. (2.17
Например, производство алкилбензолов путем конденсации хлоркеросип
с бензолом.
4. Высокотемпературный гидролиз хлоридов металлов:
MgCl2 + Н2О —- MgO + НС1, (2.1«.
2FeCl3 + ЗН2О —* Fe2O3 + 6НС1. (2.18(
В абгазном хлороводороде содержатся примеси исходного сырья, полущи
дуктов, побочных и целевых продуктов. Разнообразие состава абгазов, равны
требования к чистоте НС1 не позволяют разработать универсальный метод и
очистки от всех видов примесей. Анализ этих методов выходит за рамки паск
ящей работы. Отметим лишь, что подробная информация об очистке НС1 о
примесей приведена в [151].
Хлороводород образуется также при утилизации хлорорганических огх<
дов, как сжиганием, так и химической переработкой (гидрированием, окислс
нием, исчерпывающим хлорированием). Подробно эти процессы рассмотрен!
в главе 9.
Абгазный хлороводород либо перерабатывается в целевые продукты ме i <
дами гидрохлорирования или оксихлорирования, либо используется для пол)
чения соляной кислоты.
2.3. Ацетилен
Ацетилен СНЕ=СН был открыт в 1836 г. английским химиком Э. Дэви при
исследовании состава светильного газа. Целенаправленный синтез ацетилена
был впервые осуществлен германским химиком Ф. Велером из карбида кальция
в 1862 г. В ряду непредельных алифатических углеводородов общей формулы
С„Н2и_2 ацетилен — первый представитель, его молекулярный вес 26,04, при
обычных условиях — бесцветный газ со следующими физическими характерис-
тиками [170,171]: 1КИП= -83,8 °C, Гпл = -81 °C, 1ВОЗГ = -84,1 °C.
Тройная точка ацетилена, соответствующая устойчивому равновесию трех
фаз, характеризуется температурой -80,6 °C и давлением 128,3 кПа.
Плотность жидкого ацетилена при Ркри и 0 °C равна 0,451 г/см3.
Плотность относительная жидкости р4“83 = 0,6208.
Таблица 2.6. Давление паров
t,°C р, кПа t,°C р, МПа
-150 0,06 -80 0,138
-140 0,28 -60 0,353
-130 1,12 -40 0,773
-120 3,75 -20 1,510
-ПО 10,77 0 2,665
-100 27,33 20 4,388
-90 62,52 30 5,492
Параметры критического состояния:
давление — 6,22 МПа; температура — 35,5 °C; плотность— 231 кг/м3;
динамическая вязкость пара р = 9,35 мПа-c при 0 °C и р = 10,2 мПа-c при
20 °C.
Поверхностное натяжение:
t,°C..........-80 -70 -60 -50
о, мН/м....... 18,92 16,99 15,06 13,13
Теплоемкость с;, - 1,687 кДж/(кг-К).
Теплота испарения:
t,°C...........-80 -50 -10 0 10 20 30
q исп, кДж/кг ... 654,42 570,81 430,92 392,33 348,92 289,42 191,34
Теплота сгорания qCT = 1253,84 кДж/моль.
Теплота образования стандартная ДН298 = 226,910 кДж/моль.
Энтропия вещества в стандартном состоянии: 52д8 = 200,963 Дж/(моль-К).
)нiропия вещее। на при разных темперагурах:
Г. к.............. 100 500 700 900 1100 1 WO I >00
5п,Дж/(мольк).... 201,23 226,48 245,74 261,44 274,81 286,53 313,71
Дипольный момент жидкости в интервале от 80 до 25 °C равен пулю
Диэлектрическая проницаемость е = 1,00134 при 0 °C.
Рас творимость при 20 °C: в 1 объеме воды растворяется 1 объем ацетилен.
Пожароопасные и токсические свойства
Ацетилен — горючий, взрывоопасный газ.
Температура, °C:
самовоспламенения......................................335
горения................................................2322
Область воспламенения в воздухе, об. %...................2,5-80,1
Минимальное взрывоопасное содержание кислорода
при разбавлении ацетилено-воздушных смесей, об. %:
азотом.................................................6,5
диоксидом углерода.....................................9,0
Максимальное давление взрыва, МПа........................1,0
Ацетилен — слабый наркотик. ПДК в воздухе рабочей зоны производстве!
пых помещений 0,3 мг/м3.
Более подробно физические, горючие и взрывные свойства ацетилена р.ь
смотрены В.Н. Антоновым и А.С. Лапидусом [171], см. также [172].
Методы производства ацетилена
Старейшим методом получения ацетилена является так называемый «кщ
бидный» способ, заключающийся в воздействии воды на карбид кальция.
СаС2 + 2Н2О — С2Н2 + Са(ОН)2, ДН = -127,1 кДж/моль (2.1S
Теоретический выход сухого ацетилена из химически чистого карбида каш
ция при 0 °C и 760 мм рт. ст. составляет 372,3 л/кг. Фактический выход ацетиле
на из технического карбида кальция вследствие наличия в последнем при меге
и разложения его влагой воздуха колеблется от 230 до 310 л/кг [171].
Ниже приведен выход сухого ацетилена (С2Н2) при 20 °C и 760 мм рт. ст. г
технического карбида с разным содержанием СаС2:
СаС2,%..........80 75 70 65 60 55
С2Н2,л/кг....... 298 279 260 242 223 204
Процесс образования ацетилена часто затрудняется из-за появления корк
на кусках карбида кальция, возникающей при контакте их с гашеной швее i ы<
в которую превращается СаС2 при действии воды. Это может приводить к *н
иливанию и перегреву ацетиленовых генераторов. Для нормального протек,
ния реакции корку извести необходимо удалять.
Ацетиленовые генераторы классифицирую! по нрои!нодтелы1осги, пре
дельному давлению вырабатываемого ацетилена и главное — по способу разло
женил карбида кальция водой.
Производительность генераторов но ацетилену может быть or 0,8 до
2000 м /ч и более. В химической промышленности применяют генераторы про-
изводительностью 500-2000 м3/ч.
По способу разложения карбида кальция генераторы подразделяют на три
основные системы:
1. «Карбид в воду» (аппараты непрерывного действия). Куски карбида сбра-
сывают в воду, находящуюся в газообразователе, и там происходит разложение.
Выделяющееся тепло расходуется в основном на нагревание воды, образующий-
ся при реакции гидроксид кальция удаляется вместе с водой в виде суспензии.
2. «Вода на карбид». Воду периодически (или непрерывно) подают на кус-
ки карбида, загруженные в газообразователь. Процесс разложения регулируют,
изменяя количество подаваемой воды. В этих аппаратах можно вести «мокрый»
или «сухой» процесс газообразования. В генераторах «мокрого» типа воду по-
дают на неподвижный карбид кальция, который полностью разлагается в при-
сутствии значительного избытка воды. В «сухих» генераторах вода поступает на
непрерывно движущиеся куски карбида кальция, который находится в избытке
по отношению к количеству подаваемой воды. Отходом является порошкооб
разный гидроксид кальция (известь-пушонка).
3. Контактные генераторы. Карбид кальция периодически взаимодействует
с залитой в аппарат водой по мере расходования ацетилена. Такие генераторы
бывают только малой производительности.
Наиболее широко в химической промышленности применяют генераторы
типа «карбид в воду».
В ацетилене-сырце содержится значительное количество примесей, таких
как РН3, NH3, SiH4, AsH3, О2, N2, Н2, СО2 и других с концентрацией в пределах
0,002-0,5%, что определяет необходимость его очистки. Обрабатывают ацети-
лен обычно в следующем порядке:
- промывка водой для удаления аммиака;
- обработка раствором гипохлорита натрия для окисления фосфина, серо-
водорода и мышьяковистого водорода;
- промывка щелочным раствором для нейтрализации следов кислот;
- осушка серной кислотой или силикагелем.
В промышленных условиях достаточно распространен метод получения
ацетилена пиролизом углеводородов.
Пиролиз заключается в разложении и превращениях исходных углеводо-
родов при t > 1000 °C в адиабатических условиях за 0,5-20 мс. При Т > 1400 К
свободная энергия образования ацетилена ниже по сравнению, например, с эта-
ном или пропаном. Это свидетельствует о возможности образования ацетиле-
на непосредственно из этих углеводородов в условиях высокой температуры.
Степень равновесного превращения метана в ацетилен резко возрастает (от 0
до 99,66%) в интервале от 500 до 1800 К; повышение давления снижает степень
превращения; этот эффект, однако, заметен лишь при Т < 1600 К [173].
При 1200-1500 °C скорость разложения ацетилена значительно меньше ско-
рости его образования, что является основным кинетическим фактором, позво-
ляницим получать ацетилен. Соотношение i м»|чн ien <>(>рл «тапни н ра i/ны ।
пин ацетилена можно регулировать изменением общею давлении 11анрнмс|
при снижении давления с 20 до 4,7 кПа соотношение скорое н и потр.ц i.iri
Ipasa 1171].
Процесс пиролиза метана до ацетилена можно представил> как совоку ।
ность реакций разложения метана, этана, этилена и ацетилена 1174]:
|.2СН4^=^С2Н6+Н2, к} - 4,5-10I3exp[-380380/(RT)]> (2.21
2. С2Н6 С2Н4 + Н2, к2 = 91013exp]-288 000/(RT)], (2.2
3. С2Н4^^С2Н2+Н2, к3 = 2,57-108exp[-167000/(RT)|, (2.2.
4. С2Н22С + Н2, Jc4 = l,7-106exp[-12 500/(RT)]. (2.2.
Максимальное превращение метана в ацетилен происходит при Г > 2000 I
гак как в этих условиях отношение констант скорости реакций разложения м<
। ана и ацетилена к}: к4 > 10.
Химический состав сырья является одним из факторов, влияющих на ныхе
ацетилена; наилучшие результаты получаются при пиролизе парафинов по|
мального строения. При этом каждому виду сырья соответствуют определи
ные оптимальные параметры процесса.
Известны следующие технологические схемы пиролиза в зависимости <
способа подачи тепла в зону реакции.
Регенеративный пиролиз. Процесс идет на стационарной или движунцчк
огнеупорной насадке, периодически или постоянно нагреваемой топливным i
зом до 1800-2000 °C. После эндотермических реакций пиролиза насадка охл.о
дается до 800-900 °C, затем ее тепло используется для подогрева углеводородо
воздуха и топливного газа. При движущейся твердой насадке стадии процее
остаются такими же, но насадка последовательно перемещается через все зон
нагрева и охлаждается в реакторе.
Пиролиз в трубчатых печах. Подогретый углеводород пропускают чер
грубы, обогреваемые снаружи продуктами сгорания топливного газа.
Гомогенный пиролиз в потоке газообразного теплоносителя. В этом случ
углеводороды вдувают в струю горячих продуктов сгорания топливного ia.i
углеводороды и теплоноситель смешиваются непосредственно в зоне реакцит
Пиролиз с погружным горением. В таких процессах осуществляется нет»
редственный контакт жидких углеводородов с продуктами сгорания, образу!
щимися в погружной горелке.
Наибольшее распространение получили способы пиролиза в потоке i а <
теплоносителя, что связано прежде всего с гибкостью (возможностью варкир
вания мощностей по ацетилену и этилену), а также с удачным конструктивна
решением узлов сжигания топливного газа и смешения продуктов сгорания
парообразными углеводородами.
Распространенный промышленный метод получения ацетилена — эле
грокрекинг углеводородов. При электрическом разряде в среде газообра мн
углеводородов происходит местный разогрев газа и расщепление ею молек
на радикалы, рекомбинация которых приводит к образованию ацетилена, с
гомологов, сажи, водорода и некоторого количества этилена.
kl
Рис. 2.12. Возможные пути образования ацетилена из метана
Ацетилен и другие продукты реакции могут получаться из метана по схеме,
приведенной на рис. 2.12.
Предполагается, что образование ацетилена идет одновременно по четырем
направлениям: непосредственно из метана (константа скорости fc5); через этан,
минуя этилен (к2 и к6), через этилен, получающийся непосредственно из метана
(fci и к7), через этан и этилен (/с2, к3 и к7).
Основная часть ацетилена получается непосредственно из метана [175] и
лишь не более 10% образуется по четвертому маршруту — через этан и этилен.
При установлении оптимального режима (1500-1600 °C, продолжительность
реакции 0,5 1 мс) можно добиться максимального выхода ацетилена и подавле-
ния побочных реакций.
При технологическом оформлении процесса электрокрекинга наибольшее
распространение получили трубчатые аппараты с линейным или коаксиальным
расположением электродов. В них поток газа непрерывно проходит через зону
высоких температур. Газообразный углеводород, проходящий через дугу, при ли-
нейном ее расположении нагревается неравномерно. В центральной части дуги в
зоне нижнего электрода газ имеет наиболее высокую температуру, при которой в
основном протекают процессы деструкции и образуется сажа. Там же происходит
термический крекинг углеводорода с образованием ацетилена. По мере удаления
от горячей зоны температура углеводорода снижается; у стенки электрода темпе-
ратура газа самая низкая, и он охлаждает электрод, нагретый до 700 °C электри-
ческой дугой. В описанных реакторах дуга перемещается с большой скоростью.
При использовании для электродугового процесса природного газа соста-
ва, об. %: 92,3 метан, 1,4 этан, 0,5 пропан, 0,4 бутан, 5,4 азот крекинг-газ имеет
следующий состав, об. % [171]: ацетилен — 14,5, метилацетилен — 0,4, диацети-
лен — 0,6, винилацетилен — 0,1, диметилацетилен — 0,01, метилвинилацети-
лен — 0,04, этилен — 0,9, пропилен — 0,02, бутилен — 0,02, метан — 16,3, этан —
0,03, пропан — 0,02, бензол — 0,3, толуол — 0,02, водород — 63,46, монооксид
углерода — 0,6, азот — 2,7.
В зависимости от состава исходного сырья содержание ацетилена в крекинг-
газе может варьироваться в пределах 8-18 об. %. Крекинг-газ затем подверга-
ется специальной обработке, имеющей целью концентрирование ацетилена и
очистку от гомологов.
1 l.iiiOoaec расnpociраненным промышленным методом получения ацетиле
на m углеводородного сырья является окисли тельный нироли.1. В ном процессе
тепло, необходимое для проведения .>ндо термической реакции образования аце
пилена, получается в результате сжигания части сырья в кислороде.
Тепло, выделяющееся при неполном окислении метана, например, по реакции
СН.,+ О2^:СО + Н2+Н2О, АН = -277,5 кДж/моль, (2.21а)
используется для образования ацетилена:
2СН4 С2Н2 + ЗН2, АН ~ +380 кДж/моль. (2.216)
Кроме того, значительное количество тепла выделяется при полном окисле
нии метана:
СН4 + 2О2 СО2 + 2Н2О, ДН - -801,4 кДж/моль. (2.2-1 и)
Всего в суммарном процессе окислительного пиролиза участвует примерно
90% метана и 99% кислорода; при этом до 33% исходного метана преврантаекя
в ацетилен по реакции (2.24в) [176, 177].
Суммарное уравнение процесса окислительного пиролиза метана при on i и
мадьном времени образования ацетилена [176, 178]:
45 СН4 + 30,25 О2 —— 3,5 СО2 + 8С2Н2 + 25,5 СО + 28 Н2О + 54 Н2. (2.25)
Значения коэффициентов в этом эмпирическом уравнении зависят от ус
ловий подогрева, соотношения О2:СН4, состава пиролизуемого сырья и кисло
рода, степени конверсии метана, потерь тепла, степени сажеобразования и др.
Направленность процесса неполного окисления метана в зависимости от О2/
СН4 показана на рис. 2.13. Видно, что оптимальное количество ацетилена об
Рис. 2.13. Области окисления метана и крекинга метана до ацетилена в зависимое i и оi
состава метано-кислородной смеси:
А метан не реагирует; Б — окисление метана; В — крекинг метана до цетилена
p.i «yen я при O,/( J 11 = 0,55.0,65. Дальней шее увеличение coo i ношения способ
cinyei развитию реакций окисления метана.
При промышленном получении ацетилена методом неполного окисления
метана необходимо осуществлять три взаимосвязанных процесса: получение
гомогенной метано-кислородной смеси, неполное горение этой смеси с образо-
ванием аце тилена и закалку реакционных газов.
Основным критерием выбора реактора является его производительность.
Мощность современных реакторов превышает 10 тыс. т ацетилена в год. При
этом в зависимости от производительности реактора процесс можно осущест-
влять при атмосферном и повышенном давлении.
На рис. 2.14 представлена технологическая схема производства ацетилена
при атмосферном давлении [179].
Природный газ из сети при избыточном давлении 250 кПа поступает в ради-
ационно-конвективный подогреватель 3. Вначале газ проходит конвективную
зону, где нагревается до 350-400 °C, затем фильтр 4 для очистки от механичес-
ких примесей и поступает в радиационную зону, после которой температура
его повышается до 650 °C за счет тепла дымовых газов, получаемых в топочной
камере подогревателя при сжигании природного газа. Дымовые газы в радиа-
ционной зоне охлаждаются с 1200 до 800 °C, а в конвективной — с 800 до 300 °C.
Для нормальной работы в подогревателе поддерживается постоянное количес-
тво сжигаемого газа с коррекцией по температуре. В топке подогревателя ус-
тановлена постоянно горящая дежурная горелка (с сигнализацией о погасании
пламени) для поджигания исходной газовой смеси, выходящей из рабочей го-
релки. Кислород, сжатый турбогазодувкой 1 до избыточного давления 150 кПа,
в подогревателе 2 нагревается до 650 °C. По конструкции этот подогреватель
аналогичен подогревателю 3.
Нагретые газы в смесителе 5 образуют метано-кислородную смесь, которая
поступает в кольцевую горелку реактора 6 и на выходе из горелки загорается за
счет аутостабилизации. Для стабилизации горения в реактор отдельно подают
дополнительное количество кислорода (5 6% от основного потока).
При нормальной работе реактора газы пиролиза имеют следующий со-
став (% об. в пересчете на сухой газ): ацетилен 7,5-8,2; метан 4,0-6,0; высшие
ацетиленовые и ароматические углеводороды — 0,34-0,4; водород 54,7ч-56,0;
кислород до 0,2, монооксид углерода 26,0-27,0; диоксид углерода 3,0-4,0;
азот 1,6.
Кроме того, в газе содержатся пирогенетическая влага, смолы, нафталин, ан-
трацен (до 3-5 мг/м3 в сумме) и сажа (3-5 г/м3).
На выходе из реакционной зоны газы с температурой -1500 °C резко охлаж-
даются (до 100 °C) большим количеством воды; на «закалку» подается горячая
(50 °C) и холодная (25-27 °C) вода При этом около 45% сажи вымывается водой
и уходит с ней через гидравлический затвор 7. Частично очищенный газ на-
правляется на окончательное охлаждение и доочистку от сажи, смол и аромати-
ческих углеводородов.
Очищенные от сажи (до нескольких миллиграммов на 1 м3) газы пиролиза
при температуре около 60 °C поступают на окончательное охлаждение в пенный
аппарат 12. Там газ и вода проходят противотоком, в результате чего температу-
ра газа понижается до 30-35 °C, а вода нагревается до 50 °C.
I
к
5
5
§
s
s
3“
£
S
QJ
X
О
Q-KD
S Q
s »s
s x
Охла кдеппые и пенном аппарате 12 газы пиролиза перед с гадиеп компри
мирования и выделения ацетилена очищают от аромаiических углеводородов
и па<|)1алипа гем же селективным раствори гелем (N мегилпирролидопом или
диметилформамидом), который используется на стадии выделения и концент-
рирования ацетилена. Для этой цели применяют тарельчатые или барботажные
аппараты 13, в которых снизу вверх проходит газ пиролиза, а противотоком те-
чет абсорбент. Система форабсорбции выполнена с замкнутым циклом абсор-
бента, циркулирующим от насоса 14. Количество абсорбента выбирают с таким
расчетом, чтобы концентрация влаги, уловленной из газов пиролиза, не превы-
шала К) мае. %. Кратность орошения 20 м3 на 1000 м3газа.
За счет тепла, выделяющегося при растворении воды, температура абсор-
бента и, соответственно, давление его паров повышаются, поэтому абсорбент
охлаждают в холодильнике 15. Для улавливания брызг жидкого растворителя, а
также его паров несколько верхних тарелок в аппарате 13 орошаются водяным
конденсатом. Полученная на тарелках смесь воды и абсорбента смешивается со
всей массой абсорбента. Очищенные и охлажденные газы направляются в газ-
гольдер и далее — на выделение ацетилена.
Окислительному пиролизу может также подвергаться нефтяное сырье [171].
Принципиальная схема установки представлена на рис. 2.15.
В верхней части реактора 4, имеющего водяную рубашку, расположено со-
пло 2, через которое в реактор впрыскивается смешанная с кислородом нефть
(для лучшего распыления нефти необходимо оснащать реактор несколькими
соплами). Смесь поджигается дежурной горелкой 3, работающей на топливном
газе и кислороде. Газы после «закалки» поступают в циклон 5, где от них отделя-
ются капли нефти, и затем — на переработку. Жидкие углеводороды, собранные
в циклоне и нижней части реактора, насосом 6 вновь подаются в процесс.
Рис. 2.15. Принципиальная схема установки окислительного пиролиза сырой нефти:
1 — теплообменник; 2 — сопло; 3 — дежурная горелка; 4 — реактор; 5 — циклон; 6 — насос
Около 20% образующейся сажи ножращаегея с жидкими yi леводородамн
и реактор, а 80% — уносится с газами пиролиза. Допускается содержание са ки
н рециркулируемых продуктах 100 г на I кг, излишнее количество выводи ня
из системы. 11ачальная концентрация сажи в газе пиролиза 10 15 г на I м i аза
или примерно 65 г на 1 кг смеси ацетилена и этилена. Для отделения сажи и i
промывают нефтяными фракциями.
Для производства 20 т суммы ацетилена и этилена в сутки достаточно ре
акционного объема 1 м3, что свидетельствует о допустимых высоких тепловых
напряжениях в зоне реакции. Соотношение ацетилена и этилена в получаемом
газе может изменяться от 2:1 до 1:2. Характерный состав газов, об. %: 6,7 С>1П,
6,4 С2Н4, 4,4 СН4, 0,035 С2Н(„ 0,06 С3Н8, 1,2 С3Н6, 0,95 С4Н8, 6,97 гомологи аце-
тилена, 0,1 С6Н6, 28,1 Н2,0,8 О2, 42,27 СО, 1,5 N2.
Расходные показатели при использовании сырой нефти составляют (в рас
чете на 1 кг С2Н2) 6,7 кг нефти и 3,9 кг кислорода. Примерно 45% введенной
нефти сжигается для достижения необходимой температуры процесса, ос
тальное количество подвергается пиролизу. Расход нефти на «закалку» реак
ционных газов примерно вдвое больше, чем на стадиях горения и пиролиза.
Для выделения ацетилена из реакционных газов обычно используют ме-
тод абсорбции органическими растворителями — диметилформамидом, Ме-
танолом, N-метилпирролидоном. В качестве абсорбента может быть также
использован жидкий аммиак.
2.4. Этилен
Этилен (С2Н4, структурная формула — СН2=СН2) — бесцветный горю
чий газ со сладковатым запахом. Этилен в свободном виде в природе не встре-
чается, его получают в больших масштабах путем переработки органического
сырья. Он является основным сырьевым материалом для получения широко! о
ассортимента продуктов органического и нефтехимического синтеза, прежде-
всего полиэтилена и поливинихлорида, а также полистирола, полиэфиров, ок
сида этилена и др.
Впервые этилен был получен голландским химиком И. Дейманом в 1795 г.
отщеплением воды от этанола концентрированной серной кислотой. Сущее
твуют также указания на то, что этилен был получен Дж.Пристли в 1778 i
пропусканием паров этанола через горячую глиняную курительную трубку
первая известная гетерогенно-каталитическая реакция [15].
Основные физические свойства [180-182,194]
Гкип = -103,70 °C, tnn = -169,15 °C.
Плотность жидкости:
1, °C.......-150 -140
р, кг/м3....631,0 618,2
-130 -120 -ПО -100
604,5 591,0 577,2 563,0
Давление паров:
Г,"С ... 160 150 140 1 30 120 110 100 ВО 50
/>, к 11 а.. . ... 0,525 2,01 6,12 15,60 34,59 68,81 125 340 1060
/,°с ....-40 -20 -10 0
р. к! 1а... . 1451 2531 3242 4084
I (араметры критического состояния:
Давление Температура... 5,12 МПа 9,9 °C.
1 Ьюгность .... 220 кг/м3.
Вязкость:
f, °C .-160 -150 -140 -130 -120 -110 -100
р, мПа-с .0,508 0,387 0,307 0,251 0,211 0,182 0,160
v, мм7с .. .0,788 0,614 0,497 0,416 0,358 0,316 0,280
Поверхностное натяжение:
t,°C........-120 -110 -100
о, мН/м.....19,50 17,65 15,71
Коэффициент преломления при -103,8 °C hd - 1,36.
Теплоемкость Ср = 1,554 кДж/(кг-К).
Теплопроводность жидкости:
t, °C X, ВтДм-К).. ..-160 .. 0,2562 -140 0,2348 -120 0,2120 -100 0,1893
Теплопроводность газа:
t, °C -73,3 -28,9 -17,8 -6,7 4,4
X, Вт/(м-К).. .. 0,01125 0,01488 0,01592 0,01696 0,01817
t, °C .... 15,6 26,7 37,8 68,19
X, Вт/(м К).. .. 0,01938 0,02059 0,02181 0,02855
Теплота испарения:
Г, °C -160 -120 -100 -50 -20 -10 0
Чисп. кДж/кг .. 541,85 501,54 476,17 385,12 289,58 241,82 162,70
Теплота сгорания qcr = 1330,68 кДж/моль.
Теплота образования стандартная ДН^ = 52,321 кДж/моль (12,496 ккал/
моль).
Ьироння вещества и стандартном состоянии: S2yti = 219,61 Дж/(моль К)
Энтропия вещества:
/, К..............300 400
S'", Дж/(моль-К).... 219,86 234,01
500 800 1000 1300 1500
246,95 281,70 301,72 327,93 343,36
Дипольный момент жидкости в интервале от -80 до 25 °C равен нулю.
Диэлектрическая проницаемость при 110 °C — е = 1,00144.
Растворимость в воде незначительная: при 20 °C растворяется 0,015%.
Пожароопасные и токсические свойства
Этилен — горючий газ, горит в воздухе слабокоптящим пламенем
Температура самовоспламенения минимальная, °C............490
Область воспламенения, об. %:
в воздухе...............................................3-32
в кислороде.............................................2,9-79,9
Минимальное содержание кислорода при разбавлении
этилено воздушных смесей, об. %:
азотом..................................................10
диоксидом углерода......................................12,1
Устойчив до 350 °C, при более высокой температуре разлагается на метан и
ацетилен.
Методы производства этилена
Получение этилена из этилового спирта
Старейший способ получения этилена — дегидратация этанола:
СН3СН2ОН —- С2Н4 + Н2О (2.26)
Процесс может быть осуществлен в жидкой или газовой фазе в npncyi
ствии кислотных катализаторов (серной или фосфорной кислоты, аромати
ческих сульфокислот, кислых солей, например, сульфатов и фосфатов, хло
ридов цинка, алюминия и др.). Катализаторами дегидратации могут служи и
также оксиды алюминия и тория, фосфорный и фталевый ангидриды. При
использовании в качестве катализатора фосфорной кислоты оптимальна
температура 210-220 °C, а серной кислоты — 160-180 °C. Газофазную дегид
ратацию этанола проводят в присутствии оксида алюминия при 250 350 ”(
Этилен, получаемый в этом процессе, содержит в качестве примесей водород
метан и бутадиен.
Достаточно чистый этилен можно получить, проводя процесс на нем ie, про
питанной пирофосфорной кислотой при 280-300 °C. Выход этилена в процесс*
составляет ~90% при фактическом отсутствии примесей [183, 184].
Принципиальная схема процесса получения этилена дегидратацией этанол*
в реакторе с неподвижным слоем катализатора представлена на рис. 2.16.
Реактор Водная Щелочная
\ промывка промывка
.lianon насосом нодасия в испари гель, обогреваем1.1П паром высокого дан
лепил, и кием в реактор, заполненный активированным оксидом алюминия
Выход этилена за проход составляет 94% при конверсии этанола 99%. ПоОоч
ним продуктом является в основном диэтиловый эфир. 11овы1пение темпериту
ры выше оптимальной ведет к образованию побочных альдегидов. Кагализаю|
подвергается периодической регенерации воздухом и водным паром для удалс
ния коксовых отложений.
Реакционный газ подвергается закалке и водной промывке для удадени)
непрореагировавшего этанола, а также следов альдегидов и кисло г. После ло
го он направляется на отмывку каустической содой для связывания диоксид,
углерода.
После этого газ компримируется, охлаждается и направляется на узел ад
сорбции «тяжелых» примесей, таких, как бутан и бутадиен, с помощью актин»
рованного угля. Содержание этилена в газе после очистки превышает 99 мол.1
[185].
В случае больших (>60 тыс. т/год) мощностей предпочтительно исполню
вать реактор псевдоожиженного слоя. Процесс более технологичен, npoci в уп
равлении. Выход этилена достигает 99% [186].
Получение этилена из этанола может иметь промышленное значение в с i p.i
нах с большими запасами растительного сырья: в Бразилии, Индии, в некою
рых странах Африки. В странах с развитой нефтехимической промышленное
тью используют обратную реакцию: гидратацию этилена до этанола.
Производство этилена из продуктов переработки нефти
Практически весь этилен, производимый в мире, получают из нефтянок
сырья. В зависимости от того или иного региона и связанных с этим экопомп
ческих показателей в качестве сырья могут быть использованы этан, пропап
тяжелые парафины, а также фракции перегонки нефти: прямогонный бен шн
нафта, керосин, газойль. Получение этилена непосредственно из нефти не н >
шло мирового промышленного применения [187].
Пиролиз углеводородного сырья с целью получения этилена как целевоп
продукта проводят при 800-900 °C и времени контакта 0,2-0,5 с. Конструкции
печей и условия процесса могут существенно меняться в зависимости от госта
ва исходного сырья. Обычно процесс осуществляют в трубчатых печах.
Пиролиз углеводородов протекает по свободно-радикальному механизм)
впервые предложенному Ф.О. Райсом и К.Ф. Герцфелдом в 1934 г. [188].
Общая схема процесса для парафинов:
инициирование
С^Нэи+2 H2H1+1 + С(п_т)Н2(и-т)+1 (при п > 2, ш — 0), (2.27
продолжение цепи
^п^2н+2 Cfn H2,n+i ► CnH2n+] + Cm Н2,м+2 (при Н > 2, ГП > 0), (.t.2.8,1
СпН2п+1 Ст Н2,и + С (и-т)Н2 (и-т)+1 (при П > 2, Ш > 0), (2.28ti
оорыв цени
C8ll?„+i + i CnH2fl + Сш 112и(+2 (при н :(),ш О), (2.29а)
С„112|1+1 + С,„Н2ш+, —— С„Н2и+2 + С„, Н2„, (при п > 0, ли > 0), (2.29б)
С„Н2п+, + СшН>„,+ 1 —— С(и+ли)Н2(и+„,)+1 (при и > О, т > 0). (2.29в)
Согласно С.Б. Здонику [189], свободно-радикальный механизм описывает
первичный распад насыщенных углеводородов с образованием олефинов, мета
па и водорода, а также с достаточной точностью предсказывает распределение
углеводородов в продуктах процесса. Теория, однако, не объясняет образование
моно- и полициклических ароматических углеводородов.
Наибольший выход этилена имеет место при пиролизе этана. При промыш-
ленном осуществлении процесса выход этилена достигает 85-90% от теорети-
чески возможного. Согласно Х.К. Шатту [190] выход этилена, моль/моль про-
конвертировавшего этана, может быть описан формулой:
УС2н4 =1,67(кр/р)[(1-а2)/а2]{[1 + 1,2(р Кр)а2/1-а2]°'5-1}, (2.30)
где р — парциальное давление углеводорода, Па; Кр — константа равновесия
реакции дегидрирования этана; а — степень превращения этана.
Таблица 2.7. Выход продуктов при пиролизе нефтяных фракций, мае. % (данные по
промышленным установкам) [185]
Фракции Нафта Керосин Газойль Вакуумный газойль
Средняя Тяжелая Тяжелые
н2 0,63 0,8 0,63 0,65 0,47
сн4 11,7 15,3 11,4 10,6 7,9
С2Н2 0,3 0,75 0,37 0,35 0,2
С2Н4 24,0 29,8 23,2 24,0 19,5
С2Н6 3,9 3,75 3,4 3,25 2,83
С3Н4 0,76 1,1 0,85 1,1 0,38
С3Н6 15,75 14,1 13,8 14,45 11,3
СзН8 0,41 0,25 0,45 0,45 0,36
С4Н6 4,6 4,85 4,4 4,7 5,5
С4н8 6,85 4,2 4,0 4,5 4,88
с4н10 1,0 0,25 0,1 0,1 0,09
zc5 5,0 2,3 3,0 3,3 5,0
С6Н6 3,15 6,3 7,2 5,8 4,16
с7н8 4,4 4,9 4,0 3,15 2,5
с8н8 2,65 2,0 2,2 1,2 1,28
с8н10 0,52 0,7 0,75 0,7 0,5
Топливное масло 1,8 3,8 13,5 17,6 29,15
Скорость реакции дегидрирования папа подчппясия уравнению першио
порядка 1189, 191].
Пиролиз пропапа осложнен шачи тельным количеством побочных реакции,
включая образование кокса и тяжелых углеводородов. Ио агой причине выход
пилена снижается примерно в 1,5 раза но сравнению с пиролизом папа [ 192[.
Порядок реакции меняется от 1 до 1,5, энергия активации 205-280 кДж/моль,
нредэкспоненциальный множитель 10 -10 с '.
При пиролизе нафты и газойля (табл. 2.7) необходимо учитывать наличие
в их составе значительных количеств нафтеновых и ароматических углеводо
родов. Согласно Е.Дж. Грину [193], парафины, линейные или разветвленные,
характеризующиеся высоким соотношением С:Н, при пиролизе дают высокий
выход легких газообразных продуктов, в частности, этилена. Наибольший вы
ход тяжелых продуктов имеет место при пиролизе ароматических углеводоро
дов. Циклопарафины занимают промежуточное положение.
Зависимость выхода этилена от содержания водорода в исходном сырье
представлена на рис. 2.17.
О - промышленное производство
А - пилотная установка
Рис. 2.17. Зависимость выхода этилена от содержания водорода в исходном сырье
Вы код продуктов при пироли и* индивидуальных углеводородов, мае. %
(данные по промышленным установкам) [185]:
Углеводороды. Конверсия,% . Эган .50 60 Пропан 75 90 н Бу ган 95 Изо- бутан 92 н-Пенган 95 Изо- пентан 95
1>2 .3,06 3,55 1,08 1,29 1,0 1,39 0,8 0,9
СИ. . 2,6 4,2 18,83 24,7 21,8 23,6 17,52 20,6
с2н2 .0,12 0,25 0,17 0,33 0,4 0,46 0,55 0,42
с,н, 41,65 48,2 25,0 34,5 35,8 10,3 36,5 17,95
с2н6 .50,0 40,0 5,15 4,4 5,1 0,73 5,6 3,2
CtH4 . 0,1 0,02 0,22 0,34 0,55 2,46 0,95 1,7
с,н(, .0,89 1,11 18,86 13,96 16,4 21,2 19,8 19,0
с3н8 .0,14 0,17 25,0 10,0 0,15 0,33 0,55 0,55
С4н(, . 0,5 1,07 1,05 2,65 3,4 1,94 3,7 4,6
С|Н8 .0,25 0,21 1,28 1,0 1,7 16,7 2,6 14,38
С4НЮ .0,35 0,27 0,07 0,05 5,0 8,0 0,1 0,1
хс5 .... . 0,2 0,27 1,43 1,81 1,65 2,12 6,1 7,5
С6н6 . 0,2 0,48 1,09 2,2 2,58 3,42 1,8 4,1
С?Н8 .0,03 0,06 0,33 0,48 1,12 1,51 0,6 1,25
с8н8 0,45 0,67 0,1 0,25
С8Н|0 0,06 0,15
Топливное масло 0,3 0,85 1,7 1,99 1,09 1,5
На начало 2006 г. объем мирового производства этилена составлял
115 млн т/год, в том числе в России 2,1 млн т/год, в США 25 млн т/год. Среднего-
довой рост объемов производства составляет 4-6% и, оценочно, к 2010 г. объем
производства этилена достигнет 127 млн т/год.
В настоящее время в структуре производства этилена 64% приходится на
крупнотоннажные установки пиролиза, 17% — на малотоннажные установки
газового пиролиза, 11% составляет пиролиз бензина, 8% — пиролиз этана [183].
Соотношение методов может существенно меняться в зависимости от региона.
Так, в США из этана производят до 40% этилена. По разным оценкам использо-
вание этана для получения этилена экономически целесообразно при содержа-
нии этана в природном газе не менее 5%.
В Западной Европе этилен практически полностью получают из бензина.
В России доля этана в качестве исходного сырья составляет около 15%, до 25%
этилена получают пиролизом пропан-бутановой фракции и около 60% пироли-
зом бензина.
До 50% lipoil 1ВОДИМО! о >111/1(11.1 lit по/ll. I\< till ДЛИ IIOIIV 'll llllil I II >'l 11 • I 11 It 11.1
низкой и высокой luioiuoiiii I l.i ирон 1к<|д< iно впппл vi<>pii/ii и да i- г noun
винилхлорида поправляется U> 18% выпускаемо)о пилена
i(ринципиальпая технологическая схема получения пилена ппролп к>М
бензина приведена на рис. 2.18.
В трубчатую печь / подают нары бензина и воды для частичного копвер
тирования кокса и снижения парциального давления сырья. Печь обогреваем
ся топливом, подаваемым в специальные горелки. Испарение бензина и воды
осуществляется в теплообменниках с использованием тепла топочных газов.
Продукты пиролиза выходят из печи с температурой 800-850 °C и поступают в
закалочный аппарат 2, где за счет испарения водяного конденсата охлаждаю гея
до 650-700 °C. Тепло горячих продуктов снимается в котле-утилизаторе 3 с по
лучением водяного пара (10-12 МПа) и охлаждением их до 350-400 °C. Пиро
газ охлаждается далее в колонне 4 циркулирующим захоложенным «средним»
маслом, отбираемым в виде жидкости с одной из нижних тарелок колонны /.
Часть «среднего» масла выводится из колонны на переработку или используе i
ся в виде топлива. Из куба колонны 4 выводятся смолы и кокс, а газ пиролиза
вместе с парами воды с температурой около 110 °C из верха колонны 4 нос
тупает в водяной скруббер 5, где охлаждается циркулирующей захолаживае
мой водой. В скруббере из пирогаза конденсируется «легкое» масло, которое
отделяется от воды во флорентине 6. Вода возвращается частично на орошение
скруббера 5, частично на закалку пирогаза, частично на пиролиз после очист ки
от смолистых.
«Легкое» масло выводится на переработку для выделения ароматических
соединений (бензол, толуол, ксилолы). Пирогаз, охлажденный до 30-40 °C и
насыщенный парами воды, поступает в пятиступенчатый компрессор 7. Пос-
ле каждой ступени компримирования газ охлаждается, а выделившийся при
этом конденсат отделяют от газового потока. Конденсат с первых трех ступеней
конденсации поступает в отпарную колонну 8, где отпариваются растворимые
в нем газы и возвращаются на первую ступень компрессора, а пирокондепса!
выводится на переработку. Пирогаз после третьей ступени проходит щелочной
скруббер 9 для улавливания кислых примесей и компримируется на двух пог
ледующих ступенях до давления 3,5-4 МПа. Конденсат четвертой-пятой ступе
ней конденсации отпаривается на колонне 10. Отпаренный газ возвращается па
четвертую ступень, а кубовая жидкость, представляющая в основном углено
дороды С4 и С5, передается на ректификационную колонну 22 — депропаниза
тор. Пирогаз после компрессора 7 проходит осушитель 11, наполненный окси
дом алюминия или цеолитами и захолаживается в системе холодильников 12,
охлаждаемых за счет испарения жидкого пропилена. Охлажденный пирогаз
поступает в систему низкотемпературной ректификации. Перед поступлением
на колонну 13 — деметанизатор пирогаз дополнительно охлаждается в тепло
обменниках за счет выходящих с последующих ступеней этановой фракции и
метано-водородной фракции.
В колонне 13, работающей при самом низком холоде (-100°С) происходи!
выделение метана и водорода в виде дистиллата. Охлаждение дефлегматора /1
згой колонны происходит за счет испарения жидкого этилена последующих
ступеней, и частичного дросселирования метано-водородной фракции .пой
Рис. 2.18. Принципиальная технологическая схема получения этилена пиролизом бензина (1-24 см. текст)
колонны. KyOOB.HI ЖИДКОСТЬ КОЛОННЫ IJ, СОсКЫЩЛЛ К О< II<1I<H<1M II I yi/к подоро
доп ( 2 (. । носгунает на колонну 15 — деэгапп iarop, где происходи i выделенке
фракции С; при темпера । у ре верха около 60 "С и давлении 2,5 МПа. Дефис! мл
юр 16 >гой колонны охлаждается за счет испарения пропилена с последующие
г туиспей ректификации.
Выходящая из колонны 15 фракция С2 содержит примеси ацетилена и дл>
очистки от него подвергается гидрированию в контактном аппарате 20. Спя i in
।сила гидрирования происходит в теплообменниках 17 и 18, а разогрев га ioi
перед гидрированием в паровом подогревателе 19. Очищенный газ iiocryiiaci
и колонну 21 —этиленовую, где происходит выделение этилена при давленш
2 2,3 Ml 1а и температуре верха минус 30-35 °C, что обеспечивается охлаждени
гм дефлегматора 16 за счет испарения жидкого пропилена. С верха колонны от
поди гея водород с примесью метана и этилена и возвращается на компрессор 7
а чис । ый этилен отбирается с верхних тарелок.
II । куба этой колонны выводится жидкий этан, который возвращаю! обыч
по па пиролиз.
Кубовую жидкость колонны 15 направляют в колонну 22 — депропапи ia
гор, куда подается и легкая фракция последних отделений компримирования
В колонне 22 под давлением 2 МПа отгоняют пропан-пропиленовую фракцию
Дефлегматор 23 этой и последующей колонн охлаждается водой.
Пропан-пропиленовая фракция в случае необходимости подвергается чет
кои ректификации для выделения чистого пропилена. Кубовая жидкость колон
ны 22 поступает на колонну 24 для разделения на бутиленовую и амиленовук
фракции.
Производство этилена из этана
Наилучшим сырьем по выходу этилена и стоимости установки являете!
и ан. Однако при этом необходимо переработать максимальное количеств!
природного газа, а также найти потребителей на сжиженные газы и стабильны!
оепзин. Обе эти фракции могут быть использованы для пиролиза.
Пиролиз этана проводится в таком режиме, чтобы степень его конвереш
сос тавляла 60-65%. Непрореагировавший этан выделяется в системе газора
деления и возвращается на пиролиз. При полной конверсия этана (65% за одш
проход реактора и рецикл этана) выход продуктов пиролиза, мае. %, следую
щий: Н2 — 5,5; СН4 — 11,6; С2Н4 — 75,1; С3Н6 — 2,1; С3Н8 — 0,5; С4Н6 — 2,1
(' ,Н8 — 0,85; С5 (180 °C) — 2,2; ТЖТ — 0,05.
На рис. 2.19 показан вариант технологической схемы получения этилена пи
ролизом этана.
Сырьевой и рецикловый этан подаются в секцию конвекционной ioiii
печи П-1 для подогрева до -150 °C. Подогретый этан поступает в колонну К /
|де он насыщается парами воды при -140 °C до содержания воды -30 мае.% п<
о। ношению к этану. Паро-этановая смесь проходит по трем секциям конвекцн
(ншой зоны печи П-1 и при -670 °C поступает в радиантный змеевик, рас поло
кенный в топочной камере. Греется топочная камера собственной MCian водо
родной фракцией.
Рис. 2.19. Схема получения этилена пиролизом этана
Жидкие
углеводороды
i\K0M-1
Подпитка
р-ром
NaOH
_ Конденсат
Вывод
Р-Ра
NaOH
Из К-6
Рис. 2.19 (окончание)
Ппролиi iitiii.i проводи it. »i при 8 Ч) 85()'’( . ( iciieni. иописрчп! напаОО 65%,
II роду мы пироли т, выходящие и i радиантного висеипка, быс гро охлаждают
ся до -370 °C в аакадочно испарительном аппарате /'• /. Тепло снимается за счет
генерации пара с давлением 0,4 12 МПа. Пар с давлением > 10 МПа получали
если компрессоры приводятся от паровых турбин Полученный пар отделяется
от воды в паросепараторе С-11 и перегревается в одной из секций конвективной
зоны печи П-1. Далее пирогаз охлаждается до -200 °C в теплообменнике Т-2 за
сче т нагрева питательной воды для закалочно-испарительного аппарата, после
чего поступает в промывную колонну К-2. В ней охлаждается пирогаз до 35-
40 °C и конденсируются продукты. Охлаждающая сконденсировавшаяся вода
циркулирует от насоса Н-3 через воздушный Т-3 и водяной Т 4 холодильники
из емкости С-2. Сконденсировавшаяся жидкость состоит из воды и небольшого
количества тяжелой смолы, которые разделяются в емкости С-2.
Балансовое количество воды из емкости С-1 частично выводится из сис-
темы, а частично подается насосом Н-2 через теплообменник Т-17, где она на-
гревается до -140 °C, на орошение колонны К-1. Избыток воды возвращается в
емкость С-1.
Охлажденный пирогаз из колонны К-2 поступает на всас пяти- или шес-
тиступенчатого компрессора КОМ-1 (на схеме показаны только две ступени).
После сжатия на каждой ступени проводится охлаждение газа (теплообмен-
ники Т-17 и Т-5) и конденсация воды и жидких углеводородов. Вода возвра-
щается в емкость С-2, а жидкие углеводороды из емкости С-3 насосом Н-6
выводятся из системы. Частично они могут возвращаться в емкость С-2 для
уменьшения вязкости и плотности тяжелой смолы. Перед последней ступе
нью компрессора газ промывается в колонне К-3 раствором щелочи от сер-
нистых соединений и диоксида углерода. В верхней части колонны К-3 газ
промывается водой от следов щелочи. В последней ступени газ дожимается
до -4,0 МПа. В сепараторе С-4 отделяется вода, а газ подается в реактор гид-
рирования ацетилена Р-1. Это кожухотрубчатый аппарат с теплосъемом за
счет испарения метанола Очищенный газ охлаждается в теплообменнике Т-7
до -15 °C, после чего проходит осушитель О 1. Осушители работают перио-
дически: один — на осушке газа, другой — на регенерации горячей метано-
вой фракцией. Осушенный газ охлаждается в теплообменнике Т-13 до -30 °C
и подается в сепаратор С-5. Жидкая фаза из него поступает в колонну К-4, из
куба которой выводятся углеводороды С3+. Колонна К-4 работает при давле-
нии -2,0 МПа. В ее кубе поддерживается температура -80 °C, чтобы отдуть
углеводороды С2. Газ из сепаратора С-5 проходит многопоточный теплооб-
менник Т-8, где охлаждается до -60 °C. При этом происходит конденсация
части углеводородов, которые отделяются в сепараторе С-6. Колонна К-5 (де-
метанизатор) имеет два питания: жидкостное — из сепаратора С-6 и паро-
вое — с верха колонны К-4. Колонна К-5 работает при давлении 1,1 МПа. Из
куба ее выводится этан-этиленовая фракция. Газ из сепаратора С 6 проходит
многопоточный теплообменник Т-10, где охлаждается до -85 °C. Образовав-
шийся при этом конденсат в сепараторе С-8 стекает в сепаратор С 7. Туда же
попадает конденсат паров из колонны К-5 после холодильника Т-11, где теп-
лоносителем служит кипящий этилен с температурой -93 °C. Жидкая фаза
из сепаратора С-7 насосом Н-9 подается в виде флегмы в колонну К-5. Газ
и 11eiiap.uopa ( -7 пред», ган/uiei mciанону и> фракцию, мноран лужи i хладе
агентом в теплообменниках Г 10 и Г 8.
1 аз из сепаратора С-8 охлаждается обратными потоками и j сепаратора ( /
и miioioiioio'iiiom холодильнике / 12 до -120 °C Газовый поток h i cenapaiop
( 10 представляет водородную фракцию, которая служит хладоагентом в мп<
гоноточных теплообменниках Т-12, Т10 и Т-8. Сконденсировавшийся меч ап и
сепаратора С-10 дросселируется и, нагревшись в теплообменнике /' 12, пост)
пае г в сепаратор С-7.
В многопоточных теплообменниках Т-8 и Т-10 недостаточно холода обр.и
пых потоков. Недостающее количество холода в них подводится за счет испарс
ния этилена при -70 °C и -93 °C, соответственно.
Э ган этиленовая фракция из куба колонны К-5 насосом Н-70 прокачиваете
через теплообменник Т-14, где нагревается за счет охлаждения теплопосител>
и подается в колонну К-6. С верха колонны выводится товарный этилен, а и
куба — этан, который возвращается на пиролиз. Колонна К-6 работает при д.н
лепии 2,0 МПа. Хладоагентом в дефлегматоре Т 16 служит жидкий пропилен
1емпературой -37 °C, а теплоносителем в кипятильнике Т-15 — пары пропилен
при 0 °C.
Выход этилена при пиролизе этановой фракции может составить до 80"i
11апример, при мощности в 200 тыс. т. этилена в год расход этановой фракцп
~270 тыс. т/год.
Одновременно с данным количеством этилена будет получено, тыс. т/кд
31,2 водородной фракции (90% об. Н2), 17,0 метановой фракции, 15,5 фракци
углеводородов С3-С4, 6,3 пироконденсата и тяжелого жидкого топлива.
Водородная и метановая фракции используются для сжигания в пироли
пых печах.
Ниже приведены показатели расхода сырья и энергоресурсов на полученн
1 т этилена при использовании этана и всех углеводородов С2-С4, содержащи»
ся в природном газе (расходы пара и электроэнергии даны для случая вырабт
ки пара высокого давления ~10 МПа) и использования его для привода пирог,
ювого компрессора и компрессоров холодильных циклов):
Пиролиз........................Этана
Расход:
сырья, т......................1,35
электроэнергии, квт-ч.........65,0
теплоэнергии, ГДж.............8,8
азота, нм3....................25,0
химочищенной воды, м3.........2,5
оборотной воды, м3 .......... 300,0
воздуха, нм3 ... 18,0
Углеводородов С2-С4
1,63
85,0
10,6
50,0
3,2
380,0
38,0
Альтернативным источником сырья для получения этилена может быч
природный газ. Получение этилена из метана осуществимо тремя ну гями: черт
синтез-газ, через метанол и прямой димеризацией метана в этилен.
Превращение метана в пилен даже при очень высоких гемнерагурах (»i
'.шичивается установлением термодинамического равновесия реакции. Зпа
тельные степени конверсии достигаются только при проведении реакции с
часгием окислителей. Использование окислителя, в свою очередь, затрудняет
юдбор катализатора для этой реакции, так как катализатор, эффективно ак-
инируя метан, должен одновременно слабо активировать кислород, чтобы
предотвратить глубокое окисление метана. Это обстоятельство не позволяет
|рименяп> для данной реакции такие эффективные катализаторы активации
глеводородов как, например, металлы платиновой группы [183].
Во избежание этой сложности процесс ведут в две стадии: на первой — чис-
ый метан димеризуют с использованием кислорода катализатора, на второй —
атализатор реокисляют кислородсодержащим газом. Разработаны двухста-
,ийный периодический и одностадийный непрерывный способы окислитель-
юй димеризации метана. По одностадийной схеме процесс проводят при 700-
00 °C в присутствии диоксида тория. Конверсия метана -50%, селективность
|бразования этилена 8%.
По двухстадийной схеме процесс проводят в присутствии оксидных ката-
изаторов NaMnO4/MgO-SiO2 и Mn3O4/SiO2. Выход углеводородов С2 на этих
истемах при 800 °C составляет 13-15%. При использовании в качестве окис-
ителя гомиоксида азота и катализатора, содержащего 12,5% NaMnO4/MgO и
3H4/N2O = 1:1, выход этилена при 800 °C составляет -12%.
Перспективен также способ получения этилена и пропилена из метанола,
•заработанный фирмами «Mobile» (США) и «Vinnolit» (Германия). В присутс-
вии цеолитсодержащих катализаторов выход этилена при 600-700 °C состав-
ит 30-60%. Процесс реализован в опытно-промышленном масштабе.
По информации фирм «UOP» (США) и «Norsk Hydro» (Норвегия), ими так-
ке разработан процесс получения этилена и пропилена из метанола (так на-
ываемый МТО-процесс) с использованием силикоалюмофосфатного катали-
атора SAPO-34, причем за счет применения связки «реактор-регенератор», в
[роцессе осуществляется непрерывная регенерация катализатора. Суммарный
1ыход этилена и пропилена достигает 80-90%, соотношение их может меняться
। достаточно широких пределах в зависимости от условий.
Глава 3.
Химия и технология получения
винилхлорида
сн2 ----- -..—.........сна
3.1. Общие положения
Получение винилхлорида в промышленных масштабах впервые было осу
ществлено в Германии в конце 1920-х годов, (фирмы «BASF», «I.G.Farbenindustrie»).
В основе процесса лежала реакция гидрохлорирования ацетилена, открытая не
мецким химиком Ф. Клатте в 1912 г. [33, 195]. Не позже 1933 г. винилхлорид начал
выпускаться в США, причем масштабы его производства составили тысячи гони
в год [13]. В этот период винилхлорид использовался в основном для сополимери
зации с винилацетатом. Впервые всю промышленную технологическую цепочку
реализовала фирма «Union Carbide Chemical Company».
Создание первых производств винилхлорида в других промышленно разви-
тых странах датируется следующими сроками [10]:
- Япония, 1939 г. («Chin Nippon Chisso Hiryo»);
- Великобритания, 1940 г. («Imperial Chemical Industries»);
- Франция, 1940 г. («Pechiney-Saint-Gobain»);
- Италия, 1951 г. («Montecatini S.G.»).
Объем мирового производства винилхлорида к началу Второй мировой
войны не превышал 80 тыс. т в год, в том числе в США 45 тыс. т/г (данные за
1943 г.). Бурное развитие промышленности по получению винилхлорида и его
переработке в разные полимеры началось после 1946 г. В течение пяти-семи
лет после окончания Второй мировой войны химическая промышленность в
разных странах и прежде всего в США освоила выпуск около 20 тыс. новых хи
мических продуктов, в числе которых был и винилхлорид. Так, к 1958 г. объем
производства винилхлорида в США составил 490 тыс. т/год, а в Западной Евро
пе (Германии, Англии, Франции, Италии — более 90%) — 414 тыс. т/г. При этом
ежегодный рост производства винилхлорида в Западной Европе в 1950-х годах
составил 18-19% [13].
Основными промышленными методами производства винилхлорида в .тип
период времени были щелочное омыление дихлорэтана (ДХЭ) и гидрохлориро
вание ацетилена.
Щелочное омыление дихлорэтана — метод, основанный на проведении ре
акции дегидрохлорирования дихлорэтана в жидкой фазе с использованием ще
лочных агентов. Метод характеризуется простотой технологической схемы, что
и определило широту его использования. В середине 1950-х годов в США .ним
методом производили около 50% всего получаемого винилхлорида. К началу
1960-х годов этот метод потерял свое значение в промышленности из-за боль
шого расхода гидроксида натрия и низкого показателя полезного использова
пин хлора — побочным продукты процесса являегся рас шор хлорида натрия,
который в силу сто ыгря iiieiiiioc ги поможем бы и> квалифицированно у гили
И1 роваи 1197].
В i960 е годы вместо щелочного дегидрохлорирования в промышленности
стал использоваться метод термического дегидрохлорирования дихлорэтана.
Достоинства данного метода заключаются прежде всего в возможности выде-
ления хлороводорода с дальнейшей его утилизацией, например, в процессе гид-
рохлорирования ацетилена.
Промышленная технология гидрохлорирования ацетилена (рис. 3.1) осно-
вана на газофазном каталитическом процессе, в котором в качестве исходных
реагентов используют синтетический или абгазный хлороводород, а также кар-
бидный или пиролизный ацетилен.
Метод характеризуется высокой, близкой к 100%, конверсией реагентов,
высокой селективностью (более 98%), простой технологической схемой и, как
следствие, низкими капитальными вложениями. К недостаткам относятся ис-
пользование относительно дорогого ацетилена, низкая единичная мощность
реакторов, а также экологические проблемы, связанные с использованием
дихлорида ртути (сулемы).
Указанные недостатки процессов щелочного дегидрохлорирования дихлор-
этана и гидрохлорирования ацетилена послужили толчком к созданию новых,
более современных технологий получения винилхлорида. В начале 1960-х годов
получили распространение процессы получения винилхлорида по комбини-
рованному, этилен ацетиленовому методу. Наряду с полным использованием
хлорного сырья в процессе совместно с ацетиленом стал использоваться более
дешевый этилен [198].
Рис. 3.1. Блок-схема промышленного процесса гидрохлорирования ацетилена
Рис. 3.2. Блок-схема получения винилхлорида комбинированным методом из 1ии|пы
(легкой бензиновой фракции)
Относительно широкое распространение, прежде всего в Японии, в I960 х
годах получила другая модификация комбинированного метода на основе Леч
кой бензиновой фракции (рис. 3.2.).
Данный процесс отличается от предыдущих тем, что в состав произволе i ini
винилхлорида включена стадия термического пиролиза нафты с получением смс
си разбавленных (8-9%) ацетилена и этилена примерно в стехиометрическом со
отношении. Эта смесь без концентрирования и разделения подвергается сначала
гидрохлорированию с превращением ацетилена в винилхлорид, а оставшийся
этилен хлорируется до дихлорэтана, пиролизуемого до винилхлорида и хлорово
дорода, который возвращается на стадию гидрохлорирования ацетилена.
Технологическая схема процесса заметно сложнее по сравнению с комой
иированным процессом на основе концентрированных ацетилена и этилена;
вместе с тем она сохраняет достоинства комбинированного метода и позволяет
использовать в качестве исходного сырья бензиновую фракцию вместо этилена
и ацетилена. Определенные ограничения накладываются и на мощность таких
производств.
Начиная с середины 1960-х годов, во всем мире наблюдается резкий рос i
потребности в поливинилхлориде как универсальном материале, использую
щемся во многих отраслях промышленности. С 1964 по 1981 г. мировой обьсм
выпуска ПВХ увеличился с 2,7 до 19 млн т/год, причем средний ежегодный рос i
производства составил 12%.
Это потребовало создания многотоннажных комплексов получения винил
хлорида, мощность которых составляла сотни тысяч тонн в год. Данная нрооле
ма была решена созданием промышленного процесса окислительного хлорнро
вания (или окснхлорирования) л плена с получением дихлор.пана, ианравляе
мою далее па пиролиз. Хлорид водорода со стадии иироли та полностью пере
рабатывае тся в процессе оксихлорирования этилена. 11олучеиие винилхлорида
получается полностью сбалансированным по хлору; углеводородным сырьем
является только этилен.
С учетом того, что доля сырья в структуре себестоимости винилхлорида со-
ставляет более 70%, сбалансированный по хлору процесс получения винилхло-
рида является основным методом его производства в течение более 40 лет (по
данным на начало 2007 г.).
Блок-схема сбалансированного процесса получения винилхлорида пред-
ставлена на рис. 3.3.
Процесс оксихлорирования этилена в промышленных условиях впервые
был осуществлен компанией «B.EGoodrich» (США) в апреле 1964 г. Мощность
установки составляла 136 тыс. т дихлорэтана в год. Процесс был реализован в
псевдоожиженном слое катализатора — хлориде меди, нанесенном на микро-
сферический оксид алюминия. За эту разработку фирма «B.EGoodrich» получи-
ла премию Киркпатрика в 1965 г. [199].
Одновременно с «B.EGoodrich» промышленную технологию оксихлориро-
вания освоили еще несколько компаний. «Stauffer Chem.Co» разработала про
С2Н4
Винилхлорид
Рис. 3.3. Блок-схема сбалансированного по хлору процесса получения винилхлорида
из этилена
цесс в неподвижном слое к.иалн i.ijop.i Др) him пионером ом iixiiupiipoh.iiiioi
была компания «1’1’G Ind», разработавшая процесс в пнвдоожи ценном инн
катализатора внутри трубок. Крупнейшими нрои 1вод|целями винилхлорида
стали фирмы «Dow» (США), «Monsanto» (США), «Hoechst» (1ермаиия). « 1<>уо
Soda» (Япония) и ряд других.
В табл. 3.1 представлены данные о ведущих мировых производиiелях ни
пилхлорида (на 2006 г.).
По состоянию на 2004 г. около 90% всего выпускаемого в мире випилхло
рида производили сбалансированным методом на основе этилена. За счет ввода
в эксплуатацию в Китае в 2004-2006 гг. более 70 (!) установок, использующих в
Таблица 3.1. Производство винилхлорида и распределение по фирмам
Наименование фирмы Производство, тыс. т/год
США и Канада
Оху Vinyls 1723
Dow Chemical 1701
Georgia Gulf 1145
Formosa Plastics 1239
OxyMar 1088
Westlake 864
PPG Industries 295
PPH Monomer 227
Мексика
Pemex 405
Япония
Tosoh 1910
Kashima Vinyl 600
Kaneka Corp. 520
V Tech. 400
Tokuyama 330
Китай
Qilu Petrochemical 600
Yibin Tianyuan 520
Dalian Shide Plastics 490
Корея
LG Chemical 1090
Тайвань
Formosa Plastics 1520
Западная Европа
Ineos Vinyls 1450
Soivin 760
Vinnolit 680
Hydro Polymers 790
LVM 550
Vestolit 400
качестве исходного сырья ацетилен общей мощностью до 7 млн .1 в год, доля
винилхлорида, производимого в мире данным методом, несколько возросла.
С овершенио иная ситуация в Российской Федерации. Общая мощность
производств винилхлорида по сосюянию на 2007 г. составляет 649 тыс. т/год,
в том числе гидрохлорированием ацетилена 98 тыс. т/год, комбинированным
методом (разбавленные этилен и ацетилен) 85 тыс. т/год, по сбалансированной
схеме 430 тыс. т/год, из дихлорэтана — 36 тыс. т/год.
Первая полупромышленная установка получения винилхлорида, созданная
в СССР на дзержинском заводе «Капролактам» в 1936 г., имела в основе техно-
логию, основанную на методе щелочного омыления дихлорэтана. Промышлен-
ное производство этим же методом мощностью 30 тыс. т винилхлорида в год
впервые заработало на заводе «Капролактам» в 1949 г. [11].
Производства винилхлорида из ацетилена были в основном введены в
эксплуатацию в 1960-х годах на основе репарационных поставок из Германии
(Дзержинское ПО «Капролактам» в 1959 г.) и по отечественным проектам — в
Волгоградском ПО «Химпром», Усольском ПО «Химпром», Стерлитамакском
ПО «Каустик». Мощности этих производств составили 30-35 тыс. т винилхло-
рида в год при единичной мощности реактора 1,8-2,0 тыс. т/год. Таким образом,
технологические процессы, действующие и в настоящее время, имеют техничес-
кий уровень конца 1930-х годов.
Производства винилхлорида комбинированным методом были введены
в эксплуатацию в начале 1970-х годов, на основе закупок по импорту у фирм
«Speichim» (Франция) на Дзержинском ПО «Капролактам» и «Kureha» (Япония)
на Волгоградском ПО «Каустик».
Одно из производств винилхлорида по сбалансированной схеме закуплено
у фирмы «Uhde» (Германия) и введено в эксплуатацию в 1982 г. в АО «Саянск-
химпласт» [24].
Единственное производство винилхлорида в России, созданное по отечест-
венной технологии (ФГУП НИИ «Синтез»), пущено в 1996 г. в Стерлитамакском
АО «Каустик».
Таким образом, объемы выпуска, инфраструктура и технический уровень
производств винилхлорида в России не соответствуют современным требова-
ниям.
С учетом того, что объем производства винилхлорида составляет пример-
но 30% объема производства этилена и около 60% объема производства хло-
ра, причем это соотношение справедливо для выпуска данных продуктов как в
мире, так и в отдельных странах, в том числе в России, расширение выпуска ви-
нилхлорида должно быть тесно увязано с соответствующим увеличением про-
изводства этилена и хлора, а также с решением проблемы квалифицированного
использования каустической соды.
Рассмотрим химические и технологические аспекты промышленных про-
цессов получения винилхлорида как ранее действовавших, так и современных,
проанализировав также широкий спектр исследовательских и технологических
проработок процессов, не нашедших промышленного воплощения.
Подробное описание процесса получения винилхлорида из этилена сбалан-
сированным методом дано в последующих главах.
3.2. Щелочное омыление 1,2-дихлорэтана
Синтез винилхлорида действием спиртового раствора едкого кали на 1,2-
дихлор »ian впервые был осуществлен в 1835 г [31]:
, КОН
СН2С1—СН2С1---------СН2=СНС1 + КС1 + Н70. (3.1)
С2Н5ОН
В дальнейшем щелочной метод синтеза винилхлорида из ди хлор.) тана соиер
шенствовался рядом исследователей. Так, вместо чистого спиртового раствори
щелочи был предложен этилат натрия [200], причем винилхлорид отводился из
юны реакции по мере образования. Кроме метилового и этилового спиртов,
можно использовать бутиловый спирт, гликоли или их моноэфиры.
Возможно получение винилхлорида при помощи ионообменных смол
1201]:
СН2С1—СН2С1 + R4NOH —- СН2=СНС1 + R4NC1 + Н2О, (3.2а)
R4NC1 + NaOH —* R4NOH + NaCl. (3.26)
Реакции проводятся при 70-80 °C.
Термодинамические ограничения на реакцию дегидрохлорирования
СН2С1—СН2С1 (ж) СН2=СНС1 (г) + НС1 (г) (3.3)
накладываются в случае отсутствия щелочного агента [97]. Параметры реакции
дегидрохлорирования дихлорэтана:
1g Кр = 12,65 - 5630/Т; АН = 107,8 кДж/моль; AS = 242,1 Дж/(моль-К).
В зависимости от условий реакции 1,2-отщепление НО может протекаи>
по-разному [73]. Механизм классифицируют, как правило, в зависимости oi
очередности разрыва связей С—С1 и С—Н. Выделяют £ь £2 и £1св — карбо
пильные механизмы (индексы 1,2 показывают молекулярность определяющен
стадии, а индекс «св» означает сопряженное основание). При проведении дсч ид
рохлорирования в жидкой фазе данная реакция практически всегда конкуриру
ет с реакцией замещения по SN1 и SA4 механизмами [73]. В случае конкуренции
SW) и £, механизмов состав продуктов реакции не зависит от общей скорое ги и
способа образования катиона и определяется соотношением констант к\ и к :
I I -с. ||
н—с—с—а^=^ —н—с—с+ —*
II II
Л- >С=С< + Н+ (3.3а)
~* к, Н2О
L— >С—С+ОН2^=: —>С—СОН + Н3О++ Н2О (3.36)
При регулировании сосзава продуктов как ио SN/ и ык и ио S\2 и /'2 —
механизмам можно использовать их неодинаковую зависимость от температу-
ры. Энергия активации реакции отщепления несколько выше, но сравнению с
энергией активации реакции замещения. Поэтому с повышением температуры
следует ожидать увеличения выхода алкена и, в частности, винилхлорида. Боль-
шое значение в конкуренции реакций имеет природа нуклеофильного реагента.
Для сильных оснований, таких, как едкие щелочи и третичные амины, наибо-
лее характерны реакции отщепления, а для относительно слабых оснований,
но сильных нуклеофилов (HS\ PS , ArO , NH3, Br”, Г) преобладающим может
стать замещение. Однако использование сильных оснований может приводить
к дальнейшему дегидрохлорированию винилхлорида с образованием ацетилена
с соответствующим снижением селективности и повышением взрывоопаснос-
ти. Для снижения доли реакций замещения хлора на алкокси-группу рекомен-
дуется [97] использовать не спиртовые растворы щелочей, а водные суспензии,
например, Са(ОН)2.
Исследование кинетики реакции дегидрохлорирования дихлорэтана вод-
ным раствором гидроксида натрия при 10-80 °C показало [202], что скорость
процесса удовлетворительно описывается уравнением второго порядка:
г = А-[ОН ] [S], (3.4)
где [S] — концентрация дихлорэтана в жидкой фазе.
Параметры уравнения Аррениуса: А — 1О109 л/(моль-с); Е = 96,3 кДж/моль.
Приведенное уравнение скорости реакции описывает процесс в гомогенной
среде. Варьирование концентрации электролитов приводит к изменению кон-
центрации дихлорэтана в растворе и в итоге — к изменению скорости процесса
дегидрохлорирования, что отражается следующим уравнением:
г = к [OH’]S0-l{rX[NaOH + NaC11, (3.4а)
где So — растворимость дихлорэтана в воде; [NaOH] и [NaCl] — концентрация
электролитов, моль/л.
Условия реакции щелочного омыления дихлорэтана были подробно разра-
ботаны И.И. Остромысленским в 1916 г. Реакцию ведут в среде спирта, так как
с водным раствором едкого натра дихлорэтан не смешивается. Избыток спирта
ускоряет реакцию, при его недостатке реакция почти прекращается. Для глад-
кого протекания реакции требуется также избыток щелочи (против теорети-
ческого). Процесс ведут при 78-90 °C в реакторе с обратным холодильником.
В качестве общего растворителя для дихлорэтана и едкого натра применяют
этиловый или метиловый спирт (данные о взаимной растворимости в системе
вода-метанол-дихлорэтан [12] приведены в табл. 3.2).
Рецепт Остромысленского, проверенный и уточненный П.И. Павловичем в
1936 г. в полузаводских условиях, был положен в основу реализованного в Со-
ветском Союзе промышленного метода получения винилхлорида. До 1960-х
годов этот метод был основным промышленным методом, применявшимся в
СССР.
Исходными продуктами для получения винилхлорида по этому методу слу-
жат чистый перегнанный дихлорэтан, техническая 42%-я каустическая сода и
технический метанол.
Габлица 1.2. I’.и iiinpiiMoi 11> виды ii смесях дич/iop ii.uia i mci анолом, % m.u
Вода Дихлорэтан Метанол Вода Дихлорэтан Метано т
0,6 94,3 5,1 38,8 10,5 50.7
1,5 88,3 10,2 46,0 7,1 16.9
3,0 80,6 16,4 50,9 5,1 1 1,0
6,4 67,4 26,2 58,8 3,3 37,9
8,0 61,6 30,4 65,2 2,6 32,2
10,4 55,5 34,1 73,6 1,9 24,5
11,7 50,0 38,3 78,6 1,6 19,8
14,1 43,0 42,9 82,3 1,4 16,3
19,2 32,1 48,7 88,3 1,2 10,5
26,1 22,0 51,9 93,8 1,0 5,2
Реакция между дихлорэтаном и едким натром проводится периодически в
реакторе — вертикальном цилиндрическом сосуде с рубашкой для обогрева па
ром и мешалкой.
Количество загруженного дихлорэтана должно быть таким, чтобы объем
реакционной массы в реакторе по окончании операции не превышал 80 85%
полного объема реакционного сосуда. Для достижения полного превращения
дихлорэтана количество едкого натра должно на 15-25% превышать теореги
ческое. Метанол, для лучшего использования объема реакционного сосуда,
берут в минимальном количестве, обеспечивающем, однако, достаточную ско
рость реакции. Практически это составляет примерно 500 л 100% го метанола
на 1 т 100%-го едкого натра (дальнейшее уменьшение количества загружаемою
метанола приводит к замедлению реакции).
Строгое соблюдение установленного соотношения загружаемых комнопеп
тов является необходимым условием получения высокого выхода при мини
мальной продолжительности реакции.
Загружать исходные компоненты в реактор можно двумя способами:
1. Залить в реактор полностью спирт и едкий натр, а дихлорэтан прилива1ь
постепенно или порциями по мере протекания реакции.
2. В реактор полностью заливать спирт и дихлорэтан, а раствор едкого nai ра
приливать постепенно во время реакции.
В первом случае реакция протекает все время при избытке щелочи, во bio
ром — при избытке дихлорэтана. Наличие избытка щелочи способствует де
гидрохлорированию образующегося винилхлорида до ацетилена, содержание
которого в реакционных газах может достигать 0,6-1 об. % в начале операции
и постепенно понижаться до 0,05-0,2 об. % к концу операции. При проведении
процесса в избытке дихлорэтана содержание ацетилена в реакционных газах в
среднем на протяжении всей операции не превышает 0,2 об. %. График измене
ния содержания ацетилена в реакционных газах в течение операции для ооопх
вариантов приведен на рис. 3.4.
0 9
Рис. 3.4. Содержание ацетилена в реакционных газах при получении винилхлорида де-
гидрохлорированием дихлорэтана с добавлением дихлорэтана (I), едкого натра (2)
Процесс ведут при 85-90 °C, избыточном давлении до 0,3 МПа, с переме-
шиванием. Реакция образования винилхлорида из дихлорэтана и щелочи эк-
зотермична, но так как в реакторе одновременно испаряются винилхлорид и
другие компоненты реакционной массы, то выделяющегося тепла оказывается
недостаточно, и для поддержания необходимой температуры требуется подвод
тепла извне. Образующийся винилхлорид тотчас же отгоняется от реакционной
массы, проходя через обратный холодильник.
Наряду с основной реакцией образования винилхлорида в реакторе идет
ряд побочных процессов.
Прежде всего, часть образовавшегося винилхлорида, растворенного в реак-
ционной массе, под действием спиртового раствора едкого натра подвергается
дальнейшему дегидрохлорированию до ацетилена:
СН2=СНС1 + NaOH —— НС=СН + NaCl + Н2О. (3.5)
В качестве побочного продукта получается также ацетальдегид, образую
щийся по схеме:
НС=СН + СН3ОН —► СН2=СН—ОСН3, (3.6а)
СН2=СН—ОСН3 + Н2О —- СН3—СНО + СН3ОН. (3.66)
При наличии в дихлорэтане примесей трихлорэтана и дихлорпропана в ре-
акторе образуются:
винилиденхлорид
СН2С1—СНС12 + NaOH —— СН2=СС12 + NaCl + Н2О (3.7а)
и <//»</•</ и 6сп1ч хлорнронилены:
г-*СН,—СН==СП( 1 । NaCl । ПЛ), ( V70)
< II,—CHCI —СН2С1 + NaOH —-
L—СН3—СС1=СН, + NaCl 4 И Л). (3.7и)
Присутствие большого количества воды и высокая температура способе
niyioi в незначительной степени омылению дихлорэтана с образованием ин
/ц-ill ликоля:
CIЬС1—СН2С1 + 2Н2О —— СН2ОН—СН2ОН + 2НС1. (3.8)
I (обочные продукты, имеющие сравнительно низкие температуры кипения,
удаляются из реактора вместе с газообразным винилхлоридом. Пары после об
ратного холодильника (винилхлорид-сырец) имеют примерно следующий со
i гав, %: винилхлорид 85-90, дихлорэтан 4-6, метиловый спирт 3-4, винилиден
хлорид 0,6-1,0, вода 0,5-0,6, ацетилен 0,2-0,8.
Температура газов на выходе из обратного холодильника поддерживается
не выше 45-50 °C во избежание большого уноса паров дихлорэтана и спирта
из реактора и понижения концентрации винилхлорида в газе. Обратный холо
дильпик, как и реактор, работает под избыточным давлением до 0,3 МПа. Изме
ияя давление в реакторе, регулируют температуру, а следовательно, и скорое и.
реакции. Благодаря давлению уменьшается унос спирта и дихлорэтана, так как
паровая фаза в реакторе с повышением температуры обогащается парами ви
ни дхлорида. Давление паров винилхлорида и других компонентов реакционной
С меси в зависимости от температуры в реакторе приведено на рис. 3.5.
Рис. 3.5. Давление паров: 1 — винилхлорида, 2 — дихлорэтана, метанола и воды (a icoi
рои)над реакционной смесью
Рис. 3.6. Изменение параметров в процессе дегидрохлорирования дихлорэтана едким
натром:
1 — загрузка; 2 — нагрев до температуры, предусмотренной режимом; 3 — нормальный ход реак-
ции; 4 — отгонка винилхлорида; 5 — выдавливание реакционной массы сжатым азотом и выпуск
остаточных газов
Каждая операция дегидрохлорирования продолжается 5-6 часов. Измене-
ние параметров процесса в течение одной операции показано на рис. 3.6.
Повышение температуры на выходе из обратного холодильника при одно-
временном падении давления в реакторе (после приливания всего заданного ко-
личества дихлорэтана или едкого натра) служит признаком окончания реакции.
Конец операции определяют по содержанию дихлорэтана в пробе реакционной
массы, взятой из реактора. Реакция считается законченной, если дихлорэтан в
пробе отсутствует или содержание его не превышает 0,5 об. %.
В литературе достаточно мало упоминаний о технологии получения ви-
нилхлорида щелочным омылением дихлорэтана. Информация о типичном
процессе, в котором в качестве омыляющего агента использовали раствор
NaOH в метаноле, приведена, например, в работах М.В. Хрулева [2] и М. Ма-
медова [42].
Примерная схема получения винилхлорида дегидрохлорированием дихлор
этана едким натром в спиртовой среде приведена на рис. 3.7.
Дегидрохлорирование происходит в реакторе 4, снабженном обратным хо-
лодильником 5, где конденсируется основное количество дихлорэтана, спирта
и воды. Конденсат стекает обратно в реактор, а несконденсировавшиеся про-
дукты отводятся из верхней части холодильника. Обратный холодильник 5 —
вертикальный трубчатый теплообменник, охлаждаемый водой.
Из обратного холодильника пары винилхлорида через брызгоуловитель,
дроссельный вентиль и конденсатор 8 поступают в ректификационную колон-
ну 9. В конденсаторе 8 температура паров понижается от 45-50 до 12-15 °C.
Часть паров при этом конденсируется и конденсат сливается в сборник 10. Кон-
денсат (плотность 1,01-1,02 г/см3) содержит 45-50% дихлорэтана, 25-32% ви-
нилхлорида и 20-27% метанола.
Ректификация винилхлорида сырца проводи к» в непрерывно действую
щей насадочной колонне 9 с выносным кипятильником Кипятильник обогре
пае гея горячей водой, коюрая в свою очередь нагревается паром в трубчатом
подогревателе 12, и циркулирует в замкнутой системе: сборник 11 — паровой
подогреватель 12 — межгрубное пространство кипятильника колонны 9 —
сборник / /. Температура в кубе колонны 9 поддерживается ниже +33 °C, тем-
пература греющей воды 65-70 °C. Трубчатый дефлегматор колонны 9 охлаж-
дается рассолом (раствором хлорида кальция с температурой не выше -20 °C).
Температура верхней части колонны при давлении 0,1 МПа не должна превы-
шать -13 °C.
Сырец из конденсатора 8 поступает в газообразном состоянии в среднюю
часть ректификационной колонны 9. Из верхней части колонны 9, назначение
которой отделять от винилхлорида все высококипящие примеси, пары посту
нают в дефлегматор колонны, где они частично конденсируются. Сконденси-
ровавшийся винилхлорид возвращается в колонну в качестве флегмы, нескон-
денсировавшиеся пары поступают в секционный конденсатор, охлаждаемый
рассолом с t - -23 °C.
Жидкий винилхлорид из конденсатора 20 стекает в куб отпарной колонны,
который через змеевик обогревается горячим раствором гликоля таким обра-
зом, чтобы винилхлорид находился при -13,5 °C.
Над колонной 21 установлен обратный холодильник, охлаждаемый рассо-
лом с температурой -50 °C. Винилхлорид конденсируется и стекает в колонну,
а ацетилен и инертные газы, пройдя через огнепреградитель, выбрасываются
в атмосферу.
Освобожденный от ацетилена винилхлорид из кубовой части отпарной
колонны 21 непрерывно стекает в охлаждаемый рассолом сборник 22 гото-
вого продукта. Абгазы из конденсатора 20 и сборника 22 через конденса-
тор 15 направляются в хвостовой конденсатор 14, откуда конденсат сливает-
ся в сборник 13. Все конденсаторы периодически, через 8-10 суток работы,
промывают метанолом для освобождения от вымороженной из винилхло-
рида влаги.
В кубе ректификационной колонны 9 после ректификации сырца остается
кубовая жидкость состава, %, не более: дихлорэтан — 40, метанол — 30, ви-
нилхлорид — 25, винилиденхлорид — 0,5, цис- и транс-дихлорэтилены — 3,5,
прочие примеси — 1.
Из куба колонны эта жидкость сливается в сборник 16, обогреваемый па
ром через змеевик. В сборнике кубовую жидкость выдерживают некоторое
время при температуре до 30 °C для отделения растворенного в ней винил
хлорида, а затем передают в систему водной промывки для извлечения из нее
метанола.
Метанол из кубовой жидкости извлекается в противоточном колонном
экстракторе 17 с насадкой из колец Рашига. Кубовая жидкость из сборника 16
поршневым насосом подается в верхнюю часть экстрактора 17, заполняя его
насадочную часть. Вода поступает в экстрактор снизу вверх, противотоком к
движению кубовой жидкости, и, смешиваясь с последней, почти полностью
вымывает из нее метанол. Раствор метанола из верхней части экстрактора сли-
вается в сборник 18 и вновь возвращается в экстрактор для донасыщения.
I.Шлица 3. к < <к i ан ынтряжснных фа i в < нс геме вод.» метанол дихлор пан, м.и '*«>
Фили дихлор згана Водная фала
Вода Метанол Дихлор »ган Вода Метанол Дихлор >ган
0,1 0,3 99,6 86,5 12,1 1,1
0,1 0,7 99,1 75,1 23,1 1,8
0,3 1,6 98,1 59,2 37,5 3,3
0,4 2,3 97,3 51,0 44,0 5,0
0,5 3,9 95,6 40,4 49,9 9,7
0,8 6,3 92,6 28,2 52,1 19,7
1,4 9,5 89,1 19,9 49,2 30,9
2,3 13,8 84,0 14,7 43,8 41,6
2,7 15,4 81,9 12,4 39,8 47,8
6,2 25,9 67,9
В табл. 3.3 приводится состав растворов в системе вода-метанол-дихлор
пап.
Из нижней части экстрактора 17 в сборник 19 сливается освобожденный от
метанола дихлорэтановый раствор состава (%): дихлорэтан — 75-80, винили
дснхлорид — 10-15, винилхлорид — 3-4, метанол — 0,5-1,0, дихлорэтилены —
2,0 -5,0.
11о мере накопления этой смеси ее разгоняют в периодически действующей
ректификационной системе, выделяя фракции дихлорэтана и винилиденхло
рпда. Дихлорэтан вновь возвращают в реактор 4 для дегидрохлорирования, а
випилиденхлорид стабилизируют гидрохиноном и выпускают как товарный
продукт. Кубовые остатки уничтожают или используют для чистки цистерн.
Раствор из сборника 18 по достижении содержания в нем метанола 12- 15%
поступает в отпарную колонну 27 для отделения метанола.
В экстракционной системе одновременно с удалением метанола испаряется
винилхлорид, растворенный в кубовой жидкости. Отходящие газы из всех ан
парагов экстракционной системы выводятся через конденсатор 15 в хвостовой
рассольный конденсатор 14. Конденсат из него собирается в сборник 13, откуда
поступает на повторную ректификацию.
Жидкость, оставшаяся в реакторе после дегидрохлорирования и отгонки
винилхлорида, содержит от 3 до 10% NaOH, от 12 до 18% метанола и большое
количество поваренной соли (как в растворе, так и в виде взвеси). Горячук
жидкость из реактора передавливают сжатым азотом в сборник 26, снабжен
ный пропеллерной мешалкой, где жидкость непрерывно перемешивают, что
бы предупредить осаждение взвешенной соли. Во избежание потерь метанола
с испаряющейся горячей жидкостью сборник26 во время приема жидкост и и
реактора соединяют с атмосферой через насадочный абсорбер 23 (в осталытот
время сборник с атмосферой не сообщается). Через абсорбер 23 выводятся
в атмосферу также остаточные газы из реактора после освобождения его от
кидкости.
Метанол из отходящих газов абсорбируется водой, орошающей абсорбер 23
представляющий насадочную колонну, заполненную кольцами Рашит а. Вода но
даек» и абсорбер и i сборника 25 через холодильник 21 и циркулируй i в системе
до образования 12 15% го расшора метанола. Такой раствор присоединяют к
общему потоку жидкости, поступающей в отпарную колонну 27, а в систему
абсорбции вводя г свежую воду.
Жидкость из сборника 26 подается в верхнюю часть отпарной колонны 27;
га юное пространство сборника 26 соединяют для уравнивания давления с вер-
хней частью отпарной колонны 27.
В отпарной колонне 27 имеется семь пар конических полок (насадочная
колонна в данном случае неприменима из-за большого количества соли,
находящейся в жидкости во взвешенном состоянии). Метанол отпарива-
ют острым паром, подаваемым под нижнюю полку. По мере продвижения
по колонне острого пара снизу вверх метанол отгоняется из стекающей по
полкам жидкости. Выводимый из нижней части колонны щелок содержит
14-15% растворенной и около 18% взвешенной поваренной соли, от 2 до
5% едкого натра и не более 0,5% метанола. Температура щелока на выходе
из колонны поддерживается в пределах от 105 до 117 °C с помощью термо-
регулятора, изменяющего количество подаваемого острого пара. Темпера-
тура в верхней части колонны не должна превышать 92 °C. Выходящая из
верхней части колонны 27 смесь паров воды и метанола, содержащая около
60% СН3ОН, направляется в насадочную ректификационную колонну 32,
где концентрация метанола в парах повышается до 80-95%. После конден-
сации паров в конденсаторе 33 и охлаждения в холодильнике 34 метанол
возвращается в сборник 35 для повторной загрузки в реактор. Температуру
в кубе колонны 32 поддерживают в пределах 102-110 °C, регулируя подачу
греющего пара в кипятильник колонны. Кубовая жидкость — вода, содер-
жащая не более 0,8% метанола (нормально 0,5%) Температура в верхней
части колонны 32 поддерживается в пределах 64-70 °C. Приток флегмы на
орошение колонны регулируется изменением количества охлаждающей
воды, поступающей в трубное пространство дефлегматора колонны. Кон-
денсатор и холодильник колонны 32 охлаждаются водой. Щелоки из ниж-
ней части отпарной колонны 27 сливаются в промежуточный сборник 28
с коническим днищем, где перемешиваются мешалкой, после чего центро-
бежным насосом подаются в отстойник 29. Отстоявшаяся пульпа выводит-
ся из отстойника с помощью шнека в сборник пульпы 30, где непрерывно
перемешивается и через коническое днище поступает в горизонтальную
центрифугу 31 для выделения соли. Соль в центрифуге промывается водой,
дополнительно отжимается до содержания влаги не более 3%, срезается
ножом и транспортером передается на склад. Выделенная таким образом
соль почти не содержит примесей и может использоваться в разных про-
изводствах.
Осветленная жидкость из отстойника 29 и фильтрат из центрифуги 31 пе-
редаются для упарки в цех производства каустической соды. В этих жидкостях
содержится 30-35% едкого натра.
Таким образом, побочные продукты, получающиеся при производстве ви-
нилхлорида дегидрохлорированием дихлорэтана едким натром, могут быть ис-
пользованы почти полностью. Этот метод получения винилхлорида был доста-
точно широко распространен в промышленных условиях в 1950-60-х годах.
Расход на I । винилхлорида:
Дихлор хан ремификаг, кг..........................................1900 1950
I дкии ii.ilр (100%-ный), кг......................................745-820
Метанол (100% пый), кг............................................75 90
bicKipo >пер1ия, кВт ч.......... ................................ 90 93
Пар (0,5 МПа), МДж................................................5,8-7,5
Холод, МДж........................................................5,6
Л ют, м'......... ................................................90
За одну операцию в реакторе объемом 12 м3 можно получить 3,6-3,7 г ни
нилхлорида Средняя производительность одного реактора достигает 560 кг/ч
винилхлорида.
Аппаратурное оформление процесса несложно; химически стойкие мате
риалы не требуются, так как агрессивные среды в процессе отсутствуют. Ik
требуется также и применение высоких температур и давлений. Исходным
сырьем служит сравнительно недорогой этилен. Однако хлор и каустическая
сода используются в этом процессе нерационально. Праю гчески на каждую
тонну получаемого по этому методу винилхлорида выделяется в качестве но
(ючпого продукта 1 т поваренной соли, т.е. значительная часть хлора и едкого
натра вновь образуют продукт, из которого они были получены. Кроме того,
и процессе производства расходуется дорогостоящий метанол, который в ре
акции образования винилхлорида не участвует. Регенерация метанола услож
пяет производственный процесс и требует дополнительного расхода энергии,
большим недостатком этого метода является также периодичность процесса
11ереход к непрерывной схеме связан со сложным аппаратурным оформлены
см, так как необходимо постоянно выводить из цикла образующуюся твердую
поваренную соль.
Винилхлорид, получаемый этим способом из дихлорэтана — это бесцвет
пая прозрачная жидкость с температурой начала кипения не ниже -14,5 °C. До
температуры -10 °C выкипает не менее 99,95 об. %; содержит ацетилен — менее
10 '*%, ацетальдегид — менее 30-10“4%.
Процессы жидкофазного дегидрохлорирования можно существенно ин ген
сифицировать, используя катализаторы межфазного переноса. Данное направ
ление начало широко использоваться в исследовательской, а затем и в промыт
ленной практике в 1970-1980-х годах.
В качестве катализаторов могут быть использованы четвертичные аммони
свые и фосфониевые соединения, оксиды третичных аминов, а также цикличсс
кие полиэфиры, полиэтиленгликоли и фосфаты.
Начало изучению подобных процессов было положено работами С. Старкси
|3()0] и М. Макоши [301]. Используемые катализаторы, например, соединения
типа [R|R2R3R4N]+X (N — азот, фосфор, мышьяк, R] 4 - С| 30, X — анион opi а
нической или минеральной кислоты), имеют в составе большой органический
клион. Влияние природы центрального ядра на активность в достаточной мере
не установлено. В [302] приведены данные по сравнительной активности 21 х
ка гализаторов и показано, что в целом фосфониевые соединения по активноегп
превосходят аммониевые, хотя есть и исключения.
Ускоряющее де1К1Вие шпользуемых соединений обусловлено, нерол I по,
их поверхностно активными свойствами и способностью к распределению
между водной и органической фазами, вследсшие чего может увеличиваться
концентрация ОН в органической фазе. Согласно механизму, предложенному
С. Старксом [300|, четвертичная соль распределяется между фазами, даже если
ее растворимость в воде настолько мала, что не определяется обычными мето-
дами. Были установлены первые порядки реакции по органическому соедине-
нию и катализатору и независимость скорости от концентрации нуклеофила,
что объясняется его избытком. Отмечается резкое увеличение (на один два
порядка) константы скорости в присутствии катализаторов. Большое влияние
на скорость процесса оказывает соотношение водной и органической фаз, при
50-кратном увеличении объема водной фазы скорость реакции проходит через
экстремум при количестве водной фазы, соответствующем насыщению реаген-
том, и далее несколько снижается (примерно в 3 раза) из-за уменьшения степе-
ни гидратации ионов и, соответственно, их экстрагируемое™ в органический
слой. Однако показано, что переход аниона из одной фазы в другую не обяза-
тельно должен сопровождаться переходом органического катиона. По мнению
М. Макоши [301], отрыв протона от молекулы галогенуглеводорода и образо-
вание ионной пары происходит не в глубине органической фазы, а на границе
раздела фаз.
Основные принципы проведения процессов с использованием катализато-
ров межфазного переноса приведены в [303-305]. Применительно к проблеме
получения винилхлорида большая серия работ была выполнена С.С. Шавано-
вым и Г.А. Толстиковым [306-312]. Авторы указывают, что оптимальными ка-
тализаторами для этих целей являются четвертичные аммониевые соли (ЧАС)
вследствие своей доступности и высокой каталитической активности. При
этом выбор катализаторов конкретных реакций на основе ЧАС должен прово-
диться с учетом всех факторов, определяющих строение и свойства катиона и
аниона [306].
Изучение реакционной способности алкоксидов триэтилбензиламмония
(ТЭБА) позволило установить, что они, будучи применены в стехиометричес-
ких количествах, дегидрохлорируют полихлоралканы с высокими скоростями и
селективностью [307]. Винилхлорид из 1,2-дихлорэтана образуется при -20 °C
практически мгновенно.
-20 °С,1 мин
С2Н4С12 + (ТЭБА)ОС2Н5--------- С2Н,С1 + (ТЭБА)С1 + С2Н5ОН, (3.9а)
99,5%
-10 °C
СН2С1СНС1СН2С1 + (ТЭБА)ОС2Н5--------
98%
---- СН2=СС1СН2С1 +- (ТЭБА)С1 + С2Н5ОН. (3.9б)
Показано [308-309], что скорость реакции дегидрохлорирования 1,2-
дихлорэтана существенно зависит от строения как аниона (табл. 3.4), так и ка-
тиона бензилоксидов тетраалкиламмония (табл. 3.5), вследствие изменения его
липо-, гидрофильности и величины положительного заряда.
()6i i.i ру жена также высокая катали i и чес кая ак i и внос ь систем, пол учен пых
растворением в дихлорэтане ЧАС и спиртов |31(), 311|. Эффективноегь них
систем в большой степени определяется основностью алкоксианиона cniipia.
что позволило установить последовательность возрастания каталитической
ак тивности в зависимости от структуры алкоксианиона спирта: (СН3)(< О <
(СН3)2СНО < СН3О < СН2=С(СН3)СН2О < С2Н5О < С10Н21О < С,11,0 <
С71113О < СН2—СНСН2О“ < СНС1=СНСН2О < СН2=СС1СН2О < l’h( 11.0
<4—С1С2Н4СН2О .
При этом каталитическая система формируется в результате сольватации
дихлорэтана или другого хлорорганического субстрата комплексами ЧАС
спирт.
Фактом, подтверждающим сольватацию, является образование беи тил
р хлорэтилового эфира при термолизе растворов комплексов ЧАС ciuipi
в дихлорэтане. Было показано [306], что наибольший выход бсчиил р
Таблица 3.4. Влияние строения аниона в (C2H5)3N+CH2PhX на скорость дегидрохлори
рования 1,2-дихлорэтана (Г = 50^-55 °C; C2H4Cl2:NaOH=l:2; [NaOH] = 50%, т = 30 мин.;
[R,N+X ]=0,3 мол %)
X W • 104, моль/(л-с) Конверсия С2Н4С12, %
С1 8,6 28,5
ОСНз 13,9 45,7
ОС2Н5 16,8 56,3
ОС4Н9 20,7 68,1
ос7н15 21,7 71,6
ОСН2С6Н5 31,6 99,5
ОСН2С6Н4С1-4 37,9 99,7
он* 9,3 30,8
CI* 9,2 30,6
4 Катион — (C4Hq)4N.
Таблица 3.5. Влияние строения катиона R4N1 в R4NOCH2Ph на скорость дегидрохлори
рования 1,2-дихлорэтана
R4N+ W • 104, моль/(л-с) Конверсия C2H4C12, %
(C2H5)3NCH2Ph 31,6 99,5
(C2H5)3NCH2C6H4Cl-4 28,4 89,4
(C4H9)4N 26,2 82,4
(CH3)3NC12H25 18,0 56,3
(CH3)2(C12H25)NCH2Ph 17,3 54,5
(CH3)3NCH2CH2C1 11,0 34,4
(CH3)3NCH2CH2OH 9,0 28,3
хлор и иловою »<|>пра ||.К>л 1одасня ори о< [.ночной концепiрации дихлор
панд ?()"<> I [реддожеиа следующая схема обра (окания беи итлхлор иилоного
>фира при гермолизе.
СН2—CHCI
Cl н
: \)CH2Ph —- PhCH?OCH2CH2Cl
Et3+NCE----н
I
СН2РЬ
(3.10)
Экспериментальным доказательством образования трехкомпонентных ас-
социатов можно считать гомогенизацию водных растворов хлоридов ЧАС, на-
пример (ТЭБА)С! с дихлорэтаном и малорастворимыми в воде спиртами. Так,
гомогенные растворы образуются при смешении 40-60% растворов (ТЭБА)С1
с бензиловым, 4-хлорбензиловым и Р-хлораллиловым спиртами и дихлорэта-
ном.
Как видно из рис. 3.8а, активность каталитических систем зависит не только
от строения спирта, но и от соотношения спирта с (ТЭБА)С1.
Формирование каталитически активной системы проходит во времени, что
подтверждается существованием индукционного периода реакции (т), продол-
жительность которого зависит как от строения, так и от количества спирта, ис-
пользуемого для получения катализатора (рис. 3.86).
Характерно, что в присутствии бензилата ТЭБА, (ТЭБА)ОСН2РЬ, реакция
дегидрохлор рования дихлорэтана протекает без индукционного периода со
скоростью, несколько превышающей скорость реакции, катализируемой сис-
темой (ТЭБА)С1-РЬСН2ОН-дихлорэтан. При добавлении к бензилату ТЭБА
эквимолярного количества НС1 можно получить систему, ведущую реакцию
как упомянутый выше комплекс, но индукционный период реакции также
исчезает. После выдерживания 20 ч этой системы получается катализатор, по
всем показателям идентичный свежеприготовленному трехкомпонентному
комплексу:
НС1
(Et3N+CH2Ph) О CH?Ph----- (Et3N+CH2Ph) О CH.Ph
20ч х ;
^С1—44 <311)
1 - 4 мин т = 0
Механизм элиминирования НС1 может быть представлен в виде последо-
вательного образования ассоциатов, включающих все три компонента, а за-
тем предреакционного комплекса (Х4) с участием водного раствора щелочи,
в котором происходит отщепление НС1 [306]. Стадия образования ассоциа-
тов характеризуется упоминавшимся выше индукционным периодом реак-
Рис. 3.8. Влияние соотношения ROH:(T9BA)C1 на скорость реакции (а) и индукциоп
ный период (6) щелочного дегидрохлорирования 1,2-дихлорэтана [50%NaOH, 1 мае. %
(ТЭБА)С1, C2H4Cl2:NaOH=l:2, t = 50-55 °C]:
/ — R:C2H5OH, 2 — СН2=СС1-СН2ОН, 3 — PhCH2OH
ции. При этом сама реакция не требует фазового переноса ионных пар или
ионов и происходит как в органической фазе, так и на границе раздела фа t
(рис. 3.9).
Предложенная структура предреакционного комплекса (Х4) учитывает вл и
яние всех его составляющих на скорость реакции элиминирования. Величина
положительного заряда на атоме азота в катионе весьма сильно влияет на ката
литическую активность систем, включающих ЧАС и бензоловый спирт; силь
ное влияние оказывает соотношение ЧАС : спирт; оптимальным является cool
ношение 1:Зэ-4.
Рис. 3.9. Вероятная структура предреакционного комплекса Х4
Зависимость скорости дегидрохлорирования дихлорэтана (W-104,
моль-л'-с”1) от строения катиона (условия реакции см. табл. 3.4)
ЧАС: PhCH2OH 1:0 1:1 1:4
(CH3)2N(CH2Ph)CH2CH=CH2 2,42 45,0 76,0
(CH3)2N(CH2Ph)CH2C(CH3)=CH2 - 34,3 41,1
(CH3)2N(CH2Ph)2 5,50 26,3 46,4
(C2H5)3NCH2Ph - - 86,2
(C2H5)2N(CH2Ph)CH2CH=CH2 0,84 32,8 77,3
(CH3)2N(CH2CCH3=CH2)CH2C6H4C1 - 2,3 4,34
(CH3)3NCH2CH=CH2 4,80 5,8 12,3
(CH3)3NCH2C(CH,)=CH2 6,40 8,3 25,7
(CH3)3NCH2Ph 6,20 17,6 34,6
Элиминирование НС1 представлено шестью стадиями:
4АСОФ + РЬСН2ОНОФ X,, (I)
X,+ С2Н4С12с,Ф^Х2> (II)
№1ОНВФ ^±1 №1ОНПРФ, (III)
р^ПРФ
(IV)
Cl
+ NaOHnp<1>^^X3,
(V)
2X3^=^X4. (VI)
Стадия I соответствует образованию комплекса ЧАС-РЬСН2ОН в органи-
ческой фазе (ОФ). На стадии II происходит включение в комплекс субстрат и.
При образовании интермедиата Х3 происходит одновременное вытеснение
воды из гидратной оболочки NaOH. На стадии VI завершается формирование
предреакционного комплекса в органической фазе и на поверхности раздела
фаз(ПРФ).
Включение этой стадии в механизм элиминирования НС1 позволяет объяс
ни । ь высокие эмпирические порядки по (ТЭБА)С1 и NaOH.
Предлагаемый путь формирования предреакционного комплекса подтверж
дастся расчетными константами и энергией активации [312].
Дальнейший ход процесса дегидрохлорирования представлен следующими
с । адиями:
Х4^=^С2Н3С1ОК + Х51 (VII)
С2Н3С1ОК С2Н3С1ОФ,
HCl + NaOHIIP<I)^=^NaCl+ Н2О, (VIII)
С2Н3С1ОФ С2Н4С12ОК, (IX)
Х5 +- С2Н4С12ОК Х4. (X)
РЬСН2ОНОФ PhCH2OHOK, (XI)
Х5 + PhCH2OHOK Xg. (XII)
В сформированном предреакционном комплексе Х4 происходит синхрон
ный отрыв винилхлорида и NaCl. Стадия VII может включать: одновремен
ный или последовательный разрыв связей С—Н и С—С1 в одной из молекул
дихлорэтана, связанных в комплексе Х4; образование в клетке растворителя
винилхлорида и комплекса Х5, который расположен в органической фазе и
на поверхности раздела фаз; выход винилхлорида из области катализа (ОК) в
органическую фазу. На стадии VIII происходит взаимодействие NaOH с 11С1,
входящим в комплекс Хц. Далее следуем переход дихлор чана в м.орицуХ,, что
laBcpiii.ier обра ишание предреакциоппого комплекса X । (стадии IX, X).
( тадии XI, XII описываю! ингибирующий эффект при включении изоы гоч
ново бензилового спирта в клетку комплекса Х5 с образованием неактивного
комплекса Х().
Кинетические параметры реакции щелочного каталитического дегидро-
хлорирования дихлорэтана были использованы при выборе режима работы
промышленного реактора [306]. Было установлено, что в присутствии 0,3-0,4
мол. % катализатора, приготовленного из (ТЭБА)С1 и бензилового спирта (1:4),
процесс проходит при 50-55 °C и начальной концентрации NaOH 40-50% со
скоростью, соответствующей производительности 350 кг/ч винилхлорида в
расчете на 1 м' реакционного объема. Получаемый винилхлорид не требует до-
полнительной очистки.
Промышленная реализация процесса была осуществлена в Стерлитамакс-
ком АО «Каустик».
В литературе упоминается об использовании в качестве катализатора меж-
фазного переноса N-винилкарбазола [313], катамина АВ [314] с выходом винил-
хлорида на уровне 98-99%. Отсутствие катализаторов межфазного переноса в
растворителе КОН-бензол снижает выход винилхлорида до 5%, а ввод в систему
краун-эфиров увеличивает его до 83-95%. Природа растворителя также играет
существенную роль: замена бензола на этилбензол снижает выход винилхло-
рида до 50% [314].
Тем не менее, данный метод получения винилхлорида представляет лишь
исследовательский интерес, так как в промышленных условиях он не выдержи-
вает конкуренции со сбалансированным по хлору процессом.
3.3. Газофазное гидрохлорирование ацетилена
в неподвижном слое катализатора
Процесс получения винилхлорида гидрохлорированием ацетилена по реакции
С2Н2 + НС1^^ С2Н3С1 + 112,9 кДж/моль (3.12)
в течение 30 лет оставался основным промышленным методом получения дан-
ного мономера.
По этой причине в 1950-70-е гг. в литературе появилось значительное количес-
тво публикаций, посвященных исследованиям кинетики и механизма процесса,
подбору оптимальных каталитических систем, проблемам дезактивации катализа-
торов, а также разработке научных и технологических основ проведения процесса
в газовой и жидкой фазах. По мере снижения промышленного значения использо-
вания ацетилена в качестве сырья для производства винилхлорида в 1980-90-е гг.
количество исследований в этом направлении заметно уменьшилось.
Термодинамические расчеты реакции получения винилхлорида и основной
побочной реакции получения 1,1-дихлорэтана
С2Н3С1 + НС1^^1,1-С2Н4С12 (3.13)
представлены в работах [203-205].
Д. ( галл с соавторами [205| рассчитал кош t.inii.i равнинен и» дли реакции
образования винилхлорида и 1,1-дихлорэтана:
Koiictub iа равновесия ........Квх ^i.i-дхэ
При 30(1 К...................6,58'101" 1,06'105
» 100 К......................2,94'106 71,3
» 500 К.......................6.55103 0,877
Полученные значения хорошо согласуются с данными работы [203|, в ко
горой рассчитан состав равновесной газовой фазы, образующейся при сип геле
винилхлорида из концентрированного и разбавленного ацетилена при разных
давлениях (табл. 3.6, 3.7).
Приведенные в табл. 3.6, 3.7 данные позволяют заключить, что;
- суммарная степень превращения ацетилена в винилхлорид несколько
уменьшается с ростом температуры и очень мало зависит от повышения дав
лепил;
- количество образующегося 1,1-дихлорэтана слабо возрастает с увеличе-
нием давления. Поэтому с термодинамической точки зрения синтез винил
хлорида выгоднее проводить при низких давлениях и невысоких темпера гурах.
Однако опыт, как будет показано, свидетельствует, что выход винилхлорида
при добавлении требуемого количества катализатора почти пропорциональ
по возрастает с увеличением давления при использовании как концентриро
Габлица 3.6. Состав равновесной газовой фазы, мол. %, рассчитанный для реакции сип
и- га винилхлорида из концентрированного ацетилена (карбида)
р, МПа t,°C С2Н2 СН3СНС12 НС1 СН2СНС1
0,1 100 0,02 0,02 0,01 99,95
0,1 200 0,49 0,05 0,43 99,03
0,5 100 0,02 0,02 0,00 99,96
0,5 200 0,26 0,10 0,16 99,48
1,0 100 0,02 0,02 0,00 99,96
1,0 200 0,22 0,12 0,10 99,56
I .шлица 3.7. Состав равновесной газовой фазы, об. %, рассчитанный для реакции сип
ic ы винилхлорида из разбавленного (7,7 об. %) ацетилена
/>. МПа t,°C Инерт- ный газ С2Н2 СН3СНС12 НС1 СН2СНС1 Степень пре- вращения С2П2 в СН2СНС1,*%
0,5 100 92,31 0,00 0,00 0,00 7,63 100,00
0,5 200 92,26 0,06 0,00 0,06 7,63 99,22
1.0 100 92,31 0,00 0,00 0,00 7,69 100,00
1.0 200 92,27 0,04 0,00 0,04 7,64 99,35
ванного, гак и разбавленного ацетилена. Но.ному наиболее экономичные ус
ловим ведения процесса определяю гея наряду со стоимостью катализатора,
производственными затратами па создание и эксплуатацию компрессорных
установок.
В качестве катализатора газофазных реакций присоединения к ацетилену
молекул типа НХ использовались или предлагались соли МеХ„, нанесенные
на активный уголь (АУ) (где Me — Hg, Bi, Cd, Zn и X — СГ1, CH3COO , CN ,
ОН ).
В работах [206-210] показано, что в МеС1„ катионы располагаются по ката-
литической активности в следующий ряд:
Hg2+ > Bi3+ > Cd2+ > Zn2+.
Такие катализаторы как PdCl2/AY или РЮ12/АУ недостаточно стабиль-
ны в условиях процесса [211]. Активность нанесенных на АУ хлоридов ме-
таллов в синтезе винилхлорида коррелирует с окислительным потенциалом
(М”+ + иё-»Л1) и растет, начиная с Е° = -0,8В с увеличением последнего в ряду
[172, 212]:
Zn2+ < Bi3+ < Cu2+ < Hg2+ < Pd2+ < Pt2+.
Единственным промышленным катализатором реакции гидрохлорирова-
ния ацетилена является катализатор HgCl2/AY. Содержание сулемы в катали-
заторе составляет обычно 10% масс, для концентрированных и 15% масс, для
разбавленных смесей. Вывод о том, что именно данное содержание сулемы яв-
ляется оптимальным, впервые был сделан Д. Бартоном и М. Мигданом [213].
Полученные ими результаты показаны на рис. 3.10.
При исследовании механизмов реакции первоначально принимали [42, 214,
215], что реакция гидрохлорирования ацетилена протекает в две стадии, первой
из которых является изомеризация ацетилена (медленная стадия):
СН=СН —СН2=С<. (3.14а)
На второй стадии происходит присоединение хлорида водорода к изоацети-
лену с образованием винилхлорида:
СН2=С< + НС1 —— СН2=СНС1. (3.146)
Эта стадия из-за высокой реакционной способности изоацетилена рассмат-
ривалась как протекающая очень быстро. Указанный механизм подвергался
критике [216] из-за отсутствия экспериментальных доказательств образования
побочного изомера ацетилена, а также того, что если механизм действия ката-
лизатора сводится к изомеризации ацетилена, то присоединение любой молеку-
лы адденда независимо от природы протекало бы в одних и тех же условиях и
определялось природой катализатора.
В настоящее время общепринятой является точка зрения, согласно ко-
торой реакция гидрохлорирования ацетилена протекает через промежуточ-
ные комплексные соединения. Основанием для этого изначально служил тот
факт, что сулема легко образует с ацетиленом комплексное соединение, из-
вестное в литературе как комплекс Бигинелли (p-хлорвинилмеркурхлорид):
CHCl=CHHgCl [216, 217].
50
1’iic. 3.10. Зависимость выхода винилхлорида от количества сулемы: 1 — на 1 г HgCL;
’ — па 1 г АУ
И свестны стереоизомеры этого соединения.
С1 Н С1 Н
с—С С=С
11' ^HgCl Н HgCl+
НН НН
Хс=с/ ^^2 С=С
Cl HgCl сГ 'HgCl+
(3.15а)
(3.156)
Возможно также образование так называемых веществ Дженкинса [21 к]:
(C2H2)2HgCl2 (ClCH=CH)2Hg. (3.16)
Представление о том, что комплекс Бигинелли является собственно ка
ылиаатором процесса, было положено в основу механизма, впервые они
санного С.Л. Варшавским [219], который обнаружил, что при нагревании
комплекса Бигинелли с соляной кислотой почти количественно выделяется
винилхлорид:
С !CH=CHHgCl + НС1 СН2СНС1 + HgCl2. (3.17)
Винилхлорид образуется также при взаимодействии Р-хлорвинилмеркур
хлорида с 40%-ным раствором бромида водорода (винилхлорид и HgClBr).
Механизм, предложенный Варшавским [ 2191
снЕ=си л ngci2—- chci=ch iigi i.
Cl ICl=CI IHgCl + HCI —- CH2=CH( 1 + HgCl2
(3.1 Ha)
(3.186)
был подтвержден X. Ватанабе и М. Онозука [220], К. Мицутани и К. Ямашита
[221|.
Р.Д. Вессельхофт с сотр. [222] детально исследовал влияние парциального дав-
ления компонентов гидрохлорирования ацетилена на скорость реакции при раз-
ных условиях. Полученные зависимости представлены на рис. 3.11.
Рис. 3.11. Влияние (при 100 °C) парциального давления: а — ацетилена (рА) на началь-
ную скорость его реакции с хлороводородом; б — хлороводорода (рн)
0.03
Рис. 3.11 (окончание). Влияние (при 100 °C) парциального давления: в — винилхлорида
(/>пх) на скорость гидрохлорирования ацетилена; г — ацетилена (рА) и хлороводорода
(/>i ।) на скорость реакции с винилхлоридом
Полученные им зависимости (рис. 3.11) указывают, что хлороводород ад
। орбируется очень быстро и занимает большую часть поверхности катали <аю
ра лишь при относительно низких значениях парциального давления. Адсорб
ция ацетилена протекает медленнее по сравнению с хлороводородом. Если (>ы
ацетилен и хлороводород адсорбировались на одних и тех же активных цент
рах, и реакция шла только между адсорбированными молекулами, го скорост ь
реакции в начале опыта возрастала бы, достигнув максимального значения
Далее скорость реакции должна падать вследствие вытеснения адсорбирован
пого ацетилена хлороводородом. Авторы работы 12221 делают вывод о том, что
все |ри компонента реакции адсорбируются па разных участках катализатора.
При >том в реакцию вступают только адсорбированные молекулы ацетилена и
хлороводорода, причем между скорос тью адсорбции и десорбции них комно
центов и скоростью образования винилхлорида быстро устанавливается равно-
весие. Схема реакции следующая:
1)C2H2 + S1^=^C2H2:S1, ^2 (3.19а)
2) HC1 + S2^=^HC1:S2, Л-4 (3.196)
3) С2Н2 : Sj + НС1: S2 С2Н3С1 + Sj + S2, (3.19в)
4) С2Н3С1 + S3 С2Н3С1: S3 , (3.19г)
тде S|, S2, S3 — активные центры поверхности катализатора.
Иной подход к толкованию механизма процесса дан в работе [223]. Есть ос-
нование полагать, что соединения типа комплекса Бигинелли не промежуточ-
ные, а побочные продукты. Показано [224, 225], что анион каталитической соли
входит в состав продукта реакции. Указано [223], что при наличии акцептора
протона ацетилен способен к ионизации типа
С2Н2 С2Н + Н+ (3.20)
и реагирует далее с комплексообразователем — сулемой по типу донорно-ак-
цепторного взаимодействия, при котором ацетилен отдает, а сулема притягива-
ет пару л-электронов. Это, с одной стороны, ослабляет связь центрального иона
Hg2+ со связанным с ним анионом СГ, а с другой — облегчает отдачу ацетиле-
ном протона. Активация ацетилена заключается в его деформации частичным
оттягиванием пары п-электронов к катализатору. Катализатор должен быть
способен к комплексообразованию, как с ацетиленом, так и с присоединяемым
веществом — аддендом. При этом образуется тройной комплекс: ацетилен-ка-
тализатор-адденд.
А.И. Гельбштейн [206 209] так же, как и авторы работ [216, 223], рассматри-
вает механизм реакции как гетеролитический. Каталитическое действие солей
типа МеХ„ трактуется как действие льюисовских кислот, т.е. акцепторов или
доноров электронов. Р.М. Флид и О.Н. Темкин [216] исследовали закономер-
ности в строении электронных слоев катионов солей — катализаторов реакции
присоединения к ацетилену. У каждого из этих элементов подгруппа d предпос-
леднего слоя заполнена десятью электронами, внешними являются один-два
электрона подгруппы S (например, Hg — 5</106$2).
При взаимодействии солей таких металлов с ацетиленом возможно как об-
разование донорно-акцепторной связи (за счет пары п-электронов иона и раз-
рыхляющеи орбиты ацетилена), так и дативное в 1аимод>ткtunc ( ы ок 1 пары
</ >лек тронов иона и разрыхляющей орбиты ацетилена).
А.И. 1ельбн1 тейп [ 2091 подчеркивает, ч то именно последнее тнрает ос ионную
роль в механизме каталитического воздействия сулемы. Катион pi ути имеет 5г/
HK'Kipoiibi большой подвижности, что ведет к легкой поляризуемости катион i
и большей склонности к дативному взаимодействию.
Механизм процесса, предложенный А.И. Гельбштейном [206-209|, имеет
вид:
С
I) МсС1„ + НС1 МеС1„ • НС1; (3.21а)
Л,
2) МсСЛ,, + С2Н2 МеС1„ • С2Н2; (3.21 б)
к.
3) МеС1„ • НС1 + НС1 МеС\п 2НС1; (3.21 в)
Л'4
4) МеС1„ НС1 + С2Н2 МеС1„ • С2Н2 • HCI; (3.21 г)
к5
5) МеС1„ С2Н2 НС! + НС! —— МеС\п НС! + С2Н3С1 (3.21 д)
Лимитирующей считается пятая стадия.
Согласно Р.М. Флиду [223] хлороводород обладает меньшим сродством к
С улеме. Образующийся комплекс HgCl2-C2H2, как будет показано, является од
пои из главных причин перехода сулемы в каталитически неактивную форму.
( чой точки зрения, с целью уменьшения дезактивации сулемы ацетиленом
и для сдвига реакции в сторону образования винилхлорида, необходим избы
кж НС! как компонента, обладающего меньшим сродством к сулеме. В работе
|223| приводится значение этого избытка, равное 180, и указывается, что он
практ ически неосуществим. Мерой, препятствующей образованию комплекса
с улемы с ацетиленом, является предварительное насыщение катализатора хло
роводородом.
М.И. Силингом показано, однако, на основании адсорбционных измерении,
uni ацетилен и хлороводород сопоставимы по своему сродству к сулеме |226|.
В этом случае предварительная обработка катализатора хлороводоро
дом переводит сулему в комплексы HgCl2HCl или HgCl2-2HCl и предо
। вращает тем самым образование в промышленных условиях комплекса
I lgCl2-C2H2, способствующего дезактивации катализатора. Согласно [226|
ацетилен присоединяется к комплексу HgCl2-HCl, давая смешанный комп
леке HgCl2 C2H2 HCl.
Реакции комплексообразования HgCl2-2HCl с С2Н2 и НС1, по-видимому, ма
ловероятны, поскольку в комплексе HgCl2-2HCl ослаблена акцепторная способ
пос гь катиона ртути, а контрполяризующее действие протонов этого комплекса
гикже препятствует присоединению других молекул [223].
В монографии по химии и технологии ацетилена под ред. О.Н. Темкина [ 172]
с не гематизированы кинетические уравнения, полученные различными ангора
ми для катализаторов HgC!2/AY Эти данные приведены в табл. 3.8. Уравнения I
и 2 получены, исходя из предположения, что в реакции участвуют С2Н2 и IIC1,
адсорбированные па одних и тех же (I) или ра.шых (2) центрах поверхности.
Уравнения 3 -5 предусматривают разные варили гы реакции двойных и тройных
комплексов (HgCl2-2HCl, 1^С12С21121 1С1) с газообразными молекулами ацети
лена или НС1.
Дифференцировать эти гипотезы, используя сравнение адсорбционных
характеристик, не удается. Теплоты адсорбции С2Н2 и HCI близки и лежат
в интервале 20-50 кДж/моль [206, 231, 232]. Измерения дифференциальных
теп лот адсорбции HgCl2 на АУ показали [231], что теплоты адсорбции этого
соединения (100-110 кДж/моль) существенно превышают его теплоту субли-
мации (76 кДж/моль). Из этого следует, что связь HgCl2 — АУ имеет хими-
ческую природу, и на некоторых участках поверхности появление молекуляр-
ных аддуктов предпочтительнее образования кристаллического дихлорида
ртути. Следовательно, адсорбцию реагентов целесообразно рассматривать
как реакции образования поверхностных комплексов HgCl2-HCl, HgCl2-2HCl,
HgCl2-C2H2 и др. Адсорбция НС1 обратима вплоть до 200 °C. Для ацетилена
выделяют три вида адсорбции [206]: полностью обратимую, частично обра-
тимую (в вакууме при нагревании) и необратимую. Обратимую адсорбцию
отождествляют с образованием поверхностных л-комплексов, частично об-
ратимую — с появлением CHCl=CHHgCl, а необратимую — с продуктами
олигомеризации ацетилена.
Кинетика и механизм гидрохлорирования ацетилена на ртутном катализа-
торе в безградиентном реакторе в стационарных и нестационарных условиях
изучена X. Шенкаром и Д. Агню с сотрудниками [234]. Установлено, что при
низких температурах (< 135 °C) осуществляется взаимодействие С2Н2 и НС1, ад-
сорбированных на разных центрах катализатора (см. табл. 3.8, уравнение 2), а
при более высоких — газообразного ацетилена с адсорбированным НС1. Отме-
чено, что все кинетические уравнения (см. табл. 3.8) хорошо описывают экспе-
риментальные данные.
О.Н. Темкин [172] полагает, однако, что уравнение Г. Бремера [230], описы-
вающее процесс в более широком интервале давлений НС1, является эмпири-
ческим, поскольку образование комплексов ртути (II) HgCl2-2HCl-C2H2 с коор-
динационным числом 5, да еще на поверхности активированного угля, весьма
сомнительно.
Приведем также кинетические уравнения, достаточно широко используе-
мые при расчете промышленных реакторов. В работе [235] приведены уравне-
ния вида:
г = fll0“4exp[-b/(RT)] /с2н/на (3.22К
где a, b,c,d — параметры, зависящие от условий процесса;
а = 1,01ч-3,04; b = 2474-4-3110; с = 0,654-0,79; d = 0,254-0,38.
Для разбавленного (8%-ного) ацетилена
r = k-pc2li2,
где к = 9,55exp[-3140/(RT)]-
Таблица 3.8. Кинетические модели газофазного гидрохлорирования ацетилена на ртутном катализаторе
Источник X. Ватанабе, 1959 [220] Р.Д. Весселхофт, 1959 [222] А.И. Гельбштейн, 1963 [208, 227] Нгуен Зан, 1981 [228, 229] Г. Бремер, 1985 [230]
Условия и константы t= 140 °C, /сн = 5,32-Ю5, моль/(л-ч-МПа2), ^с2н, + i’HCi= 42,7 МПа ^c2h,ci = 48,6 МПа 1 Р = 0,1 МПа, Рна = ^с,н2 t = 100 °C, к = 1,4-104, моль/(МПа2-кг-ч), ^с2н, = 4,6 МПа ^нс! = 26,7 МПа Ьс-н,а = 38,0 МПа 1 ГЧ С S СЗ 1 2S Дёе Н 2 s 0^0 о Г"4'- Он (N И 1 о И * 2 Ь ео Ш . О 40 о, т* J II О\ 5 'L н j S s б и ¥ § £ S „ рЕс > in h ^Г 1—< о. о Ш г< СО о> । ся & й к <L><4 И ОС —< Qi ° ° Дч ч ч СС _? •Ч СО II II II и —< Г-) X kg
к X 0 Л Он о S О ЕГ S X гч ч х' и1 Ч х -У £ t g х^ £ X X -* + хГ и1 Ч о' + II ч ч и' О ч о + о и X X х U сГ х + >- ч и' ч сГ + 11 к О X с «с + х' х‘ и' б' <ь, а, X" + II О Г-4 х' А) о (J -S4 и X и X + 3 о о X ч о' + О X ч и X о ^х + ч и' 1
т—< ГН ег) Ш
Факторы, влияющие на активность катализатора
Взаимодействие каталитической соли с ацетиленом
Адсорбция ацетилена на катализаторе предшествует переходу ртути в ка-
талитически неактивную форму. Механизм адсорбции ацетилена может быть
представлен следующими реакциями комплексообразования [226]:
HgCl2 + С2Н2 zs* HgCl2 • С2Н2, (3.22а)
/с2
HgCl2 С2Н2 + С2Н2 HgCl2 (С2Н2 )2. (3.226)
11ри этом делаются следующие допущения:
1) адсорбция на единицу поверхности носителя мало отличается от адсорб
ции на единицу свободной от соли поверхности катализатора;
2) на поверхности катализатора соль располагается в виде незаконченного
монослоя.
Адсорбция ацетилена на катализаторах, рассчитанная на единицу поверх-
ности, значительно больше адсорбции на чистых носителях, на которых ацети
лен адсорбируется в основном обратимо.
Адсорбция ацетилена на каталитических солях изучена в ряде работ. Мицу-
тани [221] указывает, что адсорбция ацетилена на сулеме, нанесенной на активи-
рованный уголь, уменьшается с ростом температуры до 140 °C и увеличивается с
дальнейшим ростом температуры. Из этого следует, что ниже 140 °C адсорбция
ацетилена является преимущественно обратимой, а выше — необратимой. Со-
гласно М.И. Силингу [226], роль необратимой адсорбции ацетилена резко возрас
тает при подъеме температуры от 100 к 139 °C. Если при 100 °C адсорбция ацети-
лена почти полностью обратима в широком интервале давлений (до 15-20 кПа),
то при 139 °C она становится необратимой уже при давлении 1,5-2,5 кПа.
Обратимая адсорбция протекает быстро: равновесие устанавливается за не-
сколько минут. Процесс необратимой адсорбции продолжается 20-25 ч.
На сулеме скорость обратимой и необратимой адсорбции ацетилена намного
выше, чем на других каталитических солях. По своей адсорбционной способное-'
ти хлориды располагаются в следующей последовательности катионов [226]:
в области обратимой адсорбции Hg » Bi > Cd > Zn,
в области необратимой адсорбции Hg » Bi > Zn > Cd.
Молекула каталитической соли может присоединять до двух молекул аце-
тилена. Выражение для скорости адсорбции первой молекулы ацетилена имеет
вид [233]:
г = /срс2н2(1-е)-^6. (3.23а)
где к и к1 — соответственно константы адсорбции и десорбции;
6 = V/ Vm — степень покрытия поверхности;
V — объем адсорбционного газа;
Vm — объем газа, соответствующий полному покрытию.
При к1 = 0 (необратимая адсорбция) имеем:
г/рс2н2 = k-kVIVm. (3.236)
Хара кгорной особенностью необратимой адсорбции ацетилена являспя ю,
чго но сравнению со скоростью адсорбции одной молекулы ацетилена скорое гь
присоединения второй молекулы значительно ниже и мало зависит от количес-
тва адсорбированного ацетилена [226, 236].
Константы необратимой адсорбции первого моля ацетилена равны
0,35 моль/(моль-ч-кПа) при 139 °C и 0,50 моль/(моль-ч-кПа) при 181 "С. >тн
константы на один-два порядка ниже констант скорости обратимой адсор
бции. Энергия активации необратимой адсорбции ацетилена приближенно
оценивается в 12,5-21 кДж/моль [233]. Необратимая адсорбция, по-видимому,
не является результатом одного процесса. Можно предположить переход об
рагимо адсорбированного ацетилена в необратимо адсорбированный. Энергия
активации этой реакции для каталитических солей BiCl3 и ZnCl2 оцениваекя в
63-75 кДж/моль.
При рассмотрении вопроса о роли обратимо и необратимо адсорбиро
ванного ацетилена в процессе синтеза винилхлорида предполагается, чго при
I < 200 °C тройной комплекс HgCl2-C2H2-HCl, из которого получается конем
ный продукт, образуется и за счет обратимо адсорбированного ацетилена. При
/ > 200 °C, когда обратимая адсорбция исключена, образование винилхлорида
возможно также из ацетилена, адсорбированного необратимо [226].
Можно сделать вывод о том, что адсорбция первого моля ацетилена пред
шествует переходу соответствующих комплексных соединений в металлоор
ганические (например, в (3-хлорвинилмеркурхлорид). Это является основной
причиной перехода ртути в каталитически неактивную форму. Процесс дезак
гивации сулемы может быть упрощенно записан в брутто-виде:
ЗС2Н2 + HgCl2 —* 2С + Hg + 2С2Н3С1 (3.2-1 а)
или
ЗС2Н2 + 2HgCl2 —► 2С + Hg2Cl2 + 2С2Н3С1. (3.246)
Образующиеся при адсорбции металлоорганические соединения могу г вы
ступать в роли катализатора последующей полимеризации ацетилена [237].
Содержание влаги
Непосредственное взаимодействие между ацетиленом и сулемой — н<
единственная причина перехода ртути в каталитически неактивную форму. ( о
гласно [233] существует прямая связь между содержанием влаги в катализаторе
и степенью перехода ртутной соли в неактивную форму (рис. 3.12).
Й. Янда [238] предлагает следующий механизм:
6HgCl2 + 2С2Н2 + 2Н2О —-
HgCl HgCl
I I
— HOC—c—Hg—О—Hg—С—CHC12 + 6HC1. (3.25a)
I I
HgCl HgCl
<)<>pa tyioigniii я комплекс—ди (о,а днхлорртуп. р.р дихлордп нилдичлор
pi у । ь.щегальдс! идоксиртуп.) дает полимер сое гака:
HgCl 1 HgCl HgCl 1
1 ПОС—с—Hg— 1 —о—Hg—c-Hg— 1 —o—Hg—C—CHC12. (3.256)
HgCl HgCl HgCl n
Этот комплекс разлагается на ацетальдегид, винилиденхлорид, диоксид уг-
лерода, монохлорид ртути и элементарную ртуть.
3. Чарны [239] нашел, что при использовании газов с содержанием влаги до
2 г/м’ катализатор не теряет активности в течение месяца. При введении 25 г
воды на 1 м3 ацетилена степень превращения ацетилена падает с 98% до 75%
уже в течение часа, причем несколько повторных введений влаги приводили к
необратимой дезактивации катализатора. При повышении температуры син-
теза до 180 °C процесс дезактивации резко ускоряется. Поэтому необходима
тщательная осушка реакционных газов перед подачей их на синтез. Одним из
наиболее эффективных методов осушки ацетилена является промывка серной
кислотой.
Содержание влаги в катализаторе определяется влажностью как его, так и
реакционных газов. Влажность катализатора зависит от способа его приготов-
ления и подготовки к работе. Среди этих способов имеются и довольно слож-
ные, например, пропитка активного угля раствором хлоридов ртути и церия,
сушка катализатора в течение 8 ч при НО °C и абсолютном давлении 7 кПа, а
I
Рис. 3.12. Зависимость содержания активной фазы и скорости реакции от содержания
влаги в катализаторе HgCl2/AX t = 180 °C
тагем в течение 21 ч при 175 .’НО ( и .«><<> iiihiiom (.пик ним О к)11 | ’ 1<»| <н i 1
дение хлорида ртути на носитель в юке дномидл упн-роди при • > "( | '11|нли
азота [248]; пропитка активного угля р.ювором хлоридов ртути н парня в мс
аноле с последующей сушкой при 105 °C [2421 и т.п.
Наиболее распространенным промышленным способом притоювлення
катализатора является пропитка активного угля водным, иногда подкислен
ным раствором хлорида ртути и последующая сушка полученного катали та
гора.
Указывается при этом [240,241], что пропитка активного угля таким коли
чеством раствора сулемы, которое целиком поглощается порцией угля, — не
оптимальный метод из-за неравномерного покрытия поверхности носи теля
хлоридом ртути. Этими работами были определены условия получения ката
лизатора с необходимой степенью заполнения поверхности носителя катали
тической солью. Катализатор, полученный пропиткой носителя водным рас
твором хлорида ртути, менее активен, чем катализатор, полученный из рас
твора, содержащего некоторое количество какой-то неорганической кислоты.
Это объясняется образованием неорганической кислотой с хлоридом ртути
каталитически активного комплекса, например, HgCl2-HCl, HgCl2HClO4 и г. д.
[243, 244].
Однако присутствие неорганической кислоты в катализаторе приводи! к
образованию высококипящего азеотропа вода — кислота. Последующая сушка
такого катализатора при температуре вплоть до 140 °C не обеспечивает удовлет
верительного удаления влаги. Кроме того, добавление электролита в процессе
приготовления катализатора снижает содержание ртутной соли в катализа горе
Поэтому [233] более целесообразна пропитка активного угля водным, не со
держащим неорганических кислот раствором, причем объем раствора должен
в несколько раз превышать объем пропитываемого носителя, а концентрация
его соответствовать требуемому содержанию хлорной ртути в активном угле
Наилучшие результаты достигаются при пропитке активного угля циркулиру
ющим раствором малой концентрации (1—1,5% сулемы в воде) с большой с ко
ростью циркуляции [245].
В промышленных условиях катализатор осушается в два этапа: после про
питки носителя раствором хлорида ртути и окончательно — после загрузки в
контактный аппарат перед подачей исходной газовой смеси [246].
На первом этапе осушку ведут продувкой влажного катализатора горячим
(100—110 °C) воздухом до остаточного содержания влаги в катализаторе не выше
1 мае. %. Окончательную сушку ведут продувкой азотом с начальной влажное
тью не более 1 г влаги на 1 нм3 азота при температуре в слое катализатора 130-
140 °C до выравнивания концентрации влаги в азоте до и после контактного
аппарата [233].
Нанесение сулемы возможно также из невязких растворов. Т. Парийчак
[247] показал, что природа растворителя существенновлияетнаэффективткм. п.
диффузии в порах. При этом метанол — более эффективный растворитель но
сравнению с водой и хлороформом.
Цикл приготовления катализатора должен включать стадию так пазы вас
мого «дозревания», включающую диспергирование сулемы на поверхности
до малых кристаллов, а также мягкое окисление. Согласно А.А. Панфилову с
«о
60 90
120
150
т, сут
Рис. 3.13. Зависимость степени конверсии ацетилена от продолжительности хранения
катализатора
сотр. [245], активность катализатора после трех месяцев хранения существен-
но возрастает (рис. 3.13). Более длительный срок созревания не способствует
повышению активности контакта. Авторы [233] объясняют подобный эффект
также уменьшением восстановительных свойств угля. Установлено, что свеже-
приготовленный сулемовый катализатор имеет налет каломели, образующейся
за счет восстановления сулемы. При этом существует прямая зависимость меж-
ду уменьшением восстановительной способности и увеличением содержания
сулемы. Например, содержание сулемы в катализаторе на основе угля АГН-1
составляет 79,4% от общего количества ртути, а в катализаторе, обработанном
5%-ной азотной кислотой — 93,1%. Однако условия окисления должны быть
мягкими: в противном случае за счет накопления ионных групп унос сулемы
резко увеличивается.
Состав газов
Составы применяемых в процессе газов — ацетилена и хлороводорода
влияют как на качество получаемого продукта — винилхлорида, так и на срок
службы катализатора.
Ацетилен, получаемый из карбида кальция, содержит примеси соединений
серы, фосфора, мышьяка (в основном, сероводород, фосфин, мышьяковистый
водород и др.). Эти соединения — яды для катализатора гидрохлорирования
ацетилена. Технология производства винилхлорида предусматривает очистку
ацетилена, получаемого из карбида, до суммарного содержания примесей со-
единений серы, фосфора и мышьяка порядка 30-50 ppm. Считается, что такое
количество примесей вызывает незначительное снижение активности катали-
затора в процессе работы.
Ряд производств винилхлорида базируется на ацетилене, полученном из уг-
леводородного сырья (нафты или природного газа). В таком ацетилене содер-
жаня нежелательные для процесса примеси: метил-, лил-, винил-, диметил ,
диацетиле!!, дивинил, пропадиен [249, 250]. Исследования и производственная
практика 12351 показали, чго большинство из л их нримсссй (особенно винил
ацетилен и диацетилен) являются сильными контактными ядами и быс! ро сни
жают срок службы катализатора.
В промышленности считается допустимым суммарное содержание ука ап
пых примесей порядка 0,01 об. % при практически полном отсутствии диацети
лена. Поэтому необходима тщательная очистка ацетилена перед подачей его на
синтез винилхлорида.
Промывка серной кислотой, осуществляющаяся с целью уменьшения
влажности ацетилена, является одновременно и тонкой очисткой ацетилена
от примесей высших ацетиленовых и диеновых углеводородов [249, 250, 316|.
Хлороводород, получаемый синтезом хлора и водорода, может содержать в
виде примесей хлор, сероводород, хлорное железо и др. Свободный хлор не
сколько снижает выход винилхлорида, так как при реакции с ацетиленом д;ш ।
1,2-дихлорэтилен, но он, с другой стороны, замедляет снижение активности
катализатора, так как окисляет восстановленную часть ртути в хлорид ртути.
Поэтому целесообразно иметь в хлороводороде примесь свободного хлора в
количестве, допустимом с точки зрения экономичности процесса гидрохло
рирования и безопасности его проведения. Это количество лежит в пределах
0,4-4,0 об. % [246].
Сероводород образуется в печах синтеза хлороводорода при восстановле-
нии сернистого газа, серного ангидрида и тумана серной кислоты, поступаю
щих в печи синтеза с газообразным хлором. Хлороводород, употребляющийся
в производстве винилхлорида, должен содержать минимальное количество се-
роводорода (не более 30 мг/м3) [233].
Химический состав носителя
Вид и характер применяемого носителя существенно влияет на свойства ка
тализатора, в первую очередь, на его активность, избирательность и срок служ-
бы. В качестве носителя катализатора предлагались и испытывались едва ли не
все поверхностно-активные вещества: пемза, кокс, силикагель, активный и дрс
весный уголь, кизильгур, оксид алюминия и др. [252-260].
На практике повсеместно принято применение активного угля, который,
выступая в роли донора электронов, увеличивает способность катионов ката
литической соли к дативному взаимодействию. Кроме того, в отличие от оксид
ных носителей типа А12О3, активный уголь не содержит льюисовских кислот
ных центров, способствующих полимеризации мономера и резко сокращающих
срок службы катализаторов.
Химическая связь HgCl2-AY образуется не только за счет п-электронов ipo
магических ядер, но и в результате взаимодействия с боковыми цепями послед
них, которые обладают высокой электронодонорной активностью. При разрыве
боковых цепей в угле были выделены алифатические амины (основания Льюи
са) с высокой способностью к комплексообразованию [266, 267]. Отрицатель
ное влияние указанных групп можно компенсировать рядом приемов: добавкой
аминосоединений, метилированием или введением антиоксиданта, например.
। ндрохинона. Введение элскгронодонорных групп в с груклуру активного угля
в ходе иршотопления последнего значительно улучшает свойства катализатора.
Прежде всего эго относится к добавке азотсодержащих лигандов, способных
давать п комплексы с рту гью, например, этилендиамина, гексаметилендиамина
1268-270]. Ниже приведены результаты опытов [268] по оценке стабильности
катализатора за 20 ч непрерывной работы:
Время непрерывной работы, ч .. ..............................1 20
Конверсия ацетилена, %, при использовании катализатора:
HgCl,/АР-3 ............................................... 71,66 41,58
I IgCl, /(АР-3 + ЭДА)..................................... 73,60 52,2
HgCl,/(АР 3 + ГМДА)....................................... 71,50 60,75
Этот эффект подтверждается А.А. Панфиловым [271], по данным которо-
го катализаторы на основе угля АР-В как обычного, так и модифицированного
NHз и (NH4)2CO3 показали следующую стабильность (по величине удерживаю-
щей способности сулемы): 57, 77, 78% соответственно. Введение электронодо-
норных групп осуществляется нитрованием поверхности угля с последующим
восстановлением нитрогрупп [266]; обработкой аммиаком при t > 900 °C [272]
или щелочным раствором хлорамина [273]. В ряде патентов предлагается так-
же предварительная обработка активного угля водяным паром при 800-900 °C
[274], у-лучами б0Со [275], диоксидом углерода [276].
Активный уголь в некоторой мере также катализирует реакцию образования
винилхлорида. Согласно А.И. Гельбштейну [206-208], скорость реакции гидро-
хлорирования ацетилена в присутствии чистых носителей — активированного
угля и силикагеля составляет (8-9)40 5 и 1,64 (Г5 л/(м3-ч) соответственно. А.А. Го-
лева с соавторами [261 ] приводит уравнение скорости реакции на угле АГ-2:
'=^с2н,Рнс1, (3-26)
где к - 1,29407 ехр(-500/Т), моль/(л-ч-МПа2).
В большой степени уголь катализирует побочные реакции образования
ацетальдегида, 1,1-дихлорэтана, транс-дихлорэтилена, 1,1,2-трихлорэтана.
Было найдено [238, 243], что избирательность катализатора не зависит от при-
роды активной ртутной соли, и реакции, приводящие к образованию нежела-
тельных побочных продуктов, катализируются частью поверхности активного
угля, не покрытой каталитически активной солью. Избирательность катализа-
тора определяется, в первую очередь, содержанием в угле некоторых тяжелых
металлов, особенно железа и цинка. Состав реакционного газа, полученного
на чистом угле, качественно соответствует составу смеси, полученной на ак-
тивном угле, пропитанном хлоридом цинка и хлоридом железа [111]. Увеличе-
ние содержания этих металлов в носителе приводит к увеличению количества
высококипящих побочных продуктов реакции, таких, как 1,1-дихлорэтан, 1,2
дихлорэтилены [262].
В.Е. Попов [263] указывает, что решающая роль в дезактивации катализа-
тора принадлежит именно примесям; они влияют на процесс кристаллизации
нанесенной сулемы и равномерность ее распределения. Свой вывод автор обое-
ИОВЫ К.к I >1 > III |<Н II III < ИЛЫ III ч I ИНЫМИ III > 111)1|>ОЫ1|||Н1|Ч1|| ПНИН >1Ц| III II II I 111!
двух ОНр.| Щ IX I I 'II МЧ11ЫХ hillil'lll Lllupuli Illi 1НМЫН1М |l III IIIMI.IIIIM ylliv MilpMI
АР I iii'i'iiiiii г) пи и I । inulin |>i ini I 1 I, ун ilia до Пн I । hi i niiiBrii ibcihio.
( <>i/bn пи II II /Li lyioiny | ’<> l|, предварин/н.наи шмывка yiли АР 1д
иым p.i< i вором । iHiiiiioii ми иона при ()( i iio</1сдукицси промывкой вод
ним ко||д|-Ц( а том и сушкой приводи) к снижению содержания Ре, ( и, Mg,
Al, Si в К) '() ра । Ак ihbiiocii> огмы того катали татра не менялась 111) ч. Ав
юры |26 l| полагают, что при Z < I 10 °C дезактивация катализатора происхо
ди г । данным образом за счет хемосорбции ни ткомолекулярных соединений и
образования каталитически неактивных комплексов с примесями металлов.
При I > 140 °C главная причина дезактивации — сублимация сулемы Со
держание тяжелых металлов и органических примесей может быть снижено
отмывкой углей какой-либо неорганической кислотой, в частности, соляной
[258,265].
Унос сулемы
Важной причиной «старения» катализатора является унос сулемы с повер
хпосги активного угля. Известно большое количество работ по уменьшению
уноса каталитической соли. Их авторы стремились увеличить термическую
стойкость катализатора заменой части ртутной соли другой активной солью,
образующей с сулемой комплексную соль с более низкой упругостью пара.
Предлагалось в качестве катализаторов использовать комплексные соли хло
рида ртути и хлоридов щелочных и щелочноземельных металлов, комплекс
ные соли хлорида ртути и хлоридов церия, модифицировать ртутный ката
лизатор солями железа, применять соли одновалентной ртути и т.д. [240, 252,
253, 255].
В качестве активного начала применяли органические соединения ртути
как индивидуально, так и в смеси с хлоридом ртути, трихлоридом висмута
и др. [279]. Для уменьшения уноса хлорида ртути предлагалось наносить ее на
пористый носитель в присутствии синтетических полимерных веществ, обла
дающих клеящими свойствами: солей полиакриловой кислоты, поливинилово
го спирта, полиметакриламида, поликапролактама, полиамида из адипиновом
кислоты и гексаметилендиамина, полиаминовых смол [280, 281].
Предполагалось, что полимеры будут играть роль «склеивающего мостика»
между поверхностью носителя и хлорида ртути. В качестве клеящих аген тов и<
пользовались также декстрин, жидкое стекло, крахмал.
Увеличение срока службы катализатора решалось добавлением к нему ве
1цеств, с которыми катализатор становится достаточно активным при более
низких температурах, чем обычно применяемые в процессе гидрохлорирова
ния ацетилена, например, композиций, состоящих из хлорида ртути и оксида
металла IV группы Периодической системы (тория и титана) [282-284].
Предлагались катализаторы с более активным началом, например, хлорид
ртути на активном угле [257, 285, 286], а также катализаторы, модифицирован
ные неорганическими кислотами (фосфорной, серной или хлорной) [243, 287,
2881, причем подчеркивалось, что активность модифицированного катализат
ра возрастает в ряду H3PO4,H2SO4,HC1O4, т.е. с повышением степени диссоциа
ции кислоты. Описан метод повышения активности катали «агора nyic-м пред
вариleniiiioio облучения активного угля у лучами и последующей обработкой
облученною носителя хлоридом ртути |289].
Несмотря на огромное количество предложенных и испытанных катализа-
торов, попытки создать какой-либо модифицированный катализатор оказались
неудачными. Исследования катализатора, содержащего 14% HgCl2 и 10%ТЮ2 на
активном угле АР-3 и сравнение его свойств со свойствами ^модифицирован-
ного катализатора, содержащего 10% HgCl2 на том же носителе, показали [233],
ч го на поверхности модифицированного катализатора имеются кристалличес-
кие образования HgCl2 и ТтО2, тогда как на поверхности немодифицированного
катализатора сулема располагается монослоем.
Имеют место большая летучесть сулемы с поверхности носителя и меньшая
активность модифицированного катализатора. Эти обстоятельства свидетельс-
твуют о нецелесообразности добавки модификатора (в частности, TiO2) в про-
мышленный катализатор.
Все применяемые в промышленности катализаторы процесса газофазного
гидрохлорирования ацетилена, известные в настоящее время, представляют со-
бой активный уголь, содержащий около 10% хлорида ртути без каких-либо мо
дифицирующих добавок. Такие катализаторы достаточно активны, а проблема,
которая стоит перед производством, заключается в эффективном отводе тепла
реакции с понижением максимальной температуры в слое катализатора с целью
увеличения стабильности. )
В НИФХИ им. Карпова [233] проведены исследования уноса сулемы с
промышленного катализатора в потоке азота, хлороводорода и ацетилена
(рис. 3.14«-б). Унос исследовался при линейных скоростях потока в промыш-
ленном режиме (~3 см/с).
В первом случае происходил чисто физический унос сулемы, и довольно
значительный (рис. 3.14я). В промышленных условиях катализатор, загружен
ный в аппарат, предварительно сушится в токе азота при достаточно высоких
температурах (до 140 °C) не менее суток. Эта мера, необходимая с точки зрения
уменьшения дезактивации влагой, может, однако, сама вызвать падение содер-
жания активной соли.
Найденная энергия активации десорбции сулемы возрастает при умень-
шении количества соли, оставшейся на поверхности угля (рис. 3.15). Это сви-
детельствует о неоднородности поверхности в отношении энергии ее связи с
молекулами сулемы.
В потоке хлороводорода наблюдается ничтожно малый унос сулемы, что
свидетельствует об образовании нелетучего комплекса сулемы (см. рис. 3.146).
Очевидно, что хлороводород и в условиях реакции препятствует уносу су-
лемы.
В потоке ацетилена, в противоположность десорбции сулемы в потоке хло-
роводорода, с поверхности угля за короткое время снимается практически вся
сулема (см. рис. 3.14в). Частично она уносится в виде металлической ртути.
Описанная совокупность экспериментальных данных дает возможность
предположить, что в условиях реакции гидрохлорирования ацетилена, в резуль-
тате побочных реакций ацетилена с сулемой получают металлическую ртуть и
каломель, которые подвергаются уносу наряду с самой сулемой.
Рис к 14. Унос сулемы в токе азота (а), ацетилена (б), хлороводорода (в); уд — iiccoinhi
ч.к । ь десорбирующейся сулемы
Е, кДж/моль
0,9 0,8 Уд
Рис. 3.15. Изменение энергии активации десорбции сулемы при ее уносе
Таким образом, взаимодействие сулемы с ацетиленом не только обусловли-
вает химическую дезактивацию катализатора, но и предшествует уносу части
активной соли. X
П.Г. Романков [290] указывает, что при проведении процесса в неподвиж-
ном слое катализатора одновременно с процессом десорбции сулемы, сущес-
твенным в области «горячей точки», может происходить и ее адсорбция на
участках слоя с низкими температурами. Во всяком случае, активный уголь яв-
ляется хорошим адсорбентом для паров диэтилртути, тетрахлорида углерода
и т.д. [291].
Структура катализатора и ее изменение в процессе работы
Активные угли имеют разветвленную структуру пор, в которой микропоры
являются ответвлениями от более крупных переходных пор. Это было показано
изучением влияния осажденного непористого углерода на структуру пор и ад-
сорбционные свойства углей [292].
Согласно В. Вольфу [293, 294] и М.М. Дубинину с сотр. [295], микропоры —
это щели между графитоподобными плоскостями или отдельными кристалли-
ками угля.
Возможно, что участки около устья микропор имеют повышенную хими-
ческую активность, так как здесь много краевых атомов гексагональных угле-
родных сеток. Разумно допустить, что сулема находится в устьях микропор, а
доступ молекул реагентов в поры более крупного размера неограничен.
В работах [206-209,226] выдвинуто предположение о высокой дисперсности
сулемы на поверхности угля, оно подтверждается рядом фактов. Так, измерения
распределения объема и поверхности пор по радиусам исходного угля и катали-
затора, проведенные в НИФХИ им. Карпова [233], показывают, что 70-90% их
объема и поверхности приходится на поры с диаметром меньше 6 нм. При этом
для катализаторов объем и поверхность, приходящиеся на поры с диаметром
35
Pin. 3.16. Распределение объема пор по их радиусам для исходного активированною
yi ля и катализатора
I нм, меньше, чем для исходного угля. В то же время в области пор больших
ра 1меров соответствующие кривые распределения близки (рис. 3.16). Следона
к'лык», соль в катализаторе непосредственно перекрывает устья микропор с
диаме гром 2-4 нм.
Данные 3. Чарны [239] также свидетельствуют, что устья микропор част ич
но ыкрываются сублиматом в процессе приготовления катализатора и стаио
ня геи недоступными для реагирующих веществ.
Другое подтверждение мнения о высокой дисперсности сулемы на угле дани
1>с1ультаты электронно микроскопических и рентгенографических исследона
нии, которые не обнаружили кристаллической решетки соли на поверхности
in и и геля [226].
Кроме того, в цитируемых работах отмечалось резкое уменьшение субли
мации каталитической соли, нанесенной на уголь, по сравнению с кристаллы
ческой солью. Это объясняется взаимодействием п-электронов ароматических
колец иоликонденсированных углеводородов, на которых построен уголь, к>
i ноооднымир-орбитами катиона HgI 2 * * * * * В'. Вследствие этого также предполагается
| ’ ’(>| значительное «расползание» соли по поверхности носителя.
В работах [225,296, 297] указывается, что зависимость активност и кл али ia
юра газофазного синтеза винилхлорида от содержания сулемы проходит через
максимум, и оптимальная концентрация сулемы составляет 10-15%.
к
HgCI2, %
?ис. 3.17. Зависимость (при 140 °C) наблюдаемой константы скорости от содержания
Активной сулемы
X. Коминами, К. Мицутани [296] высказали предположение, что при
-HgCi, > Сопт. образуется полимолекулярный слой, но если CHgCi2 < Сопт соль
Должна быть расположена монослоем.
Согласно [233], существует пропорциональная зависимость наблюдаемой
константы скорости к от содержания активной соли в катализаторе вплоть до
?5 мае. %. При этом сохраняется вид кинетических закономерностей (рис. 3.17).
Во всяком случае, при работе на промышленном катализаторе, где в любое
кремя цикла CHgCl2 < 10%, допущение о пропорциональности наблюдаемой кон-
станты скорости и содержания активной соли является справедливым.
Изучение свойств катализатора, приготовленного на разных образцах ак-
тивного угля [238, 243, 258], показало, что удельная поверхность, расположе-
ние и размер пор в значительной мере определяют пригодность катализатора
для процесса гидрохлорирования ацетилена. Активный уголь должен обладать
Определенной механической прочностью, не истираться в процессе синтеза ви-
нилхлорида, иметь достаточно крупные (2-5 мм) частицы и однородный грану-
лометрический состав, чтобы не создавать значительного сопротивления пото-
ку газов.
Исследования Й. Янды [238] показали, что, чем больше удельная поверх-
ность носителя, тем активнее катализатор, но тем меньше срок его службы, что
объясняется повышением десорбции сулемы с поверхности носителя. Наибо-
лее пригодным, согласно этим исследованиям, оказался уголь с удельной повер-
хностью в пределах 500-600 м2/г: основное влияние имеет объем переходных
нор и макропор, ввиду того, что устья микропор частично закупориваются суб-
лиматом в процессе приготовления катализатора.
I I IM''ll line lull .1 III I IГII < I'oil II I II Hili >1 III I »l »\ i III Hl III Ни, II <lib I Hoi III. Ill I III Ml
|>ll 1.1Ц1 It'll .11(1'111/ICII.I l.l i *H I I ipiH у 11 IIH 11НЦ11 Ч II HIM lipilMI I III (Д1ыН< III'» «Il
1111».11(1'111/1111.1 И 1.Д.). < Mip.l lyiOIHiH'i II i Mli/inoOp.l IIII.IC 11117(11111'111111 pt IKl> yMCIII.
iii.uni HiiHcpxiHH и. и iiopiiciiii и. K.it.i/iii i.iiop.i и । ii/ii.iin iiihp.imaioi < рок его
служим |233|. Ниже нредсlan/ieiH.i некоюрыс хар.ж icpiic i ihui oip.1001.1111101 о
к.нал и загора, па котором происходило । и дрохлорирона I Hie .щешлена с содер
капнем примесей: диацстилена — до 0,1%, мегилацс'1 илеиа — до 0,2%, вини
лацешлеиа — до 0,05%, димегилацегилена — до 0,03%, бутадиена — до 0,01%,
пропадиена — до 0,07%, этилацетилена — до 0,05%:
К.пилизагор .. Свежий Отработанный, проба 1 /проба 2
( ЛОЙ Верхний Средний Нижний
(1 м) (2 м) (Зм)
I одержание, %:
сулемы общее 9,0 3,08/3,36 3,88/3,67 7,27/4,24
сулемы каталитически активной 9,0 0,07/* 0,09/* 0,52/’
смолообразных соединений - 0,076/0,070 0,273/0,080 0,338/0,070
NH/ . 0,003/0,04 0,031/0,04 0,019/0,017 0,047/0,017
SO.!2 - - - 0,006
1 IlIBCpXHOCTb, м2/г .... 519 30/40 36,6/47,0 30/45
11ористость (сумма макро и переходных пор), см ’/г .. ... .0,350 0,060/0,060 0,037/0,090 0,084/0,080
Измерения не проводились.
Удельная поверхность и пористость отработанного катализатора намного
ниже, чем свежего катализатора: 30-47 м2/г вместо 519 м2/г и 0,034-0,09 см 7г
вместо 0,35 см3/г соответственно.
В отработанном катализаторе содержится значительное количество ноли
меров (0,07-0,34%), которые можно частично извлечь бензолом.
Сокращение срока службы катализатора в значительной степени опреде-
ляется изменением его структуры, загрязнением активной поверхности осмо
лившимися веществами и т. д., а не только химической дезактивацией и уносом
сулемы. Именно поэтому не нашли производственного применения предложе
ния, направленные на регенерацию частично дезактивированного катализате»
ра: обработка катализатора с пониженной активностью хлором при высокой
температуре непосредственно в контактном аппарате [298]; применение смеси
исходных газов (ацетилена и хлороводорода), насыщенной парами ртути; не
прерывное или периодическое нанесение на катализатор небольших количеств
ртути или хлорида ртути [255, 299] и др. Немаловажными причинами отказа от
предложенных методов регенерации катализатора являются сложность и доро
говизна их реализации.
Итак, можно резюмировать, чю падение активности клали iaгора 061.
яснясчся переходом ртути в каталитически неактивную форму, уносом части
активной соли и изменением пористой структуры зерна. Взаимодействие ка-
талитической соли с ацетиленом (особенно в присутствии высших гомологов
ацетилена и влаги) является фактором, не только обусловливающим переход
ртути в неактивную форму, но и предшествующим уносу части активной соли
в виде ртути и каломели).
Технологические аспекты процесса
гидрохлорирования ацетилена
В промышленности процесс парофазного гидрохлорирования ацетиле-
на осуществляется в трубчатых контактных аппаратах с трубками диаметром
50 мм (рис. 3.18).
Авторами работы [233] исследовано влияние разных параметров на профи-
ли температур и степени превращения исходных реагентов: температуры хла-
доагента tK, °C; линейной скорости (нагрузки) реакционной смеси, V, м/с, отне-
сенной к полному сечению аппарата и приведенной к нормальным условиям;
мольного избытка хлороводорода по отношению к ацетилену, (3; давления ре-
акционной смеси р Па; длины участка с катализатором уменьшенной активнос-
ти fH, м; начальной температуры реакционной смеси tH, °C; мольного избытка
инертных газов по отношению к ацетилену у (при гидрохлорировании концен-
трированного ацетилена можно принять у — 0). 7
Повышение температуры хладоагента (рис. 3.19, 3.20) увеличивает макси-
мальную температуру в реакторе. Одновременно происходит смешение к нача-
лу трубки области высоких температур и уменьшение длины участка, на кото-
ром происходит практически полное превращение.
Рассмотрение профилей степени превращения показывает, что во всем диа-
пазоне изменения температур хладоагента полное превращение достигается не
в узкой области высоких температур (30-40 см), а на участке большей длины
(1,5-2,0 м).
Повышение нагрузки реакционной смеси (рис. 3.21) при условии неравно-
мерного распределения активности по слою катализатора сдвигает область мак-
симальных тепловыделений процесса в участок слоя с большей каталитической
активностью. Именно поэтому, несмотря на то, что процесс гидрохлорирова-
ния протекает в кинетической области, при повышении нагрузки реакционной
смеси максимальная температура в слое возрастает (иными словами, тепловы-
деление в реакторе с повышением нагрузки растет быстрее, чем теплоотвод).
При этом практически полное превращение осуществляется на большем учас-
тке трубки.
Увеличение длины участка Сн с катализатором уменьшенной активности
является естественным следствием «старения» катализатора. Со временем об-
ласть точки М (рис. 3.22), до которой находится катализатор с уменьшенной
активностью, передвигается к концу трубки. Участок трубки, на котором осу-
ществляется полное превращение, при этом расширяется. Максимальная тем-
пература снижается, так как тепловыделение происходит теперь на большем
Рис. 3.18. Промышленный реактор получения винилхлорида парофазным гидрохлори
цоканием ацетилена
Рис. 3.19. Распределение температуры и конверсии по длине реактора (по модели идс
ального вытеснения); £н = 1 м
Рис. 3.20. Распределение температуры по длине реактора (с учетом переноса вещества
и тепла):
Гх = 90 °C (1-3), 110 °C (4), 130°С (5); р = 1 (1), 0,4 (2), 0 (3-5)
участке трубки. Происходит также смещение максимальной температуры к
концу трубки (рис. 3.23).
Увеличение давления, как следует из кинетического уравнения, ведет
к увеличению скорости реакции, к росту тепловыделения и температуры
(рис. 3.24). При этом практически полное превращение должно достигаться
при меньшем времени контакта. Для промышленного реактора, в котором
лимитирующим является отвод тепла, переход на повышенное давление не-
целесообразен.
I’m. *.21, Распределение температуры по длине трубки (с учетом переноса нещеста и
li 11(1.1)
Рис. 3,22. Профиль каталитической активности по длине реактора (7н — длина уча< к
<. лом катализатора с уменьшенной активностью, м)
Изменение начальной температуры реакционной смеси от 20 °C до 120 °
практически не влияет на распределение температуры и степени превращена
но длине реактора (рис. 3.25), когда длина участка, занятого катализатором
уменьшенной активностью, не менее 1 м, так как количество тепла, идущее i
нагрев реакционной смеси, мало по сравнению с тепловыделением в промыв
ленном реакторе.
Повышение избытка хлороводорода уменьшает адиабатический ра ин ре
снижает температурный максимум, одновременно сдвигая его к концу гру
0 -I--------------1------------1-------------1-------------1------------1------------1—
О 0,5 1 1,5 2 2,5 3
I, м
1
Рис. 3.23. Изменение профиля температур по мере работы катализатора
ки (рис. 3.26). Существование избытка хлороводорода, оптимального в дан-
ном температурном режиме, отмечено в работе [282]. Расчеты показывают, что
максимальная степень превращения достигается при избытке хлороводорода,
равном 1,31-1,33 (рис. 3.27). Этот оптимум имеет место в широком диапазоне
температур и нагрузок, однако он довольно пологий и поэтому, использование
повышенного, по сравнению с обычно применяемым в производстве (1,1-1,2),
избытка хлороводорода вряд ли целесообразно.
Таким образом, наиболее существенными управляющими параметрами
процесса являются нагрузка реакционной смеси и температура хладоагента.
Рис. 3.25. Влияние изменения начальной температуры 1Н = 40 °C (I), 50 °C (II), 70°( (III).
100 °C (IV) смеси газов; р = 1,14; р = 130 кПа, tx = 90йС
Рис. 3.26. Положение температурного максимума в зависимости от избытка IICI
Промышленный процесс гидрохлорирования ацетилена проводится в га
юной фазе в присутствии катализатора. Для достижения высокой конверсии
исходных реагентов (98-99%) и селективности (> 99%) в качестве катализатора
применяется дихлорид ртути, нанесенный на активный уголь. Как нами ука <а
но, активный уголь в этой каталитической системе является не инертным носи
1 елем, а активным компонентом, и поэтому его химическая природа и струк гура
гаметно влияют на свойства катализатора. Для промышленного катали га гора
важнейшими являются экономические показатели — стабильность катали га го
ра, его производительность и селективность. Эти показатели определяются н
основном дезактивацией катализатора, связанной с уносом и восстановлением
дихлорида ртути до металлической ртути, что в определенной степени гависн i
Рис. 3.27. Изменение температурного максимума (7) и конверсии в зависимости от из-
бы гка НО на выходе из трубки реактора (2)
о г природы и структуры носителей. Срок службы катализатора в промышлен-
ных условиях колеблется от 6 месяцев до года.
Структура носителя определяется его пористостью, т.е. наличием макро-,
микро- и переходных пор. Линейные размеры участвующих в реакции гидро-
хлорирования молекул по расчетам, пм: Гс2н3с1 = 816, Л:2н2 = 581 и гнс1 = 472, а
образующийся по реакции промежуточный л-комплекс имеет линейный раз-
мер не менее 1,0—1,2 нм. Следовательно, микропоры с диаметром менее 1,0 нм
не могут участвовать в процессе гидрохлорирования. Преобладающая роль в
этом процессе принадлежит переходным порам: чем больше переходных пор,
тем активнее адсорбируется дихлорид ртути и тем активнее и стабильнее ката-
лизатор. Химическая природа носителя определяется наличием поверхностных
функциональных групп: карбоксильных, карбонильных и гидроксильных (фе-
нольного и спиртового типа) и др. Увеличение содержания карбонильных групп
понижает стабильность и активность катализатора, по-видимому, за счет спо-
собности к восстановлению дихлорида ртути вплоть до металлической ртути, а
фенольные группы могут способствовать увеличению стабильности за счет их
окисления до хинонов.
Для увеличения стабильности ртутного катализатора гидрохлорирования
ацетилена на специально приготовленный активный уголь вместе с хлоридом
ртути (II) наносят органические амины и их соли. Из за высокой активности
ртутного катализатора использование его кинетических возможностей весьма
сложно. Потому, что, с одной стороны, реакция гидрохлорирования ацетилена
весьма экзотермична, а с другой — из-за высокой летучести дихлорида ртути
максимальная температура реакции ограничена 150-180 °C.
Проблема теплосъема, который и определяет ограничение по нагрузке на
реактор, может быть решена реакцией гидрохлорирования в псевдоожижен-
ном слое ртутного катализатора, промотированного добавкой хлорида лантана
[315]. С целью исключения вредного влияния неорганических примесей (со-
единений кремния, алюминия, железа), присутствующих в активном угле, пос-
ледпии перед пропи к<>й дихлоридом ргуги отмывают минеральной кислотой
|26-1|. Однако такой процесс имеет недоспаточно высокую конверсию ацетиле
на — 80 90% за проход, по сравнению с неподвижным слоем, хотя с ьсм винил
хлорида с единицы катализатора возрастает в 5 7 раз.
Для обеспечения надежного теплосъема при проведении реакции в испод
вижном слое используют трубчатые реакторы типа теплообменника. В груб
ках помещается катализатор, а в межтрубном пространстве — иода или opi а
пический теплоноситель [317]. Число трубок в реакторе около 1000, диаметр
S0 80 мм, длина 3-6 м, что определяется конструкционными ограничениями.
Такие реакторы имеют мощность до 10 тыс. т винилхлорида в год Даже при ис-
пользовании трубок малого диаметра реакция идет в относительно узкой зоне,
которая по мере срабатывания катализатора (унос дихлорида ртути) перемета
ется вдоль трубки по ходу потока реагентов.
Предлагалось использовать в качестве контактного аппарата плоские ка
меры длиной 4 м и поперечным сечением 18x50 мм или узкие вертикальные
пространства, образованные большим количеством параллельных пластин па
небольшом расстоянии одна от другой. Эти пространства, заполненные катали
татором, пересекаются горизонтальными трубами с охлаждающей жидкос тью,
образуя так называемую «печь Фишера» [326-328]. Для более равномерного
распределения температур по длине реактора предлагалось заполнять трубки
реактора катализатором с увеличивающейся концентрацией каталитической
соли от входа в реактор к выходу из него [240]. Для увеличения выхода продук
। а и срока службы катализатора возможно изменение направления потока газов
через реактор каждые 24 ч [329].
Снижение температуры в слое катализатора возможно путем организации
процесса в несколько стадий. Катализатор располагают по высоте в несколь
ко слоев, образующих зоны реакции. Температура реакционных газов между
соседними зонами снижается посредством добавления холодного газа, в ка
честве которого может быть использован либо свежий ацетилен, либо часть
непрореагировавшего газа, выделенного после конденсации винилхлорида.
Добавление холодного газа предполагается в таком количестве, чтобы пере-
пад температуры по длине реактора был небольшим (1-3 °C). Уменьшение
активности катализатора во времени компенсируется повышением рабочей
температуры [330, 331].
Возможен также ступенчатый ввод в систему хлороводорода. Каждая сту
пень — трубчатый реактор, охлаждаемый кипящей жидкостью с постоя! и юн
температурой. При применении трехступенчатой схемы в первый по ходу газа
реактор вводится все необходимое количество ацетилена и Уз необходимого для
процесса количества хлороводорода. В каждый из последующих реакторов до-
бавляется по Уз количества хлороводорода. Температура во всех реакторах оди
наковая [332]. Другой ступенчатый способ предусматривает выделение винил
хлорида из реакционного газа после каждой ступени и поступление оставшихся
ацетилена и хлороводорода в следующий контактный аппарат. Указывается па
значительное снижение температуры реакции в таком процессе по сравнению с
одностадийным [333].
Можно организовать процесс в несколько стадий с периодической сменой
места ввода реакционной смеси в систему. Реакционная смесь проходит чсрс i
гри (как минимум) последовательно расположенные камеры Koi да конверсия
ацетилена снижается до определенно!о уровня, место подачи комноненгон пе-
ремещается от первой камеры к последующей, в конце подключается камера со
свежим катализатором, а первая камера отключается. Этим способом можно
увеличить удельную производительность до 100 г/(лкат-ч) [253].
По другому методу предварительно нагретая до 50-100 °C реакционная
смесь ацетилена и хлороводорода сначала поступает в охлаждаемую зону с
обычным катализатором (хлорид ртути на активном угле), после чего по-
падает во вторую обогреваемую зону реакции с более активным катализа-
тором (например, ванадат ртути на активном угле). В первой и во второй
зонах поддерживаются температуры 110-170 °C. Затем газы охлаждаются и
пропускаются через слой активного угля для адсорбции испарившегося ка-
тализатора [296].
Срок службы катализатора может быть увеличен посредством тщательного
регулирования температуры хладоагента. Указывается, что существуют опти-
мальные температуры хладоагента и имеется зависимость между этими темпе-
ратурами, скоростью подачи реакционной смеси и скоростью движения горя-
чей точки по слою катализатора [334].
Известны производственные установки, применявшие некоторые из
описанных способов. На ряде реакторов завода «Винилак» (Франция) был
предусмотрен секционированный отвод тепла реакции — имелись шесть
выводов хладоагента (воды) по высоте реактора. Однако опыт показал, что
регулировка подачи воды не дает возможности в данном случае воздейство-
вать на температурный режим гидрохлорирования из-за больших пропус-
ков воды через кольцевые отверстия у труб в перегородках межтрубного
пространства [233].
Американская фирма «General Tire and Rubber» применяла системы гидро-
хлорирования, состоящие из пяти реакторов, установленных по ходу газа: два
параллельно, затем еще два параллельно и в конце — один реактор Каждый
реактор — кожухотрубчатый теплообменник, состоящий из 200 труб с внут-
ренним диаметром 50 мм и длиной 5 м каждая. Указывается, что удельная про-
изводительность катализатора в такой системе в 3-^4 раза выше, чем в обычных
реакторах, работающих в одну стадию [335].
Согласно [318], при дифференцированном по зонам теплосъеме производи-
тельность реактора может быть заметно увеличена. Производительность реак-
тора может быть увеличена и в условиях изотермического режима при заполне-
нии реактора двумя слоями катализатора: первый слой содержит 15-20% дихло-
рида ртути на активном угле, а второй слой, стабилизированный хлороксидом
цинка, содержит 1-5% дихлорида ртути на силикагеле. Процесс проводится при
90-250 °C и 0,01-0,15 МПа [319]. Вместо двух слоев катализатора используется
и прием последовательного включения двух реакторов: исходная смесь вначале
реагирует не полностью при 110-150 °C в первом реакторе, заполненном ис-
пользованным катализатором, а затем при 70-115 °C во втором реакторе, запол-
ненном свежим катализатором [320]. Достижение хорошей стабильности ката
лизатора и быстрого выхода на рабочий режим возможно при подаче в реактор
предварительно нагретых реакционных газов (100-110 °C). Процесс начинается
при н 1<>ы । ке хлорида водорода или инертного ia ы, ооеснечивающнх он шмаль
IIУ К» OOl.CMIiyiO скорое I !>.
Весьма интересным приемом, позволяющим при улучшении рабочих харак
1срисгик сократить расход ртути почти в два раза, является разбавление кп
iализатора — смеси активного угля, пропитанного 8-15% дихлорида ртути, с
чис тым активным углем при соотношении объемов (2 1):(I —2), причем среднее
содержание дихлорида ртути в смеси в этом случае должно быть более 4 мае. %
[321]. Используется и разбавление катализатора инертным материалом, причем
ра давление осуществляется только в первом по ходу газа слое катализатора.
. )го снижает местные перегревы и предотвращает быструю дезактивацию ка
।ализатора. Процесс проводится при изменяющемся вдоль слоя катализатора
температурном режиме [322].
Изучение влияния концентрации дихлорида ртути на срок службы ката
лизатора показало, что начальная каталитическая активность и последующее
уменьшение степени превращения зависят от начальной концентрации дихло
рида ртути, которая должна быть 10-14% [323].
При проведении реакции газофазного гидрохлорирования ацетилена па
дихлорид-ртутном катализаторе неизбежен унос дихлорида ртути. Для очис i ки
реакционного газа от дихлорида ртути используют адсорбцию на активном угле
или абсорбцию крепкой соляной кислотой. В последнем случае использование
солянокислого раствора хлорида железа позволяет очистить реакционный газ
не только от дихлорида ртути, но и от ртути [324].
Технологическая схема процесса газофазного
гидрохлорирования ацетилена
Углеводородное сырье для синтеза винилхлорида — ацетилен — получают
из карбида кальция или высокотемпературным пиролизом природного газа
(либо углеводородов нефти). Полученный ацетилен очищают от соединении
фосфора, серы, аммиака и высших гомологов ацетилена. При необходимости
(в случае получения ацетилена пиролизом углеводородов) его концентрирую!
сольвентным способом, компримируют до 0,52 МПа и сушат до остаточно! о со
держания влаги менее 1,5 г/м3 обычно в две стадии: предварительно — путем
конденсации влаги в рассольных холодильниках и окончательно — используя
кусковое твердое едкое кали [325] либо концентрированную серную кисло!у
[251]. Второй способ более эффективен и экономичен.
Перекачивают ацетилен мокрыми ротационными компрессорами с водя
11 ы м поршнем. Схема процесса получения винилхлорида газофазным гидрохло
рированием ацетилена представлена на рис. 3.28.
Осушенный ацетилен направляют в смеситель 1, куда также подаюг пред
варительно очищенный и осушенный хлористый водород. Соотношение аце
। илен : хлороводород составляет обычно 1,0 : (1,1-М,05). Смесь газов далее пос
I упает в верхнюю часть реактора 2, представляющего собой кожухотрубчатый
1сплообменник, в межтрубном пространстве которого циркулирует теп лоно
ситель, а в трубках находится катализатор — активный уголь с нанесенным на
него (10-12%) дихлоридом ртути. Реактор изготовлен из углеродистой стали;
Кмщтршя
Глава J Химии и технологам получении euuuixaopu1
I 147
высота трубок 3-6 м, диаметр 50 80 мм Обычно в реактор 1агружаю1 6 I ’ м'
катализатора. Температура в реакционной зоне 150 180 °C.
Затем реакционные газы последовательно поступают в колонны очист
ки 1 6. После реактора газы проходят через колонну 3, орошаемую 10% и
соляной кислотой, для извлечения унесенного дихлорида ртуш. Затем реак
циопный газ промывают последовательно в двух колоннах: колонна / про
нас гея 5-7%-й соляной кислотой, образующейся в колонне 5, орошаемой во
дои. В колонне 4 получается -18 20%-я соляная кислота. Количество 30% и
соляной кислоты (колонна 3) составляет -10-15% от общего количества об
ра тующейся в процессе соляной кислоты. Колонна 6 орошается раствором
щелочи для удаления из газа хлористого водорода, ацетальдегида и диоксида
yi лерода. Нейтрализованный винилхлорид-сырец содержит многочисленные
примеси, от которых он должен быть очищен. Характер примесей зависит от
ме года получения ацетилена, применяемого для синтеза винилхлорида. Ос
полные из них: ацетилен, 1,1-дихлорэтан, транс-1,2-дихлорэтилен, ацеталь
дегид, трихлорэтилен, винилиденхлорид, хлоропрен, винилацетилен, 1,2 и
1,3 бутадиен и др. Далее газ «захолаживают» в конденсаторе 7, охлаждае
мом рассолом, для удаления влаги, компримируют в компрессоре 8 до 0,71
0,81 МПа и направляют на ректификацию. Система ректификации состоит
и i двух тарельчатых колонн: на первой из них (9) выводятся высококипящие
примеси, в основном, 1,1-дихлорэтан, на второй (10) — низкокипящие при
меси. Полученный ректификат проходит колонну осушки готового продукта
/ /, та полненную твердым гидроксидом натрия, для окончательной осушки и
ней грализации винилхлорида.
Выходящий из ректификационной системы винилхлорид по качеству при
годен для последующей полимеризации и имеет (при переработке пиролизного
ацетилена) следующий примерный состав (мае. %): винилхлорид — не менее
94,98, ацетилен — не более 3-Ю"4, ацетальдегид — не более Ю-Ю"4, хлороводо
род — не более 2-Ю 4, железо — не более 1-10 4, перекисные соединения — не
более 8,5-1 О', пропилен — не более 10-Ю’4, аллен — не более 3-10 4, метилхло
рид — не более 120-Ю"4, 1,3-бутадиен — не более 15-10 4, винилацетилен — не
более 4-Ю"4, хлористый этилен — не более 4-10"4.
1еоретический расход основного сырья (кг на 1 т готового продукта): аце ги
леи — 416, хлороводород — 584.
Реально расходные коэффициенты (кг на 1 т винилхлорида): ацетилен
125, хлороводород — 610, катализатор — 1,0.
( рок службы катализатора составляет обычно 9-12 месяцев. Замена ката
ли штора в промышленном реакторе обусловлена снижением содержания ак
in иною компонента — сулемы за счет ее уноса. По мере снижения содержа
пня сулемы температура процесса повышается со 150 до 220 °C. Катализатор
и । реактора обычно выгружают при остаточном содержании сулемы на уровне
2,5-3 мае. % (в свежем катализаторе 10-12 мае. %). Отработанный катализатор
направляют на ртутные комбинаты для переработки (сжигания) с выделением
ме галлической ртути.
11олучение винилхлорида гидрохлорированием ацетилена находит свою ре
алн 1ацию в промышленности также и в структуре комбинированных ацетилен
ннлеповых схем Ппм этом прпртЛп------------------------------
лнпыи, гак и разоавдсплыи ацетилен lexno/ioi мческие особенное ги процесса
ереработки koi щен грироваиного сырья ничем не отличаются oi отдельно язя
эго процесса, полому ниже мы приводим лишь принципиальную технологи
ескую схему процесса и ее описание.
Комбинированный процесс получения винилхлорида из концентрированных
цетилена и этилена. Он состоит из шести стадий:
I) синтез и очистка 1,2 дихлорэтана;
2) дегидрохлорирование 1,2-дихлорэтана с получением винилхлорида и
лороиодорода (химические и технологические особенности процессов синтеза
ихлорэтана и его дегидрохлорирование рассматриваются ниже в разделе «По-
учение винилхлорида по сбалансированной схеме»);
3) разделение продуктов дегидрохлорирования;
4) синтез и очистка ацетилена;
5) синтез винилхлорида из ацетилена;
6) ректификация винилхлорида.
Технологическая схема производства винилхлорида комбинированным ме-
пдом из концентрированных этилена и ацетилена приведена на рис. 3.29.
Исходные газы (этилен и хлор) влажностью не более 20 млн 1 поступа-
и в стальной цилиндрический аппарат — реактор 1, заполненный жидким
,2-дихлорэтаном. Хлорирование проводят в присутствии катализатора —
лорного железа. Образующийся 1,2-дихлорэтан вместе с 1,2 дихлорэтаном,
извращаемым со стадии термического дегидрохлорирования, подвергается
ектификации на двухколонной системе. На первой колонне 2 отделяются
изкокипящие примеси, в основном дихлорэтилены и хлористый этил, а на
торой колонне 3 отделяются высококипящие примеси, в основном полихло-
иды этана.
Полученный 1,2 дихлорэтан-ректификат подвергается дегидрохлорирова-
нию в трубчатой печи 4 змеевикового типа, обогреваемой панельными горел-
ами. Диаметр трубок 100-200 мм (определяется производительностью), мате-
риал — сталь, преимущественно ХН78Т или ОХ18Н12Б. Температура процесса
егидрохлорирования 400-500 °C. Степень превращения 1,2-дихлорэтана 50-
0%.
Более подробное описание технологических процессов хлорирования эти-
ена и дегидрохлорирования дихлорэтана приведено в разделах 6 и 7.
Образующийся пирогаз быстро охлаждается в специальной колонне 5, где
гделяются осмоленные продукты пиролиза. Далее пирогаз поступает в ректи-
>икационную колонну 6 для выделения чистого хлористого водорода, передава-
мого на стадию гидрохлорирования ацетилена. Кубовые продукты колонны 6
оступают на ректификацию в колонну 7 для выделения товарного винилхло-
ида. Кубовая жидкость этой колонны, состоящая в основном из 1,2 дихлорэта-
а, возвращается на ректификацию в колонну 2.
Хлороводород с верха колонны 6 поступает в смеситель 9, куда одновремен-
о подается предварительно очищенный, осушенный и скомпримированный
цетилен. Смесь газов поступает в реактор гидрохлорирования 10 и далее про-
водится по схеме, описанной в разделе «Гидрохлорирование ацетилена».
Винилхлорид-ректификат после колонн 7 и 17 проходит колонну осушки 8,
аполненную едким натром, и передается на склад.
Рис. 3.29. Принципиальная схема получения винилхлорида комбинированным методом из концентрированного этилена и аце-
тилена
1 — реактор синтеза ДХЭ; 2, 3, 6, 7, 16, 17 — ректификационные колонны; 4 — печь дегидрохлорирования ДХЭ; 5 — закалочная колонна;
8 — колонна осушки готового продукта; 9 — смеситель; 10 — реактор; 11, 12, 13 — колонны очистки реакционного газа: 14 — конденсатор;
15 — компрессор
Г<ч>рстический/реалы1ый расход основного ci>ipi.»i (ki на 1 i гогоиого про-
дукта): ацетилен — 208/215, этилен — 221/235, хлор — 568/590.
Болес конкурентоспособным с экономической точки зрения процессом по
лучения винилхлорида, использующим ацетилен, является комбинированный
процесс на основе легкого углеводородного сырья (нафгы, пропан-бугановой
фракции, прямогонного бензина).
Процесс получения винилхлорида комбинированным методом из бензина. Он
сос той г из девяти стадий:
1) пиролиз прямогонного бензина;
2) компримирование, очистка и осушка пирогаза;
3) гидрохлорирование разбавленного ацетилена;
4) абсорбционное улавливание винилхлорида после реактора гидрохлори-
рования;
5) хлорирование разбавленного этилена до 1,2-дихлорэтана;
6) ректификация 1,2-дихлорэтана;
7) термическое дегидрохлорирование 1,2-дихлорэтана;
8) разделение продуктов дегидрохлорирования 1,2-дихлорэтана;
9) десорбция винилхлорида из 1,2-дихлорэтана и ректификация винилхло-
рида.
Примерный состав крекинг-газа, направляемого затем на синтез винил-
хлорида, об. %: 22,0 СО2, 16,6 СО, 31,9 Н2, 9,0 СН4, 8,1 С2Н2, 8,6 С2Н4, 3,2 N2,
0,6 О2.
Гидрохлорирование разбавленного ацетилена в крекинг-газе указанного со-
става протекает селективно: при 180 °C этилен практически не вступает в реак-
цию гидрохлорирования. Это объясняется существенной разницей в значениях
констант равновесия указанных реакций: для ацетилена Rp = 105, для этилена
Кр = 3,96, а также тем, что дихлорид ртути неактивен в реакциях газофазного
гидрохлорирования олефинов, в частности, этилена.
Так как процесс основан на избирательной реакции гидрохлорирования
ацетилена в смеси газов, большое значение имеет качественный состав газов
крекинга нафты. Наличие в газах крекинга повышенного количества высших
ацетиленов приводит к резкому сокращению срока службы катализатора. При-
чиной этого являются сильные восстановительные свойства высших ацетиле-
нов. Аналогичным образом недопустимо увеличение объемной доли ацетилена
выше 1,02:1 по отношению к НС1. По этой причине при остановке реакторов
гидрохлорирования для сохранения активности катализатора необходимо от-
дуть реакционный газ азотом.
Присутствие влаги в газах крекинга нафты приводит к образованию аце-
тальдегида по реакции:
О
//
СН=СН + Н2О— СН3—С—Н, (3.27)
что также снижает активность катализатора и ухудшает качество винилхлорида.
На катализаторе и показателях процесса в целом также негативно сказыва-
ется попадание с газами крекинга тумана серной кислоты и керосина, использу-
ющихся для осушки и очистки крекинг-газа.
Рис. 3.30. Принципиальная схема получения винилхлорида комбинированным методом из бензина:
1 — смеситель; 2 — реактор-гидрохлоратор; 3 — абсорбер; 4 — холодильник дихлорэтана-абсорбента; 5 — реактор-хлоратор; 6 — конденсатор;
7, 8, 11, 12, 13, 15, 16 — ректификационные колонны; 9 — печь дегидрохлорирования дихлорэтана; 10 — закалочная колонна; 14 — щелочной
скруббер
Icxiio/ioi ическая схема получения винилхлорида комоинировапиым ме-
годом из бен шпа показана на рис. З.ЗО Исходное сырье — бензин, легкий не
||няной погон с темпера гурой кипения 30 180 °C подвергаемся пиролизу при
1000 1200 °C в аппарате специальной конструкции. Крекинг газ закаливается
водой, очищается от сажи и компримируется до 0,71 МПа.
Далее крекинг-газ очищается от высших гомологов ацетилена и олефино
вых углеводородов С3-С4 и сушится (стадия получения и очистки крекинг-газа
па схеме не показана).
Очищенный крекинг-газ, содержащий 8-10% ацетилена и столько же этиле-
на, смешивается в смесителе 1 с хлороводородом, поступающим со стадии де-
гидрохлорирования 1,2-дихлорэтана, в соотношении, обеспечивающем 1-2%-й
избыток ацетилена против стехиометрического. Смесь затем направляется в ре-
актор гидрохлорирования 2 — стальной кожухотрубный аппарат. Трубки аппа-
paia заполнены катализатором — активированным углем с нанесенной на него
сулемой в количестве до 15%. В межтрубном пространстве аппарата циркули-
рует теплоноситель, с помощью которого температура в реакторе поддержива-
ется равной 220 °C. Реакция гидрохлорирования проводится при давлении до
0,61 МПа, степень конверсии ацетилена близка к 100%.
Реакционный газ после реактора поступает в абсорбер 3, орошаемый 1,2-
дихлорэтаном, захоложенным рассолом в холодильнике 4. В абсорбере 3 из ре-
акционного газа улавливается винилхлорид. Из абсорбера 3 реакционный газ
поступает в реактор хлорирования 5 — стальной цилиндрический аппарат, за-
полненный жидким 1,2 дихлорэтаном. В реактор 5 одновременно подается хлор
в соотношении, обеспечивающем 2-3%-й избыток этилена против стехиомет-
рического.
Хлорирование осуществляется в присутствии катализатора — хлорного
железа при давлении —0,51 МПа в кипящей реакционной массе. Выходящие из
реактора газы охлаждаются в конденсаторе 6. Здесь конденсируется образовав-
шийся в результате реакции 1,2-дихлорэтан, а остаточные газы, пройдя систему
щелочной очистки (на схеме не показана), используются как топливный газ на
стадиях пиролиза бензина и дегидрохлорирования 1,2-дихлорэтана.
1,2 -Дихлорэтан-сырец после конденсатора 6 идет на ректификацию в сис-
тему двух колонн: в первой колонне 7 выделяются низкокипящие примеси, а
во второй колонне 8 — высококипящие примеси. 1,2-Дихлорэтан-ректифи-
кат со степенью чистоты не ниже 99,8% передается на дегидрохлорирование
в печь 9 — трубчатый аппарат змеевикового типа с панельными горелками;
материал змеевика — сталь 0Х18Н12Б. Дегидрохлорирование ведется при
р = 1,01 МПа и t ~ 550 °C. Степень конверсии 1,2-дихлорэтана за проход со-
ставляет около 70%.
Реакционные газы, выходящие из печи, поступают в закалочную колонну 10,
в которой отделяются смолистые вещества, и далее в колонну 11 для выделения
хлороводорода, который из колонны 11с примесью винилхлорида направляет-
ся на гидрохлорирование в реактор 2, а кубовая жидкость этой колонны вместе
с абсорбатом из абсорбера 3 поступает в колонну 12 для отделения ацетилена
и инертных газов. Отпаренные газы из колонны 12 направляются на гидрохло-
рирование в реактор 2, а кубовая жидкость — в ректификационную колонну 13
для отделения винилхлорида-сырца от 1,2-дихлорэтана.
1,2 Дихлор >i.ui кубовая жидкость и> колонны 13 — nocryiiaei частично в
абсорбер 3, а остальное количество — в систему ректификации 1,2 дихлор на
на в колонну 7. Винилхлорид-сырец через щелочной скруббер I I передается на
двухколонную систему ректификации винилхлорида: в первой колонне /5 oi
винилхлорида отделяются низкокипящие примеси, а во второй колонне /6 но
л у чается товарный винилхлорид.
Теоретический расход основного сырья (кг на 1 т готового продукта): беи
(ин прямогонный — 820, хлор — 580; расход катализатора обычно составляет
1.0 кг/т винилхлорида.
Количество жидких органических отходов составляет 80-100 кг/тВХ. Как
правило, отходы направляются на сжигание. Количество технологических с гоч
пых вод составляет 8-10 м3/тВХ.
Качество винилхлорида, получаемого комбинированным методом, анало
тично качеству продукта, полученного в процессе гидрохлорирования ацеги
лена.
X. Брэчертом и Х.Е. Конерманом [336] разработана специальная конструк
ция реактора для гидрохлорирования разбавленного ацетилена. Эскиз реакто
ра — трубчатого аппарата представлен на рис. 3.31. Ацетиленсодержащий газ
в смеси с хлороводородом поступает в реактор через сопло 2, смесь газов про-
ходит через зону предварительного нагрева 3 (с нагревательным прибором 2),
где нагревается до 50-100 °C. Нагретая смесь поступает в первую реакционную
году 4, заполненную стандартным сулемовым катализатором на основе акти-
вированного угля и снабженную рубашкой 5. Температура теплоносителя ме-
няется от 20 до 120 °C. Далее смесь поступает во вторую реакционную зону 6,
где также находится катализатор — соли предпочтительно ванадата ртути на
основе активированного угля. Температура в рубашке 7 при этом меняется в
пределах 110-170 °C. Третья реакционная зона 8 заполнена активированным уг
лем. Рубашка 9 служит для охлаждения угля.
Такая конструкция реактора вкупе с разбавлением позволяет более эффек-
(ивно поддерживать необходимую температуру процесса. Оптимально исход
ное содержание ацетилена в газе составляет 11,3% мол.
Согласно патенту [337] ацетилен-, этиленсодержащий газ образуется при
пиролизе жидких и газообразных углеводородов в водородной или метановой
плазме; при этом дальнейшая переработка газа аналогична вышеописанной.
Выход винилхлорида достигает 82% в расчете на исходные углеводороды.
Основными недостатками технологии получения винилхлорида газофаз
пым гидрохлорированием ацетилена являются:
- высокая стоимость ацетилена;
- относительно низкий срок службы катализатора, что связано с использо
ванием активных углей низкого качества (с повышенной зольностью и содер
канием железа);
- применение дихлорида ртути в качестве активной массы катализа гора,
который, обладая относительно высокой летучестью, присутствует на всех ста
днях технологической схемы, часто в виде металлической ртути вследствие вое
i 1ановления Hg(II);
- низкая единичная производительность реактора из-за необходимости ор
ганизации интенсивного теплосъема.
Ацетилен-содержащий газ + HCI
Рис. 3.31. Реактор для получения винилхлорида гидрохлорированием разбавленного
ацетилена (1-9 см. текст)
3.4. Гидрохлорирование ацетилена
в псевдоожиженном слое катализатора
Одно из возможных направлений усовершенствования процесса гидрохло
рпровапия ацетилена — повышение мощности единичного агрегата. Увеличе
пне производительности реактора гидрохлорирования ацетилена в пеподниж
пом слое лимитируется скоростью теплоотвода. В промышленных условиях
производительность катализатора составляет лишь 10-15% от кинетически
возможной. Повышение нагрузки на катализатор без эффективного теплое ье
ма приводит к перегреву катализатора, сублимации хлорида ртути, протеканию
побочных реакций и в конечном счете — к значительному сокращению срока
службы катализатора.
Использование для реакции гидрохлорирования ацетилена псевдоожижен
пою слоя катализатора позволяет даже при повышенных нагрузках обеспечить
изо гермичные условия по всей реакционной зоне и во многом снимает oi pai i иче
пня для создания высокопроизводительных агрегатов.
Успешная разработка процесса гидрохлорирования ацетилена в псевдоожи
•кепном слое прежде всего зависит от выбора катализатора, помимо высокой
активности и селективности отвечающего по совокупности свойств требова
пиям, предъявляемым к катализаторам псевдоожиженного слоя, в частности,
высокой механической прочности и наличия микросферической формы час гиц
для создания благоприятного гидродинамического режима.
Т.Д. Гужновская с соавторами [360] исследовали применительно к сулемо
вому катализатору гидрохлорирования ацетилена в псевдоожиженном слое три
условные группы носителей: активные угли, оксидные носители и так назы
ваемые «новые» носители (обуглероженные модификации оксида алюминия).
Из активных углей авторами был выбран уголь АГН-2, модифицированный
добавкой этилендиамина. Основой для такого выбора являлось то, что азотсо
держащие лиганды, способные к образованию л-комплексов с катионом ртути,
заметно улучшают активность и стабильность сулемового катализатора [26К,
269]. Авторы [360] исследовали также катализаторы на оксидных носителях
шпа А12О3, SiO2 и других, представлявших интерес для использования опыта
их широкой промышленной эксплуатации в процессах с псевдоожиженпым
слоем, например, при оксихлорировании этилена, крекинга нефтяных углево-
дородов и т.д. К группе «новых» носителей были условно отнесены разные об
разцы обуглероженных модификаций у-А12О3, сочетающие наличие активного
углерода с микросферической формой у-оксида алюминия, а также синте тичес
кие углеродные носители типа «сибунит», обладающие развитой поверхнос тыо,
преимущественно мезопористой структурой, высокой механической прочное
тью [363]. Содержание сулемы в катализаторах составляло ~10 мае.% .
Данные в табл. 3.9 показывают, что катализатор на основе у-А12О3 обладас!
наиболее высокой активностью, но уступает по стабильности катализатору па
основе активного угля (Houdry Huis и АГН-2). Два других представителя ок
сидной группы — силикагель и алюмосиликат в реакции гидрохлорировапия
ацетилена мало активны, но способствуют интенсивному саже- и смолообра
зованию. Кроме того, удерживающая способность по сулеме также крайне не
велика.
о
I.юница 3.9. Влияние природы носителя па акгииность 1улемовок> кашли шторм и ре
акции шдрочлорнронапня лцсшнена (/= 120"С,Г = 2с, I К 1 С211 = II)
Носи гель Конверсия(%) Удельная актив ность MBx/(MHga2-4) Удерживаю щая способ- ность по сулеме, % Примечание
С2Н2 HCI
Катализатор «Поиску Huis» 46,8 41,6 54,9 85,5 -
Активный уюль АГН 2 48,6 45,0 46,1 75,5 -
С иликагель КСК 1,1 1,0 7,4 23,6 Интенсивное саже- и смолообразование
Алюмосиликат 3,3 3,0 5,6 То же
у-оксид алюминия 70,5 ' 64,5 69,4 63,0 Умеренное смолообразование
Таблица 3.10. Оценка активности и стабильности сулемовых катализаторов на основе
у А12О3 и ее модификаций
Характеристика носителя Конверсия,% Удельная актив- НОСТЬ Мвх/(Мн8а2-ч) Стабильность ка- тализатора, %
с2н2 НС1
у-А12О3 56,3 62,4 128,0 10,9
у-А12О3 + 4,98% С 46,2 50,9 128,4 23,2
у А12О3 + 11,0% С 41,8 46,0 55,3 21,1
у-А12О3 + 15% С 62,4 68,7 81,7 15,7
Таблица 3.11. Проверка активных углей и углеродных модификаций в качестве носите-
ля сулемового катализатора
Характеристика носителя Конверсия, % Удельная ак- тивность MBX/(MHgCl2-4) Удерживающая способность по сулеме, %
С2Н2 НС1
Активный уголь АГН-2 48,6 45,0 46,1 75,5
Активный уголь АР-3 50,9 46,8 54,8 68,0
Активный уголь АГН-1, модифицированный 26,6 27,5 25,3 69,0
меламином
Углеродный носитель «Сибунит» 53,2 50,1 80,9 29,7
Активный уголь «ИГИ» 56,8 52,0 66,6 41,5
Обуглероживание поверхности у Al.О, гак ке не способе гвуе i гакрспленню
сулемы на поверхности (табл. 5.10). кроме того, в процессе работы обуыеро
женные модификации у А12О, подпертаимея интенсивному измельчению, i.e.
мех а 11 и чески не проч и ы.
Авторами рабо ты [360] показано также, что уголь АГН-2 является наиболее
предпочтительным с точки зрения удерживающей способности но сулеме но
сравнению с другими углеродными носителями, включая сибунит (табл. 3.11).
Как и в процессе гидрохлорирования ацетилена на неподвижном слое ката
лила гора, замена сулемы на хлориды других металлов ведет к снижению пока
газелей [360].
На рис. 3.32д и 3.326 представлены зависимости степени превращения аце
гилена от температуры (при постоянном времени контакта), и наоборот, oi
Рис. 3.32. Зависимость конверсии ацетилена от температуры при разном времени кон
такта (а) и времени контакта при разной температуре (б)
100
120
140
100
Рис. 3.33. Изменение активности катализатора HgC12/ArH-2 (т = 10 с, dp = 42 мм)
времени контакта (при постоянной температуре). Катализатор — 10% HgCK/
АГН 2. Влияние температуры особенно заметно при малом времени контакта:
1 -2 с; конверсия ацетилена при повышении температуры от 120 до 180 °C воз-
растает примерно в 2 раза. На рис. 3.326 видно, что конверсия ацетилена с уве-
личением времени контакта достигает максимального значения и далее прак-
тически от него не зависит. В изученных условиях максимально достигнутая
конверсия ацетилена составила 93-94%.
Высокая стабильность катализатора подтверждена результатами 150 ч про-
бега катализатора (рис. 3.33). Линейная скорость газов составила 6-7 см/с, что
соответствовало числу псевдоожижения на уровне 50-60. За первые 80 ч кон-
версия ацетилена упала с 98 до 88% и далее оставалась без изменений. Содержа-
ние сулемы в исходном и отработанном образцах катализатора составило 10,6 и
9,08%. В целом, испытания сулемового катализатора на основе АГН-2 в разных
условиях подтвердили его высокую стабильность.
Кинетические закономерности процесса гидрохлорирования ацетилена в
псевдоожиженом слое катализатора аналогичны неподвижному слою и лучше
всего описываются уравнением, предложенным А.И. Гельбштейном [362].
*Рс,н2 Рна
г = —7^----->
+Phci
(3.28)
где к — наблюдаемая константа скорости реакции; к' — постоянная, обратная
константе адсорбции НС1; рс2н2>рнс1 — парциальные давления ацетилена и хло-
роводорода.
В табл. 4.12 представлены основные кинетические параметры уравнения
для катализаторов «Houdry Huis» и 10% HgCl2/AFH-2. Аррениусовские зависи-
мости выражаются следующими соотношениями [360]:
/с-3,48 108 ехр
45,6 А
RT J
мвх 1° 5
МНёС12-чПа’
(3.29а)
К аде. НС1
= 1,1 ехр
5,75'
~RT;
10 5/Па.
(3.296)
Полученные кинетические данные были использованы для составления
математической модели процесса гидрохлорирования ацетилена в псевдоожи-
I .Шлица 3.12. I 1арамегры yp.iiuieiiiin < м>р<п iti p< in tun i it/ip<ivi<>pitptiii.iiiini inn ii.i
/, °C Катализатор «Iloudry I litis» l\ a tn tn la nip 11 ц< 1,/iikiiiiiHMii \1 null Al 11 2
fcMnK/MHgci> ч-МПа Л-105, Па ЛадС Ю5, Па 1 ч-Мпа A 10 , Ila Л.М.1» I Ila 1
120 2353 0,0268 3,73 2387 0,Ml 7,09
МО 4237 0,083 12,05 6250 0,350 2,86
160 - - 9940 0,310 3,23
180 - - 15870 0,66 1,52
лепным сдое, на основе которой в НИИ «Синтез» были разработаны реактор
н технологическая схема данного процесса. Единичная мощность реактора
70 гыс. т/год.
Математическая модель основана на следующих физических соображе
ни ях:
1. Часть газа, соответствующая его расходуя момент псевдоожижения, про
ходи г через слой в тесном контакте с частицами катализатора и образует плот
ную фазу. Остаток газа проходит в фазе пузырей;
2. Ike частицы катализатора находятся в плотной фазе, вследствие чего ре
акция имеет место лишь в этой фазе;
3. 1аз движется в режиме идеального вытеснения как через плотную фазу,
i.iK и через фазу пузырей;
4. Между фазами имеет место обмен реагентами и продуктами превраще-
ния: скорость пропорциональна разности их концентраций;
5. Концентрация реагентов в сечении каждой из фаз постоянна;
6. Слой считается изотермичным в обеих фазах.
Реакция гидрохлорирования ацетилена характеризуется заметным у меч и.
шением объема реакционной смеси вследствие химического превращения. Это
приводит к уменьшению линейной скорости потока при одновременном новы
пении его плотности. В таких условиях в общем случае возможно гидролина
мпческое перераспределение потока между фазами для обеспечения состояния
минимального псевдоожижения для плотной фазы. Однако при малых зпаче
пиях критерия Архимеда
Лг = ^ Рт~Рг, (3.30)
V2 Рг
характерных для псевдоожижения мелких частиц невысокой плотности как в
данном случае, для условий начала псевдоожижения Remin = const и не записи!
<>| величины Аг. Отсюда следует, что Wpr = const и доля газа, проходящего через
плотную фазу, не меняется по мере протекания реакции.
( учетом этого замечания для единственной протекающей в слое реакции
получаем математическую модель:
(l-x,)(a х.)
(l+a+5-xJ
к'+Р
«-х, Mi
,---* ,-Nix.
(l+a + 5-xJ
(3.31а)
^l = N-i-(x1-x2); (3-316)
1—q ' 27
граничные условия: при £; = О Х| = 0; х2 = 0.
Здесь X] и х2 — текущие значения степени превращения ацетилена в плот-
ной фазе и в пузырях соответственно; — безразмерная координата по объ-
ему (высота слоя) катализатора; £ - V/VKaT, где V — текущее значение и Укат —
полная величина объема катализатора, загруженного в слой, м3; а — соотно-
шение подач НС1 и С2Н2 на входе в слой; 6 — соотношение потока инертов,
поступающих с С2Н2 и НС1 к потоку С2Н2 на входе в слой; р — давление в слое,
На; К — обратная величина константы адсорбции НС1 на сулеме, к1 = 1/к3,
к3= 1,1-ехр( 1370/1,986Т) атм1; Т — температура процесса, К; q — отношение
массового (объемного) потока реагентов через плотную фазу к полной его ве-
личине. Параметры К и N — безразмерные критерии химического превраще-
ния и межфазного обмена.
К = кр 22,4CHga2 т/360()д, (3.32)
где к = 3,42 • 108 ехр(-10940/1,9867) — наблюдаемая константа скорости реак-
ции, моль BX/(MonbHgCl2-4-Ha);
CHgci2 — содержание сулемы в единице объема катализатора, моль HgCl2 /
Л кат»
т — фиктивное время контакта, равное отношению объема катализатора к
объемному расходу реакционной смеси при нормальных условиях, с;
N = ((Зт)д, где (3 — коэффициент межфазного обмена в расчете на единицу
объема загруженного катализатора, 1/с.
На выходе из слоя потоки из обеих фаз смешиваются и степень превраще-
ния ацетилена на выходе рассчитывается по формуле:
X = Xlkq + X2Jt(l-g). (3.33)
Пределы изменения основных переменных: т = 5^-30 с; t = 393-453 °C;
Р = 0,05ч-0,5 с-'.
Скорость процесса гидрохлорирования ацетилена в псевдоожиженном слое
определяется соотношением между скоростью переноса реагентов из пузырей
в плотную фазу и обратно и скоростью химического превращения на катализа-
торе. При низких значениях [3 скорость процесса лимитируется межфазным об-
меном и для интенсификации процесса следует использовать гидродинамичес-
кие факторы, увеличивающие величину (3; при высоких значениях (3 скорость
процесса определяется кинетическими факторами и для его интенсификации
следует повысить температуру процесса Т, давление в реакторе р или соотно-
шение реагентов а. Возможен процесс и в промежуточной области, где влияют
оба типа факторов.
Рис. 3.34. Влияние коэффициента межфазного обмена на степень превращения ацеги
лена X при т = 10 с, т = 30 с, t - 120 °C, Р = 130 кПа, а = 1,05
На рис. 3.34 представлена зависимость степени превращения ацетилена X
о г коэффициента межфазного обмена р при двух значениях времени кон гак га
г, = 10 с и т2 = 30 с. Из приведенных зависимостей следует, что при малых зна
чениях р скорость роста X с увеличением р достаточно велика, особенно при
высоком времени контакта. При увеличении р выше 0,2 с 1 рост степени пре
вращения замедляется как при т = 30 с, так при т = 10 с. Это свидетельствуе т о
том, что в области этого значения режим переходит из области, лимитируемом
межфазным переносом, в кинетическую область, где дальнейшее повышение
степени превращения можно обеспечить, повышая температуру и тем самым
увеличивая скорость химического превращения. Другими словами, при задан
ных условиях t - 120 °C, р = 130 кПа, а - 1,05 — высокая степень превращения
ацетилена на данном катализаторе может быть получена при т - 30 с. Для умень
шения времени контакта т и повышения съема продукта необходимо повышать
температуру процесса.
На рис. 3.35 представлено влияние температуры t на конечную степень пре-
вращения Хк при р = 0,15 с 1 и тех же значениях времени контакта т, = 10 с и
г2 = 30 с. Из этих данных следует, что при указанном значении (3 температура
процесса в интервале 120-180 °C дает заметный эффект, однако не позволят
получить степень превращения Хк > 80% при т = 10 с. Расчеты показали, что
лишь при р = 0,5 с 1 и т = 10 с в указанном интервале температуры степень пре
вращения ацетилена превышает 90%.
Р.С1.. 0,05 0,10 0,15 Значен 0,20 ие X при: 0,30 0,40 0,50
120 °C.... ... 33,1 48,3 56,7 62,0 68,0 71,4 73,5
140 °C.... ... 35,9 54,4 65,3 72,2 80,2 84,4 81,9
160 °C.... ... 37,4 57,8 70,1 77,8 86,4 90,7 93,1
Приведенные данные и рис. 3.34 и 3.35 свидетельствуют о том, что в o6n.it
ти р = 0,2 0,5 с 1 процесс протекает в переходной области, где на степень пре
вращения ацетилена заметно влияют и р, и t. Влияние давления р показано на
рис. 3.36.
Хк
О
с. 3.36. Зависимость степени превращения ацетилена Хк от коэффициента межфаз-
го обмена при т = 10 с (1, 2), 30 с (3, 4); а = 1,05, р = 0,13 МПа (1, 3) и а = 1,1, р =
1,21 МПа (2, 4)
Важная характеристика процесса — время контакта т, определяющее съем
•одукта в единицу времени с единицы объема катализатора, ВХ:
g = (62,5/22,4)3600Хк/[т(1+а)]. (3.34)
Здесь 62,5 молярная масса винилхлорида, Хк — конечная степень превра-
?ния ацетилена не ниже 0,9, исходя из необходимости обеспечить достаточно
1сокое значениеg при разных вариантах осуществления процесса, в том числе
>и рецикле непрореагировавшего ацетилена. С ростом заданной степени пре-
,ащения заметно (и непропорционально) увеличивается необходимое время
нтакта т. Из приведенного соотношения следует, что при Хк = 0,9 и т = 10 с
ем составит около 440 г ВХ/(лкат-ч), а при т = 15 с он снизится до 294 г ВХ/
кЛГ-ч). На рис. 3.37 приведено влияние времени контакта на степень превра-
;пия ацетилена Хк. Из приведенных данных следует, что как увеличением р,
к и повышением t удается снизить время контакта, необходимое для достиже-
1Я заданной степени превращения Хк. Так, X.. = 0,9 достигается при t = 120 °C,
23 с для р - 0,2 с-1 и т = 20,5 с для Р = 0,3 с ; при t - 140 °Ст = 15,5 с и т = 12 с
Pin.. 1.37. Влияние времени контакта на степень превращения ацетилена ХЕ при р =
-0,21 с (7, 3), 0,31 с (2, 4) и Т= 393 К (7, 2), 433 К (3,4)
iоот вегеI венно для Р = 0,2 и 0,3 с-1. При этом заметно возврастает съем винил
хлорида. Для указанных четырех режимов он равен соответственно: 191, 215,
.’К I и 367 гВХ/(лкат-ч). В табл. 3.13 приведены данные по влиянию температуры
на с гспень превращения для двух значений Р = 0,1 и 0,5 с *.
Из сопоставления эффекта двух факторов на степень превращения следует,
то при увеличении р возрастает и влияние температуры — особенно при ма
лом времени контакта. Так при т = 10 с и Р = 0,1 с"1, повышение температуры
па 10 °C дает приращение Хк только 9,5%, тогда как при р = 0,5 с ’, приращение
го1 гавляет 19,6%, свидетельствуя, что р = 0,1 с 1 соответствует области межфа i
пого диффузионного торможения, а значение Р = 0,5 с1 соответствует кине
iической области процесса. На рис. 3.38 приведены профили степеней превра
щения ацетилена в фазах при двух значениях р = 0,05 и 0,2 с *. Из этих данных
с ледуе г, что при очень малых Р степень превращения в плотной фазе велика уже
па начальном участке слоя даже при малых т. С ростом Р профили сближаются,
но при Р = 0,2 с 1 они еще существенно отличны, что характеризует переходную
область протекания процесса. В кинетической области разница между концеы
1.к>лица 3.13. Изменение степени превращения Хк в зависимости от времени при раз
и i.ix р и температурах
Р.С 1 7, °C Степень превращения Хк при т, с
5 10 15 20 25 30
0,1 120 28,3 48,3 62,6 72,8 80,1 85,3
140 32,8 54,4 68,9 78,6 85,2 89,7
160 35,5 57,8 72,3 81,7 87,8 91,9
0,5 120 47,7 73,5 86,2 92,5 95,7 97,4
140 64,6 86,9 94,7 97,6 98,9 99,4
160 75,4 93,1 97,7 99,2 99,7 99,9
Рис. 3.38. Профили степени превращения ацетилена в фазах при:
120 °C (а, б — плотная фаза; в, г — фаза пузырей)
трациями (или соответственно степенями превращения) в фазах мала по срав
нению с абсолютными значениями.
Как следует из изложенных результатов и выполненных расчетов, дости-
гаемая степень превращения зависит сложным образом от температуры, ко-
эффициента межфазного обмена и времени контакта. Поскольку температура
является фактором, позволяющим поддерживать заданную активность катали-
затора во времени в случае его дезактивации, при выборе исходного режима
следует ориентироваться на возможно более низкую температуру. Ниже при-
ведены расчетные результаты, характеризующие связь между выбранной тем-
пературой и необходимыми значениями Рит, обеспечивающими достижение
заданной степени превращения Хк:
• -Ml
11 1 'п 1 'И 1 1(1 1 i(i
1 Ш. 1) II I) III 1) Hl и li О, ’0 Н. И) О, .1) О 10 0,1(1 (I. >0
1м • in ."| 1) .’<) ' । .’0 is Г> ID 1 • 10
, >!<• 14.. И 1 1 1 а ' ’ll ' 41 176 220 293 293 1-17 29’ I 10
Х„-9'. %
• 1 1'0 130 140
I' 1' п. <11 П/К) 0,10 0,20 0,30 0,40 0,50 0,15 0,20 0,30 0,5(1
мш til 2 ’ 20 20 25 20 15 15 30 15 10
! 1 ИХ 1 1 176 220 220 176 220 293 293 147 293 14(1
Хк = 98%
» •( !.’() 1 ’() 140 150 160
Plf' 0, () 0, ’() 0,40 0,30 0,40 0,50 0,40 0,20 0,30 0,50
«1 30 30 25 25 20 20 30 20 15
1 «ИХ/ 1'1, ч) 1 17 176 220 220 176 220 293 293 147 293
В И1илсдпеи строке приведены значения съемов винилхлорида при соог
к туннцнх условиях. Как следует из приведенных результатов, съем в неси
..... и 1ч ином слое даже при самых «мягких» условиях (низкая температура и
in'ii.ii шачения коэффициента обмена) достигает 150 г винилхлорида с литра
। ин/in i.iiopa в час. Это значение в 3-4 раза превышает съем, достигаемым и
п< индии ином слое катализатора.
< hiiipiibie значения Хк выбраны в соответствии с разными вариантами офор
м и нии процесса в псевдоожиженном слое: Хк- 90% для реактора со свободно
1Н1Н1ЦИМ слоем и с рециклом непрореагировавшего ацетилена; Хк= 95% — дли
I ihiopa с кипящим слоем, секционированным провальными решетками, и с
нрн< । лпкой с неподвижным слоем, Хк = 98% — для реактора с организованным
и. । идоо киженным слоем. Каждый из этих вариантов имеет преимущества и
и. im 1 а гки, и выбор оптимального варианта должен выполняться с учетом гех
иппо жопомических показателей всей схемы.
Полученные данные были использованы для расчета и создания реактора
н гидоо киженного слоя в процессе гидрохлорирования ацетилена прои шоди
и п.нос । ыо 70 тыс. т ВХ/год (рис. 3.39).
Принципиальная технологическая схема. Исследования процесса гидро
хлорирования ацетилена в псевдоожиженном слое катализатора, приведенные
и НИИ «Синтез», позволили выбрать следующий режим работы реактора: ка
ia in >.пор — HgCl2/aKT. уголь АГН-2, содержание сулемы — 10 мае. %, размер
ч.и 1нц — 0,1-0,35 мм; температура — 120-140 °C; время контакта — 10 15 с;
мо п.иое соотношение — С2Н2:НС1 = 1:1,1.
Рис. 3.39. Схема реактора гидрохлорирования ацетилена в псевдоожиженном слое
Указанный режим оиссцечивас! конверсию ацетилена & VS",,, конверсию
хлороводорода 285 86%, съем винилхлорида « ИЮ 320 г/(лк11|-ч).
Один из вариантов принципиальной гехноло! ической схемы процесса гид
рохлорирования ацетилена в псевдоожиженном слое катализатора представлен
на рис. 3.40
Исходные реагенты — ацетилен и хлороводород после предвари тельной
oi ушки и очистки 1 поступают на смешение в аппарат 2 и далее смесь направ
ппетхя в реактор с псевдоожиженном слоем 3.
В реактор загружают сулемовый катализатор и псевдоожижают азотом, но
щнасмым также через блок осушки и очистки 1. Разогрев реактора и оконча
сльную сушку катализатора ведут также азотом. Смесь реагентов nocryiiaci в
реактор через специальное газораспределительное устройство, расположенное
в нижней части реактора. Для улавливания измельченных в процессе раб(.
ча< 1иц катализатора в верхней части аппарата расположена система циклопов.
1сплос ьем осуществляют теплоносителем (например, кипящей водой), подава
емым в змеевик непосредственно в слое катализатора. Конверсия ацетилена ы
проход в аппарате с псевдоожиженным слоем достигает 95%. Количественное
превращение непрореагировавшего ацетилена возможно за счет либо рецик
ы, либо установки дополнительного догидрохлоратора с неподвижным ело
гм катализатора, работающего в адиабатическом режиме 4. Схема выделения
винилхлорида из реакционного газа существенно отличается от принятой на
действующих производствах винилхлорида из ацетилена. По существующей
। хсме реакционный газ после реактора проходит последовательно отмывку от
п< прореагировавшего хлороодорода водой, нейтрализацию раствором щелочи,
компримирование, осушку, затем следуют ректификация и выделение товарно-
III винилхлорида. В предлагаемой схеме реакционный газ подается на компрес
i ор 5 и поступает в колонну выделения непрореагировавшего хлороводорода 6,
ко горы й возвращается обратно на гидрохлорирование ацетилена. Часть газа и i
верхней части колонны, во избежание накопления инертов, через санитарную
। олоппу 7 сбрасывается в атмосферу. Кубовая жидкость из колонны 6 направ
пи-Ия на ректификацию и разделение в колоны 9, 10. Кубовые передаются па
ун1лизацию, а винилхлорид после дефлегматора проходит дополнительную
п нгрализацию следов хлороводорода в аппарате 11 и поступает на склад то
и ipiioi <> продукта.
(^писанная схема выделения винилхлорида исключает стадии промывки и
н in рализации реакционного газа, его сушки и тем самым позволяет разрабо
ini. оолее рациональную и эффективную схему процесса.
Тб 8 I
Рис. 3.40. Принципиальная схема процесса гидрохлорирования ацетилена в псевдоожиженном слое катализатора:
1 — блок очистки исходных реагентов; 2 — смеситель НС1 и С2Н2; 3 — реактор; 4 — догидрохлоратор; 5 — компрессор; 6, 9, 10 — ректифика-
ционные колонны; 7 — колонна для улавливания НО; 8 — емкость для HCi; 11 — колонна нейтрализации ВХ
I,‘>. I ИД|Х>К>Н>рИ|)< >ПЛ11И<* .|Ц<* 1ИНГ1Ы
к присугс।ими инициаюр* >н
\ л i.i । рил । и иным 11 л 11 рли л < н in м р<'.| ли । uulu i л ик|ы пни о процесса i пдро
к 111 р 11 р< 11 (л > 111 > >|Ц< iH/iciia лилией» об I.CMHI.III прицеп и ирису и inmi иницн
aiopon Дос кипи i вом >uno направления являски oiK.it <н использования
кил и mi 1ЫЧ с улемовы \ к л i ли и ia торов, л i лк ке возможное i ь некогоро! о сип
) Hinn i peooit.iiiiiii к K.rieciuy исходною сырья, в частности, по содержанию
IWIlll II
Большой комплекс работ в пом направлении был проведен 10.А. Налдере
ним с corp | ИН 31||. Реакцию проводят с использованием инициирующих до
ii nioic хлора или органических пероксидов.
В ном случае инициированное гидрохлорирование ацетилена протекает
но радикально ценному механизму. В литературе описаны реакции ацетиле-
H.I, нроюкакицие по свободно радикальному механизму. Наиболее характер
нымп in них являются реакции пиролиза, полимеризации и присоединения
| »1 471.
Упоминания о реакции хлоррадикала с ацетиленом приведены в работах
| ' 18 3511, которые весьма важны для трактовки механизма инициированного
। ндрохлорирования ацетилена. Рассмотрим некоторые закономерности них
реакций.
Гидрохлорирование ацетилена, инициированное хлором
( огласно [344], в интервале 150-600 °C конверсия ацетилена растет с по
в...опием температуры до определенного значения в зависимости от концеп
। рации инициатора (рис. 3.41). До 300 °C основным продуктом реакции явлн
спи винилхлорид, побочно образуются дихлорэтилены (транс-, цис-) и 1,1,2
цшхлорэтан.
При t > 300 °C в побочных продуктах появляются, кроме перечисленных,
।ри.хлорэтилен, тетрахлорэтилен, винилиденхлорид, тетрахлорэтан, пета
хлораган, смолы, сажа.
Гак как при 150 °C конверсия ацетилена незначительна (-2%), а выше 300 "( .
i.iMci'HO снижается избирательность реакции по винилхлориду, в работе |34'1|
делается вывод о том, что реакцию целесообразно проводить в интервале 200
100 °C.
Значительно влияет на конверсию ацетилена и выход винилхлорида оба,ем
пан доля хлора в смеси (табл. 3.14). При объемной доле хлора 0,3% конверсия
ацетилена при 100%-й селективности по винилхлориду составляет 5,5-к13,5% i
1ависимости от температуры. Увеличение объемной доли хлора (2,3%) нриво
ди г к значительному возрастанию конверсии ацетилена (-50%), однако селек
I ивность реакции снижается до 70%.
Уменьшение соотношения НС1:С2Н2 до 3:1 в присутствии 1,2 об. % CI . ве
де г к снижению конверсии ацетилена и селективности реакции гидрохлори
рования. При НС1:С2Н2 = (Зз-5):1 увеличиваются конверсия ацетилена и выхо;
винилхлорида. Дальнейшее увеличение соотношения НС1:С2Н2 не приводит i
значительному повышению конверсии и селективности реакции.
Рис. 3.41. Зависимость конверсии ацетилена (1, 2, 3) и селективности реакции по пи
нилхлориду (4,5, 6) от температуры при гидрохлорировании смесей,% :
1,4 — С2Н2 = 15; С12 = 0,4; HCI = 84,6;
2,5 — С2Н2 = 15; С12 = 1,7; HCI = 83,8;
3, 6 — С2Н2 = 15; С12 = 3,2; НС1 = 81,8
Согласно [352-354] скорость образования продуктов реакции, приведенных
в табл. 3.14, может быть описана следующими выражениями:
W’bx - кък [С2Н2] [С12] 1/2[НС1]1/2, (3.35а)
^.г-дхэ-ен - *дхэ [С2Н2] [С12]3/2 [НС1] 1/2, (3.356)
^тхэ = *тхэ [С2Н2][С12]3/2 [НС1]1/2. (3.35в)
Выражение температурной зависимости эффективных констант скоро
стей брутто-реакций образования винилхлорида, 1,2-дихлорэтиленов и 1,1,2
трихлорэтана, см3/(моль-с):
квх = 1О8’2±о’2ехр[(-33,3 ± 1,7)/RT). (3.36а)
Кдхэ = 1О9,4±о’6ехр[(-34,1 ± 6,3)/RT], (З.Збо)
Ктхэ = Ю14’2±0’8ехр[(-43,7 ± 3,9)/RT]. (З.Збн)
Дробные порядки реакций образования винилхлорида, 1,2 дихлорэтане
нов и 1,1,2-трихлорэтана при гидрохлорировании ацетилена, инициированного
хлором, указывают на сложный радикально-цепной механизм процесса.
Г.К111НЦ.1 3. II. 3.1Н1Ц I1MOCI I, С ОС Г.111.1 продую ОН Пр<1ЦССС.1 1|||ИЦ1Н||>1>Н.И<||<1|<1 I пдрихш>|>11
ринапия ацетилена от концеш рации хлора
Исходная смесь, об. % Продукты реакции, об. % Конвер- сия,% Селскгип- носи* по винилхло риду, %
IIC1 С2Н2 С12 Ви- нил- хлорид 1,2-дихлор- этилен 1,1,2- трихлор- этан С2Н2 С12
транс- цис-
1р = 200 °C, тр = 1,32 с
ч з,о 6,7 0,3 0,3 - - - 5,59 2,1 юо,о
92,7 6,7 0,6 0,51 0,03 0,006 0,01 8,1 8,6 91,2
92,1 6,7 1,2 0,70 0,09 0,016 0,03 12,3 11,4 83,6
91,0 6,7 2,3 1,05 0,27 0,051 0,11 22,0 18,6 70,5
79,7 20,0 0,3 1,11 - - - 5,5 2,6 100 0
79,4 20,0 0,6 1,55 0,01 0,011 0,04 8,0 10,0 96,8
78,8 20,0 1,2 2,10 0,27 0,048 0,09 12,5 34,1 84,0
77,7 20,0 2,3 3,15 0,75 0,121 0,25 21,1 48,6 74,6
Гр = 230 °C, тр = 1,21 с
93,0 6,7 0,3 0,57 - - - 8,3 2,3 100,0
92,7 6,7 0,6 0,75 0,05 0,031 0,01 11,2 15,8 89,3
92,1 6,7 1.2 1,15 0,14 0,090 0,06 21,6 24,1 80,6
91,0 6,7 2,3 1,79 0,42 0,260 0,20 39,8 39,2 67,2
79,7 20,0 0,3 1,81 - - - 9,0 2,4 100,0
79,4 20,0 0,6 2,30 0,11 0,031 0,03 12,1 28,3 94,6
78,8 20,0 1,2 3,51 0,31 0,111 0,10 20,0 41,6 87,5
77,7 20,0 2,3 5,30 0,91 0,301 0,30 33,3 65,2 79,5
tp = 270 °C, тр = 1,14с
93,0 6,7 0,3 0,87 - - - 12,9 2,7 100,0
92,7 6,7 0,6 1,10 0,06 0,042 0,02 18,6 20,3 90,1
92,1 6,7 1,2 1,71 0,19 0,121 0,08 31,2 32,5 81,3
91,0 6,7 2,3 2,27 0,50 0,301 0,28 50,0 46,9 67,6
79,7 20,0 0,3 2,70 - - - 13,5 2,9 100,0
79,4 20,0 0,6 3,91 0,14 0,111 0,04 20,9 48,8 93,0
78,8 20,0 1,2 5,20 0,40 0,301 0,15 30,2 70,8 86,0
77,7 20,0 2,3 7,60 1,00 0,801 0,42 51,0 96,0 69,7
Инициирующая роль хлора в реакции гидрохлорирования ацетилена обус
лоплена, по мнению авторов [355], участием хлора в образовании активных час
। п ц, которые ведут цепь реакции ацетилена с хлороводородом.
I Io-видимому, в исследуемой реакции присутствие ацетилена может облеч
чип, акт зарождения радикалов по реакции:
С2Н2 + CI2
С2Н2СГ + СГ.
(3.37)
Дли объяснения наблюдаемых порядков реакции образования всех продуй
гоп предложена 13441 следующая последовательное гь:
к) 1.С2Н2 + СГ—С?Н2СГ; (3.37а)
кг 2. С2Н2СГ + НС1 —► С2Н3С12‘; (3.376)
кз 3. С2Н3С12’ —- С2Н3С1 + СГ; (3.37в)
к4 4. С2Н3С12 + С1/ —- С2Н3С13 + СГ; (3.37i)
кз 5. С2Н2С1 + С12‘ —- С2Н2С13‘; (3.37д)
6. С2Н2С13' —С2Н2С12 + СГ; (3.37е)
к? 7. С2Н2СГ + СГ С2Н2С12. (3.37ж)
Исходя из приведенного механизма и применив метод стационарных кон
центраций к активным частицам, принимающим участие в реакциях, получим:
w _ ^2^з[НС1][С2Н2] ATqAc! _С12 _________________ /ТЗЯа)
ВХ k3 + k4[Cl2] ^2[НС1]+к5[С12> ;
^дхэ - 1 ^0^1 С1 2_ (г
| к2 [НС1] + ^[^2X7
Мтхэ -
fc2*4[HCl][C2HjCl2] | ^[С12] ~
ку + fc4 [с/2 ] \ к2 [ НС1] + к5 [С12 ]к7
(3.38в)
где WBX, ШдХэ> ^тхэ — соответственно скорости образования винилхлорида,
1,2-дихлорэтиленов и 1,1,2-трихлорэтана.
В присутствии избытка НС1 в исходной смеси выход 1,2-дихлорэтиленов и
1,1,2-трихлорэтана значительно меньше выхода винилхлорида.
С учетом этого скорости образования продуктов реакции:
[с. ifДс^Пна]1'2.
\ К7 )
(I-1 V/2
^дхэ = ^5 гг [С2Н2][С12]3/2[НС1]-1/2,
v x2k7 J
(3.38 а’)
(3.38 б’)
%х>=7
А А(,А,
( » »K u‘)
Приведенные выражения объясняю! iMiiepuMein.uii.iio паО подаем ыс ио
рядки реакций по всем исходным веществам.
Изучение влияния гетерогенных факторов на < идрохлорирование ацешлс
на, инициированное хлором, показало, что с увеличением S/V (S — поверхнос i ь
стенки, V — объем реактора) скорость реакции имеет тенденцию к уменыпе
пию (рис. 3.42).
При 200 °C это уменьшение несущественно, при 300 °C увеличение S/V в
шесть раз приводит к уменьшению скорости реакции приблизительно вдвое.
И »этого следует, что увеличение температуры приводит к увеличению влияния
। етерогенного фактора в протекании реакции.
Данный эффект можно объяснить тем, что при низких температурах за
рождение и гибель радикалов происходят преимущественно на поверхности
реактора, в этом случае влияние величины S/V незначительно. При повышении
температуры увеличивается доля гомогенных реакций зарождения, имеющих
более высокую энергию активации, что приводит к большей зависимости ско
рости реакций от объема реактора.
В работах [338, 344] изучено влияние добавок кислорода и водорода на ско
рос I ь реакции гидрохлорирования ацетилена, инициированного хлором. Влия
иие кислорода изучено при температуре 200 и 230 °C. Объемную долю кислоро
да в смеси, подаваемой на гидрохлорирование, изменяли в пределах 0,006-0,8%.
Как видно из данных табл. 3.15, очень малые (0,01-0,1%) количества кислорода
ускоряют гидрохлорирование приблизительно в три раза. При увеличении он ь
емпой доли кислорода в смеси больше 0,5% скорость образования винилхло
рида падает.
Рис. 3.42. Кинетические кривые накопления винилхлорида в реакторе с S/V = I см
(I, 2) и S/V = 22 см-1 (1а, 2а); 1 — 200 °C, 2 — 300 °C (объемная доля состава, % С2112
(>.7; С12 = 0,6; НС1 = 92,7)
Гао лица 1 15. Влияние кислорода на скорость <>(>ра юпапия винилхлорида (объемная
доли смеси, %: ( 111 - 6,7; I К I — 93,3 92,1, время реакции 1,2 с, гемнер.п ура: 23(1"(' —
числитель, 300 ”( — знаменатель)
|О2|, об. % Wbx‘107, моль/(см’-с)
при [С12] = 0,3, об. % при [С12] = 1,2, об. %
0,006 0,91/1,91 1,72/3,72
0,010 0,92/1,90 1,70/3,72
0,024 1,81/2,81 2,92/5,71
0,050 2,21/3,40 3,68/6,50
0,1 2,40/3,80 3,96/7,01
0,2 2,70/4,21 4,95/7,20
0,4 2,80/4,10 5,05/7,30
0,8 2,61/4,00 3,96/7,00
Таблица 3.16. Влияние добавок водорода на гидрохлорирование ацетилена, иницииро-
ванного хлором (тр = 1,2 с)
1,°С Добавки, об. % Продукты реакции, об. % Конверсия ацетилена, %
ВХ ДХЭ-ен тхэ
Аг Н2 транс- цис-
Объемные доли С2Н2 = 6,7%, С12 = 1,2%, НС1 = = 92,1%
200 1,7 - 0,7 0,091 0,016 0,03 12,3
- 1,7 1,1 1,140 0,025 0,05 19,5
230 1,7 - 1,15 0,14 0,09 0,06 21,6
- 1,7 1,75 0,22 0,14 0,09 32,8
Объемные доли С2Н2 = 20%, С12 = 1,2%, НС1 = 77,1%
200 1,7 - 2,15 0,27 0,05 0,09 12,5
- 1,7 3,01 0,37 0,07 0,14 17,9
230 1,7 - 3,5 0,31 0,10 0,101 20,9
- 1,7 5,10 0,45 0,15 0,160 29,3
При гидрохлорировании ацетилена расходуется около 50% добавленного
кислорода, из которого образуется в основном оксид углерода, а также неболь-
шое количество диоксида углерода.
Добавки водорода в смесь, подаваемую на гидрохлорирование, также уве-
личивают скорость реакции. Так, объемная доля водорода 1,7%, добавленная в
смесь, увеличивает конверсию ацетилена в 1,5 раза (табл. 3.16).
Показано также [344], что добавки 2% оксида углерода, диоксида углерода, мета-
на не влияют на инициированное хлором гидрохлорирование ацетилена (рис. 3.43).
8
A
Рис. 3.43. Влияние добавок диоксида углерода (о), оксида углерода (х) и метана (□) на
расходование ацетилена (7, 2) и образование винилхлорида (1а, 2а) при гидрохлори
ровании объемной доли смеси, %: [С2Н2] = 6,7; [С12] = 1,2; [НС1 ] = 92,1; 1, 1а — 200 "С;
2,2а — 300 °C
Рис. 3.44. Влияние (1,6 об.%) добавки этилена (7,2) на кинетику образования винилхло
рида; 1а, 2а — без добавок; 1, 1а — 300 °C; 2, 2а — 200 °C
Авторами работы [344] обнаружено также тормозящее влияние на процесс
добавок толуола, пропилена, оксида азота.
Влияние этилена носит двойственный характер (рис. 3.44). При 200 °C до
бавки этилена значительно замедляют реакцию гидрохлорирования, а при
300 °C торможение незначительно.
Аналогичным закономерноеiям, согласно |3 I I], подчиняется процесс нпн
цииропапно! о i идрохлориронания разбавленною ацетилена, входящего в с<>
став продуктов гермоокислитсльного пиролиза природною газа и элекгрокрг
киша хлороринических отходов.
Состав, об. %, ацетиленсодержащих газов:
Компонент... С2Н2 НС1 С2н4 Н2 с2н6 сн4 СО СО2 О
1аз электрокрекинга отходов производства хлоропрена .. 18,98 44,61 0,95 32,19 1,17 2,09 — 1
Компонент С2Н2 - с2н2 н2 - СН4 со со, о,
Продукты пиролиза природного газа ...7,9 — 0,4 55 — 5,1 24,3 3,1 0,2
При инициировании процесса хлором выход винилхлорида достигает 83%.
Ю.А. Паздерским с соавторами [338,339, 343, 344] установлено, что для про
ведения реакции инициированного гидрохлорирования ацетилена можно так
же применять соляную кислоту.
Замена хлороводорода на соляную кислоту в процессе гидрохлорирования
ацетилена экономически обоснована в связи с утилизацией абгазной соляной
кислоты, в значительном избытке образующейся в процессах, связанных с пе-
реработкой хлора.
Зависимость степени превращения ацетилена от температуры при исполь
зовании кислоты разной концентрации представлена на рис. 3.45.
С повышением температуры с 448 до 540 К и увеличением объемной доли
исходной соляной кислоты до 30 об. % конверсия ацетилена и выход винилхло-
рида возрастают. Дальнейшее повышение температуры существенно не влияет
Рис. 3.45. Зависимость степени превращения С2Н2 от температуры и объемной доли
соляной кислоты (концентрация С12 = 0,83%)
I'm 3 16. Bjiiiiiiiik' концен i рации хлора на степень превращения С2Н2 (/) и выход про
i |<цц|р1 1кцпп ВХ (2); цис+гранс-1,2 ДХЭ (3). Сна = 20%; t- 250°C
Гп * 17. Зависимость степени превращения С2Н2 от температуры, концентрации щ
и।utiiihi IKI и добавки 0,2% O2 (объемная доля Cl2 = 0,83%)
на конверсию ацетилена. Выход цис-, транс-1,2-дихлорэтиленов в значительной
к ре ывисит от концентрации вводимого в реакционную смесь хлора. Записи
mix । ь влияния хлора на степень превращения ацетилена и на выход продуктов
реакции представлена на рисунке 3.46.
Добавки кислорода, как и в случае применения концентрированного хлоро
водорода, ускоряют гидрохлорирование ацетилена. На рис. 3.47 приведена зави
имос । ь степени превращения ацетилена при разных температурах и исполь »о
в.шин соляной кислоты разной концентрации при добавлении в реакционную
смесь 0,2% кислорода.
Авторы [344] делают вывод, что для инициированного хлором гидрохлори
ропалия ацетилена можно использовать растворы 20-30%-й соляной кисло!ы.
Реакцию целесооОра.шо проводип> при 500 530 К, оОьемнои доле хлора до 1%
и времени контакта 4 6с. Объемная доля добавок кислорода до 0,5% ускоряем
реакцию образования винилхлорида.
Гидрохлорирование ацетилена, инициированное
пероксидными соединениями
В качестве инициаторов реакции гидрохлорирования ацетилена можно ис-
пользовать пероксидные соединения. Радикалы, образуемые при их распаде, мо
гут быть источниками зарождения цепных реакций [357, 358]. Авторами [3591
установлена инициирующая способность пероксидных соединений в реакции
гидрохлорирования ацетилена. Установлено, что реакция гидрохлорирования
ацетилена в присутствии пероксидных соединений протекает высокоселектив
но по винилхлориду. На рис. 3.48 представлена зависимость степени превраще-
ния ацетилена от природы инициатора в интервале 225-300 °C.
Степень превращения ацетилена в винилхлорид зависит от инициирующей
способности пероксида, причем наибольшую эффективность проявляют гид-
ропероксид третбутила (ГПТБ) и надуксусная кислота.
Максимальная степень превращения ацетилена достигается при концен
грации ГПТБ в реакционной смеси 0,25-0,5%. Дальнейшее увеличение кон-
центрации ГПТБ незначительно влияет на степень превращения ацетилена
(рис. 3.49).
Реакция гидрохлорирования ацетилена начинается при t > 100 °C (рис. 3.50).
Дальнейшее повышение температуры выше 150 °C приводит к значительному
возрастанию степени превращения ацетилена, но при температуре выше 280 °C
изменение конверсии ацетилена малозаметно; при этом идут нежелательные
Рис. 3.48. Зависимость степени превращения ацетилена от природы инициатора при
разных температурах, НС1:С2Н2 = 3:1, [in] = 0,32%:
1 — ГПТБ; 2 — надуксусная кислота; 3 — (С4Н9)3—Si—ООН; 4 — (С2Н3)2—Si(OOH),; 5 — над-
пропионовая кислота; 6 — (СН3)3 — ООО—С(СН3)3; 7 — гидроперекись кумола; 8 — Н2О2
I’m, '.-19. Зависимость степени превращения ацетилена от концентрации инициатора
(Kill.)
I'Hi I.SO. Зависимость степени превращения ацетилена и выхода винилхлорида oi icm
in р.п уры (объемная доля ГПТБ = 0,32%); соотношение HCI: С2Н2:
I 1.2 —3:1
нпиочные реакции (полимеризация, смолообразование), выход винилхлорида
। ин иле гея. Оптимальной температурой для проведения реакции, по-видимому,
• нгдуст считать 230-260 °C.
Добавки водорода в реакционную смесь до 0,8% приводят к незначительно
му Boipaciанию степени превращения С2Н2, дальнейшее увеличение добавок
водорода существенно не влияет на степень превращения ацетилена.
юница 3 17. Гидрочлорировапле ацетилена, и и ипнироплп mu- I 1111
Г 1 г Подача рса ген гоп, л/ч Крем я кои гак та, с Оо ьсм пая доля ГПТБ, % Выход ВХ па пропущен ный С2П2, % Коппер сия С2Н2, % ( елскhim поси> но ВХ, %
с2н2 НС1
225 100 300 13,6 0,39 16,5 16.6 9Ч,|
1 100 700 6,8 0,37 19,7 19,8 99,1
200 600 6,8 0,35 17,1 17,2 99. 1
250 100 300 12,9 0,39 17,1 17,1 99,7
100 700 6,4 0,37 24,1 24,2 99,4
200 1000 4,3 0,30 23,4 23,5 99,5
200 1400 3,2 0,30 24,1 24,3 99,0
400 1600 2,6 0,31 16,5 16,6 99,4
275 100 500 8,2 0,38 25,9 26,2 99,3
200 1000 4,1 0,30 24,8 25,0 99,4
200 1400 3,1 0,29 22,6 22,7 99,4
400 1600 2,5 0,31 17,1 17,3 98,9
Объемная доля добавок СО и СН4 от 0,1 до 2,5% не влияет на гидрохлориро
1ание ацетилена. Объемные доли добавок кислорода до 1,3% и винилхлорида до
,0% не влияют на скорость реакции, а увеличение добавок больше указанных
количеств тормозит реакцию.
В результате изучения кинетических закономерностей реакции иницииро
данного ГПТБ гидрохлорирования ацетилена получено уравнение для опреде-
ления скорости реакции образования винилхлорида:
WBX - /свх-[С2Н2]-[НС1]-[ГПТБ]0’5 , (3.39)
де ках = 106,б2± 1,36ехр[(-16000 ± 1240)/(RT)], (л/моль)1’5^’1
Опытная проверка процесса была проведена в реакторе диаметром 57 мм.
Эсновные результаты представлены в табл. 3.17. Селективность образования
зинилхлорида превышает 99%.
Процесс гидрохлорирования ацетилена в присутствии инициаторов пред-
ставляет определенный интерес с исследовательской точки зрения. Промыш-
ленная его реализация, даже в случае возобновления интереса к процессам на
Ьснове ацетилена, маловероятна в силу невысокой конверсии ацетилена и низ-
кой технологичности.
Гидрохлорирование ацетилена, инициированное с помощью
лазерного излучения
Реакция газофазного объемного гидрохлорирования ацетилена, иниции-
рованная с помощью лазерного излучения с длиной волны 308 нм была изуче-
на в работе [570]. Поскольку хлороводород и ацетилен практически прозрачны
1'н< «Л1 I.iihk. iimoi п> степени превращения ацетилена (X) от температуры для доба
цп|< II, I об %;
/ nut ины. 2 |рихлоруксусного альдегида; 3 — метилэтилкетона; 4 — уксусного альдегида;
<|«>рм>| и.дегида С211 :11С1:Не= 1 I 15 (время контакта 84,2 с, мощность лазера 1 Вт)
I’m 3.52. Зависимость степени превращения ацетилена (X) от содержания ацетона и
нк 1 плене
для длины волны 308 нм [690], инициирование процесса с помощью лазерши о
и i лучения возможно осуществить лишь при введении в реактор добавок, пог
ло|ц.1кнцих УФ-излучение, в частности, ацетона, метилэтилкетона, формальдс-
। ид.| и др.
Па рис. 3.51 представлены зависимости степени превращения ацетилена
<н юмпературы для разного вида фотохимических добавок. В интервале 100
.'00 °C наилучшие результаты получены в присутствии ацетона. Степень пре
вращения ацетилена при этом растет по мере увеличения содержания ацетона
и ацетилене (рис. 3.52).
Автор работы 1570] у калы пае!, что скороеп> реакции сложным обратом ia
ШКИ1 or конце!! 1 рации ацетона и исходных реагентов, а также иг мощное in
лазерною излучения. Уравнение скорости реакции может быть записано следу
видим образом:
г = клрл°’5-[(СН3)2СО]0-5 -[С2Н2]0'5 [НС1]05. (3.40)
Константа скорости реакции гидрохлорирования равна;
кл = 35,3 exp[-12400/(RT)], мол°'5-л 0>5-с '-В-Г0’5. (3.41)
Предложен механизм реакции:
(СН3)2СО —* 2С‘Н3 + СО, (3.42а)
С‘Н3 + НС1 —- СН4 + СГ, (3.426)
СГ + С2Н2 —- С2Н2СГ, (3.42в)
С2Н2СГ + НС1 —С2Н3С1 + СГ, (3.42т)
или
С2Н3СГ2 —* С2Н3С1 + СГ, (3.42д)
С2Н2СГ + СГ —- С2Н2С12. (3.42е)
О реализации процесса в укрупненных (опытных или опытно-промышлсп
ных) масштабах информация отсутствует.
З.б. Жидкофазное гидрохлорирование ацетилена
Исследование процесса получения винилхлорида жидкофазным гидро-
хлорированием ацетилена имеет достаточно давнюю историю. Впервые эта
реакция была осуществлена германским химиком Ф. Клатте в 1915 г. Пик ин
тереса к этой проблеме приходится на 1950-1970-е годы, когда возможность
осуществления процесса в промышленных условиях представлялась доста
точно реальной. Данная реакция привлекала внимание многих исследовате
лей [365-370]. Основные достижения в этой области обобщены в моногра-
фии [172].
Присоединение хлороводорода к ацетилену протекает в присутствии кис-
лотно-основных или металокомплексных катализаторов. К ним относятся сис-
темы AlX3, SnX4, SbX4, SbX5 в неводных растворах и комплексы Hg (II), Си (I), Pd
(II), Pt (II) в водных и неводных растворах.
Среди гомогенно-каталитических систем синтеза винилхлорида наиболее
интересны с практической точки зрения системы на основе комплексов Hg
(II), Си (I) и Pt (II). Весьма подробно изучены ртутные и медные катализаторы
[366, 367], а также смешанные системы: HgCl2—CuCl—НС1—Н2О [371, 372].
Сравнение металлокомплексных катализаторов (табл. 3.18) показывает, что
наиболее активными являются системы CuCl—N-метилпирролидон —НС1,
HgCl2—CuCl—НС1—H2O и Pt(II) — НС1 — C2H5OH, которые в 1970-х гг.
I Hll IHIIH I I И I IIIHlH | f I UMIIII lllll.lll R4 I I >11 I >, II ' II • • III < > M
1 11 il III 1II II I h illl I 111 II Mil 1, ‘ 1 , 1 /(<! 'll N >i 1 ни*’ 1 Jhi irp.ii) pa
||).11 Hi 1 Illi №1 no ISO ’.Il 1 > I V 3. *711
llpl I t II. 1 in 1 II <> HI) 100 1Н» 1.0 l’75|
1 III 1 HI 1 II < > HO KI 1 .() 0.9 [376, 3771
1 III 1 ЦМФ lit 1 611 MI-70 0,25 |37H|
1 III 1 N mi 111Л1111ррпЛ11Д1>11 — 1 II 1 60 100 130 0,46 |37«|
Пн' 1 < hi III 1—ll.o 90 350-420 1,5-2,0 [371,3721
1*1(11) ll< 1 < 11.011 jo io 300 100 300 1379]
I и hm.i ii.ii.iu i Mi|>ui и. реакции | моль/л кл али затора в час], отнесенная к суммарной моляр
н мини nip.imiii аминных компонентов (металлов).
р и с мл । рнпались как Основа для реализации новых промышленных процессов
. нпи i.i винилхлорида.
hiiiieinha реакций гидрохлорирования исследовалась в разных сисгемах
ti.iioi < 19). кроме >миирическихуравнений (1, 3, 4, 8), получены теоретически
। н и к । и цы и 11ые кипе iические модели, учитывающие материальный баланс по ка
। uni i.iiopy, включающие концентрации (активности) компонентов раствора.
Ьппцсшрированные растворы солей, кислот и комплексов металлов Л1С1„
п(| 1.Щ.ПО1 особенностями, в частности: растворимость НО в воде и органичес
। их рас шорителях не подчиняется закону Генри [387, 388]; рост концентрации
I К I влияет на изменение растворимости ацетилена [388]; увеличение концен
। р 1ЦП11 I К I повышает закомплексованность катализатора (термодинамическая
и hi внос ib HgCl2 падает, рис. 3.53) [172].
I ложный и часто антибатный характер зависимостей разных параметров
н приводит к экстремальной зависимости [380] скорости реакции (г/ас,ц.) 1,1
। Hinn-Hi рации НО (рис. 3.53). Увеличение концентрации HgCl2 от 0 до 2 моль/л
приводит к увеличению кислотности раствора (й0) [389] и, следовательно, нц1()>
(г равнение 2, табл. 3.19). Поэтому наблюдаемый порядок реакции п по [Hg( l.|L
при пос 1ПЯННОМ парциальном давлении НО по уравнению г = k[HgCl2]xn в он
рс деленных условиях выше первого (1 < и < 2). Механизм гидрохлорированни
.ни । плена в растворах металлокомплексных катализаторов (L = СГ, Н2О, моле
куля расгворителя — л-ацетиленовый л-комплекс):
к,
ML„ + C2H2—~n + L, (3.43а)
Г
п г О ^^LnlMCH=CHCl, (3.436)
^-2
кз
I „ ,МСН=СНО + Н3О+ —- + СН2=СНО, (3.13»)
+ (3.43i)
Габлица 3.19 Кинетические уравнения р акции тндрихлирпривапия ац гнлена
Кинетическая система Кинетическое уравнение
1 HgCl2—NH.(C1 —НС1—Н2О кг = Инёа2]х |на]2рС2Н2
2. HgCl2—НС1—Н2О 2- г = (рс2н2aHgci2 ан,о+ + aci ан,о) / № аН3О+ + ^2 аС| )
з.сиС1—на—н2о 3.r = fcpc2H2[Cua]2
4 Сна—ын4а—на—н2о 4. г=(крС2н2 [Сиа]2[на]) / [NH4aj
5. cua—NH4a—на—н2о 5. г = /срс2н2 ан3о+ - ^Рс2н2 h0 аН2о
6. PdO2—Fea3—н2о 6. r= ([Pd(II)x] pc2H2)/U'i + ^гРс2н2)
7. Pda2 —HgCl2—Fea3—на—на—н2о 7.г=(Рн[На] [HgCl2]zpc2H2)/ (1 + ^Рс2н2)
8 pt(ii)—на—с2НяОн 8. г = (кр( 2н?Рна [Р^х) / (1 + b рна)
9.r=k[c2H2][Ha]3[Pth
h i’.i ini.ix жидкофа «ных t 11c гемах
Условия Константы к и нс гичгс к и х уравнений Л и।ера।ура
(><! ( 80 "( км = 4,94-10‘2, л2/(моль2 -ч-МПа) к№ = 0,19 л2/(моль2 -ч-МНа) Л Л Гаспарин, 1969 1375]
ID 90 "C £,=4,96-10 21 ехр (-3350/Т), (л-ч-МПа)/моль к2 = 2,63 10 9ехр (-4850/Т), (лч-МПа)/моль Н.Ф. Ллексеспа, 1970 [374, 380|
80 "( |l К l| = 5,5моль/л k6v = 5,7-10 7, л/моль -ч-МПа) к№ = 0,17, л/(моль-ч-МПа) Л.А. Гаспарян, 1968 [377]
80 4C к = 0,15, л/(моль-ч-МПа) Л.А. Гаспарян, 1969 [381]
80'( , |( llCl] = 11 моль /1кг H2O |NI 1,( l| + [HCI] = - 12 моль /1 кг Н2О к = 9,8-10 2, моль / (л ч-МПа) Г.К. Шестаков, 1970 [382]
ЧТ "( [ 11С1] = 3 моль/л, 111< 1] = 5 моль/л £, = 10 4,МПа-ч 1 к2 = 6,0-10 2, ч"1 С.М. Браиловский, 1983 [383]
’()"(11lgCl2] < 2 моль/л, |1 К 1| » |l.iCl] = 5 моль/л, |1\1(1,| = (05 6,7) • 10 3 моль/л, || <•( III = 1 моль/л - С.М. Браиловский, 1983 [384]
10 ’( к= 12,5-10 6,ч 1 -МПа 3 Ь = 7,5-10“2, МПа-2 Нгуен Зан, 1986 [385]
10 "( к = 5,0 л4/(моль4-ч) Г.К. Шестаков, 1985 [379, 386]
0,9
Рис. 3.53. Зависимости разных параметров от [НС1] в растворе HgCl2 (2 моль/л) — НС1
1'и<. 1.54. Влияние [СиСЬ] на скорость образования винилхлорида (гвх), дихлор ни ле! и
(!,,,) и суммарную скорость (гЕ = гвх + гдхэ):
1 * ня» ~ Гджэ» 3 Г£ — Гвх + Гдхэ
В монографии [172] рассмотрены доказательства правомерности этого ме
Hili >ма для ртутных и медных катализаторов.
< гадия образования тт-комплексов ацетилена с Hg(II) и Cu(I) доказана i
p.iooiax [366, 367]. Образование промежуточного а металлоорганическо! о со
гдпнения (ML„_1CH=CHC1) в случае ртути доказано его синтезом [366] и дан
Шамп, полученными при изучении протолиза цис- и транс ClHgCH=( Н(
| 190 191], а также специальными опытами [380] с введением окислителе!
(< п< .12) непосредственно в процесс гидрохлорирования ацетилена (рис. 3.54)
Цихлорид меди резко уменьшает скорость образования винилхлорида, при
пом в продуктах реакции появляется транс-1,2-дихлорэтилен, причем
уммарная скорость образования винилхлорида и транс-1,2-дихлор >1 и
'Н'па больше стационарной скорости образования винилхлорида, что го
нори г об обратимости второй стадии синтеза винилхлорида (см. рис. 3.53)
Аналогичный эксперимент в купрохлоридной системе [380, 391, 392] при
полит к таким же результатам, что подтверждает близость механизмов i
них системах. Отметим, что в условиях протолиза смеси цис- и транс /:
хлорвинилмеркурхлоридов введение СиС12 приводит к образованию смей
цис и транс-1,2-дихлорэтиленов. Появление 1,2-дихлорэтиленов в система.'
1 IgC 12— НС1—Н2О, CuCl—NH4C1 — НС1 — Н2О в присутствии СиС12 и в ре
акциях ClHgCH=CHCl с СиС12 является результатом окислительного деме
i пилирования транс /5-хлоровинильных производных Hg(II) и Cu(I). Обра ю
н.шие транс- 1,2-дихлорэтиленов указывает на внешнесферную атаку хлорид
попов л-комплекса ацетилена.
В стационарных условиях при предположении квазиравновесности перво!
। ыдии изложенного механизма схеме (уравнения 3.43а-г) соответствуем кипе
। ическое уравнение:
А|К1А2|Л1/|||( < . п Г||п1С(|
г =------- ---- д . (1 11)
k2H\ + ^Clio.{L]
В швисимости от соотношения констант скорости стадий, концентрации
компонентов, формы выражения скорости (функция суммарных или свобод
пых концентраций) это уравнение преобразуется в уравнения 2, 5-7 (табл. 3 19).
В соответствии с кинетическими данными (уравнение 2, табл. 3.19) в стадии ио
лучения хлоровинильного производного Hg(II) отщепляются два иона О :
Cl3Hg(C2H2) + Cl ^^ClHgCH=CHCl + 2СГ. (3.45)
В отличие от рассмотренного механизма нуклеофильного присоединения
С1 к ^ комплексу предложен электрофильный механизм гидрохлорирования в
растворах комплексов Cu(I) и Hg(II) [393, 394]. л-Комплекс ацетилена с метал
лом реагирует с протоном (НС1) с образованием стабилизированного металлом
винильного катиона £ИЛ4(СН2=С+Н), который, реагируя с СГ, превращается я
винилхлорид. Если принять во внимание, что Cu(I) и Hg(II), скорее всего, явля
ются акцепторами л-электронов ацетилена, а также информацию об электро
фильном присоединении к л-комплексам [172], предложенная схема представ
ляется весьма сомнительной. Кинетический изотопный эффект [394] кц/кр = 3
(kH/kD = 2,04-2,5 в стационарных условиях [395]) и высокая скорость обмена
протонов в ацетилене на медных системах могут быть объяснены и с других
позиций.
Сравнение кинетических изотопных эффектов в реакции гидрохлорирова
ния в растворах HgCl2 (табл. 3.20) и протолиза транс ClHgCH=CHCl в водной
НС1 (kH/kD = 2,0 [395]) показывает, что они близки. Отметим, что kH/kD про
толиза ClHgCH=CHCl в диоксане равно 1,10 [390] (1,25 при 50 °C [395]), что
объясняется предположением о лимитировании процесса стадией распада про-
тонированной формы ртутьорганического соединения.
Смешанные ртутно медные каталитические системы обладают большей
активностью, чем можно ожидать в случае аддитивного действия компонен-
тов катализатора. В работе [380] высказано предположение о возможности
обменной реакции между п-металлоорганическими соединениями ртути и
меди и, следовательно, появления нового маршрута реакции, который может
привести к дополнительному приросту скорости. Смешанная каталитическая
система отличается высокой стабильностью. Скорость накопления смол в рас-
творе при температуре обратного конденсатора 55 °C составляет 0,03 г/(л-ч).
В барботажном реакторе при конверсии ацетилена 98-100% производитель-
ность (по винилхлориду) по расчетным данным составит 350-420 г/(л-ч) при
90 °C ([CuCl] = 0,84-1 моль/л, [HgCl2] = 2,754-3 моль/л, НС1/С2Н2 = 1,14-1,3)
[371,372].
Основной побочной реакцией во всех водных каталитических системах
является образование ацетальдегида, содержание которого в продуктах ре-
акции зависит от концентрации кислоты и в оптимальных условиях не пре-
вышает 1%. Этот процесс объясняет в основном дезактивацию катализатора,
так как ацетальдегид образует продукты кротоновой конденсации и связы-
вает часть катализатора. Свободные от этих недостатков неводные каталити-
1й11|П1|й I Ml I Hill III li , I III |l*iHnlllll*ll ii|h|h h 11 I II llpnlh Hlllp'i' lii|'ll|<iili.llllin ||Ц<
i lilt Ihi
1*1 ПК 1(101 Mr i ii't'i hilbl'lll Hl Kip 1 lllipil 1СЛ1. /, < lv(>lll(l l< 1 |>.l 1(101 МОЛ1>//1 Ац/Кр
1 II ||| 1 (1 Н 1) llg(ll) Ila >(!>.(>) (>0 |llg(l..|-2 |1IC1|=5,7 1,82 f 0,1 1
1 II 1 IK 1 (|>( 1) < 4(1) II,O(1)2O) 60 [CnCI| =6 [NH|('lj =4,5 IFIC1] -4 [HCl] = 10,1 2,5 ±0,18 2,0010,18
( II । 1 It 1 (IX 1) Pt (II) C,H,OH (C2H2OD) 40 [Pt]x= 10 3 [HC1] =8 1,5 ±0,15
1 I» «IX 1(111 1) Pt (11) C,H,OD (C2H2OH) 40 [Ptk = W3 [HC1] =8 1,45 ± 0,12
( II НК 1(1X1) Pl(II) H2O (D2O) 30 [Pt]L = 10 3 [HC1] = 10 1,65 ±0,15
( 1» ИХ 1(П( 1) Pt (II) d2o (H2O) 30 [Pt]x= 10 3 |HC1] = 10 1,61 ±0,10
ml'iiiti СП lgCH=CHCl+ «1 К 1 (1X1) - H,O (D2O) 20 [HC1] = 9,7 2,00 ±0,16
III КС - Диоксан 50 [HC1] = 3,0 1,25 ±0,10
4i4 кие системы на основе Cu(I) и Hg(II) (см. табл. 3.18) описаны в многочис
лепных патентах, однако до последнего времени они не нашли практического
применения.
каталитические системы на основе PdCl2 проявляют достаточно высокую
нк । инпость, но они нестабильны из-за параллельных гидрохлорированию ре
акций олигомеризации и оксихлорирования ацетилена. Введение окислите
лей (1 еС13) повышает стабильность раствора. При этом стационарный режим
yi ыпавливается с приблизительно одинаковыми скоростями гидрохлориро
напил и окислительной олигомеризации ацетилена [383]. Механизмы про
цессон гидрохлорирования в растворах PdCl2 в медных и ртутных системах
аналогичны [383].
В 1975 г. появились сообщения о гидрохлорировании ацетилена в сиирло
пых растворах комплексов Pt(II) и Au(III). Платиновые катализаторы проявил
ioi высокую активность уже при 20-40 °C и концентрациях 0,001-0,01 моль/л и
по частоте оборотов катализатора на винилхлорид, моль/(г-атом-ч), значительно
превосходят все ранее известные катализаторы (см. табл. 3.18). Кинетическое
уравнение, полученное в работе [385], является эмпирическим (уравнение 8,
ыбл. 3.19) и не отражает специфику взаимодействия НС1 с раствори телем и
влияние НС1 на растворимость С2Н2. Исследования фазовых равновесий в бе i
иодной системе НС1-С2Н2-С2Н2ОН [Сц2о <1 мае. %] показали, что зависимое i н
[I IC1] отрНС] и [С2Н2] отрнс1 описываются уравнениями:
[С2Н2]
ncl|=- /’"‘l (, (-’ l(’)
* + ^i /’nd
(/с2 +к} к3- pHC1) • Рс2н2 ' Croh (3 47)
/ + Phci
где k'| = 32,4 МПа-1; fc2 = 0,356 МПа- ; fc3 = 0,14 МПа'1; CrOH — концентрация
спирта в системе HCI — ROH, является функцией рна и рассчитывается по и i
меренным экспериментально значениям плотности раствора и концентрациям
ПСЦ388].
Кинетическое уравнение процесса гидрохлорирования [379, 386, 388] выра
женное через концентрацию С2Н2 и НС1 (рис. 3.55), имеет вид:
r=fc[C2H2][HCl]3(Ptb (3.48)
Опыты с дейтерированным ацетиленом (C2D2) в системе
Pt(ll) — СН3СН2ОН— НС1 и с С2Н2 в системе CH3CH2OD—DC1 показали,
что атомы водорода ацетилена не обмениваются с атомами водорода среды
в растворах комплексов Pt(ll). В обоих случаях происходит шрансирисоеди
нение фрагментов молекулы НС1 к ацетилену. В этой системе при 30 °C А.ц/
/ср = 1,5 ± ± 0,15 [395]. Сравнение кинетических изотопных эффектов в раз
ных гомогенных каталитических системах (табл. 3.20) показывает, что к^/к^ в
растворах комплексов Cu(I), Hg(II) и Pt(II) довольно близки (1,5-2,5). Только
в медно-хлоридной системе наблюдается быстрый обмен ацетиленовых водо
родов на дейтерий с получением полностью дейтерированного винилхлорида.
Совокупность результатов, полученных с дейтерированными реагентами в
растворах Pt(II) и Hg(II), не противоречит рассмотренной выше схеме ме-
ханизма (уравнения 3.43а-г), постулирующей образование и протолиз соот
ветствующего трднс-0-хлорвинильного производного металла. Возможность
получения р-хлороалкенильного интермедиата в случае Pt(II) получила [397]
Рис. 3.55. Зависимость константы скорости к процесса гидрохлорирования от концен
трации кислоты Сна
под т нерждепие при изучении реакции Pt(C О)2(’12с С2К> (R=Pr) в толуоле при
комнатной температуре. Образовавшаяся цис р хлороалкепилытая группа ре
.пирует с С2П > и СО, чго приводит к димерному комплексу |RC(C1)=CRCR =
( А'(< O)l’tCI|2. Высокие порядки по НС1 при гидрохлорировании в растворах
плат и полого катализатора могут быть связаны с наличием ассоциатов хлоро
водорода в очень концентрированных по НС1 спиртовых растворах 13981 или
i явлениями специфического кислотного катализа, поскольку имеет место
< к ш иная зависимость кислотности (h0) от НС1 [399]: hp =Ь[НС1 ] .
(Обнаружена [400] возможность синтеза винилхлорида по следующей реак
) цни:
Катализатор
Bu ,N4 IC1 + С2Н2-------- Bu3N + СН2=СНС1, (3.49)
1Д< Ви — бутил.
В качестве катализатора использовали H[AuC14] (t = 90 °C, р = 0,185 МПа,
К.ч)
Авторы работы [401] на основании уравнения для скорости реакции (3.41)
(идрохлорирования ацетилена осуществляли моделирование процесса для сис-
Р'мы 1IC1 — С2Н5ОН—С2Н2. Для интервала парциальных давлений ацетилена
. 40 КПа и хлороводорода 10-70 КПа были получены следующие выражения,
. ия 1ывающие парциальные давления реагентов с их концентрацией в растворе:
с _ 66»76Рнс1 (3.50а)
НС1 1+5,811рнс1’
с = °>73?НС1 (1 + 1>27Рнс1) (3.506)
1+5,811рнс1 С'Н/
Выло показано, что при использовании каскада реакторов идеального пере
мсшивания суммарный объем системы находится в обратной зависимости от
давления реакторов на входе в систему (рис. 3.566). Для единичного реактора
ывиеимость объема реактора от давления представлена на рис. 3.56д. Общая
конверсия ацетилена превышает 96%.
Технологические аспекты жидкофазного гидрохлорирования ацетилена
рассмотрены в монографии [172]. Процесс может быть осуществлен в водной
i реде и в среде органических растворителей. В качестве катализаторов приме-
няют в основном хлориды ртути и меди (1), либо их смеси. Используют также
.меси хлорида меди с хлоридом аммония, хлоридом кальция, хлоргидратами
। ри:> таноламина, моно-, ди и триметиламинов. Катализатор может быт ь либо в
рас । воре, либо в виде суспензии.
Реакция гидрохлорирования в водной среде осложняется побочной реакци
сн образования ацетальдегида. Для устранения побочных процессов к хлори
дам ргути и меди (и др.) добавляются фосфин и алкиларилфосфины. Для предо
। вращения деактивации катализатора восстановителями, присутствующими в
щетилене в качестве примесей, в водные растворы дихлорида ртути добавляют
хлориды железа [402].
р, МПа
Рис. 3.56. Зависимости объема реактора (а) и системы реакторов (б) от давления р при
постоянных давлении в реакторе (1) и объемной скорости (2); п — количество реакторов
В качестве неводных растворителей предлагается использовать хлоргидра-
ты диметилформамида и N-метилпирролидона, а также тетрахлорид олова.
Основной сложностью при реализации процесса жидкофазного гидрохло-
рирования ацетилена является выбор конструкционных материалов для реак-
тора с обвязкой, насосов, арматуры и т.д.
Процесс жидкофазною iидрохлорироиапня ацетилена проводя! при >0
') > "( в реакторах барботажного типа. Концентрация хлороводорода в воде
должна Оми. не менее 5%. Конверсия ацетилена за проход составляет Ю 50%
в । медных кагалигшторих и 75-90% на ртутных.
(к ионным преимуществом жидкофазного процесса является относится!,
пая легкость решения проблемы теплосъема, а, следовательно, и укрупнения
реакционного аппарата. К недостаткам процесса следует отнести более низкие
конверсию ацетилена и селективность процесса, а также большую сложность
аппаратурного оформления реакционного узла и технологической схемы.
\ Один из вариантов технологического и аппаратурного оформления про
цесса жидкофазного гидрохлорирования ацетилена, по показателям близкий к
га 1о(|>азному, был предложен в [372]. Авторы показали, что при растворении
хлорида ртути в солянокислом растворе протекают следующие реакции:
HgCl2 + Cl ^=^[HgCl3]“, (3.51а)
[HgCl3] + Cl ^ [ HgCl4] г~. (3.516)
Ион [HgCl4]' не может активировать ацетилен, так как является коорди
национнонасыщенным комплексом. Хлорид двухвалентной ртути обладав!
наибольшей активностью и может способствовать протеканию побочных ре-
акций, однако в условиях избытка НС1 концентрация HgCl2 относительно неве-
лика. Избирательное гидрохлорирование обеспечивается комплексом [HgClJ .
Присутствие в катализаторном растворе второго компонента, способного к
комплексообразованию с СГ, обеспечивает увеличение концентрации [HgCl.] ,
а следовательно, и активности раствора в результате уменьшения концентра-
ции комплекса [HgCl4]2".
Повышение активности раствора способствует, в свою очередь, образова-
нию поверхностно-активных веществ. Вследствие резкого увеличения поверх
пости контакта газ/жидкость уменьшается индукционный период, обусловлен
ный диффузионным торможением, повышаются конверсия ацетилена и произ
водительность по винилхлориду.
В результате проведенных исследований для осуществления гидрохлори
рования в промышленных условиях авторы [372] рекомендовали раствор, со
держащий двухвалентную ртуть и одновалентную медь. Выход винилхлорида в
>том случае составляет 97-99% на прореагировавший ацетилен, съем С2Н3С1 в
реакторе барботажного типа — до 154 г/(л-ч). При проведении гидрохлориро
вания в реакторах других типов за счет снятия эффекта диффузионного тормо
жения возможно повышение съема винилхлорида. Так, при той же конверсии
ацетилена съем винилхлорида в реакторе типа «эрлифт» составлял 250 г/(л-ч), в
реакторе с принудительной циркуляцией — 500 г/(л-ч).
Контактный раствор, содержащий HgCl2 и BiCl3, по активности не уступает
ртутно-медному, но отличается от него нестабильностью во времени вследс-
!вие значительного смолообразования. Раствор, содержащий хлориды олова,
>акже нестабилен; кроме того, в этом случае до 15% ацетилена расходуется па
побочные реакции.
При длительной работе катализаторного раствора на основе солей ртути и
меди в нем накапливаются смолообразные продукты. Часть продуктов находи г
ся в растворе, час! в оседает па гениях реак гора В результате накопления смол
активность катализаторпогп раствора со временем падает. Данные тлементпого
анализа смолы (суммарное содержание yiлерода и водорода ~85%) позволяет
предположить, что она образуется из ацетальдегида. Скорость смолообра юва
пия непостоянна во времени и достигает максимума через 70 ч после начала
работы катализаторного раствора. В дальнейшем скорость смолообразования
не изменяется. Если выводить часть раствора на регенерацию, удастся зпачи
гельно снизить интенсивность смолообразования. Как видно из рис. 3.57, при
выводе на регенерацию 1,25% объема катализаторного раствора в 1 ч ин ген
сивность смолообразования снижается почти вдвое. Содержание примесей в
винилхлориде не превышает 0,1-0,2%.
Для поддержания стабильной работы катализаторного раствора необходи
мо поддерживать соотношение НС1:С2Н2 в пределах (1,1-1,2):1 и соблюдать ба
ланс по воде.
Исследованиями установлено, что в реакторе гидрохлорирования ацетиле
на реакция осложняется диффузией, а реальные кинетические закономернос-
1 и процесса определялись истинной кинетикой реакции и условиями массо и
теплопереноса. Анализ влияния скорости газа, его содержания и высоты бар
ботажного слоя на степень превращения ацетилена показал, что оптималь
ной областью осуществления реакции является пенный режим. На основании
имеющихся данных в работе [172] предложен вариант технологической схемы
(рис. 3.58).
Каталитический раствор готовится в аппаратах с мешалкой 1, 2, куда загру-
жаются соляная кислота, хлорид меди с медной стружкой (1) и оксид ртути (2).
Полученные растворы подаются в пустотелый реактор колонного типа 3, куда
Рис. 3.57. Зависимость скорости смолообразования W от длительности работы катали
заторного раствора (Т = 90 °C, скорость подачи ацетилена 80 ч'1, объем раствора = 400
см1, [HgCl2] = 2,75 моль/л, [CuCl] = 0,8 моль/л):
I — без вывода раствора на регенерацию 2 — 0,625%, 3 — 1,25%, 4 — 2,5% объема раствора в час
выводится на регенерацию
Соляная кислота
ф
X
Рис. 3.58. Технологическая схема процесса жидкофазного гидрохлорирования ацетилена (1-11 см. по текст'
(>ар(><»гирус1ся смесь хлороводорода и ацетилена в соотношении 1,1:1,0. В реак
юре поддерживаются концентрации HgCB 2-3 моль/л, CuCI 1,2 моль/л и icm
нература 90 °C. Часть каталитического раствора (0,2%) посюянно выводится
для регенерации через фильтр 5 и экстрактор 6, где извлекаются смолообразные
продукты путем их растворения в дихлорэтане. Потери восполняются свежим
каталитическим раствором.
Съем тепла осуществляется за счет рециркуляции реакционной массы чере i
выносной теплообменник 4, а также испарения воды, с которой уходят обра
зующийся винилхлорид, непрореагировавшие ацетилен, хлористый водород и
органические примеси. Реакционные газы поступают в промывную колонну 7,
1де отмываются водой от хлороводорода и ацетальдегида. Образующаяся со
ляная кислота, содержащая ацетальдегид, подвергается отпарке в колонне 8 и
возвращается на приготовление каталитического раствора. Отпаренная орга
ническая часть выводится через экстрактор 6, куда подается с дихлорэтаном.
Винилхлорид с верха колонны 7, проходя через осушитель 9, направляется на
ректификацию для отпарки ацетилена в колонне 10 и выделения товарного про
дукта в колонне 11.
Конверсия ацетилена за проход составляет 98-100%, селективность до
99%. Выход винилхлорида составляет 350-420 г/л реакционного объема в час.
В процессе образуется до шести примесей, общее содержание которых не пре-
вышает 0,15%. Единственным побочным продуктом, содержание которого до
ходит до 1%, является ацетальдегид. При отборе 0,25% каталитического раство
ра на извлечение осмола каталитический раствор отличается высокой стабиль-
ностью.
Весьма перспективны для жидкофазного процесса платиновые катализате
ры в водных и неводных растворах. Однако еще недостаточно изучены длитель
ность работы и методы регенерации при их использовании.
Процесс жидкофазного гидрохлорирования ацетилена так и не был ре-
ализован в промышленных условиях. В настоящее время, когда винилхлорид
повсеместно получают из этилена, вероятность осуществления этого процесса
на практике еще более снизилась. Тем не менее, когда ацетилену как источни-
ку углеводородного сырья нет альтернативы (например, ацетилен образуется
как побочный продукт при пиролизе метана, где целевым продуктом является
водород), процессы получения винилхлорида из ацетилена могут применяться
еще долгое время. Реализация жидкофазного процесса теоретически возможна
прежде всего по экологическим соображениям (отказ от использования суле-
мы); выбор метода должен быть продиктован данными технико-экономическо-
го анализа.
Глава 4.
Получение винилхлорида из этилена
по сбалансированной схеме
< Н — ... — — _ - CHCI
4.1. Принципы сбалансированности и их использование
в производстве винилхлорида
В середине 1960-х годов процессы получения винилхлорида щелочным
омылением дихлорэтана или гидрохлорированием ацетилена начали новее
м<ч ню вытесняться процессами по сбалансированной схеме на основе этиле
нового сырья. Это стало возможным благодаря реализации в промышленных
1ОВИЯХ процесса окислительного хлорирования (оксихлорирования) этилена
получением дихлорэтана. При оксихлорировании этилена происходит пран
1НЧССКИ полная утилизация хлороводорода, образующегося на стадии получе
ни» винилхлорида пиролизом дихлорэтана. Впервые реализация сбалансиро
। ninon) цикла была осуществлена компанией «B.EGoodrich» (США) с вводом
। промышленную эксплуатацию в апреле 1964 г. процесса оксихлорировапия
и плена мощностью 136 тыс. т дихлорэтана в год [403|.
Рассмотрим более подробно понятие сбалансированности в промывшей
мОм хлорорганическом синтезе. В широком смысле слова под сбалансирован
цо< |ыо понимается полное использование сырья и энергии, включая при этом
in черпывающую и квалифицированную утилизацию отходов. Это имее1 при
юс опюшение к производству большинства хлорорганических продуктов, ос
и..ншому на реакциях прямого и окислительного хлорирования, гидрохлори
ров шия и дегидрохлорирования.
В промышленном хлорорганическом синтезе под сбалансированностью
процессов принято понимать применение хлора в виде хлороводорода в реак
цпих оксихлорирования и гидрохлорирования [404].
Одной из основных проблем промышленного хлорорганического синтеза
как по эффективности переработки хлорного сырья, так и в экологическом <>
шипении является утилизация отходящего (абгазного) хлороводорода. В про
ншодстне хлорорганических продуктов в результате хлорирования и дегид
ро.члорпрования неизбежно образуется хлороводород, основным способом
in рерлботки которого до недавнего времени было его превращение в соляную
1 ш югу с последующей ее утилизацией. Однако загрязненная хлорортаничес
ними соединениями «абгазная» соляная кислота не всегда находит сбыт, а ме
1оды се очистки до необходимых требований достаточно трудоемки. В связи <
ним проблема квалифицированного использования абгазного хлороводорода
11 (ьма важна. Наиболее целесообразным при решении данной проблемы яв
нити возврат хлороводорода для получения хлорорганических продуктов, в
рг lynbiare чего производства становятся сбалансированными по хлору, т.е. весь
но ыппый хлор расходуется только на целевые продукты. Основными методами
lepepaooi кп аОга.пюго хлороводорода являются процессы I ндрохлорированпи
1 окисли тельного хлорирования. Комбинацией стадий хлорирования, ди идрп
Одорирования и окислительного хлорирования удастся создать соалапс upon.in
цяе по хлору процессы получения практически всех многогоннажпых хлору i
1еводородов.
। Существуют понятия «внутренних» и «внешних» сбалапсировапиы^с чем
1404), где хлороводород используется в одной технологической схеме, напри
liep, при производстве винилхлорида из этилена или хлорме танов иа меч апл
1 метанола («внутренняя» схема), либо в разных технологических процессах,
пшример, при гидрохлорировании ацетилена хлороводородом, отходящим oi
производства хлорметанов («внешняя» схема). Мы полагаем, что принципам
гбалансированности в большей мере отвечают «внутренние» схемы, хараме
^изующиеся единством сырья и технологии. Именно к таким схемам относ пня
Сбалансированный по хлору процесс получения винилхлорида.
На рис. 4.1 приведена упрощенная блок-схема сбалансированного по хлору
1роцесса получения винилхлорида.
Процесс включает три основные реакционные стадии:
прямое хлорирование этилена
СН2=СН2 + С12 —С1СН2—СН2С1 + 188 кДж, (4.1а)
окислительное хлорирование этилена
СН2=СН2 + 2НС1 + 0,5О2 — С1СН2—СН2С1 + Н2О + 238 кДж, (4.1 о)
НО
Рис. 4.1. Блок-схема сбалансированного процесса получения винилхлорида
иii)• । iii । iiiinnn)< цини
i li II (Illi • ’( II < ll< I i ’IK I I 1()кДк (-1 In)
I . MMiipilo
'(II ( II I ( J I (>,'.() » 2( 11 ( IK I i 11.0 (11)
I и uh inn hi приведенных схем, i гадин прямого хлорирования этилена с
1)о>цч1 inn-м дополни к'Л1.по|о количсс । на дихлор папа необходима для обесие
инпи । ыдип ом нхлорнропания нужным количеством хлороводорода (на об
। iiHiainii I мн in ДХ ) на i 1адии расходуемся 2 моля НС1).
11роц<чс получения винилхлорида но сбалансированной схеме сочетает три
и ниц । mi iiiBepiHciiiio ра nn.ie стадии. Жидкофазный процесс электрофильно
ы Ирш осдппення хлора к этилену при 50-120 °C, газофазный гетерогенноката
иtii'ii i кип процесс оксихлорирования этилена при 210-250 °C и наконец, ра
пн л'П.но цепной процесс юрмичсского дегидрохлорирования дихлорэтана при
Kai ’О ’( Отметим, что процессы прямого и окислительного хлорирования
Hinn на харзмери |уются конверсией исходных реагентов, превышающей 98%,
и 11 лгк IНННО1 । ыо образования целевого 1,2- дихлорэтана на уровне 96-99%. Се
и । iHuiioi и. образования винилхлорида на стадии пиролиза превышает 98%.
I '1/1114(4 ню хдорорганических отходов, образующихся при достижении таких
ши и кнелей процесса, не превышает 25-30 кг на тонну винилхлорида, т.е. про
ш । < и целом может быть отнесен к категории малоотходных. Химия и техноло
I ни кл к дои и «стадий сбалансированного процесса будут рассмотрены в следую
Huix ра щепах. Здесь мы приведем лишь основные особенности этих стадий.
( у щесгвует два основных варианта процесса прямого хлорирования этиле
пи ни ।ко температурное (НТХ) и высокотемпературное (ВТХ). За многие годы
p.i ши । ня у обоих процессов появился ряд модификаций.
При 11 ГХ хлорирование этилена проводят в жидком дихлорэтане, содер
к ищем небольшие концентрации трихлорида железа в качестве катализатора.
I'. в к юр снабжен внешним охлаждающим теплообменником, поддерживающим
и мперагуру реакции ниже температуры кипения ДХЭ. Соотношение между
хлором и этиленом приблизительно равно стехиометрическому. Для подавлс
нпи образования побочных продуктов, в частности трихлорэтана, в систему
1п(>.Н1ля1от небольшие количества кислорода или воздуха. Концентрация три
хлорида железа обычно ниже 100 млн *. Хлор превращается количественно с
( снек 1ИШЮСТЫ0 по ДХЭ выше 99%. Реактор обычно изготавливают из углеро
Д1Н юн стали вследствие мягких условий ведения процесса. ДХЭ, полученный
при 11 ГХ, далее направляется на очистку и ректификацию.
(Уличительная черта ВТХ: теплоту реакции хлорирования можно испоив
lon.iib для испарения и дистилляции ДХЭ из смеси реагентов. По существу,
реактор хлорирования служит испарителем ректификационной колонны. Нос
польку теплота реакции приблизительно в шесть раз выше теплоты испарения
ДХ ), ее можно использовать также для дистилляции ДХЭ, как полученного и.)
стадии оксихлорирования, так и непрореагировавшего при пиролизе. Таким
пора юм, ВТХ снижает энергетические затраты на получение винилхлорида.
Высокотемпературное хлорирование осуществляется в диапазоне темпера
। ур от точки кипения ДХЭ при атмосферном давлении (83,5 °C) до 130 °C. Жид
кая фаза состои! преимущес 1 нечто и.» ДХ.), н котором основными примесями
являются другие хлорированные углеводороды С2. Часы, peai ирующей жидкое
ги периодически удаляют, чтобы поддержать желаемый состав смеси. Как и при
II ГХ, катализатором служит грихлорид железа, концентрация которого лини,
слабо влияет на селективность. Для хлорирования можно использовать газооб
разный или жидкий хлор; этилен обычно вводят в реактор в некотором избытке
но отношению к хлору. Для подавления побочных реакций используют кисло
род или воздух. Селективность образования ДХЭ превышает 99%.
Оксихлорирование этилена может протекать в реакторах с псевдоожижен
ным или неподвижным слоем катализатора. Окисляющим агентом является
концентрированный кислород или кислород воздуха. В качестве катализатора,
как правило, применяют хлорид меди на разных носителях, отводят тепло обыч
но кипящей водой. Выбор катализатора псевдоожиженного слоя определяется
прежде всего его стойкостью к истиранию, способностью к псевдоожижению в
условиях процесса и селективностью. В системе с неподвижным слоем темпера
тура процесса непосредственно связана с активностью катализатора. Местные
перегревы устраняют путем разбавления катализатора инертным материалом
Эффективность использования этилена в процессе достигает 98%, побоч
ные продукты — оксиды углерода, а также хлорированные органические со
единения С]-С2. Дихлорэтан-сырец после промывки и осушки направляют на
ректификацию.
Пиролиз дихлорэтана с получением целевого продукта — винилхлорида
осуществляют в трубчатых печах. В большинстве установок конверсия ДХЭ
за проход составляет 50-60%. Для достижения такой конверсии температуру
газов на выходе из трубок печи поддерживают равной приблизительно 500 °C
Профиль температур по длине трубки зависит от конструкции печи и приня
того технологического режима. При высоких температурах глубина пиролиза
повышается, что увеличивает выход побочных продуктов и усиливает коксооб
разование. Для очистки трубок печи необходимо периодически проводить их
регенерацию. Большую часть ДХЭ, непрореагировавшего в печах, выделяют и
вновь возвращают в процесс. Таким образом, устанавливается оптимальное со
четание производительности по ВХ, его выхода и чистоты со стоимостью пов
торного выделения ДХЭ и простоев [199].
Термин «сбалансированность» в промышленном хлорорганическом синте-
зе должен распространяться также на использование углеводородного сырья и
энергии.
Для углеводородного сырья сбалансированность достигается полным или
максимально возможным возвратом отходов в технологический цикл. Это мо
жет быть реализовано, в частности, селективным гидродехлорированием обра
зовавшихся хлорорганических отходов в исходный углеводород и хлороводо
род.
Например, переработка хлорорганических отходов производства винилхло-
рида селективным гидродехлорированием с получением смеси этана и этиле-
на с преимущественным образованием последнего дает возможность создать
максимально возможную «внутреннюю» сбалансированную схему с рециклом
газа, содержащего этилен-этан-хлороводород, на стадию окислительного хло-
рирования.
( балансироваппо* 1ь процесса no теплу может достш ai i>c »i pa шымн мс!ода
ми Ik ноль lonaniie тепла реакции для целей выделения или разделения продук
кш является наиболее распространенным и освоенным в промышленноеги.
Анализ сбалансированного производства винилхлорида показывает, что
оно имеет ряд внутренних источников тепла: теплота реакций па стадиях
х нитрования и оксихлорирования этилена, переработки и сжигания отхо
ц»в. а также теплота отходящих реакционных и дымовых газов на стадии ни
роли ia
В действующих производствах винилхлорида сбалансированным меюдом
hi пилена теплота реакции утилизируется практически на всех стадиях. На
пример, па стадии оксихлорирования этилена образуется низкопотенциальпый
и ip, направляемый обычно в общий коллектор. Утилизация теплоты реакции
прямого хлорирования для ректификации образующегося при этом ДХЭ, а ык-
1.1 ДХЭ со стадий пиролиза и оксихлорирования реализуется использованием
ргиктора в качестве кипятильника ректификационной колонны. Особенность
। ihoi о способа заключается в том, что процесс проводится при температуре к и
п< пня ДХ ), и его отбор в паровой фазе позволяет исключить на стадии прямо) о
|<1рирования кислотную и воднощелочную отмывку дихлорэтана от катализа
Hip.i (хлорного железа) и хлороводорода, в результате чего общее количество
।очных вод в производстве сокращается в 1,5 раза. Соответственно умепыпа
юн я и все затраты на очистку сточных вод, в том числе и расход пара для от
и |рки ДХЭ из сточных вод.
11а стадии пиролиза ДХЭ теплота пирогаза может быть использована, па
пример, для подогрева исходного дихлорэтана, подаваемого на пиролиз, а теп
ноia дымовых газов — для получения пара.
При утилизации теплоты реакции в сбалансированном процессе получе
инн винилхлорида на стадиях прямого и окислительного хлорирования, а так
i t и плоты реакционных газов пиролиза ДХЭ может быть использовано более
HH ’ii шергоресурсов. ,
( ш 1ематизированный анализ материальных и тепловых потоков проит
нпде та винилхлорида по сбалансированной схеме приведен, например, в ра
h.ii.ix |405, 406].
В производствах винилхлорида, использующих традиционную технологию
1*Ж() х 1одов (оксихлорирование с использованием воздуха, низкотемпера гур
in к хлорирование, термический пиролиз), количество утилизируемого тепла
in превышает 40% тепла, выделенного на реакционных стадиях процесса. Доля
V i и in тируемого тепла материальных потоков не превышает 5%; однако она мо
। <>ы । ь увеличена до 15-25% введением ряда усовершенствований.
(>< ионные принципы в технологии получения винилхлорида сбалаисиро
винным по хлору методом сформулированы и систематизированы В.С. Гимо
|» • ным и Л.А. Серафимовым [407].
Ангоры указывают, что технология получения винилхлорида сбалаисиро
ванным по хлору методом (комбинация хлорирования и оксихлорировапия
н и лена с термическим дегидрохлорированием 1,2-дихлорэтана) выступае! од
ним in наиболее интересных примеров реализации принципов создания гех-
ни ни пи основного органического и нефтехимического синтеза. Технология не
пр< рынка. 11о химической составляющей, несмотря на наличие трех отдельных
pe.iKiopiu.ix подсистем, ее можно oiiiec ги к дпухстадииной — каждая из цсцги
химических превращении, ведущих к винилхлориду, сектой i из днух стадии
оксихлорирование + термический пиролиз и хлорирование + термический ни
ролиз.
Эти два параллельных процесса связаны, во-первых, рециркуляционным
потоком по хлороводороду, что позволяет почти полностью его утцли 1Иро
вать, а во-вторых, общей стадией термического пиролиза, использующей ди
хлорэтан как оксихлорирования, так и хлорирования этилена. Суммарные
потери хлора составляют всего 20-25 кг, а этилена 15-20 кг на 1 т товарною
винилхлорида. Большая доля потерь этилена связана с процессом его полно
го окисления на стадии оксихлорирования (6-10 кг на 1 т винилхлорида), л
хлора — на всех реакционных стадиях примерно в равном соотношении. Га
ким образом, комбинирование двух процессов в одной технологии позволяй
с использованием рециркуляции по образующемуся хлороводороду свести
потери сырья к минимуму и одновременно обеспечить эффективную защи ту
окружающей среды от хлора и хлороводорода. В данном случае реализуется
принцип организации рециркуляционных потоков по компонентам. Другой
иллюстрацией данного принципа служит рецикл по 1,2-дихлорэтану. Этот но
ток обеспечивает полную конверсию 1,2-дихлорэтана на стадии термического
пиролиза и используется из-за того, что конверсия за один проход на этон
стадии не превышает 48-55%.
Технология базируется на использовании дешевого и доступного этилена и
хлора, высокоэффективна в целом, хотя отдельные ее составляющие разнятся но
этому показателю. Например, хлорирование этилена обладает более высокой се
лективностью по сравнению с оксихлорированием и тем более с термическим
пиролизом. Стадии оксихлорирования и хлорирования имеют высокие конверсии
за один проход. Рециркуляция части реакционных газов на стадии оксихлориро
вания связана в основном с необходимостью обеспечения газодинамического и
концентрационного режимов при использовании аппарата с псевдоожиженным
слоем.
Эффективное использование теплоты (принципы разработки процессов с низким
энергопотреблением полноты использования энергии системы) в данной технологии до
стигается не только за счет ее утилизации в подсистеме ректификационного разделе
ния, но и путем теплообмена между экзотермичными (хлорирование, оксихлориро
вание) и эндотермичными (пиролиз) стадиями процесса (хотя реализация такого
теплообмена технологически достаточно сложна).
Принцип полноты выделения продуктов из реакционной смеси используется до-
статочно полно, поскольку как целевой продукт, так и 1,2-дихлорэтан, направляе-
мый на пиролиз, должны иметь высокую чистоту.
В рассматриваемой технологии используется принцип минимального расходования
воды, так как в ней практически отсутствуют промывные скрубберы, а хлороводо-
род выделяют в ректификационной колонне при повышенном давлении. Использо-
вание для хлорирования этилена совмещенного процесса позволяет, по сравнению с
реакторами низкотемпературного хлорирования, наиболее интенсивно применять
низкопотенциальное тепло хлорирования для предварительного фракционирова-
ния продуктов реакции (снижение энергозатрат на выделение 1,2-дихлорэтана на
50-70%).
Ill I Ill'll lib I >l> I Illi Uli* Il II >lli. |>I| UH HU III! Il II pl I IIII.HHHI ПрИНЦППП III'IllOIIIhl
Ih Ill" II' > ilhlllllll illHilt-H llllllliihillt II il'llh lllhll II li>ni<n .. Illlll ly III.KllKoll 11114ПЧ
IIIII III IU||.| II I Illi III null IIHII В IK |4iy hi 11’11 рсд|. I< IIU nil Illi Iilli'i III'HIII.IC I у III
iiiiinniii хлороводорода в pr lyni.i .nr <нч ii.Miiipiipiin.iiiiiii iiiinrii.i Реакционные
illll.I|>.III.I i II.HI I I III.I III Ill'll.Ml ПОДПЫМ11. Illi II p.KllUll.llblMII КО11Д1Ч11 .lllip.lMH, KO
lupl.li Д.11И1 Illi IMO I III И II. i Illi III 11. lll.ll'ipiK 1.1 Xllupiipi.lllirici. KI1X продух I OB II .11 MOI
i|u py 1.1 i'll 1 in nice hi.kuKoii i iriiciin их конденсации при пониженных темпера
ij p.tx. |1ыдснг1нн хлороводорода h i реакционной массы пиролиза проводи к и
pi । ।iii|iiiiiai(ii<’ii Hio д.1С1 во 1М<1Ж1П1СГ|> непосредственно организовать его ре
цикл пл i 1.1ДНК1 OKI ихлориропания, избежать процессов абсорбции его водой и,
Kiuiiieii 1 пенно, кислотных и солевых стоков Наконец, технология позволяет
индапап. липни отпчнои единичной мощности. Реакционные подсистемы ок-
i нчлириропапия и пироли ia и используемые в них реакционные аппараты дани
niiiMioKiHii и, их проектирования на любую требуемую производительность. Ре
.ин 1.ЩИН кого принципа для стадии хлорирования может быть осуществлена
применением параллельно работающих жидкофазных хлораторов так, чтобы
in и 1схнологическая цепочка представляла собой линию большой единичной
М1НЦП01 1И.
Ьолыпая часть материальных потоков в сбалансированной схеме произ
ППД1 ina винилхлорида приходится на дихлорэтан, как синтезируемый, гак и
рецикловый. Именно условия его синтеза и очистки определяют техники-эко
комические показатели производства в целом. По этой причине мы приводим
ш ионные свойства этого важного полупродукта в производстве винилхло
рида.
4.2. Основные свойства 1,2-дихлорэтана
1,2-Дихлорэтан (этиленхлорид, хлористый этилен, сим-дихлорэтан) —
< 11,С12, СН2С1—СН2С1, молекулярная масса 98,96 — наиболее ранний hi
к ex известных хлорированных углеводородов — впервые был синтезирован
।оллапдскими химиками И. Дейманом и П. ван Труствиком в 1795 г. [28|, и
к'чение многих лет известен под названием «масло голландских химиков».
11 рн комнатной температуре ДХЭ — бесцветная летучая жидкость со сладко
па । ым запахом: tKnn = 83,47 °C; 1ПЛ = -35,36 °C, при влажности менее 0,1% i И1
рископичен. Физические и химические свойства 1,2-дихлорэтана достаточно
подробно представлены, например, в работах [29, 30, 34].
Физические свойства
11лотность:
/. "С..... 0 15 20 30
р. г/см3.. ..1,282 1,260 1,253 1,238
Относительная плотность пара по воздуху d = 3,4.
Температурный коэффициент объемного расширения при 0-30 *’(
|1 0,00117 °C’1.
Таблица 1.1. Да и'ни нс парив
t, ”( р, кПа /, "С />, к Па р, М11а 1,°С р, МПа
44,5 0,13 5,80 4,00 83,7 0,101 183,5 1,013
-30,0 0,43 10,0 5,33 100,0 0,152 200,0 1,379
-24,0 0,67 18,2 8,00 108,1 0,203 226,5 2,026
-20,0 0,90 29,4 13,33 140,0 0,413 254,0 3,039
-13,6 1,33 40,0 21,33 147,8 0,507 272,0 4,053
-10,0 1,72 46,2 26,66 180,0 0,827 285,0 5,066
-2,4 2,67 60,0 46,66
0 3,04 5,8 53,32
Параметры критического состояния:
Давление..........5,37 МПа
Температура.......288 °C
Плотность.........0,440 г/см3
Вязкость динамическая
а) жидкости:
t,°c.............................. 0 10 15 20 25 40 50 70 80
р, мПа-с... .1,077 0,962 0,887 0,840 0,780 0,652 0,565 0,479 0,417
6) пара:
при 83,5 °C р - 16,8 мкПа-с
Поверхностное натяжение:
t,°C....-20 0 15 20 25 30 40 60 100 140
а, мН/м ... 37,2 34,1 32,9 32,2 31,8 30,8 28,3 27,4 22,5 17,5
Коэффициент преломления:
1, °C.... 15 20 25 30
nD‘°.... 1,4476 1,4448 1,4421 1,4393
Коэффициент преломления пара nD° = 1,00134.
Теплоемкость
а) жидкости:
t, °C .. -20 0 20 40 60 80 100 120 140
ср, кДж/(кг-К) .. . 1,259 1,276 1,289 1,310 6) пара: 1,335 1,372 1,410 1,469 1,569
t, °C ...0 100 200 300 400 500 600
ср, кДж/(кг-К).. .. 0,774 0,891 1,004 1,105 1,192 1,276 1,347
|||11'11>Ц|| I I > »> >1 >111 *1 I >1 II I ' Multi lit II ll.lpil
В и и III III ll.i|>
/. < X, 11|/(м h) 1. < А Hi/(m k)
(1 0.1 1 >0 0,0067
(ill 0,127 100 0,0087
100 0,121 200 0,0131
1 ’0 0,122 300 0,0184
1 10 0,120 400 0,0243
500 0,0306
leiuiora испарения:
I ( ........20 60 83,5 100 140 180 220 260
./ „ кД k/m ... 353,5 334,7 323,4 318.0 292,9 265,7 217,6 146,1
Icnaoia сгорания (пара) qcr = 1134,6 кДж/кг.
leiuiora плавления qnn - 88,4 кДж/кг.
I сило i а образования стандартная ДН^я = -129,7 кДж/моль.
)п । ропия вещества в стандартном состоянии .$298 = 308,82 Дж/(мольК).
Дипольный моментре - 5,7 1 0 30 1<л-м.
Ди »лск । рическая проницаемость жидкости:
I '( . 20 25 30
। ..10,65 10,36 9,96
Ди wick грическая проницаемость пара: е - 1,00481 при 120 °C.
Iiiii ища 4.3. Растворимость
I < C2H4CI2 в воде Вода в С2Н4С12
Расгвори- мость, % f,°C Раствори- мость, % Г, °C Раствори- мость, % f,°C Рас гвори мос ть, %
0 0,91 40 0,97 0 0,09 40 0,26
III 0,84 46,5 1,02 10 0,11 50 0,34
’ll 0,87 56,5 1,13 20 0,16 57 0,35
) i 0,86 67,5 1.29 25 0,19 69 0,52
<h 0,86 72,5 1,38 30 0,22
I Ц|1к.|п|> Нин хорошо растворяется в спирте, эфире, нефтяных углеводородах, плохо рас гни
рш 1> и и воде.
Расширимость пилена u 1,2-дихлор мане [191, 108]:
/I, MM pi. er. 50 100 200 "410 10() 500 bOO 700 («0
В 0,25 0,55 1,15 1,80 2,40 3,00 3,55 1,15 1 .0
Абсорбционный коэффициент Бунзена, т.е. объем газа при (0 "С, 760 мм рт. ст.), абсоронропэн
ного н единице объема жидкости. -х
Растворимость этилена в органических растворителях в общем виде описы
вается уравнением [409]:
lg kpv - А - ДН/(2,3 RT),
(1.2)
где к - Y/X (Y— мольная доля этилена в газовой фазе, %; X — мольная долл
этилена в растворе, %); р — общее давление, МПа; v — коэффициент летучее i и
этилена; ДИ — теплота растворения; А — константа.
По данным [409] растворимость этилена в 1,2-дихлорэтане характеризую!
следующие константы: А = 3,166; АН - 6,35 кДж/моль; (кру)20 °C = 9,7 МПа.
Растворимость этилена в 1,2-дихлорэтане следующим образом зависит oi
давления [410] (указано содержание этилена в насыщенном растворе):
f, °C 20 40 60
рЮ1, МПа... 3,03 8,07 15,4 17,3 30,4 2,84 8,11 17,4 30,7 2,84 8,13 17,6
Содержание
этилена,
% мол...... 2,8 7,8 15,4 17,9 33,7 2,0 6,0 14,0 26,7 1,5 4,8 11,6
В работе [405] приведены данные по фазовому равновесию системы ДХЭ
вода, а также по растворимости хлороводорода и винилхлорида в дихлор
этане. Эти данные важны для расчета системы осушки и ректификации ди
хлорэтана.
На рис. 4.2 представлена кривая фазового равновесия в области концен!
рации влаги 0,002-0,01 мае. %. Форма кривой указывает на сильное влияние
концентрации влаги на величину коэффициента разделения. Коэффициент раз
деления на участке концентраций 0-0,003 мае.% близок к единице, что делает
достаточно затруднительной осушку дихлорэтана до остаточного содержании
влаги на уровне 10-20 ppm.
Данные по растворимости хлороводорода и винилхлорида в дихлорэтане
приведены в [405, 411]. На рис. 4.3 представлена зависимость растворимос-
ти НС1 в ДХЭ от парциального давления. Зависимость подчиняется закону
Генри:
Хр = р/с,
(4.3)
где Кр — константа растворимости; р — парциальное давление газа; С — кон
центрация растворенного газа в растворе.
Концентрация воды
в жидкости (X), мае. %
Рис. 4.2. Фазовое равно
весие смеси ДХЭ-вода в
области малых концентра
ций последней
Рис. 4.3. Зависимость растворимости НС1 в ДХЭ от давления
11а рис. 4.4 представлены зависимости растворимости НС1 и ВХ в ДХЭ tn
н-миературы.
11а рис. 4.5 представлена зависимость IgKp для НС1 и ВХ, растворенных в
дихлорэтане.
Рис. 4.4. Зависимость растворимости ВХ и НС1 в ДХЭ от температуры
Теплота растворения HCI в ДХЭ 17380 Дж/моль, ВХ в ДХЭ 25540 Дж/моль.
Растворимость хлора в ДХЭ падает с ростом температуры (рис. 4.6) и увели-
чивается с ростом концентрации хлора в хлор-газе (рис. 4.7).
Рис. 4.6. Зависимость растворимости хлора в ДХЭ от температуры
Pm 1.7. Зависимость растворимости хлора в ДХЭ от концентрации используемого
• пор 1,1 ы
абинца -I. I. Ллеогроиные смет и, образуемые 1,2 дихлор Ианом
Второй KOMIIOIICII 1 Лзеотропнан смесь
Название 1 "С *КШР г "С ‘МНР Mil С. %
Нода 100,0 71,6 91,8
Бензол 80,4 80,0 15,0
Метиловый спирт 64,7 60,0 65,0
Трихлорэтилен 87,0 82,0 61,0
Этиловый спирт 78,0 71,0 63,0
[ройной азеотроп, содержащий 78% 1,2-дихлорэтана, 17% этанола и 5%
юды, кипит при 66,7 °C.
Удельная электропроводность при 20 °C равна 3 • 10 6 См/м (или 340 8 Ом ' см 1).
Пожароопасные и токсические свойства
1,2-Дихлорэтан — легковоспламеняющийся и токсичный продукт.
Температура вспышки, °C:
в закрытом приборе ................................13
в открытом приборе..................................9
Температурные пределы воспламенения, °C................8-31
Область воспламенения паров в воздухе, об. %...........6,2-16,9
Коэффициент диффузии пара в воздухе, м2/с..............0,7210
Порог восприятия запаха человеком, мг/м3...............25
Минимальное содержание О2, необходимое
для диффузионного горения, об. %.....................17,6
Минимальное взрывоопасное содержание О2 при разбавлении
дихлорэтановоздушной смеси диоксидом углерода, об. %.16,4
Предельно допустимая концентрация паров в воздухе
рабочей зоны производственных помещений, мг/м3........101
Предельно допустимая концентрация в атмосферном воздухе
населенных мест, мг/м3
среднесуточная.........................................I1
максимальная разовая................................З1
Предельно допустимая концентрация в водоемах
санитарно-бытового водопользования, мг/л...............21
Химические свойства
1. Галогенирование. 1,2-ДХЭ реагирует с хлором в жидкой или паровой фазе
> присутствии радикальных инициаторов с образованием 1,1,2-трихлорэтана:
СН2С1СН2С1 + С12 —- СН2С1СНС12 + НС1. (4.4)
Высокотемпературное хлорирование в псевдоожиженном слое катализато-
ра приводит к образованию перхлорэтилена [412]:
1 Данные соответствуют российским нормативам.
С112С1СН2С1 « 12—- C2( I, i ПК I. (4.5)
I 1роцесс может ишь осуществлен шкже и жидкой фазе [4 12|.
2. Дегидрохлорирование. При действии спиртовых или водных растворов
щелочей или при нагревании выше 250 °C от 1,2-ДХЭ отщепляется хлороводо
род с образованием винилхлорида:
CI12С1СН,С1 + NaOH —— СН2=СНС1 + NaCl + Н2О. (4.6)
' 1идролиз. Действие воды в присутствии кислот или щелочей при 140-
>0 "С и давлении до 4 МПа приводит к гидролизу 1,2-дихлорэтана с образов.»
пнем этиленгликоля:
( 112С1СН2С1 + 2Н2О —— СН2ОНСН2ОН + 2НС1. (4.7)
I. Аммонолиз. В водных или спиртовых средах в присутствии солей аммо
них 1,2-ДХЭ реагирует с аммиаком, давая этилендиамин:
120 °C
Cl 12С1СН2С1 + 4NH3---- CH2NH2CH2NH2 + 2NH4C1. (4.8)
11аряду с первичным амином возможно образование вторичных и третич
ных аминов, а также получение этиленимина.
5 Цианирование. При взаимодействии 1,2-ДХЭ с цианидом натрия в спир
книги среде получается сукциннитрил:
Cl 12С1СН2С1 + 2NaCN —*- CH2CNCH2CN + 2NaCl. (4.9)
6 Этерификация. 1,2-ДХЭ реагирует с ацетатом натрия с образованием эти-
ле 11 гл и кольацетата:
Cl 12С1СН2С1 + 2CH3COONa —- СН3ОСОСН2ОСОСН3 + 2NaCl. (4.10)
7. Алкилирование. В присутствии катализаторов Фриделя-Крафтса 1,2 ДХэ
|н .п прует с бензолом и его аналогами, давая соответствующие производные;
1А1СЫ
( 112С1СН2С1 + 2С6Н6---- С6Н5СН2СН2С6Н5 + 2НС1. (4.11)
8. Действие SC>2- При взаимодействии с олеумом образуется
2-хлорэтилсульфурилхлорид:
( 112С1СН2С1 + SO2 —-- CH2C1CH2OSO2C1. (4.12)
9. Действие Na2S. Полисульфид натрия реагирует с 1,2-ДХЭ, образуя каучу-
ючюдобные материалы — полиэтилентетрасульфиды (тиокол).
10. Гидрирование. Взаимодействие с водородом в газовой или жидкой <|>а ie
в присутствии I’d- или Ni-катализаторов приводит к образованию этилена или
этана в результате реакции дехлорирования:
СН2С1СН2С1 + 2Н2 —* С2Н6 + 2НС1; (4.1 За)
СН2С1СН2С1 + Н2 — С2Н4 + 2НС1. (4.1 Зб)
11. Окисление. При окислении 1,2-ДХЭ воздухом образуется монохлорук
сусная кислота с небольшим выходом [415]. В процессе окисления 1,2-ДХЭ при
200-350 °C в присутствии катализаторов (Fe, Си) образуются монохлорацеталь
дегид, хлораль, моно- и диоксид углерода в разных соотношениях.
Методы синтеза
1. Хлорирование этилена в газовой или жидкой фазе в присутствии катали
затора:
[FeCl3]
СН2=СН2 + С12----«► СН2С1—СН2С1. (4.14)
При хлорировании этилена сульфурилхлоридом [414] селективность обра
зования 1,2-ДХЭ 53,4% мол. Образуются также 1,4-дихлорбутан и 1,6-дихлор
гексан.
2. Хлорирование этана в газовой фазе:
С2Н6 + 2С12 —— СН2С1—СН2С1 + 2НС1. (4.15)
3. Окислительное хлорирование этана или этилена в газовой фазе на катали-
заторе Дикона, например:
СН2=СН2 + 2НС1 + 0,5О2 —СН2С1—СН2С1 + Н2О. (4.16)
4. Взаимодействие ацетилена с хлороводородом в присутствии диокисида
азота или с хлором в присутствии тетрахлорида углерода в паровой фазе на сме-
шанном катализаторе (AlCl3/NaCl/FeCl3):
СН=СН + 2НС1 — СН2С1—СН2С1. (4.17)
5. Хлорирование этилхлорида хлором или другими хлорирующими агента-
ми, например SbCl5:
С2Н5С1 + С12 —- СН2С1—СН2С1 + НС1. (4.18)
6. Синтез из 1,2-дибромэтана и пентахлорида сурьмы, взятой в количестве
не менее 2 моль:
CH2Br—CH2Br + SbCl5 —► СН2С1—СН2С1 + SbCl3Br2. (4.19)
7. Взаимодействие этиленгликоля с дымящей соляной кислотой, с хлорида-
ми фосфора в присутствии хлоридов цинка или с тионилхлоридом в пиридине,
например:
СН2ОН—СН2ОН + 2НС1 —- СН2С1—СН2С1 + 2Н2О. (4.20)
Н < пп i * i и i диазоме laiia и хлорида цинка в >фирпом рас 1 вире:
.'< II.N2 । /п(’1.—— ( I I.Cl —СИЛ I +-N> + Zii. (4.21)
Лабораторный метод получения
1,2 Дихлор >тан получаю! хлорированием этилена в среде 1,1,2,2-гетре
nip папа в присутствии FeCl, как катализатора. В цилиндрический стекляп
in.hi .Hiii.ip.n емкостью 0,5 л засыпают железную стружку на высоту половины
pi и н>ра и ыливают 250-300 мл сухого 1,1,2,2-тетрахлорэтана (или другого
т.н пкокипящего хлорорганического растворителя). Аппарат снабжен Парно
ч рамп для подачи хлора и этилена и обратным воздушным холодильником в
и и елочного дефлегматора высотой 20-30 см. В течение трех часов в реактор
иид inn I 50 мл/мин этилена и 140 мл/мин хлора, поддерживая в реакторе темпе
р и vpy 8 >-95 °C с помощью водяной бани.
< >нра |ук>|циеся в результате реакции пары 1,2-дихлорэтана проходят елоч
iii.ni дефлегматор, где отделяются от паров высококипящего растворителя,
»п||Д| Ik ируюгея в прямом водяном холодильнике и собираются в приемнике.
IliiuV'K пныи 1,2-дихлорэтан-сырец разгоняют на стеклянной ректификациоп
null колонке с насадкой Фенске высотой 0,5 м. В приемнике собирают фракцию
»ппнщую при ~85 °C и нормальном давлении. Выход 1,2-дихлорэтана-рекгифи
и 11 оу । а вл яет 80-85% от теоретического.
Промышленное производство
И промышленном масштабе 1,2-ДХЭ получают двумя методами: прямым
• nipiipoiiaiineM этилена в жидкой фазе в присутствии катализатора — хлори
it 11 hi ia (I II), или окислительным хлорированием этилена в паровой фа ie на
। и i ni и юре Дикона. Оба этих метода являются составными частями сбалап
пропаппий схемы производства винилхлорида и будут подробно рассмогре
Н| I ни КС.
П I1' 30 1960-х годах дихлорэтан широко использовали как растворитель в
"Il оком спектре отраслей промышленности: лакокрасочной, кожевенном, нс
фишой и газовой, текстильной, органического синтеза и др. С 1927 г. 1,2-ДХЭ
. ни и. питался в небольших количествах как фумигант. Со временем ДХ ) вы
о пи в и другими растворителями вследствие их более низкой токсичное!и и
.. lim n растворяющей способности. В 1930-1950-х годах ДХЭ являлся основ
им in ходпым соединением, использовавшимся при разработке химического
I । ни и частности, для синтеза иприта.
Н и к тящее время ДХЭ используется почти исключительно как полупро
। и Органическом синтезе. Свыше 90% полученного ДХЭ идет на пром i
* в iho винилхлорида. В небольших количествах он используется также дли
н • Ц'н ния н илендиамина, этиленгликоля, тиоколов и др., может быть также
и. iih ii. iuB.ui для получения хлорорганических растворителей: три- и пер
4.3. Окислительное хлорирование этилена
Понятие оксихлорирования
il
Окислительное хлорирование (или оксихлорирование) — процесс хлорн
рования углеводородов при участии окисляющих агентов, в качестве коюрыч
обычно используют кислород, в частности, воздуха. Хлорирующим агентом
как правило, является хлороводород, необходимость утилизации больших ко
личеств которого и послужила причиной разработки метода оксихлорирова
ния. Помимо хлороводорода источником хлора могут быть, например, хлорид
аммония или хлориды металлов переменной валентности.
В основе оксихлорирования — реакция окисления хлороводорода кислоро
дом (воздуха) в присутствии катализатора, известная как реакция Дикона:
2НС1 + 0,5О2 С12 + Н2О. (4.22)
Основные принципы этой реакции известны с 1845 г., однако первый цн
тент на осуществление такого способа получения хлора был получен английс
ким химиком Г. Диконом в 1868 г. и был им же развит в дальнейшем [416, 417|.
Предложенный Диконом катализатор — хлорид меди на носителе по настов
щее время является основой всех каталитических систем процессов оксихло
рирования.
Интерес к процессу Дикона как самостоятельному источнику получения
хлора существовал лишь в конце XIX в. и возобновился в начале 1920-х годов,
когда были получены первые патенты на процесс оксихлорирования метана и
этилена [418, 419]. С тех пор процесс Дикона рассматривается исключительно
как составная часть процессов оксихлорирования углеводородов, прежде всего
парафинового ряда, с получением широкой гаммы хлорорганических соедине
ний.
В зависимости от природы исходного углеводорода и условий проведении
процессы оксихлорирования могут протекать по совершенно разным механи i
мам. А.И. Гельбштейн [420] сформулировал четыре обобщенные схемы перено
са хлора в реакциях оксихлорирования углеводородов:
I. Сопряжение реакции окисления хлороводорода (4.22) с реакцией замес j и
тельного хлорирования предельных углеводородов:
2НС1 + 0,5О2 С12 + Н2О, (4.23)
HR,R2H + С12 —HR,R2C1 + НС1, (4.24а)
HR]R2C1 + С12 —- C1R,R2C1 + НС1. (4.240)
II. Сопряжение реакций (4.22) и (4.23) схемы (I) с реакцией дегидрохлориро
вания предельных хлорпроизводных углеводородов;
HRjR2C1 —- R!=R2 + НС1; (4.25а)
C1R,R2C1 —► R,=R2' Cl + HC1. (4.256)
III. Реакция присоединительного оксихлорирования непредельных углево
дородов:
Ri =R2 I-211C1 i 0,5O2- — <.?1R,I<.<.1 I I1O. (4.2b)
IV. Реакция заместительного оксихлорировапия непредельных углеводородов:
И,— K2]| + | К | + о^— R,=R2CI + 1I2O. (4.27)
II i схематически представленных четырех типов процессов оксихлорпро
11.Н1НЯ лишь процессы оксихлорирования предельных углеводородов (схема I),
в |ом числе при их сопряжении с реакцией дегидрохлорирования (схема II),
нк И1<1чаюг в качестве необходимого этапа предварительное образование хлора
ы с че г окисления хлороводорода по реакции (4.22).
Применительно к проблеме промышленного производства винилхлорида
«ми прежде всего будет рассмотрена схема III, согласно которой процесс ок
। п хлорирования этилена протекает при относительно низких температурах с
и |гк । ивным образованием 1,2-ДХЭ. Определенный интерес представляет 1ак
11 идди тинное оксихлорирование этилена в жидкой фазе.
Отдельно будут рассмотрены процессы получения винилхлорида замес
i (цельным оксихлорированием этана I и этилена IV, пока не реализованные в
промышленных условиях. В этих процессах, протекающих при высоких (350
। >11 ( ) температурах, начальной стадией является реакция Дикона.
Реакция Дикона: термодинамика, катализаторы, кинетика
Окисление хлороводорода до хлора — обратимая реакция, протекающая
пьычпо при 350-450 °C. Повышение температуры ведет к снижению равно
в i ного выхода хлора и, соответственно, к снижению конверсии НС1 и О2. Гак,
повышение температуры с 327 до 527 °C приводит к уменьшению IgKp с 3,12
(о (>,57. Увеличение избытка кислорода и повышение давления приводит к уве
и'ii-iiiiio конверсии НС1, а также содержания С12в реакционной смеси; тем не
кв нее при 327-427 °C, когда скорость процесса достаточно высока, равновесная
кппнерсия НС1 не достигает 90%.
11и же представлены уравнения зависимости констант равновесия от темпе
। 11 уры для реакции Дикона (4.22), полученные различными исследователями и
>||||11>|цеппые Дж. Алленом и А. Кларком [421]:
Vравнения зависимости констант равновесия от температуры Источник
ЫН(> 7 '-7,244 [422|
Ж / 1 - 1,751 1g Т- 2,8 • 10 4 Т- 1,4 [422|
•о / 1 -2,1361 1g Т- 8,57- 10 4 Т+ 6,83 10 8 7^ + 0,296 [423|
НПО / ' - 1,891 1g Т+ 5,00- 10~4 Г- 5,75- 10 8 Т2 - 1,748 [424[
УНН1.7 / '-0,930351 1g Г + 1,3704- IO 4 Т- 1,7581 10"8 Т2 - 4,1744 [425|
In 1 1 / ' - 7,0994 (менее точно) 1.1(111/ ' 0,272 1g Т- 1,704 -10 4 Г-6,06 [426|
< Хнцепринятой в настоящее время является зависимость, предложенная
I । \лленом:
Г.к>П11ц.1 1.5. Ггрмпдипамыческне параметры прицепов оыкпенни 1 К I и оксихлориро
1ЛПНИ
Реакция кДж/ моль AG, кДж/монь Кр
500 К 700 К 500 К 700 к
21ICI + 0,5О2^^ С12 + Н2О -57,1 -25,2 -12,3 4,2-102 8,3
Cl 1,! + HCI + 0,5О2 —*- СН3С1 + Н2О -156,3 -130,0 -120,0 4,4-Ю13 9,1 1()К
СП, + 2НС1 + О2 —*- СН2С12 + 2Н2О -316,2 -254,0 -235,5 4,0-1026 3,4-К)1
СИ, + 3 HCI + 1,5О2 —*- СН,С1 + + ЗН:О -273,0 -374,0 -340,0 3,6-Ю39 2,3-10 '
Cl 14 + 4НС1 + 2О2 —► СС14 + 4 Н2О -628,5 -505,0 -455,0 7,1-1052 9,8-К)"
0,11, + 2НС1 + 0,5О2 —*- С2Н4С12 + + Н2О -234,0 -139,5 -100,0 3,8-1014 2,8-10
С2Н,С12 + НС1 + О2—► С2НС13 + 2Н2О -271,5 -272,0 -271,8 2,4-1028 1,7-10 "
С2Н4С12 + 2НС1 + 1,5О2—► С2С14 + + ЗН2О 427,5 -386.5 -370,0 2,5-Ю40 3,9-10”
С,Н6 + 2НС1 + 0,5О2 —► С,Н6С12 + + Н2О -243,2 -145,3 -106,5 1,6-1015 8,5-10
С.,Н6С12 + 6НС1 + ЗО2 —- СС14 + + C2CI4 + Н2О -1038,0 -905,0 -840,0 4,4-1094 3,2-106’
IgKp = 6104,4 Т } - 7,0994. (4.28)
При совместном протекании реакций Дикона и хлорирования равновесие
вдвигается за счет расходования хлора, вследствие чего возможно практичес
и полное превращение НС1. Например, при оксихлорировании метана был.1
достигнута степень превращения хлорида водорода, соответствующая Кр при-
мерно 1 • 108. В табл. 4.5 приведены термодинамические характеристики некото
рых реакций оксихлорирования. Все рассмотренные реакции оксихлорирова
ия практически необратимы в интересующем нас интервале температур.
Таким образом, наиболее предпочтительна реализация процесса Дикона
ри возможно низких температурах. Это важно и для повышения селективное -
ги процессов хлорирования при совмещении их с реакцией Дикона. Оптималь-
ные условия проведения такого рода процессов могут быть достигнуты при ис-
пользовании активных и стабильных катализаторов.
Предложенный Диконом катализатор — хлорид меди на пемзе оказался не-
пригодным вследствие низкой активности, а также нестабильности, связанной с
высокой летучестью солей меди [420, 427, 428]. В результате исследований, про-
веденных К. Фонтана [429], а также Д. Рутвеном и К. Кеннеем [430], были пред-
тожены катализаторы на основе сочетания хлорида меди с хлоридами щелочных
и редкоземельных элементов. Было установлено, что переход к смешанным со
певым системам не только уменьшает летучесть медной соли (согласно [428] для
эквимолярной смеси СиС12—КС1 в 200 раз при 500 °C), но обеспечивает также
эначительное повышение активности катализаторов [426, 428-431]. Рост актив-
ности обычно связывают с увеличением при этом скорости сорбции кислорода.
Хлорид меди до н.п юище* о времени oiidcn.ii основным шпал или гором
ниш пения 11( I и оксихлирировапия. 1. Дикон предложил следующую схему
процесса окисления 1ICI:
2( ц( '1Х =. - Си ,С12 + С12; (4.29а)
( и ( I. + (),5О2—* СиОСиС12; (1.296)
( и С )-СиС12 + 2НС1 -л-— 2Си С12 + Н2О. (4.29в)
На основании этой схемы исследователи пытались теоретически обосно
паи. выбор катализатора окисления хлорида водорода и оксихлорировапия.
Для оценки каталитического эффекта металлов переменной валентное гн
Ф Хартер 1432] сравнил количество теплоты образования хлоридов и оксидов
in.li шеи и низшей степеней окисления, в результате чего предпочтение было
П1Д.1НО меди. Дж. Аллен [421], рассмотрел изменение свободной энергии пре
вращения оксидов в хлориды и хлоридов в оксиды для одно-, двух- и грехва
л< НП1ЫХ металлов в интервале 300-1000 К. Для двухвалентных металлов рас
м.привались реакции:
\С12 + 0,5О2 ХО + С12; (4.30)
\Ч) + 2НС1^^ХС12 + Н2О. (4.31)
I к» его мнению, лучшими катализаторами могут быть металлы с наимснь
пип разницей свободной энергии образования оксидов и хлоридов. Дж. Аллен
и А Кларк рассмотрели также термодинамику отдельных стадий превращения
меди в процессе оксихлорирования [421].
111менение стандартной свободной энергии в реакциях «оксиды меди + хло
рнподород» (4,18 КДж/моль):
1.К 400 500 600 700 800
( п<)(гв) + 2НС1(г) * СиС12(тв) + Н2О(г) -14,8 -12,2 -9,6 -7,2 4,8
< п< >( in) + 2НС1(г) СиС12(г) + Н2О(г) + 14,2 + 12,5 +11,1 +9,7 +8,3
I и .1)(гн) + 2НС1(г) — “ Си2С12(тв) + Н2О(г) -28,5 -26,2 -23,9 -21,9 -20,8’
< ill >( 1 в) + 2НС1(г) Си2С12(г) + Н2О(г) +72,4 +68,1 +64,1 +60,0 +56,1
Hi. ИИ1Ч.1Сг фазовые изменения в ходе образования Си2С1, при 703 К.
()ксиды одно- и двухвалентной меди не обладают заметной летучестью в
пределах рассматриваемого интервала температур, поэтому анализ реакций
проведен с участием только твердых оксидов. Для случаев образования хлор
кhi и хлористой меди рассмотрены также их газообразные состояния. Для хло
pin гой меди, точка плавления которой 703 К, фазовое превращение находи тся
пну । ри рассматриваемого ряда температур.
I (риведенные данные показывают возможность протекания реакций с учас
тем хлорсоединений меди. Для первой и третьей приведенных реакций огня
11иелы1ые константы равновесия быстро уменьшаются с ростом температуры,
ч in можеч несколько ограничивать равновесный выход целевых продуктов.
Данные но реакциям «хлорид меди t кислород» приведены ниже
Изменение стандартной свободной анергии Л(>, 4,18 кДж/моль:
/ ', К 400 500 600 700 80(1^
СвС12 (гв) + 0,5О2 (г) -л-— СиО (тв) + С12 (г) 7,3 6,2 5,3 4,4 3,6
2СиС12 (тв) + 0,5О2 (г) **“ СиО-СиС12 (тв) + С12 (г) 8,0 6,7 5,4 4,1 2,9
СиС12 (г) + 0,5О2 (г) -<-^-СиО (тв) + С12 (г) -21,7 -18,5 15,4 -12,5 9,5
Си2С12 (тв) + 0,5О2 (г) **“ Си2О (тв) + С12 (г) 21,0 20,2 19,6 19,2 19,7'
Си2С12 (тв) + 0,5О2 (г) -л-— СиО-СиС12 (тв) -8,7 -5,7 -2,6 0,5 3.5
Си2С12 (г) + 0,5О2 (г) — “ Си2О (тв) + С12 (г) -79,8 -74,1 -68,4 -62,8 57,3
Включает изменения при образовании СиС12 при 703 К.
Результаты экспериментальных исследований равновесия реакций «хлор
ная медь — кислород» сообщались К. Джелинеком [433], А. Корвезе [4341,
B.I4. Смирновым и А.И. Тихоновым [435]. Согласно этим данным реакция про
текает в две стадии:
СиС12 (тв) + 0,25О2 (г) 0,5CuO-CuC12 (тв) + 0,5С12 (г); (4.32а)
0,5CuO-CuC12 (тв) + 0,25О2 (г) СиО (тв) + 0,25С12 (г). (4.326)
Для обеих реакций, измеренные константы равновесия Кр = pci /ро*:
Т, К 573 623 673 723
/<р реакции: -1,42 -1,16 -1,01 -0,9 [435]
(4.32а) -1,48 -1,04 - -0,66 [433]
-1,24 -1,02 -0,83 -0,66 [434[
(4.326) - -1,53 -1,25 -0,99 [435|
— -1.65 -1,26 -0,99 [433]
На основании термодинамических расчетов Дж. Аллен и А. Кларк [421] де
лают следующие важные выводы:
1) в системе «оксид меди-хлороводород» реакции, приводящие к образова
нию газообразных хлоридов металла, могут быть исключены из рассмотрения.
Реакции с образованием твердых хлоридов меди энергетически более благопри
ятны. Во всех случаях равновесный выход хлоридов металла благоприятнее при
низких температурах и высоких давлениях;
2) при взаимодействии хлоридов меди с кислородом для твердого хлорида
меди II равновесный выход оксида меди достаточно низок, реакции с участием
твердого хлорида меди I должны быть исключены из рассмотрения. Напротив,
окисление газообразных хлоридов меди имеет лучшие условия, хотя скорость
и равновесие предшествующей стадии испарения ограничивает эту реакцию.
Благоприятная энергетика имет место для реакции:
2СиС1 (тв) + 0,5О2 (г) CuO CuC12 (тв). (4.33)
В целом равновесному выходу твердых оксидов металлов благопрннк inyioi
высокая темпера гура и низкое давление. Гем не менее, если выводы на основании
1ермоди11амических расчетов о невозможности получения значи! слыви о кашли
। ического эффекта от какого-либо из рассматриваемых металлов можно счи га i ь
окончательными, то положительный эффект должен быть проверен эксперимен
ально. Как видно на рис. 4.8, соединения магния обладают лучшим комплексом
|ермодинамических свойств, чем соединения меди, тем не менее, в процессах
Дикона и оксихлорирования особой активности они не проявляю!. Расчеты мо
। у । оказаться полезными при рассмотрении влияния промоторов и составлении
многокомпонентных систем. Например, они хорошо объясняют активирующую
роль редкоземельных элементов в катализаторах, содержащих хлориды меди.
Исследования фазового состава и структуры системы СнС12—KCI [420, 129,
I М)| показали, что смешение этих солей приводит в зависимости от их соотно
тения к образованию разных, но более легкоплавких (tnn - 320-^364 °C), чем
нс ходные соли, комплексных соединений типа KCuCl3, К2СиС14, а также биядер
пых комплексов К2Си2С16 в которых катионы меди соединены хлорными мос
। нками. Таким образом, в условиях катализа реакции Дикона, т.е. при t > 350 "С,
каталитическая солевая система находится в состоянии расплава, состоящего
и । соответствующих комплексных соединений. В случае катализаторов грегер
ною типа солевая система находится на поверхности носителя в виде пленки
или капель расплава [420].
1’нс. 4.8. Зависимости свободной энергии, реакций образования хлоридов (—) и окси
ion (--) двухвалентных металлов
A.I4. Гельбштейн [420] полагает, что механизмы процесса в расплавах и в
присутствии катализаторов на носи гелях подобны. 11ояпление в процессе хло
ридов одновалентной меди, как и введение в систему хлоридов щелочных (йе
таллов, приводит.к снижению температур плавления эвтектик в используемых
катализаторах [429].
Расплавы, по-видимому, не состоят из химически однородных ионов. Важная
особенность расплавов хлоридов металлов — неравнозначность ионов по энер-
гиям связи. Образование хлормеднокалиевых комплексов подтверждено данны-
ми фазового и рентгеноструктурного анализов. Состав комплексов зависит от
соотношения Си2+ и СР [436]. Существование анионов CuCl2 и CuClg было уста-
новлено и в катализаторах на носителях [437] в присутствии катионов щелочных
и редкоземельных металлов. Химическая неоднородность соединений меди в ка-
талитической системе СиС12—CuCl—КС1 должна влиять [430] и на равновесие
первой стадии реакции, которая может лежать в основе процесса Дикона.
Д. Рутвен и К. Кенией [430] подтвердили сильную зависимость равновесно-
го давления хлора над расплавами от соотношения K+:Cu2+:Cu , что было объ-
яснено участием меди в комплексе с хлоридами в следующих реакциях:
2CuClt = Cu2Cl3 +ЗС1 + С12; (4.34а)
Си2С1б = Си2С13" + СГ + С12. (4.346)
Основываясь на результатах адсорбции кислорода, предполагают, что ско-
рость процесса Дикона определяется скоростью окисления CuCl в СиС12 и опи-
сывается уравнением:
4ку2К2а2ро
г = ~,----(4.35)
(УК + у[р^)
где у — отношение коэффициентов активности Сн2+ и Си+; к — константа ско-
рости адсорбции кислорода; К — константа равновесия термической диссоциа-
ции хлорида меди; а — мольная доля меди в расплаве.
Схема процесса была уточнена в связи с наблюдаемым вторым порядком
зависимости скорости адсорбции кислорода от |Си+] [437]:
(быстро)
2CuC12 ~ - 2СиС1 + С12; (4.36а)
(медленно)
2СиС1 + О2 - * Си2О2С12; (4.366)
(быстро)
Cu2O2Cl2 + 4НС1 - 2СиС12+ С12 + 2Н2О. (4.36в)
Этой схеме соответствует следующее кинетическое уравнение:
г = 4кК[ CuCl2f = к" (4.37)
Pci2 Pci2
В работе | В8| выска 1ываегея мнение о координационном виимодшн iiuiii
меди с К('1 и 11(1, предшествующем медленной стадии адсорбции кислорода,
hoiopoe должно привести к уменьшению эффективного заряда катионы меди и
i ни ши» гермодинамические ограничения, присущие реакции CuCl, с кислоро
дом |421,4261. Это согласуется с данными по снижению работы выхода электро
на при переходе о г чистого СиСЬ к смесям с КС1.
В условиях наблюдаемого нулевого порядка по хлороводороду скороеп. про
цесса описывается уравнением (4.37). Цитируемый механизм был подтвержден
и работе [439], в которой показано взаимное влияние хлора и кислорода при
адсорбции и возрастание адсорбции О2 на необратимо сорбированном 1JCI.
Как было отмечено, свойства комплекса меди должны зависеть от нриро
О-i катиона соли, добавляемой к СиС12. При исследовании процесса Дикона на
онализаторах, содержащих хлориды лития, натрия, калия и цезия в мольном
in ношении к меди, равном 0,5, установлено |440. С. 19]: характер зависимости
< кчрос ги от парциальных давлений реагентов на всех катализаторах одинаков,
по наблюдаемая энергия активации (£набл-) уменьшается в ряду Li > Na > К > (. s,
Ч1о объясняется авторами уменьшением ионного потенциала и увеличением
г нособности связанных с ним ионов хлора к координации с катионами меди.
Зависимости, представленные на рис. 4.9, показывают, что каталитическая
лк । явность как функция состава солевых систем, имеет экстремальный харак
и р для хлоридов калия и цезия с максимумом у составов с МС1/СиС12 = 0,5. Для
хлоридов лития и натрия эта функция монотонно возрастает с ростом атомного
г по । ношения М/Си [420]
/WCI/CuCI, мольн.
I’m I 9. 1ависимости каталитической активности систем CuCl—MCI (М = Li, Na, К, ( ч)
•I । in i.ina при 375 °C:
i ill ( iiC12;2 —NaCl—CuCl2;3 —LiCi—CuCl2;4 —KC1—CuCl2
,1Г Naf К* Cs+
35,9 33,9 31,8 23,9
213,5 202,2 191,3 148,2
I Уменьшение наблюдаемой энергии активации при увеличении содер i .hhiii
'хлорида (цепочного металла, очевидно, обусловлено повышением с iciieiiii уч,и
тип катионов меди в комплексе:
I
{Катион.. .....
।
Ф к0...........
I
Енабл., кДж/моль
Каталитическая активность системы СиС12—КС1 является экстремально!!
^функцией ее состава [420]. Согласно Ю.М. Бакши и А.Г. Аглулину [440], мам к
мальной активностью по величине наблюдаемой константы скорости согласии
i(4.37) обладает состав с мольным отношением КС1/СиС12 = 0,5 (рис. 4.10)
Как следует из зависимости кинетических параметров реакции Дикона oi
состава системы СиС12—
КС1/СиС12........0
1:'и кДж/моль. 248,7
In Ка...........39,8
КС1 (350-450 °C) [440]:
0,1 0,5 1,0 1,5 2,5
210,3 191,0 129,2 135,4 134,6
34,0 31,8 19,5 20,0 17,2
максимальная активность солевой композиции с КС1/СпС12 = 0,5 является
следствием симбатной зависимости от состава солевой системы наблюдаемых
значений энергии активации (Ен) и предэкспоненциального множителя Ка коп
станты к" уравнения (4.37), хотя минимальному значению Ен отвечает состав с
KCl/CuCl2 = 1.
Рис. 4.10. Зависимости скоростей реакций окисления HCI {к = г • (Дсц/До2)} при разных
температурах
U lllltl.HI H I Hill Im
I < •< I < 'IK I . ( IK I 'III I. (1 18a)
< III I 'I И I < II 'I II I i I I (4.386)
i < и;n< iiu ► < n<) 'in I, (4.з«в)
I i ill) 41» 1 I ( II 'III I- - ( u< I i 2112O; (4.38r)
НН I I I) - - :ci2 I 21!.О (4.38)
iipiViyiM.iipitii.ier координационное связывание хлороводорода катионом меди,
уже шкомплектованпым ионами молекул КС1. О значительном его сродстве с
IK I 1 пндс1ел1>сгвуег большая величина теплового эффекта стадии 1, опрсде
кппая пдсорбциоппым методом (АН = -75,2 кДж/моль). Доказано [420], что
< iioinOiioi 1ь к термическому отщеплению хлора резко возрастает в прису г-
। типи хлорис того водорода. Кислород также способствует образованию хлора
и i чс г ра 1ЛОЖСНИЯ CuCI2 в его комплексе с KCL, однако в значительно меньшей
< iciieiin, чем I К I.
Д к. Аллеи и А. Кларк [421] подробно рассмотрели кинетические законо
мерное ги промежуточных реакций взаимодействия оксидов меди с хлорово
дородом, а также хлоридов меди I и II с кислородом. Согласно данным, по-
лученным К. Шалеро [441-443], хлориды образуются в стехиометрическом
< ooi ношении при реакции соответствующих оксидов с хлороводородом. При
100 "С для оксида меди устанавливается минимум скорости реакции. Ниже
нои температуры реакция тормозится парами воды, при более высоких тем
пера гурах вода практически не влияет на скорость реакции. При 400 °C хло
рид меди II переходит в хлорид меди I, а при 500 °C наблюдается сублимация
хлорида меди II [444].
Ангоры [421] исследовали кинетику реакций оксидов меди с хлороводоро
дом при t < 300 °C. Основные выводы, сделанные ими:
а) для оксидов меди конечными продуктами были соответствующие им хло
ряды. 11оследующая выдержка CuCl на влажном воздухе приводила к образова
пню смеси CuCl + Cu2(OH)3Cl;
<») кривые общей скорости реакции для системы СиО—НС1 имеют ло
। арнфмическую зависимость. Анализ начальной скорости при давлениях
- 7 мм рт. ст. (моль Н2О/с) в интервале от 130 до 307 °C показал, что начальная
। корос гь реакции может быть выражена уравнением:
г0 = 1,796 • 10 *ехр[-21 600/(RjD1Phci SCuO > (4.39а)
। i скорость реакции не зависит от парциального давления хлористого водо
рода и пропорциональна площади поверхности твердой СиО Общая скорость
реакции подчиняется уравнению, предложенному в работе [445] в логарифми
чп кой форме:
r0d[l-(l-a)1/3]=KIg(t-t0),
(4.39(5)
де 'и — исходный радиус реагирующих частиц; </ — плотность; а — фракция
Твердого реагента; К — константа; / и /(| — и тменепия времени пребывания.
Кажущаяся rinepi ия активации сос тавляет 83,2 кДж/моль;
в) реакция для системы Си2О—НС1 подобна вышеописанной реакции с ок
.идом меди; уравнение скорости (моль Н2О/с) подчиняется уравнению:
г0 = 3,42 • 10 ‘exp[-14600/(RT)]p^cl SCuO . (4 39н)
Кажущаяся энергия активации равна 54,8 кДж/моль.
Применительно к реакциям хлоридов одно- и двухвалентной меди с кисло
тодом в работе [446] сделаны следующие выводы:
а) хлорид меди II в чистом состоянии не реагирует с кислородом в уело
виях исследования. Дегазированные образцы хлорной меди почти неизбежно
.одержат небольшие количества низшего хлорида, который абсорбирует ки£
пород;
6) хлорид меди I быстро реагирует с кислородом. Например, при 350 °C про
дукг реакции через 80 минут составляет порядка 20 монослоев на поверхнщ
ги 104 см2/г. Кривые конверсии по времени показывают линейную зависимое ть
при кажущейся энергии активации — 74,0 кДж/моль.
Уравнения реакции могут быть представлены так:
Сп2С12 (тв) + 0,5 О2 (г) CuO CuCl2 (тв) (4.40а)
или
Сп2С12 (тв) + 0,5 О2 (г) СпО (тв) + СпС12 (тв). (4.40о)
Основные закономерности, которым подчиняется реакция Дикона, игр*
ют определяющую роль при высокотемпературном оксихлорировании пре
дельных и непредельных углеводородов, и в сочетании с этими процессами
будут рассмотрены в разделах, посвященных получению винилхлорида. При
низкотемпературном (200-300 °C) аддитивном оксихлорировании оксидов н,
в частности этилена, реакция Дикона не имеет места, и общим для этих про
цессов является катализ за счет окислительно-восстановительных превраще
ний меди.
Известен ряд работ [447-449], направленных на создание реакторных пи
тем, в которых процесс Дикона может быть реализован с высокой конверсией
НС1. Так, в статье [447] предложен циркуляционный реактор псевдоожижен по
го слоя (рис. 4.11).
Реактор состоит из двух секций, в одной из которых при 340-400 °C идс i
реакция окисления НС1 до С12, а во второй — регенерация катализатора (ка
тализатор при 180-200 °C реагирует с непроконвертированным НС1 из первоп
секции). Конверсия НС1 достигает -94,5%, увеличение мольного соотношения
НС1/О2 снижает конверсию НС1. В качестве катализатора авторы [447] испил .
зуют смесь хлоридов меди и натрия, нанесенную на цеолит. В работе [448] пре
вращение хлороводорода в хлор предложено осуществлять в две стадии:
хлорирование 2НС1 + СиО—► СиС12 + Н2О, (4.41а)
дехлорирование СиС12 + 0,5О2—► СиО +С12. (4.116)
Термопара
IcpMoiiapa
Рис. 4.11. Циркуляционный реактор псевдоожиженного слоя
Возможность реализации второй стадии вызывает, однако, сомнение из-за
термодинамического запрета на протекание такой реакции (см. выше). В про-
мышленных условиях эти и им подобные процессы не реализованы.
Аддитивное газофазное оксихлорирование этилена
Катализаторы оксихлорирования этилена
Большое промышленное значение процесса оксихлорирования этилена, в ко-
тором утилизируется абгазный хлороводород и, кроме того, образуется дихлор-
этан, привело к значительному количеству публикаций и патентов по разработке
и выбору оптимального катализатора. Ввиду того, что процесс аддитивного ок-
сихлорирования этилена с получением дихлорэтана протекает при относитель-
но невысоких температурах (210-260 °C), окисление хлороводорода по реакции
Дикона в этих условиях практически не идет. Отсутствие стадии окисления НС1
подтверждается отсутствием хлора в реакционной массе [450], а также тем, что
хлорид хрома, высокоактивный в процессе Дикона, — наименее эффективный
2?6
катализатор оксихлорировання. Если конверсия 11(1 при окснхлорнропанни
пилена в ирису тсгвии хлорида меди превышает 90%, то в реакции Дикона в их
же условиях выход хлора нс достигает и 1%. Следовательно, природа кагалнтц)
ческой активности в двух этих реакциях совершенно разная.
Н. Годо [451] впервые предложил механизм образования дихлорэтана при
взаимодействии этилена с СиС12, в результате которого образуется восстания
ленная форма меди:
2СиС12 + С2Н4 ** С2Н4С12 + Си2С12; (4.42а)
Cu2Cl2 + 2НС1 + 0,5О2 2СпС12 + Н2О. (1.1'0)
По мнению авторов [451], при t < 300 °C лимитирующей является первая
стадия, а выше 300 °C — вторая.
Приведенная схема подтверждается рентгеноструктурным анализом [450|.
согласно которому в процессе оксихлорирования под действием хлорной меди
на носителе в реакционной системе присутствуют одновременно хлориды как
одно-, так и двухвалентной меди, причем по мере повышения температуры
реакции содержание первой возрастает за счет падения второй. Максимум со
держания хлорной меди в зависимости от температуры совпадает по времени с
максимумом скорости реакции оксихлорирования этилена.
Была предпринята попытка связать каталитическую активность ряда хло
ридов металлов переменной валентности (MCI) с теплотой образования свя нт
М—С1 согласно приведенному механизму [452, 454]. На рис. 4.12 представле
ны экспериментальные и расчетные скорости реакции хлорирования этилен i
хлоридами соответствующего металла. Галогениды металлов с более прочной
связью металл-галоген обладают меньшей реакционной способностью при
хлорировании этилена. Для стадии регенерации катализатора была получена
обратная зависимость. Комбинируя значения двух скоростей, Н. Коминами
Рис. 4.12. Экспериментальная (1) и расчетная (2) зависимости константы скорости ре
акции от АН связи М—О
I I 11 вычислил скорость суммарной реакции и предсказал ник активное гн в
iiHih hmociii oi содержания меди. Эго нашло под| верждепие при определении
niiiiiM.i/ibiioio содержания меди в промышленных катали iaгорах, обычно со
н1/1Я1о1цего 4-7% or массы катализа гора.
1аким образом, процесс аддитивного низкотемпературно)о оксихлориро
напил и плена имеет в основе окислительно-восстановительные превращения
> ,11.1/ш ыгора, причем альтернативы хлориду меди как катализатору данного
прицепа не найдено. В связи с этим именно хлорид меди находится в ценгре
внимания специалистов и фирм, занимающихся проблемами приготовления
он । ПМ.1/1ЫЮГО катализатора и его использования в процессе. Различия заклю
ч нон и в промотирующих добавках, а также носителях.
А Кларк [421,453] исследовал термодинамику 122 возможных реакций хло
рядов меди I и II с этиленом в интервале 400-800 К. Такое число реакций мо
н 1 проткать с образованием разных хлорированных продуктов и при уч.к
inn шердых и газообразных хлоридов одно- и двухвалентной меди. В табл. 16
приводятся (ермодинамические параметры этих реакций [453] за исключением
нр< вращений:
( п( I ,(г) —* Си2С12 (г), (4.43а)
( 1ь(12(г)—► Си (тв), (4.436)
। огорые при данных температурах не могут иметь места из-за термоди нам и чес
। их oi рапичений.
1и<> ища 4.6. Изменение стандартной свободной энергии для реакций между этиленом,
< III I , I U2C12 И хлором
Реакция AG°, 4,18 кДж/моль при
400 К 500 К 600 К 700 К 800 К
(1)< 11 ,(г) + 4СиС12(тв) — 1 Н,( 1(г) + СНС13(г) + 2Си2С12(тв) -9,3 -14,2 -19,4 -24,3 30,5
( ’) < 11 ,(г) + 4СиС12(тв) — ч 11 .С|,(г)+2Си2С12(тв) -8,7 -13,8 -19,2 -24,3 10,7
(М < 11 ,(г) + 6СиС12(тв) — ч 1 К 1,(г) + ЗСи2С12(тв) + Н2(г) 5,6 -3,0 -11,8 -20,4 - 31,1
(111 11.1(г) + 8СиС12(тв) * Ч ( l|(i) + 4Си2С12(тв) + 2Н2(г) 25,3 13,6 2,1 -9,5 -24,1
(’>) 1 11 ,(г) + 5СиС12(тв) — 1 < 1,(1) г СН3С1(г) + 72Си2С12(тв) + 72Н2(г) 0,6 -5,9 -12,4 -18,8 -27,0
((>)< 1 1,(г) + 6СиС12(тв) — ** < < 1,(1) + СН2С12(г) + ЗСи2С12(тв) +Н2(г) 8,3 -0,1 -8,5 -16,9 -27,1
I 1 < II |(г) + СиС12(тв) ( ИД 1(г) + */2Си2С12(тв) + ’/2Н2(г) 3,4 1,2 -0,9 -3,0 -5,5
(К) 1 .11 |(г) + 2СиС12(тв) — (1.1 )(’ 112С12 (г) + Си2С12 (тв) + Н2(г) 6,5 2,7 -1,2 -5,0 9,6
Таблица 1.6 (n/HHitvi’in iiiic)
Реакция AG1/, 1,18 кД>к/м<»п> при
400 К 500 К 600 к 7О<) К »КК> 1\
(у) С2Н4(г) + 2CuCI2(tb) (1,1-)С2Н2С12 (г) + Си2С12 (тв) + Н2*(г) 14,2 10,4 6,5 2,7 !.'J
(10) С2Н4(г) + 2СиС12(тв) — “ (цис-1,2-)С2Н2С12(г) + Си2С12 (тв) + Н2(г) 4,7 0,8 -3,0 6,8 1(1
(11) С2Н4(г) + 2СиС12(тв) —- ** (транс 1,2-)С2Н2С12 (г) + Си2С12(тв) + Н2(г) 4,1 0,1 -4,0 8,1 l.’.l
(12) С2Н4(г) + ЗСиС12(тв) - *“ С2НС13(г) +3/2Си2С12(тв) + 3/2Н2(г) 13,3 7,5 1,8 -1,0 9.8
(13) С2Н4(г) + 4СиС12(тв) — *• С2С14(г) + 2Си2С12(тв) + 2Н2(г) 22,4 15,0 7,6 0,1 8.8
(14) С2Н4(г) + 2СиС12(тв) (1,1 )С2Н4С12 (г) + Си2С12 (тв) 14,1 -14,9 -15,7 -16,4 17,8
(15) С2Н4(г) + 2СиС12(тв) (1,2-)С2Н4С12 (г) + Си2С12 (тв) -14,3 -15,2 -16,1 -17,0 18,5
(16) С2Н4(г) + ЗСиСЬ(тв) (1,1.1-)С2Н3С13(г) + 3/2Си2С12(тв) + 1/2Н2(г) -1,6 -3,9 -6,2 -8,6 12,0
(17) С2Н4(г) + ЗСиС12(тв) (1,1.2-)С2Н3С13(г) + 3/2Си2С12(тв) + 1/2Н2(г) -7,0 -9,6 -12,3 -14,9 18,6
(18) С2Н4(г) + 4СиС12(тв) (1,1,1,2-)С2Н2С14(г) + 2Си2С12(тв) + Н2(г) 3,0 -1,2 -5,4 -9,6 -15,3
(19) С2Н4(г) + 4CuC12(tb) (1,1,2,2-)С2Н2С14(г) + 2Cu2C12(tb) + Н2(г) -0,8 -5,2 -9,5 -13,8 -19,7
(20) С2Н4(г) + 5СиС12(тв) С2НС15(г) + 5/2Си2С12(тв) + 3/2Н2(г) 12,8 6,9 1,0 -5,0 -12,8
(21) С2Н4(г) + 6СиС12(тв) — ** С2С16(г) + ЗСи2С12(тв) + 2Н2(г) 24,6 17,1 9,6 2,1 -7,6
(22) С2Н4(г) + 4СиС12(г) СН3С1 (г) + СНС13 (г) + 2Си2С12(тв) -125,4 -113,1 -102,2 -91,8 -82,8
(23) С2Н4(г) + 4СиС12(г) 2СН2С12(г) + 2Си2С12(тв) -124,8 -112,7 102,0 -91,8 -82,9
(24) С2Н4(г) + 6СиС12(г) 2СНС13(г) + ЗСи2С12(тв) + Н2(г) -168,6 -151,3 -136,0 -121,7 -109,5
(25) С2Н4(г) + 8CuCl2(r) 2СС14(г) + 4Си2С12(тв) + 2Н2(г) -207 -184,1 -163,5 -144,6 -128,6
(26) С2Н4(г) + 5СиС12(г) СС14(г)+СН3С1(г) +5/2Си2С12(тв) + 72Н2(г) -144,6 -129,5 -115,9 -103,3 -92,3
(27) С2Н4(г) + 6СиС12(г) СС14(г)+СН2С12(г) + ЗСи2С12(тв) +72Н2(г) -165,9 -148,4 -132,7 -118,2 -105,8
II И1К<1 I f< I >| I I Mil Hill I
14 ilhuiui 10(1 k >00 k <>00 k 700 k 800 k
• 11,(1) 1 ( •»< 1,(1) w " 1) 1 1(1) 1 '/ 1 II 1 1 (III) 1 '/ II (l) 25.7 -23,5 21,6 14,4 I8.0
1 11,(1 ) 1 'I III 1 (1) «« ” 1 1 )< II I 1 (1) 1 1 II 1 1. (Ill) I II.(l) -51,6 16,8 -42,6 -38,8 35,7
1 ЧП 1 11,(1 ) 1 ll< 1.(1) - — (Illi. 1 )( II ( l (l) 1 < 11 < I (ill) ♦ 1 1(1) -52.5 -47,1 -41,6 -36,1 -30,6
II H IIt(i I । ‘t in l .(ui) (ipui. 1 ' )l 111 l.(i) i (’и.С12(гв) i-H,(r) -53,1 -47,8 -42,6 -37,4 -32,1
к ll,(i) i H u< 1 (i)^--* ( l(< l,(i) i ’/< u < 1 (ib)+ ’/2Il2(r) -72,5 -64,3 -56,1 -47,9 -39,7
H)< ll,(i) i K'uCi.(r)^^- ( 1 l,(i) i 4 u.< l.(ru) +2H2(r) -93,7 -83,9 -75,2 -67,5 61,1
UH 11,(1) i 2C uCl,(r)^^ (1 1 )( 11,( L(i) + Cu2C12(ib) -72,2 -64,4 -57,1 -50,2 -44,0
(I.) I 11 ,(i) 1 2CuCl2(r) - ** 1 )l 11,( 1 (ij + Cu2C12(tb) -72,3 -64,6 57,5 -50,8 -44,6
(К. II 11,(i) t 3CtiCI2(r) 1 1 1 )( 11,( 1 ,(r) + 3/2Cu2C12(tb) + /2Н2(г) -88,6 -78,1 68,3 -59,3 -51,2
(I )( 11 ,(i) + 3CuCl2(r) — (11 )( 11 / I,(r) + 72Cu2C12(tb) + /2Н2(г) -94,0 -83,8 -74,4 -65,6 -57,8
( 18) ( 11,(1) + 4CuCl2(r)^=^ (1 1,1,2 )C2H2Cl4(r) + 2Cu2C12(tb) + H2(r) -113,3 -100,1 -88,2 -77,1 -67,5
(<•>)< >1 1 i(r) + 4CuCl2(r) (1.1,2.2 )C2H2CI4(r) + 2Cu2CI2(tb) + H2(r) -116,9 -104,0 -92,3 -81,4 -71,9
( 1(1) ) ( ;1 I |(r) + 5CuCl2(r) < 1 К 1 ,(i) + 5/2Cu2C12(tb) + 3/2H2(r) -132,3 -116,7 -102,5 -89,4 -78,1
( 11) ( ,11 ,(r) + 6CuCl2(r) ( ( 1, (i) f3Cu2CI2(tb) + 2H2(r) -149,6 -131,2 -114,6 -99,2 86,0
(1’) < H4(r) + 2Cu2Cl2(r) 1 1 (,( 1 (i) + CHCl3(r) + 4Cu(tb) -136,1 -124,4 -113,1 -100,4 -90,6
( H) ( >H,(r) + 2Cu2Cl2(r) — .’( 11.( I2 (r) + 4Cu(tb) -135,5 -124,0 -112,9 -100,4 90,8
(II) C2H4(r) + 3Cu2Cl2(r) — ’( 1 К l3 (r) + 6Cu(tb) + H2(r) -184,6 -168,2 -152,4 -134,6 -121,2
(45) C 2H4(r) + 4Cu2CI2(r) — .’( ( l,(r) + 8Cu(tb) +2Н2(г) -228,3 -206,0 -185,3 -161,7 -144,2
(16) C2H4(r) + 5/2Cu2CI2(r) ( ( 1 ,(r) + CH3C1 (r) + 5Cu(tb) + H2(r) -157,9 -143,6 -129,5 -114,0 102,1
иц<1 1.6 (n/XKlMVil lllieJ
Реакция A(ii*, 4,18 кДж/моя|> при
100 К 500 К 600 К 700 К 800 к
C,ll4(i) + 3Cu2CI2(i)Z=Z*l i(r) + Cl I2Cl2(r) + 6Cu(tb) + H2(r) -181,9 -165,3 -149,1 -131,0 117,5
<’2ll.,(i)+72Cu2CI2(r)^ ,C 1 (Г) + Cu(TB) + 72 H2(r) -28,3 -26,3 -24,3 -22,1 20,6
C,ll,(r) + Cu2Cl2(r) zi^z )C2112CI2(r) + 2Cu(tb) + H2(r) -56,9 -52,4 -48,0 -43,0 39,6
( H4(r) + Cu2CI2(r) z^z^z 1,2 )C2H2Cl2(r) + 2Cu (тв) + H2(r) -58,6 -53,7 -48,9 -44,0 -39,2
C,H4(r) + Cu2Cl2(r) ZS^Z lie l,2-)C2H2Cl2(r) + 2Cu2(tb) + H2(r) -59,1 -54,5 -49,9 -45,2 10.6
C2H4(r)+72Cu2Cl2(r)Z^±Z Ch (г) + ЗСи(тв) + 3/2Н2(г) -81,6 -74,4 -67,0 -59,8 -52,5
С2Н4(г) + 2Си2С12(г) — 11 (г) + 4Си(тв) + 2Н2(г) -104,4 -95,1 -86,1 -76,0 -68,9
С2Н4(г) + Си2С12(г) — ** )С2Н4С12 (г) + 2Си(тв) -77,5 -70,0 -62,5 -54,5 -47,9
С2Н4(г) + Си2С12(г) zsz?z )С2Н4С12 (г) + 2Си (тв) -77,7 70,2 -63,0 -55,0 -48,5
С2Н4(г) + 72Си2С12(г) ZS^Z 1 )С2Н3С13г(г)+ ЗСи(тв) + */2Н2(г) -96,7 -86,5 -76,5 -65,7 -57,1
С2Н4(г) + 3/2Си2С12(г) — "Z 2-)С2Н3С13(г)+ЗСи(тв) + */2Н2(г) -102,0 -92,2 -82,6 -72,0 -63,7
С2Н4(г) + 2СиС12(г) *" 1,2-)С2Н2С14(г)+4Си(тв) + Н2(г) -123,8 -111,3 -99,1 -85,7 -75,4
С2Н4(г) + 2СиС12(г) zsz^z 2,2 )С2Н2С14(г)+4Си(тв) + Н2(г) -127,6 -115,3 -103,2 -89,9 -79,7
С2Н4(г) + 72Си2С12 (г) zsz±z С15(г) + 5Си(тв) + з/2 Н2(г) -145,7 -130,8 -116,2 -100,1 -87,8
С2Н4(г) + ЗСи2С12(г) — 16(г) + 6Си(тв) + 2Н2(г) -165,6 -148,1 -130,9 -112,0 -97,7
С2Н4(г) + 2С12(г) zs±z Cl (г) + СНС13(г) -43,0 39,8 -36,9 -33,8 -30,7
С2Н4(г) + 2С12(г) !<С12(г) -42,4 -39,4 -36,7 -33,9 -30,9
С2Н4(г) + ЗС12(г) zs^z Cl, (г) + Н2(г) -45,0 -41,4 -38,0 -34,7 -31,4
С2Н4(г) + 4С12(г) -л 14(г)+2Н2(г) -42,2 -37,5 -32,9 -28,6 -24,5
I i<> цаца •!.(> (om»i*hi»i« )
Реакция 4,18 кДж/м<»п> при
400 К 500 К 600 К 700 К 800 К
(пи) < .Н|(г) + *ЛС12(г)^=^ ( .и,( Ki) f 72н2(г) -5,0 -5,2 -5,3 -5,4 -5,6
(г. )(,||,(1) + С12(г)^=^ (1,1 )( 2112С12 (г) + Н2(г) -10,4 -10,1 -9,9 -9,8 -9,7
(68) t .|-|4(i) + Cl2(r)^z (ци. 1,2 )С2Н2С12 (г) + Н2(г) -12,1 -11,6 -11,2 -10,7 -10,2
((.')) ('2Hi(i ) + Cl2(r)zsz±z (। рапе-1,2 )С2Н2С12 (г) + Н2(г) -12,6 -12,4 -12,1 -11,9 -11,7
( ОН Д 1 ,(г) + 72С12(г) zsz±z ( 11С1,(1) + 3/2Н2(г) -11,9 -11,2 -10,5 -9,8 -9,0
( IH .ll|(r) + 2Cl2(r)zsz^ ( .( 1,(г) + 2Н2(г) -11,4 -10,6 -9,9 -9,5 -9,0
( >)C2H4(r) + Cl2(r)zs*z (1 1 )(2Н |С12 (г) -31,0 -27,7 -24,4 -21,2 -17,9
4C2H,(r) + ci2(r)^z (1.2 )С2Н4С12(г) -31,1 -28,0 -24,9 -21,8 -18,6
( IH Н4(г) + 3/2С12(г) z^z (1 1.1 )С2Н3С13(г) + 72Н2(г) -26,9 -23,1 -19,3 -15,8 -12,2
( 1) С2Н4(г) + 72С12(г) zs±z (1.1,2 )С2Н3С13(г) + ‘/2Н2(г) -32,2 -28.8 -25,4 -22,1 -18,8
( <>) ( 2Н4(г) + 2С12(г) zsz^z (1,1.1,2-)С2Н2С14(г) + Н2(г) -30,7 -26,7 -22,9 -19,1 -15,5
( 7) С2Н4(г) + 5/2С12(г) Zi^z ( ИСЦг) + 3/2Н2(г) -29,4 -25,1 -20,9 -16,9 -13,0
( 8) С2Н4(г) + ЗС12(г) z^±z ( (-цо + гндг) -26,0 -21,2 -16,6 -12,2 -7,9
1 0) С2н4(г) + 72С12(г) zsz^z < < 1,(1) + СН3С1(г) + 72Н2(г) -41,6 -37,9 -34,3 -30,8 -27,2
(НО) С2Н4(г) + ЗС12(г) zsz?z ( ( 14(г) + СН2С12(г) + Н2(г) -42,3 -38,4 -34,8 -31,2 -27,7
(Hl)C,H4(r) + 72Cl2(r)zsz±z ( ( l4(i) + CHCl3(r) + 72H2(r) -43,6 -39,4 -35,5 -31,7 -27,9
( ’) С2Н4(г) + 72С12(г) - *" ( Н( 1,(1) + СН2С12(г) + 72 Н2(г) -43,7 -40,4 -37,4 -34,3 -31,1
(8 3) С,Н4(г) + 2С12(г) z^z (1 1.2,2-)С2Н2С14(г) + Н2(г) -34,5 -30,7 -27,0 -23,4 -19,9
Хотя боЛЫПИ||СГ11<> приведенных реакций »пергегичесм1 НЫГОДПО II условиях
процесса оксихлорирования, термодинамика не определяет ход процесса. Болес
важен выбор оптимального катализатора и его кинетические характерна пткп )
Для повышения селективности и снижения летучести дихлорида меди к ката
ди тагору добавляют соли других металлов, например, хлориды калия, натрия
или алюминия [456]. Дж. Аллен и А. Кларк [421] использовали хлорид иттрия,
показав, что его добавки повышают активность катализатора и его срок служ
бы. На основе патентной литературы они составил^ классификацию носителей
и катализаторов процесса оксихлорирования этилена:
катализаторы: оксиды, хлориды, силикаты меди, железа, хрома, магнии, се
ребра, золота, никеля, кобальта, ванадия;
носители: оксид алюминия, силикагель, бентонит, активированный уголь,
цеолит, оксид магния, оксид титана, оксид циркония;
промоторы: а) стабилизаторы (хлориды, бисульфаты: лития, натрия, калия,
аммония, цинка, кадмия, кальция, стронция); б) активаторы (оксиды, хлориды:
лантана, платины, циркония, титана, молибдена, вольфрама).
Большинство этих соединений, особенно относящихся к классу промою
ров, применяются достаточно ограниченно и практически не используются и
промышленных процессах. Эффекты как активации, так и стабилизации вы
ражены довольно слабо. Из носителей в процессах оксихлорирования этилена
от носительно часто использовались и поэтому наиболее подробно изучалис ь
оксид алюминия, силикагель, алюмосиликат.
Подробная информация о катализаторах и условиях проведения процесса
оксихлорирования этилена приведена в обзоре [457]. Мы воспроизводим здесь
эти данные в целях систематизации материала и достижения его наглядное i и
(табл. 4.7).
Т.Д. Гужновской [554] проведен комплекс исследований процесса оксихло
рирования этилена в присутствии катализаторов на основе силикагелей и алю
мосиликатов.
Она указывает, что медьхлоридные катализаторы на основе алюмосиликл
ных носителей достаточно активны при 260-280 °C, тогда как использование
катализаторов на основе силикагелей требует температур 315-320 °C.
При оксихлорировании этилена в псевдоожиженном слое катализатора при
указанных температурах в качестве катализаторов необходимо использовать
смешанные медно-калиевые системы (СиС12-КС1/носитель). Такое решение
диктуется высокой летучестью меди при повышенных температурах, а также
наличием эффекта слипания катализатора в отсутствие добавок хлоридов щс
лочных металлов.
Согласно Дж. Аллену [426], давление паров СнС12 при t < 407 °C описывае тся
выражением:
IgpMMpT CT. - -3550 Г1 + 6,55 . (4.41)
Теплота сублимации СиС12 равна 68,1 кДж/моль, что существенно ниже
энергии диссоциации СиС12, равной -171 кДж/моль. Таким образом, из всех ни
дов возможных превращений СиС12, включая диссоциацию и взаимодействие с
кислородом, сублимация является наиболее кинетически выгодной.
I 213
Источник [458,459] [460] [461] [462,463] [464] [465,466] J [467, 468] [469, 470] [471] [472,454] [473]
Продукты образования m X 5 и I и СЧ 1—1 ДХЭ 95,6; С,Н3С1 (ВХ) 2,0; СО22,4 ДХЭ 95,0 ДХЭ 99,1 ДХЭ 96,0 ДХЭ 99,6 ДХЭ 98,6; ВХ 30 ppm; СНС13190 ppm; транс- С2Н2С12 65 ppm; CHCI3 400 ppm; С2Н2С14 300 ppm ДХЭ 99,1 ДХЭ 96; С2Н5С1 3,2; ВХ 0;4; С2Н3С13 0,3; и другие хлорсодержашие 0,1 ДХЭ 99,0 ДХЭ 90,0
Тип реактора Псевдоожижен - ный Неподвижный Псевдоожижен - ный Неподвижный Псевдоожижен - ный (т = 3-;-4 с) Неподвижный 1 Псевдоожижен - ный Неподвижный С обьемной скоро- стью 300 ч"1 Неподвижный
кции ] д К ° с 7 сч 'Тг' О £ ts (3,0) (2,5) 0,58 1 0Д5 0,56 0,5 3,2 un о (2,5) 0,5 (2,5)
ювия pea НС1/ С2Н4 2,0 2,0 2,0 1 2,0 2,25 2,0 2,6 2,0 3,0 2,0 2,0 |
и о Ч-Г 270-280 300 288 216-227 285 180-300 260-280 216 400 220 О о m
Состав катализатора СиС12 (9,3%) на у-оксиде алюминия СиСЬна активированном оксиде алюминия СиС12 (10%) на оксиде алюминия 1 СиС12 (10%) на оксиде алюминия Оксиды меди и алюминия СиС12 (1,25% Си) на оксиде алюминия CuCl2 на оксиде алюминия СиС12 на микросферическом оксиде алюминия СиС12на активированном оксиде алюминия с частицами графита СиС12 на активированном древесном угле | CuCl2 (15%) на кизельгуре
234
Таблица 4.7 (продолжение)
Состав катализатора Условия реакции Тип реактора Продукты образования Источник
t, °C НС1/ С2Н4 О2 (воз- дух)/С2Н4
СиС12 на пемзе 250 0,167 NH4C1 вместо НС1 0,67 Псевдоожижен- ный (г = 18 с) ДХЭ 100,0 [474,475]
СиС12 с 7% Си на кремнеземе с диаметром частиц меньше 45А 250 2,0 (3,0) Неподвижный (т = 3 с) ДХЭ 100,0 [476]
СиС12 (0,5-5% Си) на цеолите 350 2,0 0,75 Псевдоожижен- ный (т = 2,5 с) ДХЭ 9,3; другие хлорированные продукты 34; и продукты горения 14,0 [477]
СиС12 на f|- или к-оксиде алюминия 35 м2/г (БЭТ) и 0,4 мл/г (объем пор) 200-240 2,0 0,5 Псевдоожижен- ный ДХЭ (только) [478]
СиС12 на г]- или к-оксиде алюминия. Носитель может содержать менее чем 10% у- и а-оксида алюминия 240-300 2,0 0,5 Трехступенчатый, неподвижный ДХЭ 96,0; С2Н5С1 1,2; и остальное 2,8 [479]
СиС12—КС1 на активированном оксиде алюминия 230 2,0 (4,0) Псевдоожижен- ный (ц = 0,07 м/с) ДХЭ 98,5 [480-482]
СиС12 — КС1 на кизельгуре или оксиде алюминия, кремнеземе или активированном угле 320 2,0 (2,5) Неподвижный объемная скорость 187 ч’1 ДХЭ 95,26; С2Н3С10,79; С2Н5С1 0,75; другие продукты, включая цис- и транс- С2Н2С12, С2Н3С12, С2Н3С13,С2Н2С13, С2С14, СО и СО2 [483,484]
КОНЦ. НС1) ШЛ
СиСЬ — КО на кремнеземе 500 3,5 и 0,2 ДХЭ 1,15 Неподвижный ВХ 44,5; С2НС139,3; 1,1-С2Н2СЬ13,1 и др. 33,0 [487]
СиС12—КО на флорексе 274 2,0 0,55 Неподвижный ДХЭ 97,0 [488]
СиС12 — КО на фуллеровых землях 300 1,5 1,0 Псевдоожижен- ный ДХЭ 91,3 [489, 490]
СиС12—КС1 (8% Си и 5,5% К) на очищенной глине 300 2,0 0,5 Псевдоожижен- ный ДХЭ 61,0 [491,492]
СиС12— КС1 и /или NaCl на активированном оксиде алюминия 250 2,0 0,5 Неподвижный ДХЭ 95,2 (макс) [493]
CuCl2—КС1 (5% Си и 3% К) на а-оксиде алюминия 1 м2/г (БЭТ) и пористостью 0,2-0,8 мл/г 350-400 2,3 0.65 - ДХЭ 86,9 [494,495]
СиС12 — КО на диоксиде кремния — оксиде магния 120 м2/г (БЭТ) и 0,73 мл/г объем пор 340 2,0 0,5 Псевдоожижен- ный ДХЭ 84,9 [496]
СиС12 и/или FeCl3 на активированном угле 200-220 2,0 2,5 Неподвижный ДХЭ 96,2; С2Н4и СО2 3,8 [497]
СиС12 и/или FeCl3 и LaCl3 на активированном угле 200-350 1,5 (2,5) - ДХЭ 92,0 [498]
FeCl3 (5%), КС1 (3,5%), La2O3 (0,5%) на цеолите 500 1,0 (2,5) - ВХ 46,3 [499]
СиС12 и FeCb на оксиде алюминия 230 1,0 0,6 - 1,3 кг ДХЭ с кг С2Н4 [500]
СиС12 и СаС12 на флорексе 278 2,0 0,5 Псевдоожижен- ный ДХЭ 96,9 [501, 502]
Таблица 4.7 (продолжение)
Состав катализатора Условия реакции Тип реактора Продукты образования Источник
t, °C НС1/ С2Н4 О2 (воз- дух)/С2Н4
СиС12 и КС1 — СеС13 на оксиде алюминия или пемзе или кремнеземе 600 м2/г (БЭТ) и диаметром частиц 67 А 425-525 1-10 (3,5) Псевдоожиженный или неподвижный (т = 0,5-5-15 с) ДХЭ 98,5 [503-505)
СиС12(6-15,5%), редкоземельные хлориды (0,5%) и NaCl (0,5%) на оксиде алюминия 1-250 м2/г (БЭТ) 250-350 1,0 (1,3) Псевдоожижен- ный ДХЭ 99,0 [506-508]
СиС12 (5%), дидимиум хлориды (0,5%) и КС1 (0,3%) на кремнеземе 250 2,25 0,75 Неподвижный ДХЭ 98,0 [509-510]
СиС12 (7-10%), КС1 (2,8-6%) и редкоземельные хлориды на силикагеле 60-95 м2/г (БЭТ) или оксиде алюминия; цирконии, или оксиде бериллия или сульфате кальция 340 2,26 0,8 Псевдоожижен- ный (ц = 0,5 м/с) ДХЭ 82,0; ВХ 115; 1,1- С2Н2С122,0 [511]
Хлориды Си, К и Nd на силикагеле 420 н.д. н.д. Псевдоожижен- ный (т = 38 с) Перхлорэтилены и три- хлорэтилены [512]
Си (0,5-2%), Nd (1-3%) и Cs (2%) на оксиде алюминия 7 м2/г (БЭТ) 307-311 2,0 (2:1 NH3 или 8:1 НС1) 0,5 Неподвижный ДХЭ 51-61 [513]
СиСО3, СеО2 и/или Fe2O3 и дидимиум оксид на инертном носителе NA 2,0 0,5 2:1 = Н:С - ДХЭ; 1,1-ДХЭ; С2Н5С1; и ВХ (в незначительных количествах) [514]
Си (2,4%), К (2,12%) и Се (1,94%) и дидимиум (0,42%) на силикагеле 360-500 1,7 (по С1) (3,5) С2НС13 26,5 и С2Н4 68>2 [515]
-
Ссагпаы — со* we'• жежза ‘ редкоземельных и магния и щелочи или щелочноземельные металлы на микропористом а-оксиде алюминия 20-45 А (БЭТ) с содержанием меди 7,0% пс евлоожиженный ' - з 1 ”
Шпинель: А12О3 — СиО — MgO- катализатор с 10% Си, 12,4% Mg и 13,3% А1 на инертном носителе 238 1,9 (3) Псевдоожижен- ный (объемная скорость 400 ч’1) ДХЭ 99 [518-520]
СиС12—NaCl и/или на активированном угле н.д. 2,0 (2,5) Неподвижный (объемная ско- рость 80 -150 ч-1) ДХЭ 35 [521]
Fe,O4 0,4%; FeO 2,2%; CaO 2,22%; Na2O 2,42%; K2O 0,65%; SO, 1,24%; SiO2 75,2%; A12O, 14,97% 400-450 1,0 (1,8) Неподвижный (объемная ско- рость 80-227 ч’1) ДХЭ 21,7 и ВХ 78,3 [522]
Хлориды Cu, Zn, Mg на оксиде алюминия 350 0,3 (по С1) 1,2 Неподвижный (объемная ско- рость 260 ч’1) Селективность по ВХ 81 [523]
Хлориды Си, Cd, Та, Bi, Zn, и Cr на у оксиде алюминия 310 2,0 0,5 Неподвижный (т = 4 с) ДХЭ 45,3; С2Н3С1 1,8; ВХ 36.9 [524, 525]
Хлориды меди, цинка, калия и лантанидов на а-оксиде алюминия 375 1,0 (2,4) Псевдоожижен- ный (т = 13 с) ДХЭ 12,1; ВХ 62,2 и другие хлоруглеводороды 16,7; и СО+СО2 9,0 [526]
PdCl2 (1%), CuCl2 (2%), FeCl3 (5%), СеС13 (6%) и NaCl (19%) на а-оксиде алюминия н.Д. 1,0 0,5 Неподвижный Селективность по ВХ 57,5 [527]
Таблица 4.7 (продолжение)
Состав катализатора Условия реакции Тип реактора Продукты образования Источник
t,°C НС1/ С2Н4 О2 (воз- дух)/С2Н4
Водные растворы, содержащие хлориды Pd, Си, Сг 120-200 3,5 по кон- центр, кислоте 1,0 СН2С1СНО СНС12СНО (а-хлорированные альдегиды) [528]
FeCl3, NaCl или КС1 и РС12 на оксиде алюминия 350 3,0 1,0 Неподвижный ВХ 60,2 при конверсии этилена 88,1 [528]
CuCl2, PtCl4, МоС13 на гранулированном угле 175 2,3 1,4 Неподвижный (объемная ско- рость 1200 ч’1) ДХЭ 96,2 и ВХ 1,0 [530]
Хлориды меди, церия и платины, содержащие 5% Си, 1% Pt и 3% Се на активированном оксиде алюминия 250-350 2,0 3,8 Неподвижный (объемная ско- рость 1ч1) ДХЭ 3,5 и ВХ 3,0 [531]
Соли Rh или Pt, Fe или Си и А1. Предложенная композиция: Fe 1,3%; Rh 0,05; Zn 3,3%; Li 0,37 350 1,0 (2,0) Неподвижный (объемная ско- рость 1 ч-1) ДХЭ 24,92 ВХ 38,86 цис- и транс- С2Н2С12 1,3; СО2 6,55; не конвер- тированный С2Н4 28,37 [532]
CuCL и щелочи, бисульфаты металлов и/или бисульфат аммония в мольном отношении от 1:0,1 до 1:10. Использовались также схожие промоторы ТЬ, Се и La. Носители не установлены 150-350 0,7 0,3 Неподвижный (объемная ско- рость 1ч1) ДХЭ 97,6; С2Н2С1 1,96 и ВХ 0,65 [533]
CuCl — КС1 в расплаве в соотношении 3,5:1 400 2,0 0,5 ВХ 10-20; количество ВХ в инертном газе в 3-хстадийном процессе 36,3 [534-535] W-
Кйамватар — сяяЛ . ККржаший хлориды щелочных металлов, щелочноземельных металлов и/или немного хлоридов Сг и Ni в смеси с Fe, Мп, Pd или редкоземельными металлами 25 ' 0с5 Трточатым С2НзС14ОЛ СНцСГ 2Д, цис- С2Н2С12 15,4; транс- С2Н2С12 7,2; С2НС1314,0; С2Н3С1з 1,7; С2С1414,9; ССЦ 1,0. Небольшое количество перхлорэтилена (до 3,0%) и неидентифицированные продукты 536-539]
Катализатор — расплав солей, содержащий CuCl2, CuCl, КС1, хлориды редкоземельных и хлориды Zn, Pb и Cd 300 1,0 0,5 — ВХ 83,6 [540]
Хлориды меди (1% Си) и конденсированный фосфат Na (0,5-3 моль/моль СиС12) на кизельгуре 300 3,0 2,5 Неподвижный (объемная ско- рость 300 ч"1) ВХ 99,22, когда был использован фосфат натрия [541-544]
СиС12 + МоС13 вместе с КС1 на кизельгуре 300 2,0 0,5 Неподвижный (объемная ско- рость 300 ч*1) ДХЭ 96,5 (макс) [545, 546]
CuCl2, АиС13 и КС1 в мольном отношении 0,25:0,0025:0,025 на силикагеле 215 2,0 0,5 Неподвижный (объемная ско- рость 300 ч ') ДХЭ 78,2 [547]
CuCl2, M0CI3, WC16, СгС13, AgCl, MnCl2, CdCl2 или ZnCl2 на гранулированном угле 185 2,3 1,4 Неподвижный (объемная ско- рость 1000 ч ') ДХЭ 98,2 и ВХ 1,0 [548]
Хлориды Си, Аи, К, Са и La на силикагеле 150-250 2,30 (4,0) Неподвижный (объемная ско- рость 900 ч’1) ДХЭ 99,5 и СО2 0,5 [549, 550]
240 I
Таблица 4.7 (окончание)
Источник [551] [552] [553]
Продукты образования ДХЭ 98,0 ДХЭ 98,7; С2Н5С1 0,7; и другие галогенсодержа- щие продукты 0,6 Селективность по ДХЭ 100%, выход 70%
Тип реактора Неподвижный (объемная ско- рость 900 ч ’) Неподвижный (объемная ско- рость 900 ч~‘) Псевдоожижен- ный (т = 3+20 с)
S SX Л 02 Оч S СО с с; £ О2 (воз- дух)/С2Н4 (3,3) (3,3) 0,5
11ГЭ /ОН 1,7 г-Ч 2,0
t, °C 225 250 300
Состав катализатора Хлориды Си и Cd на активированном оксиде алюминия с диаметром частиц 60 ц 340 м2/г (БЭТ) и объемом пор 0,4 мл/г Сульфаты меди или натрия на активированном оксиде алюминия СиС12, КО и Sb(OH)6 в соотношении от 1:0,5 до 2,0:1,0
Jle i учес 1i. хлорида меди може г бы п> снижена увеличением энергии актива-
ции сублимации, например, за счет применения добавок с образованием менее
летучих комплексов. В качестве именно таких добавок используют хлориды
щелочных металлов. Наличие катионов щелочных металлов заметно меняет
характер адсорбции хлорной меди на носителе. Например, в работе [555] ука-
зано, что СиС12, нанесенный на пористый носитель (А12О3), легко удаляется
промывной водой, но при наличии щелочных металлов удалить медь из пор
невозможно.
Авторы работы указывают, что для систем «хлорид меди (I и II) — силика-
гель» летучесть CuCl более, чем в 30 раз превышает летучесть СиС12; для систе-
мы же CuCl-KCl-силикагель летучесть хлорида меди снижается в 230 раз.
Добавка хлорида калия снижает температуру плавления смеси хлоридов и
на поверхности образуются твердые и частично жидкие растворы. Так, напри
мер, температура плавления эвтектической смеси CuCl2-CuCl равна 375 °C, а
тройной смеси CuCl2-CuCl-KCl — 150 °C. Поскольку в ходе реакции оксихло
рирования образуется CuCl, то на поверхности в зависимости от соотношения
CuCl2/CuCl возможно образование эвтектических смесей разного состава с
разными же температурами плавления, например, низкоплавких соединений
KCuCl3, К2СиС14 и К2СиС13.
Таким образом, реакция оксихлорирования идет в жидкой пленке рас-
плава, адсорбированной носителем. В зависимости от температуры реакции
вязкость этой пленки меняется, чем, очевидно, и объясняется слипание ката-
лизатора.
Показано [554], что при оксихлорировании в псевдоожиженном слое ка
тализатора слипание контактов на основе силикагеля наблюдается при 310-
300 °C и ниже. Этот факт, в сочетании с малой активностью катализаторов при
t <300 °C обусловливает необходимость поддержания температуры на уровне
320 °C. Показатели процесса оксихлорирования этилена в присутствии мед-
но-калиевых катализаторов на основе силикагеля (содержание меди 6 мае. %,
атомное соотношение Си:К = 1:1) характеризуются конверсиями этилена на
уровне 78-84%, хлороводорода 76-86%, при содержании дихлорэтана в смс
си хлорорганических продуктов 98,0-98,6%. Изменение размера пор от 3 до
35 нм при использовании силикагелей разных марок практически не влияет
на активность катализатора [554]. Влияние пористой структуры в большей
степени проявляется в селективности действия катализатора в отношении
глубины хлорирования: увеличение радиуса пор снижает выход побочного
1,1,2-трихлорэтана в 3,5-4 раза. Эти данные согласуются с выводами в pa6oie
[556], согласно которым мелкопористые силикагели менее избирательны, чем
крупнопористые за счет высокой концентрации продуктов реакции в порах и
возможности их дальнейшего превращения.
Существенным достоинством силикагелевых катализаторов является прак
тически отсутствие продуктов глубокого окисления (СО2 и СО) в реакционной
смеси.
В целом силикагели следует отнести к группе инертных носителей примени
тельно к условиям процесса оксихлорирования этилена, что обусловливает не
обходимость использования высоких температур и приводит к значительным
трудностям в практической реализации данного процесса.
Несколько но иному веду г себя к.и литические системы, в основе коюрых
лежат алюмосиликатные носители. Алюмосиликлы — эго смешанные оксиды
алюминия и кремния, содержащие в качестве активатора небольшое количес|
гпо воды. Наиболее подробно природа алюмосиликатов, механизм их катали
гического действия изложены в монографии В.К. Скарченко [557]. Активное гь
алюмосиликатных катализаторов в реакциях крекинга, алкилирования, и.юмс
ризации и других, в частности, оксихлорирования (при наличии, естественно,
хлорида меди) в той или иной степени связывается с их кислотностью. При этом
не все атомы, входящие в состав катализатора, равноценны по своему положе
нию в структуре. Так, по данным Х.Г. Вейса с сотр. [558], в зависимости от уело
вий активирования от 11 до 37% гидроксильных групп в катализаторе связано
с атомами алюминия, остальные — с атомами кремния. Согласно результатам
работы [559], в алюмосиликатном катализаторе в зависимости от его активное
ти от 45 до 95% имеющегося в нем оксида алюминия способны реагировать с
фторидом натрия.
Из подобных фактов можно сделать вывод о том, что разные участки по
верхности обладают разными каталитическими свойствами. При этом наличие
каталитических участков разной природы не исключает однородности повер
хности катализатора, т.е. равноценности каждого, не слишком малого участка
поверхности данного катализатора или постоянства удельной активности по
всей поверхности катализатора.
Существуют различные точки зрения на характер кислотности алюмосил и
катных катализаторов. Ряд авторов [560-562] указывает, что алюмосиликаты
являются бренстедовыми кислотами, т.е. наличие кислотных свойств вызвано у
них существованием структуры со слабо связанным протоном, способным <п
щепляться с присоединением к другим веществам, которые рассматриваются
как сопряженное с данной кислотой основание. Подвижность протона в струн
туре алюмосиликата рассматривается как результат взаимного влияния сосед
них атомов кремния и алюминия, соединенных через кислород Si—О—А1.
По К. Томасу и Л. Полингу [560, 561] атомы алюминия изоморфно замеща
ют атомы кремния в кристаллической решетке SiO2. Поскольку алюминий, и
отличие от четырехвалентного кремния, является трехвалентным, то для элек i
ронейтральности решетки требуется дополнительно один положительный ион
Таковым и является протон, способный к отщеплению вследствие непрочной
связи в решетке.
Ряд авторов [563-565] указывает на льюисовскую природу активных цеш
ров алюмосиликатов. С учетом того, что углеводороды обладают слабыми ос
новными свойствами, активирование их молекул на поверхности катализатор.»
происходит вследствие образования непрочных поверхностных промежузоч
ных комплексов между молекулой углеводорода и апротонными центрами ка
тализатора. Важна связь каталитической активности алюмосиликатов с крио
таллической фазой оксида алюминия в катализаторе: у- или х-ф<>рм [564, 566,
567]. Здесь необходимо учитывать значительную кислотность оксида алюминия
[568].
При небольшом относительном содержании оксида алюминия в алюмоси
ликатном катализаторе на каждый грамм-атом алюминия приходится 1 г эки
кислоты. Количество кислотных центров в катализаторе в зависимости от со
держания Al-О, проходи г через максимум, который cooiitri. niyei <<>jv pianino
Л1.О( от 18 до 50% 1557, 560, 568].
Применительно к процессу оксихдорирования пилена хлормеднокалн
< iliac катализаторы на основе алюмосиликата достаточно эффективны при
.'»>(> 280 °C. Т.Д. Гужновская [554], исследовавшая ряд катализаторов процесса
па основе разных алюмосиликатных носителей, характеристика которых пред
i i явлена в табл. 4.8, указывает, что во всех случаях конверсия этилена составил
< । 89-92% при содержании 1,2-дихлорэтана в смеси хлорорганических продук
ion 98,5 98,8%. Различие в поведении катализаторов в процессе определяется
прежде всего содержанием оксида алюминия в носителе: минимальный выход
побочных продуктов глубокого окисления — СО2 и СО — на уровне 0,4 0,8%
имеет место при использовании образцов с содержанием А12О34-10%, а макси
мальпый (1,3-2,2%) — при содержании А12О3 10-15%.
На рис. 4.13 представлены зависимости активности и селективности алю
мос иликатных и силикагелевых катализаторов при оксихлорировании этилена
и содержания меди в системе. Температура процесса для алюмосиликатного
(.блица 4.8. Свойства алюмосиликатов
М«|рка алю м осн /1 и ката Техни- ческие условия Химический состав, мае. % Структурная характеристика На сыпная масса, г/см’
А12О3 SiO2 Na2O Fe2O3 S, м2/г размер пор, нм V пор, см3/г
V 1МИ1ОрИСТЫЙ ПИИ1 ИВОВОЙ опомосиликат (। р.шулиро 1Ы11ПЫЙ) МРТУ 38-1- -189 65 9-15 83-89 н.б. 0,4 0,2 305 2,5-3,0 0,710
Шнрокопорис 1 l.l II аЛЮМОСИ- iiih.ii (грану ||>1р1>11.!11НЫЙ) МРТУ 38-1 188-65 8-10 86-90 0,12 0,15 260 3,5-4,0 — 0,513
Адсорбент» 1 тропленный) МРТУ 38-1- 188-65 8-10 86-90 0,12 0,15 320 2,5-3,0 — 0,630
Мвкросфсри и кии алюмо . Il'lllh.ll «А» - 4,26 -95,4 - - 509 2,62 0,665 0,554
Мпкр< нфери- '|| кип алюмо 1 llllllh.il "К» - 15,21 -84 - - 354 3,07 0,544 0.646
М in. р< кфери- 1.1 кип алюмо- IHIHKUI ВТУ 38-1- 142-67 12-15 -88 н.б. 0,8 н.б. 0,2 500- 540 1,5-2,0 - 0,640
Рис. 4.13. Зависимость активности катализатора оксихлорирования этилена от сидер
жания меди.
Алюмосиликат: 1 — конверсия НС1; 2 — содержание ДХЭ в сырце; 3 — сгорание этилена. Силикл
гель: 4 — конверсия НС1; 5 — содержание ДХЭ в сырце
катализатора 280 °C, для силикагелевого — 320 °C. Увеличение содержания меди
до 6 мае. % в целом благоприятно влияет на показатели процесса. Дальнейшее
повышение содержания меди ведет к агломерации катализатора и нарушениям
режима псевдоожижения.
Именно наличие вклада алюминия (в связанном или несвязанном виде) и
алюмосиликатном катализаторе позволяет существенно снизить температуру
процесса по сравнению с катализаторами на основе силикагеля. Процесс ок
сихлорирования этилена катализируется, очевидно, активными комплексами,
возникающими при взаимодействии хлорида меди с оксидом алюминия и су
щественно снижающими энергию активации процесса. В свою очередь, темпе
ратура 260-280 °C слишком высока для системы СиС12—А12О3, что и обуслои
ливает увеличение выхода продуктов глубокого окисления.
Каталитические системы на основе алюмосиликатных носителей не получи
ли распространения в промышленной практике процессов оксихлорирования
этилена. Причиной этого явилось прежде всего использование температур 260
280 °C, при которых в качестве конструкционного материала реактора необхо
димо применение дорогостоящих специальных сталей. Применение углеродис
той стали в этих условиях невозможно из-за высокой скорости так называемо!!
«температурной» коррозии — более 1 мм/год.
С 1970-х годов в большинстве промышленных процессов оксихлорирова
ния этилена в качестве катализатора используют системы на основе оксида алю
миния. Причиной этого является ряд факторов. К важнейшим из них относи i
ся то, что в зависимости от направления использования оксида алюминия его
LU
I'u. 1.14. Пример типичной зависимости объема пор от удельной поверхности
(у. Л. О фалы А1>О3)
ф । юный состав может существенным образом варьироваться. При этом меня
юн я такие характеристики носителя, как удельная поверхность и объем пор,
ihii пянающие большое влияние на эксплуатационные свойства катализаторов.
I Понос । рация этой взаимосвязи представлена на рисунке 4.14 |569]. В зависи
м«к in от условий (температура прокаливания) получения носителя конечным
продуктом может являться и а-А12О3 (корунд), удельная поверхность которого
in превышает 1-10 м2/г.
II iMc-нение фазового состава оксида алюминия влечет за собой изменение
। io кислотно-основных характеристик, что также является важным условием
формирования активных комплексов на поверхности катализаторов.
Рассмотрим несколько более подробно основные физико-химические спой
। । на оксидов алюминия, которые детально изложены, например, в монографии
()дпим из важных свойств оксида алюминия является его амфотерность, г.е.
и <н ионной среде он проявляет кислотные свойства, а в кислой — основные: из
in < и н алюминат натрия, в котором алюминий присутствует в анионной форме,
и хлорид алюминия, в котором он находится в виде катиона.
Цепным для носителя качеством является высокая температура плавления
hi • нда алюминия — немного выше 2000 °C. Он относится к тугоплавким ок
и ым и обладает замечательной способностью стабилизировать мелкодиспер<
т.н’ час гицы катализатора и предотвращать их (имеющих более низкую гемпе
рн} ру плавления) слипание или спекание, являясь как бы термостабили.ыго
ром к.нализатора.
< лмой примечательной особенностью оксида алюминия является много
ihp.i шс сто модификаций и наличие фазовых переходов между ними в очечн.
широком интервале температур. Эти фазы, которые обычно характеризуючея
*ис. 4.15. Плотно- и рыхлоупакованные решетки соответственно a-Al2Oi (а) и
'-А1,О3 (6)
Рис. 4.16. Схема диссоциативной адсорбции воды:
А1 — кислота Льюиса; О — основание Льюиса
[ппинельной структурой (рис. 4.15), разнятся дефектами кристаллической ре
летки. Хотя образование плотноупакованной решетки а-А12О3 термодинами
чески возможно при низкой температуре, реально оно происходит только после
некоторой перестройки кристаллических решеток переходных фаз оксида алк»
ииния, что требует высокой температуры. Этим объясняется сохранение ра i
витой поверхности оксида алюминия при температурах прокаливания 1000 и
даже 1200 °C.
Благодаря способности оксида алюминия легко образовывать гель, оксид
Алюминия и его соли очень легко поддаются экструзии или формованию в гра
дулы или сферические частицы. Оксид алюминия не уникален в этом отношс
1ии, но более других оксидов и гидроксидов пригоден для формования.
Свежепрокаленный оксид алюминия в присутствии влаги воздуха вновь
идратируется и гидроксилируется при обычной температуре. Это явление со
этветствует диссоциативной адсорбции воды (рис. 4.16), ведущей к образова
нию ОН-групп.
Образующиеся гидроксильные группы на поверхности оксида алюминия в
водной среде носят амфотерный характер [604, 605]. Это свойство приводи ! к
эазной ионизации ОН-групп в зависимости от того, является ли пропитываю
дий раствор кислым или щелочным.
Рассмотрим структуру кислотных центров оксида алюминия с бренстедо
зыми и льюисовыми видами кислотности. Как уже было показано, в резуль
on oil о II o' и о
1 ' 11.11 IK’HJIIIK' ।
O—Al—O—Al-----------O — Al — o—Al------------* O—Al — O — Al -
Рис. 4.17.11ереход льюисовых кислотных центров в бренстедовы
С
А В
Рис. 4.18. Кислотные и основные центры на катализаторе у-А12О3(+ обозначает ион А1' ‘)
и ге дегидратации на А12О3 одновременно образуются льюисовы кислотные и
основные центры. В присутствии молекулы воды льюисовы кислотные центры
переходят в бренстедовы (рис. 4.17):
Таким образом, благодаря нагреванию свойства кислотных центров могут и i
меняться, что справедливо не только для А12О3, но и для других оксидов [607].
Поверхность у-А12О3, полученного путем термообработки гидроксида алк>
миния при 800 °C, на 10% покрыта гидроксильными группами (ОН) . При пом
«уществует пять типов ОН-групп (А-Е), отличающихся своим окружением
(рис. 4.18). На ОН-группах типа А, окруженных четырьмя ионами О2 , плен
пость отрицательного заряда максимальна, и поэтому они имеют основные
i нойства; ОН-группы типа С, не окруженные ионами О , обладают максималь
ним положительным зарядом и проявляют кислотные свойства [607].
Упрощенная схема взаимодействия оксида алюминия с водным раствором,
(«•держащим ионы, не способные к хелатообразованию и осаждению, может
<>ы । ь представлена следующим образом.
В кислой среде поверхность оксида алюминия будет заряжаться положи
iciii.Ho, что приведет к адсорбции ионов противоположного знака. Обозначив
। идроксильную группу, локализованную на поверхности оксида алюминия,
< выводом S—ОН, можно представить процесс ионизации уравнением
S—ОН + Н+ = S—ОН2+. (4.45а)
В ном случае оксид алюминия выступает в качестве адсорбента анионов.
В щелочной среде оксид алюминия >аряжаегся отрицательно, чю может
<»ы и. выражено уравнением
S—ОН + ОН* = S—О + Н2О, (4.456)
и в этом случае оксид алюминия адсорбирует катионы.
На рис. 4.19 показано количественное изменение катионной и анионном
адсорбционной емкости оксида алюминия с удельной поверхностью 200 м7г в
зависимости от pH [606].
Можно отчетливо видеть амфотерную природу оксида алюминия: увеличе
ние адсорбции хлор-ионов в отсутствие адсорбции ионов натрия с увеличением
кислотности среды и увеличение адсорбции катионов Na+ по мере увеличения
щелочности среды при полном отсутствии адсорбции анионов хлора. Кроме
того, следует обратить внимание на определенный интервал pH, где оксид ал к»
миния находится с самого начала в равновесии с пропиточным раствором. Эт<н
интервал, между pH = 8,5 и 9, соответствует потенциалу нулевого заряда оксида
алюминия (рНнз) [603].
Подобные ионно-адсорбционные свойства оксида алюминия можно выгод
но использовать для атомного диспергирования предшественника металличег
кой активной фазы, которая должна быть нанесена на оксид алюминия. Пос
ле активации такого катализатора дисперсность активной фазы оказывается
близкой к 100% [606]. При использовании конкурентной адсорбции двух ионои
одного типа и ионного обмена также можно добиться оптимального распреде
пения исходного соединения металла внутри зерна оксида алюминия.
Более того, используя конкурирующие ионы со значительно большим
сродством к оксиду алюминия, чем исходное соединение наносимого металла,
можно блокировать адсорбционные центры по периферии зерна и вынуди гь
металл адсорбироваться внутри зерна (распределение типа «яичного белка»
или «желтка»).
Рис. 4.19. Адсорбция Na+ и CI на g-оксиде алюминия в зависимости от равновесного pH
)iu факты указывают, что поверхнос iiii.ic харакк pin iпкп оклнда нлюмн
ния придаю! ему очень интересные адсорбционные и ионообменные iiioiiuiiia,
которые даю г возможность готовить высокодис нерп пае кат али споры с коп i ро
лируемым распределением металлов — оно может бы п. он гимн шроиапо в юот
веге гпии с характером применения катализатора. Совершенно очевидно, что ни
inoiicTBa оксида алюминия находятся в прямой связи с его текстурными (удель
ния поверхность) и структурными характеристиками, а также с его химической
ч ис го гой, поскольку содержащиеся в носителе примеси могут привести к измене
limo его поверхности и особенно к смещению потенциала нулевого заряда |6(М|.
Поверхность оксида алюминия представляет комбинацию ионов алюми
пня и кислорода, каждый из которых может иметь меньшее координационное
число, чем в объеме. Эти поверхностные ионы являются вакантными центра
ми, которые при комнатной температуре всегда оказываются занятыми либо
। ндроксильными группами, возникающими при диссоциативной адсорбции
воды, либо координационно связанными молекулами Н2О. При этом восста
на вливается нормальное координационное состояние поверхностных ионов:
октаэдрическое или тетраэдрическое для ионов алюминия и октаэдрическое
для ионов кислорода. Дегидрирование и дегидроксилирование поверхности ок
i нда алюминия приводит к появлению координационно ненасыщенных атомов
кислорода (основные центры Льюиса) и алюминия (кислотные центры Льюи
i.i), в результате чего оксид алюминия приобретает каталитические свойства в
некоторых реакциях.
Типичные характеристики оксидов алюминия:
Анализаторы . ...F-l H-151 S-100
Л1,.О„ % ... .92,0 90,0 95,0
Na-О, % ... .0,58 1,6 0,35
‘м(),,% ....0,12 2,0 0,03
1 <-.(),» % .. .0,06 0,03 0,05
101, % .... 7,0 6,0 5,0
11.к ыиная плотность, г/см3 .. ... .0,83 0,82 0,80
( Юьемная плотность, г/см3 .. . . .0,85 0,85 0,75
1 Индий объем пор, см3/г . . .0,40 0,43 0,49
( Инцая пористость, % . . .56,3 59,4 60,6
I Ип.ем пор, см /г; диаметром, нм:
io'-io7 . . .0,0031 0,0003 0,0000
KI-106 .. .0,0045 0,0001 0,0001
ю-ю5 .. .0.0059 0,0001 0,0004
IO3-IO4 .. .0,0029 0,0004 0,0033
I(> -IO3 .. .0,0045 0,0093 0,0047
<5-102.. .. .0,0021 0,0099 0,0289
' <5 .. .0,0165 0,0232 0,0115
Роль оксида алюминия как носителя заключается к him, чтооы p.i юани i ь,
диспергировать и стабилизировать нанесенный металл. Нели количество на
несенной фазы меньше адсорбционной емкости носителя, оксид алюмиАия
служит также для макроскопического распределения кристаллитов. При этом
адекватным подбором исходного соединения металла и условий пропитки
можно получать разные профили пропитки — гомогенный, типа «яичного бел
ка», «яичного желтка» или периферический. Роль носителя заключается также
в повышении устойчивости метастабильной дисперсии мелких кристаллитом
металла на поверхности А12О3 к агломерации и спеканию.
Кроме того, благодаря внутренней пористости оксид алюминия облегчаю
диффузию реагентов и продуктов реакций к каталитически активным центрам
и от них. Это может быть очень важным в случае каталитических реакций, ли
митированных диффузией реагентов, т.е. в процессах, идущих с очень высокой
скоростью [603].
Э. Стайлз [603] суммирует позитивные свойства и положительные момен гы
использования оксида алюминия:
1) доступность в больших количествах и умеренная цена;
2) возможность получения высокоразвитой поверхности, как правило, терм и
чески устойчивой в обычно используемых условиях катализа;
3) широкий диапазон микро- и макропористости;
4) наличие как кислотных, так и основных поверхностных центров.
Все эти качества объясняют распространение, которое получил оксид адц>
миния и, по-видимому, будет иметь в будущем.
Как будет показано, в отличие от силикагеля у- и 0 А12О3 не являются инер
тными носителями, а взаимодействуют с компонентами солевой фазы. Эго
взаимодействие приводит к изменениям состава и структуры образующихся
на поверхности носителя солевых комплексов и их каталитической активное
ти. Оксихлорирование этилена становится возможным в этом случае уже при
200-220 °C.
В связи с тем, что промышленные процессы оксихлорирования этилена мо
гут протекать как в псевдоожиженном, так и в неподвижном слое катализатора,
требования к носителям, используемым для приготовления соответствующих
катализаторов, существенно различаются.
Дж. Наворски и Э. Велез [199] указывают, что для процессов, протекающих
в псевдоожиженном слое, носитель катализатора должен обладать свойствами,
способствующими хорошей флюидизации: высокой адсорбционной способное
тью в отношении пропитывающих солей, препятствующей слипанию частиц
катализатора; высокой механической прочностью, снижающей количество об
разующихся мелких частиц катализатора; распределением частиц по размерам,
удобным для флюидизации.
Ниже представлены характерные значения физико-механических парамет
ров микросферического оксида алюминия, используемого в качестве носителя
для приготовления катализатора псевдоожиженного слоя в процессе оксихло
рирования этилена:
Насыпная плотность, г/см3..........................................0,80-0,95
Удельная поверхность, м2/г.........................................120-180
Удельный об bum пор, iM '/I О 0.1’
IpaiiymtMcuipuuri кии < <>< гнав, мт %:
<20 мкм........................................................ ,ь5
2(1 40 мкм... ......... ..................... .................25 10
10 80 мкм .................................. ......... . .35-50
80 мкм............................... ............................6- 10
11огери при истирании, 5 ч, по методике «Аминко-Роллер», мае. %......1 6
( редкий радиус пор, нм.............................................. 1 6
Сочетание приведенных физико-механических характеристик носителя с
по фазовым составом и оптимальной технологией приготовления катали »аго
рл обеспечивают высокие показатели процесса в ходе промышленной зкеплуа
1.1Ц11И.
Оксид алюминия используют также как носитель для приготовления ката
ли ia торов неподвижного слоя. На практике оксид алюминия формуют в цилип
дры и шарики диаметром 2-5 мм [199, 571]. Цилиндрические таблетки готовя!
прессованием порошка в аппарате для таблетирования. Плотность таблетки
мо кно в небольших пределах варьировать, меняя давление при их прессовании.
В целях предотвращения прилипания таблеток к матрице для таблетирования
in пользуют малые количества графита или других смазочных материалов. Ци
шндры готовят, выдавливая пасту оксида алюминия через пластину с огверс
1нями соответствующего размера, почти так же, как при приготовлении мака
рои. Стержни оксида алюминия нарезаю! вращающимся ножом на цилиндры
и) иной длины. В отличие от гладкой поверхности таблеток торцы цилиндров
шероховаты. Цилиндры мягче таблеток и легче разрушаются [199].
< 'уществует много методов получения шариков оксида алюминия.
В одном из методов твердые частицы, которые называют затравкой, враща
ini или погружают в суспензию оксида алюминия, близкую по консистенции
। краске. Образовавшиеся зерна подсушивают и снова повторяют процесс до
получения шариков нужного диаметра. Шарики состоят из концентрических
i лиев оксида алюминия.
В другом методе используется изогнутая по винтовой линии вращающая
. и предка. Жидкая паста оксида алюминия медленно падает на вращающуюся
предку. Оксид алюминия накапливается и слипается в центре тарелки до <>о
p.i нныния сферы, масса которой достаточно велика, чтобы она соскользнула
। .щелки. Размер сфер регулируют, меняя угол изгиба тарелки и скорость ее
вращения.
После формования оксида алюминия его гранулы прокаливают для удале
пни влаги и повышения прочности. Удельная поверхность прокаленных носи
н леи из оксида алюминия, как правило, составляет 200-400 м2/г. Поверхнос н>
пир должна составлять определенную часть от общей поверхности, что обсч
п> ’iiinacT их доступность для молекул газообразных реагентов. По-видимому,
на in и шыпее значение имеют поры диаметром 8-60 нм [571]. Носитель катали ia
ii’p.i должен быть очень устойчив к истиранию, чтобы пропитанный катали ia
nip выдержал операции пропитки, сушки, транспортировки, загрузки в груОки
pi .и. юра и условия реакции. Размер гранул катализатора также весьма важен,
'ТП----
। а к как вл и яс i на нас ыппую плотное гь катали ia i ора в i руоках реак юра, а еле
донагелыю — на активное и., приходящуюся на единицу объема реак гора.
Существуют серьезные ограничения на содержание примесей в оксидах
алюминия, используемых для приготовления катализаторов оксихлорирова
ния. Прежде всего это относится к соединениям железа, кремния и натрия.
Ниже представлены предельные значения содержания соответствующих окси
дов в носителях:
Примесь..............Fe2O3 Na2O SiO2
Содержание, 10”4 %...200 50 350
Рассмотрим более подробно характер медьсодержащих структур на повер-
хности катализатора, определяющих его эксплуатационные свойства. Напри-
мер, в работе [572] утверждается, что комплексы хлорида меди с а-А12О3 не
возникают. Предполагается, что в нанесенных на а А12О3 системах СпС12-КС1
и CuCl2-CsCI образуются ионы C11CI4 . Н.П. Уторов с соавторами [573] ука-
зывают, что СиС12 и СиС12-КС1 (мольное соотношение 1:1) при нанесении их
на силикагель практически не изменяют своей симметрии в отличие от сис-
тем CuCl2-RbCl, CuCl2-CsCl. В работе [572] делается вывод, что образование
структур, подобных Q1CI4 , при мольном соотношении CuCl2:MCl = n < 1 воз-
можно лишь в предположении, что они являются фрагментами полимерных
цепей (CuCl3)m. На основании анализа данных электронных спектров диффу-
зионного отражения [573] делается вывод о том, что структура комплексных
ионов меди изменяется под влиянием силикагеля лишь в тех случаях, когда
не требуется большой энергии для перестройки комплекса, что подтверждает
инертность силикагеля по отношению к наиболее распространенным катали-
тическим комплексам СиС12-КС1.
Инертность хлорида меди по отношению к а-А12О3 и, в меньшей степени, к
SiO2 в отличие от гидратированного носителя у-А12О3 была также установлена
в работе [581 ]. В катализаторах на основе у-А12О3 координационная сфера Сп2+
состоит из поверхностных групп (ОН” или О при сушке и активации) Н2О и
С1 ; при этом концентрация Н2О-лигандов снижается при пропитке, сушке и
прокаливании.
Образцы СиС12-у-А12О3, прокаленные при 400 °C, показывают наличие фазы
Си2(ОН)3С1; при более высоких температурах образуется СиА12О4. Отсутствие
Си2(ОН)3С1 в образцах на основе SiO2 авторы работы [582] относят на счет кис-
лотно-основных характеристик носителей. Стабильность образующихся комп-
лексов увеличивается в ряду:
у-А12О3 > SiO2 > с-А12О3
Это также может являться причиной большей пригодности у-А12О3 для ис-
пользования в процессе оксихлорирования этилена.
Авторы [574] указывают, что для катализатора CuCl2-KCl/SiO2 добавка к Си-
С12 хлорида калия в количестве, отвечающем КС1/СиС12 <0,15 (мол.) не влияет
на активность CuCI2. При увеличении соотношения КС1/СиС12 >0,15 (мол.) зна-
чение константы скорости снижается, что, видимо, обусловлено стерическим
эффектом, т.е. усилением внешнесферного влияния катионов К+ на способность
этилена входить в координационную сферу катиона меди (II) в составе солевого
комплекса. В присутствии малых добавок КС.'I увеличнв.ин л с icik iii. дшпср
iкропания солевой фазы на носи геле [574|. При ном увеличение содержании
меди с 1,7 до 5,0 мае. % в катализаторе приводит к пропорциональному уве
пилению концеп грации каталитически активной меди. Степень диснергирона
ннн солевой фазы на поверхности носителя также существенно увеличивается
ы счет процессов комплексообразования с участием НС1, что, в свою очередь,
приводит к увеличению концентрации каталитически активной меди.
Для систем СиС12-КС!/у-А12О3 в работе [575] показано, что поверхностный
катион алюминия способен образовать мостиковую связь с комплексом меди
п влиять на его акцепторную способность. Имеются данные [576, 577] о взаи
модейсгвии у-А12О3 с комплексами меди и влиянии этого взаимодействия на
характер распределения меди на поверхности А12О3. Данные свидетельствуют,
чк> в катализаторе с содержанием меди < 2 мае. % степень диспергирования
.паевой фазы даже в отсутствие в ней добавок МС1П достигает максимальной
величины. В системе CuCI2-y-AI2O3 наряду с поверхностным взаимодейсгни
см имеет место объемное взаимодействие между катионами меди и вакантны
мп октаэдрическими и тетраэдрическими центрами решетки А12О3, ведущее к
внедрению ионов меди в объем решетки носителя и образованию при прогреве
।пюминатов меди [576]. Именно эти структуры, по-видимому, являются наибо
иге ак гивными в реакции оксихлорирования этилена.
В работе [575] в качестве моделей стадий восстановления катализатора н и
в пом и реокисления катализатора кислородом предложены следующие бру г
п> реакции:
2(ZClr2HCl) + С2Н4 = 2(ZC1-2HC1) + С2Н4С12, (4.46а)
I(ZCI-2HC1) + О2 = 2(ZClr2HCl) + 2ZC12 + 2Н2О, (4.466)
। к /С12-2НС1 и ZC1-2HC1 — соответственно окисленная и восстановленная
фирмы поверхностного активного центра, содержащего один катион меди в
ни Ыне солевого комплекса, определенным образом связанного с поверхпос
Н1ЫМП частицами А12О3 [573].
В рамках предложенной схемы важна роль HCI в формировании активных
п« и । ров на поверхности солевого катализатора с носителем у А12О3. М.П. Дми i
|чн на 1575] предлагает схему, согласно которой наряду с рассмотренным выше
। |1>ьемпым» взаимодействием СпС12 с А12О3 хлорная медь взаимодействует так
I поверхностными кислотно-основными центрами А12О3 и с группами OI I,
। ни ынпыми с поверхностными центрами этого носителя. При этом образуются
комплексы двух типов. В комплексах первого типа одним из лигандов являс!
> и поверхностный ион кислорода А12О3. Эти комплексы, кроме того, обладают
ин 1ыо с поверхностным катионом алюминия. Комплексы другого типа — гид
р>>ю охлориды меди, содержащие одну или более групп ОН в зависимости oi
г п< пи ымещения ими ионов хлора из координационной сферы катиона меди.
' >п.1 ины комплексов, как было показано в [579, 580] не обладают активностью
< р> акции оксихлорирования этилена при 200-225 °C.
В присутствии НС! солевые комплексы, адсорбированные на парных кис
«пип основных центрах А12О3, взаимодействуют с НС! с образованием новых
i > шлемов с высоким координационным числом меди [573]. Эти комплексы
111 < >>| Н ЛII l< > I ilh I ШИКИ ll> II III I1<ItII В 1.1НМОДС1К 1 ВИ» с олефином В lipill у II
। nun ll< I v величин.ic к я и число клали i ичсски активных комплексов меди ы
। че| увеличения количп ilia хлорной меди, образующейся в результате и ыим*
дет. । пня I К 1 с 1 идроксохлоридом меди:
1 K'l r Си2(ОН)С13 —► Си2С14 + Н2О . (1.1 )
В 1980-х годах впервые была показана важная роль 0-А12О3 как эффекгпв
ною носителя для катализатора оксихлорирования этилена [583]. 0 А12<»। и
совокупности с нанесенным хлоридом меди образует шпинельные структуры
алюмината меди помимо сорбированной молекулы СиС12. Структура поверх
постных гидроксохлоридов также зависит от структуры носителя, активно! и.
же катализатора является функцией пористости, содержания 0-фазы и чипы
сильноосновных центров на поверхности носителя.
Кинетика и механизм реакции оксихлорирования этилена
Представления о механизме реакции оксихлорирования этилена в прис) i
ствии хлорида меди (II), с 1960-х годов претерпели существенную эволюцию
Р. Эргенбрайт [584] впервые показал, что в псевдоожиженном слое нанесенного
дихлорида меди олефины могут подвергаться прямому хлорированию:
II II
С—С + 2CuC12 —* Cl—С—С—Cl + Cu2Cl2,
II II
(I IH)
а затем в присутствии НС1 и О2 происходит окисление Сп2С12 до начальной!
состояния катализатора
Cu2Cl2 + 2НС1 + '/гО2 —* 2СпС12 + Н2О. (4.49)
Кинетика реакции оксихлорирования этилена изучалась с 1960-х годов n.i
ряде катализаторов в разных условиях и подробно освещена в обзорах [457,58*,
586].
Промышленная реализация процесса оксихлорирования этилена, впервые
осуществленная в 1964 г., послужила толчком к детальному исследованию кп
нетики реакции. Пик этих исследований пришелся на вторую половину 19Ы) х
годов. Одно из первых кинетических исследований реакции оксихлорироваип i
этилена было проведено К. Намбургом с соавт. [587] на катализаторе Сн( I.
А12О3 при 200-300 °C в дифференциальном реакторе. Эти авторы предложили
следующее кинетическое уравнение:
fcoexp[-£o (RT)]po
г =------------------------------------— ----------------------, (4.4))
{1 +Ао ехр[-£л (K^)]pc2H4ci2, Ро2+Л) ехр[~£в (R^)]Phci£o2 }
где fc0 = 0,747 109; £0 = 0,222 105; Ао = 0,322 10 3; £А = 0,74 104; Во = 0,174 10
£в= 0,332 104.
каким образом, coi ласп<» К. I laMbypiy, реакция ок»ихлорироваппя п илена
ормо hi । с я дихлор »। аном и хлороводородом,скорость реакции при помпе »а
ни» in о) парциального давления этилена. В дальнейших исследованиях л и вы
поды в основном небыли подтверждены. Оказалось 1431,585,5881, ч то скоро» гь
реакции сложным образом зависит от парциальных давлений этилена, I В I и О
и отбывается степенным уравнением типа:
Р = ^Рс2н4 Рнс1 Ро2 • • (4 1)
11а катализаторе CuCl2-KCl/SiO2 в области ограниченных конверсий ли
'в па показатели степени в этом уравнении составляют т = 0,6; и = 0,2; / = 0,5.
111 катализаторе на основе А12О3 установлена прямая зависимость скорое i и
»и /•< .и, и Ро = 1) и слабая зависимость от рНС] (н = 0,3) в области ма
нах парциальных давлений кислорода; при pOi > 0,01МПа I = 0. Аналогичная
। .ipimia (т - 1; н = 0,2; / = 0) наблюдается при использовании катализатора
< п( I,/активированный уголь в области 130-180 °C; при этом избирательное
Hi'ipa ювание дихлорэтана возможно только в условиях избытка этилена.
I’ Карруба [431] объяснил полученные кинетические данные лимитирова
пнем реакции стадией взаимодействия этилена с кислородом на поверхности
। пализагора с образованием этиленоксида или другого этиленкислородного
। омплекса. Скорость реакции, рассчитанная по модели Лэнгмюра-Хиншельву
ы Хоутена Ватсона (LHHW), описывается выражением:
6,83-10 рс?нДро2)
г —-------------------------------------—.
1 + 0,0017рС2и4 +0,035(рОг )0,5 +0,0165рН2О (4’52)
•версия активации равна 102,4 кДж/моль.
Предложенный механизм реакции с промежуточным образованием этиле
ник» пда не нашел, однако, дальнейшего подтверждения.
( (иласно Н. Коминами [455], скорость образования дихлорэтана подчини
। к л у равнению:
r = fcpc2H4(pHCi)013- (4.53)
1\ Миаучи и Г. Сато [589] для катализатора СиС12/у А12О3 при 230 °C пред
Ki Kmili уравнение:
г= 1,72.10-2рС2н4Рнс1- (4-54)
hiepi ия активации равна 62,7 кДж/моль.
При ггом высказано предположение о том, что реакция оксихлорировапия
пилена протекает через промежуточное образование оксихлорида меди (в рам
। >н ими ли тельно-восстановительной схемы превращений):
( 11, г 2CuC12 —*- С2Н4С12 + Си2С12; (4.55а)
< и < I + УЮ2 — СиС12 СиО; (4.556)
» п( I. ( иО + 2НС1 —* 2СиС12 + Н2О. (4.55н)
II Годо с соавг. ( 1511 показал, чк» в рамках окислигелыю восстанови и нь
। схемы при малой конверсии эгилена скорое и> реакции лимитруеия i ia
и восс гаповления СнС12 этиленом, а ври более высокой конверсии — с гадн
зеокисления меди I в хлорид меди II. При этом утверждается, что при ’ь() ’ (
версия НС1 по реакции оксихлорирования превышает 99% и менее 1% 11( I
вращается по реакции Дикона.
Наиболее полное представление о кинетике и механизме аддитивного <я>
лорирования алкенов и, в частности, этилена было получено на основании
от А.И. Гельбштейна с сотрудниками, проводившихся в течение ряда лет в
1ФХИ им. Л.Я. Карпова [420, 586, 588, 590-592]. Механизм реакции окспхло
топания этилена изучался на катализаторах с разным составом солевой
шозиции и носителем; в работе [586] он передан следующей обобщенно!!
мой:
Z„C12 + 2мНС1 Z„C12 • 2мНС1;
Z„C12 • 2мНС1 + С2Н4 Z„C12 - 2мНС1 • С2Н4;
(m-1)Z„C12 2мНС1 • С2Н4 —*- (м-1) Z„C12 „ + (м-1)С2Н4С12;
медл.
(2-м) Z„C12 • 2мНС1 • С2Н4 —- (2-м) Z,,C12.„ • 2мНС1 • С2Н4С1;
(2-м) Z,,C12.„ • 2нНС1 • С2Н4С1 + (2-м) Z„C12 • 2мНС1-
2(2-m)Z„C12_„ • 2мНС1 + (2-м)С2Н4С1;
(м-1) Z„C12 „ • 2мНС1 + (н-1)О2 = (м—1) Z„C12.„O2 • 2мНС1;
(2-м) Z„C12 „ • 2мНС1 + (2-м)О2 —* (2-м) Z„CI2_„O2 • 2мНС1;
(м-1) Z„C12_„O2 2мНС1—— (m-1)Z„C14 „ (ОН)2 • 2(м-1)НС1;
(м-1) Z„C14.„ (ОН)2 • 2(м-1)НС1 + (м-1) Z„C12 „ • 2мНС1---** (m-1)Z„C I
- 2мНС1 + (m-1)ZC12 + 2Н2О;
I (2-м) Z„C12.„O2 • 2мНС1 + 3(2-м) Z„C12 „ 2мНС1-2(2-м) Z„C12 2мНС1 i
+ 2(2-м) Z„C12 + 2Н2О,
р Z„Cl2 — поверхностный активный центр, содержащий м катионов меди в
с гаве солевого комплекса;
► — соответственно равновесная, обратимая и необратимая
|Дии;
н = 1 или 2 в зависимости от состава солевой композиции и природы носи
|.я.
Для системы CuCl2-KCl/SiO2 с эквимолярным соотношением хлоридов ме
л лов и в системе СиС12/у-А12О3 м = 2 и 3 соответственно. Схеме при м = 1 огне
ег кинетическое уравнение для скорости расходования этилена [593]:
Г =----- --'--------, (4.56)
] 1 ^2?Г пДМпС1+^|) ^зРс н,
^1 Pl ICI ^1 Рн< I (^-2 + ^3 ) ^5ро, (^-2 + ^3 )
I ц А„ К| — копе гаи гы скоростей и равновесия соответствующих стадий.
Для условий, koi да реализуется нулевой кинетический порядок по хлорово-
нчроду (/>|1( । > 2к11а), уравнение (4.56) преобразуется к виду:
^2^зРс2н4 2^5Ро, . ч
Г = 7------—-------(4.57а)
(^2^зРс2Н4 +2^5Ро2 )
I ели скорость реакции оксихлорирования этилена лимитируется либо ста-
ли! й 3', либо стадией 5, то уравнение (4.57а) переходит в уравнение первого по-
рядка по этилену или кислороду соответственно:
г - К2к3 рс2нл, (4.576)
г = 2к5рО1 (4.57в)
11ростота реализаций условий, при которых выполняется уравнение (4.57б),
по шоляет использовать константу этого уравнения (к' - К2к3) в качестве крите-
рия оценки активности катализаторов оксихлорирования [586, 594].
В основе вышеприведенной стадийной схемы лежат экспериментальные
цнные, полученные, в частности, в работе [588]. На рис. 4.20«-в представле-
ны 1ависимости скорости реакции оксихлорирования этилена от парциальных
давлений реагентов. Эти зависимости указывают, в частности на то, что ре-
акция выходит при всех температурах на нулевой кинетический порядок по
I И I при относительно малых значениях pHci (2-3 кПа). Порядок по кислоро-
ду — переменная величина, зависящая не только от pOi, но и отрс,Н4. Величина
(/’oJmin не очень сильно меняется с температурой, но зависит от парциального
давления этилена, ((pi)min — величина парциального давления любого из ком-
понентов реакции (р,), выше которой ее скорость перестает зависеть от этого
параметра.)
Таким образом, величина параметра (р|)П1(п для компонентов реакции ок-
‘ II хлорирования уменьшается В ряду: (pC,H4)min > (pO2)min > (pHCl)min-
В работе [588] показано, что скорость реакции не тормозится ее продукта-
ми — дихлорэтаном и водой, что иллюстрируется, в частности, линейным ха-
рак гером зависимости скорости реакции от глубины превращения этилена при
ра шых температурах (рис. 4.21).
Образование промежуточных соединений катионов меди с С2Н4 и НС1 по
координационному механизму с участием соответствующих свободных орбит
к<1 гиона меди, неподеленных электронных пар ионов хлора и л-электронов эти-
л(| па, обусловливает возможность последующего их взаимодействия с кислоро-
дом по окислительно-восстановительному механизму.
Нулевой кинетический порядок по кислороду при рО1 > (po2)min и зависи-
мость величины (po2)min 01 парциального давления этилена указывают на то,
Рис. 4.20. Зависимость скорости реакции от pop г, (а)> рна (б) и от ро (в) при рс н
> 30 кПа:
1 — 250 °C; 2 — 263 °C; 3 — 287 °C; 4 — 287 °C при рС;п4 =7 кПа
2.0
1’iic. 4.21. Зависимость скорости реакции от степени превращения этилена (X)
Гш 1.22. Зависимость скоростей восстановления СиС12—КС1 (1:1)/А12О3 — клали
• |inpti пиленом и его реокисления при 230 °C кислородом от степени восстановлен
н><> III
по в шимодействие катализатора с этиленом в этих условиях протекает мед
>. нисс и предшествует стадии, в которой участвует кислород. Таким образом,
<• ышн имосги от соотношения между парциальными давлениями этилена и
। юрода природа лимитирующей стадии может меняться, либо наблюдаемую
»»npm и, процесса определяют обе стадии. Поскольку величина (рна)тт весьма
• ин, можно принять, что стадия с участием НС1 протекает быстро.
I i.i pin. 1.22 предс i а плен харак icp i.iihii iimoi hi । unpin i • n mn । i шиплиши
кашли затора лилсном с образованием дмхлор и иы и < in pi in и к инн мн io
родом (в обоих случаях в ирису icthmh хлорт iок» водорода) ш i hihiiii hoi i .
новленпости катализатора (мольной доли non шновленнпп u нем Mi ли) • i *
рость восстановления снижается, а скорость реокислсннп pai н i 1 у in тгн hih i
степени восстановленности, а при их равенстве (точка н< pci гчснпп) они i пип •
дают со скоростью реакции в условиях стационарного катали ы | 1?0| Ин pi нч
активации стадии восстановления, характеризующаяся первым порядком ни
этилену и по концентрации катионов Си21 и равная -92 кДж/моль npai и hi
чески совпадает с энергией активации на стадии восстановления, полученной и
работе [588] и равной - 88 кДж/моль.
Согласно А.И. Гельбштейну [420] энергия активации стадии нои шнпшн
ния (~88 кДж/моль) практически не зависит от природы олефина Hi hhhi
следует, что энергетика стадии определяется не столько разрывом днонпип
связи, сколько разрывом связи Си—С1. Ее величина меньшая, чем при кр
мическом отщеплении хлора (ИЗ кДж/моль), получает естественное пиши н<
ние, если принять, что олефин координационно связывается катионом mi hi
в форме л-комплекса. Это облегчает разрыв связи Си—CI с трансформацпгЦ
п-комплекса в о комплекс. Энергия активации стадии реокисления кашли i.iih
ра кислородом составляет ~29 кДж/моль [420].
Сходные выводы были сделаны при изучении реакции оксихлорироидппн
этилена в присутствии катализатора СиС12/у-А12Оз в интервале 210 2 <
[595]. Уравнение скорости реакции оксихлорирования этилена:
г = ^нРнс|Рс2н4Ро2_____________, (1 .Нй)
(l + bipHa)^ + ^2Рс2н4 )(1 + ^зРо2 )
где Ь], Ь2, Дз — константы адсорбции реагентов.
В предположении, что btp~Ha » 1 и Ь2С2Н4 << 1 уравнение преобразуется
к виду: /I , \
г - ш i Рс,п4Ро2 / + &зРо2). (4.581»)
Автор [595] указывает, что в условиях процесса, приближающихся к про
мышленным, порядок по кислороду близок к 0,5. Это подтверждает определен
ную роль адсорбированного кислорода в реакции реокисления одновалентно!!
меди. Предложено уравнение, описывающее скорость реакции в виде:
г = ^нРс2н4Ро2. (4.58и)
Аррениусовские зависимости для наблюдаемой константы скорости имени
вид:
кн = 106'98 exp[-57400/(RT)], пт/мопьш гкат ч. (4.59)
П. Шарп и Дж. Викерман [596] показали, что активность катализатора ок
сихлорирования этилена определяется присутствием в системе ионов меди,
при этом особо указывается на различные активные центры протекания ре
акций. Реакция оксихлорирования этилена [596] имеет первый порядок но
кислороду и небольшие отрицательные порядки по этилену и хлороводороду.
II *
I I
| ,11,1 l,/< u< I 0,1
Сдхэ - 0,427 мол/м'
-I 11 500 550
T,K
I'iii I I Количес i no ДХ », полученного па моль меди, нанесенной на катализа гор
Рис. 4.24. Зависимость скорости реакции от концентрации этилена
I oi лас по Ф. Вульфу с соавт. [597], реакция имеет первый порядок по этиле
н ) и присутствии хлоридов меди и калия с добавками серебра.
111 вышеизложенного следует важная и определяющая роль реакции взаимо
ц ч uulu п илена с хлоридом меди. Кинетика этой реакции была детально исследи
ны III Уачи и Й. Асаи [598]. Для катализатора, содержащего 5 мае. % (в пересчете
н. медь) СиСЬ на у-А12О|, показано, что в реакции хлорирования этилена эффск
hihiiii участвует не более 64% общего количества меди (рис. 4.23). Исходя из этого,
опоры делают вывод о том, что течение реакции определяется, с одной стороны,
। hoi ношением Си I и Си II, а с другой — полагают, что часть меди геометрически
и цо! । упна для этилена. Авторы [598] указывают, что скорость реакции
( 41(1- 2СиС12 —► С2Н4С12 + Си2С12 (4.60)
подчиняется уравнению первого порядка по хлориду меди. Первый порядок
но и плену имеет место лишь при низких концентрациях этилена (рис. 4.21).
I* ном случае скорость реакции описывается уравнением Лэнгмюра-Хин
ни <п.пуда:
г = К, К, (<’,<’г/(1 гК„С,), (4.01)
когда реакция протек юг между хлоридом меди (11) и молекулами .пилена, ад
сорбированными на катализаторе. С учетом того, что скорость диффузии
лена существенно превышает скорость реакции, константа скорости реакции
хлорирования этилена описывается уравнением:
КГг = 269 exp[-37,8/(RT)]. (4.62)
Константа адсорбции Ка = 0,63 и не зависит от температуры.
Некоторое снижение скорости образования дихлорэтана при увеличении
концентрации меди (рис. 4.25) авторы [598] объясняют либо блокировкой мик-
ропор, либо многослойным расположением меди. Второе предположение, по
видимому, более соответствует реальным процессам, протекающим на поверх
ности катализатора.
В работе [594] указано, что реакция хлорирования этилена хлоридом меди,
нанесенным на оксид алюминия, подчиняется уравнению первого порядка по
этилену. Энергия активации составляет 79-84 кДж/моль; при этом хлорид меди,
избыточный относительно взаимодействия с носителем, практически не прини-
мает участия в реакции. Этот вывод подтвержден в [595] результатами экспе-
риментов по взаимодействию этилена с хлоридом меди в отсутствие носителя,
согласно которым реакция начинает идти с заметной скоростью при t > 300 °C.
Авторы работ [579,595] указывают, что в рабочем интервале температур про
цесса оксихлорирования этилена (210-235 °C) СиС12, так же, как и Cu2(OH)tCl,
составляющая значительную часть медьсодержащей структуры в катализаторах,
не является реакционноспособной фазой. Каталитически активные комплексы
образуются лишь после разрушения Си2(ОН)3С1 под действием хлороводорода
и последующего взаимодействия образующегося дихлорида меди с носителем.
При этом образуется координационная связь Си—О со смещением электрон
ного заряда в сторону меди. За счет уменьшения положительного заряда на Си2*
ослабляется энергия связи Си—С1 и облегчается реакция атомов хлора с эти
Рис. 4.25. Зависимость скорости образования ДХЭ от концентрации меди при t = 200 °C,
Сс,н. = 2,34 моль/м3
^2* *4
ином, чк>. и целом, повышает реакционную спосоОпос п>. ( хемаiпчески но
п Kibp.i/K.ie к я i ак
(I Cl Cl Cl
C1C2H,C1. Cl
Cu Cu
t f (la) f I (lb)
() Al—O—Al—+ C2H4 —-Al—O—Al—O—Al--
II III
Cl Cl
I I
( u Cu
t f (1C)
\l ()—Al—o—Al— + У2О2—-
I I I
t f (lb)
—Al—O—Al—O—Al—
(2)
t f (2a)
Al—()—Al—0—Al— + 2HC1 —
f t (la)
— Al—O—Al—O—Al— +H2O
I I I
< СГМЛ 1.1
i учетом того, что Cu2Cl2 (как на носителе, так и без него) не обладает ка>а
иноческой активностью при взаимодействии с этиленом, можно полагать, что
I акция оксихлорирования протекает только в том случае, когда на активных
и трах катализатора содержится два или более атомов хлора [579]. Роль кис
'юрода и приведенной схеме сводится не только к окислению Си+ в Си“*, но он
। ж ке способствует миграции ионов хлора с образованием СиС12 (реакция (2) и
приведенной схеме).
Аюмпое соотношение С1/Си, находящееся в ряде катализаторов в пределах
и. I1’ 0,50, свидетельствует о преимущественном содержании в данном катали
ьпоре соединений типа Си2(ОН)3С1; после воздействия НС1 соотношение С1/( н
приближается к 2,0. Это согласуется с предложенным Ю.М. Бакши и Е.И. Гель
псрипым |58CS| механизмом действия хлористого водорода, в котором перпич
ным актом является адсорбция IICI па катали заторе, увеличивающая cieiicnii
диспергирования солевой фазы. »
Таким образом, эксплуатационные характеристики катализаторов в апачи
тельной степени зависят от характера поверхностных медьсодержащих соеди
нений, что, в свою очередь, зависит от используемого носителя и способа папе
сения активной фазы.
Подробные исследования закономерностей адсорбции реагентов на у-А1,(),
и CuCl (СиС12)/у-А12О3, проведенные П. Холлом [599], показали, что хотя коп
станта равновесия адсорбции этилена не зависит от температуры, активное гь
адсорбционного центра повышается с ростом температуры. Скорость реакции
может лимитироваться миграцией хлора или этилена, поскольку хлорид меди 11,
нанесенный на оксид алюминия, не может быть диспергирован в виде моносло
ев, а присутствует в виде твердых блоков, состоящих из кристаллов.
Теплота адсорбции этилена на СиС12 составляет 65 кДж/моль, что свидетель
ствует о хемосорбции. На CuCl этилен лишь физически адсорбируется с ген
лотой 13 кДж/моль. Хотя у А12О3 не является инертным носителем в процессе
оксихлорирования этилена, адсорбция этилена на нем практически отсутстпу
ет. Присутствие же хлороводорода в системе существенно снижает адсорбцию
этилена на катализаторе. Й. Кси с соавторами [6001 предложили следующую
схему взаимодействия хлорида меди с оксидом алюминия:
С1
I
О—Си2+—О
I
С1
А1 — О — А1
При этом хлороводород координируется с ионом Си24 и ингибирует образо
вание комплекса с этиленом. Указывается, что активность катализатора снижа
ется с увеличением содержания НС1 и не зависит от содержания кислорода.
Иное строение активных центров предложено в [601, 602], где указывается,
что у-А12О3 уже при низких заполнениях СиС12 не является инертной струк
турой и изменяет состояние ионов меди с образованием комплексов типа:
А12О3—О—Си—С1 или CuAO,;(OH);,.„Clz.m.
Эксплуатационные и технологические параметры
процессов оксихлорирования этилена
Все процессы оксихлорирования этилена могут быть разделены по двум oi
новным признакам: 1) способу отвода тепла реакции вследствие ее высокой эк
зотермичности, т.е. использованию псевдоожиженного или неподвижного слоя
катализатора; 2) использованию кислорода воздуха или чистого кислорода для
регенерации катализатора оксихлорирования. В табл. 4.9 указаны источники
кислорода и и способы реализации процессов оксихлорирования этилена [199|,
осуществляемых рядом фирм.
IV, Hi lO'iiniKki к шло рода и cn<icol>i.i pe.uin 1.щии процеион oki nxaopiipoii.i
1Н1Ч llll'lril.l
Фирма Источник кислорода Слои К.11.1ЛИ l.liop.l
1 'nW < III lllklllv (( 111 A) Воздух Исподни aiiii.iu
1 ilnl < nip » (( IIIA) То же То же
< Uy V Inylx» (( IIIA) Воздух, кислород
lii <il» (Я i и и i и я) Кислород _ н_
Muii-.iiiiii>.. (( IIIA) Воздух, кислород
IT!... (( IIIA) Кислород
i «illi I» (( IIIA) Воздух, кислород Неподвижный
1' 1 1 >S» (1 lipoila) Кислород Псевдоожижен и bi и
' iiuiolli» (k рмания) То же Тоже
|l качес rue катализатора используют CuCb на разных носителях, в основном
л им пде алюминия.
И промышленных условиях конверсия НС1 практически полная, а селек
пинии и. образования дихлорэтана достаточно высока при использовании ка»
in । пд<1<» киженного, так и неподвижного слоя катализатора. Выход ди хлор пап.
по и пл< пу составляет 95-98%, а по НС1 — 98-99%. Среди побочных продукт!
in шин всего 1,1,2-трихлорэтана, тетрахлорида углерода, хлороформа, хлораля
। । ih кг оксидов углерода.
Нис кольну реакция оксихлорирования весьма экзотермична (238 кДж)
• io п.), hi реакторов необходимо отводить большое количество тепла. В реак
iiipax i псевдоожиженным слоем катализатора тепло отводят с помощью ох
• । । щинцпх змеевиков, погруженных в слой [609]. Температуру реакции под
( pi.iiB.noi равной 210 250 °C. Так как псевдоожиженный слой катализатор.
.....(чески изотермичен, то условия реакции одинаковы по всему слою Он
inMani.ii.ui температура реакции достигается благодаря конструкции и режим)
1'Ннны ciicieMbi отвода тепла, поэтому активность катализатора мало влияем
h i поддержание температуры реакции в системе. Выбор катализатора основан
। ынпым образом, на его стойкости к истиранию, способности к ожижению ело*
к 1гк । нанести. Катализаторы, работающие в псевдоожиженном слое, должны
<•41 и. прочную структуру, чтобы исключить возможность образования мелки»
Ki । иц 11еобходимо также избегать слипания частиц катализатора, которое мо
। 1 нарушить режим псевдоожиженного слоя или даже полностью исключи и
. hi niip.i кшание.
В мпература катализатора в реакторах с неподвижным слоем непрерып
пи п 'меняется по их длине. Трубки с катализатором диаметром около 25 мм
в pr.iKiiipe типа кожухотрубного теплообменника окружены охлаждающе!
|р|'Цоп, обычно кипящей водой. При подаче в трубку реактора предвари
и hi.ио нагретых газов тепловыделение пропорционально скорости реакции
I । ли па входе в реактор катализатор слишком активен, то тепло выделяется
inn ipce, чем отводится через стенки реактора. Температура слоя будет по
266 Г
пытаться до iex пор, пока скорость отвода тепла через стенки не сравняек я
со скоростью тепловыделения. Профиль температур по длине трубки будет
иметь максимум в определенном месте трубки. Поэтому для обеспечений
высокой селективности, длительного срока службы катализатора и низкого
перепада давления в реакторе, необходимо регулировать его температуру. 11<*
этой причине работа реактора с неподвижным слоем катализатора больше
зависит от его активности по сравнению с системами с псевдоожиженным
слоем. Во избежание местных перегревов катализатор разбавляют инертны
ми частицами или изменяют концентрацию активного компонента по длине
реактора 1199].
Оксихлорирование этилена в псевдоожиженном слое катализатора
Общая характеристика псевдоожижения [610]. Псевдоожиженным на ты
вают состояние двухфазной системы (твердые частицы — газ или жидкое i ь),
которое характеризуется перемещением одних твердых частиц относительно
других за счет обмена энергией с каким-либо ее источником. Под псевдоо кп
жением понимают превращение слоя зернистого материала в псевдогомот ен
ную систему под воздействием ожижающего агента (газа или капельной жид
кости), двухфазную псевдогомогенную систему называют псевдоожиженным
(или кипящим) слоем. Этому слою присущи многие свойства капельных жид
костей.
Псевдоожиженный слой образуется при восходящем движении ожижатоще
го агента через слой зернистого материала в момент, когда перепад давления и
слое достигает величины, достаточной для поддержания зернистого материала
во взвешенном состоянии.
Широкое внедрение метода псевдоожижения в промышленную практику
обусловлено:
- интенсивным перемешиванием твердой фазы, приводящим к практичес
кому выравниванию температур и концентраций в объеме псевдоожижен пою
слоя. Благодаря этому, в частности, устраняется опасность локального перс
грева (или переохлаждения) твердых частиц, препятствующего оптимальному
протеканию ряда тепловых, каталитических и других процессов;
- высокими значениями коэффициентов эффективной теплопроводности и
теплоотдачи от псевдоожиженного слоя к поверхностям теплообмена (или ты
оборот), соизмеримыми с соответствующими значениями коэффициентов для
капельных жидкостей. Эта важнейшая особенность псевдоожиженного слои
позволяет не только экономить поверхности теплообмена и рабочие объемы
аппаратов, но также осуществлять химические и другие процессы с высокой
тепловой нагрузкой при тонком температурном регулировании;
- возможностью использования твердых частиц малых размеров, т.е. гнер
дой фазы с развитой удельной поверхностью, для понижения диффузионных
торможений и повышения производительности аппаратов при осуществлении
ряда сорбционных, тепловых, каталитических и других процессов. Заметим,
что применению мелких твердых частиц в аппаратах с неподвижным слоем
твердой фазы часто препятствуют неравномерность температурного ноля и
поперечных и продольных сечениях слоя, высокое гидравлическое сопротпп
ление и малоинтенсивный теплообмен (низкие коэффициенты теплоотдачи)
кроме того, н отличие ог неподвижного слоя тердых чат шц, 1 д< iyMM.ipn.ui
повсрхнос । ь последних шачительно превышает активную поперхнос. н> фа ю
кого контакта, в псевдоожиженном слое величины этих поверхности i.imciho
। олп каются;
неподвижностью («текучестью») псевдоожиженного слоя, позволяющею
hi |д.н i> аппарат ы с непрерывным вводом свежей и отводом отработанной гнер
дон <|»а н>1, используя при этом выносные устройства для теплообмена и регулы
рования температуры;
небольшим гидравлическим сопротивлением и независимостью его вели
чипы ог скорости ожижающего агента (газа, жидкости) в пределах сущее гвова
пни псевдоожиженного слоя;
широким диапазоном свойств применяемых твердых частиц и ожижаю
щнх агентов (газов, паров и капельных жидкостей), включая возможность пи
ыппя аппаратов с псевдоожиженным слоем пастообразными материалами и
। yi пепзиями;
- сравнительно простым устройством аппаратов с псевдоожиженным ело
। м. легкостью их механизации и автоматизации.
Методу псевдоожиженния свойственны также некоторые недостатки, па
нболее существенные из которых:
- невозможность противотока фаз в пределах отдельного псевдоожижении
in слоя вследствие интенсивного перемешивания и, следовательно, невозмож-
ное п> осуществления процессов химического превращения, тепло- и массооб
мена при максимальной движущей силе. Интенсивное перемешивание частиц
in епдоожиженного слоя тормозит течение химических реакций в результате
понижения рабочих концентраций реагентов и, следовательно, движущей силы
процесса. При этом влияние перемешивания проявляется тем сильнее, чем
выше порядок химической реакции и больше требуемая глубина превращения.
Процессы в отдельном псевдоожиженном слое часто характеризуются более
пн 1кой степенью превращения и большим необходимым объемом катализат
ра, чем процессы в неподвижном слое катализатора. Сказанное относится как
к простым необратимым, так и к обратимым и сложным процессам. В случае
< ложных процессов, если целевыми являются продукты неполного превраще
пнн реагентов, перемешивание понижает селективность из-за образования не
желательных конечных продуктов. Однако если конечные продукты являются
целевыми, то перемешивание частиц в псевдоожиженном слое играет положи
ильную роль;
- неравномерность времени пребывания в псевдоожиженном слое частиц
нк-рдой фазы и ожижающего агента. Одинаково возможны быстрый проскок
ч.ц 1иц и их пребывание в слое дольше среднестатистического времени;
- возможность в ряде случаев нежелательного изменения свойств твердых
4.UI иц (истирание, растрескивание, науглероживание, слипание, спекание
н ги.). Замечено, что порошкообразные материалы начинают агломерирован,
। и и псевдоожиженном слое при температурах ниже их точки плавления или
ра пчягчения;
необходимость установки мощных пылеулавливающих аппаратов па вы
ходе газов из псевдоожиженного слоя, особенно при широком гранулометри
чы ком составе твердой фазы;
>ponni аппаратуры и лоне псеидоо кпжеппого слоя, особенно лничитель
пая и случае использования частиц с высокими абразивными свойствами;
ограниченность рабочих скоростей ожижающего агента пределами, co<;i
вегсгвующими началу псевдоожижения твердой фазы и ее уносу из слоя;
- возникновение значительных зарядов статического электричества при
псевдоожижении частиц диэлектрических материалов (взрывоопасность сис
темы).
Перечисленные недостатки метода псевдоожижения не являются, как нра
вило, определяющими, а некоторые из них могут быть частично или полност ыо
устранены. Достоинства метода псевдоожижения, безусловно, превалирую!
над его недостатками.
Широкое использование метода псевдоожижения в промышленных про
цессах оксихлорирования этилена подтверждает этот тезис.
Основные принципы и понятия псевдоожижения подробно изложены, на
пример, в монографии [700]. Укажем лишь, что скорость газа (жидкости), при
которой неподвижный слой зернистого материала переходит в псевдоожижен
ное состояние, называется скоростью начала псевдоожижения (или первой
критической). Скорость газа, при которой твердые частицы выносятся из слоя,
называется скоростью уноса (или второй критической). Таким образом, дна
пазон псевдоожижения ограничен первой и второй критическими скоростями.
При относительной объемной концентрации твердой фазы в псевдоожиженном
слое не ниже ~0,3 говорят о псевдоожижении в плотной фазе, а при концентра
ции ниже 0,3 — о псевдоожижении в разбавленной фазе.
Зернистый материал для псевдоожижения может быть загружен в аппарат
единовременно (периодическое псевдоожижение) либо непрерывно вводиться
и выводиться из аппарата (непрерывное псевдоожижение).
Оксихлорирование этилена в псевдоожиженном слое катализатора с ис
пользованием катализаторов типа СиС12/А12О3 ведут при 210-250 °C, причем
оптимальный интервал температур индивидуален для каждого катализатора.
В связи с необходимостью максимально полного связывания хлороводоро
да процесс оксихлорирования этилена проводят обычно с мольным избытком
этилена и кислорода по отношению к хлороводороду. Избыток этилена варьи
руют, как правило, в интервале 2-10%, а кислорода — 2-15%. Естественно, что
снижение избытка этилена и кислорода приводит к повышению конверсии этих
реагентов и улучшает технико-экономические показатели. Селективность обра-
зования дихлорэтана при этом также увеличивается за счет снижения выхода
побочных продуктов глубокого окисления, образующихся по суммарным реак
циям:
С2Н4 + 2О2 —- 2СО + 2Н2О; (4.63а)
С2Н4 + ЗО2 —- 2СО2 + 2Н2О. (4.636)
Основные параметры и показатели процесса являются функцией как физи
ко-механических характеристик катализатора: удельной поверхности, объема
пор, гранулометрического состава, так и характера поверхностных соединений,
образующихся при взаимодействии активной фазы с носителем с учетом влия
ния промотирующих добавок.
В работе |595| исследованы laiiiiciiMoc 111 пока laieneii процесса окспхлорп
ропапия пилена oi физики механических харакгерисчик каыли i.nopoB, раз
11>иц||хс>1 точениями удельной поверхности, объема пор, а также распредели
пнем частиц по ра (мерам. Содержание меди в Kai алита торах составляло 1,5
18 мае. %.
Физико-механические характеристики носителей (у-А12О3 — числитель)
и катализаторов (CuCl2/y-AL2O3— знаменатель) оксихлорирования пилена
А Б В
< одержание меди, мае. % . . -/4,75 -/4,71 -/4,55
(дслыы>1 поверхность, м2/г .. 280/225 150/130 87/75
I хч.ем пор, см3/г . .0,42/0,34 0,34/0,30 0,30/0,21
11иту/к1меп1рический состав, мае. %:
50 мкм . 30/31 28/30 15/17
>0 мкм ... 48/50 60/64 36/35
80 мкм . .. 71/72 92/94 71/70
11а рис. 4.26-4.28 представлены зависимости конверсии хлористого подо
рода, выхода продуктов глубокого окисления и эффективности использования
о плена от температуры и соотношения исходных реагентов: Cl/C = HC1/2C J 11
(мол), ()2/С = О2/С2Н4 (мол.), указывающие, что при прочих близких значениях
фн шко-механических характеристик катализаторов разность в величинах обь
। ма пор и удельной поверхности существенно влияют на показатели процесса.
В первую очередь это относится к выходу продуктов глубокого окисления ('Ох
1’в< 1.2(>. Зависимость конверсии НС1 и эффективности использования этилена or со
.апоив min Cl/C; t = 220 °C, т = 8,9 с, О2/С = 0,70
270
!
Рис. 4.27. Зависимость выхода продуктов глубокого окисления СОХ от мольного cool
ношения Cl/C; t = 220 °C, т = 8,9 с, О2/С = 0,70
Рис. 4.28. Зависимость конверсии хлороводорода и выхода продуктов СОЛ от темпера
туры; С1/С = 0,95; т = 8,9 -9,3 с, О2/С = 0,70
СО + СО2. Так, катализатор № 1 с высоким значением удельной поверхности
достаточно активен в реакции оксихлорирования — конверсия хлороводорода
достигает 99,6%. Это, однако, сочетается с низкой селективностью образования
дихлорэтана за счет увеличения выхода продуктов СОХ.
Катализатор № 3, имеющий относительно низкую величину удельного обь
ема пор, менее активен в основной реакции оксихлорирования и одновремен
но менее селективен за счет повышенного образования продуктов СОх и от
носительно низкого содержания дихлорэтана в хлорорганических продуктах
(табл. 4.10)
Таким образом, низкая величина объема пор катализатора (катализатор
№ 3) ведет при постоянном содержании меди на уровне 4,5 мае. %, к снижению
как активности, так и селективности процесса, что выражается в более низкой
конверсии хлороводорода, увеличении выхода побочных продуктов, прежде
всего 1,1,2-трихлорэтана и оксидов углерода. Причиной этого может являться
1>|0||ИЦ.| 1.10. ( ОС ЫН ХЛОрор) .ШПЧСиКПХ продумон, MO/ll.ll. %, ОКе.HXlUipilpOD.IIItill >1 НДС
и.। и ирису re । пип кашли uiopou (I = 220 "C, Cl/C = 0,95,O2/C 0,70, i 0,2 e)
Катализа юр 1,2-ДХЭ (дихлор этан) 1,1,2-ТХЭ Хло- ро- форм 11ерхлор- .ТГИ11СН 1,1 Дихлор- этан Хло раль icipa хлорид углерода
№ 1 98,55 0,38 0,14 0,08 0,05 0,20 0.1 1
№2 98,84 0,31 0,15 0,11 0,06 0,18 0,15
№3 98,20 0,47 0,15 0,12 0,06 0,24 0,16
u i(»i,i точное содержание хлорида меди на поверхности зерна; при этом Си<12,
пс взаимодействующий с носителем, катализирует реакции как глубокого окис-
ления этилена и дихлорэтана, так и дегидрохлорирования дихлорэтана в винил
хлорид с последующим образованием 1,1,2-трихлорэтана. Определенную роль
может играть также унос меди с поверхности катализатора. Подробно э ги явле
пил будут рассмотрены ниже.
Одним из важных критериев, определяющих качество псевдоожижен по
ю слоя и, в конечном итоге, показателей процесса, является размер частиц
катализатора. Как показано в [610], однородность псевдоожиженного слоя
। вердых частиц повышается с уменьшением их размера. Однако ниже опреде
лепного его предела возрастают силы взаимодействия между частицами, чю
противодействует упорядоченному расширению слоя и способствует агломе
рации частиц и каналообразованию. На рис. 4.29 представлены зависимости
флуктуации падения давления и диаметра пузырей от вида используемого
катализатора, показывающие, что параметры псевдоожижения существенно
ухудшаются при переходе к катализатору с более крупными размерами час
|иц (катализаторы № 1 и № 3). Повышение температуры улучшает качество
псевдоожижения, хотя тенденция остается прежней. Эксперименты прово
дились в реакторе диаметром 42 мм, ожижающий агент — воздух, темпера
1 ура 20 и 180 °C.
Фиктивный коэффициент неоднородности Кн = d6() / d10,
где d60 и dI0 — диаметры отверстий, через которые проходят соответственно
60 и 10% частиц, для исследованных катализаторов равен:
для катализатора № 1 — 2,0;
для катализатора №2 — 3,2;
для катализатора № 3 — 2,2.
Как указывает М. Лева [611], смеси твердых частиц, характеризующиеся
шачениями Кн > 2,5, менее склонны к поршне- и каналообразованию. Этим
требованиям отвечает катализатор № 2.
Основные требования к катализатору оксихлорирования этилена в чаш и
его физико-механических характеристик озвучиваются большинством веду
щих производителей данного катализатора:
Удельная поверхность, м2/г............120-150
Удельный объем пор, см3/г............0,30-0,35
Насыпная плотность, г/см3............0,95-1,05
I ранулометрический состав, мае. %:
менее 20 мкм....................£5
менее 40 мкм....................30 50
менее 60 мкм....................75-95 ,
более 80 мкм....................5-15
Тем не менее, соответствие физико механических характеристик катализа
тора указанным параметрам является условием, недостаточным для обеспече
ния оптимальной работы катализатора. Большую роль в этом играет характс|
поверхностных соединений, образующихся как при приготовлении катали ы
тора, так и под влиянием реакционной среды в ходе его эксплуатации. Формп
Рис. 4.29. Зависимость флуктуации падения давления в системе (а) и диаметра типич
ного пузыря (б) от образца используемого катализатора
I пин медьхлорпдпых клади ыгоров окисли ienbiioro хлорирования пилена
л прицепе их пригогонлепия, протекающее при участии носителя но iii.iiimo
и и..... i солевой системой, определяет химический сослав и агрегатное со
। iii'Iiiik соленых <|>a i, распределение солевого компонента на поверхности [383,
|>1 '| *1|<>рмпро|1апие каталигической системы завершается в условиях реально
ы прицепа, причем одним из лимитирующих факторов является состав реак
I । в он к in среды.
Дополнительные ограничения на выбор оптимального катализатора пакля
I.H к । переход ог промышленного процесса оксихлорирования этилена с ис
мнынием воздуха к «кислородному», что также влечет изменение состава
р <||Ц1|о11пои среды.
< шла-по [613, 614], наилучшие эксплуатационные свойства в процессе
...... катализаторы псевдоожиженного слоя, содержащие минимальное
> и urn i ню меди на внешней поверхности зерна катализатора. Атомное сото
цини пне А1:( п на внешней поверхности зерна, равное 40-43, указывает, ч то до
T'i"»t меди, нанесенной на катализатор, находится внутри зерна. В работе [615|
н.п ihiiio, ч то в таких катализаторах солевой компонент находится в высоко
ни п< р. пом состоянии. Распределение медьсодержащих фаз, охарактери к»
юпики по распределению участков поверхности по теплотам адсорбции 11(1
11 пин иная доля поверхности в области высоких теплот адсорбции выше
1 । Д к/моль, что относится к адсорбции НС1 на сшитых медьсодержащих
фи|х), отличается для таких катализаторов увеличением доли поверхпос i и,
. ||п1и1Д111Н1 от солевых фаз, и значительной долей поверхности с участками, па
• 1>|<>рыч медь находится в решетке носителя в виде алюминатных структур.
Iiihnii >ффект достигается при использовании катализатора на основе 6-А12С)ъ
1< Оолыпая часть А13+ имеет тетраэдрическую координацию по кислороду, а
попы меди танимают, в основном, октаэдрические пустоты. Этот процесс эпер
I'm I 30. Зависимость конверсии НС1 и от соотношения С1/С. Катализатор CuCh/AI Ot.
I ’О' (;, т = 8,9 с, О2/С2Н4 = 0,70
274 I ---------
готически более выгоден по сравнению с замещением тот ра »дрнческих влип
сии. Часть солевого компонента проявляете»! в виде сшитых с поверхностью
неупорядоченных структур толщиной в несколько монослоев и слабо окщп.
таллизованных фаз [615].
Чакое строение катализатора позволяет проводить процесс оксихлорирова
ния этилена при минимальных (2-3%) избытках этилена и кислорода по опт
тению к хлористому водороду. На рис. 4.30 представлены зависимости конвер
сии хлороводорода и эффективности использования этилена от соотношения
С1/С для разных катализаторов. Заштрихованные участки на кривых указывай» i
на агломерацию катализатора в условиях процесса. Явление агломерации при
соотношении С1/С = 0,96ч 1,00 (мол) характерно для катализаторов, содержа
щих избыточные относительно взаимодействия с носителем медьсодержащие
фазы — CuCl2, Cu2(OH)3CI и другие на поверхности катализатора, имеющих
низкие значения соотношения А1:Си [616].
Окислительное хлорирование этилена схематично может быть представле
но в виде окислительно-восстановительной реакции, в которой активный ком
понент — медь находится в динамическом равновесии между одно- и двухва
лентным состоянием [617]:
С2Н4 х х 2СпС12 > < Н2О
С2Н4С12 * * Cu2Cl2 ' 2НС1 + 0,5О2
Для оптимальной работы катализатора необходимо соблюдение опреде
ленного баланса между Сии и Си+2; поддерживаемого как введением промоти
рующих добавок [618], так и изменением состояния реакционной среды. Реак
ционная среда вызывает перестройку катализаторов, затрагивая как солевой
компонент, так и кислотно-основные центры носителя, причем наблюдается
аморфизация солевого компонента [615, 619]. Тогда как в катализаторе с высо
ким значением А1:Си медь в основном стабилизирована в двухвалентном со
стоянии, при снижении этого соотношения до 10-15 фазы гидроксохлоридов
в условиях реакции переходят в дихлорид меди, который легче восстанавлп
вается до избыточного Си+. Кроме того, для таких катализаторов в режимах
с преимущественным содержанием хлороводорода также наблюдается разру
шение алюминатов одновалентной меди с образованием CuCl. Это приводит' к
нарушению баланса между Си 1 и Си+\ Избыточный CuCl образует нестехио
метрический комплекс с реакционной водой НСиС12- 0,5Н2О с низкой темпе
ратурой плавления [620]. На рис. 4.31 представлена дериватограмма указанно
го комплекса. Низкая температура плавления комплекса (145 °C) способствует
агломерации частиц катализатора и далее — нарушению гидродинамики псев
доожиженного слоя.
Аналогичная внешне картина наблюдается при подаче в систему хлороводо
рода в холостом режиме (в отсутствие этилена) в смеси с воздухом или инертны
или без нее. На рисунке 4.32 представлены зависимости перепада давления но
слою катализатора от парциального давления НС1 в газовой смеси. Темпера т у
ра — 453 К.
743
418
300 400 500 600 700 800 т к
Рис, 4.31. Дериватограмма комплекса НСиС12-0,5Н2О
I in 1.12. Зависимость перепада давлений в системе от парциального давлении хлоро
• (ород.|. t = 180 °C
111 швисимостей, представленных на рис. 4.32, видно, что поведение ка гал и
пироп с разным атомным соотношением А1;Си заметно разнится. Критичес
•ч пычениернсь при котором псевдоожиженный слой практически перестает
»»шее топать, находится в пределах 5-20 кПа.
( огласно рис. 4.32, катализаторы, в которых величина А1:Си = 10-И5, весьма
Иш ши гельны к увеличению концентрации хлороводорода: перепад давлении
по высоте слоя катализатора начинает заметно снижаться да,кг при нарцналь
ных давлениях 1К 21 на уровне 5-8 KI 1а. При увеличении атомного соотношения
А1:Си в катализаторе эффект агломерации частиц начинает нроявлятьсяЛ1ри
Phci >15з-20 кПа.
Как отмечалось выше, солевые комплексы, адсорбированные на парных
кислотно-основных центрах А12О3, взаимодействуют с НС1 с образованием
новых комплексов с высоким координационным числом меди [573, 621]. При
этом в присутствии НС1 идет реакция взаимодействия НС1 с гидроксохлори
дом меди:
HCI + Cu2(OH)Cl3 — Си2С14 + Н2О, (4.64)
что наиболее характерно для катализаторов, имеющих избыточное содержание
гидроксохлоридов указанного состава в своей структуре. Присутствие в систе-
ме реакционной воды и кислорода также приводит к образованию комплекса
НСиС12- 0,5Н2О, и последующей агломерации частиц катализатора. Причиной
агломерации катализатора может быть также образование полимерных струк-
тур хлорида меди (СиС12)п, однако сообщений об экспериментальном подтверж-
дении этого факта нами не обнаружено.
Восстановление нормального псевдоожижения может быть достигнуто пу-
тем обработки катализатора воздухом. В этих условиях происходит разрушение
комплекса с одновременным окислением меди до Си2+:
HCuC120,5H2O + !4О2 —- СиС12 + Н2О (4.65)
и в дальнейшем — восстановлением необходимого баланса Си2+—Си4.
Скорость восстановления псевдоожиженного слоя катализатора увеличива-
ется с повышением температуры, что подтверждается стабилизацией величины
перепада давления на первоначальном уровне через 0,5-3 часа в зависимости от
используемого катализатора. Количество кислорода в отходящих газах снижа-
ется симбатно повышению перепада давлений, и затем выходит на постоянный
уровень. Это также совпадает по времени со стабилизацией перепада давлений;
при этом повышение температуры сокращает время, в течение которого псев-
доожиженный слой возвращается в исходное состояние.
На основе данных по поглощению кислорода при обработке катализаторов
в работе [595] проведена оценка доли количества меди, связанной в комплекс
НСиС120,5Н2О. Для катализатора с величиной А1:Си = 13,5 эта величина со-
ответствует -36%, для катализатора с величиной А1:Си = 43,5-10,6%. Эти зна-
чения, вероятно, соответствуют содержанию избыточных относительно взаи-
модействия с носителем медьсодержащих фаз и, в конечном итоге, определяют
параметры процесса оксихлорирования этилена.
Для обеспечения нормальных гидродинамических условий при исполь-
зовании катализаторов с избыточными медьсодержащими фазами процесс
необходимо проводить при избытке этилена по отношению к НС1 не менее
7-10% мол., что ведет к снижению конверсии этилена, увеличению выхода
продуктов СОХ (рис. 4.33) и ухудшению технико-экономических показателей
процесса.
Показатели процесса оксихлорирования этилена являются функцией четы-
рех независимых параметров: температуры, соотношения С1/С и О2/С, а также
Рис. 4.33. Зависимость выхода продуктов СОх от С1/С
ир. »ii иного параметра X - VKaT/WHcl, где VKaT — объем псевдоожиженного слоя
• hi । in i.uopa (л); WHci— расход хлороводорода (моль/ч).
Варьирование именно этих параметров с достаточной степенью достовер
И in описывает скорость убыли хлороводорода в реакторах разной прои.шо
пн И1.ПОС1И, начиная с лабораторных и заканчивая промышленными маснпа
Памп 11о сути дела, временной параметр указывает на производительность ка
• in i.i । ора при условии, что реактор является системой идеального вытеснения
ни I а юному потоку.
Данные, представленные на рис. 4.34, в сочетании с зависимостями кон
hi р. ini IIC1 от температуры указывают на то, что для каждого катализатора
,।п<•« । нуе г свой интервал варьирования независимых параметров, внутри ко
lopnio показатели процесса остаются практически неизменными. Максимум
ни н'мпсратуре, через который проходит конверсия НС1, также индивидуален
। in каждого катализатора и может быть объяснен нехваткой кислорода при
>||ц|ч прованном соотношении О2/С, а также увеличением доли побочных ре
>п<ц|||| глубокого окисления дихлорэтана (вторичное выделение НС1 в реакци
••ши к» среду) или этилена (нехватка этилена для связывания НС1 по основной
р. 1кцни).
( оогпошение О2/С лишь слабо влияет на конверсию НС1. Ее некоторое спи
11 пне связано, очевидно, с увеличением доли реакций глубокого окисления
....пческого субстрата при повышении концентрации кислорода в реакци
niiiion смеси.
(качение временного параметра может являться основой для расчета про
1Ы1нлеппого реактора. Обычно производительность катализаторов оксихло
। пронация этилена, при которой конверсия НС1 превышает 99%, составляет
i.iii) 650 г/(л кат.-ч). Реальная производительность в промышленных условиях
in • колько ниже из-за ограничений по отводу тепла.
(акономерности образования побочных продуктов в процессе оксихло
риронания этилена
11обочные продукты в процессе оксихлорирования этилена могут быть ус
няню разделены на две группы:
Рис. 4.34. Зависимость конверсии НС1 от: а — временного параметра при разных тем
пературах, С1/С = 0,98 , О/С = 0,30; б— соотношения О/С при разных С1/С, (у кривых)
t = 225 °C, временном параметре (X) = 0,20
- побочные хлорорганические продукты, которые включают большую часть
хлорзамещенных углеводородов С|—С2 как парафинового, так и олефинового
ряда;
- продукты глубокого окисления — СО и СО2.
В связи с этим процесс может быть охарактеризован «селективностью по
хлору», учитывающей долю хлороводорода, пошедшую на образование побоч
ных хлорорганических продуктов, и «селективностью по этилену», учитываю-
щей, наряду с углеродной составляющей побочных хлорорганических продук
тов, также и этилен, пошедший (впрямую или опосредованно) на образование
оксидов углерода.
—
I'ui I IS. Зависимость скорости глубокого окисления этилена от парциальных даплс
ной пилена, кислорода и хлороводорода
Винду того, что в процессе оксихлорирования этилена на долю продуктов
। । *,( । • СО2 + СО) приходится до 70% этилена, превращающегося в побочные
продукты, исследование условий их образования является важным звеном в
у । ip.u нике промышленного процесса. В свою очередь, промышленное осущес i
.... процесса оксихлорирования этилена с концентрированным кислородом
I । 1чесгве окислителя предполагает использование рециклового газа, в кою
ром будет протекать накопление СО2 и СО в разных соотношениях. Изменение
। i.iua реакционной среды, по сравнению с «воздушным» процессом, может
и lion ь на катализатор и на показатели процесса в целом.
Кш ютические закономерности образования продуктов глубокого окисло ш я
в условиях процесса оксихлорирования этилена изучены в работе [623]. Выло
। шовлено, что скорость реакции глубокого окисления возрастает при уне
прении парциальных давлений кислорода и этилена и уменьшается с ростом
н >[ цпального давления хлороводорода (рис. 4.35). Скорость реакции окисле
инн п илена пропорциональна парциальному давлению кислорода во всем ин
и риале изменения этого параметра. Предполагается, что наиболее медленным
паном превращения этилена является его взаимодействие с молекулой кисло
рода, приводящее к деструкции олефина и последующему быстрому окислению
пора ювавшихся углеводородных осколков.
В рамках общей схемы превращений, предложенной в работе [588], авторы
|ь * 1| описывают скорость реакции окисления этилена уравнением:
гсо2 - ^п^рс2н4Ро2Рнс1/(1 + ^рна)>
(4.66а)
?яо
которое рассм.11 рипается как iсорегическое обоснование >мпирическ<ло уран
нения, предложенного К. Миаучи (589]:
гсо2 = ^Рс,н4 До, /(е + Pud) > (4.6бб)
где е — константа.
Оценка константы е показывает, что она близка к нулю (е = 0,02+0,04). Эю
указывает на высокое значение К — константы равновесия стадии взаимодейс
твия хлорида меди с хлороводородом: К = 5000+20000. Наблюдаемая энергии
активации окисления этилена равна 188 кДж/моль.
Согласно К. Миаучи [622], реакции оксихлорирования этилена и его окис -
ления протекают на одних и тех же активных центрах катализатора, а их конку
ренция объясняет вид вышеприведенного кинетического уравнения.
Близкие выводы были сделаны Дж. Борном с соавторами [624], которые иг
следовали условия одновременного оксихлорирования и окисления этилена н
присутствии Си, СиО и СиС12, нанесенных на силикагель. Процесс изучали и
псевдоожиженном слое катализатора в интервале температур 250-500 °C. При
температуре ниже 350 °C практически единственным продуктом в процессе ян
ляется дихлорэтан. Дальнейшее увеличение температуры ведет к резкому росту
выхода СО (до 10%) и в меньшей степени СО2 (2-2,5%). Растет также выход
винилхлорида и хлорметанов. Авторы предлагают обобщенную схему превра
щений этилена:
СО/СО2
(4.67)
С]С1Х/С2С1Х
Уравнение, описывающее скорость превращения этилена, выглядит следу-
ющим образом:
Г,,2,3 = *1.2,з[С2Н4] [O2]fl[HCl]b. (4.68)
Аррениусовские параметры.
а) суммарно Ig^/M'04 b)c ’г1) - 11,0 (28 ± 1,4)/(RT), (й = 0,64; b = 0,52);
б) окисление 1§(&2/М(я +fc)c“1r”1) = 16,9 • (48 ± 2)/(RT), (« = 0,90; b = 0,49);
в) оксихлорирование lg(fc3/M(,! + fc)c ’г1) = 7,96 • (20 ± 0,3)/(R7’), (а = 0,62; b =
0,57).
Авторы [623] обнаружили также первый порядок реакции по меди, что вы-
зывает сомнения, так в этом случае не учитывается характер распределения
меди на поверхности катализатора и априори принимается катализ солевой
фазой СиС12. Делается также важный вывод, что независимо от того, какое со
единение меди изначально используется для приготовления катализатора —
Си, СиО или СиС12, дихлорид меди является конечным продуктом их превра
щений в ходе процесса.
Иной точки зрения о маршрутах образования оксидов углерода придер-
живаются авторы работы [625], в которой получены следующие зависимости
1*ис. 4.36. Зависимость скорости образования оксидов углерода от парциального данле
moi НС1 (а), О2 (6), 1,2-ДХЭ (в)
скорое 1 и образования оксидов углерода (( Оч) oi парциальных давлении хлоро
водорода, кислорода и 1,2-дихлор мана (рис. 4.36). Согласно ним данным реак
ция глубокого окисления 1,2 дихлорэтана тормозится хлороводородом и имеем
переменные (от 1 до 0) порядки по кислороду и 1,2 дихлорэтану.
При этом установлено [625], что этилен тормозит окисление 1,2 дихлор на
на во всем диапазоне варьирования парциального давления хлористого водоро
да. В качестве наиболее вероятной предложена следующая схема образовании
продуктов глубокого окисления:
l. Z, + НС1 —ZpHCl;
2. Z( + О2 + С2Н4С12 Z, • С2Н4С12 • О2;
3. Z, • С2Н4С12 • О2- Z,HCI С2Н2О2 + НС1;
4. Z,HC1 С2Н2О2 —► Z!C2H2O2 + НС1;
быстро
5. Z|C2H2O2 + О2 ► Z| + Н2О + СО + СО2;
6. Z] + С2Н4 — Z[ • С2Н4;
С2Н4С12 + 2О2 = СО + СО2 + 2 НО + Н2О. (4.69)
Приведенной схеме отвечает уравнение скорости образования оксидов yi
лерода, полученное при условии пренебрежения стадией 6.
_ 2K2K3fc4popC2HC12
rCOtCO, “ / \ >
Рна Р + Ро2 Рс,Н4С12 ) + ^2^зДо, Рс2Н4С12
где К, и /с, — соответствующие константы равновесия и скорости.
Предложены следующие параметры уравнения (4.70):
In К2 ° = 14,0;
AQ2 = 42,6 кДж/моль;
In К3 °-7,79;
AQ3, = -24,7 кДж/моль;
In К4° = 33,2;
£4 = 173,0 кДж/моль.
Авторы работы [626] указывают, однако, на то, что при проведении процее
са в условиях, приближенных к реально осуществляемым в промышленное 1 и
увеличение избытка этилена и кислорода ведет к повышению выхода продук 1ов
СОХ (рис. 4.37а и 6).
В табл. 4.11 представлены экспериментальные данные по окислению этиле
на и дихлорэтана кислородом воздуха на чистом носителе — микросферичес
кой у-А12О3. Интервал температур 210-320 °C, время контакта 8,2-9,3 с.
Данные табл. 4.11 указывают, что глубокое окисление этилена и дихлор
этана в присутствии микросферического оксида алюминия начинает идти i
заметной скоростью при t > 280 °C. Из этого следует, что в рабочем ингер
Рис. 4.37. Зависимость выхода продуктов СОх от соотношений С1/С (а), О2/С (6)
пл ie температур процесса оксихлорирования этилена участки катализатора,
i вободные от солевого компонента, не являются активными центрами окис
лепил.
Сопоставление данных, представленных на рис. 4.37 и в табл. 4.11, указы
ii.u i на то, что выход продуктов глубокого окисления СОХ зависит от содер
клиня в системе и этилена и кислорода, а также на протекание окисления на
медьсодержащих активных центрах катализатора [595]. Увеличение селем ив
нос 1 и окисления с ростом избытка этилена и кислорода по отношению к 11( I,
по видимому, вызвано изменением состояния меди в составе солевого компо
tn и га. Восстановленный этиленом медьсодержащий центр при взаимодейез вин
. адсорбированным кислородом становится активным центром окисления как
адсорбированного дихлорэтана, так и этилена, поступающего из газовой <|>а па
И случае снижения конверсии хлороводорода (например, в условиях пронеде
пил процесса при незначительном избытке этилена) становится возможной де
i.iKтивация хлороводородом центра окисления, что влечет уменьшение выхода
продуктов СОХ [615].
Сказанное позволяет пренебречь влиянием активных центров носи геля на
процесс глубокого окисления по сравнению с вкладом в эту реакцию медьсо
держащих центров солевого компонента.
С этой точки зрения более выгодным является использование кагализаго
ров с высокими значениями атомного соотношения А1:Си. Более низкий выход
продуктов СОХ на таких катализаторах определяется, как указано выше, сraini
ппацией меди в алюминатных структурах и увеличением доли поверхности,
284 I
и '
Таблица 4.11. Данные по окислению пилена и 1,2 ДХ.) кислородом воздуха па микро
сферическом у — АI .О,
t,°C Концентрация 1 • К)3, моль/л ( гепеХь окисления в С()р%
Орг. комп. 2 о2 n2 СОХ
210 10,8/10,8 Эп 7,1/7,0 илен 26А/26А -/0,1 0
240 10,7/10,7 7,3/7,3 26,3/26,3 -/0,1 0
280 10,8/10,4 7,1/6,7 26,4/26,4 -/0,8 3,7
320 10,8/9,8 7,1/5,7 26,4/26,3 -/1,9 9,20
210 4,8/4,8 1,2-Дш 8,4/8,4 слорэтан 31,1/31,1 -/<0,1 0
240 4,8/4,8 8,4/8,3 31,1/31,0 -/<0,1 0
280 4,8/4,0 8,4/6,7 31,1/31,0 -/1,2 16,7
320 4,8/3,1 8,4/5,0 31,1/31,0 -/2,5 35,4
1 Числитель — на входе в реактор, знаменатель — на выходе.
2 Органических компонентов.
с корое n il реакции в мимодеис гвия 1IC1 с окисленной формой меди | реакция
( |)| схемы и реакции окисления органическою cy6cip.ua выражается пример
ио той же величиной
При высокой активности катализатора в целевой реакции оксихлорирова
ния .пилена (конверсия HCI > 99-99,5%) вуравнении (4.71а) величина А.2Сц( । I.
Уравнение (4.71а) в этом случае приобретает вид:
(1.710)
В условиях процесса порядок по кислороду близок к 0,5. Тогда уравнение
(1716) преобразуется к виду:
г - кС С0,5
гсо„ -К%О2 •
(4.71в)
В случае низкой активности катализатора (конверсия НС1 < 97%) АэСщ I ~1
и сависимость приобретает более сложный вид. Из этого следует, что более вы
с окая активность катализатора в процессе оксихлорирования этилена сопро
вождается, как правило, снижением селективности образования дихлорэтана
та счет увеличения выхода продуктов глубокого окисления.
Аррениусовские параметры реакций окисления этилена и дихлорэтана,
л” ’/(моль0,5 ткат-ч) в интервале 215-235 °C [595]:
свободной от солевых компонентов, что приводит к снижению количества ак
тивных центров окисления.
Окисление органического субстрата протекает в значительной степени
на активных центрах, содержащих избыточные медьсодержащие фазы ины
СиС12 и Си,(ОН)3С1 [595].
Принимая оксидные формы меди (СиО, СиОСиС12) в качестве актив
ных центров реакций глубокого окисления этилена и дихлорэтана, можно
полагать, что в схеме 4.1 (с. 263) реокисленная форма меди, образующаяся
по реакции (2), координационно связанная с носителем, окисляет оргапи
ческий субстрат. Реакция окисления конкурирует в этом случае с реакцией
(3) схемы 4.1, что и объясняет тормозящую роль хлороводорода в реакциях
окисления.
Поскольку, согласно этой схеме, реакция протекает с участием адсорбиро
ванного кислорода, это объясняет нелинейную зависимость между скоростью
реакций окисления как этилена, так и дихлорэтана, и концентрацией кисло
рода в системе.
Уравнение скорости реакций окисления этилена и дихлорэтана можеч
быть в общем виде записано следующим образом:
гсок
“ ^С1СО2 / Ц + ^1СО2 + ^2СНС1) ’
(4.71а)
где с, — концентрация этилена или дихлорэтана.
С учетом того, что скорость реакции оксихлорирования этилена на 1,5-2 по
рядка превышает скорость реакций окисления, можно полагать, что отношение
kRX3 = 5,6 109exp[-105000/(RT)], (4.72а)
^с2щ = 6,3 • 106ехр[-80 500/(RT)] (4.726)
Полученные зависимости указывают на то, что при стандартных темпе
р.иурах процесса оксихлорирования этилена отношение констант скорое in
окисления 1,2-дихлорэтана и этилена составляет ~2:1. Проведение процесса
при 230-255 °C способствует увеличению вклада составляющей окисления 1,2-
дпхлорэтана в образование оксидов углерода.
Полученные данные объясняют также эффект снижения селективное in
образования дихлорэтана за счет роста выхода продуктов СОХ с увеличением
н мпературы: энергия активации реакцией окисления выше энергии активации
реакций оксихлорирования на 17-30 кДж/моль в зависимости от используемо
। о катализатора.
В рамках данного подхода и в соответствии со схемой 4.1 комплекс (1в)
претерпевает дальнейшие превращения по двум конкурентным направлепи
мм Первое из них — основная реакция (1 с) с образованием монохлорида
меди, координированного с носителем. Второе протекает по схеме 4.11 с об
ратованием комплекса (1с) и СО2, с немедленной десорбцией последнего с
поверхности катализатора. Монооксид углерода, образующийся в ходе про
цесса, частично реадсорбируется с образованием комплекса Си —С=О и
последующим превращением его в СО2. Приведенная последовательное и>
превращений соответствует схеме окисления карбонильных комплексов
I и* — СО, указанной в работах [627, 628] и рассмотренной также О.В. Кры
новым и В.А. Матышаком [629]. Исходя из реального соотношения дихлор
Cl iClCJhCl Cl
Cu Cu
t f
— Al—O—Al—O —Al —
2O2
2HC1
-H2O
O6 (/'
III , III
Cl с O" C ( 1
\ I / \ I /
СдЛ Cu
—Al—O —Al—O —Al----------
O6+ O6+
III 7 III
Cl c—-o2 C Cl
\ I / \ I /
Cu Cu
------Al—O—Al—O—Al —
—Al—O—Al—O—Al---Al—O—Al—O—Al—+ CO2
III III
Схема 4.II
димому, реакция взаимодействия этилена с фрл мен ими окисленной формы
меди. Скорое п> пой реакции и 2-3 раза ниже но сравнению с реакцией окисле
ния адсорбированного 1,2-дихлорэтана.
11олученпые результаты указывают на го, что проведение процесса оксихло
рирования этилена при стехиометрических соотношениях НС1:С2Н4:О2 ведет к
снижению выхода продуктов СОХ на 20-30%, по сравнению с режимами, пред
полагающими использование 7-10%-ного избытка этилена и 10-15%-пого из
!>ы гка кислорода.
Важной составляющей процесса оксихлорирования этилена является об
ратование побочных хлорорганических продуктов. В зависимости от исполь
туемого катализатора и условий проведения процесса мольное содержание
дихлорэтана в смеси хлорорганических продуктов составляет 98,5-99,6%. Для
производительности реактора по дихлорэтану 120 тыс. т/год снижение ею со
держания в продуктах реакции на 0,1 мол. %, ведет к увеличению количества
хлорорганических отходов на 120-150 т/год, а расходный коэффициент по эти
лену увеличивается на 2-3 кг/т винилхлорида.
В процессе оксихлорирования этилена обычно образуются следующие
хлорированные углеводороды парафинового и олефинового рядов Q—С2:
1,1,2-трихлорэтан, трихлорацетальдегид (хлораль), хлороформ, тетрахло
рнд углерода, этилхлорид, винилхлорид, 1,2-дихлорэтилены, 1,1-дихлорэтап,
1,1,2,2-тетрахлорэтан и некоторые другие. В наиболее значительном количес i ве
образуются первые четыре соединения настоящего ряда, при этом особенно не
желательным является образование хлораля в связи со сложностями его даль
пейшего выделения и необходимости, вследствие этого, создания специального
у ыа для его разрушения раствором щелочи.
1а исключением этилхлорида все побочные хлорорганические продукты
образуются в результате вторичных превращении 1,2 дихлорэтана, при этом
i одержание 1,1,2-трихлорэтана в продуктах реакции выше по сравнению с дру
i ими хлорорганическими примесями. Характерная зависимость выхода: 1,1,2-
1рихлорэтана от температуры в процессе оксихлорирования этилена представ
лена на рис. 4.38.
этан/оксиды углерода в продуктах реакции, можно полагать, что скорот и.
десорбции дихлорэтана с поверхности катализатора в 30-40 раз превышает
скорость его окисления.
Окисление этилена, избыточного по отношению к хлороводороду, в процсч
се оксихлорирования протекает по иному механизму по сравнению с окислени
ем 1,2-дихлорэтана, что подтверждается видом кинетического уравнения, пред
полагающего участие в данной реакции адсорбированного кислорода. В этом
случае имеет место конкуренция реакций комплекса (2а) с хлороводородом по
реакции (3) схемы 4.1 и с этиленом в соответствии с общей схемой, предложен
ной в работах [627, 629].
Образующийся монооксид углерода также частично реадсорбируется с пос
ледующим окислением до диоксида.
Таким образом, в реакции окисления этилена участвует как адсорбирован
ный, так и молекулярный кислород. Лимитирующей стадией является, по-ви
I’m 4.38. Зависимость выхода 1,1,2-трихлорэтана от температуры при хлорировании
. । плена. CI/C = 0,96; О2/С = 0,60; т = 8,9 с
ZtiH
Oopaювапие 1,1,2 трихлорлапа к процессе оксихлориронаппя лилгпа
происходи! по следующей схеме |586|:
+2НС1+0.5О, -HCI +2НС1+0.5О,
с2 н4------------- С2Н4С12 —- С2Н3С1---------------► С2Н3С13
(Этилен) (1,2-ДХЭ) (ВХ) (1,1,2 ТХЭ)
Такой маршрут образования 1,1,2 трихлорэтана косвенно подтверждаю п и
отсутствием свободного хлора в условиях процесса оксихлорирования этилен.!
(температура 210 230 °C слишком низка для реакции Дикона — окисления хло
роводорода с получением хлора). По этой причине заместительное хлориропи
ние 1,2 дихлорэтана до 1,1,2-трихлорэтана, по-видимому, не имеет места в ходе
процесса.
Исследования в НИФХИ им. Л.Я. Карпова показали, что процессы окисли
тельного хлорирования этилена и винилхлорида с получением, соответственно,
1,2-дихлорэтана и 1,1,2-трихлорэтана подчиняются одним и тем же кинетичес
ким закономерностям. Аррениусовы параметры для данных реакций:
in А, E,/R
С2Н4 16,68 -7,59
С2Н,С1 13,85 -6,00
Поскольку окислительное хлорирование винилхлорида с получением 1,1,2
трихлорэтана в присутствии медьхлоридных катализаторов протекает со см»
ростью, сопоставимой со скоростью реакции окислительного хлорирован ни
этилена [592], можно предположить, что скорость реакции образования 1,1,2
трихлорэтана определяется концентрацией винилхлорида в системе, т.е. в ко
нечном счете скоростью дегидрохлорирования 1,2-дихлорэтана.
Е.И. Гельперин с соавторами [586] показали, что наблюдаемая кинетика ре
акции образования винилхлорида имеет следующие особенности:
- первый порядок по 1,2-дихлорэтану при приблизительно постоянных пар
циальных давлениях кислорода, этилена, хлористого водорода и воды;
торможение водяным паром при его парциальных давлениях выше 1 кГl.i
- увеличение скорости образования винилхлорида с ростом парциальною
давления этилена и кислорода.
Зависимость скорости образования винилхлорида от парциальных давле
ний 1,2-дихлорэтана и воды иллюстрируется рис. 4.39.
Скорость реакции дегидрохлорирования 1,2-дихлорэтана в условиях про
цесса оксихлорирования этилена описывается уравнением [586]:
гс2н3а “ ^с,н4с1,Рс,н4с12 /(1+ ^н,оРн2о) • (4-7')
Теоретически винилхлорид может образовываться также по маршру|у,
включающему реакцию заместительного оксихлорирования этилена
С2Н4 + НС1 + 0,5 О2 —- С2Н3С1 + Н2О, (4.7-1)
400
I'm. 1.39. Зависимость скорости образования винилхлорида от парциальных давлении
I ' дихлорэтана и воды
предо гавляющемся маловероятным: в связи с высоким значением энергии ак ги
нации (130-140 кДж/моль) реакция заместительного оксихлорирования этиле
пл начинает идти с заметной скоростью t > 350 °C.
В [595] указано, что при относительно невысоких температурах (200-215 °C)
i ьорость реакции образования винилхлорида описывается уравнением перво-
п> порядка:
гвх = ^Рс2н4С12 • (4.75а)
11ри повышении температуры до 240 °C наблюдается отклонение от линей
поп зависимости, по-видимому, за счет торможения хлороводородом, также ян
лиющимся одним из продуктов реакции. Уравнение скорости реакции в общем
виде записывается выражением (4.756):
гс2н3с1 = ^Pc2h4ci2 /(1+ М’на ) • (4.756)
В работе [595] показано, что в интервале 200-240 °C чистый носитель не ка
। ,1 л и шрует реакцию дегидрохлорирования 1,2-дихлорэтана, откуда следуе г, что
определяющую роль в процессе играют медьсодержащие активные центры па
поверхности катализатора.
1емпературная зависимость константы скорости реакции дегидрохлориро
нация дихлорэтана:
к = 2 107 ехр [(-99 500 ± 3500)/(RT)]. (1.76)
Еще один побочный продукт процесса оксихлорирования этилена — этил
хлорид образуется по реакции гидрохлорирования этилена:
С2Н4 + НС1 — С2Н5С1. (4.77а)
790Т
Рис. 4.40. Зависимость скорости образования этилхлорида от парциальных давлении
этилена, хлороводорода и водяного пара
Основные кинетические закономерности этой реакции приведены в работе
[586]. Реакция подчиняется уравнению первого порядка по этилену и хлорош»
дороду и тормозится водяным паром (рис. 4.40).
гс,н5а = ^Рс2н4Рна / + ^н2оРн2о) • (4.776)
E/R = -4,04.
Важными хлорорганическими примесями, в значительной мере определяй»
щими селективность процесса, являются также хлораль, хлороформ и тетрах
лорид углерода Наличие в этих соединениях трех — четырех атомов хлора при
одном атоме углерода позволяет сделать предположение о сходных маршрутах
их образования. В работе [626] показано, что хлораль является продуктом пар
циального окисления 1,2-дихлорэтана, причем этот процесс идет через промг
жуточное образование хлорацетальдегида.
Хлораль, в свою очередь, частично трансформируется в хлороформ и re i
рахлорид углерода. Эти зависимости представлены на рис. 4.41.
В работе [626] предложена следующая последовательность превращений и
ходе процесса:
уСНС13
С2Н4С12 ------*- С2Н3ОС1 --------•- С2НОС13 \ (хлороформ) 1. 'Н
(1,2 дихлорэтан) (хлорацетальдегид) (хлораль) СС14
(тетрахлорид
углерода)
Образование хлороформа протекает при разрушении хлораля по реакции
С2НОС13 —* СНС13 + СО. (4.79.1)
0,12
I'm I II. Зависимость выхода хлорорганических продуктов от времени коп гам а
<1 ' *0 "(( 1/С = 0,97, О2/С = 0,60):
цлираце । альдегид, 2 — хлороформ, 3 — хлораль, 4 — тетрахлорид углерода
1е । рахлорид углерода образуется более сложным путем:
( .1IOCI, + НС1 + 0,5 О2 —* СС14 + СО + Н2О. (4.796)
I хема образования хлораля в присутствии медьсодержащих катализаторов
мн ьг| быть представлена следующим образом:
( Il А I —СН2С1 + 0,5О2—СН2С1—СНО + НС1; (4.80а)
СНА I—СНО + 4СиС12 —- СС13—СНО + 2НС1 + 2Си2С12; (4.800)
и А I > + О2 —2CuOCuCl2; (4.80в)
•I nt)( uCl2 + 2НС1 —► 4СпС12 + 2Н2О. (4.80г )
11а рис. 4.42 представлены зависимости выхода хлороформа и тетрахлори
лп ычерода от температуры в условиях оксихлорирования этилена, показыва
тис что мольный выход хлороформа незначительно растет с повышением
шсрптуры (в интервале температур, допустимом для проведения процесса
и» и хлорирования этилена), тогда как рост выхода тетрахлорида углерода за
м» то увеличивается с температурой. Отношение скоростей реакций разложе
пни хлораля и его оксихлорирования может быть оценено как ~2:1 при низких
пнр.цурах (210-215 °C). Повышение температуры ведет к выравниванию
i-opoi юн вследствие более высокого, видимо, значения энергии активации ре
и> цни оксихлорирования.
Необходимо также отметить, что при образовании хлороформа и гетрах
“рндл у 1 лерода получается также эквивалентное количество оксида углеро
11 ч.к iii'iiio далее трансформирующегося в диоксид. Доля оксидов углерода
Рис. 4.42. Зависимость выхода хлороформа (1) и тетрахлорида углерода (2) на прорса
тировавший этилен. С1/С - 0,97, О2/С = 0,60, т = 8,8-?9,3 с
образующихся по данному маршруту, относительно невелика и не превышаем
10-15% (соответственно выходу хлороформа и тетрахлорида углерода).
Полученные результаты дают основание предположить следующую схему
образования оксидов углерода в процессе оксихлорирования этилена:
С2Н4 ----- С2Н4С12 ---- СН2С1СНО
I В
А ! С
-------► СО ------------------
СО2
СС13СНО
Схема 4.III
Соотношение скоростей маршрутов А, В, С равно, оценочно, 1:2:0,3 [595].
Типичный состав дихлорэтана сырца в процессе оксихлорирования этиле
на, мол. %: 1,2-дихлорэтан — 99,5-99,6, 1,1,2-трихлорэтан — 0,15-0,20, хлоро
форм —0,10 0,13, тетрахлорид углерода — 0,05-0,07, хлораль — 0,05-0,08, этил
хлорид — 0,003-0,004, 1,1-дихлорэтан — 0,008-0,010, 1,2-дихлорэтилены (цис-,
транс-) — 0,010-0,015.
В зависимости от используемого окислителя (кислород или воздух), а также
катализатора процесса приведенные выше значения могут изменяться.
Дезактивация промышленных катализаторов
оксихлорирования этилена
Изменение активности катализаторов оксихлорирования этилена можш
основываться на ряде факторов. Одним из них является изменение состава ре
акционной среды, приводящее к так называемому «закислению» катализатора.
II ном случае дезактивация определяется лишь наличием пеанвитых поверх
нт пнях комплексов, которые могут быть разрушены при проведении окси.чло
рпровапия в специально подобранных условиях. Активность катали штора при
ном полностью восстанавливается.
Дг1ак(ивация катализатора может также являться следствием поюри к.па
ни.иором механической прочности. За счет истирания и разрушения частиц
ш р< над давления в реакторе увеличивается, что также ведет к потере актив
not in и селективности [630]. Большой интерес представляют причины падения
п 1 явности катализатора под воздействием реакционной среды. В |617| ука ы
ш> чю каталитические системы типа СиС12-КС1/у-А12О3 теряют акгивносп. в
Ио ц* жсплуатации по причине изменений механической и фазовой струк туры
п риз катализатора. Авторы [617] иллюстрируют эти эффекты в табл. 4.12.
Таким образом, снижение активности катализатора связано с изменсни
• м фа юного состава, резким снижением удельной поверхности и механичсс
Кои прочности. Это является следствием разложения активной двойной соли
I < ni I ( и выделением свободного КС1, спеканием катализатора за счет местных
in pri ревов, вследствие чего возникают диффузионные ограничения для дот гу
ня кислорода к поверхности зерна. Важным является образование неактивной
|н>рмы одновалентной меди — СпА1О2, нарушающей окислительно восстапо
ни и льное равновесие Cu*'/Cu+2. Авторами [617] зафиксировано физическое
ра щеление ядра и внешней оболочки частицы, происходящее за счет измене
пня профилей концентраций солевых компонентов по сечению катализатора.
• । । веде г к потере механической прочности.
Активность катализаторов оксихлорирования этилена также существенно
шин иг от содержания меди в катализаторе. При длительной эксплуатации в
промышленных условиях для катализаторов с высоким атомным соотношением
ЛI < и ( 30) содержание меди медленно возрастает, при низких значениях А1;( и
I 10 15) — снижается. Эти зависимости проиллюстрированы на рис. 4.43.
)ффект изменения содержания меди в катализаторе имеет в основе исги
раине катализатора и унос мелких частиц из реактора. При высоких значениях
Al I и в основном истирается и уносится из реактора оксид алюминия, вследс
। ши чего относительное содержание меди в катализаторе растет. При низких
in гк пнях А1:Си наблюдается обратная картина.
I пижение содержания меди в катализаторе также является одной и.1 при
пи по дезактивации.
I in ища 4.12. Изменение механической и фазовой структуры катализатора
haia/iM.iarop Фазовый состав Удельная поверх- ность, м2/г Механические характеристики
Прочность, кг Потери при ис тирании, мае. %
< Hi Mill y-Al2O3, KCuC13. CuC122H2O 166 11,3 1,0
'Ь hik inвиpo- iidiiiiiiiii y-Al2O3, KCuCI3, KC1, CuC12’2H2O, Cu2O, CuAIO2 92 5,3 4,0
f.
с. 4.43. Зависимость содержания меди в катализаторе от времени эксплуатации при
инах соотношениях Al:Cu
В работе [595] исследованы параметры процесса оксихлорировапия ин
па в присутствии катализаторов с разным содержанием меди — от 1,98 до
Й мае. %.
На рис. 4.44а и б представлены зависимости конверсии хлороводорода и иы
да продуктов глубокого окисления от температуры для всех исследован!нам
тализаторов. Анализ этих зависимостей показывает, что уменьшение сод» р
шил меди в катализаторе с 4,15 до 1,98 мае. % ведет к снижению активно» in
и фиксированном соотношении С1/С уменьшение содержания меди на (I
> мае. % вызывает снижение конверсии НС1 в среднем на 0,5%, причем по мер»*
вышения температуры (до 230-235 °C) этот эффект уменьшается. Наоборот
сличение содержания меди до 5,84 мае. % уже не влияет существенно па ,о
вность.
Несколько иная картина наблюдается при анализе закономерностей pf.ii»
й глубокого окисления (см. рис. 4.446). Для всех катализаторов выход про
ктов СОХ незначительно растет с увеличением температуры. Умеш.нк пп<
держания меди в катализаторе также способствует снижению доли рсакцы*
гбокого окисления.
Согласно данным рентгенографических исследований [576], с ростом коп
н грации меди в катализаторе растет количество как поверхностной (Cus). i <п>
решеточной (Cug) меди. Однако с увеличением содержания меди в синем»
личество катионов Си2+, занимающих вакантные позиции в решетке, по пи
мому, растет быстрее, чем количество поверхностной меди.
В результате Cus/Cug может зависеть от содержания меди в системе эк» i р<
льно; для образцов с содержанием меди 3,19 и 4,15% соответственно, ко ш
2тво меди, связанное поверхностью, остается постоянным, а медь, соотнен
ющая разнице концентраций, диффундирует в решетку.
Эти данные, в сочетании с зависимостями, представленными на рис. I I Li
», подтверждают высказанное ранее предположение, что реакция оксихлоро
вания этилена идет в основном на медьсодержащих центрах внутри крис i 11
100
I'm. 1.44. Зависимости конверсии хлороводорода (д) и выхода продуктов СОх (б) от
и миера гуры и содержания меди, мае. % при 1,98 (+), 2,65 (х), 3,19 (ж), 4,15 (о), 5,84 (А);
11/С = 0,97,02/С = 0,60, т = 8,9-9,3 с.
янческой решетки, тогда как реакции глубокого окисления — на фрагментах
< ill L, избыточных относительно взаимодействия с носителем. Это подтверж-
дае । с я ростом конверсии НС1 с увеличением содержания меди в катализаторе.
Увеличение выхода продуктов СОХ объяснено [595J ростом количества как но
in рхпостной, так и решеточной меди вплоть до общего содержания меди в ка
.1/1И заторе на уровне 3,19%.
Особняком стоит катализатор, содержащий 5,84% Си, активность которого
< оное гавима с катализатором, содержащим 4,15% Си, а выход продуктов С Ох
. ущественно выше. В [595] высказано предположение о том, что общая коп
щчпрация меди на уровне 4,15% соответствует насыщению медью решетки
nix и геля, дальнейшее увеличение содержания меди ведет к росту избыточных
• плевых фаз.
Удельная активность катализатора возрастает с увеличением содержания
меди, достигая максимума в целевой реакции оксихлорирования (рис. 4.45).
/(пи реакций глубокого окисления максимума не наблюдается. Снижение удель
Рис. 4.45. Зависимость удельной активности катализатора от содержания меди при
I = 225 °C. Реакции: 1 — оксихлорирования этилена; 2 — глубокого окисления
ной активности катализатора в реакции оксихлорирования при общей конщи
>рации меди выше 4-4,5 мае. % объясняется, вероятно, не только низкой .и<
гивностью поверхностного СиС12, но и блокировкой избыточным дихлоридом
меди активных центров в кристаллической решетке. Исходя из того, что |»«»< i
удельной активности катализатора в реакциях глубокого окисления, iia*iuu.i>i
с содержания меди 3-3,1 мае. %, выражен достаточно слабо, можно пол.н.нь
ч го оптимальное сочетание высокой активности и селективности катал изд i оря
имеет место [Си] = 44-4,2 мае. %
Эффективность использования этилена несколько увеличивается при умеш.
шении содержания меди в катализаторе, в первую очередь, за счет некоюрои»
снижения выхода продуктов глубокого окисления. Однако это сочетается <о
снижением конверсии хлороводорода, поэтому уменьшение содержания меди и
катализаторе ниже 4 мае. % ведет в целом к ухудшению показателей процесс а
Другой тип дезактивации катализатора связан с попаданием в систему шн
горонних соединений, которые, вступая в химическое взаимодействие с ка i или
затором, блокируют его активные центры, что ведет к постепенному снижении»
активности, а в ряде случаев, и селективности процесса.
Восстановление активности при такой дезактивации достаточно затрудни
гельно, так как оно должно быть связано с удалением побочных соединении и i
катализатора. Это, в свою очередь, может приводить к необратимым изменит!
ям структуры катализатора и снижению его эксплуатационных свойств.
Дезактивация катализаторов оксихлорирования этилена соединениями
железа
Наиболее подробные исследования дезактивации катализаторов оксихло
рирования этилена соединениями железа проведено авторами работ [631, 6321
Важность понимания этих закономерностей определяется тем, что железо по
падает в катализатор, как правило, за счет эрозии стенок промышленного реп к
тора. Это может, в свою очередь, сопровождаться снижением содержания меди
1 Лхаропи и Дж. lliioap [633[ iiok.i i.i'in, чн> при 210 "< it i i.wii.iiom рсакюрс
। hopoi гь отложения металлической меди на с тиках юс ывляе! 10 i/(<м /год).
( одер каппе железа н катализаторе, очевидно, увеличивается в результате еле
дмощих реакций |364):
I с । СпС12 —FeCl2 + Си;
11 е( L + О2 + 4HCI —- 4FeCl3 + 2Н,О;
’1 eCi, + ЗН2О— Fe2O, + 6HCI.
(4.81а)
(4.816)
(4.81 в)
Реакция взаимодействия железа с хлоридом меди идет при ударе частицы
laianmaropa о стенку реактора, чему способствует осуществление процесса
окш ли тельного хлорирования в псевдоожиженном слое, поэтому включения
попов железа в частицах катализатора можно наблюдать в промышленных пар
Unix катализатора, длительно работавших в реакционной среде.
Оны1 эксплуатации промышленных процессов оксихлорирования этиле
пл указывает на постепенное увеличение содержания железа в катализаторе
(pin. 1.16) и снижение технологических показателей процесса.
Во шикают также и другие проблемы, связанные с наличием железа, и важ
ныс для эксплуатации катализатора. Это возможность нарушения динамики
ш евдоожиженного слоя при процессе, напоминающем спекание частиц, и спи
и пне эффективности катализатора в результате изменения состояния медь
(одержащих фаз в присутствии железа. При этом появление в катализаторе
лк пшиых центров, содержащих железо, способно активизировать протекание
ряда побочных реакций.
Кинетические и технологические закономерности процесса оксихлориро
luiniiii на медьсодержащих катализаторах, включающих примеси железа, изу
чеппые в [631], указывают на то, что железо, попадающее в катализатор в ходе
промышленной эксплуатации, и железо, вводимое в катализатор в ходе приго
(пиления последнего, по-разному влияют на ход и показатели процесса. При ис
пользовании модельных катализаторов, содержащих, мае. %: 4,1 Си, 1,2 и 3 1е,
и рн 1 отопленных пропиткой носителя хлоридами меди и железа (III), заме тное
Г<н. 1.16. Зависимость содержания железа в катализаторе от времени работы промыт
и иного катализатора
влияние желе ia па пока кисли процесса начинается с ею содержания па урошн
2 мае. % (рис. 1.17).
При t < 225 "( конверсии НС! для катализаторов, содержащих 1% 1е и нею
держащих железо, близки, свидетельствуя, что добавка leClj (1 мае. % 1е) Ирак
гически не влияет па активность катализатора при низких температурах.
При увеличении содержания железа до 2 и 3 мае. % активность катали ыю
ров существенно снижается при одновременном увеличении выхода ироду мои
СОХ.
Соотношение СО2/СО в реакционном газе для всех катализаторов, включал
содержащий железо, составляет 3,54-4/1, что свидетельствует о преимущес ин и
ном протекании реакции окисления органических соединений до СО2. Незпачп
тельное содержание СО в продуктах реакции указывает на высокую акт ивное и.
всех образцов катализаторов в реакции окисления СО до СО2. Этот факт су щ<ч
t,°C
Рис. 4.47. Зависимость конверсий НС1 (а) и выхода продуктов глубокого окисления (л)
от температуры. С1/С = 0,97, О2/С = 0,60, £ = 8,9-9,3 с
iiii и дли obьяспенни хорошего жендоо ки кепии катализаторов, i.ik как новы
ин иное содержание (О может приводи п> к aiномерации чаниц кл али саюра и
у чудин-нню режима псевдоожижения, по видимому, вследствиеобра ювания ле1
1 иплаикнх комплексов монооксида углерода с соединениями меди 1595»626|.
Несколько по иному влияют на процесс катализаторы с повышенным со
и р капнем железа (0,8 и 9 мае. %), выгруженные из промышленного реактора.
>iit катализаторы менее активны: в сопоставимых условиях конверсия IК 1
п । I ’"'ll ниже по сравнению с модельными системами. Для них более выраже
in icMiicpaiурная зависимость выхода продуктов СОХ, причем доля продуктов
1 ц покою окисления в побочных реакциях на промышленных образцах выра
-» и.। с ильнее по сравнению с модельными системами.
1ако11омерносги изменения скоростей реакций окси хлорирования этилена
и । цпокого окисления от содержания железа на катализаторах из промыт
и иною реактора (рис. 4.48) показывают, что увеличение содержания железа в
11 раз ведет к снижению скорости основной реакции оксихлорирования ни
in п 1 и 6 раз; скорость реакций образования продуктов глубокого окисления
) in шчиваегся в ~1,6 раза [631].
В условиях оксихлорирования этилена железо (в расчете на 1 г-атом кагио
на) примерно на порядок активнее меди в реакциях глубокого окисления.
(>оразование в промышленных образцах катализатора оксидов меди и же-
н ы, слабо связанных с поверхностью, в значительной степени ведет к росту
порос in реакций окисления и к снижению скорости основной реакции ок-
пчлирирования. По этой причине восстановление активности катализаторов,
lb i.ik । пвированных соединениями железа, трудновыполнимая задача.
Как указывает Н.М. Островский [634], при дезактивации катализаторов
ihепдоожиженного слоя возникает необходимость дополнительного управле-
1’и< . 1.18. Зависимость удельной активности катализаторов с добавками FeCl, (1) и из
примышленного реактора (2) в основной и побочной реакциях при 225 °C от содержа
инн кслеса в системе
блица •1.13. ( ih i.iu xnopopi.uiii'iciKiix ироду к ion процесса ом ихлорпроп.пшя ни •>
, мол. %
с в реак (>ре, мае. % 1,2- Дихлор этап 1,1,2- 1'рихлор этан Эги/1- хлорид Хлоро- форм Хлораль Гс1р.1хл<||'п । у)'1<-род,|
- 99,30 0,32 0,01 0,11 0,01 0,08
0 (мвд.) 99,15 0,36 0,03 0,12 0,01 0.0
0 (мод.) 99,04 0,44 0,04 0,11 0,04 0.04
0 (мод.) 98,86 0,51 0,05 0,10 0 06 О 11)
8 (пром.) 98,95 0,48 0,05 0,14 0,08 0,04
0 (пром.) 98,20 0,76 0,05 0,16 0,15 0.1 .
ежим, либо непрерывной циркуляции в системе реактор-регенера юр. Приме
цельно к катализаторам оксихлорирования этилена на практике можн <*iiai|
авизован лишь первый вариант, при котором концентрация дезакiiiiinpxki
?го агента — железа будет снижена до некоторого максимально донус шмиц
ачепия. Как определено из опыта эксплуатации промышленных upon инпн iи.
а величина соответствует 0,3-0,4 мае. % Fe [631].
В табл. 4.13 представлены данные по составу хлорорганических ироды н.«
•оцесса при использовании промышленных катализаторов в лабор.порнн»
ловиях (1 = 225 °C).
Данные табл. 4.13 подтверждают снижение селективности образование
,хлорэтана при увеличении содержания железа в катализаторе. )то прг.ь и|
его связано с ростом выхода хлораля и 1,1,2-трихлорэтана. При этом > од* |>
хние хлораля увеличивается в 6-10 раз по сравнению с катализатором п«
держащим железа. Выход 1,1,2-трихлорэтана увеличивается в 1,5-2,3 p>i i<
о связано, очевидно, с тем, что, как известно, соединения железа я вл и юн *
тфективными катализаторами реакций дегидрохлорирования хлоралh.iiui#
7]. При повышении содержания железа в катализаторе растет скорое н> р,
ции дегидрохлорирования 1,2-дихлорэтана с последующим оксихлориро
нием образующегося винилхлорида. Железо, очевидно, катализирусi но.
.* реакцию парциального окисления дихлорэтана с образованием хлора К
рез промежуточный хлорацетальдегид [626]. Заметный рост содержания
их примесей начинается при содержании железа в катализаторе па х ронин
час. % и выше.
Эффективность использования этилена падает с ростом содержания ы н к
сатализаторе на 4-6%, причем в первую очередь это происходит за счс i \ in in
ния выхода продуктов глубокого окисления.
Характер поверхностных медь- и железосодержащих структур к.и । ш
щельных, так и для промышленных катализаторов подробно ран мпip> н
И. Курляндской с соавторами [632]. В ходе комплексных исследовании v< <(•
>влено, что примесь железа в катализаторах оксихлорирования л плена < >
гственно влияет на состояние медьсодержащих фаз, в частности:
1. Попы железа конкурирую! с попами меди ia вакантные позиции в крис-
I ылической решет ке носи геля, преимущес юенпо за октаэдрические позиции в
(| фа !с Л I О3,а также за реакционноспособные медьсодержащие центры новер-
xiioi in. При этом дефектные алюминаты железа и другие железосодержащие
i I руктуры образуются количественно только в отсутствие ионов меди.
2. Ионы железа сильно воздействуют на ионы меди на электронном уровне,
it «меня» как донорно-акцепторные свойства ионов меди, так и вид их коорди-
нации. С избытком ионов меди избыточные ионы железа образуют ферриты
шпинельной структуры.
3. В катализаторах из промышленного реактора обнаружена глубокая фазо-
iijii перестройка, приводящая к появлению неупорядоченной фазы а А1.2О3уже
при сравнительно низких температурах. Такую перестройку может вызвать ка-
। ионное модифицирование ионами Си2+, возможно, без участия ионов железа.
1. В связи с образованием а-А12О3 значительная часть медьсодержащих и
железосодержащих фаз находится в виде оксидов СиО и Fe2O3, слабо связанных
1 поверхностью. При этом в процессе работы медьсодержащие фазы агрегиру-
кнея в присутствии ионов железа.
Таким образом, указанное влияние ионов железа определяет снижение ак-
нвпости медьсодержащих фаз в окислительно-восстановительном процессе,
которым является реакция окислительного хлорирования, в результате взаи-
модействия ионов железа с ионами меди на электронном уровне и фазовой пе-
рсе тройки носителя (у-А12О3 —► а-А12О3). Кроме того, присутствие ферритов
меди — шпинелей, отличающихся, как известно, легкостью перестройки струк-
। \ ры, наличием дефектов и особым механизмом электронного обмена, приво-
ди г к снижению селективности процесса за счет побочных реакций окисления.
При этом ферриты меди малоактивны в основной реакции оксихлорирования
> । плена. Тот же результат следует из присутствия в катализаторах оксидов меди
п железа, катализирующих глубокое окисление за счет поверхностных ионов
кислорода О или О2
Авторами работы [364] предложены условия частичного восстановления
активности катализатора оксихлорирования этилена, дезактивированного со-
единениями железа.
Регенерацию катализатора авторы проводили в двух направлениях: путем
। ермообработки катализатора в псевдоожиженном слое в кислородном и воз-
душном режимах, а также отмыванием катализатора в ДХЭ. Данные по изме-
нению физико-механических свойств катализатора в результате регенерации
представлены в табл. 4.14.
Некоторое снижение содержания железа в катализаторе (на 15-16%) дости-
1 ic гея при экстракционной отмывке катализатора в кипящем ДХЭ. Происходит
• го, очевидно, за счет растворения FeCl3 в дихлорэтане. Авторы [364] указыва
ни, что обработка катализатора в кислородном режиме способствует некото-
рому увеличению объема пор (табл. 4.14). Содержание железа в катализаторе
после такой регенерации изменяется незначительно.
Несколько улучшаются также эксплуатационные характеристики катали-
ы гора, подвергнутого регенерации. На рис. 4.49 представлены зависимости
•ффективности превращения С2Н4 (кривые 1, 2) и горения С2Н4 (кривые 3, 4)
и процессе синтеза 1,2-ДХЭ на регенерированном катализаторе от температу-
блНЦа 1.11. <1»H IllhO MCXailH'ICcKHC l HOIK I II.I KJ I.Kill l.llop.l ОКсНХЛОрИроН.Ill 1111 l||l li II I
и после 8 ч регенерации и но (душном и кислородном режимах
Катализатор Си (1,11), мае. % 1с (III), мае. % V, < ] см г S2wi м г И И» ГСМ Unci
11СЖИЙ 4,2 0,12 0,36 140,0 1,02 кленын
: । регенерации 4,15 0,30 0,26 74,5 1,31 KopiriiK iiiiiii
осле регенерации при: 180 °C 4,10 0,30 0,26 78,5 1,29 Icmiiu
210 "С 4,51 0,301 0,276 85,0 1,28 зелены и Зеленый
250 "С 4,585 0,30 0,286 99,0 1,27 Салл iiKiii
кстракционный в ДХЭ ти: 80 °C 4,75 0,254 0,276 101,2 1,12 Гем । in
60 "С (перемешивание) 4,58 0,255 0,269 100,0 1,12 зелен ын Зелены и
осле регенерации t)2/N2, об. %: 30/70 при 210 °C 4,18 0,31 0,27 100,5 1,27 Зеленый
40/60 при 210 °C 4,76 0,275 0,28 105,0 1,26 Светло
40/60 при 250 °C 4,77 0,26 0,32 108,0 1,25 зелен ын Зеленый
с. 4.49. Зависимости эффективности: 1,2 — превращения С2Н4(Е, %), 3, 4 — горении
Н] (Г, %), на регенерированном катализаторе от температуры регенерации катали > i
ра. Среда 1, 3 — воздух; 2, 4 — кислород
ры регенерации катали t.uopa в по>^yнпн>м и кислородном режимах. Эффек-
niiiiiocib конверсии ('2II, в ДХ.) возрастает с 92,5 до 93-94% при повышении
1смнерагуры регенерации ог 180 до 250 °C в кислороде (рис. 4.49, кривая 2).
Одновременно горение С2Н i снижается с 5 до регенерации до 2-2,5% после ре
операции катализатора при повышении температуры регенерации со 180 до
250 °C. При .ном на частично восстановленном катализаторе не увеличивает-
। я выход побочных грихлорэтана, трихлорэтилена и других хлорорганических
нродук гоп.
Дезактивация катализатора оксихлорирования этилена соединениями
серы
Для успешного осуществления процесса оксихлорирования этилена в про-
мышленных условиях необходимо соблюдать жесткие требования к качеству
исходного сырья. В первую очередь, это относится к абгазному хлороводороду,
Пос гупающему в реактор оксихдорирования с других производств. Негативный
оиы | такого рода периодически имеет место в промышленных производствах
винилхлорида по сбалансированной схеме.
Одним из примесных соединений, негативно воздействующих на катализа-
юр и на показатели процесса, является диоксид серы. В частности, при содер-
жании SO2 в хлороводороде, поступающем в промышленный реактор оксихло-
рирования этилена, на уровне 0,05 мае. %, концентрация соединений серы в ка-
шлизаторе может достигать 5-6 мае. % в пересчете на сульфаты за три месяца
жсплуатации.
Расчеты показывают, что такой скорости накопления серы в катализаторе в
условиях процесса соответствует адсорбция -20-25% общего количества диок-
с ида серы, поступающего с газовым потоком.
11а рис. 4.50 представлены зависимости конверсии хлороводорода и выхода
продуктов СОХ для модельных катализаторов с разным содержанием серы при
Содержание серы в пересчете на SO42
Гис. 4.50. Зависимость конверсии НС1 (/) и выхода продуктов СОх (2) от содержания
11 ры и катализаторе. С1/С = 0,92, О2/С = 0,70; * — образец из промышленного реактора
3UA
t = 220 °C. Данные на рис. 1.50 показываю), что увеличение содержании серы в
катализаторе ведет к резкому снижению активное нт конверсия хлороводорода
падает до 88%. Наблюдается также нарушение псевдоожижения слоя: для ка
тализаторов с содержанием серы более 3 мае. % (в пересчете на SO.C) градиеш
температур по высоте слоя достигает 12-15 °C. Несколько снижается также вы
ход продуктов СОХ, что также косвенно указывает на падение активнос ти: горе
ние уменьшается с уменьшением выхода 1,2-дихлорэтана, окисление которою
вносит существенный вклад в образование СО и СО2.
Показатели процесса при использовании катализатора, взятого из промыш
ленного реактора (на рис. 4.50 отмечено звездочкой), достаточно близки полу
ченным на модельных системах.
Увеличение избытка этилена до соотношений С1/С = 0,83-^0,85 приводит к
незначительному (на 1-2%) росту конверсии хлористого водорода. Выход про
дуктов СОХ увеличивается на 1-1,5%, по-видимому, за счет окисления этилена
на медьсодержащих центрах катализатора. Активность катализатора также су
Рис. 4.51. ИК-спектры адсорбированного на катализаторе SO2 при 20-500 °C
нс пн iiiki не меняется при увеличении icMiiepai уры процесса до 235 210 "С, а
и it н<ц ле oopaooiKit его воздухом при 200 ”<
Ин данные свидетели.iпуки о достаточной прочности серосодержащих
ipyi> । у р и кашлизаторе.
Истцом ПК-спекгроскоиии (рис. 4.51) установлено наличие в катали i.iio
1 •< > . прочно адсорбированного па оксиде алюминия, а также сульфитных и
• пи т 1ы|и11пых ионон, трансформирующихся далее в поверхностные сульфаты
|Ы (|
По тем циана юне температур не наблюдается образование сульфа та меди.
1 hit коньку сульфат меди является конечным продуктом в условиях подъема
" ип р.п уры, то и других медьсернистых соединений в катализаторе не обра
и н < )1су тсгвие CuSO4 на поверхности катализатора, обработанного SO2i и
hmi ' ic с 1см уменьшение его активности указывает, что дезактивация не спя ia
и 11 уменьшением содержания хлорида меди в результате реакции с SO2.
Полученные данные не подтверждают предложенный в работе [635] меха
ни 1м де ыкгивации катализатора, основанный на возникновении поверхнос i
и.и и сульфата меди. Этот вывод подтверждается также полученными |595| ж
in римснсальными данными по использованию в процессе оксихлорированин
ниш па катализатора CuSO4/A12O3 с содержанием 5,0 мае. % Си2. В табл. 1.15
npi (i ыилены сравнительные показатели процесса оксихлорирования этилена
и присутствии промышленного катализатора СиС12/А12О3 и катализатора Си
< Ц/Л1 О,.
Данные в табл. 4.15 указывают, что сульфат меди в ходе эксплуатации в
процессе оксихлорирования этилена практически полностью превращается
и хлорид меди. Активность катализатора при этом соответствует активное!и
niibi iiioro хлормедного катализатора. Выделяющийся в ходе замещения суль
ф и попа на хлорид-ион серный ангидрид (SO3) десорбируется с поверхносin
1 ni и ли штора и удаляется из системы с реакционными газами.
Полученные результаты подтверждают данные спектрального анализа об
• и у 111 иии сульфата меди в дезактивированном катализаторе. При этом пред
< ИП1ЛИСГСЯ сомнительным предположение, сделанное в работе [615] о том, что
> худшеиие параметров процесса связано с восстановлением сернистым апсид
luniiii^.i 4.15. Сравнительные показатели процесса оксихлорирования пилена
(I ’.’О "С, С1/С = 0,92, О2/С = 0,70, т = 9,2 с, ССи = 5 мае. %)
Длительность работы, ч CuCI2 в катали- заторе, мае. % Конверсия НС1, % Выход сох, % Эффективное гь использования этилена, %
Промышленный катализатор СиС12/12Оз
10,5 99,1 3,8 92,5
Катализатор CuSO4/A12O3
5ч 4,4 95,5 2,6 93,1
20 ч 7,1 98,0 3,0 93,0
50 ч 10,0 98,9 3,4 92,7
идом Си 1 до Си' и конкуренцией .ной реакции с пои шионлепием < и ин
еном, а какжес (нюкировкой серосодержащими ионами центров поверх ш» in
осителя, участвующих в иерее тройке катализатора в реакционной греде
В соответствии со схемой 4.1 вместо реакции (1а) взаимодействии кашли 11
ора с этиленом должна идти реакция:
С1 С1
I I
Си Си
— Al—О—Al—О —Al— + SO2( I.
хема 4.IV
и далее: SO2C12 + ViO2 —► SO3 + Cl2
Эти реакции представляются маловероятными в силу того, что в ироду к ых
роцесса оксихлорирования в присутствии катализаторов, дезактивированных
иоксидом серы, отсутствуют как хлористый сульфурил, так и молекулярный
лор, хотя бы в следовых количествах. Достаточно легкая десорбция серною
Йгидрида с поверхности катализатора должна постепенно приводить к сии кг
ию содержания серы в катализаторе в условиях процесса оксихлорироваиин
ри условии прекращения поступления соединений серы в систему. Этот н|>
ект, однако, также не имеет места в ходе процесса.
Блокировка соединениями серы центров поверхности носителя, не связан
ых с медью, также представляется маловероятной ввиду того, что свободные
г меди центры не участвуют в реакциях как непосредственно оксихлорирова
ия, так и в реакции глубокого окисления.
Рассматривая дезактивацию катализатора диоксидом серы в рамках схемы
.1, можно предположить, что первой стадией процесса является взаимодеш
вие диоксида серы с координационной связью Си—О в катализаторе, в ре
рштате чего хлорид меди как бы «вытесняется» с поверхности катализатора:
г А1—О —А1 —О —Al— + SO2 —-
I I I
CuCl2 CuCl2
o=s=oo=s=o
— Al — О—Al — О — Al—
I I I
хема 4.V
В дальнейшем происходит разрыв связи S —Си, в результате чего сера
рочно адсорбируется на поверхности, а СиС12 переходит в состояние, не свя
1нное с носителем. Как было показано, активность меди в несвязанном состо
зии существенно снижается, а наличие в системе хлороводорода ведет к его
гаимодействию с соединениями серы на поверхности и, вероятно, к слипанию
1тализатора, что и обусловливает нарушение режима псевдоожижения в ходе
роцесса.
I'm 1.52. Зависимость скорости реакции оксихлорирования этилена от концентрации
пилена при 225 °C
I >аи>ры: I — свежий промышленный; 2 — дезактивированный (конц. серы 5,3 мае %, в
in pcc'ieie на SO42; 3 — регенерированный хлоридом бария
Восстановить эксплуатационные свойства такого катализатора можно сне
циальной обработкой, в ходе которой серосодержащие структуры должны быть
у цалены из системы.
Одним из таких способов является обработка дезактивированного катали
ы । ора водным раствором хлорида бария [636,637,638]. Целью такой обработки
цилис тся разрушение поверхностного серосодержащего комплекса с образов.!
пнем гонкодисперсного порошка сульфата бария и его последующей отдувкой.
В работе [595] показано, что после обработки катализатора водным раствором
хлорида бария и отдувки тонкодисперсного порошка сульфата бария содержа
пне юры в катализаторе уменьшается в ~5,5 раза. На рис. 4.52 представлены
цини имости скорости реакции оксихлорирования от концентрации этилена
дли дезактивированного и регенерированного катализаторов в сравнении со
। иг к им катализатором.
Нвисимости на рис. 4.52 показывают, что катализатор, регенерированный
хлоридом бария, частично восстанавливает активность, хотя и не достигает ак
1НННОСГИ свежего катализатора. Скорость реакции в условиях процесса увели
шипе гея в ~2,5 раза. Это связано, очевидно, с уменьшением содержания серы в
к л । ализаторе. Известно, что наличие в катализаторе оксихлорирования этилена
и 'больших добавок хлоридов щелочных или щелочноземельных металлов (К,
< л. Mg) позитивно влияет на селективность за счет снижения выхода продук
। ни । лубокого окисления. Несколько снижается, однако, при этом и активность.
>ффект от введения в катализатор солей бария, по-видимому, аналогичен.
В отличие от свежего катализатора оптимальная температура эксплуатации
pci оперированного катализатора 233-237 °C, при этом конверсия НС1 достигает
’>•)%, |ффективность использования этилена 90,5-91%. Аррениусовы параме тры:
I .И.1ЛТ1 затор...Свежий Регенерированный
У .................ю8-97 Ю8’50
I II ..............9,22 11,05
При де idh шкации катализатора диоксидом серы сущее i пенно ухудшат!
ся качество дихлор жни сырца, в основном »а сче! увеличении выхода 1,1,*
грихлорэтапа, с 0,4 до 0,75-0,8 мол. % 11ричинон лого, видимо, я вл не геи у ней и
чение доли реакции дегидрохлорирования 1,2-дихлорэтана га счет иопыик пни
концентрации хлорида меди на поверхности катализатора.
Регенерация катализатора практически не меняет данный показатель, тн
можно, по причине дополнительного протекания реакции дегидрохлорнровл
ния в присутствии основных центров за счет бария.
Таким образом, регенерация хлоридом бария частично восстанавливав! лк
гивность катализатора оксихлорирования этилена.
Приготовление катализатора оксихлорирования этилена
Эксплуатационные свойства катализатора оксихлорирования Э1 илена в
псевдоожиженном слое зависят в основном от выбора носителя с необходимым
сочетанием структурных и физико-механических характеристик, а гак кс <н
способа нанесения активной фазы.
Решение первой задачи не является предметом настоящей работы, отменим
лишь желательность наличия в носителе 6-фазы А12О3, способствующей луч
шему диспергированию активной добавки в катализаторе, а также набора пеон
ходимых физико-механических характеристик, о которых указывалось выше
Способы решения второй задачи являются, как правило, ноу-хау производи i с
лей катализаторов, конкуренция между которыми чрезвычайно велика.
По этой причине нами рассматриваются лишь основные принципы upiiio
товления катализаторов псевдоожиженного слоя для процесса оксихлорирова
ния этилена, основанные как на немногочисленных публикациях, так и на со<к
гвенном опыте авторов данной книги.
Формирование структуры катализатора как единой каталитической сне
темы, в котором участвуют и солевой компонент, и носитель, начинается у м
на стадии приготовления катализатора и завершается в условиях реакционно!!
среды, поэтому на технологические характеристики полученного катализа горл
существенно влияют способы и параметры нанесения активного солевого ком
понента на носитель, т.е. технология его получения.
В процессе приготовления катализатора при химическом взаимодействии
хлорида меди и носителя возникают сложные многокомпонентные структуры,
строение которых зависит не только от фазового состава и кислотно-основных
характеристик носителя, но и от способа и условий контактирования хлорида
меди и носителя [581, 639, 640]. Эти структуры — предшественники катали ги
чески активных центров — определяют, в конечном счете, необходимые по i ре
бительские свойства катализатора в процессе окислительного хлорирования.
В 1970-е годы практически единственным промышленным методом получе
ния солевого нанесенного катализатора процесса оксихлорирования этилена в
псевдоожиженном слое являлась пропитка микросферического оксида алюми
ния водным раствором хлорной меди.
Согласно этому методу, раствор хлорида меди при непрерывном перемени!
вании приливают в емкость с носителем, выдерживают 30 мин, пропитанный
влажный катализатор выгружают на противни и сушат горячим воздухом при
ШН I [2 °l и течение 16 20 4. Мелкая фракции ныли мылиlampa (< 2(>мкм),
। идср канне коюрои в исходном вое тигле ик тавляет 5 10%, улавливается и
удаляс1ся. Количес гво и кон цен грацию рас i пора ак тинного ком нонен га подои
ринк такими, чтобы содержание меди в готовом катализаторе составляло 1,5-
» мае, % (в расчете на медь), a cooiношение объемов раствора хлорида меди и
нот шеля равнялось 1.
Описанный способ пропитки водными растворами характеризуется мио
ни тадийностью технологической схемы, значительными энергозатратами на
। 1.1ДИЯХ кропи тки, сушки и отдувки, низкой производительностью и периодич
ши нло процесса в целом.
Кардинальным недостатком обсуждаемого метода водной прописки явля
। к я. п час । пости, блокировка адсорбционных центров поверхности носителя
। пдр.нпым покровом, конкуренция между сильно полярным растворителем
(подои) и наносимым солевым компонентом в процессе закрепления; дифф}
HKiiiiibie затруднения при прохождении ионов (или ассоциатов молекул) соли
через । идратно-гидроксильный покров для фиксации на поверхности, что не
по 1ВОЛЯСТ наиболее эффективно использовать активные центры поверхпос in.
Кроме того, даже в случае реализации первоначально достаточно хорошего
р.п нределения солевого компонента при пропитке из раствора малой концен
(рации оно исчезает при сушке: в результате неравномерной кристаллизации
и капиллярного выноса образуется большое количество грубодисперсных фа т,
Mio, и конечном счете, ведет к снижению селективности процесса оксихлори
рования.
Вместе с тем, изменение режима водной пропитки, переход от статичного
тлоя носителя при периодическом перемешивании к интенсивному перемеши
II.IHIIIO с достаточной турбулизацией при импрегнировании в псевдоожижен
ним слое носителя, возможно, способствует получению катализатора с более
равномерным распределением наносимого активного компонента.
( ной целью фирмой «Euteco Impianti» [641] предложено импрегнирова
ине пористых микросферических частиц оксида алюминия осуществлять в
ш ендоожиженном слое при t < 50 °C раствором хлорида меди II с концентра
цнен 16-60 г/100 мл, который подается по каплям сверху на псевдоожиженный
»'ion носителя. Объем раствора для импрегнирования составляет 90% обще
io объема пор частиц оксида алюминия. Частицы катализатора сушат также в
ш ендоожиженном слое, поднимая температуру газа, поддерживающего слои,
• и скоростью 30 °С/ч до 140 °C и выдерживая при этой температуре 0,5-15 ча
ыш Авторы [641] указывают, что стадия сушки является самой важной для
получения однородного распределения меди по поверхности частиц оксида
алюминия.
I кшышенные температуры способствуют взаимодействию СиС12 с гидро
। с ильными группами поверхности оксида алюминия, например, по следующим
\ равнениям:
2СпС12 + 3>А1 — ОН —- 3>А1 —Cl + Си2(ОН)3С1; (4.82а)
( иС12 + >А1—ОН —- >А1—OCuCl + НС1. (4.826)
Однако температуру сушки не рекомендуется поднимать выше 140 °C [6411.
Иолучснпып h.ii.uin i.iiop имеет однородное распределение меди на поверх
пости с отклонением oi однородное!и менее 17%.
В патентах |б!2, 643| фирмы «Monlecatini leinologie» турбулизации слом
носителя достигается перемешиванием носителя во вращающемся мнигп
пере. При этом па носитель (микросферический оксид алюминия) медленно
распыляют водный или солянокислый раствор, содержащий СиС12 (и т р.цч» in
1-10 мае. %, меди в готовом катализаторе) и MgCl2 (0,4-1,2 молей на моль < н)
Существенно, что объем раствора для импрегнирования равен объему пор ок
сида алюминия. Затем температуру поднимают до 150 °C со скоростью 25 "< ч и
выдерживают при конечной температуре 3 ч, после чего катализатор медленно
охлаждают до 40 °C.
В полученном катализаторе W = X/Y > 1,4, где
А1 на поверхности
X - -----------------, мольное соотношение,
Си на поверхности
Общее содержание А1 в катализаторе
Y = —--------------------------------, мольное соотношение
Общее содержание Си в катализаторе
Затем катализатор сушат и активируют. Данный катализатор харак repn.iyc i
ся отличной способностью к псевдоожижению и высокими выходами дихлор
этана, однако технология его приготовления включает ряд последовательных
стадий.
Описанные выше недостатки импрегнирования, связанные с использовали
ем воды как растворителя для СиС12, в значительной мере нивелируются при
нанесении активного солевого компонента с использованием органических
растворителей.
В [644] описан способ получения катализатора оксихлорирования углсво
дородов, в частности, этилена, состоящего из компонентов CuCl2-KCl-LaCI i на
носителях, таких, как силикагель, оксид титана, а А12О3 и т.п., путем импрегни
рования носителя в два этапа. На первом этапе носитель выдерживают в рас
творе безводного СиС12 в метаноле (или CuCl в ацетонитриле) в течение К) ча
сов, затем растворитель удаляют при нагревании и вакуумировании. На втором
этапе происходит пропитка коллоидным раствором КС1 и LaCl3 в муравьиной
кислоте также в течение 10 ч. Затем катализатор сушат при 100 °C и понижен
ном давлении для удаления избытка муравьиной кислоты. Содержание хлорида
меди в катализаторе должно быть не менее 10 мае. %., что, согласно [599], дос га
точно для образования мономолекулярного слоя; содержание КО — не меш i
5 мае. %., соотношение KCl:LaCl3 = 1:1.
Ввиду того, что катализатор активен уже при t ~ 200 °C, его частицы нс
слипаются, так как при таких температурах активные солевые компоненты нс
образуют расплавов. Катализатор также обладает достаточной стабильностью
вследствие низкой летучести активных компонентов при пониженных темне
ратурах процесса оксихлорирования.
В [639] указано, что применение катализатора, приготовленного пропиткой
носителя раствором хлорида меди в этиловом спирте, по сравнению с катализ.!
тором, полученным стандартным методом, позволяет облегчить решение про
блем, связанных с загрязнением катализатора железосодержащими продуктами
коррозии. Гем не менее, при введении в н<ч каыдн i.nop около 3% Ге в виде
1сЛ)| конверсия IICI снижается при одновременном повышении скорое ги ik
iiip.iiiwii частиц катализатора.
Применение органических рас твори гелей, однако, не является целееооора j
ним прежде всего ввиду их пожаро и взрывоопасное i и Кроме того, неоохо
димос 11. применения вакуумной техники значи сельпо усложняет технологичес
кую схему.
Алыернативным способом приготовления медьхлоридного нанесенного ка
1алн гатора является нанесение активного компонента в отсутствие' какого ли
оо растворителя, например, сухим смешением хлорида меди с носи телем |б 1 [|
К.иализагор получают смешивая безводный тонко измельченный хлорид меди
< ак гнвпым оксидом алюминия, нагревая до 350 °C (и до 400 800 °C) в течение,
но крайней мере, 3 ч (и до 4-8 ч) непосредственно в реакторе оксихлорирования
или любом другом аппарате смешения. Катализатор весьма устойчив к влиянию
примеси железа и при введении 3% железа (в виде Fe2O3) сохраняет конверсию
по IК 'I на уровне 97-98% при низкой скорости истирания.
Высокие технологические показатели катализатора авторы [644] об ьясняюi
ра» положением мономерных молекул СиС12 на поверхности оксида алюминия с
<><>ра юванием связей —О — CuCl при взаимодействии хлорида меди с гидрок
i ильными группами поверхности.
1кдостатки метода вызваны прежде всего высокой температурой и дли гель
ностью смешения. Так, согласно [645], реакция
CuCl2 —► CuCl + 0,5С12 (4.83а)
протекает, хотя и с малой скоростью, уже с 210 °C. Образовавшийся хлорид
меди (I) при t > 330 °C вступает в реакцию с кислородом воздуха:
2CuCl + 0,5О2 —- Си2ОС12, (4.836)
чю в сочетании с одновременной потерей ОН-групп поверхностью носителя
п се перестройкой при высоких температурах должно сильно затруднять про
цее с образования связанных медьсодержащих фаз на поверхности носителя, г.е.
процесс нанесения в целом.
Сухое смешение разлагаемой соли меди (из группы солей щавелевой, му-
равьиной и молочной кислот) и хлорида щелочного металла (LiCl или КС I) с
ч.к типами носителя (силикагеля, оксида алюминия, кизельгура, диатомитовых
шмель и др.) описано в [646]. Через эту смесь в псевдоожиженном слое про
нуекшфт газообразные НС1 и О2 при соотношении от 0,1:1 до 10:1, температуре
* >0 550 °C и давлении от одной до нескольких десятых мегапаскалей в тече
нпе 1-24 часов. Содержание меди в катализаторе 5-20 мае. %., содержание щс
ночного металла — до 10%. При использовании этого катализатора в реакции
<нипхлорирования этилена при 305 °C, времени контакта 1,9 с, соотношении
I К I С 2Н4:О2 = 2,05:1,07:0,6, степень превращения НС1 составляет 96%, а э гиде
н.। 99%.
Данный способ характеризуется, однако, неоднородностью нанесения соле
нои составляющей по глубине гранулы вследствие кратковременного контакта
i.u 1иц солевой разлагающейся фазы и частиц носителя в псевдоожиженном
(дос, а также необходимость длительного хлорирования оксидов, образующих
312
ся при ра ши кеннп солен меди (оксалатов, формиатов, лактатов) при кыкжнч
давлениях хлороводорода
К безводным способам нанесения также можно отнести способ прошикн
заключающийся в погружении носителя в расплав хлоридов активных комно
пен сов, последующем охлаждении ниже температуры плавления и удалении и i
бытка активных компонентов [647|.
Таким образом, представленные нетрадиционные методы получения медь
хлоридных нанесенных катализаторов окислительного хлорирования пилена
могут быть классифицированы следующим образом: нанесение путем подачи
водного раствора хлорида меди в псевдоожиженный слой носителя или па но
ситель, помещенный во вращающийся контейнер; импрегнирование раствора
ми хлорида меди в неводных растворителях; пропитка погружением гранул по
сителя в расплав хлоридов меди-калия; импрегнирование при сухом смешении
носителя с разлагаемой солью меди с последующей обработкой НС1 и О2 при
прогреве; импрегнирование при сухом смешении хлорида меди с носителем.
В [648] предложен метод приготовления катализатора оксихлорироваппн
этилена методом напыления раствора СиС12 в псевдоожиженный слой носители
с совмещением операций нанесения, сушки и отдувки. Схема установки пред
ставлена на рис. 4.53.
Согласно этой схеме воздух, являющийся теплоносителем и псевдоожижаю
щим агентом, подается воздуходувкой I в калорифер 2, где нагревается до гем
пературы 220-250 °C. Из калорифера нагретый воздух поступает в распредели
тельный короб сушилки фонтанирующего слоя 3 под газораспределительную
решетку аппарата. Пройдя через решетку, воздух приводит в псевдоожиженное
Воздух
Рис. 4.53. Принципиальная технологическая схема процесса получения катализатора
оксихлорирования этилена методом напыления:
1 — воздуходувка; 2 — калорифер; 3 — сушилка; 4 — циклон; 5 — рукавный фильтр; б — венти
лятор; 7 — инжектор; 8 — емкость с мешалкой; 9 — дозировочный насос; 10 — пневматическая
форсунка; 11 — дозировочный бункер; 12 — приемный бункер
В атмосферу
Воздух
i1» iiHiiiiie слои ч.к гпц носи ген», 1мходнщ|ик и и anii.ip.iie Orpaboiaiini.ui 1101
цх с icmihpaiурон 60 80 ”(’ из сушилки попадав! и циклон /, tде очищавгси
и пыли, а ином и рукавный фильтр 5 для окончиleai.uofi очис тки, после чего
in и шля юром о выбрасывается в атмосферу. Пылевидная фракции из бункера
циклон.। ин кекгором 7 возвращается в аппарат.
Р.п шор хлорида меди в обессоленной воде с концентрацией 20-22 мае. %.,
очищенный от механических примесей, из емкости 8 дозировочным насосом 9
подле к я па пневматические форсунки 10. Для распыления раствора использу
I. и воздух давлением 0,3 МПа.
I loi птель подается в сушилку самотеком из дозировочного бункера / /, куда
предварительно загружается в количестве, необходимом для проведения одной
ши рации Готовый продукт через выгружной патрубок самотеком выводится
п । аппарата в приемный бункер 12, откуда поступает на расфасовку.
11одача носителя в дозировочный бункер и подача катализатора из приемио
н> 1ц икера в отделение фасовки могут быть осуществлены пневмотранспортом.
Дли установки мощностью 100 т катализатора в год определены оптимал >
iii.li технологические параметры процесса пропитки, определяющие стабиль
пн 11> его хода в сочетании с качеством продукта [648]:
скорость воздуха-теплоносителя у газораспределительной решетки аппа
р.п л в интервале 0,37-2,2 м/с обеспечивает стабильное и равномерное (без ла
। пятых зон) псевдоожижение при достигаемых плотностях орошения;
повышение температуры и концентрации раствора способствуют увеличе
пню производительности. Однако с увеличением концентрации раствора про
цил ки гельность напыления сначала резко падает, а затем изменяется незначи
и выю; аналогичный эффект наблюдается и при увеличении температуры..)н>
i пи i.Hio с увеличением вязкости раствора при повышении его концентрации с
дос жжением критической плотности орошения и приводит к дестабилизации
ре кима псевдоожижения и увеличению температуры слоя обрабатываегого ма
(срнала и отходящего воздуха;
в интервале температур 200-250 °C и концентрации СиС12 20-25% обеспе
чппается максимальная производительность установки при минимальном пы
лсуносе (не более 3%); содержание частиц размером до 10 мкм в образцах ката
ян штора составляет 1-2% (в исходном носителе 3-5%), что сокращает время па
Пос ледующую отдувку. Продолжительность процесса составляет 25-50 минут;
- плотность орошения по следу факела распыла форсунки, определяющая
габильность режима псевдоожижения, составляет 16-47 г/(см2- ч).
11аряду с совмещением операций пропитки, сушки и отдувки катализаюр,
полученный по предложенной технологии, характеризуется высокими значепи
ими А1:Си (> 30), что важно для достижения высоких эксплуатационных пока
la 1 елей.
Технология приготовления катализатора оксихлорирования этилена мето
дим сухой термообработки предложена в работе [649]. Схема установки пред
i 1авленанарис. 4.54. Каталитически активный компонент — хлорид меди (крис
। аллогидрат) из бункера 1 поступает в шаровую мельницу 2, где измельчается до
ра (мера частиц, соответствующих фракции 80-100 мкм (80-90%) и далее — и
бункер 3, откуда подается в реактор 5, где смешивается при 20 °C с носителем,
поступающим из бункера 4.
Рис. 4.54. Схема получения катализатора методом сухой термообработки-
Л 3,4 — бункеры; 2— шаровая мельница; 5 — реактор; б — инжектор; 7 — поглотительная колон
на; й — вентилятор; 9 — приемный бункер; 10 — емкость для раствора щелочи
При постоянной скорости перемешивания в реакторе, снабженном >лсь
трообогревом, осуществляется ступенчатый подъем температуры до >0 (
с 0,5-ч выдержкой при этой температуре, далее до 100, 500 и 200 °C с сношен
твующим выдерживанием при этих температурах. Затем реактор охлажд.1101
до 30-40 °C, прекращают перемешивание и выгружают готовый продукт черт i
приемный бункер 9 в сухую герметичную тару.
При сухой термообработке солевой компонент более активно реагнрус i
как с решеткой, так и с поверхностно-активными группами носителя. Солен ы
система становится рентгеноаморфной, образуются также алюминатные ст р\ к
туры — твердые растворы катионов Си2+ в решетке носителя. Авторы рабе.
[649] указывают на высокую эффективность (не менее 98,5%) таких катали,iai<>
ров в процессе оксихлорирования этилена.
Технологическое оформление процесса оксихлорирования этилена
в псевдоожиженном слое катализатора
Как указывалось, промышленные процессы оксихлорирования этилена мп
гут быть реализованы в двух вариантах: с использованием в качестве окисли и
ля кислорода воздуха или концентрированного кислорода.
Принципиальная технологическая схема «воздушного» процесса предо ап
лена на рис. 4.55.
Процесс оксихлорирования этилена идет при 210-230 °C и давлении 250
400 кПа. Продуктами реакции, помимо дихлорэтана и побочных хлорорглнн
ческих соединений, являются вода, оксиды углерода, непрореагировавшие ни
лен, кислород и хлороводород. В системе имеется также азот, поступают.
реактор в составе воздуха.
I’m. 1.55. Принципиальная технологическая схема «воздушного» процесса оксихлори
роп.тия пилена (1-11 см. текст)
Дли проведения процесса оксихлорирования этилена в распредели тельное
И |роис гво реактора подают смесь хлороводорода с этиленом; воздух как нс
ючпик кислорода вводится в реактор снизу под распределительную тарелку.
( мешение всех исходных реагентов происходит в специальных стаканах р.п
н редел и тельной тарелки. Этилен и хлороводород попадают через опускные наг
рупки, заглубленные в стакан на 70 мм.
Хлороводород, выделяемый из продуктов пиролиза дихлорэтана и содержа
|цин ацетилен, предварительно поступает в реактор гидрирования 1, проходиi
н pei подогреватель, смешивается с подогретым этиленом, и полученная га.к»
ii.hi смесь вводится в реактор оксихлорирования 2.
Выходящий из верхней части реактора реакционный газ поступает в низ ко
лоппы закалки 3 для охлаждения до 90-110 °C и улавливания следов ненрореа
1 пропавшего хлороводорода, а также уносимой пыли катализатора. Поступаю
mini в колонну реакционный газ барботирует через слой воды, уровень которой
поддерживается в контролируемых пределах. Небольшое количество воды но
дистся также на орошение верха колонны.
Вода, выходящая из куба колонны 3 и содержащая хлорид натрия, дихлор
и.hi, соли меди и алюминия, хлорид аммония, направляется на стадию очистки
< годных вод.
Охлажденный реакционный газ после колонны закалки поступает в кон
дснсагор 4, охлаждаемый оборотной водой, где одновременно с охлаждением
реакционного газа происходит конденсация паров дихлорэтана и воды Опра
|уи>|цаяся газожидкостная смесь поступает в сепарационную часть отделителя
дихлорэтана 5, где происходит разделение жидкой и газовой фаз. Жидкая фа за
н 1 сепарационной части стекает в разделитель, где разделяется на водный слои
и слой дихлорэтана. Верхний водный слой через вертикальную перегородку
’.11делн1ели переieK.icI it oicck дли поды, откуда насосом нодаеил па оропк ни
колонны закалки <.
Газовая фаза из верхней части сепаратора 5 поступает па дальнейшее o.vin 1
дение в конденсатор 6, где дополнительно конденсируются пары дихлор илы
и воды. Образующаяся газожидкостная смесь поступает в фазоразделнicai. ,
откуда жидкая фаза возвращается в отделитель 5, а газовая фаза направлж i я
па узел абсорбции-десорбции дихлорэтана из абгазов (колонны 8 и 9). В абсор
зере 8 дихлорэтан удаляется из абгазов при помощи высококипящего аромаш
веского растворителя — «керосина».
Очищенный газ выбрасывается в атмосферу; дихлорэтан отделяется от >м
росина» в десорбере 9 и возвращается в схему (сепаратор 5). Регенирированпып
абсорбент вновь направляется на абсорбцию.
Нижний слой дихлорэтана сырца насосом откачивается в емкости 10 и II
для водно-щелочной промывки и далее — на систему ректификации.
На рис. 4.56 представлена принципиальная технологическая схема процп » л
эксихлорирования этилена с использованием концентрированного кислорода
Согласно схеме, хлороводород поступает на окислительное хлорирование * • •
стадии пиролиза дихлорэтана. В связи с тем, что в нем содержится до 0,25% ацг i и
лена, образующегося в процессе пиролиза, хлороводород предварительно по» i у
чает в реактор гидрирования 1, в котором ацетилен гидрируется с образованием
п илена. Процесс протекает при 130-180 °C в неподвижном слое катализатора
оксиде палладия, нанесенном на экструдированный оксид алюминия. Оста точно!
содержание ацетилена в НО не превышает 0,01 %. Хлороводород после реа кто ра /
с температурой 150-180 °C поступает в смеситель, где смешивается с конценi рп
ис. 4.56. Принципиальная технологическая схема «кислородного» процесса окышю
ирования этилена:
1 и 2 — реакторы гидрирования и оксихлорирования этилена, 3 — колонна закалки, 4 кои
ценсатор, 5 — разделительная емкость, б — холодильник, 7 — компрессор, 8, 9 — емко» in ('in
промывки
рованным кислородом. Befit гему можс| i.ik кг подана i i>c я хлороводород с других
upon ।иоде ।и, отвечающий необходимым криicpiniM качс<_ гва.
В реактор вводятся также пилен под давлением 0,6 МПа и в качестве рааба
тигля — абгазы окислительного хлорирования сдавлением 0,6 МПа, нагретые
to 180 °C.
I’eaKiop окислительного хлорирования этилена 2 представляет собой ко
лонный аппарат, содержащий смесительное устройство для ввода кислорода и
хлороводорода, распределительную тарелку для ввода смеси рециклового по-
пжа и этилена (в период пуска — азота), встроенный змеевик для отвода тепла
реакции и трехступенчатый циклон для улавливания мелких фракций катали
спора.
Рециркулирующая смесь абгазов и этилен вводятся в нижнюю часть реак
юра под распределительную тарелку сферической формы, снабженную накрав
ж иными вниз трубками. Хлороводород и кислород входят в реактор через сме
< тельное устройство, также имеющее направленные вниз трубки, входящие в
н.п рубки распределительной тарелки. Это обеспечивает интенсивное псреме
шивапие кислорода, хлороводорода, этилена и рециклового потока абгазов не
ргд входом в псевдоожиженный слой катализатора. Такое решение необходимо
дли предотвращения образования взрывоопасной смеси. Катализатор подается
и реак тор с помощью пневмотранспорта.
Реакционные газы проходят снизу вверх через слой катализатора со ско-
рт 1ыо 25-50 см/с и приводят его во взвешенное состояние. Это обеспечиваем
хорошее взаимодействие между реакционными газами и частицами катализа
юра.
В связи с экзотермичностью процессов в реакторе в змеевики подается под
давлением 0,8-1,2 МПа паровой конденсат, за счет испарения 10% которого
। помается значительная часть тепла реакции.
После слоя катализатора прореагировавшая смесь газов, содержащая пы
ьвидные частицы, проходит сепарационное пространство, где происходит
улавливание—оседание основного количества увлеченного газом катализатора.
11ос ле этого газовая смесь поступает на дальнейшую очистку в расположенный
и верхней части реактора трехступенчатый циклон. Уловленный в циклонах ка
шли iaгор возвращается в псевдоожиженный слой.
Для образования хорошего псевдоожиженного слоя массовая доля частиц
p.i 1мером менее 40 мкм должна составлять 25-35%. Это достигается в процессе
работы при истирании более крупных частиц. При нормальной работе цикло
нив частицы размером более 10 мкм отделяются и возвращаются в псевдоожи-
женный слой.
Для обеспечения эффективного отвода тепла уровень псевдоожиженною
। лоя катализатора в реакторе поддерживается на такой высоте, чтобы змеевики
пыли полностью покрыты слоем катализатора.
Реакционные газы из реактора оксихлорирования 2, состоящие из дихлор
папа, воды, непрореагировавших кислорода, этилена и хлороводорода, оксида
и диоксида углерода и побочных хлорорганических продуктов, направляются в
куб колонны закалки 3, орошаемой щелочной водой для нейтрализации хлоро-
водорода, абсорбции катализаторной пыли и охлаждения продуктов реакции
ж.95 105 °C.
Реакционны!! i*i i, ii.u 1.11ЦС1И1Ы11 водой, выходи) oi верхней части колонны
закалки 3 и пос lynaei н конденсатор дихлорэтапа сырца 4, где охлаждасн и до
30 35 °C. Из нижней части колонны закалки 3 выходи т водный раствор хлорида
натрия, хлоридов аммония и меди, содержащий также дихлорэтан, и направив
ется на стадию очистки сточных вод.
Смесь газа и жидкости из конденсатора дихлоэтана-сырца поступав! в p.i i
делительную емкость 5, где газовая и жидкая фазы разделяются. Газовая фа ы
состоит из кислорода, этилена, оксида и диоксида углерода, азота, а также n.i
ров дихлорэтана и воды. Жидкая фаза образует два слоя: верхний — вода, пи i
ний — дихлорэтан-сырец.
Выходящие из разделительной емкости 5 абгазы с температурой 20 "( и
давлением 230-330 кПа поступают в холодильник 6. Сконденсировавшие в дн
хлорэтан и вода сливаются в емкость 5, а основная часть абгазов из холодил!,
ника направляется в компрессор 7, где дожимается до 0,6 МПа и после смете
ния со свежим этиленом поступает в реактор оксихлорирования 2. Небольшая
часть абгазов (50-100 нм3/ч) после конденсации в холодильнике 6 направят- к я
на сжигание. Количество и содержание СО и СО2в абгазах, а, следовательно, и
общий расход абгазов, определяются условиями процесса и типом исполь iy г
мого катализатора.
Дихлорэтан-сырец из разделительной емкости 5 направляется в емкое 1ь Н.
куда добавляются вода и 40%-ный раствор щелочи для разрушения хлораля, к»
держащегося в продуктах реакции. Водный слой из разделительной емкости 8 на
правляется на колонну закалки 3. После щелочной промывки дихлорэтан из см
кости 8 подается в емкость 9 на водную промывку и затем — на ректификацию
Анализ обеих технологических схем показывает, что «кислородный» про
цесс более рационален по сравнению с «воздушным» вследствие минимиз.1Ц11н
количества абгазов, выводимых из системы и исключением, вследствие этою
узла абсорбции—десорбции дихлорэтана из отходящих газов. «Кислород!ii.iii
процесс также технологически более гибок, так как в нем расход кислорода, а
следовательно, и выдерживание оптимальных соотношений исходных реапи
тов не зависит от расхода инертного газа, которым в «воздушном» процессе ян
ляется азот.
Органический недостаток промышленных процессов оксихлорировапнн
этилена — необходимость создания громоздких узлов закалки, конденсации и
промывки дихлорэтана-сырца. Причиной этого является наличие реакционно!!
воды, что ведет, в конечном счете, к образованию значительного количеч i1ы
сточных вод, для очистки которых необходимо использование специальным
технологических приемов.
Как было показано, стадия оксихлорирования этилена включает два реак
торных узла: вспомогательный — реактор гидрирования и основной — реак к>|
оксихлорирования этилена. Рассмотрим работу этих систем более подробно.
Необходимость установки в схеме реактора гидрирования связана с тем, ч io
ацетилен, содержащийся в хлороводороде, при попадании в реактор оксихло
рирования будет превращаться в побочные хлорорганические продукты
трихлорэтилен и перхлорэтилен по суммарным реакциям:
С2Н2 + ЗНС1 +О2
С2НС13 + 2Н2О;
(4.8 1з)
( 11 I 11 К I I 1/20,- - C.CI, I 311.U. ( I.HIh)
II uiii'HK' них нродукгов в дихлор и.inc сырце ведет к увеличению расход
in>1 mi >ффицмс шов по исходному сырью, а ьн ке к росту >иерго iaiр11 при
। и mi дпхлпр пана tin системе ректификации и, cooibcic iiieinio, к увеличе-
iiiiiii киличес nia хлорорганических отходов.
11ИЫЧШ1 ос га ।очное содержание ацетилена в НС1 после реактора гидрирова
пни н> нревышае г 0,01%, т.е. его конверсия соответствует 94 96%.
Дос ои очно подробная информация о закономерностях реакций гидрирования
ни ннк новых соединений представлена, например, в работе [172]. Мы приведем
Hinn. о< ионные результаты исследований процесса гидрирования ацетилена.
I'i акции 1 идрирования ацетилена и его производных известны более 120 лег
в in iiiii кницею времени занимают важное место в промышленной и цренара
। iHiiiihi химии В крупнотоннажном органическом синтезе основным методом
ШН1НГ к и Kaiадитическое гидрирование, в малотоннажной химии биологически
>н 11П1ПЫХ и душистых веществ, а также в синтетической органической химии
и ноль iyioi и другие методы восстановления тройной связи.
( иптел пилена гидрированием ацетилена в промышленных условиях ис-
пои, ювали в 1ермании во время Второй мировой войны как основной исгоч
ши пилена. Этот процесс исследовали также в начале 1950-х годов в Японии па
пплынои пилотной установке [368]. В настоящее время селективное гидрирова
ипс лце 1 плена до этилена и ацетиленовых углеводородов С3 и С4 до олефинов и
<1н поп представляет большой интерес как метод очистки газов пиролиза угле-
водородов (пирогаза и фракции С2, С3 и С4, синтез-газа, коксового газа) [650|.
11 и реакции гидрирования (гидроочистки) в промышленных процессах прово-
пи в 1 а ювой фазе на гетерогенных катализаторах.
Во всех реакциях гидрирования тройной связи основные проблемы связаны
пнычпо не с активностью катализаторов, а с селективностью. В случае ацетиле
пи yi гаповлено несколько превращений на разных металлах (см. схему):
н2 н2
-^С2Н4^С2Н6
2Н2
— С2Н6
QH2---Зн,
2Н, Н2 Н2 _ т,
-* с4н6 с4н8 —- с4н10
С4Н8 —С4Н10
(4.85)
(4.86)
(4.87)
(4.88)
>С4 (олиго- и полимеры, продукты
реакции гидрополимеризации)
СН4 и С
При 300 °C на Ni образуется его карбид (Ni3C). Установлено, что помимо пос-
ледовательных реакций гидрирования С2Н2 в С2Н6, С4Н6 и С4Н8 до С4Н|0 есть
и независимые маршруты образования С2Нб (4.86), С4Н6 (4.87) и С4Н8 (4.88) из
ацетилена, играющие важную роль в проблеме селективности [651].
Реакции газофазного гидрирования ацетилена проводят на металлических
п оксидных катализаторах. Изучены разные формы металлических катализато
ни 1 и полное покры i не 1 loiiepxhoi i и ацс i пленом вило) ь до очень малою » од» р
жапия ацетилена в га ie
Установлены га к же факторы, влияющие на «собственную» (.електнншн и
i.e. па изменение соотношений констант скоростей стадий, ведущих к «юра ю
нацию этана и этилена из ацетилена. Основным вредным фактором, влияющим
на этот параметр, является, как полагают [655, 658], гидрид палладия (|< I'dlI)
Установлено, что симбатно образованию P~PdH растет скорость и надаг > 11
лективность гидрирования ацетилена. Таким образом, количество гидрнди и
факторы, способствующие его образованию, контролируют собственную и
лективность процесса. Судя по всему, гидридные формы палладия учат iny ко и
эбразовании из С2Н2 интермедиатов, ведущих к этану.
Показано, что отложения углерода и полимеров способствуют увеличс пню
количества PdH. Добавки О2 повышают селективность гидрирования ia inri
ркисления гидридного водорода и углерода, а увеличение рН2 приводи! к увели
нению фазы 0-PdH. Очень существенное обстоятельство обнаружено при и iy
дении степени дисперсности палладиевых частиц [658]. Так, в случае частиц Pd
с диаметром < 0,13 нм селективность гидрирования ацетилена достигает 100'hi,
1 на частицах 0,40 нм — всего лишь 50% [659]. На малых частицах велико даплг
ние диссоциации гидрида, что резко уменьшает его количество, поэтому можно
зредположить, что с уменьшением (З-PdH понижается и скорость гидриронл
1ия этилена.
Таким образом, в условиях процесса гидрирования ацетилена, содержа щг
ося в НС1, до 80% прореагировавшего ацетилена превращается в этилен и око
ю 20% — в этан.
В промышленных условиях обычно используют реакторы высотой 5,5 м
л диаметром 3,0-3,2 м. Катализатор — PdO/Al2O3 (0,2 мае. % Pd) — предвари
гельно активируют водородом. Срок службы катализатора составляет пя гь но
:емь лет. Потерявший активность катализатор перед выгрузкой пассивирую!
шотом во избежание самовоспламенения при контакте с воздухом.
Рассмотрим более подробно работу ключевого аппарата стадии — реакпшри
эксихлорирования этилена. Один из вариантов конструкции реактора предг i ан
1ен на рис. 4.58. Основными элементами реактора являются распределительное
устройство для подачи исходной смеси газов в псевдоожиженный слой катали
штора, теплообменный змеевик, предназначенный для отвода тепла реакции н
эатарея циклонов, функция которых состоит в улавливании и возврате в ре
!ктор частиц катализатора, унесенных газовым потоком из псевдоожижен шло
:лоя.
Конструкция и параметры распределительных устройств играют важней
пую роль в обеспечении оптимального протекания процесса оксихлориронп
лия этилена.
Формирование псевдоожиженного слоя и диспергирование ожижающею
!гента происходит в зоне, непосредственно примыкающей к распределитель
юму устройству (активная зона). Именно здесь получают развитие химичес
сие реакции, а также процессы тепло- и массообмена, поэтому эффективно». 1i.
ехнологического процесса в целом во многом зависит от конструкции распре
[елительного устройства, определяющей гидродинамическую обстановку в а к
ивной зоне [610].
|,'МКЦИ<>П11ЫИ 1.1 I
Рис. 4.58. Реактор окислительного хлорирования этилена
1 IpiiMeiiincniaio к реак юру океихлорирования >1 плена ус лопнем перемени!
ия исходных рс.псн।он с помощью распределительного устройства дол кн<>
яться полное исключение образования градиента концентраций, который
[<ег привести к агломерации частиц катализатора (при локальном xi.ioi.iii>«
роводорода), а также к повышению взрывоопасности, благодаря наличию и
(темах горючих компонентов (этилена) и окислителя (кислорода).
Детальное рассмотрение конструктивных особен нос гей распредели) ел ьпых
ройств не входит в задачу настоящей книги. Подробная информация подан
iy вопросу приведена, например, в [610]. Мы рассмотрим лишь два iiapii.ni
конструкции распределительных устройств в реакторах оксихлориронанпн
депа, получивших достаточно широкое распределение в промышленное in
1 la рис. 4.59 показана схема распределительного устройства, разработан нив
(ианией «Badger» (США). Реакторы с таким распределительным устрош i ном
тлуатируются на ряде установок и в России (например, в ОАО «Саяжкхнм
|ст»).
О.Р.
Рис. 4.59. Схема распределительного устройства «Badger» (США):
ТЭ — теплообменные элементы; П. — патрубки; О.Р — опорная решетка
I'.к предел тельное yr i potic i но включае! вогнуiу io рас предел и гельву к> гарел-
> > < вваренными i низу н.п рубками, сплушеиными 1 шоским и донышками с калиб-
рованными отверстиями диаметром 11+0,1 мм для подачи воздуха («воздушный»
процесс) или циркуляционною газа в смеси с этиленом («кислородный» процесс).
II ><1 распределительной тарелкой расположено распределительное устройство
щи подачи хлороводорода и этилена/кислорода в «воздушном»/«кислородном»
процессе, сос гоящее из центральной трубы, от которой отходят горизонтальные
руны с опушенными вниз вертикальными патрубками. Смешение газов проис
Поли । в кольцевом зазоре, образованном внутренней стенкой смесительного пат-
р» пка распределительной тарелки и наружной стенкой вертикального устройства
на iu еп высоте патрубка распределительной тарелки.
Распределительное устройство такой конструкции обеспечивает достаточ-
по высокую эффективность смешения реагентов как бы сепаратно в каждом
।> । ц 1ыю взя том элементе «стакан-патрубок». Поскольку в момент смешения
р< at еп । ы еще не вступают в химическую реакцию, их концентрация в смеси on-
pt (гллегся исходным соотношением. Концентрация кислорода в данной смеси
. о< 1авляег 9-14 об. %, этилена — 18-25 об. %. Такая смесь потенциально взры-
воопасна. Ограниченный объем каждого из смесительных стаканов препятс-
। нуг । возможному распределению пламени.
как показывает опыт эксплуатации распределительных устройств такой
конструкции, при длительной работе и высоких скоростях газа происходит
ipniiui патрубков за счет абразивных свойств частиц катализатора. По этой
причине современные распределительные устройства не содержат опускных
1ьи рубков, и смешение реагентов происходит в иных условиях. Достаточно ха
р п крна конструкция распределительного устройства, предложенная [660,661]
I pin I 60), авторы которой утверждают, что эрозия конструктивных элементов,
। i.iK ке истирание частиц катализатора происходит в существенно меньшей
। пени, когда по трубам, в которые входят сопла, пропускают газовый поток
в <н повпом против потока газа, который поддерживает катализатор в псевдо-
п 1<П 14 ином состоянии.
Данное распределительное устройство содержит нижний ограничитель 2
и । । и с сидоожиженного слоя катализатора 3, распределительную трубу 4, распо-
>||| iuиную выше ограничителя 2 и внутри псевдоожиженного слоя 3 катализа-
iiip.i, с соплами 5, распределенными по всему поперечному сечению реактора.
( пила 5 входят в трубы 6, по которым подводят выходящий поток газа, в основ-
ном в противотоке с потоком газа, который создает псевдоожиженный слой ка-
। i hi штора. Устройство также содержит трубопровод 7, расположенный ниже
in рапичителя 2.
Vt. । ройство выполнено таким образом, что число труб 8 равно числу труб 6,
равномерно распределенных по всему поперечному сечению реактора 1. Бла-
| пдиря соосности каждой из труб 8 и 6 обеспечивается соответствие количеств
pi in ирующих газов из труб 8 и 6.
11ространство между верхними концами проходящих через ограничитель 2
руб Я и нижними концами труб 6, в которые входят сопла 5, образует зону сме-
шивания, имеющую размеры, при которых уже происходит смешение каждого
и । выходящих из этих труб компонентов реакции с катализатором, причем ис-
। пичае гея зона смешивания от 25 до 300 мм.
Рис. 4.60. Конструкция распределительного устройства
(1-9 см. текст; 10 — теплообменник)
Сопла 9 располагаются на таком расстоянии от верхних концов труб 8, что
направленные вверх струи газа из сопел 9 до верхнего конца трубы 8 равномер
но распределяются по соответствующему поперечному сечению труб 8.
Пространство между верхними концами труб 8 и нижними их концами 6
имеет такой размер, что отсутствует эрозия труб 4, 6, 8, а также нижнего огра
ничителя 2.
В другом варианте выполнения устройства такое же количество труб 8 и 6
расположено со взаимным смещением одних относительно других. Такая гео
чн i рпн обусловливает более низкую iponno ipyt» 6 олаюдаря восходящему по
ня. v । .1 i.i и i i руб <4. С помощью i a koi о p.k поло копия дос । ш acre я немедленный
п in посредс i пениып кон такт с катали i.пором реагентов, выходящих и i i ру(> 8
и п п шепдоо киженпый слой катали затора 3.
Необходимо огмегшь, что использование распредели тельных устройств
hihoii конструкции возможно лишь при условии эксплуатации катализаторов,
ш।шоликицих работать при соотношениях О?/НС1 — 0,52-^0,54/2 (мол), гак как
инн.ко в этом случае концентрация кислорода в газовой смеси не будет превы
пы । ь 8,5 9 об. %., что ниже порога взрываемости.
II i вязи с 31 им представляется важным рассмотреть данные по взрывоопас-
ным i пойс гнам реакционной смеси.
< о щание взрывоопасных условий в реакторе может произойти, во-первых,
। • лучае плохого перемешивания компонентов; во-вторых, при больших ено
нодных объемах, заполняемых реагирующими компонентами; в-третьих, при
наличии источников инициирования (поджигания) горючих газовых смесей.
II ipi.iiiooiiacnocTb узла оксихлорирования определяется тем, что после сме
ш< пня потоков, содержащих основные количества этилена и кислорода соог
in о ни нно, образующаяся смесь расчетного состава хотя и не взрывчата, по
пли ша к пределу взрываемости. Отклонения от расчетного состава могут при
води । ь к образованию взрывоопасных смесей.
II |рынобезопасность реактора оксихлорирования достигается за счет качесг
in iiiiiiiо аппаратурного оформления и создания псевдоожиженного слоя катали
। пора, играющего роль огнепреградителя в случае поджигания газовой смеси.
| ьцыко под слоем катализатора возникает зона свободного пространства, в ко
lopoii возможно горение взрывчатой смеси, а также его инициирование разря
1.1МИ статического электричества между витающими частицами катализатора.
(>насные пустотные зоны, заполненные взрывчатой газовой смесью, могут
пн шикнуть в газоходах подачи потоков, содержащих как кислород, так и эти
к и п случае отклонений расходов компонентов от заданных значений.
Для профилактики возникновения взрывоопасных смесей:
расходы кислородсодержащего и этиленсодержащего потоков должны
iii.i । ь кестко сбалансированы, чтобы колебания расходов одного из потоков не
м« длинно и точно компенсировались соответствующим изменением расходов и
'I’iii iiioporo потока;
необходим непрерывный контроль изменения обоих параметров, так как
при произвольном (под влиянием статической электризации) загорании смеси
и < походном пространстве реактора под слоем катализатора следует ожидать в
in м i । ационарного пламени. Его появление приведет к соответствующему разо
। реву с гонок и изменению гидравлического сопротивления реактора в целом.
( меси этилен-кислород и этилен-воздух взрывоопасны в широком диа
н । инн* концентраций этилена. Ниже приведены концентрационные пределы
и 1рынаемости двойных смесей при 20-22 °C и атмосферном давлении:
1 pi да .............Воздух Кислород
Ни кпнй предел, %.....2,80 2,80
Игрхиии предел, %.... 34,40 76,30
(’ ростом ivMiicp.ii уры и давления обла<п> взрываемое 1 и концентрации
расширяется. При .имоеферном давлении и температуре 200 "С нижнин пре
дел изрываемое।п смеси этилена с кислородом составляет 2,75%; при тон кг
темпера! уре и давлении 0,6 МПа этот предел снижается до 2,5%, а при I МПа
до 2,2%.
Зависимости верхнего предела взрываемости смесей этилен-кислород н
этилен-воздух от давления и температуры представлены на рис. 4.61.
Для предотвращения воспламенения смесь этилен-кислород и этилен во i
дух может быть разбавлена такими инертными компонентами как СО2, На»
N2, НО и т.д.
Графическая зависимость пределов взрываемости смесей этилена с впаду
хом от содержания СО2, N2, НО при атмосферном давлении и температуре 20
22 °C представлена на рис. 4.62.
Область составов, отвечающих горючим смесям, ограничена соответствую
щей критической кривой и осью ординат. Например, содержащая СО2~41% или
’ис. 4.61. Верхний предел взрываемости смеси этилена с кислородом (а, б) и воздухом
в), в зависимости от давления и температуры
35
30
25
20-
е 15
10
5
о 10 20 30 40 50
С, %
I'm I 62. Пределы взрываемости смесей этилен-воздух с хлороводородом (/), айном
и дномидом углерода (3)
I >()"< смесь этилен-воздух становится невзрывоопасной при любом соотно
...ни между горючим и окислителем.
Кри । ическое содержание кислорода в бедных смесях этилена с кислородом
и iiiicpiiibiM компонентом при атмосферном давлении и комнатной темпера
И ре ин гавляет 11,7% для СО2, 10% — для N2 (HCI). С ростом температуры и
laii'ii пня область поджигания смесей этилен—кислород—инертный компоиеи i
р и пи|ряегея.
При 300 °C и давлении 1,0 МПа критическое содержание кислорода в бед
пых ।месях этилен-кислород-азот ~7%.
Кри ।ическое содержание кислорода в богатой смеси С2Н4+О2+СО2:
I Ml la .......................... ...0,1
I* * kpu ПРИ содержании СО2, %:
II ..................................20
|п ..................
Ш ....................................-
0,5 1,0 1,5
15,7 11,7 10,3
16,0 13,6 12,1
14,5 12,5 12,1
I la рис. 4.63 представлена зависимость пределов взрываемости смесей
I 111 I ()> + N2 от давления. Повышение давления расширяет пределы взрыва
« М(н I п
На рис. 4.64 представлены сравнительные концентрационные пределы
к |рынаемости смесей этилен-кислород-азот в свободном объеме и в псевдо
< г и кепиом слое, полученные авторами работ [662, 663].
। t и (оставление кривых показывает, что взрывобезопасная область коп
>н и (раций реагентов в псевдоожиженном слое значительно больше, чем в
• 1К111ПД11ОМ объеме. Согласно [663] расширение взрывобезопасной области в
’nc. 4.63. Зависимости пределов взрываемости смесей С2Н4—О2—N2 от давлении и
одержания азота при 20 °C
'ис. 4.64. Концентрационные пределы взрываемости этилен-кислород-азотной смеси
ри 0,5 МПа, в свободном объеме (я) и в псевдоожиженном слое (б)
севдоожиженном слое обусловлено затруднением горения реакционной смет и
следствие отвода тепла от фронта пламени на нагрев частиц.
Ниже приведены усредненные составы газовых смесей на входе в реактор и
а выходе из него (после конденсации дихлорэтана и воды) для «воздушною»
роцесса, об. %:
< )мк ли (ели, горючие ьещес тиа Вход Выход
о2 К) II 1 7
С211.1 Г. 18 0,5 1,5
н2 0,5 1.0 -
СЫ.1 0,5-0,98
со - 0,5 0,7
С2Н, 0,01
Инерты:
НС1 28-33 -
N, 40-45 88-90
со. - 1,0 1,5
В работе [663] указано, что по условию материального баланса реактора
концен грация кислорода в абгазах С*к равна:
| _ Ск ~ 5(^нс1^на 0.5 + Ук (1 - S3); S3 ] (4 89)
1 —CHci—О’^Сна^нс! l,5 + (vK _ОО'$э), $э]
1 де Хц( । — конверсия НС1; Сна — исходная концентрация НС1; Ск — исходная
концентрация кислорода; С° — исходная концентрация этилена; Sa — селек
। iiiiiiocib процесса по этилену; vK= 2,7^-2,9 — стехиометрический коэффициеп i
р.ь ходования кислорода в реакциях горения, отвечающий соотношению кои
цен граций О2:СО = (Зд-9):1.
Решая уравнение (4.84) относительно С*к при Хна = 0,97, $э = 0,95 и прини
мая С'на/Ск - 1,9, имеем:
с£ =С°(о,6О-3,ЗСк) + Ск . (4.89а)
11ачальные концентрации кислорода и этилена, отвечающие допустимым
шачепиям Ск, определяются по диаграмме (рис. 4.65), из которой видно, что
для смесей с 3 < Ск S 9 об. %, выполняется и условие взрывобезопасное ги и
п< ендоожиженном слое.
11оказатели процесса оксихлорирования этилена в значительной мере онре
теляются гидродинамикой псевдоожиженного слоя. В гидродинамических рас-
четах наиболее часто используют двухфазную модель, согласно которой часть
। a ia проходит через слой со скоростью, близкой к скорости начала псевдоожи
ы-ния (плотная фаза). Остальной газ поднимается через слой в виде пузырей
(пузырьковая фаза). Реакция протекает в плотной фазе, а между фазами осу-
ществляется массообмен, интенсивность которого определяет конечную сге
нет* превращения реагентов.
Уравнения двухфазной модели для хлороводорода имеют вид:
(4.90а)
с. 4.65. Начальные концентрации этилена и кислорода, отвечающие фиксировании
содержанию, об. %., кислорода в абгазах (числа у кривых)
^ = _₽_(x_y)+JL. (|-и>.,
di 1-g l-q
Начальные условия: х = у - 0, при т = О
хк- qx + (l-q)y, при т - тк,
1 у, х — концентрации НС1 в пузырьковой и плотной фазе с соответстнеино
- текущее время (с), изменяется от 0 до тк — времени контакта; q — доля i .111
оходящего через плотную фазу (обычно 0,01-0,03); |3 — коэффициент мас< о
мена; с '; хк — конечная степень превращения НС1; ц — суммарная скорей п<
разования или расходования i-ro компонента.
Интенсивность тепло- и массообмена в псевдоожиженном слое в значится!,
й степени определяется скоростью потока газов, величина которой, в енот
ередь, лимитируется скоростью начала псевдоожижения. Скорость нами 11
евдоожижения (м/с) может быть определена, например, по формуле М Л< н i
1]:
"о=О,ОО923 -|^
\0.94
Тэ |
; d3 — эквивалентный диаметр частиц катализатора (м); v — кинетическ.ш
зкость (м2/с); у — плотность ожижающего агента; уэ — эффективная кажущ.1
я плотность твердого материала.
Для промышленных катализаторов, производимых фирмами «Siid-Cheinie
talysts», «Engelhard», эксплуатируемых в 70-80% промышленных процесиш
сихлорирования этилена в псевдоожиженном слое, характерны размеры ч.ц
ц 20-100 мкм и насыпная плотность 1,00-1,10 г/см3.
Экшпылепiцып диаметр частиц ранги для них к.иалп ыюрои 15—50 мкм.
II >।ом случае в зависимости or природы ожижающего агента (воздух или цир-
куляционный газ, отличающиеся один or другого плотностью за счет высокого
i одержания СО, — 50-60% в газе рецикла) скорость начала псевдоожижения Wo
раина 0,0035-0 0050 м/с.
Исходя из предельного значения числа псевдоожижения:
w _ wo _ 1400+5,22л/Аг
Wq 18 + 0, 617Аг
(4.92)
ранного для ламинарного режима 1400/18 - 77,7, получаем максимальное значе-
ние скорости газового потока в промышленном реакторе: 0,27-0,39 м/с в зави
и мости от природы ожижающего агента.
Увеличение линейной скорости потока в промышленном реакторе благо-
приятно сказывается на интенсивности тепло- и массопередачи1:
V. м/с .. .0,26 0,29 0,30 0,38 0,40
1'.с' .. .0,22 0,25 0,27 0,39 0,41
1>. Вг/(м2- К)... ...186 204 222 273 294
где h — коэффициент теплоотвода.
Таким образом, увеличение линейной скорости, т.е. числа псевдоожижения,
существенно интенсифицирует перемешивание твердого материала. Это под-
1верждается зависимостями [610]:
W.......... 1,09 1,45 2,5 3,12
II ........ 0,26 0,60 0,745 0,96
1де W — число псевдоожижения, П — степень перемешивания твердого мате-
риала.
Поскольку W для промышленного реактора оксихлорирования этилена су-
щественно превышает приведенные величины, можно считать промышленный
реактор системой идеального смешения по твердому материалу, а также по теп-
лу, что подтверждается изотермичностью слоя.
Более сложная картина наблюдается при выборе модели процесса по газо-
вому потоку. Рост скорости газа способствует значительному увеличению ско
рости обратного перемешивания [665]. Увеличение диаметра частиц катализа-
юра ведет, в свою очередь, к уменьшению перемешивания газового потока. Для
расчета степени перемешивания газового потока применительно к условиям
промышленного реактора оксихлорирования этилена может быть использова
па формула И.Я. Тюряева [666]:
s = l + 7,5(H0/Do)d32’85
(4.93)
ще $ — степень перемешивания, изменяющаяся от 1 (полное перемешивание)
до оо (отсутствие перемешивания); Но — высота слоя; Da — диаметр аппарата;
</, — эквивалентный диаметр частиц катализатора, мм.
1 Данные получены В А. Махлиным
В |595| ука мио, что применительно к промышленному реак гору диаме i
ром 3,3 м и Ш.1СОТОЙ псевдоожиженного слоя катализатора ~14 м crenciii.
перемешивания газового потока достаточно велика. Однако наличие в реак
торах такого типа геплосьемных элементов, занимающих 15-20% сечении,
играет существенную организующую роль и снижает продольное перемс
шивание. Это дает основание считать промышленный реактор (см. рис, I. »Н)
аппаратом, приближающимся к режиму идеального вытеснения по газовому
потоку.
Важнейшим ограничительным моментом работы промышленных реакiо
ров оксихлорирования этилена является проблема теплоотвода. Для псевдо
ожиженных систем в целом характерна высокая интенсивность переноса тепла
о г слоя к поверхности теплообмена. Благодаря высоким значениям коэффн
циента теплоотдачи от слоя к поверхности практически весь темперагурныН
напор сосредоточен в непосредственной близости к поверхности тсплоон
мена [610]. По этой причине разность температур между псевдоожиженным
слоем и поверхностью теплообмена практически равна перепаду темпера! у
ры в «пограничной пленке», примыкающей к поверхности темплообмена 1а
пределами этой «пленки» и участка стабилизации у газораспределигелыкш
решетки псевдоожиженный слой вследствие интенсивного перемешивании
твердой фазы представляет практически изотермическую систему даже при
значительных габаритах.
В силу высокой экзотермичности процесса оксихлорирования этилена
(с учетом реакций глубокого окисления) адиабатический разогрев при начал!,
ной концентрации хлороводорода, равной 28 мол. %, составляет 1400 °C. >ю
указывает на необходимость максимально возможного увеличения поверх
ности охлаждающих элементов. Так, например, для промышленного реакtop,i
диаметром 3,5 м с высотой псевдоожиженного слоя катализатора ~14 м попер
хность теплообмена составляет 840 м2, т.е. удельная поверхность теплообме
на расширенного слоя составляет 11,33 м2/м3кат. Как указано В.А. Махлипым,
это практически предельная величина для реакторов псевдоожиженного слоя,
один из вариантов которого представлен на рис. 4.58.
Коэффициент теплопередачи существенно зависит от качества псевдоо кп
жения и меняется в пределах 2090-2492 кДж/(м2-ч-°С) при отличном псевдо
ожижении до 627-836 кДж/(м2-ч-°С) при плохом. При использовании в процес с <•
низкотемпературных катализаторов оксихлорирования (210-225 °C) разнос и.
температур катализатора и хладоагента 40-45 °C. Этому соответствует коэффн
циент теплопередачи 1630-1760 кДж/(м2-ч-°С).
Производительность промышленных катализаторов оксихлорирования i и
лена, определенная на основе кинетических данных, составляет 1,1-1,4 кг ДХ.)/
(г кат. ч). Однако в реальных условиях промышленной эксплуатации низко гем
пературных катализаторов съем дихлорэтана не превышает 350 г/(г кат. ч), г.е
степень использования катализатора в промышленных условиях находился н
пределах 20-25%.
Иллюстрацией этого тезиса может служить опыт промышленной эксплуа
тации реактора оксихлорирования этилена диаметром 3,5 м и высотой псевдо
ожиженного слоя катализатора -13,7 м. Поверхность теплообмена в реак горе
составляет 840 м2. При этом доля сечения, занятая элементами теплосъема, не
up. iii.ini.ir । 20%. В pe.iKiop з,п ружае н я (>() i клали i.irop.i, мощное n> реакгора
|i. i Д.Х > в чис (~270 г/(л каг. ч). (ooiiich i вукнцпеданной производиie;n>iioi in
p«< поды реагентов (пм /ч):
ll|<i»ii<i .. «Воздушный»
• и>11
। |<||>о|>одорода ....7315
oil'll и.। ....3750
пн по x.i .........12 200
ЫН пород,) . -
pi lunoiiiiioio газа ...
«Кислородный»
7315
3750
2200
8000-10 000
При iai<nx параметрах работы реактора обеспечивается необходимое со
и иг и । вне чисел псевдоожижения (60-70) и коэффициента массообмепа
(|< п.25 i ') количеству тепла, отводимого из системы. Увеличение теплового
iiaiKip.i (At между псевдоожиженным слоем и хладоагентом) возможно либо за
. >н । повышения температуры процесса, либо за счет снижения температуры
Iпадоа! епта. Зависимость температуры хладоагента от нагрузки по хлороводо
роду для реактора мощностью 16 т ДХЭ в час представлена на рис. 4.66. Снижу
ни< । ем пера гуры хладоагента ниже 167 °C является нежелательным, вследствие
пн (можносги конденсации паров воды, что приведет к коррозионному разру-
шению внутренних элементов реактора. Как показано в [668], максимальная
i.-Miiepai ура точки росы при заданных концентрации хлороводорода и давле
пин i in гемы составляет 118 °C. На рис. 4.67. представлена зависимость крити-
4KIUHI влажности газообразного хлороводорода от температуры для разных
luiiii 1рукционных материалов [668]. Для каждого материала существует ми-
нимальная температура, превышающая точку росы на величину так называ-
। мин «коррозийной депрессии», выше которой он может быть использован в
процессе. Для углеродистой стали — основного материала, из которого изго
ншлепы практически все реакторы низкотемпературного оксихлорирования
пилена, коррозийная депрессия составляет ~50 °C, что и определяет нижний
iinpni температуры хладоагента.
Рис. 4.66. Зависимость температуры хладоагента от нагрузки по HCI
336
Рис. 4.67. Зависимость критической влажности газообразного хлороводорода о I 1 <-м 11<
ратуры (В — влажность НО, соответствующая точке росы)
Увеличение А/ за счет повышения температуры процесса для ниакотемш
ратурных катализаторов также нежелательно, так как ведет к снижению ы ы
тивности образования дихлорэтана и увеличению выхода побочных продук ши
глубокого окисления.
Таким образом, промышленным процессам оксихлорирования ни к hi
свойственна «разбалансированность» между кинетической производителю u и
тью катализаторов и реально существующими возможностями теплое ъема
Резервы увеличения производительности реактора могут заключагы и и
использовании менее активных, но более селективных катализаторов. Имев
но по этому пути в 1990-2000-х годах пошли ведущие фирмы-произноднн ш
винилхлорида. Это позволяет использовать для проведения процесса о<>/к<
высокие температуры (240-250 °C) при сохраняющемся низком выходе про
дуктов глубокого окисления. Нагрузка по исходным реагентам может in.ni
увеличена, дополнительно выделяющееся тепло снимается за счет увелн'и
ния А/.
Одним из условий увеличения производительности должно являться ик 1
использование катализаторов с более высоким значением эквивалентного ди i
метра частиц. С учетом того, что скорость начала псевдоожижения пропорци
ональна квадрату диаметра частиц катализатора, увеличение d;, с 50 до 100 мкм
ведет к росту Wp с 0,35 до 1,3 см/с. В этом случае линейная скорость газово! о но
тока может быть увеличена до 0,7-0,8 м/с. (число псевдоожижения равно ’(> till
что вписывается в пределы существования псевдоожиженного слоя).
Увеличение линейной скорости газового потока приводит, естественно к
увеличению уноса катализатора из псевдоожиженного слоя. Механизм явлении
уноса подробно рассмотрен, например, в [610]. Мы остановимся лишь на <н
новных аспектах этого явления применительно к реакторам оксихлорированни
этилена.
Иышн твердого материала происходи! co свободной новерхпос i в псевдо
i(4 ii i<i iiiioi о слоя, причем ин геж и внос и. уши .1 находи гея в прямой спя ш с ко
ш и > том мелочи вбли ш ной новерхпос in |611,6б9|. В ходе псевдоожижения
ирит ходш сепарация частиц по высоте слоя в 1ависимосги ог их ра тмеров, в
р. цлы.не ко трои более мелкие частицы накапливаются преимучцесгвеино в
рхнпх, <1 оолее крупные — в нижних зонах слоя. Минимизация продольного
pi mi inniiai 1ия способствует сепарации и. следовательно, уносу мелких час гиц.
Ни uiaiiii, что в промышленных реакторах оксихлорирования этилена полно
. и.io перемешивается твердая фаза, это способствует выравниванию размерен»
। « inn, выносимых из слоя и, таким образом, уменьшению общего количества
ши HMOIO к а i-али затора.
Bi.inoi частиц катализатора из псевдоожиженного слоя существенно тави
hi <н ра шости со - шв, где со — скорость ожижающего агента, со,, — скороегь
Н1П.111НЯ частиц, т.е. для частиц данного размера — от скорости ожижающего
du hi а 11ри использовании в процессе полидисперсных катализаторов с час ги
памп диаметром 20-100 мкм, т.е. для ламинарного режима предельный диаметр
। в г иц, уносимых из слоя, согласно [610]:
Д =4,243 < соц/ут , (4.91)
। а <п скорость ожижающего агента; ц — динамическая вязкость ожижающе-
।.. ai < п ia, Ут — кажущийся удельный вес катализатора.
Для промышленного полидисперсного катализатора оксихлорировапия
ниж на, имеющего следующее распределение частиц по размерам: <20 мкм —
H,,i< <0 мкм —15%, <45 мкм —50%, <80 мкм — 90%, <100 мкм — 98% и иредель-
IIIhi линейной скорости газового потока в реакторе 0,4 м/с, величина d' равна
и '» мкм в зависимости от состава ожижающего агента.
Ка иную роль в обеспечении оптимальной работы реактора оксихлори
рц|ыпия этилена играет высота сепарационного пространства. Эта величина
in । кпа превышать некоторое критическое значение, являющееся, в свою оче
р> щ, функцией диаметра аппарата и линейной скорости газового потока. 11а
piii I (>8 представлены зависимости, позволяющие определить критическую
вьюн у сепарационного пространства для реакторов разного диаметра |610|.
Inn реактора диаметром 3 м при линейной скорости потока газов 0,3 м/с
11, 1,5 м. Высота сепарационного пространства в промышленных реакторах
iдвляет 10-12 м. Работа в таких условиях приводит к тому, что в циклоп
и»и> систему попадают в основном частицы размером не более 50 мкм. Эф
(|нч< iявность работы циклонов для частиц размером выше 30 мкм превышав!
г1) "о, для частиц размером 10-30 мкм составляет 95% и для частиц размером
мгпее 10 мкм равна 65-70%. Усредненный фракционный состав катализа го
р । унесенного из промышленного реактора: <10 мкм — 80%, <20 мкм — 95%,
10 мкм — 99%.
И июженное указывает на значительную роль величины потерь катали та
юра при истирании (механической прочности). Современные катализаторы
имеют величину механической прочности на уровне 96-98%, что обеспечива
< । расход катализатора не выше 10 т/год при загрузке в реактор 60 т катали
киора.
Ниже представлен гранулометрический состав, мае. %, одного из промыт
ленных катализаторов — свежего, а также после 0,5 (А) и 1 (Б) года эксплуаы
ции:
Катализатор.. ..Свежий А Б
>125 мкм .... 0,7 0,5 0,5
125-90 мкм... ... 4,2 2,3 2,9
90-63 мкм.... ....18,3 16,5 3,1
63-40 мкм.... ....45,9 50,0 41,9
40-20 мкм.... ....28,1 30,6 40,3
<20 мкм ... 2,8 0,1 11,3
Таким образом, в ходе эксплуатации происходит некоторое перераспределе
ние размеров частиц катализатора в сторону преимущественного содержания
фракций размером 20-60 мкм. Периодическая подгрузка свежего катализатора
делает состав катализатора в реакторе в среднем постоянным. Средневзвешсн
ное время пребывания частицы катализатора в реакторе превышает два года
без какого-либо снижения активности и селективности. Содержание меди в ка
тализаторе практически не изменяется.
В табл. 4.17 представлены типичные данные по работе реакторов оксихлори
эования этилена в псевдоожиженном слое катализатора, эксплуатирующихся н
промышленных условиях разными фирмами. Данные приведены для «воздуш
I «'I illllll I I . I lllh.l 1.НСЛ11 НроМЫШЛСПНЫХ 11|>ОЦ1ЧИ>11 Uhl llXHOplipOll.lllllll HIUIOI.I II IKl'Il
' i ' < ином слое h.ii.iiiu i.пора
Пока ijiciiii процесса Кислород ВолдуX
А Б В Г А Б
i i. । дпчлпр >1,111,1, । ДХ.)/|-кат’ч 0,253 0,739 0,663 0,175 0,694 0,349
11| и НИ1Д11 iciii.ikk 11. по ДХЭ, г/ч 13,92 26,6 39,49 10,52 25,0 -
• и Ikil 1м1ТН/1И.1.чК>ра, т 55,0 36,0 55,0 60,0 36,0 75,3
Il Ч11О1 II. М1ПЯ1ЦС1О слоя, кг/м3 0,577 0,540 0,516 0.516 0,540 0,655
III . .О II 1 >11111, М 13,70 10,6 12,54 14,2 10,6 11,7
К, В’Д । .1 кш. им /ч: IK 1 6317 12209 16500 4828 11472 12056
И, 3180 6147 8310 2375 5902 6250
< । 1700 3277 4220 1211 - -
lloVQX - - - - 18270 19933
I', 1П1Ю1О11ЫЙ газ 10205 8000 18000 12342 - -
< >1>|ЦПП 21402 29633 47030 20757 35647 38239
l> win pal ура, "С 221 255 240 210 255 234
1.Ill'll line, Mild 5,06 3,80 5,2 2,50 3,80 3,65
llpi мн контакта, c 44,84 19,5 22,6 29,6 16,2 27,1
< ooiiitiouieuue реагентов tn хооном сырье: ll< l/l II, мол. 1,986 1,986 1,986 2,033 1,944 1,929
II 1 .11 |, мол. 0,535 0,533 0,508 0,510 0,650 0,670
Ul.-<i im, об. %: < II,. Oil % 4,92 5,20 3,6 3,50 0,55 0,69
( ( 1, oil. % 2,05 3,50 4,0 2,40 0,55 0,66
< < • . Ot’l. % 43,62 10,30 43,0 46,00 1,30 1,54
i» i Ar, об. % 1,01 1,20 1,3 0,45 4,15 4,73
Пока шмели качества: 'liii юга дихлорэтана, мол. % 99,38 99,35 99,44 99,67 99,30 99,10
i II, к|>ф. начисляй ДХЭ, мол. % 98,43 98,07 98,76 97,61 95,66 94,454
|ми(Персия НС1, % 100,00 99,30 99,70 99,20 98,82
Коннерсия С2Н4, % 99,81 99,47 99,91 99,74 98,59 98,15
ll/'HWiaHUe: А, Б, В, Г — фирмы
340 1
пых» и «кислородных» Процессов при Ис ноль loii.iiiini низко п пысокогсмнерл
гурпых кашли ыюрой.
Расход сырьеиых компонентой и катализатора при оксихлорировании ни
лена (кг/гДХЭ): л илена — 287,3; хлороводорода — 745,0; кислорода — 177.0,
катализа гора — 0,10.
Оксихлорирование этилена в неподвижном слое катализатора
Ведущими фирмами, эксплуатирующими процесс оксихлорирования этиле
на в неподвижном слое катализатора, являются «Stauffer», «Dow», «Shell». Ли
цензии на эти процессы проданы более чем 20 компаниям в разных странах
Впервые процесс оксихлорирования этилена в неподвижном слое был осущес i
влен компанией «Dow» в 1960-х годах во Фрипорте и в Плакмине (США) |67О|
Другие компании, использующие процесс в неподвижном слое катализатора, нс
вносят большого вклада в мировое производство дихлорэтана и винилхлорида
Число публикаций о технологических аспектах данного процесса очень незна
чительно. По этой причине мы приводим лишь основные закономерности про
цесса фирмы «Stauffer», опубликованные в работе [199].
Оборудование
Процесс оксихлорирования этилена в неподвижном слое катализатора обыч
но многостадийный. Это облегчает управление сильноэкзотермической реак
цией. На рис. 4.69 показана схема трехстадийной установки оксихлорировапия
с реактором для извлечения этилена. Реакторы соединены последовательно, и
каждый реактор подают воздух. По конструкции реакторы подобны большим
трубчатым теплообменникам (рис. 4.70) и содержат много узких вертикальных
трубок, вваренных сверху и снизу в трубные решетки. Внутрь трубок диамш
ром около 25 мм помещают катализатор. Диаметр трубок выбирают с таким
расчетом, чтобы температура реакции не превышала температуру, при которой
Рис. 4.69. Система оксихлорирования этилена в неподвижном слое катализатора с реак
тором для извлечения этилена
но (можно разрушение катализатора. В трубках большего диаметра теплопере-
(.1ча от центра трубки к ее стенкам замедлена, и катализатор разогревается до
(><>лее высоких температур.
В верхней и нижней частях межтрубного пространства имеются отверстия,
оисспечивающие омывание внешней поверхности каждой трубки реактора ох
л.| кдающей жидкостью, обычно водой. Тепло, выделяемое при реакции, перс
дае гея воде, а образующийся при этом пар можно использовать на других уста
попках.
Грубки реактора оксихлорирования в неподвижном слое, как правило, из
югавливают из никелевого сплава [671]. Нержавеющую сталь и сплавы с высо
ким содержанием железа обычно не применяют из-за их возможной коррозии
( тальные трубные решетки и крышки реактора никелируют. Корпус реактора,
контактирующий с водой и водяным паром, изготавливают из углеродистой
i шли.
Грубы, 1<>еД1111>п<>Щ1К* pc.ik горы, дел.hoi и i никеля или никелированной i 1.1
ли. Катали загор фиктруется снизу и сверху перфорированными никелевыми
пластинами.
Небольшая часть срубок реактора снабжена карманами для термопар
запаянными снизу никелевыми трубками диаметром 6 мм, которые введены
но оси трубок реактора на всю их длину. Термопары вводят через верхнюю
крышку реактора. Температуру измеряют либо одной подвижной термопарой,
либо чаще стационарным пучком термопар, так как это упрощает обслужи
вание. Пучок состоит из нескольких термопар, измерительные спаи которых
отстоя! один от другого на равные расстояния. Глубина погружения пучков
термопар изменяется при переходе от одного кармана к другому, чго полно
ляет измерять температуру через небольшие промежутки и строить графики
распределения температуры по длине трубок. Информация о профиле темпе
ратуры катализатора в реакторе необходима для нахождения мест nepei ревов
и управления процессом оксихлорирования за счет изменения его рабочих
параметров.
Температуры, измеренные в трубках с карманами для термопар, несколько
отличаются от температур в трубках без таких карманов. Карман для термопар
уменьшает плотность слоя катализатора и его поперечное сечение, омываемое
потоком реагентов. Поэтому скорость газа на единицу сечения потока в i руокг
с карманом для термопар выше, чем в трубке без него. Вследствие большей лн
нейной скорости потока и меныпей плотности слоя катализатора темпера гура в
трубках с карманом для термопар оказывается ниже.
Кроме того, реакторы оксихлорирования обычно снабжены устройствами,
обеспечивающими однородность газовой смеси, контроль условий работы ка
тализатора и безопасность обслуживания. Каждый реактор имеет смесиicui.
для получения однородной смеси трех реагирующих газов перед их поступлс
нием в трубки реактора. Смесители помогают также избежать образования зон
с взрывчатыми смесями. Необходим также замер перепада давления в каждом
реакторе. Повышенный перепад давления свидетельствует о физическом cia
рении катализатора. Термопары, помещенные на выходе из каждого реактора,
позволяют судить об активности катализатора. Когда активность катализатора
снижается настолько, что реакция не завершается в основной части его слоя,
температура выходящих газов увеличивается, так как реакция происходит на
конечном участке слоя. Повышение выходной температуры указывает на спи
жение активности катализатора и является сигналом к изменению рабочих ни
раметров процесса.
Газы, выходящие из последнего реактора оксихлорирования, поступают в ох
лаждаемый водой конденсатор жидких продуктов, устойчивый к коррозии. Жид
кие продукты накапливаются в емкости, служащей, кроме того, отстойником для
разделения фаз. Несконденсировавшийся газ нагревают выше точки росы и на
правляют в систему извлечения этилена. Из реактора для извлечения этилена но
ток поступает в конденсатор, в котором конденсируются практически все жидкие
продукты перед подачей отходящего газа на последующую переработку.
Исходными газами для оксихлорирования этилена являются хлорид водо
рода, этилен и воздух (или кислород). На большинстве установок весь хлорид
водорода и этилен попадают на первую стадию. На некоторых установках, чго
I < ни iii 11> перепад да пленим, ноiок IК I ра 1дсл>п<>1 между нерпой и в троп с ia
ini ш количес гво воздуха, подаваемою па каждую с гндию, iubikhi 01 гимне
। nypia реакции, эффекгивпос:iи оксихлориронапия и в ipuBooiiaciioc 1 и. Перед
поври и в peak top, особенно на первую стадию, исходные газы необходимо на
ipi и, •попы и 1(>ежагь коррозии, темпера! уру в верхней части реактора первой
I । иш поддерживают выше точки росы.
Работа реактора
Длн управления сильноэкзотермической реакцией оксихлорирования ско
р,„ н< реакции па первых секциях ограничивают, уменьшая подачу воздуха.
111 > 111и ю подавать весь НС1 и этилен на первую секцию, а воздух раздели гь меж
I) икцнями, чтобы поддерживать необходимые температуры реакции. Газы,
вы кодящие из первой секции, поступают на вход реактора второй секции, где
им । mi iiiiib.iiot с воздухом. Так же работает каждая следующая секция.
Конверсия НС1 на выходе последней секции обычно близка к 99%. Для по
ipu пня максимально высокой конверсии НС1 на большинстве установок ра
<>10.1101 с избыточной (по сравнению со стехиометрической) концентрацией
но щуха и пилена. Поток, выходящий из последней секции оксихлориронапия,
и 11ЖД.1ЮГ для конденсации ДХЭ и воды. Отходящий газ, содержащий нейро
р н пропавший этилен, вновь нагревают и подают в реактор извлечения этиле-
на ||олыпинство реакторов извлечения этилена являются реакторами катали
। ПЧ11 кого хлорирования с образованием дополнительного количества дихлор
и ilia 11огок, выходящий из такого реактора, пропускают через охлаждаемый
। ондсшагор для улавливания как можно большего количества хлорированных
Vi чсподородов. Отходящий газ содержит в основном азот (85-90%) и небольшие
количества монооксида и диоксида углерода, непрореагировавшего кислорода,
ipioii.i, п илена, ДХЭ, этилхлорида и воды.
кидкие продукты из реактора оксихлорирования и извлечения этилена
п< 111 рализуют, промывая каустиком. После разделения фаз перед подачей в ус-
।японку пиролиза ДХЭ очищают двухстадийной дистилляцией: на первой ста
him удаляют остатки воды и легкие фракции (например, винилхлорид и этил
юрид), на второй стадии от ДХЭ отделяют высококипящие примеси. Главный
компонент тяжелых фракций — 1,1,2-трихлорэтан.
Загрузка катализатора. Разбавленный катализатор
11ри оксихлорировании этилена в неподвижном слое важнейшую роль игра
। । регулирование температуры реакции. Высокие температуры повышают выход
побочных продуктов главным образом за счет увеличения скорости дегидрохло
рпронания ДХЭ в винилхлорид и в меньшей степени за счет избыточного окси
ч вирирования с образованием более хлорированных продуктов. Высокие темпе
р.п уры увеличивают также количество этилена, окисляемого в моно- и диоксид
> । лерода. Еще важнее то, что при повышенных температурах катализатор мо
г <* г дезактивироваться из за коксоотложения, истирания частиц катализатора и
। уолимации хлорида меди с поверхности носителя [672]. Наиболее интенсивно
ни явления происходят вблизи горячих пятен. Для сведения к минимуму мес
гпых перегревов предложено много мегодоп. 1 ккоюрые n.i них состоят п парь
ирокаиии cooi ношения реагентов, разбавлении сырья инертным газом, подач,
одного или двух реагентов в большом избытке, использовании трубок разных
диаметров и гранул катализатора разных размеров, большинство из вредно к< н
них методов регулирования температуры имеет определенные нреимугщч run
но наиболее широко используется разбавление катализатора.
Реакция оксихлорирования не вызывает значительного повышения темне
ратуры по длине реактора, так как быстро и интенсивно проходит на начальном
участке, где ее скорость не лимитируется ни одним из реагентов. В этой ч.и in
реактора температура может подняться так сильно, что ее трудно регулирован.
В многореакторных установках с раздельной подачей воздуха или кислорода
в каждый реактор скорость реакции лимитируется кислородом. По мере not
ребления кислорода реакция протекает все менее интенсивно и температура пн
длине слоя снижается. Поэтому для интенсификации реакции в нижних час nix
реакторов целесообразно помещать туда более активный катализатор [199|.
Надежное регулирование температуры путем варьирования активное ш
катализатора по длине слоя обеспечивается разбавлением катализатора. Для
снижения активности катализатора используют инертные разбавители с ма
лой удельной поверхностью, например плавленые оксиды алюминия, кремнии,
графит и т.п. Частицы катализатора и разбавителя смешивают в таких cooi
ношениях, чтобы концентрация катализатора вблизи входа в реактор была
очень низкой. По направлению потока концентрация катализатора возрастать
достигая максимума у днища каждого реактора; следовательно, реакторы ра i
деляются на зоны с разной концентрацией катализатора. Все реакторы запои
няют катализатором сходным образом, но концентрация катализатора возр.к
тает от предыдущего реактора к последующему. Часто в последнем реакк>|><
катализатор вообще не разбавляют [199].
Рассмотрим установку с одним реактором, в котором катализатор разбап
лен [673]. Катализатор содержит 8,5 мае. % СиС12 на активированном окси
де алюминия. Реактор разделен на четыре зоны. Ближайшая к входу в ре
актор зона содержит 7 об. % катализатора и 93 об. % разбавителя-графиы
Во второй зоне объемное отношение между катализатором и разбавителем
15:85. Третья зона содержит катализатор и разбавитель в соотношении 40.ПО. а
четвертая — только катализатор. В другом варианте вблизи входа в первын
реактор трехреакторной установки помещали слой с высокой концентрацией
катализатора [674], инициирующий реакцию. Для регулирования темпера
туры реакции следующая за ним зона содержала гораздо более разбавленпын
катализатор. Второй реактор имел три зоны одинаковой длины. Соотношение
между катализатором и разбавителем в первой зоне составляло 40:60, во in о
рой — 65:35, третья зона состояла только из катализатора. Третий реактор
либо полностью заполняли катализатором, либо в верхнюю его часть помета
ли несколько менее концентрированный катализатор.
Каскадный катализатор
Системы с разбавленным катализатором имеют ряд недостатков. Прежде
всего они требуют составления нескольких смесей катализатора и разоа
1П1НЛЯ. Для получения ОДИОрлДИОП TMClH К.1Г.1ЛИ l.liop.l It p.i Ш.111111СЛЯ CMC
пн uni необходимо проводить очень гщ.нельно Однако если ла процедура
। in it я слишком долго, слой катализатора ысоряется чаоитцми шили. В про
шипом случае получаются неоднородные смеси. В зонах реактора, содержащих
in однородные смеси разбавителя и катализатора, возникают местные нерсгрс
вы Другим недостатком разбавленных катализаторов является их склонность
к отделению разбавителя при заполнении трубок. Это особенно вероятно при
полыпой разнице плотностей между частицами катализатора и разбавителя.
I щс один недостаток — возникновение перепада давления по реактору, если
форма частиц разбавителя и катализатора обеспечивает очень плотное запои
in пне трубок. Это часто происходит, когда цилиндрические таблетки катализа
lup.i разбавляют частицами плавленого оксида кремния неправильной формы.
< »нра 1ующиеся пылевидные частицы катализатора заполняют щели между со
। сдпнми гранулами, приводя к дальнейшему росту перепада давления.
Ьолее предпочтительна каталитическая система, либо вообще не требую
пыл ра юавления, либо использующая разбавитель той же формы и плотности,
мн» и катализатор. Первое наиболее интересно, поскольку трудно найти раз
Оавитель, имеющий такую же форму и плотность, как катализатор. Форма ка
I.шиза гора (и разбавителя) должна быть такой, чтобы слой катализатора не
i.u орялся пылью.
В промышленности применяют несколько катализаторов, исключающих
или резко снижающих необходимость в разбавителе. Активность катализатора
понижают введением в носитель менее активного компонента или изменением
удельной поверхности носителя. Иногда активность катализатора изменяется
при изменении соотношения между медью и калием. Обычно поставляют ката-
ли шторы трех или четырех уровней активности. Наиболее активные содержа!
I > 20 мае.% СиС12 и менее 5 мае.% КС1. При этом форма и материал носителя
у разных поставщиков могут быть разными. Например, катализаторы фирмы
Stauffer» состоят из сферических частиц диаметром 3-7 мм с тремя разными
концентрациями активного компонента. Катализаторы приготовлены так, что
нс требуют разбавления слоя катализатора. Поэтому при заполнении реакто
p.i не происходит ни смешения, ни сегрегации частиц. Число зон в реакторе е
каскадным» катализатором обычно меньше, чем в реакторе с разбавленным
катализатором. В каждом из первых двух реакторов должно быть только две
или три зоны. Поэтому для всей установки оксихлорировання требуется набор
катализаторов трех или четырех составов.
Пример такого типа катализатора фирмы «Stauffer» и вариант его загрузки
описаны в патенте [571]. Используются три катализатора: А, Б и В (наименее
пк гивен катализатор А, наиболее — катализатор В). Рекомендуемые концен гра
ци и хлоридов меди и калия в этих катализаторах:
катализатор А Б в
( иС12, мае. % .... .... 6,0 10,0 18,0
КС '1, мае. % .... 3,0 3,0 1,8
Реакторы заполняют следующим образом. Первый разделен па две зоны —
входную и выходную. Входная составляет 60% слоя катализатора, выходная
10%. Входную зону ыпилиякн k.ii.uim задором А, выходную — катализагорим /I
В юрой реактор разделен на такие же зоны, как и первый. Входную зону заноз
ннют катализатором />, ныходную — катализатором В. Третий реактор полно
стью заполняют катализатором В.
Эта каталитическая система оказалась очень эффективной при промыт
ленном оксихлорировании. Катализаторы легко за1ружаются в реакюры и и<
вызывают большого перепада давления по реактору. Благодаря сферическоп
форме частиц свободный объем слоя катализатора достаточен для предолнрд
щения возникновения перепада давления по слою и засорения пылью. Крохи
того, благодаря меньшему перепаду давления такой катализатор может рано
тать при более высоких скоростях потока, без снижения конверсии НО. В це
лом, каскадные катализаторы позволяют достигать более высоких выходов н
селективностей, чем системы с разбавленным катализатором 1199].
Параметры процесса
Для контроля эффективности работы установки оксихлорирования с невод
вижным слоем катализатора важны фиксированные или варьируемые парами i
ры: выбор катализатора; способ загрузки катализатора; давление в реакторах;
давление в рубашке реактора; распределение воздуха; распределение HCI; с ко
рость потока реагентов; избыток воздуха и этилена; предварительный подо; реп
сырья.
Желаемую эффективность реактора можно получить, по-разному комон
нируя рабочие параметры. Хотя эти параметры, как уже указывалось, взаимо
связаны, отдельно обсудим влияние каждого из них на процесс оксихлорнро
вания.
Выбор катализатора. Выбор каталитической системы в основном опреде
ляет срок службы катализатора, оптимальную скорость потока, профиль 1ем
ператур, начальный перепад давления, скорость нарастания перепада давления,
максимальный выход и селективность. Хотя эффективность системы можно су
щественно изменить, варьируя параметры процесса, решающим является вы
бор катализатора.
Сроком службы катализатора считается обычно просто длительное 11.
работы катализатора в реакторе. Катализаторы неподвижного слоя замени
ют либо при возникновении большого перепада давления, либо при потере
активности. Срок службы катализаторов в разных реакторах установки ок
сихлорирования различен из-за отличий условий проведения процесса и со
става катализатора. Самый короткий срок службы у катализатора первого
реактора, где условия реакции наиболее жестки. Он составляет около одного
года при проектной скорости потока. Катализаторы в следующих двух реак
торах служат в 1,5-3 раза дольше, чем в первом. На срок службы катализатора
влияют такие факторы, как примеси в исходном сырье и условия процесса.
Хотя разные катализаторы имеют разный возможный срок службы, он зави
сит от условий конкретной установки, поэтому при выборе катализатора для
данной установки нужно рассчитывать его срок службы с учетом накоплении
го опыта работы на ней.
100
100 120 140 160 180
Скорость потока, г-моль / (ч-дюйм2)
Рис. 4.71. Влияние скорости потока на конверсию НС1 при оксихлорировании этилена н
неподвижном слое разбавленного (1) и каскадного (2) катализаторов
Катализаторы заметно разнятся по эффективности при повышенных ско
ростях потока. На рис. 4.71 показана зависимость конверсии НО от скорое го
потока для двух катализаторов, иллюстрирующая их чувствительность к скоро
сти потока. Для системы с разбавленным катализатором при высоких скоросг их
потока конверсия НС1 достаточно быстро снижается. Каскадный катализатор
сохраняет высокие конверсии НС1 и при повышенных расходах реагентов.
Выбор катализатора определяет начальное падение давления и, в извес гной
мере, скорость возникновения перепада давления. Например, сферический ка
шлизатор характеризуется меньшим начальным перепадом давления, чем габ
дотированный катализатор со сравнимым размером частиц. Это очень важно
в начальный период работы катализатора и дает более широкие возможное ги
регулирования температуры высокоактивного свежего катализатора путем и»
менения давления в системе.
Кроме типа катализаторов на выход и селективность сильно влияют техно
логия процесса и конструктивные особенности установок. Они определяются
числом реакторов, распределением воздуха или кислорода, температурой, дав
пением и длительностью работы катализатора. Как правило, катализаторы без
разбавителя обеспечивают несколько большие выходы и селективности j 1991.
Загрузка катализатора. Хотя операция загрузки катализатора не я вл я
ется рабочим параметром процесса, она может влиять на его эффективное и>,
особенно в системах с разбавленным катализатором. Эти системы проект
руют так, чтобы обеспечить надежное регулирование температуры, высокую
конверсию НС1 и хорошую селективность по ДХЭ. Для получения желаемой
эффективности в каждой зоне реактора нужно поддерживать определенное
соотношение между катализатором и разбавителем. Перед загрузкой в реак
гор катализатор и разбавитель должны быть тщательно перемешаны, а затем
осторожно загружены в реактор. 11ри слишком быстрой загрузке катализатора
348 I
on может o6p.iion.ui> пробки в некоторых грубках реактора Чтобы итбекаи.
мото, периодически niMepiiioi глубину слоя клали затора и проверяют, cooi
вегегвуег ли опа количеству добавленного клализатора. Образование iiponoi
и плохое смешение катализатора с разбавителем приводя! к неравномерному
распределению катализатора и снижению эффективности процесса. Сиысмы
с неразбавленным катализатором вызывают меньше затруднений при загрузке,
поскольку не требуют смешения. Если исключить образование пробок, го мн i
лизатор может обеспечивать проектную эффективность. Удовлетвори тел ынн
качество упаковки катализатора достигается и при ручной, и при механическоп
загрузке.
Давление в реакторах. Обычно оксихлорирование проводят при давлении
0,4-0,6 МПа. Давление процесса прямо связано с объемной скоростью потоков
реагентов и продуктов в реакторах. Проведение реакции при высоких да иле
ниях снижает объем газов, приводя к уменьшению линейной скорости потока
на единицу поперечного сечения. При этом уменьшается падение температуры
по слою катализатора. Снижение давления действует противоположно. Объем
газов и линейная скорость потока возрастают, горячие пятна в слое катализа го
ра охлаждаются более эффективно. Чтобы проследить это, рассмотрим тру (ж у
реактора, в начале которой реакция идет очень быстро, в 1 м от начала слоя до
стигается максимум температуры 285 °C, а в 2 м от начала температура снижа
ется до 220 °C (кривая 2 на рис. 4.72). В более активной зоне слоя катализатора
на длине 2,5 м имеется второй максимум температуры — в 241 °C. Увеличение
линейной скорости потока (при снижении давления) сдвинет точку начала ре
акции несколько дальше по слою (кривая 1 на рис. 4.72). Температура по длине
слоя будет расти медленнее, так как газы движутся по нему с большей скоро
стью. В этом случае температура первого максимума составляет только 262 °(
Температура второй горячей точки возрастает, так как через нее проходит боль
ше реагентов. В конце слоя катализатора температура по его длине убывает мед
Рис. 4.72. Влияние стационарного (1), возрастающего (2) давления на температурный
профиль реактора
и нисс, поскольку здесь псе еще прош ходи i реакция I (овышепис давления вы
u.iii.tci прямо npoiивоположиыи h|x|»cki. Реакция пачипаеня бди ке к началу
i mm катализатора, верный максимум iемпера ту ры выше и располагаете я ближе
। входу в реактор, а температура по длине слои снижается быстрее.
В промышленной практике давление варьируют, чтобы контролировать по
жькспие и температуру горячих пятен. Для инициирования быстрой реакции
пнычно в начале процесса устанавливают высокое давление. Затем его снижают,
регулируя температуру реакции. Минимально возможное давление определяет
< и давлением, необходимым для эффективной конденсации ДХЭ и транспорт и
роттки ДХЭ для дальнейшей переработки, и составляет, как правило, на выходе
п । последнего реактора 0,2-0,25 МПа. Верхний предел давления процесса, изме
репного на входе в первый реактор, определяется давлением наименее сжатого
и । исходных газов. Часто оно соответствует давлению, создаваемому воздуш
Шам компрессором.
Давление в системе регулируют на выходе из реакторов оксихлорировапия,
а входное давление меняют для компенсации перепада давления в реакторах.
I крепад давления по слою катализатора зависит от условий его работы и ско
рос ги потока. Как правило, при старении катализатора перепад давления уве
личттвается.
Давление в рубашке реактора. Тепло, выделяемое при реакции, переноси ня
or катализатора через стенки трубок реактора к кипящей в рубашке жидкости,
оьычно воде. Общей движущей силой теплопередачи является разность темпе
р.иур между слоем катализатора и охлаждающей жидкостью в рубашке. Гели
давление в рубашке возрастает, то повышается температура кипения охлажда
вицей жидкости, а скорость теплопередачи снижается. Это приводит к увеличс
пню температуры катализатора. Таким образом, давление в рубашке реактора
не является независимым параметром, который можно варьировать для регу-
лирования температуры реактора.
Влияние давления в рубашке на температурный профиль очень похоже па
оосуждавшееся выше влияние давления в реакторе. Когда давление в рубашке
возрастает, температура катализатора вблизи входа в реактор повышается, пос
кольку уменьшается движущая сила теплопередачи. В верхней части реактора
ус ганавливается новое равновесие при более высокой температуре. За сче г э го
го возрастает скорость реакции, кислород поглощается быстрее и темпера!у
ра катализатора быстрее снижается по длине реактора, так как вдали от входа
реакция идет очень медленно. В результате горячее пятно сдвигается к входу в
реактор. При промышленной эксплуатации обычно поддерживают низкое дав
пение в рубашке в течение первых нескольких месяцев работы при наивысшей
активности катализатора. Когда горячее пятно начинает смещаться ниже по ре
актору, давление в рубашке можно повысить, чтобы уменьшить это смещение
или изменить его направление без перегрева катализатора. Самое высокое дав
ление в рубашке обычно бывает в последние месяцы работы катализатора перед
его заменой.
Многие каталитические системы составлены с учетом возможности регу
пирования положения горячих пятен путем согласованного варьирования дав
ления в реакторе и в рубашке. Такое регулирование особенно важно в первые
350 I
педели р.ннны *.o e нежим, очень активным кл али ыюром. Свежий к.палиыюр
работает при относительно высокой температуре. Правильное регулировано*
температуры обеспечивает хорошие условия для работы катализатора и новы
шает срок его службы |199].
Распределение воздуха. При распределении воздуха в многореакторной у<
тановке оксихлорирования с неподвижным слоем катализатора учитываю i i pu
момента. Первый из них — безопасность эксплуатации. Необходимо исключи 11<
способы составления исходных смесей и скорости потоков, при которых noi
можно образование взрывчатых смесей. В основе безопасного распределении
воздуха между реакторами лежат данные о концентрационных пределах в ipi.i
ваемости, представленные, например, в работах [675-677] и иллюстрирующие
ся на рис. 4.73.
При одновременной подаче всего сырья в один реактор оксихлорировапии
с неподвижным слоем катализатора может образоваться взрывчатая смесь. 11рн
подаче более 50% воздуха в первый реактор, куда вводят также весь хлороно
дород и этилен, образуется смесь, близкая к пределу взрываемости. Поэтому
для установок с неподвижным слоем катализатора особое внимание уделяем i я
подаче воздуха в первый реактор.
При выборе распределения воздуха следует также учитывать образование
побочных продуктов. Подача избытка воздуха на последние стадии системы
Рис. 4.73. Треугольник взрываемости на 1-й ступени, O2/C2H4/HC1+N2
hi i нчлорнровапия приводил к обра lon.iiuiio оолыпею количесiHit ( <> и СО
I |и Ц|>му для обсг печения максимальных им ходок и селск ihihioci и нужно пода
। hi. пл перпыесюдии оксихлориропапия кик можно больше воздуха, насколько
но допускают соображения безопасности. Гретий момент, очень важный при
piUioie со снежсзагруженным катализатором, связан с гем, что распределение
(••иду ха не должно приводить к перегреву одного из реакторов. При работе со
. bi кг '.пруженным катализатором распределение воздуха выбирают исходя из
iioiMo кпос гей регулирования температуры, а не по условиям достижения он
1ПМИЛЫ1ЫХ конверсии или селективностей. Поддерживая низкую температуру
р. .и цпн, увеличивают срок службы катализатора и сводят к минимуму коксо
niipii uiii.iHHc и разрушение катализатора. Это выгодно потому, что образование
хи (кик час1иц катализатора повышает перепад давления, а коксообразовапие
ц< д< I к снижению площади поверхности катализатора и его активности. Осп»
рижная работа в начальный период службы катализатора целесообразна пре
*.цс всего по экономическим причинам.
При подаче большого количества воздуха в последние реакторы увеличила
и и выход оксидов углерода. В этом случае парциальное давление воздуха (О,)
в in рвом реакторе понижается, а парциальные давления НС1 и этилена возрас
ыкн, что способствует образованию этилхлорида за счет гидрохлорировапия
пилена. Преимущественная подача воздуха в последний реактор может повы
.nib и нем температуру катализатора. Если температура последнего реактора
। ишони гея достаточно высокой, то образующийся ДХЭ крекируется, давая ви
нплхлорид и НС1. Это обнаруживается по снижению конверсии НС1, сопровож
д>1К>|цемуся повышением температуры.
В начале эксплуатации установки, когда все три реактора загружены свежим
к .пл л и затором, распределение воздуха определяется регулированием темпера
ivpi.i Подачу воздуха в первый реактор увеличивают до достижения макси
малыш допустимой температуры. Поток воздуха, подаваемого во второй реак-
к>р. также подбирают исходя из его температуры. Оставшуюся часть воздуха
подают в последний реактор. Со свежим катализатором давление в реакторах и
р} пшиках тоже подбирают так, чтобы установка работала при как можно более
ни «кой температуре [199].
11о мере старения катализатора скорость подачи воздуха в первые два реак
юра можно периодически повышать, поддерживая их температуру на макси
малыш допустимом уровне. Это обычно делают, пока не достигаются лучшие
выходы и селективности, после чего распределение воздуха по реакторам под
дер кивают неизменным. На последних стадиях эксплуатации варьируют давле
пне в реакторе и в рубашке, чтобы свести к минимуму смещение горячего пя i на
в реакторах.
)ффективность данного реактора можно периодически определять, вычис
лил отношение числа молей образующегося в рубашке реактора пара к числу
молей воздуха, подаваемого в реактор. Если эффективность реактора достаточ
но высока, это отношение очень устойчиво. При понижении активности кала
ли ia гора отношение уменьшается. Заметное снижение этого отношения ука
и.шает на необходимость замены катализатора. Соотношение между паром и
воздухом в последнем реакторе не очень существенно, поскольку здесь воздух
нс лимитирует скорость процесса.
PiUH/HtiCJienue IK I. В установках оксихлорировапия, использующих воздух,
поток 11(1 ооычпо не разделяю г. Одной из причин лого является близость емс
си, подаваемой в первый реактор, к пределу взрываемости в случае разделения
потока I ИЛ. Другой недостаток разделения по i ока IIC1 — происходящее при лом
снижение скорости теплое ьема. Оно способствует повышению температуры ре
акции, поэтому разделение потока HCI нельзя применять в начальный период
эксплуатации свежего катализатора. Однако разделение потока НС1 дает и неко
горые преимущества, особенно в конце срока эксплуатации катализатора: умепь
шает перепад давления в первом реакторе, а за счет более низкого парциального
давления НО приводит к образованию меныпего количества этилхлорида.
Скорость потока реагентов. Скорости подачи реагентов сложным образом
влияют на процесс оксихлорирования в неподвижном слое катализатора при
использовании воздуха. Хотя при более высоких скоростях потока увеличив.!
ются выход продуктов и количество выделяемого тепла, повышение темпера
туры катализатора не обязательно. Кроме ускорения реакции и выделения теп
ла возрастает линейная скорость потока газов в реакторах, вызывающая более
эффективное охлаждение катализатора и смещение горячих пятен по трубкам
реактора. Варьирование давлений в системе и в рубашке может вернуть горячие
пятна в первоначальное положение. При этом достигается устойчивый широ
кий профиль температур, а скорость реакции поддерживается на более высо
ком уровне без увеличения температурных максимумов.
Наоборот, при снижении скорости потока уменьшается линейная скорое ь
газов. При этом горячие пятна могут смещаться ближе к входу в реактор и вы
зывать сужение температурного пика. Расширение зоны реакции и уменьшение
температуры горячих пятен и в этом случае достигаются варьированием давле
ния в рубашке и в реакторах.
Избыток воздуха и этилена. Для того чтобы достичь 100%-ной конверсии
НС1, реакцию оксихлорирования обычно проводят при концентрациях возду
ха и этилена, превышающих стехиометрические. В таких условиях отходящие
газы оксихлорирования практически не содержат НС1. Небольшие количества
непрореагировавшего НС1 растворяются в образующейся при реакции воде. Не
прореагировавшие воздух и этилен можно отделить от жидких продуктов и ис
пользовать для дальнейшей переработки.
Без достаточного избытка воздуха и этилена снижается конверсия HCI.
Обычно в таком режиме установки не работают. Работа при очень большом из
бытке этилена мало влияет на эффективность. Температура горячих пятен не
сколько уменьшается, но этот эффект незначителен, если только избыток эти
лена не слишком большой. Последний случай не применяется, так как непроре
агировавший этилен нужно выделять для последующей переработки.
Слишком большой избыток воздуха вызывает несколько эффектов. Повыша
ется температура, особенно в первом реакторе, где скорость реакции лимитирует-
ся подачей воздуха. При достаточном избытке воздуха реакция может завершить
ся уже в первых двух реакторах, снижая степень использования третьего реактора.
Еще одним отрицательным эффектом слишком большого избытка воздуха явля
ется возрастание скорости окисления этилена с образованием СО и СО2.
///>< Т>«<1/>ПШ(7||>Ш>Ш HOdO.’/X'd llc\4(>lil'l\ .’(I toil On СЛОЖНЫМ обр.пОМ ИЛИЯС1 ll.l
>ффск iintiioi u> системы оксихлорпроп.шия. I ели поступающие и первый ре
>1мор 1<ны не подогреты или подогреты слабо, го возможна коррозия крышки
реактора и верхней трубной решетки Эго mokci произойти при конденсации
пял! и, содержащейся в воздухе.
Предварительный подогрев сырья очень важен для начала реакции. Ис
модные газы оксихлорирования должны иметь определенную минимальную
icMiieparypy, ниже которой реакция не может начаться. Эта минимальная
н'мпср.и ура инициирования зависит от давления и концентраций реагентов.
In i предвари тельного подогрева или при слабом подогреве реагирующие га па
н.преваюгся до температуры инициирования в верхней части трубок рсакго
ра Реакция начинается не в верхней части слоя катализатора, а на некотором
р.п । юяпии по ходу потока. Поэтому верхняя часть катализатора использу
i ня неэффективно, а служит только для предварительного подогрева газов
Поскольку начальное горячее пятно находится ниже по слою катализатора,
ос ыеия меньшее расстояние для его смешения до замены катализатора. Для
><||фск 1 ивпого использования всего слоя катализатора газы нужно предвари
г< шин подогревать так, чтобы реакция начиналась как можно выше по слою
। л 1 ализатора.
Оксихлорирование с использованием кислорода
11рименение при оксихлорировании кислорода вместо воздуха дает то пре
имущество, что исключаются большие объемы азота на выходе из систем, ис
по п. 1ующих воздух. Поток отходящих газов из системы, работающей на кисло
роде, в 20-100 раз меньше потока отходящих газов из системы, использующем
шндух [678]. Снижение потока отходящих газов облегчает разрушение соеди
к пни, которые не разрешено выбрасывать в окружающую среду. Экономия на
i юн алии компенсирует затраты, связанные со стоимостью кислорода [679].
В системах, использующих воздух, азот играет две важные роли: растворяет
< мег ь реагентов, исключая возможность образования взрывчатой смеси, и удаляе г
п нло н 1 перегретых зон катализатора. В системах, работающих на кислороде, э i и
ыдачи решаются использованием больших избытков этилена [680]. После про
хождения через реакторы оксихлорирования большую часть этилена комприми
р\ ко и возвращают в цикл. Благодаря более высокой теплоемкости этилен лучше
нн кает температуру, чем азот. Поэтому при замене моля азота на моль этилена
и миература катализатора оказывается гораздо ниже, чем в системах оксихлори
poH.iinui, работающих на воздухе. Более низкие температуры катализатора позпо
ниш увеличить скорость потока до достижения предельного значения темперагу
ры пли давления. Согласно [678], производительность кислородных систем может
и (вне превышать производительность систем, использующих воздух.
кислородные системы обладают и другими преимуществами перед система
in работающими на воздухе. Срок службы катализатора в них больше благо
। ц>я более низким температурам эксплуатации. Выходы продукта выше за счо
। ннжепия выхода побочных продуктов, который здесь почти вдвое ниже, чем
в । иг 1смах, использующих воздух. Потери продукта в системах, применяющих
। in лород, меньше, поскольку гораздо меньше поток отходящих газов, представ
ляющих сооои небольшое количество продувочных га юн, удаляемых из цпнлц
для предотвращения накопления таких инертных газов, как оксиды углерода
Продувочные газы сушат и направляют в реактор прямого жидкофазного хло
рировапия, где этилен превращается в дихлорэтан. Кроме извлечения пилим в
жидкофазном реакторе поглощаются присутствующие в небольших количе» г
вах хлорированные углеводороды, что снижает их выброс в атмосферу.
Главным недостатком систем, использующих кислород, могут быть за трапа
на кислород и на компримирование рециркулирующего потока газов. Однако
эти затраты компенсируются более высокими выходами ДХЭ и более низкими
капиталовложениями.
Оборудование установок, использующих кислород, сходно с оборудованием
установок, применяющих воздух, за несколькими исключениями. Единствен
Расход рециклового газа, % от минимального
Рис. 4.74. Влияние расхода рециклового газа на температуру процесса (д), селекгив
ность образования ДХЭ(б) конверсию НС1 (в)
и» Miiii.in/ieiiiic — компрессор для рсцпрк)лицпп i>i»n.iini иных шинном ш
«кллщих i.i юн. Для подачи чистого мн порода необходимы < н< цп алыпл ipy
нонроноды и прокладки. 1'рубопроноды не дол кны е одержан. м.к ла и смаюк,
• прокладки должны быть изготовлены hi негорючих млкрпалон Процедура
.о >||очсппя установки должна проводи пая i.ik. чшбы кислород вымывался
ши рп1ым газом для предел вращения образования взрывчатых сменен.
i runtанпые выше рабочие параметры процесса с использованием воздуха
и о< ионном применимы к кислородным установкам. Кроме этих параметров,
• । । пук» роль в кислородных установках играют распределение 11(21, распреде
нн пилена и количество рециркулята. При разделении потоков HCI и пн
о на ме кду реакторами образуется меньше этилхлорида и понижается общий
in pi и.1Д давления.
Ггмпературу реакции можно эффективно регулировать, повышая скорос ть
। < циркулирующих газов выше уровня, необходимого для образования взрыво
in иные пой газовой смеси.
1акопомерности процесса с использованием кислорода подробно изложе
цы в работе [677]. Основные зависимости, представленные на рис. 4.74-4.78,
ил пострируют справедливость изложенных выше положений о влиянии icm
02,%
Рис. 4.75. Влияние температуры (я) и кислорода (б) на выход СОх
la:
Рис. 4.76. Влияние давления на селективность образования этилхлорида (я) и дихлор
этана (б)
пературы, давления, расхода рециклового газа и др. на показатели процесса
Увеличение расхода рециклового газа ведет к снижению температуры и, cooi
ветственно, к росту конверсии НС1 и селективности образования дихлорэтана
Это, в частности, указывает на то, что увеличение массообмена с лихвой ком
пенсирует снижение времени контакта. Автор [677] указывает, что увеличение
содержания азота в рецикловом газе ведет к росту пика температуры и снижс
нию селективности образования основной примеси — этилхлорида. В отличие
от процесса в псевдоожиженном слое катализатора рост содержания оксидов
углерода в системе практически не влияет на селективность процесса. Рост коп
версии хлороводорода ведет к снижению выхода этилхлорида и некоторому
увеличению выхода продуктов глубокого окисления (рис. 4.77.6), хотя суммар
ный их выход остается приблизительно постоянным.
Согласно данным фирмы «Shell» [677], замена воздуха на кислород позволи
ла увеличить производительность реакторов на 75% по сравнению с проектной
с существенным увеличением выхода целевого продукта — дихлорэтана, что
иллюстрируется данными, представленными в табл. 4.18.
Рост селективности образования дихлорэтана обусловлен прежде всего
существенным снижением селективности образования 1,1,2-трихлорэтана,
1’н , 1.77. Влияние конверсии НС1 на селективность образования ДХЭ (а) и на выход
п<>|»I'liii,lx продуктов (б). 1— оксиды углерода; 2 — этилхлорид
и первую очередь, за счет выравнивания профиля температур в каждом из
ipex реакторов (рис. 4.79). Автор [677] подтверждает отсутствие выбросов в
атмосферу в кислородном процессе в пусковой период в отличие от воздуш
ши о процесса.
Таким образом, использование кислорода в качестве окислителя существен
пн улучшает эксплуатационные показатели процессов оксихлорирования зги
iciiii, как в псевдоожиженном, так и в неподвижном слое катализатора, вклю
чая, в первую очередь, селективность и производительность.
Таблица I.IK. I Iok.i i.iicnii процесса оксихлорировапни фирмы «Shell» к псподпп кпо i
слое на 1 али.ыюра
Селскгииномъ превращении пилена в продукты,% «Воздушный» процесс «Кислородным» процесс
Расчетные данные Промышленное нроизнодс! но
Дихлорэтан 95,11 97,29 97,28
Этилхлорид 1,73 1,47 1,50
Оксиды углерода 1,78 0,86 0,68
1,1,2-Трихлорэтан 0,88 0,10 0,08
Другие компоненты 0,50 0,28 0,46
Конверсия НС1, % 99,13 98,64 99,83
Рис. 4.79. Температурные профили 1-й («), 2-й (б) и 3-й (в) ступеней (сплошные кри
вые — кислород, штриховые — воздух)
550
500
40 60 80 100
Высота реактора, %
I'm. -1.79 (окончание)
Сопоставление использования псевдоожиженного и неподвижного
слоя катализатора в процессе оксихлорирования этилена
В псевдоожиженном слое катализатора достигается высокая изотермич
нос гь процесса, возможно точное регулирование температуры оксихлорирова
пня 1674, 681]. Реакторы с псевдоожиженным слоем катализатора имеют более
длительный рабочий цикл, так как катализатор добавляется и удаляется без
отключения реактора. В условиях изотермичности и возможности компепси
ропать унос катализатора длительно поддерживается постоянная активность
катализатора, а, следовательно, и стабильность производительности реакюра,
качество продукта, стабильность потребления реагентов. К недостаткам тк
пользования в промышленности реакторов с псевдоожиженным слоем о тио
инея: коррозия теплосъемных элементов, более низкая, в сравнении с испод
пижным слоем, конверсия реагентов; трудная моделируемость необходимою
режима псевдоожижения. Используя ректоры с псевдоожиженным слоем ка та
лита гора, большое внимание уделяют условиям псевдоожижения и смешения
реагентов. Для исключения обратного перемешивания и проскока реагентов (в
виде пузырей) рекомендуется ограничить диаметр реактора 100-500 мм [682|.
Для создания реактора высокой единичной мощности объединяют в один ко
кухотрубчатый реактор несколько труб этого реактора. Хорошее смешение
реагентов способствует увеличению селективности, его можно проводить не
посредственно в псевдоожиженном слое [683], в предварительных смеси гелях в
шевдоожиженном слое инертных материалов [684] и т.д.
К достоинствам использования реакторов с неподвижным слоем катали <а
юра относится прежде всего высокая конверсия реагентов [456, 622] и, в пер
ную очередь, хлороводорода (>99%).
Реакторы с неподвижным слоем отличаются меньшей производитсяыюс
п.-io в сравнении с реакторами псевдоожиженного слоя, однако возможность
использования кислорода взамен воздуха позволяет достичь увеличения про
изводительности уже действующих установок. Реакторы неподвижною слоя
более сложны в регулировании температуры, возможны значительные местные
перегревы, приводящие к интенсивному протеканию побочных реакций глубо
koi <» окисления и иои*рс активности катали i.nop.i Однако coiiepiiienciiioii.iiiiH
методов контроля и управления параметрами процесса иолволяо .жснлу.инро
вать установки с неподвижным слоем катализатора не менее гибко,чем yi 1.111011
ки с псевдоожиженным слоем. Но крайней мере, coi ласпо Дж. Паворски 1|ч*)| и
развивающихся странах ожидается использование установок и с исподни киым
и с псевдоожиженным слоем катализатора. Кроме того, использование комом
нации реакторов с псевдоожиженным (первый по ходу) и неподвижным слоем
позволяет [686] создать агрегат с высокими производительностью и конпсре н< и
реагентов.
Процессы оксихлорирования этилена, не нашедшие реализации
в промышленных условиях
Прямой синтез винилхлорида заместительным
оксихлорированием этилена
Непосредственное получение винилхлорида из этилена по суммарной рсак
ции
С2Н4 + НС1 + 0,5О2 — С2Н3С1 + Н2О (4.9 I)
может быть реализовано либо использованием высоких температур (3'0
550 °C), либо за счет применения каталитических систем, альтернативных
медьсодержащим и позволяющим осуществлять процесс заместительною
оксихлорирования этилена при 200-250 °C, т.е. при температурах, характер
ных для процессов аддитивного оксихлорирования этилена с получением
1,2-дихлорэтана.
При осуществлении процессов оксихлорирования этилена при темпера!у
рах выше 350 °C необходимо учитывать вклад реакции Дикона. Источником
хлора при взаимодействии с этиленом в этом случае является молекулярнын
хлор в отличие от хлорида меди, выполняющего эту роль при более низких 1ем
пературах.
Согласно [687], при высоких температурах имеет место цепной мехапи «м
хлорирования этилена и его производных свободным хлором, образующими!
при окислении хлористого водорода. Реакция в этом случае должна идти пре
имущественно в направлении замещения водорода в молекуле этилена:
С2Н4 + С12 —* С2Н3С1 + НС1. (4.95)
В пользу протекания данной реакции, как указано в работе [688], служи i
большое значение энергия активации (-180 кДж/моль), найденное [689] для ме-
ханизма:
С12^=^2СГ; (4.96а)
С2Н4 + С1’ —► С2Н3’ + НС1; (4.966)
С2Н3 + С12 *- С2Н3С1 + С1 (4.96в)
в сравнении с энергией активации аддитивного хлорирования (-120 кДж/
моль),
C211, I ( 12— * C2II,CL. С'»)
механизм которой передается схемой:
С1,^=^2С1’; (1.97а)
С2Н4 + С1‘ С2Н4С1; (4.976)
С2Н4С1 + С12 — С2Н4С12 + СГ. (4.97н)
Скорость реакции хлорирования этилена в этом случае описывается ныра
кением [691]:
гс2н., =^с2н4Рс?н, Ра2- (4.98)
Гипотеза о заместительном хлорировании этилена при его высоко темпе
рагурном оксихлорировании не подтверждается кинетическими исследовапи
ими, проведенными А.И. Гельбштейном с сотрудниками [691, 692]. Согласно
ним данным, а также [693] этилен присоединяет хлор и при температурах,
превышающих 375 °C. Не в пользу цепного механизма процесса свидетельс
гвуют низкие значения энергии активации, характерные для каталитических
процессов [691]:
гс2н4 =Ю 40,l/RT)pC H моль/(л-с) (4.99)
Последовательность каталитических стадий выглядит следующим образом
|б94]:
равновесие
1) ZC12 + 2НС1 ~~ ~~ ~ ZC12 • 2HCI (4.100а)
2) 2ZC12 • 2НС1 2ZC12 2НС1 + С12 (4.1 000)
медленно ZC12 • 2HCI
3) ZC12 2HCI + С2Н4------- ZC1 • 2НС1 • С2Н4С1 +------
быстро
—- 2ZC1 • 2НС1 + С2Н4С12 (4.1 (Юн)
медленно 2ZC1 • 2НС1
4) 2ZC1 • 2НС1 + С)2----- 2ZO2C12 • 2НС1 +-------*
быстро
—2ZC12 • 2НС1 + 2ZC12 + 2Н2О (4.1001)
где активный центр ZC12 содержит один катион меди.
На рисунке 4.80 представлены данные по зависимости скорости превраще
пия этилена от рс2н4 и Рс1? при разных температурах [691], подтверждающие
наличие реакции аддитивного хлорирования и независимость скорости этой
реакции от парциального давления дихлорэтана.
Существенную роль в высокотемпературном оксихлорировании этилена
играют побочные реакции глубокого окисления. В работе [695] для описания
кого процесса предложено уравнение вида:
^ = к,р.рО2р^2. (4.101)
Рис. 4.80. Зависимость скорости превращения этилена от рс,н4 и Ра, при разных 1 емш
ратурах (черные точки — оксихлорирование этилена в присутствии добавок 1,2-ДХ.))
Основной вклад в образование продуктов СОХ вносят хлорированные угле-
водороды.
При высокотемпературном оксихлорировании этилена каталитические с щ
гемы в общих чертах аналогичны принятым для аддитивного оксихлорирона
ния этилена до дихлорэтана. Это, как правило, солевые композиции на носите
ле, одним из основных компонентов которых является хлорид меди. Практичен
ки во всех каталитических системах присутствует хлорид щелочного металла
(натрия, калия), роль которого сводится к понижению летучести хлорида меди
при повышенных температурах. В качестве носителей используются разнооб
разные природные и синтетические материалы разных химической природы п
структуры.
Подробное исследование основных параметров процесса высокотемпера
турного оксихлорирования этилена проведено [696].
На рис. 4.81 представлены зависимости селективности реакции оксихло
рирования этилена от температуры для хлормеднокалиевых катализаторов: па
основе силикагелей «КСК» и «МСА». Повышение температуры заметно меня
ет селективность реакции: доля образования суммы хлоруглеводородов пада
ет (кривая 1), а интенсивность побочной реакции глубокого окисления до ок
сидов углерода (кривая 2) резко возрастает. Одновременно меняется и состав
хлоруглеводородов: содержание винилхлорида (кривая 3) растет, а дихлорэтана
(кривая 4) падает; содержание прочих хлоруглеводородов, в основном, три- и
тетрахлорэтилена (кривая 5) меняется с увеличением температуры весьма не
значительно и составляет в среднем 10-15%. В области высоких температур
500-600 °C наблюдается и интенсивное смолообразование.
Из температурных зависимостей селективности реакции оксихлорирова
ния этилена, полученных на двух образцах катализаторов с разной удельной
поверхностью исходного силикагеля (220 и 69,4 м2/г), видно заметное перерасп
ределение выхода суммы хлоруглеводородов и оксидов углерода. Поэтому пред
ставлялось целесообразным более подробно изучить влияние носителя, его хи
Рт. 4.81. Зависимость селективности реакции оксихлорирования этилена ог темпера
। у ры (т = 5 с; об. соотношение СгНфНСЮг = 1:1:0,5; катализатор СиС12+КС1)
<1 — носитель силикагель «КСК» (уд. поверхность 220 м2/г); б — носитель силикагель «М( Л»
Суд. поверхность 69,4 м2/г);
1,2 — выход соответственно суммы хлоруглеводородов и оксидов углерода; 3,4,5 — содержание
и хлорорганическом продукте соответственно винилхлорида, дихлорэтана и суммы три и нерх
лор п илена.
мической природы и структуры на активность и селективность катализатора
оксихлорирования этилена в целом.
В табл. 4.19 представлены данные по сравнительной активности и селек
швности катализаторов, разнящихся как природой носителя, так и величиной
удельной поверхности, при 450 °C.
Данные табл. 4.19 показывают, что для носителей с развитой поверхностью
химическая природа заметно влияет на поведение катализатора. Но активности
и селективности испытанные носители образуют следующий ряд в порядке убы
нация: силикагель > оксид алюминия > алюмосиликат. При этом последние дна
вида — оксид алюминия и алюмосиликат — в диапазоне температур 400-500 “(
способствуют интенсивному смолообразованию. Аналогичные тенденции, хо гя
и в меньшей степени, прослеживаются на носителях той же группы, но с мало
развитой поверхностью.
Для носителя одного химического вида изменение структуры в сторону
уменьшения поверхности положительно влияет на избирательность реакции
оксихлорирования этилена, особенно в части снижения доли реакции глубоко
го окисления и увеличения выхода винилхлорида.
Более наглядно влияние структуры носителя можно проследить по данным
1 абл. 4.20, где представлены результаты сравнительных испытаний хлормедно
калиевых катализаторов на основе трех групп носителей, отличающихся струк
гурной характеристикой. Там же приведены результаты испытаний катализато
Таблица ‘1.19. Влияние химическои природы поыпеля па акипшосп. и селенппикн п>
кагализаюра оксихлорирования этилена
Носитель Sy«., м2/г Ст. К» С2Н4, % Селективность реакции, %
С2Н,С1 Сумма углеводородов СОН од
предельных непредельных
| Силикагель 220 45,5 29,2 52,6 6,8 II 1
Алюмосиликат 400 30,5 37,9 10,1 6,2 15,8
Оксид алюми- ния 150 36,9 27,0 40,3 3,4 29,3
Силикагель 13 58,5 54,3 25,2 10,0 10,6
Алюмосиликат 12 53,6 58,3 19,2 9,5 13,0
Оксид алюми- ния 10 53,3 47,4 29,6 7,2 15,5
Степень конверсии.
Примечание. Условия опыта: 450 "С, 5 с, Об. соотношение С2Н4:НС1:О2 = 1 1:0,5.
ров на основе оксидов магния и титана. В пределах одной химической группы
уменьшение удельной поверхности и соответственно увеличение размера пор
благоприятно влияет на избирательность реакции оксихлорирования этилена
степень превращения этилена, выход суммы хлоруглеводородов, в том числе и
винилхлорида, растут, а доля образования оксидов углерода падает. В такой кг
последовательности снижается доля процессов смолообразования, хотя группа
оксидов алюминия отличается наиболее интенсивным осмолом. Состав хлор
органических продуктов практически не зависит от структуры носителя и он
ределяется в основном температурой проведения реакции. Катализаторы n.i
основе оксидов титана и магния по активности и селективности не уступаюi
основным группам носителей, но малая механическая прочность, интенсивное
пылеобразование и комкование частиц в процессе работы в псевдоожиженном
слое и, как следствие этого, нарушение нормального режима псевдоожижен пл
делает использование этих носителей в процессе малоперспективным.
По совокупности свойств предпочтительными для использования в качес
тве носителей для катализаторов высокотемпературного оксихлорировапня
этилена следует считать две группы: силикагели и алюмосиликаты с уделы ши
поверхностью 10-13 м“/г. Носители с малой удельной поверхностью (2-7 м2/г)
непригодны из-за повышенной способности к слипанию частиц и нарушении
режима псевдоожижения.
Важную роль в процессе играет также состав активной фазы катализат
ра, прежде всего концентрации хлорида меди. При сохранении эквимолярною
соотношения хлоридов меди и калия и температуре 500 °C увеличение концен
трации СиС12 способствует некоторому повышению активности (например,
конверсия этилена возрастает с 60% для массовой доли Си 2%, до 65,1% для
массовой доли Си 7,8%, но при этом ухудшается состояние псевдоожижению
слоя из-за повышенной слипаемости частиц катализатора. Относительно малая
зависимость активности катализатора от концентрации хлорида меди позво
Г>-»
। по ища I 20. Влияние c i рум уры пос it lean tt.t .th iiiiiihh 1i, u селем ин inn 11> к.пали lain
| i <>к< ихлориропапня пилена
Характеристика носителя Cl. к. С2П.|, % Селем и внос и> % реакции, Условия
м /г R X V пор* см /г C.lljCI J/'X.Y. со+со2 опы га
< иликпгель 220 -68 0,92 37,1 54,5 9,9 35,6 1 = 500 "С,
69,4 30 -350 - 40,1 46,6 57,4 69.1 20,2 15,8 22,4 15,1 т = 5 с., С2Н|:|1С1;О2 = = 1 1 0,5*’
13 -2500 0,7 54,8 73,6 17,3 9,1
1,8 -5000 0,5 55,3 71,4 18,7 9,9
1 >1м ИД il 1ЮМИИИЯ 130- 150 - 0,38- 0,43 26,3 39,4 3,6 57 Те же
70 - 0,45 27,5 48,5 2,9 48,6
12 - - 44,7 65,6 19,0 15,4
0,65 1500- 2000 0,56 Проверить не удалось из-за интенсивного слипания частиц
Л/1К1МОСИЛИ 400 27 0,62 30,5 36,6 7,9 55,5 t = 450 “С,
lull 10,8 7 320 4000 0,35 0,40 53,3 58,7 47,4 57,4 37,1 33,3 15,5 9,3 т = 5 с., С2Н4:1К 1:О2 = = 1:1:0,5
< ha нд Mill ПИЯ 5,13 - - 55,3 58,5 39,6 7,9 t = 500 "С, т = 2 с.,
1 К >., I MgO 0,43 - 56,4 61,0 30,2 8,8 С2Н4:НС1:(). = = 1:1:0,5
' Прочие хлоруглеводороды.
< •!>>.<. миое соотношение.
пне। увеличить срок службы катализатора при t > 450 °C, и быстром падении
। одержания хлорида меди за счет его высокой летучести.
Указано [696], что введение промоторов в катализатор для реакции высоко
к мпературного оксихлорирования этилена не дает ощутимого эффекта и вряд
in целесообразно усложнение солевой композиции СпС12+КС1 дорогостоящи
ми добавками.
Исследование реакции высокотемпературного оксихлорирования этилены и
присутствии хлормеднокалиевых катализаторов на основе носителей с ограни
ченно развитой поверхностью (10 13 м7г) показало, что условия проведения
и. и частности, изменение времени контакта, соотношения исходных реагенте»!
пычительно влияют на показатели процесса. Согласно данным табл. 4.21, уве-
личение избытка хлороводорода увеличивает степень превращения пилена
< vmc-сгвенно (в 2-3 раза) уменьшает выход оксидов углерода и соответствен
но снижает смолообразование. На составе хлоруглеводородов изменение коп
цеп । рации хлористого водорода в исходной смеси реагентов сказывается мало
хотя несколько увеличивается выход дихлорн членов. Ин>ыгок кислорода и
исходной смеси и основном активизирует реакцию горения и сникаем выход
винилхлорида за chci резкого (в 3 раза) увеличения выхода дихлор инденов
Варьирование времени контакта в широком интервале изменения объемном
скорости реагентов 80-2500 ч 1 (н.у.) сказывается на основных показателях
(степени конверсии С2Н4, НС1, выходе винилхлорида) лишь в узком ин тервале
малых значений т (рис. 4.82). Так, конверсия С2Н4 достигает максимального ни
чения 67% при т = 1,5 с (кривая 1) и далее практически не меняется. Конверс ни
НО после максимального значения в этом же интервале монотонно убынас!
(кривая 2), что связано с увеличением выхода винилхлорида (кривая 4). 11е ша
чительно меняется и селективность реакции оксихлорирования: выход суммы
хлоруглеводородов с увеличением т от 0,5 до 14,6 с уменьшается с 96,2 до 90,3%
(кривая 3). Более существенно увеличение времени контакта сказывается па со
ставе хлоруглеводородов:
т, С. 0,5 1,0 1,5 3,0’1 5,0’1 7,О*2 ю’3 15’’
СН4С1 - 0,15 0,14 - 0,42 0,98 1,49
С2Н3С1 44,4 63,8 68,11 71,86 75,01 75,87 74,97 89,48
С,Н5С1 ... 0,2 0,03 0,04 0,06 0,09 0,19 0,25 0,28
1,1 С2Н2С12 .. .3,71 2,84 2,29 1,85 1,58 1,35 1,95 0,5
транс-С2Н2С12.. .. .8,99 5,45 5,38 4,43 3,67 3,1 3,82 2,85
цис-С2Н2С12.... ... 12,8 8,24 7,18 6,52 5,35 4,75 5,79 4,28
СНС13 .... - - 0,12 0,09 - - - -
1,2-С2Н4С12.. 19,26 13,02 10,44 7,7 4,86 3,06 1,11 0,43
С2НС13 . .4,62 1,63 2,27 3,52 4,96 7,09 8,53 0,59
С2Н3С13 . . 6,0 2,95 1,91 Сл. - - - -
С2С14 . . . Сл. 2,04 - — - 2,6 0,1
1 Незначительный осмол.
2 Усиленный осмол.
J Значительный осмол.
Если в области малых значений т содержание хлорэтанов и хлоролефипов
составляет 29 и 25%, то при т = 10 с, где более интенсивно протекают процессы
дегидрохлорирования, их содержание снижается соответственно до 1,2 и 7,8%.
Смолообразование происходит при т > 3 с.
В работе [696] указано, что стабильность хлормеднокалиевых катализаго
ров в условиях процесса достаточно высока: содержание меди в катализаторе и
активность приблизительно постоянны в течение 650 часов.
Другим направлением прямого получения винилхлорида из этилена меч о
дом оксихлорирования может являться совмещение в одном реакторе реакции
аддитивного оксихлорирования этилена и каталитического дегидрохлорирова
ния дихлорэтана. Закономерности процесса каталитического дегидрохлориро
вания дихлорэтана будут нами рассмотрены в следующих разделах. Отметим
лишь, что использование катализаторов позволяет снизить температуру реак
й? Sполихло- ридов 5,1 Ю 2,9 о| тг •—4 “П п ч 1
ержание в сыр мол. % ч к и и ч© О X© 04 40 тГ 04 то г—< о »—<
О и О Я и хо со 04 ТО тг in 40 in СП о то СП СП 04 in
вностъ ИИ, % со+со2 СП 04 т—1 •—< о иП т—< in »—4 о 4© 04 04 04 04 04 in
Сетекти реакщ Е хлоруг- ле-водоро- дов о со о\ СП О' о in то X© то О 04 С4 о 04 »—и 04 О 04 m in 04
Я * X 3 в в О 0 я сп Ш 5 40 04 со СП СП т—-< сП то 40 X© О\ 04 ТО X© 04
в и н “ Я и В ** и У, о Е с я б 1П то сп х© СП то б то ш то тГ ш СП тГ X© in in in CO X©
i — • 9 и В н иоъеммис соотношение С2Н4:НС1:О2 1П о иП о 04 т-И то о ш о г—< •—н in о 04 т—< in о in о 04 «-И in о •—< in о 04 •—<
»» и i Вид носителя Алюмосиликат (Буд. = 10,8 м2/г) б Л (5уд. = 12 м2/г) Силикагель Ns сП II со R * S 1 u (j/_w 8‘] =-ttAs)
Примечание. Условия опыта: t = 500 °C, т = 5 с, окислитель - воздух.
’ис 4.82. Зависимость показателей процесса оксихлорирования этилена от времени
.онтакта t = 500 °C; об. соотношение С2Н4:НС1:О2 = 1:1:0,5; катализатор — CuCl2 + I'
люмосиликат с уд. поверхностью 10,8 м2/г:
. 2 — степень конверсии этилена, НС1; 3,4 — выход соответственно суммы хлоруглеводородои,
ишилхлорида
щи, по сравнению с термическим процессом на 130-150 °C, и позволяет прово
1ить процесс при 330-350 °C. В работе [406) указано, что в самом сбалаисиро
шнном процессе скрыта возможность использования тепла экзотермическоп
реакции оксихлорирования этилена (238 кДж/ моль) для эндотермической ре
1кции дегидрохлорирования дихлорэтана (70 кДж/моль). При этом можно сов
иестить эти процессы в одной реакционной зоне, либо только по теплу. Однако
। обоих случаях эти процессы должны проводиться при одной температуре
Это означает необходимость разработки либо бифункционального катализа го
>а для осуществления совмещенного процесса, либо двух катализаторов: для
«низкотемпературного» дегидрохлорирования 1,2-дихлорэтана (350 °C) и «вы
окотемпературного» окислительного хлорирования этилена (350 °C).
Возможны три варианта совмещенного процесса оксихлорирования этилена
г дегидрохлорирования 1,2-дихлорэтана, разнящиеся исходными реагентами,
уть же всех трех вариантов одинакова — на активных центрах катализатора
юследовательно-параллельно идут:
оксихлорирование этилена
СН2=СН2 + 2НС1 + */2О2 —► СН2С1—СН2С1 + Н2О; (4.102)
дегидрохлорирование 1,2-дихлорэтана
СН2С1—СН2С1 —* СН2=СНС1 + НС1. (4.103)
Первый вариант. Исходные реагенты: этилен, хлороводород, кислород.
На катализаторе вначале идет только оксихлорирован^ге этилена с образова
тем 1,2-дихлорэтана. По мере увеличения концентрации последнего на основ
[ых центрах катализатора происходит реакция дегидрохлорирования с образ»
:анием винилхлорида и хлороводорода. Выделяющийся при дегидрохлориро
ании НО вновь вступает в реакцию оксихлорирования этилена и т.д. В резуль
атс получается циклический процесс, подчиняющийся брутто-уравнению:
СН2=СН2 + НС1 + %О2 — СН2=СНС1 + Н2О. (4.104а)
Нтороп вариант. Исходные pcarenibi: пилен, дихлорэтан, кислород.
К ирис у н шин данною сое тана исходных реагентов в начальный момент вре-
хп ни оке нхлорпровапие пилена не идет (пег НС1), происходит только дегидро
Iriupnpi шаппе 1,2-дихлор нала. 11о мере увеличения концентрации хлороводоро-
ы iiuip н iaer скорость оксихлорирования этилена, и процесс вновь приобретает
<нн iii'iec кип харак тер. Брутто-уравнение процесса имеет следующий вид:
( II —( 11, < CIKC1 — СН2С1 + 0,5 О2 —- 2СН2=СНС1 + Н2О. (4.1046)
Ipiniiiii вариант. Исходные реагенты: этилен, дихлорэтан, хлороводород,
< Н< Пород
»ioi вариант фактически является комбинацией первых двух вариантов,
ii'iibiKo он имеет несколько преимуществ по сравнению с первым. Наличие в
исходных реагентах и дихлорэтана, и хлористого водорода позволяет обеим
р 1кцн>1м протекать одновременно с первого момента контакта реакционной
м< । н с кашли >агором, что интенсифицирует процесс. Кроме того, становится
ио 1мо,кпым осуществление рециклов по дихлорэтану (или НС1).
При создании технологии процесс получения винилхлорида можег быть
•pi । и 11 юпап по всем трем вариантам. Каждый вариант имеет свои преимущес-
и л и псдос татки. К недостаткам данной технологии относятся необходимост ь
in ikihii 1ОН.1ПИЯ в качестве исходного реагента хлористого водорода, который
hi । in II быть получен дополнительно либо по реакции хлора с водородом, либо
пнро in юм дихлорэтана, получаемого при прямом хлорировании. В последнем
п\’ы< остается стадия пиролиза, что нарушает исходную идею исключения
। iniinii с шдии из процесса.
Но шорому варианту^ в качестве исходного реагента используется дихлор
и hi с <» с шдии прямого хлорирования. Стадии пиролиза не требуется, а непро-
pi и пропавший дихлорэтан рециркулирует в совмещенном процессе.
1 pc iin'i вариант, как уже упоминалось, является модификацией второго ва-
ри пил и используется при технологической необходимости.
Pi акции можно разбить на пять групп:
I) оксихлорирования,
’) i.iMec гительного и присоединительного хлорирования,
*) ।идрохлорирования,
I) дегидрохлорирования,
|) окисления.
К первой группе относятся реакции оксихлорирования этилена, винилхло
ри ы и дпхлорэтиленов:
< 11 ( 1Ь + 2НС1 + 0,5О2—*СН2С1—СН2С1 + Н2О; (4.105)
< 11 С 1ICI + 2НС1 + 0,5О2 —► СНС12—СН2С1 + Н2О; (4.106)
< I К 1= CHCI + 2НС1 + 0,5О2—- СНС12—СНС12 + Н2О. (4.107)
К нгп может быть отнесена и реакция Дикона в присутствии медьсодержа
ни in к пали штора при t> 300-^350 °C:
'lit 1 । 0,5О2
С12+Н2О.
(4.108)
370
Во второй।руине о।Meiпм реакции: С11,С1—С11К1 1 С12—► CUCL —ClhCI d IK 1; (4.109)
СН2=СН2 + Cl2—— СН2С1—СН2С1; (4.110)
СН2=СНС1 + С12—*- СНС12—СН2С1; (4.111)
СНС12—СН2С1 + С12—*- СНС12—СНС12 + НС1. (4.112)
В третьей группе основной интерес представляют две реакции:
СН2=СН2+ НС1—СН3—СН2С1; (4.113)
СН2=СНС1 + НС1 —- СН3—СНС12. (4.11 I)
В четвертой группе отметим реакции:
СН2С1—СН2С1 —СН2=СНС1 + НС1; (4.115)
СН2=СНС1 —СН=СН + НС1; (4.116)
СН2С1—СНС12 -— СНС1=СНС1 + НС1. (4.117)
Среди большого числа возможных реакций окисления основной интерес
представляют три реакции:
СН2=СН2 + ЗО2 —- 2СО, + 2Н2О;(4.118)
СН2=СНС1 + 2,5 О2 —* 2СО, + Н2О + НС1; (4.119)
СН2С1—СН2С1 + 2,5 О2 — 2СО2 + Н2О + 2НС1. (4.120)
Термодинамические характеристики реакций представлены в табл. 4.22.
Как видно из термодинамического анализа, только реакции гидрохлорирова
ния (4.113), (4.114) и дегидрохлорирования винилхлорида (4.116) при 600 К име-
ют AG > 0, т. е. равновесная концентрация продуктов этих реакций (побочных по
отношению к основному процессу) не будет превышать нескольких процентов.
Все остальные реакции обладают существенной «необратимостью», особен
но это относится к реакциям окисления (4.118)—(4.120), поэтому проблема се-
лективности процесса по отношению к основным продуктам — винилхлориду
и дихлорэтану, представляется весьма сложной. Она может быть решена, глав
ным образом, подбором соответствующей каталитической системы.
В работе [406] указано, что перспективной основой для создания бифунк
ционального катализатора могут являться алюминаты кальция, предложенные
Е.З. Голосманом и И.И. Курляндской. Системы CuCl2-CsCl/CaAlO2 проявляют
достаточно высокие эксплуатационные свойства в совмещенном процессе. На
рис. 4.83а-в представлены зависимости выхода продуктов реакции от времени
контакта соотношения реагентов и температуры.
Как видно из представленных данных, при повышении температуры про
цесса с 300 до 370 °C селективность образования винилхлорида увеличивает
I инпца 1.22. IcpMcvuiiiJMiriCcMli* xapiMcpiii гнкн ih iiouiii.ix реакции n uiiim< iiiciiiiiim
прпцшс получении нинплхлорида
|*< IlklUIII All, кДж/моль AS, кДжДмоль• К) Д(|, кДж/моль
298 К 600 К 298 К 600 К 298 К 600 к
1 1 105) -239,01 -239,01 300,79 311,87 -149,39 50,58
1 1 106) -230,65 -230,15 -316,30 -326,33 -136,39 -34,36
1 1 107) -211,63 -211,01 -213,97 -213,05 -147,89 83,18
( 1 108) -57,18 -58,19 -64,29 -66,88 -38,04 -18,06
( 1 109) -100,99 -99,10 -7,27 -28,84 -97,56 -81,80
( 1 110) 181,83 -180,21 -134,09 -132,76 -141,87 100,57
(1111) -173,47 -171,97 -149,64 -146,38 -128,87 84,1 1
(111?) -98,06 -104,75 -10,58 -6,56 -102,12 98,19
(’1 ИЗ) -71,65 -72,82 -142,16 -154,99 -29,30 20,19
(1 HD -72,69 -73,15 -143,50 -147,09 -29,30 15.09
( 1 115) 72,48 72,86 142,37 143,50 30,05 -13,25
(1 НО) 99,19 102,70 123,56 132,38 62,37 -23,28
I 1 117) 48,03 48,07 139,07 139,61 6,60 -35,70
(1118) -1321,72 -1320,21 -29,64 -25,87 -1312,94 -1304,58
( 1 119) -1155,18 -1154,06 26,38 29,43 -1147,33 -1136,42
(1 120) -1082,70 -1077,02 168,75 172,93 -1032,42 -977,15
сн с 11 до 46%. При этом селективность по продуктам окисления (CO+COJ
практически не меняется. Изменение времени контакта с 5 до 15 с при прочих
ранных условиях приводит к увеличению селективности по винилхлориду до
1°<>. Таким образом, использование алюминатов кальция, модифицированных
хлоридами щелочных металлов и меди, в качестве катализаторов совмещенного
процесса оксихлорирования этилена и дегидрохлорирования дихлорэтана мо
ке г являться перспективным.
Принципиальная технологическая схема совмещенного процесса, разрабо
1апного для опытной установки мощностью 1 тыс. т винилхлорида в год, пред
с гавлена на рис. 4.84.
Совмещенный процесс получения винилхлорида идет при 350 °C, под дав
лепием 0,4 МПа в кожухотрубном реакторе 1 с неподжвижным слоем катализа
юра. Объем катализатора 2,8 м3. Количество реакционных труб — 493, высота
1 Руб — 3 м. В реактор под давлением 0,4 МПа подаются подогретые до 150 "С
исходные реагенты: этилен (расход 130-150 м3/ч), хлороводород (90-100 м /ч)
н воздух (220-250 м3/ч). Перед реактором реагенты смешиваются в смеси геле
ис. 4.83. Влияние на состав продуктов: а — времени контакта (t = 350 °C, С2Н4:НС1 =
1,4:1,0), б— соотношения С2Н4:НС1 (т = 15 с., t = 350 °C); в — температуры (т = 15 с.
:2Н4:НС1= 1,4:1,0).
ДХ ) (О 11.1ДИИ ХШфИ|Ю11.1НИя
И |)СК1йфИК<1ЦИИ
промывку ДХЭ
Гт. 4.84. Принципиальная технологическая схема опытно-промышленной установки
тчмучсния винилхлорида {1-14 см. текст)
i предохранительным клапаном, срабатывающим при повышении давлении
iii.iiiie 0,5 МПа.
В ходе реакции выделяется тепло, для снятия которого в межтрубное про
। грннство подается теплоноситель — «даутерм» (35-40 м3/ч) или ПЭС-5 (35
Ю м'/ч) с температурой на входе 170 °C, на выходе 185 °C. Регенерация тепло
пт июля осуществляется в котле-утилизаторе 2, в который подается вода (или
парожидкостная смесь) под давлением 1 МПа.
Выходящие из реактора реакционные газы, содержащие органические про
думы (винилхлорид, 1,2-дихлорэтан, этилхлорид, дихлорэтилены и др.), ок
1 иды углерода, пары воды, азот и непрореагировавшие этилен, хлороводород,
ion лород, при 350 °C поступают в куб закалочной колонны 3. Температура газон
н колонне снижается с 350 °C до 100-110 °C. Для нейтрализации хлороводоро
да, содержащегося в газах, и снижения их температуры, на верхнюю тарелку
। шапочной колонны подается 3-5%-ный раствор щелочи, который пригог.ш
/пишется в смесителе 7. Для приготовления щелочного раствора в смеситель в
необходимых количествах подается концентрированная щелочь и вода. Из куна
колонны при 80 °C в емкость 6 выводится водный раствор, содержащий гид
роксид, хлорид и карбонат натрия. Раствор направляется на систему очистки
I 104пых вод.
Охлажденные и нейтрализованные газы из верха закалочной колонны пос
|упают в конденсатор 4, в котором происходит охлаждение газового потока и
час । ичная конденсация влаги и дихлорэтана. Для охлаждения в конденсаторе /
in пользуется оборотная вода. Органическая фаза из фазоразделителя 5 посту
пае г в сборник дихлорэтана-сырца 8, вода для приготовления щелочи — в смс
< и юль 7.
Г । юный no i ок, i одержи щи п пип ид хлорид, .пилен, нес кон дет iipon.uiiiiiici и
opi шические продукты, плату, инерты, пос lyiiaei в холодильник 9, охлаждас
мыи рассолом до 5 °C. При лом происходит конденсация, главным образом,
дихлорэтана, который поступает в емкость 10, а затем в сборник 8. Дихлор лап
сырец из сборника 8 направляется на систему отмывки и дальнейшей персра
ботки в производстве винилхлорида.
Несконденсировавшиеся в холодильнике 9 газы (300-350 м3/ч) проходя!
через скруббер 11, заполненный таблетированным NaOH, высушиваются до
10-20 ррм влаги и поступают в абсорбционную колонну 12, орошаемую зах<>
ложенным до -15 °C в холодильнике 12 дихлорэтаном. В качестве хладо иен
та в холодильнике 13 используется рассол или этиленгликоль с темпера гуров
25 °C. Дихлорэтан для абсорбции винилхлорида поступает со стороны. Р.п
ход дихлорэтана 1,5 м3/ч. Колонны осушки и абсорбции винилхлорида рабо
тают под давлением 200-250 кПа. Дихлорэтан с растворенным винилхлорп
дом выводится снизу абсорбционной колонны при -10...-12 °C поступаем и
емкость 14, а затем на разделение. Абгазы (250 м3/ч), содержащие (27 об. %)
этилена, (1,5 об. %) кислорода, (67 об. %) азота, следы дихлорэтана и винил
хлорида, из верха колонны 12 направляются в реактор прямого хлорирования
этилена.
Основные показатели процесса, %: селективность по винилхлориду — >1,
селективность по СО+СО? — 5, конверсия этилена — 76, хлороводорода — б(>,
кислорода — 91.
Закономерности низкотемпературного заместительного оксихлорирования
этилена изучены в [697]. Указано, что хлорид палладия на носителе, предпоч
тительно на оксиде алюминия, избирательно катализирует реакцию замещении
водорода на хлор в молекуле этилена уже при 250-280 °C.
С2Н4 + PdCl2 —- С2Н3С1 + Pd° + НС1. (4.121 а)
Восстановленная форма палладия окисляется далее смесью хлороводорода
и кислорода:
Pd° + 2НС1 + 0,50? —- PdCl2 + Н2О . (4.1216)
В отсутствие солей металлов переменной валентности, являющихся одно
временно окислителями, процесс реокисления палладия протекает с заметноп
скоростью лишь при температурах, превышающих 300 °C. Вследствие этого,
в качестве катализаторов процесса заместительного оксихлорирования эти
лена предложено [697] использовать комплексные системы PdCl2—CuCl2 и
PdCl2—FeCl3, так как медь и железо имеют более высокий окислительный по-
тенциал по сравнению с системой Pd°/Pd+2. Ввиду того, что в присутствии медь
содержащих катализаторов в условиях процесса наблюдается значительный
(30-35%) выход 1,2-дихлорэтана как продукта аддитивного оксихлорирования,
в качестве оптимальной предложена система PdCl2—FeCl3/Al2O3, содержащая
1,0 мае. % Pd и 2,0 мае. %. Fe при использовании в качестве носителя оксида алю
миния с удельной поверхностью 40-50 м2/г.
Недостатком такого рода систем является значительный выход этилхлори-
да — продукта реакции гидрохлорирования этилена, катализируемой хлоридом
железа. Общая схема превращений, предполагающая ход реакций оксихлори-
MCln • 02. C2H4 — C,H3C1 + HAfCln.! • 02-- WC1 • O, - H2O,
х
и
о
и
о
и
о
о
и
и
U
о
6
о
и
X
и
х
и
6
и
5
ос
нгэ У
ir>
о
и
£
о
к
<N
и
d
к
s
tn
s
к
гЗ
m
о
о.
S
гх
о
5
S
о
о
S и
Т"!
о
X
6
и
£
и
X
и
£
wo
и
о
и
£
б'
о
о
£
Л
о
Пч
с
’S
S
к
о
н
с
сЗ
S
о
Й
в;
сз
В
X©
о
о
ровапи» и iидрохлорпровапия через один и гот же иромежу ro4in.ni комилсм,
содержащий адсорбированный кисдороднринедена па с. 375.
Скорость образования винил и эгилхлорида в интервале 230 280 °C ап
п рокси м и руется у рав iiei i иями:
ГВХ =^нРс2н4Рнс1Ро'2 ; («1.12(>а)
гэх “КнРс2н4Рнс|Ро2- ('1.120(1)
Показатели процесса, %: конверсия НС1 — 90, С2Н4 — 90, выход винилхло
рида — 80, этилхлорида — 10, продуктов СОХ — 3-5.
Информация о технологических проработках процесса прямого син юза вп
нилхлорида в литературе отсутствует, очевидно, вследствие недостаточно вы
сокого выхода целевого продукта.
Оксихлорирование этилена в жидкой фазе
Процессы оксихлорирования этилена в жидкой фазе являлись предметы
изучения ряда исследователей в 1960-х годах. Однако ввиду серьезных огра
ничений в их практической реализации это направление не получило ра нш
тия.
М. Спектор с соавторами [698] изучали оксихлорирование этилена в водных
растворах CuCl и СиС12 при 140, 150 и 160 °C. В работе, на основании кипе i в
ческих данных, предполагается образование смешанного комплекса этилена *
моно- и дихлоридом меди:
CuCl + CuCl2 (изб.) СиСГ 2CuCl2 + CuCl2 (своб.); (4.127a)
C2H4 (г.) C2H4 (раств.); (4.1276)
С2Н4 (раств.) + СиСГ 2СиС12 + 2СиС12 (своб.) —►
С2Н4С12 + СиСГ 2СиС12 + 2СиС1. (4.127и)
Полагая, что последняя стадия является лимитирующей, авторы [698] полу
чили кинетическое уравнение вида:
аГс2н.с121
L 2 4 2j = fc(CuCl)(CuCl? -2CuCl)2. (4.128)
di
Энергия активации процесса равна 92 кДж/моль. Селективность образова
ния дихлорэтана составляет 99,0-99,6%, конверсия этилена — 5-8% при часо
вой производительности 20-50 г ДХЭ/л раствора-ч. Селективность процесс, а
увеличивается при предварительном насыщении раствора хлороводородом
Единственным побочным продуктом является этилхлорид.
X. Хейнеманн [699] показал, что хлорид меди в исходном растворе сущест
венно снижает индукционный период гетерогенной реакции.
4.4. Прямое хлорирование этилена1
Впервые хлорирование этилена с получением 1,2-дихлорэтана было осу
ще< пшено в конце XVIII в. в Голландии, дихлорэтан достаточно долго называли
«маслом четырех голландских химиков». Однако первое упоминание о промыт
лепном получении дихлорэтана из хлора и этилена относится к 1920 г. [ 1119|.
В настоящее время процесс прямого хлорирования этилена является наж
пешней составной частью любой модификации производства винилхлорида по
комбинированной или сбалансированной схемам. Именно на этой стадии обра
|уется дихлорэтан, направляемый затем на пиролиз для получения винилхло
рида. В комбинированном процессе получения винилхлорида стадия прямого
хлорирования — единственная стадия получения дихлорэтана. В сбалансиро
пнппом процессе дихлорэтан получают также на стадии окислительного хлори
рования этилена. Соотношение мощностей стадий прямого и окислительного
хлорирования обычно близко к 1:1.
Понятие прямого хлорирования
Прямое хлорирование этилена с использованием молекулярного хлора по
реакции
СН2=СН2 + С12 —* С2Н4С12 (4.129)
осуществимо как в жидкой, так и в газовой фазах.
Жидкофазное хлорирование этилена обычно проводят в среде дихлорэ тана.
Могут быть также использованы и другие растворители. В жидкость подают га
юобразные реагенты — хлор и этилен. Катализаторами процесса обычно янля
Ю1ся кислоты Льюиса.
Технология жидкофазного процесса прямого хлорирования этилена дос га
точно проста, поскольку при добавлении в жидкую реакционную массу соеди
пений, выполняющих функции катализатора основной и ингибитора побочных
реакций, конверсия реагентов и селективность близки к 100% [34, 197]. Па
раметры процесса прямого хлорирования в значительной степени завися! oi
1емпературы. В ряде случаев на процесс влияют и другие факторы: давление,
соотношение и чистота реагентов, степень диспергирования реагентов, способ
утилизации тепла реакции, длительность работы катализатора, ингибиторы в
жидкой и газовой фазах, конструкция реактора, особенности технологической
с хемы и др.
Жидкофазное хлорирование этилена относится к быстрым реакциям, иду
щим по электрофильному механизму. Единственным продуктом электрофиль
пою присоединения хлора к этилену является 1,2-дихлорэтан. Побочные со
единения, образующиеся в незначительных количествах при практической ре.т
лизации процесса, являются продуктами радикально-цепных реакций, идущих
и жидкой и газовой фазах.
Значительное выделение тепла при взаимодействии хлора с этиленом может
вызвать бурное испарение жидкой реакционной среды. При этом растворенный
1 В соавторстве с |М.Г. Аветьяном .
378 I
хлор переходи। в паровую <|>азу, :де и происходиi его реакция с га tooOp.i iiii.im
пиленом (или дихлорнаном) по радикально ценному механизму. Доля ради
кальпо-цсппых реакции в процессе может достигать 10% и более, па прямим
она не превышает 2-3%.
Теоретические основы процесса аддитивного
хлорирования этилена
Процесс жидкофазного хлорирования этилена относится к ионно-катали
гическому галогенированию. В газовой фазе он идет по радикально ценному
механизму. С появлением жидкой фазы процесс резко ускоряется, идя в рас i п< >
ре, при этом изменяется и механизм реакции [1101].
Пропуская исходные реагенты (этилен и хлор) через жидкую фазу, коюроп
обычно является продукт реакции, легко присоединяют хлор по двойной спя ш
с получением 1,2-дихлорэтана. Эта реакция идет достаточно быстро даже при
низких температурах. Но еще больше она ускоряется катализаторами гипа ни
ротонных кислот (чаще всего FeCl3)-.
FeCl3
СН2=СН2 + С12----* СН2С1—СН2С1. (4.1 10)
При достаточно низких (до 30 °C) температурах хлор присоединяется поч i и
исключительно по двойной связи, а при более высоких температурах увеличи
вается выход продуктов заместительного хлорирования. Соотношение подана
емых реагентов также влияет на выход продуктов заместительного хлорирона
ния.
Аналогично другим реакциям хлорирования равновесие реакции хлорп
рования этилена полностью сдвинуто вправо, в направлении образования дн
хлорэтана. До настоящего времени в промышленных условиях для получения
дихлорэтана обычно используют процессы, в которых хлорирование этилена
проводят в жидком дихлорэтане [966, 989]. Тепловой эффект реакции сослан
ляет 188-218 кДж/моль в зависимости от того, в каком состоянии образует и
дихлорэтан — в виде пара или жидкости [197, 1106].
Реакция протекает по типу электрофильного присоединения [978,988,1111]
При этом первой стадией, по-видимому, является образование п-комплекса ia
счет размещения пары п-электронов двойной связи на свободной орбитали
электрофила—хлора. Процесс может быть представлен следующей схемой:
С1+—С1
СН2=СН2 + Cl2 СН2=СН2 —- СН2С1—с н2 + сг —►
п-Комплекс Карбокатион
—- СН2С1—СН2С1. (4.131)
Лимитирующей стадией в этой схеме является стадия перехода п-комплекса
в ион карбония. Скорость реакции описывается уравнением второго порядка.
i = A |( 1.1 |( ll,| (I 132)
Кисло i ы JIi.iohl.i и, и час i hoc ih, хлорное желе io, способе iiiyioi oi щен ленто
oi п комплекса аниона хлора и обра loiiainno карбокатиона. При ном кислоты
Льюиса сами могут образовывать комплекс с хлором, который далее реагирует
с п иленом, образуя п-комнлекс. Процесс хлорирования этилена в присутствии
хлорного железа может быть описан следующей схемой:
С12 + FeCl, СГ —* Cl :FeCl3 (1.133)
СГ —*С1 :FeCl,
(:1' —* ClzFeCl, + СН2=СН2 СН,=СН2 (1.13-1)
или
С1+ —* СГ
С12 + СН2=СН2 СН2=СН2 , (1.1 35)
С1+ —* СГ С1+ —- Cl :FeCl3
СН2=СН2 + FeCl3 СН2=СН2 (4.136)
и далее
СГ —- Cl:FeCI3
СН2=СН2— СН2С1—С'Н2 + FeCl 4 —- СН2С1—СН2С1 + FeCl3. (4.137)
Скорость реакции в этом случае описывается уравнением третьего порядка.
г = А • [FeCl3] • |С12] [С2Н4]. (4.1.ЗЯ)
Предложена следующая модель образования дихлорэтана при взаимодейс
! вии этилена и хлора в среде жидкого дихлорэтана [1102], согласно которой
хлор и этилен раздельно растворяются в дихлорэтане, реакция протекает и
жидкости. Допускается, что реакция протекает мгновенно. Так как раствори
мость хлора в дихлорэтане примерно в семь раз выше растворимости этилена,
а их коэффициенты диффузии приблизительно равны, то предполагается, что
реакция протекает вблизи поверхности пузырьков этилена, и основная масса
раствора содержит постоянную концентрацию свободного хлора. Модель реак
ции в пленке представлена на рис. 4.85.
Этилен растворяется и диффундирует через жидкую пленку, чтобы ветре
гиться с хлором, диффундирующим с противоположной стороны. Реакционная
юна передвигается на некотором расстоянии от промежуточной поверхнос ги
газ жидкость и за очень малое время достигает равновесного положения RR'
Вблизи этого положения, с одной стороны, имеется равновесная концентрация
этилена в жидкости (Сэж,р), а с другой — хлора в жидкости (Ссж). Так как ре
R
Рис. 4.85. Схема диффузионной модели процесса жидкофазного хлорирования эгиде!ы
протекающего в пленке
(1 — газообразный этилен; II — реакционная поверхность; III — жидкая пленка; IV — жидким
дихлорэтан; V — газообразный хлор)
акция протекает почти мгновенно, то концентрации этих веществ падают до
нуля на поверхности реакции, а образовавшийся дихлорэтан диффундирует и
жидкость.
Приняв допущение, что концентрации этилена и хлора на поверхнос i и
пленки равны нулю, можно записать следующие уравнения скоростей перенос а
этилена и хлора:
N3 = к3 (Сэж’р - 0) = D3 Сэж’р/ хэ;
-Ncl - ка • (СС1Ж - 0) - Da Сс1ж/ха,
(4.1.VI)
(4.110)
где N — число молей, диффундирующих через единицу поверхности в едн
ницу времени, г-моль/(см-ч); к — коэффициент переноса массы (г-моль/ч)/
(г-моль/см ); Сж — концентрация в жидкости (г-моль/см3); Сж,р — равновесная
концентрация (г-моль/см3); D3, Da — коэффициент диффузии для этилена п
для хлора соответственно (см“/с);хэ и ха — толщина жидкой пленки со стороны
этилена и хлора соответственно (см).
<)i|Hin.we;ii>iii>iii шак N( ।о4i.i4.li*i. чшднффу ши хлора iiporcKaei н папран
лепии, которое противоположно направлению диффузии пилена. При ном
делается еще одно допущение, что в реакционную tony диффундирую! ранные
количества молей пилена и хлора. Гогда h i них уравнений на основании жни-
молярной диффу <ии следует:
= D.-C^/Ix,-(1 +/)1ГС(|Ж/Л,С,Ж’Р)], (1.1 11)
гдехь— общая толщина жидкой пленки (см.).
Из рис. 4.85 следует, что
xL = x3 + xa. (4.142)
Скорость растворения газообразного хлора в дихлорэтане при условии, ч го
толщина пленки и площадь поверхности для пузырьков реагентов одинаковы,
можно представить в следующем виде:
-N3 = N3 = D3/xL (СсГ - Сс1Ж>р). (4 1 I 3)
Объединяя последние два уравнения, получим:
N3 = D3 • Сэж’р/хь (1 + £>сг СсГ’р/ Оэ • Сэж,р - N3 • xLZ D3 Сэж’р) (4.14 la)
или
N3 = 0,5 кэ Сэж’р • (1 + Dcl • Сс1ж,р/ D3 • Сэж-Р). (4.1446)
Скорость реакции измеряется в грамм-молях реагирующих веществ на еди
ницу объема в единицу времени. Поэтому уравнение (4.1446) можно выразить
через скорость, если ввести член а, обозначающий площадь поверхности разде
ла на единицу объема (см2/см3):
г=к^-Сж'р, (4.1446')
где к'э - к3-а — коэффициент переноса массы (1/ч).
Объединяя последние уравнения, а также используя закон Генри (Сжр = р/11)
и упрощая, получим следующее выражение для уравнения скорости реакции
г = 0,5^-р3-{1/Нэ+(Пс1-рс|/Оэ-рэ)/Нс|]}> (4 145)
где Н — константа Генри, (0,1 МПа см3/г-моль); р — парциальное давление, (М11а).
Поскольку реагенты вводятся раздельно, то их пузырьки не сливаются.
Поэтому можно допустить, что их парциальные давления одинаковы и равны
0,1 МПа (рэ = pci)- Кроме того, коэффициенты диффузии практически одинако
вы, т.е. D3 ~ Da. В результате последнее уравнение принимает вид:
г = 0,5&3-(1/Нэ + l/На). (4.145а)
Для проверки этого уравнения были определены константы Генри 11 „
Ид измерением растворимости газов в дихлорэтане. Полученные значения
равны:
Н3 = 3,27 • 103 МПа-см3/г-моль; На = 0,477-103 МПа • см3/г-моль.
Тогда уравнение (4.145а) принимает вид:
г =1,2 -104Л3. (4.1456)
ну
Ко )<|><|>пцие1и переноса массы (А',) определяли гак после пас мщения жидко
го дихлор этапа хлором мере i полученный рас гпор барботировали с определен
пой скоростью пилен, при пом определяли копцеп грацию хлора в рас i поре
как функцию времени. Уравнение скорос ти реакции для растворения одшпи
этилена может бы п> выражено в виде: г = к’, (1 /Н, + СС|Ж).
Затем из этого уравнения рассчитывали коэффициент переноса массы.
Зависимость от скорости подачи этилена Уэ, см/ч, может быть выра ксна
уравнением:
/< = 0,16 Уэ2/3. (4.110)
Объединяя (4.146) с (4.1456), получим:
г = 1,92- 10 5 Уэ2/3. (4.1-17)
В работе [ 1102] также показано, что температура в интервале 32-56 °C прак
гически не влияет на скорость реакции.
Для более широкого интервала температур данное утверждение, по видп
мому, нуждается в корректировке. Так, при увеличении температуры процесса
до величины равной и выше температуры кипения дихлорэтана растет его нар
циальное давление. За счет этого происходит падение парциальных давлении
этилена и хлора. Изменится и общее давление. В результате изменения косну к л
констант Генри и коэффициентов диффузии, что может серьезно влиять на п i
менение скорости реакции.
Реакция аддитивного хлорирования этилена в жидкой фазе осложняемся
побочными реакциями заместительного хлорирования этилена, в результате
которых образуются высшие полихлориды, в основном три- и тетрахлорэтан:
СН2С1—СН2С1 + С12 —* СНС12—СН2С1 + НС1; (4.148а)
СНС12—СН2С1 + С12 СНС12—СНС12 + НС1; (4.1480)
СН2=СН2 + 2С12 —* СНС12—СН2С1 + НО; (4.1 -19)
СН2=СН2 + ЗС12 —► СНС12—СНС12 + 2НС1. (4.150)
При этом реакции заместительного хлорирования хлорпроизводных этапа
индуцируются этиленом. Если же в этих условиях обрабатывать хлором один
только продукт присоединения хлора к олефину, то замещение идет значитель-
но медленнее.
Наряду с реакциями заместительного хлорирования протекает побочная
реакция гидрохлорирования этилена с образованием этилхлорида:
СН2=СН2 + НС1 —* СН3СН2С1. (4.151)
Процесс заместительного хлорирования протекает по радикальному меха
низму и ингибируется различными соединениями (например, хлорным желе
зом, кислородом и др.), которые обрывают цепь.
Для подтверждения этих положений авторами были проведены исследо
вания процесса жидкофазного хлорирования этилена на лабораторной уста
2,5
Pm I я<>. Влияние температуры и количества хлорного железа на выход трихлорл апа
цинке с использованием реактора проточно-циркуляционного типа. В ходе
п<< лсдований концентрация хлорного железа менялась в пределах ог 0,01 до
О 10 мае. %. Соотношение этилена к хлору во всех опытах поддерживалось
рапным 1:1.
Результаты проведенной работы представлены на рис. 4.86. При рассмог
р< нии полученных данных можно сделать вывод, что повышение температуры
процесса при постоянной концентрации хлорного железа ведет к росту выхода
полихлоридов. При этом увеличение концентрации FeCl3 способствует повы-
шению селективности процесса.
Скорость процесса жидкофазного хлорирования этилена увеличивается с
емперагурой, причем температурный коэффициент реакции заместительною
хлорирования хлорпроизводных этана больше, чем у реакции аддитивного хло
рнрования.
Побочные продукты образуются по радикальному механизму и при значи
ельцом избытке этилена можно ожидать увеличения скорости побочных реак
Ц1111
к,
С2н4+ С12— С2Н4С1 + СГ; (4.152)
^2 СГ + С2Н4С12—- С’2Н3С12 + НС1; (4.153)
к, с2[ 13С12 + С12— С2Н3С13 + СГ; при к3« к2, (4.154)
С‘2Н3С12 + М —► обрыв цепи; (4.155)
। де М — молекула этилена, хлора или FeCl3
В соответствии с предложенными механизмами уравнение скорости основ
пои реакции хлорирования этилена имеет вид:
И = • [С2Н4 ] [С12]. (4.156)
384 I
Уравнение <кор<>< ih образования грихлор напа можно представит!, следу
кицим обра и>м:
г2 = к2- |С211,] • ]С1,]2. (1.157)
Сравнение этих уравнений показывает, что повышение концентрации р.к
торенного хлора в реакционной массе ведет к росту скорости образовании
грихлорэтана. Концентрация хлора в значительной степени зависит от соот
ношения подаваемых реагентов и температуры, при этом, чем больше избы i ок
этилена, тем ниже концентрация хлора и выход трихлорэтана. Рост темпера
гуры ведет к увеличению констант скорости реакций аддитивного и замени
тельного хлорирования. Рост избытка этилена способствует увеличению выхо
да высококипяидих побочных продуктов хлорирования за счет индуцирукящ
го эффекта [1145, 1146]. Но предварительно определить более полно и точно
влияние каждого отдельного параметра на выход побочных продуктов весьма
проблематично.
Именно скорость побочной реакции заместительного хлорирования ди
хлорэтана с образованием 1,1,2-трихлорэтана и высших полихлоридов обу<
ловливает селективность процесса синтеза дихлорэтана. Однако эта реакции н
присутствии добавок хлорного железа изучена недостаточно.
В работе [1114] была исследована кинетика реакции хлорирования 1,2
дихлорэтана в присутствии хлорного железа (Сресц - 4-6-10 4 %).
Эксперименты проводились в интервале 40 80 °C, а также при температуре
кипения реакционной массы (83,5 °C и выше) как в присутствие, так и в отсу и
твие кислорода (подаваемого в реактор в виде воздуха).
Согласно данным [1114], побочная реакция хлорирования 1,2-дихлорэтаиа
протекает по радикально-цепному механизму:
С12—-2СГ; (4.158)
/6
СГ + С2Н4С12 — С2НзС12 + НС1; (4.159)
к<
С’2Н3С12 + С12— С2Н3С13 + СГ; (4.160)
к<
СГ + М —► обрыв цепи, (4.161)
где М — молекула этилена, хлора или FeCl3
Следовательно, скорость реакции при к3 « к2 может быть описана уравие
нием:
Г1 = [(/с, • к2)Цк< [М4])] • [С12] • IС2Н4С12] = к' • [С12] • [С2Н4С12], (4.162)
где к' — наблюдаемая константа скорости.
Изучение растворимости хлора в дихлорэтане (рис. 4.87я) показало, что при
постоянной температуре в условиях проводимых экспериментов концентрация
хлора в течение опыта остается практически неизменной. Это свидетельствуе г
и II|«>|Ck.ll!ltlt ПрОЦСсса « KIIHCI ПЧСсКОИ OlWI.Il III ll I IO ЩО/1'Н I IH1111 <11 I > ^ | >. 11111 < 111«<
• hopoi in реакции u упрощенном виде:
г, = V'|C.,II|C1>], (1.161)
uli’A' А'|С1?].
В ном случае для периодических условий констан га скорое ги реакции:
А = (1/т) 1п(СД()/Сд), (4.16-1)
i ц । время реакции; СДо иСд — соответственно, исходная и текущая концеп
। рация дихлорэтана [978].
Как видно из рис. 4.876, температурная зависимость константы скорости до
нмиературы кипения реакционной массы описывается уравнением Аррениу
। < корост т» реакции заместительного хлорирования дихлорэтана оиисывае гея
)равнением:
г = 0,12 exp[-30500/(RT)] [С2Н4С12], моль/л-с. (4.16S)
Кинетические параметры хорошо согласуются с результатами, получеппы
мн при научении заместительного хлорирования дихлорэтана в отсутствие ип-
। поп юра [ 1144]. В частности, константа скорости неингибированного процесса
I’m.. 4.87. Температурные зависимости количества растворенного в ДХЭ хлора («) и
константы скорости заместительного хлорирования ДХЭ (6).
38'----------------------------------------------------------------------
при 60 n( равна 7,5-10 с *, гогда как определен на и в них же условиях копе ian
га скорости inn ивиропаппой реакции состапляс! всего лини. 14,710 с ', чк>
отражает замедляющее влияние хлорного железа. По этой причине наблюдай
мая энергия активации ингибированной реакции выше на 10 кДж/моль. Однако
полученное кинетическое уравнение применимо лишь при температурах пи к»
/ЬИ1| реакционной смеси.
Как видно из результатов, приведенных в табл. 4.23 и на рис. 4.87б, хлориро
вание дихлорэтана в режиме кипения реакционной массы приводит к аномаль
ному возрастанию скорости процесса.
Таблица 4.23. Заместительное хлорирование дихлорэтана в присутствии хлорного кг
леза (загрузка дихлорэтана в реактор — 62,5 г, скорость подачи хлора — 0,24 л/ч)
Время хлори- рова- ния, ч Состав реакционной массы, % 0,1 • сд, моль/л 103- •>g(C„0/C„) 107k,c 1
1,2- ди- хлор- этан 1,1,2- три- хлор- этан 1,1,1,2- тетра хлор- этан 1,1,2,2- тетра- хлор- этан Пента- хлор- этан
1 99,44 0,50 X лорирова ниепри 4 о°с 1,255 2,42 8,43
2 4 99,35 98,35 0,62 1,62 0,03 0,03 — - 1,254 1,242 2,76 7,01
8 97,55 2,11 0,14 0,16 0,03 1,232 10,56
10 96,63 2,83 0,21 0,26 0,08 1,220 14,67
16 95,52 3,18 0,45 0,49 0,35 1,206 19,69
2 98,96 0,95 X 0,09 лорирова ние при 5 о°с 1,250 4,32 9,1
3 98,16 1,40 0,44 - - 1,240 7,85
4 97,39 2,03 0,46 0,06 0,06 1,230 11,27
6 97,28 2,12 0,46 0,05 0,09 1,228 11,76
8 96,42 2,60 0,63 0,12 0,23 1,217 15,62
10 95,36 3,65 0,64 0,15 0,20 1,204 20,42
12 95,05 3,71 0,68 0,26 0,30 1,192 24,63
16 93,97 4,11 0,77 0,37 0,78 1,174 31,44
2 98,85 1,03 X 0,12 лорирова ниепри б о°с 1,248 4,80 14,7
3 96,24 3,19 0,57 - - 1,215 16,43
5 95,90 3,20 0,64 0,09 0,09 1,212 17,56
И 93,95 4,84 0,62 0,22 0,37 1,186 20,89
17 91,94 6,07 0,67 0,56 0,75 1,161 36,28
19 90,50 6,62 0,71 0,87 1,30 1,142 43,14
23 88,64 7,97 0,89 0,98 1,72 1,12 52,16
I.m ntii.i I 21(окончание)
Нргм >i Й lOpil pllllll пин. ч Cot lav реакционной m.ku.i % 0.1 сл, мо/н>/;| ю’. IO7k,t 1
1.2 ди- хлор >mn 1,1,2- три хлор этап 1,1,1,2- тстра- хлор- эган 1,1,2,2 Пеп га хлор лап
гсгра ХЛОр- 31 а и
Хлорирование при 70 "С
। 9V,9ft 0,68 0,11 - - 1,252 3,27 20.4
1 97,98 1,82 0,19 - - 1,234 9,53
f> 96,57 3,05 0,20 0,08 0,10 1,216 15,80
к 95,50 3,97 0,17 0,09 0,26 1,206 19,78
III 9-1,81 4,27 0,22 0,17 0,54 1,197 22,93
12 93,31 5,50 0,16 0,27 0,76 1,178 29,86
IS 89,25 9,30 0,16 0,44 0,84 1,127 49,18
21 85,28 12,14 0,19 0,84 1,54 1,070 69,86
з 82,74 14,42 0,23 1,02 1,58 1,040 82,10
Хлорирование при 80 °C
A 99,57 0,43 - - - 1,260 1,66 27,1
1 97,05 2,95 - - - 1,223 12,79
<> 95,41 4,39 - - - 1,210 20,20
9 94,67 5,20 - 0,07 0,05 1,195 23,57
10 92,59 7,17 - 0,12 0,14 1,170 33,22
1? 91,73 7,89 - 0,25 0,12 1,158 37,27
15 86,45 12,95 0,04 0,34 0,22 1,090 63,02
17 84,81 14,13 0,07 0,47 0,50 1,070 71,34
’0 83,00 15,80 0,09 0,59 0,51 1,048 80,70
21 75,62 22,51 0,11 0,97 0,79 0,950 121,15
\ юрирование з режиме кипения (t повышалась с 83,5 до 88,5 и С) без подачи воздуха
2 95,79 4,21 - - - 1,21 18,46 76,8
I 90,79 9,20 - 0,10 - 1,15 41,75
8 84,09 15.62 0,02 0,20 0,07 1,06 74,99
12 71,23 28,07 0,01 0,35 0,34 0,90 147,12
1ft 62,90 35,32 - 1,20 0,67 0,79 201,13
Хлорирование в режиме кипения (83,5 °C, с подачей 0,1 л/ч воздуха
3 98,39 1,61 - - 1,24 6,83 8,4-1
7 97,10 2,90 - - - 1,23 9,89
1 1 96,31 3,69 - - - 1,22 16,11
15 95,49 4,50 - - - 1,21 19,83
19 94,17 5,76 - 0,07 - 1,19 25,87
23 93,16 6,26 - 0,13 - 1,18 28,46
Увеличение rcMiicp.i i уры процесса всего лишь на 3,5 "< (с НО до 83,5 "( )
сопровождающееся, однако, переходом в режим кипения реакционной массы
приводи! к повышению константы скорости в 2,8 раза, гогда как в соотиен
гвии с полученным кинетическим уравнением опа должна возрасти в 1,1 ра ы
Это можно объяснить следующим: при переходе в режим кипения происходи!
насыщение жидкой реакционной массы парами дихлорэтана (в виде пузырен),
подаваемый в зону реакции газообразный хлор взаимодействует с парами дп
хлорэтана, и реакция заместительного хлорирования смещается в газовую <|>а iy;
поскольку ингибитор реакции — хлорное железо, вследствие малой летучее iii
в рассматриваемом диапазоне температур способен эффективно действовав,
только в жидкой фазе, скорость реакции заместительного хлорирования редко
возрастает. Очевидно, что введение в зону реакции веществ, способных nuiii
бировать радикально-цепные реакции, должно снижать скорость заместитель
него хлорирования дихлорэтана в режиме кипения. Известно, что кислород я в
ляется эффективным ингибитором газофазных реакций заместительного хло
рирования углеводородов [1113] благодаря своей способности к обрыву ценен
за счет образования малоактивных радикалов типа С^НзСЮг-
Проведенные эксперименты (см. табл. 4.23) с подачей в реакционную зону
кислорода (воздуха) подтвердили выдвинутое предположение. Введемте
=7,5% кислорода от объема подаваемого хлора вызывает снижение коне ган
ты скорости заместительного хлорирования дихлорэтана в режиме кипения
примерно в 9 раз. Следовательно, в условиях хлорирования этилена при ки
пении реакционной массы металлоорганические каталитические системы с
низкой летучестью должны быть малоэффективны как ингибиторы замести
тельного хлорирования дихлорэтана. Поэтому для достижения высокой се
лективности процесса хлорирования этилена в режиме кипения необходимо
ингибирование процесса заместительного хлорирования, как в жидкой, гак
и в газовой фазе.
Катализаторы процесса
В качестве катализаторов процесса прямого жидкофазного хлорирования
этилена используют соединения типа кислот Льюиса [1081], чаще всего хлорид
железа [948, 954, 982, 999, 1077] вследствие его доступности, а также постояв
ного присутствия в реакционной среде за счет коррозии реакторного оборудо
вания [959].
В значительном количестве работ предложено в качестве катализаторов ис-
пользовать сложные комплексные системы с целью повышения прежде всего
селективности процесса.
Так, [1088] апробирована каталитическая система для прямого хлориро
вания этилена, представляющая тройные комплексные системы, включающие
комплексы галогенидов металлов типа: FeCl2, FeCl3, SnCl4, SbCl5 с азотсодер
жащими электронодонорными соединениями: гексаметилфосфортриамидом,
пиридином, а также соединениями, ингибирующими радикальные реакции:
2,2-ди-(п-оксифенил)-пропаном; 2,6-дитретбутил-4-метилфенолом; п-нитро
фенолом. При температуре кипения дихлорэтана (84 °C) селективность его об-
разования достигает 99,85%.
I Ipn I5"C наиболее эф<|>ек i ивпым no длиichi.iioi hi h cwicmiiiiikk hi являс1
л комплексный кашли uiop let ’12 • 1смамс|ил<|>1н<|><»р|риамид/п пи i рофепол
« массовым отношением комплекса к иш ибптору ) I, а при 20 "< — катализа
iop, пк толщин из SnCJ| • пиридина / 2,2 ди (п окснфепилДщопапа с массовым
hi шипением комплекса к ингибитору 1:0,5. В ывисимоши от условий проведе
пил процесса, в частности, от изменения температуры выше или ниже точки
юпк пня дихлорэтана, состав используемого катализатора может претерпеват ь
। ущ<ч гигнные изменения.
1 (есмогря на довольно высокие выходы дихлорэтана и чистоту получаемого
продукта, процессы, проводимые в присутствии таких сложных каталигичес
• пк i пс гем, отличает ряд негативных сторон, а именно — высокая слоимое и,
। пали la гора, ограниченный срок его работы, усложнение установки и спя ш с
ппнжодимостью регенерации катализатора.
В качес гве катализатора предлагается также использовать амиды RCONR R,
। Ц| R ном водорода и/или алкилрадикал, имеющий от 1 до 4 атомов yi ле
рода |956]. Количество катализатора, необходимое для проведения процсс
। । ioc гавляет от 0,001 до 5 мол. %. Полученные результаты иллюстрируюия
|.||>л 1.24.
()днако кислород является не катализатором основной реакции, как ука гано
к ил к п ।с, а ингибитором побочных реакций в процессе прямого хлорирования
и плена. Это подтверждается и данными, приведенными в табл. 4.24. По всей
in роя । пости, катализатором является только диметилформамид.
В других работах в качестве катализатора предлагается применять диме ги л
формамид, стабилизированный гексаметилентетрамином [958, 989, 1084|.
Много разработок посвящено использованию в качестве катализаторов
«мегсп солей хлорида щелочного, щелочноземельного металла или аммония
п х lopnoro железа. Такая смесь в среде жидкого дихлорэтана в присутствии
хлора образует тетрахлорферраты типа NaFeCLj, KFeCl4, Mg(FeCl4)2, NH tFe( lf
и i и По мнению авторов [964, 1025], эти соединения — чрезвычайно актин
iii.il катализаторы и одновременно сильные ингибиторы побочных реакции
процесса.
I .|||>111ц«1 4.24. Жидкофазное хлорирование этилена в присутствии диметилформамнда
Опыт Катализатор СдмФ» МОЛ.% Выход ДХ Э, %
М 1 - 0 79,9
№2 Кислород 0 95,0
№3 ДМФ 0,030 98,0
М 4 ДМФ 0,0135 96,7
№5 ДМФ + О2 0,00135 99,7
№6 ДМФ + О2 0,0135 99,8
1*7 ДМФ + О2 0,3375 100,0
№8 ДМФ + о2 1,35 99,9
т;и ;-----------------------------------------------------------------
I’a зработан ciioioo получения дихлор папа при гсмпера гуре ею кипения, и
присутствии комплексного катализатора, состоящего из безводного хлорною
железа и соединения, включающего азот, например, соль на его основе [1()25|
Из реактора выводят дихлорэтан сырец, содержащий 99,ВО мае. %, основною
компонента.
В [964] реакция хлора с этиленом проведена в среде жидкого дихлор.п апл
при 0,11,5 МПа, 20-100 °C, но ниже температуры кипения дихлорэтана и в
присутствии каталитической смеси. Эта смесь включает эквивалентные ко
личества безводного хлорного железа, азотного основания или его соли. При
ректификации дихлорэтана катализатор, оставшийся в кубовой жидкое in.
вместе с ней рециркулирует в реактор. Количество возвращаемой кубовоп
жидкости регулируется из расчета, чтобы концентрация хлорного железа и
реакционной массе составляла 0,001-0,5 мае. %. Для упрощения проведения
процесса реактор заполняют железной насадкой. Это позволяет поддержи
вать необходимое количество хлорного железа в реакционной массе за счет
реакции выделяющегося в ходе процесса НС1 с железной насадкой.
Этот процесс неоднократно усовершенствовали. Например, в [955] было
предложено реакцию хлора с этиленом проводить в среде жидкого дихлор
этана в присутствии каталитической смеси, состоящей из галогенидов метал
лов, имеющих свойства кислот Льюиса, и галогенидов металлов 1а и/или 2а
группы Периодической таблицы при 20-200 °C, атмосферном или повышен
ном давлении. В качестве катализатора предлагается использовать безводный
натрий (NaFeCl4), калий (KFeCl4) или магний [Mg(FeCl4)2] тетрахлорферра
ты. Можно также применять в стехиометрических соотношениях соедипе
ния, которые в реакционной смеси образуют указанные тетрахлорферра! ы
Процесс проводят в присутствии кислорода или воздуха при концентрации
катализатора 0,005-0,500 мае. %, в расчете на хлорное железо по отношению к
находящемуся в реакторе дихлорэтану. Процесс иллюстрирует табл. 4.25.
В патенте [1034] дихлорэтан получают в присутствии тетрахлорферрата на
трия в количестве 0,2 мае. %. Тетрахлорферрат натрия применяют в качесгпе
катализатора и в работе [1095].
В другой модификации процесса [1036], проводимого при температуре
кипения реакционной смеси также в присутствии смеси хлорида натрия и
хлорного железа, реактор имеет выносной циркуляционный контур жидко
Таблица 4.25. Жидкофазное хлорирование этилена в присутствии тетрахлорферратов
щелочных металлов
При- мер Катали- затор Концентрация катализатора , % Состав полученного продукта, мае. %
Этил- хлорид Дихлор- этан Трихлор- этан Хлоро- водо- род Прочие
№ 1 NaFeCl4 0,14 < 0,002 99,94 0,03 0,002 0,005
№2 KFeCl4 0,24 < 0,002 99,86 0,13 0,002 0,006
В пересчете на FeCl3.
i<> дихлор ii.iii.i, в коюрып перед вводом в pc.iKiop, подаю! реагенты, что
। uni о1н । вуег предвари тельному н.п мщению дихлор п апа хлором и ноле
пом ( месь хлорида па грия и хлорпо! о желе ia неволь iye ген в мольном cooi
ношении ниже 0,5.
( ооощаегся, чго добавка в реакционную массу хлорида натрия и хлорного
। «He ta в мольном соотношении ниже 0,5 (предпоч ги гельно от 0,3 до 0,5) по >
волне г дос тичь такой чистоты дихлорэтана сырца, при которой его можно be i
рем нфикационной очистки направлять непосредственно на пиролиз |1О35,
К) <8|. 11роцесс осуществляют при 75 °C и давлении 54 кПа. Содержание хлор
ною железа в реакционной массе составляет 710 2 мае. %.
Результаты исследований:
1 1рпмср .. № 1 №2 №3 № 4
N\i( ITeClj, мол 0 0,35 0,40 0,45
( одержание в продукте, мае. %:
1 1,2 трихлорэтана .. .0,157 0,064 0,025 0,035
1 дихлорэтана .. .99,78 99,90 99,95 99,94
Из приведенных данных следует, что оптимальная величина мольного со
oi ношения хлорида натрия к хлорному железу колеблется в очень узких преде
лих — от 0,40 до 0,45.
При использовании катализатора из смеси хлоридов железа и натрия в мо
лирном соотношении от 1:1,5 до 1:2 достигается такая чистота дихлорэтана
i ырца, что его также можно без ректификационной очистки от высококипя
щич продуктов направлять непосредственно на пиролиз [1083].
Влияние соотношения NaCl и FeCl3, содержащихся в каталитическом
комплексе, на селективность процесса хлорирования этилена, а также со
поставление данных работ [1095] и [1038], приведенное на рис. 4.88 пока
1ывает, что характер кривых идентичен и данные удовлетворительно согла
суются. Полное совпадение приходится на точку, в которой соотношение
NaCl:FeCl3 =0,ЗЗн-0,35.
Образование высококипящих примесей в процессе можно минимизиро
u.iTb с помощью превращения хлорида железа в тетрахлорферратный ком
нлекс [1083]. За счет введения в реактор катализатора, который состоит нт
1меси хлоридов железа и натрия в молярном соотношении от 1:1,5 до 1:2,
достигается высокая чистота дихлорэтана-сырца. Однако в этом случае со
oiношение хлоридов железа и натрия диаметрально противоположно двум
предыдущим.
Наряду с каталитическим комплексом NaCl-FeCl3, используются катали та
юры типа HFeCl4, NH4FeCl4, NaFeCl4, а также более сложные, типа HFeCI, • о
крезолат, NaFeCl3 • о-крезолат [1052]. Отмечается, что убыль каталитической
< нс гемы из реакционной массы зависит от температуры процесса. В свою оче-
редь, температура процесса определяется температурой кипения используемо
io в качестве жидкой среды растворителя. Так, при проведении хлорирования
пилена в среде кипящего дихлорэтана, температура процесса 84 °C, а в среде
кипящего гексахлорбутадиена — 212 °C. Убыль катализатора в последнем слу
ЛП7
Рис. 4.88. Влияние состава катализатора на селективность хлорированного этилена
чае примерно на два порядка выше, чем в первом. Убыль катализатора завис и i
и от его состава. Легче всего происходит убыль хлорного железа, примерно и
два раза медленнее — разных тетрахлорферратов, еще приблизительно в два
раза медленнее — смеси тетрахлорферратов с о-крезолатом.
Наименьшее количество трихлорэтана образуется при использова
нии в качестве катализатора следующих систем: NaCl+FeCl3 + о-крезол,
CaCl2+FeCl3 + n-крезол, NaCl+FeCl3 + фенол и NaCl + BiCl3 + резорцин. При
этом каждая из указанных смесей наиболее эффективна, когда все комно
ненты находятся в равных мольных количествах. При использовании этих
систем отмечается и самое низкое содержание трихлорэтана в отгоняемом
дистиллате. Основное влияние на количество трихлорэтана и этилхлори
да в дистиллате оказывает изменение мольного соотношения компонентов
катализатора. При этом оптимальное содержание о-крезола в смеси лежт
в очень узких пределах от 0,9 до 1,1% мол. Выход дихлорэтана составляет
99,68-^99,98%.
Согласно (1025) с целью выделения катализатора из смеси последнюю про
пускают через адсорбер, где улавливается катализатор, а оставшийся жидки и
продукт подается на выделение дихлорэтана в ректификационную колонну. Ка
тализатор выводится из адсорбера и вновь возвращается в цикл. Адсорбента
ми могут быть активированный уголь, основные ионообменные смолы, оксиды
кремния или алюминия.
1,2-Дихлорэтан предлагается получать хлорированием этилена в присутс-
твии сложного катализатора, состоящего из хлоридов железа, алюминия, олова
или сурьмы, а также аминов, пиридина, различных хлоридов или хлоргидратов
аммония. Из получаемого дихлорэтана-сырца отгоняли 85-90% дихлорэтана
[1032]. Оставшуюся часть дихлорэтана с растворенной в нем каталитической
I iO ины I 2(». Хлорирование пилена в npiu у ii iiuiii 11111 >« hi 11 u »nia « i< tia in luiiipiin
При М< |> (ос bin каталитической системы (мольное соотношение компонентов) ( ОС IIIII Н<>'1) Ч.1ГМ010 НрОД) К III, Мне % Выход дихлор илы, %
Дихлор паи Ipilinitp Me цы хлорид, хлороформ
Результаты хлорирования .пилена при 33-37 "(
№ 1 1 с( 1(:пиридин = 1:1 99,95 0,01 0,04 99,9
№2 1 с('12:пиридин = 1:1 99,95 0,01 0,04 99,9
№ я 1с< 1|:гриэгиламин = 1:1 99,94 0,02 0,04 99,8
№ 1 AlCl t: 1 -2-(хлорэтил) пиридинхлорид = 1:1 99,94 0,02 0,04 99,9
I'c ivitbmumbi хлорирования этилена при температуре кипения реакционной мааы (84-85 °C)
№25 1 еС1(:пиридин = 1:1 99,92 0,03 0,04 99,9
№ 29 Sn( '12:пиридин = 1:1 99,92 0,03 0,05 99,9
№ II 1'еС13:1-(2-хлорэтил) аммонийхлорид = 1:1 99,93 0,03 0,04 99,9
i in iемой возвращали в реактор. В табл. 4.26 сведены данные опытов, в которых
пыли получены лучшие результаты.
Дихлорэтан получают в присутствии растворимых железокомплексиых
катализаторов. Хлорирование этилена проводят при 20-65 °C с отбором про
дума из жидкой фазы. Затем продукт направляют в другой реактор, работаю
iii.liи при температуре кипения реакционной массы [1053]. В реактор загружаю!
|( 1 l',NC2H4Cl]+-[FeCl4] ', используемый в качестве катализатора. Дополни гель
ные характеристики процесса приведены в табл. 4.27, в которой используются
следующие обозначения: тр — время работы; шзк — масса загруженного ката
ли затора; Срм — концентрация катализатора в реакционной массе; ш'„р — ко
личество продукта, получаемого в реакторах; шпр — общее количество полу
чеппого продукта; тк — количество катализатора, отбираемого из реакторов,
/н, и — реакционная масса, отбираемая из реактора-испарителя; Р.К —расход
катализатора на получение продукта; В — выход высококипящих примесей при
получении продукта.
Следует обратить внимание на высокое содержание катализатора (до 2,7 %
мае) в реакционной массе, особенно в реакторе, работающем при температуре
85 90 °C.
Ч'И
Таблица 4.27. Характеристики процесса хлорирования этилена до 1,2-дихлорэтана в присутствии катализатора при 35-86 °C на
лабораторных установках и при 42-90 °C на промышленных установках
В, г/кг (1-4), кг/т (5-8) 0,028-0,042 0,009-0,015 0,015-0,021 0,012-0,018 Lf) О m «п о о о о 0,012 0,020 ’Г ш ГЧ го о о о о 0,018 0028
Р.К, г/кг (1-4), кг/т (5-8) 0,303 1,0 0,09 0,055 сч rq о о о о 0Г1 6‘0 40 rq —< о о о о о о 0,039 0,012 0,010
^р-и’ г/ч (1-4), кг/ч (5-8) О 2 ° ° ох 2 о о о о 500 255 170 goo 2 qs 3 О'1 мо
тк, г/ч при О о rq а о L; £ о о о о о о О тг О LT? О 04 40 тг ос •—< 1—< »—<
Do iZl 0 0,044 0,170 0,352 о о о. о о о о 5,0 4,4 4,0 О 04 \О ос г—1 т—1 »—1
с* £ ГЧ 40 40 сг г^. О гг —< _ тГ ОО 8838 23420 884 1768 2652 8839 17678 04 LT О я £ й £ S £
т пр> г/ч (1-4), кг/ч (5-8) при о о ЧО \О чО 40 ° 40 ЧО 4419 4419 О о О' О> 04 Tji Mt4 Tf 04 04 04 тГ тГ
о о 0 44 | 170 352 О о 884 884 ООО rq rq rq тГ тГ тГ 40 40 \О rq rq rq ^"4 Ч 1—4
г ** '“'р.М > мае. % 2,0 0,1 2,0/0,1 2,0/0,1 2,0 4,6 0,09 0,11 1,0/0,11 1,7/0,10 2,3/0,09 1,0/0,10 1,6/0,12 2,7/0,15
* 00 1 « 1 ш СГ 40 о со + + О о 240 550 1 796 1592 120 + 5 207 + 5 278 + 5 120+12 190+12 324+ 12
3* О о О О g О О о g о о (Ч v (Ч (М 2000 5300 1000 2000 300 1000 2000 200 1000 2000
Пример —< rq гГ Tf №5 №6 №7 оо
Суммой представлено количество загруженного катализатора в реактор, работающим при f; = 84-86 °C (примеры 1-4), 85-90 °C (примеры
5-8), и в реакторы, работающие при t2 = 35-36, 42-48 °C соответственно.
Дробью представлены концентрации катализатора в реакторе, работающем при fb и при t2, соответственно.
Влияние некоторых факюров на процесс
прямого хлорирования этилена
В процессе получения дихлор >1.111.1 кидкофа шым хлорированием ни
дела может быть переработан хлор пр.п гически любого состава. В ташки
мости от наличия или отсутствия на преднрияiни узла сжижения хлора в
процессе хлорирования этилена может быть использован испаренный, элек
iролигический или абгазный хлор, различающиеся своими составами. Хлор,
выходящий из электролизеров, — элекгролигический хлор. Он содержит
Vo 98 об. % основного вещества, а также такие примеси, как кислород, а йн,
сиоксид углерода и немного водорода. Электролитический хлор можем быть
направлен на узел конденсации. При этом часть хлора сжижается, а часть
вместе с примесями остается в газообразном состоянии. После разделения
ы тообразного и жидкого хлора, жидкость испаряют при нагревании с по
лучением испаренного хлора с содержанием основного вещества более 99%.
Полученный при разделении фаз газообразный, так называемый, абгазный
хлор содержит от 48 до 85% мол основного вещества и является, по сути
дела, отходом хлорного производства. В производстве дихлорэтана жидко
фа птым хлорированием этилена можно использовать в качестве сырья лю
(>оп из перечисленных видов хлора.
В некоторых случаях используют содержащие хлор абгазы из других произ
иоде гв, а также хлор, получаемый в качестве побочного продукта. Например,
при электролизе расплава хлорида магния в качестве товарного продукта но
лучают металлический магний, а в качестве побочного — газообразный хлор.
В ряде случаев при хлорировании этилена используют разбавленный этилен
пли смесь газовой фракции, содержащей этилен.
Распространению процесса получения дихлорэтана способствуют следую
щие обстоятельства:
- возможность использования чрезвычайно простой технологии с примене
пнем весьма ограниченного набора несложного оборудования;
- небольшие накладные расходы;
- простота обслуживания;
- возможность использования хлора любого состава, а также концентри
рованного или разбавленного этилена без оказания существенного влияния па
качество товарного продукта.
В настоящее время жидкофазное хлорирование этилена является наибо
нс распространенным методом получения дихлорэтана в крупнотоннажных
хлорорганических производствах. Реакция хлорирования этилена в жидкой
фа те сопровождается образованием побочных высококипящих продуктов
чю ведет к снижению выхода целевого продукта и ухудшению техники .>ко
помических показателей процесса. В связи с этим, поиск путей повышения
селективности процесса жидкофазного хлорирования этилена является до
с i.iточно важным с точки зрения повышения эффективности комплекса ви
пплхлорида в целом.
На селективность процесса влияют:
- температура;
- давление;
- мольное соотношение реагентов;
качес । во <. ырья
иш ибигоры;
- дисперсность подаваемых реагентов;
- циркуляция реакционной массы и т.д.
Рассмотрим влияние на процесс жидкофазного хлорирования этилена неко
торых параметров и пути усовершенствования этого процесса.
Влияние температуры процесса
Процесс хлорирования этилена в среде дихлорэтана может осуществляться
при температуре ниже температуры его кипения [947, 1096], так называемый
«низкотемпературный» процесс. Если же процесс проводится при температуре
выше температуры кипения дихлорэтана [1036], то такой процесс называется
«высокотемпературным».
В низкотемпературных процессах тепло реакции снимается за счет ох-
лаждения реакционной массы во встроенных в реактор или выносных теп
лообменниках [34, 197, 199, 948, 982]. Выносные теплообменники могут бы п,
установлены на линии циркуляции жидкой фазы или на линии ввода в реак
тор жидкого дихлорэтана из какого-либо аппарата другой стадии процесса
За счет снятия в теплообменниках реакционного тепла процесс проводи ня
при температуре от 20 до 80 °C [999, 1035, 1038, 1099]. Однако температура
может поддерживаться и в более узких пределах, например, от 40 до 50 С
[1097].
В низкотемпературном процессе полностью теряется тепло реакции и тре
буются значительные энергетические затраты на охлаждение реакционно!!
массы. При этом дихлорэтан-сырец выводится из реактора в жидком виде и
подвергается сначала водно-кислотной промывке от катализатора — хлор
ного железа, а затем водно-щелочной промывке от хлористого водорода —
побочного продукта процесса. Расход хлорного железа при этом составляе!
0,15-0,50 кг на 1 т дихлорэтана, поскольку хлорное железо выводится из реак
тора вместе с целевым продуктом - жидким дихлорэтаном. Расход сточной
воды — результат промывки дихлорэтана — достигает 170 кг на 1т дихлор
этана. Более того, отмытый дихлорэтан-сырец необходимо подвергнуть обез
воживанию на ректификационной колонне азеотропной осушки.
Преимуществом низкотемпературного процесса является высокая селек
тивность, достигающая 99,0-99,9%.
Необходимость использования вторичных энергоресурсов, а также реше-
ния экологических вопросов (сокращение количества сточных вод) в последние
два десятилетия вызвало во всем мире переориентацию на использование вы
сокотемпературных процессов.
В высокотемпературных процессах, осуществляемых при температуре
выше температуры кипения дихлорэтана, которая колеблется в зависимости
от давления в реакторе, тепло реакции частично или полностью утилизиру
ется за счет отбора дихлорэтана из реактора в виде паров с последующим
обогревом ими одной или нескольких колонн ректификации. Для достиже
ния этого можно использовать несколько способов. Например, реактор ян
ляется своего рода кипятильником колонны ректификации образующегося
днмлорхана [ И, 197, 199, 968, 985 987, 990, 1О87|, к<нд.1 пары из реак юра
подаются п пи । колонны, а кубовая кндюн и. из колонны мекает в реактор,
(руки! способ заключается в том, ч ю нары ди к лор папа из реак гора в качес
inc i реющею агент направляю! в один или несколько кипятильников ко
ло1Н1 ректификации.
Процесс проводи гея в широком интервале температур: ог не менее 8< "(
|9Ч,| и до 95 °C |1059], 83-160 °C [994], 100 160 °C 11077], 84-102 °C |1087|, 110
Г.0 "С [1031], 75-200 °C [954, 1032], 90 160 °C [1078, 1098], 105-225 °C [ 10811,
I L”C |1082].
11меегся целый ряд разработок, в которых процесс осуществляют в двух ре
ак юрах, имеющих разную температуру. Так, в одном температура поддержива
। и я ниже температуры кипения — на уровне 50-55 °C [1005], 20-45 °C [ 10881,
*0 65 °C 11053], 20-75 °C [1032], а в других случаях поддерживается темпера i у
ра кипения реакционной среды. Жидкий дихлорэтан-сырец из низко гем пера
। \ рпого реактора переводится в высокотемпературный, из которого весь обра
вжавшийся дихлорэтан выводится в виде паров на дальнейшую переработку.
При этом катализатор не выводится с парами, а остается в жидкой фазе и вмес
1с с дихлорэтаном возвращается в первый реактор. Такая схема способст вуе i
i ни кепию расхода катализатора.
Тепло реакции можно использовать также для подогрева жидких или изо
образных продуктов, в частности, для обогрева теплообменников, в которых
веде гея нагрев и испарение воды с получением пара низкого давления.
Вместо паров дихлорэтана в кипятильники можно подавать жидкий пе-
регретый дихлорэтан из реактора. Так, в ряде случаев процесс проводя т при
давлениях, подобранных таким образом, чтобы хлорирование протекало
при 20-160 °C в жидкой, но не кипящей реакционной массе [959, 964, I 1211
Температура может составлять также 20-200 °C [955], 75-105 °C [1124] или
75-200 °C [1034]. При этом процесс проводят в нескольких реакционных зо
нах, в каждой из которых поддерживается определенная температура.
В последнее время чаще всего процесс хлорирования этилена осуществляе i
с л и среде кипящего дихлорэтана при температурах 90-110 °C. Как было ука ыпо
выше, количество тепла, выделяющегося при получении 1 г-моля дихлорэтана,
дос гаточно для испарения 6 г-молей стороннего дихлорэтана [197]. Э го обе гоя
юльство и определяет пути утилизации тепла реакции, в частности, разработа
ны и реализованы в промышленности технологические схемы процесса прямо
го хлорирования этилена с использованием тепла реакции для ректификации
образующегося дихлорэтана [998].
При правильной организации аппаратурного оформления процесса реак
ционного тепла вполне достаточно для ректификации не только дихлор на
на-сырца, но и дихлорэтана со стороны. Экономия пара может при этом до
i гигать 50%, что позволяет уменьшить издержки производства на 18 20%
[997, 1002].
Еще одно преимущество связано с тем, что дихлорэтан из реактора вы
водят в парообразном состоянии и поэтому катализатор практически пол
иостью остается в реакционной зоне. За счет этого отпадает необходимое i ь
в промывке дихлорэтана-сырца от растворенных в нем катализатора и хло
роводорода.
Однако селективное гь такого процесса, по сравнению с хлорированием при
температуре 50-70 °C, несколько снижается из-за ускорения реакций замести
тельного хлорирования дихлорэтана. В промышленности при эксплуатации
многотоннажных процессов, к которым относится синтез дихлорэтана, даже
незначительное повышение селективности процесса дает существенный поло
жительный эффект.
Авторами в лабораторных условиях было исследовано влияние температу-
ры на селективность процесса хлорирования этилена в жидкой фазе. Сначала
хлорирование этилена проводилось в отсутствие катализатора и ингибитора в
реакторе с насадкой при трех температурах (20°, 40°, 60°). Отбор производился
из жидкой фазы. Соотношение этилен : хлор составляло 1:1. Объем реактора
равнялся 250 мл.
По полученным данным был построен график зависимости выхода по-
лихлоридов от температуры реакции (рис. 4.89). Конверсия этилена в этих
опытах составляла 80-85%, а конверсия хлора практически равнялась 100%.
Из графика видно, что при повышении температуры до 60 °C содержание ди-
Рис. 4.89. Зависимость выхода полихлоридов от температуры: а — в отсутствие катали-
заторов и ингибиторов (1 — ди-, 2 — три-, 3 — тетра- и 4 — пентахлорэтан); 6 — в при-
сутствие 0,1 мас.% FeCl3
хлор ii.iu.i в реакционной массе iiaaaci с 37 'Я до 10%. При >юм содержание
। рнхдорпапа вырастает с 55 до 62% масс., а содержание высших цолихлори
цон с 7 до 29% масс.
Затем были проведены исследовании в ирису гс гвии хлорного железа, коп
ши।рации которого поддерживалась равной 0,1 мае.% , а избыток осилена —
> МОЛЫ1.%.
Данные (рис. 4.89) показали, что выход полихлоридов с повышением темпе
। >а । у ры рас те г. Гак, с увеличением температуры от 45 до 80 °C выход три хлор > га
п.। и сменяется от 0,28 до 0,68% мол.
Но расходу катализатора, электроэнергии, пара, а также наличию гехполо
। инее кич с точных вод, низкотемпературный процесс существенно проигрывае!
по сравнению с высокотемпературным. Основными недостатками последнею
являются более низкая селективность процесса за счет увеличения скорое ги
реакции заместительного хлорирования с ростом температуры и за счет хлори
рования этилена и дихлорэтана в паровых пузырях, барботирующих в жидкой
р> акционной массе, при отсутствии в них ингибитора реакций замести гельно! о
хлорирования.
Влияние давления процесса
Давление в реакторах жидкофазного хлорирования поддерживается в до
। |аючпо широких пределах. Оно может быть атмосферным [948,955, 982, 1077,
10 9| пли повышенным [955] и даже пониженным 54 кПа [1035, 1038].
'Гак например, предлагается проводить процесс при давлении 25-180 кПа
| Ю80. 1087], 80-900 кПа [1031, 1124], 0,1-1,5 МПа [964, 994, 1032, 1034, 1082| и
о..' .’,0 МПа [1081].
В ряде случаев при синтезе дихлорэтана давление в хлораторе следуе т под
церж’ивать на уровне, обеспечивающем протекание процесса при температуре
ниже температуры кипения дихлорэтана [959, 964, 1121,1098].
Повышение давления положительно сказывается на увеличении скорости
реакции и съема продукта с единицы реакционного объема. Однако при этом
происходит увеличение температуры процесса, что негативно отражае1ся па
го селективности. Данное положение было подтверждено исследованиями в
л...раторных [1105] и промышленных [1092] условиях. Тем не менее, в новей
ни х । схоологических разработках давление процесса поддерживается на у ров
пр 0,3 0,5 МПа. Этому способствует использование новых эффективных ката
пнлгоров и ингибиторов, а также разработка новых конструкций реакторов,
пн (воняющих работать с мелкодисперсными формами реагентов и паровых
ну ii.ipcii.
Влияние состава жидкой фазы, а также некоторых примесей
В к шестве жидкой фазы в процессе хлорирования этилена могут использо
пл пн я разные соединения. По методу Курме хлорирование этилена проводи!
. и под давлением в среде жидкого хлора [939, 940]. Этот метод из-за сложное
in иппаратурного оформления и низкой селективности (около 90%) не н нпел
практического применения. Жидкой фазой для хлорирования могут служи и.
хлорис Ii>in водород и/iii органические рас iнори ie;in, такие как геграхдорид yi
нерода, хлороформ, reipax/iop.xaii, пенгахлор хан [941 443, 1101], iiiiiponap.i
фиши 19441 и др.
Для повышения выхода целевого продукта процесс хлорирования миле
новых углеводородов предлагается проводить в среде три, геграхлорпропапа
и/или бутана [1079), или в жидкой среде, состоящей, главным образом, н i ди
хлорэтана, содержащего не менее 0,003% тетрахлор- и/или гексахлор 1,3 б\
тадиена.
В качестве жидкой среды можно использовать образующийся дихлорэтан и
присутствии небольшого количества хлорированных ненасыщенных углеводо
родов, например, перхлорэтилена, гексахлорбутадиена-1,3 или их смеси в коли
честве не менее 0,003% [1073, 1078]. Указанные примеси добавляют или получи
ют в качестве побочных продуктов реакции.
Предлагается проводить процесс, в котором с помощью газлифта и термо
сифонного эффекта циркулирует реакционная масса — дихлорэтан, содержа
щий три-, тетра и пентахлорэтаны [986, 987]. Авторами было отмечено, что
при проведении высокотемпературного жидкофазного хлорирования этилен.i
снижение вывода кубового продукта из реактора ведет к накоплению в жидкой
реакционной массе высококипящих примесей, в основном три- и тетрахлор на
нов. Это, в свою очередь, снижает селективность процесса.
Кроме того, процесс предлагается проводить в растворителе, в качестве ко
горого могут быть использованы, как сам дихлорэтан, так и алифатические yi
леводороды: n-гексан, изооктан, изододекан и смесь парафинов; ароматические
углеводороды: бензол, толуол, ксилолы, этилбензол, кумол; галогенсодержащпг
углеводороды: бромэтан, дибромэтан, трихлорэтан, дихлорбутан, хлорирован,
хлорбутан [1099], гексахлорбутадиен [1052].
При проведении хлорирования этилена в среде кипящей реакционной мае
сы температура процесса определяется температурой кипения используемого в
качестве жидкой среды растворителя.
В некоторых случаях хлорирование этилена в среде различных хлоруглево
дородов способствует повышению селективности процесса, но усложнение с га
дий, связанных с выделением товарного дихлорэтана, препятствует промыт
ленному распространению таких процессов.
В результате единственно используемой в производстве жидкой фазой для
хлорирования этилена является дихлорэтан, поскольку, играя роль растворите
ля, он не вносит новых компонентов в реакционную зону, затрудняющих очис i
ку получаемого продукта, и не требует использования глубокого холода.
Влияние мольного отношения этилена к хлору
Этилен в среде дихлорэтана хлорируют, варьируя мольное соотношение
этилена к хлору в очень широких пределах: на уровне (0,9-^ 1,2): 1 [948, 982, 994,
999, 1077, 1087]. Процесс можно проводить и при почти эквимолярном соотно
шении этилена с хлором [1081].
Фактор соотношения реагентов существенно влияет на селективность про
цесса. Избыток хлора повышает выход трихлорэтана и других полихлорзаме
щенных этан-этиленового ряда. При этом возможны реакции в паровой фазе,
I ii i пчаг работы с хлором, еодер к.нцнм кислород, — oiiauioiu. попирания
i nipopi .11111414mix соединений с получением <оляпои кислоты и сажи. В резуль
и нрош ходя) ыбивки ।рубок и их коррозия в холодильниках конденсаторах
и * выходе а<яазов из реактора.
11 к>ы ток пилена, казалось бы, наоборот, должен способе гвова ь снижению
|*ы*1*д<1 полихлоридов. Однако лабораторные исследования показали, что с уве
чип пнем и юыгка пилена также происходит рост выхода полихлоридов, прав
i i и ном случае по более пологой кривой, нежели при повышении избытка
юра
Данные по зависимости выхода полихлоридов от соотношения исходных
pi пипов содержатся в [1105]. Эксперименты по хлорированию этилена
нроподилп в интервале 70-120 °C при соотношении исходных компонентов
in Ui"i поп» избытка этилена до 20%-ного избытка хлора. В качестве ката-
ли ытора реакции аддитивного хлорирования и ингибитора реакции замес
in к лыки о хлорирования использовали хлорид железа (III), концентрация
i.iiii>poio в реакционной массе составляла в среднем 0,04%. Зависимости
выхода фихлорэтана от соотношения исходных реагентов представлены па
pm I 90щ б.
И 1 полученных данных видно, что во всем исследованном интервале гем
шр.п ур наблюдается увеличение выхода полихлоридов при соотношении
< 1 ( II, выше стехиометрического. Из рис. 4.90д следует также, что выход
।рнхлорэгапа несколько возрастает и при значительном избытке этилена.
< ледовагельно, при проведении процесса синтеза дихлорэтана оптимальный
и ini.iioK этилена находится в пределах 5-10%. При увеличении температуры
реакции с 83,5 до 120 °C увеличивается выход трихлорэтана (рис. 4.91) при
л юном избытке этилена. Необходимо отметить, что увеличение избытка эти
lu lu несколько ослабляет температурную зависимость выхода полихлоридов
(рис. 4.92).
В промышленных условиях были получены несколько другие результаты.
Vi ыповлено, что выход полихлоридов уменьшается при увеличении молык»
то соотношения этилен : хлор до 1,2:1 [1091, 1092]. Дальнейшее увеличение и i
бы1ка этилена ведет к проскоку этилена в парогазовую фазу. Соответственно,
происходит увеличение сброса его в атмосферу вместе с абгазами. Обычно для
предотвращения этого абгазы перед сбросом в атмосферу направляются в ап
параты термического или каталитического окисления, где сжигаются этилен,
дихлорэтан и отводимые с ними другие хлорорганические соединения. Сжи
|.шие больших количеств этилена экономически нецелеобразно. Поэтому в
производствах большой мощности абгазы из основного реактора подают во
к помогательный, так называемый дохлоратор, который обычно рассчитан па
переработку 20%-ного избытка этилена [1091, 1092].
Влияние ингибиторов
В отсутствие ингибиторов при -25 °C реакция идет с образованием в ос
ионном дихлорэтана, выход которого составляет 93%. При 0 °C образование
|рихлорэтана превышает 30%, а выход дихлорэтана уменьшается до 70%. При
20 °C продукты реакции уже содержат только 28% дихлорэтана, а содержание
Рис. 4.90. Зависимости выхода 1,1,2-трихлорэтана. а — от соотношения этилена с хло
ром при разных температурах (у кривых) время реакции — 4 ч; б — от продолжитель
ности реакции при 83,5 °C и разных мольных соотношениях этилена с хлором (у кри
вых)
трихлорэтана и полихлоридов достигает 70%. При этом выход трихлорэтана
возрастает до 53%, высших полихлоридов этана — до 19% [197].
Энергетически примерно с одинаковой степенью вероятно аддитивное
хлорирование по бимолекулярному и радикальному механизмам. Добавка
ингибиторов (хлорное железо) будет тормозить процесс аддитивного хлори
рования, протекающий по радикальному механизму и катализировать бимо
пекулярный процесс. Такой прием широко используется в практике произ
водства дихлорэтана.
В лабораторных условиях авторами было исследовано влияние хлорида же
леза (III) на хлорирование этилена в жидкой фазе.
6j
Рис. 4.91. Зависимость выхода 1,1,2-трихлорэтана от продолжительности реакции при
мольном соотношении этилен:хлор - 1,05 (й), 1,2 (б)
Рис. 4.92. Зависимости выхода 1,1,2-трихлорэтана от температуры при разных мольных
соотношениях этилена с хлором (время реакции 4ч)
Рис. 4.93. Зависимость выхода полихлоридов от концентрации FeCl3 в реакционном
массе
Рис. 4.94. Растворимость хлорида железа (III) в дихлорэтане
При всех прочих равных условиях, таких, как температура, соотношение
реагентов, подачи их и т.д. изменялась величина концентрации хлорного желе
за в растворе от 0,01 до 0,1 мас.% На основании полученных данных построен
график зависимости выхода полихлоридов от концентрации хлорного железа и
системе (рис. 4.93), из которого видно, что при низких концентрациях хлорного
железа (<0,05 мас.%) и при температурах 50 и 70 °C увеличивается выход по-
лихлоридов до 2 мае. %. При увеличении концентрации хлорного железа выход
полихлоридов снижается до 0,3% при 50 °C и остается далее практически пос-
тоянным.
При 70 °C и концентрации хлорного железа 0,1 мае. % выход трихлорэтана
достигает 0,50 мае. % Оптимальная рабочая концентрация FeCl3 — в пределах
от 0,06 до 0,1 мае. %.
Увеличение выхода полихлоридов до I ? май. % при конценiрациях IcCI,
микс 0,05 мае. % указываем на нолрае laiinc при |акич концен грациях I е( 1(еко
р<п hi побочных реакций и не’дое laioniioe и, его иш ибирующей сиосоопое tn.
1авиеимоетв рае гворимое 1 и 1с( ’1,в дихлор хане ог гемнературы показана
11.1 pile 4.91.
Обычно в жидкой реакционной массе конценграцию катализатора — хло
рнда |рехвале11гпого железа поддерживают на уровне между 0,05 и 0,10 мае. %.
11а повышение селективности процесса прямо! о хлорирования и плена, по
мнению авторов [999], влияет также количество растворенного в реакционной
мае < < хлора и соотношение между расходами потоков этилена, вводимого в i о
мок иную реакционную зону и в поток дихлорэтана, подаваемого в эту зону.
11ри хлорировании этилена в жидкой фазе при 85-150 °C [1121], в реак торе
н । нержавеющей стали с добавками ванадия, а также из никеля селективное и,
процесса достигает 99,2+99,47%. Авторы считают, что это происходил в основ
ном за счет высокого содержания в реакционной массе FeCl3. Во всех случа
их концентрация FeCl3 превышала 0,09 мае. % в отдельных опытах составила
0,2 мас.%. Тем не менее, из полученных данных нельзя вывести четкую записи
мое и, влияния количества FeCl3 на селективность процесса.
Для подавления реакции заместительного хлорирования могут быть и<
пользованы и другие ингибиторы:
- хлориды металлов — Sb, Al, Sn, Pb и др. [197, 946-948];
пигропарафины, амиды, амины, бензол и его производные [949, 958-9601;
- гетрахлор- и/или гексахлор- 1,3-бутадиен;
- толуол, ксилолы, их хлор- или спиртовые производные, фенол, крезол, пи
рокагехин, пирогаллол, или их хлорпроизводные, алкилбензол 0,001-20 мае. %.
Бели эти ингибиторы обладают электрофильными свойствами, они одно
временно выполняют и роль катализаторов аддитивного хлорирования.
Для сравнения ингибирующего действия разных каталитических систем на
процесс образования полихлоридов авторами в лабораторных условиях пыла
проведена серия экспериментов по хлорированию этилена в присутствии ката
ли кпоров при температуре кипения дихлорэтана [1055].
Были изучены:
I. Хлорное железо с поддержанием концентрации ионов Fe3+ в пределах
(1.0 1-0,07%;
2. Комплексный мелкодисперсный катализатор:
— [UN—R—N—СО—R—СО]П—
СН2
СНОН
O-FeCl3
— [СН2—СН2—O]m—Н,
|де R = С6Н4, температура размягчения катализатора 270 °C, содержание в нем
I еС13 — 7,8%; катализатор не растворяется в дихлорэтане, диоксане, хлорофор
ме, тетрахлориде углерода, загружается в реактор в количестве 0,95%;
1 K.u.uiii iaгор на основе моно идиглицидиловых |фиров олигоэтиленгли
шля (марка С 593)
R —О—[СН2 —< П2—О]„,—R,
де R = ОН, CI I С2Н5, С6Н3СН2; т = 3-20,
или
R'—N—СН2—СНОН —[СН2—СН2—O]m—R",
О
де R' = , (C2H4OH)2N; R" = ОН; т = 3-20,
NZ
Катализатор представляет бурую маслянистую жидкость. Во внешнюю его
:феру входит также FeCl3;
4. Комплекс хлорида железа (III) с диглимом:
СН3—О—|СН2—СН2—О]„—CH3:FeCl3 при и = 8-10.
Комплекс может быть представлен в виде
I Fei+ \С13
СН3 СН3
Загрузка катализатора — 0,95% от реакционной массы;
5. Металлизованные окатыши, содержащие не менее 90% металлического
келеза, 1,4% углерода, 4,1% диоксида кремния, не более 0,005% серы и не более
),015% фосфора; степень металлизации 97%. Реактор заполняется окатыша
ли на половину высоты слоя дихлорэтана;
6. Металлокомплексный катализатор, представляющий соединения в соот
штствии с формулой: MCl„Lm, где С1 — хлор; М — железо (и = 2,3), алюминий
п = 3), олово (и = 2,4), сурьма (и = 5) и L — триметиламин (т = 1,3), триэтила
лин (т = 1,3), пиридин (т = 1,2,3,4), 1-(2-хлорэтил)триметиламмоний хлорид
т = 1), 1 (2-хлорэтил)триэтиламмоний хлорид (т = 1), 1-(2-хлорэтил)пириди-
зий хлорид (т = 1), пиридинхлоргидрат (т — 1) [1032].
Катализатор хорошо растворим в дихлорэтане, загрузка его составляет
1-2,2% от количества реакционной массы.
Ингибирующая способность катализаторов оценивалась по выходу побоч-
loro продукта — 1,1,2-трихлорэтана во времени. Изменение во времени выхо-
щ трихлорэтана при хлорировании с 5%-ным избытком этилена показано на
эис. 4.95.
Было выявлено, что у хлорида железа (III), комплексного мелкодисперс-
1ого катализатора с 7,8% FeCl3 на носителе и металлизированных окатышей
шгибирующая способность по отношению к реакции заместительного хло-
зирования дихлорэтана примерно одинакова. Однако при использовании
еС13 на комплексном мелкодисперсном носителе образуется повышенное
I’ll» I 95. Изменение во времени содержания 1,1,2 трихлорэтана в реакционной смеси
при хлорировании этилена на свету (83,5 °C, 5%-й избыток этилена):
I кд । или м гор (марки С-393); 2 — комплекс FeCl3 с диглимом; 3 — FeCl3 на комплексном носи
inn /- 1сС13;5 — железные окатыши
(до 0,26% за 20 ч) количество 1,1,1,2- и 1,1,2,2-тетрахлорэтанов, тогда как
при использовании двух других каталитических систем образование тетрах-
лор п анов не отмечено.
Ингибирующая способность катализатора С-393 и комплекса FeCl3 с ди1
лимом по отношению к реакции образования высших полихлоридов значи
Н ЛЫ1О слабее. За 10 ч. работы при использовании катализатора С 393 обра
1уется 11,1% трихлорэтана, а при работе со вторым катализатором — 13,5%.
Вес но подтверждает нецелесообразность использования данных соединении
и качестве ингибиторов реакции заместительного хлорирования в процессе
прямого хлорирования этилена.
При проведении опытов в реакторе, изолированном от света, было пока
I.M1O (рис. 4.96), что ингибирующая активность FeCl3 и металлокомплекспою
катализатора в реакции хлорирования этилена практически одинакова. Ис
i ледования велись при температуре кипения реакционной смеси и также с
'.% 11ым избытком этилена. Концентрация FeCl3 в реакционной массе состав
лила в среднем 0,07%.
На опытно-промышленной установке проводились исследования но
с равнению селективности хлорного железа и металлокомплексного катали
а юра.
При использовании в качестве катализатора FeCl3 был получен усреднен
ный состав реакционной массы, мае. %:
99,848 дихлорэтана,
0,109 легкокипящих компонентов,
0,043 высококипящих компонентов.
Рис. 4.96. Изменение во времени содержания 1,1,2-трихлорэтана при хлорировании
>1 плена в темноте без добавок кислорода (1,2) и при добавлении в этилен 0,02% кисло
рода (3,4); I, 4 — металлокомплексный катализатор; 2, 3 — хлорное железо
Рис. 4.97. Изменение во времени концентрации трихлорэтана в реакционной смеси
при проведении процесса хлорирования в промышленных условиях (при температуре
кипения реакционной массы, 5-10%-м избытке этилена); стрелками показано добавле
ние новых порций катализатора
Среднестатистический состав реакционной массы при использовании ме
таллокомплексного катализатора масс. %:
99,270 дихлорэтана,
0,146 легкокипящих компонентов,
0,584 высококипящих компонентов.
Из данных рис. 4.97 следует, что в отдельных анализах концентрация ди-
хлорэтана в реакционной массе при работе с металлокомплексным катализа-
Гором coi гавляла всею 97,2 98,1 m.u % >i<> мвмч шидсicbi.i гвовагь как <>
dociohhiiom уносе mci аллокомнл<ы hoio клали iaiop.i hi реакционной зоны
( i.i 16 дней испытаний всею было гагружсно 9 И к1 катали гатора), гаи и о не
достаточной ингибирующей активности no отношению к заместительному
хлорированию.
I (ервоначальное содержание FeCI, в реакционной массе составляло около
0,1 мае. %. Замешых изменений концов грации FeCl3 в ходе процесса хлори
рованил этилена не наблюдалось, несмотря на то, что катализатор не добап
лился.
Таким образом, можно заключить, что при использовании металлокомн
лексного катализатора селективность процесса прямого хлорирования этилена
при юмпературе кипения реакционной массы снижается в среднем на 0,6% но
i равнению с процессом, в котором используется FeCl3.
I Доведенные экспериментальные исследования дают основание заключи i ь,
мт из всех проверенных катализаторов наиболее оптимальным по технологи
четкому оформлению процесса, стоимости и другим факторам является Fe( 1( в
сочетании с небольшими добавками кислорода для предотвращения реакции
ыместительного хлорирования в газовой фазе.
Введение свободного кислорода в реакционную зону резко изменяет состав
продуктов реакции, повышая селективность процесса [955, 1103].
В частности, в целях повышения качества дихлорэтана для ингибиро
вания побочных реакций, протекающих в газовой фазе, используют хлор,
который содержит 5,0-10,0 мол % кислорода [1087]. Наряду с хлоргазом, в
абсорбер перед реактором вводят воздух, чтобы растворить в дихлорэтане
кислород [1081].
В последнее время в промышленности наиболее широко используется хло
рид железа в сочетании с кислородом [1091, 1092]. При этом иногда оговарива
юг, что в качестве катализатора применяется хлорид железа, а для повышения
селективности реакции используют добавку кислорода [1077].
По аналогии с этим в качестве ингибитора предлагается применять 0,5-5%
кислорода от объема подаваемого в реактор хлора, а жидкая среда должна со
держать 0,005-0,1% масс FeCl3 [959].
Авторами работы [1055] были получены зависимости выхода полихлори
дов от температуры и содержания кислорода в реакционной смеси, представ
ленные на рис. 4.98. Было показано, что при содержании кислорода в пода
ваемом хлоре 6 мол. % и 30 °C выход полихлоридов составляет около 3%, ч го
согласуется с составом дихлорэтана-сырца, образующегося в промышленных
условиях.
Из рис. 4.98д видно, что с повышением температуры от 20 до 40 °C выход
полихлоридов увеличивается незначительно. При дальнейшем повышении гем
пературы процесса хлорирования наблюдается резкий рост выхода полихло
ридов в реакционной массе, что указывает на увеличение скорости побочных
реакций.
Из рис. 4.986 видно, что при изменении концентрации кислорода в хлоре
от 2 до 8% мол. содержание полихлоридов в реакционной массе изменяется
незначительно. При этом следует отметить, что выход полихлоридов при тем
пературе 60 °C значительно больше, чем при 20 °C и 40 °C. Это, возможно.
!а
Концентрация кислорода в хлоргазе, мольн. %
Рис. 4.98. Зависимость выхода полихлоридов от температуры («) и содержания кисло
рода в реакционной смеси (б)
объясняется уменьшением растворимости кислорода в реакционной массе с
повышением температуры. При концентрации кислорода менее 2% происхо
дит, как видно из графика, резкое увеличение количества полихлоридов. Пос-
леднее обстоятельство, по-видимому, объясняется тем, что данная концен г
рация кислорода для ингибирования реакции заместительного хлорирования
недостаточна.
Были также проведены эксперименты в присутствии хлорного железа и мс-
таллокомплексного катализатора с введением в реактор небольших количеств
воздуха (в расчете 0,02% кислорода от подаваемого этилена) [1055].
Влияние влажности |м*<1гентов
Обычно па влажность реагсчнон, осоОгнпо хлора, накладываются строгие
ограничения. Эго снизано с гем, что хлор и присутствии влаги является весьма
'Орепивным в отношении коррозии агентом. Полому на практике хлор перед
подачей в реактор обычно сушат с помощью серной кислоты до остаточного
содержания влаги 2-10 3 ppm мас.% и менее. В принципе влажность реаген
ив о|рицательно сказывается на селективности процесса. В присутствии воды
хлорное железо гидратируется с образованием комплексов, содержащих разное
количество молекул Н2О. При этом хлорное железо дезактивируется. Для спя
ывания влаги иногда применяют хлорное железо, которое образуется за счет
реакции с помещенной в реактор металлической насадкой [1000], или за счет
реакции образующегося хлористого водорода с металлической стенкой реакю
ра В некоторых случаях хлорное железо предварительно смешивают с дихлор
паном и полученную смесь подают в реактор.
Авторы работы [1061] предлагают проводить процесс при температ уре 20
НО “(’ в присутствии катализатора — кислоты Льюиса, концентрация которой в
реакционной массе поддерживается на уровне 60-200 ppm масс. Концен грация
хлора в реакционной массе должна быть на уровне 5-20 ppm масс, а концен гра
цня влаги — менее 200 ppm масс. Мольное соотношение содержащихся в реакци
он ной массе кислоты Льюиса и влаги, по крайней мере, должно быть равно 1.
Варианты технологической схемы процесса жидкофазного
хлорирования этилена
Технология и аппаратура процесса прямого хлорирования этилена,
осуществляемого при температуре ниже температуры
кипения дихлорэтана
До начала 1980-х годов в промышленности преобладал низкотемператур
ный процесс [951, 958,959,962, 964, 965,974-980]. В этом случае в произволе
1 вах небольшой мощности в качестве реактора использовался барботажнып
аппарат с мешалкой или без нее. Для съема тепла используется встроенный
или выносной циркуляционный теплообменник. Реакционная масса за c’ici
«рлифта или насоса непрерывно циркулирует либо внутри реактора, либо но
циркуляционному контуру, состоящему из реактора с восходящим потоком
и выносной трубы и циркуляционного теплообменника с нисходящим по
юком. В реакционную массу, содержащую катализатор и при необходимое
। и ингибитор, через барботеры или инжекторы вводят газообразные хлор и
пилен. Образующийся дихлорэтан выводится в жидком виде. Процесс не
дут непрерывно. Необходимая концентрация катализатора обеспечивается
либо постоянной подачей его в реакционную зону, либо за счет пропускания
части циркулирующего потока через специальную емкость с катализатором.
При использовании в качестве катализатора FeCl3 реакционная масса может
пополняться катализатором за счет коррозии стенок реакционного аппарата
и случае изготовления его из стали или содержащих железо сплавов [947,
1099, 1151].
Принципиальная технологическая схема получения дихлорэтана низко
емпературным хлорированием этилена представлена на рис. 4.99.
Сухие пилен и Vicki роли iii'ici кип хлор подаюкя и нилипдрн'кч кую
часть / реактора хлорирования в поюк циркулирующего через 1еплоо(*»меп
инк 2 дихлорэтана. Реактор выполнен из углеродистой стали. Абгазы, содержа
щие инергы, непрореагировавший этилен, хлористый водород и нары дихлор
пана, охлаждаются до -30 °C в рассольном холодильнике 3 для конденсации
дихлорэтана и далее поступают на санитарную колонну. Конденсат вместе с
реакционной массой из хлоратора собираются в сборнике сырца 4. Из этого
сборника дихлорэтан, содержащий хлориды железа и хлороводород, вместе с
водой поступает в линию всасывания насоса 5, где происходит перемешивание
дихлорэтана, свежей воды и кислой циркулирующей воды. Полученная смесь
насосом 5 подается для отстаивания в емкость 6
Кислая вода возвращается в основном в линию всасывания насоса 5, а ди
хлорэтан, промытый от хлоридов железа и хлороводорода, собирается в сборни
ке 7, откуда дихлорэтан, насыщенный кислой водой, поступает в линию всасыва
ния насоса 8, куда подается свежий раствор щелочи и циркулирующая щелочная
вода. Образовавшаяся в насосе 8 перемешанная смесь поступает для отстаивания
в емкость 9. Выделенная при этом щелочная вода возвращается в основном в ли
нию всасывания насоса 8, а частично, так же, как и часть кислой воды, выводи гея
на очистку. Нейтрализованный дихлорэтан из отстойной емкости 9 собирается
в сборнике 10. Из этого сборника насосом И дихлорэтан вместе со свежей водой
для отмывки от щелочи поступает для отстаивания в емкость 12. Из емкости 12
вода поступает в основном на циркуляцию в насос 11. Отмытый нейтральный
дихлорэтан выводится в сборник 13, откуда насосом 14 подается на двухколон
ную систему ректификации. В колонне 15 происходит азеотропная осушка ди
хлорэтана и отделение от него легкокипящих примесей. Сухой кубовый продую
этой колонны поступает в колонну 16 для выделения дихлорэтана-ректификата.
Кубовая жидкость из колонны 16, состоящая в основном из трихлорэтана, и дне
гиллат колонны 15 передаются на утилизацию, например, в производство хлор
органических растворителей или на сжигание. Дихлорэтан-ректификат в виде
дистиллата колонны 16 направляется в складскую емкость.
В работе [1171] рассмотрен промышленный процесс получения дихлорэтана
прямым хлорированием этилена при низких температурах. Использован реак
гор, конфигурация которого соответствует барботажной колонне с наружным
циркуляционным контуром. Реактор с рабочим объемом 19 м имеет прои то
дительность 54 тыс. т/год.
В патенте [ 1053 ] предложено хлорирование этилена проводить при 20 65 °C.
Жидкий продукт направляют во второй реактор, работающий при температуре
кипения реакционной массы (84-90 °C), где за счет тепла реакции дихлорэтан
испаряют, отделяя его от катализатора. При этом соотношение количества по
даваемого на испарение дихлорэтана из реактора хлорирования этилена при
20-65 °C к количеству получаемого непосредственно в реакторе хлорирования
при температуре кипения реакционной массы можно изменять в пределах <п
1 до 4,5. Накапливающийся в реакторе хлорирования при 84-90 °C катализа
тор непрерывно или периодически в виде раствора в дихлорэтане в количес
гве 2-12 мае. % от количества подаваемого в него на испарение дихлорэтана
направляют в реактор(ы) хлорирования при 20-65 °C. В случае дезактивации
катализатора этот раствор выводят на утилизацию. Испаренный дихлорэтан
[индене иpyio i. Чае i ! ci о во (вращают в реак iop хлорировании при темпера i) р<
.мнения дихлор лапа для поддержания заданного уровня реакционной маны
к гавшаяся часть дихлор лапа является целевым продуктом. Проведение про
щсса в двух реакторах, один из которых является испарителем наработанного
фодукта, позволяет снизить расход катализатора почти в пять раз.
Для обеспечения заданного времени пребывания реагентов в реакционно)!
tone цилиндрическая часть реактора имеет отношение высоты к диаметру не
зенес грех [ 1096].
С этой же целью в ряде конструкций предусматривается секционирование
«еакционной зоны тормозящими устройствами в виде тарелок со свободным
ечением 5-20% [1152].
Для интенсификации теплосъема предложена конструкция реактора в виде
шастинчатого теплообменника. Реакция идет в пленке жидкого дихлорэтана,
юторый стекает по поверхности пластинок, охлаждаемых до температуры ниже
очки росы дихлорэтана в условиях реакции [1153].
Конверсия этилена за счет использования его с некоторым избытком но oi
юшению к хлору обычно не ниже 97%, а селективность образования дихлор
[тана выше 99%.
Дихлорэтан, выходящий из реактора в виде реакционной массы, содержи!
кроме органических примесей остатки катализатора — хлорида железа. Для
Удаления хлоридов железа очистка может проводиться либо с помощью твер
1ых адсорбентов, таких, как активированный уголь, бокситы, фуллерова земля,
1ктивированный бентонит, либо промывкой водой или водными растворами
гислоты и щелочи [1128, 1155-1157]. На практике обычно применяются метод
фомывки водными растворами кислоты и щелочи.
Однако необходимость использования вторичных энергоресурсов, а также
Решения экологических вопросов (в частности, сокращения количества сточ
5ых вод) вызвало переориентацию в мировой промышленности на использова
ие высокотемпературных процессов [948, 953, 957, 960, 966, 968, 981-1000].
Технология и аппаратура процесса прямого хлорирования этилена,
осуществляемого при температуре кипения дихлорэтана
Тепло реакции хлорирования этилена можно снимать и в результате йена
эения дихлорэтана и выводить при конденсации в дефлегматоре. Коэффициеи
Ъ1 теплопередачи при этом больше, чем в случае системы жидкость-жидкость,
юэтому для такого теплосъема поверхности будут несколько меньше. Кроме
ого, снятие тепла реакции за счет испарения дихлорэтана позволяет отбирать
ютовый продукт из паровой фазы.
Количество реакционного тепла примерно в семь раз больше, чем это не-
>бходимо для испарения образующегося в ходе процесса дихлорэтана, поэто-
му есть возможность утилизации оставшегося количества тепла. Наиболее це-
1есообразно использовать его для ректификации дихлорэтана-сырца, а также
:тороннего дихлорэтана. Для этого реактор хлорирования конструктивно или
: помощью трубопроводов совмещают с колонной ректификации, и он выпол-
няет функцию кипятильника колонны. В ряде случаев отходящие из реактора
зары дихлорэтана направляют в один или несколько кипятильников ректифи-
к.щионных колонн для их обогреня *Н>8, *Ж11 II процгчих проводимых
при повышенном давлении, ДЛЯ OOOipcH.l KIIIDIIIIJIJIHKOll КОЛОНН IkHOHI.iyiOl
циркулирующий поток жидкой реакционном мт । ы
Для обеспечения надежного подавления реакции «амсч i тельного хлориро
нация в обеих зонах совмещенного агрегата применяю! комплексный ипгиби-
юр — НеС13 + О2. Кислород работает как в парогазовой фазе реактора, гак и в
колонне ректификации [960].
I (оскольку образовавшийся дихлорэтан отбирается в паровой фазе, он не со
дер кит органических или неорганических соединений, используемых в качестве
анализатора и/или ингибитора, и, следовательно, не нуждается в промывке.
Один из вариантов принципиальной технологической схемы получения ди
хдорэгана хлорированием этилена в среде кипящей реакционной массы с огбо
ром получаемого продукта в парах представлен на рис. 4.100.
.) силен и электролитический хлор с влажностью не более 2-10 3 мае. % посту-
паю! в реакционную зону I совмещенного агрегата синтеза дихлорэтана. Реак
циоппая зона заполнена дихлорэтаном, содержащим определенное количество
хлорного железа. За счет тепла реакции происходит испарение части реакцион
пой массы. При этом пары дихлорэтана и хлорорганических примесей посту-
пают в ректификационную зону 2 совмещенного агрегата синтеза дихлорэтана.
В ректификационной зоне 2 происходит очистка дихлорэтана от легкокипящих
н нысококипящих примесей. Дихлорэтан-ректификат выводится с промежу
точной тарелки зоны ректификации и через промежуточный холодильник 4
направляется в емкость-сборник. Пары дихлорэтана вместе с легкокипящими
примесями сверху ректификационной зоны поступают в дефлегматор 3 и пос-
ле конденсации в основном в виде флегмы возвращаются в ректификационную
«опу, а частично, выводятся на переработку или сжигание.
1’ис. 4.100. Принципиальная технологическая схема получения дихлорэтана хлориро
яанием этилена в среде кипящей реакционной массы с отбором получаемого продукта
и нарах (1-6 см. текст)
1 ю
Для исключения накопления в реакционной маис полихлоридов u гмолш i 1.1 х
шределе)।ное количсс изо реакционной массы ciiu.iy реакционной зоны / arpeiaia
lociynaer в колонну ректификации или перегонный ку(> осяеглигель5, где виде
i я юте я высококинящие и смолистые соединения. Лги продукты выводятся uni
ту осветлителя и направляются на сжигание. Осветленная жидкость выводи ня
верху, конденсируется в дефлегматоре 6 и возвращается в реакционную топу
для снятия тепла реакции. Часть конденсата из дефлегматора 6 подается на пере
эаботку, например, в производство хлорорганических растворителей.
При более высокой температуре хлорирования в кипящей реакционной
лассе селективность процесса несколько ниже по сравнению с низкотемпера
гурным хлорированием. Однако этот процесс обладает рядом достоинств:
- исключение из технологической схемы узла промывки, в котором обра ту
чея большое количество сточных вод, нуждающихся в обязательной очис ткг
1еред сбросом в канализацию;
- экономия пара за счет использования тепла реакции для ректификации
тбразующегося и стороннего дихлорэтана.
Технологически этот вариант процесса обладает большей суммой досто
1нств по сравнению с низкотемпературным хлорированием, и по этой причине
три создании новых производств винилхлорида в 1990-2000-х годах ему отдают
тредпочтение.
Некоторые технологические и конструктивные
особенности процесса
Вывод дихлорэтана из реактора
В промышленных условиях в зависимости от технологии используют три
варианта вывода образующегося продукта дихлорэтана из реактора или реак
дионной зоны.
Вывод в жидкой фазе осуществляется почти исключительно в низкотемпе
эатурных процессах. Образующийся дихлорэтан в жидкой фазе направляется
за дальнейшую переработку — промывку и ректификацию. В последнее время
тсе чаще появляются высокотемпературные процессы, в которых образующийся
щхлорэтан также выводится из реактора в жидкой фазе. Температура в реак
оре и циркуляционной петле 90-160 °C, а давление поддерживается на уровне,
зредотвращающем испарение дихлорэтана при данной температуре [999, 1098].
Вывод дихлорэтана в паровой фазе начали осуществлять при вводе в строй
зысокотемпературных процессов. В настоящее время в большинстве такого
>ода процессов синтезируемый дихлорэтан также выводится из системы в виде
заров [994, 1036, 1074-1078, 1081, 1115-1118, 1124].
При необходимости образовавшийся высокочистый 1,2-дихлорэтан удаляю!
юсредством испарения при снижении давления в любом месте установки [1032].
Известен способ получения дихлорэтана путем подачи этилена и хлора в
диркулирукиций дихлорэтан [1172]. Процесс проводят при интенсивном пере-
лешивании с помощью перемешивающего устройства при 65-125 °C и давле-
зии 50-320 кПа, причем давление и температуру выбирают такими, чтобы ре-
1кционная смесь кипела. Из реактора отводят часть газообразной реакционной
(меси в теплообменник, дихлорэтан конденсируют и возвращают в реактор.
|li>iii<x> н >нш>мш и « mi/ншчи ф<1 км 11рсдла1аеня luinvibioitaii. реакlop
прямого хлорирования пилена, нключающнп вертикально располо/кепный
।< рмеIH'liibiH корпус, который имеет сообщенные между собой топы — ре
н-цнонную топу с восходящим потоком дихлор мана и сепарационную зону
р < |делспия паровой и жидкой фаз дихлорэтана [ 1 165|. Реакционная топа вы
iKiiiiiciia в виде, по меньшей мере, двух отдельных вертикальных реакцион
пых секций, площадь сечения каждой из которых составляет, не менее 10%
опщеп площади сечения реакционной зоны реактора. Суммарная же площадь
• ж пил всех реакционных секций равна величине общей площади сечения
р< акционной зоны реактора. При этом гидравлическими сопротивлениями,
распределителями хлора и этилена и штуцерами их ввода снабжена каждая
р< акционная секция.
Имеются разработки, в которых хлорирование этилена предлагается про
води и. при 20 65 °C с отбором продукта из жидкой фазы. В целях отделения
и дихлорэтана растворенного катализатора его непрерывно или периодичеч
кн направляют в параллельно работающий реактор хлорирования этилена при
к мпературе кипения реакционной массы (84-90 °C), где за счет тепла реакции
дихлор пан испаряют и тем самым отделяют от катализатора [1053].
Утилизация тепла реакции
Гепло реакции можно утилизировать следующим образом. Процесс синтеза
дихлорэтана осуществляют в реакторе при температуре кипения дихлорэтана
п нормальном или повышенном давлении. Чаще всего тепло реакции иегюль
пеня для ректификации дихлорэтана-сырца [959]. Пары дихлорэтана, выхо
дящие сверху реактора, направляют в выносной циркуляционный теплообмен
ник 11095] или в кипятильник колонны, в которой происходит ректификация
дихлорэтана-сырца [1036, 1074-1076, 1078]. При этом утилизируют тепло, вы
делающееся при конденсации паров дихлорэтана [1077].
Рассмотрим наиболее интересные разработки и предложения.
Фирма «Stauffer С.Со.» предложила изготавливать реактор в виде полого ко
лонного аппарата и использовать его в качестве кипятильника ректификациоп
ион колонны [968].
Гепло реакции используют для обогрева ректификационной колонны, в ко
юрой очищают дихлорэтан-сырец, полученный прямым или окислительным
хлорированием этилена [1033]. Для этого используют специальную конструк-
цию реактора или соединяют его и ректификационную колонну трубопровода
ми для парового и жидкого потоков.
По крайней мере 85% тепла реакции можно снимать в выносном тенлооб
мепнике, через который циркулирует реакционная масса и в котором образуег-
i я водяной пар. За счет этого система ректификации дихлорэтана не зависи г о г
пара, подаваемого со стороны. Количество рекуперированного тепла достигает
.’840 кДж/ч, а чистота полученного дихлорэтана — 98,73% [1098].
Согласно [1094], реакционное тепло снимается в выносном циркуляциоп
пом теплообменнике — одновременно выносном кипятильнике ректифика
ционной колонны. В колонне можно очищать дихлорэтан-сырец, полученный
прямым или окислительным хлорированием этилена.
Для этого Moiyi гакже применяться реакторы разной конструкции. Гак, и
качестве реактора предлагают использовать кожухотрубный теплообменник со
сливной грубой но центру трубчатки. Предполагается, чго процесс хлориропа
ния будет идти в трубках, а в меж трубном пространстве будет циркулировать ку
бовая жидкость колонны ректификации. Жидкий дихлорэтан по сливной трубе
будет сливаться в нижнюю часть реактора, а оттуда вместе с кубовыми колонны
направляться в межтрубное пространство. Тепло реакции можно снимать кубо
выми колонны ректификации в трубках выносного циркуляционного тенлооб
менника, а реакционную массу циркулировать через межтрубное пространство
Предлагается еще один вариант утилизации реакционного тепла. В реакторе
устанавливается несколько ситчатых тарелок и по его оси монтируется сливная
груба, по которой предполагается подавать сливаемую реакционную масса в куб
ректификационной колонны над зеркалом жидкого слоя. Поскольку в колонне
давление на 0,3-0,5 МПа ниже чем в реакторе, реакционная масса испаряется
Кубовые остатки насосом откачивают в нижнюю часть реактора [1086].
Предлагается кипятильники колонн обогревать парами дихлорэтана, пос
тупающими из реактора прямого хлорирования [1124]. В реакторе прямое хло
рирование ведут при 75-125 °C и давлении 80-900 кПа. Пары дихлорэтана кон
денсируются, например, при 108 °C и 190 кПа.
Предложен метод ректификации дихлорэтана, содержащего высококипя
щие компоненты. Ректификация осуществляется в колонне при повышенном
давлении 50-150 кПа [1125]. Выводимый из верха колонны дихлорэтан подаю!
в компрессор для повышения давления на 50-200 кПа и температуры на 7 °C и
более с последующей утилизацией тепла этого потока. Регенерацию тепла ежа
того дихлорэтана осуществляют в кипятильниках двух из пяти колонн ректи
фикации дихлорэтана или винилхлорида.
Оба этих способа очистки дихлорэтана с помощью ректификации представ
ляют интерес лишь с точки зрения утилизации реакционного тепла процесса
прямого хлорирования этилена.
Установка получения дихлорэтана представлена на рис. 4.101 [1082]. Реак-
тор 1 и сборник 2 заполняются чистым дихлорэтаном, в котором в качестве ка-
тализатора растворено не менее 1000 мг хлорида трехвалентного железа на 1 кг
дихлорэтана с pH = 2,4. Концентрация хлора в циркулирующем потоке дихлор
этана поддерживается на уровне 2-10 2 мае. % тонким регулированием расхода
этилена в потоке. Давление в реакторе 0,8 МПа, температура на выходе из реак-
тора 132 °C. Горячий циркулирующий поток поступает в промежуточный сбор-
ник 2 для сброса давления. Доля паровой фазы регулируется вентилем 3. Разде-
ление дихлорэтан-сырца и абгазов осуществляется путем конденсации в теп-
лообменнике 4, в котором нагревается котловая вода. Циркулирующий поток,
охлажденный до 128 °C, затем охлаждается в теплообменниках 5 и 6 до 99 °C.
При этом вырабатывается насыщенный водяной пар под давлением 180 кПа с
температурой 118 °C. Процесс оценивается по следующим показателям: степень
превращения этилена 99,6%, при этом на дихлорэтан — 98,9%.
В процессе в среде дихлорэтана при температуре его кипения и атмосфер-
ном давлении используется катализатор — FeCl3 в количестве (500-^20)-10 2 мае.
%. Мольное соотношение хлора к этилену поддерживают равным (0,9<-0,95):1
[948, 982].
11роцесс проводят в реакторе, который является кубом совмещенной с ним
ректификационной колонны [982]. В колонне за счет тепла реакции ведется
очистка паров дихлорэтана-сырца. Наряду с этим, для дополнительного снятия
1снла реакции жидкость из куба реактора непрерывно циркулирует через вне
шний теплообменник.
Работа с разбавленными реагентами
При работе с разбавленными реагентами [954, 962, 978, 979, 1003] наблюди
с1ся падение скорости реакции. Для увеличения последней используется боль
той избыток этилена, что ведет к его потерям. Имеются разработки, направлен
ные на уменьшение такого рода потерь этилена [953,976, 999,1004]. В основном
1го двухреакторные системы [952, 976, 1005, 1006], где во втором реакторе про
исходит дохлорирование непрореагировавшего в первом реакторе этилена.
При работе с реагентами, содержащими кислород, или при введении в ре
актор кислорода или воздуха с целью повышения селективности процесса вы
ходящие из реактора абгазы могут иметь взрывоопасную концентрацию кисло
рода с этиленом, водородом (находится в абгазном и электролитическом хлоре)
и парами дихлорэтана. В этом случае чрезвычайно важно располагать справоч
ними или расчетными данными пределов взрываемости и величиной давлении
взрыва тех или иных смесей [1191].
Для обеспечения взрывобезопасных условно ведения процесса в нервом
реакторе работаю! с Оолыпим избытком пилена, по суммарное соотношение
реагентов но двум реакторам близко к стехиометрическому. Обычно в обоих
реак юрах поддерживают температуру на уровне 20-70 °C, но иногда в одном н i
них температуру повышают до температуры кипения реакционной среды [9 18,
952, 1005, 1006].
Способ автоматического управления технологическим процессом нодуче
ния дихлорэтана из этилена и абгазного хлора хлорного производства описан
в работе [1168]. Абгазный хлор может иметь резко меняющуюся концс1ира
цию основного компонента и примесей — инертных газов. Процесс протекает и
жидкой кипящей реакционной массе образующихся продуктов с выводом лих
продуктов в виде парогазовой фазы совместно с инертами, содержащимися в
исходном абгазном хлоре. Процесс проводится в реакторе хлорирования > i и
лена с последующим дохлорированием этилена, содержащимся в абгазах этого
реактора в дополнительном реакторе — дохлораторе. Способ характеризуем
ся тем, что коэффициент избытка этилена по отношению к 100%-ному хлору
на входе в основной реактор корректируется в зависимости от концентрации
хлора в потоке исходного абгазного хлора. Чем выше концентрация хлора, тем
ниже величина избытка этилена. Автоматически постоянно рассчитывается
величина расхода этилена в абгазах основного реактора, поступающих в до
хлоратор. Пропорционально этому расходу подается абгазный хлор. На входе
в дохлоратор корректируется избыток этилена к пересчитанному на 100%-пып
хлор, поданный в основной реактор и в зависимости от концентрации этилена
в абгазах после дохлоратора.
Устройства для ввода реагентов
Разработан реактор синтеза дихлорэтана, в котором массо- и теплообмен
значительно интенсифицированы за счет применения эжекторных смесителей
на входе хлора и этилена [ 1189]. Эжекторы хлора и этилена имеют идентичную
конструкцию и установлены один над другим. Изучена работа данного реак
тора на пилотной установке производительностью 1750 т дихлорэтана в год.
Эжекторные смесители позволяют увеличить интенсивность массообменных
процессов и сократить размеры зоны реакции. Это позволяет увеличить селек
тивность процесса выше 99%.
Предложен процесс [1036], проводимый в реакторе, который снабжен вы
носным циркуляционным контуром жидкого дихлорэтана. В последний перед
вводом в реактор подают реагенты, что способствует предварительному насы-
щению дихлорэтана хлором и этиленом. Этилен и хлор подают в поток цирку-
лирующего дихлорэтана при интенсивном перемешивании в статическом сме-
сителе [1036, 1038].
Реактор аналогичной конструкции предложен в работе [1037]. Конструк-
тивное отличие лишь в том, что один из реагентов подается не в поток цир
купирующего дихлорэтана, а непосредственно в статический смеситель внутри
реактора.
Жидкий дихлорэтан направляется в реактор через сопла, в которые через
жиклер подаются хлор и этилен [1096].
Buy I pi. p.U положенном II pc.lh IO|>C Вер! 11К.ЫЫ1ОИ I руЬы подают поток ди
хлор папа с хлором. Кроме юю, iniyipi. ipyoi.i но отдельному грубопроноду
ВН0ДЯ1 п илен. 11оток газон, паров и жидкого дихлорл апа из грубы, являющей
i я одновременно зоной смешения и первой реакционной зоной, попадает в емг
кнель, где парогазовая смесь эффективно диспергируется в жидкосги и затем
поступает во вторую основную реакционную зону [954].
Во многих реакторах реагенты вводятся в нижнюю часть по кольцевым бар
(ю герам. Так, согласно [1017], хлор вводится в нижнюю секцию по кольцевому
Оарбогеру над соплом, в которое подают циркуляционный дихлорэтан.
Предлагается около 90% реагентов подавать через основные барботеры,
около 10% их стехиометрического количества — через вспомогательные вводы
. учетом содержания непрореагировавшего этилена в абгазах [1080]. Дихлор
пан удаляют из абгазов, охлаждая их до -20 °C, а оставшиеся газы разбавляю!
а ютом.
Дихлорэтан получают в присутствии тетрахлорферрата калия при темпе
рагуре в зоне реакции от 75 до 200 °C и давлении от 100 до 1500 кПа [ 1032]
В предварительной зоне смешения растворяют хлор-газ в циркулирующем
1,2-дихлорэтане, в дополнительной реакционной зоне этилен-газ переводят в
кшкодисперсную жидкую фазу с пузырьками диаметром не более 2 мм. Jia
1опкодисперсная жидкая фаза со скоростью от 0,3 до 1 м/с, при времени реак
ции от 2,5 до 25 с, рассчитанном на жидкую фазу, проходит через реакционную
ioiiy. Образовавшийся высокочистый 1,2-дихлорэтан, который содержит менее
5-10 2 мае. % хлорированных побочных продуктов, в газообразном виде удаля
с гея посредством испарения при пониженном давлении. Затем парообразный
1,2-дихлорэтан конденсируется. Чистота полученного продукта превышаез
99,95 мае. %.
Выделяющееся при конденсации тепло может, например, использоваться
для ректификации 1,2-дихлорэтана, который получается методом оксихлори
рования. Кроме того, полностью отпадает необходимость в ректификации!!
пых колоннах, необходимых при получении 1,2-дихлорэтана по методу при
мого хлорирования, для отделения легко- и высококипящих фракций. Выход
1,2-дихлорэтана составляет 99,7% в расчете как на этилен, так и на хлор. Су
щественное значение при этом имеет тонкое диспергирование этилена. Одна
ко простого диспергирования недостаточно, так как пузырьки этилена быстро
сливаются в большие пузыри, и тогда скорость реакции сильно падает. Поэтому
необходимо обеспечить предотвращение слияния этилена в большие пузыри.
1акое тонкое диспергирование этилена достигается в так называемых «стати
ческих смесителях» [1033].
Например, согласно [1174], реагенты вводят в виде пузырьков со средним
диаметром 2 мм и со скоростью 0,2-1,5 м/с.
Одним из перспективных направлений повышения селективности процесса
является реконструкция реакторов жидкофазного хлорирования этилена, на
правленная на обеспечение необходимой температурной и гидродинамической
обстановки в аппаратах [1179]. При этом необходима информация о распре
делении локальных параметров и, в частности, локальных объемных концепт
раций дисперсной газовой фазы (локального газосодержания). Аналитический
расчет структуры газожидкостного потока очень сложен, поэтому надежную
информацию Moiyi дагь только .)кспсрименг.|лы1ыс исследовании распределе
ни» локальною га юс одержания но объему реак юра. Для и ого при меняю! г нс
циальные устройства и приемы, позволяющие основную часть газооориных
реагентов растворять в жидкой реакционной среде или добиваться создания
мельчайших пузырьков реагентов размером не более 1,0+ 1,5 мм. В результате
создается эффективный массообмен между жидкой и парогазовой фазой и спи
жается объем последней до минимума, что благоприятно сказывается на новы
шении селективности процесса.
Конструкции реакционного оборудования и варианты
его усовершенствования
Конструкции реакторов для низкотемпературного и высокотемпературного
процессов постоянно совершенствуются [959,963,1012-1016]. В основном упор
делается на создание эффективной циркуляции реакционной массы и на дости
жение чрезвычайно высокой степени диспергирования газообразных реагентов
[963,986,987, 998, 999].
В случае значительного количества в реакционном объеме жидкого дихлор
этана выделившееся тепло снимается за счет испарения последнего, что предо
твращает возможность взрыва. Однако при упущении уровня жидкого дихлор
этана в реакторе такая возможность реальна.
Гидродинамические условия проведения процесса на стадии прямого хло
рирования этилена могут существенным образом влиять на конверсии реагеп
тов и селективность образования продуктов реакции. Наиболее благоприятным
следует считать мелкодисперсное распыление газообразных реагентов в жид
ком дихлорэтане. Необходимо добиваться предотвращения слияния пузырьков
реагентов в большие пузыри. При слиянии только пузырьков этилена или толь
ко пузырьков хлора происходит лишь падение скорости реакции хлорирования
этилена и, соответственно, скорости побочных реакций. При слиянии пузырь-
ков этилена с пузырьками хлора возможны реакции в газовой фазе, особен но
при высокой температуре. В результате выделяется большее количество тепла,
что ведет к увеличению количества испаряющегося дихлорэтана и, соответс-
твенно, к повышению температуры и давления, а следовательно, к росту скоро
сти побочных реакций заместительного хлорирования.
Обычно эти факторы учитываются при создании новых конструкций реак-
торов жидкофазного хлорирования этилена.
Чаще всего реактор изготовлен в виде цилиндра из углеродистой стали.
В последнее время получили распространение реакторы в виде одной или не-
скольких петель из труб разного диаметра и даже из колонн. Температура подде-
рживается такой, чтобы образующиеся пары дихлорэтана выводились из зоны
реакции с абгазами. Поскольку в промышленности в качестве жидкой среды
используется дихлорэтан, то при проведении процесса при низком давлении
температура реакционной массы соответствует температуре кипения дихлор-
этана. В ряде случаев по технологической необходимости хлорирование этиле-
на проводят при повышенном давлении и при температуре ниже температуры
кипения дихлорэтана. Чтобы вывод дихлорэтана из реактора осуществлялся
в паровой фазе, обычно в верхней части реактора устанавливают сепаратор, в
котором давление ниже чем в рсакюре, та i чгт ною в сепараторе происходи!
вскипание реакционной массы. Пары дихлор папа выводятся па дальнейшую
переработку, а жидкость возвращается в ст н’му.
Поддержание необходимого уровня реакционной массы в реакторе обес
испивается либо возвратом части жидкого дихлорэтана после конденсации его
паров, либо за счет снятия основной части выделяющегося реакционного теп
ла во встроенном или выносном теплообменнике, через который циркулирует
реакционная масса за счет эрлифта [982]. При этом в результате регулируемого
тенлосъема количество отбираемого в парах дихлорэтана соответствует коли
чес । ву дихлорэтана, образовавшегося в результате реакции.
Реактор колонного типа
Процесс хлорирования этилена предлагается вести в цилиндрическом реак
юре с соотношением высоты к диаметру не менее 3, причем жидкую реакцион
пую смесь циркулируют с помощью насоса со скоростью 0,06-0,12 м/с [978].
Фирма «Hoechst AG» разработала установку для получения дихлорэтана
[1031]. В инжектор I (рис. 4.102), который приводится в действие циркули
рующим дихлорэтаном, поступающим по трубопроводу 2, вводится по тру
бопроводу 3 хлор, содержащий инертный газ. Этилен подается в инжектор /
под давлением по трубопроводу 4. Циркулирующий дихлорэтан, хлор, со
держащий инертный газ, и этилен поступают из инжектора 1 в среднее ци
линдрическое пространство 5 реактора 6, где хлор и этилен превращаются
в дихлорэтан при температуре реакции 110-150 °C и давлении 250-700 кПа.
Вновь образованный дихлорэтан вместе с циркулирующим стекает через вер
хний край среднего цилиндрического пространства 5 во внутреннее кольце
вое пространство 7 и далее у дна реактора 6 перетекает во внешнее кольцевое
пространство 8, в котором он поднимается вверх до уровня выше бокового
вывода из реактора 6 циркулирующего дихлорэтана. Таким образом, преодо
левается значительная разница уровней во внутреннем кольцевом прострат
гве 7 и внешнем кольцевом пространстве 8. Пространства 5, 7, 8 разделены
цилиндрическими стенками 12 и 13. Открытием вентиля 11 снижается дав
ление газа в верхней части внешнего кольцевого пространства по сравнению
с давлением газа сбоку пространств 5 и 7 на 50 кПа, что дает возможное гь
отбирать по трубопроводу 9 газообразный дихлорэтан в необходимом коли
честве. Отбором газообразного дихлорэтана посредине между измерителем
уровня 10 и вентилем 11 поддерживается постоянный уровень жидкости во
внешнем кольцевом пространстве 8. Пары дихлорэтана поступают в теплооб
менник 14 и выводятся по трубопроводу 15.
Хлоратор представляет вертикальный цилиндрический аппарат с шестью
перфорированными решетками для предотвращения вертикального переме
шивания [1087]. В нижнюю часть реактора, заполненного 1,2-дихлорэтаном, по
отдельным трубопроводам подают этилен и хлор. В результате прохождения ре
акции, начинающейся в зоне смешивания и завершающейся в реакционной зоне,
в реакторе поддерживается температура 84 °C. Избыток тепла, выделяющийся
в процессе реакции, снимается возвратным 1,2-дихлорэтаном-сырцом, который
поступает в нижнюю часть хлоратора из фазоразделитедя после охлаждения.
Рис. 4.102. Установка для получения дихлорэтана (1-15 см. текст)
Секционирование реакционной зоны с помощью тарелок позволяет исполь-
зовать их также в виде тормозящих устройств [1152].
Запатентован барботажный реактор (рис. 4.103) прямого хлорирования эти-
лена [1115] — вертикальная колонна со штуцерами ввода хлора, этилена, возврат-
ного ДХЭ, а также вывода паров ДХЭ и жидкого ДХЭ с трихлорэтаном (ТХЭ).
б]
a
i
Рис. 4.103. Реактор прямого хлорирования этилена:
ii — общий вид; б — циркуляционная труба; в — секционирующие тарелки с локализованными
перфорированными участками
Кроме roi о, реак юр содержи! расположенную вдоль вертикальной о< и циркули
ционпую трубу. Но высоте верхней части грубы установлены секционирующие
перфорированные тарелки. В нижней части реактора расположены распредели
гели возвратного дихлорэтана, хлора и этилена. Циркуляционная груба имес!
в средней части под нижней тарелкой окна для перетока в нее жидкого дихлор
этана. Кроме того, труба снабжена установленной внутри над окнами поперечном
перегородкой с центральным отверстием, к которому снизу присоединен сливной
патрубок. Отверстия каждой перфорированной тарелки локализованы на одном
ее участке, площадь которого составляет менее половины всей площади тарелки.
Эти же авторы предлагают еще несколько конструкций реактора прямо
го хлорирования этилена [1116-1118]. Во всех случаях реакторы — колонные
аппараты с циркуляционной трубой, установленной вдоль вертикальной оси
(рис. 4.104я-г).
Рис. 4.104. Реакторы прямого хлорирования этилена: а — со спиралевидной секциони-
рующей тарелкой; б — с двумя контурами циркуляции реакционной массы
Рис. 4.104 (окончание): в, г — с установленными внутри циркуляционной грубы фор
сунками для ввода реагентов и ДХЭ
Для улучшения растворимости этилена реактор снабжается установлен
ной концентрично центральной циркуляционной трубе более широкой вне
шней циркуляционной трубой. Эта труба размещена между распредели гелем
возвратного дихлорэтана и нижней перфорированной тарелкой [1117]. Me кд)
внешней циркуляционной трубой и стенками реактора имеется кольцевой ы
зор. При этом распределитель хлор-газа расположен в кольцевом прост ране пи
между обеими циркуляционными трубами, а распределитель этилена — но нне
шнем кольцевом зазоре (рис. 4.104я). Реактор аналогичной конструкции пред
ложено использовать в работе [1096].
Авторы работы [1111] предлагают реактор прямого хлорирования, пред
ставляющий секционированный барботажный аппарат. В нижнюю част ь реак
тора по раздельным барботерам поступают газовые потоки с этиленом и хло
ром. В центральной части аппарата расположена циркуляционная труба. Сек
ционирование осуществляется посредством установки четырех-пяти сигкиы»
тарелок на расстоянии 800-1000 мм одна от другой и направлено на снижен н<
продольною iiepcMriiiiiii.iiiiui газа и жидкости, а наличие внутреннем циркули
ционпой грубы 11О11ЮЛЯС1 организовать циркуляцию жидкое! и, чго соотнсп
тнует режиму идеальною смешения по жидкости и идеального вытеснения по
газу.
Иная конструкция реактора прямого хлорирования этилена предложена и
работе [ 1177]. Реактор включает цилиндрический корпус с патрубками для ии<>
да и вывода продуктов, снабженный секционирующими тарелками, разбиваю
1цими реактор по высоте на отдельные секции, и внутренней циркуляционной
трубой. Для этого верхнюю часть циркуляционной трубы перфорируют в виде
прямоугольных окон по окружности циркуляционной трубы у основания сек
ционированных тарелок или у основания секционирующей тарелки (реше i ки),
по крайней мере, одной верхней секции.
Прямое хлорирование этилена осуществляется в вертикальной колонне
|1180]. Через трубчатый распределитель вводят газообразный этилен, а после
установления вертикальной циркуляции дихлорэтана в колонне через трубча
тый распределитель вводят газообразный хлор, который равномерно раствори
ется в восходящем потоке дихлорэтана, выходящем из нижнего конца циркуля
ционной трубы. Установленная на днище колонны кольцевая решетка, состоя
щая из двух кольцевых обечаек, образующих кольцевой проход, выполняет роль
направляющего устройства для выравнивания скорости восходящего потока
дихлорэтана и обеспечивает формирование равномерного профиля скорости
восходящего потока дихлорэтана в зоне ввода хлора, что позволяет получить
однородную расчетную концентрацию растворенного хлора по всей площади
сечения колонны в зоне ввода этилена, где реакция его хлорирования протекает
без возникновения локальных зон кипения дихлорэтана. Это позволяет повы
сить выход и качество дихлорэтана и снизить удельный расход этилена.
Процесс получения дихлорэтана может быть проведен в вертикальном двух
колонном реакторе, который обеспечивает газовую циркуляцию дихлорэтана в
вертикальном циркулирующем контуре с подачей в него хлора и этилена [1181].
Местное гидравлическое сопротивление в момент движения восходящего пото-
ка дихлорэтана образуется при помощи тарелки на выходе реакционной зоны и
первой колонне. Давление восходящего потока дихлорэтана на тарелке снижа-
ют до значения давления паров дихлорэтана, которое они имеют на выходе из
реактора. Перепад давления в восходящем потоке дихлорэтана осуществляется
в зоне, расположенной выше реакционной зоны. Содержание пара в парожид-
костной смеси в зоне в процессе создания перепада давления по отношению к
подаваемой смеси составляет 0,60-0,95, предпочтительно 0,80-0,85. Разделение
потока дихлорэтана и жидкой фазы осуществляется в зоне сепарации.
Согласно [954, 978, 1182], получение дихлорэтана осуществляется прямым
хлорированием этилена в среде жидкого циркулирующего дихлорэтана. Конс-
трукция реактора позволяет обеспечить газлифтную циркуляцию дихлорэтана
в вертикальном циркуляционном контуре с нисходящим и восходящим пото-
ками. Ввод газообразных исходных реагентов — хлора и этилена осуществля-
ется в циркуляционный контур. Технология процесса позволяет поддерживать
температуру дихлорэтана в зоне реакции ниже, чем равновесная температура
его кипения при давлении в зоне реакции. Для этого зона кипения дихлорэтана
расположена над зоной реакции, в восходящем потоке вертикального цирку-
ляционного копiура На выходе и i юны реакции в циркуляционном контуре
создают местное i идравлическое сонр<ннп icnnc дни кепию восходяще!о нон»
ка дихлорэтана и снижают давление восходящею ноюка меипым )идравли
ческим сопротивлением до значения, близкого к давлению паров дихлор .папа
в месге их вывода. В зоне кипения формируют парокаиельный режим течения
восходящего потока дихлорэтана. При этом паросодержание в зоне кипения
должно составлять 0,8-0,95.
Определенный интерес представляет реактор жидкофазного хлорирования
п илена с эжекционными смесителями реагентов [1182], состоящий из эжекто
ра абсорбции хлора, эжектора хемосорбции этилена, эрлифтной трубы, сепара
юра и циркуляционной трубы. При испытаниях опытного реактора селекгии
ность процесса составляла 99,5-99,8%.
На рис. 4.105 показана схема двухколонного реактора, в котором реализу
ется вариант проведения процесса с указанием направления циркуляции жид
кого в замкнутом циркуляционном контуре и расположения технологических
юн [1100]. Процесс получения 1,2-дихлорэтана осуществляется в реак юре,
содержащем сообщающиеся по верху и низу две вертикальные колонны
Рис. 4.105. Двухколонный реактор прямого хлорирования этилена
(1,2 — см. текст; 3 — крыльчатка; 4 — ее привод)
реакционную / шн ходящего потока и циркуляционную 2 нисходяще!о шт>
ка дихлор папа В нижнюю часть колонны / через вводы и диспергирующие
устройства (не показаны) вводят в газообразном виде хлор и этилен. Меси»
ввода хлора расположено ниже уровня ввода этилена. Одновременно с хлором
в восходящий ноток дихлорэтана подают в небольшом количестве (0,5-2,5%)
газообразный кислород или воздух в качестве ингибитора побочных реакции,
протекающий в паровой фазе. Повышенный расход жидкого дихлорэтана спо
собствует интенсивной турбулизации жидкого дихлорэтана в зоне реакции,
поэтому вводимые хлор и этилен быстро и активно перемешиваются с жидким
дихлорэтаном и равномерно распределяются по сечению восходящего потока
дихлорэтана. При этом происходит быстрое их растворение в жидком дихлор
этане, а также снижение его температуры. Процесс химического взаимодейс
твия хлора и этилена заканчивается на верхней границе зоны реакции и восхо
дящий поток дихлорэтана поступает в расположенную выше зону вскипания,
где гидростатическое давление ниже, чем в зоне реакции. В составе восходя
щего потока дихлорэтана, поступающего в зону вскипания, содержится собс
твенно дихлорэтан, некоторое количество избыточного этилена и кислорода
воздуха.
При поступлении потока нагретого дихлорэтана в зону вскипания проис
ходит постепенное его вскипание, так как его равновесная температура при
снижении гидростатического давления соответственно снижается. Кипение
дихлорэтана сопровождается выделением его паров и постепенным снижением
его температуры по мере расходования тепла реакции на переход дихлорэтана
из жидкого агрегатного состояния в парообразное, в котором он и отбирае гея
для дальнейшей переработки.
Петлевидный реактор
Одним из вариантов повышения интенсивности массообмена является ис-
пользование в процессе прямого хлорирования этилена так называемого пет
левидного реактора. Впервые реактор указанной конструкции был разработан
фирмой «Stauffer» [986,987]. В реакторе с помощью газлифта и термосифонного
эффекта циркулирует реакционная масса — дихлорэтан, содержащий три-, тет
ра- и пентахлорэтаны. Эскиз такого реактора представлен на рис. 4.106.
Использование реактора такой конструкции позволяет повысить интенсив
ность массообмена. Аналогичные варианты исполнения реакторного узла пред
ложены в работах [1072, 994, 1098].
Предложен способ получения дихлорэтана также в петлевидном реакторе с
циркуляцией растворителя — дихлорэтана. Процесс проводят при 20-80 °C без
добавки катализатора при времени пребывания дихлорэтана в системе от 2 до
7 часов и факторе циркуляции от 20 до 500. В низ восходящей части трубчатого
реактора подаются реагенты. В средней части этой трубы находится холодиль
ник для снятия тепла реакции. Сверху петли отводится газовая фаза. В нисходя
щей части реактора установлен насос для создания давления в потоке дихлор-
этана, необходимого для преодоления гидравлического столба в восходящей
части реактора. После насоса часть дихлорэтана возвращается в восходящую
ветвь реактора, а часть откачивается в колонну [1099].
Рис. 4.106. Петлевидный реактор прямого хлорирования этилена
Предлагается также использовать специальный реактор, изготовленный в
виде двух сообщающихся между собой U-образных петель [963].
Реактор, выполненный в виде змеевика
В патенте [1097] предложено осуществлять процесс в трубчатом реакторе,
выполненном в виде змеевика при 40-50 °C. На выходе из реактора установлен
теплообменник для отвода тепла реакции. Этилен подают по двум трубопрово
дам. По одному вводят основную часть этилена, а по второму—не более 1,5-2,0%.
За счет этого добиваются превращения этилена на 99,99%. В циркулирующий
дихлорэтан добавляют катализатор — хлорид железа (III). В схеме предложено
использование устройства для измерения содержания свободного хлора в поп»
ке растворителя. С помощью этого устройства, соединенного с циркуляционным
трубопроводом, определяют избыток хлора и дозируют необходимое количес i во
этилена для его связывания. Технология процесса позволяет получать дихлор»
тан-сырец, в котором содержится не более 0,4 мае. % трихлорэтана.
Разработаны малогабаритные высокопроизводительные ректоры — груб
чатые турбулентные аппараты действия разной конструкции (цилиндрические,
диффузор-конфузорные, кожухотрубчатые), работающие в квазиизотерм и чес
ком режиме [1184]. Данный тип аппаратов сочетает достоинства классических
трубчатых реакторов вытеснения и объемных аппаратов смешения, харак гер
ные особенности определяют их технические и технологические преиму щес гва.
14х можно использовать, в частности, в процессе получения дихлорэтана.
Реактор, выполненный в виде теплообменника-кипятильника
В работе [1086] в качестве реактора предложено использовать кожухогруб
ный теплообменник со сливной трубой по центру трубчатки. Хлорирование
осуществляю г и ipybK.ix, а в меж группой пр<к 1 р.пц i нс организуют циркули
цию кубовой жидкое in колонны рек! ификацпи Жидкий дихлорэтан но слип
ной груби стекаез в нижнюю часть реактора, a oi 1 уда вместе с кубовыми колон
пы направляется в межтрубное пространство.
Реакционный аппарат с несколькими зонами или использование
нескольких реакторов
В последнее время наметилась тенденция улучшения показателей процесса
прямого хлорирования в отношении повышения его скорости, селективное!и
и уменьшения расхода этилена использованием двух реакторных систем при
работе с концентрированными реагентами [963, 966,976, 980, 1007].
В первой реакционной зоне хлорируется большая [976, 1006] или меньшая
[966, 1008] часть этилена соответственно с подачей его в эту зону с меньшим
или большим избытком, а во второй реакционной зоне происходит дохлориро
вание непрореагировавшего этилена.
В некоторых схемах сбалансированного процесса получения винилхлорида
содержащие непрореагировавший этилен абгазы со стадии прямого хлорирова
ния направляют в реактор оксихлорирования [969, 1013]. Однако есть и другон
подход к указанной проблеме, когда используют многоступенчатое хлорирова
ние [959].
1,2-Дихлорэтан предлагается получать хлорированием этилена в прису i
ствии сложного катализатора [1032, 1053]. Хлорирование этилена проводят в
реакторе при 20-75 °C. Затем продукт с катализатором подают в реактор хло
рирования этилена, работающий при температуре кипения реакционной массы
в присутствии того же катализатора. Сверху этого реактора отбирают дихлор
этан в парообразном состоянии. Снизу реактора выводят раствор катализатора
в дихлорэтане. Этот раствор возвращают затем в первый реактор, работающий
при 20-75 °C.
Реактор имеет несколько зон [1034]. В одной из них температуру поддержи
вают в пределах 75-200 °C, а давление 0,1-1,5 МПа. Сначала растворяют в ди
хлорэтане хлор. Затем в следующей зоне в полученный раствор подают этилен с
получением тонкодисперсной фазы с пузырьками диаметром не более 2 мм. Эта
газожидкостная смесь со скоростью 0,3-1 м/с пропускается через реакционную
зону с получением высокочистого дихлорэтана, содержащего всего 5-10“2 мае. %
хлорированных побочных продуктов. Время пребывания должно составлять
2,5-25 с из расчета на жидкую фазу. Тепло реакции снимается в выносном цирку-
ляционном теплообменнике. Образовавшийся дихлорэтан удаляют посредством
испарения при снижении давления в системе. Чистота полученного продукта
более 99,95 мае. %. Выделяющееся при конденсации тепло может, например, ис-
пользоваться для ректификации дихлорэтана, полученного методом оксихлори-
рования. Кроме того, полностью отпадает необходимость в ректификационных
колоннах для отделения низкокипящих и высококипящих побочных продуктов,
образующихся при получении дихлорэтана методом прямого хлорирования. Вы-
ход дихлорэтана в расчете на поданные хлор и этилен составляет 99,7%.
При получении дихлорэтана хлорированием этилена в нескольких реакци-
онных зонах прямоток реагентов с рециклом реакционной массы из верхней
Рис. 4.107. Реактор прямого хлорирования этилена с несколькими реакционными и»
нами
реакционной зоны в нижнюю осуществляют за счет естественной циркуляции
реакционной массы [1089]. Отличительные особенности этого способа заклю
чаются в отборе циркулирующей реакционной массы из нескольких верхних
реакционных зон с вводом циркулирующей реакционной массы, по крайней
мере, в две реакционные зоны. Способ безопасен и характеризуется высокой
интенсивностью (рис. 4.107).
Предложено применять двухреакторные системы с использованием разбив
ленных реагентов или с насыщением подаваемого в реактор дихлорэтана кис
лородом или воздухом [952, 995, 1006, 1081]. Известен также процесс, который
осуществляют в присутствии от 1 до 10 мол % инертного газа, отнесенного на
поданный хлор [1005].
Статический смеситель
В ряде случаев перед реактором устанавливают специальный «статичес
кий смеситель» (подробное описание которого приводится в работе [1033|),
позволяющий достичь тонкого диспергирования этилена в так называемых
«статических смесях». Предлагается устройство (рис. 4.108), состоящее и.|
смесителя 1 с впуском хлора 11, статического смесителя 2 с впуском этилена 5,
10-
Рис. 4.108. Схема устройства со «статическим смесителем» (1-12 см. текст)
Рис. 4.109. Схемы реакторов со «статическим смесителем»(обозначения см. текст)
<tn уда дли сброса давлении <, циркуляцнонио) о насоса о и iсилообмен
пика 7, При ном смеситель /, с laiH'ii'isuii смеси гель 2, отсеканшь 12, сосуд
дли сброса давления 3, циркуляциопнын насос 6 и теплообменник 7 сняла
ни друг с другом cooгвегс। пукицими потоками. (’осуд для сброса давлении 3
соединен через дроссель / с конденсатором 8, a oi копдспсаюра 8 отходил
ншии слива продукта 9 и сброса абгаза 10. Повышаемое в статическом смс
с н ।еле давление препятствует кипению 1,2 дихлорэтана в реакционной тоне
п, как следствие, снижает скорость реакции с одновременным увеличением
селек।ивности.
11одача этилена и хлора может быть осуществлена в поток циркулирующей о
дихлорэтана при интенсивном перемешивании в статическом смеси теле |10 И»|.
I la рис. 4.109н показаны реактор 1, смеситель 2, граница жидкого дихлорэтана 1
। рубонронод для циркуляции дихлорэтана 4, насос 5, потоки питания хлора и
пилена 6 и 7. Эффективное смешение реагентов происходит в потоке циркули
рующего дихлорэтана в статическом смесителе 2 [1033].
11а рис. 4.1096 изображена схема процесса с использованием статического
смесителя [1082]. Для абсорбции хлора во всасывающий поток дихлор папа
инжектора 3 совместно вводятся электролитический хлор 1 с содержантм
инертного газа до 5% моль и воздух 2' через трубопровод 2а . Поток смечи /
насосом 5' сжимается до давления 10 атм, в него дополнительно подастся воз
дух но трубопроводу 2 в. Полученная смесь совместно с потоком этилена 6 ,
подаваемым под давлением 1,1 МПа, поступает в статический смеси тель 8 ре
ак юра 7'. Реакция хлорирования этилена проводится в газожидкостной дис
Персии, при этом энергия плотности рассеивания составляет ~1кВт/м .
Данные промышленной эксплуатации процесса жидкофазного
хлорирования этилена
Промышленные испытания схемы
Технология процесса высокотемпературного хлорирования этилена с ih
пользованием тепла реакции для ректификации дихлорэтана впервые в России
пыла разработана авторами и реализована в промышленных условиях АО «< а
янскхимпласт». В основу процесса был положен большой массив лабораторных
исследований и опытных работ.
Реактор технологически объединялся в один агрегат с колонной рек тифи
кации дихлорэтана. Последняя с помощью материальных потоков была спя ыпа
с колонной выделения высококипящих примесей [1106]. При необходимости
в реакционную зону или в нижнюю часть совмещенной с реактором колонны
ректификации можно подавать сторонний дихлорэтан для его очистки путем
хлорирования содержащихся в нем непредельных примесей и последующей
ректификацией. Ректификация в совмещенном агрегате осуществляется за счет
тепла реакции.
Первоначально разработанная технология была апробирована на опытно
промышленной установке мощностью 15 тыс. т дихлорэтана в год в АО «('а
янскхимпласт», а затем внедрена на стадии прямого хлорирования этилена и
производстве винилхлорида мощностью 270 тыс. т/год.
11рсиму|Ц1ч ।и.। i.ikok) процесса заключаются в следующем. В реакциоп
пук» зону или в нижнюю часгь совмещенной с реак сором колонны рекгифнка
ции можно подавать сторонний дихлорэтан для его очистки хлорированием
содержащихся в нем непредельных примесей и последующей ректификацией.
Можно использовать тепло реакции для ректификации дихлорэтан т в совме-
щенном агрегате [1109, 1110]. Отпала необходимость в водно-щелочной про
мывке дихлорэтана-сырца с соответствующим снижением количества сгоч
ных вод. Уменьшился расход катализатора, так как синтезируемый дихлор-
этан выводится из реактора в паровой фазе и не содержит FeCl3 в отличие от
схемы вывода продукта в жидкой фазе. Были изготовлены два промышлен
ных реактора высотой 18,75 м и диаметром 3,6 м и осуществлена соответс-
твующая переобвязка технологической схемы стадии прямого хлорирования
этилена [1091]. Как и на опытной установке, конструкция реакторов предус-
матривала организованную структуру потоков [405, 1108, 1112]. Оба аппара
га были подсоединены к нижней части действующей колонны ректификации
дихлорэтана. В схеме был оставлен один из кипятильников этой колонны. До-
полнительно в схеме были установлены рассольные конденсаторы-абсорбе-
ры, орошаемые водой для улавливания хлороводорода. В реакторы вводился
дихлорэтан, полученный на стадии оксихлорирования, не прореагировавший
в печах пиролиза, а также отводимый с верха колонны выделения высококи-
пящих примесей. В колонне ректификации дихлорэтана происходила очистка
направляемого затем на пиролиз дихлорэтана от высококипящих примесей,
образующихся на основных реакционных стадиях процесса, также за счет
хлорирования непредельных примесей в рециркулирующем с пиролиза ди-
хлорэтане. Во избежание накопления в реакционной массе высококипящих
примесей в новой схеме, как и в ранее действующей, функционировала колон-
на выделения высококипящих компонентов. Питание этой колонны осущест-
влялось из куба колонны ректификации дихлорэтана или новых реакторов
прямого хлорирования.
Проектные нагрузки по реагентам составляли около 3000 нм3/ч на каждый
реактор. В действительности реакторы работали устойчиво в довольно широ-
ких пределах нагрузок нм3/ч.: 1200-5600 по хлору, 1250-6000 по этилену. Режим
работы осуществлялся как с избытком этилена, который достигал 20 мол %, так
и с избытком хлора, доходившим в отдельных случаях до 16 мол %. В широких
пределах менялись и режимные параметры: давление вверху реактора от 0,10 до
0,22 МПа, температура процесса в интервале 100-120 °C, содержание хлорного
железа в реакционной массе от 0,0015 до 0,2800 мае. %, а содержание растворен-
ного хлора в реакционной массе от 0 до 0,1 мае. % и более.
При работе по старой схеме расход пара на колонне ректификации дихлор-
этана составлял около 30 т/ч. При работе по новой схеме расход пара колебался
от 0 до 20 т/ч в зависимости от нагрузки работающих реакторов [1091].
Исследования, проводимые на промышленных установках
В ходе освоения схемы было исследовано влияние ряда факторов на процесс
хлорирования этилена [1091]. На рис. 4.110 представлена зависимость содер-
жания полихлоридов в реакционной массе от соотношения реагентов. Четко
Рис. 4.110. Зависимость содержания полихлоридов в реакционной массе ог соотноше-
ния п илена с хлором
прослеживается тенденция уменьшения выхода полихлоридов при увеличении
пабы гка этилена в изученных пределах.
Влияние соотношения этилена к хлору на содержание полихлоридов в реак
ционной массе может быть оценено и по наличию хлора в верхней части колон
ш>1 ректификации дихлорэтана, в частности, в потоке флегмы. Для выявления
количественной зависимости были взяты два граничных случая — полное от
су гствие хлора во флегме и присутствие его в следовых количествах и выше (до
10 ’%). При образовании полихлоридов в результате заместительного хлориро
вания выделяется хлороводород. Поэтому контрольным показателем служила
концентрация хлороводорода во флегме колонны ректификации дихлорэтана.
Исследования, проводившиеся в течение полугода, дали следующие результа гы
11ри отсутствии хлора во флегме колонны ректификации дихлорэтана содержа
п ие полихлоридов в реакционной массе составляло 7,6%, а хлороводород ! но
флегме этой колонны — 0,11%. При наличии же хлора во флегме колонны рек
гификации дихлорэтана содержание полихлоридов и хлороводорода повыша
лось и достигало 8,7% и 0,12% соответственно. Следовательно, увеличению с<»
держания полихлоридов в реакционной массе на 1% соответствует повышение
концентрации хлороводорода во флегме колонны ректификации дихлорэтана
па 0,01%. Такая разница в величинах обусловлена несколькими причинами. Во
первых, расход потока с верха колонны (по составу поток аналогичен флегме) в
6-11 раз больше количества выводимой из реакторов кубовой жидкости, сос гав
которой такой же, как и у реакционной массы. Во-вторых, на один моль обра
зующихся высококипящих продуктов выделяется немногим более моля хлоро
водорода. При этом молекулярная масса хлороводорода примерно в три раза
меньше, чем у высококипящих продуктов. В-третьих, отходящие из колонны
ректификации дихлорэтана абгазы содержат до нескольких объемных прицеп
тов хлороводорода, т.е. его содержание в абгазах примерно в 5-10 раз больше,
чем во флегме этой колонны.
Из приведенных данных следует, что необходимо поддерживать такой из
быток этилена, который бы обеспечивал отсутствие даже следов хлора во флег
ме колонны ректификации дихлорэтана.
Поскольку хлор расходуется также и на хлорирование непредельных приме
сей, ностуннощих в реакционную зону с дихлор.паном от других стадии, го в
зависимости от их количества требуется избыток хлора и 2-6 мол %. Изучение
материального баланса системы хлораторы ректификационная колонна пока
зало, что при нормальном (5 мол %) избытке этилена непосредственно в резуль-
тате прямого хлорирования последнего за счет реакций заместительного хло-
рирования образуется 0,5% три- и тетрахлорэтана. Ведение процесса при таком
соотношении реагентов, когда хлор начинает проскакивать во флегму колонны
ректификации дихлорэтана, приводит к увеличению полихлоридов, образую-
щихся только в результате заместительного хлорирования, в три раза.
В низкотемпературном процессе прямого хлорирования этилена оптималь-
ная концентрация хлорида железа (III) в реакционной массе поддерживается
обычно на уровне 0,02-0,03 мае. %. Из данных, представленных на рис. 4.111,
следует, что с ростом концентрации трехвалентного железа в реакционной мас-
се количество хлористого этила, образующегося в период пуска, может дости-
гать почти 1% (кривая 1). Кривая 2 на том же рисунке описывает аналогичную
зависимость для уже установившегося режима установки после многократного
обновления реакционной массы (с выводом железа из куба колонны выделе-
ния высококипящих примесей). В этом случае содержание хлористого этила в
дихлорэтане-ректификате значительно ниже и практически не зависит от коли-
чества железа. Таким образом, на побочную реакцию гидрохлорирования эти-
лена, в результате которой образуется хлористый этил, влияет не только коли-
чество, но и состав соединения, содержащего трехвалентное железо.
В дихлорэтане, поступающем в реактор со стадии ректификации винил-
хлорида, содержится до 0,4% последнего. Логично было бы предположить, что
Рис. 4.111. Зависимость содержания этилхлорида (1 — в начальный период; 2 — в ус-
тановившемся режиме); 1,1-ДХЭ (3) и НС1 (4) во флегме колонны ректификации ДХЭ и
содержания полихлоридов в реакционной массе (5)
ii псрвопачалы1ын период p.itroi ы upon iводе, i ва концеu i рация 1,1 ДХ ) (про
дук га гидрохлорировалия винилхлорида) во флегме колонны ректификации
дихлор.)гана, как и конценграция хлор пила, Borpaciaei с увеличением коли
честна 1е’' в реакционной массе хлоратора. Однако результаты анализов по-
катывают иногда практически полное отсутствие 1,1 ДХЭ даже при концепт
рации трехвалентного железа 0,08% и более. В установившемся режиме по ш
нисимость 3 на рис. 4.111. С ростом концентрации Fe выход 1,1-дихлор лани
ноч ги не меняется. Кривая 4 описывает зависимость количества хлороводорода
во флегме колонны ректификации дихлорэтана от концентрации FeClj в реак
ционной массе. Поскольку содержание НС1 во флегме этой колонны записи i oi
количества образующихся полихлоридов, указанная зависимость качественно
характеризует влияние концентрации FeCl3 на выход последних.
Зависимость концентрации высококипящих примесей в реакционной массе
о г содержания в ней FeCl3 описывается кривой 5.
При низких концентрациях FeCl3 содержание НС1 и полихлоридов во флс!
ме колонны ректификации дихлорэтана практически не меняется. Эю мо кеч
говорить только о заместительном хлорировании. С увеличением конценi
рации FeCl3 до 0,02% и выше скорость этой реакции, вероятно, не меняется,
поскольку количество обнаруживаемого хлороводорода остается практически
одним и тем же. Продолжающийся же рост количества полихлоридов можно
объяснить присоединением хлора к непредельным хлоруглеводородам и бен го
лу. При этом скорость последней реакции растет с повышением концен грации
катализатора — хлорного железа.
Анализ зависимостей, представленных на рис. 4.111, показывает, что опти
мальное количество катализатора в реакционной массе должно быть на уровне
0,02-0,04%. При более низкой его концентрации хлорирования непредельных
соединений практически не происходит, при более высокой — существенно
увеличивается выход полихлоридов.
На рис. 4.112 показано влияние температуры на содержание хлорис того во
дорода во флегме колонны ректификации дихлорэтана, а следовательно, и па
характер выхода полихлоридов. С ростом температуры в интервале 103 110 (
Рис. 4.112. Зависимость содержания НС1 во флегме колонны ректификации ДХ ) <п
температуры в нижней части реактора
конце»грация хлороводорода noipacjaei незначительно. Дальнейшее ее попы
шениедо 120 "С приводи । к резкому увеличению содержания IK 1 во флегме.
При проведении процесса синтеза винилхлорида по сбалансированной схе
ме возникает проблема очистки рециркулирующего дихлорэтана от образую
щихся на стадии его пиролиза легкокипящих непредельных примесей, которые
не отделяются от дихлорэтана при ректификации. Представляется целесооб-
разным возвращать непрореагировавший при пиролизе дихлорэтан на стадию
прямого хлорирования этилена и дехлорированием переводить их в высоко
кипящие продукты, которые затем легко отделяются от дихлорэтана на стадии
ректификации.
В [1105] было изучено влияние разных факторов на степень хлорирования
легкокипящих примесей в условиях высокотемпературного жидкофазного хло
рирования этилена. В качестве исследуемых примесей были выбраны винил и
денхлорид, цис- и транс-дихлорэтилены, бензол, три- и перхлорэтилен. Данные
соединения наиболее часто присутствуют в дихлорэтане, направляемом на пи
ролиз. Исследования проводили в интервале от 70 до 130 °C и времени контакта
от 25 до 100 мин. Концентрация примесей варьировалась в пределах от 0,3 до
4,5%. В реактор непрерывно подавали дихлорэтан с фиксированным количест-
вом растворенной примеси для поддержания постоянного реакционного объ-
ема.
Полученные данные показывают, что непредельные примеси в условиях
прямого хлорирования этилена переходят в высококипящие продукты по ре-
акциям:
СН2=СС12 + С12— СН2С1—СС13; (4.166)
СНС1=СНС1 + С12 —- СНС12—СНС12; (4.167)
СНС1=СС12 + С12—► СНС12—СС13; (4.168)
СС12—СС12 + С12 — СС13—СС13; (4.169)
С6Н6 + С12 —- С6Н5С1 + HCI. (4.170)
При повышении температуры степень конверсии примесей возрастает
(рис. 4.11 Зя), особенно, в интервале от 70 °C до температуры кипения дихлор-
этана. Увеличение времени пребывания также повышает степень конверсии
примесей (рис. 4.1136). Наиболее легко хлорируется бензол, степень конвер-
сии которого составляет в среднем 92%, для винилхлорида она равна 90%, для
винилиденхлорида и трихлорэтилена — 85%, а для перхлорэтилена — около
87%. Концентрация примеси в исходной смеси не влияет заметно на степень ее
конверсии. Полученные данные подтвердили возможность хлорирования не-
предельных примесей в условиях работы реактора, совмещенного с колонной
ректификации. При этом для обеспечения максимальной степени конверсии
примесей оптимальна температура 100-110 °C.
В лабораторных условиях и в опытно-промышленном реакторе, совмещен-
ном с колонной ректификации, была подтверждена возможность хлорирования
непредельных примесей, содержащихся в дихлорэтане, непрореагировавшем
Рис. 4.113. Зависимость степени конверсии примесей: а — от температуры процесса
при 1-ч контакте, 6 — от продолжительности процесса при 83,5 °C:
/ — трихлорэтилен, 2 — перхлорэтилен, 3 — бензол, 4 — транс-дихлорэтилен
при пиролизе. В первом случае суммарная конверсия непредельных соединений
в условиях получения дихлорэтана колеблется в пределах 70-90%, а во втором —
от 50 до 70%. При этом степень конверсии отдельных примесей соответственно
составляла: винилхлорид — 90 и 50%, винилиденхлорид — 85 и 50%, трихлор.»
гилен — 80 и 75%, перхлорэтилен в опытно-промышленном реакторе — 100%, и
лабораторном — не определялась [1091].
Работа совмещенного агрегата прямого хлорирования этилена в режиме,
когда в реактор не подается рецикловый дихлорэтан со стадии пиролиза, харак
теризуется высокой степенью чистоты дихлорэтана-ректификата — до 99,7%.
Схема предусматривает ввод рециклового дихлорэтана в реактор прямого хло
рирования с целью перевода содержащихся в нем непредельных легкокипящих
примесей в высококипящие. При этом степень чистоты дихлорэтана-рек гифи
ката снижалась в среднем до 99,3%, что, тем не менее, заметно выше величн
ны, получаемой в реакторе низкотемпературного хлорирования (97,5-98,01!»).
Таким образом, непредельные соединения дос । точно интенсивно хлорирую к. и
и данном реакторе. Гак, например, содержание бензола в дихлор нале ректнфн
Kaie снизилось noni и на два порядка (с 0,8 до 0,01%). 11рак гически полнос тью
хлорируются такие примеси, как хлоропрен и цис-дихлорэтилен. Степень кон
версии трихлорэтилена в среднем на уровне 50%, винилхлорида от 50 до 60%.
Остальные непредельные соединения также подвергаются хлорированию в гои
или иной степени.
Таким образом, данная схема позволяет хлорировать непредельные соеди
нения с получением высококипящих полихлоридов. Это в значительной мерс
облегчает очистку рециркулирующего дихлорэтана путем его ректификации
На рис. 4.114 представлены данные по составу реакционной массы при уело
вии постоянного вывода последней из реакторов в жидкой фазе в количестве
15 м3/ч. Можно считать, что содержание ди- и трихлорэтана находится на до
статочно стабильном уровне и характеризует выход реакторов на стабильный
режим работы. Уровень содержания высококипящих примесей, образующихся
в реакционной зоне, во-первых, при синтезе дихлорэтана, а во-вторых, в ре
зультате хлорирования содержащихся в непрореагировавшем при пиролизе ди
хлорэтане непредельных соединений обеспечивается заданным выводом кубо
вой жидкости. Естественно, что при снижении вывода последней концентрация
высококипящих примесей должна возрастать, что и было доказано специаль
ными опытами.
Рис. 4.114. Влияние продолжительности работы совмещенного реактора на состав ре
акционной массы
Длительная jkiплуагация ною реакюра, совмещенною с колонной рек
। пфикации, и процесса хлорирования и плена в целом подтвердила возмож
ность использования тепла реакции для рем пфикации как синтезируемою, так
и стороннего дихлорэтана, количество которого почти в три раза больше но
сравнению с синтезируемым, с увеличением выхода дихлорэтана-ректификата.
При этом расход потребляемого пара определяется флегмовым числом и в он
। имальном режиме равен нулю.
Основным недостатком данного процесса является более низкая селскгин
ность по сравнению с процессом, проводимым при 55-65 °C. Как указывалось,
на увеличение выхода полихлоридов существенно влияет повышение темпера
гуры. В значительной мере выход полихлоридов увеличивается при проведе
нии процесса в условиях кипения реакционной массы. При этом происходи!
насыщение жидкости парами дихлорэтана, которые формируются в виде пулы
рей. Подаваемый в зону реакции газообразный хлор взаимодействует с нарами
дихлорэтана, и реакция заместительного хлорирования смещается в парогазо
вую фазу. При использовании FeCl3 в качестве ингибитора этой реакции из ia
малой его летучести ингибирующее действие достаточно эффективно только в
жидкой фазе. Для подавления этой реакции в парогазовой фазе целесообразнее
всего ввести газообразный ингибитор, например, кислород.
По этой причине часть исследований была направлена на определение в
промышленных условиях эффективности ингибирующей способности b’cCI,
в присутствии газообразного кислорода. Исследования проводились как на
опытно-промышленной установке получения дихлорэтана, так и в промыт
ленных условиях [1092]. Установка состояла из реактора хлорирования этиле
на проектной мощностью 15 тыс. т/год, совмещенного с насадочной колонной.
Реактор выполнял функции кипятильника. В качестве хлорирующего агента
использовали абгазы хлорного производства, содержание кислорода в которых
колебалось от 5 до 8,1 мол % (в среднем около 6 мол %). Процесс осуществлял
ся при температуре кипения реакционной массы. Получаемый в этих условиях
дихлорэтан-сырец содержал 99,3-99,6% основного вещества, 0,01-0,03% легко
кипящих и 0,37-0,67% высококипящих примесей. Ректификат содержал 99,8%
дихлорэтана; 0,04-0,1% легкокипящих и примерно столько же высококипящих
примесей.
Более детально ингибирующая активность этой смеси изучалась в реак юрах
хлорирования этилена, совмещенных с колонной ректификации в произволе
тве винилхлорида мощностью 270 тыс. т/год [ 1092]. Исследования показали, ч го
при проведении процесса в условиях кипения реакционной массы в прису к
твии только одного ингибитора — 0,015-0,035% FeCl3 дихлорэтан-ректифика!
содержал 98,9-99,6% основного вещества.
Полученные данные свидетельствуют, что использование комплексного ин
гибитора FeCl3 + О2 может оказаться эффективнее, чем применение каждою из
них в отдельности.
Рис. 4.115 иллюстрирует влияние соотношения реагентов на выход полихло
ридов. Кривая 1 получена при стабильном и соответствующем регламентным
показателям выводе кубовых из реакционного агрегата. Наблюдается четкая
тенденция уменьшения выхода полихлоридов с ростом избытка этилена. Авали i
многочисленных данных дает представление, что при содержании полихлори
’ис. 4.П5. Зависимость содержания полихлоридов в реакционной массе от избытка
пилена:
! — вывод кубового продукта в соответствии с регламентом; 2 — то же и подача в реак юр
),6 мол. % кислорода; 3 — уменьшенный по сравнению с регламентным вывод кубового продук
а; 4 — то же и подача в реактор 0,6-мол. % кислорода
job в реакционной массе на уровне 10% и менее улучшается работа ректифи
сацнонной колонны, совмещенной с реакторами, и обеспечивается получение
дихлорэтана-ректификата чистотой выше 99%. 11ри проведении процесса в ана
логичных условиях, но с подачей в реактор кислорода 0,6-1,0 мол % от расхо-
да хлора содержание полихлоридов в реакционной массе несколько снижается
[кривая 2). При уменьшении вывода кубовых из реакционного агрегата в три и
эолее раза содержание полихлоридов в реакционной массе резко увеличивается
[кривая 3) даже при сравнительно высоких избытках этилена — от 4 до 8 мол %.
Ведение процесса при таком же пониженном выводе кубовых из реакцион
лого агрегата, но в присутствии кислорода, вызывает очень резкое снижение
удержания полихлоридов в реакционной массе (кривая 4); при 4%-ном избыт-
ке этилена количество полихлоридов при работе с добавкой кислорода умень-
шается почти в два раза и становится ниже 10%.
Косвенно влияние соотношения компонентов при хлорировании в при-
гутствии кислорода было оценено по наличию растворенного хлора во флегме
ректификационной колонны, совмещенной с реакторами. Поскольку наличие
хлора во флегме колебалось в очень незначительных пределах (от следов до
-20-10 3%), то для выявления количественной зависимости, как и ранее, были
взяты два граничных случая: полное отсутствие хлора во флегме совмещен-
ной с реакторами колонны; присутствие его в следовых количествах и выше.
В первом случае содержание полихлоридов в реакционной массе составляло в
среднем 6,2%, во втором — 7,7% (данные получены на основе усреднения более
чем шестимесячных замеров). В отсутствие кислорода сохраняется такая же
тенденция.
Рис. 4.116. Зависимость содержания полихлоридов в реакционной массе от содержания
кислорода в исходном хлоре
Рис. 4.117. Зависимость содержания хлороводорода во флегме совмещенной с реакiо
ром колонны ректификации от содержания кислорода в исходном хлоре при регламеп
тном (1) и пониженном (2) выводе кубового продукта из агрегата хлорирования
Влияние кислорода на процесс прямого хлорирования этилена оценивали
по выходу полихлоридов. На рис. 4.116 и 4.117 представлены зависимости со
держания полихлоридов и хлористого водорода во флегме колонны, совмещен
ной с реакторами, от количества поданного кислорода.
Из графика на рис. 4.116 видно, что наличие кислорода способствует умень
шению выхода полихлоридов. При этом добавление кислорода в количестве 1%
от объема исходного хлора вызывает снижение выхода полихлоридов почти
вдвое. Наиболее резкое снижение наблюдается при малых (~ до 0,25%) добав
ках кислорода. Дальнейшее повышение расхода кислорода до 0,8-1% от объема
хлора на выходе полихлоридов сказывается незначительно.
Кривая 1 на рис. 4.117 построена по данным, полученным при соблюдении
регламентных параметров, а кривая? — поданным, полученным при минималь
пом выводе кубовою продукта in агрег.на хлорирования. H i обоих рисунков
следует однозначный вывод о том, чго наличие кислорода положительно сказы
ваегся на пока iaгелях процесса. Определенное различие в характере кривых на
рис. 4.) 16 и 4.117, вероятно, можно объясни н> в том числе инициированием кис
породим реакций гидрохлорирования этилена и винилхлорида в паровой <|>а ie,
приводящим к снижению содержания хлороводорода во флегме, совмещенной с
реакторами колонны, с увеличением содержания кислорода в исходном хлоре.
В производстве винилхлорида функционирует ректификационная колонна
выделения высококипящих примесей из дихлорэтана. Эта колонна связана м.1
термальными потоками с колонной, совмещенной с реакторами. На рис. 4.118
показано влияние температуры на выход полихлоридов, о котором можно су
дить по их содержанию в реакционной массе. Кривая 1 получена при работе
на проектных нагрузках ректификационной колонны, совмещенной с реак го
рами, кривые 2 и 3 — при выводе из колонны выделения высококипящих в три
раза меньшем количестве кубового продукта, чем это предусмотрено по проек
ту. При этом кривая 3 построена по данным, полученным при осуществлении
процесса хлорирования в присутствии кислорода. С ростом температуры, как
и следовало ожидать, выход полихлоридов несколько увеличивается. Характер
кривых аналогичен, но присутствие кислорода резко снижает уровень содержа
ния полихлоридов в реакционной массе.
Рецикл непрореагировавшего при пиролизе дихлорэтана отрицательно ска
зывается на показателях процесса, так как возрастает выход высококипящих
примесей. Рециркулирующий дихлорэтан содержит заметные количества при
месей, в том числе непредельных соединений, которые достаточно интенсивно
подвергаются хлорированию в условиях работы реакторов прямого хлориро
вания этилена. Степень конверсии исследуемых примесей в дихлорэтане при
хлорировании практически не зависит от добавок кислорода.
Рис. 4.118. Зависимость содержания полихлоридов в реакционной массе от температу-
ры в нижней чаете реактора:
1 — вывод кубового продукта в соответствии с регламентом; 2 — пониженный втрое по сравне-
нию с регламентным вывод кубового продукта; 3 — то же и подача в реактор 0,6 мол. % О2
Поскольку хлорирование винилхлорида приводи! к oop.i lou.iiinio
рихлормана, io возможна определенная ошибка и оценке телек iiiunoi 1 и про
цесса прямого хлорирования пилена при подаче рециркулирующею дихлор
п ана непосредсгневно в реактор, совмещенный с колонной рек i ификацни. Для
проверки этого предположения был проведен авали > зависимости содержания
в реакционной массе трихлорэтана — основного компонента высококипящих
примесей от количества подаваемого в реактор рециркулирующего дихлор пана
(рис. 4.119). Видна четкая линейная зависимость содержания трихлорэтана в
продуктах хлорирования от количества подаваемого в зону реакции возвра i но
го дихлорэтана. Небольшой разброс усредненных данных может быть связан с
некоторыми колебаниями содержания винилхлорида в этом дихлорэтане.
Использование в исследуемом процессе в качестве ин гибитора 1еС13 + ()21101
полило снизить выход побочных продуктов. Введение в реактор 0,6-1,0 мол %
О? позволяет достигать 98,5%-ной селективности процесса при 4%-ном избы гке
этилена и 99,5-99,7%-ной селективности при 6-8%-ном его избытке. Для дос ги
жения селективности на уровне 98,5% без добавки кислорода необходим 8-12%
ный избыток этилена. В свою очередь, это облегчило очистку дихлорэтана oi
высококипящих примесей на совмещенной с реакторами колонне рекгифика
ции. В результате при работе на проектной нагрузке и даже при ее превышении
до 20% потребность в подаче пара в кипятильник совмещенной колонны огпа
ла, поскольку ректификация полностью осуществлялась за счет тепла реакции.
Кроме того, повышение селективности сказалось и на количестве жидких
отходов — кубового продукта колонны выделения высококипящих примесей
Установлено, что количество этих отходов снизилось почти в 3 раза, по срапне
нию с их количеством, образующимся при использовании в качестве ингиби го
ра только РеС1з-
Таким образом, применение указанного комплексного ингибитора за
местительного хлорирования этилена позволяет повысить степень чисто гы
дихлорэтана-ректификата, исключить подачу пара в совмещенную с реактора
ми колонну ректификации и уменьшить количество жидких хлорорганических
отходов.
Рис. 4.119. Зависимость содержания полихлоридов в реакционной массе от количества
возвращенного в цикл ДХЭ
При cp.iinieiiiiii да.. полученных при обследовании промышленных
процессов ни iKOieMiiep.il урною прямого хлорирования пилена и пысокотем
пераIурною хлорирования, было установлено, что и степени превращения ре
агентов, и наличие в дихлорэтане-сырце легкокинящих примесей в этих про
цессах примерно на одном уровне. В низкотемпературном процессе наблюдя
ется несколько меньший выход высококипящих примесей. Гак, их содержание
в дихлорэтане-сырце колеблется в пределах 0,25-0,35 мае. %. Тем не менее, вы
сокотемпературный процесс характеризует несомненное преимущество — воз
можность утилизации тепла реакции. Данные, полученные после обследования
реконструированной стадии прямого хлорирования производства винилхло
рида, позволили сопоставить разные процессы жидкофазного хлорирования
этилена.
Сравнительные характеристики трех промышленных процессов прямою
хлорирования: А — низкотемпературного; Б — высокотемпературного без ути
лизации тепла реакции; В — высокотемпературного, в котором тепло реакции
используется для ректификации образующегося дихлорэтана-сырца [1109,11101:
Хлорирование этилена А Б В
Г, °C .... 55-65 83-86 83-110
Расход пара на ректификацию дихлорэтана, МДж/т ДХЭ ....1555,3 1117,9 602,9
Расход промывной воды, м3/т ДХЭ .... 0,160 0 0
Выход высококипящих примесей, % ... 0,3-0,4 0,35-0,42 0,114
Селективность процесса, % .. 99,5-99,8 99,3-99,6 >99,8
Электрохимическое жидкофазное хлорирование этилена
Среди известных способов получения дихлорэтана вызывает определенный
интерес неординарный электрохимический метод. Была показана возможность
получения дихлорэтана при пропускании этилена через подвергающийся элек-
тролизу раствор соляной кислоты [1148-1150]. Организованный таким обра-
зом процесс дает возможность использовать абгазную соляную кислоту.
Проведено изучение электрохимического хлорирования этилена в проточ-
ном режиме на графитовом электроде в среде НО при температурах от 10 до
70 °C [1175]. Методами вольтамперометрии и циклической вольтамперомет-
рии показано, что этилен влияет на электрохимическое поведение графитово-
го анода в среде НО. На основании полученных результатов предполагается,
что наряду с гетерогенным процессом хлорирования этилена на графите при
электрохимическом хлорировании этилена на графите в НО протекает процесс
свободно-радикального хлорирования с инициированием цепи реакции на по-
верхности электрода.
Однако этот метод не получил промышленного внедрения, поскольку для
больших мощностей необходимы десятки электролизеров, которые должны бу-
дут работать в одном и том же режиме, а также специальные устройства для
подачи в них этилена и отвода образовавшихся дихлорэтана и водорода.
Предложен способ получения дихлорэтана прямым хлорированием пиле
на с использованием газообразного хлора, получаемого в электрохимическом
электролизере из безводного хлороводорода [1176], который подают в анодную
часть электрохимического электролизера, снабженного катионообменной мем
браной (анод контактирует с одной стороной мембраны, а катод — с другой).
Напряжение в электролизер подается так, что потенциал на аноде выше, чем па
катоде. На аноде происходит окисление безводного НО с образованием газо
образного хлора и протонов. Хлор выводят из анодной части элект роди юра, а
протоны проходят через катионообменную мембрану, попадая па катод. ( жн
женный хлор и рециркулирующий хлор поступают в реактор прямого хлориро-
вания, где взаимодействуют с этиленом, образуя дихлорэтан.
Газофазное хлорирование этилена
Первые исследования, показавшие возможность замещающего хлорирова
ния олефинов, были выполнены еще в 1884-1885 гг. [1192, 11931. Усгановле
но, что количество хлора, вступающего в реакцию замещения, увеличивается
при повышении температуры.
Первый патент на получение хлорпроизводных олефинов mciодами
высокотемпературного хлорирования был опубликован в 1937 г. |1194)|.
Предлагалось олефины и хлор, предварительно нагретые до 200-500 °C, сме-
шивать в реакционной трубке. Время пребывания реагентов в трубке подде
рживается около 1 с. Несколько позднее был опубликован патент, согласно
которому хлорирование проводится при избытке этилена и температуре
200-700 °C в присутствии катализатора или без него, пропуская реагенты со
скоростью, превышающей скорость распространения пламени [1195]. Этилен
и хлор должны быть предварительно нагреты до 200-700 °C. Тепло реакции
отводится разбавлением реакционных газов. Диаметр реакционной трубки —
44,4 мм, длина — 554 мм.
Позднее были опубликованы более обстоятельные исследования по хлори
рованию газообразных алкенов [1266]. Была обнаружена закономерность, со
гласно которой процесс хлорирования олефинов в зависимости от их ст рук у
ры имеет определенный критический температурный интервал.
Ниже приведены критические температуры, °C, при которых у отдельных
олефинов нормального строения наступает переход от реакции присоединения
к заместительному хлорированию:
Изобутилен и другие третичные олефины .... <40
Пентен-2 .............................. 125-200
Бутен-2 ............................... 150-225
Пропилен............................... 200-350
Этилен................................... 250-350
В приведенных температурных интервалах реакция протекает с образованием
продуктов присоединения по двойной связи. Выше этих температур увеличит»
ется скорость реакций заместительного хлорирования. Из приведенных данных
пидио, чго реакция шмсс i и гельпо) о хлорирования шпчитслыю i рудпее протека
ег у этилена и пропилена. У пилена она ускоряется только при I > 350“С.
Согласно данным о заместительном хлорировании пилена, впервые онуб
линованным в 1938 г. 11361|, в процессе хлорирования этилена при 400 °C па ка
тализаторах — хлоридах алюминия или меди — реакционная смесь содержала
21,9% винилхлорида, 45,9% этилхлорида и 3,7% метилхлорида.
В результате газофазного хлорирования этилена образуются, главным обра
зом, два продукта. Реакция присоединения дает дихлорэтан:
С2Н4 + С12 —С2Н4С12 + 180 кДж. (4.171а)
Продуктом реакции замещения является винилхлорид:
С2Н4 + С12 —- С2Н3С1 + НС1 + 109 кДж. (4 1716)
Реакция присоединения начинается при комнатной температуре и протека-
ет с выделением большого количества тепла.
Скорость этой реакции возрастает с увеличением температуры. Однако рост
температуры способствует протеканию и реакции замещения с постоянным
преобладанием над аддитивным хлорированием. Поэтому выход дихлорэтана
проходит через максимум, который, согласно [1199], соответствует 275-315 °C,
т.е. интервал температур несколько уже по сравнению с приведенным выше.
По другим данным, критическая температура для хлорирования этилена
t = 270-350 °C [ 1195, 1200, 1202].
Соотношение между этими двумя реакциями зависит от их энергии актива-
ции. В реакции присоединения она ниже, поэтому реакция замещения ускоря-
ется с ростом температуры и, в конце концов, становится основной.
Возможен также маршрут, согласно которому сначала протекает реакция
присоединения, а затем идет дегидрохлорирование полученного дихлорэтана с
образованием винилхлорида:
С2Н4С12—С2Н,С1 + НС1 - 71 кДж. (4.171в)
Последняя реакция ускоряется с ростом температуры, что способствует
снижению выхода дихлорэтана с соответствующим увеличением выхода ви-
нилхлорида.
Значительное количество тепла, выделяющегося в ходе реакции хлориро-
вания, приводит к деструктивному хлорированию углеводородов и хлоругле-
водородов с выделением большого количества сажи и хлороводорода. Реакция
деструктивного хлорирования особенно интенсивно протекает при использо-
вании исходной газовой смеси в концентрированном состоянии.
Для устранения деструктивного хлорирования предложен ряд условий: раз-
бавление исходной газовой смеси инертными газами; использование этилена в
большом избытке; проведение процесса в среде расплавленных солей; добавка
разных ингибиторов.
Механизм и кинетика процесса
Первое серьезное исследование газофазного хлорирования этилена провели
В. Воган и Ф. Раст [1199]. Они предположили, что в зависимости от ряда условий
реакция может идеи либо но ценному, undo но бимолекулярному мех.нт 1му.
Гакос кажущееся противоречие пощнее было обьяснемо единым радикально
ценным механизмом |357, 120()| В ном случае ырождепие цепи идет на стены
реактора, а обрыв протекает как на с iciikc, ык и в объеме.
Впервые о возможности протекания реакции но гегерогеппо гомогеп
пому механизму сообщается в работах 11289, 1290]. Позднее это обосновано
ермодипамически [357, 1291). Были проведены специальные исследования,
которые показали, что реакция протекает в объеме, а зарождение цепи — па
поверхности реактора [1203, 1208]. Цепной характер реакции заместителюю
то хлорирования этилена подтверждается ингибирующим действием ne.iiia
чи тельного количества кислорода, обычно содержащегося в хлоре. При этом
развитие цепи происходит пока активные центры, зародившиеся на поверх
пости, не погибнут в результате столкновения с молекулами кислорода. По
ному, чем меньше кислорода в смеси, тем дальше от поверхности идет рас
просгранение цепи. В результате зона развития реакции становится шире.
Доказательством гетерогенно-гомогенного механизма являются также опы i ы
с изменением общего давления, поскольку расстояние, на которое активный
центр может отойти от стенки, зависит от общего давления и концентрации
кислорода [1203].
Авторы работ [1199, 1202-1204, 1208] также подтверждают радикально
цепной характер процесса, основываясь на ингибирующем воздействии гетра
и илсвинца. При этом кислород, введенный в систему, на реакцию не расходу
е тся. В присутствии кислорода цепи зарождаются и обрываются на поверхнос
ги; в отсутствие кислорода цепи зарождаются на поверхности, а обрываются
в объеме.
Стенка реактора не только способствует зарождению активных цен трон, по
и может вызвать гетерогенное замедление гомогенной реакции [1292] в резуль
гате захвата радикала твердой поверхностью [357, 1293], на которой образуется
пленка продуктов, прочно адсорбированных за счет валентных и коордипаци
онных связей [357, 1294]. Обрыв же цепей зависит от сорбции свободных ра
дикалов, что определяется энергией активации образования связи радикала с
поверхностью. Эта энергия зависит от природы поверхности и составляет 37,7-
41,9 кДж/моль [1295]. Свободные радикалы, возникшие на поверхности, веду i
к разрыву связи С1—С1 или R—С1 с десорбцией атома хлора в объем 1129^,
1296].
Если энергия активации небольшая, то поверхность при сравнительно низ
ких температурах инициирует хлорирование этилена [ 1208].
Рост температуры увеличивает скорость поверхностного обрыва, гак как
эта реакция обладает более высокой энергией активации. Зарождение и обрыв
цепи находятся в динамическом равновесии, зависящем от температуры. Одна
ко при высокой температуре увеличение поверхности, как отмечалось, не ока
зывает значительного эффекта. Повышенная (более 300 °C) температура ено
собствует гомогенному зарождению цепей за счет диссоциации молекул хлора
в объеме [1203]. Однако сильно развитая поверхность, например, стеклянная
вата, ускоряет реакцию и при высоких температурах, хотя и в меньшей степени
[ 1199]. Еще более развитая поверхность (активированный уголь) катализируем
процесс образования полихлоридов за счет того, что образующиеся первич
иые моиохлориропанкые продукты адсорбирую ня на аминной поверхнос)и
лстче, чем пилен. Лдсор(»ция в ном случае также снижает энергию разрыва
связей, чао способствует последовательному хлорированию на поверхности.
Накопление же атомов хлора в молекуле, содержащей два атома углерода, нс
дет к ослаблению связи С—С. Более тою, распад этой связи облегчается на
активной поверхности; в результате при 500-560 °C образуется тетрахлорид
yiлерода [1204, 1208].
Поверхность сосуда или насадка в ходе процесса обуглероживается. Угле
родистая пленка имеет состав Si — О — R и не содержит атомов хлора. Поэтому
рост скорости реакции по мере покрытия поверхности пленкой объясняется
уменьшением адсорбции радикалов [1297]. В ходе процесса эта пленка утолща
ется, что ведет к замедлению скорости реакции [1298].
Расчеты энергии активации бимолекулярных и радикально-цепных реак
ций между этиленом и галогенами показали, что энергетически более выгоден
радикально-цепной механизм [689].
Известно [357], что процессы газофазного хлорирования протекают по не
разветвленному цепному механизму, причем активными центрами служат ато-
марный хлор и радикалы хлорируемого продукта. Неразветвленный характер
цепей приводит к стационарности процесса и постоянству концентрации актив-
ных центров при изотермическом режиме. Процесс нестационарен только не-
посредственно после впуска или подачи компонентов в нагретый реактор, т.е.
на стадиях выравнивания температуры и начального генерирования активных
центров [1391].
В дальнейшем при изотермическом режиме концентрация атомарного хлора
заметно изменяться не будет, поскольку в процессе развития неразветвленной
цепной реакции он генерируется в том же количестве, что и расходуется. Гете-
рогенная и гомогенная рекомбинация атомарного хлора, т.е. реакции обрыва
цепи, не приводят к уменьшению величины [СГ], так как рекомбинация компен-
сируется генерированием такого же количества СГ. По аналогичной причине
невозможно и накопление активных центров. Поэтому протекание хлориро-
вания, не возмущаемого внешними воздействиями, не будет изменять концент-
рацию атомарного хлора по сравнению с его концентрацией в нереагирующей
смеси аналогичного состава. Данное предположение справедливо при обрыве
цепи на СГ. При этом [СГ] не будет отличаться от равновесной для спонтанной
диссоциации:
[СГ] =/сс12[С12]0’5 (4.172)
гДе ^а2 — константа равновесия диссоциации [1393]. Пересчет данных позволя-
ет получить для температурной области, в которой обычно проводят процессы
хлорирования, kCi2 = /соехр(-1448О/Т), к0 - 2,6 моль1/2/см3/2.
На возможность определения скорости хлорирования на основе предпо-
ложения о равновесной диссоциации хлора было впервые указано в работе
[1392].
В силу стационарности неразветвленного цепного процесса концентрация
радикалов хлорируемого продукта [R’] также остается постоянной, однако толь-
ко в отсутствие возмущений. Если в реакционной среде присутствуют ингибито-
ры, способные реагировать с R', стационарный режим соответствует более низко-
му уровню | R'|. К i.i ком уже peiyni.i.ny mokci приводи i i> парная рекомопнацня
радикалов R’ или перекрестия R' I ( Г В г плу общеп стационарноеш умет,
шепие [R"| будет приводи и. и к уме о |( Г|, при ном равновесие диссоцн
ации хлора будет нарушаться, [СГ] < К( । [С|2|“'’. Возможен и o6paiin>in >ффсм
в случае побочных реакций радикалов R* с исходными компонентами, приводя
|цими к дополнительному образованию конечных продуктов без уменьшения
общего числа радикалов.
Если влияние описанных возмущений не очень велико, а константа скоро
с i n kt элементарной реакции СГ и хлорируемого продукта известна по данным
независимых измерений, возможно приближенное априорное определение сум
мирной скорости хлорирования.
Для многих процессов хлорирования значения к,, а также к2 — констан гы с ко
рос ги реакции R" + СГ были определены независимыми от исследования хлорн
рования методами [1395]. Использованию описанной системы благоприя тс i нус i
го обстоятельство, что энергии активации элементарных реакций атомарною
хлора во всех известных случаях не превышают 23-28 кДж/моль, а чаще тиа
чи гельно меньше.
Также невелика разница между значениями предэкспоненциальных множи
гелей для констант скорости к}. Оба эти обстоятельства позволяют даже в тех
случаях, когда экспериментальные значения не были определены, оценивать
их с удовлетворительной точностью по известным к1 для реакций СГ с продукта
ми, имеющими родственную изучаемому химическую природу.
При хлорировании олефинов или неполностью замещенных хлоролефипов
возможна двоякая природа взаимодействия атомарного хлора с хлорируемым
продуктом: либо с присоединением хлора по двойной связи, либо с отрывом а го
ма водорода и его последующим замещением другим атомом хлора. Для этилена
возможны реакции
fc.(I)
С2Н4 + СГ —- С2Н4СГ; (I)
fci(ll)
С2Н4 + СГ —- С2Н3* +НС1. (II)
Дальнейшие реакции в первом случае приводят к образованию хлоралкапов,
во втором — хлоролефинов.
Разница величин и /сДП) будет сказываться на соотношении выходов
дихлорэтана и винилхлорида, но не на скорости брутто-реакции.
В работе [1203] были измерены скорости реакции в эквимольной смеси
С2Н4 + С12 путем измерений изменения давления р при начальных давлении
Ро= 0,026 МПа и t0— 150+250°C, в работах [1283,1279] р = 0,1 МПа, f0= 320+380 °( ,
в работе [1396] Рс= 0,016 МПа, t0= 250+385 °C. В [1205] и [1206], скорость реакции
определяли по изменению состава смеси 98% С2Н4 + 2% С12, проходящей через
кварцевый реактор, с ее последующим анализом, в работе [1396] по изменению
состава смесей С2Н4 + С12 + Аг, первоначально содержащих 4,6-19% С12, 20 95%
С2Н4 и 0-7% Аг. (концентрацию недостающего компонента смеси — хлора —
определяли фотометрически). Результаты расчета и экспериментальные данные
представлены на рис. 4.120.
Рис. 4.120. Зависимость keff от температуры по данным:
1 — расчетным [1395]; 2— [1203]; 3 — [1283]; 4 — [1205]; 5 — [1204].
Сопоставление к, и кех показывает, что кех < к,.
А.И. Розловский [1391] указывает, что разность величин кех и kt может быть
значительней и достигать 1,7 порядка при минимальной их разности 10% кеа. Ав
тор полагает, что этот эффект обусловлен связыванием активных центров без их
регенерации. Эффект ингибирования обусловлен не только влиянием посторон
них примесей, но и ингибированием цепного процесса конечными продуктами,
способными взаимодействовать с активными центрами без их регенерации.
Для реакции присоединения предложена схема:
М+С12 —-2СГ, (4.173а)
С2Н4 + СГ —— С2Н4СГ,
С2Н4СГ + С12 ‘ ► С2Н4С12 + СГ и т.д.,
(4.1736)
(4.173в)
для которой энергия активации равна 119,3 кДж/моль. Эта величина пример-
но в 1,6 раза отличается от энергии активации, найденной экспериментально
(71,2 кДж/моль) [1300].
Для реакции замещения предложена схема [689]:
С2Н4 + СГ —- С2Н3’ + НС1,
С2Н3‘ + С12 —С2Н3С1 + СГ и т.д.,
(4.174а)
(4.1746)
для которой энергия активации составляет 181,7 кДж/моль. Так как реакция за-
мещения имеет большую энергию активации и малое значение предэкспонент-
ного множителя, то она значительно ускоряется с ростом температуры.
Однако винилхлорид можс! i>l»p.i ioiu.ih.ii 1.1 н и в ре тулы.пс де! идрохлори
рования продукта присоединения — дихлор мана:
С,Н|С12 + (. Г— * C2lltCb’ + IK I; (1.175a)
C2H3C12* —* C2H tCI + СГ. (1 175(>)
Этот процесс хорошо изучен и достаточно полно отображен в работах [763,
105].
Дегидрохлорирование дихлорэтана в присутствии даже небольших коли
честв хлора, который является инициатором этой реакции, идет при темпера
гуре 350-450 °C и времени пребывания 3-12 с с заметной скоростью — степень
превращения составляет от 20 до 100%. Таким образом, вполне вероятно, что
образование винилхлорида протекает одновременно по двум реакциям- »аме
щсния и дегидрохлорирования. Кинетика процесса свидетельствует, что с ко
рость последней реакции значительно ниже по сравнению с первой, однако, по
лучение винилхлорида за счет пиролиза дихлорэтана может иметь сущей вен
ное значение в общем процессе [1205, 1208, 1209]. Подтверждением протекания
процесса пиролиза дихлорэтана служит то обстоятельство, что малые досыпки
кислорода в количестве 0,5-3% инициируют этот процесс [1208]. Однако, как
было показано выше, кислород с концентрацией до 0,5% инициирует и реакцию
замещения, а повышение концентрации кислорода ингибирует эту реакцию.
11оэтому все доводы в пользу того или другого механизма недостаточно досто
верны, а доказательства их существования имеют косвенный характер.
По кинетическим кривым изменения концентрации хлора по времени паи
дено, что скорость термического хлорирования в интервале 320-380 °C онисы
вается уравнением 1-го порядка по хлору [1208], а константа скорости, с ’, со
гласно [1205, 1208, 1283]:
к = 7,2-Ю12 ехр[-33700/(RT)L (4.176)
В реакции высокотемпературного хлорирования этилена существенны про
цессы зарождения цепей на стенках реактора. Об этом свидетельствует сравни
тельно низкая величина энергии активации процесса (141 кДж), тогда как при
гомогенном зарождении цепей по реакции (4.173а) расчет энергии активации
по схемам (4.1736, в) или по схемам (4.174а, б) показывают более высокое зпаче
ние (167,5 кДж) [1205].
Кажущаяся энергия активации реакции термического хлорирования этиле
на в присутствии кислорода несколько падает и составляет примерно 113 кДж/
моль [1208].
Образование НС1 по реакции замещения также имеет 1-й порядок. Автор
[ 1208] считает, что протекание побочных процессов замещения невозможно, i а к
как в опытах использовалась исходная смесь в соотношении этилен:хлор 50:1.
Процесс описывается уравнением:
к" = —— In —=----------=-, (4.177а)
т-т0 1-[ВХ]/[С12]0
где т0 — период индукции, с;
ICI Jd и IB\| — нача/илыл концентрации хлора и текущая концентрация ни
пипхлорида, моль/л.
Для температур 320-380 °C константа скорости, с 1 [1205, 1208|:
£" = l,210llexp[-29800/(R7)]. (4.177b)
Температурная зависимость константы скорости расходования хлора и
процессе хлорирования винилхлорида, л/(моль-с), при 320-380 °C описывается
уравнением [90, 1208]:
к = 1,5-Ю12 exp[-25900/(RT)]. (4.177в)
Общая скорость реакции хлорирования этилена выше скорости реакции
хлорирования винилхлорида на 0,5-1 порядок [ 1205], так как связь С—С1 более
стабильна, чем С—Н. Кроме того, связь С—С1 обладает двоесвязным харакге
ром и сопряжена с двойной связью, что повышает термическую устойчивос ть
винилхлорида. Таким образом, атом хлора в молекуле винилхлорида тормози г
последующее замещение атомов водорода хлором, т.е. винилхлорид менее реак-
ционноспособен, чем этилен. Хлорирование этилена в интервале 150-250 °C под
чиняется уравнению 2- го порядка. Энергия активации составляет 79,5 ± 8,4 кДж/
моль, а в присутствии кислорода — 146,5 ± 4,2 кДж/моль [1203].
Была найдена эффективная энергия активации: 72 кДж/моль при 250-345 °C
и 125,6 кДж/моль при 345-385 °C [ 1202]. Процесс имеет первые порядки по ком-
понентам.
В присутствии же кислорода при проведении процесса в интервале 270-
320 °C порядки реагентов меняются и составляют соответственно для этилена,
хлора и кислорода — 0,5, 2,0 и 1,4, а эффективная энергия активации достигает
213,5 кДж/моль.
При сравнении энергий активации, полученных разными исследователями,
видно, что при 320-380 °C она колеблется в допустимых пределах 125,6 кДж/
моль [1202] и 141,7 кДж/моль [1205, 1208, 1283]. Аналогично при более низких
температурах (-250 °C), когда £акт = 41,9 ± 8,4 кДж/моль [1203] и 72 кДж/моль
[1202]. Однако в присутствии кислорода значения энергий активации, получен-
ных разными последователями, существенно разнятся.
Для высокотемпературного хлорирования этилена была составлена матема-
тическая модель процесса [1303]. Согласно расчетам, при 327 °C £акт = 165,9 кДж/
моль, а при 527 °C £акт = 154,9 кДж/моль, что удовлетворительно согласуется с
опытными данными [1202, 1208]. Кроме того, расчеты показали [1303], что ско-
рость реакции замещения не может обеспечить выход винилхлорида, получен-
ный экспериментально, т.е. образование его идет одновременно двумя путями
по реакциям (4.1716, в).
При этом увеличение соотношения С12:С2Н4 способствует образованию по-
лихлоридов и хлороводорода, а не продуктов замещения.
На основании ряда исследований и термодинамических расчетов был пред-
ложен следующий многостадийный механизм этого процесса [1239].
I. Радикальное присоединение хлора к этилену, протекающее по реакциям
(4.173я-в), а также по реакциям:
< 11 ,< Г - ► ( .11, । СГ; (1.178а)
С2Н4С12 *• СГ—С2П,(Т t cij. (1.1780)
11. Последовательное хлорирование дихлорэтана, включающее реакцию
(4.175а), а также:
С2Н3С12' + С12 — С2Н3С1, + СГ; (1.179а)
С2Н,С12' + НС1 — С2Н4С12 + СГ; (4.1790)
С2Н3С13 + СГ— С2Н3С1/ + С12. Ill. Реакция обрыва: (1.179в)
2СГ + М — С12 + М; (4.180а)
С2 Н4СГ + СГ— С2Н4С12; (4 I80O)
2С2Н4С1 * —► продукты; (4.180в)
С2Н3С12* + СГ— С2Н3С13; (4.180г)
2С2Н3С12’ —продукты; (4.180Д)
С2Н3С12’ + С2Н4СГ —продукты. (4.180с)
IV. Реакции замещения, включающие реакции (4.174а, б), и, кроме того:
С2Н3* + НС1 — С2Н4 + СГ; (4.181а)
С2Н3С1 + СГ — С2Н3" + С12. V. Реакции последующего замещения в молекуле винилхлорида: (4.1816)
С2Н3С1 + СГ — С2Н2СГ + НС1; (4.182а)
С2Н2СГ + С12 —- С2Н2С12 + СГ; (4.1820)
С2Н2СГ + НС1 — С2Н3С1 + СГ; (4.182в)
С2Н2С12 + СГ — С2Н2СГ + С12. (4.1821)
VI. Присоединение хлора к винилхлориду:
С2Н3С1 + СГ — С2Н3С12'. VII. Обрыв цепи в реакции замещения: (4.183)
С2Н3* + СГ — С2Н,С1; (4.184а)
2С2Н3’—•- продукты. VIII. Радикальное разложение дихлорэтана по реакции (4.1756): (4.1810)
IX. Радикальное* разложение грихлор.пана
( Jhdj + < Г-— С2Н2С1; + IIC1; (4.185а)
С2Н3С1/ —*С2Н2С12 + СТ. (4.1856)
X. Обрыв цепи в реакции разложения трихлорэтана:
С2Н2С13 + С1 —*- С2Н2С14; (4.186а)
2С2Н2С13‘ —•- продукты; (4.1866)
С2Н2С13* + R —•- продукты, (4.186и)
где R — частица со свободной валентностью.
XI. Образование 1,1-дихлорэтана:
С2Н4СГ + HR —- С2Н5С1 + R’; (4.187а)
С2Н5С1 + СГ —► СНзСНСГ + НС1; (4.1876)
СНзСНСГ + СГ —- СН3СНС12. (4.187b)
Обращает на себя внимание, что ряд реакций в предложенной схеме ма
ловероятен. Так, например, реакции (4.1796), (4.181а), (4.182в) практически
невозможны, так как энергия разрыва связи Н — О в молекуле хлороводоро
да несоизмеримо больше энергии разрыва связи С1—С1 в молекуле хлора, и
даже энергии разрыва связи С—С во всех хлорзамещенных углеводородах.
Кроме того, реакции (4.178), (4.179в) и (4.1816) являются обратными реак-
циям (4.1736, в), (4.1746), (4.179а), и протекание их маловероятно, поскольку
они обладают более высокой энергией активации. Реакции (4.1816) и (4.183)
также маловероятны, так как, по данным [1205], винилхлорид гораздо менее
реакционноспособен, нежели этилен. Реакции (4.185) обладают значительно
более высокой энергией активации, чем реакции (4.175). Поэтому речь о них,
а также о реакциях (4.186), может идти, если процесс проводится при очень
высокой температуре (порядка 450-500 °C). Это подтверждают и эксперимен-
тальные данные [1206, 1211].
Термическая устойчивость хлорэтильного радикала намного выше по срав-
нению с этильным радикалом, поэтому реакции (4.187) возможны лишь при
высоких температурах [1206].
Несколько упрощенный механизм в виде уравнений (4.173б)-(4.174) был
предложен в [1200].
На основании известных из литературы данных в [1303] представлена схе-
ма этого процесса, состоящая, последовательно, из реакций: (4.180г), (4.1756),
(4.183), (4.173а), (4.180а), (4.180в), (4.1876), а также (4.1736), (4.1806), (4.173в),
(4.1786), (4.1796, в):
С2Н4С1 * С1 + С2Н4; (4.188а)
С2Н4СГ + НС1 —*- С2Н5С1 + СГ; (4.1886)
C21I|( 12 t ( Г — ( 21I,( I/ i ll( I;
C211,CI/ > Cl2 — C-l hCI, I CT;
C2Ut('l + (T—*C2I1(CI2’.
(I.188b)
(1.188г)
(•!.188д)
В этом случае реакции (4.188а), (4.1876)» (4.1786) и (4.183) являются, ио су ги,
обратными реакциям (4.1736), (4.1886), (4.173в), (4.188в, г), (4.179б, и) и (4.1756),
поэтому их ход, как указывалось, маловероятен. Реакции (4.1886) и (4.1786) i.u
же практически невозможны, поскольку, как указано выше, энергия разрыва
связи Н—С1 в молекуле хлороводорода много больше энергии разрыва связи
С1— С1 в молекуле хлора. В рассматриваемых схемах не приводятся стадии, по
которым происходит образование тетрахлорида углерода.
Важные данные о закономерностях процесса газофазного хлорирования
этилена получены М.В. Хрулевым [2] при опытно промышленных работах.
Основные условия процесса были следующими:
Содержание, %:
этилена в этиленовой фракции........98
хлора в испаренном газе.............100
Температура, °C:
этилена перед смесителем............400
хлора перед смесителем..............20
газов в зоне реакции................ 475-490
газов на выходе из реактора.........385
Скорость, м/с:
газов в хлорном и этиленовом соплах.. . 200
потока в зоне реакции...............8-10
Результаты производственных опытов приведены в табл. 4.28.
Таблица 4.28. Результаты производственных опытов
Расход, м3/ч Объемное отношение С2Н4:С12 Состав реакционных газов, об. % Выход: ВХ/(ВХ + дихлорэтилсны), % от геор., в расчете
С2Н4 (100%- ный) С12 (100%- ный) С2Н4 НС1 ВХ Дихлор- этилены Дхэ на хлор на ЭТИ/IVII
66 8,8 7,5:1 78,6 10,8 8,41 1,18 1,01 70,6/80,6 79,4/90,0
66 8,8 7,5:1 80,1 9,91 6,73 1,59 1,67 56,2/69,3 70,5/87,1
66 8,8 7,5:1 78,6 10,7 7,12 1,81 1,77 59,2/74,4 63,7/79,4
66 8,8 7,5:1 78,6 10,8 8,35 1,26 0,99 70,0/80,4 78,9/90,7
66 9,4 7,0:1 76,8 11,35 7,25 2,04 2,61 56,7/72,6 55,5/71,1
66 10,0 6,6:1 74,5 14,4 6,62 4,02 0,46 50,0/80,7 52,2/84.0
66 12,0 6,5:1 71,9 16,3 6,3 5,03 0,43 40,8/73,2 47,2/85,3
66 12,0 5,5:1 72,1 16,4 5,5 5,40 0,6 35,6/70,5 42,6/81,6
) 1ИЛГ1 I
Рис. 4.121. Схема высокотемпературного хлорирования этилена:
I — буферная емкость для этилена; 2 — электроподогреватель этилена; 3 — испаритель жидкого хло
ра; 4 — буферная емкость для хлора; 5 — подогреватель газообразного хлора; 6 — смеситель; 7 — ре
акционная печь
Принципиальная схема установки изображена на рис. 4.121. Этилен через
буфер 1 и ротаметр поступает в электроподогреватель 2, где нагревается до
температуры 400 °C. Хлор из испарителя 3 через буфер 4, ротаметр и подогрева
тель 5 подается в хлорное сопло Хлор и этилен через смеситель газов 6 направ
ляются в реакционную печь 7. Прореагировавшие газы подаются из реактора на
конденсацию и разделение.
Процесс получения винилхлорида хлорированием этилена обладает рядом
недостатков. Есть трудности и технологического оформления, в частности, в
реализации удовлетворительной теплопередачи.
Процесс весьма осложняется деструктивным хлорированием этилена с
выделением углерода (сажи), а также образованием продуктов дальнейшего
хлорирования этилена и этана. В некоторой степени эти недостатки устраня-
ются [1196] при хлорировании этилена в среде расплавов, например, смеси
КС1 + СаС12, плавящейся при 206 °C и имеющей теплоемкость 1,25 кДж/кг. Более
низкая температура плавления достигается добавлением к смеси КС1 и СаС12
других солей.
Как видно из приведенных ниже данных, в этих условиях при 200 °C образу-
ются преимущественно предельные хлорпроизводные, а при 400 °C преоблада-
ют непредельные продукты хлорирования этилена:
1, °C.......
Выход, %:
ДХЭ
ВХ..........
200 284 400 500
83,4 50 16,7 7
16,6 50 83,3 93,0
В pe.iy/ii.raie лабораторных in г ледов.1111111 пыл pa ip.1601.111 режим, при
котором винилхлорид может бы и. получг н с выходом 65-67% 11 19б|.
Состав расплава, %:
КС1.................................35.9
СаС12................................64,1
Концентрация, %:
этилена..............................>98
хлора................................98-100
кислорода в реакционной смеси......0,2
Температура процесса, °C............... 390-400
Этилен : хлор..........................2,5:1
Скорость газа (на 1 л расплава солей), л/ч .. 600
Одновременно с винилхлоридом в этом процессе образуются винили
денхлорид (10-12%, или 150 кг на 1 т винилхлорида), дихлорэтан (7-10%, или
100 кг на I т винилхлорида) и высшие полихлориды (7-10%). Для получения
1 т винилхлорида этим методом расходуется 1,9 т хлора и 0,6 т этилена. 11ри
этом получается 3 т соляной кислоты. На рис. 4.122 приводится возможная iex
нологическая схема производства, предложенная в [1196].
Процесс в среде расплавленных солей дает возможность значительно спи
зить не только соотношение этилена и хлора, но и степень деструктивного хло
рирования.
Рассмотрим более подробно влияние разных факторов на показатели про
цесса газофазного хлорирования этилена.
Рис. 4.122. Схема получения винилхлорида хлорированием этилена в среде рас
плавленных солей:
1 — воздуходувка; 2 — реактор; 3 — конденсатор; 4 — газоотделитель; 5 — сборник хлорор
ганических продуктов; 6, 8, 9 — скрубберы; 7 — сборник соляной кислоты; 10 — отбойник
71 — буферная емкость; 12— компрессор; 13— теплообменник; 14 — конденсатор; 15— 1.1:10
отделитель (0,4 МПа); 16 —сборник; 17 — холодильник.
Влияние температуры
11роцесс изучался и довольно широким диаца юнс температур oi 150 [ 1 Л0'|
до 560 °C 11201]. Выход винилхлорида увеличивается с ростом температуры до
определенной величины, а затем начинает падать. Максимальный выход раз.
ми исследователями зафиксирован при 275 315 °C [1199], 440 °C (1205], 450 "С
(1206], 400-460 °C [1207], 440-480 °C [1208], 440-500 °C [1204] и т.д. Естественно,
такое расхождение обусловлено прежде всего некоторыми различиями разра
ботанных авторами методик проведения исследований, анализов реагентов и
продуктов реакции, а также точностью определения и точностью измеритель
ных приспособлений.
Опыты проводились как в статических [1203, 1204], так и в динамических
условиях [1199, 1209]. При одинаковой температуре показатели процесса хло
рирования особенно сильно зависели от длительности пребывания в реакциоп
ной зоне. Тем не менее, обобщая приведенные данные, можно утверждать, что
выход винилхлорида максимален при 400-460 °C. Дальнейший рост температу-
ры до 500-560 °C приводит к разложению целевого продукта по реакции [1204,
1206]:
С2Н3С1 —- С2Н2 + НС1 (г) (4.189)
и образованию полихлоридов, метана [1206] и сажи [1204, 1205, 1208, 1211].
Имеются данные, что увеличение температуры реакции от 390 до 450 °C вле-
чет рост образования сажи примерно в 20 раз [1210]. В продуктах реакции
были найдены также дихлорэтилены, образование которых связывается с
протеканием в этих условиях инициированного пиролиза 1,1,2-трихлорэтана
[1213].
Кроме того, проведение процесса при высоких температурах связано с
еще одной весьма сложной проблемой. Высокая экзотермичность процесса
(для реакции присоединения АН = 171,5 кДж/моль, а для реакции замещения
АН = 96,2^113,0 кДж/моль [1214-1218]) требует значительного теплоотвода, в
противном случае возможно горение олефина в токе хлора, что ведет к глубо-
кому разложению продуктов замещения и, как следствие, к забивкам реакцион-
ной системы [1208].
Большое значение придается температуре, с которой реагенты вводятся в
хлоратор. Поскольку реакция замещения протекает при более высокой темпе-
ратуре, чем реакция присоединения, то для повышения выхода винилхлорида
рекомендуется, чтобы температура хлора и этилена была близка к температуре
процесса [1191, 1272, 1285, 1287]. Разницу этих температур желательно иметь в
пределах 0-120 °C [1239].
Влияние критического температурного интервала на характер реакции хло-
рирования иллюстрируют следующим: если при 308 °C содержание продукта
реакции присоединения хлора к этилену — 1,2-дихлорэтана достигает 64%, ви-
нилхлорида ----20,8% дихлорэтиленов — 4,6%, то при 346 °C содержание ди-
хлорэтана снижается до 20%, а содержание винилхлорида и дихлорэтиленов,
т.е. продуктов реакции заместительного хлорирования, резко увеличивается
(соответственно 52,7 и 14,8%).
Влияние соотношения реагентов
Нс ГОЛГКО ПЛОХОЙ 1С11ЛООГНОД (.IKKOOi lliyci горению исходных продукюв
и реакционной tone. Воспламенение moiu-i n.riaii.oi при подаче в реактор зги
лена и хлора в соотношении 1:1 даже при ~215 °C, причем процесс сгаповиим
I рудно управляемым [ 11991. Поэтому лини, применение значи тельного изоьпка
олефина позволяет вести реакцию при высоких температурах [ 1195, 1219, 1220].
При увеличении соотношения олефин:хлор для получения таких же больших
степеней превращения, как в случае использования эквимолярных соогноше
ний, необходимо повышение температуры [1199, 1220]. Относительно влия
ния соотношения реагентов на выход винилхлорида имеются противоречивые
сведения. По одним данным [1199], выход винилхлорида повышается в случае
уменьшения этого соотношения. По другим [1195,1211,1220], выход винилхло
рида растет с повышением соотношения этилентхлор.
Очевидно, что при хлорировании этилена, кроме винилхлорида, могут об
разовываться и другие хлорпроизводные этилена, например, СНС1=С1К '1,
СН2=СС12, СНС1=СС12, СС12=СС12. Бесспорно, образование этих сосдипс
ний будет сопровождаться выделением хлороводорода. Кроме того, следует
ожидать, что в процессе реакции какое-то количество этилена будет подпер
гаться деструктивному хлорированию, которое также сопровождается oopa.io
ванием хлороводорода. При этом уменьшение соотношения может привес ги к
росту деструктивных процессов [1204, 1205].
Данные о влиянии избытка этилена [ 1195] на реакцию заместительного хло
рирования:
С2Н4:С12, моль/моль........................... 7,7 7,7 8,9 4,1 4,6 6,6
Скорость подачи хлора, моль/л-ч............... 0,38 0,38 0,88 1,26 1,30 4,30
Время прохождения потока через реактор, с..... 0,02 0,02 0,06 0,04 0,05 0,021
Температура предварительного подогрева, °C.... 200 255 300 300 350 100
Средняя температура реакции, °C............... 198 254 314 326 361 100
Количество хлора, не вступившего в реакцию, % 19,0 0 0 1,3 0,7 0.6
Выход винилхлорида, %. ... 0 21,6 47,3 78,2 9,03 98,5
Максимальный выход (98,5%) винилхлорида достигается при соотношении
этилена и хлора 6,6:1 и t = 400 °C. Примечательно, что авторы во всех случаях
исходную газовую смесь предварительно нагревали примерно до температуры
реакционной зоны.
Избыток хлора вообще крайне нежелателен, так как это может ин теней
фицировать реакции, связанные с уплотнением углеродного скелета [12211.
При соотношении этилена с хлором <2 процесс становится неустойчивым,
о чем свидетельствуют вспышки в реакторе. В этом случае [1204], как и при
проведении процесса при высокой температуре, недостаточен теплоотвод с
продуктами реакции, поскольку начальная скорость тепловыделения очень
велика [1222].
OiMC'i.ieiLH, чго (.оо.ш получаемых продукта практически идешичеи при
соотношениях > шлеп: хлор 55 11201, 1208|. При избытке же этилена менее 5 пы
ход основных продуктов— винилхлорида и дихлорэтана падает, напротив, вы
ход смолистых, состоящих из полихлоридов и сажи, повышается.
По другим данным 11204, 1206, 1207] следует, ч го выход винилхлорида имел
максимум в области отношений этилен:хлор = 2+4:1. Повышение соотношения
до 8:1 приводит к сглаживанию этого максимума. Наиболее широкий ин горная
соотношений исследован в работе [1209], в которой показано, что выход винил
хлорида с ростом отношения этилен:хлор от 4:1 до 99:1 падает очень сильно
Наибольшее уменьшение выхода винилхлорида происходит в интервале соог
ношений примерно от 10:1 до 99:1. Поэтому авторы делают вывод, что макси
мальный выход винилхлорида, достигнутый в работах [1206, 1223, 1224, 1240]
при соотношениях 4+10:1 связан с образованием винилхлорида в результате
реакции дегидрохлорирования дихлорэтана, полученного по реакции (4.171а).
Дальнейшее же разбавление реакционной смеси этиленом ведет к снижению в
ней содержания свободного хлора — инициатора реакции дегидрохлорирова-
ния, вследствие чего выход винилхлорида уменьшается. Рост избытка этилена
соответственно повышает выход дихлорэтана и понижает выход полихлоридов
[1206]. По мнению авторов, это происходит вследствие того, что при сильном
разбавлении реакционной смеси этиленом вероятность столкновения его с хло
ром растет, а хлора с хлорированным продуктом падает. В результате скорость
реакции замещения уменьшается, а реакции присоединения увеличивается.
Влияние разбавителей
Самым простым решением является использование в качестве разбавителя
олефина [1195, 1204, 1208, 1223] или соответствующего ему парафина [1224-
1229]. Правда, в последнем случае одновременно идет хлорирование этого па-
рафина, что может привести к усложнению системы разделения реакционной
смеси. Более того, имеются сведения [1199, 1230], что парафины ингибируют
реакцию заместительного хлорирования. Например, введение -15% этана в ре-
акционную смесь, находящуюся при 327 °C, приводит к снижению доли реак-
ции замещения с 95 до 13%.
В качестве разбавителей можно использовать азот [1208, 1211, 1231, 1219],
диоксид углерода [1208, 1231, 1219], водяной пар [1233], метан [1234], хлоро-
водород [1235] и хлорпроизводные этилена и этана, в частности, этилхлорид
[1236, 1237], 1,1,2-трихлорэтан [1238], 1,2-дихлорэтан [1236, 1239-1250, 1257,
1258], смеси двух последних соединений [1238, 1252] и винилхлорид [90, 1208,
1253], смеси винилхлорида с 1,2-дихлорэтаном [1255]. Наибольший эффект
дает разбавление исходной смеси дихлорэтаном. При этом тепло, выделяемое
при взаимодействии хлора с этиленом расходуется на пиролиз добавляемого
дихлорэтана [см. (4.171в)]. В результате облегчается теплосъем, процесс стано-
вится легче в управлении, а главное — выделяется дополнительное количество
винилхлорида без внесения энергозатрат.
Было показано [1208], что выход винилхлорида зависит от избытка этилена,
но не зависит от природы разбавителя, в качестве которого использовались азот,
диоксид углерода и винилхлорид. Оказалось, что выход винилхлорида растет при-
Таблица 4.29. Влияние i renciiii ра in.пик itini ш ходиых ы ion n.i замш инсльпос хлорн
ронание этилена при 275 "(
Подано, мл/мин llpopcai пропало хлора, % Доля С12 = р.пр/всего, % t 1 °C
С12 С2Н4 N2 или СО2 X всего *1 р.пр
35 35 230 300 22 20 91 315
50 50 200 300 33 29 88 310
50 100 150 300 57 49 86 288
75 75 150 300 58 43 74 275
1 В реакции присоединения.
Температура, при которой получено максимальное количество продуктов реакции нрисосди
нения.
мерно с 30 до 90% при увеличении концентрации этилена в исходной смеси с 25 до
55%. При дальнейшем повышении избытка этилена выход винилхлорида ос гас ня
на одном уровне. По мере разбавления реакционной смеси азотом доля продук гон
заместительного хлорирования в продуктах реакции повышается [1211].
В качестве разбавителя использовался либо чистый дихлорэтан, либо
его смеси с азотом, диоксидом углерода, винилхлоридом и хлороводоро
дом [1239]. Были установлены оптимальные соотношения этилешхлор = 3 4,
разбавителыхлор » 5.
В других аналогичных работах указывают оптимальные отношения:
этилешхлор = 0,5-ь10, разбавителыхлор = 0,2-40 [1238-1240, 1243, 1244, 1252,
1255-1258].
В работе [1199] изучена реакция заместительного хлорирования этилена и
области 200-500 °C при разбавлении реагентов азотом или углекислым газом
[ 1199]. Полученные данные приведены в табл. 4.29.
Как видно из данных табл. 4.29, с увеличением концентрации исходной га
зовой смеси количество хлора, участвующего в реакции присоединения, умень
шается. При хлорировании газовой смеси, состоящей из этилена, хлора, а юга
(50:50:200 мл/мин), были получены хлорпроизводные продукты реакции, следу
ющего состава, моль %:
Температура, °C.................................................. 308 346
Винилхлорид...................................................... 20,7 52,7
1,1-, 1,2-Дихлорэтилены.......................................... 4,6 14,8
1,2-Дихлорэтан.................................................... 64 20,2
1,1,2-Трихлорэтан................................................ 40,3 9,9
Тетрахлорэтаны................................................... 6,4 2,4
Количество прореагировавшего хлора, 57 77
в том числе в реакции:
заместительного хлорирования................................... 37 25
присоединения.................................................. 20 52
Влияние времени пребывания
Снедения но ному iiapaMcipy процесса также противоречивы. С одной сто
ропы, утверждается 11207), что в диапазоне от 0,7 до 8 с выход винилхлорида
и хлороводорода остается без изменения. С другой стороны [1209], при срап
нении данных, полученных ст — временем пребывания 0,2 и 1 с, при прочих
равных условиях, оказалось, что чем больше т, тем больше выход винилхлорида
и меньше выход дихлорэтана. Особенно четко эта зависимость прослеживалась
при большом избытке этилена (примерно в 50 100 раз). Кроме того, имеются
данные [1204], что в диапазоне времени пребывания примерно от 1 до 11 с, вы
ход винилхлорида имеет максимум при т - 2з-4 с, который становится более
выраженным с повышением температуры.
Увеличение времени пребывания ведет к повышению выхода хлороводо-
рода и сажи. Объясняется это явление усилением процессов деструктивного
разложения хлорпроизводных, но при этом оговаривается, что вероятность
дегидрохлорирования винилхлорида весьма мала. Лишь при 540-550 °C в ре-
акционных продуктах были обнаружены следы ацетилена. В других работах
[1205,1208] утверждается, что количество хлора, участвующего в реакциях за-
мещения, с ростом времени пребывания увеличивается. Количество же хлора,
взаимодействующего в реакциях присоединения, имеет максимум, который с
ростом температуры смещается в сторону уменьшения времени пребывания.
На основании этого делается вывод, что наряду с образованием винилхлорида
в результате реакции замещения идет его дополнительное выделение за счет
дегидрохлорирования продукта реакции присоединения — дихлорэтана.
Влияние давления
В основном все исследования этого процесса проводились при общем ат-
мосферном давлении. Однако парциальное давление реагентов в каждом слу-
чае было неодинаково из-за разного соотношения этилена с хлором и добавок
разбавителей.
Относительно общего давления есть разные суждения. С одной стороны, ряд
авторов высказываются за пониженное давление, порядка 40-66 кПа, что, по их
мнению, способствует повышению выхода продуктов замещения [1259-1261].
С другой стороны, в качестве оптимального варианта рекомендуется давление
350-700 кПа [1262].
Очевидно, значительного влияния давления в таком малом диапазоне на
процесс высокотемпературного хлорирования этилена не происходит. Можно
предположить [1262], что рост давления увеличивает скорость реакции присо-
единения, которая идет с уменьшением объема. Однако повышение давления
может ускорить реакцию хлорирования дихлорэтана, которая также протекает
с понижением объема.
Влияние инициирующих и ингибирующих добавок
На реакцию заместительного хлорирования положительно влияет присутс-
твие в зоне реакции таких соединений, как дихлорэтан [1368], тетраэтилсвинец
[1199].
Добавки к-1 pa н и лиши на в колпчеч i вс до 0,002 мол. % способе i пуки с ниже
пик) темпера гуры процесса примерно до 200 "< 11199, 1261]. Гетра и илевипец
реагируя с молекулярным хлором, даш аллильные радикалы, инициирующие
цепи. Однако при ном ускоряются как реакции смещения, гак и присоеднне
ния Данное обстоятельство свидетелвс'гвуег, что обе ли реакции имски ра
локально ценной механизм и тесно взаимосвязаны. Последнее подтверждаете»
(ем, что каждой температуре соответствует определенное отношение иродук
юв реакции.
Влияние кислорода на данный процесс разными авторами трактуете»
неоднозначно. Так, имеются данные [1203], что кислород при хлорированы!
>1 плена является сильным ингибитором, добавление которою даже в коли
честве 0,5% значительно сказывается на скорости реакции. Анало! ичныс дан
ные приведены и в других работах [1199, 1264, 1265]. В случае, когда исход
пая смесь содержит 70% кислорода, скорость реакции снижается примерно i
200 раз [1203].
Ряд исследователей [1264, 1265] считают, что кислород тормози! реакции
смещения и не влияет на реакцию присоединения.
Напротив, в некоторых работах [1204, 1208] утверждается, чю введение
малых добавок кислорода (порядка 0,3-0,5%) увеличивает скороен> процесса
причем особенно возрастает скорость реакции замещения.
По мнению других авторов [1199, 1219], инициирующим эффектом реак
ции замещения обладает добавка кислорода до 3 об. %. Так, при введении в ис
ходную смесь 0,5% кислорода доля продуктов замещения выросла с 16 до 91%
С ростом температуры необходимо несколько увеличить количество кислород»
для достижения аналогичного эффекта [1204, 1208, 1219].
Объяснение такого действия кислорода вытекает из результатов исследова
ний [1203]. Выявлено, что скорость реакции с увеличением добавок кислород,
проходит через максимум, который приходится на 0,5%. Ускоряющее дейсгвш
малых добавок кислорода резко возрастает с увеличением температуры. I [иже
220-270 °C любые количества кислорода ингибируют процесс.
Данные о зависимости [1367] реакции заместительного хлорирования oi
присутствия кислорода:
Содержание кислорода в общей смеси газов, % Прореагировало хлора, %: 0,33 0,50 2,00
всего 39 88 91 13
в реакции заместительного хлорирования 14 76 87 4
в реакции присоединения 25 12 4 9
Согласно приведенным данным, в присутствии кислорода количество хло
ра, участвующего в реакции, возрастает, причем такая закономерность сохрани
ется до определенного содержания кислорода (0,5%). С дальнейшим увеличен!!
ем количества кислорода расход хлора резко уменьшается. Увеличение расход,
хлора приводит к росту количества продуктов реакции заместительного хлорн
рования.
Небольшие добавки водяного пара, сернистого газа и сероводорода нс влия
ют на хлорирование этилена [1208]. Введение в реакционную систему инертны»
i.ioii (Лг, I le, (. I |) inn iionpyioi пот процесс 11202] ( оедипения ины сероподо
ода, селеноводорода и теллуроводорода тормозят деструктивное хлориронп
ис 11195, 1211].
Влияние поверхности
Значительную роль в процессе высокотемпературного хлорирования эти
сна играет поверхность. При низкой температуре (примерно до 240 °C) изме
ение отношения поверхности реактора к его объему (S/V) с помощью насадки
рактически не влияет на скорость процесса [1199]. С повышением темперагу
ы до 300 °C увеличение этого отношения с 2,9 см-1 до 8,43 см 1 тормозит про
есс. Дальнейший рост S/V до 12,44 см"1 практически не сказывается на ско-
ос1 и процесса. Однако при заполнении реактора стеклянной ватой скорость
роцесса становится значительно выше, особенно при низких температурах.
)тмечено, что увеличение S/V ослабляет ингибирующее действие кислорода
1199, 1203].
В работе [1202] наоборот указывается, что увеличение S/ V существенно сни
тает скорость реакции в присутствии малых добавок кислорода.
Исследовался процесс в реакторах с разными отношениями поверхности к
бъему [1203]: незаполненном с S/V = 1,3 см"1, заполненном стеклянными труб-
ами с S/V = 4,4 и 9,3 см"1, а реакторе, заполненном капиллярными трубками
/V = 4,1 см В случаях, когда реактор был заполнен стеклянными трубками,
есмотря на значительную разницу в отношении S/V скорость реакции превы-
ила скорость реакции в полом реакторе, примерно на одну и ту же величину.
1ри заполнении реактора стеклянными капиллярами, несмотря на сравнитель-
ю низкое отношение S/V, скорость реакции превышала скорость реакции в
лораторе, заполненном трубками.
Имеются данные [1204, 1208], что развитая поверхность приводит к росту
>бразования полихлоридов. Так, при заполнении реактора шамотом основным
|родуктом реакции был дихлорэтан, причем его выход соответствовал выхо-
ду дихлорэтана, полученному при заполнении стеклянными трубками и битым
|теклом. При хлорировании в псевдоожиженном слое пемзы [1207], по сравне-
нию с реакцией в объеме ] 1220], выход винилхлорида уменьшается и составля-
т 50-60% [1211] и 66% [1207] с одновременным ростом выхода полихлоридов.
1 другом случае [1206] указывается, что в тех же условиях (1 = 450 °C) выход
шнилхлорида достигает 80%. Однако при этом выход полихлоридов остается
(остаточно высоким.
Несколько лучшие результаты дает хлорирование этилена в присутствии
»ксида алюминия, графита и кизельгура, хотя выход винилхлорида даже при
>00 °C составляет 81%, что ниже, чем при проведении процесса в объеме
1267].
Использование псевдоожиженного слоя контакта (силикагеля) способству-
ет повышенному образованию полихлоридов [1220].
Применение активированного угля еще более усиливает глубокое хлориро-
>ание с получением значительного количества полихлоридов [1204, 1208, 1220].
1асть полихлоридов при высокой температуре в присутствии активированного
тля подвергается хлоринолизу с получением тетрахлорида углерода. Так, при
>00 и 560 "(’ в продуктах реакции содержится с осн нс* ic i нечто 1,9 и 1,2 мае. %
iciрахлорида yi лерода и 65-72% полихлоридов При 120 1б0"( количес тво rei
рахлорида углерода снижалось до 0,1 0.1%
Кроме того, рост icMiiepaiуры веде1 к увеличению скорости реакции нос
ледова тельного хлорирования и деструкции, что сказывается на образовании
дополнительных количеств хлороводорода
При умеренных температурах активированный уголь подавляет реакцию
амещеиия [1204, 1208].
Основной причиной деструктивного хлорирования является цепной ха
рак । ер реакции, сопровождающейся выделением большого количества тепла.
11ри своевременном отводе избытка тепла можно в какой-то мере уменьши i ь
степень деструктивного хлорирования. В этом отношении проведение про
цесса в псевдоожиженном слое теплоносителя должно дать положительные
результаты. На примере низших газообразных алканов было показано 11 370,
I 371], что в условиях псевдоожиженного слоя теплоносителя степень дес i рук
гивного хлорирования резко снижается. Это было подтверждено при замес
тигельном хлорировании этилена в псевдоожиженном слое контакта [I 372|.
Опыты проводились в кварцевом реакторе при разных соотношениях этиле
па и хлора и температуре 300-500 °C. В качестве теплоносителя исполь гона
ли пылевидную пемзу и кварцевый песок. Полученные данные приведены в
табл. 4.30, 4.31.
Согласно данным табл. 4.30 и 4.31, на обоих теплоносителях винилхлорид
получается с приемлемым выходом.
При использовании пемзы хлорорганические продукты реакции содержат
55-70%, кварцевого песка — 35-75% винилхлорида. Хлорорганические продук
гы реакции содержат около 60-70% винилхлорида. Остальная часть этих про
дуктов состоит из хлорзамещенных этиленов, количество которых колеблется
в пределах от 1 до 27%. Оптимальный выход винилхлорида в обоих случаях
достигался при температуре 450 °C и соотношении С2Н4:С12 в первом случае
4:1, во втором — 6:1. Степень деструктивного хлорирования на кварцевом песке
была меньше, чем на пемзе.
Влияние катализаторов
Применение катализаторов в неподвижном слое существенно не меняет
характер процесса [1266]. Реакция идет при более низкой температуре, чго
способствует повышению выхода продуктов присоединения. При процессе
в присутствии гранулированного хлорида кальция выход винилхлорида при
275 и 375 °C составляет соответственно 20 и 38% [1268]. Лучшие результаты
были получены в случае использования в качестве катализатора угля, про
питанного хлоридом кальция или стронция. В качестве катализаторов — не
реносчиков хлора — предложено применять также расплавы солей, главным
образом, хлоридов [1196, 1271]. Хлорирование в расплаве имеет примерно те
же закономерности, что и хлорирование в объеме. Выход винилхлорида до
стигает 60-70%. Однако использование расплава дает некоторые преимущее
тва. Благодаря хорошему теплоотводу, можно работать с большими концепт
рациями хлора (С2Н4:С12 = 2,5:1) без существенного сажеобразования [1196,
Таблица 4.30. Состав продуктов реакции хлорирования этилена
Опыты №1 №2 №3 №4 №5 №6 №1 №2 №3 №4 №5 №6 №7 №8 .V9 .¥> 10
в псевдоожиженном слое пемзы в псевдоожиженном слое кварцевого песка
Температура, °C 450 450 450 450 450 450 400 450 450 400 400 450 450 450 350 350
Объемное соотношение этилена с хлором 3 1 4:1 4:1 4:1 4,6:1 4,6:1 3-1 3:1 3:1 6:1 6:1 6:1 6:1 6:1 6:1 6:1 I
Подано, г
этилена 137,5 137,6 137,6 124,0 207,1 207,1 217 208,6 209,8 152,5 225,9 228,5 225,9 231,6 229,5 225,9
хлора 116,3 87,1 87,1 74,1 112,5 112,5 174,9 174,9 174,9 63,9 95,9 96,0 96,0 96,0 96,0 96,0
Получено, г
этилена 104,9 99,4 109,0 95,0 178,4 181,7 171,2 181,0 179,4 137,6 203,5 208,5 201,25 213,05 209,0 203,1
хлороводорода 62,0 40,0 44,0 29,0 64,4 73,0 105,2 99,2 100,2 34,5 61,0 57,52 60,7 66,0 56,5 54,0
винилхлорида 54,6 48,6 42,2 51,6 41,5 31,1 46,7 25,7 39,0 20,6 21,2 28,7 39,9 34,6 27,9 32,6
хлорэтила 2,2 0,5 2,9 1,9 7,2 3,4 2,1 4,0 1,9 2,0 2,3 3,9 2,8 1,7 0,9 0,9
винилиденхлорида 11,4 9,0 7,1 6,7 6,4 5,8 8,6 8,8 15,1 3,1 3,5 2,4 1,8 1,1 4,5 4,5
дихлорэтиленов 4,7 1,9 6,6 2,1 4,2 4,2 9,5 9,2 7,8 3,8 4,3 3,1 2,3 1,3 5,0 5,0
трихлорэтилена 1,4 4,0 4,1 4,4 2,4 2,4 5,9 21,8 23,6 5,5 6,2 7,8 5,7 3,4 2,5 2,5
дихлорэтана 4,7 4,7 3,9 4,1 9,8 9,5 19,9 3,7 5,0 5,5 6,3 3,7 2,8 1,6 1,5 14,9
трихлорэтана 1,4 0,8 0,7 0,7 1,6 1,5 3,1 6,4 9,1 2,1 2,4 2,1 1,5 0,9 1,5 1,5
К! иод I
2 РОС о
п я -о с гс
О я р X н гс д ГС TJ СО О р о Я W _ s Й 3 * * й И g х 2 я о я Е Й Я <Т) g ГС й £ Н -г ° X ТЗ ± pl г*
н тз S Е ' £ п о г—’ X 40 ТЗ Р О
со ГС ГС g Й
н 2 1— гс
So® X g гс к S н ~ гс гс Н- Гс ЧО ГС чо ГС
О н х т* *
2 £ § о * 5 я s § NJ со О W У1 2 S »— СО
О 'С О Е S 2 о 2 00 Й
гс sq <—1 гс
я • qq X
- I S X я
s гс чО CD со
Я 2 “ я X
75 2 _ о о о « Я гс
я 1 * о о** “ я 2 я гс Я 75 £ “ Я И Я
2 р и я fa 5 J—। со Я § Р Я - м -1 75 Я Л * й s со
я “ 72 о 2
н я л g g 2 5 Т5 ГС
4^ g Н я § гс X й гс -О Р S Я р Н Й о
Ж Ж о 75 Я гс О Ж
о\ о р
я
о
я
Я
о
73
Я
fa
о
03
X
тз
р
и
fa
р
я
ТЗ
о
fa
X
и
ж
й
S
S
г>
о
fa
п>
о
*
р
н
я
Т
S
и
н
о
2
W
я
р
я
S
а
ОГ
ж
о
го
•X
о
№ 10 №9 N»8 №7 №6 №5 №4 №3 №2 № 1 tn №6 №5 №4 СЮ ZqM 1 оМ fa Опы 1
180,7 183,7 185,3 180,7 182,6 180,7 122,02 167,9 166,9 173,4 166,0 166 0 110,1 99,2 141,1 110 О N) I llpunyi
30,27 30,27 30,27 30,27 30,27 30,27 20,2 55,04 55,05 55,05 35,5 35,5 nj СП 23,4 35,1 Сю СЛ Q KJ С 5
СЛ н-* 1-9 6:1 1=5 СЛ >— 6:1 >—* СЮ сю СЮ 4,6:1 4,6:1 3:1 х;
198,5 198,5 198,5 198,5 198,5 198,5 198,5 206,3 206,3 206,3 188,5 191,7 169,0 0'691 174,1 180,0 ь
0,01543 0,01543 0,01543 0,01543 0,01543 0,01543 0,01543 0,0160 0,0160 0,0160 0,01447 0,01491 0,0154 0,0154 0,0154 0,016 м/с т:
3,0 3,0 3,0 СЮ о 3,0 3,0 3,0 сю о 3,0 3,0 3,0 3,0 2,0 ‘4 NJ СЮ 2,0 -
60,0 54,1 49,1 65,2 52,8 59,3 59.1 44,0 40,0 66,4 Сл NJ 64,6 83,3 98,3 81,7 71,2 с2и.| 7^
100 100 100 100 100 100 001 100 100 100 001 100 100 100 100 100 1<ИЭ<1.1
350 350 450 450 450 400 400 450 450 400 450 450 450 450 450 У. ч
1'09 61.0 ое л. О' 42,4 62,1 57,7 — 45 6 55,2 64 9 ’-с ОС 75,1
Скорость хлорирования и реакторе на стекла «пирекс» была выше, чем в
кварцевом реакторе с гем жеобьемом [90, 1205, 1208|. При покрытии реакци
окном новерхпос!и продуктами разложения (обуглероживании стенок реакго
ра) скорость процесса стабилизируется, и можно получи ть воспроизводимые
результаты. Однако при продолжительном ведении процесса происходит угол
щение пленочного покрытия реактора, что ведет к снижению скорости хлори
рования [1204].
Заместительное хлорирование преобладает в случае, когда в качестве мате-
риалов при изготовлении реактора используют кварц, графит или сплав монель
11195]. Применение хромистой стали [1260] или покрытие поверхности реакто
ра никелем [1272] снижают выход полихлоридов.
При покрытии стенок хлоратора слоем парафина было обнаружено падение
скорости реакции в 1200 раз [1273].
Эксплуатация в течение трех лет нержавеющего реактора, в котором про-
цесс протекал в псевдоожиженном слое кварцевого песка, показала незначи-
тельную коррозию отдельных участков реактора [1206].
Смешивать реагенты рекомендуется в специальных соплах-жиклерах [1208,
1239,1240, 1256, 1274, 1275] со скоростью выше скорости распространения пла-
мени. При этом точки ввода реагентов одна относительно другой располагают-
ся на равном расстоянии, в зависимости от скорости подаваемых компонентов,
температуры в реакционной зоне и т.д. Уменьшение времени смешения при
употреблении реакторов в виде сферы без мертвого пространства ведет к по-
вышению выхода продуктов замещения с одновременным снижением выхода
насыщенных хлоридов [1276].
Основная особенность конструкции реактора для проведения этого про
цесса заключается в повышенном теплоотводе из реакционной зоны. Обычно
реактор выполняется в виде охлаждаемой трубчатки [1199, 1208, 1240, 1256],
предпочтительно малого диаметра.
Чистота исходного сырья
Практически во всех работах [1199, 1203, 1204, 1205, 1208, 1283] сырье очи-
щалось по специальным методикам почти до 100%-ной чистоты, осушалось и
хранилось в специальных приспособлениях. Проводились опыты и с использо-
ванием технических реагентов, предварительно осушенных с использованием
хлорида кальция [1209]. Кроме того, хлор осушали дополнительным барботи-
рованием через серную кислоту. По немногочисленным сведениям [1206] осуш-
ка хлора и этилена никак не сказывается на процессе.
Исследование процесса в объеме
Авторами исследован процесс газофазного высокотемпературного хлори-
рования этилена в лабораторных условиях. Использовался кварцевый реактор
дифференциального типа с мешалкой и капиллярным смесителем хлора и эти-
лена. Объем реактора составлял 73 мл. Давление соответствовало атмосфер-
ному.
Время пребывания во всех опьнах поддерживалось un уровне I с Реакция
исследовалась в интервале 250 10(1. Мольное cooi ношение пилена с хлором
составляло о г 0,6 до 2.
При исследовании влияния гемпературы на течение га.юфа иного хло
рирования этилена мольное cooiношение реагентов поддерживалось близ
ким к двум, оно было выбрано не случайно: с одной стороны, э то но июля
ст иметь максимальное превращение реагентов при минимальном paio.ni
лении исходной смеси. Кроме того, при малом времени пребывания такое
соотношение способствует пониженному горению этилена и сравнигельно
небольшому закоксовыванию реакционной зоны, особенно узла смешения
этилена с хлором.
Первоначальные опыты показали, что при времени пребывания, равном
примерно 3 с и соотношении этилена с хлором, близком 2:1, идет сильное ю
рение этилена с выделением значительного количества кокса в зоне смешения
реагентов. Это также подтверждают данные работы [1204].
Снижение времени пребывания до 1 с и разбавление исходной смеси этиле
ном (С2Н4:С12 = 5:1) также не дало положительных результатов. Горение продол
жалось. В дальнейшем узел смешения реагентов был выполнен в виде нерпеп
дикулярных капилляров диаметром 0,5-1,0 мм. Это позволило вести процесс и
нормальных условиях.
Результаты эксперимента сведены в табл. 4.32.
Полученные результаты подтверждают тезис о том, что низкая темпера
тура способствует реакции присоединения. Так, содержание дихлориапа и
конденсате при проведении процесса при 250 °C достигает 55%, тогда как
содержание винилхлорида на уровне 27%, т.е. практически в два раза ниже
Кроме того, при этой температуре образуется значительное количество
трихлорэтана, что можно объяснить последовательным хлорированием ди
хлорэтана и присоединением хлора к винилхлориду, а также реакцией поры
ва (*) (табл. 4.32).
В этих условиях конверсия исходных реагентов сравнительно низкая. ) i и
лен реагирует лишь на 30%, а хлор на уровне 94%.
Повышение температуры процесса способствует повышению степени коп
версии. Полностью хлор реагирует при 400 °C. Степень конверсии этилена и
зависимости от температуры имеет размытый максимум в районе 350 °C и до
стигает порядка 52%. Увеличение температуры до 400 °C несколько снижао
степень конверсии этилена. Содержание винилхлорида в конденсате продукт)
Таблица 4.32. Параметры реакции
Реакция Е, кДж/моль 1g А
С2Н4С12 + СГ —•- С2Н3С12' + НС1 52,8 10,4
С2Н3С1+СГ —— С2Н3С12* 0 10,2
С2Н3С12* +С12 —•- С2Н3С13 + СГ 17,6 8,8
(*)С2Н3С12- +СГ—► С2Н3С13 0 11,3
Таблица 4.33. Влияние температуры реакции на состав реакционных продуктов (конденсата), мае. %
рсия, % X О Ш |> OS 00 00 Qs П к "Г. об °° ~ 4 об О in rq —rq |< М Tf 't 1Г Ш 4 Tf
и X о и ОчОсПГЧГЧоЯй О сП сП -и О 4 4 \огчг<(<о\о\23 СТ' СГ' Ch 04 Ch 04 Хн ~
Е J г э X н la—<гчо^спспгх г'14^.4.и7и7г14 О О г-< ? ГЧ -4
Тетра- хлорэтан la о rq 1 i 4 Ч i 1 Iй! о о о
Трихлор- этан 4 4 la сп ст о 4 Ч. о —< чо । rq ст
Перхлор- этилен \D т т оо оо тг । । о о ко rq о о о о о о
1,2- Дихлор- этан z г 2 £ £ £ Й Ж й S 'О °
Трихлор- этилен 2,52 2,90
Дихлор- этилены —’OOl>.mo0 4004(N Lrl ч О о с<? т сч ептб сг
Винили- денхло- рид ЧОТ^ОегсД40гПО Ч 1 ч 00сг’б'сг?обг^40
Винил- хлорид СО—<400Tfcr>00\ rl°).l/7.rlcr?.4£?ciQ^ обчО4ОСТ?1>Сс\С^СП rq rq la m о co оо
с2н4 С12 crLnen^rqi^r-.rq O —<00404.04 cn^rqrq—? —7 —7
и о о о о о LA О LA О rq т cn
реакции <. ростом температуры реакции до Г>(1"( резко pac iei и ыгсм Иракi и
чески не меняе тся.
Аналогичная картина имеет меч го по содержанию пинилиденхлорида. faro
содержание и конденсате ироду к юн реакции дихлор пана и грихлор и а па ре «ко
падает, чго говорит о преобладании при чих температурах реакции замещения.
Кроме того, следует отметить, что рост гемперптуры приводит к увеличению
выхода ос1альных хлора генов.
Таким образом, максимальный выход винилхлорида достигаегся ври гимне
ратуре в реакционной зоне 350-400 °C. Такая температура способствует также
повышенной степени превращения реагентов.
Исследовалось также влияние соотношения исходных реагентов па про
цесс газофазного хлорирования этилена. Опыты проводились при темпера гуре
400 ”С. Полученные результаты сведены в табл. 4.34.
Как видно из приведенных данных, изменение соотношения этилена к хлору
в пределах от 0,5 до 2 практически не сказывается на изменении доли реакции
замещения в процессе. Последняя явно преобладает над реакцией присоедине
ния. Выход 1,2-дихлорэтана во всех опытах незначителен. Количество получи
емого винилхлорида с уменьшением соотношения этилен : хлор резко падле г,
одновременно увеличивается количество получаемых хлорэтенов, в первую
очередь винилиденхлорида. Так, при соотношении С2Н4:С12 = 0,53^0,61:1 ко
личество винилхлорида и винилиденхлорида в конденсате практически одипа
ково и достигает примерно 30%. Одновременно растет выход трихлор пилена
Количество этого продукта при изменении указанного соотношения с 2 до 0,53
возрастает примерно на порядок и в весовом отношении приближается к коли
честву образующегося винилхлорида.
Такая картина может быть объяснена следующим. Уменьшение соогпоше
ния этилена к хлору повышает долю последующего замещения в молекуле пи
нилхлорида:
С2Н3С1 + СГ — С2Н2СГ + НС1; (4.190а)
С2Н2СГ +С12 — С2Н2С12 + СГ (4.1900)
И Т.д.
Степень конверсии хлора с уменьшением соотношения этилена к хлору
от 2 до 0,5 падает со 100% до -90%, тогда как степень конверсии этилена, па
против, возрастает с -45% до ~95%.Таким образом, оптимальное соотношение
C2H4:C12 = 2. При этом выход винилхлорида имеет максимальное значение, а
хлор реагирует полностью.
Оптимальный режим проведения процесса высокотемпературного хлори
рования этилена: температура исходного сырья, подаваемого в реак юр, 100
400 °C; температура в реакционной зоне — 400-450 °C; время пребывания —
1-2 с; давление атмосферное; соотношение этилена с хлором (2ъЮ):1, предпоч
тительно (3-н4):1; желательно использовать разбавитель, в качестве которого
целесообразнее применять избыток этилена или дихлорэтана; соотношение
разбавителя с этиленом и хлором примерно 5:5:1; процесс осуществлять с ог
ношением реакционной поверхности к объему реактора не ниже 4 см , одна
ко высокое отношение S/Vотрицательно сказывается на выходе полихлоридов,
Таблица 4.34. Влияние соотношения этилена с хлором на состав продуктов реакции (конденсата), мае. %
узел смешения хлора с .пиленом должен (или. выполнен ык, чтобы скорость
смешения значи гелыю превышала скорое 1ь par прос i ранения пламени; и к.шее
гве материала для изготовления реак iори следyei применять монельме галл или
КС-стали с повышенным содер капнем никеля
Реализация данною режима в промышленных условиях теоретически пол
воляет получать винилхлорид н одну стадию без применения каких либо ката
лизаторов, используя для этого несложное технологическое оборудование. Более
того, балансируя этот процесс по неиспользованному этилену и образующемуся
в результате процесса хлороводороду с высокотемпературным окислительным
хлорированием этилена, можно получать винилхлорид по двухстадии нои схе-
ме. При этом упраздняются сразу три стадии, присущие традиционному про
изводству винилхлорида по сбалансированной схеме: получения дихлорэтана,
ректификации дихлорэтана и пиролиза дихлорэтана. Однако низкая селек i ив
ность образования винилхлорида в данном процессе делает его промышленпу ю
реализацию маловероятной.
4.5. Пиролиз 1,2-дихлорэтана
Целевой продукт сбалансированного процесса — винилхлорид — образу
ется на стадии дегидрохлорирования (пиролиза) дихлорэтана. Именно прове-
дение процесса в газовой фазе дает возможность получения безводного хлоро
водорода, который затем утилизируется на стадии оксихлорирования этилена.
Процесс дегидрохлорирования дихлорэтана может быть чисто термическим с
температурой 480-520 °C; инициированным с температурой 400-450 °C; ка га
литическим с температурой 330-450 °C.
Термический пиролиз
В промышленности наибольшее распространение получил процесс терми
ческого пиролиза, осуществляемый при давлении 2,0-2,5 МПа. Конверсия дн
хлорэтана в этих условиях составляет 45-55%.
Пиролиз дихлорэтана — эндотермический процесс, протекающий по сум
марной реакции:
С2Н4С12 —- С2Н3С1 + НС1 - 73 кДж/моль. (4.191)
Ниже 350 °C скорость этой реакции крайне низка, а при 250 °C — 1,2
дихлорэтан не разлагается вообще. Пиролиз 1,2-дихлорэтана — цепной свобод
но-радикальный процесс, включающий:
1. Инициирование цепи
СН2С1—СН2С1 —► СН2С1—С"Н2 + СГ; (4.191а)
(дихлорэтан)
2. Развитие и рост цепи
СГ + СН2С1—СН2С1— СН2С1—C’HCl + НС1; (4.1910)
(дихлорэтан)
СИЛ 1- С‘ IK I - ► ( IL=: CHCI t СГ;
(пипинхлорид)
3. Обрыв цени
( 1.191b)
СН,С|—С НС 1 + СГ — СН2С1—СНС12; (1,1,2-трихлорэ ган) (4 191г)
2СН2С1 — С*НС1 — СН2С1 — CHCI—СНС1 — СН2С1; (4.191д)
(1,2,3,4-тетрахлорбутан)
2СН2С1—С‘НС1 — СНС1=СНС1 + СН2С1—СН2С1 и др., (4.191е)
(дихлорэтилен)
СГ + стенка —► обрыв. (4.191ж)
Таким образом, реакция начинается разрывом связи С—С1 в молекуле ди-
хлорэтана, за которым следует развитие цепи — отрыв атома Н радикалом СГ
от молекулы дихлорэтана и молекулярный распад 1,2-дихлорэтильного радика-
ла, обрыв цепи происходит гомогенно при рекомбинации радикалов. Радикал
СГ ведет цепные превращения дихлорэтана, чем объясняется, в частности, ус-
корение или замедление реакции различными добавками, которые, участвуя в
цепи, способствуют ее зарождению или обрыву.
Побочные продукты образуются в основном в случае перегревов выше
500 °C. Особенно нежелателен процесс терморазложения образовавшегося ви-
нилхлорида. Эта реакция при 500 °C идет с заметной скоростью с образованием
ацетилена:
СН2=СНС1 —► НС=СН + НС1.
(ацетилен)
(4.192)
Далее из ацетилена образуется целый ряд побочных продуктов
(4.193)
2НС=СН — нс=с—СН=СН2 (винилацетилен) (4.194)
нс=сн + СН2=СН—С1 — н2с=сн—с=сн2. (4.195)
С1
хлоропрен (1 и 2-изомеры)
При наличии солей тяжелых металлов хлоропрен (изомеры) образуется из
двух молекул винилхлорида
2СН2=СН—С1 — Н2С=СН—С=СН2 + НС1;
I
С1
хлоропрен (1 и 2-изомеры)
(4.196а)
( IK I
II
2CH2=CII —Cl—- CD — ('ll :=C1I2 i IK I. (4.1966)
(1 -хлорбу 1.1ДИС||)
1,3 бутадиен образуется из этилена и винилхлорида
СН2=СН2 + СН2=СНС1 —► Н2С=СН—СН=СН2+ НС1. (4 197)
(1,3 бутадиен)
При более сильных перегревах (600-^700 °C) возможно расщепление спя ш
углерод-углерод и образование хлористого метила и подобных ему соединений
(хлороформ, дихлорметан, тетрахлорид углерода).
СН2С1—СН2С1—2С‘Н2С1; (4.198а)
СН2С1—СН2С1 + С’Н2С1 —► СН3С1 + СН2С1—С* НС1; (4.1986)
(метилхлорид)
С' Н2С1 + СГ — СН2С12. (4.198II)
(метиленхлорид)
Пиролиз примесей, поступающих с 1,2 дихлорэтаном, также приводит к об
разованию ряда продуктов:
СНС12—СН2С1 — СС12=СН2 + НС1; (4.199а)
(винилиденхлорид)
СНС12—СН2С1— СНС1=СНС1 + НС1. (4.1996)
(цис, транс-дихлорэтилены)
При глубоком распаде 1,1,2-трихлорэтана образуются метиленхлорид и хло
реформ:
СНС12—СН2С1 —► С'НС12 + С‘Н2С1; (4.200а)
СНС12 + СНС12 —СН2С1 — СН2 С12 + С‘НС1 — СН2С1; (4.2000)
метиленхлорид
С'НС12 + СГ —- СНС13. (4.20011)
хлороформ
При пиролизе хлороформа образуется перхлорэтилен:
2СНС13 — СС12=СС12 + 2НС1. (4.201)
тетрахлорэтен
(перхлорэтилен)
Из 1,1,2,2-тетрахлорэтана получается трихлорэтилен.
СНС12—СНС12 — СНС1=СС12 + НС1. (4.202)
трихлорэтилен
Распад хлороформа может проходить через образование дихлоркарбена:
СНС13 —► :СС12 + НС1. (4.203)
Последний образуем наряду с нерхлор пиленом еще и геграхлорнд угле
рода:
2 XX :12 —* СС12=СС12; (4.203а)
псрхлорзтилен
:СС1, + С12 —СС14. (4.2036)
тетрахлорид
углерода
Таким образом, селективность процесса пиролиза дихлорэтана является од-
ной из определяющих характеристик, влияние на которую оказывают различ-
ные параметры и, прежде всего, температура. Изменение температуры и дав-
ления может существенным образом влиять на равновесный состав продуктов
пиролиза. Термодинамические расчеты процесса пиролиза приведены в работах
[204, 762-763]. В табл. 4.35 представлены данные по равновесному выходу ви-
нилхлорида, ацетилена, 1,1-дихлорэтана и составы равновесной газовой смеси
в интервале 298-900 К и давлений 0,1-4,0 МПа 1763]. Ацетилен и 1,1-дихлорэтан
представлены в таблице как продукты вторичных превращений винилхлорида
по реакциям:
С2Н3С1 —* С2Н2 + НС1;
(4.204)
С2Н3С1 + HCI —— 1,1 С2Н4С12. (4.205)
Дальнейший распад ацетилена до углерода и водорода в расчетах не учиты-
вался, так как в пределах выбранных температурных условий константа ско-
рости этой реакции весьма мала и равна 3,2-10 5 с”1 при 700 К, 3,3-10 4 с-1 при
800 К, 2,0 -10 3 с1 при 900 К |764].
При увеличении давления от 0,1 до 4,0 МПа равновесное превращение
1,2-дихлорэтана несколько уменьшается, однако при Т > 600 К даже при давле-
нии 4,0 МПа равновесное превращение дихлорэтана остается выше 90%. Выход
винилхлорида и ацетилена с увеличением давления несколько уменьшается, а
1,1-дихлорэтана несколько увеличивается.
С увеличением температуры выход винилхлорида вначале увеличивается,
а затем, после достижения максимума, начинает уменьшаться. С увеличением
давления максимальный выход винилхлорида смещается в область более высо-
ких температур: если р = 0,1 МПа максимальный выход (более 99%) возможен
при 500-600 К, 0,5 МПа — 600 К, 2-4 МПа — 700 К. В этих условиях равновес-
ный выход ацетилена менее 1%.
Данные, представленные в табл. 4.35, показывают, что для условий, обеспе-
чивающих технологически приемлемые параметры процесса (Т - 600-^-800 К,
р — 1,5-3,0 МПа), термодинамические ограничения отсутствуют.
Кинетика реакций термического дегидрохлорирования 1,2-дихлорэтана
изучалась рядом исследователей [43, 762-764, 766-768]. Д. Бартон и К. Хаулетт
[43,766] исследовали процесс в проточном трубчатом реакторе из тугоплавкого
стекла диаметром 15 мм и длиной 750 мм в интервале 362-485 °C.
Габлицц 4.35. I’aiiiionei ный выход iiiiiHiJix>n>pii/i.i ацсiплена и 1 I дихлор h.iii.i и coci.i
вы равновесной газовой смеси
р’ МПа Г, °C Конверсия ДХЭ, % Выход, % Сое тв, мол. %
ВХ 1,1- Дхэ ‘ 2112 1,2 ДХЭ 1,1- ДХЭ ВХ IICI С2Н2
0,1 298 13,5 18,16 81,84 0 84,44 10,78 2,39 2,39 0
400 57,9 87,99 12,01 0 27,88 4,61 33,76 33,76 0
500 97,2 99,39 0,57 0,04 1,40 0,28 49,13 49,17 0,019
600 99,7 99,68 0,06 0,26 1,30 0,03 49,72 49,99 0,1 1
700 99,95 93,67 0,01 6,32 0,02 0,005 45,39 51,51 3,06
800 100 65,17 0 34,83 0,004 0 27,75 57,41 14,83
900 100 35,02 0 64,98 0,004 0 13,22 62,26 24,52
0,5 298 12,3 8,95 91,05 0 86,76 11,07 1,09 1,09 0
400 36,2 70,82 29,18 0 50,84 8,40 20,38 20,38 0
500 88,5 97,35 2,65 0 6,20 1,26 46,27 46,27 0
600 98,7 99,66 0,29 0,05 0,65 0,14 49,56 49,62 0,020
700 99,8 98,54 0,06 1,4 0,11 0,029 48,89 50,28 0,69
800 99,9 87,43 0,02 12,58 0,028 0,007 41,13 52,93 5,9(1
900 100 60,82 0 39,18 0,008 0 25,43 58,19 16,38
1,0 298 12,0 6,42 93,58 0 87,33 11,14 0,76 0,76 0
400 30,0 61,41 38,59 0 59,13 9,77 15,55 15,55 0
500 80,7 95,13 4,87 0 10,93 2,22 43,42 43,42 0
600 97,5 99,41 0,57 0,02 1,28 0,28 49,08 49,10 0,012
700 99,5 99,17 0,12 0,71 0,23 0,057 49,33 50,03 0,35
800 99,9 92,88 0,04 7,08 0,063 0,017 44,83 51,67 3,42
900 100 72,29 0,01 27,70 0,022 0,006 31,74 56,06 12,16
2,0 298 11,8 4,66 95,34 0 87,72 11,19 0,55 0,55 0
400 25,4 51,59 48,41 0 65,90 10,89 11,61 11,61 0
500 70,6 91,53 8,47 0 17,86 3,63 39,25 39,25 0
600 95,2 98,87 1,12 0,01 2,47 0,54 48,25 48,24 0,006
700 99,1 99,41 0,23 0,36 0,46 0,11 49,45 49,80 0,18
800 99,8 96,13 0,07 3,80 0,12 0,035 47,13 50,85 1,86
900 99,9 82,15 0,03 17,82 0,04 0,013 37,68 54,07 8,20
3,0 298 11,7 3,76 96,24 0 87,90 11,22 0,44 0,44 0
400 23,4 45,96 54,04 0 69,15 11,42 9,71 9,71 0
500 64,2 88,64 11.36 0 22,85 4,65 36,25 36,25 0
600 93,1 98,32 1,68 0 3,61 0,79 47,80 47,80 0
700 98,6 99,42 0,34 0,24 0,69 0,17 49,39 49,63 0,12
800 99,6 97,30 0,11 2,59 0,19 0,052 47,96 50,52 1,28
900 99,9 86,68 0,04 13,28 0,66 0,020 40,62 53,07 6,22
4,0 298 11,7 3,34 96,66 0 88,00 11,23 0,39 0,39 0
400 22,2 42,02 57,98 0 71,19 11,76 8,53 8,53 0
500 59,6 86,20 13,80 0 26,70 5,43 33,94 33,94 0
600 91,1 97,86 2,14 0 4,89 10,31 47,14 47,14 0
700 98,2 99,37 0,45 0,18 0,91 0,25 49,30 49,48 0,089
800 99,5 97,90 0,13 1,97 0,25 0,07 48,40 50,18 0,975
900 99,8 89,34 0,06 10,60 0,09 0,03 42,40 52,42 5,022
В результа и* напсримешон в реакторе, стенки xoiopoi<> были покрыты yi
леродной пленкой, ангоры получили следующее выражение константы скоро
сти реакции, с ’:
ку = 6,4-101 "ехр |-196 500/( R71) ], (4.206а)
эго выражение заметно отличается от выражения константы скорости, с ’, по-
лученного Д. Бартоном для реактора с чистыми стенками:
кч = l,59-106exp[-112900/(RT)]; (4.2066)
ку/кч = 0,08 при 758 К, ку/кч = 6-10 3 при 635 К.
Таким образом, при покрытии стенок углеродистой пленкой инициирую-
щая способность поверхности сильно уменьшается.
Л. Дорейсвами с сотр. [762] изучали кинетику реакции термического дегид-
рохлорирования 1,2-дихлорэтана в интервале 400-500 °C, используя проточный
реактор.
Принимая первый порядок реакции, автор получил формулу константы
скорости реакции, с1, для степеней превращения менее 80%:
fc= l,49-102°exp[-59000/(RT)]. (4.206в)
В отличие от Д. Бартона, Л. Дорейсвами проводил опыты с неочищенным
ДХЭ (пределы перегонки последнего 82-84 °C при 0,1 МПа) и в своих расчетах
не учитывал влияние увеличения объема реакционной смеси, что ставит под
сомнение справедливость полученных данных.
Согласно данным, М. Китабатакэ [767], константа скорости реакции, с-1,
первого порядка в интервале 470-530 °C описывается зависимостью:
к= 1,15-107ехр[-114500/(RT)]. (4.206г)
Весьма близкие результаты получил Т. Такахаши [768]:
к = 3,3-107ехр[-121800/(RT)]. (4.206д)
Авторы [768] не обнаружили заметного влияния на скорость реакции изме-
нения давления от 0,1 до 0,8 МПа, тогда как отношение поверхности к объему
(S/V) заметно влияло на скорость: скорость реакции пропорциональна S/V в
степени Уг. При изучении продуктов реакции было установлено, что с увеличе-
нием давления выход ацетилена уменьшается, а выход 1,1-дихлорэтана резко
возрастает.
Э.В. Сонин [763] при исследовании процесса в безградиентном стеклянном
реакторе получил следующее выражение для константы скорости реакции, с-1:
к = 6,47-109ехр[-154 100/(RT)]. (4.206е)
Таким образом, все исследователи сходятся на том, что реакция термическо-
го пиролиза дихлорэтана подчиняется уравнению первого порядка. Разница в
предэкспоненциальных множителях и энергиях активации может быть объяс-
нена рядом факторов: диаметром и материалом реактора, чистотой используе-
мого дихлорэтана, степенью обуглероживания стенок реактора и др. Типичная
Рис. 4.123. Зависимость конверсии ДХЭ от температуры термического пиролиза
кривая зависимости конверсии дихлорэтана от температуры для процесса гер
мического пиролиза представлена на рис. 4.123.
Инициированный пиролиз
Снижение температуры пиролиза при сохранении высокой скороеги про
цесса, а следовательно, конверсии дихлорэтана может быть достигнуто за счо
использования инициирующих добавок, например, хлора, тетрахлорида угле
рода, гексахлорэтана, других соединений (табл. 4.36).
Поскольку реакция дегидрохлорирования 1,2-дихлорэтана протекает но
радикально-цепному механизму, присутствие инициаторов существенно уне
личивает ее скорость. Одним из наиболее эффективных инициаторов является
хлор, у которого энергия разрыва связи на 60-80 кДж/моль ниже энергии раз
рыва связи С—С1 в хлорэтанах.
Так, в присутствии незначительного количества хлора конверсия дихлор
этана резко увеличивается [769]. Это наглядно видно из приведенных ниже дан
ных и на рис. 4.124.
Влияние хлора на конверсию дихлорэтана (Кдхэ) в зависимости от темпера
туры реакции:
С12, мае. %........................... - 0,5 0,5 0,5
tp,°C................................ 400 370 350 300
Кдхэ,%................................ 2 70 50 30
Таблица 4.36. Энергии разрыва связей С — С1 и С1—С1 для дихлорэтана и некогоры
инициаторов
Добавки Образующийся радикал или молекула Энергия разрыва связи, кДж/моль
Тетрахлорид углерода С‘С13 297
Гексахлорэтан СС13С*С12 290
1,2-Дихлорэтан СН2С1С’Н2 335,1
Хлор СГ 242,4
Рис. 4.124. Зависимость конверсии дихлорэтана от концентрации хлора
Как видно, в отсутствие хлора конверсия дихлорэтана при 400 °C достигает
всего лишь 2%, а при наличии 0,5% хлора уже при 370 °C — 70%.
Конверсия дихлорэтана возрастает лишь в пределах 300-400 °C. Повышение
температуры от 300 до 400 °C приводит к возрастанию конверсии дихлорэтана
почти в 20 раз.
Были исследованы инициирующие действия и других галогенов. Так,
З.С. Смолян [770] расчетным путем определил, что инициирующие способнос-
ти F, Cl, Br, I существенно разнятся не только из-за разных степеней диссоциа-
ции, но и по причине прочности связи Н—Hal. Наибольшую инициирующую
активность проявляют хлор и бром. Активность йода очень низка
Аналогичные исследования проведены Д. Бартоном [766]:
Галоген, 0,5 мае. %... Cl Br I
КДХЭ,%................ 44 33 1
Процесс газофазного дегидрохлорирования 1,2-дихлорэтана, инициирован
ный хлором, изучался рядом исследователей [763, 766, 768, 770]. Д. Бартон [766]
и независимо от него Т. Такахаши [768] пришли к выводу, что скорость реакции
описывается кинетическим уравнением вида:
0,5
г = к Синиц. Сдхэ. (4.207)
т.е. реакция имеет половинный порядок по инициатору и первый по дихлор-
этану.
Согласно Д. Бартону выражение для константы скорости, с \
A=l,l-103exp[-51000/(RT)]- (4.208)
По Т. Такахаши константа скорости, с-1:
к -3,3- 103схр[-51 800/(RT)]. (4.208а)
С ними авторами полемизируем ) В I опии |7(>3|, согласно коюрому см»
росгь реакции дегидрохлорирования 1,2 дихлорнапа, инициированной хло
ром, моль/(л-с):
r = 1,30-107ехр| 6650O/(R7)| Сс|2СдХ;-). (-1.20X6)
Расхождение в виде кинетических уравнений влечет различную трактовку
авторами механизма процесса. При гомогенном дегидрохлорировании реакция
может протекать или по мономолекулярному, или по цепному свободно ради
кальному механизму. Дегидрохлорирование хлорпроизводного идет по молеку
лярному механизму, если само хлорпроизводное или продукты его разложения
являются ингибиторами цепей.
Д. Бартон и К. Хаулетт [771-774] разделили изученные ими хлоралкапы па
две группы.
В первую были включены хлориды, распад которых замедляется добавка
ми пропилена, ацетальдегида и ускоряется добавками хлора и, следовательно,
идет по радикально цепному механизму. В эту группу входят 1,2-дихлор пап.
1,1,1-трихлорэтан, 1,1,2-трихлорэтан, 1,1,2,2-тетрахлорэтан и 1,1,1,2-rcipa
хлорэтан.
Во вторую группу входят хлориды, скорость распада которых не чувсши
гельна к добавкам пропилена, хлора и т.д. В эту группу входят эгилхлорид,
1,1 дихлорэтан, 1,2-дихлорпропан, 2-хлорпропан, пропилхлорид, бутил хлорид
и изобутилхлорид. Авторы полагали, что эти хлориды распадаются по иглиц
ному мономолекулярному механизму непосредственно на олефин и хлоро
водород, так как при реакции атома хлора с молекулой такого хлорида с па
иболылей вероятностью образуется радикал, не способный к превращению в
олефин выбрасыванием атома хлора. В этом случае цепной распад затруднен, и
преимущество получает молекулярный распад. Например, при взаимодействии
1,1-дихлорэтана с атомом хлора с большей вероятностью образуется радикал
С‘С12—СНз, который не способен к выбросу атома хлора.
Наоборот, 1,2-дихлорэтан в этих условиях дает преимущественно радикал
СН2С1—C'HCl, который может отдать атом хлора и образовать винилхлорид,
способствуя развитию цепной реакции.
Аналогичные взгляды на общий механизм дегидрохлорирования хлоралка
нов высказаны Н.Н. Семеновым [357].
К. Хаулетт предложил следующую схему цепного свободно-радикальною
механизма дегидрохлорирования 1,2-дихлорэтана:
к,
СН2С1—СН2С1—► С'Н2—СН2С1 + СГ; (4.209а)
ki СГ + СН2С1—СН2С1 —- C'HCl—СН2С1 + НС1; (4.2096)
кз C'HCl—СН2С1 — СН2=СНС1 + СГ; (4209»)
к4 С'Н2 —СН2С1 + Cl' —* СН2=СНС1 + НС1; (4.209г)
А4
Cl KI- Cl) Cl I ( r- ► ( 112=( IK I i IK I ('1.209л)
I la основании кого механизма с использованием метода стационарных со
стояний было выведено уравнение скорости цепной реакции дегидрохлориро
вания 1,2-дихлорэтана:
w = (^M^ACHjCl—СН2С1]. (4.210)
Рассматривая механизм реакции инициированного дегидрохлорирования
1,2-дихлорэтана различные авторы [768, 770, 776] пришли к выводу, что на-
иболее вероятной стадией зарождения цепи является реакция термического
распада инициатора. Т. Такахаши предложил схему механизма инициирован-
ного дегидрохлорирования 1,2-дихлорэтана, которая описывается следующим
образом:
А/
С12—«►2СГ; (4.211а)
С
сг + сн2с1—сн2с1 —* сна—сн2а + на; (4.2116)
А,
сна—сн2а —— сн2=сна + сг; (4.211в)
А.;
СГ + СГ—-С12. (4.211г)
Выведенное на основании этого механизма уравнение скорости цепной ре-
акции инициированного дегидрохлорирования 1,2-дихлорэтана имеет вид
ш = (АЛ2^4)°’5[а2]°’5[сн2а—сн2а] (4.212)
и показывает, что реакция имеет общий полуторный порядок (в частности, по-
ловинный порядок по хлору и первый — по дихлорэтану).
Большинство авторов, изучавших распад хлоралканов и, в частности,
1,2-дихлорэтана, полагали, что цепная реакция зарождается и обрывается
в объеме, а тормозящее действие ингибиторов объясняется обрывом на них
цепей в объеме. Н.Н. Семенов [357] показал, что реакция зарождается на
стенках, а развивается в объеме. Обрыв же цепей в основном идет на стенке,
но частично и в объеме. Н.Н. Семенов отмечал, что при отсутствии обрыва в
объеме цепная реакция дегидрохлорирования 1,2-дихлорэтана должна про-
текать по первому порядку, если обрыв цепи определяется захватом стенкой
радикала C'HCl — СН2С1, а зарождение цепи на стенке происходит по реак-
ции:
СН2С1 — СН2С1 + стенка —► С1арсор6 + С'Н2—СН2С1. (4.213)
Сравнение предложенных механизмов термического и инициированного
дегидрохлорирования 1,2-дихлорэтана показывает, что стадии развития цепи у
них одинаковы. Принципиально не отличаются и стадии зарождения цепи, но
стадии обрыва разнятся: если в случае инициированного дегидрохлорирования
обрыв цепи предполагает регенерацию молекулы хлора (или в общем виде —
молекулы инициатора), го и случае icpMirin кою д«ч ндрохлориронаиия пропс
ходт рекомбинация радикалов хлора и нилхлорнда с обра нжанием Петра ль
пых молекул.
Такая разница в механизмах обрыва цепи подвергается критике в |763| с
предположением, что механизм реакции термического дегидрохлорирования
1,2-дихлорэтана должен быть следующим:
к,
СН2С1 —СН2С1 — С*Н2—СН2С1 + СТ; (4.21 1а)
fc,
С'Н>—СН2С1 —* СН2=СН2 + СГ; (4.2J Ю)
к,
СН2С1—СН2С1 + СГ —► С‘НС1—СН2С1 + НС1; (4.21 1 в)
кз
C’HCl — СН2С1 — СНС1=СН2 +СГ; (4.21 li)
fc4
C'HCl—СН2С1 + СГ —- СНС1=СН2+ НС1. (4.21 |д)
Этот механизм аналогичен предложенному К. Хаулеттом, но несколько <>i
личается от предложенного Д. Бартоном: стадия зарождения цепи рассматри
кается как термический распад молекулы дихлорэтана на два радикала, что на
иболее вероятно в пристеночном слое, и в энергетическом отношении более вы
годно, чем предлагаемый Д. Бартоном мономолскулярный распад дихлор этана
на этилен и молекулу хлора [43].
Обрыв цепи идет только за счет рекомбинации радикалов хлора и хлорэги
да. Это было доказано специальными опытами по термическому дегидрохлори
рованию 1,2-дихлорэтана, показавшими, что в продуктах реакции отсутствуем
свободный хлор, в то же время было найдено заметное количество дихлорэти
ленов (~5-10 3 молей на моль винилхлорида) [763].
Даже если предположить, что свободный хлор все же присутствует в про
дуктах реакции, но в количествах меньших, чем чувствительность метода ею
определения, то и тогда вероятность протекания реакции обрыва по предло
женной схеме будет несравнимо больше, чем по схеме с образованием молеку
лярного хлора. Поэтому последней реакцией можно пренебречь.
При допущении, что к3 » А:4[СГ], уравнение скорости реакции термичес
кого дегидрохлорирования 1,2-дихлорэтана, отвечающее предложенному меча
низму, имеет следующий вид:
гвх = (2^fc2fc3/fc4)0’5[CH2Cl—СН2С1]. (4.215)
Для реакции инициированного дегидрохлорирования дихлорэтана Э.В. ( о
ниным [763] предложен следующий механизм:
М—*ГГ + СГ; (4.216а)
кг
СН2С1—СН2С1 + СГ —- СН2С1—С*НС1 + НС1; (4.2160)
С1ЬС1— (. ‘HCI—*- С112=( 11(1 + ( Г; (4.216»)
и СН,С1—СНС12; (4.216г)
СН2С1— C’HCl + СГ Q
снс1=снс1 + на, (4-216д)
где М — молекула хлора, гексахлорэтана или другого инициатора.
В соответствии с предложенным механизмом обрыв цепи происходит глав-
ным образом в результате взаимодействия хлора с дихлорэтильным радикалом,
а не в результате рекомбинации радикалов хлора с образованием молекулы хло-
ра.
Исходя из предложенного механизма, методом стационарных состояний
было получено при допущении, что к4-[СГ] « к3 » к4-[СГ]:
w = (fc1fc2fc3/fc4)(,’5|CH2Cl—СН2С1]0’5 [М]0,5 (4.217)
где к4 = Ау + к4; [М] — молекула хлора, гексахлорэтана или 1,2-дихлорэтана.
Этот механизм позволяет также рассматривать реакцию дегидрохлориро-
вания 1,2 -дихлорэтана с одних и тех же позиций независимо от того, идет она
в присутствии инициатора или без него. Поскольку при термическом дегидро-
хлорировании 1,2-дихлорэтана роль инициатора, дающего начало цепи, выпол-
няет сам дихлорэтан, то уравнение (4.217) может быть представлено в следую-
щем виде:
w = К [СН2С1—СН2С1] °’5 [СН2С1—СН2С1] °-5, (4.218)
которое после простого преобразования превращается в (4.215) для реакции
термического дегидрохлорирования 1,2-дихлорэтана.
Хотя экспериментально описываются оба механизма, общий первый поря-
док для реакций как термического, так и инициированного дегидрохлорирова-
ния 1,2-дихлорэтана представляется более логичным и достоверным, чем пред-
лагаемое Д. Бартоном, К. Хаулеттом и японскими исследователями изменение
общего порядка реакции с первого на полуторный при использовании иници-
ирующих добавок.
С практической точки зрения важно определить минимальное количест-
во хлора, необходимое для осуществления инициирования. Это объясняется
тем, что хлор, вводимый в зону реакции, образует, в конечном счете, продук-
ты хлорирования и, следовательно, несколько снижает селективность процес-
са пиролиза.
Зависимости конверсии дихлорэтана от количества вводимого хлора по
нижнему и верхнему пределам, полученные в [763], представлены на рис. 4.125.
Представленные зависимости показывают эффект инициирования в интерва-
ле концентраций хлора 0,3-1,1 мол. %. При содержании хлора в системе менее
0,3 мол. % инициирование практически отсутствует.
Кинетические закономерности превращения хлора в процессе иницииро-
ванного пиролиза дихлорэтана в предположении, что хлор помимо иницииро-
Хлор, %
I’m 1125. Зависимость степени превращения ДХЭ от количества вводимого хлора:и
на pxniiii, б — нижний пределы
пиния расходуется в основном на реакцию заместительного хлорирования дп
хлор пана могут быть записаны следующим образом [763]:
г = 3,72-102ехр[-41 400/(RT)]Cci2°'5 Сдхэ0’5 , моль/(л-с). (4.219
Отношение констант скорости реакций дегидрохлорирования дихлор наш
и превращения хлора:
^деГ/Лхл = 3,5-104exp[-2500/(RT)]. (4.220
( увеличением температуры скорость реакции дегидрохлорирования ди
хлорэтана растет быстрее по сравнению с реакцией превращения хлора 11рт
й)0 °C к№Г превосходит кхп на ~2 порядка, при 500 °C — уже на ~3 порядка
1см не менее, скорость реакции превращения хлора также растет с новы
шсиием температуры, что может негативно сказываться на селективное 11
процесса.
Достаточно перспективными инициаторами процесса пиролиза 1,2
дихлорэтана являются перхлорзамещенные предельные углеводороды С, и (
iciрахлорид углерода и гексахлор.нан. При гемперагурах выше 400 "С гекса
хлорэтан способен распадаться но схеме:
** С 2С13 "* С| ’
(4.221а)
С’2С15 — С2С14 + СГ .
(4.221а)
Согласно Э.В. Сонину [763], кинетическое уравнение реакции дегидрохло-
рирования 1,2-дихлорэтана, инициированной гексахлорэтаном, имеет вид:
г = 1,32409ехр[-107000/(К7ЖГХЭ°'5 Сдхэ°'5 > моль/(л-с). (4.222)
Г. Шильтц и П. Голдфингер [777] рассчитали величину энергии активации
данной реакции: -167 кДж/моль. Разница, согласно Н.Н. Семенову и Г.А. Кап
раловой [778], а также М. Бриджу [779] объясняется каталитическим влиянием
обуглероженной стенки реактора. При пиролизе углеводородов и их производ-
ных в углеродистом отложении на стенках реактора существуют свободные ра-
дикалы, концентрация которых достигает величины 1015 спин на грамм отложе-
ния [779]. Наличие этих радикалов облегчает распад молекулы гексахлорэтана
на перхлорэтилен и два атома хлора.
Процесс пиролиза дихлорэтана, инициированный тетрахлоридом углерода,
может быть представлен следующей схемой:
С
СС14—* - С’С13 + СГ, (4.223а)
СН2С1— *1 СН2С1 + СГ —*- СН2С1—C’HCl + НС1; (4.2236)
СН2С1— ^2 C’HCl — СН2=СНС1 + СГ; (4.223в)
СН2С1—C’HCl + СГ
СН2С1—СНС12;
СНС1=СНС1 + НС1,
к?
С’С13 + СГ —- СС14.
(4.223г)
(4.223д)
(4.223е)
М.Г. Аветьяном [405] получено кинетическое уравнение:
г = 3,02-108exp[-127700/(RT)] Сдхэ0’5 СТХу0,5 , моль/(л-с). (4.224)
Зависимость конверсии тетрахлорида углерода (ТХУ) от температуры
приведена на рис. 4.126. Отметим, что даже при 560 °C конверсия ТХУ ниже
100%. Поэтому при работе с ТХУ в промышленных условиях за счет рецик-
ла непрореагировавшего ДХЭ, содержащего неразложившийся инициатор,
расход последнего может быть несколько снижен. Конверсия ТХУ при этом
растет по мере увеличения его содержания в подаваемом на пиролиз ДХЭ
(рис. 4.126).
[a]
Рис. 4.126. Зависимость конверсии ТХУ от температуры (а) и его содержания (б) при
пиролизе ДХЭ в присутствии ТХУ
Авторами работы [780] показано, что инициирующее воздействие смеси
гексахлорэтана (ГХЭ) и тетрахлорида углерода на пиролиз дихлорэтана более
сильно по сравнению с наиболее активным ее составляющим — гексахлор па
ном. С ростом температуры увеличивается инициирующее действие как смеси,
так и каждого компонента в отдельности (рис. 4.127я). Зависимость конверсии
дихлорэтана от состава смеси носит экстремальный характер при разных гем
пературах (рис. 4.1276). При этом максимум приходится на смесь, содержащую
75% гексахлорэтана.
На рис. 4.127е показано влияние количества инициатора, добавляемо! о
к дихлорэтану, на показатели процесса пиролиза. При 420 °C рост конверсия
дихлорэтана практически прямо пропорционален повышению концентрации
инициатора в исходном дихлорэтане. При 480 °C конверсия резко возрасте i до
содержания инициатора в дихлорэтане на уровне 0,8 мае. %. Дальнейшее уве
личение концентрации инициатора на скорости реакции практически не ска
зывается.
При этом увеличение соотношения ГХЭ.ТХУ ведет к увеличению скорое i и
реакции.
t,°c
Рис. 4.127. Зависимость конверсии ДХЭ от: а — температуры при его пиролизе в при-
сутствии 1% разных инициаторов, 6 — смеси ГХЭ+ТХУ в подаваемом на пиролиз ДХЭ,
в — содержания в нем комплексного инициатора ГХЭ+ТХУ
Вер<»1ГИ.1Я схема превращения инициирующих добавок 1105|:
(1) CCI, — - CCI , + СТ, АИ=297 кД к,
(2) CCJ, + СС14 - * СС13—СС13 । < 1', (3) СС13—СС13 —- СС1,—С*С12 + <Т, А/ / = 290 кДж; (4) СС13—С*С12 — СС12=СС12 + СГ, АН = 318 кДж; (5) СС13 —СС13 —- 2С'С13, АН = 264 кДж; (6) СС13—СС13 + СС13 —* СС13—С'С12 + СС14; > (4.225)
(7) СС13 —СС13 + СГ СС13—С'С12 + С12; стенка (8) СГ + СГ - С12; стенка (9) С'С13 + С*С13 СС13—СС13; стенка (10) С'С13 + СГ *- СС14.
Из схемы (4.225) следует, что одним из промежуточных продуктов превра
щения ТХУ является ГХЭ. При этом процесс распада ГХЭ протекает в основном
через реакцию (5). Доля реакции (3) при низких температурах невелика и с рос
том температуры увеличивается. Объяснить более высокую активность смеси
ТХУ и ГХЭ можно тем, что последний, начиная распадаться при более ни »кон
температуре, инициирует распад ТХУ при этой же температуре. Кроме гого
можно предположить, что при распаде смеси имеет место более оптимальное
соотношение активных радикалов, которое, в свою очередь, зависит от cool по
шения компонентов в смеси.
Сильное инициирующее действие на пиролиз дихлорэтана оказывает также
кислород. Конверсия ДХЭ резко повышается только при низких концентрациях
(до 0,5%) кислорода (рис. 4.128). Дальнейшее повышение концентрации приво
дит к снижению конверсии ДХЭ.
Присутствие кислорода дает возможность вести реакцию при более низких
температурах (250-300 °C), благодаря чему почти исключается образование по
бочных продуктов пиролиза за счет, правда, увеличения выхода продукюв ре
акций взаимодействия кислорода с органическими соединениями.
Сравнительные инициирующие действия кислорода и хлора в зависимое in
от температуры приведены на рис. 4.129. Из представленных данных видно, чн>
хлор является более сильным инициатором пиролиза дихлорэтана, чем кисло
род и, кроме того, отсутствие инициаторов расщепления дихлорэтана происхо
дит при значительно более высоких температурах.
Отдельно следует рассмотреть влияние на процесс пиролиза дихлорэтана
таких элементов как кислород и водород, так как при определенных условиях
возможно их использование не только как веществ, оказывающих влияние на
Рис. 4.128. Зависимость конверсии дихлорэтана от концентрации кислорода
Рис. 4.129. Влияние температуры на конверсию дихлорэтана 1 — без инициатора, в при-
сутствии инициатора: 2,3 — с добавкой кислорода (2), хлора (3)
скорость процесса, но и для снижения коксообразования путем окисления или
восстановления коксовых отложений.
Влияние кислорода на кинетику радикально-цепных процессов на примере
дегидрохлорирования 1,2-ДХЭ исследовано в работе [781]. Скорость реакции,
моль/(л-с), описывается кинетическим уравнением:
г =1,05-10"схр|-57300/(КГ)]рдхэрО2’4 > (4-226)
Авторы работы предположили, что в реакции дегидрохлорирования 1,2-
дихлорэтана причиной ускоряющего действия кислорода является его адсорб-
ция на активных центрах стенки. Разложение дихлорэтана протекает по извес-
тному механизму с перекрестным обрывом цепи. Возможно, процессу обрыва
цепи предшествует адсорбция радикалов на стенке (S):
(5 + СГ) + (5 + С*2Н4С1) —* [Cl] [С2Н4С1] [S]2. (4.227)
Кислород, адсорбируясь на стенке, уменьшает активную поверхность S и,
следовательно, приводит к понижению скорости обрыва цепи, увеличению
Рис. 4.130. Зависимость конверсии ДХЭ от концентрации поданного кислорода и oi
температуры
длины цепи и скорости процесса в целом. Дробный порядок по кислороду спи
детельствует, что его адсорбция протекает в соответствии с уравнением Деи
гмюра. Наблюдаемая энергия активации, вычисленная как разность энер! ни
активации бескислородного процесса (154,1 кДж/моль) и удвоенной теплоты
адсорбции кислорода на поверхности стенки (2-54,3 кДж/моль), равна 45,4 кДж/
моль и близка к экспериментальной.
М.Г. Аветьян [405] изучил влияние кислорода на процесс пиролиза дихлор
этана при разных температурах в области как его инициирования, так и ипги
бирующего действия. Из рис. 4.130 видно, что в области 370-400 °C зависимое । ь
конверсии дихлорэтана от количества добавляемого кислорода носит экстре
мальный характер со смещением максимума при повышении температуры в
сторону большего количества подаваемого кислорода. При более высоких 1ем
псратурах конверсия дихлорэтана падает с увеличением количества кислоро
да более плавно, чем в области низких температур. В области, где количес ню
кислорода не превышает 1,5-1,8 мае. %, он ускоряет реакцию пиролиза дихлор
этана; в области, где количество кислорода составляет более 2 мае. %, он прояв
ляст себя как ингибитор. Кинетические уравнения для каждого из paccMoi реп
ных случаев, определяющие г, моль/(л-с):
для инициирующего действия кислорода:
г = 1,19-101|ехр[-28900/(КТ)]р°1;хэРо243 5 (4.228а)
для ингибирующего действия кислорода:
г — 3,80-10^'ехр [ — 141 100/(КТ)]р°д^р^5. (4.2286)
Пиролиз, инициированный кислородом, сопровождается образованием
продуктов горения углеродсодержащих соединений (рис. 4.131). Увеличение
концентрации кислорода в системе в целом снижает селективность образова
ния винилхлорида в процессе (рис. 4.132).
Рис. 4.131. Зависимости: а — скорости горения продуктов; б — селективности реакции
пиролиза ДХЭ от температуры и количества поданного кислорода
содержания метилхлорида в продуктах реакции от степени
Рис. 4.132. Зависимость
превращения дихлорэтана
В работах [786,1703] показано, что за исключением оксидов углерода новые,
в том числе кислородсодержащие, продукты в инициированном кислородом
процессе, по сравнению с термическим пиролизом, не образуются. Набор про-
дуктов обоих процессов одинаков; этан, этилен, ацетилен, бутадиен, метилхло-
рид, винилхлорид, этилхлорид, 1,1- и 1,2-дихлорэтилены, 1,1-дихлорэтан, хло-
ропрен, бензол, хлорбензол, 1,1,2-трихлорэтан, тетрахлорэтан, перхлорэтилен.
Выход побочных продуктов в процессах дегидрохлорирования 1,2-ди-
хлорэтана, инициированном кислородом (числитель) и термическом (знамена-
тель):
t,°C.................... 400 450 500
Кдхэ, %............... 38/- 60/17 65/50
Е примесей, %.......
1,0/- 1,4/0,3 1,5/1,4
H i приведенных данных видно, чю i уммлриос количес гио примесей нс ia
виси г ог присутствия кислорода, но uoip.K iaci с увеличением темпера iypi.i и
степени превращения дихлор этана При 110*1111 одинаковых степенях копвер
сии дихлорэтана суммарное количество примесей равно для обоих процессов.
Но видимому, эго указывает, что образование большинства примесей связано
с глубокими превращениями винилхлорида, образующегося в результате реак
ции при повышенных температурах.
Качество винилхлорида в значительной степени определяется содержанием
в готовом продукте таких примесей, как метилхлорид и бутадиен. Э го сияла
по, в частности, со сложностями очистки винилхлорида от примесей на стадии
ректификации. На рис. 4.132 показана зависимость содержания метилхлорида
в винилхлориде от степени превращения дихлорэтана. Содержание бутадие
на в винилхлориде составляет (8-н Ю)-10 4%, слабо увеличиваясь с ростом кон
версии дихлорэтана. Наличие кислорода в системе не влияет на выход данных
примесей.
Наличие оксида углерода в системе как продукта взаимодействия кислорода
с дихлорэтаном оказывает незначительное ингибирующее действие на скорое 1 ь
основной реакции (рис. 4.133). Из этого следует, что инициирующий тффем
воздействия кислорода осуществляется не через СО.
Скорость радикально-цепного процесса в общем виде:
г = г, I, (4.229)
где г; — скорость инициирования; I — длина цепи.
Таким образом, скорость реакции может быть увеличена либо за счет скор< >
сти инициирования, либо за счет длины цепи. Все традиционные инициаюры
(С12, ТХУ, ГХЭ) ускоряют процесс именно за счет увеличения скорости иници
ирования. Кислород же является, по видимому, единственным инициатором,
способствующим увеличению длины цепи [786]. Как указано, кислород, адсор
бируясь на активных центрах углеродистого отложения, блокирует эти центры,
препятствуя обрыву цепей.
Рис. 4.133. Зависимость степени превращения дихлорэтана от температуры:
1 — без добавки; 2 — с добавкой 0,23 мае. % СО
Процесс можно опшагь совокупностью элемеп ирных стадий:
a) инициирования
кп
СН,С1—СН2С1 —* СН2С1—С'Н2 + СГ;
б) продолжения цепи
к,
СН2С1—СН2С1 + СГ —* СН2С1—C’HCl + НС1;
к2
СН2С1—C'HCl —* СН2=СНС1 + СГ;
в) обрыва
кт.
СН2С1—C'HCl + S —•- обрыв,
где 5 — поверхность реакционного сосуда, покрытая пироуглеродом.
Согласно методу стационарных концентраций, общепринятому для нераз-
ветвлснных радикально-цепных реакций, скорость зарождения и обрыва цепи
равны:
k0 [СН2С1—СН2С1] = к3 [СН2С1—C'HCl] • [S], (4.230)
где [S] — концентрация активных центров на поверхности углеродистого отло-
жения, на которых происходит обрыв дихлорэтильного радикала.
В соответствии с предположением об ускоряющем действии кислорода на
данный процесс концентрация активных центров должна быть пропорцио-
нальна общей величине поверхности углеродистого отложения и обратно про-
порциональна концентрации адсорбированного кислорода:
[S] = S/[O2]aflc. (4.231)
В свою очередь, концентрация адсорбированного на поверхности углеро-
дистого отложения кислорода в соответствии с уравнением Лэнгмюра-
[О2]адс = каяс • [О2]/(1 + Ь[О2]), (4.232)
где кадс — константа адсорбции кислорода на поверхности пироуглерода; b —
коэффициент адсорбции; [О2] — концентрация кислорода в реакторе.
С учетом этих трех уравнений общая скорость процесса дегидрохлорирова-
ния дихлорэтана в присутствии кислорода:
Г - (ко к2 кадс/к3 S) • ([С2Н4С12][О2]/(1 + 6[О2]), (4.233а)
при £>[О2] « 1
г = (к0 к2 кадс/к3 S) [С2Н4С12] [О2]. (4.2336)
В пользу предложенного механизма реакции свидетельствует, что иници-
ирующее действие кислорода проявляется, только когда стенки реактора пок-
роются пироуглеродом. На рис. 4.134 приведены зависимости конверсии ДХЭ
от времени для термического (1) и инициированного (2) кислородом (1 мае. %)
2
I’ik. 4.134. Зависимости (1,2 — см. текст) степени превращения дихлорэтана or с iciicuii
ortyi лсроженности реактора
процессов, полученные в необуглероженных реакторах. В течение первых 5 ми
пуг обе зависимости совпадают, именно в этот период происходит отложение
пироуглерода на стенках реактора [789].
Усредненный элементный состав пироуглерода из промышленной печи пи
ролиза: углерод — 93,9-94,6%, водород — 2,0-2,5%, хлор — 2,6-2,7%, железо
0,5-2,0%. Удельная поверхность пироуглерода — 1,8-2,5 м2/г.
Образцы пироуглерода, обработанные кислородом, имеют удельную повер
х ность на уровне 40-50 м2/г, что свидетельствует, во-первых, об увелимиi по
щейся активности углеродистых отложений в процессе пиролиза и, во-вторых,
о возможности автоматического удаления углеродистых отложений в виде ок
с идов углерода.
Рассмотрим процесс коксообразования более подробно. Основные нробле
мы, возникающие при эксплуатации печи пиролиза по мере увеличения коксо
ооразования, могут быть подразделены на три группы. Первая — слой кокса на
с 1снках змеевика ведет к снижению коэффициента теплопередачи, что i ребуса
повышения температуры при сжигании топлива для достижения требуемой
конверсии дихлорэтана. Вторая — слой кокса постепенно уменьшает площадь
поперечного сечения труб, что приводит к увеличению перепада давлений и, в
конечном счете, остановке печи для проведения регенерации. Третья — час i н
цы кокса, содержащиеся в газовом потоке, должны быть удалены из жидкое гн
после колонны закалки во избежание загрязнения других аппаратов технологи
ческой схемы.
В обобщенном случае суммарный процесс образования углистых продук гои
в паровой фазе, характерный для термического пиролиза углеводородов в про
мышленности, А.К. Бухаркин [911] представляет в виде следующих стадий:
- расщепление углеводородов в газовой фазе, накопление ненасыщенных и
ароматических соединений, их конденсация и/или полимеризация;
- образование полиацетиленов и аналогичных им соединений с последую
щим замыканием циклов;
- рос г углеродных чаыиц (ядер) и рашо кение углеводородов па поверхнос
ги л их ядер;
- ai номерация пора дующихся ядер и их дезактивация;
- образование на стенках «кокса».
В [865] указывается, чго бензол промотирует образование «зародышей» при
пиролизе, а в образовании последних с разной скоростью и активностью учас-
твуют;
Аллен —► 1,3-Бутадиен —► Ацетилен.
Соединения, в которых винильная группа сопряжена с электроноакцептор-
ной группой (например, винилацетилен), при температурах 300-500 °C образу
юг димеры и полимеры, которые затем превращаются в полициклические аро-
матические углеводороды и коксовые отложения [765].
В работе [674] высказано предположение о том, что природа новообразова-
ния в процессе пиролиза дихлорэтана и процессах пиролиза нехлорированных
углеводородов имеет сходные причины. Л. Олбрайт с соавторами в цикле работ
по пиролизу углеводородов [932-934] показал, в частности, что кокс образует-
ся непосредственно из ацетилена и толуола как интермедиатов при получении
этилена. Кокс подразделяется на каталитический, формирующийся из углево-
дородов на металлических поверхностях, и некаталитический, образующийся
из капель смолы [931].
Каталитический кокс имеет волокнистую структуру, промежуточными со-
единениями при его образовании являются карбиды металлов [935]. Каталити-
ческий кокс имеет более высокую плотность по сравнению с некаталитическим.
Общие принципы коксообразования — три параллельных механизма [933]:
1. Образование волокнистого кокса, катализируемое металлами. Промежу-
точными соединениями являются гидриды металлов. Волокнистый кокс имеет
форму игл или волокон. Углеродные нити получаются на каталитически актив-
ных участках поверхности, содержащих атомы металлов группы железа. Длина
нитей такого углерода на несколько порядков превышает их диаметр;
2. Образование пироуглерода из полиядерных ароматических вязких ка-
пель, которые превращаются в кокс при контакте с горячей поверхностью, при-
чем морфология кокса зависит от вязкости капель;
3. Реакция олефиновых, диеновых, ацетиленовых соединений, а также сво-
бодных радикалов со свободными радикалами на закоксованной поверхности.
Слой кокса, образующийся по механизмам 1 и 2, растет посредством механиз-
ма 3.
Экспериментальные исследования [674, 936, 937] кокса, образующегося
при пиролизе дихлорэтана, показали, что образование волокнистого углерода
в процессе незначительно. Наиболее высокая скорость коксообразования кор-
релируется с высоким содержанием хлоропрена в реакционной смеси. В ходе
пиролиза образуются также хлориды металлов, преимущественно FeCl3j кото-
рый, в свою очередь, заметно катализирует дальнейшее коксообразование [937].
Авторы [674] рекомендуют минимизировать количество источников железа, с
которыми может соприкасаться дихлорэтан, поступающий в печь.
Влияние водорода на процесс пиролиза 1,2-дихлорэтана было исследовано
в работах [786, 790, 791]. На рис. 4.135 представлена зависимость конверсии ди-
Рис. 4.135. Зависимость степени превращения дихлорэтана от концентрации водорода
хлорэтана от концентрации водорода [786]. Полученные данные показыпанн,
то водород является ингибитором исследуемой реакции, возможно, ia с меч
рекомбинации радикалов Н' и СГ . При изменении концентрации водорода н
пределах 0,1-2% наблюдается резкое снижение скорости реакции, особенно
при низких температурах. При дальнейшем увеличении содержания водорода
в реакционной смеси скорость реакции практически не меняется. При этом не
сколько меняется характер кривых, описывающих данные зависимости: с по
кишением температуры кривые постепенно сглаживаются, практически с iaiio
вясь от 530 °C прямыми.
Скорость реакции пиролиза дихлорэтана с добавками водорода, моль/(л с)
г =3,6-107exp[-157500/(R'T)] [ДХЭ] • [Н2]-1. (4.234)
Ингибирующее действие водорода автор [786] сравнивает с эффектом спи
жения температуры пиролиза, что приводит как к снижению степени превра
щения дихлорэтана, так и к снижению выхода побочных продуктов.
Это подтверждается данными [791]. При увеличении концентрации подо
рода в реакционных газах до 0,3% содержание ацетилена в продуктах пироли ia
надает с 0,39 до 0,06%. Влияние концентрации водорода на содержание меч ил
хлорида относительно невелико (выход последнего возрастает от 0,2 до 0,4%)
Зависимость содержания бутадиена от концентрации водорода экстремальна
Максимальное количество бутадиена (0,11%) наблюдается при добавке 0,2%
Н2. Увеличение концентрации водорода до 0,55% приводит к росту содержа
ния этилена от 0,14 до 5,73%. Дальнейшее повышение концентрации водорода
до 1,2% ведет к некоторому снижению содержания этилена — до 5,02%. Кроме
того, показано, что повышение концентрации водорода в реакционной смеси
вызывает постепенное снижение содержания хлоропрена, хлороформа, хлор
бензола с последующим их исчезновением, а также рост выхода пропана, про
пилена и изобутилена[791].
В целом добавка водорода в систему пиролиза дихлорэтана способна пред
восхитить метод переработки хлорорганических отходов путем их селективно
го гидрирования с получением углеводородов, в частности, этилена, и визира
том их в технологический цикл.
-пгг
В табл. 4.37 140 предс гавлены данные по инициирующей ак гивпос ги неко
горых соединений на пиролиз дихлорэтана при 375-500 °C. Из представленных
данных видно, что хлор, кислород и хлористый нигрозин являются наиболее
активными инициаторами |406|. Активность ни грозилхлорида также объясня-
ется его способностью диссоциировать на радикалы [782]:
NOC1 —*- NO' + СГ. (4.235)
Введение в систему инициаторов, наряду с увеличением скорости процесса,
способно существенно влиять на его селективность. В табл. 4.41 представлены
данные по содержанию легко-(числитель) и высококипящих (знаменатель) при-
месей в дихлорэтане под воздействием разных инициаторов при 425 °C.
Таким образом, использование хлора как инициатора ведет к образованию
наибольшего количества примесей за счет побочных реакций хлорирования
дихлорэтана, винилхлорида и побочных продуктов.
В [570] рассмотрены основные закономерности процесса пиролиза дихлор-
этана при действии лазерного излучения. Результаты, полученные автором в
Таблица 4.37. Инициирующая активность некоторых соединений при 375 °C в проточ-
ном реакторе
Инициатор, мае. % Кдхэ> % Инициатор, мае. % Кдх.'ь %
Без инициатора (термический пиролиз), 0,0 0,8 Гексахлорэтан + хлор, 2,5 + 0,1 32,0
Оксид азота, 1,0 1,8 Оксид азота + хлор, 0,5 + 0,5 55,9
Диоксид азота, 1,0 3,6 Хлор, 0,5 56,1
Тетраэтилсвинец, 0,1 8,4 Хлор, 1,0 63,7
Тетраэтилсвинец + хлор, 0,1 +0,1 8,5 Нитрозилхлорид, 1,0 66,3
Гексахлорэтан, 1,0 12,4 Кислород, 1,0 67,9
Диоксид азота + хлор, 0,5 + 0,5 20,5 Кислород, 1,5 71,0
Гексахлорэтан, 2,5 25,0 Хлор, 1,5 72,0
Таблица 4.38. Инициирующая активность некоторых соединений при 450 °C в диффе-
ренциальном реакторе
Инициатор, мае. % Кдхэ, % Инициатор, мае. % Кдхэ> %
Без инициатора (термический пиролиз) 23,3 Тетрахлорид углерода + гексахлорэтан, 0,75 + 0,25 43,5
Тетрахлорид углерода, 1,0 32,5 Тетрахлорид углерода + гексахлорэтан, 0,50 + 0,50 44,7
Гексахлорэтан, 1,0 39,5 Тетрахлорид углерода + гексахлорэтан, 0,25 + 0,75 58,0
1.|<>лпц| >1.39. I Ini иьнрующлл .ikiiiiiikx n> nchoiopi.ix соединений при >00 "( н диффс
репциальпом реакгоре
Ингиби гор, мае. % Кд.х.ъ 11 hi пои гор, мае. % Кдх »%
Вез ингибитор । (термический пиролиз) 41,2 1,2,1 । рихлорнропии, 0,1 28.0
1,1 дихлорэтан, 0,1 21,2 Бензол, 0,1 29,0
1,2-дихлорпропан, 0,1 24,2 Аллилхлорид, 0,1 31,0
бензол, 0,3 27,5 Бензол, 0,5 32,5
Таблица 4.40. Влияние некоторых соединений при 480 °C в дифференциальном реак
юре
Соединение, мае. % Кдхэ> %
.Пилен, 0,4 62,6
Зги лен, 1,1 21,7
Этилен, 1,9 19,9
Цис- и транс-дихлорэтилены, 1,0 +1,0 16,0
Винилиденхлорид, 2,0 50,1
1аблица 4.41. Влияние инициаторов на селективность процесса при 425 °C
Инициатор Кдхэ, % Непрореагировавший ДХ;), мае. %
Хлор 82,0 0,39/1,77
Кислород 79,0 0,29/0,10
Гексахлорэтан 65,4 0,17/(0,01 +2,19ГХЭ)"2
1 В числителе легко-, в знаменателе высококипящие примеси.
2 Непрореагировавший
проточном реакторе объемом 208 см3 и соотношении S/V = 1,9, приведены в
табл. 4.42.
Представленные в табл. 4.42 данные иллюстрируют увеличение степени пре
вращения 1,2-дихлорэтана при действии лазерного излучения по сравнению с
термическим процессом, причем при меньших температурах увеличение более
значительно. В [570, 1702] указано, что общая скорость процесса может быт ь
записана уравнением:
г = /ердхэ кпр дхэ> (4.236)
где к — константа скорости термического процесса, с кп — константа скоро
сти лазерного процесса, моль’/(ли’ с).
Гаилицл 1.42. 3uiuu iimoi ri. X степени иренр.нц ни» 1.2 дихлор пана 01 температуры и
проточном реак1 ори
г, с 1,2-ДХ ), ммоль/с т» с Х,%
Термический процесс
400 0,2315 16,3 2,88
450 0,2315 15,1 13,22
500 0,2385 13,7 36,06
530 0,2385 13,2 59,09
При действии лазерного излучения
350 0,2104 19,3 4,84
400 0,2315 16,3 10,59
450 0,2175 16,1 26,60
500 0,2315 14,2 50,63
530 0,2315 13,6 63,38
Подача.
Энергия активации лазерного дегидрохлорирования 1,2-дихлорэтана равна
64,4 кДж/моль, предэкспоненциальный множитель А = 93,0 моль0, л °’5с-1.
На рис. 4.136 представлена зависимость степени превращения 1,2-дихлор-
этана от энергии в лазерном импульсе. С увеличением энергии наблюдается
рост степени превращения 1,2-дихлорэтана. Это, однако, сочетается со сниже-
нием величины квантового выхода, который падает в степени (-0,5) с ростом
мощности лазерного излучения.
При 400 °C и степени превращения 1,2-дихлорэтана 0,451 автором работы
[570] в продуктах процесса не обнаружены примеси, наиболее характерные для
Рис. 4.136. Зависимость степени превращений 1,2-дихлорэтана от энергии в лазерном
импульсе
кармическою процесса: бензол, хлороформ. I I бутадиен, ацетилен Чистота
пи и пл хлорида W.V1-».
Ингибирование процесса пиролиза
Дихлорэтан, поступающий в промышленных условиях на пиролиз, содержи i
как правило, ряд органических соединений, оказывающих в юм числе и иш ибн
рующее влияние на процесс. На рис. 4.137 показано влияние дихлорзамещенных
тгана и пропана на конверсию исходного дихлорэтана. Представленные записи
мости указывают на сильное ингибирующее действие 1,2-дихлорпронаиа. Обра
зующийся в процессе при инициировании хлором 1,1,2-трихлорэтан не обладает
и ш ибирующим действием. На рис. 4.138я показано влияние побочных ироду к гоп
взаимодействия с инициатором на конверсию дихлорэтана при 400 °C. Ди хлор»
гилены ингибируют реакцию, причем наибольшим ингибирующим >ф<|тек1ом
обладает винилиденхлорид. При этом с увеличением содержания дихлор.ниле
нов выше 0,5 мае. % падение конверсии прекращается. Такой характер кривых,
по видимому, связан с насыщением части активных центров зарождения цепи пл
стенке более стабильными радикалами дихлорэтиленов до некоторого равнове-
сия, когда количества сорбируемых и десорбируемых радикалов равны.
Влияние ингибирующих примесей на процесс термического пиролиза 1,2
дихлорэтана показано на рис. 4.1386 [783]. Ингибирующая активность умепь
шается в ряду соединений: аллилхлорид >1,2,3,-трихлорпропан >1,2-дихлор
пропан >1,1-дихлорэтан. Влияние бензола на процесс термического пироли ia
дихлорэтана представлено на рис. 4.138в. В этом случае кривая конверсии носи i
экстремальный характер, т.е. бензол сначала является ингибитором, а затем не
рестает ингибировать процесс.
Действие этих соединений на пиролиз 1,2-дихлорэтана можно объяснить
тем, что они дают более стабильные радикалы, чем дихлорэтан. При этом у ал
Рис. 4.137. Зависимость конверсии ДХЭ от температуры при его пиролизе без доо.1
вок (1) и в присутствии ингибирующих примесей: 1,2-дихлорпропана — 0,1 мае. % (2),
1,1-дихлорэтана — 1,0 мае. % (3), 1.2-дихлорпропана — 1,0 мае % (4), в прису гсгнии
1 мае. % С12
|а
Рис. 4.138. Зависимость конверсии ДХЭ от концентрации примесей при его пиролизе
в присутствии 1 мае. % С12 при 400 °C (а) и термическом пиролизе при 500 °C (б, в):
1 — 1,1,2-трихлорэтан; 2 — сумма симметричных дихлорэтиленов; 3 — винилиденхлорид; 4 —
1,1-дихлорэтан; 5 — 1,2-дихлорпропан; 6 — 1,2,3-трихлорпропан; 7 — аллилхлорид
лилхлорида и бецюли обративши' с i.i<ni/ii п<и<> радикала происходи! присо
единением радикала хлора, на что требуется и.цельно меньше шсрти, чем
на образование стабильных радикалом из хлонрсишиои путем разрыва чини
С—Н. Интересно, что ит 1,2 дихлорнронана и аллилхлорида может обрито
паться один и тот же стабильный радикал; С II ( I - ( 1IC1—Cllj.
Однако аллилхлорид является более сильным ингибитором именно hi за
своей большей реакционной способности при образовании стабильною ради
кала.
Экстремальный характер ингибирования реакции дегидрохлорирования
1,2 дихлорэтана бензолом может быть объяснен так: поскольку бензол являет
ся реакционноспособным соединением, ввод его в малых количествах ера >у же
приведет к связыванию радикалов С1»; постепенное увеличение копцеп i рации
бензола ведет к уменьшению концентрации этого радикала до тех пор, пока ш.
наступает равновесие между количеством образующихся и связывающихся ра
дикалов С1»; дальнейшее добавление бензола может приводить к его реакции с
дихлорэтаном с образованием монохлорбензола, что способствует некоторому
увеличению конверсии дихлорэтана. В частности, в работе [784] было пока кто
слабое ингибирующее действие бензола на термический пиролиз дихлор.наиа.
На процесс пиролиза 1,2-дихлорэтана могут существенно влиять такие со
единения как этилен, вода и хлорид железа, примеси которых всегда ирис у тс
твуют в дихлорэтане-ректификате, направляемом на стадию пиролиза.
Данные о влиянии этилена на процесс пиролиза дихлорэтана достаточно
противоречивы. Д. Бартон и К. Хаулетт [43] указывают, что этилен облвдпег
средней ингибирующей способностью: при концентрации этилена в реакциям
ной смеси, равной 0,01-0,1%, скорость снижается на 20% по сравнению с не
ингибируемой реакцией. В противоположность этому авторы работы [785| yi
верждают, что наличие в системе этилена с концентрацией -2,5% увеличивав!
скорость дегидрохлорирования дихлорэтана почти в два раза.
Результаты, полученные автором работы [786], указывают на ингибироиа
ние этиленом реакции пиролиза (рис. 4.139).
Рис. 4.139. Зависимость конверсии дихлорэтана от концентрации этилена
ИшиОирующпп >ффск I наблюдается при конценI рации пилена до 1%;
дальнейшее уиеличепие содержания пилена нс влияе! на скорость реакции.
I leperno на кривых сохраняется при всех температурах (400-510 °C). Уравнение
скорости реакции:
г = 4,2-105ехр [-105 400/( R7')] [ ДХЭ] [С2Н4] 1. (4.237)
Предполагаемый механизм можно описать совокупностью следующих эле-
ментарных стадий:
СН2С1—СН2С1 —» СН2С1—С’Н2 + СГ; (4.238а)
СН2С1—СН2С1 + СГ —*- СН2С1—C’HCl + НС1; (4.2386)
СН,С1—C’HCl — СН2=СНС1 + СГ; (4.238в)
СН2С1—C’HCl + Cl’ —* СН2С1—СНС12; (4.238г)
СН2=СН2 + СГ —- С’Н=СН2 + НС1. (4.238д)
Ингибирующее действие этилена состоит в том, что он связывает радикал
СГ, генерируя радикал С’Н=СН2, не способный к продолжению цепи.
Наличие этилена в дихлорэтане, поступающем на пиролиз, весьма влияет
на состав образующихся продуктов [788]. Так, при пиролизе дихлорэтана, не
содержащего этилен, при 500 °C образуется -4-10 3 мае. % бутадиена. Добав-
ление -0,1% этилена вызывает повышение содержания бутадиена до 11-10 3%,
т.е. почти в три раза. Количество образующегося бутадиена с увеличением со-
держания этилена от 0 до 0,25% возрастает по линейной зависимости от 4-10 3
до 21-103%. Дальнейшее увеличение содержания этилена до 0,36% приводит к
резкому повышению скорости образования бутадиена. Концентрация его до-
стигает 53-10 3%.
Возможны два варианта объяснения этого явления. Первый — протекание
реакции конденсации этилена с ацетиленом, образующимся при пиролизе ви-
нилхлорида.
СН2=СН2 + СН=СН —- СН2=СН—СН=СН2.
(4.239)
Второй — ингибирование этиленом основной реакции дегидрохлорирова-
ния дихлорэтана, что приводит к локальным перегревам и увеличению за счет
этого доли побочных реакций.
Влиянию воды и соединений железа на процесс пиролиза посвящено лишь
небольшое количество публикаций.
Э.В. Сонин [763] показал, что увеличение содержания хлорида железа (III) в
дихлорэтане более 1 -10 4% ведет к снижению конверсии дихлорэтана.
Влияние содержания FeCl3 на конверсию ДХЭ при дегидрохлорировании,
инициированным хлором, t - 365 °C:
С12 в ДХЭ, мае. %.............
FeCl3 в ДХЭ, 10 4 мае. %......
Хдхэ> %.......................
1,54 1,20 0,96
38,4 6,8 1,0
49,5 57,0 65,0
М Маме доп |12| у калы вас I. ч 10 прш } н 1 шин дпхлор > 1 апс не шачн icubiioi о
количества воды (даже в пределах р.п iворпкнм. in) режо спи каем степень коп
версии, причем ингибирующее дейс i вис воды Оолес о)чс) л и во проявляв ня при
относи гелыю низких темпера турах. ( ГИ)"( присутствие воды ускоряет реак
цик). Авторы работы [787] полагаю), однако, чю скорость дегидрохлорирова
нил не зависит от содержании влаги в исходном дихлор.пане при температуре
процесса 500 °C и варьировании концентрации влаги в интервале от 0,25-10 до
15-10 2 мае. %. Тем не менее, в промышленных условиях в дихлорэтане не долж
но содержаться более 0,001% влаги по технологическим соображениям.
В связи с тем, что цепная реакция термического разложения дихлорэтана
зарождается и обрывается на стенке реактора, а развитие цепи происходи i и
объеме, отношение площади поверхности реактора к его объему (57\ ) долж
по влиять на процесс. По мнению японских исследователей [767], скорость
реакции дегидрохлорирования дихлорэтана с увеличением диаметра реакци
онной трубки, т.е. с уменьшением S/V несколько уменьшается. Аналтн ичн<» о
мнения придерживался Н.Н. Семенов [357]. Однако Д. Бартон (7661 пока тал.
что скорость реакции уменьшается при ее проведении на стеклянной насад
ке, т.е. при увеличении S/V. Это представляется достаточно естественным,
так как при использовании насадки относительная величина поверхпосiи
резко возрастает, и в этих условиях стенка начинает ингибировать ценной
процесс, поскольку на поверхности происходит не только зарождение, но и
обрыв цепей.
Согласно данным [763], результаты, полученные с использованием реакци
онных трубок диаметра 21, 50 и 60 мм не показывают четкой зависимое ги коп
версии при одинаковых значениях линейной скорости потока.
Э.В. Сонин [763] указывает, что скорость реакции возрастает в основном за
счет увеличения скорости газового потока, характеризуемого критерием Рей
нольдса до Re = (0,8-е-1)-105. Дальнейшее увеличение скорости потока практичес
ки не влияет на скорость реакции.
В связи с этим при расчете и создании промышленных печей пиролиза ра i
ной производительности соответствующим образом меняют и диаметр груб
реакционного змеевика.
Реализация пиролиза дихлорэтана в промышленных условиях
Промышленные печи пиролиза обычно представляют стальные, пер in
кальные, зауженные вверху камеры, футерованные огнеупорным материалом.
Схема типичной печи производительностью 135 тыс. т в год представлена па
рис. 4.140. На верхней части печи расположена дымовая труба. Общая высо
та печи составляет 40 м. В печи размещен змеевик из 88 горизонтальных труп,
который по ходу дихлорэтана условно делится на зоны подогрева, испарения,
перегрева паров и реакции. В зонах подогрева и испарения дихлорэтан нагрева
ется отходящими дымовыми газами.
По ходу дымовых газов печь пиролиза разделена на зоны излучения (ради
антную), переходную (шоковую) и конвекции. Зона излучения находится в рас
ширенной части печи пиролиза, где расположены в шахматном порядке 128 го
релок (64 на каждой стенке в 5 рядов по высоте).
Вход
Встроенный подогреватель
0114,3x4(10)
оребренные
шт.
17
16
15
14
18
19
12
20
13
Верхняя зона подогрева
0114,3x4 (10)-20 шт.
оребренные
Зона конвекции
35
Нижняя зона подогрева
0114,3x4 (10)-20 шт.
Г №26
43
44
41
45
48
47
46
49
50
Зона испарения
0168,3x9,5 (10]
14
шт.
54
53
52
Шоковая зона илi
переходная зона
5 ряд
4 ряд
Зряд
2 ряд
1 ряд
62
т. №28
Зона перегрева паров
0168,3x9,5 (10) -8ш
т. №30
т. №32
Зона излучения
Радиантная зона
т.
т. №36,
90
'67
76
.80
82
,84
Выход
89
т. №41 ,
Зона реакции
0168,3x9,5 ('
26
шт
8
7
8
9
Y
3
4
6
5
Общая длинна змеевика 1308 м
Рис. 4.140. Распределение зон печи по ходу ДХЭ и дымовых газов
В верху расширенной части находится переходная или шоковая зона. Зона
конвекции размещается в самой верхней части печи, которая заужена для уве-
личения скорости дымовых газов и улучшения теплообмена.
В конвекционной зоне дихлор или ii.iipeiuicu я до 200 260 ( . Гемнср.нура
паров после шоковой юны ин i.uuuiei MIO МЮ С, и зоне излучении, где пропс
ходш непосредственно пиролиз, темпе р.п у р.1 180 520 u( .
Обогрев печи пиролиза осущсс пишется, как правило, с помощью га юных
юрелок. Дымовые газы с обьемиои долей СО2 9,5-11,5% и кислорода 1,5 1%, с
температурой не более 385 °C через трубу рассеивания выбрасываются в ашос
фсру.
Процесс пиролиза сопровождается коксообразованием, скорость которою
1ависи г от ряда факторов: оптимального профиля температур, чистоты исход
пою дихлорэтана и т.д. Явлению коксообразования подвержены в основном
юны испарения и шоковая зона. Рост количества коксовых отложений в змее
вике ведет к увеличению перепада давления по длине змеевика, вследс типе чего
становится необходимой его регенерация.
Критерии остановки печи пиролиза для проведения регенерации:
- температура стенок труб змеевика печи пиролиза выше 650 °C;
- увеличение входного давления дихлорэтана при заданной нагрузке па 1,0
1,1 МПа.
Обычно регенерацию осуществляют 1 раз в 9-12 месяцев. При несоблюде
пии оптимальных условий ведения процесса межрегенерационный цикл умепь
шается до 3-4 мес.
Обычно в промышленных условиях конверсия дихлорэтана составляет
48-55% при селективности процесса по винилхлориду на уровне 98,5-98,7%.
Оставшиеся 1,5-1,3% включают примеси, содержащиеся в дихлорэтане, подана
емом на пиролиз (бензол, хлороформ, перхлорэтилен и др.), а также побочные
реакции превращения самого дихлорэтана с образованием коксовых отложе-
ний, ацетилена, бутадиена, хлоропрена и других примесей.
Содержание в дихлорэтане, подаваемом на пиролиз, мае. %: >99,8 основ
кого вещества; < 0,1 легкокипящих примесей, в том числе <0,05 бензола; 1 0,05
хлоропрена; <0,1 высококипящих примесей, в том числе <0,03 перхлорэгилспа;
<1-10 4 влаги; <0,5-10“4 железа.
Расходный коэффициент по дихлорэтану обычно принимается 1,6 г/1 ни
нилхлорида.
Регенерацию проводят, подавая в змеевик водяной пар и воздух при 650' (
Критериями ее окончания является достижение объемной доли СО, ниже
0,03% в регенерационном газе.
По состоянию на 2006 г. в России и Украине в составе производств винил
хлорида по сбалансированной схеме в эксплуатации находились печи пироли за
мощностью 84, 120 и 135 тыс. т в год. Данная производительность была опреде
лена по реализации чисто термического процесса пиролиза. В работе [792, 1704,
1705] рассмотрены пути интенсификации промышленных реакторов дегидро
хлорирования дихлорэтана за счет введения инициирующих добавок.
Возможность интенсификации процесса дегидрохлорирования изучали
на примере реактора мощностью 84 тыс. т в год. Реакционная зона состои 1 и i
28 труб длиной 11 м каждая, т.е. общей длиной (с учетом соединительных кала
чей) 326 м. Наружный диаметр труб — 152,4 мм, толщина стенки — 11 мм. Кро
ме реакционной зоны, размещенной в радиантной камере печи, имеется зона
подогрева и испарения ДХЭ (40 труб), размещенная в конвективной камере
печи. Ila выходе hi >1011 юны температура паров ДХ.) ин ганляе! около 240 °C,
давление — около 2,5 Ml 1а. В зоне реакции происходи г перегрев паров сырья до
температуры начала реакции — 100 °C и собственно химическое превращение.
В .ной зоне температура потока монотонно возрастает. При инициированном
дегидрохлорировании в случае неизменной интенсивности теплоподвода ввод
инициатора приводит к быстрому снижению температуры процесса из-за по-
вышения скорости химического превращения; эндотермическое превращение
идет частично за счет внутренней энергии самого потока, и его температура
снижается. После полного израсходования инициатора на взаимодействие с
ДХЭ температура потока монотонно возрастает.
Результаты расчетов процессов перегрева паров дихлорэтана и его превра-
щения приведены в табл. 4.43. Шоковая зона длиной 93,6 м расположена на
переходе из конвективной камеры печи в радиантную и соответствует части
змеевика, в которой происходит перегрев паров ДХЭ от 240 °C до температу-
ры, указанной в табл. 4.43. Радиантная зона длиной 232,4 м является продол-
жением шоковой. Температура в точке а соответствует температуре потока
на выходе из шоковой зоны, в точке б — температуре потока на выходе из
реактора, в точке в — максимальной температуре стенки. Как следует из дан-
ных табл. 4.43, в случае инициированного пиролиза производительность печи
достигает проектного значения для термического пиролиза при температуре
на выходе из реактора на 100 °C ниже и при меньшей (на 24%) нагрузке. Более
того, одна и та же производительность в условиях инициированного процесса
может быть достигнута при еще более низких температурах (еще на 50 °C) и
при меньших значениях теплонапряженности зон. Общее потребление тепла
снижается за счет выхода реакционного потока из змеевика при более низкой
Таблица 4.43. Результаты расчетов перегрева паров дихлорэтана и его превращения
Теплонапряже- ние, кДж/(м2-ч), в зоне Расход хлора Г, °C, в точке Кон- версия ДХЭ, % Произво- дитель- ность, тыс.т/год Приме- чание
шоко- вой радиант- ной в точке, кг/ч суммар- ный, % а б В
50240 133980 80 0,95 326 443 547 68,7 84,7 Нагрузка
41870 125600 80 0,95 313 415 515 65,9 81,2 по ДХЭ
41870 125600 90 1,07 313 395 495 69,3 85,3 25,2 т/ч
46060 129790 85 1,01 320 421 513 68,7 84,7
46060 129790 90 1,07 320 411 514 70,3 86,6
50240 129790 90 1,07 326 413 516 71,0 90,3 А
50240 133980 67 0,60 306 401 492 51,6 84,3 Подача
50240 133980 90 0,81 306 355 448 59,0 96,2 ДХЭ
50240 133980 100 0,90 306 332 430 62,2 101,2 33,25 т/ч
62800 167470 77,5 0,70 322 460 552 60,5 98,9
62800 167470 90 0,81 322 443 553 63,3 103,4
54430 146540 100 0,90 312 372 472 63,3 103,4
ii-Miicp.i । у ре, хоти c ii'ueiib Koiiiwpc mi Д\ ) n no ip.u i ,n-1 нследс rune увеличен
пой подачи хлора.
Полученные данные показы паю г, чю шачтельное снижение темпера) уры
процесса <а счет ввода хлора при уменьшении выхода смол, кокса и продуктов
деструкции или расхода топлива сули i определенный жопомический эффект
Однако дополнительные расходы на хлор, повышение количества образующе-
гося трихлорэтана и некоторое усложнение конструкции реактора и системы
управления удорожают процесс. Необходима его оптимизация посредс твом ре i
кого увеличения производительности реактора при малом расходе хлора.
Анализ данных табл. 4.43 показывает, что с увеличением подачи хлора тем
псратура процесса понижается, степень конверсии ДХЭ и производительное i ь
реактора возрастают. С увеличением количества подводимого тепла темпера
гура процесса, степень конверсии сырья и производительность реактора новы
шлются. Однако при регламентной для действующего реактора нагрузке дос i ичь
70%-ной степени конверсии увеличением подачи хлора и теплонапряжепия при
равномерном ее распределении в радиантной зоне не удается в связи с гем, ч ю
в случае эндотермических реакций скорость процесса определяется количеством
подводимого тепла, а оно лимитировано температурой нагрева трубы imccihi
ка. В табл. 4.43 два варианта расчетов при q = 167470 кДж/(м2-ч) показываю!,
что превышены предельно допустимые температуры потока (420 °C) и стенки
(520 °C). Увеличение производительности в определенных пределах при таком
профиле теплонапряжений возможно за счет снижения плотности теплового
потока и одновременного увеличения подачи хлора в каждую точку (два пос
ледних варианта).
Определенный резерв роста производительности реактора позволяет увс
личить теплонапряжение в шоковой зоне. В табл. 4.44 приведены результаты
математического моделирования процесса в более жестких условиях. В ряде
Таблица 4.44. Производительность реактора при повышенных теплонапряженинх и
зоне перегрева паров ДХЭ
Нагруз- ка по ДХЭ, т/ч Тепло- напряжение, кДж/(м2-ч), в зоне Расход хлора t, °C, в точке Сте- пень кон- версии ДХЭ, % Проилво* ди гель- нос гь, тыс. т/год
шоко- вой радиан- тной в точке, кг/ч суммар- ный, % а б В
39,9 125600 146 1,10 373 356 505 75,4 147,4
39,9 174590 119 0,89 415 408 536 74,8 146,2
46,5 125600 Я74 590 140 0,90 356 334 477 67,0 152,9
46,5 174590 140 0,90 396 345 443 70,8 162,0
45,0 174590 120 0,80 400 379 478 68,1 150,8
33,25 62800 167470 90 0,81 322 443 553 63,3 103,4
33,25 104670 142350 110 0,99 372 374 452 70,7 115,2
33,25 121420 138160 110 0,99 390 371 504 72,6 118,3
I, м
Рис. 4.141. Профили температуры потока и стенок, давления, степени конверсии и тепло-
напряжения по длине реактора при термическом (а) и инициированном (б) пиролизе
ДХЭ (я, в, и с — точки ввода хлора)
расчетов использована предельная теплой а пряжен ность, достигаемая в анало-
гичной промышленной печи пиролиза.
Результаты математического моделирования показывают, что интенсифика-
ция зависит от возможностей увеличения количества подводимого тепла. Пос-
кольку увеличить теплонапряженность в шоковой зоне пока нельзя, а общее ко-
личество подводимого тепла в действующих агрегатах ограничено, исследовалась
возможность дальнейшей интенсификации процесса реализацией в радиантной
зоне не постоянного, а кусочно-постоянного профиля теплонапряжений.
Промышленная печь оснащена четырьмя рядами беспламенных горелок, ус-
танавливаемых на каждой из боковых поверхностей радиантной камеры печи.
Так как каждый ряд горелок допускает независимое регулирование подачи
топлива, то для участков змеевика реактора, отнесенных к соответствующим
рядам горелок, может бин. задан.i uu»i i<'ii/t<>ii.ni|»i kciiiiocti., а нееь профиль
по длине радиан1 нои час i и гмеевпка приближенно аппроксимирован кусочно
Пос голпиой функцией При гаком подходе для р.нсм»принаемо)о реактора был
подобран профиль распределении геплонаприжепнн, обеспечивающий совпадс
пне расчетных температур с измеряемыми (н реакционной части змеевика ус га
новлено пять термопар) и степени конверсии ДХЭ в регламентированном для
термического пиролиза режиме работ ы реактора.
На рис. 4.141п показаны подобранный профиль теплонанряжений по дли
не промышленного реактора, а также изменение температуры потока и стенки,
давления и степени конверсии ДХЭ по длине реактора. Нагрузка но ДХ.) со
ставила 33,25 т/ч, температура потока на выходе из реактора — 500 °C, степень
конверсии — 50,1%, производительность печи — 81,8 тыс. т винилхлорида
Для этого же профиля теплонапряжений при указанной нагрузке и подаче
хлора в три точки в суммарном количестве 1% моделировали процесс ипици
ированного пиролиза ДХЭ. Полученные результаты приведены на рис. 1.1 Ио.
Максимальная температура потока в реакторе составила 388 °C, степень коп
версии ДХЭ достигла 72,6%, производительность печи — 116,3 тыс. т винил
хлорида в год. Эти результаты следует признать практически значимыми. Опп
показывают, что при одинаковом количестве подводимого тепла и при более
низкой температуре процесса возможно увеличение производительное 1 и реак
тора на (116,3-81,8) х 100/81,8 = 42%. При этом ограничения на максимально
допустимые температуры потока и стенки не нарушаются. В то же время при
веденные в табл. 4.44 режимы 3-5 указывают на еще большие потенциальные
возможности повышения производительности реактора.
Резервы для интенсификации процесса пиролиза дихлорэтана могут заклю
чаться в «ультрапиролизе» — снижении времени контакта, т.е. в увеличении
объемной скорости в сочетании с использованием более высоких температур.
Авторы работы [938] предлагают проводить процесс при t < 750 °C и времени
контакта 0,01-0,5 с. Конверсия дихлорэтана при этом может быть увеличена до
70-75%, однако растет также выход побочного ацетилена. Процесс «улырапн
ролиза» не реализован в промышленных условиях.
Каталитическое дегидрохлорирование дихлорэтана
Альтернативным вариантом реализации процесса газофазного дегидрохло
рирования 1,2-дихлорэтана может являться его осуществление в присутствии
гетерогенных катализаторов. Целью разработки таких процессов является мак
симально возможное снижение температуры, что должно привести к уменыне
нию расхода энергоресурсов, а также образования нежелательных побочных
продуктов — ацетилена, бутадиена, хлоропрена, смол и кокса, присущего про
цессу термического пиролиза дихлорэтана.
Хотя, как указывалось в табл. 4.35, термодинамические ограничения даже
для относительно невысоких температур отсутствуют, по технологическим
соображениям процесс дегидрохлорирования 1,2-дихлорэтана должен нрово
диться при давлении не менее 1,5 МПа, температура процесса должна быть нс
ниже 600 К (327 °C). Следует при этом указать, что катализаторы, описанные в
литературе, активны именно при t > 300 °C.
Каталитические еш темы для реакций до идрохлорирования, как правили,
представляют соли (чаще хлориды) металлов I и II групп на носителях с раз
витой поверхностью (активированный уголь, SiC>2> АКО, и др.). Однако при
высокой начальной активности все описанные катализаторы в процессе 3-4 ч
работы дезактивируются и, как правило, необратимо. Это обстоятельство явля
ется главным препятствием для внедрения процесса каталитического дегидро
хлорирования в промышленную практику.
Значительное количество работ [261, 731—735] посвящено исследованиям
процесса дегидрохлорирования 1,2-дихлорэтана в присутствии катализаторов
на основе активированных углей. Авторы [261] приводят ряд активности углей
разных марок при 240-320 °C:
АГ-2 > АГ-3 > АГ-5 > АР-3 > БАУ > СКТ. (4.240)
Скорость реакции дегидрохлорирования описывается уравнением первого
порядка:
г=/снСдхэ> (4.241)
где кн — наблюдаемая константа скорости.
В работе [732] показано, что порядок реакции по дихлорэтану близок к еди-
нице, а энергия активации составляет 120-140 кДж/моль. По мнению авторов,
механизм реакции основан на образовании адсорбированного активированно-
го комплекса дихлорэтана на поверхности угля.
Влияние исходного сырья и способа приготовления угля на его каталитичес-
кую активность в реакции разложения 1,2-ДХЭ изучено в работе [735]. Установ-
лено, что угли, полученные из ПВХ и сахарозы, малоактивны. Напротив, уголь,
приготовленный из торфа, обладает высокой активностью. Описан способ де-
гидрохлорирования 1,2-ДХЭ на активированном угле, импрегнированном хло-
ридами меди и калия, в присутствии кислорода [733]. При 400 °C конверсия
дихлорэтана составляет 58%. В данном случае, по-видимому, имеет место не ка-
тализ на активированном угле, а инициирование процесса молекулярным хло-
ром, образующимся за счет окисления хлороводорода в присутствии хлорида
меди по реакции Дикона.
Описан способ дегидрохлорирования 1,2 ДХЭ при 200-300 °C на активиро-
ванном угле, пропитанном гидрохлоридом третичного амина. Однако эффек-
тивность этого способа вызывает сомнения ввиду инертности гидрохлоридов
аминов в реакциях дегидрохлорирования [734].
Вообще активированный уголь является одним из старейших катализаторов
процесса газофазного дегидрохлорирования дихлорэтана. На первых промыш-
ленных установках получения винилхлорида использовали пемзу и древесный
уголь [734]. Компания «Worker-Chemie» применяла в качестве катализатора
крекинга хлорид бария, нанесенный на активированный уголь [737].
Соли и оксиды ряда металлов существенно влияют на условия процесса.
Промотирующее влияние ряда добавок иллюстрируется в табл. 4.45, согласно
которой уже при умеренных температурах (240-300 °C) конверсия дихлорэтана
составляет 65-78% [741, 742].
Максимальный выход винилхлорида составляет 88,2% при температуре
500 °C на катализаторе — yronb/SnO2+FeCl2 (табл. 4.46) [742].
1.Н>лица 1.15. Влияние eoncii и icMiiip.i 1 уры niipu/ni 1.1 il.i Miimipi пн» дпхлор i.iii.i
Соли 1, X/(\ ). %
VCl2, VOBr, V2O„ 1аВг3, BiCI3,CcO( h, Sbl „ Aslj 300 70
NiSO.„ NiCI2, CoCi,, PlCI, 2-10 70
( rCLCrF,, MoC!5, WBr5, Wl4,TeCl„ fcO., SeCI, SeO2 300 65
MnBr2, МпСЬ, MnF2, Mn2O,, ReF4, Re,O 300 70
NaCl, КС!, NaBr, CuCl2, Cu2Cl2, Cu2I2 300 67
CaCl,, MgCI2, BaCl2, ZnCl2, CdCl2, MgBr2, BeF,, ZnO 300 65
Si2I6, Snl4, SnF4, SnBr2, SnCl2, PbCl2, PbO, TiO 300 65
Таблица 4.46. Условия процесса дегидрохлорирования дихлорэтана на равных кашли
за юрах
r,°c Активирован- ный уголь Уголь/Си2С12 (10 мае. %) Уголь/СиС12 + + ТЮ (10 мае. %) Уголь/СиС12 + + SnO (10 мае. %) Уголь/СиС12 + + FeCl2 (10 мае. %) Уголь/SnOj + к 1е( 12 (10 масс. %)
Vo6> (Лкат*ч) Ввх> % Кб, л/(Лкат’Ч) Ввх, % Кб, л/(лкат-ч) Ввх> % Кб, л/(лкат*ч) ®вх> % Кб, л/ (Лкат*1^) Ввх> % V А л/ (Лка1’Ч) «ИХ. %
340 240 30,2 360 38,2 510 45 540 45,8 600 47,5 650 18,1
380 420 46,4 550 53,2 690 59,8 720 60,2 780 61,5 800 62,5
420 640 61,2 780 69,6 840 71,8 860 72,4 960 73,9 1080 71,8
460 840 70,3 900 78,4 1020 79,6 1140 80,3 1220 81,4 1260 82,6
500 920 78,8 1020 84,1 1140 85,5 1200 85,9 1300 86,8 1380 88,2
Значительное количество работ посвящено исследованиям процесса дегид
рохлорирования дихлорэтана в присутствии катализаторов на основе оксида
алюминия. Авторы работы [738] для дегидрохлорирования 1,2-дихлор >1.111,1
предлагают нанесенный на оксид алюминия металлический Ni-Ni/AI2O3. При
проведении реакции в интервале 350-400 °C, скорости подачи дихлорэтана I ч 1
и времени контакта 30 с конверсия 1,2-дихлорэтана достигает 98%.
Для увеличения срока службы катализатора без окислительной регенерации
теми же авторами предложено введение в катализатор 0,2-0,4% фтора. Резуль
таты приведены в табл. 4.47.
В работе [739] предлагается проводить дегидрохлорирование 1,2-ДХЭ на
твердом катализаторе MgCI2/AI2O3, при соотношении Mg:AI = (0,24-0,5):! в гс
чение 2 ч при атмосферном давлении, 400 °C и скорости подачи 1,2-ДХЭ 13 ч
при времени контакта 5-7 с. Выход винилхлорида при этом составляет 56,3%.
В [740] исследование реакции дегидрохлорирования дихлорэтана осуществи»!
ли в присутствии катализатора СоС12/А12О3 в интервале 350-400 °C при време
1<1опица 4.17. Дегидрохлорирование 1,2-дихлор >1,111.1 пл Л1 < >( k.ii.uih i.nop.ix с добив
пением фтора
Катализатор т» ч 1, °C ХдХ >• % СОцх > %
Ni на фторированном А12О3 15 350 98,0 98,9
Ni на А12О3 10 400 94,1 98,1
Селективность образования.
Таблица 4.48. Дегидрохлорирование 1,2-ДХЭ на А12О3-катализаторах с добавлением Ni
Пример t,°C ДХЭ: инерт, моль/моль Удхэ> г/(гкат-ч) Хдхэ» % совх, %
№ 1 400 - 0,8 94,1 98,1
№2 420 1 : 0,2 1.0 97,1 98,8
№3 450 1 :9,5 1,5 96,1 98,3
* Селективность образования.
ни контакта 5-7 с в течение 4 ч с последующей регенерацией воздухом в течение
2 ч. Степень превращения 1,2-дихлорэтана в винилхлорид составляет 50-60%.
Малое время контакта при относительно высокой температуре и регенера-
ция катализатора продувкой воздухом позволяют снизить образование смол и
кокса.
Известны способы получения винилхлорида дегидрохлорированием 1,2-
ДХЭ в присутствии оксида алюминия, промотированного платиной и элемен-
тарным фтором [743]. Процесс проводили при 350-450 °C при разбавлении 1,2-
ДХЭ инертным газом в отношении 1:1 и скорости подачи до 8,4 мл/(г-ч). Выход
винилхлорида достигал 97-99%. С целью замены дорогостоящей платины авто-
ры предлагают проводить процесс в присутствии катализатора на основе окси-
да алюминия, в состав которого введено 3-10% Ni. Процесс исследовали как с
разбавлением сырья инертными газами, так и без них (табл. 4.48).
Предложенная авторами [743] каталитическая система обеспечивает выход
винилхлорида на уровне 92-96%.
Использование добавки водорода при каталитическом дегидрохлорирова-
нии 1,2-ДХЭ предлагается в [744]. Процесс проводили при 325-350 °C, мольном
соотношении Н2:ДХЭ = (0,02-0,25):! на катализаторе, содержащем Pt или Pd на
у-А12О3. Такие условия позволяют существенно снизить температуру и тем са-
мым уменьшить количество образующихся смол и сажи, а также расход энер-
горесурсов за счет активации катализатора при введении в реакционную зону
оптимальных количеств водорода.
Дегидрохлорирование на цеолите ZSM-5 при 325 °C характеризуется кон-
версией дихлорэтана 50%, при этом в продуктах реакции наблюдается в соизме-
римых с винилхлоридом количествах этилен [745], что свидетельствует о реак-
ции дехлорирования дихлорэтана.
Авторами [746] предложены к использованию относительно недорогие,
доступные оксидные железо-хромовые катализаторы (85-95% Fe2O3 и 5-15%
1а(»лица 1.19. Де|ндрохлорнропаннс 1,2 днх к>р >i ill i II i кслс ulo хромоных K.il.oiiiii
inp.ix (lllk,ir = 15 4)
jyo /, °C У"дх ь г/ (г-кат. ч) Катализатор, мае. % Пропущено ДХЭ, 1 Подучено i.iiooopa.t пых II род у к гои» г Хд* »> мае. % ОрДХЛ моль/моль
1 500 1,5 Fe,O,—85,1 Сг2О3— 14,9 45 (за 2 ч) 44,7 99,0 0,5
2 450 0,8 Fe2O3 —90,4 Сг2О3 — 9,6 2 (за 2 ч) 23,8 99,0 0,1
3 500 0,5 Fe2O3— 94,1 Сг2О3 — 5,9 22,5 (за 3 ч) 22,2 99,0 0,1
Cr2O3). Процесс лучше вести при 400 500 °C в присутствии кислорода и со
отношении О2:ДХЭ = (0,1 -5-0,5): 1,0 и скорости подачи дихлорэтана 0,5 1,5 г/1
катализатора. Степень конверсии 1,2-ДХЭ при этом составляет 99%. Па
ряду с винилхлоридом образуется 1,1-дихлорэтилен, содержание его и про
дуктах реакции возрастает с увеличением подачи в зону реакции кислорода
(габл. 4.49).
Выход, мае. %, продуктов реакции дегидрохлорирования дихлорэтана на
железо-хромовых катализаторах:
Катализатор........... №1 №2 №3
ВХ................... 77,1 86,3 64,7
1,1-ДХЭ-ен........... 22,7 11,8 31,4
Прочие.............. 0,2 1,9 3,9
Из представленных данных видно, что при практически полной коннерспн
дихлорэтана, наибольший выход винилхлорида (86,3%) наблюдается при нс
пользовании катализатора 90,4% Fe203 с 9,6% Сг2О3 при 450 °C.
Для очистки выбросов, утилизации токсичных соединений и получен пи
полезных продуктов, в частности, переработки 1,2-дихлорэтана в винилхло
рид, процесс, по мнению авторов [747], можно проводить на гетерогенном
катализаторе при 350 490 °C, времени контакта 0,1-5,0, пропуская нары ди
хлорэтана с инертным газом — аргоном или азотом в объемном соотноше
нии Ь42:ДХЭ - (0,95-2,0): 1,0 при соотношении объем катализатора и реактора
(0,15-ь1,0):1,0. Периодически подачу 1,2-дихлорэтана заменяют продувкой реак
тора водородом: через каждые 0,5-10 ч подачи дихлорэтана — 1ч подачи подо
рода. В качестве катализатора используют углеграфитовый материал, в котором
слои углерода ориентированы в пространстве в виде граней многогранника с
выходом на поверхность базальных плоскостей с межплоскостным рассгояпи
ем J002 = 0,34э-0,349 нм, размером микрокристаллитов по направлению «а» —
La - 3,0-5-12 нм, по направлению «с» — Lc = 3,0-5-8 нм, удельной поверхностью
12-650 м2/г. При этом достигается практически количественный выход винил
хлорида.
В работе [7181 ыявлеп способ получения инпилхлорида дегидрохлорирона
нием 1,2-дихлор нала и присутствии катализатора хлорида цезия, нанесенного
на силикагель в количестве 5 11% или смеси хлоридов цезия и меди в количес-
тве 5-11 и 0,5-11%. 11роцесс проводили при 379-390 “С, конверсия дихлорэтана
была на уровне 73,77-83,04%, а выход винилхлорида — 73,48 82,98%.
Исследования вели при непрерывной подаче дихлорэтана в реактор со ско-
ростью 10 г/ч. Активность сохранялась 100 ч. При 400 °C и 2 ч работы конверсия
составляла 83,22 мае. %, селективность — 99,89 мае. %, при 8 ч работы конвер
сия — 82,97 мае. %, селективность — 99,28 мае. %, при 100 ч работы конверсия —
83,04 мае. %, селективность — 99,31 мае. %. Результаты приведены в табл. 4.50
Управление процессом дегидрохлорирования дихлорэтана с учетом разде-
ления его на гомогенную и гетерогенную составляющие может быть реализо-
вано расчетом сферы катализа [749-752]. Катализатор, при протекании про-
цесса по гетерогенно-гомогенному механизму, может усиливать гетерогенную
составляющую благодаря участию его поверхностных активных центров в ре-
акциях продолжения цепи. Катализатор может также увеличивать гомогенную
составляющую. В этом случае роль активных центров заключается в генерации
в газовую фазу дополнительного количества радикалов. Вокруг гранулы ката-
лизатора возникает зона с повышенной концентрацией радикалов — «сфера
катализа», так как именно она определяет вклад катализатора в процесс [750].
Вне сферы катализа, в газовом объеме между гранулами катализатора, проте-
кает обычный термический пиролиз. Размер сферы катализа зависит от усло-
вий протекания процессов (температуры, давления) и от природы реагентов и
ведущих процесс радикалов. Ее размером, в свою очередь, определяется опти-
мальная величина гранул используемого катализатора и способ его размеще-
ния в реакторе [751].
Таблица 4.50. Дегидрохлорирование 1,2-дихлорэтана на CsCl-CuCl2/SiO2-Kara/iH3aTope
Пример t,uC Катализатор, % от массы носителя тк,с Удхэ» % СО*Вх> % Выход ВХ, %
CsCl СиС12
№ 1 370 11 11 87,40 78,12 99,71 77,89
№2 380 11 11 90,00 79,97 99,62 79,67
№3 400 11 11 94,50 83.22 99,39 82,71
№4 390 И 11 92,76 83,40 99,50 82,98
№5 380 11 - 90,00 74,32 99,59 74,02
№6 380 5 - 90,00 73,77 99,61 73,48
№7 380 11 0,5 90,00 76,22 99,59 75,91
№8 360 11 11 85,71 57,41 99,83 57,31
№9 380 4 - 90,00 56,57 99,60 56,34
№10 380 12 - 90,00 68,46 99,57 68,17
Селективность образования.
В работе |752| расчетным путем пока i.iiio, 410 для реакции дегидрохлори
рования дихлорэтана время киши .нома хлора, диффундирующего в га швом
объеме с поверхности акгиппою щчнра, кк 1авляе> 8,0810 с. Расстояние, на
которое атом хлора диффундирует в га юную фа iy, составляет 3,810 1 мкм. )к>
расстояние и определяло бы размер сферы катализа, если бы длина цепи при
пиролизе равнялась единице. Однако эта величина может достигать значении
106 и, поскольку катализатор увеличивает концен грацию радикалов, ведущих
процесс, можно ожидать, что в условиях каталитического дегидрохлорирова
ния дихлорэтана длина цепи резко сократится. Авторы [752] указывают, ч го для
каждого конкретного катализатора значение длины цепи нужно определи i ь ж
снериментально. От длины цепи зависит время жизни свободной вален i пос i и,
уродившейся на поверхности и принадлежащей атомам хлора а, следова голыш,
и радиус сферы катализа:
г = (6£>//)0,5, (4.2-12)
1де D — коэффициент диффузии (при атмосферном давлении и 700 К ко >ффи
циент диффузии атомов хлора в среде дихлорэтана D = 0,295 см2/с); I — длина
цепи
Учитывая, что на 1 см2 поверхности находится ~10L атомов, можно оцени 11
стационарную концентрацию активных центров [752]:
/. 1 103 104 I05
*1 г , мкм 3,8 120 380 1200
S2, 2,2 ат .... 10“7 Ю10 10 11 10 12
R 3, см .... 0,13 0,40 1,10
V4, % — 25 21 3,6
Расчетный радиус сфер катализа.
Концентрация активных центров.
1 Оптимальный радиус гранул катализатора.
4 Оптимальная степень заполнения реактора гранулами диаметром 1 мм.
При использовании гранул меньшего размера, чем рекомендуемый, ангоры
[752] указывают на необходимость соблюдения условия примерного равенсгпа
свободного объема между гранулами и суммарного объема сфер катализа.
При подборе катализаторов дегидрохлорирования ди- и полихлорэганоц
важную роль играет соотношение кислотных и основных центров на их попер
хности. В [754] на примере 1,1,2-трихлорэтана показано, что одним из наиболее
активных катализаторов процессов дегидрохлорировании является хлорид це-
зия, нанесенный на силикагель. При этом селективность образования винили
денхлорида и симметричных цис- и транс-дихлорэтиленов зависит от cooiпо
шения основных и кислотных центров катализатора. Авторы предполагают, ч го
с повышением способности катализатора к прочной адсорбции хлороводорода
усиливается дезактивация основных центров по сравнению с кислотными. 11ри
этом поверхность частично дезактивированного «хлорированием» (замещенн
ем ОН-групп на С1) [754] катализатора может быть представлена кластерами,
содержащими как остаточные группы ОН , так и группы вида:
(I (Ml Cl OCs
I I ” I I •
M—O — Si M—o — Si
где M — атом кремния или модификаторы.
Хлорирование поверхности носителя приводи г к возникновению протонной
кислотности, обусловленной индуктивным влиянием галогена на Si-OH группу
и одновременно — к снижению электронодонорной способности соседней OCs-
группы. Это приводит, с одной стороны, к блокировке первичных кислотных
центров, с другой — к генерированию новых кислотных центров и дезактивации
основных центров.
Адсорбция хлороводорода на катализаторе может быть как обратимой, так и
необратимой В обоих случаях катализатор теряет активность, однако в первом
случае она может быть частично восстановлена термической регенерацией, т.е. сла-
бо адсорбированный хлороводород блокирует основные центры (ОН-группы).
В работе [755] показано, что использование сильно кислотных катализа-
торов в процессе дегидрохлорирования 1,2-дихлорэтана уже при 100-175 °C
приводит к значительному коксообразованию. Основной продукт реакции —
1,1-дихлорэтан, конверсия 1,2-дихлорэтана 48%. Предполагаемый механизм
реакции: взаимодействие 1,2-дихлорэтана с кислотным центром образует ион
хлорэтилкарбония, который, потеряв протон, образует винилхлорид и хлоро-
водород. Наличие кислотных центров катализирует присоединение хлорово-
дорода непосредственно к винил двойной связи по правилу Марковникова с
образованием 1,1-дихлорэтана.
Введение металлов в систему также уменьшает кислотность координиро-
ванных центров для алкенов, поэтому присоединение хлороводорода к винил-
хлориду по правилу Марковникова не катализируется.
Введение металлов активирует водород, предотвращая коксообразование.
Результаты экспериментов с исследованием палладиевых, родиевых и рутение-
вых катализаторов представлены в табл. 4.51. Наилучшие результаты получены
на катализаторе с добавками рутения. Катализатор на основе рутения селекти-
Та6лица4.51. Гидродехлорирование и дегидрохлорирование 1,2-дихлорэтана
Г, °C Катализатор Продукт, мкмоль/мин
Этан ЭХ* ВХ
150 PdCl2 (SG)„ А1С12 1,7 1,5 0,5
125 RhCl3 (SG)„ A1C12 2,4 0,5 -
150 RuC13 (SG)„ A1C12 0,6 0,5 2,6
125 K2PdCl4 (SG)„ AIC1, 1,0 - -
150 PdCl2 RhCl3 (SG)„ A1C12 0,2 3,5 1,6
150 K2PdCl4 RuCl, (SG)„ A1C12 2,5 1,2 -
150 Z«C12 RhCl3 (SG)„ A1C13 8,8 1,3 2,1
125 ZnCl2 RuCl3 (SG)„ A1C12 0,12 0,59 3,3
Этилхлорид.
ш и и реакции <>(>pa loii.iiimi in111»iич>i<>|>iiд.' но < равнению с палладиевым н p<>
диено цинковым катали спорами, коюрые пвляюкя наиболее аминными, ш
пора (ующими преимущественно насы щепные yi ленодороды. Введение ру lenii
приводи) к получению винилхлорида как основного продукта, селективное i
обра говаиия которого составляет 70% при конверсии дихлор папа 36%, катали
iaiop показывает хорошую устойчивое п> к коксоонра товапию и высокую ста
нильпоегь.
Невысокая в целом стабильность катали заторов дегидрохлорировании дп
хлор пана имеет в основе такие дезактивирующие факторы, как прочная адсор
оцпя хлороводорода и коксовые отложения, причем для разных катали тически
i ih (ем л и факторы также проявляются по-разному. По мнению В.В. Чесноко
на с со 1 рудниками [756-759], процесс разложения углеводородов па металла
подгруппы железа идет по механизму «карбидного цикла» в соогнетс i вин
которым в начале реакции углеводород адсорбируется и разлагается па метал
лпческой частице с образованием нестабильных поверхностных карбидов. 1’а ।
л.паясь, карбиды выделяют атомарный углерод, который диффундирует чере
ооьем металлической частицы к местам образования фазы графита. Таким о(>
разом, при разложении углеводородов, не содержащих хлора, на металлах под
। руины железа образуются углерод и водород. Принципиальная схема ною мс
хани ма включает каталитическое разложение углеводородов с обра ювап пег
нсусюйчивых карбидоподобных интермедиатов (М—С) на поверхности и и
последующий распад с выделением атомарного углерода [756]:
M + CnHm— IM—С/ + Н2 —-М + С (4.213
1______________________________I
Такое разложение происходит предпочтительно на некоторых гранях ме
1аллических кристаллов, названных «лобовыми». Выделяющейся углерод днф
фундирует через массу металлической частицы к граням, названным «тыльны
ми». На них происходит процесс отложения углерода в виде графитовых пи ici
с разной морфологией и широким спектром свойств, зависящих oi режим
процесса, природы металлической частицы и углерода. Это справедливо и дл
хлоруглеводородов, при разложении которых, наряду с графитовыми ни тями i
водородом, образуется также и хлороводород [759].
В работе [758] исследованы закономерности разложения дихлорэтана npi
его дегидрохлорировании на алюмоникелевом, алюмокобальтовом и алюмо ке
лезном катализаторах.
На рис. 4.142 приведены зависимости скорости прироста количеста угле
рода от температуры разложения 1,2 дихлорэтана, разбавленного арюном
мольном соотношении Аг:С2Н4С12 = 9:1 при 450-550 °C.
При 450 и 475 °C происходит дезактивация катализатора, но начиная
500 °C количество углерода увеличивается практически линейно, и работа ка
гализатора стабилизируется. При 550 °C достигается наибольшая скорое гь об
разования углерода, и реакция протекает даже с ускорением, что може т бы i
связано с диспергированием частиц никеля в процессе зауглероживания ка
гализатора. Дезактивация катализатора обусловлена, очевидно, отравление!
активной поверхности металлических частиц хлороводородом, образующим
ся в процессе разложения дихлорэтана. В связи с этим авторами [758] был
Рис. 4.142. Образование углерода из 1,2-дихлорэтана, разбавленного аргоном в моль-
ном соотношении Аг:С2Н4С12 = 9:1, на алюмоникелевом катализаторе при разных тем-
пературах
проведена термодинамическая оценка устойчивости металлов подгруппы же-
леза к действию НС1. Для этого рассчитали зависимости термодинамических
параметров от температуры восстановления хлорида металла водородом:
МС12 + Н2 М + 2НС1, (4.244)
где М - Ni, Со, Fe.
Константа равновесия реакции для любого металла определяется уравне-
нием:
Кр = [HCI]2/[Н2] - exp[-AG°T/(Rr)L (4.245)
где AG°t — изменение свободной энергии Гиббса.
Полученные графики зависимости Кр от t для разных металлов представ
лены на рис. 4.143, из которого следует, что для никеля Кр = 1 при 430 °C, для
кобальта Кр - 1 при 570 °C, а для железа даже при 700 °C константа равновесия
составляет всего 0,1. Таким образом, термодинамический расчет показывает,
что наибольшую стабильность в реакции разложения хлорпроизводных угле-
водородов должен проявлять алюмоникелевый катализатор.
При разложении 1,2-дихлорэтана в среде аргона алюмокобальтовый и алю-
можелезный катализаторы подвергаются дезактивации хлороводородом при
400-600 °C. Это согласуется с результатами термодинамического анализа. Рен-
тгенофазовый анализ образца дезактивированного катализатора показал нали-
чие в его составе фазы хлорида металла, что свидетельствует о хлорировании
поверхности металла как причине дезактивации.
При добавлении водорода в реакционную смесь С2Н4С12+Аг равновесие ре-
акции смещается (4.244), способствуя очищению поверхности металла. В этом
случае скорость отложения углерода на катализаторе значительно снижается
t,°C
Рис. 4.143. Температурная зависимость константы равновесия реакции
МС12 + Н2 = М + 2НС1, где М = Ni (1), Со (2) или Fe (3)
Рис. 4.144. Образование углерода на катализаторе Кт4 при 550 °C в среде аргона и но
дорода:
1 — Ar:C2H4C 12 = 9:1; 2 — Н2: Ar:C2 Н4С12 = 50:9:1
(рис. 4.144, кривая2), так как водород, адсорбируясь на поверхности никеля, гид
рирует промежуточные продукты каталитического разложения 1,2-дихлор >ы
на и уменьшает, таким образом, скорость накопления углерода.
В табл. 4.52 приведены результаты определения количества углерода, пора
зукицегося в реакции разложения 1,2-дихлорэтана на катализаторе (Кр) в ин гер
Таблица 1.52. I’a ни» кепие 1,2 дихлорэтана, ра толил» 111101 о арюпом и водородом, па
алюмопиксленом кашли шоре (время реакции 100 мин)
Ооисмная скорое 11>, д/ч Г, "С С*, % от Кт
С2П4С12 Аг н2
1 9 — 550 630,9
1 9 2 550 350,1
1 9 5 475 25,2
1 9 5 500 131,5
1 9 5 550 325,4
1 9 10 550 238,8
1 9 20 550 86,3
0,2 1,8 10 550 13,0
' Доля углерода
вале 475-550 °C при разных соотношениях С2Н4С12:Аг:Н2. Скорость накопления
углерода возрастает с повышением температуры и снижается при увеличении
мольной доли водорода в реакционной смеси.
Авторы [758] делают вывод о том, что разложение 1,2-дихлорэтана может
идти по двум независимым маршрутам. В условиях недостатка водорода пре-
валирует механизм «карбидного цикла», и процесс можно описать следующей
схемой:
4Ni + С2Н4С12 —- 2[Ni —С] + 2[Ni — С1] + 2Н2; (4.246а)
[Ni—С] — Ni + С; (4.2466)
2 [Ni — С1] + Н2 —► 2Ni + 2НС1. (4.246в)
Молекула 1,2-дихлорэтана адсорбируется на частице никеля, после чего про-
исходит разрыв связей С—С1, как самых слабых в молекуле. Выделяющийся в
ходе разложения водород связывает на поверхности никеля хлор, образуя НС1,
а также частично гидрирует поверхностный карбид с образованием СН4. В дан-
ном случае процесс идет преимущественно по пути образования углерода.
В условиях избытка Н2 преобладает процесс гидродехлорирования:
н2
С2Н4С12 + 2Ni —- 2[Ni — С] + 2НС1 + 2Н2; (4.247а)
[Ni —С] + 2Н2 — Ni + СН4; (4.2476)
С + 4Н — СН4. (4.247в)
Процесс гидрирования отлагающегося углерода также катализируется ни-
келем. Таким образом, поверхностный карбид и образующийся углерод прак-
тически полностью гидрируются водородом, а связь С—С1 подвергается гид-
рогенолизу. В этом случае основным продуктом реакции (кроме НС1) является
метан. При 350-450 °C наблюдается также заметное образование этана.
Габлица 4.55. ( <к ын продуктн п|к>ц<<< a ыы/шигич к<н<> до идрохлорнронанни 1,2
дихлор >ijii.1 (opr.iiiiriccK.ui <|>а 1.0
Karaлилагор /, С т, с Ад\ >. % зд*а С,Н2 с2н.| С211,С13 С.11.(1.
С ибупит 300 40,6 26,8 96,ч 0.01 2,45 0,15 0.35
330 41,4 31,6 96,1 0,01 2,80 0,20 0,55
370 43,5 41,5 94,0 0,01 3,35 0,25 0,80
400 42,8 50,1 91,1 0,03 4,40 0,30 1,25
CsCl/сибунит 300 41,0 32,5 99,0 - 0,55 0,15 0,25
330 42,3 45,0 98,6 0,01 0,70 0,20 0,10
370 40,8 54,6 97,5 0,01 1,15 0,25 0,60
400 41,6 60,4 96,6 0,01 1,40 0,30 0,90
Хотя целью работ [758, 759J являлось определение не условий образования
целевого продукта — винилхлорида, а закономерностей образования углерода
в процессе дегидрохлорирования, приведенные в этих работах результаты про
ливают дополнительный свет на механизм процесса.
Одним из маршрутов каталитических превращений дихлорэтана является ei о
дехлорирование с образованием этилена и хлора. В работе [760] рассмотрено ой
разование молекулярного хлора при разложении дихлорэтана в присутствии ра i
ных металлов, нанесенных на смесь диоксида титана и силикагеля. По данным ап
горов, введение Pt, Pd, Rh, Ru, Cu, Cr в структуру носителя TiO2-SiO2 сущее i пенно
увеличивает конверсию дихлорэтана (при 350 °C с 29,5 до 44-53%, при 400 °C с 31
до 82-99%). При этом отмечается, что с 400 °C одним из продуктов процесса я вл я
ется хлор. Наиболее активны в реакции дехлорирования рутений (селективное и*
по хлору достигает 53,5% при 400 °C; при дальнейшем повышении температуры
снижается) и медь (селективность по хлору монотонно растет с повышением гем
пературы, достигая 45% при 650 °C и полной конверсии дихлорэтана).
Реакция дехлорирования дихлорэтана при его каталитическом разложении
отмечена также в работе [595], в которой процесс дегидрохлорирования и луча
ли с использованием катализаторов на основе сибунита. В табл. 4.53 предс тав
лены составы продуктов процесса для катализаторов сибунит и CsCl/сибуни i
при разных температурах.
Данные табл. 4.53 указывают, что в присутствии сибунита селективное п>
образования винилхлорида существенно снижается за счет увеличения выхода
побочных этилена, 1,1,2-трихлорэтана, а также сим-1,2-дихлорэтиленов. Такой
состав продуктов реакции свидетельствует, что в условиях процесса протека
ет побочная реакция дехлорирования 1,2-дихлорэтана с образованием л плена
и хлора. Этилен десорбируется с поверхности катализатора, а хлор вступай i в
реакцию с адсорбированным винилхлоридом (или 1,2-дихлорэтаном) с обра к»
ванием 1,1,2-трихлорэтана и его последующим дегидрохлорированием. Общая
схема превращений:
С2Н3С13
С2Н2С12 (цис-, транс)
(4.2-18)
Рис. 145. Корреляция между выходом винилхлорида и количеством адсорбированного
аммиака при 350 °C:
I — АГН 3; 2 — сибунит; 3 — CsCl/АГН-З; 4 — CsCl/сибунит; 5 — CsCl/SiO2
Отметим, что доля реакции дехлорирования в общем балансе расходования
1,2-дихлорэтана существенно снижается при модифицировании катализатора
хлоридом цезия. Очевидно этот эффект наступает под влиянием изменения по-
верхностных свойств катализаторов при введении модифицирующих добавок.
На рис. 4.145 представлена зависимость выхода винилхлорида от количества
адсорбированного аммиака как зонда кислотных центров катализатора для ис-
следованных катализаторов.
Зависимость, представленная на рис. 4.145, показывает, что выход винил-
хлорида снижается с увеличением количества адсорбированного аммиака, т.е.
с увеличением количества кислотных центров на поверхности катализатора.
В силу того, что снижение выхода винилхлорида определяется, в первую оче-
редь, наличием вторичных продуктов реакции дехлорирования, можно пола-
гать, что при прочих сопоставимых характеристиках наличие кислотных цен-
тров способствует протеканию реакции дехлорирования и, соответственно,
снижению образования винилхлорида.
В работе [595] установлено, что скорость реакции дегидрохлорирования
1,2-дихлорэтана в присутствии катализатора CsCl/сибунит описывается урав-
нением, моль/(л-с):
г — 9,1 • 107 exp[-115000/(RT)]- (4.249а)
При использовании в качестве катализатора сибунита скорость реакции де-
гидрохлорирования 1,2-дихлорэтана, моль/(л-с):
г = 1,25-107 ехр[-110000/(RT)], (4.2496)
а скорость реакции дехлорирования, моль/(л-с):
г = 3,15-104 exp[-125000/(RT)]. (4.249в)
Указано 1акле, что цезинхлоридныс Kaiа/ш споры на основе споунша нс
сколько более активны но сравнению с силикагелевыми системами Опиши
ние констант скорости равно 1,8 2 < учетом достаточно высокой сгабнль
поспи (3 4 тыс ч) обеих систем, оо» клали спора перспективны для исиоль
ювапия в промышленном процессе каталитического дсчидрохлорирования
1,2 дихлор.) тана.
При возможной реализации процесса в промышленных условиях наиболее
целесообразным является использование трубчатого реактора с применением
высокотемпературного теплоносителя (например, даутерма) для эффективно) о
подвода тепла.
Глава 5.
Технологические
и экологические аспекты
производства винилхлорида
по сбалансированной схеме
сн2 сна
5.1. Принципиальная технологическая
схема производства винилхлорида
сбалансированным методом
На рис. 5.1 представлен вариант сбалансированной блок-схемы производ-
ства винилхлорида, который был широко распространен в мировой промыш-
ленной практике в I960-1970-х годах. Основные стадии процесса (без учета
переработки хлорорганических отходов и очистки сточных вод): окислитель-
ное и прямое хлорирование этилена, азеотропная осушка дихлорэтана-сырца,
ректификация дихлорэтана; ректификация винилхлорида.
HCI
Хлорорганические
отходы
Рис. 5.1. Блок-схема производства винилхлорида
Таблица 5.1. Типичный материальный баланс производства по сбалансированной схеме (т на 1 т ВХ). Процесс 19”0-1980-х годов
1 Винил- хлорид j 000*1 000*1 '
Газовые выбросы осталь- ное 0,0003 | 0,0001 | 1000*0 0,0045 0,0024 1 0,0075
с окси- хлори- рова- нием 0,0116 0,0032 0,5779 0,0214 о о 0,0017 г юо‘о 0,0025 1 I 0,6609 1
с прямо- го хлори- рования 0,0001 0,0025 9100*0 0,0001 0,0003 | 0,0045 1
Потоки щелоч- ные 0,0030 0,0008 0,0038
вод- ные 0,1166 0,0014 0811*0
Продукты побочные тяже- лые 0,0012 г юо'о 0,0023 0,0047
лег- кие 0,0017 0,0008 0,0017 | 0,0042
о гч к 0,1438 СО о
промежуточные НС1 0,6036 0,6036
ДХЭ 1,6370 1,6370
Сырьевые материалы Воз- дух £000‘0 0,5782 0,1537 0,0171 0,7493
гч с 10,5871 0,5871
к ГЧ о 0,4656 0,0002 0,4658
Компоненты со2 оэ ГЧ Z гч О гч о НС1 О, к NaOH NaCl Я и Углеводороды ДХЭ X и Легкокипягцие Высококипящие Сумма
Мы нс приводим здесь нодроОного описании данного варианта процесса.
Огмегим лишь наиболее характерные технологические решения.
На с гадии окисли тельного хлорирования этилена в качестве окислителя ис-
пользуется кислород воздуха, что ведет к необходимости улавливания дихлор-
этана из отходящих газов с помощью ароматического абсорбента. На стадии
прямого хлорирования этилена процесс проводился, как правило, при темпера-
туре ниже (кип дихлорэтана (50-60 °C) с отбором продукта в жидкой фазе и пос-
ледующей кислотно-щелочной промывкой. На стадии пиролиза дихлорэтана
межпрожиговый пробег печей не превышал 3-4 мес из-за недостаточно высо-
кого качества дихлорэтана, поступающего в процесс (99,1-99,2 мае. %).
Типичный материальный баланс данного варианта сбалансированного про-
цесса представлен в табл. 5.1 [13].
Усовершенствования производства винилхлорида по сбалансированной
схеме в мировой промышленной практике 1990-х годов включали, в первую
очередь, замену воздуха на кислород и использование высокотемпературных
катализаторов на стадии оксихлорирования этилена; реализацию процесса вы-
сокотемпературного хлорирования этилена с утилизацией тепла реакции для
ректификации дихлорэтана; хлорирование легкокипящих примесей в рецик-
ловом дихлорэтане с целью повышения качества дихлорэтана, подаваемого на
пиролиз; утилизацию тепла пирогаза и дымовых газов печи пиролиза.
Эти технические решения позволили достичь весьма высокой степени ис-
пользования сырья и энергоресурсов в сбалансированном процессе получения
винилхлорида, что дает основания считать его одним из наиболее эффектив-
ных химико-технологических процессов основного органического синтеза в
промышленности.
Принципиальная технологическая схема производства винилхлорида по
сбалансированной схеме представлена на рис. 5.2а-е постадийно, с целью бо-
лее четкого понимания технологических решений, осуществленных в данном
процессе.
Стадия окислительного хлорирования этилена
Принципиальная технологическая схема стадии оксихлорирования этилена
приведена на рис. 5.2п.
Современные решения в технологии связаны с использованием в процессе
концентрированного кислорода как окислителя и рециклового газа как псевдо-
ожижающего агента. Другая важная роль газа-рецикла сводится к обеспечению
взрывобезопасных условий ведения процесса. Реализация данной технологии
позволяет:
- снизить количество абгазов, выводимых из системы, по сравнению с «воз-
душным» процессом, в несколько десятков раз. Вследствие этого появляется
возможность отказаться от эксплуатации узла абсорбции ДХЭ из абгазов и рез-
ко уменьшить потери ДХЭ и этилена;
- используя высокотемпературный катализатор повысить производитель-
ность в 1,5-1,7 раза.
Рецикловый поток содержит значительное количество (25-70 об. %) диокси-
да углерода, обладающего более высокой теплоемкостью — 44,7 Дж/(моль-град)
Рис. 5.2л. Принципиальная технологическая схема «кислородного» процесса омк/ш
1с*лы1ого хлорирования этилена (1-12 см. текст, 64 — рис. 5.2е)
по сравнению с азотом (29,3). Это позволяет добиться лучшего тенлосъсма к
реакторе и возможности работы при более низкой температуре.
В условиях использования рециклового газа становится реальной работа
при минимальном избытке этилена в реакторе оксихлорирования. Это, наряду
с понижением температуры, позволяет значительно уменьшить горение в про
цессе оксихлорирования (с 2-3 до 0,8-1,3%), снизить выход побочных хлору!
леводородов и, соответственно, повысить содержание дихлорэтана в сырце до
99,5-99,6%. В этих условиях эффективность использования этилена возрас iaei
с 95 до 98-99%.
Процесс окислительного хлорирования этилена протекает в реакторе псов
доожиженного слоя с использованием концентрированного кислорода. Кисло
род поступает на установку с концентрацией 95-99,8% (концентрация опреде-
ляется возможностями кислородного модуля).
Основные аппараты технологической схемы стадии оксихлорировапия (см.
рис. 5.2й): реакторы гидрирования 1 и оксихлорирования этилена 2; колонна ia
калки реакционного газа 3; конденсаторы дихлорэтана и воды из реакционного
газа 4, 5; отделитель конденсированных продуктов реакции (6); емкости ра аде
ления и нейтрализации дихлорэтана и воды 7, 8; колонна отпарки дихлор пана
из сточных вод 9; компрессор рециклового газа / 0.
На стадии оксихлорировапия эксплуатируются аппараты узла под!отопки,
загрузки и выгрузки катализатора, узла приготовления охлаждающей воды для
реактора оксихлорирования, ряд емкостей и насосов.
Реактор оксихлорирования этилена — цилиндрический аппарат, в кою
ром находится распределительное устройство для смешения исходных рса
гентов — этилена, кислорода, хлороводорода и рециклового потока, встроен
ное теплообменное устройство, предназначенное для снятия тепла основной
и побочных реакций за счет испарения до 15% парового конденсата, а также
грехеryncii'iaiый циклон для улавливания ча< гнц катализатора размером бо
лее 10 мкм.
В качестве катализатора процесса используется высокотемпературный ката
лизатор, оптимальные показатели работы которото достигаются при 235 250 °C
и давлении 250 400 кПа. Продуктами реакции, помимо дихлорэтана и побоч-
ных хлорорганических соединений, являются вода, оксиды углерода, непроре-
агировавшие этилен, кислород и хлороводород. В системе имеется также азот,
поступающий в реактор, в основном с кислородом.
Этилен, хлороводород, кислород, рецикловый поток перед смешением по-
догреваются до 140-160 °C.
Хлороводород, выделяемый в колонне 64 (рис. 5.2е) из продуктов пироли-
за дихлорэтана смешивается с водородом и направляется в реактор 1, где идет
процесс гидрирования содержащегося в НС1 ацетилена. Такое решение обес-
печивает практически отсутствие в ДХЭ-сырце три- и перхлорэтилена, являю-
щихся продуктами реакции оксихлорирования ацетилена. Основные продукты
гидрирования — этилен, утилизирующийся в процессе оксихлорирования, и
этан, являющийся инертом применительно к условиям процесса. Вышедший из
реактора 1 поток смешивается с кислородом и поступает в реактор оксихлори-
рования 2.
При прохождении газовой смеси исходных реагентов через слой катализа-
тора идет реакция оксихлорирования этилена с образованием дихлорэтана и
воды, сопровождающаяся побочными реакциями окисления этилена и дихлор-
этана с образованием воды и оксидов углерода. В процессе оксихлорирования
образуется также небольшое количество побочных хлоруглеводородов, основ-
ным из которых является трихлорэтан.
Выходящий из верхней части реактора 2 реакционный газ поступает в низ
колонны закалки 3 для охлаждения до 90 110 °C и улавливания следов непроре-
агировавшего хлороводорода, а также уносимой пыли катализатора. Поступаю-
щий в колонну 3 реакционный газ барботирует через слой воды, уровень кото-
рой поддерживается в контролируемых пределах. Небольшое количество воды
(или водного раствора щелочи) подается также на орошение верха колонны.
Вода, выходящая из куба колонны 3 и содержащая хлорид и карбонат на-
трия, дихлорэтан, соли меди и алюминия, направляется на стадию очистки
сточных вод.
Охлажденный реакционный газ после колонны закалки поступает в кон-
денсатор 4, охлаждаемый оборотной водой, где одновременно с охлаждением
реакционного газа до 30-50 °C происходит конденсация паров дихлорэтана и
воды. Образующаяся газожидкостная смесь поступает в сепарационную часть
отделителя дихлорэтана 6, где происходит разделение жидкой и газовой фаз.
Жидкая фаза из сепарационной части стекает в разделитель, где разделяется на
водный слой и слой дихлорэтана-сырца. Верхний водный слой через вертикаль-
ную перегородку разделителя перетекает в отсек для воды, откуда подается на
орошение колонны закалки 3.
Газовая фаза из верхней части сепаратора 6 поступает на дальнейшее ох-
лаждение до 5-15 °C в конденсатор 5, где дополнительно конденсируются пары
дихлорэтана и воды. Образующаяся газожидкостная смесь поступает в фазо-
разделитель 11, откуда жидкая фаза возвращается в отделитель 6, а газовая фаза
n<*< I) ii.ii-i на in.к компрессора /0. oiкуда с давлением до (),(> Ml la iioiiipaiiiac
। и и реактор оксихлорирования Рецикл пред< i.ibbhci (мл i. оксидов углероде
(в основном ( О >), ипергон, пилена, кислорода и дихлор п апа < ос гаи рецп
ливою газа определяется содержанием ннерюн и Исходных реагентах, доно
пи 1СЛ1.ПЫМ окислением СО до СО в слое катализатора, а также1 окислением и
дорода до поды, вследствие чего и системе не происходит его накопление. Час
рецикла выводится из схемы и подается па узел термообезвреживания. Кол
чес । по выводимых из схемы газов примерно равно сумме введенных в реак к
и in рюв и образовавшихся в процессе оксидов углерода. Сброс абгазов при и
ноль топании 93%-ного кислорода примерно в 30 раз меньше, чем при работе
но тдухом.
1111ЖПИЙ слой дихлорэтана-сырца насосом откачивается в емкости 7 и 8 Д1
водно щелочной промывки и далее направляется через танки на колонну a.i
oi ровной осушки, в которой происходит отгонка воды и легкокипящих прим
сен
В условиях использования концентрированного кислорода конверсии ре
। еп гов на стадии оксихлорирования достигают следующих значений: этилен
99,6%, хлороводород — 99,5%, кислород — 99,7%. Выход хлорорганических о
ходов составляет 5,0 кг/т ДХЭ.
Расход на производство 1 т ДХЭ (окси): 287 кг этилена, 745 кг I K.I, 177
кислорода; 1 т ВХ: 230 кг этилена, 141 кг кислорода.
Экономия этилена, по сравнению с «воздушным» процессом, сосгавлж
(> 8 кг/т ВХ.
Стадия осушки дихлорэтана-сырца
Принципиальная технологическая схема стадии изображена па рис. 5.2
Дихлорэтан-сырец, поступающий со стадии оксихлорирования этилена, пре.
парительно нагревается до 85-90 °C в подогревателе 13, после чего подастся i
осушку и отгонку легкокипящих примесей в колонну 14.
Паровой поток, выходящий из верха колонны 14, поступает в систему ко
дснсации, включающую: холодильники-конденсаторы 15,16, охлаждаемые о(>
ротной водой; эжектор 17, подпитываемый потоком азота; разделительную сч
кость 18; сборную емкость 19; разделительную емкость 20.
В холодильниках-конденсаторах 15, 16 производится конденсация осповп
го количества парового потока до температуры конденсата на выходе К) И)
Несконденсированная парогазовая смесь поступает в эжектор 17 за счет »жс
ционного эффекта, создаваемого при истечении азота из смесительной камер
через сопло.
При истечении через сопло эжектора происходят охлаждение парогазов
го потока и частичная конденсация паров. Образовавшийся конденсат сепар
руется в разделительной емкости 20 и стекает в сборную емкость 19. Лбгаз
содержащие не более 10-15 кг/ч 1,2-дихлорэтана, направляются в санигарпу
колонну (на схеме не показана).
Влажный конденсат, образовавшийся в холодильниках-конденсаторах /5
16, поступает в разделительную емкость 18, где происходит его расслаиваю
на водную и органическую фазы. Водная фаза направляется на стадию очис
Рис. 5.26. Принципиальная технологическая схема стадии осушки дихлорэтана-сырца
(13-24 см. текст)
ки сточных вод. Органическая фаза направляется в рекуперативный подогре-
ватель 21, где подогревается с 40-50 °C до 75-80 °C за счет тепла парогазового
потока, выходящего из реакторов прямого хлорирования этилена. Подогретая
органическая фаза частично подается на флегмирование колонны 14, частично
подается в колонну 23 на извлечение легкокипящих примесей. Подогрев захоло-
женного конденсата органической фазы позволяет снизить расход тепла в испа-
ритель 22 на 1,9-2,1 ГДж/ч, по сравнению с традиционной схемой.
Для снижения потерь целевого компонента с фракцией легкокипящих ком-
понентов, выводимой на уничтожение, часть подогретой органической фазы
из емкости 18 направляется в колонну 23, в которую также направляется кон-
денсат из емкости 19. В колонне 23 производится извлечение легкокипящих
компонентов из отбираемой фракции и их экономичный вывод на уничтоже-
ние за счет снижения содержания 1,2-дихлорэтана в легкокипящих отходах до
10-15 мае. %.
Паровой поток, выходящий с верха колонны 23, поступает в холодильник-
конденсатор 24, охлаждаемый оборотной водой. Конденсат подается на флег-
мирование колонны 23. Несконденсировавшийся в теплообменнике 24 паровой
поток направляется в эжектор 17 на дополнительную конденсацию. Кубовый
отбор колонны 23 с содержанием 1,2-дихлорэтана не менее 98,5-99,0 мае. % воз-
вращается в колонну 14.
Стадия прямого хлорирования этилена и ректификации ДХЭ
Для получения дихлорэтана применяется технология процесса прямого
хлорирования этилена в режиме полного испарения ДХЭ (так называемого вы-
< (минемнер.и у pi юго хлорнронпним). I гипс» p< .ищи и нс ноль |уекя для рек ик|>и
к.щин дихлор ti.iii.i как синтезируемого 11.1 поп (. гадин гак и получаемого на
< |.1дии оксихлорировапия пилена Кроме юю. исключается образование на
noil стадии загрязненных сточных под.
Используется прием двухступепчаюго хлорирования, заключающийся в
дохлорировании этилена, не прореагировавшего в основном реакторе. 5а счег
ного суммарная конверсия этилена дос гигаег 97,8% при селскз инпос ги (ио и п
лену) 99,6%.
Узел хлорирования рециклового дихлорэтана позволяет преврати и> легко
кипящие примеси непредельных соединений и бензола в высококипящие поли
хлор.паны и хлорбензол. В результате этого повышается эффективность рек
। ификационной очистки рециклового дихлорэтана, повышается качество ДХ ),
подаваемого на пиролиз, и снижается количество жидких хлорорганических
отходов производства, образующихся при ректификации ДХЭ.
Серьезную роль играет перераспределение функции получения целевою
продукта на все ректификационные аппараты.
Использование такой схемы ректификации дихлорэтана позволяет, по с ран
пению с традиционными техническими решениями, в 1,5-2 раза снизить содер
/капие этилена в дихлорэтане, направляемом на пиролиз. Это, в свою очередь,
позволит уменьшить количество образующегося при пиролизе бутадиена.
Для снижения затрат тепловой энергии на процесс ректификационной очис
тки дихлорэтана схемой предусматривается:
- утилизация избыточного тепла потока рециклового дихлорэтана;
- частичное использование тепла парогазового потока, выходящего из ре-
акторов прямого хлорирования этилена 25, для подогрева очищенного рецик
лового дихлорэтана, выходящего из реактора 49, перед подачей в ректификаци
о иную колонну 26;
- частичное использование тепла парогазового потока, выходящего из реак
гора 25, для подогрева флегмы колонны 26.
Технологическая схема узла прямого хлорирования этилена и совмещсппоп
(или сопряженной) с ним колонны ректификации дихлорэтана приведена па
рис. 5.2в.
Процесс хлорирования этилена проводят в реакторе 25, в который подаю i
этилен и хлор с избытком этилена 5-10 мол. %. Реактор 25 заполнен жидком
кипящей реакционной массой, состоящей главным образом из дихлорэтана и
в меньшей степени трихлорэтана, являющегося продуктом побочной реакции
хлорирования ДХЭ. Для повышения селективности образования ДХЭ исполь
зуется комплексный ингибитор заместительного хлорирования в виде хлорного
железа и кислорода.
Реактор хлорирования 25 представляет собой барботажный секциоииро
ванный тарелками газлифтный аппарат с внутренней циркуляционной i рубои
обеспечивающей вертикальную циркуляцию жидкой реакционной массы пну i
ри реактора. Назначение секционирующих тарелок — интенсифицировать мае
сопередачу и одновременно обеспечить градиент концентраций реагентов по
высоте за счет уменьшения продольного перемешивания.
Реактор 25 совмещен с колонной ректификации 31с помощью трубонроно
дов. Из реактора 25 в нижнюю часть колонны ректификации 31 поступают пары
дп\лор пана и образующихся побочных хлорорганичсс них соединений, a i пк ке
испрореш пропавший и плен и и нер па, i (>д< рк.ицпеся и ш ходпы.х ре.н ен гах.
Часть реакционной массы и> реак юра 25 периодически подаю I в дохлора
юр 27 для подпитки хлорным железом реакционно)! массы дохлорагора.
Параметры процесса хлорирования л плена в реакторе 25:
давление хлора и этилена на входе в реактор, MIU ... до 0,37
Iшлепие вверху реактора, МПа до 0,27
ем пера I ура, °C.................. 115 ±5
концен । рация исходного хлора, об % . .. 99,6-99,0
конверсия этилена, %...................... 94,0
селективность (по этилену), %.... 99,5-99,6
Абгазы системы хлоратор 25 — ректификационная колонна 37, содержащие
непрореагироававший (абгазный) этилен, направляются на всас компрессора
(газодувки) 28, а затем в реактор дехлорирования (дохлоратор) 27.
Дохлоратор представляет газлифтный секционированный тарелками реак
юр барботажного типа с внешним циркуляционным контуром для естествен
пои циркуляции реакционной массы. В этот контур монтируется холодильник,
охлаждаемый оборотной водой. Абгазы после компрессора 28 и хлор подаюц я
в нижнюю часть дохлоратора.
Параметры процесса дехлорирования:
температура, °C...........................60 ± 5
давление верха, МПа........................0,165
конверсия этилена, % ........................62,9
селективность (по этилену)................ 99,8-99,9
Абгазы, выходящие из верха дохлоратора 27, последовательно охлаждаются
до 40-50 °C в водяном холодильнике 29 и далее — до температуры -10 °C в холо
дильнике 30, в который подается рассол. Конденсат дихлорэтана, образующий
ся в холодильниках 29 и 30, частично возвращается в дохлоратор и час гично
откачивается в хлоратор 25.
Несконденсировавшиеся в холодильнике 30 абгазы направляются в сани
тарную колонну, из которой выводятся на рассеивание.
Технологическая схема узла ректификации дихлорэтана включает три рек
। ификационных аппарата — массообменные колонны 26, 31 и 32 с соотве i с i ву
ющим теплообменным и емкостным оборудованием.
Колонна 26 предназначена для очистки хлорированного рециклового ди
хлорэтана от высококипящих примесей с получением ДХЭ-ректификаы, ис
пользуемого в качестве сырья на стадии пиролиза. Предусмотрена возмож
ность подачи части кубового отбора колонны 31. Колонна оборудована иена
рителями 33, в которые подается водяной пар низкого давления (р = 0,5 МПа,
t- 158 °C). Для снижения тепловых затрат на процесс ректификации колонна 2ft
оборудуется третьим испарителем 34, для обогрева которого используе гея и «
быточное тепло потока рециклового дихлорэтана, подаваемого из куба колон
ны 65 с температурой 160 °C.
Хлорированный рецикловый дихлор и ан выходи! из реактора /9 (рис. 5.2.’)
с температурой 60 "( в колонну 26, нодотревается до 90 95 °C за счет тепла па-
ротазового потока реактора 25 в подогревателе 35.
Значи тельное снижение содержания легкокипящих примесей в хлорирован-
ном рецикловом дихлорэтане позволяет повысить качество дихлорэтана-рек-
тификата, получаемого в колонне 26, до кондиций, позволяющих использовать
получаемый продукт в качестве сырья на стадии пиролиза.
Паровой поток, выходящий из верха колонны 26 поступает в систему кон-
денсации, включающую:
- аппараты воздушного охлаждения (АВО) 36;
- холодильник-конденсатор 37, охлаждаемый оборотной водой;
- флегмовую емкость 38.
В АВО 36 конденсируется основное количество парового потока до темпе-
ратуры конденсата на выходе 50-60 °C. Несконденсированные пары поступают
в холодильник-конденсатор 37 на хвостовую конденсацию.
Товарный продукт колонны 26 — дихлорэтан-ректификат — выводится из
флегмовой емкости 38 на пиролиз, остальной конденсат направляется:
- по балансу на подпитку реакторов прямого хлорирования этилена 25;
- на флегмирование колонны 26.
Перед возвратом в колонну флегмовый поток направляется в рекуператив-
ный подогреватель 39, где подогревается с 50-60 °C до 85-90 °C за счет тепла
парогазового потока реактора 25.
Кубовая жидкость колонны 26, содержащая высококипящие примеси, из-
влеченные из хлорированного рециклового дихлорэтана, выводится в колон-
ну 32.
Колонна 31 совмещена с узлом прямого хлорирования этилена и предна-
значена для очистки от высококипящих примесей дихлорэтана, синтезирован-
ного:
1) на стадии оксихлорирования, подаваемого из куба колонны азеотропной
осушки 14;
2) в реакторе 25, подаваемого с парогазовым потоком под низ колонны;
3) в дохлораторе 27, подаваемого с парогазовым потоком из реактора 25 с
получением дихлорэтана-ректификата, используемого в качестве сырья, на ста-
дии пиролиза.
Колонна 31 технологически соединена с реактором прямого хлорирования
этилена 25: поток синтезируемого в реакторах ДХЭ и испаренной реакторной
жидкости, содержащий примеси газов, легкокипящих и высококипящих ком-
понентов, подается под нижнюю тарелку. Работая в нормальном режиме, ре-
актор 25 выполняет роль испарителя, обеспечивающего необходимую паровую
нагрузку колонны 31. Для обеспечения пускового режима совмещенного агре-
гата, а также при работе реактора 25 на пониженных нагрузках используется
испаритель 40 для дополнительного подвода тепла
Паровой поток, выходящий из верха колонны 31, поступает в систему кон-
денсации, включающую:
- аппараты воздушного охлаждения 41;
- холодильник-конденсатор 42, охлаждаемый оборотной водой;
- флегмовую емкость 43.
В ABO // копдепсируепя огионное колпч<ч mo n.ipoiioio потока. HciKoii
депсироваппая парота юная «.меч. (ПК) iioiiynaci и холодильник кондеи
гатор 12 на хвостовую конденсацию, искондсш ирующисся абгазы после ген
лообменника 12 — на всас компрессора 2N н направляются в дохлор.нор 27.
Копдепса!, образующийся в АВО •// и холодильнике конденсаторе 12, пос
гунает во флегмовую емкость, откуда направляемся на ф/iei мировапие колон
вы 31.
Коварный продукт колонны 31 — дихлорэтан ректификат — выводится с
промежуточной тарелки и направляется на пиролиз.
Кубовая жидкость колонны, содержащая высококипящие примеси, часиш
по возвращается в реактор 25, частично выводится в колонну 32.
Колонна 3/ обеспечивает получение ДХЭ-ректификата, содержащею не бо
лее 0,25 мае. % высококипящих примесей.
Колонна 32. В ректификационную колонну 32 подаются кубовые о тборы и >
колонн 26 и 31, обогащенные высококипящими компонентами. Колоппа пред
назначена для:
- дальнейшего концентрирования высококипящих примесей и вывода их из
технологической схемы с минимальными потерями целевого компонента — 1,2-
дихлорэтана;
- очистки подаваемых технологических потоков от высококипящих ком
понентов с получением ДХЭ-ректификата, используемого в качестве сырья па
стадии пиролиза.
Паровой поток, выходящий из верха колонны 32, поступает в систему коп
денсации, включающую:
- холодильники-конденсаторы 44, охлаждаемые оборотной водой;
- флегмовую емкость 45.
Для работы колонны 32 под вакуумом используется вакуумная система.
В холодильниках-конденсаторах 44 производится конденсация парового по го
ка с подачей образовавшегося конденсата в емкость 45. Из флегмовой емкости
конденсат направляется:
- на пиролиз;
- на флегмирование колонны 32.
Кубовая жидкость колонны 32, содержащая 70-90% высококипящих комно
нентов (высококипящие отходы), выводится на утилизацию.
Колонна 32 обеспечивает получение дихлорэтана-ректификата, содержаще
го не более 0,40% высококипящих примесей.
Узел хлорирования рециклового дихлорэтана
Назначение этого узла — очистка дихлорэтана от примесей легкокипя
щих непредельных соединений (винилхлорид, хлорэтилены, бензол, хлоро
прен) методом их хлорирования с образованием высококипящих полихло
ридов.
Использование данного приема существенно повышает эффективность
ректификационной очистки дихлорэтана.
Технологическая схема узла хлорирования рециклового дихлорэтана пред
ставлена на рис. 5.2г.
Рис. 5.2г. Принципиальная технологическая схема узла хлорирования рециклового ди
хлорэтана (46-49 см. текст)
Рецикловый дихлорэтан, выводимый из куба колонны ректификации 65
(рис. 5.2е), дросселируется и в виде парожидкостной смеси поступает в разде-
лительную емкость 46, где происходит разделение паровой и жидкой фаз. Отде-
лившиеся пары ДХЭ, обогащенные легкокипящими примесями, конденсируют-
ся в водяном холодильнике 47.
Образующийся конденсат через смеситель, в который вводится хлор, пода-
ется в реактор 48, где происходит хлорирование легкокипящих примесей. Вы
ходящий из реактора 48 частично прохлорированный конденсат паровой фазы
объединяется в трубопроводе с жидкой частью рециклового дихлорэтана из ем
кости 46 и полученная смесь через смеситель, в который вводится хлор, посту-
пает в реактор 49, где происходит окончательное хлорирование непредельных
легкокипящих примесей. Хлорированный рецикловый дихлорэтан, выходящий
из реактора 49, направляется в ректификационную колонну 26 (рис. 5.2е) для
очистки от высококипящих примесей.
Реакторы 48 и 49 имеют одинаковую конструкцию и размеры. Это цилин-
дрические аппараты колонного типа, внутри которых на поддерживающих ре-
шетках расположен слой стальных колец. При взаимодействии стальных колец
с хлором образуется хлорное железо, необходимое для ингибирования побоч
ной реакции хлорирования дихлорэтана.
Параметры процесса:
температура, °C. 60
давление, кПа 400
очищенный ДХЭ, мае. %:
содержание ЛКК.................0,2
содержание бензола.............<0,01
11а стадии прямого хлорирования пилена и рекiпфикации дихлорэтана
расход сырья составляет (на 1 i ВХ): 251, м пилена (100%-ного), 587 кг хлора
(100% пою), и том числе 58-1 ki — на прямое хлорирование .пилена, 3 ki — па
хлорирование рециклового ДХЭ.
Стадия пиролиза 1,2-дихлорэтана
Современные процессы пиролиза дихлорэтана обеспечивают максимальное
снижение коксообразования, полное выделение смол и кокса, а также хараме
ризуются минимальными затратами энергоресурсов и минимально возможны
ми потерями целевых и промежуточных продуктов.
Реализованные в промышленных условиях технические решения позволяю г:
— утилизировать тепло продуктов пиролиза и дымовых газов;
— увеличить межпрожиговый пробег печи пиролиза;
— уменьшить потери хлористого водорода и винилхлорида.
На выходе из змеевиков испарения дихлорэтана устанавливается сепара
гор. Это позволяет предотвратить попадание жидкой органической фазы в юлу
змеевика с температурой стенки более 300 °C и тем самым снизить образование
кокса. Кроме того, образовавшиеся в зонах нагрева и испарения дихлорэтана
кокс и смолистые не откладываются на стенках змеевика в этих зонах, а вымы
ваются жидким дихлорэтаном в сепаратор, из которого отводятся непосредс-
твенно в закалочную колонну.
Использование сепаратора между зоной испарения и зоной реакции в печи
пиролиза позволяет увеличить время непрерывной работы печи в 1,5-2 раза.
Тепло продуктов реакции пиролиза утилизируется высокотемпературной за
калкой в закалочных колоннах специальной конструкции, что позволяет совмес
тить процесс закалки с процессом ректификации и выделить из реакционных га
зов примеси с температурой кипения выше температуры кипения дихлорэтана.
Ректификация в закалочной колонне осуществляется за счет высокого тенлосо
держания пирогаза без использования других источников тепла. Метод высоко
температурной «медленной» закалки позволит в 8-10 раз снизить образование
бензола по сравнению с низкотемпературной «быстрой» закалкой.
В ряде промышленных процессов реализовано использование тепла реак
ционных газов пиролиза, выходящих из колонны закалки, для подогрева ди
хлорэтана, подаваемого в печь пиролиза.
Включение в технологическую схему узла отпарки хлороводорода и винил
хлорида из кубовых продуктов закалочной колонны позволяет минимизиро
вать потери этих целевых продуктов с абгазами.
Использование узла осветления кубовых продуктов колонны закалки с вы
водом на сжигание сконцентрированных смол и кокса дает возможность сущее
твенно увеличить срок непрерывной работы кипятильников ректификациоп
ных колонн.
Принципиальная технологическая схема стадии пиролиза дихлорэтана
представлена на рис. 5.2д.
Исходный ДХЭ-ректификат нагревается до 110-130 °C в теплообменнике
рекуператоре 50 и подается в змеевик подогрева, расположенный в конвекгип
ной зоне печи пиролиза 51 и далее в змеевик испарения. Из змеевика испарения
- ПГС
— ВОП
56
HCI газ
на емкости
В колонну 64
Вода
Высококипящие
на сжигание
В колонну 64
В колонну 64
В колонну 64
Абгазы
в колонну 64
Рис. 5.23. Принципиальная технологическая схема стадии пиролиза дихлорэтана (50-63
см. текст)
ДХЭ ректификат
НО газ на стади.
оксихлорирования
В колонну 64
парожидкостная смесь дихлорэтана поступает в сепаратор 52. Из нижней части
сепаратора 52 жидкая смесь дихлорэтана, кокса и смол самотеком отводится в
закалочную колонну 53.
Пары дихлорэтана из сепаратора 52 поступают сначала в шоковую, а затем в
реакционную зону змеевика печи пиролиза 53.
Продукты реакции пиролиза дихлорэтана (пирогаз) поступают в кубовую
секцию колонны закалки 53.
Утилизация тепла дымовых газов печи пиролиза осуществляется в змеевике
парообразования. В змеевик парообразования подается котловая вода, превра-
щающаяся в водяной пар с давлением 0,5 МПа.
Параметры технологического режима:
температура пирогаза на выходе из печи пиролиза, °C. 485-505
давление пирогаза на выходе из печи пиролиза, МПа..<1,6
степень конверсии ДХЭ, %............................50-55
В кубовую секцию колонны закалки 53 подаются: газы пиролиза ДХЭ из
печи пиролиза 51; жидкий дихлорэтан из сепаратора 52.
Колонна работает при давлении вверху колонны 1,4 1,6 МПа.
При барботаже горячих газов через слой закалочной жидкости температура
газов пиролиза снижается до 175-185 °C. В колонне 53 происходит очистка про-
дуктов пиролиза от кокса, смолистых и высококипящих соединений, которые
концентрируются в кубе колонны.
Парогазовый поток, выходящий из верха закалочной колонны 53, поступает
в систему конденсации, включающую:
- подо) репа i ели (реку пера юры) ди хлор папа рек шфпкага 50;
- аппараты воздушного охлаждения >/
В сие геме конденсации кондепе ируюи я ос ионные количес run ди хлор пана
п винилхлорида
Обра «гнавшаяся газожидкос ищи смесь и i воздушною конденсатора54 пос
iyiiaer в фазоразделительную емкость 55. Жидкая фази и> сепаратора 55 час
I ично во шращается на флегмировапне гакалочпой колонны 53, часi ичио выпо
ди гея в ректификационную колонну 64 (см. рис. 5.2с).
11арогазовая смесь, выходящая из сепаратора55, последовательноохлаждас-1
с я в теплообменниках-конденсаторах 56 (хладоагент — вода) и 57 (хладоагент —
хлористый водород из колонны 64) и разделяется в емкости 58 на газовые и жид
кие компоненты, которые подаются в ректификационную колонну 64.
Часть кубовой жидкости из закалочной колонны 53 (температура 170
190 °C) выводится в узел выделения высококипящих примесей и осмолов, i дс
предварительно подается в отпарную тарельчатую колонну отпарки хлористого
водорода и винилхлорида 59.
Для подогрева флегмовой жидкости, подаваемой из емкости 60 на верхнюю
ырелку колонны 59, используется обогреваемый паром теплообменник 61. ()>
паренная в колонне 59 смесь хлористого водорода и винилхлорида направляем
ся в ректификационную колонну 64.
Из куба колонны 59 выходит смесь дихлорэтана, смол и высококипящих со
единений, которая подается на осветление в тарельчатую ректификационную
колонну 62. Из куба колонны 62 выводится смесь высококипящих соединении,
смол и кокса, содержащая до 10% дихлорэтана и являющаяся отходом произ
водства, направляемым на сжигание.
11аровая фаза, выходящая сверху из колонны 62 с температурой 90 "С, на
правляется на конденсацию в холодильник-конденсатор 63, охлаждаемый
оборотной водой. Конденсат из конденсатора 63 поступает во флегмовую ем
кость 60, где происходит частичное выделение газовой фазы, которая удаляс i с я
в санитарную колонну или на рассеивание.
Конденсат из емкости 60 частично возвращается на флегмировапие ко
лонн 59 и 62, а часть конденсата выводится в колонну 64 (см. рис. 5.2е).
Стадия ректификации винилхлорида
Технические решения на стадии пиролиза дихлорэтана и закалки реакциоп
пых газов пиролиза, максимально снижающие коксообразование и обеспечн
кающие наиболее полное выделение кокса и продуктов осмоления из тсхиоло
гических потоков, поступающих на стадию ректификации винилхлорида, по г
воляют снизить до минимума действие фактора, осложняющего эксплуатацию
аппаратов стадии ректификации винилхлорида в части загрязнения тарелок
колонны выделения хлороводорода и загрязнения трубного пространства ис
парителей.
Принципиальная технологическая схема стадии ректификации винилхло
рида представлена на рис. 5.2е. Процесс получения товарного продукта — вп
пилхлорида-ректификата — организуется в трех последовательно соединенных
узлах:
Рис. 5.2е. Принципиальная технологическая схема стадии ректификации винилхлорида
(64-71 см. текст)
1) узел отпарки хлороводорода: ректификационная колонна 64; холодильная
фреоновая установка; теплообменное, емкостное и насосное оборудование;
2) узел выделения винилхлорида-сырца: ректификационная колонна 65;
теплообменное, емкостное и насосное оборудование;
3) узел выделения винилхлорида-ректификата: ректификационная колон-
на 66; теплообменное оборудование.
Узел отпарки хлороводорода
В узел отпарки хлористого водорода из технологической схемы стадии пи-
ролиза дихлорэтана поступает пять технологических потоков:
1) газовая фаза из емкости 58;
2) жидкая фаза из емкости 58;
3) конденсат из емкости 55;
4) конденсат из 60;
5) паровой поток из колонны 59.
Кроме того, в колонну 64 подают газообразный винилхлорид из колонны 66.
Сверху из колонны 64 выходит поток газообразного хлористого водорода,
подаваемого в холодильник-конденсатор 67, охлаждаемый испаряющимся жид-
ким хладоном.
В холодильнике-конденсаторе происходит частичная конденсация НС1-газа
в количестве, требующемся для обеспечения необходимого технологического
режима работы колонны 64.
Парожидкостный поток из холодильника-конденсатора 67 поступает в
разделительную емкость 68, где происходит сепарация паровой и жидкой фаз.
Несконденсированный поток хлористого водорода из емкости 64 поступает в
чи ||>дн и. и н к конце) ил iop 57 (pili. 5.?<>) у ui.i и роме к) io'i noil конце! нац и и, ное
и hoiopoi о 11< I гл । паправляен я пл 1ыдню <нм и хлорирован и я и плена. ( кон
В in проплпнын хяоровоцороц и । ем мп i и 6.4 направляси я па <|inei мировлпие и
I О |< >1111 у о /
ку новый отбор и j кояоппы б/, соцер к.пцнп винилхлорид, 1,2 дихлор пап, .
। и- не ле! компппцие и высококипящие хлороргапические примеси, подается в
। олоппу 65.
Узел выделения винилхлорида-сырца
И у юл выделения винилхлорида сырца из колонны 64 поступает кубовый
niiiiip, содержащий винилхлорид, 1,2-дихлорэтан и хлорорганичсские приме
и II 1н1>олес близкокипящей к винилхлориду примесью является 1,2-бугади
। п количес! во которого в потоке питания достаточно мало, чтобы повлия 1ь па
пп кепие качества получаемого винилхлорида-ректификата. Поэтому техполо
। iriei кии режим работы колонны 65 должен обеспечивать четкое разделение по
точеной парс компонентов ВХ-1,2-ДХЭ.
( верху из колонны 65 выходит поток газообразного винилхлорида, подана
( moi о в холодильники-конденсаторы 69, охлаждаемые оборотной водой.
конденсат винилхлорида-сырца из холодильников-конденсаторов 69 пос ту
ii.tr 1 в емкость 70, откуда направляется:
па флегмирование колонны 65;
в колонну 66 на отгонку остаточного количества хлористого водорода.
кубовый отбор колонны 65, содержащий 1,2 ДХЭ, остаточное количество
винилхлорида и хлорорганические примеси (как легкокипящие относительно
1,2 ДХЭ, так и высококипящие), направляются в узел хлорирования рецикло
пою дихлорэтана.
Узел выделения винилхлорида-ректификата
В узел выделения винилхлорида-ректификата из колонны 65 подается ВХ
i ырец, содержащий целевой продукт с примесью хлористого водорода. Техно
ло| ический режим работы колонны 66 должен обеспечивать отпарку хлорис ю
। о водорода из ВХ-сырца.
Колонна 66 работает без возврата флегмы — ВХ-сырец из колонны 65 пода
ется на верхнюю тарелку отпарной колонны.
Отпарка хлороводорода производится посредством частичного испарения
кубовой жидкости подачей греющего водяного пара в испаритель 71 Пары ви
пилхлорида, содержащие отпаренный хлороводород, выходят сверху из колон
пы 66 в колонну 64.
Кубовый отбор колонны 66 — винилхлорид-ректификат — поступает па
башни сушки.
Нормы расхода сырья, кг, на производство I т винилхлорида
( тадия оксихлорирования:
этилен (100%)......................................... 230,33
кислород (100%).........................................141,00
(тидии прямого хлорирования
этилен (100%)......................................... 23-1,37
хлор (100%)............................................. 584,05
Узел хлорирования рециклового дихлорэтана:
хлор (100%)..................................................2,95
Стадия пиролиза:
1,2-дихлорэтан (100%).....................................1618,27
Суммарно:
этилен (100%)............................................. 464,70
хлор (100%)............................................. 587,00
кислород................................................ 141,00
Технико-экономические показатели производства винилхлорида
Расходные нормы:
этилен...............................................464,7 кг/т ВХ
хлор.................................................587 кг/т ВХ
кислород.............................................141 кг/т ВХ
пар..................................................0,87 Гк/т ВХ
топливо..............................................106 кг/т ВХ
электроэнергия.......... ............................212 кВт-ч/т ВХ
(с учетом
получения кислорода)
вода оборотная.......................................180м3/тВХ
Отходы:.......................................................
высококипящие со стадий ректификации и пиролиза ДХЭ .. .28,2 кг/т ВХ
легкокипящие со стадии осушки ДХЭ..................1,6 кг/т ВХ
абгазы со стадии оксихлорирования.................19,8 кг/т ВХ
5.2. Производственные выбросы в окружающую среду
Современные производства винилхлорида характеризуются минимальны-
ми значениями выбросов вредных веществ в окружающую среду. Это является
следствием высокой степени полезности использования исходного и вспомо-
гательного сырья, а также квалифицированной очистки и утилизации отходов
производства.
Виды отходов, образующихся в ходе эксплуатации производства винилхло-
рида: твердые — кокс и полимеризационные отложения; жидкие хлороргани-
ческие; сточные воды; газовые выбросы.
Твердые отходы
Полимеризационные отложения представляют твердые и смолообразные
продукты темно-коричневого цвета, содержащие небольшие примеси дихлор-
этана. Образуются, как правило, в системе обезвоживания дихлорэтана за счет
полимеризации непредельных легкокипящих хлорорганических соединений.
Количество их составляет -0,01 кг/т винилхлорида, т.е. для производства мощ-
поыыо 250 цис. i/год 1одо|юе к<>;1пче< ino полпмсрп ыцпоиных О1локепияй co
c гапляст ~2,5 r.
Коксовые отложения предсш идя lift гвсрдыс асфпльтообразпые продукты
icmiio коричневого цвета, содержащие неin.rinтельное колпчесто оргаппчес
ки связанного хлора. Коксовые огло копия образуются практически па всех с га
днях гехнолотическо!о процесса и удаляются чисткой оборудов шия (рсктифи
кационные колонны и кипятильники), а также печи пиролиза дихлорэтана.
Количество кокса на стадии ректификации дихлорэтана — около 0,01 кг/г
винилхлорида. Приблизительно столько же кокса образуется на стадии пиро
ли «а, причем около 50% извлекается из печей пиролиза и фильтров, остальной
кокс выделяется на стадии ректификации винилхлорида.
Для мощности производства винилхлорида 250 тыс. т/год количес тво кокс i
с оставляет до 5 т/год.
Твердые отходы направляются обычно на захоронение в полигонах, специ
алыю оборудованных для этих целей.
Жидкие хлорорганические отходы
Жидкие хлорорганические отходы (~30 кг/т винилхлорида) направляются,
как правило, на термообезвреживание, либо на переработку химическими сно
собами. (Подробно о методах переработки жидких хлорорганических отходов
см. гл. 7.)
Сточные воды
Технологические сточные воды образуются в производстве винилхлорида в
основном на стадиях оксихлорирования этилена, прямого хлорирования ни
лена (при условии эксплуатации низкотемпературного процесса), а также па
стадии азеотропной осушки дихлорэтана. Сброс этих стоков на очистку осу
ществляется постоянно.
Примерный состав сточных вод, направляемых на очистку со стадии ок
сихлорирования этилена, мг/л:
CuCI2(Cu(OH)2)..................................... .. 100
А12О3.............................. ...................600
хлорорганические соединения,
и основном 1,2-дихлорэтан.............................4000
NaCl...................................................9500
NaCOOH.................................................1500
Na2CO3...............................................<10 000
Количество сточных вод со стадии оксихлорирования этилена сосгавляш
0,4-0,5 м3/т винилхлорида.
На стадии прямого низкотемпературного хлорирования этилена сточные
воды образуются на узле кислотно-щелочной промывки дихлорэтана-сырца.
Усредненный состав кислых сточных вод, мг/л:
НС1 ..............................................4500
FeCl3............ ................................1200
ДХЭ............................................... 10 000
550 1
Усредненный сое i.iii щелочных с сочных под, мг/л:
N.1OII . ... ... .14 000
NaCIO.............................................27 000
NaCl..............................................22 000
Na2CO3............................................И 0 000
ДХЭ...............................................8000
Количество сточных вод со стадии прямого низкотемпературного хлориро
вания этилена -0,15 м3/т винилхлорида.
Технологический поток загрязненной воды, также отбираемый в постоян-
ном режиме со стадии азеотропной осушки дихлорэтана, содержит в качест-
ве загрязнений дихлорэтан, содержание которого достигает 8 г/л. Количество
воды в этом потоке составляет -0,004 м3/т винилхлорида.
На очистку поступают также сточные воды, образующиеся при чистке обо-
рудования, включении в работу санитарной колонны, предназначенной для аб-
сорбции хлорсодержащих газов, промывные воды от сборника кокса и др. Все
эти сбросы периодические. Количество промывных сточных вод оценочно рав-
но 0,1-0,15 м3/т винилхлорида.
Таким образом, общее количество сточных вод, поступающих на очистку,
0,6-0,7 м3/т винилхлорида.
Очистка сточных вод — процесс многостадийный. Вначале удаляют ди-
хлорэтан из воды обработкой ее острым паром. Затем разрушают гипохлорит
натрия обработкой сульфитом или формиатом натрия. Следующая стадия —
последовательное осаждение тяжелых металлов (медь, железо, алюминий) из-
менением pH среды.
Для достижения уровня солесодержания, не превышающего экологически
допустимых величин, при очистке воды могут быть использованы обратноос-
мотические мембраны.
Средневзвешенное содержание примесей в сточной воде после очистки:
- концентрация солей — <4 г/л;
- доля дихлорэтана — <0,0001 мае. %;
- доля тяжелых металлов, в частности, меди — <10 5 мае. %;
- активный хлор — нет.
Очищенные воды сбрасываются в канализацию.
Газовые выбросы
Проблема газовых выбросов в производстве винилхлорида имеет первосте-
пенное значение в тех случаях, когда в процессе оксихлорирования этилена в
качестве окисляющего агента используется кислород воздуха. Объем газовых
выбросов на этой стадии достигает 800 м3/т винилхлорида, т.е. для мощнос-
ти производства 250 тыс. т/год часовой объем газовых выбросов составляет
25000 м3.
В работе [13] приведен типичный состав абгазов (табл. 5.2) со стадии ок-
сихлорирования мощностью 240 тыс. т дихлорэтана в год (соответствует мощ-
ности установки по винилхлориду 320 тыс. т в год).
Несмотря на то, что дихлорэтан, уносимый потоком абгазов, в значитель-
ной мере улавливается путем абсорбции с использованием высокотемпера-
I .>0/1111 Vi 5-2. I ип ичп i.iii i <>i i.iii о i ходи щи x i i ion n i модули океи хлорировании мощти
1ыо24() пас. г/год но дихлор пану
Компопеи г < )О'11 .111 флу к 1 унция содержания, мол. % ( редин и расход
кми/и»/ч К1 /ч
Диоксид углерода 0,8-35 36,3 1598
Монооксид углерода 0,6 1,3 15,7 111
Ajo г 82-95 1881,5 52 706
Кислород 0,5-7,5 92,6 2962
Меган 0-5,0 8,1 1 30
Ггилеп 0,2-0,8 11,4 318
> । .in 0-3,8 14,5 135
Дихлорэтан 0,07-0,75 4,6 •154
)гилхлорид 0-0,75 6,0 386
Винилхлорид 0-0,25 1,6 101
Ароматический 0-0,1 0,7 71
рас гворитель
|урного ароматического растворителя, его выбросы в атмосферу для мши о
гоннажных установок составляют десятки и даже сотни килограммов в ч.к
В атмосферу также выбрасывается значительное количество ароматически!
абсорбента.
В [13] приведена сравнительная экономическая оценка вариантов модер
низации «воздушного» процесса оксихлорирования этилена, где показано, 41
наиболее экономичной является замена воздуха на концентрированный кмелс
род. Это более выгодно по сравнению с термообезвреживанием и катали гичес
ким окислением отходящих газов, соотношение затрат при реализации каждш
из этих вариантов равно соответственно 1:1,83:2,61.
С середины 1980-х годов практически во всех вновь вводимых процессм
оксихлорирования этилена в качестве окислителя используется копцешрт
рованный кислород. Это позволяет снизить объем абгазов в 70-100 раз. и
сравнению с «воздушным» процессом, и направлять отходящие газы на ежг
гание.
Реализация такого варианта технологической схемы практически полп<
стью исключает выброс продуктов процесса оксихлорирования в атмосферу.
Выбросы абгазов с других узлов технологической схемы незначительны и i
превышают 2-5% от выбросов на стадии оксихлорирования (с использование
воздуха).
В атмосферу выбрасываются также дымовые газы печей пиролиза в кош
честве 1500 м/т винилхлорида. Состав газов: N2, СО2, О2, Н2О, незначигелык
количество SO2.
Вентиляционные выбросы (для установок, работающих в закрытых пом
щениях) в атмосферу осуществляются постоянно. Содержание в них не цреш
шает: дихлорэтана — 10 г/м3, винилхлорида — 1 г/м3, хлороводорода — 5 i/m '
Авторы работы [13], проанализировав данные по эксплуатации восьми у
тановок по производству винилхлорида в США, провели оценку уровня выбр<
сов винилхлорида в атмосферу из разных точек технологической схемы.
и,б
Суммарное производство ВХ, %
Рис. 5.3. Зависимость эмиссии винилхлорида в атмосферу от суммарного его прои.з
водства
Указано, что периодические выбросы винилхлорида в атмосферу могут
иметь место в зонах загрузки и хранения, а также через предохранительные кла
паны, в точках отбора проб, фильтрах, насосах и при вскрытии резервуаров.
Оценена величина фугитивных выбросов. Эта составляющая выбросов
невелика — не превышает 0,4-1,5 г ВХ/кг продукта На рис. 5.3 приведена за
висимость эмиссии винилхлорида в атмосферу от суммарного производства
винилхлорида в процентах. Авторы [13], оперируя данными о работе промыт
ленных установок в США в 1974 г„ показали, что суммарная эмиссия винил
хлорида в атмосферу составила 5200 т/год при общем объеме производства
~2,5 млн т/год. Они указывают, что производительность большинства устано-
вок в течение года остается на фиксированном уровне, а на управление вы-
бросами винилхлорида не влияют ни температура окружающей среды, ни тем-
пература охлаждающей воды, поэтому эмиссия винилхлорида в течение года
практически не изменяется.
5.3. Характеристика готового продукта
Винилхлорид, предназначенный для полимеризации, не должен содержать
примесей, превышающих значения, указанных в [22].
Типичные уровни примесей в мономере винилхлорида, мг/кг:
ацетилен......................................................0,5-2,0
кислотность по НО, мае. %.....................................0,1-1,0
ацетальдегид..................................................0,4-1,0
щелочность по NaOH, мае. %......................................0,25
1,3-бутадиен. 8,12
этилхлорид................................................. .... 35
дихлорэтан.......................................................10
железо, мае. %................................................0,15-0,4
mi । и и хлорид, . <>0 75
loiitiinaneiai .. .10
подл.......................................................................... 10(1
iii-’ieryiiie соединения........................................................ ..25 50
нищее колнчес гно neiLici.iincmii.ix <', .... 10
hin пород п паровом прострапс me, после laipy.ihii................................200 1000
|1||нн>1хиорнд, мае. %............................................................99,6 99,98
Минимальное содержание винилхлорида.
Глава б.
Получение винилхлорида из этана
сн2 —..- CHCI
Этан — наиболее естественный источник углеводородного сырья для полу
чения винилхлорида, в том числе и по сравнению с этиленом. До 40% этилена и
мире производят именно из этана в качестве исходного сырья (в США из этап.»
получают -60% этилена [16]). Этан дешевле этилена в 3,5-4 раза, что может слу
жить серьезным аргументом в пользу создания производств винилхлорида на
его основе.
Пик исследований и технологических разработок процесса получения ви
нилхлорида из этана пришелся на 1970-1980 е годы. В промышленных масш
табах процесс так и не был реализован из-за достаточно жестких условий его
проведения с образованием большого количества отходов по сравнению с «эти
леновым» процессом. В начале XXI в. наблюдается возобновление интереса к
данной проблеме, что оставляет вероятность создания промышленных процес-
сов получения винилхлорида из этана, правда, не ранее 2010-2015 гг.
Этан ввиду большей, чем этилен, насыщенности требует более сложной схе-
мы переработки, и только его более низкая стоимость может в определенной
степени компенсировать эту сложность.
Маршруты превращения этана в винилхлорид могут быть представлены не-
сколькими схемами:
Дегидрирование Хлорирование Крекинг
а) С2Н6-------------- С2Н4 ------------ С2Н4С12-------------- С2Н3С1;
Оксихлорирование
Хлорирование Крекинг
б)С2Н6 -------------- С2Н4С12-------------С2Н3С1;
Оксихлорирован ие
Хлорирование
в) С2Н6 ------------- С2Н3С1.
Оксихлорирование
Рассмотрим в основном маршрут «в», поскольку это способ непосредствен-
ного получения винилхлорида из этана.
Для прямого получения винилхлорида из этана могут использоваться два
основных метода: окислительного хлорирования этана и хлорирования этана с
помощью переносчиков хлора.
Газофазный каталитический процесс окислительного хлорирования этана
до винилхлорида по суммарному уравнению:
С2Н6 + НС1 + О2
С2Н3С1 + 2Н2О
(6.1)
прсдсзапаяет совокупноеiь двух последов.цельных. кинетически пиi.uiikими
протекающих реакции — окисление хлороводорода до хлора (реакция Дикона)
и хлорирование этапа.
Окислительное хлорирование папа — сло кпыи, многоступенчатый про
цесс, представляющий сопряжение реакции окисления хлороводорода до хлора
последовательно параллельными реакциями хлорирования, дегидрохлори
ропания, гидрохлорирования и окисления исходного лапа и промежуточных
хлоралканов и хлоралкенов [420|. Оксихлорирование этана обычно протекает
при 350 600 "С в зависимости от того, какие продукты являются целевыми.
В отличие от низкотемпературного процесса оксихлорирования этилена при
оке и хлорировании этана бесспорны факты образования хлора по реакции Ди
копа и участия его в процессах превращения углеводородов [457, 691, 701|. < о
пшено данным, полученным в НИФХИ им. Л.Я. Карпова [420, 6911, окисление
хлороводорода до хлора идет с одинаковой скоростью в присутствии и без yi
леводорода. Стадия хлорирования может протекать и безучастия катали штора
по для протекания последующих реакций дегидрохлорирования катали тор
вновь необходим. По этой причине подбор оптимальной каталитическом сиси
мы определяется составом целевых продуктов в данном процессе. Прак i ичес кп
все катализаторы процесса оксихлорирования этана, описанные в ли тературе,
содержат хлорид меди. А. Эллсворт [702] при использовании системы Си< 12
КС 1/фуллерова земля при 520 °C достиг селективности образования винил
хлорида 60%. Дж. Баркли [703], используя смесь СиС12-СаС12-СеС12 на оксиде
алюминия, при 530 °C в псевдоожиженном слое катализатора достиг конвер
< ии этана 98,1% при селективности образования этилена 38,3% и винилхлорида
10,1 %. Применение в качестве катализатора фосфата железа на силикагеле 170-11
ведет к образованию в основном этилхлорида с селективностью 86%. Г. Ригель
|705| предложил проводить процесс в расплаве, содержащем 70% СиС12 и И)%
Кс I при 470 °C Суммарный выход этилена и винилхлорида — 31,5%, этилхло
рида и дихлорэтана — 59,5% указывает, в частности, на то, что данная система
малоактивна в реакциях дегидрохлорирования хлоралканов. Предложено 17061
использовать соли меди в виде оксида, сульфата, нитрата, фосфата, хромат »
молибдата и др. в смеси с соединениями РЗЭ. Однако в условиях оксихлориро
нация можно ожидать постепенного перехода их в хлориды, в связи с чем реаль
пая каталитическая система не будет отличаться от традиционной.
Таким образом, катализатор Дикона — хлорид меди — обязательная сое гав
ляющая каталитических систем, используемых в процессе оксихлорировапия
эгана. Обычно в катализаторе содержатся также хлориды щелочных или ще
лочноземельных металлов для уменьшения уноса меди с поверхности катали
затора.
В условиях оксихлорирования смесь хлоридов меди, щелочных металлов и
ряда промоторов, как правило, находится на поверхности носителя в состоя
нии расплава [420, 438, 708]. Появление в системе одновалентной меди гак же,
как и введение в систему хлоридов щелочных металлов, приводит к снижению
температуры плавления эвтектики. При этом образуются комплексы ра изо
го химического состава — KCuCl3, K2CuCl4, K2CuCl3, K2Cu2Cl4, K2Cu2Clft |429|
Значительно снижают температуру плавления хлорной меди и хлориды редко
земельных металлов [572].
Рпспланактивной массы хлоридов меди и калия па носи телях разной хими
ческой природы с разной удельной поверхностью образует в развитой системе
нор носи теля изолированные включения толщиной 10-200 молекулярных сло-
ев [707, 708, 709].
В качестве носителя используются пористые вещества с удельной поверх-
ностью в интервале 0,5-500 м2/г [97, 450, 457]. Его выбор достаточно специфи-
чен. Как правило, для осуществления процесса в псевдоожиженном слое ката-
лизатора используют носители с более развитой поверхностью по сравнению
с носителями для неподвижного слоя. Это связано с тем, что широкопористые
носители с ограниченной поверхностью, используемые для снижения скорости
побочных реакций, плохо удерживают расплав, что приводит к слипанию кон-
такта и значительно ухудшает показатели процесса [710]. В работах [438, 695]
показано, что окисление хлороводорода происходит в объеме расплава солей.
Хлорирование до 425 °C идет в основном по гетерогенному механизму на по-
верхности расплава солей, а при более высокой температуре — преимуществен-
но по гетерогенно-гомогенному пути. Г. Капрара с соавторами [712] полагают,
что хлорирование идет в основном гомогенно, в газовой фазе.
Важным фактором в выборе контакта оксихлорирования является миними-
зация побочных реакций глубокого окисления алканов и хлоралканов до окси-
дов углерода. А.Е Аглулин [713] считает реакцию окисления каталитическим
процессом, при этом, по данным И.И. Курляндской и Т.Ф. Кудрявцевой [707,
714], за эту реакцию ответственны как свободная от солевого расплава поверх-
ность носителя, так и поверхность солевых включений.
Л.М. Карташовым с соавторами [715] показано, что использование носи-
телей и стабилизирующих хлорную медь добавок позволяет сохранять актив-
ность контакта в течение 400-500 суток непрерывной работы. Особенно остро
проблема потери активных компонентов стоит при проведении процесса в не-
подвижном слое, когда имеют место локальные перегревы катализатора. Уле-
тучивание хлоридов меди может привести к изменению селективности катали-
затора во времени за счет изменения соотношения меди с калием в активной
массе солей.
Кинетика и механизм процесса окислительного хлорирования этана под-
робно изучена в Н14ФХ14 им. Л.Я. Карпова [438, 716, 718, 692]. В присутствии
катализатора CuCl2-KCl/SiO2 авторами предложена следующая совокупность
последовательных и параллельных радикально-цепных и гетерогенно-катали-
тических реакций:
Гомогенно
2
Гомогенно
с2н6-------------С2Н5С1
Гомогенно
С2Н4
| Гетерогенно С2Н2С12
1ЛС2Н4С12 g | Гетерогенно | ]Q
С2Н3С1 9 -С2Н3С13
1,2-С2Н4С12------7—*
Гетерогенно
Гомогенно
Hi схемы для упрощения ис ключе при оор.нопания оксидов углерода
Опа включает реакции |120(
- замести тельного хлорирования num на и хлор панов (1, 2, 4, 8);
- Дегидрирования этапа и хлор панов;
- дегидрохлорирования хлорэгапов (3, 6, 7, /());
- аддитивного хлорирования пилена и хлоралканов (5, 9);
- заместительного хлорирования .пилена и хлоралканов.
Установлено [701, 713|, что при t > 400 "С скорости расходования всех про
дуктов оксихлорирования этана, за исключением 1,1-ДХЭ, подчиняются уран
пению:
г, = , (6.2)
। де i — условное обозначение реагента; п ~ 0,5.
Величины наблюдаемых энергий активации реакций превращения иредель
пых соединений составляют 125-137 кДж/моль [420].
Скорости превращения предельных соединений слабо зависят oi выбран
него для нанесения на сорбент хлорида металла, а также от величины удельной
поверхности сорбента [713]. Согласно [420, 701], этилхлорид является едиш
твенным продуктом, образующимся непосредственно из этана (рис. 6.1 и 6.2).
В [691] указано, что наблюдаемая константа скорости в уравнении (6.2) за
висит от природы углеводорода и для предельных соединений уменьшается в
ряду С2Н6 > С2Н5С1 > С2Н4С12.
При этом наблюдаемая константа скорости хлорирования этилена больше,
чем для винилхлорида.
Величины наблюдаемых энергий активации превращения предельных и не
предельных соединений существенно разнятся (130 и 45 кДж/моль cooiвен
твенно). Как следствие, с повышением температуры скорости образования не
предельных соединений возрастают быстрее, чем скорости их превращения,
что и приводит к увеличению их выхода [691]. Предельные углеводороды об
разуются и превращаются по параллельно-последовательной схеме череi cooi
ветствующие непредельные соединения, причем молекулярный хлор, ускоряя
образование последних, участвует в реакциях дегидрохлорирования.
Рис. 6.1. Зависимость избирательности по этилхлориду от рс,щс1/рс2н6
Рис. 6.2. Зависимость скорости превращения этилхлорида от Pq2 и Pc2h5ci> в том числе
при оксихлорировании этана (светлые точки), t = 375-525 °C
Закономерности превращений 1,1-ДХЭ в условиях процесса оксихлориро-
вания этилена отличаются по сравнению с другими хлорэтанами. Зависимости,
представленные на рис. 6.3, показывают, что скорость превращения 1,1-ДХЭ не
зависит от парциального давления хлора и подчиняется уравнению 1-го порядка
Гщ-дхэ = 4,9 • 105[-83,8/(RT)], (6.3)
т.е. энергия активации и частотный фактор реакции превращения 1,1-ДХЭ су-
щественно ниже, по сравнению с другими предельными соединениями. Единс-
твенный продукт превращения 1,1-ДХЭ — винилхлорид [692].
/’i ,n,i I!P< ji.
PC,H,CI / PCJH,
Рис. 6.4. Зависимость избирательности по оксидам углерода от ро2>Рс2щ и Pi ,11,1.1 •ф11
разных температурах: S — избирательность по оксидам углерода
При анализе кинетических закономерностей процесса оксихлориронапия
этана необходимо учитывать вклад реакций глубокого окисления с образова
нием моно- и диоксида углерода. В работах [691, 695] показано, что скорость
реакций глубокого окисления описывается уравнением:
Г, =kipipo2pc\2 (6z1)
Зависимость избирательности по оксидам углерода от парциальных давле
ний кислорода, этилена и винилхлорида при 400, 450, 525 °C представлена па
рис. 6.4.
Значения констант скорости реакций глубокого окисления этилена (числи
тель) и винилхлорида (знаменатель), рассчитанные по (6.4):
t, °C........................... 400 450 525
к,- 10 5, моль/(л кат-ч-атм2’5).0,0115/0,077 0,0436/0,295 0,268/2,18
Скорость окисления винилхлорида в 7-8 раз выше по сравнению с этиле
ном. Величины наблюдаемой энергии активации для этих реакций близки и со
ставляют - ПО кДж/моль. Дальнейшее увеличение содержания хлора в молеку-
ле уменьшает сгорание хлоралкенов.
Таким образом, в интервале 350-525 °C реакции предельных и непредель
ных соединений имеют нулевой порядок по хлороводороду. Кислород нс или
яет на превращения предельных соединений и преимущественно расходуется
в реакциях окисления хлороводорода и аддитивного хлорирования непредель
ных соединений по окислительно-восстановительной схеме. Катализатор ус
коряет реакции окисления хлороводорода, диссоциации молекулярного хлора,
аддитивного хлорирования непредельных соединений и дегидрохлорирования
1,1-дихлорэтана.
Рис. 6.5. Зависимость степени конверсии этана (1), хлороводорода (2) и селективности
образования суммы хлоруглеродов (3), С2Н4 (4), оксидов углерода (5) от температуры
(т = Зс, объемное соотношение С2Н6:НС1:О2 = 1:2:1)
Рис. 6.6. Изменение состава хлорорганических продуктов с изменением температуры
процесса:
1 — С2Н.,С1; 2 — С2Н,С1, 3 — С2Н,С1„ 4 — С2Н4С12,5 — С2НС13 + С2С14
На рис. 6.5 и 6.6 представлены зависимости степени конверсии этана и хло-
роводорода, селективности образования продуктов реакции оксихлорирования
этана (хлорпроизводных этана и этилена, оксидов углерода, непредельных угле-
водородов), а также состава продуктов хлорирования от температуры в диапа-
зоне 300-600 °C. Нетрудно заметить, что с повышением температуры направ-
ленность процесса оксихлорирования этана заметно меняется: при сравнитель-
но умеренных для этой реакции температурах (до 375-400 °C) в основном про
ick.uoi реакции umi’i i и ic/ibiioio хлорировании г oop.i lonaiincM хнор иапон,
суммарный выход которых составляв! около 95%. Соотношение получаемых
при одной и гой же температуре хлор rraiioii, в свою очередь, определяется i лу
пипой превращения СЛ1(,. При повышении температуры более -100 "С характер
процесса усложняется, и наряду с реакциями ааместигельного хлорирования
идут реакции дегидрохлорирования с образованием .пилена и хлоролефипов,
адди тинного хлорирования непредельных хлоруглеводородов и глубокого окис
лепил с образованием оксидов углерода. Начиная с 550 °C наблюдается также
образование ацетилена. Содержание винилхлорида в продуктах хлорирования
с 10% при 400 °C возрастает до 90% при 600 °C, суммарный выход хлороргапи
ческих соединений при этом снижается главным образом за счет значи тельного
увеличения выхода свободного этилена (с 18% при 450 °C возрастает до 70%
при 600 °C). В области t > 450 °C заметно возрастает и скорость реакций глупо
кого окисления этана и хлоруглеводородов до оксидов углерода, однако выход
последних в интервале 450-600 °C изменяется незначительно. Степень коппер
сии этана с повышением температуры непрерывно увеличивается и при 550 "(
составляет 95-97%, степень конверсии хлороводорода достигает максимума,
чго свидетельствует об интенсификации процессов дегидрохлорирования при
I > 400 °C. Ниже приведены наиболее типичные составы продуктов хлорирова
ния и степень конверсии этана для двух температур, иллюстрирующие измене
ния направленности процесса оксихлорирования:
/, °C................................. 325 550
Степень конверсии этана, %............. 40 95
Состав, %:
С2Н5С1.............................. 70 3-5
С2Н3С1............................ Сл. 65-70
С2Н2С12......................... 15 3,5
C2H3CI3............................. 15 Сл.
С2НС13 + С2СЦ...................... Сл. 20
Природа и состав катализатора значительно влияют на скорость превраще
пия этана и селективность реакции. Следует отметить, что для солевых систем
эго прежде всего относится к исходному носителю.
В табл. 6.1 представлены данные по сравнительной активности и селекгив
пости образцов медьсодержащих катализаторов на основе различных исход
пых носителей, испытанных в одинаковых условиях. Из приведенных данных
видно, что в одинаковых температурных условиях степень конверсии этапа и
селективность образования продуктов реакции в значительной мере определи
ются природой носителя.
Исследование реакции оксихлорирования этана на разных катализа юрах
в широком диапазоне варьирования показателей процесса (времени кон гак га,
концентрации и соотношения исходных реагентов) показало, что они сущее
гвенно влияют главным образом на селективность реакции. Так, увеличение
концентрации НС1 в исходной смеси приводит к подавлению реакций дегидро
I .Шпица 6.1. IbiUHiiiK- природы носи гели на акипикн и. и селем инпоегь ка гализатора
оксихлориронапия >iana
Ката- лиза- тор Удельная повер- хность носителя, м2/г Хс н , С2Н6 % Селективное ть реакции, %, по Примечание
С2Н3С1 сумме хлор- углеводородов С2Н4 + С2Н2 СО + со2
пре- дель- ных непре- дель- ных
Температура процесса 300 °C
1 115 29 — 98 - - 2,0 т = 5 с,
2 220 25 - 100 - - - объемное соотношение
3 180 40 - 100 - - - С2Н6:НС1:О2 = 1:1:1
Температура процесса 450 °C
1 115 64 16,4 12,7 4,9 38 28
2 220 52 15,3 8,8 5,9 40 30
3 180 77 15,1 15,1 5,9 47 16,9
4 80 79,6 26 28,4 11,2 25 9,4 То же
5 26 79 20 31,7 5,8 30,3 12,2
6 3,5 80 19,9 38,2 6,0 25 10,9
Температура процесса 550 °C
1 115 85 6,5 0,8 0,7 70 22
2 220 91 6,7 1,1 1,2 72 19 т = 3 с,
3 180 96 20,6 4,2 7,2 54 14 объемное
7 6 97 35 3 9,0 46 7 соотношение С2Н6:НС1:О2 =
8 4 98 34 3,8 9,2 46 7 1:2:1
9 0,9 95 25,8 2,4 5,8 55 11
хлорирования и глубокого окисления, при этом выход хлорорганических про-
дуктов возрастает. Повышение концентрации кислорода в смеси в основном
благоприятствует реакции глубокого окисления (табл. 6.2).
Варьирование времени контакта в интервале изменения нагрузки реаген-
тов 50-1600 л на 1 л катализатора в час (здесь и далее объем газа приведен
к нормальным условиям) также существенно меняет характер селективности
оксихлорирования этана, хотя глубина превращения С2Н6 при этом меняется
аблица 6.2. Влияние с ос iana их ходпоп х м< in па хслек иноки 11> реакции окхихлориро
панн» папа
Состав исходной смеси , об. % Cc/icixi iihiiox 11. реакции. %>
С2н„ IIC1 О2 n2 сумме хлор угленодородон C.II, со+со2 Примечание
14,7 14,7 14,7 55,9 63,9 16,4 19,7
12,8 25,6 12,8 48,8 83,0 10,7 6,3
14,7 14,7 14,7 55,9 21,2 65,0 13,8 Катализатор 3.
12,8 25,6 12,8 48,8 31,5 56,8 11,7 (см. табл. 6.1),
11,4 34,2 11,4 43,0 33,3 56,6 10,7 т= 1,4 с, 150'4
14,7 14,7 14,7 55,9 36,1 47,0 16,9
11,5 11,5 16,1 60,8 33,3 39,4 27,3
незначительно (рис. 6.7). Во всей исследованной области при умеренных гем
нературах увеличение времени контакта приводит к возрастанию выхода но
лихлорпроизводных. При повышении температуры до 450 °C с увеличением
времени контакта селективность процесса меняется в сторону образования
.п илена и хлоролефинов. Однако при дальнейшем ее повышении до 550 °(.‘ та
висимость селективности от времени контакта приобретает более сложный
характер: при постоянной степени конверсии этана, близкой с 95-97%, наблю
дается увеличение выхода непредельных углеводородов (наряду с этиленом об
разуется и ацетилен) и оксидов углерода и уменьшение выхода винилхлорида.
Это свидетельствует о вторичных превращениях продуктов оксихлорирова
ния этана при большом времени контакта и малых концентрациях кислорода
Оценка факторов масштабирования на показатели процесса оксихлорирова
ния этана приведена в работе [720]. Эскиз реактора диаметром 200 мм привс
ден на рис. 6.8.
Известно, что с увеличением диаметра аппарата существенно меняется и
структура псевдоожиженного слоя вследствие изменения интенсивности об
ратного перемешивания в слое, газораспределения и т.д. На рис. 6.9. приведены
зависимости степени конверсии этана и селективности образования основных
продуктов реакции (этилена, суммы хлорорганических продуктов и оксидов yi
лерода) от времени контакта для реакторов диаметром 42 и 200 мм. С увеличе-
нием диаметра аппарата для достижения высокой степени конверсии этапа грс
буется значительно увеличить время контакта. Так, в опытном реакторе макх и
мальная степень конверсии этана, равная 90%, достигается лишь при времени
контакта 22 с, тогда как в лабораторном реакторе степень конверсии 95-97%
достигается через 0,7-1,5 с. Характер зависимости селективности процесса or
времени контакта в изученных условиях при переходе к аппарату ббльшего диа
метра остается практически без изменений.
Авторами работ [726, 727] предложено использовать в качестве катализат
ров цементсодержащие системы (алюминаты кальция) с введением в структуру
кристаллической решетки хлоридов меди и цезия. Установлено, что катализа
торы такого типа особенно эффективны в реакциях дегидрохлорирования хло
ралканов, благодаря чему суммарный выход винилхлорида и этилена достигает
80% уже при 400 °C. Выход продуктов глубокого окисления не превышает 3 1%.
I В|
Рис. 6.7. Зависимость показателей процесса оксихлорирования этана от времени кон-
такта (катализатор 3, С2Н6:НС1:О2 = 1:2:1) при разных температурах (а-вУ.
1 — степень конверсии этана; 2-7 — селективность образования С2Н5С1 (2), C2H4CI2 (3), С2Н4 (4),
С2Н,С1_, (5), СО + СО2(6), С2Н2 (7)
Рис. 6.8. Реактор оксихлорирования этана:
1 — катализатор; 2 — гильза для термопары; 3 — футеровка из бетона; 4 — рубашка; 5 — электро
обогрев; 6 — фильтры; 7 — крышка реактора; 8 — насадка; 9 — газораспределитель
Активность катализаторов (конверсия этилена 88 92%, хлороводорода 32-36%,
кислорода 88 91%) остается постоянной на протяжении 1500 ч работы.
Аналогичные результаты достигнуты в работе [595], где добавка хлорида цс
зия дает схожий эффект при использовании меднокалиевых катализаторов па
основе силикагеля в псевдоожиженном слое контакта.
Показано, что наиболее эффективными являются катализаторы с низкой
удельной поверхностью, не превышающей 15 м2/г.
Рис. 6.9. Зависимость показателей процесса оксихлорирования этана от времени кон-
такта в реакторе диаметром 42 мм (сплошные кривые) и 200 мм (штриховые) (при
555 °C, С2Н4:НС1:О2 = 1:2:1):
1 — степень конверсии этана; 2-4 — селективность образования соответственно С2Н4, суммы
хлорорганических продуктов, СО + СО2
Рис. 6.10. Зависимость конверсии этана (д), суммарного выхода винилхлорида и эти-
лена (б), продуктов глубокого окисления (в) от температуры. С2Н6:НС1:О2 = 1:2:1 (мол).
Катализатор CsCl-CuCl2-KCl/SiO2, 5,5% Си-
1 — 1% CsCl; 2 — 5,4% CsCl; 3 — 10,5% CsCl; Си:К = 1
Необходимо отметить, что добавление 1% CsCl к стандартной системе Си-
С12-КС1 практически не изменило показателей процесса. Замена хлорида калия
на хлорид цезия приводит к агломерации частиц катализатора и поэтому не мо-
жет быть рекомендована.
На рис. 6.10 представлены зависимости показателей процесса от темпера-
туры в присутствии катализаторов с разным содержанием цезия, указывающие
на то, что увеличение содержания цезия в катализаторе практически не влияет
на конверсию этана; суммарный выход непредельных соединений — винилхло-
рида и этилена — увеличивается, снижается также выход продуктов СОХ.
11 iMeiieiiiie выхода непредельных соединении при увеличении содержания
цезия в катализаторе указывает на (о, что реакции окисления и дегидрохлори
рования протекают па различных активных центрах. При ном хлорид меди, в
том числе в виде расплава с хлоридом калия, катали тирует реакции окисления
хлористого водорода, а также реакции окисления папа, пилена и их хлорнро
и.тводных, в основном этилхлорида и дихлорэтана.
Наличие в системе цезия практически не ока тывает влияния па активность
катализатора — конверсия л апа нс изменяется — приводит к увеличению вы
хода винилхлорида и этилена и одновременно к снижению выхода продуктов
СОХ. С учетом данных, полученных A.I4. Гельбштейном [420], что реакции де
। идрирования этана и этилхлорида с образованием, соответственно, этилена и
винилхлорида не имеют места в условиях процесса, можно полагать, что хлорид
цезия увеличивает скорость реакции дегидрохлорирования с одновременным
юрможением скорости реакций окисления как предельных, так и непредель
ных соединений.
Установлено [595], что в присутствии Cu-K-Cs-катализатора процесс ок
сихлорирования этана можно проводить при относительно невысоких темпе
ратурах (430-470 °C) без снижения выхода целевых непредельных соединении
При этом следует ожидать относительного увеличения выхода винилхлорида
в сочетании с соответствующим снижением выхода этилена. Этот аспект я в
ляется достаточно важным при разработке технологической схемы процесса и
балансировании ее по хлору.
В присутствии катализатора 10,5%CsCl-ll,5%CuC12-6,5%KCl/SiO2 было ис
следовано влияние времени контакта и соотношения исходных реагентов па
показатели процесса. В табл. 6.3 представлены данные по конверсии исходных
реагентов и выходам основных продуктов процесса в зависимости от времени
контакта.
Согласно данным табл. 6.3, увеличение времени контакта способствует уве
личению конверсии этана и выхода этилена. Суммарный выход хлорорга ни чес
ких продуктов снижается, однако, содержание винилхлорида в них проходи i
через максимум. Наблюдается также рост выхода продуктов глубокого окисле
ния СОХ.
Таблица 6.3. Влияние времени контакта на основные показатели процесса оксихлори
рования этана
№ Время контакта, с Конверсия реагентов, % Выход в расчете на превращенный этан, %
1,1- C2H4C12 1,2- C2H4C12 С2Н3С1 С2Н4 сох
с2н6 НС1 О2
1 1,5 83,2 38,2 93,4 8,4 16,5 33,8 35,4 2,5
2 3,6 89,6 35,4 91,7 6,0 14,3 36,5 36,8 2,9
3 5,8 91,2 35,0 90,9 4,4 12,1 41,6 37,5 3,0
4 7,9 94,5 33,8 89,2 3,1 10,0 40,5 38,0 4,7
5 10,0 96,1 32,9 88,7 2,0 6,7 38,4 39,2 6,5
Катализатор CsCl-CuC^-KCl/SiC^. t = 450 °C. Соотношение С2Н6:НС1:О2= 1:2:1 (мол)
Рис. 6.11. Зависимость степени конверсии винилхлорида (1), выхода оксидов углерода
(2) и суммы ацетилена и этилена (3) от температуры (я) и времени контакта (6) при вза-
имодействии винилхлорида с водяным паром (СиС12 в катализаторе — 10,5%, С2Н3С1 в
исходной смеси — 8 об. %, Н2О — 30 об. %)
Важным параметром, существенно влияющим на показатели процесса, яв-
ляется соотношение исходных реагентов. Установлено [595], что избыток хло
роводорода благоприятно влияет на селективность процесса в части умень-
шения выхода продуктов глубокого окисления. Позитивным является также
уменьшение зауглероживания катализатора. Увеличение избытка НС1 по отно-
шению к этану до 3:1 ведет к снижению выхода непредельных углеводородов за
счет торможения хлороводородом реакций дегидрохлорирования дихлорэтана
и этилхлорида. Увеличение избытка кислорода способствует повышению кон-
версии этана, но главным образом за счет увеличения его окисления — выход
оксидов углерода увеличивается в 1,8-2 раза. Поэтому для обеспечения мак-
симального выхода целевых продуктов оптимальным является соотношение
С2Н6:НС1:О2 = 1:2:1 (мол).
К аналогичным выводам пришли авторы работы [728] при использовании
меднокалиевого катализатора на основе А12О3.
Вторичные превращения продуктов процесса оксихлорирования этана изу-
чены в работе [719]. На рис. 6.11 представлены зависимости степени конверсии
винилхлорида и выхода продуктов его взаимодействия с водяным паром от тем-
пературы и времени контакта. Анализ полученных данных показывает, что уже
при 400 °C начинается, хотя и в незначительной степени, образование этилена,
ацетилена и оксидов углерода. При повышении температуры до 550 °C степень
конверсии винилхлорида возрастает до 34%, а суммарный выход ацетилена и
этилена и оксидов углерода достигает соответственно 17,7 и 13,7% (рис. 6.11а).
Увеличение времени контакта также способствует превращению винилхлорида
(рис. 6.116). При 550 °C и времени контакта 7,5 с степень конверсии винилхло-
рида составляет 55%, суммарный выход ацетилена и этилена и оксидов углерода
соответственно 28,6 и 22%.
Таким образом, приведенные данные подтверждают возможность взаимо-
действия винилхлорида с водяным паром на солевом медьсодержащем ката-
лизаторе, что до некоторой степени объясняет изменение характера селектив-
пос i n реакции оке ихлорнровапня мана при увеличении продолжи)СЛЫ)ос гн
кои гак ।а выше 0,7 с.
Образование .пилена, но видимому, происходи г в результате воссгановле
пия винилхлорида при его вшимодейс мши с водяным паром.
Катализатор реакции восстановления водяным паром — CuCl образуется
при восстановлении хлорида меди в процессе хлорирования соответствующего
углеводорода.
Следовательно, для осуществления предполагаемой реакции воссгаповле
ния винилхлорида в условиях оксихлорирования этана также необходим CuCl
Как упоминалось, в качестве катализатора оксихлорирования этана иснользу
егся эквимолярная смесь хлоридов меди и калия, в которой медь находи ня в
окисленной форме. В соответствии с общепринятым механизмом реакции ок
сихлорирования алканов [420], первоначально происходит диссоциация хлори
да меди (II) с образованием элементарного хлора и CuCl, который далее с но
мощью кислорода вновь регенерируется до исходного хлорида. В ходе реакции
оксихлорирования этана при 550 °C и времени контакта 0,7 с степень нревра
щения кислорода, как правило, составляет 95-97%, соответственно концеш ра
ция непрореагировавшего кислорода в реакционном газе не более 0,5-0,8 об %.
Следовательно, в этом случае можно предположить вероятность сущее шона
ния на носителе восстановленной формы меди.
Согласно рис. 6.12, селективность взаимодействия винилхлорида с водяным
паром в значительной мере зависит от концентрации кислорода: с увеличен и
ем содержания последнего степень восстановления С2Н3С1 до этилена резко
снижается, при значении 2,6% процесс практически не идет, тогда как при кон
центрации кислорода менее 0,5 об. % селективность по этилену достигает 53%.
Таким образом, правильность предположения, что реакция восстановления
винилхлорида протекает лишь в присутствии восстановленной формы меди,
подтверждается.
Изменение концентрации водяных паров и хлороводорода в смеси с винил
хлоридом практически не влияет на селективность реакции восстановления.
В составе реакционного продукта оксихлорирования этана, а также про
дукта взаимодействия винилхлорида с водяным паром наряду с этиленом
присутствует ацетилен, образование которого следует отнести к вторичным
превращениям винилхлорида и, в первую очередь, за счет его дегидрохлори
рования. В соответствии с термодинамическими расчетами, в области 550
равновесный выход ацетилена по реакции дегидрохлорирования винилхло
рида составляет 58%. В присутствии хлормеднокалиевого катализатора при
550 °C и времени контакта 3 с дегидрохлорирование винилхлорида иде г с до
статочной скоростью, выход ацетилена составляет 18%. На рис. 6.13 при веде
ны зависимости селективности образования этилена и ацетилена в условиях
оксихлорирования этана при 550 °C и разном времени контакта. С его уве
личением выход этилена возрастает, а ацетилена снижается. Следовательно,
ацетилен в условиях оксихлорирования этана, в свою очередь, подвергается
дальнейшим превращениям. По-видимому, он взаимодействует с водяным
паром [370]:
С2Н2 + 2Н2О —- 2СО + ЗН2. (6.5)
Рис. 6.12. Зависимость степени конверсии винилхлорида (1) и селективности образо-
вания этилена (2), ацетилена (3) и оксидов углерода (4) от концентрации кислорода в
смеси при взаимодействии винилхлорида с водяным паром (t = 550 °C, время контакта
4 с; СиС12 в катализаторе 10% С2Н3С1 в смеси 8 об. %, Н2О об. 35%)
Рис. 6.13. Зависимость селективности образования этилена (1), ацетилена (2), окси-
дов углерода (3) и винилхлорида (4) от времени контакта при оксихлорировании этана
(t = 550 °C, время контакта 4 с; содержание CuCl в катализаторе 8,5%, мольное соотно-
шение С2Н6: НО =1:2
Правильность этого предположения подтверждает и одновременное увели-
чение выхода оксида углерода (см. рис. 6.13, кривая 3).
Анализируя результаты проведенных экспериментов и имеющихся литера-
турных данных, можно предположить следующую схему вторичных превраще-
ний продуктов реакции оксихлорирования этана при 550 °C и т > 0,7 с, т.е. в
области высоких степеней превращения исходных реагентов (более 95-97%):
С2Н3С1 ------
с +Н,О
х --------------- С2Н4 + СО + НС1
С2Н2 --------
(6.6)
11|>иьсдсн11.1Я схема вторичных превращении ноет упрощенный xapauiep,
поскольку в реальных условиях указанным превращениям могут подвергаться
все хлорсодержащие углеводороды; при ном выход хлорпрои.людных с уве
личепием времени контакта снижается, а пилена увеличивается. Такая схема
объясняет изменение селективности процесса оксихлориронапия этапа при
высоких температурах и большом времени контакта, что позволяет выбрать
условия, обеспечивающие максимальную степень превращения реагентов, по
ограничивающие последовательные вторичные превращения винилхлорида и
других продуктов реакции.
Процесс оксихлорирования этана может проводиться в газовой фазе, в нс
подвижном или псевдоожиженном слое катализатора, или в их комбинации;
основная проблема — отвод тепла реакции в условиях сильной коррозионное ги
среды за счет высоких температур.
Этот вопрос решается либо использованием теплосъемных элемен тов из
материалов, стойких в условиях реакции, либо созданием условий адиабаты
веского проведения процесса. В последнем случае используют прием р тзоавле
ния реакционной смеси исходными компонентами или продуктами реакции
Для этого осуществляют рецикл непрореагировавшего этана и образующихся
хлорпроизводных после выделения винилхлорида в количествах, обеспечив.!
ющих снятие выделяющегося тепла за счет разогрева рецикла до температуры
реакции. В качестве разбавителя исходных реагентов могут использоваться и
водяные пары.
При высокотемпературном оксихлорировании этана образуются зпачи
тельные количества этилена, т.е. наряду с оксихлорированием идет и реакция
оксидегидрирования этана. Выход этилена в значительной степени при прочих
равных условиях определяется составом катализатора. Фосфаты и силикаты
железа, а также хлориды марганца, кобальта и лития увеличивают выход эгиде
на, а хлориды меди увеличивают выход винилхлорида.
Образовавшийся этилен может быть превращен в винилхлорид через ди
хлорэтан по обычной схеме. Практического осуществления процесс газофазпо
го окислительного хлорирования этана пока не получил, несмотря на опреде
ленный практический интерес.
Хлорирование этана с помощью переносчиков хлора
Хлорирование углеводородов с помощью переносчиков хлора, в основном
хлорида меди — хорошо известный процесс, в котором используется хлор, об
разующийся при диссоциации хлорида меди. Схема возможных превращении
этана в этом случае включает совокупность последовательно-параллельных ре
акций хлорирования и дегидрохлорирования, и аналогична превращениям на
на при его окислительном хлорировании на хлормеднокалиевом катализа торе,
который также протекает через диссоциацию хлорида меди.
Отличие от процесса окислительного хлорирования заключается в отсу гс
твии побочных реакций глубокого окисления.
Поскольку в процессе хлорирования СнС12 превращается за счет отщенле
ния хлора в CuCl, необходимым элементом этого процесса является регепера
ция переносчика хлора.
Хлорирование с помощью хлорида меди мо кег нестись как на твердом ка
гадизагоре, так и в расплаве солей. В свою очередь, процесс с твердым ката
лизагором можно осуществлять в псевдоожиженном или в неподвижном слое.
Твердый катализатор — смесь хлоридов меди и калия, нанесенных на пористый
носитель — пемзу, диатомовую землю и др. с удельной поверхностью до 10 м2/г.
Реакция может проводиться в секционированном аппарате. Этан подается в
зону, где находится контакт с хлоридом меди. За счет диссоциации хлорида меди
в этой зоне при 350-415 °C происходит хлорирование этана. Отработанный
контакт эрлифтом подается в зону регенерации, где вначале обрабатывается
при 210-250 °C воздухом для получения оксихлорида меди, а в следующей сек-
ции при 320-360 °C оксихлорид реагирует с хлороводородом, идущем из зоны
хлорирования, и/или хлором, поступающим со стороны, с получением хлорида
меди. Конверсия этана за проход — на уровне 50%, в продуктах реакции в ос-
новном полихлориды (тетрахлорэтан, перхлорэтилен и дихлорэтилен) и только
около 3 мол. % винилхлорида и 23 мол. % дихлорэтана.
Хлорирование этана можно проводить и в расплаве солей, содержащих пере-
носчик хлора, — смеси хлоридов алюминия, натрия и меди или хлоридов цинка,
Ееди и калия, или одно и двухвалентных хлоридов меди и калия. Хлориды ще-
очных металлов используются главным образом для снижения температуры
лавления расплава. В качестве активных добавок к расплаву используются и
ругие хлориды, в частности, хлориды железа, марганца, хрома, никеля, палла-
дия, кадмия, свинца и РЗЭ. Однако наиболее интересна в практическом отно-
шении система, состоящая из хлоридов одно- и двухвалентной меди и хлорида
алия. Эта тройная система впервые была описана в 1950-х годах и применена
для хлорирования в расплавленом состоянии углеводородов [429]. Расплав со-
держит 20-40% хлорида калия, а отношение дихлорида меди к монохлориду от
1:4 до 4:3.
Такая же тройная смесь была использована фирмой «Lummus Со» для раз-
эаботки процесса «Transcat» [13], широко разрекламированного в 1970-х годах,
зо так и не реализованного в промышленных условиях.
Процесс получения винилхлорида хлорированием этана в расплаве солей,
как и окислительное хлорирование, основан на совокупности последовательно-
параллельных реакций хлорирования и дегидрохлорирования при прохожде-
нии газовых пузырей этана и его хлорпроизводных через расплав солей СиС12-
Zu2CI2-KCl. Схема возможных превращений этана и его хлорпроизводных была
эписана ранее. С определенной степенью достоверности можно утверждать, что
эеакция происходит как в объеме газовых пузырей, так и на границе раздела
раз газ/расплав.
Источником хлора является реакция диссоциации СиС12, который затем ре-
генерируется с помощью кислорода и хлороводорода или хлора. При регенера-
ции с помощью кислорода и хлороводорода определяющей стадией является
реакция окисления. При этом кислород реагирует с CuCl на границе раздела
£аз, поскольку его растворимость в расплаве крайне мала. Образовавшийся ок-
сихлорид меди затем вступает в реакцию с хлороводородом и превращается в
слорид меди (II). Такой механизм объясняет причину повышенной концентра-
ции дихлорида меди на контактной поверхности расплава по сравнению с его
концентрацией в объеме, т.е. степень окисления монохлорида меди смесью хло-
ронодорода к кислорода iie.iaiiiKiii 01 1 <и ына р.п плана u глубине, а окисление
является реакцией нулевою норядк i
Суммируя, можно нредсгапп11. н<нледова! ел ы юс11. химических реакции
для процесса хлорирования папа расплавом медных солей (ограничиваясь пре
вращениями до винилхлорида):
4CuC12 —- 2Cu2CI2 + 2С12 - 155,64 кДж; (6.7а)
С2Н6 + С12 —- С2Н5С1 + HCI + 120,08 кДж; (6.76)
С2Н5С1 —- С2Н4 + НС1 - 72,38 кДж; (6.7н)
С2Н4 + С12 —*- С2Н4С12 + 194,14 кДж; (6.7г)
С2Н4С12— С2Н3С1 + НС1 - 70,71 кДж; (б.7д)
1,5Си2С12 + 0,75О2 —► 1,5CuO-CuC12 + 106,69 кДж; (6.7е)
1,5CuOCuC12 + ЗНС1 3CuCl2 + 1,5Н2О + 95,81 кДж; (6.7ж)
0,5Си2С12 + 0,5С12 —► СиС12 + 38,91 кДж (6.7з)
или суммарно:
С2Н6 + 0,5С12 + 0,75О2 —* С2Н3С1 + 1,5Н2О + 256,9 кДж. (6.7)
Использование для хлорирования этана расплава медных солей определи
ет основные аспекты этой технологии. Прежде всего необходимо придать рас
плаву достаточную подвижность, что достигается снижением его вязкое ги ia
счет повышения температуры. Высокая подвижность расплава необходима, с
одной стороны, для облегчения контакта реакционных газов и хлорировании и
регенерации расплава, а с другой — для облегчения его транспортировки между
реакционными зонами.
В то же время повышение температуры увеличивает скорость диссоциации
дихлорида меди, что полезно при хлорировании и мешает при регенерации.
Следовательно, необходимо проводить хлорирование и регенерацию в разде
ленных зонах, обеспечивая в зоне регенерации минимально возможную гемне
ратуру с учетом скоростей реакции окисления и нейтрализации, а также диссо
циации дихлорида меди. Разделение зон необходимо для исключения кош акта
углеводородных компонентов с кислородом.
Крайне высокая коррозионная активность расплава по отношению к метал
лам требует применения керамических или других стойких материалов. Приме
нение керамики, однако, усложняет проблему теплосъема сильно экзотермич
ного процесса в целом.
Даже этот далеко не полный перечень весьма противоречивых технологи
ческих условий, необходимых для осуществления процесса хлорирования на
на расплавом медных солей, показывает большие трудности его практической
реализации.
Процесс «Transcat» предназначался для получения винилхлорида, трихлор >
тилена и перхлорэтилена из этана, а также мог быть использован для получения
хлорметанов из метана, т.е. был рассчитан для проведения реакций, где есп»
Рис. 6.14. Принципиальная схема процесса «Transcat» (1-5 и 8-11 — см. текст; 6,7,13 —
холодильники; 12 — насос)
необходимость в переработке значительных количеств хлороводорода. Послед-
нее делало этот процесс конкурентом процессу окислительного хлорирования,
обладающим при этом тем достоинством, что благодаря исключению контакта
окислителя и углеводорода отсутствовала реакция горения и, следовательно,
повышалось полезное использование углеводородного сырья.
В этом процессе основные химические реакции хлорирования и дегидро-
хлорирования углеводородных компонентов и регенерации расплава были сба-
лансированы единой реакционной системой, объединяющим компонентом ко-
торой является циркулирующий поток расплавленных солей меди и калия.
Процесс был отработан на опытной установке; выход винилхлорида по эта-
ну составлял около 80%, по хлору — почти 100% [ 17, 18].
Принципиальная схема процесса «Transcat» представлена на рис. 6.14.
Реакционный агрегат 1, разделенный, по крайней мере, на три реакционные
зоны, заполнен расплавленной смесью, содержащей 60-80% хлоридов одно- и
двухвалентной меди и 20-40% хлорида калия.
Этан поступает в зону хлорирования 1/2, куда из зоны регенерации 1/1 пос
гупает регенерированный расплав. В эту же зону поступают этилен и непро-
эеагировавший этан со стадии разделения продуктов хлорирования. В зоне
хлорирования 1/2 при 370-590 °C происходит хлорирование углеводородов с
эбразованием смеси, содержащей винилхлорид, дихлорэтилены, три- и пер-
клорэтилены, этилхлорид, дихлорэтан, а также полихлорэтаны и этилен. Кон-
версия этана за проход находится на уровне 60%. Состав продуктов аналогичен
приведенному ранее для процесса окислительного хлорирования этана. Реак-
ционные газы из зоны хлорирования поступают в колонну 2.
Отработанный расплав поступает в зону дегидрохлорирования 1/3, куда
:о стадии разделения поступает фракция хлорэтанов, содержащая в основном
щхлорэтан и этилхлорид. За счет тепла, аккумулированного расплавом в зоне
итерирования, происходит дегидрохлорирование хлорэтанов с получением со-
ответствующих непредельных соединений. Одновременно в некоторой степени
происходи) и реакции хлорирования iu <’ц t днхлорида меди. Реакционные г.) на
из ной зоны также поступаю) и реюнфикацпоппую колонну 2. Из зоны де) и
дрохлориронания 1/3 расплав ia сче) >рлнф>а, например, кислородом и (азами
сжигания хлорорганических отходов, пос lynaci и юну регенерации 1/1. Винчи
ле монохлорид меди за сче) кислорода нревращаена к оксихлорид, а татем об
разевавшийся оксихлорид нейтрализуется хлороводородом, поступающим со
стадии разделения и хлором. Температура расплава в зоне регенерации состав
ляет 315 480 °C. Остаточные газы после зоны регенерации 1/1 направляются на
санитарную очистку, а регенерированный расплав снова в зону хлорирования
1/2. Тепловой баланс в реакционном агрегате обеспечивается сочетанием реак
ций хлорирования и дегидрохлорирования и соответствующим рециклом ре
акционных продуктов. Реакционные продукты, поступившие из реакционного
агрегата 1 в закалочную колонну 2, охлаждаются за счет охлаждаемого в холо
дильнике 4 конденсата, рециркулируемого насосом 3. Охлажденные продую ы
поступают в ректификационную колонну 5, где выделяется в виде дистилла i а
хлороводород, направляемый в зону регенерации. Несконденсировавшиетя
газы, содержащие, в основном, этилен и непрореагировавший этан, направля
ются в зону хлорирования 1/2 реакционного агрегата.
Кубовая жидкость колонны 5 поступает в колонну 8, где выделяется винил
хлорид, который при необходимости подвергается ректификационной очистке
по общепринятой схеме.
Реакционные продукты после выделения винилхлорида поступают в ко
лонну 9, где выделяются легкокипящие компоненты, а также перхлорэтилен.
Фракция легкокипящих компонентов, состоящая в основном из этилхлорида
и дихлорэтана, направляется в зону дегидрохлорирования 1/3. Перхлорэтилен
выводится из колонны 9 в виде целевого продукта, а кубовые остатки направ
ляются на сжигание. После сжигания хлорорганических остатков в печи 10 и
закалки их в колонне 11 реакционные газы, содержащие хлороводород, направ
ляются в реакционный агрегат, где используются для регенерации расплава.
Основной особенностью этого процесса является наличие оригинальною
агрегата, содержащего расплав. Эскиз реактора представлен на рис. 6.15 [729|.
Сложность его технологического исполнения, связанная с крайне высокой
коррозионностью среды и высокой температурой, явилась, по-видимому, ос
новной причиной, по которой процесс не нашел практического применения.
Интерес к промышленной реализации процесса получения винилхлорида
из этана вновь возобновился в 1990-х годах. Одной из причин этого явился рос i
цен на нефтяное сырье.
В 1995 г. компания «EVC» запатентовала ряд технических решений, реализо
ванных в опытно-промышленном масштабе в 1998 г. [721-724]. Процесс оспо
ван на сочетании реакций:
С2Н6 + НС1 + О2 —С2Н3С1 + 2Н2О; (6.8а)
С2Н6 + С12 + 0,5 О2 —- С2Н3С1 + НС1 + Н2О. (6.86)
Суммарно:
2С2Н6 + С12 + 1,5 О2 —2С2Н3С1 + ЗН2О. (6.8в)
Рис. 6.15. Эскиз реактора
фирмы «Lummus Со»
Однако количество реально идущих реакций существенно превышает чис-
ло вышеприведенных. По этой причине для процесса требуются три реактора,
каждый из которых несет определенные функции. Наиболее важным узлом яв-
ляется реактор оксихлорирования, в котором этан реагирует с хлороводородом,
хлоруглеводородами или хлором с образованием винилхлорида и побочных
продуктов. Утверждается, что для повышения селективности необходимо ми-
нимизировать образование оксихлоридной формы катализатора, представляю-
щего смесь хлоридов меди, калия и лантана на носителе. Этого достигают повы
шением температуры, а для уменьшения горения и коррозии стенок реактора
используют избыток хлороводорода.
Рис. 6.16. Принципиальная технологическая схема процесса фирмы «EVC» (1-11 ал. гекс i)
Принципиальная технологическая схема процесса представлена на рис. 6.16.
Этан, подаваемый в реактор оксихлорирования 1, предварительно смеши нас гея
с рецикловым хлористым водородом и далее направляется в псевдоожиженный
слой катализатора, где смешивается с кислородом (или с воздухом, обогащен
ным кислородом) и со сторонними насыщенными хлоруглеводородами.
Реакционный i.i i чаегичпо конденсируется и направляется и фа юра щели
гель 2, где разделяется па три потока: влажный паровой поток, влажный поток
жидких хлоруглеводородов, водный поток, содержащий хлористый водород.
Водный поток смешивается с раствором хлорида кальция и направляется
на стриппинг 3. Сухой хлористый водород, выходящий сверху колонны стрип
пинга 3, возвращается в реактор оксихлорирования 1, чистая вода используется
для получения пара, а хлорид кальция смешивается с водным потоком из фазо-
разделителя.
Влажный поток жидких хлоруглеводородов из фазоразделителя 2 направ-
ляется на колонну обезвоживания 5. Кубовый продукт колонны 5 поступает в
колонну отгонки легкокипящих 7, а головной продукт возвращается в фазораз
делитель 2.
Влажный паровой поток из фазоразделителя 2 направляется на колонну
осушки 4, где он в противоточном режиме контактирует с холодной (-20 °C)
соляной кислотой. Осушенный паровой поток в компрессоре 6 компримирует-
ся и направляется в колонну легкокипящих 7. Небольшое количество соляной
кислоты из куба колонны 4 направляется в колонну осушки НС1 3.
Осушенные жидкий и газовый потоки смешиваются, предварительно нагре-
ваются путем контакта с головным продуктом колонны легкокипящих 7 и пос-
тупают в эту колонну. Кубовый продукт этой колонны подается в продуктовую
колонну винилхлорида 10, а кубовый продукт, содержащий ненасыщенные со-
единения, гидрируется в струйном реакторе 11 и направляется затем в реактор
оксихлорирования 1.
Головной продукт колонны легкокипящих 7, содержащий этилен, направля-
ется в реактор прямого хлорирования 8. Продукты хлорирования возвращают-
ся в реактор оксихлорирования 1. Тепло реакции хлорирования утилизируется.
Небольшие количества реакционной смеси, содержащие, например, тетрахло-
рид углерода, подаются на узел угольной адсорбции 9 для выделения дихлор-
этана. Абгазы направляются на сжигание. Хлороводород со стадии сжигания
также может быть возвращен в процесс.
Расходные коэффициенты на 1 т винилхлорида:
Этан, кг.............................520
Хлор, кг...........................665,4
Кислород, кг ........................469
Водород, кг..........................4,6
Электроэнергия, кВт-ч............200
Пар (1,4 МПа), кг................150
Расходный коэффициент по этану, равный 520 кг/т винилхлорида, пред-
ставляется нам излишне оптимистичным Это означает, что лишь около 8% эта-
на превращается в побочные продукты, включая оксиды углерода, что близко
к показателям этиленового процесса. Для температур, превышающих 400 °C,
реальность таких показателей вызывает определенные сомнения.
На основании лабораторных исследований фирма «EVC» пустила в эксплуа-
тацию пилотную установку по производству винилхлорида мощностью 1000 т/
год из этана [725].
(KI i.и
Рис. 6.17. Блок-схема получения винилхлорида из этана
Руководство компании заявило, что катализатор на пилотной установке
испытывался более 18 мес. В конечном счете, конверсия этана составила 90%,
однако ее можно увеличить до 92-95%. В рекламируемых планах фирмы одну
чивалось создание промышленного производства винилхлорида из этана мощ
ностью 300-500 тыс. т в год, однако они так и не были реализованы.
На рис. 6.17 представлена блок-схема процесса получения винилхлорида из
этана, разработанного в российском Институте хлорной промышленное ти при
непосредственном участии авторов настоящей книги [595].
Оксихлорирование этана осуществляется в псевдоожиженном слое катали
затора при 450-480 °C и давлении 0,2 МПа. Катализатор — силикагель, upon и
тайный хлоридами меди, калия и цезия. Содержание меди 4-6 мае. %. Реактор
оксихлорирования — вертикальный цилиндрический аппарат с конической
нижней частью, включающий узел распределения исходных газов, узел филь
трации реакционных газов и узел ввода охлаждающего агента — 35%-ной соля
ной кислоты.
Исходные компоненты — газообразный хлороводород со стадий пиролиза
дихлорэтана, гидродехлорирования отходов и стриппинга, а также этан иода
ются под распределительную решетку. Кислород воздуха вместе с концентриро
ванной соляной кислотой подается непосредственно в псевдоожиженный слой
с помощью форсунки. Теплосъем в реакторе осуществляется за счет испарения
35%-ной соляной кислоты, поступающей в виде рецикла в аппарат.
Реакционный га i выводи гея из верхней час ги реак гора через сие гему фи л в
гров и, пройдя конденсаторы, поступает па абсорбцию. ()бразующаяся соляная
хислога возвращается в реактор.
Газ из абсорбера направляется на стадию жидкофазного хлорирования эги-
нена, а смесь хлорорганических продуктов с. абсорбентом проходит через сис-
тему колонн десорбции, где происходит разделение этой смеси. Абсорбент воз-
вращается на орошение колонны абсорбции. Легкая фракция — винилхлорид,
)тилхлорид и дихлорэтилены — поступает на ректификационную колонну для
наделения винилхлорида. Тяжелая фракция — смесь дихлорэтанов, трихлор-
тгана, три- и перхлорэтилена направляется на стадию гидродехлорирования
pi я получения этана (этилена) и хлороводорода.
Хлорирование этиленсодержащего газа со стадии оксихлорирования этиле-
на осуществляется в среде кипящего дихлорэтана при 95-105 °C в присутствии
(анализатора FeCl3. Хлоратор — аппарат колонного типа, в котором совмещены
реакционная и ректификационная части. Образующийся дихлорэтан поступает
за систему ректификации и далее — на стадию пиролиза.
Пиролиз дихлорэтана идет при 480-520 °C в печах специальной конструк-
дии, давление в системе 2,0-2,5 МПа. Степень превращения дихлорэтана —
50%. Продукты пиролиза подвергаются закалке с последующим выделением
оюроводорода и рециклового дихлорэтана. Выделенный винилхлорид-сырец
:оединяется с винилхлоридом-сырцом со стадии оксихлорирования этана и на-
травляется на ректификацию для выделения товарного продукта.
Высоко- и легкокипящие хлорорганические отходы направляются на пере-
работку методом каталитического гидродехлорирования. Процесс протекает в
неподвижном слое катализатора при 320-380 °C. Реакционная смесь поступает
далее на систему конденсации, после которой газовая фаза — этан и хлорово-
дород — возвращается в реактор оксихлорирования, а непрореагировавшие
<лорорганические продукты направляются на стадию сжигания. Образующая-
ся соляная кислота направляется на установку стриппинга, где выделяется хло-
роводород, который возвращается на стадию оксихлорирования этана.
Расходные коэффициенты, кг/т ВХ:
Этан. 676
Кислород...........647
Клор...............630
Научное издание
Флид Марк Рафаилович
Трегер Юрий Анисимович
ВИНИЛХЛОРИД: ХИМИЯ И ТЕХНОЛОГИЯ
В 2-х книгах
Книга 1
Редактор А.В. Савенков
Корректор М.Б. Почаева
Дизайн и верстка Д. Б. Гавриленко
Подготовка иллюстраций М.В. Ворокова
Дизайн обложки Е.Р. Аронова
Подписано к печати 13.03.2008
Формат 70 х 100 'Ле, Гарнитура Миньон
Печать офсетная
Усл. печ. л. 36,5 Тираж 700 экз. Тип. зак.
Издательство «Калвис» тел/факс (495) 913-80-94
www.kalvis.ru; e-mail: info@kalvis.ru
Типография «Стратим» г. Рыбинск